/































































































































































































































































































































































































































































































































































































































































































































































































































































































































































































Text
ПРОИЗВОДСТВО
ЛЕКАРСТВЕННЫХ СРЕДСТВ
КОНТРОЛЬ КАЧЕСТВА
И РЕГУЛИРОВАНИЕ
аГАЛАХИМ
ГРУППА КОМПАНИЙ ПР ФЕССИЯ
PHARMACEUTICAL
MANUFACTURING
HANDBOOK
Regulations and
Quality
SHAYNE COX GAD, PH.D., D.A.B.T.
Gad Consulting Services
Cary, North Carolina
vWILEY-INTERSCIENCE
A JOHN WILEY & SONS, INC., PUBLICATION
Ш. К. Гэд (ред.)
ПРОИЗВОДСТВО
ЛЕКАРСТВЕННЫХ СРЕДСТВ.
Контроль качества
и регулирование
Практическое руководство
Перевод с английского языка
под редакцией Береговых В. В.
Санкт-Петербург
2013
ОБРАЗОВАТЕЛЬНЫХ
ПРОГРАММ
ПРОФЕССИЯ
ББК 35.66я2
УДК 661.12.01/09
П78 Ш. К. 1Ъд (ред.)
Производство лекарственных средств. Контроль качества и регулирование.
Практическое руководство : пер. с англ. / [Ш. К. Гэд и др.] ; под ред. В.В. Бе-
реговых. — СПб. : ЦОП «Профессия», 2013. — 960 с., ил.
ISBN 978-5-91884-046-7
ISBN 978-0-8031-7001-8 (англ.)
В практическом руководстве раскрываются все основные вопросы по контролю качества
и регулированию в производстве лекарственных средств — надлежащая производственная
практика (GMP), процессно-аналитические технологии (РАТ), валидация методик, стабиль-
ность лекарств и др. В отдельных разделах рассмотрены важнейшие вопросы загрязнения
лекарств и его контроль, обучения персонала, введения систем контроля качества и аудита.
Руководство предназначено сотрудникам производственных и аналитических лаборато-
рий фармацевтических производств, R&D подразделений фармкомпаний, испытательных
центров и надзорных органов, осуществляющих выпуск, контроль, испытания и обращение
лекарственных средств.
ББК 35.66я2
УДК 661.12.01/09
Copyright ©2008 by John Wiley & Sons, Inc.
All rights reserved. Autorised translation from the English language edition published by John Wiley & Sons, Inc.
Responsibility for the accuracy of the translation rests with EPC "Professiy" and is not the responsibiblity of John
Wiley & Sons, Inc. Not part of this book may be reproduced in any form without the written permission of the original
copyright holder, John Wiley & Sons, Inc.
Все права защищены. Никакая часть данной книги не может быть воспроизведена в какой бы то ни
было форме без письменного разрешения владельцев авторских прав.
Информация, содержащаяся в данной книге, получена из источников, рассматриваемых издатель-
ством как надежные. Тем не менее, имея в виду возможные человеческие или технические ошибки,
издательство не может гарантировать абсолютную точность и полноту приводимых сведений и не несет
ответственности за возможные ошибки, связанные с использованием книги.
ISBN 978-0-470-25959-7 (англ.)
ISBN 978-5-91884-046-7
© John Wiley & Sons, Inc., 2008
© ЦОП «Профессия», 2013
© Перевод, оформление: ЦОП «Профессия», 2013
Группа Компаний «ГалаХим» рада участвовать в издании книги Pharma-
ceutical Manufacturing Handbook: Regulations and Quality на русском языке
и представить это уникальное издание вниманию российских читателей.
«ГалаХим», много лет поставляя на российский рынок всё, что необходи-
мо для контроля качества в фармацевтической отрасли, отмечает растущий
интерес к проблеме соответствия международным стандартам со стороны
российских предприятий.
Текущий уровень развития фармацевтической отрасли в нашей стране
предполагает строгое регулирование всех этапов производства лекарствен-
ных средств и тщательный контроль качества продукции на каждом этапе.
Для этой цели как нельзя лучше подходят международные правила Феде-
рального управления США по контролю за пищевой продукцией и лекар-
ствами (FDA). Издание носит энциклопедический и прикладной характер.
В книге обсуждаются все основные аспекты фармацевтического производ-
ства: надлежащая производственная (GMP) и лабораторная практика (GLP),
методы и создание систем контроля качества, аналитические технологиче-
ские процессы (РАТ), валидация методик, стабильность лекарственной про-
дукции. Материал изложен ясно, последовательно и детально, что делает его
незаменимым помощником и практическим руководством для широкого
круга специалистов.
Мы надеемся, что наша совместная работа принесет пользу как участ-
никам фармацевтического рынка, так и потребителям лекарственных пре-
паратов. jy у/"
Группа Компаний «ГалаХим».
Директор по развитию
Андресюк Алексей Николаевич
Оглавление
Авторы статей.............................................................21
Предисловие к русскому изданию............................................24
Предисловие ..............................................................26
Часть!
НОРМАТИВНОЕ РЕГУЛИРОВАНИЕ ПРОИЗВОДСТВА
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Бгава 1.1. Надлежащая производственная практика и требования FDA..........29
1.1.1. Требования FDA. законы и рекомендации.....................29
1 1.2. Том 21 CFR, разделы 210 и 211: Текущая надлежащая производственная
практика для готовой фармацевтической продукции.............30
1.1.2.1. Общие положения....................................30
1.1.2.2. Организация и персонал.............................31
1.1.2.3. Помещения и оборудование...........................32
1.1.2.4. Оборудование.......................................34
1.1.2.5. Контроль компонентов фармацевтической продукции,
ее контейнеров, а также используемых укупорочных
приспособлений.............................................36
1.1.2.6. Технический контроль и управление производственным
процессом..................................................38
1.1.2.7. Контроль упаковки и маркировки.....................40
1.1.2.8. Хранение и распределение...........................45
1.1.2.9 Лабораторный контроль...............................45
1.1.2.10. Записи и протоколы................................49
1.1.2.11. Возвращенные и переработанные фармацевтические
препараты..................................................54
1.1.3. Руководство для промышленности «Системный подход к обеспечению
качества и нормативы Текущей надлежащей практики производства
фармацевтической продукции».......................................54
1.1.3.1. Текущая надлежащая производственная практика
и концепции современных систем управления качеством.........55
1.1.3.2. Модель систем менеджмента качества.................56
1.1.4. Руководство для промышленности «РАТ — система инновационного
развития фармацевтического производства и обеспечения качества» ... .57
1.1.4.1. Система РАТ........................................58
1.1.5. Руководство для промышленности «Часть 11. Электронные записи,
электронные подписи: возможности и применение»....................60
1.1.5.1. Записи, регламентированные разделом 11 тома 21 CFR.61
1.1.5.2. Подход FDAк особым требованиям раздела 11 тома 21 CFR ... .61
1.1.6. Руководство для промышленности и FDA «Текущая надлежащая
производственная практика для комбинированных продуктов»..........62
1.1.7. Руководство для промышленности «Порошковые смеси и готовые
единицы дозирования: стратифицированный отбор проб и оценка
качества единиц дозирования в процессе производства».............63
Оглавление
7
1.1.7.1. Валидация однородности серии порошкообразной смеси...64
1.1.7.2. Верификация производственных критериев...............65
1.1.8. Руководство для промышленности «Масштабирование производства
твердых пероральных дозированных форм с немедленным
высвобождением и порядок внесения пострегистрационных изменений
(SUPAC): химия, производство и контроль, документирование
исследований растворимости in vitro и биоэквивалентности in vivo»...67
1.1.8.1. Измененияв компонентах (вспомогательныхвеществах)
и составах............................................67
1.1.8.2. Изменение производственной площадки..................71
1.1.8.3. Изменения размеров производственных серий............72
1.1.8.4. Изменения в производственном процессе................73
1.1.9. Другие руководящие документы, связанные с GMP...............76
Глава 1.2. Внедрение текущей надлежащей производственной практики...........78
1.2.1. Введение....................................................78
1.2.2. Органы регулирования........................................80
1.2.3. Методы регулирования PDA....................................81
1.2.З.1. Инспекции............................................81
1.2.З.2. После инспекции: Форма 483 ..........................83
1.2.З.З. Отзыв................................................85
1.2.З.4. Письмо-предупреждение................................87
1.2.4. Судебное правоприменение без письма-предупреждения..........88
1.2.4.1. Введение.............................................88
1.2.4.2. 1ражданские иски.....................................91
1.2.4.З. Уголовное преследование..............................95
1.2.5. Заключение.................................................101
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных
изменений (SUPAC).................................................102
1.3.1. Введение....................................................102
1.3.2. Научное и юридическое обоснование масштабирования
производства и пострегистрационных изменений (SUPAC).... 104
1.3.2.1. Документация, обосновывающая изменения, а также
масштабы последних.........................................104
1.3.2.2. Документация, обосновывающая внесение изменений
в спецификации.............................................105
1.3.2.3. Протоколы сравнения.................................105
1.З.2.4. Требования по проведению испытаний in vitrowin vivo.106
1.3.3. Регулирующие органы и руководства..........................107
1.З.З.1. Правила SUPAC(FDA)..................................107
1.З.З.2. Руководство по SUPACФармацевтического союза ЕС......122
1.З.З.З. Регулирующие указания по SUPACНационального агентства
по контролю в области здравоохранения (Бразилия).....126
1.3.4. Гармонизация...............................................126
1.3.5. Аспекты GMP: контроль изменений и валидация процесса.......129
1.З.5.1. Контроль изменений..................................129
1.З.5.2. Валидация процесса..................................132
1.3.6. Заключение.................................................134
Литература .......................................................135
8
Оглавление
Eiaea 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток
человека в соответствии с правилами GMP...................................137
1.4.1. Введение....................................................137
1.4.2. Сокращения и определения....................................139
1.4.2.1. Мезенхимальные стромальные клетки...................139
1.4.2.2. Клеточная терапия соматическими стволовами клетками.140
1.4.2.З. Надлежащая производственная практика................141
1.4.2.4. Лекарственные препараты для клеточной терапии.......141
1.4.2.5. Лизат тромбоцитов человека..........................141
1.4.3. Подходы.....................................................142
1.4.3.1. Приверженность принципам GMP в процессе
доклинической разработки.....................................142
1.4.3.2. Эффективное стандартизованное культивирование MSC
с низкой плотностью посева...................................143
1.4.3.3. Более активная пролиферация клеток в питательных средах
с лизатом тромбоцитов человека по сравнению
с питательными средами с фетальной бычьей сывороткой.........143
1.4.3.4. Возможность минимизации риска контаминации при
использовании обоснованных (надлежащих) процедур
культивирования MSC.........................................144
1.4.4. Методики испытаний..........................................146
1.4.4.1. Безопасность и эффективность препаратов для клеточной
терапии на доклинической стадии.............................146
1.4.4.2. Контроль качества в процессе культивирования клеток
(внутрипроизводственный контроль) и критерии выпуска
готового продукта...........................................146
1.4.4.З. Исследование функциональности и биологической
активности стволовых клеток.................................147
1.4.5. Заключение..................................................153
Благодарность......................................................153
Литература ........................................................153
Часть 2
МЕЖДУНАРОДНЫЕ ПРАВИЛА НАДЛЕЖАЩЕЙ ПРОИЗВОДСТВЕННОЙ
ПРАКТИКИ
Глава 2.1. Национальные правила и требования GMP, международные требования GMP
и руководящие документы: совпадения и различия...................159
2.1.1. Введение...................................................159
2.1.2. Национальные правила и требования GMP......................159
2.1.2.1. Соединенные Штаты Америки...........................159
2.1.2.2. Канада..............................................160
2.1.2.3. Европейский Союз....................................162
2.1.2.4. Страны Восточной Азии...............................165
2.1.2.5. Индия...............................................166
2.1.2.6. Австралия...........................................167
2.1.2.7. Новая Зеландия......................................170
2.1.2.8. Южная Африка........................................170
2.1.3. Международные требования GMP и их координация...............171
2.1.3.1. Всемирная организация здравоохранения (ВОЗ).........171
Оглавление
9
2.1.3.2. Программа сотрудничества фармацевтических инспекторатов
(PIC/S)................................................173
2.1.3.3. Международная конференция по гармонизации (ICH)......175
2.1.3.4. Ассоциация государств Юго-Восточной Азии (ASEAN).....178
2.1.3.5. Общий рынок государств Южной Америки (MERCOSUR)......178
2.1.1. Соответствие правил GMP США требованиям и правилам GMP
других стран.......................................................178
2.1.4.1. Общие вопросы.......................................178
2.1.4.2. Организация и персонал..............................179
2.1.4.З. Здания и помещения..................................181
2.1.4.4. Оборудование........................................184
2.1.4.5. Контроль качества компонентов, первичной упаковки
и укупорочных средств.................................186
2.1.4.6. Производство и контроль процесса....................189
2.1.4.7. Упаковка и итоговый контроль продукта...............192
2.1.4.8. Хранение и поставки.................................195
2.1.4.9. Контроль качества...................................195
2.1.4.10. Документация.......................................198
2.1.4.11. Возвращенные некачественные лекарственные препараты
и лекарственные препараты с нарушением условий хранения . .201
Литература ........................................................202
Часть 3
КАЧЕСТВО
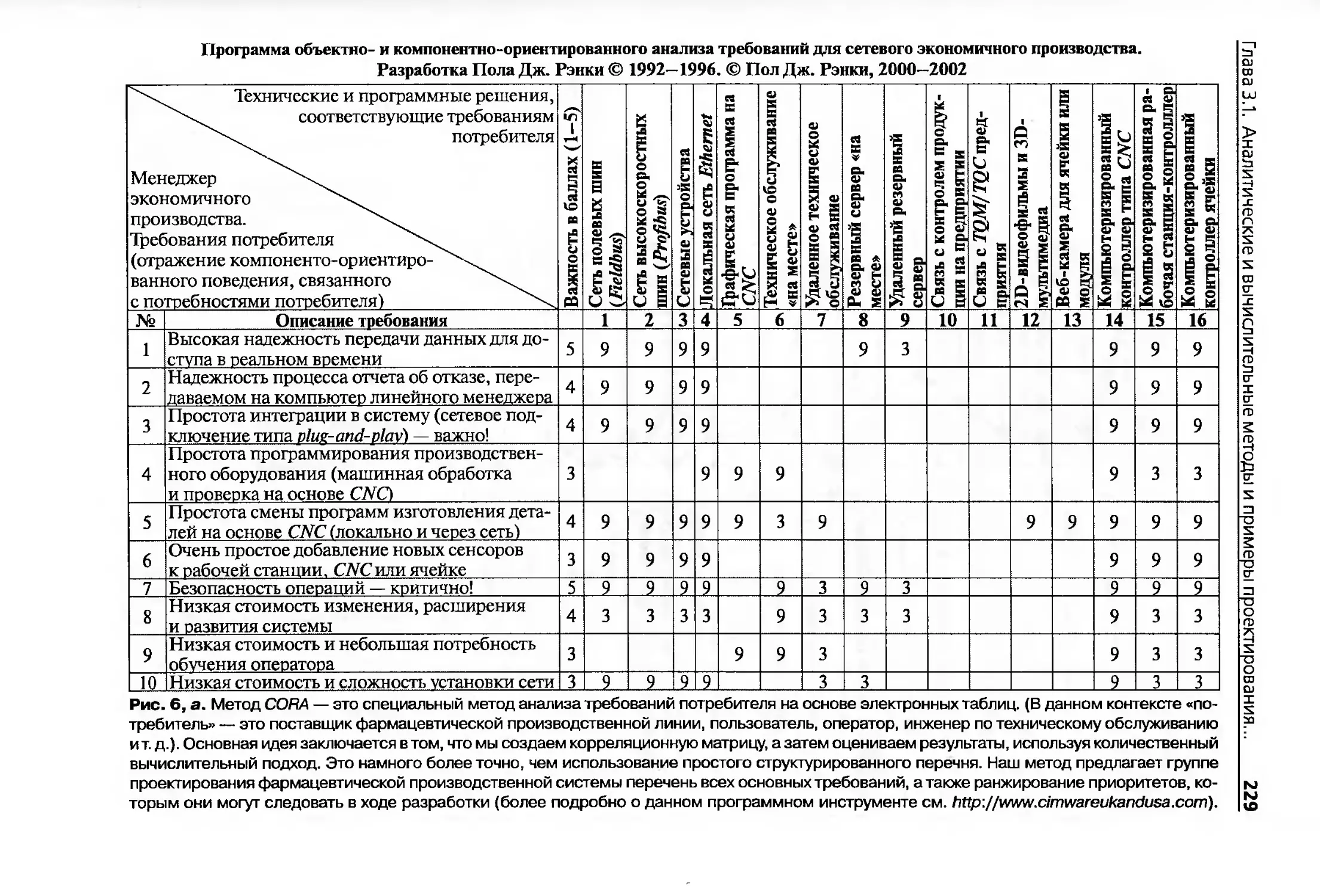
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования
и контролирования фармацевтических систем общего управления качеством.209
3.1.1. Введение..................................................209
3.1.2. Проектирование гибкой фармацевтической производственной
и сборочно-упаковочной системы..............................211
3.1.3. Модель гибкого производства, интегрированная с проектированием.. .213
3.1.4. Операционный контроль в режиме реального времени..........214
3.1.5. Инновационное проектирование..............................216
3.1.6. Открытая инновационная архитектура........................218
3.1.7. Типовой объектно-ориентированный метод моделирования
инновационного процесса и образцовая модель.................221
3.1.8. Системный подход к управлению фармацевтической
производственной системой...................................223
3.1.9. Анализ требований при проектировании инновационной системы
продукта, процесса и обслуживания...........................227
3.1.10. Анализ инновационных рисков, метод возможностей и программное
обеспечение для фармацевтических производственных систем..........228
3.1.11. Мультимедийные трехмерные и программные статистические
приложения из открытых источников для инноваций фармацевтических
производственных систем и взаимодействия при работе с проектом... .232
3.1.12. Приложения RFID...............................................234
3.1.13. Примеры RFID..................................................235
3.1.14. Модели интеграции RFID в товаропроводящие сети
компьютеризированных фармацевтических производственных
и сборочно-упаковочных систем.....................................236
10
Оглавление
3.1.15. Оценка результатов имитации работы сети....................241
3.1.16. Заключение.................................................243
3.1.17. Дополнительные видеоматериалы на DVD.......................243
Литература ........................................................244
Diaea 3.2. Значение систем обеспечения качества и аудитов
в фармацевтическом производстве.............................................249
3.2.1. Требования cGMP.............................................250
3.2.1.1. Обязанности подразделения по контролю качества
в правилах cGMP...............................................251
3.2.2. Деятельность по обеспечению качества........................252
3.2.3. Концепция систем качества...................................253
3.2.4. Ответственность руководства.................................256
3.2.5. Ресурсы.....................................................258
3.2.6. Производственные операции...................................260
З.2.6.1. Проектирование, разработка и документирование процесса
и продукта...................................................261
3.2.6.2. Входы...............................................263
3.2.6.3. Выполнение и мониторинг операций....................264
3.2.6.4. Рассмотрение несоответствий.........................265
3.2.7. Оценка деятельности.........................................266
3.2.7.1. Анализ основной тенденции (тренда)..................266
3.2.7.2. Проведение внутренних аудитов.......................267
3.2.7.3. Управление рисками качества.........................271
3.2.7.4. Корректирующие и предупреждающие действия...........273
3.2.7.5. Стимулирование улучшений............................273
3.2.8. Переход к модели системы качества...........................274
3.2.9. Перечень вопросов для аудита в фармацевтической
промышленности...............................................276
З.2.9.1. Инструкция по применению перечня вопросов для аудита.276
Литература...................................................290
Diaea 3.3. Создание системы менеджмента качества и управление ею............292
3.3.1. Введение....................................................292
3.3.2. Понимание системы менеджмента качества......................293
3.3.2.1. Определение термина «система менеджмента качества»..294
3.3.2.2. Целое и часть целого................................297
3.3.2.3. Система и процесс...................................298
3.3.2.4. Преимущества для коммерческой деятельности организации
при установлении надежной системы менеджмента качества.. .300
3.3.2.5. Ожидания промышленности и ретуляторных агентств......305
3.3.3. Руководство и персонал: лидерство и поддержка...............307
3.3.3.1. Описание выгод для организации.......................308
3.3.3.2. Общение на языке менеджмента........................309
3.3.3.3. Объяснение выгод для персонала......................310
3.3.3.4. Обеспечение поддержки персонала и лидерство руководства.. .310
3.3.3.5. Ловушки, которых следует избегать....................311
3.3.4. Установление области применения системы менеджмента качества ... .313
З.З.4.1. Определение требований коммерческой деятельности.....314
3.3.4.2. Интеграция системы менеджмента качества в планы
качества.......................................................315
Оглавление
11
3.3.4.3. Определение требований по декомпозиции процесса......316
3.3.4.4. Масштабируемость процессов на предприятии............318
3.3.5. Владение системами и процессами: функции и ответственность...319
3.3.5.1. Владение и управление системой менеджмента качества..319
3.3.5.2. Владение процессом...................................321
3.3.5.3. Выбор владельца процесса.............................321
3.3.5.5. Право принятия решений...............................323
3.3.5.6. Отраслевой опыт......................................324
3.3.5.7. Регуляторные инспекции и аудиты......................325
3.3.5.8. Технические эксперты.................................326
3.3.5.9. Владение показателями................................326
3.3.5.10. Владение документацией..............................327
3.3.5.11. Обучение............................................327
3.3.5.12. Управление рисками..................................328
3.3.5.13. Непрерывное улучшение и управление проектами........328
3.3.5.14. Несоответствия, корректирующие и предупреждающие
действия (САРА) и владение планируемыми отклонениями... .329
3.3.6. Управление изменениями и распространение информации..........330
3.3.6.1. Управление организационными изменениями..............331
3.3.6.2. Распространение информации...........................332
3.3.6.3. Обратная связь и корректировка программы.............333
3.3.6.4. Обучение.............................................335
3.3.7. Измерение успеха с помощью информативных показателей
эффективности.......................................................336
3.3.7.1. Разработка показателей эффективности.................336
3.3.7.2. Анализ показателей...................................337
3.3.7.3. Модель зрелости системы..............................338
3.3.7.4. Выполнение требований к зрелости процесса............340
3.3.8. Обеспечение постоянного улучшения: проекты...................341
3.3.8.1. Улучшение процессов..................................341
3.3.8.2. Предложения по улучшению процессов...................342
3.3.8.3. Задача и проект......................................343
3.3.8.4. Показатели проекта...................................344
3.3.9. Обеспечение постоянного успеха...............................344
3.3.9.1. Установка совместных целей...........................345
3.3.9.2. Награды и признание..................................346
3.3.9.3. Обеспечение совместимости действующей программы......347
3.3.9.4. Привыкание к программе...............................347
Литература....................................................348
Каава 3.4. Улучшение качества процесса.......................................349
3.4.1. Диагностика процесса.........................................349
3.4.1.1. Введение.............................................349
З.4.1.2. Основные инструменты диагностики процесса............349
3.4.2. Стабилизация и улучшение процесса............................354
3.4.2.1. Введение.............................................354
3.4.2.2. Контрольные карты для качественных данных............356
3.4.2.3. Контрольные карты для количественных данных..........359
3.4.2.4. Специальные контрольные карты........................364
3.4.3. Повышение эффективности процесса.............................367
12
Оглавление
З.4.З.1. Введение............................................367
3.4.3.2. Изучение возможностей процесса и его улучшение......369
Литература...................................................372
Часть 4
ПРОЦЕССНО-АНАЛИТИЧЕСКИЕ ТЕХНОЛОГИИ
Lhasa 4.1. Аргументы в пользу процессно-аналитической технологии: юридические
и промышленные перспективы.................................................375
4.1.1. Введение...................................................375
4.1.2. Основы процессно-аналитической технологии..................375
4.1.2.1. Процессно-аналитическая химия.......................376
4.1.2.2. Управление качеством................................377
4.1.2.З. Ресурсосберегающее производство.....................380
4.1.3. Исторические факторы, ограничивающие внедрение РАТ.........383
4.1.3.1. Реальные и субъективно воспринимаемые технологические
барьеры.....................................................384
4.1.З.2. Отсутствие экономических стимулов...................385
4.1.3.3. Юридические препятствия.............................389
4.1.4. Инициатива FDA по cGMP двадцать первого века...............389
4.1.4.1. Концепция инициативы................................390
4.1.4.2. Ориентация с учетом рисков..........................392
4.1.4.З. Системы обеспечения качества........................393
4.1.4.4. Стратегия, основанная на научном подходе............395
4.1.4.5. Международное сотрудничество........................397
4.1.5. Развитие РАТ в фармацевтическом производстве...............401
4.1.5.1. Понимание процесса..................................402
4.1.5.2. Принципы и инструменты РАТ..........................406
4.1.5.3. Стратегия внедрения.................................408
4.1.6. Процесс внедрения РАТ......................................410
4.1.6.1. Подготовка..........................................411
4.1.6.2. Оценка..............................................412
4.1.6.З. Анализ..............................................413
4.1.6.4. Контроль............................................413
4.1.6.5. Философия выпуска продукции.........................413
4.1.6.6. Оптимизация.........................................413
4.1.6.7. Перспективы внедрения РАТ...........................414
Благодарность.....................................................416
Литература .......................................................416
Lhasa 4.2. Процессно-аналитическая технология (РАТ)........................420
4.2.1. Основные принципы и эффект от внедрения....................420
4.2.1.1. Определение.........................................420
4.2.1.2. Что способствовало появлению РА7?...................420
4.2.1.3. Анализ корневых причин и процессный контроль........421
4.2.1.4. Когда следует внедрять РАТ..........................421
4.2.1.5. РАТ способствует углублению понимания процесса......422
4.2.1.6. Изменение действующей практики при помощи РАТ........423
4.2.1.7. Продвижение физической фармакологии и фармацевтики ... .424
4.2.1.8. Глубинный анализ данных..............................425
4.2.1.9. Хранение данных......................................425
Оглавление
13
4.2.1.10. Методы глубинного анализа данных применительно
к фармацевтическим процессам................................427
4.2.1.11. Практика глубинного анализа данных................428
4.2.1.12. Комментарии к глубинному анализу данных...........429
4.2.1.13. Методы РАТ........................................430
4.2.1.14. Заключение........................................430
4.2.2. Колебательная спектроскопия................................432
4.2.2.1. Введение...........................................432
4.2.2.2. Теория ИК-спектроскопии............................435
4.2.2.3. Механическая модель ИК-колебаний...................436
4.2.2.4. Квантово-механическая модель.......................438
4.2.2.5. Ангармоничность....................................441
4.2.2.6. Применение спектроскопии в среднем ИК-диапазоне
для структурных исследований................................442
4.2.2.7. Расширение области применения спектроскопии в среднем
ИК-диапазоне................................................443
4.2.2.8. Рамановская спектроскопия..........................445
4.2.2.9. Введение в БИК-спектроскопию.......................448
4.2.2.10. Преимущества БИК-спекгроскопии....................448
4.2.2.11. Введение в химическую визуализацию методом
спектроскопии в БИК- и средней ИК-области...................451
4.2.2.12. Типы инструментов для спектроскопии в среднем
ИК-диапазоне................................................452
4.2.2.13. Заключение........................................456
4.2.3. Хемометрика................................................457
4.2.3.1. Введение...........................................457
4.2.3.2. От одномерной регрессии к многомерной..............459
4.2.3.3. Качество проб и погрешность данных.................460
4.2.3.4. Предварительная математическая обработка данных
спектроскопии...............................................462
4.2.3.5. Предварительная обработка данных БИК-спектроскопии..463
4.2.3.6. Математическая обработка и преобразования..........464
4.2.3.7. Метод главных компонент............................466
4.2.3.8. Применение метода главных компонент
в БИК-спектроскопии.........................................469
4.2.3.9. Распознавание паттернов............................471
4.2.3.10. Классификация SIMCA...............................472
4.2.3.11. Регрессия.........................................473
4.2.3.12. Множественная линейная регрессия..................474
4.2.3.13. PCR и PLS регрессии...............................475
4.2.3.14. Практика построения регрессий в БИК-спектроскопии.476
4.2.3.15. Некоторые «подводные камни».......................480
4.2.3.16. Примеры аналитического применения БИК-спектроскопии.. .481
4.2.3.17. Заключение........................................484
Литература .......................................................485
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты
процессно-аналитической технологии.........................................487
4.3.1. Введение...................................................487
4.3.2. Построение гиперспектральных изображений...................487
14
Оглавление
4.3.3. Оборудование для получения гиперспектральных изображений.....489
4.3.3.1. Принципы получения гиперспектральных изображений...489
4.3.3.2. Спектроскопическое оборудование....................490
4.3.4. Применение хемометрики для построения изображений............492
4.З.4.1. Предварительная обработка данных...................492
4.3.4.2. Классификация пикселей.............................494
4.3.5. Практика химической визуализации в режимах at-line и on-line.497
4.З.5.1. Практические инструменты анализа карт распределения..497
4.3.5.2. Выбор длины волны и химическая интерпретация.........499
4.3.5.3. Классификация «без обучения» для поисков сбоев процесса... 502
4.3.5.4. Классификация «с обучением», БИК-визуализация
и планирование процесса.....................................504
4.3.5.5. Перспективные разработки: анализ гиперспектральных
изображений в реальном времени.............................506
4.3.6. Выводы.....................................................507
Благодарность.....................................................508
Литература .......................................................509
Часть 5
ПЕРСОНАЛ
1)тава5.1. Обучение персонала, занятого в фармацевтическом производстве...513
5.1.1. Обзор .....................................................513
5.1.1.1. Общая часть........................................513
5.1.1.2. Требования к обучению..............................513
5.1.1.3. Надлежащая практика обучения в фармацевтическом
производстве...............................................516
5.1.1.4. Понятие обучения, основанного на компетенциях......516
5.1.1.5. Почему так важно обучение, основанное на компетенциях? ...517
5.1.2. Разработка плана обучения: стратегия обеспечения соответствия
требованиям к обучению на фармацевтическом предприятии1...........517
Раздел 1. Организация обучения..............................519
Раздел 2. Программы обучения................................525
Раздел 3. Разработки учебных протрамм.......................528
Раздел 4. Реализация программ обучения......................530
Раздел 5. Ведение записей, относящихся к обучению...........531
Литература .......................................................533
Часть 6
КОНТАМИНАЦИЯ И ЕЕ КОНТРОЛЬ
Става 6.1. Источники контаминации.........................................537
6.1.1. Введение...................................................537
6.1.2. Внутренние источники контаминации..........................538
6.1.2.1. Исходные материалы.................................539
6.1.2.2. Вспомогательные вещества...........................548
6.1.2.З. Продукты разложения веществ, входящих в состав продукта.. .556
6.1.3. Внешние источники контаминации.............................564
6.1.3.1. Остаточные органические растворители...............564
6.1.3.2. Контейнеры.........................................565
6.1.3.3. Системы доставки...................................596
Оглавление
15
6.1.3.4. Механические включения..............................605
6.1.4. Заключение.................................................615
Литература .......................................................616
Пгава 6.2. Количественное определение маркеров грамотрицательных и грамположительных
эндотоксинов при мониторинге производственной рабочей среды и контроле
микробной контаминации фармацевтических продуктов методом ГХ-МС/МС... .620
6.2.1. Введение..........................................................620
6.2.2. Анализ маркеров для липополисахаридов и пептидогликанов
методом ГХ-МС/МС и его применение............................623
6.2.3. Рабочие параметры..........................................626
6.2.4. Заключение.................................................628
Литература .......................................................629
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического
производства ..............................................................631
6.3.1. Введение...................................................631
6.3.2. Международные правила и регуляторные руководства в отношении
контроля микробиологической чистоты в нестерильном
производстве.....................................................633
6.З.2.1. Правила СМРрля готовых продуктов и их компонентов...633
6.3.2.2. Правила GMP для действующих фармацевтических веществ
(фармацевтических субстанций).......................635
6.3.2.3. Правила GMPдля фармацевтических вспомогательных
веществ.....................................................636
6.3.3. Фармакопейные требования в отношении микробиологических
аспектов нестерильного производства..............................636
6.3.3.1. Фармакопея США......................................637
6.3.3.2. Европейская фармакопея..............................639
6.3.3.3. Японская фармакопея.................................640
6.3.4. Действующие ожидания регуляторных органов в отношении
контроля микробиологической чистоты при нестерильном
производстве.....................................................640
6.3.5. Промышленные перспективы контроля микробиологической
чистоты для нестерильного фармацевтического производства.........642
6.3.5.1. Результаты опроса...................................643
6.3.5.2. Рекомендации........................................643
6.3.6. Контроль микробиологической чистоты в течение срока годности
фармацевтических продуктов.......................................644
6.3.7. Заключение и выводы........................................645
Литература .......................................................645
Часть 7
СТАБИЛЬНОСТЬ ЛЕКАРСТВ
1)гава 7.1. Стабильность и сроки годности фармацевтических продуктов.......649
7.1.1. Введение...................................................649
7.1.2. Требования к стабильности в нормативах/руководствах по GMP.651
7.1.2.1. Готовые препараты...................................651
7.1.2.2. Вспомогательные материалы...........................652
7.1.2.З. Активные фармацевтические ингредиенты...............652
7.1.3. Требования к стабильности вспомогательных веществ..........653
16
Оглавление
7.1.4. Требования к стабильности лекарственных веществ (API).......654
7.1.4.1. Выбор серий и первичной упаковки....................654
7.1.4.2. Условия хранения и частота тестирования.............654
7.1.4.3. Стресс-испытание и методы оценки стабильности
при анализе стабильности API..................................656
7.1.4.4. Оценка результатов исследования стабильности API....657
7.1.4.5. Обязательства продолжать испытания стабильности.....657
7.1.4.6. Формулировка условий хранения и указание длительности
хранения в маркировке........................................658
7.1.5. Требования к стабильности лекарственных препаратов..........658
7.1.5.1. Выбор серий и первичной упаковки....................658
7.1.5.2. Брекетинг и построение матриц.......................659
7.1.5.3. Выбор условий хранения и частоты тестирования.......659
7.1.5.4. Стресс-испытание и методы оценки стабильности при анализе
стабильности лекарственной продукции.........................662
7.1.5.5. Оценка результатов исследования стабильности........663
7.1.5.6. Обязательства продолжать испытания стабильности.....663
7.1.5.7. Формулировка условий хранения и надписей в маркировке .. .664
7.1.6. Исследование фотостабильности...............................664
7.1.6.1. Фотостабильность API................................666
7.1.6.2. Фотостабильность лекарственных препаратов...........667
7.1.7. Оценка данных исследований стабильности и определение срока
хранения.....................................................668
7.1.7.1. Определение сроков хранения лекарственных веществ или
лекарственных препаратов, предназначенных для хранения
при комнатной температуре....................................668
7.1.7.2. Определение сроков хранения лекарственных веществ или
лекарственных препаратов, предназначенных для хранения
в холодильнике...............................................670
7.1.7.З. Определение сроков хранения лекарственных веществ
и препаратов, предназначенных для хранения в морозильной
камере.......................................................671
7.1.7.4. Оценка сроков хранения лекарственных веществ
или лекарственных препаратов, предназначенных
для хранения при температуре ниже — 20 °C.....................671
7.1.8. Рекомендованные условия хранения в исследованиях стабильности
с учетом установленных климатических зон............................671
7.1.9. Исследуемые параметры (характеристики) при изучении
стабильности различных лекарственных форм...........................672
7.1.9.1. Характеристики, проверяемые при изучении стабильности
всех лекарственных форм......................................673
7.1.9.2. Характеристики, проверяемые при исследовании
стабильности определенных лекарственных форм.................673
7.1.10. Заключение и выводы........................................674
Литература ........................................................675
Diaea 7.2. Стабильность лекарств............................................677
7.2.1. Общие вопросы стабильности..................................677
7.2.1.1. Введение............................................677
7.2.1.2. Требования регуляторного органа ..7.................678
7.2.1.3. Стабильность и срок годности........................679
Оглавление
17
7.2.1.4 Краткосрочные и долгосрочные исследования стабильности.. .679
7.2.1.5. Статистическая обработка...............................680
7.2.2. Планирование исследований стабильности.........................684
7.2.2.1. Введение..............................................684
7.2.2.2. Основные принципы планирования исследования...........685
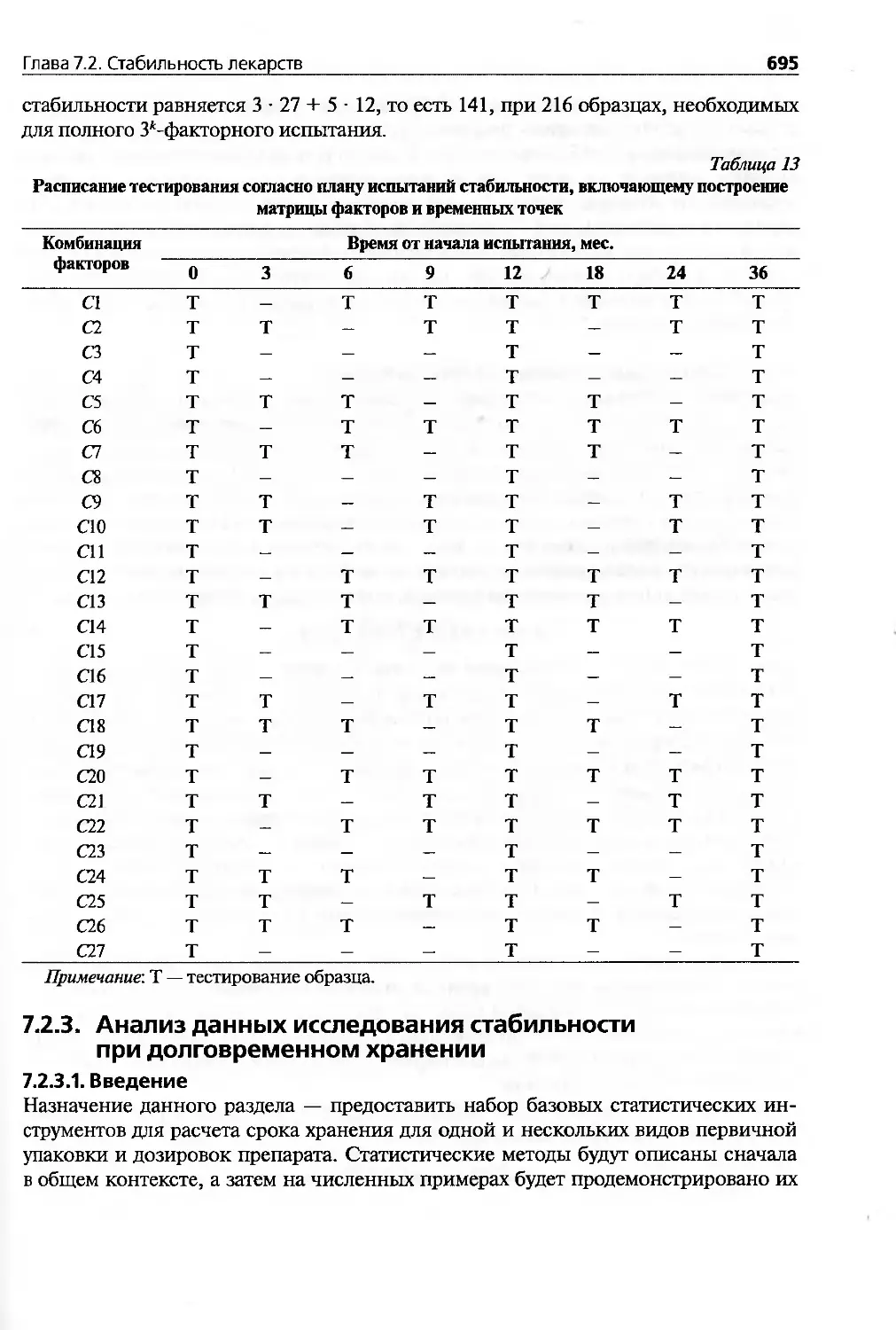
7.2.2.3. Методы планирования исследований стабильности.........686
7.2.3. Анализ данных исследования стабильности при долговременном
хранении.............................................................695
7.2.3.1. Введение..............................................695
7.2.3.2. Сроки хранения одной партии лекарств..................696
7.2.3.3. Расчет срока годности по данным изучения стабильности
нескольких серий препарата....................................699
7.2.3.4. Определение срока хранения с учетом множества факторов... .713
7.2.4. Анализ данных исследования стабильности при краткосрочном
хранении..............................................................722
7.2.4.1. Введение...............................................722
7.2.4.3. Оценка скорости процесса разложения по данным
исследования стабильности при ускоренном хранении..............724
7.2.4.4. Предварительные расчеты срока хранения на основании
данных стрессовых испытаний...................................729
7.2.5. Заключительные замечания......................................730
Компьютерные программы...............................................731
Литература ..........................................................736
Diaea 7.3. Влияние упаковки на стабильность фармацевтических субстанций
и лекарственных препаратов....................................................738
7.3.1. Введение......................................................738
7.3.3.1. Основные проблемы, связанные со стабильностью
лекарственных средств..........................................738
7.3.2. Факторы, влияющие на стабильность лекарственных средств.......741
7.3.2.1. Влажность, гидролиз и pH..............................741
7.3.2.2. Кислород и окисление..................................744
7.3.2.3. Свет..................................................745
7.3.2.4. Температура...........................................747
7.3.2.5. Микроорганизмы........................................748
7.3.2.6. Активные фармацевтические ингредиенты и наполнители....750
7.3.3. Упаковка для лекарственных средств и упаковочные материалы....753
7.3.3.1. Введение..............................................753
7.3.3.2. Материалы, применяемые при изготовлении компонентов
упаковки......................................................754
7.3.4. Влияние упаковки на стабильность лекарственных препаратов.....757
7.З.4.1. Введение..............................................757
7.3.4.2. Твердые лекарственные формы...........................757
7.3.4.3. Нестерильные жидкие лекарственные формы...............761
7.3.4.4. Стерильные жидкие лекарственные формы.................762
7.3.4.5. Влияние веществ, выделяющихся из материала упаковки,
на стабильность лекарственного препарата......................766
7.3.4.6. Биотехнологические препараты..........................770
7.3.4.7. Влияние упаковки на стабильность лекарственных
препаратов: взгляд в будущее..................................773
Литература ..........................................................780
18
Оглавление
Пгава 7.4. Стабильность фармацевтических препаратов.......................789
7.4.1. Введение..................................................789
7.4.2. Кинетические уравнения и срок годности....................790
7.4.2.1. Уравнение скорости реакции........................790
7.4.2.2. Определение порядка реакции.......................792
7.4.2.3. Прогнозирование срока хранения....................792
7.4.2.4. Уравнение Аррениуса и ускоренное испытание
стабильности................................................793
7.4.3. Механизмы разложения лекарственных средств................795
7.4.4. Химическое разложение.....................................795
7.4.4.1. Сольволиз.........................................795
7.4.4.2. Окисление.........................................796
7.4.4.3. Фотолиз...........................................797
7.4.4.4. Дегидратация......................................798
7.4.4.5. Рацемизация.......................................798
7.4.5. Физическое разложение.....................................798
7.4.5.1. Полиморфизм.......................................798
7.4.5.2. Испарение.........................................799
7.4.6. Микробиологическое разложение.............................799
7.4.7. Руководства и требования в отношении стабильности.........799
7.4.8. Руководства ICHпо качеству................................800
Литература ......................................................801
Пгава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств:
исследования кинетики разложения с переменными параметрами................804
7.5.1. Введение..................................................804
7.5.2. Теоретические положения...................................805
7.5.3. Экспериментальная часть...................................810
7.5.3.1. Компьютерное моделирование........................810
7.5.3.2. Устройства для достижения условий переменных
параметров..................................................812
7.5.3.3. Аналитические приборы.............................814
7.5.3.4. Программное обеспечение для обработки
экспериментальных данных...................................815
7.5.4. Примеры кинетических экспериментов с переменными
параметрами......................................................816
7.5.4.1. Кинетические эксперименты с переменным параметром
температуры................................................816
7.5.4.2. Кинетические эксперименты с переменным параметром
концентрации...............................................819
7.5.4.3. Кинетические эксперименты с переменным параметром
ионной силы................................................824
7.5.5. Заключение................................................825
Литература ......................................................825
Часть 8
ВАЛИДАЦИЯ
Пгава 8.1. Валидация аналитических методик: принципы и подходы.............831
8.1.1. Введение...................................................831
8.1.2. Цели валидации аналитических методик.......................831
Оглавление
19
8.1.3. Действующие правила надлежащей производственной практики
в двадцать первом веке............................................831
8.1.4. Цикл аналитической методики.................................832
8.1.5. Валидационные характеристики аналитической методики.........833
8.1.5.1. Правильность........................................833
8.1.5.2. Прецизионность методики.............................833
8.1.5.3. Специфичность.......................................835
8.1.5.4. Предел обнаружения..................................836
8.1.5.5. Предел количественного определения..................837
8.1.5.6. Линейность..........................................838
8.1.5.7. Диапазон............................................839
8.1.5.8. Робастность.........................................840
8.1.6. Процесс валидации аналитической методики....................841
8.1.7. Необходимая для аналитической методики информация...........843
8.1.8. Валидация методик на стадии разработки......................843
8.1.9. Верификация методики........................................845
8.1.10. Ревалидация методики.......................................846
8.1.11. Заключение.................................................847
Литература ........................................................847
Diaea 8.2. Валидация аналитических методик и обеспечение качества...........848
8.2.1. Введение....................................................848
8.2.2. Прослеживаемость и неопределенность измерений...............849
8.2.2.1. Введение: качество аналитических результатов........849
8.2.2.2. Роль валидации методики в прослеживаемости
и неопределенности измерений..................................850
8.2.2.3. Руководства по прослеживаемости и погрешности
результатов...................................................852
8.2.2.4. Концепция прослеживаемости..........................854
8.2.2.5. Концепция неопределенности измерений................857
8.2.2.6. Различные рабочие определения неопределенности
измерения...................................................859
8.2.2.7. Подходы к нахождению неопределенности измерений.....860
8.2.2.8. Значение прослеживаемости и неопределенности
измерения.....................................................863
8.2.2.9. Заключение..........................................864
8.2.3. Валидация методики и обеспечение качества...................865
8.2.3.1. Роль валидации методики в обеспечении аналитического
качества....................................................865
8.2.3.2. Руководства и рекомендации по обеспечению
аналитического качества.....................................867
8.2.3.3. Подходы к оценке приемлемых методов анализа.........867
8.2.3.4. Рабочие характеристики методик и подход оценки
по критериям..................................................869
8.2.3.5. Обеспечение аналитического качества.................891
8.2.4. Заключение..................................................898
Литература ........................................................898
Глава 8.3. Валидация лабораторного оборудования.............................904
8.3.1. Введение....................................................904
8.3.2. Область применения..........................................905
20
Оглавление
8.3.3. Классификация лабораторного оборудования..................905
8.3.4. Этапы валидации...........................................906
8.3.4.1. Этап планирования и определения требований........906
8.3.4.2. Этап квалификации (проверки)......................910
8.3.4.3. Этап функционирования.............................918
8.3.4.4. Завершение жизненного цикла.......................922
8.3.5. Заключение................................................922
Литература ....................................................922
Пгава 8.4. Принципы валидации фармацевтического производства..............924
8.4.1. Введение..................................................924
8.4.2. Область применения процессов валидации....................926
8.4.3. Сводный план валидации (VMP)..............................926
8.4.4. Валидационные протоколы и отчеты..........................928
8.4.4.1. Валидационные протоколы...........................928
8.4.4.2. Валидационные отчеты..............................931
8.4.5. Валидация производственных зданий.........................931
8.4.5.1. Общие положения...................................931
8.4.5.2. Проектирование производственных зданий............932
8.4.6. Валидация процесса производства...........................937
8.4.7. Аналитические методики....................................940
8.4.8. Комплексы оборудования и компьютерные системы.............942
8.4.8.1. Комплексы оборудования............................942
8.4.8.2. Компьютерные системы..............................945
8.4.9. Валидация очистки.........................................953
8.4.10. Заключение...............................................954
Литература ....................................................956
Список сокращений.........................................................957
АВТОРЫ СТАТЕЙ
Андреас Райншп, Медицинский университет Граца (1рац, Австрия). Культиви-
рование мультипотентных мезенхимальных стромальных клеток человека в соответ-
ствии с правилами GMP
Б. Сараменто, Факультет фармацевтики, Университет Порто (Порто, Португа-
лия). Принципы валидации фармацевтического производства
Гарри Родригес, Cordis LLC, Johnson & Johnson Company, Сан-Хуан, Пуэрто-Рико.
Стабильность лекарств
Герман Лэм, Wild Crane Horizon, Inc. (Скарборо, Онтарио, Канада). Валидация ла-
бораторного оборудования
Грегори Н. Рэнки, Общественный исследовательский университет Нью-Джерси,
(Ньюарк, Нью-Джерси). Аналитические и вычислительные методы и примеры про-
ектирования и контролирования фармацевтических систем общего управления каче-
ством
Д. С. Феррейра, Факультет фармацевтики, Университет Порто (Порто, Португа-
лия). Принципы валидации фармацевтического производства
Дениз Борер, Федеральный университет Санта Мария (Санта Мария, Бразилия).
Источники контаминации
Джеймс М. Барквест, Ground Zero Pharmaceuticals, Inc. (Ирвин, Калифорния).
Значение систем обеспечения качества и аудитов в фармацевтическом производстве
Д жеймс Р. Харрис, James Harris Associates, Inc. (Дарем, Северная Каролина). Над-
лежащая производственная практика (GMP) и требования FDA
Джузеппе Алибрацди, Университет Мессины (Мессина, Италия). Альтернатив-
ные ускоренные методы изучения стабильности лекарств: исследования кинетики раз-
ложения с переменными параметрами
Дирк Струнк, Медицинский университет Граца (Грац, Австрия). Культивирование
мультипотентных мезенхимальных стромальных клеток человека в соответствии с
правилами GMP
Дэввд А. Гэллап, Training and Communications Group, Inc. (Беруин, Пенсильвания).
Обучение персонала, занятого в фармацевтическом производстве
Е. Б. Соуто, Свободный университет Берлина (Берлин, Германия); Факультет
фармацевтики, Университет Порто (Порто, Португалия). Принципы валидации фар-
мацевтического производства
Ева Роде, Медицинский университет Граца (Грац, Австрия). Культивирование
мультипотентных мезенхимальных стромальных клеток человека в соответствии
с правилами GMP
Жи-хон Ванг, Университет Род-Айленда (Кингстон, Род-Айленд). Улучшение ка-
чества процесса
Ив Рогго, F. Hoffman-La Roche Ltd. (Базель, Швейцария). Процессно-аналитическая
технология; Химическая визуализация и хемометрика: полезные инструменты
процессно-аналитической технологии
22
Авторы статей
Изабель Таверньерс, Институт исследований в области сельского хозяйства и ры-
боводства (ILVO), Научно-исследовательский институт Фламандского сообщества
(Мерелбек, Бельгия). Валидация аналитических методик и обеспечение качества
Катарина Шалмазер, Медицинский университет Граца (Грац, Австрия). Культи-
вирование мультипотентных мезенхимальных стромальных клеток человека в соот-
ветствии с правилами GMP
Катрин В. Доменик, Training and Communications Group, Inc. (Беруин, Пенсильва-
ния). Обучение персонала, занятого в фармацевтическом производстве
Катрина Нордстрем, Технологический университет Хельсинки (Хельсинки, Фин-
ляндия). Национальные правила и требования GMP, международные требования GMP
и руководящие документы: совпадения и различия
Кеннет Дж. Нолан, Nolan & Auerbach (Форт Лодердейл, Флорида). Внедрение те-
кущей надлежащей производственной практики
Кристина Бартманн, Медицинский университет Граца (Грац, Австрия). Культи-
вирование мультипотентных мезенхимальных стромальных клеток человека в соот-
ветствии с правилами GMP
Л. Антонио Эстевес, Университет Пуэрто-Рико, (Маягуэс, Пуэрто-Рико). Ста-
бильность лекарств
Мардж Гйллис, Training and Communications Group, Inc. (Беруин, Пенсильвания).
Обучение персонала, занятого в фармацевтическом производстве
Марк де Лууз, Институт исследований в области сельского хозяйства и рыбовод-
ства (ILVO), Научно-исследовательский институт Фламандского сообщества (Ме-
релбек, Бельгия). Валидация аналитических методик и обеспечение качества
Марко Нярхи, Технологический университет Хельсинки (Хельсинки, Финлян-
дия). Национальные правила и требования GMP, международные требования GMP и
руководящие документы: совпадения и различия
Мишель Е. Доулинг, Amgem, Inc. (Саузенд Оукс, Калифорния). Создание системы
менеджмента качества и управление ею
Мишель Ульмшнайдер, F. Hoffman-La Roche Ltd. (Базель, Швейцария). Процессно-
аналитическая технология; Химическая визуализация и хемометрика: полезные ин-
струменты процессно-аналитической технологии
Назарио Д. Рамирес-Белтран, Университет Пуэрто-Рико (Маягуэс, Пуэрто-
Рико). Стабильность лекарств
Пол А. Фрэнкел, Amgem, Inc. (Саузенд Оукс, Калифорния). Создание системы ме-
неджмента качества и управление ею
Пол Дж. Рэнки, Технологический университет Нью-Джерси (Ньюарк, Нью-
Джерси); Аналитические и вычислительные методы и примеры проектирования и кон-
тролирования фармацевтических систем общего управления качеством
Пунит Шарма, Университет Окленда (Окленд, Новая Зеландия). Правила мас-
штабирования производства и внесения пострегистрационных изменений (SUPAC)
Ранга Велагалети, BASF Corporation (Флорэм Парк, Нью-Джерси). Микробиоло-
гические аспекты нестерильного фармацевтического производства; Стабильность и
сроки годности фармацевтических продуктов
Авторы статей 23
Ричард Г. Рэнки, Технологический университет Нью-Джерси (Ньюарк, Нью-
Джерси). Аналитические и вычислительные методы и примеры проектирования и кон-
тролирования фармацевтических систем общего управления качеством
Роберт П. Когдилл, Центр фармацевтических технологий Университета Дьюкей-
на, (Питтсбург, Пенсильвания). Аргументы в пользу процессно-аналитической техно-
логии: юридические и промышленные перспективы
Санджей Гарг, Университет Окленда (Окленд, Новая Зеландия). Правила мас-
штабирования производства и внесения пострегистрационных изменений (SUPAC)
Т. Васконселос, Лаборатория фармацевтических разработок, BIAL (Мамеде де
Коронадо, Португалия); Факультет фармацевтики, Университет Порто (Порто,
Португалия). Принципы валидации фармацевтического производства
Чунг Чжон Чан, Azopharma Contract Pharmaceutical Services (Мирамар, Флорида).
Валидация аналитических методик: принципы и подходы
Шринивас Ганта, Университет Окленда (Окленд, Новая Зеландия). Правила мас-
штабирования производства и внесения пострегистрационных изменений (SUPAC)
Эван Б. Сигель, Ground Zero Pharmaceuticals, Inc. (Ирвин, Калифорния). Значение
систем обеспечения качества и аудитов в фармацевтическом производстве
Эдвард Р. Арлинг, Amgen, Inc. (Саузенд Оукс, Калифорния). Создание системы ме-
неджмента качества и управление ею
Элвин Фокс, Университет Южной Каролины (Колумбия, Южная Каролина).
Количественное определение маркеров грамотрицательных и грамположительных эн-
дотоксинов при мониторинге производственной рабочей среды и контроле микробной
контаминации фармацевтических продуктов методом ГХ-МС/МС
Эммануэль О. Акала, Фармацевтический факультет университета Говарда (г. Ва-
шингтон, округ Колумбия). Влияние упаковки на стабильность фармацевтических
субстанций и лекарственных препаратов
Эндрю Вебстер, Фармацевтический факультет имени Мак-Хортера, Сэмфорд-
ский университет (Бирмингем, Алабама). Стабильность фармацевтических препа-
ратов
Эрик ван Бокстель, Институт исследований в области сельского хозяйства и ры-
боводства (ILVO), Научно-исследовательский институт Фламандского сообщества
(Мерелбек, Бельгия). Валидация аналитических методик и обеспечение качества
Эшли Джон, Технологический университет Нью-Джерси (Ньюарк, Нью-Джерси).
Аналитические и вычислительные методы и примеры проектирования и контролирова-
ния фармацевтических систем общего управления качеством
Предисловие к русскому изданию
Настоящее издание является трудом большого коллектива авторов — специалистов
по промышленной фармации. В нем рассмотрены вопросы организации производ-
ства и системы обеспечения качества лекарственных средств. Материалы книги ре-
комендуется использовать при обучении специалистов фармацевтической отрасли
как на этапе вузовского, так и послевузовского образования.
Федеральное управление США по контролю за пищевой продукцией и лекарства-
ми (FDA) издает руководства, представляющие собой инструкции для производителя.
Правила cGMP пришли на смену старым системам контроля качества, сводив-
шимся к изъятию тех лекарств, несоответствие которых спецификациям было уже
подтверждено. Новые же Правила были призваны исключить поступление в про-
дажу некондиционных лекарственных средств. Таким образом, американских про-
изводителей обязали соблюдать Надлежащие производственные практики, раз-
работанные для предотвращения загрязнений фармацевтических продуктов, их
ненадлежащей биодоступности или эффективности.
Конгресс США аргументировал необходимость внедрения cGMP к фармацевти-
ческое производство следующим образом: «Производство лекарственной продук-
ции требует привлечения высококвалифицированного и специально обученного
персонала, наличия специализированной лаборатории и тщательного контроля
производства, упаковки и маркировки. Выполнение этих требований необходимо
для гарантирования безопасности лекарственного продукта для потребителя, а так-
же соответствия таким заявленным параметрам, как идентичность, содержание ак-
тивного компонента, качество, чистота и эффективность».
Целью cGMP является интеграция системы менеджмента качества в планиро-
вание и производство фармацевтической продукции, гарантирующая, что рецеп-
турные лекарственные средства, не соответствующие стандартам, не будут угрожать
здоровью потребителей.
Правила cGMP требуют от производителей наличия соответствующего произ-
водственного оборудования, специально обученного персонала, точного контроля
производственных процессов, надлежащего лабораторного контроля, ведения пол-
ных и точных записей и протоколов, надлежащего исследования готовой продукции
и т. д. Не претендуя на роль эталонных процедур, эти Правила скорее устанавливают
пороговые, или минимальные, стандарты, обязательные для фармацевтических про-
изводственных операций.
Внедрение процессно-аналитической технологии (А47) происходит в период
наиболее интенсивных изменений в фармацевтическом производстве, наблюдаю-
щийся последние три десятилетия. Главной движущей силой этих изменений явля-
ется инициатива FDA по внедрению современных, основанных на оценке рисков,
способов контроля и надзора за фармацевтическим производством. В книге опи-
сано историческое развитие процессной аналитики; дано общее представление о
применении РАТъ фармацевтической промышленности и бизнес-стимулах, спо-
собствующих внедрению изменений; изложена сущность новой инициативы FDA и
требований РАТ, а также представлен базовый план внедрения РАТ.
Пятая глава описывает общие требования к обучению персонала в условиях
фармацевтического производства; далее описание сосредотачивается на методах
планирования и реализации стратегии обучения, которое гарантирует соответствие
Предисловие к русскому изданию
25
фармацевтических предприятий законам, внесенным в Свод федеральных норма-
тивных актов (CFR — Code of Federal Regulations), а также Правилам надлежащей
практики обучения (GTP).
В соответствии с требованиями закона и этическими нормами, компании, про-
изводящие фармацевтические продукты, должны гарантировать эффективность и
безопасность своей продукции. Уверенность в том, что персонал, занятый в фарма-
цевтическом производстве, обладает компетенцией, необходимой для правильного
и эффективного выполнения своих обязанностей, критически важна для обеспече-
ния безопасности и эффективности производственного процесса. При обучении в
окружении, ориентированном на соблюдение правил надлежащей производствен-
ной практики, критически важно совершенствовать необходимые для работы навы-
ки, углублять знания и прививать персоналу ответственное отношение к труду.
Шестая глава рассматривает проблемы контаминации фармацевтических про-
дуктов. Несмотря на то, что источники контаминации легко можно выявить, ис-
пользуемые в этой области определения можно рассматривать с нескольких
позиций. Во-первых, контаминация фармацевтического продукта может рассма-
триваться в рамках терминов «родственные вещества» и «технологические приме-
си». При этом родственные вещества по своей структуре близки к действующему
веществу, а технологические примеси вносятся в ходе технологического процесса или
обработки. Эти два термина включают все типы загрязнений, но не содержат их
полного описания.
Фармакопея США с Национальным формуляром содержит определение терми-
нов, связанных с контаминацией продукта, в статьях примеси в фармакопейных про-
дуктах и обычные примеси.
Поскольку нормативы GMP требуют оценки параметров стабильности фарма-
цевтического лекарственного препарата, то в главе 7 приведен их краткий обзор.
Особое внимание авторы обращают на отбор проб для изучения стабильности.
Пробы, отобранные для оценки стабильности лекарственного препарата, должны
храниться в упаковке, аналогичной той, в которой препарат поступает на рынок.
Условия хранения проб, предназначенных для оценки стабильности, должны со-
ответствовать пробам, определенным в спецификации. Результаты тестирования
стабильности используют для определения надлежащих условий хранения и сроков
хранения препарата.
В восьмой главе рассмотрены вопросы валидации аналитических методик. Вали-
дация аналитической методики представляет собой доказательство того, что харак-
теристики методики соответствуют требованиям предполагаемого применения. Все
аналитические методики, предназначенные для анализа любых клинических образ-
цов, должны быть отвалидированы. Валидация аналитических методик — важная,
но трудоемкая процедура, требующая детального понимания возможностей опти-
мального использования аналитических ресурсов испытательной лаборатории.
Научный перевод глав 3, 4, 6 выполнен Алдашевой Жанной Игоревной, гла-
вы 7 — Беляевым Василием Викторовичем. Благодарю моих коллег Алдашеву Жанну
Игоревну и Пятигорскую Наталью Валерьевну за помощь в редактировании книги.
Редактор русского издания, член-корреспондент РАМН,
профессор, лауреат премии Правительства РФ в области образования
В. В. Береговых
ПРЕДИСЛОВИЕ
Настоящее практическое руководство охватывает все нормативно-правовые аспек-
ты и требования, регулирующие производство лекарственных препаратов. В 25 гла-
вах раскрывается широкий ряд вопросов: от связанных с ранними стадиями про-
изводственного процесса (когда количество и сложность используемых материалов
невысоки) до проблем воспроизводимости и непрерывного производства больших
объемов продукции высокой сложности, от задач определения состава и способа
доставки разрабатываемого лекарственного препарата до идентификации источни-
ков загрязнения и оценки стабильности.
Авторы книги попытались рассмотреть все возможные подходы, направленные
на изготовление лекарства, которое отвечало бы нормативным требованиям, соот-
ветствовало точному описанию, входило в состав высокоэффективного фармацев-
тического продукта и производилось оптимальным способом.
Благодаря усилиям Михаэля Левенталя книга из 25 глав, каждая из которых на-
писана ведущими мировыми специалистами, освещает все основные подходы к ре-
шению фундаментальных нормативно-правовых проблем, с которыми приходится
сталкиваться, добиваясь успешного производства новых лекарств в стабильных и
удобных для введения в организм формах.
Часть I
НОРМАТИВНОЕ РЕГУЛИРОВАНИЕ
ПРОИЗВОДСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
Глава 1.1. НАДЛЕЖАЩАЯ ПРОИЗВОДСТВЕННАЯ
ПРАКТИКА И ТРЕБОВАНИЯ FDA
Джеймс Р. Харрис
James Harris Associates, Inc. (Дарем, Северная Каролина)
1.1.1. Требования FDA: законы и рекомендации
Закон всегда обязателен. В США все федеральные законы собраны в своды, обе-
спечивающие максимальное удобство поиска конкретного правового документа.
Свод федеральных нормативных актов (CFR) представляет собой совокупность всех
федеральных законов, внесенных в Федеральный регистр исполнительными орга-
нами власти. Этот кодекс состоит из 50 томов, рассматривающих различные сфе-
ры федерального регулирования. Каждый том состоит из разделов1, посвященных
более узким областям. Изменения и дополнения в первую очередь публикуются
в Федеральном регистре. Для ознакомления с последними версиями законов сле-
дует воспользоваться и Кодексом законов, и Федеральным регистром. Все законы,
связанные с пищевыми продуктами и лекарственными средствами, входят в 21-й
том CFR. Каждый том CFR проходит ежегодную редакцию. Обновления 21-го тома
публикуются 1 апреля.
Поскольку эти законодательные акты обычно представляют собой лишь требо-
вания к лекарственной продукции, не подкрепленные никакими рекомендациями,
Федеральное управление США по контролю за пищевой продукцией и лекарствами
(FDA) издает руководства, представляющие собой инструкции для производителя.
Эти руководства не имеют нормативной силы. Производители не обязаны приме-
нять подходы и методики, изложенные в руководящем документе: по заверениям
представителей FDA, руководства выполняют лишь рекомендательную функцию.
Следование предписаниям, содержащимся в руководящем документе, поощряется,
но при этом не исключаются и любые другие решения. Подобная политика не пре-
пятствует внедрению инноваций. Вместе с тем производитель должен быть готов
предоставить доказательства того, что разработанные им методы позволяют достичь
желаемых результатов.
Для стимулирования генерации новых идей FDA практикует проведение про-
мышленных конференций. К последним относятся, например, семинары, спон-
сируемые FDA, а также встречи промышленных групп, таких как Ассоциация фар-
мацевтических производителей (РМА), Ассоциация парентеральных препаратов
(PDA) и Международное общество инженеров фармацевтической промышленности
(ISPE). Еще раз стбит подчеркнуть, что хотя публикуемые комментарии отражают
существующую на данный момент точку зрения FDA, они представляют собой не
более чем рекомендации.
1 В рамках данного издания принят следующий перевод рубрикации C£R: Title — том,
Chapter — раздел, Part — глава, Section — параграф, Paragraph — пункт. — Примеч. отв. ред.
30 Часть I. Нормативное регулирование производства лекарственных средств
Некоторые промышленные группы также выпускают комментарии, издают ру-
ководящие документы и т. д. Эти публикации весьма интересны и зачастую содер-
жат ценную информацию. Тем не менее важно помнить, что они не включают в себя
нормативных требований и даже не входят в сферу официальной документации. Если
фирма следует рекомендациям, есть высокая вероятность того, что она делает пра-
вильный выбор. Вместе с тем неофициальный характер таких рекомендаций требует
научного обоснования принятых на их основе решений.
1.1.2. Том 21 CFR, разделы 210 и 211: Текущая надлежащая
производственная практика для готовой
фармацевтической продукции
Разделы 210 и 211 21-го тома CFR представляют собой законы, определяющие
принципы Надлежащей производственной практики (GMR) для готовой фармацев-
тической продукции. Этим предписаниям должны следовать все производители,
намеренные продавать свою продукцию на территории США. Если фирма пода-
ет заявку на продажу продукта в США по процедуре NDA1 (New Drug Application),
ANDA(Abbreviated New Drug Application), BLA (Biologic License Application) и т. д., од-
ним из последних этапов процесса одобрения заявки является предшествующая
принятию решения инспекция производственного предприятия. Главная задача
такой инспекции — проверка исполнения предписаний GMP. Инспекция, предше-
ствующая одобрению заявки, является обязательной. Таким образом, если фирма
ожидает рассмотрения 10 заявок, то ей придется принять 10 инспекций. Тот факт,
что на производственном предприятии уже побывала инспекция, не отменяет не-
обходимости повторных проверок.
Кроме того, FDA правомочно инспектировать любые производственные пред-
приятия, изготавливающие продукцию, реализуемую в США. Эти инспекции
проводятся без предварительного уведомления. Производитель обязан принять
инспектора, когда он появляется на предприятии, и должен сделать это без необо-
снованных задержек.
Ниже обсуждаются требования GMP к производителям фармацевтических дози-
рованных форм. Не являясь точным изложением закона, представленная информа-
ция дает общее представление о его содержании, поясняя ряд конкретных норма-
тивных предписаний. Для ознакомления с первоисточником следует обратиться
к CFR, а затем свериться с Федеральным регистром на предмет возможных изме-
нений.
1.1.2.1. Общие положения
1. Этот параграф регулирует производство лекарственной продукции, предна-
значенной для человека или животных.
1 Заявка на регистрацию нового фармацевтического препарата. — Примеч. отв. ред.
2 Заявка на сокращенную процедуру регистрации нового фармацевтического препарата. —
Примеч. отв. ред.
3 Заявка на регистрацию биологического препарата. — Примеч. отв. ред.
Глава 1.1. Надлежащая производственная практика и требования FDA 31
2. Лекарства, отпускаемые без рецепта врача (англ. ОТС — over-the-counter),
и биологически активные добавки (БАД) к человеческой пище не подпадают
под действие обозначенных правил, за исключением особых случаев.
1.1.2.2. Организация и персонал
1. Функции отдела контроля качества:
1) данный отдел является непременной частью любого фармацевтического
предприятия;
2) он должен обладать полномочиями по одобрению или отклонению лекар-
ственной продукции, а также ее компонентов, контейнеров, укупорочных
приспособлений, сырья, упаковочного материала и этикеток, как и пра-
вом ознакомления с производственной документацией;
3) необходимо наличие соответствующей лабораторной базы для исследова-
ния, одобрения или отклонения вышеперечисленных материалов;
4) отдел несет ответственность за одобрение или отклонение всех процедур
или спецификаций, способных повлиять на подлинность активного веще-
ства и качество лекарственного средства, а также на содержание активного
компонента в последнем;
5) область ответственности и процедуры, применяемые для контроля каче-
ства, должны быть представлены в письменном виде.
2. Квалификация персонала:
1) каждый участник процесса производства, обработки, упаковки или хране-
ния лекарственной продукции должен быть дипломированным специали-
стом, прошедшим необходимую подготовку и обладающим определенным
опытом работы. Это требование распространяется и на выполнение кон-
кретных операций в области текущей GMP (cGMP). Тренинги по требова-
ниям GMP проводятся квалифицированными преподавателями и с регу-
лярной частотой;
2) уровень образования и профессиональной подготовки лиц, отвечающих за
контроль качества, должен гарантировать безопасность активного компо-
нента, его подлинность и надлежащее содержание, а также необходимые
качество и эффективность лекарственного средства;
3) количество квалифицированного персонала определяется спецификой
решаемых задач.
3. Ответственность персонала:
1) работники обязаны носить чистую технологическую одежду, соответству-
ющую исполняемым ими обязанностям. При необходимости следует но-
сить защитную одежду;
2) соблюдение санитарных норм и правил личной гигиены строго обязательно;
3) право входа в зоны ограниченного доступа предоставляется исключитель-
но службой контроля;
4) все работники с явными признаками заболеваний или открытыми по-
вреждениями кожи отстраняются от прямого контакта с лекарственной
продукцией, ее компонентами либо контейнерами.
32
Часть I. Нормативное регулирование производства лекарственных средств
4. Консультанты, дающие рекомендации по производству, обработке, упа-
ковке и хранению лекарственной продукции, должны обладать доста-
точной компетенцией в своей профессиональной области, что опять же
подразумевает наличие соответствующего диплома и определенный опыт
работы. Производитель обязан вести записи, где фиксирует имя, адрес
и квалификацию каждого консультанта, а также тип оказываемой послед-
ним услуги.
1.1.2.3. Помещения и оборудование
1. Требования к планировке:
1) размеры производственных помещений и планировка здания должны обе-
спечить возможность проведения очистки, технического обслуживания
и прочих необходимых операций;
2) пространство помещения должно способствовать упорядоченному рас-
положению оборудования и материалов, предполагающему предотвраще-
ние смешивания различных компонентов, контейнеров и упаковок для
лекарственной продукции, этикеток, перерабатываемых материалов или
готовой лекарственной продукции и т. п.;
3) необходимо предусмотреть оптимальный маршрут передвижения компо-
нентов лекарственной продукции или ее самой внутри здания, исключаю-
щий вероятность загрязнения перемещаемых объектов;
4) операции следует проводить в специально отведенных зонах, оборудован-
ных адекватными системами контроля для предотвращения загрязнения
или внесения примесей во время таких процедур, как:
а) получение, идентификация, хранение и размещение компонентов, не
допущенных в производство до утверждения отделом контроля каче-
ства, контейнеров для лекарственной продукции, упаковки, укупороч-
ных систем, а также маркировка, хранение соответствующих выборок
аналитических проб, анализ и отправка материалов в производство или
упаковка;
б) хранение не допущенных в производство материалов, которые пере-
числены в предыдущем абзаце;
в) хранение отпускаемых компонентов, контейнеров для лекарственной
продукции, а также маркировка;
г) хранение материалов в процессе технологической обработки;
д) операции по изготовлению и обработке продукции;
е) операции упаковки и маркировки;
ж) карантинное хранение лекарственной продукции перед ее отправкой;
з) хранение готовой лекарственной продукции;
и) лабораторные и контрольные операции;
к) асептическая обработка, включающая:
—гарантирование гладкой и твердой поверхности полов, стен и потол-
ков, легко поддающейся очистке;
Глава 1.1. Надлежащая производственная практика и требования FDA 33
—контроль температуры и влажности;
—подачу воздуха, профильтрованного через высокоэффективные воз-
душные фильтры нагнетательной системы, осуществляемую как для
ламинарного, так и для турбулентного потоков;
—с истему мониторинга условий окружающей среды;
—с истему очистки и дезинфекции помещения и наличие оборудова-
ния, обеспечивающего асептические условия;
—с истему технического обслуживания оборудования, применяемого
для контроля асептических условий;
5) операции, относящиеся к изготовлению, обработке и упаковке антибио-
тиков, должны проводиться на отдельном оборудовании, не применяемом
для изготовления других лекарственных средств, предназначенных для че-
ловека1.
2. Во всех зонах необходимо обеспечить адекватное освещение.
3. Отопление, вентиляция и кондиционирование воздуха:
1) во всех зонах необходимо обеспечить адекватную вентиляцию;
2) при изготовлении, обработке, упаковке или хранении лекарственной про-
дукции там, где это целесообразно, следует установить оборудование для
адекватного контроля давления воздуха, количества микроорганизмов,
пыли, влажности и температуры;
3) в некоторых случаях следует проводить фильтрацию воздуха, подаваемого
в рабочую зону: это исключит вероятность загрязнения, в том числе пере-
крестного;
4) системы очистки воздуха при изготовлении, обработке и упаковке анти-
биотиков следует полностью отделить от аналогичных систем для другой
лекарственной продукции, предназначенной д ля человека.
4. Водопроводно-канализационная сеть:
1) питьевая вода должна поступать непрерывно через нагнетательную систе-
му, в которой отсутствуют дефекты, способные привести к загрязнению
лекарственной продукции;
2) питьевая вода должна соответствовать стандартам, предписанным Основ-
ными требованиями к качеству питьевой воды Федерального управления
США по охране окружающей среды (ЕРА), изложенными в разделе 141
тома 40 CFJR;
3) у дренажного отверстия должен быть адекватный размер. Если система
стока соединена непосредственно с канализационной системой, необхо-
дим воздушный зазор или другое приемлемое механическое устройство
для предотвращения обратного засасывания.
5. Сточные воды, мусор и прочие отходы, находящиеся внутри здания, а так-
же вынесенные из него или из близлежащих зданий, подлежат утилизации
безопасным и гигиеничным способом.
1В контексте требований GMP FDA признаёт цефалоспорины антибиотиками.
34 Часть I. Нормативное регулирование производства лекарственных средств
6. Следует обеспечить наличие всего необходимого для умывания, в том числе
горячей и холодной воды, мыла или моющего средства, фенов-сушилок или
одноразовых полотенец, а также чистых туалетов, доступных во всех рабочих
зонах.
7. Санитарное состояние:
1) все помещения, используемые для изготовления, обработки или хранения
лекарственной продукции, требуется содержать в чистоте. Там не должны
обитать грызуны, птицы, насекомые и другие животные-вредители;
2) мусор и органические отходы следует хранить и своевременно утилизиро-
вать с соблюдением санитарных условий;
3) график уборки должен включать описание оборудования и материалов,
применяемых для уборки помещений и очистки оборудования, с указани-
ем ответственности за санитарное состояние;
4) необходимо наличие письменных инструкций, описывающих примене-
ние соответствующих родентицидов, инсектицидов, фунгицидов, фуми-
гантов, очищающих и дезинфицирующих реагентов; эти инструкции не-
обходимо соблюдать. Процедуры должны быть спланированы так, чтобы
предотвратить загрязнение оборудования, компонентов, контейнеров для
продукции, укупорочных устройств, упаковочных материалов, этикеток
или лекарственной продукции. Запрещено пользоваться незарегистриро-
ванными агентами, а также теми, способ применения которых отличен от
изложенного в Федеральном законе об инсектицидах, фунгицидах и ро-
дентицидах (7 ГОС 135');
5) все санитарные процедуры в равной степени обязательны для выполнения
как подрядчиками и временными работниками, так и штатными служа-
щими.
8. Все помещения, подпадающие под регламентацию GMP, должны быть хоро-
шо отремонтированы.
1.1.2.4. Оборудование
1. Конструкция оборудования, его размеры и расположение должны соответ-
ствовать его целевому назначению, а также обеспечивать удобство операций
по его очистке и техническому обслуживанию.
2. Конструкция оборудования:
1) его поверхности, соприкасающиеся с компонентами, перерабатываемыми
материалами или фармацевтическими препаратами, не должны обладать
реакционной способностью, аддитивными или поглощающими свойства-
ми, способными изменить параметры безопасности, подлинности, содер-
жания активного компонента, качества или чистоты лекарственного сред-
ства, установленные в официальных или других утвержденных требованиях;
1 Рубрикация USCта же, что и CFR, за тем исключением, что «параграфы» могут перево-
диться как «статьи». Приведенная здесь сокращенная запись, даваемая согласно американ-
ской традиции, означает 135-й параграф (статью) 7-го тома. — Примеч. отв. ред.
Глава 1.1. Надлежащая производственная практика и требования FDA
35
2) все вещества, применяемые в технологических процессах, например сма-
зочные материалы или хладагенты, не должны соприкасаться с лекар-
ственными средствами, контейнерами и т. д., поскольку это может из-
менить такие параметры фармацевтического препарата, как его безопас-
ность, подлинность, эффективность, качество и чистота, закрепленные
в официальных или других утвержденных требованиях.
3. Очистка и техническое обслуживание оборудования:
1) следует регулярно проводить очистку, техническое обслуживание и сани-
тарную обработку оборудования и вспомогательных приспособлений для
предотвращения сбоев в работе или возникновения загрязнений, способ-
ных изменить характеристики лекарственного средства, установленные
в утвержденных требованиях;
2) для очистки и технического обслуживания оборудования и вспомогатель-
ных приспособлений, используемых при производстве фармацевтическо-
го препарата, необходимо наличие письменных инструкций. Эти инструк-
ции содержат, помимо прочего, следующую информацию:
а) указание лица, ответственного за очистку и техническое обслуживание
оборудования;
б) графики очистки и технического обслуживания, включая график сани-
тарной обработки там, где это необходимо;
в) подробное описание методов, оборудования и материалов, применяе-
мых в рамках операций очистки и технического обслуживания, а также
методики разборки и сборки оборудования в процессе очистки и тех-
нического обслуживания;
г) удаление следов продуктов предыдущей серии;
д) защита чистого оборудования от загрязнения перед применением;
е) проверкачистотыоборудованиянепосредственнопередиспользованием;
ж) ведение записей по техническому обслуживанию, очистке, санитарной
обработке и проверкам всего технологического оборудования.
4. Автоматическое, механическое и электронное оборудование:
1) всё оборудование такого типа, включая компьютеры или связанные
с ними системы, применяемые в регулируемых GMP процессах, необходи-
мо регулярно калибровать, инспектировать или проверять в соответствии
с особой программой, предназначенной обеспечивать соответствующие
эксплуатационные характеристики. Любые из перечисленных действий
должны быть письменно зафиксированы;
2) необходимо следить за тем, чтобы изменения в основных производствен-
ных и контрольных записях (или аналогичных записях) производились
исключительно авторизованным персоналом. Нужно проверять точность
данных, вводимых в систему, а также данных, выводимых из последней;
3) следует создавать резервную копию файла данных, вводимых в компью-
теризованную систему, за исключением случаев, когда определенные дан-
ные, такие как результаты расчетов, связанных с лабораторным анализом,
36 Часть I. Нормативное регулирование производства лекарственных средств
уничтожаются в ходе вычислительного или иного автоматизированного
процесса. В этом случае записи программы необходимо вести вместе с за-
писями данных по валидации.
5. Волокна из фильтров, применяемых для жидкостной фильтрации в процессе
изготовления, обработки или упаковки составляющих медицинских инъек-
ций, не должны попадать в производимую продукцию. Применение подоб-
ных выделяющих волбкна фильтров ограничено лишь случаями, когда невоз-
можно изготовить лекарство без их участия. В этой ситуации после фильтра-
ции через фильтр, выделяющий волокна, проводят дополнительную фильтра-
цию через не выделяющий волокна фильтр с максимальным размером пор
0,22 мкм. Применение асбестосодержащих фильтров допустимо лишь при
наличии заключения соответствующего бюро FDA о том, что использование
фильтров, не выделяющих волокна, может нарушить безопасность и эффек-
тивность лекарственного средства.
1.1.2.5. Контроль компонентов фармацевтической продукции,
ее контейнеров, а также используемых укупорочных приспособлений
1. Общие требования:
1) необходимы письменные инструкции и методики, достаточно подробно
описывающие получение, идентификацию, хранение, пробоотбор, тести-
рование, подтверждение качества или отбраковку компонентов* продук-
ции, контейнеров и укупорочных приспособлений. (Разумеется, все эти
процедуры строго обязательны. К сожалению, далеко не все организации
следует своим же письменным инструкциям и методикам);
2) хранение и обработка всех вышеперечисленных компонентов должны
предотвращать загрязнение;
3) компоненты, поставляемые в мешках или коробках, следует хранить на
полу. Их размещение не должно препятствовать уборке помещения или
инспекции;
4) каждый контейнер, содержащий компоненты, необходимо маркировать
специальным кодом или номером подсерии, соответствующими серии
данного препарата. Даже если следующая серия имеет тот же номер подсе-
рии поставщика, производитель обязан присвоить ей новый идентифика-
ционный номер. Маркировка подсерий должна соответствовать их статусу
(карантинный, утвержденный или отбракованный).
2. Получение и хранение непроверенных компонентов:
1) получаемые контейнеры с компонентами подвергаются обязательному
визуальному осмотру, включающему проверку правильности маркировки,
отсутствия повреждений и загрязнения каждого контейнера;
2) перед тем как компоненты пройдут контроль качества и будут допущены
к использованию, они должны находиться на карантинном хранении.
1В контексте данного обсуждения термин «компоненты» относится к ингредиентам про-
дуктов, контейнерам, крышкам и т. д.
Глава 1.1. Надлежащая производственная практика и требования FDA
37
3. Проверка компонентов, их апробация или отбраковка:
1) из каждой подсерии компонентов необходимо отобрать пробы и провести
анализ; использование компонентов в производстве допустимо лишь по-
сле допуска отдела контроля качества;
2) при каждом получении партии компонентов необходимо проводить отбор
репрезентативных проб. Число проб или количество компонента определя-
ются внешним видом последнего, уровнем статистической достоверности,
историей взаимоотношений с поставщиком, требуемым для анализа количе-
ством компонента, а также числом резервных проб там, где это необходимо;
3) процедура отбора проб:
а) грязные контейнеры, содержащие компоненты, нужно подвергнуть
очистке;
б) контейнеры следует открыть, отобрать пробы, затем вновь закрыть так,
чтобы предотвратить загрязнение проб и содержимого;
в) там, где это необходимо, используются стерильные приспособления
и асептические методы отбора проб;
г) если пробы отбирают из разных частей контейнера, не стоит объеди-
нять их в единую пробу для анализа;
д) контейнеры, из которых были отобраны пробы, маркируются с целью
обозначить, что пробы были взяты;
4) осмотр и анализ проб:
а) для всех партий компонентов проводится как минимум один анализ на
подлинность;
б) каждый компонент проверяется на соответствие всем письменным
спецификациям по чистоте и содержанию активного компонента; для
ингредиентов обязательно соответствие качеству, а для контейнеров
и крышек — спецификациям;
в) упомянутая проверка производителем (покупателем) может быть заме-
нена анализом, выполняемым поставщиком, если по меньшей мере один
специфический анализ компонента на идентичность проводил произ-
водитель (покупатель) или если последний подтвердил достоверность
анализа, сделанного поставщиком, в ходе валидационной процедуры;
г) при необходимости компоненты осматривают под микроскопом;
д) каждую подсерию компонента, подверженную загрязнению землей,
инфестации насекомыми или другими внешними примесями, следует
проверить на соответствие установленным спецификациям по указан-
ным видам загрязнений;
е) каждую подсерию компонента, целевое использование которого может
быть затруднено микробиологическим загрязнением, следует подвер-
гать предварительному микробиологическому исследованию;
5) Если подсерия компонента отвечает письменным спецификациям, можно
разрешить ее использование. Все подсерии, качество которых не отвечает
письменным спецификациям, следует отбраковывать.
38 Часть I. Нормативное регулирование производства лекарственных средств
4. Утвержденные компоненты (включая контейнеры для лекарственной про-
дукции и крышки) следует использовать в порядке их утверждения (принцип
«первый пришел, первый ушел»).
5. Длительно хранимые компоненты, а также испытавшие воздействие атмос-
ферного воздуха, нагревания или иных факторов, способных повлиять на их
характеристики, необходимо подвергнуть повторному анализу и/или осмотру.
6. Отбракованные компоненты идентифицируют и хранят в специальной ка-
рантинной системе, исключающей их участие в процессе производства или
переработки.
7. Контейнеры и упаковка:
1) контейнеры и упаковка не должны обладать реакционной способностью,
аддитивными или поглощающими свойствами, ведущими к несоответ-
ствию характеристик лекарственного средства официально установлен-
ным требованиям;
2) системы укупоривания контейнера обязаны обеспечивать адекватную за-
щиту от воздействия внешних факторов, чреватых разложением или за-
грязнением лекарства;
3) контейнеры и укупорочные приспособления должны быть чистыми, а при
необходимости — стерильными и прошедшими процедуру удаления пиро-
генов;
4) обязательны наличие письменных стандартов или спецификаций, а также
описание методик анализа и, при необходимости, процедур стерилизации
и депирогенизации, которые следует строго соблюдать.
1.1.2.6. Технический контроль и управление производственным процессом
1. Письменные инструкции методики анализа и отклонения от них:
I) требуется наличие письменных инструкций и методик анализов техниче-
ского контроля и управления производственным процессом; все эти пред-
писания необходимо соблюдать. Разработка этих документов направлена
на обеспечение заявленной подлинности, содержания активного компо-
нента, качества и чистоты лекарственного средства. Данные процедуры
должны соответствовать всем представленным ниже требованиям и со-
ставляются, анализируются и утверждаются заинтересованными произ-
водственными подразделениями; итоговый анализ и утверждение прово-
дятся отделом контроля качества;
2) выполнение обозначенных выше инструкций обязательно фиксируется на
бумаге. Любые отклонения от нормативных процедур должны быть описа-
ны и обоснованы.
2. Регламентация производственного процесса и контрольных процедур, разра-
ботанная для обеспечения соответствия фармацевтической продукции всем
стандартам и спецификациям, должна включать следующие пункты:
1) серия препарата составляется так, чтобы количество активного ингреди-
ента не уступало указанному на этикетке;
Глава 1.1. Надлежащая производственная практика и требования FDA 39
2) взвешивание, дозирование и разделение используемых компонентов про-
исходит согласно установленным правилам. Если компонент извлекают
из одного контейнера и помещают в другой, на последний наклеивают
этикетку со следующей информацией:
а) наименование компонента и/или код продукта;
б) приемочный или контрольный номер;
в) вес или количество материала в новом контейнере;
г) кодовое число для производственной серии или подсерии, из которых
был извлечен компонент, включающее наименование продукта, содер-
жание активного вещества и номер подсерии;
3) за операциями взвешивания, отмеривания или разделения компонентов
необходимо осуществлять надзор. Каждый контейнер с компонентами,
поступающий в производство, осматривается другим лицом, подтверж-
дающим:
а) факт того, что компонент был одобрен отделом контроля качества;
б) соответствие веса или количества компонента данным протокола, со-
провождающего производственную серию;
в) правильность маркировки контейнеров (в частности, соответствие ко-
личества содержащегося внутри продукта данным этикетки);
4) добавление каждого компонента, проводимое одним лицом, обязательно
проверяется другим работником.
3. Реальный выход и процент от теоретически возможного выхода следует опре-
делять по завершении соответствующей фазы процесса производства, обра-
ботки, упаковки или хранения. Эти расчеты, производимые одним лицом,
должны быть подтверждены другим специалистом.
4. Идентификация оборудования:
1) все контейнеры для хранения и смешивания компонентов, а также тех-
нологические линии и основное оборудование, используемое в процессе
производства лекарственных средств, непрерывно маркируются для опре-
деления содержимого и/или фазы производственного процесса;
2) основное оборудование маркируется исходя из его спецификации. Если
на предприятии используется только один вид оборудования, маркировка
может ограничиться лишь наименованием последнего.
5. Отбор проб и анализ материалов в ходе технологического процесса:
1) для подтверждения однородности и целостности серии необходимо следо-
вать письменным инструкциям и методикам анализа, регламентирующим
контроль технологического процесса, а также процедуры анализа или
осмотра проб материалов. Протекание этих процедур фиксируется в спе-
циальных протоколах. Это необходимо для мониторинга результата и ва-
лидации тех производственных процессов, что могут быть ответственны
за вариабельность характеристик изготавливаемой продукции. Объектами
контрольных процедур являются:
а) варьирование веса таблетки или капсулы;
40 Часть I. Нормативное регулирование производства лекарственных средств
б) время разложения;
в) адекватное смешивание или измельчение, обеспечивающее однород-
ность и гомогенность;
г) время и скорость растворения;
д) чистота растворов;
е) pH растворов и др.;
2) параметры технологического процесса должны обеспечивать итоговые
спецификации фармацевтических препаратов для всех характеристик
и разрабатываются исходя из средних данных по предыдущим процессам,
в ходе которых был произведен приемлемый продукт, а также данных по
изменчивости процесса;
3) обрабатываемые в ходе технологического процесса материалы следует
проверять на подлинность, содержание действующего вещества, качество
и чистоту. Они апробируются или бракуются отделом контроля качества,
после чего производственный процесс возобновляется;
4) отбракованные материалы необходимо идентифицировать и хранить в ка-
рантинной системе, предназначенной для предотвращения их использо-
вания в производственных операциях, для которых они признаны непри-
годными.
6. Каждая фаза производственного процесса может быть ограничена опреде-
ленным сроком, изменение которого допускается, только если не приводит
к ухудшению качества продукта. Всякий выход за пределы предписанного
срока должен быть документирован и обоснован.
7. Контроль микробиологического загрязнения:
1) для предотвращения роста нежелательных микроорганизмов в не требую-
щей стерильности продукции следует разработать надлежащие письмен-
ные инструкции;
2) если стерилизация является частью описанного выше процесса, то по-
следний подлежит валидации.
8. Переработка:
1) необходимо составить письменные инструкции, описывающие системы,
применяемые для переработки серий, не соответствующих установлен-
ным стандартам;
2) не следует проводить переработку до анализа и одобрения отделом кон-
троля качества.
1.1.2.7. Контроль упаковки и маркировки
1. Осмотр материалов и критерии возможности их использования:
1) необходимо разработать и утвердить письменные инструкции, подробно
описывающие получение, идентификацию, хранение, обработку, пробо-
отбор, осмотр и/или анализ материалов, применяемых для маркировки
и упаковки, и следовать этим инструкциям;
2) все материалы, не вполне отвечающие критериям приемки, следует отбра-
ковывать;
Глава 1.1. Надлежащая производственная практика и требования FDA
41
3) при получении каждой отдельной серии материала для упаковки и этике-
ток необходимо делать запись, удостоверяющую факт получения материа-
ла, констатирующую результаты осмотра (или анализа) последнего и со-
держащую вывод о приемке или отбраковке;
4) этикетки и маркировочные материалы, указывающие наименование для
каждого отдельного лекарственного средства, содержание активного ком-
понента, форму дозирования или количество содержимого, следует хра-
нить отдельно, снабдив их надлежащей идентификационной информаци-
ей. Доступ в зону хранения должен быть ограничен;
5) просроченные и испорченные этикетки, материалы для этикеток и упа-
ковки подлежат карантинному хранению и последующему уничтожению;
6) запрещается использование этикеток, отпечатанных на дублирующем пе-
чатающем устройстве (этикетки в рулоне), этикеток для разных лекарств
или фармацевтических препаратов, содержащих неодинаковое количество
активного компонента, а также для препаратов, отличающихся весом нет-
то. Единственное исключение из этого правила представляет собой случай,
когда этикетки, отпечатанные на дублирующем устройстве, различаются
по размеру, форме или цвету, что гарантирует недопущение их смешивания;
7) при использовании разрезанных этикеток инструкции упаковки и марки-
ровки должны включать одну или несколько следующих процедур контроля:
а) выделение специальной маркировочной и упаковочной линии для
каждого лекарственного средства с конкретным содержанием актив-
ного компонента;
б) применение соответствующего электронного или электромеханиче-
ского оборудования для проведения сплошной проверки правильно-
сти маркировки во время конечной операции или после завершения
последней;
в) визуальный контроль при проведении сплошной проверки правиль-
ности маркировки. В рамках визуального контроля проверку прово-
дит один сотрудник, а второй независимо подтверждает полученные
результаты;
8) чтобы гарантировать тождественность маркировки той информации, что
указана в протоколе на производственную серию лекарственного сред-
ства, следует вести мониторинг работы печатающих устройств, установ-
ленных на производственной линии или связанных с ней, применяемых
для нанесения маркировки на единицу фармацевтического препарата или
единичную упаковку.
2. Изготовление этикеток:
1) за изготовлением этикеток для маркировки лекарственной продукции
осуществляется строгий контроль;
2) этикетки, предназначенные для маркировки произведенной серии, следу-
ет тщательно проверять на предмет соответствия маркировке, указанной
в сопутствующем протоколе;
42
Часть I. Нормативное регулирование производства лекарственных средств
3) необходимо наличие письменных инструкций, регламентирующих учет
количества изготовленных, использованных, испорченных и возвращен-
ных этикеток. Процедура должна включать выявление причин расхожде-
ний между количеством готовых упаковок лекарственной продукции
и числом изготовленных этикеток, если расхождение превышает заранее
ограниченные пределы. Последние устанавливают на основании ранее по-
лученных технико-эксплуатационных данных. Выявления расхождений не
требуется при использовании этикеток в рулонах, разрезанных этикеток,
а также при проведении сплошной проверки правильности маркировки;
4) излишек этикеток с номером серии или контрольным номером подлежит
уничтожению;
5) возвращенные этикетки следует хранить так, чтобы предотвратить вероят-
ность их смешивания.
3. Требования к этикеткам, маркировочным и упаковочным материалам содер-
жатся в специальных инструкциях, направленных:
1) на предотвращение смешивания и перекрестного загрязнения, достигае-
мое физическим или пространственным разделением операций с разными
лекарственными средствами;
2) идентификацию заполненных контейнеров с лекарствами, хранящихся
без этикеток в ожидании маркировки. Подобные инструкции гарантируют
правильную маркировку на индивидуальных упаковках, сериях или под-
сериях фармацевтических препаратов. Не требуя обязательной идентифи-
кационной маркировки на каждой индивидуальной упаковке, инструкция
должна обеспечить беспроблемное определение наименования продукта,
содержания и количества активного вещества, номера серии или кон-
трольного номера на каждом контейнере;
3) идентификацию лекарственного средства по номеру серии или контроль-
ному номеру, раскрывающим историю производства данной серии и про-
хождения контроля последней;
4) проверку материалов для упаковки и маркировки на пригодность к уча-
стию в производственных и упаковочных операциях. Эти проверки долж-
ны быть зафиксированы в протоколе к производственной серии;
5) проверку упаковочного и маркировочного оборудования непосредственно
перед использованием, проводимую в целях контроля удаления всех ле-
карственных средств и маркировочных материалов, оставшихся от преды-
дущих операций. Результаты проверки следует зафиксировать в протоколе
на производственную серию лекарственного средства.
4. Требования к упаковке с индикацией признаков вскрытия для лекарственных
средств, предназначенных для человека и отпускаемых без рецепта врача:
1) продукт, отпускаемый без рецепта врача (за исключением дерматологи-
ческих препаратов, средств по уходу за зубами, инсулина или таблеток),
предназначенный для розничной продажи, признается фальсифициро-
ванным и/или неверно маркированным, если его упаковка не содержит
индикации признаков вскрытия;
Глава 1.1. Надлежащая производственная практика и требования FDA
43
2) требования к упаковке с индикацией признаков вскрытия:
а) все фармацевтические препараты (с вышеперечисленными исключе-
ниями), доступ к которым открыт покупателям (к примеру, выстав-
ленные для продажи на витринах аптек), содержатся исключительно
в упаковке с контролем вскрытия. Последняя должна обладать одним
или несколькими индикаторами либо барьерами, отсутствие или по-
вреждение которых явно свидетельствует о вскрытии упаковки. Упа-
ковка с контролем вскрытия может включать первичный контейнер
и систему укупорки, вторичный контейнер (как правило, картонную
упаковку) или комбинацию вышеперечисленных систем, предназна-
ченных для визуальной индикации целостности упаковки. Индикатор
целостности одноразовой упаковки должен оставаться неповрежден-
ным при разумном обращении с продуктом в процессе его производ-
ства, распределения (продажи) или демонстрации;
б) вышеописанное требование к обязательной индикации целостности
упаковки относится к капсулам из твердого желатина, состоящим из
двух частей; они должны быть изготовлены с применением соответ-
ствующей технологии, обеспечивающей защиту от вскрытия;
3) маркировка:
а) чтобы привлечь внимание потребителей к специфическим признакам,
указывающим на целостность упаковки, каждая упаковка фармацев-
тического препарата, отпускаемого без рецепта врача и подпадающего
под данное требование, должна нести информацию, которая:
— указывает на все признаки целостности упаковки и соблюдения тех-
нологии запечатывания капсул;
— хорошо заметна на упаковке;
— размещена на упаковке так, что остается неповрежденной, если ин-
дикатор нарушения целостности отсутствует или поврежден;
б) если на выбранном индикаторе признаков вскрытия указана иденти-
фикационная характеристика, то последняя должна быть отражена
в маркировке. Например, маркировка на бутылке, запечатанной стя-
гивающей лентой, может включать в себя надпись «Для вашей безопас-
ности бутылка запечатана с контролем первого вскрытия»;
4) производитель или упаковщик могут обратиться с просьбой об освобожде-
нии от выполнения требования о наличии упаковки с индикацией призна-
ков вскрытия. Просьбу следует представить в виде заявления; на конверте
должно быть указано «Просьба об освобождении от выполнения правила
об упаковке с индикацией признаков вскрытия». Просьба должна содер-
жать следующую информацию:
а) наименование лекарственного средства (или, если просьба относится
к классу лекарств, наименование конкретного класса и список входя-
щих в последний фармацевтических продуктов);
б) причины, по которым для данного лекарственного средства не являет-
ся необходимым или не может быть осуществлено требование о нали-
44
Часть I. Нормативное регулирование производства лекарственных средств
чии упаковки с индикацией признаков вскрытия или соответствующей
маркировки;
в) описание возможных или уже предпринятых подающей ходатайство
организацией альтернативных шагов, направленных на уменьшение
вероятности того, что лекарственное средство или класс лекарств ока-
жутся объектом намеренной фальсификации;
г) другую информацию, обосновывающую освобождение;
5) владельцы утвержденных заявок на производство новых лекарственных
средств, отпускаемых без рецепта врача, должны оповещать FDA об из-
менениях в упаковке и маркировке, осуществляемых для достижения
соответствия требованиям данного раздела. Эти модификации упаковки
и маркировки можно проводить до одобрения FDA. Изменения в техно-
логии производства запечатанных капсул требуют предварительного одо-
брения FDA;
6) данный раздел не затрагивает требований к «специальной упаковке», ко-
торые регламентирует Закон о безопасной упаковке ядовитых веществ
(Poison Prevention Packaging Act) 1970 г.
5. Инспекция фармацевтической продукции:
1) при проведении заключительных операций упакованные и маркирован-
ные лекарственные средства необходимо осматривать для проверки пра-
вильности маркировки контейнеров и упаковок;
2) по окончании заключительной операции репрезентативные пробы единиц
продукта отбираются, после чего проводится их визуальный контроль для
проверки правильности маркировки;
3) результаты проверок заносят в досье к производственной серии.
6. Данные о сроке годности лекарственного средства:
1) на всех упакованных лекарствах указывается срок годности, устанавливае-
мый соответствующим анализом их стабильности;
2) проставляемый срок годности должен соответствовать рекомендуемым
условиям хранения, определяемым в ходе исследования стабильности
продукта и указываемым на этикетке;
3) если лекарственное средство подлежит отпуску в восстановленной форме,
на этикетке должна присутствовать информация о сроке годности для вос-
становленной и не восстановленной форм;
4) дата истечения срока годности, указываемая на маркировке, должна соот-
ветствовать требованиям, изложенным в этом разделе;
5) требования настоящего раздела не распространяются на гомеопатические
лекарственные средства, а также аллергенные экстракты с маркировкой
No U. S. Standard of Potency,
6) новые фармацевтические препараты, предназначенные для клинических
исследований, не подлежат регулированию со стороны FDA при условии,
что они отвечают соответствующим стандартам или спецификациям по
результатам исследования стабильности в рамках клинических исследо-
Глава 1.1. Надлежащая производственная практика и требования FDA
45
ваний. Если новые лекарственные средства, предназначенные для иссле-
дований, подлежат отпуску в восстановленной форме, то их маркировка
должна содержать информацию о сроке годности восстановленного ле-
карственного средства;
7) согласно правилам рассмотрения ходатайства об освобождении от дей-
ствия норматива, опубликованным в Федеральном регистре 29 сентября
1978 г., требования данного раздела не распространяются на лекарствен-
ные средства, предназначенные для человека, если их маркировка не
указывает пределов дозирования и они сохраняют стабильность на про-
тяжении по меньшей мере трех лет, что подтверждено соответствующими
исследованиями.
1.1.2.8. Хранение и распределение
1. Процедуры складирования:
1) необходимо наличие письменных инструкций, описывающих хранение
лекарственных препаратов на складе. Эти инструкции, в частности, пред-
писывают:
а) карантинное хранение лекарственных средств до получения разреше-
ния отдела контроля качества;
б) особый режим хранения лекарственных средств, предполагающий
определенные показатели температуры, влажности и освещения.
2. Распределение:
1) необходима четкая регламентация отпуска фармацевтической продукции
со склада, предусматривающая:
а) принцип очередности, предполагающий отпуск той производственной
серии, что была одобрена прежде всего. Нарушения данного принципа
возможны лишь в отдельных случаях и при наличии достаточных осно-
ваний;
б) систему документирования распределения, составленную так, чтобы
можно было легко определить серию (или часть серии) лекарственного
средства, которую следует отозвать, если возникнет такая необходи-
мость.
1.1.2.9 Лабораторный контроль
1. Общие положения:
1) утверждение любых спецификаций, стандартов, планов отбора проб, ин-
струкций проведения испытаний или других механизмов лабораторного
контроля, описанных в данном разделе документа, в том числе любых из-
менений в вышеперечисленных процедурах, должны быть затребованы
соответствующим отделом организации, проанализированы и утвержде-
ны отделом контроля качества. Все действия документируются во время
их осуществления, а любые отклонения от предписанного инструкциями
порядка фиксируются и подлежат обоснованию;
46
Часть I. Нормативное регулирование производства лекарственных средств
2) лабораторный контроль подразумевает утверждение надлежащих научно
обоснованных спецификаций, стандартов, планов отбора проб и методов
проведения испытаний, обеспечивающих соответствие всех материалов
определенным стандартам по идентичности, количеству активного веще-
ства, качеству и чистоте. Лабораторный контроль включает в себя:
а) оценку соответствия письменным спецификациям для приемки каж-
дой серии в каждой партии сырья. Спецификации обязательно содер-
жат описание отбора проб и процедур проведения испытаний. Пробы
должны быть репрезентативными и правильно маркированными. Эти
процедуры могут потребовать повторного исследования любого мате-
риала, подверженного разложению;
б) оценку соответствия письменным спецификациям, инструкцию отбо-
ра проб и исследования перерабатываемых материалов;
в) калибровку инструментов, аппаратуры, измерительных приборов и за-
писывающих устройств через определенные временное интервалы со-
гласно утвержденной письменной программе, содержащей специфиче-
ские указания, графики, пределы точности и прецизионности, а также
рекомендации по устранению неисправностей. Устройства, не соответ-
ствующие утвержденным спецификациям, использовать нельзя.
2. Контроль и отпуск для реализации:
1) для установления соответствия каждой подсерии лекарственного средства
итоговым спецификациям проводится лабораторное исследование, под-
разумевающее проверку подлинности всех активных ингредиентов и их
содержания. Там, где необходима стерильность и/или исследование на на-
личие пирогенов в короткоживущих радиофармацевтических препаратах,
серии можно отпускать до окончания данного анализа при условии, что
анализ будет завершен в максимально короткие сроки;
2) каждая серия продукта, который не должен содержать нежелательных ми-
кроорганизмов, следует подвергать особому исследованию;
3) все планы отбора и анализа проб отражаются в письменных инструкциях, со-
держащих методики пробоотбора и число исследуемых единиц продукции;
4) критерии выборки и приемки при отборе проб и их анализе, проводимы-
ми отделом контроля качества, должны способствовать оптимальному
соответствию исследуемой серии всем спецификациям. Статистические
критерии для контроля качества включают в себя уровни приемки и/или
отбраковки;
5) точность, чувствительность, специфичность и воспроизводимость приме-
няемых методик анализа должны быть установлены и документированы.
Валидация и документация проводятся согласно данному нормативу;
6) лекарственные средства, не отвечающие установленным стандартам или
спецификациям, а также любым соответствующим критериям качества,
следует отбраковывать. Тем не менее, перед приемкой и использованием
может быть проведена переработка исходного материала, который также
Глава 1.1. Надлежащая производственная практика и требования FDA 47
должен отвечать всем стандартам, спецификациям и другим соответству-
ющим критериям.
3. Исследование стабильности:
1) необходимо наличие письменной программы исследований, разработан-
ной для оценки параметров стабильности каждого лекарственного сред-
ства. Исходя из результатов этих исследований определяются надлежащие
условия хранения фармацевтических препаратов и сроки их годности.
Письменная программа должна содержать следующую информацию:
а) размер пробы и интервалы тестирования, основанные на статистиче-
ских критериях, для каждой исследуемой характеристики;
б) условия хранения проб перед проведением анализа;
в) надежные, обоснованные и специфичные методики анализа;
г) результаты анализа продукта в той же системе контейнер — крышка,
что поставляется на рынок;
д) результаты анализа лекарственных средств, подлежащих восстановле-
нию во время отпуска, до и после процедуры восстановления;
2) для установления срока годности исследуют определенное количество се-
рий каждого лекарственного продукта, занося все результаты на бумагу.
Для предварительной оценки срока годности можно проводить экспресс-
исследования, подкрепленные базовой информацией о стабильности
компонентов и лекарственного продукта в системе контейнер — крышка;
предполагаемый срок годности близок к тому, что определяется в ходе ис-
пытаний годности продукта при хранении. Вместе с тем необходимы ис-
следования стабильности, включающие анализ продукта через определен-
ные временное интервалы и проводимые вплоть до подтверждения пред-
полагаемого срока годности;
3) к гомеопатическим лекарственным средствам предъявляют следующие
требования:
а) необходимо наличие письменной оценки стабильности лекарства,
основанной на анализе или исследовании последнего на совместимость
ингредиентов, а также на маркетинговом опыте, подтверждающем от-
сутствие разложения данного фармацевтического препарата в течение
обычного или ожидаемого срока его годности;
б) оценка стабильности проводится в той же системе контейнер — крыш-
ка, что поставляется на рынок;
4) требования данного раздела не распространяются на аллергенные экс-
тракты с маркировкой No U. S. Standard of Potency.
4. Особые требования к испытаниям:
1) каждая серия лекарственного средства, заявленная как «стерильная» и/или
«не содержащая пирогенов», должна быть подвергнута соответствующему
лабораторному анализу, направленному на подтверждение соответствия
данным требованиям. Процедуры испытаний составляются в письменном
виде и подлежат строгому соблюдению;
48
Часть I. Нормативное регулирование производства лекарственных средств
2) каждую серию офтальмологической мази необходимо проверять на со-
ответствие спецификациям по содержанию чужеродных частиц, твердых
и абразивных веществ. Процедуры испытаний составляются в письмен-
ной форме, их необходимо соблюдать;
3) все серии лекарственного средства, содержащегося в дозированной фор-
ме с замедленным высвобождением, следует подвергать лабораторному
испытанию на соответствие спецификациям на скорость высвобождения
каждого активного ингредиента. Процедуры испытаний составляются
в письменной форме и подлежат строгому соблюдению.
5. Архивные образцы:
1) необходимо сохранять маркированные архивные образцы, репрезента-
тивные для каждой серии; это относится ко всем активным ингредиентам.
Архивные образцы должны содержать количество материала как минимум
вдвое большее того, что необходимо для проведения всех испытаний на
соответствие активного ингредиента заявленным характеристикам, за ис-
ключением испытаний на стерильность и содержание пирогенов. Период
обязательного хранения архивных образцов тех активных ингредиентов
лекарственного средства, что не подпадают под описанные ниже пункты 2
и 3, составляет год по истечении срока годности последней серии данного
фармацевтического препарата;
2) для активного ингредиента в составе радиоактивного лекарственного
средства (за исключением нерадиоактивных наборов реагентов) срок хра-
нения архивных образцов составляет:
а) 3 мес. после истечения срока годности последней серии лекарственно-
го средства, содержащего подсерию исследуемого активного ингреди-
ента, если срок годности данного лекарства не превышает 30 сут;
б) 6 мес. после истечения срока годности последней серии лекарственно-
го средства, содержащего подсерию исследуемого активного ингреди-
ента, если срок годности данного лекарства превышает 30 сут;
3) для активного ингредиента в составе лекарственного средства, отпускае-
мого без рецепта врача и освобожденного от нанесения маркировки со
сроком годности, резервную пробу следует хранить в течение трех лет по-
сле распределения последней подсерии данного лекарства, содержащей
исследуемый активный ингредиент;
4) правильно маркированный архивный образец, репрезентативный для
каждой серии лекарственного средства, хранят согласно предписаниям,
указанным в маркировке данного лекарства. Архивный образец содер-
жится в первичной системе контейнер — крышка, аналогичной или очень
близкой той, в которой продукт поставляется на рынок. В архивном об-
разце должно быть по меньшей мере двойное количество материала, необ-
ходимого для проведения всех испытаний, предназначенных для опреде-
ления соответствия активного ингредиента заявленным характеристикам,
за исключением испытаний на стерильность и содержание пирогенов.
Архивные образцы из репрезентативных проб, отобранных из произвол-
Глава 1.1. Надлежащая производственная практика и требования FDA 49
ственных серий по надлежащей статистической процедуре, следует под-
вергать визуальному осмотру. Такой осмотр проводится не реже раза в год
и служит для выявления признаков разложения; от него отказываются
лишь в тех случаях, когда он грозит нарушением целостности архивного
образца. Любые признаки разложения последнего требуют исследования.
Результаты осмотров следует документировать и соотносить с данными по
стабильности рассматриваемого лекарственного средства. Периоды хра-
нения архивного образца составляют:
а) для не относящегося к вышеперечисленным исключениям лекарствен-
ного средства — один год после истечения срока годности последнего;
б) для радиоактивного фармацевтического препарата (за исключением
нерадиоактивных наборов реагентов) — 3 и 6 мес. после истече-
ния срока его годности, составляющего соответственно не более 30 сут
для первого случая и более 30 сут — для второго;
в) для лекарственного средства, отпускаемого без рецепта врача и осво-
божденного от нанесения маркировки со сроком годности, — три года
после полного распределения его последней подсерии.
6. Животные, участвующие в исследованиях компонентов, материалов в про-
цессе обработки или лекарственных средств, должны получать соответствую-
щий уход. Этих животных маркируют, а историю их использования отражают
в специальных протоколах.
7. При угрозе перекрестного загрязнения антибиотиком фармацевтического
препарата, не содержащего антибиотик, проводится соответствующий ана-
лиз на наличие последнего. Лекарственное средство нельзя выпускать на ры-
нок, если при анализе по методике, изложенной в руководстве «Процедуры
обнаружения и измерения содержания антибиотика в лекарствах», ссылка на
которое присутствует в настоящем издании, находят антибиотик на уровне
предела обнаружения.
1.1.2.10. Записи и протоколы
1. Общие положения:
1) записи, освещающие производство, контроль или распределение серии ле-
карственного средства, хранятся как минимум год после истечения срока
годности для тех лекарств, что отпускаются по рецепту врача, и в течение
трех лет после полного распределения серии — для фармацевтических пре-
паратов, отпускаемых без рецепта врача и не имеющих сроков годности;
2) сроки хранения записей, относящихся к компонентам, контейнерам,
крышкам и этикеткам, аналогичны представленным в предыдущем абзаце;
3) все сохраняемые записи или их копии должны предоставляться государ-
ственной инспекции по первому ее требованию в течение всего перио-
да хранения. Записи хранятся непосредственно там, где производились
описанные в них действия. Инспекторы имеют право на копирование
записей;
50
Часть I. Нормативное регулирование производства лекарственных средств
4) записи могут храниться как в оригинальном виде, так и в форме копий,
точно воспроизводящих оригинал (фотокопий, микрофильмов, диами-
крокарт и т. п.);
5) записи, подлежащие сохранению, следует вести так, чтобы содержащиеся
в них данные можно было использовать для оценки стандартов качества
каждого лекарственного средства; это выявит степень устаревания специ-
фикаций лекарства, производственных или контрольных инструкций либо
методик анализов. Анализ такого рода следует проводить по меньшей мере
ежегодно. Должны быть утверждены письменные инструкции проведения
подобных оценок, включающие следующие действия:
а) анализ репрезентативного числа серий, одобренных или отбракован-
ных, и записей, связанных с ними;
б) анализ рекламаций, отзывов, случаев возврата или переработки лекар-
ственных средств, а также исследования, проводимые в соответствии
с § 211.192 Правил СМРцля каждого фармацевтического препарата;
6) необходимо утвердить процедуры, направленные на гарантирование пись-
менного уведомления ответственных официальных лиц обо всех рассле-
дованиях, регламентируемых параграфами 211.198, 211.204 либо 211.208,
о рекламациях, докладах по замечаниям инспекций, публикуемых FDA,
или о любых регуляторных действиях, осуществляемых FDA, которые име-
ют отношение к GMP.
2. Записи операций очистки, технического обслуживания (за исключением ру-
тинного технического обслуживания) и применения основного оборудова-
ния вносят в отдельный журнал с указанием точного времени производимых
операций, наименования продукта и номера подсерии для каждой произ-
водственной серии. Лица, выполняющие очистку оборудования и производ-
ственные операции, а также двойную проверку, должны поставить в журнале
дату и собственную подпись с расшифровкой. Записи в журнале следует вести
в хронологическом порядке.
3. Записи, относящиеся к компонентам, контейнерам для лекарственных
средств, укупорочным приспособлениям и маркировке, должны содержать
следующую информацию:
1) маркировку и количество продукта в каждой подсерии компонентов, кон-
тейнеров для лекарственной продукции, укупорочных приспособлений
и этикеток. Кроме того, требуются сведения о поставщике, номер(-й) се-
рий последнего, код и дата получения, а также название и адрес первично-
го производителя в том случае, если он не является поставщиком;
2) результаты всех проведенных анализов и осмотров и заключения, сделан-
ные на их основе;
3) отдельные инвентаризационные записи для каждого компонента и сведе-
ния об использовании каждой подсерии последнего. Инвентаризационная
запись должна позволять идентифицировать каждую товарную партию
или подсерию лекарственного средства, имеющего отношение к данному
компоненту;
Глава 1.1. Надлежащая производственная практика и требования FDA 51
4) документированные результаты осмотра и проверки этикеток и маркиров-
ки на соответствие установленным спецификациям;
5) местонахождение отбракованных материалов.
4. Основные записи производственных и контрольных операций (досье на се-
рию). Для каждой серии лекарственного средства оформляют производствен-
ные и контрольные записи, которые должны содержать:
1) исчерпывающее воспроизведение соответствующих основных записей по
производственным и контрольным операциям. Копию необходимо про-
верить на точность, указать дату и поставить подпись;
2) документацию, подтверждающую надлежащее осуществление всех значи-
мых этапов производства, обработки, упаковки и хранения серии, вклю-
чающую:
а) точную дату;
б) идентификационную информацию для всех единиц основного исполь-
зованного оборудования, в том числе упаковочных линий;
в) полную и специфическую идентификационную информацию для каж-
дой серии компонентов или материалов в процессе обработки;
г) вес и меру для компонентов, применявшихся в технологическом про-
цессе;
д) результаты лабораторного контроля и контроля в процессе производ-
ства;
е) проведение инспекций зон упаковки и маркировки до и после соответ-
ствующих работ;
ж) документированныеданные по реальному выходупродуктаипроцентуот
теоретически возможного выхода на критических этапах производства;
з) подробные записи по контролю маркировки, включая образцы или ко-
пии всех использованных этикеток (маркировочных записей);
и) описание контейнеров и укупорочных приспособлений для лекар-
ственных средств;
к) информацию обо всех произведенных отборах проб;
л) указание конкретных лиц, осуществлявших непосредственный надзор
за существенными этапами производственных операций;
м) все выполненные исследования;
н) результаты проведенных осмотров.
5. Все производственные и контрольные записи, относящиеся к фармацев-
тической продукции, включая записи по упаковке и маркировке, должны
быть рассмотрены отделом контроля качества на предмет соответствия всем
утвержденным письменным процедурам. Эта апробация проходит до посту-
пления серии в продажу. Необходимо тщательно расследовать все случаи не-
объясненных несоответствий серии или ее компонентов любым утвержден-
ным спецификациям. Под такое расследование подпадают и другие серии
того же лекарственного средства (и даже других фармацевтических препара-
тов), потенциально связанного(-ых) с конкретным сбоем или несоответстви-
52
Часть I. Нормативное регулирование производства лекарственных средств
ем. Результаты расследования отражаются в соответствующей записи, содер-
жащей указание последующих действий.
6. Лабораторные записи:
1) лабораторные записи представляют собой исчерпывающие данные по ре-
зультатам всех анализов, подтверждающих соответствие установленным
спецификациям и стандартам, в том числе по результатам осмотров и ис-
пытаний. Записи должны содержать:
а) описание анализируемых проб (в частности, место пробоотбора, коли-
чество материала, номер серии или другого отличительного кода, даты
отбора проб и получения последних лабораторией для анализа);
б) отличительные характеристики каждой методики, применяемой для
анализа проб. Здесь необходимо давать ссылку на данные, подтвержда-
ющие соответствие методик анализа проб существующим стандартам
точности и достоверности. (Если используемый метод внесен в теку-
щую редакцию Фармакопеи США (U. S. Pharmacopeia, USP), Нацио-
нального свода правил {National Formulary, NF) или в другой общепри-
знанный сборник стандартов либо подробно изложен в утвержденной
заявке на новые лекарственные вещества, то описания не требуется);
в) указание веса (или меры) пробы, отобранной для каждого анализа;
г) подробные записи по всем данным, сохраненным в процессе каждо-
го анализа, включая все графики, диаграммы и спектры, полученные
с помощью лабораторных приборов, а также идентификацию каждого
отдельного анализируемого компонента и подсерии;
д) записи всех расчетов, сделанных в процессе анализа и произведенных
согласно утвержденным стандартам идентичности, содержания актив-
ного вещества, качества и чистоты исследуемого компонента;
е) изложение результатов анализа и сравнение их с утвержденными стан-
дартами идентичности, содержания активного вещества, качества и чи-
стоты исследуемого компонента;
ж) фамилию или подпись лиц, производивших каждый анализ, а также
дату проведения последнего;
з) фамилию или подпись второго лица, проводившего проверку ориги-
нальных записей на точность, полноту и соответствие установленным
стандартам;
2) при внесении любых изменений в утвержденную методику анализа следу-
ет вести подробные записи, содержащие как обоснование произведенной
модификации, так и доказательство надежности полученных результатов.
Последние должны быть по меньшей мере столь же точными и достовер-
ными, как и те, что были бы получены для материалов, анализируемых по
утвержденной методике;
3) необходимо детально фиксировать проведение любых анализов и стандар-
тизацию лабораторных стандартных образцов, реагентов и стандартных
растворов;
Глава 1.1. Надлежащая производственная практика и требования FDA
53
4) при проведении периодических калибровок лабораторных приборов, обо-
рудования, измерительных приборов и записывающих устройств также
ведутся подробные записи;
5) необходимо подробно описывать проведение любых исследований ста-
бильности, регулируемых § 211.166 нормативного документа.
7. В записях распределения продукции указываются наименование лекарства
и содержание в нем активного компонента. Кроме того, дается описание
форм дозирования, сообщаются наименование и адрес грузополучателя, дата
события, количество отправленного фармацевтического препарата, его кон-
трольный номер и номер его серии. Для медицинских препаратов, представ-
ляющих собой газы под давлением, указание номера подсерии или контроль-
ного номера необязательно.
8. Рассмотрение претензий:
1) необходима четкая регламентация разбора письменных или устных претен-
зий, предполагающего составление соответствующего документа. Послед-
ний обязательно должен содержать положение о том, что рассмотрение
всех претензий, указывающих на возможное несоответствие лекарствен-
ного средства каким-либо спецификациям, проводится отделом контроля
качества, который выносит решение о проведении расследования. Кроме
того, обязательно наличие указаний, позволяющих безошибочно относить
ту или иную претензию к неожиданному и серьезному неблагоприятному
воздействию лекарства, о котором следует доложить FDA,
2) факты жалоб следует вносить в специальный журнал, предназначенный
для регистрации претензий к продукции. Журнал может храниться где
угодно при условии, что будет предоставлен инспекции по первому ее
требованию. Письменные отчеты, относящиеся к определенному лекар-
ственному средству, следует хранить в течение как минимум одного года
по истечении срока годности этого лекарства либо одного года после даты
получения претензии (когда бы она ни была получена). Для некоторых
лекарственных средств, отпускаемых без рецепта врача и не содержащих
маркировку с датой истечения срока годности из-за нераспространения на
них данного требования, записи следует хранить в течение трех лет после
поступления этих лекарств в розничную сеть. Запись должна содержать
следующую информацию:
а) наименование лекарственного средства, содержание в последнем ак-
тивного вещества, номер серии, вид претензии, ее суть и ответ ее со-
ставителю;
б) результаты расследования в случае его проведения, а также предпола-
гаемые дальнейшие действия. Запись (или ее копию) следует хранить
в том же месте, где проводилось расследование;
в) причины, по которым расследование не было признано необходимым,
и имя ответственного лица, принявшего такое решение.
54
Часть I. Нормативное регулирование производства лекарственных средств
1.1.2.11. Возвращенные и переработанные фармацевтические препараты
1. Возвращенные лекарственные средства следует маркировать как «возвращен-
ные» и надлежащим образом хранить. Если при хранении лекарственных
средств, их транспортировке в процессе возврата или до него были допу-
щены нарушения, ставящие под сомнение их безопасность, идентичность,
долю активного компонента, качество, чистоту, целостность их упаковки или
маркировки, такие возвращенные фармацевтические препараты подвергают
специальным исследованиям, подтверждающим их соответствие установ-
ленным стандартам. Не прошедшие подобную проверку лекарства подлежат
уничтожению. По возвращенной фармацевтической продукции ведутся со-
ответствующие записи. Они содержат наименование активного компонента
в лекарстве, его дозу, номер серии, причину и дату возврата, количество воз-
вращенного препарата и окончательное решение по нему. При необходимо-
сти исследуют всю серию возвращенного препарата. Необходимо наличие
письменных процедур удерживания, анализа и переработки возвращенных
лекарственных средств.
2. Переработанные лекарственные средства. Фармацевтические продукты, хра-
нившиеся в ненадлежащих условиях, связанных с чрезмерными колебаниями
температуры, влажности, давления, с воздействием дымов, испарений, ради-
ации, пожаров, не подлежат переработке и не могут быть возвращены на ры-
нок. При любом подозрении на то, что продукт подвергался подобным воз-
действиям, его переработку осуществляют лишь при условии: а) проведения
лабораторных анализов и исследований, подтверждающих его соответствие
всем установленным стандартам по параметрам идентичности, содержания
активных веществ, качества и чистоты; б) гарантии того, что лекарственное
средство, а также его упаковка не подвергались неправильному хранению,
обусловленному стихийными бедствиями или авариями. При этом органо-
лептические исследования приемлемы лишь в качестве дополнительных
свидетельств соответствия лекарственных средств стандартам по параметрам
идентичности, содержания активного вещества, качества и чистоты. Необ-
ходимо вести записи, содержащие наименование лекарственного средства,
номер его серии и место хранения.
1.1.3. Руководство для промышленности «Системный
подход к обеспечению качества и нормативы Текущей
надлежащей практики производства фармацевтической
продукции»
Составляя этот руководящий документ, FDA предполагало помочь производителям
в деле внедрения современных и отвечающих требованиям нормативов GMP систем
менеджмента качества (СМК) и новых подходов, связанных с управлением рисками.
В Руководстве описаны системы, которые ЛХ4 считает моделями систем всестороннего
управления качеством, атакже способы их внедрения. Не возлагая новых обязательств
на производителей, этот документ не задумывался FDA и как замена правил GMP.
Глава 1.1. Надлежащая производственная практика и требования FDA
55
Подобно другим руководящим документам, Руководство не устанавливает юри-
дической ответственности, а скорее отражает существующую на данный момент
позицию FDA. Таким образом, его следует рассматривать лишь как свод рекомен-
даций — за исключением тех случаев, когда оно содержит ссылки на нормативные
документы.
Цель Руководства — описать модель СМК и продемонстрировать, где и как
элементы данной модели могут соответствовать требованиям cGMP. Принцип, ле-
жащий в основе данного документа, можно сформулировать так: качество фарма-
цевтического препарата всегда должно быть высоким; для его обеспечения нельзя по-
лагаться на одни испытания.
1.1.3.1. Текущая надлежащая производственная практика и концепции
современных систем управления качеством
Согласно предписаниям FDA, для современных СМК, относящихся к производству
фармацевтических дозированных форм, критически важен ряд ключевых концеп-
ций. Это, прежде всего:
> Качество. В контексте рассматриваемого Руководства это понятие означает
достижение идентичности, а также таких параметров качества (содержание
активного компонента, чистота и т. д.), которые обеспечили бы безопасность
и эффективность лекарственного средства.
> Качество через планирование и разработку. Подразумевает разработку такого
производственного процесса, который позволит обеспечить последователь-
ное достижение заранее определенных параметров качества лекарственной
продукции.
> Управление рисками качества. Эта составляющая СМК может помочь в уста-
новлении технических условий и параметров производственного процесса
при производстве дозированных форм, оценке и снижении рисков измене-
ния процесса или технических условий, а также в определении масштабов
расследования несоответствий и корректирующих действий.
> Корректирующие и профилактические действия (САРА). Это концепция нор-
мативных действий, предусматривающая расследование любых отклонений,
а также их коррекцию в целях дальнейшего предотвращения. Модель САРА
состоит из трех отдельных составляющих:
• из корректирующих действий, направленных на решение выявленной
проблемы;
• анализа ключевых причин отклонений, сопровождаемого выбором необ-
ходимых корректирующих действий;
• профилактических действий, призванных предотвратить возникновение
схожих проблем.
> Контроль изменений. Этот процесс направлен на предотвращение неконтро-
лируемых изменений.
> Отдел качества. В то время как в GMP упоминается единый отдел качества,
в cGMP область ответственности данного отдела разделена между двумя его
подразделениями:
56
Часть I. Нормативное регулирование производства лекарственных средств
• по контролю качества, что подразумевает: а) оценку стабильности ис-
ходных компонентов и готовых продуктов; б) отслеживание параметров
производственного процесса; в) принятие решения о пригодности каждой
серии для отпуска и распределения;
• обеспечению качества, состоящему: а) из анализа и одобрения всех проце-
дур, связанных с производством и техническим обслуживанием; б) анали-
за записей; в) проведения аудитов и трендового анализа.
> Модель инспекции шести систем. Принятая в FDA инструкция для проводящих
расследование лиц разработана на основе комплексного подхода к инспекции
и выделяет шесть взаимосвязанных компонентов: 1) СМК, охватывающую
все прочие системы; 2) систему материалов; 3) продукции; 4) упаковки и мар-
кировки; 5) технологической части и оборудования; 6) систему лабораторного
контроля. По мнению специалистов FDA, использование подобного комплекс-
ного подхода поможет фирмам в достижении более высокого уровня контроля.
1.1.3.2. Модель систем менеджмента качества
В этой части Руководства описана модель СМК, обеспечивающая стабильное про-
изводство приемлемой фармацевтической продукции. Стержневыми составляющи-
ми этой модели являются:
> ответственность управления;
> ресурсообеспечение;
> производственные операции;
> анализ.
Ответственность руководства. Согласно руководящим документам FDA, плани-
рование надежной СМК, ее внедрение и управление ею осуществляются непосред-
ственно руководством.
Ресурсообеспечение. Создание надежной СМК, соответствующей требованиям
GMP, требует грамотного ресурсообеспечения. Эти функции возложены на опреде-
ленных должностных лиц, которые разделяют ответственность за их выполнение
с высшим руководством.
Технологическая часть и оборудование. Технические эксперты, обладающие зна-
нием фармацевтической науки, осознающие факторы риска и понимающие про-
изводственный процесс, связанный с изготовлением лекарственных средств, несут
ответственность за определение специфических технических условий и требований
к оборудованию. Последнее должно отвечать специальным требованиям, быть со-
ответствующим образом откалибровано, очищено и отремонтировано для предот-
вращения загрязнений и попадания примесей в продукт. Следует иметь в виду,
что GMP уделяет технологическому оборудованию столько же внимания, сколько
и аналитическому, в то время как в фокусе внимания большинства СМК находится
исключительно аналитическое оборудование.
Контрольные операции, выполняемые третьими лицами. Руководство предприятия
может заключать с внешними поставщиками контракты по созданию СМК. В этих
контрактах должны быть четко прописаны сущность услуги, ответственность за ка-
чество последней и механизмы коммуникации.
Глава 1.1. Надлежащая производственная практика и требования FDA 57
Производство. Между отдельными элементами СМК и требованиями GMP для
производственных операций существует определенное сходство. Это обусловлено
тем, что программы контроля и инспекций, осуществляемые FDA, исходят из тре-
бований GMP. В FDA считают, что производственная СМК должна учитывать сле-
дующие ключевые факторы:
1) планирование, разработку и документирование продукции и процессов;
2) анализ входящих материалов;
3) осуществление и мониторинг производственных операций;
4) рассмотрение рекламаций.
Аналитическая работа включает следующие виды деятельности:
1) анализ данных для выявления трендов;
2) проведение внутренних аудитов;
3) управление рисками качества;
4) корректирующие действия;
5) профилактические действия;
6) содействие внедрению усовершенствований.
1.1.4. Руководство для промышленности «РА Т — система
инновационного развития фармацевтического
производства и обеспечения качества»
Целью данного Руководства является описание системы нормативов (процессно-
аналитической технологии (РАТ) по терминологии FDA). Эта система стимулирует
добровольное внедрение новых разработок, развитие фармацевтического произ-
водства и обеспечение качества. Данное Руководство предназначено для широкой
аудитории из различных организационных подразделений. Одной из главных его
тем является обсуждение принципов РАТ, выявляющее возможности развития нор-
мативной базы, которая способствовала бы инновациям.
Традиционное фармацевтическое производство обычно представляет собой се-
рийное производство с лабораторным контролем аналитических проб на разных
этапах процесса в целях оценки качества продукта. Согласно FDA, существуют воз-
можности усовершенствования производства и обеспечения качества через внедре-
ние инноваций в разработку продуктов и технических процессов, через управление
процессами и аналитическую работу.
Как правило, фармацевтическая промышленность чуждается новшеств. Это свя-
зано с опасением, что новый подход не будет одобрен FDA. Такое неодобрение неиз-
бежно привело бы к задержкам и связанным с ними финансовым потерям, а значит,
к необходимости пересмотра технологий. Поэтому предприятия не хотят идти на
риск. Вместе с тем официальная позиция FDA на сегодня состоит в том, что подоб-
ная нерешительность, «тормозящая» внедрение полезных инноваций, не отвечает
интересам общественного здоровья. Согласно правилам FDA, любое фармацевти-
ческое производство должно исходить из следующих принципов:
> разработка результативных производственных процессов;
58 Часть I. Нормативное регулирование производства лекарственных средств
> спецификация продукта и технические характеристики процесса, которые
основаны на понимании того, как состав материалов и параметры процесса
влияют на технические характеристики продукта;
> непрерывное обеспечение качества в режиме реального времени;
> надлежащая политика управления и применение юридических процедур, со-
ответствующих современному уровню научных знаний;
> подход к оценке рисков, основанный на признании того, что:
• уровень научного осмысления сущности процесса, состава исходных ма-
териалов и параметров процесса влияет на качество и технические харак-
теристики продукта;
• правильная стратегия контроля производственных процессов может пре-
дотвратить или уменьшить риск изготовления продукта низкого качества.
Задача данного Руководства — стимулировать прогресс в области произ-
водства лекарственных средств. Цели, заявленные FDA, еще далеко не достигнуты.
Представители FDA выражают озабоченность тем, что промышленность не торо-
пится с внедрением инноваций. Однако затраты на внедрение последних делают их
не столь привлекательными для производителей.
1.1.4.1. Система РАТ
Качество фармацевтической продукции должно стать ее неотъемлемой характери-
стикой, что достигается через всестороннее понимание:
> предполагаемых терапевтических задач, отличительных характеристик раз-
личных групп конечных потребителей, путей введения лекарства и его фар-
макокинетических характеристик;
> химических, физических и биофармацевтических характеристик лекарства;
> состава фармацевтического продукта, а также выбора его компонентов и упа-
ковки исходя из его характеристик;
> планирования производственного процесса с применением принципов ин-
женерии, материаловедения и обеспечения качества, что позволит получить
качественное лекарственное средство с приемлемыми и воспроизводимыми
характеристиками, сохраняющее заявленные свойства в течение всего срока
годности.
Осмысление процесса. Процесс считается хорошо изученным, если все крити-
ческие источники погрешности идентифицированы и объяснены, изменчивость
процесса хорошо контролируется, а характеристики качества продукта могут быть
точно предсказаны.
Принципы и инструменты. Зачастую фармацевтическое производство представ-
ляет собой последовательность отдельных операций, каждая из которых изменяет
определенные свойства обрабатываемого материала. Для гарантирования приемле-
мости и воспроизводимости этих изменений следует не упускать из виду параметры
качества исходных материалов, а также пригодность последних для применения
в конкретной операции. Большинство современных фармацевтических процессов
имеет определенные временное ограничения (например, указание «Смешивать
в течение десяти минут»). В отдельных случаях такие указания не учитывают эффект
Глава 1.1. Надлежащая производственная практика и требования FDA
59
физических различий в исходных материалах. Трудности в процессе переработки,
приводящие к несоответствию продукта установленным спецификациям, мотуг
возникнуть даже тогда, когда исходные материалы полностью отвечают последним.
Применение инструментов и принципов РАТ поможет раскрыть важные физиче-
ские, химические и биологические характеристики, что позволит улучшить кон-
троль за процессами, привлечет внимание к указаниям, связанным с временнйми
ограничениями, и повысит эффективность производства.
Инструменты РАТ. Существует множество инструментов, способствующих бо-
лее углубленному пониманию процессов. Эти инструменты, применяемые в рам-
ках системного подхода, могут оказаться действенным средством получения новой
информации, необходимой для непрерывного развития стратегии уменьшения ри-
сков. Эти инструменты подразделяются на следующие категории:
> различные средства для планирования, получения данных и анализа;
> анализаторы процессов;
> инструменты контроля процессов;
> постоянное совершенствование и управление знаниями.
Стратегия внедрения. Для успешного внедрения РЛТчрезвычайно важна взаимо-
связь разработчиков данной системы с производителями. В FDA считают, что суще-
ствующая нормативная база достаточно широка и позволяет проводить внедрение
этой стратегии. В процессе введения системы РАТ производители могут пожелать
оценить приемлемость ее инструментов на экспериментальном и/или производ-
ственном оборудовании либо в технологических процессах. До установки инстру-
ментов системы рекомендуется провести анализ рисков, способных повлиять на
качество продукта. Это можно сделать посредством системы управления качеством
работы данного оборудования без предварительного уведомления агентства. Дан-
ные, собранные с помощью экспериментальных инструментов, считаются экспери-
ментальными. Если эксперимент проводится на производственном оборудовании,
следует задействовать систему контроля качества работы последнего. Инспекция
экспериментальных данных, собранных на существующем производстве реального
продукта в целях оценки приемлемости экспериментальных инструментов РАТ, не
входит в планы FDA. Рутинная инспекция производственного процесса, включаю-
щего инструменты РАТ, используемые для исследовательских задач, будет основана
на текущих нормативных стандартах.
Большой объем информации по программе РА ^доступен на сайте http://www.fda.
gov/cder/ops/pat.htm.
Все маркетинговые заявки, поправки, дополнения к заявкам следует представ-
лять в соответствующее подразделение Центра оценки и исследований лекарствен-
ных средств (CDER) или Центра ветеринарной медицины (СУМ). Планы внедрения
РАТ нужно соотносить с анализом рисков. Федеральное управление США по кон-
тролю за пищевой продукцией и лекарствами предлагает следующие планы внедре-
ния системы:
> РАТ может быть внедрена в рамках собственной СМК. Инспекции cGMP,
проводимые группой РАТ или инспектором, уполномоченным последней,
могут предшествовать внедрению РАТ или следовать за ним;
60 Часть I. Нормативное регулирование производства лекарственных средств
> приложения («Ожидаемые изменения» (СВЕ), «Изменения, ожидаемые в те-
чение 30 дней» (СВЕ-30) или «Предварительно утвержденное приложение»
(PAS)) могут быть представлены в FDA перед внедрением; при необходимости
может быть проведена предварительная инспекция, осуществляемая группой
РЛТили уполномоченным ею инспектором.
> В FDA может быть направлен протокол совместимости, который содержит
общее описание стратегии исследований, валидации и внедрения РАТ, а так-
же срок реализации последней. После одобрения данного протокола можно
выбрать рациональный путь внедрения системы.
Для содействия внедрению или одобрению РАТ производители могут подать
запрос на проведение предэксплуатационного анализа работы производственного
оборудования и процессов с системой РАТ и РЛТ-группой. Для этого им необхо-
димо связаться с Группой процессно-аналитических технологий FDA (электронный
адрес PAT@cder.fda.gov). Следует отметить, что при реализации некоторых планов
внедрения РАТ не затрагиваются ни текущий процесс, ни спецификации к нему.
В подобных случаях перед производителем открывается целый ряд возможностей.
Оптимальный вариант выбирается по согласованию с FDA.
1.1.5. Руководство для промышленности
«Часть 11. Электронные записи, электронные подписи:
возможности и применение»
Из всех нормативных документов, изданных FDA, больше всего вопросов вызыва-
ет раздел 11 тома 21 CFR. Вместо обзора самогб нормативного документа, который
еще может быть пересмотрен, целесообразнее будет рассмотреть Руководство для
промышленности, опубликованное FDA в августе 2003 г. с целью снять многие на-
копившиеся вопросы. Несмотря на то что этот руководящий документ далеко не
достиг тех целей, что преследовали его авторы, содержащиеся в разделе 11 норма-
тивные требования всё равно подлежат выполнению.
Согласно Руководству, в основе подхода FDA лежат три принципа:
> Норматив следует толковать ограничительно.
> Записи, которые будут сочтены подпадающими под регламентацию упомя-
нутого 11-го раздела, подлежат свободному толкованию в отношении требо-
ваний к валидации, проведения аудитов, сохранения и копирования записей.
Системы, которые эксплуатировались до даты появления данного норматив-
ного документа, также подпадают под его юрисдикцию.
> Все нормативные акты, включая требования к ведению и сохранению запи-
сей, приобретают силу закона.
Далеко не все положения 11-го раздела имеют обязательную силу. Выполнению
подлежат только следующие требования:
> доступ в систему, открытый лишь для уполномоченных лиц;
> обязательны проведение оперативных проверок системы, полномочий
и средств контроля;
Глава 1.1. Надлежащая производственная практика и требования FDA 61
> лица, занимающиеся разработкой, обслуживанием и использованием элек-
тронных систем, должны иметь соответствующее образование и опыт работы,
а также обязаны пройти обучение, позволяющее выполнять порученные им
задания;
> необходимо письменное положение, налагающее меру ответственности на
лиц, которые поставили под соответствующими актами свою электронную
подпись;
> обеспечение надлежащего контроля системной документации;
> установление контроля открытых систем, соответствующего контролю за-
крытых систем;
> регламентация скрепления документов электронными подписями.
1.1.5.1. Записи, регламентированные разделом 11 тома 21 CFR
Согласно ограничительному толкованию FDA, под регулирование 11-го раздела
подпадают следующие записи и подписи в электронных документах:
1) записи, которые требуется вести по предписанным правилам, сохраняемые
в электронном формате, заменяющем бумажный формат;
2) записи, которые требуется вести по предписанным правилам, сохраняемые
в электронном формате в дополнение к бумажному, используемые при осу-
ществлении мероприятий, регулируемых нормативными документами;
3) записи, представляемые в FDA в электронном формате согласно установлен-
ным требованиям (вместе с тем запись, которую не представляют, а исполь-
зуют при составлении представляемого документа, не регламентируется 11-м
разделом);
4) факсимиле, подписи инициалами и другие требуемые виды подписей.
1.1.5.2. Подход FDA к особым требованиям раздела 11 тома 21 CFR
1. Валидация. При рассмотрении особых требований 11-го раздела допускается
их свободное толкование, предполагающее, однако, соответствие всем при-
меняемым при валидации нормативным правилам. Валидация исходит из
документированной оценки рисков и определения способности системы по-
влиять на качество продукта, безопасность и сохранность записей.
2. Журнал аудита. Право на свободное толкование распространяется и на осо-
бые требования, относящиеся к электронным журналам аудита с проставлен-
ными датами внесения записи, как и на любые соответствующие требования
11-го раздела. При этом необходимо соблюдение всех нормативных правил,
регулирующих время и последовательность регистрируемых событий, а также
процедур обновления записей, гарантирующих сохранность нужной инфор-
мации.
3. Унаследованные системы. Свободному толкованию подлежат и требования
11-го раздела для систем, которые были в эксплуатации до 20 августа 1997 г.
Таким образом, FDA не настаивает на строгом соответствии подобных систем
всем требованиям 11-го раздела, если конкретная система:
1) находилась в эксплуатации до даты проверки;
62 Часть I. Нормативное регулирование производства лекарственных средств
2) соответствовала всем применяемым утвержденным правилам до даты про-
верки;
3) отвечает всем действующим правилам;
4) помимо всего перечисленного, подходит для использования по назначе-
нию, что подтверждается документированными свидетельствами.
4. Копии записей. Право на свободное толкование может применяться к особым
требованиям раздела 11, регламентирующим копирование записей. Инспек-
тор должен иметь постоянный доступ к записям. Все записи, которые ведет
производитель, подлежат инспектированию.
5. Хранение документации. Требования раздела 11, регламентирующие защиту
записей, также подлежат свободному толкованию, если это способствует бы-
строму и точному поиску записей в течение всего периода их хранения.
1.1.6. Руководство для промышленности и FDA
«Текущая надлежащая производственная практика
для комбинированных продуктов»
Данный документ обсуждает применение принципов GMPno отношению к комби-
нированным продуктам согласно определению пункта 3.2(e) тома 21 CFR. Произ-
водитель должен гарантировать, что продукт не фальсифицирован, обладает адек-
ватным содержанием активного ингредиента, хорошим качеством, идентичностью,
чистотой и соответствием утвержденным стандартам. Данное Руководство не рас-
сматривает технических методов производства и не дает рекомендаций по выбору
производственного оборудования.
Комбинированный продукт представляет собой сочетание вспомогательных ве-
ществ с активной фармацевтической субстанцией, биологическим продуктом либо
тем и другим; смешивание может производиться в разном порядке.
Для регулирования качества комбинированных продуктов их представляют
в филиал FDA или в любую альтернативную организацию, обладающую первичной
компетенцией по предпродажному анализу и регулированию. Рассматриваемая там
лекарственная продукция проверяется на соответствие определенным правилам
GMP. Последние могут представлять собой:
> нормативы GMP для готовой фармацевтической продукции (21-й том CFR,
разделы 210 и 211);
> нормативы СМК для компонентов (раздел 820 тома 21 CFR);
> нормативы для биологической продукции (разделы 600—680 тома 21 CFR),
применяемые лишь для лекарств, имеющих биологическую природу.
Особых нормативов GMP для комбинированных продуктов не существует. До
опубликования этих нормативов производители каждой составной части продукта
руководствуются нормативами для отдельного компонента.
Для получения консультаций производители могут обращаться в Бюро комбини-
рованной продукции {Office of Combination Products) по телефону (301)427-1934 или
электронной почте combination@fda.cov. С последними версиями документов можно
ознакомиться на сайте Бюро http//:www.fda/gov/oc/combination.
Глава 1.1. Надлежащая производственная практика и требования FDA 63
1.1.7. Руководство для промышленности
«Порошковые смеси и готовые единицы дозирования:
стратифицированный отбор проб и оценка качества
единиц дозирования в процессе производства»
Задача данного Руководства — помочь производителям в достижении соответствия
требованиям GMP, относящимся к смешиванию компонентов для однородных об-
рабатываемых смесей и готовых единиц дозирования.
Стратифицированный пробоотбор. В этом процессе отбор проб единичных дози-
рованных форм производят через определенные временное интервалы. Репрезента-
тивные пробы отбирают на специально выбранных точках пробоотбора в процессе
операций наполнения-сжатия; это позволяет изготавливать единицы дозирования
с экстремальными концентрациями лекарства.
Данное Руководство описывает методы пробоотбора, позволяющие продемон-
стрировать однородность лекарственного ингредиента. Оно не имеет нормативной
силы и не исчерпывает все возможные способы подтверждения правильности сме-
шивания порошкообразных компонентов.
Оценка однородности порошкообразной смеси. В рамках данной задачи рекомен-
дованы следующие процедуры:
1. Анализ смешивания в производственных сериях, производимый путем экс-
тенсивного отбора проб из смесителя и/или промежуточных контейнеров для
объемных смесей.
2. Определение времени смешивания и диапазона скоростей, «мертвых зон»
в смесителе и мест сегрегации в промежуточных контейнерах для объемных
смесей.
3. При разработке методики, способной определить истинную однородность
смеси, необходимо оценить влияние объема пробы (последний может превы-
шать величину единицы дозирования в 1—10 раз). Оптимальное отношение
объема отбираемой смеси к единице дозирования не должно быть меньше
трех.
4. Составление плана отбора проб смеси и оценка их с использованием соот-
ветствующих методов статистического анализа.
5. Количественное измерение любой вариабельности среди анализируемых
проб. Вариабельность объясняется недостаточной однородностью или по-
грешностью пробоотбора. Значительная вариабельность данных при отборе
проб в определенной точке пробоотбора может указывать на неправильное
смешивание компонентов, погрешность пробоотбора и/или агломерацию.
Высокая вариабельность данных по пробам из разных точек пробоотбора ча-
сто свидетельствует об ошибках, допущенных в ходе операции смешивания.
Корреляция данных по однородности порошкообразной смеси и данных анализа при
стратифицированном отборе единиц дозирования в ходе производственного процесса.
При оценке корреляции рекомендуется:
1. Проводить периодический отбор проб и анализ единиц дозирования в про-
цессе обработки пробы. Такой пробоотбор осуществляется в определенных
64
Часть I. Нормативное регулирование производства лекарственных средств
точках через определенные временное интервалы в ходе процессов сжатия
или наполнения. Отбор единиц дозирования во время производственного
процесса проводят как минимум в 20 точках при надлежащем пространствен-
ном распределении точек пробоотбора. В каждой точке отбирают не менее
семи проб. Таким образом, минимальное общее количество отобранных проб
должно составить 140 единиц.
2. Отобрать по семь проб с каждой дополнительной точки для более полной
оценки существенных стадий процесса, таких как наполнение или опустоше-
ние загрузочных воронок либо промежуточных контейнеров для объемных
смесей, начало и завершение процессов наполнения или сжатия, а также от-
ключение оборудования.
3. Учитывать возможные изменения при переходе от одной серии к другой.
4. Подготовить перечень итоговых данных и провести анализ и корреляцию
данных анализа при стратифицированном пробоотборе в ходе процесса сме-
шивания.
5. Сравнить однородность порошкообразной смеси с данными анализа проб,
отобранных в ходе процесса согласно описанным выше принципам.
6. Изучить все случаи расхождений между данными по порошкообразной смеси
и единицам дозирования и установить их основные причины. Существует по
меньшей мере один доступный способ, помогающий справиться с этой зада-
чей. Возможные коррекционные действия могут варьироваться от пересмо-
тра состава смеси вплоть до оптимизации процесса. Проблемы пробоотбора
можно решить и с помощью альтернативных методик, таких как отбор и ана-
лиз проб in situ в режиме реального времени.
Корреляция данных анализа при стратифицированном пробоотборе в ходе производ-
ственного процесса с данными по готовой продукции. Рекомендованные этапы пред-
ставляют собой:
1. Проведение анализа на однородность содержимого готового продукта с при-
менением соответствующей процедуры или согласно указаниям ANDA либо
NDA для утвержденных продуктов.
2. Сравнение результатов анализа проб при стратифицированной выборке
в ходе производственного процесса с данными по однородности содержимо-
го готовых единиц дозирования по предыдущему этапу. Этот анализ следует
проводить без коррекции по весу.
3. Подготовку сводки итоговых данных и результатов анализа, подтверждаю-
щей, что данные, полученные с помощью стратифицированного пробоотбора
в ходе производственного процесса, позволяют гарантировать однородность
содержимого в готовом продукте.
1.1.7.1. Валидация однородности серии порошкообразной смеси
Пятая глава Руководства описывает отбор проб и анализ порошкообразной смеси
из демонстрационных и валидационных серий, используемых при внедрении мето-
да стратифицированной выборки.
Глава 1.1. Надлежащая производственная практика и требования FDA
65
Согласно Руководству, в процессе производства демонстрационных и валидаци-
онных серий целесообразно провести оценку характеристик однородности: 1) по-
рошкообразной смеси; 2) единиц дозирования, используемых в ходе производствен-
ного процесса; 3) готового продукта. Каждую характеристику следует определять
независимо. Перед производством демонстрационной и/или валидационной серии
для идентификации точек пробоотбора и критериев приемки рекомендуется:
1. Тщательно определить по меньшей мере 10 точек пробоотбора в смесителе,
чтобы выявить потенциальные зоны недостаточного перемешивания. На-
пример, в смесителях барабанного типа (V-образных смесителях, двойных
конусных смесителях или барабанных миксерах) пробы следует отбирать на
расстоянии, равном как минимум двум значениям глубины по оси смесите-
ля. В случае конвективных смесителей (например, ленточно-винтовых) отбор
объемных проб на однородность смеси представляет особую важность: точки
пробоотбора должны учитывать углы и зону выгрузки (минимальное коли-
чество точек, рекомендованных для надлежащей валидации конвективных
смесителей, равно 20).
2. Из каждой точки пробоотбора следует отобрать не менее трех репликатных
проб. Результаты анализов должны соответствовать следующим требованиям:
1) анализ одной пробы с каждой точки пробоотбора (число проб равно 10,
а для ленточно-винтового смесителя — 20);
2) относительное стандартное отклонение (RSD) индивидуальных результа-
тов составляет 5%;
3) колебание значений конкретных результатов относительно среднеариф-
метического не должно превышать 10%.
Внедрение представленных в Руководстве методов рекомендуется лишь при
условии соблюдения обозначенных выше трех условий.
Ошибки могут происходить при отборе проб из отдельных смесей, при использо-
вании некоторых приспособлений для пробоотбора или методик, которые в резуль-
тате оказываются неподходящими для оценки пригодности смеси исключительно
по данным о смешивании. В этих случаях для оценки однородности смеси рекомен-
дуется использовать данные по единицам дозирования в процессе производства,
а также данные, полученные при анализе проб смеси.
При непосредственном отборе проб некоторых порошкообразных смесей часто
возникает неоправданный риск получения неверных данных. В таких случаях могут
использоваться альтернативные процедуры, в рамках которых данные, полученные
при непрямом пробоотборе, сопоставляются с данными по единицам дозирования,
отобранным в ходе производственного процесса. Это дает более-менее точное пред-
ставление об однородности порошкообразной смеси. Анализ данных, подтверж-
дающих возможность применения упомянутых альтернативных процедур, должен
быть описан в итоговом отчете, составленном на производстве.
1.1.7.2. Верификация производственных критериев
Оценка однородности порошкообразной смеси и процедура анализа корреляции
с данными, полученными при стратифицированном отборе проб в ходе произвол-
66
Часть I. Нормативное регулирование производства лекарственных средств
ственного процесса, должны быть завершены до того, как будут установлены кри-
терии и процедуры контроля для рутинного производства. Кроме того, получаемые
данные рекомендуется оценивать на соответствие закону нормального распределе-
ния. При определении RSD следует исходить из результатов анализа проб, отобран-
ных в ходе производственного процесса по принципу стратифицированной выбор-
ки. Значение RSD используется для классификации результатов анализа: readily pass
(критерий приемлемости 1 -го уровня, критерий полного прохождения) соответствует
четырехпроцентному RSD, marginally pass (критерий приемлемости 2-го уровня, кри-
терий достаточного прохождения) — шестипроцентному, a inappropriate (неприемле-
мый) используется для демонстрации гомогенности материала серии при RSD >6,0%.
По завершении вышеописанных процедур следует определение адекватности со-
става порошкообразной смеси и однородности содержимого готовых дозированных
форм. Федеральное управление США по контролю за пищевой продукцией и ле-
карствами рекомендует следующую процедуру оценки рутинных производственных
серий:
1. Метод стандартных критериев (SCM). Этот метод рекомендован в сле-
дующих случаях:
1.1. По итогам установленных предварительных критериев есть соответствие
классификации readily pass (критерию полного прохождения).
1.2. Результаты анализа по методу допустимых критериев (МСМ) позволяют
перейти к SCM.
1.2.1. Стадия 1 анализа. Для ее проведения следует отобрать как минимум три
единицы дозированных форм в каждой точке пробоотбора. Далее иссле-
дуют одну единицу дозирования с каждой точки пробоотбора, после чего
корректируют результаты по весу и подвергают их проверке на предмет
соответствия того, что:
1.2.1.1. Показатель RSD всех отдельных результатов составляет менее 5%.
1.2.1.2. Среднее значение по всем результатам составляет 90—110% от целевого
значения.
Если результаты отвечают всем упомянутым требованиям, идентичность смеси
и однородность содержимого единичной дозированной формы считаются приемле-
мыми и для следующей серии можно применять SCM. Если результаты анализа не
соответствуют критериям стадии 1, то требуется расширенный анализ, т. е. стадия 2.
1.2.2. Стадия 2 анализа. Для ее проведения необходимо исследовать две остав-
шиеся с каждой точки пробоотбора единицы дозирования и рассчитать
среднее значение и RSD по комбинированным данным, полученным
в рамках обеих стадий. Полученные результаты проверяют на соответ-
ствие следующим критериям:
1.2.2.1. Для всех отдельных результатов RSD должно быть менее 5%.
1.2.2.2. Среднее значение по всем результатам составляет 90—110% от целевого.
Если результаты демонстрируют соответствие вышеуказанным критериям, то
состав смеси и однородность содержимого серии являются приемлемыми и для
анализа следующей серии можно воспользоваться процедурой, описанной в пун-
кте 1.2.1. При негативных результатах применяют описываемый далее метод МСМ.
Глава 1.1. Надлежащая производственная практика требования FDA 67
2. Метод допустимых критериев можно применять, если налицо одно из
следующих условий:
2.1. Результаты предварительного установления критериев соответствуют
квалификации marginally pass.
2.2. Результаты предварительного установления критериев соответствуют
квалификации readily pass.
2.3. Серию тестировали согласно SCM, а результаты анализов не прошли по
критериям обеих стадий.
Если применим любой из двух вышеуказанных критериев, следует использовать
результаты, полученные на стадии 2 методом SCM, скорректированные по весу,
и сравнить их с критериями МСМ (для отдельных результатов RSD должно быть ме-
нее 6%, а среднее значение по всем результатам — составлять 90,0—110,0% от целе-
вого значения).
Допустйм переход к SCM, если пять последовательных серий проходят по крите-
риям МСМ, a RSDрезультатов меньше 5,0%.
1.1.8. Руководство для промышленности
«Масштабирование производства твердых пероральных
дозированных форм с немедленным высвобождением
и порядок внесения пострегистрационных
изменений (SUPAQ: химия, производство и контроль,
документирование исследований растворимости in vitro
и биоэквивалентности in vivo»
Данное Руководство представляет собой рекомендации спонсорам NDA и ANDA,
намеренным внести изменения в утвержденный продукт. Под изменениями понима-
ются любые модификации компонентов, перемены в составе продукта, смена места
производства, увеличение-уменьшение размеров серии и/или масштабов производ-
ственного процесса и/или оборудования при производстве лекарственных составов
с немедленным высвобождением.
1.1.8.1. Изменения в компонентах (вспомогательных веществах) и составах
Руководство не рассматривает такие показатели, как изменение количества лекар-
ственного вещества или его источника. Изменения в компонентах или составах,
такие как добавление нового вспомогательного вещества или разбавление старого,
регламентированы в правилах изменений третьего уровня.
1. Изменения первого уровня.
1.1. При внесении изменений первого уровня выявляемое изменение каче-
ства и характеристик состава маловероятно.
1.2. Разрешенные изменения (изменения, осуществление которых допустимо
без предварительного одобрения FDAl) представлены в табл. 1. Предпола-
гается, что содержание лекарственного вещества в продукте должно пол-
ностью соответствовать информации на маркировке. Чтобы изменения
были оценены как соответствующие первому уровню, общий ад цитив-
68
Часть I. Нормативное регулирование производства лекарственных средств
ный эффект от изменений во всех вспомогательных веществах не должен
превышать 5 %масс. относительно целевого значения веса дозированной
формы.
Таблица 1
Нормативные диапазоны изменений первого уровня
Вспомогательное вещество Доля наполнителя к суммарному целевому весу дозированной формы, %масс.
Наполнитель Дезинтегрант: крахмал другие Связующий агент Смазывающий агент: ±5 ±3 ±1 ±0,5
стеарат кальция или магния другие Глидант (агент, облегчающий скольжение): ±0,25 ±1 ±1
тальк ±0,1
другое Пленочное покрытие ±1
1.3. Аналитическая документация.
1.3.1. Химия: нормативные требования (или требования соответствия к высво-
бождению) и исследование стабильности. Для одной серии проводится
исследование стабильности при долгосрочном хранении, данные кото-
рого публикуются в ежегодном отчете.
1.3.2. Регистрационная документация: вся информация (в том числе данные по
стабильности при долгосрочном хранении) должна быть включена в еже-
годный отчет.
2. Изменения второго уровня.
2.1. Изменения второго уровня могут существенно повлиять на качество
состава и его характеристики. Исследования таких изменений и соот-
ветствующая регистрационная документация зависят от трех факторов:
1) терапевтического диапазона; 2) растворимости; 3) проницаемости.
Терапевтический интервал определяют как «узкий» или «расширенный»,
растворимость или проницаемость лекарства — как «высокую» или «низ-
кую». Изменения в содержании вспомогательных веществ, превышаю-
щие величины, представленные для первого уровня, однако попадающие
в указанные ниже процентные диапазоны, соответствуют изменениям
второго уровня (табл. 2).
Данная оценка процентных значений исходит из предположения, что со-
держание лекарственного вещества в готовом продукте составляет 100%
от указанной в маркировке величины. Общий аддитивный эффект от из-
менений во всех вспомогательных веществах не должен превышать 10%.
Глава 1.1. Надлежащая производственная практика и требования FDA
69
Таблица 2
Нормативные диапазоны изменений второго уровня
Вспомогательное вещество Доля наполнителя к суммарному целевому весу дозированной формы, %масс.
Наполнитель ±10
Дезинтегрант:
крахмал ±6
другие ±2
Связующий агент ±1
Смазывающий агент:
стеарат кальция или магния ±0,5
другие ±2
Глидант (агент, облегчающий
скольжение): ±2
тальк ±0,2
другое ±2
Пленочное покрытие
Всем компонентам состава должны соответствовать цифровые целевые зна-
чения, представляющие номинальный состав продукта. Допустимые изме-
нения состава должны исходить из одобрения целевого состава, а не соста-
ва, основанного на предыдущих изменениях первого или второго уровней.
2.2. Аналитическая документация.
2.2.1. Химия.
2.2.1.1. Нормативные требования (или требования соответствия) к высвобождению
и протоколы серий.
2.1.1.2. Исследованию на стабильность подвергают две производственные се-
рии: одну — в течение 3 мес. для получения дополнительных данных
ускоренного испытания на стабильность и другую — при долгосрочном
хранении.
2.2.2. Растворимость.
2.2.2.1. Лекарства с высокими показателями проницаемости и растворимости
должны растворяться не менее чем на 85% за 15 мин в 900 мл 0,1 N НС1.
Если лекарственный продукт не соответствует этому критерию, следует
провести испытание согласно пунктам 2.2.2.2 или 2.2.2.3.
2.2.2.2. Для лекарств с низкой проницаемостью и высокой растворимостью
строят многоточечные профили (кривые) растворения через промежут-
ки в 15,30,45,60 и 120 мин или вплоть до достижения асимптоты. Про-
фили растворения предложенного и используемого в настоящее время
составов должны быть одинаковы.
2.2.2.3. Для лекарств с высокой проницаемостью и низкой растворимостью
строят пять кривых растворения: в воде, 0,1 N НС1, а также в буферной
фармакопейной среде при pH 4,5, 6,5 и 7,5. Адекватный пробоотбор
должен производиться через промежутки в 15, 30, 45, 60 и 120 мин до
70 Часть I. Нормативное регулирование производства лекарственных средств
растворения 90% действующего вещества из лекарственного продукта
либо до достижения асимптоты. При необходимости можно исполь-
зовать сурфактант (поверхностно-активное вещество). Профили рас-
творения предложенного и используемого в настоящее время составов
должны быть одинаковы.
2.2.3. Для второго уровня документирование испытаний биоэквивалентности
необязательно. Если продукт не соответствует критериям первого уров-
ня (см. табл. 1), см. пункт 2.3.
2.2.4. Регистрационная документация представляет собой предварительно
одобренное приложение, содержащее все данные, включая результаты
ускоренного исследования стабильности. Эти изменения должны быть
представлены в ежегодном отчете наряду с данными исследования ста-
бильности при долгосрочном хранении.
2.3. Изменения третьего уровня.
2.3.1. Изменения третьего уровня, как правило, существенно затрагивают ка-
чество лекарственного продукта и характеристики его состава. Испыта-
ния и регистрационная документация меняются в зависимости от трех
факторов: терапевтического диапазона, растворимости и проницаемо-
сти. Примерами изменений третьего уровня являются:
2.3.1.1. Любые качественные и количественные изменения во вспомогательных
веществах, входящих в состав лекарства узкого терапевтического диа-
пазона, выходящие за рамки показателей, определенных в табл. 1.
2.3.1.2. Все прочие лекарства, не соответствующие критериям второго уровня.
2.3.1.3. Изменения диапазонов вспомогательных веществ в составе лекарств
с низкой растворимостью и малой проницаемостью, в дополнение к пе-
речисленным для первого уровня.
2.3.1.4. Изменения диапазонов вспомогательных веществ в составе всех ле-
карств, в дополнение к перечисленным в табл. 2.
2.3.2. Аналитическая документация.
2.3.2.1. Химия.
2.3.2.1.1. Нормативные требования (или требования соответствия) к высвобож-
дению и протоколы к серии:
— информация доступна: для одной серии в приложении представляют
данные ускоренного исследования стабильности в течение 3 мес.,
а для другой — данные по исследованию стабильности при долго-
срочном хранении, отражаемые в ежегодном отчете;
— информация недоступна: до трех серий в течение 3 мес. исследуют на
стабильность по процедуре ускоренного испытания (данные пред-
ставляют в приложении); одну серию исследуют на стабильность при
долгосрочном хранении (данные представляют в ежегодном отчете).
2.З.2.1.2. Документация по исследованиям растворения.
2.З.2.1.З. Документация исследований эквивалентности: полное исследование
биоэквивалентности. Выполнение этого требования можно отложить
Глава 1.1. Надлежащая производственная практика и требования FDA 71
при условии надлежащей верифицированной корреляции in vivo либо
in vitro.
2.2.3.2. Регистрационная документация: предварительно одобренное прило-
жение, включающее данные ускоренного исследования стабильности
и ежегодный отчет, содержащий данные по стабильности при долго-
срочном хранении.
1.1.8.2. Изменение производственной площадки
Под изменением местоположения понимается переезд производства. Изменение
местоположения не подразумевает масштабирования производства, смены произ-
водственного оборудования или производственного процесса, перемен в окружа-
ющей среде, а также модификаций стандартных операционных процедур (СОП),
представляющих собой документально оформленные инструкции по выполнению
рабочих процедур. Каждое изменение производственной площадки должно рассма-
триваться отдельно.
1. Изменения первого уровня связаны с переездом производства в пределах
одного здания без замены оборудования, СОП, климат-контроля или
персонала. Кроме того, они не предполагают изменений протоколов
к производственным сериям, кроме записей о перестановках оборудова-
ния или новых административных решениях.
1.1. Необходимая документация исчерпывается обычными документами, со-
держащими нормативные требования или требования соответствия. До-
кументация по исследованиям биоэквивалентности не требуется.
1.2. Регистрационная документация представляет собой ежегодный отчет.
2. Изменения второго уровня — это переезд производства в пределах близ-
лежащих зданий или кварталов с сохранением прежних оборудования,
СОП, климат-контроля и персонала. Изменения второго уровня не пред-
полагают никаких правок протоколов к производственным сериям, кро-
ме фиксации фактов перестановок оборудования или новых администра-
тивных решений.
2.1. Необходимая документация.
2.1.1. Химия процессов отражается в специальных документах, содержащих но-
вый адрес производства и обновленные протоколы к производственным
сериям. Документы должны содержать основные параметры процесса
высвобождения либо нормативные (фармакопейные) требования к нему;
новой документации не требуется, однако одна производственная серия
должна быть исследована на стабильность при долгосрочном хранении,
а полученные данные — представлены в ежегодном отчете. Кроме обыч-
ной информации, подтверждающей соответствие требованиям к высво-
бождению, не нужны ни данные по растворению, ни отчет об исследова-
ниях биоэквивалентности in vivo.
2.1.2. Регистрационная документация. Следует сохранять приложение, под-
тверждающее эффективность нововведений. В ежегодный отчет включа-
ются данные исследований стабильности при долгосрочном хранении.
72
Часть I. Нормативное регулирование производства лекарственных средств
3. Изменения третьего уровня связаны с переносом производства на пло-
щадку, находящуюся в другом комплексе производственных зданий.
При этом оборудование, СОП и климат-контроль остаются прежними, а
персонал меняется. В протоколы к производственным сериям не вносят
никаких изменений, кроме административной информации, указания
нового местоположения производства либо перевода на другой язык там,
где это необходимо.
3.1. Документация.
3.1.1. Химия процессов отражается в специальных документах, содержащих но-
вый адрес производства и обновленные протоколы к производственным
сериям.
3.1.2. Стабильность.
3.1.2.1. Если доступен значительный массив данных, в приложении представля-
ют результаты трехмесячных испытаний стабильности одной из серий.
Другую серию исследуют на стабильность при долгосрочном хранении;
результаты фиксируют в ежегодном отчете.
3.1.2.1. При недостаточном объеме имеющихся данных в приложение заносят
результаты ускоренных испытаний стабильности для трех производ-
ственных серий. До трех серий испытывают на стабильность при долго-
срочном хранении; результаты отражают в ежегодном отчете.
3.1.3. Растворимость. Многоточечный профиль растворения строят через про-
межутки в 15, 30, 45, 60 и 120 мин либо до достижения асимптоты. Эти
данные отражаются в ежегодном рапорте.
3.1.4. Документация по исследованиям биоэквивалентности in vivo. Необязательна.
3.2. Требования к регистрационной документации. Все изменения заносят
в специальное приложение. Данные по исследованию стабильности при
долгосрочном хранении представляют в ежегодном отчете — в досье на
регистрацию.
1.1.8.3. Изменения размеров производственных серий
Корректировка уже утвержденных размеров серии продукта, предназначенного
для клинических испытаний, должна быть отражена в приложении. Сокращение
масштабов производства до менее чем 100 000 единиц дозирования не подпадает
под регламентацию данного Руководства. Вместе с тем любое увеличение произ-
водственных серий должно пройти валидацию, а при необходимости — инспекцию
уполномоченным персоналом FDA.
1. Изменения первого уровня связаны с не менее чем десятикратным увели-
чением (или сокращением) размера производственной серии по сравне-
нию с пилотной серией. При этом производитель должен обеспечить вы-
полнение следующих условий: 1) необходимо сохранить принцип функ-
ционирования применяемого оборудования и конструкцию последнего;
2) производство продукта должно полностью отвечать требованиям GMP-,
3) процедуры приготовления и производства состава лекарственных про-
дуктов, а также СОП и процедуры контроля должны быть тождественны
Глава 1.1. Надлежащая производственная практика и требования FDA 73
тем, что применялись в процессе производства пилотной производствен-
ной серии.
1.1. Документация по химии включает в себя: 1) основные параметры процес-
са либо нормативные (фармакопейные) требования к высвобождению;
2) уведомление об изменениях, представляемое в FDA, а также обнов-
ленные протоколы к сериям в ежегодном отчете; 3) результаты проверки
одной серии на стабильность при долгосрочном хранении, представлен-
ные в ежегодном отчете.
1.2. Документация по растворимости отраничивается требованиями к заявке
либо нормативными предписаниями к высвобождению.
1.3. Биоэквивалентность in vivo. Не требуется.
1.4. Досье на регистрацию представляет собой ежегодный отчет, содержащий
данные исследований стабильности при долгосрочном хранении.
2. Изменения второго уровня — это более чем десятикратное увеличение (или
сокращение) размера производственной серии по сравнению пилотной,
предполагающее: 1) сохранение принципов функционирования и кон-
струкции применяемого оборудования; 2) полное соответствие лекар-
ственной продукции требованиям GMP-, 3) применение тех же процедур
приготовления и производства состава, а также тех же СОП и процедур
контроля, что и при производстве пилотной серии.
2.1. Документация по химии содержит: 1) основные параметры процесса либо
нормативные (фармакопейные) требования к высвобождению; 2) уве-
домление об изменениях, представляемое в FDA, а также обновленные
протоколы к сериям в ежегодном отчете. Для одной серии следует прове-
сти экспресс-испытание на стабильность при хранении, для еще одной —
на стабильность при долгосрочном хранении.
2.2. Растворимость: основные параметры процесса либо нормативные (фар-
макопейные) требования к высвобождению; дополнительной докумен-
тации не требуется.
2.3. Биоэквивалентность in vivo. Не требуется.
2.4. Досье на регистрацию должно содержать все изменения, изложенные
в приложении. Данные исследований стабильности при долгосрочном
хранении отражаются в ежегодном отчете.
1.1.8.4. Изменения в производственном процессе
К данным изменениям относятся модификации производственного процесса или
оборудования, участвующего в последнем.
1. Оборудование.
1.1. Изменения оборудования первого уровня предполагают переход от неавто-
матизированного и немеханизированного оборудования к автоматизиро-
ванному и механизированному, позволяющему перемещать ингредиенты,
а также переход на альтернативное оборудование прежней конструкции,
но с другой (или той же) производительностью.
74
Часть I. Нормативное регулирование производства лекарственных средств
1.1.1. Документация по химии включает в себя: 1) основные параметры процес-
са либо нормативные (фармакопейные) требования к высвобождению;
2) уведомление об изменениях, представляемое в FDA, а также обнов-
ленные протоколы к сериям в ежегодном отчете; 3) результаты проверки
одной серии на стабильность при долгосрочном хранении, представлен-
ные в ежегодном отчете.
1.1.2. Документация по растворимости ограничивается требованиями к заявке
либо нормативными предписаниями к высвобождению.
1.1.3. Документация по биоэквивалентности in vivo. Не требуется.
1.1.4. Досье на регистрацию представляет собой ежегодный отчет с данными ис-
следований стабильности при долгосрочном хранении.
1.2. Изменения оборудования второго уровня включают в себя переход на обору-
дование другой конструкции и/или иных принципов функционирования.
1.2.1. Документация по химии включает в себя: 1) основные параметры про-
цесса высвобождения либо нормативные (фармакопейные) требования
к нему; 2) уведомление об изменениях и обновленные протоколы к сери-
ям в ежегодном отчете; 3) результаты проверки одной серии на стабиль-
ность при долгосрочном хранении.
1.2.1.1. Если доступен значительный массив данных, в приложении представ-
ляют результаты трехмесячных испытаний стабильности для одной из
серий. Другую серию исследуют на стабильность при долгосрочном хра-
нении; результаты фиксируют в ежегодном отчете.
1.2.1.2. Если доступа к большому массиву данных нет, в приложение заносят ре-
зультаты экспресс-испытаний стабильности для трех производственных
серий. До трех серий испытывают на стабильность при долгосрочном
хранении; результаты отражают в ежегодном отчете.
1.2.2. Документация по растворимости. Многоточечный профиль растворимо-
сти исследуют в надлежащей и фармакопейной средах через 15, 30, 45,
60, 120 мин или до достижения асимптоты. Профили растворения лекар-
ственного продукта, произведенного на используемом и предложенном
оборудовании, должны совпадать.
1.2.3. Документация по биоэквивалентности in vivo. Не требуется.
1.2.4. Досье на регистрацию представляет собой предварительно одобренное
приложение, содержащее обоснование необходимости внесения изме-
нений. Данные исследований стабильности при долгосрочном хранении
включают в ежегодный отчет.
2. Изменения процесса.
2.1. Изменения процесса первого уровня включают изменения времени смеши-
вания и рабочей скорости в рамках диапазонов применения/валидации.
2.1.1. Документация по химии содержит основные параметры процесса высво-
бождения либо нормативные (фармакопейные) требования к нему.
2.1.2. Документация по растворимости представляет собой требования к заявке
либо нормативные требования к высвобождению.
Глава 1.1. Надлежащая производственная практика и требования FDA
75
2.1.3. Документация по биоэквивалентности in vivo. Не требуется.
2.1.4. Досье на регистрацию. Ежегодный отчет.
2.2. Изменения процесса второго уровня связаны с такими параметрами про-
цесса, как время смешивания и рабочие скорости, выходящие за рамки
диапазонов применения или валидации.
2.2.1. Документация по химии содержит основные параметры процесса высво-
бождения либо нормативные (фармакопейные) требования к нему, а так-
же уведомление об изменениях и предоставление протоколов к сериям
с обновлениями. Одну серию исследуют на стабильность при долгосроч-
ном хранении.
2.2.2. Документация по растворимости. Многоточечный профиль растворимо-
сти исследуют в надлежащей и фармакопейной средах через 15,30,45,60,
120 мин или же до достижения асимптоты. Профили растворения лекар-
ственного продукта, произведенного на используемом и предложенном
оборудовании, должны совпадать.
2.2.3. Документация по биоэквивалентности in vivo. Не требуется.
2.2.4. Досье на регистрацию представляет собой предварительно одобренное
приложение, содержащее разъяснение необходимости изменений. Дан-
ные исследований стабильности при долгосрочном хранении включают
в ежегодный отчет.
2.3. Изменения процесса третьего уровня затрагивают тип процесса, применя-
емого при производстве продукта. Примером такого изменения является
переход от влажного гранулирования к осевому сжатию.
2.3.1. Документация по химии содержит основные параметры процесса высво-
бождения либо нормативные (фармакопейные) требования к нему, а так-
же уведомление об изменениях и предоставление протоколов к сериям
с обновлениями. Одну серию исследуют на стабильность при долгосроч-
ном хранении. Исследования стабильности зависят от величины массива
доступных данных:
2.3.1.1. Если доступен значительный массив данных, в приложении представ-
ляют результаты трехмесячных испытаний стабильности для одной из
серий. Другую серию исследуют на стабильность при долгосрочном хра-
нении; результаты фиксируют в ежегодном отчете.
2.3.1.2. Если доступа к большому массиву данных нет, в приложение заносят ре-
зультаты экспресс-испытаний стабильности для трех производственных
серий. До трех серий испытывают на стабильность при долгосрочном
хранении; результаты отражают в ежегодном отчете.
2.3.2. Документация по растворимости. Многоточечный профиль растворимо-
сти исследуют в надлежащей и фармакопейной средах через 15,30,45,60
и 120 мин либо до достижения асимптоты. Профили растворения лекар-
ственного продукта, произведенного на используемом и предложенном
оборудовании, должны совпадать.
76
Часть I. Нормативное регулирование производства лекарственных средств
2.3.3. Документация исследований биоэквивалентности. Выполнение этого тре-
бования можно отложить при условии надлежащей верифицированной
корреляции in vivo или in vitro.
2.3.4. Досье на регистрацию. Предварительно одобренное приложение следует
хранить вместе с объяснением необходимости изменений. Данные ис-
следований стабильности при долгосрочном хранении включают в еже-
годный отчет.
1.1.9. Другие руководящие документы, связанные с GMP
В этом разделе перечислены нормативные требования cGMPn некоторые из наибо-
лее важных руководящих документов. Существует ряд дополнительных руководя-
щих документов, связанных с GMP и изданных FDA Все эти документы выложены
на сайте FDA. Ниже перечислены их названия и электронные адреса.
> Текущая надлежащая производственная практика для комбинированных
продуктов: http://www.fda.gov/cder/guidance/OCLoveldft.pdf.
> Формальное разрешение конфликтных ситуаций. Научные и технические во-
просы, связанные с cGMP в фармацевтической промышленности: http://www.
fda.gov/cder/guidance/588Cfhl.pdf.
> Вопросы и ответы о сСМРцля. лекарственных средств: http://www.fda.gov/cder/
guidance/cGMPs/default.htm.
> Порошковые смеси и готовые формы дозирования. Стратифицированный
отбор проб единиц дозирования в процессе производства и оценка данных:
http://www.fda.gov/cder/guidance/583ldft.pdf.
> Стерильные лекарственные продукты, изготовленные методом асептической
переработки. Текущая надлежащая производственная практика: http://www.
fda.gov/cder/guidance/5882fnl.pdf
> Текущая надлежащая производственная практика при производстве меди-
цинских газов: http://www.fda.gov/cder/guidance/3823dft.pdf
> Общие принципы валидации процессов: http://www.fda.gov/cder/guidance/pv.htm.
> SUPAC-IR. Твердые формы дозирования с немедленным высвобождением
для перорального введения: масштабирование производства и пострегистра-
ционные изменения. Химия, производство и формы контроля; документа-
ция по исследованию растворения in vitro и биоэквивалентности in vivo', http://
www.fda.gov/cder/guidance/cmc5.pdf.
> SUPAC-IR/MR. Приложение к производственному оборудованию для изго-
товления твердых дозированных форм для перорального введения с немед-
ленным высвобождением и контролируемым высвобождением: http://www.
fda.gov/cder/guidance/1721fnl.pdf
> SUPAC-MR. Масштабирование производства твердых дозированных форм
для перорального введения с контролируемым высвобождением и постреги-
страционные изменения. Химия, производство и формы контроля; докумен-
тация по исследованиям растворения in vitro и биоэквивалентности in vivo:
http://www.fda.gov/cder/guidance/1214fhl.pdf.
Глава 1.1. Надлежащая производственная практика и требования FDA
> SUPAC-SS. Нестерильные полутвердые дозированные формы; масштабиро-
вание производства и пострегистрационные изменения. Химия, производ-
ство и формы контроля; документация по исследованиям растворения in vitro
и биоэквивалентности in vivo: http://www.fda.gov/cder/guidance/1447fnl.pdf.
> SUPAC-SS. Приложение к производственному оборудованию для изготовле-
ния нестерильных полутвердых дозированных форм: http://www.fda.gov/cder/
guidance/1722dft.pdf.
Глава 1.2. ВНЕДРЕНИЕ ТЕКУЩЕЙ НАДЛЕЖАЩЕЙ
ПРОИЗВОДСТВЕННОЙ ПРАКТИКИ
Кеннет Дж. Нолан
Nolan & Auerbach, (Форт-Лодердейл,Флорида)
1.2.1. Введение
Право Федерального управления по контролю за пищевой продукцией и лекар-
ственными средствами (FDA) устанавливать производственные стандарты закре-
плено в Законе о пищевых продуктах, медикаментах и косметической продукции
(FDCA) (том 21 USC, § 301 и далее, а также § 351(a)(2)(B)). Соответствующие норма-
тивы собраны главным образом в разделах 210 и 211 21-го тома Свода федеральных
нормативных актов (CFR) и носят название правил Текущей надлежащей производ-
ственной практики (cGMP).
Правила cGMP пришли на смену старым системам контроля качества, сводив-
шимся к изъятию тех лекарств, несоответствие которых спецификациям было уже
подтверждено. Новые же Правила были призваны исключить поступление в про-
дажу некондиционных лекарственных средств. Таким образом, американских про-
изводителей обязали соблюдать Надлежащие производственные практики, раз-
работанные для предотвращения загрязнений фармацевтических препаратов, их
ненадлежащей биодоступности или эффективности.
Конгресс США аргументировал необходимость внедрения cGMP в фармацевти-
ческое производство следующим образом1:
«Производство лекарственной продукции требует привлечения высококвалифициро-
ванного и специально обученного персонала, наличия специализированной лаборато-
рии и тщательного контроля производства, упаковки и маркировки. Выполнение этих
требований необходимо д ля гарантирования безопасности лекарственного продукта
для потребителя, а также соответствия таким заявленным параметрам, как идентич-
ность, содержание активного компонента, качество, чистота и эффективность».
Целью cGMP является «интеграция системы менеджмента качества в планиро-
вание и производство фармацевтической продукции»2, гарантирующая, что рецеп-
турные лекарственные средства, не соответствующие стандартам, не будут угрожать
здоровью потребителей.
Правила cGMP требуют от производителей наличия соответствующего произ-
водственного оборудования, специально обученного персонала, точного контроля
производственных процессов, надлежащего лабораторного контроля, ведения пол-
1 Н. R. Rep. No. 2464, 87th Cong., 2d Sess. 2 (1962). См. также 1962 U. S. Cong. And Admin. News,
p. 2884.
2 FDA, cGMP на фармацевтическом производстве XXI в.: подход, основанный на оценке
рисков. Rockville, MD, 21 августа 2002 г.
Глава 1.2. Внедрение текущей надлежащей производственной практики 79
них и точных записей и протоколов, надлежащего исследования готовой продукции
и т. д. Не претендуя на роль эталонных процедур, эти Правила скорее устанавливают
пороговые, или минимальные, стандарты, обязательные для фармацевтических про-
изводственных операций.
За период с 1963 по 2002 г. Правила cGMP редактировались только раз — в 1978 г.
Поправки были связаны с появлением новых технологий и были направлены на
более подробное и конкретное изложение Правил. Вместе с тем за последние де-
сятилетия производственная и инженерная наука ушла далеко вперед. Инновации
коснулись, в частности, разработки усовершенствованных систем менеджмента ка-
чества (СМК). Этот прогресс, а также желательность координации производствен-
ных стандартов на фоне глобализации промышленного производства создали пред-
посылки для внесения новых поправок в cGMP.
В августе 2002 г. FDA объявило о всестороннем пересмотре правил фармацевти-
ческой cGMP. Данная инициатива получила название «Правила организации про-
изводства лекарственных средств в XXI в.: подход, основанный на анализе рисков»
и преследовала следующие цели:
> координация программы анализа подаваемых на рассмотрение заявок и пла-
на инспекций;
> последовательное применение юридических и производственных стандар-
тов;
> оптимальное использование ресурсов FDA, обеспечивающее наиболее эф-
фективное и оперативное решение вопросов, связанных с существенными
рисками д ля здоровья человека.
Один из основных результатов инициативы по cGMP был опубликован FDA
в сентябре 2006 г. в рамках документа «Руководство для промышленности. Си-
стемы менеджмента качества: подход к правилам cGMP для фармацевтического
производства»1. Основной задачей Руководства было описание модели всеобъем-
лющей СМК, позволяющей улучшить контроль качества и обеспечить соответствие
требованиям cGMP.
Согласно нормативам FDCA, лекарства, произведенные без соблюдения тре-
бований cGMP, включая требование о контроле качества и управлении качеством,
признаются «некондиционными». Так, в соответствии со статьей 351 тома 21 USC,
«...лекарство признается «некондиционным», если методы, оборудование или сред-
ства контроля, применяемые для его производства, обработки, упаковки или хране-
ния, не соответствуют cGMP либо применяются без учета требований Текущей над-
лежащей производственной практики, обеспечивающей соответствие производимого
лекарства требованиям закона по параметрам безопасности, подлинности и содержа-
нию активного вещества, а также заявленным характеристикам качества и чистоты,
которыми лекарство должно обладать».
1 Руководящие документы FDA содержат пояснения, оговаривающие, что они не устанав-
ливают юридической ответственности и должны рассматриваться как рекомендации. Ис-
ключения составляют лишь случаи, когда они содержат ссылки на конкретные нормативные
документы или законы.
80
Часть I. Нормативное регулирование производства лекарственных средств
1.2.2. Органы регулирования
Очевидно, что FDA является одним из самых влиятельных регулирующих органов
США. Кроме того, можно утверждать, что FDA — наиболее авторитетная в мире ор-
ганизация, защищающая права потребителей. Ее решения, в частности по утверж-
дению лекарств, оказывают непосредственное влияние на тестирование, одобре-
ние, допуск и распределение рецептурных лекарственных препаратов в целом ряде
стран. Как регулирующий орган, Управление занимается вопросами фармацевти-
ческой науки и проблемами, связанными с доступностью лекарств по всему миру.
В качестве научной организации FDA привлекает к работе медиков, фармацевтов,
биологов, биохимиков, инженеров, биостатистиков и других высокообразованных
и прошедших специальное обучение профессионалов.
Кроме того, в сферу ответственности FDA входят важные функции отслежива-
ния исполнения законов. В Управлении трудятся специалисты по гражданским
и уголовным расследованиям, аудиторы, адвокаты и другие юристы. Среди многих
функций контроля, осуществляемых FDA, — проведение расследований, коррекци-
онных действий, а также преследование за нарушение требований cGMP.
Региональные подразделения FDA работают под патронажем Отдела нормативно-
правового регулирования {Office of Regulatory Affairs, ORA) и насчитывают пять от-
делений: Северо-Восточное, Центральное, Юго-Восточное, Юго-Западное и Ти-
хоокеанское. В структуру каждого регионального отделения входят окружные
отделения, 20 из которых являются общегосударственными. Как правило, окруж-
ные отделения включают три-четыре подразделения, в том числе по оценке соот-
ветствия {compliance branch) или принудительному правоприменению {enforcement
branch). Эти подразделения осуществляют первичный правовой надзор в округах,
являясь «глазами и ушами» FDA.
Отдел криминальных расследований {Office of Criminal Investigations, OCI) зани-
мается профилактикой уголовных правонарушений, в частности связанных с по-
тенциальным нарушением правил cGMP. Инспекторы OCIпроводят расследования
так, как считают нужным, иногда в сотрудничестве с другими федеральными след-
ственными органами, включая ФБР и Управление генерального инспектора Мини-
стерства здравоохранения и социальных служб. Если OCIне находит оснований для
того, чтобы рекомендовать Министерству юстиции США {DOJ)1 начать уголовное
разбирательство в отношении того или иного лица, то окружное отделение име-
ет право предъявить предполагаемому правонарушителю административный или
гражданский иск.
Хотя в составе FDA есть Отдел генерального юрисконсульта {Office of General
Counsel), занимающийся как гражданскими, так и уголовными делами, случаи
судебного вмешательства контролируют атторнеи — помощники федерального
прокурора США. Атторнеи являются региональными представителями DOJ. Их
назначает и контролирует президент после одобрения сенатом. Общее число атгор-
1 Министерство юстиции США возглавляет генеральный атторней. В контексте настоящей
главы его задача состоит в том, чтобы обеспечить выполнение федеральных законов. Мини-
стерство юстиции расследует случаи правонарушений, на которые указывают FDA и другие
федеральные службы.
Глава 1.2. Внедрение текущей надлежащей производственной практики
81
неев — 93. Каждый атторней возглавляет контроль за исполнением федеральных
законов США в своем округе.
Атторней является главным представителем правительства США на судебных
разбирательствах. При этом решающее слово всегда принадлежит, разумеется, суду
и экспертизе. Тщательность рассмотрения того или иного случая нарушения тре-
бований cGMP зависит от возможностей конкретного представительства атторнея
США, от опыта его сотрудников и от других причин.
Устранить подобные недочеты призвана Служба рассмотрения потребительских
исков (Office of Consumer Litigation, ОСЬ), подконтрольная DOJ. Эта Служба занима-
ется проведением судебных преследований, инициированных FDCA. Она пользует-
ся большим авторитетом и обладает значительной свободой действий при принятии
решения о возбуждении судебного преследования или отказе от него. Многие раз-
новидности гражданских исков требуют обязательного одобрения ОСЬ, что сводит
к минимуму последствия возможных ошибок представительств атторнеев США.
1.2.3. Методы регулирования FDA
1.2.З.1. Инспекции
Федеральное управление США по контролю за пищевой продукцией и лекарства-
ми имеет право инспектировать производственное оборудование на предмет его
соответствия требованиям законодательства. Задача подобных инспекций состо-
ит в том, чтобы «минимизировать риск отравления потребителя некондиционной
продукцией»1.
Статья 374 тома 21 USC устанавливает, что FDA имеет право доступа на любое
предприятие и может «инспектировать в разумные сроки, в разумных пределах
и разумным путем... всё применяемое оборудование, готовые и перерабатывае-
мые материалы, контейнеры и маркировку» на производственном или связанном
с производством предприятии. Кроме того, закон устанавливает право инспекции
«изучать все материалы, относящиеся к делу (включая записи, картотеки, докумен-
тацию, процессы, контроль и оборудование)», поскольку подобная информация
может указывать на потенциальную фальсификацию или неправильную маркиров-
ку2, а также на другие нарушения правил FDCA.
Закон лишает FDA права подвергать инспекции финансовую документацию,
а также запрашивать информацию о персонале (за исключением данных по квали-
фикации технического и профессионального персонала, выполняющего производ-
ственные задачи). Не подлежит инспектированию документация по ценам, объемам
продаж, а также некоторая другая.
Инспекторы должны уведомить компанию о предстоящем визите; впрочем, цели
последнего они раскрывать не обязаны. Инспекторы могут отбирать пробы и делать
любые фотографии, имеющие отношение к предмету инспекции. Недопущение ин-
1 Compliance Program Guidance Manual for FDA Staff: Drug Manufacturing Inspection Program.
7356.002. (Документ доступен по адресу www.fda.gov.)
2 Неправильная (незаконная) маркировка фармацевтического продукта содержит инфор-
мацию, вводящую в заблуждение. См. 21 USC 331(fc).
82 Часть I. Нормативное регулирование производства лекарственных средств
спекторов FDA и других официальных лиц, надлежащим образом подготовивших
проведение инспекции, является уголовным преступлением (21 USC331(f)).
В дополнение к инспекциям по конкретному основанию, FDCA предоставляет
FDA право проводить регулярные инспекции производственных предприятий на
предмет выполнения требований cGMP. Подобные инспекции могут проводиться
каждые два года. Действие 21-го тома распространяется на отечественные и зару-
бежные предприятия, производящие лекарства, продаваемые на территории США1.
На практике временной интервал между инспекциями на одном предприятии за-
частую составляет более двух лет. Это связано с недостаточными людскими ресурса-
ми окружных управлений FDA, ответственных за проведение подобных инспекций.
Инспекции промышленных объектов, проводимые FDA, подразделяются 1) на
надзорные и 2) инспекции на соответствие правилам cGMP. Надзорные инспекции
носят периодический характер. Сроки проведения подобных инспекций на кон-
кретном производственном предприятии определяются исходя из разных сообра-
жений, в том числе из показаний аналитической модели для выявления объектов
высокого риска. В конце 2004 г. FDA опубликовало доклад, озаглавленный как «Ме-
тодика определения очередности инспекций фармацевтических производственных
предприятий по контролю за соблюдением правил cGMP, основанная на оценке
рисков: пилотная модель оценки рисков». Данная методика позволяет ранжиро-
вать риски несоответствия производственных предприятий требованиям cGMP по-
средством анализа, состоящего из следующих этапов: 1) постановки вопроса о воз-
можных рисках; 2) выявления потенциальных рисков; 3) обнаружения факторов,
которые можно использовать в качестве переменных для количественной оценки
рисков; 4) проведения математического комбинирования переменных для оценки
общего показателя риска {riskscore). Со временем новая методика пополнилась еще
одним этапом: учетом данных о неблагоприятных побочных явлениях. Предполага-
ется, что надзорные инспекции включают аудит двух или более систем2 с обязатель-
ным аудитом СМК3.
Инспекции по контролю соответствия проводятся для оценки результатов кор-
ректирующих действий, а также для подтверждения правильности выбора послед-
них. Само собой, такая оценка дается уже после выявления проблемы и проведе-
ния всех необходимых действий. Объектами инспекций по контролю соответствия
выступают участки, признанные несоответствующими нормативам и требующими
корректирующих действий. Одной из разновидностей подобных инспекций являет-
ся инспекция по конкретному основанию, направленная на расследование опреде-
ленной проблемы, попавшей в фокусе внимания FDA. Причинами проведения ин-
спекции по контролю соответствия могут быть экстренные сообщения, претензии
к предприятиям или отзывы продукции.
1 Другая базовая стратегия проверки соблюдения правил cGMP состоит в отборе проб ле-
карств в процессе производственного контроля, а также отборе и анализе лекарственных
продуктов в процессе распределения.
2 В правилах cGMP FDA выделяет шесть систем: менеджмента качества, аппаратуры и обо-
рудования, продукции, материалов, упаковки и маркировки, а также лабораторного контроля.
3 Compliance Program Guidance Manual, 7356.002, 1 февраля 2002 г.
Глава 1.2. Внедрение текущей надлежащей производственной практики
83
В течение 2005-го финансового года местными подразделениями FDA было про-
ведено 1437 инспекций по контролю за соблюдением требований cGMP. Их резуль-
татом стали 15 писем-предупреждений, шесть судебных постановлений и одно на-
ложение ареста. Эти действия по принуждению к исполнению закона обсуждаются
далее. Данные по 2000—2005 гг. представлены на рис. 1 и 2.
Внутренние и внешние инспекции
Рис. 1. Данные по результатам инспекций CDER
(Центра оценки и исследований лекарственных средств) с 2000 по 2005 г
Инспекции
Н Внутренние пробы
И Импортные пробы
Рис. 2. Надзорная деятельность (источник: FDA)
1.2.3.2. После инспекции: Форма 483
Если инспектор находит отклонения от нормативов cGMP, он заполняет форму
/ВЛ-483 «Наблюдения инспекции», где детально описывает все нарушения. Список
обнаруженных нарушений отправляют производителю, которому предоставляется
возможность ответить на предъявленные претензии. Ниже приведен ряд ключевых
положений Формы /ВЛ-483.
Данный документ включает перечисление замечаний, сделанных представите-
лем(-ями) FDA во время инспекции вашего предприятия. Эти замечания представля-
ют собой наблюдения инспекции и не содержат итогового решения Управления от-
носительно соответствия вашего предприятия нормативным требованиям. Если у вас
есть возражения по замечаниям либо если вы уже провели (или планируете провести)
соответствующие корректирующие действия, вы можете обсудить ваши возражения
или действия с представителем(-ями) FDA в ходе инспекции либо представить всю
необходимую информацию в FDA по вышеуказанному адресу. Для разрешения кон-
кретных вопросов свяжитесь с FDA письменно или по телефону (адреса и номера теле-
фонов указаны выше).
84 Часть I. Нормативное регулирование производства лекарственных средств
Большинство производителей представляют письменный ответ на FD/-483, где
либо оспаривают замечания, либо сообщают, какие корректирующие действия они
намерены предпринять. Переговоры, как правило, длятся месяцами и даже годами
и завершаются либо снятием всех выявленных инспекцией проблем, либо решени-
ем FDA о принуждении производителя к соблюдению закона. Управление оставляет
за собой право применения любых принудительных мер, если придет к заключению
о существенном риске нанесения вреда потребителям лекарств, причем действия по
принуждению тем более вероятны, чем выше такая возможность.
В дополнение к Форме FZM-483 инспекторы FDA подготавливают отчет о про-
верке предприятия (EIR — establishment inspection report)). Отчет отсылают в штаб-
квартиру FDA, где его анализируют и при необходимости определяют перечень кор-
ректирующих действий. Далее инспекции классифицируют по результату: «не было
выявлено отклонений»; «были выявлены нежелательные факты, но они сочтены
незначительными»; «предписаны административные меры». Отчет EIR содержит
гораздо больше подробностей, чем Форма 483, и не предоставляется производителю
вплоть до окончания инспекции.
Согласно материалам последних отчетов по Форме FZM-4831, чаще всего сооб-
щалось о следующих двух нарушениях:
1) более половины отчетов содержали замечание о нарушении §211.100(b) 21-го
тома CFR (несоблюдение процедур контроля продукции и производственных
процессов и/или неправильное ведение документации);
2) 42% отчетов содержали замечание о нарушении § 122(d) 21-го тома CFR (от-
сутствие надлежащих документов с перечислением ответственных лиц, а так-
же отсутствие письменных процедур для отдела контроля качества или их не-
соблюдение).
Далее перечислены еще восемь нарушений в порядке убывания частоты их ре-
гистрации:
> отсутствие письменных процедур контроля фармацевтической продукции
и производственных процессов;
> отсутствие тестирования лекарственного средства перед отпуском, подтверж-
дающего удовлетворительное соответствие готового продукта итоговым спе-
цификациям, его подлинность, а также содержание в нем каждого активного
ингредиента;
> протоколы к производственным сериям и контрольные записи не были под-
готовлены или оказались неполными;
> процедуры контроля, направленные на мониторинг готовой продукции
и/или валидацию параметров производственного процесса, способных стать
причиной вариабельности характеристик лекарственного средства, не были
утверждены;
> персонал не прошел надлежащего обучения;
1 Аналитические данные были собраны FDA по материалам отчетов 614 Turbo EIR, прово-
дившихся с 2001 по 2003 г. (Инспекторы FDA вводят свои замечания в систему Turbo EIR FDA.
Электронный формат Turbo позволяет инспектору выбрать конкретное рассматриваемое на-
рушение правил cGMP, а затем объяснить факты, выявленные в ходе инспекции.)
Глава 1.2. Внедрение текущей надлежащей производственной практики 85
> лабораторный контроль не включает утверждения соответствующих научно
обоснованных спецификаций (стандартов, планов пробоотбора, аналитиче-
ских процедур);
> производство лекарственного продукта и контрольные записи не заверены
отделом контроля качества для подтверждения соответствия всем утвержден-
ным и одобренным письменным процедурам перед отпуском или распределе-
нием производственной серии;
> процедуры рассмотрения письменных и устных претензий к лекарственному
средству не утверждены или не соблюдаются.
1.2.3.3. Отзыв
В главе 7 Руководства по применению регулирующих процедур (Regulatory Procedures
Manual', опубликован в марте 2007 г., доступен на сайте www.fda.gov) представлены
подробные инструкции для персонала FDA об отзыве продукции. Согласно FDCA,
FDA не правомочно просто «приказать» производителю отозвать фармацевтический
препарат1.
Вместе с тем производителей или дистрибьюторов лекарственных средств поо-
щряют к добровольному отзыву продукции для исполнения обязательств по защите
потребителей. Нередко компании, обнаружив дефекты в одном из своих продуктов,
отзывают его по собственной инициативе; в противном случае FDA информирует
компанию о том, что определенный продукт имеет недостатки, и просит о его от-
зыве2.
На рис. 3,4 представлены сведения по отзывам на 2005 г.
• Класс I 18
• Класс II 314
• Класс III 170
• Всего 502
Рис. 3. Отзывы за 2005 г. по классам (источник: Национальный отчет CDER)
При инициации добровольного отзыва FDA обычно следует следующему прото-
колу:
1. Классификация отзыва. Специалисты FDA анализируют соответствующую
информацию, после чего классифицируют отзыв согласно уровню риска для
здоровья потребителя.
> Отзывы класса I относятся к лекарственным средствам, способных при-
вести к серьезным проблемам со здоровьем и даже к летальному исходу.
1 Впрочем, в ряде случаях FDA всё же имеет право отзыва лекарств (например, это каса-
ется препаратов, предназначенных для грудных детей, а также биологических продуктов и
устройств, «представляющих серьезную опасность для здоровья», но не включающих фарма-
цевтических продуктов.
2 Если компания никак не отреагировала на эту просьбу, то FDA может потребовать судеб-
ного разбирательства.
86
Часть I. Нормативное регулирование производства лекарственных средств
Число отзывов
|г_ Лекарства, отпускаемые
I без рецепта
Лекарства, отпускаемые
по рецепту
Рис. 4. Отзывы фармацевтических продуктов. На одну из фирм приходится более
100 отзывов за 2005 г., что явилось причиной высокого количества отзывов в указанном
году (источник: Национальный отчет CDER)
> Отзывы класса II относятся к лекарственным средствам, дефекты которых
вызывают только временные проблемы со здоровьем или же вероятность
возникновения серьезных заболеваний из-за которых невелика.
> Отзывы класса III относятся к лекарственным средствам, при употребле-
нии которых маловероятно неблагоприятное воздействие на здоровье, но
изготовление и маркировка которых не соответствуют требованиям FDA.
2. Мониторинг и аудит отзывов. Специфика надзора за отзывом лекарственных
средств определяется в зависимости от риска последних для здоровья. Для
отзывов класса I FDA проводит проверки, направленные на гарантирование
полного отзыва дефектного продукта. Напротив, при отзывах класса III над-
зор FDA может быть ограничен выборочной проверкой.
Согласно статистике FDA, основными причинами отзыва лекарственных про-
дуктов на 2005 г. стали:
> различные нарушения требований сбЛ/Р(отличные от нижеперечисленных);
> несоответствие нормативам теста Фармакопеи США на растворимость;
> загрязнение нестерильных продуктов микроорганизмами;
> недостаточная эффективность;
> наличие примесей и/или продуктов разложения;
> недостаточные гарантии стерильности;
> недостаточная стабильность продукта;
> неверное указание активного вещества в маркировке;
> неправильная маркировка: рекламные материалы с неподтвержденными
лечебными свойствами;
Глава 1.2. Внедрение текущей надлежащей производственной практики
87
> правильно маркированный продукт в несоответствующих картонаже или
упаковке.
3. Извещение и публичное предупреждение. Отзывы класса I почти всегда со-
провождаются пресс-релизом для средств массовой информации. Сообще-
ния в прессе об отзывах классов II и III не обязательны, однако включаются
в еженедельный отчет FDA о мерах принуждения, публикуемый на сайте ww.
fda.gov/opacom/Enforce.html.
4. Окончание. Федеральное управление США по контролю за пищевой продук-
цией и лекарствами представляет письменное уведомление производителю,
осуществляющему отзыв продукта, о сроках окончания отзыва.
5. Несоответствие. При необходимости FDA предпринимает все предусмотрен-
ные законом действия принудительного характера, если производитель отка-
зывается от отзыва или же не успевает завершить его в назначенный срок.
1.2.З.4. Письмо-предупреждение
Цель письма-предупреждения — оповещение производителей о нарушении пра-
вил, документированных FDA в ходе инспекций или расследований. Письмо-
предупреждение сообщает ответственным лицам и/или фирмам о том, что один или
несколько продуктов, практик, процессов или других видов деятельности не соот-
ветствует требованиям cGMP. Письма-предупреждения следует рассылать при нару-
шениях, требующих принудительных действий, если документированные наруше-
ния не скорректированы адекватно и в срочном порядке. Письмо-предупреждение
является одним из основных инструментов FDA, позволяющим на добровольной
основе и в короткие сроки обеспечить соответствие фармацевтической продукции
всем существующим требованиям.
Примеры ситуаций, в которых FDA может выслать производителю письмо-
предупреждение, включают в себя:
> распределение серий активного фармацевтического ингредиента (АФИ), не
соответствующих утвержденным спецификациям;
> намеренное смешивание серий АФИ в целях разведения либо сокрытия вред-
ной примеси или загрязнителя, а также отсутствие оценки реального выхода
продукта и процента от теоретически возможного выхода;
> загрязнение лекарств токсичными химическими веществами, остаточными
количествами фармацевтических продуктов, содержащимися в воздухе за-
грязнителями или посторонними веществами;
> несоответствие характеристикам, указанным в заявке на регистрацию лекар-
ства;
> объединение серии, не соответствующей критически важным параметрам,
с серией, которая им соответствует;
> отсутствие доказательств пригодности воды, применяемой в процессе произ-
водства;
> отсутствие валидации систем водоснабжения;
> отсутствие надлежащим образом оформленной письменной программы ва-
лидации процесса производства АФИ;
88
Часть I. Нормативное регулирование производства лекарственных средств
> отсутствие подтверждения однородности продукта после итоговых операций
смешивания;
> отсутствие надлежащих протоколов к серии;
> отсутствие надлежащим образом оформленных процедур внесения измене-
ний в систему контроля процесса на местах;
> использование непригодных и не прошедших валидации лабораторных ана-
литических методов;
> применение процессов упаковки и маркировки, существенно повышающих
риск нанесения неправильной маркировки;
> отсутствие анализа на содержание остаточных количеств органических или
неорганических растворителей, способных попадать в АФИ;
> неполное исследование стабильности для определения параметров стабиль-
ности АФИ в течение предполагаемого периода применения последних.
Письма-предупреждения с подробным описанием нарушений требований cGMP
обычно завершает абзац следующего содержания:
Продукг(-ы) [НАИМЕНОВАНИЕ ЛЕКАРСТВА] является недоброкачественным в со-
ответствии с определением пунктов 501(a)(2)(B) и 351(a)(2)(B) 21 USC, поскольку ме-
тоды, оборудование или способы контроля, применяемые для его производства, обра-
ботки, упаковки или хранения, не соответствуют требованиям cGMP (21 CFR210,211).
Количество писем-предупреждений, рассылаемых FDA относительно рецептур-
ных и отпускаемых без рецепта врача лекарств, варьируется год от года: так, в 2000 г.
их число составило 130, а в 2005-м — только 79.
Следует различать письмо-предупреждение и уведомление о нарушении,
так называемое письмо без заглавия (untitled letter). Письмо без заглавия содер-
жит перечисление нарушений, недостаточно значительных для отсылки письма-
предупреждения. В отличие от последнего письмо без заглавия не содержит
предупреждения о том, что отсутствие срочных корректирующих действий может
привести к мерам принудительного характера со стороны FDA. Кроме того, письмо
без заглавия содержит скорее просьбу, чем требование, предоставить письменный
ответ в разумные сроки.
1.2.4. Судебное правоприменение без письма-предупреждения
1.2.4.1. Введение
В определенных обстоятельствах FDA обходится без отправки письма-преду-
преждения. Согласно главе 4 Руководства по применению регулирующих процедур, по-
добные случаи являются следствием нарушения, которое:
1) отражает историю систематических схожих нарушений, о которых произво-
дитель получал уведомления;
2) является намеренным или тяжким;
3) несет существенную угрозу здоровью потребителя вплоть до летального ис-
хода;
4) предварялось надлежащими уведомлениями, однако было оставлено без вни-
мания производителем;
Глава 1.2. Внедрение текущей надлежащей производственной практики 89
5) ретулируется статьей 1001 тома 18 USC и является сознательным и умышлен-
ным действием, которое не подлежит исправлению. (При таких тяжких нару-
шениях также не предусмотрено предварительное уведомление. Таким обра-
зом, в письма-предупреждения не включают нарушения, регламентируемые
18 КУС, 1001.)
Порядок ведения записей регламентируют параграфы 211.180—211.208 CFR, наи-
более важные из которых представлены ниже.
§ 211.182. Журнал учета очистки и эксплуатации оборудования
В индивидуальные журналы учета эксплуатации оборудования, фиксирующие дату,
время, наименование продукта и номер подсерии каждой производственной серии,
вносятся записи по проведению очистки и техническому обслуживанию (за исключе-
нием рутинных операций, таких как смазка и поверка).
§ 211.184. Записи по компонентам, контейнерам для лекарственных продуктов, укупо-
рочным приспособлениям и маркировкам
Эти записи должны включать:
(а) идентификационную информацию с указанием количества для каждой транс-
портной партии каждой серии компонентов, контейнеров для лекарственных средств,
укупорочных приспособлений и маркировочных материалов; наименование постав-
щика; номер серии поставщика (если информация известна); код получения, оформ-
ленный согласно § 211.80; дату приемки. Желательно указывать наименование и место-
положение первичного производителя, если они отличаются от данных поставщика;
(Ь) результаты всех проведенных анализов и осмотров (в том числе проведенных
согласно требованиям § 211.82(a), § 211.84(d) или § 211.122(a)), а также выводы, сде-
ланные на их основе.
§ 211.186. Основные производственные и контрольные записи
Для обеспечения однородности параметров различных серий должны быть подго-
товлены основные производственные и контрольные записи для каждого лекарствен-
ного средства, включающие размер каждой серии, дату производства и подпись того,
кто оформлял записи. Все эти данные независимо проверяются и датируются вторым
лицом, тоже ставящим свою подпись. Процесс подготовки основных производствен-
ных и контрольных записей должен быть письменно зафиксирован.
§ 211.188. Протоколы производства серии и контрольные записи
Протоколы производства серии и контрольные записи оформляются для каждой
произведенной серии лекарственного средства. Они должны включать полную инфор-
мацию по производству и контролю.
§ 211.194. Лабораторные записи
Лабораторные записи содержат полную информацию, полученную по данным всех
анализов, необходимых для обеспечения соответствия всем утвержденным специфи-
кациям и стандартам, включая испытания и осмотры <...>.
§ 211.198. Учет претензий
Следует вести записи всех претензий к продукции в специальном журнале.
Кроме того, сознательное введение FDA в заблуждение, включая предоставление
ложной информации в записях или отсылаемых письмах, почти гарантирован-
90
Часть I. Нормативное регулирование производства лекарственных средств
но влечет за собой применение юридических мер принуждения. Эти нарушения,
в частности, представляют собой:
1) приемку лекарственных средств, не соответствующих утвержденным стандар-
там или спецификациям, а также любым другим критериям контроля каче-
ства (таким как скорость растворения, однородность содержимого, чистота,
эффективность), с последующей записью ложных данных, свидетельствую-
щих о соответствии фармацевтических продуктов вышеуказанным критери-
ям, стандартам и спецификациям;
2) утверждение параметров стабильности некондиционных лекарственных
средств с последующим внесением ложных записей, свидетельствующих
о соответствии стандартам и критериям;
3) наличие записей об осмотре и проверке маркировки, в то время как в дей-
ствительности никакого осмотра не производилось (результатом чего стало
распределение лекарств с неточной маркировкой);
4) внесение ложной информации в основные производственные и контрольные
записи;
5) внесение ложной информации в протоколы к производственным сериям
и в контрольные записи;
6) подделка записей по использованию аналитических методик;
7) отсутствие точных записей по всем устным и письменным претензиям к ле-
карственному продукту и/или выдача сертификата о проведении исследова-
ний, в то время как исследования не проводились; выдача документа, удосто-
веряющего, что наблюдения инспекции не подтвердились, в то время как они
подтвердились, ит. д.;
8) подделка записей, касающихся изменений в производственном процессе, ко-
торые требуют одобрения FDA,
9) предоставление ложной информации в письмах, направленных в FDA в от-
вет на замечания о нарушениях, содержащихся в отчете инспектора по Фор-
ме 483.
При возбуждении судебного преследования со стороны FDA или DO/суды США,
как правило, принимают решение не в пользу производителя.
«Репрессивные» механизмы FDA зачастую носят ступенчатый характер. Пока-
зательный пример подобной схемы представляет собой случай с Glaxo SmithKline
(GSK). В июле 2002 г. FDA направило этой компании письмо-предупреждение, вы-
званное обнаружением многочисленных существенных нарушений правил cGMP
инспекцией, проводившейся с февраля по апрель 2002 г. В отправленном письме
содержались просьба исправить все нарушения и предупреждение о том, что от-
сутствие корректирующих действий может привести к мерам принудительного
характера, включая арест продукции и/или ее конфискацию. Несмотря на то что
эта инспекция FDA, проведенная «по следам» предыдущей инспекции в октябре
2002 г., отметила ряд улучшений, последующие инспекции в ноябре-декабре 2003 г.
и сентябре-ноябре 2004 г. выявили факт продолжающихся серьезных нарушений
правил cGMP. При этом сама GSK осуществила отзыв некоторых (хотя и не всех)
серий двух видов своей продукции. 4 марта 2005 г. из-за возрастающей озабочен-
Глава 1.2. Внедрение текущей надлежащей производственной практики
91
ности качеством производимой GSK продукции FDA и DOJ инициировали арест
двух серий лекарственных средств. Эти действия были вызваны тем, что нарушения
производственных стандартов в GSK могли привести к появлению на рынке лекар-
ственных средств низкого качества, представляющих риск для здоровья потребите-
лей. 28 апреля 2005 г. FDA объявило о подписании мирового соглашения, обязавше-
го GSK к исправлению нарушений в промышленном производстве на предприятии
Cidra в Пуэрто-Рико. Соглашение было призвано устранить возможные нарушения
производственных стандартов компанией GSK.
1.2.4.2. Гражданские иски
Наложение ареста. Если инспектор FDA имеет основания полагать, что то или иное
лекарство является некондиционным, то он может потребовать ареста серии на раз-
умный срок, не превышающий 20 сут (если FDA не применяет действий, предусмо-
тренных пунктом 334(a), или не вводит судебный запрет, разрешающий удержива-
ние продукта на более длительный период).
Закон о пищевых продуктах, медикаментах и косметической продукции дает
прямое разрешение на административный арест на основе разумного доверия одной
из сторон (21 USC 334 (g)). Арест материально-производственных запасов лишает
компанию как капитальных вложений, так и потенциальных доходов.
Если FDA возбуждает иск о судебной защите без задержания, то государство США
может предъявить претензию на конфискацию, предписывающую своему федераль-
ному маршалу (U. S. marshall) арестовать фармацевтическую продукцию (или всту-
пить во владение ею, или поместить ее на юридическое хранение по решению суда).
Основанием для подобной претензии {complaint for forfeiture) служит некондицион-
ность самого фармацевтического продукта. В таких случаях правительство обращает-
ся в суд с просьбой исполнить статью закона, объявив о конфискации. При регистра-
ции претензии чиновник обязательно выписывает ордер. Таким образом, FDA может
получить ордер без рассмотрения претензии судебным должностным лицом или
даже до выявления вероятной причины нарушения требований законодательства.
Существуют три типа ареста продукции: массовый, открытый и арест конкрет-
ной партии. Объектом массового ареста выступает вся продукция на территории
организации или предприятия, подпадающая под регулирование правил FDA. Мас-
совые аресты могут проводиться, когда все продукты производятся в одинаковых
условиях (например, с нарушениями требований cGMP). Открытый арест {open-
ended seizure) налагается на все единицы конкретной продукции, предположительно
не отвечающие нормативным требованиям, независимо от номера серии. Открытый
арест может быть произведен, если под подозрение попадают все партии или серии
продукта, но не вся продукция предприятия.
После ареста лекарственных средств у производителя есть три варианта действий.
Во-первых, он может ничего не предпринимать; в этом случае лекарства будут уни-
чтожены. Во-вторых, он может пойти на мировое соглашение, признав нарушения,
согласившись оплатить все издержки и изыскав возможность уничтожения или реа-
билитации продукта. Выражение согласия обычно предусматривает: 1) признание
лекарственного средства непригодным; 2) уплату штрафа, приблизительно в два
раза превышающего розничную стоимость арестованной партии; 3) оплату стоимо-
92
Часть I. Нормативное регулирование производства лекарственных средств
сти хранения арестованной партии и деятельности федерального маршала, а также
надзора FDA перед отпуском продукта; 4) приведение лекарственного средства в со-
ответствие требованиям FDA.
В-третьих, производитель может опротестовать арест. В этом случае дело рас-
сматривается наряду с другими гражданскими делами, а правительство должно
доказывать свою правоту. Это подразумевает представление свидетельств в пользу
обвинения, включая факты перемещения лекарства или его компонентов на терри-
торию других штатов. Помимо сотрудников FDA, показания могут давать и сторон-
ние эксперты (речь идет, например, об оценке значительности несоответствия тре-
бованиям cGMP). При вынесении решения не в пользу производителя (после суда
или в результате мирового соглашения) суд может издать прямое распоряжение об
уничтожении продукта.
Представленная ниже выдержка из Руководства по применению регулирующих
процедур касается некоторых случаев возвращения {disgorgement) лекарственных
средств.
Основные мировые соглашения, недавно зарегистрированные в результате процес-
сов: правительство США против Abbot labs., постановление о выражении согласия на
бессрочный судебный запрет, зарегистрировано 2 ноября 1999 г.; правительство США
против Wyeth-Ayerst Labs., постановление о выражении согласия на признание про-
дукта некондиционным и бессрочный судебный запрет, зарегистрировано 4 октября
2000 г.; правительство США против корпорации Schering-Plough, постановление о вы-
ражении согласия на бессрочный судебный запрет, зарегистрировано 20 мая 2002 г.
Чтобы не ввести производителей в заблуждение разрешением сохранить на рынке
продукты, признанные произведенными с нарушением требований cGMP, FDA вводит
три различных типа выплат. Первый тип представляет собой единовременную выплату
(Abbot — 100 млн долл.* 2, Wyeth — 30, Schering— 500). Если же коррекционные действия
не были осуществлены в обозначенные в соглашении сроки, предусматривается либо
выплата процентов с продаж (Abbot— 16%, Wyeth — 18,5, Schering— 24,6%), либо еже-
дневная выплата фиксированной суммы. И та и другая выплачиваются до достижения
соответствия фармацевтической продукции всем необходимым требованиям.
Судебные запреты. Закон о пищевых продуктах, медикаментах и косметической
продукции наделяет суды правом пресекать любые действия, нарушающие § 331
тома 21 USC. В частности, это подразумевает и запрещение некондиционной ле-
карственной продукции. Применение судебного запрета правомочно в следующих
случаях:
1) при наличии сохраняющейся явной угрозы здоровью потребителей или при
массовом обмане последних;
2) наличии больших количеств продукции, изготовленной с нарушениями пра-
вил и размещенной в различных местах, которую производитель отказался
добровольно отозвать или же масштабы отзыва которой недостаточны для
' Руководство по применению регулирующих процедур, подраздел 6-1-11, март 2007. Здесь
подразумевается, что арест конкретного продукта(-ов) представляет собой единичный слу-
чай. Более сложные варианты мирового соглашения описаны ниже.
2 Здесь и далее имеются в виду доллары США. — Примеч. отв. ред.
Глава 1.2. Внедрение текущей надлежащей производственной практики
93
того, чтобы защитить потребителя — при том, что арест такого продукта эко-
номически неэффективен;
3) наличии постоянных нарушений, не представляющих угрозы здоровью по-
требителя и не являющихся массовым обманом последнего, однако не ис-
правленных добровольно либо с помощью других нормативных подходов1.
Иск с требованием судебного запрета обычно подкрепляется ходатайством
о предварительном судебном запрете2.
Для определения необходимости предварительного судебного запрета суд на-
значает дату судебных слушаний и в короткие сроки проводит рассмотрение дела.
На этом начальном этапе основной целью государства является доказательство «су-
щественной вероятности» того, что ответчик производил некондиционные лекар-
ственные продукты с нарушением 21 USC 331 и при значительных несоответствиях
требованиям cGMP. При этом по возможности представляется свидетельство о том,
что ответчик и ранее нарушал требования FDCA и связанные с ними нормативы.
Американские суды вольны осуществлять превентивные действия по пресечению
возможного несоблюдения положений статьи 331, если FDA предоставило доказа-
тельства того, что случаи соответствующих правонарушений имели место или могут
произойти. Вероятность дальнейшего несоблюдения закона определяется с учетом:
1) степени осознания ответчиком последствий нарушений; 2) частоты последних;
3) признания ответчиком неправомерности своего образа действий; 4) искренности
заверений ответчика о недопущении будущих нарушений; 5) серьезности наруше-
ний; 6) соблюдения ответчиком всех рекомендаций государственных органов.
Добросовестность производителя, как и его возможное разорение, не являются
аргументами против судебного запрета.
Судебное решение на основании мирового соглашения и возврат средств. Решение
на основании мирового соглашения выносится судом при достигнутом согласии
сторон. При этом ответчик берет на себя обязательство прекратить любую незакон-
ную деятельность. При получении одобрения суда арест продукции снимается, а ре-
гулирующие меры сводятся к отслеживанию нарушений соглашения, требующих
принудительных действий.
Решения на основании мирового соглашения исходят из согласия ответчика на
исправление всех несоответствий путем, удовлетворяющим FDA, в установленные
соглашением сроки. Кроме того, такие решения могут предусматривать периоди-
ческое привлечение эксперта-консультанта для составления подробных отчетов,
удостоверяющих, что производственное предприятие полностью соответствует
правилам cGMP и обладает надлежащими средствами контроля неблагоприятных
событий, персонал надлежащим образом обучен, а процедура отзыва соответствует
всем требованиям и строго соблюдается. Кроме того, соглашение может обязывать
производителя к уплате денег в Федеральное казначейство.
1 Руководство по применению регулирующих процедур, март 2007 г.
2 Государство может также потребовать применения временного запретительного судеб-
ного приказа (temporary restraining order, TRO) о немедленном временном запрете (на срок до
10 сут, который может быть дополнительно продлен еще на 10 сут) до судебных слушаний
о предварительном судебном запрете. FDA обычно рекомендует TRO, когда полагает, что на-
рушение настолько серьезно, что ситуация должна быть взята под контроль немедленно.
94
Часть I, Нормативное регулирование производства лекарственных средств
Разновидностью судебных действий является назначение выплаты возмещения
по представлению FDA. Подобные выплаты лишают правонарушителя полученной
нечестным путем прибыли, а также удерживают его от совершения противоправных
действий. Размер возмещения не всегда напрямую связан с реституцией. На прак-
тике назначаемая сумма выплат обычно достаточно велика для того, чтобы ответчик
воспринял ее всерьез, но не предполагает полного изъятия прибыли, полученной
с помощью небезупречного лекарственного средства.
Закон «О фальсификации правопритязаний» (False Claims Act). Закон Гражданского
кодекса США «О фальсификации правопритязаний», том 31 USC, § 3729 и далее,
является основным инструментом взыскания ущерба вследствие мошенничества
с государственных подрядчиков. Закон касается нарушений cGMP, поскольку США
(вкладывающие средства в Фонд медицинского страхования, государственные про-
граммы Medicaid, Ведомство по делам ветеранов, программу TRICARE и т. д.) явля-
ются самым крупным в мире потребителем лекарств.
Любые ограничительные действия правительственных органов, связанные
с нарушением данного закона, должны сопровождаться выдвижением соответ-
ствующего судебного иска. Это связано с тем, что исполнительная власть облада-
ет множеством других инструментов принуждения (таких как уголовные штрафы
и взыскания), при помощи которых можно получить компенсацию за ущерб, на-
несенный недобросовестными производителями.
В настоящее время многие граждане выступают инициаторами процессов qui
tam'. Поскольку Закон о фальсифицикации правопритязаний возлагает финан-
совую ответственность на любого правительственного подрядчика, умышленно
предоставляющего необоснованные правопритязания к государству (в целях неза-
конного обогащения, связанного с подделкой документов, продажей некачествен-
ной продукции и т. п.) или использующего фальшивые документы для получения
необоснованных выплат, то каждый производитель фармацевтической продукции,
который который своими действиями или бездействием способствует нарушению
правил cGMP, приравнивается к любому подрядчику, обязанному соблюдать условия
1 Qui tarn — сокращение латинской фразы qui tarn pro domino rege quam pro se ipso in hac
parte sequitur — буквально «тот, кто предъявляет иск от имени короля, как если бы действо-
вал в своих интересах». Процессы qui tarn появились в Англии в XIII столетии. Они служили
средством, позволяющим частным лицам предъявлять обвинение в суде от имени короля и
таким образом получить доступ в королевские суды.
Положения qui tarn, содержащиеся в федеральном законе о фальсификации правопри-
тязаний, позволяют любому гражданину, узнавшему об обмане, направленном против го-
сударства, подать гражданский иск в федеральный суд от имени США. Такой гражданин
приобретает право на долю от выплат в возмещение убытков. Положения qui tarn поощряют
осведомленных граждан на выявление преступлений, заключающееся в предоставлении пра-
вительственным органам реальных и подробных свидетельств, на основании которых можно
начать расследование и возбудить судебное преследование.
В 1986 г. Конгресс США ввел поправки к Закону о фальсификации правопритязаний, по-
вышавшие денежное вознаграждение за подобные услуги. Это решение было продиктовано
ожиданиями, что большинство исков qui tarn будет направлено против фирм, выполняющих
заказы военного ведомства. Однако за последнее десятилетие большинство подобных дел ка-
салось нарушений в промышленности, связанной со здравоохранением.
Глава 1.2. Внедрение текущей надлежащей производственной практики
95
контракта или нормативные стандарты. Согласно Закону о фальсификации право-
притязаний, ответственность производителя объясняется наличием фальшивых за-
писей, приводящих к возникновению ложных правопритязаний к лекарствам, ко-
торые оплачивает государство США1. Денежные штрафы являются следствием того,
что плательщик (в данном случае государство США) отвечает за некондиционные
лекарства, изготовленные с нарушением правил cGMP.
Это резонно: правила cGMP представляют собой набор требований, гарантирую-
щих надлежащее производство лекарств, которые отвечают требованиям безопасно-
сти для потребителя, обладающих указанным содержанием активного компонента
и соответствующих заявленным параметрам чистоты. Основные правительственные
программы оказания медицинской помощи, финансируемые из федерального бюд-
жета (Medicare и Medicaid), формулируют принцип своей работы следующим обра-
зом: оплачиваются только медицинские услуги и продукты, являющиеся «приемле-
мыми и необходимыми». Небезопасные и неэффективные лекарственные средства
не являются ни приемлемыми, ни эффективными. Соответственно, теоретически
государство США несет финансовые потери, если программы Medicare и Medicaid
оплачивают небезопасные и недостаточно эффективные продукты. Эти и другие
программы оказания медицинской помощи, финансируемые из федерального бюд-
жета, тратят на фармацевтическую продукцию миллиарды долларов каждый год.
Фальсификация записей, относящихся к малым или техническим нарушениям, не
является основой для финансовой ответственности по FCA. Реализация фармацевти-
ческих препаратов, не соответствующих требованиям cGMP (однако сопровождаемых
фальшивой документацией, подтверждающей их соответствие), не всегда означает
появление на рынке небезопасных (или недостаточно эффективных) лекарств. Вместе
с тем сокрытие существенных нарушений cGMP может подпадать под регулирование
Аннотированного свода федеральных законов (Federal Code Annotated, FCA). Поэтому
любое нарушение рассматривают на предмет того, является ли оно настолько серьез-
ным, что лекарство при попадании к потребителю может оказаться существенно ме-
нее безопасным и эффективным в сравнении с изготовленным по правилам cGMP.
1.2.4.3. Уголовное преследование
Введение. Уголовная ответственность за нарушения FDCA призвана обеспечить
охрану здоровья потребителя. Исторически сложилось так, что FDA не выдвигает
уголовных обвинений, если ответчик не совершает постоянных нарушений норма-
тивных требований; исключение составляют случаи умышленного пренебрежения
законодательством, мошенничества, а также производство лекарственных средств,
представляющих опасность для здоровья потребителей2.
1 Даже в этом случае неизбежны вопросы по фактическим обстоятельствам, например та-
кие: 1) было ли принуждение к направлению иска? 2) если бы FDA знало о нарушениях, за-
претило бы оно производителю продолжать выпуск продукции? 3) какая ложная запись или
утверждение сделали правопритязания необоснованными?
2 Авторы обзора недавних принудительных действий, предпринятых FDA, пришли к за-
ключению, что деятельность последнего по принуждению к исполнению закона является
недостаточно эффективной. См. Prescription for Harm: The Decline in FDA Enforcement Activity.
House Committee on Government Reform, June 2006.
96
Часть I. Нормативное регулирование производства лекарственных средств
Наиболее частое обвинение в нарушении FDCA, связанное с cGMP, выдвигает-
ся по статье 21 USC 331(a), которая, в частности, содержит запрет на торговлю не-
кондиционным1 лекарством между фирмами, находящимися в различных штатах.
Статья 331(e) запрещает отказ в допуске к целому ряду записей, a 331(f) — недопу-
щение инспекции на территорию производственного предприятия (21 USC 374).
Совершение любых действий, запрещенных статьей 331, относится к катего-
рии федеральных мисдиминоров2 (21 USC 333). Тем не менее, нарушения пункта
331(a) могут рассматриваться как тяжкие уголовные преступления (felonies), если
нарушение включает намерение совершить мошенничество либо попытку ввода
в заблуждение или же если ответчик ранее признавался виновным в совершении
преступления из категории мисдиминоров согласно FDCA (21 USC 333(b)). Соглас-
но федеральному законодательству, дела о мисдиминорах обычно рассматриваются
мировыми судьями, а тяжкие преступления — федеральными районными судьями.
Ицдивидуальная и корпоративная финансовая ответственность. Допуск неконди-
ционного продукта к торговле по всей территории США является преступлением,
влекущим штрафы и возмещение ущерба. Ответственность несут как отдельные
лица, так и компания в целом. Кроме того, виновными могут быть признаны лица,
не предпринявшие адекватных мер по предотвращению нарушения cGMP.
Письма-предупреждения обычно направляют президентам компаний или их
исполнительным директорам (Chief Excecutive Officer, CEOs), а также в адрес юри-
дических лиц; возможны и другие способы коммуникации. В постановлении Вер-
ховного суда США3, вынесенного в 1964 г. (через два года после выхода FDCA в той
редакции, которая известна нам), в частности, сказано:
Законодательство по пищевым продуктам и лекарствам, охраняющее жизнь и здоро-
вье людей в обстоятельствах, которые могут не позволить человеку самостоятельно
защитить себя, зачастую расходится с традиционным определением криминального
поведения, подразумевающего осознание противоправного действия. В интересах
общественного блага оно возлагает ответственность за действия в подобной «угрожа-
ющей ситуации в отношении общественной опасности» на человека в других обстоя-
тельствах невиновного...
В 1975 г. Верховный суд пояснил, насколько важна личная ответственность4:
Закон о пищевых продуктах, медикаментах и косметической продукции обязывает
производителя не только выявлять и исправлять обнаруженные нарушения cGMP, но
и принимать превентивные меры, гарантирующие отсутствие таких нарушений. По-
добные требования, возлагаемые на ответственных корпоративных агентов, являются,
вне всякого сомнения, весьма затруднительными, однако их строгость оправдана тем,
что общественность имеет право требовать ответственности тех, кто добровольно бе-
рет на себя задачу управления торгово-промышленными предприятиями, продукция
которых влияет на здоровье и благосостояние потребителя.
1 Невыполнение требований cGMP является самой распространенной формой нарушения
запрета на междуштатную торговлю некондиционным лекарством.
2 Категория наименее опасных преступлений, граничащих с административными право-
нарушениями (калька с англ, misdemeanour). — Примеч. отв. ред.
3 United States v. Wiesenfield Warehouse Co., 376 U.S. 86, 91 (1964).
4 United States v. Park, 421 U.S. 658(1975).
Глава 1.2. Внедрение текущей надлежащей производственной практики 97
Таким образом, руководящий состав промышленных предприятий берет на
себя бремя огромной финансовой ответственности. Правительство должно лишь
доказать, что индивидуальный ответчик не предпринял действий, которые могли
бы предотвратить нарушение или исправить его. Поэтому виновным может быть
признан даже тот ответчик, который не имел намерений нарушить закон. Главный
вопрос, обсуждаемый на судебных заседаниях, состоит в том, обладал ли руково-
дящий работник полномочиями для предотвращения или недопущения действий,
ставших источниками жалоб, а также то, мог ли он предотвратить всё это в одиноч-
ку. Принятие решения не становится легче, от того что нормативы cGMP открыты
для различных толкований или оттого что технологии меняются постоянно.
Судопроизводство по статье 305. Сущность процесса инспекции такова, что ком-
пания, подозреваемая FDA в нарушении cGMP, не должна удивляться, получив
письмо-предупреждение или уведомление о применении более строгих мер при-
нуждения. Как правило, FDA присылает формальное уведомление о предстоящем
предъявлении уголовных обвинений согласно так называемому уведомлению по ста-
тье 305.
Согласно статье 305 тома 21 USC, перед сообщением в DO Jo любых нарушениях
FDCA, влекущих уголовное преследование, ответчик должен получить надлежащее
уведомление и возможность изложить свою позицию относительно предполагаемо-
го преследования в устной или письменной форме. Правда, Верховный суд США
зачастую игнорирует это положение, полагая, что уведомление по статье 305 не яв-
ляется необходимым по закону предварительным условием инициации правитель-
ством судебного преследования.
Таким образом, на практике FDA при рассмотрении мисдиминоров лишь ино-
гда присылает Уведомление 305 и проводит слушания по соответствующей статье.
Процесс обычно носит неформальный характер; производитель может привлекать
защиту лишь в том объеме, который укажет юрисконсульт. Такая ситуация обуслов-
лена множеством доводов «за» и «против» в вопросе представления правительству
всех аргументов защиты на этом этапе.
Образец Уведомления 305 представлен на рис. 5.
Рассмотрение дела расширенной комиссией присяжных (Большим жюри). При
возбуждении уголовного преследования по обвинению в тяжком преступлении в от-
ношении производственного предприятия или отдельных лиц дело рассматривает-
ся расширенной комиссией присяжных. Согласно Пятой поправке к американской
конституции, обвинения по всем преступлениям, предусматривающим длительное
тюремное заключение или смертную казнь, должны рассматриваться расширенной
коллегией присяжных. В практике судопроизводства это означает, что все тяжкие
федеральные преступления требуют вердикта Большого жюри.
Рассмотрение обстоятельств дела, как и принятие решения Большим жюри, про-
ходят при закрытых дверях1.
Строгое соблюдение положения о секретности работы Большого жюри необхо-
димо для предотвращения давления на присяжных, для привлечения свидетелей,
недопущения раскрытия личности последних, а также защиты прав обвиняемых
1 См. Rule 6(e) Fed. Crim. Pro.
98
Часть I. Нормативное регулирование производства лекарственных средств
В ответе ссылайтесь на
Образец №
Продукт
Название фирмы и ответственное лицо Дата
Адрес:
Город, штат, почтовый индекс:
Расследование, проведенное FDA, выявило Вашу ответственность за нарушения
FDCA, а также других федеральных законов, описанных в приложенном обвинении.
[Далее следует описание конкретных нарушений cGMP]
Заседание, на котором у Вас будет возможность представить Вашу точку зре-
ния по обозначенным вопросам, назначено на [день, дата, время] и будет проходить
[место проведения заседания]. Прилагаемый ИНФОРМАЦИОННЫЙ ЛИСТ объясняет
цель и сущность заседания, а также регламентирует характер Ваших объяснений.
При неполучении ответа до назначенной даты заседания наше решение о передаче
дела в DOJ для возбуждения судебного преследования будет исходить из имеющих-
ся в нашем распоряжении свидетельств.
По распоряжению секретаря Министерства здравоохранения и социальных
услуг:
Должностное лицо по обеспечению соответствия
Приложения:
Документ о правовом статусе (3 экз.)
Обвинительный лист
Информационный лист
Нормативные требования
Рис. 5. Форма Уведомления 305
(в особенности несправедливо обвиненных), выражающейся в нераспространении
информации о том, что они находились под следствием. Кроме государственного
обвинителя, на слушаниях Большого жюри разрешено присутствовать свидетелям,
переводчикам (при необходимости), секретарю суда и лицам, получившим специ-
альныей допуск.
Задача Большого жюри — определить, существуют ли основания предполагать,
что конкретное физическое либо юридическое лицо (или лица) совершило феде-
ральное преступление1. Обвинителям разрешено выступать перед Большим жюри
и практически вести процесс. Как правило, решение о том, кто из свидетелей будет
вызван и какое именно свидетельство будет предъявлено Большому жюри, прини-
мает прокурор. Он же задает вопросы свидетелю, после чего присяжные могут до-
просить свидетеля прямо или через обвинителя.
1 Система Большого жюри в настоящее время не используется за пределами США. Вели-
кобритания, Новая Зеландия, Канада и Австралия, например, отказались от системы Боль-
шого жюри (http://enwikipedia.org/wiki/GrandJury).
Глава 1.2. Внедрение текущей надлежащей производственной практики 99
В процессе рассмотрения Большим жюри обвинений в нарушении правил cGMP
и/или фальсификации продукции присяжные могут выслушивать свидетельства не
только от лиц, ведущих расследование (федеральных агентов и федеральных ин-
спекторов), но и от бывших сотрудников компании (или от нынешних хранителей
документации) и/или экспертов в области фармацевтического производства. Эти
лица признаются свидетелями. Последних обычно вызывают повесткой, явка по
которой обязательна.
Свидетелю не разрешается приводить адвоката в комнату заседаний Большого
жюри. Тем не менее, свидетелю позволено консультироваться со своим адвокатом вне
комнаты заседания, даже если для этого нужно прервать свои показания1. Как прави-
ло, компании—производители фармацевтической прод укции обеспечивают адвокат-
ской помощью всех своих сотрудников, вызванных в суд повесткой Большого жюри.
Объект иска — это лицо, находящееся в центре расследования Большого жюри,
которому, по всей вероятности, будет вынесен обвинительный приговор. Компания
или конкретное лицо могут получить от Большого жюри специальное письмо {target
letter), содержащее как вызов в суд, так и официальное уведомление о риске уголов-
ной ответственности.
Если объектом расследования является производитель, правительство вызывает
в суд свидетелей и стремится предоставить материалы, которые помогут доказать
виновность ответчика. Как правило, повестка о явке в суд содержит предложение
представить корреспонденцию, записи и служебные письма за определенный пери-
од времени, относящиеся к конкретному делу.
Большое жюри может выслать повестку ответственному за хранение производ-
ственных записей и затребовать документацию на конкретную продукцию. К за-
прошенным документам могут относиться постановления организации, заявления,
изложение принципов производственной политики или схемы процедур, еже-
дневные записи, содержимое электронной почты, производственные журналы, до-
рожные чеки, финансовые документы и постановления, корреспонденция, записи
переговоров и любая другая документация, имеющая отношение к производству
определенных лекарств. Кроме того, суд может запрашивать все записи (любого
типа) и описания, находящиеся во владении компании, на ее хранении или под ее
контролем, имеющие отношение к расследуемому уголовному преступлению. Как
правило, запрашиваются записи, сделанные от руки, напечатанные машинописным
способом, распечатанные на принтере, а также протоколы или расшифрованные
записи, включая магнитные ленты и компьютерные диски.
Одна из задач обвинения — доказать, что преступление было совершено в том
округе, где проводится расследование: Большое жюри рассматривает лишь те пре-
ступления, что были совершены в округе, где проходит заседание2. При рассмотре-
нии дел о фальсификации лекарств суды исходят из того, что заседания должны
происходить в том округе, из которого ответчик произвел незаконную отправку
продукции в торговую сеть — даже в случае, если физическая доставка продукции
осуществлялась из другого округа.
1 См. Rule 6 (d) Fed. R Crim. Pro.-, 28 U.S.C. secs. 515, 542, 547.
2 Cm. Rule 18 Fed. R Crim. Pro.
100
Часть I. Нормативное регулирование производства лекарственных средств
Штрафы и издержки. При успехе обвинения присяжные Большого жюри выносят
обвинительное заключение. Иногда случается так, что первоначальное расследова-
ние, объектом которого является исключительно изготовление некондиционной
лекарственной продукции, «обрастает» дополнительными пунктами обвинения.
Это происходит, к примеру, если обвиняемый пытался ввести в заблуждение чле-
нов Большого жюри. В итоге ответчику, в дополнение к обвинениям в изготовлении
некондиционных лекарств, может быть предъявлено обвинение в лжесвидетельстве
и создании препятствий расследованию FDA или DOJ.
Обвинительное заключение включает описание сущности преступления, а также
места и времени его совершения. Каждое правонарушение излагается в отдельной
статье. Ответчик, признанный виновным, имеет право потребовать разбирательства
дела обычной коллегией присяжных. Это право распространяется на любое уголов-
ное преступление, предусматривающее тюремное заключение сроком более 6 мес.1
Если дело возбуждается по правонарушению из категории мисдиминоров, то
обвинитель выдвигает обвинение by information. Такую форму обвинения называют
«жалобой». Как и обвинительное заключение, упомянутая «жалоба» является все-
го лишь заявлением, содержащим обвинение ответчика в совершении преступле-
ния. Различие между «жалобой» и «предъявлением обвинения» (indictment) состоит
в том, что прокурор может зарегистрировать «жалобу» и возбудить судебное пресле-
дование без участия Большого жюри либо без установления вероятных оснований.
Что до «предъявления обвинения», то оно подлежит обязательному утверждению
Большим жюри2.
Наказание за нарушение статьи 21 USC 331(b), где описывается нарушение пра-
вил cGMP, которое повлекло за собой появление некондиционных лекарств более
чем в одном штате США, предусмотрено также статьей 21 USC 333(a). Каждая из
этих статей по нарушению cGMP (при обвинении в совершении мисдиминора) уста-
навливает наказание в виде возможного тюремного заключения сроком не более
одного года, либо в виде штрафа, не превышающего 1000 долл., либо и того и друго-
го. При выдвижении обвинения в тяжком преступлении за нарушение правил cGMP
наказание может предусматривать тюремное заключение не срок не более трех лет,
или штраф, не превышающий 10 000 долл., или и то и другое вместе. Разумеется, об-
винения могут предполагать и другие наказания — более или менее строгие, а также
не связанные с изготовлением некондиционных лекарств.
Тем не менее, новые федеральные законы могут устанавливать другие размеры
штрафа (но не длительность тюремного заключения) и, соответственно, аннулиро-
вать действие предыдущих статей. Статья 3571 тома 18 USC предполагает гораздо
бблыпие штрафы по сравнению с теми, что предусмотрены FDCA. Для производи-
теля, обвиненного в фелонии, штраф может достигать 500 000 долл., при мисди-
миноре — 200 000 долл. Штрафы, налагаемые на отдельных лиц, могут доходить
1 См. Шестую поправку к конституции США
2 Иногда государственные обвинители вступают в переговоры с ответчиком и его адво-
катом на стадии расследования. Если переговоры касаются обвинения в тяжком уголовном
преступлении и проходят успешно, то ответчик может отказаться от своего права на рассмо-
трение дела Большим жюри, и государственный обвинитель ограничивается обвинением by
information.
Глава 1.2. Внедрение текущей надлежащей производственной практики 101
до 150 000 долл, при фелонии и до 100 000 долл. — при мисдиминоре. Кроме того,
закон предусматривает наложение штрафа, в два раза превышающего денежных
доход, полученный в результате правонарушения. И FDA, и DOJ могут повышать
взыскиваемые с производителей денежные суммы за несоблюдение cGMP в соот-
ветствии с положением гражданского законодательства о возвращении незаконно
полученной прибыли. В последние годы суммы возмещения достигали сотен мил-
лионов долларов; многие из таких сумм согласовывались в ходе переговоров по до-
стижению мирового соглашения.
1.2.5. Заключение
Принуждение к соблюдению Надлежащей производственной практики — крае-
угольный камень системы, обеспечивающей безопасность фармацевтических про-
дуктов в США. И конгресс, и суды, и (в первую очередь) производители ожидают
от этой системы определенной адекватности, соответствующей столь действенному
и важному для общественного здоровья закону, каким является FDCA. Если суще-
ствует практика последовательного, эффективного и беспристрастного принужде-
ния к исполнению закона, то последний будет не только защищать общество, но
и обеспечит единство требований к производителям, придерживающимся правил
cGMP.
Глава 1.3. ПРАВИЛА МАСШТАБИРОВАНИЯ
ПРОИЗВОДСТВА И ВНЕСЕНИЯ
ПОСТРЕГИСТРАЦИОННЫХ ИЗМЕНЕНИЙ
(SUPAQ
Пунит Шарма, Сринивас Ганта, Санджей Гарг
University of Auckland (Новая Зеландия)
1.3.1. Введение
Фармацевтическая разработка заключается в придании действующему веществу
пригодной лекарственной формы. Перенос технологии производства фармацев-
тической продукции из исследовательской лаборатории в производственный цех
с одновременным увеличением объема серии выпускаемой продукции обычно на-
зывают масштабированием с увеличением. Иначе говоря, масштабирование с увели-
чением — это процедура увеличения объема производственной серии. Напротив,
масштабирование с уменьшением означает уменьшение объема серии в ответ на
падение рыночного спроса.
Изменение масштабов производства при переносе технологического процесса из
исследовательской лаборатории или опытного производства в производственный
цех зачастую вызывает ряд проблем. Основной причиной их появления является
использование разного производственного оборудования в исследовательской ла-
боратории и производственном цехе. Проблемы могут усугубляться недостаточной
осведомленностью о работе оборудования, различными требованиями к контролю
процессов, сложностью последних, отсутствием точных сведений о свойствах ин-
гредиентов при использовании их в различных количествах, а также применени-
ем метода проб и ошибок. Каждый продукт, поступающий из экспериментального
производства, должен быть технологичным, а процесс должен надежно воспро-
изводиться в условиях производственного цеха. Это означает, что при разработке
лекарственной продукции критически важны вопросы переноса технологий и мас-
штабирования. После успешного переноса технологии и валидации продукт обычно
выпускается на промышленном оборудовании (т. е. в промышленных, коммерче-
ских масштабах) в штатном порядке. После регистрации лекарственного средства
в утвержденный процесс его производства могут вноситься изменения. Последние
способны затрагивать состав исходных материалов, а также процессы, оборудова-
ние, производственную площадку или размеры производственной серии — все то,
что влияет на параметры качества фармацевтического продукта. Воздействие этих
изменений на качество лекарства или готового продукта необходимо предвидеть
и тщательно оценивать. К факторам, вынуждающим производителя вводить такие
изменения, относятся, в частности, колебания рыночного спроса (влияющие на
размер производственных серий), новые источники сырья, модификации произ-
Глава 1.3. Правила масштабирования производства и внесения пострегисграционных... 103
водственного процесса, совершенствование упаковочных материалов и переход на
новую методологию анализа процесса или продукта.
Степень риска негативных последствий, вызванных конкретным изменением,
определяется типом лекарственной формы. Например, изменение неактивного ин-
гредиента, выходящее за рамки определенного интервала, сильнее повлияет на ле-
карственную форму с модифицированным высвобождением (англ, modified-release,
MR), чем на лекарственную форму с немедленным высвобождением (immediate-
release, IR), у которой биодоступность не лимитируется скоростью процесса. Схо-
жим образом, изменение первичной упаковки жидких препаратов для парентераль-
ного введения может оказать более выраженное влияние на их эффективность, чем
это могло бы быть в случае твердых лекарственных форм. Следовательно, требова-
ния регулирующих органов к документации по обоснованию изменений могут раз-
личаться в зависимости от масштаба изменений или их неблагоприятного воздей-
ствия на критические параметры лекарственной формы.
В течение своего жизненного цикла фармацевтический продукт может претер-
певать множество изменений. Эти изменения способны неблагоприятно повлиять
на общую безопасность и эффективность такого лекарства. Следовательно, дан-
ные, предоставляемые регулирующим органам для одобрения изменения, должны
содержать сравнение характеристик лекарственного средства с внесенными из-
менениями с теми, которые были утверждены изначально. Документацию, обо-
сновывающую внесение каких бы то ни было изменений в зарегистрированный
фармацевтический продукт, подают в контролирующие органы для экспертизы;
изменения утверждаются на основании оценки соотношения «риск — польза».
Объем документации, подаваемой в регуляторный орган, зависит от значимости
изменений.
Органы надзора, такие как Федеральное управление США по контролю за пи-
щевой продукцией и лекарствами (FDA), Европейская комиссия, Национальное
агентство по контролю в области здравоохранения (Бразилия) (ANVISA) и др., тре-
буют от производителей фармацевтической продукции в соответствующих странах
соблюдать положения Руководств по масштабированию (с увеличением) производ-
ства и внесению пострегистрационных изменений (SUPAC), чтобы обеспечить тре-
буемое качество фармацевтической продукции. Время от времени эти руководства
пересматривают с тем, чтобы они соответствовали современному уровню техноло-
гических достижений; новые руководства направлены на сокращение накладных
расходов фармацевтической промышленности и ретуляторных органов. Кроме упо-
мянутых руководств, существуют и другие ориентиры, с помощью которых про-
мышленность гарантирует производство качественной продукции. Примерами та-
ких ориентиров являются контроль изменений и процедуры валидации, которые
подробно обсуждаются далее в настоящей главе. Эти действия контролируются
в соответствии с принципами Надлежащей производственной практики (GMP),
установленными регуляторными органами.
В данной главе рассматриваются нормативные документы, изданные различ-
ными органами надзора, а также мероприятия в рамках фармацевтической про-
мышленности, направленные на гарантирование качества лекарственных средств.
Подробно анализируются руководства FDA. Другие руководства обсуждаются лишь
104
Часть I. Нормативное регулирование производства лекарственных средств
в общих чертах, но тем читателям, кто нуждается в более детальной информации,
предлагаются ссылки на веб-сайты регуляторных органов.
1.3.2. Научное и юридическое обоснование масштабирования
производства и пострегистрационных изменений (5UPAQ
Руководства, регулирующие пострегистрационные изменения, классифицируют
их в соответствии с различными категориями исходя из степени того влияния, ко-
торое конкретное изменение оказывает на качество и эффективность лекарствен-
ного средства. Независимо от терминологии, используемой различными органами
надзора, изменения в целом можно охарактеризовать как незначительные, умерен-
ные и существенные-, объем обосновывающей изменения документации зависит от
категории такого изменения. Например, руководство FDA «Изменения, внесенные
в зарегистрированные оригинальные или воспроизведенные лекарственные пре-
параты» описывает упомянутые изменения как 1) незначительные, которые можно
внедрять немедленно и указывать в следующем периодическом отчете; 2) умерен-
ные, которые можно внедрять немедленно; 3) умеренные, которые требуют подачи
уведомления за 30 дней до внедрения; 4) существенные, требующие утверждения
FDApo внедрения [1]. Аналогичным образом в Европейском Союзе (ЕС) все изме-
нения типа I (L4 и 15) и типа II необходимо подавать до выпуска измененного препа-
рата на рынок [2]. Управление по контролю медикаментов (Австралия) (Therapeutic
Good Administration, TGA) подразделяет пострегистрационные изменения на три ка-
тегории: 1) не подлежащие оценке; 2) оцениваемые самостоятельно и 3) изменения,
требующие предварительного утверждения органом надзора.
1.3.2.1. Документация, обосновывающая изменения, а также масштабы
последних
Согласно руководящим документам FDA, изменения содержания вспомогательных
веществ в составе препарата, не превышающие 5 %масс., считаются незначительны-
ми, и вся информация о них предоставляется в ежегодном отчете. Однако при на-
личии изменений, способных значительно повлиять на качество и эффективность
лекарственного средства, необходимо получить утверждение изменения до выпуска
на рынок, для чего следует представить соответствующее заявление с приложением
документации, содержащей как данные по растворению in vitro и in vivo, так и ре-
зультаты исследований стабильности (при ускоренном и естественном хранении),
включенные в ежегодный отчет [4]. Схожего принципа придерживаются и руковод-
ства ЕС: так, менее чем десятикратное увеличение (или уменьшение) объема про-
изводственной серии готового продукта по сравнению с исходным размером, ука-
занным в регистрационном досье, определяется как изменение типа L4 и требует
предоставления данных (в виде сравнительных таблиц) как минимум по одной из
производственных серий, выпущенной в одобренном и предполагаемом масштабах.
Данные по двум следующим полным производственным сериям должны быть до-
ступны по запросу; держатель регистрационного удостоверения должен представить
их в отчете с указанием предполагаемых действий. Однако при изменениях типа
15 (более чем 10-кратные изменения) в дополнение к данным, упомянутым выше,
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 105
необходимо предоставить либо копию утвержденных спецификаций (по выпуску
в обращение и при хранении) с номерами серий (не менее трех), использовавшихся
в процессе валидационного исследования, либо протокол валидации (программу),
подкрепленный указанием числа производственных серий, использованных в про-
цессе исследования стабильности.
1.3.2.2. Документация, обосновывающая внесение изменений
в спецификации
Изменения в спецификациях любого типа должны быть обоснованы соответствую-
щей документацией. Во всех руководящих документах смягчение требований (пока-
затель качества) или исключение из спецификации любой части классифицируется
как существенное изменение, а значит, требует достаточно большого документаль-
ного обоснования. Необходимая документация может включать в себя как подачу
в FDA соответствующего дополнения к досье для предварительного утверждения,
так и сравнительные таблицы действующих и предлагаемых спецификаций, а так-
же подробное изложение любого нового аналитического метода, данных валидации
или результатов анализа производственной серии по всем показателям качества для
двух производственных серий готового продукта согласно новой спецификации
в ЕС. Спецификации являются ориентирами при сравнении пригодности для при-
менения любого продукта. Например, требования по однородности количественно-
го содержания ограничивают отклонение результатов анализа каждой единицы про-
дукции в пределах 90—110% от заявленного в спецификации содержания активного
ингредиента в таблетке сильнодействующего вещества массой 20 мг (средняя мас-
са), что указывает на необходимость особого контроля стадии смешивания для обе-
спечения однородности изготавливаемых таблеток, содержащих столь малые дозы
активного ингредиента. Любое смягчение требований, содержащихся в специфика-
ции на такой сильнодействующий препарат, должно быть обосновано с помощью
достаточного количества документов, свидетельствующих о качестве препарата
с внесенными изменениями. Однако ужесточение требований к качеству считается
незначительным изменением и не должно оказывать серьезных негативных воздей-
ствий на подлинность продукта, а также на другие показатели качества, включая
чистоту или количественное содержание (биологическую активность).
1.3.2.3. Протоколы сравнения
Федеральное управление США по контролю за пищевой продукцией и лекарствами
ввело концепцию протоколов сравнения, призванных ускорить процесс утверждения
пострегистрационных изменений по конкретному изменению [5]. Протокол охва-
тывает предполагаемые изменения, которым может подвергаться продукт в течение
срока его годности. В недавно опубликованном проекте руководства «Протоколы
сравнения: информация по качеству, производству и контролю» {СМС, от англ.
Chemistry, Manufacturing, and Controls) были описаны принципы составления про-
токолов сравнения. Этот документ апробирует менее строгую категорию измене-
ний, подлежащих представлению в ретулирующий орган, для любых предстоящих
изменений там, где это возможно. Кроме того, Руководством оговорено, что при
наличии подробного протокола сравнения снижается вероятность того, что FDA за-
106 Часть I. Нормативное регулирование производства лекарственных средств
просит дополнительные документы для обоснования изменений в целях сравнения
пред- и пострегистрационных изменений. Далее, такой протокол облегчает внедре-
ние конкретных изменений в СЛ/Си, следовательно, ускоряет поставку измененно-
го продукта на рынок. Согласно правилам FDA,
...протокол сравнения представляет собой четкий, подробный письменный план оцен-
ки влияния конкретных изменений в СМСв отношении подлинности, количественно-
го содержания и других показателей качества, включая чистоту и фармакологическую
активность конкретного лекарственного препарата, поскольку эти критерии связаны
с безопасностью и эффективностью продукта. Протокол сравнения описывает изме-
нения, для которых он был разработан, и содержит перечень испытаний и исследова-
ний, которые предстоит провести (это, в частности, методики анализа, которые будут
использованы, а также критерии приемки (нормы), соответствие которым должно по-
казать, что определенные изменения в СМС не оказывают неблагоприятного воздей-
ствия на продукт). Предоставление протокола сравнения не является обязательным.
Протокол сравнения может подаваться вместе с NDA (досье на оригинальный
препарат) или ANDA (досье на воспроизведенный препарат) либо являться допол-
нением к этим досье.
Протоколы сравнения могут описывать сразу несколько изменений, если по-
следние не связаны друг с другом и имеют четкую спецификацию для оценки из-
менения.
1.3.2.4. Требования по проведению испытаний in vitro v\ in vivo
Стабильность лекарственного препарата, испытание на растворение in vitro и оценка
биоэквивалентности in vivo являются краеугольными параметрами его эффективно-
сти и играют ключевую роль при определении качества препарата в рамках внедрения
пострегистрационных изменений. Любое существенное изменение (например, пере-
ход от сухого гранулирования к влажному) может повлиять на биодоступность и ста-
бильность фармацевтического продукта. Тщательный подбор условий растворения
может помочь избежать дорогостоящего исследования биоэквивалентности. В руко-
водствах FDA [4] и ANVISA [6] при выборе критерия растворимости для конкретного
лекарственного средства (с немедленным или модифицированным высвобождени-
ем) учитываются его растворимость и проницаемость; в то же время руководства
ЕС и TGA рекомендуют предоставлять сравнительные данные по определенному
числу производственных серий, изготовленных до и после внесения изменений.
При определении категории изменения или отклонения по значимости воздей-
ствия следует учитывать те характеристики лекарственного препарата, которые спо-
собны повлиять на его биодоступность. Критически важные характеристики, такие
как размер частиц активного ингредиента или вспомогательного вещества, смачива-
емость поверхности лекарственного средства, а также свойства последнего в твердом
состоянии, могут изменяться при измененной технологии и оказывать неблагопри-
ятное влияние на фармацевтико-технологические характеристики готового продук-
та, искажая профиль его растворения. Более критическими могут стать изменения
в лекарственных препаратах, которые содержат активный компонент, обладающий
плохой растворимостью, что может существенно снизить биодоступность. В руко-
Глава 1.3. Правила масштабирования производства и внесения посгрегистрационных... 107
водствах FDA учтены рекомендации Биофармацевтической системы классификации
(Biopharmaceutic Classification System, BCS) в отношении растворимости и проницае-
мости для определения необходимости какого-либо исследования биоэквивалент-
ности in vivo наряду с исследованием растворения in vitro. По тому же принципу
ANVISA разделяет лекарства на три категории для твердых лекарственных форм:
> категория А — активные вещества с высокой проницаемостью и хорошей рас-
творимостью;
> категория В — активные вещества с низкой проницаемостью и хорошей рас-
творимостью;
> категория С — активные вещества с высокой проницаемостью и плохой рас-
творимостью.
Категория А. «Необходимая документация должна включать технический отчет
и оценку результатов теста на растворение, выполняемого согласно Фармакопее
Бразилии, а при их отсутствии — согласно другим кодексам, использование кото-
рых допустимо действующим законодательством. Высвобождение по меньшей мере
85% активного вещества в 900 мл 0,1 МНС1 должно занимать не более 15 мин. При
несоответствии этому критерию следует выполнить тесты, предусмотренные для ка-
тегорий Били С».
Категория В. «В необходимую документацию должны быть включены техниче-
ский отчет и оценка результатов исследования профиля растворения, выполняемого
в фармакопейных условиях, при удалении проб из среды в определенные моменты
времени до достижения плато профиля. Полученный профиль растворения должен
быть аналогичен профилю растворения для неизмененного состава».
Категория С. «В необходимую документацию должны быть включены техниче-
ский отчет и оценка результатов исследования профиля растворения в пяти различ-
ных условиях: в дистиллированной воде, в 0,1 МНС1 и фосфатном буфере с пока-
зателями pH 4,5, 6,5 и 7,5 для предложенного и исходного составов. Отбор проб из
среды растворения необходимо проводить в определенные моменты времени до до-
стижения или высвобождения 90% активного вещества либо плато профиля раство-
рения. Применение поверхностно-активных веществ должно быть оправданным.
Полученный профиль растворения должен соответствовать профилю растворения
для неизмененного состава».
Кроме того, для изменений уровня 2 не требуется дополнительного исследова-
ния биоэквивалентности, если предложенное изменение соответствует категориям
А, В и С. Однако при наличии любых отклонений необходимо представить доку-
ментацию, содержащую результаты испытаний и оценку биоэквивалентности и/или
биодоступности нового состава (если не была установлена соответствующая корре-
ляция данных in vitro/in vivo (ivivc)) в условиях, описанных для изменений уровня 3.
1.3.3. Регулирующие органы и руководства
1.33.1. Правила SUPAC(FDA}
Закон о модернизации FDA (The Food and Drug Administration Modernization Act,
FDAMA) был опубликован 21 ноября 1997 г. К Закону о пищевых продуктах, медика-
108 Часть I. Нормативное регулирование производства лекарственных средств
ментах и косметической продукции (FDCA) были добавлены пункты 506Л и 314.70
(21 CFR 314.70), содержащие рекомендации по категориям (с использованием
утвержденной терминологии) для любых типов изменений технологического про-
цесса для зарегистрированных лекарственных препаратов (NDA или ANDA). Вскоре
FDA опубликовало «Руководство для промышленности: внесение изменений в заре-
гистрированные NDA или ANDA», окончательно завершенное в 2004 г. Это руковод-
ство представляет собой действующий стандарт для фармацевтических производи-
телей, регламентирующий как внесение изменений (и описание последних) в досье
на зарегистрированный препарат, так и поставки продукции, произведенной после
введения изменений.
Руководство SUPAC-IR «Твердые лекарственные формы для приема внутрь с не-
медленным высвобождением. Масштабирование производства и пострегистра-
ционные изменения в досье по качеству, производству и контролю, исследование
растворения in vitro и документация исследования биоэквивалентности in vivo», опу-
бликованный в 1995 г., стал первой попыткой дать фармацевтической промышлен-
ности четкое руководство, регламентирующее порядок предоставления в регулиру-
ющий орган документации по пострегистрационным изменениям. Появление этого
руководства явилось следствием: а) семинара по масштабированию (с увеличением)
производства лекарственных препаратов с немедленным высвобождением, прове-
денного Американской ассоциацией совместно с Фармакопеей США и FDA; б) ис-
следований в области СМС лекарственных препаратов с немедленным высвобож-
дением, проведенных Университетом Мэриленда в Балтиморе; в) исследований по
проницаемости лекарственных средств разных категорий, проведенных университе-
тами Мичигана и Упсалы; г) работы специальной комиссии по SUPAC, учрежденной
Комитетом по координации исследований СЛ/Спри CDER (Центре оценки и иссле-
дований лекарственных средств). Опубликованное руководство сразу стало ориен-
тиром для промышленности. Были опубликованы еще два подобных руководства: по
SUPACjvm лекарственных препаратов с модифицированным высвобождением (опу-
бликовано в 1997 г.) [7] и для нестерильных полутвердых лекарственных препаратов
(опубликовано в 1997 г.) [8]. «Руководство по изменениям в зарегистрированных NDA
и ANDA» отменяет действие предыдущих руководств в отношении изменений, под-
лежащих подаче в регуляторный орган, не соответствующих данному руководству.
Руководство для промышленности: изменения в зарегистрированных NDA или ANDA
В данном руководстве определены категории различных пострегистрационных из-
менений. Оно смягчило некоторые требования, которые были сочтены минимально
влияющими (или не влияющими) на характеристики лекарственного средства [1].
Это позволило освободить от лишней работы и органы надзора, и производителей.
В данном руководстве представлены четыре категории изменений, подлежащие по-
даче в регуляторный орган:
1. Изменение, требующее предварительного утверждения. Для согласования су-
щественных изменений (т. е. тех, что могут оказать существенное влияние на
качество и действие лекарственного средства) соответствующее дополнение
1 Аббревиатура словосочетания Scale Up and Post-Approval Changes, т. e. «масштабирование
(с увеличением) и пострегистрационные изменения». — Примеч. отв. ред.
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 109
к регистрационному досье должно быть предварительно подано в FDA для
утверждения до выпуска в обращение измененного продукта. Кроме того, су-
ществует процедура «Рассмотрение дополнений в досье, требующих утверж-
дения: запрос об ускоренном рассмотрении». Такая процедура предусмотрена
для случаев особых нужд национального здравоохранения, а также для тех
ситуаций, когда отсрочка утверждения может вызвать существенные финан-
совые последствия для заявителя.
2. Дополнение в досье «Изменения, вводимые в действие через 30 дней после уведом-
ления». При внесении умеренных изменений (средний риск воздействия на
качество и действие) дополнение в досье следует предоставить FDA за 30 дней
до того, как продукт, произведенный с учетом таких изменений, будет выпу-
щен в обращение.
3. Дополнение в досье «Изменения, вводимые в действие одновременно с уведомлени-
ем (Одней)». Для некоторых изменений необходимо предоставить дополнение
в регистрационное досье в FDA\ при этом допускается одновременно начать
выпуск в обращение продукта, произведенного с учетом изменений.
4. Ежегодный отчет. При внесении незначительных изменений (минимальная
вероятность влияния на качество и действие) вся информация должна быть
предоставлена в FDA в очередном ежегодном отчете; продукт, произведенный
с учетом таких изменений, может поступать в обращение.
Согласно данному руководству, все три типа изменений поделены на следующие
категории:
1. Изменения компонентов и состава. Любые качественные и количественные
изменения компонентов и состава лекарственного средства рассматриваются
как существенные. В действующем документе «Руководство для промышлен-
ности: изменения в зарегистрированные NDA или ANDA» об этом не гово-
рится подробно из-за сложности подходов, используемых в рекомендациях.
Поэтому необходимо соблюдать требования SUPAC пля любых подобных из-
менений и требований, касающихся документации, предоставляемой в регу-
ляторный орган.
2. Изменения производственных площадок. Для смены одобренной при регистра-
ции производственной площадки (осуществляющей производство, упаковку,
маркировку фармацевтических препаратов, контроль качества компонентов,
контейнеров для лекарственных средств, укупорочных средств, упаковочных
материалов), входящей в состав компании или используемой по контракту,
требуется предварительное согласование с CDER. Если планируется исполь-
зование площадки, которая не получила удовлетворительной оценки инспек-
ции по соблюдению cGMP для производимой операции, в FDA подается допол-
нение, требующее предварительного утверждения. Кроме того, считается,
что изменение производственных площадок, задействованных в таких опе-
рациях, как маркировка, упаковка в потребительскую (вторичную) упаковку
и выполнение лабораторных испытаний, вне зависимости от лекарственной
формы оказывает влияние на продукт. Это означает, что изменения любых
таких производственных площадок будут отнесены к одной категории. Вме-
110
Часть I. Нормативное регулирование производства лекарственных средств
сте с тем считается, что влияние изменений производственных площадок, за-
действованных в производстве лекарственной формы и фасовке, зависит от
лекарственной формы, поэтому дополнения к досье могут различаться.
3. Изменения производственного процесса могут оказывать существенное влияние
на подлинность формацевтического препарата, на его количественное содержа-
ние и другие показатели качества, включая чистоту или действие; кроме того, из-
менение эффективности лекарственного средства может иметь место даже при
аналитическом подтверждении его соответствия утвержденной спецификации.
4. Изменения спецификаций. Спецификации, нормы и обязательные аналитиче-
ские процедуры входят в каждое регистрационное досье, подаваемое в регу-
ляторный орган. Спецификации представляют собой стандарты (критерии
приемки), регламентирующие предельные значения показателей специфи-
кации, а обязательные аналитические процедуры используются для проведе-
ния испытаний на соответствие норм, утвержденных регуляторным органом.
Наряду с основной аналитической процедурой, в заявке может быть указана
альтернативная аналитическая процедура.
5. Изменения первичной упаковки (с укупорочными средствами). Степень влияния
изменений первичной упаковки главным образом зависит от пути введения
препарата, технологической операции фасовки и характера взаимодействия
продукта с упаковкой. В отдельных случаях эти последствия могут иметь ме-
сто несмотря на соответствие лекарственного средства утвержденной специ-
фикации.
6. Изменения маркировки. Изменения содержания листка-вкладыша (инструк-
ции по применению препарата для пациента) и этикетки на вторичной упа-
ковке относятся к изменениям маркировки; в таких случаях заявитель должен
немедленно произвести соответствующую корректировку всех рекламных
образцов препарата и рекламных материалов в соответствии с утвержденной
маркировкой.
7. Прочие изменения включают те, что касаются протокола стабильности, срока
годности или протокола сравнения.
8. Множественные взаимосвязанные изменения. Одно изменение может повлечь
за собой намеренное или ненамеренное внедрение другого: например, из-
менение производственной площадки может привести к изменениям про-
изводственного оборудования и производственного процесса, а изменения
упаковочного материала — к изменениям протокола стабильности. Центр
экспертизы и изучения лекарственных средств (CDER) рекомендует подавать
дополнение в досье для таких комбинированных изменений в соответствии
с наиболее жесткими требованиями для отдельных изменений.
Масштабирование производства (с увеличением) и пострегистрационные измене-
ния: лекарственные формы с немедленным высвобождением и модифицированным вы-
свобождением (SUPACIR1). Руководство по SUPAC классифицирует пострегистра-
1 Scale-up and Postapproval Changes: Immediate-Release and Modifled-Release Dosage Forms. —
Примеч. отв. ped.
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 111
ционные изменения по отдельным «уровням» [4]. В зависимости от степени риска
неблагоприятного воздействия на лекарственный состав различают три уровня.
Уровень 1 означает, что итоговое влияние изменения на качество будет минималь-
ным; как следствие, объем документации, предоставляемой в ежегодном отчете для
FDA, тоже будет минимальным. Изменения, соответствующие уровню 2, могут ока-
зать существенное влияние на качество и эффективность лекарственной формы. Ве-
роятность негативного влияния изменений уровня 3 на качество и эффективность
лекарственной формы наиболее велика; в этом случае выпуск в обращение изме-
ненного продукта может осуществляться только после их утверждения FDA. Кро-
ме описания характеристик уровней изменений, Руководство содержит рекомен-
дации по исследованиям растворения in vitro, биоэквивалентности in vivo и объему
документации по СМС. В каждом разделе Руководства («Компоненты и составы»;
«Изменения производственных площадок»; «Изменение масштабов производ-
ства (увеличение и уменьшение)»; «Производственное оборудование и процессы»)
представлена классификация изменений по этим трем уровням. При определении
уровня каждого конкретного изменения также рассматриваются терапевтический
диапазон, растворимость и проницаемость лекарства. Согласно Руководству, при
исследовании растворения выделяют три варианта (табл. 1). Кроме того, изменение
содержания вспомогательных веществ для лекарств узкого терапевтического диа-
пазона, предусматривающее превышение пределов, установленных для уровня 1,
рекомендуется рассматривать как изменение уровня 3, для обоснования которого
требуется исчерпывающая документация. В руководстве SUPAC для лекарствен-
ных форм с модифицированным высвобождением изменения классифицировались
в соответствии с этими же уровнями [7].
Таблица 1
Различные варианты и соответствующие условия испытания на растворение для твердых
лекарственных форм с немедленным высвобождением
Вариант Л*
Вариант 2F
Вариант I?
Высвобождение 85% в 900
мл 0,1 7VHC1 в течение 15
мин. Если лекарственный
препарат не проходит дан-
ное испытание, заявитель
должен провести тесты,
описанные для вариантов
Вили С
Многоточечный профиль рас-
творения необходимо постро-
ить при растворении в указан-
ной в регистрационном досье
или фармакопейной статье
среде растворения отбором
проб через 15, 30, 45, 60 и 120
мин или до достижения асим-
птоты
Многоточечный профиль растворения
необходимо исследовать при растворении
в воде, 0,1 N НС1 и буферной фармако-
пейной среде при pH 4,5; 6,5 и 7,5 (пять
различных профилей) для предложенных
и утвержденных составов лекарственного
препарата. Адекватный пробоотбор следу-
ет проводить через 15,30,45,60 и 120 мин,
или до высвобождения 90% активного ве-
щества из препарата, или до достижения
асимптоты. Применение поверхностно-
активных веществ должно быть обосно-
вано
’Лекарства с высокой проницаемостью и хорошей растворимостью.
'Лекарства с низкой проницаемостью и хорошей растворимостью.
•Лекарства с высокой проницаемостью и плохой растворимостью.
Однако условия растворения лекарственных форм соответственно с пролонги-
рованным или замедленным высвобождением несколько различны (табл. 2). При
112
Часть I. Нормативное регулирование производства лекарственных средств
составлении отчета о любом изменении уровня 3 необходимо представить или ре-
зультаты ускоренного трехмесячного исследования стабильности по трем производ-
ственным сериям (значительный массив данных недоступен), или результаты уско-
ренного трехмесячного исследования стабильности по одной серии (значительный
массив данных доступен). Эти данные заносят в приложение вместе с включенны-
ми в ежегодный отчет результатами исследования стабильности при долгосрочном
хранении для одной производственной серии. В Руководстве значительный массив
данных определяется как «доступность достаточной информации о стабильности
лекарственного средства» (данные по стабильности по результатам исследования
пяти коммерческих партий). В табл. 3—8 представлено сравнение требований руко-
водств для лекарственных форм с немедленным и модифицированным высвобож-
дением.
Таблица 2
Условия растворения для лекарственных форм с модифицированным высвобождением
Пролонгированное высвобождение
Замедленное высвобождение
В дополнение к указанным в досье/Фармакопее
требованиям испытания растворения, следует ис-
следовать многоточечные кривые растворения
в трех различных средах: например, в воде, 0,1
N НС1 и буферной среде (по Фармакопее США)
при pH 4,5 и 6,8 для измененного лекарственного
средства и серии, использованной в клинических
исследованиях, или коммерческой серии (лекар-
ственного средства, произведенного без измене-
ний). Адекватный пробоотбор следует произво-
дить, например, через 1,2 и 4 ч и через каждые 2 ч
далее до высвобождения 80% активного вещества
из препарата или до достижения асимптоты. При-
менение поверхностно-активных веществ должно
быть обосновано
В дополнение к указанным в досье/Фармакопее
США требованиям испытания растворения, сле-
дует провести исследование растворения в 0,1 А
НС1 в течение 2 ч (кислотная стадия) с последую-
щим растворением в буферной среде (по Фармако-
пее США), в интервале pH 4,5—7,5 (буферная ста-
дия) в стандартных (согласно досье/Фармакопее)
условиях растворения и дополнительно при двух
различных скоростях перемешивания с помощью
прибора для растворения, указанного в досье/ру-
ководствах (три дополнительных варианта условий
исследования). Многоточечные профили раство-
рения должны быть получены на буферной стадии
исследования. Адекватный пробоотбор следует
проводить через 15,30,45,60 и 120 мин (после по-
мещения лекарственной формы в буферный рас-
твор) до высвобождения 80% активного вещества
из препарата или достижения асимптоты. Выше-
описанное исследование растворения следует про-
водить с использованием лекарственного сред-
ства, произведенного с учетом изменения, а также
серии, участвовавшей в клинических исследова-
ниях, или коммерческой серии (лекарственного
средства, произведенного без изменений)
1. Изменения компонентов и составов рассмотрены в табл. 3. Руководство по вне-
сению изменений в зарегистрированные NDA или ANDA не содержит подробного
описания этих изменений; таким образом, для получения информации и составле-
ния требуемой для подачи в FDA документации необходимо следовать указаниям
по SUPAC. Дополнения в регистрационное досье, касающиеся изменения содер-
жания вспомогательных веществ, подают для утверждения до их введения в дей-
ствие (с представлением результатов исследования стабильности при ускоренном
старении); в то же время данные о любом изменении содержания красителей или
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 113
ароматизаторов представляют в ежегодном отчете (данные исследования стабиль-
ности при естественном старении). Для лекарственных форм с модифицирован-
ным высвобождением эти изменения были логически разделены: 1) на изменения
содержания вспомогательных веществ, не влияющие на профиль высвобождения;
2) изменения содержания вспомогательных веществ, влияющие на профиль вы-
свобождения. При изменениях уровня 2 для лекарственных средств с немедленным
высвобождением либо лекарственных препаратов широкого терапевтического диа-
пазона с модифицированным высвобождением следует представить на утверждение
дополнение в регистрационное досье. Это дополнение должно включать в себя дан-
ные исследования стабильности при трехмесячном ускоренном старении для одной
серии (для лекарственных форм с модифицированным высвобождением с узким
терапевтическим диапазоном — данные на трех сериях); для ежегодного отчета не-
обходимы данные исследования стабильности при естественном старении на одной
серии. Дополнительно для лекарственных форм с замедленным высвобождением
из веществ с узким терапевтическим диапазоном необходимо предоставить дан-
ные многоточечного профиля растворения для измененного и выпускаемого про-
дуктов с использованием среды растворения, указанной в регистрационном досье
или Фармакопее. Для лекарственных форм с пролонгированным высвобождением
из веществ с узким терапевтическим диапазоном строится многоточечный профиль
растворения для средства, выпущенного с учетом изменений, и коммерческого про-
дукта с использованием среды растворения, указанной в регистрационном досье
или Фармакопее.
2. Изменения производственной площадки (см. табл. 5), осуществляющей произ-
водство либо упаковку (или контрактной производственной площадки), одобрен-
ные FDA в исходном регистрационной досье, подлежат оценке с точки зрения их
влияния на качество и эффективность лекарственного средства. Эти изменения
подробно описаны в действующем руководстве по внесению изменений в зареги-
стрированные NDA или ANDA.
Новый адрес площадки обязательно сообщается FDA. При умеренных изме-
нениях любого типа следует предоставить в дополнении к регистрационному до-
сье, вводимому в действие одновременно с подачей в FDA, данные исследования
стабильности при ускоренном хранении на одной производственной серии (англ.
changes being effected, СВЕ) для лекарственных форм с модифицированным вы-
свобождением; данные по исследованию стабильности в условиях естественного
старения на одной производственной серии предоставляются в ежегодном отчете
для лекарственных форм как с немедленным, так и с модифицированным высво-
бождением. Кроме того, дополнение к регистрационному досье, вводимое в дей-
ствие одновременно с подачей в FDA, представляется при осуществлении любых
умеренных изменений. Требования по представлению данных по стабильности при
любых существенных изменениях аналогичны упомянутым в предыдущем разделе,
т. е. представляют данные по исследованию стабильности в условиях трехмесячно-
го ускоренного старения на трех производственных сериях (значительный массив
данных не доступен) или на одной производственной серии (значительный массив
данных доступен); все эти данные включаются в дополнение к регистрационному
досье, требующему утверждения в FDA. Также подается ежегодный отчет, где со-
114
Часть I. Нормативное регулирование производства лекарственных средств
Изменения составов и компонентов,
Уровень Классификация Терапевтический диапазои/тип препарата
1 Полное или частичное удаление красителя/ ароматизатора. Замена чернил, гравировки. Изменения вспомогательных веществ соглас- но SUPAC IR (уровень 1). Отсутствие других изменений Все лекарства
2 Изменение технической категории и/или спецификаций. Изменения вспомогатель- ных веществ, выходящие за пределы, уста- новленные для уровня 1, но не достигающие уровня 2. Отсутствие других изменений. Из- менение технической категории и/или спец- ификаций выше определенного для SUPAC IR (уровень 1). Другие изменения отсутствуют Все лекарства с модифицированным вы- свобождением. Зависит от терапевтического диапазона, растворимости и проницаемости (согласно BCS) для лекарственных средств с немедленным высвобождением
3 Изменения вспомогательных веществ, пре- вышающие изменения, определенные в пра- вилах SUPAC IR для уровня 2 (с модифици- рованным и немедленным высвобождением), а также изменения, выходящие за рамки уровня 1, для лекарств с плохой растворимо- стью и низкой проницаемостью Все препараты с модифицированным высво- бождением и все препараты, не выдержавшие испытание на растворение для уровня 2 для лекарственных препаратов с немедленным высвобождением
1 Имеются в виду исследования биоэквивалентности. — Примеч. перев.
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных.., 115
Таблица 3
не контролирующих высвобождение
Документация по испытаниям Подаваемая в регуляторный орган документация
Стабильность. Требования согласно досье/руководству. Клинического’ исследо- вания не требуется Ежегодный отчет
Модифицированное высвобождение (пролонгированное) Уведомление и досье на про- мышленные серии, изготов- ленные с учетом изменений. Стабильность. Требования согласно досье/Фармакопее плюс многоточечные про- фили растворения в трех различных средах (таких как вода, 0,1 N НС1 и буферная среда (по Фармакопее США) при pH 4,5 и 6,8) до высво- бождения 80% лекарства или достижения требуется асимптоты. При сравне- нии профилей растворения применяется определенный статистический критерий (тест fl). Клинического ис- следования не требуется Модифицированное высвобождение (замедленное) Уведомление досье на про- мышленные серии, произве- денные с учетом изменений. Стабильность. Требования согласно заявке/Фармако- пее, многоточечные профи- ли растворения и дополни- тельное исследование в бу- ферной среде (например, в буферной фармакопейной при pH 4,5—7,5) в стандарт- ных условиях и в условиях интенсивного перемеши- вания до растворения >80% лекарства или достижения асимптоты. При сравнении профилей растворения при- меняется определенный ста- тистический критерий (тест /2). Клинического исследо- вания не требуется Немедленное высвобождение Уведомление и досье на промышленные серии, произведенные с учетом изменений. Стабильность. Требо- вания к растворению: категория А, катего- рия В или категория С. При сравнении профилей раство- рения применяется определенный стати- стический критерий (тест fl). Клиниче- ского исследования не требуется Требуется утверж- дение дополнения к досье до введения изменения в дей- ствие
Досье на измененные производственные серии. Требова- ния согласно досье/руководству (профиль) и описанию для уровня 2. Стабильность. Клиническое исследование или ivivc Досье на промыш- ленные серии, вы- пущенные с учетом изменений. Профили растворения (соглас- но уровню 2). Ста- бильность. Клини- ческое исследование или ivivc Тоже
116
Часть I. Нормативное регулирование производства лекарственных средств
Изменения состава и компонентов,
Уро- вень Классификация Терапевтический д иапазон
1 Изменения < 5 %масс. от общего количества вспомогательного вещества, контролирующего высвобождение (например, поли- мера или пластификатора). Отсутствие других изменений Все лекарства
2 Изменение технической категории и/или спецификаций. Более 10 %масс. от общего количества вспомогательного вещества, контролирующего высвобождение (например, полимера или пластификатора). Отсутствие других изменений Широкий
Узкий
3 Изменения >10 %масс. от общего количества вспомогательного вещества, контролирующего высвобождение (например, поли- мера или пластификатора) Все лекарства
держатся данные исследования стабильности при естественном хранении на одной
производственной серии.
3. Масштабирование производства (см. табл. 6). При изменении объема про-
мышленной серии лекарственного средства (уменьшение или увеличение) вполне
вероятны некоторые изменения в рабочих параметрах. Это может неблагоприятно
сказаться на качестве продукта.
4. Изменения производственного оборудования (см. табл. 7) и процессов (см. табл. 8).
К ним относятся любые изменения производственного оборудования и процессов.
Например, замена восьмигранного смесителя на двойной конусный или изменение
процесса гранулирования (переход от влажного гранулирования к сухому) требует
предоставления надлежащих вал идационных документов на утверждение в FDA. Все
эти изменения, включая содержание документации, подаваемой в FDA, описаны
в действующем руководстве по изменениям в зарегистрированных NDA и ANDA.
Процедура «биовейвер» (исключение исследований in vivo). Для оценки биодо-
ступности и биоэквивалентности обычно используют подходы in vitro и in vivo. Ис-
следование растворения относится к методам in vitro и применяется для контроля
качества лекарственных средств. В некоторых обстоятельствах тест растворение in
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 117
Таблица 4
контролирующих высвобождение
Документация по испытаниям Подаваемая в регуляторный орган документация
Стабильность. Требования согласно досье/Фармакопее. Клинического исследо- вания не требуется Ежегодный отчет
Модифицированное высвобождение (про- лонгированное) Уведомление и досье на промышленные серии, произведенные с учетом измене- ний. Требования согласно досье/Фар- макопее плюс многоточечные профили растворения в трех различных средах (например, в воде, 0,1 JVHC1 и буферной фармакопейной среде при pH 4,5 и 6,8) до высвобождения > 80% лекарства либо достижения асимптоты. При сравнении профилей растворения применяется определенный статистический критерий (тест fl). Клинического исследования не требуется Модифицированное высвобождение (замедленное) Уведомление и досье на промышлен- ные серии, произведенные с учетом изменений. Стабильность. Требова- ния согласно досье/Фармакопее плюс многоточечные профили растворения и дополнительное исследование в бу- ферной фармакопейной среде (напри- мер, при pH 4,5—7,5) в стандартных условиях и при интенсивном переме- шивании до высвобождения > 80% ле- карства либо достижения асимптоты. При сравнении профилей растворе- ния применяется определенный ста- тистический критерий (тест fl). Кли- нического исследования не требуется Требуется утверж- дение дополнения к досье до введения в действие
Досье на измененные промышленные серии. Требования согласно заявке/Фар- макопее. Клиническое исследование или ivivc Тоже
Досье на измененные промышленные серии. Требования согласно заявке/Фарма- копее. Клиническое исследование или ivivc
vitro может также служить суррогатным маркером для исследования биоэквивалент-
ности in vivo, позволяя определить биоэквивалентность in vitro и in vivo.
Руководство CDER для промышленности «Процедура “вейвер” для исследо-
ваний биодоступности и биоэквивалентности in vivo для твердых лекарственных
форм с немедленным высвобождением на основании Системы биофармацевтиче-
ской классификации (BCS)» рекомендует исключение клинического исследования
в определенных обстоятельствах. Например, допускается исключение клинических
исследований для одной или нескольких более низких дозировок препарата при на -
личии корреляционных данных и данных по биоэквивалентности in vivo, получен-
ных для более высокой дозировки при условии, что все дозировки препарата про-
порционально эквивалентны по составу. Иногда для подтверждения безопасности
продукта (как в случае с таблетками митразапина) может потребоваться исследова-
ние биоэквивалентности более низкой дозировки. В подобных случаях исключение
этого исследования для более высоких дозировок препарата приемлемо, если фар-
макокинетические параметры линейны во всем диапазоне доз, составы препарата
с различными дозировками пропорциональны, а данные сравнительных исследова-
ний растворения имеются для всех дозировок препарата.
Таблица 5
Изменения производственной площадки
Уровень Классификация Терапев- тический диапазон Документация по испытаниям Подаваемая в регуляторный орган документация
1 Одно здание. Общий пер- сонал. Другие изменения от- сутствуют Все ле- карства Требования согласно досье/Фармакопее. Клинического исследования не требу- ется Ежегодный отчет
2 Один объеди- ненный произ- водственный комплекс. Об- щий персонал. Другие измене- ния отсутству- ют Тоже Модифицированное высвобождение (пролонгированное) Идентификация и описание изменений площадки и досье на серию, выпущенную на новой площадке. Уведомле- ние об изменении площадки. Стабильность. Требования согласно досье/Фармакопее плюс многоточечные про- фили растворения в трех раз- личных средах (например, в воде, 0,1 N НС1 и буферной фармакопейной среде при pH 4,5 и 6,8) до высвобождения > 80% лекарства или дости- жения асимптоты. При срав- нении профилей растворения применяется определенный статистический критерий (тест fl). Клинического ис- следования не требуется Модифицированное высвобождение (замедленное) Идентификация и описание изменений площадки и до- сье на серию, выпущенную на новой площадке. Уведомле- ние об изменении площадки. Стабильность. Требования согласно досье/Фармакопее плюс многоточечные профили растворения и дополнитель- ное исследование в буферной среде (например, буферной фармакопейной среде при pH 4,5—7,5) в стандартных усло- виях или при интенсивном перемешивании до высвобож- дения > 80% лекарства либо достижения асимптоты. При сравнении профилей раство- рения применяется опреде- ленный статистический кри- терий (тест fl). Клинического исследования не требуется Немедленное высвобождение Идентификация и описание измене- ний площадки и до- сье на серию, вы- пущенную на новой площадке. Уведом- ление об изменении площадки. Стабиль- ность. Требования согласно досье/Фар- макопее. Клиниче- ского исследования не требуется Изменения вводятся одновременно с подачей дополнения в досье. (Ре- зультаты исследования стабильности в условиях ускоренного старения для лекарственных форм с мо- дифицированным высво- бождением. Результатов по оценке стабильности для лекарственных форм с немедленным высво- бождением не требуется.) Ежегодный отчет (дан- ные по стабильности при естественном старении для лекарственных форм с модифицированным или немедленным высво- бождением)
Часть I. Нормативное регулирование производства лекарственных средств
3 Другой произ- водственный комплекс. Дру- гой персонал Уведомление об изменении площадки. Досье на серию, выпущенную на новой площадке. Требования согласно досье/Фармакопее (профили) такие же, как для уровня 2. Стабильность. Клиническое исследование или ivivc Уведомление об из- менении площадки. Досье на серию, вы- пущенную на новой площадке. Исследо- вание растворения по варианту В, как при изменении вспо- могательных веществ (уровень 2). Стабиль- ность. Клинического исследования не тре- буется Требуется утверждение дополнения к досье до введения изменения в действие (данные ис- следования стабильности при ускоренном старе- нии) для лекарственных форм с модифицирован- ным или немедленным высвобождением. Изме- нения вводятся одновре- менно с подачей допол- нения в регистрационное досье Ежегодный отчет
Глава 1.3. Правила масштабирования производства и внесения посгрегистрационных... 119
Таблица 6
Изменение объема производственной серии: масштабирование с увеличением/уменьшением
Уровень Классификация Изменение Аналитическая документация Подаваемая в регуляторный орган документация
1 Масштабирование (с уве- личением) серий, исполь- зованных в исследованиях биоэквивалентности, или опытно-промышленных се- рий для клинических иссле- дований. Другие изменения отсутствуют Не менее де- сятикратно- го увеличе- ния (для всех лекарств) Досье на измененную серию. Стабильность. Требования согласно до- сье/Фармакопее. Клинического исследования не требуется Ежегодный отчет
2 Тоже Тоже Модифицированное высвобождение (пролонгированное) Досье на промышлен- ную серию, произведен- ную с учетом изменений. Требования согласно досье/Фармакопее плюс многоточечные профи- ли растворения в трех различных средах (на- пример, в воде, 0,1 N НС1 и буферной фарма- копейной среде при pH 4,5 и 6,8) до высвобож- дения > 80 % лекарства либо достижения асим- птоты. При сравнении профилей растворения применяется опреде- ленный статистический критерий (тест fl). Кли- нического исследования не требуется Модифицированное высво- бождение (замедленное) Досье на промышленную серию, произведенную с учетом изменений. Ста- бильность. Требования со- гласно досье/Фармакопее плюс многоточечные про- фили растворения и допол- нительное исследование в буферной фармакопей- ной среде (например, при pH 4,5—7,5) в стандартных условиях и при интенсив- ном перемешивании до высвобождения > 80% ле- карства либо достижения асимптоты. При сравне- нии профилей растворе- ния применяется опреде- ленный статистический критерий (тест fl). Клини- ческого исследования не требуется Немедленное высвобождение Досье на промыш- ленную серию, произведенную с учетом изменений. Стабильность. Ис- пытание на рас- творение для ва- рианта В, как при изменении вспо- могательного ве- щества (уровень 2). Клинического ис- следования не тре- буется Изменения вводятся одновременно с по- дачей дополнения в досье (результаты исследования ста- бильности при уско- ренном старении). Ежегодный отчет (данные по стабиль- ности при естествен- ном старении)
120______Часть I. Нормативное регулирование производства лекарственных средств
Таблица 7
Изменения в производственном процессе: оборудование
Уровень Классифи- кация Изменение Документация по испытаниям Подаваемая в регуляторный орган документация
1 Изменения оборудова- ния. Другие изменения отсутствуют (для всех лекарств) Альтернативное оборудование той же конструкции и того же принци- па действия. Авто- матизированное оборудование Досье на серию, произведенную с учетом изменений. Стабильность. Требования согласно досье/Фармакопее. Клинического исследования не требуется Ежегодный отчет
2 Тоже Переход на обо- рудование дру- гой конструкции и иного принци- па действия Модифицированное высвобождение (пролонгированное) Досье на промышленную се- рию, произведенную с учетом изменений. Стабильность. Требования согласно досье/ Фармакопее плюс многото- чечные профили растворения в трех различных средах (на- пример, воде, 0,1 N НС1 и бу- ферной фармакопейной среде при pH 4,5 и 6,8) до высвобож- дения > 80% лекарства либо достижения асимптоты. При сравнении профилей раство- рения применяется опреде- ленный статистический кри- терий (тест fl). Клинического исследования не требуется Модифицированное высвобождение (замедленное) Досье на промышленную серию, произведенную с учетом измене- ний. Стабильность. Требования согласно досье/Фармакопее плюс многоточечные профили раство- рения и дополнительное исследо- вание в буферной среде (например, буферной фармакопейной среде при pH 4,5—7,5) в стандартных условиях и при интенсивном пере- мешивании до высвобождения £ 80% лекарства либо достижения асимптоты. При сравнении про- филей растворения применяется определенный статистический критерий (тест fl). Клинического исследования не требуется Немедленное высвобождение Досье на про- мышленную серию, произ- веденную с уче- том изменений. Стабильность. Испытание на растворение по варианту С, как при изме- нении вспо- могательного вещества (уро- вень 2). Кли- нического ис- следования не требуется Требуется утверж- дение дополнения к досье до введения в действие изменения (результаты исследо- вания стабильности при ускоренном хра- нении). Ежегодный отчет (результаты ис- следования стабиль- ности при естествен- ном хранении)
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 121
122 Часть I. Нормативное регулирование производства лекарственных средств
Система биофармацевтической классификации подразделяет лекарства на четы-
ре класса:
> класс I — хорошая растворимость, низкая проницаемость;
> класс II — плохая растворимость, высокая проницаемость;
> класс III — хорошая растворимость, низкая проницаемость;
> класс IV — плохая растворимость, низкая проницаемость.
Растворение, растворимость и проницаемость — три фактора, регулирующие
биодоступность действующего вещества для лекарственных препаратов с немед-
ленным высвобождением. Если неактивное вспомогательное вещество не участвует
в регулировании или контроле высвобождения и всасывания активного вещества, от
проведения исследования биоэквивалентности можно отказаться. Согласно Руко-
водству, класс растворимости определяется для количества действующего вещества,
содержащегося в препарате с максимальной дозировкой, для которого запраши-
вается процедура «биовейвер». Если вещество в количестве, содержащемся в пре-
парате с максимальной дозировкой, растворяется в 250 мл (или менее) воды при
значениях pH 1,0—7,5, то вещество считается легко растворимым. Для определения
класса проницаемости применяются различные способы анализа in vivo (например,
метод баланса масс, метод определения абсолютной биодоступности и метод перфу-
зии тонкой кишки), а также методы in vitro, такие как исследование проницаемости
с использованием изолированной ткани или монослоя культивируемых эпители-
альных клеток. Если степень абсорбции (всасывания) превышает 90% от введенной
человеку дозы, считается, что лекарство обладает высокой проницаемостью. При
исследовании растворения высвобождение лекарства следует исследовать в трех
средах: 1) в 0,1 N НС1 или описанном в USP «искусственном желудочном соке» без
ферментов; 2) буферном растворе с pH 4,5; 3) буферном растворе с pH 6,8 или опи-
санном в USP «искусственном кишечном соке» без ферментов. К лекарственным
препаратам с быстрым высвобождением относятся те, из которых высвобождается
более 80% действующего вещества в среде объемом 900 мл (из числа вышеупомяну-
тых сред) менее чем за 30 мин при использовании прибора для растворения USP I
и скорости перемешивания 100 об/мин (либо при использовании прибора USP II
и скорости 50 об/мин).
Процедура «биовейвер» может быть запрошена для продуктов, изготовленных
с учетом изменений класса I BCS, а также обладающих профилем быстрого раство-
рения. Она применяется при совпадении кинетики растворения продукта до и после
изменений (что определено с помощью теста fl) во всех трех средах. Для лекарств
класса II BCSсущественная корреляция (уровень корреляции А, Вили С) между вы-
свобождением лекарства in vitro и всасыванием in vivo может также являться основа-
нием для проведения процедуры «биовейвер». Методы деконволюционного анализа
используют для прогнозирования процессов растворения и всасывания in vivo.
1.3.3.2. Руководство по 5(УР/4СФармацевтического союза ЕС
Европейский фармацевтический рынок — один из крупнейших в мире. Для сти-
мулирования рынка и обеспечения надлежащего качества лекарственных средств
в странах ЕС фармацевтический сектор Комиссии ЕС инициировал серию ин-
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 123
формационных и коммуникационных проектов, в целом называемых проектами
компетентных органов стран ЕС, регулирующих лекарственные средства (European
Union Drug Regulatory Authorities, EUDRA). Кроме того, подразделение EUDRALEX
фармацевтического сектора отвечает за формирование фармацевтического законо-
дательства ЕС, руководящих документов и указаний для заявителей [9]. В разделе С
тома 2 регуляторного руководства «Фармацевтическое законодательство: указания
для заявителей» EUDRALEXимеется «Руководство по требованиям к досье для уве-
домлений об изменениях типов L4 и 1В» [2].
Указанные правила были разработаны с целью как снизить административную
нагрузку на регулирующие органы, так и упростить процедуры внесения постре-
гистрационных изменений без обсуждения отдельных параметров качества лекар-
ственного препарата [10]. Кроме определения типов L4 и IД Правила формулировали
четкие термины для расширения регистрационного досье, параллельного/последо-
вательного уведомления об изменениях, а также срочных изменений, связанных
с безопасностью. Для внедрения этих Правил было подготовлено четыре документа:
1) руководство для стран-участниц (страны, зарегистрировавшей препарат,
и стран, осуществивших признание регистрации) и заявителей по процедуре
подачи уведомлений об изменениях в рамках процедуры взаимного призна-
ния регистрации;
2) руководство для заявителей по процедуре подачи уведомлений/изменений
при централизованной процедуре регистрации;
3) единая форма заявления, которую можно использовать при составлении уве-
домлений для изменений типа L4 и IB либо изменений типа II как при цен-
трализованной процедуре, так и в рамках процедуры взаимного признания
регистрации;
4) указания по предоставляемой документации для уведомлений об изменениях
типа L4 и 1В.
Для всех стран — участниц ЕС существуют единые правила внесения изменений
в зарегистрированный медицинский препарат. Согласно Руководству, выделяют три
типа изменений: изменения типа I, классифицируемые на типы L4 и IB, и изменения
типа II. Руководство описывает некоторые определенные изменения, относящиеся
к типам L4 или IB. Кроме того, оно содержит анализ определенных данных, необ-
ходимый для присвоения изменению соответствующего типа, а также перечисляет
документацию, которую необходимо предоставлять в регуляторные органы. Любые
изменения, не упомянутые в данном разделе, относят к изменениям типа II.
В соответствии с Постановлением Комиссии ЕС № 1084/2003, изменение типа I
определено следующим образом: «Незначительное изменение типа L4 или IB пред-
ставляет собой изменение, указанное в Приложении I и соответствующее услови-
ям, в нем указанным». Приложение I Постановления содержит список изменений
и условий (которым необходимо соответствовать), которые рассматриваются как
изменения типа L4 и IB, а в приложении II перечислены изменения, попадающие
в категорию расширенного регистрационного досье. Кроме того, представлена
категория «срочных изменений, связанных с безопасностью». К ним относятся
временные или предварительные изменения в инструкции по медицинскому при-
124
Часть I. Нормативное регулирование производства лекарственных средств
Изменения в производственном
Уровень Классификация Изменение
1 Изменения в процессе производства, влияющие на вспомогательные вещества, не контролирующие/кон- тролирующие высвобождение для лекарств с модифи- цированным высвобождением. Изменения в границах диапазона валидации (лекарства с немедленным вы- свобождением). Другие изменения отсутствуют Корректировка рабочих параметров оборудования (время смешива- ния, рабочие скорости) в границах утвержденного диапазона
2 Изменения в процессе производства, влияющие на вспомогательные вещества, не контролирующие/ контролирующие высвобождение. Изменения, выхо- дящие за границы диапазона валидации, для лекарств с немедленным высвобождением. Другие изменения отсутствуют Корректировка рабочих параметров оборудования (время смешивания, рабочие скорости), выходящих за границы утвержденного диапазона
3 Изменения в процессе производства, влияющие на вспомогательные вещества, не контролирующие/ контролирующие высвобождение Изменение типа процесса (напри- мер, переход от влажного гранули- рования к сухому)
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 125
Таблица 8
(технологическом) процессе
Документация по испытаниями Подаваемая в регуляторный орган документация
Досье на измененную производственную серию. Требования согласно досье/ Фармакопее. Клинического исследования не требуется Ежегодный отчет
Модифицированное высвобождение (пролонгированное) Досье на промышлен- ную серию, произве- денную с учетом изме- нений. Стабильность. Требования согласно досье/Фармакопее плюс многоточечные профили растворения в трех различных сре- дах (например, воде, 0,1 N НС1 и буферной фармакопейной среде при pH 4,5 и 6,8) до высвобождения > 80% лекарства либо дости- жения асимптоты. При сравнении профилей растворения приме- няется определенный статистический крите- рий (тест/2). Клиниче- ского исследования не требуется Модифицированное высвобождение (замедленное) Досье на промышленную серию, произведенную с учетом изменений. Ста- бильность. Требования согласно досье/Фармако- пее плюс многоточечные профили растворения и дополнительное иссле- дование в буферной сре- де (например, буферной фармакопейной среде при pH 4,5—7,5) в стан- дартных условиях и при интенсивном перемеши- вании до высвобождения > 80% лекарства либо достижения асимптоты. При сравнении профи- лей растворения при- меняется определенный статистический крите- рий (тест fl). Клиниче- ского исследования не требуется Немедленное высвобождение Уведомление об измене- нии. Досье на промышлен- ную серию, произведен- ную с учетом изменений. Стабильность. Испытание на растворение по вариан- ту В, как при изменении вспомогательных веществ (уровень 2). Клинического исследования не требуется Изменения вводятся одновременно с пода- чей дополнения в до- сье (результаты ис- следования стабиль- ности при ускоренном старении для лекарств с модифицирован- ным высвобождени- ем). Ежегодный отчет (результаты исследо- вания стабильности при естественном хра- нении для лекарств с модифицированным и немедленным вы- свобождением)
Досье на измененную промышленную серию. Стабильность. Требо- вания согласно досье/ Фармакопее (профи- ли). Клиническое ис- следование или ivivc Досье на измененную промышленную серию. Стабильность. Испыта- ние на растворение по варианту В, как при из- менении вспомогатель- ных веществ (уровень 2). Клинического исследо- вания не требуется Требуется утверждение до- полнения к досье до введе- ния изменения в действие (результаты исследования стабильности при ускорен- ном старении для лекарств с модифицированным вы- свобождением). Ежегод- ный отчет (результаты ис- следования стабильности при естественном старении для лекарств с модифици- рованным и немедленным высвобождением)
126
Часть I. Нормативное регулирование производства лекарственных средств
менению препарата (краткой характеристике лекарственного препарата), такие
как показания, дозирование, противопоказания, особые указания, особые группы
пациентов, а также указания при отмене препарата, обусловленные новой инфор-
мацией, существенно изменившей представления о безопасности медицинского
препарата [11].
Обо всех изменениях, являющихся следствием первичного изменения, регуля-
торные органы следует уведомлять отдельно. Последовательные (взаимосвязанные)
изменения являются частью одного уведомления, а параллельные — нет. Последо-
вательные изменения по типу L4 могут быть лишь другими изменениями типа L4,
в то время как последовательные изменения по типу IВ могут представлять тип L4
или тип IB. Все другие изменения должны подаваться как изменения типа II
«Руководство по требованиям к досье для уведомлений об изменениях типа L4
и 12?» содержит полный список изменений и необходимых условий для конкретных
изменений, а также документацию, требующуюся для регуляторных органов [10].
13.3.3. Регулирующие указания по 5№4СНационального агентства
по контролю в области здравоохранения (Бразилия)
Национальное агентство по контролю в области здравоохранения (Бразилия)
(ANVISA) формирует лекарственную политику Бразилии в области дженериков
(воспроизведенных лекарственных препаратов). Согласно отраслевому законода-
тельству, Резолюция RE 93 от 29 мая 2003 г. включена в «Руководство по внесению
пострегистрационных изменений, дополнений и подаче уведомлений» [6]. Это ру-
ководство описывает изменения после регистрации как собственно «изменения»
и «дополнения», а также содержит перечень документации и исследований, ко-
торые необходимо провести для обоснования изменений любых видов. Согласно
Руководству, каждое изменение (или дополнение) перед внедрением должно быть
отдельно представлено на одобрение ANVISA. В табл. 9 приведены некоторые при-
меры изменений для каждой категории.
Для всех категорий определены соответствующие требования перед внедрением
изменения. Например, увеличение объема производственной серии должно пред-
варяться уведомлением, если увеличение является более чем десятикратным. При
этом в регуляторный орган следует предоставить документацию, включающую: ори-
гинал документа об оплате пошлины или освобождении от оплаты; копию сертифи-
ката соответствия Правилам надлежащего производства и контроля лекарственных
средств (от англ, certificate of good manufacturing and control practices, CBPFC), выдан-
ного ANVISA; техническое обоснование; производственные протоколы и протоко-
лы анализа по одной производственной серии для каждой дозировки; технический
отчет; технический отчет вместе с оценкой профиля растворения.
1.3.4. Гармонизация1
Важно, чтобы оценка безопасности и качества новых или измененных лекарствен-
ных средств проводилась до их поступления на рынок. Однако необходимость уста-
1 Калька с английского harmonization, сохраненная здесь по инициативе переводчика и на-
учного редактора. — Примеч. отв. ред.
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 127
Таблица 9
Примеры различных категорий
Пострегистрационные Пострегистрационные Пострегистрационные Аннулирование/отмена
изменения дополнения уведомления регистрации
Изменения маркировки Включение новой фор- мы выпуска Временная приостанов- ка производства Отмена регистрации представления лекар- ства по запросу
Изменение фирменного Включение новой упа- Возобновление произ- Отмена регистрации
наименования Изменения даты окон- чания срока годности Изменение условий хранения Изменение пути синте- за лекарства Смена производителя фармацевтической субстанции Смена производствен- ной площадки Изменение вспомога- тельного вещества ковки Включение новой концентрации, уже одо- бренной в стране Включение новой лекар- ственной формы, уже одобренной в стране Введение нового тера- певтического показания в стране Введение производ- ственной площадки Включение производи- теля фармацевтической субстанции Включение дополни- тельного объема произ- водственной серии водства лекарства
новления отдельных руководств была признана в разных странах в разное время.
Например, в США трагический инцидент с детским парацетамолом стал поводом
для составления Руководства по регистрации медицинских препаратов. Евро-
пейские страны выразили озабоченность этими проблемами после трагедии из-
за талидомида в 1960-х гг. С тех пор было издано значительное количество руко-
водств, посвященных оценке медицинских препаратов в отношении их качества,
безопасности и эффективности [12]. Однако в связи с тем, что фармацевтическая
промышленность становится международной отраслью, существующей в рамках
глобального рынка, налицо тенденция к принятию руководящих документов и си-
стем регистрации, признанных в международном масштабе. Чтобы обеспечить воз-
можность выхода медицинских препаратов на международный рынок, компании
вынуждены дублировать многие трудоемкие анализы и исследования, требующие
длительного времени.
В табл. 10 представлены примеры документации, которую запрашивают в раз-
ных странах при внесении пострегистрационных изменений. При изменениях
в спецификациях вспомогательных веществ Администрация по медицинской
продукции Австралии ('EGA) и Европейское агентство по оценке медицинских
продуктов (European Agency for Evaluation of Medicinal Products, ЕМЕА) требуют пре-
доставления разных документов. Это может означать, что преимущества патента/
препарата не будут оценены в глобальном масштабе. Кроме того, возрастает веро-
128 Часть I. Нормативное регулирование производства лекарственных средств
Таблица 10
Изменения в спецификациях вспомогательных веществ (добавление нового аналитического
предела): сравнение руководств
Руководство Необходимая документация Тйп изменения
TGA Требуется описание деталей аналитического метода. Необходимо собрать соответствующие данные валида- ции для аналитического метода. Предложенные пределы основаны на аналитических данных по производственной серии и соответствуют официальным стандартам и/или соответствующим принятым руководствам там, где они применимы Изменения, оцениваемые самостоятельно
ЕМЕА Сравнительная таблица текущих и предложенных спецификаций. Результаты всех анализов двух производственных серий согласно новой спецификации. При необходимости приводятся данные сравнения профилей растворения готового продукта по меньшей мере из одной пилотной серии, содержащей вспомо- гательное вещество, в соответствии с существующей и предложенной спецификациями. Для растительных лекарственных средств допустимо предоставление сравнительных данных по измельчению продуктов Малые изменения типа 1В требуют утверждения
ятность ошибки. Например, отзыв вагинальных таблеток клотригексала (100 мг)
из аптек Новой Зеландии в 2003 г. был вызван тем, что клотригексал был упакован
согласно указаниям TGA. Подобный отзыв с рынка вызвал недоумение среди па-
циентов и медиков и проблемы с поставками. Гармонизация представляет собой
процесс принятия единых для всех производителей фармацевтической продукции
в мире законов и правил. Она направлена на обеспечение безопасности, качества
и эффективности медицинских препаратов в глобальном масштабе. Главная зада-
ча гармонизации — обозначить и минимизировать разницу в специальных требо-
ваниях различных регуляторных органов разных стран к разработке медицинской
продукции.
Основным направлением деятельности по гармонизации является сокращение
типов исследований, выполняемых производителями фармацевтической продук-
ции для регистрации медицинского продукта в разных странах. Кроме того, гармо-
низация заключается в упрощении и сокращении количества протоколов, которым
необходимо следовать при проведении упомянутых исследований, методологий,
используемых для валидации данных, а также методов оценки рисков.
Гармонизация сокращает дублирование и наработку излишних серий при реги-
страции новых и измененных продуктов. В 1980-х гг. в ряде европейских стран были
проведены исследования, которые продемонстрировали как пользу гармонизации,
так и возможность ее практической реализации. По итогам работ по гармонизации
в ЕС в 1990 г. была создана Международная конференция по гармонизации {the
International Conference on Harmonization, ICH) [13]. В табл. 11 представлены некото-
рые «гармонизированные» правила, разработанные в рамках ICH.
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 129
Таблица 11
Примеры гармонизации руководств по качеству в рамках ICH
Руководства по качеству Пример руководящего документа
Q1: стабильность <22: валидация аналитической процедуры Q3: исследование на наличие примесей Q4: фармакопеи Q5: качество биотехнологических продуктов Q6: спецификации на новые лекарственные вещества и препараты Q7: СМРдля фармацевтических компонентов Q1R исследование фотостабильности Q2A: методология Q3A: примеси в новых лекарственных веществах Q4: гармонизация фармакопей С5А: оценка вирусной безопасности биотехнологических продуктов Q6A: критерии приемлемости новых лекарственных веществ (77: GMP для активных фармацевтических компонентов
1.3.5. Аспекты GMP\ контроль изменений и валидация
процесса
В производстве изменения неизбежны. Производители вносят их на определен-
ных этапах производства в ходе и по окончании процесса регистрации препарата.
Тем не менее, неизменное качество лекарственного средства можно гарантировать
только с помощью тщательно разработанных процедур валидации. Когда в процесс
производства фармацевтического препарата вносится изменение, ответственность
спонсоров заключается как в оценке влияния любых изменений на безопасность,
эффективность, качество, стабильность и действие лекарственного препарата, так
и в гарантии того, что на эти свойства не повлияли вышеупомянутые изменения.
На производственном предприятии различные службы (отделы продаж, маркетин-
га, медицинский отдел и отдел регуляторных вопросов, производственные, элек-
трические, технические службы и т. д.) работают сообща. Поэтому любое изменение
в одной из областей может оказать непосредственное влияние на другие области.
Каждая компания должна иметь процедуру по работе с изменениями. Отделы кон-
троля и обеспечения качества обычно отслеживают изменения в сфере, регулируе-
мой правилами GMP. Поэтому при оценке влияния любых изменений и принятии
соответствующих действий по их контролю не обойтись без должным образом под-
готовленного персонала. Требуется собрать соответствующие данные и провести их
оценку, позволяющую подтвердить необходимость (или отсутствие необходимости)
дальнейших клинических или доклинических исследований.
1.3.5.1. Контроль изменений
При внесении изменений на производстве важно оценить их масштаб. Поскольку
изменение может потребовать подачи документации в регуляторный орган, затро-
нуть производственные параметры, спецификации и работу технических подраз-
делений, необходимо учитывать цели и задачи всех затронутых областей, что воз-
можно только при наличии тщательно разработанных стандартных операционных
процедур, устанавливающих порядок валидации изменения, его оценки и после-
дующего внедрения. Без детальной регламентации оценки изменений с привлече-
нием специально обученного персонала невозможно гарантировать изготовление
продукции надлежащего качества (рис. 1).
130
Часть I. Нормативное регулирование производства лекарственных средств
Комментарии
Рис. 1. Цикл контроля изменений при внесении последних в производственный процесс
Производитель должен подготовить протоколы, где оценивается масштаб пред-
полагаемых изменений. Следовательно, «контроль изменений» чрезвычайно ва-
жен. Внедрение контроля может быть эффективным лишь при наличии тщательно
разработанных стандартных рабочих процедур. Главная задача «контроля измене-
ний» — осуществление действенного систематического контроля для точной оценки
масштаба вариаций при помощи определенных испытаний. Другая цель контроля —
оценка влияния предполагаемого изменения на качество, безопасность и эффек-
тивность лекарственного средства до его введения в действие. Контроль изменений
и оценка их результатов в соответствующей документации должны включать [14]:
1) описание и цель изменения;
2) информацию от научно-исследовательского отдела;
3) этапы оценки влияния изменений (пример: оценка стабильности, требования
по необходимости проведения валидации и биоэквивалентности in vivo) -,
4) необходимость оформления документации для подачи в регуляторный орган,
ее объем и утверждение;
5) план — график внедрения;
6) исчерпывающий перечень лиц, ответственных за одобрение изменений;
7) протокол мониторинга внедрения изменения и периодический анализ влия-
ния изменений.
Вслед за неформальным предложением о внесении изменений ответственный
инициатор проводит его анализ, после чего составляет формальное предложение
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 131
[15]. Последнее должно содержать четкое описание как изменений, так и процеду-
ры их валидации, а также указание срока их внедрения. Конечный вариант фор-
мального предложения оценивается всеми вовлеченными в процесс функциональ-
ными подразделениями. Если изменение получает одобрение, его можно внедрять,
после чего цикл завершается. На рис. 2 обозначена сфера ответственности различ-
ных подразделений фармацевтической компании в рамках процедуры контроля из-
менения.
Обеспечение качества
Обеспечение качества,
контроль качества,
производство, организация
производственного процесса,
технические службы, отдел по
регуляторным вопросам
Инициирование контроля
изменений
[I
Проверка
Соответствие GMP
и применимость к другим
системам
Анализ и утверждение
Обеспечение качества,
контроль качества,
производство, организация
производственного процесса,
технические службы, отдел по
регуляторным вопросам
Обеспечение качества
Эффект на регуляторные аспекты
и стратегия глобальной регистрации
Отдел нормоконтроля
Валидация
Обеспечение качества
Рис. 2. Ответственность различных подразделений фармацевтической компании в процедуре
контроля изменения согласно [15]
Так как СОП (стандартные операционные процедуры) контроля изменений яв-
ляются важной частью любого аудита GMP, крайне важно, чтобы внедрением изме-
нений занимался квалифицированный персонал соответствующих подразделений.
Изменения, прошедшие одобрение всеми функциональными группами на про-
изводстве, можно немедленно внедрять, если нет необходимости в предварительном
согласовании таких изменений регуляторными органами. Однако если изменение
подпадает под область нормативного регулирования, оно требует предварительного
утверждения регуляторным органом.
132
Часть I. Нормативное регулирование производства лекарственных средств
1.3.5.2. Валидация процесса
Валидация процесса — важная часть внедрения пострегистрационных изменений.
Она обеспечивает получение документированного свидетельства соответствия
фармацевтической операции спецификациям. «Руководство по общим принципам
валидации процесса», изданное FDA, подробно описывает принципы и практику
валидации процесса, а также перечисляет документацию, требуемую регуляторным
органом [ 13]. В общем, валидация процесса определяется как процедура, в ходе ко-
торой собирается информация, гарантирующая достаточную уверенность в том, что
определенная производственная операция проводится надлежащим образом и по-
зволяет производить лекарственные средства, соответствующие спецификациям.
Валидация подразделяется на четыре вида: перспективную, ретроспективную,
сопутствующую и ревалидацию (повторную валидацию). Перспективная валида-
ция проводится перед выпуском фармацевтических продуктов в обращение либо
после внесения изменений в производство лекарственного средства, способных по-
влиять на его качество и характеристики. Ретроспективная валидация применяется
для давно выпускаемого лекарственного препарата, процесс производства которого
стабилен. Такая валидация нужна для гарантии того, что текущие фармацевтиче-
ские операции ведутся в соответствии с протоколами (регламентами производства)
и спецификациями с получением продукта удовлетворительного качества. Сопут-
ствующая валидация проводится с помощью мониторинга критических параметров
в ходе производственного процесса и тестирования готового продукта и направлена
на подтверждение соответствия параметров текущего производственного процесса
спецификациям по внутрипроизводственному контролю. Ревалидация проводится
после внедрения изменений в зарегистрированный лекарственный препарат и при-
звана гарантировать отсутствие неблагоприятного влияния на качество и действие
лекарственного средства [16].
В ходе валидации продукты и технологические процессы испытывают в экс-
тремальных условиях на пределе установленных рабочих параметров и норм и оце-
нивают на соответствие установленным критериям приемлемости. Меняя разные
параметры различных фармацевтических операций, записывают и оценивают ха-
рактеристики получаемых продуктов (рис. 3). Если обнаруживают, что необходима
корректировка, то необходимые действия предпринимают с учетом рекомендаций
научно-исследовательского отдела. Для обеспечения высокого уровня достоверно-
сти при оценке качества обычно сравнивают данные по валидации трех производ-
ственных серий.
Для процесса валидации очень важно систематическое документирование влия-
ния вариации параметров производственного процесса на характеристики продук-
та. Любое изменение параметров процесса, как и окончательная редакция любого
протокола или отчета по валидации, требуют утверждения со стороны разработчи-
ков продукта, инженерных и технических служб, а также производственного под-
разделения и подразделения по регуляторным вопросам. В зависимости от уровня
изменения или степени произведенного эффекта определяют необходимый объем
валидации.
На основании валидационных требований пробы отбирают на различных ста-
диях и предоставляют для анализа согласно протоколу валидации. Окончательные
Глава 1.3. Правила масштабирования производства и внесения посгрегистрационных... 133
Параметры процесса
Характеристики продукта
Влажное гранулирование
Время предварительного
смешивания, время внесения
связующего вещества, скорость
мешалки/чоппера
Твердость гранул
и распределение
по размерам
Номер сита, направление
и скорость перемещения
ножей/молотков
Температура воздуха на входе,
температура слоя, скорость
потока воздуха, частота
выгрузок
Содержание влаги в гранулах,
остаточное количество
растворителя
Распределение по размерам,
насыпная плотность/плотность
после уплотнения гранул
Смешивание
Объем смесителя, время
смешивания, скорость
смешивания
Однородность по содержанию
в смесителе и барабане,
плотность получившейся
смеси
Таблетирование
Скорость прессования, сила
прессования(прочность на
излом и толщина таблетки),
массатаблетки
Масса таблетки, прочность на
излом, толщина, однородность
по содержанию, прочность
на истирание, растворение,
распадаемость, количественное
содержание/активность
Нанесение оболочки
Емкость камеры, температура
на входе/выходе, скорость
вращения камеры, сила
распыления,скорость потока
воздуха, температура слоя
Растворение, распадаемость,
масса полученной оболочки,
неравномерное окрашивание,
содержание/активность
Рис. 3. Различные параметры производственного процесса и характеристики продукта,
исследуемые в ходе валидации обычной таблетки с оболочкой
134
Часть I. Нормативное регулирование производства лекарственных средств
данные заносят в отчет о валидации. Систематически составляемые протокол ва-
лидации и отчет о валидации являются фундаментом валидационного процесса.
В табл. 12 продемонстрированы ключевые компоненты любых работ по валидации
[16]. Эти протоколы и отчеты должны быть проверены и утверждены (согласованы)
соответствующими подразделениями.
Отдельные изменения зачастую вносят в производственный процесс без предва-
рительного уведомления регуляторного органа, поэтому можно посоветовать про-
водить ревалидацию с заранее определенной частотой (или по факту наблюдения
необычных эффектов).
При приобретении нового оборудования или изменении производственной пло-
щадки в качестве этапа валидации проводятся работы по квалификации. Квалифи-
кация любого оборудования или производственных помещений (при их установке,
функционировании и эксплуатации) представляет собой процесс в экстремальных
условиях, включающий тестирование, проверку и документирование и призванный
гарантировать соответствие оборудования или помещений спецификациям и опре-
деленным стандартам, заявленным организацией-поставщиком и требуемым про-
изводственным и инженерным персоналом [14].
Таблица 12
Ключевые составляющие работ по валидации
Протокол валидации Отчет о валидации
Цель исследования Ответственность персонала Критические этапы процесса Критические параметры процесса Критические параметры продукта План отбора проб План испытаний Критерии приемлемости Задача исследования Список исходных материалов, используемых в ис- следовании Перечень производственного оборудования Изученные критические этапы Собранные данные и их анализ Оценка критериев приемлемости Статистический анализ Рекомендации отдела валидации
1.3.6. Заключение
Мировая фармацевтическая индустрия развивается в условиях быстро меняющейся
среды систем здравоохранения. Ежегодно на рынке появляются новые лекарства
и системы доставки. Для гарантирования качества новых и существующих на рын-
ке лекарств и технологий доставки существует регламентация фармацевтических
операций, осуществляемая с помощью руководств регуляторных органов. Разра-
ботка руководящих документов направлена в первую очередь на охрану здоровья
и безопасности человека путем производства качественных лекарственных средств.
Следование этим указаниям и систематическое отслеживание влияния постреги-
страционных изменений документированным способом жизненно важны для пред-
упреждения любых возможных отказов в системе. Контроль изменений и валидация
гарантируют отсутствие негативного влияния на характеристики лекарственного
средства. Ожидаемые изменения, включенные в протоколы сравнения, снижают
высокий риск возникновения непредсказуемых неблагоприятных эффектов и по-
могают сократить сроки внедрения препарата. Если эффект является ожидаемым,
Глава 1.3. Правила масштабирования производства и внесения пострегистрационных... 135
его следует обсудить со специалистами научно-исследовательских отделов, со служ-
бой разработки производственных процессов, а также с другими заинтересованны-
ми подразделениями на предмет подачи соответствующих документов в регулятор-
ный орган согласно установленным требованиям. При надлежащем соблюдении
регуляторных руководств можно гарантировать качество и эффективность лекар-
ственного продукта.
Литература
1. U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug
Evaluation and Research (CDER), Guidance for industry: Changes to an approved NDA or ANDA,
available: http://www.fda.gov/cder/guidance/3516fiil.pdf, accessed Apr. 15,2006.
2. European Commission, Guideline on dossier requirements for type LA and type IB notifi cations:
Pharmaceuticals: Regulatory framework and market authorizations, available: http://ec.europa.eu/en-
terprise/pharmaceuticals/eudralex/vol-2/c/gdvartypiab_rev0_200307.pdf, accessed Apr. 20, 2006.
3. Department of Health and Ageing, Therapeutic Goods Administration, Australian regulatory guide-
lines for prescription medicines. Appendix 12: Changes to the quality information of registered medi-
cines: Notifi cation. Self-assessment and prior approval, available: http://www.tga.gov.au/pmeds/
argpmapl2.pdf, accessed Apr. 12,2006.
4. Food and Drug Administration, Centre for Drug Evaluation and Research (CDER) , Guidance for in-
dustry: SUPAC-IR: Immediate-release solid oral dosage forms: Scale-up and post-approval changes:
Chemistry, manufacturing and controls, in vitro dissolution testing, and in vivo bioequivalence docu-
mentation, available: http://www.fda.gov/cder/guidance/cmc5.pdf, accessed May 11, 2006.
5. Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), Guidance for in-
dustry: Comparability protocols — Chemistry, manufacturing, and controls information (draft), avail-
able: http://www.fda.gov/cder/guidance/5427dft.pdf, accessedApr. 15, 2006.
6. Brazilian Sanitary Surveillance Agency (ANVISA), Resolution: Guide for making post-registration
alterations, inclusions and notifi cations of drug products, Brazil’s generic drug policy, industry leg-
islation, available: http://www.anvisa.gov.br/hotsite/genericos/legis/resolucoes/893_03re_e.htm, ac-
cessedApr. 11,2006.
7. U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug
Evaluation and Research (CDER), Guidance for industry: SUPAC-MR: Modified release solid oral
dosage forms scale - up and postapproval changes: Chemistry, manufacturing, and controls; in vitro
dissolution testing and in vivo bioequivalence documentation, available: http://www.fda.gov/cder/
guidance/1214firl.pdf, accessed May 5, 2006.
8. U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug Eval-
uation and Research (CDER), Guidance for industry: SUPAC-SS: Nonsterile semisolid dosage forms;
scale-up and post-approval changes: Chemistry, manufacturing and controls; in vitro release testing
and in vivo bioequivalence documentation, available: http://www.fda.gov/cder/guidance/1447fiil.pdf,
accessedApr. 24, 2006.
9. Pharmaceutical Unit of European Commission at EUROPA, available: http://ec.europa. eu/enterprise/
pharmaceuticals/pharmacos/docs/brochure/pharmaeu.pdf, accessed May 21, 2006 .
10. Variations. Pharmaceuticals: Regulatory framework and market authorizations, Chapter 5, in
Procedures for Marketing Authorisation, Vol. 2A, European Commission, available: http://ec.europa.
eu/enterprise/pharmaceuticals/eudralex/vol-2/a/v2a_chap5_r l_2004-02.pdf, accessedApr. 16, 2006.
11. Commissionregulation(EC)No.l084/2003ofJune3,2003,concemingtheexaminationofvariationstothe
termsofamarketingauthorisationformedicmalproductsforhumanuseandveterinarymedicinalproducts
granted by a competent authority of a member state (Official Journal L159,27/6/2003, pp. 1-23), avail-
able: http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homevl.htm, accessed Apr. 10, 2006.
12. The ICH process for harmonisation of guidelines, available: http://www.ich.org/cache/compo/276-
254-l.html, accessed May 15, 2006 .
136 Часть I. Нормативное регулирование производства лекарственных средств
13. Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), Guideline on gen-
eral principles of process validation, available: http://www.fda.gov/cder/guidance/pv.htm , accessed
Apr. 26, 2006.
14. Willig, S. H. (2001), Production and process controls, in Swarbrick, J., Ed., Good Manufacturing
Practices for Pharmaceuticals: A Plan for Total Quality Control from Manufacturer to Consumer,
Marcel Dekker, New York, pp. 99-138.
15. Waterland, N. H., andKowtna, С. C. (2003), Change control and SUPAC, in Nash, R. A., and Wachter,
A. H., Eds., Pharmaceutical Process Validation, Marcel Dekker, New York, pp. 699-748 .
16. Ahmed, S. U., Naini, V., and Wadgaonkar, D. (2005), Scale-up, process validation and technology
transfer, in Shargel, L., and Kanfer, I., Eds., Generic Drug Product Development: Solid Oral Dosage
Form, Marcel Dekker, New York, pp. 95-136 .
Глава 1.4. КУЛЬТИВИРОВАНИЕ МУЛЬТИПОТЕНТНЫХ
МЕЗЕНХИМАЛЬНЫХ СТРОМАЛЬНЫХ
КЛЕТОК ЧЕЛОВЕКА В СООТВЕТСТВИИ
С ПРАВИЛАМИ GMP
Ева Роде, Катарина Шалмазер, Кристина Бартман, Анреас Райниш
и Дирк Струнк Medical University of Graz (1рац, Австрия)
1.4.1. Введение
Терапия при помощи соматических стволовых клеток (SCT) является стремительно
развивающимся направлением, открывающим широкий спектр терапевтических
возможностей. Концепция регенеративной SCT исходит из предположения, что
трансплантация зрелых стволовых клеток человека способствует регенерации орга-
нов, регулирует иммунитет и гемопоэз. Трансплантация гемопоэтических стволовых
клеток, полученных из костного мозга, для регенерации кроветворной и иммунной
систем применяется в клинической практике в течение почти 40 лет. Наличие де-
тектируемого числа мезенхимальных и эндотелиальных клеток-предшественников
в крови и костном мозге позволяет использовать легко культивируемую гемопоэти-
ческую ткань в качестве источника стволовых клеток для негемопоэтической реге-
неративной SCT (рис. 1).
В настоящее время проводится ряд клинических исследований с целью оценить
терапевтическое воздействие мультипотентных мезенхимальных стромальных кле-
ток (MSC) (www.clinicaltrials.gov). Эти негемопоэтические клетки впервые были опи-
саны в работе Friedenstein et al. по результатам исследования колониеобразующих
единиц фибробластов (КОЕ-Ф) в культурах костномозговых адгезивных клеток при
низкой плотности посева [1—3]. Альтернативные источники MSC описаны в ряде
исследований, где показано успешное выделение клеток — предшественников фи-
бробластов из крови пуповины, плаценты, ткани пуповины, амниотической жид-
кости и жировой ткани [4—13]. В настоящее время основной экспериментальный
и клинический опыт связан с применением MSC костного мозга [14—21]. Проли-
ферация ex vivo этих редких составляющих костного мозга (представляющих менее
1% аспирированных ядросодержащих клеток костного мозга) является предвари-
тельным этапом получения дозы MSC (количество, равное по меньшей мере 2 • 106
Л/Л’С/кг веса реципиента). В настоящее время большинство процедур культивиро-
вания клеток основано на использовании фетальной бычьей сыворотки (FBS), из-за
чего возникает риск ксеноиммунизации и переноса как неизвестных патогенов, так
и известных (например, прионов — возбудителей губчатой энцефалопатии крупно-
го рогатого скота). Этого можно избежать разработкой методик (протоколов) выра-
щивания MSCc использованием альтернативных сред на основе крови человека.
138
Часть I. Нормативное регулирование производства лекарственных средств
Рис. 1. Гемопоэтические стволовые клетки, полученные из ткани, и клетки-предшественники.
Гемопоэтическая ткань содержит мезенхимальные (а) и эндотелиальные (б) клетки в дополнение
к гемопоэтическим (в) клеткам-предшественникам: а — MSC, полученные из костного мозга
взрослого человека, окрашены для визуализации актинового цитоскелета, митохондрий и ядер;
б — периферия колонии эндотелиальных клеток-предшественников (ЕРС), полученных из
пуповинной крови, имеет типичную морфологию: характерные плотные скопления клеток, внешне
напоминающие булыжную мостовую. Вся колония выращена из одной клетки, что свидетельствует
о значительном потенциале пролиферации (более 70 тыс. клеток получено культивированием
колоний, выращенных из единственной ЕРС, а значит, завершено не менее 16 циклов удвоения
популяции). Для выращивания достаточного количества клеток для терапевтических целей
достаточно менее 10 мл костного мозга взрослого человека (а, в), но не менеее 40 мл пуповинной
крови (в)
Доклиническая разработка медицинских препаратов, как правило, усложняется
из-за отсутствия установленных регламентов. Длительные манипуляции с медицин-
скими препаратами для клеточной терапии могут увеличить риски возникновения
нежелательных эффектов в процессе культивирования клеток ex vivo. Безопас-
ность клинического применения MSC, выращенных ex vivo, требует материально-
технического обеспечения, гарантирующего соблюдение установленных правил
GMP, внедренной в высокоэффективную систему качества. Подтверждение ста-
бильности производства и продукта достигается тщательным и обоснованным вну-
трипроизводственным контролем. Критерии качества готового продукта в идеале
определяются процессом его успешной разработки, по возможности включают
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток.., 139
стерильность, показатели безопасности, чистоты, подлинность и биологическую
активность1. Методики испытаний должны быть быстрыми, чувствительными,
надежными и достаточно гибкими. Сложность и функции различных препаратов
для клеточной терапии предусматривают применение ряда аналитических проце-
дур, направленных на адекватную оценку отдельного препарата (количественные
определения биологической активности). Персонализированные препараты для
клеточной терапии (только для конкретного пациента) отличаются от промышлен-
но производимых фармацевтической отраслью серий лекарственных средств тем,
что по сравнению с определением биологической активности готового продукта бо-
лее практичным может оказаться процессно-ориентированный ее анализ. Согласно
действующим в США требованиям, «...испытания на биологическую активность
должны включать тесты in vivo или in vitro (либо оба типа тестов), специально раз-
работанные для каждого препарата, позволяющие определить его биологическую
активность способом, соответствующим определению активности согласно 21 CFR
600.3(s)». Функциональный анализ, сопровождающий разработку процесса культи-
вирования клеток, позволяет получить полную характеристику продукта, а также
оптимизировать производственный процесс. Этот анализ является обязательным
условием производства безопасного и эффективного препарата для клеточной те-
рапии.
В этом разделе будет показано, что быстрое и стандартизированное (воспроизво-
димое) культивирование MSC, обеспечивающее получение достаточное количество
клеток в дозе препарата (например, > 2 • 106 на килограмм веса тела для человека
весом 75 кг соответствует > 1,5 • 108 MSC), возможно менее чем за четыре недели.
Замена FBS на HPL {лизат тромбоцитов человека) является одним из способов по-
лучения более безопасного клеточного препарата (рис. 2). Надлежащие доклиниче-
ские разработки, проведенные в соответствии с принципами GMP, позволят умень-
шить риски безопасности при проведении последующих клинических испытаний
MSC как терапевтического агента.
1.4.2. Сокращения и определения
1.4.2.1. Мезенхимальные стромальные клетки
Адгезия мононуклеарных клеток из аспирата костного мозга человека {BM-MNC)
к поверхности культурального пластика и удаление свободных клеток в течение
первых дней культивирования позволяют отобрать популяцию пролиферирующих
веретенообразных фибробластоподобных негемопоэтических мультипотентных
MSC. Мезенхимальные стромальные клетки можно также получить из крови или
ткани пуповины, из плаценты, жировой ткани и некоторых фетальных тканей. Ми-
нимальные критерии приемлемости для MSC определены в меморандуме Между-
народного общества по клеточной терапии {International Society for Cellular Therapy,
ISCT), опубликованном в 2006 г. [22]. Мезенхимальные стромальные клетки облада-
ют высоким потенциалом самообновления и способны к дифференцировке in vitro
в клетки-предшественники с остео-, хондро- и адипоцитарным фенотипом.
‘Законодательство США: 21 CFR210,21 CFR211,21 07(312.21,21 СИ? 312.22(a) и 21 CFR
312.23(a)(7)(i).
140
Часть I. Нормативное регулирование производства лекарственных средств
Рис. 2. Выращивание MSC человека в соответствии с правилами GMP. Краткое описание
двухэтапной процедуры выращивания MSC (число посеянных и выращенных BM-MNC
и итоговое число MSCHPL в сравнении с числом MSCFBS) [23]: а-МЕМ — минимальная питательная
среда (а-МЕМ); HPL — лизат тромбоцитов человека (от human platelet lysate}-, BM-MNC —
мононуклеарные клетки аспирата костного мозга человека (от mononuclear cells from human bone
marrow aspirates}-, 7225 — флакон для клеточных структур площадью поверхности 225 см2; CF —
система культифирования клеток
1.4.2.2. Клеточная терапия соматическими стволовами клетками
Концепция регенеративной SCT основана на экспериментальных данных и ран-
них клинических наблюдениях, показывающих, что применение стволовых клеток
взрослого человека может способствовать регенерации органов после ишемиче-
ских, токсических или метаболических нарушений. Костный мозг содержит гемо-
Глава 1.4, Культивирование мультипотентных мезенхимальных стромальных клеток... 141
поэтические и мезенхимальные стволовые клетки, а также эндотелиальные клетки-
предшественники и является легкодоступным (но не единственным) источником
клеток, способных ускорить восстановление органа при системном или местном
применении. Регулирование гемопоэза и иммуномодуляция представляют собой
два общепринятых способа SCTаутологическими и аллогенными стволовыми клет-
ками и клетками-предшественниками.
1.4.2.З. Надлежащая производственная практика
Надлежащая производственная практика является частью системы менеджмента
качества {Quality management system, QMS), регулирующей производство и контроль
качества лекарственных средств, применяемых в медицине и ветеринарии. Система
регламентирует порядок оформления документации при производстве фармацев-
тической продукции, затрагивая также вопросы обучения персонала, планировки
помещений, контроля оборудования и производственного процесса.
1.4.2.4. Лекарственные препараты для клеточной терапии
Для общего описания лекарственных препаратов, содержащих жизнеспособные
клетки, используется термин «лекарственные препараты для клеточной терапии»
{cell-based medicinal products, СМВР). Данный термин не распространяется на пре-
параты, содержащие нежизнеспособные клетки или клеточные фрагменты. Пред-
полагается, что препараты для клеточной терапии будут способны излечивать за-
болевания, до сих пор считавшиеся неизлечимыми. Эти препараты разнородны
по происхождению и типам клеток, а также по сложности состава. Клетки могут
представлять собой самообновляющиеся стволовые клетки, более детерминиро-
ванные клетки-предшественники или полностью дифференцированные клетки,
проявляющие специфические регенеративные функции. Клетки могут иметь как
аутологическое, так и аллогенное происхождение. Их можно применять отдельно
или в комбинации с биомолекулами, химическими веществами или структурными
материалами, которые предположительно усиливают их ожидаемый эффект.
1.4.2.5. Лизат тромбоцитов человека
Лизат тромбоцитов человека {HPL) можно получить из лейкотромбоцитарного слоя
обогащенной тромбоцитами плазмы. Фракцию тромбоцитов отделяют от плаз-
мы и фракций лейкоцитов и эритроцитов центрифугированием и концентриру-
ют до плотности по меньшей мере 1 • 109 тромбоцитов на миллилитр. Тромбоциты
можно активировать тромбином или лизировать проведением повторных циклов
замораживания-оттаивания. И то и другое приводит к высвобождению факторов
роста и митогенов, присутствующих в неповрежденных тромбоцитах. Медиаторы,
высвобождающиеся из тромбоцитов, включают, среди прочего, эпидермический
фактор роста {epidermis growth factor, EGF), основной фактор роста фибробластов
{basic fibroblast growth factor, bFGF), фактор роста тромбоцитов {platelet-derived growth
factor, PDGF), трансформирующий фактор роста {transforming growth factor, TGF-wl)
и инсулиноподобный фактор роста {insulinelike growth factor, IGF) [23,24]. Вероятно,
HPL может заменить FBSvo многих системах клеточных культур, традиционно ори-
ентированных на FBS.
142
Часть I. Нормативное регулирование производства лекарственных средств
1.4.3. Подходы
1.4.3.1. Приверженность принципам GMPb процессе доклинической
разработки
Стандартное культивирование MSC необходимо проводить в соответствии с хорошо
спланированной, последовательно документируемой и оптимизированной проце-
дурой, которая также снижает риск микробной контаминации, контаминации ме-
ханическими включениями и пирогенными веществами уменьшением количества
этапов и длительности обработки. Согласно действующему европейскому законода-
тельству1, принципы GMP следует применять уже при изготовлении лекарственных
препаратов для клеточной терапии, предназначенных для клинических исследова-
ний первой фазы у человека. Согласно законодательству США2, эти требования не
распространяются на лекарственные препараты для клеточной или тканевой тера-
пии, применяемые в клинических исследованиях первой фазы, как и на продукты,
проходящие стадию доклинической разработки. Если предполагается, что резуль-
таты, полученные на доклиническом этапе, позволят быстро перейти к клиниче-
ским исследованиям, можно рекомендовать разработку технологического процесса,
удовлетворяющего требованиям GMP, на фазе доклинической разработки любого
клеточных препарата. Это даст гарантию того, что продукты стабильно производят-
ся и контролируются на соответствие стандартам качества, корреспондирующим
с их предполагаемым предназначением, или спецификации на продукт. Требования
GMP подробно описаны в Руководстве по надлежащей производственной практики
для медицинских продуктов Схемы взаимодействия фармацевтических инспектора-
тов (PIC/S) и включают внедрение эффективно работающей системы менеджмента
качества, создание специальных участков для производства стерильных лекарствен-
ных средств в соответствии с правилами GMP, наличие квалифицированного и обу-
ченного персонала, надлежащего оборудования, пригодных для использования
материалов, упаковочных и этикетировочных материалов, утвержденных процедур
и инструкций, надлежащих помещений и систем для хранения и транспортировки,
а также системы ведения записей, позволяющей полностью проследить историю
лекарственного препарата (см. http://www.picscheme.org). Проведение доклинических
исследований и разработки в соответствии с правилами GMP— сложная задача, по-
скольку процедуры зачастую требуют намного больше затрат труда и времени, чем
обычные исследования лабораторного масштаба. Как следствие, либо ускоряется
прогресс в исследовательских разработках за счет снижения стандартов качества,
либо наоборот. Поэтому решения о том, насколько строго будут соблюдаться стан-
дарты GMP, принимаются отдельно по каждому конкретному случаю, в зависимо-
сти от временного графика (более или менее строго) по планируемым клиническим
исследованиям препарата для клеточной терапии.
Европейское законодательство: директивы 65/65£Е'С, 75/318££'С, 75/318/£ЕС, Комис-
сия взаимодействия по вопросам авторизации медицинской продукции на европейском
рынке (98/С229/03); Директива 2001/20/£С, £ЛЖД/СШ/Р/410869/2006.
Законодательство США: 21 CFR 210, 21 CFR 211, 21 CFR 312.21, 21 CFR 312.22(a) и 21
CER312.23(a)(7)(i).
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 143
1.4.З.2. Эффективное стандартизованное культивирование MSCc низкой
плотностью посева
Для дальнейшего проведения клинических исследований препарата MSC может по-
требоваться очень большое его абсолютное количество. Этого требует получение
большего количества доз для пациентов (> 5 • 106 на килограмм веса тела челове-
ка) по сравнению с количеством, использующемся в экспериментальных моделях
на мелких животных in vivo [20]. Поэтому желательно разработать протоколы куль-
тивирования MSC в большом объеме, позволяющие получать до (5—10) 108 MSC из
ограниченного исходного объема первичного материала.
Плотность посева клеток критически важна для величины скорости пролифе-
рации MSC, она должна быть определена для первичного посева и последующих
пассажей (т. е. этапов субкультивирования). Большинство описанных до настояще-
го времени экспериментальных и клинических процедур культивирования клеток
предусматривают изначально высокую плотность посева: более 1 105 BM-MNC/сы1
[2, 14, 16]. Первые исследования показали, что при последующем субкультивирова-
нии очень низкая плотность посева (от 5 до 10 МСУ/см2) позволяет получить быстро
пролиферирующую субпопуляцию рециклирующих стволовых клеток, так называе-
мых BS-клеток [25—28]. Эта плотность посева, называемая клональной плотностью,
требует теоретической площади роста приблизительно от 100 000 до 2 000 000 см2
(от 10 до 200 м2) для получения клинического количества > 1 108 MCSws исходного
количества 1 106 за один пассаж. Поэтому чашечный посев с плотностью 30—100
МУС/см2 представляет собой разумный компромисс: площадь роста составляет от
10 000 до 25 000 см2 (1,0—2,5 м2). Авторы недавно доказали, что первичный посев
лишь 10 мл аспирата костного мозга на площадь роста, приблизительно равную
0,2 м2, и культивирование в течение двух недель (первый этап культивирования:
костный мозг вносят в питательную среду сразу же после аспирации, без разделения
по градиенту плотности; удаление свободных клеток проводится на третий день)
с последующим пассажем на площадь 2,5 м2 (второй этап) позволяет стабильно ге-
нерировать по меньшей мере 1,5 • 108 MSC в среде, содержащей FBS, менее чем за
четыре недели (см. рис. 2) [29]. Это исследование дало дополнительное подтвержде-
ние ранее полученным данным об обратной корреляции между величинами плот-
ности посева и пролиферации МУС (рис. 3) [25—28].
1.4.3.3. Более активная пролиферация клеток в питательных средах
с лизатом тромбоцитов человека по сравнению с питательными
средами с фетальной бычьей сывороткой
Самыми распространенными питательными средами, применяемыми для выращи-
вания MSC, являются минимальная питательная среда а-МЕМ и модифицирован-
ная по Дульбекко среда Игла с низким (1 г/л) содержанием глюкозы и добавками
Д-глутамина, антибиотиков и 5—20 % FBS [14, 16, 19, 24, 25, 30]. Опыт авторов по
культивированию MSC касается использования питательной среды а-МЕМ, содер-
жащей FBS или HPL. В отличие от HPL, лишь недавно признанного эффективной
добавкой к питательной среде [24], FBS является традиционной основной добав-
кой к питательной среде для клеточных культур и применяется в течение более чем
144
Часть I. Нормативное регулирование производства лекарственных средств
1 -й день
2-й день
5-й день
10-й день
Рис. 3. Обратная корреляция между величинами плотности посева и пролиферации MSC.
MSC костного мозга, полученные после второго пассажа, посеяны с логарифмическим шагом
уменьшения плотности посева: 1000, 100, 10 и 1 см 2. Фотографии были сделаны через 1, 3, 5
и 10 дней культивирования в среде а-МЕМ / 10% FBS (исходное увеличение 40х). При посеве
MSC с плотностью 100 и 1000 клеток/см2 из-за образования сливного слоя клеток возникла
необходимость трипсинизации между пятым и десятым днями, с последующим пересевом
с плотностью 100 и 1000 клеток/см2 соответственно, обозначенным как Р + 1
50 лет [31]. Традиционное использование FBS при культивировании MSC в качестве
источника факторов роста и митогенов сопряжено с риском переноса известных
и неизвестных патогенов, а также риском ксеноиммунизации к бычьим патогенам.
Поэтому применения FBS следует избегать при изготовлении клеточных продуктов,
предназначенных для клинического использования [32, 33].
В ходе недавнего исследования авторы изучали возможность замены FBS на HPL
при культивировании больших объемов (для клинических исследований) MSC.
Было показано, что MSC, культивируемые в присутствии HPL, обладают более вы-
раженной пролиферативной активностью, чем клетки, культивируемые в присут-
ствии FBS [23]. На рис. 4 представлены высокая пролиферативная активность MSC
при низкой плотности посева и более высокая скорость удваивания популяции
в присутствии HPL при длительности культивирования менее 14 дней.
1.4.3.4. Возможность минимизации риска контаминации при
использовании обоснованных (надлежащих) процедур
культивирования MSC
Культивирование клеток проводят, как правило, в соответствии с методиками
(«протоколами»), требующими значительных затрат усилий и времени, в открытых
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 145
FBS
HPL
чем*
1б/см2 0/см2
MSC, культивируемые в течение 13 дней
Рис. 4. Пролиферативная активность MSC зависит от плотности посева в присутствии
ксеногенного FBS и HPL. Обратная корреляция между величинами пролиферации MSC и плотности
посева выразилась в образовании сливных слоев MSC в культурах с исходной плотностью
посева 1 MSC / см2 в а-МЕМ /10% FBS и культурах с исходной плотностью посева 1 MSC / см2
в а-МЕМ / 10% HPL, но не в культурах с изначальной высокой плотностью посева; длительность
культивирования составила менее двух недель. Представлены рассчитанные по результатам
репрезентативного эксперимента (сбор клеток на 13-й день эксперимента) значения кратности
увеличения количества клеток и соответствующего числа удвоений популяции
системах, повышающих риск контаминации микроорганизмами или механически-
ми частицами. Чтобы избежать подобных проблем, в питательные среды добавляют
мощные антибиотики. Отказ от применения пенициллина в ходе культивирования
клеток в опытно-промышленном масштабе (для клинических исследований) объ-
ясняется необходимостью снижения риска сенсибилизации и анафилактической
преципитации. По этой же причине следует избегать использования и других анти-
биотиков при культивировании MSC в соответствии с требованиями GMP. Один
из подходов, направленных на минимизацию риска возможной контаминации,
заключается в строгом ограничении манипуляций с MSC до минимального коли-
чества абсолютно необходимых этапов. Наш опыт показывает, что традиционный
этап центрифугирования в градиенте плотности может быть исключен перед пер-
вичным посевом клеток аспирата костного мозга. Немедленное внесение ограни-
ченных объемов (например, 10—20 мл) гепаринизированного аспирата костного
мозга в среду а-МЕМс добавками для прямого посева клеток не приводит к умень-
шению выхода MSC [23]. Кроме того, вышеупомянутый посев с низкой плотностью
и увеличение площади роста, наряду с использованием HPL, согласно упрощенной
процедуре фактически позволяет эффективно получать большое число MSCaa один
или два пассажа культуры. Сравнительно короткий период пролиферации клеток ех
vivo (менее 3—4 недель) позволяет уменьшить общий риск их контаминации.
146
Часть I. Нормативное регулирование производства лекарственных средств
1.4.4. Методики испытаний
1.4.4.1. Безопасность и эффективность препаратов для клеточной терапии
на доклинической стадии
На этапе доклинической разработки следует подробно изучить показатели качества
препарата для клеточной терапии. Необходимо определить критерии выпуска про-
дукции и установить разумные временное рамки, позволяющие гарантировать вы-
сокую степень безопасности и соответствие готового клеточного продукта высоким
стандартам качества. С другой стороны, организация работ по контролю готового
продукта должна обеспечить быстрый выпуск клеточного препарата (в пределах не-
скольких часов), поскольку предполагается, что срок годности большинства кле-
точных продуктов небольшой. Необходимо провести оценку чистоты клеток, сте-
рильности, а также убедиться в отсутствии пирогенных веществ и эндотоксинов,
поскольку все эти показатели качества чрезвычайно важны. Характерной особен-
ностью препаратов для клеточной терапии является то, что спецификации продук-
та должны быть адаптированы для его индивидуального (персонифицированного)
применения. На стадии доклинической разработки необходимо найти удовлетвори-
тельные ответы на все вопросы относительно типа клеток, источника, дозы и спо-
соба применения для лечения определенного заболевания. Таким образом, каждый
клеточный препарат должен удовлетворять критериям как внутрипроизводствен-
ного контроля, так и выпуска готового продукта. Поскольку многие препараты
для клеточной терапии представляют собой персонифицированное лекарственное
средство, определение биологической активности должно проводиться для выбран-
ных репрезентативных продуктов (например, перед началом исследования и впо-
следствии один раз в год).
1.4.4.2. Контроль качества в процессе культивирования клеток
(внутрипроизводственный контроль) и критерии выпуска готового
продукта
Общая безопасность. Согласно требованиям FDA, препараты для клеточной терапии
не подлежат исследованию на общую безопасность (21 CER610.11(g)(l)).
Содержание клеток в дозе. На доклинической стадии можно определить мини-
мальное эффективное количество жизнеспособных и функциональных клеток
и максимальное переносимое количество клеток. Оптимальную дозу вводимых
в организм клеток предстоит определить впоследствии [20].
Жизнеспособность MSC может быть легко оценена сразу же после трипсиниза-
ции. Это достигается исключением клеток, окрашенных трипановым синим или
7-амино-актиномицином-Т). Согласно требованиям, разработанным авторами по
результатам исследований клеточных культур, жизнеспособность должна быть на
уровне более 90%. В исключительных случаях допустимо более низкое предельное
количество собранных жизнеспособных клеток (70%).
Микробиологические испытания. Испытание на стерильность, позволяющее
выявить контаминацию грибами, анаэробными и аэробными бактериями, а также
микоплазмой, следует проводить после каждого критического этапа обработки при
культивировании MSC, во время которого может произойти микробиологическое
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 147
загрязнение [34]. Решающую проверку отсутствия бактериальной флоры, проводи-
мую в конце последнего этапа — сбора клеток, невозможно выполнить перед при-
менением продукта in vivo, если MSC нужно использовать сразу после культивиро-
вания из-за ограниченного срока существования культуры. Результаты испытания
на присутствие микоплазмы методом полимеразной цепной реакции (ПЦР) можно
получить в день сбора клеток, спустя менее 6 ч. Результаты теста MycoAlert® доступ-
ны уже через час после сбора клеток. Однозначные результаты, позволяющие ис-
ключить контаминацию микоплазмой, можно получить в течение двух-трех недель,
поэтому данный подход неприменим для клеточных препаратов с коротким сроком
годности, которые предполагается использовать сразу же после культивирования.
Испытание на содержание бактериальных эндотоксинов и пирогенных веществ. Ис-
пытание на содержание эндотоксинов с использованием лизата амебоцитов Limulus
(ЛАЛ-тест) обычно является альтернативой испытанию на пирогенность на ранних
фазах испытаний. Для всех препаратов для парентерального введения, за исклю-
чением вводимых интратекально, FDA рекомендует установить верхний предел со-
держания эндотоксинов на уровне пяти эндотоксиновых единиц (ЭЕ) на килограмм
массы тела в дозе. ЛАЛ-тест можно использовать для оценки безопасности биоло-
гических препаратов согласно существующим нормативам1. Авторы используют
ЛАЛ-тест как замену длительного микробиологического анализа готового продукта
для получения данных перед его клиническим применением менее чем через 2 ч
после сбора клеток.
Фенотипическая подлинность MSC. В дополнение к морфологической идентифи-
кации микроскопическим методом, иммунофенотипическую характеристику MSC
можно получить с помощью широкого ряда конъюгированных с флуоресцеином
антител. В настоящее время специфического маркера, однозначно определяющего
MSC, не существует. Поэтому для демонстрации экспрессии определенных марке-
ров и исключения контаминации клетками с профилями экспрессии других мар-
керов используется получение фенотипического профиля клеток. Для подтвержде-
ния положительной окраски МУС на CD73, CD90 и CD105 и негативной реакции на
HLA-DR, CD]A, CD3\, CD34 и CD45 (рис. 5) ISCTрекомендует использовать метод
проточной цитометрии [22, 23]. Более детальный фенотипический анализ прово-
дили без получения дополнительной информации о типе или функции MSC [35].
Можно надеяться на то, что получение профиля по экспрессии генов улучшит иден-
тификацию МУС человека [35—42].
1.4.4.З. Исследование функциональности и биологической активности
стволовых клеток
Клоногенный количественный анализ. Способность клеток к самообновлению и долю
пролиферирующих клеток в гетерогенной смеси клеток можно оценить определе-
нием количества колониеобразующих единиц (КОЕ). Метод тканевой культуры,
1 Испытание на содержание эндотоксинов (ЛАЛ-тест), в соответствии с Европейской
фармакопеей (раздел 2.6.14) и Руководством по валидации испытания с лизатом амебоцитов
Limulus в качестве испытания готовых парентеральных лекарственных средств для медицин-
ского и ветеринарного применения, биологических продуктов и медицинских изделий на
содержание эндотоксинов в 1987 г., разделы 1—IV; http://www.fda.gov/cber/gdlns/lal.pdf.
148
Часть I. Нормативное регулирование производства лекарственных средств
FBS
HPL
Рис. 5. Иммунный фенотип MSC человека. Для определения реактогенности антител (гистограммы
закрашены серым цветом) по сравнению с рядом контрольных разведений с радиоактивной
меткой (черная линия) использовали проточный цитометрический анализ по меньшей мере
10 тыс. жизнеспособных MSC. Фенотипические критерии требуют позитивной реакции (> 90%)
на СВ73, CD90 и CD105 и отрицательной (< 2%) на HLA-DR, CD14 (или CD11b), CD19 (или CD79a),
CD34 и CD45. Отсутствие (CD3 + 7")-клеток может оказаться желательным при терапии реакции
«трансплантат против хозяина» (РТПХ, англ, graft-versus-host disease, GvHD). В зависимости от
условий культивирования у MSC, как и у клеток нейробластомы, меланомы и мелкоклеточного
рака легкого, определяется реактогенность с антидисиалоганглиозидными антителами GD2
позволяющий проводить подсчет колоний клеток, был впервые описан в 1956 г.
[43]. Внедрение методов определения КОЕ в клетках костного мозга привело к от-
крытию гемопоэтических стволовых клеток [44]. Предшественники фибробластов,
присутствующие в гемопоэтической системе, также были обнаружены специфиче-
ским методом подсчета колониеобразующих единиц фибробластов, предложенного
Friedenstein в 1974 г. (КОЕ-Ф) [2]. Авторы анализировали клоногенную активность
MSC определением КОЕ-Ф. На рис. 6 показаны внешние различия колониеобра-
зующих фибробластов в MSC, полученных в присутствии HPL и FBS. При исследо-
вании нативного костного мозга для определения количества КОЕ-Ф необходимо
соответствующее разведение (рис. 7). Для анализа обогащенных MSC рекомендо-
ванная плотность посева для клоногенного количественного анализа варьируется
от 1 до 5 MSC/cm2 [2, 29].
Остео-, хондро- и адипоцитарная дифференцировка. В 1999 г. было показано, что
изолированные MSC, полученные из костного мозга, могут дифференцировать по
множественным мезенхимальным линиям [45]. Полученные данные позволяют
предположить, что MSC могут также экспрессировать фенотипические характери-
стики эндотелиальных и нервных клеток, клеток гладкой мускулатуры, скелетных
миобластов и сердечных миоцитов [46]. Образующиеся при дифференцировке MSC
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 149
Рис. 6. Морфологическая оценка MSC. Колониеобразующие фибробласты в MSC, полученных
в присутствии HPL и FBS, различаются по размеру, морфологии и плотности (масштабная полоса
обозначает увеличение на верхней панели; фотографии колоний сделаны на 12-й день, исходное
увеличение — 40х)
11-й лень
0,18 ’О3 I.89 3 4,4g 10: 2,23-104
Число посеянных MNC на 1 см2
Рис. 7. Количество КОЕ-Ф MSC зависит от плотности посева клеток костного мозга. Соответствующее
разведение гепаринизированного аспирата костного мозга необходимо для точного подсчета
частоты образования первичных КОЕ-Ф. Как показано в этом репрезентативном эксперименте, весь
объем гепаринизированного аспирата костного мозга был посеян согласно измеренному количеству
мононуклеарных клеток (BM-MNC) на квадратный сантиметр площади роста, после чего его культивировали
в течение 11 дней при 37 °C во влажной атмосфере, содержащей 3% О2 и 5% СО2. Неадгезивные
клетки удалялись на третий день. КОЕ-Ф детектировались окрашиванием гематоксилином по Харрису
150 Часть I. Нормативное регулирование производства лекарственных средств
клетки-предшественники по остеоцитарным, хондроцитарным и адипоцитарным
линиям подробно описаны во многих публикациях [47]. Такое определение биоло-
гической активности можно проводить регулярно, если предполагается восстанов-
ление костной или соединительной ткани, в то время как временное границы не
позволяют немедленно выпустить продукт.
Иммуномодулирующее действие. Мезенхимальные стромальные клетки ингибиру-
ют аллореактогенность Т-клеток в смешанных культурах лимфоцитов или пролифе-
рацию лимфоцитов, индуцированную митогенами, такими как фитогемагглютинин
(ФГА) или конканавалин А [29.48—51]. Следует отметить, что в высоких концентра-
циях (10—40 MSC на 100 отвечающих лимфоцитов) MSC оказывают ингибирующий
эффект, в то время как низкие концентрации MSC (0,1—1%) могут стимулировать
пролиферацию лимфоцитов в смешанных культурах лимфоцитов [50]. Если MSC
применяют для иммуносупрессивной терапии, то эти данные могут означать, что
высокие дозы MSC необходимы для ингибирования пролиферации Г-клеток у па-
циентов с реакцией «трансплантант против хозяина», возникшей после аллогенной
трансплантации костного мозга. Применение низких доз MSCможет стимулировать
пролиферацию лимфоцитов in vivo и, следовательно, привести к развитию нежела-
тельной реакции «трансплантант против хозяина», которая будет являться побоч-
ным эффектом (нежелательная лекарственная реакция) терапии MSC. До сих пор
неясно, следует ли определять точное количество Г-клеток в каждом трансплантате
MSC для исключения возможного развития аллогенных реакций. Для определе-
ния иммуномодулирующего действия (активности) используют клетки, меченые
N-сукцинимидиловым эфиром карбоксифлуоресцеиндиацетата (CFSE), что позво-
ляет количественно оценить пролиферацию в ответ на аллогенную или митогенную
стимуляцию [52]. Авторы анализировали снижение интенсивности флуоресценции
CFSE, указывающее на пролиферацию клеток, методом проточной цитометрии по-
сле культивирования мононуклеарных клеток (MNC), меченых CFSE, при наличии
либо отсутствии различных количеств MSC [53]. Иммунорегуляторную способность
MSC, полученных в присутствии HPL или FBS, изучали измерением аллогенной
пролиферации MNC после совместного попарного культивирования мононуклеар-
ных клеток от трех различных доноров с двумя независимыми параллелями MSC
и MSC, полученных в присутствии HPL и FBSсоответственно (рис. 8).
Регулирование гемопоэза. Регулирование поведения гемопоэтических клеток-
предшественников {НРС) можно оценивать при совместном культивировании
культур MSC-HPC in vitro [54]. В жидких культурах очищенных СЛ34' (НРС) с пред-
варительно подготовленным питающим слоем MSC происходит увеличение коли-
чества СЛ34+/38+ (НРС) и CD34 '/38 гемопоэтических стволовых клеток. При этом
создаются условия для роста предшественников всех зрелых гемопоэтических ядро-
содержащих клеток (TNC) (рис. 9).
Генетическая стабильность и канцерогенность. Методики генетического анализа MSC
человека недостаточно отработаны. Использование метода дифференциального
окрашивания стандартной метафазной хромосомы G ограничено из-за малого ко-
личества метафаз, подходящих для стандартных анализов. Успехи, связанные с при-
менением метода флуоресцентной гибридизации in situ с разной окраской (FISH)
и высокоразрешающей аппаратуры на матричной основе, могут привести к появле-
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 151
★ -к
।-------------------------------------------------
★
I
г
I a MNC без ФГА
□ MNC + MSC без ФГА
(MNC + ФГА) к MSC =10:1
(MNC + ФГА) к MSC = 100:1
□ Только MNC (+ ФГА)
Рис. 8. Иммуномодулирующее действие MSC. Аллогенная пролиферация MNC (среднее
количество клеток плюс-минус стандартная ошибка среднего (Standard Error of the Mean, SEM)
измерена после совместного попарного культивирования MNC от трех разных доноров с двумя
независимыми параллелями MSC, полученных в присутствии HPL, и двумя параллелями MSC,
полученных в присутствии FBS (см. диаграмму). Для оценки влияния MSC на пролиферацию
активированных фитогемаглютинином (ФГА) MNC в культуру вносили MSC в соотношении 1:10 (от
3 104доЗ 105 MSC на лунку; [(MNC + ФГА) к MSC = 10:1] или 1:100 [(MNC + ФГА) к MSC = 10:1)],
чтобы оценить их влияние на ФГА-активированную пролиферацию MNC. Количество MNC
измерялось методом проточной цитометрии с использованием трубок BD Truecount™. В качестве
контролей использовались уровень пролиферации MNC, активированных митогеном, без
добавления MSC (только MNC + ФГА), и уровень фоновой пролиферации в отсутствие ФГА (MNC
без ФГА). Мезенхимальные стромальные клетки не индуцировали пролиферацию MNC (MNC+MSC
без ФГА). Значительные отличия помечены знаком * (*р < 0,05 и **р < 0,01) [53]
нию в ближайшем будущем практических диагностических способов, позволяющих
оценивать безопасность клеточных препаратов, применяемых в регенеративной ме-
дицине [55].
Генетическая нестабильность может быть редким явлением, иногда возникающим
после продолжительного культивирования MSC мышей и человека в среде, содер-
жащей FBS [56, 57]. Для оценки канцерогенности in vivo MSC, полученные в резуль-
152
Часть I. Нормативное регулирование производства лекарственных средств
а)
Только CD34
CD34 +
+ MSC(FBS)
CD34 +
+ MSC (HPL)
300
250
200
150
100
50
0
Рис. 9. Регулирование гемопоэза MSC: а — клетки CD34', полученные из пуповинной крови,
выращенные в питательной среде, содержащей цитокины-1640 (RMPI, от Roswell Park Memorial
Institute), 10% FBS, гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF),
интерлейкин-3 (IL-3), фактор стволовых клеток, а также лиганд FMS-подобной тирозинкиназы-3
(Flt-3L) при наличии либо в отсутствие полученных для клинических исследований MSC. Серые
колонки показывают количество всех собранных ядросодержащих клеток (TNC №), а черные
колонки — кратность увеличения количества TNC № по отношению к исходному количеству клеток
CD34’; б — число полученных клеток CD34+ (серые колонки) и кратность увеличения (черные
колонки) клеток CD34* после культивирования в жидкой культуре при наличии либо в отсутствие
MSC; в — число полученных клеток (серые колонки) и кратность увеличения (черные колонки)
CD347CD38- гемопоэтических стволовых клеток после культивирования клеток CD34* в жидкой
среде в присутствии MSC (при FBS) или MSC (при HPL) по отношению к количеству клеток,
полученных при культивировании в жидких культурах, содержащих цитокины, в отсутствие MSC
(контроль, среднее плюс-минус стандартная ошибка по двум независимым параллелям) [53]
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 153
тате краткосрочного культивирования в оптыно-промышленном масштабе (для
клинических исследований) в питательных средах, содержащих FBS или HPL, под-
кожно вводили иммунодефицитным бестимусным мышам. Предполагаемое фор-
мирование опухоли оценивали с помощью гистологического анализа через 3 мес.
после инъекции 2 • 106 и 2 • IO4 MSC. Результаты сравнивали с данными контроль-
ной группы животных, получавших инъекцию за 48 ч перед эвтаназией. Первичное
накопление клеток наблюдалось визуально сразу же после инъекции и через 48 ч,
и MSC можно было оценить с помощью традиционного микроскопирования. Од-
нако ни у одного из 12 экспериментальных животных не было выявлено макроско-
пической или видимой под микроскопом опухоли по окончании 90-дневного на-
блюдения [23]. В этой ситуации генетическое исследование может помочь получить
проспективные данные, однако не считается в настоящее время обязательным при
выпуске продукта для проведения клинических испытаний MSC.
1.4.5. Заключение
Существуют ограничения в использовании традиционных фармакологических ме-
тодик, применяемых для определения безопасности и эффективности препаратов
для клеточной терапии на доклинической стадии. Традиционные методы, исполь-
зуемые в фармацевтической промышленности для построения фармакологических
профилей и оценки острой токсичности лекарств у животных, а также токсиколо-
гические исследования не всегда можно непосредственно переносить на клеточные
продукты, произведенные ex vivo. Как бы то ни было, доклинические исследования
и разработку клеточных продуктов необходимо проводить согласно индивидуаль-
ным или согласованным спецификациям и определениям, которые будут постоян-
но совершенствоваться. Этот подход обещает быть полезным при разработке эф-
фективных терапевтических клеточных агентов.
Благодарность
Данную работу частично финансировали Фонд исследований стволовых клеток
взрослого человека и Фонд грантов для молодых исследователей Министерства об-
разования, науки и культуры Австрии. Программа «Австрийская наноинициатива»
софинансировала эту работу как часть проекта «Наноздоровье» (субпроект NANO-
STEM, финансируемый Австрийским научным фондом).
Литература
1. Luria, Е. A., Panasyuk, А. Е, and Friendenstein, А. У. (1971), Fibroblast colony formation from
monolayer cultures of blood cells, Transfusion, 11 (6), 345-349.
2. Friedenstein, A. J., Deriglasova, U. F., Kulagina, N. N., et al. (1974), Precursors for fi broblasts in
different populations of hematopoietic cells as detected by the in vitro colony assay method, Exp.
Hematol., 2 (2), 83-92.
3. Kuznetsov, S. A., Friedenstein, A. J., and Robey, P. G. (1997), Factors required for bone marrow
stromal fi broblast colony formation in vitro, Br. J. Haematol., 97 (3), 561-570.
4. Erices, A., Conget, P., and Minguell, J. J. (2000), Mesenchymal progenitor cells in human umbilical
cord blood, Br. J. Haematol., 109 (1), 235-242.
154 Часть I. Нормативное регулирование производства лекарственных средств
5. Kogler, G., Sensken, S., Airey, J. A., et al. (2004), Anew human somatic stem cell from placental cord
blood with intrinsic pluripotent differentiation potential, J. Exp. Med., 200 (2), 123-135.
6. Bieback, K., Kern, S., Kluter, H., and Eichler, H. (2004), Critical parameters for the isolation of mes-
enchymal stem cells from umbilical cord blood, Stem Cells, 22 (4), 625-634.
7. Romanov, Y. A., Svintsitskaya, V. A., and Smirnov, V. N. (2003), Searching for alternative sources
of postnatal human mesenchymal Stem Cells: Candidate MSC-like cells from umbilical cord, Stem
Cells, 21 (1), 105-110.
8. Miao, Z., Jin, J., Chen, L., et al. (2006), I solation of mesenchymal Stem Cells from human placenta:
Comparison with human bone marrow mesenchymal stem cells, Cell Biol. Int., 30 (9), 681-687.
9. In‘t Anker, P. S., Scherjon, S. A., Kleijburg-van der Keur, C., et al. (2004), Isolation of mesenchymal
Stem Cells of fetal or maternal origin from human placenta, Stem Cells, 22 (7), 1338-1345.
10. In‘t Anker, P. S., Scherjon, S. A., Kleijburg-van der Keur, C., et al. (2003), Amniotic fluid as a novel
source of mesenchymal stem cells for therapeutic transplantation, Blood, 102 (4), 1548-1549.
11. Gronthos, S., Franklin, D. M., Leddy, H. A., Robey, P. G., Storms, R. W., and Gimble, J.
M. (2001), Surface protein characterization of human adipose tissue-derived stromal cells,
J. Cell Physiol., 189 (1), 54-63.
12. Zuk, P. A., Zhu, M., Ashjian, P., et al. (2002), Human adipose tissue is a source of multipotent stem
cells, Mol. Biol. Cell, 13 (12), 4279^4295.
13. Kern, S., Eichler, H., Stoeve, J., Kluter, H., and Bieback, K. (2006), Comparative analysis of mes-
enchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue, Stem Cells, 24 (5),
1294-1301.
14. Lazarus, H. M., Haynesworth, S. E., Gerson, S. L., Rosenthal, N. S., and Caplan, A. I. (1995), Ex
vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells
(mesenchymal progenitor cells): Implications for therapeutic use, Bone Marrow Transplant., 16 (4),
557-564.
15. Кос, О. N., Peters, C., Aubourg, P., et al. (1 995), Bone marrow-derived mesenchymal Stem Cells
remain host-derived despite successful hematopoietic engraftment after allogeneic transplantation in
patients with lysosomal and peroxisomal storage diseases. Exp. Hematol., 27 (11), 1675-1681.
16. Кос, О. N., Gerson, S. L., Cooper, B. W., et al. (2000), Rapid hematopoietic recovery after coinfusion
of autologous-blood stem cells and culture-expanded marrow mesenchymal Stem Cells in advanced
breast cancer patients receiving high-dose chemotherapy, J. Clin. Oncol., 18 (2), 307-316.
17. Lee, S. T., Jang, J. H., Cheong, J. W., et al. (2002), Treatment of high-risk acute myelogenous leukae-
mia by myeloablative chemoradiotherapy followed by co - infusion of T cell-depleted haematopoietic
Stem Cells and culture-expanded marrow mesenchymal Stem Cells from a related donor with one
fully mismatched human leucocyte antigen haplotype, Br. J. Haematol., 118 (4), 1128-1131.
18. Кос, О. N. Day, J., Nieder, M., Gerson, S. L., Lazarus, H. M., and Krivit, W. (2002), Allogeneic
mesenchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler
syndrome (MPS-IH), Bone Marrow Transplant., 30 (4), 215-222.
19. Le Blanc, K., Rasmussen, L, Sundberg, B., et al. (2004), Treatment of severe acute graftversus-host
disease with third party haploidentical mesenchymal stem cells, Lancet, 1, 363 (9419), 1439—1441.
20. Lazarus, H. M., Кос, О. N., Devine, S. M., et al. (2005), Cotransplantation of HLA - identical sibling
culture-expanded mesenchymal Stem Cells and hematopoietic Stem Cells in hematologic malignancy
patients, Biol. Blood Marrow Transplant., 11 (5), 389—398.
21. Ringden, O., Uzunel, M., Rasmusson, I., et al. (2006), Mesenchymal Stem Cells for treatment of
therapy-resistant graft-versus-host disease, Transplantation, 81 (10), 1390-1397.
22. Dominici, M., Le Blanc, K., Mueler, I., et al. (2006), Minimal criteria for defi ning multipotent mesen-
chymal stromal cells. The International Society for Cellular Therapy position statement, Cytotherapy,
8(4), 315-317.
23. Schallmoser, K., Bartmann, C., Rohde, E., et al. (2007), Human platelet lysate can replace fetal bovine
serum for clinical scale expansion of functional MSC, Transfusion. 2007 Aug; 47 (8), 1436-1446.
24. Doucet, C., Emou, I., Zhang, Y. Z., et al. (2005), Platelet lysates promote mesenchymal stem cell
expansion: A safety substitute for animal serum in cell-based therapy applications. J. Cell. Physiol.,
205 (2), 228-236.
Глава 1.4. Культивирование мультипотентных мезенхимальных стромальных клеток... 155
25. Digirolamo, С. М., Stokes, D., Colter, D., Phinney, D. G., Class, R., and Prockop, D. J. (1999),
Propagation and senescence of human marro stromal cells in culture: A simple colony-forming assay
identifi es samples with the greatest potential to propagate and differentiate, Br J. Haematol., 107 (2),
275-281.
26. Colter, D. C., Class, R., DiGirolamo, С. M., Prockop, D. J. (2000), Rapid expansion of recycling Stem
Cells in cultures of plastic-adherent cells from human bone marrow, Proc. Nat. Acad. Sci. USA, 97
(7), 3213-3218.
27. Colter, D. C., Sekiya, I., and Prockop, D. J. (2001), Identifi cation of a subpopulation of rapidly self-
renewing and multipotential adult Stem Cells in colonies of numan marrow stromal cells, Proc. Nat.
Acad. Sci. USA, 98 (14), 7841-7845.
28. Sekiya, I., Larson, B. L., Smith, J. R , Pochampally, R., Cui, J. G., and Prockop, D. J. (2002), Expan-
sion of human adult Stem Cells from bone marrow stroma: Conditions that maximie the yields of early
progenitors and evaluate their quality, Stem Cells, 20 (6), 530-541.
29. Bartmann, C., Rohde, E., Schallmoser, K., et al. (2007), Two steps to functional MSC for clinical ap-
plication, Transfusion. 2007 Aug; 47 (8), 1426-1435.
30. Horwitz, E. M., Gordon, P. L., Koo, W. K., et al. (2002), Isolated allogeneic bone marrow-derived
mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implica-
tions for cells therapy of bone, Proc. Natl. Acad. Sci. USA, 99 (13), 8932-8937.
31. Pollard, J. W. (1989), Basic cell culture, in Pollard, J. W., Ed., Animal Cell Culture, Human, Clifton,
NJ, pp. 1—2.
32. WHO (1997), Medicinal and other products and human and animal transmissible spongiform enceph-
alopathies: Memorandum from a WHO meeting, Bull. World Health Org., 75 (6), 505-513.
33. European Union (2004), Note for guidance on minimising the risk of transmitting animal spongiform
encephalopathy agents via human and veterinary medicinal products (EMEA/410/01 Rev. 2 — Octo-
ber 2003) adopted by the Committee for Proprietary Medicinal Products (CPMP) and by the Commit-
tee for Veterinary Medicinal Products (CVMP), Off. J. Europ. Union, C24/26-C24/19.
34. Schallmoser, K., Rosin, C., Vormittag, R., et al. (2006), Specifi cities of platelet autoantibodies and
platelet activation in lupus anticoagulant patients: A relation to their history of thromboembolic dis-
ease, Lupus, 15 (8), 507—514.
35. Wagner, W., Wein, E, Seckinger, A., et al. (2005), Comparative characteristics of mesenchymal Stem
Cells from human bone marrow, adipose tissue, and umbilical cord blood, Exp. HematoL, 33 (11),
1402-1416.
36. Ramalho-Santos, M., Yoon, S., Matsuzaki, Y, Mulligan, R. C., and Melton, D. A. (2002), “Sternness”
: Transcriptional profi ling of embryonic and adult Stem Cells, Science, 298 (5593), 597-600.
37. Boquest, A. C., Shahdadfar, A., Fronsdal, K., et al. (2005), Isolation and transcription profi ling of pu-
rifi ed uncultured human stromal stem cells: Alteration of gene expression after in vitro cells culture,
Mol. Biol. Cell, 16 (3), 1131-1141.
38. Shahdadfar, A., Fronsdal, K., Haug, T., Reinholt, F. P., and Brinchmann, J. E. (2005), In vitro expan-
sion of human mesenchymalstem cells: Choice of serum is a determinant of cell proliferation, dif-
ferentiation, gene expression, and transcriptome stability, Stem Cells, 23 (9), 1357—1366.
39. Silva, W. A., Jr., Covas, D. T, Panepucci, R. A., et al. (2003), The profi le of gene expression of human
marrow mesenchymal stem cells, Stem Cells, 21 (6), 661-669.
40. Panepucci, R. A., Siufi, J. L., Silva, W. A., Jr., et al. (2004), Comparison of gene expres-
sion of umbilical cord vein and bone marrow-derived mesenchymal stem cells, Stem Cells,
22 (7), 1263-1278.
41. Jeong, J. A., Hong, S. H., Gang, E. J., et al. (2005), Differential gene expression profi ling of hu-
man umbilical cord blood-derived mesenchymal stem cells by DNA microarray, Stem Cells, 23 (4),
584-593.
42. Monticone, M., Liu, Y, Tonachini, L., et al. (2004), Gene expression profi le of human
bone marrow stromal cells determined by restriction fragment differential display analysis,
J. Cell. Biochem, 92 (4), 733—744.
156 Часть I. Нормативное регулирование производства лекарственных средств
43. Puck, Т. Т., and Marcus, Р. I. (1956), Action of х - rays on mammalian cells. J. Exp. Med., 103 (5),
653-666.
44. Tepperman, A. D., Curtis, J. E., and McCulloch, E. A. (1974), Erythropietic colonies in cultures of
human marrow, Blood, 44 (5), 659-669.
45. Pittenger, M. E, Mackay, A. M., Beck, S. C., et al. (1999), Multilneage potential of adult human mes-
enchymal stem cells, Science, 284 (5411), 143-147.
46. Pittenger, M. E, and Martin, В. J. (2004), Mesenchymal Stem Cells and their potential as cardiac
therapeutics, Circ. Res., 95 (1), 9—20.
47. Delorme, В. C. S., and Charbord, P. (2006), The concept of mesenchymal Stem Cells, Regenerative
Med., 1 (4), 497-509.
48. Di Nicola, M., Carlo-Stella, C., Magni, M., et al. (2002), Human bone marrow stromal cells suppress
T-lymphocyte proliferation induced by cellular or nonspecifi c mitogenic stimuli, Blood, 99 (10),
3838-3843.
49. Bartholomew, A., Sturgeon, C., Siatskas, M., et al. (2002), Mesenchymal stem cells suppress lympho-
cyte proliferation in vitro and prolong skin graft survival in vivo, Exp. Hematol., 30 (1), 42—48.
50. Le Blanc, K., Tammik, L., Sundberg, B., Haynesworth, S. E., and Ringden, O. (2003), Mesenchymal
Stem Cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently
of the major histocompatibility complex, Scand. J. Immunol., 57 (1), 11-20.
51. Tse, W. T, Pendleton, J. D., Beyer, W. M., E galka, M. C., and Guinan, E. C. (2003), Suppression of
allogeneic T-cell proliferation by human marrow stromal cells: Implications in transplantation, Trans-
plantation, 75 (3), 389-397.
52. Muller, I., Kordowich, S., Holzwarth, C., et al. (2006), Animal serum-free culture conditions for isola-
tion and expansion of multipotent mesenchymal stromal cells from human BM, Cytotherapy, 8 (5),
437-444.
53. Reinisch, A., Bartmann, C., Rohde, E. S. K., Bjelic-Radisic, V., Lanzer, G., Linkesch, W., and Strunk,
D. (2007), A humanized system to propagate cord blood - derived mesenchymal Stem Cells for clini-
cal application, Regen. Med. 2007 Jul; 2 (4), 371-382.
54. Robinson, S. N., Ng, J., Niu, T., et al. (2006), Superior ex vivo cord blood expansion following co-
culture with bone marrow - derived mesenchymal Stem Cells, Bone Marrow Transplant., 37 (4),
359-366.
55. Speicher, M. R., and Carter, N. P. (2005), T he new cytogenetics: Blurring the boundaries with mo-
lecular biology, Nat. Rev. Genet., 6 (10), 782-792.
56. Rubio, D., Garcia-Castro, J., Martin, M. C., et al. (2005), Spontaneous human adult stem cell trans-
formation, Cancer Res., 65 (8), 3035—3039.
57. Tolar, J., Nauta, A. J., Osbom, M. J., et al. (2007), Sarcoma derived from cultured mesenchymalstem
cells, Stem Cells, 25 (2), 371-379.
Часть 2
МЕЖДУНАРОДНЫЕ ПРАВИЛА НАДЛЕЖАЩЕЙ
ПРОИЗВОДСТВЕННОЙ ПРАКТИКИ
Глава 2.1. Национальные правила и требования
GMP, международные требования GMP
и руководящие документы: совпадения
и различия
Марко Нярхи, Катрина Нордстрём
Технологический университет Хельсинки (Хельсинки, Финляндия)
2.1.1. Введение
Впервые правила производства продукции и требования к качеству, впоследствии
трансформировавшиеся в Правила надлежащей производственной практики, были
опубликованы в США Федеральным управлением США по контролю за пищевой
продукцией и лекарствами (FDA) в 1940-х гг. [1]. На общем собрании Всемирной
организации здравоохранения (ВОЗ), созванном в 1969 г., Всемирная ассамблея
здравоохранения выпустила рекомендации по внедрению правил надлежащей про-
изводственной практики (GMP—good manufacturing practice) [2]. С тех пор в странах
с наиболее развитой промышленностью изданы законы о процедурах контроля, не-
обходимых при изготовлении лекарственной продукции. В некоторых странах GMP
встроены в национальные законодательства в виде законов или правил регулирова-
ния производства, распределения, маркетинга и применения лекарственных про-
дуктов (руководства GMP). В других странах GMP представляют собой отдельные
руководящие документы, не включенные в национальное законодательство по ле-
карственной продукции (Правила GMP). Кроме того, в дополнение к националь-
ным GMP, некоторые международные организации и торговые союзы выпустили
собственные международные правила СМРрля. гармонизации требований к лекар-
ственной продукции, существующих в разных странах. Однако, несмотря на причи-
ны появления правил GMP, их основная задача состоит в обеспечении соответствия
параметров безопасности, идентичности, эффективности и качества произведен-
ных лекарственных продуктов заявленным значениям [3]. Для достижения данной
цели большинство правил GMP, как правило, охватывают вопросы, связанные с си-
стемой управления качеством, персоналом, недвижимостью, оборудованием, доку-
ментацией, управлением материалами, производственным контролем и контролем
технологических процессов, упаковкой и маркировкой промежуточных и готовых
продуктов, лабораторным контролем, валидацией и контролем изменений [4].
2.1.2. Национальные правила и требования GMP
2.1.2.1. Соединенные Штаты Америки
В Соединенных Штатах производство лекарственной продукции регламентирует
Федеральный закон о пищевых продуктах, лекарствах и косметике (Food, Drug and
Cosmetic Act). Закон устанавливает, что лекарственный продукт считается неконди-
160
Часть 2. Международные правила надлежащей производственной практики
ционным, если методы или оборудование, или система контроля, используемая для
его производства, обработки, упаковки или хранения не соответствуют правилам
GMP, не функционируют, или не применяются в соответствии с текущими прави-
лами GMP [5]. Действующие правила GMP представлены в виде части Свода феде-
ральных нормативных актов (CFR — Code of Federal Regulations) и, таким образом,
являются федеральными законами. Действующий свод правил GMP основан на
пересмотренных в 1978 г. [6, 7] изначальных требованиях GMP, впервые опубли-
кованных в 1963 г. Правила GMP пересматривают ежегодно в апреле [8]; одна-
ко с 1978 г. существенных изменений внесено не было. FDA публикует в качестве
дополнения к правилам GMP также другие руководящие документы, связанные
с GMP, относящиеся к различным аспектам производства лекарств [9]. С другой
стороны, несмотря на то, что эти документы отражают текущее видение проблем
и ожидания Управления, они лишь представляют собой руководство по принципам
и практикам, которое не является сводом законных нормативов [1]. Как страна-
участник Международной конференции по гармонизации технических требова-
ний к регистрации фармацевтических продуктов, используемых человеком (ICH—
International Conference on Harmonization of the Technical Requirements for Registration
of Pharmaceuticals for Human Use), США приняла руководящий документ ICH QI —
«Руководство по надлежащей производственной практике для активных фармацев-
тических ингредиентов». Он был опубликован в качестве руководящего документа
для промышленности США [10].
Правила GMP США содержатся в двух главах CFR: 210 и 211. Глава 210 «Теку-
щая надлежащая производственная практика в производстве, обработке, упаковке
и хранении лекарств — Общая часть» представляет общие подходы [6], а глава 211
«Текущая надлежащая производственная практика для готовых фармацевтических
продуктов» формулирует действующие требования. Далее, глава 211 делится на
11 подглав, охватывающих требования к персоналу, недвижимости, оборудованию,
контролю материалов, контролю продукции и технологических процессов, упаков-
ке и маркировке, хранению и распределению, лабораторному контролю, докумен-
тации, а также возвращенным повторно выпущенным в обращение лекарственным
продуктам [7]. Содержание главы 211 представлено в табл. 1.
2.1.2.2. Канада
В Канаде производство лекарственной продукции (лекарств) регулирует закон
о пищевых продуктах и лекарствах, который устанавливает, что дистрибьюторы
и импортеры не имеют права продавать лекарственный продукт, если он не про-
изведен в соответствии с требованиями GMP. Принципы GMP изложены в разде-
ле 2 (Division 2) части С (Part С) Нормативных требований к пищевым продуктам
и лекарствам, являющейся частью Закона о пищевых продуктах и лекарствах [11].
Отраслевой инспекторат по медицинским и пищевым продуктам также выпустил
руководящий документ (Правила GMP), подготовленный для оказания помощи
в разъяснениях преписаний GMP. Действующие в настоящее время Правила GMP
Канады были опубликован в 2002 г. и с тех пор не пересматривались. Они были раз-
работаны с целью согласования со стандартами GMP других стран и международных
организаций (ВОЗ, Программа сотрудничества фармацевтических инспекгоратов
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 161
Таблица 1 Содержание главы 211 правил GMP США
Раздел Предмет
Подглава А Подглава В Подглава С Подглава D Подглава Е Подглава F Подглава G Подглава Н Подглава I Подглава J Подглава К Общие вопросы Организация и персонал Здания и помещения Оборудование Контроль компонентов, первичной упаковки и укупорочных средств Производство и контроль процесса Упаковка и итоговый контроль продукта Хранение и поставки Контроль качества Документация Возвращенные некачественные лекарственные препараты и лекарственные препараты с нарушением условий хранения
(PIC/S— Pharmaceutical Inspection Cooperation Scheme), ICH). Органы здравоохране-
ния Канады также опубликовали несколько приложений к основному своду Правил
GMP, охватывающему такие темы, как СМРцля медицинских газов, биологические
лекарственные продукты, препараты крови и производство новых медицинских
препаратов для клинических исследований. В дополнение к Правилам GMP и их
приложениям канадские власти выпустили несколько других специальных руко-
водств, освещающих вопросы, относящиеся к GMPvi способам производства [12].
Таблица 2
Содержание Правил GMP Канады [12]
Введение
Управление качеством
Глоссарий терминов
Нормативы
Здания и помещения
Оборудование
Персонал
Санитарный надзор
Анализ исходных материалов
Контроль производственного процесса
Отдел контроля качества
Тестирование упаковочных материалов
Анализ готовой продукции
Записи
Образцы
Стабильность
Стерильные продукты
Медицинские газы
Приложение А: Международные гармонизированные требования к сертификации серий
Приложение В: Заявка на хранение архивных образцов
Приложение С; Ссылки
162 Часть 2. Международные правила надлежащей производственной практики
Как показано в табл. 2, в Правилах GMP Канады можно выделить четыре главы
и приложения. Первые три главы посвящены общим вопросам, таким как границы
и область применения Правил GMP, определение используемых терминов, а также
вопросы, относящиеся к управлению качеством и общим требованиям GMP. Ин-
струкции GMP и их применение изложены в четвертой главе (нормативы), разде-
ленной на 14 подразделов (subchapters), описывающих требования к зданиям и по-
мещениям, оборудованию, персоналу, санитарным условиям, анализу компонентов
и упаковочных материалов, анализу готовой продукции, производственному кон-
тролю, отделу контроля качества, документации, архивным образцам, исследова-
нию стабильности и производству стерильных лекарственных продуктов и меди-
цинских газов. Каждый раздел содержит нормативы, соответствующие положениям
раздела 2 (Division 2) [ 11 ], снабженные разъяснением для применении. Приложения
включают требования к сертификации производственных серий, форму заявки на
хранение архивных образцов и ссылки, например гиперссылки на законы Канады
о лекарственных продуктах и другие национальные и международные руководства,
связанные с предписаниями GMP [12].
2.1.2.3. Европейский Союз
Производство лекарственной продукции (медицинской продукции) в Европей-
ском Союзе (EU) регулируется директивой 2001/83/ЕС Европейского парламента
и Совета, которая устанавливает: владелец лицензии на производство медицинской
продукции должен обеспечить соответствие правилам надлежащей производствен-
ной практики в соответствии с Законом Европейского Сообщества [13]. Принци-
пы и правила GMP для медицинской продукции изложены в директиве Комиссии
2003/94/ЕС, обеспечивающей законодательную базу для GMPv, ЕС [14]. Действую-
щие требования GMP с подробным описанием существующих процедур опубли-
кованы в томе 4 Правил, регулирующих медицинскую продукцию в Европейском
Союзе (The Rules Governing Medicinal Products in the European Union). Действующие
требования Правил GMP EC были впервые введены в 1989 г.; они состоят из девяти
глав, охватывающих общие требования GMP, и одного приложения, относящего-
ся к производству стерильных лекарственных продуктов. С тех пор Правила GMP
ЕС были неоднократно пересмотрены и к ним было добавлено несколько новых
приложений [15]. В дополнение к Правилам GMPEC опубликовал также несколь-
ко других руководств, посвященных вопросам качества лекарственной продук-
ции в Правилах, регулирующих медицинскую продукцию в Европейском Союзе,
том ЗА [16].
Как показано в табл. 3—5, Правила GMP ЕС представлены двумя частями основ-
ных правил и 18 приложениями. Часть I (Part I) «Основные требования к меди-
цинской продукции» охватывает принципы GMP при производстве лекарствен-
ных продуктов. Она состоит из девяти глав (chapters), описывающих требования
к управлению качеством и контролю, персоналу, недвижимости, оборудованию,
производству, обслуживанию по контракту, претензиям, отзывам продукта и само-
контролю. Часть II «Основные требования к активным веществам, используемым
в качестве исходных материалов» охватывает правила GMP для активных веществ,
используемым в качестве исходных материалов. Нормативы основаны на докумен-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 163
Таблица 3
Содержание части I Правил GMPTC, описывающей основные требования к производству
лекарственных продуктов [15]
Раздел________________________________Предмет______________________________
Введение
Глава 1 Управление качеством
Глава 2 Персонал
Глава 3 Здания и помещения
Глава 4 Документация
Глава 5 Производство
Глава 6 Контроль качества
Глава 7 Контрактное производство и анализ
Глава 8 Претензии и отзыв продукции
Глава 9 Самоинспекция
Глоссарий
Таблица 4
Содержание части II Правил GMP ЕС, описывающей основные требования к производству
активных веществ, используемых в качестве исходных материалов [15]
Раздел Предмет
1 Введение
2 Управление качеством
3 Персонал
4 Здания и помещения
5 Производственное оборудование
6 Документация и записи
7 Управление материалами
8 Производство и оперативный производственный контроль
9 Упаковка и идентификационная маркировка API и промежуточных
продуктов
10 Хранение и распределение
11 Лабораторный контроль
12 Валидация
13 Контроль изменений
14 Отбраковка и повторное использование материалов
15 Претензии и отзывы
16 Производители, привлеченные по контракту (включая лаборатории)
17 Агенты, брокеры, коммерсанты, дистрибьюторы, переупаковщики
и перемаркировщики
18 Особые указания для API, произведенных путем культивирования клеток/
ферментации
19 APTдля применения в клинических испытаниях
20 Глоссарий
164 Часть 2. Международные правила надлежащей производственной практики
Таблица 5
Приложения к Правилам GMP ЕС, относящиеся к конкретным руководящим документам
Раздел Предмет
Приложение 1 Приложение 2 Производство стерильных медицинских продуктов Производство биологических медицинских продуктов, предназначенных для человека
Приложение 3 Приложение 4 Производство радиофармацевтических препаратов Производство ветеринарных медицинских продуктов (за исключением иммунологических ветеринарных продуктов)
Приложение 5 Приложение 6 Приложение 7 Приложение 8 Приложение 9 Приложение 10 Производство иммунологических ветеринарных медицинских продуктов Производство медицинских газов Производство растительных медицинских продуктов Отбор образцов исходных и упаковочных материалов Производство жидкостей, кремов и мазей Производство аэрозольных препаратов под давлением с системой дозирования для ингаляций
Приложение 11 Приложение 12 Компьютеризованные системы Применение ионизирующего излучения при производстве медицинских продуктов
Приложение 13 Приложение 14 Производство медицинских препаратов для клинических исследований Производство продуктов, полученных из крови или плазмы крови человека
Приложение 15 Приложение 16 Приложение 17 Приложение 18 Квалификация и валидация Сертификация уполномоченным лицом и выпуск серии в обращение Выпуск по параметрам Стандартные и архивные образцы
те ICH Q1 «Руководство по надлежащей производственной практике для актив-
ных фармацевтических ингредиентов» (Good Manufacturing Practice Guide for Active
Pharmaceutical Ingredients), первоначально введенном в действие в 2001 г. в виде
Приложения 18 Правил GMPEC. В пересмотренных Правилах б-Л/РЕС, опубли-
кованных в октябре 2005 г., Приложение 18 заменили частью II. Часть II состо-
ит из 19 глав, охватывающих основные вопросы, связанные с GMP, относящиеся
к управлению качеством, персоналу, недвижимости, оборудованию, документа-
ции, материалам, контролю продукции и технологических процессов, упаковке
и маркировке, хранению и распределению, лабораторному контролю, валидации,
контролю изменений, претензиям, отзывам, контрактным службам, сотрудникам,
активным фармацевтическим ингредиентам (API— active pharmaceutical ingredient),
произведенным путем культивирования/ферментации и API, применяемым в кли-
нических испытаниях. В приложениях представлены специфические требования
к производству стерильных и биологических лекарственных продуктов, радиофар-
мацевтических препаратов, ветеринарных лекарственных продуктов, медицин-
ских газов, растительных лекарственных продуктов, жидкостей для перорального
применения, препаратов для наружного применения (кремов, мазей), аэрозолей,
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 165
новых медицинских препаратов для клинических исследований, а также крови
и препаратов крови. Они относятся также к отбору проб материалов, компьюте-
ризованным системам, применению ионизирующих излучений, квалификации
и валидации, выпуску производственных серий, выпуску по параметрам, ссылкам
и архивным материалам [15].
2.1.2.4. Страны Восточной Азии
Япония. В Японии производство лекарственной продукции (лекарств) регулирует-
ся Законом о фармацевтике {PAL — Pharmaceutical Affairs Law), которым установ-
лено, что все производители лекарств, планирующие производство лекарственных
продуктов для продажи в Японии, должны обладать японской лицензией на про-
изводство лекарств и обеспечить соответствие правилам GMP Японии. Правила
GMPn Японии были впервые введены в 1974 г. под названием Стандарты производ-
ственного контроля и контроля качества {The Standards for Manufacturing Control and
Quality Control). В 1979 г. PAL был частично пересмотрен, и правила GMPприобрели
статус закона [2].
Контроль за соблюдением PAL и принуждение к его соблюдению осуществля-
ется путем административных распоряжений и замечаний, представляющих со-
бой подробные нормативы, подготовленные правительством Японии. Требования
к объектам недвижимости, используемым для производства лекарственных про-
дуктов, представлены в указе Министерства здравоохранения, труда и социального
обеспечения {MHLW— Ministry of Health, Labor and Welfare) № 73, 2005 г. «Норма-
тивы для зданий и помещений фармацевтического назначения и т. д.» (изначально
указ MHLW№ 2, 1961 г.) [17]. Правила производства и контроля — в указе MHLW
№ 95, 2003 г. «Правила производственного контроля и контроля качества лекарств»
(изначально указ MHLW№ 3, 1994 г.). Как страна — участник ICHЯпония приня-
ла руководящий документ ICH Q1 — «Надлежащая производственная практика для
активных фармацевтических ингредиентов». Этот документ был опубликован как
уведомление генерального директора Бюро безопасности пищевых и фармацевти-
ческих продуктов {Pharmaceutical and Food Safety Bureau) № 1200,2001 г., озаглавлен-
ное «Руководство по GMP для лекарственных веществ, устанавливающее требования
для производства API». Требования, касающиеся импортируемых лекарственных
продуктов, представлены в указе MHLW№ 97, 2003 г. «Руководство по импорту/
розничной торговле и контролю качества лекарств и парамедицинских продуктов»
(изначально указ MHLW№ 62, 1999 г.). Требования, относящиеся к производству
препаратов для клинических исследований, представлены в Уведомлении № 480,
1997 г. «Продукты и стандарты для зданий и помещений производственных пред-
приятий по изготовлению препаратов для клинических исследований {GMP пре-
паратов для клинических исследований)» [2].
Южная Корея. Производство лекарственной продукции (лекарств) в Южной Ко-
рее регулирует Закон о фармацевтике, впервые введенный в действие в 1953 г.; с тех
пор этот закон неоднократно пересматривался [18]. Регистрация новых лекарств
и связанные с этим процедуры во многом аналогичны существующим в США
и Японии. GMP Кореи (зачастую называемые KGMP) появились в 1984 г. и стали
обязательными в 1995 г. [19]. Производитель лекарств, намеренный выпускать ле-
166 Часть 2. Международные правила надлежащей производственной практики
карственный продукт для продажи в Корее, должен получить одобрение уполно-
моченного Управлением по контролю за пищевой продукцией и лекарствами Кореи
(KFDA). Чтобы получить лицензию, производитель должен доказать соответствие
производственных стандартов правилам KGMP [20].
Китай. Китай регулирует производство лекарственных продуктов (лекарств)
при помощи Drug Administration Law — Закона Китайской Народной Республики об
управлении лекарственными средствами, который устанавливает, что производитель
должен вести производство в соответствии с правилами GMP для фармацевтиче-
ских продуктов, составленными Департаментом регулирования лекарств при Го-
сударственном Совете на базе Закона об управлении лекарственными средствами
[21]. В июне 2004 г. правила GMP стали обязательными для исполнения в Китае,
и государственное Управление по лекарственным продуктам объявило, что местные
организации, занимающиеся производством лекарств, не имеющие утвержденных
сертификатов соответствия GMP, не получат разрешения на продолжение произ-
водства лекарственной продукции [22].
2.1.2.5. Индия
Производство лекарственной продукции (лекарств) в Индии регулируют Прави-
ла производства лекарств и косметики (утверждены в 1945 г., последние поправки
внесены в 2005 г.), согласно которым требуется, чтобы держатель лицензии на про-
изводство лекарств обеспечил соответствие производства требованиям GMP, изло-
женным в Программе М (Schedule М) [23]. Программа М представляет собой часть
Правил производства лекарств и косметики, включающую правила GMP Индии
[24], основанные на правилах GA/PBO3 версии 1982 г. [25].
Как показано в табл. 6—8, Правила GMPИндии состоят из восьми частей (parts)-.
I, IA, IB, IC, ID, IE, IF и II. Часть I охватывает общие правила GMP. Она разделена
на 29 глав (chapters), в которых описаны требования к персоналу, объектам недвижи-
мости, оборудованию, санитарному состоянию, контролю продукции и технологи-
ческих процессов, материалам, документации, управлению качеством, валидации,
архивным образцам, отзывам, претензиям и самоконтролю. В частях IA—IE опи-
саны специфические требования к производству различных дозированных форм,
относящиеся к объектам недвижимости (зданиям и помещениям), оборудованию
и методам. Часть IA описывает требования к производству парентеральных пре-
паратов; часть IB — требования к производству твердых дозированных форм для
перорального применения, таких как таблетки и капсулы; часть IC — требования
к производству жидкостей для перорального применения, таких как сиропы, элик-
сиры, эмульсии и суспензии; часть ID — требования к производству препаратов
для наружного применения, таких как кремы, мази, пасты, эмульсии и лосьоны;
часть IE — требования к производству препаратов для ингаляции. В части IF изло-
жены специфические требования к производству API (активных фармацевтических
ингредиентов), относящихся к зданиям и помещениям, системам инженерного обе-
спечения, оборудованию, контролю и контейнерам. Часть II Правил GMP Индии
состоит из подробных рекомендаций по технологическому оборудованию, приме-
няемому при производстве различных дозированных форм и требований к разделе-
нию производственной зоны [24].
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 167
Таблица 6
Содержание части I Правил GMP Индии, относящихся к надлежащей производственной
практике для объектов недвижимости и материалов [24]
Раздел Предмет
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 Общие требования Складские территории Зона производства Вспомогательные зоны Зона контроля качества Персонал Здоровье, одежда работников и санитарные требования к работникам Производственные операции и контроль Соблюдение санитарных требований в производственных помещениях Исходные материалы Оборудование Документация и записи Этикетки и прочие печатные материалы Обеспечение качества Самоинспекция и аудит качества Система контроля качества Спецификация Записи по производственному регламенту Записи по операциям упаковки Записи по упаковке серии Записи по обработке производственной серии Стандартные операционные процедуры — СОП {standard operating procedures) и записи
23 24 25 26 27 28 29 Стандартные образцы Повторная обработка и восстановление Записи по распределению Валидация и валидация процессов Отзыв продукции Претензии и сведения о побочных эффектах Досье производственного участка
2.1.2.6. Австралия
В Австралии производство лекарственной продукции (лекарств) регулируется За-
коном о продукции терапевтического назначения, наделяющим министра здраво-
охранения и социального обеспечения правом утверждать письменные принципы,
включая правила GMP, которые следует соблюдать при производстве лекарствен-
ных продуктов, предназначенных для употребления человеком [26]. В изданном ми-
нистром приказе № 2 от 2002 г. о товарах терапевтического назначения (принципы
производства) установлено, что лекарственные продукты должны производиться
168 Часть 2. Международные правила надлежащей производственной практики
Таблица 7
Содержание частей ТА, IB, IC, ID, IE и IF Правил GMP Индии, описывающих специфические
руководящие указания
Раздел________________________________Предмет______________________________
Часть IA Специфические требования к производству стерильных продуктов,
парентеральных препаратов (составов для инъекций в малых объемах и составов
для парентерального введения в больших объемах), а также стерильных
офтальмологических препаратов
Часть ТВ Специфические требования к производству твердых дозированных форм
(таблеток и капсул)
Часть ТС Специфические требования к производству жидких лекарственных форм для
перорального применения (сиропов, эликсиров, эмульсий и суспензий)
Часть ID Специфические требования к производству продуктов для местного
применения, т. е. препаратов для наружного применения (кремов, мазей, паст,
эмульсий, лосьонов, растворов, порошков и т. п.)
Часть ТЕ Специфические рекомендации по производству дозированных порошковых
ингаляторов (MDI— metered-dose inhaler)
Часть IF Специфические требования к зданиям, помещениям и материалам для
производства активных фармацевтических ингредиентов (нерасфасованных
лекарств)
Таблица 8
Содержание части II Правил GMP Индии, охватывающих требования к производственному
предприятию и оборудованию [24]
Раздел________________________________Предмет________________________________
I Препараты для наружного применения
2 Жидкие препараты для перорального применения
3 Таблетки
4 Порошки
5 Капсулы
6 Хирургические перевязочные материалы
7 Офтальмологические препараты
8 Пессарии и суппозитории
9 Препараты для ингаляций
10 Переупаковка лекарств и фармацевтических веществ
11 Парентеральные препараты
в соответствии с Правилами Австралии о надлежащей производственной практике
для медицинских продуктов, датированным 16 августа 2002 г. [27]. Содержание дей-
ствующих Правил GMP Австралии полностью основано на руководящем документе
GMP Р/С/5 версии PH 1/97, который был опубликован с небольшими изменениями
в 2002 г. [28].
Как показано в табл. 9, Правила GMP Австралии состоят из 9 глав (chapters)
и 13 приложений. В главах представлены общие правила СЛ/Рпри производстве
лекарственных продуктов, требования к управлению качеством и контролю каче-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 169
ства, персоналу, объектам недвижимости, оборудованию, документации, произ-
водству, контрактным службам, претензиям, отзывам продукции и самоконтролю.
В приложениях представлены специфические требования, которые необходимо со-
блюдать при производстве стерильных лекарственных продуктов, биологических
лекарственных продуктов, радиофармацевтических препаратов, растительных ле-
карственных продуктов, жидкостей для перорального введения, препаратов для на-
ружного применения (кремов и мазей), аэрозолей, новых медицинских препаратов
для клинических исследований, крови и препаратов крови. Кроме того, главы охва-
тывают вопросы, связанные с отбором проб материалов, компьютеризованными
системами, применением ионизирующего излучения, квалификации, валидации
и выпуска по параметрам [28].
Таблица 9 Содержание Правил GMP Австралии [28]
Раздел Предмет Введение Разъяснения
Глава 1 Глава 2 Глава 3 Глава 4 Глава 5 Глава 6 Глава 7 Глава 8 Глава 9 Приложение 1 Приложение 2 Управление качеством Персонал Здания и помещения Документация Производство Контроль качества Контрактное производство и анализ Претензии и отзыв продукции Самоинспекция Производство стерильных медицинских продуктов Производство биологических медицинских продуктов для медицинского применения
Приложение 3 Приложение 6 Приложение 7 Приложение 8 Приложение 9 Приложение 10 Производство радиофармацевтических продуктов Производство медицинских газов Производство лекарственных препаратов растительного происхождения Отбор проб исходных и упаковочных материалов Производство жидкостей, кремов и мазей Производство герметических дозированных аэрозольных препаратов для ингаляций
Приложение 11 Приложение 12 Компьютеризованные системы Применение ионизирующего излучения при производстве медицинских продуктов
Приложение 13 Приложение 15 Приложение 17 Производство лекарственных препаратов для клинических исследований Квалификация и валидация Выпуск по параметрам Глоссарий
170 Часть 2. Международные правила надлежащей производственной практики
Австралия не приняла Приложений 4, 5, 14, 16 и 18 руководящего документа
PIC/S GMP. Приложения 4 и 5 относятся к производству ветеринарных лекарствен-
ных продуктов. Приложение 14, относящееся к производству продуктов, изготав-
ливаемых из крови или плазмы крови человека, не вошло в Правила GMP Австра-
лии. Приложение 16 является специфичным для Правил GMP ЕС, а Приложение 18
представляет собой руководящий документ ICH по GMP при производстве API, ко-
торый Австралия приняла отдельно в качестве принципа производства [28].
2.1.2.7. Новая Зеландия
Производство лекарственной продукции (лекарств) в Новой Зеландии регулируется
Законом о медицине 1981 г., который устанавливает, что производитель лекарств не
имеет права изготавливать лекарственные продукты без лицензии на производство,
выдаваемой лицензирующими органами. Чтобы получить лицензию на производ-
ство заявитель должен доказать лицензирующим органам, что производственные
помещения и оборудование, которыми он располагает, являются приемлемыми
и адекватными для производства лекарств. Также заявитель должен показать, что
созданы надлежащие условия для ведения и обеспечения сохранности надлежащих
записей, относящихся к лекарственным продуктам, которые планируется произ-
водить [29]. Управление по контролю безопасности применения лекарственных
средств и медицинского оборудования Новой Зеландии {Medsafe) требует, чтобы все
производители лекарств, планирующие производство лекарственных продуктов для
продажи в Новой Зеландии, представляли свидетельства соответствия производ-
ственной площадки требованиям GMP. Копии надлежащих сертификатов, лицен-
зии на производство или рапорты органов нормативно-правового регулирования,
компетенция которых признана Medsafe, принимаются в качестве доказательств со-
ответствия Правилам СЛ/Р[30].
Как показано в табл. 10, собственные Правила GMP Новой Зеландии состоят из
пяти частей (parts). Первая часть посвящена производству лекарственных продук-
тов, а вторая часть — производству препаратов крови. Часть 3 описывает процес-
сы смешивания и измельчения, включая смешивание стерильных лекарственных
продуктов. Часть 4 посвящена оптовой торговле, а часть 5 — отзывам продукции.
Части 4 и 5 представляют собой единый документ [31].
Таблица 10
Содержание Правил GMP Новой Зеландии [31]
Раздел________________________________Предмет______________________________
Часть 1 Производство фармацевтических продуктов
Часть 2 Производство крови и препаратов крови
Часть 3 Смешивание и измельчение
Часть 4 Оптовая торговля лекарствами и медицинскими изделиями
Часть 5 Универсальная процедура отзыва лекарств и медицинских устройств
2.1.2.8. Южная Африка
Производство лекарственной продукции (лекарств) в Южной Африке регулирует
Закон о контроле над лекарственными средствами и связанными с ними вещества-
ми (Закон 101,1965 г.). Закон устанавливает, что Совет по контролю лекарств может
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 171
выдать производителю лекарств лицензию на производство лекарственного про-
дукта на условиях обеспечения производителем принципов обеспечения качества
и правил GMP — решение о соответствии принимает Совет [32].
Заявка на получение лицензии включает раздел, в котором производитель дол-
жен представить надлежащие документальные доказательства способности обеспе-
чить соответствие Правилам GMP—решение о соответствии принимает Совет [33].
Действующие Правила GMP Южной Африки, принятые Советом, целиком осно-
ваны на руководящем документе PIC/S по GMP версии РЕ 009—2, опубликованном
с небольшими изменениями в 2004 г. [34].
Как показано в табл. 11, Правила GMP Южной Африки состоит из 9 глав
(chapters) и 17 приложений. В главах представлены общие правила СЛ/Рпри произ-
водстве лекарственных продуктов, охватывающие требования к управлению каче-
ством и контролю качества, персоналу, объектам недвижимости, оборудованию, до-
кументации, производству, контрактным службам, претензиям, отзыву продукции
и самоконтролю. В приложениях описаны специфические указания, относящиеся
к производству стерильных лекарственных продуктов, биологических лекарствен-
ных продуктов, радиофармацевтических препаратов, ветеринарных лекарственных
продуктов, медицинских газов, растительных лекарственных продуктов, жидко-
стей для перорального введения, препаратов для наружного применения (кремов,
мазей), новых медицинских препаратов для клинических исследований, а также
крови и препаратов крови. Они описывают также отбор проб материалов, компью-
теризованные системы, применение ионизирующего излучения, квалификацию
и валидацию, организацию, персонал и выпуск по параметрам. Оригинальное При-
ложение 16, специфичное для Правил GMPEC, в Правилах Южной Африки заме-
нено приложением, относящимся к организации и персоналу. Кроме того, Южная
Африка не приняла Приложения 18, охватывающего руководящие указания ICH,
относящиеся к GMP при производстве API, поскольку оно было принято отдельно
в качестве принципа производства [34].
2.1.3. Международные требования GMPu их координация
2.1.3.1. Всемирная организация здравоохранения (ВОЗ)
В 1948 г. была создана ВОЗ в качестве специализированного агентства Организа-
ции Объединенных Наций (ООН). Задача агентства — управление и координация
международных действий, связанных с охраной здоровья человека и общественного
здоровья. Одной из главных функций ВОЗ является предоставление объективной
и достоверной информации и рекомендаций, связанных со здоровьем человека; эта
задача частично выполняется через публикации ВОЗ [35]. Первый проект текста
ВОЗ, посвященного GMP, был подготовлен в 1967 г., а исправленная версия была
опубликована в 1968 г. в виде приложения кдвадцать второму рапорту комиссии экс-
пертов ВОЗ по спецификациям фармацевтических препаратов. В течение несколь-
ких лет ВОЗ выпустила несколько версий правил GMP, а также других указаний,
связанных с GMP и вопросами качества производства продукции терапевтическо-
го назначения. Последняя версия Руководства по GMP от ВОЗ была опубликована
в 2003 г. в виде приложения к Техническому докладу ВОЗ № 908 [36].
172__________Часть 2. Международные правила надлежащей производственной практики
Таблица 11
Содержание Правил GMP Южной Африки
Раздел Предмет
Введение Разъяснения
Глава 1 Управление качеством
Глава 2 Персонал
Глава 3 Недвижимость и оборудование
Глава 4 Документация
Глава 5 Производство
Глава 6 Контроль качества
Глава? Контрактное производство и анализ
Глава 8 Претензии и отзыв продукции
Глава 9 Самоинспекция
Приложение 1 Производство стерильных медицинских продуктов
Приложение 2 Производство биологических медицинских продуктов для употребления
человеком
Приложение 3 Производство радиофармацевтических продуктов
Приложение 4 Производство ветеринарных медицинских продуктов, отличных от иммунологических продуктов
Приложение 5 Производство иммунологических ветеринарных медицинских продуктов
Приложение 6 Производство медицинских газов
Приложение 7 Производство растительных медицинских продуктов
Приложение 8 Отбор проб исходных и упаковочных материалов
Приложение 9 Производство жидкостей, кремов и мазей
Приложение 10 Производство герметичных дозированных аэрозолей для ингаляций
Приложение 11 Компьютеризованные системы
Приложение 12 Применение ионизирующего излучения при производстве медицинских продуктов
Приложение 13 Производство лекарственных препаратов для клинических исследований
Приложение 14 Производство препаратов крови и плазмы крови человека
Приложение 15 Квалификация и валидация
Приложение 16 Организация и персонал
Приложение 17 Выпуск по параметрам Глоссарий
Как показано в табл. 12, Руководство ВОЗ по GMP разделено на пять частей:
введение, общие принципы, глоссарий, управление качеством в фармацевтической
промышленности, ссылки. Действующие требования GMP представлены в четвер-
той части, состоящей из 17 глав, охватывающих требования к обеспечению и кон-
тролю качества, персоналу, недвижимости, оборудованию, санитарному состоянию,
материалам, валидации, документации, производству, контрактным службам, пре-
тензиям, отзывам и самоконтролю [36]. В дополнение к этим указаниям, излагаю-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 173
Таблица 12
Содержание Руководства ВОЗ по GMP, описывающее общие правила GMP при производстве
лекарственных продуктов [36]
Введение
Общие принципы
Глоссарий
Управление качеством в фармацевтической промышленности: философия и основные
элементы___________________________________________________________________
Раздел_______________________________Предмет_______________________________
1 Обеспечение качества
2 Надлежащая производственная практика при производстве фармацевтических
продуктов
3 Санитария и гигиена
4 Квалификация и валидация
5 Претензии
6 Отзыв продукции
7 Контрактное производство и анализ
8 Самоинспекция и аудиты качества
9 Персонал
10 Обучение
11 Гигиена персонала
12 Здания и помещения
13 Оборудование
14 Материалы
15 Документация
16 Надлежащая производственная практика
17 Надлежащая практика контроля качества
Ссылки
щим основные принципы GMP, ВОЗ опубликовала несколько руководящих доку-
ментов, описывающих специфические требования к компонентам, качеству воды
для применения в фармацевтических целях, API, вспомогательным веществам,
стерильным лекарственным продуктам, биологическим лекарственным продуктам,
препаратам для клинических исследований, растительным лекарственным продук-
там и радиофармацевтическим препаратам (табл. 13).
2.1.3.2. Программа сотрудничества фармацевтических инспекторатов
(Р/С/5)
Конвенция по фармацевтическим инспекциям (PIC — Pharmaceutical Inspection
Convention), являющаяся предшественницей PIC/S, создана в 1970 г. в Европейской
зоне свободной торговли (EFTA — European Free Trade Area). Первоначально в состав
конвенции входили 10 стран — участниц EFTA. С самого начала одной из основных
целей организации стало согласование правил GMP, а также продвижение процес-
са взаимного признания инспекций и установления единообразия инспекционных
174 Часть 2. Международные правила надлежащей производственной практики
Таблица 13
Документы ВОЗ, содержащие специфические требования, связанные с GMP
________Документ______
7X9929, Приложение 2 [37]
7X9823, Приложение 1 [38]
TRS885, Приложение 5 [39]
TRS9C1, Приложение 6 [40]
7719834, Приложение 3 [41]
77J5863, Приложение 7 [42]
7X9863, Приложение 8 [43]
7X5'908, Приложение 3 [44]
7X9929, Приложение 3 [45]
_______________________Предмет_____________________
Правила отбора исходных материалов
Активные фармацевтические ингредиенты
(нерасфасованные лекарственные вещества)
Фармацевтические вспомогательные вещества
Стерильные фармацевтические продукты
Биологические продукты
Фармацевтические продукты для клинических испытаний
Растительные медицинские продукты
Радиофармацевтические продукты
Вода для использования в фармацевтике
систем путем усовершенствования системы обучения инспекторов, улучшения об-
мена информацией и роста взаимного доверия [46]. Первоначально PIC являлась
формальным договором между странами — участницами, и в таком виде также об-
ладала статусом закона. Когда неевропейские страны изъявили желание присоеди-
ниться к PIC, оказалось, что согласно европейскому законодательству отдельные
страны ЕС, участвующие в PIC, не имеют права подписывать соглашения с неевро-
пейскими странами. Только Европейской Комиссии, которая сама по себе не являет-
ся участницей PIC, было разрешено подписывать соглашения. Поэтому была пред-
ложена более гибкая и менее формальная PIC/S, позволившая продолжить работу
PIC. PIC/S, действующая с ноября 1995 г., представляет собой неформальное согла-
шение, не обладающее статусом закона, между нормативно-правовыми органами (а
не странами — участницами). PICvt PIC Scheme, работающие совместно как PIC/S,
обеспечивают активное и конструктивное сотрудничество в области GMP [47].
В настоящее время странами — участницами PIC/S являются Австралия, Ав-
стрия, Бельгия, Великобритания, Венгрия, Германия, Греция, Дания, Ирландия,
Исландия, Испания, Италия, Канада, Латвия, Лихтенштейн, Малайзия, Нидер-
ланды, Норвегия, Польша, Португалия, Румыния, Сингапур, Словакия, Финлян-
дия, Франция, Чехия, Швейцария и Швеция. Кроме того, в качестве наблюдате-
лей в деятельности PIC/S принимают участие Эстония, Европейское агентство по
оценке медицинских продуктов {ЕМЕА), UNICEF (Детский фонд ООН) и ВОЗ [48].
Многие другие нормативно-правовые органы также заинтересованы в присоедине-
нии к PIC/S, в частности, среди них органы, представляющие такие страны, как
Аргентина, Болгария, Бразилия, Индонезия, Израиль, Кипр, Литва, Оман, Россия,
Словения, США, Таиланд, Украина, Филиппины, Эстония и Южная Африка [49].
Для присоединения к PIC/S нормативно-правовой орган, желающий присоеди-
ниться к конвенции, должен пройти подробную оценку для доказательства, что
орган обладает возможностями и компетенцией, необходимых для создания ин-
спекционной системы, эквивалентной инспекционной системе, существующей
в странах — участницах PIC/S. Для подтверждения того, что вновь присоединив-
шиеся и прежние участники выполняют одни и те же требования, на регулярной
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 175
основе проводится переаттестация всех участников. Одной из важнейших функций
PIC/S является составление руководящих документов по GMP, которые организа-
ция разрабатывает в тесном сотрудничестве с ЕС и соответствующими его агент-
ствами. Сотрудничество позволяет обеим сторонам принимать документы друг дру-
га, сводя таким образом к минимуму повторные усилия при разработке документов,
связанных с GMP. Среди прочих руководств, содержащих обширную информацию
по различным аспектам GMP и вопросам качества [49], PIC/S издала собственное
Руководство по GMP (Руководство по надлежащей производственной практике для
медицинских продуктов), гармонизированное с Правилами GMP ЕС.
Последняя по времени пересмотренная версия Руководства по GMP от PIC/S
(версия РЕ009—3) была опубликована в январе 2006 г. Как показано в табл. 14, Руко-
водство состоит из 9 глав {chapters) и 16 приложений. В главах представлены общие
правила GMP при производстве лекарственных продуктов, описывающие требова-
ния к управлению качеством и контролю, персоналу, недвижимости, оборудова-
нию, документации, производству, контрактным службам, претензиям, отзыву про-
дукции и самоконтролю. В приложениях представлены специфические указания,
относящиеся к производству стерильных лекарственных продуктов, биологических
лекарственных продуктов, радиофармацевтических препаратов, ветеринарных ле-
карственных продуктов, медицинских газов, растительных лекарственных продук-
тов, жидкостей для перорального введения, препаратов для наружного примене-
ния (кремов, мазей), аэрозолей, новых препаратов для клинических исследований,
а также крови и препаратов крови. Кроме того, отдельные приложения посвящены
отбору проб, компьютеризованным системам, применению ионизирующего излу-
чения, квалификации и валидации, а также выпуску по параметрам [50].
Несмотря на то что Руководство по GMP PIC/S гармонизировано с Правилами
GMP ЕС, и их содержания аналогичны, между ними существуют небольшие раз-
личия. Вместо термина квалифицированный персонал в Руководстве /7С/5употре-
блен термин авторизованный персонал. Кроме того, из Руководства PIC/S убраны
все ссылки на директивы ЕС. Следует отметить, что PIC/S не приняла Приложе-
ния 16 и 18 Правил GMP ЕС. Приложение 16 является специфичным для Правил
GMP ЕС и относится к статусу квалифицированного сотрудника при выпуске про-
изводственной серии, а Приложение 18 является требованием ICH из Руководства
по GMP при производстве API, принятого Комиссией PIC/S в виде отдельного до-
кумента (РЕ007) [50].
2.1.3.3. Международная конференция по гармонизации (ICH)
Организация ICH была создана в 1990 г. Главная ее задача состояла в увеличении
эффективности процесса разработки лекарств и регистрации новых лекарственных
продуктов в странах-участницах путем гармонизации национальных руководящих
документов. Эта совместная инициатива предусматривает равное партнерство регу-
ляторных органов и производителей. Основателями и действующими участниками
ICH, представляющими нормативно-правовые органы и промышленные предпри-
ятия, занимающиеся научными исследованиями стран-участниц, являются ЕС, Ев-
ропейская Федерация фармацевтической промышленности и ассоциаций {EFPIA),
МНЕ W, Ассоциация фармацевтических производителей Японии (JPMA), FDA и Ас-
176 Часть 2. Международные правила надлежащей производственной практики
социация фармацевтических производителей и исследователей Америки (PhRMA).
Кроме стран — действительных членов, в организации работают наблюдатели, пред-
ставляющие связующее звено между странами и регионами, участвующими и не уча-
ствующими в ICH. Действующими наблюдателями являются ВОЗ, EFTA, Swissmedic
(представляющая Швейцарию) и Health Canada (представляющая Канаду) [51].
Содержание Руководства GMPPIC/S [50]
Таблица 14
Раздел Предмет
Введение
Глава 1 Глава 2 Глава 3 Глава 4 Глава 5 Глава 6 Глава? Глава 8 Глава 9 Приложение 1 Приложение 2 Управление качеством Персонал Недвижимость и оборудование Документация Производство Контроль качества Контрактное производство и анализ Претензии и отзыв продукции Самоинспекция Производство стерильных медицинских продуктов Производство биологических медицинских продуктов для употребления человеком
Приложение 3 Приложение 4 Производство радиофармацевтических препаратов Производство ветеринарных медицинских продуктов, отличных от иммунологических
Приложение 5 Приложение 6 Приложение 7 Приложение 8 Приложение 9 Приложение 10 Приложение 11 Приложение 12 Производство иммунологических ветеринарных медицинских продуктов Производство медицинских газов Производство растительных медицинских продуктов Отбор проб исходных и упаковочных материалов Производство жидкостей, кремов и мазей Производство герметических дозированных аэрозолей для ингаляций Компьютеризованные системы Применение ионизирующего излучения при производстве медицинских продуктов
Приложение 13 Приложение 14 Приложение 15 Приложение 17 Производство медицинских препаратов для клинических исследований Производство продуктов из крови и плазмы крови человека Квалификация и валидация Выпуск по параметрам Глоссарий
Среди других руководств ICHопубликовала Руководство по GMP для API (QT.
Надлежащая производственная практика для активных фармацевтических ингреди-
ентов). Руководство описывает требования GMP при производстве API и помогает
обеспечить соответствие API заявленным нормативам чистоты и качества. Руковод-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 177
ство охватывает API, произведенные путем химического синтеза, экстракции, куль-
тивирования клеток/ферментации, извлечения из природных источников или путем
комбинирования этих процессов. Исключение составляют вакцины, медицинские
газы, лекарственные продукты в крупной фасовке, радиофармацевтические препа-
раты, цельные клетки, цельная кровь и плазма, производные крови и плазмы M.API
для генной терапии. Тем не менее, в Руководство включены API, изготовленные
с использованием крови или плазмы в качестве исходных материалов [52]. Руковод-
ство приняли все страны — участницы ICH: ЕС в ноябре 2000 г., Япония в ноябре
2000 г. и США в сентябре 2001 г. [53]. Кроме того, документ принят несколькими
странами, не входящими в ICH, такими как Австралия [28] и Южная Африка [34].
Базовая структура Руководства ICH по GMP при производстве API представле-
на в табл. 15. Руководство состоит из 19 глав (chapters), описывающих требования
к управлению качеством, персоналу, недвижимости, оборудованию, документации,
материалам, контролю продукции и технологических процессов, упаковке и марки-
ровке, хранению и распределению, лабораторному контролю, валидации, контролю
изменений, претензиям, отзывам, контрактным службам, сотрудникам, API, изго-
тавливаемым путем культивирования клеток/ферментации, и API, применяемым
в ходе клинических испытаний [52].
Таблица 15
Содержание Руководства ICHпо GMP при производстве API [52]
Раздел________________________________Предмет______________________________
1 Введение
2 Управление качеством
3 Персонал
4 Недвижимость и оборудование
5 Технологическое оборудование
6 Документация и ведение записей
7 Управление материалами
8 Контроль готовой продукции и контроль в процессе обработки
9 Упаковочная и идентификационная маркировка АР1и полупродуктов
10 Хранение и распределение
11 Лабораторный контроль
12 Валидация
13 Контроль изменений
14 Отбраковка и повторное использование материалов
15 Претензии и рекламации
16 Производители, работающие по контракту (включая лаборатории)
17 Агенты, брокеры, трейдеры, дистрибьюторы, переупаковщики
и перемаркировщики
18 Специальные правила производства API, произведенных путем
культивирования клеток/ферментации
19 API, предназначенные для клинических испытаний
20 Глоссарий
178 Часть 2. Международные правила надлежащей производственной практики
2.1.3.4. Ассоциация государств Юго-Восточной Азии (ASEAN)
Организация ASEAN была основана в 1967 г. Индонезией, Малайзией, Сингапуром,
Таиландом и Филиппинами. В настоящее время в число стран-участниц входят
также Бруней и Даруссалам (присоединились в 1984 г.), Вьетнам (присоединился
в 1995 г.), Лаос и Мьянма (присоединились в 1997 г.), а также Камбоджа (присо-
единилась в 1999 г.). Цели и задачи ASEAN включают укрепление сотрудничества
в экономической, социальной, культурной, технической, образовательной и других
областях [54]. Среди прочих программ сотрудничества страны ASEAN разработали
свои собственные Правила GMP, опубликованные в 1984 г. [55].
2.13.5. Общий рынок государств Южной Америки (MERCOSUR)
Оганизация MERCOSUR (Mercado Сопит del Sur—Общий рынок государств Южной
Америки) была основана в 1991 г. Аргентиной, Бразилией, Парагваем и Уругваем для
развития общего рынка между странами-участницами. В настоящее время в число
стран-участниц входят также Боливия и Чили (присоединились в 1996 г.). Одной
из первичных задач организации являлась гармонизация фармацевтических зако-
нодательств стран-участниц. В рамках деятельности по гармонизации MERCOSUR
разработала собственные требования GMP, основанные на рекомендациях ВОЗ.
В дополнение к руководству по GMP, MERCOSUR изданы другие руководства по
GMP, связанные с инспекциями, требованиями к оборудованию и контролю каче-
ства [56].
2.1.1. Соответствие правил G/V7PCLLIA требованиям
и правилам GMP других стран
В следующих разделах рассматриваются сходства и различия между правилами GMP
США, Правила GMP Канады, Правилами GMP ЕС и Руководством по GMP ВОЗ.
Поскольку Правила GMP ЕС гармонизированы с Руководством по GMP PIC/S, со-
ответствие между Правилами GMP ЕС и правилами GMP США обусловливает также
соответствие между правилами GMP США и Руководством по GMP PIC/S, а также
другими национальными GMP, основанными на Руководстве по GMPPIC/S. Разли-
чия между Правилами GMPEC и Руководством по GMP Р/С/А представлены в раз-
деле 2.1.3.2.
2.1.4.1. Общие вопросы
В правилах GMP США общие вопросы, связанные с соблюдением и применимо-
стью правил GMP, рассмотрены в главе 210 [6], состоящей из §§ 210.1, 210.2 и 210.3
и в подглаве A (Subpart А) главы 211 [7], состоящей из §§ 211.1 и 211.3. Содержание
главы 210 и подглавы А главы 211 представлено в табл. 16. Положение, изложен-
ное в § 210.1, определяет статус, в § 210.2 — применимость, а § 211.1 устанавливает
границы действия правил. Определения терминов, используемых в положениях,
даны в § 210.3 и в § 211.3, который устанавливает, что определения, представленные
в § 210.3, применяются также в главе 211.
Соответствия в Правилах GMP Канады. В законодательстве Канады общие во-
просы, связанные с соблюдением и применимостью требований и правил GMP,
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 179
охвачены во введении к Правилам GMP [12] и в разделах 1А [57] и 2 части С Правил
для продуктов питания и лекарств [11]. Определения из требований GMP представ-
лены в положении С.01А.001 раздела 1А {Division 1А) [57] и в положении С.02.002
раздела 2 [11]. Определения из кодекса Правил GMPпредставлены в глоссарии тер-
минов [12].
Таблица 16
Содержание части 210 и раздела А части 211 правил GMP США, описывающей общие вопросы,
связанные с соблюдением и применимостью правил [6, 7]
Параграф______________________________Предмет____________________________
CFR 210.1 Статус действующих правил надлежащей производственной практики
CFR 210.2 Применимость действующих правил надлежащей производственной практики
CFR 210.3 Определения
CFR 211.1 Область применения
CFK211.3 Определения
Соответствия в Правилах GMP ЕС. В европейском законодательстве общие во-
просы, связанные с соблюдением и применимостью требований GMP и правил,
охвачены Директивой Комиссии 2003/94/ЕС [14] и представлены во введении
к Правилам GMP [15]. Определение терминов, используемых в Директиве, даны
в главе 2 {Article 2) Директивы [14], а определения, используемые в Правилах, пред-
ставлены в глоссарии к Правилам GMP [15].
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] общие
вопросы, связанные с соблюдением и применимостью правил GMP, рассмотрены
в разделе «Общие вопросы». Определения, используемые в Руководстве по GMP,
даны в глоссарии к Руководству.
2.1.4.2. Организация и персонал
В правилах GMP США вопросы, связанные с организационной структурой и пер-
соналом, рассмотрены в подглаве В {Subpart В) [7], которая состоит из §§ 211.22,
211.25,211.28 и 211.34. Содержание подглавы В представлено в табл. 17. Положение,
закрепленное в § 211.22, устанавливает ответственность руководства отдела контро-
ля качества, включая требования к ресурсам. Положение § 211.25 устанавливает
требования к квалификации персонала, в том числе к образованию и опыту работы,
а также требования к специальному обучению персонала. В § 211.28 устанавлива-
ются обязанности персонала, в том числе требования к одежде и другим защитным
приспособлениям, личной гигиене и здоровому образу жизни, а также к состоянию
здоровья персонала. Кроме того, положение устанавливает требования к автори-
зации при ограниченном доступе. В § 211.34 описаны требования к образованию,
специальному обучению и опыту работы консультантов, в том числе требования
к документам.
Соответствия в Правилах GMP Канады. В Правилах GMP Канады [12] вопро-
сы, связанные с организацией и персоналом, рассматриваются, главным образом,
в разъяснении положения С.02.006 (персонал). Частично эти вопросы рассматрива-
ются в разъяснениях к положениям С.02.004 (недвижимость), С.02.008 (санитарное
180
Часть 2. Международные правила надлежащей производственной практики
состояние), С.02.011 (контроль производства), С.02.013 (отдел контроля качества),
С.02.015 (отдел контроля качества) и С.02.024 (записи). Соответствия в положении,
закрепленном в § 211.22, рассмотрены в пунктах 1—5 {Sections 1—5) и в пункте 2 разъ-
яснения положения С.02.013.
Таблица 17
Содержание подглавы В {Subpart В) главы 211 правил GMP США, относящихся
к организационной структуре и персоналу [17]
Параграф Предмет
СИ? 211.22 СЕЛ 211.25 CFR 211.28 СЕЛ 211.34 Ответственность отдела контроля качества Квалификация персонала Ответственность персонала Консультанты
В пунктах 1—5 разъяснения положения Правил С.02.015 описаны обязанно-
сти отдела качества (отдела контроля качества), а в пункте 2 разъяснения положе-
ния С.02.013 указаны требования к ресурсам. Нормативы, соответствующие по-
ложению § 211.25, устанавливающему требования к образованию, специальной
подготовке и опыту персонала, представлены в пунктах 1—5 разъяснения положе-
ния С.02.006. Нормативы, соответствующие положению § 211.28, представлены
в пункте 6.3 разъяснения положения С.02.004, в пунктах 1—2 разъяснения положе-
ния С.02.008, в пункте 8 разъяснения положения С.02.011 и пункте 4 разъяснения
положения С.02.013. Пункт 1 разъяснения положения Правил С.02.008 описывает
требования к состоянию здоровья персонала, пункт 2 — требования к одежде и дру-
гим средствам защиты, а также к личной гигиене. Пункт 6.3 разъяснения положения
С.02.004, пункт 8 разъяснения положения С.02.011 и пункт 4 разъяснения положе-
ния С.02.013 описывают требования, связанные с ограничением доступа. Норма-
тивы, соответствующие положению § 211.34, представлены в пункте 6 разъяснения
положения С.02.006 и подпункте {Subsection) 1.3.2 разъяснения положения С.02.024.
Пункт 6 разъяснения положения С.02.006 содержит требования к образованию, спе-
циальной подготовке и опыту работы консультантов и подрядчиков, а подпункт 1.2.3
разъяснения положения С.02.024 описывает требования к документации.
Соответствия в Правилах GMP ЕС. В Правилах GMPEC [15] вопросы организа-
ции и персонала изложены, в основном, в главе 2 {Chapter!) (персонал) и частично
в главах 3 (недвижимость и оборудование), 5 (производство) и 6 (контроль каче-
ства). Нормативы, соответствующие предписаниям, изложенным в § 211.22, пред-
ставлены в разделах {Subchapters) 2.6, 2.7, 6.1 и 6.2. Разделы 2.6 и 2.7 определяют
ответственность заведующего отделом контроля качества, а 6.2 — ответственность
отдела контроля качества в целом. Требования к ресурсам описаны в разделе 6.1.
Нормативы, соответствующие положению 211.25, представлены в разделах 2.1, 2.4
и 2.8—2.12. Разделы 2.1 и 2.4 описывают требования к персоналу, а разделы 2.8—
2.12 — требования к специальному обучению персонала. Нормативы, соответству-
ющие положению § 211.28, представлены в разделах 2.15, 2.16, 3.5, 3.21, 5.16 и 6.4.
Раздел 2.15 определяет требования к состоянию здоровья персонала, а 2.16 — тре-
бования к одежде и защите. Вопросы, связанные с ограничением доступа, освеща-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 181
ются в разделах 3.5, 3.21, 5.16 и 6.4. В Правилах GA/PEC отсутствует соответствие
предписанию, представленному в § 211.34 и охватывающему требования, связанные
с привлечением консультантов. Тем не менее, в главе 7 Правила регулируют общие
вопросы, связанные с контрактными службами.
Соответствие в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] вопро-
сы, связанные с организационной структурой и персоналом, описаны главным об-
разом в главе 9 (Chapter 9) (персонал) и частично в главах 10 (обучение), 11 (личная
гигиена), 16 (надлежащая практика производства) и 17 (надлежащая практика кон-
троля качества). Нормативы, соответствующие положению § 211.22, представлены
в разделах (Subchapters) 9.8,9.10,17.3 и 17.4. Разделы 9.8 и 9.10 устанавливают ответ-
ственность заведующего отделом контроля качества, а раздел 17.4 — ответственность
отдела контроля качества в целом. Раздел 17.3 описывает требования к ресурсам.
Нормативы, соответствующие правилу § 211.25, представлены в разделах 9.2,9.4,9.7
и 10.1—10.4. Разделы 9.2,9.4 и 9.7 устанавливают требования к персоналу, связанные
с образованием и опытом работы, а разделы 10.1—10.4 — требования к специально-
му обучению. Нормативы, соответствующие § 211.28, изложены в разделах 11.1—
11.8, 9.5 и 16.7. Разделы 11.1—11.5 устанавливают требования к состоянию здоровья
и личной гигиене, а разделы 11.6—11.8 — требования к одежде и другим средствам
защиты. Разделы 9.5 и 16.7 освещают вопросы, связанные с ограничением доступа.
Нормативы, соответствующие § 211.34, изложены в разделе 10.6, регламентирую-
щем привлечение консультантов.
2.1.4.З. Здания и помещения
В США требования GMP в отношении производственных зданий и помеще-
ний изложены в подглаве С (Subpart С) [7], которая включает §§ 211.42, 211.44,
211.46, 211.48, 211.50, 211.52, 211.56 и 211.58. Содержание подглавы С представле-
но в табл. 18. Положение § 211.42 относится к характеристикам планировки и осо-
бенностям конструкции с учетом требований к размеру, конструкции и размеще-
нию зданий производственного назначения. Кроме того, положение устанавливает
требования к размещению оборудования, а также потоку материалов и продуктов,
а также специфическим операциям, которые необходимо проводить в раздельных
или выделенных зонах для предотвращения загрязнения или смешивания. В прило-
жении изложены также специальные требования к помещениям для асептического
производства, а также системам и помещениям для производства пенициллина. По-
ложение § 211.44 устанавливает требования к освещению, а § 211.46 — к вентиля-
ции, включая требования к системам контроля и установкам для обработки воздуха.
Далее, оно устанавливает специальные требования к вентиляции при производстве
пенициллина. В § 211.48 описываются требования к санитарно-техническому обо-
рудованию, в том числе требования к водопроводно-канализационной сети, дрена-
жу и качеству питьевой воды. Положение § 211.50 регулирует вопросы, связанные
со сточными водами, мусором и другими отходами, устанавливая требования по их
ликвидации. § 211.52 содержит требования к душевым и туалетам. Правило § 211.56
относится к санитарному состоянию, в нем устанавливаются требования к состоя-
нию, которое необходимо поддерживать на производственных площадках. Кроме
того, оно содержит требования к ликвидации мусора и органических отходов. Так-
182 Часть 2. Международные правила надлежащей производственной практики
же оно устанавливает требования к письменным процедурам, описывающим сани-
тарные операции и применение биоцидов, фумигацию, очистку и дезинфицирую-
щие агенты. Это правило также определяет требования к применению биоцидов
и проведению санитарных процедур. Положение § 211.58 устанавливает требования
к эксплуатации зданий производственного назначения.
Таблица 18
Содержание подглавы С главы 211 правил GMP США, относящихся к зданиям и помещениям
Параграф Предмет
CFR211.42 CFR2U.44 С/К 211.46 CFR211.48 CFR 211.50 С7К211.52 CFR 211.56 CFR 211.58 Планировка и особенности планировки конструкции Освещение Вентиляция, фильтрация, нагревание и охлаждение воздуха Система водоснабжения Сточные воды и отходы Душевые и туалеты Санитарное состояние Эксплуатация помещений
Соответствия в Правилах GMP Канады. В своде Правил GMP Канады [12] во-
просы производственных зданий и помещений в основном освещаются в разъяс-
нениях положения С.02.004 (недвижимость) и частично в разъяснениях положе-
ния С.02.005 (оборудование), С.02.007 (санитарное состояние), С.02.009 (анализ
исходных материалов), С.02.011 (контроль производства) и С.02.029 (стерильные
продукты). Нормативы, соответствующие правилу, представленному в § 211.42, из-
ложены в пунктах (Sections) 1, 2, 2.3, 6, 6.2 и 6.4 разъяснения положения С.02.004,
в пункте 15 разъяснения положения С.02.011, а также в пункте «Недвижимость»
разъяснения положения С.02.029. В пунктах 1 и 2 разъяснения положения С.02.004
рассматриваются требования к размеру, конструкции и расположению зданий, ис-
пользуемых в производственных целях. Пункт 6.2 разъяснения положения С.02.004
и пункт 15 разъяснения положения С.02.011 устанавливают требования к разме-
щению оборудования. Требования к потоку материалов и продуктов представлены
в пункте 6 разъяснения положения С.02.004, а операции, которые нужно проводить
в раздельных или выделенных зонах, описаны в пунктах 2.3 и 6.4 разъяснения пра-
вила С.02.004. Требования к помещениям для асептического производства изложе-
ны в пункте «Здания и помещения» разъяснения положения С.02.029, а требования
к помещениям для производства пенициллина — в пункте 11.1 разъяснения поло-
жения С.02.004. Нормативы, соответствующие правилу 211.44, устанавливающие
требования к освещению, изложены в пункте 6.5 разъяснения положения С.02.004.
Нормативы, соответствующие положению § 211.46, представлены в пунктах 3.6 и 4
разъяснения положения С.02.004. Пункт 3.6 разъяснения положения С.02.004 уста-
навливает требования к системам обработки воздуха, пункт 4 — требования к кон-
тролю температуры и влажности. Специфические требования, относящиеся к про-
изводству пенициллина, представлены в пункте 11.1. Нормативы, соответствующие
§ 211.48, содержатся в пунктах 3.5 и 7 разъяснения положения С.02.004, пункте 3.7
разъяснения положения С.02.005, пункте 4 разъяснения положения С.02.009 и пун-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 183
кте «Системы водоочистки» разъяснения правила С.02.029. Пункт 7 разъяснения
положения С.02.004 устанавливает требования к коммунальным и вспомогательным
системам, включая систему снабжения очищенной водой. Пункт 3.7 разъяснения
положения С.02.005 устанавливает требования к функционированию оборудования
для очистки воды, хранению и поставкам. Требования к качеству воды изложены
в пункте 4 разъяснения правила С.02.009 и в пункте «Системы водоочистки» разъ-
яснения правила С.02.029. Требования к дренажным системам изложены в пун-
кте 3.5 разъяснения положения С.02.004. Нормативы, соответствующие § 211.50,
устанавливающие требования к обработке стоков и отходов, изложены в пун-
кте 2.6 разъяснения положения С.02.007. Нормативы, соответствующие § 211.52,
содержащие требования к душевым и туалетам, изложены в пункте 5 разъясне-
ния положения С.02.004. Нормативы, соответствующие § 211.56, определяющие
требования к санитарному состоянию, изложены в пунктах 1 и 2 разъяснения по-
ложения С.02.007. Правила GMP Канады не устанавливают никаких отдельных тре-
бований к обработке органических отходов. Общие требования к обработке отходов
материалов изложены в пункте 2.6 разъяснения положения С.02.007. Нормативы,
соответствующие § 211.58, в котором устанавливаются требования к эксплуатации
недвижимости, изложены в пункте 9 разъяснения положения С.02.004.
Соответствия в Правилах ЕС по GMP. В Правилах ЕС по GMP [15] вопросы, от-
носящиеся к зданиям и помещениям, изложены главным образом в главе 3 (здания
и помещения) и частично в Приложении 1 (производство стерильных медицинских
продуктов). Нормативы, соответствующие § 211.42, изложены в предисловии к гла-
ве 3 (СЛор/егЗ) и в разделах (Subchapters) 3.6—3.8,3.13, 3.22, 3.23, 3.26 и 3.33. Требова-
ния к размеру, конструкции и расположению производственных зданий, представ-
лены в предисловии к главе 3. Раздел 3.8 устанавливает требования к размещению
оборудования, а раздел 3.7 — к потоку материалов и продуктов. Операции, которые
необходимо проводить в отдельных или выделенных зонах, перечислены в разде-
лах 3.6, 3.13, 3.22, 3.23, 3.26 и 3.33. В Приложении 1 описаны требования к помеще-
ниям для асептического производства, а в разделе 3.6 — требования к помещениям
для производства пенициллина. Нормативы, соответствующие § 211.44, изложены
в разделах 3.3 и 3.16, устанавливающих требования к освещению. Нормативы, соот-
ветствующие § 211.46, изложены в разделах 3.3 и 3.12, устанавливающих требования
к вентиляции. Специфические требования к производству пенициллина изложены
в разделе 3.6. Нормативы, соответствующие § 211.48, представлены в разделах 3.10
и 3.11 главы 3 и в подразделах (Subsection) 35 и 44 Приложения 1. Раздел 3.10 устанав-
ливает требования к санитарно-техническим системам, а раздел 3.11— требования
к дренажным системам. Подраздел 35 Приложения 1 описывает требования к мони-
торингу источников воды и оборудованию для водоочистки. Дополнительные указа-
ния по качеству воды даны в руководящем документе ЕС «Примечания к руководству
по качеству воды для использования в фармацевтических целях» [58]. Правила GMP ЕС
не содержат нормативов, соответствующих § 211.50, устанавливающих требования
к обработке стоков и других отходов. Нормативы, соответствующие § 211.52, пред-
ставлены в разделе 3.31, устанавливающем требования к душевым и туалетам. Нор-
мативы, соответствующие § 211.56, представлены в разделах 3.2, 3.4, 3.43 главы 3
и 4.26 главы 4. Разделы 3.2 и 3.4 охватывают требования к условиям эксплуатации
184
Часть 2. Международные правила надлежащей производственной практики
производственных помещений. В разделе 4.26 описываются процедуры очистки
и дезинфекции, а в разделе 3.43 — требования к санитарному состоянию водопро-
водных труб. Правила GMP ЕС не выделяют отдельных требований по обработке
органических отходов. Нормативы, соответствующие § 211.58, представлены в раз-
деле 3.2, в котором описаны требования к эксплуатации производственных зданий.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] вопро-
сы, связанные со зданиями и помещениями, изложены главным образом в главе 12
{Chapter 12) (недвижимость) и частично в главах 3 (санитария и гигиена), 14 (ма-
териалы) и 15 (документация). Нормативы, соответствующие положению § 211, 42
представлены в разделах {Subchapters) 12.1, 12.2, 12.4, 12.5, 12.10, 12.14, 12.17, 12.19,
12.22—12.26 и 12.33. Требования к размеру, конструкции и размещению зданий пред-
ставлены в разделах 12.1, 12.4 и 12.5. Разделы 12.2 и 12.26 охватывают требования
к размещению оборудования, а разделы 12.10 и 12.25 — требования к потоку ма-
териалов и продуктов. Операции, которые необходимо проводить в отдельных или
специально выделенных зонах, перечислены в разделах 12.14, 12.17, 12.19, 12.22—
12.24 и 12.33. Требования к помещениям для асептического производства изложены
в главе 9 Приложения 6 TRS 902 ВОЗ, а требования к помещениям для производства
пенициллина — в разделе 12.24. Нормативы, соответствующие § 211.44, представле-
ны в разделах 12.8 и 12.32, устанавливающих требования к освещению. Нормативы,
соответствующие § 211.46, представлены в разделах 12.8 и 12.30, устанавливающих
требования к вентиляции. Специфические требования к производству пеницилли-
на описаны в разделе 12.24. Нормативы, соответствующие § 211.48, представлены
в разделах 12.28,12.29 и 14.6 и в Приложении 3 TRS929 ВОЗ [45]. В разделе 12.28 из-
ложены требования к санитарно-техническим системам, а в разделе 14.6 — к каче-
ству воды, используемой при производстве лекарственных продуктов. Дополнитель-
ные указания по качеству воды даны в Приложении 3 TRS929 ВОЗ [45]. Требования
к дренажным системам установлены в разделе 12.29. Нормативы, соответствующие
§ 211.50, представлены в разделах 14.44 и 14.45, устанавливающих требования к об-
работке стоков и других отходов. Нормативы, соответствующие § 211.52, представ-
лены в разделах 3.1,12.7,12.9,14.44 —14.46 и 15.48. Разделы 12.7 и 12.9 устанавлива-
ют требования к условиям, которые необходимо поддерживать в производственном
помещении, а раздел 3.1— общие требования к санитарии и гигиене. В Руководстве
ВОЗ по GMP нет отдельных указаний по обработке органических отходов. Общие
требования к обработке отходов изложены в разделах 14.44 и 14.45. Раздел 15.48
устанавливает требования к письменным процедурам, описывающим операции по
санитарной обработке, а раздел 14.46 регламентирует применение родентицидов,
инсектицидов, фумигаторов и дезинфицирующих материалов. Нормативы, соот-
ветствующие § 211.58, представлены в разделе 12.6, устанавливающем требования
к эксплуатации зданий, используемых при производстве лекарственной продукции.
2.1.4.4. Оборудование
В правилах GMP США вопросы, связанные с оборудованием, изложены в подгла-
ве D [7] {Subpart D), включающей положения, представленные в §§ 211.63, 211.65,
211.67, 211.68 и 211.72. Содержание подглавы D представлено в табл. 19. В § 211.63
устанавливаются требования к производственному оборудованию, в том числе
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 185
к дизайну, размеру и расположению. Положение § 211.65 содержит требования
к конструкции оборудования, в том числе к свойствам применяемых материалов,
и специальные требования к структуре оборудования. § 211.67 относится к очист-
ке, эксплуатации и дезинфекции оборудования и приспособлений, в нем описа-
ны требования к процедурам очистки и технического обслуживания. В положении
§ 211.68, относящемся к автоматическому, механическому и электронному обору-
дованию, изложены требования к калибровке и проверке оборудования, включая
требования к документации проверок и инспекций. Кроме того, положение охва-
тывает требования к контролю компьютерных или связанных с ними систем, вклю-
чая требования к архивным данным. § 211.72 включает требования к фильтрам для
жидкостной фильтрации, применяемым при производстве продуктов для инъек-
ций, включая специфические требования к использованию фильтров, выделяющих
волокна, и фильтров, содержащих асбест.
Таблица 19
Содержание подглавы D главы 211 правил GMP США, относящихся к оборудованию [7]
Параграфы Предмет
CER 211.63 СИ? 211.65 сиг 211.67 сиг 211.68 сиг 211.72 Дизайн, размер и размещение оборудования Конструкция оборудования Очистка и эксплуатация оборудования Автоматическое, механическое и электронное оборудование Фильтры
Соответствия в Правилах GMP Канады. В Правилах GMP Канады [12] вопросы,
относящиеся к оборудованию, изложены главным образом в разъяснениях прави-
ла С.02.005 (оборудование) и частично в разъяснениях правила С.02.007 (санитарное
состояние) и С.02.024 (записи). Нормативы, соответствующие § 211.63, в отноше-
нии требований к дизайну, конструкции и размещению оборудования, применяе-
мого при производстве лекарственных продуктов, изложены в пунктах (Sections)
1 и 5 разъяснения положения С.02.005. Нормативы, соответствующие § 211.65, в ко-
торых устанавливаются требования к конструкции оборудования, изложены в пун-
ктах 2.1—2.3 разъяснения правила С.02.005. Нормативы, соответствующие § 211.67,
устанавливающие требования к конструкции оборудования, изложены в пунктах
1, 2 и 3 разъяснения положения С.02.007. Нормативы, соответствующие § 211.68,
изложены в пункте 5.4 разъяснения положения С.02.005 и в предисловии к разъяс-
нению положения С.02.024. Пункт 5.4 разъяснения положения С.02.005 определяет
требования к использованию автоматического, механического и электронного обо-
рудования, включая компьютеризованные системы, и в предисловии к разъяснению
положения С.02.024 — требования к резервному копированию данных. Правила
GMP Канады не содержит соответствия § 211.72, в котором устанавливаются тре-
бования к фильтрам для жидкостной фильтрации, применяемым при производстве
продуктов для инъекций. Кроме того, он не охватывает требований к фильтрам, вы-
свобождающим волокна, и фильтрам, содержащим асбест.
Соответствия в Правилах G7WPEC. В Правилах GMP ЕС [15] вопросы, связанные
с оборудованием, освещаются в основном в главе 3 (Chapter 3) (здания и помещения)
186 Часть 2. Международные правила надлежащей производственной практики
и частично в главе 4 (документация) и Приложении 1 (производство стерильных ме-
дицинских продуктов) и 11 (компьютеризованные системы). Нормативы, соответ-
ствующие § 211.63, изложены в разделе 3.34 (Subchapter 3.34), устанавливающем тре-
бования к дизайну и размещению оборудования, применяемого при производстве
лекарственных продуктов. Нормативы, соответствующие § 211.65, представлены
в разделах 3.38 и 3.39, устанавливающих требования к конструкции оборудования.
Нормативы, соответствующие § 211.67, представлены в разделах 3.36, 3.37 и 3.43,
охватывающих требования к очистке и дезинфекции производственного оборудо-
вания. Нормативы, соответствующие § 211.68, представлены в разделах 3.41 и 4.9
ив Приложении 11. Раздел 3.41 устанавливает требования к техническому обслужи-
ванию измерительного, весового, записывающего и контрольного оборудования,
а раздел 4.9 — требования к использованию электронных систем обработки данных
и резервному копированию данных. Дополнительные указания по применению
компьютеризованных систем даны в Приложении 1. Нормативы, соответствую-
щие § 211.72, в которых устанавливаются требования к фильтрам для жидкостной
фильтрации, применяемым для стерильной фильтрации, изложены в пунктах 84—87
Приложения 1. Правила GMP ЕС не содержат отдельных указаний по использова-
нию фильтров, высвобождающих волокна, и фильтров, содержащих асбест.
Соответствия в Руководстве ВОЗ по GMP. Вопросы оборудования в Руковод-
стве ВОЗ по GMP [36] изложены преимущественно в главе 13 (Chapter 13) (обору-
дование) и частично в главах 14 (материалы), 15 (документация) и 16 (надлежащая
производственная практика). Нормативы, соответствующие § 211.63, содержатся
в разделах (Subchapters) 13.1 и 13.2, устанавливающих требования к дизайну, раз-
мещению и установке оборудования, применяемого при производстве лекарствен-
ных продуктов. Нормативы, соответствующие § 211.65, приведены в разделах 13.9
и 14.3, устанавливающих требования к конструкции оборудования. Нормативы, со-
ответствующие § 211.67, описаны в разделах 13.6, 13.8, 13.12, 16.17, 16.18 и 16.22,
устанавливающих требования к очистке и дезинфекции оборудования. Нормативы,
соответствующие § 211.68, представлены в разделах 16.23 и 15.9. Требования ктехни-
ческому обслуживанию измерительного, весового, записывающего и контрольного
оборудования и инструментов, изложены в разделе 16.23. Раздел 15.9 устанавлива-
ет требования к использованию электронных систем обработки данных, включая
требования к ведению архива резервных данных. Нормативы, соответствующие
§ 211.72, предписывающие требования к использованию фильтров, представлены
в разделах 7.6—7.9 Приложения 6 TRS902 ВОЗ [40]. Раздел 7.6 охватывает требова-
ния к фильтрам, содержащим асбест.
2.1.4.5. Контроль качества компонентов, первичной упаковки
и укупорочных средств
В США вопросы контроля качества компонентов, первичной упаковки и укупороч-
ных средств для лекарственной продукции, изложены в подглаве Е (Subpart Е) [7],
включающей §§ 211.80, 211.82, 211.84, 211.86, 211.87, 211.89 и 211.94. Содержание
подглавы Е представлено в табл. 20. Положение, закрепленное в § 211.80, определя-
ет требования к процедурам контроля качества компонентов, первичной упаковки
и укупорочных средств. Оно устанавливает также требования к их обработке, хра-
Глава 2.1, Национальные правила и требования GMP, международные требования GMP... 187
нению и маркировке. В § 211.82 описываются требования к получению и хранению
компонентов, не прошедших тестирования, первичной упаковки и укупорочных
средств. Таблица 20
Содержание подглавы Е главы 211 правил СЛГРСШЛ, устанавливающих требования
к контролю качества компонентов, первичной упаковки и укупорочных средств для
лекарственной продукции [7]
Параграф Предмет
СИ? 211.80 СИ?211.82 Общие требования Получение и хранение компонентов, не прошедших тестирования, первичной упаковки и укупорочных средств для лекарственной продукции
СИ?211.84 Тестирование, одобрение или отбраковка компонентов, первичной упаковки и укупорочных средств для лекарственной продукции
СИ? 211.86 Использование одобренных компонентов первичной упаковки и укупорочных средств для лекарственной продукции
СИ?211.87 Повторное тестирование одобренных компонентов, первичной упаковки и укупорочных средств для лекарственной продукции
СИ? 211.89 Отбракованные компоненты, первичной упаковки и укупорочных средств для лекарственной продукции
СИ? 211.94 Первичная упаковка и укупорочные средства для лекарственной продукции
Положение § 211.84 относится к тестированию и одобрению или отбраковке
компонентов, первичной упаковки и укупорочных средств и устанавливает требо-
вания к отбору проб, тестированию и выпуску. В § 211.86 описывается применение
одобренных компонентов, первичной упаковки и укупорочных средств для лекар-
ственной продукции, устанавливаются требования к ротации склада. Положения,
представленные в § 211.87, устанавливают требования к повторному тестированию
одобренных компонентов, первичной упаковки и укупорочных средств. В § 211.89
устанавливаются требования к обращению с отбракованными компонентами, пер-
вичной упаковкой и укупорочными средствами. Правила, изложенные в § 211.94,
относятся к первичной упаковке для лекарственной продукции и укупорочным
средствам; в этом параграфе изложены требования к материалам и чистоте первич-
ной упаковки и укупорочных средств. Кроме того, в этом параграфе изложены тре-
бования к системам укупорки, стандарты и методики.
Соответствия в Правилах GMP Канады. В Правилах GMP Канады [12] вопросы,
связанные с контролем компонентов, а также первичной упаковки и укупорочных
средств для лекарственной продукции, изложены в разъяснениях правила С.02.009
(анализ исходных материалов), правила С.02.010 (анализ исходных материалов),
правила С.02.011 (производственный контроль), правила С.02.014 (отдел контро-
ля качества), правила С.02.016 (тестирование упаковочных материалов) и С.02.017
(тестирование упаковочных материалов). Нормативы, соответствующие § 211.80,
устанавливающие общие требования к обработке, хранению и маркировке компо-
нентов (исходных материалов), упаковке для лекарственной продукции и укупороч-
ных приспособлений (упаковочных материалов) представлены в пунктах (Sections)
188
Часть 2. Международные правила надлежащей производственной практики
1, 20 и 21 разъяснения положения С.02.011. Нормативы, соответствующие § 211.82,
устанавливающие требования к получению, анализу и хранению компонентов до
анализа, а также к первичной упаковке и укупорочным средствам изложены в пун-
ктах 16, 18 и 19 разъяснения положения С.02.011. Нормативы, соответствующие
§ 211.84, устанавливающему требования к анализу и одобрению компонентов,
первичной упаковки и укупорочных средств для лекарственной продукции, изло-
жены в пунктах 6 и 7 разъяснения положения С.02.009, пунктах 1—8 разъяснения
положения С.02.010, пунктах 1 и 2 положения С.02.016, в пункте 4 его разъяснения
и пункте 1 разъяснения положения С.02.017. Разъяснения 6 и 7 правила С.02.009
и разъяснения 1—8 правила С.02.010 описывают требования к компонентам. Пун-
кты 1 и 2, а также разъяснение 4 положения С.02.016 и разъяснение 1 положения
С.02.017 устанавливают требования к первичной упаковке и укупорочным сред-
ствам для лекарственной продукции. В Правилах GMP Канады нет соответствий
положению 211.86, описывающему требования к ротации склада. Нормативы, соот-
ветствующие § 211.87, устанавливающему требования к повторному тестированию
одобренных компонентов, первичной упаковки и укупорочных средств, представ-
лены в пунктах 1—8 разъяснения положения С.02,009. Свод Правил GMP Канады не
содержит указаний по повторному тестированию емкостей и крышек емкостей для
лекарственной продукции. Нормативы, соответствующие § 211.89, устанавливают
требования к обработке отбракованных компонентов, первичной упаковки и уку-
порочных средств для лекарственной продукции, представлены в пункте 14 разъ-
яснения положения С.02.011 и в пункте 5 разъяснения положения С.02.014. Кодекс
Правил G'MP Канады не содержит соответствий § 211.94, в котором приводятся тре-
бования к первичной упаковке и укупорочным средствам для лекарственной про-
дукции.
Соответствия в Правилах GMP ЕС. Вопросы, связанные с контролем компо-
нентов, первичной упаковки и укупорочных средств для лекарственной продукции
в Правилах GMP ЕС [15], изложены в главе 5 (Chapter 5) (продукция) и частично
в главе 6 (контроль качества). Нормативы, соответствующие § 211.80, изложены
в разделах (Subchapters) 5.2, 5.7, 5.10, 5.29 и 5.40—5.42, описывающих требования
к обработке, хранению и маркировке компонентов (исходных материалов), первич-
ной упаковки и укупорочных приспособлений (первичных упаковочных материа-
лов). Нормативы, соответствующие § 211.82 представлены в разделах 5.5, 5.27 и 5.40,
устанавливающих требования к получению, тестированию и хранению до тестиро-
вания компонентов, первичной упаковки и укупорочных средств для лекарствен-
ной продукции. Нормативы, соответствующие § 211.84, сообщаются в разделах 5.31,
5.40 и 6.11—6.22 и Приложении 8. Общие требования к отбору проб и тестированию
изложены в разделах 6.11—6.22. Дополнительные указания по отбору проб содер-
жатся в Приложении 8. Требования к одобренному использованию компонентов,
первичной упаковки и укупорочных средств для лекарственной продукции приве-
дены в разделах 5.31 и 5.40. Нормативы, соответствующие § 211.86, представлены
в разделе 5.7, который устанавливает требования к условиям хранения и ротации.
Нормативы, соответствующие § 211.87, изложены в разделах 5.29 и 5.40, связанных
с повторным тестированием компонентов, первичной упаковки и укупорочных
средств для лекарственной продукции. Нормативы, соответствующие § 211.89, со-
Глава 2.1. Национальные правила и требования GMP. международные требования GMP... 189
держатся в разделе 5.61, устанавливающем требования к обработке отбракованных
компонентов, первичной упаковки и укупорочных средств для лекарственной про-
дукции. Нормативы, соответствующие § 211.94, приводятся в разделе 5.48, устанав-
ливающем требования к первичной упаковке для лекарственной продукции и уку-
порочным приспособлениям.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] во-
просы, связанные с контролем компонентов, первичной упаковки и укупорочных
средств для лекарственной продукции, изложены в главах (Chapters) 14 (материа-
лы), 16 (надлежащая производственная практика) и 17 (надлежащая практика кон-
троля качества). Нормативы, соответствующие § 211.80, сообщаются в разделах
(Subchapters) 14.5, 14.13, 14.14, 14.19—14.21 и 16.2, устанавливающих требования
к обработке, хранению и маркировке компонентов (исходных материалов), первич-
ной упаковки и укупорочных приспособлений (первичным упаковочным материа-
лам). Нормативы, соответствующие § 211.82, представлены в разделах 14.4, 14.9—
14.11 и 14.9, устанавливающих требования к получению, тестированию, маркировке
и хранению компонентов до тестирования, первичной упаковки и укупорочных
средств для лекарственной продукции. Нормативы, соответствующие § 211.84, при-
ведены в разделах 14.12, 14.15 и 17.7—17.17. Требования к отбору проб и анализу (те-
стированию) представлены в разделах 17.7—17.17 и 14.2. Раздел 14.15 устанавливает
требования к использованию одобренных компонентов, первичной упаковки и уку-
порочных средств для лекарственной продукции. Нормативы, соответствующие
§ 211.86, изложены в разделе 14.5, устанавливающем требования к условиям хра-
нения и ротации склада. Нормативы, соответствующие § 211.87, содержатся в раз-
деле 14.13, устанавливающем требования к повторному тестированию одобренных
компонентов. Руководство ВОЗ по GMP не охватывает требований к повторному
тестированию первичной упаковки и укупорочных средств для лекарственной про-
дукции. Нормативы, соответствующие § 211.89, приводятся в разделе 14.28, уста-
навливающем требования к обработке первичной упаковки и укупорочных средств
для лекарственной продукции, прошедших повторное тестирование. Нормативы,
соответствующие § 211.94, изложены в разделе 16.94, устанавливающем требования
к первичной упаковке и укупорочным средствам.
2.1.4.6. Производство и контроль процесса
В правилах GMP США вопросы, связанные с производством и контролем за тех-
нологическими процессами, изложены в подглаве F (Subpart F) [7], включающей
§§211.100, 211.101, 211.103,211.105, 211.НО, 211.111, 211.113 и 211.115. Содержание
подглавы F представлено в табл. 21. Положение § 211.100 устанавливает требования
к процедурам, связанным с контролем производства и процессов, включая реко-
мендации к документации и действиям при обнаружении отклонений. В § 211.101
указаны требования к загрузке компонентов. В § 211.103 описываются требования
к определению выхода продукта. В § 211.105 приводятся требования к идентифи-
кации производственного оборудования, такого как контейнеры, технологические
линии и основного оборудования, используемого в производственном процессе.
В § 211.110 устанавливаются требования к контролю в процессе производства, вклю-
чая тестирование и одобрение обрабатываемых материалов, и распоряжение отбра-
190 Часть 2. Международные правила надлежащей производственной практики
кованными обрабатываемыми материалами. Правила, представленные в §211.111,
содержат требования к временным ограничениям в производстве, включая действия
при отклонениях, превышающих установленные предельные значения. В§211.113
описывается контроль микробиологических загрязнений. В § 211.115 устанавли-
ваются требования к переработке производственных серий, не соответствующих
стандартам и спецификациям.
Таблица 21
Содержание подглавы F главы 211 правил GMP США, относящихся к производству и контролю
процессов [7]
Параграф Предмет
CFK211.100 CFR211.101 CFK 211.103 CFK211.105 CFK211.110 Письменные процедуры, отклонения Загрузка компонентов Расчет выхода Идентификация оборудования Отбор проб и тестирование промежуточной продукции и лекарственных продуктов
CFK211.111 CFK211.113 CFK211.115 Временные ограничения в производственных процессах Контроль микробиологических загрязнений Переработка
Соответствия в Правилах GMP Канады. Вопросы, связанные с контролем про-
изводства и технологических процессов, в кодексе Правил GMP Канады [12]
в основном изложены в разъяснениях положения С.02.011 (контроль производства)
и частично в разъяснениях положения С.02.005 (оборудование), С.02.014 (отдел
контроля качества) и С.02.029 (стерильные продукты). Нормативы, соответствую-
щие § 211.100, изложены в пунктах 1—5 разъяснения положения С.02.011. Разъяс-
нения 1—4 устанавливают требования к производственным процессам, а разъяс-
нение 5 — к отклонениям. Нормативы, соответствующие § 211.101 устанавливают
требования к загрузке компонентов (charge-in components), они представлены в пун-
кте (Section) 22 разъяснения правила С.02.011. Нормативы, соответствующие
§ 211.103, устанавливающие требования к определению выхода продукции, вклю-
чая действия при отклонениях от ожидаемого выхода, представлены в пунктах 6 и 7
разъяснения правила С.02.011. Нормативы, соответствующие § 211.105, устанавли-
вающие требования к идентификации (маркировке) трубопроводов, контейнеров,
оборудования и помещений, используемых при производстве лекарственных про-
дуктов, изложены в пункте 3.5 разъяснения положения С.02.005 и пункте 13 разъ-
яснения положения С.02.011. Нормативы, соответствующие § 211.110, изложены
в пунктах 11 и 14 разъяснения правила С.02.011 и в пункте 5 разъяснения прави-
ла С.02.014. В пункте И разъяснения положения С.02.011 изложены требования
к контролю процесса производства. Свод Правил GMP Канады не содержит тре-
бований к тестированию материалов в процессе обработки. Обращение с отбрако-
ванными материалами описано в пункте 14 разъяснения положения С.02.011 и в
пункте 5 разъяснения положения С.02.014. Нормативы, соответствующие § 211.111,
относящиеся к требованиям по ограничениям во времени в ходе производствен-
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 191
ного процесса, изложены в пункте 24.7 разъяснения правила С.02.011. Нормати-
вы, соответствующие § 211.113, изложены в разъяснениях указания С.02.029, от-
носящегося к производству стерильных продуктов. Нормативы, соответствующие
§211.113, устанавливающие требования к переработке производственных серий, не
соответствующих спецификациям, представлены в пунктах 7—9 разъяснения поло-
жения С.02.014.
Соответствия в GMP ЕС. В Правилах GMP ЕС [15] вопросы, относящиеся к про-
изводству и контролю технологических процессов главным образом сообщаются
в главе 5 (Chapter 5) (производство) и частично в главе 3 (недвижимость и обору-
дование), 4 (документация) и 6 (контроль качества). Нормативы, соответствующие
§ 211.100, представлены в разделе {Subchapter) 5.2, 5.15 и 5.22—5.24. В разделе 5.15
установлены требования к действиям при отклонении от инструкций или проце-
дур. Нормативы, соответствующие § 211.101, изложены в разделах 5.28—5.34, уста-
навливающих требования к загрузке компонентов. Нормативы, соответствующие
§ 211.103, представлены в разделах 5.8 и 5.39, устанавливающих требования к опре-
делению выхода, включая действия при отклонениях от ожидаемого выхода. Нор-
мативы, соответствующие § 211.105, приведены в разделах 3.42 и 5.12, устанавли-
вающих требования к идентификации трубопроводов, контейнеров, оборудования
и помещений, используемых для производства лекарственных продуктов. Нормати-
вы, соответствующие § 211.110, изложены в разделах 3.17,4.10,4.12,5.38, 5,61 и 6.18.
Требования к контролю в процессе производства представлены в разделах 3.17, 5.38
и 6.18. Разделы 4.10 и 4.12 устанавливают требования к спецификациям материалов
в процессе обработки (полупродуктов), а раздел 5.61 — к обращению с отбракован-
ными материалами. Правила GMP ЕС не содержат отдельных указаний по тести-
рованию и одобрению материалов в процессе обработки. Общие указания по от-
бору проб и тестированию даны в разделах 6.11—6.22. Нормативы, соответствующие
§211.111, описывающие ограничения времени в ходе производственного процесса,
представлены в главе 4.15. Нормативы, соответствующие § 211.113, освещены в раз-
деле 5.10 и Приложении 1; они относятся к контролю микробиологических загрязне-
ний. Нормативы, соответствующие § 211.115, изложены в разделах 5.62 и 5.64, уста-
навливающих требования к переработке отбракованных производственных серий.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] во-
просы, связанные с контролем производства и технологических процессов, изложе-
ны в главах 13 (оборудование), 14 (материалы), 15 (документация), 16 (надлежащая
производственная практика) и 17 (надлежащая практика при контроле качества).
Нормативы, соответствующие § 211.100, содержатся в разделах 16.1—16.3. Разде-
лы 16.1 и 16.2 устанавливают требования к производственным операциям, а раз-
дел 16.3 — к действиям при отклонении от инструкций или процедур. Нормативы,
соответствующие § 211.101, приведены в разделах 14.12—14.18, устанавливающих
требования к загрузке компонентов. Нормативы, соответствующие § 211.103, опре-
делены в разделах 16.4 и 16.20, устанавливающих требования к определению выхо-
дов, включая действия при отклонениях от ожидаемого выхода. Нормативы, соот-
ветствующие § 211.105, представлены в разделах 13.3, 13.4 и 16.6, устанавливающих
требования к идентификации трубопроводов, контейнеров, оборудования и про-
изводственных помещений. Нормативы, соответствующие § 211.110, изложены
192 Часть 2. Международные правила надлежащей производственной практики
в разделах 14.28, 15.20, 16,9, 16.16 и 17.8. В разделах 16.9, 16.16 и 17.8 описаны тре-
бования к контролю в процессе производства, а раздел 15.20 — к спецификациям
материалов в процессе обработки (полупродуктам). Требования к обращению с от-
бракованными материалами изложены в разделе 14.28. Нормативы, соответствую-
щие § 211.111, относящиеся к ограничениям времени производственных процес-
сов, освещены в главе 15.23. Нормативы, соответствующие § 211.113, представлены
в разделах 16.10—16.14 и в Приложении 6 7R5902 ВОЗ [40]. Разделы 16.10—16.14
охватывают общие требования к предотвращению перекрестного загрязнения
и бактериального загрязнения в процессе производства, а Приложение 6 — общие
рекомендации по производству стерильных лекарственных продуктов. Нормативы,
соответствующие § 211.115, содержатся в разделах 14.29, 14.31 и 15.40, устанавли-
вающих требования к переработке отбракованных производственных серий.
2.1.4.7. Упаковка и итоговый контроль продукта
В правилах GMP США вопросы упаковки и итогового контроля продукции изло-
жены в подглаве G [7], содержащей §§ 211.122, 211.125, 211.130, 211.132, 211.134
и 211.137. Содержание подглавы G представлено в табл. 22. Положение, представ-
ленное в § 211.122, относится к исследованию материалов и критериям применения,
в нем содержатся рекомендации по приему, идентификации, хранению, обработке,
отбору проб, тестированию и одобрению маркировки и упаковочных материалов,
включая документацию. Положение включает также требования к контролю мар-
кировки, правила обращения с устаревшими этикетками и этикетками с истекшим
сроком годности и особые требования к применению различных способов марки-
ровки. В § 211.125 устанавливаются требования к выпуску этикеток, включая те-
стирование маркировочных материалов, контроль за расхождением между числом
этикеток выпущенных, использованных и возвращенных, а также за обращени-
ем с возвращенными или выпущенными в избыточном количестве этикетками.
В § 211.130 описываются требования к операциям упаковки и маркировки в изло-
жении письменных процедур. В § 211.132 определяются требования к упаковкам
с контролем первого вскрытия. В § 211.134 устанавливаются требования к провер-
кам упакованной продукции и продуктов с нанесенной маркировкой, включая тре-
бования к выборке (отбору проб), осмотру и документации. В § 211.137 приводятся
требования к срокам годности, включая освобождение от требований.
Таблица 22
Содержание подглавы G главы 211 правил GMP США, относящихся к упаковке и итоговому
контролю продукции [7]
Параграф Предмет
CPR211.122 Осмотр материалов и критерии применимости
CFR 211.125 Выпуск этикеток
CFK 211.130 Операции упаковки и маркировки
сиг 211.132 Требования к упаковке с контролем первого вскрытия для безрецептурных лекарственных продуктов для медицинского применения
сиг211.134 Проверка (инспекция) лекарственных продуктов
сиг211.137 Датировка срока годности
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 193
Соответствия в Правилах GMP Канады. В своде Правил GMP Канады [12] во-
просы, связанные с упаковкой и итоговым контролем продукции, изложены в разъ-
яснениях положений С.02.011 (производственный контроль), С.02.016 и С.02.017
(тестирование упаковочного материала), С.02.019 (тестирование готового продук-
та) и С.02.027 (стабильность). Нормативы, соответствующие § 211.122, представле-
ны в пунктах (Section) 1, 8 и 9 разъяснения правила С.02.017, а также в пунктах 1
и 4—7 разъяснения правила С.02.016. В разъяснении правила С.02.011, в пунктах 1,
16, 43 и 48 описываются общие требования к обращению с упаковочным материа-
лом и этикетками, включая требования к приемке и хранению. Пункт 8 разъясне-
ния указания С.02.017 устанавливает требования к идентификации упаковочных
и маркировочных материалов. Требования к тестированию упаковочных и марки-
ровочных материалов представлены в пунктах 1 и 9 разъяснения положения С.02.17.
В пунктах 1 и 4 разъяснения положения С.02.016 и пунктах 6 и 7 разъяснения по-
ложения С.02.017 описываются требования к одобрению упаковочных и марки-
ровочных материалов. Разъяснения правила С.02.011 (пункты 44—47) описывают
требования к применению этикеток в рулонах, разрезных этикеток, дублирующих
печатных устройств и мониторингу процесса печати. Требования к обращению
с устаревшими и вышедшими из употребления упаковочными и маркировочными
материалами представлены в пункте 40 разъяснения положения С.02.11 и пункте 5
разъяснения положения С.02.016. Нормативы, соответствующие § 211.125, пред-
ставлены в пунктах 39 и 42 разъяснения положения С.02.011 и в пункте 8 разъ-
яснения указания С.02.017. Пункт 8 разъяснения правила С.02.017 устанавливает
требования к анализу упаковочных и маркировочных материалов. Требования
к контролю и действия при обнаружении несоответствия между количеством эти-
кеток выпущенных, использованных и возвращенных описаны в пункте 42, а тре-
бования к обращению с неиспользованными упаковочными и маркировочными
материалами, содержащими код производственной серии, освещены в пункте 39
разъяснения положения С.02.011. Нормативы, соответствующие § 211.130, в кото-
ром устанавливаются требования к операциям упаковки и маркировки, изложены
в пунктах 29—38 разъяснения положения С.02.011. В кодексе Правил GMP Кана-
ды нет соответствий § 211.132, в котором устанавливаются требования к этикеткам
с контролем первого вскрытия. Нормативы, соответствующие § 211.134, устанавли-
вающие требования к проверкам (инспекциям) упаковочного или маркировочного
материала, представлены в пункте 1 разъяснения положения С.02.019. Нормативы,
соответствующие § 211.137, устанавливающие требования к срокам годности, из-
ложены в правиле С.02.027 и в пункте 1 его разъяснения.
Соответствия в Правилах GMP ЕС. В Правилах GMP ЕС [15] вопросы упаковки
и итогового контроля продукции освещаются главным образом в главе 5 (произ-
водство) и частично в главах 4 (документация) и 6 (контроль качества). Нормати-
вы, соответствующие § 211.122, представлены в разделах 4.11, 4.19, 4.21—4.23, 5.2,
5.40—5.43 и 5.50—5.52. Разделы 4.19, 4.21—4.23, 5.2 и 5.40—5.42 охватывают требова-
ния к приобретению, обработке, контролю, хранению и идентификации упаковоч-
ных и маркировочных материалов. Спецификации упаковочных и маркировочных
материалов представлены в разделе 4.11, а требования к обращению с устаревши-
ми и вышедшими из употребления упаковочными и маркировочными материала-
194 Часть 2. Международные правила надлежащей производственной практики
ми — в разделе 5.43. В разделе 5.51 изложены требования к применению разрез-
ных этикеток, этикеток в рулонах и нанесению печати на тару в режиме офлайн
off-line overprinting. Требования к контролю операций печати и нанесения марки-
ровки установлены в разделах 5.50 и 5.52. Нормативы, соответствующие § 211.125
представлены в разделах 5.2, 5.56 и 5.57. Раздел 5.2 устанавливает общие требования
к обращению с упаковочными и маркировочными материалами. Требования к кон-
тролю несоответствий между количеством этикеток выпущенных, использованных
и возвращенных, представлены в разделе 5.56, а требования к обращению с неис-
пользованными упаковочными и маркировочными материалами, содержащими
код производственной серии, изложены в разделе 5.57. Нормативы, соответствую-
щие § 211.130, содержатся в разделах 5.2 и 5.44—5.49. Раздел 5.2 устанавливает об-
щие требования к обращению с упаковочными и маркировочными материалами,
а разделы 5.44—5.49 представляют требования к операциям упаковки и маркировки.
Правила GMP Европейского Сообщества не содержит соответствий § 211.132, со-
держащему рекомендации к упаковкам с контролем первого вскрытия. Нормативы,
соответствующие § 211.134, изложены в разделах 5.54 и 6.3, устанавливающих тре-
бования к контролю упакованных продуктов и продуктов с нанесенной маркиров-
кой. Правила GMPEC не содержат соответствий § 211.137, в котором описываются
требования к указаниям сроков годности.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] во-
просы, относящиеся к упаковке и итоговому контролю продукции, представлены
в главах 6 (отзыв продукции), 12 (недвижимость), 14 (материалы), 15 (документа-
ция), 16 (надлежащая производственная практика) и 17 (надлежащая практика при
контроле качества). Нормативы, соответствующие § 211.122, представлены в раз-
делах 12.21, 14.19-14.23, 15.18, 16.2, 17.14 и 17.16. Разделы 6.2, 12.21, 14.19-14.21,
14.23 и 17.16 содержат требования к приобретению, обработке, контролю, хранению
и идентификации упаковочных и маркировочных материалов. Требования к одо-
брению упаковочных и маркировочных материалов приводятся в разделах 17.14
и 15.18. Раздел 14.20 устанавливает требования к применению этикеток в рулонах
и разрезанных этикеток, а раздел 14.22 — требования к обращению с устаревшими
и вышедшими из употребления этикетками и упаковочными материалами. Норма-
тивы, соответствующие § 211.125, представлены в разделах 16.2, 16.34 и 16.35. Раз-
дел 16.2 излагает общие требования к обработке упаковочных и маркировочных
материалов, а раздел 16.34 — требования к действиям при несоответствии между
количеством этикеток выпущенных, использованных и возвращенных. Требования
к обращению с неиспользованными упаковочными и маркировочными материала-
ми, содержащими код производственной серии, представлены в разделе 16.35. Нор-
мативы, соответствующие § 211.130, сообщаются в разделах 16.25—16.30, устанав-
ливающих требования к операциям упаковки и маркировки. Руководство ВОЗ по
GMPnc содержит соответствий нормативу, закрепленному в§211.132,в котором
изложены требования к упаковке с контролем первого вскрытия. Нормативы, соот-
ветствующие § 211 134, представлены в разделе 16.32, устанавливающем требования
к контролю упакованных продуктов и продуктов с нанесенной маркировкой. Нор-
мативы, соответствующие § 211.137, приведены в разделе 17.24, устанавливающем
требования к определению сроков годности и спецификаций по условиям хранения.
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 195
2.1.4.8. Хранение и поставки
В руководстве по GMP США вопросы, относящиеся к хранению и поставкам ле-
карств, представлены в подглаве Н [7], включающей §§ 211.142 и 211.150. Содер-
жание подглавы Н представлено в табл. 23. Положение, изложенное в § 211.142,
устанавливает требования к процедурам складского хранения, включая карантин
и хранение на складе, ав§211.150 описываются требования к процедурам поставок,
включая порядок поставок и отзывы продукции.
Таблица 23
Содержание подглавы Н главы 211 правил GMP США, относящихся к хранению
и распространению лекарств [7]
Параграф Предмет
CER211.142 Процедуры складского хранения
CFR211.150 Процедуры поставок
Соответствия в Правилах GMP Канады. В кодексе Правил GMP Канады [12] во-
просы, относящиеся к хранению и поставкам лекарств, представлены в разъясне-
ниях положений С.02.004 (недвижимость), С.02.011 (производственный контроль),
С.02.012 (производственный контроль) и С.02.019 (тестирование готовой продук-
ции). Нормативы, соответствующие § 211.142, устанавливающие требования к ка-
рантину и хранению продуктов, описываются в пунктах 1 и 49 разъяснения пра-
вила С.02.011, пункте 11.4 разъяснения правила С.02.004 и пункте 2 разъяснения
правила С.02.019. Нормативы, соответствующие § 211.150, устанавливающие требо-
вания к распространению и отзыву продукции, представлены в пункте 1 разъясне-
ния указания С.02.011 и пункте 1 разъяснения указания С.02.012.
Соответствия в Правилах GMPEC. В Правилах GMP EC [15] вопросы, связан-
ные с хранением и поставками лекарств, изложены в главах 4 (документация),
5 (производство) и 8 (претензии и отзыв продукции). Нормативы, соответствующие
§ 211.142, представлены в разделах 5.2, 5.58 и 5.60, устанавливающих требования
к хранению и карантину продукции. Нормативы, соответствующие § 211.150, со-
держатся в разделах 4.25, 5.2 и 8.8—8.15, устанавливающих требования к распростра-
нению и отзыву продукции.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] вопро-
сы, связанные с хранением и поставками лекарств, представлены в главах 6 (отзыв
продукции), 14 (материалы), 15 (документация) и 16 (надлежащая производствен-
ная практика). Нормативы, соответствующие § 211.142, приведены в разделах 14.4,
14.26 и 16.2, устанавливающих требования к хранению и карантину продукции.
Нормативы, соответствующие § 211.150, содержатся в разделах 6.1—6.86 15.45 и 16.2,
устанавливающих требования к распространению и отзыву продукции.
2.1.4.9. Контроль качества
В правилах GMP США [7] вопросы, связанные с контролем качества, изложены
в подглаве I, в которую входят инструкции, представленные в §§ 211.160, 211.165,
211.166, 211.167, 211.170, 211.173 и 211.176. Содержание подглавы I показано
в табл. 24. В § 211.160 устанавливаются требования к организации контроля ка-
чества, такие как спецификации, стандарты, планы отбора образцов и аналити-
196
Часть 2. Международные правила надлежащей производственной практики
ческие процедуры. Кроме того, в нем содержатся требования, установленные для
калибровки инструментов, аппаратуры, измерительных приборов и записывающих
устройств. В § 211.165 устанавливаются требования к лабораторному анализу про-
изводственных серий перед отпуском продукции, включая требования к отбору об-
разцов, анализу и одобрению.
Таблица 24
Содержание подглавы I главы 211 правил GMP США, относящихся к лабораторному контролю
Параграф Предмет
C/R211.160 CFR211.165 CFR 211.166 C/R211.167 CER211.170 CFK211.173 СИ? 211.176 Общие требования Испытания и выпуск в обращение Исследование стабильности Особые требования к испытаниям Архивные образцы Лабораторные животные Контаминация пенициллином
Также в § 211.165 устанавливаются требования к обращению с отбракованными
лекарственными продуктами. В § 211.166 определяются требования к исследова-
нию стабильности, включая требования к определению сроков годности и требо-
вания к исследованию стабильности гомеопатических лекарственных продуктов.
Положение § 211.167 относится к особым аналитическим требованиям, включаю-
щим анализ стерильных продуктов, офтальмологических мазей и дозированных
форм с контролируемым высвобождением. В § 211.170 устанавливаются требова-
ния к архивным образцам, включая идентификацию, количество, период хранения
и условия хранения. Кроме того, положение описывает требования к исследовани-
ям разложения продукта. Правила § 211.173 относятся к лабораторным животным,
включая требования к их содержанию и контролю. В § 211.176 устанавливаются тре-
бования к анализу на загрязнение пенициллина и правила обработки лекарственно-
го продукта, загрязненного пенициллином.
Соответствия в Правилах GMP Канады. В своде Правил GMP Канады [12] во-
просы, связанные с контролем качества, изложены в разъяснениях правил С.02.004
(здания и помещения), С.02.009 (анализ исходных материалов), С.02.011 (про-
изводственный контроль), С.02.014 (отдел контроля качества), С.02.015 (отдел
контроля качества), С.02.016 и С.02.017 (тестирование упаковочных материалов),
С.02.018 (тестирование готовой продукции), С.02.025 (пробы), С.02.026 (пробы),
С.02.027 и С.02.028 (стабильность). Нормативы, соответствующие § 211.160, уста-
навливающие общие требования к лабораторному контролю, представлены в указа-
нии С.02.009 и пунктах 1—3, 5—6 его разъяснения, в указании С.02.016 и пунктах 1—3
его разъяснения, в пункте 1 разъяснения указания С.02.017, в указании С.02.018
и пунктах 1—5 его разъяснения и в пункте 6.4 разъяснения С.02.015. Нормативы,
соответствующие § 211.165, устанавливающие требования к отпуску продукта для
распределения, включая анализ готовых лекарственных продуктов и обращение
с отбракованными лекарственными продуктами, представлены в пунктах 7, 14
разъяснения положения С.02.011, в пунктах 2, 5 разъяснения положения С.02.014,
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 197
в пункте 3 разъяснения положения С.02.015 и в пункте 2 положения С.02.018, а так-
же в пунктах 1, 4 его разъяснения. Нормативы, соответствующие § 211.166, уста-
навливающие требования к исследованию стабильности, представлены в пункте 1
разъяснения правила С.02.027 и пунктах 1 и 2 разъяснения правила С.02.028. Ка-
надский кодекс Правил GMP не содержит отдельных требований к исследованию
стабильности гомеопатических лекарственных продуктов. Нормативы, соответству-
ющие § 211.167, в котором установлены требования к испытанию на стерильность,
представлены в пунктах 1—4 разъяснения положения С.02.029 (стерильные про-
дукты). Правила GMP Канады не содержат указаний, относящихся к тестированию
офтальмологических мазей и дозированных форм замедленного высвобождения.
Нормативы, соответствующие § 211.170, устанавливающие требования к архивным
образцам, представлены в пункте 1 положения С.02.025 и в положении С.02.026
и пунктах 1 и 3—5 их разъяснения. Нормативы, соответствующие правилу § 211.173,
относящемуся к лабораторным животным, изложены в пункте 2.4 разъяснения по-
ложения С.02.004. Кодекс Правил GMP Канады не содержит соответствия §211.176,
в котором описываются требования к анализу загрязнения пенициллином и дей-
ствиям в случае обнаружения загрязнения пенициллином.
Соответствия в Правилах СТИР ЕС. В Правилах GMPEC [15] вопросы, связан-
ные с контролем качества, представлены в главах 1 (управление качеством), 4 (до-
кументация), 5 (производство) и 6 (контроль качества), а также в Приложениях 1
(производство стерильных медицинских продуктов), 9 (производство жидкостей,
кремов и мазей) и 19 (стандартные и архивные образцы). Нормативы, соответству-
ющие § 211.160, содержатся в разделах 1.4, 4.2, 4.20—4.13, 5.15, 6.7 и 6.18, устанав-
ливающих общие требования к контролю качества. Нормативы, соответствующие
§ 211.165, приведены в разделах 4.22,4.23, 5.61, 5.62, 6.3, 6.11 и 6.15, устанавливаю-
щих требования к отпуску продукции для распределения, анализу готовых лекар-
ственных продуктов и обращению с отбракованными лекарственными продуктами.
Правила GMP ЕС не содержат соответствий положению § 211.166, устанавливаю-
щему требования к исследованию стабильности. Тем не менее, существует специ-
альное руководство «Исследование стабильности активных ингредиентов и готовых
продуктов» [59], содержащее указания по исследованию стабильности. Далее, раз-
делы 6.23—6.33 охватывают требования к программе постоянных исследований ста-
бильности. Нормативы, соответствующие § 211.167, изложены в Приложениях 1 и 9.
Пункт 93 Приложения 1 устанавливает требования к испытаниям на стерильность,
а Приложения 9 — требования к мазям. Правила GMP ЕС не содержат указаний
по анализу дозированных форм с контролируемым высвобождением. Нормативы,
соответствующие § 211.170, представлены в разделах 1.4 и 6.12 и в Приложении 19,
устанавливающем требования к архивным образцам. Нормативы, соответствующие
§ 211.173, приведены в разделах 3.33 и 6.22, устанавливающих требования к содер-
жанию животных. Правила GMP ЕС не содержат соответствия положению 211.76,
в котором изложены требования к контролю загрязнения пенициллином и действи-
ям в случае обнаружения загрязнения пенициллином.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] вопро-
сы, связанные с контролем качества, изложены в главах 14 (материалы), 15 (доку-
ментация), 16 (надлежащая производственная практика) и 17 (надлежащая практи-
198 Часть 2. Международные правила надлежащей производственной практики
ка при контроле качества). Нормативы, соответствующие § 211.160, представлены
в разделах 15.14—15.16, 15.18—15.21,16.3 и 16.23, устанавливающих общие требова-
ния к лабораторному контролю. Нормативы, соответствующие § 211.165, сообща-
ются в разделах 14.28,14.29,15.13,15.42,17.7—17.13,17.19 и 17.20, устанавливающих
требования к отпуску продукции для распространения, включая анализ готовой ле-
карственной продукции и обращение с отбракованными лекарственными продук-
тами. Нормативы, соответствующие § 211.166, приведены в разделах 17.23—17.26,
устанавливающих рекомендации к исследованию стабильности. Руководство ВОЗ
по GMP не содержит отдельных рекомендаций по исследованию стабильности го-
меопатических лекарственных продуктов. Нормативы, соответствующие § 211.167,
представлены в Приложении 6 TRS 902 ВОЗ [40], устанавливающие требования
к исследованию стабильности. Руководство ВОЗ по GMPws содержит указаний, от-
носящихся к анализу офтальмологических мазей и дозированных форм с контроли-
руемым высвобождением. Нормативы, соответствующие § 211.170, изложены в раз-
деле 17.22, устанавливающем требования к архивным образцам. Руководство ВОЗ
по GMPwe. содержит соответствий положению §211.173, в котором представлены
требования к содержанию лабораторных животных. Кроме того, оно не имеет соот-
ветствий положению § 211.176, описывающему требования к контролю загрязнения
пенициллином.
2.1.4.10. Документация
В США вопросы, связанные с учетной документацией, представлены в подглаве J
(Subpart J) [7], включающей §§ 211.180, 211.182, 211.184, 211.186, 211.188, 211.192,
211.194, 211.196 и 211.198. Содержание подглавы J представлено в табл. 25.
Таблица 25
Содержание подглавы J главы 211 правил GMP США [7], относящихся к документации
Параграф Предмет
CFR 211.180 CFR 211.182 CER 211.184 Общие требования Журнал очистки и эксплуатации оборудования Записи, относящиеся к компонентам, первичной упаковке, укупорочным средствам и маркировке
CFR211.186 CFR211.188 CFR211.192 CFR 211.194 CFR 211.196 CFR211.198 Регламент производства Записи по производству и контролю серий Анализ производственных записей Лабораторные записи Отчетность о поставках Рассмотрение претензий
В § 211.180 устанавливаются общие требования к документации, включая ве-
дение документации, время хранения записей и доступность записей. Кроме того,
положение устанавливает требования к ежегодной оценке стандартов качества.
В § 211.182 предписываются требования к индивидуальным журналам эксплуата-
ции оборудования. В § 211.184 определяются требования к записям по компонен-
там, первичной упаковке для лекарственной продукции, укупорочным средствам
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 199
и маркировке. В § 211.186 содержатся требования к основным записям по произ-
водственным операциям и процедурам контроля. В § 211.188 сообщаются требо-
вания к протоколам производственных серий и записям, связанным с контролем
продукции. В § 211.192 устанавливаются требования к анализу производственных
и контрольных записей, в том числе требования к расследованию всех необъяснен-
ных несоответствий. В § 211.194 приводятся требования к лабораторным записям,
включая требования к документированию модификаций. Кроме того, в § 211.194
описывается требование к документации по анализу и стандартизации эталонных
образцов, реагентов и стандартных растворов; калибровке лабораторных приборов
и записывающих устройств и исследований стабильности. Положение § 211.196
устанавливает требования к отчетности по распространению лекарственной про-
дукции. Положение § 211.198 содержит требования к работе с претензиями, вклю-
чая ведение и время хранения журнала регистрации претензий.
Соответствия в Правилах GMP Канады. В кодексе Правил GMP Канады [12]
вопросы, относящиеся к документации, в основном содержатся в положениях
С.02.021, С.02.022, С.02.023 и С.02.024 (записи) и в их разъяснениях, и частично
в разъяснениях положений С.02.005 (оборудование), С.02.010 (анализ исходных
материалов), С.02.011 и С.02.012 (производственный контроль), С.02.014 (отдел
контроля качества) и С.02.017 (тестирование упаковочного материала). Нормати-
вы, соответствующие § 211.180, устанавливающие общие требования к ведению за-
писей, включая периодическую оценку качества (самоинспекция), и время хране-
ния записей изложены в положениях С.02.021, С.02.022, С.02.023 и С.02.024 и их
разъяснениях, а также в пункте 2 разъяснения положения С.012. Нормативы, со-
ответствующие § 211.182, в котором устанавливаются требования к индивидуаль-
ным журналам эксплуатации оборудования, приведены в пункте 5.5 разъяснения
положения С.02.005. Нормативы, соответствующие § 211.184, в котором устанав-
ливаются требования к ведению записей, относящихся к компонентам, первичной
упаковке, укупорочным средствам и маркировочным материалам для лекарствен-
ной продукции, представлены в пунктах 4 и 5 разъяснения положений С.02.20—24,
в пункте 5 разъяснения положения С.02.010 и в пункте 7 разъяснения положения
С.02.017. Нормативы, соответствующие § 211.186, устанавливающие требования
к основным производственным и контрольным записям (мастер-формулы произ-
водственных и упаковочных операций), представлены в пунктах 23—25 разъяснения
положения С.02.011 и в пункте 1.1 разъяснения положений С.02.020—24. Норма-
тивы, соответствующие § 211.188, в котором определяются требования к прото-
колам производственных серий и контрольным записям (протокол производства
и упаковки продукции для производственной серии), описываются в пунктах 26,
27,29 и 30 разъяснения положения С.02.011 и в пункте 1.2 разъяснения положений
С.02.020—24. Нормативы, соответствующие § 211.192, предписывающие требования
к анализу и одобрению производственных и контрольных записей, включая рассле-
дование случаев отклонений в производственных сериях, представлены в пункте 2
разъяснения правила С.02.014. Нормативы, соответствующие § 211.194, в котором
сообщаются требования к лабораторным записям, представлены в пунктах 6.4, 6.6
и 6.7 разъяснения положения С.02.015. Нормативы, соответствующие § 211.196,
в котором устанавливаются требования к отчетности по распространению лекар-
200 Часть 2, Международные правила надлежащей производственной практики
ственной продукции, представлены в пункте 1.6 разъяснения указания С.02.012
и в пункте 2.1 разъяснения указания С.02.0120—24. Нормативы, соответствующие
§ 211.198, в котором содержатся требования к ведению журналов регистрации пре-
тензий, включая время их хранения, представлены в пункте 4 разъяснения прави-
ла С.02.015, в пункте 3.1 разъяснения правила С.02.020—24 и в правиле С.02.023.
Соответствия в Правилах 6Л/РЕС. В Правилах GMPEC [15] вопросы, связанные
с ведением документации, в основном освещаются в главе 4 (документация) и ча-
стично в главах 1 (управление качеством), 5 (производство), 6 (контроль качества),
8 (претензии и отзыв продукции) и 9 (самоинспекция). Нормативы, соответствую-
щие § 211.180, изложены в разделах 4.1—4.9, 6.8 и 9.1—9.3, устанавливающих общие
требования к ведению записей, включая периодическую оценку качества (самоин-
спекция) и время хранения записей. Нормативы, соответствующие § 211.182, пред-
ставлены в разделах 4.28 и 4.29, устанавливающих требования к индивидуальным
журналам эксплуатации оборудования. Нормативы, соответствующие 211.184,
приведены в разделах 4.19 и 4.20, устанавливающих требования к ведению запи-
сей, связанных с получением компонентов, емкостей для лекарственной продук-
ции, крышкам емкостей и маркировочных материалов. Нормативы, соответствую-
щие § 211.186, представлены в разделах 4.14—4.16, устанавливающих требования
к основным производственным и контрольным записям (производственная ре-
цептура manufacturing formula, инструкции по обработке и упаковке). Нормативы,
соответствующие § 211.188, содержатся в разделах 4.17 и 4.18, устанавливающих
требования к протоколам производственных серий и контрольным записям (про-
токол производства и упаковки серии). Нормативы, соответствующие § 211.192, со-
общаются в разделах 1.4,4.3,4.24,5.8 и 5.39, устанавливающих требования к анали-
зу и одобрению производственных и контрольных записей, включая расследование
случаев необъясненных несоответствий. Нормативы, соответствующие § 211.194,
представлены в разделах 3.41, 6.7, 6.17, 6.20 и 6.21, устанавливающих требования
к лабораторным записям. Нормативы, соответствующие § 211.196, изложены в раз-
деле 4.25, устанавливающем требования к ведению отчетности по распространению
лекарственной продукции. Нормативы, соответствующие § 211.198, приведены
в разделах 4.26 и 8.1—8.8, устанавливающих требования к работе с жалобами.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] во-
просы, связанные с документацией, изложены в главах 5 (претензии), 8 (самоин-
спекция и аудиты качества), 13 (оборудование), 14 (материалы), 15 (документация),
16 (надлежащая производственная практика) и 17 (надлежащая практика при кон-
троле качества). Нормативы, соответствующие § 211.180, представлены в разде-
лах 8.1—8.6 и 15.1—15.9, устанавливающих общие требования к ведению записей,
включая периодическую оценку качества (самоинспекция) и время хранения за-
писей. Нормативы, соответствующие § 211.182, изложены в разделах 15.46 и 15.47,
устанавливающих требования к индивидуальным журналам эксплуатации оборудо-
вания. Нормативы, соответствующие § 211.184, приведены в разделах 15.32 и 15.33,
устанавливающих требования к ведению записей, связанных с получением компо-
нентов, емкостями для лекарственной продукции, крышками емкостей и маркиро-
вочными материалами. Нормативы, соответствующие § 211.186, содержатся в раз-
делах 15.22—15.24, устанавливающих требования к основным производственным
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 201
и контрольным записям (мастер-формула и инструкции по упаковке). Нормативы,
соответствующие § 211.188, представлены в разделах 15.25—15.30, устанавливающих
требования к протоколам производственных серий и контрольным записям (про-
токолы обработки и упаковки серии). Нормативы, соответствующие § 211.192, из-
ложены в разделах 16.4,16.20 И17.21, устанавливающих требования к анализу и одо-
брению производственных и контрольных записей, включая расследование случаев
необъясненных несоответствий. Нормативы, соответствующие § 211.194, описаны
вразделах 13.5,14.34, 14.35, 14.41, 15.12, 15.42, 15.43 и 16.23, устанавливающих тре-
бования к лабораторным записям. Нормативы, соответствующие § 211.196, пред-
ставлены в разделе 15.45, устанавливающем требования к ведению отчетности
по распространению лекарственной продукции. Нормативы, соответствующие
§ 211.198, приводятся в разделах 5.1—5.10, устанавливающих требования к работе
с претензиями.
2.1.4.11. Возвращенные некачественные лекарственные препараты
и лекарственные препараты с нарушением условий хранения
В правилах GMP США [7] вопросы, связанные с возвращенной некачественной ле-
карственной продукцией и препаратами с нарушением условий хранения, изложе-
ны в подглаве К, включающей §§ 211.204 и 211.208. Содержание подглавы К пред-
ставлено в табл. 26.
Таблица 26
Содержание подглавы К главы 211 правил GMP США, относящихся к возвращенным
некачественным лекарственным препаратам и лекарственным препаратам с нарушением условий
хранения [7]
Параграф Предмет
CFP 211.204 Возвращенные лекарственные продукты
СН? 211.208 Лекарственные продукты с нарушением условий хранения
Положение § 211.204 устанавливает требования к обращению с возвращенны-
ми лекарственными продуктами, включая вопросы переработки и документации.
В § 211.208 устанавливаются правила, относящиеся к лекарственным продуктам
с нарушением условий хранения.
Соответствия в Правилах GMP Канады. В своде Правил GMP Канады [12] вопро-
сы, связанные с возвращенными лекарственными продуктами и лекарственными
продуктами с нарушением условий хранения, изложены в разъяснениях положе-
ния С.02.014 (отдел контроля качества). Нормативы, соответствующие § 211.204,
устанавливают требования к обращению с возвращенными лекарственными про-
дуктами, представлены в пункте 4 разъяснения правила С.02.014. Кодекс правил
GMP Канады не содержит соответствий § 211.208, в котором изложены правила, от-
носящиеся к лекарственным продуктам с нарушением условий хранения.
Соответствия в Правилах GMP ЕС. В Правилах GMP ЕС [15] вопросы, связан-
ные с возвращенными лекарственными продуктами и лекарственными продуктами
с нарушением условий хранения, изложены в главах 4 (документация) и 5 (произ-
водство). Нормативы, соответствующие § 211.204, изложены в разделах 4.26 и 5.26,
202 Часть 2. Международные правила надлежащей производственной практики
устанавливающих требования к обращению с возвращенными лекарственными
продуктами. Правила GMP ЕС не имеют соответствий § 211.208, в котором описы-
ваются правила, относящиеся к лекарственным продуктам с нарушением условий
хранения.
Соответствия в Руководстве ВОЗ по GMP. В Руководстве ВОЗ по GMP [36] вопро-
сы, связанные с возвращенными лекарственными продуктами и лекарственными
продуктами с нарушением условий хранения, содержатся в главе 14 (материалы).
Нормативы, соответствующие § 211.204, приведены в разделе 14.33, устанавливаю-
щем требования к обращению с возвращенными лекарственными продуктами. Ру-
ководство ВОЗ по GMPw. включает соответствий § 211.208, содержащему предпи-
сания относительно лекарственных продуктов с нарушением условий хранения.
Литература
1. Immel, В. К. (2001), A brief history of the GMPs for pharmaceuticals, Pharm. Technol. No. Am., 25
(7), 44-48.
2. Anonymous Pharmaceutical Administration and Regulations in Japan, Japan Pharmaceutical Manu-
facturers Association, available: http://www.jpma.or.jp/english/library/pdf72005.pdf.
3. Vesper, J. L. (2003), So what are GMPs, anyway? BioProcess Int., 1 (2), 24-29.4. Rosin, L. J. (2006),
Regulatory affairs: If you didn’t write it down, it didn’t happen, BioProcess Int., 4 (3, Suppl), 16-23.
5. Anonymous (2002), Sec. 351: Adulterated drugs and devices, in United States Code, Title 21, Chapter
9, Subchapter V, Part A, U.S. Government Printing Offi ce, Washington, DC, available: http://fiwe-
bgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=browse_usc&docid=Cite:+21USC351. TABLE 26
Contents of Subpart К of Part 211 of U. S. GMP Regulations Covering Returned and Salvaged Drug
Products [7] Section Subject CFR 211.204 Returned drug products CFR 211.208 Drug product sal-
vaging
6. Anonymous (2005), Part 210: Current good manufacturing practice in manufacturing, processing,
packing, or holding of drugs: General, in Code of Federal Regulations, Title 21, Chapter I, U.S. Gov-
ernment Printing Offi ce, Washington, DC, pp. 118—119, available: http://www.access.gpo.gov/nara/
cfr/waisidx_05/21 cfr210 05.html.
7. Anonymous (2005), Part 211: Current good manufacturing practice for fi nished pharmaceuticals, in
Code of Federal Regulations, Title 21, Chapter I, U.S. Government Printing Offi ce, Washington, DC,
pp. 120-141, available: http://www.access.gpo.govnara/cff/waisidx_05/21cff211_05.html.
8. Grazal, J. G., and Earl, D. S. (1997), EU and FDA regulations: Overview and comparison, Qual. As-
sur. J., 2, 55-60.
9. Anonymous (2006), Guidance page, available: http://www.fda.gov/cder/guidance/index. htm. 10.
Anonymous (2001), Guidance for industry: Q7A Good manufacturing practice guidance for active
pharmaceutical ingredients, Offi ce of Training and Communications, available: http://www.fda.gov/
cber/gdlns/ichactive.pdf.
11. Anonymous (2005), Division 2: Good manufacturing practices, in Consolidated Statutes and Regula-
tions, Food and Drugs Act, Food and Drug Regulations, Part C, Department of Justice, Canada, avail-
able: http://laws.justice.gc.ca/en/f-27/c.r.c.-c.870/230079.html.
12. Anonymous (2002), Good manufacturing practices guidelines, Version 2, Health Products and Food
Branch Inspectorate, available: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/compli-
conformZ2002v2_e.pdf.
13. Anonymous (2001), Directive 2001/83/EC of the European Parliament and of the Council, Off. J.
Eur. Union, 44 (L311), 67-128, available: http://europa.eu.int/eur - lex/pri/en/oj/dat/2001/l_311/
l_3112OO11128enOO67O128.pdf.
14. Anonymous (2003), Commission directive 2003/94/EC, Off. J. Eur. Union, 46 (L262), 22-26, avail-
able: http://europa.eu.int/eurlex/pri/en/oj/dat/2003/l_262/l_26220031014en00220026.pdf.
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 203
15. Anonymous (2005), EU guidelines to good manufacturing practice, in The Rules Governing Medici-
nal Products in the European Union, Vol. 4, European Commission Enterprise and Industry Director-
ate-General, available: http://pharmacos.eudra.org/F2/eudralex/vol-4/home.htm.
16. Anonymous (1998), Quality and biotechnology, in The Rules Governing Medicinal Products in the
European Union, Vol. ЗА, European Commission Directorate General III, available: http://pharma-
cos. eudra.org/F2/eudralex/vol3/home.htm.
17. Anonymous (2005), Regulations for Buildings and Facilities of Pharmacies, etc., MHLW Ministerial
Ordinance, Yakuji Nippon Ltd., Tokyo, No.73.
18. Kim, Y.-O., Ha, K.-W., and Choi, K.-S. (2001), Safety evaluation for new drug approval in Korea,
Drug Info. L, 35 (1), 285-291.
19. Shin, S.-G. (1998), Current status of clinical trials in the Republic of Korea, D rug Info. J., 32 (Suppl),
1217S-1222S.
20. Anonymous (2000), Competition in the Pharmaceutical Industry—Republic of Korea, Working Party
No. 2 on Competition and Regulation, Committee on Competition Law and Policy, OECD Publish-
ing, Paris.
21. Anonymous (2001), Drug Administration Law of the People’s Republic of China, Order of the Presi-
dent of the People’s Republic of China No. 45, available: http://www.sfda.gov. cn/cmsweb/webportal/
W45649037/A48335975.html.
22. Deng, R., and Kaitin, К. I. (2004), The regulation and approval of new drugs in China, Drug Info. J.,
38 (1), 29-39.
23. Anonymous (2005), The Drugs and Cosmetics Act and Rules, Ministry of Health and Family Welfare,
Department of Health, available: http://cdsco.nic.in/html/Drugs&CosmeticAct.pdf.
24. Anonymous (2005), Schedule M: Good manufacturing practices and requirements of premises, plant
and equipment for pharmaceutical products, in The Drugs and Cosmetics Act and Rules, Ministry of
Health and Family Welfare, Department of Health, pp. 386-436, available: http://cdsco.nic.in/html/
Drugs&CosmeticAct.pdf.
25. Venkateswarlu, M. (2006), Why do we need revision of schedule M, available: http://www. pharm-
abiz.com, 2006.
26. Anonymous (2006), Section 36: Manufacturing principles, in Therapeutic Goods Act 1989, Offi
ce of Legislative Drafting and Publishing, pp. 96-97, available: http://www.tga.gov.au/legis/index.
htm#instruments.
27. Slater, T. (2002), Therapeutic goods (manufacturing principles) determination no 2 of 2002, Com-
monwealth Austral. Gaz., GN 34, 2306-2307.
28. Anonymous (2002), Australian code of good manufacturing practice for medicinal products, Thera-
peutic Goods Administration, available: http://www.tga.gov.au/docs/pdf/gmpcodau.pdf.
29. Anonymous (2005), Medicines Act 1981, Parliamentary Counsel Offi ce, available: http://www.legis-
lation.govt.nz/browse_vw.asp?content-set=pal_statutes. 30. Anonymous (2001), Guidance notes for
applicants for consent to distribute new and changed medicines and related products, in New Zealand
Regulatory Guidelines for Medicines, Vol. 1, 5th ed., MedSafe, available: http://www.medsafe.govt.
nz/downloads/voll .doc.
31. Anonymous (2005), New Zealand code of good manufacturing practice for manufacture and distribu-
tion of therapeutic goods, available: http://www.medsafe.govt.nz/Regulatory/Guideline/code.htm.
32. Anonymous (2002), Medicines and Related Substances Control Act 101 of 1965, Medi-
cines Control Council, available: http://www.mccza.com/showdocument. asp?Cat=27
&Desc=Acts%20and%20Regulations.
33. Anonymous (2003), General regulations made in terms of the Medicines and Related Substances Act
1965 (Act no. 101 of 1965) as Amended, Government Notice, Department of Health, available: http://
www.mccza.com/showdocument.asp?Cat=27&Desc=Acts%20and%20Regulations.
34. Anonymous (2005), Guide to GoodManufacturing Practice for Medicines in SouthAfrica, Medicines Con-
trol Council, available: http://www.mccza.com/showdocument. asp?Cat=21&Desc=Guidelines%20 -
%20Good%20Manufacturing%20Practices.
204 Часть 2, Международные правила надлежащей производственной практики
35. Anonymous (2002), WHO Expert Committee on Specifi cations for Pharmaceutical Preparations:
37th Report, WHO Technical Report Series 908, World Health Organization, Singapore, available:
http://whqlibdoc.who.int/trs/WHO_TRS_908.pdf.
36. Anonymous (2002), Annex 4: Good manufacturing practices for pharmaceutical products: Main prin-
ciples, in WHO Expert Committee on Specifi cations for Pharmaceutical Preparations: 37th Report,
WHO Technical Report Series 908, World Health Organization, Singapore, pp. 36-89, available:
http://whqlibdoc.who.int/trs/WHO_TRS_908.pdf.
37. Anonymous (2005), Annex 2: Good manufacturing practices: Requirement for the sampling of start-
ing materials (Amendment), in WHO Expert Committee on Specifi cations for Pharmaceutical Prepa-
rations: 39th Report, WHO Technical Report Series 929, World Health Organization, Singapore, pp.
38—39, available: http://whqlibdoc.who.int/trs/WHO_TRS_929_eng.pdf.
38. Anonymous (1992), Annex 1: Good manufacturing practices for pharmaceutical products: Good man-
ufacturing practices for active pharmaceutical ingredients (bulk drug substances), in WHO Expert
Committee on Specifi cations for Pharmaceutical Preparations: 32th Report, WHO Technical Report
Series 823, World Health Organization, Geneva, pp. 72-79, available: http://whqlibdoc.who.int/trs/
WHO_TRS_823.pdf.
39. Anonymous (1998), Annex 5: Good manufacturing practices: Supplementary guidelines for the man-
ufacture of pharmaceutical excipients, in WHO Expert Committee on Specifi cations for Pharma-
ceutical Preparations: 35th Report, WHO Technical Report Series 885, World Health Organization,
Madrid, Spain, pp. 50-71, available: http://whqlibdoc. who.int/trs/WHO_TRS_885.pdf.
40. Anonymous (2002), Annex 6: Good manufacturing practices for sterile pharmaceutical products,
in WHO Expert Committee on Specifi cations for Pharmaceutical Preparations: 36th Report, WHO
Technical Report Series 902, World Health Organization, Singapore, pp. 76-93, available: http://
whqlibdoc.who.int/trs/WHO_TRS_902.pdf.
41. Anonymous (1993), Annex 3: Good manufacturing practices for biological products, in WHO Expert
Committee on Specifi cations for Pharmaceutical Preparations: 33th Report, WHO Technical Report
Series 834, World Health Organization, Geneva, pp. 20-30, available: http://whqlibdoc.who.int/trs/
WHO_TRS_834.pdf.
42. Anonymous (1995), Annex 7: Good manufacturing practices: Supplementary guidelines for the man-
ufacture of investigational pharmaceutical products for clinical trials in humans, in WHO Expert
Committee on Specifi cations for Pharmaceutical Preparations: 34th Report, WHO Technical Report
Series 863, World Health Organization, Geneva, pp. 97-108, available: http://whqlibdoc.who.int/trs/
WHO_TRS_863_(pl - p98).pdf (pp. 97-98); http://whqlibdoc.who.int/trs/WHO_TRS_863_(p99-
pl94).pdf (pp. 99-108).
43. Anonymous (1995), Annex 8: Good manufacturing practices: Supplementary guidelines for the man-
ufacture of herbal medicinal products, in WHO Expert Committee on Specifi cations for Pharma-
ceutical Preparations: 34th Report, WHO Technical Report Series 863, World Health Organization,
Geneva, pp. 109-113, available: http://whqlibdoc.wbo int/trs/WHO_TRS_863_(p99-pl94).pdf.
44. Anonymous (2002), Annex 3: Guidelines on good manufacturing practices for radiopharmaceutical
products, in WHO Expert Committee on Specifi cations for Pharmaceutical Preparations: 37th Re-
port, WHO Technical Report Series 908, World Health Organization, Singapore, pp. 26-35, available:
http://whqlibdoc.who.int/trs/WHO_TRS_908.pdf.
45. Anonymous (2005), Annex 3: WHO Good manufacturing practices: Water for pharmaceutical use,
in WHO Expert Committee on Specifi cations for Pharmaceutical Preparations: 39th Report, WHO
Technical Report Series 929, World Health Organization, Singapore, pp. 40-58, available: http://
whqlibdoc.who.int/trs/WHO_TRS_929_eng.pdf.
46. Anonymous (2006), Background to PIC, available: http://www.picscheme.org/indexnofl ash.
php?p=backg.
47. Anonymous (2006), Introduction, available: http://www.picscheme.org/indexnofl ashphp?p=intro.
48. Anonymous (2006), List of PIC/S participating authorities (& observers), available: http://www.pic-
scheme.org/indexnofl ash.php?p=members.
Глава 2.1. Национальные правила и требования GMP, международные требования GMP... 205
49. Brunner, D. (2004), Pharmaceutical inspection co-operation scheme (PIC/S), Qual. Assur. J., 8, 207-
211.
50. Anonymous (2006), Guide to good manufacturing practice for medicinal products, PE 009-3, Phar-
maceutical inspection co - operation scheme, available: http://www.picscheme. org/guides.php#.
51. Anonymous (2006), Structure of ICH, available: http://www.ich.org/cache/html/510-272-l.html.
52. Anonymous (2000), Good manufacturing practice guide for active pharmaceutical ingredients Q7,
ICH harmonised tripartite guideline, ICH Steering Committee, available: http://www.ich.org/LOB/
media/MEDIA433 .pdf.
53. Anonymous (2006), Quality guidelines, available: http://www.ich.org/cache/compo/363- 272-1.html.
54. Anonymous (2006), The founding of ASEAN, available: http://www.aseansec.org/7069.htm.
55. Anonymous (2006), Pharmaceuticals, available: http://www.aseansec.org/8657.htm.
56. Vemengo, M. J. (1998), Advances in pharmaceutical market integration in MERCOSUR and other
Latin American countries, Drug Info. J., 32 (3), 831-839.
57. Anonymous (2005), Division IA establishment licences, in Consolidated Statutes and Regulations,
Food and Drugs Act, Food and Drug Regulations, Part C, Department of Justice Canada, available:
http://laws.justice.gc.ca/en/f-27/c.r.c.-c.870/230049.html.
58. Anonymous (2002), Note for guidance on quality of water for pharmaceutical use, СРМР/
QWP/158/01, Committee for Proprietary Medicinal Products, Quality Working Party, London, avail-
able: http.7/www.emea.eu.int/pdfs/human/qwp/015801en.pdf.
59. Anonymous (1998), Stability testing on active ingredients and fi nished products, in The Rules Gov-
erning Medicinal Products in the European Union, Vol. ЗА, European Commission Directorate Gen-
eral III, pp. 143
Часть 3
КАЧЕСТВО
Глава 3.1. Аналитические и вычислительные
методы и примеры проектирования
и контролирования фармацевтических
систем общего управления качеством
Пол Дж. Рэнки
Технологический университет Нью-Джерси (Ньюарк, Нью-Джерси),
Грегори Н. Рэнки
Общественный исследовательский университет Нью-Джерси
(Ньюарк, Нью-Джерси),
Ричард Г. Рэнки и Эшли Джон
Технологический университет Нью-Джерси (Ньюарк, Нью-Джерси)
3.1.1. Введение
Всеобщее управление качеством (TQM, total quality management) и операционный
контроль крайне важны при проектировании системы фармацевтического про-
изводства. Ориентированное на TQM конструирование системы фармацевтиче-
ского производства включает постоянное удовлетворение требований заказчика
при наименьших затратах путем объединения усилий всех сотрудников компании.
Обеспечение качества означает поддержание в рабочем состоянии системы, пре-
дотвращающей появление дефектов. Такая система включает контроль качества
и технический контроль. Контроль качества означает установление и поддержание
соответствующих стандартов качества продукта. Технический контроль — это раз-
работка и проведение испытаний, позволяющих оценить качество продукта и соот-
ветствие принятым критериям.
В данной главе объясняется важность сокращения числа различных отклонений
при внедрении системы общего качества в каждый процесс фармацевтического ди-
зайна и производства. Кроме того, в главе обсуждаются модульный продукт, про-
цесс, проектирование обслуживания, реализация, а также управленческий подход
при внедрении в практику различных методов, инструментов и технологий TQM
и вопросы управления ими на разнообразных мелких, средних и крупных предпри-
ятиях при проектировании и контроле систем фармацевтического производства.
Особая важность этих вопросов была четко показана при установлении Феде-
ральным управлением США по контролю за пищевой продукцией и лекарствами
(FDA, Food and Drug Administration) трехуровневой классификации медицинских
продуктов:
• продукты класса I — пассивные изделия, не поступающие в организм больно-
го или контактирующие только с кожей;
• продукты класса II — активные изделия или изделия, предназначенные для
введения жидкостей в организм больного;
• продукты класса III — изделия, имплантируемые в организм больного.
210
Часть 3. Качество
FDA прекрасно осознает степень сложности задач при проектировании фарма-
цевтических систем. Для облегчения этой деятельности созданы несколько про-
граммных продуктов, которые помогают разработчикам систем, продуктов и про-
цессов достичь указанных выше целей.
Следует отметить, что FDA при подаче некоторых регистрационных досье рас-
сматривает также результаты валидации проектирования. В частности, это относит-
ся к медицинским изделиям классов II и III. Регулирующий орган ожидает, что ре-
зультаты такого анализа будут совпадать с результатами, полученными с помощью
других хорошо известных методов. На сегодняшний день имеется различное про-
граммное обеспечение, в том числе метод конечных элементов, имитация движения
и запуска, вычислительная гидродинамика (CFD, computational fluid dynamics) вме-
сте с системой автоматизированного проектирования (CAD, computer-aided design),
используемые для создания самих проектов, и другие продукты, помогающие со-
временным разработчикам фармацевтических, медицинских, производственных
и сборочно-упаковочных систем обеспечивать выполнение комплексных требова-
ний и отрасли, и FDA. (В данном случае ключевым является принятие важнейшего
принципа, что к проектированию фармацевтического производства, сборки и даже
процесса упаковки должен применяться интегрированный подход.)
Основные проблемы, возникающие при применении традиционной философии
менеджмента качества к любой задаче в области фармацевтического проектирова-
ния, производства и сборки, заключаются в следующем:
• эта философия сфокусирована в большей степени на исправлении ошибок
после их совершения, чем на их предупреждении;
• эта философия допускает совершение ошибок. На самом деле она встраивает
ошибки в каждый элемент системы, что обычно обходится в 20% оборотной
стоимости;
• эта философия допускает возможность жертвовать качеством при росте объе-
мов и производительности;
• как было показано экономистами, эта философия является дорогостоящим
дополнением к элементам стоимостной цепочки.
Тем не менее, современное представление основывается на том, что поскольку
TQM вовлекает каждого сотрудника, все элементы и оборудование компании, оно
требует всеобщих обязательств в области качества. Это не означает подход «обна-
ружь и исправь». Это предупреждающая система, пронизывающая каждый аспект
создания промышленной организации, оказывающей услуги мирового уровня,
включающая проектирование продукта, производственный процесс и управление
(и даже в экономических терминах стоящая менее, чем традиционные системы ка-
чества, — обычно около 10% оборотной стоимости).
Фундаментальная цель TQMvt всеобщего контроля качества (TQC, total quality
control) состоит в программировании, измерении и поддержании изменчивости про-
цесса под контролем. В этой главе обсуждаются, в частности, следующие аспекты
достижения этой глобальной цели:
• методы и инструменты проектирования фармацевтических производствен-
ных систем;
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 211
• моделирование процессов для проектирования и эксплуатации фармацевти-
ческих производственных систем;
• моделирование анализа требований для фармацевтических производствен-
ных систем;
• моделирование анализа рисков для фармацевтических производственных си-
стем;
• динамическое моделирование и сетевая эмуляция для фармацевтических
производственных систем с глобальным распределением, а также другие ме-
тоды и инструменты.
3.1.2. Проектирование гибкой фармацевтической
производственной и сборочно-упаковочной системы
Гибкая фармацевтическая производственная и сборочно-упаковочная систе-
ма (FMS/FAS1, FMS — производственная, FAS — сборочно-упаковочная; flexible
manufacturing/assembly system) представляет собой высокоавтоматизированную рас-
пределенную систему с контролем обратной связи, включающую данные, инфор-
мацию и физические элементы, такие как оборудование с компьютерным и ручным
управлением, производственные модули и ячейки, рабочие станции и роботы, ко-
торые зачастую должны принимать решения в режиме реального времени. Это воз-
можно только в том случае, когда все устройства обработки информации (включая
человеческие ресурсы такой системы) являются «хорошо информированными», не-
перегруженными информацией и гибкими, то есть получают четкую информацию
в нужное время, в требуемых формате и режиме, что позволяет им принимать ответ-
ственные решения в течение установленного ограниченного временного интервала.
Следует отметить, что эта концепция проектирования системы фундаментально от-
личается от разработанной для крупномасштабного производства концепции пере-
даточного конвейера, работающего ограниченный временной цикл [1—8].
При проектировании гибкой производственной и сборочно-упаковочной систе-
мы группа разработчиков должна выполнить следующие шаги:
1. Собрать все текущие и вероятные будущие пользовательские и системные тре-
бования.
2. Провести анализ системы (т. е. выяснить ограничения обработки данных, обо-
рудования FMS/FASu программного обеспечения).
3. Спроектировать пригодную структуру и базу данных для описания устройств
и их ресурсов: машин, роботов и станков, а также, при необходимости, роботизиро-
ванных рук, щупов, инструментов-сенсоров для операционного контроля, сбороч-
ных инструментов и т. д.
1В РФ «гибкая производственная система» — это управляемая средствами вычислитель-
ной техники совокупность технологического оборудования, состоящая из разных сочетаний
гибких производственных модулей и/или гибких производственных ячеек, автоматизиро-
ванной системы технологической подготовки производства и системы обеспечения функ-
ционирования, обладающая свойством автоматизированной переналадки при изменении
программы производства изделий, разновидности которых ограничены технологическими
возможностями оборудования (см. ГОСТ 26228—90). — Примеч. перев.
212
Часть 3. Качество
4. Подготовить спецификации и проекты программ, шаблоны запросов и диа-
логи, которые будут обращаться к этой базе данных и связываться с системой пла-
нирования производства и контроля FMS/FAS, работающей в режиме реального
времени.
5. Разработать и интегрировать систему с другим оборудованием и программным
обеспечением, включая руководства on-line, обучающие и тренировочные материа-
лы преимущественно в интерактивном мультимедийном техническом формате.
6. Осуществлять поддержку системы и постоянно изучать возможности дальней-
шего повышения эффективности существующих и проектируемых систем.
Как правило, перед началом проектирования такой системы должны быть по-
лучены ответы на самые важные вопросы: кто будет использовать эту систему? для
каких целей она нужна? с какими данными она будет работать? как она будет ис-
пользоваться?
К примеру, обрабатываемые в FMS данные могут использоваться персоналом
предприятия, а также подсистемами:
• планирования производства;
• контроля процесса;
• программирования детали;
• предварительной настройки инструментов и их обслуживания;
• инструментальной сборки (ручной или роботизированной);
• складского контроля и хранения материалов.
С помощью указанных подсистем система планирования производства должна
получать информацию в режиме реального времени о наличии инструментов на
складе, а также о текущем состоянии журналов, в которых ведется учет инструмен-
тов для станков (в случае проектирования FAS— сведения о роботизированных ру-
ках из журналов насадок для рук); в противном случае она не сможет создать пра-
вильный производственный график.
Следует отметить, что условие о работе в режиме реального времени является
критичным, поскольку замена инструментов производится с их обязательным уче-
том в журналах конкретных станков (или производственных модулей и ячеек) не
только вследствие износа, но также потому, что для разных программ по изготов-
лению деталей могут требоваться разные наборы инструментов. (Сама операция по
замене инструмента в большинстве случаев выполняется или манипуляторами, или
роботами. Процедура заполнения инструментального журнала, как правило, вы-
полняется оператором-человеком, иногда роботом или специализированным меха-
низмом, например сменщиком инструмента.)
Системы контроля процесса и планирования производства должны обновляться
и функционировать в режиме реального времени; в противном случае работа систе-
мы может быть нарушена.
С позиций управления и оснащения инструментами в системе FMS/FAS особое
значение должно быть уделено связям между САО-системой, в которой разраба-
тываются детали (с помощью проектирования на основе технологических прин-
ципов), и системой автоматизированного проектирования САМ {computer-aided
manufacturing), в которой пишутся регламенты (инструкции) по изготовлению де-
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 213
талей в рамках FMS. Как правило, разработчик деталей FMS анализирует выход-
ные данные из CAD (т. е. дизайн готовой формы фармацевтического продукта, ко-
торый будет производиться и собираться в FMS), крепежную оснастку, различные
установочные задачи (т. е. установку обрабатываемой детали), а также необходимые
и альтернативные операции, требуемые инструменты и, наконец, предварительный
перечень ресурсов (т. е. предполагаемые обрабатывающие станции, ячейки, модули
или станки).
Базы данных и компьютерные системы, действующие в режиме реального вре-
мени, также важны, поскольку они обеспечивают получение отчетов и информации
о статусе, необходимых для бесперебойного функционирования FMS (в частности,
следует обращать особое внимание на ее динамический электронный планировщик
и подсистемы, подобные подсистеме обслуживания инструментов) [4,9—14].
3.1.3. Модель гибкого производства, интегрированная
с проектированием
Выходные данные С4Л/-системы представляют собой производственную базу пра-
вил1. Это информация, необходимая FMS для производства каждого фармацевти-
ческого продукта. В этой производственной базе правил, кроме прочего, каждой
операции приписаны инструменты. Инструментальные коды, содержащиеся в базе
данных инструментов, выбираются планировщиком процесса в FMS или автомати-
чески присваиваются системой планирования процессов.
Перечень требуемых инструментов посылается через сеть на участок или стан-
цию подготовки инструментов, где инструменты соответствующим образом подго-
тавливаются (т. е. собираются и устанавливаются) и хранятся таким образом, что
система управления материалами FMS может их забрать [12—21].
Станция подготовки инструментов выполняет и другие действия, среди которых
наиболее важными являются:
• обработка и обслуживание инструментов;
• сборка инструментов согласно заказам (при необходимости замена изношен-
ных инструментов);
• наладка инструментов, их проверка и регулировка;
• забор инструментов и их транспортировка в режиме реального времени, орга-
низованные для выполнения задач FMS, функционирующей также в режиме
реального времени.
Станция подготовки инструментов получает заказы, которые изначально фор-
мируются системой CAD обработки данных, затем пересылаются через сеть FMS
и технически описываются системой САМ в виде производственной базы правил.
Другие данные, поступающие на станцию подготовки инструментов, включают:
• заказы деталей (состоят из кодов деталей и их количеств); следует отметить,
что это очень важный набор данных также и для динамического планиров-
щика задач FMS в режиме реального времени;
1 База правил — термин, используемый в компьютерных экспертных системах. База пра-
вил содержит элементарные выражения, называемые в теории искусственного интеллекта
продукциями. — Примеч. перев.
214
Часть 3. Качество
• уведомление о физической готовности деталей для обработки в FMS, содер-
жащее дату подготовки инструментов;
• приоритетный заказ (обратите внимание, что он может меняться вследствие
изменений в режиме реального времени в системе, и, следовательно, станция
должна быть способна выполнять также это измененное задание);
• часть производственной базы правил, описывающую требования по подго-
товке инструментов.
Станция подготовки инструментов находится в постоянном контакте с системой
FMS, а также с другими системами, отправляя им с помощью обратной связи важ-
ные данные, относящиеся к инструментальной оснастке:
• отчеты о запасах (в отношении инструментов);
• отчет о статусе FMS (в отношении инструментов);
• отчеты о приоритетном статусе детали (в случае необходимости проведения
динамических изменений в FMS, что влияет на потребности в инструментах
и точные даты их подготовки).
3.1.4. Операционный контроль в режиме реального времени
Часть системы операционного контроля и управления FMS, работающая в режиме
реального времени, должна:
• контролировать использование инструментов для множества процессов, как
описано в производственной базе правил и назначено в режиме реального
времени динамическим планировщиком заданий соответствующим ресурсам
FMS/FAS-,
• предоставлять данные для контроля транспортировки инструментов и журна-
лов учета инструментов в рамках FMS;
• предоставлять информацию для выполнения и контролирования смены ин-
струментов и изменений в журналах учета инструментов на всех уровнях;
• получать извещения о результатах проверки инструментов (например, если
во время процедуры проверки она обнаруживает изношенный инструмент,
то она должна генерировать команду системе обновления журнала учета ин-
струментов по замене данного инструмента в соответствующем журнале учета
инструментов);
• предоставлять информацию в случае чрезвычайных ситуаций;
• предоставлять необходимые интерфейсы и данные для проведения операций
по диагностике и восстановлению, желательно с использованием экспертных
диагностических систем.
Наконец, следует подчеркнуть важность петли обратной связи, начинающейся
в системе, действующей в режиме реального времени и заканчивающейся на стан-
ции подготовки инструментов, которая содержит данные о статусе инструментов
в режиме реального времени, их состоянии и информацию о приоритете детали.
Эти данные часто используются сотрудниками и/или системными компьютерными
системами, которые занимаются созданием производственной базы правил. Также
они представляют собой очень информативный набор данных для разработчиков
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 215
. FMS, поскольку большинство данных, которые ранее терялись, таким способом бу-
дут сохранены.
Наиболее важные действия при операционном контроле в FMS/FAS выполня-
ются на трех уровнях, на каждом из которых требуется проведение имитационного
моделирования и оптимизации перед производством или в процессе производства
детали в системе FMS/FAS:
1 . Уровень предприятия или бизнес-уровень, управляемый бизнес-системой
автоматизированной системы управления производством (С1М, computer integrated
manufacturing) или, даже более широко, системой управления ресурсами предпри-
ятия.
2 . Уровень FMS off-line, представляющий действия по планированию, имитаци-
онному моделированию и оптимизации до загрузки серии или отдельного компо-
нента в FMS (иногда управляется С4Л/-системой, иногда компьютером FMS, про-
граммирующим деталь).
3 . Уровень контроля в режиме реального времени, управляемый системой опера-
ционного контроля FMS/FAS, динамическим планировщиком с интегрированным
управлением инструментами и мультимедийной поддержкой, представляющий си-
туацию, когда детали уже существуют и физически, и логически в среде, контроли-
руемой в режиме реального времени.
Вследствие такой сложности при проектировании производственной базы пра-
вил требуется применение по-настоящему интегрированного подхода, чтобы предо-
ставить описание работы для динамического планировщика FMS. Так как динами-
ческая система в значительной мере зависит от базы знаний, представленной базой
правил, то чрезмерно ограниченная база правил приводит к неэффективным, ино-
гда даже неправильным решениям. Другими словами, такая структура должна пред-
ставлять все многоуровневые взаимодействия и их возможные правила действий,
связанные с планированием производственного процесса и выработки решений
в FMS. Это реально сложная задача.
Следует подчеркнуть, что применение мультимедийных технологий на этом уров-
не чрезвычайно полезно при подготовке программ изготовления деталей, для обуче-
ния и тренингов операторов по регулировке деталей, приборов и инструментов, для
действий при исправлении неполадок, для регулярного обслуживания, при програм-
мировании на уровне компьютерного числового программного управления (CNC,
computer numerical control), программировании роботов и программировании разме-
щения станков, программировании логических контроллеров (PLC, programmable
logic controller), контроле качества, техническом обслуживании и других задачах.
Большинство FMS обладают способностями к частичной буферизации. Это
может быть обусловлено не графиком, а технологическими процессами, други-
ми словами, связано с планированием процессов (например, деталь должна быть
охлаждена перед проведением адекватной процедуры проверки). Некоторая сте-
пень буферизации полезна и необходима для обеспечения надежности. (Актуальное
количество буферных хранилищ должно быть установлено по результатам имитаци-
онного моделирования и опыта.)
Ячейки и модули тоже часто обладают буферной емкостью. Это обусловлено тем,
что постановка детали в очередь на вход в определенную ячейку непосредственно
216
Часть 3. Качество
перед окончанием изготовления предыдущей детали в ячейке обеспечивает наибо-
лее эффективную эксплуатацию ячейки, так как время затрачивается только на за-
мену детали. Другой важный элемент — хорошо спроектированные буферные зоны
для деталей, обеспечивающие прямой доступ для погрузочно-разгрузочного обору-
дования, что делает процесс ожидания в очереди короче, проще и динамичнее [18,
19,21-27].
3.1.5. Инновационное проектирование
Основная цель данной главы — описать типовой и систематический метод проекти-
рования фармацевтической производственной и сборочно-упаковочной системы,
включающий продукт, процесс, обслуживающие системы, а также особенности ар-
хитектуры управления инновационным проектом в этих системах.
Архитектура такой системы должна быть одновременно новой и в то же время
соответствовать отраслевым руководствам и руководствам института управления
проектами PMI (Project Management Institute), принятым в отношении проектиро-
вания продукта и процесса в отрасли и основанным на стандартах качества се-
рии 9000:2000 Международной организации по стандартизации ИСО (International
Organization for Standardization). Наше протестированное программное решение
по проектированию фармацевтической производственной системы включает
объектно-ориентированное моделирование процесса, анализ требований и ри-
сков, статистические методы, дизайн экспериментов, трехмерные (3D) интерак-
тивные мультимедийные методы и инструменты, которые на 100% совместимы
с интернетом. Более того, наши методы и инструменты программного обеспече-
ния являются типовыми, то есть они могут применяться не только к системам,
используемым в фармацевтической отрасли или производстве автомобилей, но
также к процессам (например, в нефтяной отрасли) или к услугам (например, об-
разовательным).
При проектировании фармацевтической производственной системы требуется
значительный уровень инноваций. В наиболее широком понимании инновация —
это действие по внедрению чего-то нового: продукта или процесса — в общество
или отдельную группу лиц. Инновацию часто путают с изобретением, которое на-
целено на определенные объекты. В фармацевтической отрасли инновация может,
таким образом, включать введение новых бизнес-структур в рамках компании и но-
вых производственных процессов, контроль качества лекарственных средств и ис-
ходных материалов. Улучшения процессов и обслуживания также можно считать
инновациями, но следует помнить, что в этом случае обслуживание часто рассма-
тривается как процесс [4, 13, 21, 23, 28—32].
Научные открытия, такие как составление карт новых планет, нахождение их
масс или изучение новых форм жизни, не рассматриваются как инновации, по-
скольку эти объекты существовали до того, как были обнаружены человечеством.
Но когда новые виды организмов внедряют в общество и находят им определенное
применение, это считается инновацией. В фармацевтической отрасли таким при-
мером может быть антибиотик пенициллин. Несмотря на то что и до своего откры-
тия он существовал в природе в виде продукта жизнедеятельности грибов, только
в прошлом столетии он стал использоваться для уничтожения в организме человека
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 217
патогенных бактерий. Однако при проведении ранних исследований не считалось,
что это вещество будет сохраняться в организме человека достаточно долго, чтобы
оказать соответствующее воздействие.
Способность распознать альтернативные возможности использования существу-
ющих процессов или инструментов является неотъемлемым свойством инновации.
Это трудная задача, так как неожидаемые изменения в системе обычно считаются
ошибками или аномалиями. В качестве примера можно привести разработку само-
клеющихся блоков для записей Post-It. Изначальной целью разработчиков было
создание высокоэффективного адгезивного вещества, но в результате случайно
было получено вещество с исключительно слабыми адгезивными свойствами. Тем
не менее, вместо его уничтожения как обычных отходов возможности применения
полученного нового вещества были изучены специалистами и менеджерами, что
позволило получить легко заменимые листки для напоминаний для каждодневного
использования. Ультраслабое адгезивное покрытие позволяет удалять стикеры Post-
It без повреждения поверхности, к которой их прикрепляют. В настоящее время
стикеры выпускают в большом ассортименте цветов и размеров.
Приведенный пример является примером радикальной инновации не только по-
тому, что это привело к значительным изменениям в индустрии бумаги для записей,
но и потому, что инновация была абсолютно неожиданной. Расширение ассорти-
мента стикеров по различным размерам и цветам является примером поэтапной ин-
новации, которая заключается в пошаговых изменениях и улучшениях существую-
щих продуктов или процессов. Радикальные инновации происходят намного реже,
хотя их эффект на общество и историю намного более глубок.
Одной из общих тенденций развития общества является осознанное распозна-
вание и управление инновациями, частично путем комбинирования науки и техно-
логии. Часто создавались определенные процессы, при этом даже не понимались
полностью их эффекты или принципы. Это четко видно в истории развития метал-
лургии. Например, железо, бронза и золото использовались веками до того време-
ни, когда стало возможным видеть и изучать структуру молекул. Следует отметить,
что хотя наши предки и не могли описать химический состав сплавов, эти металлы
успешно выполняли свои многочисленные функции. В подобных случаях цель ин-
новаций является исключительно прагматичной, и успешные решения сохраняют-
ся и передаются следующим поколениям через обучение.
Те, кто вводит новшества, могут учиться на примере жизнеспособных решений,
чтобы набирать собственный опыт. Те, кто постоянно имеет дело с ограниченным
набором разработок, должны хотеть экспериментировать, проводить испытания
и разнообразить свою деятельность, чтобы не отстать, поскольку инновации не
только позволяют выжить, но и приводят к процветанию.
Способность к нововведениям также связана с обучением на прошлых ошиб-
ках, и не только собственных. Ошибки и погрешности в деятельности могут быть
одновременно дорогостоящими и опасными, но если установить их причину, то
появление их в будущем можно предотвратить. Это довольно-таки трудно, так как
сценарий «почти ошибка» (near-miss) может рассматриваться или как нечастое со-
бытие, или как предотвращаемая опасность. Первая реакция на ошибку, как прави-
ло, заключается в продолжении текущей деятельности без внесения изменений, что
218
Часть 3. Качество
приводит к возникновению подобных ошибок в дальнейшем. Обучение на ошибках
требует участия всех уровней организации и создания коммуникационных каналов
для эффективного введения инновации, так как неспособность делиться опытом
обусловливает отсутствие новых возможностей [13, 29—40].
3.1.6. Открытая инновационная архитектура
Инновация как процесс, а также управление связанным с ней научно-исследо-
вательским и опытно-конструкторским проектом {R&D — исследования и проек-
тирование, research-and-development) считаются наиболее сложными информаци-
онными системами и техническими структурами вследствие большого количества
свойств, процессов и динамически изменяющихся программных проектов, которые
сопровождают их жизненный цикл.
Мы рассматриваем каждый инновационный процесс и проект как систему, со-
стоящую из объектов и классов объектов. Затем изучаем пути взаимодействия
компонентов этих систем друг с другом. Как только становится понятным их взаи-
модействие, мы применяем интегрированный системный подход в отношении си-
стемы управления проектами, рассматривая ее как процессы и учитывая требования
заказчика и возможные риски.
Далее помещаем модель этой системы в программную оболочку с инструментами
статистического анализа и интерактивной мультимедийной 3D-средой (рис. 1). Мы
используем статистические методы для выявления состояния процессов, близких
к неконтролируемому, а также для анализа трендов, что предоставляет большие воз-
можности для инноваций, и применяем интерактивную мультимедийную ЗД-среду
и методы 3D-визуализации для коммуникации через интернет с членами глобаль-
ной группы по инновации. Важность сотрудничества в современной конкурентной
фармацевтической среде делает необходимым применение этих виртуальных сред
для облегчения взаимодействия группы. (Обратите внимание на то, что действую-
щие кодированные электронные таблицы и 3D-o6beKTbi, упоминаемые в этой главе,
являются частью электронной библиотеки Рэнки (Ranky) и мотуг быть получены по
адресу http\//www. cimwareukandusa. сот).
Для иллюстрации важности «открытости» нашей архитектуры рассмотрим со-
временные инструменты имитационных испытаний и анализа, разработанные
корпорацией параметрических технологий {Parametric Technology Corporation, PTC)
(рис. 2), и инструменты управления жизненным циклом продукта {PLM, product life-
cycle management), например инструменты IBM/Dassault Systems Delmia для проек-
тирования и моделирования фармацевтических производственных систем (рис. 3)
с сенсорной обратной связью (рис. 4).
Поскольку проектирование, редактирование, эксплуатация и управление этими
моделями возможны даже при наличии ограниченных мощностей, они могут быть
крайне ценными источниками для моделирования в цифровой форме, анализа про-
цессов, моделирования требований, анализа рисков и даже сбора статистических
данных и моделирования отказов сложных систем.
Рассмотрим метод конечных элементов на рис. 2, а. Это торсионный тест детали
фармацевтического производственного оборудования на сборочной линии нового
упаковочного цеха для лекарственных препаратов. Линия пока еще находится на
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
219
Анализ
процессов
Анализ рисков
Анализ
.требований
Статистический анализ, дизайн эксперимента,
интерактивная мультимедийная ЗО-среда
на интернет-платформе, полноэкранные видео
на DVD и видео «точно в срок» на iPod для
управления знаниями
Рис. 1. При проектировании неизбыточной («стройной») и гибкой фармацевтической
производственной и сборочно-упаковочной системы необходимо провести анализ требуемых
процессов, требований заказчика и пользователя, требований к обслуживанию, качеству,
надежности, гибкости, неперегруженности и структуре, а также анализ рисков, связанных
с перечисленными процессами. Все анализы проводятся в статистическом программном
обеспечении. Обратите внимание, что наши интерактивная мультимедийная ЗО-среда и база
для имитационных испытаний поддерживают интегрированный цифровой дизайн и принципы
проектирования цифровых производственных систем, то есть сначала должны быть проведены
испытания всех конструкций и систем на экране, и только после удовлетворительных результатов
испытания проходят в реальном режиме
стадии проверки и наладки в виртуальном окружении, что существенно ускоряет
процесс ее регулировки. Как видно из распределения напряжения по фон Мизесу1,
необходимо закруглить острые углы вокруг оси. При этом также будет увеличено
распределение этого напряжения и, таким образом, уменьшено количество красных
зон (высокое напряжение) за счет их перехода в синие или даже зеленые (низкое на-
пряжение). Без использования виртуальной сборочной линии для предварительной
проверки будущих нагрузок неожиданный отказ этой детали мог бы создать простой
в производстве, контаминацию продукта или даже травму оператора линии.
Как можно видеть, цифровые инструменты проектирования фармацевтических
производственных и сборочно-упаковочных систем и предприятий включают не
только оборудование, но и сенсоры, включатели, приборы контроля, системы об-
работки материалов, маркировочные машины и даже эргономически реалистич-
ные модели людей и операторов, выполняющих такую же работу, как в физическом
мире, в близких к реальности моделях предприятий. Такие имитационные систе-
мы являются не только хорошими моделями — они позволяют также экономить
значительные фонды, так как предприятие не строят, пока не будет получена удо-
влетворяющая всем требованиям модель. Следует помнить, что внесение измене-
ний в физически существующее производство стоит денег, времени и, может быть,
эффективности производства, даже если проводится только проверка возможного
улучшения. Виртуальные модели могут одновременно тестироваться тысячи раз
в течение нескольких дней, с подбором сотен оптимизируемых параметров до вы-
бора подходящей комбинации [30—36, 40—44].
1 Трехмерные напряжения и нагрузки образуются в нескольких направлениях. Обычно эти
многонаправленные напряжения суммируются для получения эквивалентного напряжения,
которое также называется напряжением по фон Мизесу. — Примеч. науч. ред.
220
Часть 3 Качество
а)
Stress von Mises (WCS)
(Ibf / in"2)
Deformed
Scale 5.5773E+OI
Loadset :LoadSetl
|Э.В00е»е<
в.ие&ке*
r—j Г.00&ЧВ1
— B.00tetB4
График распределения
напряжения по фон Мизесу
Displacement Mag (WCS)
Deformed
Max Dlsp +4.66I8E-O3
Scale 5.5773E+OI
LoadsetiLoadSell
4.500c-03
4.00Be-03
э.5еве-ез
3.B00C-03
2.500C-03
2.000e-03
1.500e-03
1.000e~03
5 ‘W0e 0И
График перераспределенного
напряжения
max_disp_mag (mm)
P_Pass
Scale 1.0000E + 00
PLoop Pass
—о— max_disp_mag
strain_energy
(mm N)
P_Pass
Scale 1.0000E + 00
max_strees_vm
(N/mmA2)
P_Pass
Scale 1.0000E + 00
Loadset: LoadSetl
PLoop Pass
—о— strain_energy
Рис. 2. Торсионный тест методом конечных элементов детали фармацевтического производ-
ственного оборудования на сборочной линии нового упаковочного цеха для лекарственных пре-
паратов. Линия пока еще находится на стадии проверки и наладки в виртуальном окружении, что
существенно ускоряет процесс ее регулировки. Как видно из распределения напряжения по фон
Мизесу (см. примечание в тексте), необходимо закруглить острые углы вокруг оси. При этом также
будет увеличено распределение этого напряжения и, таким образом, уменьшено количество крас-
ных зон (высокое напряжение) за счет их перехода в синие или даже зеленые (низкое напряжение)
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
221
Рис. 3. Современный инструмент имитации, анализа и управления жизненным ци-
клом продукта IBM/Dassault Systems Delmia для моделирования и проектирования
фармацевтических производственных систем. Польза при их использовании огром-
на, поскольку система может быть построена и протестирована в цифровом виде
(предоставлено IBM/DassaultSystems Delmia, Inc.)
3.1.7. Типовой объектно-ориентированный метод
моделирования инновационного процесса и образцовая
модель
Чтобы оставаться в лидерах, для каждой бизнес-структуры жизненно необходимо
понимать, моделировать и затем применять на практике новые процессы и процеду-
ры в соответствии с передовым опытом в области процессов. «Инновационный про-
цесс» в фармацевтических производственных системах не является исключением.
Большое количество международных стандартов по проектированию продук-
тов и процессов, написанных и рассмотренных тысячами ведущих исследователей
и компаний по всему миру, помогают создать модель для такой сложной проблем-
ной задачи, как инновации. В связи с этим в данном разделе обсуждаются два из
восьми основных положений менеджмента качества, изложенных в международном
стандарте качества ИСО 9000:2000, и пути применения этих правил при проектиро-
вании фармацевтических производственных систем.
Мы делаем это с целью развития систематических инноваций (вместе с со-
ответствующим опытом моделирования проектов) и воспроизводимых прове-
ренных процессов проектирования фармацевтических систем и используем объ-
ектно-ориентированный метод моделирования CIMpgr. Принципы метода транс-
формированы в Г/Л/£-модели {Unified Modeling Language — универсальный язык
222
Часть 3 Качество
Рис. 4. Высокотехнологичные сенсоры, работающие в фар-
мацевтической сборочной системе, помогают операционному
контролю и системе обеспечения качества в режиме реального
времени проводить проверку каждой единицы продукции. (Та-
кой подход часто называют бездефектной политикой, встро-
енной в систему.) Сенсор STEALTH-UV был разработан для
обнаружения невидимых флуоресцентных материалов, содер-
жащихся или добавляемых во многие продукты. С его помо-
щью пользователи могут обнаружить наиболее сложные объ-
екты, включая прозрачные наклейки для контроля вскрытия,
прозрачные этикетки и невидимые регистрационные отметки.
Этот уникальный сенсор также идеально подходит для реше-
ния многих современных трудных задач в логистике продуктов,
их проверке и верификации (предоставлено TRI-TRONICS Со.,
Inc., www.ttco.com)
моделирования в информационных технологиях) и компилированы с международ-
ными стандартами моделирования процессов, используемыми в программных сре-
дах моделирования сложных систем.
Прежде всего мы обсудим несколько важных определений, которые тесно свя-
заны со стандартным принципом № 4, приведенным в стандарте ИСО 9000:2000
(моделирование процесса):
• Процесс, или деятельность, может быть определен как передаточная функ-
ция с одним или более входами, выходами, контрольными значениями пока-
зателей и ресурсами, которые все вместе обеспечивают присвоение значений
переменным после чего функция выполняется.
• Передаточные функции при выполнении создают процесс трансформации.
Процесс трансформации внутри программного проекта состоит из методов,
шагов, задач и различных алгоритмов и процессов, которые получают данные
и манипулируют ими, а затем передают их на выход (выходы) системы. Сле-
дует отметить, что данными на входе можно считать материалы, человеческие
знания, технологические установки, экономические данные и т. п.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 223
• Выход процесса — это продукт, который состоит из определенных техниче-
ских и/или социальных продуктов и услуг, которые соответствуют требовани-
ям заказчика.
• В рамках управления проектами по качеству процессы обладают видимостью
(идентификацией), документацией и прослеживаемостью.
• В этом контексте под видимостью понимается, что мы знаем и четко видим
(или имеем графическое представление), какие задействованы методы и при-
емы, шаги системного процесса и технологии при создании требуемого выхо-
да. Знаем ли мы последовательность этих шагов и взаимосвязи с возможными
параллельными процессами? Как один процесс влияет на другой?
• Документация означает, что методы, шаги, процессы и технологии определе-
ны и записаны в соответствии со стандартными согласованными специфика-
циями.
• Прослеживаемость означает, что шаги процесса, а также выход (выходы)
можно проследить в обратном направлении вплоть до требований соответ-
ствующего заказчика.
• Мощность процесса можно определить как способность производственного
процесса выдерживать требования и допуски, установленные в спецификации.
• Несоответствие процесса — это отклонение параметров процесса от специ-
фикаций.
• Вариабельность процесса — это зависимость в размерных или других изме-
ряемых характеристиках выхода от производственного процесса. (Следует от-
метить, что абсолютная задача любого проекта — оставаться в предваритель-
но установленных пределах вариабельности процесса и, если это возможно
и целесообразно, снижать вариабельность процесса, поскольку при этом, как
правило, уменьшаются риски.)
• Вариабельность может быть выражена в виде среднего диапазона стандартно-
го отклонения.
• Переменная процесса — это параметр процесса, характер изменений которо-
го представляет случайную переменную, и который, следовательно, требуется
контролировать.
• Управление процессом означает перенесение деятельности и процедур, ко-
торые высокопрофессиональные и опытные менеджеры выполняют в уме,
в формализованный процесс путем создания подробно описанной модели,
часто называемой моделью процесса [40—46].
3.1.8. Системный подход к управлению фармацевтической
производственной системой
Идентификация, понимание и управление взаимосвязанными процессами в виде
системы делают более эффективной деятельность организации и помогают дости-
жению поставленных целей. (Следует отметить, что каждый из ключевых факторов,
приведенных далее, связан с одной и более возможностями инноваций!)
224
Часть 3. Качество
Важнейшие факторы и достигаемые выгоды включают следующее:
• Процессы, которые обеспечивают получение желаемых результатов, будут
лучше интегрироваться и налаживаться.
• Администрация и владельцы процессов будут иметь возможность сосредото-
чить свои усилия на основных процессах.
• Поскольку слаженность взаимодействий в организации, ее эффективность
и производительность будут расти, доверие к организации заинтересованных
сторон и партнеров также возрастет.
• Структурирование системы и тонкая наладка сделают возможным достиже-
ние организацией своих целей наиболее эффективным путем.
• Понимание взаимозависимостей процессов в системе приведет к получению
хороших результатов.
• Структурированное (и объектно-компонентно-ориентированное) моделиро-
вание обеспечивает получение реально гармонизированных и интегрирован-
ных процессов. Персонал будет понимать и выполнять их, и, соответственно,
будут снижены потери и увеличено качество в каждом процессе.
• Сопротивление, созданное межфункциональными барьерами, будет умень-
шено, и тем самым будет обеспечено лучшее понимание ролей и ответствен-
ности, необходимые для достижения общих целей.
• Организационные возможности и установление ограничений ресурсов до на-
чала работы будут лучше восприниматься всеми вовлеченными сторонами
(и прежде всего теми, кто принимал участие в создании модели).
• Целеполагание и установление требований по выполнению соответствующей
деятельности в рамках системы станет реальностью.
• Постоянное улучшение системы через измерения и оценку контролируемой
обратной связи становится возможным благодаря аналитическому и изме-
ряемому подходу в моделях процессов. (Следует отметить, что на последнем
уровне развития это приведет к появлению предприятия, контролируемого
по обратной связи в режиме реального времени, которое способно реагиро-
вать на динамически изменяющиеся потребности рынка.)
После этого введения рассмотрим компоненты нашей объектно-ориентированной
системы, выполняющей вышеописанные принципы ИСО 9000:2000, и изучим, как
с их помощью можно моделировать сложные инновационные процессы и связан-
ные с ними процессы управления проектами [40—54].
В качестве простого примера предположим, что вы осуществляете упаковку фар-
мацевтического продукта с использованием линии, выполняющей различные этапы
процесса. На рис. 5, а показан один из этих этапов. У него есть вход(ы), выход(ы),
контрольный(ые) показатель(и) и ресурс(ы). Эти типы данных помогают опреде-
лить, в каких условиях процесс должен быть выполнен фармацевтической произ-
водственной системой. (Определения приведены на диаграммах.) Мы также можем
видеть, каким образом CIMpgr трансформирует карты процесса в С/Л/£-диаграмму
(рис. 5, б). Это важно, так как UML является языком моделирования, на котором
ИТ-специалисты программируют жизненные циклы продуктов и системы контроля
для линий. На рис. 5, в мы видим, как различные процессы будут взаимодействовать
в разработанной нами фармацевтической сборочной системе.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 225
а) Это сторона контроля. С этой стороны данные каким-либо образом лимитируют или контролируют
процесс. (В качестве примера регуляторов представьте международные требования по контролю
выбросов, выполнение которыхдолжны предусмотреть разработчики автомобилей.) Мы можем обозначить
каждый регулятор именем переменной вида С1А0
ЦАО: С1А0: С2А0: СЗАО: С5А0: С5А0:
Это входная
сторона
процесса.
С этой стороны
данные
поступают
в процесс.
Мы можем
обозначить
входы именами
переменных
вида И АО
С6А0:
12А0:
I3A0:
I4A0:
ШАО:
I6A0:
Этот прямоугольник представляет
процесс. Мы можем обозначить
процесс, назвав его АО (если это
верхний, родительский уровень),
а его подчиненные, дочерние про-
цессы можем назвать Al, А2 и т. д.
Модель процесса CIMpgr—АО
♦ О1А0:
♦ О2А0:
♦ О4А0:
» О5А0:
Эта выходная
сторона
нашего
процесса.
Мы можем
обозначить
выходы
именами
переменных
вида О1 АО
(РВ1 АО:)
Это обозначает хранилище
данных, файл или базу дан-
ных для процесса
R1A0: R2A0: R3A0: R4A0: R5A0:
Это сторона ресурсов нашего процесса. С этой стороны данные описывают
имеющиеся ресурсы: людские, компьютеры и серверы, программное
обеспечение и другие имеющиеся ресурсы для выполнения процесса. Мы
можем обозначить каждый ресурс именем переменной вида R1A0
Это административный раздел нашей модели
Цель: почему мы это делаем? какова основная цель этой модели?
Точка зрения: «как есть» — системный аналитик, разработчик системы; «будет» — системный аналитик,
разработчик системы
Члены группы разработчиков: Рэнки, совместно
Контактное лицо: Пол Дж. Рэнки, Email: cimware@iearthink.net. Тел. в США: (201 )4930521
Заказчик: Компания ABC Inc.
Дата: 21 января 2004; версия: ver. 1.0
Конфиденциально! Выпуск в обращение: ДА. Наследование объектов и классов: вкл.
6)
Рис. 5. Объектно-ориентированный метод моделирования процесса (CIMpgr) применительно
к проектированию фармацевтической производственной и сборочно-упаковочной системы
Связи:
имя файла с моделью анализа требований:
имя файла с моделью анализа рисков:
В)
I1A1: новые С1А1:
требования \
от покупателяг \
или заказчика |
проекта I
С2А1:
Длительность проекта, бюджет и контроль качества (обратите внимание, что существует много других типов контроля,
связанных с охраной окружающей среды, определенным дизайном, материалами, производственным процессом, сборкой, —..
испытанием, обслуживанием, ИТ и другими процессами и подсистемами)
I2A1:
I3A1:
I4A1:
I5A1:
I6A1:
С5А1:
С6А1:
Концепция, фаза
концепции, фаза анализа
требований — процессА!
02А1: спецификация
на проект вида
^что? когда? сколько?»
I7A1:
( DBI_A1: \
/здесьхранятся)
результаты
анализа
требований,
связанных
с процессом А1
(т. е. результаты
, исследования ,
\ CORA) J
Ibldri Q_., П,1Г инмолю»!
Н2АГ. консультант
группа по анализу
требований
DBIA2:
здесь хранятся
документы
и данные
по анализу
системы
01А1: результаты работ по анализу требований (этот набор данных содержит полезную информацию
для будущих проектов, для извлечения данных и управления знаниями)
С2А2:
Определение, плани- I
рование, фаза анализа
системы — процесс А2 I
О2А2: подробная
спецификация
I на проект
С1АЗ:
01А2: результаты спецификации проекта (это важный набор данных в случае множественных проектов с ограничениями
по приоритету; также набор данныхсодержит полёзную информацию для извлечения данных и для управления знаниями)
С2АЗ:
модели проекта,
программное
обеспечение
для оптимальной
динамической
имитации и опытный
консультант /
по планированию
•ратная связь «проект-анализ».
Обратная связь «проект-требования»
Типовая модель управления проектом CIMprg для процессов А1-А5. \ \
Контактное лицо: Пол Дж. Рэнки, Email: cimware@earthlink.net. \ \
Тел. в США: (201 )4930521. \
Заказчик: Компания ABC Inc. Дата: 09 июня 2004. Версия: ver. 5.0. \
Встроенный шаблон электронной книги для управления \
детализированным проектом.
Наследование объектов и классов: вкл.
Примечание: надписи вида «Концепция, фаза концепции, фаза анализа
требований — процесс А1» означают, что данный процесс обычно называют одним
из перечисленных терминов, но для нас все они имеют одинаковое значение.
Проектирование,
разработка, фаза
' здесь хранятся
данные
по запуску
системы,
внедрению
,и тестированию
0(
01АЗ: результаты разработки проекта (этот набор данных содержит полезную
О2АЗ:
детальное
проектирование
\ проекта
С1А4:
информацию для извлечения данных и для управления знаниями)
С2А4:
а (этот набор данных
Функционирование,
интегрирование, фаза
запуска и тестирования
системы — процесс А4
\O1A4: результаты внедрения проек
содержит полезную информацию для извлечения данных,
и для управления знаниями} -
2А4:
226 ______________________________________Часть 3. Качество
С1А5:
I1A4:
С2А4: опер
С2А5:
R2A4:
R1A4: программное
обеспечение
для управления
длительностью
и бюджетом проекта
и опытная группа
по внедрению
проекта
01А5:
результаты
тестов ь
Обратная связь «внедрение-проект»
>ратная связь «длительное тестирование
и поддержка-проект»
/ DBLA5: \
г здесь хранятся'
данные после-
проектного ана-
лиза, основанного
на результатах
длительного
. тестирования /
\ и поддержки j
О2А5:
Послепроектный анализ, ?
длительное тестирование j
системы, фаза поддержки —,
процесс А5
R1A5: группа
постоянного
улучшения
Рис. 5. Окончание
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 227
3.1.9. Анализ требований при проектировании инновационной
системы продукта, процесса и обслуживания
Процессы в успешном инновационном проекте должны соответствовать требова-
ниям рынка и заказчиков, а также идеям самого изобретателя. Принято считать, что
анализ требований является одним из наиболее важных параметров любого инно-
вационного проекта фармацевтической производственной системы, так как если он
проведен на должном профессиональном уровне, он помогает установить, исследо-
вать и разработать параметры и процессы, необходимые потребителям.
В наших примерах инновационных проектов мы сфокусировались на типовых
потребностях и требованиях, а нашими предполагаемыми «потребителями» явля-
ются члены группы фармацевтических НИОКР (R&D), менеджеры и операторы
различных отраслей.
В рамках исследовательского подхода мы использовали проверенный метод:
проанализируйте потребности и требования, выполнение которых должно быть
обеспечено или предположительно обеспечивается указанными процессами, ме-
тодами и системами, и если будет обнаружен «пробел», значит вы нашли возмож-
ность для инновации. Следует помнить, что когда вы ищете этот «пробел», он будет
одновременно показан в нашей модели CIMprgB виде отсутствующего процесса или
в виде существующего процесса с недостающими атрибутами, а также в виде тре-
бования в нашей СОЛ4-модели (модели компонентно-ориентированного анализа
требований; component-oriented requirements analysis).
• Проведите анализ имеющихся актуальных методов. Найдите ключевые мето-
дологии, математические модели и базовое техническое и/или другое научное
обоснование.
• Проведите анализ используемых технологий. (Каким образом наука превра-
щается в практическое решение, а также в инженерную и/или компьютерную
технологию?) Есть ли потребность в новой оригинальной технологии, кото-
рая еще не была создана или не применялась в этой области?
• Проведите анализ и обзор действующих процессов и способов обеспече-
ния целостности процессов. (Используйте объектно-ориентированный ме-
тод анализа процесса, т. е. от концепции к продукту.) Обратите внимание на
атрибуты процесса. Помните, что добавляя новый атрибут, вы создаете новые
типы данных с новой информацией, и если ваш процесс может выполняться
новым способом — добавляете новое знание; таким образом, ваша комбини-
рованная CIMprg- и t/ML-модель становится также моделью представления
нового знания. Это важно, так как этим способом осуществляется формализа-
ция инновации, и она может обсуждаться глобальными группами участников.
• Проведите анализ возможных альтернативных решений. (Современная фар-
мацевтическая производственная и сборочно-упаковочная система должна
быть очень гибкой вследствие быстро меняющихся требований потребителей
и даже условий работы.)
• Проведите анализ преимуществ и недостатков каждого процесса и решения.
• Разработайте альтернативные методы и процессы на основе вашего опыта,
наблюдений и знаний.
228
Часть 3. Качество
• Спроектируйте интегрированную систему на основании полученных резуль-
татов анализа.
• Работайте в мультидисциплинарной группе и обменивайтесь идеями.
• Изучите ограничения и огромный потенциал новых идей и разработок на
базе данного примера (помните — чтобы выжить и победить, вы должны до-
бавлять стоимость) [54—57].
После этого короткого введения мы продемонстрируем решение, полученное ме-
тодом CORA в виде электронной таблицы, на реальном примере из жизни (рис. 6).
3.1.10. Анализ инновационных рисков, метод возможностей
и программное обеспечение для фармацевтических
производственных систем
Наш анализ рисков отказа и метод возможностей вместе с итеративным програм-
мным обеспечением, являющиеся частью библиотеки инструментов NPPI (New
Product & Process Innovation — инновации для новых продуктов и процессов), по-
вышают эффективность систематического сотрудничества и ориентированного на
рабочие группы технического обсуждения при проектировании нового продукта
или процесса в фармацевтической производственной системе. Мы называем его
«методом возможностей», поскольку большинство рисков, если не вообще все, от-
крывают новые возможности д ля инноваций. Метод основан на нашем общем ана-
лизе рисков отказа процесса, который может применяться практически к любому
процессу, включающему в себя риски, — а инновация представляет собой очень ри-
скованный процесс.
При анализе рисков, связанных с объектами, компонентами и их атрибутами,
мы следуем методу, основанному на системе правил. Эти не требующие дополни-
тельных доработок правила могут не совпадать для различных объектов, областей
исследований и отраслей. Они создаются и стандартизируются для различных сек-
торов экономики, способствуя применению аналитического подхода, системной
стандартизации и получению точных и предсказуемых результатов.
Наш метод анализа рисков и инструменты помогают группе технического управ-
ления ответить на следующие вопросы:
• Какие проблемы могут возникнуть с процессами, задействованными в инно-
вационном проекте?
• Насколько серьезными могут быть эти проблемы, и каковы будут при этом
финансовые потери?
• Какие процессы и операции имеют наибольший риск при работе над иннова-
циями и проектами, касающимися продуктов, процессов и услуг?
• Что необходимо сделать, чтобы предупредить отказы?
• Какие процессы необходимо изменить, чтобы снизить риск отказов?
• Какие инструменты и оснастка требуются для предупреждения отказов и сни-
жения рисков?
• Какое обучение необходимо для участников, разработчиков, инженеров
и владельцев процессов, таких как линейные менеджеры и операторы, чтобы
снизить вероятность или предупредить отказы?
Программа объектно- и компонентно-ориентированного анализа требований для сетевого экономичного производства.
Разработка Пола Дж. Рэнки © 1992—1996. © Пол Дж. Рэнки, 2000—2002
Технические и программные решения, соответствующие требованиям потребителя Менеджер экономичного производства. Требования потребителя (отражение компоненто-ориентиро- ванного поведения, связанного с потребностями потребителя) Важность в баллах (1-5) Сеть полевых шин (Fieldbus) с^еть высокоскоростных шин (Profibus) с S J S э а U а U V J Локальная сеть Ethernet Графическая программа на CNC Техническое обслуживание «на месте» Удаленное техническое обслуживание Резервный сервер «на месте» Удаленный резервный сервер Связь с контролем продук- ции на предприятии Связь с TQM/ TQC пред- приятия 2В-видеофильмы и 3D- мультимедиа । Веб-камера для ячейки или модуля Компьютеризированный контроллер типа CNC компьютеризированная ра- бочая станция-контролллер Ко мпьютеризир ованный контроллер ячейки
№ Описание требования 1 2 3 4 5 6 7 8 9 10 и 12 13 14 15 16
1 Высокая надежность передачи данных для до- ступа в реальном времени 5 9 9 9 9 9 3 9 9 9
2 Надежность процесса отчета об отказе, пере- даваемом на компьютер линейного менеджера 4 9 9 9 9 9 9 9
3 Простота интеграции в систему (сетевое под- ключение типа plug- and- plav) — важно! 4 9 9 9 9 9 9 9
4 Простота программирования производствен- ного оборудования (машинная обработка и проверка на основе CNO 3 9 9 9 9 3 3
5 Простота смены программ изготовления дета- лей на основе CNC (локально и через сеть) 4 9 9 9 9 9 3 9 9 9 9 9 9
6 Очень простое добавление новых сенсоров к рабочей станции, CNCwm ячейке 3 9 9 9 9 9 9 9
7 Безопасность операций — критично! 5 9 9 9 9 9 3 9 3 9 9 9
8 Низкая стоимость изменения, расширения и развития системы 4 3 3 3 3 9 3 3 3 9 3 3
9 Низкая стоимость и небольшая потребность обучения оператора 3 9 9 3 9 3 3
10 Низкая стоимость и сложность установки сети 3 _2_ _2_ JL 3 3 _2_ 3
Рис. 6, а. Метод CORA — это специальный метод анализа требований потребителя на основе электронных таблиц. (В данном контексте «по-
требитель» — это поставщик фармацевтической производственной линии, пользователь, оператор, инженер по техническому обслуживанию
и т. д.). Основная идея заключается в том, что мы создаем корреляционную матрицу, а затем оцениваем результаты, используя количественный
вычислительный подход. Это намного более точно, чем использование простого структурированного перечня. Наш метод предлагает группе
проектирования фармацевтической производственной системы перечень всех основных требований, а также ранжирование приоритетов, ко-
торым они могут следовать в ходе разработки (более подробно о данном программном инструменте см. http'.//www.cimwareukandusa.com).
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 229
2Q Доступность в сети базы данных по истории системы 5 5 3 3 3 3 3 3 9 9 9 3 3 9
Целевые значения (перечислите здесь параметры, точно определяющие технические решения; если вы не знаете диапазон приемлемых величин, используйте программу Тагучи «Калькулятор для проектирования эксперимента») В течение 27 мс В течение 27 мс В течение 27 мс В течение 27 мс Графический интерфейс пользо- вателя, иконки, мультимедиа Менее 3 мин Менее 24 ч О-секундное переключение Переключение менее чем за 30 с Очистка памяти каждые 2 мин Обновление каждые 2 мин 320x240 пикселей или лучше 320x240 пикселей или лучше Win, Linux, Solaris или OSX Ответ в течение 12 мс Ответ в течение 24 мс
о » Наш продукт 5 - Продукт конкурентам 3 —а— Продукт конкурента В Продукт конкурента С п. — Г*—1 £ — С « - ' л н-— — h LV— —
*
*1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 -
Абсолютная важность 416 416 416 489 207 147 111 186 107 144 135 87 87 567 381 588 0 0 0 0
Относительная важность 9,3 9,3 9,3 11 4,6 з,з 2,5 4,1 2,4 3,2 3 1,9 1,9 13 8,5 13 0 0 0 0
Введите баллы для нашего продукта (1 — низкий, 5 — высокий) 4 5 5 4 5 3 4 3 4 5 5 5 4 5 4 5
Введите баллы для конкурента А (1-5) 3 4 4 5 4 3 2 2 3 3 4 4 3 4 4 4
Введите баллы для конкурента В (1-5) 4 4 4 4 4 5 4 4 3 2 3 2 3 4 3 4
Введите баллы для конкурента С <Ь5) 3 2 3 3 2 2 1 1 2 3 4 3 4 3 2 3
230 Часть 3. Качество
Рис. 6, б
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 231
После этого введения мы рассмотрим компоненты системы анализа рисков, удо-
влетворяющей упоминавшимся ранее принципам ИСО 9000:2000, и наши возможно-
сти по моделированию рисков управления сложным проектом с использованием этих
принципов (рис. 7). Обратите внимание, что активные закодированные электронные
таблицы и ЗЛ-объекты являются частью электронной библиотеки Рэнки [50—55].
Рэнки 11 1601/DFRA_Ver.5 Код процесса демонтажа RankyPC DisassyCode: 07.05.1997
16.11.2001 Дата технической публикации методологии процесса 07.05.1997
19.09.2001 Тип демонтируемого объекта Электромеханический
Rev.2.1.3 проведена Рэнки Классификатор группы продукта Настольный персональный компьютер (ПК)
Дата технического выпуска про- дукта Приблизительно 1993 г.
Укажите или идентифи- цируйте детали и компо- ненты, извлекаемые на каждом этапе процесса демонтажа Длитель- ность про- цесса, с Стоимость процесса, 16,40, USD Суммарная стоимость процесса, USD Группа DFRA описывает и показывает виды и последствия возможных отказов при монтаже и риски отказов
ID Виды отказов и последствия
Окрашенный металлический корпус ПК (файл 3DMetalCover. mov) 45 0,21 0,21 /01.1 Металлический корпус оцарапан соскользнувшей отверткой
ID 1.2 При удалении корпуса внутренние детали получают царапины
ID 1.3
Дисковод и жесткий диск, прикрепленные к прочной стойке из листового металла внутри ПК (файл 3DFIoppyHDassy. mov) 137 0,62 0,83 /D2.1 Извлечение дисковода может повредить материнскую плату
ID 2.2
ID2.3 Видеофильм, показывающий риски при монтаже и извлечении дисковода и жесткого диска
Дисковод (файл 3DFIoppyDrive.mov) 35 0,16 0,99 ID ЗА Может быть поврежден дисковод, если конструкцию уронят
ID 3.2 Может быть поврежден жесткий диск, если конструкцию уронят
ID 3.3
Жесткий диск (файл 3DHDobj.mov) 65 0,30 0,28 ID 4.1 Может быть поврежден жесткий диск, если конструкцию уронят
/04.2
ID 4.3
Металлическая стойка, держащая дисковод и жесткий диск (файл 3DFIoppyHDBracket.mov) 12 0,05 1,34 ID 5 A
ID 5.2
ID 5.3
а)
Рис. 7. Инструмент для анализа рисков отказа процесса (PFRA, process failure risk analysis)
представляет собой аналитический программный продукт, использующий базы правил для
оценки рисков процесса. Это идеальный метод и инструмент для уменьшения дорогостоящих
отказов. (Более подробно об этом программном продукте см. http://www.cimwareukandusa.com.)
232
Часть 3. Качество
Этот отчет DFRA подготовлен Пол Дж. Рэнки, NJIT/MERC
Группа DFRA Группа DFRA, NJIT/MERC
Ответственная организация или подразделение NJIT/MERC
Примечания
ID Тяжесть отказа . Вероятность выявления отказа Вероятность возникновения отказа Степень риска Максимальный риск Фактор оборудования Фактор крепежа и оснастки Фактор опыта Фактор, определенный вами Суммарная степень риска Связанный риск
в баллах от 1 до 10 0,12,1 = 100%
1.1 3 2 3 18 20 1,40 1,00 1,20 1,00 33,60 Низкий
1.2 5 4 1 20
1.3 0
2.1 5 9 2 90 90 1,60 1,40 1,40 1,00 282,24 Высо- кий
2.2 0
2.3 0
3.1 8 2 1 16 48 1,20 1,20 1,40 1,00 96,77 Низкий
3.2 8 2 3 48
3.3 0
4.1 9 2 3 54 54 1,40 1,20 1,40 1,00 127,01 Тоже
4.2 0
4.3 0
5.1 0 0 1,00 1,00 1,00 1,00 0,00 »
5.2 0
5.3 0
б)
Рис. 7 (продолжение)
3.1.11. Мультимедийные трехмерные и программные
статистические приложения из открытых источников
для инноваций фармацевтических производственных
систем и взаимодействия при работе с проектом
Так как мы применяем аналитический, количественный и открытый вычислитель-
ный подход, наши методы управления фармацевтическим продуктом, процессом
и проектом, а также программное обеспечение представлены в виде электронных
таблиц MS-Excel (открываемых в интернет-браузерах), связанных с помощью гипер-
ссылок с базой правил и дополнительными 2Л-видеоматериалами и 3D-объектами
в виртуальной реальности для визуализации.
Это обусловлено тем, что мы хотим предложить нашим пользователям возмож-
ность не только понимать методы и закодированную логику, но также получать
удовольствие от интерактивной 3D-графики, цифровых видеоматериалов, цветных
иллюстраций и, что наиболее важно, пользоваться активными кодированными
электронными таблицами. Вместе с любой другой доступной визуализацией это по-
зволит обрабатывать и исследовать свои собственные данные.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
233
В отношении статистических методов следует отметить, что наша библиотека
инструментов NPPI содержит несколько инструментов для статистического ана-
лиза, позволяющих выявлять инновационные возможности в процессах, которые
могут приближаться к установленному уровню допусков (верхнему или нижнему)
и могут выходить из-под контроля, или в процессах, которые выполняются со слу-
чайным отказом.
Для выявления таких критических возможностей для инноваций и улучшения
процесса мы используем набор контрольных карт для анализа дрейфа данных, ме-
тоды Тагучи проектирования эксперимента для разработки требуемого перечня па-
раметров для технических решений в нашем методе анализа требований и методы
Вейбулла для оценки надежности процесса. По мере развития мы планируем ввести
в библиотеку инструментов NPPI дополнительные статистические и другие про-
граммы [56—70].
234
Часть 3. Качество
3.1.12. Приложения RFID
В Соединенных Штатах Америки технологии радиочастотной идентификации
(RFID, radio-frequency identification) активно внедряются в управление фармацевти-
ческим производством, сборкой и упаковкой, производством в целом, складиро-
ванием, распределением и глобальной товаропроводящей сетью. Ожидается, что
объем рынка для этой технологии возрастет с 500 млн долл. США в 2005 г. до при-
близительно 4 млрд долл. США в 2010 г. В данном разделе мы рассматриваем не-
которые из основных областей применения RFID, в частности в фармацевтической
отрасли.
Мы также обсудим возможности для исследований и проектирования (R&.D)
и результаты моделирования некоторых фармацевтических производственных си-
стем с технологией RFID. Более того, мы предлагаем ЛЕН)-интеграцию типовой
промышленной конструкции с отслеживающей цифровой моделью — наиболее
сложную задачу, с которой столкнулись разработчики производственных систем,
промышленные инженеры и ИТ-эксперты, если учесть смешанные режимы ре-
ального времени, глобальную прослеживаемость и проблемы обработки сигналов,
которые возникают с ЛТШ-мечеными деталями и поставками. Возможности техно-
логии RFID огромны, так как при наличии соответствующей ИТ-инфраструкгуры
она помогает крупным дистрибьюторам и производителям, а также другим логи-
стическим операторам, в том числе учреждениям здравоохранения, оборонной от-
расли и др., взаимодействовать с комплексными глобальными товаропроводящими
сетями, в которых продукты и их поставки должны отслеживаться и идентифици-
роваться бесконтактным беспроводным способом с помощью компьютерной сети,
что обусловлено стоимостью, секретностью или безопасностью этих продуктов или
тем, что изделия могут подвергаться коррозии, а лекарственные средства могут те-
рять качество.
Всем этим требованиям удовлетворяет метод автоматической идентификации
с использованием датчиков для беспроводного считывания данных, предлагающий
больше функциональных возможностей и значительно более «интеллектуальный»,
чем хорошо известные штрихкоды или универсальный код товаров (UPC, unified
product code).
RFID-метки выпускаются в виде пассивных и/или активных чипов с возможно-
стью считывания и записи данных (однократной или многократной), что актуально
для относительно больших площадей (например, д ля больших складских логических
центров, контейнеровозов и т. д.), при этом все операции выполняются автомати-
чески, контролируются компьютерами, а обмен данными осуществляется беспро-
водным способом через защищенные локальные интранет-сети. Данная технология
привлекательна для менеджеров фармацевтического сборочного предприятия или
товаропроводящей сети тем, что когда RFID-сетъ встроена в информационные си-
стемы управления складскими запасами предприятия, точная информация по всем
изделиями с Л/71)-метками может быть получена практически в режиме реального
времени с любого участка товаропроводящей сети. Эта информация касается гло-
бально распределенных предприятий и включает сведения об изделиях и сборных
конструкциях в процессе поставок, включая транзит. Поэтому метод RFID очень
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 235
интересен для исследований и технологий и предоставляет огромные коммерческие
возможности.
В данной главе мы познакомимся с некоторыми из наиболее важных принципов
управления техническими и информационными системами и проблемами, о кото-
рых следует помнить разработчикам, специалистам по внедрению и пользователям
технологии RFID при проектировании таких систем и/или планировании таких
приложений, как, например, модель интеграции RFID в автоматически управляе-
мое предприятие на языке UML [60—64, 70, 73].
3.1.13. Примеры/?/7Р
Давайте представим себе большое складское помещение для хранения множества
лекарственных средств или фармацевтический логистический центр с тысячами ко-
робок, изделий и сборных конструкций со стоимостью от минимальной до самой
высокой, а возможно, даже с высокотехнологичными или скоропортящимися ле-
карственными формами, которые должны храниться в определенных условиях при
фиксированных температуре, влажности или давлении на протяжении всей транс-
портировки и/или производственных и упаковочных операций.
Принцип доставки «точно в срок» в данной отрасли означает, что для выпол-
нения складом заказа, включающего поставку разнообразных препаратов или ис-
ходных веществ, каждый компонент должен быть на месте в нужное время и в хоро-
шем состоянии, что бывает крайне сложно выполнить.
Понятно, что современная сеть поставок является глобальной, и транспорти-
ровка обычно осуществляется различными способами, включая авиаперевозку,
морской, автомобильный и железнодорожный транспорт. Во всех случаях могут
возникнуть проблемы вследствие погодных условий, интенсивности трафика, про-
мышленных забастовок или других причин. Высокая сложность таких товаропро-
водящих систем обусловлена неопределенностью в поставках, потерей или кражей
изделий и материалов, повреждением товаров при транспортировке, большим ко-
личеством международных портов и таможен, где проверка поставок может за-
нять непредсказуемое время из-за различных уровней безопасности и секретности,
и многими другими причинами.
Существует множество доводов, по которым беспроводные компьютерные ме-
тоды, инструменты и технологии идентификации изделий с помощью сенсоров
должны постоянно исследоваться и внедряться в различные отрасли. Области при-
менения этих технологий и их возможности огромны. Ключевой принцип заключа-
ется в том, что даже в хаотичном широко распределенном и более стохастическом,
чем детерминированном, деловом мире адаптивные организации и предприятия
должны быстро реагировать на запросы, иначе их место займет конкурент. Следова-
тельно, они должны снижать издержки и повышать эффективность во всех областях
деятельности. Одним из важных аспектов этой стратегии является получение точ-
ных данных о наличии изделий на своих складах, точном их месторасположении,
степени законченности их сборки или состоянии готовности. Кроме того, основные
дистрибьюторы, взаимодействуя со сложной глобальной товаропроводящей сетью,
должны иметь возможность детально отслеживать свои отгрузки, что связано со
236
Часть 3. Качество
стоимостью, секретностью, безопасностью, возможным ухудшением качества (как
в случае компонентов или препаратов, чувствительных к температуре, влажности
и/или свету) и с другими причинами.
Как уже говорилось, технология RFID вместе с соответствующей ИТ-
инфраструкгурой помогает крупным дистрибьюторам и производителям, а также
другим логистическим операторам, в том числе учреждениям здравоохранения,
оборонной отрасли и др., взаимодействовать с комплексными глобальными това-
ропроводящими сетями, в которых продукты и их поставки должны отслеживаться
и идентифицироваться бесконтактным беспроводным способом с помощью ком-
пьютерной сети.
RFID-метка содержит блок данных с уникальным идентификатором. Напри-
мер, 64-битная метка класса 0, предлагаемая поставщиком, имеет 64 бита общей
памяти в самой метке (чипе), включая уникальный серийный номер. Этот номер
кодируется производителем и позволяет ввести уникальную идентификацию до
264 = 18446744073709551616 предметов с метками.
Метод RFID очень интересен для исследований и технологий и предоставляет
огромные коммерческие возможности. Чтобы показать это, представим такие ис-
следовательские задачи, как удаленное сканирование и отслеживание:
1) продуктов и изделий в коробках на грузовом судне, которое приближается
к суверенной акватории из международных вод;
2) изделий, которые могут подвергнуться коррозии и используются в сельскохо-
зяйственном или военном оборудовании;
3) поддельных медицинских препаратов, которые были переупакованы, а затем
отгружены и нелегально импортированы в другие страны;
4) ноутбуков, которые случайно уронили и повредили.
Очевидным признаком коммерческих возможностей данного метода служит то,
что согласно опубликованной презентации Министерства обороны США экономия
товаропроводящей сети, использующей RFID, достигла более 460 млн долл. США
в 2004 г., и по проектам превысит 4 млрд в 2010 г.
3.1.14. Модели интеграции RF/D в товаропроводящие
сети компьютеризированных фармацевтических
производственных и сборочно-упаковочных систем
В американском производстве и сборочно-упаковочной промышленности целью
многих пилотных 7?/77)-проектов является достижение 100%-ного уровня считы-
вания на скоростях, установленных для широко распространенной технологии
штрихового кодирования. В задачи этих проектов входит поиск адекватного место-
расположения меток на коробках и паллетах, а также подбор конфигурации паллет,
позволяющей выполнять 100%-ное считывание. Это вопрос огромной важности
в фармацевтическом производстве и сборке в борьбе против попадания на рынок
поддельных продуктов и упаковок! (См. рис. 8—15.)
На основе описанного выше анализа требований, планирования сети и с учетом
баланса сервера, для наших целей мы решили выбрать простую, но мощную архи-
тектуру сети, в которой предусмотрели подсети. В соответствии с порядком работы
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
237
с подсетями в OPNETIT-Guru, подсеть содержит другие сетевые объекты и объеди-
няет их в один объект. Подсеть может включать набор узлов и связей в виде физи-
ческой группировки объектов (например, локальная сеть станков с ЧПУ на основе
CNC или роботов с компьютеризованными контроллерами), или она может содер-
жать другие подсети (например, включая линейный контроль системы управления
материалов) [32, 34, 63, 66, 69—77].
Подсети внутри других подсетей формируют иерархическую модель сети. Эта
иерархия может быть затем расширена в зависимости от требований модели струк-
туры сети. Подсеть считается родителем объектов внутри нее, а объекты являются
дочерними объектами подсети. Подсеть наивысшего уровня в иерархии всей сети
не имеет родителя и соответственно является верхней, или глобальной, подсе-
тью. Подсети могут создаваться и быть связаны внутри этого верхнего уровня или
в других подсетях. Подсети представляют собой мощный механизм для управления
сложными сетями, использующий абстракцию для упрощения сложности сети.
Поскольку в наших имитационных моделях фармацевтической сети мы имеем
дело с пакетами информации, необходимо привести некоторые пояснения относи-
тельно их форматов. Пакеты содержат информацию и передаются между отправи-
vOemendjype SVmo
C6tart_dete DMe
«шом .&свг Fie
DArluai ttnr tire tart
Рис. 8. Сегмент UML-модели, иллюстрирующий путь, которым исходный файл интегрируется
с файлами маршрутизации и инструментальной оснастки при условии, что все детали и все
инструменты снабжены RF/D-метками. Такие UML-модели, как эта, следует использовать перед
любой работой по внедрению RFID, чтобы оценить требования, потребности в технологии
и проблемы, которые могут возникнуть с остальной ИТ-структурой предприятия при внедрении RFID
tX>der_mmber Irteger
cOrder Jype iriege;
oMerarf&cU«ing_method String
<»«nfemjm_send_ehew_quertiy нвди
0Sp«jacKr Double
<*tochme_Wne OaiHe
ОТ<кй_С ОМ_Т<ю1 JTte
*»ieOM_DeHwnd_Fie
OQueueJime OrWte
d₽tanoed_eekpjkne. Coude
□Plaoned. labour fine Double
oHove time DoUbte
<XMrtendrg_t)ueri1ty Double
ocwnponeriдмИ_гил*>ег OM_Frodua_smeure_Fae
uKrish_dete Drte
dPartjMMtoer OM_part_Mwter_Fle
ooverlapjime. tx-jue
OM_Work_Certre_Ffe
O>derjauntier • CAt_Or<ier_Fte
eArtuol_seh<sJme Doo*
3Mn„open*or>Jine Doutts
6Mex_number_uf_spBs. Integer
c6cbecMed_st8rt_oae
OM_partJrtsstw_F»e
ottttcency DoutJe
OMJ*«rt_Cs«re_Fle
cM*jrk_certre_desa(jbori String
c6et_up_cost_rate Doubfe
twne_ot_me&9ure Integer
<*Vxk_i'J!rtrs_name • swig
OWcuk_certre_coife: rteger
OAts»nt*we_wort._cerire UM Wjk_€enlie_Ffe
coverageJianspotUVne - DouWe
rerirr faction String
dWxk_eerire_code.
C*tr«num_Bifch. true • Double
oRmiirig jnitiber Integer
OL utxur tme _r*werf Double
c6knriiage_factor Dottie
oCo<Tponert_p«t_F<jrr*)er. OM_Product_Structure_Fie
CPart_rtir*er OM_Parl_«ist«_Fte
<Xestr^ofjner«ft«l^l>ioperAon' String
cJHrtenoete Date
OM_pcx±ngJ^e
238
Часть 3. Качество
Рис. 9. Имитационная сеть для распределенных фармацевтических производственных систем
и их складов в США, Европе, Индии и Азии. Модель создана для оценки управления информацией
и данными, а также выполнения серверами задачи по отслеживанию фармацевтического продукта
и RF/D-данных в международной базе. В качестве инструмента моделирования мы используем
OPNET — профессиональный инструмент для имитации сети
Рис. 10. Сегмент имитационной модели, иллюстрирующий штаб-квартиру корпорации в Нью-
Йорке. Там мы имеем основные серверы в нашей распределенной модели. Инструмент
моделирования — OPNET
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 239
Рис. 11. Сегмент имитационной модели, иллюстрирующий сеть на площадке в Нью-Дели. Здесь
расположена группа по бизнес-процессам, выполняемым по договору, и вспомогательные
серверы в нашей распределенной модели. Инструмент моделирования — OPNET
Рис. 12. Порталы фармацевтической компании в виде беспроводной сети фармацевтической
производственной системы. Мощность модели такова, что мы можем имитировать запрос,
комментарий или предупреждение производственного подразделения через всю международную
сеть глобально распределенной фармацевтической компании со всеми важными функциями
и процессами. Это означает, что перед физическим построением любой фармацевтической
производственной системы мы можем смоделировать всю систему в цифровом виде, уменьшая
расходы и сберегая время. Инструмент моделирования — OPNET
240
Часть 3 Качество
Колебания задержки АГМ-ячейки
Рис. 13. Диаграмма имитации работы системы, иллюстрирующая и подтверждающая, что
с точки зрения соотношения «колебание АГМ-время отклика» дизайн сетевой системы может
выдерживать требуемую нагрузку. Инструмент моделирования — OPNET
телями и получателями. В нашем примере пакеты могут содержать программы для
роботов, когда загружаются с серверов офиса по проектированию или программи-
рованию на роботизированные линии, а затем в индивидуальные CNC, роботы или
их части, если существует необходимость в обновлении, редактировании, контроле
качества, производственном контроле, обслуживании и т. д. (Пакеты могут вклю-
чать критические для выполнения работы, «аварийные» данные, генерируемые в ре-
жиме реального времени, при обмене информацией между компьютеризированным
контроллером робота и линейными серверами.)
Пакеты представляют собой структуры данных, состоящие из областей хране-
ния, называемых полями, и могут быть форматированными или неформатирован-
ными. Форматированные пакеты имеют поля, сформированные согласно форма-
ту пакета, который устанавливает название полей пакета, типы данных, размеры
и значения по умолчанию. Форматированные пакеты могут распознаваться только
соответствующими коммуникационными протоколами. Неформатированные па-
кеты не имеют предварительно определенных полей. В программном обеспечении
IT-Guru форматы пакетов предустановлены и, как правило, называются по модели,
в которой их предполагается использовать.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
241
Сервер-2 БД в Нью-Йорке
Рис. 14. Диаграмма имитации работы системы, иллюстрирующая и подтверждающая, что по
параметрам баланса серверов дизайн сетевой системы может выдерживать требуемую нагрузку.
Инструмент моделирования — OPNET
3.1.15. Оценка результатов имитации работы сети
Основная цель большинства имитационных сценариев заключается в оценке не-
скольких аспектов работы системы или ее работоспособности, в количественном
выражении, часто с применением статистических методов, результатов и последу-
ющем использовании полученных результатов для принятия решений. Для таких
испытаний требуется имитационная среда с программными инструментами, обе-
спечивающая взгляд изнутри на динамическую работу системы.
На основе детального анализа средствами IT-Guru аналитик по проектированию
сети фармацевтической производственной системы может получить статистические
данные о конкретном объекте, об объектах в сценарии и общую статистику.
• Статистика по объекту собирается по отдельным объектам. Эти данные по-
зволяют аналитику по проектированию сети оценить работу определенных
узлов сети или связей (задержку в одном хабе интранет-сети или изменение
баланса сервера, как в нашем примере).
• Статистика по объектам сценария собирается по всем соответствующим объ-
ектам в сети (например, задержка в интранет-сети по каждому узлу). Эти дан-
242
Часть 3. Качество
HTTP. Время ответа объекта, с
Рис. 15. Диаграмма имитации работы системы, иллюстрирующая и подтверждающая, что дизайн
системы сети при оценке по параметру «изменение объекта-время ответа» сможет выдерживать
требуемую нагрузку. Например, очень важно, если линейный менеджер фармацевтической
производственной системы в Индии хочет с целью оценки качества обратиться к менеджеру
в Нью-Йорке, послав ему графическое изображение, или звуковой файл, или мультимедийное
изображение станка в линии. Инструмент моделирования — OPNET
ные позволяют аналитику по проектированию сети с легкостью наблюдать за
работой всех объектов определенного типа.
• Общая статистика собирается по всей сети. Эти данные представляют резуль-
таты, которые применяются к сети как к единому целому (например, гло-
бальная задержка от одного конца сети до другого) и позволяют проектантам
и администрации анализировать общие параметры работы сети.
• IT-Guru предлагает следующие типы статистических данных при анализе
сетей:
— размер очереди;
— доступное место;
— частота переполнения;
— задержка;
— длительность интервала между двумя последовательными входами;
— размер пакетов;
— пропускная способность;
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 243
— эксплуатация;
— частота ошибок;
— конфликты;
— статистика, специфичная для приложения и определяемая проектиров-
щиком модели.
Так как существует много разных статистических данных, файлы данных могут
быстро увеличиваться и становиться неинформативными, если программа имита-
ции будет регистрировать их всех. Поэтому перед проведением имитационных ис-
пытаний аналитик всегда должен выбрать ту определенную статистику, которая бу-
дет информативна для этого конкретного исследования [71—79].
3.1.16. Заключение
В этой главе мы представили основные, основанные на стандартах всеобщего каче-
ства принципы проектирования аналитической и одновременно автоматизирован-
ной «стройной» и гибкой фармацевтической производственной системы. Мы обсу-
дили, почему данный подход необходим при проектировании фармацевтического
продукта, процесса и производственной системы.
Как было показано на основе результатов имитационных испытаний, с помощью
распечаток графиков и скриншотов администрация может легко оценить различные
альтернативные проекты, модели поведения оборудования и людей, контрольные
системы, обработку сенсорной обратной связи и необходимость сбалансирован-
ной архитектуры серверов и даже исследовать в дальнейшем сценарий «что будет,
если...» без выполнения крупных капиталовложений.
Мы уверенно заявляем, что пришло время, когда фармацевтические производ-
ственные системы могут быть спроектированы и построены полностью в цифровой
форме, что существенно уменьшит финансовые и другие связанные затраты и одно-
временно увеличит качество.
3.1.17. Дополнительные видеоматериалы на DVD
Для иллюстрациипримеров проектирования существующих в реальном мире вы-
сокотехнологичных продуктов, процессов, производственных, сборочных и упако-
вочных систем мы не всегда можем использовать статичные печатные книги. Для
таких случаев мы создали дополнительные видеоматериалы с высоким разрешени-
ем и поместили их на DVD. Профессионально созданные видеодиски поддержива-
ют материалы этой главы в качестве независимой самостоятельной публикации,
показывающей проектирование высокотехнологичных фармацевтических и меди-
цинских препаратов, процессов и производственных систем, соответствующие про-
цессы и решения по обеспечению качества и другие материалы вместе с объяснени-
ями отраслевых экспертов. Для получения более детальной информации см. Ranky,
Р. G., Ranky, G. N. and Ranky, R. G. (2006), Design Principles and Examples of Pharm-
aceutical Manufacturing Systems (Product, Process, Lean and Flexible Manufacturing,
Assembly and Packaging System Designs) — видео на DVD, доступное на сайте www.
cim wareukandusa. com.
244
Часть 3. Качество
Литература
1. Ranky, Р. G., A generic, analytical method to assess process-related risk with case studies, The Project
Management Institute (PMI) Risk SIG and the Institute for International Research (HR), paper pre-
sented at the Annual US National Project Risk Symposium, Houston, TX, May 22-May 26, 2006.
2. Ashley, S. (2004), Penny-wise smart labels, Sci. Am. 291 (2), 30-31.
3. Ashton, P., and Ranky, P. G., (1999, Feb.), An advanced concurrent engineering research toolset and
its application at Rolls-Royce motor cars, ADAM (Adv. Des. Manufacturing), available: http://www.
cimwareukandusa.com, listed and indexed by the Association of Research Libraries, Washington, DC,
and the Edinburgh Engineering Virtual Library, United Kingdom, Vol. 1.
4. Bradbrook, R. (2004), Wal-Mart and RFID, folding carton industry, Printing News, 31 (4), 30-33
5. Bradbrook, R. (2004), Procter and Gamble aim to be among the world leaders in RFID implementa-
tion, Int. Paper Board Ind., 47 (8), 20-23.
6. Brzozowski, C. (2004), Tags, tickets & labels: New technologies emerge, Printing News, 153 (3).
7. Ranky, P. G., An integrated PM approach, including: Process modeling, requirements analysis, risk
analysis, statistical tools and 3D multimedia, presented at the Project Management Institute
(PMI), New Jersey Chapter. Jan. 2006.
8. Ranky, P. G., Focus on RFID (radio frequency identifi cation) methods, technologies and education,
presented as part of the NCME Mission (National Center for Manufacturing Education), sponsored
by NSF (National Science Foundation, USA) and industry, Jan. 2006.
9. Flaherty, M., Ranky, P. G., Ranky, M. F., Sands, S., and Stratful S. (1999, Mar.), An engineering
multimedia approach to servo pneumatic positioning, ADAM (Adv. Des. Manufacturing), available:
http://www.cimwareukandusa.com, listed and indexed by the Association of Research Libraries,
Washington DC, and the Edinburgh Engineering Virtual Library, United Kingdom, Vol. 1.
10. Glidden, R., Bockorick, C., etal. (2004), Design of ultra-low-cost UHF RFID tags for supply chain
applications, IEEE Commun. Mag. 42 (8), 140-151.
11 Graham- Rowe, D. (2004), Tags to banish forgetfulness, New Scientist, 183 (2460), 19.
12. Graham-Rowe, D. (2004), Who ’ s keeping tabs on your tags? New Scientist, 183 (2462), 22.
13. Ho, K. L., and Ranky, P. G. (1999, Mar.), An object oriented approach to fl exible conveyor system
design, ADAM (Adv. Des. Manufacturing), available: http://www.cimwareukandusa.com, listed and
indexed by the Association of Research Libraries, Washington DC, and the Edinburgh Engineering
Virtual Library, United Kingdom, Vol. 1.
14. Knights, P. F., Henderson, E., and Daneshmend, L. K. (2004), Drawpoint control using radio fre-
quency identifi cation systems, CIM Bull., 89 (1003), 53—58.
15. Loose, D. C., and Ranky; P. G. (2007), A Case-Based Introduction to IBM ’ s Telematics Solutions,
interactive multimedia eBook with 3D objects, text, and videos in a browser-readable format on CD-
ROM/intranet, available: http://www.cimwareukandusa.com, CIMware USA, Inc., and CIMware
Ltd., United Kingdom; multimedia design and programming by P. G. Ranky and M. F. Ranky.
16. Mongeon, D. G. (2005, Feb.), RFID: A tool for the 21st century distribution, USA Department of
Defense Conference Presentation, RFID Media Briefi ng, Washington DC.
17. Nadler, S. E, Ranky, P. G., and Ranky, M. (2002-2003), A 3D multimedia approach to the diagno-
sis of low back pain (Vol. 1, 18-and 40-year-old males), interactive 3D multimedia presentation on
CD-ROM with off-line Internet support (650 Mbytes, approx. 150 interactive screens, 50 minutes of
digital videos, 3D internal and external body tour, animation, and 3DVR objects), by CIMware (LEE
and IMechE Approved Professional Developer); also in Multimedia design and programming by P. G.
Ranky and M. F. Ranky.
18. Ranky, G. N., and Ranky, P.G. (2005), Japanese service robot R & D trends and examples, Ind. Robot,
32 (6), 460-467.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования... 245
19. Ranky, Р. G., Caudill, R. J., Limaye, К., Alli, N., ChamyVelumani, S., Bhatia, A., and Lonkar, M.
(2002, May), A Web-enabled virtual disassembly manager (web-VDM) for electronic product/process
designers, disassembly line managers and operators, a UML (Unifi ed Modeling Language) model of
our generic digital factory, and some of our electronic support system analysis tools, ADAM with IT
(Adv. Des. Manufacturing), available: http://www.cimwareukandusa.com, listed and indexed by the
Association of Research Libraries, Washington DC, and the Edinburgh Engineering Virtual Library,
United Kingdom, Vol. 3.
20. Ranky, P. G. (2006), Introduction to RFID — Radio frequency identifi cation methods and solutions,
Assembly Automation, 26 (1), 28-33.
21. Ranky, P. G. (1999, Apr.), New trends in fl exible, lean and agile manufacturing cells and systems,
ADAM (Adv. Des. Manufacturing), available: http://www.cimwareukandusa.com, listed and indexed
by the Association of Research Libraries, Washington DC, and the Edinburgh Engineering Virtual
Library, United Kingdom, Vol. 1.
22. Ranky, P. G. (2002), A method for planning industrial robot networks for automotive welding and
assembly lines, Ind. Robot: Int. J., 29 (6), 530-537.
23. Ranky, P. G. (2003), A real-time manufacturing/assembly system performance evaluation and control
model with integrated sensory feedback processing and visualization, Assembly Automation.
24. Ranky, P. G. (2003), A simulation method and distributed server balancing results of networked indus-
trial robots for automotive welding and assembly lines, Ind. Robot: Int. J., 30 (2), 192-197.
25. Ranky, P. G. (2002), Advanced digital automotive sensor applications, Sensor Rev.: Int. J., 22 (3),
213-217.
26. Ranky, P. G. (2003), Advanced machine vision systems and application examples, Sensor Rev.: Int.
J., 23 (3), 242-245.
27. Ranky, P. G. (2003), Collaborative synchronous robots serving machines and cells, Ind. Robot: Int.
J., 30 (3), 213-217.
28. Ranky, P. G. (2004), Digital, Internet-enabled assembly line and factory modeling, Assembly Auto-
mation, 24 (3), 247-253.
29. Ranky, P. G. (2004), Novel automated inspection methods, tools and technologies, Assembly Automa-
tion: Int. J., 23 (3), 252-257.
30. Ranky, P. G. (2003), Reconfi gurable robot tool designs and integration applications, Ind. Robot: Int.
J., 30 (4), 338-344.
31. Ranky, P. G. (2002), Smart sensors, Sensor Rev.: Int. J., 22 (4), 312-318.
32. Ranky, P. G. (1999, Jan.,) Some generic algorithmic solutions to the problem of dynamic scheduling
in fl exible manufacturing systems that operate globally, ADAM (Adv. Des. Manufacturing), avail-
able: http://www.cimwareukandusa.com, listed and indexed by the Association of Research Libraries,
Washington DC, and the Edinburgh Engineering Virtual Library, United Kingdom, Vol. 1.
33. Ranky, P. G. (2001), Trends and R & D in virtual and robotized product disassembly, Ind. Robot, 28
(6), 454-456.
34. Ranky, P. G. (2000, May), Some analytical considerations of engineering multimedia system design
within an object oriented architecture, Int. J. CIM, 13 (2), 204—214.
35. Ranky, P. G., and ChamyVelumani, S. (2003), A method, a tool (CORA), and application examples for
analyzing disassembly user interface design criteria, Int. J. CIM, 16 (4-5), 317-325.
36. Ranky, P. G., and ChamyVelumani, S. (2003), An analytical approach, a tool (DFRA) and application
examples for assessing process-related failure risks, Int. J. CIM, 16 (4-5), 326-333.
37. Ranky, P. G., and Nadler, S. F., A novel multimedia approach to low back pain diagnosis with internal
and external 3D interactive body tours, paper presented at the 29th Annual Northeast Bioengineering
Conference, New Jersey Institute of Technology, University Heights, Newark, NJ, Mar. 2003.
246
Часть 3. Качество
38. Ranky, Р. G., and Nadler, S. E, A new, Web- enabled multimedia approach with 3D virtual reality
internal and external body tours to support low back pain diagnosis, paper presented at the 4th Annual
Faculty Best Practices Showcase in Kean University, NJ, Mar. 2003.
39. Ranky, P. G., and Ranky, M. F. (2000), A Dynamic Operation control algorithm with multimedia ob-
jects for fl exible manufacturing systems, Int. J. CIM, 13 (2), 245—263.
40. Ranky, P. G., Lonkar, M., and ChamyVelumani, S. (2003), eTransition models of collaborating design
and manufacturing enterprises, Int. J. CIM, 16 (4—5), 255—266.
41. Ranky, P. G., Morales, C., and Caudill, R. J., Lean Disassembly line layout, process and network
simulation models and cases, based on real-world data, paper presented at the IEEE (USA) Interna-
tional Symposium on Electronics and the Environment and the IAER Electronics Recycling Summit,
Boston, MA, May 19-22,2003.
42. Ranky, P. G., Ranky, G. N., and Ranky, R.G. (2006), Examples of pharmaceutical product/process/
manufacturing/assembly and packaging system designs, video on DVD, available: www.cimwareu-
kandusa.com.
43. Ranky, P. G., Subramanyam, M., Caudill, R. J., Limaye, K., and Alli, N., A dynamic scheduling and
balancing method and software tool for lean and reconfi gurable disassembly lines, paper presented
at the IEEE (USA) International Symposium on Electronics and the Environment and the IAER Elec-
tronics Recycling Summit, Boston, MA, May 19-22, 2003.
44. Ranky, P. G., 3D engineering multimedia cases. A customizable 3D Web-enabled library with reus-
able objects, paper presented at the ASEE (American Society of Engineering Educators) Mid-Atlantic
Conference, Kean University, NJ, Apr. 2003.
45. Ranky, P. G., A 3D multimedia approach to biomedical engineering: Low back analysis, paper pre-
sented at the ASEE, American Society of Engineering Educators, U.S. National Meeting, Biomedical
Engineering Division, Nashville, TN, June 2003.
46. Ranky, P. G., Ranky, G. N., and R anky, R. G. (2006), Design principles and examples of pharmaceuti-
cal manufacturing systems (product, process, lean & fl exible manufacturing, assembly and packaging
system designs), video on DVD, available: www.cimwareukandusa.com.
47. Ranky, P. G. (2001-2006), A 3D multimedia case: Component oriented disassembly failure risk analy-
sis, an interactive multimedia publication with 3D objects, text and videos in a browser-readable
format on CD-ROM/intranet available: http://www.cimwareukandusa.com, CIMware USA, Inc., and
CIMware Ltd., United Kingdom; Multimedia design and programming by P. G. Ranky and M. F.
Ranky (published 6 volumes of this main title with different risk analysis challenges explained).
48. Ranky, P. G. (2001-2005), A 3D multimedia case: component oriented disassembly user requirements
analysis, an interactive multimedia eBook publication with 3D objects, text and videos in a browser-
readable format on CD-ROM/intranet available: http://www.cimwareukandusa.com, CIMware USA,
Inc., and CIMware Ltd., United Kingdom, Multimedia design and programming by P. G. Ranky and
M. F. Ranky (published 7 volumes of this main title with different requirements analysis challenges
explained).
49. Ranky, P. G., A 3D Web collaborative concurrent automotive engineering Method Based on our “
distributed digital factory ” and “ digital car ” models, paper presented at the Society of Automotive
Engineers World Congress, Detroit, Ml, Mar. 2003.
50. Ranky, P. G. (2003), A 3D Web-enabled, case based learning architecture and knowledge documenta-
tion method for engineering, information technology, management, and medical science/biomedical
engineering, Int. J. CIM, 16 (4—5). 346-356.
51. Ranky, P. G., A Biomedical Engineering case with 3D lower back interactive virtual anatomy tours
inside and outside the human body with automated post-test student assessment, paper presented at
the ASEE (American Society of Engineering Educators) Mid-Atlantic Conference, Kean University,
NJ, Apr. 2003.
Глава 3.1. Аналитические и вычислительные методы и примеры проектирования...
247
52. Ranky, Р. G., A new approach for teaching and learning about engineering process failure risk analysis
with IE (industrial engineering) case studies, paper presented at the ASEE, American Society of Engi-
neering Educators, US National Meeting, Industrial Engineering Division, Nashville, TN, June 2003.
53. Ranky, P. G., A novel 3D Internet-based multimedia method for teaching and learning about engineering
management requirements analysis, paper presented at the ASEE, American Society of Engineering Ed-
ucators, US National Meeting, Engineering Management Education Division, Nashville, TN, June 2003.
54. Ranky, P. G., An interactive 3D multimedia problem-based library for manufacturing engineering tech-
nology education with Internet support, paper presented at the ASEE, American Society of Engineer-
ing Educators, US National Meeting, Engineering Technology Division, Nashville, TN, June 2003.
55. Ranky, P. G. (2003-2005), An introduction to alternative energy sources: Hybrid & fuel cell vehicles;
an interactive multimedia eBook publication with 3D objects, text, and videos in a browser-readable
format on CD ROM/intranet, available: http://www.cimwareukandusa.com, CIMware USA, Inc., and
CIMware Ltd.; United Kingdom, Multimedia design and programming by P. G. Ranky and M. F.
Ranky, (2003-2005), Customer needs, wants & requirements analysis: Automotive exterior rearview
mirror, an interactive multimedia eBook publication with 3D objects, text, and videos in a browser-
readable format on CD-ROM/intranet, available: http://www.cimwareukandusa.com, CIMware USA,
Inc., and CIMware Ltd., United Kingdom.
56. Ranky, P. G. (2003-2005), An introduction to digital factoiy & digital telematic car modeling with R
& D and industrial case studies, an interactive multimedia eBook publication with 3D objects, text,
and videos in a browser-readable format on CD-ROM/intranet, available: http://www.cimwareukan-
dusa.com, CIMware USA, Inc. and CIMware Ltd., United Kingdom, Multimedia design and pro-
gramming by P. G. Ranky and M. F. Ranky.
57. Ranky, P. G. (2005), An introduction to RFID, radio frequency identifi cation methods and applica-
tions, DVD video, available: www.cimwareukandusa.com (approximately 30 min).
58. Ranky, P. G. (2005-2006), An introduction to RFID, radio frequency identifi cation methods and ap-
plications with a total quality management and control focus, interactive browser-readable 3D eBook,
available: www.cimwareukandusa.com, (approximately 30 min).
59. Ranky, P. G. (1999, Apr.), An object oriented system analysis and design method (CIMpgr) and an R
& D case study, Adv. Des. Manufacturing, available: http://www.cimwareukandusa.com, listed and
indexed by the Association of Research Libraries, Washington DC, and the Edinburgh Engineering
Virtual Library, United Kingdom, Vol. 1. 60. Ranky, P. G., Computerized engineering assessment
method based on 3D interactive multimedia, That students enjoy, paper presented at the ASEE, Amer-
ican Society of Engineering Educators, US National Meeting, Continuing Professional Development
Division, Nashville, TN, June 2003. 61. Ranky, P. G. (1999), Design, manufacturing and assembly
automation trends and strategies in China, Assembly Automation, 19 (4), 301-305.
62. Ranky, P. G. (2003, Feb.), Designing a lean infrastructure; advanced machining cell design concepts,
methods, architectures and cases, Manuf. Eng., J. IEE, 22—24. 63. Ranky, P. G. (2000), Engineering
multimedia in CIM (computer integrated manufacturing) , Int. J. CIM, 13 (2), 169-171.
64. Ranky, P. G. (2003), eTransition in the multi-lifecycle CIM (computer integrated manufacturing) con-
text, Int. J. CIM, 16 (4-5), 229-234. 65. Ranky, P. G., Interactive 3D multimedia cases for engineering
education with Internet support, ASEE, American Society of Engineering Educators, paper presented
at the U.S. National Meeting, Computers in Education Division, Nashville, TN, June 2003.
66. Ranky, P. G., Interactive 3D multimedia cases for manufacturing engineering education with Inter-
net support, paper presented at the ASEE, American Society of Engineering Educators, US National
Meeting, Manufacturing Engineering Education Division, Nashville, TN, June 2003.
67. Ranky, P. G. (2002, Dec.), Introduction to concurrent engineering, an NSF (National Science Founda-
tion, USA) sponsored Gateway Coalition streamed multimedia narrated web presentation, New Jersey
Intitute of Technology, Public Research University, Newark, New Jersey.
248
Часть 3. Качество
68. Ranky, Р. G. (2003-2005), Key R & D and eTransition trends in US and international collaborative
design & manufacturing enterprises, an interactive multimedia eBook publication with 3D objects,
text, and videos in a browser-readable format on CD-ROM/intranet, available: http://www.cimwareu-
kandusa.com, CIMware USA, Inc., and CIMware Ltd., United Kingdom; Multimedia design and pro-
gramming by P. G. Ranky and M. F. Ranky. 69. Ranky, P. G. (2000, Jan.), Modular fi eldbus designs
and applications, Assembly Automation, 20 (1), 40-45.
70. Ranky, P. G. (2003), Network simulation models of lean manufacturing systems in digital factories
and an intranet server balancing algorithm, Int. J. CIM, 16 (4-5), 267-282.
71. Ranky, P. G., Rapid prototyping cases for integrated design and manufacturing engineering education
with 3D Internet support, paper presented at the ASEE, American Society of Engineering Educators,
US National Meeting, Design in Engineering Education Division, Nashville, TN, June 2003.
72. Roman, H. T, and Ranky, P. G. (2003-2005), A case-based Introduction to Service robotics, an inter-
active multimedia eBook publication with 3D objects, text, and videos in a browser-readable format
on CD-ROM/intranet, available: http://www.cimwareukandusa.com, CIMware USA, Inc., and CI-
Mware Ltd., United Kingdom. Multimedia design and programming by P. G. Ranky and M. F. Ranky.
73. Romero, C., Department of logistics passive RFID initial implementation, paper presented at the USA
Department of Defense Conference, RFID Media Briefi ng, Washington DC, Feb. 2005.
74. Sangoi, R., Smith, C. G., et al. (2004), Printing radio frequency identifi cation (RFID) tag antennas
using inks containing silver dispersions, J. Dispersion Sci. Technol. 25 (4), 513-521.
75. Smith, K., Enabling the supply chain, paper presented at the USA Department of Defense Conference,
RFID Media Briefi ng, Washington, DC, Feb. 2005.
76. Sugimoto, M., Kusunoki, F., Inagaki, S., Takatoki, K., and Yoshikawa, A. (2004), A system for sup-
porting collaborative learning with networked sensing boards, Syst. Comput. Jpn., 35 (9), 39-50.
77. Wilke, P., and Braunl, T. (2001), Flexible wireless communication network for mobile robot agents,
Ind. Robot, 28 (3), 220-233.
Глава 3.2. Значение систем обеспечения качества
и аудитов в фармацевтическом
производстве
Эван Б. Сигель и Джеймс М. Барквест
Ground Zero Pharmaceuticals, Inc. (Ирвин, Калифорния)
Согласно нормативно-правовым актам, практической деятельности и здравому
смыслу обеспечение качества представляет собой критическую функцию в фарма-
цевтическом производстве. Крайне важным является наличие независимого под-
разделения, осуществляющего аудит и оценку надлежащего выполнения стандарт-
ных операционных процедур, записей в досье на серию, процедур, одобренных
в регистрационных досье, а также надлежащего функционирования подразделения
по контролю качества. Эта деятельность помогает обеспечить надежность произ-
водства продуктов, соответствующих утвержденным спецификациям, и соблюде-
ние правил текущей производственной практики (cGMP, current good manufacturing
practice) в соответствии с действующими нормативными правовыми актами как
в целом на предприятии, так и для каждого производимого продукта.
Сотрудники подразделения по обеспечению качества должны пройти необхо-
димое обучение и тренинги, обладать требуемым опытом, знать производствен-
ный участок и производимые продукты, обладать официально утвержденной неза-
висимостью от управляющих производством структур и возможностью оценивать
степень выполнения процедур и правил и соответствие принятым подходам при
производстве качественных лекарственных средств. Это позволяет создавать про-
изводственную среду и производимый продукт, которые могут выдержать проверки
PDA (Федеральное управление США по контролю за пищевой продукцией и лекар-
ствами, Food and Drug Administration) и поддерживать репутацию предприятия, вы-
пускающего качественную продукцию.
Нормативные правовые акты по cGMP устанавливают требования, выполнение
которых должно обеспечить достижение высокого уровня уверенности в том, что
выпускаемый фармацевтический продукт будет удовлетворять требованиям к ко-
личественному содержанию, чистоте, активности и другим показателям качества,
установленным для готового продукта, обеспечивающим его пригодность для ис-
пользования. Производители должны организовать подразделение по контролю
качества, которое ответственно за выполнение целого ряда работ, связанных с каче-
ством и требующихся согласно нормативным правовым актам. Эти акты, по боль-
шому счету, не обновлялись с 1978 г.1 С тех пор научные и практические подходы по
обеспечению качества значительно расширились: для более надежного обеспечения
1 Авторы писали этот текст раньше принятия руководств ICH Q8 и Q10 регуляторными
агентствами США, ЕС и Японии, и соответственно, на сегодняшний день имеются очень
современные правила СЛ/Рдля фармацевтических субстанций, включающих, в частности,
управление рисками. Также предложена модель современной СМК, охватывающая практи-
чески весь жизненный цикл продукта. См. www.ich.org. — Примеч. перев.
250
Часть 3. Качество
качества продукта и его пригодности для использования стали применять системы
менеджмента качества и инструменты управления рисками. Постоянно растет ин-
терес производителей фармацевтической продукции к всесторонней системе ме-
неджмента качества (СМК/ QMS, quality management system) и методам управления
рисками, так как, по мнению производителей, более современные принципы ме-
неджмента качества позволят повысить эффективность работ по обеспечению ка-
чества и лучше координировать их с усиливающимися международными регулятор-
ными требованиями к системам качества. FDA не изменила акты по cGMP, однако
в своей программе «Фармацевтическая cGMP для инициативы XXI века» поощряет
использование систем качества для удовлетворения требованиям cGMP.
В данной главе приводятся и обсуждаются основные положения и правила, от-
носящиеся к деятельности по обеспечению качества и соответствующему струк-
турному подразделению, структура, функции, положение и использование этого
подразделения в фармацевтическом производстве. Кроме того, рассматриваются
дополнительные работы, которые могут появиться при переходе производителей на
концепцию систем качества, включающую действующие модели систем качества,
с целью дальнейшего улучшения качества и координации с международными тре-
бованиями к системам качества.
Также описываются обоснование и проведение аудитов обеспечения качества,
включая подготовку, ключевые элементы и типовой перечень вопросов для само-
го аудита, корректирующие и предупреждающие действия после проведения аудита
и предлагаемые меры по обеспечению успешного функционирования подразделе-
ния.
3.2.1. Требования cGMP
Нормативные правовые акты по cGMP для производства фармацевтических про-
дуктов содержатся в главах 210 и 211 тома 21 CFR (Свод федеральных нормативных
актов, Code of Federal Regulations) [1]. Эти нормативные акты, а также рекоменда-
тельные документы и другие документы FDA, относящиеся к регулированию и ин-
спекторским проверкам FDA производителей фармацевтической продукции, можно
найти на официальном сайте FDA по адресу www.fda.gov. В главе 210 указаны область
действия и применимость правил cGMP и приводятся определения терминов, ис-
пользованных в нормативных актах. Там же указано, что правила устанавливают
«минимальные» требования cGMP и что продукты, производящиеся не в условиях
cGMP, являются фальсифицированными. Фальсифицированные продукты и лица,
ответственные за фальсификацию продукции, подлежат юридическому преследо-
ванию со стороны FDA.
В главе 211 содержатся частные требования GMP к готовым лекарственным пре-
паратам. Эта глава поделена на подглавы А—К:
А. Область применения.
В. Организация и персонал.
С. Здания и помещения.
D. Оборудование.
Е. Контроль компонентов, первичной упаковки и укупорочных средств.
Е Производство и контроль процесса.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 251
G. Упаковка и итоговый контроль продукта.
Н. Хранение и поставки.
I. Контроль качества.
J. Документация.
К. Возвращенные некачественные лекарственные препараты и лекарственные
препараты с нарушением условий хранения.
Правила GMP посвящены первичным потенциальным причинам вариабель-
ности продукта. В подглаве В определена обязательность наличия на предприятии
подразделения по контролю качества и его обязанности, приведены требования
к персоналу и описаны правила поведения персонала (например, санитарные пра-
вила), предназначенные для снижения вероятности контаминации продукта. В под-
главах С и D определены требования к зданиям и производственным помещениям,
оборудованию, используемому при производстве, фасовке, упаковке и хранении ле-
карственных препаратов. В подглавах Е—Нуказаны требования к контролю основ-
ных технологических процессов производства и упаковки лекарственных средств,
подготовленных для отправки пользователям. Определена необходимость контроля
качества исходных материалов и компонентов, технологического процесса, упаков-
ки, маркировки, хранения и поставок готового, упакованного, промаркированного
и разрешенного к выпуску в обращение лекарственного препарата. Подчасть /тре-
бует установления научно обоснованных и приемлемых спецификаций, стандартов,
планов отбора образцов и процедур проведения испытаний; наличия спецификаций
на оборудование и проведение его калибровки; проведения испытаний партии или
серии продукта и разрешения серии к выпуску в обращение. В подразделе /установ-
лены требования к документации предприятия, включая основные записи и форми-
рование досье на серию. В подразделе ^рассматриваются контроль и уничтожение
возвращенных потребителями лекарственных препаратов и вводятся ограничения
на последующее использование лекарственных средств, которые хранились в не-
надлежащих условиях (например, при наличии дыма, жары, огня, влаги).
3.2.1.1. Обязанности подразделения по контролю качества в правилах cGMP
Правила cGMP определяют функции подразделения по контролю качества. Требует-
ся, чтобы подразделение имело обязанности и права по запрещению использования
в производстве (в смысле признания дефектными) любых компонентов, первичной
упаковки и укупорочных средств, полупродуктов, упаковочных материалов, этике-
ток и лекарственных препаратов, имело право проверять производственные записи,
чтобы обеспечить отсутствие отклонений или, при наличии отклонений, обеспечить
их всестороннее изучение. В обязанности подразделения должно входить одобрение
или запрещение лекарственных препаратов, произведенных, расфасованных, упа-
кованных или хранящихся субподрядными производителями. Организация должна
обеспечить наличие у подразделения по контролю качества адекватных лаборатор-
ных мощностей для проведения испытаний и одобрения или запрещения компо-
нентов, первичной упаковки для лекарственных препаратов, укупорочных средств,
упаковочных материалов, полупродуктов и лекарственных препаратов.
В дополнение к обязанностям, связанным с одобрением материалов и готовых
продуктов, подразделение также отвечает за одобрение или отклонение любых про-
252
Часть 3. Качество
цедур и спецификаций, влияющих на подлинность, количественное содержание,
качество и чистоту лекарственного препарата. В эти функции входит рассмотрение
и согласование процедур и инструкций по внутрипроизводственному контролю,
включая внесение любых изменений в эти документы. Эти процедуры, а также обя-
занности и процедуры, относящиеся к подразделению по контролю качества в рам-
ках организации, должны быть зафиксированы в письменном виде и выполняться.
Все спецификации, стандарты, планы по отбору, процедуры проведения испыта-
ний и другие механизмы лабораторного контроля, включая любые изменения в них,
должны быть изложены в письменном виде, проверяться и согласовываться подраз-
делением по контролю качества.
Должны быть установлены процедуры, описывающие обращение со всеми пись-
менными или устными жалобами в отношении лекарственных препаратов. Подраз-
деление по контролю качества несет ответственность за рассмотрение любой жа-
лобы, в том числе о возможном несоответствии лекарственного препарата любому
показателю в его спецификации, и для таких препаратов принимает решение о не-
обходимости проведения расследования на соответствие требованиям cGMP. При
рассмотрении нужно установить, является ли жалоба информацией о серьезной
и неожидаемой реакции на применение лекарственного препарата, о чем должно
быть извещено FDA. Письменная запись каждого обращения должна сохраняться
в папке с жалобами.
3.2.2. Деятельность по обеспечению качества
Термин «качество» используется во многих отраслях промышленности и в повсед-
невной жизни и может иметь различные значения в зависимости от контекста.
В этой главе под качеством мы будем понимать требования к продукту или параме-
тры, отражающие определенные требования к продукту. Работами по обеспечению
качества являются процессы и действия, проводимые для обеспечения постоянно-
го соответствия продукта или услуги установленным требованиям и обеспечения
пригодности продукта для использования. В фармацевтическом производстве это
означает работы, выполняемые для обеспечения соответствия фармацевтического
продукта требованиям подлинности, количественного определения, чистоты, ак-
тивности и другим показателям качества, перечисленным в утвержденной специ-
фикации.
В США требования к сСМРъ области производства лекарственных средств были
установлены нормативными правовыми актами в 1978 г. и существенно не меня-
лись с того времени. В то же время научные и практические аспекты обеспечения
качества значительно расширились вследствие включения в них разработок систем
качества [2, 3] и методов управления рисками [4] для лучшего обеспечения качества
продукта и его пригодности для использования. Постоянно растет интерес произ-
водителей фармацевтической продукции к внедрению этих подходов, так как они
позволяют производителям применять более современные принципы управления
качеством, что, по мнению производителей, повышает эффективность работ по
обеспечению качества и позволяет в большей степени соответствовать усиливаю-
щимся международным регуляторным требованиям к системам качества.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 253
3.2.3. Концепция систем качества
Концепция систем менеджмента применительно к качеству включает скоорди-
нированный подход к управлению деятельностью, связанной с качеством, в виде
управления процессами, которые взаимодействуют между собой, обеспечивая по-
лучение продукта, соответствующего установленным требованиям. В эту концеп-
цию входят:
• обязательства руководства в области качества, которые доводятся до сведения
всех сотрудников организации;
• определение требований к качеству с использованием управления рисками
и других пригодных для этого методов;
• разработка политики, планов и целей в области качества;
• установление организационной структуры с определенными обязанностями
и правами, позволяющими достичь поставленных целей в области качества;
• предоставление ресурсов, необходимых для достижения целей в области ка-
чества;
• разработка требуемых систем и процессов;
• установление методов для регулярной объективной оценки эффективности
систем и процессов, включая аудиты качества;
• инициирование корректирующих и предупреждающих действий, в случае не-
обходимости, для обеспечения постоянного и надежного достижения целей
в области качества.
Применение методов управления рисками для определения требований к про-
дукту, установление процессов и методов контроля и мониторинга, оценивание
данных по качеству, определение подходящих корректирующих и предупреждаю-
щих действий для решения проблем с качеством и другими связанными процессами
могут повысить общую эффективность и работоспособность СМК.
FDA признала ценность этих методов и поощряет применение СМК, исполь-
зующих управление рисками, в производстве фармацевтической продукции. Эта
позиция отражена в документе FDA «Фармацевтическая cGMP для инициативы
XXI века». Помимо этого FDA опубликовала отчеты и руководящие документы, со-
держащие информацию, которая может быть использована производителями ле-
карственных средств при внедрении СМК и управления рисками для выполнения
правил cGMP [5—8]. При введении этой инициативы FDA ясно определила, что эта
программа не устанавливает новые регуляторные требования к производителям.
Агентство предоставило информацию и руководства, предназначенные для созда-
ния связи между правилами от 1978 г. и современными СМК, и разъяснило, каким
образом производители, внедряющие эти системы, могут при этом полностью соот-
ветствовать правилам cGMP. Этот подход отличается от подхода, использованного
FDA при обновлении правил cGMPjxnn. медицинских изделий с применением прин-
ципов СМК. Постановление по СМК от 1996 г. изменило требования GMP к про-
изводителям готовых медицинских изделий с целью снижения риска неадекватной
проектировки изделий и согласования требований с международными стандартами
по СМК, действовавшими в то время [9]. Эти стандарты за прошедший период были
254
Часть 3. Качество
пересмотрены [10]; при этом нормативно-правовое регулирование по системам ка-
чества изделий по-прежнему согласуется с современными моделями СМК.
В современной системе административное подразделение, ответственное за ка-
чество в рамках организации, играет ключевую роль в разработке и управлении всей
системой. Эта деятельность включает контроль качества, обеспечение качества,
планирование в области качества и улучшение качества. Правила cGMPtie содержат
определений и не используют перечисленные термины, однако обязанности, уста-
новленные этими правилами для подразделения по контролю качества, попадают
в рамки этих определений [2, 8, И].
Современные модели СМК включают деятельность в области качества и исполь-
зуют термины, не указанные в правилах cGMP. Более того, качество развивается как
профессиональная дисциплина. Следовательно, организация, собирающаяся ис-
пользовать концепцию СМК, должна однозначно определить термины и концеп-
ции качества, которые будут использованы, и включить эти определения в обучаю-
щие программы для всех сотрудников организации, привлекаемых к выполнению
работ в области качества. Это поможет обеспечить эффективное взаимодействие
внутри организации, с поставщиками и другими сторонами (например, регулятор-
ными агентствами, аудиторами третьей стороны1, которые взаимодействуют с орга-
низацией по вопросам качества. Должны быть приняты определения, используемые
в нормативно-правовых актах, а использование нестандартных или устаревших тер-
минов по возможности следует исключить. Может быть полезным включение тер-
минов и определений со ссылками на соответствующие стандарты и руководящие
документы FDA. Руководство FDA по концепции СМК применительно к правилам
фармацевтических cGMP (руководство по фармацевтической системе качества) со-
держит следующие определения:
• обеспечение качества {quality assurance, QA) — упреждающие и ретроспектив-
ные действия, направленные на создание уверенности, что требования вы-
полнены;
• контроль качества {quality control, QC)2 — действия, выполняемые в процессе
производства продукта или оказания услуги, обеспечивающие соответствие
продукта и услуги требованиям и их воспроизводимость;
• менеджмент качества {quality management, QM) — ответственность за успеш-
ное внедрение системы качества;
• система (менеджмента) качества {quality system, QS) — формализованные
бизнес-правила, которые определяют ответственность руководства за орга-
низационную структуру, процессы, процедуры и ресурсы, необходимые для
выполнения требований к продукту и услуге, удовлетворения потребителей
и постоянного улучшения;
• подразделение по качеству {quality unit, QU) — группа, созданная в структуре
организации с целью продвижения качества в деятельности организации.
1 Аудиты, проводимые третьей стороной, осуществляются внешними независимыми ор-
ганизациями. — Примеч. перев.
2 Словарь ГОСТ Р ИСО 9000—2008 переводит термин quality control как управление каче-
ством. См. ГОСТ Р ИСО 9000—2008. — Примеч. перев.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 255
В своем руководящем документе по фармацевтической системе качества FDA
отмечает, что многие современные концепции СМК очень близки правилам cGMP
и что действия, требуемые правилами, в целом совместимы с концепцией СМК.
В этом и других руководящих документах для описания структурного подразделе-
ния, ответственного за деятельность по качеству, FDA использует термин «подразде-
ление по качеству», а не термин «подразделение по контролю качества», установлен-
ный в правилах cGMP. В современных моделях СМК эта деятельность по качеству
может быть шире требований, установленных правилами cGMP, но необязательно
с ними несогласованной.
Использование термина «подразделение по качеству» согласуется с современны-
ми моделями СМК [2, 10], которые предназначены для обеспечения уверенности
в том, что различные действия, связанные со всеми системами, планируются, одо-
бряются, выполняются и мониторируются надлежащим образом, а правила cGMP
специально наделяют подразделение по качеству правами по созданию, монито-
рингу и внедрению системы качества. FDA предупреждает, что такая деятельность
не замещает и не исключает обычную ответственность производственного персо-
нала по встраиванию качества в продукт. FDA специально указывает, что избыточ-
ные принципы, формулируемые в правилах cGMP и в устойчивых к ошибкам со-
временных системах качества, заключаются в необходимости встраивания качества
в продукт и что уверенность в качестве продукта не может базироваться только на
проведении испытаний.
Другие обязанности подразделения по качеству, установленные в правилах
cGMP, согласуются с концепциями СМК и включают:
• обеспечение уверенности в том, что контрольные параметры установлены
и успешно выдерживаются в ходе технологических операций;
• обеспечение уверенности в том, что разработанные процедуры и специфи-
кации являются адекватными и выполняются, в том числе документы, ис-
пользуемые компанией, для которой производитель выполняет работу по
контракту;
• одобрение или запрещение использования входящих материалов, полупро-
дуктов и лекарственных препаратов;
• рассмотрение производственных записей и расследование любых незаплани-
рованных отклонений.
FDA обращает внимание, что выпуск руководящего документа по фармацевти-
ческой системе качества не является введением новых регуляторных требований
к производителям, но поощряет их принимать концепцию СМК с целью соответ-
ствия правилам cGMP для получения потенциальных преимуществ. Разработанная
надлежащим образом и внедренная СМК позволяет:
• сократить количество отзывов продукции (или вообще предотвратить их); со-
кратить объемы возвращенной некачественной продукции и пришедшей в не-
годность продукции; уменьшить попадание дефектной продукции на рынок;
• привести в соответствие, насколько это возможно, правила cGMP с другими
широко использующимися СМК, что желательно, учитывая глобализацию
фармацевтического производства и увеличивающееся количество комбини-
256
Часть 3. Качество
рованных продуктов, содержащих синтетический или биологический лекар-
ственный препарат и медицинское изделие;
• управлять изменениями помещений, оборудования и процессов без пред-
варительной подачи в регуляторное агентство заявлений на изменения при
условии совмещения СМК с системой управления знаниями о производ-
ственных процессах и продуктах и при эффективном применении инстру-
ментов управления рисками;
• потенциально сократить количество и длительность инспекторских проверок
FDA благодаря снижению риска производственных проблем;
• создать необходимую основу для внедрения концепции «качество, заложен-
ное при проектировании» (QbD, quality by design — качество, заложенное
в продукт на стадии фармацевтической разработки и поддерживаемое в тече-
ние жизненного цикла продукта) и для постоянного улучшения и управления
рисками в процессе производства лекарственных средств.
Можно предположить, что даже не внося изменения в правила cGMP, FDA может
рассматривать их с учетом «новой» перспективы систем качества. Правила содержат
термины «адекватный» и «соответствующий», которые могут интерпретироваться как
«удовлетворяющий современным техническим и научным возможностям и научно-
техническому прогрессу». По мере развития науки и техники понимание того, что
является адекватным или соответствующим, может также меняться. На практике
большинство производителей стремятся внедрить методы, которые могут улучшить
качество и безопасность фармацевтических продуктов, поскольку в долгосрочной
перспективе это является экономически выгодным [11], но при этом могут сомне-
ваться, боясь получить заключение инспектора о несоответствии внедряемых методов
правилам cGMP. Предпринимаемые в настоящее время усилия FDA в этом направле-
нии должны способствовать ослаблению сомнений производителей по этому поводу.
Основные элементы модели системы качества, описанной в руководящем до-
кументе по фармацевтической системе качества, полностью согласуются с суще-
ствующими стандартами СМК. Они включают:
• ответственность руководства;
• ресурсы;
• производственные операции;
• оценку деятельности.
3.2.4. Ответственность руководства
Современные модели систем качества отьсдят руководству ведущую роль в развер-
тывании и функционировании успешной системы качества. В таких системах от-
ветственность высшего руководства включает:
• демонстрацию лидерства путем установления своих обязательств в области
качества, что поддерживается на всех уровнях организации и доводится до
сведения всех сотрудников;
• создание организационной структуры, в которой четко определены обязан-
ности и права по выполнению функций в области качества, связанных с до-
стижением поставленных целей;
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 257
• построение и документирование системы качества, соответствующей опреде-
ленным регуляторным требованиям, в том числе в области качества, и позво-
ляющей достичь поставленных целей в области качества;
• установление политики и целей в области качества, а также планов по каче-
ству, согласованных со стратегическими планами организации, которые до-
водятся до сведения сотрудников организации;
• анализ системы путем установления соответствующих форм отчетности в рам-
ках организации для мониторинга и предоставления руководству данных по
качеству и состоянию системы, а также обеспечение выполнения и докумен-
тирования надлежащих корректирующих и предупреждающих действий при
появлении проблем с качеством с помощью эффективных процедур контроля
изменений.
Правила cGMP отдельно не определяют ответственность руководства по данным
действиям; вместе с тем действия как таковые законодательством требуются. Эта
взаимосвязь показана в табл. 1 из руководящего документа по фармацевтической
системе качества.
Таблица 1
Правила cGMP, том 21 CFR, в отношении ответственности руководства
Элемент системы качества Ссылка на пункт правил
1. Лидерство 2. Структура Установление функций по качеству: § 211.22(a); также см. определение в § 210.3(Ь)(15). Уведомление: § 211.180(f)
3. Построение СМК Процедуры подразделения по качеству: § 211.22(d). Процедуры и спецификации подразделения по качеству: § 211.22(c), с усилением в § 211.100(a), 211.160(a). Контрольные действия подразделения по качеству: § 211.22(a), с усилением в § 211.42(c), 211.84(a), 211.87,211.101(c)(1), 211.110(c), 211.115(b), 211.142,211.165(d), 211.192. Обеспечение качества, проверки и расследования: § 211.22(a), 211.100(а—Ь), 211.180(f), 211.192, 211.198(a). Контроль записей: § 211.180(а—d), 211.180(c), 211.180(d), 211.180(e), 211.186,211.192,211.194, 211.198(b)
4. Установление политики, целей и планов Процедуры: § 211.22(с—d), 211.100(a)
5. Анализ системы Анализ записей: § 211.100,211.180(e), 211.192, 211.198(b)(2)
258
Часть 3. Качество
После перехода к всесторонней системе качества подразделение по качеству
будет выполнять более широкую и более значимую роль в рамках организации
с большей ответственностью и взаимодействием с высшим руководством. Подраз-
деление по качеству в идеале должно быть независимым от других структурных под-
разделений организации, чтобы обеспечить четкое разграничение ответственности
и полномочий и предупредить возможные конфликты. В определенных случаях,
например при аудите, независимость и объективность являются критичными для
эффективности всего процесса аудита, при этом аудиторы не должны нести прямую
ответственность за проверяемый участок.
Правила cGMP не указывают, каким образом подразделение по качеству должно
быть включено в общую структуру организации, но, как правило, структура этого
подразделения должна отражать серьезность обязательств руководства в области
качества и способствовать достижению целей в области качества. Структура (т. е.
организационные взаимосвязи с другими структурными подразделениями и по-
дотчетность) должна отражать четкие границы прав и обязанностей, которые под-
держивают деятельность по производству, качеству и менеджменту, необходимую
для достижения целей в области качества. В различных организациях это достига-
ется различными способами; однако опыт показывает, что помещение функции по
качеству на одинаковый уровень в административной иерархии с другими основ-
ными подразделениями (например, с производством) обеспечивает внутри и вне
организации четкое понимание серьезности обязательств руководства в области
качества.
Правила cGMP требуют выполнения действий, связанных с качеством, на всех
этапах производственного процесса: от приемки исходных материалов до выпуска
серии в обращение, упаковки и маркировки. Правила также требуют, чтобы все со-
трудники, в том числе задействованные в деятельности по обеспечению качества,
обладали достаточным образованием, навыками и опытом или их комбинацией,
позволяющими выполнять их служебные обязанности. При применении концеп-
ции систем качества с учетом соответствия cGMP функции персонала подразде-
ления по качеству могут существенно расшириться, включив в себя проведение
внутренних аудитов по качеству, расширенное рассмотрение и анализ данных по
качеству, расследование несоответствий, анализ основных причин несоответствий
и отказов, анализ рисков и другие действия, связанные с качеством. Большинство
этих работ, вероятно, будет выполняться сотрудниками других структурных под-
разделений, таких как производственные подразделения, контроль материалов,
инженерно-технические службы и подразделения по разработке продукции. Со-
трудники подразделения по качеству должны обладать значительными научными
и техническими знаниями и навыками (например, владеть статистическими мето-
дами и анализом рисков), знанием продукта и технологического процесса, чтобы
выполнять отведенные им функции и при необходимости компетентно взаимодей-
ствовать с сотрудниками других структурных подразделений.
3.2.5. Ресурсы
Грамотное распределение ресурсов крайне важно для любого предприятия, и это
особенно критично для фармацевтической отрасли. Неадекватные численность
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 259
персонала, обучение, производственное оборудование и помещения, контроль
окружающей среды, аналитическое оборудование и другие ресурсы могут быть ис-
точниками изменений, приводящих к производству продукта, не соответствующего
требованиям и спецификации. Современные стандарты по системам качества спе-
циально обращают внимание на вопрос ресурсов, устанавливая обязанность орга-
низации определить и выделить персонал, инфраструктуру и рабочее пространство
для функционирования системы качества. Правила cGMP в отношении ресурсов
включают требования по обеспечению пригодности персонала (включая консуль-
тантов), производственных мощностей (включая контрактные производственные
площадки), оборудования и лабораторных мощностей. В этом смысле на подраз-
делении по качеству лежит значительная ответственность.
FDA в своем руководстве по фармацевтической системе качества обсужда-
ет необходимость наличия адекватных ресурсов при разработке, внедрении
и управлении системой качества, что соответствует правилам cGMP. Руководство
организации несет ответственность за определение потребностей в ресурсах и со-
ответственно за их предоставление, включая предоставление обучения, отвечаю-
щего назначенной области деятельности. Сотрудники должны понимать влияние
своих действий на поставленные цели и знать требования правил cGMP и систему
качества организации. Это совпадает с общепринятой идеей о том, что культура
качества в рамках организации обязывает сотрудников понимать концепции ка-
чества, цели организации в области качества и государственного регулирования,
вклад их повседневной работы в достижение этих целей и ее место в общей системе
качества. Руководство должно создать рабочее пространство, способствующее ре-
шению проблем и улучшающее коммуникации для выявления и устранения про-
блем качества. Если предоставление ресурсов, как правило, является обязанностью
руководства, то подразделение по качеству и другие структурные подразделения
должны привлекаться крещению задач по идентификации ресурсов, необходимых
для достижения целей в области качества, включая соответствие регуляторным
требованиям, оценку адекватности имеющихся ресурсов, изучение влияния изме-
нений персонала, производственных мощностей, продукта, процессов, регулятор-
ных требования и других факторов на потребности в ресурсах, и обычно должны
представлять руководству информацию, необходимую для принятия правильных
решений по ресурсам.
Для разработки адекватных требований к системам качества современные моде-
ли таких систем используют подходы, основанные на управлении рисками и управ-
лении данными. FDA отмечает, что правила cGMP обращают больше внимания на
технологическое оборудование как на испытательное и содержат специальные тре-
бования по квалификации, калибровке, очистке и техническому обслуживанию
производственного оборудования, что является более жестким подходом по срав-
нению с требованиями к моделям нефармацевтических систем качества. Предпри-
ятия всегда должны учитывать, что хотя FDA поощряет внедрение универсальных
систем качества, любая разработанная система должна отвечать требованиям пра-
вил cGMP.
В рамках модели системы качества разработка требований и спецификаций на
помещения и оборудование может осуществляться техническими экспертами (на-
260
Часть 3. Качество
пример, инженерами, научно-техническими специалистами), обладающими зна-
ниями фармацевтики, производственных процессов и факторов риска, связанных
с продуктом и его производством. Правила cGMP требуют, чтобы подразделению
по качеству вменялось в обязанности рецензирование и согласование всех базо-
вых критериев дизайна, процедур, относящихся к производственным помещениям
и оборудованию, и всех последующий изменений. Эти требования не являются вза-
имоисключающими: хотя правила устанавливают окончательную ответственность
по согласованию и утверждению этих работ за подразделением по качеству, в них не
запрещается проведение согласований с разработчиками и квалифицированными
сотрудниками из различных подразделений организации. И правилами cGMP, и в
современных моделях систем качества установлено, что рецензирование и согласо-
вание должно осуществляться сотрудниками, обладающими соответствующей ква-
лификацией на основании образования, тренингов и опыта работы.
При контроле работ по контракту правила cGMP требуют, чтобы подразделение
по качеству одобряло или отклоняло продукты или услуги, выполняемые по кон-
тракту. В рамках современных моделей систем качества организация должна осу-
ществлять формализованный процесс по квалификации поставщиков услуг или
продуктов по контракту и подтверждать путем инспекции или другими подходящи-
ми способами, что поставщик способен выполнить требования организации. Для
выполнения установленных регуляторных требований эти действия должны произ-
водиться подразделением по качеству.
В табл. 2 сопоставлены основные элементы концепции систем качества в отно-
шении ресурсов с соответствующими требованиями правил cGMP.
Таблица 2
Правила cGMP, том 21 CFR, в отношении ресурсов
Элемент системы качества Ссылка на пункт правил
1. Общие положения 2. Управление персоналом Квалификация: § 211.25(a). Количество сотрудников: § 211.25(c). Обучение сотрудников: § 211.25(а—Ь)
3. Помещения и оборудование Здания и помещения: § 211.22(b), 211.28(c), 211.42-211.58, 211.173. Оборудование: § 211.63—211.72,211.105, 211.160(b)(4), 211.182. Лабораторные мощности: § 211.22(b)
4. Контроль операции, производимых по контракту Консультанты: § 211.34. Работы по контракту (аутсорсинг): § 211.22(a)
3.2.6. Производственные операции
Имеется значительное сходство требований к производственным операциям, уста-
новленных в современных моделях систем качества, таких как ИСО 9001—2000,
и правил cGMP. Управление FDA определило четыре основных элемента систем ка-
чества в отношении производственных операций. Они приведены в табл. 3 в срав-
нении с требованиями cGMP.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 261
Таблица 3
Правила cGMP, том 21 CFR, в отношении производственных операций
Элемент системы качества Ссылка на пункт правил
1. Проектирование и разработка продукта и процессов 2. Изучение входов (в процессы) Производство: § 211.100(a) Материалы: § 210.3(b), 211.80-211.94,211.101, 211.122,211.125
3. Выполнение и мониторинг операций Производство: § 211.100,211.103, 211.110, 211.111,211.113. Критерии контроля качества: § 211.22(а—с), 211.115(b), 211.160(a), 211.165(d), 211.188. Контрольные точки для контроля качества: § 211.22(a), 211.84(a), 211.87,211.110(c)
4. Работа с несоответствиями Расследование отклонений: § 211.22(a), 211.100,211.115, 211.192, 211.198. Отзывы с рынка: том 21 CFR, глава 7
З.2.6.1. Проектирование, разработка и документирование процесса
и продукта
В производственной среде современных систем качества критические параметры
планируемого к производству продукта должны быть определены и подтвержде-
ны как соответствующие требованиям от стадии проектирования до поставки, при
этом должен осуществляться контроль всех изменений. Это совпадает с требовани-
ями правил cGMP, которые устанавливают, что все производственные и связанные
с качеством операции и процедуры, а также изменения в них должны быть уста-
новлены, одобрены и проконтролированы. Концепция контроля проектирования
продукта и процесса согласуется с идеями, включенными в документ FDA «Фар-
мацевтическая cGMP для инициативы XXI века», о том, что для обеспечения без-
опасности продукта необходимо охватить весь его жизненный цикл. Никакое ко-
личество последующих проверок и испытаний не сможет компенсировать ошибки
при проектировании, результатом которых становится неспособность продукта или
производственного процесса выдерживать требования, необходимые для обеспече-
ния безопасности и эффективности продукта для предназначенного применения.
Требуемая документация может включать:
• использованные ресурсы и мощности;
• процедуры для выполнения процесса;
• идентификацию владельца процесса, который будет под держивать и обнов-
лять процесс по мере необходимости;
• идентификацию и контроль важных переменных параметров;
• меры по контролю качества, сбор необходимых данных, мониторинг и кон-
троль продукта и процесса;
• воздействия на связанные процессы, функции или персонал.
Правила cGMP включают особый контроль процессов упаковки (расфасовки)
и маркировки, поэтому требования к этим процессам, их описание и способы про-
262
Часть 3. Качество
верки должны быть включены в установленный в системе качества порядок проек-
тирования и разработки продукта и процесса.
Производители и FDA выражали опасения, что существующие регуляторные тре-
бования (например, необходимость прохождения регуляторного процесса подачи
документов для внесения изменений в производственный процесс) могут оказаться
недостаточно гибкими и не способствующими внедрению инноваций, вне зависи-
мости от возможной пользы. FDA признала, что нежелание заниматься потенциаль-
но инновационными изменениями в фармацевтическом производстве может быть
нецелесообразным с позиций общественного здоровья, и опубликовала рекоменда-
ции по технологии анализа процессов (А471*), которые предназначены для решения
данного вопроса путем поощрения использования аналитических инструментов для
получения знаний о производственном процессе и удовлетворения регуляторным
требованиям к валидации и контролю производственных процессов [7].
Руководство по РАТ описывает добровольно принимаемый производителями
подход к проектированию, анализу и контролю производственных процессов, в ко-
тором с целью обеспечения качества готового продукта в режиме реального времени
(т. е. во время процесса) проводится измерение критических показателей качества
и функциональных параметров исходных материалов, полупродуктов и процессов.
Термин анализ в названии РАТ имеет широкое значение, включающее интегриро-
ванное применение химического, физического, микробиологического, математи-
ческого анализа и анализа рисков при необходимости. Одной из целей РА/является
проектирование и разработка хорошо изученных процессов, которые будут посто-
янно обеспечивать установленное качество на выходе процесса. Это соответствует
целям системы качества. В идеале РА /должна инициироваться на стадии разработ-
ки, при этом предполагается ее встраивание в существующие регуляторные процес-
сы при постоянном общении с FDA, что имеет ключевое значение. FDA опублико-
вала этот рекомендательный документ и другую относящуюся к РАТ информацию
на своем сайте www.fda.gov. Компании, заинтересованные в методах РАТ, должны
связаться с FDA. По мнению FDA внедрение РАТ включает следующее:
• использование группы РА Тпри рассмотрении данных раздела регистрацион-
ного досье по химическому и производственному контролю и при инспекци-
ях на соответствие правилам cGMP’,
• совместное обучение РАТ и сертификация персонала FDA, занятого в экспер-
тизах досье, инспекциях и оценке соответствия;
• научная и техническая поддержка РА Тдля персонала, занятого в экспертизах
досье, инспекциях и оценке соответствия.
Технология анализа процесса совпадает с принципами систем качества в том,
что она основана на научно-технических принципах оценки и снижения рисков,
связанных с низким качеством продукта и процесса. В руководстве по РАТ управле-
ние FDA указывает, что желаемое состояние фармацевтического производства мо-
жет быть описано следующим образом:
1 Термин PAT (process analytical technology) произошел от термина process analysis, т. е. «ана-
лиз процесса», поэтому мы перевели этот термин на русский язык, как «технологию анализа
процесса». Более подробно РАТ рассматривается в главе 4. — Примеч. перев.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 263
• Качество и эффективность1 продукта обеспечиваются путем разработки эф-
фективного и результативного производственного процесса.
• Спецификации на продукт и процесс основаны на механистическом пони-
мании влияния состава продукта и параметров процесса на эффективность
продукта.
• Постоянное обеспечение качества в режиме реального времени.
• Выполнение регуляторных требований и процедур обеспечивает применение
наиболее современного уровня научных знаний.
• Регуляторные подходы на основе управления рисками учитывают:
— уровень научного понимания влияния состава препарата и параметров
процесса на эффективность продукта;
— способность стратегий контроля процесса предупреждать или снижать
риск получения продукта несоответствующего качества.
Технология анализа процессов согласована с подходом по обеспечению соответ-
ствия правилам cGMP в современных системах качества, основанных на управлении
рисками и управлении данными.
3.2.6.2. Входы
Современные модели СМК используют процесс-ориентированный подход к про-
ектированию и управлению СМК, то есть рассматривают СМК как систему взаи-
мосвязанных процессов, в каждом из которых есть входы и выходы и которые спро-
ектированы для функционирования определенным образом. Выходы некоторых
процессов являются входами для других процессов. Эта концепция легко приме-
нима и понятна производителям, поскольку они также процесс-ориентированы.
К входам в производственные процессы относятся все материалы, которые при-
сутствуют в готовом продукте, включая материалы, покупаемые у поставщиков,
и полупродукты. Производственные операции, как правило, включают множество
процессов, выполняемых установленным способом для получения готового продук-
та. Каждый процесс имеет ряд входов и один или более выходов, которые, в свою
очередь, могут быть входами для следующего процесса. Спецификации на входы
вводят для соответствия готового продукта установленным требованиям. Надежная
система качества будет гарантировать, что все входы производственных процессов
пригодны для использования с помощью установления контроля качества на всех
1 За рубежом широко распространено понятие «работоспособности, действенности» пре-
парата (performance). При одновременном использовании этого термина вместе с термином
«качество» (не упоминая требования) имеют в виду химическое, микробиологическое и био-
логическое качество: содержание действующего вещества, чистоту и подлинность, пироген-
ность. Параметры спецификации, характеризующие лекарственную форму, также называют
функциональными или технологическими параметрами (например, распадаемость, раство-
римость и т. д.). В теории существует ситуация, когда включенные в спецификацию показа-
тели не могут гарантировать получение требуемого действия препарата в организме челове-
ка. Поэтому мы использовали термин «эффективность» в смысле оказания эффекта, а не в
смысле «терапевтической эффективности» (т. е. показания к применению). — Примеч. перев.
264
Часть 3. Качество
входах при наличии квалифицированных поставщиков, имеющих систему обеспе-
чения качества производства, хранения и использования продукции.
Для разрешения использования материалов в производстве правила cGMP тре-
буют или проведения испытаний, или использования сертификатов (протоколов)
анализа, а также дополнительно испытания на подлинность. Кроме того, модель
системы качества устанавливает проведение первичной квалификации постав-
щика, основанной на объективной оценке, и периодический аудит поставщиков,
основанный на оценке рисков, с целью подтверждения адекватности систем каче-
ства поставщика. При аудите производитель может наблюдать за проведением ис-
пытаний или исследований, проводимых поставщиком, чтобы помочь определить
надежность сертификатов анализа поставщика. В модели СМК подразделение по
качеству, как правило, несет ответственность за аудит поставщиков в рамках общей
ответственности за качество материалов.
Контроль изменений включает методическую оценку предлагаемых изменений,
чтобы определить их влияние на выходы процесса и, в конечном итоге, на готовый
продукт, и является важным элементов современных моделей систем качества. Пра-
вила cGMP содержат требования об одобрении спецификации со стороны подраз-
деления по качеству и рецензировании и одобрении им определенных изменений.
В рамках модели системы качества изменения в материалы (например, уточнение
спецификации, смена поставщика или изменения обработки материала) должны
вноситься через формализованную систему контроля изменений, включающую до-
кументированное компетентное рецензирование и одобрение предлагаемого измене-
ния до его введения в действие, а также распространение информации об изменениях
в организации в требуемом объеме. Производители должны продумать, как наилуч-
шим образом обеспечить выявление и оценку изменений, которые поставщики мог-
ли внести в свои материалы и которые могут влиять на качество готового продукта.
Такие условия должны включаться в договора с поставщиками, если это возможно.
3.2.6.3. Выполнение и мониторинг операций
Правила cGMP и модели систем качества содержат требования по мониторингу кри-
тических процессов, которые могут вызывать вариабельность в ходе производства.
Правила cGMP требуют наличия письменных процедур по производственным опе-
рациям и их контролю и определяют виды работ по контролю процессов, которые
должны выполняться и документироваться. Современные модели систем качества
также требуют наличия письменных процедур, валидации и верификации процесса,
установления соответствующих мер и документации по контролю процесса. Мето-
ды анализа рисков и данные по проектированию и разработке могут быть использо-
ваны для установления требований к контролю и мониторингу процессов. Концеп-
ция систем качества позволяет производителю более эффективно и результативно
валидировать, выполнять и мониторировать производственные операции и обе-
спечивать научную обоснованность и пригодность контрольных процедур. Произ-
водственный процесс и его контроль должны быть спроектированы таким образом,
чтобы обеспечить соответствие готовых продуктов заявленным подлинности, до-
зировке, качеству и чистоте. При системном подходе будут рассмотрены все источ-
ники изменчивости от входов, производственного процесса, упаковки, маркировки
Глава 3.2, Значение систем обеспечения качества и аудитов в фармацевтическом производстве 265
и транспортировки, чтобы обеспечить соответствие требованиям к качеству про-
дукта, поставляемого потребителю.
Одним из важных аспектов системного подхода является постоянный сбор
и анализ данных по качеству, проводимый для постоянной оценки эффективности
системы качества. Архивные данные, знания о процессе и методы анализа рисков
могут использоваться для идентификации потребности в определенных данных.
Метод анализа основной тенденции (анализ трендов) и другие методы анализа
данных могут помочь при выявлении реальных и потенциальных источников не-
соответствий, чтобы можно было предпринять соответствующие корректирующие
и предупреждающие действия в соответствии с установленными процедурами кон-
троля изменений.
В системе качества должен рассматриваться весь жизненный цикл продукта пу-
тем установления механизмов мониторинга и постоянного улучшения. Даже хоро-
шо изученные или старые производственные процессы могут «дрейфовать» вслед-
ствие массы факторов, включая старение оборудования и помещений, изменения
или вариабельность исходных материалов, перепады в энергосистемах и изменения
окружающей среды. Таким образом, валидация процессов является не однократным
событием, а деятельностью, которая регулярно осуществляется на протяжении жиз-
ни продукта. Одной из основных целей систем качества должна быть идентифика-
ция возникающих проблем с качеством до возникновения несоответствий. Анализ
периодически собираемых данных по состоянию окружающей среды может, напри-
мер, выявить медленное, но устойчивое увеличение содержания в воздухе частиц,
которое, если не вмешаться, при сохранении данной тенденции могло бы привести
к превышению внутренних стандартов компании по окружающей среде и негативно
сказаться на продукте. Раннее выявление таких проблем позволяет инициировать
расследование для идентификации причины, чтобы выполнить соответствующие
корректирующие и предупреждающие действия в соответствии с установленными
процедурами контроля изменений. После выявления изменения его эффективность
должна быть объективно подтверждена, а затронутые процессы должны подвер-
гнуться повторной валидации, если это окажется необходимым.
3.2.6.4. Рассмотрение несоответствий
Ключевой компонент в любой системе качества — это правильная реакция на несо-
ответствия (т. е. отклонения от требований, установленных в системе качества для
полупродуктов или показателей качества готового продукта, контрольных параме-
тров процесса, записей, процедур и др.). Несоответствия могут быть обнаружены на
любой стадии производственного процесса или в ходе выполнения работ по кон-
тролю качества. Правила cGMP требуют проведения расследования и документи-
рования самого расследования, сделанных заключений и последующих действий.
Основной целью любой системы качества на производстве является предотвраще-
ние изготовления несоответствующего продукта и его поставки. Полная реакция на
несоответствия должна быть основана на управлении рисками и может включать
следующие компоненты:
• оценку возможного влияния несоответствия на качество готового продукта
(т. е. установление, привело ли несоответствие или могло привести к тому,
266
Часть 3. Качество
что продукт не соответствует установленным требованиям к чистоте, количе-
ственному содержанию и другим параметрам качества);
• определение любых действий, необходимых для гарантии того, что продукт,
не отвечающий требованиям, не производится, и что приняты соответству-
ющие меры по отношению к любому произведенному несоответствующему
продукту, обеспечивающие отсутствие ущерба для потребителей и выполне-
ние регуляторных требований;
• определение причины несоответствия;
• идентификацию действий, необходимых для устранения причины и преду-
преждения повторных появлений несоответствий;
• документирование расследования, обнаруженных фактов и последующих
действий;
• оценку эффективности последующих действий;
• повторение цикла при необходимости.
Несоответствие может не приводить к возникновению брака готового продук-
та; однако расследование несоответствия может выявить процесс или недостатки
системы качества, нуждающиеся в особом внимании. Например, небольшое, но
неожиданное отклонение от требований к контролируемому параметру процесса
(например, температуре или времени смешения) может не превышать допустимые
пределы, на которые изначально процесс был отвалидирован, и следовательно, от
данного отклонения не ожидают негативного воздействия на готовый продукт, од-
нако это несоответствие может стать свидетельством формирующейся проблемы
в контроле процесса или оборудовании, которая, если не будет исправлена, может
в дальнейшем привести к возникновению несоответствий продукта. Аналогично,
несоответствия в виде ошибок или пробелов в рабочих записях или отклонения от
письменных процедур не всегда могут привести к несоответствию продукта, но мо-
гут свидетельствовать о проблемах в обучении персонала, проектировании процес-
са или других проблемах, на которые следует обратить внимание. Таким образом,
ответ на несоответствия должен не только ограничиваться определением прямого
воздействия на готовый продукт, но включать также рассмотрение его последствий
для общей эффективности системы качества.
3.2.7. Оценка деятельности
Оценочный компонент СМК предназначен для предоставления объективной ин-
формации и данных, которые позволяют организации оценить соответствие про-
дукта установленным требованиям, провести анализ эффективности своей системы
качества, поддерживать и улучшать ее эффективность [10]. Правила cGMP также
требуют проведения оценки деятельности, как показано в табл. 4.
З.2.7.1. Анализ основной тенденции (тренда)
Правила cGMP устанавливают, что обзор и анализ определенных данных по каче-
ству должны выполняться по крайней мере 1 раз в год. Современные модели систем
качества делают упор на применении механизма принятия решений на основании
анализа данных с помощью подходящих методов статистического анализа [2, 11].
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 267
Правила cGMP, том 21 1 CFR, в отношении оценки деятельности Таблица 4
Элемент системы качества Ссылка на пункт правил
Анализ данных на наличие тренда Проведение внутренних аудитов Оценка рисков Корректирующие действия Предупреждающие действия Стимулирование улучшений Годовой отчет: § 211.180(e) Расследование отклонений: § 211.22(a), 211.192 §211.100
Анализ основной тенденции (тренда) — это статистический инструмент, особо
рекомендованный FDA в руководстве по фармацевтической системе качества, ко-
торый может быть очень полезным при мониторинге процессов и эффективности
системы качества для выявления формирующихся проблем и проведения оценки
эффективности действий по улучшению. Традиционный статистический контроль
процесса и другие методы также представляют определенную ценность для прове-
дения объективного и текущего анализа данных по качеству и могут помочь при
внедрении практики обеспечения качества в режиме реального времени, как реко-
мендуется FDA [7].
3.2.7.2. Проведение внутренних аудитов
Правила cGMP1 не требуют специального проведения внутренних аудитов, однако
производители традиционно используют внутренние аудиты в качестве инструмента
самооценки и для подготовки к инспекциям FDA. Уже в течение некоторого време-
ни FDA признает полезность внутренних аудитов и поощряет компании проводить
аудиты, обращая внимание компаний на то, что не изучает результаты внутреннего
аудита при проведении инспекций [12].
Современные модели систем качества оговаривают обязательность проведения
аудитов через запланированные временные интервалы с целью оценки эффектив-
ности внедрения и поддержания системы качества и проверки соответствия про-
цессов и продуктов установленным параметрам и спецификациям. Международные
стандарты содержат рекомендации по проведению аудитов [13]. Процедуры аудитов
должны быть разработаны и документированы, чтобы обеспечить при разработке
графика планируемых аудитов учет относительных рисков различных работ в си-
стеме качества. К факторам, которые могут учитываться при планировании частоты
и области аудита с применением управления рисками, относятся [6]:
• существующие обязательные требования (например, правила cGMP)',
• общее состояние дел по достижению соответствия требованиям и архивные
данные компании или площадки;
• надежность деятельности компании по управлению рисками качества;
• сложность производственной площадки;
• сложность производственного процесса;
1 Авторы обсуждают требования cGMP, установленные в США. — Примеч. перев.
268
Часть 3. Качество
• сложность продукта и его терапевтическая значимость;
• количество и уровень тяжести дефектов по качеству (например, отзывов про-
дукции);
• результаты предыдущих аудитов и инспекций, к которым относятся резуль-
таты предыдущих внутренних аудитов и результаты регуляторных инспекций
(например, инспекция штата, федеральная или другого регуляторного агент-
ства) и аудитов третьей стороны;
• значительные изменения зданий, оборудования, процессов и ключевого пер-
сонала;
• производственный опыт по продукту (например, частота, объем серии, коли-
чество серий);
• результаты испытаний в государственных контрольных лабораториях.
В принципе, аудиторы не должны нести прямую ответственность за вопросы,
по которым проводится аудит. Аудиторы должны пройти обучение методам аудита
и обладать достаточными техническими знаниями, чтобы провести оценку прове-
ряемых систем с использованием объективных критериев аудита [14]. Критерии ау-
дита могут быть основаны на применимых регуляторных требованиях, стандартах,
которым должна соответствовать система качества (например, ИСО 9001—2000),
и специальных требованиях к проверяемой системе качества, указанных в докумен-
тах по ней. Критерии аудита должны быть установлены до начала проверки.
В зависимости от целей проверки и проверяемой области могут использовать-
ся различные подходы. При нисходящем (сверху вниз) подходе сначала оцени-
вается общая структура системы качества и ее подсистем. Для оценки могут быть
выбраны отдельные подсистемы. Системы, выделенные и разработанные FDA для
6-системной модели инспектирования производителей лекарственных средств [15],
включают:
• всю систему качества;
• помещения и оборудование;
• систему материалов;
• систему производства;
• упаковку и маркировку;
• лабораторный контроль.
Подсистемы должны быть характерны для проверяемой определенной системы
и могут совмещать основные элементы стандарта, на соответствие которому пред-
полагается проверить систему качества, или основные элементы, перечисленные
в руководства FDA по фармацевтической системе качества. При использовании
нисходящего подхода аудитор сначала проводит обзор каждой подсистемы, чтобы
определить, выполняются ли требования, применяемые к подсистеме (например,
регуляторные требования, требования стандарта), при определении, документи-
ровании и внедрении соответствующих процедур. Убедившись в наличии обя-
зательных процедур, аудитор будет изучать относящиеся к этому записи и другие
документы для проверки того, что процедуры выполняются, а факт выполнения
регистрируется и что система качества эффективно функционирует в соответствии
с проектом и отвечает регуляторным требованиям и стандартам. Данный подход по-
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 269
зволяет провести системную оценку каждой подсистемы и может быть настолько
детализирован, насколько это необходимо.
Восходящий подход может использоваться для расследования определенной
проблемы с качеством, выявленной при анализе тренда, несоответствиях продук-
та, нежелательных лекарственных реакциях, жалобах потребителей или из других
источников данных по качеству. Начиная с записей по качеству, связанных с про-
блемой, аудитор будет двигаться в своей работе через систему качества, изучая про-
цессы качества, относящиеся к рассматриваемой проблеме. Данный подход полезен
при выявлении недостатков системы качества, которые могут быть связаны с опре-
деленными проблемами, но при его использовании нелегко провести оценку всей
системы.
Также может использоваться комбинированный подход, совмещающий элемен-
ты нисходящего и восходящего подходов. Это позволяет провести на некотором
уровне анализ эффективности всей системы качества с одновременным рассмотре-
нием причин конкретных проблем качества.
Аудиторы должны выбрать метод, наиболее пригодный для установленных целей
аудита. Для первичных аудитов системы качества или регулярных плановых ауди-
тов наиболее подходящим будет нисходящий подход, а для аудитов, проводимых
в рамках анализа причины лучшим выбором будет восходящий метод. FDA исполь-
зует похожий подход для своих инспекций. При регулярных плановых двухлетних
инспекциях наиболее вероятно применение нисходящей методологии проверки.
При внеплановых инспекциях, проводимых в ответ на проблемы с определенным
препаратом, такие как отзыв с рынка, наиболее вероятным будет применение вос-
ходящего подхода. Инспекторы FDA могут применять комбинированный подход
при двухлетних инспекциях, если имеют информацию об определенных проблемах
с качеством, которые они хотят включить в инспекцию.
Аудит, как описано в моделях СМК, проводится для оценки эффективности си-
стемы качества в целом, как она была спроектирована, и для соответствия приме-
няемым стандартам. Вся система качества не может быть охвачена в рамках одного
аудита. Производители могут использовать так называемый «скользящий» под-
ход, при котором отдельные подсистемы выбираются для проверки в соответствии
с одобренным графиком аудита. Разработка планов по аудиту должна обеспечить
эффективное выполнение этой оценки.
Соответствие требованиям сСЛ/Ртакже является основной задачей аудита, и при
его планировании должна быть включена оценка соответствия требованиям cGMP
и готовность к инспекции FDA. Существующие рекомендации FDA и руководства
по методикам оценки соответствия содержат описание инспекционных подходов
агентства и могут быть использованы в качестве справочной литературы при раз-
работке планов аудитов [15—17]. Может быть полезным включение в общий поря-
док проведения аудитов имитации инспекции FDA. Некоторые компании предпо-
читают использовать внешних аудиторов для имитационных аудитов, чтобы лучше
имитировать процесс инспектирования FDA. Имитационные аудиты также полезны
в обучающих целях при подготовке организации для инспекции FDA.
План аудита должен согласовываться с письменными процедурами по проведе-
нию аудита качества, включенными в руководство по качеству или другую докумен-
270
Часть 3. Качество
тацию системы качества. План должен включать или иметь ссылки на объективные
критерии, которые используются при оценке соответствия требованиям. Также план
должен включать или содержать ссылки на другие документы, которые будут ис-
пользованы в ходе аудита, включая отчеты ранее проведенных аудитов. Если в объ-
ем работ по аудиту входит изучение серии или производственных записей, такое
изучение должно быть проведено в соответствии с определенным планом отбора
проб или другим пригодным статистически обоснованным способом, как указано
в процедурах компании по системе качества.
Производители, внедряющие систему качества, соответствующую имеющемуся
стандарту, могут счесть полезным создание таблицы или другого документа, в ко-
тором были бы указаны взаимосвязи между требованиями cGMP, требованиями
стандарта и элементом или элементами системы качества производителя. Такой
инструмент может помочь учесть все необходимые требования в проекте системы
качества и обеспечить включение в проекты планов по аудиту проведения оценки
всех соответствующих требований.
Так как современные модели систем качества используют системный подход,
при аудите может быть полезным использовать перечень вопросов (чек-лист) для
аудита, формат которого приведен в табл. 5. Форма может включать соответствую-
щую информацию для контроля документа, такую как сведения об идентифика-
ции, пересмотре и утверждении. Компания также может включить в форму ссыл-
ки на информацию, использованную при планировании аудита, например, отчеты
предыдущих аудитов, заполненную форму 483 FDA «Наблюдения при инспекции»,
отчеты об аудите третьей стороной и связанные с аудитом документы системы каче-
ства (например, процедуры по проведению аудита). В зависимости от целей аудита
подсистемы могут корреспондировать с шестью подсистемами, выделенными FDA
Таблица 5
Пример перечня вопросов для аудита
[Название компании] Перечень вопросов по аудиту системы качества
Форма: Версия: Дата: Утверждено:
Дата(ы) аудита: Ссылки:
Аудитор: Название: Подпись:
Требование Ссылка на раздел cGMP Соответствует (да/нет/не применимо) Объективные доказательства и комментарии
Подсистема 1
Требование 1.1
Требование 1.2
Подсистема 2
Требование 2.1
Требование 2.2
Подсистема 3
Требование 3.1
Требование 3.2
Подсистема N
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 271
для инспекторов при проведении проверок на соответствие правилам cGMP (т. е.
качество, производство, помещения и оборудование, лабораторный контроль, ма-
териалы, упаковка и маркировка), или с основными элементами стандарта по си-
стемам качества. Могут быть включены перекрестные ссылки между элементами
используемого стандарта и соответствующими разделами правил cGMP, если тако-
вые имеются. Форма для аудита должна допускать внесение в нее информации о со-
ответствии или несоответствии каждому требованию и иметь достаточно места для
описания соответствующих наблюдений.
Модели СМК требуют проведения периодических аудитов, но не устанавливают
частоту проведения. Частота проведения должна определяться на основании ана-
лиза риска, связанного с соответствующими областями аудита, и других факторов,
включая результаты предыдущих аудитов и другие данные по качеству. Периоди-
ческие аудиты должны проводиться в течение всего жизненного цикла продукта,
и последующие аудиты проводятся, при необходимости, для подтверждения того,
что ранее выявленные проблемы качества были исправлены в соответствии с при-
меняемыми регуляторными требованиями и требованиями к системе качества.
3.2.7.3. Управление рисками качества
FDA включило управление рисками качества в общую концепцию систем качества
для обеспечения соответствия правилам cGMP и достижения общих целей систе-
мы качества [6]. Методологии управления рисками позволяют руководству устано-
вить приоритеты д ля деятельности в целом или для конкретных действий на основе
оценки рисков, учитывающей вероятность возникновения вреда и его тяжесть.
Внедрение управления рисками качества включает определение риска, выбор
и внедрение методов контроля для управления рисками, соразмерных с уровнем ри-
ска, и оценку результатов мероприятий по управлению рисками. В производствен-
ных системах качества управление рисками используется как инструмент при раз-
работке спецификаций на продукт и критических параметров процесса. Управление
рисками качества, совмещенное со знаниями о процессе, помогает производителям
эффективно управлять и контролировать изменения.
Формализованный процесс управления рисками состоит из нескольких компо-
нентов:
• определение риска;
— идентификация риска;
— анализ риска;
— оценка риска;
• контроль риска;
— снижение риска;
— принятие риска;
• распространение информации о риске;
• обзор рисков.
Определение риска начинается с идентификации риска, систематического ис-
пользования имеющейся информации для идентификации опасностей (т. е. собы-
тий или каких-либо условий, которые могут вызывать вред). Информация может
272
Часть 3. Качество
получаться из разнообразных источников, включая основные заинтересованные
стороны, архивные и литературные данные и результаты математического или
научного анализа. Затем проводится анализ рисков для определения уровня ри-
ска, связанного с выявленными опасностями. Уровень устанавливается на основе
схожести частоты опасностей и результирующей тяжести вреда. В некоторых ин-
струментах управления рисками способность выявлять опасности также рассма-
тривается. Если опасность выявляется с трудом, то это может включаться в виде
дополнительного фактора при общем определении рисков. При оценке рисков на
основе установленных критериев определяют, приемлем ли данный риск. В систе-
ме качества эти критерии будут включать влияние на общую эффективность си-
стемы качества и показатели качества готового продукта. Ценность определения
риска зависит от оценки надежности данных, использованных в процессе опре-
деления. При определении рисков должны приниматься во внимание допущения
и допустимые источники неопределенности. Работы по определению риска долж-
ны документироваться.
Контроль риска начинается с его снижения, что включает любые действия, пред-
принятые для исключения или снижения риска. Предпринятые действия должны
быть сопоставимы со значимостью риска. Если риск был снижен до приемлемо-
го уровня, то может быть принято положительное решение о допустимости риска
(принятие риска). При этом следует задать вопрос, не появились ли новые риски
в результате контроля идентифицированных рисков. Мероприятия по контролю
риска, как правило, следует проводить в соответствии с процедурами контроля из-
менений и документировать.
Распространение информации о рисках включает донесение соответствующей
информации о риске до заинтересованных сторон (например, тех, кто вовлечен или
на кого влияет система качества, в том числе руководство, пользователей, регуля-
торные агентства). Предоставление информации о рисках должно документиро-
ваться. Представляемая информация может быть связана с характером и формой
рисков, вероятностью, тяжестью, приемлемостью, контролем, управлением, вы-
являемостью и другими аспектами рисков качества. Предоставление информации
должно быть обоснованным, при этом нет необходимости в информировании каж-
дого участника процесса и во всех случаях принятия риска.
Обзор рисков следует проводить для оценки результатов процесса управления
рисками и повторять по мере необходимости, с учетом новых данных по качеству
или изменений в процессах или продукте.
Руководство Международной конференции по гармонизации ICH (International
Conference on Harmonisation) Q9 «Управление рисками качества» [6] перечисляет ряд
инструментов по управлению рисками, которые могут использоваться производи-
телями, включая анализ видов, последствий и критичности отказов (FMECA',.failure
mode effects and criticality analysis), анализ рисков и критические контрольные точки
(ХАССП1 2, НАССР, hazard analysis and critical control points), предварительный анализ
1 Описан в ГОСТ Р 51901.12—2007. — Примеч. перев.
2 Описан в ГОСТ Р ИСО 22000—2007 и для пищевой промышленности в ГОСТ 51705.1—
2001. — Примеч. перев.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 273
опасностей (PHA, preliminary hazard analysis), и предоставляет примеры применения
управления рисками качества в менеджменте качества, разработке, управлении ма-
териалами, производстве и в других операциях внутри организации.
3.2.7.4. Корректирующие и предупреждающие действия
Корректирующие и предупреждающие действия (САРА, corrective and preventive
action) — это термин, обычно использующийся для описания подсистемы в общей
системе качества, посвященной систематическим расследованиям, изучению и реа-
гированию на проблемы качества, включая несоответствия. Корректирующее или
предупреждающее действие может быть инициировано на основании обзора и ана-
лиза данных по качеству из различных источников, включая нежелательные лекар-
ственные реакции, жалобы, результаты аудитов качества, инспекций FDA и ауди-
тов третьей стороной, сообщения о несоответствующих материалах, информацию
о контроле процессов, анализы тренда и другие источники.
Корректирующее действие инициируется с целью исправления причины уста-
новленного несоответствия и для предупреждения его повторного возникновения
или схожего с ним несоответствия. Это действие может включать первоначальные
и последующие шаги (например, выполняемые после анализа дерева причин). Со-
временные модели систем качества и правила сСЛГРтребуют выполнения корректи-
рующих действий и их документирования. В современных моделях систем качества
предупреждающие действия включают действия, выполняемые в ответ на данные
по качеству и направленные на устранение причины возможных несоответствий
для предупреждения их возникновения. Следовательно, эффективная система САРА
включает активные и проактивные компоненты. Эффективность корректирующих
и предупреждающих действий должна оцениваться, если это возможно, с использо-
ванием объективных критериев, а проведение оценки должно регистрироваться.
Система САРА организации и ее процессы должны быть разработаны для систе-
матического анализа и реагирования на вопросы, связанные с качеством, сораз-
мерно риску. Система должна предусматривать подтверждение или валидацию кор-
ректирующих и предупреждающих действий для обеспечения их эффективности
и проверки того, что эти действия не оказывают негативного эффекта на готовый
продукт. Также система должна обеспечивать по мере необходимости надлежащее
распространение информации, связанной с САРА, внутри организации для гаран-
тированного эффективного функционирования системы качества и для анализа со
стороны руководства.
3.2.7.5. Стимулирование улучшений
Постоянное улучшение — это требование существующих моделей систем качества,
таких как стандарт ИСО 9001—2000, в соответствии с которым организация обязана
постоянно улучшать эффективность СМК путем использования политики в обла-
сти качества, целей в области качества, результатов аудитов, анализа данных, кор-
ректирующих и предупреждающих действий и анализа со стороны руководства. При
адаптации стандарта ИСО 9001—2000 к регуляторному стандарту по СМК в произ-
водстве медицинских изделий разработчики ИСО стандарта 13485 незначительно
изменили изложение требования, указав, что организация обязана «идентифици-
Часть 3. Качество
274
ровать и внедрить любые изменения, необходимые для обеспечения и поддержания
постоянной пригодности и эффективности СМК путем использования политики
в области качества, целей в области качества, результатов аудитов, анализа данных,
корректирующих и предупреждающих действий и анализа со стороны руководства».
Слово «улучшение» было из текста удалено, так как оно не входит в цели действую-
щих регуляторных стандартов, однако концепция постоянного мониторинга функ-
ционирования системы качества и соответствующего реагирования на данные по
качеству была сохранена.
Правила cGMP специально не оговаривают требования по постоянному улучше-
нию, при этом правила содержат очень детальные указания по отбору и проведе-
нию испытаний полупродуктов и готового продукта, и отсутствие обоснованных
действий для сокращения выявленных источников изменений может вызвать у
инспекторов FDA сомнения в выполнении правил. В руководстве по фармацев-
тической системе качества FDA рекомендует организациям стимулировать улуч-
шения с помощью деятельности системы качества и отмечает, что крайне важным
в этом процессе является участие высшего руководства. Улучшение процесса вместе
с улучшением внутрипроизводственного контроля может сделать производствен-
ный процесс более продуктивным и надежным. Конечный результат может приве-
сти к снижению расходов, а в дальнейшем к предотвращению отказов, связанных
с продуктом, и дефектов.
3.2.8. Переход к модели системы качества
Правила cGMP устанавливают значительную ответственность структурного подраз-
деления организации, отвечающего за деятельность в области качества. Организа-
ции, внедряющие модель системы качества, будут обязаны осуществлять дополни-
тельные работы, связанные с качеством, включая, но не ограничивая, проведение
аудитов, анализ данных по качеству, определение рисков и выполнение предупре-
ждающих действий, основанных на обзоре и анализе данных по качеству и направ-
ленных на предупреждение возникновения несоответствий продукта. Дополнитель-
но от руководства требуется осуществлять необходимое лидерство путем активного
участия в системе качества и обеспечения надлежащего функционирования систе-
мы качества. Это достигается посредством установления политики в области каче-
ства и связанных с ней целей, планирования качества, создания соответствующей
организационной структуры с установленными правами и обязанностями для над-
лежащего выполнения требований системы качества, предоставления необходимых
ресурсов и обучения, периодического анализа информации и данных по качеству
и обеспечения реагирования на них организации соответствующим образом.
Структурное подразделение, ответственное за деятельность, связанную с каче-
ством, будет, очевидно, играть еще большую роль внутри организации, а роли и от-
ветственность других подразделений также, вероятно, изменятся. Для обеспечения
плавного перехода без негативных последствий для качества продукта потребуется
тщательное планирование. Ниже приведены некоторые вопросы, рассматриваемые
при планировании перехода.
• Создание группы по переходу на новую модель. Должна быть создана межфунк-
циональная группа с участием ключевых руководителей и сотрудников ор-
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 275
ганизации для разработки плана и выполнения работ по переходу. Группа по
переходу должна иметь четкое понимание своей задачи и организационных
целей, связанных с переходом
• Обучение группы по переходу. Решение по выполнению перехода должно исхо-
дить от руководства, и руководство обязано обеспечить, чтобы все участники
группы прошли соответствующее обучение требованиям системы качества,
управлению рисками и рекомендованной FDA концепции систем качества.
• Разработка плана перехода. Группой по переходу должен быть разработан
план перехода, основанный на четко сформулированных целях.
• Идентификация требований по персоналу. Переход, по всей видимости, по-
влечет изменения должностных инструкций, распределение и создание до-
полнительных обязанностей, поэтому будут необходимы перераспределение
сотрудников, найм новых сотрудников и предоставление требующегося обу-
чения всем сотрудникам, затронутым переходом.
• Определение потребности в других ресурсах. В план следует включить описание
требуемых ресурсов для планирования и выполнения плана.
• Определение функций и ответственности. В плане должны быть четко опреде-
лены роли и ответственность всех сотрудников, отвечающих за разработку
и выполнение плана по внедрению системы качества, а также функции со-
трудников и ответственность в рамках системы качества.
• Рассмотрение требований к организационной структуре. Для надлежащего вы-
полнения работ лица, ответственные за деятельность по качеству, должны
иметь установленные обязанности и права, информация о которых надлежа-
щим способом доведена до сведения сотрудников организации.
• Проведение анализа различий (gap analysis). В план должно быть включено
проведение анализа различий, который выявляет эффективный путь встраи-
вания выбранной модели системы качества в существующие процессы для
создания системы, удовлетворяющей целям организации в области качества,
регуляторным требованиями и согласующейся с другими требованиями орга-
низации. В концепции систем качества предусмотрена их относительная гиб-
кость при использовании и подстройка под специфические организационные
требования. Для надлежащего функционирования система качества должна
быть эффективно интегрирована в организацию таким образом, чтобы она
не выглядела в виде дополнительной нагрузки или набора дополнительных
требований, мешающих выполнению реальной работы.
• Использование бенчмаркинга. По возможности следует организовать встречи
с организациями, которые успешно осуществили переход, изучить их систему
качества и обсудить проблемы при переходе и опыт их решения.
• Консультации с экспертами. Дополнительно к бенчмаркингу очень полез-
ной может оказаться помощь лиц, знакомых с системами качества, особенно
в случаях, когда имеющийся персонал имеет относительно небольшой опыт
работы с системами качества. Может оказаться полезным приглашение одно-
го или нескольких внешних экспертов для работы с группой по переходу в ка-
честве тренеров или кураторов.
276
Часть 3. Качество
• Регулярное распространение информации. Регулярный обмен четкой информа-
цией между сотрудниками группы по переходу крайне необходим для эффек-
тивного координирования работ по выполнению плана, подготовки отчетов
о ходе и результатах работ, решения проблем и выявления увеличения потреб-
ности в ресурсах.
• Реклама системы. Для успешного внедрения системы качества требуется ак-
тивное и информированное участие большинства сотрудников организации.
Намерения руководства должны быть ясно доведены до сведения сотрудни-
ков, и все задействованные сотрудники должны пройти обучение для по-
нимания основных принципов системы качества и роли самих сотрудников
в этой системе.
• Валидация системы.
• Обеспечение соответствия регуляторным требованиям.
3.2.9. Перечень вопросов для аудита в фармацевтической
промышленности
Перечень вопросов, приведенный в табл. 6 [15], предназначен для помощи в систе-
матическом СМР-аудите производства, выпускающего фармацевтические субстан-
ции или готовые продукты.
Пригодность любой процедуры — это факт, устанавливаемый аудитором. По-
этому авторы не несут никакой ответственности за любые последующие замечания
государственных инспекторов или действия, произошедшие в результате использо-
вания этого контрольного списка для аудита.
3.2.9.1. Инструкция по применению перечня вопросов для аудита
Разработайте план аудита до его выполнения на месте. Ознакомьтесь с прошедши-
ми аудитами, отметьте указания о возможных проблемных зонах и темах, которые
были причинами корректирующих действий в предыдущем аудите. Если вы еще не
знакомы с этим производством, изучите тип выпускаемой продукции, организацию
в нем персонала и функций. Что ожидает узнать ваш «потребитель», то есть ваш ру-
ководитель или высшее руководство производства, с помощью этого аудита?
1. Перечень вопросов следует использовать вместе с журналом, в котором дела-
ются записи в ходе аудита.
2. Перечень вопросов предназначен для направления действий аудитора, но не
заменяет правила GMPu не отменяет необходимость знать эти правила.
3. Несмотря на то что по каждому проверяемому требованию можно задать один
вопрос, ответ, как правило, должен быть многоплановым, поскольку аудитор обя-
зан определить все действия по нескольким продуктам, которые могут иметь раз-
личный состав. Вносите детали в свой журнал и указывайте перекрестные ссылки
на вопросы.
4. Тщательный анализ по крайней мере трех выбранных промышленных серий
должен включать рассмотрение: а) прослеживаемости всех компонентов или мате-
риалов, использованных в анализируемых сериях; б) документации по испытаниям
исходных материалов или компонентов, полупродуктов и готового продукта для
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 277
трех анализируемых серий; в) складских записей по хранению и отгрузке, так как
они могут быть связаны с возможным отзывом с рынка.
5. Ответы, вносимые в перечень вопросов, должны быть единообразно оформ-
лены. Крестик «х» рекомендуется для ответа «нет», галочка «V» — для ответа «да»;
«н/п» — для ответа «не применимо» для вопросов, которые некорректны для дан-
ного аудита. Звездочка и страница журнала должны быть указаны в контрольном
списке, чтобы идентифицировать месторасположение зафиксированных соответ-
ствующих описаний или вопросов в вашем журнале.
6. Используемый для записей журнал должен быть журналом лабораторного типа
с прошитыми страницами. На журнале должно быть четко указано, что он предна-
значен для аудита, проставлены дата и автор(ы). Многие аудиторы предпочитают
использовать отдельный журнал для каждого аудита, чтобы его можно было архиви-
ровать вместе с перечнем вопросов и окончательным отчетом.
7. Ссылки на параграфы правил GMP приведены для вашего удобства при воз-
никновении вопросов. В некоторых случаях два и более параграфа правил GMP мо-
гут содержать требования по определенному вопросу. Заголовки в правилах GMP
обычно содержат определенную информацию об описываемых областях.
8. Общая рекомендация для проведения успешного аудита — тратить ббльшую
часть вашего времени на основные вопросы и меньшую — на менее значительные.
У вас Moiyr быть наблюдения, которые вы захотите сообщить руководящему пер-
соналу для привлечения их внимания, но не будете включать в отчет об аудите, по-
скольку они относительно несущественные. Также справедливо и то, что слишком
большое количество мелких вопросов свидетельствует о тенденции к несоответ-
ствию, и, следовательно, требует внимания. При их цитировании будьте предметны.
Таблица 6
Перечень вопросов для аудита
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
1.0 Общий контроль
Производство и его структурные подразделения работают как установлено в правилах GMF!
1.1 Ответственность организации и руководства
1.101 В данном подразделении или филиале используется корпоративная или собственная политика в области качества?
1.102 § 211.22(a). Отдел (управление) по обеспечению качества (ООК) выделен в самостоятельное структурное образование?
1.103 § 211.22(a). ООК имеет права и обязанности для одобрения или отклонения любых компонентов, контейнеров и укупорочных средств для лекарственных препаратов, полупродуктов, упаковочных и этикетировочных материалов и готовых лекарственных препаратов?
1.104 § 211.22. ООК проводит повседневную проверку производственных записей, чтобы обеспечить выполнение и надлежащее документирование процедур?
278
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
1.105 § 211.22(b). Достаточно ли помещений, оборудования и квалифицированного персонала для проведения требуемых испытаний?
1.106 Если часть испытаний выполняется по контракту, ООК проверял производственную площадку подрядчика и подтвердил приемлемость помещений, оборудования, квалифицированного персонала и процедур?
1.107 Дата последнего аудита —
1.108 § 211.22(c). Все процедуры ООК имеются в письменном виде?
1.109 § 211.22(c). Все обязанности ООК имеются в письменном виде?
1.110 Все письменные процедуры ООК действующие и утверждены? (Проверьте журнал учета процедур)
1.111 Все процедуры выполняются? (Проверьте постоянное ведение записей, которые надлежащим образом документируют выполнение испытаний)
1.112 § 211.25. Все процедуры ООК имеются в письменном виде?
1.113 § 211.25. Другой персонал ООК (т. е. химики, аналитики, лаборанты) прошел соответствующее обучение и имеет опыт (требуемую квалификацию)?
1.2 Программа контроля документов
1.201 § 211.22(a). В ООК есть сотрудник или подразделение, специально уполномоченное следить за проектированием, пересмотром и получением одобрения для производственных и лабораторных процедур, форм и записей?
1.202 § 211.22(d). Имеется письменная стандартная операционная процедура (СОП), которая для каждой существующей записи или формы указывает, как заполняется форма, кто подписывает и подтверждает подпись?
1.203 § 211.65(а)(Ь)(с). Записи о производстве серии и результаты испытаний выходного контроля проверяются на правильность и полноту до выпуска в обращение серии или партии готового продукта?
1.3 Введение сотрудника в должность, внимание к вопросам качества и обучение выполняемой работе
1.301 Обведите кружком документы и процедуры, предусмотренные для каждого нового сотрудника при введении в должность: 1) буклет с информацией о компании; 2) литература, описывающая правила GMPvi подчеркивающая важность точного выполнения инструкций; 3) обучение на рабочем месте для каждой выполняемой функции {проводимое до разрешения выполнять такие задачи); 4) что-либо другое — отметьте в журнале.
1.302 § 211.25(a). Повторное обучение каждого сотрудника СОП (процедурам) проводится при внесении в процедуру значительных изменений?
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 279
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
1.303 Укажите, как осуществляется постоянное регулярное обучение GMP
1.304 § 211.25. Все документы по обучению имеются в письменном виде с указанием даты обучения, типа обучения и подписями обучаемого и инструктора?
1.305 § 211.25. Все ли записи по обучению доступны для просмотра, чтобы можно было определить, когда сотрудник проходил обучение, какие сотрудники обучались выполнению определенной процедуры или прошли определенную обучающую программу?
1.306 Инструкторы по GMP имеют соответствующую квалификацию, включая опыт и обучение?
1.307 § 211.25(a). Старший персонал проинструктирован не допускать к работе любого сотрудника, который вследствие состояния здоровья (как определено медицинским осмотром или отмечено старшим персоналом) может оказать негативное воздействие на безопасность или качество лекарственного препарата при наличии прямого контакта с любым компонентом препарата или первичной упаковкой для готового продукта?
1.308 § 211.28(d). Установлено требование, чтобы все сотрудники сообщали старшему персоналу о любом состоянии здоровья или заболевании, которое может оказать негативное воздействие на безопасность и эффективность лекарственного препарата?
1.309 § 211.25(a). Временные сотрудники проходят такое же введение в должность, как постоянные?
1.310 § 211.34. У консультантов, нанимаемых для обсуждения любых аспектов производства, технологии, упаковки или хранения, разрешения на выпуск продукции в обращение, запрашивают подтверждение их образования, обучения и опыта?
1.311 § 211.34. Имеются письменные записи, содержащие ФИО, адрес, квалификацию консультанта, даты предоставления услуги и типы предоставляемых услуг?
1.4 Техника безопасности работ и охрана предприятия
1.401 Имеется на производственной площадке корпоративная или собственная программа по охране труда?
1.402 Процедуры по технике безопасности зафиксированы в письменном виде?
1.403 Процедуры по технике безопасности действующие?
1.404 Сотрудники проходят вводный инструктаж по технике безопасности до начала работы на предприятии?
1.405 Имеются документы по проведению инструктажа по технике безопасности, в которых указаны фамилия сотрудника, тип инструктажа, дата проведения инструктажа, фамилия инструктора и подписи инструктора и обучаемого сотрудника?
280
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
1.406 Имеет предприятие официальную политику по мерам безопасности, оформленную в письменном виде?
1.407 Ограничен доступ на предприятие?
1.408 Опишите, каким образом контролируется доступ на территорию и ограничен ли он?
1.409 Охрана имеется в течение 24 часов?
1.5 Программа GMP-аудитов и внутренних аудитов качества
1.501 Имеется в данном подразделении или филиале политика в области качества, оформленная в письменном виде?
1.502 Копия данной политики в области качества предоставлена каждому сотруднику?
1.503 Если на предыдущий вопрос был ответ «да», то когда эта копия была предоставлена?
1.504 Проводится обучение улучшению качества?
1.505 В обязанности ООК официально включена функция по проведению аудита?
1.506 В письменной СОП по проведению аудита указано, кто будет проводить аудиты, а также их квалификация (образование, обучение и опыт)?
1.507 В письменной СОП указаны область и частота проведения аудитов и порядок их документирования?
1.508 В письменной СОП указан порядок рассылки отчета об аудите?
1.6 Программа затрат на качество
1.601 В данном подразделении проводится периодический и формализованный анализ затрат на качество?
1.602 Имеется в данном подразделении необходимые персонал, программное обеспечение и бухгалтерские данные, чтобы выявлять и учитывать затраты на качество?
1.603 Данное подразделение осуществляет целенаправленные работы по снижению затрат на качество?
2.0 Контроль проектирования Не входит непосредственно в область государственного регулирования лекарственных средств
3.0 Контроль производственных мощностей
3.1 Проектирование и планировка предприятия
3.101 § 211.42(a). Конструкция всех частей производственного комплекса обеспечивает их пригодность для производственного процесса, проведения испытаний и хранения лекарственных средств?
3.102 § 211.42(b). Площадь комплекса достаточна для выполнения производимого типа работ и стандартного объема продукции?
3.103 Планировка и организация работ предупреждают контаминацию?
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 281
Таблица б (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
3.2 Программа контроля окружающей среды
3.201 Здания НЕ расположены на участке, на котором сотрудники или продукт могут подвергнуться загрязнению механическими включениями, испарениями или паразитами?
3.202 Стоячие воды на земельном участке отсутствуют?
3.203 § 211.44. Освещение надлежащее во всех зонах?
3.204 § 211.46. Вентиляция надлежащая?
3.205 § 211.46. Контроль давления воздуха, пыли, влажности и температуры осуществляется надлежащим образом для производства и обработки, хранения и проведения испытаний лекарственных средств?
3.206 § 211.46. Если используются воздушные фильтры, имеется ли письменная процедура, в которой указана частота проверок и замен фильтров?
3.207 Достаточно канализации и повседневных процедур уборки для предупреждения образования стоячих вод внутри зданий?
3.208 § 211.42(d). На предприятии существуют раздельные вентиляционные системы, если они вообще требуются, для предупреждения контаминации? (Обязательны при производстве препаратов пенициллина!)
3.3 Программа технического обслуживания и уборки помещений
3.301 § 211.56(a). В помещениях отсутствуют грызуны, птицы, насекомые и паразиты?
3.302 § 211.34(c). В подразделении имеются письменные процедуры по безопасному использованию пригодных (т. е. зарегистрированных в надлежащем порядке) зооцидов, инсектицидов, фунгицидов и фумигантов?
3.303 В помещениях поддерживается чистота и надлежащее санитарное состояние?
3.304 В этом подразделении имеются письменные процедуры, детально описывающие график и методы уборки, соответствующее оборудование и материалы?
3.305 В этим подразделении имеются письменные процедуры для безопасного и правильного использования чистящих и дезинфицирующих средств?
3.306 § 211.58. Все части производственного комплекса находятся в хорошем состоянии?
3.307 § 211.52. Утилизация сточных вод, сливов, отходов и других непригодных материалов осуществляется безопасным и адекватным с точки зрения промышленной санитарии способом (с достаточной частотой)?
282
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
3.4 Программа контроля внешних подрядчиков
3.401 § 211.56(d). Установлены требования к подрядчикам и временным сотрудникам о выполнении работ в требуемых санитарных условиях?
3.402 Квалифицированы ли подрядчики, которые на основе своего опыта или обучения выполнять работы, могут влиять на производство, упаковку или хранение лекарственных средств?
4.0 Контроль оборудования
4.1 Конструкция оборудования и размещение
4.101 § 211.34. Все оборудование, используемое для производства, обработки и хранения лекарственных средств, имеет надлежащую конструкцию и размер согласно предназначению?
4.102 Пригодны следующие единицы оборудования для своего назначения: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.103 Пригодны следующие единицы оборудования по своему размеру и емкости: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.104 Пригодна конструкция у следующих единиц оборудования: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.105 Следующие единицы оборудования размещены в пригодных зонах: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.106 Следующие единицы оборудования установлены надлежащим образом: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.107 Имеется достаточно места для указанного оборудования: смеситель(и), транспортер(ы), таблеточные прессы, машины для заполнения капсул, аппараты для розлива в бутылки, другое (укажите)?
4.108 § 211.65(a). Поверхность оборудования, контактирующая с материалами или готовыми продуктами, сделана из инертного, неабсорбирующего и невыделяющего материала, чтобы не оказывалось влияния на продукт?
4.109 § 211.65(b). Конструкция оборудования и меры предосторожности при эксплуатации обеспечивают ОТСУТСТВИЕ контакта смазки или охладителей с компонентами лекарственного средства или готовым продуктом?
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 283
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
4.110 § 211.72. В производстве инъекционных препаратов НЕ используют фильтры, выделяющие волокна?
4.111 § 211.72. В производстве НЕ используют фильтры, содержащие асбест?
4.112 Каждая простаивающая единица оборудования промаркирована указанием «Нуждается в очистке» или «Чистое, готово к использованию»?
4.113 Очистка оборудования проводится сразу после использования?
4.114 Неиспользуемое оборудование хранится в выделенной зоне?
4.115 § 211.67(a)(6). Имеются письменные процедуры для каждой единицы оборудования, используемого в производстве и обработке или при хранении компонентов, полупродуктов и готового продукта?
4.116 Инструкции по очистке включают операции по разборке и осушению, обеспечивающие отсутствие в оборудовании остатков чистящего раствора или промывных вод?
4.117 Процедура по очистке или процедура запуска оборудования обеспечивает проведение систематической и тщательной очистки?
4.2 Идентификация оборудования
4.201 § 211.105. Все единицы оборудования четко идентифицированы с помощью легко различимой маркировки?
4.202 § 211.105(b). На всех единицах оборудования также имеется идентификационный номер, совпадающий с записями в журнале оборудования?
4.203 Для каждой единицы оборудования имеются письменные инструкции по техническому обслуживанию, содержащие график обслуживания?
4.204 Журнал технического обслуживания для каждой единицы оборудования хранится на оборудовании или рядом с ним?
4.3 Техническое обслуживание оборудования и очистка
4.301 § 211.67(b). Письменные процедуры по очистке технического оборудования и инструментов установлены?
4.302 Эти процедуры соблюдаются?
4.303 § 211.67(b)(1). Письменная процедура устанавливает ответственность за очистку и техническое обслуживание оборудования?
4.304 § 211.67(b)(2). Письменный график технического обслуживания и очистки оборудования был установлен и выполняется?
4.305 Процедура очистки была провалидирована надлежащим способом?
4.306 § 211.67(b)(2). Оборудование дезинфицируется согласно письменной процедуре для данной обработки? (если применимо)
284
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
4.307 § 211.67(b)(3). Для очистки и технического обслуживания каждой отдельной единицы оборудования была разработана достаточно детализированная процедура с указанием всех требуемых разборок и сборок?
4.308 § 211.67(b)(3). В процедуре указано, удалить или стереть информацию о производстве серии из каждой единицы оборудования при ее очистке?
4.309 Оборудование очищается сразу после использования?
4.310 На чистом оборудовании имеется четкая маркировка «Чистое» с указанием даты очистки?
4.311 § 211.67(b)(5). До использования чистое оборудование защищено надлежащим образом от контаминации?
4.312 § 211.67(b). Оборудование проверяется непосредственно перед использованием?
4.313 § 211.67(c). Письменные записи по очистке, дезинфекции и техническому обслуживанию хранятся возле или на каждой единице оборудования?
4.4 Программа калибровки измерительного оборудования
4.401 § 211.68(a). На производстве имеются письменные процедуры по проверке и калибровке каждой единицы измерительного оборудования? (Проверьте процедуру и журнал на каждую единицу оборудования, отметьте исключения в журнале аудита с указанием перекрестных ссылок)
4.402 § 211.68(a). Записи о калибровках и проверках ведутся таким образом, чтобы действия можно было легко проверить?
4.5 Программа квалификации оборудования
4.501 § 211.63. Подтвердите, что все единицы оборудования, используемого в производстве, упаковке и обеспечении качества, способны генерировать достоверные результаты
4.502 § 211.68(a). При использовании компьютеров для автоматизации производства или контроля качества, компьютер и программное обеспечение были провалидированы?
4.503 Для квалификации оборудования были использованы тесты или испытания на месте в виде последовательных производственных циклов?
4.504 Для обеспечения надежных результатов тесты повторялись достаточное количество раз?
4.505 § 211.63. На каждой единице оборудования указаны его минимальные и максимальные емкость и рабочая скорость для получения достоверных результатов?
4.506 Рабочие характеристики определены для каждой единицы оборудо- вания? (Могут быть представлены производителем оборудования, но должны быть подтверждены в типовых рабочих условиях)
Глава 3.2, Значение систем обеспечения качества и аудитов в фармацевтическом производстве 285
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
4.507 Пределы и допуски для рабочих режимов были установлены из рабочих характеристик?
5.0 Контроль материалов и компонентов
5.1 Спецификации на материалы и компоненты и контроль закупок Хотя закупка специально не оговаривается в действующих правилах GMP, в обязанности потребителя компонентов и материалов входит обеспечение качества продукта, материала или компонента
5.101 Каждый поставщик или производитель материалов или компонентов был проинспектирован или подвергся аудиту для оценки надлежащего контроля производства? (Просмотрите поставщиков и аудиты, внесите названия поставленных материалов и даты последнего аудита в журнал)
5.2 Приемка материала или компонента, проверка, отбор проб и лабораторный контроль
5.201 § 211.80(a). На предприятии имеются действующие письменные процедуры по приемке и отказу лекарственных средств, контейнеров, укупорочных средств, упаковочных и этикетировочных материалов? (Перечислите выбранные материалы и компоненты в журнале и проверьте процедуры)
5.202 § 211.80(d). Каждой партии в каждой поставке материала или компонента присваивается уникальный код, обеспечивающий возможность их отслеживания на протяжении всего производственного процесса и при отгрузке готовой продукции?
5.203 § 211.82(a). Проверка начинается с визуального исследования каждого транспортного контейнера на наличие соответствующей маркировки, признаков повреждений или контаминации?
5.204 § 211.82(b). Количество репрезентативных образцов, отбираемых из контейнера или от партии, основано на статистических критериях и опыте работы с каждым типом материала или компонента?
5.205 § 211.160(b). Техника отбора проб изложена письменно и соблюдается для каждого типа отбираемых проб?
5.206 Количество отобранных образцов достаточно для анализа и резерва в случае проведения повторного контроля или подтверждения результатов?
Подтвердите, что указанные далее операции включены в письменные процедуры, если не выполняются более специфические процедуры:
5.207 § 211.84(c)(2). Контейнеры очищают до извлечения проб
5.208 § 211.84(c)(4). Отдельные образцы не смешиваются для анализа
5.209 § 211.84(c)(5). Контейнеры, из которых были отобраны образцы, маркируются с указанием даты и приблизительного количества отобранных проб
286
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
5.210 Контейнер с каждым образцом имеет четкую маркировку с указанием материала или названия компонента, номера партии, даты отбора проб, фамилии сотрудника, отобравшего пробу, и идентификационных данных контейнера, из которого отобрали пробу
5.211 § 211.84(d)(l)(2). По крайней мере, проводится одно испытание для подтверждения подлинности сырья (фармацевтической субстанции или химического вещества), если поставщиком предоставляется сертификат анализа, принимаемый ООК
5.212 Если сертификат анализа на партию материала не принимается, то проводятся дополнительные испытания в соответствии с письменным протоколом в целях определения пригодности для использования
5.213 § 211.84(d)(6). Микробиологический анализ выполняется, если требуется
5.3 Хранение и обращение с материалами и компонентами
Верификация материалов и компонентов и недопущение загрязнения (контаминации) и смешения
5.301 § 211.42(b). Все входящие материалы и компоненты находятся в карантине, пока не будут одобрены для использования?
5.302 Со всеми материалами обращаются надлежащим способом, чтобы предупредить контаминацию?
5.303 Никакие материалы не хранятся на полу?
5.304 Размещение материалов позволяет проводить очистку и проверку?
5.305 §211.122(d). Этикетировочные материалы для различных препаратов, дозировок, лекарственных форм и др. хранятся по отдельности с пригодной идентификацией?
5.306 Доступ в зону хранения этикетировочных материалов разрешен только специальному персоналу?
5.307 § 211.89. Забракованные компоненты, материалы и контейнеры находятся в карантине с четкой маркировкой, предотвращающей их использование?
5.4 Программа контроля запасов
5.401 § 211.142. Письменные процедуры по контролю запасов имеются?
5.402 В программе указаны даты уничтожения материалов, компонентов, упаковочных материалов с истекшим сроком годности или устаревших?
5.403 § 211.159(a). Запасы перекладываются, чтобы обеспечить использование в первую очередь продуктов или материалов, одобренных для использования прежде других?
5.404 §211.184(e). Уничтожение материалов документируется с четкой идентификацией уничтоженного материала и даты уничтожения?
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 287
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
5.5 Программа контроля производителей (поставщиков)
5.501 Производители периодически инспектируются в соответствии с письменной процедурой?
5.502 Процедура по подтверждению результатов контроля производителя оформлена в письменном виде и выполняется?
6.0 Операционный контроль
6.1 Проверка, хранение и обращение с материалами, компонентами и этикетировочными средствами
6.101 § 211.87. Письменные процедуры устанавливают период хранения, по истечении которого компоненты, контейнеры и укупорочные средства должны быть повторно проверены перед использованием?
6.102 § 211.87. Разрешение на использование материала, прошедшего повторный контроль (retest), четко маркируется?
6.103 Дополнения с информацией по повторному контролю предоставляются в оригинале?
6.104 Письменные процедуры описывают операции по отпуску материалов в производство?
6.105 Эти процедуры включают: 1) разрешение со стороны подразделения по качеству; 2) документирование правильного веса или измерения объема; 3) надлежащую идентификацию контейнеров?
6.106 Второй сотрудник наблюдает за взвешиванием, отмериванием и отпуском и подтверждает правильность второй подписью?
6.107 § 211.101(c). Дополнение каждого компонента документируется сотрудником, добавляющим материал в ходе производства?
6.108 § 211.101(d). Второй сотрудник наблюдает за каждым добавлением материала, а документация подтверждается второй подписью?
6.109 § 211.125(a). Письменная процедура указывает, кто имеет право выдавать этикетки?
6.110 § 211.125(a). Письменная процедура указывает, как этикетки выдаются, используются и согласуются с производством (по количеству), возвращаются, если не были использованы, а также определяет операции по оценке любых расхождений?
6.111 § 211.125(d). Письменные процедуры указывают на необходимость уничтожения лишних этикеток с проставленным номером серии или контрольным номером?
6.2 Уборка, подготовка и очистка оборудования, линии и зоны
6.201 § 211.67(b)(5). Письменные процедуры подробно описывают порядок проверки оборудования непосредственно перед использованием на чистоту, удаление каких-либо этикеток и этикетировочных материалов из линий до начала печатных операций?
288
Часть 3. Качество
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
6.202 § 211.67(b)(3). Письменные процедуры подробно описывают проведение необходимых разъединений или разборок для подтверждения готовности к очистке?
6.3 Валидация производственного процесса и контроль изменения порядка выполнения технологического процесса
6.301 Производственные процедуры были провалидированы? (Проверьте выбранные процедуры на наличие документации по валидации. Она адекватная?)
6.302 § 211.100(a). Контроль технологических процессов охватывает все вопросы, чтобы обеспечить подлинность, количественное содержание компонентов, качество и чистоту продукта?
6.303 § 211.101(a). Процедура включает указание состава препарата с письменным расчетом выхода продукта с содержанием не менее 100% от указанного на этикетке количества активных ингредиентов?
6.304 § 211.101(c). Все операции взвешивания и измерения проводятся одним уполномоченным сотрудником и контролируются вторым сотрудником?
6.305 § 211.101(d). Записи подтверждают выполнение вышеуказанного правила? Две подписи имеются?
6.306 § 211.103. Реальный выход продукта рассчитывается по завершении соответствующих стадий и в конце производственного цикла?
6.307 § 211.103. Расчеты производятся одним сотрудником? Независимое подтверждение проводится вторым сотрудником?
6.4 Внутрипроизводственный контроль, отбор проб и лабораторный контроль
6.401 § 211.110(a). Установлены письменные процедуры по мониторингу выхода и валидации эффективности производственных процедур, которые могут вызывать изменчивость параметров полупродуктов и готовых лекарственных средств?
6.402 § 211.110(c). Полупродукты подвергаются контролю на соответствующих стадиях на подлинность, количественное содержание компонентов, качество, чистоту и одобряются или отклоняются отделом контроля качества?
6.403 § 211.160(b). Лабораторный контроль включает процедуры отбора проб и проведения испытаний для обеспечения соответствия компонентов, контейнеров, укупорочных материалов, полупродуктов и готового продукта спецификациям?
6.5 Переработка и уничтожение материалов
6.501 § 211.115(a). Письменные процедуры содержат описание действий в отношении перерабатываемых серий?
6.502 §211.115(Ь). Требуется рассмотрение и разрешение отдела контроля качества на переработку любого материала?
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 289
Таблица 6 (продолжение)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
6.503 Испытаниями подтверждается соответствие переработанных серий установленным спецификациям?
6.504 Письменная процедура описывает необходимые действия для переработки возвращенных лекарственных средств (если можно определить, что такие продукты не подвергались ненадлежащему хранению)?
6.505 ООК оценивает переработанные возвращенные продукты и проводит испытания таких материалов для подтверждения соответствия спецификациям до выдачи разрешения на повторный выпуск в обращение?
7.0 Контроль готового продукта
7.1 Верификация готового продукта, хранение и введение в обращение
7.101 § 211.30. В письменных процедурах имеется указание, как и кто подтверждает правильное использование контейнеров и упаковок д ля готового продукта на конечной стадии производства?
7.102 § 211.134(a). Кроме того, письменные процедуры требуют проведения визуальной оценки репрезентативной выборки произведенной продукции для подтверждения правильности маркировки по окончании процесса упаковки?
7.103 § 211.137(a). Дата окончания срока годности штампуется_или печатается на этикетках?
7.104 § 211.137(b). Сроки годности связаны с условиями хранения, указанными на этикетке?
7.105 § 211.142(a). Все готовые продукты хранятся в карантине до окончания отделом контроля качества посерийного проведения испытаний и выдачи разрешения на продажу продукта?
7.106 § 211.142(о). Готовый продукт хранится в надлежащих условиях по температуре, влажности, освещенности и другим параметрам?
7.2 Проверка готового продукта, отбор образцов, проведение испытаний и выпуск в обращение
7.201 § 211.166. Состав каждого продукта был проверен на стабильность в соответствии с письменным протоколом? (Контейнеры должны быть такими же, как используемые для упаковки готового продукта)
7.202 § 211.166. Письменные процедуры по отбору проб и проведению испытаний с установленными нормами имеются для каждого продукта, чтобы обеспечить соответствие готового продукта спецификациям?
7.203 § 211.170(a). Количество отобранных образцов, сохраняемых в качестве архивных, равно по крайней мере двукратному количеству, необходимому для проведения выходного контроля?
7.204 § 211.167(a). Испытания на стерильность и пирогенность выполняются в соответствии с требованиями?
290
Часть 3. Качество
Таблица б (окончание)
Вопрос Инструкции и вопросы (отмечайте любое исключение и комментарии в журнале) Да, нет или не применимо
7.205 §211.167(b).В испытания офтальмологических мазей входят специальные испытания на наличие посторонних включений или твердых частиц?
7.206 § 211.167(c). Продукты с контролируемым или замедленным высвобождением подвергаются определенным испытаниям для подтверждения соответствия спецификации в отношении длительности высвобождения?
7.3 Контроль оттрузки
7.301 § 211.150(a). Письменная процедура определяет порядок складирования, чтобы обеспечить отгрузку в первую очередь продукта, разрешенного к реализации прежде всех других?
7.302 §211.150(a). Отклонения от указанного выше правила документируются?
7.703 § 211.150(a). Письменная процедура указывает действия, выполняемые в случае необходимости произвести отзыв с рынка?
7.304 Политика в отношении отзыва с рынка действующая и адекватная?
7.4 Контроль продаваемой продукции
7.401 Действующее законодательство не содержит как таковых требований по контролю продаваемой продукции, кроме того, что все готовые продукты должны удовлетворять требованиям своих спецификаций
7.5 Программа работы с жалобами и по удовлетворению потребителей
7.501 § 211.198(a). Получаемые в устной или письменной форме жалобы регистрируются письменно и хранятся в соответствующем файле?
7.502 § 211.198(a). Жалобы рассматриваются своевременно отделом контроля качества?
7.503 § 211.198(b)(1). Действие, предпринимаемое по каждой жалобе, документируется?
7.504 § 211.198(b)(3). Решения не проводить расследования жалоб также документируются с указанием ответственного лица?
7.505 § 211.198(b)(2). Расследования жалоб документируются, причем с описанием стадий расследования, наблюдений и последующих действий, если таковые потребуются? По каждой рабочей записи указываются даты?
Литература
l. U.S. Code of Federal Regulations (CFR), Title 21, Part 211, Current good manufactur-
ing practice for finished pharmaceuticals, available: http://www.accessdata.fda.gov/scripts/
cdrh/cfdocs/cfcfr/CRFSearch.cfm?CFRPart=211, accessed Dec. 5,2006.
2. American National Standards Institute (ANSI) (2000), Quality management system — Re-
quirements, ANSI/ISO/ASQ Q9001-2000, ANSI, New York.
3. American National Standards Institute (ANSI) (2000), Quality management system —
Fundamentals and vocabulary, ANSI/ISO/ASQ Q9000 - 2000, ANSI, New York.
Глава 3.2. Значение систем обеспечения качества и аудитов в фармацевтическом производстве 291
4. International Organization for Standardization (ISO), Application of risk management of
medical devices, ISO 14971:2000, ISO, Geneva.
5. U.S. Department of Health and Human services, U.S. Food and Drug Administration, Phar-
maceutical cGMPs for the 21st century—Arisk-based approach, Final Report—Fall 2004,
September2004, available: http://www.fda.gov/cder/gmp/gmp2004/GMP_finalreport2004.
htm, accessed Dec. 5, 2006.
6. U.S. Department of Health and Human Services (DHHS), Food and Drug Administra-
tion (2006, June), Guidance for industry: Q9 Quality risk management, DHHS, Rockville,
MD.
7. U.S. Department of Health and Human Services (DHHS), Food and Drug Administration
(2004, Sept.), Guidance for industry: PAT — A framework for innovative pharmaceutical
development, manufacturing, and quality assurance, DHHS, Rockville, MD.
8. U .S. Department of Health and Human Services (DHHS), Food and Drug Administration
(2006, Sept.), Guidance for industry: Quality systems approach to pharmaceutical CGMP
regulations, DHHS, Rockville, MD.
9 U .S. Code of Federal Regulations (CFR), Title 21, Part 820, Quality system regulation
for medical devices, available: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/
CFRSearch.cfm?CFRPart=820, accessed Dec. 5, 2006.
10. International Organization for Standardization (ISO), (2003), Medical devices — Quality
management systems —Requirements for regulatory purpose, ISO 13485:2003, ISO, Ge-
neva.
11. Juran J. M., and Godfrey, A. B., Eds. (1999), Juran’s Quality Handbook, 5th ed., McGraw-
Hill, New York.
12. FDA compliance policy guide section 130.000, FDA access to results of quality assurance
program audits and inspections (CPG 7151.02), available: http://www.fda.gov/ora/compli-
ance_ref/cpg/cpggenl/cpgl 30-300.html, accessed Dec. 5,2006.
13. International Organization for Standardization (ISO), (2002), Guidelines for quality and/or
environmental management systems auditing, ISO 19011:2002, ISO, Geneva.
14. The Global Harmonization Task Force, SG4, Training requirements for auditors (guide-
lines for regulatory auditing of quality systems of medical device manufacturers — Part 1:
General requirements — Supplement 3), available: http://www.ghtf.org/sg4/inventorysg4/
trainingfi nal.pdf, accessed Dec. 5,2006.
15. FDA compliance program guidance manual for FDA staff: Drug manufacturing inspec-
tions program (7356.002), 2/1/ 2002, available: http://www.fda.gov/cder/dmpq/compli-
ance_guide.htm, accessed Dec. 5,2006.
16. U.S. Department of Health and Human Services (DHHS), Food and Drug Administration
(2004, Sept.), Guidance for industry: Sterile drug products produced by aseptic process-
ing — Current good manufacturing practice, DHHS, Rockville, MD.
17. U.S. Department of Health and Human Services (DHHS), Food and Drug Administration
(2001, Aug.), Guidance for industry: Q7A good manufacturing practice guidance for active
pharmaceutical Ingredients, DHHS, Rockville, MD.
Глава 3.3. Создание системы менеджмента качества
и управление ею
Эдвард Р. Арлинг, Мишель Е. Доулинг и Пол А. Фрэнкел
Amgen, Inc., (Саузенд Оукс, Калифорния)
3.3.1. Введение
Население планеты растет, а ожидаемая средняя продолжительность жизни уве-
личивается. По мере роста численности населения возрастает спрос на фармацев-
тические и биофармацевтические продукты, в частности на новые и специали-
зированные лекарственные препараты для лечения распространенных и редких
заболеваний. От отрасли требуется быстрое обнаружение и вывод на рынок продук-
тов для удовлетворения возникающих новых потребностей здравоохранения и для
противодействия растущим угрозам, так как мутации вирусов приводят к появле-
нию новых заболеваний, угрожающих стабильности известного нам мира.
В то же время глобальный рынок продолжает расширять свои требования к ин-
дустрии. Правительства, потребители и дистрибьюторы усиливают давление на про-
изводителей по снижению цен. И регуляторы, и потребители ожидают соответствия
более высоким стандартам. Растет конкуренция со стороны производителей вос-
произведенных лекарственных препаратов, биоаналогов и фальсифицированной
продукции. Развивающиеся страны с более низкой стоимостью накладных расходов
наращивают экономичные производственные возможности. При этом стоимость
исследований и разработок возрастает.
В данной главе будут рассмотрены концепции, преимущества и практические
действия по разработке всесторонней системы менеджмента качества (СМК), под-
держивающей фармацевтические и биофармацевтические производственные про-
цессы. Представленный материал является универсальным — он применим для ма-
лых и крупных предприятий, для научно-технических и коммерческих компаний.
СМК — это проактивный структурированный подход к обеспечению разработки
и технологических процессов. СМК включает все процессы и показатели, контроль
со стороны руководства и деятельность по постоянному улучшению. Работоспособ-
ность СМК, как описано в этой главе, поддерживается в дальнейшем с помощью
программ управления активными изменениями и годовых планов по качеству для
обеспечения устойчивого развития действующей системы.
Хорошо спланированная СМК со «зрелыми» развитыми процессами предо-
ставляет требуемую инфраструктуру и поддержку, необходимые для эффективных
производственных операций. Интегрированные процессы, управляемые проактив-
но и допускающие быструю модификацию для удовлетворения меняющихся ком-
мерческих и регуляторных требований, будут обеспечивать выполнение текущих
производственных операций и давать конкурентное преимущество. В этой главе
приведены рекомендации по созданию и управлению надежной СМК, которая обе-
спечивает выполнение производственных операций в фармацевтической и биофар-
мацевтической отрасли.
Глава 3.3. Создание системы менеджмента качества и управление ею
3.3.2. Понимание системы менеджмента качества
293
Каждая научно-техническая, испытательная, производственная, упаковочная,
складская или дистрибьюторская структура обладает собственной уникальной ро-
лью при производстве материала или продукта для использования потребителем
где-то в фармацевтической или биофармацевтической товаропроводящей сети.
Каждое предприятие и организация имеют несколько различных критических про-
цессов, которые, функционируя взаимосвязанно, производят желаемый выход.
Выживаемость организации и ее прибыльность напрямую зависят от эффектив-
ности проектирования, выполнения, работоспособности этих процессов, а также
от их взаимосвязанных параметров. В рамках жизненного цикла продукта от изо-
бретения и разработки до масштабирования, клинических исследований, передачи
технологии продукта, регистрации, разрешения на выпуск1, коммерциализации и,
в конечном итоге, прекращения выпуска препарата, надежные процессы являются
основанием успешного предприятия.
Процессы обеспечения производства разделены по своим выходам, но взаимо-
связаны по общему эффекту. Слабые или плохо определенные процессы оказывают
общий снижающий эффект на предприятие и выпускаемый им продукт. Это прояв-
ляется в увеличении переделываемой работы, бракуемых материалах, увеличенной
длительности производственных циклов, задержках разрешений на выпуск, боль-
шом количестве несоответствий, жалоб, отзывов с рынка и других фактов неспо-
собности удовлетворить запросы потребителя или требования рынка. Всесторонняя
СМК может охватывать все процессы, поддерживающие разработку и производство.
Она включает стандарты, методики и процедуры, необходимые для оценки эффек-
тивности и зрелости этих процессов. Она предоставляет показатели эффективно-
сти, необходимые руководству для установления приоритетов на основе управления
рисками и позволяющие сконцентрировать ресурсы для улучшения коммерческой
деятельности и выполнения регуляторных требований.
Для владельцев надежных процессов должны быть установлены функции, от-
ветственность и подотчетность. Владельцы процессов должны быть полностью по-
гружены в свои процессы. Они должны знать возможности своих процессов и отно-
сящиеся к ним ожидания, знать взаимосвязь между своими и другими процессами
и управлять ими как своей «коммерческой деятельностью». Функциональный ме-
неджмент (персонал, управляющий функциями) должен поддерживать владельцев
процессов, а руководство должно понимать и руководить СМК как постоянной
программой, относясь к ней как к интегральной части коммерческой деятельности,
каковой она и является.
СМК — это организационный подход, объединяющий в себе людей, взаимо-
связанные процессы, входы и выходы процессов и программы структурированного
анализа, которые вместе обеспечивают постоянное непрерывное улучшение. Для
процессов такой сложности необходима упорядоченная структура и управление для
достижения эффективного взаимодействия компонентов системы. Чтобы получить
1В США некоторые препараты выпускаются в обращение только после разрешения FDA.
Разрешение выдается на каждую серию. Объем проверок зависит от типа препарата. — При-
меч. перев.
294
Часть 3. Качество
организационные преимущества, ожидаемые от хорошо управляемых процессов,
потребуется структурное подразделение, отвечающее за СМК, и оно должно быть
одним из первых элементов, создаваемых при запуске программы.
СМК и составляющие ее процессы не являются принадлежностью только функ-
ции качества или одной функциональной группы. По своей природе эти процессы
не имеют границ внутри организации. Концепция качества должна быть осознана
и выполняться всем персоналом — от высшего руководства до сотрудников самого
низкого звена. Мышление, связанное с качеством, должно быть свойственно каж-
дому сотруднику, участвующему в разработке, производстве, упаковке, испытаниях,
складском хранении и отгрузке продукта или материала. Культура качества и по-
нимание процесса, в котором участвует сотрудник, крайне важны для продвижения
СМК, чтобы максимально увеличить преимущества организации и оставаться кон-
курентоспособным.
Использование при создании СМК целостного подхода, обеспечивающего вы-
полнение технологических процессов, позволит удовлетворить и превысить ожи-
дания заказчиков, пациентов, акционеров и сотрудников. Для этого необходимо
использование межфункциональных групп, проактивное управление всеми про-
цессами, связанными с производством, включая функциональные связи разра-
ботки, производства, аналитического контроля, инженерных работ и обеспечения
качества. Разработка и поддержание в рабочем состоянии проверенной надежной
СМК требует затрат времени и ресурсов. В некоторых случаях могут уйти годы на
изменения процессов и организационной структуры, прежде чем концепция будет
внедрена полностью и станет приносить выгоду, но это стоит затрачиваемых усилий
и времени. К серьезным вопросам, возникающим в СМК, нельзя относиться как
к одноразовым проблемам. Напротив, действия, предпринимаемые для исправле-
ния неэффективного процесса, должны рассматриваться как долговременные ис-
правления, направленные на устранение основных причин отказов процесса, чтобы
в дальнейшем эти отказы повторно уже не мешали организации.
Окончательная ответственность за надежную функционирующую СМК лежит
на высшем руководстве. Организация следует за лидером, и, следовательно, лидер
должен поддерживать СМК, разработанную специально для организации, следить
за развитием системы и ее вкладом в деятельность организации, непрерывно под-
держивать, направлять и использовать систему. Такая позиция высшего руководства
организации обеспечит жизнеспособность СМК, а СМК, в свою очередь, обеспечит
высшее руководство данными и рекомендациями, необходимыми для эффективно-
го управления.
3.3.2.1. Определение термина «система менеджмента качества»
Термин «система» или «система качества» используется с поразительной несогла-
сованностью повсеместно в фармацевтической и биофармацевтической отрасли
и государственными регуляторами. Даже в рамках одной компании или управления
термины могут иметь расплывчатую интерпретацию и противоречивое использова-
ние. Термин «система» часто используется для описания отдельного процесса или
деятельности подразделения. Иногда этот термин применяется в очень узком значе-
нии — для описания отдельного правила, стандарта или даже отдельной процедуры.
Глава 3.3. Создание системы менеджмента качества и управление ею 295
Недавние инициативы таких международных организаций, как ИСО (Меж-
дународная организация по стандартизации, ISO, International Organization for
Standardization, www.iso.org) и ICH(Международная конференция по гармонизации,
International Conference on Harmonization, www.ich.org), направлены на согласование
и стандартизацию концепции и определений СМК. В 2004 г. Организация по схеме
взаимодействия фармацевтических инспекций (PIC/S, Pharmaceutical Inspection Co-
Operation Scheme, www.picscheme.org) издала рекомендации по требованиям к систе-
мам качества фармацевтических инспекторатов. Федеральное управление США по
контролю за пищевой продукцией и лекарствами (FDA, Food and Drug Administration)
инициировало использование в надзорных проверках подходов, основанных на
СМК организации, что является еще одним источником определений и толкования.
Несмотря на имеющуюся несогласованность в понимании терминов и ожиданий,
все-таки существует прогресс в попытках минимизировать разночтения и достичь
глобальной гармонизации в отношении структуры и терминологии по системам ка-
чества.
В словаре Вебстера система определена как постоянно взаимодействующая или
взаимозависимая группа объектов, образующая единое целое; группа взаимодей-
ствующих тел под воздействием связанных сил; совокупность веществ, которые
находятся в состоянии равновесия или стремятся к нему; группа органов, которые
совместно выполняют одну или более жизненных функций; организация, образую-
щая сеть, особенно для распределения чего-то, или служащая общей цели; органи-
зованный набор доктрин, идей или принципов, обычно предназначенный для объ-
яснения устройства или работы единого целого [1].
Терминология и определения, используемые в этом разделе, подразумевают под
системой менеджмента качества объединение всех процессов, необходимых для
производства, упаковки, испытаний, выпуска и распределения активного фарма-
цевтического ингредиента (АФИ) или лекарственного препарата. Данное опреде-
ление согласуется с программой оценки соответствия 7356.002 Центра по оценке
и исследованиям лекарств FDA (CDER, Center for Drug Evaluation and Research), из-
данной для проведения инспекций фармацевтических и биофармацевтических про-
изводственных предприятий (www.fda.gov/IOM7356.QQ2). Инспекционная програм-
ма CDER разделяет процессы, составляющие СМК, на шесть подсистем: качество,
помещения и оборудование, производство, контроль материалов, лабораторный
контроль, упаковка и маркировка.
В требованиях CDER нет указаний, к какой подсистеме какие процессы относят-
ся; однако можно легко определить, какие процессы будут проверяться в ходе офи-
циальной инспекции, используя приведенную в главе 211 тома 21 CFR (Свод феде-
ральных нормативных актов, Code of Federal Regulations) структуру правил, которые
применяются в производстве лекарственных средств для человека. FDA разделяет
все процессы, составляющие СМК, на шесть подсистем, чтобы обеспечить полно-
ценный и варьированный анализ в ходе проверки (рис. 1). Использование такой же
организационной структуры процессов и такой же терминологии, какую применя-
ют регуляторы, дает предприятию преимущества по более эффективной подготовке
к проверкам и позволяет исключить неверное понимание инспекторов в ходе и по-
сле официальных проверок.
296
Часть 3. Качество
Рис. 1. Взаимосвязь подсистем и функции управления
Таблица 1
Процессы и подсистемы системы менеджмента качества
Качество Аудиты и инспекции Анализ со стороны руководства Управление рисками Организация и персонал Обучение Управление документацией Контроль изменений Несоответствия Корректирующие и предупреждающие действия Отклонения для биологических продуктов Валидация Производство Производственный процесс Мониторинг процессов Мониторинг окружающей среды и производственной одежды Внутрипроизводственный контроль Производственная одежда Помещения и оборудование Конструкция помещений и оборудования Техническое обслуживание оборудования Очистка оборудования Калибровка Контроль материалов Управление качеством поставщиков Отбор проб и проверка Получение, складирование и хранение Управление запасами Возврат продукции и спасенная продукция Лабораторный контроль Лабораторные испытания Управление образцами и планы отбора проб (выборки) Программа по стабильности Упаковка и маркировка Контроль и утверждение маркировки Разработка упаковки
Глава 3.3. Создание системы менеджмента качества и управление ею
297
Установленная CDER структура подсистем позволяет регуляторам и руководству
направить свое внимание на особые функциональные области. В табл. 1 приведен
пример процессов, организованных в соответствующих подсистемах, обеспечиваю-
щих типовую операцию по фасовке АФИ или лекарственного препарата. Эти под-
системы построены в соответствии с требованиями, установленными в правилах,
применяемых инспекторами при проведении инспекций (главы 210, 211 тома 21
CFR), деятельностью подразделений и поддерживающими процессами, необходи-
мыми для производства.
Один подход не может подойти ко всем ситуациям. У каждого предприятия есть
обязательство и свобода создания СМК, удовлетворяющей его собственным потреб-
ностям. Даже производственные комплексы с очень похожими производственными
операциями мохуг нуждаться в разных процессах для обеспечения своей деятель-
ности. Каждая производственная организация должна иметь свой набор процес-
сов, которые будут составлять ее СМК. Группа управления, ответственная за СМК,
должна иметь возможность выявить и обосновать процессы, составляющие систему.
Не существует универсального набора процессов, которые можно было бы приме-
нить ко всем операциям, так как у каждой организации свои уникальные способы
ведения коммерческой деятельности, особенности производства продукта, админи-
стративная структура, культура, а также национальные и глобальные регуляторные
требования и требования потребителей.
Процессы, определенные как часть СМК, мотуг быть организованы в указанные
CDER подсистемы, чтобы обеспечить соответствие с методологией, используемой
при официальных инспекциях. Это также дает руководству возможность определе-
ния сильных областей или возможностей для улучшения в рамках СМК. Регулято-
ры всегда будут концентрировать внимание на процессах, входящих в подсистему
«качество». Другие подсистемы будут проверяться в ходе инспекций в зависимости
от типа инспекций и архивных данных о соответствии предприятия установленным
требованиям. Дополнительную информацию о том, как FDA проводит инспекции,
основанные на концепции системы качества и на структуре подсистем, можно най-
ти на сайте FDA (www.fda.gov) или в статьях по этой теме [2].
Для получения максимального эффекта от СМК ее следует спроектировать мас-
штабируемой и переносимой в рамках предприятия, простой для восприятия и вы-
полнения.
3.3.2.2. Целое и часть целого
С позиций системы целое всегда будет больше, чем сумма его частей. Системы
основаны на взаимодействии нескольких процессов. У отдельного процесса есть
ограниченное значение само по себе, вне зависимости от достигнутого уровня раз-
вития. Процессы вносят вклад в полезность системы через комбинирование с дру-
гими процессами.
В начале XX века исследователи начали находить взаимозависимые связи и ор-
ганизационные структуры среди на вид совершенно отдельных частей. Именно
взаимосвязи позволяют частям функционировать как единое целое. «Единое це-
лое» и есть система. Системное мышление рассматривает части в контексте этого
целого.
298
Часть 3. Качество
Постулаты системного мышления:
• Все в системе связано со всем, имеющимся в системе.
• Части системы функционируют совместно для достижения общей цели всей
системы.
• Помимо непосредственных эффектов действия возникнут еще и другие по-
следствия, которые волнообразно распространятся по системе.
• Каждое изменение несет преимущества и последствия.
• Изменение или усиление структур и взаимосвязей в системе необходимо для
достижения целей системы так же, как и изменение или сохранение частей
системы.
• Системы являются «живыми» структурами, которые поддерживают свою
жизнедеятельность с помощью саморегулируемого динамического равнове-
сия и организованы для реагирования на вносимое извне изменение.
Использование такого видения СМК полезно для высшего руководства органи-
зации и менеджмента, отвечающего за систему. Такое мышление позволяет видеть
перспективу общего воздействия на организацию, производимого индивидуальны-
ми процессами по отдельности, а также то, что может быть получено и укреплено
при активном управлении и взаимодействии этих процессов.
3.3.2.3. Система и процесс
В традиционной производственной парадигме существует Департамент качества,
отвечающий за качество, и Департамент производства, отвечающий за производ-
ство продукта. Вследствие борьбы функциональных приоритетов в этой модели
существует неизбежный конфликт. При встраивании концепций качества и ответ-
ственностей в технологические процессы, отвечающие за производство, качество
распространяется по всей организации. Оба подразделения, таким образом, имеют
общую цель по обеспечению получения высококачественного продукта путем эф-
фективного выполнения своих процессов.
Исторически очень немногие процессы рассматривались как «системы качества»
и соответственно относились к Департаменту качества. Эти «системы» в действи-
тельности были плохо описанными и несвязанными процессами, использовавши-
мися для мониторинга или выявления индивидуальных действий и работ, выпол-
няющихся в ходе производства. Эти системы были основаны на ответственности
типа «контроль качества», то есть на достижении качества продукта путем проведе-
ния многочисленных лабораторных испытаний. Примерами таких процессов были
испытания сырья, полупродуктов и готового продукта, анализ несоответствующих
материалов, мониторинг рабочей среды, выпуск продукта и отгрузка. Некоторые из
них были взаимосвязаны с другими процессами, которые активно управлялись ме-
неджментом или анализировались руководством в отношении эффективности или
соответствия регуляторным требованиями.
Процессы мониторинга контроля качества, описанные выше, если и управля-
лись, были ограничены в возможности обеспечивать улучшение и могли привести
только к действиям, реактивным по своей сути (т. е. осуществляемым только в ответ
на проблему). Интеграция процессов в такой модели низкая или вообще отсутству-
Глава 3.3. Создание системы менеджмента качества и управление ею
299
ет. Ни зрелость и развитость процесса, ни проактивное управление системой при
этом не могут быть достигнуты. В прошлом расширение СМК рассматривалось как
затратный механизм и не считалось относительным вкладом в цепочку создания
ценности продукта. Теперь сознательный менеджмент понимает — благодаря ре-
гуляторным санкциям, штрафам и взысканиям, задержкам разрешений на выпуск,
отзывам с рынка и другим неприятностям — что установление всесторонней СМК
жизненно необходимо для выживания в современной регуляторной среде и сохра-
нения конкурентоспособности в коммерческой среде.
По мере развития принципов и концепций обеспечения качества в конце про-
шлого века СМК становилась все более и более проактивной с включением кон-
троля изменений, проведением внутренних аудитов и аудитов поставщиков, управ-
лением рисками, сбором и анализом запаздывающих и опережающих показателей1
процессов. Анализ репрезентативных показателей стал основой для предупреждаю-
щих действий и программ постоянного улучшения. Современная конкурентная сре-
да вынуждает ведущих производителей и организации мирового уровня применять
проактивное системное мышление, расширять область применения системы на все
процессы, обеспечивающие качество продукта вне зависимости от стадии разработ-
ки или производства. Раннее внедрение соответствующих процессов обеспечивает
выполнение концепций и практики «качество, заложенное при проектировании»
в рамках СМК и гарантирует качество во всех процессах, создает основу для хоро-
ших исследований и постоянного улучшения.
СМК должна состоять из всех процессов, поддерживающих коммерческую дея-
тельность организации, и включать эффективный анализ со стороны менеджмента
показателей эффективности процессов. Менеджменту необходимо знать и пони-
мать эффективность процессов с помощью программ анализа структурированных
показателей эффективности, чтобы предпринимать соответствующие действия,
предоставлять ресурсы и финансы для улучшения СМК. Эта иерархия показана на
Рис. 2. Иерархия системы менеджмента качества
1 Термины «запаздывающие» и «опережающие показатели» используются в концепции
ключевых показателей эффективности (KPI) в системе сбалансированных показателей. За-
паздывающие показатели измеряют результаты в конце установленного временного интер-
вала; их также называют ключевыми индикаторами результатов (KRJ). Пример — количество
серий продукта, произведенных предприятием за день, месяц, квартал и т. д. Опережающие
показатели — это показатели деятельности, для которой надо выявить тенденцию, оценить
«поведение» запаздывающих показателей; их используют для промежуточных процессов и
работ. Также см. раздел З.З.7.1. — Примеч. перев.
300
Часть 3. Качество
Обеспечение процессов и их применимость к фармацевтическому и биофарма-
цевтическому производству легко определяются при изучении потребностей ком-
мерческой деятельности организации и регулирующих их правил. Тщательно спро-
ектированная СМК будет рассматривать потребности компании в целом, а также
операции каждого структурного подразделения, составляющего компанию. Если
конструкция СМК продумана всесторонне, то она будет представлять огромную
ценность для местного и глобального руководства. Она будет помогать сотрудни-
кам путем стандартизации процессов, требований и ожиданий и будет обеспечивать
руководство информативными и сопоставимыми показателями эффективности си-
стемы и процессов. При необходимости модификации процесса можно будет легко
разработать и внести изменения. Единообразное представление процессов регу-
ляторам вызовет у них уверенность в том, что предприятие способно производить
продукт, на который получена регистрация.
3.3.2.4. Преимущества для коммерческой деятельности организации
при установлении надежной системы менеджмента качества
Конкурентная природа фармацевтической коммерческой деятельности требует на-
личия в компании работоспособных и результативных процессов, обеспечивающих
создание, разработку, передачу технологии и масштабирование, промышленное
производство и отгрузку. Выполнение результативных процессов — основа для до-
стижения успеха новыми и действующими компаниями. Это основа эффективного
производства и фундамент, на котором менеджмент и регуляторы могут определять
уровень возможностей компании. Снабжение больных необходимыми лекарствен-
ными средствами своевременно и по рентабельным ценам, без задержек, связанных
с производством или проблемами с соответствием, должно стать основной движу-
щей силой развития фармацевтической и биофармацевтической отрасли.
Высшее руководство может задать вопрос: «Зачем надо внедрять СМК?». Ответ
заключается в том, что хорошо спроектированная система необходима для установ-
ления контролируемого состояния, гарантирующего получение и доступность для
больных высококачественного, безопасного и эффективного препарата. Системы
качества, как указано во вступающем в силу руководстве ICH 010, — это логиче-
ское дополнение к ее предшественникам, ICH 08 «Разработка продукта» и ICH Q)
«Управление рисками» (yvww.ich.org). Эти три руководства встроены друг в друга от
работ по принципу «качество, заложенное при проектировании» до участия в пол-
ном жизненном цикле продукта. При совместном применении достигается макси-
мальная полезность этих руководств для предприятия, обусловленная лучшим по-
ниманием процессов, менее скрупулезным надзором регуляторов и увеличением
свободы действий. Комплексное выполнение требований этих руководств обеспе-
чивает более эффективное управление жизненным циклом продукта от открытия до
разработки и промышленного производства.
Неэффективные операции обходятся бизнесу неисчислимыми количествами
финансовых и людских потерь. Плохо спроектированная система вместе с неэф-
фективными процессами может привести к переделке работ по проектированию
и коммерциализации, проблемам целостности данных, неэффективному исполь-
зованию ресурсов и задержкам при регистрации. Плохо разработанные процессы
Глава 3.3. Создание системы менеджмента качества и управление ею
301
также могут привести к потере будущих прибылей и коммерческих партнеров и соз-
данию негативных регуляторных последствий.
В недавнем исследовании, проведенном Исследовательским проектом в фарма-
цевтическом производстве — совместным предприятием Университета Джорджтау-
на и Университета Вашингтона в бизнес-школах Сент-Луиса, — были собраны дан-
ные по 42 производственным комплексам, принадлежащим 19 компаниям, с целью
определения факторов, влияющих на эффективность отрасли. Окончательный от-
чет по бенчмаркингу содержал оценку эффективности в терминах длительности
производственных циклов, частоты отклонений от промышленных регламентов,
причин отклонений, практических выходов продукции и коэффициентов улучше-
ния для этих показателей.
Исследование определило, что улучшения производственного процесса могут
принести отрасли экономию производственных затрат более чем в 50 млрд долл.
США, что, по мнению исследователей, может привести к снижению цен на лекар-
ственные средства и лучшему финансированию НИОКР. Исследование не было
профинансировано ни отраслью, ни государством [3].
Высшее руководство получает и проблемы, и выгоды, под держивая разработку
надежной СМК. С одной стороны, это отнимает время и ресурсы, необходимые для
проектирования и разработки всесторонней программы. Немедленный возврат этих
инвестиций, как правило, не предвидится. Менеджмент обычно находится под дав-
лением вследствие требований предоставить самые позитивные результаты за ко-
роткое время, что нелогично и не способствует тщательному планированию и раз-
работке на протяжении определенного времени. С другой стороны, проактивная
формализация и под держка надежной СМК позволит в долгосрочной перспективе
обеспечить свободу действий в работе (соответствие обязательным требованиям)
и повысит эффективность коммерческой деятельности.
В фармацевтической и биофармацевтической промышленности восприятие
качества резко изменилось в течение нескольких последних лет, и потеря рыноч-
ной капитализации может прямо коррелировать с этим восприятием. Большие
фармацевтические компании прошли путь от всеобщего восхищения их успехами
и прибылями до потерь значительного процента своей стоимости вследствие ново-
го отношения потребителей, СМИ и инвесторов к вопросам качества и этики. Вы-
ступая недавно на совместной конференции по регуляторным вопросам Ассоциа-
ции производителей парентеральных препаратов (PDA, Parenteral Drug Association)
и управления FDA, Дэниел Диермейер (Daniel Diermeier), профессор по регулятор-
ной и конкурентной практике Северо-Западного университета, отмеченный IBM за
безупречность, заявил: «Восприятие качества в фармацевтической цепочке форми-
рования ценности продукта значит больше, чем в других отраслях (автомобилестро-
ении, производстве мебели и др.). Больные не могут оценить качество лекарствен-
ных препаратов так, как они это делают при покупке машины или выборе комнаты
в гостинице. В здравоохранении предложение ценности1 стоит больше, чем в других
1 Предложение ценности — часть предпринимательской стратегии организации; экономи-
ческая категория, используемая при описании ценообразования продуктов и услуг; это обо-
снование того, что товар, обладающий ценностью для потребителя, должен стоить «опреде-
ленных» денег (соответствовать цене). — Примеч. перее.
302
Часть 3. Качество
отраслях, и система [менеджмента] качества является критичным элементом этого
восприятия» [4]. Д-р Диермейер пошел дальше, предложив включить в СМК про-
цессы для решения и определения дальнейшей защиты предложения ценности пред-
приятия.
Предприятия, не имеющие собственных работоспособных процессов, подвер-
гаются массе негативных воздействий по всей организации. Это справедливо для
процессов, обеспечивающих научные исследования, разработку, производство или
продажи. Недавние примеры свидетельствуют, что стоимость наказаний, нало-
женных регуляторами вследствие неудовлетворительных процессов СМК, увели-
чивается1 (табл. 2). Эти расходы свидетельствуют только о самом взыскании и не
включают потерянную прибыль, стоимость консультаций по исправлению ситуа-
ции, сниженную ценность для акционеров, потерю морального духа и поддержки
персонала. Эти затраты, как правило, составляют столько же или даже больше, чем
наложенное взыскание.
Таблица 2
Возможные финансовые эффекты
Компания Выявленные несоответствия установленным требованиям Тип наказания Стоимость для бизнеса (млн долл. США)
А Невыполнение процедуры. Неудовлетворительное обучение Многочисленные наблюдения по форме 483 <1
В Неудовлетворительное определение процесса, неудовлетворительный контроль и надзор Письмо- предупреждение >1
С Повторные наблюдения: прямой ущерб продукту; невыполнение обязательств по письму- предупреждению Согласованное решение суда >100
D Остановка завода Прямые штрафы. Снятие продукта с рынка >500
Частое заблуждение фармацевтических и особенно небольших биофармацевти-
ческих компаний заключается в том, что кроме промышленного производства не
требуется внедрение СМК в других областях деятельности компании. Небольшие
биотехнологические новые компании (стартапы) также имеют обыкновение откла-
дывать внедрение тщательно спроектированного процесса, пока они не прибли-
зятся к стадии завершения регистрационного процесса, и концентрируют внима-
ние организации вместо этого на регистрации продукта или продаже. Это может
оказаться затратным просчетом, так как скорость продвижения продукта до рынка
1 Следует отметить, что FDA, т. е. регуляторы, имеет право накладывать штрафы в опре-
деленных ситуациях, установленных законодательством. Максимальный размер штрафа —
10 тыс. долл. США. Денежные взыскания по представлению FDA накладывает судья. — При-
меч. перев.
Глава 3.3. Создание системы менеджмента качества и управление ею
303
и ограниченный капитал требуют наличия процессов, обеспечивающих эффектив-
ную разработку, успешные клинические исследования и процесс регистрации, ко-
торые бы выполнялись с минимальными издержками и практически безошибочно.
Хотя обычно СМК связывают с промышленным производством, она критична
для установления параметров процесса и для научных исследований, разработок,
передачи технологии и масштабирования, изучения процесса, аналитической ме-
тодологии и валидации. Разработка продукта выполняется более продуктивно при
применении надежных процессов и в конечном итоге становится основой для на-
дежного производственного процесса. Несостоятельные исследования при разра-
ботке, неадекватные протоколы сравнений, необходимость проведения повтор-
ных клинических исследований или слабо обоснованные аналитические методики
и характеристики технологического процесса приводят к отсроченной подаче ре-
гистрационного досье, которое к тому же оказывается скудным, и неудачам при
инспектировании. Выявление процессов, обеспечивающих эти виды деятельности,
идентификация владельцев процессов, отчетность и поддержка обеспечат успеш-
ную деятельность предприятия и снизят тревожность и неопределенность, которая
неизбежна при разработках и общении с регуляторным агентством.
У фармацевтических и биофармацевтических предприятий существует несколько
возможностей улучшить эффективность и экономические показатели, что в конеч-
ном итоге подтвердит пользу программы и поддержит высшее руководство в дости-
жении своих финансовых целей. Производство лекарственных средств традиционно
находится под жестким государственным регулированием и является очень «анти-
рисковым». Совокупность этих двух условий предлагает бесчисленные возможно-
сти для улучшения нерезультативных и плохо определенных процессов, уточнения
области действия процессов, определения подотчетности и ответственности и ис-
ключения дублирования процессов или пробелов. Выполнение нерезультативных
процессов с непродуманным соблюдением регуляторных требований, или попытки
защитить плохо описанные процессы без адекватных данных и их интерпретации
саморазрушительны для отрасли. Неудовлетворительная расстановка приоритетов
в работе, плохая взаимосвязь процессов, небрежность или вмешательство функцио-
нального менеджмента также могут вносить свой вклад в отсутствие результатив-
ности работы. Персонал нуждается в процессах, которые легко выполнять, которые
хорошо интегрированы и выражаются в результативной деятельности. Этого можно
достичь только с помощью проектирования и выполнения эффективных процес-
сов, взаимосвязанных между собой и приносящих ценность предприятию, владель-
цам процессов и заинтересованным подразделениям.
Примером надежного процесса является проектирование, разработка и выпол-
нение процесса управления несоответствиями. Правила требуют наличия опера-
ционного процесса по выявлению, документированию и исправлению несоответ-
ствий, возникающих в лицензированных фармацевтических производственных
комплексах для зарегистрированных продуктов. Компании тратят значительные че-
ловеческие ресурсы для выявления, документирования и отслеживания несоответ-
ствий. Но как много делается на самом деле для исправления этих несоответствий?
Могут ли несоответствия быть связаны с ранее завершенной разработкой или ис-
следованиями? Связан ли процесс управления несоответствиями с эффективным
304
Часть 3. Качество
процессом для корректирующих или предупреждающих действий (САЛ4, corrective
or preventive action)'! Эффективно взаимосвязаны предупреждающие действия с про-
цессом контроля результативных изменений для обеспечения соответствия предла-
гаемых изменений с регистрационными документами? Хорошо спроектированная
СМК обеспечит наличие поддерживающих процессов и установление функцио-
нальных взаимосвязей. Внедрение системы предусматривает использование целост-
ного подхода, который одновременно обеспечивает и построение эффективных от-
дельных процессов, и взаимосвязи этих процессов, чтобы максимально увеличить
их воздействие на коммерческую деятельность организации, создавая эффективную
и научно обоснованную деятельность.
Постоянное соответствие правилам надлежащей производственной практики
(GMP, good manufacturing practice) жизненно важно для фармацевтических и био-
фармацевтических компаний. Результаты несоответствий выражаются в дорогосто-
ящих наказаниях, потерях прибыли, более высоких накладных расходах, задерж-
ках регистраций и плохом восприятии компании потребителями и регуляторами.
Нарушения правил связаны с неадекватно спроектированной СМК, в которой не
хватает процессов и анализа со стороны руководства, необходимого для поддержки
деятельности предприятия. Процессы, обеспечивающие соответствие, включают
самоинспекции, контроль изменений, пересмотр и согласование документации,
программы обучения персонала. Регулярный анализ этих процессов со стороны ру-
ководства обеспечит выделение ресурсов для соответствующих инициатив, и тогда
при официальных инспекциях не будет никаких сюрпризов. Хорошо спроектиро-
ванная СМК должна предупреждать отрицательные регуляторные последствия.
Результативные и соответствующие требованиям процессы обеспечат «стройную»
производственную деятельность1 с помощью документации и понимания процес-
сов. Анализ этих процессов со стороны руководства предусматривает, что благодаря
информированности и поддержке высшего руководства организация предпримет
соответствующие действия, когда потребуется.
На рис. 3 и 4 показан процесс рецензирования документов и числовые показа-
тели процесса по длительности цикла рецензирования на протяжении двух лет: се-
рьезные задержки на ранних стадиях процесса обусловлены отсутствием владельца
процесса, четкого определения процесса и анализа со стороны руководства. Эта си-
туация представляла риски для соответствия организации требованиям и привела
к низкой эффективности коммерческой деятельности. Улучшение было достигну-
то путем назначения владельца процесса, которые описал и улучшил весь процесс,
разработал информативные числовые показатели процесса и представил эти дан-
ные руководству. Руководство было проинформировано об эффективности процес-
са, осознало риски для соответствия и предприняло соответствующие действия по
направлению усилий персонала на выполнение требований процесса. Результатами
стали улучшенный цикл согласования документов, проактивное соответствие вну-
тренним процедурам и регуляторным требованиями и удовлетворение от понима-
ния, что для достижения лучших результатов коммерческой деятельности и соот-
ветствия регуляторным требованиям не требуется дополнительных усилий.
1 О «стройных» производственных системах см. главу 3.1. — Примеч. перев.
Глава 3.3. Создание системы менеджмента качества и управление ею
305
кв.1/05 кв.2/05 кв.3/05 кв.4/05 кв.1/06 кв.2/06 кв.3/06 кв.4/06
Рис. 3. Процесс рецензирования документов в течение двух лет
—Фактическое значение
---Целевое значение
Рис. 4. Длительность цикла рецензирования документов
3.3.2.5. Ожидания промышленности и регуляторных агентств
Хотя в действующих правилах FDA, применяемых для фармацевтических и био-
фармацевтических производств, отсутствуют требования к «системе качества», ре-
гуляторные агентства и отраслевые торговые организации постепенно признают
важность надежно функционирующих систем качества при обеспечении мирового
производства медицинских продуктов. FDA признает, что все принципы качества
приведены в действующих правилах СМРцпя лекарственных средства (CFR, том 21,
глава 211), которые пересматривались последний раз в 1978 г.
Вопросы системы менеджмента качества и их связь с управлением рисками часто
обсуждаются на коммерческих и регуляторных семинарах и конференциях. Недав-
но изданные руководства FDA, такие как «Применение концепции систем качества
к действующим правилам надлежащей производственной практики», имеющееся
на сайте FDA, и часть инициативы FDA, называемой «GMPдля XXI века», были на-
писаны для дополнения существующих правил. Хотя руководство FDA может из-
306
Часть 3. Качество
меняться или даже стать избыточным с введением в действие руководства ICH Q10,
имеется единое намерение отрасли и государства содействовать внедрению СМК.
Согласно Джо Фамуларе (Joe Famulare), директору DMPQ, FDA, «FDA хотела опи-
сать всестороннюю модель системы качества, которая бы поддерживала и корре-
лировала с правилами cGMP. Руководство согласуется с определением контроли-
руемого состояния [производства], способствует работам по качеству, контролю
изменений, внедрению концепции „качество, заложенное при проектировании"
и управлению рисками» [4].
При обсуждении систем качества на недавно прошедшей отраслевой конфе-
ренции по GMP Крис Джонекис (Chris Joneckis) из CBER FDA (Центр по экспер-
тизе и исследованиям биологических препаратов, Center for Biological Evaluation and
Research) сказал следующее: «Надежная система менеджмента качества создает на-
дежную основу для качественного продукта. Это выигрыш для больного, отрасли
и регуляторов Она приносит пользу передаче технологий, контролю процессов,
мониторингу, возможностям производства, улучшает производство, уменьшает ве-
роятность несоответствий и повышает качество исследований. Регуляторные пре-
имущества включают улучшенную экспертизу по химическим, производственным
и контрольным аспектам, контроль изменений и подачу пострегистрационных из-
менений» [5].
Регуляторные и отраслевые рекомендации были разработаны в поддержку раз-
работки и организации систем качества. В конце 90-х годов прошлого века инспек-
ционный подход, основанный на системах, был официально введен Центром по
изделиям и радиологической безопасности (CDRH, Center for Devices & Radiological
Health) FDA [6]. Эти правила получили название QSR (Quality Systems Regulations),
правила систем качества, и были включены в главу 820 CFR.
CDER и CBER вскоре приняли подходы CDRH и издали собственные докумен-
ты — руководства по программе оценки соответствия 7356.002 [7] и 7345.848 [8]
соответственно, взяв за основу правила систем качества CDRH. CDER и CBER от-
вечают за обеспечение инспекций фармацевтических и биофармацевтических про-
изводственных комплексов, проходящих каждые два года. Указанные руководства
используются инспекторами при проведении проверок. Владельцы процессов и за-
интересованные подразделения, а также менеджмент и высшее руководство должны
быть знакомы с этими руководствами по оценке соответствия и планами инспекто-
ров по их использованию при проверках.
Действующая инспекторская надзорная практика FDA, основанная на описан-
ных выше моделях, требует от инспекторов оценивания процессов в рамках под-
систем, определенных в правилах систем качества, для определения соответствия
установленным требованиям и рисков для безопасности больных. Имеется не-
значительное в начале, но существенное в итоге преимущество для регуляторных
агентств и регулируемых компаний при использовании системного подхода, так
как при этом инспекции становятся более короткими и охватывают разные типы
продуктов в рамках одной инспекции. Компании с хорошей историей соответствия
требованиям могут получить преимущества при плановых инспекциях, в то время
как компании с проблемами в прошлом будут подвергнуты более пристальному
изучению и, возможно, регуляторным действиям.
Глава 3.3. Создание системы менеджмента качества и управление ею 307
Инициатива промышленных организаций, таких как ИСО, старающейся обе-
спечить многие отрасли признанными стандартами, также получила свое отраже-
ние в системном подходе с публикацией и сертификацией стандартов ИСО серии
9000 и позднее ИСО 2000:9004 (www.iso.org), основанном на создании СМК и по-
стоянном улучшении после стабилизации процессов.
ICH, совместная инициатива промышленности и регуляторных органов по меж-
дународной гармонизации разработки и регистрации лекарственных средств, также
признает ценность и значимость систем качества, разработав свое руководство по
данной теме (ICH 010). Фармацевтическая и биофармацевтическая промышлен-
ность и регуляторные агентства активно сотрудничают, чтобы завершить разработку
этого документа приблизительно в 2008 г. ICH 010 посвящено фармацевтическим
продуктам и предназначено для дополнения требований GMP с учетом концепции
систем качества. Оно будет применимо к фармацевтическим субстанциям и лекар-
ственным препаратам, продуктам из больших и малых молекул и обеспечит единый
подход к системам качества. Оно также будет комплементарно руководствам ICH
08 и ICH Q). ICH 010 содержит фармацевтические аспекты и при этом обращает
особое внимание на всесторонний подход; описанные ключевые элементы — реаги-
рование руководства и постоянное улучшение. Несколько руководств /СЯуже при-
няты регуляторными агентствами, в частности ICH Qla для производства АФИ. Как
только данные руководства принимаются, они становятся основой для ожиданий
регуляторных агентств и инспекций.
3.3.3. Руководство и персонал: лидерство и поддержка
Все производственные операции в какой-то степени оперируют элементами и ком-
понентами СМК. Эти элементы и процессы могут быть не выделены и не управ-
ляться, так как они являются неотъемлемой частью более крупной системы, и могут
быть изначально реактивными по своей природе. Значительное время и ресурсы
требуются для изменений культуры и практики организации, чтобы перенести дей-
ствующие элементы из фрагментированной реактивной программы в заданную
структуру, которая управляется проактивно. Степень, до которой программа управ-
ляется проактивно и поддерживается высшим руководством, напрямую зависит от
преимуществ, которые получает организация.
Для успешного внедрения программы СМК требуется три различных уровня под-
держки: высшим исполнительным руководством, функциональным менеджментом
и операционным персоналом. Все три уровня организации должны прикладывать
усилия для достижения успеха. Программа, обеспечивающая понимание каждым
сотрудником системы в целом и получаемой от нее пользы, должна стать одной из
приоритетных задач. Мотивация персонала и руководства с помощью объяснения
преимуществ и результатов коммерческой деятельности крайне важна для поддер-
жания постоянного функционирования программы.
Высшее руководство нуждается в способных и преданных сотрудниках для про-
ектирования и поддержания динамической программы СМК. Высшее руководство
должно запустить эту программу и осуществлять ее поддержку в рамках организа-
ции. Функциональный менеджмент должен понимать программу, чтобы поддер-
308
Часть 3. Качество
живать ее и руководить своим персоналом по ее выполнению. Персонал должен
понимать значение для него этой программы, испытывать ее и видеть пользу от ее
применения, чтобы осознанно поддерживать ее.
Подразделение по качеству должно рассматриваться как партнер в обеспечении
качества продукта, а не как управление, которое распространяет качество. В рамках
СМК владельцем определенных процессов является функция качества, так же как
производство, инженерная служба, разработки, техническая поддержка и помеще-
ния владеют процессами в рамках системы. Все функциональные группы должны
иметь заданные функции и ответственность для обеспечения качества производи-
мого продукта. Межфункциональная поддержка и установление ответственности
гарантирует, что качество встроено в каждый процесс, а владелец каждого процес-
са в конечном итоге отвечает за выход своего процесса. Высшее руководство, по-
нимающее и использующее эту концепцию, будет поддерживать и распространять
культуру качества в организации, максимально повышая вероятность успеха и кон-
курентные преимущества.
Высшее руководство организации, менеджмент, персонал и группа по программе
СМК должны работать согласованно, чтобы разработать и развивать СМК. Успеш-
ная программа должна подробно описать ожидаемую пользу для всех заинтересо-
ванных сторон в организации и предоставлять текущие результаты, отражающие
функционирование и использование системы.
3.3.3.1. Описание выгод для организации
Установление формализованной структурированной СМК в организации требу-
ет одобрения высшего руководства, ресурсов и капитала. Поддержка и одобрение
высшего руководства — это точка инициирования программы, гарантирующая под-
держку всем усилиям по выполнению программы и соответствие предлагаемой си-
стемы потребностям коммерческой деятельности. Это включает наличие выделен-
ных ресурсов, которые будут направлены на разработку и управление программой,
исполнение и управление процессами.
Высшее руководство имеет представление о потребностях бизнеса, состоянии
бюджета, видение и понимание будущего организации. Для удовлетворения суще-
ствующих и будущих потребностей проект СМК должен быть надежным и результа-
тивным. Анализ пробелов в имеющихся бизнес-процессах может помочь высшему
руководству понять, где имеются возможности для улучшения процессов. Эти про-
белы могут быть определены путем анализа целей организации и ее способности
получать качественные результаты вовремя и в рамках бюджета.
Изучение производственных зон на предмет улучшения операций включает
рассмотрение соответствия регуляторным требованиям, наблюдений при аудите,
переработок, несоответствий, изменений в документах, нарушения сроков постав-
ки, полученных жалоб, имеющихся запасов, отказов оборудования, длительности
производственного цикла, текучести кадров и возможностей обучения. Допол-
нительные зоны, предназначенные для улучшения, могут появиться в результате
бенчмаркинга при сравнении основных производственных параметров с лучши-
ми показателями в отрасли. Результаты анализа пробелов начинают обсуждение
эффективности процесса и необходимости его улучшения. Высшее руководство
Глава 3.3. Создание системы менеджмента качества и управление ею
309
должно быть убеждено в возможности получения финансовых и конкурентных
преимуществ, а получаемые от программы выгоды должны превосходить объем ин-
вестируемых ресурсов для оперирования СМК.
Высшее руководство организации должно понимать и поддерживать стратегию
систем качества и руководить внедрением такой системы на предприятии. Как пра-
вило, при этом требуется трансформация коммерческой деятельности до опреде-
ленного уровня, изменение культуры и поведения организации и принятие опре-
деленного уровня риска. Риск, связанный с внедрением изменений, минимален
в сравнении с риском отсутствия надежной системы, как было описано в этом разделе.
3.3.3.2. Общение на языке менеджмента
Без руководящего наблюдения высшего руководства по внедрению систем каче-
ства менеджмент среднего звена не будет поддерживать усилия, выделять требуемое
время, корректировать культуру производства, которые необходимы для внедрения
и поддержания процессов в рабочем состояния. Высшее руководство должно быть
осведомлено обо всех эффектах и последствиях, которые возникнут, если система
не будет внедрена, и четко и последовательно показывать поддержку программы пу-
тем частых последовательных сообщений об этом менеджменту и персоналу. Также
необходимо показывать полученные материальные и нематериальные выгоды выс-
шему руководству для убеждения его в правильности и результативности проводи-
мых работ, и регулярно сообщать результаты персоналу.
Материальные выгоды должны включать показатели эффективности и улучше-
ния, демонстрирующие уменьшение себестоимости процессов и системы, соответ-
ствие при инспекциях и аудитах со стороны потребителей, более быструю регистра-
цию продуктов и сокращение длительности производственных циклов, снижение
количества бракованных материалов, уменьшение проблем несоответствий, более
результативное постоянное улучшение и внедрение проекта. К нематериальным вы-
годам относятся улучшение коллективного духа, более быстрое и точное принятие
прозрачных решений, меньшая текучка кадров, повышенная отчетность персона-
ла и культура качества в организации. «Ощущения», пронизывающие организацию
с реактивным управлением и «в состоянии постоянного стресса» без хорошо спро-
ектированных процессов, существенно отличаются от проактивной организации
с простыми процессами, которые легко и эффективно выполняются обученным
персоналом.
Системный подход позволяет выполнять принятие решений и управление про-
цессами на уровне владельцев процессов, а не на уровне функционального управле-
ния (т. е. управления функцией). Для многих организаций это серьезный культурный
сдвиг, который, однако, имеет много преимуществ. Более быстрое принятие реше-
ний специалистами по конкретным вопросам — это ценное качество для организа-
ций. Это может принести пользу как для повседневной работы, так и предоставить
длительные стратегические преимущества для высшего руководства. Избавление
от излишней нагрузки функционального менеджмента и определение ответствен-
ности владельцев процессов позволяет функциональному менеджменту управлять
ресурсами, решать вопросы персонала и не разрывать свое внимание и время между
ресурсами, персоналом, процессами и техническими вопросами.
310
Часть 3. Качество
3.3.3.3. Объяснение выгод для персонала
Так же, как необходимо убедить высшее руководство и менеджмент в получаемых
преимуществах при внедрении и функционировании СМК и обеспечить понима-
ние СМК, для принятия культурных изменений необходимо добиться понимания
со стороны персонала, что принесет в организацию системный подход. Как только
программа была инициирована, материальные и нематериальные выгоды должны
быть определены и приняты персоналом, чтобы сотрудники постоянно поддержи-
вали программу. Поддержка сотрудников, получаемая с помощью осознания вы-
год и управления со стороны менеджмента, обеспечит выполнение программы, что
в конечном итоге даст ожидаемые коммерческие результаты.
Превращение разномастных процессов в процессы, которые просты для пони-
мания, легки в исполнении и дают чувство завершенности, — одно из обязательств
менеджмента перед сотрудниками. Интересы сотрудников заключаются в возмож-
ностях успешного выполнения своей работы, участии в постоянном улучшении
и достижении разумного баланса между работой и жизнью в свободное от работы
время. Наконец, сотрудники хотят должны иметь возможность карьерного роста
Хорошо спроектированная СМК может помочь в предоставлении этих возможно-
стей для сотрудников.
Выгоды для персонала должны быть учтены при проектировании СМК. Опи-
сание ожидаемых выгод должно быть представлено сотрудникам, чтобы получить
их поддержку создаваемой системы. Достижения должны рекламироваться и воз-
награждаться. Внедренные хорошо спроектированные процессы обеспечивают во-
влечение и участие сотрудников, их вклад в деятельность организации. Это укре-
пляет культуру качества в организации и открывает пути участия персонала в этой
работе.
3.3.3.4. Обеспечение поддержки персонала и лидерство руководства
В ответственность менеджмента входит предоставление персоналу надежных ин-
струментов и процессов для эффективного выполнения поручаемой работы. Слож-
ные, отсутствующие и фрагментированные процессы не позволяют сотрудникам
выполнять требуемые действия с легкостью и в приемлемые сроки и могут привести
к производству продукта низкого качества или его переделке. Этот тип рабочей сре-
ды быстро перестает удовлетворять сотрудников и приводит к «низкому» коллек-
тивному духу, низкой результативности и, в конечном итоге, потере интереса к ра-
боте и увольнению сотрудников.
Расширение полномочий сотрудников приводит к появлению гордости за вы-
полняемую работу. Хорошо спроектированные системы четко показывают сотруд-
никам, кто имеет право принимать решения и ответственен за процесс, содержат
четкие требования к процессу и владельцам процесса и предоставляют персоналу
четкий путь развития.
Точно идентифицированные атрибуты процессов дают организациям больше,
чем «знание племени», которое передается владельцу следующего процесса. Си-
стемы предоставляют четкую структуру, процессы и другие атрибуты, критичные
для постоянного успеха производства. Организация начинает больше полагаться на
Глава 3.3. Создание системы менеджмента качества и управление ею
311
свою систему и процессы, а не на знания людей, которые могут быть потеряны со
сменой кадров.
Для обеспечения поддержки со стороны высшего руководства и сотрудников
требуется хорошо разработанный план, который доводится до сведения персонала.
Перспективный план, разработанный на несколько лет, может помочь организации
сохранять перспективы и управлять ожиданиями. Годовой план по качеству должен
охватывать все аспекты СМК и содержать подробные цели и задачи на этот период.
Прогресс выполнения плана по качеству должен отмечаться и пропагандироваться.
Информирование о наградах и достижениях в небольших группах и больших коллек-
тивах должно быть включено в программу коммуникаций и управления изменениями.
Таблица 3
Перспективное стратегическое видение
1-й год 2-й год З-й год 4-й год 5-й год
Получить Внедрить программу Включить Сконцентрировать Адаптироваться
поддержку менеджмента Обучить менеджмент, владельцев процесса, отдел обеспечения качества и поддерживающий остающиеся процессы внимание на ключевых к меняющейся коммерческой
Создать подразделение по СМК Определить на местах процессы и ресурсы Разработать план коммуникаций и управление изменениями в программу процессах, среде
Документировать и распространять информацию по сокращению опирающихся на документы по СМК и анализ со стороны руководства Обеспечить текущее обучение, получение информации и управление изменениями и регуляторным требованиям Обеспечить лидерство
персонал Сконцентрировать внимание на зрелости процессов с высоким риском или влиянием Выявлять затрат и экономии ресурсов Приступить к интеграции процессов в организации в отрасли на основе парадигмы СМК
и награждать за работы в СМК
В табл. 3 представлен пример перспективного видения и цели для СМК. Пер-
спективная долговременная стратегия дает высшему руководству, менеджменту
и персоналу понимание программы и ожидаемые временные рамки для внедрения
системы и ожидания выгод. Годовые планы по качеству представляют собой описа-
ние краткосрочных стратегических этапов, необходимых для достижения перспек-
тивного стратегического видения.
3.3.3.5. Ловушки, которых следует избегать
При внедрении формализованной СМК возникает несколько сложных вопросов
и требований. Основное требование — наличие опытной группы, которая пони-
мает потребности организации, требования государства и потребителя. У группы
должны быть навыки, опыт и экспертные знания, чтобы спроектировать надежную
систему, выявить процессы, которые обеспечивают деятельность предприятия. Не-
312
Часть 3. Качество
совпадение опыта группы с потребностями предприятия может привести к полу-
чению нежизнеспособной системы, которая не поддерживается высшим руковод-
ством и персоналом, что приведет к ошибкам системы и со временем к отказу по ее
использованию.
Проект СМК должен быть тщательно продуман и протестирован. Пилотные
программы являются критичными для надежности и устойчивости тестируемой си-
стемы и принятия системы менеджментом, а также для способности производить
желаемые результаты. Время, затраченное на проектирование системы, будет опла-
чено сторицей в последующие годы, увеличит ее под держку персоналом и обеспе-
чит требуемое количество персонала и ресурсов предприятия, участвующих в работе
программы. Следует избегать внедрения любой системы или процесса, проект ко-
торых не был тщательно проработан, не имеет входов от заинтересованных сторон,
использующих систему, или которые не были апробированы до полномасштабного
внедрения. Как правило, можно осуществить только одну попытку внедрить новую
программу до того, как персонал и менеджмент или примут, или отвергнут ее идеи
и концепцию. Изменять сложившееся мнение и восстанавливать доверие к неудач-
ной системе сложно. Очень важно принимать соответствующие меры по правиль-
ному внедрению системы в самом начале этой деятельности.
Управление изменениями — другой очень важный вопрос при внедрении СМК,
так как от организации требуется изменение культуры. Некоторые ресурсы могут по-
мочь в управлении изменениями, и они должны быть включены в проект системы.
Важно понимать, что для успешного внедрения требуются изменения на всех трех
уровнях организации: высшего руководства, функционального менеджмента и ис-
полнительного персонала. На каждом из уровней необходимы различные сообще-
ния, поддержка, вознаграждения и выгоды. Следует обсудить, как ознакомить всех
заинтересованных сотрудников с материальными и нематериальными выгодами.
Требуется поддержка высшего руководства наиболее высокого уровня организа-
ции. Менеджмент среднего звена не будет выполнять работы, которые не под дер-
живаются их руководителями. Высшее руководство должно оказать твердую под-
держку, не посылать смешанных сигналов, продолжать рекламировать и отмечать
успехи программы, оказывать поддержку в сложное время. Совпадение слов и дел
менеджмента поддерживает понимание и приемлемый риск, предпринимаемый ме-
неджментом и персоналом.
Функциональный менеджмент также должен поддерживать работу по СМК
и долговременные стратегии, чтобы сотрудники, ключевые для выполнения про-
цессов, знали, что от них ожидается поддержка и работа. Функциональный менед-
жмент должен посылать значимые сигналы своим сотрудникам о том, что их под-
держка долговременного плана и ежегодных планов по качеству крайне важны.
Конкретные цели системы, включенные в цели высшего руководства, менеджмента
среднего звена и исполнительного персонала, укрепляют приверженность системе
и помогают добиться успешного выполнения программы.
Потребности системы должны быть гибкими. Наличие долговременного плана
и перспективное видение необходимы для формирования плана действий на буду-
щее. Этот план может пересматриваться, так как вследствие изменений коммерче-
ской среды предприятие будет нуждаться в корректировках. Долговременный план
Глава 3.3. Создание системы менеджмента качества и управление ею
313
и видение следует писать с таким уровнем детализации, который обеспечит лишь
незначительные изменения, а требуемая гибкость будет достигаться путем подго-
товки годовых планов по качеству, соответствующих потребностям текущей ком-
мерческой деятельности.
До внедрения какой-либо инициативы СМК следует четко определить требо-
вания высшего руководства, менеджмента, персонала и потребителей. При про-
ектировании системы крайне важно определить, какие процессы необходимы для
обеспечения потребностей потребителей, и влияние этих процессов друг на друга.
Для достижения успеха жизненно необходимо развитие владельцев процессов, ко-
торые понимают свои функции, и устранение ограничений, мешающих им решать
поставленные задачи. Владельцы процессов должны понимать продукт и приорите-
ты процессов, чтобы можно было получать существенные выгоды от системы. Эти
важные моменты при проектировании системы и процессов, поддерживающих дея-
тельность организации, являются информативными показателями эффективности,
свидетельствующими о прогрессе работ. Рассмотрение этих вопросов предотвраща-
ет неудачи с системными инициативами и изменяет мнение о системе как о допол-
нительной нагрузке для производства, которое и без того перегружено и как об из-
лишнем усложнении требований внутри организации.
3.3.4. Установление области применения системы
менеджмента качества
Во многих фармацевтических и биофармацевтических производственных опера-
циях имеется дублирование одних процессов и пробелы в других. Часто не ясны
точные границы или область применения процесса, ожидаемые продукты про-
цесса (выходы), нет четких указаний, кто является потребителем процесса, кто —
владельцем, а кто несет ответственность за постоянное улучшение. Дублирование
нерезультативно и затратно. Примером этого является наличие разных слоев орга-
низации, осуществляющих анализ данных в виде документации или информации,
движущейся по цепочке создания ценности. Предоставление данных по валидации
аналитических методик в регистрационном досье — конкретный пример дублиро-
вания. Первичные данные могут проверяться в лаборатории, руководителем под-
разделения, подразделением по обеспечению качества, группами по соответствию
и по взаимодействию с регуляторными органами. С другой стороны, в процессе
могут иметься проблемы, когда каждая функциональная группа, перечисленная
выше, предполагает, что проверка данных осуществляется другой группой, и в итоге
возникают пробелы в целостности и правильности данных. В этом случае результат
может быть невероятно дорогостоящим, если при официальной инспекции будут
обнаружены ошибки и окажется, что проблемы правильности данных имеются во
всем досье.
В этом разделе будет рассмотрена важность определения требований коммер-
ческой деятельности, чтобы обеспечить, что спроектированные процессы, состав-
ляющие СМК, поддерживают работу предприятия, встроены в планы по качеству,
описаны достаточно детально, допускают перенос и масштабирование на уровень
предприятия.
314 Часть 3. Качество
3.3.4.1. Определение требований коммерческой деятельности
СМК и составляющие ее процессы должны быть спроектированы специально для
потребностей коммерческой деятельности. Универсальный набор не может быть
пригоден для всех ситуаций. Требования предприятия могут быть различными для
разных площадок и разных фаз жизненного цикла продукта. Всесторонняя система
будет обеспечивать целостный программный подход в своей поддержке деятельно-
сти предприятия. Но это не означает, что в каждой фазе жизненного цикла продук-
та (исследования, разработка, промышленное производство) будут использоваться
все процессы, составляющие систему. Также не требуется, чтобы на всех произ-
водственных площадках обязательно были внедрены все процессы. Тем не менее
система обеспечит единую платформу и ожидания для всех процессов, владельцев,
программ анализа данных, работ по постоянному улучшению и др., когда все они
будут внедрены.
На первом этапе проектирования СМК определяют потребности коммерческой
деятельности и процессы, необходимые для обеспечения деятельности предпри-
ятия. Особое внимание следует обратить на то, чтобы все процессы оценивались.
Оценка должна охватывать все виды деятельности, которые могут влиять на каче-
ство продукта: высшего руководства, сотрудников производственных площадок,
дистрибьюторов, субподрядчиков или совместных предприятий. Процессы, контро-
лирующие поступающие от поставщиков материалы, лабораторные работы, работы
по договору и другие входы, также должны быть включены в первичную оценку.
На следующем этапе, после выявления процессов, требуемых для обеспечения
деятельности предприятия, должно быть четко определено, что находится в области
применения процесса, а что не включено в эту область. Создание карт всех процес-
сов и их взаимоотношений с другими процессами поможет определить, имеются
ли в системе дублирования или пробелы. Пробелы между процессами должны быть
заполнены. Например, процесс работы с несоответствиями должен иметь прямую
связь с процессами корректирующих действий. Эффективно функционирующий
процесс работы с несоответствиями без активного взаимосвязанного процесса кор-
ректирующих и предупреждающих действий не обеспечит получение существенной
пользы для организации, и усилия, затраченные на выполнение процесса работы
с несоответствиями, будут формальными в отношении их положительного влияния
на коммерческую деятельность предприятия.
Такой всесторонний подход позволяет обеспечить эффективную интеграцию
процессов, различных фаз жизненного цикла продукта и различных площадок в то-
варопроводящей сети. Эта интеграция делает более эффективной связь владельцев
процессов друг с другом посредством своих потребностей и ожиданий. Дублирова-
ние работ исключается, и результативность повышается. Качественные продукты
одного процесса становятся надежными входами в следующем процессе. Менед-
жмент и высшее руководство будут иметь доступ и информацию по соответствию
всех процессов требованиям, по инфраструктуре процессов и показателям эффек-
тивности на сопоставимой основе. Такой подход позволяет высшему руководству
производить распределение ресурсов на основе анализа рисков по соответствую-
щим областям предприятия.
Глава 3.3. Создание системы менеджмента качества и управление ею 315
Создание карт процессов для удовлетворения требований предприятия по по-
ставке продукта позволяет проектировать процессы, требуемые в системе. Участие
персонала, менеджмента и высшего руководства в формулировании потребностей
коммерческой деятельности дает дополнительную информацию о параметрах про-
цессов.
3.3.4.2. Интеграция системы менеджмента качества в планы качества
Требование наличия плана по качеству имеется в правилах, регулирующих меди-
цинские изделия (QSR), но такие планы могут быть легко использованы в каче-
стве полезного инструмента в фармацевтических и биофармацевтических произ-
водственных операциях. План по качеству представляет собой письменный план
и цели по укреплению и развитию СМК. В него можно включить принципы, требо-
вания и цели организации, миссию, продукт и бизнес-практику, использующиеся
для производства качественного продукта. План по качеству может детализировать
процессы, составляющие СМК, уровень зрелости, необходимый для каждого про-
цесса, организационную структуру и другие требования, необходимые для дости-
жения цели организации. В план по качеству включают элементы коммерческой
деятельности, такие как месторасположение, объемы, продукты и другие показа-
тели. Он также содержит структуру, вспомогательные функции, ценности и другие
характеристики организации.
Ежегодный план по качеству может быть детализированным планом перспек-
тивного видения СМК организации. Он указывает персоналу и менеджменту огра-
ничения и ставит цели по улучшению СМК. Он позволяет сотрудникам увидеть всю
ситуацию целиком и узнать, насколько они подходят организации и ее ожиданиям.
В рамках плана по качеству должны быть описаны характеристики СМК, включая
ответственность функционального менеджмента. Затем план становится основани-
ем для дальнейшего определения процесса, для анализа деятельности менеджмента
и его ответственности и для программ постоянного улучшения. Подготовка плана
по качеству начинается с определения того, что предполагается знать на каждом
из трех уровней организации. Это механизм обеспечения выполнения требований
и исключения пробелов в организации.
План по качеству может описывать долгосрочные (на несколько лет) и кратко-
срочные цели организации по улучшению качества продукта на основании управ-
ления рисками. Это основа для производственной структуры и поддерживающих
процессов. План по качеству предусматривает интеграцию сотрудников, их квали-
фикацию, требования к продукту, СМК и инфраструктуру по соответствию регу-
ляторным и иным требованиям. Пример основных положений плана по качеству
приведен в табл. 4. Следует провести анализ со стороны руководства и утверждение
плана по качеству с целью обеспечения согласованности плана с глобальными це-
лями организации, учета области применения, ожиданий и разделения труда в ор-
ганизации.
В более крупных организациях могут разрабатываться планы по качеству пло-
щадок или подразделений, чтобы обеспечить масштабирование СМК для всех эле-
ментов предприятия. Планы отдельных площадок позволяют сконцентрировать
316
Часть 3. Качество
внимание на процессах, которые для этой площадки более важны, чем для других,
вследствие различий в коммерческой среде и установленных требований. Хотя от-
дельные планы акцентируют внимание на целях, основанных на приоритетных за-
дачах площадки, они также связывают членов организации с миссией более круп-
ной СМК, как показано на рис. 5.
Элементы ежегодного плана СМК
Таблица 4
Элемент Определение
Введение План Цели Проекты Показатели эффективности Одобрение Цель плана и определения для однозначного понимания Запланированная деятельность на календарный год Специфичные и многоуровневые цели площадки Основные проекты для поддержки целей Ключевые показатели эффективности с установленными целевыми значениями Менеджмент площадки или завода
Рис. 5. Масштабирование СМК в планах по качеству площадок
3.3.4.3. Определение требований по декомпозиции процесса
Высшее руководство ожидает экономически эффективных достоверных результатов
от своих производственных операций. Менеджменту нужны квалифицированные
кадры, оборудование, производственные помещения и материалы для производства
продукта. Сотрудникам требуются надежные процессы, которые они могут легко
Глава 3.3. Создание системы менеджмента качества и управление ею 317
осуществлять для выполнения своих обязанностей. Все это необходимо учитывать
при проектировании процессов, составляющих СМК.
Возможно, потребуется управление сложными процессами как раздельными
подпроцессами, чтобы предоставить возможность владельцам процессов осущест-
влять свою работу с установленными четкостью и квалификацией. Менеджменту
и высшему руководству могут требоваться данные и показатели эффективности по
определенным областям процесса, которые не будут доступны, если процесс слиш-
ком сложный и большой. Декомпозиция процесса на более простые, более управ-
ляемые процессы также позволяет выполнять масштабирование и перенос внутри
организации.
После того как необходимость процесса для организации установлена, следует
определить допустимую сложность процесса. Для этого оценивают способность
владельца процесса управлять им и исполнять требования процесса. Другими фак-
торами в этом определении являются данные и показатели эффективности, не-
обходимые менеджменту и руководству по этому процессу. Примером сложного
процесса, который приносит выгоды организации при управлении раздельными
подпроцессами, является валидация.
Валидация — регуляторное требование и отраслевой стандарт по получению
гарантий в том, что продукт постоянно и целиком отвечает показателям качества
и регуляторным требованиям. Валидационные требования пронизывают всю произ-
водственную цепочку, охватывая многие различные подпроцессы. Валидационный
процесс управляется наилучшим образом при декомпозиции его на управляемые
подпроцессы. Это обеспечивает эффективное управление и исполнение подпро-
цессов, а показатели эффективности, собираемые по этим подпроцессам, являются
информативными и специфическими. На рис. 6 показана возможная организация
валидационных подпроцессов. Подпроцессами внутри валидационной системы
могли бы быть очистка, компьютерное обеспечение, автоматизированные линии,
аналитические методики, упаковка, технологические процессы, валидация транс-
портировки и т. д. Производство — это еще один пример большого комплексного
Рис. 6. Валидационные подпроцессы
318
Часть 3. Качество
процесса, который, возможно, лучше разделить на подпроцессы для лучшего управ-
ления и более информативных показателей для менеджмента.
Выполняя декомпозицию более крупного процесса на управляемые и особые
подпроцессы, менеджмент может выделить соответствующих специалистов в опре-
деленных областях для развития каждого подпроцесса и управления им. Показате-
ли, измеряющие эффективность подпроцесса, могут анализироваться особым обра-
зом, оцениваться и сопоставляться с показателями похожих подпроцессов на других
площадках или других компаний. Могут быть проведены информативные полезные
сравнения эффективности процесса и подпроцесса, которые в другом случае были
бы «слепыми» или размытыми, если бы суммировались с показателями процесса
более высокого уровня.
Дополнительным преимуществом выделения подпроцессов является возмож-
ность быстрой ассимиляции и переноса подпроцесса на другие площадки пред-
приятия. Примером может служить сравнение производственного комплекса по
производству продукта in bulk с дистрибьюторским центром. На обеих площадках
должны быть внедрены элементы подпроцесса «валидация транспортировки», од-
нако дистрибьюторский центр не нуждается во внедрении других подпроцессов,
таких как валидация упаковки или технологического процесса. Как только подпро-
цесс «валидация транспортировки» разработан и внедрен на одной площадке, бы-
стро выполняется передача его инфраструктуры и базы знаний на другую площадку,
что исключает дублирование работ. Обмен информацией и ожиданиями между дву-
мя площадками становится общим принципом работы. Менеджмент может, таким
образом, сравнивать потребности в валидации транспортировки и уровень зрелости
процессов между площадками равным образом.
После окончания работ по определению всех процессов и подпроцессов, обеспе-
чивающих операцию, может быть проведен также анализ пробелов, чтобы убедиться
в отсутствии упущений и включении всех требуемых процессов и подпроцессов для
поддержания коммерческой деятельности в область применения СМК. Для просто-
ты проведения такого анализа перечисляют все основные элементы коммерческой
деятельности, связанные с операцией, и сравнивают их с установленными процес-
сами. Необходимо письменно определить процесс и область применения подпро-
цесса. Заинтересованные стороны, владельцы и пользователи процессов должны
быть привлечены в эту работу, чтобы обеспечить однозначное определение и по-
нимание области применения процесса. При этом должны быть получены ответы
на следующие вопросы: все ли потребности коммерческой деятельности учтены?
все ли наши работы и операции были включены в оценку? имеется ли дублирова-
ние в ожиданиях процессов? существуют ли пробелы между входами и выходами
процессов? После выполнения такой оценки можно легко определить, были ли ис-
ключены из рассмотрения какие-нибудь существующие процессы и требуется ли
дальнейшая модификация системы.
3.3.4.4. Масштабируемость процессов на предприятии
Хорошо спроектированные процессы и подпроцессы должны быть масштабируе-
мы на предприятии. Всесторонний проект позволит копировать и сравнивать про-
цессы и подпроцессы на разных участках и площадках. Это обеспечивает быстрое
Глава 3.3. Создание системы менеджмента качества и управление ею
319
внедрение новых технологий, обмен практическим опытом и сравнение похожих
показателей эффективности для определения степени соответствия регуляторным
требованиям, инфраструктуры и эффективности. Всесторонняя система позволя-
ет каждой операции подразделения или площадки в рамках предприятия обладать
гибкостью для применения соответствующих процессов и подпроцессов и при этом
функционировать в рамках определенной структуры СМК. Например, производ-
ственная площадка может использовать почти все процессы, упомянутые в валида-
ционном процессе, в то время как дистрибьюторская площадка может использовать
только процесс транспортировки. При этом на обеих площадках могут быть внедре-
ны одинаковые структуры для процесса транспортировки, что позволяет произво-
дить информативное сравнение данных и показателей процесса и быстро осущест-
влять любые требуемые изменения в процессе.
Хорошо спроектированная СМК поддерживает структурированный естествен-
ный рост компании и применяется при оценке и интеграции возможностей при-
обретенных производств. Коммерческий и производственный менеджмент долж-
ны использовать СМК и ее стандарты при оценке внешних производственных
комплексов для определения стоимости, одобрения, интеграции или расширения.
Информативные показатели, получаемые от СМК, предоставляют стандарт для
принятия важных решений, влияющих на разные внутренние или внешние произ-
водственные возможности.
Структура документированных процессов обеспечивает быстрое привыкание
сотрудников при переводе с одного участка на другой. Новые сотрудники, заменяя
владельцев существующих процессов, способны быстро осваивать данные процес-
сы благодаря сокращенной длительности обучения, что происходит при наличии
хорошо определенных и задокументированных процессов. Системы, спроектиро-
ванные так, как описано в этой главе, предоставляют высшему руководству инфор-
мативные и сопоставимые численные показатели для оценки прогресса, соответ-
ствия и эффективности.
3.3.5. Владение системами и процессами: функции
и ответственность
Хорошо спроектированная СМК и составляющие ее процессы нуждаются в компе-
тентном владении с установленными ролями и ответственностями для успешного
выполнения программы. Такая комбинация будет гарантировать, что система и про-
цессы установлены, поддерживаются в рабочем состоянии, улучшаются и остаются
действующими в согласии с имеющимся в отрасли опытом и коммерческими ожи-
даниями. При операционном исполнении СМК и ее процессов будут задействованы
заинтересованные подразделения, менеджмент и высшее руководство, будут полу-
чены требуемые коммерческие результаты, поддержка и обеспечение соответствия
регуляторным требованиям.
3.3.5.1. Владение и управление системой менеджмента качества
Наилучшим является случай, когда системой менеджмента качества владеют на
самом высоком уровне организации. Как минимум, это должен быть уровень над
320
Часть 3. Качество
производством и подразделением по качеству. Основная ответственность владельца
системы — поддержка программы и обеспечение требуемой ориентации организа-
ции. Государственные инспекторы ожидают, что процессы, обеспечивающие произ-
водство, полностью включены в СМК. Также они ожидают, что высшее руководство
хорошо знает операции и будет взаимодействовать с инспекторами в ходе проверки,
показывая необходимую степень знаний процессов, обеспечивающих производ-
ство. В заключение по результатам инспекции регуляторы оформляют наблюдения,
сделанные в ходе проверки, и при необходимости накладывают наказание на само-
го главного члена группы высшего руководства. Только при активном участии руко-
водства самого высокого уровня и владении им системой программа СМК и пред-
приятие будут успешными.
Как указывалось ранее, лучше всего поручить управление СМК группе, выде-
ленной специально под эту программу. Подразделение программы по СМК должно
иметь установленные функции и ответственность. В правилах FDA (том 21 CFR, па-
раграф 211.22) описана ответственность подразделения по качеству. Это единствен-
ная функциональная группа в производственной организации, обязанности и ра-
боты которой установлены федеральными правилами. Эти обязанности не должны
противоречить или размывать ответственность по обеспечению качества других
функциональных групп в организации. Все функциональные группы, связанные
с производством, должны выполнять свою деятельность в соответствии с GMP. Ре-
гуляторы ожидают, что управление по качеству осуществляет надзор и одобрение
всех процессов, влияющих на качество продукта. Чтобы выполнить цели програм-
мы, необходимо установить управление программой для обеспечения координации
и подотчетности, обобщения информации индивидуальных процессов, долговре-
менной стратегии системы, ежегодных планов по качеству и задач. Эти работы не
могут быть выполнены владельцами отдельных процессов.
Управление программой СМК может осуществляться в виде управления проек-
том с предварительно заданными критериями, сбором показателей эффективности,
анализом со стороны менеджмента, который завершается применением управления
рисками для постоянного улучшения программ. Эти показатели и деятельность по
улучшению должны быть одобрены и разрешены руководством по итогам анализа
СМК, чтобы обеспечить регулирование деятельности системы в рамках всей орга-
низации.
Возможное распределение ролей и ответственности в подразделении по про-
грамме СМК показано в табл. 5. С помощью определения ролей и ответственности
в этом подразделения устанавливается точка контакта и отчетности по выполне-
нию программы. Она обеспечивает надежную связь и контроль достижения целей
программы владельцами процессов, обучающим персоналом, функциональным
менеджментом и высшим руководством. Аналогичные структуры по управлению
программой требуются на производственных площадках, и для получения макси-
мальной пользы корпорация функционирует, устанавливая общие и частные задачи,
дает возможность для обмена опытом и знаниями. По мере увеличения сложности
организации может потребоваться дополнительное управление, чтобы обеспечить
интеграцию, функционирование всех элементов СМК и получение ожидаемых ре-
зультатов.
Глава 3.3. Создание системы менеджмента качества и управление ею
321
Таблица 5
Функции и ответственность подразделения по программе СМК
_________________Эксперт по определенному вопросу в программе СМК_______________
Разработка и выполнение плана распространения информации.
Первичное и текущее обучение.
Обеспечение процесса анализа со стороны менеджмента.
Выявление задач по повышению зрелости процесса и соответствующих показателей
эффективности.
Разработка долговременного стратегического видения.
Создание и выполнение ежегодного плана действий
3.3.5.2. Владение процессом
Проектирование СМК, которая определяет и распределяет владение процесса-
ми среди назначенных лиц, представляет собой важное стратегическое решение
для внедрения успешной СМК. Оно обеспечивает результативность, экспертные
знания, безраздельное внимание к процессу и концентрацию владельца на доку-
ментации, улучшении, бенчмаркинге и соответствии. Без установленного и назна-
ченного владения функциональный менеджмент де факго становится владельцем
процесса. Это создает проблемы, так как функциональный менеджмент и без того
перегружен вопросами управления персоналом и коммерческой деятельности и не
имеет возможности сконцентрировать внимание на одном вопросе и обеспечить
выполнение требований к владельцу процесса, существующих в современной про-
изводственной среде. Как правило, под одного функционального менеджера орга-
низованы несколько процессов, что еще более размывает концентрацию, внимание
и контроль, если на функциональный менеджмент полагаются как на владельцев
процесса.
3.3.5.3. Выбор владельца процесса
Выбор владельцев процесса требует от руководителей программы установления
определенных критериев для процесса выбора. Критерием является, в частности,
способность выполнять функции владельца процесса и нести ответственность,
включая саморазвитие и принятие решений. Наделенные полномочиями владель-
цы процессов отвечают за поддержание и выполнение процессов, на которые опи-
рается менеджмент для получения коммерческих результатов. Такая подотчетность
показвывает, что персонал, менеджмент и высшее руководство знают, кто отвеча-
ет за вопросы и проблемы, связанные с процессом. Это также приводит к лучше-
му информированию регуляторов, клиентов и потребителей. Активный владелец
с установленной ответственностью добивается результативности и поддерживает
действующие тренды.
Критериями выбора также могут являться особенности технического опыта,
межличностного общения и опыта управленческой деятельности. Способности,
необходимые для разных процессов, могут быть различными и должны быть учте-
ны при выборе владельца. По окончании процесса выбора функциональный менед-
жмент и владелец процесса могут обсудить включение функций и ответственности
322
Часть 3. Качество
в должностные обязанности владельца процесса. Личные задачи и деятельность по
разработке должны быть направлены на улучшение возможностей владельца управ-
лять процессом и на подготовку владельцев будущих процессов с помощью про-
грамм активного наблюдения и развития талантов.
Необходимо, чтобы владельцы процессов имели все возможности по управлению
своими процессами. Они должны иметь необходимые полномочия и отчитываться
за параметры, указанные в их функциях и ответственности. Владельцы процессов
могут обладать более чем одним процессом и иметь другие должностные обязанно-
сти, но в организации должно быть четко определено, кто имеет полную власть над
каждым конкретным процессом.
Необходимо установить для владельца процесса определенный набор обязан-
ностей по поддержанию работоспособности и результативности процесса, поддер-
живающего показатели качества продукта. Наличие определенных функций и обя-
занностей предоставляет владельцам структуру и параметры, которые должны быть
эффективными. В качестве примеров обязанностей владельцев можно привести
идентификацию заинтересованных подразделений, право принятия определенных
решений, владение документацией, владение несоответствиями, знание норматив-
ных правовых актов и отраслевых тенденций, экспертизу по установленному вопро-
су, содержание обучения, владение показателями эффективности и представление
процесса внутренним аудиторам и внешним инспекторам. Выявление, обучение
и развитие владельца процесса для выполнения им своих функций можно сравнить
со сбором головоломки. Если отсутствует один элемент, то результативность вла-
дельца процесса будет минимальной (рис. 7). Создание обоснованной методологии
Владение
несоответствия ми
и корректи-
рующими
действиями
Экспертное I
знание /
процесса
внутри
компании
и в отрасли
Содержание
документов
и обучающих
материалов
Улучшение
процесса
и показателей
его
эффективности
Право
принятия
решений
Точка
контакта при
инспекциях
и аудитах
Рис. 7. Функции и обязанности владельца процесса
Подотчетное
владение
Менеджмент
заинтересо-
ванного
подразделения
Глава 3.3. Создание системы менеджмента качества и управление ею
323
для выбора владельцев процессов обеспечивает объективность и является жизненно
важной для успеха программы. Краткое обсуждение каждой из функций и обязан-
ностей владельца процесса приводится далее.
Владельцы процессов должны определить подразделения, заинтересованные
в этих процессах, и обеспечить соответствие конструкции и продукта процесса по-
требностям заинтересованных подразделений. Постоянный обмен информацией
с заинтересованными сторонами, взаимодействие и поддержка обеспечиваются
посредством запланированных совещаний для обсуждения состояния процесса
и улучшений. Любые изменения процесса согласовываются с группой представи-
телей заинтересованных подразделений. Как правило, к заинтересованным под-
разделениям относится подразделение по обеспечению качества, которое отвечает
за рецензирование и согласование компонентов процесса, поставщиков процесса
и получателей продуктов процесса (т. е. владельцев других процессов, которые взаи-
модействуют с данным процессом), пользователей, менеджмента и высшего руко-
водства. Функции и обязанности каждого заинтересованного подразделения также
требуют определения.
Включение основных заинтересованных подразделений в процессы принятия
решений относительно конструкции процесса или внесения в него изменений обе-
спечивает правильность работ владельца процесса. Надежность процесса зависит от
удовлетворения потребителей и коммерческих результатов, а владельцы процессов
нуждаются в участии и поддержке заинтересованных подразделений.
3.3.5.5. Право принятия решений
У каждого владельца процесса должен быть определенный уровень прав на при-
нятие решений. Этот уровень устанавливает границы решений, в рамках которых
организация разрешает владельцу процесса принимать решения. Коммерческие
требования и оценка риска должны быть встроены в процесс принятия решений
владельцем процесса в рамках данной ему компетенции. В табл. 6 приведен при-
мер матрицы прав принятия решений владельцем процесса. Для результативности
такой схемы требуется под держка межфункционального менеджмента.
Подготовка матрицы прав принятия решений, которая разрабатывается и со-
гласуется с группой из представителей заинтересованных подразделений и функ-
ционального менеджмента, гарантирует быстрое принятие решений и доведение
их до сведения соответствующих лиц. Такой процесс принятия решений снижает
нагрузку функционального менеджмента по принятию решений по каждому тех-
ническому процессу. Важно установить функции владельца процесса при принятии
решений и условия расширения его полномочий. Эффективное управление процес-
сом достигается, когда культура организации сможет поддерживать схему матрицы
решений и не полагаться постоянно на функциональный менеджмент. Если функ-
циональный менеджмент продолжает оставаться ответственным за технические
процессы и рассматривается как группа лиц, принимающих решения, то эффект
и прогресс работы владельца процесса будут формальными. Культура организации
должна поддерживать каждого владельца процесса на всех уровнях компании, что-
бы владелец процесса был успешным.
324
Часть 3. Качество
Таблица 6
Матрица прав принятия решений
Категория решений Определение Лицо, принимающее решение Лица, от которых требуется поддержка решения Лица, которые информируются о решении
Стандарты компании Глобальные стандарты на процессы, относящиеся ко всем производственным площадкам Корпоративный владелец процесса Владельцы процессов на площадках. Куратор из подразделения по обеспечению качества на площадках Подразделения, заинтересованные в процессе. Задействованный персонал
Стандартные операционные решения СОП, связанные с определенным процессом Владелец процесса на площадке Заинтересованные в процессе подразделения. Куратор из подразделения по обеспечению качества на площадках Аналитики со стороны руководства. Корпоративный куратор из подразделения по обеспечению качества. Задействованный персонал
Обучение Обучение процессам или процедурам Корпоративный владелец и владелец процесса на площадке Эксперт по техническим вопросам системы Подразделения, заинтересованные в процессе. Корпоративный куратор из подразделения по обеспечению качества. Задействованный персонал
Проекты на площадке Все проекты, связанные с существующим или проектируемым процессом Руководитель площадки Владелец процесса. Группа высшего руководства. Менеджер портфеля проектов на площадке Подразделения, заинтересованные в процессе. Корпоративный куратор из подразделения по обеспечению качества. Подразделение по СМК
3.3.5.6. Отраслевой опыт
Буква с в аббревиатуре cGMP (current good manufacturing practice) означает указание на
действующую производственную практику. Владельцы процессов должны работать
на существующем в отрасли уровне и с учетом регуляторных тенденций, влияющих
Глава 3.3. Создание системы менеджмента качества и управление ею 325
на их процессы, на СМК в целом и на коммерческую деятельность. Крайне важно
обладать знанием возможностей процесса1 и возможностью их сравнения с другими
похожими процессами внутри фармацевтической и биофармацевтической отрасли
и вне ее. Регуляторы будут сравнивать процесс владельца с другими похожими про-
цессами, по которым у них есть опыт, при вынесении оценочных суждений. Бенч-
маркинг похожих процессов предоставляет владельцам данные, необходимые для
определения адекватности собственных процессов в сравнении с однородными от-
раслевыми группами.
Целесообразность улучшения технологии, результативности, эффективности
или степени соответствия процесса должна рассматриваться грамотным и инфор-
мированным владельцем процесса. Функциональный менеджмент не может отсле-
живать изменения, происходящие со всеми процессами, обеспечивающими произ-
водство. Выделение владельцам процессов достаточного времени для постоянной
актуализации внешней информации о событиях, связанных с их процессом, будет
гарантировать успешность процесса. Это может включать обзоры периодической
отраслевой литературы, посещение семинаров и конференций регуляторных орга-
нов, а также повседневную самооценку и бенчмаркинг родственных процессов.
Часто наилучшие примеры эффективности процессов можно найти в других от-
раслях, вне фармацевтической и биофармацевтической отрасли. Другие области,
такие как электроника, космическая отрасль и программное обеспечение, развили
свои системы документации, обучения, качества и контроля изменений до стату-
са «лучших в своем классе». Эти отрасли подвержены более жестким временным
ограничениям по поставкам продукта на рынок и часто обладают высокоэффек-
тивными производственными процессами и очень быстрыми процессами приня-
тия решений. Владельцы процессов могут расширять свои знания путем исследо-
вания опыта других отраслей, искать наилучший опыт и применять его для своих
процессов.
3.3.5.7. Регуляторные инспекции и аудиты
Владельцы процессов играют очень важную роль при регуляторных проверках
и аудитах со стороны потребителей. Владельцы процессов лучше всех могут пред-
ставить проверяющим информацию о параметрах процесса и его эффективности.
Они играют для государственных инспекторов и аудиторов роль информирован-
ного квалифицированного источника представлений о процессе и отвечают на
подробные вопросы. Владелец процесса должен обладать знаниями об истории
процесса, требованиях, операциях, исключениях, изменениях и несоответствиях.
Обладая подробными знаниями об операциях, составляющих процесс, его статусе
соответствия, владелец процесса имеет возможность защитить его при проверке.
Предоставление с первого раза четкого точного ответа на вопросы инспекторов
и аудиторов является жизненно необходимым для создания доверия и демонстра-
ции компетентности.
Каждый владелец процесса должен тесно взаимодействовать со своим курато-
ром из подразделения по обеспечению качества. Тесное взаимодействие гарантиру-
1 Про возможности процесса см. главу 3.4. — Примеч. перев.
326
Часть 3. Качество
ет четкое рассмотрение и одобрение вопросов конструкции и функционирования
процесса представителями подразделения по обеспечению качества и оправдывает
регуляторные ожидания. Куратор из обеспечения качества должен знать процесс,
понимать документацию по процессу и быть способным объяснить, какое одобре-
ние департамент по качеству выразил в отношении процесса, а также значение это-
го одобрения. У куратора также должны быть определены функции и обязанности
по процессу.
При совместной работе владелец процесса и куратор из подразделения по обе-
спечению качества произведут благоприятное впечатление на государственных ин-
спекторов, смогут объяснить все операции, включенные в процесс, соответствую-
щую документацию и любой выполняемый проект или улучшения процесса. Эта
пара является наиболее пригодной для оценки и рассмотрения любого отклонения
процесса или рекомендаций по постоянному улучшению.
В большинстве случаев владелец процесса и куратор из обеспечения качества
будут обладать ббльшим объемом информации о процессе, чем инспекторы, и смо-
гут защитить конструкцию процесса и его операции. При наличии у инспекторов
предложений по структуре процесса или его функциональности владелец процесса
сможет обсудить их. Любые рекомендации или наблюдения, сделанные государ-
ственными инспекторами или аудиторами, могут быть включены в конструкцию
процесса. Однако владелец процесса и заинтересованные подразделения должны
провести оценку предлагаемых изменений, чтобы исключить избыточные обяза-
тельства менеджмента, которые могут быть опасными для эффективности работ
и продукта процесса.
3.3.5.8. Технические эксперты
Для предприятия более результативно сконцентрировать экспертные функции у
определенных лиц, обладающих правами и подотчетностью, описанными в этом
разделе, чем распределять их по организации, тем самым повышая вероятность не-
надлежащего исполнения обязанностей и неудачного управления процессами.
В качестве технического эксперта по процессу у владельца есть возможность
предоставлять результаты, указанные в перечне обязанностей владельца, обучать
будущих владельцев процесса, помогать в развитии персонала и давать точные ре-
комендации менеджменту в отношении стратегии, связанной с процессом. Само-
развитие владельца процесса по расширению экспертных знаний о своем процессе
является необходимым при анализе результатов работы процесса и разработке на-
правлений дальнейших стратегических изменений процесса.
3.3.5.9. Владение показателями
В обязанности владельцев процесса входит определение пригодных показателей
эффективности своих процессов. Эти показатели должны включать запаздываю-
щие и опережающие показатели1, информативные для владельца процесса и менед-
жмента для оценки эффективности, соответствия и инфраструктуры процесса.
1 См. примечание редактора в разделе 3.3.2.3 относительно терминов «запаздывающие»
и «опережающие» показатели и раздел 3.3.7.1.
Глава 3.3. Создание системы менеджмента качества и управление ею 327
Владелец процесса должен представлять и объяснять эти показатели высшему
руководству организации. Численное измерение выхода процесса — фундамент, на
котором строятся действия менеджмента и высшего руководства по использованию
ресурсов и согласованию проектов по постоянному улучшению. Основные опера-
ционные параметры, такие как количество несоответствий и наблюдений, обнару-
женных при регуляторных инспекциях в отношении процесса, должны отслежи-
ваться и учитываться при оценке зрелости процесса.
Каждый владелец процесса обязан основывать свой план постоянного улучше-
ния процесса на показателях, характеризующих выход процесса. Показатели долж-
ны быть разработаны таким образом, чтобы помогать при принятии этих решений
и быть легко доступными для анализа, представления и интерпретации.
3.3.5.10. Владение документацией
Владельцы процессов, как правило, являются владельцами всей документации, ка-
сающейся их процессов. Владение документами включает или прямое владение,
или контролирующее влияние на руководящую и исполнительную документацию,
такую как корпоративные методики и стандарты, локальные требования и стан-
дартные операционные процедуры (СОП), журналы и записи.
Для владельца производственного процесса это означает владение регламентами
производства и протоколами произведенных серий (досье на серию), СОП, исполь-
зование журналов и связанных с процессом документов по обучению. Комбини-
рование ответственностей за управление процессами и владение процессом при-
водит к истинной подотчетности для владельца процесса. Это также способствует
прогрессу и постоянному улучшению СМК. Решение вопросов ответственности
и подотчетности обеспечивает взаимосвязь между требованиями (стандартами, ме-
тодиками и процедурами) и исполнением (обучением, результативностью и доку-
ментацией).
3.3.5.11. Обучение
Обеспечение надлежащего обучения для пользователей процесса является важной
обязанностью владельца процесса. Владельцы процессов должны четко понимать
требования предъявляемые к своим процессам и их функционированию. При этом
пользователи должны быть способны понимать и применять обучение. Сложные
процессы в совокупности с неоднозначным обучением приведут к путанице и не-
возможности выполнять процесс надлежащим образом, что обернется потерями для
организации. Простой процесс с легкими для понимания этапами, согласованными
с инструкциями и требованиями документации, обеспечит достижение успеха, сни-
зит производственные издержки, минимизирует случаи возникновения несоответ-
ствий и принесет сотрудникам удовлетворение от работы.
Владельцы процессов являются техническими экспертами, поэтому должны
влиять на обучение процесса и оказывать соответствующие консультации. Они
также могут участвовать в проведении обучения. Надлежащее обучение является
одним из основных шагов для достижения эффективности системы и обеспечения
соответствия регуляторным требованиям. Владельцы процессов, способные объяс-
328
Часть 3. Качество
нить порядок выполнения требований к процессу, увеличат эффективность обуче-
ния пользователей процесса. Программы личного развития владельцев процессов
должны включать приобретение эффективных навыков по созданию презентаций
и проведению тренингов.
3.3.5.12. Управление рисками
Владельцам процессов необходимо знать основные положения управления риска-
ми и их применение к конструкции процесса и расстановке приоритетов при по-
стоянном улучшении. Существует несколько отраслевых и регуляторных докумен-
тов, таких как руководство ICH (79, содержащих основные положения по анализу
рисков, идентификации, контролю, методологии и общему процессу управления
рисками. Владельцы процессов должны быть знакомы с методами и инструмента-
ми управления рисками и должны применять их при управлении своими процес-
сами, при проектировании, выполнении и управлении изменениями своих про-
цессов.
Управление рисками особенно важно при представлении менеджменту пред-
ложений по улучшению процесса, когда необходимо выделение ресурсов. Коли-
чественная оценка риска и демонстрация преимуществ от постоянного улучшения
будут востребованы для одобрения проекта и выделения ресурсов. Анализ рисков,
управление ими и представление рисков составляют данные для высшего руковод-
ства для работы над требуемыми вопросами в нужный момент.
3.3.5.13. Непрерывное улучшение и управление проектами
Введение работ по «качеству, заложенному при проектировании» на ранних ста-
диях проектирования процесса должно исключить необходимость последующих
значимых улучшений процесса. Однако со временем вследствие коммерческих
требований, изменений законодательства или технологических улучшений потре-
буется внесение в процессы некоторого рода изменений для обеспечения соответ-
ствия регуляторным требованиям или увеличения результативности процессов.
В рамках своих работ по исполнению требований и управлению процессами вла-
дельцы обязаны собирать и предоставлять менеджменту и персоналу показатели
эффективности процессов. Эти показатели неизбежно привлекут внимание к воз-
можностям по улучшению, которое потребует вложения средств и человеческих
ресурсов. Благодаря глубокому знанию процесса и отчетности по выходу процесса
владельцы процессов являются лучшими руководителями проектов по постоян-
ному улучшению.
Для руководства проектом по постоянному улучшению владелец процесса дол-
жен обладать знаниями по управлению проектами и навыками управления труппой.
Как правило, для проектов по улучшению требуется создание межфункциональной
группы и экспертные знания в таких областях, как информационные системы,
управление проектами, производство, инженерные системы и разработка. Чтобы
обеспечить соответствие требуемого результата проекта потребностям владельца
процесса, заинтересованных подразделений и предприятия в целом, необходимо
назначить руководителем проекта владельца процесса. Но бывает так, что об успехе
сообщают, так и не получив нужного результата.
Глава 3.3. Создание системы менеджмента качества и управление ею
329
По завершении проекта по постоянному улучшению в обязанности владельца
процесса включают мониторинг изменений, внесенных в процесс, для оценки вли-
яния улучшений. Показатели эффективности, отражающие изменения процесса до
и после внесения изменений, должны быть включены в существующие показатели
эффективности и отчеты при регуляторном анализе со стороны менеджмента.
3.3.5.14. Несоответствия, корректирующие и предупреждающие действия
(САРА) и владение планируемыми отклонениями
Важным показателем результативности процесса является количество несоответ-
ствий, предпринятых корректирующих и предупреждающих действий и плани-
руемых отклонений, инициированных для данного процесса. Эти типы дефектов
процесса должны быть известны и управляться владельцем процесса и заинтересо-
ванными подразделениями. Владелец процесса должен анализировать эти показате-
ли процесса и рассматривать целесообразность внесения изменений в конструкцию
процесса, обучение, документацию и результативность.
Несоответствия могут попасть в категории человеческих ошибок, отказов обо-
рудования, дефектов материалов и т. д. Наличие сотрудников, не выполняющих
процедуры или неспособных исполнять требуемые операции процесса, свидетель-
ствует о плохо спроектированном процессе, нуждающемся в модификации и/или
улучшении обучения. Отказы оборудования часто свидетельствуют о ненадлежа-
щих программах квалификации, валидации, калибровки или технического обслу-
живания. Неожиданные результаты или продукты означают неудовлетворительную
конструкцию процесса, характеристик процесса или наличие разрыва между про-
цессами.
Хотя планируемые отклонения не приветствуются многими специалистами
в отрасли и регуляторами, существуют ситуации, когда для поддержки коммерче-
ской деятельности организации в используемый процесс должны быть внесены
временные изменения. Постоянные изменения следует вносить путем формально-
го процесса контроля изменений. При необходимости внесения планируемого от-
клонения затрагиваемый этим отклонением владелец процесса должен знать о нем
и контролировать его. Такой подход предоставляет владельцу информацию о дли-
тельности и значимости изменения, вносимого в процесс, и обеспечивает данны-
ми по возможному последующему рассмотрению целесообразности внесения по-
стоянного изменения. Ситуации с планируемым отклонением должны быть редки
и тщательно наблюдаться, так как отклонение затрагивает установленные стандар-
ты, ожидания и обучение.
Владельцы процессов должны быть способны оценивать и интерпретировать
влияние несоответствий и планируемых отклонений на свои системы. Для обеспе-
чения внедрения надлежащих исправлений и улучшений владельцы процессов мо-
гут проводить оценку потребностей и руководить работами по корректирующему
или предупреждающему действию. Эффективная СМК гарантирует, что отклонения
от утвержденных процессов имеют владельцев, расследуются надлежащим образом
владельцем процесса и, в конечном итоге, согласуются с его куратором в подраз-
делении по обеспечению качества. База знаний по этим случаям является основа-
нием для оценки владельцами процессов с помощью методов управления рисками
330 Часть 3. Качество
необходимости внесения изменений в процесс, документацию или обучение, или
оправданности работ по постоянному улучшению.
Хорошо спроектированная СМК будет включать назначенных владельцев про-
цессов с определенными функциями и ответственностью. Владельцам процессов
необходима поддержка со стороны менеджмента, потребителей процессов, подраз-
деления обеспечения качества и других заинтересованных подразделений. Подот-
четность и параметры принятия решений расширят полномочия владельцев про-
цессов, что обеспечит надлежащее выполнение процессов и их улучшение, которое
выразится в получении ожидаемых коммерческих результатов. Без этих прав и под-
держки будут получены минимальные результаты, а функциональный менеджмент
будет перегружен техническими задачами и ответственностью за принятие реше-
ний, что и должно быть поручено подходящим владельцам процессов.
3.3.6. Управление изменениями и распространение
информации
Для внедрения и поддержания в работоспособном состоянии СМК, как описано
в этой главе, потребуется значительный сдвиг в культуре организации. Многие со-
трудники и функциональный менеджмент решат, что трансформация деятельности
по определению процессов, назначение владельцев, делегирование прав и ответ-
ственности за эффективность процессов в рамках СМК представляют значитель-
ное изменение в порядке ведения деятельности. Наиболее значительное изменение
обусловлено переходом контроля в экспертизе процесса и права принятия решения
от функционального менеджмента к владельцу процесса. Также может произойти
существенное изменение коммерческой деятельности организации вследствие пе-
редачи ответственности и подотчетности владельцу процесса и поддержки со сто-
роны менеджмента владельцев процессов, которые являются основным фактором
постоянного улучшения процессов.
В книге «Вторая американская революция» Рокфеллер описывает консерватизм,
присущий организациям: «Организация — это система со своей собственной логи-
кой, и вся во власти традиций и инерции. И все в ней устроено в пользу опробован-
ного и проверенного способа делать дела и против принятия рисков и поиска новых
путей» [9]. Если организация еще не использует принципы делегирования полно-
мочий, владения процессами, сбора установленных показателей эффективности,
анализа со стороны менеджмента и постоянного улучшения, то необходимо будет
обратить внимание на барьеры, существующие в организации, и разрушить их, что-
бы установить новый образ действий. Барьеры, которые надо устранить, будут су-
ществовать внутри функций и между ними, между функциональным менеджментом
и персоналом, и возможно, между компаниями и регуляторными агентствами.
Хотя ожидаемые преимущества при внедрении СМК значительны, а конечный
результат желателен для сотрудников и менеджмента, описание ожидаемого со-
стояния организации и мотивация персонала к изменениям и внедрению нового
образа действий представляет большие сложности. Успешная трансформация дея-
тельности организации потребует надежного управления изменениями и плана рас-
пространения информации, который необходим для поддержки всех сотрудников,
которых затронут изменения.
Глава 3.3. Создание системы менеджмента качества и управление ею 331
3.3.6.1. Управление организационными изменениями
Объединение опытных сотрудников по управлению персоналом и обучению с груп-
пами управления изменениями способно в значительной мере усилить работы по
изменениям организации и внедрению новой культуры. Часто инструменты по со-
ставлению профиля личности оказываются эффективными для определения пред-
почтений организации, стилей обучения и тенденций усвоения информации. Ис-
пользование этих типов инструментов следует предусмотреть в общей программе
управления изменениями и в модификациях программы, выполняемых в зависимо-
сти от полученных результатов.
Первым и самым критическим этапом в разработке успешного плана управ-
ления изменениями является получение предварительной поддержки со стороны
высшего руководства организации и функционального менеджмента. Без этой под-
держки СМК никогда не станет жизнеспособной и не сможет обеспечить достиже-
ние желаемых эффектов и проведение необходимых изменений. Чтобы получить
эту под держку, группа по внедрению должна создать очень убедительное технико-
экономическое обоснование, соответствующее потребностям и пожеланиям выс-
шего руководства. Технико-экономическое обоснование должно включать оценку
рисков в отношении соответствия регуляторным требованиям и описание выгод
в финансовых терминах. Важно быть честными, рассмотреть реальное состояние
действующей системы, учесть будущие требования и выработать долговременную
стратегию в отношении затрат и пользы. План управления изменениями должен
включать частое распространение информации на всех уровнях организации о за-
тратах и пользе, об ожидаемых и полученных успешных результатах программы.
Для успешной реализации новой программы, в том числе СМК, также необхо-
дима поддержка функционального менеджмента. Во всех случаях, когда сотрудника
просят принять новую функцию или ответственность, он нуждается в поддержке со
стороны функционального менеджмента так же, как и со стороны высшего руко-
водства. Ресурсы корпорации всегда ограничены, и необходимо постоянно опре-
делять приоритеты при их распределении. Сотрудники будут с пониманием отно-
ситься к новым функциям и принимать на себя новую ответственность, только если
будут видеть поддержку функциональным менеджментом успешного выполнения
своих непосредственных задач. Количественно выражаемая поддержка со стороны
высшего руководства и функционального менеджмента может быть прямо пропор-
циональна успеху или провалу программы СМК.
Значительная часть работ по внедрению СМК связана с обучением новых вла-
дельцев процессов, функциональных менеджеров, высшего руководства, вспомо-
гательных служб и с осуществлением их нового образа действий. Для управления
новыми функциями и сохранения эффективности процессов должна быть внедрена
система, поддерживающая владельцев процессов, заинтересованные подразделения
и менеджмент. Предпочтительно, чтобы эта поддерживающая система внедрялась
с помощью выделенной группы, которая будет полностью удовлетворять все по-
требности участников. Отсутствие единого центра, руководящего работами, может
привести к несогласованным толкованиям и ошибкам при внедрении, что ослабит
различные функции программы и ее выполнение на разных площадках, и ее эффек-
тивность и результаты будут существенно ниже ожидаемых.
332
Часть 3. Качество
Определение структуры, управляющей внедрением системы, очень важно. Для
этой структуры потребуются управление, стандарты и параметры, схожие с теми,
которыми характеризуется отдельный процесс по качеству. Для нее должны быть
установлены функции и ответственность, определены показатели эффективности,
которые собираются, анализируются и используются для принятия решений, и по-
лучены признание и поддержка со стороны менеджмента и высшего руководства.
Лучше всего организовывать программу по СМК в виде функции департамента по
качеству и рассматривать ее как постоянно действующую программу, а не кратко-
срочный проект или работы, выполняемые в течение ограниченного периода вре-
мени. Группа должна управляться компетентными лицами, знающими концепции
качества и их применение, ожидания инспекторов и регуляторные требования, по-
требности предприятия и при этом обладающими хорошими навыками общения
и влияния на людей, гибкостью и терпением.
3.3.6.2. Распространение информации
Попытка довести до сотрудников информацию об альтернативном будущем являет-
ся коммуникативной задачей абсолютного другого порядка, чем организация их для
выполнения краткосрочного плана. Это напоминает разницу между играющим по-
мощником футбольного тренера (квотербеком), рассказывающим игрокам планы
на следующие две или три игры, и им же, пытающимся объяснить команде абсолют-
но новый подход к игре, который должен быть реализован во второй части игрового
сезона. Регулирование организации для принятия персоналом системного подхода
требует осторожного распространения сообщений и поддержки менеджмента.
Для того чтобы сообщения возымели действие, недостаточно лишь их понима-
ния сотрудниками. Большой проблемой для высшего руководства является кредит
доверия людей к полученным сообщениям. Совпадение слов и дел обеспечивает
реальную ценность сообщений и доверие к ним. Сотрудники на своем опыте убеж-
даются, что даже если они правильно воспринимают важные внешние изменения
и выполняют соответствующие действия, они могут оказаться беззащитными перед
каким-нибудь более высокопоставленным сотрудником, которому не нравится их
работа. Выговоры могут иметь много форм: «Это не соответствует нашей полити-
ке», или «Мы не можем позволить это», или «Замолчите и делайте, как вам было
сказано» [10].
При наличии специальной группы, которая обеспечивает управление всей
программой, крайне важно, чтобы группа создала стратегический план и пред-
ставила его высшему руководству. Без стратегического видения и долгосрочно-
го плана, поддерживаемого высшим руководством предприятия, инициативы по
созданию системы качества станут труднореализуемыми. По своей природе план
должен быть всесторонним и достаточно общим, чтобы донести до понимания на
достаточно высоком уровне целей, миссии, получаемых преимуществ и при этом
получить одобрение. Такой план дает структуру и направления для высшего руко-
водства и группы управления программой. Он также помогает группе управления
программой при разработке ежегодных целей и планов по качеству, согласующихся
с общей стратегией, и указывает основные этапы работ и итоговые результаты для
организации.
Глава 3.3. Создание системы менеджмента качества и управление ею 333
Ежегодные планы по качеству, подготавливаемые структурой по программе
СМК, должны отвечать долгосрочной стратегии и предусматривать вероятность по-
явления промежуточных целей, возникающих по мере внедрения программы. В го-
довой план по качеству должны быть включены обучение, изменения регулятор-
ных требований, проекты по достижению показателей эффективности и изменения
процесса, требующиеся в особых обстоятельствах, например при внедрении новой
технологии или программы.
Долгосрочная стратегия и годовые планы по качеству требуют поддержки и одо-
брения со стороны менеджмента и высшего руководства. Эти документы долж-
ны быть сначала согласованы и обсуждены с высшим руководством организации
и уточнены для удовлетворения требований коммерческой деятельности и регули-
рующих органов, а затем полностью поддержаны менеджментом верхнего звена по-
сле соответствующих согласований. Таким образом, цели устанавливаются высшим
руководством и менеджментом, а не какой-то отдельной группой в организации.
После подписания программы высшим руководством она может быть распростра-
нена по организации различными способами.
Если высшее руководство сможет поддержать долгосрочную стратегию и еже-
годные планы по качеству, будет создан фундамент для управления изменениями
и сдвига в культуре организации. Высшему руководству придется постоянно об-
суждать необходимость внедрения системы с различными аудиториями. Это бу-
дут, в том числе, совещания руководящего персонала, совещания с менеджментом
среднего звена и с рядовыми сотрудниками. Важность поддержки высшего руко-
водства нельзя недооценивать. Без его поддержки и надлежащего менеджмента
владельцы процессов и персонал вернутся к прежнему образу действий, возмож-
но, нарушат взаимосвязи между интегрированными процессами. Высшее руко-
водство должно требовать включения параметров программы в годовые задачи
функционального менеджмента с указанием определенных результатов и их оцен-
кой. В свою очередь, функциональный менеджмент должен требовать включения
соответствующих параметров и целей программы по СМК в свои индивидуальные
планы и работы.
3.3.6.3. Обратная связь и корректировка программы
Управление изменениями, требуемое для полного внедрения СМК, может вклю-
чать несколько форм распространения информации и обратной связи. Предметный
годовой план по распространению информации может помочь группе СМК в уста-
новлении целевых групп, методов и частоты распространения информации, типа
сообщений и механизмов обратной связи, необходимой для мониторинга прогрес-
са и определения целесообразности модификации программы. В табл. 7 приведен
пример ежегодного плана распространения информации, который обеспечивает
информированность сотрудников, вовлеченность и единение.
Каждая целевая аудитория требует определенных сообщений, связанных с их
потребностями. Невозможность получить соответствующее сообщение, то есть ин-
формацию о том, что дает программа этим людям, будет сводить к минимуму под-
держку программы. В план информирования следует включить распространение
устной (лицом к лицу) и письменной информации, указав различные аудитории
334
Часть 3. Качество
и типы средств связи. Устное распространение информации заключается в прове-
дении презентаций для советов директоров и акционеров, владельцев процессов
и функциональных подразделений, и также оно может происходить на всех собра-
ниях персонала. Письменное информирование включает рассылки сообщений по
всей организации, изготовление плакатов (постеров) и информационные письма.
Сообщения должны быть обращены ко всей аудитории («Что в этом для меня?»).
Темами сообщений могут быть обязательства по СМК высшего руководства (пря-
мые цитаты или предпринятые действия), достигнутые успехи (реальные истории
от владельцев процессов), влияние на площадку (улучшение процесса или сниже-
ние риска) и информация о прогрессе (достижения и показатели эффективности).
Не следует опасаться, что информации о прогрессе и достижениях СМК может быть
слишком много — такого просто не бывает.
Другим полезным инструментом по обмену информацией и для определения
необходимости модификации программы является использование обратной связи
в виде проведения опросов. Если опрос подготовлен правильно и проводится в за-
интересованной стороной, то он может дать полезную внутреннюю информацию
о том, как персонал и менеджмент относятся к программе и ее прогрессу, а также
позволит получить предложения по модификации программы. Если опросные ли-
сты распространяются электронным образом и предполагается односторонний об-
мен информацией, то получаемая от опроса польза может быть ограничена, так как
респондентам не предоставляется возможность полностью описать свои впечатле-
ния или предложить эффективную обратную связь. Электронные опросы с обрат-
ной связью могут быть первым приемлемым этапом в понимании представлений
и тревог заинтересованных лиц.
Таблица 7
План распространения информации
Способ распространения Ъш распространения Частота Даты
Совещание по функциональным показателям эффективности Устное Ежемесячно Первая неделя месяца
Интервью руководителей среднего звена Устное Ежегодно В течение января
Информационные письма по СМК Письменное Ежеквартально Первая неделя квартала
Собрания всего коллектива Презентация 1 раз в полугодие Март и сентябрь
Вывешивание плакатов (постеров) Письменное и устное Ежегодно Июль
Другим вариантом или действием, следующим за электронным опросом, яв-
ляется использование метода фокус-групп, в котором происходит взаимодействие
с интервьюерами от программы. В этом методе используется двустороннее обще-
ние — вербальный диалог, обеспечивающий более глубокое понимание программы
участниками и устанавливающий обратную связь, более информативную для адми-
нистраторов программы. Следует набирать фокус-группы из представителей разных
Глава 3.3. Создание системы менеджмента качества и управление ею 335
уровней организации, включая владельцев процессов, сотрудников заинтересован-
ных подразделений, пользователей системы, представителей высшего руководства,
функционального менеджмента и общего персонала. Использование фокус-групп
может дать ценную информацию, о которой администраторы программы могли не
знать, и показать необходимость внесения исправлений.
Предложения по программе должны быть рассмотрены, после чего в нее вклю-
чают идеи и модификации, которые следует внедрить в производственные процес-
сы. Вносимые изменения должны обсуждаться и оцениваться с участием членов
фокус-группы, чтобы показать, что их время было потрачено не зря, усилия были
востребованы, а предложения услышаны. Это наилучший способ распространения
информации о программе СМК и получения поддержки от рядовых сотрудников.
3.3.6.4. Обучение
С целью определения потребностей персонала и задействованных функциональных
областей, поддержка которых необходима для успешного внедрения СМК, должен
быть разработан план по обучению. В обязанности компании входит предоставле-
ние персоналу надлежащего обучения и инструментов, когда предполагается наде-
ление сотрудников новыми функциями, обязанностями или введение нового образа
действий. План по обучению должен включать целевое обучение рядовых сотрудни-
ков, владельцев процессов и функционального менеджмента.
Все сотрудники должны быть ознакомлены, как минимум, с целями, задачами
и требованиями СМК. Для получения поддержки программы со стороны сотруд-
ников обучение должно быть максимально простым и должно разъяснять важность
программы и риски в случае неудачи с внедрением СМК. Такое обучение проводит-
ся или инструктором, или в электронном виде с помощью обучающих модулей на
основе Жей-технологий в зависимости от размеров компании.
Для владельцев процессов требуется обучение более сложных уровней, чтобы
обеспечить полное понимание их функций и ответственности в рамках програм-
мы. Обучение владельцев процессов должно включать основные концепции и ин-
струменты, которые понадобятся владельцам для оценки и обеспечения своих про-
цессов. Такое обучение может проводиться поэтапно, чтобы поддержать развитие
и рост процесса в рамках выбранной организацией модели зрелости СМК.
При обучении функциональных менеджеров, руководящих владельцами про-
цессов, должно быть достигнуто абсолютное понимание ими СМК. Обучение долж-
но включать обсуждение новых функций и ответственности персонала, временных
затрат, требуемых от владельцев процессов, общих сроков выполнения программы
и влияния на функциональные области. Правильное восприятие программы и ее
поддержка со стороны функционального менеджмента — критический фактор для
успешного внедрения СМК.
Для эффективного управления организационными изменениями требуется под-
робная письменная стратегия, набор навыков и ресурсы, которые обеспечат пони-
мание изменений, происходящих при внедрении системы, их поддержку и защи-
ту. Фундаментом для понимания и поддержки со стороны менеджмента являются
самые общие представления о конструкции системы, получаемых преимуществах
и временное сроки для ее внедрения. Подробные ежегодные планы по качеству мо-
336
Часть 3. Качество
гут использоваться в качестве тактического инструмента для внедрения программы.
Жизненно необходима поддержка со стороны высшего руководства, выражающаяся
в виде понимания и утверждения ежегодных планов по качеству, включения целей
программы в задачи руководящего персонала, а также частных вербальных и визу-
альных поддерживающих сообщений. Построение инфраструктуры программы —
это важное мероприятие. Всестороннее обучение, распространение информации
и план по управлению изменениями должны быть встроены в общие задачи про-
граммы, должны регулярно оцениваться и представляться на рассмотрение.
3.3.7. Измерение успеха с помощью информативных
показателей эффективности
Успешность внедрения всесторонней СМК можно оценивать с помощью про-
граммы информативных показателей эффективности. У программы показателей
двойное назначение: во-первых, она позволяет организации оценивать прогресс
работ по выполнению поставленных задач объектно ориентированным способом
с информационным типом управления (основанным на данных), и во-вторых, обе-
спечивает мониторинг эффективности каждого процесса, чтобы гарантировать по-
стоянное улучшение. Осуществляя анализ показателей эффективности СМК и ее
процессов, компания получает знания и понимание общего состояния своей систе-
мы и процессов и может разработать стратегии, основанные на управлении риска-
ми, для постоянного улучшения СМК и ее процессов.
Как только программа показателей введена в действие, показатели эффектив-
ности системы и процессов должны быть доступными и понятными владельцам
процессов, руководящему звену и заинтересованным подразделениям. Владель-
цы процессов должны понимать тенденции показателей, возникающие проблемы
и связанные риски. Заинтересованные подразделения вместе с владельцем процесса
выявляют возможности улучшений и готовят предложения по их реализации. Выс-
шее руководство обязано понимать возникающие проблемы и связанные с ними
риски и, соответственно, выделять ресурсы для корректирующих действий.
З.З.7.1. Разработка показателей эффективности
Информативные показатели эффективности работ по качеству и коммерческой дея-
тельности также должны регулярно анализироваться. К ним относятся, например:
• показатели эффективности работ по качеству — способность удовлетворять
требования стандартов и процедур;
• показатели поставок — способность удовлетворять потребность рынка;
• показатели издержек — экономия средств и эффективность затрат;
• показатели безопасности — случаи типа «почти ошибка» (near misses) и про-
исшествия, связанные с процессом.
Установление собираемых и анализируемых показателей эффективности осу-
ществляется на основе главной задачи: возможности делать заключения о стабиль-
ности системы или процесса. Все показатели должны разрабатываться совместно
с заинтересованными подразделениями с учетом запросов потребителей. Это вклю-
чает точки контакта нисходящих процессов системы качества. Без этого участия
Глава 3.3. Создание системы менеджмента качества и управление ею 337
и понимания разработанные показатели окажутся изолированными (несвязанными
с данными других процессов и функций) и малоинформативными, вызывая чув-
ство неудовлетворенности у высшего руководства и исполнителей. Без надлежаще-
го проектирования показатели могут стать только индикатором деятельности, что
приводит к отсутствию поддержки или минимальной поддержке работы владельца
процесса со стороны менеджмента.
Показатели эффективности могут быть «запаздывающими» индикаторами или
«опережающими». Оба типа показателей важны для владельца процесса и менедж-
мента. «Запаздывающие» индикаторы — это показатели, которые информируют
о способности процесса давать результаты или продукты. Они характеризуют эф-
фективность системы в прошедшем времени. Такие индикаторы могут помочь вла-
дельцам процессов, менеджменту и высшему руководству при определении степени
выполнения задач, достижения целей или соответствия существующим стандартам
и ожиданиям. «Опережающие» показатели фокусируются на входах процесса и по-
ставщиков процесса. Эти показатели являются важными индикаторами, дающими
возможность владельцам процессов и менеджменту предпринимать действия про-
активно, до возникновения несоответствий стандарту или отклонений от цели. Эф-
фективный владелец процесса будет понимать взаимосвязь «опережающих» показа-
телей и их влияние на «запаздывающие» показатели процесса. Показатели должны
разрабатываться исходя из потребностей организации, быть простыми для отслежи-
вания и представления и регулярно анализироваться.
3.3.7.2. Анализ показателей
Игнорирование системы качества и принципов улучшения процессов приводит
к неэффективности производства, ненадлежащему выполнению регуляторных тре-
бований и низкому моральному духу сотрудников. В надлежащую практику ведения
бизнеса входит регулярный анализ показателей процессов для оценки состояния
и результатов работы системы и процессов, обеспечивающих развитие организации.
Владельцы процессов должны знать все показатели, влияющие на их процессы,
и иметь доступ к вышестоящему руководству для сообщений о критических пока-
зателях. В отрасли имеются примеры, когда владельцы процессов, ответственные за
их исполнение, не знают, какие показатели эффективности собираются по их про-
цессу и собираются ли они вообще, а также не имеют фактических данных для обо-
снования адекватности своего процесса или его эффективности.
Регуляторные агентства возлагают ответственность за исполнение операций
в организации на руководителей. Владельцы процессов имеют доверительную от-
ветственность за результаты и эффективность операций, разделяемую с менеджмен-
том, и имеют возможность объяснять и интерпретировать полученные показатели.
В обязанности менеджмента входит знание операций, их результативности и ини-
циирование действий, необходимых для обеспечения соответствия со стандартами
и правилами, устанавливаемыми государством, отраслью и компанией.
Правила для фармацевтических продуктов требуют проведения ежегодного ана-
лиза продукта для определения и оценки внесенных в процессы изменений, кото-
рые могли повлиять на качество продукта. Однако принятая в отрасли надлежащая
практика может установить необходимость проведения ежеквартального или еже-
338
Часть 3. Качество
месячного анализа для более быстрого выявления проблем, принятия решений
и выполнения корректирующих действий. В анализе должны быть учтены ключе-
вые операционные параметры и критические показатели качества, важные для обе-
спечения безопасности и эффективности продукта. Некоторые другие ключевые
показатели коммерческой деятельности также помогают оценить преимущества
организации и должны быть включены в программу анализа показателей эффек-
тивности. Собираемые показатели должны демонстрировать владельцу процес-
са и менеджменту непосредственные признаки контролируемости процесса и его
способность получить желаемый результат. В противном случае владелец процесса
должен представить менеджменту предложение о возможностях постоянного улуч-
шения и описать изменения, необходимые для улучшения процесса.
3.3.7.3. Модель зрелости системы
Модель зрелости — это эффективный инструмент управления для определения
статуса процесса и установления стандарта, по которому оцениваются процессы.
Она предоставляет возможность оценки надежности и развития процесса в целом
и помогает в установлении приоритетов при распределении ресурсов. При оценке
степени развития процесса модель формирует базовую структуру для применения
управления рисками. Например, разработка систем, подобных асептической фасов-
ке, где имеется высокий риск и для больных, и для коммерческой деятельности,
должна предусматривать развитие процессов до зрелости более высокого уровня,
чем зрелость процессов, имеющих меньший риск несоответствия регуляторным
требованиям и незначительный риск для больных. Коммерческие условия ведения
бизнеса, в которые поставлена фармацевтическая и биофармацевтическая отрасль,
ограничивают ресурсы для разработки, повышения качества и производства, тре-
буют продуманного направления этих ресурсов в области, которые могут дать наи-
большие преимущества для организации.
Пример модели зрелости системы приведен на рис. 8. Использование такой мо-
дели позволяет группе по программе СМК и высшему руководству оценивать про-
цессы на основе объективного стандарта. Выделены пять основных уровней зре-
лости: от неформализованного, неструктурированного до «лучшего в классе». Для
каждого уровня в модели установлены определенные критерии, при выполнении
которых считается, что процесс достиг данного уровня. Эту модель также можно
разделить на отдельные подкатегории, например инфраструктуру, эффективность
и соответствие, что отражено в табл. 8. Каждая подкатегория может быть спроек-
тирована так, чтобы предоставлять информативные данные владельцам процессов
и менеджменту. Модель зрелости является отличным показателем эффективности
для измерения степени развития СМК и концентрации ресурсов, выделяемых выс-
шим руководством.
Подкатегории модели с помощью специально определенных параметров по-
казывают отдельные области, в которых требуется надежный процесс. Категория
инфраструктуры включает наличие способного владельца процесса и выделенно-
го куратора из подразделения обеспечения качества; владелец процесса обладает
глубоким знанием хода процесса, области его применения, границ процесса, по-
ставщиков, потребителей, а также функций и обязанностей. Задача — разработать
Глава 3.3. Создание системы менеджмента качества и управление ею
339
Рис. 8. Общее представление модели зрелости (источник: «Интеграция модели зрелости
возможностей», www.sei.cmu.edu, с изменениями)
Таблица 8 Пример модели зрелости
Категории Уровень 1 Уровень 2 Уровень 3 Уровень 4 Уровень 5 (формализованный (процесс (проактивное (постоянное (лучший подход отсутствует) определен) управление) улучшение) в классе)
Соответствие Инфраструктура Эффективность
Источник: «Интеграция модели зрелости возможностей», www.sei.cmu.edu, с изменениями.
в высокой степени интегрированный процесс, который полностью масштабируем
и универсален (может переноситься на другие площадки, подразделения и т. д.).
Соответствие — это ключевой параметр процесса в фармацевтической и био-
фармацевтической отрасли. Зрелость процесса зависит от степени соответствия
требованиям и наличия документации, такой как стандарты и СОП, от количества
негативных наблюдений, зарегистрированных при внутреннем аудите, аудите по-
ставщиков и государственных инспекциях, от количества несоответствий, оценки
риска процесса в отношении безопасности больных и, конечно, от эффективности.
Также должны иметься программы обучения, которые рассматриваются как одно
из условий соответствия процесса. Негативные наблюдения, сделанные при аудите
и инспекциях, являются основными показателями зрелости процесса. Процессы,
соответствующие критериям зрелости высокого уровня, представляют собой хоро-
шо контролируемый процесс, который постоянно производит однородный, отве-
чающий требованиям качественный продукт.
Параметры подкатегории эффективности определяют эффективность процес-
са — желательно по заранее установленным стандартам или ожиданиям. Показатели
340
Часть 3. Качество
эффективности должны служить индикаторами состояния и надежности системы.
В показатели эффективности могут быть включены длительность выполняемого
цикла, время подготовки к новому циклу после завершения производства, оцен-
ка риска в отношении факторов коммерческой деятельности. Задача — повысить
целевые показатели эффективности, развивая стратегический подход, снижая ва-
риабельность и улучшая результативность. Результаты, полученные при повышении
эффективности процесса, вносят значительный вклад в удовлетворение коммерче-
ских потребностей.
К преимуществам модели зрелости относится полезная методология, исполь-
зующаяся группой по программе СМК и высшим руководством, которая помогает
оценить и классифицировать процессы, а также предоставляет владельцам процес-
сов цели по развитию процессов. Также, используя положения управления риска-
ми, высшее руководство может оценить все процессы системы, определить области
вложения ресурсов и требуемый уровень зрелости для каждого процесса, чтобы обе-
спечить эффективную деятельность компании.
Задачи, определяемые уровнями зрелости, лучше всего выполняются владельца-
ми процессов, группой по программе СМК и высшим руководством. Рекомендуется
проведение оценки рисков каждого процесса в отношении рисков для потребителей
и коммерческой деятельности. Это позволяет СМК присвоить процессам приори-
тетность и определить, какие процессы следует поднять на более высокий уровень
в модели зрелости. Результаты оценки рисков должны быть проанализированы выс-
шим руководством, чтобы определить правильность установленных приоритетов
и соответствие задачам компании. Такое обсуждение при наличии обратной связи
обеспечит поддержку владельцев процессов высшим руководством при проведении
работ по развитию процесса до более высокого уровня зрелости. Хорошо спроек-
тированная СМК будет обеспечивать двусторонний обмен информацией между
высшим руководством и владельцами процессов. Следует подчеркнуть важность
доведения установленных высшим руководством приоритетов до сведения владель-
цев процессов, а также необходимость информирования руководства владельцами
процессов о серьезных проблемах и затруднениях. Это улучшит процедуру согласо-
вания приоритетов между высшим руководством и владельцами процессов. Такая
интеграция обеспечивает работу компании над соответствующими вопросами в со-
ответствующее время соответствующими людьми.
3.3.7.4. Выполнение требований к зрелости процесса
Должна быть создана специальная группа или комиссия для анализа и одобрения
результатов процесса после достижения им желаемого уровня зрелости. Основная
задача этой экспертной комиссии — проверка соответствия всех получаемых резуль-
татов нужному уровню качества и наличия требуемой документации. Эта комиссия
может предоставлять в качестве обратной связи информацию владельцам процесса
или группе по программе СМК для распространения полученного опыта.
Хорошо спроектированная программа анализа показателей жизненно необходи-
ма для успешного внедрения СМК. Анализ должен включать изучение показателей
СМК, оценку уровня зрелости процесса и рассмотрение его эффективности, ин-
фраструктуры и соответствия. Эти показатели являются основными данными для
Глава 3.3. Создание системы менеджмента качества и управление ею 341
оценки прогресса работ по СМК и в долгосрочной перспективе, и в отношении еже-
годных планов по качеству. Показатели снабжают высшее руководство и владельцев
процессов определенными и объективными данными для определения достижения
поставленных целей. Высшее руководство сможет иметь информативные и сопо-
ставимые данные об эффективности процессов на разных площадках и участках
и данные по оценке рисков для обоснования распределения ресурсов, согласующе-
гося с потребностями коммерческой деятельности.
3.3.8. Обеспечение постоянного улучшения: проекты
Фармацевтические и биофармацевтические компании испытывают значительное
напряжение из-за обязательств по соблюдению однородности качества поставляе-
мого продукта, а также требований по снижению общей стоимости продукта. Целью
внедрения руководств ICH £?8, Q9 и, в конечном итоге, СЮ является установление
характеристик процессов на основании оценки рисков и улучшение их с помощью
хорошо спроектированной СМК. Существуют регуляторные и коммерческие спо-
собы постоянного улучшения процессов СМК, встраивающие качество в процес-
сы и улучшающие их результативность. В настоящее время регуляторные агентства
сосредоточивают внимание на наличии в компаниях эффективных систем, защи-
щающих общественное здравоохранение путем гарантий безопасности и эффек-
тивности продуктов. Понимание производственных процессов с помощью хорошо
спроектированных исследований характеристик процесса является одним из наи-
более эффективных и результативных методов обеспечения успешности и качества
процесса. Для выполнения требований коммерческой деятельности и потребите-
лей, а также ожиданий и рекомендаций регуляторных органов от фармацевтиче-
ских и биофармацевтических компаний ожидается внедрение методов постоянного
улучшения на основе оценки рисков производственного процесса.
3.3.8.1. Улучшение процессов
Процесс СМК должен соответствовать стандарту «шесть сигм», который устанав-
ливает жизненный цикл улучшения процесса и включает следующие этапы: опре-
деление (процессов и показателей), измерение и контроль (выявление проблем
и вопросов), анализ (проблем и результатов) и улучшение (внедрение), которое за-
мыкается на измерение и контроль [11]. Пример жизненного цикла улучшения про-
цесса представлен на рис. 9.
Основополагающим элементом постоянного улучшения является владелец
процесса, который полностью понимает процесс и обладает знаниями о том, ка-
ким образом процесс влияет на другие процессы в рамках СМК. Для понимания
причинно-следственных взаимосвязей между процессами требуется тесное взаимо-
действие между владельцами процессов и заинтересованными подразделениями.
Такая интеграция критична на протяжении всего жизненного цикла процесса: от
проектирования до разработки и управления.
Процессы, которые можно подвергать улучшениям, предварительно должны
быть тщательно определены и при этом стабильны (предсказуемы). Это означает не
то, что процесс или продукт процесса имеет желаемое качество, а то, что он хорошо
342
Часть 3. Качество
Рис. 9. Процесс постоянного улучшения
изучен и предсказуем. Используя показатели, тенденции и оценку рисков, следует
выявить явные проблемы и затруднения. Владельцы процессов могут использовать
совещания менеджмента, на которых обсуждаются результаты анализа, для пред-
ставления предложений по улучшению процессов.
3.3.8.2. Предложения по улучшению процессов
Если проблема или изменение требует установления приоритета из-за необходимо-
сти получения от компании инвестиций или дополнительных ресурсов, владелец
процесса вместе с заинтересованными подразделениями должен представить на
рассмотрение предложение об улучшении процесса. Предложение должно содер-
жать по крайней мере описание проблемы или необходимых изменений, оценку
влияния проблемы на площадку или участок, основанную на анализе риска, и опи-
сание необходимого действия и/или проекта, включая стоимость проекта и требую-
щихся ресурсов.
Владелец процесса должен рассмотреть целесообразность привлечения к раз-
работке предложения технических экспертов для подготовки описания проблемы,
проведения анализа рисков и экономической оценки снижения затрат или исклю-
чения неэффективных затрат. Как правило, основная компетенция владельца про-
цесса согласуется с процессом, однако у владельца могут быть недостаточные на-
выки коммерческой деятельности или управления проектами. Возможно, владельцу
процесса потребуется помощь в изложении, понятном высшему руководству, какие
выгоды будут получены при реализации предлагаемых изменений в сравнении с ри-
сками при отказе от предложения.
Имеется достаточное количество инструментов оценки рисков, например девя-
тиблочная матрица анализа рисков (табл. 9) или анализ вида отказов и их послед-
ствий (FMEA, failure mode and effect analysis), которые владелец процесса может ис-
пользовать при оценке процесса или проблемы, чтобы лучше понять и обосновать
вероятность отказов и тяжесть проблемы процесса. Результаты этого оценивания
помогают при установке приоритетов проблем и выявлении конкретных действий,
Глава 3.3. Создание системы менеджмента качества и управление ею
343
Таблица 9 Девятиблочная матрица анализа рисков
Частота Ущерб малый средний большой
Высокая Низкая Одиночные случаи Умеренный риск Значительный риск Значительный риск Низкий риск Умеренный риск Значительный риск Низкий риск Низкий риск Умеренный риск
необходимых для снижения риска или определения плана действий в непредвиден-
ных обстоятельствах при возникновении проблем, риски которых снизить нельзя.
Для фармацевтической и биофармацевтической отрасли важно, чтобы все оценки
рисков завершались оценкой влияния на эффективность и безопасность продукта,
а также на факторы коммерческой деятельности. О любом этапе процесса и любой
проблеме, которые могут оказать негативное влияния на эффективность и безопас-
ность продукта, должно быть немедленно сообщено высшему руководству, после
чего проблема должна быть немедленно рассмотрена. Если выдвинутому предло-
жению необходимо установить приоритет, владелец процесса должен четко указать
возможности сокращения или исключения затрат, используя модель затрат на ка-
чество с последующим привлечением вышестоящего руководства. Комбинирова-
ние рисков и затрат — это эффективный способ привлечь внимание и поддержку
высшего руководства.
Реакция высшего руководства на предложение по улучшению процесса заклю-
чается в осознании проблемы или возможности, понимании связанного риска(ов),
одобрении или возврате на доработку предлагаемого действия или проекта и предо-
ставлении соответствующего финансирования и/или ресурсов. После одобрения
и инициирования проекта и предложенных действий следует на регулярной основе
наблюдать за ходом их выполнения, чтобы обеспечить необходимый прогресс.
3.3.8.3. Задача и проект
Улучшения процесса могут проводиться с помощью завершения задачи или проек-
та. Задача — это деятельность, которая может быть выполнена владельцем процесса
с минимальными затратами и/или ресурсами в течение короткого периода времени.
Проект определяется как временная работа по предоставлению продукта или услуги,
которые находятся вне поддержки владельца процесса. В целом проект требует бо-
лее одного эквивалента полной занятости (full-time equivalent, РТЕ/’ЭПЗ)', пересека-
ется с различными функциями в организации и имеет большую длительность работ.
Статус улучшения, актуализация данных и проблемы должны обсуждаться регуляр-
но на совещаниях менеджмента или управляющего комитета. Задачам и проектам
обязательно присваивают приоритет на основании результатов анализа рисков в от-
ношении безопасности больных, эффективности и соответствия процесса.
1 Эквивалент полной занятости — единица измерения рабочего времени, соответствующая
времени, отрабатываемому за определенный период на должности с полной занятостью. —
Примеч. перев.
344
Часть 3. Качество
Если улучшение процесса соответствует критериям проекта, то должен быть на-
значен руководитель проекта. Формальное управление проектом позволяет при-
менить холистический и интегрированный подход к изменению. Руководитель
проекта не должен заменять владельца процесса, но обязан контролировать, что
проблемы установлены, приоритеты присвоены, ресурсы выделены, основные эта-
пы выполняются в поставленные сроки, вопросы поднимаются и решаются, о про-
грессе сообщается. Необходимо, чтобы владелец процесса участвовал в руководстве
проектом вместе с заинтересованными подразделениями или в управляющем коми-
тете, предоставляя поддержку и рекомендации. Это позволяет владельцу процесса
сосредоточить внимание на проблемах и улучшениях (его основная компетенция),
а управляющему проектом — методически продвигать выполнение проекта. При
выполнении проекта критичным является определение и измерение успехов про-
екта.
3.3.8.4. Показатели проекта
Показатели проекта должны быть определены, чтобы сравнивать реальную полу-
чаемую прибыль от произведенного изменения с запланированными результатами.
Очень часто компании внедряют изменение и переходят к следующему проекту без
полного понимания, получили ли они желаемый результат. Проект, который не при-
нес ожидаемой пользы, может привести к нерезультативному процессу, конфликтам
в точках контакта с другими процессами и к неудовлетворенности персонала и по-
требителей.
Применение системного подхода к постоянному улучшению СМК и использова-
ние инструментов формального управления рисками повышают общую эффектив-
ность организации. Осуществление постоянного улучшения входит в полномочия
и обязанности владельцев процессов. Для выявления тенденций, проблем и воз-
можностей используются численные показатели. Заинтересованные подразделения
задействованы в процессе, а менеджмент участвует в установлении приоритетов
и выделении сотрудников для решения задачи или выполнения проекта. Процес-
сы непрерывно управляются и оцениваются. Постоянные улучшения, основанные
на анализе рисков, позволяют организации выделять ресурсы и финансы для вы-
полнения наиболее важных проектов, которые окажут наибольшее влияние. После
внедрения улучшений сотрудники получат выгоду от предсказуемого «стройного»
процесса, который позволит им сосредоточиться на проактивной по своей природе
работе, в отличие от высокострессового реагирования на каждодневные проблемы.
Владелец процесса заслужит доверие, если он демонстрирует образ действий, при
котором своевременно принимаются соответствующие решения, и улучшения
процесса направлены на системное решение проблем, а не на поверхностное, при-
водящее к повторному появлению тех же проблем.
3.3.9. Обеспечение постоянного успеха
Построение структуры для внедрения и поддержания в рабочем состоянии СМК
требует ресурсов и решимости высшего руководства и персонала. Современная ми-
ровая фармацевтическая и биофармацевтическая отрасль и регуляторные органы
Глава 3.3. Создание системы менеджмента качества и управление ею
345
требуют от организации проведения работ по созданию и поддержанию в рабочем
состоянии надежной системы и процессов, соответствующих требованиям по про-
изводству качественного продукта. Дальнейшее усиление конкуренции, сокращение
времени, отводимого для запуска продукта на рынок, необходимость в результатив-
ной разработке и прохождении регистрации с первой попытки усиливают потреб-
ность в надежных устойчивых процессах. Глобальный рынок продолжает усиливать
давление на отрасль по доставке жизненно необходимых препаратов и средств, вли-
яющих на образ жизни, в более короткие сроки и по более низким ценам.
3.3.9.1. Установка совместных целей
Компании, спроектировавшие, разработавшие и внедрившие СМК и соответствую-
щие процессы, легкие для исполнения и понимания и обеспечивающие получение
коммерческих результатов и выполнение регуляторных требований, будут обладать
конкурентным преимуществом над другими такими же предприятиями. Они будут
быстрее и эффективнее применять новые технологии, поглощать новые организа-
ции путем слияний и приобретений, будут способны выделить требуемые ресурсы
на соответствующие коммерческие потребности и, что наиболее важно, смогут бы-
стро измениться и адаптироваться под рыночные требования. Зависимость от че-
ловеческого фактора, фрагментированность процедур и «родоплеменные знания»,
в отличие от интегрированных функциональных процессов, приведут к нежелатель-
ным результатам на всех уровнях организации.
Для обеспечения постоянного успеха прежде всего требуется установление взаи-
мосвязанных целей для организации. Эти цели должны соответствовать потребно-
стям коммерческой деятельности, сотрудников и акционеров. Хорошо спроекти-
рованные процессы с подотчетным владением, которые внедряются при помощи
обсуждений, проектирования и поддержки со стороны высшего руководства, функ-
ционального менеджмента, линейных заинтересованных подразделений и рядового
персонала, формируют фундамент для общих совместных потребностей (рис. 10).
Если не учитывается мнение хотя бы одной из этих групп, можно ожидать лишь
формальной поддержки и, как следствие, несостоятельности программы.
Необходимо зафиксировать эти совместные цели в документации программы.
К ним относятся основные долгосрочные цели программы, выгоды, которые долж-
ны получить заинтересованные подразделения, и ежегодные планы по качеству для
достижения основных промежуточных целей. Успех программы зависит от под-
держки со стороны высшего руководства, надежности конструкции системы, над-
лежащего обучения сотрудников, наличия информативных показателей для изме-
рения эффективности и от работ по постоянному улучшению.
Для поддержания постоянной работоспособности СМК следует предусмотреть
механизмы обратной связи, передающие информацию от заинтересованных под-
разделений по восприятию программы, ясности в понимании и возможностям
улучшения. Одним из методов получения информации такого типа является соз-
дание фокус-групп. Другая возможность заключается в регуляторных инспекциях
и аудитах потребителями. Надлежащие действия по внедрению изменений и рас-
ширений с признанием роли сотрудников обеспечат поддержку и участие в про-
грамме заинтересованных подразделений.
346
Часть 3. Качество
Рис. 10. Модель поддержки владельца процесса
Совместные цели будут способствовать успеху программы, усиливать понимание
необходимости системного подхода и выгод, которые он даст. Не бывает ситуации
лучше, чем такая, когда вся организация выстроена вокруг бизнес-проекта и рабо-
тает над его исполнением при постоянной поддержке сотрудниками друг друга.
3.3.9.2. Награды и признание
Владелец процесса имеет важные обязанности. Необходимо выбирать владельцев
на основании предварительно установленных критериев, которые по своей приро-
де являются селективными. Владельцы процессов управляют производственными
операциями, и, следовательно, необходимо отмечать их особые усилия и высокую
ответственность. Такое признание может выражаться в различных формах. Значи-
тельное различие в базовых квалификациях и премии являются важным стимулом
для занятия места владельца процесса. Другим стимулом и наградой для владель-
цев является постоянное развитие. В дополнение к материальным и финансовым
поощрениям еще одной формой награды и признания является выражение дове-
рия руководства к владельцу процесса. Процессы неизбежно сопровождаются не-
нужными тратами и включают неэффективные операции, что побуждает владельца
процесса улучшить его и получить за это признание.
Публичное признание системной программы и достижений владельца процесса
крайне важно. Такое признание можно выражать в ходе регулярных оперативных
совещаний, совещаний по анализу показателей, на собраниях коллектива пред-
приятия и планерках, с помощью плакатов, информационных писем и на собра-
ниях подразделения. Простая благодарность и небольшие подарки высоко ценятся
сотрудниками, усиливают поддержку менеджмента и приверженность программе.
Владельцы процессов и заинтересованные подразделения представляют собой наи-
более влиятельную группу, мнение которой о полезности программы следует рас-
пространять и которую надо развивать для обеспечения постоянного успеха.
Глава 3.3. Создание системы менеджмента качества и управление ею 347
Исследования показывают, что одни лишь финансовые вознаграждения не удо-
влетворяют персонал и не могут удержать сотрудников. Большая текучесть кадров
означает для компании огромные финансовые и конкурентные издержки. Боль-
шинство сотрудников, получив предложение о равной или более высокой оплате
труда, но не имея удовлетворяющей их работы, перейдут в другую компанию или на
другую должность. Плохо интегрированная СМК с излишне усложненными проце-
дурами часто является причиной такой неудовлетворенности. Повторение работы,
потеря ценного времени или получение нестандартного продукта не удовлетворяют
современных хорошо образованных и конкурирующих друг с другом сотрудников
в фармацевтической и биофармацевтической промышленности. Затраты на поиск
кадров, перемещения и удержания сотрудников достаточно высоки. Исключение
этих затрат может быть одним из аргументов для поддержки программы.
3.3.9.3. Обеспечение совместимости действующей программы
Достижения всесторонней программы СМК должны разделяться между площадка-
ми и быть совместимыми. Совместимость обеспечивает общее компетентное выс-
шее руководство предприятия. Непротиворечивая программа СМК также позволя-
ет переводить сотрудников с одной площадки на другую практически без затрат на
обучение и ознакомление с требованиями. Различия в путях развития будут снижать
поддержку СМК и эффективность ее работы. Гибкость в выполнении важна, одна-
ко должны быть приняты меры по уменьшению расхождений в терминологии, ин-
терпретации понятий и философии. За небольшое время исполнения всесторонней
программы будет быстро достигнута необходимая результативность. Обеспечение
совместимости также увеличивает количество пользователей процессов с одинако-
вым опытом, оказывает большее воздействие на улучшение процесса и, следова-
тельно, усиливает поддержку.
Регуляторы и потребители требуют гарантий в совместимости фармацевтических
и биофармацевтических производственных операций. Современные промышлен-
ные цепочки используют многочисленные площадки с разным местонахождением
для производства продукта. Системы качества должны рассматриваться как неотъем-
лемая часть цепочки создания ценности. Для этого требуется, чтобы все площадки
соответствовали регуляторным требованиям по всем своим процессами и системам.
Сильные подсистемы одной площадки не компенсируют слабые или отсутствую-
щие подсистемы на другой. На компанию налагают штрафы, и бизнес несет убытки
из-за проблем на отдельной площадке или в самом слабом звене в товаропроводя-
щей цепи. У менеджмента должен быть механизм для измерения своих процессов,
и всесторонняя СМК — это механизм демонстрации способностей организации.
3.3.9.4. Привыкание к программе
Со временем происходит привыкание к программе. На всех уровнях организации
необходимо признавать и подтверждать, что подход в виде СМК является образом
ведения бизнеса. Такой способ осуществления коммерческой деятельности стано-
вится частью культуры организации и своего рода привычкой для высшего руковод-
ства, менеджмента и персонала. Регуляторы и потребители, акционеры и больные
увидят в этом преимущества организации.
348 Часть 3. Качество
Литература
1. Webster s New Collegiate Dictionary, ninth edition, 1986, p. 1199.
2. Arling, E. R. (2004), Integrating QSIT into quality plans, Biopharm. Int., June, 44-46,48,
50-52.
3. Drug Industry Daily, Oct. 12,2006, Vol. 5, No. 200, Washington Business Information.
4. 2006 PDA/FDA joint regulatory conference, Sept. 11,2006, Washington, DC.
5. Joneckis, C. (2006), Ph.D. presentation at 11th Annual GMP by the Sea, Aug. 28-30, Cam-
bridge, MD.
6. Quality systems regulations CDRH, available: www.fda.gov.
7. CPGM 7356.002, CDER, available: www.fda.gov.
8. IOM biological inspections 7345.848 CBER, available: www.fda.gov.
9. Rockefeller, J. D., Ill (1973), The Second Revolution, Harper-Row, New York.
10. Kotter, J. P. (2001), Leadership insights, Harvard Bus. Re., p. 29.
11. George, M. (2004), Lean Six Sigma Pocket Tool Book, McGraw-Hill Professional, New
York, p. 4.
Глава 3.4. Улучшение качества процесса
Жи-хон Ванг
Университет Род-Айленда (Кингстон, Род-Айленд)
3.4.1. Диагностика процесса
З.4.1.1. Введение
Улучшение качества процессов начинается с проведения диагностики для выявле-
ния проблем. Затем выполняют корректирующие действия, после чего за процес-
сом ведется постоянное наблюдение. Как правило, при проведении диагностики
анализируются симптомы, формулируются и проверяются гипотезы и определяют-
ся причины проблем. В табл. 1 указаны основные инструменты для проведения диа-
гностики. Их описание приведено в разделе 3.4.1.2.
Таблица 1
Основные инструменты улучшения качества процесса при его диагностике
Основные инструменты для улучшения качества процесса
Основные действия для диагностики причины о i - | = | S Ш £ к и g S се s ° се си sB « s s се & о в се » S = « Е = S uvvvnnn» График g = 1 s fl S a. а в 2 S £ & = М О м о © с § = се К[ сх © S £ и се s 6Г & с « | & ! се м & 1
с 5 * tl а <=> о о к се & 1© и и о
Анализ симптомов • • • • • • о • о
Формулирование • о • о • о о • о
гипотез
Проверка гипотез • • о • • о • о •
Определение • • • • о о о о •
причины (причин)
Примечание: • — основной; о — вспомогательный.
3.4.1.2. Основные инструменты диагностики процесса
Причинно-следственная диаграмма. Причинно-следственная диаграмма1 связывает
возможные причины проблемы с их эффектами. Этот инструмент может быть очень
полезен для диагностики процесса. Он позволяет исследовать возможные причины
определенной проблемы структурированным и систематическим образом. Для по-
строения диаграммы рекомендуется выполнить следующие действия:
1. Определить проблему (эффект).
2. Записать формулировку проблемы на правой стороне листа и провести к ней
стрелку с левой стороны листа.
1 Другое название — диаграмма (схема) Исикавы. — Примеч. перев.
350
Часть 3. Качество
3. Обдумать основные категории причин, вызывающих проблемы, и нарисовать
основные боковые стрелки для каждой причины по направлению к основной
стрелке.
4. Для каждой основной боковой стрелки указать уточненные причинные фак-
торы (подфакторы) в виде основных боковых ответвлений.
5. Подписать факторы следующего уровня, влияющие на подфакторы, на боко-
вых ответвлениях от основных ответвлений.
6. Убедиться, что все возможные элементы, которые могут вызвать проблему,
указаны на диаграмме.
На рис. 1 показана причинно-следственная диаграмма, которая используется для
выявления причин, приведших к проблеме в биофармацевтическом производствен-
ном процессе, и указаны возможные основные причины и сопутствующие им фак-
торы. После выявления причин применяются другие инструменты для определения
значимости разных причин для наблюдаемого эффекта. Затем предпринимаются
действия по исключению или снижению влияния этих причин.
Диаграмма Парето. Принцип Парето предполагает, что проблема (эффект) может
быть обусловлена относительно небольшим количеством причин. В количествен-
ных терминах это выражается так: 80% проблем обусловлены 20% причин (обору-
дование, исходные материалы, операторы и т. д.) и, следовательно, усилия, направ-
ленные на решение 20% основных причин, могут решить 80% проблем. Диаграмма
Парето имеет три основных элемента: 1) причины проблемы, расположенные по
степени значимости; 2) частота каждой причины; 3) кумулятивная сумма значимо-
сти причин (в %). На рис. 2 приведен пример диаграммы Парето, показывающий
ошибки, сделанные в аптечной сети в течение 1 месяца.
Гистограмма. Гистограмма — это графическое представление вариабельности
в наборе данных. Данные группируются в категории, а значения для всех категорий
откладываются на графике в виде ряда столбцов. В табл. 2 приведены 40 наблюде-
ний по сроку годности определенного препарата и распределение частот. На рис. 3
показана гистограмма для указанных данных по сроку годности препарата.
Рис. 1. Причинно-следственная диаграмма
Глава 3.4. Улучшение качества процесса
351
% 48,1 29,8 12,4 6,3 2,1 1,2
Кумулятивный 481 780 д04 дб7 988 1000
суммарный эффект, %
Рис. 2. Диаграмма Парето, показывающая ошибки в аптеках
Таблица 2
Срок годности препарата (дни)
102,2 104,1 103,5 104,5 103,2 103,7 103,0 102,6
103,4 101,6 103,1 103,3 103,8 103,1 104,7 103,7
102,5 104,3 103,4 103,6 102,9 103,3 103,9 103,1
103,3 103,1 103,7 104,4 103,2 104,1 101,9 103,4
104,7 103,8 103,2 102,6 103,9 103,0 104,2 103,5
Диапазон значений Медиана Частота Кумулятивная сумма, %
101,5 <х< : 102,0 101,75 2 5,00
102,0 <х< : 102,5 102,25 2 10,00
102,5 <х< : 103,0 102,75 5 22,50
103,0 <х< : 103,5 103,25 15 60,00
103,5 <х< : 104,0 103,75 8 80,00
104,0 х< : 104,5 104,25 6 95,00
104,5 <х< : 105,0 104,75 2 100,00
Диаграмма рассеяния. Диаграмма рассеяния является основным инструментом
для выявления возможной связи между двумя переменными. Диаграммы рассеяния
похожи на линейные диаграммы тем, что в них используются оси абсцисс и ординат
для расположения данных в виде точек. Однако у них другое назначение: диаграм-
мы рассеяния показывают, насколько одна переменная влияет на другую. Взаимо-
связь между двумя переменными называется корреляцией. Чем больше расположе-
ние точек-данных на графике похоже на прямую линию, тем выше корреляция двух
переменных. Если точки-данные формируют прямую линию от близких к нулю
352
Часть 3. Качество
Срок годности
Рис. 3. Гистограмма для срока годности препарата
значений до бблыпих значений х и у, то говорят о наличии положительной корре-
ляции двух переменных. Если линия расположена от бблыпих значений на оси у
к большим значениям на оси х, то имеет место отрицательная корреляция перемен-
ных. На рис. 4 приведены несколько примеров диаграмм рассеяния.
График нормального распределения. График нормального распределения — это
графический метод оценки, определяющий, подчиняется ли набор данных закону
нормального распределения. Данные наносятся на график для сравнения с теоре-
тическим нормальным распределением таким образом, что точки формируют при-
близительно прямую линию. Отклонения от этой прямой линии свидетельствуют
об отклонении от нормального распределения. График нормального распределе-
ния является важным инструментом для улучшения качества процесса, поскольку
многие другие инструменты требуют допущения о нормальном распределении дан-
ных. График нормального распределения для срока годности препарата по данным
табл. 2 приведен на рис. 5. Как видно из приведенного графика, данные группиру-
ются вдоль прямой линии и находятся в пределах 95%-ного доверительного интер-
вала. Таким образом, можно сказать, что срок годности данного препарата подчи-
няется закону нормального распределения.
Другие инструменты
Столбчатый график. Данный график применяется для анализа структуры дан-
ных. Он показывает несколько важных характеристик: медиану, вариабельность,
отклонение от симметрии и наличие выпадающих значений.
Диаграммы потоков. Диаграмма потока процесса может использоваться для изу-
чения и понимания процесса.
Сбор данных. Данные необходимы для проведения надлежащей оценки действу-
ющего процесса. Инструментами для сбора данных являются перечень вопросов
и листы данных.
Стратификация (расслоение). Этот метод используется для разделения данных на
группы на основе категорий или характеристик. Метод используется или для пред-
Глава 3.4. Улучшение качества процесса
353
у
х
Срок годности
Рис. 5. График нормального распределения срока годности препарата с 95%-ным
доверительным интервалом
354
Часть 3 Качество
варительной обработки данных с последующим применением других инструмен-
тов, или вместе с другими инструментами анализа данных, например диаграммами
рассеяния.
3.4.2. Стабилизация и улучшение процесса
3.4.2.1. Введение
Основные положения концепции контрольных карт. Контрольные карты — это один
из важнейших инструментов улучшения качества процессов. Они используются для
определения природы изменчивости процесса и для облегчения прогнозирования
и управления процессом. Значения параметров качества откладывают на графике
относительно номера пробы или времени, как показано на рис. 6. Центральная ли-
ния обозначает среднее значение параметра процесса. Верхняя и нижняя контроль-
ные границы UCL и LCL (upper control limit и lower control limit) обычно устанавли-
ваются на расстоянии трех стандартных отклонений выше и ниже средней линии,
и, таким образом, они могут использоваться для обнаружения ситуаций «вне зоны
контроля», причем не создавая ложных тревог. О наличии ситуации «вне зоны кон-
троля» обычно сигнализирует точка, нанесенная вне контрольных пределов, или
группа точек (кластер), образующая аномальную конфигурацию.
Точки, вносимые в контрольную карту, обычно основаны на данных, получае-
мых при отборе проб в ходе процесса. После отбора достаточного количества образ-
цов и внесения данных в контрольную карту можно провести оценку стабильности
процесса. Стабильный процесс находится «под контролем», то есть в управляемом
состоянии, в то время как процесс «вне зоны контроля» является нестабильным
и находится в неуправляемом состоянии. В зависимости от типа параметра качества
контрольные карты можно разделить на две группы: контрольные карты для коли-
чественных данных и контрольные карты для качественных данных1. Контрольные
карты для количественных данных применяются для мониторинга параметров каче-
Верхняя контрольная граница (UCL)
' <АА/"—
Нижняя контрольная граница (LCL)
Номер образца
Рис. 6. Контрольная карта на основе нормального распределения
1В стандартах ГОСТ Р по контрольным картам и методам статистического управления ка-
чеством такие карты называются контрольными картами для альтернативных данных. Карты
для количественных данных часто называют картами для непрерывных переменных. — При-
меч. перев.
Глава 3.4. Улучшение качества процесса
355
ства, которые из-за своей природы постоянно изменяются; контрольные карты для
качественных данных используются для мониторинга параметров качества, кото-
рые не измеряются численно. Определение центральной линии и контрольных ли-
ний описано в разделах 3.4.2.2 и 3.4.2.3 с учетом различных типов контрольных карт.
Применение контрольных карт. Контрольные карты предназначены для направле-
ния внимания менеджмента на особые (неслучайные) причины отклонений в про-
цессе при их появлении. При оценке контрольных карт следующие признаки могут
указывать на неуправляемость процесса:
• Выпадающее значение. Одна или более точек, которые лежат вне контрольных
линий.
• Серии. Серия точек выше или ниже центральной линии.
• Тренд. Непрерывное возрастание и падение точек на графике.
• Цикл. Периодически повторяющееся особое расположение точек.
Как правило, при разработке контрольной карты и ее применении необходимо
выполнить следующие действия:
• Определить «базовый период» для разработки первоначальной контрольной
карты.
• Собрать пробные данные за базовый период.
• Рассчитать параметры для контрольной карты, то есть центральную линию
и контрольные линии.
• Нанести собранные пробные данные в виде точек на график с центральной
и контрольными линиями.
• Определить, могут ли параметры карты использоваться для мониторинга про-
цесса; пересмотреть параметры, если это необходимо.
• Собрать действующие образцы и продолжать мониторинг процесса с исполь-
зованием разработанной контрольной карты.
• Проводить периодические проверки параметров контрольной карты.
Контрольные карты для количественных данных широко применяются во мно-
гих производственных и непроизводственных образованиях. Они могут исполь-
зоваться для мониторинга, например, внутреннего диаметра корпуса самолета,
содержания влаги в таблетированных лекарственных препаратах, массы нетто фар-
мацевтического продукта, длительности обработки телефонных запросов и уровня
удовлетворенности потребителей. Последние два примера относятся к непроизвод-
ственным приложениям.
Контрольные карты для качественных данных используются реже, чем карты
для количественных данных. Как правило, контрольные карты для качественных
данных применяют, когда невозможно или непрактично измерение показателей ка-
чества продукта. Примерами использования таких карт являются мониторинг доли
несоответствия в производстве определенного датчика, количества дефектных дио-
дов в электронной сборке, количества дефектов в текстильном производстве, доли
дефектных серий в биомедицинском промышленном производстве и количества
ошибок, обнаруженных в аптеке.
В большинстве случаев выбор между контрольными картами для количествен-
ных и качественных данных абсолютно ясен, но иногда этот выбор не будет таким
356
Часть 3. Качество
очевидным. Например, если показатель качества представляет мягкость предмета,
например в производстве подушек, то может использоваться или фактическое из-
мерение, или классификация мягкости. Менеджеры по качеству и инженеры будут
обязаны рассмотреть несколько факторов при выборе контрольной карты, включая
стоимость, трудоемкость, чувствительность и размер пробы. Контрольные карты для
количественных данных обычно предоставляют больше информации для анализа, но
стоят дороже при внедрении и использовании. Контрольные карты для качествен-
ных данных являются менее чувствительными и менее дорогостоящими, однако для
получения определенной статистической значимости требуется больше образцов.
3.4.2.2. Контрольные карты для качественных данных
Контрольные карты для качественных данных включаютр-карты, пр-карты, с-карты
и и-карты. Первые два типа карт применяются, когда внимание направлено на долю
или количество несоответствий, другие два типа карт используются для изучения
самих несоответствий. Большинство фармацевтических промышленных произво-
дителей применяют одну или несколько таких карт.
Контрольные р-карты. Эти карты могут использоваться для отслеживания доли
дефектной продукции в процессе. Долей дефектов называют отношение количества
дефектных единиц в популяции к общему количеству единиц в этой популяции.
В фармацевтическом производстве единица считается дефектной, если она не вы-
держивает требований стандартов по одному или более показателям, например по
номинальному объему флаконов, влажности, прочности на излом и растворимости.
Давайте предположим, что была отобрана и исследована случайная выборка п
единиц из процесса с постоянной долей дефектов р и было найдено D дефектных
единиц, не соответствующих требованиям; тогда D — случайная величина, подчи-
няющаяся биномиальному закону распределения с параметрами пир. Если истин-
ная доля несоответствий р известна, то параметры р-карты будут следующими:
l/CL = p+3 1Р^~Р\
N п
центральная линия =р,
(1)
UCL=p-3 J-(1—-,
V п
В реальных условиях доля дефектов р неизвестна в большинстве случаев и поэто-
му должна быть рассчитана по данным пробы. Рассчитанная доля р. может быть
вычислена для z-й отобранной пробы, и тогда среднее значение р может быть по-
лучено как среднее арифметическое индивидуальных значений pt, найденных по т
пробам:
- е:,л
пгп т
(2)
Затем р может быть подставлено вместо р в выражения (1). Следует отметить,
что значение р должно периодически оцениваться, чтобы обеспечить его репрезен-
тативность среднему значению доли дефектов процесса.
Глава 3.4. Улучшение качества процесса 357
Контрольные лд-карты. Эти карты используются для мониторинга количества де-
фектных единиц, произведенных процессом. Параметры яр-карт очень похожи на
параметры р-карт:
U CL =рп+Зх/«р(1-р),
центральная линия = пр, (3)
LCL - пр-Зу/прЦ-р).
Так же, как для р-карт, если фактическое значение р недоступно, в расчетах мо-
жет использоваться р.
Контрольные с-карты. Эти карты могут использоваться для мониторинга коли-
чества несоответствий (дефектов) на проверяемую единицу. Проверяемая единица
может быть единицей продукта, серией разных продуктов или определенным из-
меренным объемом (массой) продукта. Многие фармацевтические производствен-
ные процессы являются серийным производством, когда исходные материалы или
полупродукт передается из одного процесса в другой процесс. Например, таблетка
с ненадлежащей оболочкой в процессе ее нанесения может считаться несоответ-
ствием (дефектом), а проверяемая единица при этом может быть определена как
1 кг таблеток.
Предположим, что проверяемая единица определенного продукта выбрана и ис-
следована для процесса, работающего со стабильной долей несоответствий с в про-
веряемой единице, и было найдено X несоответствий. Следовательно, X — случай-
ная величина, имеющая распределение Пуассона с параметром с. Если истинный
уровень несоответствия с известен, то параметры с-карты будут следующими:
UCL=c+3-Jc,
центральная линия = с, (4)
LCL =c-3-Jc.
Если истинный уровень несоответствий с неизвестен, то его можно рассчитать,
используя средние значения с, полученные из т проверяемых единиц, отобранных
в базовый период:
Затем в приложениях полученное значение с может быть подставлено в форму-
лы (4) вместо с. По этим формулам можно получить отрицательное значение для
LCL, в этом случае для нижней контрольной линии следует брать нулевое значе-
ние.
Контрольные «-карты. Этот вид карт используется для мониторинга доли несоот-
ветствий. Доля несоответствий и — это количество несоответствий х в проверяемой
единице по отношению к количеству п проверенных физических единиц (т. е. 100
футов трубы, 100 единиц в серии). Так же, как и в с-картах, параметры «-карты бу-
дут следующими:
358
Часть 3. Качество
tZCZ=w+
центральная линия = и,
(6)
LCL-u-3
Если истинное значение и недоступно, в уравнении (6) может быть использова-
но и .
Пример 1. Производитель медицинских изделий озабочен количеством дефект-
ной продукции и количеством несоответствий (дефектов) в продукции, произво-
димой на новой производственной линии. На этой линии случайным образом были
отобраны 20 партий медицинского изделия. В каждой партии 100 единиц продук-
ции. Каждая единица продукции в выбранных партиях была проконтролирована
и определена как «соответствующая» или «дефектная». При проверке также под-
считывалось количество выявленных дефектов. Полученные данные приведены
в табл. 3.
Таблица 3
Дефектная продукция и количество дефектов, обнаруженных в 20 партиях медицинского
изделия
Номер партии 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Количество дефектных единиц 3 2 4 2 5 2 1 2 0 5 2 4 1 3 6 0 1 2 3 2
Количество дефектов 9 7 13 8 6 8 10 10 7 10 12 9 11 15 8 12 11 8 7 15
По данным таблицы средняя доля р несоответствующих единиц продукции со-
ставляет 2,5%; среднее количество дефектов на партию с = 9,8; среднее количество
дефектов на единицу продукции й = 0,098, Полученные контрольные карты при-
ведены на рис. 7—10. Эти карты показывают, что процесс является управляемым,
и, соответственно, вычисленные параметры могут быть использованы для монито-
ринга последующей продукции.
-г 0,08-
UCL = 0,07184
р =0,025
LCL = 0
1 3 5 7 9 11 13 15 17 19
Номер пробы
Рис. 7. р-карта для примера с производством медицинского изделия
Глава 3.4. Улучшение качества процесса
359
8-1
х
L/CL = 7,184
пр = 0,025
LCL = 0
1 3 5 7 9 11 13 15 17 19
Номер пробы
Рис. 8. пр-Карта для примера с производством медицинского изделия
Номер пробы
Рис. 10. и-Карта для примера с производством медицинского изделия
3.4.2.3. Контрольные карты для количественных данных
К контрольным картам для переменных данных выборки относятся х- и 5-карты.
При рассмотрении численно выражаемых параметров качества обычно применя-
ются х -карты для мониторинга среднего значения параметра процесса и s-карты
для мониторинга изменчивости процесса. Если для каждого образца имеется одно
наблюдение, то для мониторинга среднего значения и изменчивости процесса
360
Часть 3. Качество
используют карты индивидуальных значений (/-карты) и карты скользящего раз-
маха (ЛЖ-карты). Следует отметить, что вследствие неудовлетворительного объема
выборки /-карты и Л//?-карты являются менее чувствительными для обнаружения
неуправляемого процесса, чем х -карты и 5-карты.
Контрольные х-карты. Карты этого вида используются для мониторинга среднего
значения параметра процесса. В фармацевтическом производстве х-карту обычно
применяют, когда разнородные единицы отбираются в одну выборку (пробу) (на-
пример, выборка разных таблеток, полученных методом сухого прессования или из
влажного гранулята). Вследствие риска контаминации и стоимости отбираемых об-
разцов (включая потери продукта из-за небольших объемов и трудозатраты на лабо-
раторные испытания) объем выборки обычно небольшой.
Среднеарифметические значения проб х наносятся на х -карту. Предположим,
что отобраны и оценены случайные выборки п продуктов из стабильного процесса
со средним значением ц и стандартным отклонением о. Тогда х может рассматри-
ваться как случайная величина, подчиняющаяся нормальному_закону распределе-
ния со среднеарифметическим и стандартным отклонением s, где
о
(7)
у1п
Если истинное среднее значение ц и стандартное отклонение о известны, то у
х-карты будут следующие параметры:
UCL = ]i-+3ax =[1 + 3-^=,
у/п
центральная линия = цж=ц, (8)
LCL-^-Зо- = ц-3-^=.
у/п
Так как ц и о обычно неизвестны, их расчетные значения можно получить исхо-
дя из среднеарифметических значений (х) и стандартных отклонений (s)m выборок,
сделанных в течение базового периода:
Em __
X,
расчетное значение ц = л = —; (9)
расчетное значение о = —-------= —.
4(и-1)/4и-3 с4
Параметры х-карты, полученные по расчетным значениям, будут такими:
UCL = х+зф = +Л35,
yjn
центральная линия = 5с, (10)
LCL = f-зф = х - Л35.
Общие значения коэффициентов с, и А3 приведены в табл. 4 для выборок объ-
емов от 2 до 10. Как и для других контрольных карт, значения и s должны пе-
Глава 3.4. Улучшение качества процесса
361
риодически подтверждаться, чтобы обеспечить их пригодность для получения рас-
четных стандартных значений для среднего значения и стандартного отклонения
процесса.
Таблица 4
Значения коэффициентов в контрольной карте для количественных данных
п АЗ с4 вз В4 О1 d3
2 2,659 0,798 0 3,267 1,128 0,853
3 1,954 0,886 0 2,568 1,693 0,888
4 1,628 0,921 0 2,266 2,059 0,880
5 1,427 0,940 0 2,089 2,326 0,864
6 1,287 0,952 0,030 1,970 2,534 0,848
7 1,182 0,959 0,118 1,882 2,704 0,833
8 1,099 0,965 0,185 1,815 2,847 0,820
9 1,032 0,969 0,239 1,761 2,970 0,808
10 0,975 0,973 0,284 1,716 3,078 0,797
Контрольные s-карты. Эти карты используются для мониторинга изменчивости
процесса. Так как одинаково важно оценивать и среднее значение процесса, и сте-
пень его изменчивости, то обычно s-карту используют вместе с х-картой. Стандарт-
ные отклонения выборки откладываются на s-карте. Будем считать s случайной ве-
личиной со средним значением ps и стандартным отклонением ст,. Тогда параметры
s-карты можно определить по следующим уравнениям:
UCL = и + Зег,
• S S'*
центральная линия = gs,
(И)
LCL = ц — Зо.
rS S
На практике параметры s-карты можно рассчитать с помощью s следующим об-
разом:
UCL = ?+= B.s,
С4
центральная линия = s, (12)
LCL = s - 3—Jl-c* = B3s.
Если при вычислении LCL получают отрицательное значение, полагают LCL
равным нулю.
Пример 2. При производстве таблеток Пет (витамины для домашних животных)
фармацевтический производитель использует мельницы и машины для тонкого из-
мельчения для превращения исходных материалов в хорошо измельченные части-
цы. Затем подготовленные частицы смешивают и обрабатывают в смесителях. Сме-
шанные компоненты прессуются в таблетки, высушиваются и расфасовываются
362
Часть 3. Качество
в первичную упаковку. Мониторируется показатель качества — влажность таблетки,
подчиняющийся нормальному закону распределения. В ходе технологического про-
цесса каждый час отбираются п = 4 таблетки. Данные, полученные по 25 выборкам,
приведены в табл. 5.
По этим данным получено: х =10,254 и У = 0,926. По формулам (10) и (12) рас-
считаны параметры х - и s-карты:
_____________________х -карта s-карта
UCL 11,761 2,098
Центральная линия 10,254 0,926
LCL 9,747 0
Полученные контрольные карты показаны на рис. 11. Они свидетельствуют, что
процесс управляем и установленные параметры могут быть использованы для мо-
ниторинга последующей продукции.
Таблица 5
Влажность таблеток Пет, определенная по 25 пробам, %
Номер пробы Результаты испытания X 5
1 2 3 4
1 7,84 11,01 10,14 9,41 9,600 1,343
2 10,51 9,1 9,52 10,83 9,990 0,814
3 9,74 10,39 9,62 11,16 10,228 0,708
4 10,71 11,41 10,71 8,63 10,365 1,203
5 9,93 10,95 8,99 10,73 10,150 0,889
6 9,94 10,27 9,35 9,42 9,745 0,438
7 12,11 9,72 8,89 9,75 10,118 1,387
8 9,61 8,93 11,12 8,75 9,603 1,077
9 9,17 10,87 9,97 10,79 10,200 0,798
10 11,41 10,39 8,83 12,19 10,705 1,451
11 8,43 9,48 10,56 10,2 9,668 0,939
12 9,92 10,13 9,66 8,21 9,480 0,868
13 8,39 9,94 10,4 8,69 9,355 0,967
14 10,42 10,27 10,94 10,91 10,635 0,341
15 10,98 12,57 11,14 8,97 10,915 1,481
16 9,73 10,05 12,82 12,43 11,258 1,592
17 11,36 8,91 10,08 10,55 10,225 1,024
18 9,42 11,12 9,01 10,52 10,018 0,973
19 10,15 10,08 10,12 9,88 10,058 0,122
20 11,73 11,1 10,75 9,94 10,880 0,746
21 11,52 9,И 9,88 11 10,378 1,087
22 11,29 10,43 11,6 11,74 11,265 0,588
23 9,39 12,96 11,42 10,28 11,013 1,541
24 10,26 9,59 9,33 9,26 9,610 0,456
25 11,25 10,65 11,06 10,63 10,898 0,307
Глава 3.4. Улучшение качества процесса
363
UCL =2,098
2,0-
а> о
1,5-
0,5-
1,0-
s = 0,926
0,0
1 3 5 7 9 11 13 15 17 19 21 23 25
Номер пробы
Рис. 11. Карты х и s для примера с производством таблеток Пет
LCL = 0
05
Карты ивдиввдуальных значений. В некоторых химических и биофармацевтиче-
ских производствах имеются длительные и дорогостоящие процессы, для которых
нецелесообразно формировать выборку более чем из одного образца, поскольку
в момент отбора имеется только один продукт или одна серия. В тех случаях, когда
объем пробы, используемой для статистического мониторинга процесса, ограни-
чен одним образцом, необходимы карты индивидуальных значений — /-карты и
Л/Р-карты.
/-Карты выполняют те же цели, что х-карты, за исключением того, что теперь
х — это значение индивидуального измерения. Предположим, что х подчиняется
закону нормального распределения со средним у. и стандартным отклонением о.
Тогда теоретические параметры /-карты будут следующими:
17С/ = у + Зо,
центральная линия = у, (13)
LCL = у — Зо.
Среднее значение у может быть вычислено с помощью х следующим образом:
р=х-^1Х|. (14)
т
Поскольку имеются только данные индивидуальных измерений, для вычис-
ления стандартного отклонения о должны быть рассчитаны скользящие размахи.
Скользящий /^-точечный размах MRk может быть вычислен по формуле
MRk = max(x, ..., xi+l) - mm(x.,.... x.+Jt). (15)
364 Часть 3. Качество
Для т индивидуальных измерений получают т —к величин MR*, и рассчитыва-
ют стандартное отклонение процесса о по формуле
а «К «"..Л"-*) (16)
d2 d2
Рассчитанные среднее значение и стандартное отклонение могут быть исполь-
зованы для вычисления практических параметров для Z-карты по формулам (13).
Коэффициент d2 определяется значением к и может быть взят из табл. 3, в которой
следует положить п равным к. Обычно значения к находятся в диапазоне от 2 до 5.
MR-карта применяется для мониторинга вариабельности процесса. Рассматри-
вая MRk как случайную переменную со средним значением и стандартным от-
клонением <3MRk, теоретические параметры MR-карты можно определить следую-
щим образом:
UCL — liMRt
центральная линия = Ммд t, (17)
LCL = — Зо^.
Поскольку Мд/д 4 и смкк обычно неизвестны, их рассчитывают в виде
Мл/я, ~MRk и Од/я, = (18)
Значение коэффициента также можно взять из табл. 3. Если при расчетах по-
лучают отрицательное значение LCL, то вместо него используют нулевое значение.
3.4.2.4. Специальные контрольные карты
Контрольные карты, описанные выше, очень информативны при диагностике каче-
ства процесса и изучении возможностей его улучшения. Их можно использовать для
стабилизации процесса, выявляя ситуации, когда он неуправляем. После стабили-
зации процесса и возвращения в контролируемое состояние дальнейшее улучшение
процесса может быть достигнуто с помощью специальных контрольных карт, та-
ких как контрольная карта накопленных сумм (ССбТОИ-карта, cumulative sum control
chart) и контрольная карта экспоненциально взвешенного скользящего среднего
(УГИ^ЛМ-карта, exponentially weighted moving average control chart). Эти контрольные
карты могут использоваться при изучении «небольших сдвигов» в процессе.
Контрольные карты CVSUM. Эти карты позволяют эффективно выявлять незна-
чительные сдвиги среднеарифметической величины процесса (<1/2о). Для выяв-
ления более выраженных сдвигов (>1/2ст) обычно используются х -карты. CUSUM-
карта включает информацию, содержащуюся в последовательности точек пробы
(выборки). Она отслеживает накопительную сумму отклонений между каждой точ-
кой пробы (средним значением пробы) и целевым значением. В отличие от х -кар-
ты, которая обычно обосновывает заключение о неуправляемости процесса только
на самых последних отобранных пробах, карта CUSUM, рассчитанная для точки
пробы, содержит «историю» до этой пробы. Например, последовательность точек
Глава 3.4. Улучшение качества процесса
365
пробы над центральной линией может стать сигналом тревоги, хотя все точки рас-
положены существенно ниже значения UCL х -карты.
Существует две формы CUSUM-карт — в виде таблиц и в виде ^диаграмм.
В промышленных приложениях более предпочтительна табличная форма, так как
она более практична. Табличная CUSUM-карта. накапливает отклонения от целевого
значения (или известного среднего для процесса цс). Отклонения выше целевого
значения накапливаются как односторонние верхние значения CUSUM (С), а от-
клонения ниже целевого значения собираются как односторонние нижние значе-
ния CUSUM(C-):
С* =шах[0,х( -(ц0 + йог)+С,+_1],
С,. =тах[О,(цо-Ахтг)-х,+С/Ч],
(19)
где Со+=С-=О.
Параметр к называется допущением и обычно определяется как амплитуда сдви-
га, который следует выражать в терминах ох. Если или С,+, или С/ превышают интер-
вал решения й, то делают заключение о неуправляемости процесса. Другими слова-
ми, значение й рассматривается как UCL, а значение —й — как LCL. Центральная
линия всегда равна 0. Приемлемым значением й считается пятикратное стандартное
отклонение процесса о.
Контрольные карты EWMA. Карта этого типа содержит график значений взве-
шенного скользящего среднего для различных переменных. Фактор взвешивания
выбирается пользователем для определения, насколько более ранние точки влияют
на значение среднего показателя в сравнении с более поздними данными. Посколь-
ку ЕГ/ЛМ-карта использует информацию всех образцов, она является хорошей аль-
тернативой С USUM-картам для определения меньших сдвигов процесса.
Значение EWMA для образца i (z) наносится на карту и определяется как
Zi = Xxi + (1 — Х)^_р где Zq = ц0. Коэффициент X определяет вес, назначенный этому
образцу (0 < X < 1), а 1 — X — вес, назначенный более ранним образцам. Параметры
ЕГКИЛ-карты будут следующими:
UCL = ц0 +£о-А_[1_(1-А)2/],
V z—л
центральная линия = ц0,
(20)
LCL = р0 -£о Ц-[1-(1-Х)2'],
у 2 — Л
где L — это назначенный параметр, определяющий расстояние между контрольны-
ми линиями. Приемлемым считается сочетание L = 3 и 0,05 < X < 0,25.
Контрольные линии раздвигаются при увеличении номера пробы i и, в конеч-
ном итоге, достигают постоянных значений
/ А
UCL=Vl0+LgsA—
° V2-X
центральная линия = ji0,
(21)
366
Часть 3. Качество
LCL — jTq Lg~
2-Х'
Пример 3. Теперь проведем анализ данных из примера 2 с помощью CUSUM-
и ТРИЛМ-карты. В табл. 6 показаны вычисленные значения CUSUM и EWMA.
Значение h в CUSUM выбрано равным 5-кратному стандартному отклонению
х (а, = 0,5027), значение к выбрано равным 0,5. Показатели С,+ и С? вычисляются
на основе целевого значения ц0 = 10. С1/5С/ЛТкарта показана на рис. 12. Значение
X для EWMA выбрано равным 0,2; L = 3. UCL и LCL для индивидуальных проб по-
казаны в табл. 6, а Е^Л/Л-карта приведена на рис. 13. Несмотря на то, чтох-кар-
та и 5-карта на рис. 6 свидетельствуют об управляемости процесса, на обеих картах
CUSUM и EWMA имеются тревожные сигналы на пробе номер 22. Видно, что не-
большой сдвиг процесса произошел после отбора пробы 21.
Таблица 6
Значения карт CUSUM и EWMA для примера с таблетками Пет
Номер пробы CUSUM EWMA
X с; с- Z, UCL LCL
1 9,600 0,000 0,149 9,920 10,302 0,608
2 9,990 0,000 0,000 9,934 10,386 9,614
3 10,228 0,000 0,000 9,993 10,432 9,568
4 10,365 0,114 0,000 10,067 10,458 9,542
5 10,150 0,012 0,000 10,084 10,475 9,525
6 9,745 0,000 0,004 10,016 10,485 9,151
7 10,118 0,000 0,000 10,036 10,491 9,509
8 9,603 0,000 0,146 9,950 10,495 9,505
9 10,200 0,000 0,000 10,000 10,498 9,502
10 10,705 0,454 0,000 10,141 10,500 9,500
11 9,668 0,000 0,081 10,046 10,501 9,499
12 9,480 0,000 0,350 9,933 10,501 9,498
13 9,355 0,000 0,744 9,817 10,502 9,498
14 10,635 0,384 0,000 9,981 10,502 9,498
15 10,915 1,047 0,000 10,168 10,502 9,498
16 11,258 2,054 0,000 10,386 10,502 9,498
17 10,225 2,027 0,000 10,354 10,502 9,498
18 10,018 1,794 0,000 10,286 10,502 9,498
19 10,058 1,600 0,000 10,241 10,502 9,498
20 10,880 2,229 0,000 10,368 10,502 9,498
21 10,378 2,355 0,000 10,370 10,503 9,497
22 11,265 3,369 0,000 10,549 10,503 9,497
23 11,013 4,130 0,000 10,642 10,503 9,497
24 9,610 3,489 0,139 10,435 10,503 9,497
25 10,989 4,135 0,000 10,528 10,503 9,497
й = 2,513 Х = 0,2
к = 0,5 £=3
Глава 3.4. Улучшение качества процесса
367
Рис. 12. CUSUM-карта для примера с производством таблеток Пет
Номер пробы
Рис. 13. EW/VM-карта для примера с производством таблеток Пет
3.4.3. Повышение эффективности процесса
3.4.3.1. Введение
Основные концепции. После проведения диагностики процесса, исправления и полу-
чения статуса статистически управляемого процесса возникает следующий вопрос:
«Как можно повысить эффективность процесса?». Для ответа на данный вопрос ме-
неджерам по качеству и инженерам необходимо сначала измерить эффективность
имеющегося процесса. Это измерение можно произвести с помощью изучения воз-
можностей1 процесса, т. е. возможности производить продукты в соответствии со
спецификацией. Процесс может достичь статуса статистически управляемого, но
по-прежнему проявлять неудовлетворительные возможности вследствие своей из-
менчивости. Следовательно, для улучшения возможностей процесса необходимо
снижение его изменчивости. Спроектированные эксперименты, основанные на
статистических принципах, могут помочь в решении вопроса о снижении изменчи-
1 В литературе английский термин capability переводится по-разному. В данной книге тер-
мин соответствует ГОСТ Р 50779.44—2001 «Статистические методы. Показатели возможно-
стей процессов. Основные методы расчета». — Примеч. перев.
368
Часть 3. Качество
вости и оптимизации процесса. Использование спроектированных экспериментов
позволяет внести преднамеренные изменения в различные области процесса. Ре-
зультаты, полученные в этих экспериментах, могут привести к дальнейшему улуч-
шению процесса и вывести его на новый уровень. В данном разделе представлены
наиболее часто используемые методы изучения возможностей процесса. Методы
планирования экспериментов1 могут быть найдены и в этой книге, и во многих дру-
гих.
Нормы спецификации, контрольные показатели и естественные предельные допу-
ски. Для проведения исследования возможностей процесса очень важно различать
понятия «нормы спецификации» продукта, «контрольные показатели» процесса,
производящего продукт, и «естественные предельные допуски» продукта. В целом
нормы спецификации устанавливаются потребителями и определяются инженера-
ми по внутренней разработке перед началом производства. Продукт, не соответству-
ющий нормам спецификации, является дефектным, не отвечающим требованиям.
Контрольные показатели обычно определяются на образцах, отбираемых в ходе
технологического процесса в течение базового периода. Показатель (точка) пробы,
который вышел за пределы контрольных линий, будет сигнализировать о неуправ-
ляемом состоянии процесса; однако продукт, произведенный при неуправляемом
состоянии процесса, не всегда является дефектным. Следует также отметить, что
точка пробы, наносимая на карту, обычно представляет статистический показатель
пробы, например среднеарифметическое значение показателя в пробе. Другими
словами, выпадение одного продукта за пределы контрольных линий не будет обу-
словливать неуправляемость процесса, а также не приведет к его несоответствию.
Изменчивость производимых продуктов обычно называют естественными предель-
ными допусками. Обычно приемлемыми считаются предельные допуски ±3 стан-
дартных отклонения от среднего показателя процесса.
Пример 4. Продолжаем рассмотрение ситуации, описанной в примере 2. Для рас-
сматриваемого показателя в спецификации установлены нормы 10 ± 2,00, то есть:
1) номинальное или целевое значение ц0 = 10,00;
2) верхний допустимый предел для спецификации USL (upper specification limit)
равен 10,00 + 2,00 = 12,00;
3) нижний допустимый предел для спецификации LSL (lower specification limit)
равен 10,00 — 2,00 = 8,00.
Контрольные параметры для х -карты:
1) центральная линиях=10,254;
2) верхняя контрольная линия UCL =11,761;
3) нижняя контрольная линия LCL = 8,747.
Используя естественные предельные допуски х ± Зо, получаем:
1) среднее значение процессах =10,254;
2) верхний естественный предельный допуск UNTL (upper natural tolerance limit)
равен 13,270;
1В данном случае автор ссылается на научную дисциплину — планирование эксперимен-
тов (Design of experiments), занимающуюся разработкой различных методов планирования
экспериментов и моделей, например статистических и рандомизированных экспериментов,
регрессионных моделей, линейных и т. д. — Примеч. перев.
Глава 3.4. Улучшение качества процесса
369
3) нижний естественный предельный допуск LNTL (lower natural tolerance limit)
равен 7,238.
Взаимосвязи между тремя наборами предельных параметров показаны на рис. 14.
Как видно из рисунка, действующий процесс не отцентрирован относительно но-
минального значения и его нормы в спецификации более жесткие, чем его есте-
ственные предельные допуски. Из этого следует, что часть производимого продукта
(~ 5,4%) не будет соответствовать спецификации.
LSL ц0 USL
LNTL LCL х UCL UNTL
Рис. 14. Нормы спецификации, контрольные показатели и естественные предельные допуски
для примера с производством таблеток Пет
3.4.3.2. Изучение возможностей процесса и его улучшение
Показатели возможности процесса. Показатели возможности процесса предоставля-
ют количественную меру для оценки возможности процесса производить продукты,
отвечающие спецификации. Наиболее часто использующийся индекс воспроизво-
димости процесса Ср может быть вычислен по формуле
„ USL-LSL
с'=—<22>
где USL — верхний допустимый предел для спецификации; LSL — нижний допусти-
мый предел для спецификации; о — стандартное отклонение процесса. Поскольку
о обычно не известно, его оценивают по формуле ст = s/ c4. Значение С = 1 озна-
чает, что процесс просто воспроизводим. Если процесс отцентрирован по своему
номинальному значению, он будет производить 2700 дефектных единиц продукции
на 1 млн (ррт). Целевое значение С обычно устанавливают равным 1,33 для имею-
щегося процесса и 1,50 для нового процесса.
Следует отметить, что значение Ср не может свидетельствовать о надлежащей
воспроизводимости, если процесс неотцентрирован, так как С не учитывает, где
находится среднее значение процесса относительно спецификации. Чтобы уточ-
нить данную информацию, используют другой индекс воспроизводимости — Ср1:
С. = min(C , С,),
рк v ри1 pl' ’
где
_USL-[l с P-Z5Z (23)
Зо " Зо
370
Часть 3. Качество
Значение ц может быть рассчитано по х, а значение о — так, как описывалось
выше. В целом процесс считается «центрированным» по номинальному значению
спецификации, если С = Срк, и «неотцентрированным», если С < Срк. Взаимосвязь
между С и Срк более детально показана на рис. 15, где среднее значение процесса
сдвинуто от ц0 до ц0 + 2ст и до ц0 + 4ст. Как видно из рисунка, Ср остается одинако-
вым вне зависимости от сдвига, в то время как Срк значительно снижается.
Если используются спецификации с одним пределом («не более» или «не ме-
нее»), то воспроизводимость для процесса с одним пределом также может быть
определена по уравнению (23), где Сри вычисляется для верхнего предела специфи-
кации, а С (для нижнего.
Интерпретация результатов и улучшение возможностей процесса. Оценка и интер-
претация возможностей процесса представляют собой важный этап в улучшении
качества процесса. Источник нестабильности процесса должен быть удален до того,
как процесс будет подвергаться улучшению. Результаты, полученные в исследова-
ниях возможностей процесса, помогают определить стабильность процесса и со-
ответствие его спецификациям. Следует отметить, что достоверное исследование
Рис. 15. Взаимосвязь между Ср и Срк
Глава 3.4. Улучшение качества процесса
371
возможностей процесса основано на предположении о подчинении параметров
процесса нормальному закону распределения. Истинность этого предположения
следует проверить перед переходом на следующий этап.
Заключения о том, является ли процесс центрированным в отношении целевого
значения и соответствует ли он спецификациям, могут быть сделаны по результатам
изучения возможностей процесса. Если С = Срк, то процесс центрирован. Если Ср
имеет значение 1,0 и более, то процесс способен производить продукцию, соответ-
ствующую спецификациям; в противном случае он не способен производить такую
продукцию.
Пример 5. По ситуации, описанной в примере 2, рассчитаны Си Срк.
USL-LSL 12-8
бет 6-1,0054
С k = min(C u, Ср) = min(0,5790; 0,7476) = 0,5790,
где
За 3-1,0054
p-Z5Z = 10,254 -8 =
Зо 3-1,0054
На рис. 16 приведена гистограмма данных, связанных со спецификацией. Как
видно изх- и 5-карты на рис. 11, процесс статистически управляем. Однако, по-
скольку С < Срк, процесс не центрирован. При значении С к, равном 0,579, следует
ожидать 53 711 дефектных единиц на 1 млн таблеток Пет, произведенных на этой
производственной линии.
Для повышения возможностей процесса сначала необходимо центрировать
процесс. Обычно при этом регулируют параметры (установки) ведения процесса.
Для поиска причин, вызывающих «отклонение от центра», можно использовать
причинно-следственную диаграмму, диаграмму Парето и другие инструменты, опи-
l_SL Целевое значение (jgi_
Рис. 16. График возможностей процесса для примера с производством таблеток Пет
372
Часть 3. Качество
санные ранее в этом разделе. После приведения процесса к его номинальному по-
казателю (10) общее количество дефектных таблеток Пет снизится до 44 673 ррт.
Это по-прежнему не соответствует уровню 2700 ррт, который свидетельствовал бы
о достижении статуса «просто воспроизводимого» процесса (Ср = 1), не говоря уже
о целевом значении 63 ррт при С = 1,33. Планирование экспериментов — это си-
стемный подход, который позволяет инженерам и менеджерам вносить намерен-
ные изменения в некоторые параметры ведения процесса и оценивать эффект этих
изменений. Для данного примера проводились эксперименты с изменением не-
скольких ключевых параметров ведения процесса, таких как длительность сушки,
длительность стадии смешения и изменение температуры. По результатам серии
экспериментов были найдены оптимальные параметры для указанных параметров
ведения процесса, и изменчивость процесса была снижена на 50%. Благодаря сни-
жению изменчивости процесса новый индекс воспроизводимости Ср составил 1,265
с 694 ррт дефектных таблеток. Возврат процесса в центрированное состояние при-
вел к уменьшению «выпадения» процесса из спецификации на 13,1%. С помощью
планируемых экспериментов изменчивость процесса была уменьшена наполовину,
а «выпадение» процесса из зоны спецификации было уменьшено на 98,5%, то есть
очень значительно.
Литература
Aft, L. S. (1997), Fundamentals of Industrial Quality Control, 3rd ed., CRC Press, Boca Raton, FL.
DeVbr, R. E., Chang, T. H., and Sutherland, J. W. (2006), S tatistical Quality Design and Control, 2nd ed.,
Prentice-Hall, Upper Saddle Brook, NJ.
Gitlow, H., Gitlow, S., Oppenheim, A., and Oppenheim, R. (1989), T ools and Methods for the Improve-
ment of Quality, Irwin, Boston.
Grant, E. L., and Leavenworth, R. S. (1996), Statistical Quality Control, 7th ed., McGraw- Hill, New
York.
Ishikawa, K. (1982), Guide to Quality Control, 2nd ed., Asian Productivity Organization, Tokyo, Ja-
pan. Juran, J. M., and Godfrey, A. B. (1998), Juran’s Quality Handbook, 5th ed , McGraw-Hill, New
York.
Ledolter, J., and Burrill, C. W. (1998), Statistical Quality Control: Strategies and Tools for Continual
Improvement, Wiley, New York.
Montgomery, D. C. (2004), Design and Analysis of Experiments, 6th ed., Wiley, New York.
Montgomery, D. C. (2001), Introduction to Statistical Quality Control, 4th ed., Wiley, New York.
Ryan, T. P. (2000), Statistical Methods for Quality Improvement, 2nd ed., Wiley, New York.
Smith, G. M. (2003), Statistical Process Control and Quality Improvement, 5th ed., Prentice-Hall, Upper
Saddle Brook, NJ.
Summers, D. C. S. (2006), Quality, 4th ed., Prentice-Hall, Upper Saddle Brook, NJ.
Tague, N. R. (2005), Quality Toolbox, ASQ Quality Press, Milwaukee.
Thompson, J. R., and Koronacki, J. (2001), S tatistical Process Control: The Deming Paradigm and Be-
yond, 2nd ed., Chapman & Hall, New York.
Часть 4
ПРОЦЕССНО-АНАЛИТИЧЕСКИЕ ТЕХНОЛОГИИ
Глава 4.1. Аргументы в пользу процессно-
аналитической технологии: юридические
и промышленные перспективы
Роберт П. Когдилл
Duquesne University, Center for Pharmaceutical Technology (Питтсбург,
Пенсильвания)
4.1.1. Введение
Внедрение процессно-аналитической технологии (РАТ) происходит в период наи-
более интенсивных изменений в фармацевтическом производстве, наблюдающий-
ся последние три десятилетия. Многие технологические, нормативно-правовые
и экономические факторы в последние пять лет сошлись воедино, открыв новые
возможности для инноваций в области развития и управления процессами фарма-
цевтического производства. Главной движущей силой изменений является ини-
циатива Федерального управления США по контролю за пищевой продукцией
и лекарствами (FDA) по внедрению современных, основанных на оценке рисков,
способов контроля и надзора за фармацевтическим производством [1]. Цель дан-
ной главы — описать историческое развитие процессной аналитики; дать общее
представление о применении РАТв фармацевтической промышленности и бизнес-
стимулах, способствующих внедрению изменений; изложить сущность новой ини-
циативы FDA и требований РАТ [2], а также представить базовый план внедрения
РАТ. В данной главе основное внимание уделяется РАТ, однако следует иметь в виду,
что процессно-аналитические технологии являются важной частью более обшир-
ной концептуальной модели на основе анализа рисков, порожденной инициативой
двадцать первого века — внедрением текущей надлежащей производственной прак-
тики (cGMP).
4.1.2. Основы процессно-аналитической технологии
Вводимая FDA система РАТ (и ее требования) начала формироваться ещё до появ-
ления в 2001 г. инициативы по внедрению cGMP двадцать первого века. Однако,
как известно, некоторые ее основные идеи начали применяться несколько десяти-
летий тому назад в других отраслях промышленности, таких как промышленность
тонкого органического синтеза, полупроводниковое производство, нефтяная про-
мышленность и производство потребительских товаров. Главным отличием РАТ от
традиционных промышленных фармацевтических отраслей (к которым относят-
ся фармация и материаловедение, химия и инженерные дисциплины) являются
процессно-аналитическая химия (РАС) и передовые наукоемкие технологии (рис. 1).
В рамках данного обсуждения термин «наукоемкие технологии» обозначает на-
уку и технологию производства, основанные на инновациях в области разработки
технологических процессов и управления ими. Поскольку нет ни возможности, ни
376
Часть 4. Процессно-аналитические технологии
Рис. 1. Междисциплинарные компоненты системы РАТ
в фармацевтическом производстве
необходимости подробно излагать все аспекты современных фармацевтических
наукоемких технологий, в следующих параграфах обсуждаются две специфические
темы, часто поднимаемые в профессиональных промышленных дискуссиях, однако
не описанные подробно в фармацевтической литературе: системы управления каче-
ством и «ресурсосберегающее производство (lean manufacturing).
4.1.2.1. Процессно-аналитическая химия
В целом процессно-аналитическая химия охватывает науку и технологию, связан-
ные с проведением лабораторных измерений при помощи сенсоров и приборов,
расположенных в непосредственной близости от места проведения технологиче-
ских операций. Несмотря на то что промышленные процессные анализаторы ис-
пользуются уже более 60 лет [3], современный этап развития РЛС начался с орга-
низации Центра процессно-аналитической химии (СРАС) в 1984 г. [4]. Согласно
Кэллису (Callis), Иллману (Ulman) и Ковальски (Kowalsky) [5], задачей РАС является
«предоставление количественной и качественной информации о химическом про-
цессе» с целью мониторинга, контроля и оптимизации процесса. Они выделяют
пять этапов развития РАС: 1) вне линии (off line), 2) у линии (at line), 3) на линии
(online), 4) встроенные в линию инструменты (inline) и 5) неинвазивные методы, по-
явившиеся вследствие эволюции сенсорных технологий. Кроме того, они рассмо-
трели важные вопросы, выходящие за рамки химического анализа, такие как отбор
проб, извлечение информации из имеющихся данных (хемометрика), интеграцию
с управлением производственным процессом, а также социологические аспекты
внедрения Л4Г(например, завоевание доверия рекламодателей).
Движение по внедрению РАС в промышленность было поддержано успехами,
достигнутыми за последние два десятилетия в таких отраслях как материаловеде-
ние, электроника и хемометрика. С момента организации СРАС темпы развития
инновационных сенсоров, приборов и аналитических методик существенно ускори-
лись. Разработки более надежных чувствительных материалов для фотодетекторов,
микроэлектромеханических систем (microelectromechanical system), а также волокон-
ной оптики наряду с постоянным прогрессом компьютерных мощностей (что было
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...377
предсказано законом Мура), позволили повысить эффективность и одновременно
снизить стоимость систем РАС. В результате РАС в настоящее время представляет
собой критически важную часть рутинных операций в сфере химической промыш-
ленности. Вопросам, связанным с РАС (и А47), посвящено множество опублико-
ванных обзорных статей [6—10]. Серии публикаций по вопросам РЛС регулярно пу-
бликуются в Analytical Chemistry.
В первом обзоре перечислены работы, опубликованные между 1987 и 1992 г.,
в которых наряду с проблемами будущего РАС освещаются семь специфических
тем: общие вопросы РАС, хроматография, оптическая спектроскопия, волокон-
ная оптика, масс-спектрометрия, хемометрика и проточно-инжекционный анализ.
В первом обзоре содержались ссылки на 507 источников. Последующие обзоры
были опубликованы в 1995 [12], 1999 [13], 2001 [14], 2003 [15] и 2005 г. [16]. Обзор-
ные серии являются значительным ресурсом для ученых, ищущих информацию по
специфическим методам РАС; всего авторы предоставили 2650 источников, охваты-
вающих более 16 тем.
В настоящее время существует три основных консорциума, привлекающих
университетских, правительственных и промышленных партнеров: СРАС, Центр
технических измерений и контроля (Measurement & Control Engineering Center),
Центр теоретических и прикладных исследований в области контроля (Control
Theory and Applications Center). Они работают совместно с ежегодной конференци-
ей — Международным форумом по процессно-аналитической химии (IFPAC —
International Forum on Process Analytical Chemistry) и многочисленными интеренет-
ресурсами, посвященными вопросам процессной аналитики [16]. Параллельно
с распространением инициативы FDA термин процессно-аналитическая химия
в профессиональной проиводственной среде постепенно замещается термином
«процессно-аналитическая технология». Это отражает расширение сферы РАТ по
мере признания важности описания физических характеристик, анализа рисков
и производственной науки.
4.1.2.2. Управление качеством
Многие задачи, связанные с улучшением качества и внедрением РАТ в фармацев-
тической промышленности, были успешно решены компаниями, работающими
в других областях, таких как автомобилестроение и бытовая электроника, в резуль-
тате принятия принципов управления качеством. Современная система управления
качеством берет начало в появившейся в середине 1920-х гг. работе Вальтера Шу-
харта — статистика из лаборатории Белла (Bell Laboratories) [17]. Он заметил, что
статистический анализ размеров промышленных продуктов, проводимый в течение
определенного времени, можно использовать для контроля качества продукции; это
наблюдение заложило основы применения современных карт контроля. Шухарта
считают основателем статистического контроля процессов (statisticalprocess control);
в его работе представлены первые признаки перехода от контроля качества продук-
та (путем анализа) к концепции контроля качества процессов [18,19].
Методология Шухарта была принята и расширена В. Эдвардсом Демингом
(W. Edwards Deming) [20] и Джозефом М. Джураном (Joseph М. Jurari) [21], которых
считают создателями системы TQC — «тотального контроля качества», впервые при-
378
Часть 4. Процессно-аналитические технологии
мененной в Японии после Второй мировой войны. К движению за тотальный кон-
троль качества присоединились последователи систем целевого управления (МВО —
management by objectives) (1960-е гг.); движение Кросби за бездефектное производство
(ZD — zero defects) (1970-е гг.); Американское движение за тотальное управление ка-
чеством (TQM) (1970—1980-е гг.); кружки качества (1970-е гг.); движение Технология
развертывания функций качества (QFD — Quality Function Deployment) (1980-е гг.);
ISO — Международная организация стандартизации серии 9000 (1987) и организа-
ция Национальная премия качества Малькольм Бридж (1987). Последняя из поя-
вившихся методологий управления качества называется Six Sigma «Шесть сигм» (6о)
[22,23]. Впервые была внедрена в компании Motorola и стала чрезвычайно популяр-
на, поскольку многие генеральные директоры корпораций (такие как Томас Галвин,
Джек Уэлч) открыто высказались за внедрение инициативы 6g в своих фирмах с це-
лью существенного улучшения качества готовой продукции. Однако в основе всех
движений за качество [24], так же, как и в основе РАТ, лежат принципы Шухарта,
Деминга, Джурана, Кросби, Тагучи [25, 26] и др. Все системы основаны на система-
тических методах выявления источников отклонений в производственных процес-
сах и сведению к минимуму их неблагоприятного влияния на качество продукции.
Так называемая методология DMAIC (define, measure, analyze, improve, control —
выявить, измерить, анализировать, исправить, контролировать) является обще-
принятым подходом, который используют команды по улучшению качества во
многих областях индустрии, применяющие концепции управления качеством для
систематического выявления и устранения первопричин ухудшения качества. Ва-
риация DMAIC, называемая DMADV(выявить, измерить, анализировать, планиро-
вать и проверять), иногда используется, когда процесс или операция требует полной
реструктуризации для необходимого улучшения качества, и является центральной
концепцией движения DFSS (design for six sigma — разработанный для концепции
«Шесть сигм»). Происхождение концепций DMAIC, DMADV, DFSS и других кон-
цепций управления качеством прослеживаются до «цикла Шухарта»: 1) планиро-
вать, 2) сделать, 3) изучать и 4) действовать [24].
Вероятно, самыми важными аспектами управления качеством для РАТ являют-
ся принципы количественной оценки показателей процесса при помощи индек-
сов пригодности, являющихся универсальными идентификаторами, создающими
основу этапов «измерения» и «анализа» в модели DMAIC. Индексы пригодности
процесса отражают одновременно и изменчивость, и спецификации процесса, что
позволяет определить, является ли процесс «пригодным» [27]. Процесс считается
пригодным, если результаты измерения характеристик качества практически для
всех проб попадают в установленный интервал.
Общеупотребительный индекс пригодности процесса Срк рассчитывается по
формуле
. FtASZ-p ц-САйЛ
C„t=min --------------- ,
рк L Зо Зо J
где р. и о представляют собой среднее и стандартное отклонение; USL и LSL — верх-
ний и нижний установленный предел, соответственно, при измерении характери-
стик качества.
Глава 4.1. Аргументы в пользу процессно-аналитической технологи и...379
Индексы пригодности процесса полезны на этапе усовершенствования процес-
са, поскольку они трансформируют различные измеренные характеристики каче-
ства (например, вес, концентрацию, скорость процесса) в безразмерные единицы,
что позволяет исследователю выявить основные причины отклонений в процессе
(операции с минимальными значениями С ) там, где задействовано множество из-
мерительных систем и получены большие массивы данных по качеству.
Индекс пригодности процесса С соотносится с размахом процесса так, что бст
процесс соответствует С = 2,00 или 2,0 дефектных единицы на биллион (РРВ), при-
нимая — 7V(0, о) распределение отклонений характеристик качества (альтернативный
расчет размаха процесса дает оценку 3,4 дефектных единицы на миллион для процес-
са бст). Примеры соотношений между С к, размахом процесса и долей дефектных еди-
ниц для распределения — N(0, ст) представлены на рис. 2. Пригодность процесса (на
основании наблюдаемого выхода) в фармацевтическом производстве по результатам
некоторых сравнительных исследований равняется приблизительно 0,7 (2,1ст) [28].
В то время как промышленные оценки четко указывают на то, что фармацев-
тические производители обладают большими возможностями улучшения контроля
качества, прямое сравнение с другими отраслями промышленности может ввести
в заблуждение. В противовес таким отраслям промышленности, как полупроводни-
ковое производство, где дефект зачастую становится очевидным в определенной
точке товаропроизводящей цепи (т. е. устройство, изготовленное из дефектной де-
тали, не будет работать), для лекарственных продуктов характерна неоднозначность
характеристик качества.
Например, спецификации выпускаемой готовой продукции, такие как параме-
тры однородности содержимого, редко коррелируют с данными клинических испы-
Концентрация дефектов
Рис. 2. Графическая иллюстрация соответствия между числом дефектных единиц на миллион
случаев (DPMO), пригодностью процесса (Срк) и размахом процесса (process sigma) (с учетом
предположения о нормальном распределении отклонений характеристик качества)
380 Часть 4. Процессно-аналитические технологии
таний; скорее, их устанавливают в соответствии с фармакопейными стандартами.
Функциональные соотношения между характеристиками материалов в процессе
обработки и качеством готового продукта редко замечают на высоком уровне управ-
ления; следовательно, измеренные характеристики отдельных операций техноло-
гического процесса могут вызвать переоценку или недооценку истинного уровня
пригодности процесса. По мере повышения общего уровня понимания технологи-
ческих процессов в фармацевтической промышленности, теоретические и опытные
разработки спецификаций для процесса обработки и выпуска продукта позволят
увеличить достоверность С к как инструмента описания процесса.
Для получения дополнительной информации можно воспользоваться замеча-
тельными источниками общей информации и технических деталей по управлению
качеством — Справочником по инженерной статистике Handbook of Engineering
Statistics, выпущенный NIST/SEMATECH, имеющемся в свободном доступе on-line
[23], а также сайтом Американского общества качества (www.ASQ. org).
4.1.2.3. Ресурсосберегающее производство
В отличие от систем управления качества, для которых прослеживаются четкие па-
раллели с РАТ (т. е. уменьшение колебаний характеристик качества), связь между
РАТ и ресурсосберегающим производством менее очевидна. Фактически системы
управления качеством занимаются анализом колебаний характеристик качества
процесса. Направление ресурсосберегающего управления занимается анализом вре-
мени как одного из параметров производственного процесса. Кроме того, ключе-
вые принципы ресурсосберегающего производства обеспечивают технологическую
платформу, которую можно использовать в фармацевтической промышленности
для получения выгоды за счет повышения эффективности производства вследствие
внедрения РАТ. Если бы РАТ не влияла на эффективность производства (например,
на окупаемость инвестиций (ROI— return on investment) вследствие внедрения РАТ),
то стимулы для добровольного внедрения РЛТбыли бы весьма слабыми. В следую-
щих параграфах будет представлено краткое введение в ресурсосберегающее («бе-
режливое») производство; в последней части этой главы будут обсуждаться бизнес-
стимулы, способствующие внедрению РАТ.
Концепцию ресурсосберегающего производства зачастую понимают неправиль-
но (так же, как концепции всеобщего управления качеством (TQM — total quality
management) или бст). Для некоторых людей инициатива ресурсосберегающего биз-
неса аналогична тактике радикальных мер, направленных на сокращение рабочей
силы или отказ от низко продуктивных операций. На самом же деле, бережливое
производство можно описать как «сочетание методологий, в том числе организа-
ции промышленного производства, принципа ЛТ — “just in time” («точно вовре-
мя») (Osada) 5-S, TQS, непрерывного улучшения качества (CQI — continuous quality
improvement), визуального контроля, тотальной поддержки производства (ТРМ —
total productive maintenance — тотальное обслуживание оборудования), кружков ка-
чества и концепции Kaizen» [24].
Происхождение системы ресурсосберегающего производства зачастую связыва-
ют с производственной системой Тойота (TPS — Toyota Production System), внедрен-
ной в корпорации Toyota Motors. Однако история ресурсосберегающего произвол-
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...381
ства началась с промышленных разработок, появившихся более 150 лет тому назад.
Основание современной системы производства было заложено Эли Уитни в 1878 г.
Уитни более всего известен как изобретатель волокноотделителя, применявшего-
ся в хлопковом производстве; однако его изобретение взаимозаменяемых деталей
и идея унифицированного производства произвели настоящую революцию в сфере
массового производства (www.EUWhitney.org).
Почти век спустя Фредерик У. Тэйлор ввел понятия хронометража и стандартных
операций и ввел в обращение термин научное управление. Лишь в 1908 г., когда Генри
Форд запустил производство Модели Т, значение ресурсосберегающего производства
стало общепризнанным во всем мире. Считается, что Генри Форд первым осуще-
ствил принцип ЛТ (точно вовремя); более того, некоторые исследователи полагают,
что его производственная система вдохновила создателей TPS [29]. Позже в компа-
нии Ford Motor была разработана модернизированная версия оригинальной системы
Генри Форда — производственная система Форд [24], многое воспринявшая от TPS.
Ресурсосберегающее производство является частью наукоемких технологий
и представляет собой техническую философию, сосредоточенную на уменьшении
семи видов потерь (куда — японское слово, которое означает потери, отходы, то
есть любую деятельность, которая потребляет ресурсы, но не создает ценности)
в процессе производства: перепроизводство, ожидание, транспортировка, ненуж-
ная обработка, лишние запасы, избыточное перемещение и дефекты. Преобразо-
вание процесса в ресурсосберегающую операцию («лин»-операцию) требует при-
менения ряда инструментов и стратегий. Вероятно, самым важным механизмом
перемен является переход от традиционной системы «производства по прогнозу»
или «выталкивающей» системы к «вытягивающей» стратегии, такой как стратегия
канбан. Принципы ресурсосбережения (принципы «лин») были успешно примене-
ны в производстве и сфере услуг, а также в органах государственной власти. Кон-
цепции ресурсосберегающего производства, так же как и системам управления ка-
чеством, посвящены сотни книг, описывающих различные инструменты и способы
применения данной методологии. Общество инженеров-технологов (www.SME.org)
поддерживает технические сообщества, выпускает публикации, устраивает конфе-
ренции, посвященные управлению производством, и является хорошим первоис-
точником дополнительной информации по ресурсосберегающему производству.
Фармацевтические производители относительно поздно (по сравнению с други-
ми отраслями) восприняли систему ресурсосберегающего производства; в резуль-
тате периоды фармацевтических циклов чрезвычайно велики по сравнению с про-
изводственными циклами в других областях промышленности [30, 31]. Сравнивая
отношение суммарной себестоимости реализованной продукции (COGS — cost of
goods sold) к стоимости товарно-материальных запасов по 22 ведущим копаниям,
торгующим патентованными лекарственными препаратами, дженериками и био-
технологической продукцией, с аналогичными показателями для других отраслей
промышленности, можно сделать приблизительный вывод о том, насколько низка
эффективность управления цепями поставок фармацевтических производителей
(рис. 3). Для большинства ученых, работающих в области фармацевтической про-
мышленности, было бы нетрудно привести примеры любого из «семи видов потерь»
на типичном фармацевтическом производстве.
382 Часть 4. Процессно-аналитические технологии
Общеизвестно, что существуют определенные производственные ограничения,
которые не позволяют фармацевтическим производителям управлять цепями снаб-
жения и эффективностью производства на мировом уровне. Кроме того, оказалось,
что применение инструментов ресурсосберегающего производства и управления
качеством в фармацевтическом производстве является достаточно проблематич-
ным. Недавний обзор данных 1500 фармацевтических производителей показал, что
хотя более половины компаний сообщают о внедрении систем ресурсосберегающе-
го производства, 6о или производственной эффективности, только менее половины
внедренных программ привели к получению удовлетворительных результатов [32].
Эти данные как будто позволяют предположить, что система ресурсосберегаю-
щего производства не подходит для фармацевтической промышленности. Однако
следует учитывать, что большинство принципов ресурсосберегающего производства
(например, TPS) было разработано для производств с большими объемами одно-
родной продукции. Несмотря на то что многие лекарства-«лидеры продаж» произ-
водятся на специальных предприятиях, специализирующихся на выпуске несколь-
ких препаратов, для фармацевтической промышленности производство множества
продуктов на одном предприятии с большой долей унифицированного оборудова-
ния не является исключительной ситуацией. В производственной среде с широкой
номенклатурой продукции внедрение традиционных методов ресурсосберегаю-
щего производства, таких как карты канбан, происходит с трудом. Чтобы обойти
данные ограничения, разработаны инновационные компьютерные алгоритмы для
«управления потоками материалов» [33], позволяющие стимулировать, планиро-
вать и оптимизировать процессы фармацевтического производства в соответствии
с принципами «лин» (ресурсосберегающего производства).
Кроме того, эффективность ресурсосберегающего производства ограничена из-
менчивостью времени цикла {С/Т) для индивидуальных типовых процессов, а так-
же риском производства неудачной серии; эти события не зависят от сложности по-
тока материалов или степени унификации оборудования. Прежде чем приступить
к стандартным технологическим операциям, фармацевтические производители
справляются с такими рисками, накапливая большие запасы незавершенной про-
дукции (WIP — work in process). Хотя это помогает улучшить использование мощ-
ностей и увеличить общук эффективность оборудования {ОЕЕ — overall equipment
effectiveness), в то же время снижается эффективность за счет потребления оборот-
ных средств и повышения интенсивности вспомогательных операций (необлоди-
мость финансирования транспортировки и складирования WIP). Чтобы получить
прибыль от внедрения системы ресурсосберегающего производства, фармацевтиче-
ские производители должны в первую очередь минимизировать изменчивость С/ Т
и риски, связанные с качеством продукции.
Наконец, хорошо известно, что в типовом производстве значительную долю С/ Т
составляет время ожидания между стандартной операцией, отбором проб, анали-
зом, отчетом и выпуском полупродукта или готового продукта. В отдельных сце-
нариях РАТ позволяет производителям выпускать готовую продукцию на рынок
немедленно, без задержки на ручное тестирование в режиме off-line, это так назы-
ваемый выпуск в режиме реального времени (RTR — real time release), являющийся
преимуществом РАТ. В отсутствии РАТ и RTR эффективность стратегии бережли-
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
383
Рис. 3. Отношение суммарной COGS к стоимости товарно-материальных запасов. Соотношение
COGS/стоимость товарно-материальных запасов является приблизительным индикатором
скорости логистической цепочки. Большая величина отношения указывает на то, что
материально-технические запасы невелики по сравнению с COGS и скорость их оборота велика.
Например, в Toyota Motors, известной эффективным управлением логистической цепочкой,
величина отношения COGS к материальным запасам больше, чем в General Motors
вости (стратегии «лин») в снижении С/ Т будет ограничена максимальной скоро-
стью тестирования продукта и его выпуска. Таким образом, для фармацевтических
производителей критически важно параллельное внедрение РАТ и «лин», если они
хотят добиться решения реальных проблем, связанных с эффективностью процесса.
Концепция «лин-РЛТ» аналогична концепции «лин-бст», или «fusion management —
управлению сопряженными системами» [24].
4.1.3. Исторические факторы, ограничивающие внедрение РА Т
Несмотря на доказательства финансовых и конкурентных преимуществ использо-
вания процессно-аналитических систем, полученные в различных областях про-
мышленности, усилия фармацевтических компаний по внедрению РАТ откро-
венно сдерживаются. На самом деле фармацевтическая промышленность отстала
от смежных отраслей настолько, что статья в Wall Street Journal, опубликованная
в 2003 г., охарактеризовала состояние производства лекарственных продуктов как
«значительное отставание от уровня производства картофельных чипсов и моющих
384
Часть 4. Процессно-аналитические технологии
средств» [34]. Несмотря на то что это заявление шокировало многих, оценка оказа-
лась точной. Однако прежде чем обвинять руководителей промышленности в гру-
бой небрежности, необходимо рассмотреть различные факторы, которые привели
к современному состоянию дел.
За определенное количество лет накопилось множество причин, оправдываю-
щих отсутствие производственных инноваций; некоторые из этих причин хорошо
известны и описаны в ряде публикаций [35]. Для упрощения факторы, ограничи-
вающие внедрение РАТ, можно разделить на три категории: реальные и субъектив-
но воспринимаемые технологические барьеры, отсутствие экономических стимулов
и нормативно-правовые препятствия.
4.1.З.1. Реальные и субъективно воспринимаемые технологические
барьеры
Несмотря на то что спектроскопия в ближней инфракрасной области (БИК) в тече-
ние десятилетий применяется в промышленности [36], принять «новую» процессно-
аналитическую технологию измерений как равноценную или превосходящую тра-
диционные методики непросто. Например, если наблюдаются расхождения между
результатами БИК-спектроскопии в режиме реального времени и лабораторных
анализов, эталонные методики с разрушением пробы редко рассматриваются в ка-
честве источника погрешности, хотя БИК-спекгроскопия зачастую является более
прецизионным методом. Недоверие к более современным, многомерным инстру-
ментам (вероятно, более сложным для непосредственного восприятия) — препят-
ствие для прогресса во внедрении РАТ.
Аналогичные проблемы возникают в отношении хемометрики (многомерного
анализа данных), информационных технологий (IT) и упреждающего регулирова-
ния. Одна из причин может заключаться в практике калибровки и валидации сен-
соров РАТ путем корреляции их сигналов при помощи традиционных эталонных
лабораторных методик и в описании процесса в терминах ошибки прогнозирова-
ния [37—39]. Статистикой признано, что независимо от точности и чувствительно-
сти сенсора РАТ, измеряющего изменение параметров качества, уровень точности
измерения всегда ограничен параметрами эталонной методики. Намного более
точное описание характеристик сенсоров РАТ в сравнении с эталонными методи-
ками получают путем сравнения показателей надежности, таких как соотношение
сигнал/шум (S/N) или аналитическая чувствительность, напрямую связанных с чув-
ствительностью измерения [40,41].
Несмотря на то что восприятие инструментов РАТ стало более позитивным, ком-
пании обеспокоены возможными негативными последствиями глубокого анализа
параметров качества, достигаемого при помощи сенсоров РАТ, такими как увели-
чение количества инспекций и расследований. Иначе говоря, во многих компаниях
опасаются «того, что они обнаружат», если начнут анализировать свои технологиче-
ские операции более подробно. До внедрения быстродействующих неразрушающих
средств мониторинга качества существовало несколько альтернативных методов
эффективного обеспечения качества, в дополнение к методам, основанным на кри-
териях выпуска производственных серий, таких как хорошо известный протокол
Фармакопеи США (USP) (905), основанный на строго ограниченной выборке об-
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
385
разцов для анализа (т. е. исследование 10 индивидуальных дозированных единиц из
образца, состоящего из 30 единиц на серию производственного масштаба).
Несмотря на то что кривая рабочих параметров (ОС — operational charateristics)
при анализе в соответствии с USP (905) гарантирует, что перед тем как возникнет
вероятность отбраковки производственной серии, значительная доля продукции
в каждой серии будет иметь низкое качество [42,43], компании готовы мириться со
своими трудностями. Средства процессно-аналитического мониторинга, такие как
БИК-спектроскопия, способные с высокой скоростью проводить отбор и анализ
проб в режимах in-line (в линии), on-line (на линии) или at-line (у линии), восприни-
маются как дополнительная нагрузка, замедляющая скорость выпуска серии.
Тем не менее, отказываясь от внедрения систем тотального мониторинга каче-
ства в режиме реального времени, компании потенциально теряют значительные
средства по меньшей мере тремя путями. Во-первых, без непрерывного мониторин-
га остается мало шансов на внедрение RTR: задержки, связанные с анализом off-
line (вне линии) при выпуске производственной серии являются одним из самых
значимых факторов, ограничивающих скорость цепи поставок в фармацевтическом
производстве. Во-вторых, несмотря на то что существует вероятность обнаружения
большего числа серий, не соответствующих критериям выпуска, статистические
модели показывают, что потенциально меньшее количество серий будет отбракова-
но при увеличении объема выборки. Иначе говоря, с учетом неточности измерения
и истинного распределения характеристик качества при применении традицион-
ных методик анализа выпускаемых серий существует определенный риск отбра-
ковки удовлетворительных серий (которые могли бы пройти контроль), поскольку
ограниченный объем выборки не дает адекватного представления о характеристи-
ках совокупности (рис. 4). Наконец, самый важный фактор — традиционные мето-
дики отбора и анализа проб являются реальными барьерами на пути непрерывного
совершенствования; знания основ статистической теории позволяют показать, что
для обнаружения нарастающих изменений в воспроизводимости процесса необ-
ходимо исследовать, по меньшей мере, несколько сотен элементов совокупности
(рис. 5). Следовательно, если даже компания захотела бы исследовать потенциаль-
ные возможности улучшения эффективности процессов, то лишь маловероятные
изменения технических характеристик процесса можно было бы признать со стати-
стической достоверностью.
4.1.3.2. Отсутствие экономических стимулов
Среди производителей распространено мнение, что инвестиции в процессный ана-
лиз или модернизацию производственных технологий не настолько окупаются, что-
бы оправдать расходы. До известной степени это весомый аргумент. Исторически
сложилось, что большинство отраслей промышленности, в которых значительные
инвестиции в процессный анализ оправдали себя, внедрили систему непрерывного
производства; эффективно контролировать непрерывный процесс (по сравнению
с серийным производством) без процессного анализа в режиме реального времени
намного труднее [35]. Поэтому в то время как в фармацевтической индустрии су-
ществовала возможность выбора, другие производители вынуждены были внедрять
систему РАТ в свои производственные операции.
386
Часть 4. Процессно-аналитические технологии
98 96 94 92 90 88 98 96 94 92 90 88
Доля партий производственной серии, оцененных правильно, %
Рис. 4. Сравнение кривых, построенных по методу Монте-Карло, описывающих рабочие
характеристики (ОС) стратегий выпуска, основанных на USP (905) (а) и РАТ (б). Кривая ОС
USP (а) построена на основании предположения о точности измерений, соответствующей 2%
RSD (relative standard deviation — относительное стандартное отклонение); кривая ОС РАТ (б)
охватывает результаты анализа 800 таблеток методом БИК, точность измерения 0,9%. Обе
кривые построены на основании данных по одной и той же генеральной совокупности, состоящей
из 1 млн таблеток с различным уровнем однородности по качеству. Каждая кривая состоит из
четырех зон: зоны над и под сигмовидным изгибом соответствуют долям производственных
серий, явно прошедшим контроль или отбракованным на основании критериев выпуска. Вдоль
сигмовидного изгиба находятся зоны, соответствующие количеству ошибочных отбраковок
серий (ниже кривой) и ошибочных приемок серий (выше кривой). Зубчатая форма кривых связана
с ограниченным числом итераций. Наклон кривых демонстрирует повышенную специфичность
(или «приспособляемость») испытаний при выпуске, оптимизированных для систем РАТ
Детектируемые изменения параметров пригодности процесса
(95% достоверность) как функция частоты выборки
Рис. 5. Взаимосвязь между частотой выборки и эффективностью вывода о пригодности процесса.
Кривая основана на ширине доверительных интервалов для оценок среднего и отклонения.
Представленное соотношение не учитывает влияния точности эталонных измерений, которое
снижает дополнительно способность распознавать изменения в технических характеристиках
процесса
Глава 4.1. Аргументы в пользу процессно-аналитическои технологии...387
Поскольку инвестиции в РАТ продолжают оставаться для большинства фарма-
цевтических компаний скорее возможностью, чем приоритетной необходимостью,
аргументы, оправдывающие расходы на РАТ, конкурируют с аргументами в поль-
зу расходов капитала, связанных с другими инициативами. В каждом цикле пла-
нирования управляющие компаний должны принять решение о дополнительном
вложении капитала, выбрав среди множества направлений, таких как расширение
научно-исследовательских разработок {R&D), усовершенствование производствен-
ных мощностей или дополнительные вложения в продажи и маркетинг (например,
продажные, общие и административные расходы {SG&A)). Для любого конкретного
проекта, который необходимо профинансировать, ожидаемая прибыль от инвести-
руемого капитала не просто должна превышать стоимость инвестированного капи-
тала компании (т. е. взвешенную среднюю стоимость капитала {weighted average cost
of capital)-, для проектов-победителей может быть выдвинуто требование превыше-
ния ожидаемой окупаемости инвестированного капитала {return on invested capital),
или, по меньшей мере, ожидаемая рентабельность должна превысить прибыль от
альтернативных инвестиций. Недавнее академическое исследование — практиче-
ский анализ потенциальной окупаемости инвестиций {ROI — return on investment)
в РАТ и ресурсосберегающее производство в фармацевтической промышленности
показало, тем не менее, что для большинства фармацевтических компаний усовер-
шенствование производственных технологий может, в конечном итоге, принести
огромную выгоду [44].
К сожалению, сторонники внедрения РАТ только начинают разрабатывать ме-
тодики количественного анализа потенциальной доходности инвестиций. Далее
важно учитывать относительный уровень риска, связанного с инвестициями в РАТ
(в сравнении с альтернативными возможностями). В отличие от инвестиций в про-
дажи или маркетинг, в промышленности остается значительная неопределенность
в части вероятности достижения планируемой прибыли на инвестиционный капи-
тал или возможности появления новых проблем, вызванных инвестициями в РАТ.
Поэтому руководство обычно считает, что проще обосновать вложения в R&.D
и маркетинг по сравнению с РАТ к модернизацией технологий.
Кроме обеспокоенности вероятностью и величиной прибыли от инвестиций
в РАТ, часто встречается мнение, что технология и оптимизация стоимости про-
дукции не являются приоритетами для промышленности; производство зачастую
рассматривается как источник затрат, а не как компонент, создающий стоимость.
В качестве свидетельства в пользу этой теории представляется распределение расхо-
дов корпорации (рис. 6). На основании ежегодных деклараций о доходах по 2005 г.
средние расходы на R&D и SG&.A в ведущей десятке брендовых фармацевтических
компаний (по рыночной капитализации; данные на 7 ноября 2006 г.) почти вдвое
превышали стоимость проданных товаров согласно отчетам. Еще одним следствием
этой теории является то, что коллективные и индивидуальные инвесторы (владею-
щие фармацевтическими компаниями и инвестирующие капиталы в их деятель-
ность), а также избранные ими советы директоров компаний благожелательно от-
носятся к инвестициям в R&.D и маркетинг, в то же время их политика в отношении
производственных технологий весьма близорука. Иногда говорят, что Wall Street
поощряет фармацевтические компании за инновации в открытиях и повторы в об-
388
Часть 4. Процессно-аналитические технологии
а)100
90
Чистая прибыль
80
по кредитам
50
«Ж
л
40
30
20
Торговые общие и административные
расходы
70
60
.учнс 1ИСС.. ЛИНИЯ
и разраб тки
Компания (тикерный символ)
Торговые, общие
и административ-
ные расходы
25%
ч Торговые, общие
чистая и административные Чистая
ппмКипк расхода Прибыль
33%
10
°JNJ
Компания (тикерный символ)
прибыль
Прочее
12%
Научные
исследования
и разработки
15%
товаров
25%
Научные
ледования
и разработки
9%
Стоимость проданных
Джинерик то^ов
Прочее
10%
Патент
Стоимость
проданных
Чистая прибыль
19%
Прочее
14%
Стоимость
проданных
товаров
16%
Научные
исследования
и разработки
23%
Торговые, общие
адми н истративные
расходы
28%
Биотех.
Рис. 6. Распределение компонентов дохода (согласно отчетам по итогам 2005 финансового
года) у производителей патентованных препаратов (а), дженериков (б) и биотехнологической
лекарственной продукции (в). Компании перечислены в порядке величин рыночной капитализации
(по данным на ноябрь 2006 г.)
ласти технологий [45]. Не случайно совпадение в том, что, например, назначение
вице-президента по производству Ричарда Т. Игарка исполнительным директором
компании Merck ъ мае 2005 г., по словам финансовых обозревателей, «разочаровало
инвесторов», которые определенно предпочли бы видеть на этом посту человека,
«занимавшегося научно-исследовательской деятельностью» [46]. Назначение со-
впало с началом падения рыночной капитализации почти на 25% за последующие
шесть месяцев.
В то время как причины равнодушного отношения фармацевтических про-
изводителей к системе РАТ и к реформированию производства выглядят весьма
основательными, они вторичны по отношению к реальным и воспринимаемым
субъективно рискам, связанным с нормативно-правовой неопределенностью, со-
провождающей производственные инновации. Например, хорошо известно, что
многие компании начали использовать инструменты РАТ задолго до появления
инициативы FDA, что позволяет предположить: внутреннее осознание экономиче-
ской выгоды от внедрения процессной аналитики произошло уже довольно давно.
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...389
В ответ на опасения, связанные с тем, что использование новых технологий может
спровоцировать дополнительные инспекции со стороны FDA, отдельные из ком-
паний действовали по отношению к РАТ по принципу “Don’t use, or don’t tell”— не
пользуйся, или не говори [что пользуешься] [45].
4.1.3.3. Юридические препятствия
Реальные и субъективно воспринимаемые опасения законодательного несоответ-
ствия, вероятно, являются важнейшими факторами, объясняющими сопротивле-
ние промышленности производственным инновациям [1, 2]. В то время как первые
25 лет существования фармацевтических GMP оказались эффективными для обе-
спечения безопасности лекарств, отпускаемых по рецепту для потребителей, эф-
фективность быта достигнута за счет инноваций и гибких подходов. При невозмож-
ности регулировать процессы путем изменения материалов, условий работы или
уровня понимания процесса, процессная аналитика почти бесполезна, поскольку
отсутствует способность реагировать на новую информацию (за исключением от-
браковки материала/производственной серии).
Кроме того, компании, осмелившиеся внести изменения или внедрить новые
технологии, усовершенствовать технологические процессы, ввести новые стандарт-
ные операции или процессный анализ, сталкивались с требованиями предоставле-
ния обширной дополнительной документации инспекциям FDA и в итоге — с ри-
ском остановки производства. В конце концов, потенциальные законодательные
действия гасят желание производителей внедрять технологии, которые могут по-
казаться необычными, например, анализ в режиме реального времени или хемоме-
трику. И наконец, без пользы, приносимой рекомендациями РАТ и основанной на
анализе рисков инициативой cGMP, у производителей редко находятся стимулы для
формального анализа рисков устоявшихся процессов, так как они опасаются, что
информация, которую они могут обнаружить, будет использована против них путем
применения нормативных или законодательных мер.
4.1.4. Инициатива FDA по cGMP двадцать первого века
Состояние cGMP к началу двадцать первого века тормозило инновации в фарма-
цевтической промышленности. Это не осталось незамеченным в FDA, где также
увидели новые возможности в модернизации основ регулирования. Поскольку
многие изменения, включая малые оперативные модификации, требовали пред-
варительного санкционирования всех этапов — от организации до внедрения,
сотрудники нормативно-правовых органов были завалены документами, кото-
рые тысячами ежегодно поступали к ним. Средства тратились на рассмотрение
документов-дополнений, анализ и утверждение новых производственных мощно-
стей, процессов, документации и экспертизу. Всё это время FDA сдерживали внеш-
ние ограничения на рост бюджета (рис. 7).
В 2001 г. сотрудники FDA были столь загружены, что не могли осуществлять обя-
зательную инспекцию по соблюдению правил GMP, которая должна проводиться
один раз в два года. В конце концов, рассмотрение дополнений, анализ и инспекти-
рование стали препятствием на пути продвижения на рынок новых методов фарма-
цевтической технологии.
390
Часть 4. Процессно-аналитические технологии
1000
Регистрации новых препаратов по сокращенной процедуре
2001 2002 2003 2004 2005
Рис. 7. Тенденции изменений объема работ и кадровых ресурсов FDA. (Перепечатано из L.X.Yu,
Implementation of quality-by-desigm Question-based review, Drug Information Association (DIA) 42nd
Annual Meeting, Philadelphia, PA, 2006.)
4.1.4.1. Концепция инициативы
FDA инициировало общественный диалог, посвященный состоянию фармацевтиче-
ского производства и правилам FDA, в ходе дискуссий с Консультативной комисси-
ей по фармацевтической науке (ACPS—Advisory Committee for Pharmaceutical Science)
в июле 2001 г.; за которыми последовало обсуждение в рамках собраний Научного
совета FDA в ноябре 2001 г. и апреле 2002 г. [47]. В значительной мере обсуждения
сосредоточились на влиянии нормативно-правовой базы на инновации, качество
и эффективность, а также на возможности внедрения изменений. Начала оформ-
ляться новая парадигма, основанная на анализе рисков, предусматривающая по-
ощрение производителей, внедряющих инновации, путем предоставления «помощи
со стороны органов регулирования»; в новой парадигме вытесняется представление
о «соответствии нормативам» как о движущей силе инноваций. Новая парадигма
дала преимущества FDA еще и в том, что она позволила устанавливать приорите-
ты в проведении инспекций и распределять ресурсы в соответствии с рисками, что
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
391
снижает нагрузку на FDA. Эти изменения дали начало развитию отношений «FDA —
промышленность», ранее внешне напоминавших противостояние, в направлении
более тесного сотрудничества.
Термин РАТ в отношении фармацевтического производства был введен фор-
мально в ходе этих совещаний [48], однако значительная часть концепций, опреде-
ляющих ключевые принципы РАТ в фармацевтической науке, была представлена
специалистами-технологами и академическими учеными, многие из которых ра-
ботали над этими проблемами в своих организациях многие годы. Промышлен-
ные и научные презентации включали такие темы, как всеобщее управление каче-
ством [49], новые технологии фармацевтического производства [50], подход QbD
{quality by design — качество, заложенное при проектировании) [51] и др.
В августе 2002 г. FDA объявило об инициативе под названием «сСЛ/Рв фарма-
цевтическом производстве XXI века», с которой началась реализация двухгодич-
ной программы, осуществляемой междисциплинарными рабочими группами FDA,
а также комитетом управления cGMP {cGMP steering committee). Целью программы
являлась оценка действующей нормативно-правовой структуры и формирование
нового взгляда на управление производственными рисками и качеством продук-
ции. Новая инициатива, направленная на модернизацию требований FDA к фар-
мацевтическому качеству предназначенных для людей, ветеринарных и отдельных
биологических продуктов, пыталась реформировать фармацевтические програм-
мы также, как химические, производственные и управленческие {СМС — Chemistry,
Manufacturing, Controls) со следующими целями:
• поддерживать раннее внедрение новых технологий в фармацевтическую про-
мышленность;
• содействовать промышленному применению современных методов управле-
ния качеством, включая реализацию подходов систем качества ко всем аспек-
там фармацевтического производства и обеспечения качеством;
• содействовать внедрению подходов, основанных на анализе рисков, фокуси-
рующих внимание производителей и FDA на критических областях;
• обеспечить гарантии того, что проверка регламента, его соблюдение и мето-
дики проведения инспекций будут основаны на современных достижениях
фармацевтической науки;
• усилить согласованность и координацию программ FDA по управлению каче-
ством лекарственных продуктов, в частности, путем дальнейшего интегриро-
вания систем повышенного качества в бизнес-процессы FDA и за счет мето-
дик управления, связанных с аналитической деятельностью и инспекциями.
Оценки, сделанные рабочими группами, позволили разработать новые принци-
пы, включенные в cGMP двадцать первого века, а также в связанные с ними доку-
менты, такие как руководство по РАТ. В процессе исследований и разработок и на
последующем этапе реализации инициативы сохранялся набор руководящих прин-
ципов:
• ориентация с учетом рисков;
• стратегия и стандарты, основанные на научных представлениях;
• ориентация на интегрированные системы качества;
392
Часть 4. Процессно-аналитические технологии
• международное сотрудничество;
• крепкая система общественного здравоохранения.
Итоговый отчет о результатах инициативы и планах на будущее был опублико-
ван в сентябре 2004 г. Отчет рационально описывает мотивы, происхождение, про-
цесс развития и механизмы внедрения и оценки эффективности инициативы; он
представлен на веб-сайте Управления по фармацевтической науке (OPS — Office of
Pharmaceutical Science) Центра оценки и исследований лекарств (CDER — Center for
Drug Evaluation and Research) (http./fwww.fda.gov/cderOPS/).
Поскольку в точном изложении всех важных аспектов отчета в рамках этой гла-
вы нет необходимости, в следующих разделах будут представлены отдельные кон-
цепции и основные принципы инициативы, особо важные для понимания РАТ.
Это краткое изложение описывает отдельные принципы, положенные в основание
с GMP двадцать первого века; в нем нет намерения отразить структуру документа-
ции FDA, связанной с cGMP, во всей ее полноте. Всем активно вовлеченным в фар-
мацевтическое производство или заинтересованным в РАТ, предлагается прочитать
итоговый отчет [1], который следует считать первичным источником указаний.
4.1.4.2. Ориентация с учетом рисков
Выбор FDA курса на управление с учетом рисков является самым важным аспек-
том концепции cGMPдвадцать первого века. Общим заблуждением является то, что
инициатива Управления состоит в создании нового свода практических рекоменда-
ций для индустрии. На самом деле задача FDA — содействовать инновациям в про-
мышленности, в то время как инициатива cGMP двадцать первого века полностью
сосредоточена на изменении нормативно-правовой основы: качество и инновации
должны поощряться ослаблением надзора. Сейчас, когда Управление приступило
к этапу внедрения инициативы, многие из ранее существовавших барьеров исчезли.
Иначе говоря, в настоящее время фармацевтические компании могут добровольно
принимать решение о внедрении инновационных перемен, таких как система РАТ,
в свое развитие, функционирование и обеспечение качества технологических про-
цессов.
Установление очередности инспекций с учетом рисков. Механизм, при помощи
которого FDA намерено содействовать внедрению новых методов в промышленное
производство, обеспечивается за счет применения алгоритма установления очеред-
ности инспекций по соблюдению cGMP. Отметим кстати, что выбор объекта на
основании анализа рисков происходит по тому же механизму, который позволяет
Управлению оптимально распределять свои ограниченные ресурсы по надзору для
достижения максимального положительного влияния на общественное здоровье.
Высокая оперативность является основным компонентом планов FDA на будущее.
Ключевым аспектом программы выбора объекта инспекции с учетом анализа ри-
сков является построение модели классификации рисков, действующей в качестве
пилотной модели с начала 2005 финансового года.
Модель основана на методике иерархического ранжирования и фильтрации ри-
сков, в то время как потенциал риска объекта (SRP — site risk potential) оценивается
как функция взвешенных потенциалов для каждого из трех компонентов риска выс-
шего уровня — продукт, оборудование и процесс (рис. 8).
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
393
Рис. 8. Схематическое изображение пилотной модели оценки рисков, предложенной FDA
для расчета потенциального риска для объекта
Потенциал риска для каждого из трех компонентов высшего уровня рассчиты-
вается как функция выбранных факторов риска, связанных с компонентом (спе-
цифичны для объекта). Набор субкатегорий определяется для каждого компонента
высшего уровня; каждая категория состоит из индивидуальных факторов риска.
Исходные веса рисков в модели (фактические оценки рисков на самом низком
уровне) оптимизированы при помощи комбинации эмпирических свидетельств
и экспертных суждений. Примеры потенциальных факторов риска для каждого
компонента высшего уровня (и связанных с ним субкатегорий) были приведены
в отчете, подробно описывающем первую итерацию пилотной модели ранжирова-
ния рисков [52].
Результаты, полученные после первой итерации модели ранжирования рисков,
продемонстрировали способность модели распространять баллы SRP на фильтра-
цию рисков. Дальнейшие итерации в модели ранжирования рисков генерируют
путем корреляции прогнозируемых потенциалов риска (SRP) и коррекции весов
факторов риска для максимального увеличения эффективности прогноза SRP
(аналогично многомерной линейной регрессии). Отбор факторов риска в первой
итерации модели включает объединяющие факторы, такие как системы непрерыв-
ной оценки характеристик процесса как индикаторов уровня понимания и кон-
троля процесса на объекте. Разумеется, по мере обновления модели для фиксации
преимуществ новых лучших производственных практик, таких как РАТ, ранжи-
рование рисков начнет эффективно стимулировать производителей к внедрению
инноваций.
4.1.4.3. Системы обеспечения качества
Согласно руководству для персонала FDA [53], система качества представляет со-
бой «набор формальных и неформальных бизнес-практик и процессов, сосредото-
ченных на нуждах потребителей, видении лидерства, вовлечении персонала, посто-
янном совершенствовании, принятии информированных решений на основании
данных, полученных в режиме реального времени, и взаимовыгодных отношениях
с внешними партнерами по бизнесу, необходимых для достижения организацион-
ных результатов». На основании данного описания РАТ следует рассматривать как
394
Часть 4. Процессно-аналитические технологии
важный инструмент поддержки системы управления качеством. Как сказано ранее,
одной из задач FDA, связанных с инициативой, стало интегрирование систем каче-
ства и подходов, связанных с управлением рисками, в существующие программы
для содействия внедрению современных и инновационных технологий в промыш-
ленность, включая размещение на промышленных предприятиях систем управле-
ния качеством, аналогичных описанным в этой главе (например, ISO 9000).
В сентябре 2006 г. FDA выпустило «Руководство для промышленности: Систе-
мы качества в правилах с GMP в фармацевтическом производстве» [54]. Руководство
предназначено для «оказания помощи производителям во внедрении современных
систем качества и подходов, связанных с управлением рисками, для обеспечения
соответствия требованиям FDA по cGMP», в частности, частей 210и211.В процессе
работы группа по Разработке практического руководства по системам качества (QS)
выявила и отобразила отношения между правилами cGMP и различными моделями
систем качества, как внутренними, так и внешними по отношению к FDA. В резуль-
тате построена всесторонняя модель, позволяющая производителям, желающим
внедрить свои собственные системы управления качеством, быстро идентифици-
ровать аспекты качества, соответствующие (и не соответствующие) cGMP.
Руководство по системам качества начинается с описания основных концеп-
ций современных систем качества, включая разработки по качеству, подход QbD
(качество, заложенное при проектировании) и планирование продукции, управ-
ление качеством с учетом рисков, корректирующие и профилактические действия
(G4A4 — corrective and preventative action), контроль изменений, «отдел качества»
и инспекционная модель шести систем (six-system inspection model). Обсуждение во-
просов, связанных с отделом качества, включает описание его отношение к концеп-
циям контроля качества (QC — quality control) и обеспечения качества (QA — quality
assurance) и взаимоотношения между отделом качества и другими отделами органи-
зации, занимающейся фармацевтическим производством. Инспекционная модель
шести систем описывается как шаблон, по которому должны быть организованы
инспекции по соответствию с учетом новых подходов, связанных с системами ка-
чества. Кроме того, эта модель является образцовой при проведении внутренней
верификации соответствия в рамках фармацевтических организаций, внедряющих
системы управления качеством (рис. 9).
Большая часть руководства по QC посвящена описанию основных компонентов
современных систем качества, включая описание четырех главных факторов, на ко-
торые следует обратить внимание: ответственность управления, ресурсы, производ-
ственные операции и аналитическая деятельность. Для каждого фактора представ-
лено подробное разъяснение, включающее аспекты, пересекающиеся с правилами
cGMP (для каждого фактора соответствующие цитаты из нормативных документов
сведены в таблицу). В частности, в главе, посвященной производству, представлены
аспекты систем качества (и соответствующие правила cGMP), имеющие непосред-
ственное отношение к РАТ, в том числе анализ исходных материалов, мониторинг
производственных операций и процедуры, описывающие действия при обнаруже-
нии несоответствий. Наконец, руководство содержит множество важных ссылок
и соответствующих руководящих документов, с которыми должны ознакомиться
компании, заинтересованные во внедрении систем управления качеством.
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
395
Рис. 9. Модель инспекции FDA «Шесть систем»
4.1.4.4. Стратегия, основанная на научном подходе
Непрерывное совершенствование, называемое в FDA «существенным элементом
современной системы качества», направлено на повышение эффективности путем
«оптимизации процесса и уменьшения излишних затрат в процессе производства»
[1]. Одним из последствий существования регуляторной системы (до появления но-
вой инициативы) стало ненамеренное подавление практически всех возможностей
постоянного обновления производства после того, как фармацевтический продукт
был одобрен для рынка. Изменения в материалах и процессах необходимо было
оправдывать с рассмотрением их воздействия на качество продукта, зачастую про-
цедура внесения дополнений после утверждения оказывалась весьма длительной.
Во многих отраслях современной промышленности производители (большинство
из которых сталкивается с равными или превосходящими рисками для обществен-
ной безопасности в сравнении с существующими в фармацевтической отрасли)
ввели в практику постоянную корректировку и регулирование производственных
операций для достижения максимального качества и эффективности. Фармацевти-
ческие производители, напротив, были вынуждены относиться к устоявшимся тех-
нологиям, как к священным текстам, высеченным в камне.
Можно признать, что есть некоторые логические обоснования для ограничения
пределов и темпов изменений в процессах. Однако очевидно ошибочна идея о том,
что первая одобренная технология операции по производству лекарственного про-
дукта окажется оптимальной, особенно с учетом громадного финансового и мораль-
ного давления на команды разработчиков процессов, которые должны как можно
быстрее выпустить на рынок продукт новой лекарственной терапии. Осознание
этого побудило FDA приступить к разработке стратегий и стандартов содействия
инновациям, основанных на научных подходах. Действующий документ включает три
396 Часть 4. Процессно-аналитические технологии
обновленных руководящих документа: «Стерильные лекарственные препараты,
произведенные методом асептической обработки — cGMP» [55], руководство по
РА Ти проект руководства по протоколам совместимости. Каждый руководящий до-
кумент поощряет добровольное внедрение новых технологий в фармацевтическое
производство при помощи современных правовых механизмов, основанных на на-
учных подходах, позволяющих производителям вводить стратегические усовершен-
ствования, обеспечивающие соответствие нормативам.
Протоколы совместимости. В действительности фармацевтические произво-
дители всегда имели возможность исследовать технологические процессы на соб-
ственных предприятиях. Разница между старой нормативно-правовой моделью
и инициативой по cGMP двадцать первого века состоит в том, что после внедрения
инициативы производители, заинтересованные в повышении качества и эффектив-
ности технологических процессов, смогут вводить изменения быстрее при значи-
тельном уменьшении ресурсов на поддержание соответствия техническим услови-
ям. Ключ к достижению этих преимуществ — обоснованное понимание процесса
и предстоящих изменений, а также осознание того, что риск для потребителя, свя-
занный с внедрением усовершенствований, очень мал.
Новый механизм введения изменений в технологические процессы, отражающий
поворот к стратегии, основанной на научном подходе, подробно изложен в проекте
руководящего документа FDA «Протоколы совместимости — информация по хи-
мии, технологии и контролю (СМС)». Протокол совместимости (СР — comparability
protocol) представляет собой «четко изложенный подробный письменный план
оценки влияния специфических изменений СМС на идентичность, содержание ак-
тивных компонентов, качество, чистоту и активность конкретного лекарственного
препарата, поскольку эти факторы имеют отношение к безопасности и эффектив-
ности препарата» [56]. Предоставление СР производителем желательно и может
способствовать изменениям в технологических процессах, аналитических процеду-
рах, технологическом оборудовании или аппаратуре, системах контейнерной упа-
ковки, а также внедрению РАТ.
Выгода для производителей, предоставляющих СР, состоит в том, что после одо-
брения СР «FDA может назначить там, где это возможно, пониженную категорию
отчетности для будущих отчетов по изменениям СМС, описанных в одобренных
СР». Например, изменения, которые в противном случае потребовали бы предостав-
ления, анализа и утверждения дополнений после утверждения (PAS — postapproval
supplement) могут быть отнесены к изменениям, представляемым в ежегодном отче-
те (annual report), если они предусмотрены в одобренном СР. СР представляет собой
один из механизмов, при помощи которых FDA собирается уменьшить количество
документов-дополнений, требующих рассмотрения. Кроме того, СР введен для со-
действия свободному обмену информацией с Управлением, снижая, таким образом,
риск того, что изменения в технологических процессах приведут к неожиданной
остановке или задержке производства.
Валидация процесса. В соответствии со стратегией, основанной на научном под-
ходе, FDA притупило к пересмотру «Руководства по общим принципам валидации
процессов» 1987 г. и в марте 2004 г. выпустило пересмотренное Руководство по стра-
тегии соответствия (CPG — compliance policy guide) (ст. 490.100) «Требования к ва-
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...397
лидации процессов при производстве лекарственных продуктов и активных фар-
мацевтических ингредиентов для одобрения перед выпуском продуктов на рынок»
[52]. Современные переработки планировались для поддержания непрерывных
усовершенствований технологии содействие непрерывному улучшению технологий
и замене требования валидации с использованием образцов из трех производствен-
ных серий. В CPG изложена концепция, заключающаяся в том, что после иденти-
фикации и установления контроля всех основных источников погрешности, для де-
монстрации того, что при нормальных условиях и нормальных рабочих параметрах
технологический процесс позволяет получить приемлемый продукт, подготавлива-
ют представительную производственную серию {conformance batch). Тем не менее,
в СР(7не указано необходимое количество представительных производственных се-
рий; производителю предлагается представить «разумное обоснование» процедуры,
которую они выбирают для демонстрационной валидации.
Может показаться, что неопределенность пересмотренных правил (CGP) озна-
чает, что производителям понадобится проводить даже более интенсивные валида-
ционные испытания, в то время как CGP содержит указания, предлагающие путь
продвижения серии к распределению на рынке, совпадающий с изготовлением
нескольких исходных представительных серий или единой представительной се-
рии [57]:
Принципы передовой фармацевтической науки и производства и технологии произ-
водственного контроля помогают обеспечить высокий уровень понимания процес-
са и возможностей для контроля. Применение передовых принципов и технологий
контроля помогает обеспечить высокое качество путем непрерывного мониторинга,
оценки и внесения поправок в процесс производства каждой серии при помощи про-
шедших валидацию измерений, анализов, процедур контроля и предельных параме-
тров процесса. Для производственных процессов, разработанных и контролируемых
таким образом, возможно, у фирмы не возникнет необходимости производить мно-
жество согласованных серий перед окончательным распределением.
Интерпретация CPG предполагает, что важным аргументом в пользу РАТ может
стать ускорение валидации производственного процесса. Наконец, достойная ци-
тирования интерпретация новой модели, основанной на научном подходе, предпо-
лагает, что (вместо валидации процесса) производители должны «контролировать
процесс и проводить валидацию процедур контроля». Ожидается, что в дополнение
к пересмотренному CPGb ближайшем будущем FDA выпустит проект руководящего
документа по валидации технологических процессов, который будет приведен в со-
ответствие с принципами РАТ, концепцией QbD и другими, связанными с cGMP
двадцать первого века.
4.1.4.5. Международное сотрудничество
Признавая реалии действующего глобального рынка, PDA сделало координацию
деятельности с международными партнерами по нормативно-правовому регулиро-
ванию приоритетом инициативы по cGMPдвадцать первого века. Путем углубления
сотрудничества с международными партнерами по охране здоровья и нормативно-
правовой деятельности FDA может более эффективно использовать ресурсы путем
398 Часть 4. Процессно-аналитические технологии
интенсификации обмена информацией и координации деятельности. Междуна-
родная конференция по гармонизации технических требований к регистрации
фармацевтических продуктов, предназначенных для человека (ICH — International
Conference on Harmonization) (www.ich.org) стала важнейшим механизмом междуна-
родного сотрудничества между органами нормативно-правового регулирования
стран Европы, Японии и США.
Выражением единодушных взглядов стал проект концепции задач ICH по гар-
монизации усилий нормативно-правовых органов при введении систем качества
на производстве, представленный в июле 2003 г. на совещании ICH. «Разработать
гармонизированную систему качества для фармацевтического предприятия, приме-
нимую в течение жизненного цикла продукта, уделяя особое внимание управлению
рисками качества и научному подходу».
Три согласованных руководства определяют основные принципы привлечения
ICH к гармонизации фармацевтических систем качества — Q8: Фармацевтические
разработки; <29: Управления рисками качества и СЮ: Системы качества в фарма-
цевтическом производстве (кроме того, каждый руководящий документ включает
фрагменты из Q6A. Спецификации: Процедуры контроля и критерии приемлемо-
сти для новых лекарственных веществ и новых лекарственных продуктов: Химиче-
ские вещества).
Q8: Фармацевтические разработки. В соответствии с руководством ICH Q8 [58],
задачей фармацевтического планирования является «разработка качественного про-
дукта и технологического процесса, позволяющего получить продукт с воспроизво-
димыми характеристиками». В то время как руководство не содержит отдельного
упоминания о концепции QbD, в задачу экспертной рабочей группы (EWG — expert
working group) ICH по созданию (98 входило описание системы, обеспечивающей
стимулы для производителей к включению аспектов QbD и системы непрерывного
совершенствования в течение всего жизненного цикла продукта. Для достижения
поставленной цели они включили содержание статьи 3.2.3.2 нормативного доку-
мента в общий технический документ (CTD — common technical document) ICH —
M4 [59] и электронный общий технический документ FDA (eCTD — electronic common
technical document) [60].
Статьи CTD, посвященные фармацевтическим разработкам и общим аспектам
качества (QOS— quality overall summary) (рис. 10), предлагают ученым-фармацевтам
специальные каналы для представления в нормативно-правовые органы информа-
ции, связанной со специальными знаниями и пониманием процесса, собранной во
время разработки нового продукта. (Информацию можно обновлять по мере на-
копления новых знаний, полученных во время жизненного цикла продукта после
его одобрения.) Информация, изложенная в этих главах, содержит важные сведе-
ния, связанные с понижением уровня потенциального риска для объекта (т. е. SRP,
если речь идет об инспекции, основанной на анализе рисков) и содействием эф-
фективному, основанному на вопросах обзору (QbR — question-based review) химико-
фармацевтических свойств, процессов производства и контроля [61]. Анализ, осно-
ванный на вопросах, представляет собой еще один механизм, при помощи которого
FDA намерено рационализировать регуляторные процессы, а также поощрять про-
изводителей к внедрению лучших практик управления качеством.
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
399
Кроме содействия контролю, основанному на анализе рисков, содержание ста-
тей, посвященных фармацевтическим разработкам и QOSb СТО, включает инфор-
мацию, важную для внедрения систем непрерывного совершенствования и гибких
технологий. Информация, изложенная в этих статьях, представляет научный подход
к пространству проектных параметров производства, спецификаций обрабатывае-
мых и выпускаемых материалов и производственному контролю.
Согласно руководству Q8, пространство проектных параметров представляет со-
бой «многомерную комбинацию и взаимодействие исходных переменных и параме-
тров процесса, которые, как показано, обеспечивают гарантии качества». Посколь-
ку производственный контроль осуществляется в пределах пространства проектных
параметров, оперативные параметры можно регулировать для улучшения качества
продукта или эффективности производства. На основании действующего опреде-
ления, функционирование за пределами установленного пространства проектных
параметров инициирует регуляторный процесс внесения изменений после утверж-
дения. Таким образом, полная и точная передача информации, связанной с про-
странством проектных параметров жизненно важна для компании, заинтересо-
ванной в максимальном повышении эффективности при сохранении соответствия
нормативам. Далее, при наличии новых путей передачи информации, у компаний
появляются стимулы к проведению технологических исследований до утвержде-
ния продукта на рынке при необходимости расширения пространства параметров
процесса или обновления спецификаций и процедур контроля. Кроме того, при
разработке нового продукта при необходимости может быть использован опыт,
полученный в процессе разработки (и производства) аналогичных лекарственных
Рис. 10. Схематическая иллюстрация общего технического документа (CTD — common technical
document) ICH M4; содержание модулей «Общий обзор по вопросам качества» (2.3) и «Качество»
(3) больше всего связано с РАТ
400
Часть 4. Процессно-аналитические технологии
69: Управление рисками качества. Задача второй рабочей группы (ICH Q9 EWG)
состоит в более четком определении принципов интегрирования управления риска-
ми в процесс принятия решений, касающихся качества, в том числе соответствия
требованиям cGMP, органами регулирования и производителями. В ноябре 2005 г.
Q9 EWG опубликовала версию руководства по 69 «Этап 4», в которой определены
два приоритетных принципа управления рисками качества, представлена модель
процесса управления рисками качества (рис. 11), а также терминология и инстру-
менты оценки риска и управления рисками. Кроме этого, документ включает крат-
кий список ссылок для получения более подробной информации по методикам
управления рисками, таким как FMECA (failure mode effect and criticality analysis) —
анализ видов и критичности отказов, являющихся важными инструментами при-
оритетного внедрения РАТ. Руководство не претендует на роль подробного спра-
вочника по управлению рисками; тем не менее руководство 69 является ценным
источником информации для компаний, заинтересованных во встраивании систем
управления качеством в производство [62].
Рис. 11. Схема процесса управления рисками, представленная в ICH 09
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
401
СЮ: Фармацевтические системы качества. В то время как документ «Этап 2» для
третьей, последней части руководства «010: Фармацевтические системы качества»
еще не опубликован, итоговая статья с изложением концепции в целом доступна
с 2005 г. [63]. Аналогично тому, как в руководстве FDA по системам качества обозна-
чена взаимосвязь между cGMP и другими промышленными системами управления
качеством, в документе 010 переброшен мост между подходами к системам каче-
ства, принятыми в различных региональных руководящих документах, что способ-
ствует глобальной гармонизации систем качества. Предполагается, что руководство
усилит и дополнит изложение вопросов, затронутых в 06Л, 08 и 09, а также пред-
ставит принципы фармацевтических систем качества, основанных на элементах из
стандартов ISO 9001 и 9004. Кроме того, предполагается, что в руководстве будут
представлены гармонизированные определения спорных вопросов, важных для си-
стем РАТ, в том числе для непрерывного совершенствования, методов глубинного
анализа данных и принципов валидации измерительных систем.
4.1.5. Развитие РА Тв фармацевтическом производстве
Несмотря на то что представляется заманчивым охарактеризовать /МТкак револю-
ционное изменение в фармацевтическом производстве, история, вероятно, покажет,
что началом внедрения инициативы по cGMP двадцать первого века и разработкой
руководства по РАТ знаменуется начало периода быстрой эволюции в фармацевти-
ческом производстве, которая продолжится в будущем. Хотя инициатива по cGMP
двадцать первого века является более масштабной (по отношению к изменению
взаимоотношений между FDA и фармацевтической промышленностью), интерес
к руководству по РАТ и возможностям, которые предоставляет система, изначально
был намного больше. Позднее (видимо, одновременно с некоторыми изменениями
в руководстве FDA) произошел заметный сдвиг интереса в сторону системы QbD,
которая практически не упоминалась в большинстве документов по cGMP двадцать
первого века. Важно иметь в виду, что в то время, как во многих областях промыш-
ленности в течение ряда лет (со времени опубликования первых методов Шухарта)
проходил «парад» новых систем качества, принципы, на которых основаны системы
РАТ и QbD, такие как планирование робастных технологий, мониторинг качества
и эффективный контроль, остаются неизменными, независимо от названия ини-
циативы. Кроме того, как и PAT, QbD не является новой концепцией. В действи-
тельности, доктор Геничи Тагучи, считающийся создателем системы QbD, начал
применять QbD в фармацевтическом производстве, когда работал консультантом по
статистике в компании Morinaga Pharmaceuticals, в Японии в 1947—1949 гг. [25].
Уникальность руководства по РАТ ъ сравнении с обычными руководящими до-
кументами FDA состоит в том, что оно не является инструктивным или ограничи-
вающим документом; скорее, руководство представляет принципы и инструменты,
на основании которых строится система РАТ, с целью «выявления возможностей
и развития регуляторных процессов, способствующих инновациям». FDA при раз-
работке руководства по РАТ ставило задачу уменьшить угрозу регуляторной неопре-
деленности, рассматриваемой как основной фактор, ограничивающий инновации
в фармацевтической промышленности. Руководство представляет действующие
402
Часть 4. Процессно-аналитические технологии
нормативы; оно создавалось в соответствии с инициативой FDA по cGMP двадцать
первого века. Кроме того, руководство подчеркивает, что решение части произво-
дителей работать при внедрении системы РАТ совместно с FDA является доброволь-
ным. Поскольку руководство по своей сути не ориентировано на перспективу, в нем
нет указаний «как внедрить РАТ», а также нет описания практик или технологий,
«одобренных для системы РАТ».
4.1.5.1. Понимание процесса
FDA считает, что РАТ представляет собой «систему планирования, анализа и кон-
троля технологического процесса при помощи периодических измерений крити-
ческих параметров качества и эффективности исходных и находящихся в процессе
переработки материалов, а также технологических процессов с целью обеспечения
качества готового продукта». На основании этого определения можно было бы счи-
тать РАТ расширенным вариантом РАС (Process analytical chemistry — процессно-
аналитическая химия); система РА Т включает такие аспекты РАС, как измерения
и контроль, делая дополнительный акцент на QbD и понимание процесса.
Согласно РАТ считается, что хорошее понимание процесса, в общем, достигну-
то, если соблюдены следующие условия:
1. Идентифицированы и объяснены все критические источники вариабельно-
сти.
2. Вариабельность процесса управляема.
3. Параметры качества продукта предсказуемы точно и достоверно в простран-
стве проектных параметров, установленных для используемых материалов,
параметров процесса, технологии, окружающей среды и других условий.
Кроме того, согласно руководству, возможность прогноза «отражает высокую
степень понимания процесса».
Наличие одной лишь прогностической модели (для характеристик качества про-
дукта) не всегда, тем не менее, отражает понимание процесса. Относительно об-
щим примером является прогноз характеристик материала или продукта при по-
мощи многомерного анализа, например прогноз скорости растворения таблетки
при помощи БИК-спектроскопии (ближней ИК-спектроскопии). Многие иссле-
дователи продемонстрировали, что (в отдельных случаях) можно прогнозировать
высвобождение лекарства из таблеток in vitro при помощи неразрушающей БИК-
спектроскопии путем построения калибровочной модели для скорости растворе-
ния. Без механического понимания физико-химических характеристик процесса (с
корреляцией по скорости растворения), измеренных при помощи БИК, калибро-
вочная модель поможет лишь в распознавании паттерна (рис. 12) [64]. В то время
как такая калибровка может быть полезной, без более глубокого понимания ее как
основы для корреляции, она, вероятно, не может демонстрировать понимание про-
цесса.
Пространство проектных параметров и качество, заложенное при проектирова-
нии (QbD). Концепция многомерного пространства приемлемых операционных
условий, или пространства проектных параметров, является одним из важнейших
аспектов инициативы по cGMP двадцать первого века, способствующих непрерыв-
Глава 4.1. Аргументы в пользу процессно-аналитическом технологии...
403
Планирование
продукта и процесса
(кри~ .ч эские параметры)
Причинная
связь >
Данные PAT-ci сора
(например, БИК)
Понимание метода
РАТ
Причинная
\ связь
Корреляция
(например, модель калибровки)
Параметры, харак • ризую-
щие лекарственный продукт
(например, растворение)
Рис. 12. Иллюстрация: аспекты понимания методов, необходимые для подтверждения
измерений характеристик продукта при помощи непрямого и/или неразрушающего анализа
ному совершенствованию. В среде, где внедрена система РАТ, в пространстве про-
ектных параметров процесса должно прослеживаться наличие QbD [65], и должен
существовать математический аппарат, при помощи которого выявляется связь
между пониманием процесса и решениями по контролю в режиме реального вре-
мени (рис. 13).
Действующее определение пространства проектных параметров (JCH 08), к со-
жалению, содержит мало указаний по отношению к пространству проектных пара-
метров процесса, необходимых для внедрения. В результате множество интерпре-
таций понятий, описывающих «пространство приемлемых проектных параметров»
с недавних пор распространилось среди производителей. Одним из самых распро-
страненных заблуждений является то, что пространство эффективных проектных
параметров функционирования процессов или стандартных операций может быть
определено при помощи обычной траектории измерений параметров РАТ (т. е.
«сигнатуры процесса») для производственных серий, обладающих приемлемыми
характеристиками качества (т. е. golden path — «золотой путь»). Такие данные по-
лезны для мониторинга, однако они не являются чем-то большим, чем современ-
ная версия валидации процесса по трем сериям. «Золотые пути» или траектории
процессов недостаточны для контроля, поскольку 1) сам путь не обязательно пред-
сказуемый и 2) такой контроль предполагает, что процесс ограничен исторически
сложившимся путем в пространстве проектных параметров. Изначально термин
«сигнатура процесса» был определен как многомерный анализ процесса, например,
БИК-спектр, несущий характеристики, полезные для описания влияния обработки
на химические и физические аспекты обрабатываемого материала [38].
В то время как, вероятно, слишком рано устанавливать окончательный стандарт
разработки пространства проектных параметров фармацевтического процесса, сле-
дует определить следующие минимальные критерии пространства проектных пара-
метров процесса, который можно контролировать:
• пространство проектных параметров процесса должно быть представлено
в виде математической модели, устанавливающей количественную взаимо-
связь между эффективностью процесса, качеством исходных материалов
и оперативными параметрами процесса;
• соответствующие характеристики продукта, связанные с качеством, должны
быть учтены в модели пространства рабочих параметров (например, однород-
ность содержимого, биодоступность, стабильность);
404
Часть 4. Процессно-аналитические технологии
Процесс
Пространство
проектных
параметров
РАТ
Характеристики
продукта
Предназначена для не-
прерывного обеспече-
ния качества в режиме
реального времени
Пространство проектных пара-
метров устанавливает границы
для составов и переменных
процесса, в которых достигает-
ся качество и заданные харак-
теристики продукта
Рис. 13. Взаимосвязь между пространством проектных параметров, РАТ и контролем процесса
в системе производства, основанной на качестве через проектирование. (Источник: R. С. Lyon,
Process monitoring of pilot-scale pharmaceutical blends by near-infrared chemical imaging and
spectroscopy. Eastern Analytical Symposium, Somerset. NJ. 2006.)
• перефразируя известную цитату Альберта Эйнштейна, можно сказать: мо-
дель (пространства рабочих параметров) должна быть настолько сложной,
насколько это необходимо (для точного прогноза), но не более того;
• характеристики продукта, являющиеся избыточными или не критическими
для качества, не должны учитываться моделью пространства проектных па-
раметров (тем не менее не должно быть штрафов за мониторинг этих параме-
тров);
• перед внедрением необходимо провести валидацию способности модели
пространства проектных параметров прогнозировать качество готовой про-
дукции, точно так же, как в процедуре «биовейвер» при внесении измене-
ний после утверждения необходимо подтвердить корреляцию in vivo—in vitro
(IVIVC);
• точность модели пространства проектных параметров нельзя установить
а priori со статистической достоверностью в части гиперпространства пара-
метров; операции в таких режимах должны инициировать дополнительную
активность по обеспечению (инспекции) качества до того, как модель про-
странства проектных параметров можно будет обновить и провести повтор-
ную валидацию;
• если в зоне пространства проектных параметров, где ожидается приемлемое
качество продуктов, наблюдается неприемлемое качество продукта, про-
странство следует считать непригодным для контроля процесса (из-за смеще-
ния или появления новых факторов в пространстве параметров) до иденти-
фикации пропущенного фактора (факторов) и включения их в модель с по-
следующей повторной валидацией модели.
Если пространство проектных параметров, построенное на основании модели,
содержит существенную часть факторов вариабельности характеристик качества
продукции, то можно включить в проект пространство контроля для обозначения
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
405
границ нормальных операций. На основании этих определений алгоритм модели
контроля для каждой операции технологического процесса будет построен исходя
из подмножества пространства контроля, охватывающего параметры качества ма-
териалов и параметры технологического процесса, влияющие на данную операцию.
Каждая модель контроля стандартной операции включает периодическое регули-
рование параметров процесса в ответ на изменения в исходных материалах (упре-
ждающий контроль) или готовых продуктах (обратная связь). Иначе говоря, следует
контролировать процесс и проводить валидацию процедур контроля.
Математическая связь между моделями пространства проектных параметров,
процесса и контроля позволяет проводить непрерывную оптимизацию качества
продукта путем поиска оптимальной точки пространства контроля. По мере повы-
шения уровня понимания процесса или при изменении условий технологического
процесса факторы могут быть добавлены или убраны из пространства проектных
параметров, а модели процесса и контроля обновлены. При учете таких факторов,
как выход, эффективность или С/ Ткак функции переменных, которые охватывают-
ся пространством проектных параметров процесса, процесс может быть оптимизи-
рован одновременно по качеству и экономичности.
Вероятно, многие операции фармацевтического производства не поняты на-
столько, чтобы полностью описать вариации характеристик качества продукта
в форме функций (например, функций преобразования); достижение такого уровня
понимания технологии должно стать целью для промышленности. Использование
в качестве базиса для пространства проектных параметров функциональных пред-
ставлений понимания процесса в отличие от предполагаемых характеристик про-
являет множество оперативных преимуществ:
• Эффективная разработка процесса. В то время как действующее определение
пространства проектных параметров не препятствует включению знаний,
накопленных при производстве других продуктов и применении других про-
цессов, представления знаний на основе модели предлагают более робастные
рамки включения внешней информации или информации a priori. Несмотря
даже на то, что ожидаемый при конкретной комбинации входных параметров
и параметров процесса уровень качества не переносится, по всей вероятности,
на новый продукт или процесс (в абсолютном исчислении), функциональные
соотношения, прогнозирующие качество, могут быть аналогичными. Кроме
того, разработка пространства проектных параметров на основе определен-
ной модели дает возможность непосредственного объединения основных
принципов и механистических знаний, что позволяет значительно снизить
сложность экспериментальных проектов, необходимых для разработки про-
цесса, поскольку важнейшие условия могут быть определены в процессе ком-
пьютерного моделирования эксперимента.
• Качество, заложенное при проектировании. Объединение функциональных
соотношений между входными параметрами и качеством продукта (или эф-
фективностью), по определению включающее магнитуду и направленность,
позволяет использовать пространство проектных параметров как инструмент
многокритериальной оптимизации процесса. Далее, представление знаний
на основе модели совместимо с концепцией управления рисками, так как
406 Часть 4, Процессно-аналитические технологии
позволяет проводить более гибкие операции, поскольку риск в соединении
с экстраполяцией может быть прогнозирован.
• Разработки систем контроля. Разработка пространства проектных параме-
тров, основанная на определенной модели, представляет собой плавный пе-
реход между планированием процесса и контроля. Буквально, пространство
проектных параметров, построенное на основании модели, представляет ша-
блон для разработки моделей управления с упреждающим контролем. Кроме
того, построение пространства проектных параметров при помощи опреде-
ленной базовой модели облегчает валидацию системы контроля и идентифи-
кацию научно обоснованных спецификаций материалов в процессе обработ-
ки и спецификаций при выпуске продукции.
• Изменение масштаба производства и перенос технологий. В действующей си-
стеме разработки процесса обычно применяют планирование эксперимента
{DOE — design of experiment), где некоторые входные переменные параметры
специфичны для конкретного продукта (например, степень чистоты вспомо-
гательных материалов) или же специфичны параметры процесса для конкрет-
ного оборудования (например, скорость измельчителя, угол увлажнителя —
damper angle). В парадигме, основанной на построении модели, пространство
проектных параметров процесса в идеале строится на основании единиц, не
зависящих от продукта и оборудования, имеющих более фундаментальный
физический смысл (таких как вязкость, энергия или работа). При проектиро-
вании и описании процесса производства в фундаментальных терминах или,
возможно, в стандартизированных безразмерных единицах облегчается за-
дача изменения масштаба производства и переноса пространства проектных
параметров и моделей контроля процесса на аналогичные производственные
процессы, основанные на тех же самых физических принципах.
Проходящие в настоящее время академические исследования развивают кон-
цепцию пространства проектных параметров, основанную на модели. Работаю-
щие в пределах действующей системы, производители, которые способны проде-
монстрировать понимание процесса или готовы инвестировать в систему РАТ, для
облегчения понимания процесса могут использовать инструменты и положения
системы для внедрения инноваций и непрерывного совершенствования с более
эффективным регуляторным контролем (это означает возможность внесения из-
менений без дополнительного анализа). Система РАТ состоит из двух компонентов:
1) ряд научных принципов и инструментов, поддерживающих инновации и 2) стра-
тегия внедрения нормативно-правовой системы, приспособленной к инновациям.
В следующих главах подробно описаны отдельные аспекты обоих компонентов.
4.1.5.2. Принципы и инструменты РАТ
Центральным принципом системы РАТ является признание того, что некоторые
физические и механические характеристики фармацевтических ингредиентов не
всегда хорошо поняты, и даже процессы, понимание которых расценивается как
высокое, подвержены ограниченным стохастическим вариациям. Таким образом,
сердцевина руководства по РАТ посвящена описанию принципов и инструментов,
таких как процессные анализаторы и анализ рисков, которые производители могут
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...407
использовать для углубления понимания процессов и оценки латентных рисков для
качества продукции.
Инструменты РАТ. Руководство описывает четыре категории инструментов РАТ.
• многофакторные инструменты для проектирования, сбора данйых и анализа;
• процессные анализаторы;
• инструменты контроля процессов;
• непрерывное совершенствование и инструменты управления знанием.
Поскольку каждая из четырех категорий основана на методиках и технологиях,
уже устоявшихся в других областях, таких как РАС, обсуждение каждой категории
в рамках руководства фокусируется на аспектах, уникальных или существенных для
фармацевтического производства, таких как сигнатура процесса [2]. В соответствии
с сущностью руководства как катализатора инноваций, FDA попыталось избежать
упоминания в итоговой версии руководства по РАТ каких бы то ни было конкрет-
ных инструментов или технологий. Статья руководства, посвященная инструмен-
там РАТ включает перекрестные ссылки на соответствующие части действующих
нормативов, которые следует учитывать производителям, разрабатывающим стра-
тегию или систему РАТ.
Ставдарты применения РАТ в фармацевтическом производстве. На ранних этапах
разработки структуры системы РАТ, в FDA существовало понимание того, что отсут-
ствие международных стандартов — серьезное препятствие на пути к координации
нормативно-правовых аспектов РАТ а внедрению системы в мировом фармацевти-
ческом производстве. В 2003 г. группа PATFDA работала вместе с ASTMInternational
(American Society for Testing and Materials — Американское общество (специали-
стов) по испытаниям и материалам) с целью создания технического комитета Е55
по применению РАТ в фармацевтическом производстве. Комитет Е55 занимался
проблемами, связанными с контролем, проектированием и техническими харак-
теристиками процессов, а также анализами, связанными с оценкой/обеспечением
качества в фармацевтической промышленности. В работе комитета участвовали
производители фармацевтической продукции и фармацевтического оборудования,
федеральные агентства, проектировщики, профессиональные сообщества, торгово-
промышленные ассоциации, финансовые организации и академические ученые
(www.ASTM.org).
В середине 2006 г. существовало три подкомитета Е55: по управлению системой
РАТ, по внедрению и практическому применению системы РАТм. по терминологии,
связанной с РАТ. Задача группы РАТ представлена в комитетах Е55 как обеспечение
согласования разработанных стандартов с руководством по РАТ и их приемлемости
для FDA. На данный момент опубликован один активный стандарт, в то время как
предложено 16 дополнительных стандартов. ASTM International представляет еще
одно поле для международного сотрудничества (в соответствии с инициативой по
cGMP двадцать первого века). Определения РАТ (в руководстве FDA и ASTM Е55), а
также другие концепции включены в руководство ICH Q8.
Выпуск в режиме реального времени (RTR). Руководство по РАТ определяет RTR
как «возможность оценивать и обеспечивать приемлемое качество продукта в про-
цессе обработки и/или готового продукта на основании параметров процесса». В то
время как готовые продукты обычно выпускают на рынок только после проведения
408 Часть 4. Процессно-аналитические технологии
выборки для анализа, обследования (т. е. проверки качества в лаборатории) и осви-
детельствования, внедрение систем RTR позволяет проводить выпуск готовой про-
дукции одновременно с завершением технологических операций. С практической
точки зрения R TR представляет собой одну из самых существенных и осязаемых
выгод для производителей, внедривших систему РАТ, поскольку она может содей-
ствовать резкому снижению параметра С/Тпроцесса.
Руководство считает, что выпуск в режиме реального времени сопоставим с аль-
тернативными аналитическими процедурами, проводимыми при выпуске готовой
продукции, и определяется в руководстве как расширение выпуска по параметрам.
Отличительной чертой RTR является то, что учитывается одновременно степень из-
мерения и контроля характеристик материалов и параметров процесса при произ-
водстве. Подразумевается не просто установка системы быстрых измерений в конце
технологического процесса; подобное использование РА Тбыло бы равносильно про-
цедуре освидетельствования и никак не помогло бы улучшить управление качеством.
Тем не менее, руководство предполагает, что вполне реально внедрение RTR без
мониторинга качества готовой продукции при применении «комбинированных из-
мерений параметров процесса и других аналитических данных, собранных в ходе
производственного процесса». Аналогичные выводы сделаны в общих замечаниях
USP (USPgeneral notices), где предполагается, что данные, полученные в процессе
«валидационных исследований и при контроле по ходу процесса, могут обеспечить
большую уверенность в том, что производственная серия соответствует требованиям
отдельной монографии, чем аналитические данные, полученные при исследовании
готовых единиц продукции, выбранных из этой серии». Было бы не трудно создать
систему, более способную к обнаружению изменений качества, чем действующие
методы, основанные на освидетельствовании. Недавний статистический анализ
[42] показал, что для определения качества производственной серии традиционный
метод USP (905) (проверки однородности материала) может иметь статистическую
мощность, не многим большую, чем метод подбрасывания монеты, когда более
5% продукта превышает пределы спецификации (соответствует Срк внутри серии
на уровне приблизительно 0,65, что лишь немного хуже результата, наблюдаемого
в недавнем бенчмаркинговом исследовании в промышленном производстве [28]).
С другой стороны, размещение системы RTR без мониторинга готовой продук-
ции потребовало бы от производителя очень высокого уровня понимания процес-
са на основании, например, предоставления собственных построений пространств
проектных параметров и/или модели процесса, прошедшей тщательную валида-
цию. Несмотря на возможность внедрения RTR без мониторинга в конце процесса,
тщательно спроектированная система РАТ, как правило, будет включать в какой-
либо форме мониторинг качества готового продукта, который позволит уменьшить
латентный риск и создаст стратегически избыточный контроль процесса и явится
дополнительным инструментом, способствующим пониманию процесса.
4.1.5.3. Стратегия внедрения
Одна из задач FDA, связанная с руководством по РАТ, сформулирована следующим
образом: «приспособить обычную проверку соблюдения нормативов, проводимую
FDA, к нуждам инновационных технологий, основанных на системе РАТ, позво-
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...409
ляющих 1) укрепить научную основу установления нормативных спецификаций,
2) содействовать непрерывному совершенствованию и 3) совершенствовать произ-
водство при сохранении или повышении текущего уровня качества продукции».
Для достижения этой цели требуется исключительно тесное взаимодействие
между регулирующими органами и производителями, заинтересованными во вне-
дрении РАТ, поэтому была разработана стратегия внедрения интегрированных си-
стем. Задача стратегии внедрения — содействовать четкому, эффективному и со-
держательному обмену информацией между FDA и производителями, например,
в форме заседаний или неформальных встреч.
На практике стратегия ломает традиционные пути коммуникации между FDA
и производителями. Когда дело касается РАТ, ожидается, что управляющий пер-
сонал будет напрямую обмениваться информацией с учеными-фармацевтами
и инженерами-технологами, связанными с разработкой и эксплуатацией систем
РАТ, через департамент нормативно-правового регулирования. Компоненты стра-
тегии регулирования, предложенной FDA, включают:
• командный подход к РАТ для анализа СМС и работы с инспекциями по
cGMP;
• совместное обучение и сертификацию персонала, занятого анализом работы
РАТ, инспекциями и обеспечением соответствия;
• научную и техническую поддержку персонала, занятого анализом работы
РАТ, инспекциями и обеспечением соответствия;
• рекомендации, содержащиеся в руководстве по РАТ.
Командный подход к РАТ. Формирование команды по внедрению РАТ в FDA стало
одним из самых значимых стимулов для промышленности к внедрению производ-
ственных инноваций согласно инициативе по cGMP двадцать первого века и руко-
водству по РАТ. Цель создания команды РАТ — обеспечение рационального и точ-
ного применения РАТ в промышленности специалистами, владеющими самыми
современными методами РАТ. В какой-то момент команда РАТ состояла из 20 уче-
ных, включая исследователей, специалистов по внутреннему контролю, аналити-
ков, координаторов обучения и команды разработки стратегий. В последнее время
FDA предприняло шаги, направленные на роспуск команды РАТ, обязанности кото-
рой, в конце концов, перейдут к персоналу FDA, специально обученному для рабо-
ты с системами РАТ. Всесторонняя научная программа обучения была разработана
для команды РАТъ соответствии с указаниями ACPS подкомитета. В январе 2006 г.
приступили к обучению, при этом планируется продолжить курс с привлечением
преподавательского состава университетов Дьюкейн и Делавер [47].
Предоставление экспериментальных данных. FDA при разработке руководства
по РАТ признало, что даже при наличии руководства производители, заинтересо-
ванные в оценке приемлемости или потенциальной ценности новых технологий
процессного контроля, могут колебаться, полагая, что данные станут предметом
проверки инспекции по cGMP, и у них появятся дополнительные обязательства,
связанные с действиями органов регулирования. Чтобы развеять подобные опасе-
ния, FDA включило в документ положение, относящееся экспериментальному вне-
дрению новых технологий:
410
Часть 4. Процессно-аналитические технологии
Данные, собранные при помощи экспериментальных инструментов, следует считать
экспериментальными данными. Если исследование проводится на производственном
оборудовании, то на нем должна действовать собственная система качества... FDA не
намерено проверять экспериментальные данные по существующему продукту с це-
лью оценки пригодности экспериментального процессного анализатора или другого
инструмента РАТ. Рутинная инспекция FDA по проверке технологических процессов,
включающих инструменты РАТ для исследовательских задач, должна быть основана
только на действующих нормативных стандартах (например, результатах анализов по
утвержденным на настоящий момент или приемлемым методам контроля). Решение
FDA о проверке экспериментальных данных должно приниматься в исключительных
ситуациях, аналогичных описанным в Руководстве по стратегии соответствия (CPG),
ст. 130.300. Эти данные, обосновывающие валидацию или представляемые в органы
регулирования, подлежат обычной инспекционной проверке.
4.1.6. Процесс внедрения РА Т
Руководство по РАТ выделяет три возможных плана для компаний, заинтересован-
ных во внедрении РАТ:
• РАТ может быть внедрена в рамках собственной системы качества предприя-
тия; cGMP инспекции, проводимые командой РЛТили сертифицированным
инспектором по РАТ, могут предшествовать внедрению РАТ или следовать за
ним;
• внесенные изменения (СBE — changes being effected), СВЕза 30 дней (СВЕ 30)
или дополнение, внесенное до одобрения (PAS — prior approval supplement),
могут быть представлены в FDA до внедрения, и при необходимости инспек-
ция может быть проведена командой РАТ или сертифицированным инспек-
тором по РАТ перед внедрением;
• протокол совместимости (СР), содержащий исследования РАТ, стратегии
валидации и внедрения систем, а также временные границы, может быть
представлен в FDA. Вслед за одобрением этого протокола в FDA для внедре-
ния системы может быть выбран один из путей управления (или их комби-
нация).
Руководство по РАТ не носит предписывающего характера. Это отражается
в том, что все три плана внедрения, в сущности — части руководства, описывающие
«как сделать». Это налагает на производственных (и академических) ученых и тех-
нологов ответственность за принятие решения о выборе наилучшего пути разверты-
вания системы РАТ. Несмотря на то что отдельные передовые компании приступи-
ли к внедрению элементов РАТъ технологические процессы задолго до появления
инициативы FDA по cGMP двадцать первого века, в подходах к внедрению системы
сохраняются значительные различия. В то время как эта неопределенность (относи-
тельно лучшего пути внедрения) несколько замедлила введение РАТ, в перспективе
разнообразие предпочтительно, поскольку оптимальный путь внедрения системы,
вероятно, будет уникальным для большинства предприятий.
Производителям фармацевтической продукции представлен перечень из 10 кон-
трольных вопросов, на которые должны ответить компании, заинтересованные
в одобрении своих планов [10, 66]:
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...411
1. Является ли внедряемая система системой РА П
2. Включает ли она аспекты планирования, измерений и контроля производ-
ства?
3. Применяются ли в ней принципы и инструменты РАТ?
4. Какие именно инструменты применяются для контроля производства?
5. Как осуществляется непрерывное совершенствование и управление знаниями?
6. Какой из подходов к управлению рисками существует в компании — оценка,
предотвращение или управление?
7. Каким образом интегрированы системы РАТ!
8. Какой способ RTR предложен или применяется?
9. Какой способ нормативного регулирования предусмотрен?
а) Могут ли системы качества компаний управлять изменениями РАТ!
б) Являются ли предложения, представленные для рассмотрения, приемле-
мыми и оправданными?
10. Какие критические аспекты будут оцениваться в ходе посещений /инспекций
объекта?
Основываясь на модели DMAIC, а также на подходах, основанных на управлении
рисками и системах качества, поддержанных инициативой FDA по cGMP двадцать
первого века, специалисты Центра фармацевтических технологий университета
Дьюкейн {Duquesne University Center for Pharmaceutical Technology) предложили ше-
стифазный повторяющийся цикл улучшения процесса, базирующийся на системе
Л4Г(рис. 14).
Разумеется, существует множество приемлемых вариантов стратегии, и о неко-
торых из них сообщают на конференциях и в литературе по технологиям, в то же
время всякое успешное размещение РАТ — крупно- или маломасштабное, скорее
всего, связано с комбинированием этих элементов. Кроме того, каждая фаза про-
екта обязательно включает один или несколько образовательных модулей. Нако-
нец, несмотря на то что фазы проекта изображены как дискретные, некоторые фазы
в определенной степени перекрываются друг с другом. В частности, к определению
целей контроля, стратегий выпуска и планированию непрерывного совершенство-
вания следует приступать на самым ранних фазах цикла, одновременно с управле-
нием закупками.
4.1.6.1. Подготовка
Фаза подготовки является, вероятно, самым критическим этапом на пути к внедре-
нию системы РАТ. По своей сути проекты процессно-аналитических технологий
являются междисциплинарными и требуют согласований и поддержки от подраз-
делений корпорации, которые иногда имеют разные цели и применяют разные про-
цедуры. Важнее всего, чтобы заинтересованные во внедрении системы РАТ полу-
чали поддержку руководства на самом высоком уровне корпоративной структуры
для обеспечения доступности достаточного количества ресурсов и гарантии при-
верженности компании политике положительных изменений. В фазе подготовки
необходимо создать команду РАТ, обладающую обширными знаниями и практиче-
412
Часть 4. Процессно-аналитические технологии
ВЫПУСК
АНАЛИЗ
^Критерии выпуска
"^Информационная
инфраструктура
"^Параллельный мониторинг
"^Валидация
^Накопление данных
^Описание процесса
КОНТРОЛЬ
г "Ф’Модель процесса
^Стратегия контроля
"^Планирование расхождений
"Ф"Оценка
Рис. 14. Цикл внедрения РАТ с примерами соответствующих действий для каждой фазы
скими навыками, и приступить к формальному планированию проекта, включая
отбор продукта и процесса. В идеале диалог FDA с командой РАТ должен начаться
на начальном этапе подготовки.
4.1.6.2. Оценка
В руководстве по РАТ четко указано, что промышленные системы должны быть
основаны на оценке рисков. Вскоре после формирования команды РАТ и иденти-
фикации целей следует приступить к формальной оценке рисков. При оценке ри-
сков следует сосредоточиться на идентификации и классификации видов повреж-
дений, представляющих собой риск для качества продукта; результат оценки риска
позволит определить приоритеты размещения ресурсов РАТ и базисную линию для
анализа влияния РАТ на уменьшение рисков для качества.
Глава 4.1, Аргументы в пользу процессно-аналитической технологии...413
4.1.6.З. Анализ
Фаза «анализа» (от англ, analyze — «анализируй») проекта состоит из действий, ко-
торые обычно связаны с РАС, включая идентификацию и оценку потенциальных
сенсорных технологий, разработку методик, квалификацию и валидацию. Кроме
того, можно проводить планирование экспериментов {DOE) или анализ данных для
углубления понимания процесса или для достижения целей РАТ. Следует принять
во внимание проблемы, связанные с информационной инфраструктурой, методи-
ками пробоотбора и разработкой процедур контроля.
4.1.6.4. Контроль
Введение контроля начинается с момента внедрения каждой новой аналитической
методики или технологии. Контроль может быть весьма простым, таким как, на-
пример, автоматическое прерывание стандартной операции в критической точке.
При углубленном понимании процесса можно применять более сложные формы
контроля, такие как управление с обратной связью (например, контроль силы уда-
ра в процессе прессовки таблетки, контроль температуры или потока воздуха при
обработке в кипящем слое) или упреждающий контроль (например, регулирова-
ние параметров процесса на основании данных о качестве исходных материалов).
При разработке и внедрении методик контроля следует предусмотреть процедуры
действий при управлении в неблагоприятной ситуации и инициировать повторную
оценку рисков для определения пригодности методик контроля.
4.1.6.5. Философия выпуска продукции
Проекты РАТ, включающие введение RTR или модификацию существовавшего ра-
нее механизма для одобренного процесса, потребуются дополнительные разработки
методик и процедуры валидации. Решение о выпуске в режиме реального времени
обычно определяется моделью процесса, которая может представлять собой матема-
тическое уравнение или алгоритм в системе контроля. Кроме того, передача данных
как система связи должна точно передавать решение о выпуске и сопутствующие
данные для последующих операций (т. е. складского хранения, логистике) и пред-
шествующих операций (т. е. производственного планирования, отчетности) или
в производственный архив предприятия. Встроенная система связи и научные ком-
поненты требуют интегрированного системного подхода к разработке, валидации,
размещению и производственным операциям. Наконец, внедрение систем РАТпо-
зволяет пересмотреть критерии приемлемости выпускаемой продукции по качеству;
задача идентификации устойчивых критериев выпуска, приемлемых для больших вы-
борок, например, требует продолжения исследований характеристик продукта [43].
4.1.6.6. Оптимизация
Фаза оптимизации проекта предоставляет возможность оценить характеристики
системы РАТъ соответствии с целями проекта, а также уровнем латентных рисков
системы. В идеале, при наличии системы РАТ, уровень понимания процесса будет
углубляться, если для каждой производственной партии будет собрано большее ко-
личество данных. Дополнительные знания о производственных операциях могут
выявить новые возможности для улучшения качества продукции и повышения эф-
414 Часть 4. Процессно-аналитические технологии
фекгивности производства, а также для решения похожих проблем при производ-
стве другого продукта. Ключ к успеху на стадии оптимизации — осознание того, что
эта стадия является лишь началом процесса непрерывного совершенствования.
4.1.6.7. Перспективы внедрения РАТ
РАТ и инициатива по cGMP двадцать первого века определенно придали импульс
развитию фармацевтической промышленности и связанным с ней отраслям инду-
стрии. Значительные потоки капитальных вложений потекли в новых направлени-
ях навстречу благоприятным возможностям и задачам, поставленным переменами.
Некоторые специалисты, работающие в этой отрасли, сомневаются, будет ли влия-
ние нового подхода продолжительным; при этом они перечисляют ряд начинаний,
представленных как «новая эра в индустрии», которые, как оказалось, не принесли
ничего нового. Вместе с тем некоторые наблюдения позволяют без труда увидеть,
что на этот раз ситуация иная.
Всего лишь 10 или 15 лет назад лишь немногие могли предвидеть, что совре-
менная фармацевтическая промышленность окажется в столь трудном положении.
Скорость одобрения новых лекарств-«хитов продаж» продолжает падать, а создание
и разработка новых лекарств неуклонно дорожает. Несмотря на то что рынок лекар-
ственных продуктов сейчас обширен как никогда ранее, прибыли производителей
лекарств сокращаются; в то же время потребители считают, что прибыли фарма-
цевтических компаний неоправданно высоки, и их отношение к отрасли в целом
постоянно ухудшается. По результатам недавнего обзора, опубликованного Фондом
семьи Кайзер, в рейтинге общественного мнения фармацевтические компании на-
ходятся между нефтяными и табачными, а также органами здравоохранения (health
management organizations) [67]. Увеличивается давление влиятельных лиц в государ-
стве и частном секторе с целью получения все больших компенсаций со стороны
промышленности. Не иссякает поток публикаций из промышленных и финансовых
источников, в которых представлены хроники бед фармацевтической индустрии
[34,45,68, 69].
Преимущество фармацевтической промышленности состоит, вероятно, в том,
что она идет по следам (а не является лидером) других отраслей промышленно-
сти во внедрении систем автоматизированного контроля, процессной аналитики,
управления качеством и ресурсосберегающего производства. Положение фарма-
цевтических компаний относительно ориентиров производителей мирового класса
предоставляет им простор к усовершенствованиям. Если бы уровень фармацевти-
ческой промышленности в целом мог хотя бы приблизиться к уровню производства
мирового класса (через внедрение РАТ), то возвращенные производителям и акцио-
нерам прибыли были бы огромны (рис. 15).
Наконец, рентабельность инвестиций в РАТ не ограничена основными продук-
тами.
Оценки, основанные на недавних отчетах, позволяют предположить, что по-
средством преобразований процессов, основанных на внедрении систем РАТ и ре-
сурсосберегающего производства («лин»), обычные мелкие или средние производи-
тели фармацевтических продуктов могут увеличить операционную рентабельность
до 600 базисных пунктов [44].
Глава 4.1. Аргументы в пользу процессно-аналитической технологии...
415
Общая скорость оборачиваемости складских запасов,
оборотов/год
Рис. 15. Потенциальные финансовые прибыли от размещения систем РАТ и «лин» —
ресурсосберегающего производства. Кривые рассчитаны на основании суммарной COGS (cost
of goods sold — себестоимость реализованной продукции) и складских запасов по данным
ежегодных отчетов за 2005 г. 16 крупнейших производителей патентованных фармацевтических
препаратов и дженериков (в соответствии с рыночной капитализацией). Важно иметь в виду, что
экономия оборотного капитала является разовой выгодой, в то время как стоимость качества,
финансирование запасов и экономия накладных расходов обеспечивают постоянный прирост
прибыли на инвестиции. По представленным кривым можно переоценить экономию из-за
неточностей в исходных данных или ограничений возможности внедрения РАТ, тем не менее, они
не отражают других многочисленных потенциальных путей получения прибыли от РАТ, таких как
повышение мощности, увеличение производительности труда, снижение расходов на QC или
сокращение времени на доставку товара на рынок
Силы, выходящие за рамки управления отраслью, дают больше, чем когда-либо
ранее, оснований добиваться эффективности фармацевтического производства,
и FDA выполняет свои обязанности по разъяснению пути. В то время как фармацев-
тическая промышленность, вероятно, несправедливо объявлена виновником фи-
416
Часть 4. Процессно-аналитические технологии
нансового кризиса в здравоохранении США, у нее есть широкие возможности для
перемен на благо пациентов, а также инвесторов.
Благодарность
Автор благодарит рецензентов за существенный вклад, повлиявший на качество
этой публикации: Джеймса К. Дреннена III {James К. Drennenio, III), Ph. D., ди-
ректора Центра фармацевтической технологии Университета Дьюкейн, старшего
консультанта по стратегии процессного контроля технологических процессов; Роб-
би С. Лайона {Robbe С. Lyon), Ph. D., заместителя директора Отдела исследования
качества продуктов FDA/CDER-, Д. Кристофера Уоттса {D. Christopher Watts) Ph. D.,
руководителя рабочей группы Standards & Technology FDA/ CDER/ OPS и Тома Найта
{Tom Knight), основателя и главного исполнительного директора Invistics Corp.
Литература
1. U.S. Department of Health and Human Services (2004), Pharmaceutical CGMPs for the 21st century
—A risk-based approach, Final report, Food and Drug Administration, Rockville, MD.
2. U.S. Department of Health and Human Services (2004), Guidance for industry: PAT—A framework
for innovative pharmaceutical development, manufacturing, and quality assurance, Food and Drug
Administration, Rockville, MD.
3. Clevett, K. J. (1986), Process Analyzer Technology, Wiley, New York. 4. Ulman, D. L. (1986), CPAC:
An industry — university cooperative research center for process analytical chemistry, TrAC Trends
Anal. Chem., 5, 164.
5. Callis, J. B., Ulman, D. L., and Kowalski, B. R. (1987), Process analytical chemistry, Anal. Chem., 59,
624A.
6. Balboni, M. L. (2003), Process analytical technology: concepts and principles, Pharm. Technol.
7. Koch, M. V. (2006), Optimizing the impact of developments in micro-instrumentation on process
analytical technology: a consortium approach, Anal. Bioanal. Chem., 384, 1049.
8. Kiippers, S., and Haider, M. (2003), Process analytical chemistry — future trends in industry, Anal.
Bioanal. Chem., 376, 313.
9. Kiippers, S., and Haider, M. (2006), Process analytical chemistry, Anal. Bioanal. Chem., 384,1034.
10. Hinz, D. C. (2006), Process analytical technologies in the pharmaceutical industry: the FDA’s PAT
initiative, Anal. Bioanal. Chem., 384, 1036.
11. Beebe, K. R., et al., (1993), Process analytical chemistry, Anal. Chem., 65, 199R.
12. Blaser, W. W, et al., (1995), Process analytical chemistry, Anal. Chem., 67, 47R.
13. Workman, J. J., et al., (1999), Process analytical chemistry, Anal. Chem., 71, 121R.
14. Workman, J. J., et al., (2001), Process analytical chemistry, Anal. Chem., 73, 2705.
15. Workman, J. J., Koch, M. V, and Veltkamp, D. J. (2003), Process analytical chemistry,
Anal. Chem., 75, 2859.
16. Workman, J. J., Koch, M. V., and Veltkamp, D. J. (2005), Process analytical chemistry,
Anal. Chem., 77, 3789.
17. Gluckman, P., Roome, D. R., Deming, W. E., and Delavigne, K. (1993), Everyday Heroes of the
Quality Movement: From Taylor to Deming, the Journey to Higher Productivity, 2nd ed., Dorset
house, New York.
18. Shewhart, W. A. (1980), Economic Control of Quality of Manufactured Product, ASQC Quality Press,
Milwaukee, WI.
19. Shewhart, W. A. (1986), in Deming W. E. ed., Statistical Method from the Viewpoint of Quality
Control, Dover, New York.
Глава 4,1. Аргументы в пользу процессно-аналитической технологии...417
20. Scherkenbach, W. W. (1991), The Deining Route to Quality and Productivity: Road Maps and Road-
blocks, ASQC Quality Press, Milwaukee, WL
21. Juran, J. M. (2003), Architect of Quality: The Autobiography of Dr. Joseph M. Juran, McGraw-Hill,
New York.
22. Bhote, К R. (2002), What is Six Sigma? The Ultimate Six Sigma. Vol 1. New York : AMACOM ;
9-14.
23. Galvin, R.W. (2002), Forward. The Ultimate Six Sigma. Vol 1. New York : AMACOM ; xxi-xii.
24. Marash, S. A., Berman, P., and Flynn, M. (2004), Fusion Management: Harnessing the Power of Six
Sigma, Lean, ISO 9001:2000, Malcom Baldridge, TQM and Other Quality Breakthroughs of the Past
Century, QSU Pub., Fairfax, VA.
25. Ealey, L.A. (1988), Quality by design. Taguchi Methods and U.S. industry. Dearborn, MI: ASI
Press.
26. Taguchi, G., Chowdhury, S., and Wu, Y. (2005), Taguchi’s quality engineering handbook. Hoboken,
NJ : John Wiley & Sons.
27. e-Handbook of Statistical Methods. NIST/SEMATECH. Available at: http://www.itl.nist. gov/div898/
handbook/.
28. Macher, J., and Nickerson, J. (2006), Pharmaceutical manufacturing research project: Final bench-
marking report, Georgetown University, McDonough School of Business and Washington University
Olin School of Business, Washington DC.
29. Levinson, W. A. (2002), Henry Ford s Lean Vision, Productivity Press, New York.
30. Leiper, K. (2006), paper presented at the The Heidelberg PAT Conference, Heidelberg, Germany.
31. Lewis, N. A. (2006), A tracking tool for lean solid-dose manufacturing, Pharm. Technol. 30, 94-108.
32. Roumeliotis, G. (2006), Lean proves mean in drug manufacturing, available: in-PharmaTechnologist.
com.
33. Gerecke, G., and Knight, T, Improving performance and reducing cycle time using flow path manage-
ment: A case study, Pharm. Eng. 21.
34. Abboud, L., and Hensley, S. (2003), Factory shift: New prescription for drug makers: Update the
plants, Wall Street J., Sept. 3, p. 1.
35. Cooley, R. E., and Egan, J. C. (2004), The impact of process analytical technology (PAT) on pharma-
ceutical manufacturing, Am. Pharm. Rev., 7, 62-68.
36. Cogdill, R. P. (2006), in Brittain, H. G., Ed., Spectroscopy of Pharmaceutical Solids, Taylor & Francis
Group, New York, pp. 313-412.
37. Cogdill, R. P., et al., (2005), Process analytical technology case study, Part II: Development and vali-
dation of quantitative for tablet API content and hardness, AAPS Pharm. Sci. Tech., 6, Article 38.
38. Cogdill, R. P., et al., (2005), Process analytical technology case study, Part I: Feasibility studies for
quantitative NIR method development, AAPS Pharm. Sci. Tech., 6, Article 37.
39. Cogdill, R. P., Anderson, C. A., and Drennen, J. K. (2005), Process analytical technology case study,
Part III: Calibration monitoring and transfer, AAPSPharm. Sci. Tech., 6, Article 39.
40. Lorber, A. (1986), Error propagation and figures of merit for quantifi cation by solving matrix equa-
tions, Anal. Chem., 58, 1167.
41. Braga, J. W. B., and Poppi, R. J. (2004), Figures of merit for the determination of the poly-
morphic purity of carbamazepine by infrared spectroscopy and multivariate calibration ,
J. Pharm. Sci., 93, 2124.
42. Lunney, P., and Drennen, J. К. I. (2005), A prevention based strategy for quality control using PAT,
NIR News, 16, 7.
43. Sandell, D., Diener, M., Vukovinsky, K., Hofer, J., and Pazdan, J. (2006), Development of a content
uniformity test suitable for large sample sizes, Drug. Info. J., 40, 337.
44. Cogdill, R. P., Knight, T. P., Anderson, C. A., and Drennen, J. K. (2007), The fi nancial returns on
investments in process analytical technologies and lean manufacturing: Benchmarks and case study,
J. Pharm. Innov., 2(1—2), 38 — 50.
418 Часть 4, Процессно-аналитические технологии
45. du Pre Gauntt, J. (2005), Quality manufacturing: A blockbuster opportunity for pharmaceuticals,
Economist Intelligence Unit.
46. Steyer, R. (2005), Merck names dark CEO, available: TheStreet.com, accessed May 5, 2005.
47. Watts, D. C. (2006), PAT—An FDA paper presented at the The Heidelberg PAT Conference, Heidel-
berg, Germany.
48. Hussain, A. S. (2001), Emerging science issues in pharmaceutical manufacturing: Process analytical
technologies, paper presented at the Science Board Presentations to FDA, Rockville, MD.
49. Chisholm, R. S. (2001), TQMS, statistically based in-process control with real time quality assurance,
the AstraZeneca total quality management strategy, paper presented at the Science Board Presenta-
tions to FDA, Rockville, MD.
50. Raju, G. K. (2001), Pharmaceutical manufacturing: New technology opportunities, paper presented at
the Science Board Presentations to FDA, Rockville, MD.
51. Scherzer, R. H. (2002), Quality by design: A challange to the pharma industry, paper presented at the
Science Board Presentations to FDA, Rockville, MD.
52. U.S. Department of Health and Human Services (2004), Risk-based method for prioritizing cGMP
inspections of pharmaceutical manufacturing sites — A pilot risk ranking model, Food and Drug Ad-
ministration, Rockville, MD.
53. U.S. Department of Health and Human Services (2006), SMG 2020 — FDA quality system frame-
work for internal activities, Food and Drug Administration, Rockville, MD.
54. U.S. Department of Health and Human Services (2006), Guidance for industry: Quality systems ap-
proach to pharmaceutical cGMP regulations, Food and Drug Administration, Rockville, MD.
55. U.S. Department of Health and Human Services (2004), Guidance for industry: Sterile drug products
produced by aseptic processing — Current good manufacturing practice, Food and Drug Administra-
tion, Rockville, MD.
56. U.S. Department of Health and Human Services (2003), Guidance for industry: Comparability proto-
cols — Chemistry, manufacturing, and controls information, draft guidance, Food and Drug Admin-
istration, Rockville, MD.
57. U.S. Food and Drug Administration (FDA) (2004, Mar.), Sec. 490.100 Process validation require-
ments for drug products and active Pharmaceutical ingredients subject to pre-market approval (CPG
7132c.O8), FDA, Rockville, MD.
58. International Conference on Harmonization (ICH) (2004), Q8: Pharmaceutical development, Interna-
tional Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals
for Human Use, ICH, Geneva.
59. International Conference on Harmonization (ICH) (2004), M4: Organisation of the common techni-
cal document for the registration of pharmaceuticals for human use, International Conference on
Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH,
Geneva.
60. U.S. Department of Health and Human Services (2006), Guidance for industry: Providing regulatory
submissions in electronic format — Human pharmaceutical product applications and related submis-
sions using the eCTD specifi cations, Food and Drug Administration, Rockville, MD.
61. Yu, L. X. (2006), Implementation of quality-by-design: Question-based review, paper presented at the
Drug Information Association 42nd Annual Meeting, Philadelphia, PA.
62. International Conference on Harmonization (ICH) (2005), Q9: Quality risk management, Interna-
tional Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals
for Human Use, ICH, Geneva.
63. International Conference on Harmonization (ICH) (2005), Q10: Pharmaceutical quality systems, fi
nal concept paper, International Conference on Harmonization of Technical Requirements for Regis-
tration of Pharmaceuticals for Human Use, ICH, Geneva.
64. Hussain, A. S. (2006), Quality by design and bioequivalence/bioavailability assessment, paper pre-
sented at the The Heidelberg PAT Conference 2006, Heidelberg, Germany.
65. Lyon, R. C., and Hammond, S. (2006), Process monitoring of pilot-scale pharmaceutical blends by
Глава 4,1, Аргументы в пользу процессно-аналитической технологии...419
near-infrared chemical imaging and spectroscopy, paper presented at the Eastern Analytical Sympo-
sium, Somerset, NJ.
66. D’Sa, A. (2005), Process analytical technology (PAT): regulatory process, review and inspection,
paper presented at the 19th International Forum on Process Analytical Technology— IFPAC 2005,
Arlington, VA.
67. Views on prescription drugs and the pharmaceutical industry, The Kaiser Family Foundation, 2005.
68. Arlington, S., Barnett, S., Hughes, S., Palo, J., and Shu, E. (2002), Pharma 2010: The threshold of
innovation, IBM Business Consulting Services, Somers, NY.
69. Arlington, S., et al., (2005), The metamorphosis of manufacturing, IBM Business Consulting Ser-
vices, Somers, NY.
Глава 4.2. Процессно-аналитическая технология {РА 7)
Мишель Ульмшнайдер и Ив Рогго
F. Hoffinan-La Roche Ltd. (Базель, Швейцария)
4.2.1. Основные принципы и эффект от внедрения
4.2.1.1. Определение
Процессно-аналитическая технология (-R47) является одной из целей, стремление
к которой провозглашает Инициатива по cGMP в фармацевтическом производстве
двадцать первого века, опубликованная Федеральным управлением США по кон-
тролю за пищевой продукцией и лекарствами (FDA). В соответствии с руководством
FDA можно кратко определить РАТ как систему планирования, анализа и контроля
фармацевтического производства через измерение критических параметров каче-
ства и технических характеристик. Измерения, проводимые на исходных и обраба-
тываемых материалах, или измерения процессных параметров, способствуют повы-
шению качества готовой продукции.
Процессно-аналитическая технология облегчает технологические инновации,
в частности, внедрение в фармацевтическую промышленность новых аналитиче-
ских методик, предназначенных для углубления понимания и контроля производ-
ственных процессов. Эксперты, работающие в FDA и в промышленности, ожидают
от РАТ следующих преимуществ в сравнении с традиционными производственны-
ми практиками: повышения качества готовой продукции, повышения эффективно-
сти производства, снижения производственных расходов, повышения пригодности
процесса и снижения количества бракованной продукции. Соответственно ожида-
ются фундаментальные изменения в нормативно-правовой сфере. В будущем фар-
мацевтическое производство потребует инновационных подходов к производству
и наукоемких технологий. Внедрение РАТ приведет к углублению сотрудничества
между подразделениями, занимающимися научно-исследовательскими разработ-
ками (R&.D) и производственными отделами внутри компаний и повысит общую
эффективность производства. Органы, отвечающие за одобрение заявок и проведе-
ние инспекций, будут все больше сосредотачиваться на научных и технологических
принципах. В результате регулирующие органы будут возлагать на новые продукты
большие ожидания сразу после запуска продуктов в производство.
4.2.1.2. Что способствовало появлению РАТ?
Предварительное обсуждение принципов РАТ между FDA и отдельными фарма-
цевтическими компаниями, уже проявившими активность в данной области, отно-
сится к концу 1990-х гг. В сентябре 2004 г FDA выпустило документ для промыш-
ленности, озаглавленный «Руководство по РАТ для промышленности: Рамочная
программа инновационного развития фармацевтического производства, техноло-
гий и обеспечения качества». РА ^закрепилась в корпоративной культуре FDA.
Глава 4.2. Процессно-аналитическая технология (Я47)
421
Фармацевтические компании сталкиваются с растущей необходимостью по-
вышения производительности труда и снижения стоимости производства. Кроме
того, им приходится учитывать ужесточение стандартов качества лекарственных
средств и ожидания потребителей. В то же время серьезную проблему представляет
поиск новых активных веществ. Уменьшение отсева среди отобранных лекарств-
кандидатов позволит большему числу новых лекарств попасть на рынок. С точки
зрения рынка лекарственных продуктов, задача состоит в усовершенствовании со-
става лекарственных препаратов и предложении пациентам инновационных и бо-
лее эффективных решений, что приведет к коммерческому успеху или крупным
достижениям. Целью введения стратегии РАТ путем придания приоритета научно-
обоснованной разработки и внедрению новых или модернизированных технологий
при накоплении большего числа критических данных в течение жизненного цикла
лекарства является направление фармацевтической промышленности на путь, ве-
дущий к достижению этих основных целей.
Многие существующие методы исследований и производственных процессов
считаются устоявшимися, поскольку они применяются в течение многих лет. Счи-
тается, что они создают незначительную погрешность и вносят лишь небольшой
вклад в изменчивость процесса. Благодаря длительности их использования эти ме-
тоды продолжают широко применяться при разработках лекарств. Усовершенство-
вание существующих технологий всегда возможно и происходит постоянно. Однако
это затрудняет рассмотрение или выбор потенциальных альтернативных технологий
при отсутствии критического анализа или волевого решения управления о замене
устоявшихся методик. FDA отмечала, что почти во всех недавних разработках ле-
карств отсутствовала возможность расширения или улучшения процесса с помощью
более новых или альтернативных технологий. Точнее говоря, FDA хотела подтол-
кнуть производителей лекарств к более активному внедрению инноваций и улучше-
нию управления рисками при выпуске новых лекарственных продуктов на рынок.
4.2.1.З. Анализ корневых причин и процессный контроль
Когда в современном производстве возникают проблемы, связанные с качеством
продукции, все труднее становится выявлять их корневые причины. На пути к глу-
бокому пониманию процесса и характеристик продукта зачастую встречаются барье-
ры, связанные со знанием (knowledge barriers), возникающие из-за проблем с увели-
чением количества документации, нехватки времени или профессиональных кадров.
Задача РАТ состоит в усилении контроля и понимания процесса, чтобы процедуры
можно было осуществлять различными путями и с большей эффективностью. Ини-
циатива РАТ содействует и облегчает введение инновационных подходов. Она де-
лает возможным переход от валидации к непрерывной верификации. Следующий
этап — эффективный выпуск продукта в режиме реального времени при непрерыв-
ной переработке в качестве альтернативы традиционному серийному производству.
4.2.1.4. Когда следует внедрять РА Т
О встраивании качества в фармацевтический продукт следует помнить с самого на-
чала жизненного цикла продукта. Важным предварительным условием является
равная вовлеченность в процесс подразделений R&D и технологов, а также посте-
422
Часть 4. Процессно-аналитические технологии
янная связь между ними. Одна из задач РАТ— обеспечить мотивацию для встраива-
ния качества в продукт с самого начала его жизненного цикла. Поэтому так важно
применить принципы РАТ на этапе научно-исследовательской работы. Если тре-
бования к качеству продукта хорошо поняты и принимаются во внимание с самого
начала, то анализ корневых причин сбоев в качестве или технологическом процессе
после перехода к производству в промышленных масштабах должен быть намного
проше. Поэтому РАТ должна играть еще более важную роль в планировании и ана-
лизе производственных процессов, что позволит проводить контроль параметров
процесса, основываясь на периодических измерениях хорошо описанных критиче-
ских характеристик процесса.
Следует также принимать во внимание проблемы, связанные с обработкой дан-
ных, в контексте общей стратегии процессного анализа для обеспечения соответ-
ствия возрастающим требованиям к скорости и объему собранных данных. Анализ
в режиме реального времени вместе с управлением знаниями требует сбора и хране-
ния всей информации о производственных сериях, например, путем создания объ-
единенных баз данных. Таким образом, стратегия управления данными в системе
РАТ, основанная на процессном анализе в режиме on-line или глубинном анализе
данных, может быть запущена задолго до накопления больших массивов результа-
тов измерений. Анализ исторических данных связан с разработкой метода, вали-
дацией и постоянным мониторингом процесса, а также с рутинными результатами
данного производственного процесса.
4.2.1.5. РА Тспособствует углублению понимания процесса
Процессно-аналитическая технология может значительно углубить понимание
процесса. Фактически, внедрение РАТ может оказаться ключевым стимулом к луч-
шему пониманию процесса. При внедрении РАТ ожидается осуществление следу-
ющих этапов: сбор данных по критическим параметрам в режимах on-line, in-line
и at-line (рис. 1), извлечение информации и анализ индикаторов состояния процес-
са, и замыкает петлю контроля динамическое управление процессом. Инновации
в процессе разработки, применение перспективных технологий и моделирование
In-line On-line At-line
Технологический маршрут
Рис. 1. Измерения параметров процесса в режимах in-line, online и at-line
Глава 4.2. Процессно-аналитическая технология (РАТ)423
процессов там, где это возможно, являются частью более фундаментального иссле-
дования научной подоплеки процесса. Важно понимать, что РАТ— не просто вве-
дение в процесс дополнительных аналитических методик, но и разработка методов
прогнозирования хода процесса на основании заданного набора критических пара-
метров. Это равносильно способности прогнозировать качество готового продукта.
Например, при внедрении процесса важно исследовать все источники вариации
компонентов, а также их влияние на готовый продукт с целью выбора параметров
качества (т. е. характеристик продукта), которые необходимо измерять для достиже-
ния оптимального и реалистичного контроля процесса.
Научные, производственные и контрольные технологии могут обеспечить очень
высокий уровень понимания процесса и возможности контроля. Хорошее понима-
ние процесса достигнуто, если идентифицированы и объяснены все критические
источники вариабельности. Для управления вариабельностью процесс должен быть
достаточно робастным. Ожидается также, что критические показатели качества мож-
но прогнозировать с достаточной точностью и достоверностью в соответствующем
пространстве параметров ведения процесса, когда возникают неожиданные вариа-
ции других переменных (например, при смене поставщика исходных материалов).
4.2.1.6. Изменение действующей практики при помощи РАТ
Подход, объединяющий R&D и производство, позволит углубить понимание про-
цесса и сделать возможным управление приемлемыми рисками. Путем построения
переносимых моделей процесса достигается возможность разработки и внедрения
адекватных технологий измерений, скорее способствующих протеканию процес-
са, чем наоборот. Более эффективные и экономичные переносы технологий со-
действуют пониманию процесса, непрерывной верификации процесса и соответ-
ствию нормативам, что позволяет улучшить качество готового продукта. Лучшее
понимание процесса позволяет проводить непрерывную верификацию процесса
вместо валидации по трем производственным сериям. Выбор методики измерения
и интеграция происходит на ранних этапах. Доступ к накопленным знаниям легко
осуществим при применении методик глубинного анализа данных для управления
и контроля технологических процессов. Ряд замкнутых динамических петель кон-
троля/соответствия нормативам на этапах технологического процесса, определяе-
мых как критические, повышает уверенность в качестве готовой продукции. Кроме
того, знания, накопленные за определенный период времени, обеспечивают базис
для немедленного и быстрого вмешательства при обнаружении отклонений или
сбоев в ходе процесса.
Типичной иллюстрацией подхода РАТ к повышению качества является приме-
нение спектроскопии в ближней инфракрасной области (БИК-спектроскопии) для
оценки качества вспомогательных веществ и активных компонентов перед их пере-
работкой, например, перед отпуском в производство. Как будет описано в следую-
щем разделе главы, БИК-спекгры являются информативными источниками данных
о структуре продукта и общей оценки качества. Поскольку диапазоны характери-
стик качества вспомогательных веществ были когда-то исследованы и включены
в базы спектров для калибровки, БИК-анализ может обеспечить одновременное
подтверждение значений основных физических и химических параметров неразру-
424
Часть 4. Процессно-аналитические технологии
тающем способом. Это эффективный метод уменьшения неопределенностей, свя-
занных с вероятными причинами сбоев в технологии или плохого качества готовой
продукции. Каждый раз, когда определенное вспомогательное вещество не соот-
ветствует требованиям качества в момент его использования, следует предпринять
немедленные корректирующие действия. Контроль возможен до того, как риск на-
рушения начнет возрастать. Такой подход дополняет идентификацию материалов
по маркировке контейнера при передаче на складское хранение.
4.2.1.7. Продвижение физической фармакологии и фармацевтики
Процессно-аналитическая технология предполагает подход к фармацевтическим
процессам, основанный на научном знании. В действительности, система РЛТпод-
черкивает наблюдаемую слабость формальных знаний о физических явлениях, ле-
жащих в основе фармацевтических процессов. Физика процесса менее ясна, чем
химия. Традиционная физика становится областью, интересующей инженеров
и технологов. Формальные принципы не сформулированы. Вследствие этого боль-
шой объем ценных знаний о физических явлениях распылен между различными
научными дисциплинами. Экспертиза физики процесса зачастую является чисто
технологической, а не формализованной процедурой, интегрированной в специ-
альную дисциплину.
Когда-то границы физики и химии были размыты, и появилась физическая хи-
мия; точно также в наше время появилась возможность объединения научных зна-
ний, являющихся частью различных дисциплин. Задача заключается в углублении
понимания процесса путем глубокого исследования физических явлений, лежащих
в основе фармацевтических процессов. Эта цель побуждает к развитию физической
фармацевтики, позволяющей углубить понимание процесса путем изучения основ
теоретической физики.
Важнейшими вопросами, требующими изучения, являются научные знания
и технологии твердых частиц и порошкообразных веществ: характеристики, анализ
форм и размеров частиц, понимание процессов и т. д. Среди других аспектов — во-
просы, связанные с образованием частиц и выделением твердых частиц из жидко-
сти, стабильностью смеси, а также пониманием и моделированием динамики по-
рошкообразных смесей. Например, степень компактирования порошкообразных
веществ и смесей может быстро изменяться в зависимости от времени и условий
хранения. Не всегда удается контролировать сроки их использования, вследствие
чего могут произойти неконтролируемые изменения. Перемешивание двух свобод-
нотекучих порошков, содержащих частицы разного размера, может привести не
к улучшению качества смеси, а наоборот, к ее разделению. Параметры текучести
порошков зависят не только от свойств составляющих материалов, таких как рас-
пределение частиц по размерам, форма частиц и свойства их поверхностей, но и от
внешних условий, например, от влажности и степени компактирования. Другие
сферы интереса включают описание поведения капель жидкости, эмульсий и кол-
лоидных систем, пузырьков и полимеров, а также характеристики поверхностей,
анализ состояния поверхности, явления на поверхности раздела и электростатиче-
ские взаимодействия, химическая активность поверхности, физические характери-
стики мокрых химических процессов и растворимость.
Глава 4.2. Процессно-аналитическая технология (РАТ)425
4.2.1.8. Глубинный анализ данных
За время применения комплексных процессов накапливаются большие массивы
данных. По мере увеличения объема накапливаемой и сохраненной информации
быстро растет разрыв между легкодоступными знаниями и скрытой информацией,
если не принимаются специальные меры. Глубинный анализ данных позволяет из-
влекать новые знания из накопленных наблюдений, обеспечивая базис для приня-
тия решений и соответствующих действий. Как лучше использовать интерпретацию
скрытых сведений? Как осуществлять оперативную обратную связь, извлекая су-
ществующие и, тем не менее, скрытые эмпирические знания? Подобные вопросы
возникают при осуществлении глубинного анализа данных.
Междисциплинарная методика глубинного анализа данных возникла на грани-
це статистики, математики и вычислительной науки. Она представляет собой со-
вокупность методов обнаружения регулярностей и паттернов, а также извлечения
сведений из массивных баз данных при помощи традиционных и передовых инстру-
ментов анализа. Другой подход к глубинному анализу данных заключается в много-
мерном моделировании реального окружения на базе многомерных накопленных
исторических данных. Таким образом, глубинный анализ данных сродни экспло-
ративному анализу. К нему побуждают сами данные. Тем не менее, он отличается
от традиционного статистического анализа огромными объемами обрабатываемых
данных, намного превышающими мегабайтовый уровень. Свыше этого критиче-
ского уровня объема баз данных для большинства традиционных статистических
пакетов предел оперативных возможностей оказывается превышенным. Глубинный
анализ данных может быть проведен без помощи профессиональных статистиков.
Его проводят по полуавтоматическим процедурам, что делает его более привлека-
тельным и применимым в промышленной среде.
Подобные ситуации характерны для фармацевтических процессов, для которых
накоплено большое количество исторических данных без учета релевантности ин-
формации. Накопление информации является систематическим и избыточным.
Однако перекрестные связи между источниками данных или типами не всегда могут
быть установлены, что приводит к неоправданной и не выявленной статистической
неопределенности системы. Надежность накопленных данных не устанавливается
четко в течение некоторого времени, и вариации могут остаться не выявленными.
4.2.1.9. Хранение данных
В 1990-х гг. происходило развитие технологий информационных хранилищ. Иде-
альным хранилищем данных является собрание архивных данных, упорядоченных
во времени, организованных тематически, интегрированных в уникальную базу
данных и сохраняемых путем, облегчающим принятие решений (рис. 2).
Для управления хранилищами данных необходимо осуществление трех дей-
ствий. Во-первых, к собранным данным должен быть альтернативный путь доступа,
например, через ранее существовавшие базы данных или файлы. Во-вторых, управ-
ление хранилищем информации требует применения специальных инструментов
управления и контроля. Только после этого может быть применено третье действие,
а именно анализ данных с целью принятия решения и получения новых знаний.
Специальные инструменты управления информацией помогают переносить внеш-
426
Часть 4. Процессно-аналитические технологии
Рис. 2. Схематическая структура хранилища данных
ние, оперативные и исторические данные в хранилище документов. Компоненты
управления принятием решений используют для извлечения и визуализации дан-
ных из хранилища информации. При обработке данных в режиме on-line {OLAP —
online data processing) происходит анализ в режиме реального времени и визуализа-
ция исторических данных. Глубинный анализ данных включает извлечение правил
и моделей на основании собранных данных.
Аналитическая обработка в режиме on-line представляет, главным образом, ин-
терактивное исследование многомерных массивов данных, или кубов данных, при
помощи операций матричной алгебры, таких как slice-and-dice (анализ «вдоль и по-
перек»), roll-up (свертывание) и drill-down (детализация). Эффективность компью-
терной обработки зависит от объема хранилища данных и от их качества, например,
наличия пропущенных, неточных и избыточных данных. Для извлечения суще-
ственной информации и отбора результатов для сохранения и визуализации крити
чески важна многомерность обработки.
Инструменты глубинного анализа, встроенные в большинство коммерческих
программных пакетов, представляют собой набор методик и алгоритмов исследо-
вания больших баз данных для поиска семантических линий, имеющих отношение
к объяснению события и приобретению новых знаний. Более общей целью глу-
бинного анализа данных является генерирование правил и моделей, существенных
для понимания связей и облегчающих принятие решений. Существует множество
сфер применения управляемых хранилищ информации: анализ рисков, производ-
ственные тренды, управление исходными материалами, эксплуатация, валидация
процессов, разработки, контроль качества и так далее. Идея глубинного анализа
состоит во введении закономерностей (или предположении об их наличии), свя-
занных с коэффициентами подобия (likehood coefficients), установленными по боль-
шим массивам существующих (т. е. исторических) данных. Применяемые методы
заимствуются из разработок искусственного интеллекта и анализа статистических
и цифровых данных. К ним относятся, например, функциональное моделирование,
Глава 4.2. Процессно-аналитическая технология (РАТ)427
обучающие машины, нейронные и байесовские сети, метод опорных векторов, мо-
делирование ассоциаций и пояснительные правила, классификации и сегментации.
Сложность обработки определяется резким переходом с уровня базы данных на
уровень хранилища данных (от мега-базы к пета-базе, т. е. 106—1015).
4.2.1.10. Методы глубинного анализа данных применительно
к фармацевтическим процессам
Хранилище данных представляет собой центральный репозиторий данных, нако-
пленных за некоторое время из различных источников: контроль качества, обе-
спечение качества, производство, разработки и т. п. Накопленные данные — это
потенциальная золотая жила, предоставляющая пользователю конкурентные преи-
мущества за счет углубленного понимания фармацевтического процесса и оптими-
зации его при помощи скрытых эмпирических знаний.
Глубинный анализ применяется для извлечения ранее не использовавшихся дан-
ных и знаний. Его потенциал в приобретении знаний и генерировании поясняющих
правил может компенсировать потерю данных или недостаточное использование
накопленных данных. Существует два способа действия. Первый способ — прямой
или направленный, например, проверка гипотезы. Предполагается наличие кон-
кретных групп или характеристик и проводится верификация или подтверждение
их идентичности. Второй способ — реагирующий или непрямой путь, состоящий
из простого исследования данных. Группы неизвестны, характеристики не выявле-
ны или скрыты, паттерны не идентифицированы. Представленные подходы опи-
сывают альтернативные термины — управляемое и неуправляемое обучение, соот-
ветственно. Анализы сверху вниз и снизу вверх дополняют друг друга. Например,
инструменты подтверждения при управляемом обучении можно использовать для
верификации и сертификации качества открытий, сделанных в процессе исследо-
вания.
Чего можно достичь при помощи инструментов глубинного анализа данных?
Здесь представлен короткий список достижимых целей:
• характеризация данных с целью извлечения или определения дескрипторов
или инидикаторов, например, путем обобщения, суммирования или группи-
ровки;
• установление ассоциативных или поясняющих правил;
• классификация (управляемое обучение) единиц или объектов по классам
в соответствии с определенной вероятностью;
• выделение кластеров единиц данных (не управляемое обучение) и отнесение
их к классам после индуктивного определения границ классов в существую-
щих наборах данных;
• обнаружение аналогий во временных сериях;
• распознавание паттернов.
Данные из внешних и внутренних источников объединены, собраны в совокуп-
ности или связаны в хронологические серии. Единицы данных могут содержать
ошибки, данные могут отсутствовать, быть нечеткими (unsharp), избыточными или
противоречивыми. Операторы и переменные являются компонентами языка, ко-
428
Часть 4. Процессно-аналитические технологии
торый необходим для построения моделей. Кроме того должны быть определены
уровни валидации при помощи соответствующих критериев оптимизации и вали-
дации. Далее необходим метод поиска с целью извлечения данных из хранилища
и подготовки их для анализа.
Таким образом, глубинный анализ данных является операцией, проводимой
в три этапа. Перед каким бы то ни было анализом собранные данные подвергают-
ся предварительной обработке для интеграции в хранилище; для сохранения уров-
ня данных проводится определенная верификация: например, интегрирование,
объединение в кластеры или группы данных из различных внутренних и внешних
источников. Затем данные отбирают и проводят глубинный анализ с применением
соответствующих алгоритмов или моделей. Результаты визуализируют или интер-
претируют эксперты в данной области.
4.2.1.11. Практика глубинного анализа данных
Глубинный анализ данных является частью процесса, называемого цепочкой орга-
низации бизнес-процессов (рис. 3).
Цепочка организации бизнес-процессов
Рис. 3. Место глубинного анализа в цепи принятия решений
Глубинный анализ — гибкое решение периодически возникающей задачи: как
извлечь знания из массивов данных? Источником для глубинного анализа являются
большой, хотя и скрытый набор данных. Анализ соответствующих данных представ-
ляет собой интеллектуальный метод, работающий только тогда, когда он интегри-
рован в текущий операционный процесс. Проверка гипотез, приобретение знаний
и генерирование поясняющих правил управляется при активном сотрудничестве
между различными участниками процесса. Глубинный анализ данных — командная
работа, требующая экспертных знаний в различных областях, таких как информа-
ционные технологии (IT), управление хранилищами данных и анализ данных. Тем
не менее, методики доступны в составе коммерческих пакетов и, возможно, не тре-
буют экспертизы со стороны статистиков, работающих традиционными методами.
Распознавание паттернов или идентификацию правил или характеристик осущест-
вляет компьютер. В итоге глубинный анализ данных представляет собой логическую
петлю, включающую следующие шаги:
• понимание бизнеса;
• точное планирование проекта по глубинному анализу данных, например:
— постановка реальных задач;
— определение области обрабатываемой информации;
— инвентаризация доступных или используемых данных;
Глава 4,2. Процессно-аналитическая технология (Я47)
429
• подготовка данных:
— извлечение из внешних или внутренних источников;
— верификация и коррекция;
— предварительная обработка;
• построение хранилища информации;
• моделирование, например:
— описание и визуализация;
— группировка по сходству;
— правила ассоциаций, поясняющие правила;
— кластерный анализ;
— классификация;
— оценка;
— прогноз;
• оценка и сравнение моделей;
• документация и представление результатов;
• использование для принятия решения;
• петля замыкается — возвращение к пониманию бизнеса.
4.2.1.12. Комментарии к глубинному анализу данных
Глубинный анализ данных позволяет провести разъясняющий анализ по подтверж-
дающему анализу {confirmatory analysis). Он предназначен для извлечения макси-
мальной информации из доступных источников, таких как накопленные данные
любого типа. Но для максимальной эффективности анализа необходим крити-
ческий взгляд специалиста-эксперта на знания, реально скрытые в базах данных
и хранилищах информации. Цель исследования данных состоит в получении до-
ступа к скрытой информации для достижения знаний, позволяющих давать объяс-
нения, делать прогнозы и производить оценки. Поэтому глубинный анализ требует
объединенных усилий по предоставлению данных специалистами, пользователями,
информационными технологами и специалистами в соответствующей области зна-
ний (в нашем случае — в фармацевтическом производстве). Кроме того, необходи-
ма поддержка высшего руководства организации. Глубинный анализ данных явля-
ется компонентом надлежащей практики согласно установленным правилам, и в то
же время представляет собой вызов для создателей инновационных математических
методик. Не все паттерны или правила, выявленные в ходе глубинного анализа, ин-
тересны, но результаты должны оставаться логичными и применимыми на прак-
тике специалистами в соответствующей области знания. Поскольку применяемые
алгоритмы являются довольно сложными, а объемы данных огромны, важнейшими
являются вопросы, связанные с программным обеспечением и уровнем информа-
ционных технологий.
Глубинный анализ данных управляется накопленными данными, однако он
всегда направлен на решение проблем, связанных с процессом, бизнесом и научно-
исследовательской работой. Результаты представляют так, чтобы облегчить поста-
новку диагноза или принятие решения. Вероятно, результаты полезны исключи-
430
Часть 4. Процессно-аналитические технологии
тельно в контексте: т. е. они представляют собой не только цифры и графики, но
и помощь экспертам в соответствующей области знания. Кроме того, ни одна из
существующих методик глубинного анализа данных не является универсальной.
Следует принимать во внимание ряд различных методик или алгоритмов, посколь-
ку ни одна методика не может работать одинаково хорошо или превосходить другие
методики при решении всех проблем. Также ценность результатов анализа не может
превосходить ценности данных, на которых основан анализ.
4.2.1.13. Методы РАТ
Практически все существующие аналитические методы могут соответствовать зада-
чам РАТ. Уже существует множество способов применения аналитических методов
в режиме on-line. Применение новых методик, таких как визуализация результатов
БИК-спектроскопии или времяпролетная масс-спектрометрия с лазерной ионизаци-
ей/десорбцией из матрицы (MALDI- ТОР) влечет за собой определенные технологи-
ческие проблемы, связанные с выполненными on-line или in-line аналитическими ме-
тодиками. Внедрение аналитического оборудования в процесс или параллельное его
использование не всегда способствует лучшему пониманию процесса. Неинформа-
тивные параметры не следует измерять вовсе; они не должны усложнять ход процесса.
Применение различных методик (табл. 1) зависит от требований пользователя
к технологическому процессу. Технологический прогресс или появление новых ана-
литических методик ставит вопрос о надежности определенной методики или обо-
снованности ее применения. Инновации постоянно требуют оптимизации общих
показателей производственного процесса.
4.2.1.14. Заключение
Процессно-аналитическую технологию можно рассматривать как совокупность,
в которой большее или меньшее внимание уделяется определенным функциям,
в зависимости от текущей проблемы или ситуации (рис. 4).
Не существует описанных правил и прямых путей достижения успеха посред-
ством РАТ. Необходимы опыт и экспертные знания вместе с прекрасным знанием
фармацевтической науки и технологии. Когда фармацевтическая компания при-
нимает решение о внедрении РАТ, важна постоянная поддержка со стороны руко-
водства деятельности, связанной с РАТ. Стратегически необходимым шагом к вне-
дрению РАТ является поощрение, стимулирование и побуждение к сотрудничеству
в научной области, а также поддержка соответствующих программ образования
и специального обучения. Существует необходимость в улучшении понимания хи-
мических и фармацевтических процессов, а также разработки передовых измери-
тельных приборов и методов анализа данных.
Итак, перечислим преимущества РАТ:
• немедленное действие при нарушении качества;
• улучшение контроля и понимания процесса;
• уменьшение числа неконтролируемых отклонений и потерь продукции;
• улучшение качества продуктов и его стабильности;
• сбор данных и углубленный анализ архивных данных.
Таблица 1
Аналитические методы, применяемые в РАТ
Метод Описание Применение в режиме on-line Химическая идентификация Примеры применения в фармацевтическом производстве
ИК-, БИК- и рамановская спектроскопия Колебательная спектроскопия (обсуждается в данной главе) X X Мониторинг реакции Полиморфизм Определение состава Мониторинг процесса (сушка, гранулирование, измельчение)
Визуализация ги- перспектральных данных Колебательная спектроскопия в сочетании с пространственным анализом (см. главу о химической визуализации) X Распределения химических соединений Обнаружение контрафактов
Спектроскопия УФ и видимого диапазона Фотоэлектронная спектроскопия X X Измерение окрашенности Исследование растворения Валидация очистки (обнаружение на уровне ррт)
Терагерцовая спектроскопия ИК-спектроскопия дальней области, 3D-визуализация X Полиморфизм Целостность и толщина покрытия Возможно, распределение API
Лазерно-искровая Генерирование плазмы лазерным импульсом эмиссионная и детектирование испускаемого света спектрометрия (разрушение пробы) X Разработка лекарств Поиск сбоев в процессе
Лазерная дифракция Взаимодействие лазерного луча с частицами и детектирование рассеянного света X Определение размера частиц
Оценка тепловой инерции Комбинация методов, основанных на измерении теплопроводности, плотности и теплоемкости X Мониторинг смешивания, измельчения. Гранулирования
Акустические методы Активные или пассивные X Твердые, полутвердые образцы; образцы с высокой вязкостью Мониторинг гранулирования с большим усилием сдвига Мониторинг кристаллизации
Глава 4.2. Процессно-аналитическая технология {РАТ}
432
Часть 4. Процессно-аналитические технологии
Фармацевтическая
физика
Хемометрика
Сенсорные
технологии
РАТ
Планирова.
экспериментов
Аналигичегхие
метод™
Фармац, ,ти ж»..'
науки
Анализ рисков
Фармаиевтич'эские
технологии
Глубинный анализ
Рис. 4. Система РАТ
Применение процессно-аналитической технологии постоянно повышает каче-
ство продукции, способствует расширению информационной базы для новых про-
ектов и сокращает время продвижения продуктов на рынок.
4.2.2. Колебательная спектроскопия
4.2.2.1. Введение
Современная спектроскопия в инфракрасной области (ИК, IR — infrared) представ-
ляет собой универсальный инструмент, применяемый для качественного и количе-
ственного анализа молекулярных частиц всех типов. Спектроскопические методики
можно разделить на три категории на основании анализируемых диапазонов спек-
тра. ИК-спектроскопия в средней области спектра (MIR — Mid-IR) применяется
чаще всего; как для качественного, так и для количественного анализа исследуют
спектры поглощения, отражения и испускания. Ближний диапазон спектра — БИК
(в английском варианте NIR — Near-IR) используют для рутинного количествен-
ного анализа сложных проб; метод представляет интерес для сельского хозяйства,
производства продуктов питания и кормов для животных, и в последнее время для
фармацевтической промышленности. Как правило, в основе аналитических мето-
дов лежат измерения параметров диффузного отражения в необработанных твер-
дых или жидких образцах, или, в отдельных случаях, исследования пропускания.
Дальняя ИК-спектроскопия (FIR — far IR) применяется, в основном, для анализа
поглощения в неорганических и органометаллических образцах.
Глава 4.2. Процессно-аналитическая технология (РАТ)433
В электромагнитном спектре (рис. 5) ИК-диапазон составляет от 12 800 до 10 см-1
или от 0,78 до 1000 мкм. Обычно ИК-область разделяют на ближний (БИК), средний
(MIR) и дальний (FIR) диапазоны, для которых установлены следующие границы:
ближний 0,78—2,5 мкм 4000—12 800 см-1;
средний 2,5—50 мкм 200—4000 см-1;
дальний 50—500 мкм 20—200 см-1.
Методики и их применение различаются в зависимости от исследуемого ИК-
диапазона. Академические ученые и химики-аналитики обычно рассматривают ди-
апазон средний диапазон (MIR) как представляющий по умолчанию наибольший
интерес. Современные приборы для спектроскопии в средней ИК-области спектра
полностью отличаются от традиционных спектрофотометров с дифракционной
решеткой. Широкое распространение Фурье-спектрофотометров (FT — Fourier
transform) в начале 1980-х гг. и снижение стоимости приборов позволило расширить
область и разнообразие применения среднего диапазона ИК, в частности, благо-
даря применению интерферометров для улучшения параметров соотношения сиг-
нал/шум и пределов обнаружения. Первоначально применение ИК-спектроскопии
было ограничено качественным органическим анализом. Практически с самого
начала спектроскопия поглощения в средней ИК-области применялась для иссле-
дования структуры образцов. Химиков-органиков обучали визуальной и прямой
интерпретации спектров в среднем ИК-диапазоне. В наше время спектроскопия
в средней ИК-области спектра (MIRS — mid -IR spectroscopy) считается полезным
инструментом количественного анализа сложных образцов методами абсорбцион-
ной и эмиссионной спектрометрии, которые могут потребовать предварительной
калибровки и обработки результатов.
Анализ в ближнем ИК-диапазоне можно проводить аналогично анализу с по-
мощью спектрофотометров, предназначенных для работы в ультрафиолетовом (УФ)
или видимом диапазоне. Исторически наиболее важной областью применения ме-
тода стал количественный анализ при производстве продуктов питания и кормов
для животных. Лишь недавно спектроскопией в БИК-диапазоне заинтересовались
химическая и фармацевтическая промышленность. Главная причина такой задерж-
ки заключается в типе получаемой информации. Все наблюдаемые линии соответ-
ствуют обертонам или комбинации обертонов, возникающих в основном среднем
ИК-диапазоне спектра. Поскольку этот аналитический метод является неразруша-
ющим, перед проведением измерений практически не требуется специальной обра-
0,1 0,4 0 в 2,5 50 500 3000 мкм J
УФ Види- мый ИК Микро- волновой
Ближний Средний Дальний
105 25 000 12 500 4000 200 20 3,3 см-1
Рис. 5. Границы и обозначение спектроскопических диапазонов
434 Часть 4, Процессно-аналитические технологии
ботки образцов. Спектры БИК содержат информацию о физических и химических
характеристиках образца. Возможность прямой интерпретации данных ограничена
или вовсе невозможна; это означает, что для извлечения релевантной информации
требуется многомерный анализ данных. Это привело к тому, что большинство хи-
миков игнорировало возможности БИК-спектроскопии. Вплоть до начала 1990-х гг.
преобладали БИК-спектрофотометры дисперсионного типа с дифракционными
решетками. Последующий технологический прогресс привел к появлению Фурье-
спекгрофотометров и приборов на основе диодных матриц. Приборы со встроен-
ными светофильтрами (для снижения уровня рассеянного света) применяются для
сверхбыстрого измерения состава материалов при производстве продуктов питания
и кормов для животных.
Считалось, что находящийся на краю ИК-спектра, /Ж-диапазон обладает мень-
шим потенциалом для применения в промышленности. Причиной этого отчасти
являются нерешенные экспериментальные и технологические проблемы. Иссле-
дования в дальнем диапазоне ИК-излучения позволяет получить релевантную ин-
формацию, однако усилия непропорциональны результату. Рутинное применение
методов спектроскопии в дальнем ИК-диапазоне в сфере фармацевтики в ближай-
шем будущем не предвидится, поэтому мы не будем обсуждать их в дальнейшем.
Самые современные разработки в области ИК/БИК технологий включают ви-
зуализацию больших поверхностей образцов, неразрушающий анализ твердых ве-
ществ методом нарушенного полного внутреннего отражения (ATR — attenuated total
reflectance) и фотоакустические измерения. Технические характеристики приборов
продолжают совершенствоваться, в особенности это касается надежности и мо-
дульного принципа конструирования прибора. Уменьшение размеров спектроме-
тров, скорость измерений и мобильность прибора больше не являются сложными
задачами. Однако реально расширить сферу применения исследований методом
и проводить измерения во всем диапазоне БИК позволило непрерывное увеличение
мощности компьютерных систем. Сфера применения ИК-спектроскопии расши-
ряется до возможностей количественного анализа сложных образцов в различных
режимах. Именно такие образцы типичны для фармацевтической индустрии. Не-
инвазивный спектральный отбор образцов с использованием световых датчиков,
в конце концов, делает привлекательными аналитические методики in situ, напри-
мер, для проведения измерений в режиме реального времени.
Инфракрасную микроскопию стали применять с начала 1980-х гг. Комбиниро-
вали два микроскопа, обычный оптический микроскоп и ИК-Фурье-прибор с отра-
жающей оптикой. Оптический микроскоп применяют для визуальной локализации
интересующего исследователя участка. Затем на пятно направляют пучок ИК- или
БИК-излучения. Существует множество сфер применения неинвазивного анали-
за, включая обнаружение загрязнителей, частиц, мини-дефектов и идентификации
волокон. Усовершенствованной методикой является система химической визуали-
зации (CIS — chemical imaging systems). Спектры снимаются на всех прилегающих
зонах (пикселях) большой площади поверхности. На практике прорыв в области
визуализации стал возможным после отхода от принципов сканирования «pixel-
after-pixel» (пиксель за пикселем). Гибкость и скорость получения информации при
помощи CIS увеличились при появлении новых детекторов, например, детекторов
Глава 4,2. Процессно-аналитическая технология (РАТ)
435
на основе фокально-плоскостных матриц (FPA — focal plane array). Многочислен-
ные ИК/БИК спектры (более тысячи) сканируются с поверхности образца в один
этап. При наличии алгоритмов для анализа изображений и быстродействующих
компьютеров современные средства визуализации данных 7Д/БИК анализа являют-
ся многообещающими методами, способствующими решению проблем качества.
4.2.2.2. Теория ИК-спектроскопии
В типичном ИК-спектре поглощения органического вещества (рис. 6) ось ординат
отражает параметры пропускания, а ось абсцисс — длину волны.
Линейная шкала волновых чисел предпочтительна из-за линейной зависимости
между волновым числом и энергией и частотой. Частота поглощенного излучения
соответствует собственной частоте колебаний молекулы.
Поглощение, испускание и отражение ИК-излучения молекулярными образ-
цами можно объяснить переходами с одного вращательного или колебательного
энергетического уровня на другой. Энергии ИК-излучения недостаточно для того,
чтобы вызвать переход электронов, аналогичный переходу, вызываемому УФ, види-
мым или рентгеновским излучением. Поглощение ИК-излучения ограничено мо-
лекулярными частицами с небольшой разностью энергии на разных вращательных
и колебательных уровнях. Чтобы стало возможным поглощение ИК-излучения,
в молекуле должно произойти изменение (net change) дипольного момента вслед-
ствие ее колебательного или вращательного движения. При этом происходит взаи-
модействие молекулы с переменным электрическим полем, что вызывает измене-
ния амплитуды ее движения. Дипольный момент определяется величиной разности
436
Часть 4. Процессно-аналитические технологии
зарядов и расстоянием между двумя центрами зарядов. Кроме того, происходят
регулярные флуктуации дипольного момента и устанавливается поле, взаимодей-
ствующее с электрическим полем, связанным с падающим излучением. Если часто-
та излучения полностью соответствует собственной частоте колебаний молекулы,
происходит перенос энергии, изменяющий амплитуду колебаний молекулы, и в
результате происходит поглощение. Точно так же вращение асимметричных моле-
кул вокруг их центров масс вызывает периодические флуктуации диполей, взаи-
модействующих с излучением. Гомонуклеарные частицы не могут поглощать ИК-
излучение.
Количество энергии, необходимое для изменения энергетического уровня, при-
близительно соответствует излучению с длиной волны от 100 см-1 и менее. Относи-
тельное расположение атомов в молекуле подвержено постоянным флуктуациям,
вокруг молекулярных связей возможны колебания и вращения различных типов.
Точный анализ всех типов движения становится невозможным для молекул, состоя-
щих из нескольких атомов. Более сложные колебательные движения свойственны
более крупным молекулам, кроме того, следует учитывать взаимодействия между
различными центрами колебаний. Колебания могут быть валентного и деформа-
ционного типа. Валентные колебания заключаются в постоянном изменении меж-
атомного расстояния по оси связи между атомами. Деформационные колебания
характеризуются изменением угла между двумя связями и бывают пяти типов: нож-
ничные, маятниковые, веерные и крутильные колебания. В молекуле, содержащей
более двух атомов, возможны колебания всех типов. Кроме того, возможны взаимо-
действия и соединение колебаний, если колебания включают связи с единым цен-
тральным атомом с изменением характеристик рассматриваемых колебаний.
4.2.2.3. Механическая модель ИК-колебаний
При поглощении света органическими молекулами регистрируют ИК-спектры.
Простейшим способом описания теоретических основ метода колебательной спек-
троскопии является изучение изолированных колебаний на механической модели,
так называемого гармонического осциллятора. Валентные колебания атомов можно
приблизительно описать при помощи механической модели, представляющей со-
бой две массы /и, и т2, соединенные идеальной пружиной. Смещение одной из масс
вдоль оси пружины приводит к возникновению гармонического движения. Мно-
гие собственные частоты можно рассчитать, исходя из предположения, что полосы
спектра соответствуют энергиям, возникающим в результате колебаний в идеальном
двухатомном гармоническом осцилляторе (рис. 7), и подчиняются закону Гука, т. е.
где и — частота колебаний; к — коэффициент возвращающей силы; и — приведен-
ная масса двух атомов (и = +т2)).
Модель представляет хорошее описание поведения истинно двухатомных мо-
лекул, и довольно близка к средней модели двухатомных систем, содержащихся
в многоатомной молекуле. Соответствующая кривая потенциальной энергии пред-
ставляет собой обычную параболу, представленную на рис. 8.
Глава 4,2. Процессно-аналитическая технология (ДДТ)
437
Лх2
равновесие
Рис. 7. Идеальный двухатомный гармонический осциллятор
Рис. 8. Энергетическая диаграмма идеального двухатомного осциллятора
Это приближение позволяет рассчитать среднюю частоту колебаний связи. На-
пример, приведенные массы для С—О, О—Н и N—Н соответствуют 0,85, 0,89 и 0,87.
Цифры близки по значению, поэтому и частоты будут довольно близки. Тем не
менее, донорные и акцепторные свойства соседних атомов в молекулах влияют на
наблюдаемый диапазон интенсивности, длины волны и частоты. Среднее значе-
ние мало употребимо в структурном анализе, а эти различия вызывают изменение
реального спектра. Силовая постоянная к является мерой жесткости химической
связи и величиной, эквивалентной коэффициенту возвращающей силы пружины
в гармонической модели. Значения А: широко варьируют и являются причиной раз-
личных значений энергий, которые можно рассчитать и использовать при интер-
претации спектров. При помощи ИК-спектроскопии удалось рассчитать отдельные
силовые константы для различных типов химической связи. Как правило, значе-
ние к попадает в диапазон от 3 • 102 н/м для большинства простых связей (среднее
значение — 5 102 н/м). Значения А: для двойных и тройных связей в два и три раза
превышают это среднее значение. На практике эти значения можно использовать
для расчета волновых чисел пиков собственного поглощения, т. е. пиков, соответ-
ствующих переходу из основного состояния в первое возбужденное состояние, для
различных типов связи.
438
Часть 4. Процессно-аналитические технологии
Классическая механика не применима для описания явлений на уровне атомов,
поскольку не учитывает квантовой природы энергий колебания молекул. Таким об-
разом, в отличие от обычной механики, где колебательные системы могут обладать
любой потенциальной энергией, энергии квантово-механических колебательных
систем могут принимать лишь определенные дискретные значения. Переходы на
новые энергетические уровни колебаний Moiyr быть вызваны поглощением излуче-
ния при условии, что энергия излучения точно совпадает с разностью энергий уров-
ней квантовых состояний колебательной системы, и при условии, что колебания
вызывают флуктуации в диполе.
4.2.2.4. Квантово-механическая модель
В отличие от классической модели пружинного осциллятора для описания коле-
баний молекул квантово-механическая модель не предусматривает бесконечного
множества энергетических уровней. Вместо энергетического континуума существу-
ют дискретные энергетические уровни, описанные квантовой теорией. Уравнение
Шредингера, не зависимое от времени, решают при помощи колебательного Га-
мильтониана для двухатомной молекулы. Значения для основного состояния (и = 0)
и возбужденных состояний можно рассчитать путем решения уравнения (рис. 8).
Поглощение фотона определенной энергии может вызвать изменение колебатель-
ных энергетических уровней молекулы. При комнатной температуре значительная
населенность наблюдается только на основном уровне; при этом переходы, вы-
званные поглощением при этих температурах, происходят из основного состояния.
Переходы из основного состояния на энергетический уровень 1 соответствуют соб-
ственному поглощению, если это приводит к изменению дипольного момента мо-
лекулы. Переходы между основным состоянием и энергетическим уровнем 2 или
более дают обертоны. Могут происходить переходы между кратными энергетиче-
скими уровнями, что приводит к образованию комбинационных полос.
Упрощенный вариант энергетических уровней может быть записан для энерге-
тических уровней двухатомной молекулы:
и = 0, 1,2,...
k 2;2пуи
в котором можно увидеть условия закона Гука. Уравнение, переписанное с учетом
квантового условия hV =(h/2n)Jk/u принимает вид:
Е =| и+- о = 0, 1, 2, ...
° I 2)
В случае многоатомных молекул количество энергетических уровней становится
весьма многочисленным. В идеале можно рассматривать такую молекулу как ряд
двухатомных, независимых, гармонических осцилляторов; в этом случае уравнение,
представленное выше, можно обобщить:
ЗЛГ-6Г 1 \ _
£(ор о2, о3, ...)= Ч+т Ш Р,- и2, о3,... = 0, 1, 2, 3, ...
i=i к 2)
Любой энергетический переход с уровня 0 на 1 во всех колебательных состояниях
(ир и2, и3,...) является основным (фундаментальным) и разрешен правилами отбора.
Глава 4.2. Процессно-аналитическая технология {РАТ)439
Если переход происходит из основного состояния к и. = 2,3 и т. д., а все остальные
равняются нулю, он называется первым обертоном, вторым обертоном и т. д. Пере-
ходы из основного состояния в состояние, для которого и. = 1 и vy = 1 одновремен-
но, являются комбинациями. Кроме того, возможны другие комбинации, такие как
о(. = 1, и = 1, = 1 или о. = 2, иу = 1 и так далее. Обычно БИК спектры включают эти
обертоны и комбинации, производные от основных колебаний, которые присут-
ствуют в средней ИК-области спектра. Обертоны и комбинации не разрешены, од-
нако появляются в виде слабых полос из-за ангармоничности или резонанса Ферми.
Как правило, обертоны появляются на одной второй или одной трети длины волны
собственного поглощения или на двух- или трехкратной частоте. Большая часть пи-
ков обертонов возникают по причине R—Н валентных и изгибных колебаний, из-за
большого дипольного момента: О—Н, С—Н, S—Н и N—Н сильно поглощают БИК
и формируют большинство БИК-полос. Поскольку большая часть поглощения по-
вторяется в БИК-диапазоне, эта область, так же, как и средний диапазон (MIR), ис-
пользуется для идентификации молекул. ИК-полосы традиционно используют для
идентификации функциональных групп, обладающих характерными частотами.
БИК-спекгры перекрываются и, несмотря на то что полосы можно идентифициро-
вать, их нельзя соотнести с остальной молекулой. Поэтому БИК-спекгры использу-
ют, главным образом, для подтверждения идентичности вещества.
Согласно квантово-механическим уравнениям энергия перехода с энергетиче-
ского уровня 1 на уровень 2 или с уровня 2 на 3 должна быть идентичной энергии
перехода с 0 на уровень 1. Далее, квантовая теория устанавливает правило: возмож-
ны только те квантовые переходы, при которых, согласно квантовой теории коле-
баний, колебательное квантовое число изменяется на единицу. Это так называемое
правило отбора.
Итак, мы описали классическую и квантово-механическую модель гармониче-
ского осциллятора. Потенциальная энергия колебательной системы изменяется
периодически по мере изменения расстояния между массами. Тем не менее, для
оценки количественных параметров молекулярных колебаний эта модель является
несовершенной. Например, при сближении двух атомов в дополнение к связываю-
щей силе возникает кулоновское отталкивание между двумя атомными ядрами; при
этом можно ожидать более быстрого роста потенциальной энергии по сравнению
с ростом, предсказанным гармонической моделью. В другой экстремальной точке
осцилляции уменьшается возвращающая сила при приближении межатомного рас-
стояния к значениям, при которых происходит разрыв связи.
Теоретически квантово-механические волновые уравнения можно использовать
для построения близких к реальным кривых потенциальной энергии молекулярных
колебаний. К сожалению, математическая сложность этих уравнений препятствует
их применению для количественного анализа во всех системах, приходится ограни-
чиваться простейшими системами. Согласно качественной оценке, кривые должны
быть ангармоническими по форме. Подобные кривые отклоняются от гармониче-
ской кривой в различной степени, в зависимости от природы связи и атомов. Тем
не менее, гармонические и ангармонические кривые почти идентичны при низких
потенциальных энергиях, что указывает на корректность примененных методов ап-
проксимации.
440 Часть 4. Процессно-аналитические технологии
Ангармоничность вызывает отклонения двух типов. При высоких квантовых
числах Л£ уменьшается и правило отбора становится менее строгим; в результате
наблюдаются переходы Д ± 2 или Д ± 3. Подобные трансформации вызывают по-
явление линий обертонов на частотах, в 2 или 3 раза превышающих частоту фунда-
ментальных колебаний; интенсивность обертонного поглощения зачастую низка,
пики могут не наблюдаться. Колебательные спектры усложнены дополнительно
тем, что в молекуле могут взаимодействовать две колебательные системы, что при-
водит к образованию пиков поглощения на частотах, приблизительно равных сумме
или разности их собственных частот. Отметим вновь: интенсивности пиков сумм
и разностей фундаментальных частот, как правило, низки.
Обычно оказывается возможным оценить число и типы колебаний в простой
двухатомной или трехатомной молекуле и определить, участвует ли в колебаниях
несколько типов атомов и связей; для подобных молекул множество вероятных ко-
лебаний приводит к формированию ИК-спектров, которые очень трудно, а иногда
и невозможно анализировать. Число вероятных колебаний в многоатомной моле-
куле можно рассчитать следующим образом. Для описания положения точки в про-
странстве необходимо задать три координаты; чтобы описать положение N точек,
следует задать ЗА координат. Каждая координата соответствует одной степени сво-
боды одного из атомов многоатомной молекулы, поэтому молекула, состоящая из
.У атомов, обладает ЗА степенями свободы. Молекула характеризуется тремя типа-
ми движения. Во-первых, это движение целостной молекулы в пространстве; во-
вторых, вращательное движение целостной молекулы вокруг центра ее тяжести; и,
в-третьих, колебательное движением каждого из атомов относительно других ато-
мов. Поскольку все атомы в составе молекулы перемещаются в пространстве со-
вместно, описание поступательного движения требует трех из ЗА степеней свобо-
ды. Еще три степени свободы необходимы для описания вращательного движения
молекулы в целом. Остальные ЗА — 6 степени свободы включают внутриатомное
движение и, следовательно, представляют число возможных колебаний в молекуле.
В линейной молекуле для описания вращательного движения достаточно 2 степе-
ней свободы. Таким образом, число колебаний для линейной молекулы составляет
ЗА — 5. Каждое из ЗА — 6 или ЗА — 5 колебаний представляет нормальную моду
(нормальное колебание). Для каждой нормальной колебательной моды существует
уравнение потенциальной энергии. Кроме того, в той степени, в которой поведение
колебаний приближено к гармоническому, разности между энергетическими уров-
нями определенных колебаний одинаковы; то есть каждое колебание с изменением
диполя сопровождается единичным актом поглощения.
Тем не менее, может наблюдаться меньшее количество экспериментальных пи-
ков по сравнению с количеством, ожидаемым исходя из теоретического количества
нормальных мод. Число пиков может оказаться меньше ожидаемого, когда симме-
трия молекул такова, что при специфических колебаниях отсутствуют изменения
диполя. Энергии двух или нескольких колебаний могут быть идентичными, или
почти идентичными. В некоторых случаях интенсивность поглощения столь мала,
что его невозможно обнаружить традиционными методами. Кроме того, колеба-
тельная энергия может относиться к диапазону длин волн, не воспринимаемому
данным инструментом.
Глава 4.2. Процессно-аналитическая технология (РАТ)441
И наоборот, число наблюдаемых пиков может превысить число ожидаемых ис-
ходя из числа нормальных мод. Эта ситуация характерна для БИК-области. Ино-
гда регистрируют пики обертонов на частотах, в два или три раза превышающих
частоту фундаментального пика, или дополнительные комбинационные полосы
на частотах, соответствующих приблизительно сумме или разности двух фунда-
ментальных частот. На энергию колебания и на длину волны, соответствующую
пику поглощения, могут влиять другие колебательные системы в составе молеку-
лы. Степень влияния определятся рядом факторов. Связанные колебания (vibration
coupling) представляют распространенное явление. В результате положение пика по-
глощения, соответствующего определенной органической функциональной группе,
не всегда может быть определено точно. Эффекты, вызванные взаимодействием,
могут явиться причиной неопределенности при идентификации функциональных
групп, входящих в состав соединения, в то же время именно этот эффект придает
ИК-спектрам характеристические черты, важные для положительной идентифика-
ции соединения в целом.
4.2.2.Б. Ангармоничность
Идеальный гармонический осциллятор представляет собой ограниченную модель.
При очень тесном сближении осциллирующих масс реальные силы сжатия — не
учтенные в расчетах — противодействуют дальнейшему сжатию пружины. Когда
пружина растягивается, она достигает точки, в которой теряет форму и не может
возвратиться к исходной форме спирали. Идеальный случай представлен на рис. 9.
К барьерам с обеих сторон цикла приближаются равномерным и упорядоченным
путем. Точно так же в молекулах соответствующие электронные облака двух свя-
занных атомов ограничивают сближение ядер на этапе сжатия, создавая энергети-
ческий барьер. При растяжении связь, в конце концов, разрывается, когда уровень
колебательной энергии превышает энергию разрыва связи.
Барьер быстро возрастает при уменьшении расстояния, в то же время барьер при
большом растяжении медленно приближается к нулю (рис. 9). Форма кривой по-
тенциальной энергии типична для ангармонического осциллятора.
Энергетические уровни ангармонического осциллятора не одинаковы, хотя они
несколько сближаются при повышении энергии. Это явление можно описать сле-
дующим уравнением
Рис. 9. Энергетическая диаграмма ангармоничного двухатомного осциллятора
442
Часть 4. Процессно-аналитические технологии
( ( 1Y
Е = от— \hW - ин— WX+higher terms,
и I 2 У е I 2 у и е ° 1
где higher terms — члены более высокого порядка; We /м)1/2 — частота ко-
лебаний; WX — постоянная ангармоничности; к — ангармоническая силовая по-
стоянная; и — приведенная масса двух атомов.
На практике ангармоничность составляет от 1 до 5%. Таким образом, первый
обертон серии основных колебаний, например, на 3500 нм равняется
и = ^~-+(3500 [0,01; 0,02; ...]).
В зависимости от структурных или пространственных условий величина мо-
жет варьировать от 1785 до 1925 нм в данном примере. Тем не менее, как правило,
значение составляет 3500/2 плюс относительно малый сдвиг в сторону увеличения
длин волн. Вследствие запрещенных переходов интенсивность обертонов от 10 до
1000 раз меньше, чем фундаментальных колебаний. Таким образом, полоса, возни-
кающая вследствие вращения или изгибных колебаний будет в третьем или четвер-
том обертоне, наблюдаемом в БИК-области спектра. Например, фундаментальному
валентному колебанию на 1750 см-1 или 5714 нм соответствует первый обертон при-
близительно на 3000 нм, более слабый второй обертон — приблизительно на 2100 нм
и третий, очень слабый обертон — на 1650 нм. Четвертый обертон, приблизительно
на 1370 нм, настолько слаб, что не может быть полезен. Представленные цифры
основаны на иллюстративной 5%-ной константе ангармоничности.
Подробное исследование спектров простых молекул представляет прямой источ-
ник определения частотных БИК-характеристик выбранных колебательных мод.
К количественному и качественному анализу существует требование: необходимо
интерпретировать как можно больше информации, содержащейся в БИК-спекгре.
Хотя интерпретация спектров способом, аналогичным способу интерпретации спек-
тров в среднем ИК-диапазоне, не приемлема, попытки определения и категориза-
ции наблюдаемых БИК-частот предпринимаются. Примеры частот для алифатиче-
ских углеводородов, представленных в литературе, включены в следующий список:
8547 см-1
8474 см-'
7700-9000 см-1
8696 см-1
8285 см-1
1080-1140 см-1
7692, 8237, 8576 см"1
С—Н второй обертон в CH = СН
С—Н группа в щ/с-олефинах
С—Н второй обертон
второй обертон СН2 антисимметричного
валентного колебания
второй обертон СН2 симметричного
валентного колебания
второй обертон олефина
второй обертон С—Н валентного колебания в СН2
4.2.2.6. Применение спектроскопии в среднем ИК-диапазоне
для структурных исследований
Спектроскопия поглощения и отражения в средней ИК-области спектра обычно
применяется для определения структуры органических и биохимических образцов.
Глава 4,2. Процессно-аналитическая технология (РАТ)443
При использовании в комбинации с другими аналитическими методами, такими
как масс-спектроскопия, ядерный магнитный резонанс и элементный анализ, ИК-
спектроскопия обычно позволяет провести положительную идентификацию об-
разцов. Спектры снимают после подготовки образца, как правило, включающей
разбавление аналитического вещества. Обработка образца является трудоемкой
и времязатратной частью аналитической процедуры. В органических образцах ре-
гистрируют ряд ИК-пиков поглощения, которые используют для подтверждения
качественной оценки структуры. Во-первых, предположительные функциональные
группы идентифицируют путем исследования диапазона частот приблизительно от
3600 до 1200 см*1. Ранее упоминалось, что частота, на которой органическая функ-
циональная группа поглощает излучение, может быть оценена приблизительно по
атомным массам и энергии связи между ними. Эти групповые частоты не являются
полностью инвариантными из-за взаимодействий с другими колебаниями. Однако
подобные эффекты взаимодействия невелики, и можно определить диапазоны ча-
стот, в которых с высокой вероятностью можно обнаружить пик поглощения дан-
ной функциональной группы. Групповые частоты перечислены в корреляционных
диаграммах, которые служат исходной точкой процесса идентификации.
Во-вторых, спектр неизвестного соединения сравнивают со спектрами эталон-
ных соединений, содержащих все функциональные группы, обнаруженные на пер-
вом этапе. Область спектра от 1200 до 600 см-1 (отпечаток) содержит чрезвычайно
полезную информацию, поскольку небольшие различия в структуре и составе вызы-
вают значительные изменения в форме и распределении пиков поглощения в этой
области спектра. Большинство простых связей дают полосы поглощения на этих
частотах. Поскольку их энергии примерно одинаковы, между смежными связями
существует сильное взаимодействие. Таким образом, полосы поглощения объеди-
няют различные взаимодействия и зависят от общей структуры углеродного скелета
молекула. Из-за сложности спектров точная интерпретация этой области спектра,
как правило, невозможна. С другой стороны, именно сложность картины определя-
ет уникальность характеристик спектра и последующую информативность этой об-
ласти для итоговой идентификации. Близкое соответствие двух спектров в области
отпечатка указывает на почти полную идентификацию соединения.
При анализе групповых частот важно проводить анализ и соотнесение полного
спектра, а не только малой изолированной его части. Диаграммы корреляции слу-
жат лишь руководством к дальнейшим более тщательным исследованиям. Каталоги
ИК-спектров, применяемые для количественной идентификации путем сравнения
и соотнесения эталонных спектров большого числа чистых соединений, можно
приобрести в электронном виде. Оптимизированные системы поиска, предназна-
ченные для идентификации соединений по базам данных ИК-спектров, и алгорит-
мы этапа соотнесения, могут иметь успех как быстрые и надежные методы.
4.2.2.7. Расширение области применения спектроскопии в среднем
ИК-диапазоне
Органические и неорганические молекулярные частицы (за исключением гомону-
клеарных молекул) поглощают излучение в ИК-области. ИК-спектроскопия обла-
дает потенциалом идентификации необыкновенно большого количества веществ.
444 Часть 4. Процессно-аналитические технологии
Более того, уникальность спектроскопии в среднем ИК-диапазоне {MIRS) состо-
ит в специфичности, равной или превышенной сравнительно небольшим числом
других аналитических методов. Благодаря специфичности принцип был применен
при разработке методик количественного анализа спектров ИК-поглощения. Одна-
ко они отличаются от методик количественного анализа, использующих УФ и ви-
димую область спектра: для MIRS характерна большая сложность спектров, более
узкие полосы поглощения; кроме того, существуют технические ограничения при-
менения ИК-приборов. Количественные результаты, полученные путем анализа
ИК-спектров, обычно уступают в качестве и надежности результатам, полученным
посредством изучения спектров в УФ и видимой области и БИК-спектроскопии.
Далее, построение одномерных или линейных калибровочных графиков требуют
скрупулезного внимания ко множеству деталей. Одна из причин неудач — часто
встречающееся несоответствие закону Бэра, вызванное сложностью, присущей ИК-
спектрам, что выражается в наложении пиков поглощения или влиянием рассеян-
ного излучения. Аналитические неопределенности не удается понизить до уровня,
сравнимого с уровнем неопределенности других методов, несмотря на значитель-
ные усилия и старания аналитиков.
Диффузионно-отражательная спектроскопия в среднем ИК-диапазоне нашла
применение в анализе труднообрабатываемых (hard-to-handle) твердых проб, таких
как полимерные пленки, волокна или твердые дозированные формы. Спектры от-
ражения в средней ИК-области не идентичны соответствующим спектрам поглоще-
ния, однако достаточно близки по уровню предоставляемой информации. Спектры
отражения можно использовать как для качественного, так и для количественного
анализа. В общем, отражение излучения бывает четырех типов: зеркальное, диф-
фузное, внутреннее и нарушенное полное внутренне отражение (ATR).
Зеркальное отражение происходит, когда отражающая среда представляет собой
гладкую блестящую поверхность. Угол отражения идентичен углу падения пучка из-
лучения. Если поверхность способна к поглощению ИК-излучения, относительная
интенсивность отражения меньше для поглощаемых частот излучения, чем для не по-
глощаемыхчастот излучения. Таким образом, график зависимости отражения R, опре-
деляемого отношением энергии отраженного падающего излучения к длине волны
(или волновому числу), оказывается аналогичным для спектра пропускания образца.
Спектры диффузного отражения получают непосредственно при исследовании
порошкообразных образцов после минимальной подготовки. Кроме преимущества,
связанного с экономией времени, анализ является неразрушающим и оставляет об-
разец неповрежденным для последующего анализа. Широкое применение методов
диффузного отражения стало возможным после появления Фурье-спектрометров.
Отраженное излучение от порошкообразных веществ слишком мало и не может
быть измерено при посредственном уровне разрешения и неадекватном соотноше-
нии сигнал-шум. Диффузное отражение (рис. 10) происходит, когда пучок излуче-
ния падает на поверхность тонкоизмельченного порошка. В образцах такого типа
зеркальное отражение происходит на всех плоских поверхностях. Тем не менее, та-
ких поверхностей множество, и они ориентированы случайно, отражение излуче-
ния происходит во всех направлениях. Интенсивность отраженного излучения не
зависит от угла визирования. В то время как в спектрах отражения и пропускания
Глава 4.2. Процессно-аналитическая технология (РАТ)
445
Диффузное пропускание
Рис. 10. Измерение отражения, трансфлекции
(поглощение при двукратном прохождении излучения через образец) и пропускания
пики расположены идентично, относительные высоты пиков различаются значи-
тельно. Например, малые пики в спектре пропускания, как правило, больше в спек-
тре отражения.
Спектроскопия внутреннего отражения применяется для получения ИК-
спектров для труднообрабатываемых и трудноподготавливаемых образцов (hard-
to-handle или hard-to-prepare), таких как твердые вещества с ограниченной раство-
римостью, пленки, пасты, клеи и порошки. Отражение происходит, когда пучок
излучения переходит из среды с большей плотностью в среду менее плотную. От-
ражаемая часть падающего луча повышается по мере увеличения угла падения. По-
сле прохождения определенного критического угла отражение становится полным.
В процессе отражения пучок проникает на небольшое расстояние в среду с меньшей
плотностью до того, как происходит отражение. Глубина проникновения варьирует
от доли длины волны до нескольких длин волн. Она зависит от длины волны падаю-
щего излучения, коэффициентов преломления двух материалов и угла падения луча
относительно поверхности раздела.
Спектроскопия нарушенного полного внутреннего отражения (МНПВО, в ан-
глийском варианте ATR — attenuated total reflectance) является наиболее часто при-
меняемым методом измерения отражения. Спектры МНПВО не могут быть сравни-
мы со спектрами поглощения. На обоих видах спектров наблюдаются аналогичные
пики, в то же время их относительные интенсивности существенно различаются.
Величины поглощения зависят от угла падения и не зависят от толщины образца,
поскольку излучение проникает вглубь образца лишь на несколько микрометров.
Главным преимуществом МНПВО спектроскопии является простота применения
при анализе множества различных твердых проб. Спектры можно получить при ми-
нимальной подготовке образца: образец прижимают к МНПВО кристаллу высокой
плотности. Пластики, резины, упаковочные материалы, пасты, порошки, твердые
вещества и дозированные формы, такие как таблетки, можно непосредственно под-
готавливать к анализу аналогичным способом.
4.2.2.8. Рамановская спектроскопия
Когда излучение проходит через прозрачную среду, часть пучка рассеивается во всех
направлениях. Малая часть рассеянной радиации отличается от падающего луча по
длине волны за счет влияния химической структуры молекул среды. Дискретные
изменения колебаний того же типа связаны с ИК-поглощением, разность длин
волн падающего и рассеянного излучения соответствует длинам волн средней ИК-
области спектра. Спектры рамановского рассеяния и спектры ИК-рассеяния для
446
Часть 4. Процессно-аналитические технологии
Рис. 11. Примеры двух рамановских спектров
(две полиморфные формы фармацевтического наполнителя)
одинаковых образцов весьма схожи. На рис. 11 представлен типичный рамановский
спектр.
Как правило, выбор делают в пользу ИК-спектроскопии, однако рамановская
спектроскопия в некоторых случаях позволяет получить больше информации об
отдельных типах органических соединений. Например, метод чувствителен по от-
ношению к информации, связанной с конформациями и окружением. Наложение
пиков в сложных смесях менее вероятно, что облегчает количественный анализ.
В частности, метод позволяет проводить точный количественный анализ очень ма-
лых образцов. Несмотря на эти преимущества, рамановская спектроскопия до сих
пор не применяется широко, что связано с довольно высокой стоимостью оборудо-
вания.
Есть различия в типах групп, которые поглощают ИК-излучение, и таких, кото-
рые активны в спектре рамановского рассеяния. Части рамановского и ИК-спектров
комплементарны, и каждая часть ассоциирована с разными наборами колебатель-
ных мод в молекуле. Другие колебательные моды могут обладать активностью как
в спектре рамановского рассеяния, так и в ИК-спектре. Интенсивность (мощность)
пика рамановского спектра связана сложной зависимостью с поляризуемостью мо-
лекулы, энергией источника и концентрацией активных групп, а также с другими
факторами. Интенсивность рамановских пиков, как правило, прямо пропорцио-
нальна концентрации активных веществ.
В рамановской спектроскопии используется возбуждающее излучение с длиной
волны, отстоящей от пиков поглощения аналитического вещества. Механизм обра-
зования рамановского спектра отличается от механизма образования спектра сред-
Глава 4.2. Процессно-аналитическая технология (РАТ)447
него ИК-диапазона, хотя зависит от тех же колебательных мод. ИК-поглощение
требует изменения дипольного момента или соответствующего распределения
заряда. Только в этом случае излучение той же частоты может взаимодействовать
с молекулой и способствовать переходу на возбужденный колебательный уровень.
Напротив, при рассеянии происходит моментальная деформация электронного об-
лака, распределенного вокруг связи в молекуле, с последующим вторичным излуче-
нием при возвращении связи в нормальное состояние. При деформации молекула
временно поляризуется. Моментально возникает диполь, который исчезает после
релаксации и вторичного излучения. Поэтому активность в спектре рамановского
рассеяния определенной колебательной моды может существенно отличаться от ее
ИК-акгивности. Например, гомонуклеарная молекула не имеет дипольного момен-
та в состоянии равновесия или в растянутом состоянии, при этом ИК-поглощение
излучения на возбуждающей частоте невозможно. С другой стороны, поляризуе-
мость связи между двумя атомами такой молекулы варьирует периодически вместе
с валентными колебаниями связи, достигая максимума в момент наибольшего раз-
деления и минимума при наибольшем сближении. Происходит рамановский сдвиг
частоты, соответствующий частоте колебательной моды. Величина рамановского
сдвига не зависит от длины волны возбуждающего излучения. Таким образом, пат-
терны сдвигов оказываются идентичными, независимо от типа лазера, применяе-
мого для возбуждения.
В спектроскопии в средней ИК-области наличие воды всегда вызывает интер-
ференцию, чего не происходит при рамановском рассеянии. Поэтому рамановские
спектры можно получать непосредственно из водных растворов. Кроме того, мож-
но использовать стеклянные или кварцевые ячейки. Развитие рамановской спек-
троскопии было тесно связано с доступностью несложных в применении лазерных
устройств. Рамановские спектры получают при облучении образца при помощи
лазерного источника видимого или БИК монохроматического излучения. Во вре-
мя облучения при помощи соответствующего устройства фиксируют рассеянное
излучение под определенным углом (например, 90 °). Интенсивность рамановских
линий составляет 0,001% от интенсивности источника (или менее), и более трудны
для детектирования, чем ИК-спекгры. Анализ рамановских спектров может быть
затруднен из-за флуоресценции или примесей, содержащихся в образце. Эта про-
блема отчасти решается за счет применения лазерных источников БИК-излучения,
работающих на больших длинах волн. Образцы можно облучать мощным излуче-
нием, не вызывая фоторазложения или нагревания. БИК лазеры недостаточно
мощны, чтобы в большинстве молекул возникло значительное число возбужденных
энергетических состояний, вызывающих флуоресценцию. Флуоресценция в этом
случае менее интенсивна или практически незаметна.
Существует три типа рассеянного излучения: стоксово, антистоксово и рэлеев-
ское излучение. Длина волны рэлеевского излучения идентична длине волны от
источника возбуждения; интенсивность его намного превышает интенсивность
излучения двух других типов. Условно в рамановских спектрах по оси абсцисс от-
ложена разность в волновых числах между наблюдаемым излучением и излучением
источника. Поскольку линии антистоксова излучения менее интенсивны по срав-
нению с соответствующими стоксовыми линиями, для анализа используют только
448
Часть 4. Процессно-аналитические технологии
эту часть спектра. Тем не менее, когда возникает флуоресценция, мешающая на-
блюдению стоксовых линий, антистоксова часть спектра несет полезную информа-
цию, несмотря на меньшую интенсивность линий.
4.2.2.Э. Введение в БИК-спектроскопию
До недавнего времени БИК-область спектра считалась недостаточно информатив-
ной для спектрального анализа органических соединений. Комбинационные по-
лосы и полосы обертонов молекулярных колебаний наблюдаются в сравнительно
узком диапазоне 750—3000 нм, по сравнению с фундаментальными полосами, на-
блюдаемыми в диапазоне 2800—50 000 нм. Это считалось недостатком. Кроме того
наблюдали сильное перекрывание БИК-полос, возникали трудности с разрешением
полос, а при разрешении — трудности с интерпретацией. Если образцы не высуше-
ны перед БИК-анализом, интерпретацию нередко перекрывающихся спектральных
линий затрудняют изменения водородных связей, на которые влияют температура
образца, ионная сила и концентрация аналитического вещества. Изменения водо-
родных связей вызывают сдвиг полосй, а также изменение их формы — сглажива-
ние или расширение полос. Полосы обертонов и полосы комбинационного моле-
кулярного поглощения в БИК-области намного менее интенсивны по сравнению
с линиями собственного ИК-поглощения. Поэтому изменения поглощения в БИК-
области довольно малы по отношению к изменениям концентрации. Относительно
небольшие коэффициенты экстинкции, связанные с комбинационными линиями
и линиями обертонов, сильно ограничивают допустимые уровни шума и требу-
ют стабильности БИК инструмента, применяемого для количественного анализа.
Представление образца (samplepresentation) и относительно прямое измерение отра-
жательной способности включает аспекты, которых не предполагала традиционная
ИК-спектроскопия.
Если полученная информация отличается лучшим разрешением в ИК-области,
то почему химики-аналитики интересуются БИК-спектроскопией? Многочислен-
ные трудности, связанные с использованием БИК-спектроскопии для качествен-
ного анализа, вызвали неприятие метода со стороны химиков-аналитиков. Карл
Норрис, инженер Департамента сельского хозяйства США, показал потенциальную
ценность измерений в БИК-области для количественных оценок, применив метод
для исследования сельскохозяйственной продукции в 1960-х гг. Основная идея за-
ключалась в обеспечении сельского хозяйства различной научной и производствен-
ной аппаратурой, способной проводить в режиме on-line БИК-спектрометрические
измерения. Метод БИК-спектроскопии распространился в химической и фарма-
цевтической промышленности; в специализированных журналах постоянно публи-
куется информация о практическом применении метода, и количество публикаций
продолжает расти.
4.2.2.10. Преимущества БИК-спектроскопии
БИК-область спектра представляет большой интерес с точки зрения применения
в фармацевтическом производстве. БИК-спектроскопия является быстрым, нераз-
рушающим и экономически эффективным методом. Не требуется предварительной
подготовки проб; образцы остаются неповрежденными и доступными для дальней-
Глава 4.2. Процессно-аналитическая технология (РАТ)449
шего анализа. Метод БИК-спектроскопии можно осуществлять в режимах in-line,
on-line и off-line. Кроме того, для удаленного анализа можно использовать стекло-
волоконную оптику, позволяющую подвести излучение непосредственно к образ-
цу. Анализ практического применения БИК-спектроскопии в фармацевтическом
производстве при решении конкретных задач позволяет обнаружить многие другие
преимущества метода.
Как упоминалось ранее, полосы поглощения в этой области представляют обе-
ртоны или комбинационные полосы основных валентных связей в диапазоне
3000—1700 см-1 (рис. 12). Обычно в процесс вовлечены связи С—Н, N—Н и О—Н.
Поскольку полосы представляют обертоны и комбинации, молярные коэффициен-
ты поглощения низки, а пределы обнаружения составляют около 0,1%.
Обработка пробы упрощена, поскольку для изготовления окон, линз и всех про-
чих оптических компонентов можно использовать стекло. Кроме того, излучение
от лазерного источника легко сфокусировать на малой площади образца. Образцы
очень малого размера можно исследовать без времязатратной подготовки, также воз-
можна передача излучения от источника через оптические волокна. Оптоволокон-
ный зонд можно привести в контакт с образцом или погрузить в него. Зонд состоит
из подводящих (входных) волокон, окруженных несколькими собирающими волок-
нами, через которые рассеянное излучение передается в монохроматор. Это позво-
ляет получать спектры немедленно при сравнительно неблагоприятных условиях.
В отличие от спектроскопии в среднем ИК-диапазоне, БИК находит применение
в рутинном количественном анализе образцов, таких как вода, белки, углеводороды
и жиры, например, в продуктах питания и кормах для животных, а также в нефтехи-
мической и химической промышленности. На рис. 13 представлены спектральные
данные образца фармацевтического продукта с варьирующим содержанием воды.
Рисунок показывает, что возможно применение метода в количественном анализе
при изменении определенного физического параметра, которое можно специаль-
но смоделировать впоследствии. Распространению методов БИК в фармацевтиче-
ской промышленности способствовали успехи в области компьютерных технологий
и обработки данных. Находят применение анализ диффузного отражения и пропу-
скания (рис. 10), хотя диффузное отражение применяется намного шире благодаря
простоте методики. Многие спектрометры были специально сконструированы для
измерений в БИК-диапазоне. Приборы эти весьма разнообразны, и разнообразие
их намного шире, чем для среднего ИК-диапазона. Самыми сложными являются
Длина волны,нм
Рис. 12. Типичный БИК-спектр
450
Часть 4. Процессно-аналитические технологии
Рис. 13. Совокупность БИК-спектров. Четко видна варьирующая
полоса воды приблизительно на 1950 нм
двухлучевые спектрометры с дифракционной решеткой, приборы с диодной матри-
цей и Фурье-спектрометры. До сих пор доступными ценными инструментами оста-
ются более простые приборы, снабженные фильтрами. Эти приборы можно считать
аналогичными приборам, применяемым для анализа в УФ и видимой области спек-
тра. Вольфрамово-галоидные лампы с кварцевыми окнами служат источниками из-
лучения. Применяются детекторы на основе сульфида свинца (PbS) или арсенида
галлия (AsGa), многие приборы сконструированы для работы в диапазоне от 180 до
2500 нм.
БИК-спектры менее информативны при решении задач идентификации и более
полезны при количественном анализе соединений, содержащих функциональные
группы, в состав которых входит водород, связанный с углеродом, азотом или кис-
лородом. Правильность и точность анализа таких соединений эквивалентна скорее
спектроскопии в УФ и видимой области, чем в среднем ИК-диапазоне. В настоящее
время метод часто применяется для определения содержания воды в образцах раз-
личного типа. Отражательная БИК-спектроскопия стала надежным инструментом
количественного определения состава твердых веществ. Первым применением этой
быстрой методики стал анализ белка, влаги, крахмала, масла, липидов и целлюлозы
в продуктах питания и кормах для животных. Способ обработки пробы, процедура
анализа и обработки данных устанавливались в ходе первых применений методики.
Обычно при БИК-спектроскопии твердый образец облучают в одном или несколь-
ких узких диапазонах, или в полном диапазоне излучения от 1 до 2,5 мкм. Происхо-
дит диффузное отражение — излучение проникает через поверхностный слой частиц,
возбуждает колебательные моды молекул аналитического вещества, а затем рассеи-
вается во всех направлениях. Таким образом, получают спектр, зависящий от состава
образца. В этом случае ордината представляет собой логарифм величины, обратной
/?, log (1/А), где R — отношение интенсивности излучения, отраженного от образца,
к отражению от стандартного отражателя, такого как тонко измельченный сульфат
Глава 4.2. Процессно-аналитическая технология (РАТ)
451
бария или оксид магния. Типичная полоса отражения на 1940 нм представляет со-
бой пик воды, используемый для определения влажности, как показано на рис. 13.
В настоящее время доступно множество диффузно-отражательных приборов.
В некоторых приборах используют несколько интерференционных фильтров для
получения узких полос излучения. Другие приборы оборудованы монохроматора-
ми с диффузионными решетками. Как правило, существуют строгие требования
к калибровке, поскольку образцы для калибровки должны состоять из материала,
содержащего аналитическое вещество в концентрациях, с которыми, вероятно,
придется работать аналитикам. Возможно, полезным окажется измельчение твер-
дых образцов до порошков с воспроизводимым размером частиц. Для анализа вы-
ведены и используются уравнения. По завершении разработки и валидации мето-
дики, твердые образцы можно анализировать за несколько минут. Сообщают, что
правильность и точность методики составляют 1 и 2% соответственно.
Кроме эмпирической и довольно трудоемкой разработки методик применения
ВИКУ, необходимо предоставить точную документацию. БИК методики должны
соответствовать правилам надлежащей производственной практики (cGMP), дей-
ствующим в фармацевтической промышленности. Следует тщательно учитывать все
нормативные аспекты. Например, применение БИК-спектроскопии для классифи-
кации, идентификации или количественного анализа требует построения и валида-
ции полной модели процесса, изучения влияния рисков из-за возможных ошибок,
определение переменных и измеряемых параметров модели, а также полный анализ
данных. Далее, для проведения рутинных анализов необходимо четко определить
рабочие процедуры и провести обучение персонала. Общие нормативные требо-
вания предусматривают ведение достоверной документации в течение всего срока
применения модели БИК, спектрометра, компьютера и т. п.
4.2.2.11. Введение в химическую визуализацию методом спектроскопии
в БИК- и средней ИК-области
Гомогенность смесей химических соединений является серьезной проблемой для
фармацевтических твердых дозированных форм. Классический спектрометр ин-
тегрирует информацию о пространственном распределении веществ. При анализе
твердых форм использование усредненного спектра на поверхности может оказать-
ся недостатком. Например, в фармацевтической промышленности важно получить
карту распределения активных ингредиентов и вспомогательных веществ в таблет-
ке, чтобы выявить физическое взаимодействие между компонентами и решить
проблемы, связанные с однородностью продукта. Спектроскопические приборы,
способные строить изображения, визуализирующие распределение химических
компонентов, представляют большой интерес для производителей фармацевтиче-
ских продуктов.
Самой современной разработкой, объединяющей химическую информацию,
полученную при помощи спектроскопического анализа, с информацией о про-
странственном строении образца, является визуализация колебательных гипер-
спекгров. В принципе, получить гиперспектральное изображение можно при помо-
щи обычных детекторов, т. е. путем классического сканирования или картирования
при помощи микроскопов. Появление на рынке детекторов на основе фокально-
452
Часть 4. Процессно-аналитические технологии
плоскостных матриц {FPA) сделало визуализацию более информативным аналити-
ческим методом. Матричные многоэлементные детекторы регистрируют все пик-
сели анализируемой поверхности одновременно, что резко снижает время анализа
и обеспечивает однородность фона, что позволяет улучшить соотношение сигнал/
шум. Полный спектр получают для каждого пикселя, что указывает на то, что гипер-
спектральное изображение фактически является кубом данных, то есть трехмерной
(3D) матрицей. Гиперспектральные изображения предоставляют пространственную
и спектральную информацию, а также информацию о качественном и количествен-
ном составе образца.
Методы построения изображений обычно являются эффективными при при-
менении к таблетированным лекарственным продуктам с целью изучения количе-
ственных аспектов или визуализации процессов растворения, распределения по-
лиморфных форм, при определении содержания влаги, локализации и описания
активного фармацевтического ингредиента {API — active pharmaceutical ingredient),
однородности содержимого, измельчения и гранулирования. Подобную информа-
цию невозможно получить при помощи традиционной спектроскопии. Путем ви-
зуализации можно провести идентификацию химического соединения или опреде-
ление размера частиц. Выбор метода построения изображения обусловлен рядом
критериев, таких как пространственное и спектральное разрешение, время измере-
ния и диапазон длин волн.
Популярность систем химической визуализации {CIS) быстро растет, увеличива-
ется их надежность по мере расширения сферы применения. Это связано с построе-
нием изображений поверхности путем многоточечного сканирования и преобразо-
вания данных. Показано, что гиперспектральное изображение может быть полезно
для количественного анализа фармацевтических продуктов, и его можно использо-
вать, когда информация о пространственном строении имеет отношение к анализу
образцов. Если даже вопросы применения методик в режиме on-line и нормативной
валидации требуют дальнейших разработок, то значение CIS в системах контроля
качества и РАТ не нуждается в дополнительном подтверждении, независимо от диа-
пазона длин волн или метода сбора спектров.
4.2.2.12. Типы инструментов для спектроскопии в среднем ИК-диапазоне
На рынке представлены ИК-приборы различных типов: спектрофотометры с дис-
персионной решеткой, Фурье-анализаторы и недисперсионные фотометры. До
1980-х гг. и появления более надежных интерферометров, чаще всего пользовались
интерферометрами дисперсионного типа. Во многих случаях предпочтительно при-
менение Фурье-спектрофотометров благодаря их скорости, надежности и удобству.
Они заменили многие другие приборы в аналитических лабораториях. Излучение
от источника разделено на два пучка, для которых можно варьировать длину пути
периодически для получения интерференционных паттернов. Затем для обработки
данных используется алгоритм обратного преобразования Фурье. Первые Фурье-
спектрометры требовали частых настроек оптических частей, однако их уникаль-
ные характеристики, такие как быстрота, высокое разрешение, чувствительность,
прецизионность и точность по длине волны, сделали эти приборы незаменимыми.
Приборы более поздних конструкций намного более надежны и просты в настрой-
Глава 4.2. Процессно-аналитическая технология (РАТ)
453
Источник
излучения
Рис. 14. Интерферометр Майкельсона
ке. Обычно Фурье-спектрометры основаны на применении интерферометра Май-
кельсона (рис. 14), хотя иногда в них используют и другие оптические системы.
Лабораторные Фурье-спектрометры обычно представляют собой однолучевые
приборы. Для получения спектров пропускания или поглощения на этих приборах
необходимо сперва получить стандартную интерферограмму, например, для возду-
ха, и сохранить ее. Анализируемый образец затем помещают на пути лучей света
и повторяют процесс. Пропускание на различных частотах получают путем вычис-
ления соотношения параметров спектров образца и стандартных спектров.
В большей части среднего ИК-диапазона спектра Фурье-спектрофотометры об-
ладают на порядок лучшим соотношением сигнал/шум, чем дисперсионная аппара-
тура высокого качества. С увеличением соотношения сигнал/шум может, конечно,
конкурировать быстрое сканирование, позволяющее получать спектры за несколь-
ко секунд. Интерферометрические приборы также обладают высоким разрешени-
ем (менее 0,1 см '), высокой точностью и воспроизводимой частотой определений.
Фурье-спектрометры обеспечивают пропускание намного большего излучения по
сравнению с дисперсионными приборами, в которых светосила ограничена узкой
входной щелью. Однако это преимущество отчасти сглаживается низкой чувстви-
тельностью быстродействующих детекторов, необходимых для интерферометрии.
Для получения изображения поверхности образца Фурье-спектрометры мож-
но объединять с микроскопом или микрокамерой, оснащенной FPA детектором.
Системы химической визуализации (CIS) выпускаются для рамановской, БИК-
и М/Д-спектроскопией. На рис. 15 представлена оптическая система для химиче-
ской визуализации.
Конструкция прибора зависит от того, как проводятся измерения (рис. 10).
В идеальном случае, на приборе можно получать как спектры пропускания, так
и отражения. Например, анализ отражения позволяет проникнуть лишь на 1 —4 мм
в поверхность твердых образцов. Малая глубина проникновения излучения в об-
разец вызывает большую изменчивость данных при анализе неоднородных образ-
цов по сравнению с методами пропускания. При измерении пропускания весь путь
света через образец включен в измерение, что уменьшает погрешность, связанную
454
Часть 4. Процессно-аналитические технологии
Рис. 15. ИК-микроскоп, соединенный с FPA детектором
с неоднородностью образца. Метод пропускания пригоден для анализа плотных
образцов, например цельные таблетки, однако поверхностное рассеяние вызывает
потерю пропускаемого излучения, что в общем приводит к уменьшению отноше-
ния сигнал/шум. В некоторых обстоятельствах размеры частиц настолько малы, что
большая часть энергии, падающей на образец, рассеивается. Если размеры частиц
достаточно малы, то не удается пропустить достаточно энергии сквозь образец, что-
бы детекторы зарегистрировали сигнал. При пропускании для увеличения глубины
проникновения излучения в образец используют большие частоты (800—1400 нм).
Однако энергия излучения большей частоты более подвержена поверхностному
рассеянию по сравнению с энергией низкой частоты. Поэтому измерения параме-
тров пропускания должны быть оптимизированы по отношениям частот, применяе-
мых для измерений, поверхностному рассеянию и длине пути излучения в образце.
В первых БИК-спектрометрах был использован принцип наклонного фильтра
{tiltingfilter). Ему на смену пришли вращающиеся системы с рядом фильтров, вмон-
тированных в диск датчика для большей точности позиционирования (воспроиз-
водимости длин волн) и большей достоверности. Интерференционные фильтры
применяются в конструкции приборов другого типа, для которых производятся ин-
терференционные фильтры для дискретных длин волн с предварительно установ-
ленными спецификациями. Фильтры устанавливаются в турель {turret) и ее медленно
поворачивают, в то же время проводят аналитическое сканирование. Эти приборы
достаточно надежны при наличии твердо установленных калибровок, специфичных
для измеряемого аналитического вещества. При неправильном выборе интерферен-
ционных фильтров для конкретного применения успешная калибровка невозмож-
на. Конфигурацию современных систем составляют от 6 до 44 интерференционных
фильтров для дискретных длин волн; в частности, они нашли рутинное применение
в анализе продуктов питания и кормов для животных.
Дисперсионные, решеточные, сканирующие БИК-спекгрометры появились на
рынке с конца 1970-х гг. Эти приборы различались оптическими схемами, однако
все включали в качестве источника вольфрамово-галогеновую лампу, одиночный
монохроматор с голографической дифракционной решеткой и неохлаждаемые де-
текторы на основе сульфида свинца. Эта конструкция относится крайним 1980-м гг.
Глава 4.2. Процессно-аналитическая технология (РАТ)455
На рис. 16 представлена типичная конструкция прибора на основе предваритель-
ного монохроматора, где свет рассеивается перед падением на образец, с системой
детектирования диффузного отражения. Монохроматический луч света освещает
образец при 0 (нормальное падение), а детекторы собирают отраженный свет под
углом 45°. Можно установить от двух до шести детекторов, как правило, детекторов
на основе сульфида свинца, для измерений в диапазоне от 1100—2500 нм. Для обра-
ботки данных, калибровки и хранения данных необходим компьютер. Спектр пред-
ставляет собой разность между величиной исходного отраженного излучения для
образца и величиной исходного отраженного излучения для эталонного материала.
При использовании метода отражения поглощение рассчитывают как log 1/R, так
как поглощение эквивалентно — log10 (отражения). Исходное пропускание преобра-
зуют в поглощение при помощи выражения log 1/Т.
Для измерения пропускания применяется оптическая система, позволяющая
пропускать больше энергии в единичную оптоволоконную нить или в пучок свето-
проводящих волокон. Излучение передается по оптоволоконной нити или по пучку
светопроводящих волокон, затем падает на образец и возвращается к рассеивающе-
му элементу, т. е. к решетке. После попадания на решетку свет разделяется на два
пучка с различными длинами волн, а потом попадает в детектор (детекторы).
Интегрирующая сфера до сих пор является общепринятой геометрией измере-
ний при БИК-спектроскопии. Интегрирующие сферы (фотометрические шары)
были применены в первых коммерческих спектрофотометрах. Их главным преиму-
ществом является то, что излучение, попадающее в детектор, расположенный на
измерительном порте сферы, не теряет энергии. Однако по мере развития техно-
логии изготовления детекторов, преимущества интегрирующей сферы для нако-
пления энергии уже не имеют решающего значения. В отдельных особых случаях
все еще необходимо использовать сферы, соединенные с пучками оптоволоконных
нитей. Использование сферы позволяет проводить внутреннюю фотометрическую
калибровку — подобие двухлучевого прибора. Применение Фурье-спектрометров
Рис. 16. Решеточный спектрофотометр с предварительным монохроматором для измерений
диффузного отражения
456
Часть 4. Процессно-аналитические технологии
для БИК-спектрометрии началось в конце 1980-х гг. В настоящее время распростра-
нены Фурье-БИК анализаторы для спектроскопии пропускания и диффузионно-
отражательной спектроскопии. Как уже было упомянуто, Фурье-спектрометры
отличаются от сканирующих спектрометров тем, что регистрируемый сигнал пред-
ставляет собой интерферограмму (детали см. в разделе данной главы, посвященном
спектроскопии в средней ИК-области). В приборах других моделей используются
детекторы на основе диодной матрицы (рис. 17) и диодные источники, испускаю-
щие БИК. Акустооптические перестраиваемые фильтры (AOTF— acoustooptic tunable
filters) представляют собой устройства, основанные на дифракции. БИК-фильтр
фактически является прозрачным кристаллом, в котором генерируется ультразву-
ковое поле. Таким образом, выбранная длина волны является функцией напряжен-
ности поля. Скорость AOTF сканирования измеряется в микросекундах. Спектро-
метр такого типа считается быстродействующим, однако требует дополнительных
усилий для надежной работы в фармацевтической производственной среде. Доступ-
ны и другие приборы, такие как сверхбыстрые вращающиеся интерференционные
фильтры (ultrafast-spinning interference filter wheels), интерферометры с неподвижны-
ми зеркалами и перестраиваемые лазерные источники.
4.2.2.13. Заключение
В заключение отметим, что приборы для колебательной спектроскопии можно раз-
делить на категории по способу оптимизации оптических схем для образцов кон-
кретного типа и решения различных аналитических проблем. Для решения иссле-
довательских задач выбор, как правило, делают в пользу одного прибора с широким
спектром возможностей. Если четко определена аналитическая задача, достоверные
результаты могут быть получены при помощи прибора, специально приспособлен-
ного для измерений образцов специфической геометрии или определенного типа.
Тем не менее, на данном этапе следует отметить, что из-за специфики оптических
схем и способа измерений, перенос калибровок или аналитических методик с одно-
го прибора на другой может оказаться весьма проблематичным. Например, вопрос
переноса калибровок периодически возникает при БИК-спектроскопии и относит-
ся к приборам всех конструкций. Это важно при планировании проведения изме-
Глава 4.2. Процессно-аналитическая технология (РАТ)457
рений определенной характеристики образца в различных локациях различными
приборами, например, в лаборатории или в производственном процессе. Пробле-
ма становится серьезной при работе со сложными фармацевтическими образцами,
в которых сигнал аналита или измеряемая характеристика сильно зависит от значе-
ний других параметров.
Выбор типа спектроскопии — в средней ИК-области, БИК- или спектроско-
пии Рамана, имеет большое значение для совершенствования мониторинга и углу-
бления понимания фармацевтических процессов. В табл. 2 представлены главные
характеристики методик спектроскопии — в средней ИК-области, БИК- или ра-
мановской спектроскопии. Наиболее подходящую стратегию выбирают, исходя из
необходимости и требований технологических или аналитических операций. При
выборе методики спектроскопического исследования следует обратить внимание
на такие критерии, как химический состав образца, место проведения измерений,
скорость или возможность измерения в режиме реального времени, трудоемкость,
простота применения, способ подготовки образца, необходимость разрушения об-
разца и т. и. Многокомпонентный анализ требует уровней точности, сравнимых
с аналогичными уровнями для первичных стандартных методик. В наше время су-
ществует большое количество технологий и множество производителей. Имея это
в виду, при выборе оборудования следует учитывать наиболее существенные кри-
терии, необходимые для решения интересующих исследователей аналитических
проблем; в то же время следует помнить о пределах измерений или ограничениях
в применении.
Таблица 2
Сравнение методик ЛЯК/БИК, рамановской спектроскопии
Спектроскопия БИК ИК Рамановская
Спектральный диапазон (см~’) 12 000-4000 4000-400 4000-50
Интенсивность сигнала ++ +++ +
Микроскопический анализ Нет Да Да
Оптоволоконный интерфейс Да Да (ограничение длины) Да
Анализ через стекло Да Нет Да
Качественный анализ Да Нет Да
Количественный анализ Да Затруднено Да
Надежность прибора +++ + ++
Химическая интерпретация Хемометрика Прямая Прямая
4.2.3. Хемометрика
4.2.3.1. Введение
Термин «хемометрика» описывает химическую дисциплину, привлекающую мате-
матические методы и компьютерные технологии для извлечения информации из
данных различного типа, происхождения и степени сложности. Типичные спосо-
бы применения включают соотнесение величин концентрации анализируемого
458
Часть 4. Процессно-аналитические технологии
вещества, найденных в образце, со спектральными данными, или определение не-
которых физических или химических характеристик образца. Несмотря на то что
эффективное использование хемометрики невозможно без компьютера, базовые
принципы многомерных математических расчетов известны с начала двадцатого
века.
Ближняя ИК-спекгроскопия — типичная область применения хемометрики, на-
пример, для соотнесения БИК-спектров содержимого закрытой стерильной ампу-
лы, полученных без особых затрат, с концентрациями конкретного вещества в по-
рошкообразной смеси, полученной стандартным методом без открытия ампулы.
Чтобы достичь этого, следует найти взаимосвязь между двумя матрицами данных
и рассчитать количественную калибровку. Решение этой задачи имеет некоторые
особенности, в зависимости от того, как были получены данные: генерированы
при помощи статистически обоснованного планового эксперимента (т. е. спроек-
тированные данные — designed data) или просто собраны, более или менее случай-
но, из данной популяции (т. е. неспроектированные данные — nondesigned data).
Матрицы спроектированных данных ортогональны по определению. Традицион-
ные статистические методы, такие как вариационный анализ (ANOVA — analysis of
variance) и метод множественной линейной регрессии (МЛР, в английском варианте
MLR — multiple regression method), хорошо подходят для построения регрессионной
модели на таблицах ортогональных данных. Матрицы неспроектированных дан-
ных редко являются ортогональными, однако они более или менее коллинеарны.
В этих обстоятельствах наиболее вероятно, что MLR может оказаться не примени-
мой, и рекомендуется использовать проекционные методы, такие как регрессия на
главные компоненты (PCR — principal component regression) или проекция на латент-
ные структуры (ранее назывался методом частичных наименьших квадратов — PLS,
partial least squares). Регрессионная модель затем может быть использована для
прогнозирования характеристик новых, т. е. неизвестных образцов. Методы про-
гнозирования полезны, поскольку их можно использовать вместо дорогостоящих
и времязатратных измерений.
Другой пример — неразрушающая идентификация образцов по БИК спектрам
поглощения. Классификация просто представляет собой выяснение вопроса: при-
надлежат ли новые пробы к классам образцов, которые использовали для построе-
ния модели. Если новые образцы практически полностью соответствуют конкрет-
ной модели, считается, что они являются членами этого класса. К этой категории
относятся многие аналитические задачи. Например, исходные материалы, такие
как вспомогательные вещества, можно рассортировать на материалы «хорошего»
и «плохого» качества, готовую продукцию классифицировать на «категории А, В,
С» и так далее. Метод главных компонент (РСА — principal component analysis) —
типичная математическая процедура для разложения подобных наборов данных
в ортогональные компоненты, линейные комбинации которых приближаются
к оригинальным данным с требуемой степенью точности. Каждая из рассчитанных
последовательных компонент максимально объясняет остаточную дисперсию в на-
боре данных. При БИК-спектрометрии данные обычно представляют собой боль-
шие серии зарегистрированных спектров, и число компонент будет меньше или
равно числу известных переменных или количеству спектров.
Глава 4.2. Процессно-аналитическая технология (РАТ)
459
4.2.3.2. От одномерной регрессии к многомерной
В спектроскопии простейший метод количественной калибровки основан на одной
независимой переменной, например, длине волны, поскольку характеристики об-
разца, такие как концентрация анализируемого вещества, является линейной функ-
цией поглощения на определенной длине волны. Моделирование концентрации
требует подгонки по методу наименьших квадратов. Прямую линию проводят через
информационные точки для минимизации суммы квадратов отклонений между оце-
ночными и известными информационными точками. При данном подходе длину
волны выбирают при высокой степени корреляции между концентрацией и погло-
щением. Корреляция является индикатором критерия согласия между концентра-
цией и поглощением и того, насколько хорошо калибровка описывает набор дан-
ных. Линейные отношения имеют практическое преимущество в том, что позволяют
проводить прямую и визуальную оценку критериев согласия, что укрепляет доверие
аналитика к собранным данным. Если речь идет о фармацевтических образцах,
линейный подход оказывается весьма ограниченным, и требуется другой подход.
Множественная линейная регрессия позволяет провести калибровку методом
линейной регрессии для одной длины волны методом наименьших квадратов с ис-
пользованием нескольких длин волн. Сложность исследуемых образцов должна
учитываться при данном подходе. Для объяснения набора данных метод МЛР тре-
бует наличия независимых переменных. Поскольку фармацевтические образцы об-
ладают сложным составом, в котором молекулы в разной мере взаимодействуют
в БИК- диапазоне, невозможно выбрать подходящие длины волн. Наблюдаемые ве-
личины поглощения связаны и могут описывать связанные характеристики в набо-
ре спектральных данных. Для БИК-спектров смесей типична коллинеарность среди
длин волн, а метод МЛР никогда не даст приемлемой линейной калибровки.
При методе проекции на латентные структуры с многомерной регрессией (PLC-
регрессия) используется множество длин волн без ограничения по коллинеарности.
Фактически коллинеарность делает PLSметодом выбора при рассмотрении сегмен-
тов или полного БИК-спектра. PLS рассматривает данные составляющих элемен-
тов и спектральные данные на одной основе, без предварительных статистических
условий о стандартном распределении или неколлинеарности. В обоих наборах дан-
ных погрешности оценивают одинаково. Спектральные данные и данные составля-
ющих элементов моделируют одновременно согласно повторяющемуся алгоритму.
Этот этап обучения необходим для того, чтобы модель могла прогнозировать иссле-
дуемые характеристики образцов. Для каждой итерации часть спектральных данных
и часть соответствующих данных составляющих элементов комбинируют до опти-
мального описания набора данных. Необъясненная часть набора данных состоит
из остатков, являющихся мерой качества модели. Исходные данные комбинируют
в множители или основные компоненты. Рассчитывают коэффициенты — нагруз-
ки для спектральных данных и счета для данных составляющих элементов — чтобы
указать степени участия исходных данных в вычислении каждого фактора. Нако-
нец, моделированную дисперсию, т. е. объясненную часть данных, максимизируют
для каждого фактора, а остатки минимизируют.
Исходные данные обычно подвергают предварительной обработке перед PLS
калибровкой, как показано далее. Критическим этапом построения PLSмодели яв-
460
Часть 4. Процессно-аналитические технологии
ляется выбор числа факторов. Слишком малое число факторов приведет к неадек-
ватному объяснению вариабельности обучающеей системы, в то же время слишком
большое число факторов является причиной чрезмерно близкой подгонки и неста-
бильности полученной калибровки. Оптимальное число факторов определяется при
валидации калибровки, являющейся частью PLS алгоритма. Используют дополни-
тельный и независимый набор данных (внешняя валидация в один вычислительный
этап) или же разделяют обучающий набор данных на группы для постоянной вну-
тренней валидации на каждом итерационном шаге (кросс-валидация). Кроме того,
необходимо доказать адекватность полученной PLS калибровки д ля прогноза пара-
метров неизвестных образцов. Хорошей практикой является проверка полученной
калибровки, увеличивающая доверие к будущим результатам. PLS калибровка ни
в коей мере не является черным ящиком, несмотря на то что по сравнению с базо-
вой линейной регрессией ее визуализация далека от очевидной.
4.2.3.3. Качество проб и погрешность данных
В идеальном случае результирующее калибровочное уравнение позволяет получить
значение концентрации для неизвестного анализируемого вещества исключительно
в терминах измеряемых величин поглощения. Для калибровки необходимо знать
только концентрации исследуемого вещества. В измерении значений других со-
ставляющих образца нет необходимости, хотя для получения правильного калибро-
вочного уравнения вариации этих величин тоже должны быть распределены равно-
мерно. В действительности, в отдельных случаях даже число других компонентов
в калибровочных образцах может быть не известно. При помощи пошаговой мно-
жественной регрессии характеристики спектра, наиболее близко коррелирующие
с концентрацией анализируемого вещества, выбирают для конкретного набора дан-
ных. После расчета оптимальных калибровок БИК инструмент можно использовать
для прогноза количества исследуемого компонента в неизвестных образцах. Таким
образом, анализ регрессии представляет собой метод выявления соотношений (т. е.
составления калибровочного уравнения регрессии) между рядом спектральных ха-
рактеристик и исследуемой составляющей. Для измерений диффузного отражения
влияния внешних физических явлений накладываются на данные поглощения, и их
следует принимать во внимание. Можно составить уравнения, учитывающие эти
явления, неявно включающие поправки на их влияние на калибровочные коэффи-
циенты.
Одна из проблем хемометрики состоит в том, что она не учитывает методики
выборки или подготовки проб. Тщательная выборка образцов повышает вероят-
ность извлечения полезной информации из спектральных данных. Когда есть воз-
можность экспериментировать с выбранными переменными или параметрами,
качество результатов повышается. Критически важной частью является принятие
решений, о том, какие переменные можно изменять и какие установить пределы
для вариаций. Понимание того, какие параметры могут влиять на данные, зависит,
главным образом, от квалификации оператора БИК, но это тема обсуждения для
специалистов. Каждая проблема генерирует и включает специфический набор более
или менее приемлемых данных. Ключ к успеху лежит в получении опыта в решении
ряда разнообразных проблем. Экспериментальные модели могут идеально помочь
Глава 4.2. Процессно-аналитическая технология (РАТ)
461
в генерировании данных, указывающих на влияние параметров или исходных пере-
менных на получаемый сигнал или конечные переменные.
Понимание взаимодействий между исходными переменными при определении
оптимальных условий извлечения релевантной информации должно в принципе
минимизировать количество необходимых экспериментов, что снижает стоимость
исследования. Программа для планирования эксперимента предлагает соответству-
ющие методы планирования эксперимента и способствует внедрению надлежащей
исследовательской практики путем проведения небольшого числа полезных экспе-
риментов, включающих важные вариации. Здесь не будет обсуждаться планирова-
ние эксперимента, а лишь только способ сбора и организации данных на практике
для построения многомерной регрессии. Как правило, при работе со спектральны-
ми данными проблематично обнаружить, какие переменные действительно суще-
ственны для вариаций в матрицах данных. Типичной задачей при определении ко-
нечной переменной становится поиск переменных, необходимых для адекватного
описания образцов, поиска аналогичных образцов и групп образцов внутри набора
данных. Хороший способ поиска информации состоит в разложении матрицы спек-
тральных данных на структурированную часть и шум, как правило, с применением
РСА. Другая проблема — построение модели регрессии между матрицей спектраль-
ных данных и матрицей переменных отклика.
Перед проведением многомерного анализа данных, полезным для проверки
качества данных может оказаться статистический анализ значений реакции об-
разца или спектральных данных. Описательная статистика суммирует распреде-
ление одной или нескольких переменных одновременно. Предполагается, что они
не раскрывают подробную информацию о структуре данных, но они полезны для
быстрого рассмотрения каждой отдельной переменной перед началом анализа.
Одностороннюю статистику, т. е. определение среднего, стандартного отклонения,
колебаний, медианы, минимума, максимума, нижнего и верхнего квартиля, можно
использовать для исследования набора данных и обнаружения величин вне диапа-
зона, аномального разброса или асимметрии. Статистика выявляет сомнительные
моменты в таблицах данных и указывает на необходимость преобразований. Дву-
сторонняя статистика, например, корреляция, показывает, как вариации двух пере-
менных могут быть связаны в таблице данных. Проверка статистики также полезна
для выявления величин, выпадающих из заданного диапазона, и выбросов.
Практический опыт БИК-спектроскопии показывает, что критические шаги для
успешного применения варьируют в зависимости от каждой конкретной проблемы.
Тем не менее, следующая процедура рекомендована при анализе незапланированных
данных (nondesignated data). В первую очередь нужно исследовать происхождение
и доступность данных. При формулировании изучаемой проблемы следует опреде-
лить точную цель сбора данных и предполагаемые результаты анализа. Собранные
данные должны охватывать соответствующую вариацию (вариации) или характе-
ристику (характеристики). Если доступные данные не включают предполагаемых
вариаций, приготовить или проанализировать образцы с соответствующими экспе-
риментальными характеристиками. Возможно, потребуется трансформация и ма-
тематическая обработка исходных спектральных данных. Калибровку и валидацию
модели следует проводить при помощи РСА или PLS. Исследование и испытание спо-
462 Часть 4. Процессно-аналитические технологии
собов калибровок проводится на реальных образцах или данных, например, валида-
ция метода согласно современным требованиям и фармацевтическому применению.
Существует три типа погрешности данных: случайная погрешность значений
эталонных образцов в лаборатории, случайная погрешность оптических данных
и систематическая погрешность соотношений между двумя величинами. Надлежа-
щий подход к погрешностям данных зависит от того, являются ли поврежденные
переменные параметры эталонными величинами или спектроскопическими данны-
ми. Калибровки обычно проводятся эмпирически и являются специфическими для
конкретной проблемы. В этой ситуации вопросы, связанные с погрешностью дан-
ных, приобретают важное значение. Однако трудно решить, является ли спектро-
скопическая погрешность больше, чем погрешность эталонной лабораторной мето-
дики, или наоборот. Уровень шума у применяемого БИК прибора обычно меньше,
чем практически любой другой фактор при калибровке. Суммарная погрешность
спектроскопических данных включает погрешности, вносимые образцом, которые
могут значительно превышать уровень шума прибора. Подобные погрешности вы-
званы влиянием размера частиц, вариациями плотности размещения компонентов
в порошкообразных образцах, влиянием примесей и эффектом, вызванным изме-
нением физических характеристик пробы (например, кристаллического строения).
Практический опыт показывает, что во многих случаях погрешность оптических
данных, привнесенная образцом, не велика, что указывает на то, что эмпирическая
оценка погрешности оптических данных всегда меньше, чем погрешность лабора-
торного стандартного образца.
4.2.3.4. Предварительная математическая обработка данных
спектроскопии
Как сказано ранее, самым большим источником погрешности при калибровке
БИК-спектроскопического оборудования обычно является погрешность эталонной
лабораторной методики, неоднородность образца и нерепрезентативная выборка
в обучающем множестве или популяции набора калибровочных данных. Качество
оборудования и выбор статистической модели лишь частично ответственны за ва-
риации и погрешность, связанные с БИК-аналитическими приборами при текущем
рутинном применении.
При отражении излучения от твердой поверхности происходит наложение рас-
сеянного и отраженного излучений. Энергия диффузно отраженного излучения за-
висит от углов падения и падения, а также от плотности упаковки образца, кристал-
лической структуры, показателя преломления, распределения частиц по размерам
и адсорбционных характеристик. Таким образом, на практике идеальная диффузно
отражающая поверхность существует лишь в приближении, даже при тончайшем
измельчении материала проб. Всегда существуют когерентно отражающие зоны
поверхности, действующие как элементарные зеркала, отражение от которых под-
чиняется формулам Френеля. Излучение, возвращающееся к поверхности образца
извне можно считать в приближении изотропическим, что соответствует требова-
ниям закона Ламберта—Бэра. Предположим, что излучаемая энергия постоянно
отводится от падающего пучка БИК-излучения и преобразуется в термическую
колебательную энергию атомов и молекул. Уменьшение интенсивности диффузно
Глава 4.2. Процессно-аналитическая технология (РАТ)
463
рассеянного света зависит от коэффициента поглощения образца. Коэффициент
поглощения (К), представленный в виде отношения К/S, где Л1— коэффициент рас-
сеяния, пропорционален количеству поглощающего материала в образце. Согласно
теории Кубелки—Мунка, величины отражения (R), поглощения (К) и коэффициен-
та рассеяния (5) связывает уравнение
S 2R
Диффузное отражение R является функцией соотношения К/S и пропорцио-
нально количеству поглощающих веществ в отражающей среде образца. В практике
БИК-спектроскопии величину абсолютного отражения R заменяют отношением
интенсивности излучения, отраженного от образца, к интенсивности излучения,
отраженного от эталонного материала, то есть керамического диска. Таким образом,
R зависит от концентрации анализируемого вещества. Предположение о том, что
диффузное отражение падающего излучения прямо пропорционально количеству
поглощающих веществ, взаимодействующих с падающим излучением, основано
на этих соотношениях. Подобно закону Бэра, уравнение Кубелки—Мунка ограни-
чено для слабых поглощений, таких как поглощения, наблюдаемые в БИК диапа-
зоне. Тем не менее, на практике нет необходимости в предположении о линейных
отношениях между данными БИК-спектроскопии и концентрацией компонента,
поскольку преобразования или предобработка данных используются для линеари-
зации данных по отражению. Чаще всего осуществляют линейные преобразования
через log 1/R и математическую предобработку по методу Кубелки—Мунка. До не-
которой степени PCR, PLSh полилинейная регрессия компенсируют нелинейность.
Калибровочные уравнения можно вывести для компенсации в некоторой мере не-
линейности отношений между концентрациями анализируемого вещества и log 1/Д
или данными, преобразованными по алгоритму Кубелки—Мунка. Если матрица по-
глощает на длинах волн, отличных от длин волн, соответствующих анализируемому
веществу, метод Кубелки—Мунка может оказаться полезным методом линеаризации
спектроскопических данных. Если матрица поглощает на той же длине волны, что
и анализируемый материал, практика показывает: выбор следует сделать в пользу
преобразования log 1/R, которое лучше отражает зависимость отражения от кон-
центрации. Это преобразование, в основном, хорошо изучено на БИК-спектрах
диффузного рассеяния большинства смесей с поглощающими матрицами. Графи-
ки зависимости F(R) от концентрации менее линейны, чем графики зависимости
log 1/R от концентрации.
4.2.3.5. Предварительная обработка данных БИК-спектроскопии
При выводе калибровочных уравнений при помощи образцов известных составов
независимую переменную представляют спектроскопические данные (т. е. log 1/R)
на определенных длинах волн, в то время как концентрация исследуемого вещества,
для определения которой используют традиционные лабораторные методики, явля-
ется зависимой переменной.
Распределение или форма исходных спектральных данных может иметь рас-
пределение или форму, не оптимальную для анализа. Влияние фона, сдвиг базовой
464
Часть 4. Процессно-аналитические технологии
линии, измерения в различных условиях, различные колебания взаимозависимых
переменных и т. п. могут усложнить извлечение информации с использованием ме-
тодов многомерного анализа. Важно минимизировать уровень шума, создаваемый
подобными эффектами. Предварительная обработка данных включает такие опера-
ции, как центрирование, взвешивание и многочисленные математические преоб-
разования. Центрирование заключается в вычитании среднего спектра из каждого
индивидуального спектра. Эта операция обеспечивает возможность интерпретации
всех результатов с точки зрения вариации вокруг среднего, рассматриваемого в ка-
честве центра модели. Это полезно при работе с моделями, в которых график ли-
нейной зависимости между спектральными данными и данными отклика проходит,
как предполагается, через начало координат. В зависимости от типа информации,
которую нужно извлечь из спектральных данных, для градуировки можно восполь-
зоваться весами измеренных величин, которые вычисляют на основании значений
стандартного отклонения (т. е. квадратный корень из дисперсии, где дисперсия вы-
ражена в тех же единицах, что и исходная переменная). Эта операция может являть-
ся типичной предобработкой при РСА, PLS или PCR калибровках, которые являют-
ся проекционными методами, основанными на поиске направлений максимальных
вариаций, и зависят, таким образом, от относительной дисперсии переменных.
Одним из способов взвешивания является 1/5D стандартизация, при которой дис-
персия всех переменных одинакова и составляет 1. В этом случае все переменные
в равной степени влияют на оценку компонентов. Этот путь можно рекомендовать,
если переменные измеряются в разных единицах, имеют разные ранги или относят-
ся к разным типам. Возможно также фиксировать постоянный вес для каждой пере-
менной вручную. Взвешивание включает расширение и сжатие данных путем из-
мерения положений относительно экстремумов в реальной таблице спектральных
данных. При стандартизации спектров вариации в наборах данных оцениваются от-
носительно экстремумов в таблицах данных. Тем не менее, эта процедура усиливает
относительное влияние недостоверных или связанных с шумом характеристик.
4.2.3.6. Математическая обработка и преобразования
Перед анализом спектральных данных их трансформируют; существует ряд алго-
ритмов преобразования данных. Главная цель преобразований — сделать скрытые
переменные доступными полнофункциональному анализу. Одним из наиболее
широко используемых является логарифмическое преобразование, особенно при-
годное для получения более симметричного распределения асимметричных пере-
менных. Кроме того, такие преобразования показаны, когда ошибка измерения
переменной увеличивается пропорционально уровню данной переменной. Лога-
рифмирование позволит добиться равномерной прецизионности во всем интервале
вариаций. Этот вид преобразований называется также стабилизацией дисперсии.
При наличии ограниченной асимметрии достаточно извлечения квадратного кор-
ня. Сглаживание подходит для переменных, которые сами являются функциями
соответствующих переменных, например, времени. Это также одна из первых опе-
раций, проводимых с данными БИК-спектров. При этом удаляется максимальный
объем шума без нарушения важного информационного контента. Сглаживание
методом скользящей средней (МРА — moving point average) — классический метод
Глава 4,2. Процессно-аналитическая технология (РАТ}
465
сглаживания, в котором каждый результат наблюдения заменен усредненной ве-
личиной смежных результатов наблюдений, включая рассматриваемый результат.
Число усредняемых наблюдений является переменным параметром. Полиноми-
альное сглаживание, называемое также сглаживанием Савицкого—Голея, включает
подбор полиномиального уравнения по методу наименьших квадратов для окна из
п последовательных информационных точек спектральных данных. Если порядок
полинома меньше, чем число информационных точек, полином не проходит через
все выбранные информационные точки, а подгонка по методу наименьших квадра-
тов дает сглаженное приближение к исходному окну. Нормализация — семейство
преобразований, которые рассчитываются по отношению к образцу. Здесь ставит-
ся задача улучшения данных по специфическим характеристикам. Нормализация
по среднему является классическим алгоритмом. Метод заключается в делении
каждого ряда матрицы данных на его среднее значение, при этом нейтрализуется
влияние возможных скрытых факторов. Операция эквивалентна замене исходных
переменных профилем, центрированным вокруг 1. Для описания образца исполь-
зуются только относительные значения переменных, а информацию, которую несут
их абсолютные значения, опускают. Преобразование показано в отдельном случае,
где все переменные измерены в одинаковых единицах, и их значения пропорцио-
нальны некому фактору, который невозможно непосредственно принять в расчет
при анализе. Альтернативной процедурой является нормализация по максимуму —
деление каждого ряда на максимальное абсолютное значение. Максимальное полу-
ченное значение становится +1, а минимальное —1. При нормализации по рангам,
каждый ряд делят на соответствующий ранг, т. е. разность максимального и мини-
мального значения, тогда размах кривой (curve span) становится равным 1.
Более специфическими для спектроскопических данных преобразованиями яв-
ляются преобразования поглощения в отражение, отражения в поглощения и по-
глощения по алгоритму Кубелки—Мунка. Мультипликативная коррекция рас-
сеяния (MSC — Multiplicative scatter correction) является дополнительным методом
преобразования данных, применяемым для компенсации аддитивных и/или муль-
типликативных эффектов в спектральных данных. Изначально метод MSC был
разработан исключительно для решения задачи мультипликативного рассеяния.
Однако MSC с успехом применяется для решения аналогичных задач, связанных
с влиянием длины пути, сдвигами смещения и интерференцией. Принцип MSC за-
ключается в удалении двух эффектов — амплификации, являющейся мультиплика-
тивным эффектом, и смещения, аддитивного эффекта — из таблиц спектральных
данных для предотвращения их доминирования над информационным контентом
таблиц. Дифференцирование обычно применяется для обработки спектральных
данных, являющихся функцией основной переменной, влияющей на поглощение
при разных длинах волн. Дифференцирование является простой и эффективной
методикой выявления тонкой структуры в структуре исходного спектра, что харак-
терно для БИК-спектроскопии. При повышении порядка дифференцирования,
увеличивается разрешение полос. Недостатком метода является увеличение уровня
сигнал-шум, которое не рассматривается в частном случае БИК-спектра, посколь-
ку сглаживание при дифференцировании уменьшает влияние шума. С другой сто-
роны, при этом могут оказаться скрытыми некоторые более слабые спектральные
466
Часть 4. Процессно-аналитические технологии
характеристики. Главным преимуществом дифференцирования второго порядка
состоит в сохранении структуры полосы: максимум пика не сдвигается, в то время
как форма пика и разрешение улучшаются. Алгоритм Савицкого—Голея позволяет
проводить дифференцирование высших порядков, включая фактор сглаживания,
определяющий, какое количество смежных переменных следует использовать для
полиномной аппроксимации при дифференцировании. Альтернативным алгорит-
мом для вычисления только первых производных является получение производной
Норриса. Алгоритм коррекции фоновой линии — метод стандартного нормального
распределения (SNV — standard normal variate), не влияющий на общую спектраль-
ную картину. Усреднение образцов при наличии репликатов или переменных, под-
лежащих сокращению, например, для уменьшения числа скрытых переменных, яв-
ляется методом получения более стабильных результатов, которые легче поддаются
обработке. На рис. 18 представлен эффект от выбранных комбинированных преоб-
разований на массив спектральных данных.
4.2.3.7. Метод главных компонент
Большие таблицы данных содержат частично скрытую информацию, посколь-
ку сложность данных не позволяет легко их интерпретировать. Это типично для
данных по БИК-спекграм. Метод главных компонент {РСА — principal component
analysis) представляет собой проекционный метод, применяемый для визуализации
всей информации, содержащейся в таблицах данных. Метод можно использовать
для демонстрации характеристик, по которым образцы отличаются друг от друга;
выявления переменных, вносящих наибольший вклад в эти различия; определе-
ния направления воздействия этих переменных, выявление наличия корреляций
между переменными или их независимости. Кроме того, метод позволяет распозна-
вать паттерны или группы образцов. Также метод определяет количество полезной
информации, в противовес шуму или незначительным вариациям, содержащимся
в таблице данных. Главные компоненты определяются только для набора данных,
по которому они были рассчитаны. Они могут оставаться актуальными для других
наборов данных аналогичного типа, однако это нельзя гарантировать, и, разумеет-
ся, не соответствуют наборам данных другого типа.
Моделирование РСА формирует основу для нескольких классификаций и мето-
дов регрессионного анализа. Основной принцип метода — замена сложного много-
мерного набора данных на более простую версию с меньшим числом измерений,
которая, тем не менее, достаточно близка к исходному набору данных и может счи-
таться близкой аппроксимацией. Извлечение информации из таблицы данных за-
ключается в изучении вариаций между образцами, т. е. поиска характеристик, по
которым образцы различаются или, наоборот, совпадают друг с другом. Два образца
можно считать одинаковыми, если большинство переменных для них имеют близкие
значения. С точки зрения геометрии, в случае близости координат в многомерном
пространстве переменных можно сказать, что две точки находятся в одной области.
Аналогично, два образца считаются разными, если их значения сильно различают-
ся, по меньшей мере, по нескольким переменным. В многомерном пространстве
они выглядят как две точки с разными координатами, расположенными далеко друг
от друга. Иллюстрация геометрических принципов РСА представлена на рис. 19.
Глава 4.2. Процессно-аналитическая технология (РАТ)
467
SNVD + вторая производная
Стандартное нормальное
распределение (SNV)
Рис. 18. Примеры комбинированной предобработки спектральных данных
Основной принцип РСА состоит в поиске пространственных направлений —
главных компонент (PC — principal components), по которым информационные точ-
ки отстоят друг от друга максимально. Для этого необходимы линейные комбина-
ции исходных переменных, вносящих наибольший вклад, в отличие образцов друг
от друга. PC вычисляют итеративно, причем первая PC несет наибольший объем
информации, т. е. является наиболее объясненной переменной; вторая PC несет
наибольшую часть остаточной информации, не учтенной первой PC, и т. д. Про-
цесс может продолжаться до тех пор, пока число рассчитанных PC не сравняется
с числом потенциальных переменных в таблице. В этой точке будут учтены все ва-
риации между образцами, а PC образуют новую систему координат, обладающую
двумя преимуществами по сравнению с первой. Во-первых, PC ортогональны по
отношении друг к другу. Во-вторых, они ранжированы так, что каждая последую-
щая компонента несет больше информации, чем любая из последующих. Таким об-
разом, устанавливают приоритеты интерпретации, начиная с первой PC. Этот метод
преобразований позволяет перейти к новой системе координат, более пригодной
для интерпретации структуры данных. Обычно только первая PC несет релевант-
ную информацию, последующие PC с большей вероятностью описывают шум. На
практике исследуют только первую PC вместо всей таблицы исходных данных: это
менее сложно; кроме того, это гарантирует, что шум не будет ошибочно принят за
релевантную информацию. Если бы были исследованы все PC, то это означало бы
468
Часть 4. Процессно-аналитические технологии
с
Рис. 19. Геометрическая иллюстрация РСА. Каждый спектр нанесен на график в виде точки,
координаты которой представляют величины интенсивности, измеренные на трех разных длинах
волн. Для данной совокупности спектров первой главной компонентой является направление
в пространстве, охватывающее самую большую вариацию соответствующего набора данных
отсутствие аппроксимации и упрощения. Выбор числа компонентов, исследуемых
в модели РСА, является компромиссом между простотой, полнотой и эффективно-
стью. Модель РСА является всего лишь приближением к реальной картине.
Каждый компонент модели PG4 характеризуется тремя комплементарными на-
борами характеристик, т. е. дисперсией, нагрузками и счетами соответственно. Зна-
чимость данной PC выражается через дисперсию. Нагрузки описывают отношения
между переменными, а счета — свойства образцов. Дисперсия представляет меру
погрешности, которая показывает, какой объем информации учтен в последующих
PC. Их вариации вместе с числом компонентов можно изучать, чтобы определить,
насколько сложной должна быть модель. Остаточная дисперсия указывает на вари-
ации данных, остающиеся не объясненными при рассмотрении определенной PC,
в то время как объясненная дисперсия, часто измеряемая в виде процентной доли от
общей дисперсии данных, измеряет долю вариаций в данных, учтенных рассматри-
ваемой PC. Эти дисперсии можно учитывать как для одной переменной или одного
образца, так и для данных в целом. Их рассчитывают как среднеквадратичные от-
клонения, скорректированные для числа оставшихся степеней свободы.
Нагрузки описывают структуру данных в терминах переменной корреляции.
Каждая переменная несет нагрузку на каждую главную компоненту. Она отража-
ет вклад переменной в данную PC, и то, насколько хорошо PC отражает вариации
Глава 4.2. Процессно-аналитическая технология (РАТ}
469
рассматриваемой переменной. В терминах геометрии нагрузка представляет собой
косинус угла между переменной и рассматриваемой PC, со значениями в диапазоне
от —1 до +1. Чем меньше угол, тем больше связь между переменной и PC, и тем
больше величина нагрузки. Переменные с большими нагрузками (то есть близкими
к +1 или —1) для определенной PC вносят большой вклад в значение этой конкрет-
ной PC. Таким образом при анализе корреляций между переменными нагрузки ука-
зывают на соответствующие им углы в многомерном пространстве. Например, если
две переменные имеют высокие нагрузки вдоль одной PC, то образуемый ими угол
мал, что означает, что между двумя переменными существует высокая корреляция.
Если обе нагрузки имеют одинаковый знак, то корреляция положительна; если уве-
личивается одна переменная, то же происходит и с другой переменной. Если корре-
ляция негативна, то переменные антикоррелированы.
Счета описывают структуру данных в терминах паттернов образцов и подчерки-
вают сходства и различия. Каждому образцу соответствует счет в каждой PC, кото-
рый представляет координату образца в PC. Если информация, которую несет PC,
интерпретирована при помощи нагрузок, для описания характеристик данного об-
разца можно воспользоваться счетом образца вдоль данной PC. Счет характеризует
основные свойства образца относительно переменных с высокими нагрузками по
той же PC. Образцы с близкими счетами вдоль одних и тех же PC считаются анало-
гичными, если близки их значения для соответствующих переменных. И наоборот,
образцы с сильно различающимися счетами значительно отличаются друг от друга
по значению этих переменных.
4.2.3.8. Применение метода главных компонент в БИК-спектроскопии
Как уже упоминалось, любой многомерный анализ включает валидацию, т. е. фор-
мальное тестирование возможности экстраполяции модели на новые аналогичные
данные. Эта операция требует двух раздельных этапов при вычислении компонен-
тов для каждой модели: калибровки, заключающейся в поиске новых компонентов,
и валидации, проверяющей, насколько адекватно рассчитанные компоненты опи-
сывают новые данные. Для каждого этапа необходим собственный набор образцов:
калибровочные или обучающие образцы и валидационные или контрольные образ-
цы. Расчет главных компонент спектроскопических данных основан исключитель-
но на оптических данных. Не существует явных или формальных отношений между
PC и составом образцов в наборах, где измерялись спектры. Кроме того, PC счи-
таются выше по сравнению с исходными спектральными данными, полученными
непосредственно при помощи БИК-инструмента. Поскольку несколько первых PC
содержат полосы шума, они представляют реальные вариации спектров, в большей
части вызванные физическими или химическими явлениями. Поэтому PC можно
рассматривать как латентные переменные, в противовес непосредственно измеряе-
мым явным переменным.
Удобную аналогию для понимания смысла латентных переменных представля-
ет реконструкция спектра из смеси спектров чистых химических веществ, содер-
жащихся в смеси. Спектры этих чистых веществ являлись бы латентными пере-
менными измеренного спектра, поскольку в смеси спектров их нельзя наблюдать
непосредственно. Тем не менее, PC не обязательно являются спектрами чистых
470
Часть 4. Процессно-аналитические технологии
химических веществ в смесях, представляющих образцы. PC представляют все не-
зависимые явления, воздействующие на спектры образцов, составляющих калибро-
вочный набор. Если одна составляющая образца варьирует полностью независимо
от всех других составляющих и имеет свой собственный спектр, то одна из PC будет
представлять спектр этой составляющей. Вариации составляющей, абсолютно неза-
висимые от вариаций любой другой переменной, весьма необычны. Определенная
корреляция между различными составляющими набора данных обязательно суще-
ствует, и любая PC будет представлять суммарный эффект этих коррелированных
составляющих. Даже если достигнута полная независимость, то зависимость со-
стоит в том, что сумма всех составляющих должна равняться 100%. Следовательно,
PC, представляющая источник независимой вариабельности, будет выглядеть как
разность между исследуемой составляющей и всеми другими составляющими об-
разцов. Спектр рассматриваемой составляющей можно было бы вычесть математи-
чески, но PC не будут в точности представлять спектр чистой составляющей.
После отбора образцов и получения соответствующих БИК-спектров, сохраняе-
мых в надлежащих файлах, построение и использование модели РСА проводится
в три этапа: выбор надлежащей процедуры (процедур) предварительной обработки
данных, подбор алгоритма РСА и диагностика модели, интерпретация графиков на-
грузок и счетов. Когда модель построена, важно оценить ее качество перед приме-
нением для интерпретации данных. Диагностика модели проводится в два этапа.
Необходимо проверить дисперсии для определения числа компонентов, которые
должна включать модель, и оценки объема информации, которую должны учиты-
вать выбранные компоненты. Важно провести верификацию дисперсий, рассчи-
танных в процессе валидации. Далее рекомендуется провести поиск выбросов, не
соответствующих общему паттерну.
Суммарная объясненная дисперсия показывает, какую часть исходной вариации
данных описывает модель. Она отражает долю структуры, обнаруженной в данных
при помощи модели. Суммарные остаточная и объясненная дисперсии показывают,
насколько хорошо модель описывает данные. Модели с малой суммарной остаточ-
ной дисперсией (близкой к 0) или суммарной объясненной дисперсией, близкой
к 100%, объясняют большую часть вариаций в данных. В простых моделях остаточ-
ная дисперсия падает до нуля после выделения нескольких компонент. Если тако-
го падения не происходит, это означает, что в данных присутствует большое коли-
чество шума. Альтернативно, это может означать, что структура данных слишком
сложна и не может быть описана лишь несколькими компонентами. Переменные
с малой остаточной дисперсией и большой объясненной дисперсией для определен-
ного компонента хорошо объяснимы с помощью релевантной модели. Переменные
с большой остаточной дисперсией для всех или нескольких первых компонентов
мало или умеренно связаны с другими переменными. Если остаточные дисперсии
некоторых переменных намного превышают остаточные дисперсии других пере-
менных для всех компонентов или первых компонентов, то их можно исключить из
новых расчетов. Это поможет построить модель, более легкую для интерпретации.
Калибровочная дисперсия основана на соответствии калибровочных данных моде-
ли. Валидация дисперсии рассчитывается путем тестирования модели по данным,
не используемым при построении модели.
Глава 4.2. Процессно-аналитическая технология (РАТ)
471
Иногда большая остаточная дисперсия может быть вызвана выбросами. Вы-
бросом является образец, который отличается от других настолько, что либо плохо
описывается моделью, либо слишком сильно влияет на модель. На практике ис-
тинные выбросы в спектральных данных в первую очередь рассматриваются как
образцы, спектральные характеристики которых не представлены в определенном
наборе образцов. По меньшей мере один из компонентов модели может сфоку-
сироваться на попытках описания только этого конкретного образца, даже если
это не относится к более важным структурам, присутствующим в других образцах.
В PG4 выбросы можно обнаружить путем использования различных графиков
или анализом. Например, графики счетов демонстрируют паттерны образцов по
одному или двум компонентам. По ним легко выделить компонент, лежащий в от-
далении от других. Такой образец, вероятно, представляет собой выброс. Остат-
ки являются мерой того, насколько образцы и переменные соответствуют модели,
определяемой компонентами. Образцы с высокими остатками плохо описывают-
ся моделью, которая, тем не менее, описывает другие образцы довольно хорошо.
Подобные образцы выделяются среди образцов, хорошо описываемых моделью,
и могут считаться выбросами. Решение об исключении или сохранении подоб-
ных спектральных выбросов, основанное исключительно на математических кри-
териях, не всегда правильное. Выброс можно считать образцом, не относящимся
к группе, составляющей калибровочный набор. Тем не менее, такие выбросы мо-
гут указывать на дополнительные характеристики, не принятые во внимание при
первоначальном выборе образцов.
4.2.3.9. Распознавание паттернов
Метод распознавания паттернов можно классифицировать в соответствии с разли-
чиями между методами классификации «с обучением» и «без обучения». Методы
«без обучения», такие как кластерный анализ, классифицируют данные без кали-
бровки и основаны только на собранных данных по образцам. Классификации «с
обучением» используют спектральные данные и информацию о принадлежности
к классу. Поэтому на первом этапе математические модели рассчитывают при по-
мощи калибровочного набора, включающего информацию о спектрах и классе об-
разцов. Затем данную модель применяют для прогнозирования новых классов об-
разцов. Методы выделения характерных признаков, такие как РСА или рекурсивное
сжатие зачастую применяют перед кластерным анализом. РСА является ценным ин-
струментом выделения характерных признаков или визуализации набора данных.
Другое преимущество РСА — снижение числа длин волн. Здесь применяются мно-
гие алгоритмы кластерного анализа. Неиерархические методы включают модели
гауссовых смесей, алгоритм К-средних и нечетких С-средних, каждый из которых
включает формальные и содержательные методы кластеризации. Например, при
формальной кластеризации по алгоритму Л'-средних один данный объект будет от-
несен к только одному классу (кластеру), в то время как при содержательной кла-
стеризации по алгоритму нечетких С-средних будет определена частичная степень
принадлежности объекта к каждому кластеру.
В настоящее время существует множество методов распознавания паттернов
«с обучением». Типичными являются методы линейного дискриминантного ана-
472 Часть 4. Процессно-аналитические технологии
лиза (LDA — linear discriminant analysis), основанные на расчете расстояния, фор-
мальное независимое моделирование аналогий классов (SIMCA — soft independent
modeling of class analogy), сосредоточивающееся на схожести объектов внутри класса,
РГУ-дискриминационный анализ (дискриминационный анализ с помощью ре-
грессии на латентные структуры) (PLS-DA: PLS-discriminant analysis) — метод по-
строения регрессий между спектрами и принадлежностями к классу. Более передо-
вые методы, такие как метод нейронных сетей, основаны на нелинейных подходах.
Применение параметрических и непараметрических методов вычисления — допол-
нительные отличительные признаки разных подходов. В параметрических методах,
таких как LDA, статистические параметры нормального распределения образцов
используются в правилах принятия решений. Подобных ограничений не содержат
непараметрические методы, такие как метод SIMCA, более эффективно применяе-
мый при обработке массивов данных БИК-спектроскопии.
4.2.3.10. Классификация SIMCA
Классификация полезна, если рассматриваемый отклик является категориальной
переменной, которую можно интерпретировать в терминах классов, к которым
может принадлежать образец. Главная задача классификации — отнести новые об-
разцы к существующим классам с достаточной достоверностью, однако результаты
классификации можно также использовать в качестве инструмента диагностики
для идентификации самых важных переменных, которые следует сохранить в мо-
дели, или для поиска выбросов. Метод применяется для прогноза соответствия
фармацевтического продукта требованиям качества (в этом случае результат пред-
ставляет собой просто бинарный отклик), или, в более общем случае, в качестве
тестирования или подтверждения идентичности вещества. При БИК-анализе для
решения подобных задач широко применяются РСА или дискриминантный анализ.
SIMCA — метод многомерного анализа, оптимизированный для анализа данных
БИК-спектроскопии, объединяет модели РСА для каждого определенного класса
обучающего набора. Такой подход можно применить к решению проблем более
общего характера, чем простая классификация, например, к идентификации. РСА
проводится на наборе данных, требующих количественного анализа. После выде-
ления главных компонент вычисляют счета и используют результаты вычислений
для количественного анализа путем построения поверхности, окружающей каждую
область многомерного пространства, содержащую счета каждой группы. Умозри-
тельно эту ограничивающую поверхность можно смоделировать в виде эллипсоида,
а расстояния до облаков данных можно рассчитать, исходя из параметров Махало-
нобиса, основанных на счетах PC.
На практике оптимальное число главных компонент (PC) следует выбирать для
модели каждого класса отдельно, согласно соответствующей схеме валидации.
Каждую модель проверяют на наличие вероятных выбросов и максимально со-
вершенствуют, как и любую другую модель РСА. Перед применением моделей для
прогнозирования принадлежности новых образцов к определенным классам следу-
ет проверить специфичность моделей, т. е. проверить наличие наложений классов
и адекватность расстояния между классами. После построения моделей всех классов
и подтверждения того, что наложение классов не является избыточным, в каждую
Глава 4.2. Процессно-аналитическая технология (РАТ)
473
модель вводят новые образцы. Неизвестные образцы сравнивают с моделями клас-
сов и приписывают к классам, в зависимости от сходства с обучающими образцами.
Стадия моделирования предполагает, что определена принадлежность к классам
для достаточного количества образцов, что позволяет строить достоверные моде-
ли. Точное описание образцов также требует достаточного количества переменных.
В настоящее время классификация основана на статистическом анализе расстоя-
ний Махаланобиса между образцом и моделью. Для каждого неизвестного образца
все значения переменных величин рассчитывают с помощью счетов и нагрузок мо-
дели перед сравнением с реальными значениями. Затем остатки объединяют в меру
расстояния между объектом и моделью. Счета также используют для измерения
расстояния от образца до центра модели, так называемого рычага. Наконец, при
определении класса, к которому принадлежит образец, в расчет принимаются оба
параметра — расстояние от объекта до модели и рычаг. Любой образец, принадле-
жащий к определенному классу должен находиться на малом расстоянии от модели
класса.
4.2.3.11. Регрессия
Исторически сложилось так, что условием внедрения БИК-спектроскопии ста-
ла разработка быстрого и неразрушающего метода количественного анализа. Для
достижения этой цели недостаточно выделить PC из массива данных. Необходи-
мо применить метод регрессии, связывающий эти PC с составляющим компонен-
том, анализируемым веществом или физическим свойством, для которого строят
калибровку. Все методы подгонки модели к наблюдаемым данным включают по-
строение регрессии. Подобранную модель можно использовать для описания соот-
ношений между двумя группами переменных или для прогнозирования значений
неизвестных образцов. Если Хи У представляют собой две матрицы данных, вовле-
ченных в регрессию, задача состоит в построении модели Y=f(X), которая пытается
объяснить или прогнозировать вариации переменной (переменных) У в зависимо-
сти от вариации переменных X. Связь между Хи У исследуют через общий набор
образцов, из которого отобраны и хорошо известны величины Xи У. Построение
модели регрессии включает накопление значений переменных для выбранных об-
разцов и установление математических соотношений, описывающих соответствую-
щие спектральные данные. Например, спектроскопические измерения проводятся
в растворах с известными концентрациями определенного соединения. Построе-
ние регрессии направлено на установление связи концентрации со спектральными
данными. Когда построена модель регрессии, неизвестные концентрации новых
образцов можно прогнозировать непрямым путем с использованием спектроско-
пических данных в качестве предикторов. Преимущество очевидно, если учесть
неразрушающий характер измерений методом БИК-спектроскопии. Если прямое
измерение концентрации затруднительно или экономически невыгодно, можно
воспользоваться спектроскопическим анализом — альтернативным, намного более
экономичным аналитическим методом. Таким образом, использование регрессии
в качестве инструмента прогнозирования рекомендуется, поскольку метод позво-
ляет проводить быстрый и экономичный анализ, являясь альтернативой более до-
рогостоящему и времязатратному методу.
474 Часть 4, Процессно-аналитические технологии
Возможно, перед построением и применением регрессии потребуется соответ-
ствующая обработка данных. За этапом калибровки следует этап валидации; это
означает, что эффективность модели должна быть проверена путем прогнозирова-
ния независимых данных. После выбора числа компонентов на основании диспер-
сий, определенных при калибровке и валидации, проводится диагностика модели
путем интерпретации графиков нагрузок и счетов (для PCR и PLS), графиков взве-
шенных нагрузок (для PLS) и коэффициентов В (РСК), и исследования для пред-
сказания новых данных.
4.2.3.12. Множественная линейная регрессия
В классической одномерной регрессии используется один предиктор, которого
обычно не достаточно для моделирования достаточно сложных образцов. Много-
мерная регрессия учитывает несколько предиктивных переменных одновременно
для увеличения точности модели. Цель построения многомерной регрессионной
модели заключается в извлечении релевантной информации из доступных данных.
Данные наблюдений, как правило, содержат шум и включают некоторую нереле-
вантную информацию. Шумом можно считать случайные вариации, возникающие
под влиянием факторов, изначально не учитываемых моделью. Например, спектры
БИК-поглощения, в дополнение к информации о концентрации аналитического
раствора, могут нести информацию, относящуюся к растворителям, способу пере-
работки, состоянию прибора, световым зондам и т. п. Качественная регрессионная
модель должна выбирать только релевантную информацию, удаляя информацию
нерелевантную.
Множественная линейная регрессия является методом, основанным на обыч-
ной регрессии по методу наименьших квадратов. Метод заключается в инверсии
матрицы, которая быстро приводит к вопросам коллинеарности, если переменные
не являются линейно независимыми. В модели MLR все переменные Xучаствуют
независимо друг от друга, и их ковариации не принимаются в расчет. Дисперсия
X в данном контексте значения не имеет. Важным предварительным условием яв-
ляется независимость переменных. Далее, для проведения инверсии MLR требует,
чтобы число образцов превышало число предикторов и чтобы в таблице данных не
было пропущенных значений. Если таблица данных отвечает этим условиям, MLR
аппроксимирует значения откликов линейной комбинацией значений предикто-
ров, рассчитав коэффициенты регрессии, известные как ^-коэффициенты. Другие
результаты — прогнозированные значения Y, остатки с погрешностями и AN OVA.
Следует заметить, что MLR является единственным многомерным методом, для ко-
торого доступны формальные статистические проверки значимости коэффициен-
тов регрессии. Для оценки качества модели инструменты диагностики ассоциируют
с коэффициентами регрессии. Стандартная погрешность представляет собой оцен-
ку прецизионности данного коэффициента. Значения t критериев Стьюдента можно
рассчитать и сравнить со стандартным ^-распределением, которое, в свою очередь,
обозначает уровень достоверности или р-значение. P-значение показывает вероят-
ность того, что величина t окажется равной или превысит наблюдаемую величину,
если истинное значение коэффициента регрессии равняется 0. Прогнозируемые
значения У рассчитываются для каждого образца приложением уравнения модели
Глава 4.2. Процессно-аналитическая технология (РАТ)475
с рассчитанными Б коэффициентами к наблюдаемым величинам. Для каждого об-
разца остаток представляет собой разность между наблюдаемой величиной У и про-
гнозированной величиной Y. Единственную релевантную меру качества модели
MLR представляют дисперсии Y. Остаточная У-дисперсия — это дисперсия остатков
У Она выражает остаточную вариацию в наблюдаемом отклике после выделения
моделированной части. Это общая мера несоответствия, т. е. погрешность, внесен-
ная, когда соответствующие величины У рассчитывались как функции значений X.
4.2.3.13. PCR и PLSрегрессии
Метод многомерной регрессии устанавливает соотношение между спектроскопиче-
скими данными и анализируемой переменной. Регрессия на главные компоненты
(PCR — PC regression) является двухэтапной процедурой: во-первых, проводят раз-
ложение матрицы X методом РСА, затем подгонку модели MLR с использованием
PC вместо исходных данных в качестве предикторов на этапе регрессии. MLR и PCR
моделируют одну переменную У единовременно, в то время как PCR модель, ис-
пользующая все PC, дает то же решение, что и MLR. Метод частичных наимень-
ших квадратов или проекции на латентные структуры (PLS) моделирует как X, так
и У матрицы одновременно для поиска латентных или скрытых переменных в X,
что лучше всего позволит прогнозировать латентные или скрытые переменные в Y.
Различие между PCR и PLS состоит в алгоритме. Компоненты PLS аналогичны PC
и также именуются PC. PLS1 рассматривает только один переменный отклик еди-
новременно (как MLR и PCR). PLS1 обрабатывает несколько откликов одновре-
менно. PCR и PLS, как и РС4, являются проекционными методами. Компоненты
модели выделяют так, чтобы большую часть информации несла первая PC, затем
вторая PC и т. д. В определенной точке вариация, моделированная новой PC, пред-
ставляет, в основном, шум. Остаточные дисперсии используются для определения
оптимального числа PC, моделирующих полезную информацию, избегая чрезмер-
ной подгонки. PLS использует как независимые, так и зависимые переменные для
поиска регрессионной модели. Метод попеременно применяют для матриц X и У
Для оптимального решения PLS обычно требует меньшего числа PC, чем PCR, по-
скольку фокусируется на зависимых переменных. Результаты РСД-моделирования
представляют в виде счетов, нагрузок, прогнозированных значений У, остатков, из-
мерений погрешностей и ^-коэффициентов. Результаты РЕЛ'-моделирования пред-
ставляют в виде Т- и [/-счетов, Р- и 2-нагрузок, весов нагрузок, прогнозированных
значений У, остатков и меры погрешности.
PLS счета интерпретируют так же, как и счета РСА, поскольку они являются
координатами образцов вдоль компонентов модели. Дополнительная характери-
стика PLS: рассматриваются два различных набора компонентов, суммируются
вариации в X- или У-пространстве. Р£5-нагрузки выражают связь каждой X- или
Y переменной с компонентой модели. Т-счета представляют координаты информа-
ционных точек, расположенных в пространстве X, описывающие часть структуры X,
максимально предсказательную для У [/-счета суммируют часть структуры У, объ-
ясненную У вдоль данной компоненты модели. Соотношение между Т- и [/-счетами
представляет модель отношения между X и У вдоль определенной компоненты
и с целью диагностики его можно визуализировать. Из этого следует, что нагрузки
476
Часть 4. Процессно-аналитические технологии
интерпретируют по-разному в пространстве X и пространстве Y. Р-нагрузки ана-
логичны РС4-нагрузкам. Они отражают вклад, вносимый каждой Х-переменной
в определенный компонент модели. Направления, определенные проекциями
переменных X, используются для интерпретации локации проекции информаци-
онной точки на график Т-счетов в терминах вариаций X. 0-нагрузки выражают
прямые соотношения между У-переменными и Т-счетами. Таким образом, направ-
ления, определенные проекциями У-переменных при помощи 0-нагрузок можно
использовать для интерпретации локации проекции информационной точки на
график Т-счетов в терминах вариаций образцов матрицы У При нанесении на один
график Р- и Q-нагрузок можно интерпретировать Т-счета, учитывая вариации как
в X, так и в У В отличие от РС4-нагрузок, Р/А-нагрузки не нормализованы, так
что Р- и 0-нагрузки различаются по масштабам. Таким образом, интерпретировать
можно только направления, а не длины проекций. Остатки должны иметь случай-
ное распределение и не содержать систематических трендов. Самую полезную ин-
формацию несут графики зависимости У-остатков от прогнозов У- и У-остатков от
счетов.
При наличии более чем одной У-переменной для одновременной интерпрета-
ции всех переменных применяют PLS2. Зачастую доказывают, что PLSY или PCR
являются лучшими предикторами, Обычно это утверждение справедливо, если
в данных присутствует сильная нелинейность; в этом случае моделирование каж-
дой У-переменной по отдельности в соответствии с ее собственными нелинейны-
ми характеристиками может оказаться более эффективным, чем попытки постро-
ить модель, общую для всех переменных У С другой стороны, если У-переменные
содержат избыточный шум, но при этом сильно коррелированны, PLS1 позволить
включить в модель всю информацию, исключив больше шума.
Так же, как в РСА, на моделирование могут повлиять выбросы — их следует выяв-
лять. В регрессионной модели есть много способов выделения образцов, определяе-
мых как выбросы. Возможны выбросы по X- или У-переменным исключительно,
или по тем и другим одновременно. Кроме того, возможно, что образец не является
выбросом для одного из отдельных наборов переменных, однако становится выбро-
сом для (X, У) регрессии.
4.2.3.14. Практика построения регрессий в БИК-спектроскопии
Калибровка является стадией подгонки модели: основной набор данных, содержа-
щий только наборы калибровочных образцов, используют для расчета параметров
модели, таких как PC, коэффициенты регрессии и т. п. Модели должны пройти ва-
лидацию для проверки, насколько эффективна регрессионная модель для прогноза
параметров новых неизвестных образцов. Для этого используют контрольный на-
бор, состоящий из образцов с известными значениями переменных. В модели учте-
ны только те образцы X, значения откликов которых прогнозируемы и сравнимы
с известными, истинными значениями откликов. Модель выдерживает валидацию,
если прогнозируемые остатки малы. Валидация модели — это проверка эффектив-
ности модели на реальных новых данных. Поскольку регрессионная модель обычно
строится для прогноза параметров будущих неизвестных образцов, валидация долж-
на включать оценку неопределенности прогноза. Если неопределенность достаточ-
Глава 4.2. Процессно-аналитическая технология (РАТ)
ATI
но мала, модель может считаться приемлемой. Этапы построения полной модели
представлены на рис. 20.
На рис. 21 представлен конечный результат типичной хемометрической обра-
ботки, включая калибровку и валидацию.
Независимая (внешняя) валидация по контрольному набору и кросс-валидация
являются самыми современными методами оценки погрешности прогноза. Метод
внешней валидации по контрольному набору заключается в проверке модели на
подгруппе доступных образцов, которые не будут использованы для расчета компо-
нентов модели. Например, общую таблицу данных делят на две подгруппы. Набор
калибровочных данных включает все образцы, используемые для расчета компо-
нентов модели, при помощи значений как Х-, так и У-величин. Контрольный набор
содержит все оставшиеся образцы, для которых 2Г-величины учитываются в модели
при расчете новых компонентов. Затем прогнозированные У-величины сравнива-
ют с наблюдаемыми У величинами, получая остатки прогноза, по которым можно
рассчитать остаточную дисперсию валидации, или меру неопределенности будущих
прогнозов, так называемый среднеквадратичный остаток прогноза (RMSEP — root
mean square error of prediction). Данную величину, указывающую на среднюю не-
определенность, ожидаемую при прогнозировании У величин для новых образцов,
выраженную в тех же единицах, что и переменные У, рассчитывают для каждого
моделированного отклика. Далее (табл. 3) представлена формула расчета RMSEP.
Результаты прогнозов в этом случае можно представлять как прогнозированное
значение +RMSEP. Этот результат является достоверным при условии, что новые
образцы аналогичны применяемым для калибровки. В противном случае ошибка
Калибровочной набор
БИК-сканирование
Независимый
валидационный
набор
Рис. 20. Типичная рабочая схема построения PLS калибровки по БИК-спектрам
478
Часть 4. Процессно-аналитические технологии
Рис. 21. Типичные графики PLS-калибровки и валидации. SEC (standard error of calibration) —
стандартная ошибка калибровки; SEP (standard error of prediction) — стандартная ошибка
прогноза; Bias — погрешность измерения
прогноза может оказаться намного больше. При построении модели не было сде-
лано предположения о статистической оценке распределения погрешности. Вслед-
ствие этого ошибка прогноза не может быть представлена в виде соответствующей
оценки статистического интервала (например, двукратное стандартное отклонение,
и т. д.). RMSEP представляет практическую среднюю погрешность прогноза. Если
и калибровочный, и валидационный набор образцов репрезентативны для прогноза
параметров будущих образцов, то RMSEP представляет собой адекватную оценку
погрешности.
При кросс-валидации для оценки модели и тестирования используют одни
и те же образцы. Кросс-валидация является альтернативным путем валидации
при малом или умеренном количестве образцов. Метод заключается в выделении
нескольких образцов из калибровочного набора данных и калибровке модели по
оставшимся информационным точкам. Значения выделенных образцов затем про-
гнозируют и рассчитывают соответствующие остатки прогноза. Процесс повторяют
с другой подгруппой калибровочного набора и т. д. до тех пор, пока каждый объект
не окажется единожды выделенным. Все остатки прогноза комбинируют и вычис-
ляют остаточную дисперсию валидации и RMSEP, при полной кросс-валидации вы-
деляют лишь по одному образцу единовременно, при сегментированной — группу
образцов единовременно. Сегментированная кросс-валидация — более быстрый
метод, однако следует быть внимательными при выборе сегмента, поскольку он
должен содержать уникальную информацию. Например, образцы, которые можно
считать репликами, не должны оказаться в разных сегментах.
При использовании независимого контрольного набора файл должен содержать
20—40% полного набора данных. Калибровочный и контрольный наборы должны
максимально охватывать одинаковые популяции образцов. Репликатные измерения
не должны присутствовать в калибровочном и контрольном наборе одновременно.
Этот риск возникает при случайном выборе, предлагаемом современным програм-
мным обеспечением БИК-исследований. Простейший способ выбора контрольного
Глава 4.2. Процессно-аналитическая технология (РАТ)479
набора — предоставить выбор компьютеру. Ручной выбор рекомендован, поскольку
предполагает полное управление выбором образцов для контрольного набора.
Программы для расчета многомерной регрессии также рассчитывают вспомо-
гательные статистические данные, помогающие оценить соответствие калибровки
набору данных, а также качество ожидаемого прогноза параметров будущих образ-
цов. Например, две из этих статистических величин — стандартная погрешность
калибровки (SEC) и коэффициент многомерной корреляции (R). SEC (называемая
также стандартной погрешностью оценки или остаточным стандартным отклоне-
нием) и коэффициент многомерной корреляции показывают, насколько хорошо
калибровочное уравнение описывает данные. Статистические данные рассчитыва-
ются по формулам (табл. 3), где — эталонное значение для образца j, у'} — про-
гнозируемое значение для образца j,tn — число образцов в калибровке, п — число
образцов в валидации, д — число PC.
Таблица 3
Дескрипторы, применяемые для оценки эффективности калибровки
0,-11 £(У,-У)2 >1 cov(y',y) = \ J=i = cov(y’,y)
т-1 т-1
SEC = \ Eb'y-J'/)2 >i SEP = \ EO'y-J'-)2 j=i Bias=— n
т-1-д n
SEC измеряется в тех же единицах, что и зависимые вариации, и отражает раз-
ность между измеренным прибором значением параметров исследуемого аналити-
ческого раствора и опорным значением, измеренным в лаборатории. Она отражает
погрешность моделирования и не может быть использована для оценки будущих
ошибок прогнозов. В отсутствии приборной ошибки SEC является лишь мерой по-
грешности эталонного измерения, выполненного в лаборатории. Она показывает,
будет ли расчет при помощи калибровочного уравнения достаточно точно соответ-
ствовать целям, для которых было сформулировано уравнение. Практически SEC
менее точна, чем погрешность эталонного лабораторного метода даже в отсутствии
погрешности прибора, если интервал длин волн, используемых при калибровке в ка-
честве независимых переменных, не описывает всех взаимодействий образцов или
влияния других физических явлений. Предел обнаружения и чувствительность (или
соотношение сигнал/шум), определяет эффективность применения прибора в кон-
кретном случае. Пределы обнаружения для любого метода БИК-спектроскопии
(конкретной методики) можно определить приблизительно как значение SEC,
умноженное на три. Чувствительность количественных БИК-методов обычно оце-
нивают по наклону калибровочного графика зависимости концентрации исследуе-
мого анализируемого вещества (ось у) от изменения оптического отклика (ось х)
для образцов различных концентраций. Чувствительность с чисто инструменталь-
ной точки зрения выражается соотношением сигнал/шум или отношением высот
480
Часть 4. Процессно-аналитические технологии
пиков для конкретного соединения, в сравнении с уровнем шума (пик к пику) при
определенной величине поглощения (обычно 0). Однако в практическом смысле
вышеописанные соображения не так уж важны для БИК-спектроскопии. Причи-
на заключается в том, что способы применения, разработанные для практической
БИК-спектроскопии, основаны, главным образом, на эмпирических методах кали-
бровки, а калибровка специфична для конкретной решаемой проблемы. Характер-
ной особенностью калибровочных уравнений, использующих математические мно-
гомерные методы, является компенсация частых вариаций в химических образцах
с высоким уровнем шума и несовершенных инструментальных измерений. Поэтому
надлежащим образом рассчитанные и прошедшие валидацию калибровочные моде-
ли для БИК-спектроскопии являются робастными и работают очень хорошо.
Коэффициент многомерной корреляции (R) является безразмерной величиной,
обозначающей, насколько хорошо калибровка соответствует данным. Значение R
может изменяться от —1 до +1, однако при калибровке возможны только положи-
тельные значения. Близость величины к нулю означает, что калибровка не соотно-
сит спектры с калибровочными величинами. По мере повышения коэффициента
корреляции, спектры становятся все лучшими предикторами референтных вели-
чин. Поскольку коэффициент многомерной корреляции не имеет размерности,
он удобен при сравнении данных или результатов, измеренных в разных единицах,
которые трудно сравнивать другими способами. Тем не менее, его значение не от-
ражает ожиданий относительно того, насколько хорошо калибровочное уравнение
применимо для прогноза параметров будущих образцов.
4.2.3.15. Некоторые «подводные камни»
Этапом, предшествующим любому многомерному анализу, является отбор образ-
цов. Выборка или подготовка набора калибровочных образцов является критически
важным моментом. Например, аналитик должен отобрать или подготовить образцы,
охватывающие весь интервал концентраций, расположенных с максимальной рав-
номерностью. При случайной выборке обычно строят модели, максимально близ-
кие к средним значениям концентраций. Образцы с высокими или низкими уров-
нями концентраций не влияют на наклон и точки пересечения с координатными
осями в случае многомерной регрессии. В идеальном случае равномерное распре-
деление концентраций позволит модели минимизировать остатки в точках экстре-
мумов и в центре с относительно равными весами. Калибровочные наборы должны
не только равномерно заполнять весь интервал составляющих данных, они должны
также состоять из правильно распределенных множеств типов образцов. Например,
в идеале калибровочные наборы состоят более чем из 10—15 образцов для каждой
аналитической характеристики. В идеале составы этих проб равномерно распреде-
лены по всему калибровочному интервалу. И наконец, отметим, что спектроско-
пические измерения для калибровочных и рутинных образцов следует проводить
в максимально близких условиях. Постоянной проблемой является влияние влаги,
содержащейся в твердых или порошкообразных образцах. Наличие или отсутствие
воды в образце влияет на количество водородных связей, которое влияет и на поло-
жение, и на ширину полосы в полном БИК-диапазоне. Если калибровочная модель
строят по образцам, содержащим исследуемый компонент в широком диапазоне,
Глава 4.2. Процессно-аналитическая технология {РАТ)
481
и влагу в узком диапазоне, то модель окажется полезной только для образцов с со-
держанием влаги в упомянутом узком диапазоне. Такая калибровка может оказаться
недостаточно надежной для рутинного применения. В итоге отметим, что каждая
модель калибровки акцентирует несколько разные аспекты проблемы.
Очевидно, что следует обращать внимание на более тонкие детали, поскольку
неожиданные источники погрешностей могут повлиять на качество расчетных мо-
делей. Ошибки возникают, например, при подготовке и анализе образца. Параме-
тры потенциального влияния включают разность температур образцов или инстру-
ментов при регистрации данных, нестабильность калибровочных образцов, шум
и дрейф инструмента, изменения настроек инструмента по длинам волн, нелиней-
ность, эффект рассеянного света, различия в размерах частиц, зависящие от кон-
центрации различия цвета, взаимодействие с остатками растворителя и неоднород-
ность образцов. Возможно, стандартный метод не измеряет те же компоненты, что
и спектроскопический метод. Контроль этих аспектов может показаться излишне за-
труднительным и может первоначально охладить энтузиазм к БИК-спектроскопии.
Тем не менее, многокомпонентные проблемы являются комплексными и требуют
управления несколькими переменными одновременно, чтобы построить приемле-
мую калибровку. Конечная цель успешной калибровки состоит в расчете математи-
ческой модели по калибровочным образцам, наиболее чувствительным к изменени-
ям моделируемого параметра и менее чувствительным ко всем другим факторам, не
связанным с калибровкой, таким как физические, химические и инструментальные
переменные. Каждый случай следует оценить с точки зрения химических и физиче-
ских данных, свойственных калибровочным образцам, а также информации, кото-
рую хочет получить аналитик.
4.2.3.16. Примеры аналитического применения БИК-спектроскопии
Предположим, что БИК-спектры получены, проведен анализ фармацевтических
образцов высокопрецизионным аналитическим стандартным методом, концентра-
ции исследуемого анализируемого вещества четко определены или подтверждена
идентичность данного соединения; теперь полученные данные можно объединить
в обучающий набор для генерирования калибровки для последующих прогнозов.
Для построения приемлемого обучающего набора образцы должны равномерно за-
крывать интервал концентраций исследуемого вещества или ожидаемые колебания
качества. Очевидная трудность состоит в построении калибровок, в которых ис-
пользованы наборы данных с неравномерным распределением составляющих или
слишком узким интервалом вариаций исследуемой характеристики. В этом случае
модель ближе всего соответствует доминирующим образцам калибровочного набо-
ра. Калибровка будет высоко взвешена по средней величине и будет плохо отражать
вариации в отдаленных образцах. И наоборот, идеально равномерный калибровоч-
ный набор взвешивает калибровочную модель в равной степени во всем интервале
концентраций. Правильно построенная модель калибровки такого рода будет точно
соответствовать образцам с высокими и низкими концентрациями.
Несмотря на проблемы, связанные с выборкой, весьма важные для БИК-
спектроскопистов, работающих в фармацевтической промышленности, потенциал
метода БИК-спектроскопии не ставится под вопрос. БИК-спекгроскопия — бы-
482
Часть 4. Процессно-аналитические технологии
стрый, неразрушающий метод, требующий незначительной подготовки образца,
или не требующий подготовки вовсе. Он позволяет проводить мониторинг концен-
траций нескольких химических веществ и физических параметров одновременно.
При выборе аналитического метода или метода мониторинга производственного
процесса главными преимуществами этого метода являются его скорость и про-
стота. Он позволяет работать с материалами любого типа: твердыми или жидкими
исходными материалами (например, наполнителями), активными фармацевтиче-
скими ингредиентами (API) промежуточными продуктами синтеза, порошкообраз-
ными полупродуктами, суспензиями, гранулами или шариками, а также с готовыми
лекарственными формами, такими как капсулы, таблетки или лиофилизированные
вещества. БИК-измерения можно проводить вблизи образца, или при непосред-
ственном контакте с образцом, как в лаборатории, так и на производстве в режи-
мах on-line или in-line, что позволяет быстро получать аналитическую информацию
и экономить время. Анализ спектров пропускания стал альтернативой традицион-
ной отражательной спектроскопии фармацевтических продуктов. Важное различие
методов состоит в том, что при БИК-спектроскопии пропускания происходит объ-
емный анализ образца, в то время как при БИК-спектроскопии отражения — толь-
ко поверхностный анализ твердых образцов. В результате получают более репрезен-
тативные значения для образцов с меньшей однородностью, таких как таблетки или
капсулы. С другой стороны, следует уделить повышенное внимание поведению об-
разцов в рассеянном световом излучении и при рассеянии света.
Традиционно фармацевтические вспомогательные вещества характеризуют в ла-
бораториях по таким параметрам, как вязкость, pH в дисперсиях/растворах, содер-
жание воды, зольность, содержание составляющих компонентов, размер частиц
и т. д. Кроме того, есть и другие неявные характеристики, не охваченные этими
параметрами, которые тем не менее влияют на свойства лекарственных составов.
Информацию о многих из них можно извлечь из БИК-спектров отражения в допол-
нение к идентификационному исследованию другими методами (рис. 22).
Спектры исходных материалов и результаты, полученные другими аналитиче-
скими методами, можно комбинировать для прогнозирования влияния вариаций
качества исходных материалов на качество готовой производственной серии. Записи
по ряду партий исходных материалов известного качества и расчет диапазона при-
емлемости вокруг среднего спектра помогают провести анализ соответствия при по-
мощи БИК-спектроскопии. Исходный материал квалифицируется как пригодный,
если его характеристики попадают в область, ограниченную пороговыми величина-
ми диапазона приемлемости для каждой длины волны. Это исследование поможет
отбраковать некачественные материалы с высоким содержанием примесей и воды.
Большая часть фармацевтической продукции изготавливается сериями: это от-
носится как к синтезу активных соединений, так и к производству фармацевтиче-
ских составов. БИК-спектры помогают провести анализ производственной серии
готовой продукции в терминах спектрального сходства с помощью SIMCA, спек-
тральной корреляции (spectral correlation) или спектрального расстояния (spectral
distance). Кроме того, можно проводить мониторинг производства индивидуальной
серии с применением РСА. Несколько параметров одного продукта можно опреде-
лять количественно с использованием РЛУ-моделей. Текущий базис для сравнения
Глава 4.2. Процессно-аналитическая технология (РАТ)
483
_____4008,000 4764,000 5520000 6276.000 7032.000 ?788СС6 8544000 9300.000
Орасйу (15j fcfcSl (76) SOS (50, SusrU-' <= CeUufose HZ) I кдо *61»
Рис. 22. Библиотека спектров вспомогательных веществ с результатом РСА
для проверки подлинности
включает время переработки в виде переменной Y в модель PLS, построенную на
основе нескольких серий, признанных приемлемыми к выпуску. Далее можно рас-
считать пределы приемлемости серии и сравнить с измерениями БИК, полученны-
ми в процессе переработки серии в режиме on-line.
Сушка лекарственных веществ является важным этапом в процессе производства
активных материалов. Обычно материалы сушат в вакуумной сушилке с перемеши-
ванием при тщательном контроле для обеспечения адекватной просушки продукта
и минимизации риска пересушки, которая может привести к повреждению доро-
гостоящего материала. Некоторые лекарственные вещества являются гидратами,
для которых существуют специальные требования к контролю условий сушки. Если
удалить кристаллизованную воду, то производственная серия потребует переработ-
ки или даже может быть испорчена окончательно. Традиционные средства мони-
торинга и управления операцией сушки состоят в небольших изменениях темпера-
туры сушилки для определения конечной точки процесса с последующим отбором
484
Часть 4. Процессно-аналитические технологии
проб летучих веществ для лабораторного анализа. Зачастую явной температурной
конечной точки не существует. Некоторые продукты будут содержать остаточный
растворитель в составе кристаллов. Отбор проб из вакуумной сушилки затруднен
из-за возможности нарушения вакуума, а лабораторный анализ может занять не-
сколько часов. Применение оптоволоконного зонда для получения БИК-спектров,
введенного непосредственно в сушилку заводского масштаба, позволяет провести
анализ содержимого сушилки в режиме реального времени и, таким образом, может
являться ценной альтернативой. Различия физических и химических характеристик
полиморфных форм активного соединения (например, растворимости, скорости
растворения, химической реакционной способности, устойчивости к разложению,
биодоступности) имеют большое значение для фармацевтической промышленно-
сти. Требуются эффективные методы не только для контроля содержания активного
компонента, но и для детектирования и количественного анализа нежелательных
форм. В определенных обстоятельствах БИК-спектроскопию можно использовать
для подтверждения низкого содержания нежелательных кристаллических форм
в аморфной форме соединения. Соответствующие методы пригодны как для каче-
ственного (например, классификационного), так и для количественного анализа.
Простота, скорость и надежность БИК-спектроскопии делает ее многообещающим
инструментом контроля полиморфной чистоты аморфной фазы.
4.2.3.17. Заключение
Во многих фармацевтических компаниях отделы контроля качества уже приме-
няют БИК-спектроскопию для идентификации составов. На рис. 23 представле-
на калибровка по методу PLS для определения содержания активного компонента
в таблетке с низким его содержанием. После прохождения теста на идентичность
следующим шагом является определение содержания активного компонента в це-
лых таблетках. Здесь качественный и количественный анализ может быть проведен
при получении одного БИК-спекгра для каждого образца. Две аналитических ме-
тодики заменены на один неразрушающий анализ БИК. Для решения этих задач
ИК-спекгроскопия ближней области спектра является быстрой и эффективной
альтернативой традиционному анализу, который остается необходимым лишь в ка-
честве стандартного аналитического метода.
Жизнеспособность аналитических методик, объединяющих принципы колеба-
тельной спектроскопии (в основном, БИК-спектроскопии) и хемометрики, при
условии прохождения валидации, может быть доказана множеством примеров. Тем
не менее, доминирующей областью применения хемометрики является не фарма-
цевтическая промышленность, основной накопленный опыт относится к сельско-
хозяйственной и пищевой индустрии. Практика применения хемометрики не связа-
на с конкретной аналитической проблемой или сферой деятельности. Для решения
комплексных фармацевтических проблем при помощи ряда спектроскопических
методов с одновременной оптимизацией подготовки пробы, настоятельно реко-
мендуется применять алгоритмы многомерного анализа данных. Таким образом,
обучение химиков, фармацевтов и аналитиков принципам хемометрики позволит
с самого начала оценить пользу от применения многомерного анализа примени-
тельно к спектроскопическим данным.
Глава 4.2. Процессно-аналитическая технология (Д47)
485
Рис. 23. Количественный анализ содержания API в таблетке и представление соответствующей
PLS регрессии. SECV (standard error of cross validation) — стандартная ошибка кросс-валидации;
SEC (standard error of calibration) — стандартная ошибка калибровки; SEP (standard error
of prediction) — стандартная ошибка прогноза; Bias — погрешность измерения; SEP(C) —
стандартная ошибка прогноза для концентрации С; Вга1 — среднеквадратичный остаток прогноза
Литература
Раздел 4.2.1
Bakeev, К. A., Ed. (2005), Process Analytical Technology, Blackwell, London.
Giodici, P. (2003), Applied Data Mining. Statistical Methods for Business and Industry, Wiley, Hoboken,
NJ.
Initiative for Pharmaceutical cGMPs for the 21st Century, FDA, Washington, DC.
Martin, A. (1993), Physical Pharmacy, 4th ed., Lippincot Williams & Wilkins. Philadelphia, PA.
Раздел 4.2.2
Colthup, N. B., Daly, L. H., and Wiberley, S. E. (1990), Introduction to Infrared and Raman Spectroscopy,
3rd ed., Academic.
The Proceedings of NIR-97, J. Near-Infrared Spectres., 6 (1-4), NIR Publications, 1998.
Rouessac F., and Rouessac, A. (2000), Chemical Analysis. Modem Instrumentation Methods and
Techniques, Wiley-VCH, New York.
Siesler, H. W., Ozaki, Y., Kawata, S., and Heise, H. M. Eds (2002), Near-Infrared Spectroscopy. Principles,
Instruments, Applications, Wiley-VCH, New York.
Williams, P., and Norris, K. (2001), Near-Infrared Technology in the Agriculture and Food Industries, 2nd
ed. American Association of Cereal Chemists, St Paul, MN.
486 Часть 4. Процессно-аналитические технологии
Раздел 4.2.3
Brereton, R. G. (2003), Chemometrics: Data Analysis for the Laboratory and Chemical Plant, Wiley,
Hoboken, NJ.
Kramer, R. (1998), Chemometric Techniques for Quantitative Analysis, Marcel Dekker, New York.
Manly, B. F. J. (2000), Multivariate Statistical Methods. A Primer, 2nd ed., Chapman & Hall/CRC, New
York.
Mark, H. (1991), Principles and Practice of Spectroscopic Calibration, Wiley, New York.
Martens, H., and Martens, M. (2001), Multivariate Analysis of Quality: An Introduction, Wiley, New
York.
Massart, D. L., Vendeginste, B. G. M., Buydens, L. M. C., De Jong, S., Lewi, P. J., and Smeyers-Verbeke,
J. (1997/1998), Handbook of Chemometrics and Qualimetrics, Parts A and B, Elsevier, Amsterdam.
Naes, T., Isaksson, T., Feam, T., and Davies, T. (2002), A User-Friendly Guide to Multivariate Calibration
and Classifi cation, NIR Publications, Chichester.
Статьи Peer Review
Chalus, P., Roggo, Y., Walter, S., and Ulmschneider, M. (2005), Near-infrared determination of active
substance content in intact low-dosage tablets, Taianta, 66, 1294—1302.
Inschneider, A. (2002), Validierung von chemometrischen Methoden am Beispiel multivariater Datena-
nalyse in der Nah-Infrarot Spektroskopie, in Handbuch der Validierung, Kromidas, S. Ed., Wiley-
VCH, Weinheim, pp. 302-307.
Roggo, Y., Edmond, A., Chalus, P., and Ulmschneider, M. (2005a), Infrared imaging for qualitative analy-
sis of pharmaceutical solid forms and trouble shooting, Anal. Chim. Acta, 535, 79-87.
Roggo, Y, Jent, N., Edmond, A., Chalus, P., and Ulmschneider, M. (2005b), Characterizing process ef-
fects on pharmaceutical solid forms using near-infrared spectroscopy and infrared imaging, Eur. J.
Pharm. Biopharm., 61 (1—2), 100—110.
Roggo, Y, Roeseler, C., and Ulmschneider, M. (2004), Near-infrared spectroscopy for qualitative com-
parison of pharmaceutical batches, J. Pharm. Biomed. Anal., 36, 777-786.
Russell, E, and Ulmschneider, M. (2004), Dissolution testing by means of NIR transmittance spectros-
copy, Am. Pharm. Rev., July/August.
Sukowski, L., and Ulmschneider, M. (2005), In-line process analytical technology on qualitative NIR
modelling: An innovative approach for the pharmaceutical quality control, Pharmlnd, 67 (7), 830-
835.
Ulmschneider, M., Barth, G., Reder, B., Vogel, A., and Schilling, D. (2000a), A transferable basic library
for the identifi cation of active substances using near-infrared spectroscopy, Pharm. Ind., 62 (4),
301-304.
Ulmschneider, M., Barth, G., and Trenka, E. (2000b), Building transferable cluster calibrations for the
identification of different solid excipients with near-infrared spectroscopy, Pharm. Ind., 62 (5), 374-
376.
Ulmschneider, M., and Penigault, E. (2000a), Assessing the transfer of quantitative NIR-calibrations from
a spectrometer to another one, Analusis, 28, 83—87.
Ulmschneider, M., and Penigault, E. (2000b), Direct identifi cation of key-intermediates in containers us-
ing Fourier-Transform near-infrared spectroscopy through the protective polyethylene primary pack-
aging, Лиа/иж, 28,136-140.
Ulmschneider, M., and Penigault, E. (2000c), N on-invasive confirmation of the identity of tablets by
near-infrared spectroscopy, Analusis, 28, 336-346.
Ulmschneider, M., Wunenburger, A., and Penigault, E. (1999), Using near-infrared spectroscopy for the
non invasive identifi cation of five pharmaceutical active substances in sealed vials, Analusis, 27,
854-856.
Глава 4.3. Химическая визуализация и хемометрика:
полезные инструменты процессно-
аналитической технологии
Ив Рогго и Мишель Ульмшнайдер
E.Hoffman-La Roche Ltd (Базель, Швейцария)
4.3.1. Введение
Серьезной проблемой для обеспечения качества твердых лекарственных форм явля-
ется гомогенность химических соединений. Классический спектрофотометр [1—3]
предоставляет интегрированную пространственную информацию. Тем не менее,
получение смешанного спектра с поверхности образца может оказаться недостат-
ком при исследовании твердых лекарственных форм. Например, для фармацевти-
ческого производства важна информация о распределении активных ингредиентов
и вспомогательных веществ в таблетке, поскольку она выявляет физическое взаи-
модействие между компонентами и решить проблемы, связанные с однородностью
продукции, поэтому растет количество исследований, посвященных построению
изображений по спектральным данным, в частности, визуализации однородного
распределения химических компонентов [4—9].
Построение гиперспектральных изображений по данным колебательной спек-
троскопии (химическая визуализация) представляет собой современную разработ-
ку, объединяющую химическую информацию, полученную спектроскопическим
методом, с пространственной информацией. В принципе, можно получить гипер-
спекгральные изображения при помощи обычных детекторов, т. е. путем классиче-
ского микроскопирования. Однако матричные детекторы позволяют получить ин-
формацию обо всех пикселях одновременно, сократить время анализа, обеспечить
однородность фона и улучшить соотношение сигнал/шум [4]. Для каждого пикселя
получают полный спектр; это означает, что набор гиперспектральных данных фак-
тически представляет собой трехмерную (3D) матрицу.
Эта глава начинается с определения понятия гиперспектралъное изображение.
Далее приводится подробное описание оборудования и процесса построения изо-
бражения; в заключение представлено описание инструментов хемометрического
анализа изображений на примерах из практики фармацевтического производства.
4.3.2. Построение гиперспектральных изображений
В этой главе рассмотрены цифровые изображения трех типов. Первый тип — би-
нарные (или черно-белые) изображения, где значение пикселя может равняться О
или 1. Второй тип — монохроматическое изображение (например, серая шкала),
которое можно представить в виде двухмерной (2D) X х Yматрицы, описывающей
распределение интенсивности света, где Хи Y— число шагов оцифровки (т. е. пик-
селей) вдоль двух пространственных направлений. Каждому пикселю приписано
488
Часть 4. Процессно-аналитические технологии
значение от 0 (черный) до 255 (белый). Третий тип — цветное изображение, которое
можно описать в красно-зелено-синем пространстве (RGB — red-green-blue), на-
пример, в трех плоскостях (красной, зеленой и синей, соответственно) в виде 3D
X х Y хЗ матрицы. Значение каждого пикселя составляет от 0 до 255 для красной,
зеленой и синей плоскости, что генерирует 2553 возможных цветов в подобных изо-
бражениях (известных как 24-битные изображения).
По аналогии гиперспектральные данные определяют по меньшей мере 50 пло-
скостей; для каждой длины волны определенного спектрального диапазона состав-
ляют карту поглощения. Если число длин волн меньше 50, используют термин по-
строение мультиспектралъного изображения.
При химической визуализации возникает необходимость анализа структуры
данных нового типа. В эксперименте по химической визуализации строят 3D ма-
трицу X х Y х X или трехмерные данные, где X и Y представляют собой простран-
ственные измерения, а X — спектральное измерение. Регистрируют один спектр на
пиксель и на выбранной длине волны получают картину поглощения излучения об-
разцом [10] (рис. 1).
Рис. 1. Структура данных химического изображения
Трехмерные данные объединяет спектральную и пространственную информа-
цию и содержат необходимую статистику для спектральных классификаций. Тем не
менее для интерпретации результатов химической визуализации требуются новые
хемометрические стратегии.
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...489
4.3.3. Оборудование для получения гиперспектральных
изображений
4.З.З.1. Принципы получения гиперспектральных изображений
В этом разделе обсуждаются три способа получения изображений: картирование,
матричное детектирование и оптоволоконные системы.
Картирование. Исторически картирование [11] стало первым методом получения
трехмерных гиперспектральных данных, в частности, для анализа данных раманов-
ской спектроскопии и инфракрасной микроскопии. Изображение создают от пик-
селя к пикселю пошаговым способом: измеряют спектр в одной точке образца, за-
тем образец перемещают в другую измерительную позицию и снимают следующий
спектр. Процесс повторяют для всех позиций в зоне, определяющей изображение.
Недостатком метода является длительность проведения измерений, зависящая от
числа пикселей. Поэтому производители спектрометров разработали метод линейно-
го картирования, при котором образец сканируют от линии к линии, что позволяет
сократить время получения изображения. Подобные устройства были разработаны
для рамановской, ИК- или БИК- (NIR-) спектроскопии (с диодноматричными де-
текторами). Тем не менее из-за наличия стадии движения этот принцип построения
изображений применим лишь для работы с отобранными образцами в лаборатории
(в режиме at-line).
Детекторы с фокально-плоскостной матрицей. Оптические детекторы с фокально-
плоскостной матрицей (FPA — focal plane array) состоят из нескольких тысяч инди-
видуальных детектирующих элементов, образующих матрицу из пикселей. Как ясно
из названия, они размещены в фокальной плоскости спектрофотометра. Элементы
могут быть чувствительны к излучению в ультрафиолетовой (УФ) и видимой обла-
стях, БИК- или ИК-области. Современные разработки в области оптики предлага-
ют охлаждаемые или не охлаждаемые /РД-детекторы с различным числом пикселей
от 64x64 до 1024x1024 и различными спектральными диапазонами детектирования
(от 1000 до 12 000 нм). Разные типы FPA включают матрицы, изготовленные на
основе антимонида индия (InSb), силицида платины (PtSi), арсенида индия-галлия
(InGaAs) и теллурида ртути-кадмия (HgCdTe) [12]. Детектор на основе теллурида
ртути-кадмия (МСТ— mercury cadmium telluride) лидирует среди инфракрасных FPA
детекторов благодаря широкому диапазону детекции.
Оптоволоконные системы. Оптоволоконные системы используют для построения
рамановских изображений. Несколько оптических волокон соединены в пучок, каж-
дый из которых анализирует конкретную область образца [13]. 3D трехмерные дан-
ные собираются при помощи детектора на основе 2D устройства с зарядовой связью
(CCD — charge-coupled device). В месте объединения волокна собраны в круговой пат-
терн или квадратную матрицу для анализа определенных областей образца (рис. 2).
В точке детектирования волокна выстроены в линию для детектирования сигна-
ла: первое измерение CCD детектора применяется для получения пространственной
информации, а второе — для спектрального диапазона. Техническая трудность со-
стоит в соотнесении позиции конечного сигнала детектора с расположением пиксе-
ля в изображении. Главное преимущество данной методики — возможность быстро-
го построения изображения.
490
Часть 4, Процессно-аналитические технологии
Рис. 2. Принцип оптоволоконных пучков. (Адаптировано из A. D. Gift, J. Ма, К. S. Haber,
В. L. McClain, andD. Ben-Amotz, Journal of Raman Spectroscopy, 30,757-765,1999.)
4.3.3.2. Спектроскопическое оборудование
Детекторы, формирующие изображение через картирование точек, линейное ска-
нирование или матричные детекторы можно соединять со спектрометрами различ-
ных типов. Типы инструментов классифицируют по способу выбора длин волн на
Фурье-спектрометры (FT — Fourier transform} с возможностью визуализации данных
и спектрометры с перестраиваемым фильтром (TF — tunable filter) — обсуждение
обоих типов приборов приведено далее, и дисперсионные спектрометры. Фурье-
спектрометры с возможностью визуализации являются классическими лаборатор-
ными приборами, в то время как /’/'-спектрометры представляют собой компакт-
ные и надежные системы химической визуализации.
Фурье-спектрометры. Спектрометры этого типа (рис. 3) отличаются от сканиру-
ющих тем, что в них регистрируемый сигнал представляет собой интерферограмму
[14] (см. главу 4.2). Их можно совместить с микроскопом или макровидеокамерой
с /ТИ-детектором. Оснащенные Фурье-спектрометрами системы химической ви-
зуализации (CIS — chemical imaging system) выпускаются для рамановской, БИК-
и ИК-спектроскопии. Тем не менее, они относятся к приборам, применяемым
лишь для научных исследований. Например, большинство ИК-систем визуали-
зации представляют собой Фурье-спектрометры, совмещенные с микроскопами.
Спектрометры такого типа позволяют получать спектры в режимах отражения, на-
рушенного полного внутреннего отражения (НПВО) или пропускания.
TF-системы. TF (tunable filter — перестраиваемый фильтр) представляет собой
устройство, для которого спектральное пропускание можно контролировать прило-
жением напряжения или акустического сигнала. TF-устройства делятся на два типа:
акустооптические (AOTF — acousto-optical tunable filter), основанные на дифракции,
и жидкокристаллические (LCTF — liquid crystal tunable filter), основанные на двой-
ном лучепреломлении.
AOTFпредставляет собой прозрачный кристалл, в котором создается ультразву-
ковое поле, т. е. выбор длины волны зависит от интенсивности поля. LCTFсостоит
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...
491
Рис. 3. ИК-микроскоп, соединенный с ЕВА-детектором
Рис. 4. Фильтр Лайота; LCTFэлемент.
(Адаптировано из N. Gat, Proceedings SPIE, 4056. 50-64, 2000)
из нескольких фильтров Лайота (рис. 4). Входящий свет поляризуется на одном эле-
менте фильтра Лайота, затем при прохождении через двулучепреломляющий кри-
сталл появляется фазовый сдвиг (5) между лучами света, и, наконец, свет проходит
через второй поляризатор — «анализатор», который выбирает длину пропускаемой
волны. Недостатком LCTF в сравнении с AOTFявляется скорость измерений: по-
рядок скорости сканирования для AOTF составляет микро- и миллисекунды для
£С7У[15].
С помощью TF-систем образцы можно сканировать последовательно на разных
длинах волн. Изображения для каждой определенной длины волны затем группи-
руют в трехмерные данные. Для сокращения времени получения данных можно
выбрать специфические (например, специфические для конкретных химических
соединений) длины волн. Коммерческие 7У-устройства (рис. 5) выпускаются для
492
Часть 4. Процессно-аналитические технологии
FPA-детектор
«-- Выбор длины волны
*--- Оптические части (объективы)
Перестраиваемые
фильтры
__________________I
Источники излучения
Образцы
Рис. 5. Получение изображение с помощью LCTF БИК-системы.
(Адаптировано из N.Lewis, J. Schoppelrei, Е. Lee and L. Kidder, in Process Analytical
Technology, Editor K. A. Bakeev Blackwell, London., 2005, pp. 187-225)
визуализации данных рамановской и БИК-спектроскопии. TF системы визуализа-
ции применимы в процессно-аналитической технологии (Л47), так как они могут
работать в режиме on-line благодаря быстрому, простому и надежному способу по-
строения изображения.
4.3.4. Применение хемометрики для построения изображений
Системы визуализации гиперспектральных данных генерируют большие массивы
данных, которые необходимо обработать для выявления релевантной информации.
Поэтому для построения изображений, подтверждающих однородность химических
компонентов («карт распределения») необходимо разработать методы извлечения
данных. Анализ трехмерных данных (рис. 6), основные этапы которого последова-
тельно представлены далее, начинают с предварительной обработки данных (для
улучшения качества информации), затем проводят классификацию (для идентифи-
кации основных химических соединений для каждого пикселя и построения карт
распределения) и заканчивают применением инструментов анализа карт распреде-
ления.
4.3.4.1. Предварительная обработка данных
Предварительная обработка позволяет выявлять больший объем химической ин-
формации, а также удаляет шум и эффект рассеяния [ 14]. Специфика обработки со-
стоит в том, что трехмерные данные можно подвергать предварительной обработке
как в направлении длин волн, так и в пространственном направлении.
Классическая обработка спектральных данных, описанная в разделе 4.3.1, заклю-
чается в нормализации, сглаживании данных и коррекции фона. В некоторых CIS
интенсивность изображения соответствует величине отражения. Поэтому данные
преобразуют в поглощение. Другой тип предварительной обработки спектральных
данных может быть дополнительно проведен на основании существующих химиче-
ских знаний: выбор длины волны. Менее информативные спектральные диапазоны
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...
493
Рис. 6. Последовательность анализа изображения: цепочка обработки данных
494
Часть 4. Процессно-аналитические технологии
можно удалить для сокращения времени вычислений и улучшения конвергенции хе-
мометрических алгоритмов.
Кроме того полезны методики предварительной обработки изображений. С их
помощью можно удалить плохие пиксели (т. е. участки изображения без сигнала
или выпадающие данные) и сгруппировать вместе несколько ЗД-наборов данных
для одновременного анализа с целью упрощения сравнения. При обработке гипер-
спектрального изображения можно использовать маску для выделения исключи-
тельно интересующих исследователя зон. В учебниках описаны другие методики
предварительной обработки изображений, такие как пространственное сглажива-
ние, усиление контрастности, устранение размытости и фильтрация [16].
4.3.4.2. Классификация пикселей
После предварительной обработки данных приступают к классификации — крити-
чески важному этапу построения карты распределения. При этом можно применять
различные алгоритмы. Несмотря на то, что исчерпывающий их список не вписыва-
ется в рамки этой книги, здесь будет представлен обзор способов решения задачи по
обработке трехмерных данных (рис. 6). Хемометрические методики сгруппированы
по противопоставлениям следующих критериев: одномерный и многомерный ана-
лиз, простые и комплексные TV-мерные методы, методы многомерного разрешения
кривых и методы распознавания паттернов, а также классификации «с обучением»
или «без обучения».
Одномерные и многомерные методы. Одномерные методы используются в клас-
сической спектроскопии: в зависимости от химической структуры соединения
для расчета карты распределения выбирают определенную длину волны. Для по-
строения псевдо-цветного изображения, в котором наименьшие значения вели-
чин показаны синим или черным цветом, а наибольшие — белым или красным,
используют параметры высоты пика, площади и отношения между двумя пиками.
Одномерные методы — простейший путь построения карт распределения. Тем не
менее трудности могут возникнуть при выборе длин волн, специфичных для данно-
го соединения, особенно в случаях, когда образцы представляют собой комплекс-
ные смеси. Преимуществом гиперспектрального изображения является то, что пол-
ные спектры доступны в виде трехмерных данных. Извлечение всей информации,
содержащейся в наборах данных, вместо информации, полученной на нескольких
длинах волн, улучшает качество химической карты. В этом случае применяемые
методы описывают термином «многомерные». Многомерный анализ изображений
{MIA — multivariate image analysis), основанный на хемометрике, как правило, по-
могает в процессе построения карты распределения. Несколько типов методов MIA
представлено далее: комплексный TV-мерный {strong N-way) метод, метод разреше-
ния многомерной кривой и методы распознавания паттернов.
Многомерный анализ изображений: комплексные и простые многомерные мето-
ды. Комплексные и простые TV-мерные методы используют для анализа 3D- и 2D-
матриц, соответственно. Для описания трехмерной структуры гиперспектральных
данных используется хемометрическая терминология [17]. Двумерная матрица, та-
кая как классический набор БИК-спектрометрических данных, имеет две формы:
О — объекты (строки матрицы данных) и V— переменные (колонки матрицы дан-
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...
495
ных). Трехмерные гиперспектральные данные обладают двумя формами объектов
и одной формой переменных; это можно записать как матрицу данных ОО V из-за
наличия двух пространственных направлений.
Комплексные многомерные методы. Комплексные многомерные методы позво-
ляют анализировать трехмерные данные непосредственно, без преобразования
матрицы, в то время как простые многомерные методы требуют предварительного
шага развертывания. Примеры комплексных многомерных методов, применяемых
для анализа гиперспектральных изображений, включают параллельный факторный
анализ (PARAFAC — parallel factor analysis) и Tucker 3 (трехмерный метод главных
компонент (РСА)) [18]. Их главным преимуществом является то, что в них при-
нимаются во внимание корреляции между пикселями изображения в ОО формах:
В методах развертывания не используется пространственное прилежание пикселей,
приводящее к утере части информации, содержащейся в трехмерных данных.
Несмотря на то, что комплексные JV-мерные методы используются для удаления
шума в изображении, сжатия данных и улучшении визуализации трехмерных дан-
ных, простые многомерные методы применяются чаще, поскольку делают возмож-
ной классификацию с помощью классических одноточечных спектров.
Классические хемометрические методы, т. е. классификация и регрессия, опи-
санные в разделе 4.3.1, также применяются при анализе гиперспектральных изо-
бражений. Однако матрицы X* УхХ необходимо развернуть в матрицы (X*. YyxX перед
обработкой. Иначе говоря, трехмерная OOVматрица разворачивается в классиче-
скую двухмерную ОИматрицу.
Простые многомерные методы. На рис. 7 представлены три этапа простого
JV-мерного анализа: развертывание трехмерных данных, применение выбранных
хемометрических методов и свертывание матрицы для построения карты распреде-
ления. Существует два основных варианта простого JV-мерного анализа:
Разрешение многомерной кривой (MCR — multivariate curve resolution) — общепри-
нятый метод аналитической химии, применяющийся в течение 30 лет (высо-
коэффективная жидкостная хроматография (ВЭЖХ, в английском варианте
HPLC — high-performance liquid chromatography), ИК-Фурье-спектроскопия
(FTIR — Fourier transform infrared), УФ-, БИК-, рамановская спектроско-
пия). Он относится к автомодельному анализу смеси. Для построения гипер-
спектральных изображений из множества разнообразных методов наиболее
успешно применяются простой интерактивный автомодельный анализ сме-
сей (SIMPLISMA — simple-to-use interactive self-modeling mixture analysis) [19]
и MCR методом чередующихся наименьших квадратов (MCR-ALS — MCR
alternating least squares) [20]. Их целью является определение спектральных
профилей химических соединений (так называемые «чистые» спектры) и по-
строение профилей концентраций д ля многокомпонентных систем. Их преи-
муществом является отсутствие необходимости калибровки, т. е. не требуется
предварительной информации.
Распознавание паттернов. Методы распознавания паттернов можно классифи-
цировать по нескольким параметрам. Ниже обсуждается лишь альтернатива
«с обучением»—«без обучения», поскольку она представляет два разных под-
496
Часть 4. Процессно-аналитические технологии
хода к анализу гиперспектральных трехмерных данных. Методы «без обуче-
ния» (кластерный анализ) классифицируют пиксели изображения без кали-
бровки исключительно по спектрам, в отличие от классификаций «с обуче-
нием». Методы выделения характерных признаков [21], такие как РСА (метод
главных компонент) или рекурсивное сжатие (wavelet compression) зачастую
применяются перед кластерным анализом.
РСА применяют для выделения характерных признаков и визуализации данных.
Самый важный результат применения РСА — уменьшение числа длин волн.
Примеры неиерархических методов кластерного анализа [22] включают модели
гауссовых смесей, методы A-средних и нечетких С-средних. Они подразделяются
на методы формальной и содержательной кластеризации. Методы формальной
классификации, такие как метод ^-средних, определяют принадлежность пикселя
к одному из кластеров, в то время как методы содержательной классификации, та-
кие как метод нечетких С-средних, определяют частичную степень принад лежности
пикселя к каждому кластеру.
В классификации «с обучением» используется информация о спектрах и принад-
лежности к классам [23]. Математические модели, в первую очередь, рассчитывают
по калибровочному набору, содержащему информацию о спектрах и классах. Далее,
модель используют для прогноза новых классов образцов. Алгоритмы распознавания
паттернов «с обучением» делятся на три основные категории по парам противопо-
ставлений. К первой категории относятся методы, основанные на дискриминации
(например, линейный дискриминантный анализ (LDA — linear discriminant analysis))
или методы, выявляющие аналогии внутри класса (например, методы формального
независимого моделирования аналогий классов (SIMCA — soft independent modeling
of class analogy)). Вторая категория включает линейные и нелинейные методы, та-
Гиперспектральные
трехмерные данные
D
Матрица
спектральных
данных
Рис. 7. Применение простых А/-мерных методов: предварительная
с развертыванием матрицы (б); свертывание (в). Методы разрешения многомерных кривых
и распознавания паттернов
обработка (а); анализ
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...497
кие как методы нейронных сетей. К третьей категории относятся параметрические
и непараметрические методы. В параметрических методах, таких как LDA, правила
решений используют статистические параметры нормального распределения об-
разцов. В общем, к классическим методам классификации «с обучением» относятся
методы, опирающиеся на расстояния между объектами, LDA, SIMCA и дискри-
минантный анализ (PLS-DA — PLS-discriminant analysis), при котором находят ре-
грессию между спектральными данными и принадлежностью к классам.
Выбор эталонного спектра критически важен при применении методов распо-
знавания паттернов «с обучением». Первый подход к решению задачи — использо-
вать спектры чистых соединений в качестве эталонных. Недостатком этого подхода
является то, что спектры смесей в трехмерных данных зачастую отличаются от эта-
лонных спектров. Поэтому применение модели может дать неправильные результа-
ты. Второй подход, применимый в отдельных исследованиях, заключается в выборе
пикселей изображения, соответствующих только одному соединению, для получе-
ния калибровочных наборов.
4.3.5. Практика химической визуализации в режимах at-Une
и оп-Ппе
После обзора методов, мы проиллюстрируем их применение в фармацевтическом
производстве, уделяя особое внимание одномерным методам, РСА и классифика-
ции «с обучением» {PLS-DA).
4.3.5.1. Практические инструменты анализа карт распределения
Сравнение изображений. Одним из способов анализа карт распределения является
сравнение изображений. В изображение могут быть включены эталоны, такие как
чистые вещества или оригинальные образцы. Например, эталоны и образцы можно
измерять одновременно, если поле обзора достаточно велико; в противном случае
два набора трехмерных данных можно конкатенировать, т. е. сгруппировать вместе.
После построения карты распределения, ее можно интерпретировать простым срав-
нением изображений. В примере, представленном на рис. 8, задача состояла в об-
наружении фальсифицированных продуктов. Оригинальные образцы сравнивали
с с образцами возможно фальсифицированного продукта. После проведения РСА
были обнаружены четкие различия между двумя группами данных. В контрафакт-
ном продукте отсутствовал активный фармацевтический ингредиент {API — active
pharmaceutical ingredient) и имелись другие наполнители. Далее, было проведено ав-
токалибрующееся сравнение с БИК-изображением для быстрого детектирования
контрафактной продукции.
Распределение пикселей и распределение частиц по размеру. Для анализа кар-
ты распределения можно рассчитать количественные параметры. Первый способ
(рис. 9) — визуализация гистограммы пикселей и расчет классических статисти-
ческих параметров, таких как среднее, стандартное отклонение и параметров нор-
мального распределения, т. е. сдвига и эксцесса (остроты пика распределения). На-
пример, большее значение среднего можно объяснить повышенным содержанием
химического соединения, а меньшее стандартное отклонение может свидетельство-
498
Часть 4. Процессно-аналитические технологии
О 10 20 30 40 50 60 70 80
Рис. 8. Автокалибрующееся сравнение изображений для
идентификации контрафактного продукта: отображение счетов
(белый — высокий счет, черный — низкий счет)
вать о большей однородности. Второй способ заключается в расчете размера частиц
и получении информации об их пространственном распределении (рис. 10). Полу-
чают несколько количественных параметров, таких как количество частиц, размер
частиц и процентную долю поверхности, покрытой частицами. Таким образом, ста-
тистические параметры распределения представляют меру однородности образца.
а)
Пиксели
Рис. 9. Изображение и соответствующая гистограмма пикселей (белый — интенсивное
поглощение, черный — слабое поглощение)
б)
3000
2500
2000
1500
1000
500
о
-5 -4 -3 -2 -1
0 1
Пиксели
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...
499
б)
Статистика размеров частиц:
- число частиц 209
- процентная доля покрытой площади 8,7
- средняя площадь 0,2 мм2
- стандартное отклонение площади 0,06 мм2
Статистика распределения частиц
- среднее расстояние до ближайшей частицы 1,35 мм
- координаты центра массХ= 1,5; Y = 1
Рис. 10. Расчет размера частиц методами визуализации: БИК-изображение порошка (а)
(в изображении частицы черные); статистика (б)
4.3.5.2. Выбор длины волны и химическая интерпретация
Общей целью в представленных ниже примерах применения рамановской и
БИК-спектроскопии было представление расположения API и вспомогательных
веществ на поверхности таблетки с целью оценки однородности распределения
и описания характеристик твердого состояния API.
Пример 1. Построение карты распределения API и рамановская спектроскопия.
Метод
Анализ твердых дозированных форм проводили при помощи рамановского ми-
кроскопа (Renishaw, лазер 785 нм, спектральный диапазон 800—100 см-1), детектор
с отображением линий (21 пиксель/линия). Размер изображения — 105x88 пиксе-
лей, то есть 325x270 мкм, время получения изображения составило около 40 мин.
Спектры сглажены и нормализованы. Определены высоты пиков для трех основных
компонентов — API, лактозы и целлюлозы (рис. 11) — с целью построения карт рас-
пределения.
Обсуждение результатов
Полосы рамановского излучения узкие и выбор специфических пиков не сло-
жен. Здесь может быть с успехом применен один из простейших методов классифи-
кации — одномерная классификация по высоте пика. Недостатками рамановской
спектроскопии являются длительность измерения и эффект линейного сканирова-
ния в изображениях (см. рис. 11). Другую проблему представляет собой флуорес-
ценция. Некоторые химические соединения флуоресцируют (например, целлю-
лоза), затемняя соответствующий химический сигнал, что затрудняет построение
карты распределения.
Таким образом, рамановские изображения являются полезным инструментом
при детектировании мелких частиц API на поверхности твердой лекарственной
формы. Этот метод может оказаться наиболее приемлемым способом химической
визуализации при построении карт распределения API благодаря низкому про-
странственному разрешению (до 0,5 мкм/пиксель) и полиморфизму спектральной
информации.
500
Часть 4. Процессно-аналитические технологии
Рамановские стандартные спектры Рамановское изображение
0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
Рамановский сдвиг, см-1
Рис. 11. Рамановские изображения на специфических длинах волн
и стандартные спектры (размер изображения 325x270 мкм; белый —
интенсивное поглощение, черный — слабое поглощение)
Пример 2. Построение модели таблетки методом визуализации данных БИК-
спектроскопии.
Метод
Специальным ножом с одной из поверхностей таблетки удаляли оболочку таким
образом, чтобы получить плоскую поверхность (рис. 12). Испытуемый и стандарт-
ные образцы анализировали при помощи БИК-спектрометра с системой химиче-
ской визуализацией {Sapphire, Malvern) со следующими параметрами: детектор —
матрица 320x256, спектральный диапазон — 1100—2450 нм и пространственное раз-
решение 40 мкм/пиксель. Длительность сканирования — около 5 мин.
Обсуждение результатов
После взятия второй производной можно выбрать длины волн, на которых полу-
чают контрастные изображения, визуализирующие распределение маннитола, API
и кросповидона (рис. 13,14) с помощью БИК-изображений, полученных на опреде-
ленных длинах волн. В этом примере хемометрика помогает в построении карт кон-
центраций, поскольку может использовать информацию, присутствующую во всех
Глава 4,3, Химическая визуализация и хемометрика: полезные инструменты...
501
а)
Маннитол
2080 нм
API
2260 нм
Кросповидон
1930 нм
Рис. 12. Образец и держатель для срезания оболочки
Длина волны,нм
Рис. 13. Изображения на длинах волн, специфичных для компонентов (а), и спектр пикселей
интенсивного поглощения (вторая производная) (размер изображения: 1,02x1,3 см; белый:
интенсивное поглощение, черный: слабое поглощение (б)
502
Часть 4. Процессно-аналитические технологии
Рис. 14. Реконструкция поверхности таблетки. Серый (обычно
красный) — АР1\ темно-серый (обычно синий) — маннитол;
светло-серый (обычно зеленый) — кросповидон; черный —
другие. Размер изображения 1,02 * 1,3 см
спектрах, а не только на определенных длинах волн. Например, метод MCR-ALS
помог выявить пять соединений, в то время как на выбранных длинах волн удалось
выявить только три компонента из-за наложения пиков в БИК-диапазоне. Тем не
менее, более простые методы также являются быстрыми и удобными в применении.
Поскольку ставилась задача определения локализации API, метод выбранной дли-
ны волны позволил получить ожидаемые результаты.
Были построены изображения для определенных длин волн (метод высоты пи-
ков). Изображения были бинаризованы путем, аналогичным преобразованию изо-
бражения в шкале серого цвета в черно-белое изображение. В результате было полу-
чено цветное изображение (рис. 14). Красный канал был ассоциирован с API, синий
канал — с маннитолом, а зеленый — с кросповидоном. После этого стала возмож-
ной реконструкция изображения таблетки. Здесь проявляется главное преимуще-
ство спектроскопии: расширенная область анализа, а это означает, что изображения
более репрезентативны для данного образца.
4.3.5.3. Классификация «без обучения» для поисков сбоев процесса
В данном примере ставились задачи сравнения таблеток в терминах «пло-
хих» и «хороших» характеристик растворения с помощью визуализации данных
ИК-спектроскопии и выявления основных причин плохого растворения [24].
Методология. Анализировали шесть образцов: три выдержали испытание на
растворение (хорошие образцы), другие три — не выдержали (плохие образцы).
Несколько ингредиентов использовали в качестве стандартных образцов: авицел
(целлюлоза), API, стеарат магния и полоксамер. Растворение оценивали после
получения визуальных данных БИК-спектроскопии для получения увлажнен-
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...503
ных химических стандартных образцов. Для получения ИК-спекгров в диапазоне
от 3900 до 900 см-1 при разрешении 16 см-1 (то есть 376 информационных точек)
с продувкой азотом использовали спектрометр Equinox 55, соединенный с микро-
скопом Hyperion 300, с детектором МСТ FPA 64x64 {Вгикег, Эттлинген, Германия;
www.bruckertopicd.com). Для улучшения сигнала применили функцию группировки
пикселей {binning function)-, в итоге получили изображение 16x10 пикселей. Число
сканирований составило 20, а площадь, анализируемая с помощью FPA, составила
270x270 мкм. Для каждой таблетки было получено по два УРЛ-изображения, то есть
изображение размером 32x16 пикселей для каждой таблетки. Три набора трехмер-
ных данных по хорошим образцам были конкатенированы в измерение У для по-
лучения изображения 32x48. Такое же изображение 32x48 было построено для пло-
хих образцов. Два набора хороших и плохих образцов были затем конкатенированы
в измерение X для получения изображения 64x48. Итоговые трехмерные данные
представляли собой матрицу 6x48x376.
Обсуждение результатов. В результатах РСА (рис. 15, 16) интерпретировали на-
грузки с помощью стандартных спектров исходных материалов. Первую нагрузку
соотнесли с полоксамером, вторую — со стеаратом магния (рис. 16), третью — с API,
а четвертую — с авицелом. Тем не менее, другие ингредиенты также могли внести
вклад в нагрузки, поскольку PG4 нагрузки не относятся к спектрам чистых ком-
понентов. РСА выделяет ортогональные сигналы, не относящиеся к стандартным
спектрам. В частности, нагрузка РСЗ, соотнесенная с API, могла быть контамини-
рована другими компонентами, в особенности стеаратом магния. Это подтверждает
недостатки методов извлечения признаков.
Исследование отображения результатов РСА показывает разницу между двумя
наборами данных, т. е. произошло разделение образцов с хорошими и плохими ха-
рактеристиками растворения. Основные различия возникли из-за распределения
стеарата магния и API. Различий в пространственном распределении полоксамера
и авицела выявлено не было. Стеарат магния является гидрофобным веществом,
поэтому центральная часть таблетки защищена от влаги, что замедляет процесс рас-
Рис. 15. Изображение счетов РСА: РС2 — стеарат магния; РСЗ — API: РС4 — авицел; РС1 —
полоксамер не дифференцирован, отсутствует на изображении (белый — интенсивное
поглощение; черный — слабое поглощение)
504
Часть 4. Процессно-аналитические технологии
Рис. 16. РСА нагрузки и сравнение со стандартными спектрами: РС2, стеарат
магния (а); РСЗ, активный ингредиент(б); РС4, авицел (в)
творения. Когда активный ингредиент в большем количестве присутствует на по-
верхности образца, показатели растворения увеличиваются.
Метод анализа высот пиков является классическим методом интерпретации
ИК-спектров. Главное его преимущество состоит в возможности выбора опреде-
ленных длин волн. Недостатком является то, что вследствие наложения полос на-
хождение специфических областей оказывается проблематичным. РСА разрешает
проблему выбора длины волны. Основное его преимущество состоит в том, что он
снижает число переменных, т. е. число анализируемых изображений. Недостатки
кроются в интерпретации нагрузок, отличных от спектров чистых веществ. Интер-
претацию может затруднить тот факт, что несколько химических образцов могут
вносить вклад в одну нагрузку.
43.5.4. Классификация «с обучением», БИК-визуализация и планирование
процесса
Целью данного исследования являлась визуализация БИК-данных для разрешения
проблем при гранулировании в рамках разработки нового лекарственного препа-
рата. Нежелательные агломераты порошка образуются на этапе гранулирования
(рис. 17). Для описания характеристик агломерированных структур был применен
метод визуализации.
Метод. Образец содержал крахмал, API, авицел, кросповидон и натрия лаурил-
сульфат. Испытуемый и стандартные образцы анализировали триплетами с помо-
щью построения изображения БИК-данных {Spectral Dimensions, 20 coadds, спек-
тральный диапазон 1100—2450 нм). Полный размер изображения составил 320x256
пикселей или 4,1x3,3 мм. БИК-изображения интерпретировали, картировали ис-
ходные материалы по образцам при помощи РДУ-классификации с пятью нагруз-
ками (на основании стандартных спектров крахмала, API, авипела, кросповидона
и натрия лаурилсульфата).
Обсуждение результатов. ДЦУ-модель идентифицировала все пять химических
веществ. Многомерный Р£5-анализ показал (рис. 18), что ядро таблетки содержит
Авицел и АРР, на периферии обнаруживается крахмал и кросповидон. Было пред-
ложено решение проблем, возникающих в процессе гранулирования, — чтобы из-
бежать агломерацию, необходимо добавить этап предварительного перемешивания.
Показано, что построение БИК-изображения является полезным инструментом
улучшения понимания процесса.
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты. ..
505
Авицел
Рис. 17. Агломераты порошка: визуально
Натрия лаурил сульфат
API
Крахмал
Кросповидон
мм
Изображение 1 Изображение 2 Изображение 3
Рис. 18. PLS классификация изображений
(три репликатных образца и пять химических
соединений). Имитация процесса гранули-
рования
506 Часть 4. Процессно-аналитические технологии
Преимущество классификации «с обучением» состоит в том, что в ней отсут-
ствует этап выбора длины волны или интерпретации РС4-нагрузки. Она позволяет
также быстро выделить несколько химических компонентов. Этими методами по-
лучают точные результаты при условии, что спектры образцов аналогичны стан-
дартным спектрам. В нашем случае агломераты порошка были гетерогенными, слои
содержали большое количество наполнителя, что сделало возможным применение
классификации «с обучением».
4.3.5.5. Перспективные разработки: анализ гиперспектральных
изображений в реальном времени
Два основных способа применения гиперспектральных изображений в режиме ре-
ального времени относятся к определению конечной точки процесса смешивания
(рис. 19) и контролю процесса капсулирования (рис. 20). Возможны и другие спо-
собы применения, такие как проверка однородности содержимого, определение ха-
рактеристик растворения и количественный анализ содержания воды, однако они
не описаны в этой главе.
При контроле процесса смешивания основным преимуществом визуализации
перед точечной спектроскопией является способность к анализу большей площади.
Мы изучали смеси трех компонентов (см. рис. 19), выбирая определенные длины
волн для авицела, API и лактозы. В начале эксперимента в образце присутствовал
только авицел. Авицел и API были выявлены после трех оборотов смесителя; две
Процесс смешивания
Рис. 19. ИК-изображение мониторинга процесса
смешивания (г— оборот смесителя; белый — интенсивное
поглощение, черный — слабое поглощение)
Глава 4.3. Химическая визуализация и хемометрика: полезные инструменты...
507
Рис. 20. Контроль капсул путем построения БИК-изображений
(размер изображения 33x41 мм). Имитация наличия пустой
капсулы в блистере
карты были комплементарными. Все смеси компонентов стали однородными по-
сле 27 оборотов. Таким образом, анализ временных карт распределения является
инструментом мониторинга процесса смешивания. В нашем примере представлены
ИК-изображения. Тем не менее, в литературе описаны примеры весьма успешного
применения построения БИК-изображений [25].
При контроле процесса капсулирования БИК-излучение проникает через обо-
лочку, что позволяет проверить наполнение капсулы. В нашем примере (рис. 20) ис-
следовали синие непрозрачные капсулы. Однако БИК-изображение позволяет об-
наруживать незаполненные капсулы и проводить мониторинг содержимого капсул,
включая состояние пеллет.
4.3.6. Выводы
Гиперспектральное изображение предоставляет спектральную и пространственную
информацию, которая является как качественной, так и количественной. Изобра-
жение отражает распределение химического соединения и размеры частиц веще-
ства. Подобную информацию невозможно получить, пользуясь методами классиче-
ской спектроскопии. Мы применили методы визуализации для решения проблем,
связанных с контролем качества и процессами, влияющими на качество фармацев-
тических таблеток, такими как растворение, распределение полиморфных форм,
определение содержания влаги, распределение API и его характеристики API, одно-
родность содержимого, смешение и гранулирование. Выбор метода визуализации
основан на нескольких критериях, таких как пространственное и спектральное раз-
решение, длительность измерения и диапазон длин волн. Основные преимущества
и недостатки различных методов приведены в табл. 1 и 2.
Построение спектральных изображений — сложная междисциплинарная область.
Появление новых FPA -детекторов делает метод мощным и привлекательным ин-
508
Часть 4. Процессно-аналитические технологии
Таблица 1
Сравнение типов оборудования
Параметры /Т-системы Рассеивающие Жидкокристаллические перестраиваемые фильтры
Спектроскопия БИК-, ИК-, рамановская Рамановская, БИК- Рамановская, БИК-
Получение изображения Картирование, детектор FPA Картирование Детектор FPA
Режим измерений Отражение, пропускание, НПВО Отражение, пропускание Отражение
Размер изображения Микроскопическое, макроскопическое Микроскопическое Микроскопическое, макроскопическое
Длительность сбора данных Долго Долго Быстро
Таблица 2
Сравнение методов химической визуализации
Параметры Рамановское изображение ИК-изображение БИК-изображение
Надежность оборудования LCTF: +++ Другие: + + +++
Специфичность спектральной информации +++ +++ 1 1
Построение карты составляющих Легко Легко Необходимо применение хемометрики
Примеры Скрининг Идентификация Реконструкция поверх-
применения полиморфных форм, неизвестных ности таблетки, однород-
в фармацевтическом производстве детектирование мелких частиц API частиц ность при смешивании, поиск сбоев в процессе, идентификация фальси- фицированной продукции
струментом. Доказан потенциал метода для качественного фармацевтического ана-
лиза; его можно использовать там, где информация о пространственном строении
становится существенной в случае аналитического применения. В то время как ис-
пользование метода в режиме реального времени и валидация в качестве регулятор-
ного метода требуют дальнейших исследований, потенциальное значение визуали-
зации для контроля качества и PATwe нуждается в дополнительных подтверждениях.
Благодарность
Мы хотим поблагодарить Антона Фишера, руководителя службы контроля качества
(QC manager) Hoffman-La Roche и Рольфа Альтерматта, руководителя секции (Section
Head), Hoffman-La Roche за предоставление ресурсов, позволивших провести данное
исследование, а также Кристель Гендрин, соискателя Ph. D. Hoffman-La Roche за ее
неоценимую помощь.
Глава 4.3, Химическая визуализация и хемометрика: полезные инструменты...509
Литература
1. Roggo, Y., Roeseler, С., and Ulmschneider, М. (2004), Near infrared spectroscopy for qualitative
comparison of pharmaceutical batches, J. Pharm. Biomed. Anal., 36, 777—786.
2. Chalus, R, Roggo, Y., Walter, S., and Ulmschneider, M. (2005), Near-infrared determination of active
substance content in intact low-dosage tablets, Taianta, 66, 1294—1302.
3. Roggo, Y, Duponchel, L., and Huvenne, J.-R (2003), Comparison of supervised pattern recognition
methods with McNemar’s statistical test: Application to qualitative analysis of sugar beet by near-
infrared spectroscopy, Anal. Chim. Acta, 477, 187-200.
4. El-Hagrasy, A. S., Morris, H. R., D’Amico, E, Lodder R. A., and Drennen, J. K. (2001), Near-infra-
red spectroscopy and imaging for the monitoring of powder blend homogeneity, J. Pharm. Sci., 90,
1298-1307.
5. Beleites, C., Steiner, G., Sowa, M. G., Baumgartner, R., Sobottka, S., Schackert, G., and Salzer, R.
(2005), Classification of human gliomas by infrared imaging spectroscopy and chemometric image
processing, Vibrat. Spectrosc., 38, 143-149.
6. Chalmers, J. M., Everall, N. J., Schaeberle, M. D., Levin, I. W., Neil Lewis, E., Kidder, L. H., Wilson,
J., and Crocombe, R. (2002), FT-IR imaging of polymers: An industrial appraisal, Vibrat. Spectrosc.,
30, 43-52.
7. Juan, A. D., Tauler, R., Dyson, R., Marcolli, C., Rault, M., and Maeder, M. (2004), Spectroscopic im-
aging and chemometrics: A powerful combination for global and local sample analysis, TrAC Trends
Anal. Chem., 23, 70-79.
8. Lewis, E. N., Schoppelrei, J., and Lee, E. (2004), Near-infrared chemical imaging and the PAT initia-
tive, Spectroscopy, 19, 26-36.
9. Lewis, E. N., Lee, E., and Kidder, L. H. (2004), Combining imaging and spectroscopy: Solving prob-
lems with near infrared chemical imaging, Microscopy Today, 12, 8-12.
10. Lewis, N., Schoppelrei, J., Lee, E., and Kidder, L. (2005), Near-infrared chemical imaging as a pro-
cess analytical tool, in Bakeev, K. A., Ed., Process Analytical Technology, Blackwell, London, pp.
187-225.
11. Lewi, R J. (2005), Spectral mapping, a personal and historical account of an adventure in multivariate
data analysis, Chemomet. Intell. Lab. Syst., 77, 215-223.
12. Tran, C. D. (2003), Infrared multispectral imaging: Principles and instrumentation, Appl. Spectrosc.
Rev., 38, 133-153.
13. Gift, A. D., Ma, J., Haber, K. S., McClain, B. L., and Ben-Amotz, D. (1999), Near-infrared Raman
imaging microscope based on fiber-bundle image compression, J. Raman Spectrosc., 30, 757-765.
14. Bums, D. A., Ciurczak, E. W. (2001), Handbook of Near-Infrared Analysis, 2nd ed., rev. and ex-
panded, CRC Press, New York.
15. Gat, N. (2000), Imaging spectroscopy using tunable fi Iters: A review, Proc. SPIE, 4056, 50-64.
16. Russ, J. (2002), The Image Processing Handbook, 4th ed., CRC Press, London. 17. Huang, J., Wium,
H., Qvist, К. B., and Esbensen, К. H. (2003), Multi-way methods in image analysis — Relationships
and applications, Chemomet. Intell. Lab. Syst., 66, 141—158.
18. Bro, R. (2003), Multivariate calibration: What is in chemometrics for the analytical chemist? Anal.
Chim. Acta, 500, 185—194.
19. De Braekeleer, K., and Massart, D. L. (1997), Evaluation of the orthogonal projection approach
(OPA) and the SIMPLISMA approach on the Windig standard spectral data sets, Chemomet. Intell.
Lab. Syst., 39, 127-141.
20. Tauler, R. (1995), Multivariate curve resolution applied to second order data, Chemomet. Intell. Lab.
Syst., 30, 133-146.
21. Naes, T., and Martens, H. (1991), Midtivariate Calibration, Wiley, New York.
22. Massart, D. L., Vandeginste, B. G. M., Buydens, L. M. C., Jong, S. D., Lewi, P. J., and Smeyers-
Verbeke, J. (1997), Handbook of Chemometrics and Qualimetrics : Part A, Data Handling in Science
and Technology, 20A, Elsevier, Amsterdam.
510 Часть 4, Процессно-аналитические технологии
23. Massart, D. L., Vandeginste, В. G. М., Deming, S. М., Michotte, Y, and Kaufman, L. (2003),
Chemometrics: A Textbook, Data Handling in Science and Technology, 2, Elsevier, Amsterdam.
24. Roggo, Y, Edmond, A., Chalus, P., and Ulmschneider, M. (2005), Infrared hyperspectral imaging for
qualitative analysis of pharmaceutical solid forms, Anal. Chim. Acta, 535, 79-87.
25. Lyon, R. C., Lester, D. S., Lewis, E. N., Lee, E., Yu, L. X., Jefferson, E. H., and Hussain, A. S. (2002),
Near-infrared spectral imaging for quality assurance of pharmaceutical products: Analysis of tablets
to assess powder blend homogeneity, AAPS PharmSciTech [Electronic Resource], 3, E17.
Часть 5
ПЕРСОНАЛ
Глава 5.1. Обучение персонала, занятого
в фармацевтическом производстве
ДэвидА. Гэллап, Катрин В. Доменик, Мардж Гиллис
Training and Communication Group, Inc., (Беруин, Пенсильвания)
5.1.1. Обзор
5.1.1.1. Общая часть
В соответствии с требованиями закона и этическими нормами, компании, про-
изводящие фармацевтические продукты, должны гарантировать эффективность
и безопасность своей продукции. Гарантия того, что персонал, занятый в фарма-
цевтическом производстве, обладает компетенцией, необходимой для правильного
и эффективного выполнения своих обязанностей, имеет решающее значение для
обеспечения безопасности и эффективности производственного процесса. Совер-
шенствование необходимых для работы навыков, углубление специальных знаний
и формирование нравственного и ответственного подхода к работе имеют решаю-
щее значение при обучении в условиях, ориентированных на соблюдение правил
надлежащей производственной практики.
Данная глава относится к следующим типам организаций, связанным с фарма-
цевтическим производством:
• организации — производители активных фармацевтических ингредиентов
(API — active pharmaceutical ingredient) или производители нерасфасованных
продуктов;
• производители биотехнологических продуктов;
• производители готовых лекарственных форм — жидких и твердых дозирован-
ных форм;
• производители вакцин.
В этой главе рассматриваются общие требования к обучению персонала в усло-
виях фармацевтического производства; также описываются методы планирования
и реализации стратегий обучения, которые гарантируют соответствие фармацевти-
ческих предприятий требованиями, внесенным в Свод федеральных нормативных
актов (CFR — Code of Federal Regulations), проекту руководящего документа Феде-
рального управления США по контролю за пищевой продукцией и лекарствами
(FDA — Food and Drug Administration) по системам качества, а также правилам над-
лежащей практики обучения (GTP—good training practice).
5.1.1.2. Требования к обучению
Требования к обучению персонала, работающего в фармацевтическом производ-
стве, изложены в 21 CFR 211.25 [1]:
а) Весь персонал, вовлеченный в процессы производства, обработки, упа-
ковки и хранения лекарственных продуктов, должен иметь образование,
пройти обучение и иметь опыт работы, позволяющие работнику осу-
514
Часть 5. Персонал
ществлять предписанные функции. Необходимо проводить обучение по
выполнению конкретных операций, которые осуществляет работник,
и правилам действующей надлежащей производственной практики (в том
числе нормативам действующей надлежащей производственной практи-
ки, описанным в этой главе, а также письменным процедурам, включен-
ным в эти нормативы) в той мере, в какой они связаны с функциями, вы-
полняемыми работником. Обучение правилам действующей надлежащей
производственной практики должно проводиться квалифицированными
преподавателями на непрерывной основе и с необходимой частотой, обе-
спечивающей осведомленность персонала о требованиях cGMP, связан-
ных с их профессиональными обязанностями.
Ь) Весь персонал, ответственный за контроль процессов производства, об-
работки, упаковки или хранения лекарственного продукта, должен иметь
образование, пройти обучение и иметь опыт работы, позволяющие работ-
нику осуществлять предписанные функции таким образом, чтобы обеспе-
чить безопасность, подлинность, количественное содержание, другие по-
казатели качества и уровень чистоты, установленные для производимого
лекарственного средства.
с) Для осуществления процессов производства, обработки, упаковки или
хранения всех лекарственных продуктов и руководства за этими процесса-
ми на производстве должно работать надлежащее количество квалифици-
рованных работников.
Таким образом, в Своде федеральных нормативных актов установлено, что пер-
сонал, работающий на фармацевтическом производстве, должен быть обучен в сле-
дующих областях:
1) выполнение своих конкретных обязанностей;
2) выполнение правил действующей надлежащей производственной практики
(cGMP);
3) выполнение письменных инструкций (согласно 21 CFR 211.80).
Нормативы также содержат требования к обучению, которое должно
1) проводиться квалифицированными преподавателями;
2) проводиться на постоянной непрерывной основе.
Наконец, определено, что лица, осуществляющие руководство производствен-
ными процессами, должны проходить такое же обучение, что и «квалифицирован-
ный персонал».
Однако нормативные требования не содержат указаний о том, как должно про-
водиться обучение.
Как заявил Джон В. Левчук (John W. Levchuck), представитель FDA в то время:
«FDA не публиковало руководящих указаний, устанавливающих надлежащие про-
цедуры обучения персонала, и не планирует подобных публикаций в будущем.
Кроме того, FDA не устанавливает строгих требований к обучению» [2]. Возмож-
но, заявление Левчук уже не вполне актуально. Последние указания по обучению
персонала, занятого в фармацевтическом производстве, содержатся в проекте ру-
ководящего документа FDA «Руководство для промышленности: Принципы систем
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве 515
качества в действующих Правилах надлежащей производственной практики» [3].
Руководящий документ, опубликованный в 2004 г., в разделе, относящемся к обуче-
нию персонала, устанавливает:
В рамках системы качества высшее руководство обязано поддерживать в организа-
ции культуру взаимодействия и решения проблем. Руководители обязаны поощрять
информационное взаимодействие, создавая среду, в которой ценят предложения со-
трудников и реагируют на предложения по улучшению производства. Кроме того,
руководству следует создавать междисциплинарные смешанные группы для обмена
идеями по улучшению процедур и процессов.
В рамках системы качества рекомендуется, чтобы сотрудники были специально обу-
чены для выполнения операций, входящих в их профессиональные обязанности,
с учетом сущности их деятельности и потенциальных рисков качества. В соответствии
с требованиями системы качества руководство обязано определить соответствующий
уровень квалификации для каждой должности, чтобы обеспечить соответствие ква-
лификации работника возложенным на него обязанностям. Кроме того, персонал
должен осознавать меру влияния своей деятельности на качество готового продукта
и отношения с заказчиком (эти параметры системы качества также отражены в пра-
вилах cGMP, которые указывают на необходимость наличия определенных квалифи-
каций, т. е. образования, обучения и опыта работы, или любых их комбинации; см.
21 CFR 211.25 (а) & (Ь)). В соответствии с требованиями системы качества непрерыв-
ное обучение имеет решающее значение для гарантии соответствия квалификации
персонала выполняемым обязанностям, а также для понимания ими правил cGMP.
Обычно обучение в рамках системы качества направлено на изучение политик, про-
цессов, процедур и письменных инструкций, связанных с производственной дея-
тельностью, продукцией/обслуживанием, системой качества, желательным уровнем
культуры труда (т. е. командная работа, информационное взаимодействие, управление
изменениями, стиль поведения).
В соответствии с требованиями системы качества (и правилами cGMP) обучение
должно сосредоточиться как на определенных должностных обязанностях, так и на
соответствующих требованиях cGMP.
В соответствии с требованиями системы качества руководство должно утвердить про-
граммы обучения, включающие следующие пункты:
• определение задач обучения;
• предоставление обучения, способствующего решению этих задач;
• оценка эффективности обучения;
• документация по обучению и/или переподготовке.
Там, где функционирует надежная система качества, важно, чтобы органы надзора
контролировали применение навыков, полученных в ходе обучения, в ежедневной
практике.
В проекте руководства по системам качества усилены требования к обучению,
содержащиеся в 21 C/R211.25, и добавлено несколько ключевых пунктов, включаю-
щих определение потребности в обучении, оценку эффективности обучения и тре-
бования к документации. Тем не менее, в документе вновь не содержится описания
того, как нужно планировать, проводить, или оценивать обучение. Кроме того, ру-
516
Часть 5. Персонал
ководящий документ не содержит подробного описания способов сбора и хранения
конкретной информации, связанной с обучением, а также длительности хранения
документации.
В настоящее время руководители признают обучение критически важным ком-
понентом бизнеса, помогающим улучшить эффективность производства и достичь
соответствия нормативным требованиям; в то же время у них могут быть вопро-
сы, связанные с управлением процесса обучения. В отсутствии четких указаний по
обучению многие производители интерпретируют комментарии FDA и результаты
аудита в качестве обоснования подходов к обучению, основанных на компетенциях,
с утвержденными и надежными программами обучения, позволяющими получить
измеряемые результаты. Ответственность исполнителей и их задачи изложены в та-
ких документах, как стандартные рабочие процедуры, рекомендации, протоколы
производственных серий, должностные инструкции и протокольные записи. Обу-
чение должно обеспечить осведомленность всех сотрудников о существовании этих
документов, возможности доступа к ним и способах их использования для управ-
ления рабочим процессом. Далее, «квалифицированные сотрудники» должны про-
демонстрировать, что они прочитали и поняли эти документы, и могут проводить
работы в соответствии с изложенными в них указаниями.
5.1.1.3. Надлежащая практика обучения в фармацевтическом
производстве
Надлежащая практика и концепция обучения, основанные на компетенциях, веро-
ятно, впервые была введена в фармацевтическую промышленность в 1961 г., когда
была издана брошюра Роберта Маджера {Robert Mager) [4]. В этой работе Маджер
показывает преподавателям и инструкторам, как применять системный подход
к разработке обучающих материалов и программ, чтобы студенты и обучающиеся
понимали, какими компетенциями они должны обладать после прохождения кур-
са обучения. Идея разработки системы обучения, основанного на компетенциях,
в фармацевтической промышленности впервые изложена в статье, опубликованной
в 1982 г. Рональдом Тетслаффом {Ronald Tetzlaff), представителем FDA в то время
[5], основанной на работах Маджера и др., в области обучения. В этой статье ав-
тор объясняет, что системный подход к составлению обучающих программ является
лучшим способом организации эффективного последовательного обучения работ-
ников фармацевтической промышленности.
5.1.1.4. Понятие обучения, основанного на компетенциях
Обучение, основанное на компетенциях, представляет собой обучение, направлен-
ное на обеспечение достижения определенных компетенций (знаний и навыков).
Обучение, основанное на компетенциях, позволяет измерить степень усвоения
материалов с помощью тестов или демонстрации навыков (или и того, и другого)
для гарантии приобретения учащимися соответствующих компетенций. Например,
программа обучения, основанная на компетенциях, для операторов оборудования
по производству таблеток должна быть составлена так, чтобы операторы могли про-
следить за всеми стадиями работы оборудования: запуском, работой, остановкой,
а также сбоями в работе. Проверочный тест на приобретение этих компетенций
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве
517
в процессе обучения может включать проверку знания схемы оборудования и де-
монстрацию навыков работы: запуск, работу, отключение оборудования, а также
действия при сбое в работе.
5.1.1.5. Почему так важно обучение, основанное на компетенциях?
Важность обучения, основанного на компетенциях, обоснована его соответствием
рекомендациям FDA относительно обучения, и одновременно требованиям бизнеса.
Компании нужны обученные, компетентные сотрудники, способные изготавливать
продукцию эффективно и качественно. Насколько адекватно отвечает фармацевти-
ческая промышленность современным потребностям9 Согласно одному из отчетов,
«недостаточное количество обученного и опытного персонала в ближайшие пять лет
отрицательно скажется на деятельности более половины мировых биофармацевти-
ческих предприятий-разработчиков и производителей, работающих по контракту,
и повлияет на их способность удовлетворять спрос потребителей (по материалам
обзора, подготовленного с участием 100 международных биофармацевтических
производителей и производственных контрактных организаций)» [6].
С точки зрения соответствия нормативным требованиям, интерес к обучению
также начинает возрастать: «FDA США обращает повышенное внимание на про-
граммы обучения, принятые фармацевтическими производителями в рамках си-
стемного подхода к инспекциям качества. В литературе, посвященной фармацевти-
ческой GMP, вопросы обучения персонала не являются самыми острыми, однако,
как часто отмечают в наблюдениях инспекторы FDA, эта проблема поднимается
в большом числе писем-предупреждений и за последние годы несколько раз ока-
зывалась объектом надзорных действий FDA» [7]. Таким образом, обучение, осно-
ванное на компетенциях, оправдано как с точки зрения потребностей бизнеса, так
и с точки зрения соответствия нормативным требованиям. Хорошо продуманный,
тщательно выполненный план обучения не только помогает фармацевтическим
компаниям в обеспечении соответствия установленным требованиям, но и, как по-
казывают отчеты, способствует возврату средств, инвестированных в обучение.
5.1.2. Разработка плана обучения: стратегия обеспечения
соответствия требованиям к обучению
на фармацевтическом предприятии1
Обучение, соответствующее нормативным требованиям, на фармацевтическом про-
изводственном предприятии требует составления и реализации подробного плана
обучения. Правильно разработанный и выполненный план должен соответствовать
всем требованиям к обучению, включенным в документы:
• 21 CFR 211.25;
• «Руководство для промышленности: принцип системы качества в действую-
щих правилах надлежащей производственной практики»;
• правила надлежащей практики обучения.
1 Материалы, представленные в этом разделе, первоначально были опубликованы в 1999 г.
в PDA Journal [8]. Информация обновлена и расширена; представлены примеры обсуждаемых
материалов.
518
Часть 5. Персонал
Во многих организациях руководители понимают, какой должна быть система
обучения на их предприятии, однако лишь немногие имеют четкий письменный
план обучения, который мог бы помочь им добиться поставленной цели. План обу-
чения — «дорожная карта», направляющая и обобщающая процесс обучения в рам-
ках предприятия. Этот план помогает предприятию обеспечить постоянное движе-
ние к поставленной цели в соответствии с его стандартами качества, производства,
стоимости и безопасности. План должен стать наряду с другими корпоративными
директивами полновесным и авторитетным документом, пользующимся поддерж-
кой высшего руководства, поскольку обучение является важным компонентом для
производства безопасных и эффективных медицинских продуктов.
Почему необходимо разрабатывать план обучения вместо стандартной операци-
онной процедуры (СОП, в английском варианте SOP— standard operating procedure),
описывающей обучение работников? Некоторые руководители полагают, что такая
процедура «обязательна» [9]. Однако следует помнить, что разработка СОП по обу-
чению (или любой СОП) означает, что обучение будет проходить только по данной
СОП. В идеале план обучения должен охватывать область более обширную в срав-
нении с областью, которая обычно охвачена в СОП. План обучения представляет
собой документ, направляющий разработки обучающих программ и материалов,
а также пути внедрения упомянутых программ и материалов. План обучения вклю-
чает принципы обучения и выходит за рамки процедуры. Поскольку план не явля-
ется процедурой, нет необходимости в использовании его в качестве обучающего
материала.
План обучения может соответствовать схеме, представленной на рис. 1. План со-
стоит из пяти разделов.
Рис. 1. Схема плана обучения
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве 519
Примечание. На предприятии должен существовать общий план обучения, охватываю-
щий все предприятие в целом. Каждый отдел должен разработать свой собственный
план обучения, включающий необходимую подготовку персонала для выполнения
всех рабочих функций. План на уровне отдела должен включать график обучения
и переподготовки персонала для обеспечения достаточного числа квалифицирован-
ных работников для постоянного выполнения всех рабочих функций.
Контрольный перечень всех компонентов плана обучения представлен на
рис. 2.
_____ 1. Организация обучения
2ZZZZ] 1 -1. Принятие философии обучения
1.2. Разработка общей концепции обучения
_____ 1.3. Разработка организации обучения
ZZZZj 1.4. Создание системы поддержки обучения
2. Программы обучения
2.1. Идентификация всех учебных программ/персонала, который должен пройти обучение
2.2. Разработка путей повышения персональной квалификации работников
2.3. Материалы к учебным программам
3. Разработка программ обучения
2ZZZ3 3.1. Разработка модели конструирования учебной программы
3.2. Обеспечение разработки валидных учебных программ
3.3. Разработка методологии контроля изменений
4. Реализация программ обучения
4.1 Разработка графика обучения
4.2. Утверждение процедуры реагирования на неудовлетворительные результаты обучения
5. Ведение записей, относящихся к обучению
5.1. Создание системы ведения и хранения записей
5.2. Требования к записям, относящимся к обучению
Рис. 2. Контрольный перечень плана обучения
Раздел 1. Организация обучения
Этот раздел плана обучения посвящен развитию философии обучения и постановке
задач. Они обеспечивают общую основу для организации стабильной обучающей
системы. Кроме того, в обучающей системе должны работать люди, поддерживаю-
щие концепцию обучения на предприятии в целом.
1.1. Развитие философии обучения. После принятия решения о разработке плана
обучения сотрудникам, привлеченным к этой работе, необходимо сформулировать
основные принципы обучения. В большинстве организаций философия обучения
строится вокруг производства, качества, затрат, безопасности и других аспектов
деятельности. Спросите работника, в чем состоит философия компании в терминах
производственных норм, и он (или она), вероятно, сможет дать ответ в нескольких
словах. Но сколько организаций имеет четко проработанную философию обуче-
ния? Совсем немного.
Философия обучения, признающая обучение критически важным компонен-
том в достижении целей корпоративного бизнеса, является ключевым элементом
становления надежной системы обучения в организации. Философия обучения от-
ражает приверженность организации идеям обучения и сообщает работникам ком-
пании, поставщикам и другим заинтересованным сторонам о приверженности ком-
520
Часть 5. Персонал
палии системе обучения, гарантирующей наличие у сотрудников знаний и навыков,
необходимых для сохранения конкурентоспособности в условиях возрастающей
конкуренции. Изложенные принципы обучения должны содержать констатацию
факта: обучение проводится не только для обеспечения соответствия нормативным
требованиям, но и потому, что адекватно обученный персонал вносит вклад в каче-
ство технологических операций и финансовый успех компании. На рис. 3 представ-
лен пример изложения философии обучения.
Как отмечено в общей концепции деятельности нашей компании, мы являемся при-
верженцами стабильной организации обучения. Наша философия обучения включа-
ет следующие элементы:
• Приверженность принципам подготовки всех сотрудников к компетентному
и эффективному выполнению профессиональных обязанностей для про-
изводства продукции, всегда соответствующей требованиям потребителя
к безопасности и эффективности или превосходящей их.
Признание необходимости постоянного обучения всех сотрудников органи-
зации для того, чтобы они могли полностью раскрыть свой потенциал.
Рис. 3. Пример изложения философии обучения
1.2. Разработка общей концепции системы обучения. Философия обучения пред-
лагает основу для общей концепции системы обучения. Общая концепция опреде-
ляет основные цели обучения и акцентирует важность установленных стандартов
квалификации персонала. В общей концепции обучение имеет решающее значение
для качества выпускаемой продукции. Представители всех отделов должны прини-
мать участие в подготовке текста общей концепции обучения в компании, чтобы
в ней нашли отражение их интересы. Образец общей концепции обучения пред-
ставлен на рис. 4.
Наша концепция обучения основана на философии обучения. Мы обязуемся:
• способствовать решению всех корпоративных задач, упомянутых в бизнес-
плане, путем предоставления качественных программ обучения для всех ра-
ботников;
• оценивать влияние этих программ на развитие бизнеса;
• способствовать развитию и профессиональному росту наших сотрудников
во всех сферах жизни, включая технические сферы и межличностное взаи-
модействие;
• способствовать поддержанию статуса организации как лидирующей компа-
нии высшего уровня, способной адаптироваться к переменам в постоянно
изменяющейся нормативно-правовой среде;
• организовать непрерывный процесс обучения для обеспечения соответствия
персонала корпоративным и нормативным требованиям;
• обеспечить ресурсы для профессионального роста сотрудников, одновре-
менно продвигаясь к целям, поставленным организацией, — качеству, про-
изводительности, безопасности и финансовому успеху.
Рис. 4. Образец общей концепции обучения
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве
521
После выработки философии обучения и принятия общей концепции, необхо-
димо довести их до сведения всех сотрудников, чтобы получить поддержку и нала-
дить обратную связь. При распространении информации следует сосредоточиться
на политике компании в области обучения, а также на роли руководства в обеспече-
нии эффективности обучения в организации. Четко изложенная философия и об-
щая концепция обучения представляют основу для разработки плана внедрения
обучения во все сферы профессионального развития персонала.
1.3. Разработка организации обучения. Если система обучения на предприятии
еще не сформирована, то ее необходимо создать усилиями руководства корпорации
или производственного подразделения. Наш опыт показывает, что обучение наибо-
лее эффективно, когда обучающая система или отдел функционирует в рамках про-
изводственного подразделения — на одном уровне с другими основными отделами
производства. Типичная организационная структура отдела обучения в рамках фар-
мацевтического производственного предприятия представлена на рис. 5.
Рис. 5. Примерная структура отдела обучения
Распределение функций и ответственностей должно быть представлено в плане.
В зависимости от размера предприятия некоторые функции могут быть объединены.
Руководитель подразделения по обучению и развитию персонала. Поскольку цель
обучения состоит в обеспечении уверенности персонала в способности выполнять
свои профессиональные обязанности и, в конечном итоге, стабильно производить
качественную продукцию, обучение должно способствовать выполнению всех про-
чих функций предприятия и поддерживаться всеми подразделениями. Руководитель
подразделения по обучению и развитию персонала должен взаимодействовать со
всеми функциональными подразделениями, включая обеспечение качества, безо-
пасности, производство, информационные системы и лабораторию. Несмотря на то
что руководство многих организаций в принципе соглашается с этим, на практике
все выглядит несколько иначе.
Руководитель подразделения должен направлять, мониторировать и поддержи-
вать мероприятия по обучению во всей организации. Это должностное лицо долж-
но проводить регулярные встречи с руководством производственных площадок для
анализа текущего плана обучения и оценки его выполнения для обеспечения ре-
шения задач бизнеса и соответствия регуляторным требованиям. Он или она также
руководит работой координаторов обучающих программ, помогает осуществлять
522
Часть 5. Персонал
взаимодействие и разрабатывать планы обучения и процедуры итоговых оценок для
структурных подразделений для проверки эффективности внедряемых программ.
Эта деятельность должна распространяться на внутреннее обучение в структур-
ных подразделениях, чтобы обеспечить наличие в каждом подразделении системы,
отвечающей их специфическим требованиям по обучению. Руководитель подраз-
деления по обучению и развитию персонала должен разработать указания для всех
отделов для обеспечения соответствия политике обучения, принятой в корпорации.
В этих указаниях должны быть перечислены координаторы обучения; составите-
ли учебных программ; инструкторы, работающие в подразделениях, и технические
эксперты, а также лица, ответственные за составление расписания и ведение запи-
сей по обучению.
Руководитель подразделения по обучению и развитию персонала несет ответ-
ственность за исполнение следующих обязанностей:
• разработку, поддержание в рабочем состоянии и осуществление плана обуче-
ния на предприятии;
• определение потребностей каждого подразделения в обучении, необходимом
для осуществления целей бизнеса и обеспечения соответствия установлен-
ным нормативным требованиям;
• разработку графиков и планов обучения, соответствующих потребностям ор-
ганизации в обучении;
• анализ планов обучения и графиков, обеспечивающих поддержку обучающих
программ всех отделов;
• разработку методик измерения эффективности обучения;
• руководство и контроль координаторов обучения при выполнении планов
обучения.
Инструктор (инструкторы) по безопасности. Инструкторы по безопасности —
лица, проводящие занятия по технике безопасности для работников предприятия.
Инструкторы по безопасности несут ответственность, по меньшей мере, за испол-
нение следующих обязанностей:
• работа с составителями учебных программ при разработке программ обуче-
ния технике безопасности;
• проведение необходимого инструктажа по технике безопасности в соответ-
ствующих отделах по таким вопросам, как:
— действия в случае неисправностей {lockout/tagout),
— доступ в закрытые зоны,
— защита органов слуха,
— защита органов дыхания,
— инфекции, передающиеся через кровь;
• определение текущих потребностей в обучении технике безопасности.
Инструктор (инструкторы) по обучению менеджменту. Инструкторы по обуче-
нию менеджменту — лица, ведущие программы обучения «социальным» навыкам
работы с людьми для сотрудников фармацевтических производственных предпри-
ятий. В сферу ответственности инструкторов по обучению руководящих входит ис-
полнение следующих обязанностей:
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве 523
• работа с составителями учебных программ при разработке программ обуче-
ния менеджменту;
• представление программ развития управленческих навыков по таким пробле-
мам, как:
— лидерство,
— решение проблем и принятие решений,
— коучинг (индивидуальное обучение) и консультирование,
— организация рабочего времени.
Инструктор (инструкторы) по GXP. Инструкторы по GXP, в первую очередь,
несут ответственность за представление программ обучения действующим норма-
тивным требованиям, существующим в фармацевтической промышленности. Ин-
структоры по GXP отвечают за исполнение следующих обязанностей:
• работа с составителями учебных программ при разработке программ обуче-
ния GXP;
• начальное обучение основам GXP новых сотрудников;
• представление постоянных аспектов GXP в сфере:
— надлежащей производственной практики,
— надлежащей практики документирования,
— надлежащей лабораторной практики.
Составители учебных программ. Составители учебных программ разрабатывают
программы обучения в направлениях, указанных координаторами обучения. Со-
ставители учебных программ являются связующим звеном между координаторами
обучения, руководителем подразделения по обучению и развитию персонала, ли-
цами, осуществляющими контроль на уровне отдела и техническими экспертами.
К обязанностям, за исполнение которых отвечают составители учебных программ,
относятся следующие:
• подбор соответствующих технических экспертов для программ обучения при
согласовании с координаторами обучения на уровне отделов;
• разработка программ обучения, основанных на компетенциях, в соответствии
с установленными принципами системы разработки таких программ;
• разработка методик оценки эффективности обучения;
• разработка программ обучения для инструкторов, работающих в подразделе-
ниях.
1.4. Создание системы поддержки обучения. Для того чтобы отдел обучения мог
функционировать должным образом, необходимо создать систему поддержки обу-
чения во всех функциональных подразделениях производственного предприятия.
В каждом подразделении должны быть определены три роли, исполнение которых
необходимо для поддержки программ обучения. Иногда эти роли может исполнять
один сотрудник; зачастую они распределены между сотрудниками отдела. Как пра-
вило, для выполнения этих обязанностей не нужно выделять сотрудника на полную
занятость. В табл. 1 приведен список отделов и поддерживающих обучение ролей,
необходимых для каждого отдела; возможно, в этот список могут быть внесены до-
полнения. Далее представлено описание каждой из этих функций.
524
Часть 5. Персонал
Таблица 1
Отделы организации и функции, поддерживающие обучение
Координатор^) Технические Инструкторы
________________________________обучения________эксперты______подразделений
Административный
и финансовый
Зданий и территорий
Производственных помещений
и инженерных служб
Управления персоналом
Информационных систем
Производственный
Нормативно-правового
регулирования
Научно-исследовательский
Безопасности и охраны труда
Получения и отгрузки (склад)
Координаторы обучения. Координаторы обучения — посредники между функци-
ональными подразделениями и отделом обучения. Они отвечают за взаимодействие
с руководителем обучения и развития, доводя до его сведения информацию о не-
обходимости обучения на уровне подразделений. Координаторы обучения также
являются связующим звеном между техническими экспертами и инструкторами на
уровне отделов и составителями учебных программ. Их задача — определение необ-
ходимости в обучении и разработка учебных программ в рамках плана, составлен-
ного руководителем подразделения по обучению и развития персонала, одобренно-
го руководством предприятия. Кроме того, они несут ответственность за поддержку
планов обучения в рамках подразделений. Координаторы обучения могут обслужи-
вать несколько структур предприятия. Координаторы обучения отвечают, по мень-
шей мере, за исполнение следующих обязанностей:
• консультации с руководителями подразделений с целью определения потреб-
ности в обучении;
• доведение информации о потребности подразделений в обучении до руково-
дителя отдела по обучению и развитию;
• разработка планов обучения на уровне отделов и обеспечении специализа-
ции программ обучения в соответствии с указаниями руководителя обучения
и развития, а также руководства отдела;
• руководство составителями учебных программ при работе над программами
обучения, а также разработке методик оценки эффективности обучения.
Технические эксперты. Технические эксперты — лица, обладающие обширными
знаниями в сфере своей деятельности. Обычно это сотрудники, работающие дли-
тельное время в компании и подразделении, как правило, более пяти лет. К обязан-
ностям, за которые отвечают эксперты в предметной области, относятся следую-
щие:
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве 525
• работа с координаторами обучения для определения потребности в обуче-
нии;
• работа с составителями учебных программ при планировании и разработке
программ обучения;
• работа с инструкторами подразделений по реализации программ обучения.
Инструкторы подразделений. Инструкторов подразделений следует выбирать из
числа сотрудников, обладающих определенными навыками, интересом к препода-
ванию и способности к выполнению данной работы. Некоторые технические экс-
перты могут стать инструкторами отделов при условии, что они изъявляют желание
учиться мастерству преподавания. В круг обязанностей, за которые несут ответ-
ственность инструкторы отделов, как правило, входят следующие:
• работа с составителями учебных программ при планировании и разработке
программ обучения;
• обучение новых сотрудников и должностных лиц специальным навыкам ра-
боты и инструктаж по нормативно-правовым вопросам.
Для обеспечения соответствия требованиям 21 CFR 211.25 инструкторы подраз-
делений должны пройти программы обучения инструкторов. Список тем, обычно
включаемых в программы обучения инструкторов, представлен в табл. 2.
Таблица 2
Примерная схема обучения инструкторов
№ Название темы
1 Введение
2 Задачи программы обучения
3 Ответственность инструктора
4 Процесс обучения: характеристики обучающихся
5 Процесс обучения: теория обучения взрослых
6 Процесс обучения: что делает обучение успешным?
7 Что позволяет инструктору добиваться успеха?
8 Методики обучения
9 План
10 Подготовка
11 Презентация
12 Обратная связь и коучинг
13 Использование перечня контрольных вопросов для обучения на рабочем месте
14 Практическое занятие 1 — обучения на рабочем месте выполнению СОП
15 Практическое занятие 2 — перечень контрольных вопросов для обучения на рабочем
месте
Обзорная и заключительная часть
Раздел 2. Программы обучения
В этом разделе представлен список персонала, который должен пройти обучение,
а также перечень учебных программ, необходимых для производственного пред-
526
Часть 5. Персонал
приятия. Кроме того, обсуждаются пути повышения квалификации сотрудников
и приводятся соответствующие примеры.
2.1. Составление списка всех учебных программ/персонала, который должен пройти
обучение. В табл. 3 и 4 представлены примерные списки персонала, который дол-
жен пройти обучение, и учебных программ, которые они должны освоить для того,
чтобы эффективно работать на фармацевтическом производственном предприятии:
в эти списки по желанию могут быть внесены дополнения.
Таблица 3
Учебные программы для руководства
Учебные программы Персонал, который должен пройти обучение
Лидерство Коучинг и консультирование Решение проблем/принятие решений Коучинг и консультирование Руководители отделов и выше Тоже » »
Учебные программы для сотрудников Таблица 4
Учебные программы Персонал, который должен пройти обучения
Гемоконтакгные патогены Проведение эффективных исследований Правила маркировки лекарств Инспекции FDA Электронные подписи и протоколы серий Нормативы оповещения об опасности (Hazcom) Составление письменных СОП Контроль и мониторинг Работа с научно-исследовательскими организациями по контракту Концепции валидации Соответствующий персонал Тоже » » » » » » » »
2.2. Разработка персональных путей повышения квалификации. Надлежащая прак-
тика бизнеса, а также 21 CFR 211.25, устанавливают требование: каждый работник
должен иметь образование, пройти обучение и получить опыт, позволяющий ему
(или ей) осуществлять функции, соответствующие должностным обязанностям,
безопасным и эффективным образом. Учебный план должен отражать политику,
позволяющую персоналу демонстрировать свою квалификацию. Это относится как
к персоналу, проводящему обучение и осуществляющему контроль, так и к работ-
никам, непосредственно занятым в производственных операциях.
Схема повышения квалификации должна включать все учебные программы, не-
обходимые для выполнения работниками своих производственных функций. При-
мер персонального пути повышения квалификации операторов для процесса табле-
тирования представлен в табл. 5.
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве
527
Таблица 5
Примерная схема повышения квалификации персонала
Отдел: производственный
Название должности:
оператор по таблетированию
Производственные задачи _, , Дата начала Дата окончания Обязанности работника обу,!СНИЯ обучения
Заполнение документации Проведение очистки оборудования Маршрутные карты (master batch record) Мелкая чистка оборудования Большая чистка съемных частей Большая чистка рабочих центров
Эксплуатация основного производственного оборудования Таблеточный пресс фирмы Fette Таблеточный пресс фирмы Killian Таблеточный пресс фирмы Coutroy
Эксплуатация высокотехнологичного производственного оборудования Эксплуатация вспомогательного оборудования Теория прессования Эксплуатация и очистка тестера истираемости таблеток Эксплуатация и очистка тестера таблеток на прочность (на излом) Эксплуатация и очистка микрометра Эксплуатация и очистка металлодетекторов Эксплуатация автоконтроллеров массы
Перемещение материалов Перемещение мешков на поддоны (паллеты) Эксплуатация и обращение с контейнерами для транспортировки таблеток (внутри предприятия) Перемещение контейнеров/ мешков с таблетками/пакетов для покрытия оболочкой
528
Часть 5. Персонал
2.3. Материалы к учебным программам. Существует три базовых метода обуче-
ния — занятия в классах, самостоятельная учеба, а также комбинации этих методов.
В любом случае для обучения необходимо принять следующие методики:
1. Методика передачи знаний или желательных установок. Этот тип обучения
обычно применяется при занятиях в классах или в системе самостоятельного
обучения (на основе бумажных или электронных материалов).
2. Методика оценки степени передачи знаний или установок, т. е. определен-
ную форму оценки.
Если целью обучения является приобретение работниками квалификации
в определенных навыках или выполнении конкретных задач/операций, то возника-
ет необходимость разработки учебных материалов, таких как:
3. Структурированный перечень, позволяющий разным инструкторам демон-
стрировать каждому обучающемуся одинаковый способ выполнения кон-
кретной операции.
4. Контрольный список вопросов, который использует инструктор (предпо-
лагается, что инструктор, проводящий первичное обучение, не занимается
оценкой компетенций) для определения компетенции обучающихся для вы-
полнения предписанных обязанностей.
Периодичность обучения. Как правило, в проведении обучения или перепод-
готовки на рутинной основе нет необходимости. Отдельные учебные программы
обязательны для повторения. Это относится к курсам по технике безопасности,
а необходимость переподготовки устанавливают федеральные или местные власти
или правительство штата. Повторения отдельных программ, таких как программа
предотвращения сексуальных домогательств на рабочем месте, может потребовать
руководство компании. Переподготовка может понадобиться, если сотрудник (или
сотрудница) длительно отсутствовал на рабочем месте из-за болезни, отпуска по
беременности, другого рабочего назначения и т. д. В этом случае компания может
потребовать переподготовку после перерыва в выполнении профессиональных обя-
занностей. Кроме того, необходимость обучения или переподготовки может воз-
никнуть в следующих ситуациях:
• продукция не соответствует спецификациям (проблема качества);
• наличие избыточных отходов;
• снижение производительности труда.
Необходимость обучения может также быть выявлена в ходе периодических
аудитов. Однако наш опыт показал, что необходимость переподготовки, в основ-
ном, связана с ситуациями, аналогичными вышеупомянутым.
Раздел 3. Разработки учебных программ
Работникам необходимо специальное обучение в области охраны окружающей сре-
ды, СОП, техники безопасности, правил GMP и технических знаний и навыков.
В учебном плане должен быть применен системный подход к разработке и внедре-
нию обучения, основанного на компетенциях, во всех программах и учебных мате-
риалах.
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве 529
3.1. Определение модели планирования учебной программы. Эффективное обуче-
ние, основанное на компетенциях, возможно в результате применения системного
подхода к планированию учебных программ. Процесс планирования включает чет-
ко выраженные этапы разработки учебной программы, отвечающей требованиям
как обучающихся, так и организации. Самым надежным методом обучения сотруд-
ников безопасным и эффективным методам работы является модель конструирова-
ния систем обучения. Одна из моделей конструирования систем обучения — TPDM
(trainingprogram design model), разработанная Гэллопом и Гриффином, представлена
на рис. 6.
Рис. 6. Модель конструирования учебной программы
3.2. Разработка валидных программ обучения. Валидность — мера соответствия
того, насколько методы и результаты способствуют решению поставленных задач.
Учебная программа считается валидной, если она точно передает знания, необходи-
мые для выполнения профессиональных обязанностей. Как можно провести вали-
дацию учебной программы? Валидацию программы можно обеспечить, если следо-
вать модулю конструирования программы и следующему процессу:
1. Провести анализ задач, идентифицируя отдельные фрагменты информации
и специфические компетенции, требующиеся для выполнения работы; завизиро-
вать завершенный анализ работы у технического эксперта — это будет означать, что
отдельные фрагменты информации и специфические задачи, необходимые для вы-
полнения работ, идентифицированы.
2. Определить и записать измеряемые задачи обучения, по которым можно точ-
но определить, что должен знать обучающийся по окончании курса обучения; зави-
530
Часть 5. Персонал
зировать завершенный список задач обучения у технического эксперта — это будет
означать, что они подтверждают приемлемость установленного уровня обучения.
3. Разработать методики оценки, соответствующие задачам обучения; завизи-
ровать завершенный анализ способов оценки у технического эксперта — это будет
означать, что эксперты согласны с тем, что методики оценки адекватно измеряют
компетентность обучающихся.
Оценка знаний и навыков обучающихся производится по утвержденным мето-
дикам; следует убедиться, что оценивание всех групп работников проводится оди-
наковым путем.
3. 3. Разработка принципов и процедур контроля изменений. Политика контроля
изменений помогает обеспечить готовность персонала к эффективному внедрению
новых и пересмотренных технологических принципов и процедур. Эффективная
политика контроля изменений должна содержать указания по проведению регуляр-
ного анализа и пересмотра учебных программ и материалов, а также графики обу-
чения новых и действующих работников выполнению всех соответствующих СОП.
Кроме того, план должен включать процедуру отклика на новые разработки в об-
ласти операций, процессов и ведения документации.
Раздел 4. Реализация программ обучения
4.1. Составление графиков проведения обучения. План должен включать расписание,
необходимое для гарантии того, что обучение начинается при поступлении на ра-
боту и продолжается непрерывно. В расписании должны быть указаны временное
рамки завершения обучения для введения в должность, и предусмотрены курсы
обучения, которые проводятся при изменениях в процессе, или появлении новых
или усиленных требований к эффективности труда. Разработку плана внедрения
программ обучения можно завершить, добавлением его в таблицу, представленную
в разделе 2, пункт 2.1 «Составление списка всех учебных программ/персонала, ко-
торый должен пройти обучение». Исправленный вид плана показан в табл. 6.
4.2. Разработка процедуры реагирования на неудовлетворительный результат обу-
чения. План обучения должен содержать описание процесса работы с персоналом,
не получившим удовлетворительных оценок при испытаниях. В описание должны
быть включены шаги по устранению причины неудач, которые могут быть связаны
как с недостатками учебной программы, так и с неверным подбором кандидатов для
работы. План должен включать процедуры, описывающие действия в случае неудо-
влетворительных результатов испытаний. Типичная последовательность действий
при неудовлетворительных результатах испытаний представлена на рис. 7. После
прохождения работником курса обучения по программе, направленной на усвоение
знаний для выполнения конкретной задачи, выставляется оценка. Если оценка удо-
влетворительная, работник переходит к следующему этапу обучения — выработке
навыков. По окончании этого этапа происходит проверка навыка. Если обучаемый
демонстрирует хорошие навыки, он проходит аттестацию или получает квалифи-
кацию, необходимую для выполнения данной технологической операции. Если
обучаемый не проходит проверку на наличие навыков, следует повторить обучение.
Допустимое количество повторений курса обучения и испытаний должно быть ука-
зано в процедуре реагирования на неудовлетворительный результат обучения.
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве
531
Примерная схема обучения персонала Таблица 6
Программы обучения специалистов
Учебные программы Персонал, который должен пройти обучение Трафик обучения
Инфекционные заболевания, Соответствующий По необходимости
передаваемые с кровью персонал
Проведение эффективных расследований Тоже Тоже
Правила маркировки лекарств »
Инспекции FDA » »
Электронные подписи и протоколы серий )» »
Hazcom (Hazard Communication) — нормативы оповещения об опасности » »
Составление СОП » »
Контроль и мониторинг »
Работа с контрактными » »
исследовательскими организациями Концепции валидации »
Рис. 7. Базовая процедура реагирования на неудовлетворительный результат обучения
Раздел 5. Ведение записей, относящихся к обучению
FDA не устанавливает требований в Своде федеральных нормативных актов к веде-
нию документации, связанной с обучением, однако эти требования установились
в промышленности на основе прецедентов, формирующих дальнейшую практику.
Обсуждения с руководителями учебных программ фармацевтических предприя-
тий, которые были проинспектированы FDA, позволяют предположить, что аудит
системы обучения на предприятии FDA проводит через документацию по обучению.
Обычно это происходит одним из трех следующих вариантов:
532
Часть 5. Персонал
1. Аудиторы запрашивают СОП и наблюдают за выполнением процедуры работ-
ником. В зависимости от качества выполнения процедуры работником, аудиторы мо-
гут запросить документы или записи, относящиеся к обучению данного работника.
2. При изучении производственной документации аудиторы обнаруживают от-
клонения от нормативов или несоответствие спецификациям. Это может, в свою
очередь, привлечь внимание к работе оператора и проверке соответствия его ква-
лификации выполняемой работе. Аудиторы могут поинтересоваться, какое допол-
нительное обучение прошел работник, и как проводилось обучение. Для этого они
мыут запросить записи, подтверждающие, что обучение проводилось.
3. Аудиторы могут выявить процессы или рабочие практики, осуществляемые
некорректно, в ходе рутинного осмотра производственного предприятия. Аудиторы
могут запросить учебные материалы или записи, относящиеся к обучению, связан-
ные с персоналом, работающим в данной зоне.
Также руководители обучения отмечают: если отдел обучения в состоянии бы-
стро и организованно представить запрашиваемую документацию, аудиторы склон-
ны оценивать систему обучения как эффективно работающую. В то же время там,
где важную документацию, относящуюся к обучению, не могут представить доста-
точно быстро, аудиторы могут провести более глубокий анализ системы обучения
[10]. Поэтому ведение записей имеет решающее значение для демонстрации эф-
фективно работающей системы обучения на фармацевтическом производственном
предприятии.
5.1. Организация системы ведения записей. Любая система ведения записей долж-
на отвечать требованиям «организованного хранения и быстрого представления за-
писей».
На фармацевтическом производственном предприятии обычно используются
три способа ведения (хранения) документации, относящейся к обучению: ведение
записей на бумажных носителях; на электронных носителях с копиями на бумаж-
ных; исключительно электронные системы. В некоторых фармацевтических произ-
водственных компаниях используется система ведения документации в бумажной
форме. Электронные системы с бумажными копиями, известные также как систе-
мы управления обучения (LMS— learning management system), варьируют от сложных
программных разработок, таких как IsoTrain, SAP, Plateau и Registar до баз данных
отделов в Microsoft Access. Как правило, в виде бумажных копий сохраняют, по мень-
шей мере, списки персонала, проходящего обучение, и иногда методики оценива-
ния. /.Л/Л'является, главным образом, базой данных, содержащей множество таблиц
для хранения дискретных единиц информации. В таблицах может быть представ-
лена информация о сотрудниках, пройденных курсах обучения, оценках и учебных
материалах. Эти таблицы можно объединять или использовать по отдельности для
составления отчетов. Такие отчеты позволяют пользователям сравнивать, анализи-
ровать и решать конкретные вопросы планирования обучения.
5.2. Требования к ведению записей, относящихся к обучению. Любая система веде-
ния записей, относящихся к обучению, должна фиксировать следующую обязатель-
ную информацию:
• имя работника;
• идентификация работника (табельный номер);
Глава 5.1. Обучение персонала, занятого в фармацевтическом производстве
533
• персональный путь повышения квалификации, в том числе программы обу-
чения, которые необходимо пройти и намеченные даты завершения обуче-
ния;
• результаты по завершенным учебным программам.
Кроме того, материалы к учебным программам, включая руководства для обуча-
ющихся и методики оценки результатов, должны быть легко доступны в электрон-
ном или бумажном виде.
Литература
1. U.S. Food and Drug Administration (FDA) (2003), Current good manufacturing practice for fi nished
pharmaceuticals, Code of Federal Regulations, Title 21, Part 211.25, FDA, Rockuille, MD.
2. Levchuck, J. W. (1991), Training for GMPs, J. Parenter. Sci. Technol., 45 (6), 270-275.
3. U.S. Food and Drug Administration (FDA) (2004), Guidance for industry: Quality systems approach
to pharmaceutical good manufacturing practice regulations, FDA, Rockuille, MD.
4. Mager, R. (1962), Preparing Instructional Objectives, Center for Effective Performance, Atlanta,
GA.
5. Tetzlaff, R. (1982), A systematic approach to GMP training, Pharm. Technol., 6 (11), 42-51.
6. Langer, E. S. (2004), Training day for International Biopharma Manufacturers, Contract-Pharma, 6
(2), 46.
7. Morris, W. (2006), Personnel training: A growing compliance concern, PDA Newslett., XLII (4),
1-38.
8. Gallup, D. A., Beauchemin, K. A., and Gillis, M. (1999), A comprehensive approach to compliance
training in a pharmaceutical manufacturing facility, PDA J. S. Technol., 53 (4), 163-167.
9. Vesper, J. L. (2000), Defi ning your GMP training program with a training procedure, BioPharm., 13
(7), 28-32.
10. Gallup, D. A., Beauchemin, K. A., Gillis, M., Altopedi, D. and Manor, J. (2003), Selecting
a training documentation/recordkeeping system in a pharmaceutical manufacturing environment,
PDA J. Sci. Technol., 57 (1), 49-55.
Часть 6
КОНТАМИНАЦИЯ И ЕЕ КОНТРОЛЬ
Глава 6.1. Источники контаминации
Дениз Борер
Федеральный университет Санта Мария (Санта Мария, Бразилия)
6.1.1. Введение
Существует много подходов к рассмотрению проблемы контаминации фармацевти-
ческих продуктов. Несмотря на то что источники контаминации легко могут быть
выявлены, используемые в этой области определения можно рассматривать с не-
скольких позиций.
Прежде всего, контаминация фармацевтического продукта может рассматри-
ваться в рамках терминов «родственные вещества» и «технологические примеси».
При этом родственные вещества по своей структуре близки к действующему веще-
ству, а технологические примеси вносятся в ходе технологического процесса или об-
работки. Эти два термина включают все типы загрязнений, но не содержат их пол-
ного описания.
Фармакопея США и Национальный формуляр (USP-NF 27, U.S. Pharmacopeia
and National Formulary) [1] содержат определения терминов, связанных с контами-
нацией продукта, в статьях <1086> «Примеси в фармакопейных продуктах» и <466>
«Обычные примеси». В этих статьях примеси классифицируют на неорганические,
органические, биохимические, изомерические и полимерные соединения, а опре-
деления формулируют в терминах посторонних примесей, токсичных примесей,
сопутствующих компонентов, сигнальных (контролируемых) и обычных примесей.
Согласно представленным в Фармакопее определениям, посторонние примеси1 —
это примеси, появившиеся не при синтезе действующего вещества или производ-
стве препарата, а вследствие контаминации или подделки. Сопутствующие компо-
ненты являются характерными компонентами исходных химических продуктов и
не считаются «истинными» примесями с фармакопейной точки зрения. Обычные
примеси — вещества, присутствующие в исходных химических веществах, не оказы-
вающие существенного нежелательного биологического действия и не являющиеся
токсичными в определенных количествах.
В отличие от посторонних примесей, которые в принципе должны отсутствовать
в продукте, и, следовательно, не принимаются во внимание при выборе фармако-
пейных испытаний и количественных определений, токсичные примеси и сигнальные
(контролируемые) примеси могут образовываться при синтезе, приготовлении или
разложении фармакопейных веществ и, в отличие от обычных примесей, могут обла-
дать нежелательным биологическим действием даже в незначительных количествах.
Исходя из такого представления примесей, должны быть рассмотрены следующие
вопросы:
1 Обращаем внимание читателей, что в России под термином «посторонние примеси»
обычно понимают родственные вещества (родственные примеси), а примеси, о которых
идет речь, часто называют подмесями. — Примеч. перев.
538
Часть 6. Контаминация и ее контроль
• происхождение действующего вещества — природное, синтетическое, био-
технологическое;
• соотношение примесь/действующее вещество и токсикологические эффекты
примеси;
• фармакологические эффекты примеси.
При установлении предельно допустимого содержания примесей в фармацевти-
ческой субстанции также должны быть приняты во внимание следующие факторы:
• путь введения лекарственного препарата;
• дозы;
• целевая группа больных;
• длительность лекарственной терапии.
К примесям, имеющим внешнее происхождение, но не включенным в катего-
рию технологических примесей, относятся примеси, образующиеся при восстановле-
нии препарата или смешивании, а также при введении больному. В этой категории
примесей крайне важными являются механические включения в препаратах для
внутривенного введения, главным образом в растворах для инфузий, что обуслов-
лено большими объемами вводимых растворов.
На рис. 1 приведены основные источники образования примесей (как внутрен-
ние, так и внешние) и стадии, на которых происходит контаминация. На рисунке
также показано, каким образом «происхождение контаминации» будет рассматри-
ваться в этой главе: источники контаминации разделены на внутренние и внеш-
ние. Первый раздел — внутренние источники — посвящен воде, поскольку это
основной исходный материал, и даже если вода не входит в состав лекарственной
формы, она все равно используется на всех стадиях ее производства. После воды
будет рассмотрена контаминация, возникающая из исходных материалов, с учетом
наличия сопутствующих веществ (естественных примесей, содержащихся в исход-
ных материалах) и побочных продуктов, образующихся в процессе синтеза. Также
большое значение среди внутренних примесей имеют примеси, образующиеся при
случайном разложении лекарственного препарата. Далее будут рассмотрены внеш-
ние источники контаминации — вспомогательные материалы, используемые при
приготовлении действующего вещества или лекарственной формы, контейнеры и
системы доставки.
6.1.2. Внутренние источники контаминации
Примеси, возникающие от внутренних источников, могут присутствовать уже в ис-
ходных материалах в виде побочных продуктов синтеза или образовываться вслед-
ствие реакций с другими компонентами лекарственной формы. Образование при-
месей в лекарственной форме после получения и упаковки обычно вызывается
воздействием внешних агентов, таких как свет, ультрафиолетовое излучение, высо-
кая температура или воздух, вследствие неожидаемых реакций между компонента-
ми лекарственной формы.
Фармакопеи устанавливают проведение нескольких испытаний для каждой се-
рии исходных материалов для определения уровня содержащихся примесей. Обыч-
но из сопутствующих компонентов фармацевтических субстанций определяют тя-
Глава 6.1. Источники контаминации
539
Стадия
жизненного цикла
Процедура
Внутренние
источники примесей
Внешние
источники примесей
Рис. 1. Схема общего представления об источниках примесей
желые металлы, хлориды или сульфатную золу. Присутствие побочных продуктов
синтеза зависит от исходного материала. В качестве примеров можно привести не-
токсичные вещества, например гидрофосфат, присутствующий в качестве примеси
в солях дигидрофосфатов, и глицин, являющийся примесью для субстанции алани-
на, и токсичные вещества, такие как классический энантиомер ^талидомида.
6.1.2.1. Исходные материалы
Используемые в фармацевтической отрасли исходные материалы могут иметь два
различных источника происхождения. Они могут представлять собой натуральные
вещества, встречающиеся в природе, или получаться синтетическим путем. К на-
540
Часть 6. Контаминация и ее контроль
туральным веществам относятся действующие вещества, получаемые из экстрак-
тов растений и тканей животных, химические вещества и биотехнологические про-
дукты. Так же, как разнообразны различные исходные материалы, разнообразен и
спектр примесей. В данном разделе будут рассмотрены примеси, уже присутствую-
щие в исходных материалах в виде сопутствующих веществ, и примеси, образующи-
еся в ходе синтеза действующего вещества. К сопутствующим веществам относятся
обычные примеси, рассматриваемые фармакопеями, такие как тяжелые металлы и
мышьяк, предельное содержание которых определено во всех фармакопейных ста-
тьях. Присутствие в качестве примеси побочных продуктов синтеза может стать,
а может и не стать проблемой.
Вода. Вода — основной исходный материал в фармацевтических лекарствен-
ных формах. Она представляет собой наиболее часто используемый растворитель,
поскольку является основным компонентом тела человека. Во многих продуктах
вода — это основной компонент препарата, который должен присутствовать даже
в веществах, нерастворимых в воде. В зависимости от продукта и пути введения
лекарственной формы липофильные препараты готовят в виде водомасляных
эмульсий.
Количество и уровень содержания примесей или контаминирующих агентов
в воде для фармацевтического применения зависит от ее назначения. Поскольку вода
используется во всех отраслях народного хозяйства и научной работе, международные
и национальные органы по стандартизации установили требования к качеству воды
для всех типов ее применения. Стандарты по воде, направленные на охрану здоро-
вья, издаются такими организациями, как Всемирная организация здравоохранения
(ВОЗ) [2], в Соединенных Штатах Америки — Агентством по защите окружающей
среды ЕРА (Environmental Protection Agency) [3] и Американским обществом по стан-
дартизации испытаний и материалов ASTM (American Society for Testing and Materials
Standards) [4], а также фармакопеями в случаях, когда использование воды непо-
средственно связано с производством лекарственных и ветеринарных препаратов.
Стандарты качества для воды практически одинаковы во всех фармакопеях
(Фармакопея США [1], Британская фармакопея (ВР) [5], Немецкая фармакопея
(DAB) [6], Европейская фармакопея (ЕФ) [7], Международная фармакопея (IP) [8]),
имея лишь незначительные отличиям по допустимому уровню содержания хими-
ческих примесей. Фармакопеи классифицируют воду на три основных категории:
очищенную воду, высокоочищенную воду и воду для инъекций. Каким бы ни был про-
цесс очистки воды, исходным материалом для него всегда служит питьевая вода.
Параметры качества питьевой воды, установленные государственными регулятор-
ными агентствами, включают большое количество химических компонентов. В этот
перечень входят не только химические вещества, попадающие в воду естественным
путем, но и ряд веществ, которые могут в ней присутствовать вследствие антропо-
генного воздействия на окружающую среду, включая бензол, этилендиаминтетра-
уксусную кислоту (ЭДТА) и кадмий. В табл. 1 приведены рекомендации по качеству
питьевой воды, принятые ВОЗ [2].
Процесс очистки воды, используемый фармацевтическими производителями,
должен обеспечивать получение воды, соответствующей требованиям, приведен-
ным в табл. 2.
Глава 6.1. Источники контаминации 541
Таблица 1
Предельные уровни содержания веществ в питьевой воде, приведенные в руководстве ВОЗ
и национальных стан дартах ЕРА для питьевой воды [2,3]
Примесь Нормы, мг/л
Международные стандарты
Акриламид 0,0005
Алахлор 0,002
Асбест (волокна >10 мкм) 7 млн волокон/л
Атразин 0,003
Барий 2
Бензол 0,005
Бенз(я)пирен (ПАУ) 0,0002
Бериллий 0,004
Бор 0,5
Броматы 0,010
Винилхлорид 0,002
Галогензамещенные уксусные кислоты (Я4Л5) 0,060
Гексахлорбензол 0,001
Гексахлорциклопентадиен 0,05
Гептахлор 0,004
Гептахлора эпоксид 0,0002
Глифосат 0,7
Далапон 0,2
2,4-ДБ (пестицид) 0,07
1,2-Дибром-З-хлорпропан (DBCP) 0,0002
Дикват (реглон) 0,02
Диносеб 0,007
о-Дихлорбензол 0,6
л-Дихлорбензол 0,075
Дихлорметан 0,005
1,2-Дихлорпропан 0,005
1,2-Дихлорэтан 0,005
1,1 -Дихлорэтилен 0,007
транс-1,2-Дихлорэтилен 0,1
цис-1,2-Дихлорэтилен 0,07
Ди-2-этилгексил адипат 0,4
Ди-2-этилгексил фталат 0,006
Кадмий 0,005
Карбофуран 0,04
Ксилены (сумма) 10
Линдан 0,0002
Медь 1,3
542__________________________________________Часть 6. Контаминация и ее контроль
Таблица 1 (продолжение)
Примесь Нормы, мг/л
Метохлор 0,04
Мышьяк 0,010
Никель 0,02
Нитраты (в пересчете на азот) 10
Нитриты (в пересчете на азот) 1
Оксамил (Видат) 0,2
Пентахлорфенол 0,001
Пиклоран 0,5
Полихлорированные бифенилы (ПХБ) 0,0005
Ртуть (неорганическая) 0,002
Свинец 0,015
Селен 0,05
Симазин 0,004
Стирол 0,1
Сурьма 0,006
Таллий 0,002
2,3,7,8-Тетрахлордибензо-л-диоксин (2,3,7,8- TCDD) 0,00000003
Тетрахлорметан (тетрахлорид углерода) 0,005
Тетрахлорэтилен 0,005
Токсафен 0,003
Толуол 1
Тригалометаны (ТГМ) (сумма) 1
1,2,4-Трихлорбензол 0,07
1,1,1 -Трихлорэтан 0,2
1,1,2-Трихлорэтан 0,005
Трихлорэтилен 0,005
Уран 0,030
Формальдегид 0,9
Фториды 4,0
Хлор (в виде С12) 4,0
Хлора диоксид (в виде С1О2) 0,8
Хлорамины (в виде С12) 4,0
Хлорбензол 0,1
Хлордан 0,002
Хлорит 1,0
Хром (общее содержание) 0,1
Цианид (как свободный цианид) 0,2
ЭДТА 0,6
Эндотал 0,1
Эндрин 0,002
Глава 6.1. Источники контаминации
543
Таблица 1 (окончание)
Примесь Нормы, мг/л
Эпихлоргидрин 0,0004
Этилбензол 0,7
Этилена дибромид 0,00005
2,4,5- ТР (Силвекс) 0,05
Национальные стандарты
Алюминий От 0,05 до 0,2
Железо 0,3
Магний 0,05
Медь 1,0
Общее количество нерастворившихся частиц 500
Пенообразователи 0,5
Серебро 0,10
Сульфаты 250
Фториды 2,0
Хлориды 250
Цинк 5
Таблица 2
Качество воды для фармацевтического применения (фармакопейные стандарты)
Параметр Категория воды
Очищенная Высокоочшценная Для инъекций
Электропроводность 4,3 1,1 1,1
(при 20 °C), мкСмсм-1
Общий органический 0,5 0,5 0,5
углерод (ГОС), мг/л
Нитраты, ррт 0,2 0,2 0,2
Алюминий, мкг/л 10 10 10
Тяжелые металлы, ррт* 0,1 0,1 0,1
Хлориды Выдерживает/ 0,5 ррт
Сульфаты не выдерживает Выдерживает/ Выдерживает/
Аммоний, ррт не выдерживает 0,2 — не выдерживает 0,2
Кальций, магний Выдерживает/ Выдерживает/
Остаток после выпаривания, мг/100 мл не выдерживает 1 — не выдерживает 0,4 (объем <10 мл), 0,3 (объем >10 мл)
* Измеряется по свинцу.
544
Часть 6. Контаминация и ее контроль
На получение воды требуемого качества влияет ряд факторов, в том числе:
• концентрация веществ в исходной воде;
• происхождение исходной воды;
• процесс обработки.
Следует отметить, что, например, несмотря на отсутствие установленных требо-
ваний по содержанию пестицидов, они не должны присутствовать в воде для фар-
мацевтического применения.
Существующие способы очистки воды, доступные для фармацевтических про-
изводителей, должны выбираться исходя из требуемого уровня чистоты. Способы
очистки приведены в табл. 3 с учетом степени сложности процесса; чем выше уро-
вень чистоты, тем сложнее процесс.
Таблица 3
Уровень сложности процессов очистки воды от химических соединений
Уровень сложности Процесс
1 Дистилляция
2 Ионный обмен
3 Обратный осмос
4 Мембранная фильтрация
В табл. 4 приведены параметры качества воды, получаемой при помощи процес-
сов, перечисленных в табл. 3.
Таблица 4
Требования к качеству воды и различные процессы очистки
( < ( 1 Процесс очистки ( ( < 1 с 1 £ | Е i Е | S w- “ § s S g a 1 £ E f § s s § В = G « ! sis S hg g Is ii f I ! J i Ц 1g B 1 g a I °‘I ft & о H g
Без очистки
(водопроводная вода в качестве примера) 240 10 <10 <200 1 1 35 1
Дистилляция (однократно) 10,2 0,03 — - 0,5-1 0,01 1-3 0,5-1
Дистилляция (двукратно) 2,1 0,06 — - 0,1-0,8 0,01 0,3-0,1 0,1-0,7
Ионный обмен 2-30 — — — <0,01 — — 1
Обратный осмос 10-25 0,03 — <0,04 0,4 1,6 0,1
Мембранная фильтрация 0,056 0,01 — <0,01 <0,01 — <0,01
* Измеряется по свинцу.
Глава 6.1. Источники контаминации
545
Эффективность каждого процесса удаления примесей, указанных в табл. 1 и 2,
показана в табл. 5 [2].
Таблица 5
Эффективность методов очистки воды по удалению примесей
Примеси Метод очистки
Дистилляция, % Ионный обмен, % Обратный осмос, % Мембранная фильтрация, %
Ионы >70 >80 Моновалентные > 95 Поливалентные > 97 >80
Органические вещества >80 Не эффективен >99 >80
Механические частицы >80 Не эффективен >99 >99
Даже когда вода в качестве исходного материала выдерживает установленные
требования, после превращения в фармацевтический продукт она может содержать
некоторые примеси. В табл. 6 представлен уровень содержания примесей, найден-
ных в воде для инъекций (WFI). Поскольку исходные материалы должны соответ-
ствовать установленным требованиям, содержание этих примесей было ниже пре-
дельно допустимой концентрации или они были внесены в продукт после упаковки.
Наиболее вероятно, что примеси, внесенные в продукт после упаковки, происходят
из упаковочных материалов. В разделе 6.1.3.2 обсуждаются контейнеры (первичная
упаковка) в качестве источника примесей.
Таблица 6
Примеси, найденные в воде для инъекций
Показатель Образец Содержание, мкг/л Источник данных
Алюминий Стерильная вода, «Эбботт» <5 [10]
Стерильная вода, «МакГау» <5 [10]
Стерильная вода, «Травенол» <5 [10]
Вода для инъекций, «Браун», 50 мл 1 [11]
Мышьяк Вода д ля инъекций, «ЭМС» (EMS) 39,3 [12]
Вода для инъекций, «Гейер» 30,9 [12]
Цинк Вода стерильная 13,9 ИЗ]
Силикаты Вода стерильная, «Гейер» 280 ±13 [14]
Подводя итог, следует отметить, что вода может быть источником примесей. Если
исходный материал (питьевая вода) соответствует требованиям к качеству, установ-
ленным регуляторным органом, остающиеся примеси могут быть удалены с помо-
щью процессов очистки, доступных фармацевтическим производителям. Процессы
дистилляции и обратного осмоса обеспечивают получение воды, удовлетворяющей
требованиям к очищенной воде и высокоочищенной воде, но для получения воды для
инъекций, как правило, применяют метод мембранной фильтрации (вместе с дру-
гим процессом очистки), что обусловлено не столько химической контаминацией,
сколько требованиями к стерильности.
546
Часть 6. Контаминация и ее контроль
Сопутствующие вещества. Сопутствующие вещества могут считаться примеся-
ми, присутствующими в природных, несинтетических исходных материалах. Они
могут обладать токсическим действием, как мышьяк, или быть нетоксичными, как
хлорид-ионы. Сводный перечень обычных сопутствующих веществ и предельный
уровень их содержания, установленный в фармакопеях, приведен в табл. 7.
Таблица 7
Основные неорганические примеси, указанные в фармакопеях, и предельные уровни их содержания
Примесь Норма
Алюминий 0,2—1 ррт
Аммоний 200 ррт
Бромиды 50 ррт
Железо 2—100 ррт
Кадмий 5—10 ррт
Медь 0,1 ррт
Мышьяк 1—4ррт
Никель 0,2—1 ррт
Оксалаты 100—350 ррт
Свинец 0,1—50 ррт
Серебро 250 ррт
Сульфатная зола 0,01-1%
Сульфаты 50—400 ррт,
0,1-0,6%
Сульфиты 15 ррт
Тяжелые металлы 1—50 ррт
Фосфаты 25—400 ррт
Фториды 3 ррт
Хлориды 10—500 ррт
Хром 0,05—10 ррт
Цинк 10—30 ррт
Примечание. Некоторые примеси связаны только с определенными продуктами. Например,
предельное содержание серебра устанавливается для цисплатина, сульфитов — для сахаров.
Хотя присутствие этих сопутствующих веществ в фармацевтических продуктах
неизбежно, оно может и не стать проблемой. Присутствие магния в солях кальция
очень часто обусловлено сходностью этих катионов: оба являются щелочноземель-
ными металлами с очень близкими химическими свойствами. Вследствие этого
в большинстве исходных материалов, содержащих кальций, также содержатся не-
большие количества магния. То же наблюдается для катионов натрия и калия, по-
этому норма содержания калия в качестве примеси в хлориде натрия (согласно Бри-
танской фармакопее ВР) составляет 550 ррт, или 0,55 мг калия на 1 г соли натрия.
Если одни вещества, такие как натрий, калий, хлориды и сульфаты, хорошо пе-
реносятся человеком в достаточно больших количествах, то для других, обладаю-
щих токсичностью, установлены более жесткие нормы содержания. Например, для
мышьяка и свинца предельное содержание в фармакопеях обычно устанавливается
Глава 6.1. Источники контаминации
547
в диапазоне от 1 до 10 ррт- на самом деле указанное предельное содержание — вы-
сокое для таких токсичных веществ.
Исследования по определению примесей в исходных материалах проводятся не
часто, главным образом из-за того, что качество сырья сертифицируется официаль-
ными гарантиями качества и предполагается, что продукты соответствуют требова-
ниям, указанным в официальной гарантии.
Было проведено два исследования исходных материалов, используемых для про-
изводства парентеральных препаратов, по определению в них содержания алюми-
ния и мышьяка [15, 16]. Как видно на рис. 2 и 3, алюминий и мышьяк присутство-
вали во всех исследованных материалах. Наблюдались различия в контаминации
фармацевтических субстанций. Так, в солях, в частности NaCl и КС1, отмечался
низкий уровень содержания алюминия, а в фосфатах, глюконате и лимонной кис-
лоте — относительно высокий. Авторы объясняют эту разницу сродством алюминия
к этим веществам. Содержание мышьяка было более однородным. За исключением
аминокислоты тирозин, концентрация мышьяка в субстанциях была менее 1 мкг/г,
что не превышает пределы, указанные в фармакопеях.
Поскольку испытания на наличие примесей в фармацевтическом продукте, как
правило, проводят на готовом препарате1, невозможно объяснить наличие приме-
ню
6.0
з.о
2,0
1.0
0,5
о
Рис. 2. Наличие алюминия в качестве примеси в фармацевтических субстанциях, используемых
для производства препаратов для парентерального питания [15] (acetyl — ацетил; albumin —
альбумин; biotin — биотин; caproic ас. — капроевая (гексановая) кислота; caprylic ас. — каприловая
кислота; Cystine — цистин; folic ас. — фолиевая кислота; gluconate — глюконат; glucose — глюкоза;
glycerol — глицерол; НАс. — гиалуроновая кислота; heparin — гепарин; lecithin — лецитин; malic
ас. — малеиновая кислота; mannitol — маннитол; molybdate — молибдат; pantothenic ас. —
пантотеновая кислота; riboflavin — рибофлавин; selenite — селенит; sorbitol — сорбитол; Vit. —
витамин; xylitol — ксилитол)
AI, мкг/г
Crt-1» cuso4
rvtosoA h^so,
gluconate
Ha2HCO,
ZnSO4
CuCI,
Na Ac №CI3
molybdate
HeF
NaCl KCI
Cystine
Туг
NaOH
biotin
Vit.C
HaH3P04
KH,PO4
ZnCL
selerrte
KI
Cys
malic ас
Arg
Glu
Lys Orn
Try
Asp
xylitol
sorbitol
mannitol
glycerol
ribofla/m
folic ac.
Ala Asn , Pro .. . . _
lie Thrc Leu " «Wl-Tyr
Met Ser Phe
albumin
Vrt.B6
\frt.B1 pantothenic ac
Vrt.B12 Vft.B5
HCI HAc. „ glucose
hepann “
Wrt.E Vrt.D lecithin Vrt.A
caproic ac. caprylic ac.
1 Описываемая автором ситуация не соответствует концепции «качество, заложенное при
проектировании», широко используемой зарубежными производителями в настоящее вре-
мя, а также принципам стандартизации фармацевтических субстанций и входного контро-
ля, принятым в Российской Федерации. — Примеч. перев.
548
Часть 6. Контаминация и ее контроль
As, мкг/г
2,01---------
Туг
1,6
1,2 -
Na2HCO3
08__________NaaHPCU______
KH2pd4 k2hpo4
NaCI MgCI2
gluconate NaH2PO4
KCI CaCI2
0,4
О
NaAc
MgSO4
Leu
Om cystine
Thr
Arg Vai Ser His lie Gly
Met Asn Asp „
Phe Pro
Glu ..
_ Ala I ,,c
N-acetyl-Tyr LVS
_ sorbitol________________
Vit. B2 folic ас.
ascorbic ас.
Vit. B5 glucose
mannitol heparin
malic ас. хУ|Ко1
Рис. 3. Содержание мышьяка в фармацевтических субстанциях, используемых для производства
препаратов для парентерального питания [16] (acetyl — ацетил; ascorbic ас. — аскорбиновая
кислота; Cystine — цистин; folic ас. — фолиевая кислота; gluconate — глюконат; glucose — глюкоза;
heparin — гепарин; malic ас. — малеиновая кислота; mannitol — маннитол; sorbitol — сорбитол;
Wf. — витамин; xylitol — ксилитол
сей за счет исходных материалов. Однако независимо от того, проверяются ли ис-
ходные материалы, существует много других источников, которые могут влиять на
накопление примесей в готовом продукте.
Побочные продукты синтеза. Побочные продукты синтеза, вероятно, являются
наиболее сложными для обобщения примесями, так как у каждого действующего
вещества есть свои побочные продукты, которые могут проявляться в качестве при-
месей. Они синтезируются вместе с действующими веществами и достаточно слож-
но отделяются вследствие их схожести с получаемым веществом. Большинство из
них являются изомерами, отличающимися друг от друга присутствием только одной
маленькой группы (радикала) или только позицией атома водорода. Более сложно
разделить и, следовательно, очистить энантиомеры (хиральные изомеры). Совре-
менные лекарственные вещества, содержащие только один энантиомер в качестве
действующего вещества, еще более трудны для очистки. Фармакопеи обычно ука-
зывают в статьях побочные продукты синтеза, которые мыуг присутствовать в ка-
честве примеси действующего вещества.
6.1.2.2. Вспомогательные вещества
Вспомогательные вещества — это все компоненты, входящие в состав лекарствен-
ного препарата, отличные от действующего вещества. Хотя эти вещества можно
классифицировать на наполнители и носители (наполнители — для твердых лекар-
ственных форм, носители — для жидких), существует ряд других агентов, исполь-
зуемых при приготовлении лекарственных форм, с определенными функциями.
К ним относятся консерванты, подсластители, покрытия, красители, антиоксидан-
Глава 6.1. Источники контаминации
549
ты, поверхностно-активные вещества, эмульсификаторы и отдушки. Поскольку они
представляют собой широкий спектр продуктов, в данном разделе будут обсуждены
вспомогательные вещества для приготовления лекарственных форм для внутренне-
го применения [17, 18].
В табл. 8 приводятся вспомогательные вещества и возможные примеси, которые
они могут содержать. Нормы содержания этих примесей взяты из спецификаций
производителей. Для составления этой таблицы были выбраны продукты категории
«для фармацевтического применения» (Фармакопея США, ВР, ЕФ, DAB) от квали-
фицированных поставщиков («Мерк», «Алдрич», «Сигма», «Флюка», «ЭМС», «Рие-
дел де-Хаен»).
Таблица 8
Вспомогательные вещества для лекарственных форм, примеси и нормы их содержания согласно
производителям («Мерк», «Алдрич», «Сигма», «Флюка», «ЭМС», «Риедел де-Хаен»)
Вспомогательное вещество Примеси Нормы
Растворители Этанол Ацетон и изопропиловый спирт <0,01%
Ацетальдегид и ацеталь <0,001%
Бензол <0,0002%
Метанол <0,01%
Общее содержание примесей <0,03%
Глицерол Хлориды <0,0010%
Сульфаты <0,0010%
Галогениды (в виде О) <0,0030%
Тяжелые металлы <0,0005%
Мышьяк <0,0001%
Кальций <0,0001%
Кадмий <0,0001%
Ртуть <0,0001%
Аммоний <0,0005%
Свинец <0,0002%
1,2,4-Бутантриол <0,2%
Остаточные органические растворители 3-го класса* <0,5%
Альдегиды <10 ррт
Сульфатная зола <0,01%
Полиэтиленгликоль Диоксан <10 ррт
Этиленгликоль и диэтиленгликоль <0,4%
Этиленгликоль (класс 2) <620 ррт
Этилена оксид <1 ррт
Формальдегид (НСНО) <30 ррт
Тяжелые металлы <20 ррт
Сульфатная зола <0,2%
550__________________________________________Часть 6. Контаминация и ее контроль
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Консерванты
Бензиловый спирт Пероксидное число* <5
Бензальдегид <0,15%
Циклогексилметанол <0,10%
Бензол <0,0002%
Хлорбензол <0,01%
Толуол <0,01%
Бензойная кислота Сульфаты <0,02%
Тяжелые металлы <0,001%
Мышьяк <0,0001%
Кадмий <0,0010%
Медь <0,0010%
Ртуть <0,0001%
Свинец <0,0005%
Цинк <0,0010%
Галогениды (в виде С1) <0,01%
Толуол <890 ррт
Хлорбутанол Хлориды <0,01%
Хлороформ <60 ррт
Остаточные растворители 3-го класса' <0,5%
Сульфатная зола <0,1%
Метилпарабен Тяжелые металлы <0,001%
Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0005%
Цинк <0,001%
Метанол <0,3%
Сульфатная зола <0,05%
Натрия метилпарабен Хлориды <0,03%
Сульфаты <0,03%
Тяжелые металлы <0,001%
Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0005%
Цинк <0,001%
Глава 6.1. Источники контаминации
551
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Натрия пропилпарабен Хлориды <0,03%
Сульфаты <0,03%
Тяжелые металлы <0,001%
Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0005%
Цинк <0,001%
1-Пропиловый спирт <0,5%
Сульфатная зола 34-36%
Пропилпарабен Тяжелые металлы <0,001%
Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0005%
Цинк <0,001%
Остаточные органические растворители <0,5%
3-го класса*
Сульфатная зола <0,05%
Калия сорбат Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0002%
Цинк <0,001%
Альдегиды (в пересчете на ацетальдегид) Остаточные органические растворители <0,15% <0,5%
3-го класса*
Натрия бензоат Хлориды <0,02%
Сульфаты <0,01%
Общий хлор <0,03%
Тяжелые металлы <0,001%
Мышьяк <0,0001%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0002%
Цинк <0,001%
552
Часть 6. Контаминация и ее контроль
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Сорбиновая кислота Тяжелые металлы <0,0010%
Мышьяк <0,0003%
Кадмий <0,001%
Медь <0,001%
Ртуть <0,0001%
Свинец <0,0002%
Цинк <0,001%
Альдегиды (в пересчете на ацетальдегид) <0,15%
Остаточные органические растворители 3-го класса' <0,5%
Сульфатная зола <0,2%
Антиоксиданты Аскорбиновая кислота Тяжелые металлы <0,001%
Остаток после сжигания <0,05% (как SO4)
Хлориды <50 мг/кг
Сульфаты <20 мг/кг
Медь <5 мг/кг
Железо <2 мг/кг
£(+)-аскорбилпальмитат Тяжелые металлы <0,001%
Мышьяк <0,0003%
Медь <0,0025%
Свинец <0,001%
Цинк <0,0025%
Сульфатная зола <0,1%
Остаточные органические растворители 3-го класса* <0,5%
Бугилгидроксианизол Мышьяк <0,0003%
Тяжелые металлы <0,001%
Свинец <0,0005%
Ртуть <0,0001%
3-трете-Бутил-4-метоксифснол <10%
Гидрохинон <0,2%
Сульфатная зола <0,01%
Бутилгидрокситолуол Мышьяк <0,0003%
Тяжелые металлы <0,001%
Свинец <0,0005%
Ртуть <0,0001%
Остаточные органические растворители 2-го класса (МеОН)* <0,2%
Сульфатная зола <0,002%
Глава 6.1. Источники контаминации
553
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Натрия формальдегида Железо <0,0025%
сульфоксилат Натрия сульфит <5,0%
Остаточные органические растворители 2-го класса (МеОН)* <0,3%
Фосфорная кислота Летучие кислоты (в пересчете на СН3СООН) <0,001%
Хлориды <0,0005%
Фториды <0,0010%
Нитраты <0,0003%
Фосфиты и гипофосфиты (в пересчете на Н3РО3) <0,02%
Сульфаты <0,005%
Тяжелые металлы <0,001%
Мышьяк <0,0002%
Кадмий <0,00010%
Медь <0,002%
Железо <0,005%
Ртуть <0,0001%
Калий <0,005%
Натрий <0,03%
Свинец <0,0010%
Цинк <0,002%
Натрия бисульфит Мышьяк <0,001%
Тяжелые металлы <0,003%
Железо <0,005%
Натрия метабисульфит Хлориды <0,01%
Тяжелые металлы <0,001%
Тиосульфат <0,02%
Мышьяк <0,0002%
Железо <0,001%
Ртуть <0,0001%
Свинец <0,0005%
Селен <0,0006%
Натрия тиосульфат Сульфаты и сульфиты (в пересчете на SO4) <0,2%
Тяжелые металлы <0,001%
Токоферол Тяжелые металлы <0,001%
Мышьяк <0,0003%
Медь <0,0025%
Ртуть <0,0001%
Свинец <0,0005%
554
Часть 6. Контаминация и ее контроль
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Токоферол Цинк <0,0025%
Метанол <3000 ррт
Пиридин <200 ррт
Толуол <890 ррт
Наполнители/носители Сульфатная зола <0,1%
Целлюлозы порошок Вещества, растворимые в эфире <0,15%
Вещества, растворимые в воде <1,0%
Тяжелые металлы <0,001%
Сульфатная зола <0,3%
Желатин Диоксид серы (SO2) <0,004%
Тяжелые металлы <0,001%
Мышьяк <0,00008%
Хром <0,001%
Железо <0,003%
Цинк <0,003%
Пероксиды (в пересчете на Н2О2) <0,001%
Зола <2,0%
Лактоза Тяжелые металлы <0,0005%
Мышьяк <0,0001%
Медь <0,0025%
Свинец <0,00005%
Цинк <0,0025%
Сульфатная зола <0,1%
Крахмал Восстанавливающие вещества <0,7%
(в пересчете на мальтозу)
Сульфатная зола <0,4%
Сорбитол Хлориды <0,002%
Сульфаты <0,006%
Тяжелые металлы <0,0005%
Мышьяк <0,00013%
Никель <0,0001%
Свинец <0,00005%
Родственные вещества (маннитол) Восстанавливающие сахара после <2,0%
гидролиза/общие сахара (в пересчете на глюкозу) <0,5%
Восстанавливающие сахара (в пересчете <0,11%
на глюкозу)
Сульфатная зола <0,02%
Глава 6.1. Источники контаминации
555
Таблица 8 (продолжение)
Вспомогательное вещество Примеси Нормы
Сахароза Хлориды <0,0035%
Сульфаты <0,005%
Сульфиты (в пересчете на SO2) <0,0010%
Тяжелые металлы <0,0005%
Мышьяк <0,0001%
Свинец <0,00005%
Сульфатная зола <0,02%
Остаточные органические растворители <0,5%
3-го класса*
Тальк Тяжелые металлы <0,004%
Алюминий <2,0%
Мышьяк <0,0003%
Кальций <0,9%
Железо <0,25%
Свинец <0,0005%
Асбест (согласно ЕФ) Не обнаружен
Цинка оксид Хлориды <0,005%
Сульфаты <0,02%
Мышьяк <0,0005%
Кадмий <0,001%
Железо <0,001%
Свинец <0,005%
Хелатообразующие агенты ЭДТА Тяжелые металлы <0,001%
Кальций <0,001%
Железо <0,001%
Магний <5 ррт
Нитрилотриуксусная кислота (Трилон А) <0,1%
Сульфатная зола <0,1%
Буферы Борная кислота Сульфаты <0,04%
Тяжелые металлы <0,0015%
Динатрия гидрофосфат Хлориды <0,02%
Фториды <0,001%
Сульфаты <0,05%
Тяжелые металлы <0,001%
Мышьяк <0,0002%
Кадмий <0,0001%
Железо <0,002%
Ртуть <0,0001%
Свинец <0,0004%
556
Часть 6. Контаминация и ее контроль
Таблица 8 (окончание)
Вспомогательное вещество Примеси Нормы
Натрия дигидрофосфат Хлориды <0,005%
Фториды <0,001%
Гидрофосфат (НРО4) <0,5%
Сульфаты <0,01%
Тяжелые металлы <0,0005%
Мышьяк <0,0002%
Кадмий <0,0001%
Железо <0,001%
Свинец <0,0004%
Ртуть <0,0001%
* Остаточные органические растворители — см. раздел 6.1.3.1; пероксидное число — ммоль
пероксида/л.
6.1.2.З. Продукты разложения веществ, входящих в состав продукта
Наиболее характерной контаминацией внутреннего происхождения, возникающей
при разложении компонентов состава лекарственной формы, является образование
пероксидов в витаминах, аминокислотах и жировых эмульсиях под действием света
и воздуха, то есть кислорода.
Липидные эмульсии являются важными компонентами препаратов для паренте-
рального питания. Однако из-за присутствия полиненасыщенных жирных кислот
(ПНЖК) в них возможно возникновение химической деградации с образованием
гидропероксидов.
Под воздействием комнатного света и облучения (при фототерапии) в отделе-
ниях неонатальной интенсивной помощи в течение 24 часов в липидных эмульсиях
уровень содержания гидропероксидов увеличивался почти в 60 раз [19]. В смесях
липидов, хранившихся в мешках из этилвинилацетата, было обнаружено значитель-
ное количество липидных пероксидов. При хранении в течение 1 месяца в стрессо-
вых условиях (в газопроницаемых контейнерах при 40 °C) пероксидное число (ПЧ,
т. е. число миллимолей пероксидов в 1 литре) в испытуемых образцах оказалось в 450
раз выше, чем в контрольных (в стеклянных бутылках, заполненных в атмосфере
азота) [20]. Результаты, приведенные в табл. 9, показывают, что по сравнению с ли-
пидными смесями, хранившимися в закрытых под атмосферой азота стеклянных
контейнерах, во всех полимерных мешках наблюдалось выраженное образование
пероксидов, коррелирующее с длительностью хранения. Более того, в липидных
препаратах, хранившихся в мешках из V90 (полипропилен-полиамид 7:3, двойной
слой), было более низкое значение ПЧ, чем у препаратов в мешках из этилвинил-
ацетата (одинарный слой), что свидетельствует о связи пониженной проницаемости
двуслойного материала для кислорода и стабилизации ПЧ, хотя медленное увеличе-
ние ПЧ наблюдалось в образцах Интралипида, хранившихся и в мешках из V90.
Была обнаружена разница значений ПЧ между образцами Интралипида (тригли-
цериды с длинной цепью) и Липофундина (триглицериды со средней длиной цепи).
Начальное значение ПЧ у Интралипида (0,02 ммоль/л) было ниже, чем у Липофун-
Глава 6.1. Источники контаминации
557
Таблица 9 Влияние материала первичной упаковки, света и температуры на перекисное окисление липидов
В препаратах Интралипид 20% и Липофундин МСТ 20% при хранении
Длительность воздействий, ДНИ Мешки из Нутримикс 2/3 (этилвинилацетат), 20—27 °C, дневной свет" Мешки из Нутримикс 2/3 (этилвинилацетат), 20-27°С, защита от света Т)>ехкамерные мешки (V90), 20-27 °C, дневной свет* Контрольные образцы. Закрытые стеклянные бутылки, 20—27 °C, дневной свет*
Интралипид 20%
1 0,02 ± 0,006*
5 0,47±0,0166 0,06±0,00046>в 0,33 ±0,011
8 0,02 ±0,005
9 0,84 ±0,010 0,05 ±0,007 0,50 ±0,017
14 1,46 ±0,020 0,17 ±0,002 0,60 ±0,014
15 0,02 ±0,001
19 1,87 ±0,027 0,27 ±0,017 0,76 ±0,018
22 2,48 ±0,038 0,40 ±0,013 0,92 ±0,025
28 0,02 ±0,005
29 2,93 ±0,052 0,52 ±0,023 1,24 ±0,032
Липофундин МСТ 20%
1 0,11 ±0,006"
5 0,57±0,0186 0,19±0,017г 0,64 ±0,014
8 0,10 ±0,018
9 0,88 ±0,024 0,35 ±0,002 0,69 ±0,010
14 1,31 ±0,048 0,54 ±0,0025 0,64 ±0,021
15 0,08 ±0,003
19 1,59 ±0,064 0,61 ±0,018 0,70 ±0,054
22 1,99 ±0,076 0,84 ±0,007 0,69 ±0,054
28 0,10 ±0,032
29 2,48 ± 0,040 0,99 ±0,044 0,69 ±0,032
а’6Р< 0,001 (Нутримикс 2/3 с защитой от света).
"г Р < 0,002 (Нугримикс 2/3 с защитой от света в сравнении с контрольными образцами).
Источник-. [20].
дина (0,10 ммоль/л), в то же время у последнего отмечена более медленная скорость
увеличения ПЧ. Это объясняется удвоенным содержанием ПНЖК в Интралипиде.
Ненасыщенные жирные кислоты окисляются ферментативным и нефермента-
тивным способами. Неферментативное окисление представляет собой перекисное
окисление (пероксидацию) под действием свободных радикалов. Это цепная реак-
ция, обеспечивающая постоянное образование свободных радикалов, которые ини-
циируют дальнейшее распространение перекисного окисления. Весь процесс может
быть представлен следующим образом [21]:
RH + X‘->R‘ +ХН
R’ + O2->ROO'
558
Часть 6. Контаминация и ее контроль
ROO’ + RH > ROOH + R-
Реакция инициируется любым имеющимся свободным радикалом (Х‘), светом
или ионами металла, которые реагируют с липидами (RH) с образованием перокси-
радикала (ROO’) и, впоследствии, пероксида липида (ROOH). Помимо гидроперок-
сидов, также могут образовываться малондиальдегид, этан и пентан, если ПНЖК
имеет три и более двойных связи и если в продукте присутствуют ПНЖК омега-3 и
омега-6 соответственно [14]. Формирование пентана (2 мкмоль/л) и малондиальде-
гида (10 мкмоль/л) вместе с гидропероксидами наблюдалось в липидной эмульсии
(Интралипид 10%) [22].
Малондиальдегид также был обнаружен в смесях для парентерального питания
для новорожденных. В 12 проанализированных образцах его содержание варьиро-
валось от 1632 до 14,679 нмоль/л [23]. Пирони (Pironi) с соавт. [24] также находили
малондиальдегид в жировых эмульсиях. В исследовании сравнивались три 20%-ные
липидные эмульсии, содержавшие разное количество ПНЖК и а-токоферола, че-
рез 24 часа после смешивания и получения растворов «всё в одном». Авторы сделали
вывод, что образование пероксидов липидов непосредственно связано с содержа-
нием ПНЖК и находится в обратной связи с соотношением а-токоферол/ПНЖК
в эмульсии. Кроме того, в этих образцах также образовывался малондиальдегид. Его
содержание было более высоким в эмульсиях, содержащих соевое масло, в срав-
нении с эмульсиями с оливковым маслом. Результаты исследования приведены
в табл. 10.
Таблица 10
Концентрация пероксидов липидов и малондиальдегида в липидных эмульсиях
Пероксиды липидов, мкмоль/л
Малондиальдегид, мкмоль/л
Эмульсии
л = 6
Соевое масло Соевое масло + 28,8 ±29,9 <2 <2 11,3 ±15,0 8,8 ±3,1 4,9 ±0,8 5,0 ±0,3 6,9 ±1,0
триглицериды 1,7 ±0,5 <2 <2 5,2 ±8,8 3,7 ±0,4 3,4 ±0,4 4,1 ±0,3 7,8 ±1,8
средней длины Оливковое масло 4,1 ±2,8 <2 <2 5,5 ±6,4 0,7 ±0,1 5,4 ±0,3 5,3 ±0,3 3,3 ±0,4
Примечание. Значения даны в формате: среднее ± стандартное отклонение результатов по
6 мешкам (одна проба из мешка, трехкратное определение). Смесь-70 («все в одном») ана-
лизировали сразу после приготовления липидной эмульсии; смесь- 724 — через 24 часа после
добавления липидной эмульсии. Источник'. [24].
Образование пероксидов также было отнесено на счет прооксидантного дей-
ствия витамина Е на ПНЖК с учетом того, что витамин Е является другим компо-
нентом липидных эмульсий [19, 25]. Окисление липидов, происходящее несмотря
на высокие концентрации витамина Е (токоферола), может быть неожиданным,
Глава 6.1. Источники контаминации
559
поскольку общепризнано, что этот витамин является наиболее эффективным жи-
рорастворимым антиоксидантом, обрывающим цепную реакцию. Однако исследо-
вания показали, что для эффективного выполнения витамином Е функции анти-
оксиданта в изолированной липопротеиновой эмульсии требуется подходящий
«ко-антиоксидант», например витамин С [27, 28]. Этот ко-антиоксидант удаляет
радикал витамина Е, который образуется при взаимодействии витамина с иници-
ирующими окисление радикалами и может оказывать «обратное» действие, способ-
ствуя развитию реакции перекисного окисления.
Измерение содержания гидропероксидов триглицеридов в образцах Интралипи-
да, хранившихся в стеклянных и полимерных шприцах, подвергнутых воздействию
света или обернутых алюминиевой фольгой, показало, что Интралипид легко окис-
ляется при хранении в обычных условиях больницы, несмотря на наличие в нем ви-
тамина Е (^60 мкмоль/л у-токоферола и по 20 мкмоль/л а- и 5-изомеров).
Проблема особенно очевидна в ситуации с новорожденными, получающими
фототерапию. Липиды широко используются для снабжения недоношенных ново-
рожденных калориями, при этом мешки и передаточные линии подвергаются в те-
чение длительного периода времени интенсивному облучению лампами вследствие
медленной скорости инфузионных вливаний растворов для новорожденных. На
рис. 4 показана скорость формирования гидропероксидов триглицеридов в 20%-ной
эмульсии Интралипида в различных условиях светового облучения. Из рисунка
видно, что окисление липидов происходит несмотря на высокие концентрации ви-
тамина Е в образцах. Разложение липидов можно предотвратить путем обертывания
контейнеров и трубок систем для вливания в алюминиевую фольгу или с помощью
добавления в инфузию аскорбата.
Пероксиды липидов также способны вступать в реакцию с другими компонен-
тами смесей для парентерального питания (микроэлементами), вызывая снижение
Рис. 4. Скорость накопления гидропероксидов триглицеридов в Интралипиде, подвергнутом
воздействию светом и облучению в условиях, имитирующих условия в отделениях неонатальной
интенсивной терапии. Раствор Интралипида 20% был расфасован в стеклянные резервуары
с полимерными трубками (3), с алюминиевой фольгой (4) или в полимерные резервуары после
добавления раствора 1 ммоль/л натрия аскорбата (5), после чего подвергался световому
облучению длительностью до 24 ч. Для всех образцов мощность облучения составила 11,7-
25,9 мкВт/см2 на нанометр в течение 24 ч. В установленные периоды времени аликвоты отбирались
и экстрагировались, и в гексановых экстрактах определялось присутствие гидропероксидов
триглицеридов и а-токоферола, как описано в разделе «Методы» в [19]
560
Часть 6. Контаминация и ее контроль
pH с последующим возможным нарушением физико-химической стабильности
смеси [29]. В табл. 11 представлены пероксидные числа (ПЧ) и снижение pH в чи-
стой липидной смеси и в смеси «все в одном» с липидами, хранившихся в мешках из
этилвинилацетата при различных воздействиях температуры и различном световом
облучении в присутствии и отсутствии микроэлементов.
Таблица 11
Содержание пероксидов и снижение pH в чистой липидной эмульсии и смеси «все-в-одном»,
хранившихся в мешках из этилвинилацетата в различных температурных условиях и при
различном световом облучении в присутствии и отсутствии микроэлементов
Образец Условия хранения Определявшиеся параметры Без микро- элементов С микро- элементами
Смесь «все 2—8 °C, защита от света ПЧ, ммоль/л 0,04 0,19
В одном» 29 дней Снижение pH 0,01 0,02
Смесь «все 20—30 °C, световое ПЧ, ммоль/л 0,52 1,92
В одном» облучение 29 дней Снижение pH 0,03 0,11
Интралипид 40 °C, защита от света ПЧ, ммоль/л 2,77 18,04
20% 14 дней Снижение pH 0,77 1,54
Образование пероксидов также наблюдалось в поливитаминных растворах для
парентерального питания. Лавойе (Lavoie) с соавт. [30] изучали воздействие света,
воздуха и состава на стабильность поливитаминных препаратов и общих смесей
для парентерального питания, содержащих и не содержащих витамины и жирные
кислоты. Они оценивали образование пероксидов в растворах поливитаминов и
парентеральных смесях для взрослых и новорожденных. Анализ растворов поли-
витаминов для энтерального введения выявил присутствие пероксидов при первом
вскрытии бутылок. Их содержание в препарате Поли-Ви-Сол (витамины A, D, С,
Вр рибофлавин и никотинамид) было выше, чем в препарате Три-Ви-Сол (витами-
ны A, D и С) (рис. 5) [30].
Авторы объяснили разницу в содержании пероксидов в препаратах наличием ри-
бофлавина. Этот витамин служит катализатором индуцируемой фотонами реакции
между аскорбатом и кислородом, которая приводит к образованию пероксида водо-
рода. Снижение концентрации пероксида водорода с течением времени, наблюдав-
шееся в препарате Поли-Ви-Сол, объясняется преобразованием этого пероксида
в более реактогенные молекулы, которые, в свою очередь, могли вступать в реакцию
с другими компонентами препарата. И напротив, увеличение содержания каталаза-
устойчивых пероксидов в препарате Три-Ви-Сол предполагает более выраженное
действие воздуха на образование пероксидов. При сравнении с препаратами для
парентерального введения содержание пероксидов в энтеральных растворах было
в 100 раз выше, при этом основную массу пероксидов составляли каталазаустойчи-
вые пероксиды. Для изучения влияния воздуха и света на образование пероксидов
в поливитаминах для общей парентеральной смеси авторы включили в эксперимент
только смеси, не содержащие жирные кислоты, поскольку липиды также способны
образовывать пероксиды [31]. В растворах для новорожденных концентрация пе-
роксидов составляла от 190 от 300 мкмоль/л в комплектах без защиты от света и от
Глава 6.1. Источники контаминации
561
Рис. 5. Общее количество пероксидов (круги), каталазаустойчивых пероксидов (квадраты) и
Н2О2 как их разницы (треугольники) было измерено в трех сериях поливитамина для приема
внутрь без рибофлавина (Три-Ви-Сол) и в трех сериях поливитамина для приема внутрь,
содержащего 0,6 мг рибофлавина (Поли-Ви-Сол), в момент вскрытия бутылок. В поливитамине,
содержащем рибофлавин (Поли-Ви-Сол), содержание Н2О2 было изначально выше (Р < 0,01)
в сравнении с препаратом, не содержащем рибофлавин, и его уровень снижался со временем
(Р < 0,05). В обоих препаратах поливитаминов содержание каталазаустойчивых пероксидов
и общее содержание пероксидов увеличивалось (Р < 0,05) до 8-го дня исследования. Данные
представлены в формате: среднее + относительное стандартное отклонение. Незначительные
отклонения от среднего не показаны [30]
60 до 130 мкмоль/л — в защищенных от света. В препаратах для взрослых уровень
содержания пероксидов составил менее одной десятой от значений, определенных
в растворах для новорожденных (рис. 6). Авторы объяснили это разницей в составе
препаратов, так как препараты для взрослых содержат в 4 раза меньше витаминов и
больше аминокислот и глюкозы в конечном разведении. Аминокислоты и глюкоза
влияли на снижение концентрации пероксидов [32].
Как видно на рис. 6, защита от воздействия света и воздуха снижает образова-
ние пероксидов в растворах. Однако вследствие высокой концентрации пероксидов
в растворах для новорожденных защита от воздействия воздуха в течение инфузион-
ного введения является неэффективной.
Также этими исследователями изучалось поведение витаминов в присутствии
липидов [33]. Сравнивалось образование пероксидов в разных парентеральных
смесях, хранившихся в темном месте и при дневном свете в течение 6 часов. Ана-
лизировались четыре различных препарата, в составы которых и включались, и не
включались витамины и липиды. Результаты исследования, приведенные на рис. 7,
позволили сделать вывод, что в сравнении с витаминами липиды оказывают важ-
ный, но небольшой дополнительный эффект на образование пероксидов. Попада-
ние в смесь воздуха при смешивании вызывало индуцируемое фотонами образова-
ние пероксидов в растворах смесей. Однако защита раствора от света была более
эффективной, чем предупреждение контакта с воздухом.
Поскольку аскорбат снижал фотонное окисление липидных эмульсий и препара-
тов поливитаминов (рис. 4) [19], Лавойе с соавт. [34] изучили образование побочных
продуктов окисления витамина С в поливитаминах, подвергавшихся воздействию
света. Они установили, что уменьшение содержания аскорбиновой кислоты в облу-
ченных светом препаратах поливитаминов было связано с образованием продуктов,
562
Часть 6. Контаминация и ее контроль
б) Ш Свет/воздух
□ Свет/нет воздуха
И Нет света/воздух
Нет света/нет воздуха
Рис. 6. (а) Концентрация пероксидов в предназначенных для новорожденных растворах для
парентерального питания, не содержащих липидов, подаваемых через четыре различных набора
для вливаний: с воздушным клапаном и без него; с защитой и без защиты от света (люксы).
Наличие защиты от света обусловило наиболее низкое содержание пероксидов (отмечено *;
Р < 0,05), при этом защита от света обеспечивала более эффективное предупреждение
формирования пероксидов, чем защита от воздуха, (б) Концентрация пероксидов в растворах
для парентерального питания для взрослых, не содержащих липиды, подаваемых через четыре
различных набора для вливаний: с воздушным клапаном и без него; с защитой и без защиты от
света (люксы). Концентрация пероксидов — в 10 раз ниже, чем в растворах для новорожденных.
Наличие защиты от света и воздуха не оказало значительного влияния на суммарный эффект
образования пероксидов. При этом через 20 и 24 часа инфузии защита от света и воздуха
приводила к значительному уменьшению образования пероксидов (отмечено *; Р < 0,05), в то
время как эффект защиты только от света проявлялся лишь при 24-часовой инфузии (отмечено +;
Р < 0,05). Результаты показаны в формате: среднее ± относительное стандартное отклонение;
л = 3 (ТБГ — трет-бутилгидропероксид) [31]
Глава 6.1. Источники контаминации
563
Длительность, ч
—•— ПП + липиды + ПВ, с защитой от света
-О - ПП + липиды + ПВ, с воздействием света
—▲— ПП + ПВ, с защитой от света
- - ПП + ПВ, с воздействием света
Рис. 7. Влияние липидной эмульсии и дневного света на содержание пероксидов в свежепри-
готовленных растворах для парентерального питания, содержащих поливитамины (ПП + ПВ и
ПП + липиды + ПВ, где ПП — парентеральное питание, ПВ — поливитамины). Данные представ-
лены в формате: среднее ± относительное стандартное отклонение; п = 3; отклонения от средне-
го значения, не указанные на рисунке, малы по размеру по отношению к использованным гра-
фическим символам. Содержание пероксидов существенно увеличивалось с течением времени
(Р < 0,001), и дневной свет оказывал статистически значимый эффект на образование пероксидов
(Р < 0,001) (ТБГ — трет-бутилгидропероксид) [33]
отличных от дегидроаскорбата и 2,3-дикетоглукуроновой кислоты, которые обычно
являются продуктами окисления витамина С. Авторы показали, что пероксид водо-
рода в концентрациях, обнаруженных в парентеральных смесях, вызывал преобра-
зование дегидроаскорбата в новые биологически активные компоненты, влияющие
на метаболизм липидов. Авторы считают, что эти вещества обладают свойствами
пероксидов и альдегидов [35].
Поскольку воздух (кислород) является одним из факторов, вызывающих об-
разование пероксидов в липидных эмульсиях и растворах витаминов, Бале (Balet)
с соавт. [36] сравнили влияние многослойных и однослойных мешков из этилви-
нилацетата на окисление растворов для парентерального питания. В 24 паренте-
ральных растворах они измеряли ПЧ, содержание а-токоферола и аскорбиновой
кислоты в момент смешивания и через 6 и 14 дней. В смесях в многослойных меш-
ках наблюдалось менее выраженное окисление, чем в смесях в однослойных мешках
из этилвинилацетата. При этом не выявлено существенной разницы в уровнях со-
держания а-токоферола, однако после 6-го дня хранения в мешках из этилвинила-
цетата не были обнаружены аскорбиновая и дегидроаскорбиновая кислоты.
Натрия метабисульфит является антиоксидантом, широко использующимся
в фармацевтических препаратах для снижения или предупреждения реакций окис-
ления. Однако имеется ряд работ, в которых показано, что метабисульфит в особых
условиях может обладать опосредованными окислительными свойствами. Бейкер
(Baker) с соавт. [37] показали, что при имитации внутривенной инфузии в эмульсии
564
Часть 6. Контаминация и ее контроль
пропофола с сульфитом, в отличие от эмульсии пропофола с ЭДТА, происходили
химические изменения. Увеличение продуктов окисления пропофола свидетель-
ствовало, что сульфит, образованный из метабисульфита, при введении воздуха соз-
давал мощную окислительную среду. Лавойе с соавт. [38] показали, что сульфит спо-
собен вызывать реакции окисления липидов. Реакция сульфита с кислородом может
привести к образованию нескольких окисленных производных сульфита [39].
Вследствие окисления до сульфата (SO’“ -> SO’" + 2 е) сульфит может подвергать-
ся окислению одним электроном, что приводит в образованию реактогенного ради-
кала сульфита (SO’" -> SO'" + е“), который вступает в реакцию с кислородом с об-
разованием двух сильных окислителей: сульфита пероксид (SO'" + О2-> SO3OO- )
и радикала сульфата (SOj~ + О’-> SO'"). Эти вещества и собственно радикал суль-
фита ведут себя как сильные окислители, превращая натрия метабисульфит в про-
оксидантный агент, в зависимости от ситуации.
Перекисное окисление и образование свободных радикалов должны рассматри-
ваться как важный аспект фармацевтической стабильности и качества препаратов
для парентерального питания и внутривенного введения. Перекисное окисление
и образование свободных радикалов зависит от факторов окружающей среды, таких
как условия хранения и материал первичной упаковки, а также от состава препарата
или вспомогательных веществ, таких как токоферолы и метабисульфит. Поскольку
образование этих опасных веществ происходит в основном при применении препа-
ратов, контроль качества производителя не может обнаружить их присутствие.
6.1.3. Внешние источники контаминации
6.1.3.1. Остаточные органические растворители
Остаточные органические растворители представляют собой летучие органические
соединения, которые остаются в действующих и вспомогательных веществах, а так-
же фармацевтических продуктах после технологического процесса. Несмотря на
свои токсические свойства, растворители играют важную роль в фармацевтическом
производстве, при синтезе, выделении и очистке, и избежать их использования не-
возможно. Растворители, входящие в эту категорию, не включают растворители,
использующиеся в качестве вспомогательных веществ.
Остаточные органические растворители — это летучие органические раствори-
тели, которые не полностью удалены из продукта имеющимися технологиями про-
изводства и, следовательно, могут присутствовать в нем в виде примесей.
Международная конференция по гармонизации технических требований для ре-
гистрации лекарственных средств, предназначенных для людей (ICH, International
Conference on Harmonisation) [40], приняла руководство по примесям остаточных
растворителей, которое регламентирует пределы содержания растворителей в про-
дуктах для фармацевтического применения.
Органические растворители разделены на три класса. Растворители 1-го клас-
са — это растворители с установленными токсическими эффектами, использование
которых в производстве фармацевтических субстанций и вспомогательных веществ
следует избегать. Растворители 2-го класса обладают менее серьезной токсично-
стью, чем 1-го класса, а растворители 3-го класса имеют настолько несущественную
токсичность, что ограничения по их потреблению не устанавливаются. В табл. 12
Глава 6.1. Источники контаминации
565
приведены общие характеристики растворителей, включенных в каждый класс, а в
табл. 13 перечислены растворители и их допустимое содержание в фармацевтиче-
ских продуктах.
Таблица 12
Общие характеристики органических растворителей
Класс Действие Характеристика
1 Растворители, использование которых следует избегать Вещества с доказанной канцерогенностью для человека и опасные для окружающей среды
2 Растворители, использование которых должно быть ограничено Вещества с негенотоксичной канцерогенностью для животных и растворители, которые могут вызвать другие необратимые токсические эффекты, например, обладающие нейротоксичностью или тератогенностью
3 Растворители со слабым токсичным действием Установление предельного уровня потребления, связанного со здоровьем, не требуется. Растворители с разрешенной суточной дозой потребления 50 мг и более в день
Источник-. [40].
6.1.3.2. Контейнеры
Основное назначение первичной упаковки — обеспечить адекватную защиту фар-
мацевтического продукта. В фармакопеях установлены требования к контейнерам
в зависимости от характеристики лекарственной формы. Так, например, требова-
ния к контейнерам для капсул и таблеток обычно связаны с его конструкцией (на-
пример, контейнеры должны быть плотно укупорены, хорошо закрываться). Для
инъекционных, офтальмологических препаратов и препаратов для ингаляций так-
же предъявляются требования к материалу упаковки, так как совместимость с пер-
вичной упаковкой является очень важным вопросом для этих лекарственных форм.
С учетом целей настоящей главы здесь будут рассмотрены только упаковочные ма-
териалы, взаимодействие которых с препаратами может привести к образованию
примесей.
Компоненты упаковки для фармацевтических продуктов обычно изготавлива-
ются из стекла и полимерных материалов, таких как пластмассы и резины1. Не-
смотря на такую простую классификацию, стекло, полимеры и резины являются не
примитивными материалами, а скорее, достаточно сложными смесями.
Оценка химической стабильности компонентов упаковки зависит от вероятно-
сти взаимодействия компонента упаковки с препаратом и обычно проводится путем
воздействия на образец упаковки соответствующим растворителем при повышен-
ных температурах. Полученные экстракты должны анализироваться на содержание
высвобождаемых из упаковки веществ. Так, образцы стекла должны анализировать-
ся на высвобождаемые щелочные вещества, полимеры и резины, с учетом их по-
лучения, — на ряд экстрагируемых веществ. Повышенные температуры применяют
1 Хотя для фармацевтических продуктов также используется упаковка из металла, ее ис-
пользование ограничено поддержкой блистеров, при отсутствии контакта с препаратом,
и тубами для мазей.
566
Часть 6. Контаминация и ее контроль
Таблица 13
Растворители, включенные в каждый класс, и их предельно допустимая концентрация
в фармацевтических продуктах
Растворитель Предельные нормы содержания (ррт) Растворитель Предельные нормы содержания (ppni)
Класс 1 Класс 3
Бензол 2 Анизол —
1,2-Дихлорэтан 5 Ацетон —
1,1-Дихлорэтен 8 1-Бутанол —
1,1,1-Трихлорэтан 1500 2-Бутанол —
Четыреххлористый 4 Бутилацетат —
углерод /ирети-Бутилметиловый —
эфир
Класс 2 Гептан —
Ацетонитрил 410 Диметилсульфоксид —
Гексан 290 Изобутилацетат —
А.А-Диметилацетамид 1090 Изопропилацетат —
N,N-Диметил формамид 880 Кумол —
1,2-Диметоксиэтан 100 Метилацетат —
1,4-Диоксан 380 З-Метил-1-бутанол —
Дихлорметан 600 2- Метил-1 -пропанол —
1,2-Дихлорэтан 1870 Метилэтилкетон —
Ксилол 2170 Муравьиная кислота —
Метанол 3000 Пентан —
Метилбутилкетон 50 1-Пентанол —
N- Метилпирролидон 530 1-Пропанол —
Метилциклогексан 1180 2-Пропанол —
2-Метоксиэтанол 50 Пропилацетат —
Нитрометан 50 Тетрагидрофуран —
Пиридин 200 Уксусная кислота —
Сульфолан 160 Этанол —
Тетралин 100 Этилацетат —
Толуол 890 Этиловый эфир —
1,1,2-Трихлорэтан 80 Этил формиат —
Формамид 220
Хлорбензол 360
Хлороформ 60
Циклогексан 3880
Этиленгликоль 620
2-Этоксиэтанол 160
Примечание. «—» — ограничений нет. Источник: [40].
Глава 6.1. Источники контаминации
567
с целью увеличения скорости экстракции и имитации в течение более короткого
временного интервала взаимодействий с материалом упаковки при комнатной тем-
пературе, происходящих за более длительное время. Используемый для испытания
на стабильность растворитель должен демонстрировать такой же тип взаимодей-
ствия с материалом упаковки, как и лекарственная форма. Хотя предпочтительнее
использовать в испытании саму лекарственную форму, фармакопеи предписывают
проведение испытания со стандартными растворителями, такими как очищенная
вода, носитель и изопропиловый спирт.
Даже после определения профиля примесей и продуктов разложения фармацев-
тической субстанции и подтверждения соответствия контейнеров установленным
требованиям во время стерилизации или при хранении могут возникать некоторые
неожиданные взаимодействия препаратов и первичной упаковки.
В данном разделе рассматриваются наиболее важные материалы для контейне-
ров: стекло, пластмасса и эластомеры — а также вопросы контаминации инъекци-
онных препаратов.
Стеклянные контейнеры. Стеклянные контейнеры для упаковки фармацевтиче-
ских препаратов должны выдерживать требования по стабильности, которые связа-
ны с наличием в структуре стекла определенных компонентов, с типом лекарствен-
ной формы и способом ее введения. Согласно фармакопейным требованиям для
хранения фармацевтических препаратов могут быть использованы контейнеры из
стекол четырех типов. В табл. 14 приведена классификация стеклянных контейне-
ров в соответствии с характером продукта и фармацевтическим назначением.
Таблица 14
Типы стекла согласно фармакопеям и их применение в фармацевтической промышленности1
Ъш стекла Общее описание Применение
I Высокоустойчивое боросиликатное стекло Парентеральные препараты
II Обработанное натриево- Парентеральные препараты
кальциево-силикатное стекло с кислой или нейтральной средой
III Натриево-кальциево-силикатное Не для парентеральных
стекло препаратов
нп Натриево-кальциево-силикатное Для препаратов д ля наружного
(не парентеральное) стекло применения или приема внутрь
Хотя основным компонентом стекла является диоксид кремния, разные
типы стекла получают добавлением или удалением определенных компонентов.
В табл. 15 перечислены основные компоненты типов стекла, используемых в фар-
мацевтической промышленности, основанные на критериях, указанных в табл. 14.
Для повышения устойчивости стекло I типа должно содержать не менее 10% окси-
1 Европейская фармакопея 7.0 выделяет три типа стекла — I, II, III. При этом контейнеры
из стекла II типа могут быть использованы и для непарентеральных препаратов, III типа —
рекомендуется использовать для неводных парентеральных препаратов, порошков (кроме
лиофилизатов) и для непарентеральных препаратов (более подробно см. ЕФ, раздел 3.2.1
«Стеклянные контейнеры для фармацевтического применения»). — Примеч. перев.
568 Часть 6. Контаминация и ее контроль
да бора и более высокую концентрацию оксида алюминия, чем обычное натриево-
кальциево-силикатное стекло. Сниженное количество оксида натрия уменьшает
растворимость стекла I типа в воде. Стекло II типа получают из обычного натриево-
кальциево-силикатного стекла после процесса химической обработки (деалкалини-
зации) внутренней поверхности при производстве. В этом процессе, называемом
серной обработкой, диоксид серы вдувается в свежеотформованные бутылки по
мере выхода из формовочной машины и вступает во взаимодействие с поверхно-
стью стекла с образованием сульфата натрия согласно уравнению
SO, + V2O, + Na,О---------> Na,SO,
2 ' 2 2 стекло 2 4
Контейнер покрывается взвесью сульфата натрия, которая смывается перед на-
полнением.
Таблица 15
Тйпичный состав стеклянных контейнеров согласно фармакопейной классификации
Компонент Тйп I, боросиликат, % Тйпы II, III, IV, натрий-кальций-силикат, %
SiO, 70 73
в,о3 10 —
Na,O 9 14
А12О3 6 2
ВаО 2 —
К2О 1 —
СаО 1 7
MgO 0,5 4
ZnO 0,5 —
Источник', каталог «Корнинг. Науки о жизни: техническая информация, описание стекла,
используемого для приготовления лабораторной посуды Корнинг».
Испытание на стабильность стеклянных контейнеров, предписанное фармако-
пеями, ограничено воздействием воды на поверхность стекла с последующим из-
мерением выделившейся щелочи («водная атака»). После автоклавирования кон-
тейнера, наполненного водой, при температуре 121 °C в течение 60 минут титруют
100 мл полученного экстракта 0,02 н. раствором серной кислоты. Объем израсходо-
ванного кислого титранта не должен превышать установленное значение, рассчи-
танное по емкости контейнера. Например, на титрование экстракта от контейне-
ров из стекла I и II типов емкостью менее 10 мл должно потребоваться менее 2,0 мл
титранта, а для стекла III типа — 20 мл. Для контейнеров емкостью от 20 до 500 мл
расход титранта должен составить от 0,8 до 0,2 мл — для стекол I и II типов, от 8,0
до 2,2 мл — для стекла III типа. Для стекла НП (не парентеральное) пределы расхода
титранта не установлены.
Оценки количества высвободившейся щелочи, однако, недостаточно для демон-
страции реальной стабильности стеклянного контейнера. Стекло не является таким
инертным, каким кажется, а вода — не единственное вещество, которое взаимодей-
ствует с поверхностью стекла. Несмотря на это, в Фармакопее США в единственном
испытании на наличие токсичных примесей в стекле указывается только мышьяк
Глава 6.1. Источники контаминации 569
(<661 > «Контейнеры», USP 27); контейнеры из стекла I типа должны оцениваться
согласно процедуре «водной атаки», а уровень содержания мышьяка не должен пре-
вышать 0,1 ррт. Хотя мышьяк не указан в составе стекла, он может использоваться
в качестве осветляющей добавки в стекольном производстве (см. далее) и, следова-
тельно, может присутствовать в структуре стекла.
Исследования показали, что даже стекла I типа не являются полностью инерт-
ными. Бохрер {Bohrer) с соавт. [41] показали, что гидролитическая устойчивость
стеклянных контейнеров уменьшается в присутствии некоторых веществ, обычно
имеющихся в инфузионных растворах. В табл. 16 представлено количество высво-
бодившейся щелочи и концентрации некоторых компонентов стекла (диоксида
кремния, натрия, бора, алюминия) в водных экстрактах, полученных после испы-
тания на гидролитическую устойчивость1. Эти данные показывают что, несмотря
на соответствие фармакопейным требованиям, стекло может высвобождать входя-
щие в него компоненты под действием ионов в растворах и даже чистой воды. Ще-
лочные растворы гидрокарбоната и глюконата вызывали наибольшее содержание
компонентов стекла в экстрактах, подтверждая способность щелочных растворов
атаковать и растворять структуру стекла. Растворы глюкозы и лимонной кисло-
ты взаимодействовали с поверхностью стекла, избирательно экстрагируя не толь-
ко алюминий, но и медь со свинцом (их следовые количества также присутствуют
в стекле). Это избирательное действие цитрата и глюкозы может быть связано с их
свойством образовывать комплексы с металлами.
Присутствие в качестве примеси мышьяка в фармацевтических продуктах кон-
тролируется фармакопейными стандартами. Практически все фармакопейные ста-
тьи содержат требования по предельному содержанию мышьяка от 0,1 до 3 ррт. Бох-
рер с соавт. показали, что мышьяк является примесью, повсеместно встречающейся
в исходных материалах [16] (см. обсуждение по воде в разделе 6.1.2.1), и присутству-
ет в готовых продуктах в концентрациях, значительно превышающих концентра-
ции в соответствующих исходных материалах [12]. Согласно данным этих авторов,
основным источником мышьяка в инъекционных препаратах являются стеклянные
контейнеры [16]. Как уже упоминалось ранее, стекло может содержать мышьяк, так
как оксид мышьяка (III) используется в качестве осветлителя для улучшения про-
зрачности стекла [42] — крайне важного параметра контейнеров для растворов для
внутривенного введения, которые должны проверяться визуально до использования
[43]. Считают, что оксид мышьяка (III) взаимодействует с нитратом калия жидкого
стекла с высвобождением кислорода и оксидов азота. Эти газы образуют большие
пузыри, которые быстро поднимаются на поверхность, перемешивая содержимое
печи и увлекая за собой пузырьки, образованные разложением сырья. В табл. 17
приведены концентрации мышьяка, определенные этими авторами в растворах для
парентерального питания. Наиболее контаминированными были растворы гидро-
карбоната натрия и глюконата кальция; уровень содержания мышьяка в них превы-
шал даже норму содержания для инфузионных растворов — 0,1 мг/мл.
1 Испытание на гидролитическую устойчивость, описанное в этой главе, предназначе-
но для испытания стекла в виде порошка, а не для «испытания водной атакой», как указано
выше; см. приложение <661> USP21 [1].
Таблица 16
Объем H2SO4, затраченный на титрование растворов до и после нагревания при контакте со стеклянной массой в течение 30 минут
при температуре 121 °C, и элементы, экстрагированные из стекла при выполнении испытания по USP со стеклянным порошком для
чистых ампул в присутствии различных растворов
Образец Объем 0,02 н. раствора H2SO4, мл Концентрации элементов, мг/л, ± стандартное отклонение (и = 3)
до нагревания после нагревания Na Кремния диоксид Борат А1 Си РЬ
NaCl а 0,56 + 0,01 — 18,30 ±2,91 2,22 ±0,11 0,15 ±0,01 0,09 ±0,02 0,03 ±0,01
КС1 а 0,69 + 0,01 14,42 ±1,51 9,96 ±1,71 1,58 ±0,03 0,19 ±0,01 0,14 ±0,08 0,04 ±0,02
СаС12 а 0,45 + 0,02 13,41 ±1,33 7,31 ±2,08 2,00 ±0,11 0,18 ±0,01 0,06 ±0,01 0,02 ±0,01
MgCl2 а 0,13 ±0,01 15,25 ±1,64 6,54 ±0,53 3,25 ±0,21 0,16 ±0,01 0,09 ±0,03 0,02 ±0,01
Натрия глюконат 2,68 ±0,22 1,83 ±0,05 — 264,0 ±18,7 1,91 ±0,09 2,04 ±0,01 0,49 ±0,07 0,05 ±0,01
Натрия гидрофосфат а 10,22 ±0,91 — 10,21 ±3,41 2,66 ±0,09 4,93 ±0,01 0,05 ±0,01 0,05 ±0,02
Калия гидрофосфат а 8,78 ±0,95 18,38 ±2,34 45,32 ±3,76 1,73 ±0,06 4,30 ± 0,07 0,13 ±0,04 0,04 ±0,02
Натрия гидрокарбонат 24,3 ±0,41 12,68 ±1,03 — 271,7 ±23,2 5,98 ±0,14 5,22 ±0,24 0,88 ±0,09 0,06 ±0,03
Лимонная кислота а 1,92 ±0,04 19,82 ±1,90 16,46 ±0,87 1,44 ±0,10 6,36 ±0,53 0,40 ±0,03 0,10 ±0,05
Глюкоза а 1,08 ±0,05 13,86 ±1,22 11,10 ±0,8 3,18 ± 0,17 6,34 ±0,33 0,28 ±0,01 0,15 ±0,03
“рН<7.
Примечание. Концентрации растворов: 0,01 моль/л [41].
570______________________________Часть 6. Контаминация и ее контроль
Глава 6.1. Источники контаминации
571
Таблица 17 Наличие примесей мышьяка в коммерческих парентеральных препаратах
Продукт Общий мышьяк, As (V), As (III),
мкг/л мкг/л ± RSD мкг/л ± RSD
КС119,1% (10)а 41,3 41,3+1,6 н/о
КС110% (4) 23,6 23,6 + 0,8 н/о
NaCl 20% (10) 43,8 40,9 + 5,7 2,9 ±0,2
NaCl 20% (4) 15,9 15,9 ±3,2 н/о
Натрия ацетат 2 мэкв./мл (1) 46,1 41,8 + 2,0 4,3 ±0,6
Натрия фосфат 0,5 моль/л (10) 37,7 36,7 ±2,5 н/о
Натрия гидрокарбонат 8,4% (5) 248,6 227 ±0,5 20,8 ±2,0
Натрия гидрокарбонат 8,4% (9) 198,3 147,2 ±1,2 51,1 ±0,8
Кальция глюконат 10% (11) 73,1 46,8 ±4,1 26,3 ±2,3
Кальция глюконат 10% (9) 92,7 72,8 ±0,5 19,9 ±1,9
Кальция глюконат 10% (4) 239,6 176 ±7,2 63,6 ±2,0
Магния сульфат 50% (11) 15,7 15,7 ±0,2 5,7 ±0,2
Магния сульфат 50% (10) 33,5 12,2 ±1,2 21,3 ±1,8
Магния сульфат 50% (9) 53,8 39,9 ±5,6 13,9 ±3,0
Глюкоза 25% (6) 21,8 16,5 ±2,6 5,3 ±0,6
Глюкоза 25% (6) 18,6 14,0 ±2,2 4,6 ±0,7
Витамины (Тиаминоз6) 103,7 86,1 ±0,9 17,6 ±3,2
Витамины (Декстровитаз’) 10,2 10,2 ±0,2 3,8 ±0,1
Витамины (Фрутовенаг) 61,7 21,5 ±1,7 40,2 ±6,7
Аминокислоты 10% (3) 2,5 2,5 ±0,1 н/о
Аминокислоты 10% (1) 15,4 15,4 ±2,3 н/о
Аминокислоты 10% (2) 41,0 41,0 ±0,9 н/о
Аминокислоты 8% (2) 21,1 16,7 ±1,2 4,4 ±0,4
Аминокислоты 8% (2) 94,7 87,9 ± 5,7 6,8 ±1,0
Липидная эмульсия 10% (2) 0,9 0,9 ±0,3 н/о
Липидная эмульсия 20% (2) 1,7 1,7 ±0,6 н/о
Гепарин 5000 МЕ/мл (7) 56,7 17,2 ±2,8 39,5 ±2,3
Гепарин 5000 МЕ/мл (8) 79,4 79,4 ±4,9 23,3 ±1,2
а Номера в скобках означают производителя препарата: (1) — «Б. Браун», (2) — «Фрезени-
ус», (3) — «Бакстер», (4) — «Халекс Истар», (5) — Японская фармакопея, (6) — «Мерк», (7) —
«ЭМС», (8) — «Элкинс Синн», (9) — «Эристон», (10) — «Гейер», (11) — «Типолабор».
6 В 10 мл: глюкозы 3 г, аскорбиновой кислоты 0,25 г, тиамина гидрохлорида 0,015 г.
• В 20 мл: глюкозы 2 г, аскорбиновой кислоты 2 г, пиридоксина гидрохлорида 20 мг, нико-
тинамида 30 мг, рибофлавин.
г В 20 мл: фруктозы 2 г, аскорбиновой кислоты 1 г, пиридоксина гидрохлорида 20 мг, на-
трия пантотената 10 мг, рибофлавина 4 мг.
Примечание. RSD — относительное стандартное отклонение; н/о — не обнаружено (ниже
предела обнаружения). Источник: [16].
572 Часть 6. Контаминация и ее контроль
Другие химические элементы, такие как хром, барий и цинк, также были обна-
ружены в растворах для парентерального питания; хотя это не указано, источником
их происхождения, вероятно, было стекло. Поскольку стекло — неорганический
материал, оно может быть источником неорганических примесей. Исключениями
могут быть цинк и барий, которые присутствуют не только в стекле I типа, но и в до-
бавках к полимерам. В табл. 18 представлены химические элементы, присутствую-
щие в виде примесей в различных инфузионных растворах, и их соответствующие
концентрации.
Данные по содержанию алюминия представлены отдельно вследствие его осо-
бых свойств и токсичности. С момента открытия в 1976 г. Алфреем (Alfrey) с соавт.
[48] факта, что алюминий может накапливаться в организме больных со сниженной
почечной функцией, вызывая токсические эффекты и неврологические заболева-
ния, было проведено много работ по выявлению и сокращению источников кон-
таминации алюминием. В настоящее время алюминий также считается токсичным
для больных с нормальной почечной функцией, которые получают препараты для
парентерального питания, — главным образом для недоношенных новорожденных
[49]. К признанным источникам контаминации алюминием относят воду, исполь-
зуемую для диализа, растворы для полного парентерального питания и компонен-
ты для приема внутрь, содержащие алюминий. Хотя вспомогательные материалы
и фармацевтические субстанции также являются источниками контаминации,
наиболее важный источник примеси алюминия в растворах для парентерального
питания — это хранение парентеральных препаратов в стеклянных контейнерах.
На рис. 8 приведено содержание алюминия в растворах различных типов, хранив-
шихся в полимерных и стеклянных контейнерах в течение трех месяцев. В то время
как из контейнеров из полиэтилена в раствор перешло не более 50 мкг/л, в раство-
рах, хранившихся в стеклянных контейнерах, концентрация алюминия превышала
3000 мкг/л, подтверждая, что стекло является бесспорным и постоянным источни-
ком контаминации алюминием.
Кроме того, те же авторы показали, что хотя стекло и является источником кон-
таминации алюминия, его высвобождение зависит от природы вещества, контакти-
рующего с поверхностью [51, 52]. В эксперименте со стеклом и ионообменником,
содержащим прикрепленный алюминий, комплексообразующие агенты и амино-
кислоты были способны экстрагировать элемент из обоих источников, но степень
экстракции зависела от сродства лиганда к металлу. Результаты этого взаимодей-
ствия, измерявшегося в течение 2 месяцев, приведены на рис. 9. На основе этих вы-
водов существенная контаминация алюминием растворов глюконатов и фосфатов
может быть объяснена сродством этих веществ к алюминию. Они способны изби-
рательно вымывать алюминий из структуры стекла при контакте со стеклянными
контейнерами (табл. 19).
На рис. 10 показаны результаты хранения различных аминокислот, компонентов
парентеральных растворов в контейнерах из стекла I типа в течение 1 года. Не все
растворы были контаминированы алюминием, а только те из них, которые содер-
жали аминокислоты, имеющие сродство к этому химическому элементу.
В табл. 20 приведено содержание алюминия в парентеральных препаратах, опре-
деленное различными авторами в разных странах. Не все авторы упоминают в каче-
Таблица 18
Примеси металлов, обнаруженные в коммерческих растворах для парентерального питания
Образец Концентрация элемента, мкг/л Источник
Zn Сг Fe РЪ As Ge Си Cd
Мп Ba Sn
Мешки с полной смесью для парентераль- ного питания 9,1 — — — — ИЗ]
Аминокислоты 60-470 н/о — — — [44]
Z-цистеин НС1 32000-86000 110-230 — — — — — — — — — [44]
NaCl, КС1, ацетат Na 350-560 20-230 — — — — — — — — — [44]
Глюконат Са 280-2380 — — — — — — — — — — [44]
Фосфат 901-2330 390-440 — — — — — — — — — [44]
Глюконат Са 47-244 — 237-655 — — — — — — — — [45]
Полная смесь для парентерального питания 233-703 — 84 — — — — — — — — [45]
Стандартная смесь для парентерального питания для взрослых, «Бакстер» — 86,8 — 0,6 288,0 309,9 11,0 0,5 5,5 — — [46]
Стандартная смесь для парентерального питания для взрослых, «Эбботт» — 25,8 — 1,1 65,0 109,9 16,0 0,4 5,5 — — [46]
Стандартная смесь для парентерального питания д ля взрослых — 139,8 — 0,4 65,9 299,9 22,0 1,4 15,9 — — [46]
Стандартная смесь для парентерального питания для взрослых, «Бакстер» — 15,8 — 0,7 61,0 47,9 73,0 0,6 9,2 — — [46]
Стандартный раствор аминокислот для взрослых, «Фрезениус» 131,2 — — 4,99 — — — — — 2,96 0,35 [47]
Стандартный раствор аминокислот для взрослых, «Б. Браун» 88,92 — — 16,80 — — — — — 6,66 4,37 [47]
Стандартный раствор аминокислот для взрослых, «Бакстер» 1,39 — — 4,39 — — — — — 40,81 0,71 [47]
Примечания, н/о — не обнаружено;«—» — не измерялась.
Глава 6.1. Источники контаминации_______________________________________573
574
Часть 6. Контаминация и ее контроль
□ Контрольный образец
Рис. 8. Алюминий, экстрагированный из стеклянных контейнеров типа I и из полиэтиленовых
контейнеров под действием растворов NaCl, KCI, альбумина, глюкозы, гепарина, HCI и NaOH на
30-й и 60-й день хранения при комнатной температуре. Растворы альбуминов: А — бычий («Мерк»),
В — бычий («Реаген»), С — овальбумин («Сигма») [50]
стве источника контаминации алюминием стеклянную упаковку, но, по-видимому,
это был основной источник металла. Эти данные также подтверждают вывод, что
свойства компонентов препарата играют ключевую роль в избирательном вымыва-
нии алюминия из стекла.
Показанные результаты свидетельствуют о наличии существенных различий
в содержании алюминия в продукте в зависимости от компонентов препарата, про-
изводителя и, главным образом, природы материала первичной упаковки.
Полимерные контейнеры. Материалы, используемые для производства таких кон-
тейнеров, представляют собой органические полимерные вещества. Полимер — это
Глава 6.1. Источники контаминации
575
Рис. 9. Кривые содержания алюминия в экстрактах из стеклянных частиц (18 меш) и ионообменника
с AI (18 меш), полученных при воздействии некоторых аминокислот и комплексообразующих
агентов, в зависимости от времени воздействия. Концентрация лигандов 0,05 моль/л [51]
Рис. 10. Зависимость концентрации алюминия, высвобождаемого из стекла аминокислотами,
от длительности воздействия при комнатной температуре. Концентрация аминокислот
0,028 моль/л [53]
большая молекула, состоящая из повторяющихся небольших и простых химиче-
ских фрагментов. Повторяющийся фрагмент полимера обычно идентичен или поч-
ти идентичен мономеру или исходному материалу, из которого образован полимер.
Структурные фрагменты полимеров, наиболее часто использующиеся для произ-
водства полимерных контейнеров, и применение этих контейнеров в фармацевти-
ческих целях приведены в табл. 21.
Современные технологии получения полимеров основаны на катализе, и катали-
тические методы широко используются в производстве полимеров. Катализаторы,
поскольку они только способствуют протеканию реакции, не считаются компонен-
тами полимера, однако могут присутствовать в полимерных материалах в качестве
примесей. В табл. 22 перечислены распространенные катализаторы, которые исполь-
зуются в реакциях полимеризации упомянутых выше полимеров и могут быть обна-
ружены в качестве примесей в препаратах, хранящихся в полимерных контейнерах.
576
Часть 6. Контаминация и ее контроль
Таблица 19
Примесь алюминия в коммерческих парентеральных растворах и содержание алюминия
в контейнерах
Препарат А1 в растворе ± стандартное отклонение, мкг/л Контейнер А1 в контейнере, %
NaC120% 149 + 10 Стеклянная ампула 1,43
13±4 Полиэтилен 0,04
КС110% 68±6 Стеклянная ампула 1,25
23 ±5 Полиэтилен 0,06
Магния сульфат 50% 560 ±85 Стеклянная ампула 1,25
380 ±288 Стеклянная ампула 1,43
Натрия ацетат 2 мэкв/мл 45 ±7 Стеклянная ампула 2,14
17±8 Полиэтилен 0,05
Калия фосфат 2 мэкв/мл 988 ± 76 Стеклянная ампула 1,98
1325 ±142 Стеклянная ампула 2,45
Натрия фосфат 0,5 М 933 ± 88 Стеклянная ампула 1,65
879 ± 203 Стеклянная ампула 2,05
Кальция глюконат 10% 5621 ±1165 Стеклянная ампула 1,51
5960 ±62 Стеклянная ампула 2,21
Натрия гидрокарбонат 833 ±141 Стеклянная бутылка 0,99
8,4% 922 ±102 Стеклянная бутылка 1,03
Микроэлементы’ 1129 ±33 Стеклянные ампулы 2,14
Микроэлементы6 1854 ±744 Стеклянная ампула 2,21
Аминокислоты 10% 164 ±6 Стеклянная бутылка 0,82
Резиновая пробка 3,91
Аминокислоты 10% 116 ±30 Стеклянная бутылка 0,76
Резиновая пробка 4,23
Аминокислоты 10% 93 ±23 Стеклянная бутылка 0,84
Резиновая пробка 5,34
Аминокислоты 10% 65 ±13 Стеклянная бутылка 0,89
Резиновая пробка 5,70
Аминокислоты 10% 23 ±8 Полимерный мешок 0,01
Глюкоза 50% 13 ± 1 Полиэтилен 0,04
293 ± 14 Стеклянная ампула 1,87
Глюкоза 25% 9±3 Полиэтилен 0,08
370 ±23 Стеклянная ампула 1,87
Альбумин 20% 644 ±58 Стеклянный флакон 0,67
Резиновая пробка 4,06
149 ±23 Стеклянный флакон 0,66
Резиновая пробка 3,99
Гепарин 5000 МЕ/мл 732 ±23 Стеклянная ампула 2,88
738 ± 54 Стеклянная ампула 3,03
а Состав: 22,0 мг ZnSO4, 6,3 мг CuSO4,2,46 мг MnSO4, 102,53 мкг СгС13 в ампуле.
6 Состав: 8,8 мг ZnSO4,1,60 мг CuSO4,123,04 мкг MnSO4,20,50 мкг СгС13 в ампуле.
Примечание. Все растворы были в пределах установленного срока годности. Поставщики:
«Эбботт», «Эристон», «Эстер», «Бакстер», «Беринг», «Б. Браун», «Дарроу», «Фрезениус», «Фуд-
жисава», «Гейер», «Халекс Истар», «Типофарма», «Сантиса», «Рош», «Зеналб». Источник-. [52].
Глава 6.1. Источники контаминации
577
Таблица 20
Содержание алюминия в коммерческих растворах для парентерального введения
Продукт Спецификация Производитель Концентрация А1, мкг/л Источник
Электролиты
NaCl 10% Эбботт 3 [Ю]
10% Коммершиал Польфа 14 [54]
5,85% Браун 1 [П]
1М Каби 22 [Н]
10% Эбботт 43 ±8' [15]
10% Элкинс Синн 78 + Т [15]
КС1 15,0% Коммершиал Польфа 18 [54]
— Эбботт 4(5-11) [Ю]
— Элкинс Синн 3 [Ю]
7,45% Браун <0,6 [Н]
7,45% Каби 12 [Н]
7,45% Браун 33 + 5' [15]
10% Эбботт 97 ±8' [15]
10% Эстер 23 + 5' [15]
Хлорид Са — Элкинс Синн 15 (12-19) [Ю]
— Эбботт 5 [Ю]
0,5 М Берингер 27 [Н]
1н Каби 224 [И]
Ацетат Na 2 мэкв./мл Браун 17 ±8' [15]
Ацетат К — Эбботт <5 [Ю]
Сульфат Mg 50% Эбботт 5 [Ю]
50% Браун 606 [И]
— Тёйер 560 + 85' [15]
— Эристон 380 + 288' [15]
Фосфат Na — Инвенекс 2236 (2026-2370) [Ю]
0,5 М Эбботт 933 + 88' [15]
0,5 М Гейер 430+177' [15]
Фосфат К — Инвенекс 92 [Ю]
— Эбботт 2189 (2069-2301) [Ю]
— Браун 188 [П]
— Каби 2826 [И]
2 мэкв./мл Фрезениус 1021 ± 188' [15]
2 мэкв./мл Браун 988 ±76' [15]
2 мэкв./мл Эстер 332 ±27' [15]
Глюконат Са 10% Элкинс Синн 3973 (1095-5565) [Ю]
10% Лифомед 2245 (2000-2586) [Ю]
10% Браун 4734 [П]
Часть 6. Контаминация и ее контроль
578
Таблица 20 (продолжение)
Продукт Спецификация Производитель Концентрация А1, мкг/л Источник
Глюконат Са 10% Фрезениус 6549 НИ
10% Фарма Хамельн 4421 [Н]
10% Коммершиал Польфа 4400 [54]
10% Элкинс Синн 3987 + 993* [15]
10% Браун 4530 ±1072* [15]
10% Эристон 5960 + 62* [15]
10% Халекс Истар 6781 ±1842* [15]
Микроэлементы (МЭ)
Цинка хлорид — Эбботт 99 (81-123) [Ю]
Трацитранс (МЭ) — Фрезениус 994 [П]
Пед-эл (МЭ детские) — Фармация 3000 [54]
Педитрас (МЭ — Фармация 120 [54]
детские)
Пед-элемент (МЭ — Дарроу 1423 ±68* [15]
детские)
Эд-элемент (МЭ) — Дарроу 3574 + 237* [15]
Трацитранс (МЭ) — Фрезениус 5712 + 988* [15]
Аминокислоты
Фреамин 8,5% МакТоу 12 (5-24) [Ю]
Травасол 10% Травенол 7 (6-8) [Ю]
ГепатАмин 8% МакТоу 22 [Ю]
Аминоплазмаль 10% Браун 55 [П]
Аминопед 10% Каби 38 [И]
Аминостерил 8% Фрезениус 17 [П]
Примен 10% Тлинтек 120 [54]
Аминомел 12,5% Тлинтек 121 [54]
Ваминолакт 6,5% Фармация 30 [54]
Аминоплазмаль £10 — Браун 160 ±30* [15]
Педиамино 10% Браун 116 + 30* [15]
Аминопед 10% Фрезениус 195 ± 27* [15]
Нефроамино АЕН — Браун 272 ±66* [15]
Примен 10% Бакстер 65 ±13* [15]
Углеводы
Глюкостерил 70% Фрезениус 9 [Н]
Глюкоза 40% Фрезениус 20 [П]
50% Шива/Гормонхеми 18 [Н]
20% Шива/Гормонхеми 3 [П]
10% Шива/Гормонхеми <0,6 [И]
Глава 6.1. Источники контаминации
579
Таблица 20 (окончание)
Продукт Спецификация Производитель Концентрация А1, мкг/л Источник
Декстроза 10, 20, 50% Эбботт <5 [Ю]
10, 50% МакТоу <5 [Ю]
5,10, 50% Травенол <5 [10]
Глюкоза 20% Коммершиал Польфа 16 [54]
25% Дарроу 9 + 3’ [15]
Эквиплекс 9±2' [15]
50% Фрезениус 15 ±3* [15]
Дарроу 17 ±3' [15]
Дж. П. Инд. Фарм 8 ±2' [15]
Б. Браун Лаб. 13 +Г [15]
Эристон 11 ±4' [15]
Липиды
Интралипвд 10% Каби Витрум <5 [Ю]
20% Каби Витрум <5 [Ю]
10% Пфриммер Каби 5 [П]
20% Пфриммер Каби 7 [И]
Липофундин 20% Браун 35 [П]
20% Браун 14 [П]
Интралипид 20% Фармация 30 [54]
Липофундин 20% Браун 180 [54]
10% Браун 29 + 6' [15]
20% Браун 34 + 7" [15]
Витамины
Витамин С 500 мг ЭМС 3443 + 233' [15]
Витамин В12 1 мг Бункер 92 + 23' [15]
В-комплекс — Гиполабор 1089 +127' [15]
В-комплекс — Эристон 709 ±65' [15]
Поливитамины MVI — АйСиЭн 588 + 63’ [15]
Другие
Альбумин 20% — 190,4 [55]
20% Беринг 235 ± 12' [15]
Гепарин 1000 Ед./мл — 211,7 [55]
5000 Ед./мл Фуджисава 732 + 23' [15]
5000 Ед./мл Кристалиа 72 ±6' [15]
* Среднее значение ± стандартное отклонение по трем пробам одной серии.
Примечание. «—» — не указано.
580
Часть 6. Контаминация и ее контроль
Таблица 21
Полимерные материалы, их структурные фрагменты и применение в фармацевтических
продуктах
Полимер Мономеры Применение в биотехнологических продуктах
Полиэтилен (РЕ, ПЭ) Этилен Твердые лекарственные формы. Неинъекционные водные растворы. Водные инфузии для внутривенного введения
Полипропилен (РР, ПП) Пропилен Твердые лекарственные формы. Неинъекционные водные растворы. Водные инфузии для внутривенного введения
Поливинилхлорид (РУС, ПВХ) Винилхлорид Твердые лекарственные формы. Неинъекционные водные растворы. Водные инфузии для внутривенного введения. Кровь и компоненты крови. Трубки для систем переливания крови
Полиэтиленвинилацетат (EVA, ПЭВА) Этилен и винилацетат Водные инфузии для внутривенного введения. Трубки для систем вливания препара- тов для парентерального питания
Полиэтилентерефталат Терефталевая кислота Твердые лекарственные формы для
(РЕТ, ПЭТ) или диметилтерефталат и этиленгликоль приема внутрь. Жидкие лекарственные формы для приема внутрь
Источники'. [5] (ВР) и [56] (FDA).
Таблица 22
Катализаторы, используемые в реакциях полимеризации материалов, указанных в табл. 21
Полимер Катализаторы
ПЭ TiCl4 + А1(С2Н5)3; CrOj/SiO2; МоО3/А12О3
ПП a-TiCl3 + А1(С2Н5)3
ПВХ —
ПЭВА —
ПЭТ —
Источник: [57].
Другие примеси, источником которых могут быть полимерные контейнеры, —
это добавки, необходимые для превращения полимерного сырья в требуемые кон-
тейнеры. В то время как полиэтилен может использоваться без каких-либо добавок,
другие полимеры практически бесполезны в чистом виде, но могут быть превраще-
ны в исключительно удобные продукты при их комбинировании с другими веще-
ствами или материалами. Наиболее часто в полимерах, используемых в фармацев-
тической промышленности, находят антиоксиданты, стабилизаторы температуры,
смазывающие агенты, пластификаторы, наполнители и красители. Эти добавки
Глава 6.1. Источники контаминации
581
могут быть в жидком, твердом виде или в форме мелких частиц, их используют в ко-
личествах от 1 до 50% и более от полимерной массы. Добавки, необходимые для
каждого вида полимера, описаны в табл. 23.
Таблица 23
Общие сведения о добавках, обычно используемых в производстве полимеров
Добавки
Полимер f Антиоксидант Стабилизатор температуры Смазывающий агент Пластифи- катор Напол- нитель Краситель
ПЭ X — X — X X
ПП X — X — X —
пвх — X X X X X
ПЭВА X — X — X X
ПЭТ — — X — X X
Источник: [58].
Добавки, разрешенные в производстве полимеров для фармацевтического при-
менения, и предельное содержание их в полимерной массе (согласно ВР и ЕФ)
обобщены в табл. 24.
Таблица 24
Добавки, разрешенные для полимерных материалов для фармацевтического применения,
согласно ВРи ЕФ и их предельные содержания1
Номер добавки Название добавки Полимер Нормы, %
Пластификаторы
1 Ди(2-этилгексил)фталат ПВХ 40
2 Цинка октаноат ПВХ 1
3 А^-диацетилэтилендиамины ПВХ 1
4 Эпоксидированное соевое масло ПВХ 10
5 Эпоксидированное льняное масло Антиоксиданты ПВХ 10
7 Бутилгидрокситолуол ПЭ, ПП, ПЭВА 0,125
8 Этилен-бис [ 3,3-бис [ 3- (1,1 -диметилэтил)-4- гидроксифенил] бутаноат] ПЭ, ПП 0,3
9 Пентаэритритил тетракис [ 3 - (3,5-ди-лпрет-бутил-4- ПЭ, ПП 0,3
гидроксифенил)пропионат] ПЭВА 0,2
10 2,2',2",6,6',6''-гекса-/ирет-Бутил-4,4',4"-[(2,4,6- ПЭ,ПП 0,3
триметил-1,3,5-бензолтриил)трисметилен]трифенол ПЭВА 0,2
И Октадецил 3-(3,5-ди-/лретп-бутил-4-гидроксифенил) ПЭ, ПП 0,3
пропионат ПЭВА 0,2
12 трис(2,4-ди-/иретл-Бутилфенил)фосфит ПЭ.ПП 0,3
ПЭВА 0,2
13 1,3,5-трис(3,5-ди-лгрет-Бутил-4-гидроксибензил)-я- триазин-2,4,6(Ш,3//;5/Г)-трион ПЭ,ПП 0,3
1 Названия добавок приведены не по требованиям ИЮПАК, а в виде общеупотребимых
синонимов. — Примеч. ред.
Часть 6. Контаминация и ее контроль
582
Таблица 24 (окончание)
Номер добавки Название добавки Нормы, Полимер %
14 2,2'-бис(октадецилокси)-5,5'-спироби[1,3,2- диоксафосфинан] пэ,пп 0,3
15 Диоктадецилдисульфид пэ,пп 0,3
16 Дидодецил 3,3'-тиодипропионат пэ,пп 0,3
17 Диоктадецил 3,3'-тиодипропионат пэ,пп 0,3
18“ 2,4-бис( 1,1 -Диметилэтил)фенилбифенил-4,4'- пэ,пп 0,1
19 20 дииддифосфонит. 2,4-бис( 1,1 - Диметилэтил)фенилбифенил- 3,4'- диилдифосфонит. 2,4-бис(1,1-Диметилэтил)фенилбифенил-3,3'- диилдифосфонит. 2,4-бис( 1,1 -Диметилэтил)фенилбифенил-4- илфосфонит. 2,4-бис(1,1-Диметилэтил)фенилфосфит. 2,4-бис( 1,1-Диметилэтил)фенил 4'- [бис[2,4-бис( 1,1- диметилэтил)фенокси] фосфанил] бифенил-4- илфосфонат. 2,4-бис( 1,1 -Диметилэтил)фенол Смазывающие вещества и наполнители Стеариновая кислота Олеамид ПЭВА 0,5
21 Эрукамид ПЭВА 0,5
22“ Сополимер диметилбутандиоата и 1-(2- ПЭ, ПП 0,3
23 гвдроксиэтил)-2,2,6,6-тетраметилпиперидин-4-ола Гидроталцит пэ,пп 0,5
24 Алканамиды пэ.пп 0,5
25 Алкенамиды пэ,пп 0,5
26 Натрия алюмосиликат пэ,пп 0,5
27 Диоксид кремния ПЭ, ПП 0,5
28 Натрия бензоат пэ,пп 0,5
29 Эфиры жирных кислот пэ,пп 0,5
30 Натрия фосфат ПЭ, ПП 0,5
31 Жидкий парафин* 1 пэ.пп 0,5
32 Цинка оксид ПЭ, ПП 0,5
33 Магния оксид пэ.пп 0,5
34 Кальция стеарат пэ,пп 0,5
35 Цинка стеарат пэ.пп 0,5
36 Тальк ПП 0,5
37 Красители Титана диоксид пэ,пп 4
38 Ультрамарин синий2 пвх н/и
“ Только для непарентеральных препаратов.
Примечание. ПЭ и ПП для парентеральных и офтальмологических контейнеров могут со-
держать не более трех антиоксидантов; «н/и» — нет информации.
1 Синоним — вазелиновое масло. — Примеч. перев.
1 Добавка 6 в Европейской фармакопее. Нумерованных добавок в ЕФ всего 22, остальные
указаны без номеров. — Примеч. перев.
Глава 6.1. Источники контаминации
583
Поскольку добавки вместе с полимерами образуют достаточно большой спектр
веществ, и вымываемость этих компонентов не может быть предсказана априори,
фармакопеи установили процедуры изучения биологических и физико-химических
свойств полимеров. Вообще говоря, испытания включают анализ не полимерного
материала или самих добавок, а биологической активности (скрининг токсичности)
их экстрактов. Экстракты обычно получают путем автоклавирования полимерного
контейнера, заполненного водой, при температуре 121 °C в течение 30 минут или
при температуре 100 °C в течение 2 часов. Для контейнеров для парентеральных
и офтальмологических препаратов установлены более жесткие требования, так как
риски наличия у них токсического действия являются более значимыми, и поэто-
му количественное определение полимерных добавок предусмотрено в некоторых
фармакопейных статьях [5].
Регуляторные руководства по полимерам для фармацевтических контейнеров
также устанавливают предельное содержание примесей, отличных от добавок. Оце-
ниваются неорганические примеси (металлы и неметаллы), которые мшуг при-
сутствовать в полимерных контейнерах, и их определение служит критерием каче-
ства для полимерного материала. Они могут быть связаны и не связаны с любой
из добавок, указанных в табл. 24. Например, определение содержания алюминия
в ПЭ и ПП обусловлено использованием катализатора, содержащего алюминий,
для получения полимеров; примесь бария в полимерном контейнере свидетель-
ствует о присутствии стабилизатора гидроксида бария, вероятно, использованного
при смешивании полимера. Испытания на примеси, включая определение остатка
сульфатной золы и тяжелых металлов, приведены в табл. 25 с соответствующими
нормами содержания каждого элемента в различных полимерных материалах.
Несмотря на отсутствие обязательных требований, проводятся многочисленные
исследования экстрактивности добавок при контакте с различными фармацевти-
ческими препаратами, преимущественно для парентерального применения. Ис-
следования сконцентрированы на экстрактивности фталатных пластификаторов,
в основном ди-2-этилгексилфталата (DEHP, ДОФ) из ПВХ, в кровь, компоненты
крови и инфузионные растворы. Целью этих исследований является изучение его
до сих пор полностью не выясненных опасных эффектов на организм человека. Ко-
личество добавки (40% м/м), необходимое для превращения твердого ПВХ в гибкий
материал, и отсутствие химических связей между полимером и пластификатором
обусловливают возможность его присутствия в экстрактивных примесях.
Предельно допустимое содержание ДОФ в препаратах, хранящихся в мешках
из ПВХ, в фармакопейных стандартах не установлено. В Британской фармакопее
установлено, что результаты стандартного испытания на количество ДОФ, извле-
каемого воздействием эфира на мешки из ПВХ, не должны превышать 40% массы
полимера. Данное испытание предписано для ПВХ любого назначения: для твер-
дых лекарственных форм, неинъекционных водных растворов, водных растворов
для внутривенного введения, крови, компонентов крови и трубок, используемых
в системах для сбора и переливания крови и компонентов крови. С другой стороны,
Федеральное управление США по контролю за пищевой продукцией и лекарства-
ми (FDA, Food and Drug Administration) выпустило рекомендации, не имеющие ста-
туса обязательных, под названием «Оценка безопасности ди-2-этилгексилфталата,
584
Часть 6. Контаминация и ее контроль
Таблица 25
Неорганические примеси и нормы их содержания в экстрактах из полимерных контейнеров для
фармацевтических препаратов
№ п/п Примесь Полимер Назначение полимера Нормы
1 Тяжелые ПЭ, ПП Инъекционные, офтальмологические препараты 2,5 ррт
металлы ПВХ Водные парентеральные препараты, кровь, трубки систем для вливания 50 ррт
2 Сульфатная ПЭ,ПП Инъекционные, офтальмологические препараты 0,2%, 1%
зола ПЭВА Контейнеры и трубки для вливания парентеральных препаратов 1,2%
ПЭТ Лекарственные формы для приема внутрь 0,5%
3 Алюминий ПЭ,ПП Инъекционные, офтальмологические препараты 1 ррт
ПЭТ Лекарственные формы для приема внутрь 1 ррт
4 Хром ПЭ,ПП Инъекционные, офтальмологические препараты 0,05 ррт
5 Титан ПЭ,ПП Инъекционные, офтальмологические препараты 1 ррт
ПЭТ Лекарственные формы для приема внутрь 1 ррт
6 Ванадий ПЭ,ПП Инъекционные, офтальмологические препараты 0,1 ррт
7 Цинк ПЭ, ПП Инъекционные, офтальмологические препараты 1 ррт
пвх Водные парентеральные препараты, кровь 0,2%
ПЭТ Лекарственные формы для приема внутрь 1 ррт
8 Цирконий ПЭ Инъекционные, офтальмологические препараты 0,1 ррт
9 Барий ПВХ Водные парентеральные препараты, кровь, трубки систем для вливания 5 ррт
ПЭТ Лекарственные формы для приема внутрь 1 ррт
10 Кадмий ПВХ Водные парентеральные препараты, кровь, трубки систем для вливания 0,6 ррт
11 Кальций пвх Водные парентеральные препараты, кровь 0,07%
12 Олово пвх Водные парентеральные препараты, кровь, трубки систем для вливания 20 ррт
13 Аммоний пвх Кровь 2 ррт
14 Хлориды пвх Кровь 0,4 ррт
15 Сурьма ПЭТ Лекарственные формы для приема внутрь 1 ррт
16 Кобальт ПЭТ Лекарственные формы для приема внутрь 1 ррт
17 Германий ПЭТ Лекарственные формы для приема внутрь 1 ррт
18 Марганец ПЭТ Лекарственные формы для приема внутрь 1 ррт
Источник: адаптировано из Европейской фармакопеи [7].
высвобождающегося из медицинских изделий из ПВХ» [59], в которых подробно
обсуждаются проблемы токсичности данного вещества. Цель этой публикации —
предоставить менеджерам по рискам информацию, необходимую для принятия
решений о безопасности воздействия ДОФ, высвобождающегося из медицинских
изделий. Результаты этого исследования были получены путем расчета дозы ДОФ,
получаемой больными при различных медицинских процедурах.
Глава 6.1. Источники контаминации 585
Для фармацевтической отрасли наиболее важны вопросы о присутствии ДОФ
в растворах и препаратах для внутривенных инфузий и парентерального питания,
так как эти препараты хранятся в полимерных мешках и вводятся больным через
системы для вливаний из ПВХ. Из перечисленных в документе заключений следу-
ет, что риск для больного, получающего дозу ДОФ, высвобождающегося из ПВХ-
мешков, отсутствует или минимален при введении солевых растворов (например,
физиологического раствора или раствора Рингера с лактатом). Однако имеется
небольшой риск при высвобождении ДОФ из ПВХ-мешков, использованных для
хранения и введения препаратов, требующих наличия фармацевтического носителя
(растворителя) для солюбилизации. Рассчитанная доза ДОФ, получаемая взрослы-
ми больными при введении обшей смеси для парентерального питания, будет мень-
ше допустимого потребления (77)1, в связи с чем вопросов о возможных эффектах
ДОФ у этих больных возникнуть не должно. Доза ДОФ, получаемая новорожденны-
ми при введении общих смесей для парентерального питания, не определена. Ис-
ходя из данных, использованных для расчета отношения допустимого потребления
к получаемой дозе для взрослых, новорожденные могут находиться в группе риска
в отношении развития нежелательных эффектов, связанных с ДОФ.
При этом исследования показали, что в зависимости от состава препарата коли-
чество экстрагируемого ДОФ может быть значительно выше ожидаемого. Нашего
знания природы полимера и его добавок, а также стандартной миграции веществ
существенно недостаточно для прогнозирования взаимодействий контейнера с пре-
паратом. Миграция — это уникальный феномен, и прогнозы, основанные только на
структуре и физических или химических свойствах материалов, могут оказаться не-
состоятельными для решения проблем взаимодействия. В табл. 26 приведено содер-
жание ДОФ в различных типах парентеральных препаратов, хранившихся в мешках
из ПВХ. Очевидно, что количество ДОФ, попадающего в препарат, зависит от при-
роды его компонентов. В солевых растворах это количество не превысило 24 мкг/л
при хранении в течение 14 месяцев; в масляных растворах содержание ДОФ соста-
вило более 300 мкг/л уже через 2 часа хранения, что совпадает с результатами, полу-
ченными FDA. Вследствие липофильных свойств миграция ДОФ из упаковки в ги-
дрофильный препарат ограничена случайным распределением на контактирующей
поверхности. С другой стороны, в липофильных препаратах, таких как липидные
эмульсии, ДОФ свободно может покидать поверхность полимера и переходить в ли-
пофильную среду в процессе растворения.
Существуют препараты, взаимодействие которых с мешками и системами для
вливания из ПВХ настолько интенсивное, что в их маркировке должны быть указа-
ны меры предосторожности при использовании ПВХ-мешков. К таким препаратам
относятся противоопухолевые лекарственные средства: паклитаксел, доцетаксел,
такролимус и тенипозид — и другие, включая ципрофлоксацин, цефоперазон на-
трия, флуконазол, метронидазола гидрохлорид, циметидин и пропофол [64,65].
1 Значения допустимого потребления для ДОФ приведены в стандарте Международной
организации по стандартизации ISO/DIN 10933-17 «Методы установления допустимых пре-
делов д ля экстрактивных веществ».
586
Часть 6. Контаминация и ее контроль
Таблица 26
Концентрация ДОФ в инфузионных растворах, хранившихся в полимерных контейнерах
Образец Объем, мл Длительность хранения ДОФ, мкг/л Источник
Раствор хлорида натрия 0,9%-ный 1000 5 месяцев 7 [60]
14 месяцев 24
500 0,5 месяца 13 [60]
3 месяца <4
100 6 месяцев 8 [60]
1000 — 8 [60]
Раствор глюкозы 5%-ный 1000 5 месяцев 4 [60]
12 месяцев 4
500 0,5 месяца 34 [60]
3 месяца 7
ЛСО-мешки 100 12 месяцев 7 [61]
Мешки «Травенол» 100 12 месяцев 5 [61]
Поливитамины 48 ч(45 °C) 21 [62]
Общая смесь для парентерального питания — <2200 24 ч 330 [63]
3,85% липидов <2200 1 неделя (4 °C) 450 [63]
Общая смесь для парентерального питания — <2200 24 ч 230 [63]
2,50% липидов >2200 1 неделя (4 °C) 260 [63]
Общая смесь для парентерального питания — 650 24 ч 300 [63]
1,85% липидов <650 1 неделя (4 °C) 360 [63]
Общая смесь для парентерального питания — <800 24 ч 240 [63]
1,00% липидов <800 1 неделя (4 °C) 270 [63]
Раствор глюкозы 5%-ный — раствор 1ч 6600 [64]
паклитаксела 5%-ный 2ч 18 500 [64]
Раствор глюкозы 5%-ный — раствор 1ч 29400 [64]
паклитаксела 10%-ный 2ч 56600 [64]
Несмотря на то что разница в протоколах исследований, результаты которых
приведены в табл. 26 (длительность контакта, объем раствора, температура и др.),
не позволяет проводить прямых сравнений, эти данные говорят о том, что экстрак-
тивность ДОФ в значительной степени зависит от свойств состава препарата. Во-
прос миграции ДОФ в растворы актуален не только для липофильных носителей,
но и для таких препаратов, как паклитаксел и этопозид, которые способны экстра-
гировать ДОФ в количествах, значительно превышающих количества, экстрагируе-
мые простыми липофильными эмульсиями.
Попытки отказаться от использования ПВХ-контейнеров показали, что поли-
мерные контейнеры из других материалов также могут содержать в составе пласти-
фикаторы, которые приводят к высвобождению некоторого количества ДОФ в пре-
парат. В исследовании, проведенном Саутоу-Мирандой (Sautou-Miranda) с соавт.
[65], было установлено, что ДОФ быстро проникает из спрессованных 3-слойных
Глава 6.1. Источники контаминации
587
трубок систем для внутривенных вливаний в инфузионный раствор этопозида (см.
подробные результаты в разделе 6.1.3.3).
Хотя большинство исследований посвящено ДОФ, это не единственная добавка,
которая может экстрагироваться из полимерных материалов. Скалиа (Scalia) с со-
авт. [66] изучали миграцию антиоксидантов из полиолефиновых полимеров в мас-
ляные растворы. В работе рассмотрены два антиоксиданта, Ирганокс 1010 и Ир-
гафос 168, используемые в производстве полимеров ПЭ и ПП1. В исследовании
полоски полимеров, содержащих 0,15% антиоксидантов, погружали в масляную
смесь из 5 веществ (каприловой кислоты, каприлтриглицерида, циклометикона,
дикаприлилового эфира, изогексадекана и С1215-алкилбензоата), широко исполь-
зуемых в фармацевтических продуктах в качестве носителя, и хранили масляную
смесь в бутылках из полимеров. Полученные результаты показали, что количество
экстрактивных антиоксидантов, проникших в смесь, значительно различается в за-
висимости от полимерного материала и уменьшается в ряду ЕР > RACO > ПП >
полиэтилен высокой плотности2 (ПЭНД или ПЭВП, HDPE)-, RACO и ЕР— этилен-
пропиленовые сополимеры. В табл. 27 показано процентное содержание антиокси-
дантов, оставшихся в полиолефиновых бутылках, заполненных масляной смесью
и хранившихся при температуре 25 °C в течение 1 года. Авторы сделали вывод, что
миграция обоих антиоксидантов зависит от кристалличности и структуры полиме-
ра: чем выше кристалличность, тем меньше высвобождение добавки. Они предпо-
ложили, что ПП и ПЭНД пригодны для использования в производстве масляных
растворов.
Таблица 27
Процентное содержание антиоксидантов, оставшихся в полиолефиновых бутылках,
заполненных масляной смесью и хранившихся при температуре 25 °C в течение 1 года
Полиолефиновые бутылки Оставшиеся антиоксиданты. %
Ирганокс 1010 Иргафос 168
ПЭНД 92,7 ± 5,7 47,8 ±5,2
ПП 76,3 ±7,1 47,0 ±4,1
RACO 57,9 ±6,3 36,4 ±4,7
ЕР 0 0
Примечание. ЕР — смесь этилен-пропиленового аморфного сополимера; RACO — этилен-
со-пропиленовый случайный сополимер. Источник: [66].
Укупорочные средства из эластомеров. Укупорочные средства для фармацевтиче-
ских продуктов, как правило, изготавливают из полимерных материалов, которые
могут быть или синтетическими, или натуральными. В то время как твердые уку-
порочные средства, такие как закручивающиеся крышки, производят из обычного
термопластика, укупорочные средства из эластомеров делают из сложных смесей
большого количества ингредиентов. Так как из эластомеров или резин можно по-
1 Пентаэритритил тетракис[3-(3,5-ди-/ире/п-бутил-4-гидроксифенил)пропионат] (Ирга-
нокс 1010)итрис(2,4-ди-/лре«-бутилфенил)фосфит(Иргафос 168)—добавки 9 и 12 в табл. 24.
2 Также широко используется другое название материала — полиэтилен низкого давле-
ния. — Примеч. ред.
588
Часть 6. Контаминация и ее контроль
лучать почти бесчисленное количество разнообразных изделий, сохраняющих по-
стоянную форму, и через эти материалы легко проникают иглы, а место прокола
затягивается после извлечения иглы, из этих материалов изготавливают пробки для
флаконов для парентеральных препаратов, а также многодозовых препаратов, пред-
назначенных для многократного использования.
Для производства твердых укупорочных средств используются практические те
же полимерные материалы, которые были рассмотрены в разделе, посвященном
полимерным контейнерам (6.1.3.2). Следовательно, в этих компонентах упаковки
следует ожидать наличия тех же примесей. С другой стороны, хотя эластомерные
укупорочные средства и производят из полимерных материалов, у них другая струк-
тура. При производстве резины эластомер, основной компонент, комбинируется
с другими химическими веществами для получения материала с определенными
свойствами, наподобие вышеупомянутой способности затягивать место прокола
для повторного использования. В табл. 28 представлен перечень наиболее распро-
страненных эластомеров, используемых в фармацевтической отрасли, и их струк-
турные мономеры.
Вещества, перечисленные в табл. 28, соответствуют основному компоненту уку-
порочных средств из эластомеров. Другие компоненты в составе резин — это вулка-
низирующие агенты, ускорители, активаторы, консерванты, пластификаторы, на-
полнители и красители. Наиболее часто используемые добавки, входящие в состав
резины для фармацевтической отрасли, приведены в табл. 29. Количество каждого
компонента может отличаться в разных марках резин, а для некоторых компонен-
тов, например наполнителей, может превышать 50% общей массы продукта. В тоже
время ускорители используются в количествах лишь около 1% общей массы.
В табл. 30 показаны четыре типичных состава резины: на основе натурального
эластомера, эластомеров галобутила, этиленпропилендиена (EPDM) и силикона.
Наличие такого разнообразия компонентов делает укупорочные средства из эла-
стомеров потенциальным источником примесей. Полимерные материалы являют-
ся относительно инертными, однако вероятность попадания других компонентов
в препарат подлежит оценке. Более того, эти ингредиенты, хотя и предназначены
для фармацевтического применения и, соответственно, удовлетворяют фармако-
пейным требованиям, все равно могут иметь собственные примеси. Углеродная (га-
зовая) сажа — обычно неочищенный материал, она может содержать многоядерные
ароматические углеводороды [68], которые могут выделяться в упакованный препа-
рат. Глины содержат примеси металлов, которые также могут выделяться в препарат
или даже реагировать с содержимым.
Фармакопейные статьи не устанавливают предельные содержания для добавок,
как для полимеров, и испытания резиновых укупорочных средств (ВР, дополнение
XIX, £ЖГ<381>) ограничены определением содержания серы, сульфатной золы, ле-
тучих сульфидов, экстрагируемых цинка, аммония и тяжелых металлов.
Экстрактивные тесты, установленные другими регуляторными агентствами,
такими как FDA и PDA (Ассоциация парентеральных препаратов, Parenteral Drug
Association), для укупорочных средств [69,70], также ограничены определением экс-
трактивных остатков или испытаниями in vivo по оценке эффекта экстрактивного
остатка, если материал не выдерживает испытание in vitro.
Таблица 28
Наиболее распространенные эластомеры, используемые в фармацевтической отрасли
Тривиальное название__________Химическое название
Бутиловая резина Полиизобутилен-изопрен
(бутилкаучук)
Структура
CHj
50
с=си--СИ,-
СЕ,
СН3
J л
Галобутиловая резина
(галобутиловый каучук)
Галогенированный полиизобутилен-
изопрен
Этиленпропиленовая резина Полиэтилен-пропилен
(этиленпропиленовый
каучук)
Этиленпропилендиеновая Полиэтилен-пропилен-диен
резина
(этиленпропилендиеновый
каучук)
СН3
65
СИ,
CH,
X = О или Вт
сн3
_1 и
—t СН,---СН2-)НСН2---СИ—)— диен -
СН3
Глава 6.1. Источники контаминации___________________________________589
Тривиальное название Химическое название
Силиконовая резина (силиконовый каучук) Полидиметилсилоксан
Уретановая резина (полиуретан) Сложный эфир этиленгликоля с эдиповой кислотой
Фторэластомеры (фторсодержащие эластомеры) Политетрафторэтилен
Натуральная резина (каучук) иис-2,4-Полиизопрен
Полиизопреновая резина цис-1,4-Полиизопрен
(синтетический каучук)
Таблица 28 (окончание)
Структура
CH,
Jz»
НО4- сн2->,
F F
—c—c—
F F
—CH2— C = CH—CH2—
CH3
—CH2— C = CH-CH2—
590_____________________________Часть 6. Контаминация и ее контроль
Неопреновая резина
(неопрен)
Полихлоропрен
Бутадиен-стирольная резина Полибутадиен-стирол
(бутадиен-стирольный
латекс)
Нитрильная
резина (бутадиен-
акрилонитрильный каучук)
Полибутадиен-акрилонитрил
Полибутадиен
Полибутадиен
Источник: [67].
—сн2—с =сн —сн2—
-(-СН,—СН=СМ—CRj-I-CUj—сн- •
лава 6.1. Источники контаминации
Ч-СН2— СН=СН — CI ]2^СНт— сн^
CN
—сн3—сн=сн—сн,—
п
1Л
Таблица 29
Компоненты, отличные от эластомеров, используемые для получения резин для фармацевтического применения
Присадки Ускорители Активаторы Стабилизаторы (антиоксиданты) Пластификаторы Наполнители Красители (пигменты)
Сера Серосодержащие соединения Пероксиды Оксид кадмия Оксид магния Оксид цинка Гексаметилена Оксид тетрамин цинка Дитиокарбаматы Стеариновая кислота Сульфонамиды Тиурам Тиазол Амины Блокированные фенолы Воски Парафиновые воски Силиконовое масло Парафиновые масла Нафтеновые масла Органические фосфаты Углеродная (газовая) сажа Алюмосиликаты (глины) Сульфат магния (тальк) Сульфат бария Оксид цинка Диоксид кремния (кварц) Углеродная (газовая) сажа Диоксид титана Оксид железа Оксид хрома Органические красители
Источник: [67]. Таблица 30
Состав четырех типичных резин, используемых для производства укупорочных средств для фармацевтических препаратов, на основе
натурального, галобутилового, этиленпропилецдиенового и силиконового эластомеров
Ингредиент Тип резины
Красная Серая Серая Черная
Эластомер Натуральная резина Галобутиловая резина Диметилполисилоксановый полимер EPDM
Наполнитель Алюмосиликат Алюмосиликат Диоксид кремния Углеродная сажа
Пластификатор Парафиновое масло Нафтеновое масло — Нафтеновое масло
Краситель Оксид железа Оксид титана. Углеродная сажа Углеродная сажа —
Активатор Оксид цинка. Стеариновая кислота — — Оксид цинка. Стеариновая кислота
Ускоритель Тиурам. Тиазол Тиурам — Тйурам. Дитиокарбамат цинка
Стабилизатор Бутилированный гидрокситолуол Бутилированный гидрокситолуол — —
Присадки Сера Оксид цинка Пероксид 1,4-дихлорбензоила Сера
Источник-. [67].
Глава 6.1. Источники контаминации 593
Однако эти испытания не являются ни качественными (т. е. не показывают
спектр извлекаемых веществ), ни специфически количественными, так как показы-
вают только общее количество экстрактивных веществ в виде остатка. Статья USP
<381> «Укупорочные средства из эластомеров для инъекционных препаратов», на-
пример, рекомендует рассчитывать массу остатка после выпаривания растворителя
(очищенная вода, растворитель для препарата или изопропиловый спирт), исполь-
зованного для экстракции. Испытания in vivo рекомендованы только в случае, если
материал не выдерживает испытания in vitro.
Эти испытания, однако, не способны показать возможную контаминацию го-
тового продукта, поскольку экстрактивность также зависит от взаимодействия
контейнера с компонентами препарата. Несмотря на возможные взаимодействия,
крайне мало известно о проникновении веществ из укупорочных средств при их
прямом контакте с препаратами.
Меннермаа (Меппегтаа) с соавт. [71] определяли состав трех различных типов
резиновых пробок, использовавшихся для укупорки парентеральных растворов.
После погружения пробок в 0,9%-ный раствор хлорида натрия и автоклавирования
при температуре 121 °C в течение 15 минут проводился анализ водных экстрактов
методом протон-индуцированного рентгеноэмиссионого анализа. В табл. 31 приве-
дено содержание элементов в резиновых пробках, определенное в микрограммах на
грамм в пересчете на сухую массу. Разница в концентрации цинка в разных образцах
составляла до 4000 раз (5—20 000 ррт). Во всех пробках были обнаружены титан,
железо и бром, а в одном экстракте был найден даже свинец (2 ррт).
Таблица 31
Элементный состав резиновых пробок, основанный на концентрации элементов в водных
экстрактах
Количество, мкг/г
Силиконизированная пробка
Элемент Образец 1. Бромолутил, старый состав Образец 2. Бромолутил, новый состав Образец 3. Хромолутил, новый состав
Т1 500 1600 200
Fe 7500 8000 4500
Си 10 10 —
Zn 50 5 20 000
Br 100 7500 50
Pb 2 — —
Источник'. [71].
Шоенмейкерс (Schoentnakers) с соавт. [72] анализировали две репрезентативные
коммерческие резины методом газовой хроматографии с масс-спектрометрическим
детектором (ГХ-МС) и обнаружили более 100 различных соединений. Резины, сме-
си изобутилена и изопрена, исследовались после криогенного измельчения и про-
ведения двух различных процедур экстракции: по Сокслету с рядом растворителей и
в статическом устройстве для ввода парогазовой фазы, в которое помещали образцы
в герметичных флаконах объемом 20 мл при температуре 110 °C на 5, 20 и 50 минут.
Хотя выбранные условия не совпадали с условиями, в которых находятся фармацев-
594
Часть 6. Контаминация и ее контроль
тические препараты, результаты могли дать информацию о соединениях, выделения
которых из этих материалов можно было бы ожидать. В экстрактах были найдены
остаточные мономеры, изобутилен в димерной или тетрамерной форме и соедине-
ния, образующиеся при разрыве цепей. В табл. 32 представлены идентифицирован-
ные соединения, найденные в экстрактах полимеров, полученные методом Сокслета
и при парофазной газовой хроматографии. При этом жидкофазная экстракция по-
зволила извлечь менее летучие соединения, а парофазная — определить в экстракте
присутствие соединений с низкой молекулярной массой. Результаты полного скри-
нинга экстрактивных веществ показаны на рис. 11. На диаграмме можно видеть
изобилие соединений различных классов, обнаруженных в экстрактах обоих резин
с разными растворителями. В экстрактах, преимущественно от резины Р1, были об-
наружены не только олигомеры, но и фталаты, фенолы и кислотные соединения.
Таблица 32 Природа соединений, идентифицированных методом ГХ-МС в парогазовой фазе и экстрактах Сокслета i лвух исследованных резинах
Соединения, идентифицированные Система ввода парогазовой Метод Сокслета
в экстрактах фазы {headspace)
Алканы С5 и выше С —С ^"9 ^30
Олигомеры 7^ (элюирование) = 235 °C 7^ (элюирование) = 280 °C
Ароматические соединения 92 < М. м. < 132 168 < М. м. < 182
Жирные кислоты Нет 228 < М. м. < 352
Эфиры Нет Да
2,6-ди-тпре/и-Бутил-л-крезол Да Да
2,6-ди-тре/п-Бутил-л- Да Да
бензохинон
Кетон М. м. = 198 М. м. = 198
Примечание. М. м. — молекулярная масса. Источник: [72].
Дженке (Jenke) [73] изучал экстрактивность анилина, дифенилгуанидина, де-
дензиламина и трииизопропаноламина из синтетического полиизопрена, похожего
на материал, используемый для фармацевтического назначения. Образцы резины
автоклавировались (температура 121 °C) в контакте с водой или 0,9%-ным раство-
ром хлорида натрия в течение 1 часа. В табл. 33 приведена концентрация каждого
соединения в растворе после процедуры экстракции, в которой использовали по 2 г
материала. Полученные концентрации находились в диапазоне от 1,64 до 3,73 мг/л,
за исключением дифеншпуанидина, выход которого достигал 11,76 мг/л.
Таблица 33
Накопление экстрактивных веществ из синтетического полиизопрена после автоклавирования
в течение 1 ч
Соединение Концентрация в растворе, мг/л
Анилин 1,64
Дифенилгуанидин 11,76
Дедензиламин 2,12
Триизопропаноламин 3,73
Источник: [73].
Глава 6.1. Источники контаминации
595
а)
б)
Рис. 11. Влияние растворителя, использованного для экстракции по Сокслету соединений из
двух разных коммерческих резиновых пробок, на площади хроматографических пиков соединений
различных классов, обнаруженных методом ГХ-МС [72]: а — резина Р1; б — резина Р2
596 Часть 6. Контаминация и ее контроль
6.1.3.3. Системы доставки
Фармацевтические препараты вступают в прямой контакте полимерными материа-
лами при их введении больному через системы доставки, передаточные трубки и
устройства, а также в ходе их производства, то есть с прокладками и фильтрами, и
при транспортировке.
Даже при отсутствии в фармакопеях специальных указаний о проведении ис-
пытаний данных материалов, они должны обладать такими же характеристиками,
какие установлены для материалов контейнеров и укупорочных средств. Для пере-
даточных линий, которые также производят из эластомерных материалов, рекомен-
довано проведение испытаний на экстрактивность, аналогичных испытаниям для
укупорочных средств, главным образом для трубок из ПВХ.
Проникновение примесей из полимерных материалов для трубок охарактеризо-
вать намного сложнее, чем для контейнеров или укупорочных средств, поскольку
оно зависит не только от свойств полимерного материала, но и от скорости потока,
температуры, растворителя и длительности контакта продукта с трубкой.
Исследования, проведенные различными авторами по изучению высвобожде-
ния химических соединений из медицинских изделий, в основном использующих-
ся для вливания инфузионных растворов, показали, что эти изделия являются по-
тенциальными источниками контаминации фармацевтических препаратов. Одним
из наиболее изученных соединений является диэтилгексилфталат — тот же пласти-
фикатор, который применяют в мешках из ПВХ для придания им гибкости. Те же
проблемы, которые возникают при использовании мешков из ПВХ для хранения
липидов и липофильных препаратов, имеются и в отношении трубок.
В табл. 34 приведено содержание ДОФ, мигрирующего из стандартных систем
для вливания инфузий, изготовленных из ПВХ, длиной 2,25 м, выдержанных при
температуре 27 °C. Образец объемом от 8 до 140 мл пропускали через трубки в те-
чение 24 часов и собирали для анализа. Как видно из таблицы, аминокислоты не
увеличивают миграцию ДОФ из ПВХ-трубок, в то время как липофильные липид-
ные эмульсии и пропофол способствуют выделению ДОФ в препараты в большом
количестве.
Таблица 34
ДОФ, мигрировавший в инфузионные растворы после воздействия на них ПВХ-трубок систем
для вливания
Образец Объем, мл Номер образца ДОФ, мкг/мл Диапазон найденных концентраций ДОФ, мкг/мл
Глюкозо- аминокислотная смесь 140 06 0,31 ±0,56 0,0-1,05
Липидная эмульсия 24 10 422,78 ±47,39 329,15-490,0
Мидазолам 24 03 0,90 + 0,38 0,55-1,30
Инфузия пропофола 10 10 654,87 ±96,49 423,85-736,10
Фентанил 28,8 20 3,63 ±0,92 1,95-5,05
Имипенем 8 03 0 -0,10-0,05
Источник'. [74].
Глава 6.1. Источники контаминации 597
Изучение экстрактивности ДОФ из ПВХ-трубок в разные фармацевтиче-
ские препараты, используемые для различных целей, было проведено Хаишимой
(Haishima) с соавт. [75]. Авторы выделили пять групп препаратов для внутривенного
введения в соответствии со свойствами входящих в состав активных компонентов
и вспомогательных веществ. В группу 1 были включены препараты, нерастворимые
или практически нерастворимые в воде и содержащие такие вспомогательные ве-
щества, как поверхностно-активные вещества, масло, глицерин, этанол или бен-
золовый спирт. В группу 2 вошли препараты также нерастворимые в воде, но рас-
творимые в растворах кислот и щелочных металлов. Препараты, мало растворимые
в воде, были включены в группу 3, а препараты, легко и очень легко растворимые
в воде, были отнесены к группам 4 и 5. Различие между последними группами за-
ключалось в наличии в составе препаратов группы 4 вспомогательных веществ, вы-
зывающих миграцию ДОФ. Исследуемые препараты выдерживались в трубке из
ПВХ длиной 10 см и внутренним диаметром 2,13 мм в течение 1 часа при комнат-
ной температуре при постоянном покачивании. Как показано в табл. 35, в растворах
препаратов группы 1 наблюдалось высвобождение значительного количества ДОФ,
за исключением инсулина и динопроста, вероятно, вследствие отсутствия в их со-
ставе маслянистых компонентов. Значительного высвобождения ДОФ в растворы
препаратов групп 2—5 не отмечено, за исключением растворов альбумина человека
и антитромбина III (оба из группы 4) и раствора фенитоина (группа 2), вероятно,
вследствие наличия в их составе пропиленгликоля и этанола. Эти результаты под-
тверждают сродство ДОФ к липофильным средам.
Таблица 35
ДОФ, высвобожденные из ПВХ-трубок при контакте с препаратами для внутривенного
введения в течение одного часа при комнатной температуре
Действующее вещество Концентрация „ к ДОФ. Добавки ^/л’ Стандартное отклонение, мкг/л
Группа 1
Циклоспорин 500 мкг/мл Полиоксиэтиленовый эфир 27 363,9 касторового масла, этанол 384,8
Такролимус гидрат 10 мкг/мл Абсолютный этанол, HCO-6Q 4091,9 31,9
Пропофол 10 мг/мл Соевое масло, 19 451,2 концентрированный глицерин, яичный лецитин, эдетат 852,5
Флурбипрофена аксетил 10 мг/мл Соевое масло, 17 838,5 концентрированный глицерин, яичный лецитин 821,6
Жирорастворимые витамины Все количество Цитрат натрия, пиросульфат 1157,1 сорбита было натрия, тиогликолят натрия, смешано с PN- НСО-60, бензиловый спирт, Твин 2 (2,2 л) полисорбат 80 5,1
Менатетренон 5 мг/мл Аминоэтилсульфоновая 8457,5 кислота, кунжутное масло, соевый лецитин, D-сорбитол, концентрированный глицерин 62,9
598
Часть 6. Контаминация и ее контроль
Таблица 35 (продолжение)
Действующее вещество Концентрация Добавки ДОФ, мкг/л Стандартное отклонение, мкг/л
Инсулин человека 40ЕД/мл Концентрированный глицерин, jw-крезол 281,6 6,0
Динопрост 2 мг/мл — 185,8 17,3
Миконазол 1 мг/мл ЯСО-60 30 098,3 423,3
Диазепам 5 мг/мл Пропиленгликоль, этанол, бензиловый спирт, бензоат натрия, бензойная кислота 2008,8 257,6
Преднизолона 10 мг/мл Карбонат натрия, гидрофосфат 915,6 182,3
натрия сукцинат натрия, дигидрофосфат натрия
Преднизолона 1 мг/мл Карбонат натрия, гидрофосфат 407,1 2,4
натрия сукцинат натрия, дигидрофосфат натрия
Группа 2
Фамотищин 20 мг/мл L-Аспарагиновая кислота, Я-маннитол 166,0 0,9
Дроперидол 2,5 мг/мл л-Оксиметилбензоат, л-оксипропилбензоат 171,0 0,6
Дроперидол 50 мкг/мл л-Оксиметилбензоат, л-оксипропилбензоат 167,4 24,6
Сивелестата натрия 1 мг/мл Я-Маннитол, гидрохлорид 885,7 10,6
гидрат натрия, пропиленгликоль, этанол
Фенитоин' 50 мг/мл
Метотрексат 0,2 мг/мл Хлорид натрия, гидрохлорид натрия 372,8 6,8
Галоперидол 5 мг/мл Глюкоза, молочная кислота, гидрохлорид натрия 50,6 2,5
Эпинефрин 0,25 мг/мл Хлорбутанол, гидросульфит натрия, хлороводородная кислота, хлорид натрия 290,3 24,6
Группа 3
Метилэргометрина 0,2 мг/мл — 462,7 4,2
малеат
Векурония бромид 2 мг/мл Я-Маннитол 192,7 1,5
Панипенем бетамипрон 5 мг/мл — 237,0 1,2
Миноциклина гидрохлорид 1 мг/мл — 150,0 8,9
Никардипина 0,1 мг/мл Я-Сорбитол 211,6 24,0
гидрохлорид Бромгексина гидрохлорид 2 мг/мл Ппокоза 174,9 23,7
Глава 6.1. Источники контаминации
599
Таблица 35 (продолжение)
Действующее вещество Концентрация Добавки ДОФ, мкг/л Стандартное отклонение, мкг/л
Цефтазидим 10 мг/мл Карбонат натрия 301,0 0,5
Флуконазол 1 мг/мл — 210,5 0,2
Аспоксициллин 50 мг/мл Хлорид натрия 296,7 2,6
Карбазохрома 0,05 мг/мл Гидросульфит натрия, 246,1 3,0
натрия сульфонат D-сорбитол, пропиленгликоль
Группа 4
Окситоцин 0,01 ЕД/мл Хлорбутанол 423,1 0,8
Гидроксизина 0,05 мг/мл Бензиловый спирт 430,8 144,4
гидрохлорид Ранитидина гидрохлорид 0,1 мг/мл Фенол 197,9 29,5
Иммуноглобулин G 50 мг/мл D- Сорбитол 243,9 14,3
человека
Пантенол 250 мг/мл Бензиловый спирт 412,1 18,2
Альбумин человека 250 мг/мл Натрия ТУ-ацетилтриптофан, каприлат натрия, гидрокарбонат натрия 10080,8 84,1
Антитромбин III 25 ЕД/мл Хлорид натрия, цитрат натрия, 2008,2 21,8
человека D-маннитол
Нитроглицерин 0,5 мг/мл D-Маннитол 267,6 8,9
Сульпирин 2,5 мг/мл Бензиловый спирт 302,8 3,8
Эритромицина лакгобионат 2,5 мг/мл Бензиловый спирт 92,2 0,7
Клиндамицина фосфат 3 мг/мл Бензиловый спирт 274,9 4,0
Группа 5
Имипенем и циластатин натрия 5 мг/мл Гидрокарбонат натрия 205,1 1,6
Раствор глюкозы 5%-ный 50 мг/мл — 284,6 4,8
Оксида железа 0,4 мг/мл — 244,5 5,5
сахаринат Мальтоза, натрия хлорид, магния — — 262,8 5,0
хлорид, калия дигидрофосфат, натрия ацетат Атропина сульфат 0,5 мг/мл 200,7 5,1
Ампициллин натрия 10 мг/мл — 262,3 6,8
Аминофиллин 0,5 мг/мл Этилендиамин 301,1 4,0
600
Часть 6. Контаминация и ее контроль
Таблица 35 (окончание)
Действующее вещество Концентрация Добавки ДОФ, мкг/л Стандартное отклонение, мкг/л
Фосфомицин натрия 20 мг/мл Раствор глюкозы 289,6 6,7
Кальция глюконат 85 мг/мл — 179,4 4,3
Цефазолина натрия гидрат 10 мг/мл — 215,1 0,9
Аминокислоты, электролиты — Гидросульфит натрия 328,5 5,0
Суксаметония хлорид 2 мг/мл — 228,6 2,1
Иоверсол 320 мг/мл — 404,0 79,5
/-Изопреналина гидрохлорид 1 мкг/мл Гидросульфит натрия, Г-цистеина гидрохлорид 326,3 8,6
* Данные приведены по оригиналу. — Примеч. перев.
Источник'. [75].
Вследствие принципов использования трубок большинство исследований, по-
священных им, являются кинетическими, а длина трубки, длительность контакта,
скорость потока и температура — важными параметрами исследования.
Камбиа (Kambid) с соавт. [76] изучали кинетику миграции ДОФ в растворы для
полного парентерального питания из трубок ПВХ. Они определяли количество
ДОФ, проникавшего в эмульсии двух типов, через 24 часа после приготовления рас-
творов, хранившихся при температуре 4 °C. Авторы сделали вывод, что экстракция
зависела от содержания липидов в составе препарата и скоростей потока. На рис. 12
показана концентрация ДОФ в зависимости от времени, определенная из мешков
и трубок в течение 10—12-часовых инфузий растворов для полного парентерального
питания, после 24-часового хранения при температуре 4 °C.
Для решения проблемы проникновения ДОФ в парентеральные препараты, со-
держащие липофильные компоненты, при изготовлении трубок систем для влива-
ния начали использовать трехслойные материалы. Внешний слой таких материалов
изготовлен из ПВХ, внутренний слой — из инертного полиэтилена. Однако было
показано, что, несмотря на такие меры в зависимости от препарата ДОФ из внеш-
него слоя может проникать в инфузионные растворы.
Саутоу-Миранда с соавт. [77] установили, что под воздействием инфузионного
раствора этопозида ДОФ быстро мигрирует из трубок для вливаний, изготовленных
из ПВХ, экструдированного полимера и трехслойного материала. В табл. 36 при-
ведены концентрации ДОФ, мигрировавшего из трубок в инфузионные растворы
этопозида, в зависимости от д лительности контакта, определенные для двух трубок
разной длины. Результаты исследования показывают, что имеет место быстрая и
значительная экстракция ДОФ, даже когда компонент из ПВХ не вступает в пря-
мой контакт с раствором. Наибольшая концентрация ДОФ была обнаружена для
трубок из ПВХ, но миграция также наблюдалась и из экструдированного полимера
(ПЭ + ПВХ) и из трехслойных трубок — несмотря на заявление производителей об
Глава 6.1. Источники контаминации
601
Длительность инфузии, ч
Рис. 12. Сравнительная кинетика проникновения ДОФ в раствор для общего парентерального
питания при имитации инфузионного вливания через 24 часа после приготовления растворов
(л = 2 мешка) [76]: а — препарат 1: инфузионный раствор 2200 мл (скорость потока 177 мл/ч,
концентрация липидов 3,85%); б — препарат 2: инфузионный раствор 650 мл (скорость потока
46 мл/ч, концентрация липидов 1,85%)
отсутствии миграции ДОФ. Авторы заключили, что или ДОФ присутствовал во вну-
треннем слое из ПЭ, или он быстро мигрировал через другие полимерные слои.
Кинетика экстракции ДОФ из трехслойных трубок изучалась этими авторами
с использованием трубок различной длины, скорости потока и концентрации пре-
парата. На рис. 13 показано количество ДОФ, проникшего в растворы разного объ-
ема. Как и предполагалось, более высокая концентрация препарата и длина трубки
увеличивали количество высвобождаемого ДОФ. Чем ниже была скорость потока,
тем больше была миграция добавки в раствор.
602
Часть 6. Контаминация и ее контроль
Таблица 36
Концентрации ДОФ, проникшего из трубок в инфузионные растворы этопозвда, в зависимости
от длительности контакта
Длина трубки и Концентрация ДОФ, мг/мл (среднее значение + стандартное отклонение)
тип материала 0ч 1ч 2ч Зч 4ч 6ч
25 см
ПЭа <0,5 <0,5 <0,5 <0,5 <0,5 <0,5
ПВХ*-6 <0,5 22,59 ±4,21 35,05 ±2,51 49,88 ±4,78 58,57 ±1,03 73,51 ±3,51
Трехслойная трубка* <0,5 18,92 ±2,23 31,49 ±3,78 44,19 ±5,38 54,83 ±4,28 61,99 ±1,25
ПВХ* <0,5 19,93 + 1,90 33,01 ± 1,87 46,82 ±1,98 55,46 ±3,15 66,10 ±1,23
Экструдиро- ванный полимер* 50 см <0,5 18,64 ±1,38 28,77 ±2,33 39,37 ± 3,47 48,13 ±2,21 54,60 ±2,53
ПЭ* <0,5 <0,5 <0,5 <0,5 <0,5 <0,5
ПВХа-6 <0,5 51,67 ±3,63 82,00 ±2,65 112,69 ±4,98 131,67 ±3,65 155,22 ±2,35
Трехслойная трубка* <0,5 39,85 ±0,49 59,73 ± 3,04 81,51 ±1,57 85,01 ±2,17 98,72 ±2,33
ПВХ* <0,5 45,38 ±2,08 73,27 ±0,96 94,65 ±0,88 117,53 ±3,43 143,41 ±11,39
Экструдиро- ванный полимер* <0,5 39,98 ± 0,74 59,61 ±1,49 78,03 ±0,52 82,47 ±3,56 107,83 ±9,68
а Произведено Vycon, Есоиап, Франция.
6 Длина трубки 80 см.
Состав: внешний слой — ПВХ, средний слой — ПЭВА, внутренний слой — ПЭ.
г Произведено Cair, Civrieux d’Azergues, Франция.
* Трубки из ПВХ с ПЭ.
Источник: [77].
Как упоминалось в разделе о полимерных контейнерах (6.1.3.2), ДОФ является
не единственной добавкой, которая может мигрировать в препарат из полимерных
материалов. Дженке с соавт. [78] изучали состав экстрагированных веществ из поли-
мерных трубок, использующихся в производственных помещениях на фармацевти-
ческом производстве. Восемь материалов для труб, изготовленных из силиконовой
и неопреновой резины, были охарактеризованы по их экстрактивным веществам.
Авторы изучали органические и неорганические экстрактивные вещества. Экстрак-
ты получали в ходе статического эксперимента с использованием воды и этанола.
Трубка разрезалась и автоклавировалась с водой при температуре 121 °C в течение
1 часа, или отрезки трубок наполнялись 100%-ным этанолом и выдерживались при
температуре 55 °C в течение 24 часов. Определение металлов проводилось методом
атомной эмиссионной спектроскопии с индуктивно-связанной плазмой (ИСП-
АЭС), который включает 29 элементов. Помимо элементов, указанных в табл. 37,
также измерялось содержание Be, Со, Cd, Se, V, Ge, Pb и Bi, но они обнаружены не
были, так как или присутствовали в концентрациях, не отличавшихся от концен-
Глава 6.1. Источники контаминации
603
а)
—*—90 МЛ/Ч
—«-60МЛ/Ч
-*-30 мл/ч
600
Объем инфузии, мл
в)
Рис. 13. Накопленное содержание ДОФ, мигрировавшего из трубок длиной 50 и 80 см из
различных материалов после инфузии раствора этопозида: а — из чистого ПВХ, инфузия 0,4 мг/мл
при различных скоростях потока; б — из трехслойного материала, инфузия 0,2 и 0,4 мг/мл при
скорости потока 30 мл/ч; в — из трехслойного материала, инфузия 0,4 мг/мл со скоростью потока
30 мл/ч [77]
траций в контрольном растворе, или их концентрации были ниже предела обнару-
жения.
Важно отметить, что ИСП-АЭС — не очень чувствительный метод. Пределы об-
наружения для элементов составляют от 0,01 до 0,1 мг/л, что является достаточно
высоким пределом обнаружения для таких примесей, как Cd или Be. Следователь-
604
Часть 6. Контаминация и ее контроль
Таблица 37
Содержание общего углерода (органического и неорганического), кремния и металлов,
экстрагированных из материалов для трубок водой или этанолом при автоклавировании
при 121 °C в течение 1 часа
Материал Общий неорг. углерод, мкг/г Общий орг. углерод, мкг/г Кремний, этанольный экстракт, мкг/г Кремний, водный экстракт, мкг/г Концентрация металла в водном экстракте, мкг/мл
<0,5 0,5-1,0 >1
Силикон 1 0 14,0 756 101 В, Mg, Zn
Силикон 2 0,9 38,1 1360 <0,2 Са B, Mg, Mn, Mo, Ti, Zr, Sn, Zn, Sb, Li, Ag, Ni
Силикон 3 (с металличе- ским слоем) 2,3 250 1860 66,0 Са, Ва, Мп, Mg.Al, Си — В, Fe, Zn
Силикон 4 0,2 34,0 1120 130 Са, В, Мп, Fe, Mg, Al, Zn — —
Силикон 5 (с металличе- ским слоем) 0 49,9 1300 87,3 Са, В, Мп, Fe, Mg, Zn, Sb — —
Силикон 6 0 46,9 739 <0,2 Mn, Fe, Mg, Zn, Си, Sb, Ni в Ca
Сантопрен 1 10,1 16,6 а <0,2 Ca, Ba, B, Mg, Zn — —
Сантопрен 2 4,6 13,5 Не определяли 0 Ca, Mo, U, Zr, Mn, Zn, Li, Ag, Ni — Mg
"Дезинтеграция трубки.
Источник-. [78].
но, эти элементы могли присутствовать в экстрактах и при этом быть не обнару-
женными использовавшимся методом. Все исследованные материалы для трубок
содержали металлы, которые извлекались или водой, или этанолом; при исполь-
зовании воды извлекались большие количества ионов металлов, что обусловлено
бблыпим сродством воды к металлам. Все материалы для трубок содержали экстра-
гированные Са, Mg, Zn и В. Увеличенное количество других металлов также было
извлечено из образцов 3 и 5 — вероятно, вследствие наличия внутреннего металли-
ческого слоя.
Кроме того, авторы измеряли содержание кремния и углерода — органического
и неорганического. В то время как неорганический углерод указывает на наличие
карбонатов (карбонаты щелочно-земельных металлов используются в качестве до-
бавок), органический углерод связан с органическими экстрактивными веществами.
Глава 6.1. Источники контаминации
605
Как и предполагалось, в экстрактах Сантопрена имела место более высокая концен-
трация неорганического углерода и небольшое, если вообще не нулевое, содержа-
ние экстрагируемого кремния. С другой стороны, содержание кремния в экстрактах
силиконовых полимеров было очень высоким. Из данных табл. 37 видно, что эти
материалы, хотя и изготовлены все из силикона, значительно отличаются друг от
друга. Так, в водном экстракте образца 1 был обнаружен Si в количестве 100 мкг/г,
а в аналогичных экстрактах образцов 2 и 6 его не нашли. Как ожидалось, содержа-
ние экстрагируемого кремния было выше в этанольных экстрактах, а содержание
739 мкг/г Si в образце 6 было самым низким среди образцов.
Анализ хроматограмм водных и этанольных экстрактов, полученных методами
жидкостной хроматографии с масс-спектрометрическим детектором (ЖХ-МС) и
ГХ-МС, показал, что все силиконовые материалы содержали практически одинако-
вые пики, из чего следует, что были извлечены одинаковые компоненты. Однако их
распределение в образцах отличалось. Первичные органические экстрактивные ве-
щества из силикона составляли гомологичные серии олигомеров силикона со струк-
турной формулой [(CH3)2SiO]n. Прямое сравнение с библиотекой МС-спектров по-
зволило идентифицировать олигомеры с п от 5 до 25. Однако если на хроматограмме
образца 3 пики соответствовали относительно коротким олигомерам, то в образце 2
распределение олигомеров было смещено к веществам с более высокой молеку-
лярной массой, которые обладают меньшей растворимостью, и, соответственно,
меньшей экстрагируемостью. На рис. 14 показаны различия этанольных экстрактов
материалов 2 и 3.
Результаты, полученные при исследовании экстрактов Сантопрена, значительно
отличаются от результатов образцов силиконовых материалов. На хроматограммах
водных экстрактов трубок из Сантопрена, полученных методом ГХ-МС, наблюдали
пики, соответствующие кислотам С8 и С9, фталатам и другим органическим веще-
ствам. На рис. 15 приведены ГХ-МС-хроматограммы статичных этанольных экс-
трактов материала Сантопрен для трубок, а в табл. 38 перечислены компоненты,
идентифицированные на рис. 14 по соответствию пиков МС-спектров.
6.1.3.4. Механические включения
Контаминация механическими включениями по источнику происхождения может
быть разделена на внешнюю и внутреннюю. Внутренняя контаминация возникает
в производственном процессе, в процессе упаковки, при транспортировке и хране-
нии растворов; внешняя контаминация происходит главным образом при восста-
новлении препарата и при введении его больному.
Присутствие механических включений, нежелательное в любом фармацевтиче-
ском препарате, становится особой проблемой для внутривенных и офтальмологи-
ческих препаратов. Наиболее часто механическими включениями во внутривенных
препаратах являются фрагменты стекла, образующиеся при открытии стеклянных
ампул, частицы резиновых пробок и оборудования для внутривенных вливаний,
а также частицы из полимерных шприцов; при этом следует учитывать, что мани-
пуляции с такими растворами проводятся в контролируемой чистой зоне. Воздух
в контролируемой зоне, в непосредственной близости от используемых стерили-
зованных контейнеров и укупорочных средств и во время операций по розливу
606
Часть 6. Контаминация и ее контроль
Распределение
(по молекулярной
массе)
500000
450 000
400 000
350 000
300 000
250 000
200 000
150000
100000
50 000
О
Материал 2
IS
6
11 • 12»
8*
9* 10
8 10 12
Распределение 6 •
(по молекулярной
массе)
6000 000
5 000 000 5.
4000 000
3 000 000
2 000 000
1 000 000
-> 8
14 16 18 20 22 24 26 28 30
Время удерживания
Материал 3
Время удерживания
Рис. 14. ГХ-МС-хроматограммы статических этанольных экстрактов силиконовых материалов
для трубок (после обработки данных). Хроматограммы образцов всех силиконовых материалов
были похожими (одинаковые пики с разными относительными размерами), поэтому показаны
хроматограммы двух образцов. Большинство пиков соответствует олигомерам силикона.
Пики, отнесенные к циклическим олигомерам, были идентифицированы по количеству
повторяемых единиц п, например [(CH3)2SiO3]n. Пики, обозначенные *, относятся к компонентам,
идентифицированным по прямому совпадению с масс-спектрами из библиотеки, а пики,
обозначенные #, соответствовали ожидаемой группе веществ из библиотеки (циклическим
олигомерам), но не совпадали с пиком определенного олигомера. Небольшие пики в моменты
времени 7,95, 10,32 и 11,93 мин соответствуют 5-7-членным линейным олигомерам силикона. IS
(внутренний стандарт) — диметилфталат [78]
и укупориванию, является пригодным, если он содержит в 1 м3 не более 3520 частиц
размером 0,5 мкм. Это соответствует классу 100 чистоты воздуха, и это является ре-
гуляторным требованием к зонам, в которых осуществляются операции со стериль-
ными препаратами.
В фармакопеях зафиксировано требование по проверке наличия механических
включений в растворах для инъекций и инфузий. Механическими включениями
считается наличие посторонних подвижных нерастворимых частиц, отличных от
пузырьков газа, которые случайно могут попасть в препарат. Фармакопеи ограни-
чивают допустимое количество механических включений в зависимости от их раз-
мера и объема препарата в упаковке.
Глава 6.1. Источники контаминации
607
Распределение
(по молекулярной
массе)
750 000
700 000
650 000
600 000
550 000
500 000
450 000:
400 ооо:
350 000:
300 000
250 ооо:
200 ооо:
150 000:
юо ооо:
50 000:
о
Материал 2
13
3 I
5 !
9
* г vM 41 г
12 14 16
Распределение
(по молекулярной
массе)
1 000 000
900 000
800 000
700 000
600 000
500 000
400 000:
300 000:
200 000
100 000
0
10 12 14 16 18 20 22 24 26 28 30
Время удерживания
Рис. 15. ГХ-МС-хроматограммы статических этанольных экстрактов материала Сантопрен
для трубок (после обработки данных). Хроматограммы этих образцов материала Сантопрен
значительно отличались от хроматограмм силиконовых материалов (рис. 14). /S (внутренний
стандарт) — диметилфталат. См. табл. 38 для предполагаемой идентификации пиков. На
хроматограммах образцов отмечены только пики, совпадающие с масс-спектрами библиотеки,
хотя по временам удерживания и характеру пиков может быть проведена идентификация
некоторых дополнительных пиков [78]
Как правило, механические включения удаляются с помощью фильтрации, од-
нако требования, описываемые термином «удаление» или даже концепцией «прак-
тического отсутствия», сформулированными в некоторых фармакопеях1, — это
требования, которые практически невозможно выполнить. На практике соответ-
ствие фармакопейным требованиям ограничивается удалением частиц, превышаю-
щих определенный размер, поскольку по мере уменьшения размера частиц их уда-
ление становится более сложным. Сложность удаления, по-видимому, возрастает
экспоненциально с уменьшением размера частиц.
Фармакопейные требования к допустимому количеству частиц в парентераль-
ных препаратах приведены в табл. 39. Нормы зависят от метода, используемого для
обнаружения частиц, а также от размера образца. Для оценки предлагаются два раз-
1 Данное требование установлено в Европейской фармакопее для механических включе-
ний видимых частиц. — Примеч. перев.
608
Часть 6. Контаминация и ее контроль
Таблица 38
Идентификация пиков на хроматограммах этанольных экстрактов трубок из Сантопрена
для образцов 7 и 8
Номер пика Предполагаемая идентификация компонента Содержание в материале
3 2,4-ди-/-Бутилфенол 7,8
5 4-( 1,1,3,3-Тетраметилбутил)фенол 8
6 Изомер октилфенола (TMS) 8
7 Гексадекан 7
8 Изомер октилфенола (TMS) 8
9 4-Метил-б-тпрет-октилфенол 8
10 Изомер децилфенола 7,8
11 Изомер нонилфенола (TMS) 8
12 Гептадекан 7
13 Изомер ондецилфенола 7,8
14 Октадекан 7
15 Изомер децилфенола (TMS) 8
16 Нонадекан 7
17 Изомер ондецилфенола (TMS) 7,8
18 Этиловый эфир дексадекановой кислоты 7
19 Циклогексадекан, генэйкозан 7
20 Этиловый эфир оксадекановой кислоты 7
21 Докозан 7
22 Тетратриконтан, 9-метил-нонадекан 7,8
23 Тетракозан 7
24 Пентакозан 7,8
25 Гексакозан, нонадекан 7,8
26 Гептакозан 8
27 Ирганокс 1076 7,8
Источник'. [78].
личных метода1: метод подсчета частиц по светопропусканию и метод подсчета ча-
стиц при помощи микроскопа, поскольку ни один из них не является пригодным
для всех видов образцов.
Имеется возможность графического отображения взаимосвязи между размером
частиц и их количеством. Гроувз (Groves) [79] сообщил о логарифмической связи
между количеством частиц в растворе и их диаметром. На рис. 16 приведен возмож-
ный график зависимости, соответствующий нормам содержания частиц, установ-
ленным ВР.
Автор, однако, считает, что если распределение количество/размер частиц яв-
ляется случайным явлением, как графически показано на рис. 16, то это не может
считаться контаминацией. С другой стороны, если раствор не содержит случайных
частиц, а содержит определенные частицы, например зерна крахмала из состава
1 Это методы определения количества невидимых частиц. — Примеч. перев.
Глава 6.1. Источники контаминации
609
Таблица 39
Нормы допустимого содержания механических включений в инфузионных растворах,
установленные фармакопеями
Фармакопея Объем Размер частиц, мкм Допустимое количество для метода по светопропусканию, не более Допустимое количество для микроскопического метода, не более
ВР <100 мл >10 6000/контейнер 3000/контейнер
>25 600/контейнер 300/контейнер
>100 мл >10 25/мл 12/мл
>25 3/мл 2/мл
USP <100 мл >10 6000/контейнер —
>25 600/контейнер —
>100 мл >10 25/мл 12/мл
>25 3/мл 2/мл
IP Все — Одна или более частиц —
растворы более чем в одном
контейнере1
Примечания. Нормы зависят от метода, используемого для подсчета частиц. ВР — Британ-
ская фармакопея; USP — Фармакопея США; IP— Международная фармакопея.
Рис. 16. Полулогарифмический график зависимости количества частиц ненормируемого
размера (oversize) (в 1 мл) от диаметра частиц в соответствии с экстраполируемыми нормами ВР
1 Эти нормы Международной фармакопеи приведены для видимых частиц (объем выбор-
ки п = 20). В новом 4-м издании IP установила такие же нормы, как и ВР, и для тех же лекар-
ственных форм. — Примеч. перев.
610
Часть 6. Контаминация и ее контроль
Диаметр, мкм
Рис. 17. Совокупное распределение частиц по размеру в 5%-ных растворах декстрозы для
инъекций: □ — продукт 2AL, упакованный в стеклянные контейнеры по 500 мл, укупоренные
покрытыми лаком резиновыми пробками; О — продукт 2G, упакованный в полимерные мешки по
500 мл; гладкая кривая — экстраполяция спецификации ВР [80]
пробки, стеклянные частицы из расслоившегося контейнера, фрагменты кожи или
волокна с одежды, может наблюдаться «неслучайное» поведение, которое и сле-
дует определить как «контаминацию». Другими словами, только положительное
отклонение полулогарифмической кривой распределения частиц по размеру, по-
строенной с использованием, например, фармакопейных норм, будет считаться
контаминацией. На рис. 17 графически изображены частицы, найденные в 5%-ном
растворе декстрозы. Гладкая кривая представляет экстраполированные нормы ВР
и две другие — образцы. Один продукт соответствует спецификации (26), а дру-
гой — нет (2AL). Согласно автору, в образце 2G частицы были идентифицированы
как частицы черного углерода с размером от 1 до 3 мкм, а в образце 2AL, помимо
черных частиц углерода (1—5 мкм), были найдены другие частицы: зерна крахмала,
чешуйки лака и резины [80].
Методики подсчета механических включений позволяют различать частицы
даже меньших размеров, чем указано в фармакопейных нормах; хотя соответствую-
щие требования не установлены, возможно получить характеристики частиц по их
составу. В табл. 40 приведено количество частиц, найденных в растворах для инфу-
зий, с учетом их размера.
Поскольку в табл. 40 приведены результаты разных исследований, они достаточ-
но разнородны. Тем не менее, во всех растворах присутствуют механические вклю-
чения в качестве примесей, и возможно подтвердить взаимосвязь между их количе-
ством и размером в виде экспоненциальной кривой.
Форони (Foroni) с соавт. [81] определяли количество и состав инертных частиц в от-
дельных растворах и в окончательной смеси, приготовленной в асептических уело-
Таблица 40
Средние значения и диапазоны размеров частиц, обнаруженных в смешанных при использовании и несмешанных растворах для
парентерального питания (по группам в зависимости от размера)
Образец Объем, мл п Размер частиц Источник
>1,3 мкм >5 мкм >10 мкм >25 мкм >50 мкм
Средн. Диап. Средн. Диап. Средн. Диап. Средн. Диап. Средн. Диап.
Раствор декстрозы 30%-ный 25 3 452 +127 н/о' — — — — — — — [81]
Раствор декстрозы 50%-ный 25 3 2831 ±278 — ±90 — — — — — — [81]
Аминокислоты 25 3 3715 ±184 — ±58 — — — — — — [81]
Раствор для полного парентерального питания («все в одном») для взрослых6 1 20 3,47 ±1,24 1,40 +0,73 0,96 ±0,45 1,06 +0,39 — — [82]
Раствор для парентерального питания («два в одном») для детей6 1 20 7,59 ±2,56 11,77 ±7,38 1,43 ±1,09 0,38 +0,26 — — [82]
Эмульсия липидов (преднаполненный шприц) 1 20 16,72 ±10,85 8,98 ±4,63 1,27 +1,23 0,76 ±0,34 — — [82]
Несмешанные образцы 10-60 7 62,7 8-146 1,70 0-4 0,4 0-2 — — 0 183]
Смешанные образцы' 10-60 192 960,9 30-9539 42,8 0-587 6,4 1-146 — — 0,09 0-1 [83]
Растворы, смешанные с растворами из стеклян- ных ампул класса 1—3' 10-60 29 862,1 30-5707 31,3 0-176 4,4 0-24 — — 0,1 0-1 [83]
Растворы, смешанные с растворами из стеклян- ных ампул класса 4-13' 10-60 63 1163,4 142-9539 66,2 4-587 10,6 1-146 — — 0,08 0-1 [83]
• н/о — не обнаружены.
6 Отбирались первые миллилитры.
' Отбирался остаточный объем.
Глава 6.1. Источники контаминации
612
Часть 6. Контаминация и ее контроль
виях. Результаты приведены в табл. 40 и на рис. 18. Содержание частиц с размером 2
и 5 мкм различается более чем в 50 раз. Гистограмма на рис. 18 показывает количе-
ство частиц различных компонентов (рис. 18а) и количество частиц после переноса
компонентов в мешок из ПЭВА (рис. 186). Полученные результаты свидетельству-
ют, что контаминация механическими включениями 10%-ного раствора КС1, 30%-
ного раствора декстрозы, раствора аминокислот и липидной эмульсии была значи-
тельно больше, чем других компонентов (рис. 18а). Также было установлено, что
концентрация частиц в компонентах, упакованных в стеклянные ампулы, растворах
КС1 и натрия гидрофосфата была также выше, чем в других растворах. В исследова-
нии проводилась идентификация механических включений. В растворе КС1 были
найдены частицы, состоящие из кремния, алюминия и натрия, во всех растворах
в мешках — частицы, содержащие серу и силикат. Авторы считают, что присутствие
серы может быть связано с частицами резины, а присутствие силиката вызвано кон-
тактом с тальком из перчаток при манипуляциях в ходе технологического процесса.
Камия (Katniya) с соавт. [83] оценивали контаминацию механическими включе-
ниями в 199 образцах смешанных и несмешанных растворов для парентерального
питания в мешках, отобранных в 10 больницах в Японии. Семь образцов использо-
вались в качестве контрольных, так как не были смешаны с растворами ни из ам-
пул, ни из флаконов (несмешанные образцы). Размер и количество частиц измеря-
лись с помощью счетчика частиц, идентификация элементов проводилась методом
сканирующей электронной микроскопии с энергодисперсионной спектроскопией.
Авторы отбирали остаточный объем образцов (10—60 мл) после их использования.
Результаты приведены в табл. 40.
На рис. 19 показаны изображения двух типов частиц, полученные сканирующей
электронной микроскопией фильтра (мембранный фильтр с размером пор 0,22 мкм)
а)
Мешок из ПЭВА
Раствор КО 10%-ный
Фосфат
Электролиты
Микроэлементы
Растаорд1коет
Раствор декстрозы
50%-ныи
Аминокислоты
Липидная эмульсия
20 000
30 000
Общее количество частиц размером >5 мкм
Фосфат
Электролиты
Микроэлементы
Раствор декстрозы
30%-ный
Раствор декстрозы
50%-ный
Аминокислоты
Липидная эмульсия
Концентрация частиц размером >5 мкл/мл
в компоненте смеси
Общее количество частиц
размером >10 мкм
.1
I
►
100 200 300 400 500
О
Концентрация частиц
размером >10 мкл/мл в компоненте смеси
Рис. 18. Общее количество частиц в каждом компоненте приготовленной смеси, определенное на
фильтре методом микроскопии (а); концентрация частиц в 1 мл каждого компонента, определенная
на фильтре методом микроскопии (б)
Глава 6.1. Источники контаминации
613
после фильтрации 50 мл из смеси, содержащей 1700 мл глюкозы, электролитов
и аминокислот с добавлением раствора поливитаминов из 1 ампулы (Малтамин),
раствора пантола из двух стеклянных ампул по 1 мл (Пантол), раствора хлорида на-
трия из двух стеклянных ампул по 20 мл (Конклит-Na), раствора метоклопрамида
из двух стеклянных ампул по 2 мл (Примперан) и раствора £-аспартата из двух сте-
клянных ампул по 10 мл (Аспара К). На рис. 19 также приведена идентификация
этих частиц. Их состав предполагает, что механическими включениями являются
частицы стекла (рис. 19, а) и частицы резины (рис. 19, б).
Аналогичное исследование было проведено Баллем (Ball) с соавт. [82] в Новой
Зеландии и Великобритании. Авторы изучили 20 образцов приготовленных смесей
для парентерального питания для взрослых и 20 образцов приготовленных смесей
для парентерального питания для детей, собирая первую и вторую фракцию из ин-
фузионной системы. Количество частиц с размером более 5 мкм было в 50 раз боль-
ше, чем количество частиц с размером более 40 мкм во всех растворах (табл. 40).
Анализ механических включений позволил определить включения как частицы ре-
зины и стекла (табл. 41).
Роузман (Roseman) с соавт. [84] анализировали хлопьевидные частицы, найден-
ные в растворах, хранившихся в стеклянных ампулах (ампулы из стекла типа I), с по-
а)
б)
Рис. 19. Идентификация двух типов частиц, проведенная с помощью сканирующего электронного
микроскопа с энергодисперсионной спектроскопией: а — предположительно частицы стекла;
б — предположительно частицы резины [83]
614
Часть 6. Контаминация и ее контроль
Таблица 41
Химические элементы в механических включениях, обнаруженных в растворах для
парентерального питания
Наиболее часто присутствующие в частицах элементы Частота присутствия Вероятное происхождение Источник
Кислород 1 Стекло, резина [82, 83]
Кремний 1 Стекло, резина [81, 82, 83]
Углерод 3 Резина [82, 83]
Алюминий 1 Стекло [81, 82, 83]
Магний 2 Тальк [81, 82]
Натрий 2 Стекло, резина [81, 83]
Хлор 3 Резина [83]
Калий 3 Стекло [83]
Примечание. Частота присутствия: 1 — очень часто; 2 — часто; 3 — редко.
мощью сканирующей электронной микроскопии. Они установили, что все частицы
имели одинаковые характеристики (бесцветные, плоские, толщина менее 1 мкм),
но их размер варьировал от нескольких микрометров до 100 мкм в длину. Результаты
элементного анализ четырех частиц приведены в табл. 42. Так как в частицах име-
лись такие же элементы, как и в стекле, авторы разбили стеклянную ампулу и про-
вели анализ полученных фрагментов (результаты также приведены в табл. 42). По-
скольку составы частиц и фрагментов ампулы были похожи, авторы сделали вывод,
что источником образования частиц являются стеклянные ампулы, подвергнув-
шиеся воздействию какого-то химического вещества. Было показано, что наряду
с фтороводородной кислотой и гидроксидами щелочных металлов другие вещества,
например ЭДТА и другие органические кислоты, в частности лимонная кислота,
а также соли фосфорной кислоты, способны воздействовать на стекло (см. раздел
6.1.3.2). Присутствие механических включений в растворах в стеклянных ампулах
было объяснено процессом расслоения, которое могло произойти непосредственно
в основании ампулы — области термального стресса, так как для формирования дна
ампулы применяются очень высокие температуры.
Основная проблема, связанная с контаминацией механическими включениями
инфузионных растворов, обусловлена не составом препарата (поскольку частицы
являются, главным образом, элементами контейнера, и, соответственно, нетоксич-
ными элементами), а с возможностью каждой частицы вызывать нежелательные
лекарственные реакции. Вероятность развития нежелательной реакции прямо про-
порциональна количеству (и размеру) частиц, введенных в систему кровоснабже-
ния. Турко (Turco) с соавт. [85] предполагают, что к реакциям организма на введение
с инфузией механических включений относятся физическая окклюзия сосудов, вос-
палительные реакции, неопластические реакции и реакции на антиген. Исследова-
ния на животных показали, что распределение частиц в тканях связано с диаметром
частиц. Частицы размеров более 8 мкм застревают в капиллярах легких, диаметром
3—6 мкм — скапливаются в лимфатических узлах селезенки и печени, а частицы
диаметром 1 мкм — в печени [86].
Глава 6.1. Источники контаминации
615
Таблица 42
Элементный анализ частиц, обнаруженных в растворах, хранившихся в стеклянных ампулах,
и фрагментов стекла, полученных из разбитой стеклянной ампулы
Элемент Состав, %
Частица 1 Частица 2 Частица 3 Частица 4 Фрагменты стекла*
Кремний 25-30 35-45 30-35 25-30 30-40
Алюминий 10-12 3-5 8-10 10-11 2-5
Калий 4-6 н/о 3-5 3-4 н/о
Кальций 1-2 н/о н/о 1-2 н/о
Бор 3-8 2-6 3-8 Присутствует 4-10
Натрий 1-3 1-2 1-3 1-2 4-6
а Среднее значение для четырех образцов.
Примечание. Все ампулы были изготовлены из стекла типа I. «н/о» — не обнаружен.
Источник: [84].
6.1.4. Заключение
Безопасность лекарственной терапии тесно связана с качеством лекарственных
препаратов. Все этапы технологического процесса могут повлиять на увеличение
присутствия посторонних веществ в продукте. И хотя в руководствах установлены
соответствующие требования к действующим веществам, вспомогательным веще-
ствам, остаточным растворителям, контейнерам и укупорочным средствам, это не
является гарантией отсутствия контаминирующих агентов в продукте.
Несмотря на то что испытания контейнеров на биологическую реактивность яв-
ляются информативными индикаторами токсичности экстрагируемых веществ, име-
ются вещества, которые не вызывают острый токсический ответ, а способствуют раз-
витию хронических реакций, как, например, фталаты (ДОФ) и металлы, например,
алюминий. Более того, существуют вещества, экстрагируемые из упаковочных ма-
териалов только под воздействием компонентов препарата, и, следовательно, отсут-
ствующие в экстрактах, полученных стандартными фармакопейными методиками.
Разложение компонентов препарата вследствие перекисного окисления липидов
и витаминов является проблемой контаминации, не рассматриваемой фармакопея-
ми, поскольку оно происходит при воздействии на растворы воздуха и света, что
приводит к образованию пероксидов.
Контаминация механическими включениями была обнаружена в растворах для
парентерального питания и других препаратах и растворах для внутривенного вве-
дения. Введение частиц в организм человека вместе с инфузионными растворами
может привести к развитию нежелательных лекарственных реакций. Вероятность
развития этих реакций увеличивается пропорционально вводимому количеству
жидкости.
Присутствие примесей, нежелательное для препаратов любого типа, является
очень критичным для парентеральных препаратов. Больные, нуждающиеся в ин-
тенсивной или длительной терапии парентеральными препаратами, больные с на-
рушением иммунного ответа, новорожденные и дети грудного возраста мотуг иметь
повышенную восприимчивость к негативному воздействию примесей.
616 Часть 6. Контаминация и ее контроль
Литература
1. U.S. Pharmacopeia (2005), USP 27, U.S. Pharmacopeial Convention, Rockville, MD.
2. World Health Organization (WHO) (2004), Guidelines for Drinking-water Quality, Vol'. 1 : Recom-
mendations, WHO, Geneva.
3. Water programs, Environmental Protection Agency, National Interim Primary Drinking Water Regu-
lations, Code of Federal Regulations, Part 141, (1985).
4. ASTM International (2005), ASTM Book of Standards, Vol. 11.02: Water and Environmental
Technology: Water (II), ASTM International, West Conshohocken, PA.
5. BP (2003), British Pharmacopoeia Commission, London.
6. Deutsche Arzneibuch (DAB) (2005), Deutschen Apotheker Verlag, Stuttgart.
7. European Pharmacopoeia (Ph.Eur.) (2004), 4th ed., European Directorate for the Quality of Medi-
cines (EDQM), Ph.Eur., Strasbourg.
8. The International Pharmacopoeia (IP) (2003), 3rd ed., World Health Organization, Geneva.
9. Kiister, F. W., and Thiel, A. (1985), Rechentafeln fiirdie Chemische Analytik, 103 Auflage, Walter de
Gruyter, Berlin, p. 258.
10. Koo, W. W, Kaplan, L. A., Horn, J., Tsang, R., and Steichen, J. (1986), Aluminum in parenteral nutri-
tion solutions — sources and possible alternatives, J. Parenteral Enteral Nutr., 10, 591-595.
11. Recknagel, S., Bratter, P., Chrissafi dou, A., Gramm, H.-J., Kotwas, J., and R о sick, U. (1994), Paren-
teral aluminum loading in critical care medicine. Part I: Aluminum content of infusion solutions and
solutions for parenteral nutrition, Infusionsther. Transfusionsmed., 21,266-273.
12. Bohrer, D., do Nascimento, P. C., Becker, E., de Carvalho L. M., and Dessuy, M. (2005), Arsenic spe-
cies in solutions for parenteral nutrition, J. Parenteral Enteral Nutr., 29,1—7.
13. PluhatorMurton, M. M., Fedorak, R. N., Audette, P. J., Marriage, B. J., and Yatscoff, R. W. (1996),
Extent of trace-element contamination from simulated compounding of total parenteral nutrient solu-
tions, Am. J. Health-Syst. Pharm., 53,2299-2303.
14. Bohrer, D., Bortoluzzi, F., Nascimento, P. C., Carvalho, L. M., and Ramirez, A. G. (2008), Silicate
release from glass for pharmaceutical preparations, Int. J. Pharm., in press.
15. Bohrer, D., do Nascimento, P. C., Binotto, R., Becker, E., and Pomblum, S. G. (2002), Contribution of
the raw material to the aluminum contamination in parenterals, J. Parenteral Enteral Nutr., 26, 382—388.
16. Bohrer, D., Becker, E., do Nascimento, P. C., de Carvalho L. M., and Marques, M. S. (2006), Arse-
nic release from glass containers by action of intravenous nutrition formulation constituents, Int. J.
Pharm., 315,24-29.
17. Thompson, J. E., and Davidow, L. (2003), A Practical Guide to Contemporary Pharmacy Practice,
Lippincott Williams & Wilkins, Philadelphia.
18. Wade, A., and Weller, P. J. (1994), Handbook of Excipients, 2nd ed., Pharmaceutical Press, London.
19. Neuzil, J., Darlow, B. A., Inder, T. E., Sluis, К. B., Winterboum, С. C., and Stocker, R. (1995), Oxi-
dation of parenteral lipid emulsion by ambient and phototherapy lights: Potential toxicity of routine
parenteral feeding, J. Pediatr., 126, 785-790.
20. Steger, P. J. K., and M ii hlebach, S. F. (1997), In vitro oxidation of IV lipid emulsions in different All-
in-One admixture bags assessed by an iodometric assay and gas-liquid chromatography, Nutrition, 13,
133-140.
21. Murray, R. K., Granner, D. K., Mayes, P. A., and Rodwe, V. W. (2000), Harper’ Biochemistry, 25th
ed., McGraw-Hill, New York, p. 169.
22. Helbock, H. J., Motchnik, P. A., and Ames, B. N. (1993), Toxic hydroperoxides in intravenous lipid
emulsions used in preterm infants, Pediatrics, 91, 83—87.
23. Picaud, J. C., Steghens, J. P., Auxenfans, C., Barbieux, A., Laborie, S., and Claris, O. (2004), Lipid
peroxidation assessment by malondialdehyde measurement in parenteral nutrition solutions for new-
born infants: A pilot study, Acta Paediatr., 93,241—245.
24. Pironi, L., Guidetti, M., Zolezzi, C., Fasano, M. C., Paganelli, F., Merli, C., Bersani, G., Pizzoferrato,
A., and Miglioni, M. (2003), Peroxidation potential of lipid emulsions after compounding in all-in-
one solutions, Nutrition, 19, 784—788.
25. Steger, P. J. K., and M ii hlebach, S. F. (1998), Lipid peroxidatoin of IV lipid emulsions in TPN bags:
The influence of tocopherols, Nutrition, 14,179-185.
Глава 6.1. Источники контаминации 617
26. Burton, G. W., and Ingold, К. U. (1986), Vitamin E as an in vitro and in vivo antioxidant, Ann. NY
Acad. Sci., 570, 7-22.
27. Bowry, V. W., Ingold, K. U., and S tocker, R. (1992), Vitamin E in human low-density lipoprotein.
When and how this antioxidant becomes a pro-oxidant, Biochem. J., 288, 341—344.
28. Bowry, V. W, and Stocker, R. (1993), Tocopherol - mediated peroxidation. The pro-oxi-
dant effect of Vitamin E on the radical-initiated oxidation of human low-density lipoprotein,
J. Am. Chem. Soc., 115, 6029-6040.
29. Steger, P. J. K., and M ii hlebach, S. F. (2000), Lipid peroxidatoin of intravenous lipid emulsions and
all-in-one admixtures in total parenteral nutrition bags: The influence of trace elements, J. Parenteral
EnteralNutr., 24, 37—41.
30. Laborie, S., Lavoie, J-С., Rouleau, T., and Chessex, P. (2002), Multivitamin solutions for enteral
supplementation: A source of peroxides, Nutrition, 18, 470-473.
31. Laborie, S., Lavoie, J-С., Pineaut, M., and Chessex, P. (2000), Contribution of multivitamins, air
and light in the generation of peroxides in adult and neonatal parenteral nutrition solutions, Ann.
Pharmacother, 34, 440-445.
32. Helbock, H. J., Motchnick, P. A., and Ames, B. N. (1993), Toxic hydroperoxides in intravenous lipid
emulsions used in preterm infants, Pediatrics, 91, 83—88.
33. Lavoie, J-С., Belanger, S., Spalinger, M., and Chessex, P. (1997), Admixture of a multivitamin prepa-
ration to parenteral nutrition: The major contributor to in vitro generation of peroxides, Pediatrics, 99,
61-70.
34. Lavoie, J. C., Chessex, P., Rouleau, T., Migneault, D., and Comte, B. (2004), Light-induced byprod-
ucts of vitamin C in multivitamin solutions, Clin. Chem., 50, 135-140.
35. Knafo, L., Chessex, P., Rouleau, T., and Lavoie, J-C. (2005), Association between hydrogen peroxide-
dependent byproducts of ascorbic acid and increased hepatic acetyl-CoA carboxylase activity, Clin.
Chem., 51, 1462-1471.
36. Balet, A., Cardona, D., Jane, S., Molins-Pujol, A. M., Sanchez Quesada, J. L., Gich, L, andMangues,
M. A. (2004), Effects of multilayered bags vs ethylvinyl-acetate bags on oxidation of parenteral nutri-
tion, J. Parenteral Enteral Nutr., 28, 85—91.
37. Baker, M., Gregerson, M. S., Martin, S. M., and Buettner, G. R. (2003), Free radical and drug oxida-
tion products in an intensive care unit sedative: Propofol with sulfite, Crit. Care Med, 31, 787-792.
38. Lavoie, J-С., Lachance, C., and Chessex, P. (1994), Antiperoxide activity of sodium metabisulfi te. A
double-edged sword, Biochem. Pharmacol., 47, 871-876.
39. Hayon, E., Treinin, A., and Wilf, J. (1972), Electronic spectra, photochemistry, and autoxidation:
Mechanism of the sulfi te-bisulfi te-pyrosulfi te systems. The SO2", SO3 , SO4“ radicals, J. Am. Chem.
Soc., 94,47-57.
40. International Conference on Harmonization (ICH) (1997), Guideline on residual solvents, ICH, Ge-
neva.
41. Bohrer, D., do Nascimento, P. C., Becker, E., Bortoluzzi, F., Depoi, F., and de Carvalho, L. M. (2004),
Critical evaluation of the standard hydrolytic resistance test for glasses used for containers for blood
and parenteral formulations, PDA J. Pharm. Sci. Technol., 58, 96-105.
42. Scholze, H. (1988), Gias, Natur, Struktur und Eigenschaften, 3rd ed., Springer, Heidelberg, pp. 128,
324.
43. Bacon, F. R. (1986), Glass containers for parenterals, in Avis, К. E., Lachman, L., and Lieberman, H.
A., Eds., Pharmaceutical Dosages Forms: Parenteral Medications, Vol. 2, Marcel Dekker, New York,
pp. 55-110.
44. Hak, E. B., Storm, M. C., and H elms, R. A. (1998), Chromium and zinc contamination of parenteral
nutrient solution components commonly used in infants and children, Am. J. Health-Syst. Pharm., 55,
150-154.
45. Borg, C., Constant, H., Fusselier, M., and Aulagner, G. (1994), Zinc, copper and iron in total paren-
teral nutrition mixtures: A contamination study, Nutrition, 13, 325-326.
46. Buchman, A. L., Neely, M., Grossie Jr, B., Truong, L., Lykissa, E., and Ahn, C. (2001), Organ heavy-
metal accumulation during parenteral nutrition is associated with pathologic abnormalities in rats,
Nutrition, 17, 600-606.
618 Часть 6. Контаминация и ее контроль
47. do Nascimento, Р. С., Marques, М. S., Hilgemann, М., de Carvalho L. М., Bohrer, D., Pomblum, S.
G., and Schirmer, S. (2006), Simultaneous determination of cadmium, copper, lead, and zinc in amino
acid parenteral nutrition solutions by anodic stripping voltammetry and sample digestion by Uvirra-
diation, Anal. Lett., 39, 1-14.
48. Alfrey, A. C., Le Gendre, G. R., and Kaenhy, W. D. (1976), The dialysis encefalophathy syndrome,
possible aluminum intoxication, N. Eng. J. Med., 294,184-188.
49. Driscoll, W. R., Cummings, J. J., and Zorn, W. (1997), Aluminum toxicity in preterm infants, 2V. Eng.
J. Med., 337, 1090-1091.
50. Bohrer, D., do Nascimento, P. C., Binotto, R., and Pomblum, S. G. (2001), Influence of the glass pack-
ing on the contamination of pharmaceutical products by aluminium. Part I: Salts, glucose, heparin and
albumin, J. TraceElem. Med. Biol., 15, 95-101.
51. Bohrer, D., do Nascimento, P. C., Martins, P., and Binotto, R. (2002), Availability of aluminum from
glass and an Al-form exchanger in presence of complexing agents and amino acids, Anal. Chim. Acta,
459, 267-276.
52. Bohrer, D., do Nascimento, P. C., Binotto, R., and Becker, E. (2003), Influence of the glass packing on
the contamination of pharmaceutical products by aluminium. Part Ш: Interaction container-chemicals
during heating for sterilisation, J. Trace Elements Med. Biol., 17, 107—115.
53. Bohrer, D., do Nascimento, P. C., Binotto, R., and Carlesso, R. (2001), Influence of the glass packing
on the contamination of pharmaceutical products by aluminium. Part II: Amino acids for parenteral
nutrition, J. Trace Elements Med. Biol., 15, 103—108.
54. Popinska, K., Kierkus, J., Lyszkowska, M., Socha, J., Pietraszek, E., Kmiotek, W., and Ksiazky, J.
(1999), Aluminum contamination of parenteral nutrition additives, amino acid solutions, and lipid
emulsions, Nutrition, 15,683-686.
55. Baydar, T., Aydin, A., Duru, S., Isimer, A., and Sahin, G. (1997), Aluminum in enteral nutrition for-
mulas and parenteral solutions, Clin. Toxicol., 35, 277—281.
56. Guidance for industry, container closure systems for packaging human drugs and biologies, chemis-
try, manufacturing and control documentation, Food and Drug Administration, 1999.
57. Ullmann s Encyclopedia of Industrial Chemistry, Vol. A5, 5th ed., VCH, Weinheim, 1993, p. 334.
58. Solomon, D. D., Jurgens, R. W., and Wong, K. L. (1986), Plastic containers for parenterals, in
Avis, К. E., Lachman, L., and Lieberman, H. A., Eds., Pharmaceutical Dosages Forms: Parenteral
Medications, Vol. 2, Marcel Dekker, New York, pp. 111-153.
59. U.S. Food and Drug Administration (FDA) (1999), Safety assessment of di-2-ethylhexylphthalate
(DEHP) released from PVC medical devices, FDA, Center for Devices and Radiological Health,
Rockville, MD.
60. Arabin, A., and 6 stelius, J. (1980), Determination by electron-capture gas chromatography of mono(2-
ethylhexyl) phthalate and di(2-ethylhexyl) phthalate in intravenous solutions stored in poly(vinyl
chloride) bags, J. Chromatogra. B, 193,405—412.
61. Arabin, A., Jacobsson, S., Hagman, A., and Ostelius, J. (1986), Studies on contamination of intrave-
nous solutions from poly(vinyl chloride) bags with dynamic headspace gas chromatography-mass
spectrometry and gradient liquid chromatography diode array techniques, Int. J. Pharm., 28, 211—
218.
62. Faouzi, M. A., Khalfi, E, Dine, T., Luyckx, M., Brunet, C., Gressier, B., Goudaliez, F., Cazin, M.,
Kablan, J., Belabed, A., and Cazin, J. C. (1999), Stability, compatibility and plasticizer extraction of
quinine injection added to infusion solutions and stored in polyvinyl chloride (PVC) containers, J.
Pharm. Biomed. Anal., 21, 923—930.
63. Kambia, K., Dine, T., Gressier, B., Bah, S., Germe, A.-F., Luyckx, M., Brunet, C., Michaud, L., and
Gottrand, F. (2003), Evaluation of childhood exposure to di(2-ethylhexyl) phthalate from perfusion
kits during long-term parenteral nutrition, Int. J. Pharm., 262, 83—91.
64. Allwood, M. C., and Martin, H. (1996), The extraction of diethylhexylphthalate (DEHP) from poly-
vinyl chloride components of intravenous infusion containers and administration sets by paclitaxel
injection, Int. J. Pharm., 127, 65-71.
65. Sautou-Miranda, V, Brigas, F., Vanheerswynghels, S., and Chopineau, J. (1999), Compatibility of pa-
clitaxel in 5% glucose solutions with ECOFLAC low-density polyethylene containers-stability under
different storage conditions, Int. J. Pharm., 178, 77—82.
Глава 6.1. Источники контаминации 619
66. Marcato, В., Guerra, S., Vianello, М., and Scalia, S. (2003), Migration of antioxidants additives from
various polyolefi nic plastics into oleaginous vehicles, Int. J. Pharm., 257, 217-225.
67. Smith, E. J., and Nash, R. J. (1986), Elastomeric closures for parenterals, in Avis, К. E., Lachman, L.,
and Lieberman, H. A., Eds., P harmaceutical Dosages Forms: Parenteral Medications, Vol. 2, Marcel
Dekker, New York, pp. 155—215.
68. Accardi-Dey, A., and Gschwend, P. M. (2003), Reinterpreting literature sorption data considering both
absorption into organic carbon and adsorption onto black carbon, Environ. Sci. Technol., 37, 99-106.
69. U.S. Food and Drug Administration (FDA) (1999), Guidance for industry, container closure systems
for packaging human drugs and biologies, FDA, Rockville, MD.
70. Parenteral Drug Association (PDA) (1998), Pharmaceutical package integrity, Technical Report 27,
PDA Bethesda, MD.
71. Mannermaa, J. P., Raisanen, J., Hyvonen-Dabek, M., Spring, E., and Yliruusi, J. (1994), Use of pro-
ton-induced X-ray emission (PIXE) analysis in the evaluation of large volume parenteral rubber stop-
pers, Int. J. Pharm., 103, 125—129.
72. Delaunay-Bertoncini, N., van der Wielen, F. W. M., De Voogt, P., Erlandsson, B., and Schoenmak-
ers, P. J. (2004), Analysis of low-molar-mass materials in commercial rubber samples by Soxhlet and
headspace extractions followed by GC-MS analysis, J. Pharm. Biomed. Anal., 35, 1059-1073.
73. Jenke, D. R. (1997), Utilization of extraction profi les to estimate the accumulation of extractables
from polymeric materials, J. Appl. Polym. Sci., 63, 843-848.
74. Loff, S., Kabs, F., Witt, K., Sartoris, J., Mandi, B., Niessen, К. H., and Waag, K. L. (2000), Polyvy-
nilchloride infusion lines expose infants to large amounts of toxic plasticizers, J. Pediatr. Surg., 35,
1775-1781.
75. Haishima, Y., Seshimo, E, Higuchi, T., Yamazaki, H., Hasegawa, C., Izumi, S., Makino, T., Naka-
hashi, K., Ito, R., Inoue, K., Yoshimura, Y, Saito, K., Yagami, T., Tsuchiya, T., and Nakasawa, H.
(2005), Development of a simple method for predicting the levels of DEHP migrated from PVC medi-
cal devices into pharmaceutical solutions, Int. J. Pharm., 298, 126-142.
76. Kambia, K., Dine, T., Gressier, B., Germe, A.-E, Luyckx, M., Brunet, C., Michaud, L., and Gottrand,
F. (2001), High-performance liquid chromatographic method for the determination of DEHP in total
parenteral nutrition and in plasma, J. Chromatogr. B, 755, 297-303.
77. Boithias, S-В., Sautou - Miranda, V, Bourdeaux, D., Tramier, V, Boyer, A., and Chopineau, J. (2005),
Leaching of DEHP from multilayer tubing into etoposide infusion solutions, Am. J. Health-Syst.
Pharm., 62, 182-188.
78. Jenke, D. R., Story, J., and Lalani, R. (2006), Estractables/leachables from plastic tubing used in prod-
uct manufacturing, Int. J. Pharm., 315, 75-92.
79. Groves, M. J. (1991), Particulate contamination in parenterals: Current issues, Boll. Chim. Farma.,
130, 347-354.
80. Bikhazi, A. B., Shiatis, J. A., and Haddad, A. E (1977), Quantitative estimation of particulate matter
in pharmaceutical preparations intended for intravenous administration, J. Pharm. Sci., 66, 181-186.
81. Foroni, L. A., Rochat, M. H., Trouiller, P., and Calop, J. Y. (1993), Particle contamination in a ternary
nutritional admixture, J. Parenteral Sci. Technol., 47, 311—314.
82. Ball, P. A., Bethune, K., Fox, J., Ledger, R., and Barnett, M. (2001), Particulate contamination in
parenteral nutrition solutions: Still a cause for concern? Nutrition, 17, 926-929.
83. Oie, S., and Kamiya, A. (2005), Particulate and microbial contamination in in-use admixed parenteral
nutrition solutions, Biol. Pharm. Bull., 28, 2268—2270.
84. Roseman, T. J., Brown, J. A., and Scothom, W. W. (1976), Glass for parenteral products:
A surface view using the scanning electron microscope, J. Pharm. Sci., 65, 22—29.
85. Turco, S. J., and Davis, N. M. (1971), Detrimental effects of particulate matter on the pulmonary
circulation, J. Am. Med. Assoc., 217, 81-82.
86. Hearse, D. J., Sonmez, B., Saldanha, C., Braimbridge, M. V, Maxwell, M. P., and Erol, C (1986),
Particle-induced coronary vasoconstriction in the rat heart: Pharmacological investigation of underly-
ing mechanisms, Thorac. Cardiovasc. Surg., 34, 316-325.
Глава 6.2. Количественное определение маркеров
грамотрицательных и грамположительных
эндотоксинов при мониторинге производ-
ственной рабочей среды и контроле ми-
кробной контаминации фармацевтических
продуктов методом ГХ-МС/МС
Элвин Фокс
Университет Южной Каролины (Колумбия, Южная Каролина)
6.2.1. Введение
Эндотоксины — это вещества, содержащиеся в оболочке бактериальной клетки. Их
наличие в фармацевтических продуктах вызывает пирогенные реакции, иногда при-
водящие к смертельному исходу. Токсичность эндотоксинов обусловлена их хими-
ческой структурой. Однако жизнеспособность организма при этом несущественна,
так как эндотоксины выделяются как из живых, так и из убитых микроорганизмов.
Классический эндотоксин — это липополисахарид. Пептидогликан также облада-
ет свойствами, сходными с эндотоксинами. Липополисахарид находят только во
внешней мембране грамотрицательных бактерий, в то время как пептидогликан на-
ходится в клеточной стенке и грамположительных, и грамотрицательных бактерий.
Испытание с лизатом амебоцитов Litnulus (ЛАЛ) широко используется для биоло-
гического количественного определения уровня содержания липополисахаридов,
но практически не обладает чувствительностью в отношении определения пепти-
догликанов. Структуры, определенные в липополисахаридах и пептидогликанах
(3-гидроксижирные кислоты (3-ОН-ЖК) и мурамовая кислота соответственно),
редко встречаются где-либо в природе и могут служить химическими маркерами.
Оба маркера для липополисахаридов и пептидогликанов можно определить мето-
дом газовой хроматографии — тандемной масс-спектрометрии (ГХ-МС/МС). Этой
технологии и посвящен данный раздел.
Недавно опубликованная книга [1] содержит подробный обзор проблем в фарма-
цевтической отрасли, связанных с контаминацией эндотоксинами (присутствующи-
ми как в жизнеспособных, так и в нежизнеспособных бактериях), высвобождаемых
клеточными стенками структур, а также жизнеспособными бактериями. Следует
изучить содержимое рабочей среды (т. е. воздух в помещениях) и сами фармацев-
тические продукты. Эти данные содержат информацию, соответственно, о возмож-
ных источниках микробной контаминации и о чистоте готового промышленного
продукта (или чистоте полупродуктов на разных стадиях производства продукта).
В некоторых случаях крайне важно определить, какие именно виды бактерий стали
причиной контаминации. Культуральные методы относятся к стандартным микро-
Глава 6.2. Количественное определение маркеров...
621
биологическим методам, они были рассмотрены Хименезом (Jimenez) [1]; эти мето-
ды далее обсуждаться не будут. Любая контаминация (например, эндотоксинами)
вне зависимости от источника происхождения является вопросом первостепенной
важности (например, при определении безопасности серии антибиотиков для вну-
тривенного введения). Такая контаминация оптимально определяется некульту-
ральными методами.
Эндотоксиновые реакции возникают в ходе воспаления в ответ на культивиру-
емые и некультивируемые организмы. Таким образом, при применении традици-
онных культуральных методов риск присутствия эндотоксинов часто значительно
недооценен. Культуральные и некультуральные методы мониторинга существенно
отличаются по своим характеристикам. Следует отметить, что на результаты культу-
ральных методов сильно влияет изменчивость условий культивирования (например,
используемая питательная среда, длительность культивирования и температура),
в связи с чем сложно стандартизовать протоколы количественной оценки контами-
нации, а определить возможно только те организмы, которые растут в выбранных
условиях. Однако использование некультуральных методик однозначно не исклю-
чает необходимость в информации, которую предоставляет полученная культура
микроорганизмов.
Методы полимеразной цепной реакции (ПЦР) в режиме реального времени
стали основной альтернативой микробиологическим методам, так как позволяют
измерить уровень характерных генов, полученных от определенных видов микро-
организмов (вне зависимости, культивируемые они или нет). Было доказано, что
ПЦР является высокоэффективным методом в клинической микробиологиче-
ской лаборатории, но ее использование для более разнообразного мониторинга
окружающей среды или контроля фармацевтического продукта может быть более
сложным. Конечно, ПЦР не обнаружит эндотоксин. ПЦР ферментативным путем
амплифицирует (размножает) определенный участок гена с характерной последова-
тельностью дезоксирибонуклеиновой кислоты (ДНК); набор праймеров (коротких
участков комплементарной ДНК) направляет фермент на интересующий участок
ДНК. В классической ПЦР проводят определение полученного ПЦР-продукта ме-
тодом электрофореза в геле, что является достаточно длительной процедурой. Ме-
тод ПЦР в режиме реального времени отличается от классической ПЦР отсутствием
необходимости проведения электрофореза, и продукт ПЦР определяется просто по
увеличению флуоресценции, что может быть выполнено с предварительным куль-
тивированием организма или без него. Такой подход имеет много направлений ис-
пользования, но метод характеризуется изменчивостью, зависящей от среды образ-
ца (т. е. в некоторых случаях наблюдается полное ингибирование реакции ионами
металлов или гуминовыми кислотами). Оценка общей бионагрузки (т. е. измерение
содержания химических маркеров для бактериальных эндотоксинов методом ГХ-
МС/МС) намного меньше подвержена влиянию среды образца и более легко стан-
дартизуется.
Обзор относительного преимущества (и недостатков) современных некульту-
ральных методов относительно друг друга и в сравнении с культуральным методом
также представлен Хименезом [1], включая измерения аденозинтрифосфата (АТФ),
622
Часть 6. Контаминация и ее контроль
цитометрию или микроскопию активированных флуоресцентным красителем кле-
ток, методы молекулярной биологии (например, ПЦР и АНК-чипирование), имму-
нологические методы. Раздел, посвященный эндотоксинам, описывает метод био-
логического количественного определения с помощью ЛАЛ, который по-прежнему
является наиболее используемым методом для измерения контаминации такого
рода; также обсуждаются современные альтернативные методы, включая биологи-
ческое количественное определение цитокинов и аналитические химические мето-
ды (ГХ-МС). Определение с помощью ЛАЛ измеряет биологическую активность,
которая может изменяться в связи с незначительными отличиями в структуре эн-
дотоксинов, что оказывает влияние на результаты; это исключено при химическом
анализе [2]. Более того, биологические количественные определения, включая
ЛАЛ-метод, часто могут давать ложноположительные результаты вследствие пере-
крестной реактивности с другими примесями. Как было указано ранее, ЛАЛ-метод
определяет только эндотоксины грамотрицательных бактерий и не определяет эн-
дотоксинподобные пептидогликаны грамположительных бактерий. Этим и обу-
словлено введение достаточно сложного, но удобного метода ГХ-МС/МС, который
быстро заменяет ГХ-МС в анализе следовых количеств веществ в фармацевтиче-
ской отрасли и в других областях.
Данный раздел будет посвящен принципам использования ГХ-МС/МС для вы-
явления 3-ОН-ЖК (маркеры присутствия липополисахаридов) и мурамовой кисло-
ты (маркер присутствия пептидогликанов), которые хорошо изучены [3—8]. Также
в качестве маркера контаминации грибами используется эргостерол, определяемый
методом ГХ-МС/МС [9].
Токсический эффект эндотоксинов обусловлен взаимодействием компонентов
клеточной стенки бактерий (например, липополисахаридов или пептидоглика-
нов) с поверхностными рецепторами клеток, входящих в состав неспецифической
иммунной системы (т. е. толл-подобными рецепторами лейкоцитов). В результате
происходит образование цитокинов (например, интерлейкина 1 (ZZL-1) или фак-
тора некроза опухолей (TNF, tumor necrosis factor)), которые являются частью вну-
триклеточной ферментативной каскадной системы и могут привести к серьезно-
му повреждению тканей. Соответственно, биологические или иммунохимические
количественные методы анализа позволяют определить такие реакции. Как было
отмечено ранее, наиболее широко используется ЛАЛ-тест. Лизат амебоцитов ме-
чехвоста (Limulus) содержит ферментативную каскадную систему гелеобразования,
которая активируется минимальными концентрациями липополисахаридов (нано-
граммы или даже меньше). Имеются различные способы проведения этого испыта-
ния, позволяющие определить присутствие липолисахаридов, но они не так широ-
ко используются. Как было указано ранее, в других биологических количественных
методах применяются клеточные линии, реагирующие на присутствие липополи-
сахаридов или пептидогликанов. К сожалению, биологические методы довольно
часто дают ложноположительные результаты вследствие веществ, дающих такую
же реакцию, или ложноотрицательные результаты вследствие присутствия ингиби-
рующих факторов (примесей в образце) [10]. Подробное обсуждение этих методов
находится вне тематики данной главы, и эту информацию можно найти в других
источниках [1].
Глава 6.2. Количественное определение маркеров...623
6.2.2. Анализ маркеров для липополисахаридов
и пептидогликанов методом ГХ-МС/МС
и его применение
Группы, обладающие опытом анализа следовых количеств химических марке-
ров для бактерий, в настоящее время исследуют образцы исключительно методом
ГХ-МС/МС. Методики количественного определения этим методом в настоящее
время хорошо разработаны, и с их помощью было проведено исследование разно-
образных клинических лабораторных образцов и образцов объектов окружающей
среды (экологический мониторинг). Однако применение этих методик в фармацев-
тической отрасли требует дальнейшего изучения. Например, мурамовая кислота вы-
свобождается при гидролизе и определяется как ацетат алдитола; 3-ОН-ЖК после
обработки метанолом превращаются в дериваты метилтриметилсилила. Подробное
описание пробоподготовки описано во многих источниках — для мурамовой кис-
лоты [3,4,7, 8,11] и для 3-ОН-ЖК [2, 5,12,13]. Анализируемые вещества содержат
активные группы (например, ОН и СО2Н), которые взаимодействуют с хромато-
графическими колонками для ГХ. Для приведения образцов в пригодную для ана-
лиза форму необходимо проведение дериватизации. Очистка до и после деривати-
зации обеспечивает получение четких хроматограмм. Для количественной оценки
используют формы маркеров со стабильной радиоизотопной меткой (например,
пС-меченая мурамовая кислота, полученная из меченых синезеленых водорослей).
Определение 3-ОН-ЖК широко используется при оценке содержания липопо-
лисахаридов в воздухе рабочей среды. Однако при исследовании тканей и жидкостей
организма было установлено, что в них присутствуют эндогенные 3-ОН-ЖК в ма-
лых количествах в качестве продуктов метаболизма млекопитающих (р-окисление
жирных кислот в митохондриях). Учитывая вышесказанное, определение 3-ОН-ЖК
методом ГХ-МС/МС не рекомендуется использовать в качестве общего маркера по
определению следовых количеств липополисахаридов в клинических лабораторных
образцах [14]. При этом в определенных ситуациях оценка 3-ОН-ЖК была успеш-
но применена, например, для диагностики хронического перитонита [15]. Имеются
значительные возможности по использованию 3-ОН-ЖК в качестве маркеров кон-
таминации липополисахаридами в фармацевтических продуктах, состав которых
часто является намного менее сложным.
Доказано, что химический анализ содержания мурамовой кислоты является
эффективным и для клинических образцов, и для образцов для экологического
мониторинга, поскольку мурамовая кислота не синтезируется эукариотическими
клетками. Например, она легко определяется в инфицированных биологических
средах человека, синовиальной жидкости больных стафилококковыми артритами
и спинномозговой жидкости больных пневмококковой пневмонией [7,16]. Однако
наиболее часто использующаяся методика ее определения в виде ацетата алдитола
является очень длительной. Для разработки альтернативных подходов были прове-
дены испытания на большом количестве дериватов. К сожалению, предел обнару-
жения этих альтернативных методик не был оптимальным [17,18].
Мономер маркера химически конвертируют в летучую форму, пригодную для
пропускания через газовый хроматограф. Образцы затем проходят на тандем-
624
Часть 6. Контаминация и ее контроль
ный масс-спектрометр, который определяет ионизированные молекулы. Метод
ГХ-МС/МС включает газохроматографическое разделение, совмещенное с ис-
ключительной селективностью МС/МС. В методе МС (в режиме мониторинга
и количественного определения) фоновые пики не учитываются. МС/МС скани-
рует эти пики во второй раз, тем самым существенно снижая предел обнаружения.
Напротив, в режиме идентификации (МС/МС) химический «отпечаток пальцев»
анализируемого вещества позволяет провести его уверенную идентификацию.
Примером является анализ мурамовой кислоты: естественная 12С-мурамовая кис-
лота сначала высвобождается из пептидогликанового полимера (присутствует
в виде незначительного компонента в сложной среде образца) с помощью гидро-
лиза. Перевод 12С-мурамовой кислоты в летучий дериват, мурамицитола лактама
пентаацетат с молекулярной массой (М. м.) 445, критичен для анализа методом
ГХ-МС/МС.
Максимальная селективность при анализе методом ГХ-МС/МС требует мони-
торинга выбранных ионов. В режиме мониторинга выбранных ионов детектируют-
ся только один или более основных ионов, характерных для искомого компонента,
а фоновые или посторонние ионы не учитываются. Аналогичным образом, макси-
мальная избирательность анализа методом ГХ-МС/МС обеспечивается двукратным
мониторингом выбранных ионов или мониторингом выбранных реакций (MRM,
multiple-reaction monitoring). В квадрупольном масс-спектрометрическом детекторе
первая стадия сканирования выбранных реакций включает избирательный перенос
молекулярного иона из первого масс-спектрометра в ячейку столкновений. Такая
инструментальная очистка иона удаляет все другие ионы с другими молекулярными
массами, образовавшиеся при первоначальной ионизации. Затем выбранный ион
фрагментируется вследствие столкновений с инертным газом (например, аргоном).
Во втором масс-детекторе выбранный фрагментированный ион мониторирует-
ся. Если мешающие факторы повлияли на чувствительность или специфичность
методики, то ухудшается предел обнаружения. Метод ГХ-МС/МС предоставля-
ет существенно более высокую специфичность, чем ГХ-МС. При использовании
метода ГХ-МС было невозможно достоверно визуально различить хроматограммы
образцов с минимальным количеством мурамовой кислоты от хроматограмм от-
рицательных контрольных образцов (включая растения и грибы). Хроматограммы
пыли, полученные с помощью ГХ-МС/МС, были всегда легко отличимы от хрома-
тограмм контрольных образцов. Однако эти анализы достоверно выполнялись при
имеющихся пределах чувствительности для ГХ-МС/МС. Современные достижения
в технологии масс-спектрометрии могут привести к существенному увеличению
чувствительности данного метода.
Тандемная масс-спектрометрия включает две стадии. Например, при анализе
мурамовой кислоты на первой стадии выделяется практически неизмененная моле-
кула с М. м. 403 вследствие потери кетена (М. м. 42). Совместное элюирование мо-
лекул с разной молекулярной массой практически, но не полностью, исключается;
таким образом, только молекулы с М. м. 403 будут анализироваться на следующей
стадии. На этой второй стадии молекулы с М. м. 403 разрушаются на характерные
фрагменты, включая фрагмент, содержащий оригинальный лактам с М. м. 198. Та-
ким образом, на второй стадии определяются молекулы только с М. м. 198, а осталь-
Глава 6.2. Количественное определение маркеров...625
ные молекулы с другой молекулярной массой, которые могут элюировать вместе
с определяемыми молекулами, исключаются из анализа.
Использование аналога мурамовой кислоты, меченого стабильным изотопом
(13С), в каждом случае обеспечивает отсутствие ложноположительных результатов.
При этом предупреждаются потери мурамовой кислоты во время пробоподготовки
или проблемы с детекцией при настройках прибора. Хотя у 12С- и |3С-мурамовых
кислот на ГХ-хроматограммах будет одинаковое время удерживания, их можно
различить с помощью тандемного масс-спектрометрического детектора. Методи-
ка определения 13С-мурамовой кислоты методом ГХ-МС/МС такая же, как и для
12С-мурамовой кислоты. Однако молекулярные массы детектируемых молекул на
первой и второй стадиях анализа будут больше — 412 и 205 соответственно. Тан-
демный спектрометр позволяет вести мониторинг одновременно в двух окнах,
в одном — 13С-мурамовой кислоты (верхнее окно), в другом — натуральной мурамо-
вой кислоты (нижнее окно). Точное количественное определение достигается путем
сравнения отношения площадей пиков 13С- и 12С-мурамовых кислот (рис. 1).
Получаемый химический «отпечаток пальцев» — это отпечаток продуктов рас-
щепления, проведенного в ходе ГХ-МС/МС-анализа, характерный для анализируе-
мого вещества. Родительская молекула имеет М. м. 403. В каждом случае наблюда-
ются одинаковые основные фрагменты с массами 361, 301, 258, 240, 198, 156 и 138.
Молекулы с массой 361 получаются вследствие потери кетена (уменьшение массы
на 42), с массой 301 — вследствие последующей потери уксусной кислоты (умень-
шение массы на 60). Разрыв связи С,—С5 (уменьшение массы на 145) приводит к об-
разованию молекул с М. м. 258, а дальнейшая потеря уксусной кислоты (уменьше-
ние на 60) — к образованию фрагмента с М. м. 198. Отделение кетена или уксусной
кислоты от молекулы с М. м. 198 приводит к образованию фрагментов с массами
156 и 138 соответственно (рис. 2).
403,4—► 197,9
100
-1--1—I---1—г
24 26 28
30 32 34 36 38 40
Время удерживания
Рис. 1. Хроматограмма мониторинга молекул при ГХ-МС/МС-анализе. Сверху — для внутреннего
стандарта (,3С-мурамовой кислоты), снизу — для натуральной ,гС-мурамовой кислоты,
выделенной из пыли. Площади пиков в каждой хроматограмме нормализуются по отношению
к наиболее высокому пику. Примеры клинического анализа выглядят аналогично
626
Часть 6. Контаминация и ее контроль
Рис. 2. Химические «отпечатки пальцев» при ГХ-МС/МС-анализе (спектр полученных ионов).
Сверху — для стандартного образца мурамовой кислоты (общее содержание в образце — 2 нг);
снизу — для мурамовой кислоты, выделенной из пыли. Примеры клинического анализа выглядят
аналогично
6.2.3. Рабочие параметры
Каждый продукт, находящийся на рынке, содержит допустимое количество эндо-
токсинов, основанное на минимальной пирогенной дозе и количестве препарата,
вводимого больному, как описано в руководстве FDA [19]. Однако для таких науко-
емких методов химического анализа, которые описываются в этом разделе, руко-
водства отсутствуют. На данный момент имеется несколько специализированных
университетских лабораторий, обладающих опытом по следовому химическому
анализу липополисахаридов и пептидогликанов. Коммерческих испытательных ла-
бораторий не существует. Очевидно, упрощение и автоматизация сделают эти мето-
ды более широкодоступными.
В настоящее время рекомендуется использовать в анализе твердый образец мас-
сой 1—10 мг. В рабочей среде, не являющейся сильно загрязненной пылью из воз-
духа, сбор образца занимает несколько дней. В неоснащенном или минимально
оснащенном помещении 1 мг пыли, необходимый для анализа, собирают в течение
72 ч. В максимально оснащенном помещении количество пыли резко возрастает,
и сбор образца занимает около 6—8 ч. Для сбора пыли предпочтительно исполь-
зовать химически инертные фильтры (например, из тефлона), поскольку они не
изменяются при нагревании в серной кислоте, что является первой стадией хими-
ческого анализа.
Глава 6.2, Количественное определение маркеров...
627
Большое количество пыли (от миллиграммов до грамма) может быть легко собра-
но из кондиционеров (воздуха) или с поверхностей. Концентрация пыли в воздухе
может быть различной: от микрограммов до миллиграммов в 1 кубическом метре.
Низкие концентрации пыли характерны для офисных зданий и лабораторий, а вы-
сокие более характерны для таких пыльных помещений, как коровники или пти-
цефермы. Поскольку обычно используемые для сбора насосы собирают несколько
литров в минуту, за исключением сильно загрязненных рабочих сред, собрать мож-
но только миллиграммы пыли, если не увеличивать длительность отбора воздуха.
Определяемые химические маркеры являются только одним из компонентов бакте-
рий, а бактерии составляют только незначительную долю пыли. Мурамовая кислота
присутствует в количествах от 5 до 50 нг/мг пыли (что соответствует приблизитель-
но от 50 до 500 нг пептидогликанов на 1 мг пыли) в обычных домах и в пыли из
кондиционеров. Иногда находили более высокое содержание мурамовой кислоты
в пыли из воздуха (более 100 нг/мг). Липополисахариды присутствуют в количестве
от 500 до 5000 нг/мг пыли. Твердые продукты (например, фармацевтические) ана-
лизировать намного проще, при этом обычно не требуется предварительная про-
боподготовка. Для оптимального уровня чувствительности в анализе используют
около 10 мг образца.
Как было отмечено ранее, на основе методов ГХ-МС/МС были разработаны
чувствительные и избирательные методики определения 3-ОН-ЖК и мурамовой
кислоты. Определение мурамовой кислоты является альтернативой классическо-
му ЛАЛ-тесту для оценки содержания липополисахаридов, в то время как обяза-
тельное испытание для оценки пептидогликанов не установлено. Описываемые
химические методы воспроизводимы и обеспечивают проведение количественно-
го точного определения уровня микробной биоконтаминации. В настоящее время
масс-спектрометрическое определение липополисахаридов и пептидогликанов раз-
работано настолько хорошо, что может использоваться для повседневной оценки
качества воздуха. Был проведен анализ многочисленных медицинских продуктов и
проб окружающей среды. Однако использование этих методов для фармацевтиче-
ской продукции остается ограниченным.
Масс-спектрометрические методики обладают прецизионностью в диапазо-
не, характерном для большинства клинических анализов (т. е. до 5—15%). Наи-
лучшим выбором для внутреннего стандарта является форма определяемого ве-
щества со стабильной радиоактивной меткой (предпочтительно 13С) — например,
₽-гидроксимиристиновая кислота или мурамовая кислота. Избирательное детек-
тирование следовых количеств химических маркеров в сложных смесях (матрицах)
требует использования соответствующих отрицательных контролей. Часто описы-
ваются методики, в которых не используется масс-спектрометрический детектор,
и часто сообщается о ложноположительных результатах. Простой анализ чистых
фильтров или контрольных образцов воды не является достаточным, так как «хими-
ческий шум», создаваемый испытуемым образцом, намного выше и не может быть
адекватно оценен таким видом контроля достоверности результатов.
Как было отмечено ранее, основным испытанием на содержание бактериальных
эндотоксинов является ЛАЛ-тест. В этом методе оценивается активация каскадно-
го механизма образования гель-тромба в лизате амебоцитов (мечехвоста). ЛАЛ-тест
628
Часть 6. Контаминация и ее контроль
определяет в основном наличие липополисахаридов, а чувствительность детекти-
рования пептидогликанов этим методом очень низкая. Биологическое определение
бактериальных эндотоксинов с помощью ЛАЛ-теста и масс-спектрометрическая
оценка содержания липополисахаридов иногда имеют низкую корреляцию. Это
объясняется тем, что ЛАЛ-тест определяет биологическую активность веществ, в то
время как химический метод — их общее содержание. Таким образом, эти два метода
не являются прямо сопоставимыми. Однако некоторые различия (между результа-
тами, полученными ЛАП-тестом и ГХ-МС/МС) могут быть обоснованы значитель-
но более высокой избирательной способностью тандемной масс-спектрометрии.
Следует отметить, что ГХ-МС/МС может дать определенную информацию о попу-
ляции грамотрицательных бактерий, находившихся в образце, поскольку распреде-
ление гидрокси-ЖК отличается у разных видов бактерий. Так, 2- и 3-ОН-жирные
кислоты с 10—18 атомами углерода всегда присутствуют в органической пыли.
6.2.4. Заключение
Удобные для пользователя коммерческие приборы для ГХ-МС/МС стали выпу-
скаться с середины 1990-х годов. К сожалению, наблюдается ограниченное разви-
тие приборов, обеспечивающих автоматизацию обработки образцов для анализа,
хотя сообщалось о создании прототипа со встроенной автоматизированной дери-
ватизацией образцов [20]. Фармацевтическая отрасль активно использует другие
типы масс-спекгрометрических исследований для высокоскоростного анализа ле-
карственных средств (например, жидкостную хроматографию с тандемным масс-
спектрометром (ЖХ-МС/МС), которая исключает необходимость проведения де-
риватизации образцов после гидролиза путем использования масс-спектрометрии
с ионизацией электроспреем). К сожалению, эти методы не представляют большого
интереса для определения маркеров липополисахаридов или пептидогликанов [21].
Таким образом, анализ маркеров эндотоксинов остается технически сложным, что
препятствует его широкому применению вне пределов нескольких специализиро-
ванных лабораторий. Существует, однако, большой потенциал в использовании ме-
тодов ГХ-МС/МС для оценки контаминации грамположительными и грамотрица-
тельными бактериями в фармацевтической отрасли. Тандемные масс-спектрометры
по-прежнему являются дорогостоящими. Тем не менее, такие приборы широко ис-
пользуются в фармацевтической промышленности для общего анализа лекарствен-
ных средств. Наиболее современные приборы управляются программным обеспече-
нием на основе Windows. Таким образом, они мотут легко управляться операторами
с небольшим предыдущим опытом масс-спектрометрии или даже аналитической
химии, после соответствующего обучения.
Благодарность
Исследование, описанное в данном разделе, было спонсировано компаниями
Philip Morris USA, Inc., и Philip Morris International.
Глава 6.2. Количественное определение маркеров...
629
Литература
1. Jimenez, L. (2004), Microbial Contamination Control in the Pharmaceutical Industry, Marcel Dekker,
New York.
2. Saraf, A., Larsson, L., Burge, H., and Milton, D. (1997), Quantifi cation of ergosterol and 3-hydroxy
fatty acids in settled house dust by gas chromatography-mass spectrometry: Comparison with fun-
gal culture and determination of endotoxin by a Limulus amoebocyte lysate assay, Appl. Environ.
Microbiol., 63, 2554-2559.
3. Fox, A., Wright, L., and Fox, K. (1995), Gas chromatography tandem mass spectrometry fortrace detec-
tion of muramic acid, a peptidoglycan chemical marker in organic dust, J. Microbiol. Meth. ,22,11-26.
4. Fox, A., Krahmer M., and Harrleson, D. (1996), Monitoring muramic acid in air (after alditol acetate
derivatization) using a gas chromatograph-ion trap tandem mass spectrometer, J. Microbiol. Meth.,
27, 129-138.
5. Saraf, A., and Larsson, L. (1996), Use of gas chromatography ion trap tandem mass spectrometer for
the determination of microorganisms in organic dust, J. Mass Spectrom., 31, 389-396.
6. Larsson, L., and Saraf, A. (1997), Use of gas chromatography-ion trap tandem mass spectrometry
for the detection and characterization of microorganisms in complex samples, Mol. Biotechnol., 7,
279-287.
7. Kozar, M., Krahmer, M. T., Fox, A., and Gray, В. M. (2000), Failure to detect muramic acid in normal
rat tissues but detection in cerebrospinal fluid from patients with pneumococcal meningitis, Infect.
Immun., 68, 4688—4698.
8. Kozar, M., Laman, J. D., and Fox, A. (2002), Muramic acid is not generally present in human spleen
as determined by gas chromatography—tandem mass spectrometry, Infect. Immun., 70, 741—748.
9. Sebastian, A., and Larsson, L. (2003), Characterization of the microbial community in indoor envi-
ronments: A chemical-analytical approach, Appl. Environ. Microbiol., 69, 3103—3109.
10. Kaneko, T., Goldman, W. E., Mellroth, P., Steiner, H., Fucase K., Kusumoto, S., Harley, W., Fox, A.,
Golenbock, D., and Silverman, N. (2004), Monomeric and polymeric Gram-negative peptidoglycan
but not purifi ed LPS stimulate the Drosophila IMD pathway, Immun., 20, 1-20.
11. Fox, A., Harley, W., Feigley, C., Salzberg, D., Toole, C., Sebastian, A., and Larsson, L. (2005), Large
particles are responsible for elevated bacterial marker levels in school air upon occupation, J. Environ.
Monit., 7, 450-456.
12. Mielniczuk, Z., Mielniczuk, E., and Larsson, L. (1993), Gas chromatography-mass spectrometry
methods for analysis of 2- and 3-hydroxylated fatty acids: Application for endotoxin measurement,
J. Microbiol. Meth., Yl, 91—102.
13. Mielniczuk, Z., Mielniczuk, E., and Larsson, L. (1995), Determination of muramic acid in organic
dust by gas chromatography-mass spectrometry, J. Chromatogr. B, 670,167—172.
14. Sponazr, B., Norin, E., Midvedt, T., and Lairson, L. (2002), Limitations in the use of 3-hydroxy fatty
acids to determine endotoxin in mammalian tissues, J. Microbiol. Meth., 50, 283—289.
15. Ferrando, R., Szponar, B., Sanchez, A., Larsson, L., and Vaero-Guillen, P. L. (2005), 3-Hydroxy fatty
acids in saliva as diagnostic markers in chronic peridontitis, J. Microbiol. Meth., 62, 285—291.
16. Fox, A., Fox, K., Christensson, B., Krahmer, M., and Harrelson, D. (1995), Absolute identifi cation
of muramic acid at trace levels in human septic fluids in vivo and absence in aseptic fl uids, Infect.
Immun., 64, 3911—3955.
17. Kozar,M.,andFox,A.(2002),Analysisofastablehalogenatedderivativeofmuramicacidbygaschroma-
tography—negative ion chemical ionization tandem mass spectrometry, J. Chromatogr., 946,229-238.
18. Sebastian, A., Harley, W., Fox, A., and Larsson, L. (2004), Evaluation of the methyl ester O-methyl
acetate derivative of muramic acid for the determination of peptidoglycan in environmental samples
by ion-trap GC-MS-MS, J. Environ. Monit., 6, 1—6.
19. U.S. Department of Health and Human Services (DHHS) (1987), FDA guidelines on validation of the
Limulus amoebocyte lysate test as an end product test for human and animal parenteral drugs, biologi-
cal products and medical devices, DHHS, Rockville, MD.
630
Часть 6. Контаминация и ее контроль
20. Steinberg, Р., and Fox, А. (1999), Automated derivatization instrument: Preparation of alditol acetates
for analysis of bacterial carbohydrates using gas chromatography—mass spectrometry, Anal. Chetn.,
'll, 1914-1917.
21. Shahgholi, M., Ohorodnik, S., Callahan, J., and Fox, A. (1997), Trace detection of underivatized
muramic acid in environmental dust samples by microcolumn liquid chromatography — electrospray
tandem mass spectrometry, Anal. Chem., 69, 1956-1960.
Глава 6.3. Микробиологические аспекты
нестерильного фармацевтического
производства
Ранга Велагалети
BASF Corporation (Флорэм Парк, Нью-Джерси)
6.3.1. Введение
Для соответствия исходных материалов и готовых продуктов основным микробио-
логическим показателям качества для нестерильных лекарственных средств, а также
имеющимся частным фармакопейным статьям или спецификациям на лекарствен-
ные препараты производители должны выполнять правила надлежащей производ-
ственной практики {GMP, good manufacturing practice), в разных разделах которых
имеются особые указания в отношении контроля контаминации. Например, в пра-
вилах GMP, установленных Федеральным управлением США по контролю за пище-
вой продукцией и лекарствами {FDA, Food and Drug Administration), для готовых про-
дуктов в Своде федеральных нормативных актов (CFR, Code of Federal Regulations),
в томе 21, §211.113, указано: «Необходимо разработать и выполнять соответствую-
щие письменные процедуры для предупреждения контаминации нежелательными
микроорганизмами нестерильных лекарственных средств» [1]. Для выполнения
данного регуляторного требования следует проводить мониторинг и необходимый
контроль микробиологической чистоты производственной среды помещений (воз-
духа, стен и полов), где производятся нестерильные фармацевтические исходные
материалы и готовые продукты, производственного оборудования, а также готовых
исходных материалов и лекарственных продуктов, чтобы обеспечить приемлемую
микробиологическую чистоту и отсутствие нежелательных микроорганизмов. Из-
давна микробная контаминация нестерильных лекарственных средств была опре-
деленной проблемой, и ряд продуктов отзывался с рынка вследствие микробной
контаминации нежелательными микроорганизмами, которые, как предполага-
лось, представляют риск для здоровья пациента. Уже в далеком 1979 г. Данниган
{Dunnigan) из Медицинского бюро FDA в своих комментариях допустимых уровней
содержания микроорганизмов и микробной контаминации выражал озабоченность
в отношении препаратов для наружного применения, содержавших грамотрица-
тельные бактерии, которые представляют риск средней или тяжелой степени для
здоровья потребителей [2].
В Фармакопее США {USP, U. S. Pharmacopeia) в недавно пересмотренной гла-
ве <1111> (585) «Микробиологическая оценка нестерильных продуктов: критерии
приемлемости для фармацевтических препаратов и веществ для фармацевтического
применения» сказано: «Присутствие определенных микроорганизмов в нестериль-
ных препаратах может в принципе привести к снижению или даже блокированию
терапевтического эффекта продукта и представлять риск для жизни пациента. Про-
632
Часть 6 Контаминация и ее контроль
изводители, следовательно, должны обеспечить низкий уровень микробной кон-
таминации готовых лекарственных форм путем выполнения действующих правил
бМРпри производстве, хранении и поставках фармацевтических продуктов» [3].
Фармацевтические производители и их профессиональные ассоциации начали
принимать предупреждающие меры в отношении микробной контаминации в не-
стерильной производственной среде. В статье мартовского номера за 1997 г. жур-
нала «Фармацевтическая технология» рабочая группа по мониторингу производ-
ственной среды Ассоциации фармацевтических производителей и разработчиков
{PhRMA, Pharmaceutical Research and Manufacturers Association of America) представи-
ла реалистичную оценку необходимости изучения и контроля микробной конта-
минации, обусловленной исходными материалами, компонентами первичной упа-
ковки, производственным процессом и производственной средой. Рабочая группа
особо подчеркнула в этой статье, что хотя для большинства фармацевтических
продуктов, за исключением стерильных препаратов, не установлено и не ожида-
ется требование быть стерильными, эти препараты тем не менее вводятся людям,
которые больны и имеют ослабленные организмы, и, следовательно, содержание
микроорганизмов должно быть минимальным [4]. Ассоциация австралийских
фармацевтических производителей {АРМА, Australian Pharmaceutical Manufacturers’
Association) опубликовала руководство по нестерильным медицинским лекарствен-
ным средствам, в котором подчеркивается необходимость контроля микробной
контаминации и важность выполнения правил GMPksio. контроля уровня содержа-
ния микроорганизмов [5].
Помимо правил GMPpjm готовых лекарственных средств (которые также вклю-
чают обязательные требования к вспомогательным и действующим веществам, пер-
вичной упаковке и укупорочным средствам, используемым для готовых продуктов),
изданными FDA [1], руководства по GA/Р также были разработаны Международной
конференцией по гармонизации {ICH, International Conference on Harmonization) [6]
(бЛ/Рдля активных веществ), властями Австралии [7, 8] (фармацевтические про-
дукты), Всемирной организацией здравоохранения (ВОЗ) [9] (фармацевтические
продукты), Международным Советом по фармацевтическим вспомогательным ве-
ществам {IPEC, International Pharmaceutical Excipients Council) [10] (вспомогательные
вещества) и USP [11]. Во всех этих руководствах имеются требования к производ-
ству фармацевтических продуктов с допустимой микробной контаминацией и с от-
сутствием нежелательных микроорганизмов.
В этой главе описаны регуляторные микробиологические аспекты нестерильно-
го фармацевтического производства, включая производственную среду, исходные
материалы и готовые продукты. Также рассмотрен контроль микробной контами-
нации фармацевтических субстанций и продуктов с учетом типа лекарственной
формы и дана оценка требований к микробной контаминации в нестерильном
фармацевтическом производстве с точки зрения фармацевтической отрасли. Об-
суждение микробиологических аспектов фармацевтической производственной
среды при изготовлении стерильных лекарственных средств и ожидаемое качество
стерильных продуктов в отношении микробиологических показателей в этом раз-
деле отсутствует.
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 633
6.3.2. Международные правила и регуляторные руководства
в отношении контроля микробиологической чистоты
в нестерильном производстве
Для иллюстрации требований правил GMP к готовым лекарственным средствам,
действующим веществам (фармацевтическим субстанциям) и вспомогательным
веществам будут рассмотрены соответственно правила GMP для готовых лекар-
ственных средств и компонентов готовых лекарственных средств, изданные FDA
[1], руководство по 6Л/Рдля активных фармацевтических веществ (Q7A) /СН[6] и
руководство по СМРдля. вспомогательных веществ USP [11]. В контексте этого об-
суждения правил в отношении микробиологических аспектов нестерильного произ-
водства под «контаминацией» подразумевается «нежелательное внесение примесей
химической или микробиологической природы, либо посторонних частиц, в или
на исходные материалы, полупродукты, фармацевтические субстанции при произ-
водстве, отбора проб, упаковке или переупаковке, хранении или транспортировке»
[6]. Такое же определение контаминации применимо и к лекарственным продук-
там. Микробиологическая чистота обозначает уровень содержания и тип микроор-
ганизмов, которые могут присутствовать в продукте. Лекарственный продукт, его
компоненты, производственное оборудование или производственная среда могут
считаться контаминированными, когда микробиологическая чистота превышает
нормы, установленные в спецификациях, или допустимые пределы содержания.
6.З.2.1. Правила б/МРдля готовых продуктов и их компонентов
FDA осуществляет надзор за выполнением действующих правил надлежащей про-
изводственной практики (cGMP и GMP — см. CFR, том 21, главы 210 и 211) для ле-
карственных средств, производимых на территории США (также они применяются
к лекарственным средствам и их компонентам, производимым за рубежом и ввози-
мым на территорию США), согласно Закону о пищевых продуктах, лекарственных
и косметических средствах (FD&CAct; Food, Drug, and Cosmetic Act). Согласно пункту
501(a)(2)(B) этого закона при несоответствии любому требованию правил GMP про-
изводственный продукт считается фальсифицированным, и этот продукт, а также
лицо, ответственное за невыполнение требований, будут подвергнуты законному
преследованию [1].
Очень важно понимать регуляторные определения готового лекарственного
средства, действующего или вспомогательного вещества [1] в контексте приводимо-
го в этом разделе обсуждения. Согласно правилам GMP, том 21 CFR, пункт 210.3(b)
(4), под лекарственным продуктом понимается готовая лекарственная форма, на-
пример таблетки, капсулы и растворы, которая содержит действующее вещество,
как правило, но необязательно, вместе со вспомогательными веществами. В это по-
нятие также входит готовая лекарственная форма, которая не содержит действую-
щего вещества, но предназначена для использования в качестве плацебо. Плацебо
используется специально при исследовании эффективности и безопасности в ходе
разработки лекарственного средства, в доклинических и клинических исследова-
ниях. Под действующими веществами (также их называют активными фармацевти-
ческими субстанциями или ингредиентами) понимают любой компонент, который
634
Часть 6 Контаминация и ее контроль
предназначен для получения фармакологического или другого прямого воздей-
ствия на причину заболевания, для излечения, уменьшения страдания, лечения или
предупреждения заболевания или для оказания влияния на структуру или любую
функцию организма человека или животного (CFR, том 21, пункт 210.3(b)(7)). Вспо-
могательные вещества (иногда их еще называют неактивными веществами) — это
любые компоненты, отличающиеся от действующих веществ (CFR, том 21, пункт
210.3(b)(8)).
Одним из важных аспектов правил GMP является контроль качества в ходе про-
изводства для обеспечения отсутствия в продукте нежелательных микроорганизмов.
Правила GMP [1] требуют, чтобы персонал, участвующий в производстве лекар-
ственного средства, был одет в защитную одежду, закрывающую голову, лицо и руки
(целиком верхние конечности) для защиты производимого лекарственного средства
от контаминации (глава 211, пункт 211.28(a)). В отношении проектирования пото-
ков компонентов, первичной упаковки и укупорочных средств, этикетировочных
материалов, полупродуктов и лекарственных продуктов в правилах также установ-
лено требование, которое должно предупреждать контаминацию (211.42(b)).
Предупреждение контаминации также включает выделение изолированных или
особых зон или других подобных контрольных систем для технологических опера-
ций, которые необходимы для предупреждения контаминации (211.42(c)). Для адек-
ватного контроля за микроорганизмами требуется соответствующее оборудование
(211.46(b)). Надлежащие вытяжные системы или другие системы, обеспечивающие
требуемый контроль контаминантов, необходимы в местах, где может произойти
контаминация воздуха в ходе технологического процесса (211.46(c)).
Вода используется во многих стадиях фармацевтических производственных
процессов. Правила GMP (211.48(a)) требуют, чтобы поступающая на производ-
ство водопроводная вода соответствовала правилам по первичной питьевой воде
Федерального управления США по охране окружающей среды (ЕРА, Environmental
Protection Agency) (CFR, том 40, глава 141), в которых установлено проведение ми-
кробиологического контроля водопроводной воды. Правилами GMP требуется раз-
работка и выполнение письменных процедур по предупреждению контаминации
оборудования, компонентов, первичной упаковки и укупорочных средств, упако-
вочных и этикетировочных материалов или лекарственных продуктов (CFR, том 21,
пункт 211.56(c)). Очистка и техническое обслуживание оборудования также необ-
ходимы для предупреждения контаминации (CFR, том 21, пункт 211.67(a)). С ком-
понентами препарата, первичной упаковкой и укупорочными средствами следует
обращаться и хранить их таким образом, чтобы предупредить контаминацию (CFR,
том 21, пункт 211.80(b)). Также правилами GMP требуется проведение испытаний
и выдача разрешения на использование в соответствии с пунктом 211.84(d)(6) тома
21 CFR, в котором установлено, что каждая партия (серия) компонента, первичной
упаковки или укупорочных средств, которые могут подвергаться микробной конта-
минации, нежелательной исходя из их предназначения, подлежит испытаниям на
микробиологическую чистоту. Должны быть разработаны и выполняться соответ-
ствующие письменные процедуры по предупреждению присутствия нежелательных
микроорганизмов в лекарственных средствах, к которым не применяется требова-
ние стерильности (CFR, том 21, пункт211.113(a)). В пункте 211.165(b) тома 21 CFR
Глава 6,3, Микробиологические аспекты нестерильного фармацевтического производства 63S
правилами GA/P установлено требование по проведению надлежащих лабораторных
испытаний каждой, при необходимости, серии лекарственного продукта, если уста-
новлено требование отсутствия нежелательных микроорганизмов.
6.3.2.2. Правила GMP для действующих фармацевтических веществ
(фармацевтических субстанций)
Руководство ICH Q7A по GMP для действующих фармацевтических веществ [6] со-
держит предпочтения GMPb отношении микробиологического контроля при произ-
водстве фармацевтических субстанций. В руководстве ICHуказано, что если в спе-
цификациях на полупродукты, получаемые в ходе технологического процесса, или для
самой субстанции установлены требования к микробиологической чистоте, то произ-
водственные помещения для этих процессов должны быть спроектированы так, что-
бы ограничить возможное попадание нежелательных микробиологических агентов,
если это требование вообще применимо (ICH Q7A, параграф 4.1). Было предложено
использовать надлежащую вентиляцию, фильтрацию воздуха и вытяжные системы
для минимизации риска контаминации микроорганизмами (JCH Q7A, параграф 4.2).
Если необходимы более жесткие требования к воде, используемой в производ-
стве фармацевтических субстанций, то должны быть установлены соответствующие
спецификации, содержащие требования к общему количеству микроорганизмов,
нежелательным микроорганизмам и/или содержанию бактериальных эндотокси-
нов (ICH CZ4, параграф 4.2). В руководстве также указано, что если производитель
нестерильной фармацевтической субстанции или заявляет о пригодности этого ве-
щества, или предназначает его для дальнейшей обработки в производстве стериль-
ных лекарственных средств, то вода, используемая на последних стадиях выделения
и очистки субстанции, должна мониторироваться и контролироваться на общее со-
держание бактерий, нежелательных микроорганизмов и бактериальных эндотокси-
нов (ICH Q7A, параграф 4.3).
Руководство рекомендует проводить очистку оборудования, предназначенно-
го для непрерывного производства или используемого в цикличном производстве
последовательных серий одного полупродукта или готовой фармацевтической суб-
станции, в надлежащие промежутки времени для предотвращения достижения или
переноса нежелательного количества микроорганизмов (JCH Q7A, параграфы 5.2
и 8.5). В разделе по лабораторному контролю руководство ICHрекомендует в случае
наличия в спецификации на субстанцию требований к микробиологической чисто-
те разработать нормы по общему количеству бактерий, нежелательным микроор-
ганизмам и содержанию бактериальных эндотоксинов и обеспечить соответствие
этим нормам (JCH Q7A, параграф 11.1). Испытания на соответствие микробиоло-
гическим показателям должны проводиться для каждой серии полупродукта или
готовой фармацевтической субстанции, если они внесены в спецификацию (ICH
Q7A, параграф 11.2). В руководстве указано, что практическая оценка очистки или
дезинфекции оборудования должна включать оценку присутствия микроорганиз-
мов и бактериальных эндотоксинов при производстве продуктов, для которых не-
обходимо снизить общее количество микроорганизмов или бактериальных эндо-
токсинов в субстанции, например, когда нестерильная субстанция используется для
производства стерильных продуктов (ICH Q7A, параграф 12.7).
636 Часть 6 Контаминация и ее контроль
При производстве фармацевтических субстанций, получаемых из культур клеток
методами ферментации, следует учитывать, что микробной контаминации могут
быть подвергнуты исходные материалы: питательные среды и компоненты буферов.
В таких случаях, в зависимости от предназначения фармацевтических субстанций
или полупродуктов, может быть необходим контроль микробиологической чисто-
ты, вирусной контаминации и/или контаминации бактериальными эндотоксинами
в ходе технологического процесса, а также мониторинг процесса на соответствую-
щих стадиях (ICH Q7A, параграф 18.1). Если микробная контаминация может по-
влиять на качество фармацевтической субстанции, все манипуляции с открытыми
сосудами должны проводиться в ламинарах или аналогичным образом контроли-
руемых зонах (JCH Q7A, параграф 18.3).
6.3.2.3. Правила G/ИРдля фармацевтических вспомогательных веществ
В USP, в общей главе < 1078>, приведены правила GMPpjui. фармацевтических вспо-
могательных веществ [И]. В руководстве указано, что здания и помещения долж-
ны быть спроектированы таким образом, чтобы выполняемые в них операции не
влияли на имеющуюся или возможную контаминацию вспомогательного вещества.
Рекомендуется разработать и выполнять эффективную и регулярную программу
очистки используемого оборудования с целью удаления остатков продукта и гря-
зи, которые также могут содержать микроорганизмы и быть источниками конта-
минации. Далее, руководство указывает, что все оборудование, которое находилось
в контакте с контаминированным материалом, должно быть тщательно очищено
и подвергнуто дезинфекции до использования в операциях с изготавливаемым
продуктом. Может быть необходимо обеспечение контролируемой производствен-
ной среды для предупреждения микробной контаминации. Водопроводная вода,
используемая в производстве вспомогательных веществ, должна соответствовать
химическим и микробиологическим стандартам, включая отсутствие патогенных
микроорганизмов. Очищенная вода, также используемая в производстве вспомога-
тельных веществ, и системы получения очищенной воды из водопроводной (деио-
низаторы, устройства ультрафильтрации или системы обратного осмоса) могут быть
источниками роста микроорганизмов. Рекомендуется установить в спецификации
требования в отношении химического и микробиологического качества воды, ис-
пользуемой в производстве, а также проводить периодическую проверку ее соот-
ветствия этой спецификации. Если в спецификации на вспомогательное вещество
установлены требования в отношении содержания бактериальных эндотоксинов
или апирогенности, требуется проведение валидации систем получения очищенной
воды в отношении их способности производить апирогенную воду.
6.3.3. Фармакопейные требования в отношении микробиоло-
гических аспектов нестерильного производства
В Фармакопее США, Европейской и Японской фармакопеях описаны требования,
спецификации и испытания по мониторингу микробиологической чистоты не-
стерильных фармацевтических продуктов. Хотя имеются определенные различия
в фармакопейных требованиях, общие требования по мониторингу микробиологи-
ческой чистоты являются одинаковыми, как будет видно из дальнейшего текста.
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 637
6.З.З.1. Фармакопея США
В общей главе «Микробиологическая оценка нестерильных продуктов» USP [3] со-
держится руководство по микробиологической оценке различных нестерильных
лекарственных форм (табл. 1). Определение общего количества аэробных бактерий,
грибов и дрожжей в продуктах проводится методиками, описанными в общей гла-
ве USP <61> [12], а испытания на определение присутствия отдельных микроорга-
низмов (Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, Salmonella sp.,
и Candida albicans) выполняются с помощью методик, описанных в общей главе USP
<G2> [13]. Также в фармакопее приведено допустимое содержание микроорганиз-
мов (критерии приемлемости) в нестерильных лекарственных формах для различ-
ных путей введения. Дополнительно в частных фармакопейных статьях указывают-
ся определенные требования, если это вообще применимо, к микробиологической
чистоте вспомогательных или действующих веществ и лекарственных препаратов.
Общие правила надлежащей лабораторной микробиологической практики описа-
ны в общей главе USP <1117> [14]. В этих правилах описано, как внедрить данные
стандарты в лаборатории при проведении испытаний различных по составу образ-
цов. Могут использоваться также альтернативные методики, отличные от описан-
ных в USP, при условии их надлежащей валидации. Процедуры валидации альтер-
нативных методик описаны в общей главе СЖР<1223> [15].
В Фармакопее также приведено руководство по сокращенным испытаниям на
микробиологическую чистоту при выпуске продукта в обращение и при оценке
стабильности, когда лекарственные препараты имеют уменьшенную, значительно
ниже 0,75, водную активность (отношение давления водяного пара в продукте (Р)
к давлению водяного пара чистой Н2О (Ро) при одинаковой температуре). Вода не-
обходима для роста микроорганизмов в фармацевтических продуктах [16]. Однако
наиболее устойчивые микроорганизмы, включая спорообразующие Clostridium sp.,
Bacillus sp., Salmonella sp. и мицелиальные грибы, которые могут не расти в лекар-
ственном препарате с низкой водной активностью, могут присутствовать в продукте
не в вегетативном состоянии. Например, для роста грибов Xeromuces bisporus требу-
ется водная активность 0,61, для роста дрожжей Zygosaccharomyces rouxii — 0,62, в то
время как для большинства других грибов и дрожжей для роста необходима среда
с водной активностью более 0,75. Уровень водной активности 0,75 достаточен для
роста бактерий Halobacterium halobium, но для роста большинства бактерий требует-
ся более высокая водная активность.
В руководстве также приведена стратегия испытаний на микробиологическую
чистоту лекарственных препаратов с различными путями введения с учетом водной
активности. Например, для таблеток и мягких капсул с репрезентативной водной
активностью соответственно 0,36 и 0,30 рекомендуются сокращенные испытания,
а для кремов для наружного применения и назальных спреев с водной активностью
соответственно 0,97 и 0,99 — проведение испытаний на общее содержание аэроб-
ных бактерий, суммарное содержание дрожжей и грибов, присутствие 5. aureus
и Р. aeruginosa. В состав нестерильных лекарственных форм могут быть введены
антимикробные консерванты, которые препятствуют росту микроорганизмов или
обладают бактерицидным действием, обеспечивая защиту препарата от микроорга-
638
Часть 6 Контаминация и ее контроль
Таблица 1
Допустимое содержание микроорганизмов в нестерильных лекарственных формах
Лекарственные формы по нуги введения Общее содержание аэробных бактерий, КОЕ/г, КОЕ/мл Суммарное содержание грибов и дрожжей, КОЕ/г, КОЕ/мл Отдельные микроорганизмы, в 1 гили 1мл
Неводные препараты для приема внутрь 1000 100 Е. coli отсутствует
Водные препараты для приема внутрь 100 10 Е. coli отсутствует
Препараты для ректального применения 1000 100
Препараты для применения на слизистых и в ротовой полости (гингивальные, на- кожные, назальные, ушные) 100 10 5. aureus и Р. aeruginosa отсутствуют
Препараты для вагинального применения 100 10 5. aureus, Р. aeruginosa, С. albicans отсутствуют
Трансдермальные пластыри (один пластырь, включая адгезивный слой и наружную поверхность) 100 10 5. aureus и Р. aeruginosa отсутствуют
Препараты для ингаляций (кроме препаратов для небулайзеров) 100 10 5. aureus, Р. aeruginosa и желчеустойчивые грамотрицательные бактерии отсутствуют
Источник: [3].
низмов, случайно попавших в продукт в ходе производственного процесса или при
последующих операциях.
В общей главе USP <51> описано проведение испытания на антимикробную
активность консервантов [17]. В ней подчеркивается, что антимикробные кон-
серванты не должны использоваться в качестве замены выполнения правил GMP
или исключительно для снижения жизнеспособной популяции микроорганизмов
в нестерильном препарате. Рекомендуется обязательно проверять антимикроб-
ную активность консервантов для таких лекарственных форм, как препараты для
наружного применения или приема внутрь, предназначенные для многократного
использования, для офтальмологических и назальных препаратов, препаратов для
промывания, как описано в общей главе USP о лекарственных формах [18].
В USP также приведены рекомендации по воде для фармацевтического исполь-
зования [19]. В рекомендациях указывается, что основным внешним источником
микробной контаминации нерасфасованных продуктов является исходная вода.
Исходная вода (водопроводная) должна удовлетворять требованиям к качеству пи-
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 639
тьевой воды, в которой контролируется уровень содержания бактерий кишечной
группы. Другие микроорганизмы, присутствующие в исходной воде, хотя и не яв-
ляющиеся'по своей природе патогенными, могут оказать негативное влияние на по-
следующие стадии очистки. Микроорганизмы, присутствующие в исходной воде,
могут осаждаться на углеродных фильтрах, резиновых наполнителях для ионизации
и мембранных фильтрах (используемых при получении очищенной воды) и ини-
циировать образование биопленки. Микроорганизмы биопленки адаптируются
преимущественно к обедненному питательными веществами окружению. Также
микроорганизмы биопленки могут переноситься с потоком воды, когда отделяются
от существующей биопленки, и переноситься в другие части системы водоснабже-
ния. Подробная оценка образования биопленки в трубопроводах, методы выявле-
ния и количественной оценки биопленок описаны Олсоном (Olson, 1997) [20]. Кро-
ме того, микробная контаминация может возникнуть вследствие незащищенных
отверстий (входных и выходных), пришедших в негодность воздушных фильтров,
поврежденных пластин в системе водоснабжения, а также вследствие обратного по-
тока из контаминированных зон.
Несколько категорий микроорганизмов могут быть проблемными для систем
подготовки фармацевтической воды и вследствие этого для фармацевтических про-
дуктов, в производстве которых была использована вода из этих систем. К этим
категориям относятся патогенные и условно-патогенные микроорганизмы, не-
патогенные микроорганизмы, свидетельствующие о наличии необнаруженных
патогенных микроорганизмов, микроорганизмы, которые могут обладать устой-
чивостью к антимикробному консерванту, входящему в состав препарата, и микро-
организмы, вызывающие разложение действующих или вспомогательных веществ,
входящих в состав препарата. Для осуществления ранних предупреждающих дей-
ствий и предупреждения несоответствия получаемой воды для фармацевтического
применения спецификациям рекомендуется установить нормы содержания микро-
организмов для уровня тревоги и уровня действия и осуществлять мониторинг ми-
кробиологического качества воды на соответствие этим уровням согласно графику
периодической проверки. Для очищенной воды, например для уровня действий,
максимальным допустимым содержанием микроорганизмов считается 100 коло-
ниеобразующих единиц микроорганизмов в 1 миллилитре (КОЕ/мл) [19].
6.3.3.2. Европейская фармакопея
Европейская фармакопея описывает планы отбора проб и испытания для оценки
количественного содержания в лекарственных препаратах бактерий и грибов, ко-
торые могут расти в аэробных условиях [21]. Оценка содержания энтеробактерий
и некоторых других грамотрицательных бактерий, Е. coli, Salmonella sp., Р. aeruginosa,
S. aureus, Clostridia и Clostridium perfringens [22] описана в испытаниях на присутствие
отдельных видов микроорганизмов. В общем разделе, посвященном микробиоло-
гическому качеству лекарственных средств, все лекарственные формы разделены на
три категории. К категории 1 относятся стерильные препараты, которые не рассма-
триваются в данной главе. К категориям 2 и 3 относятся различные нестерильные
лекарственные формы. Например, для препаратов категории 2 (препаратов для на-
ружного применения) выполняются испытания на общее содержание аэробных бак-
640
Часть 6 Контаминация и ее контроль
терий и грибов, энтеробактерий и других грамотрицательных бактерий, Р. aeruginosa
и S. aureus. Для препаратов категории ЗА (препаратов для приема внутрь и ректаль-
ных) выполняются испытания на общее содержание аэробных бактерий и Е. coli,
в то время как для препаратов категории ЗВ, если они содержат вещества природ-
ного происхождения, проводятся испытания на общее количество жизнеспособ-
ных аэробных бактерий, нитробактерий и других грамотрицательных бактерий,
Salmonella sp., Е. coli и S. aureus. Особые требования к микробиологической чистоте,
если они необходимы, также детально описываются в каждой частной фармако-
пейной статье. Рекомендуется проведение работ по оценке эффективности анти-
микробных консервантов везде, где они используются, например, в многодозовых
препаратах, если имеется риск микробной контаминации, в водных препаратах или
препаратах для местного применения, которые также подвержены риску микроб-
ной контаминации из-за большого содержания воды в этих лекарственных фор-
мах. В общем разделе по оценке эффективности антимикробных консервантов [24]
описаны принципы их использования и испытания по оценке их эффективности.
6.3.3.3. Японская фармакопея
В общем разделе по микробиологическим показателям качества нестерильных ле-
карственных препаратов Японская фармакопея указывает, что присутствие микроб-
ных агентов в нестерильной продукции [25] может уменьшать или инактивировать
терапевтический эффект препарата и может оказывать нежелательное воздействие
на состояние больных, и рекомендует производителям обеспечить по возможности
минимальное содержание микроорганизмов в готовой продукции. Установлено
допустимое содержание микроорганизмов в исходных материалах (общее количе-
ство аэробных микроорганизмов и общее количество дрожжей и плесневых грибов)
и в лекарственных формах. Для препаратов для ингаляций, назальных препаратов
и препаратов для местного применения рекомендовано проведение испытаний на
общее количество аэробных микроорганизмов, общее содержание дрожжей и плес-
невых грибов, Р. aeruginosa и S. aureus. Для вагинальных препаратов рекомендовано
проведение испытаний на общее количество аэробных микроорганизмов, общее
содержание дрожжей и плесневых грибов, Е. coli, S. aureus и С. albicans, в то время
как для жидких и твердых лекарственных форм для приема внутрь рекомендуется
проведение испытаний на общее количество аэробных микроорганизмов, общее со-
держание дрожжей, плесневых грибов и Е. coli. Требуется проведение микробиоло-
гической оценки антимикробных консервантов, входящих в состав препарата, с це-
лью контроля микробиологической чистоты. Тестовые микроорганизмы и методы,
используемые для оценки эффективности антимикробных консервантов, описаны
в общем разделе по испытаниям их эффективности [26].
6.3.4. Действующие ожидания регуляторных органов
в отношении контроля микробиологической чистоты
при нестерильном производстве
Действующие ожидания регуляторных органов в отношении микробиологической
чистоты нестерильных лекарственных средств были изучены с использованием ру-
ководства EDA по инспектированию микробиологических лабораторий ОКК [2],
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 641
систем получения очищенной воды [27], производств препаратов для местного при-
менения [28] и растворов и суспензий для приема внутрь [29].
В руководстве по инспектированию микробиологических лабораторий ОКК [2]
рассмотрен ряд проблем, связанных с микробной контаминацией препаратов для
местного применения, назальных растворов и препаратов для ингаляций. Предста-
витель FDA Данниган {Dunnigan) подчеркивал, что препараты для местного приме-
нения, контаминированные грамотрицательными микроорганизмами, представля-
ют вероятный риск от средней до тяжелой степени для здоровья больных.
В руководстве [28] по инспектированию производства препаратов для местного
применения указано, что деионизаторы воды являются, как правило, отличным ме-
стом для роста микроорганизмов, и на их рост влияют скорость потока, температу-
ра, площадь поверхности картриджей и микробиологические показатели исходной
воды. Поскольку препараты для местного применения (например, кремы и мази)
обычно содержат очищенную воду, микробиологическое качество продукта зависит
от микробиологической чистоты очищенной воды. В руководстве также указано,
что необходимо проводить оценку значимости микробной контаминации, включая
идентификацию найденных микроорганизмов и определение их количества. Высо-
кое содержание непатогенных микроорганизмов может оказать негативное воздей-
ствие на эффективность и/или физическую и химическую стабильность продукции.
Кремы для местного применения имеют тенденцию к расслоению при хранении. Для
препаратов для местного применения должны иметься данные, подтверждающие
эффективность антимикробных консервантов на всем протяжении срока годности.
Для производства нестерильных препаратов, как правило, используется очищен-
ная вода. Руководство по инспектированию систем получения высокоочищенной
воды [27] в основном посвящено микробиологическим аспектам; в нем указано,
что для очищенной воды нормы содержания микроорганизмов более 100 КОЕ/мл
для уровня действия являются недопустимыми. Цель установления норм для уров-
ня действия — обеспечение контроля системы водоподготовки. Очищенная вода,
используемая в производстве лекарственных средств, не должна содержать «неже-
лательные микроорганизмы», под которыми подразумеваются микроорганизмы,
способные привести к развитию инфекционного процесса при применении лекар-
ственного средства по показаниям. Это определение также включает микроорга-
низмы, которые способны расти в препарате.
Микроорганизмы могут присутствовать в системах водоподготовки и быть сво-
бодно перемещающимися с потоком воды или зафиксированными на стенках труб
и емкостей (биопленка). Из биопленки микроорганизмы постоянно высвобождаются
и поступают в восходящий или нисходящий потоки воды в системе. В руководстве под-
черкивается, что при установлении допустимых норм содержания микроорганизмов
в системах подготовки воды, используемой для производства нестерильной продук-
ции, необходимо учитывать назначение продукта, состав (наличие антимикробных
консервантов) и производственный процесс. При производстве антацидных препа-
ратов, не содержащих эффективных антимикробных веществ, требуется установле-
ние нормы содержания микроорганизмов менее 100 КОЕ/мл для уровня действия.
Также в этом руководстве подчеркнуто, что оборудование, используемое в систе-
мах получения очищенной воды, может быть подвержено контаминации. Насосы,
642
Часть 6 Контаминация и ее контроль
используемые в системах водоподготовки, при отсутствии непрерывной эксплуа-
тации могут содержать «стоячую» воду, которая является местом роста микроорга-
низмов. Сообщалось, что источником контаминации системы водоподготовки бак-
терией Pseudomonas sp. был насос, который эксплуатировался лишь периодически.
Также источниками микробной контаминации в системах водоподготовки являют-
ся тупиковые части труб и места соединений. В фармацевтических системах водо-
подготовки не должны использоваться резьбовые соединения труб, поскольку они
способствуют росту микроорганизмов. Еще одним источником контаминации яв-
ляются фильтры, так как микроорганизмы имеют тенденцию накапливаться и расти
на их поверхности. Хотя использование фильтра с диаметром пор 0,2 мкм в точках
разбора может маскировать уровень микробной контаминации, он не всегда может
препятствовать накоплению эндотоксинов, поскольку токсины таким фильтром не
задерживаются. Для предупреждения контаминации следует производить частую
замену фильтров.
При контроле качества воды управление FDA обнаружило присутствие
Pseudomonas sp., и эти же виды бактерий были найдены FDA в гормональном пре-
парате для местного применения [27]. В результате проверки продукт был отозван
с рынка, a FDA выпустило письмо-предупреждение. Было установлено, что основной
причиной микробной контаминации являлась незамкнутая система водоподготов-
ки, в которой для контроля микробной контаминации использовалось ультрафио-
летовое облучение, при этом УФ-облучатель включался только при получении воды,
а когда он не работал, стерилизация была неэффективной. В результате «стоячая»
вода стала источником роста микроорганизмов. Также в рассматриваемом случае
дополнительным источником роста микроорганизмов был гибкий трубопровод, ис-
пользовавшийся в этой системе, для которого было сложно провести дезинфекцию.
В руководстве по инспектированию производства растворов и суспензий для
приема препаратов внутрь [29] указано, что микробная контаминация некоторых
жидких препаратов для приема внутрь может представлять значительный риск для
здоровья. Например, микробная контаминация грамотрицательными микроорга-
низмами недопустима для некоторых препаратов, особенно если они предназна-
чаются для новорожденных детей и больных с иммунодефицитными состояниями.
В жидких препаратах, таких как антацидные препараты, не допускается присутствие
Pseudomonas sp. В целом контаминация любых препаратов грамотрицательными
бактериями нежелательна. Такая контаминация может свидетельствовать о неудо-
влетворительном технологическом процессе, неадекватной системе антимикроб-
ных консервантов и возможном использовании контаминированных исходных ма-
териалов.
6.3.5. Промышленные перспективы контроля
микробиологической чистоты для нестерильного
фармацевтического производства
В мартовском номере за 1997 г. журнала «Фармацевтическая технология»
(Pharmaceutical Technology) рабочая группа по мониторингу рабочей среды PhRMA
опубликовала статью по микробиологическому мониторингу условий рабочей сре-
ды для нестерильного производства [14]. Эта публикация представляет собой ком-
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 643
пиляцию результатов опроса производителей с нестерильным производством на
территории США и рекомендации, сделанные на основе этого опроса, В данном
разделе рассматриваются обобщенные результаты опроса и рекомендации, опу-
бликованные в этой статье, и приводятся комментарии автора исходя из его опыта
в этой области.
6.З.5.1. Результаты опроса
Большинство производителей мониторировали рабочую среду нестерильного фар-
мацевтического производства. Компании, производящие препараты для местного
применения, жидкие формы и аэрозоли, как правило, имели более обширные про-
граммы мониторинга, чем компании, производящие таблетки или другие твердые
дозированные лекарственные формы. В программы мониторинга нестерильных
фармацевтических производств входили: воздух, технологическое оборудование
и вода. Качество воздуха оценивалось с помощью приборов для отбора проб с цен-
тробежным насосом, импакторов и т. д. Поверхности оборудования, вступающие
в контакт с продуктом, мониторировались с использованием методов мазка и кон-
тактных пластин; при этом метод мазка считается более предпочтительным, так
как обеспечивает личный доступ к неплоским поверхностям. Поскольку исполь-
зующиеся в этом методе тампоны подвергаются предварительной стерилизации,
они в меньшей степени могут стать источником контаминации для проверяемой
поверхности, в то время как контактные пластины, содержащие питательную сре-
ду с агаром, могут способствовать росту микроорганизмов после отбор пробы, если
проверяемая поверхность не была тщательно обработана этанолом или другим сте-
рилизующим раствором. Однако контактные пластины проще использовать для
плоских поверхностей, и, кроме того, они обеспечивают получение дополнитель-
ной информации для оценки микробиологической картины in situ.
Компании, имеющие программы микробиологического мониторинга, установи-
ли нормы для уровня тревоги и уровня действия. Содержание микроорганизмов на
уровне тревоги может свидетельствовать об отклонении от обычных установленных
операций, но может не требовать проведения каких-либо корректирующих меро-
приятий, в то время как содержание микроорганизмов на уровне действия пред-
полагает существенное отклонение от нормальных установленных операций и тре-
бует проведения корректирующих мероприятий для контроля дальнейшего роста
микроорганизмов.
6.3.5.2. Рекомендации
Особые рекомендации. Рабочая группа рекомендовала не устанавливать обязатель-
ным повседневный микробиологический мониторинг рабочей среды нестериль-
ных производств, но определять его необходимость в зависимости от типа произ-
водимой нестерильной лекарственной формы. Процедуры уборки и дезинфекции,
графики генеральных уборок, тщательно разработанные программы внутрипроиз-
водственного контроля, качество исходных материалов, планировка производства
и его контроль, а также обучение персонала, участвующего в производственном
процессе, может помочь в обеспечении требуемого уровня микробиологической
чистоты. Установленная программа мониторинга может обеспечить получение дан-
644
Часть 6 Контаминация и ее контроль
них в долгосрочной перспективе, а такие данные могут помочь в идентификации
отклонений, анализе первопричин проблем, решении проблем и принятии ранних
и эффективных мер. По степени критичности в отношении микробиологической
чистоты среди нестерильных лекарственных форм наиболее критичными являют-
ся препараты для ингаляций, затем жидкие препараты и препараты для местного
применения, а наименее критичными — твердые лекарственные формы для при-
ема внутрь. При введении программ мониторинга отбор проб должен включать те
зоны, вероятность контаминации которых наиболее высока, например поверхности
оборудования, контактирующие с продуктом, системы вентиляции, газы, исполь-
зуемые в технологическом процессе, очищенная вода, системы водоподготовки.
Частота отбора проб может быть определена на основании выявленных тенденций
в архивных данных и типа производимых лекарственных форм с учетом чувстви-
тельности выпускаемых препаратов к микробной контаминации. Технологический
процесс, очистка, уборка и валидация вспомогательных систем должны включать
отбор проб для микробиологических испытаний с целью контроля микробиологи-
ческой чистоты. Допустимое время хранения в ходе технологического процесса, на-
пример, растворов для покрытия и влажного гранулята должно быть подтверждено
с помощью отбора и испытаний данных объектов на микробиологическую чистоту
в ходе валидации и последующего производства.
Общие рекомендации. Соответствие требованиям правил GMP, приведенных
в главе 211 тома 21 CFR [1], является наиболее эффективным способом контроля
микробиологической чистоты рабочей среды нестерильного производства (как
описано в разделе 6.3.2.1). Должны быть введены соответствующие письменные
процедуры по контролю микробиологической чистоты и предупреждению конта-
минации нежелательными микроорганизмами. Микробиологи должны быть хоро-
шо обучены процедурам отбора проб и методикам контроля рабочей среды фарма-
цевтического производства с целью исключения контаминации образцов продукта
или проб при мониторинге рабочей среды в ходе отбора проб и во время испыта-
ний. Идентификация выделенных микроорганизмов должна проводиться, если это
требование установлено, когда содержание микроорганизмов находится на уровне
действия (или выше установленных норм) при выполнении испытаний на микро-
биологическую чистоту.
6.3.6. Контроль микробиологической чистоты в течение срока
годности фармацевтических продуктов
Правилами GMP требуется проведение работ по стабильности лекарственных
средств для оценки неизменности продукта на протяжении всего срока годности
[1, 6, 11]. В руководстве ICH Q1A (R2) «Оценка стабильности фармацевтических
субстанций и лекарственных препаратов» [30] подчеркивается, что работы по ста-
бильности должны включать оценку, если применимо, физических, химических,
биологических и микробиологических показателей качества. Дополнительно в ис-
следованиях стабильности лекарственных препаратов, в состав которых входят ан-
тимикробные консерванты, должно оцениваться содержание консервантов в тече-
ние срока хранения.
Глава 6.3. Микробиологические аспекты нестерильного фармацевтического производства 645
6.3.7. Заключение и выводы
В производстве нестерильных препаратов обеспечение требуемого уровня микро-
биологической чистоты с помощью адекватной планировки производственных по-
мещений и выполнения правил GMP поможет обеспечить надлежащее качество ис-
ходных материалов и готовых лекарственных препаратов по микробиологическим
показателям. Правила GMP для готовых препаратов и их компонентов (действую-
щих и вспомогательных веществ) обсуждались в этой главе с целью рассмотрения
ожиданий в отношении контроля контаминации в ходе производства. Для выпол-
нения ожиданий, связанных с выполнением правил GMP в отношении контроля
контаминации, и проведения испытаний, необходимых для осуществления такого
контроля, фармакопеи США, Европы и Японии содержат описания методов ис-
пытаний и действий по определению микробиологической чистоты; эти методы
и действия здесь также были кратко перечислены. Производители лекарственных
средств с помощью своих профессиональных ассоциаций приняли проактивный
подход к обеспечению уровня микробиологической чистоты в нестерильном про-
изводстве, опубликовав результаты опроса компаний-членов ассоциаций и разра-
ботав общие рекомендации по внедрению контроля микробиологической чистоты
для этих компаний. В главе приведено краткое описание результатов опроса, про-
веденного рабочей группой, по действующей отраслевой практике и выработанные
общие рекомендации. Также внимание читателей обращено на обязательное требо-
вание мониторинга микробиологической чистоты на протяжении срока годности
лекарственных средств.
Литература
1. U.S. Food and Drug Administration (FDA) (2007), Current good manufacturing practice regulations,
21 CFR Parts 210 and 211, FDA, Rockville, MD.
2. U.S. Food and Drug Administration (FDA) (1993), Guide to inspections of microbiological pharma-
ceutical quality control laboratories, FDA, Rockville, MD.
3. U.S. Pharmacopeia (USP) (2007), <1111> Microbiological examination of nonsterile products: Ac-
ceptance criteria for pharmaceutical preparations and substances for pharmaceutical use, U.S. Phar-
macopeial Convention, Rockville, MD.
4. Pharmaceutical Research and Manufacturers Association of America (PhRMA) (1997, March), Mi-
crobiological monitoring of environmental conditions for nonsterile manufacturing, Pharm. Technol.,
58-74.
5. Australian Pharmaceutical Manufacturers’ Association (АРМА) (1990), The Control of Microbial
Contamination in Nonsterile Pharmaceutical Products for Human Use, АРМА, Canberra, Austra-
lia.
6. International Conference on Harmonization (ICH) (2001), Guidance for industry. Q7A good manu-
facturing practice guidance for active pharmaceutical ingredients, ICH, Brussels, Belgium.
7. Therapeutic Goods Administration Laboratories of Australia (TGAL) (1990), Guidelinesfor Assessing
the Results ofMicrobiological Tests on Nonsterile Pharmaceuticals for Human Use, TGAL, Canber-
ra, Australia.
8. Tang, S. (1998), Microbial limits reviewed — Basis for unique Australian regulatoiy requirements for
microbial quality of nonsterile pharmaceuticals, PDA, J. Pharm. Sci. Technol., 52, 100-109.
9. World Health Organization (WHO) (2003), Good manufacturing practices for pharmaceutical prod-
ucts, main principles, WHO Technical Report Series No. 908, WHO, Geneva, Switzerland.
646 Часть 6 Контаминация и ее контроль
10. International Pharmaceutical Excipients Council (IPEC) (2006), Good Manufacturing Practices,
IPEC, Washington DC.
11. U.S. Pharmacopeia (USP) (2006), Good manufacturing practices for bulk pharmaceutical excipients,
general chapter <1078>, U.S. Pharmacopeial Convention, Rockville, MD.
12. U.S. Pharmacopeia (USP) (2007), Microbiological examination of nonsterile products: Microbial
enumeration tests <61>, U.S Pharmacopeial Convention, Rockville, MD.
13. U.S. Pharmacopeia (USP) (2007), Microbiological examination of nonsterile products: Tests for
specifi ed microorganisms <62>, U.S. Pharmacopeial Convention, Rockville, MD.
14. U.S. Pharmacopeia (USP) (2007), Microbiological best laboratory practices, general chapter <1117>,
U.S. Pharmacopeial Convention, Rockville, MD.
15. U.S. Pharmacopeia (USP) (2007), Validation of alternative microbiological methods, general chapter
<1223>, U.S. Pharmacopeial Corn ention, Rockville, MD.
16. U.S. Pharmacopeia (USP) (2007), Application of water activity determination to nonsterile pharma-
ceutical products, general chapter <1112>, U.S. Pharmacopeial Convention, Rockville, MD.
17. U.S. Pharmacopeia (USP) (2007), Antimicrobial effectiveness testing, general chapter <51>, U.S.
Pharmacopeial Convention, Rockville, MD.
18. U.S. Pharmacopeia (USP) (2007), Pharmaceutical dosage forms, general chapter <1151>, U.S. Phar-
macopeial Convention, Rockville, MD.
19 U.S. Pharmacopeia (USP) (2007), Water for pharmaceutical purposes, general chapter <1231>, U.S.
Pharmacopeial Convention, Rockville, MD.
20. Olson, P. W. (1997), Biofilms in the pipeline and in the patient, J. Pharm. Sci. Technol., 51 (6),
252-261.
21. European Pharmacopoeia (Eur. Ph.) (2005), Microbiological examination of nonsterile products (total
viable aerobic count), general chapter 2.6.12, Council of Europe, Strasbourg Cedex, France.
22. European Pharmacopoeia (Eur. Ph.) (2005), Microbiological examination of nonsterile products
(tests for specifi ed microorganisms), general chapter 2.6.13, Council of Europe, Strasbourg Cedex,
France.
23. European Pharmacopoeia (Eur. Ph.) (2005), Microbiological quality of pharmaceutical preparations,
general chapter 5.1.4, Council of Europe, Strasbourg Cedex, France.
24. European Pharmacopoeia (Eur. Ph.) (2005), Effi cacy of antimicrobial preservation, general chapter
5.1.3, Council of Europe, Strasbourg Cedex, France.
25. Japanese Pharmacopoeia (JP) (2001), Microbiological attributes of nonsterile pharmaceutical prod-
ucts, general information chapter 7, Society of Japanese Pharmacopoeia, Tokyo.
26. Japanese Pharmacopoeia (JP) (2001), Preservatives — Effectiveness tests, general information chap-
ter 12, Society of Japanese Pharmacopoeia, Tokyo.
27. U.S. Food and Drug Administration (FDA) (1993), Guide to inspections of high purity water systems,
FDA, Rockville, MD.
28. U.S. Food and Drug Administration (FDA) (2000), Guide to inspections of topical drug products,
FDA, Rockville, MD.
29. U.S. Food and Drug Administration (FDA) (1994), Guide to inspection of oral solutions and suspen-
sions, FDA, Rockville, MD.
30. International Conference on Harmonization (ICH) (2003), Guidance for industry, Q1A (R2) stability
testing of new drug substances and products, ICH, Brussels, Belgium.
Часть 7
СТАБИЛЬНОСТЬ ЛЕКАРСТВ
Глава 7.1. Стабильность и сроки годности
фармацевтических продуктов
Ранга Велагалети
BASF Corporation (Флорэм Парк, Нью-Джерси)
7.1.1. Введение
Лекарственные препараты (например, таблетки, капсулы, кремы, инъекционные
препараты и препараты для ингаляций), а также компоненты лекарственного пре-
парата, то есть активный фармацевтический ингредиент (API, фармацевтическая
субстанция, действующее вещество) и неактивные ингредиенты (вспомогатель-
ные вещества), должны сохранять стабильность в составе лекарственного препара-
та в течение указанного срока его хранения и срока хранения его индивидуальных
компонентов. Срок хранения (дата повторного тестирования или истечения срока
годности) — период времени, в течение которого вспомогательные вещества, API
и лекарственные препараты должны сохранять соответствие спецификации при
условиях хранения, указанных на упаковке. Условия стабильности и хранения вспо-
могательных веществ, API и лекарственных препаратов определяют путем оценки
параметров качества в течение определенного времени при воздействии различных
факторов окружающей среды, таких как температура, влажность и свет.
Термин «стабильность» применим кхимическим, физическим, микробиологиче-
ским, терапевтическим и токсикологическим характеристикам. Фармакопея США
(USP) [1] определяет стабильность как меру того, насколько продукт сохраняет ха-
рактеристики, которыми он обладал на момент выпуска, в течение срока годности
и применения (т. е. shelf-life — срока хранения) в пределах, указанных в специфика-
ции. Например, химическая стабильность подразумевает, что все активные ингре-
диенты лекарственного препарата сохраняют химическую целостность и заявленное
содержание активного вещества в пределах, указанных в спецификации, в течение
всего срока хранения. Физическая стабильность подтверждается, если исходные
физические характеристики, включая внешний вид, вкусовые качества, однород-
ность, растворение и суспендируемость, сохраняются в течение срока хранения.
Микробиологическая стабильность подтверждается, если стерильность или устой-
чивость к росту микроорганизмов соответствует требованиям, изложенным в спе-
цификации, а антимикробные консерванты, входящие в состав препарата, сохраня-
ют эффективность в установленных пределах. Если препарат является стабильным
в течение срока его хранения, то он должен сохранять свой терапевтический эффект
и не должно происходить значительного роста его токсичности.
В правилах надлежащей производственной практики (GMP) и ряде руководств
содержатся упоминания о важности стабильности и соблюдения сроков хранения
фармацевтических препаратов. Правила СМРрля готовой продукции, опубликован-
ные Управлением по контролю пищевых и лекарственных препаратов США (FDA),
требуют проведения исследования лекарственных препаратов для оценки параме-
650
Часть 7. Стабильность лекарств
тров стабильности; результаты исследований используют для определения надле-
жащих условий хранения и сроков годности [2]. Руководство FDA по GMP для API
требует утверждения документированной постоянной программы тестирования,
направленной на мониторинг параметров стабильности API. Полученные результа-
ты следует использовать для подтверждения приемлемости условий хранения, срока
повторного тестирования и срока годности препарата [3]. В правилах GMP, изло-
женных в Фармакопее США (USP), касающихся неупакованных фармацевтических
вспомогательных веществ, предлагается разработать документированную програм-
му тестирования, предназначенную для оценки параметров стабильности вспомо-
гательных веществ. Результаты тестирования следует использовать для определения
приемлемости условий хранения, срока повторного тестирования и срока годности
препарата [4]. В правилах GMP, изложенных в Руководстве Всемирной организации
здравоохранения (ВОЗ) [5], также упоминается о проверке стабильности готовых
препаратов и исходных материалов.
В дополнение к требованиям GMP опубликован ряд указаний, относящихся не-
посредственно к фармацевтической стабильности. Организации с международными
полномочиями, такие как ВОЗ [6] и Международная конференция по гармонизации
ICH, объединяющая, главным образом, страны Европейского Союза (ЕС), Японию
и США [7], опубликовали руководства по оценке стабильности. В руководство ВОЗ
[6] по оценке стабильности включен пакет требований к стабильности фармацевти-
ческой продукции. В дополнение к API и лекарственным препаратам, руководство
ВОЗ упоминает о необходимости изучения стабильности вспомогательных матери-
алов, которые могут содержать или накапливать реакционно-способные продукты
разложения. Руководство Q1A(R2) ICH, опубликованное FDA [7], устанавливает, что
целями тестирования стабильности является получение данных о том, как качество
фармацевтической субстанции или лекарственного препарата изменяется со време-
нем под влиянием ряда факторов окружающей среды, таких как температура, влаж-
ность и свет, и установление срока повторного тестирования фармацевтической
субстанции или срока хранения лекарственного препарата, а также рекомендации
по условиям хранения. ICH опубликовала полные серии руководств по исследова-
нию стабильности, отражающих разные аспекты стабильности: например, ICH Q1A
(R2), «Тестирование стабильности новых лекарственных веществ и препаратов» [7];
ICH QIB «Тестирование фотостабильности новых лекарственных веществ и препа-
ратов» [8]; ICH QIC, «Тестирование стабильности новых дозированных форм» [9];
ICH Q1D «Брекетинг и построение матриц при тестировании стабильности новых
фармацевтических субстанций и препаратов» [10]; ICH Q1E, «Оценка данных по
стабильности» [11]. Некоторые из этих руководств признаны и опубликованы FDA
(см. литературу в конце главы).
В этой главе речь идет о стабильности и сроках хранения фармацевтических
вспомогательных материалов, API и готовых лекарственных препаратов с учетом
информации, содержащейся в правилах и руководствах по GMP, а также в техни-
ческих и нормативных руководящих документах, освещающих вопросы стабиль-
ности. В них обсуждается значение исследований фотостабильности для разработки
методик определения стабильности, вопросы выбора соответствующей упаковки,
обеспечивающих стабильность лекарственного средства, поступившего на рынок,
Глава 7.1, Стабильность и сроки годности фармацевтических продуктов
651
в течение всего срока хранения. Далее обсуждаются вопросы, связанные с анализом
при оценке стабильности и оценкой полученных результатов для определения сро-
ка хранения продукции. Для оценки фармацевтической субстанции по показателю
«Количественное содержание» и количества продуктов разложения и примесей, на-
копленных в течение срока хранения, подчеркивается необходимость применения
аналитических методик, пригодных для определения стабильности и прошедших
валидацию. Обсуждаются рекомендуемые условия хранения продукции для различ-
ных климатических зон. Определены параметры/характеристики, применяемые для
оценки стабильности различных лекарственных дозированных форм. Механизмы и
пути разложения фармацевтических препаратов в данной главе не обсуждаются.
7.1.2. Требования к стабильности в нормативах/руководствах
по GMP
7.1.2.1. Готовые препараты
Правила GMP требуют оценки параметров стабильности лекарственного препарата.
Принципы оценки представлены в 21 CFR 211.166 (а)(Ь) [2]. В данном параграфе
приведен их краткий обзор. Пробы, отобранные для оценки стабильности лекар-
ственного препарата, должны храниться в системе упаковка/укупорочное средство
в первичной упаковке, аналогичной той, в которой препарат поступает на рынок.
Должны быть выбраны соответствующие условия хранения образцов, предназна-
ченных для оценки стабильности. Применяемые аналитические методики должны
быть надежными, избирательными и информативными. Если лекарственный пре-
парат применяется в восстановленной форме согласно указаниям на этикетке, необ-
ходимо проводить тестирование материала на соответствие параметрам стабильно-
сти, указанным в спецификации, до и после восстановления препарата. Результаты
тестирования стабильности используют для определения надлежащих условий хра-
нения и сроков хранения препарата. Оценки стабильности препарата должны ба-
зироваться на статистической оценке каждого параметра стабильности и быть до-
стоверными. Срок хранения, дата истечения срока годности и условия хранения,
определенные для лекарственного препарата, нужно подтверждать исследованием
стабильности при долгосрочном хранении и тестированием соответствующего ко-
личества производственных серий. Когда заявки на регистрацию лекарственных
веществ представляют в FDA, исследования стабильности при долгосрочном хра-
нении могут продолжаться. Возможно, что данные, подтверждающие длительность
полного предполагаемого срока хранения, дату истечения срока годности или усло-
вия хранения, еще не доступны. В подобных случаях возможно определение пред-
полагаемых дат истечения сроков годности на основании ускоренных исследований
стабильности, в ходе которых скорость химического разложения или физических
изменений повышается за счет изменений условий хранения — температуры и
влажности. Кроме того, при определении срока хранения препарата важно собрать
всю доступную информацию о параметрах стабильности известных физических и
химических характеристик вспомогательных материалов, API и систем упаковка/
укупорочное средство. Установленные таким образом предполагаемые даты истече-
ния срока годности должны быть подтверждены исследованиями стабильности при
долгосрочном хранении.
652 Часть 7. Стабильность лекарств
7.1.2.2. Вспомогательные материалы
В правилах GMP для коммерческих фармацевтических вспомогательных материалов
[4] подчеркивается: стабильность вспомогательных веществ является важным фак-
тором, вносящим вклад в стабильность готовой дозированной формы. Изменения
исходных материалов в процессе технологической обработки вспомогательных ве-
ществ могут повлиять на их стабильность. Кроме того, упаковка (система упаковка/
укупорочное средство) вспомогательных материалов весьма разнообразна и вклю-
чает без ограничения металлические и пластиковые цилиндрические контейнеры,
пластиковые бутыли и автоцистерны, которые могут повлиять на стабильность пре-
парата, находящегося внутри. Характеристики стабильности вспомогательного ма-
териала следует оценивать при надлежащих условиях хранения через определенные
интервалы времени; тестирование необходимо проводить для определения соответ-
ствия параметрам стабильности, указанным в сертификате. Результаты упомянутого
тестирования используют для определения надлежащих условий хранения и сроков
повторного контроля (ретеста) или дальнейшего тестирования. Пробы необходимо
исследовать в назначенный день повторного тестирования или накануне, для того
чтобы подтвердить соответствие продукта спецификации и, таким образом, при-
годность для дальнейшего использования. Программа исследования стабильности
вспомогательного материала должна быть непрерывной, документированной про-
токолом тестирования стабильности, включающим номера серий, тестированных
в течение года, размеры проб, интервалы тестирования, условия хранения (напри-
мер, температура, влажность) и методики тестирования стабильности препарата.
Материалы, из которых изготовлены упаковки и укупорочные устройства, приме-
няемые для хранения проб, предназначенных для исследований стабильности (если
упаковки и укупорочные устройства меньше, чем применяемые для упаковки пре-
парата, поставляемого на рынок), должны быть аналогичны материалам упаковки
продукта, поставляемого на рынок. Время и условия хранения должны соответство-
вать условиям, которые, вероятно, будут преобладать во время предполагаемого хра-
нения. Для вспомогательных материалов различных марок (категорий), например,
полимеров различной молекулярной массы или различными соотношениями мо-
номеров, или выпускаемых в виде различных смесей с другими вспомогательными
веществами, или в разной степени измельчения, может быть применен модельный
подход. Данные по стабильности продукта, полученные при помощи модели, мож-
но использовать для оценки теоретической стабильности аналогичных продуктов.
Кроме того, для определения сроков хранения, повторного тестирования и условий
хранения вспомогательных материалов, присутствующих на рынке в течение долго-
го времени, можно использовать ранее полученные данные [4].
7.1.2.З. Активные фармацевтические ингредиенты
В правилах GMPpjis.API [3] содержится требование наличия программы докумен-
тированного тестирования параметров стабильности API при хранении на соответ-
ствие параметрам, указанным в утвержденных спецификациях. Результаты иссле-
дований стабильности используются для определения условий хранения, а также
сроков повторного тестирования и даты истечения срока годности. Когда речь идет
об API, термин «дата повторного тестирования» применяется чаще по сравнению
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
653
с термином «дата истечения срока годности». Кроме того, в руководстве по GMP
подчеркивается, что методики анализа (определение лекарственных препаратов и
продуктов разложения) проб, предназначенных для исследования стабильности,
должны пройти валидацию и достоверно выявлять стабильность веществ. Методика
количественного определения, выявляющая стабильность, определяется как «мето-
дика количественного анализа, прошедшая валидацию, при помощи которой мож-
но выявлять изменения соответствующих характеристик действующего вещества и
точно измерять его содержание без мешающего действия со стороны продуктов раз-
ложения, примесей, привнесенных в ходе технологического процесса, или других
потенциальных примесей» [12].
Руководство по GMPb части, посвященной API, также подчеркивает, что пробы,
предназначенные для исследований стабильности, должны храниться в упаковках
(система компонентов упаковки, содержащей и защищающей API или лекарствен-
ные формы из него), подобных системам упаковка/укупорочное средство, в кото-
рых продукт поставляется на рынок, и в условиях хранения, определенных в руко-
водстве FDA по стабильности [7]. Руководство по GMP требует, чтобы первые три
коммерческие производственные серии были включены в программу тестирования
стабильности для подтверждения даты повторного тестирования или истечения
срока годности. Если предполагается, что API остается стабильным в течение, по
меньшей мере, двух лет, то разрешается исследовать менее трех производственных
серий. Программа исследования стабильности должна быть постоянно действую-
щей: по меньшей мере, одну серию API, произведенную в течение года, тестируют
ежегодно для подтверждения стабильности; тестирование API с меньшими сроками
хранения проводится чаще.
Правила GMP ВОЗ для лекарственных средств [5] требуют, чтобы стабильность
готовых лекарственных средств и при необходимости исходных материалов и полу-
продуктов оценивалась службой контроля качества. Согласно данному руководству,
необходимо разработать письменную программу оценки стабильности и реализо-
вать ее на практике для получения данных по стабильности лекарственных средств.
Даты окончания срока годности и требования по условиям хранения должны быть
основаны на данных по стабильности, полученных при определенных условиях хра-
нения. Руководство ВОЗ также устанавливает, что стабильность следует определять
до поступления продукции на рынок и после внесения существенных изменений,
например, в технологический процесс, оборудование и упаковочные материалы.
7.1.3. Требования к стабильности вспомогательных веществ
В отсутствии специальных руководств FDA и ICH (и ограниченности руководства
ВОЗ) по оценке стабильности вспомогательных материалов для анализа требова-
ний к стабильности используют руководство Фармакопеи США (USPno GMP) для
коммерческих фармацевтических вспомогательных веществ [4]. Описания требова-
ний к стабильности вспомогательных материалов представлено в параграфе 7.1.2.2.
Принципы, положенные в основу оценки данных по стабильности и определения
срока хранения (даты повторного тестирования) вспомогательных материалов, ана-
логичны принципам, описанным для API и лекарственных препаратов в разделе 7.1.7.
654 Часть 7. Стабильность лекарств
7.1.4. Требования к стабильности лекарственных веществ (API)
В руководстве для промышленности Q1A (R2) [7] содержатся требования о подаче
данных по стабильности API вместе с заявкой на регистрацию лекарства в странах
ЕС, Японии и США.
7.1.4.1. Выбор серий и первичной упаковки
Данные по стабильности должны быть собраны путем анализа образцов по мень-
шей мере из трех первых серий, произведенных в опытно-промышленном масшта-
бе с использованием того же пути синтеза и при помощи тех же технологических
процессов, что и производственные серии. Качество API, исследуемых согласно
формальной программе тестирования стабильности, должно быть аналогичным ка-
честву материала, который предполагается производить в коммерческом масштабе.
Упаковка должна быть аналогична или похожа на упаковку, предложенную для хра-
нения и поставки продукта на рынок.
7.1.4.2. Условия хранения и частота тестирования
Руководство ICH QIA (R2) [7] представляет информацию по условиям хранения и
частоте тестирования API. Выделяют четыре варианта условий хранения API: 1) об-
щие случаи, 2) продукты, предназначенные для хранения в холодильнике, 3) про-
дукты, предназначенные для хранения в морозильной камере и 4) лекарственные
вещества, предназначенные для хранения при температуре ниже — 20 °C (для этого
способа хранения специальные требования отсутствуют; указания сводятся к реко-
мендации рассматривать каждый случай в отдельности). Условия хранения пред-
ставлены в табл. 1.
Таблица 1
Условия хранения при испытании стабильности API [7]
Вид испытания Условия хранения Минимальный период, охватываемый данными при подаче заявки, мес.
При комнатной температуре (общий случай)
Долгосрочное (25 ± 2) °C, (60 ± 5) % RH или (30 ± 2) °C, (65 ± 5) % RH 12
Промежуточное (30 ± 2) °C, (65 ± 5) % RH 6
Ускоренное В холодильнике (40 ± 2) °C, (75 ± 5) % RH 6
Долгосрочное (5 ± 3) °C 12
Ускоренное В морозильной камере (25 ± 2) °C, (60 ± 5) % RH 6
Долгосрочное (- 20 ± 2) °C 12
Поставляемые на рынок API, предназначенные для хранения при комнатной темпе-
ратуре. Условиям хранения, определенным в руководящем документе [7] как общий
случай, определения в руководстве не дано. В данной главе автор интерпретирует
этот термин как хранение API, поставляемых на рынок, при комнатной темпера-
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
655
туре (в противоположность хранению в холодильнике или в морозильной камере).
Условия хранения (температура и влажность), а также продолжительность хранения
при исследовании стабильности должны соответствовать длительности стадий, сле-
дующих за производством: складское хранение, транспортировка и последующее
использование. На момент подачи заявки на регистрацию лекарства должны быть
доступны данные, охватывающие как минимум 12 мес. исследования трех первич-
ных серий. Однако тестирование должно продолжаться в течение предполагаемого
срока хранения или до даты повторного тестирования. При ускоренном исследо-
вании или, в случае необходимости, при промежуточных условиях хранения (при-
меняемых с целью умеренного повышения скорости химического разложения или
физических изменений), продукция хранится в особых условиях в течение 6 мес.
В этом случае долгосрочное хранение проводится при (25 ± 2) °C, относительной
влажности (65 ± 5) % RH или (30 ± 2) °C, (65 ± 5) % RH. Если исследование ста-
бильности при долгосрочном хранении проводится при (30 ± 2) °C, (65 ± 5) % RH,
хранения в промежуточных условиях не требуется. Если исследование стабильности
при долгосрочном хранении проводится при (25 ± 2) °C, (60 ± 5) % RH, и при этом
наблюдаются существенные изменения результатов при каждом тестировании в те-
чение 6 мес. в условиях ускоренного хранения (40 ± 2) °C, (75 + 5) % RH, следует
провести тестирование на соответствие утвержденным параметрам стабильности
в промежуточных условиях хранения — (30 ± 2) °C, (65 ± 5) % RH — и оценить ре-
зультаты по критериям значимых изменений. Руководящий документ [7] определя-
ет значимое изменение для API как изменение, приводящее к признанию препарата
не соответствующим спецификации. При необходимости проведения исследования
в промежуточных условиях хранения вместе с заявкой следует представить данные,
охватывающие как минимум 6 мес. с момента начала исследования.
Частота отбора проб и тестирования в ходе исследований стабильности при
долгосрочном хранении должны быть определены так, чтобы обеспечить накопле-
ние достаточного числа данных для построения кривой стабильности API. Соглас-
но руководству [7], тестирование рекомендуется проводить каждые 3 мес. в тече-
ние первого года испытаний; каждые 6 мес. в течение второго года испытаний и
впоследствии ежегодно в течение всего срока хранения в условиях долгосрочного
испытания. В условиях 6-месячного ускоренного испытания рекомендуется прово-
дить отбор и анализ проб через 0, 3 и 6 мес. Если в условиях ускоренного испытания
наблюдаются значимые изменения в сравнении с утвержденной спецификацией,
рекомендуется перейти к 12-месячному испытанию в промежуточных условиях,
с отбором проб и тестированием через 0, 6, 9 и 12 мес.
Поставляемые на рынок API, предназначенные для хранения в холодильнике. Если
в условиях ускоренного испытания API, предназначенных для хранения в холо-
дильнике — (5 ± 3) °C, происходят значимые изменения между 3-м и 6-м меся-
цами хранения в условиях ускоренного старения ((25 ± 2) °C, (60 ± 5) % RH)), то
предложенный период до повторного тестирования следует определять, основыва-
ясь на данных, полученных в режиме реального времени в условиях долгосрочного
хранения ((5 ± 3) °C) [7]. С другой стороны, если значимые изменения происходят
в течение 3 мес. в условиях ускоренного испытания, следует обсудить рассмотреть
возможные эффекты краткосрочных нарушений предлагаемых условий хранения,
656
Часть 7. Стабильность лекарств
указанных на этикетке, возможных при обработке или транспортировании про-
дукции. Когда значимые изменения происходят в течение первых 3 мес. в услови-
ях ускоренного хранения, в тестировании и учете последующих изменений в этих
условиях нет необходимости. Тем не менее, исследование одной производствен-
ной серии API, длящееся менее 3 мес., с более частым отбором проб в течение этих
3 мес., может оказаться необходимым для сужения периода, за который можно про-
демонстрировать стабильность API [7].
Поставляемые на рынок АРД предназначенные для хранения в морозильной ка-
мере. Для API, предназначенных для хранения в морозильной камере ((—20 ±5) °C),
определение периода до повторного тестирования должно базироваться на данных,
полученных в ходе испытания в реальном времени, полученных при долгосрочном
хранении при (—20 ± 5) °C [7]. Поскольку для API, хранящихся в морозильной ка-
мере, не предложено условий ускоренного хранения [7], рекомендуется провести
на одной производственной серии изучение стабильности при хранении при по-
вышенных температурах (+5 ± 3) или (+25 ± 2) °C в течение подходящего перио-
да времени, чтобы оценить влияния кратковременных выходов за рамки условий
хранения, указанных на маркировке, возможных при обработке или транспортиро-
вании продукции. Прочие условия хранения можно учитывать с предоставлением
объяснения причин их выбора.
7.1.4.З. Стресс-испытание и методы оценки стабильности при анализе
стабильности АР!
Следует тестировать физические, химические, биологические и микробиологиче-
ские характеристики API, которые, вероятно, подвержены изменениям в процес-
се хранения и могут повлиять на качество, безопасность и эффективность лекар-
ства. Анализ, проводимый для оценки стабильности API, должен характеризовать
стабильность в соответствии с описанными выше требованиями GMP для API [3].
Аналитические методики, применяемые для испытания стабильности вещества,
должны точно измерять содержание активного ингредиента без помех со сторо-
ны продуктов разложения, привнесенных в ходе технологического процесса при-
месей или других потенциальных примесей. Для разработки методики описания
стабильности материала необходимо провести испытания API в условиях стресс-
испытания. Стресс-испытание проводится на материале одной производственной
серии лекарственного вещества. Условия стресс-испытания могут включать кислот-
ный (например, 0,2 N НС1) и основной (например, 0,2 N NaOH) гидролиз, повыше-
ние температуры (от 40 °C — температура при ускоренном исследовании стабильно-
сти — шагами по 10 °C, например, 50, 60 °C), влажности (75% RH и выше), фотолиз
(см. раздел 7.1.6) и окисление API (например, 10% Н2О) [7]. Гидролиз представляет
собой процесс химического превращения, в котором органическая молекула RX
вступает в реакцию с водой (Н2О), в результате происходит замещение X группой
ОН. Среди различных вышеописанных условий стресс-испытания гидролиз чаще
других приводит к разложению API, поскольку хорошо известно, что амиды, слож-
ные эфиры и соли слабых кислот и сильных оснований подвержены гидролизу [13].
Фотолиз представляет собой процесс, в ходе которого химические вещества изме-
няются непосредственно под воздействием излучения, или косвенно в результате
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
657
взаимодействия с препаратами, образовавшимися под действием излучения [13].
Карбонилы, нитроароматика, N-оксид функциональные группы, группы, содержа-
щие двойную связь С—С, группы со слабой связью С—Н, сульфиды, алкены, по-
лиены и фенолы являются функциональными группами, которые могут вступать во
взаимодействие со светом [14]. Процессы окисления определяются рядом факто-
ров, в том числе температурой, светом, pH, концентрацией кислорода, примесями
(в том числе ионов металлов, например, меди, железа) и наличием компонентов
молекулы, способных к окислению [15].
Стресс-испытания должны показать, что примеси и продукты разложения ак-
тивного ингредиента не мешают количественному определению API [12]. Стресс-
испытание API, в дополнение к валидации способности аналитического метода
к описанию характеристик стабильности, может также помочь в определении путей
разложения и собственной стабильности молекулы [7].
7.1.4.4. Оценка результатов исследования стабильности АР!
Если вариабельность данных по стабильности для всех трех производственных се-
рий по измерениям проб, отобранных через различные интервалы времени, с на-
чала исследования не велика, API можно считать стабильными, и в этом случае
статистического анализа данных не требуется. Если данные указывают на разложе-
ние API с течением времени, следует определить момент, в который усредненная
кривая разложения (при односторонней доверительной вероятности 95%) пере-
секает линию критерия приемлемости. Когда в результатах наблюдаются неболь-
шие вариации между сериями, рекомендуется объединить данные для суммарной
оценки путем применения статистических тестов обработки углов наклона линий
регрессии и свободных членов уравнения регрессии (величины отрезка, отсекаемые
на оси у, zero time intercept — точка пересечения линии регрессии с координатной
осью в нулевой момент времени) для индивидуальных производственных серий.
Оценка длительности периода до повторного тестирования должна базироваться
на минимальном времени, в течение которого можно предполагать, что параметры
производственной серии соответствуют критериям приемлемости. Для тестирова-
ния уровня совпадения данных по всем сериям и при необходимости соответствия
данных по комбинированным сериям предполагаемой линии или кривой регрессии
следует использовать надлежащие статистические методы [7]. Оценка стабильности
должна охватывать содержание API, степень разложения препарата и другие соот-
ветствующие атрибуты.
7.1.4.5. Обязательства продолжать испытания стабильности
Если в комплект документов, подаваемых вместе с заявкой на регистрацию ле-
карственного продукта, входят результаты долгосрочных испытаний трех про-
изводственных серий фармацевтической субстанции на стабильность продукта,
охватывающих предполагаемый период до повторного тестирования, пострегистра-
ционного обязательства по изучению стабильности не требуется. С другой сторо-
ны, если в момент регистрации данные испытаний стабильности при долгосрочном
хранении на материале первичных производственных серий не охватывают пред-
полагаемого гарантированного периода до повторного тестирования, следует при-
658
Часть 7. Стабильность лекарств
нять обязательство по продолжению испытаний после регистрации для того, чтобы
установить точно длительность периода до повторного испытания. Если пакет реги-
страционных документов содержит данные испытаний менее трех серий, то следует
принять обязательства по продолжению этих испытаний в течение предполагаемого
периода до повторного тестирования и включить в испытание дополнительные се-
рии так, чтобы число их было не менее трех, и накапливать данные в течение всего
предполагаемого периода до повторного тестирования. Если при регистрации дан-
ные по стабильности производственных серий не были представлены, то следует
принять обязательство по проведению испытаний на стабильность при долгосроч-
ном хранении первых трех производственных серий в течение предполагаемого пе-
риода до повторного тестирования.
7.1.4.6. Формулировка условий хранения и указание длительности
хранения в маркировке
Формулировку условий хранения готовых API следует подготовить в соответствии
с национальными и региональными требованиями. Утвержденный период до по-
вторного тестирования, обоснованный данными по стабильности, должен быть
указан на сертификате анализа (СОА) и, соответственно, на маркировке упаковки.
7.1.5. Требования к стабильности лекарственных препаратов
В руководстве для промышленности Q1A (R2) [7] описаны требования к пакету
данных по стабильности, включаемых в документы, подаваемые при регистрации
лекарственных препаратов в странах ЕС, Японии и США. Информация о химиче-
ских свойствах молекулы API и путях ее разложения, полученная в ходе испытаний
стабильности вещества, должна помочь в разработке программы испытания ста-
бильности лекарственного препарата, поскольку одним из главных параметров при
определении срока хранения является стабильность API, входящего в состав лекар-
ственного препарата.
7.1.5.1. Выбор серий и первичной упаковки
Данные по стабильности препарата должны накапливаться на материале, по мень-
шей мере, трех первичных серий, при этом технологический процесс должен ими-
тировать процесс изготовления производственных серий продукции, состав и спе-
цификации качества которой аналогичны составу и качеству производственных
серий, предназначенных для рынка. В трех сериях лекарственного препарата, по
возможности, следует использовать различные серии лекарственного вещества; по
меньшей мере, две из трех серий должны быть произведены в пилотном масштабе
(серия должна быть изготовлена по процедуре, полностью репрезентативной и ими-
тирующей процедуру, применяемую при производстве полномасштабной серии);
третья серия может быть меньшего размера, если это оправдано [7].
Лекарственный препарат, предназначенный для испытаний стабильности, дол-
жен быть упакован в ту же систему упаковка/укупорочное средство, что и лекар-
ственный препарат, поставляемый на рынок; упаковки всех размеров, содержащие
лекарственные препараты со всеми предлагаемыми на рынке количествами актив-
ных ингредиентов, помещают на хранение для испытаний стабильности, если не
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
659
используется брекетинг и матричный план (построение матриц — исследование
стабильности по упрощенному варианту) в соответствии с руководством ICHпо ис-
пытанию стабильности [10].
7.1.5.2. Брекетинг и построение матриц
При испытаниях стабильности можно использовать брекетинг или построение ма-
триц [10] для исследования по сокращенной схеме, в то же время эти методы позво-
ляют получить достаточно данных по стабильности для оценки срока хранения.
При брекетинге эксперимент включает сокращенные планы хранения, исследо-
вания лекарств с различным содержанием активного вещества или конфигураций
системы упаковка/укупорочное средство. Например, исследование стабильности
на материале трех серий испытанием доз активного ингредиента 50,75 и 100 мг в 15-,
100- и 150-миллилитровых упаковках из полиэтилена высокой плотности (HDPE)
может быть заменено на исследование доз по 50 и 100 мг в 15- и 150-миллилитровых
упаковках без тестирования дозы 75 мг.
Матричный план [10] заключается в сокращении испытания путем исключения
временных точек измерения параметров образца, исследуемого для оценки стабиль-
ности. Например, сокращение количества временных точек наполовину означает,
что из каждых двух точек одна исключается из плана испытания, а при сокращении
на одну треть исключается одна точка из трех. Однако такой план должен включать
полное тестирование в исходной, 12-месячной и конечной точке 36-месячного ис-
пытания в целях оценки стабильности при хранении [10].
7.1.5.З. Выбор условий хранения и частоты тестирования
Руководство по лекарственным препаратам Q1A (R2) [7] содержит информацию
об условиях хранения и частоте тестирования лекарственных препаратов для ше-
сти предполагаемых условий хранения: 1) общий случай (комнатная температура),
2) лекарственные препараты, упакованные в непроницаемые упаковки, 3) лекар-
ственные препараты, упакованные в полупроницаемые упаковки, 4) лекарственные
препараты, предназначенные для хранения в холодильнике, 5) лекарственные пре-
параты, предназначенные для хранения в морозильной камере и 6) лекарственные
препараты, предназначенные для хранения при —20 °C (по этому способу хранения
не существует специальных указаний, кроме рекомендации рассматривать каждый
случай индивидуально). Условия хранения представлены в табл. 2.
Лекарственные препараты, предназначенные для хранения при комнатной темпе-
ратуре. Условия хранения, определенные в руководящем документе [7] как общий
случай, не описаны в руководстве; в этой главе их интерпретируют как хранение
представленного на рынке лекарственного препарата при комнатной температуре
(в отличие от хранения в холодильнике или в морозильной камере). Условия хране-
ния (температура и влажность) и длительность хранения при испытаниях стабильно-
сти должна быть достаточной для описания следующих стадий, наступающих после
изготовления препарата: хранение, транспортировка и последующее использование.
При подаче заявки на регистрацию лекарственного препарата срок долговре-
менного хранения должен составлять минимум 12 мес.; испытание проводится,
по меньшей мере, на материале трех первичных серий. Первичные серии исполь-
660
Часть 7. Стабильность лекарств
Таблица 2
Условия хранения при испытании лекарственных препаратов на стабильность [7]
Минимальный период,
Вид испытания Условия хранения охватываемый данными при подаче заявки, мес.
При комнатной температуре: -
долгосрочное (25 ± 2) °C, (60 ± 5) % RH 12
или (30 ± 2) °C, (65 ± 5) % RH 12
промежуточное (30 ± 2) °C, (65 ± 5) % RH 6
ускоренное В полупроницаемых упаковках: (40 ± 2) °C, (75 ± 5) % RH 6
долгосрочное (25 ± 2) °C, (40 ± 5) % RH или (30 ± 2) °C, (35 ± 5) % RH 12
промежуточное (30 ± 2) °C, (65 ± 5) % RH 6
ускоренное В холодильнике: (40 ± 2) °C, не более 25% RH 6
долгосрочное (5 ± 3) °C 12
ускоренное В морозильной камере: (25 ± 2) °C, (60 ± 5) % RH 6
долгосрочное (- 20 ± 5) °C 12
зуют для формального испытания стабильности, из которого получают данные по
стабильности, представляемые в заявке на регистрацию лекарственного препарата
с целью определения длительности периода до повторного тестирования или сро-
ка хранения/даты истечения срока годности. Тем не менее, при необходимости ис-
пытание должно продолжаться в течение всего предполагаемого срока хранения
и периода до повторного тестирования. В условиях ускоренного или, при необходи-
мости, промежуточного хранения испытание должно проводиться в течение 6 мес.
В этом случае долгосрочное хранение проводится при (25 ± 2) °C, относительной
влажности (60 ± 5) % RH или (30 ± 2) °C, (65 ± 5) % RH. Если исследование ста-
бильности при долгосрочном хранении проводится при (30 ± 2) °C, (65 ± 5) % RH,
хранения в промежуточных условиях не требуется. Если исследование стабильности
при долгосрочном хранении проводится при (25 ± 2) °C, (60 ± 5) % RH, и при этом
наблюдаются значимые изменения результатов при каждом тестировании в течение
6 мес. в условиях ускоренного хранения ((40 ± 2) °C, (75 ± 5) % RH), следует про-
вести тестирование на соответствие утвержденным параметрам стабильности в про-
межуточных условиях хранения ((30 ± 2) °C, (65 ± 5) % RH) и оценить результаты по
критериям значимых изменений (см. табл. 2).
Согласно руководящему документу [7], значимое изменение для лекарственного
препарата отвечает одному (или нескольким) из следующих критериев: 1) 5%-ное
отклонение результата анализа от первоначального значения или несоответствие
критериям приемлемости по активности лекарственного вещества (при примене-
нии биохимических или иммунологических процедур); 2) наличие любого из про-
дуктов разложения в количестве, не соответствующем критерию приемлемости;
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов 661
3) несоответствие критериям приемлемости по внешнему виду, физическим свой-
ствам и функциональным характеристикам, таким как, например, цвет, разделение
фаз, ресуспендируемость, спекание, твердость и доставка дозы при приеме; в усло-
виях ускоренного хранения можно ожидать некоторых изменений физических
свойств, например, размягчения суппозиториев, таяние или возможное разделение
фаз в кремах; 4) несоответствие критериям приемлемости по pH; 5) несоответствие
критериям приемлемости при растворении 12 единиц дозирования.
При необходимости испытания при хранении в промежуточных условиях дан-
ные по 6 мес. (как минимум) этого испытания должны быть представлены вместе
с заявкой.
Частота пробоотбора и тестирования при испытаниях стабильности должна быть
направлена на накопление данных в количестве, достаточном для построения про-
филя стабильности лекарственного препарата. При испытании в условиях ускорен-
ного хранения руководство [7] рекомендует проводить тестирование каждые 3 мес.
в течение первого года, 6 мес. в течение второго года и далее ежегодно в течение
предполагаемого срока хранения лекарственных препаратов, если предполагаемый
срок хранения составляет, по меньшей мере, 12 мес.
В условиях 6-месячного ускоренного испытания рекомендуется проводить отбор
и анализ проб через 0, 3 и 6 мес. Если в условиях ускоренного испытания вероятны
значимые изменения в сравнении с утвержденной спецификацией, рекомендуется
перейти к испытанию с включением четвертой точки пробоотбора. Если в условиях
ускоренного испытания определенно наблюдаются значимые изменения в сравне-
нии с утвержденной спецификацией, рекомендуется перейти к 12-месячной про-
грамме испытаний с пробоотбором и тестированием в 0, 6, 9 и 12 мес.
Лекарственные препараты, упакованные в непроницаемые упаковки. Для лекар-
ственных препаратов, упакованных в непроницаемые упаковки, создающие посто-
янный барьер, потеря жидкости или растворителя не является проблемой; для таких
препаратов испытания стабильности можно проводить в любых условиях контроли-
руемой или нормальной влажности окружающей среды.
Лекарственные препараты, упакованные в полупроницаемые упаковки. Испытания
стабильности лекарственных препаратов на водной основе, упакованных в полу-
проницаемые упаковки (упаковки, пропускающие растворитель, как правило, воду,
предотвращая потерю растворенного вещества), следует проводить в условиях отно-
сительно низкой влажности и температуры, описанных в табл. 2. Оцениваются па-
раметры стабильности, такие как потенциальная потеря воды, а также физическая,
химическая, биологическая и микробиологическая стабильность.
Если при исследовании стабильности при долгосрочном хранении, проводи-
мом в условиях (25 ± 2) °C, (40 ± 5) % RH, а при ускоренном хранении ((45 ± 2) °C,
(75 ± 5) % RH) в течение 6 мес. происходят значимые изменения, отличные от поте-
ри воды, следует провести тестирование на соответствие утвержденным в специфи-
кации параметрам стабильности в условиях промежуточного хранения ((30 ± 2) °C,
(65 ± 5) % RH) для оценки эффекта воздействия температуры 30 °C (см. табл. 2).
Сама по себе значительная потеря воды (5%-ное отклонение от первоначальной ве-
личины) в условиях ускоренного хранения не влечет необходимости тестирования
образцов в условиях промежуточного хранения, в то же время следует проводить
662
Часть 7. Стабильность лекарств
мониторинг потери воды в течение всего предполагаемого срока хранения для под-
тверждения того, что не произошло значимой потери воды в лекарственном препа-
рате при долговременном хранении при (25 ± 2) °C, (40 ± 5) % RH.
Лекарственные препараты, присутствующие на рынке, предназначенные для
хранения в холодильнике. Для лекарственного препарата, предназначенного для
хранения в холодильнике, при значимых изменениях между 3-м и 6-м месяцем ис-
пытания при ускоренном хранении ((25 ± 2) °C, (60 ± 5) % RH) предполагаемый пе-
риод до повторного тестирования следует рассчитывать на основании данных, по-
лученных в режиме реального времени при долгосрочном хранении при (5 ± 3) °C.
С другой стороны, если значимое изменение происходит в течение 3 мес. в условиях
ускоренного хранения, следует обсудить эффект кратковременных выходов за рам-
ки условий хранения, указанных в маркировке, возможных при транспортировке и
обработке. Если значимое изменение происходит в течение первых 3 мес. в условиях
ускоренного хранения, в дальнейшем хранении и тестировании нет необходимости.
Тем не менее, исследование одной производственной серии API, длящееся менее
3 мес., с более частым отбором проб в течение этих 3 мес. может оказаться необхо-
димым для сужения периода, за который можно продемонстрировать стабильность
лекарственного препарата.
Лекарственные препараты, представленные на рынке, предназначенные для хра-
нения в морозильной камере. Для лекарственного препарата, предназначенного для
хранения в морозильной камере, оценка периода до тестирования должна бази-
роваться на данных, полученных в режиме реального времени при долгосрочном
хранении при (— 20 ± 5) °C. Поскольку для API, предназначенных для хранения
в морозильной камере, не предложено условий ускоренного хранения, следует учи-
тывать результаты, полученные при хранении и испытании единственной серии при
повышенных температурах (5 ± 3) или (25 ± 2) °C в течение определенного периода
времени, для оценки эффекта кратковременных выходов за рамки условий хране-
ния, указанных в маркировке, возможных при транспортировке и обработке. При
соответствующем обосновании для оценки можно выбрать результаты, полученные
в других условиях хранения.
7.1.5.4. Стресс-испытание и методы оценки стабильности при анализе
стабильности лекарственной продукции
Необходимо исследовать физические, химические, биологические и микробиоло-
гические характеристики, содержание антимикробных консервантов и антиокси-
дантов и провести функциональное тестирование дозы (например, системы дози-
рования) лекарственного препарата, которая, вероятно, подвержена изменениям
в процессе хранения и может повлиять на качество, безопасность и эффективность
лекарства. Методика анализа, проводимого для оценки стабильности лекарствен-
ного препарата, должна пройти валидацию и характеризовать стабильность [12].
Методика исследования стабильности точно измеряет содержание активного ин-
гредиента в лекарственном препарате без помех со стороны продуктов разложения,
примесей, накопленных в ходе технологического процесса, вспомогательных ве-
ществ или других потенциальных примесей. Для разработки методики исследования
стабильности необходимо проводить стресс-испытания. Стресс-испытание (иссле-
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов 663
дования, предпринятые для оценки стабильности лекарственного вещества) можно
выполнять на лекарственном препарате, аналогично испытанию API, описанному
в разделе 7.1.4. Информация о разложении, полученная при стресс-испытании ак-
тивного ингредиента лекарственного препарата, должна демонстрировать специ-
фичность анализа, то есть примеси и продукты разложения активного ингредиента
и вспомогательных фармацевтических веществ не должны мешать количественно-
му анализу активного ингредиента в составе лекарственного препарата [12].
7.1.5.5. Оценка результатов исследования стабильности
Данные о стабильности лекарственных препаратов должны быть представлены для
всех интервалов тестирования; необходимо провести анализ физических, химиче-
ских и микробиологических характеристик, эффективности противомикробных
консервантов и антиоксидантов, а также тестирование функциональности для до-
зированных форм лекарственного препарата и сравнение рассматриваемых харак-
теристик с параметрами, утвержденными в спецификациях.
Если вариабельность данных по стабильности для всех трех производственных
серий по измерениям проб, отобранных через различные интервалы времени, с на-
чала исследования не велика, лекарственный препарат можно считать стабильным,
и в этом случае статистического анализа данных не требуется. Если данные указыва-
ют на изменение количественных характеристик {API и количество продуктов раз-
ложения, скорость растворения), следует определить момент времени, в который
усредненная кривая разложения (при односторонней доверительной вероятности
95%) пересекает линию критерия приемлемости. Когда в результатах наблюдаются
небольшие вариации между сериями, рекомендуется объединить данные для сум-
марной оценки путем применения статистических тестов к наклонам линий ре-
грессии и свободным членам уравнения регрессии (величина отрезка, отсекаемого
на оси у, zero time intercept — точка пересечения линии регрессии с координатной
осью в нулевой момент времени) для индивидуальных производственных серий.
Оценка длительности периода до повторного тестирования должна базироваться
на минимальном времени, в течение которого можно предполагать, что параметры
производственной серии соответствуют критериям приемлемости. Для тестирова-
ния уровня совпадения данных по всем сериям и, при необходимости, соответствия
данных по комбинированным сериям предполагаемой линии или кривой регрессии
следует использовать надлежащие статистические методы [7]. При оценке стабиль-
ности следует учитывать уровни содержания продуктов разложения и других со-
ответствующих характеристик. Следует проверить баланс масс, то есть проверить,
равняется ли сумма полученных аналитическим методом величин и содержания
продуктов разложения 100% первоначального значения, с учетом предельной по-
грешности применяемой аналитической методики. Подробно анализ данных по
стабильности обсуждается в разделе 7.1.7.
7.1.5.6. Обязательства продолжать испытания стабильности
Если в комплект документов, подаваемых вместе с заявкой на регистрацию лекар-
ственного препарата, входят результаты долгосрочных испытаний трех производ-
ственных серий на стабильность препарата, охватывающих предполагаемый пери-
664
Часть 7. Стабильность лекарств
од до повторного тестирования, пострегистрационного обязательства по изучению
стабильности не требуется. С другой стороны, если в момент регистрации данные
испытаний стабильности при долгосрочном хранении на материале первичных
производственных серий не охватывают предполагаемого гарантированного перио-
да до повторного тестирования (ускоренное 6-месячное испытание не проводит-
ся), следует принять обязательство по продолжению испытаний при долгосрочном
хранении после регистрации для того, чтобы установить точно срок хранения пре-
парата. Если при регистрации представляют данные по испытаниям стабильности
производственных серий, число которых меньше трех (серии изготовлены в про-
мышленном масштабе на технологическом оборудовании согласно спецификации,
включенной в заявку), следует принять обязательство о продолжении долгосроч-
ных испытаний в течение всего предполагаемого срока хранения и ускоренным
6-месячным испытаниям на материале трех производственных серий. Если в заявку
не включены данные по стабильности производственных серий, следует принять
обязательство провести испытания трех первичных производственных серий на ста-
бильность при долгосрочном хранении в течение предполагаемого срока хранения
и ускоренном хранении в течение 6 мес. Если значимые изменения происходят
в первичных сериях при хранении в промежуточных условиях, стабильность серий,
предназначенных для испытаний стабильности согласно обязательству, можно ис-
следовать в ускоренных или промежуточных условиях хранения. С другой стороны,
если значимые изменения наблюдаются в сериях, предназначенных для испытаний
стабильности согласно обязательству, в условиях ускоренного хранения, проведе-
ние испытаний стабильности в сериях, исследуемых согласно обязательству, также
необходимо. Формальный протокол стабильности необходим для первичных серий
и серий, исследуемых согласно обязательству [7].
7.1.5.7. Формулировка условий хранения и надписей в маркировке
Формулировка надлежащих условий хранения готовых лекарственных препаратов
должна соответствовать национальным и региональным требованиями и основы-
ваться на заключении по результатам испытаний стабильности. Для лекарственных
препаратов, которые нельзя подвергать замораживанию, следует подготовить спе-
циальные указания. Установленная дата истечения срока годности (срок хранения)
должна быть указана в сертификате анализа и, по возможности, на маркировке упа-
ковки [7].
7.1.6. Исследование фотостабильности
Руководство по тестированию стабильности лекарственных веществ и лекарствен-
ных препаратов [7] предлагает сделать испытание фотостабильности неотъемлемой
частью стресс-испытания. В руководстве по испытанию фотостабильности [8] ска-
зано, что внутреннюю стабильность новых лекарственных веществ и лекарственных
препаратов необходимо оценивать, чтобы показать, что воздействие света не приво-
дит к неприемлемым изменениям. Далее в руководстве [8] рекомендован системный
подход к испытанию стабильности, включающий испытание воздействия светового
излучения непосредственно на лекарственное вещество, а также испытание воз-
действия света на лекарственный препарат, извлеченный из первичной упаковки.
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
665
Материал первичной упаковки находится в контакте с лекарственным веществом
или лекарственным препаратом; на первичной упаковке присутствует маркировка.
Если изменения лекарственного препарата, хранившегося без первичной упаковки,
под воздействием света являются неприемлемыми, то следует продолжить испы-
тание препарата в первичной упаковке. Если изменения препарата, упакованного
в первичную упаковку после воздействия света, неприемлемы, следует провести ис-
пытания фотостабильности препарата, упакованного в потребительскую упаковку
(marketingpack) (комбинация первичной и вторичной, например, картонной пачки).
Если изменения препарата в рыночной упаковке под воздействием света неприем-
лемы, то конфигурацию упаковки необходимо поменять или же изменить состав
препарата, включив в процесс изменения проведение испытания фотостабильности.
Руководство по испытанию фотостабильности [8] представляет рекомендации
по выбору источников света, воздействующего на API или лекарственный препарат.
Предложено два способа экспонирования. Способ 1-й включает воздействие свето-
вого излучения, описанного в документе Международной организации по стандар-
тизации (ISO) 10977, соответствующим стандартам для наружного дневного света
(D 65) или внутреннего непрямого света (ID 65). Источники света в 1-способе —
флуоресцентные лампы искусственного дневного света с комбинированным вы-
ходом в УФ- и видимой области, ксеноновые или металлогалогенные лампы. Со-
гласно 2-му способу рекомендуется подвергать образец облучению светом белой
холодной флуоресцентной лампы, а также лампы, излучающей в ближней УФ-
области. Белая холодная флуоресцентная лампа должна соответствовать параме-
трам, указанным в ISO 10977. Рекомендуется, чтобы флуоресцентная лампа, излу-
чающая в ближней УФ-области со спектральным распределением от 320 до 400 нм
с максимальной эмиссией между 350 и 370 нм, использовалась со значительной
долей УФ в двух полосах, а именно 320—360 и 360—400 нм. Для подтверждающих
испытаний образцы подвергают воздействию света от источника, обеспечивающе-
го общую освещенность не менее 1,2 млн люкс/ч и общий поток энергии в ближ-
ней УФ-области не менее 200 В/м2. Рекомендовано использование химической
актинометрической системы, прошедшей валидацию, для подтверждения того,
что образцы подвергают желательному световому облучению путем использования
актинометрических растворов, расположенных рядом с лекарственным веществом
или лекарственным препаратом. Световое облучение также следует измерять при
помощи калиброванных радиометров или фотометров. Рядом с облучаемыми об-
разцами следуем поместить «темный контроль» — образец, накрытый алюминие-
вой фольгой для защиты от светового излучения. В списке литературы приведены1
важные ссылки на химические актинометры [16], межлабораторные испытания,
проводимые в промышленности и FDA [17], и мнения научных сотрудников FDA
на перспективы исследования фотостабильности [18] фармацевтических препара-
тов. Кроме того, представлена ссылка на работу Фармера с соавт. (Farmer [et al.])
[19], проводивших испытание стабильности субстанции ибупрофена и изготовлен-
ных из нее таблеток в стрессовых условиях (включавших и воздействие света) и
1 Руководство FDA никаких указанных ссылок не содержит — оно 1997 г. По-видимому,
авторы имеют в виду список литературы к разделу. — Примеч. перев.
666
Часть 7. Стабильность лекарств
показавших различия процессов разложения ибупрофена в виде чистого API и ибу-
профена в ассоциации со вспомогательными веществами в составе лекарственного
препарата. Кроме того, представлена ссылка на исследование, изучавшее влияние
различных источников света и интенсивности освещения на разложение светочув-
ствительного соединения валерофенона [20].
7.1.6.1. Фотостабильность АР!
Изучение фотостабильности лекарственного вещества может помочь получить об-
щую оценку чувствительности лекарственного вещества к свету. Поскольку уровень
воздействия согласно вышеописанным способам 1 и 2 может привести к самым се-
рьезным последствиям (форсированная деградация или light stress), вероятно, будет
наблюдаться усиленное разложение, в особенности если лекарственное вещество
содержит функциональные группы, чувствительные к фотоокислению. Если разло-
жение API происходит в ходе испытания фотостабильности, можно воспользовать-
ся возможностью разработки методики оценки стабильности. Фотостабильность
лекарственного вещества может быть оценена при исследовании его чистой формы
или растворов с образцами, помещенными в химически инертные и прозрачные
упаковки для экспонирования (например, из кварцевого стекла). В зависимости
от известных данных по фоточувствительности вещества (например, основанных
на наличии функциональных групп, подверженных окислению, или известных па-
раметрах поглощения, поскольку в диапазоне 290—800 нм, вероятно, происходит
разложение соединений [13]), можно регулировать интенсивность света и длину
волны при испытании в условиях форсированного разложения. Кроме того, важно
различать влияние температуры на разложение лекарственного вещества и свето-
вые эффекты, поскольку повышение температуры, сопровождающее воздействие
света, может независимо ускорить разложение. Необходим надлежащий темпера-
турный контроль. Изменения физического состояния, такие как плавление, субли-
мация и испарение, следует минимизировать. Для испытаний фотостабильности
рекомендуется использовать емкости, изготовленные из кварцевого стекла. Твер-
дое API помещают в емкость в количестве, достаточном для формирования слоя
высотой 3 мм. Жидкости можно экспонировать в специальных плотно закрытых
емкостях. Следует применять соответствующий «темный контроль» (контейнеры
или емкости, обернутые алюминиевой фольгой и помещенные рядом с облучаемы-
ми образцами). Анализируют как экспонированные образцы, так и «темный кон-
троль», причем анализ следует проводить одновременно. Изучаемые параметры
могут включать физические свойства, такие как внешний вид и цвет для твердых
веществ или, если экспонируют раствор, прозрачность и цвет раствора. Исследова-
ние и определение продуктов разложения следует проводить при помощи методов
оценки стабильности, прошедших валидацию, позволяющих четко отделить про-
дукты разложения и примеси от лекарственного вещества на хроматограммах об-
разцов.
Следует запланировать подтверждающие испытания, позволяющие определить
меры предосторожности для защиты API от света во время разработки или изготов-
ления лекарственного препарата, особенно если обнаруживается, что лекарствен-
ное вещество склонно к разложению под воздействием света. Эти испытания также
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов 667
помогают определить подходящий первичный и вторичный упаковочный материал
для коммерческой упаковки API.
7.1.6.2. Фотостабильность лекарственных препаратов
Лекарственный препарат подвергают воздействию светового излучения, как опи-
сано выше. Тестирование проводится последовательно: сначала полностью экспо-
нированный лекарственный препарат, далее препарат в первичной упаковке, а за-
тем препарат в коммерческой упаковке [8]. Результат испытания фотостабильности
должен продемонстрировать, что лекарственный препарат надлежащим образом за-
щищен от света во время хранения. В испытание должна быть включена одна серия
на этапе разработки с последующим подтверждением параметров фотостабильно-
сти на материале одной серии. Дополнительные испытания на материале двух про-
изводственных серий следует проводить, если результаты подтверждающего испы-
тания одной серии аналогичны результатам, полученным на этапе разработки.
В ходе подтверждающих испытаний необходимо установить характеристики
фотостабильности лекарственного препарата в стандартных условиях. Результаты,
полученные на этих этапах, позволяют: 1) определить необходимые меры предосто-
рожности, которые надо соблюдать при производственных и упаковочных опера-
циях; 2) определить конструкцию упаковки/укупорочного устройства для защиты
препарата от света и 3) описать условия хранения и способы защиты от света во
время хранения препарата, представленного на рынке. Наряду с обеспечением по-
стоянного уровня облучения, в течение экспозиции необходимо поддерживать со-
ответствующую температуру образцов, чтобы наблюдаемые продукты разложения
были вызваны только воздействием света.
Следует проводить мониторинг физических характеристик образцов в условиях
испытания, чтобы подтвердить, что изменения физического состояния лекарствен-
ного препарата минимальны. «Темный контроль» (неэкспонированные образцы)
следует размещать для тестирования рядом с образцами, подвергающимися воздей-
ствию светового излучения. Максимального воздействия света при прямом экспо-
нировании для твердых дозированных форм, предназначенных для перорального
введения, таких как таблетки и капсулы, можно достичь, распределив их в одном
слое. Для прямого экспонирования образцов рекомендуется использовать упаковки,
изготовленные из кварцевого стекла. Основываясь на результатах, полученных при
прямом облучении лекарственного препарата, делают вывод о необходимости испы-
тания препарата в первичной и коммерческой упаковке. Если такие испытания не-
обходимы, образцы размещают так, чтобы обеспечить равномерное распределение.
Среди ключевых физических параметров анализируют внешний вид, прозрач-
ность и цвет растворов, растворение и разрушение образца. Количественное содер-
жание лекарственного вещества, продуктов разложения лекарственного вещества
(известных и неизвестных), примесей и вспомогательных веществ, содержащихся
в образцах лекарственного препарата после испытания фотостабильности, следует
проводить с помощью методик определения стабильности, прошедших валидацию.
Важна репрезентативная выборка экспонированных образцов и образцов «темного
контроля» для получения реальной оценки фотостабильности различных дозиро-
ванных форм. Растворы, суспензии и кремы следует проверять на предмет разделе-
668
Часть 7. Стабильность лекарств
ния фаз; следует убедиться в том, что проводится анализ однородных и гомогенных
образцов.
7.1.7. Оценка данных исследований стабильности
и определение срока хранения
Требования к оценке данных по стабильности и определение срока годности опи-
саны в руководстве для промышленности ICH Q1E [11], описывающем способы
оценки данных по стабильности, подаваемых вместе с заявкой на регистрацию мо-
лекул новых лекарств и связанных с ними лекарственных препаратов. Кроме того,
в нем представлены рекомендации по определению периодов до повторного тести-
рования и сроков хранения лекарственных веществ и лекарственных препаратов,
предназначенных для хранения при комнатной температуре, в холодильнике или
морозильной камере и при температуре —20 °C. В дополнение к руководящему до-
кументу следует ознакомиться с исследованиями [21,22], в которых заложены осно-
вы проведения испытаний и оценки данных по стабильности.
7.1.7.1. Определение сроков хранения лекарственных веществ или
лекарственных препаратов, предназначенных для хранения
при комнатной температуре
Оценка данных по стабильности лекарственных веществ или лекарственных препа-
ратов, предназначенных для хранения при комнатной температуре, должны вклю-
чать рассмотрение всех значимых изменений в различных временных точках в про-
цессе испытания в условиях ускоренного хранения и в условиях промежуточного
хранения, если они применяются, а также для оценки данных по стабильности при
долгосрочном хранении. Представлены различные планы испытаний для определе-
ния периода до повторного тестирования и сроков хранения в условиях ускоренно-
го, промежуточного или долгосрочного хранения [11], как описано ниже.
Данные по стабильности при долгосрочном и ускоренном хранении свидетельству-
ют об отсутствии или наличии незначительных изменений с течением времени; вариа-
бельность данных незначительна (или отсутствует). Лекарственное вещество или ле-
карственный препарат могут считаться стабильными при условии, что данные по
стабильности при долгосрочном или ускоренном хранении свидетельствуют об от-
сутствии или наличии незначительных изменений; вариабельность данных незначи-
тельна (или отсутствует). В этом случае статистический анализ данных стабильности
фармацевтических субстанций (или API) или лекарственных препаратов не прово-
дится, однако в обоснование выбранных условий и сроков должно быть включено
обсуждение закономерности изменений или вариабельности данных, или отсут-
ствия изменений и вариабельности данных, дополнительных данных исследования
стабильности при ускоренном хранении, баланса массы лекарственного вещества
в образцах и/или другие дополнительные (поддерживающие заключение) данные.
В таких случаях срок хранения до повторного тестирования или срок годности мо-
жет в два раза превышать (но не более чем на 12 мес.) период, охваченный данными
по стабильности при долгосрочном хранении. Однако можно предложить экстра-
поляцию срока хранения до повторного испытания или срока годности за рамки
периода, охваченного данными по стабильности при долгосрочном испытании.
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов
669
Данные по стабильности при долгосрочном и ускоренном хранении свидетельствуют
о наличии изменений с течением времени; наблюдается вариабельность данных. В этом
случае статистический анализ данных долгосрочного испытания может оказаться
полезным для определения периода до повторного тестирования или срока хране-
ния. Когда отмечаются различия, например, между сериями или разными дозиров-
ками, размерами упаковки и/или их извлекаемыми объемами, или любыми другими
вариантами выпуска препарата, не позволяющими объединять данные, предполага-
емый период до повторного тестирования или срок хранения не должны превышать
минимальный период, обоснованный положительными данными долгосрочных ис-
пытаний любой из серий, другими факторами или комбинациями факторов. Тем не
менее, если выявленные различия касаются только разных дозировок лекарствен-
ной формы, то для разных дозировок препарата могут быть установлены разные
сроки годности с надлежащим обсуждением накопленных данных. Если проведен
статистический анализ данных по стабильности при долгосрочном хранении, и ста-
тистические и прочие релевантные данные являются подтверждающими, то можно
провести экстраполяцию и сделать предположение о том, что период до повторного
тестирования или срок хранения в два раза превышает (но не более чем на 12 мес.)
период, охваченный данными по стабильности при долгосрочном хранении. Если
данные по стабильности при долгосрочном хранении не подлежат статистическому
анализу, то предполагаемый период до повторного тестирования или срок хранения
может в полтора раза (но не более чем на 6 мес.) превышать период, охваченный
данными по долгосрочному хранению.
Данные по стабильности при ускоренном хранении существенно изменяются с тече-
нием времени. Если в условиях ускоренного хранения происходят существенные из-
менения данных, то период до повторного тестирования и срок хранения препарата
определяется в условиях промежуточного и долгосрочного хранения. Руководство
[11] предлагает два исключения для наблюдений, сделанных в условиях ускоренно-
го хранения: физические изменения, такие как размягчение суппозиториев, тем-
пература плавления которых, согласно разработке, составляет 37 °C, — если точка
плавления четко обозначена, и несоответствие критериям приемлемости по раство-
рению 12 единиц желатиновых капсул или таблеток с гелевым покрытием, — если
выявлена однозначная связь с перекрестным связыванием желатина. Эти исключе-
ния не относятся к таким физическим изменениям, как разделение фаз в полутвер-
дых лекарственных формах, кремах или другим изменениям в условиях ускорен-
ного хранения. При появлении подобных изменений следует провести испытания
в условиях промежуточного хранения.
Если в условиях промежуточного хранения значимых изменений не наблюдает-
ся, то можно предложить проведение экстраполяции1 за рамки периода, охвачен-
ного данными по стабильности в условиях долгосрочного хранения, при условии,
что данные статистического анализа и релевантные данные подтверждают возмож-
ность такой экстраполяции. Предполагаемый период до повторного тестирования
1 Речь идет о том, чтобы экстраполировать имеющиеся данные на более длительный пери-
од (т. е. зная, как ведет себя продукт в течение 12 мес., можно предположить, что он так же
будет себя вести в следующие 12 мес. ). — Примеч. перев.
670
Часть 7. Стабильность лекарств
или срок хранения может в полтора раза (но не более чем на 6 мес.) превышать пе-
риод, охваченный данными по долгосрочному хранению. Если данные по стабиль-
ности при долгосрочном хранении не подлежат статистическому анализу, однако
доступны релевантные подтверждающие данные, то предполагаемый период до по-
вторного тестирования может максимум на 3 мес. превышать период, охваченный
данными по долгосрочному хранению.
Если значимые изменения происходят в условиях промежуточного хранения,
период до повторного тестирования или срок хранения, более короткий по сравне-
нию с периодом, охваченным данными по стабильности при долгосрочном хране-
нии, является приемлемым, однако он не должен превышать период, охваченный
данными по долгосрочному хранению.
7.1.7.2. Определение сроков хранения лекарственных веществ или
лекарственных препаратов, предназначенных для хранения
в холодильнике
Данные по стабильности при долгосрочном и ускоренном хранении свидетельствуют об
отсутствии изменений или наличии незначительных изменений в течение времени; ва-
риабельность данных отсутствует или незначительна. В этом случае устанавливаемый
разработчиком период до повторного тестирования или срок годности может в пол-
тора раза (но не более чем на 6 мес.) превышать период, охваченный данными по
длительному хранению, подтвержденными статистически анализом.
Данные по стабильности при долгосрочном и ускоренном хранении свидетельствуют
о наличии изменений в течение времени; наблюдается вариабельность данных. В та-
ких случаях предлагаемый период до повторного хранения или срок годности может
в полтора раза (но не более чем на 6 мес.) превышать период, охваченный данными
по долгосрочному хранению при подтверждении статистическим анализом и ре-
левантными данными. С другой стороны, предполагаемый период до повторного
тестирования не может превышать 3 мес. сверх периода, охваченного данными по
долгосрочному хранению, если данные по долгосрочному хранению подлежат ста-
тистическому анализу, но статистический анализ не проводился; или данные по
долгосрочному хранению не подлежат статистическому анализу, однако существу-
ют данные, подтверждающие предполагаемую дату повторного тестирования и срок
хранения.
Данные по стабильности при ускоренном хранении существенно изменяются с тече-
нием времени; наблюдается вариабельность данных. Если значимые изменения в про-
цессе испытания на стабильность в условиях ускоренного хранения происходят
между 3 и 6 мес., то оценка предполагаемого периода до повторного тестирования
или срока хранения должна быть основана на данных долгосрочного испытания,
а экстраполяция считается неприемлемой. Приемлемым считается период до по-
вторного тестирования или срок хранения, меньший по сравнению с подтверж-
денными данными по стабильности при долгосрочном хранении. Если отмечается
вариабельность данных по стабильности при долгосрочном хранении, необходи-
ма верификация предполагаемого периода до повторного тестирования или срока
хранения при помощи статистического анализа. Если значимые изменения при
испытании на стабильность при ускоренном хранении происходят в течение пер-
Глава 7.1, Стабильность и сроки годности фармацевтических продуктов 671
вых 3 мес., предполагаемый период до повторного тестирования или срок хранения
должен быть определен на основании данных долгосрочного испытания, экстра-
поляция считается неприемлемой; приемлемым считается период до повторного
тестирования или срок хранения, меньший по сравнению с подтвержденными дан-
ными по стабильности при долгосрочном хранении. При таком развитии событий
следует провести испытание на стабильность в условиях ускоренного хранения про-
должительностью до 3 мес. с более частым отбором проб и анализом на материале,
по меньшей мере, одной серии.
7.1.7.3. Определение сроков хранения лекарственных веществ
и препаратов, предназначенных для хранения в морозильной
камере
В этом случае период до повторного тестирования или срок хранения должен быть
определен на основании данных долгосрочного испытания.
7.1.7.4. Оценка сроков хранения лекарственных веществ
или лекарственных препаратов, предназначенных для хранения
при температуре ниже -20 °C
В этом случае период до повторного тестирования или срок хранения следует опре-
делять на основании данных долгосрочного испытания.
7.1.8. Рекомендованные условия хранения в исследованиях
стабильности с учетом установленных климатических
зон
В июне 2004 г. FDA выпустило руководящий документ для промышленности, где
определен комплект данных по стабильности, предоставляемый вместе с заявкой
на регистрацию, для климатических зон III и IV [23] (из руководства ICH Q\F). Ру-
ководство содержит обновления, относящиеся к условиям хранения, для клима-
тических зон I и II и новые указания для климатических зон III и IV, основанные
на рекомендациях, содержащихся в руководстве ВОЗ по испытаниям стабильности
[24]. Руководство ВОЗ [24] описывает рекомендации по испытанию стабильно-
сти и условиям хранения для всех четырех климатических зон с учетом рекомен-
даций Grimm [25] для климатических зон III и IV. Руководство QIF [23] уточняет
условия долгосрочного хранения для климатических зон I и II: (25 ± 2) °C, (60 ± 5)
% RH, а также условия промежуточного хранения для климатических зон I и И:
(30 ± 2) °C, (65 ± 5) % RH. Установлено также [23], что условия хранения (30 ± 2) °C,
(65 ± 5)% RH могут быть приемлемой альтернативой условиям долгосрочного хра-
нения (25 ± 2) °C, (60 ± 5) % RH для климатических зон I и II; в этом случае нет
необходимости в определении условий промежуточного хранения. Для климати-
ческих зон III и IV руководство Q\F [23] предлагает условия (30 ± 2) °C, (65 ± 5)
% RH для долгосрочного (12 мес.) хранения препаратов, предназначенных для хра-
нения при комнатной температуре (в руководстве эти условия определены как «об-
щий случай»); условия ускоренного хранения — (40 ± 2) °C, (75 ± 5) % RH (6 мес.);
по условиям промежуточного хранения для климатических зон III и IV новых ре-
комендаций нет. Для лекарственных препаратов на водной основе, упакованных
672
Часть 7. Стабильность лекарств
в полупроницаемые упаковки, руководство рекомендует следующие условия: для
долгосрочного хранения (12 мес.) — (30 ± 2) °C, (35 ± 5) % RH и для ускоренного
хранения (6 мес.) — (40 + 2) °C и не более (25 ± 5) % RH. На сороковом совещании
Комитета экспертов ВОЗ по спецификациям фармацевтических препаратов, состо-
явшегося в Женеве (Швейцария) в октябре 2005 г., рекомендовано разделить клима-
тическую зону IV на зону IV А, для которой рекомендованы условия долгосрочного
хранения 30 °C, 65% RH, и климатическую зону IV В с условиями долгосрочного
хранения 30 °C, 65% RH [26]. В Приложении 1 к рабочему документу 045/06.179
(2006) [26] ВОЗ выделила пять климатических зон и предложила условия испыта-
ния при долгосрочном хранении для каждой зоны; условия представлены в табл. 3.
После публикации руководства ВОЗ [26] в 2006 г. руководство QIF FDA [23] было
отозвано и, возможно, будет пересмотрено в будущем.
Таблица 3
Климатические зоны и рекомендованные условия долгосрочного хранения [26]
Климати- ческая зона Определение Критерии Условия долгосрочного испытания
Среднегодовые температуры, измеренные на открытом воздухе, °C Парциальное давление паров, гПА Температура, °C RH,%
I Умеренный климат <15 <11 21 45
II Субтропический и средиземно- морский климат > 15-22 > 11—18 25 60
III Жаркий и сухой климат >22 <5 30 35
ГУЛ Жаркий и влажный климат >22 > 15-27 30 65
ГУЙ Жаркий и очень влажный климат >22 >27 30 75
7.1.9. Исследуемые параметры (характеристики)
при изучении стабильности различных
лекарственных форм
Параметры испытания, опубликованные ВОЗ в Приложении 2 к рабочему доку-
менту Q45/06.179 в 2006 г. [27], составляют основу информации, представленной
суммарно в этом разделе для различных дозированных форм. Несмотря на то что
перечисленные здесь параметры содержат указания для различных дозированных
форм, фармакопейные и другие одобренные регуляторными органами параметры,
представленные в заявках на регистрацию лекарств, следует принимать во внима-
ние при применении информации, описанной ниже, для различных дозированных
форм.
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов 673
7.1.9.1. Характеристики, проверяемые при изучении стабильности всех
лекарственных форм
Описание, количественное определение и содержание продуктов разложения, со-
держание консервантов и антиоксидантов по возможности следует проверять у всех
лекарственных форм. Для стерильных дозированных форм необходимо контроли-
ровать стерильность и проводить испытание в соответствии с фармакопейными
и/или внутренними спецификациями. Микробиологическую чистоту нестериль-
ных дозированных форм следует контролировать, проводя надлежащий отбор и ис-
пытание образцов.
7.1.9.2. Характеристики, проверяемые при исследовании стабильности
определенных лекарственных форм
Таблетки-, растворение (если допустимо — распадаемость), содержание воды, твер-
дость/истираемость.
Твердые желатиновые капсулы: ломкость, растворение (если допустимо — рас-
падаемость), содержание воды, микробиологическая чистота.
Мягкие желатиновые капсулы: растворение (если допустимо — распадаемость),
микробиологическая чистота, pH, целостность капсул и образование налета.
Эмульсии: разделение фаз, pH, вязкость, микробиологическая чистота/стериль-
ность, средний размер капель и распределение диспергированных капель по раз-
меру.
Растворы и суспензии для приема внутрь: образование осадка, прозрачность (для
растворов), pH, вязкость, микробиологическая чистота, экстрагируемые вещества
и полиморфные превращения, если применимо; дополнительные испытания для
суспензий включают определение редиспергируемости, реологических параметров,
средних размеров и распределения частиц по размеру.
Порошки для приготовления растворов и суспензий для перорального введения: со-
держание воды, время восстановления; восстановленные растворы и суспензии
следует тестировать так же, как растворы и суспензии для перорального введения
(описано выше).
Местные офтальмологические и ушные препараты: прозрачность, однородность,
pH, ресуспендируемость (для примочек lotions), густота, вязкость, микробиологиче-
ская нагрузка и потеря в массе при высушивании; дополнительные характеристики
включают стерильность, содержание механических включений и экстрагируемых
веществ.
Суппозитории: диапазон плавления, растворение при 37 °C.
Трансдермальные пластыри: скорость высвобождения in vitro, целостность слоев,
микробиологическая чистота/стерильность, а также адгезивные свойства и сила
прилипания.
Дозирующие ингаляторы и назальные аэрозоли: однородность по содержанию,
аэродинамическое распределение частиц, микроскопия частиц, содержание воды,
герметичность, микробиологическая чистота, однородность дозирования устрой-
ства, экстрагируемые вещества, экстрактивные вещества из компонентов, изготов-
ленных из полимеров и эластромеров.
Часть 7. Стабильность лекарств
674
Назальные спреи', прозрачность, микробиологическая чистота, pH, механические
включения, однородность дозирования (по количественному определению), рас-
пределение капель и/или частиц по размеру, масса содержимого, количество доз
в упаковке, микроскопия частиц), экстрагируемые вещества, экстрактивные веще-
ства из компонентов упаковки (в данном случае это контейнер или баллон) и дози-
рующего клапана, изготовленных из полимеров и эластромеров.
Препараты для парентерального введения малого объема', цветность, прозрачность
растворов, механические включения, pH, стерильность, содержание бактериальных
эндотоксинов. Порошки для приготовления растворов для инъекций: прозрачность
и цветность востановленного раствора, время растворения и содержание воды, pH,
стерильность, содержание бактериальных эндотоксинов/пирогенность и механиче-
ские включения. Суспензии для инъекций: дополнительные испытания на распре-
деление частиц по размерам, ресуспендируемость и реологические характеристики.
Эмульсии для инъекций: разделение фаз, вязкость, средний размер капель и рас-
пределение по размеру диспергированных капель.
Препараты для парентерального введения большого объема: цветность, прозрач-
ность, механические включения, pH, стерильность, содержание бактериальных эн-
дотоксинов/пирогенность и извлекаемый объем.
7.1.10. Заключение и выводы
Исследования стабильности фармацевтических продуктов позволяют получить ин-
формацию о том, как качество вспомогательных веществ, API и лекарственных пре-
паратов изменяется с течением времени под влиянием различных факторов окру-
жающей среды, таких как температура, влажность и свет, и помогает определить
срок хранения и рекомендуемые условия хранения в течение всего жизненного цик-
ла продукции. Правила надлежащей производственной практики и руководства по
бЛ/Ртребуют проведения испытаний стабильности фармацевтических препаратов и
определения сроков хранения на основании результатов испытаний стабильности.
Ускоренные исследования стабильности при повышенной температуре и влажности
и долгосрочные испытания стабильности при более мягких условиях температуры
и влажности проводятся при разработке лекарства и в процессе регистрации для
прогнозирования срока хранения фармацевтических препаратов. Затем предпола-
гаемый срок хранения подтверждают долгосрочным испытанием, длящимся в те-
чение срока хранения и более. Стресс-испытания с воздействием кислоты, щелочи,
температуры, окисления и освещения проводятся для прогноза образования про-
дуктов разложения, которые могут образовываться в процессе ускоренного и/или
долгосрочного испытания, и с целью разработки методов, необходимых для оцен-
ки стабильности. Исследования фотостабильности и испытания с температурным
воздействием помогают определить надлежащую конфигурацию упаковки, а также
предоставить рекомендации по условиям хранения в течение срока хранения. Для
реалистической оценки условий хранения в различных климатических условиях
выделено пять климатических зон; условия долгосрочного хранения для испытаний
с целью определения срока хранения продукции в этих климатических зонах осно-
ваны, главным образом, на характеристиках температуры и влажности, вероятно,
Глава 7.1. Стабильность и сроки годности фармацевтических продуктов 675
превалирующих в этих климатических зонах. Параметры качества, применяемые
для оценки стабильности фармацевтических препаратов, зависят от химической
природы анализируемого активного ингредиента, а также от формы дозирования
лекарственного препарата.
Литература
1. U. S. Pharmacopeia (2006), Stability considerations in dispensing practices, general chapter (1191),
U. S. Pharmacopeial Convention, Rockville, MD.
2. U. S. Food and Drug Administration (FDA) (2007) Current good manufacturing practice regulations,
21 CFR Parts 210 and 211, FDA, Rockville, MD.
3. U. S. Food and Drug Administration (FDA) (2001), Guidance for industry, Q7A Good manufacturing
practice guidance for active pharmaceutical ingredients, FDA, Rockville, MD.
4. U. S. Pharmacopeia (2007), Good manufacturing practices for bulk pharmaceutical excipients, gen-
eral chapter (1078), U S. Pharmacopeial Convention, Rockville, MD.
5. World Health Organization (WHO) (2003), Good manufacturing practices for pharmaceutical prod-
ucts: Main principles, WHO Technical Report Series 908, WHO, Geneva.
6. World Health Organization (WHO) (2006), Stability testing of active substances and pharmaceutical
products, Working Document QAS/06.179, WHO, Geneva.
7. U. S. Food and Drug Administration (FDA) (2003), Guidance for industry, ICH QI A (R2), Stability
testing of new drug substances and products, FDA, Rockville, MD.
8. U. S. Food and Drug Administration (FDA) (1997), Guidance for industry, ICH Q1B, Photostability
testing of new drug substances and products, FDA, Rockville, MD.
9. International Conference on Harmonization (ICH) (1997), Guidance for industry, ICH QIC, Stability
testing of new dosage forms, ICH, Brussels.
10. International Conference on Harmonization (ICH) (2003), Guidance for industry, ICH Q1D, Bracket-
ing and matrixing designs for stability of drug substances and drug products, ICH, Brussels.
11. U. S. Food and Drug Administration (FDA) (2004), Guidance for industry, ICH Q1E, Evaluation of
stability data, FDA, Rockville, MD.
12. U. S. Food and Drug Administration (FDA) (2000), Guidance for industry, Analytical procedures and
methods validation — Chemistry, manufacturing, and controls documentation, FDA, Rockville, MD.
13. Lymann W. J. , Reehl W. E, Rosenblatt D. H. (1982), Handbook of Chemical Property Estimation
Methods, McGraw - Hill, New York.
14. Albini A., Fasani E. (1998), Photochemistry of Drugs: An Overview of Practical Problems, Drugs,
Photochemistry and Photostability, The Royal Society of Chemistry, London.
15. Carstensen J. T. (1990), Drug Stability—Principles and Practices, Marcel Dekker, New York.
16. Drew H. D., Brower J. E, Juhl W. E., Thornton L. K. (1998), Quinine photochemistry: A proposed
chemical actinometer system to monitor UV exposure in photostability studies of pharmaceutical
drug substances and drug products, Pharmacopeial Forum, 24 (3), 6334.
17. Drew H. D., Thornton L. K., Juhl W. E., Brower J. F. (1998), An FDA/PhRMA interlaboratory study
of the International Conference on Harmonization’s proposed photostability testing procedures and
guidelines, Pharmacopeial Forum, 24 (3), 6317.
18. Sager N., Baum R. G., Wolters R. J., Layloff T. (1998), Photostability studies of pharmaceutical prod-
ucts, Pharmacopeial Forum,24 (3), 6331.
19. Farmer S., Anderson P., Bums P. K., Velagaleti R. (2002, May), Forced degradation of ibuprofen in
bulk drug and tablets and determination of specifi city, selectivity, and the stability-indicating nature
of the USP ibuprofen assay method, Pharm. Technol., 28-42 .
20. Farmer S., McCauslin L., Bums P. K., Velagaleti R. (2004, Aug.), Photosensitivity of internal standard
valerophenone used in USP ibuprofen bulk drug and tablet assay and its effect on quantitation of ibu-
profen and its impurities, Pharm. Technol., 68—74.
676 Часть 7. Стабильность лекарств
21. Grimm W. (1985), Storage conditions for stability testing — Long term testing and stress tests (part
I), Drugs Made in Germany, 28,196—202.
22. Grimm W. (1986), Storage conditions for stability testing — Long term testing and stress tests (part
II), Drugs Made in Germany, 29, 39—47.
23. U. S. Food and Drug Administration (FDA) (2004), Guidance for industry, ICH Q1F, Stability data
package for registration applications in climatic zones III and IV, FDA, Rockville, MD.
24. World Health Organization (WHO) (2001), Stability testing of pharmaceutical products containing
well established drug substances in conventional dosage form, WHO Technical Report Series 863,
Annex 5, WHO, Geneva.
25. Grimm W. (1998), Extension of International Conference on Harmonization tripartite guideline for
stability testing of new drug substances and products to countries of climatic zones HI and IV, Drug
Dev. Ind. Pharm., 24, 313-325.
26. World Health Organization (WHO) (2006), Assignment of climatic zones and recommended storage
conditions, Working Document QAS/06.179, Annex 1, WHO, Geneva.
27. World Health Organization (WHO) (2006), Testing parameters (for dosage forms), Working Docu-
ment QAS/06.179, Annex 2, WHO, Geneva.
Глава 7.2. Стабильность лекарств
Назарио Д. Рамирес-Белтран, Л. Антонио Эстевес
(оба из университета Пуэрто-Рико (Маягуэс, Пуэрто-Рико))
и Гарри Родригес {Cordis LLC, Johnson & Johnson Company
(Сан Джерман, Пуэрто-Рико))
7.2.1. Общие вопросы стабильности
7.2.1.1. Введение
Для каждого лекарственного препарата, представленного на рынке, требуется ука-
зание даты истечения срока годности в маркировке первичной упаковки [1]. Тре-
бования Федерального управления США по контролю за пищевой продукцией
и лекарствами (FDA) и Международной конференции по гармонизации (ICH) уста-
навливают, что фармацевтические компании должны представлять фактические
свидетельства, подтверждающие сроки хранения как существующих, так и новых
препаратов. Для того чтобы лекарственный препарат стал эффективным лекар-
ством, чрезвычайно важно наличие надлежащего количества активного ингредиен-
та в фармацевтическом составе. Как правило, для измерения содержания активного
ингредиента, изменяющегося с течением времени, используют точную методику,
а накопленные данные анализируют для определения срока хранения. Затем рас-
считывают дату истечения срока хранения. Таким образом, указанная дата истече-
ния срока годности дает потребителю уверенность в том, что лекарственный препа-
рат сохраняет идентичность, содержание активного компонента, качество и чистоту
в течение всего периода до истечения срока годности.
Главные задачи этой главы — описание практических приемов планирования ис-
пытания стабильности, а также обсуждение и иллюстрация методов определения
сроков хранения лекарственного препарата. В данном разделе представлен общий
обзор испытаний стабильности и рассмотрение действующих нормативных тре-
бований. Испытание стабильности характеризует не только разложение активных
ингредиентов лекарственного препарата с течением времени, но и предоставляет
основу для расчета срока хранения препарата. В этом разделе содержится краткое
описание краткосрочных и долгосрочных испытаний стабильности, а также важные
статистические требования, изложенные в руководствах FDA и ICH. В разделе 7.2.2
описаны основы планирования испытания стабильности. При планировании дан-
ного испытания ставится задача установления процедуры накопления надежных
данных для определения срока хранения, основанной на тестировании ограничен-
ного числа серий лекарственного препарата, который можно будет применить ко
всем будущим сериям лекарственного препарата, произведенным в аналогичных
условиях. Планирование долгосрочных испытаний стабильности обычно включает
определение таких факторов, как число серий, содержание активного компонента,
конфигурация упаковки и условия хранения. При испытаниях стабильности, глав-
ным образом, применяются брекетинг и матричное планирование. В разделе 7.2.3
представлено описание традиционной методики определения срока хранения,
установленной на основании данных долгосрочного испытания стабильности, про-
678
Часть 7. Стабильность лекарств
водящегося при нормальных условиях хранения. Будет представлено обсуждение
методики расчета срока хранения лекарственного препарата для единственной се-
рии и для множественных серий, обсуждение будет проиллюстрировано примера-
ми. Для выполнения расчетов параметров стабильности в Приложении к главе 7.2
включен набор компьютерных программ на языке Matlab. В разделе 7.2.4 обсуж-
дается методика ускоренного испытания стабильности, которую часто используют
для предварительной оценки срока хранения. На ранней стадии разработки лекар-
ственного препарата первичной задачей испытания является определение скоро-
стей химических и физических превращений и их связи с условиями хранения, та-
кими как температура, влажность, освещенность и др. Краткосрочное исследование
стабильности проводится в условиях стресс-испытания для повышения скорости
химического и физического разложения лекарственного препарата. В этом разделе
обсуждается кинетика химической реакции и методика статистической предвари-
тельной оценки срока хранения препарата. Раздел охватывает методики построения
регрессий для оценки параметров уравнения Аррениуса.
7.2.1.2. Требования регуляторного органа
FDA требует от производителей фармацевтической продукции утверждения про-
грамм испытания стабильности для каждого лекарственного препарата [1—3].
Matthews [4] предоставляет обзор ситуации с нормативно-правовым регулировани-
ем, сложившейся в Европе. Целями программы испытания стабильности являются
разработка надлежащей процедуры, направленной на накопление данных, и расчет
срока хранения для определения даты истечения срока годности лекарственного
препарата. Программа должна быть изложена в письменном протоколе, включаю-
щем все требования, установленные в нормативном документе. FDA устанавливает
следующие требования к испытанию стабильности [1]:
1. Объем выборки и интервалы между испытаниями для каждого исследуемого
параметра необходимо уточнить с учетом статистических обоснований.
2. Условия хранения образцов должны быть указаны точно. Условия хранения
должны совпадать с условиями, указанными на маркировке лекарственного
препарата.
3. Все методики испытаний для каждого лекарственного препарата должны от-
вечать требованиям адекватности и достоверности.
4. Образцы, предназначенные для испытания стабильности, следует хранить
в упаковке той же конфигурации, что используется при поставке готового
лекарственного препарата на рынок.
5. Если необходимо восстановление лекарственного препарата, то следует про-
вести анализ лекарственного препарата до и после восстановления.
6. Число серий, исследуемых согласно программе испытаний стабильности для
нового лекарственного препарата, составляет по меньшей мере три. Для ис-
пытаний следует выбирать разные серии лекарственного вещества.
7. На предприятии фармацевтической промышленности необходимо вести за-
писи всех данных, протоколов и отчетов, относящихся к испытаниям ста-
бильности.
Глава 7.2. Стабильность лекарств
679
7.2.1.З. Стабильность и срок годности
Срок хранения (срок годности) — период времени, в течение которого лекарствен-
ный препарат гарантированно соответствует требованиям к подлинности, коли-
чественному определению, чистоте и другим показателям качества при хранении
в условиях, указанных в маркировке. Дата окончания срока годности — это конец
срока хранения. Срок хранения и дату окончания срока годности лекарственного
препарата определяют при помощи испытания стабильности. Стабильность лекар-
ственного препарата оценивают тестированием всех параметров, необходимых для
выпуска препарата на рынок. Испытания проводят через определенные промежутки
времени в течение определенного периода. Дату окончания срока годности обычно
указывают в маркировке лекарственного препарата.
7.2.1.4. Краткосрочные и долгосрочные исследования стабильности
Краткосрочное испытание стабильности (другими словами, исследование при уско-
ренном хранении) проводится в экстремальных условиях окружающей среды для
повышения скорости химического разложения. Данные, полученные в результате
краткосрочного испытания стабильности, обычно используют для оценки долговре-
менных химических эффектов в условиях неэкстремального хранения, однако они
также помогают при оценке влияния кратковременных отступлений от рекомендо-
ванных условий хранения, которые могут возникнуть во время транспортировки.
Однако результаты, полученные в ходе краткосрочного испытания стабильности,
не всегда можно использовать для прогноза физических изменений в лекарствен-
ном препарате. Рекомендуется проводить анализ как минимум три раза в течение
шестимесячного исследования, включая начальную и конечную временную точку.
Если прогнозируются значимые изменения, следует провести дополнительный или
более частый анализ, наряду с исследованием стабильности в промежуточных усло-
виях. Реальные экстремальные или промежуточные условия зависят от рекомендуе-
мых условий хранения лекарственного препарата, например, при комнатной темпе-
ратуре или в холодильнике. Изменение считается значимым, если отвечает одному
или нескольким из следующих критериев:
1. Пятипроцентное изменение измеряемой величины от ее исходного значения
или несоответствие критериям приемлемости по эффективности при приме-
нении биологических или иммунологических методик.
2. Наличие любых примесей на уровне, превышающем критерий приемлемо-
сти.
3. Несоответствие критериям приемлемости по внешнему виду, физическим
свойствам или при тестировании функциональности (такие параметры, как
цветность, разделение фаз, ресуспендируемость, спекание, твердость или
однородность выпускаемой клапаном дозы). Однако некоторых изменений
физических характеристик (например, размягчение суппозиториев, плавле-
ние кремов) можно ожидать в условиях ускоренного хранения.
4. Несоответствие критериям приемлемости по pH.
5. Несоответствие критериям приемлемости при испытании «Растворение» 12
единиц лекарственной формы.
680
Часть 7. Стабильность лекарств
Долгосрочное испытание проводится в рекомендуемых условиях хранения.
Конфигурация упаковки должна соответствовать той упаковке, в которой лекар-
ственный препарат поставляется на рынок и хранится в течение срока хранения,
указанного в маркировке препарата. Рекомендуемая частота тестирования — каж-
дые 3 мес. в течение первого года, каждые 6 мес. в течение второго года и после
этого ежегодно. Общие условия хранения при испытаниях стабильности в условиях
долгосрочного, промежуточного и краткосрочного хранения для климатических ре-
гионов I и II, включающих Европу, Японию и США [5, 6], представлены в табл. 1.
При испытаниях стабильности в условиях долгосрочного, промежуточного и крат-
косрочного хранения можно использовать альтернативные условия хранения, если
это оправдано.
Таблица 1
Условия хранения при испытаниях стабильности
Условия испытания Условия хранения Минимальный период времени, охваченный испытаниями при подаче заявки (мес.)
При комнатной температуре:
долгосрочное (25 ± 2) °C, (60 ± 5) % RH 12
промежуточное (30 ± 2) °C, (60 ± 5) % RH 6
ускоренное В холодильнике: (40 ± 2) °C, (75 ± 5) % RH 6
долгосрочное (5 ± 3) °C 12
ускоренное В морозильной камере (25 ±2) °C, (60 ±5) % RH 6
долгосрочное (-20 ± 5) °C 12
Примечание: RH — относительная влажность.
7.2.1.5. Статистическая обработка
Для анализа данных, накопленных в процессе исследования стабильности, рекомен-
дуется использовать соответствующий статистический метод. Целью применения
Статистических подходов является определение с высокой степенью достоверности
срока хранения, то есть периода, в течение которого количественное значение па-
раметра лекарственного препарата соответствует критериям приемлемости для всех
серий, которые будут произведены, упакованы и будут храниться в аналогичных
условиях. Испытания стабильности дороги и затрачивают много времени, и с этой
точки зрения статистические оценки могут оказаться полезными. Принципы стати-
стического планирования можно применить для снижения числа анализов [7].
Срок хранения для единственной серии обычно рассчитывается с помощью
метода регрессии. Правильный подход к определению срока хранения с помо-
щью регрессионного анализа заключается в расчете самой ранней временной точ-
ки, в которой 95%-ный предел достоверности для среднего значения пересекается
с предложенным критерием приемлемости [8]. Подробное описание расчета срока
хранения изложено в разделах 7.2.3. и 7.2.4.
Глава 7.2. Стабильность лекарств
681
Традиционным статистическим инструментом определения принадлежности
кривых скорости разложения нескольких серий к одной популяции является кова-
риационный анализ. Если кривые скорости разложения статистически различны,
для определения срока хранения текущих и будущих серий используют минималь-
ный критерий. В соответствии с минимальным критерием срок хранения будущих
серий соответствует кратчайшему из сроков хранения для всех протестированных
серий. С другой стороны, если серии принадлежат к одной популяции, регрессион-
ный анализ применяется для построения единой кривой скорости разложения на
основании совокупности данных на материале протестированных серий.
Краткосрочное испытание стабильности проводится в стрессовых условиях для
повышения скорости химического и физического разложения лекарственного пре-
парата. Классическая статистическая методика предварительного установления
срока хранения основана на методе регрессии, который используется для расчета
параметров уравнения Аррениуса.
Для определения приемлемости экстраполяции за рамки периода, охваченно-
го данными испытания стабильности при долгосрочном испытании, следует при-
менить системный подход к оценке данных по стабильности. Подход заключается
в оценке всех значимых изменений в условиях ускоренного хранения и, при не-
обходимости, в условиях промежуточного хранения, в соответствии с указаниями
руководства о том, как устанавливать срок хранения методом экстраполяции. Не-
обходимые релевантные подтверждающие данные и обязательство о помещении
серий на испытание стабильности, длящееся до конца экстраполированного срока
хранения, должны быть представлены в регуляторные органы. Релевантные под-
тверждающие данные включают удовлетворительные данные долговременного ис-
пытания экспериментальных серий препарата, близкого по составу, изготовленного
в малом масштабе и хранящегося в упаковке, конфигурация которой аналогична
упаковке первичных серий, проходящих испытание стабильности. Оценка данных
проводится следующим образом [8].
Препарат, предназначенный для хранения при комнатной температуре. Степень экс-
траполяции будет зависеть от оценки результатов, полученных по данным кратко-
срочного, промежуточного и долгосрочного испытаний. Если в условиях ускоренно-
го (краткосрочного) испытания не наблюдается значимых изменений, оцениваются
данные, полученные в условиях долгосрочного испытания, на предмет отклонений
и вариабельности:
• Данные долгосрочного испытания с малыми отклонениями (или без отклонений)
с течением времени и малой вариабельностью (или при отсутствии вариабельно-
сти). В этом случае можно признать отсутствие необходимости в статистиче-
ском анализе, однако должно быть представлено обоснование. Можно пред-
ложить провести экстраполяцию, и в итоге срок хранения может равняться
удвоенному периоду, охваченному данными по стабильности, полученными
при долгосрочном хранении, однако он не может превышать период испыта-
ния более чем на 12 мес.
• Данные долгосрочного испытания с отклонениями с течением времени и вариа-
бельностью. В этом случае для определения срока хранения лекарственного
682
Часть 7. Стабильность лекарств
препарата можно воспользоваться статистическим анализом данных долго-
временного испытания. Если наблюдаются статистически значимые различия
среди серий, факторов или комбинаций факторов, то провести группировку
данных невозможно; в этом случае срок хранения соответствует кратчайшему
из всех сроков хранения, полученных на основании данных по всем сериям,
факторам или комбинациям факторов. Если статистические различия связа-
ны с конкретным фактором (например, величиной дозы или упаковкой), раз-
личные сроки хранения мотуг быть определены для каждого уровня значения
данного фактора. Срок хранения, полученный с помощью экстраполяции,
зависит от возможности применения статистического анализа к данным по
стабильности:
1. Данные не подлежат статистическому анализу. Предполагаемый срок
хранения может в 1,5 раза (но не более чем на 6 мес.) превышать период,
охваченный испытанием стабильности в условиях долгосрочного хране-
ния. Для того чтобы показать, что лекарственный препарат отвечает по
всем параметрам критериям приемлемости, потребуются релевантные
подтверждающие данные, относящиеся к концу предполагаемого срока
хранения.
2. Данные, подлежащие статистическому анализу. Если статистический ана-
лиз можно применить к данным долгосрочного испытания, но он не был
проведен, необходимо обоснование; срок хранения, определенный с по-
мощью экстраполяции, тот же, что и в случае, когда статистический ана-
лиз не применим. Если статистический анализ проведен, срок хранения
может до двух раз (но не более чем на 12 мес.) превышать период, охвачен-
ный долгосрочным испытанием стабильности.
Если значимые изменения происходят в любой момент в течение
6-месячного периода в условиях ускоренного (краткосрочного) испыта-
ния, то требуется испытание стабильности в промежуточных условиях,
а срок хранения, полученный с помощью экстраполяции, зависит от ис-
хода испытания в условиях как долгосрочного, так и промежуточного хра-
нения.
• Данные без значимых изменений в промежуточныхусловиях. В этом случае мож-
но предложить экстраполяцию данных долговременного испытания, а срок
хранения, определенный с помощью экстраполяции, зависит от того, под-
лежат ли данные статистическому анализу.
1. Данные не подлежат статистическому анализу. Предполагаемый срок хра-
нения можетна 3 мес. (максимум) превышать период, охваченный данными
долгосрочного испытания стабильности. Здесь потребуются релевантные
подтверждающие данные для демонстрации соответствия критериев при-
емлемости по всем параметрам к концу предполагаемого срока хранения.
2. Данные, подлежащие статистическому анализу. Если статистический
анализ применим к данным, полученным в условиях долгосрочного хра-
нения, но не был проведен, необходимо обоснование, а срок хранения,
определенный с помощью экстраполяции, тот же, что и в случае, когда
Глава 7.2. Стабильность лекарств
683
статистический анализ не применим. Если статистический анализ прове-
ден, срок хранения, определенный с помощью экстраполяции, может на
6 мес. (максимум) превышать период, охваченный долгосрочным испы-
танием стабильности. Здесь потребуются релевантные подтверждающие
данные для демонстрации соответствия критериев приемлемости по всем
параметрам к концу предполагаемого срока хранения.
• Данные со значимыми изменениями в промежуточных условиях. В этом случае
экстраполяция данных долгосрочного хранения не допустима, а срок хране-
ния не должен превышать период, охваченный испытанием стабильности
в условиях долгосрочного хранения.
Препарат, предназначенный для хранения в холодильнике. Для препаратов, предна-
значенных для хранения в холодильнике, применяют такой же подход, как и к пре-
паратам, предназначенным для хранения при комнатной температуре, но с больши-
ми ограничениями по сроку хранения, определенному с помощью экстраполяции,
если нет иных указаний.
Если в условиях ускоренного (краткосрочного) испытания не наблюдается зна-
чимых изменений, то данные долгосрочных испытаний оценивают на предмет из-
менений и вариабельности:
• Данные долгосрочного испытания с малыми изменениями (или без изменений)
с течением времени и малой вариабельностью (или при отсутствии вариабельно-
сти). Можно предложить определить срок хранения с помощью экстраполя-
ции, и этот срок может быть до 1,5 раз (но не более чем на 6 мес.) превышать
период, охваченный испытанием стабильности в условиях долгосрочного
хранения.
• Данные долгосрочного испытания с изменениями во времени и вариабельно-
стью. Срок хранения, полученный с помощью экстраполяции, зависит от
возможности применения статистического анализа к данным по стабильно-
сти:
1. Данные не подлежат статистическому анализу. Предполагаемый срок хра-
нения может на 3 мес. (максимум) превышать период, охваченный данны-
ми долгосрочного испытания стабильности.
2. Данные, подлежащие статистическому анализу. Если статистический
анализ применим к данным, полученным в условиях долгосрочного хра-
нения, но не был проведен, необходимо обоснование, а срок хранения,
определенный с помощью экстраполяции, тот же, что и в случае, когда
статистический анализ не применим. Если статистический анализ прове-
ден, срок хранения, определенный с помощью экстраполяции, может до
1,5 раз (но не более чем на 6 мес.) превышать период, охваченный долго-
срочным испытанием стабильности.
Если отмечается значимое изменение в любой момент времени в течение
6-месячного периода ускоренного (краткосрочного) хранения, то экстра-
поляция считается неприемлемой. Если в данных по долгосрочному хра-
нению наблюдается вариабельность, предполагаемый срок хранения мо-
жет быть подтвержден статистическим анализом.
684 Часть 7. Стабильность лекарств
Препарат, предназначенный для хранения в морозильной камере. Для таких пре-
паратов срок хранения определяется на основании данных по стабильности при
долгосрочном хранении; экстраполяция не разрешена.
7.2.2. Планирование исследований стабильности
7.2.2.1. Введение
Руководство FDA, во-первых, устанавливает требование о наличии протокола, опи-
сывающего организацию и проведение испытания стабильности, и, во-вторых,
приводит описание статистических методов, которые могут быть использованы
для анализа данных. Задача планирования исследования стабильности состоит
в установлении даты окончания срока годности. Дата окончания срока годности
устанавливается на основании испытания на материале ограниченного числа се-
рий лекарственного препарата, а результаты этих испытаний применяют к сериям
лекарственного препарата, которые будут произведены в аналогичных условиях.
Поэтому планирование испытания должно способствовать уменьшению система-
тической погрешности, идентификации и контролю всех ожидаемых и неожидае-
мых источников вариабельности. Надлежащее планирование исследования обеспе-
чивает высшую точность и прецизионность при определении срока хранения.
Испытание стабильности должно охарактеризовать процесс разложения актив-
ного ингредиента с течением времени и направлено на определение срока хране-
ния, используемого для расчета даты истечения срока годности лекарственного
препарата. Срок хранения — это максимально допустимый период времени хране-
ния лекарственного препарата в окончательной упаковке, в течение которого сохра-
няется терапевтическое количество активного фармацевтического вещества (API).
Дата окончания срока годности — конец периода (срока) хранения. Дату истечения
срока годности определяют, отсчитывая срок хранения от даты производства лекар-
ственного препарата. Определенный срок хранения приписывается всем сериям,
которые будут изготовлены в дальнейшем в условиях, аналогичным условиям про-
изводства серий, использованных в испытаниях стабильности.
Планирование испытания стабильности должно быть основано на знании
свойств и характеристик лекарственного вещества, полученных в ходе исследова-
ний стабильности, и данных, полученных на этапе клинической разработки. Ис-
пытание стабильности проводится согласно утвержденному протоколу, где долж-
ны быть указаны все аспекты, которые следует учесть (например, размер образца,
аналитические методики и критерии приемлемости) при выполнении испытания.
Важно соблюдение всех указаний FDA (или ICH).
Планирование испытания стабильности базируется на факторном планирова-
нии эксперимента, где систематическая процедура применяется для определения
влияния различных факторов и их комбинаций на переменную отклика. Линейная
модель используется для представления соотношений между факторами и комбина-
циями факторов и переменной отклика. Если план эксперимента утвержден, прово-
дят испытания и накапливают данные по стабильности для итогового определения
срока хранения.
Как правило, планирование испытаний стабильности включает исследование
числа производственных серий, содержания активного вещества, конфигурации
Глава 7.2. Стабильность лекарств
685
упаковки и условий хранения. Типичной переменной отклика в испытаниях явля-
ется содержание API. Однако следует учитывать и все другие переменные отклика
(характеристики лекарственного препарата), подверженные изменениям в процес-
се хранения, которые могут повлиять на качество, безопасность и эффективность
лекарственного препарата (например, внешний вид, стерильность, скорость высво-
бождения лекарства, примеси и препараты разложения). Процедура анализа долж-
на включать, по необходимости, оценку физических, химических, биологических
и микробиологических характеристик, содержание консервантов (например, анти-
оксидантов или антимикробных консервантов), а также тестирование функцио-
нальных характеристик (например, для системы доставки дозы).
В разделе 7.2.2 описаны практические указания по планированию испытаний
стабильности, соответствующих требованиям руководства для промышленности,
опубликованного FDA [9]. В зависимости от характеристик лекарственного пре-
парата и при наличии соответствующего научного обоснования можно применить
другие подходы.
1.2.2.2. Основные принципы планирования исследования
При планировании испытания стабильности, следует принять во внимание следую-
щие аспекты:
• Предварительные данные по стабильности, накапливающиеся на этапе раз-
работки лекарственного препарата, используют для выбора окончательной
конфигурации упаковки и условий хранения, а также для описания харак-
теристик препарата. Такие данные могут помочь в разработке качественного
плана испытаний стабильности, при помощи которого можно получить мак-
симальное количество информации из минимального количества данных.
Кроме того, предварительные данные по стабильности могут представить на-
учное обоснование выбору плана испытания стабильности.
• Характеристики лекарственного препарата, способные повлиять на его каче-
ство, должны быть включены в план испытания стабильности. Тестирование
этих характеристик следует проводить во всех временных точках; таким обра-
зом, следует запланировать отбор необходимого количества проб для анализа
в каждой временной точке.
• Спецификации на лекарственный препарат для каждой его характеристики не-
обходимы для определения критериев приемлемости (норм) при испытании
стабильности.
• Методики анализа, применяемые в ходе испытаний стабильности, должны
быть предварительно валидированы.
• Данные предварительных испытаний. Все данные, накопленные на этапах раз-
работки и клинических испытаний лекарства, могут стать полезными при
определении ожидаемой вариабельности производственного процесса. Ва-
риабельность процесса является важным фактором, определяющим план от-
бора проб, который будет использован в ходе испытаний стабильности.
• Идентификация факторов планирования критически важна при разработке
плана испытания стабильности. Важно идентифицировать факторы плани-
686
Часть 7. Стабильность лекарств
рования, способные повлиять на стабильность лекарственного препарата во
время хранения. Неправильная идентификация этих факторов может значи-
тельно отсрочить завершение испытания стабильности и представление дан-
ных в регуляторное агентство, например, в FDA.
• Полный и сокращенный план. Полный план требует испытания лекарствен-
ного препарата при воздействии комбинации факторов и во всех периодах
времени. Испытание по полному плану позволяет получить максимальное
количество информации для определения срока хранения лекарственного
препарата. Однако необходимое число комбинаций увеличивается экспонен-
циально с увеличением количества факторов. С другой стороны, согласно со-
кращенному плану тестирование лекарственного препарата проводится при
воздействии лишь части комбинаций факторов. Сокращенный план требует
меньше усилий, однако в нем заключается потенциальный риск — для пре-
парата может быть определен более короткий срок хранения из-за меньшего
массива накопленных данных.
7.2.2.3. Методы планирования исследований стабильности
Полный план испытания стабильности. Предположим, что испытание стабильности
охватывает три фактора: серию, дозировку (обозначены как 51, S2 и 53 и т. д.), а так-
же форму выпуска (обозначены как Pl, Р2 и РЗ). Каждое значение фактора описыва-
ется как уровень. Поэтому при наличии четырех вариантов формы выпуска фактор,
называемый вариант формы выпуска, имеет четыре уровня. Следует отметить, что
FDA требует испытания по меньшей мере трех серий; следовательно, фактор серии
имеет три уровня. Если два других фактора имеют по три уровня каждый, то чис-
ло экспериментов равняется 27. В табл. 2 представлены 27 экспериментов, которые
требуется провести в каждой временной точке. Число комбинаций С можно легко
рассчитать умножением числа уровней для каждого фактора:
С= LRLRLRLFLF,
1 2 3 4 л’
где Ы\ — число уровней фактора 1; LF2 — число уровней фактора 2; LFZ — число
уровней фактора 3; LFn — число уровней фактора п.
Таблица 2
Комбинации факторов для трех факторов с тремя уровнями для одного препарата
Серия - 51 S1 S3
Р1 Р2 РЗ Р1 Р1 РЗ РР1 Р2 РЗ
1 С1 С2 сз СЮ СП С12 С19 С20 С21
2 С4 С5 С6 С13 С14 С15 СП С23 С24
3 С7 С8 С9 С16 С17 С18 С25 С26 СП
Когда определены комбинации факторов, можно найти число образцов, необ-
ходимых для исследования. Предположим, что согласно плану исследования ста-
бильности (табл. 2), единственным необходимым тестом является количественное
определение, а для анализа требуется одна упаковка лекарственного препарата; чис-
ло образцов, подлежащих тестированию, составляет 27 упаковок в каждый период
Глава 7.2. Стабильность лекарств 687
времени. Соответственно, в план исследования включают по одной упаковке лекар-
ственного препарата в каждой из 27 комбинаций для каждого периода времени.
Если предполагаемый срок хранения препарата составляет 36 мес., тестирование
следует проводить в следующих временных точках (Г): 0, 3, 6, 9, 12, 18, 24 и 36 мес.
Если планируется исследование по полному плану, представленному в табл. 2, для
проведения всех этапов анализа потребуется 216 образцов. Для получения данных
для t = 0 в начале испытания анализируют 27 образцов. Все прочие образцы поме-
щают в камеру искусственного климата, где поддерживают температуру и влажность
в соответствии с протоколом испытания стабильности. При каждом тестировании
27 образцов достают из камеры, как показано в табл. 3. Анализ данных по стабиль-
ности обсуждается в разделе 7.2.3.
Таблица 3
Программа испытаний согласно плану полного испытания стабильности
Комбинация факторов Время от начала испытания, мес.
0 3 6 9 12 18 24 36
С1 Т т т т т т т т
С2 Т т т т т т т т
а Т т т т т т т т
С4 Т т т т т т т т
С5 Т т т т т т т т
05 Т т т т т т т т
С7 Т т т т т т т т
С8 Т т т т т т т т
сэ Т т т т т т т т
сю Т т т т т т т т
С11 Т т т т т т т т
С12 Т т т т т т т т
С13 Т т т т т т т т
С14 Т т т т т т т т
С15 Т т т т т т т т
С16 Т т т т т т т т
С17 Т т т т т т т т
С18 Т т т т т т т т
С19 Т т т т т т т т
С20 Т т т т т т т т
С21 Т т т т т т т т
СП Т т т т т т т т
С13 Т т т т т т т т
С2А Т т т т т т т т
С25 Т т т т т т т т
С26 Т т т т т т т т
С27 т т т т т т т т
Примечание: Т — тестирование образца.
688 Часть 7. Стабильность лекарств
План сокращенного испытания стабильности. Б отдельных ситуациях получение
количества образцов, требуемых для выполнения полного плана исследования ста-
бильности, невозможно. Иногда следует просто сократить количество образцов,
предназначенных для анализа. В этих случаях выбирают план сокращенного ис-
пытания. Для реализации этого плана необходимо сделать несколько допущений,
которые должны быть обоснованы; эти обоснования должны быть представлены
в регуляторные агентства. Как правило, сокращенный план используется, когда
проводится предварительное испытание стабильности.
Брекетинг и построение матриц — два вида сокращенного планирования, ре-
комендованные FDA [9]. Каждый из этих методов применим в разных ситуациях.
Применение обоих методов одновременно может затруднить определение срока
хранения, поскольку могут быть нарушены комбинации факторов из-за эффекта
наложения одного сокращения комбинаций на другое [10].
Брекетинг заключается в анализе только двух уровней каждого фактора — макси-
мального и минимального — и требует демонстрации того, что выбранные уровни
представляют экстремумы диапазона значений фактора. Если стабильность на экс-
тремальных уровнях различна, то ожидается, что стабильность на любом из про-
межуточных уровней должна быть выше, чем стабильность наименее стабильного
экстремума. В табл. 4 представлен брекетинг-план для полного испытания, описан-
ного в табл. 2.
Таблица 4
Комбинация факторов для брекетинга трех факторов с тремя уровнями
Серия S1 S1 S3
Р1 Р1 РЗ т Р2 РЗ Р1 Р2 РЗ
1 С1 — сз — — — С19 — 021
2 С4 — Об — — — СП — С24
3 С1 — С9 — — — С15 — СП
Заметим, что брекетинг не был применен по отношению к фактору серии, по-
скольку нормативы FDA требуют испытания по меньшей мере трех серий для опре-
деления срока хранения лекарственного препарата. Даже в этом случае количество
образцов, необходимых для исследования согласно брекетинг-плану, существенно
уменьшилось и составило 12 на каждую временную точку. Это существенная эко-
номия, учитывая, что для проведения полного испытания в соответствии с планом
необходимо было проводить испытания 27 образцов образцов. В результате число
образцов, необходимых для всего исследования стабильности, составляет 8 12, или
96, в то время как при полном испытании требуется 216 образцов. Процедура вы-
полнения тестирования стабильности по полной и по сокращенной программам
аналогична представленной в табл. 5.
Построение матриц. План сокращенного испытания с построением матриц за-
ключается в выборе части общего числа возможных комбинаций, включенных
в полное испытание. Каждый раз тестируют различные части комбинации факто-
ров. Основанием для такой схемы является предположение, что стабильность об-
разцов каждой части комбинации репрезентативна по отношению к стабильности
Глава 7.2. Стабильность лекарств
689
Таблица 5
Программа испытаний согласно брекетинг-плану
Комбинация Время от начала испытания, мес.
факторов 0 3 6 9 12 18 24 36
С1 т т Т Т т Т Т т
сз т т Т Т т т Т т
С4 С5 С6 т т Т т т т Т т
т т Т т т т Т т
С7 т т Т т т т Т т
С9 СЮ СИ С12 С13 С14 С15 С16 С17 С18 С19 т т Т т т т Т т
т т Т т т т Т т
С21 т т Т т т т т т
С22 т т Т т т т т т
VsZ.J С24 т т Т т т т т т
С25 rnz т т Т т т т т т
l^ZD С27 т т Т т т т т т
Примечание'. Т— тестирование образца.
образцов всех комбинаций факторов в данный момент времени. Можно построить
матрицу факторов или матрицу временных точек плана испытания стабильности.
Степень сокращения (например, одна вторая или одна треть) от объема полного ис-
пытания зависит от количества учитываемых факторов. Чем больше число факторов
и уровней включено в полное испытание, тем выше степень сокращения, которую
можно применить.
При построении матрицы исключительно по временным точкам все комбинации
факторов (полный факторный план) необходимо протестировать в начальной и ко-
нечной точках времени, в то время как часть комбинаций согласно полному фак-
торному плану тестируют в промежуточных временных точках. Если полные данные
долгосрочного испытания стабильности, необходимые для оценки предполагаемо-
го срока хранения, не доступны для анализа перед представлением в регуляторное
690
Часть 7. Стабильность лекарств
агентство, испытание по полному факторному плану следует проводить в течение
12 мес. и в последней временной точке перед представлением данных. Поэтому
нормативы FDA требуют испытаний по полному факторному плану во временных
точках 0, 12 и 36 мес. для построения матрицы. Кроме того, данные тестирования,
по меньшей мере в трех временных точках, включая исходную точку, должны быть
представлены для каждой комбинации в результатах по первым 12 мес. испытания.
Для построения матрицы для ускоренного хранения следует предусмотреть тести-
рование как минимум в трех временных точках, включая исходную и конечную, для
каждой комбинации факторов.
План, включающий составление матрицы, должен быть максимально сбалан-
сирован, чтобы каждая комбинация факторов была в равной степени исследована
в ходе испытания. Матричный план испытания приемлем, если подтверждающие
данные указывают на прогнозируемую стабильность препарата и малую вариабель-
ность данных. Статистическое обоснование для применения матричного плана ис-
пытания может быть основано на оценке предложенного плана с точки зрения его
способности выявлять различия в скоростях разложения при каждом факторе или
его уровня надежности для установления срока хранения.
Построение матрицы факторов, отличной от матрицы временных точек, как
правило, дает оценку срока хранения с меньшей точностью. Определенный таким
образом срок хранения оказывается меньше того, что был определен с помощью ис-
пытания по полному плану, из-за эффектов смешивания и наложения (confounding
and aliasing effects) [10]. Метод построения матрицы может оказаться недостаточно
эффективным для выявления главных факторов или эффекта взаимодействия фак-
торов. Поэтому избыточное уменьшение числа комбинаций факторов может при-
вести к недостоверной оценке срока хранения из-за пропущенных комбинаций.
С другой стороны, план, включающий построение матриц по временным точкам,
зачастую оказывается столь же эффективным, как и полный план испытания для
выявлении различий скорости развития изменений и установления достоверного
срока хранения. Это связано с тем, что полное испытание всех комбинаций факто-
ров проводится как в начальной, так и в конечной временной точке испытания. Для
сравнения можно разработать планы с матрицами по временным точками, по фак-
торам и их сочетаниям. Читателю следует внимательно подойти к выбору метода
при разработке плана конкретного испытания стабильности и иметь в виду необхо-
димость последующего предоставления подтверждающих данных и обоснований.
Построение матрицы по временным точкам. При построении матрицы по вре-
менным точкам часть комбинаций выбирают согласно плану дробного факторного
эксперимента. Отметим, что представленный выше полный план эквивалентен Зк
факторному плану, где к — число факторов, а 3 — число уровней для каждого фак-
тора. Общее число комбинаций для трех факторов и трех уровней равняется З3, то
есть 27. Предположим, что мы хотим сократить испытание на одну треть с помощью
матричного планирования; в этом случае необходимое число образцов на период
времени равняется 18, в сравнении с 27 для полного плана. В результате число об-
разцов, необходимых для полного испытания стабильности, равно 5 • 18 + 3 + 27, то
есть 171, в то время как для испытания по полному плану требуется 216 образцов.
В табл. 6 представлено расписание реализации матричного плана с построением
Глава 7,2. Стабильность лекарств 691
матриц по временным точкам, рассчитанного на 36 мес. испытания стабильности.
Представлен полный план испытаний в точках 0,12и36ив двух третях оставшихся
точек. Выборка спланирована для соблюдения надлежащего баланса.
Таблица 6
Программа тестирования при плане испытаний стабильности, включающем построение матрицы
по временным точкам
Комбинация факторов Время от начала испытания, мес.
0 3 6 9 12 18 24 36
С1 Т — т Т Т — Т т
02 Т т — Т Т т — т
СЗ Т т т — Т т т т
С4 Т т — т Т т — т
С5 Т т т — т т т т
Об Т — т т т — т т
С7 Т т т — т т т т
08 Т — т т т — т т
09 Т т — т т т — т
СЮ Т т т — т т т т
СН Т — т т т — т т
С12 Т т — т т т — т
С13 Т — т т т — т т
С14 Т т — т т т — т
015 Т т т — т т т т
С16 Т т — т т т — т
С17 т т т — т т т т
С18 т — т т т — т т
С19 т т — т т т — т
020 т т т — т т т т
021 т — т т т — т т
С22 т т т — т т т т
023 т — т т т — т т
024 т т — т т т — т
025 т — т т т — т т
026 т т — т т т — т
С27 т т т — т т т т
Примечание’. Т — тестирование образца.
Если к одному из факторов (содержание активного компонента или размер упа-
ковки) был применен брекетинг или план включал только два уровня для этих фак-
торов, то полный план испытания сокращается (табл. 7).
В табл. 8 представлен матричный план сокращения испытания на две трети
с тремя уровнями содержания активного компонента и двумя уровнями размеров
упаковки. Таким образом, 18 комбинаций факторов включены в расписание тести-
692
Часть 7. Стабильность лекарств
рования во временных точках 0, 12 и 36 мес. и 12 комбинаций для оставшихся вре-
менных точек.
Для плана с построением матрицы общее число образцов, необходимых для
исследования стабильности в целом, равно 3 • 18 + 5 • 12, т. е. 114, при 216 образцах,
необходимых согласно 3*-факгорному плану. Если содержание активного вещества
и размер упаковки и только два уровня применены к двум факторам (или же при-
менен брекетинг), полный план эксперимента сокращен (табл. 9).
В этом случае общее число комбинаций факторов равняется 12 в сравнении
с 27 комбинациями при полном плане исследования. В табл. 10 представлен план,
включающий брекетинг и матричное планирование. Брекетинг применяется по от-
ношению к факторам, а матричное планирование — к временным точкам. Данные
табл. 10 показывают, что существуют 12 комбинаций факторов для временных точек
Таблица 7
Комбинации факторов с учетом двух уровней для упаковки
Серия S1 82 S3
Р1 Р1 Р1 Р2 Р1 Р1
1 С1 С2 С1 С8 С13 С14
2 сз С4 С9 СЮ С15 С16
3 С5 С6 СП С12 С17 С18
Таблица 8
Расписание тестирования согласно плану испытаний стабильности, включающему построение
матрицы по временным точкам (два уровня по размерам упаковки)
Комбинация факторов Время от начала испытания, мес.
0 3 б 9 12 18 24 36
С1 Т — т Т Т — Т т
С2 Т т т — Т т Т т
СЗ Т т — т Т т — т
С4 Т — т т Т — т т
С5 Т т т — т т т т
Об Т т — т т т — т
С7 Т т т — т т т т
С8 Т т — т т т — т
С9 Т — т т т — т т
СЮ Т т т — т т т т
СИ Т т — т т т — т
С12 Т — т т т — т т
С13 Т т — т т т — т
С14 Т — т т т — т т
С15 Т т т — т т т т
С16 Т т — т т т — т
С17 Т — т т т — т т
С18 Т т т — т т т т
Примечание: Т — тестирование образца.
Глава 7.2. Стабильность лекарств
693
Таблица 9
Комбинация факторов с учетом двух уровней для размеров упаковки и содержания активного
компонента
Серия 51 52
Р1 Р2 Р1 Р1
1 С1 С1 С7 С8
2 СЗ С4 С9 СЮ
3 С5 С6 СП С12
О, 12 и 36, и 8 комбинаций факторов для оставшихся временных точек. Таким об-
разом, для матричного планирования такого типа число образцов, необходимых для
испытания стабильности в целом, равняется 3-12 + 5-8, или 76 (в сравнении с 216
образцами, необходимыми для полного 3*-факторного испытания).
Таблица 10
Расписание тестирования согласно плану испытаний стабильности, включающему построение
матрицы по временным точкам (два уровня по размерам упаковки и содержанию активного
вещества)
Комбинация факторов 0 Время от начала испытания, мес. 36
3 6 9 12 18 24
С1 т — т Т Т — Т т
С2 т т т — Т т Т т
СЗ т т — т Т т — т
С4 т — т т Т — т т
С5 т т т — т т т т
С6 т т — т т т — т
С7 т т — т т т — т
С8 т — т т т — т т
С9 т т т — т т т т
СЮ т т — т т т — т
С11 т — т т т — — т
С12 т т т — т т т т
Примечание: Т — тестирование образца.
Построение матрицы факторов. При планировании с построением матрицы фак-
торов комбинации факторов сокращаются систематическим путем, представлен-
ным в табл. 11. В результате не все комбинации факторов тестируют в процессе ис-
пытаний стабильности. Подобный план можно использовать, когда сокращенная
комбинация проявляет свойства, аналогичные свойствам других комбинаций в про-
цессе испытания. Схема реализации матричного плана представлена в табл. 12. Все
комбинации факторов тестируют во временных точках 0, 12 и 36 мес.; 18 комбина-
ций факторов тестируют в остальных временных точках. Для этого матричного пла-
на число образцов, необходимых для испытания в целом, равняется 3-27 + 18-5,
или 171; при 216 образцах, необходимых для полного З^-факторного испытания.
Составление матриц факторов и временных точек представляет собой комбина-
цию двух вышеупомянутых способов матричного планирования; схема испытания
694
Часть 7. Стабильность лекарств
Таблица 11
Составление матриц при сокращении комбинаций факторов
Серия - S1 S1 S3
Р1 Р2 РЗ Р1 Р2 РЗ Р1 Р2 РЗ
1 — а сз сю С11 — С19 — С21
2 С4 — С6 — С14 С15 С22 С23 —
3 С7 С8 — С16 — С18 — С26 С27
Расписание тестирования согласно плану испытаний стабильности, включающему построение
матрицы факторов
Комбинация ______________________Время от начала испытания, мес._________________
Таблица 12
факторов 0 3 6 9 12 18 24 36
С1 т — — — т — — т
С1 т т т т т т т т
сз т т т т т т т т
С4 т т т т т т т т
С5 т — — — т — — т
С6 т т т т т т т т
С7 т т т т т т т т
С8 т т т т т т т т
С9 т — — — т — — т
СЮ т т т т т т т т
СП т т т т т т т т
С12 т — — — т — — т
С13 т — — — т — — т
С14 т т т т т т т т
С15 т т т т т т т т
С16 т т т т т т т т
С17 т — — — т — — т
С18 т т т т т т т т
С19 т т т т т т т т
С20 т — — — т — — т
С21 т т т т т т т т
С22 т т т т т т т т
С23 т т т т т т т т
С24 т — — — т — — т
€25 т — — — т — — т
€26 т т т т т т т т
С27 т т т т т т т т
Примечание-. Т — тестирование образца.
представлена в табл. 13. Согласно этой схеме все комбинации факторов тестируют
во временных точках 0, 12 и 36 мес., а дробный факторный эксперимент проводят
в оставшихся временных точках. Общее число экспериментов в этом испытании
Глава 7.2. Стабильность лекарств 695
стабильности равняется 3 • 27 + 5 12, то есть 141, при 216 образцах, необходимых
для полного 3*-факгорного испытания.
Таблица 13
Расписание тестирования согласно плану испытаний стабильности, включающему построение
матрицы факторов и временных точек
Комбинация факторов Время от начала испытания, мес.
0 3 6 9 12 18 24 36
С1 Т — т Т Т т Т т
С2 Т т — Т Т — Т т
СЗ Т — — — Т — т
С4 Т — — — Т — — т
С5 Т т т — Т т — т
С6 Т — т т Т т т т
С7 Т т т — т т — т
С8 Т — — — т — — т
С9 Т т — т т — т т
СЮ Т т — т т — т т
С11 Т — — т * — — т
С12 Т — т т т т т т
С13 Т т т — т т — т
С14 Т — т т т т т т
С15 Т — — — т — — т
С16 Т — — — т — — т
С17 Т т — т т — т т
С18 Т т т т т т
С19 Т — — — т — — т
С20 Т — т т т т т т
С21 т т — т т — т т
С22 т — т т т т т т
С23 т —- — — т — — т
С24 т т т — т т — т
С25 т т — т т — т т
С26 т т т — т т — т
С27 т — — — т — — т
Примечание. Т — тестирование образца.
7.2.3. Анализ данных исследования стабильности
при долговременном хранении
7.2.3.1. Введение
Назначение данного раздела — предоставить набор базовых статистических ин-
струментов для расчета срока хранения для одной и нескольких видов первичной
упаковки и дозировок препарата. Статистические методы будут описаны сначала
в общем контексте, а затем на численных примерах будет продемонстрировано их
696
Часть 7. Стабильность лекарств
применение в конкретных случаях. В Приложении к настоящей главе (7.2) пред-
ставлен набор компьютерных программ, упрощающих процедуры анализа резуль-
татов исследования стабильности. Chen [et al.] [11] показал, что такой анализ, как
правило, включает три этапа. Первый этап заключается в накоплении результатов
исследования образцов, помещенных на хранение в определенных условиях и под-
вергшихся испытаниям через определенные временные интервалы. Второй этап —
выбор надлежащей модели описания соотношений между результатами испытания
и временем отбора и анализа проб. Третий этап состоит в определении (расчете)
срока годности препарата. Первый этап описан в разделе 7.2.2, второй и третий эта-
пы описаны в разделе 7.2.3.
7.2.3.2. Сроки хранения одной партии лекарств
Здесь будет представлен расчет срока хранения лекарственного препарата в одном
виде упаковки при условии, что результаты получены при анализе образцов, отно-
сящихся к одной производственной серии. Руководство FDA устанавливает, что це-
лью испытания является определение времени, по истечении которого усредненная
кривая разложения (односторонний доверительный интервал 95%) пересекает допу-
стимый нижний предел согласно спецификации, который, как правило, определя-
ется в FDA как 90% от содержания, заявленного на этикетке (LC-label claim). Пред-
положив, что концентрация действующего вещества в лекарственном препарате
уменьшается линейно с течением времени, можно описать процесс уравнением
у. = а + (Зх. + е. (г=1,..., л), (1)
где у. — содержание действующего вещества в определенный момент времени х.
для z-го образца, выраженный в процентах от количества, указанного на этикетке
(результат испытания на количественное определение); х(. — время, по истечении
которого тестировали i-й образец; аир — коэффициенты регрессии (коэффици-
ент а соответствует процентной доле от содержания, указанного в маркировке, ког-
да х. = 0 (так называемый эффект серии — batch effect), коэффициент р описывает
скорость разложения, а произведение рх. представляет собой потерю стабильности
с течением времени). Предполагается, что случайная переменная е. обладает слу-
чайным распределением вокруг нулевого среднего и постоянной дисперсией о2;
п — общее число образцов. В разделе 7.2.4 представлены различные кинетические
модели разложения. В данном контексте уравнение (1) описывает разложение ну-
левого порядка.
Дата истечения срока годности для одной производственной серии указывает на
то, что с 95%-ной вероятностью средние значения характеристик лекарства в до-
зированных единицах из данной серии соответствуют спецификациям до истече-
ния срока, обозначенного датой истечения срока годности. На рис. 1 представлена
нижняя граница предела 95%-ного доверительного одностороннего интервала для
усредненной линии разложения.
Значение 95% доверительного интервала может быть выражено в виде следую-
щего выражения доверительной вероятности:
Л^/0>05.я_2) = 0,95, (2)
Глава 7.2. Стабильность лекарств
697
где te 05 л_2 — значение процентили (квантили) распределения Стьюдента при одно-
стороннем уровне значимости а = 0,05 и степенями свободы п — 2. Предположив,
что величина процентной доли от заявленного содержания (£С%)описывается нор-
мальным распределением, статистический параметр / определяют как:
t = z = [у—(а + Рх)]/Уо2[1/и + (X - х)2Дхх ]
4x1,1т
где 5хх = £(х-х)2
>/5S£/o2[(n-2)]
5SE=X(y,-y)2 (4)
i=i
где SSE — сумма квадратов остатков регрессионной модели (из уравнения 1); Z—
случайная переменная, следующая закону стандартного нормального распределе-
ния; х2т — квадрат случайной переменной, следующий х2-распределению с т степе-
нями свободы; у — теоретическое (расчетное) значение содержания действующего
вещества (в процентах от заявленного) во временнбй точке х; а и b — оценки пара-
метров аир соответственн; х — усредненное время отбора проб (среднее значение
значений временных точек в течение 1 мес).
Для исключения неизвестного параметра о2 использовали преобразования, до-
пустимые при распределении Стьюдента. Таким образом, уравнение (1) можно за-
писать в следующем виде:
у-(а+Рх).
(5)
Jmse -+(* х)2 >
V L п Sxx
SSE
MSI^-^2’
(6)
где MSE (mean square error) — среднеквадратичная ошибка регрессии.
Рис. 1. Графическая иллюстрация определения срока хранения
698
Часть 7. Стабильность лекарств
Теперь уравнение (2) можно записать в виде
р( У (а Рх) .< f 'I — 0 95 m
с —‘о,о5,л-21 — (/)
После проведения ряда математических преобразований уравнение (7) прини-
мает следующий вид:
р(у - 'о,о5,л-2М «+₽*) = 0,95.
(8)
Исходя из этого граница нижнего предела 95%-ного доверительного односто-
роннего интервала составляет
ОД = У-4.О5,л-25У = Я-^_/'о,О5.л-2^Б ’ (9)
Точки пересечения Цх) с нижним пределом приемлемости 8 могут быть опреде-
лены путем вычисления корней уравнения 8 — £(х) = 0. Следует указать, что данное
уравнение может быть записано в виде
f(x) = [8-(fl+8x)]2-rUn-2
MSE
'1 ! (*~*)2'
_И
= 0.
(10)
Квадратное уравнение (10) имеет два корня; срок хранения рассчитывают вы-
числением корня уравнения (10), значение которого меньше контрольной точки,
определяемой так:
fco-8
—т—
-61
(Н)
Ниже представлен численный пример, иллюстрирующий шаг за шагом процеду-
ру расчета срока хранения.
Пример 1. Срок хранения для отдельной серии. Результаты испытания для одной
серии лекарственного препарата представлены в табл. 14. На основании этих ре-
зультатов определите срок хранения данной серии.
Таблица 14
Результаты испытания одной производственной серии, % LC
Время испытания, мес. 0 3 6 9 12 18 24 36
Процентная доля от заявленного содержания 103,5 97,3 97,2 94,2 94,8 94,1 91,3 88,7
Решение. Во-первых, для предварительного определения линии разложения вос-
пользуемся методом регрессии [12]. Уравнение регрессии выглядит следующим об-
разом:
У = 99,64 - 0,3335х.
Хорошо известно, что традиционные компьютерные программы для статисти-
ческих расчетов, такие как MINITAB, STATGRAPHICS или SAS, проводят подгонку
модели и позволяют получить дополнительную информацию:
Глава 7.2. Стабильность лекарств
699
^о,о5,л—2= 'о,05,6= 1>943,MSE= 4,1665, п = 8,х = 13,5, 5п = 1008.
Для удобства читателя компьютерные программы, проводящие подгонку модели
регрессии и выполняющие все статистические расчеты, необходимые для определе-
ния срока годности, представлены в Приложении.
Таким образом, нижняя граница одностороннего 95%-ного достоверного интер-
вала для усредненной скорости разложения описывается выражением
I Г1 (х-13 5)2'
Цх) = 99,64 - 0,3335х — 1,943 J4,1665 -
V Lо 100о
Следовательно, срок хранения можно определить при помощи следующего урав-
нения:
/(х) = [90 -(99,64 = 0,3335х)]2 —1,9432(4,1665)
1
.8
(х-13,5)2
1008 .
= 0.
(12)
Прежде чем приступать к определению срока хранения, необходимо рассчитать
значение контрольной точки:
^99,64-90
ге/ -/>! 0,3335
Таким образом, срок хранения соответствует корню, который меньше 28,90.
Простым и практичным инструментом расчета корней уравнения (12), вероятно,
является решение следующей эквивалентной проблемы. Следует найти х, соответ-
ствующий минимальному значению Дх). Корень находят при помощи алгоритма
квазиньютоновского линейного поиска {QNLS, quasi-Newton line search) [13]. Ком-
пьютерная программа требует расчета начальной точки; мы рекомендуем использо-
вать значение
x(0)=xRe/-J, (13)
где d — положительная величина, которую ищут методом проб и ошибок, пока про-
грамма не находит локальный минимум. Обычно значение d попадает в интервал
от 0 до 10. Например, если d = 0, метод QNLSрассчитывает xR = 23,6989. Срок хра-
нения xL представляет собой целую часть корня xR, и в этом случае временная точка
истечения срока годности данной серии соответствует х£ = 23 мес.; отсчет ведется
от/(хЛ) = 0 и xR < xref Метод QNLS включен в программу Matlab', компьютерная про-
грамма также представлена в Приложении.
7.2.3.3. Расчет срока годности по данным изучения стабильности
нескольких серий препарата
Анализ возможности группировки данных по сериям. Руководство FDA устанавливает,
что по меньшей мере три производственные серии необходимо использовать для
определения вариабельности между сериями. Таким образом, если статистическая
процедура представляет свидетельство того, что три серии принадлежат к одной
популяции, единый срок хранения для всех серий определяется с помощью груп-
700
Часть 7. Стабильность лекарств
пировки данных по всем сериям. Также руководство устанавливает, что сходство
линий разложения для отдельных серий можно оценить сравнением углов наклона
и размером отрезков, отсекаемых на оси у (интерсептов) для индивидуальных серий.
Предположим, что разложение описывается линейной зависимостью во времени
(разложение нулевого порядка); тогда мы можем представить его следующей моде-
лью, аналогичной уравнению (1):
у_=а.+ Рх, + е..; i = 1,..., I j = 1,..., п. (14)
где ytj — результат испытания (% LC) i-й серии лекарственного препарата (отбор
пробы и тестирование проведено в момент времени х„); п. — число отборов проб из
i-й серии; I— общее число серий; а. и р. — точка пересечения с осью у (интерсепт)
и наклон линии разложения для i-й серии соответственно; е.. — случайная перемен-
ная с нулевым средним и постоянной дисперсией. Следует отметить, что а, рассма-
тривается как эффект серии, а р,— как скорость разложения для i-й серии.
В руководстве FDA содержится следующее указание: анализ равенства накло-
нов и точек пересечения должен проводиться на уровне значимости 0,25, как было
предложено Bancroft [14]. Было бы желательно исследовать серии на принадлеж-
ность к единой популяции, и, если результат исследования положителен, вывести
модель с единой точкой пересечения оси у и наклоном линии разложения для всей
популяции серий. Таким образом, для определения принадлежности к единой попу-
ляции используется анализ возможности группировки данных, который проводится
в два этапа: 1) анализ равенства наклонов и 2) анализ равенства точек пересечения.
Традиционная методика проверки гипотез заключается в анализе ковариации для
полностью рандомизированного плана [15, 16]. Альтернативный метод, который
легко обобщить для изучения многофакторных систем, состоит в применении мо-
дели с индикаторными переменными [17].
Анализ ковариации (ANCOVA) при оценке равенства наклонов. Первый этап состо-
ит в проверке сходства скоростей разложения для всех серий. Проводится анализ
следующей гипотезы:
Но: р,= Ру. для всех i * J, i = 1,..., I, j = 1,I, (15)
где I— число производственных серий.
Чтобы рассчитать /'-статистику и провести проверку вышеупомянутой гипоте-
зы, необходимо рассчитать разные суммы квадратов и векторных произведений для
% LC, атакже для временной точек тестирования [15,16,18]. Общая сумма квадра-
тов для разных времен выборки определяется так:
I
ZXX=TJSXX(f)> (16)
1=1
г«е Sxx(i) = -х,.,)2 = £х2 (17)
>1 7=1 П/
Л - х,
*..=2Л *.-= •
м п.
Глава 7.2, Стабильность лекарств
701
Общая сумма квадратов % LC определяется так:
z„=ZX(o, (18)
1=1
гДе (0 = -У,.)2 = IX > <19>
7-1 7-1 П1
V - yt.
У-=~Г-
7-1 П1
Общая сумма векторных произведений между % LCw. временами взятия выбор-
ки представлена в следующем виде:
(20)
1=1
где ^(0 = Х(^-х,О2(уй-у,-.) = Хх-.у..-^^. (21)
>1 7=1 ni
Таким образом, суммарная среднеквадратичная погрешность, основанная на об-
щих суммах, рассчитывают по выражению
Z2
SSE.=Z„-^. (22)
Общая суммарная квадратичная погрешность для каждой серии, которая также
необходима для вычислений, рассчитывается так:
SSE=^SSE{i), (23)
1=1
где 5Ж(0=^-^- (24)
Среднеквадратичная погрешность с учетом вклада погрешности наклона и сум-
марные среднеквадратичные погрешности определяются по формулам
SSE-SSE
MS^oPe=----JTj--> (25)
MSE= SSE , где N = Yni- (26)
N-21 ы
В итоге выражение для проверки гипотезы по /'-статистике можно записать сле-
дующим образом:
F(slope) = MSs'0,,e.
MSE (27)
Следует отметить, что гипотеза (15) опровергается, если F{slope) > /025 21 N 2P
где /025 j j N2J — верхний процентиль /'-распределения с /— 1 иЛг- 2/степенями
702 Часть 7. Стабильность лекарств
свободы. Если гипотеза (15) не опровергается (т. е. наклоны линии одинаковы),
можно переходить к следующему этапу — проверке равенства положения точек пе-
ресечения с осью у линии разложения для разных серий, проходящих испытание.
Методика вычислений на этом этапе представлена ниже.
ANCOVA при анализе положения точек пересечения линии разложения с осью у. За-
дача второго этапа — анализ положения точек пересечения индивидуальных линий
разложения с осью у при условии равенства наклонов линий разложения серий,
проходящих испытание. Исследуемая гипотеза может быть записана в виде
Но: а.=а. длявсех z#/, z=l,j = (28)
Поскольку не выявлено различий в наклонах линий, модель (14) сокращается до
модели, описываемой единой скоростью разложения, то есть:
у^а. + рх.+ в.., j = (29)
Отрезки, отсекаемые кривой разложения на оси у, можно разложить на два эле-
мента: отрезок, общий для всех линий, и эффект серии. То есть а + т., где а = ц - рх_
представляет собой общее слагаемое, а т. — эффект серии; р — ожидаемое значение
y.j, а х_— среднее значение xff, определенное уравнением (32). Следует отметить, что
отклонение каждого интерсепта от общего слагаемого называется эффектом серии.
В этом случае модель описывается выражением
У{,=ц+т,.+р(х..-х_)+£... (30)
Модель, не учитывающую эффекта серии, сравнивают с моделью, учитывающей
эффект серии, чтобы измерить интерсепт-эффект. Уравнение (30) учитывает эффект
серии, его можно назвать уравнением полной модели. Модель, не учитывающая эф-
фекта серии, называется сокращенной моделью и выражается уравнением:
У1?=М+₽(^-^_)+£<,- z = l, ...,7 у = 1, ...,л,- (31)
Сумма квадратов, векторных произведений для сумм и погрешности для сокра-
щенной модели рассчитываются по уравнениям [12,18]:
I nt I ni г2 1 I щ
^xx = XX(xv-^--)2 =ZX^7--T7’ ^ = —YYxu x- = Nx. (32)
i=l/=l i=l/=l /V 7Vi=iy=i
i щ i ni v2 i i щ
Куу = ХХ(Уч-У-У = = y- = Ny„ (33)
i=lj=l i=l/=l /V /V i=i;=i
/ И/ 1 ni Y V
Rxy = - x..)(yij - y..) = XX^y.7 —— ’ (34)
i=lj=l i=l/=l /V
где /^определено уравнением (26).
Глава 7.2. Стабильность лекарств 703
Сумму квадратов для полной модели можно рассчитать следующим образом:
^=Еп,.(х,..-х..)2 = х— 1=1 /=1 И,- W (35)
(36)
‘ х..у.. TXy = Lni(xi. О(к у..) = х ; >=1 .=1 «i N 1 Hi (37)
Ехх = £Ё(х0 - х-)2 = R** - ‘=4=1 1 (38)
Еуу = - У;-)2 = Ryy ~ТУУ’ <=i/=i i «i (39)
Exy = XXUy - X..)(y,7 - y„) = Rxy - Txy <=i;=i (40)
Погрешность суммы квадратов для сокращенной модели можно рассчитать по
формуле
п2
SSE^R^-f-. (41)
Rxx
Погрешности суммы квадратов для полной модели можно рассчитать по фор-
муле
F2
SSE = Е —(42)
е УУ Е
XX
Среднеквадратичные погрешности интерсепт-эффекта и среднеквадратичные
погрешности полной модели можно записать в виде
MSE^^-8^ MSE^-j^. (43)
И наконец, для подтверждения сходства или различия интерсептов между серия-
ми по F-статистике можно записать следующее выражение:
F(int) =
MSEM
MSEC
(44)
Гипотеза (28) опровергается, если F(int) > F0251_j И1, где F025 7 , N_11 — верхний
процентиль F-распределения с I— 1 и N— / —1 степенями свободы. Далее, если ну-
левые гипотезы (15) и (28) не опровергнуты на уровне значимости 0,25, серии могут
быть отнесены к единой популяции или группе; в этом случае рассчитывается еди-
ный срок хранения на основании данных для серий, проходящих испытание. Тогда
уравнение модели (14) сокращается до выражения
704
Часть 7. Стабильность лекарств
yv= а + Рх.. + е.; i= 1,I, J -1,.... nr (45)
где аир — соответственно равные наклоны и интерсепты для модели (45).
Принцип расчета срока хранения единичной серии можно применить при обра-
ботке сводных данных по стабильности по всем сериям. Таким образом, срок хране-
ния рассчитывают по минимальному корню уравнения (10), где а и b соответствуют
значениям соответственно а и р в уравнении модели (45); х— усредненное время
выборки, a определяется исходя из уравнения (4) и учитывается для всех времен
выборки. Если гипотеза (15) не опровергнута, а гипотеза (28) опровергнута, то урав-
нение модели (14) принимает вид
ytj = а. + Рх. + е..; 4=1,..., I, j = 1,..., пг (46)
Предположим, что у разных серий с одинаковым наклоном линии разложения
различаются интерсепты; в этом случае даты истечения срока годности рассчитыва-
ют для каждой серии индивидуально. Датой истечения срока годности лекарствен-
ного препарата становится дата, соответствующая минимальному сроку хранения
для отдельной серии.
Третья возможность возникает, если опровергается гипотеза (15); в этом случае
делают вывод о том, что серии не относятся к одной популяции; тогда срок хра-
нения необходимо рассчитывать индивидуально для каждой отдельной серии. Ми-
нимальному сроку годности отдельной производственной серии соответствует срок
хранения лекарственного препарата.
Пример 2. Наклоны и интерсепты равны. Этот пример иллюстрирует применение
анализа возможности группировки данных для анализа сходства производствен-
ных серий. Данные испытаний трех производственных серий, выраженные в % ЕС,
представлены в табл. 15. Требуется подтвердить или опровергнуть принадлежность
серий к одной популяции.
Таблица 15
Результаты испытаний (% LC)
Время замера, мес. 0 3 6 9 12 18 24 36
Серия 1 102,4 98,1 99,2 97,5 95,0 96,1 — —
Серия 2 101,1 101,2 99,0 97,2 96,4 95,5 94,3 —
Серия 3 104,1 102,1 99,5 98,1 95,7 94,1 94,0 93,5
Решение. Анализ возможности объединения данных проводится в два этапа.
Этап 1. Во-первых, следует провести анализ, чтобы подтвердить или опровер-
гнуть равенство наклонов линий разложения для трех производственных серий. Из
табл. 15 можно извлечь следующую информацию: 1= 3, — 6, п2 = 7, п3 = 8 и N= 21.
Первая гипотеза, подлежащая проверке, описывается следующим выражением:
Яо: р1 = р2 = рз = р. (47)
При помощи уравнений (16)—(21) рассчитывают суммы квадратов и векторных
произведений для % ЕС и временных точек, в которые проводились испытания об-
разцов:
Глава 7.2. Стабильность лекарств
705
6 Л
Ml) = 2>у ~ =З2 + 62 +... +182= 210;
j=l б б
7 г? 7?2
М2) = Х4 - 4г = З2 + 62 +... + 242 - = 429,43;
м / /
8 г 2 1 ПО2
М3) = Е4 -— = З2 + б2 +... + 362 -— = 1008;
Ml) = iyb ~- = Ю2,42 + 98,12 +... + 96,12 - = 33,65;
>1 6 6
М2) = iyij ~—=Ю1,12 +101,22 +... + 94,32 - = 43,75;
/=1 7 7
М3) = J Уз/ -— = 104,12 +102,12 +... + 93,52 -= 111,98;
>1 8 8
Ml) = ЕЗД/ " = 3<98>1)+6("’2)+• • • +18(96,1) - ^f8»3) = -69,6;
/=1 б 6
М2) = £ x2iy2j - = 3(101,2) + 6(99)+... + 24(94,3) - 72(684’7) = -131,23;
7=1 7 7
М3) = £зд>" = 3(102,1) + 6(99,5)+... + 36(93,5) - --°-8(781)1) = -294,45.
7=1 8 8
Рассчитывают полные суммы квадратов:
Z„ = £М0 = 210+429,43+1008 = 1647,43;
Zyv = ^Syy(t) = 33,65+43,75+111,98 = 189,38;
Zxy = YS*y(i) = -69,6 -131,23 - 294,45 = -495,28.
По уравнению (22) вычисляют общую сумму квадратов погрешностей:
SSE =Zyy-^-^ 189,38 - 495,282 = 40,48.
° уу Zxx 1647,43
706
Часть 7. Стабильность лекарств
Сумму квадратов погрешностей для каждой серии рассчитывают по уравнению
(24):
4s£w=s>’(i)-O)=33’65’^=i0’58:
SSE(2) = Syy(2) - = 43,75 - = з,65;
5^(2) 429,43
S2 63) 294 452
SSE(3) = 5W(3) —2^4 = 111,98 - ' = 25,97.
S„(3) 1008
По уравнению (23) вычисляют общую погрешность сумм квадратов:
з
SSE = ^SSE(i) = 10,58 + 3,65 + 25,97 = 40,2.
i=i
Таким образом, среднеквадратичная погрешность за счет наклона рассчитывает-
ся по уравнению (25):
.^-5^^4038^^
71 31
Среднеквадратичная ошибка модели ретрессии (14):
MSE=
SSE
N-21
40,2
21-2(3)
= 2,68.
Окончательное уравнение для проверки равенства скоростей разложения по
F-статистике (гипотеза (47)):
F (slope) -
MSsbpt 0,14
MSE 2,69
= 0,052.
Критическое значение Fnpn анализе наклонов составляет F0 25 / 1 JV_2/ = F0 25 2 15=
= 1,52. Поскольку F (slope) < F0 25 2 15, гипотеза (47) о равных скоростях разложения
не может быть опровергнута на уровне значимости 0,25. Вывод будет таким: значи-
тельной разницы между величинами наклонов согласно модели (14) нет, т. е. мо-
дель, представленная уравнением (14), сокращается до модели (29).
Этап 2. Второй этап заключается в анализе значений интерсептов для линий раз-
ложения для разных серий. Для расчета статистических параметров, позволяющих
подтвердить или опровергнуть соответствующую гипотезу, требуется рассчитать
следующую сумму и векторные произведения для % ЕСи времена замеров. Анализу
подлежит следующая гипотеза:
Яо: а, = а2 = а3 = а.
(48)
Сумму и векторные произведения рассчитывают исходя из уравнений (32)—(40):
Глава 7.2. Стабильность лекарств
707
Йг2 ?782
х?-— = 32 + 62 + ... + 362- —= 1754,5;
N 21
з и, v2 905412
Л™ = У У J.7 -— = 102,42 + 98,12 +... + 93,52 - ’ = 189,97
УУ N 21
= ИъУц - ^ = 3(98,1)+6(99,2) +... + 36(93,5) - 228Р054Д) = _503 06;
i=ij=i /V Z1
^^^T^IO^,^
“ йл, N 6 7 8 21
2 у2 yi. 588,32 684,72 781,12 2054,12
уу hn, N 6 7 8 21
Лх,.у,. х_у„ 48(588,3) 72(684,7) 108(781,1) 228(2054,1)
У £1 п,, N 6 7 8 21
£хх = - X,) = Rxx -Т„ = 1754,5 -107,14 = 1647,36;
3 в,
Еуу = У У (№ - к)2 = Куу - Туу = 189,97 - 0,59 = 189,38;
<=1/=1
3 в,
Еху = LLU/ - *>)(№ - У..) = Ку ~Тху= -503,06 + 7,78 = -495,28.
i=i/=i
Погрешности сумм квадратов для сокращенной и полной модели представлены
уравнениями (41) и (42) соответственно:
SSEr =R------^ = 189,97- ’ =45,73;
' п 1754,5
Е2 495 282
^^-#=W8-^=40,47.
XX ’
Среднеквадратичные погрешности измерений интерсепт-эффекта рассчитыва-
ют с помощью уравнения (43):
SSEr-SSEc 45,73-40,47 _
МЕЕм =-------;—- = ———— = 2,63;
3-1
MSEC=^^ = ^- = 2,3S.
с N-1-Л 21-3-1
708
Часть 7. Стабильность лекарств
Окончательно F-статистика для оценки равенства интерсептов описывается
уравнением (44) и рассчитывается по формуле
F(int) = MSEb* = =1,105.
MSEC 2,38
Критической величиной при анализе равенстве интерсептов является 1Л NI l =
= Fo 25 217 = 1,51. Поскольку F(inf) < FQ 25 2 17, отрицать нулевую гипотезу, выраженную
уравнением (47) при 25%-ном уровне значимости, нет оснований. Можно сделать
заключение о статистическом равенстве интерсептов.
В итоге нулевая гипотеза о равенстве наклонов интерсептов не опровергнута на
уровне достоверности 0,25 и, следовательно, все производственные серии относятся
к одной популяции. Поэтому период срока годности препарата можно рассчитать
с помощью модели (45).
После объединения данных по трем сериям уравнение линии регрессии можно
записать как
у = а=Ьхв = 100,93 - 0,2867хй.
Стандартная подпрограмма расчета параметров регрессии позволяет получить
следующие данные: MSE = 2,407, х = 10,8571 и = 1754,6. Таким образом, для
определения срока хранения следует воспользоваться следующим уравнением:
fix) = [90 - (100,93 - 0,2867х)]2 - 1,729]2(2,407)
(х-10,86)2
1754,6
= 0.
Для расчета срока хранения необходимо найти корень данного уравнения, зна-
чение которого меньше, чем контрольная величина, рассчитываемая по формуле
а-90 100,93-90
' . =----=-----------=38,11.
-Ь 0,2867
Период до окончания срока годности должен составлять менее 38,11 мес. Из-
вестно положение исходной точки х(0) = хге/— d = 38,11 — 8. С помощью QNLSал-
горитма получаем значение xR = 32,801. Таким образом, период до окончания срока
годности для производственных серий, проходящих испытание, составляет 32 мес.
Принцип минимума при испытании нескольких серий. Если гипотеза о равенстве
наклонов опровергнута на уровне значимости 0,25, следует применить принцип ми-
нимума, поскольку линии разложения для индивидуальных серий нельзя считать
схожими из-за различия скоростей разложения. В данных условиях руководство
FDA предписывает: общая длительность периода до истечения срока годности долж-
на быть такова, чтобы параметры препарата находились в приемлемых пределах не-
зависимо от их происхождения (производственной серии). В этом случае рассчиты-
вают срок хранения для каждой серии, дату истечения срока годности определяют
исходя из минимального срока хранения по данным для всех производственных се-
рий. Математически это можно выразить так:
min{x£(l),.... А", х£(Л)}. (49)
где xL — срок хранения i-й серии; I— общее число производственных серий.
Глава 7.2. Стабильность лекарств 709
Поскольку минимальный период до истечения срока годности представляет
кратчайший из сроков хранения, определенных для всех серий, эта оценка обеспе-
чивает 95%-ную гарантию того, что содержание активного вещества в лекарствен-
ном препарате остается выше нижнего предела приемлемости, указанного в спе-
цификации.
Пример 3. Наклоны и интерсепты различны. В ходе испытания стабильности по-
лучены результаты, представленные в табл. 16. На основании данной информации
определите срок хранения лекарственного препарата, прошедшего испытания.
Таблица 16
Результаты испытания, %LC
Время замера, мес. 0 3 6 9 12 18 24
Серия 1 99,2 97,1 96,1 95,2 93,8 93,1 92,4
Серия 2 98,7 97 96,2 95,1 94,2 93,3 —
Серия 3 102,5 98,9 97,1 95,6 94,1 93,1 —
Решение. Традиционный подход к определению срока хранения требует анали-
за наклонов и интерсептов прямых линий, соответствующих разложению серий,
проходящих испытание. Кроме того, для анализа сходства наклонов и интерсептов
применяют метод ANCOVA.
Стратегия заключается в анализе нулевой гипотезы равенства наклонов, которая
описывается уравнением (14). Для того чтобы подтвердить или опровергнуть эту ги-
потезу, необходимо рассчитать статистические параметры по уравнению (27):
MSE(slope) 4,0507
F(slope) =------——-=—--------=5,04.
MSE 0,8032
Нулевая гипотеза, выраженная уравнением (15), опровергается на уровне значи-
мости 0,25, поскольку критическое значение F025 213 = 1,55 в этом испытании мень-
ше, чем F (slope). Отсюда следует вывод: наклоны линий разложения для серий,
проходящих испытания, различны, вследствие чего следует применить принцип
минимума. Таким образом, рассчитываются сроки хранения для всех серий, про-
ходящих испытания, и минимальный срок хранения применяют ко всем производ-
ственным сериям.
Для первой производственной серии срок хранения рассчитывают по корню
уравнения, значение которого меньше контрольного значения:
[90-(98,0461-0,2898х)]2-2,0152(0,6689)
1 (х-10,28)2
7 + 429,43
=0.
Контрольная точка для данного уравнения
= д-8 98,0461-90
п1~ -Ь ~ 0,2698
= 29,82.
Исходная точка х(0) = х^ — 8, корень уравнения хЛ(1) = 24,93, и срок хранения
для первой серии составил х£(1) = 24 мес.
710
Часть 7. Стабильность лекарств
Используя данные, полученные для второй серии, составляют уравнение
[90-(98,1157-0,2957х)]2 - 2,13182(0,2328)
1
-+
6
(х-8)2'
210 .
= 0.
Контрольная точка для данного уравнения хп = 27.44, а исходной точкой для за-
вершения преобразований является хге/— 5. Соответствующий корень хА(2) = 23,44,
а срок хранения для серии 2 равняется х£(2) = 23 мес.
Соответствующее уравнение для серии 3 имеет вид
[90—(100,91—0,5033х)]2—2,13182(1,5415)
1
-+
.6
(х-8)2
210
= 0.
Контрольная точка для этого уравнения хгс/ = 21,67, а отправная точка конвер-
генции — хг^— 5. Корень уравнения хд(3) = 17,59, а срок хранения серии составляет
х£(3) = 17 мес.
Таким образом, срок хранения для будущих производственных серий составляет
min {24 23 17} = 17 мес.
Пример 4. Равные наклоны и различные интерсепты. Результаты испытания ста-
бильности представлены в табл. 17. На основании данной информации определите
срок хранения лекарственного препарата.
Таблица 17
Результаты испытаний, %LC
Время замера, мес. 0 3 6 9 12 18 24 36
Серия 1 98,4 96,1 94,2 93,5 90 89,1 89,2 87,3
Серия 2 99,1 97,2 96,3 95,2 93,4 91,5 90,3 —
Серия 3 104,1 102,1 99,5 98,1 95,7 94,1 94,0 93,5
Решение. Метод ANCOVA используют для подтверждения или опровержения ги-
потезы о равенстве наклонов линий разложения для различных серий. Статистика,
определяющая возможность подтверждения или опровержения данной гипотезы,
представлена уравнением (27):
MSE(slope) 0,9891 n
F(slope)=----1= —-----------= 0,3645.
MSE 2,7134
Нулевая гипотеза, выраженная уравнением (15), не может быть опровергнута на
уровне значимости 0,25, поскольку критическое значение Ео 25 2,7 = 1,51 для данного
испытания больше, чем F (slope). Вывод: наклоны линий разложений для этих се-
рий совпадают.
Далее необходимо проверить, совпадают или не совпадают интерсепты серий,
проходящих испытания. Статистические параметры для проверки совпадений рас-
считывают по уравнениям (43) и (44), в результате получают:
= = 172,19-48,10 =
1,11 7-1 2
Глава 7.2. Стабильность лекарств
711
с N-I-1 19
F(int) = MSEa± = = 24,51.
MSEC 2,53
Нулевая гипотеза, выраженная уравнением (28), отвергается на уровне значимо-
сти 0,25, поскольку Fo 25 217 = 1,49 для данного испытания больше, чем /(int). Вывод:
интерсепты линий разложений для этих серий различаются на уровне значимости
0,25, сроки хранения оценивают для каждой отдельной серии, а минимальная вели-
чина срока хранения приписывается всем производственным сериям.
Модель, описывающая линию разложения, представлена уравнением (46), где
7= 3, nt = 8, пг = 7 ил3 = 8. Для оценки общего наклона и трех различных интерсеп-
тов в модели (46) ее следует записать в развернутом виде следующим образом:
Уп = а1 + ₽Хц + Еп
У12 = <Х1 + Р-Т12 + Ей
У18 = а1 + Рх18 + Е18
У21 = a2 + Рх21 + е21
У22 = a2 + Р-Т22 + ^22
У27 = «2 + РХ27 + ^27
Уз1 = а3 + Рх31 + £з1
У32 = «3 + рх32 + £32
(50)
Узе — а3 + РХ38 + Е38
Матричное представление модели (50) выражается уравнением
у = ХЬ + е,
где элементы у, X, b и е представлены в табл. 18.
Для оценки параметров модели (51) используется метод регрессии. Интерсепты
для серий 1, 2 и 3: Oj = 96,369, о2 = 97,871 и а3 = 101,78 соответственно; общий на-
клон b = — 0,3069. Все значенияр в моделях регрессии близки к нулю, что указывает
на высокую значимость параметров интерсептов для серий, проходящих испыта-
ние; коэффициент множественной детерминации очень высок, Т?2 = 0,9996, что сви-
детельствует об очень хорошей подгонке модели.
Период до истечения срока годности оценивают путем решения соответствую-
щего квадратного уравнения (10) для каждой производственной серии. Основной
трудностью, связанной с квадратными уравнениями, является оценка MSEpjin каж-
дой серии. Значение MSE для каждой производственной серии можно рассчитать
при помощи анализа регрессии для каждой серии; этот подход может оказаться не-
712
Часть 7. Стабильность лекарств
Таблица 18
Матрица и векторы модели (50)
Вектору Матрица X Вектор Ь Векторе
Ун “1 еп 0 *ч “1 еп
^12 «2 Е12 0 “2 612
• “з I • “з •
T'l. ₽ е1« 0 *i. Р еи
^21 0 621 0 *21 Е21
Уц 0 622 0 *22 622
• - • • •
^27 0 627 0 *27 е22
Уз1 0 ел 1 *31 Е31
У32 0 632 1 *32 £32
а а а
а а а а а
• • • • • •
Узь 0 638 1 *38 638
Таблица 19
Информация, необходимая для квадратного уравнения
Серия SSE MSE X ".
1 18,8860 3,1476 13,5 1008 8
2 3,0331 0,6061 10,28 429,43 7
3 26,187 4,3646 13,5 1008 8
корректным, поскольку нет гарантии того, что во всех регрессиях равны величины
наклонов. Поэтому мы рекомендуем применить следующий подход. Используйте
результаты анализа регрессии согласно модели (51) и рассчитайте остатки для мо-
дели в целом. Выявив соответствующие погрешности, рассчитайте ЛЖЕдля каждой
серии с помощью уравнения (4). Используйте полученный результат для расчета
MSE для каждой серии с помощью уравнения (6) и объема выборки для соответ-
ствующей серии. Кроме того, следует выделить соответствующие значения ху для
каждой серии и рассчитать х и Л^с помощью уравнения (4). Полученные результаты
представлены в табл. 19.
Квадратное уравнение для серии 1 можно записать в виде:
[90
-(96,369-0,3069х)]2-1,94322(3,1476)
1
----1"
8
(х-13,5)2
1008 .
= 0.
Контрольная точка для данного уравнения хге/ = 20,7492, а исходной точ-
кой для завершения преобразований является х — 6. Соответствующий корень
хл(1) = 216,62, а срок хранения для серии 1 равняется х£(1) = 16 мес.
Аналогичным способом составляют квадратное уравнение для серии 2:
[90-(97,871-0,3069х)]2-2,0152(0,6061)
1 (х-10,2857)2
7+ 429,43
= 0.
Глава 7.2. Стабильность лекарств
713
Значение контрольной точки для данного уравнения равняется х == 25,6451,
а положение исходной точки для схождения к нулю соответствует х^ — 6. Корень
этого уравнения хя(2) = 22,1394, а срок хранения серии 2 составляет х£(2) = 22 мес.
Соответствующее квадратное уравнение для серии 3:
[90-(Ю1,78-0,3069х)]2-1,94322(4,3646)
1
.8
(х-13,5)2
1008 .
Значение контрольной точки для данного уравнения равняется х^= 38,38, а по-
ложение исходной точки для схождения к нулю соответствует х^— 5. Корень этого
уравнения хЛ(3) = 30,0519, а срок хранения серии 3 составляет х£(3) = 30 мес.
Таким образом, срок хранения для всех произведенных серий составляет:
min {16 22 30} = 16 мес.
7.2.3.4. Определение срока хранения с учетом множества факторов
Большинство лекарственных препаратов выпускаются в более чем одной дизровке
и форме выпуска, и, соответственно, исследование стабильности должно быть про-
ведено для каждой комбинации дозировки и упаковки. Например, предположим,
что на рынке препарат доступен в двух дозировках и упаковках четырех размеров.
Таким образом, можно проанализировать восемь наборов данных по 2 х 4 комбина-
циям доз и размеров, и для расчета срока хранения лекарственного препарата при-
дется учесть восемь разных значений сроков хранения.
Общая модель анализа ковариаций при испытаниях стабильности для несколь-
ких серий и упаковок можно выразить в виде:
= + + j = к = 1,...,К, (52)
где уек — результат испытания в к-й временной точке z-й серии и j-й упаковки лекар-
ственного препарата; x.Jk — время замера, в результате которого был получен резуль-
тат уйк, ае — интерсепт линии разложения z-й серии и j-й упаковки; — случайная
погрешность, обладающая независимым нормальным распределением вокруг нуле-
вого среднего и постоянной дисперсией ст2.
Chen et al. [11] показали, что существует 16 различных моделей уравнения (52);
тем не менее число этих моделей можно сократить до 9. Общая методика состоит
в определении класса модели, ассоциированной с полученной в ходе испытания
информацией. После того, как определена модель, при помощи соответствующей
методики рассчитывают срок хранения лекарственного препарата. Chen et al. [11]
разработали методику расчета срока хранения.
Определение возможности применения ковариационной модели. Здесь представ-
лена общая процедура на основе регресионного анализа, определения возможно-
сти использования для расчета срока годности модели ковариационного анализа
для данного набора результатов испытания с целью определения срока хранения
препарата. Мы называем эту процедуру моделью регрессии с индикаторными пе-
ременными для анализа возможности объединения данных по сериям и упаков-
кам.
714 Часть 7. Стабильность лекарств
Для разъяснения фундаментальных принципов и углубления понимания рас-
смотрим вначале простой случай, затем общую модель и, наконец, методологию на
примере.
Предположим, что проводится испытание трех серий, а соответствующая модель
с индикаторными переменными выглядит следующим образом:
у. = + 8.и. + 37м, + х..(Р, + Д.м, + Д,и,) + е., (53)
где — результат испытания в у-й временной точке z-й серии лекарственного пре-
парата; х.. — время замера исследуемого образца у.^ и и2 — индикаторные перемен-
ные, составляющие бинарный код для каждой комбинации факторов; ро — часть
интерсепта линии регрессии; Р, — часть наклона линии регрессии.
Следует отметить, что интерсепт модели (53) описывает выражение Ро + Д,^ + б2и2,
а наклон — Р, + \и2 + Д2ы2
Параметры 8 и Д оценивают, исходя из результатов испытаний; е. представляет
собой случайную погрешность, обладающую независимым нормальным распреде-
лением вокруг нулевого среднего и постоянной дисперсией а1.
Предположим, что Б1, В2 и ВЗ — коды, соответствующие сериям 1,2 и 3 соответ-
ственно. Индикаторные переменные используются для обозначения серии (уровня
или фактора), к которой приписана переменная отклика. Например, переменная
отклика, ассоциированная с В1, представлена значениями и2 = 1 и и2 = 0. Аналогич-
но, переменная отклика, ассоциированная с В2, представлена и, = 0 и ы2 = 0. Таким
образом, значения их и и2 определяют уникальную комбинацию факторов. Индика-
торные переменные для данной модели представлены в табл. 20.
Бинарное кодирование индикаторных переменных Таблица 20
Серия М1 «2 Результат испытания
В1 1 0 J'u
В2 0 1
ВЗ 0 0
Переменную отклика для серии 1 (после замены значений индикаторных пере-
менных в модели (53)) можно записать следующим образом:
Уи = Ро + 81 + *n(Pi + Ai)+еп>
У12 = Ро + 8i + Xn(Pi + Д1)+е)2;
У1И1 = Ро + 8j + х1Л1(Р1 + Д1) + ещр
У21 = Ро + 82 + ^2i(Pi + Дг) +
У12 = Ро + 82 + Хгг(Р1 + А2) + ^22 >
(54)
У2л2 — Ро + 82 + Х2Л2 (Р1 + Д2) + e2n2i
Уз1 = Ро + Х31Р1 + «зь
Глава 7.2. Стабильность лекарств
715
У32 — Ро + X3zPl + g32
Уз„3 =Ро + ^злзР1+е3из,
где ц — число точек замера в i-й серии.
Следует отметить, что система линейных уравнений (54) представляет перемен-
ные отклика по трем сериям. Для того чтобы линии трех серий имели одинаковый
интерсепт, необходимо соблюдение условия 8t = б2 = 0. Три серии будут иметь оди-
наковый наклон, когда Л, = Л2 = 0 (и только в этом случае). Таким образом, пробле-
ма анализа возможности объединения данных сводится к подгонке модели регрес-
сии (54) и анализу следующей гипотезы: h(. 53 = 52 = 0 и й2: Л, = Д2 = 0. Если нулевая
гипотеза Л2 не опровергается на уровне значимости 0,25, это означает, что наклоны
линий трех серий равны, т. е. рз = Р2 = Р3 = р. Аналогично, если нулевая гипотеза hx
не опровергнута на уровне значимости 0,25. Это означает, что интерсепты линий
трех серий равны. То есть а2 = а2 = а3 = а. В этом случае срок хранения определяет-
ся моделью с одним интерсептом и одним наклоном.
Эта методика применена к данным из примеров 2 и 3, а результаты регрес-
сионного анализа представлены в табл. 21.
Таблица 21
Анализ возможности объединения данных серий
Пример 2: наклоны и интерсепты равны* Пример 3: наклоны и интерсепты различны1
Параметр Оценка /-Статистика Значение р Параметр Оценка /-статистика Значение р
₽о 101,58 112,20 0,0000 ₽о 100,91 163,99 0,0000
А, -0,87 -0,61 0,5513 А, -2,86 -3,44 0,0044
а2 -0,62 -0,46 0,6543 а2 -2,79 -3,21 0,0068
Р. -0,29 -5,66 0,0000 Р. -0,50 -8,14 0,0000
А, -0,04 -0,32 0,7559 А. 0,23 3,09 0,0085
А^ -0,01 -0,14 0,8883 А, 0,21 2,37 0,0337
Пример 2:
“йр 8t = 62 = 0. Значения р в данной таблице показывают, что гипотеза не опровергнута при
уровне значимости 0,25, и интерсепты линий серий одинаковы.
й2: Д1 = Д2 = 0. Гипотеза не опровергнута при уровне значимости 0,25, наклоны линий се-
рии одинаковы.
Пример 3:
bht: 8х = 82 = 0. Гипотеза опровергнута на уровне значимости 0,25, и линии серий имеют
разные интерсепты.
й2: Д[ = Д2 = 0. Гипотеза опровергнута при уровне значимости 0,25, наклоны линий серий
различны.
Модель индикаторных переменных для анализа возможности объединения
данных для двух факторов (упаковки и серии) может быть выражена в следующем
виде:
Уцк = Ро + РЛ,* + 81Щ + 82м2 +... + + ф! Vi + ф2г2 +
+... + ф, г, + Х/ДАл + А2н2 +... + А,п, + ФЛ+ (55)
716
Часть 7. Стабильность лекарств
+ Ф2У2 + • • + + ©ilMiVi + C0i2UiV2 +... +
+ со„игу5)+ец*
где i = 1,.... I,j= 1,Jh к = 1,и?; I— число серий; J— число упаковок; —
размер выборки из z-й серии и / й упаковки; и, и иг — индикаторные переменные,
определяемые согласно табл. 22.
В первой колонке табл. 22 показаны комбинации факторов. Например, В1Р1
представляет первую серию и первую упаковку, В1Р2 — первую серию и вторую упа-
ковку, BIPJ— последнюю серию и последнюю упаковку. Индексы гид определяют-
ся как г = 1— \,s=J— 1.
Таблица 22
Бинарные коды комбинаций факторов иг «а ”1 ”2 ... У*
В1Р1 1 0 0 1 0 • ж. 0 yllt
0 1 0 1 0 ... 0 Уги
• . ж ж - ж ж ж
• * Ж ж ж ж Ж ж ж
• • • • • • • • • •
BrPi 0 0 1 1 0 ... 0 Ум
BIP1 0 0 0 1 0 ... 0 Уш
В1Р2 1 0 0 0 1 ... 0 Уш
В2Р2 0 1 0 0 1 ... 0 Уггк
* • . ж ж ж « ж ж
* • ж ж ж ж а ж ж
• • • • • • • •
BrPl 0 0 1 0 1 ... 0 У ЯШ
BIP1 0 0 0 0 1 0 Уш
. Ж ж ж . ж
ж ж • ж ж ж ж ж
• • • • • • • • •
B1PJ 1 0 0 0 0 ... 1 Уш
B2PJ 0 1 0 0 0 ... 1 Уш
ж ж ж ж ж ж
• ж Ж Ж ж ж ж Ж ж
• • • • • • •
BrPJ 0 0 1 0 0 ... 1 Уш
BIPJ 0 0 0 0 0 1 Уш
Анализ возможности объединения данных требует подгонки модели регрессии
с непрямыми переменными к данным исследования и проверки четырех возмож-
ных гипотез. В табл. 23 представлены гипотезы, требующие проверки; соответству-
ющая интерпретация — критерии подтверждения или опровержения гипотезы; код
модели и модель, утвержденная в FDA [8, 11]. В табл. 23 буквами AwR обозначены
гипотезы, подтвержденные и опровергнутые.
Правила определения срока хранения. После идентификации модели анализа
ковариаций следует применить набор правил для расчета срока хранения. Данный
параграф описывает правила, которым необходимо следовать при определении пе-
риода до истечения срока хранения для каждой из девяти репрезентативных моде-
лей, описанных в предыдущем параграфе [8,11]:
Глава 7,2. Стабильность лекарств
717
Таблица 23
Процедура идентификационного анализа модели ковариаций
Код Гйпотеза A R Интерпретация Модель ANCOVA
1 СО: MQa Я1:8,= 82 = .. . = 8, = 0 X Интерсепты всех серий равны, «и=%=-=«4 = а/ у.., = а.+ Рх, + е.., •'Цк j ijk ijk
2 Cl: Ml Я2:Д1 = Д2=, “11 = “12 = - ... =Ar=0, = <о =0 rs X Наклоны для всех серий равны, Pv = ₽^= • = ₽,= ₽, У^=а!+^№+е^
2 Cl: М3 Я,иЯ2 X Все серии имеют равные интерсепты и равные наклоны, «V=«2,= - = a4 = «i PU=PV=-- = ₽4?=₽, ^=а.+ РЛ+е»
1 CO: MOb Я3:ф1 = ф2 = . .. = ф, = 0 X Интерсепты для всех упаковок равны, ал = ас = ... = ав = а, yet=«, + P^+e(*
3C2:M2 Я4:Ф, = Ф2 = “11 = “12 = - ... = Ф,= 0, = со =0 /3 X Наклоны для всех упаковок одинаковы, Pi.i = pc = ... = ₽fl = P( yfk=ay+^X№+evk
3 C2:M4 Я3иЯ4 X Интерсепты и наклоны для всех упаковок равны, ая = «0=- = «0 = а1 ₽я = ₽«=- = ₽« = ₽. ^=«,+ Рлда+е^
3C3:M5 Я2иЯ4 X Равные наклоны для всех серий и упаковок, Pv=PV=- - = Рх/=₽> рл=р0=•••=₽«=₽. р,=р,=р ^=а„+₽^+е^
4C3:M6 ЯрЯ2иЯ4 X Наклоны для всех серий и упаковок равны, интерсепты для всех серий равны, «ц = «2,= - = аЛ=аУ Р1.= Р2/.= ... = Р4=РУ РЙ = РП = - = РЯ = Р, Р. = Р,= Р ^=а.+ рх№+е(,г
4C3:M7 Я2,Я3иЯ4 X Равны наклоны для всех серий и упаковок, интерсепты для всех упаковок равны а1 = аа = ... = ал = а/ Ри=Р,/= - = Р«=₽> рл=рй=-=ря=р, р(=р,=р уда = а+рх^+е.г
718
Часть 7. Стабильность лекарств
Таблица 23 (окончание)
Код Пшотеза A R Интерпретация Модель ANCOVA
4 С8: Л/8 Я, Я, Я. и Я. X Наклоны и интерсепты для всех серий и упаковок равны av=av=... = a^=a, ап = ап = ... = аа = а, а.= а.= а ₽>4=-=₽,=р> р..=рй=-=₽а=₽, р,=р,=р у^а + рх^+е^
1 СО Я,Я,ЯиЯ4 1’ 2 3 4 X Интерсепты и наклоны для серий и упаковок различны У,Лс=ау + ^+ецк
1. СО: период до истечения срока годности рассчитывают отдельно для каждой
комбинации серии и упаковки. Минимальный срок хранения индивидуаль-
ных серий (по меньшей мере трех) из одинаковых упаковок представляет срок
хранения для данной упаковки.
2. С1: данные по индивидуальным сериям комбинируют для каждой упаковки:
• Ml: общий наклон и разные интерсепты для разных серий; рассчитывают
сроки хранения для индивидуальных серий. Наименьший из сроков хра-
нения индивидуальных серий определяет дату истечения срока годности
для данной упаковки;
• Л/3: наклоны и интерсепты для всех серий равны; рассчитывают единый
срок хранения и определяют дату истечения срока годности для данной
упаковки.
3. С2: объединяются данные для всех упаковок и серий:
• М2: наклоны линий равны, а интерсепты различны для различных упа-
ковок. Для каждой серии рассчитывают сроки хранения для индивиду-
альных упаковок. Минимальный срок хранения для индивидуальных
серий в одинаковых упаковках используют для определения срока год-
ности для данной упаковки;
• Л/4: для всех упаковок наклоны и интерсепты линий равны. В этом
случае рассчитывается единый срок хранения для каждой серии. Ми-
нимальный срок годности для индивидуальных серий является сроком
годности для любой из упаковок.
4. СЗ: Данные д ля всех упаковок и серий объединены:
• Л/5: равные наклоны и разные интерсепты для всех комбинаций серий
и упаковок. Минимальный срок годности для индивидуальных серий из
одинаковых упаковок является сроком годности для всех упаковок;
• Мб: равные интерсепты для всех серий и равные наклоны для всех ком-
бинаций серий и упаковок. Рассчитывают единый срок хранения для всех
упаковок, который является сроком годности данной упаковки;
Глава 7.2. Стабильность лекарств
719
• ЛГ7: равные интерсепты для всех упаковок и равные наклоны для всех ком-
бинаций серий и упаковок. Периоды до истечения срока годности рассчи-
тывают для каждой серии. Минимальное значение для индивидуальных
серий является сроком годности всех упаковок;
• Л/8: равные наклоны и равные интерсепты для всех комбинаций серий
и упаковок; рассчитывают единую дату истечения срока годности, являю-
щуюся сроком хранения для всех упаковок.
Пример 5. Многофакторное испытание стабильности. Хорошо известный при-
мер приводят Shao и Chow [19], который иллюстрирует расчеты срока хранения при
проведении многофакторного испытания стабильности. В ходе испытаний иссле-
довали таблетки лекарственного препарата массой 300 мг для установления срока
хранения для двух типов упаковки: флакона и блистера. Результаты представлены
в табл. 24. Каждый тип упаковки представлен пятью сериями. Активность таблеток
анализировали во временных точках 0, 3, 6, 9, 12 и 18 мес. Определите срок хране-
ния на основании данных по стабильности.
Таблица 24
Результаты испытания (% Z.Q
Упаковка Серия Результаты по временным точкам, мес.
0 3 6 9 12 18
Флакон 1 104,8 102,5 101,5 102,4 99,4 96,5
2 103,9 101,9 103,2 99,6 100,2 98,8
3 103,5 102,1 101,9 100,3 99,2 101,0
4 101,5 100,3 101,1 100,6 100,7 98,4
5 106,1 104,3 101,5 101,1 99,4 98,2
Блистер 1 102,0 101,6 100,9 101,1 101,7 97,1
2 104,7 101,3 103,8 99,8 98,9 97,1
3 102,5 102,3 100,0 101,7 99,0 100,9
4 100,1 101,8 101,4 99,9 99,2 97,4
5 105,2 104,1 102,4 100,2 99,6 97,5
Решение. На основании этих данных мы можем получить информацию, необ-
ходимую для построения модели регрессии с индикаторными переменными: 1=5
серий, J=2 типа упаковки, г=4,5=1ил = 6; временное точки замеров для всех се-
рий: 0, 3, 6, 9, 12 и 18 мес. Индикаторные переменные представлены в табл. 25. Мо-
дель индикаторных переменных в данном случае выглядит следующим образом:
Учк = ₽о + ₽ 1x-ijk + SjMi + S2u2 + 83u3 + 84м4 + фз Vi +
+ XytCAjKi + Д2и2 + Д3И3 + Д4м4 + Ф1Г1 + сОцЩГ! + (56)
+ CO21M2V1 + сОз^зГз + (o4u4Vi) + eijk,
где индекс i соответствует серии, индекс j — типу упаковки, а индекс к — времени
замера.
720
Часть 7. Стабильность лекарств
Таблица 25
Определение индикаторных переменных
Комбинация факторов “1 «2 "з «4 ”1
51Р1 1 0 0 0 1 З'н*
В2Р1 0 1 0 0 1 -У?!*
/?ЗЛ 0 0 1 0 1 •^31*
ЖР1 0 0 0 1 1 З41Л
55Р1 0 0 0 0 1 •^51*
£1Р2 1 0 0 0 0
£2Р2 0 1 0 0 0 У 11k
ВЗР2 0 0 1 0 0 У Ilk
В4Р2 0 0 0 1 0 Упк
£5Р2 0 0 0 0 0 ^52*
Для оценки параметров модели, описываемой уравнением (56), был приме-
нен традиционный пакет компьютерных программ для регрессионного анализа
(табл. 26). Оценка параметров модели индикаторных переменных Таблица 26
Параметр Оценочное значение /статистика Значение р
₽о 104,5 185,76 0,0000
8, -1,63 -2,23 0,0306
»2 -1,33 -1,83 0,0747
8з -2,83 -3,88 0,0003
»4 -3,64 -4,99 0,0000
Ф1 0,44 0,96 0,3408
Р> -0,43 -6,91 0,0000
Л. 0,15 1,73 0,0901
0,07 0,88 0,3823
Лз 0,31 3,62 0,0007
0,26 3,01 0,0043
Ф! -0,01 -0,18 0,8557
<Вц -0,06 -0,65 0,5161
®21 0,05 0,56 0,5770
®31 -0,02 -0,21 0,8313
Ю41 0,04 0,43 0,6660
Значения р из табл. 26 показывают, что гипотезы Н3: ф = 0 и Н^.
Ф[ = сои = 0)2| = го31 = со41 = О не опровергнуты на уровне значимости 0,25; таким
образом, в соответствии с табл. 23, интерсепты и наклоны равны для всех типов
упаковок, т. е. ап = ai2 = ар а Рп = Ри = Рг Поэтому анализ ковариационной модели
следует применить к результатам испытаний под кодом 3 С2: Л/4. Уравнение модели
имеет вид
Глава 7.2. Стабильность лекарств
721
у... — а. + Вх... + £....
'ук i *1 ук у к
Поскольку различий между упаковками нет, приемлема простая модель анализа
ковариаций. Согласно правилам расчета срока годности, изложенным в параграфе
7.2.3.3, следует рассчитать срок хранения для каждой серии и использовать мини-
мальный критерий для определения даты истечения срока годности для соответ-
ствующих серий лекарственного препарата. Таким образом, подгонка регрессион-
ной модели должна проводиться по модели 3 С2: М4, а результаты используются для
построения квадратного уравнения
[90-(103,54-0,3233х)]2 -1,81252(1,48)
1 (х-8)2
= 0.
Контрольная точка для данного уравнения хге/= 41,89, а исходная точка конвер-
генции хге/— 7. Корень данного уравнения хЛ(1) = 33,25, а срок хранения для серии
составляет х£(1) = 33 мес. Поскольку время замеров одинаково для всех серий, сле-
дующие величины остаются инвариантными для всех серий: л = 12, х = 8,/^ = 420,
a /ом 10 = 1,8125. Значения согласно квадратному уравнению и соответствующие
сроки хранения представлены в табл. 27. Компьютерная программа для расчетов
дана в Приложении.
Следовательно, срок хранения, предлагаемый для этих и последующих серий,
составляет
min{33 32 54 49 31} = 31 мес.
Этот подход дает консервативную оценку общего срока хранения, поскольку
предлагает более чем 95%-ный уровень достоверности для всех серий, за исклю-
чением серии, по которой производится оценка. Следует отметить, что «принцип
минимума» подвергается критике со стороны некоторых ученых. Например, Chow
и Shao [20] указывали на то, что принцип минимума статистически не обоснован.
Ruberg и Stegeman [21], Ruberg и Hsu [22] описывают недостатки данной методологии.
Тем не менее, Chen et al. [ 11 ] показывают, что методика FDA достаточно эффективна
для данных типичного испытания по трем сериям. Методика FDA основана на пред-
положении, что эффекты серий постоянны. Если анализ показывает, что различия
между сериями невелики, то полезно объединить все данные для получения одной
общей оценки.
Таблица 27
Параметры, необходимые для расчета срока хранения
Серия а b MSE X f nf XR Срок хранения
2 103,84 -0,3429 1,47 40,37 32,49 32
3 102,34 -0,1429 1,24 86,4 54,26 54
4 101,54 -0,1671 0,74 69,02 49,84 49
5 105,17 -0,4426 0,45 34,28 31,08 31
Если анализ свидетельствует о наличии различий между сериями, то, согласно
указаниям FDA, используют минимальное оценочное значение, полученное для ин-
дивидуальной серии, основываясь на предположении о том, что период до истече-
722
Часть 7. Стабильность лекарств
ния срока годности может зависеть от минимального времени, в течение которого
можно ожидать, что параметры серии остаются в приемлемых пределах. Это озна-
чает, что чем меньше колебания значений экспериментальных данных, тем больше
период годности. Таким образом, FDA проявляет консерватизм, когда число серий
велико при малой изменчивости в серии. FDA устанавливает, что уровень значимо-
сти 0,25 используется для компенсации ожидаемой низкой эффективности схемы
испытания из-за относительно ограниченного объема выборки при типичном ис-
пытании стабильности [8]. Мы поддерживаем аргументы FDA в пользу уровня зна-
чимости 0,25; описанная в этом справочнике процедура получила одобрение FDA.
7.2.4. Анализ данных исследования стабильности
при краткосрочном хранении
7.2.4.1. Введение
Подтверждение приемлемой стабильности лекарств остается проблемой для про-
мышленности. Как было упомянуто в параграфе 7.2.2.1, концепция стабильности
включает классический аспект проблемы — разложение API, а также накопление
препаратов разложения в количестве, представляющем опасность для пациента [23].
Для подтверждения соответствия коммерческого лекарственного препарата необ-
ходимым спецификациям проводятся два испытания стабильности: краткосрочное
и долгосрочное. Краткосрочное представляет собой ускоренное испытание, т. е.
исследование стабильности в стрессовых условиях. Основной задачей ускоренного
испытания стабильности является не только исследование кинетики химических
реакций, но и определение предварительной даты истечения срока годности в усло-
виях коммерческого хранения.
Стабильность лекарственного препарата зависит от условий хранения — темпе-
ратуры и влажности. Влияние условий окружающей среды на разложение зависит
от конфигурации упаковки и химических свойств лекарственного препарата. Разло-
жение лекарственного препарата вызвано, главным образом, химической реакцией
API со вспомогательными веществами или веществами, присутствующими в окру-
жающей среде (атмосферным кислородом и влажностью), приводящей к уменьше-
нию результатов анализа с течением времени. За период, соответствующий полному
сроку хранения, API в составе лекарственного препарата до некоторой степени раз-
лагается. Другие характеристики качества (например, внешний вид, стерильность,
скорость высвобождения лекарства, примеси) могут также изменяться со временем,
нарушая функциональность, эффективность или чистоту лекарственного препара-
та. Колебания этих параметров за время срока хранения ни в коем случае не должно
представлять риск для пациента.
Скорость химической реакции зависит от температуры и концентрации при-
сутствующих реагентов. Поэтому для изменения скорости реакции, необходимо
изменить температуру реакции или концентрацию реагентов. Температуру реакции
можно легко изменить, варьируя температуру хранения лекарственного препарата.
Реагентами в составе лекарственного препарата являются API, вспомогательные ве-
щества, а также окружающий воздух и влага. Изменить можно только окружающий
Глава 7,2. Стабильность лекарств 723
воздух и влажность, поскольку API и вспомогательные вещества в составе лекар-
ственного препарата указаны в спецификации и изменению не подлежат.
Для фармацевтической промышленности важно изучать параметры разложения
лекарственных препаратов в условиях ускоренного хранения для оценки их стабиль-
ности при долгосрочном хранении в неэкстремальных условиях при кратковремен-
ных отклонениях от рекомендуемых условий хранения, возможных при транспор-
тировке. Краткосрочное испытание стабильности представляет собой метод оценки
разложения лекарственного препарата в условиях ускоренного хранения. В качестве
примера приведем исследование Gil-Alegre et al. [24], изучавших кинетику разложе-
ния митонафида, антинеопластического агента, в интервале температур от 60 до
90 °C с интервалами 10 °C. Это лекарство очень стабильно, поэтому для обнару-
жимых изменений концентраций требуются довольно высокие температуры. Как
правило, ускоренное испытание проводится при постоянной температуре. Тем не
менее, к постепенному повышению температуры прибегают при неизотермических
испытаниях [25]. Данный подход может улучшить прогноз значения низкотемпе-
ратурной кинетической константы и, следовательно, периода годности препарата
[23].
Кинетика химической реакции. Чтобы понять, как обрабатывать данные, следует
вспомнить основы кинетики химических реакций. Принципы химического кон-
струирования изложены в учебниках по химическому машиностроению или кон-
струированию реакторов [26]. Химическая реакция представляет собой процесс,
в ходе которого один или несколько компонентов превращаются в один или не-
сколько других компонентов. Скорость реакции — это скорость преобразования
компонентов в ходе химического процесса. Для химической реакции
А+ В—>Е
скорость реакции описывается уравнением
— = к\А}п\В\т,
dt
(57)
(58)
где С — молярная концентрация исследуемого компонента; [Л] [Д] (квадратные
скобки обозначают концентрацию реагентов); к — константа скорости реакции.
Скорость реакции положительна относительно препарата и отрицательна отно-
сительно реагента. Порядок реакции — это сумма экспонент (п + т) в уравнении
(58). Исходя из этого, реакция нулевого порядка описывается уравнением
dC
dt
= -к0.
(59)
После интегрирования получаем выражение
C = Co-kot, (60)
где С — молярная концентрация в любой момент времени t; Се — концентрация
в момент времени t = 0; к0 — константа скорости реакции нулевого порядка; t —
время.
724 Часть 7. Стабильность лекарств
Для реакции первого порядка скорость реакции можно выразить как
dC
-^ = ~к^С- (61)
После интегрирования получаем:
(С \
—1 = -Л1Г или С = Со - Сое~к1‘. (62)
Со /
Константа скорости сильно зависит от температуры. Этот эффект описывает
уравнение Аррениуса:
k = Ae~E/(RT\ (63)
где А — стерический фактор, выраженный в тех же единицах, что и к; Е— энергия
активации; R — газовая постоянная; Т — абсолютная температуры в градусах Кель-
вина.
Уравнение Аррениуса в логарифмической форме принимает вид:
17 Е . । л (64)
1пк =-----+1пА. v 7
RT
7.2.4.3. Оценка скорости процесса разложения по данным исследования
стабильности при ускоренном хранении
Знание кинетики химической реакции помогает оценить данные испытаний в усло-
виях ускоренного хранения и прогнозировать данные испытаний лекарственного
препарата в нормальных условиях на периоды более длительные в сравнении с пред-
полагаемым сроком хранения. Применение такого подхода ограничено, поскольку
кинетика реакций с участием лекарственных препаратов зачастую весьма сложна.
В следующем примере представлена методика расчета концентрации API в разные
моменты времени для лекарственного препарата, хранящегося в нормальных усло-
виях, когда соответствующие данные еще не доступны.
Пример 6. Описание скорости процесса разложения по данным ускоренного испы-
тания: реакция первого порядка. Рассмотрим данные исследования разложения,
представленные в табл. 28, полученные в условиях нормального, промежуточного
и ускоренного хранения. Все данные выражены в процентах от заявленного значе-
ния (%£С) без учета данных двух первых колонок. То есть по данным, полученным
при повышенных температурах (30 °C и выше), рассчитайте значения для 25 °C как
функцию времени.
Примечание. В этом примере используются искусственные данные, предназна-
ченные только для демонстрации методики расчета. Данные, полученные в нор-
мальных условиях, представлены в первых двух колонках (25 °C). Как правило, при
подаче заявки на регистрацию нового лекарства (7VZL4) эти данные еще не доступ-
ны, поэтому методика расчета, представленная здесь, весьма важна.
Решение. Анализ проводится исходя из предположения, что речь идет о реакции
первого порядка (обычная ситуация в реальности) или нулевого порядка (более
простой анализ без существенной погрешности на разложение, составляющей ме-
нее 10%). Здесь рассмотрены оба случая.
Глава 7.2. Стабильность лекарств
725
Таблица 28
Характеристика процесса разложения по данным испытаний лекарственного препарата по
показателю «Количественное определение»
Время г, мес. Количественное определение, хранение при 25 °C Время г, мес. Количественное определение, хранение Г при 30 °C Количественное определение, хранение при 40 °C Количественное определение, хранение при 50 °C
0 99,9 0 99,9 99,9 99,9
3 99,4 2 99,4 98,0 95,6
6 98,2 4 98,7 95,9 91,2
9 97,8 6 97,4 95,1 87,4
12 97,4
18 95,6
24 94,5
36 91,7
Для реакций, описываемых кинетикой первого порядка (см. уравнение (61)),
первым этапом анализа является расчет натуральных логарифмов концентраций
(табл. 29). Далее эти значения наносят на график как функцию времени; график
зависимости должен представлять прямую линию (рис. 2). Наклон каждой линии
соответствует значению к{, что показано в нижней части табл. 29.
Затем для трех значений Л, строят графики зависимости 1п(Л,) от 1/Гпо одному
для каждой температуры (рис. 3). Отметим, что здесь используют значения абсолют-
ной температуры. Согласно уравнению (64) значение Л, можно экстраполировать на
Т= 25 °C (1/7'= 0,003354Х-1) и далее прогнозировать результат анализа лекарствен-
ного препарата в зависимости от времени. Значения наклонов и интерсептов пока-
заны на вставке в рис. 3; расчет выглядит следующим образом:
1пЛ25= -8237,4- 0.003354 + 21,639 = -5.990;
к25=0,00250 мес.-1.
Рис. 2. Данные исследования разложения при рассмотрении кинетики первого порядка
726
Часть 7. Стабильность лекарств
Таблица 29
Анализ данных при допущении о кинетике первого порядка
Время (0, мес. In (результата) при 30 °C In (результата) при 40 °C In (результата) при 50 °C
0 4,60 4,60 4,60
2 4,59 4,58 4,55
4 4,58 4,55 4,50
6 4,57 4,55 4,46
К 0,00415 0,00847 0,02224
1п(^) -5,483 -4,771 -3,798
1/Т 0,00330 0,00319 0,00309
1/7
Рис. 3. График Аррениуса для реакции разложения первого порядка
Когда рассчитана скорость реакции, то результат анализа лекарственного пре-
парата можно прогнозировать с помощью уравнения (62). Результаты представлены
в табл. 30 наряду с реальными данными из табл. 28.
Таблица 30
Прогнозированные результаты анализа, полученные при анализе графика Аррениуса и кинетики
первого порядка
Время (/), мес. Результат Прогноз
0 99,0 100,0
3 98,5 98,3
6 97,3 97,5
9 96,9 96,8
12 96,5 96,1
18 94,7 94,6
24 93,6 93,2
36 90,8 90,5
Пример 7. Оценка данных исследования ускоренного разложения: реакция нулевого
порядка. Повторить этапы примера 6, приняв предположение о реакции нулевого
порядка.
Решение. Для кинетики нулевого порядка (см. уравнение (60)) исходные кон-
центрации можно представить как функцию времени. По точкам можно провести
Глава 7.2, Стабильность лекарств
727
прямые линии, даже если порядок реакции разложения в действительности равен 1
(рис. 4). Наклон каждой линии соответствует значению к0. Эти величины представ-
лены в табл. 31. Затем строят график зависимости 1п(Л0) от 1/7’для трех значений к0
по одному для каждой температуры (рис. 5).
Таблица 31
Данные исследования разложения при допущении о реакции первого порядка
30 °C 40 °C 50 °C
ко 0,410 0,825 2,095
1п(*0) -0,892 -0,192 0,740
1/Т 0,00330 0,00319 0,00309
Рис. 4. Данные исследования разложения при рассмотрении кинетики нулевого порядка
1/T
Рис. 5. График Аррениуса для реакции разложения нулевого порядка
Как и в примере 6, величину кй можно экстраполировать на Т = 25 °C
(1/7’= 0,003354а-1); далее с помощью уравнения (64) прогнозировать результат ана-
лиза лекарственного препарата в зависимости от времени. Значения наклонов и ин-
терсептов показаны на вставке в рис. 5; расчет выглядит следующим образом:
728
Часть 7. Стабильность лекарств
1пЛ23= -7974,8 0.003354 + 25,369 = -1.379;
кг = 0,2519.
Когда рассчитана скорость реакции, результат анализа лекарственного препа-
рата можно прогнозировать с помощью уравнения (60). Результаты представлены
в табл. 32.
Таблица 32
Прогнозированные результаты анализа, полученные при анализе графика Аррениуса и кинетики
первого порядка
Время t, мес. Результат анализа Прогноз
0 99,0 100,0
3 98,5 99,2
6 97,3 98,5
9 96,9 97,7
12 96,5 97,0
18 94,7 95,5
24 93,6 94,0
36 90,8 90,9
Срок хранения 41 39
Сравнение моделей. Для сравнения прогнозов обеих моделей (первого и нулевого
порядка) представлены табл. 33 и рис. 6.
Таблица 33
Реальные и прогнозированные значения
Время t, мес. Результат анализа Прогноз результата
Первый порядок Нулевой порядок
0 99,9 100,0 100,0
3 99,4 99,3 99,2
6 98,2 98,5 98,5
9 97,8 97,8 97,7
12 97,4 97,0 97,0
18 95,6 95,6 95,5
24 94,5 94,2 94,0
36 91,7 91,4 90,9
Как и ожидалось, оба прогноза дали аналогичные результаты. Модель первого
порядка позволяет получить лучшие результаты, что свидетельствует в пользу того,
что реально разложение описывается кинетикой первого порядка. Результаты обоих
прогнозов аналогичны, поскольку реакции разложения протекают довольно мед-
ленно, вследствие чего временные рамки этих измерений очень малы по сравне-
нию, например, с периодом полураспада вещества (или временем, в течение кото-
рого результат анализа достигает 50% LC). Несмотря на то что обсуждение проблемы
расчета периода полураспада лежит за рамками данного руководства, достаточно
Глава 7.2. Стабильность лекарств
729
сказать, что на основании результатов из примеров 6 и 7 период полураспада для
кинетики первого порядка составляет около 23 лет, а для кинетики нулевого по-
рядка (вероятно, оценка менее точна) около 17 лет. Эти значения примерно в 40 раз
превосходят временные рамки, в которых протекает ускоренное (краткосрочное)
испытание, длящееся 6 мес. (0,5 года). Таким образом, любая кинетическая модель
работает достаточно хорошо.
7.2.4.Д. Предварительные расчеты срока хранения на основании данных
стрессовых испытаний
Предварительный срок хранения можно рассчитать на основании результатов уско-
ренного испытания. Метод регрессии, описанный в разделе 7.2.3, применим для
оценки результатов ускоренных испытаний, как показано в примерах 6 и 7. Методи-
ка расчета описана в разделе 7.2.3, т. е. необходимо записать квадратное уравнение
и определить срок хранения по всем наблюдаемым и прогнозируемым результатам
испытания.
Квадратное уравнение, определяющее срок хранения в данном исследовании,
имеет вид
[90 -(99,884 - 0,2275х)]2 -1,94322(0,0408)
1
.8
(х-13,5)2
1008
= 0.
Контрольная точка для данного уравнения хге/ = 43,44, а исходной точкой для
завершения преобразований является хге/— 1,5. Корень уравнения хл(оЬб) = 41,78,
а срок хранения по результатам испытания составил x£(obs) = 41 мес. Эта величина
используется для сравнения. Квадратное уравнение для прогнозируемого испыта-
ния при предположении о первом порядке химической реакции имеет вид:
[90 - (99,95 - 0,2393х)]2 -1,94322 (0,0032)
1
---h
8
(х-13,5)2
1008
= 0.
Контрольная точка для данного уравнения хге/= 41,60, а исходной точкой для за-
вершения преобразований является хге/— 0,4. Корень уравнения xR (первый) = 41,17,
а срок хранения по результатам испытания составил xL (первый) = 41 мес. Далее,
730
Часть 7. Стабильность лекарств
приняв предположение о нулевом порядке химической реакции, составляют ква-
дратное уравнение для прогнозируемого испытания:
[90—(99,99—0,2515х)]2 —1,94322(0,0013)
(х-13,5)2
1008 .
= 0.
1
—h
8
Контрольная точка для этого уравнения xref= 39,74, исходная точка конверген-
ции хге/— 0,3.
Корень этого уравнения хд(0) = 39,49, а срок хранения по прогнозированным
результатам при допущении о нулевом порядке химической реакции х£(0) = 39 мес.
Итак, сроки хранения, рассчитанные по реальным результатам и для кинетики
первого порядка составляет 41 мес., а срок, прогнозируемый по кинетике нулевого
порядка — 39 мес. Все три значения близки. Однако результаты показывают, что
в действительности реакция разложения лекарственного препарата описывается
кинетикой первого порядка.
7.2.5. Заключительные замечания
На примерах представлено краткое изложение основ конструирования моделей
химических реакций и применение этих знаний для оценки сроков хранения ле-
карственных препаратов. Эти методики имеют большое значение при составлении
заявок на регистрацию новых лекарственных препаратов (NDA) в регуляторные ор-
ганы, такие, как FDA. Как правило, в момент представления заявки на регистрацию
нового лекарства данные по стабильности лекарственного препарата при низких
температурах оказываются недоступными в достаточном объеме, поскольку долго-
срочные испытания длятся годами. Изложенные здесь методы представляют аль-
тернативу, одобренную FDA и ICH.
Глава 7.2. Стабильность лекарств
731
Приложение
Компьютерные программы
Данное Приложение включает 4 компьютерные программы на языке Matlab, кото-
рые пользователь может применить для проведения типичных вычислений, связан-
ных с определением срока хранения.
Данная программа рассчитывает линию разложения по результатам испытания
одной серии для ввода их в программу расчета срока хранения:
% Regression Analysis for a single batch
clear, clc
close all
batch=3 ;
i=3; % the number of the batch to be computed
nl(1)=7;
nl(2)=6;
nl(3)=6;
N=nl(i);
х=[0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36] ;
у=[99.2 97. 1 96.1 95 .2 93.8 93.1 92.4 0
98.7 97 96.2 95.1 94.2 93.3 0 0
102.5 98.9 97.1 95.6 94.1 93.1 0 0] ;
xl =[x(i,1:nl (i)) ] ;
yl=[y(i,l:nl(i))];
xl=xl';
X=[ones(N,l) xl] ;
Y=yl';
b=inv(X'*X) *X'*Y;
y_est=X*b;
e=Y-y_est;
SSE=e'*e;
SST=Y'*Y-N* (mean(Y)A2) ;
SSR=SST-SSE;
t=tinv(0.95,N-2)
R2=SSR/SST
MSE=SSE/(N-2)
av_x=mean(xl)
Sxx=xl'*xl-N* (ау_х)л2
vec= [b' N MSE av_x Sxx]
save res vec
Программа производит расчет линии разложения после объединения данных по
нескольким сериям, имеющим одинаковые наклоны и интерсепты:
732
Часть 7. Стабильность лекарств
% Regression Analysis for pooled data from several batches
clear, clc
close all
batch=3;
x=[0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36];
y=[102.4 98.1 99. 2 97.5 95 96.1 95.2 94.3
101.1 101.2 99 97.2 96.4 95.5 94.3 94.8
104.1 102.1 99 .5 98.1 95.7 94.1 94 93.5];
n(l)=6;
n(2)=7;
n(3)=8;
xl=[x(l,l:n(l)) x(2,l:n(2)) x(3,l:n(3))];
yl=[у(1,1: n (1) ) y(2,l:n(2)) у (3,1: n (3) ) ] ;
xl=xl';
N=sum(n);
X=[ones(N,l) xl] ;
Y=yl';
b=inv(X'*X) *X'*Y;
y_est=X*b;
e=Y-y_est;
SSE=e'*e;
SST=Y'*Y-N* (mean(Y)A2) ;
SSR=SST-SSE;
R2=SSR/SST
MSE=SSE/(N-2)
av_x=mean(xl)
Sxx=xl'*xl-N* (av_x)A2
vec= [b' N MSE av_x Sxx]
save res vec
Эта программа рассчитывает сроки хранения по данным, введенным в програм-
му 1 или 2:
clear, clc
close all
delete it* % para borrar archives anteriores display('1:
f = (90-(bO - bl*x))A2-tA2*MSE*[1/n + (x- av_x)A2/Sxx] ')
flag_fun=input ('Enter the number of the function ')
load res
b=vec(1:2);
n=vec(3) ;
MSE=vec(4);
Глава 7.2. Стабильность лекарств
733
av_x=vec(5);
Sxx=vec(б)
d=-5; % expiration date
reference=(b(1) - 90)/(-b(2))
if flag_fun==l
xO=(b(l) - 90)/(-b(2))+d; % expiration date lower value
end
options = optimset ('LargeScale','off') ;
cont=0;
save contador cont
[x, fval, exitflag, output] = fminunc (@ (x)
obj_fun_expiration_date(x,flag_fun),xO,options)
Эти рутинные данные затребованы программой 3, определяющей срок хранения
лекарственного препарата:
function f = obj_fun_expiration_date (x,flag_fun)
load contador
cont=cont+l
save contador cont
load res
b=vec(1:2);
n=vec(3) ;
MSE=vec(4) ;
av_x=vec(5);
Sxx=vec(6);
t=tinv(0.95,n-2);
if flag_fun==l
f = abs((90-(b(1)+b(2)*x))a2-(tA2)*MSE*(l/n+(x-av_x)
a2/Sxx));
end
save ( ['it' num2str (cont) ] ,'f','x','flag_fun')
Эта программа проводит статистический анализ данных для подтверждения или
опровержения равенства наклонов и интерсептов для линий разложения по не-
скольким сериям:
% ANCOVA
clear, clc
close all
batch=3;
х=[0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36
0 3 6 9 12 18 24 36] ;
у=[99.2 97 . 1 96.1 95. .2 93.8 93.1 92.4 0
98.7 97 96.2 95.1 94.2 93.3 0 0
102 . 5 98 .9 97. 1 91 5.6 94.1 93.1 0 0];
734
Часть 7. Стабильность лекарств
п(1)=7;
п(2)=6;
п(3)=6;
N=sum(n);
%%%%%%%%%%%%%%%%%%%
% testing slopes
%%%%%%%%%%%%%%%%%%
for i=l:batch
sy=0;
sx=0 ;
sxy= 0;
for j=l:n(i)
sy=sy+y(i,j)a2;
sx=sx+x(i,j)a2;
sxy=sxy+x(i,j)*y (i,j);
end
Syy (i) =sy- (sum(y (i, 1 :n(i) ) )) a2/length(y (i, 1 :n(i) ) ) ;
Sxx(i)=sx-(sum(x(i,1:n(i))))a2/length(x(i,l:n(i)));
Sxy(i)=sxy-(sum(y(i,1:n(i))))*(sum(x(i,1:n(i))))/
length(y(i,1:n(i)));
end
SxxW=sum(Sxx);
SyyW=sum(Syy);
SxyW=sum(Sxy);
SSEW=SyyW-SxyWA2 /SxxW;
for i=l:batch
sse(i)=Syy (i)-Sxy(i)A2/Sxx(i);
end
SSE=sum(sse);
SS_slope=SSEW - SSE;
k=batch-l;
MS_slope=SS_slope/(batch-1);
MSE=SSE/(N-2*batch);
F_slope=MS_slope/MSE;
F_slope
df_num=batch-1
df_den=N-2 *batch
F_cri=finv(0.75,df_num,df_den)
%%%%%%%%%%
% testing intercepts
%%%%%%%%%%
sy= 0 ;
syl=0;
sx= 0;
Глава 7.2. Стабильность лекарств
735
sx=0;
sxl=0;
sxy= 0;
for i=l:batch
for j=l:n(i)
sy=sy+y(i,j)л2;
sx=sx+x{i,j)л2;
sxy=sxy+x(i,j)*y(i,j);
end
syl=syl+sum(y(i,1:n(i)));
sxl=sxl+sum(x(i,1:n(i)));
end
sy;
SYl=sylA2/N;
SYY=sy-SYl;
sx;
SX1=sx1a2/N;
SXX=sx-SX1;
sxy;
SXYl=sxl*syl/N;
SXY=sxy-SXYl;
tx=0;
ty=0;
txy=0;
for i=l:batch
ty=ty+(sum(y(i,1:n(i))))a2/length(y(i,1:n(i)));
tx=tx+(sum(x(i,1:n{i))))a2/length(x(i,1:n(i)));
txy=txy+(sum(x(i,1:n(i))))*(sum(y(i,l:n(i))))/
length(y (i, 1: n (i) ) ) ;
end
my=sylA2/N;
mx=sxlA2/N;
mxy=sxl*syl/N;
Tyy=ty - my
Txx=tx - mx
Txy=txy - mxy
Eyy=SYY-Tyy
Exx=SXX-Txx
Exy=SXY-Txy
SSEr=SYY-SXYA2/SXX
SSEc=Eyy-ExyA2/Exx
SSbO=SSEr - SSEc
MSbO = SSbO/(batch-1)
MSEc=SSEc/(N-batch-1)
736
Часть 7. Стабильность лекарств
F_bO=MSbO/MSEc
d f_num=batch-1
df_den=N-batch-l
F_cri_inter=finv (0.75, df_num, df_den)
Литература
1. Expiration Dating, Code of Federal Regulations, Title 21, Vol. 4, Part 211,21CFR211.137.
2. American Medical Association (AMA) (2001), Report 1 of the Council on Scientific Affairs (A— 01),
Pharmaceutical expiration dates, available: http://www.ama-assn.org/ama/pub/category/print/13652.
html, accessed on Feb. 19,2007.
3. Bennett B., Cole G., Eds. (2003), Pharmaceutical Production. An Engineering Guide, Institution of
Chemical Engineers (IChemE), Rugby, Warwickshire, United Kingdom.
4. Matthews B. R. (1999), Regulatory aspects of stability testing in Europe, Drug. Dev. Ind. Pharm., 25,
831-856.
5. Dietz R., Feilner K., Gerst F., Grimm W. (1993), Drug stability testing: Classifi cation of countries
according to climatic zone, Drugs Made in Germany, 36, 99-103.
6. U.S. Food and Drug Administration (FDA) (2001), Stability testing of new drug substances and prod-
ucts, ICH QI A, FDA, Washington, DC.
7. Fairweather W. R., Lin T. Y., Kelly R. (1995), Regulatory, design and analysis of complex stability
studies, U.S. Food and Drug Administration, J. Pharm. Sci., 84,1322-1326.
8. U.S. Food and Drug Administration (FDA) (2004), Evaluation of stability data, ICH Q1E, FDA,
Washington, DC.
9. U.S. Food and Drug Administration (FDA) (2003), Bracketing and matrixing designs for stability
testing of new drug substances and products, ICH Q1D, FDA, Washington, DC.
10. Montgomery D. G. (2004), Design and Analysis ofExperiments, 6th ed., Wiley, New York.
11. Chen J. J., Ahn H., Tsong Y. (1997), Shelf-life estimation for multifactor stability studies, Drug
Inform. J, 31, 573-587.
12. Montgomery D. C., Peck E. A., Vining G. G., (2007), Introduction to Linear Regression Analysis, 3rd
ed., Wiley, New York.
13. Reklaitis G. V, Ravidran A., Ragsdell К. M. (1983), Engineering Optimization, Methods and
Applications, Wiley, New York.
14. Bancroft T. A. (1964), Analysis of inference for incompletely specifi ed models involving the use of
preliminary test(s) of significance, Biometrics, 20,427-442.
15. Snedecor G. W, Cochran W. G. (1989), Statistical Methods, 8th ed., Iowa State University Press,
Ames, IA.
16. Wang S. G., Chow S. C. (1993), Advanced Linear Model: Theory and Applications, Marcel Dekker,
New York.
17. Ramirez-Beltran N. D., Olivares L. (1998), Drug Shelf-life Estimation Using Clustering Techniques.
Proceeding of the Computing Research Conference CRC’98, edited by the University of Puerto Rico,
Electrical and Computer Engineering Department. Mayaguez Puerto Rico, pp. 70-73.
18. Chow S.-С., Liu J.-P. (1995), Statistical Design and Analysis in Pharmaceutical Science, Marcel
Dekker, New York.
19. Shao J., Chow S-Ch. (1994), Statistical inference in stability analysis, Biometrics, 50 (3), 753—763.
20. Chow S. C., Shao J. (1991), Estimating drug shelf-life with random batches, Biometrics, 41, 1071-
1079.
21. Ruberg S., Stegeman J. W. (1991), Pooling data for stability studies: Testing the equality of batch
degradation slopes, Biometrics, 47, 1059-1069.
22. Ruberg S., Hsu J. (1992), Multiple comparison procedures for pooling batches in stability studies,
Technometrics, 34,465-472.
Глава 7.2. Стабильность лекарств 737
23. Waterman К. С., Adami R. С. (2005), Accelerated aging: Prediction of chemical stability of pharma-
ceuticals, Int. J. Pharm., 293, 101-125.
24. Gil-Alegre M. E., Bemabeu J. A., Camacho M. A., Torres-Suarez A. I. (2001), Statistical evaluation
for stability studies under stress storage conditions, Il Farm., 56, 877-883.
25. Oliva A., Llabres M., Farina B. (2006), Data analysis of kinetic modeling used in drug stability stud-
ies: Isothermal versus nonisothermal assays, Pharm. Res., 23 ,2595—2602.
26. Levenspiel O. (1998), Chemical Reaction Engineering, 3rd ed., Wiley, New York.
Глава 7.3. ВЛИЯНИЕ УПАКОВКИ НА СТАБИЛЬНОСТЬ
ФАРМАЦЕВТИЧЕСКИХ СУБСТАНЦИЙ
И ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Эммануэль О. Акала
Фармацевтический факультет университета Говарда
(г. Вашингтон, округ Колумбия)
7.3.1. Введение
Стабильность — важнейшая характеристика лекарственных препаратов, о чем сви-
детельствует множество национальных и международных документов, публикуе-
мых в форме руководств. Устанавливаются требования к испытаниям стабильно-
сти, которые должны продемонстрировать физическую и химическую стабильность
лекарственных веществ и препаратов в различных условиях окружающей среды,
таких как температура, влажность и освещенность [1]. Кроме того, в руководствах
Международной конференции по гармонизации {ICH) содержатся указания по
проведению испытаний стабильности [2, 3]. Фармацевтические и биофармацевти-
ческие компании прилагают значительные усилия для разработки и производства
лекарственных препаратов с адекватными профилями стабильности. Все эти меры,
предпринимаемые правительственными органами, а также фармацевтическими
и биофармацевтическими компаниями, направлены на решение следующих задач
[4]: снабжение конечного потребителя (пациента) качественными (эффективными
и безопасными) лекарственными препаратами; обеспечение соответствия норма-
тивам, выработанным регуляторными органами; гарантии производителям, что их
лекарственные препараты будут использованы по назначению; наконец, создание
базы для будущих разработок лекарств.
7.3.3.1. Основные проблемы, связанные со стабильностью лекарственных
средств
Потеря эффективности. Эффективность любого лекарственного препарата (лекар-
ственной формы) зависит от его способности доставить необходимое количество ак-
тивного терапевтического компонента к биофазе (месту воздействия) на желатель-
ный период времени для достижения целей терапии. Потеря активного ингредиента
в дозированной форме, обусловленная химическим разложением, может привести
к снижению эффективности лекарственного препарата (т. е. концентрация дей-
ствующего вещества в сыворотке крови может не достичь минимальной концентра-
ции, вызывающей желательный терапевтический отклик). Не предполагается, что
эффективность какого бы то ни было лекарственного препарата может оставаться
неизменной бесконечно. Тем не менее ожидается, что качество лекарственного пре-
парата (стабильность активных фармацевтических ингредиентов {API) среди других
характеристик) сохраняется вплоть до получения его пациентами или до истечения
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...739
срока годности, обозначенного на маркировке первичной упаковки, в течение ко-
торого препарат должен сохранять соответствие спецификации [5]. Период, в те-
чение которого существует гарантия эффективности, безопасности и эстетично-
го внешнего вида лекарственного препарата, зачастую определяется как срок его
хранения. Если обнаруживается, что химическое разложение API вносит основной
вклад в процесс разложения лекарственного препарата, то сроком хранения счита-
ется время, проходящее до снижения содержания лекарственного препарата ниже
90% от заявленного содержания — при условии, что лекарственный препарат хра-
нится в соответствии с требованиями, указанными в маркировке [4, 6].
Токсичные продукты разложения. Если содержание API в лекарственном препа-
рате значительно превышает необходимые 90% (как указано выше), образование
токсичных продуктов разложения (способных вызвать нежелательные побочные
эффекты у пациентов) во время хранения может явиться основанием для пересмо-
тра срока хранения и назначения другой даты истечения срока годности или отзыва
лекарственного препарата. Вследствие этого фармацевтическую промышленность
зачастую интересует как количество, так и природа продуктов разложения. Обра-
зование этих продуктов представляет проблему, в частности для лекарств на основе
белков, которые могут сохранять терапевтическую активность после постепенной
модификации или нарушения молекулярной структуры в домене, отделенном от
домена, который ассоциируется с терапевтической активностью, однако при этом
могут стимулировать иммуногенез [4]. Обсуждения лекарств, продукты разложе-
ния которых являются более токсичными, чем исходные лекарства, представлены
Guillory и Poust [7]. Известно, что озабоченностью потенциальным повышением ко-
личества токсичных продуктов разложения объясняется нежелание регуляторных
агентств регистрировать новые лекарственные препараты с существенным превы-
шением срока годности (в сравнении с представляемыми экспериментальными
данными производителя) [4].
Низкая биодоступность. Обычно биодоступность лекарственного препарата опи-
сывают в терминах количества API, попадающего в кровь (концентрация в плазме),
и скорости его доставки. При всех путях введения лекарства (кроме интраваску-
лярного) очень важна его степень поглощения, поскольку не всё введенное лекар-
ство попадает в плазму и переносится в биофазу. Одной из важнейших проблем,
связанных с поглощением лекарства и оральной биодоступностью, является невоз-
можность доставки лекарства (в форме раствора) к участкам желудочно-кишечного
тракта, где происходит активное поглощение, за определенное время (т. е. проблема
перевода лекарства в раствор). Эта проблема может возникнуть вследствие измене-
ний параметров качества лекарственного препарата при хранении (в особенности
это касается скорости растворения) — несмотря на то, что содержание активного
вещества остается приемлемым, а токсичные продукты не образуются. Изменение
параметров качества может быть связано с изменением физико-химических свойств
вспомогательных материалов с течением времени. Результатом может стать неэф-
фективность лекарственного препарата при приеме.
Контаминация микроорганизмами. В настоящее время микробиологическому
качеству всех разрабатываемых лекарственных препаратов уделяется большое
740
Часть 7. Стабильность лекарств
внимание. В прошлом применялся несколько иной подход, при котором озабо-
ченность вызывали в основном лекарственные препараты, которые должны оста-
ваться стерильными: т. е. парентеральные и офтальмологические. Контроль общей
бионагрузки и отсутствие патогенных микроорганизмов считаются желательными
[4]. Для поддержания микробиологического качества лекарственных препара-
тов необходимо контролировать и подтверждать качество исходных материалов
и производственного оборудования. Далее при оценке приемлемости системы
первичной упаковки одним из важнейших соображений должна стать защита ее
содержимого. Таким образом, первичная упаковка должна защищать лекарствен-
ный препарат от воздействия факторов, способных вызвать разложение, таких
как свет, температура, потеря растворителя, кислород, пары воды и загрязнение
микроорганизмами [8]. Целостность упаковки не должна нарушаться на всем пути
от производителя через каналы распределения до конечного потребителя — паци-
ента.
Изменения внешнего вида лекарственного препарата. Некоторые физические из-
менения могут повлиять на эффективность отдельных лекарственных препаратов.
При этом для других препаратов содержание активного вещества может остаться
прежним, несмотря на физические изменения. Однако эти физические изменения
могут нарушить фармацевтическую эстетику, в результате чего препарат становит-
ся неприемлемым для пациента. Потери эмульгатора могут привести к разрушению
эмульсии (разделению фаз), а слеживание суспензии может нарушить возможность
восстановления первоначальной суспензии при встряхивании. Эти изменения мо-
гут нарушить однородность дозирования согласно предписанному режиму. Появле-
ние пятен на таблетках не всегда влияет на активность лекарства, но такие таблетки
неприемлемы для пациентов, вследствие чего может быть нарушен режим дози-
рования. Далее взаимодействия между лекарством и вспомогательным веществом
могут привести к изменениям внешнего вида лекарственного препарата; пример —
взаимодействие между лактозой и функциональной аминогруппой лекарства, кото-
рое часто приводит к пожелтению поверхности таблеток с течением времени. Ак-
тивность лекарства сохраняется, но таблетки становятся непривлекательными для
пациентов.
Потеря функциональной стабильности. Важно разработать эффективный лекар-
ственный препарат, однако не менее важно доставить его потребителю в соответ-
ствующей упаковке, не нарушив условий хранения лекарственного препарата. Это
означает, что эффективность упаковки для лекарственных препаратов (определяе-
мая как способность функционировать в соответствии со своим назначением, т. е.
возможность доставки дозированной формы в надлежащем количестве или со ско-
ростью, указанной на вкладыше первичной упаковки [8]), является важным компо-
нентом стабильности лекарственного препарата В отчетах сообщают, что при при-
менении первых трансдермальных пластырей возникали проблемы с прилипанием
к коже (способность к адгезии уменьшалась со временем, и пластырь отпадал от
кожи пациента) [4]. Для следующих лекарственных препаратов постоянство пара-
метров доставки необходимо контролировать в течение всего срока хранения: пред-
варительно заполненные шприцы, градуированные пробирки, капельницы, спреи,
порошковые ингаляторы и ингаляторы с дозирующими устройствами.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций...
741
7.3.2. Факторы, влияющие на стабильность лекарственных
средств
Общий принцип разработки и производства лекарственных препаратов состоит
в том, что качество (определяемое как физические, химические, микробиологи-
ческие, биологические параметры, а также характеристики стабильности, которые
должен сохранять лекарственный препарат, предназначенный для терапевтическо-
го или диагностического применения [8]) должно быть встроено во все процессы
или стадии фармацевтического производства, а не достигаться путем многочилен-
ных испытаний готового продукта, предназначенного для потребителей. Следова-
тельно, очень важно знать факторы, способные вызвать в течение срока хранения
нестабильность разного вида у фармацевтических субстанций и лекарственных
препаратов, включая химическое разложение (образование новых химических ве-
ществ), физическое разложение (уменьшение количества действующего вещества
без явного образования новых химических веществ) и биологическое разложение
(самым важным является микробиологическое разложение, хотя следует признать
опасность, связанную с немикробиологическими факторами, например муравьями
или крысами). Информация об этих факторах поможет не только на стадиях пред-
варительных исследований и разработки лекарственного препарата, но и на этапах
выбора и оценки защитной способности первичной упаковки для рассматриваемо-
го лекарственного препарата.
7.3.2.1. Влажность, гидролиз и pH
Жидкие лекарственные формы. Чаще всего гидролитическое разложение происходит
при определенной влажности. Гидролиз, являющийся процессом разложения ле-
карства, важен для всех типов дозированных форм: жидких, твердых, полутвердых
и газообразных. Большое значение также имеет влияние на гидролиз показателя pH.
Выявление химических групп, подверженных гидролизу, поможет специалисту, за-
нимающемуся разработкой лекарственных составов, заранее определить возможные
пути разложения и принять необходимые меры для получения лекарств с макси-
мальной стабильностью. Примеры таких химических групп включают сложноэфир-
ные соединения (ацетилсалициловая кислота, прокаин, теракаин и физостигимин),
амиды (цинхокаин, эргометрин и хлорамфеникол), лактамы (пенициллины, цефа-
лоспорины, нитразепам и хлордиазепоксид), имиды (глутетимид и этосукцимид)
и лактоны (пилокарпин и спиронолактон) [7, 9]. Скорость гидролиза в любой из
вышеназванных химических групп зависит от химической среды, окружающей хи-
мическую группу в молекуле лекарства. Электронное строение, пространственные
характеристики, а также способность групп-заместителей к связыванию водорода
могут усиливать склонность химических групп к гидролитическому разложению.
Фактически модификация химической структуры лекарства для контроля стабиль-
ности лекарства (следует соблюдать осторожность, чтобы не повлиять на терапевти-
ческую эффективность) при помощи соответствующих заместителей используется
для решения проблем, связанных со стабильностью. Сообщалось, что концепция
линейной зависимости свободных энергий Гамметта, применяемая для описания
влияния заместителей на скорости реакций в боковых цепях ароматических соеди-
нений, таких как гидролиз сложных эфиров, использована для подбора лучших за-
742
Часть 7. Стабильность лекарств
местителей аллилбарбитуровых кислот для достижения оптимальной стабильности
[9,10].
Лабораторные эксперименты, наряду со знанием химической кинетики, можно
использовать для создания лекарственного состава в виде раствора, где pH прида-
ет оптимальную стабильность (при помощи соответствующих буферов) лекарству,
гидролитическое разложение которого катализирует ион водорода (специфический
кислотный катализ) или гидроксил-ион (специфический основной катализ); в то
же время необходимо учитывать известное влияние компонентов буфера (общий
кислотно-основной катализ) в лекарственном препарате. Отсутствие полной оцен-
ки стабильности лекарства — определения каталитических коэффициентов для
специфического кислотного и основного катализа и каталитических коэффициен-
тов компонентов буфера — приводит к ошибкам при определении условий макси-
мальной стабильности лекарств в растворах. Кроме того, при определении профиля
«pH-скорость разложения» для окончательного определения условий максималь-
ной стабильности необходимо учитывать эффект ионизации лекарства. Другие
подходы, применяемые для стабилизации лекарственных препаратов в растворе,
включают использование неводных растворителей, таких как моно- и полиатомные
спирты (например, этанол глицерин и пропиленгликоль), способных изменить ди-
электрическую постоянную системы. Мицеллярная солюбилизация при примене-
нии сурфактантов и снижение растворимости (пример — повышение стабильности
пенициллина в прокаина пенициллине при наличии таких веществ, как цитраты,
декстроза, сорбитол и глюконат) способны уменьшить гидролиз действующих ве-
ществ в растворах [9].
При изучении всех видов лекарственных форм следует учитывать проницае-
мость упаковочных материалов для газов и жидкостей, поскольку она может при-
вести к разложению лекарственных препаратов из-за воздействия кислорода и ги-
дролиза. В случае растворов вымываемые из упаковки вещества могут изменить
предварительно установленную стабильность жидкой лекарственной формы. На-
пример, сообщают о попадании диоктил фталата — пластификатора, используемого
в поливинилхлоридных пластиках (ПВХ), в растворы для внутривенных вливаний,
содержащие сурфактанты [И, 12]. Потенциальный эффект попадания соединения
в раствор носит двойной характер: он угрожает как безопасности пациента, так
и стабильности лекарства, вводимого внутривенно. В соответствии с указаниями
Федерального управления США по контролю за пищевой продукцией и лекарствен-
ными средствами (FDA), критериями приемлемости для предполагаемого примене-
ния любых видов упаковочных систем в первую очередь являются совместимость
(компоненты упаковки считаются совместимыми с дозированной формой, если не
наблюдается взаимодействий, достаточных для того, чтобы вызвать неприемлемые
нарушения качества дозированной формы или компонента упаковки, т. е. разложе-
ния, вызванного препаратами вымывания, осаждения или изменения pH) и безо-
пасность (компоненты должны быть изготовлены из материалов, не выделяющих
существенных количеств вредных или нежелательных веществ, воздействию кото-
рых будет подвергаться пациент в ходе лечения данным лекарственным препаратом)
[8]. Четко указано, что лекарственные препараты, предназначенные для инъекции,
ингаляции, офтальмологического или трансдермального введения, должны пройти
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
743
всестороннее испытание. Последнее должно включать в себя как исследование экс-
тракции компонентов упаковки с целью выявления химических веществ (и их кон-
центраций), способных мигрировать в дозированную форму, так и токсикологиче-
ское исследование этих веществ для определения безопасного уровня воздействия
при введении способом, указанным в маркировке [8]. Если вымываемое соединение
представляет собой электролит, оно может изменить ионную силу раствора и, сле-
довательно, скорость разложения лекарства, что можно прогнозировать, применив
модель Брёнстеда—Бьеррума [9].
Жидкие лекарственные формы, представляющие собой коллоидные (т. е. вклю-
чающие микросферы, наночастицы и мицеллы) суспензии и эмульсии, зачастую со-
держат консерванты, такие как метил-, этил-, пропил- и бутиловые сложные эфиры
парагидроксибензойной кислоты в различных комбинациях. Типичным примером
являются антацидные суспензии с высокими величинами pH, из-за которых слож-
ные эфиры, содержащиеся в консервантах, подвержены гидролизу. Одним из спо-
собов решения этой проблемы является использование комбинаций нескольких
консервантов с надеждой на то, что некоторое количество консерванта сохранится
и защитит суспензию от микробиологической атаки. Представлен отчет о количе-
ственном содержании четырех эфиров и исходной кислоты (один из продуктов раз-
ложения) в лекарственных препаратах, где были использованы все указанные кон-
серванты [13].
Полутвердые лекарственные формы. Природа основы (носителя), используемого
для изготовления полутвердых дозированных форм, влияет на их гидролитическую
стабильность. Сообщают о повышении интенсивности разложения натрий бензил-
пенициллина в гидрогелях различных природных и полусинтетических полимеров
[14]. Кроме того, при pH 6 в гидрогелях Carbopol процентная доля неразложивше-
гося пилокарпина в состоянии равновесия зависит от эффективной вязкости среды
[15].
Твердые лекарственные формы. Наличие влаги может нарушить химическую,
а также физическую стабильность твердых дозированных форм. Отчеты показыва-
ют, что количество влаги, поглощаемой таблетками в блистерных упаковках, увели-
чивается при повышении влажности, в результате чего уменьшается механическая
прочность таблеток [16]. Хранение таблеток преднизона и эритромицина в упаков-
ках, проницаемых для влаги, изменило параметры высвобождения лекарства из та-
блеток [17, 18]. На высвобождение лекарства из таблеток с кишечным покрытием
или сахарной оболочкой влияние влаги сказывалось сильнее, чем на высвобожде-
ние из таблеток с пленочным покрытием. Например, при хранении таблеток с са-
харным покрытием изменилось время разложения, что приводило к повышению
или понижению скорости разложения [19]. При хранении двух капсул хлорамфени-
кола в условиях высокой влажности увеличилось время разложения и уменьшилась
скорость высвобождения [20]. Кроме того, сообщалось об уменьшении скорости
высвобождения лекарства из капсул ампициллина при хранении в условиях высо-
кой влажности; как полагают, это вызвано агломерацией частиц лекарства, вызван-
ной повышенной влажностью [21]. Отмечено существенное уменьшение скорости
растворения таблеток ацетаминофена при хранении в условиях повышенной влаж-
ности (30 °C, 80% RH); эффект удалось смягчить добавлением панкреатина в сре-
Часть 7. Стабильность лекарств
744
ду, где происходило растворение. Полагают, что воздействие панкреатина связано
с расщеплением поперечно-связанного желатина [22].
Когда твердые лекарственные формы, такие как таблетки, поглощают влагу, ле-
карство, находящееся у поверхности, начинает растворяться (если оно раствори-
мо). Лекарство в растворе на поверхности таблетки подвергается гидролитическому
разложению; на процесс влияет pH раствора. Сообщают, что увеличение давления
паров воды значительно повышает интенсивность разложения аминосалициловой
кислоты [23]. Исследование проникновения воды через резиновые крышки инъек-
ционных флаконов показывает, что резиновые крышки с малой проницаемостью
зачастую могут впитывать значительное количество воды [24]. Поэтому высокоги-
гроскопичные препараты, высушенные путем сухой заморозки и активно взаимо-
действующие с водой, должны быть адекватно защищены от проникновения воды
для обеспечения химической стабильности активного ингредиента во флаконе.
7.3.2.2. Кислород и окисление
Полутвердые и твердые лекарственные формы. При любом механизме окислитель-
ного процесса разложения — автоокислении (некаталитическая реакция, медленно
протекающая при участии молекулярного кислорода) или цепном процессе, вклю-
чающем инициирование, распространение и завершение реакции, велико значе-
ние молекулярного кислорода. Поэтому нужно приложить усилия для определе-
ния и контроля концентрации кислорода в водном растворе. Florence и Attwood [9]
представили схему окисления как цепную реакцию. Описание схемы представлено
для того, чтобы проиллюстрировать влияние взаимодействий между молекулярным
кислородом, компонентами раствора и вымываемыми из упаковки материалами
на процесс окисления. Полагают, что инициализация происходит под действием
свободных радикалов, образовавшихся из органических соединений при воздей-
ствии света, тепла или переходных металлов (например, меди и железа), в следо-
вых количествах присутствующих в буферном растворе. Свободные радикалы лег-
ко соединяются с молекулярным кислородом на этапе распространения реакции
с образованием пероксирадикала, который затем извлекает водород из молекулы
органического соединения с образованием гидроксипероксида; при этом образу-
ется другой свободный радикал. Завершение реакции наступает, когда свободные
радикалы разрушаются ингибиторами или происходит нарушение цепи за счет по-
бочных реакций.
Известно, что присутствие определенных функциональных групп повыша-
ет склонность некоторых лекарств к окислительному разложению [7, 9]. Феноль-
ная функциональная группа в составе стероидов чувствительна к окислению. Со-
общают, что эфирная группа подвержена окислению в составе нитрата эконазола
и нитрата миконазола. Окислению подвержены катехоламины, такие как допамин
и изопротеренол. В состав фенотиаксинов входит тиоэфирная функциональная
группа, которая окисляется до сульфоксида в присутствии воды. Многие молекулы
лекарств включают двойные связи углерод-углерод, с которыми легко взаимодей-
ствуют пероксильные радикалы, вызывая окислительное разложение. Фактически
двойная связь в высшей степени подвержена взаимодействию с атомарным кисло-
родом (сильный окислитель); считается, что атомарный кислород образуется при
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...745
возбуждении из основного триплетного состояния кислорода под воздействием
света. Следовательно, общий термин «окисление» описывает не только простое воз-
действие кислорода на молекулу лекарства, подверженную окислению, но и воздей-
ствие условий, способствующих окислению, таких как фотолиз [6]. Амфотерицин
представляет собой полиеновый антибиотик с семью сопряженными двойными
связями. Он окисляется пероксильными радикалами с потерей активности и агре-
гированием [25]. Добавление пероксильных радикалов к симвастатину может при-
вести к окислению с образованием полимерных пероксидов [26]. Функциональная
группа карбоновой кислоты — еще одна группа, в большой степени подверженная
окислению: скорость окисления аскорбиновой кислоты зависит от концентрации
кислорода [27]. Фармацевтическая литература изобилует свидетельствами окисли-
тельного разложения лекарств в растворах. 5-Аминосалициловая кислота окисляет-
ся, продукт реакции окисления образует полимерные соединения [28]. Кроме того,
сообщалось, что морфин [29], также, как и гидрокортизон [30], подвержен окисли-
тельному разложению в растворе.
Полутвердые и твердые лекарственные формы. Поскольку большинство реакций
окисления протекают в растворах, исследований эффекта окислительного разложе-
ния в твердых дозированных формах было немного. Описано автоокисление тетра-
зепама в таблетках [31].
7.3.2.3. Свет
Фотохимическое разложение — важный процесс химического разложения лекарств.
Общее описание или прогнозирование влияния света на лекарство весьма затруд-
нительно, поскольку параметры разложения сильно зависят от спектральных харак-
теристик лекарства, а также от спектрального распределения излучения источника
света [6]. Тем не менее существуют определенные тенденции, которые помогают
оценить влияние света на определенные молекулы лекарства, что позволяет решить
эту проблему на ранней стадии разработки лекарства. Молекулы с насыщенными
связями не способны к взаимодействию с видимым или ультрафиолетовым излу-
чением в ближней области спектра. Молекулы с л-элекгронами — ароматические
углеводороды (нитроароматические и арилгалогенидные функциональные груп-
пы), гетероциклические ароматические соединения, альдегиды, кетоны, сульфиды,
алкены и полиены — поглощают свет в диапазоне видимого и ближнего ультрафио-
летового излучения и весьма подвержены фотохимическому разложению. Солнеч-
ный свет потенциально может изменить параметры лекарств, способных к погло-
щению света на волнах менее 280 нм; лекарства, сильно поглощающие на длинах
волн более 400 нм, могут разлагаться под воздействием солнечного света или при
комнатном освещении [7, 32].
Исследователи подчеркивают важность выбора надлежащих наполнителей, при-
меняемых при разработке лекарственных препаратов, с точки зрения возможности
фотохимического разложения [9]. Фотохимическое разложение лекарств может
происходить при прямом воздействии света определенной длины волны на лекар-
ство (первичная фотохимическая реакция). Кроме того, наполнители могут погло-
щать световое излучение и переносить энергию в молекулу лекарства (которое не
может поглощать падающий свет), вызывая реакцию разложения. Такие вещества
746
Часть 7. Стабильность лекарств
называются фотосенсибилизаторами. В Гармонизированном руководстве (ICH) по
испытаниям стабильности (в 3 частях) в части «Испытания фотостабильности новых
лекарственных веществ и препаратов» [33] признано влияние, которое наполнители
в готовых лекарственных препаратах могут оказывать на фотостабильность, и дана
рекомендация проводить испытания фотостабильности в следующем порядке: ис-
пытания лекарственного вещества; испытания экспонированного лекарственного
препарата, извлеченного из первичной упаковки; при необходимости, испытания
лекарственного препарата в первичной упаковке и испытания лекарственного пре-
парата в коммерческой упаковке. Как сказано ранее, окислительное разложение мо-
жет инициировать свет; все факторы, влияющие на стабильность лекарств и лекар-
ственных препаратов, не действуют изолированно. Меры по стабилизации лекарств
по отношению к фотохимическому разложению зачастую включают применение
окрашенных упаковок (темное стекло не пропускает излучение с длиной волны ме-
нее 470 нм и может оказаться хорошим средством защиты лекарств, чувствительных
к ультрафиолетовому излучению) и хранение в темноте. Добавление поглотителей
ультрафиолетового излучения в полимерную пленку для покрытия таблетки являет-
ся еще одним методом защиты лекарств от фотохимического разложения [34].
Жидкие лекарственные формы. Нитропруссид натрия в водном растворе для инъ-
екций остается стабильным до одного года, если раствор защищен от света; тем не
менее, его срок хранения составляет около 4 ч при обычном комнатном освещении
[35]. Сообщают, что мочевая кислота повышает фотостабильность сульфатиазо-
ла натрия в растворах [36]. Кроме того, ^//-метионин повышает фотостабильность
аскорбиновой кислоты в растворе [37]. Сообщают о влиянии света на стабильность
молсидомина в жидкостях для инфузий [38]. Доказано, что фотохимическое раз-
ложение нифедипина зависит от интенсивности света (источник света: ртутная
лампа высокого давления, солнечный свет и флуоресцентная лампа) при комнат-
ной температуре: количество нифедипина, подвергшегося фотохимическому раз-
ложению, оказалось пропорциональным числу падающих фотонов [39]. Сообщают
о стабилизации процесса фотохимического разложения даунорубицина в растворе
(на 290—700 нм) при добавлении различных колорантов: Scarlet GN, амаранта, Пон-
со 6R и тартразина [40].
Твердые лекарственные формы. Было обнаружено сложное соотношение между
скоростью обесцвечивания сульфисомида в таблетках, подвергнутых облучению
ртутной лампой, и интенсивностью ультрафиолетового излучения [41]. Функцио-
нальные характеристики капсул, такие как скорость высвобождения лекарства,
могут измениться из-за взаимодействия красителей и желатина из оболочки кап-
сулы под влиянием света. Есть сведения о том, что изменения возрастают при воз-
действии света и влажности [42]. Показано, что твердофазное фоторазложение ле-
карства третиноинатокоферила сильно зависит от температуры. Обнаружено, что
температурная зависимость константы скорости фоторазложения третиноинато-
коферила подчиняется уравнению Аррениуса [43]. Показано, что при добавлении
диоксида титана к желатиновой капсуле происходит стабилизация окрашивания
индометацина [44]. Далее сообщают, что введение синтезированных оксидов железа
привела к стабилизации таблеток нифедипина и соривудина по отношению к фото-
разложению [45].
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...747
7.3.2.4. Температура
Температурная зависимость константы скорости химической реакции зачастую
описывается уравнением Аррениуса:
£ = Ле^ЛГ- (1)
где к — кинетическая константа скорости; А — преэкспоненциальный фактор; Еа —
энергия активации; R — идеальная газовая постоянная; Т — температура.
Уравнение показывает, что при повышении температуры увеличивается ско-
рость реакции. Следовательно, температура является важным фактором стабиль-
ности лекарств и лекарственных препаратов. Далее, если зависимость Аррениуса
является линейной, скорости разложения, полученные при определенных тем-
пературах, можно использовать для прогноза скоростей разложения при дру-
гих температурах. Это уравнение строго достоверно для реакций, протекающих
в растворах; предполагается, что изменения механизма разложения или порядка
реакции не происходит. Уравнение применяют для прогнозирования стабильно-
сти лекарственных форм. Для твердых лекарственных форм влияние температуры
на стабильность представляет сложную зависимость из-за вероятных изменений
характеристик наполнителей под воздействием температуры, способных нарушить
стабильность активного ингредиента. Несмотря на то что уравнения, сходные
с уравнением Аррениуса, используются для моделирования зависимости скорости
реакции от температуры для твердых дозированных форм, следует ожидать, что
в этом случае значение энергии активации будет отличаться от значения, получен-
ного для растворов лекарств [9].
Там, где параметры процесса разложения приближаются к равновесным значе-
ниям, как показано на примере витамина Е в таблетках [9, 46], равновесные кон-
центрации препаратов разложения и реагентов получены при ряде температур. За-
тем строят график зависимости логарифма константы равновесия К от обратной
температуры в соответствии с уравнением Вант-Гоффа для моделирования зависи-
мости распада лекарства от температуры:
Infc = —EH/RT + const. (2)
Жидкие лекарственные формы. Уравнение Аррениуса используют для моделиро-
вания процесса обесцвечивания жидкого препарата, в составе которого было не-
сколько сульфатов, и разложения жидких мультивитаминных составов [47—49]. Со-
общалось о разложении кодеина сульфата в растворах при различных температурах
[50]. Кроме того, изучено влияние pH на графики Аррениуса для реакции гидролиза
циклозидомина [51]. Стабильность эмульсии клофибрида для перорального вве-
дения снижалась (происходила агрегация эмульсии) при увеличении температуры
хранения с 25 до 40 °C. Кроме того, хранение при температуре 4 °C привело к разде-
лению фаз в вышеупомянутой эмульсии [52]. Предполагают, что стабильность вак-
цины можно прогнозировать, оценив потерю антигенных свойств после длитель-
ных периодов хранения при различных температурах в ходе ускоренных испытаний
стабильности. Несмотря на то что в определенных условиях для некоторых вакцин
уравнение Аррениуса можно использовать для прогнозирования реальной стабиль-
ности по данным ускоренного испытания стабильности, при высоких температурах
748
Часть 7. Стабильность лекарств
многие процессы разложения осложняют развертывание биомолекулы, структур-
ные и конформационные изменения. Ниже точки замерзания скорость разложе-
ния может внезапно возрасти или уменьшиться, при этом линейной корреляции
с температурой может не наблюдаться [53]. Далее константа скорости разложения
к является не единственным фактором, определяющим остаточную антигенность
вакцины Pt. Кроме того, имеют значение время хранения вакцины при заданной
температуре t и исходная антигенность вакцины Ро, что можно выразить уравнени-
ем [53, 54]:
k=(J>0-P)/t. (3)
Полутвердые лекарственные формы. При нагревании происходят быстрые фа-
зовые изменения в полутвердых лекарственных препаратах; следовательно, про-
водить испытания при высоких температурах для изучения кинетики разложения
и последующего прогноза стабильности нецелесообразно. Поэтому стабильность
часто оценивают при температуре хранения лекарственного препарата. Эта мето-
дика зачастую является времязатратной; проблемы, связанные со стабильностью,
могут не выявиться в ходе испытания, длящегося несколько месяцев и даже лет [55].
Температура хранения может нарушить затвердевание суппозиториев, при этом из-
меняется время, необходимое для плавления суппозитория. Доказано, что эффект
затвердевания усиливается с повышением температуры до 25 °C, однако ослабевает
при дальнейшем повышении температуры, что связано с частичным плавлением
основы суппозитория [6].
Твердые лекарственные формы. Модель разложения аспирина в таблетках с осно-
вой из микрокристаллической целлюлозы построена в соответствии с кинетикой
первого порядка и константами скорости, полученными по уравнению Аррениуса
[56]. Показано, что изменяется скорость высвобождения лекарства из таблетки, со-
держащей поливинилпирролидон (в качестве разрыхлителя), при повышении тем-
пературы хранения с 23 до 65 °C [57]. Изменение скорости растворения таблеток
гидрохлортиазида при комнатной температуре оценивали по изменениям, наблю-
даемым при 37, 50 и 80 °C [58]. Сообщают о взаимодействии некоторых фуроевых
кислот при таблетировании с микрокристаллической целлюлозой с образованием
моноксида углерода [59]. Взаимодействие происходило быстро при 55 °C и приво-
дило к крошению таблеток, однако было менее выражено при комнатной темпера-
туре.
7.3.2.5. Микроорганизмы
Одним из факторов, влияющих на надежность лекарственного препарата, явля-
ется его устойчивость к микробной контаминации. Центр биологической оценки
и исследований лекарств [5] изучил вопросы, связанные с микробиологическим
качеством препаратов, возникающие при планировании испытаний стабильности
и интерпретации его результатов. Четко показано, что лекарственные препараты,
содержащие консерванты, регулирующие микробную контаминацию, должны про-
ходить контроль содержания консервантов — по меньшей мере в начале и конце
планируемого периода годности препарата. Кроме того, адекватность системы кон-
сервации следует учитывать при применении упаковок многоразового использо-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...749
вания. Далее микробные препараты, требующие контроля микробиологического
качества, не содержащие консервантов, следует тестировать через определенные
промежутки времени в течение планируемого периода годности в соответствии со
спецификацией по бионагрузке. Препараты для местного применения также следует
проверять на наличие патогенов, которые могут считаться потенциально опасными.
Эти правила следует помнить при рассмотрении вопросов, связанных с пригодно-
стью системы упаковки: Система упаковка/укупорочное средство должна адекватно
защищать дозированную форму от факторов, вызывающих разложение; микробная
контаминация входит в список таких факторов [8].
Фармакопея США/Национальный формуляр USP2K/NF23 [1] определяет ста-
бильность лекарственного препарата как степень его соответствия параметрам
в определенных пределах и способность сохранять в течение всего периода хра-
нения и применения те свойства и характеристики, которыми препарат обладал
во время его изготовления. Микробиологическая стабильность (определяемая
как сохранение стерильности или сопротивление росту микроорганизмов в со-
ответствии с указанными требованиями; при этом присутствующие антимикроб-
ные агенты сохраняют эффективность в указанных пределах) является одним
из основных типов стабильности, признанных в USP 28/NF 23. Таким образом,
в дополнение к усилиям, направленным на стабилизацию лекарственных препа-
ратов по отношению к химическому и физическому разложению, вызываемому
факторами окружающей среды, обсуждавшимися выше, жидкие и полутвердые
лекарственные препараты должны быть при необходимости защищены от микро-
биологической контаминации. Антимикробные консерванты, добавленные в фар-
мацевтические препараты, должны быть оценены в ходе испытаний стабильности
химическими методами или путем микробной провокационной пробы. Микроб-
ная провокационная проба рекомендована в последней временной точке испыта-
ния стабильности.
Жидкие лекарственные формы. Важны как стерильные (с учетом сохранения сте-
рильности, а также изменения эффективности консервации), так и нестерильные
препараты (возможность пролиферации микроорганизмов). Офтальмологические
и парентеральные жидкие препараты зачастую изготавливают в условиях стериль-
ности, тем не менее в состав часто добавляют надлежащие консерванты для сохра-
нения стерильности в течение периода хранения, распределения и применения.
Кроме того, другие лекарственные препараты, к которым не предъявляется требо-
вание стерильности, вследствие чего их не стерилизуют в процессе производства,
могут содержать ингредиенты, способствующие росту микроорганизмов. Такие
лекарственные препараты также защищают при помощи антимикробных консер-
вантов [60]. Примерами являются препараты на водной основе, такие как эмульсии
и суспензии. Ранее упоминалось о применении метиловых, этиловых, пропиловых
и бутиловых эфиров парагидроксибензойной кислоты в различных комбинациях
в составе лекарственных препаратов [13]. Желание оставить определенное количе-
ство консервантов в суспензии для консервации препарата зачастую заставляет ис-
пользовать комбинации консервантов.
Полутвердые лекарственные формы. FDA требует, чтобы все офтальмологиче-
ские мази были стерильными. Кроме того, необходимо добавлять надлежащий
750
Часть 7. Стабильность лекарств
консервант или смесь консервантов, предотвращающих рост числа микроорганиз-
мов, в офтальмологические мази, упакованные в упаковки многоразового исполь-
зования. В качестве консервантов в мази часто добавляют хлорбутанол и метил-
и пропил-иоря-гидроксибензойную кислоту [61]. Поскольку стерилизация готовых
офтальмологических мазей затруднена (затруднения в основном связаны с наруше-
нием стабильности компонентов), для соблюдения требований стерильности обыч-
но применяются методы асептического производства [62]. К мазям для местного
применения не выдвигают требования стерильности, тем не менее они должны от-
вечать определенным стандартам по содержанию микроорганизмов [1]. В состав
кремов и гелей обычно вводят соответствующие антимикробные консерванты, по-
скольку высокое содержание воды способствует росту микроорганизмов.
Твердые лекарственные формы. На первый взгляд может показаться, что твердые
лекарственные формы не подвержены микробной контаминации. Однако исследо-
вания показывают, что контаминация таблетки может произойти через исходные
материалы. Были проведены исследования, оценивающие влияние производства,
окружающей среды, технологии и микробиологического качества исходных мате-
риалов на микробиологическую нагрузку на разных стадиях изготовления таблеток
[63]. Высокие уровни микробной контаминации отмечали при влажном гранулиро-
вании; уровень существенно снижался в процессе сушки.
7.3.2.6. Активные фармацевтические ингредиенты и наполнители
Физико-химические свойства API. Гигроскопичность представляет собой количе-
ство влаги, поглощенной порошкообразным веществом из атмосферы с известной
относительной влажностью. В ходе предварительных исследований необходимо
использовать накопленную информацию для принятия решения о том, можно
ли применять конкретную соль исследуемого лекарственного вещества при про-
изводстве дозированной формы. Есть сведения, показывающие, что флуразепам
чаще применяют в виде моносульфата, чем дисульфата, который тем не менее об-
ладает рядом желательных характеристик. Однако он настолько гигроскопичен,
что поглощает воду из твердой оболочки капсулы, которая становится очень хруп-
кой [64]. Сведения о гигроскопичности API могут помочь при выборе упаковочных
материалов.
Кристаллическое или аморфное состояние и полиморфизм. Твердые частицы ле-
карственного вещества существуют в виде кристаллических веществ определенной
идентифицируемой формы или в виде аморфных частиц, не имеющих определенной
структуры. Энергия, необходимая для того, чтобы молекула лекарственного веще-
ства покинула кристаллическую решетку, намного превышает энергию активации
в аморфном порошкообразном веществе. Лекарства кристаллической структуры
обладают низкой реакционной способностью. Обнаружено, что между твердофаз-
ной константой скорости разложения различных производных витамина А и вели-
чиной, обратной точке плавления, существует линейное соотношение [65]. Более
того, исследования соотношений между скоростью разложения и кристаллично-
стью р-лактамовых антибиотиков, таких как цефазолин, выявили тенденцию: ле-
карство с малой кристалличностью имеет пониженную химическую стабильность
[66].
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
751
Полиморфизм представляет собой явление, заключающееся в возможности су-
ществования частиц твердого вещества в различных кристаллических формах, на-
зываемых полиморфами. Лекарственные соединения образуют кристаллы разных
форм в зависимости от условий (температура, растворитель, время), в которых про-
исходила кристаллизация. В разных полиморфных формах молекул лекарств на-
блюдается разное пространственное расположение молекул и строение кристалли-
ческой решетки. Следует подчеркнуть: полиморфизм существует только в твердом
состоянии вещества. Только одна форма чистого лекарственного вещества является
стабильной при данных температуре и давлении; в то же время другие формы (так
называемые метастабильные) со временем переходят в стабильную кристалличе-
скую форму. Для метастабильной формы лекарственного вещества переход в дру-
гую полиморфную форму даже в готовом фармацевтическом составе представляет
обычное явление, хотя время, необходимое для полного перехода, может превы-
шать срок хранения самого лекарства. Несмотря на то что лекарства в разных фор-
мах химически неразличимы, полиморфные формы существенно различаются по
ряду физических свойств, таких как плотность, точка плавления, растворение, ста-
бильность и параметры растворения, что следует учитывать при разработке дозиро-
ванных форм данного лекарства. Обнаружено, что гидролиз карбамазепина в фор-
ме высоко упорядоченных игольчатых кристаллов протекает быстрее, чем гидролиз
кристаллов в форме пучка и призмы, а активность карбамазепина по отношению
к свету сильно зависит от кристаллической формы лекарства [67, 68]. Аналогичные
сведения представлены по фоторазложению фуросемида [69].
Модификация химической структуры лекарства. Ссыпки на использование прин-
ципа линейного соотношения свободных энергий (уравнения Гаммета) для изуче-
ния влияния заместителей на скорость реакций в боковых цепях ароматических
соединений, таких как гидролиз эфиров, присутствуют в ранних исследованиях, на-
правленных на достижение оптимальной стабильности [9,10]. Разложение эритро-
мицина в условиях кислой среды ингибируют замещением метокси-группы на С-6
гидроксил; этот эффект отвечает за кислотную стабильность клатромицина, кото-
рая в 340 раз превышает стабильность эритромицина [70].
Давление паров. Активные фармацевтические ингредиенты с достаточно высоким
давлением паров могут испаряться из упаковок; при этом нарушается стабильность
и однородность содержимого. Впоследствии такие соединения могут взаимодей-
ствовать с другими молекулами лекарств и компонентами упаковки [71]. Извест-
но, что такими свойствами обладает нитроглицерин, поэтому для распределения
сублингвальных таблеток нитроглицерина необходимо использовать специальные
материалы. При хранении нестабилизированных сублингвальных таблеток нитро-
глицерина в закрытых стеклянных упаковках высокая летучесть лекарства приво-
дила к перераспределению нитроглицерина среди таблеток, что сопровождалось
нарушением однородности таблеток, находящихся на хранении [72].
Фармацевтические наполнители. Лекарства, предназначенные для употребления
человеком, чаще всего представляют собой составы, содержащие лекарственное
вещество и вспомогательные материалы, называемые наполнителями, которые до-
бавляют в составы с различными целями для обеспечения адекватных свойств ле-
карственных препаратов. Однако некоторые наполнители могут нарушить стабиль-
752
Часть 7. Стабильность лекарств
ность лекарственных препаратов. Сообщают, что на разложение кодеина оказывает
влияние буфер: константа гидролиза кодеина в фосфатном буфере при pH 7,0 при-
мерно в 20 раз выше, чем в подбуференном растворе при таком же pH [9]. Далее
было обнаружено, что различные фосфаты усиливают разложение бензипеницилли-
на [73], цефадроксила [74], карбенициллина [75] и спиронолактона [76]. Некоторые
лекарства не способны к прямому поглощению света, возбуждающему фотолиз, од-
нако наполнители могут поглощать световое излучение и переносить поглощенную
энергию в лекарство, вызывая разложение [9]. Кроме того, переходные металлы,
такие как медь и железо, присутствующие в буфере в следовых количествах, спо-
собны инициировать цепную реакцию окислительного разложения. Консерванты
могут нарушить стабильность дозированных форм. Консервация инсулин-цинка
(в мультидозовом флаконе) при помощи фенола может привести к физической не-
стабильности суспензии. Все исходные материалы, применяемые для изготовления
офтальмологических фармацевтических препаратов, должны быть высшего клас-
са качества. Исходя из этого некоторые производители лекарств при изготовлении
офтальмологических составов используют воду для инъекций.
При выборе надлежащей мазевой основы стабильность API, входящих в состав
основы, является фактором первостепенной важности [60]. Исследования показали,
что стабильность гидрокортизона в мази, изготовленной на основе полиэтиленгли-
коля, очень мала [77, 78]. Полутвердые препараты зачастую со временем приобре-
тают желтую или коричневую окраску из-за окислительного разложения, затра-
гивающего основу (в особенности природные жиры и масла), входящие в состав
препаратов [55]. Активное окисление природных жиров, так называемое прогорка-
ние, связано с появлением неприятного запаха. Полагают, что различные фазовые
переходы, кристаллизация, реакции трансэтерификации в основах суппозиториев
являются причиной затвердевания суппозиториев при хранении, что может небла-
гоприятно сказаться на качестве препарата, например, затруднить высвобождение
активных ингредиентов [6]. Кроме того, обнаружено, что аспирин разлагается в по-
лиэтиленгликолях, являющихся компонентами основы суппозитория; разложение
объяснили реакцией трансэтерификации, в результате которой образуются салици-
ловая кислота и ацетилированный полиэтиленгликоль [79]. Разложение аспирина
наблюдали, когда в качестве основы суппозитория использовали масло какао [80].
Сложные эфиры полиэтиленгликоля и индометацина обнаружены в суппозиториях,
находящихся на хранении, изготовленных на полиэтиленгликолевой основе [81].
Сообщают, что стеарат магния (смазывающее вещество) ускоряет процесс
обесцвечивания таблеток, содержащих амины и лактозу [82]. Наполнители, такие
как связующие вещества (например, повидон) и разрыхлители, такие как кросспо-
видон, содержащие фенольные примеси, мозут отрицательно повлиять на фото-
стабильность таблеток, вступив в реакцию с участием свободных радикалов [83].
Сообщают об усиленном окислении фенилбутазона красителями через продуциро-
вание атомарного кислорода, участвующего в цепных реакциях [84]. Наполнители
с высокой влажностью могут усилить разложение лекарств путем повышения коли-
чества влаги, вступающей во взаимодействие с лекарством. Согласно публикации
[85], процентная доля разложившейся аминосалициловой кислоты увеличивалась
с повышением давления водяных паров.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
753
7.3.3. Упаковка для лекарственных средств и упаковочные
материалы
7.3.3.1. Введение
Система упаковки, зачастую называемая системой упаковка/укупорочное средство,
состоит из компонентов, комбинация которых обеспечивает хранение и защиту
препарата [8]. Традиционно говорят о двух типах упаковочных компонентов лекар-
ственной продукции: компоненты первичной упаковки (находящиеся в непосред-
ственном контакте с лекарственным препаратом), и компоненты внешней упаковки
(обычно серия упаковок), зачастую называемые вторичными упаковочными ком-
понентами, включающие картонные коробки, гофрированную тару и паллеты [86].
Опровергая традиционное мнение о том, что к компонентам внешней упаковки
лекарственной продукции не предъявляются специальные требования (за исклю-
чением маркировки), исследования показывают, что внешняя упаковка может су-
щественно продлить срок годности лекарственных препаратов: препарат в блистер-
ной упаковке без вторичной упаковки, хранившийся при 37 °C, 90% RH, пришел
в негодность через 21 день (предполагалось, что блистер может быть непригоден
для упаковки продукции). Тем не менее, когда такую же блистерную упаковку поме-
стили в картонную пачку, которую в свою очередь завернули во внешнюю обертку
и хранили в тех же условиях на складе компании, соответствие препарата специфи-
кации сохранялось после 6 лет хранения [87].
Фармацевтическая упаковка представляет собой систему, содержащую лекар-
ственный препарат, способную сохранять его так, что он остается безопасным
и активным в течение всего срока хранения. От этого традиционного определения
переходят к другому определению, в котором признаётся важность упаковки для
обеспечения эффективности лекарственного препарата. Так, в настоящее время
пользуются следующим определением: «Фармацевтическая упаковка представляет
собой комбинацию компонентов, необходимых для хранения, консервации, защи-
ты и доставки безопасного и эффективного лекарственного препарата» [87]. Факти-
чески это определение отвечает утверждению из руководства для промышленности
«Системы упаковка/укупорочное средство для упаковки лекарств и биологических
препаратов, предназначенных для употребления человеком» [8] о том, что система
упаковка/укупорочное средство должна отвечать своему назначению (она должна
адекватно защищать дозированную форму; быть совместимой с дозированной фор-
мой и быть изготовленной из материалов, безопасных при использовании вместе
с дозированной формой и путем введения). Это утверждение расширяет определе-
ние через следующее указание: если система упаковки должна выполнять функции,
дополнительные к хранению препарата, то необходимо продемонстрировать, что
в собранном виде система упаковка/укупорочное средство функционирует правиль-
но. Такие лекарственные препараты, как предварительно заполненные шприцы,
трансдермальные пластыри, ингаляторы с дозирующими устройствами и назальные
спреи содержат лекарственные составы, успешная доставка которых к конечному
потребителю (пациенту) зависит от правильного функционирования упаковочной
системы. Требования к параметрам защиты, совместимости, безопасности и эффек-
тивности упаковочных систем относительно пригодности для использования по
754
Часть 7. Стабильность лекарств
назначению варьируются в зависимости от типа дозированной формы и пути вве-
дения. Существует таблица, являющаяся руководством для фармацевтической про-
мышленности, в которой представлены данные по приемлемости систем упаковки
для традиционных классов лекарственных препаратов [8].
7.3.3.2. Материалы, применяемые при изготовлении компонентов
упаковки
Наука об упаковочных материалах, применяемых для фармацевтических и косме-
тических препаратов, весьма обширна. Выбор упаковки начинается с изучения ее
физических и химических свойств, необходимости защиты и требований рынка.
Выбранные материалы должны обладать следующими характеристиками: защищать
состав от различных разрушающих факторов (физических, климатических, химиче-
ских и биологических) [87]; не должны вступать в реакцию с препаратом (согласно
правилам надлежащей производственной практики {cGMP) [88]), изменять его вкус
или запах; должны быть нетоксичными; должны получить одобрение FDA (следует
отметить, что FDA одобряет лишь материалы, использованные в данной упаковке,
а не в упаковках вообще, FDA опубликовало список веществ, признанных отно-
сительно безопасными; а также отвечать требованиям защиты от фальсификаций
и быть адаптированными к традиционному высокоскоростному упаковочному обо-
рудованию [89].
Стеклянные упаковки. Стекло является общепризнанным фармацевтическим
упаковочным материалом, поскольку обладает исключительными защитными
свойствами и является экономичным материалом. Легкодоступны стеклянные упа-
ковки любых форм и размеров. Они в основном являются химически инертными,
непроницаемыми, жесткими и прочными; их применение одобрено FDA. Стекло не
разрушается со временем; при наличии надлежащей системы укупорки оно являет-
ся отличным барьером, защищающим от всех разрушающих факторов, кроме света.
Окрашенное стекло, в особенности темное, может при необходимости защитить от
воздействия света. Основными недостатками стекла как упаковочного материала
являются его хрупкость и вес.
USP/NF [1] признаёт четыре типа стеклянных упаковок. Тип I — упаковка из
боросиликатного стекла, которая используется для составов для парентерального
введения. Стеклянные упаковки типа II, изготовленные из натриево-кальциево-
силикатного стекла, деалканизированного надлежащим образом, обычно исполь-
зуют для упаковки кислых и нейтральных составов для парентерального введения
(стеклянные упаковки типа I можно также использовать для этих целей, однако
его чаще используют для упаковки щелочных парентеральных составов). Упаковки
типа III, изготовленные из натриево-кальциево-силикатного стекла, не использу-
ют для парентеральных составов, если в данных по стабильности не указано иное.
Упаковка IV категории (стекло типа NP) предназначена для непарентеральных пре-
паратов (т. е. препаратов, предназначенных для перорального введения и местного
применения).
Пластиковые упаковки. Доказано, что применение пластиков для упаковки
оправдано легкостью изготовления, высоким качеством, возможностью различно-
го дизайна. Кроме того, пластиковые упаковки чрезвычайно прочны, что делает их
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
755
безопасными для потребителя, и снижают потери за счет поломки упаковки на всех
этапах распределения и применения [89]. Стеклянные флаконы с пластиковым по-
крытием используются в качестве аэрозольных упаковок для предотвращения вы-
лета стеклянных фрагментов в случае разрушения стекла; вдобавок пластиковое
покрытие вокруг горла флакона поглощает напряжение, возникающее в процессе
обжимания колпачков, и уменьшает риск разрушения стекла у горлышка флакона.
Пластиковые упаковки для фармацевтических препаратов в основном изготав-
ливают из следующих полимеров:
1. Полиэтилен является хорошим барьером, препятствующим проникновению
влаги, однако довольно слабо защищает от кислорода и других газов; большин-
ство растворителей не разрушает полиэтилен, он не повреждается сильными кис-
лотами и щелочами; в качестве упаковочных материалов используют полиэтилены
высокой и низкой плотности; полиэтилен обладает длинной полимерной цепью.
Плотность полиэтилена определяет четыре основные физические характеристики
упаковок, изготовленных выдувным формованием: жесткость, влаго- и паропро-
ницаемость, растрескивание под действием напряжения и прозрачность или свето-
проницаемость. Эти характеристики обусловливают пригодность полиэтилена для
изготовления упаковок для лекарственных препаратов. Факторы, определяющие
пригодность полиэтилена, перечислены в USP/NF [1]: проницаемость для кисло-
рода и влаги, модуль эластичности, точка плавления, сопротивление растрескива-
нию под воздействием факторов окружающей среды и степень кристалличности
после отливки.
2. Полимеры полипропилена являются длинноцепочечными полимерами, ко-
торые синтезируют из пропилена или пропилена и других олефинов при контроле
температуры и давления с добавлением катализаторов. Пропилен не растрескива-
ется под действием напряжения независимо от условий. За исключением горячих
ароматических или галогенированных растворителей, размягчающих полипропи-
лен, этот полимер хорошо выдерживает воздействие всех прочих типов химических
веществ, включая сильные кислоты, щелочи и большинство органических мате-
риалов. Высокая температура плавления делает его пригодным для изготовления
упаковок для кипячения препаратов и для препаратов, подлежащих стерилизации.
Малая прозрачность является недостатком, однако проблема частично решается
путем изготовления упаковки с более тонкими стенками. Полипропилен являет-
ся отличным барьером, препятствующим проникновению газа и паров. Его низ-
кая проницаемость сопоставима с проницаемостью полэтилена высокой плотно-
сти и полиэтилена линейной структуры (или несколько ниже нее), и превосходит
проницаемость полиэтилена низкой плотности или полиэтилена с разветвленной
структурой [1, 89]. Основным недостатком полипропилена является его хрупкость
при низких температурах.
3. Полиэтилентерефталат (ПЭТ) является конденсационным полимером, кото-
рый обычно образуется при взаимодействии терефталевой кислоты или диметилте-
рефталата с этиленгликолем в присутствии катализатора. Полиэтилентерефталата
гликолевые смолы представляют собой высокомолекулярные полимеры, получае-
мые путем конденсации этиленгликоля с диметилтерефталатом или терефталевой
кислотой и 1,4-циклогександиметанолом в количестве 15—34 молярных процентов.
756
Часть 7. Стабильность лекарств
ПЭТ- и ПЭТГ-смолы и другие ингредиенты, используемые при изготовлении этих
флаконов, удовлетворяют требованиям соответствующих разделов Свода федераль-
ных нормативных актов (CFR), том 21, относящихся к материалам, контактирую-
щим с пищевыми и алкогольными напитками: ПЭТ- и ПЭТГ-флаконы взаимоза-
меняемы и пригодны для упаковки жидких дозированных форм, предназначенных
для перорального применения [1]. Среди других полимеров, рекомендованных или
применяемых для упаковки лекарственных препаратов, отметим поликарбонат, по-
ливинилхлорид и полистирол; реже применяют полиметилметакрилат, полиэтилен-
терефталат, политрифторэтилен и полиамиды [89].
Исследования различных параметров, которые необходимо учитывать при под-
боре пластика для упаковки лекарственного препарата, длятся годами. Характери-
стики пары «лекарство—пластик» разделены на пять категорий: проницаемость,
вымывание, сорбция, химическая активность и нарушение физических свойств
пластика или препарата [89].
Металлы. Металлы используют для изготовления гибких туб и в аэрозольных
баллонах. Самыми распространенными металлами для изготовления фармацевти-
ческих упаковок являются олово, алюминий и свинец. Олово — самый дорогой из
металлов, свинец — самый дешевый. Ламинат из свинца с оловянным покрытием
придает упаковке эстетичный внешний вид и обеспечивает защиту от воздействия
кислорода, равноценную защите, которую обеспечивает чистое олово, по более
низкой цене [89]. Из всех металлов, применяемых для изготовления складываю-
щихся туб (трубок), наиболее инертным химически является олово. Оно обеспечи-
вает эстетичный внешний вид и совместимость с большим количеством препара-
тов. Алюминиевые тубы столь же привлекательны, как и оловянные, однако цена
их несколько ниже. Свинец — самый дешевый из всех металлов, применяемых для
изготовления туб; последние широко используются для упаковки непищевых пре-
паратов, например клеев. Тем не менее, свинцовые тубы с внутренним покрытием
используют для упаковки таких препаратов, как зубные пасты, содержащие фто-
риды. Если препарат несовместим с незащищенным металлом, то внутреннюю по-
верхность покрывают воскообразным составом или растворами смол.
Алюминий используют для упаковки некоторых аэрозольных лекарственных
препаратов. Трехчастная упаковка, изготовленная из стали с тонким оловянным
покрытием, применяется для аэрозольных препаратов местного применения; для
уменьшения проблем совместимости зачастую наносят внутреннее органическое
покрытие [90].
Резина. Пробки, прокладки и части пипеток, используемые в фармацевтических
упаковках, изготавливают из резины. Резиновые пробки используют в основном
для укупорки мультидозных флаконов и одноразовых шприцев. Среди полимерных
резин чаще всего используют натуральную, неопреновую и бутиловую. Поскольку
к качеству резины предъявляют определенные требования, в резиновые укупороч-
ные средства обычно вводят добавки: например, вулканизирующий агент, ускори-
тель/активатор вулканизации, эластичный наполнитель, активный наполнитель,
умягчитель/пластификатор, антиоксидант, пигмент и воски. Сложный состав резин
требует осторожности, особенно при применении для упаковки парентеральных
препаратов: при контакте резиновой пробки с парентеральным раствором может
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...757
произойти поглощение активного ингредиента, антибактериального консерван-
та или других материалов, в то же время может произойти экстракция в жидкость
одного или нескольких ингредиентов резины [89].
7.3.4. Влияние упаковки на стабильность лекарственных
препаратов
7.3.4.1. Введение
Целью испытания стабильности является исследование изменений лекарственного
препарата (в системе упаковка/укупорочное средство и вне ее) с течением времени
под влиянием различных разрушающих факторов, таких как температура, влажность
и свет, для установления срока хранения лекарственного препарата и рекоменда-
ций по условиям хранения. В общем пригодность упаковки определяется четырь-
мя свойствами: защитой, безопасностью, совместимостью и эксплуатационными
качествами (функционирование и/или доставка лекарства). При оценке системы
упаковка/укупорочное средство необходимо учитывать следующие факторы: мате-
риалы конструкции упаковка/укупорочное средство; обработка поверхности и/или
технологические добавки; активные ингредиенты и наполнители, входящие в со-
став дозированной формы; стерилизация и/или другие подобные процессы; нако-
нец, условия хранения. Указания изложены в официальных руководствах по оценке
систем упаковка/укупорочное средство. Во-первых, в руководствах даны указания
по оценке упаковки или упаковочных материалов: физико-химические и биологи-
ческие испытания стеклянных или пластиковых бутылок, металлических крышек,
укупорочных средств из эластомеров, гибких и блистерных материалов, компонен-
тов шприцев и аэрозольных упаковок. Во-вторых, представлены указания по пла-
нированию испытаний стабильности (ускоренные, краткосрочные и долгосрочные
испытания), позволяющих оценить влияние компонентов упаковки на препарат.
В общем считается, что полностью инертная система упаковка/укупорочное сред-
ство невозможна, поэтому испытания направлены на идентификацию, описание
и мониторинг взаимодействий, что важно для получения безопасного, нефальсифи-
цированного, стабильного и эффективного лекарственного препарата [86]. В конце
испытания системы упаковка/укупорочное средство в фармацевтической промыш-
ленности составляют технический отчет, в котором излагают принципы, результаты
и выводы.
7.3.4.2. Твердые лекарственные формы
Испытание стабильности таблеток лозартана/гидрохлортиазида проводили в три
этапа [91]. Во-первых, проводили стресс-испытания (исследование форсированно-
го разложения): без первичной упаковки таблетки выдерживали в жестких условиях
хранения (50 °C, 80% RH). Через четыре недели был сделан вывод, что таблетки
чувствительны к воздействию влаги. На втором этапе проводили предварительное
исследование для выбора упаковочной системы. Исследовали две упаковочные си-
стемы с различными барьерными свойствами. Результаты показали, что блистеры
из поливинилхлорида 250 мкм — полиэтилена 25 мкм — поливинилиденхлори-
да 60 г/м2 — алюминиевой фольги (PVC-PE-PVdC-AI) способны частично защи-
тить таблетки от паров воды и газов, а блистеры из ориентированного полиамида
758
Часть 7. Стабильность лекарств
25 мкм — алюминиевой фольги 45 мкм — поливинилхлорида 60 мкм — алюминие-
вой фольги (OPA-AL-PVC-AI) способны обеспечить абсолютную защиту таблеток.
Проводился контроль состава лекарства, содержания примесей, распадаемости
и внешнего вида таблеток. Результаты, полученные в течение 6 мес. хранения при
40 °C, 75% RH, показали, что блистеры PVC-PE-PVdC-Al недостаточно защищают
таблетки лозартана/гидрохлортиазида от влаги, и их следует упаковывать в ОРА-
AL-PVC-Al-блистеры. Наконец, провели испытание OPA-AL-PVC-Al-блистеров по
утвержденной методике. Проводили мониторинг таких параметров, как содержание
API, примеси, растворение, распадаемость, твердость, содержание воды, внешний
вид и микробиологическое качество. Результаты, полученные в ходе 6-месячного
ускоренного испытания и 12-месячного долгосрочного испытания стабильности,
показали, что таблетки лозартана/гидрохлортиазида, упакованные в OPA-AL-PVC-
Л/-блистеры, сохраняли химическую, физическую и микробиологическую стабиль-
ность; предложенный срок хранения составил 24 мес.
Исследования проводились для накопления данных о стабильности лекар-
ственного препарата и характеристиках упаковки (равновесное содержание влаги
в таблетке, скорость разложения неупакованного препарата, влагостойкость упа-
ковки) в целях выбора и последующего подтверждения упаковочного материала,
обеспечивающего адекватную стабильность соединения, чувствительного к влаге
(PGE-7762928) [92]. Физическую и химическую стабильность (исследование мето-
дом высокоэффективной жидкостной хроматографии (ВЭЖХ)) препаратов иссле-
довали через 2, 4, 6, 8, 12 и 24 недели в условиях, предписанных ICH. Через 6 мес.
хранения при 40 °C, 75% RH процентная доля активного компонента составила 84%
в блистерах из поливинилхлоридов, 91% в блистерах из циклических олефинов, 97%
в блистерах из Аклара (полихлортрифторэтилена), 100% в блистерах, изготовлен-
ных из алюминия методом холодной формовки, и 99% во флаконах, изготовленных
из полиэтилена высокой плотности с термозапечатываемым фольгой отверстием.
Результаты испытаний стабильности для упакованного препарата хорошо согла-
суются с прогнозами, основанными на чувствительности препарата к влаге и вла-
гостойкости соответствующей упаковки. На основании результата анализа, пока-
завшего, что содержание API в таблетке составило 90% от заявленного значения
через 6 мес. исследования, блистерные упаковки из алюминия холодной формов-
ки и Аклара, а также флаконы, изготовленные из полиэтилена высокой плотности
с термозапечатанным фольгой отверстием, обеспечивают стабильность таблетки
PGE-7762928. Несмотря на то что упаковка лекарственной продукции в инертной
атмосфере является общепринятой практикой в парентеральном секторе фарма-
цевтического производства, примеров твердых дозированных форм, упакованных
в атмосфере с пониженным содержанием кислорода, относительно мало. Модель-
ные гранулированные составы, содержащие лекарство, подверженное окислитель-
ному разложению, были помещены в укупоренные стеклянные флаконы, которые
хранили в условиях различного содержания кислорода в свободном пространстве
флакона, различной относительной влажности в свободном пространстве флакона
и температуре 40 °C. Количественно окислительное разложение являлось функцией
времени, а данные выявили зависимость параметров окислительного разложения от
концентрации кислорода в свободном пространстве флакона, относительной влаж-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
759
ности, лекарственной нагрузки и времени. Было рекомендовано помещать погло-
тители кислорода во флаконы; кроме того, использование упаковочной блистерной
линии фольга-фольга в инертной атмосфере является способом изготовления фар-
мацевтических упаковок с пониженным содержанием кислорода [93].
Было проведено исследование in vitro влияния упаковки и условий хранения
на характеристики качества таблеток карбамазепина. В испытании изучали Тегре-
тол и египетский генерик Теграл. Оба препарата были упакованы в контурные ячей-
ковые упаковки из ПВХ и алюминия, помещенные в картонную пачку; немецкий
препарат инлепсин — во флаконы по 50 таблеток. Характеристики препаратов in
vitro оценивали проведением теста на растворение; в то же время химическую ста-
бильность карбамазепина исследовали методом ВЭЖХ [94]. Показано, что характе-
ристики препарата Теграл не изменились при стрессовых условиях хранения: упа-
кованные в блистеры и хранившиеся при 50 или 60 °C и 75% RH таблетки легко
распадаются и показывают быстрое высвобождение действующего вещества. Хра-
нение при 40 °C, 97% RH в течение 6 мес. вызывало изменения, аналогичные из-
менениям при хранении в течение 1 мес. при 40 °C, 97% RH; таблетки затвердевали
и хуже растворялись, чем свежеизготовленный препарат. Извлечение таблеток Те-
грала из первичной упаковки негативно сказалось на растворении: высвобождение
действующего вещества составило только 7% за 60 мин. Для инлепсина влияние
97% RH при 40 °C было более выраженным, чем 97% RH при 25 0С, однако и в тех
и в других условиях произошло снижение скорости высвобождения, степень кото-
рого зависит от расположения таблетки во флаконе. Тем не менее, во всех препара-
тах, подвергшихся хранению в стрессовых условиях, не было выявлено изменений
химической стабильности таблеток карбамазепина в любых стрессовых условиях.
Модель, учитывающая характеристики массопереноса в упаковочных материа-
лах и химическую стабильность, использована для описания диффузии воды через
поливинилхлоридную блистерную упаковку. Обнаружено, что эффект поглощения
влаги таблеткой влияет на прочность при раздавливании, а степень влияния зависе-
ла от лекарственного состава. Сделан вывод о необходимости учета характеристик
состава при выборе упаковки [95]. Информацию о влагостойкости прессованных
таблеток и влагопроницаемости упаковки использовали для разработки физиче-
ской модели, позволяющей прогнозировать изменения прочности таблетки при
раздавливании в различных условиях хранения и упаковки. Прогнозы полезны при
установлении методик долгосрочных испытаний стабильности. Кроме того, они по-
лезны при определении типов таблеток, хранящихся в блистерных упаковках, по-
ставляемых на рынок, для которых возможны кратковременные и долговременные
колебания условий хранения. Теоретические прогнозы и экспериментальные ре-
зультаты свидетельствуют о важности совместимости характеристик дозированных
составов с упаковочным материалом, а также необходимости соблюдения условий
испытания. В этом случае можно ожидать более рационального выбора упаковоч-
ного материала и методик испытания [96].
Известно, что перенос жидкости сквозь упаковку может повредить твердые же-
латиновые капсулы двумя путями: изменить характеристики оболочек либо свой-
ства содержимого капсул. При недавнем исследовании капсул изучали изменения
как скоростей растворения, так и активности двух брендов капсул амоксициллина,
760
Часть 7. Стабильность лекарств
упакованных в ПВХ-блистерную упаковку (ПВХ-блистер имеет толщину 0,27 мм
и алюминиевую основу толщиной 27 мкм) и ламинатную (ламинатная упаковка
включает три слоя: внешнее нитроцеллюлозное покрытие, промежуточный слой —
мягкую алюминиевую пленку; внутренний слой из полиэтилена). Два бренда амок-
сициллина хранили вне упаковки при 76, 80 и 92% RH и в упаковке при 92% RH;
существенных различий в профилях растворения не было выявлено. Хранение кап-
сул двух брендов вне упаковок при 80 и 92% RH привело к существенной потере
активности амоксициллина. Упаковка, изготовленная из ламинатного материала,
обеспечивала лучшую защиту в сравнении с ПВХ-алюминиевой блистерной упа-
ковкой. После 20 недель хранения в условиях 92% RH при комнатной температуре
потеря активности амоксициллина в капсулах, упакованных в ламинатный матери-
ал, составила только 6,4% в сравнении с потерей в 51,8% в капсулах из блистерной
упаковки [97].
Исследовали влияние условий хранения (50 °C, 50% RH и 40 °C, 90% RH) на ха-
рактеристики (твердость, разрушение, вес таблетки, размеры, скорость растворения
и содержание API) двух брендов (А и В) таблеток эритромицина стеарата с пленоч-
ным покрытием и одного бренда (С) таблеток эритромицинового основания с ки-
шечнорастворимым покрытием. Таблетки хранили в бумажных пакетах, пластико-
вых флаконах с дозаторами и стеклянных бутылках. Кроме повышения твердости
в условиях хранения при 40 °C, 90% RH для таблеток бренда С и уменьшения вре-
мени разрушения для таблеток бренда А в тех же условиях изменений физических
свойств выявлено не было. Тем не менее, для всех таблеток при хранении в двух
типах условий скорости растворения значительно снизились. Стеклянные упаковки
лучше защищали таблетки, вследствие чего они сохраняли более высокие скорости
растворения по сравнению с таблетками, которые хранились в пластиковых или бу-
мажных упаковках [98].
Проведено исследование поглощения нитроглицерина термопластичными поли-
мерами и стабильности прессованных таблеток нитроглицерина в стрип-упаковках.
Исследованные полимеры сильно различались по сродству к нитроглицерину со
следующим порядком уменьшения сродства: винилы > полиэтилен низкой плот-
ности > иономеры > полиэтилен высокой плотности. При правильном подборе
упаковки прессованные таблетки нитроглицерина, стабилизированные повидоном,
упакованные в стрип-полоски в виде единичных дозированных форм, до 2 лет со-
храняли приемлемую активность при температуре 26 °C. Кроме того, было изучено
химическое разложение нитроглицерина через гидролиз. Повидон ускорил разло-
жение нитроглицерина; при высокой температуре оно оказалось существенным
фактором, влияющим на стабильность таблетки, в случае таблеток, содержащих по-
видон [99]. Исследовали жидкую дисперсную систему, содержащую этодолак (20%)
и Гелуцир 44/14 (D-a-токоферил полиэтиленгликол 1000 сукцинат (витамин Е,
TPGS)) (80%) в различных соотношениях. Провели испытания стабильности со-
става для наполнения капсул в различных условиях температуры и влажности в со-
ответствии с руководством ICH. За период хранения при комнатной температуре
и при 4 °C, 0% RH физические и химические свойства дисперсии не изменились.
Однако относительная влажность и длительность хранения повлияли на поведение
этодолака при растворении. Был сделан вывод: изменения параметров растворения
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
761
после хранения в условиях повышенной влажности и температуры, возможно, свя-
заны с образованием микрокристаллов этодолака и поглощением воды носителем
в процессе хранения, вследствие чего необходимо применение влагостойкой упа-
ковки [100].
7.3.4.3. Нестерильные жидкие лекарственные формы
Сообщают об исследованиях влияния упаковочных материалов на стабиль-
ность эмульсий, предназначенных для защиты от ультрафиолетовых (УФ) А, УФ
В и инфракрасных солнечных лучей, при хранении в упаковках из различных ти-
пов упаковочных материалов (стеклянные и пластиковые флаконы; пластиковые
и металлические тубы) [101]. Образцы (эмульсии, содержащие бензофенон-3,
октилметоксициннамат и фикокораил) выдерживали при температурах 10, 25, 35
и 45 °C и анализировали репрезентативные пробы по прошествии 2, 7, 30, 60 и 90
дней. Полученные данные показали, что эмульсии, выдержанные при различных
температурах, имели близкие реологические характеристики в течение 3 мес. хране-
ния; существенных изменений физической и химической стабильности эмульсий,
хранившихся в различных упаковках, не наблюдалось. Подтверждена пригодность
таких упаковочных материалов, как стекло и пластик для хранения подобных пре-
паратов.
Результаты исследований показали, что хлорохин связывается со стеклом, но не
образует связей с отдельными типами пластиков. Эта информация важна для лабо-
раторных испытаний, где существенное снижение концентрации может произойти
при изготовлении и хранении лекарства в стеклянных упаковках [102]. Далее под-
буференные растворы хлорохиндифосфата различных концентраций при разных
pH выдерживали в контакте с натриевым или боросиликатным стеклом. При хране-
нии в контакте с натриевым стеклом наблюдали снижение исходной концентрации
лекарства до 60 и 97% при хранении в аналитических пробирках и в контакте со
стекловолокном соответственно. Связывания лекарства с боросиликатным стеклом
не наблюдалось. Высочайший уровень связывания отмечен при физиологических
значениях pH. Поэтому при хранении, анализе или проведении испытаний на чув-
ствительность возбудителей малярии к лекарству необходимо пользоваться бороси-
ликатным стеклом во избежание потери хлорохина из раствора [103].
Кислород часто используют в испытаниях, направленных на выявление склонно-
сти конкретных веществ к окислительному распаду. Методика предусматривает вы-
держивание лекарственного препарата в виде раствора в ампулах, заполняемых под
кислородом, с последующим сравнением их скоростей распада в сравнении с ана-
логичными растворами, хранящимися в атмосфере азота. Лекарственные составы,
подверженные окислению, можно стабилизировать, заменяя кислород в упаковках
для хранения азотом или диоксидом углерода; можно добавлять антиоксидант или,
например, выбирать упаковку, изготовленную из материала, не содержащего ионов
металла [9]. При хранении фотолабильных лекарственных препаратов (растворов,
полутвердых и твердых препаратов) следует использовать упаковки, непроницае-
мые для УФ излучения. Полагают, что излучение в этом диапазоне длин волн — са-
мая частая причина фоторазложения препарата. Темное стекло является очень эф-
фективным материалом, предотвращающим или уменьшающим фоторазложение,
762
Часть 7. Стабильность лекарств
поскольку оно не пропускает излучение с длиной волны менее 470 мм [9]. Можно
рекомендовать хранить фотолабильные лекарства в темноте.
7.3.4.4. Стерильные жидкие лекарственные формы
Применение предварительно заполненных шприцев в условиях больниц постоянно
растет благодаря преимуществам от их использования и существующего интереса
к лекарствам в единичной упаковке. Известно, что в продаже нет всех доз и комби-
наций лекарств, назначаемых пациентам в больницах, поэтому многие больничные
аптеки разрабатывают и реализуют программу централизованного изготовления
смесей для парентерального введения. Упаковка лекарственных препаратов требует
внимания к соблюдению стерильности и наличию твердых частиц в препарате, за-
полняющем шприц, а также к совместимости между лекарствами и компонентами
шприца. Несмотря на то что пластиковый шприц имеет ряд преимуществ, контакт
между пластиковой упаковкой и лекарством может создавать проблемы, связанные
с вымыванием, сорбцией, проницаемостью, химической активностью полимера
и изменениями физических характеристик пластика. Было проведено исследование
широко применяемой в качестве преанестетического средства лекарственной ком-
бинации (гидроксизина гидрохлорида, меперидина гидрохлорида и атропина суль-
фата) в целях сравнения возможных различий стабильности смесей, хранившихся
в стеклянных и пластиковых упаковках. Комбинации лекарств хранили в стеклянных
и пластиковых шприцах при 25 и 3 °C в течение 10 дней. В процессе хранения прово-
дили исследования, включая визуальный осмотр, определение pH, анализ спектров
УФ-поглощения и газохроматографический анализ. Ярко выраженного разложения
содержимого шприцев или появления новых веществ ни в одном случае обнару-
жено не было. Таким образом, хранение подобных препаратов как в стеклянных,
так и в пластиковых шприцах существенно не сказалось на их стабильности [104].
В отчете показано влияние типа упаковки на стабильность цефтазидима в со-
ставе растворов для внутривенного введения. Для испытания в пакеты из полипро-
пилена (ПП) и ПВХ (по 100 мл в каждый), а также в стеклянный флакон емкостью
100 мл поместили растворы 5%-ной декстрозы или 0,9%-ного хлорида натрия, со-
держащие цефтазидим (40 мг/мл). Эти растворы хранили при 20 и 35 °C. Пробы
объемом 1 мл отбирали из каждой упаковки в моменты времени 0 и 24 ч и подвер-
гали анализу. Уровни содержания пиридина (основного препарата разложения) уве-
личивались при хранении и были выше в пакетах из полипропилена (ПП) и ПВХ
по сравнению со стеклянными флаконами в обоих растворителях. Стабильность
растворов, хранившихся в ПП пакетах, оказалась выше стабильности растворов
в пакетах из ПВХ. Предположили, что стеклянные флаконы являются более адек-
ватными упаковками для хранения растворов цефтазидима [105]. Были проведены
исследования, нацеленные на оценку влияния типа растворителя, условий хране-
ния и материала упаковки на стабильность восстановленного парекоксиба натрия
для инъекций) [106]. Результаты исследования показали, что раствор парекоксиба
натрия, восстановленный изотоническим раствором хлорида натрия, бактериоста-
тическим изотоническим раствором хлорида натрия, 5%-ным раствором декстрозы
для инъекций и 5%-ным раствором декстрозы с гипотоническим раствором хлорида
натрия для инъекций, отвечает требованиям по описанию и практически не разла-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
763
гается в условиях хранения. Существенных различий в характеристиках растворов,
хранившихся в стеклянных флаконах и преднаполненных шприцах из ПП и стекла,
не выявлено. Тем не менее, во многих флаконах раствора парекоксиба натрия, вос-
становленного раствором Рингера с лактатом для инъекций и раствором Рингера
с лактатом и с 5%-ным раствором декстрозы для инъекций, наблюдали заметный
осадок; результаты подтверждены данными количественного определения методом
ВЭЖХ во всех временных точках.
Проведено исследование стабильности растворов офтальмологического пре-
парата унопростона изопропила (аналог простангландина, выпускающийся под
торговым названием Рескула, применяемый для лечения повышенного внутри-
глазного давления у пациентов с первичной открытоугольной глаукомой или оку-
лярной гипертензией) в двух типах упаковочных материалов, полипропилен (ПП)
и полиэтилен низкой плотности (ПНП). Осуществляли мониторинг концентраций
унопростона изопропила и продуктов его разложения в зависимости от времени.
Было обнаружено, что скорость уменьшения содержания лекарства, хранившего-
ся в бутылках из ПНП, выше, чем при хранении в бутылках из ПП. Дальнейшие
исследования показали, что пониженная стабильность, связанная с применением
упаковок из ПНП, вызвана главным образом поглощением унопростона изопро-
пила упаковочным материалом и в меньшей степенени химическим разложением.
Обнаружено, что интенсивность поглощения зависит от температуры: снижение
температуры привело к уменьшению поглощения, что позволило увеличить срок
хранения препарата [107].
В USPустановлено, что для обеспечения физической стабильности эмульсий ли-
пидов для инъекций предельное содержание капель крупных размеров, выраженное
в процентной доле общего количества капель, размеры которых намного превыша-
ет 5 мкм (или PFATJ, не должно превышать 0,05%. Недавние исследования показа-
ли, что в эмульсиях для инъекций, упакованных в недавно появившиеся на рынке
пластиковые упаковки, PFATS превышает установленные в USP предельные значе-
ния. Как следствие, стабильность эмульсий существенно уменьшается в процессе
имитированной инфузии, выполняемой с помощью шприца. Несмотря на то что
умеренный рост жировых капель большого диаметра наблюдали для липидов в сте-
клянной упаковке, их количество в течение всего исследования не выходило за
пределы, установленные в {АУР для крупных капель. Таким образом, есть основания
полагать, что липиды в стеклянной упаковке являются более стабильной дозиро-
ванной формой и потенциально более безопасным способом доставки лекарства
через шприц в организм новорожденного ребенка, находящегося в критическом со-
стоянии [108].
В последние годы значительный интерес вызывали способы клинического при-
менения нитроглицерина, вводимого внутривенно, для лечения инфаркта миокарда
и при хирургических операциях на открытом сердце. При внутривенном введении
поглощение лекарства ПВХ-пакетами признано проблемой: для процесса поглоще-
ния, контролируемого стадией диффузии, полупериод относительного поглощения
составляет 3,2 ч при 30 °C [109]. Более того, в ходе недавних исследований было
показано, что набор для внутривенного вливания является источником потери ни-
троглицерина из раствора [110—112]. Сообщают о количественных исследованиях
764
Часть 7. Стабильность лекарств
потерь нитроглицерина в пластиковых трубках для внутривенных вливаний. Было
показано, что потеря нитроглицерина из физиологических растворов поваренной
соли в ПВХ-трубках за короткие периоды времени происходит за счет адсорбции,
скорость которой была велика и количественно представляла собой процесс перво-
го порядка. Период полупотери составил 2,6 мин [113]. Недавно предложена мо-
дель, описывающая потерю нитроглицерина из раствора, помещенного в пластико-
вые пакеты, как быструю адсорбцию на пластиковой поверхности с последующим
распределением в объеме пластика [114]. Механизм потери нитроглицерина при
хранении в пластиковых и стеклянных упаковках изучали, применяя кинетический
и равновесный подходы. Данные показали, что потеря нитроглицерина из водного
раствора происходила за счет поглощения лекарства материалом пластиковой упа-
ковки. Потеря нитроглицерина не могла происходить за счет гидролиза, поскольку
растворы, хранившиеся в стеклянных упаковках, при pH 5,7 и 35 °C сохраняли ак-
тивность в течение по меньшей мере 48 ч, несмотря на то что экспериментальные
данные подтверждают предположение о том, что потеря нитроглицерина проис-
ходила за счет миграции лекарства в матрицу пластика, что не исключает возмож-
ности адсорбции лекарства на поверхности: скорость адсорбции может намного
превышать скорость абсорбции, в результате интерпретация любых проявлений ад-
сорбции может вызвать затруднения [115].
Было проведено исследование с целью описания кинетики и механизма взаи-
модействия между различными лекарствами и полимерными мешками для инфу-
зионных растворов для определения критериев, которые можно использовать для
прогноза подобных взаимодействий. Изучали поглощение натрия варфарина, раз-
личных бензодиазепинов и других лекарств с инфузионными пакетами, изготов-
ленными из ПВХ Кроме того, исследовали поглощение отдельных соединений
мешками для инфузионных растворов, изготовленных из ПП. Кинетика поглоще-
ния варфарина и диазепама может быть описана диффузионной моделью, согласно
которой потеря лекарства определяется, главным образом, диффузией соединения
в матрицу пластика. Скорость и степень поглощения варфарина выявила зависи-
мость процесса от pH, что можно объяснить в терминах ионизации лекарственно-
го вещества: происходило поглощение исключительно неионизированной формы.
Было найдено упорядоченное отношение между исходной скоростью поглощения
полимерным пакетом и коэффициентами распределения соединений в системе
гексан-вода. Таким образом, коэффициент распределения в системе гексан-вода
может оказаться полезным для прогнозирования взаимодействий между лекар-
ственным веществом и инфузионными пакетами, изготовленными из ПВХ. Погло-
щение соединений инфузионными пакетами, изготовленными из ПП, оказалось
незначительным, за исключением поглощения высоколипофильного медазепама.
Таким образом, использование мешков из ПП, возможно, является более безопас-
ным в сравнении с пакетами из ПВХ [116].
Нестабильные в воде лекарственые препараты, вводимые парентеральным пу-
тем, зачастую предлагаются потребителю в виде твердых веществ, подлежащих
восстановлению. Подобные препараты могут быть упакованы во флаконы или кар-
триджи, состоящие из двух отделений, где порошкообразное вещество находится
в одном отделении, а растворитель — в другом. Резиновые крышки используют-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...765
ся в качестве основного упаковочного материала благодаря их уникальным свой-
ствам, таким как эластичность при прокалывании и самогерметизация, которые
сохраняются в условиях лиофилизации. Кроме того, укупорочные средства для
флаконов, изготовленные из бутиловой или галогенированной бутиловой резины,
обладают низким уровнем проницаемости для влаги и газов. При хранении пре-
паратов резиновые пробки находятся в непосредственном контакте с содержимым
флакона и являются потенциальным источником загрязнения препарата: выделе-
ния механических частиц, миграции бензотиазидов, вымывания ионов металлов,
антиоксидантов и олигомеров, силиконового масла, серы и парафинового воска.
В восстановленных растворах с водными носителями наблюдается помутнение,
возникающее из-за образования осадков в пересыщенных растворах, полимериза-
ции разложившихся лекарств и высвобождения неполярных летучих соединений
(насыщенных углеводородов, хлорированных и нехлорированных олефинов, ал-
килбензолов и полидиметилсилоксанов низкого молекулярного веса) из резиновых
укупорочных средств [117]. Исследования показали, что большая площадь поверх-
ности препарата является основным фактором, способствующим адсорбции лету-
чих соединений, высвобождаемых из резиновых пробок (у препаратов с большими
площадями поверхности наблюдают высокий уровень мутности растворов после
восстановления). Попытки воспрепятствовать помутнению включают применение
солюбилизаторов и таких укупорочных средств, как крышки (пробки) из бромбути-
ловой резины (вместо бутиловой и хлорбутиловой резины) либо резиновые пробки
с тефлоновыми вставками. Уменьшения миграции летучих веществ можно достичь,
подвергая резиновые пробки воздействию высоких температур или вакуума в про-
цессе сложно чередующихся циклов промывки [118].
Было проведено исследование влияния упаковок для инфузионных препаратов
из полимерных материалов и стекла и полимерных комплектов для инфузионных
вливаний (со встроенными в линию фильтрами и без фильтров) на концентрацию
диазепама в растворе. Визуальной несовместимости отмечено не было, и значе-
ние pH оставалось постоянным. Растворы, хранившиеся в стеклянных бутылках
и вводившиеся через полимерные комплекты, сохраняли более 90% диазепама (ис-
ходного содержания активного компонента) через 4 ч. В растворах, хранившихся
в течение 2 ч в полимерных капельных камерах комплектов и вводившихся через
инфузионный комплект, отмечалось снижение концентрации диазепама более чем
на 38% от исходной. Потери происходили, главным образом, в пластиковой каме-
ре и увеличивались с возрастанием концентрации лекарства и с течением време-
ни. На доступность лекарства не повлияли тип жидкости, вводимой внутривенно,
pH, скорость потока или фильтрация. При инфузии диазепама было рекомендо-
вано разбавлять его в соотношении по меньшей мере 1:10 в стеклянных бутылках
для внутривенных вливаний и вводить через инфузионные комплекты (системы),
не имеющие полимерных капельных камер. Возможно встраивание фильтров в ли-
нию (0,45 мкм) без потери эффективности [119]. Аналогичные исследования прово-
дил Morris [120]. Было обнаружено, что раствор диазепама для инъекций визуально
совместим и химически стабилен в 5%-ном растворе декстрозы в воде, в растворе
Рингера для инъекций, растворе Рингера с лактатом и 0,09%-ном растворе хлорида
натрия при хранении в бутылках для внутривенных вливаний в течение 24 ч. Одна-
766 Часть 7. Стабильность лекарств
ко подобное испытание с использованием полимерных мешков для внутривенных
вливаний вместо стеклянных бутылок показало, что потеря активности (содержа-
ния активного компонента) составила более 24% через 30 мин после разбавления.
Уменьшение процентного содержания диазепама в растворе ускорилось с увеличе-
нием концентрации и с течением времени [121].
Лиофилизированные лекарственные препараты для инъекций зачастую произ-
водят упакованными во флаконы. Резиновая пробка является критически важным
защитным барьером. Защита препарата от влаги или кислорода сильно зависит от
качества и функциональных характеристик этого барьера. На самом деле поступали
сообщения о том, что резина не только защищает от влаги, но и может оказаться
источником воды [122]. Для лучшего понимания основных механизмов, опреде-
ляющих качество защитного барьера, и поиска несложного метода выбора из не-
скольких типов резин были проведены исследования скорости поглощения воды,
насыщения и, наконец, проницаемости различных типов резиновых укупорочных
средств. Использовали 13-миллиметровые пробки одинаковых размеров: Helvoet
FM 157 (серая) и /71/257 (серая); Pharma Gummi PH 701/45 (красная), PH 701/45
(красная) и ///4104/40 (серая); West Company 1/888 (серая). Все резины относились
к типу бромбутиловой резины, за исключением 1/888, состоящей из смеси галобу-
тил/полиизопрен. Обнаружено, что проницаемость укупорочных средств трудно
прогнозируема из-за концентрационной зависимости коэффициента диффузии D,
который сильно возрастает с повышением RH. Показано, что барьерные характери-
стики новых типов резин можно оценивать по профилям поглощения в стрессовых
условиях: например, при 40 °C и 95% RH. Этот способ позволяет сделать выбор, не
прибегая к трудоемким экспериментам по определению проницаемости [123].
7.3.4.5. Влияние веществ, выделяющихся из материала упаковки,
на стабильность лекарственного препарата
Из упаковок и изделий для введения лекарственных и биологических препаратов
в препарат могут попадать высвобождающиеся из них низкомолекулярные сое-
динения. С точки зрения токсикологии это вызывает озабоченность, так как эти
соединения могут нарушить эффективность и биологическую безопасность лекар-
ственного препарата. Упаковочные материалы, соприкасающиеся с лекарством
(синтетические полимерные составы), могут включать антиоксиданты, красители,
агенты, облегчающие скольжение, и пластификаторы.
Вопросы, связанные с экстракцией и вымыванием соединений, попадающих в ле-
карственные препараты, являются предметом активного обсуждения в сообществе
фармацевтических производителей. Между тем в последние годы регуляторные агент-
ства опубликовали указания, освещающие эту проблему. Руководство FDA по упаков-
ке определяет экстрагируемые и вымываемые вещества следующим образом [124]:
• экстрагируемые (экстрактивные) вещества — это соединения, которые могут
быть экстрагированы из эластомерных или полимерных компонентов упа-
ковки под воздействием растворителя;
• вымываемые (высвобождаемые) вещества — это соединения, которые вымы-
ваются (высвобождаются) из эластомерных или полимерных компонентов
упаковки и присуствуют в лекарственном препарате.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...767
Таким образом, термин «экстрагируемые вещества» относится к перемещению
соединения из системы упаковка/укупорочное средство в условиях экстракции под
воздействием растворителей в условиях нестандартной или нетипичной обработки
препарата с использованием различных растворителей и/или в стрессовых услови-
ях, не использующихся в процессе производства лекарственного препарата (силь-
ные органические растворители или растворы кислот/щелочных и щелочноземель-
ных металлов, а также повышенные температура и давление). Термин «вымываемые
соединения» описывает перемещение соединения из системы упаковка/укупороч-
ное средство в нестрессовых условиях, способствующих солюбилизации (взаимо-
действие лекарственного вещества с поверхностями, а также условия, связанные
с определенным специфическим технологическим процессом) [125]. Различие
между двумя терминами связано с процессом и не относится к фундаментальным
химическим свойствам соединения. Экстрагируемое или вымываемое соединение
накапливается при распределении данного соединения между двумя фазами: твер-
дой (система упаковка/укупорочное средство) и жидкой (лекарственный препарат
для вымываемых соединений или растворитель для экстрагируемых соединений).
Необходимо провести анализ содержания экстрагируемых и/или вымываемых
веществ из каждой упаковки или приспособления с последующей токсикологиче-
ской оценкой потенциальной опасности. Оценка токсикологического риска вклю-
чает исследование токсичности компонентов упаковки и конкретных экстрагируе-
мых и вымываемых веществ. Токсикологические параметры включают токсичность
единичной, повторной и хронической дозы, генотоксичность, канцерогенность,
иммуносенсибилизацию, раздражающее действие и совместимость с кровью [126].
Действия регуляторных органов (FDA), озабоченных наличием вымываемых соеди-
нений в лекарственных препаратах, связаны непосредственно с контролем конкрет-
ных форм дозирования и путей введения [8]. Наибольшую озабоченность вызывают
лекарственные препараты для ингаляций и назальные лекарственные препараты
[124,127,128], включая дозированные препараты для ингаляций, находящиеся под
давлением (MDI), порошки для ингаляций (DPI), растворы и спреи для ингаляции,
а также назальные спреи. В FDA считают, что вымываемые вещества представля-
ют угрозу для безопасного использования препаратов для ингаляций. Вымывае-
мые вещества могут представлять угрозу для растворов и суспензий для инъекций,
стерильных порошков и порошков для инъекций, офтальмологических растворов
и суспензий, а также трансдермальных мазей и пластырей. Меньшую озабоченность
вызывают жидкие и твердые лекарственные формы для приема внутрь [8].
Существует множество проблем (в основном научного характера), связанных
с попаданием в лекарственные препараты вымываемых и экстрагируемых соеди-
нений, решение которых требует создания программ и формирования групп для
фармацевтических исследований. Вопросы, которые предстоит решить, могут быть
сформулированы в следующем виде: на каком уровне содержания вымываемые
соединения, попадающие в лекарственный препарат, угрожают безопасности по-
требителя; какие процедуры квалификации безопасности можно применить в от-
ношении вымываемых соединений; как разработать стратегию контроля (включая
спецификации и критерии приемлемости) вымываемых соединений; можно ли ре-
гулировать содержание вымываемых соединений в лекарственном препарате путем
768
Часть 7. Стабильность лекарств
контроля потенциально вымываемых (или экстрагируемых) соединений [128]. В от-
чете рабочей группы по исследованию вымываемых и экстрагируемых соединений
Института исследования качества лекарственных препаратов (PQRI) предложен
полный план фармацевтических разработок препаратов для ингаляций и назальных
препаратов, учитывающий проблемы вымываемых и экстрагируемых соединений
[129], основанный на собственных научных разработках и более ранних предложе-
ниях Международного консорциума фармацевтических аэрозолей по науке и регу-
лированию [130]. В рекомендациях PQRI обсуждаются также критерии выбора си-
стемы упаковка/укупорочное средство для препаратов для ингаляций и назальных
препаратов, конструкция которой исключает потенциальное попадание вымывае-
мых соединений, представляющих угрозу для безопасности, и минимизирует число
и содержание других потенциально вымываемых веществ. Полагают, что рекомен-
дации выходят за рамки контроля качества, поэтому предлагается проанализиро-
вать всю систему понятий и встраивать качество в процесс разработки системы упа-
ковка/укупорочное средство для препаратов для ингаляций и назальных препаратов
(парадигма — качество через дизайн) [ 128]. Испытание на вымывание и экстракцию
рекомендуется даже в тех ситуациях, когда упаковки или укупорочные средства от-
вечают фармакопейным критериям приемлемости.
Кроме токсикологических рисков, которые несет наличие вымываемых и экс-
трагируемых соединений в лекарственном препарате, другой проблемой, связан-
ной с вымываемыми веществами, является стабильность, поскольку вымываемые
вещества могут быть химически активными и вступать во взаимодействие с лекар-
ственным препаратом. Наиболее распространенными вымываемыми химически ак-
тивными веществами являются переходные металлы, свободные радикалы — ини-
циаторы или пропагаторы, органопероксиды и химически активные нуклеофилы
или электрофилы, такие как амины и альдегиды [131]. Среди основных источников
вымываемых металлов — стеклянные упаковки и оборудование, соприкасающеся
с препаратом в процессе производства, в особенности оборудование из непасси-
вированной нержавеющей стали. Вымываемые металлы зачастую вызывают раз-
ложение через окисление, где металл катализирует образование короткоживущего
перокси-радикала, который впоследствии вступает в реакцию с лекарственным
веществом [132]. Как правило, в процессах участвуют переходные металлы, такие
как Рел+ и Мпл+, и окисление протекает в соответствии с каталитическим циклом
Фентона [133]. В экспериментальных внутривенных дозированных формах наблю-
дали разложение там, где ионы металлов, вымываемые из стеклянных флаконов
и упаковок из нержавеющей стали, катализировали процесс окисления с переходом
одного электрона. Интересно, что антиоксиданты, включенные в состав (тиогли-
церол, аскорбиновая кислота или бисульфит натрия), участвовали в реакциях Фен-
тона, приводящих к окислению [134]. Для фоточувствительного лекарственного
препарата применение флаконов из темного стекла привело к появлению других
проблем, связанных со стабильностью: повышенный уровень содержания вымы-
ваемых веществ интенсифицировал процесс окисления лекарств, катализируемый
металлами [135]. Вымываемые металлы в жидких составах могут вызвать образова-
ние нерастворимых комплексов с фармацевтически активным веществом или дру-
гими ингредиентами состава. Сообщают о том, что ускоренное испытание стабиль-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...769
ности при повышенных температурах может не выявить этих проблем, поскольку
осадки образуются и растут при пониженных температурах нелинейным образом
[136]. Ионы алюминия, вымываемые из стекла СЖРтипа I и некоторых пластиковых
упаковок, таких как LDPE и резиновые укупорочные средства, являются основным
источником образования твердых частиц. Вымываемые ионы Л13+ накапливаются
в растворах на уровнях от 45 до 6 ррт в зависимости от таких факторов, как наличие
буферов, pH раствора и автоклавная обработка [137].
Органопероксидные радикалы могут присутствовать в пластиковых упаковоч-
ных материалах из-за наличия остаточных свободных радикалов от диссоциации
пероксидов, образовавшихся во время полимеризации или процесса плавления
пластика, или из долгоживущих радикалов, образующихся в полимере в результате
радиолиза полимерной цепи, индуцированного гамма-излучением. Изучение ми-
грации продуктов радиолиза из пластиков, стерилизованных гамма-излучением или
пучком электронов, показало, что фрагменты молекул низкого молекулярного веса
образуются при радиолизе вносимых в пластик добавок и остаточных олигомеров
[138,139]. Сообщают, что высоко реакционноспособные вещества, такие как альде-
гиды, реагируют непосредственно с лекарственными препаратами у нуклеофильных
центров, таких как первичные или вторичные амины. Формальдегид может образо-
вываться в незначительном количестве в качестве побочного продукта окисления
длинноцепочечных спиртов и из других источников, таких как акрилаты. В сте-
рильном составе, содержащем экспериментальный противоинсультный препарат,
упакованный в стеклянные флаконы с резиновыми пробками, был обнаружен про-
дукт разложения ад дукта формальдегида на уровне 2% через 13 недель хранения при
30 °C. Был найден источник формальдегида, находившийся в резиновой пробке
(формальдегид образовался из упрочняющего агента, входившего в состав пробки)
[140]. Стратегия минимизации риска, вызванного вымываемыми веществами, для
стабильности лекарства предусматривает тщательный анализ способа стерилизации
и выбора упаковки. Применение стеклянных флаконов с силиконовым покрытием,
заполнение азотом верхнего объема упаковки, применение пластиков и введение
хелатных агентов уменьшает побочные эффекты влияния вымываемых веществ
на стабильность.
Недавно внимание FDA привлекла агрегация белка из-за ряда инцидентов, в ко-
торых проблемы со стабильностью были вызваны веществами, вымываемыми/экс-
трагируемыми из материала упаковки, или заменой наполнителей, как предположи-
ли в случае с препаратом Эпрекс (эпоэтин-альфа, Johnson & Johnson, New Brunswicks,
NY). Поскольку агрегация белка может индуцировать иммуногенные реакции с по-
тенциально серьезными последствиями, FDA требует, чтобы производители уделяли
больше внимания этой проблеме [141]. Анализ упаковочных материалов на нали-
чие потенциально вымываемых или экстрагируемых веществ чрезвычайно важен
для гарантии целостности лекарственного препарата и обеспечения соответствия
Своду федеральных нормативных актов (CFR), том 21, параграф 211.65. Согласно
CFR, оборудование должно быть сконструировано таким образом, чтобы поверх-
ности, с которыми контактируют компоненты, материалы в процессе переработки
или лекарственные препараты, не обладали химической активностью, аддитивны-
ми или поглощающими свойствами. Это позволит гарантировать, что изменения
770 Часть 7. Стабильность лекарств
параметров безопасности, качества или чистоты лекарственных препаратов не бу
дуг выходить за рамки, установленные официальными или другими требованиями
[142]. Закон «О пищевых продуктах, лекарственных и косметических средствах»
США устанавливает также, что лекарственный препарат следует считать фальсифи-
цированным, если его упаковка в целом или частично состоит из токсичных или
опасных веществ, выделяющих компоненты, способные нанести вред здоровью
[143]. Представлен обзор научных и правовых точек зрения на проблему испытаний
на наличие экстрагируемых и вымываемых веществ [144]. Для облегчения анали-
за экстрагируемых соединений (обнаружения, идентификации и количественного
определения органических экстрагируемых веществ) с недавних пор применяет-
ся масс-спектрометрия. С помощью хроматографической системы Agilent G 1888
Network Headspace, оборудованной 680А GC или масс-селективным детектором
серии 5975 inert, было проведено исследование нескольких наиболее распростра-
ненных упаковочных материалов. Представлены данные по двум типам материа-
лов (бутылки из HDPE и мягкие эластомерные прокладки внутри завинчивающей-
ся крышки). С помощью метода многоступенчатой парофазной экстракции были
определены максимально достижимые концентрации экстрагируемых веществ в ле-
карственном препарате. Обнаружено, что система для газовой хроматографии с па-
рофазным устройством ввода пробы и масс-спектрометрическим детектором серии
5975 inert (ГХ/МС) обладает высокой чувствительностью и позволяет получать до-
стоверные аналитические результаты [145].
Проведена оценка результатов обработки стеклянных флаконов из церий-
оксидного стекла сульфатом аммония после воздействия ионизирующего излуче-
ния. Общая химическая композиция облученного церий-оксидного стекла осталась
неизмененной, несмотря на временный эффект появления коричневого оттенка.
Стабильность при воздействии щелочных вымываемых веществ из внутренней
кремниевой матрицы были усилена обработкой сульфатом аммония. За исключени-
ем глинозема (А12О3 и Na2O), стерилизация облучением производила ограниченное
нарушение химии поверхности церий-оксидного стекла, обработанного сульфатом
аммония [146].
7.3.4.6. Биотехнологические препараты
Несмотря на попытки доставки биотехнологических препаратов (пептидов и бел-
ков) с помощью новых систем, основным способом доставки остается инъекция.
Единицей лекарственной формы для многих инъекционных биотехнологических
препаратов являются флаконы с одной дозой лекарственного препарата и предна-
полненные шприцы. Препарат часто выпускают в виде раствора или лиофилизата,
который необходимо восстановить и ввести инъекционно с помощью шприца. Упа-
ковка представляет собой первый защитный барьер для всех видов лекарственных
препаратов, защищающий препарат от внешней среды и наоборот. В то же время
упаковка должна быть полностью совместима с лекарственным препаратом [147].
Таким образом, требования к чистоте, активности и сроку хранения требуют соблю-
дения высоких стандартов качества упаковки лекарства для инъекций.
Условия проведения упаковочных операций влияют на стабильность белковых
препаратов. Лекарства, содержащие белки и пептиды, представляют собой высоко-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...771
молекулярные соединения с уникальными физико-химическими свойствами. Они
чрезвычайно чувствительны к условиям окружающей их микросреды: нагреванию,
освещению, pH, химическим примесям и т. д. Следовые количества металлов, пла-
стификаторов и других веществ, содержащихся в упаковке, способны деактивиро-
вать или денатурировать терапевтические пептиды и белки. Кроме того, существует
тенденция к поглощению пептидов и белков поверхностью систем упаковка/уку-
порочное средство, что приводит к удалению практически всего активного веще-
ства из лекарственного состава. Даже когда происходит десорбция лекарства об-
ратно в раствор, взаимодействие может привести к потере активности лекарства.
Лиофилизированные биофармацевтические препараты могут быть повреждены
влагой, если укупорочное средство недостаточно эффективно предотвращает по-
падание влаги в упаковку. Таким образом, упаковка оказывается критически важ-
ным элементом готового лиофилизированного препарата. Флаконы, специально не
предназначенные для лиофилизации (с выпуклыми, а не плоскими донышками),
могут сделать процесс лиофилизации менее эффективным. Резиновые укупороч-
ные средства также затрудняют сушку с замораживанием, если не обеспечивается
достаточная вентиляция в процессе сублимации. Резиновые пробки поглощают
и десорбируют влагу с различными скоростями; в условиях хранения из пробок, не
прошедших надлежащей дегидратации, в лиофилизированный препарат может вы-
свобождаться вода.
Другим источником нестабильности биофармацевтических препаратов, связан-
ным с упаковкой, является силиконовое масло, традиционно применяемое для сма-
зывания эластомерных пробок в процессе итоговой операции заполнения/укупор-
ки для облегчения вхождения пробки во флакон. Известно, что силиконовое масло
инактивирует белок путем образования конгломератов белка вокруг капель масла.
Проблему удается решить нанесением на пробки фторэластомерного покрытия,
обеспечивающего необходимую смазку в дополнение к химической инертности,
защитному барьеру и безопасности [148]. Фторэластомерные пленки уменьшают
поглощение лекарства пробкой, обеспечивают смазывание укупорочного приспо-
собления флакона и, сверх того, снижают возможность миграции экстрагируемых
веществ из резиновой пробки в препарат. На рынке товаров для инъекций большую
популярность приобретают шприцы с предварительным заполнением. Отдельные
проблемы, которые предстоит решить, связаны с совместимостью и стабильностью
биотехнологических препаратов. Биотехнологические препараты могут вступать
в реакции с силиконом в форме масла, применяемого для смазывания скользящих
компонентов шприца. Полагают, что склонность силикона к взаимодействию с со-
ставом зависит от концентрации силикона в шприце и его химической активно-
сти, определяющейся числом концевых гидроксильных групп, которое тем больше,
чем короче цепь силиконового полимера. Сообщают, что термообработка силикона
(включающая нагревание шприца с силиконовым покрытием до соответствующей
температуры в течение определенного времени) приводит к образованию более
длинных цепей, плотнее прилегающих к поверхности, на которую они нанесены.
Таким образом, снижаются концентрация силикона в шприце и его химическая ре-
акционная способность, что приводит к увеличению стабильности препарата [149].
Другой выгодой от термообработки силикона является снижение частоты возникло-
772
Часть 7. Стабильность лекарств
вения эффекта отрыва, наблюдаемого, когда резиновое укупорочное средство вну-
три шприца расширяется так, что в конце концов смещает силиконовое покрытие,
снижающее трение, и входит в прямой контакт с внутренней стеклянной поверхно-
стью. Другая проблема заключается в предотвращении нежелательного изменения
pH, которое иногда происходит в жидкостях в предварительно заполненных шпри-
цах. Изменение pH происходит, поскольку стекло USP типа I, применяемое при
производстве предварительно заполняемых шприцев, является боросиликатным
стеклом, которое подвергается различным температурным воздействиям в процессе
изготовления стеклянных трубок. Во время хранения ионы натрия высвобождаются
в препарат и повышают концентрацию гидроксид-иона. Проблему решают распы-
лением сульфата аммония в стеклянную заготовку шприца перед началом процесса
термообработки при изготовлении шприцев.
Фактор VIII (/VIII) представляет собой показатель свертывания крови, который
служит кофактором в комплексном каскадном процессе свертывания крови. Де-
фицит /VIII является причиной гемофилии (типа А), наследственного нарушения
свертывания крови, опасного для жизни [150]. Показано, что эффект влияния упа-
ковки на стабильность /VIII зависит от температуры. При 4—8 °C для 2 из 15 при-
готовленных концентратов /VIII лучшая стабильность наблюдалась в пластиковых
упаковках, а 2 концентрата показали лучшую стабильность в стеклянных упаковках.
При 20—23 °C большинство концентратов проявили лучшую стабильность в пласти-
ковых упаковках; при 37 °C все концентраты показали равную или лучшую стабиль-
ность в пластиковых упаковках [151]. Был сделан вывод: в целях сравнения различ-
ных типов упаковок испытания стабильности следует проводить при температуре
хранения препарата. Из-за ограниченной диффузии молекул в твердом состоянии
нет оснований предполагать, что упаковки будут играть критически важную роль
при хранении лиофилизированных /VIII-препаратов, если воздухопроницаемость
упаковок и/или проникновение воздуха через укупорочные средства не различа-
ются существенно. Показано, что воздух ускоряет инактивацию рекомбинантного
/VIIISQ не только в растворах, но и в лиофилизированном состоянии [152].
Известно, что взаимодействие белков с поверхностями упаковки является по-
тенциально серьезной проблемой биотехнологии. Амфипатическая природа белко-
вых молекул приводит к их поглощению поверхностями различных типов и может
вызвать их потерю или дестабилизацию. Отчеты показывают, что при поглощении
лекарства, включающего белок/пептид, поверхностью упаковки молекула лекар-
ства переходит от взаимодействия с раствором к взаимодействию с поверхностью
там, где свободная энергия перехода отрицательна [153]. Аналогично, исследова-
ния показали, что некоторые сурфактанты способны уменыпать/устранять погло-
щение лекарства, включающего белок/пептид, стеклом и ПП. В присутствии сур-
фактантов там, где энергия взаимодействия сурфактант — поверхность больше,
чем энергия взаимодействия поверхность — белок/пептид, уровень поглощения
лекарства уменьшается или сводится к нулю. При поглощении белка/пептида сте-
клом, где преобладает электростатическое взаимодействие (взаимодействие между
положительно заряженным пептидом/белком и отрицательно заряженной поверх-
ностью стекла), только наиболее гидрофобные сурфактанты (полисорбат 20 и хло-
рид бензалкония) достаточно эффективно уменьшали адсорбцию на поверхностях,
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...773
что показано на примере кальцитонина лососевых рыб и бычьего сывороточного
альбумина (БСА). Доказано, что неионный сурфактант полисорбат 20 наиболее эф-
фективно уменьшает адсорбцию как на пластиковых, так и на стеклянных поверх-
ностях. Обнаружено, что преобладающим механизмом адсорбции белка/пептида
ПП (пластиковой поверхностью) является гидрофобная дегидратация [154]. Была
проведена оценка влияния Полоксамера 407 (Pluronic F-127), неионного сурфак-
танта, на адсорбцию гранулоцитарного колониестимулирующего фактора (Г-КСФ)
поверхностью ПВХ. Обнаружено, что Полоксамер 407 в концентрации 0,05 %масс.
может стать перспективной добавкой, позволяющей минимизировать адсорбцию
ПВХ поверхностью [155].
Проведена количественная оценка поглощения ряда белков с молекулярными
массами в диапазоне от 6,5 до 670 кДа и изоэлектрическими точками (р!) от 4,3 до
10,5 поверхностями упаковок, изготовленных из традиционных материалов (сте-
клянные флаконы: необработанные, с силиконовым покрытием, обработанные се-
рой или полимерной пленкой Пуркоат; пластиковые флаконы: полиэстер + 0,3%,
полиэстер 5x0, ПП и нейлон). В каждый флакон поместили раствор белка объемом
5 мл, соотношение поверхность/объем составило 2,4 см2/мл. Корреляции между по-
глощенным количеством белка, его молекулярной массой и изоэлектрической точ-
кой обнаружено не было, однако оказалось, что стеклянные поверхности связывали
большее количество белка в условиях испытания [156].
Пациентам, получающим полные питательные растворы парентеральным пу-
тем, зачастую необходим экзогенный инсулин для полного усвоения введенной
глюкозы. Проведено исследование с целью определения процентной доли инсули-
на, поглощенного стеклом и ПВХ при добавлении в инфузионную систему с пи-
тательным раствором. Исследованы следующие параметры, влияющие на доступ-
ность инсулина из парентеральных питательных растворов: д лительность инфузии,
концентрация инсулина, аминокислота или источник полипептидов, электролиты
и витамины, встроенные фильтры, стеклянные и ПВХ упаковки для инфузий, а так-
же альбумин человека. Результаты исследования показали, что из базовых раство-
ров аминокислот и гидролизатов белка в декстрозе с 30 ЕД инсулина, не удавалось
доставить около 44—47% добавленного инсулина. Варьирование концентрации ин-
сулина имело небольшое, но статистически значимое влияние на степень потери
инсулина. Добавление альбумина или электролитов и витаминов уменьшало потери
инсулина [157].
7.3.4.7. Влияние упаковки на стабильность лекарственных препаратов:
взгляд в будущее
Новые системы доставки лекарств. Публикации показывают, что развитие новых
систем доставки лекарств для различных путей введения (орального, назального,
пульмонарного, трансдермального, безыгольного) и разработка новых биотехноло-
гических лекарств привели к необходимости усиленной защиты от таких факторов,
как влага, свет, кислород и механическое воздействие, а также сделали упаковку
неотъемлемой частью системы доставки лекарства [158]. Новые системы доставки
лекарств вызвали необходимость разработки специальных упаковок, поскольку не
все новые лекарства можно упаковывать во флаконы или стандартные блистеры.
774
Часть 7. Стабильность лекарств
Зачастую упаковка должна содержать единичную дозу и быть неотъемлемой частью
технологии доставки лекарства, поскольку ее материал и конструкция в значитель-
ной мере определяют стабильность, срок хранения и эффективность лекарства
и системы доставки лекарства при его применении. Фактически, старое уравнение,
связывающее новую химическую молекулу (NCE), систему доставки лекарства (DD)
и лекарственный препарат (DP) (т. е. NCE + DD = DP) в настоящее время заменили
уравнением NCE + DD + упаковка = DP. В табл. 1 представлены соображения, кото-
рые следует учитывать при конструировании упаковки для новых систем доставки
лекарств [158].
Проблемы стабильности систем для трансдермальной доставки лекарства зача-
стую связаны с разложением составных частей приспособлений: лекарств, усили-
телей проницаемости, материала матрицы и других компонентов, входящих в со-
став приспособления. Разложение вызывает нежелательный распад компонентов,
а также приводит к обесцвечиванию и появлению постороннего запаха в транс-
дермальной системе. Приспособления, подверженные разложению, не обладают
длительным сроком хранения, что вызывает практические проблемы при их рас-
пределении. Для решения этой проблемы в лекарственный состав, содержащийся
в системе для трансдермального введения, вводят антиоксидант, такой как ВНТ
[159]. Кроме того, в запечатанный карман системы для трансдермального введения
вводят влагопоглотитель. Например, трансдермальная система введения эстрадиола
Climara упакована и продается в запечатанном кармане, содержащем поглотитель
воды для защиты эстрадиола от гидролиза [ 160].
Проведена оценка влияния экстремальных температур на доставку лекарства из
двух дозированных препаратов албутерола сульфата для ингаляций, находящихся
под давлением (MDI), с гидрофторалканом (НЕА). Три образца препарата Провен-
тил с НЕА и три образца препарата Вентолин с НЕА хранились при комнатной тем-
пературе и служили контрольными образцами, в то время как по три образца каж-
дого препарата были помещены в багажник автомобиля в Тусконе, штат Аризона.
Мониторинг температуры в багажнике осуществляли в течение 6 мес. Эксплуатаци-
онные характеристики каждого ингалятора оценивали при комнатной температу-
ре. Дополнительное исследование проведено с целью исследования характеристик
двух препаратов при нажатии дозирующего клапана при 4, 22,47 и 60 °C. В течение
недели после помещения в багажник все баллоны претерпели физические дефор-
мации. У препарата Провентил вздутия наблюдались у дна баллона, в то время как
у препарата Вентолин — вокруг дозирующего клапана. Эти деформации были со-
чтены следствием повышения давления паров, вызванного изначально высокими
температурами. После воздействия экстремальных температур отмечено увеличе-
ние скорости утечки пропеллента у обоих препаратов, однако не было обнаруже-
но практически никаких изменений в размерах частиц, высвобождаемой дозы при
нажатии на дозирующий клапан, доставляемой (респирабельной) массы действу-
ющего вещества и массы действующего вещества, оседающего на мундштуке. Не-
смотря на толерантность обоих составов к экстремальным температурам, доставка
действующего вещества была нарушена, когда ингаляторы подвергались воздей-
ствию температур за пределами рекомендуемого диапазона условий хранения [161].
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
775
Таблица 1 Требования к упаковке и факторы учета д ля новых технологий доставки лекарств
Путь доставки Названия компании и систем Характери- стики Природа Требования к упаковке и описание
Оральный CIMA, OralSolvRP Scherer, Zydis Biovail, Flashdose EthyPharm, Flashtab, Yamanouchi, wowTab, Elan, FASTMelt, Eurand, Ziplets Быстрора- створимые или распа- дающиеся в полости рта лекарственные формы Гигроскопич- ные (с по- лисахаридной или белковой основой, ма- скированный вкус, хрупкие) Защита от механическо- го воздействия: жест- кая упаковка. Peelable opening. Сверхпрочный барьер для защиты от влаги: например, Aclar или фольга для под- держания стабильности при хранении в течение 2—3 лет
Пульмонар- Inhale/Inhance", Доставка Может пред- Средняя либо высо-
ныщ Aradigm/AERxP, Alkermes/AIR*, BatellePharma, Therapeutics/EHD pulmonary delivery через ингаля- цию глубоко в легкие в виде порошка или жидкости ставлять собой порошок с большой площадью по- верхности или водный/нево- дный состав кая барьерная защита. Механические свойства зависят от конструкции устройства для доставки Барьер для защиты сте- рильности. Химическая инертность, в особен- ности для больших жидких доз. Прозрач- ный барьер позволяет провести проверку QA как производителем, так и конечным потре- бителем
Трансдер- ЗМ/ Latitude", Noven/ Отложенная В зависимости Защита активного ве-
мальный DOT, Matrix", Alza/D-Tran^ доставка/ поглощение через кожу от вида лекар- ства/состава щества от воздействия факторов окружающей среды. Прозрачный барьер обеспечивает эстетику. Пакет — пер- вичная/вторичная упаковка. Барьерные свойства структуры носителя — критически важны. Химическая инертность
Трансмуко- Cephalon, Actiq, Atrix Отложенная Чувствитель- Для защиты от влаги.
зальный Labs, BEMA CIMA Labs, Dravescent доставка через слизистую оболочку ность к влаге Прозрачный материал позволяет проводить проверку QA. Эстетика
Часть 7. Стабильность лекарств
776
Одной из альтернатив дозирующему ингалятору, находящемуся под давлением,
является ингалятор для порошков, активируемый дыханием. Для достижения ста-
бильного и предсказуемого терапевтического отклика требуется стабильная достав-
ка дозы лекарства посредством ингалятора в течение всего его жизненного цикла
и постоянство дозы при переходе от одного ингалятора к другому. Признавая эти
требования, регуляторные органы, включая Европейскую Фармакопею (ЕР) [162]
и FDA [163], разработали методики по оценке однородности доз, вводимых с помо-
щью ингаляторов. Конструкция порошкового ингаялтора, в особенности геометрия
мундштука, крайне важна для того, чтобы пациент мог обеспечить интенсивность
воздушного потока, достаточного для доставки лекарства из дозирующей камеры,
разрушения агломератов в турбулентном потоке воздуха и доставки дозы лекар-
ства в легкие в виде терапевтически эффективных мелкодисперсных частиц [164,
165]. Поток, генерируемый вдохом, непосредственно определяет скорость частиц
и, следовательно, легкость их деагломерации. Материалы, применяемые при из-
1 готовлении порошковых ингаляторов, и свойства препарата влияют на накопление
электростатического заряда. Некоторые препараты, а также материалы, из которых
изготовлен ингалятор, накапливают и удерживают электростатический заряд силь-
нее других; это влияет как на удерживание лекарства внутри ингаляторов, так и на
свойства доставляемого в легкие пациента аэрозоля [166].
Пульмозим®—лекарственный ингаляционный препарат для местного применил
в легких, содержащий рекомбинантную дорназу одного человека (дорназу-альфа).
Первоначально дорназу-альфа помещали в стеклянные флаконы для клинических
испытаний. Было проведено прямое сравнение стабильности между препаратом,
помещенным в стеклянные флаконы, и препаратом, помещенном на автоматиче-
ской линии типа BFS (выдувание — наполнение — герметизация) в полимерные
ампулы из полиэтиленовых смол низкой плотности, проницаемых для газов. Про-
блемы, связанные с газопроницаемостью, решали путем упаковки пластиковых
ампул в газонепроницаемый пакет из фольги, который можно заполнить азотом.
Обнаружено, что скорость деамидирования дорназы альфа была одинаковой, при
хранении при 2—8 °C в ампулах, защищенных и не защищенных фольгой, однако
существенно отличалась в образцах от аналогичного параметра для белка, хранив-
шихся в стеклянных флаконах. Это различие в скорости деамидирования было объ-
яснено разницей в pH растворов, составившей 0,5 единицы, для белка, хранившего-
ся в стеклянной упаковке (из-за вымывания ионов натрия с поверхности стекла), по
сравнению с препаратом в пластиковых ампулах. Полученные результаты позволи-
ли обосновать выбор компонента упаковки для аэрозолей дорназы-альфа [167].
Усовершенствование конструкции pMDI позволило расширить ее применение,
обеспечив возможность введения макромолекул, таких как пептиды и белки. Пред-
полагают, что среда HFA внутри pMDI является инертной и почти не содержит вла-
ги, что способствует сохранению стабильности макромолекул. Кроме того, усовер-
шенствование конструкции клапана и активатора помогло обеспечить стабильную
и эффективную доставку лекарства, что является важным аргументом, принимая во
внимание стоимость и эффективность биотехнологических лекарственных веществ
[168]. Компания ЪМ(3>МСото/шиу) усовершенствовала конструкцию первичной упа-
ковки для биофармацевтических препаратов. Одна из этих модернизаций предусма-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...777
тривала нанесение покрытия на внутреннюю поверхность упаковки, что позволило
уменьшить риск осаждения внутри нее активного лекарства Кроме того, ЗМ ис-
пользует новый компонент системы — полупроницаемую мембрану для улучшения
воспроизводимости дозирования суспензий [169]. Разрабатываются и новые кон-
струкции дозирующих клапанов (быстро заполняемых и быстро опустошаемых) для
улучшения однородности дозирования, а также для избавления пациентов от не-
обходимости заправлять систему, если ее используют от случая к случаю [170—172].
Методы прогнозирования влияния упаковки на стабильность лекарственных препа-
ратов. Испытания стабильности лекарственных препаратов зачастую предусматри-
вают исследование множества серий для подтверждения того, что параметры пре-
парата неизменно остаются в пределах, указанных в спецификации, до истечения
срока годности препарата. Испытания обычно проводятся на лекарственных пре-
паратах в аналогичных упаковках или в упаковках, включающих различные дозы
лекарства. В настоящее время считается, что принципы разумного статистического
планирования позволяют уменьшить количество необходимых анализов. Принци-
пы, изложенные в публикации FDA «Требования к документации по стабильности
лекарств и биологических препаратов, предназначенных для человека», расширены
для возможности определения сроков годности препаратов в более сложных случаях
[173]. В качестве заменителя стекла при изготовлении первичной упаковки для раз-
личных лекарственных препаратов можно использовать ПП. Однако из-за диффу-
зии воды через ПП-стенки зачастую накапливается значительное ее количество при
долгосрочном хранении. Нередко именно этот параметр ограничивает срок хране-
ния химически стабильных водных растворов в ПП-флаконах. Проведено исследо-
вание скорости диффузии воды в распространенное рентгеноконтрастное вещество
Визипак™ в полпропиленовых флаконах и контрастное вещество для магнитно-
резонансного обследования Омнискан™ в полипропиленовых преднаполненных
шприцах с целью разработки математического метода расчета константы скорости
диффузии воды через ПП-стенку как функции следующих переменных: темпера-
тура, влажность, площадь поверхности, толщина стенки, концентрация активно-
го ингредиента и объем заполнения. Влияние переменных оценивали с помощью
метода регрессии частичных наименьших квадратов. Способность моделей, про-
шедших кросс-валидацию, к прогнозированию для двух активных агентов оказа-
лась хорошей. Модели использовали для прогноза срока годности соответствующих
комбинаций температуры и влажности для четырех климатических зон. Метод пре-
доставляет возможность оценить влияние переменных, нарушающих стабильность
лекарства [174].
Для выявления факторов, влияющих на стабильность лекарств, оценки срока
годности, выбора новых серий для участия в дальнейших испытаниях стабильно-
сти и оценки изменений параметров новых серий была разработана многомерная
модель процесса. С помощью этой модели удалось прогнозировать значение кон-
станты скорости как функцию температуры хранения, pH, концентрации препарата
и объема упаковки [175]. Другие исследователи сообщают о применении статисти-
ческого анализа в испытаниях стабильности лекарственных препаратов. Времен-
ную зависимость изменений параметров препарата анализировали с помощью ре-
грессии наименьших квадратов и дисперсионного анализа. Это позволило изучить
Часть 7. Стабильность лекарств
778
возможности группирования серий и увеличить точность оценки срока хранения
[176]. Байесовский подход использовали для количественной оценки неопределен-
ности прогноза срока хранения по уравнению Аррениуса как функции стабильно-
сти, включающей различные распределения погрешности [177]. Моделирование по
методу Монте-Карло использовали для проверки эффективности дисперсионного
анализа (ANOVA) на матричных данных и единичных данных для оценки срока хра-
нения. Модель показала, что большой объем данных при матричном планировании
позволяет получить более точную оценку срока хранения [178].
Зачастую в упаковку, содержащую препараты, чувствительные к влаге, добав-
ляют влагопоглотитель. Модели сорбционно-десорбционного переноса жидкости
(SDMT) с успехом применялись для прогноза параметров переноса жидкости меж-
ду твердым препаратом и влагопоглотителем, находящимся внутри запечатанной
упаковки. Далее теоретические имитации расширили применение модели SDMT,
позволив учесть характеристики влагопроницаемости упаковки [179]. Кроме того,
модель SDMTиспользовали для прогнозирования влияния количества влагопогло-
тителя, количества таблеток и исходного содержания влаги в таблетке на относи-
тельную влажность внутри флаконов из полиэтилена высокой плотности (HDPE),
содержащих препарат, чувствительный к влаге — таблетки роксибифана. Обна-
ружено, что на стабильность лекарства влияют количество влагопоглотителя, вес
таблетки и изначальное содержание влаги перед операцией упаковки. Показано,
что теоретические расчеты по модели SDMT полезны для понимания требований
к упаковке препарата. Расчетные данные по относительной влажности подтверж-
дают экспериментальные данные относительно влияния исследуемых переменных
на стабильность таблетки [180]. Известно, что хрупкость желатиновой капсулы
зависит от содержания влаги. Проведено исследование, показавшее, что пустая
капсула становится хрупкой при относительной влажности менее 40%. Капсулы,
заполненные мекситилом, обладали аналогичным профилем. ЯИЛТ-модель ис-
пользовали для оценки окончательного значения относительной влажности в си-
стеме мекситил/желатиновые капсулы; результаты исследования показывают, что
модель SMDTв общем пригодна для прогнозирования появления проблем с хруп-
костью капсул и природы состава, способного предотвратить появление хрупкости
(охрупчивание) [181].
Поглощение двух слабых кислот (варфарина и тиопентона) и двух слабых осно-
ваний (хлорпромазина и дилтиазема) инфузионными пакетами, изготовленными из
ПВХ, было описано с помощью модели постоянного распределения. Коэффициен-
ты распределения в системе ПВХ—вода были получены тремя различными метода-
ми: по равновесным величинам поглощения ПВХ-мешками, с помощью соотноше-
ний между поглощением и pH и распределения в ПВХ-полосках. Данные сравнили
с аналогичными величинами, полученными при исследовании распределения в си-
стеме жидкость—жидкость с различными органическими растворителями (октанол,
дихлорметан, четыреххлористый углерод и гексан). Предпочтительным растворите-
лем является октанол; предложено использовать данные по распределению в систе-
ме октанол—вода для прогноза сорбционных характеристик [182].
Исследования показали, что изменения твердости таблеток, включающих лак-
тозу и кукурузный крахмал, происходили из-за вариаций содержания влаги. Да-
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...779
лее показано, что твердость таблеток, помещенных в полупроницаемую для влаги
упаковку (например, в контурную безъячейковую упаковку (стрип) и контурную
ячейковую упаковку, обернутую облегающей пленкой, или без нее), можно прогно-
зировать путем итеративных вычислений по математической модели, основанной
на физико-химических свойствах таблеток и влагопроницаемости упаковочных
материалов [183]. В другом исследовании, проведенном той же группой ученых,
содержание влаги и температура использовались для прогноза срока хранения
упакованных таблеток. Были учтены флуктуации температуры и относительной
влажности в процессе длительного хранения. Исследовали таблетку с сахарным
покрытием и сердцевиной, содержащей аскорбиновую кислоту, цвет которой из-
менился под влиянием влаги и температуры окружающей среды. Обнаружено, что
изменение цвета зависит от содержания влаги и температуры окружающей среды;
кроме того, изменение цвета таблетки в полупроницаемых для влаги пакетах можно
прогнозировать путем итеративных вычислений по математической модели, осно-
ванной на кинетике изменения цвета и влагопроницаемости упаковочных материа-
лов [184].
Полагают, что сочетание упаковка/состав совместимо, если величина потери
ингредиента не выходит за приемлемые пределы в течение всего срока хранения
препарата. В этой связи был разработан подход к описанию взаимодействия лекар-
ства с пластиковыми инфузионными ПВХ-пакетами. Данный подход позволяет
устанавливать соотношение между коэффициентами распределения и константой
диссоциации (там, где это необходимо) растворенного вещества, физическими раз-
мерами упаковки и pH раствора с отдельными параметрами, определяющими про-
филь сорбции. Для определения равновесного уровня сорбции упаковки из ПВХ
были рассчитаны коэффициенты корреляции между частичным связыванием рас-
творенного вещества с коэффициентами распределения в системах гексан—вода
и октанол—вода. Предполагается, что вычисления, основанные на коэффициентах
распределения в одной системе, менее эффективны с точки зрения имитации по-
ведения ПВХ. Для построения профиля поглощения (зависимости частичного свя-
зывания от времени) устанавливают соотношение коэффициентов распределения
и частичного связывания в определенный момент времени через единичный пара-
метр, называемый числом поглощения. Равновесное частичное связывание и про-
фили поглощения для различных лекарств, хранящихся в упаковках из ПВХ, рас-
считывают по моделям. Профили поглощения хорошо согласуются с описанными
в литературе данными [185]. Соотношение между числом поглощения (параметром,
определяющим исходное поглощение растворенного вещества инфузионными
ПВХ-пакетами) и коэффициентом распределения растворенного вещества в систе-
ме октанол—вода изучали по динамике поглощения лекарств материалом инфузи-
онных ПВХ-пакетов, предварительно рассчитанной с помощью диффузионной мо-
дели с предположением о том, что действие пластика эквивалентно бесконечному
стоку. Обнаружено, что модель пригодна для оценки сроков хранения релевантных
клиническому применению лекарств и позволяет описать величину поглощения
в определенный момент времени с помощью единственного параметра, называе-
мого числом поглощения. Этот параметр был определен с помощью коэффициента
распределения в системе пластик—раствор для инфузии, коэффициента диффузии
780
Часть 7. Стабильность лекарств
в пластике, неионизированной фракции в растворе, объема раствора для инфузии
и площади поверхности пластика. Число поглощения можно определить экстра-
поляцией для прогнозирования влияния времени, площади поверхности пласти-
ка, объема раствора и pH раствора при частичной потере растворенного вещества.
Была установлена разумная корреляция между логарифмом этого параметра и лога-
рифмом коэффициента распределения различных растворенных веществ в системе
октанол—вода. Модель позволяет провести оценку доли растворенного вещества,
остающегося в пластиковом инфузионном пакете после определенного перио-
да хранения, по коэффициенту распределения растворенного вещества в системе
октанол—вода и другим легко доступным данным [186].
Литература
1. U.S. Pharmacopeia/National Formulary (2005), The Official Compendia of Standards, USP 28/NF23,
U.S. Pharmacopeial Convention, Rockuille, Maryland.
2. International Conference on Harmonization (ICH) Steering Committee (1994), ICH harmonized tri-
partite guideline, Stability *estmg of new drug substances and products, ICH, Geneva.
3. International Conference on Harmonization (ICH) Steering Committee (1998), Draft ICH harmo-
nized tripartite guideline, Stability testing of new drug substances and products, ICH, Geneva.
4. Rhodes С. T. (2000), Reasons for stability testing in Carstensen, J. T. and Rhodes С. T. Eds., Drug
Stability, Principles and Practices, Marcel Dekker, New York, p. 11.
5. Center for Drugs and Biologies, U.S. Food and Drug Administration (FDA), Department of Health
and Human Services (1987), Guideline for submitting documentation for the stability of human drugs
and biologies, CDB, Washington, DC.
6. Yoshioka S., Stella V. J. (2000), Stability of Drugs and Dosage Forms, Kluwer Academic, New
York.
7. Guillory J. K., Poust R. I. (1996), Chemical kinetics and drug stability in Banker G. S., Rhodes С. T.,
Eds., Modem Pharmaceutics , 3rd ed., Marcel Dekker, New York, p. 179.
8. Center for Drug Evaluation and Research and Center for Biologies Evaluation and Research, U.S. Food
and Drug Administration (FDA), Department of Health and Human Services (1999), Guidance for in-
dustry: Container closure systems for packaging human drugs and biologies, FDA, Washington, DC.
9. Florence A. T., Attwood D. (2006), Physicochemical Principles of Pharmacy, 4th ed., Pharmaceutical
Press, London, p. 94.
10. Cartensen J. T., Serenson E. G., Vance J. J. (1964), Use of Hammett graphs in stability programs , J.
Pharm. Sci., 53, 1547—1548.
11. Moorhatch P., Ciou W. L. (1974), Interactions between drugs and plastic intravenous fluid bags. II.
Leaching of chemicals from bags containing various solvent media, Am. J. Hosp. Pharm., 31,149-152.
12. Venkataramanan R., Burckart G. J., Ptachcinski R. J., Blaha R., Logue L. W, Bahnson A., Giam C.,
Brady J. E. (1986), Leaching of diethyl phthalate from polyvinyl chloride bags into intravenous cy-
closporine solution, Am. J. Hosp. Pharm., 43,2800-2802.
13. Scheiffer G. W, Palermo P. J., Pollard-Walker S. (1984), Simultaneous determination of methyl,
ethyl, propyl, and butyl 4-hydroxybenzoates and 4-hydroxybenzoic acid in liquid antacid formula-
tions by gas chromatography, J. Pharm. Sci., 73,128.
14. Ullmann E., Thoma K., Zelfel G. (1963), The stability of sodium penicillin G in the presence of ionic
surfactants, organic gel formers, and preservatives, Pharm. Acta. Helv., 38, 577—586.
15. Testa B., Etter J. C. (1975), Hydrolysis of pilocarpine in carbopol hydrogels, Can. J. Pharm. Sci., 10,
16-20.
16. Amidon G. L., Middleton K. R., (1988), Accelerated physical stability testing and long-term predic-
tions of changes in the crushing strength of tablets stored in blister packages, Int. J. Pharm., 45,
79-89.
Глава 7.3, Влияние упаковки на стабильность фармацевтических субстанций ...781
17. Taborsky-Urdinola С. J., Gray V. A., Grady L. Т. (1981), Effects of packaging and storage on the dis-
solution of model prednisone tablets, Am. J. Hosp. Pharm., 38, 1322—1327.
18. Gouda M. W., Moustafa M. A., Molokhia A. M. (1980), Effect of storage conditions on eiythromycin
tablets marketed in Saudi Arabia, Int. J. Pharm., 5, 345—347.
19. Barrett D., Fell J. T. (1975), Effect of aging on physical properties of phenylbutazone tablets, J.
Pharm. Sci., 64, 335-337.
20. Khalil S. A., Ali L. M., Abdel-Khalek M. M. (1974), Effects of aging and relative humidity on drug
release. Pharmazie, 29, 36-37.
21. Georgarakis M., Htzipantou P., Kountourelis J. E. (1988), Effect of particle size, content in lubricant,
mixing time, and storage relative humidity on drug release from hard gelatin ampicillin capsules,
Drug Dev. Ind. Pharm., 14, 915-923.
22. Dahl T. C., Sue I. T., Yum A. (1991), The effect of pancreatin on the dissolution performance of
gelatin-coated tablets exposed to high-humidity conditions, Pharm. Res., 8,412-414.
23. Komblum S. S., Sciarrone B. J. (1964), Decarboxylation of p-aminosalicylic acid in the solid state, J.
Pharm. Sci., 53, 935.
24. Vromans H., van Laarhoven J. A. H. (1992), A study on water permeation through rubber closures of
injection vials, Int. J. Pharm., 79 (1—3), 301-308.
25. Lamy-Freud M. T., Ferreira V. F. N., Faljoni-Alario A., Schreiter S. (1993), Effect of aggregation on
the kinetic of autoxidation of the polyene antibiotic amphotericin B, J. Pharm. Sci., 82,162—166.
26. Smith G. B., DiMichele L., Colwell L. F. et al. (1993), Autooxidation of simvastin, Tetrahedron, 49,
4447-4462.
27. Khan M. M. T., Martel A. E. (1967), Metal ion and metal chelate catalyzed oxidation of ascorbic acid
by molecular oxygen. I. Cupric and ferric ion catalyzed oxidation, J. Am. Chem. Soc., 89,4176-4185.
28. Jensen J., Cornett C., Olsen С. E., Tjomelund J., Hansen S. H. (1992), Identifi cation of major deg-
radation products of 5-aminosalicylic acid form in aqueous solutions and in pharmaceuticals, Int. J.
Pharm., 88,177-187.
29. Yeh S., Lach J. L. (1961), Stability of morphine in aqueous solution Ш. Kinetics of morphine degrada-
tion in aqueous solution, J. Pharm Sci., 50, 35-42.
30. Pitman I. H., Higuchi T., Alton M., Wiley R. (1972), Deutrium isotope effects on degradation of hy-
drocortisone in aqueous solution. J. Pharm. Sci., 61, 818-820.
31. Boccardi G., Deleuze C., Gachon M., Palmisano G., Vergnaud J. P. (1992), Autoxidation of tetraze-
pam in tablets: Prediction of degradation impurities from oxidative behavior in solution, J. Pharm.
Sci., 81, 183-185.
32. Greehill J. V., McLelland M. A. (1990), Photodecomposition of drugs, Prog. Med. Chem., 27,51—121.
33. International Conference on Harmonization (ICH) (1996), Harmonized tripartite guideline: on stabil-
ity testing: Photostability testing of new drug substances and products, Q1B, 1996 ICH, Geneva.
34. Matsuda Y., Inouye H., Nakanishi R. (1978), Stabilization of sulfi somidine tablets by use of fi Im
coating containing UV absorber: Protection of coloration and photolytic degradation from exagger-
ated light, J. Pharm. Sci., 67, 196-201.
35. Frank M. J., Johnson J. B., Rubin S. H..(1976), Spectrophotometric determination for sodium nitrop-
russide and its photodegradation products, J. Pharm. Sci., 65,44.
36. Asker A. E, Larose M. (1987), Influence of uric acid on photostability of sulfathiazole sodium solu-
tions, Drug Dev. Ind. Pharm., 13, 2239.
37. Asker A. F., Canady D., Cobb C. (1985), Influence of DL-methionine on the photostability of ascorbic
acid solutions, Drag Dev. Ind. Pharm., 11, 2109.
38. Vandenbossche G. M., deMuynk C., Colardyn F., Remon J. P. (1993), Light stability of molsidomine
in infusion fluids, J. Pharm. Pharmacol., 45, 486.
39. Akimoto K., Kurosaka K., Nakagawa H., Sugimoto I. (1988), A new approach to evaluating photo-
stability of nifedipine and its derivatives in solution by actinometry, Chem. Pharm. Bull., 36, 1483—
1490.
782 Часть 7. Стабильность лекарств
40. Thoma К., Klimek R. (1991), Photostabilization of drugs in dosage forms without protection from
packaging materials, Int. J. Pharm., 67, 169-175.
41. Matsuda Y., Minamida Y. (1976), Stability of solid dosage forms: II. Coloration and photolytic deg-
radation of sulfi somide tablets by exaggerated ultraviolet irradiation, Chem. Pharm. Bull., 24, 2229-
2236.
42. Murthy K. S., Reisch R. G., Fawzi M. B. (1989), Dissolution stability of hard-shell capsule products:
Part I: The effects of exaggerated storage conditions, Pharm. Technol., 13 (3), 72-86.
43. Teraoka R., Konishi Y, Matsuda Y (2001), Photochemical and oxidative degradation of the solid-state
tretinoin tocoferil, Chem. Pharm. Bull., 49 (4), 368—372.
44. Matsuda Y, ItookaT., Mitsuhashi Y. (1980), Photostability of indomethacin in model gelatin capsules:
effects of film thickness and concentration of titanium dioxide on the coloration and photolytic degra-
dation, Chem. Pharm. Bull., 28, 2665—2671.
45. Desai D. S., Abdelnasser M. A., Rubitski В A., Varia S. A. (1994), Photostabilization of uncoated
tablets of sorivudine and nifedipine by incorporation of synthetic iron oxides, Int. J. Pharm., 103,
69-76.
46. Carstensen J. T., Johnson J. B., Spera D. C., Frank M. J. (1968), Equilibrium phenomena in solid dos-
age forms, J. Pharm. Sci., 57, 23.
47. Garrett E. R., Carper R. F. (1955), Prediction of stability in pharmaceutical preparations I. Color sta-
bility in a liquid multisulfa preparations. J. Am. Pharm Assoc. Sci. Ed., 44, 515-518.
48. Garrett E R. (1956), Prediction of stability in pharmaceutical preparations II. Vitamin stability in
liquid multivitamin preparations. J. Am. Pharm. Assoc. Sci. Ed., 45, 171—178.
49. Garrett E. R. (1956), Prediction of stability in pharmaceutical preparations III Comparison of vitamin
stabilities in different multivitamin preparations. J. Am. Pharm. Assoc. Sci. Ed., 45, 470-473.
50. Powell M. F. (1986), Enhanced stability of codeine sulfate: Effect of pH, buffer and temperature on
the degradation of codeine in aqueous solution, J. Pharm. Sci., 75, 901.
51. Carney C. F. (1987), Solution stability of ciclosidomine, J. Pharm. Sci., 76, 393.
52. Magalhales N. S. S., Cave G., Seiler M., Benita S. (1991), The Stability and in vitro release kinetics
of clofibride emulsion, Int. J. Pharm., 76, 225—237.
53. Shi L., DeHaven P. A., Burke C. J. (2005), Biopharmaceutical stability studies: stable vaccine dosage
form development for commerical use, Am. Pharm. Rev., 8 (6), 86-92.
54. Tydeman M. S., Kirkwood T. B. L. (1984), Design and analysis of accelerated degradation tests for
the stability of biological standards. I. Properties of maximum likelihood estimators, J. Biol. Stand.,
12, 195-206.
55. Flynn G. L. (1996), Cutaneous and transdermal delivery: Processes and systems of delivery, in Banker
G. S., Rhodes С. T., Eds., Modern Pharmaceutics, 3rd ed., Marcel Dekker, New York, p. 239.
56. Carstensen J. T. (1974), Stability of solids and solid dosage forms, J. Pharm. Sci., 63 (1), 1—14.
57. Horhota S. T., Burgio J., Lonski L., Rhodes С. T. (1976), Effect of storage at specified temperature
and humidity on properties of three directly compressible tablet formulations, J. Pharm. Sci., 65,
1746-1749.
58. Alam A. S., Parrott F. L. (1971), Effect of aging on some physical properties of hydrochlorothiazide
tablets, J. Pharm. Sci., 60, 263—266.
59. Carstensen J.T., Kothari R. (1983), Solid-state decomposition of alkoxyfuroix acids in the presence of
microcrystalline cellulose, J. Pharm. Sci., 72, 1149.
60. Ansel H. C., Allen L. V., Popovich N. G. (1999), Pharmaceutical Dosage Forms and Drug Delivery
Systems. 7th ed., Lippincott Williams and Wilkins, Baltimore, MD, p. 98.
61. Hecht G., Roehs R. R., Lang J. C., Rodheaver D. P., Chowhan M. A. (1996), Design and evaluation of
ophthalmic pharmaceutical products, in Banker G.S., Rhodes C.T., Eds., Modern Pharmaceutics, 3rd
ed., Marcel Dekker, New York, p. 489.
62. Lee J. Y. (1992), Sterilization control and validation for topical ointments, Pharm. Tech., 16, 104-
110.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...783
63. Ibrahim Y. К. Е., Olurinola Р. R. (1991), Comparitive microbiological contamination levels in wet
granulation and direct compression methods of tablet production, Pharm. Acta Hel., 66, 298.
64. Carstensen J. T. (1996), Preformulation in Banker G. S., Rhodes С. T., Eds., Modern Pharmaceutics,
3rd ed., Marcel Dekker, New York, p. 213.
65. Guillory J. K., Higuchi T. (1962), Solid state stability of some vitamin a compounds, J. Pharm. Sci.,
51, 100-105.
66. Pikal M. J., Lukes A. L., Lang J. E., Gaines K. (1978), Quantitative crystalline determinations for b-
lactam antibiotics by solution calorimetry: Correlation with stability, J. Pharm. Sci., 67,161-T13.
67. Krahn F. U., Mieck J. B. (1989), Effect of type and extent of crystalline order on chemical and physi-
cal stability of carbamazepine, Int. J. Pharm., 53, 25-34.
68. Matsuda Y, Akazawa R., Teraoka R., Totsuka M. (1994), Pharmaceutical evaluation of carbam-
azepine modifi cations: Comparative study of photostability of carbamazepine polymorphs by us-
ing fourier-transformed reflection-absorption infrared spectrocopy and colorimetric measurement, J.
Pharm. Pharmacol., 46, 162—167.
69. DeVilliers M. M., van der Watt J. G., Letter A. P. (1992), Kinetic study of the solid-state pho-
tolytic degradation of two polymorphic forms of furosemide, Int. J. Pharm., 88, 275—283.
70. Nakagawa Y, Itai S., Yoshida T., Nagai T. (1992), Physicochemical properties and stability in the
acidic solution of a new macrolide antibiotic, clarithromycin, In comparison with erythromycin,
Chem. Pharm. Bull., 40, 725—728.
71. Pikal M., Lukes A. L. (1976), Kundsen vapor pressure measurements on pure materials and solutions
dispersed in porous media: Molded nitroglycerin tablets, J. Pharm. Sci., 65,1269.
72. Fusari S. A. (1973), Nitroglycerin sublingual tablets П: preparation and stability of a new stabilized
sublingual molded nitroglycerin tablet, J. Pharm. Sci., 62,2012.
73. Finholt P., Jurgensen G., Kristiansen H. (1965), Catalytic effect of buffers on degradation of penicillin
G in aqueous solution, J. Pharm. Sci., 54, 387—393.
74. Tsuji A., Nakashima E., Deguchi Y., Nishide K., Shimizu T., Horiuchi S., Ishikawa K, Yamana T.
(1981), Degradation kinetics and mechanism of aminocephalosporins in aqueous solution: Cefadroxil,
J. Pharm. Sci., 70, 1120-1128.
75. Zia H., Teharan M., Zargarbashi R. (1974), Kinetics of carbenicillin degradation in aqueous solutions,
Can. J. Pharm. Sci., 9,112-117.
76. Pramar Y, Gupta V. D. (1991), Preformulation studies of spironolactone: Effect of pH, two buffer
species, ionic strength and temperature on stability, J. Pharm. Sci., 80, 551—553.
77. Allen A. E., Das Gupta V. (1974), Stability of hydrocortisone in polyethyleneglycol ontiment base, J.
Pharm. Sci., 63, 107-110.
78. Das Gupta V. (1978), Effect of vehicles and other active ingredients on stability of hydroertisone, J.
Pharm. Sci., 67, 299-302.
79. Jun H. W., Whitworth C. W, Luzzi L. A. (1972), Decomposition of aspirin in polyethyleneglycols, J.
Pharm. Sci., 6, 1160-1162.
80. Whitworth C. W, Luzzi L. A., Thompson В. B., Jun H. W. (1973), Stability of aspirin in liquid and
semi-solid bases. II. Effect of fatty additives on stability in a polyethyleneglycol base, J. Pharm. Sci.,
62,1372-1374.
81. Ekman R., Liponkoski L., Kahela P. (1982), Formation of indomethacin esters in polyethylene glycol
suppositories, Acta Pharm. Suec., 19, 241—246.
82. Castello R. A., Mattocks A. M. (1962), Discoloration of tablets containing amines and lactose, J.
Pharm. Sci., 51, 106—108.
83. Tonnesen H. H. (2001), Formulation and stability testing of photolabile drugs, Int. J. Pharm., 225,1-14.
84. Baugh R., Calvert R. T., Fell J. T. (1977), Stability of phenylbutazone in the presence of pharmaceuti-
cal colors, J. Pharm. Sci., 66, 733-735.
85. Komblum S. S., Sciarrone B. J. (1964), Decarboxylation of p-aminosalicylic acid in the solid state, J.
Pharm. Sci., 53, 935.
784 Часть 7. Стабильность лекарств
86. Liebe D. С. (1996), Packaging of pharmaceutical dosage forms in Banker G. S., Rhodes С. T., Eds.,
Modern Pharmaceutic, 3rd ed., Marcel Dekker, New York, p. 681.
87. Dean D. A. (2000), Packaging, package evaluation, stability and shelf-life in Carstensen J. T., Rhodes
С. T., Eds., Drug Stability, Principles and Practices, Marcel Dekker, New York, p. 483.
88. Code of Federal Regulations, Food and drugs, Title 21, Part 211, Current good manufacturing prac-
tice for finished pharmaceuticals, U.S. Government Printing Office, Washington, DC, rev. Apr. 1,
2001.
89. Croce С. P., Fischer A., Thomas R. H. (1986), Packaging materials science in Lachman L., Lieber-
man H. A., King J. L., Eds., The Theory and Practice of Industrial Pharmacy, 3rd ed., Lea & Febiger,
Philadelphia, p. 711.
90. Pharmaceutics, 3rd ed., Marcel Dekker, New York, p. 547.
91. Lusina M., Cindric T., Tomaic J., Peko M., Pozaic L., Musulin N. (2005), Stability study of losartan/
hydrochlorothiazide tablets, Int. J. Pharm., 291, 127-137.
92. Allinson J. G., Dansereau R. J., Sakr A. (2001), The effects of packaging on the stability of a moisture
sensitive compound, Int. J. Pharm., 221,49-56.
93. Mahajan R., Templeton A., Harman A., Reed R. A., Chem R. T. (2005), The effect of innert atmo-
spheric packaging on oxidative degradation in formulated granules, Pharm. Res., 22 (1), 128-140.
94. Al-Zein H., Riad L. E., Abd-Elbary A. (1999), Effect of packaging and storage on the stability of
carbamazepine tablets. Drug Dev. Ind. Pharm., 25 (2), 223—227.
95. Veillard M., Bentejac R., Duchene D., Carstensen J. T. (1979), Moisture transfer tests in blister pack-
age testing, Drug Dev. Ind. Pharm. 5, 227-244.
96. Amidon G. E., Middleton K. R. (1988), Accelerated physical stability testing and long-term predic-
tions of changes in the crushing strength of tablets stored in blister packages, Int. J. Pharm., 45,79-89.
97. Khalil S. A. H., Barakat N. S., Boraie N. A. (1991), Effects of package type on in vitro release and
chemical stability of amoxycillin in capsules, Pharm. Ind., 53 (7), 698-701.
98. Gouda M. W., Moustafa M. A., Molokhia A. M. (1980), Effect of storage conditions on erythromycin
tablets marketed in saudi arabia, Int. J. Pharm., 5, 345-347.
99. Pikal M. J., Bibler D. A., Rutherford B. (1977), Polymer sorption of nitroglycerin and stability of
molded nitroglycerin tablets in unit-dose packaging, J. Pharm. Sci., 66 (9), 1293—1297.
100. Barakat N. S. (2006), Etodolac-liqui-filled dispersion into hard gelatin capsules: An approach to
improve dissolution and stability of etodolac formulation, Drug Dev. Ind. Pharm., 32, 865-876.
101. Santoro M. I. R. M., Oliveira D. A. G. D. С. E., Kedor-Hackmann E. R., Singh A. K. (2005), The
effect of packaging materials on the stability of sunscreen emulsions, Int. J. Pharm., 297, 197-203.
102. Geary T. G., Akkod M. A., Jensen J. B. (1983), Characteristics of cheloroquine binding to glass and
plastic, Am. J. Trop. Med. Hyg. 32, 19-23.
103. Yahya M. A., McElnay J. C., D’Arcy P. F. (1985), Binding of chloroquine to glass, Int. J. Pharm.,
25, 217-223.
104. Stanaszek W. E, Pan I. H. (1978), Comparison of drug stability in glass versus plastic containers:
Analysis of prefilled syringe admixtures, Proc. Okla. Acad. Sci., 58, 102-105.
105. Arsene M., Favetta P., Favier B., Bureau J. (2002), Comparison of ceftazidime degradation in glass
bottles and plastic bags, J. Clin. Pharm. The., 27 (3), 202—209.
106. Crane I. M., Mulhern M. G., Nema S. (2003), Stability of reconstituted parecoxib for injection with
commonly used diluents. J. of Clin. Pharm. Ther., 28, 363-369.
107. Wong M., Marion R, Reed K., Wang Y. (2006), Sorption of unoprostone isopropyl to packaging
materials, Int J. Pharm., 307, 163—167.
108. Driscoll D. F., Ling P. R., Bistrian B. R. (2007), Physical stability of 20% lipid injectable emul-
sions via simulated syringe infusion: effects of glass versus plastic product packaging, J Parenteral
Enteral Nutr.,31 (2), 148-153.
109. Yuen P. H., Denman S. L., Sokoloski T. D., Burkman A. M. (1979), Loss of nitroglycerin from aque-
ous solution into plastic intravenous delivery systems. J. Pharm. Sci., 68, 1163—1166.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...
785
ПО. BaaskeD. М., Amann А. Н., Wagenknecht D. М., Carter J. Е., HoytH. J., Stoll R. G. (1980), Nitro-
glycerin compatibility with intravenous fluid filters, containers and administration sets, Am. J. Hosp.
Pharm., 37, 201-205.
111. Cossum P. A., Roberts M. S., Galbraith A. J., Boyd G. W. (1978), Loss of nitroglycerin from intrave-
nous infusion sets, Lancet., 2, 34—350.
112. Crouthamel W. G., Dorsch B., Shangraw R. (1978), Loss of nitroglycerin from plastic intravenous
bags, New Engl. J. Med., 299, 262.
113. Sokoloski T. D., Wu С. C., Burkman A. M. (1980), Rapid adsorptive loss of nitroglycerin from aque-
ous solution to plastic, Int. J. Pharm., 6, 63-76.
114. Malick A. W, Amann A. H., Baaske D. M., Stoll R. G. (1981), Loss of nitroglycerin from solutions
to intravenous plastic containers: A theoretical treatment. J. Pharm. Si., 70, 798-800.
115. Yuen P. H., Denman S. L., Sokoloski T. D., Burkman A. M. (1979), Loss of nitroglycerin from aque-
ous solution into plastic intravenous delivery systems, J. Pharm. Sci., 68 (9), 1193-1166.
116. Ilium L., Bubdgaard H. (1982), Sorption of drugs by plastic infusion bags, Int. J. Pharm., 10, 339-
351.
117. Pikal M. J., Lang J. E. (1978), Rubber closures as a source of haze in freeze-dried parenterals: Test
methodology for closure evaluation, J. Parent. Drug. Assoc., 32 ,162-173.
118. Jahnke R. W. O., Kreuter J., Ross G. (1991), Content/container interactions: The phenomenon of
haze formation on reconstitution of solids for parenteral use, Int. J. Pharm., Tl, 47-55.
119. Parker W. A., MacCara M. E. (1980), Compatibility of diazepam with intravenous fluid containers
and administration sets, Am. J. Hosp. Pharm., 37, 496-500.
120. Morris M. E. (1978), Comaptibility and stability of diazepam injection following dilution with in-
travenous fluids, Am. J. Hosp. Pharm., 35, 669-672.
121. Parker W. A., Morris M. E., Shearer C. A. (1979), Incompatibility of diazepam injection in plastic
intravenous bags, J. Am. Hosp. Pharm., 36, 505—507.
122. Van Amerongen G. J. (1964), Diffusion in elastomers, Rubber Chem. Technol., 37,1065-1152.
123. Womans H., van Laarhoven J. A. H. (1992), A study on water permeation through rubber closures
of injection vials, frit. J. Pharm., 79, 301-308.
124. U.S. Food and Drug Administration (FDA) (1998, Oct.), Guidance for industry: Metered dose in-
haler and dry powder inhaler drug products, FDA Washington, DC.
125. Castner J., Anderson J., Benites P. (2007), Strategy for development and characterization of HPLC
methods to investigate extractables and leachables, Am. Pharm. Rev., 10 (3), 10.
126. Northup S. J. (2005), Assesing the biological safety of extractable and leachable chemicals in phar-
maceutical and medicial products, Am. Pharm. Rev., 8 (4), 38—43.
127. U.S. Food and Drug Administration (FDA) (2002, July), Nasal spray and inhalation solution, sus-
pension and spray drug products.: Chemistry, manufacturing and controls documentation, Guidance
for industry. FDA, Washington, DC, 1-45.
128. Norwood D. L. (2007), Understanding the challenges of extractables and leachables for the pharma-
ceutical industry—Safety and requlatory enviroment for pharmaceuticals, Am. Pharm. Rev., 10 (2),
32.
129. U.S. Food and Drug Administration (FDA), Extra 6: Safety thresholds and best practices for extract-
ables and leachables in orally inhaled and nasal drug products, PQRI Leachables and Extratables
Working Group, available: http://www.pqri.org/pdfs/ LE_Recommendations_to_FDA_09-26-06.
pdf.
130. IPAC-RS (1991, Mar.) Extra 7: Leachables and extractables testing: points to consider, available:
http://www.ipacrs.com/leachables.html.
131. Allan L., Wang Q. (2007), Impact of package on the stability of pharmaceutical products, Am.
Pharm. Rev., 10 (4), 38—44.
132. Waterman К. C., Adami R. C., Alsante К. M., Hong J., Landis M. S., Lombardo E, Roberts C. J.
(2002), Stabilization of pharmaceuticals to oxidative degradation, Pharm. Dev. Tech., 7 (1), 1-32.
786 Часть 7. Стабильность лекарств
133. Reed R. A., Harmon Р., Manas D., Wasylaschuk W, Galli C., Biddell R., Bergquist P. A., Hunke W.,
Templeton A. C., Ip D. (2003), The role of excipients and package components in the photostability
of liquid formulations, PDA J. Pharm. Sci. Technol., 57 (5), 351-368.
134. Hong J., Lee E., Carter J. C., Masse J. A., Oksanen D. A. (2004), Antioxidant-accelerated oxidative
degradation: A case study of transistion metal ion catalyzed oxidation in formulation, Pharm. Dev.
Tech., 9 (2), 171-179.
135. Quarry M. A., Sebastian D. S., Diana F. (2002), Investigation of 4,5-epoxymorphian degradation
during analysis by HPLC, J. Pharm. Biomed. Anal., 30, 99—104.
136. Nakamura K., Yokohama S., Sawada M. T., Sonobe T. (2003), A new stressed test to predict the
foreign matter formation ofminodronic acid in solution, Int. J. Pharm., 251, 99-106.
137. Bohere D., Nascimento P. C., Binotto R., Becker E. (2003), Influence of the glass packing on the
contamination of pharmaceutical products by aluminium. Part Ш. Interaction of the container-chem-
icals during heating for sterilization, J. Trace Elem. Med. Biol., 17 (2), 107-115.
138. Shen F. W., McKellop H. A. (2002), Interaction of oxidation and crosslinking in gamma-irradiated
ultrahigh molecular-weight polyethylene, J. Biomed. Mater. Res.,61 (3), 430-439.
139. Welle F. (2005), Migration of radiolysis products from radiation-sterilized plastics, Pharm. Ind., 67
(8), 970-972.
140. Nassar M. N., Nesarker V. V., Lozano R., Yande H., Palaniswany V. (2005), Degradation of a lyo-
philized formulation of BMS-204352: Identification of degradants and the role of elastomeric clo-
sure. Pharm. Dev. Tech., 10 (2), 227—232.
141. Glaser V. (2005), Addressing stability of biological drugs: forced degradation studies predict effects
on bioproducts in drug development and manufacture, GEN Genetic eng. news, 25 (6), 1—5.
142. Code of Federal Regulations, Food and drugs, Title 21, Part 211.65, U.S. Government Printing Of-
fice, Washington, DC, rev. Apr. 1, 2004.
143. Federal Food, Drug and Cosmetic Act as amended through Dec. 21, 2000, Chapter V, Drugs and
devices, Section 501(a)(3), U.S. Government Printing Office, Washington DC, 2001.
144 Taborsky C. J., Sheinin E. B. (2006), A critical approach to the evaluation of packaging components
and the regulatory and scientific considerations in developing a testing strategy, Am. Pharm. Rev., 9
(4), 10-15.
145. Gudat A. E., Firor R. L. (2006, Sept.), The determination of extractables and leachables in pharma-
ceutical packaging materials using headspace GC/MS, Application Note, Agilent Technology.
146. Janimak J. J., Marteleur M. (2004), On the stability of cerium oxide glass for terminal radiation
sterilization, Rad. Phys. Chem., 71, 195-198.
147. DeGrazio F. L. (2005), Parenteral packaging concerns for drugs: Early understanding of drug
to package compatibility lowers the cost in the long run. GEN: Genetic Eng. News (Bioprocess.
Channel), 25 (21), 2.
148. DeGrazio F. L. (2005), Parenteral packaging concerns for drugs: Early understanding of drug
to package compatibility lowers the cost in the long run. GEN: Genetic Eng. News (Bioprocess.
Channel), 25 (21), 2.
149. Romacker M. (2005), Prefilled syringes: Why new developments are important in injectable de-
livery today in Roessling G., Ed., Prefi lied Sringes: Innovations That Meet the Growing Demand,
ONdrugDelivery, Sussex, United Kingdom.
150. Wang W, Wang Y. J., Kelner D. N. (2003), Coagulation factor VIII: Strucuture and stability, Int. J.
Pharm., 259, 1-15.
151. Schulman S., Gitel S., Martinowitz U. (1994), Stability of factor VIII concentrates after reconstitu-
tion, Am. J. Hematol., 45, 217—223.
152. Osterberg T., Fatouros A., Neidhardt E., Warne N., Mikaelsson M. (2001), B-domain-deleted recom-
binant factor VIII formulation and stability, Semin. Hematol., 38,40—43.
153. Andrade J. D., Hlady V. (1986), Protein adsorption and materials biocompatibility: Atutorial review
and suggested hypothesis, Adv. Polym. Sci., 79, 1-63.
Глава 7.3. Влияние упаковки на стабильность фармацевтических субстанций ...787
154. Duncan М. R., Lee J. М., Warchol М. Р. (1995), Influence of surfactants upon protein/peptide ad-
sorption to glass and polypropylene, Int. J. Pharm., 120, 179-188.
155. Wang P. L., Udeani G. O., Johnston T. P. (1995), Inhibition of granulocyte colony stimulating factor
(c-csf) adsorption to polyvinyl chloride using a nonionic surfactant, Int. J. Pharm., 114, 177-184.
156. Burke C. J., Steadman B. L., Volkin D. B., Tsai P. K., Bruner M. W., Middaugh C. R. (1992), The
adsorption of proteins to pharmaceutical container surfaces, Int. J. Pharm., 86, 89-93.
157. WeberS. S., Wood W. A., Jackson E. A. (1977), Availability of insulin from parenteral nutrient solu-
tions, Am. J. Hosp. Pharm., 34, 353-357.
158. Weeren R. V., Gibboni D. J. Barrier packaging as an integral part of drug delivery, available: http://
www.drugdeliverytech.com/cgi-bin/articles=52 (accessed June 1, 2007).
159. U. S. Patent 5,028,431: Franz T. J., ShahK. R., Kydonieus A. Article for the Delivery to Animal Tis-
sue of a Pharmacological Active. July 2,1991; U. Patent 5,242,433: Smith J. A., Murphy B. Packag-
ing System with In-Tandem Applicator Pads for Topical Drug Delivery. Decemeber 7, 1992.
160. U. S. Patent 5,698,217: Wilking S. L. Transdermal Drug Delivery Device Containing a Dessicant.
December 16, 1997.
161. Hoye W. L., Mogalian E- M., Myrdal P. B. (2005), The effects of extreme temperatures on drug
delivery of albuterol sulfate hydrofluoroalkane inhalation products, Am. J. Health-Syst. Pharm., 62
(21), 2271-2277.
162. European Pharmacopoeia (EP) (2001), Preparations for inhalation, EP supplement 2001 2.9.18, EP,
Strasbourg, France.
163. U.S. Food and Drug Administration (FDA) (1998), Guidance for industry: Metered dose inhaler
(MDI) and dry powder inhaler (DPI) drug products, FDA CDER, Washington, DC.
164. Petersson G., Wiren J. E. (1980), The brochodilator response from inhaled terbutaline is influenced
by the mass of small particles: A study on a dry powder inhaler (Turbuhaler), Eur. Respir., 2, 253—
256.
165. Newman S. P., Busse W. W. (2002), Evolution of dry powder inhaler design, formulation and per-
formance, Respir. Med., 96,293-304.
166. Carter P. A., Rowley G., Fletcher E. J., Sylianopoulos V. (1998), Measurement of electrostatic charge
decay in pharmaceutical powdersand polymer materials used in dry powder inhaler devices, Drug
Dev. Ind. Pharm., 24, 1083-1088.
167. Shire S. J. (1996), Stability, characterization and formulation of recombinant human deoxy-
ribonuclease (Pulmozyme®, Domase Alpha), in Pearlman R., Wang Y. J., Eds., Formulation,
Characterization and Stability of Protein Drugs, Plenum Press, New York, pp. 393—426.
168. Amum P. V. (2007), 3M drug delivery systems advances inhalation and transdermal drug delivery,
Pharm. Technol. Online, available http://www.pharmtech.com, Jan. 31.
169. Jinks P., Marsden S. (1999), The development and performance of a fluoropolymer lined can for
suspension metered dose inhaler product, in Fradley G., Ed., Proceedings of Drug Delivery to the
Lungsa X, The Aerosol Society, Portshead, United Kingdom, pp. 177-180.
170. Wilby M. (2005), Increasing dose consistency of pMDis, DrugDeliv. Technol., 5 (9), 59-65.
171. Wilby M. (2006), Novel valve designs to eliminate loss of prime, in Dalby X. R. N. et al., Eds., Pro-
ceedings of Respiratory Drug Delviery. Vol. 2, Davis Healthcare, Boca Raton, FL, pp. 373—376.
172. Jinks P., Hunt K. (2006), Improving suspension MDI dose consistency in patient use by incorpora-
tion of a novel semi-permemable system component, in Van Amum P., Ed., Proceedings of Drug
Delivery to the Lungs XVII, The Aerosol Society, Portshead, United Kingdom, pp. 172-175.
173. Fairweather W. R., Lin T. Y. D., Roswitha K. (1995), Regulatory, design and analysis aspects of
complex stability studies, J. Pharm. Sci., 84 (11), 1322—1326.
174. Dyrstad K., Veggeland J., Thomassen C. (1999), A multivariate method to predict the water vapour
diffusion rate through polypropylene packaging, frit. J. Pharm., 188, 105-109.
175. Dyrstad K., Thomassen C., Eivindvik K. (1999), An opportunistic stability strategy: simulation with
real data, Int. J. Pharm., 188, 97-104.
788 Часть 7. Стабильность лекарств
176. Ruberg S. J., Hsu J. С. (1991), Multiple comparison procedures for pooling batches instability stud-
ies, Technometrics, 34, 465—472.
177. Su X. Y., Po A. L. W., Yoshioka S. (1994), A bayesian approach to arrhenius prediction of shelf-life
, Pharm. Res., 11, 1462—1466.
178. Yoshika S., Aso T., Kojima K. (1996), Statistical evaluation of shelf-life of pharmaceutical products
estimated matrixing, Drug Stab,. 1, 147—151.
179. Kontny M. J., Koppenol S., Graham E. T. (1992), Use of the sorption-desorption moisture transfer
model to assess the utility of a desiccant in a solid product, Int. J. Pharm., 84, 261-271.
180. Badway S. I. F., Gawronski A. J., Alvarez F. J. (2001), Application of sorption-desorption moisture
transfer modeling to the study of chemical stability of a moisture sensitive drug product in different
packaging configurations, Int. J. Pharm., 223 (1-2), 1-13.
181. Kontny M. J., Mulski C. A. (1998), Gelatin capsule brittleness as a function of relative humidity at
room temperature, Int. J. Pharm., 54 (1), 79-85.
182. Ilium L., Bundgaard H., Davis S. S. (1983), A constant partition model for examining the sorption
of drugs by plastic infusion bags, Int. J. Pharm., 17, 183-192.
183. Nakabayashi K., Shimamoto T., Mima H. (1980), Stability of packaged solid dosage forms. I.
shelf-life prediction for packaged tablets liable to moisutre damage, Pharm. Chem. Bull., 28,1090-
1098.
184. Nakabayashi K., Shimamoto T., Mima H. (1980), Stability of packaged solid dosage forms. II.
Shelf-life prediction for packaged sugar-coated tablets liable to moisture and heat damage, Pharm.
Chem. Bull., 28, 1099-1106.
185. Jenke D. R. (1993 , Modeling of solute sorption by polyvinyl chloride plastic infusion bags J.
Pharm. Sci, 82 (11), 1134-1139.
186. Roberts M. S., Kowaluk E. A., Polack A. E. (1991), Prediction of solute sorption by polyvinyl chlo-
ride plastic infusion bags, J. Pharm. Sci., 80 (5), 449—455.
Глава 7.4. Стабильность фармацевтических
препаратов
Эндрю Вебстер
Фармацевтический факультет имени Мак-Хортера,
Сэмфордский университет (Бирмингем, Алабама)
7.4.1. Введение
Стабильность фармацевтического препарата можно определить как способность
конкретного состава к сохранению соответствия физическим, химическим, микро-
биологическим, терапевтическим и токсикологическим спецификациям при хране-
нии в определенной упаковке. Набор достоверных данных по состоянию лекарства
при хранении в специально подобранной упаковке обеспечивает уверенность в том,
что препарат сохраняет стабильность в течение всего указанного срока хранения.
Зачастую стабильность лекарства определяют как время начиная от даты изго-
товления и упаковки, в течение которого химическая и биологическая активность
сохраняется на определенном уровне от заявленного значения без значительного
изменения физических характеристик. Для большинства лекарств 90% от заявлен-
ного значения обычно признают минимальным приемлемым уровнем содержания
активного компонента.
Существует ряд факторов, способных нарушить стабильность фармацевтическо-
го препарата. Факторы включают стабильность активных лекарств, взаимодействие
между активными и неактивными ингредиентами, форму дозирования, технологи-
ческий процесс, систему упаковки и параметры окружающей среды, в которой про-
водятся процессы транспортировки, обработки и хранения.
Фармакопея США 29/Национальный формуляр 24 (USP19/NF2A) определяет
стабильность как способность к сохранению параметров препарата в установлен-
ных пределах и сохранение в течение периода хранения и применения (т. е. срока
хранения) тех же свойств и характеристик, которыми препарат обладал в момент
изготовления [1]. Далее USP29/NF2A идентифицирует пять общепризнанных типов
стабильности:
• химическая стабильность — все активные ингредиенты сохраняют химиче-
скую целостность и заявленное значение содержания активного компонента
в определенных границах;
• физическая стабильность — исходные физические свойства, в том числе
внешний вид, вкусовые качества, однородность, растворение и суспендируе-
мость (могут быть нарушены);
• микробиологическая стабильность — стерильность или резистентность к ро-
сту микробов сохраняется в соответствии с определенными требованиями;
добавленные антимикробные агенты сохраняют эффективность в определен-
ных пределах;
790 Часть 7. Стабильность лекарств
• терапевтическая (фармакологическая) стабильность — терапевтический эф-
фект не нарушен;
• токсикологическая стабильность — не происходит существенного увеличе-
ния токсичности.
Достоверные даты истечения срока годности фармацевтического препара-
та определяют в ходе тщательно спланированного по правилам науки испытания
с применением достоверных, информативных и специфичных методик, соответ-
ствующих статистических подходов и компьютерного анализа полученных данных
[2]. Исчерпывающий обзор, охватывающий все аспекты стабильности фармацевти-
ческого препарата, представлен в публикации Lintner [3] и в более поздней работе
Connors et al. [4].
7.4.2. Кинетические уравнения и срок годности
Рассмотрим реакцию:
аА + ЬВ ->рР + #Q, (1)
где А и В — вступающие в реакцию вещества; Р и Q — продукты реакции; а, Ь, р,
q — стехиометрические коэффициенты, отражающие относительное количество
участвующих в реакции веществ. Скорость изменения концентрации любого из ве-
ществ С выражается так:
dCA dCB dCP dCQ
dt ' dt ’ dt ’ dt
Концентрации веществ уменьшаются co временем — отсюда знак «минус».
С другой стороны, количество продуктов увеличивается со временем, поэтому ско-
рость изменения концентрации продукта реакции записывают со знаком «плюс».
Скорости уменьшения количества веществ А и В и скорости накопления Ри Q свя-
заны между собой уравнениями, учитывающими стехиометрию реакции:
1 dCA 1 dCB 1 dCp 1 dCQ (2)
a dt b dt p dt q dt
7.4.2.1. Уравнение скорости реакции
Выражение скорости реакции представляет собой математическое описание скоро-
сти реакции в любой момент времени t в терминах концентрации (концентраций)
молекулярных веществ, присутствующих в системе в этот момент времени.
Если упростить уравнения (1) до вида
А + В -> Продукты. (3)
то скорость реакции можно записать следующим образом:
dC,
dt
ос са Сь
’-'АЮ’-'ВИ
(4)
Глава 7.4. Стабильность фармацевтических препаратов 791
Уравнение (4) устанавливает, что скорость изменения концентрации вещества А
в момент времени / равна соответствующей скорости изменения концентрации ве-
щества В, и все эти изменения в момент времени / пропорциональны произведению
концентраций реагентов, возведенных в соответствующие степени. Отметим, что
С и CB(t) являются переменными, зависящими от времени. По мере протекания
реакции величины СА(1)и CB(t) уменьшаются. Для упрощения обозначим эти концен-
трации САи Св соответственно:
=_^£в (5)
dt dt А в
где к — коэффициент пропорциональности, традиционно называемый константой
скорости реакции. Как правило, формат выражения скорости включает лишь вели-
чины концентраций реагентов и очень редко — концентрации продуктов. В послед-
нем случае выражение описывает ситуацию, когда продукты участвуют в реакции
с момента ее инициирования.
Порядок реакции п можно определить как п = а + Ь. В общем случае порядок
реакции представляет собой сумму экспонент членов, выражающих концентрации,
в выражении скорости. Таким образом, если а — b = 1, то вышеописанная реак-
ция является реакцией второго порядка в целом, но первого порядка по веществу А
и первого порядка по веществу В. В принципе, численное значение а или b может
быть как целым, так и дробным.
Иногда скорость реакции очевидно не зависит от концентрации одного из ве-
ществ, несмотря на то что оно расходуется в процессе реакции. Например, для ре-
акции между сложным эфиром и водой (реакции гидролиза) в преимущественно
водной среде выражение теоретической скорости по эфиру можно записать в тер-
минах концентраций эфира (С£) и воды (С^):
-^ = С£СЖ. (6)
Если исходная концентрация эфира не превышает 0,5 М, при полном гидролизе
эфира соответствующее уменьшение концентрации воды составит 0,5 М или менее.
При исходной концентрации воды около 55 М для водного раствора относительное
уменьшение содержания воды в процессе реакции будет несущественно малым, что
позволяет считать величину Cw постоянной в течение всего реакционного процесса.
Исходя из этого
—= (7)
где кг = kCv.
Реакция описывается кинетикой первого порядка по эфиру и кинетикой нуле-
вого порядка по воде. Такие реакции в целом называют реакциями псевдопервого
порядка, а кт — константой псевдопервого порядка.
Кинетика псевдопервого порядка описывает ситуацию, в которой концентрация
одного из реагентов остается постоянной либо из-за существенно избыточной ис-
ходной концентрации, либо благодаря быстрому восполнению одного из реагентов.
Если одним из реагентов является ион водорода или гидроксид-ион, его концен-
792
Часть 7. Стабильность лекарств
трация (вероятно, малая по сравнению с концентрацией лекарства) может под-
держиваться на постоянном уровне при протекании реакции путем использования
буферов в растворе. Концентрацию нестабильного лекарства в растворе можно под-
держивать на постоянном уровне путем использования суспензии, которая обеспе-
чивает избыток твердого вещества в равновесии с лекарством в растворе.
7.4.2.2. Определение порядка реакции
Порядок реакции можно определить несколькими способами:
Метод подстановки. Накопленные в ходе исследования кинетики значения мож-
но подставлять в интегрированные формы различных уравнений, описывающих
порядки реакции. Когда значения к при одной из итераций остаются постоянными,
считают, что формула соответствует реальному порядку реакции.
Графический метод. Для подтверждения порядка реакции можно использовать
график данных. Если график зависимости концентраций от времени представляет
прямую линию, то он соответствует реакции нулевого порядка. Прямая линия гра-
фика зависимости log (а — х) от времени соответствует реакции первого порядка;
для реакции второго порядка график зависимости 1/(л — х)2от времени представля-
ет собой прямую линию (при условии равенства исходных концентраций).
Метод полураспада. Для реакции нулевого порядка период полураспада (11/2) про-
порционален исходной концентрации. Период полураспада для реакции первого
порядка не зависит от исходной концентрации, в то время как для реакции второго
порядка период полураспада пропорционален обратному значению исходной кон-
центрации.
7.4.2.3. Прогнозирование срока хранения
Определение срока хранения лекарственного состава основано на точных физико-
химических законах и статистических принципах получения достоверных оценок.
McMinn и ЕдШлегразработали информационную систему обработки данных испыта-
ний стабильности лекарственного препарата [5]. Эта система позволяет экономить
время разработчиков в процессе анализа и интерпретации данных по стабильно-
сти планируемого препарата. Такие препараты, как, например, витамины, должны
иметь большой срок хранения; в таком случае статистическая часть этой передовой
методики помогает производителю создать формулу композиции с желательным
и экономически оправданным сроком хранения.
Система сохраняет как физические, так и химические данные и извлекает ин-
формацию в трех разных форматах (один из которых предназначен специально
для представления данных в регуляторные органы). Система позволяет проводить
анализ данных при одной температуре испытаний через анализ ковариаций и ре-
грессионный анализ. Данные при множестве температур испытаний анализируют
во взвешенном или невзвешенном виде с помощью соотношений Аррениуса. Этот
метод позволяет оценить срок хранения препарата с надлежащим доверительным
интервалом, распечатать карту запроса, используемую для записи результатов соот-
ветствующего анализа и ввода данных в систему, разработать основное расписание
испытаний на 5 лет, а также периодические расписания предстоящих исследований
на 14 дней.
Глава 7.4. Стабильность фармацевтических препаратов
793
Анализ данных испытаний, полученных при одной температуре, основан на ли-
нейной модели (нулевого порядка):
У Р X .а +в , (8)
где У п — процентная доля от заявленного значения в n-м испытании стабильности
zn-ro лота, Хтп — время (в месяцах) проведения замера, при котором было получено
значение У n, Рт и ат — наклон и интерсепт соответственно линии регрессии для
т-го лота и втл — случайная ошибка ассоциированная с Улл. Предполагается, что
случайные ошибки являются идентичными переменными с нормальным распреде-
лением с нулевым средним и общей дисперсией о2.
Результаты регрессионного анализа для каждого индивидуального лота и для
комбинации лотов вместе с результатами анализов ковариаций и отклонений от ре-
грессии выдает компьютер.
Поскольку данные испытаний стабильности индивидуальных серий группиру-
ются, проверка достоверности этих данных проводится с помощью F-теста. Среднее
значение квадрата коэффициента регрессии делят на среднеквадратическое откло-
нение внутри лотов; аналогично, скорректированное среднее (у-интерсепт) делят
на общий средний квадрат для получения значений соответствующих F-уровней.
Полученные значения сравнивают с критическими 5%-ным значениями F. Если
рассчитанные значения У меньше критических значений У, то данные можно объе-
динить и провести анализ сгруппированных данных.
7.4.Z.4. Уравнение Аррениуса и ускоренное испытание стабильности
Целью испытаний стабильности является оценка влияния температуры, влажности,
освещенности и других факторов окружающей среды на качество лекарственного
вещества или препарата. Полученные наборы данных используют для определения
условий хранения, периодов переконтроля (период до проведения повторного ис-
пытания), потерь при хранении для обоснования избыточных критериев по сроку
хранения, закладываемых в препарат из соображений сохранения стабильности.
Самым полезным уравнением, устанавливающим соотношение между температу-
рой и скоростью реакции, является уравнение Аррениуса:
dink _ Еа
dT ~~R1*
Уравнение можно переписать в виде
k=Ae-E-/RT
(9)
(Ю)
(И)
y^T2 Tj
где Ев является константой, а индексы 1 и 2 обозначают два различных типа темпе-
ратурных условий. График зависимости In к от 1/У, так называемый график Арре-
ниуса, представляет линейную зависимость в соответствии с уравнением (10), если
Еа не зависит от температуры. Это наблюдение позволило проводить кинетические
эксперименты при повышенных температурах и получать данные по константам
794
Часть 7. Стабильность лекарств
скорости реакции при низких температурах путем экстраполяции графика Арре-
ниуса. Этот метод, известный как ускоренное испытание стабильности, наиболее
полезен для расчета реакций, протекающих при комнатной температуре, когда ско-
рость реакции слишком мала для мониторинга традиционными способами, а Еа от-
носительно велика. Например, для реакции с Еа = 25 ккал/моль при повышении
температуры от 25 до 45 °C константа скорости возрастает примерно в 14 раз. На-
против, для реакции с £ = 10 ккал/моль при таком же повышении температуры
скорость реакции возрастает только в 3 раза. Наклон графика Аррениуса позволяет
рассчитать значение Еа. Энергия активации реакции гидролиза обычно составляет
10—30 ккал/моль, а реакции окисления и фотолиз протекают с меньшими энергия-
ми активации [6].
Наиболее часто исследование проводят при температурах 40,50 и 60 °C в сочета-
нии с обычной влажностью окружающей среды. Иногда используют более высокие
температуры. Образцы, хранящиеся при максимальных температурах, анализируют
еженедельно, контролируя физические и химические изменения. Если наблюда-
ют значительные изменения, анализируют образцы, хранящиеся при более низких
температурах. Если после 30 дней хранения при 60 °C не наблюдается изменений,
то прогнозируемая стабильность оценивается как отличная. Следует получить под-
тверждающие данные путем мониторинга образцов с более длительными периода-
ми хранения при более низких температурах.
Основное допущение уравнения Аррениуса состоит в том, что механизм реакции
не меняется в зависимости от температуры. Поскольку ускоренное испытание ста-
бильности фармацевтических препаратов обычно проводят в узком температурном
диапазоне, выявление нелинейности графика Аррениуса, построенного по экспери-
ментальным данным, зачастую оказывается затруднительным, несмотря на то, что
нелинейность ожидается на основании представлений о механизме реакции [7].
Поведение фармацевтических систем зачастую отклоняется от параметров, опи-
сываемых уравнением Аррениуса [8]. Это может быть связано с возможным испа-
рением растворителя, множественными путями протекания реакций и изменением
физической формы состава при изменении температуры реакции [9]. Нелиней-
ность графика Аррениуса зачастую наблюдают при анализе зависимости разложе-
ния белка от температуры. Механизмы разложения белков часто изменяются при
изменении температуры [10]. Агрегация интерлейкина 10 (/£-10) в водном растворе
описывается кинетикой первого порядка при 60 °C до 30% оставшегося лекарства.
При 55 °C и ниже агрегация отклоняется от первого порядка и становится двухфаз-
ной [И].
Значительный интерес проявляется к ускоренным испытаниям стабильности
в одном режиме — в условиях повышенной температуры и влажности. При сокра-
щенной процедуре регистрации новых лекарств (ANDA) в руководстве FDA по ис-
пытанию стабильности предложено следующее: предварительный срок годности
лекарственного препарата, составляющий 24 мес., может быть гарантирован, если
представлена документация, подтверждающая удовлетворительные результаты ис-
пытаний стабильности в стресс-условиях — при 40 °C и 75% относительной влаж-
ности [12]. Простота такого подхода привлекает, поскольку позволяет сократить
время продвижения лекарственного препарата на рынок [7].
Глава 7.4. Стабильность фармацевтических препаратов 795
7.4.3. Механизмы разложения лекарственных средств
Многие механизмы разложения аналогичны реакциям разложения органических
соединений, описанных в учебниках по органической химии. Наиболее часто раз-
ложение лекарства инициируется при реакции с водой, кислородом или под воздей-
ствием света. Пути разложения большинства органических соединений включают
гидролиз, окисление, фотолиз и рацемизацию. Химическое разложение происхо-
дит, когда в процессе разложения лекарства образуется новое химическое вещество.
Если при уменьшении содержания лекарства не происходит образования новых хи-
мических веществ, речь идет о физическом разложении [13]. Избежать разложения,
протекающего по различным механизмам, можно путем снижения температуры
хранения и/или буферизации pH до уровня оптимальной стабильности.
7.4.4. Химическое разложение
7.4.4.1. Сольволиз
Сольволиз представляет собой процесс разложения активного лекарства с участи-
ем присутствующего растворителя. Если растворителем является вода, то процесс
называется гидролизом. Самыми распространенными реакциями сольволиза явля-
ются реакции с участием карбонильных соединений, таких как эфиры и р-лакгамы
(табл. 1).
Таблица 1
Функциональные группы, подверженные гидролизу
Функциональная группа Примеры
Сложные эфиры Аспирин, прокаин, алкалоиды, эстрон сульфат, дексометазон натрий фосфат
Лактамы Амиды Лактоны Оксимы Имиды Малоновые мочевины Хлорметины Азометины Пенициллины, цефалоспорины Хлорамфеникол, тиацинамид Пилокарпин Стероидные оксимы Глутетимид Барбитураты Мелфалан Бензодиазепины
Аспирин гидролизуется до уксусной и салициловой кислоты в присутствии вла-
ги, однако в условиях пониженной влажности этим гидролизом можно пренебречь.
Самыми сильными катализаторами гидролиза являются неблагоприятные значения
pH и некоторые соединения (например, декстроза, медь и другие ионы металлов).
Интенсивность гидролиза зависит от pH и температуры. Существует эмпирическое
правило: при увеличении температуры на 10 °C скорость реакции возрастает экспо-
ненциально. Реальный фактор повышения скорости зависит от параметров химиче-
ской связи и состава лекарства.
Гидролиз приводит к уменьшению активности лекарства и повышению содер-
жания ов разложения. Влияние этих изменений на скорость реакции зависит от
796
Часть 7. Стабильность лекарств
порядка реакции. Для реакций нулевого порядка скорость разложения не зависит
от концентрации лекарства. При гидролизе нулевого порядка отмечают уменьше-
ние процентного уровня разложения для более концентрированных растворов по
сравнению с растворами меньшей концентрации. В отличие от реакций нулевого
порядка, при гидролизе, описываемом кинетикой первого порядка, уровень изме-
нений прямо пропорционален концентрации вступающего в реакцию ингредиента.
Поэтому изменения концентрации активного ингредиента не влияют на процент-
ную концентрацию.
Структурные модификации лекарства могут помочь замедлить гидролиз. Чаще
всего рассматривают реакции гидролиза с участием эфира. На скорость этой реак-
ции могут оказывать влияние заместители. Hansch и Taft [14], а также Hammet [15]
опубликовали отличные обзоры, посвященные данной теме. Кроме того, соедине-
ние можно стабилизировать, уменьшив его растворимость. Это можно осуществить
путем добавления липофильных заместителей в боковые цепи ароматических ко-
лец. Зачастую для улучшения стабильности препарата используют его менее раство-
римые соли или эфиры.
Скорость гидролиза зависит от ионной силы (общей концентрации) растворен-
ных электролитов. В общем случае константа скорости гидролиза прямо пропор-
циональна ионной силе ионов аналогичного заряда (катионы лекарства и катионы
наполнителя) и обратно пропорциональна ионной силе ионов противоположного
заряда.
7.4.4.2. Окисление
Окисление является основной причиной нестабильности. Обычно процесс окис-
ления заключается во взаимодействии с кислородом или потере атома водорода.
Автоокисление — это окислительное разложение, как правило, опосредованное
реакцией с кислородом воздуха. Большинство реакций автоокисления протекает
с участием свободных радикалов. Подверженность лекарства автоокислению опре-
деляют испытанием стабильности препарата в атмосфере с высоким содержанием
кислорода (как правило, 40%). Результаты сравнивают с полученными в инертной
атмосфере или при температуре окружающей среды [16]. Перечисление отдельных
функциональных групп, подверженных окислению, представлено в табл. 2.
Таблица 2
Функциональные группы, подверженные окислению
Функциональная группа Примеры
Фенолы Катехоламины, морфин
Сопряженные диены Витамин А
Тиоэфиры Хлорпромазин
Нитриты Амилнитрит
Альдегиды Паральдегид, флаворанты
Амины Клозапин
Карбоновые кислоты Жирные кислоты
Тиолы Димерцепрол
Эфиры Диэтиловый эфир
Глава 7.4. Стабильность фармацевтических препаратов ______________________797
Традиционные антиоксиданты Таблица 3
Водные системы Системы на основе масел Хелатные агенты
Сульфит натрия Метабисульфит натрия Бисульфит натрия Тиосульфат натрия Аскорбиновая кислота Аскорбил пальмитат Гидрохинон Пропил галлат Нордигидрогваяретовая кислота Бутилированный гидрокситолуол Бутилированный гидроксианизол а-Токоферол Этилендиамин тетрауксусная кислота Дигидроэтилглицин Лимонная кислота Винная кислота Глюконовая кислота Сахарные кислоты
Процессы окисления катализируют кислоты, основания, pH, превышающий
оптимальные значения, поливалентные ионы металлов, пероксиды, гидроперок-
сиды, воздействие кислорода и ультрафиолетового (УФ) излучения. Из-за этих ре-
акций может возникнуть необходимость применения антиоксидантов, инертной
атмосферы и непрозрачной упаковки. Хелатные агенты, добавляемые в воду, по-
зволяют связать тяжелые металлы. Парентеральные составы не должны соприка-
саться с ионами тяжелых металлов в процессе производства, упаковки и хранения
продукции [17].
Антиоксиданты очень эффективно стабилизируют препараты, вступающие
в цепные реакции с участием свободных радикалов. Эти препараты обладают более
низкими окислительными потенциалами по сравнению с активными лекарствами.
В идеале антиоксиданты стабильны в широком диапазоне pH и сохраняют раство-
римость в окисленной форме, являются бесцветными и нетоксичными веществами.
Список традиционно применяемых антиоксидантов представлен в табл. 3.
Зачастую возможно визуальное обнаружение продуктов окисления благодаря
возникновению конъюгатов, однако эти изменения не всегда заметны в растворах
низкой концентрации.
7.4.4.3. Фотолиз
Когда молекулы поглощают энергию, они переходят на высший энергетический
уровень, где в ходе химической реакции высвобождается энергия, и молекула вновь
возвращается в исходное состояние. Когда энергия активации соединения дости-
гается путем поглощения световой энергии, реакция разложения носит название
фотолитической реакции. Активированные вещества мотуг высвобождать энергию
в виде светового излучения различной частоты (флуоресценция), либо в ходе ре-
акции разложения (фотолиз). Комнатное освещение или воздействие солнечных
лучей может привести к разложению лекарства, вызывая фотоокисление и фотолиз
ковалентных связей. В соединениях, подверженных фотолизу, обычно содержащих
w-электроны, в ходе реакции образуются промежуточные свободные радикалы, ко-
торые могут поддерживать цепные реакции.
798
Часть 7. Стабильность лекарств
В целом лекарства, поглощающие УФ излучение менее 280 нм, подвержены раз-
ложению под воздействием солнечного света, в то время как соединения, поглоща-
ющие излучение выше 400 нм, потенциально могут разлагаться как на солнечном
свете, так и при комнатном освещении. Реакции, инициированные светом, обыч-
ны для стероидов [18]. Фоторазложение нитропруссида натрия в водных растворах
остается классическим примером фотолитического разложения [19].
7.4.4.4. Дегидратация
Реакции дегидратации — это химические реакции, в ходе которых происходит по-
теря воды. При дегидратации тетрациклина катализируемой кислотой образуется
токсичный продукт эпиангидротетрациклин [20]. Физическое разложение теофил-
лина гидрата и ампициллина тригидрата приводит к изменению кристаллической
структуры лекарства [21].
7.4.4. Б. Рацемизация
Рацемизация — это процесс перехода оптически активного соединения в рацеми-
ческую смесь. Одна из ранних дискуссий, посвященных роли стереспецифичности
в лекарственном действии, представлена в работе Pfeiffer [22].
Из лекарств, подверженных рацемизации, наиболее широко известны тетраци-
клин, эпинефрин [23], пилокарпин [24] и эрготамин. В тетрациклине реакция про-
текает быстро, когда pH среды, в которой растворено лекарство, становится боль-
ше 3; в результате происходит изменение локации диметиламино группы [25].
Рацемизация протекает в соответствии с кинетикой первого порядка, а скорость
ее зависит от температуры, системы растворителей, катализаторов и наличия или
отсутствия освещения. Наблюдается тенденция к ускорению рацемизации через
резонансную стабилизацию под влиянием заместителей, примыкающих к центру
асимметрии.
7.4.5. Физическое разложение
7.4.5.1. Полиморфизм
Многие фармацевтические твердые вещества могут существовать в различных фи-
зических формах. Зачастую полиморфизм определяют как способность лекарствен-
ного вещества к существованию в двух (или более) кристаллических фазах, раз-
личающихся конформацией молекул и/или структурой кристаллической решетки
[26]. Полиморфные формы лекарственного вещества могут обладать разными хи-
мическими и физическими свойствами, включая температуру плавления, химиче-
скую активность, условное насыщение раствора, скорость растворения, оптические
и механические свойства, давление паров и плотность вещества. Эти характеристи-
ки могут непосредственно определять способы переработки и/или производства
лекарственного вещества и лекарственного препарата, а также оказывать влияние
на стабильность, растворение и биодоступность лекарственного препарата. Таким
образом, из-за полиморфизма могут быть нарушены безопасность и эффективность
лекарственного препарата [27]. В аморфных твердых веществах молекулы располо-
жены беспорядочно, кристаллической решетки явно не обнаруживается. Сольваты
Глава 7.4. Стабильность фармацевтических препаратов
799
являются кристаллическими твердыми аддуктами, содержащими стехиометриче-
ское или нестехиометрическое количество растворителя, включенного в кристал-
лическую структуру. Если таким растворителем является вода, то сольваты обычно
называют гидратами. Изменения температуры, влажности, процессы распыления
при гранулировании, измельчения и прессования могут вызвать полиморфные пре-
вращения. Степень конверсии, как правило, зависит от относительной стабильно-
сти полиморфных форм, кинетических барьеров фазовых превращений и прило-
женного напряжения [28]. Известно, что сульфонамиды, барбитураты и стероиды
склонны к образованию полиморфных форм [29].
7.4. Б.2. Испарение
Для некоторых лекарств и наполнителей давление паров при комнатной темпера-
туре настолько велико, что испарение является для них путем потери лекарства из
состава. Низкомолекулярные спирты и соединения, применяемые для придания
лекарству приятного запаха, могут испаряться из лекарственного препарата. Часто
в качестве примера потери лекарства через испарение приводится нитроглицерин.
FDA опубликовало специальное руководство по типам упаковок, которые могут
быть использованы для нитроглицерина в таблетках [30]. Добавление макромоле-
кул, таких как микрокристаллическая целлюлоза, позволяет изготовить стабилизи-
рованные сублингвальные таблетки нитроглицерина [31].
7.4.6. Микробиологическое разложение
Микроорганизмы потенциально могут контаминировать или вызывать разложе-
ние лекарственных препаратов. Эти организмы могут размножаться в нормальных
условиях хранения или в процессе их применения пациентами, особенно в много-
дозовых препаратах. Препараты, содержащие воду, более подвержены контами-
нации. К таким препаратам относятся растворы, суспензии и эмульсии, а также
стерильные мультидозовые составы для инъекций и офтальмологические препа-
раты.
Для предотвращения или замедления роста микроорганизмов используются ан-
тимикробные консерванты. Факторы, влияющие на наблюдаемый уровень эффек-
тивности консерванта, включают химическую структуру консерванта, физические
и химические характеристики фармацевтического препарата, концентрацию кон-
серванта, а также тип и исходный уровень контаминации. Консерванты ни в коей
мере не являются альтернативой надлежащей производственной практике.
7.4.7. Руководства и требования в отношении стабильности
Требования FDA, относящиеся к испытаниям стабильности, изложены в Своде фе-
деральных постановлений (CFR), часть 21, раздел 211.166 — действующая надле-
жащая производственная практика для готовых лекарственных препаратов. В них
установлена необходимость программы испытаний с целью оценки характеристик
стабильности лекарственных препаратов. Результаты испытаний стабильности
следует использовать для определения надлежащих условий и сроков хранения
препаратов. В данной секции CFR представлены указания по объемам выборки
800
Часть 7. Стабильность лекарств
и промежуткам между испытаниями, условиям хранения образцов и специальным
аналитическим методам [32].
Указания в разделе 211.166 содержат предписания об адекватном числе серий
каждого лекарственного препарата, проходящих испытания с целью определения
срока хранения. Ускоренные испытания в сочетании с базовой информацией по
стабильности компонентов, лекарственных препаратов и системы упаковка/укупо-
рочное средство можно использовать для подтверждения предварительно установ-
ленных сроков годности, если результаты полного исследования сроков хранения
недоступны. Если данные ускоренного испытания используют для предваритель-
ной оценки срока годности, выходящего за пределы, установленные реальными ис-
пытаниями, необходимо провести испытания стабильности, включающие анализ
лекарственного препарата через определенные промежутки времени вплоть до под-
тверждения предварительно установленного срока годности или эксперименталь-
ного определения соответствующего срока годности.
Дополнительно установлены требования к резервным пробам [33], установле-
нию даты истечения срока годности [34] и лабораторным записям [35].
7.4.8. Руководства /СНпо качеству
Международная конференция по гармонизации технических требований к ре-
гистрации фармацевтических препаратов, предназначенных для человека (ICH),
представляет собой проект, объединяющий регуляторные органы Европы, Япо-
нии и Соединенных Штатов Америки и экспертов из трех регионов, работающих
в фармацевтической индустрии, для обсуждения научных и технических аспектов
регистрации препарата. Задача организации — предоставление рекомендаций по
достижению соглашений в интерпретации и применении технических указаний
и требований к регистрации препарата для уменьшения необходимости в дублиру-
ющих испытаниях при научных исследованиях и разработке новых лекарств. Цели
подобной гармонизации заключаются в более экономичном использовании чело-
веческих, животных и материальных ресурсов, уничтожении препятствий на пути
глобального развития и доступности новых лекарств при сохранении требований
к качеству, безопасности и эффективности, а также нормативных обязательств по
защите общественного здоровья [36].
Руководство представляет ряд рекомендаций по протоколам испытаний стабиль-
ности, включая температуру, влажность и длительность испытания [ QIA(R2)]; базо-
вые протоколы испытаний по оценке светочувствительности и стабильности новых
действующих веществ и лекарственных препаратов (015); указания по испытаниям
стабильности новых составов уже зарегистрированных лекарственных препаратов
(QI С). Описания различных указаний ICHпо испытаниям стабильности представ-
лены в табл. 4.
Кроме того, ICHпредоставляет указания по аналитической валидации [02(51),
примесям (серии Q3), фармакопейным требованиям (серии Q4), качеству биотехно-
логических препаратов (серии Q5), спецификациям (серии Q6), надлежащей произ-
водственной практике (серии Q7), фармацевтическим разработкам (08) и управле-
нию рисками (Q9) ].
Глава 7.4. Стабильность фармацевтических препаратов
801
Таблица 4
Коды документов ICHи указания по испытаниям стабильности
Код ICH Заглавие Описание
Q1A(P2) Испытание стабильности новых лекарственных веществ и препаратов Протоколы испытания стабильности, включающие температуру, влажность и длительность испытания
Q1B Испытания стабильности: испытания фотостабильности новых лекарственных веществ и препаратов Основной протокол испытания, проводимого для оценки светочувствительности и стабильности новых лекарств и препаратов
QIC Испытания стабильности новых дозированных форм Расширенное руководство по испытаниям стабильности новых составов ранее зарегистрированных лекарств; изложение обстоятельств, в которых могут быть приняты данные сокращенных испытаний стабильности
Q1D Брекетинг и построение матриц при испытании стабильности новых лекарственных веществ и препаратов Общие принципы сокращенных испытаний стабильности и примеры брекетинга и построения матриц
Q1E Оценка данных испытаний стабильности Разъясняет возможные ситуации, в которых возможно определение периодов переконтроля/сроков хранения путем экстраполяции за рамки данных реальных испытаний; примеры статистического подхода к анализу данных испытаний стабильности
QIF Объем данных по стабильности для регистрации препаратов в климатических зонах III и I В дополнение к предложениям по условиям хранения в ходе долгосрочных и ускоренных испытаний стабильности, даны указания по данным, полученным в условиях повышенной температуры и экстремальной влажности; в ссылках содержится информация по классификации стран в соответствии с климатическими зонами
Литература
1. U.S. Pharmacopeia (USP), Stability considerations in dispensing practice, USP 29/NF 24, USP, Rock-
ville, MD, p. 3029.
2. Guillory P., Poust R. (2002), Chemical kinetics and drug stability, in Banker and Rhodes, Eds., Drugs
in the Pharmaceutical Sciences, Vol. 121, Marcel Dekker, New York, pp. 139-166.
3. LintnerC.C. (1973), Quality Control in thePharmaceuticallndustry, Vol. 2, Academic, New York, p. 141.
4. Connors K. A., Amidon G. L., Stella J. V. (1986), Chemical Stability of Pharmaceutical, 2nd ed., Wi-
ley, New York.
5. McMinn C. S., Lintner C. J. (1973, May), paper presented at the American Pharmaceutical Associa-
tion Academy of Pharmaceutical Sciences Meeting, Pharmaceutical Technical Section, Chicago, IL.
802 Часть 7. Стабильность лекарств
6. Lachman L., DeLuca В, Akers J. J. (1986), Kinetic principles and stability testing, in Lachman L.,
Lieberman H. A., Kanig J. L., Eds., The Theory and Practice of Industrial Pharmacy, 3rd ed., Lea &
Febiger, Philadelphia, p. 766.
7. Fung H.-L., King S.-Y. (1983), in Pharmaceutical Technology Conference’ 83 Proceedings, Aster
Publishing, Springfield, OR.
8. Pikal M. J., Lukes A. L., Lang J. E. (1977), J. Pharm. Sci., 66,1312.
9. Woolfe J., Worthington H. E. C. (1974), Drug Dev. Commun., 1, 185.
10. Wang W. (1999), Instability, stabilization and dormulation of liquid protein pharmaceuticals,
International Journal of Pharmaceutics, 185, 129-188.
11. Gu L. C., Erdos E. A., Chang H.-S. (1991), Pharm. Res. 8,485.
12. (a) Center for Drugs and Biologies, U.S. Food and Drug Administration (FDA) (1987, Feb.), Guide-
line for submitting documentation for the stability of human drugs and Biologies, FDA, Rockville,
MD. (b) Center for Drug Evaluation and Research and Center for Biologies Evaluation and Research,
U.S. Department of Health and Human Services, FDA (1998, June), Guidance for industry: Stability
testing of drug substances and drug products: Draft guidance, FDA, Rockville, MD.
13. U.S. Pharmacopeia (USP), Pharmaceutical stability, USP29/NF 24, USP, Rockville, MD. p. 2994.
14. Hansch A. L., Taft R. E. (1991), A survey of Hammett substituent constants and resonance and field
parameters, Chem. Rev., 91, 165.
15. Hammett L. P. (1970), Physical Organic Chemistry, 2nd ed., McGraw-Hill, New York.
16. Johnson D. M., Gu L. C. (1998), Autooxidation and antioxidants, in Swarbrick J., Boylan J. C. Eds.,
Encyclopedia of Pharmaceutical Technology, Marcel Dekker, New York, pp. 415—449.
17. Vadas E. B. (2000), Stability of pharmaceutical products, in Remington: The Science and Practice of
Pharmacy, 20th ed, Lippincott Williams and Wilkins, Baltimore, MD, p. 989.
18. Ogata M., Noro Y, Yamada M., Tahara T, Nishimura T. (2000), Photo-degradation products of meth-
ylprednisolone sulphanate in aqueous solution — Evidence of a bicyclo[3.1.0]hex-3-en-2-one inter-
mediate, J. Pharm. Sci., 87 (1), 91—95.
19. Hauser U., Oestreich V, Rohrweck H. D. (1977), On optical dispersion in transparent molecular sys-
tems, Zeits. Phys., 280, pp. 17-25.
20. Yuen P. H., Sokoloski T. D., (1977), Kinetics of concomitant degradation of tetracycline to epitetracy-
cline, anhydrotetracycline and epianhydrotetracycline in acid phosphate solution, J. Pharm. Sci., 66
(11), 1648-1650.
21. Shefter E., Fung H.-L., Mok O. (1973), Dehydration of crystalline theophylline monohydrate and
ampicillin trihydrate, J. Pharm. Sci., 62, 791.
22. Pfeiffer С. C. (1956), Optical isomerism and pharmacological action, a generalization, Sci., 123,
3210, 29-31.
23. Schroeter L. C., Higuchi T. (1958), Racemization of epinephrine, J. Am. Pharm. Assoc. (Baltimore),
47, 6, 426, 30.
24. Nunes M. A., Brochmann-Hanssen E. (1974), Hydrolysis and epimerization kinetics of pilocarpine in
aqueous solution, J. Pharm. Sci., 63, 716.
25. Sheberstova N. V., Perel’son M. E., Kuzovkov A. D. (1975), Study of the epimerization of tetracycline
by the NMR method, Chem. Nat. Comp., 10 (1), 61—65.
26. Haleblian J., McCrone W. (1969), Pharmaceutical applications ofpolymorphism, J. Pharm. Sci., 58,911.
27. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug
Evaluation and Research (CDER), U.S. Food and Drug Administration (FDA) (2004, Dec.), Guidance
for industry ANDAs: Pharmaceutical solid polymorphism chemistry, manufacturing, and controls
information, FDA, Rockville, MD.
28. Vippagunta S. R., Brittain H. G., Grant D. J. W. (2001), Crystalline solids, Adv. DrugDeliv. Rev., 48,
3-26.
29. Kuhnert-Brandstatter M. (1971), Thermomicroscopy, in The Analysis of Pharmaceuticals, Pergamon,
Oxford, pp. 37-42.
Глава 7.4. Стабильность фармацевтических препаратов 803
30. Federal Register, 37,15959 (1972).
31. Fung H.-L., Yap S. K., Rhodes С. T. (1974), Development of a stable sublingual nitroglycerin tablet.
I. Interaction of nitroglycerin with selected macro-molecules, J. Pharm. Sci., 63,1810.
32. Code of Federal Regulations, Title 21, Food and drugs, Part 211, Current good manufacturing prac-
tice for finished pharmaceuticals, Subpart I, Laboratory controls, Section 211.166, Stability testing.
33. Code of Federal Regulations, Title 21, Food and drugs, Part 211, Current good manufacturing prac-
tice for finished pharmaceuticals, Subpart I, Laboratory controls, Section 211.170, Reserve samples.
34. Code of Federal Regulations, Title 21, Food and drugs, Part 211, Current good manufacturing prac-
tice for finished pharmaceuticals, Subpart G, Packaging and labeling control, Section 211.137, Expi-
ration dating.
35. Code of Federal Regulations, Title 21, Food and drugs, Part 211, Current good manufacturing practice
for fi nished pharmaceuticals, Subpart J, Records and reports, Section 211.194, Laboratory records.
36. International Conference on Harmonisation of Technical Requirements for Registration of Pharma-
ceuticals for Human Use, available: http://www.ich.org/cache/compo/363-272-l.html.
Глава 7.5. Альтернативные ускоренные методы
изучения стабильности лекарств:
исследования кинетики разложения
с переменными параметрами
Джузеппе Алибранди
Университет Мессины (Messina, Италия)
7.5.1. Введение
Испытания стабильности лекарств играют ключевую роль в описании физико-
химического состава и свойств веществ [1—6]. Чрезвычайно важно знать длитель-
ность периода i, в течение которого лекарственное вещество сохраняет химическую
идентичность в различных условиях окружающей среды, не утрачивая терапевтиче-
ского действия, и пути его разложения (п). Информация по обоим вопросам жиз-
ненно важна для фармацевтической индустрии. Ответ на первый вопрос необходим
для принятия решения о том, стоит ли продолжать исследование молекулы или нет.
Нестабильность лекарства может уменьшить его активность до малых долей про-
цента; в результате разложения могут накапливаться, приводящие к нежелательным
побочным эффектам. Ответ на второй вопрос необходим для понимания возмож-
ности (или невозможности) решения проблемы стабильности, например, путем
синтеза нового лекарства аналогичной структуры с пониженной химической актив-
ностью или применения надлежащего носителя, способного стабилизировать моле-
кулу и доставить ее к желательной мишени.
Температура, pH, ионная сила, концентрация ионов металла и другие параметры
окружающей среды влияют на химическую реакцию; вариации этих величин могут
свидетельствовать о существенных изменениях скорости, механизма или направ-
ления реакции в зависимости от конкретной ситуации. Поэтому изучение количе-
ственного влияния физических параметров на химическую активность занимает
в испытаниях подобного рода большую часть времени.
Обычный способ состоит в выделении и описании изучаемой реакции, а так-
же определении константы скорости в условиях реакции первого или псевдо-
первого порядка для различных значений исследуемых параметров. Результатом
является многомерное представление константы скорости как функции этих пара-
метров, которое может оказаться чрезвычайно полезным в конкретных ситуациях.
В частности, помогает в планировании эксперимент, на основе которого строится
исследование, задача которого состоит в выяснении механизма реакции. Фактиче-
ски зависимость kobs от параметров является следствием их влияния на этапе, опре-
деляющем скорость реакции; подгонка кинетических данных к приемлемым мате-
матическим моделям дает информацию, позволяющую построить схему реакции,
выбрать между кинетически эквивалентными механизмами и определить значения
элементарных кинетических констант [1—4, 7, 8].
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств... 805
Во множестве примеров, представленных в литературе, подчеркивается важ-
ность получения данных о кинетике процесса [9—11]. Историческое исследование
Эдвардса, с которого начались исследования механизма гидролиза аспирина, по-
требовало проведения сотен кинетических экспериментов [12, 13]. Ряд примеров
представлен в обзоре Carstensen [1], где в дополнение к обширной информации по
определению профилей pH-скорость реакции рассмотрено влияние температуры,
ионной силы, концентрации буфера и диэлектрической постоянной.
Построение физико-химических профилей связано с определением основных
свойств молекул, представляющих интерес для фармацевтики, которые могут быть
использованы в последующих клинических испытаниях и способствовать быстрой
идентификации соединений, непригодных с точки зрения физико-химических
и фармакокинетических характеристик. Новые методы ускоренного построения
физико-химических профилей приобрели особое значение из-за появления мно-
жества новых химических веществ, синтезируемых благодаря новым стратегиям
синтеза. Ежегодно возникает необходимость составления профилей тысяч соеди-
нений, и традиционные технологии являются барьером на пути разработок новых
лекарств. Разработаны высокопроизводительные методы построения физико-
химических профилей для таких параметров, как растворимость, проницаемость,
рКа, липофильность, стабильность и целостность; к этим вопросам сохраняется
большой интерес со стороны фармацевтических производителей и приборострои-
телей [5, 6].
Среди физико-химических характеристик лекарств-кандидатов возрастающее
внимание привлекает стабильность. К сожалению, оценка стабильности требует
длительных испытаний; построение профилей для множества молекул было бы
практически невозможным без новых компьютерных технологий. Приветствуются
любые усилия, направленные на ускорение первой части фармацевтического иссле-
дования, поскольку в итоге они приводят к существенному уменьшению стоимости
испытаний.
Здесь представлена общая картина, описывающая усилия, направленные
на ускоренное определение характеристик химической активности в растворах
молекул-кандидатов. В частности, рассматривается метод исследования кинетики
с переменными параметрами (VPaK) — новый способ, потенциально позволяющий
сэкономить время и химические реактивы в процессе исследования стабильности
лекарств.
7.5.2. Теоретические положения
Рассмотрим процесс разложения молекулы А на препараты
А->Р, (1)
скорость которого, в соответствии с законами кинетики первого или псевдопервого
порядка, описывается уравнением (2), где С — молярная концентрация исходного
вещества, а £о(и (Par 1, Par 2, ...) — наблюдаемая константа скорости, являющаяся
функцией различных параметров:
~^ = kots(Parl,Par2,...)C. (2)
at
806
Часть 7. Стабильность лекарств
В традиционных кинетических экспериментах все эти параметры должны быть
строго постоянными, чтобы экспериментальные данные C-t, т. е. кинетический
профиль, полученный путем измерения концентрации с течением времени, можно
было легко представить в дифференциальной форме уравнения первого порядка (2)
или в интегрированной экспоненциальной форме [уравнение (3)] или логарифми-
ческой форме [уравнение (4)] для получения оптимизированных значений
с=Соехр(-^/); (3)
1пС= — к. t + 1пСп.
obs 0
(4)
Зависимость коЬз от одного из параметров (температуры Т, давления Р, pH, ион-
ной силы In т. д.), т. е. kobs (Par) профиль, можно построить, выполнив ряд экспе-
риментов при различных значениях этого параметра. При этом накапливаются дан-
ные kobs — Par.. При наличии аналитической модели, описывающей эту зависимость,
на втором этапе обработки — подгонке данных можно получить величины, регули-
рующие эту зависимость. Например, для параметра температуры (7) зависимость
может описывать уравнение Эйринга (5), а членами уравнения, регулирующими за-
висимость, являются энтропия активации ДА* и энтальпия активации АТТ* (к — по-
стоянная Больцмана, h — постоянная Планка, R — газовая постоянная) [1, 7, 8]:
Д5*
, кТ
JeXP(j
ATT* А
RT J
(5)
R
Обычно уравнение Эйринга записывают в логарифмической форме (6), чтобы
получить кинетические данные в виде линейного графика. Интерсепт соответствует
значению ДА*, а наклон — значению АН*:
Т h R RT
(6)
На рис. 1 представлен пример подобного исследования, в котором для опреде-
ления параметров активации разложения пяти молекул потребовалось провести 25
кинетических экспериментов при постоянной температуре. Когда параметром яв-
ляется концентрация вещества, присутствующего в реакционной среде (Н+, ОН-,
Ме”+, нуклеофил, катализатор и т. д.), зависимость можно представить в виде урав-
нения
к =Лпн[ОН-]. (7)
Подгонка данных kobs — [ОН-], полученных в нескольких экспериментах, прово-
димых при постоянной концентрации [ОН-], позволяет определить Лон (рис. 2). Эти
функции зависимости, в которых присутствуют концентрации, могут быть весьма
сложными, но они несут очень важную информацию, необходимую для понимания
механизма реакции [1, 3,7, 8].
Когда параметром является ионная сила, функция зависимости выражается
уравнением Брёнстеда—Бьеррума (8) [1, 7, 8, 15], где к0 — константа скорости при
нулевой ионной силе, а — константа (для воды при 25 °C а = 0,50925 л^/моль14 [16])
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...
807
Рис. 1. Графики Эйринга (сплошные линии) для реакций гомологических серий из пяти молекул,
построенные по результатам кинетических экспериментов, проводимых при постоянной
температуре (геометрические маркеры) [14]
и 2^ZB — произведение зарядов реагирующих частиц на этапе, определяющем ско-
рость реакции:
^=Ло-1О (8)
Даже в этом случае традиционно для обработки кинетических данных исполь-
зуют логарифмическую форму уравнения [уравнение (9)]. Интерсепт графика ли-
нейной зависимости (рис. 3) определяет значение к0, а наклон — значение ZAZB. Эта
информация весьма полезна для различения кинетически эквивалентных механиз-
мов ионных реакций:
log kBbs = logk02aZAZB (9)
Кинетические эксперименты с постоянными параметрами (СРаК) очень время-
затратны. В наше время применение персональных компьютеров и аналитических
приборов нового поколения позволяет сделать шаг вперед в области накопления
кинетических данных.
Кинетика переменных параметров VPaK [17] представляет собой часть химиче-
ской кинетики, описывающую эксперименты, в которых параметр окружающей
среды является регулируемой переменной величиной. Целью эксперимента явля-
ется построение по результатам одного опыта зависимости наблюдаемой константы
скорости от упомянутого параметра за время в 10—100 раз меньшее по сравнению со
временем, которого требуют традиционные методы. Это можно реализовать путем
подгонки кинетического профиля, полученного в процессе реакции, к соответству-
ющей математической модели, аналитически описывающей данную реакцию.
Кинетику переменных параметров (VPaK) можно считать обобщением неизо-
термической кинетики [18, 19], применяемой главным образом при термическом
анализе [20—25] твердофазных систем. Ранее сообщалось о нескольких экспери-
ментах с переменным параметром температуры, относящихся к химии растворов.
808
Часть 7. Стабильность лекарств
Рис. 2. koijs ([ОН') профили (сплошные линии) для гомологических серий из пяти молекул,
построенные по результатам 25 кинетических экспериментов (геометрические маркеры),
проводимых при постоянной концентрации ОН" (моделированные данные)
Рис. 3. Графики Бренстеда-Бьеррума (сплошные линии), полученные при кинетических
экспериментах (геометрические маркеры) для гомологических серий из 5 соединений; реакции
протекают по одинаковому механизму (одинаковые значения ZAZB = + 1), собственная химическая
активность соединений различна (разные значения к0) (моделированные данные)
В зависимости от области научных интересов авторов работ (неорганическая, орга-
ническая, металлоорганическая химия или фармацевтика) и доступных в то время
приборов, применялись различные подходы к исследованиям [14, 26—51]. В публи-
кациях представлены несколько примеров кинетических экспериментов с перемен-
ными параметрами концентрации (VCK) [31, 35, 52, 53]. К настоящему моменту по-
ступило лишь одно сообщение об эксперименте с переменным параметром ионной
силы [54]. Здесь представлен простой подход со ссылками на важнейшие аналогич-
ные исследования.
Глава 7,5. Альтернативные ускоренные методы изучения стабильности лекарств...
809
Математическим выражением, описывающим ЕРаА'-эксперимент, является
уравнение
«о»
где С— концентрация контролируемых веществ, вступающих в реакцию. В данном
уравнении величина kobs не является постоянной, поскольку зависит от параметра
(Par), изменяющегося во времени. В таком случае kobs [Azr(01 является функцией
функции: kobs(Par) представляет функцию зависимости (D), описывающую зави-
симость константы скорости от параметра i, a Par(t) — модулирующую функцию
(М), описывающую изменение параметров с течением времени, при поддержании
постоянства других параметров. В каждой точке экспериментального кинетиче-
ского профиля содержится информация о значении константы скорости в данный
момент времени, что видно из уравнения (10), записанного в виде уравнения (11).
Отношение производной концентрации к значению концентрации определяет ве-
личину kobs. Теперь профиль kobs(Par) можно построить по одному циклу кинетиче-
ского эксперимента:
(»>
Далее, зная математическую форму функции зависимости, единственный шаг
подгонки к уравнению (10) или к его интегрированной форме позволяет получить
параметры, регулирующие зависимость. Например, в кинетических экспериментах
с переменным параметром температуры (VTK) функция зависимости, как сказано
выше, представляет собой уравнение Эйринга, а членами уравнения являются пара-
метры активации ДА* и ЛРР. Если температура изменяется линейно, то есть
М: Т= То + at,
где Тй — исходная температура, а а — градиент температуры, то модель изменяется,
и ее описывает уравнение
dC k(T0 + at) ГД5П ( АН* V
dt h Ч R ) Ч R(T0 + at)J
Или в соответствующей интегральной форме:
Г г о + «г) ГД5Ч ( АЯ* V"
С = Соехр --------ехр ----- ехр------------\dt .
Lj h Ч * J Ч R(T0 + at) J J
(12)
(13)
Подгонка экспериментальных данных С— t, то есть кинетического профиля, по-
лученного в ходе одного цикла кинетического эксперимента, к одному из этих урав-
нений позволяет получить оптимизированные значенияАА1 и АТА.
Для эксперимента, в ходе которого исследуют зависимость скорости реакции
от концентрации катализатора Y, [Y], молярная концентрация катализатора может
изменяться линейно, то есть М: [Y] = рг, где р — градиент концентрации. Если из-
вестна функция зависимости, например, Я: kobs = ку[Y], математическая модель при-
нимает вид
810
Часть 7. Стабильность лекарств
-|A:Ypr2J;
(15)
= fcYp/C. (14)
at
Один шаг подгонки экспериментальных данных С— t к одному из этих уравне-
ний позволяет получить оптимизированное значение к0.
Если функция зависимости не известна, необходимо воспользоваться уравнени-
ем (16):
С = Со ехр
-f^=*U[Y](')}> do
С at
а профиль величины kobs [¥] можно получить, разделив производную кинетическо-
го профиля на значение, соответствующее профилю. Этот метод особенно важен,
когда недоступна информация о зависимости константы скорости от концентраций
веществ, присутствующих в реакционной среде, участвующих в этапе, определяю-
щем скорость реакции. В данном случае результатом ИСХ-эксперимента является
эмпирическое соотношение, позволяющее сформулировать гипотезу о функции за-
висимости.
7.5.3. Экспериментальная часть
Для удобства выполнения VPaK экспериментов следует выделить базовые пункты
эксперимента:
1. Компьютерное моделирование.
2. Устройства для создания условий переменных параметров.
3. Аналитические приборы.
4. Программное обеспечение для обработки экспериментальных данных.
Все эти пункты предусматривают использование персональных компьютеров.
Важно подчеркнуть, что именно доступность недорогих быстродействующих ком-
пьютеров сделала возможным проведение ИРдК-экспериментов, что позволяет эко-
номить время и обеспечить приемлемый уровень достоверности данных.
7.5.3.1. Компьютерное моделирование
Компьютерное моделирование может принести большую пользу на этапе плани-
рования ИРдК-эксперимента. В сравнении с профилями СРаК, где скорость всегда
уменьшается экспоненциально с течением времени [уравнение (3)], форму про-
филей VPaK зачастую трудно себе представить. В VPaK скорость реакции в каж-
дый момент времени t определяется произведением двух факторов: РоЬ[Раг(0] и С.
Концентрация всегда уменьшается со временем, однако значение Рог,ДРдг.(0] может
варьироваться, в зависимости от вида как функции зависимости, так и модули-
рующей функции. Поиск подходящих параметров эксперимента может привести
к потере драгоценного времени. При помощи персонального компьютера можно
за несколько минут построить приемлемый профиль, получив некоторые предва-
рительные данные при помощи тестов, которые всегда выполняются перед кине-
тическим исследованием [55]. Можно использовать любую программу, способную
строить графики функций и, при необходимости, рассчитывать численное значение
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...
811
производной или интеграла. Мы обнаружили, что программа MicroMath SCIENTIST
[56] универсальна, легка в применении и позволяет использовать дифференци-
альную форму уравнения скорости (10) без его интегрирования. С помощью это-
го инструмента при таких входных данных, как функция зависимости и функция
модулирования, можно немедленно получить кинетический профиль для каждого
из экспериментальных параметров. На рис. 4 представлена типичная программа
SCIENTIST, где модулирующая функция, функция зависимости и дифференциаль-
ное уравнение первого порядка, используемые для моделирования VTK, помечены
стрелками.
//VTK_SIM
IndVars. t
DepVars: С. V. K.TE
Params: ДН, AS, CO. kb. h. R. TEo. a
- > TE = TEo + a*t
- > К = (kb*TE/h)’exp(AS /R)*exp(-AH /(R*TE)
- > C’ = -K*C
V = -C’
H Pa ram eter values
AH =118000
AS = 61
- » TEo = 298
- » a = 0.0033
CO = 1 e-3
kb = 1.3806e-16
h =6.6262e-27
R = 8.314
П Initial conditions
t = 0
C =C0
Рис. 4. Модель SCIENTIST для моделирования VTK-эксперимента. Первые три стрелки указывают
соответственно на модулирующую функцию, функцию зависимости и кинетическое уравнение
первого порядка. Последние две стрелки указывают на экспериментальные параметры Гои а
812
Часть 7. Стабильность лекарств
На рис. 5 представлен профиль, полученный при вводе в программу в качестве
экспериментальных данных значений То = 298 К и а = 0,0033 К/с. Кроме того, при
разовой имитации возможно получение нескольких профилей, относящихся к раз-
личным значениям членов уравнения, их обуславливающих. На рис. 6, например,
представлена многопараметрическая имитация профилей КСК-реакции основно-
го гидролиза с функцией зависимости kobs = Лон[ОН-] и модулирующей функцией
[ОН-] = а/, где 10-3 х аМ с-1 = 1; 2,5; 4, 5,5; 7.
Рис. 5. Моделированный кинетический профиль УТХ-эксперимента, полученный при помощи
программы SCIENTIST
Рис. 6. Моделированные кинетические VCK-профили, полученные при помощи
многопараметрической модели
7.5.3.2. Устройства для достижения условий переменных параметров
Для достижения условий эксперимента VPaK внутри реакционного сосуда необхо-
димо специальное устройство. Изменение параметра должно происходить однород-
но и когерентно с модулирующей функцией без нарушения величин прочих факто-
ров, важных с точки зрения процесса реакции и аналитического мониторинга.
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств... 813
Параметр может изменяться в реакционном сосуде, являющемся частью анали-
тического прибора, например, в спектрофотометрической ячейке для измерений
в УФ-видимом диапазоне [39, 41, 45, 14, 47, 48], инфракрасной (ИК) ячейке [42,
46] или флуориметрической ячейке [45, 51] или поляриметрической трубке [27, 29].
Параметр можно изменять в реакционном сосуде, где аналитический сигнал может
быть прочитан, например, при помощи оптоволоконной ячейки для спектрометрии
[52—54] или кондуктометрической ячейки [16, 34, 40]. Другая возможность заклю-
чается в переносе раствора из реакционного сосуда в аналитический прибор с по-
мощью перистальтического насоса [38]. Если альтернативные аналитические мето-
дики неосуществимы, можно отбирать пробы и анализировать их вне реакционной
системы [29, 31, 35, 39,43, 50].
Любая система, удовлетворяющая вышеописанным условиям, может быть ис-
пользована в ЕРаК-эксперименте. Для VTK очень удобны бани с программирова-
нием температуры (с возможностью внешней циркуляции) и устройства для про-
граммирования температуры, основанные на эффекте Пельтье. Всегда необходимо
следить за однородностью температуры путем измерения ее внутри реакционного
сосуда с возможным вводом данных измерений в компьютер. Линейный подъем
температуры достаточно легко осуществим, но другие модулирующие функции
следует контролировать [33, 35, 47]. Однако в будущих экспериментах применение
компьютеров с надлежащим программным обеспечением позволит без труда гене-
рировать профили T(t) различных форм.
Термическое расширение реакционного раствора и сосуда может нарушить со-
гласованность кинетической модели, поскольку с этими величинами связаны кон-
центрации веществ, вступающих в реакцию. Лучшим способом решения проблемы
является ограничение интервала температур значениями 20—25 °C; в противном
случае необходимы поправки, что усложняет процедуру Кроме того, ограничение
колебаний температуры обеспечивает постоянство параметров активации в ходе
экспериментов.
В VCK-экспериментах обычно применяются автоматические бюретки, высво-
бождающие концентрированный раствор веществ, так или иначе влияющих на ход
реакции. Градиент концентрации а (в молях в секунду) внутри сосуда выражается'
уравнением
где g (л/с) — скорость высвобождения; М (моль/л) — молярная концентрация до-
бавленного раствора; Ув (л) — исходный объем реакционной смеси.
Химическая однородность обеспечивается за счет хорошего перемешивания.
Проблема возникает, когда добавленный раствор имеет значительный объем (в про-
центном отношении), которым нельзя пренебречь в математической модели. В та-
ком случае приемлемые условия помогает подобрать компьютерная имитация [55].
Предпринимались отдельные попытки введения поправок в кинетическое уравне-
ние [31]. Вещества, высвобождаемые в реакционную среду, должны влиять на реак-
цию исключительно в соответствии с математическим описанием, представленным
функцией зависимости.
814
Часть 7. Стабильность лекарств
Уравнение (10), в общем верно для любого параметра при условии доступности
надлежащей аппаратуры, обеспечивающей изменяющиеся условия эксперимента.
В отдельных случаях это весьма нелегкая задача. Например, насколько нам извест-
но, никто еще не сообщал о проведении кинетических экспериментов с перемен-
ным параметром давления.
7.5.3.3. Аналитические приборы
Теоретически любой прибор, применяемый для отслеживания хода реакции в тра-
диционных кинетических экспериментах с постоянными параметрами, можно ис-
пользовать в ЕРаТС-экспериментах. Очевидно, что в некоторых случаях применение
традиционных методов может оказаться проблематичным. Например, довольно
легко менять температуру внутри ячейки спектрофотометра для УФ и видимыхл-
бластей; в то же время изменение концентрации веществ внутри трубки спектро-
метра для ядерно-магнитного резонанса (ЯМР) может оказаться весьма затрудни-
тельным.
Если вместо концентраций реагирующих веществ изменяется параметр физи-
ческого свойства вещества, возникает ряд последствий. Во-первых, в то время как
уравнения (2), (3) и (4), используемые в СРаА'-экспериментах, изменяются в соот-
ветствии с уравнениями
= ^№,Par2,...)(X-XJ; (18)
^-^=а0-^)ехр(-^/); (19)
1п(Х-Х„)=^+1п(Х0-Л„). (20)
уравнение (10) и его интегральная форма изменяется в соответствии с уравне-
ниями:
= <21>
Л.=(А.-Х„)ехр -\коЬз[Раг^]<И +Х„, (22)
где X — физическая величина, связанная с соединениями, участвующими в реакции
в момент времени /; Хо и — ее значения соответственно в начале и конце реакции.
Параметр X может представлять собой поглощение, электропроводность, опти-
ческое вращение, площадь ЯМР-пика и т. д.
Чем выше прецизионность прибора и чем больше точек в профиле на едини-
цу времени, тем лучше результат подгонки экспериментальных данных. Поэтому
весьма предпочтительны приборы с малой погрешностью измерения, соединяемые
с компьютером для автоматической и непрерывной регистрации данных. Спектро-
фотометр для УФ и видимой областей является, несомненно, самым распростра-
ненным прибором в экспериментах по изучению химической кинетики. Он имеет
хорошую чувствительность и надлежащим образом контролирует температуру. Он
соединен с компьютером (или может быть без труда подсоединен к компьютеру)
и установлен практически в любой лаборатории. Поглощение очень мало зависит
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...
815
от температуры, поэтому в применяемом диапазоне температур этим изменением
можно пренебречь при проведении ЕТК-экспериментов.
В ходе VCK-экс пери ментов обычная 10-миллиметровая кювета может оказаться
непригодной в качестве реакционного сосуда; в этом случае можно использовать
внешний реактор, в котором параметры поглощения можно считывать с помощью
сенсорного датчика для УФ и видимой областей или воспользоваться проточной
ячейкой. Проблема может возникнуть при добавлении реагента, поглощающего
в том же диапазоне, что и вещества, вступающие в реакцию. Если этого нельзя из-
бежать, необходимо провести коррекцию кинетического профиля с помощью ска-
нирования контрольного раствора [53].
К сожалению, не все молекулы поглощают излучение, поэтому приходится ис-
пользовать другие физические свойства и другие аналитические приборы.
Для реакций, сопровождающихся изменением оптического вращения, хорошим
решением является выбор в пользу поляриметра. Он обладает высокой чувствитель-
ностью, и его можно подключить к компьютеру для автоматической регистрации
аналитических данных. Легко осуществимы изменение и контроль температуры [49].
Влияние температуры на оптическое вращение невелико. О ЕСАГ-экспериментах
с использованием подобной техники сообщений не поступало, однако их можно
осуществить, например, с помощью внешнего реактора, соединенного с проточной
ячейкой.
С успехом применяется инфракрасная [42, 46], флуориметрическая [45, 51]
и кондуктометрическая [16, 34, 40] аппаратура. Часто в фармацевтических иссле-
дованиях как для аналитических, так и для кинетических испытаний применяются
методы газожидкостной хроматографии (ГЖХ) и высокоэффективной жидкостной
хроматографии (ВЭЖХ). К сожалению, они не идеальны для ЕРаК-экспериментов.
Полученный кинетический профиль строится по небольшому числу точек (для
каждой точки требуется времязатратный хроматографический анализ), и подгонка
к модели может привести к большой погрешности в оценке членов уравнения. Все
же поступают сообщения о подобных экспериментах с приемлемыми результатами
[38, 39,43, 50]. Если требуется большое число рутинных измерений, можно автома-
тизировать анализ [38] и воспользоваться соответствующим программным обеспе-
чением для быстрой компьютерной обработки полученных хроматограмм.
7.5.3.4. Программное обеспечение для обработки экспериментальных
данных
Когда ЕРаАГ-профиль построен и введен в компьютер, для быстрой обработки дан-
ных необходимо соответствующее программное обеспечение. В то же время в недав-
нем прошлом обработка кинетических данных, полученных при постоянной темпе-
ратуре, была не сложна даже при отсутствии компьютеров. В ЕРаК-экспериментах
применение компьютеров критически важно из-за сложности математической мо-
дели, которая не требует времязатратных расчетов, и с точки зрения достоверности
полученных результатов. Для подгонки экспериментальных данных к уравнению
(21) или (22), конкретная форма которых зависит от исследуемого параметра и ис-
пользуемого аналитического прибора, необходима разработка соответствующих ал-
горитмов. Кроме того, алгоритмы необходимы для оценки производной и иногда
816
Часть 7. Стабильность лекарств
интеграла (например, в уравнении (11), невозможно выразить интеграл в элемен-
тарных функциях, поэтому нужно рассчитать его численное значение). Для кустар-
ных персонализированных программ можно использовать любой язык, однако для
облегчения задачи в продаже имеются прикладные программы. Примером является
программа MicroMath SCIENTIST, упомянутая при описании моделирования с це-
лью подбора экспериментальных условий. Для подгонки полученных данных ис-
пользуют те же модели, что и для моделирования профилей, с той разницей, что
члены выражения, заданные при моделировании, в данном случае являются вход-
ными величинами, которые предстоит оптимизировать. SCIENTIST использует ме-
тод оптимизации Пауэлла в модификации Марквардта для подгонки данных и (по
умолчанию) пакет ряда методов (общее название — Episode) для решения диффе-
ренциальных уравнений [57].
Модулирующая функция известна всегда, поскольку значение параметра вво-
дится в программу и/или контролируется сенсором. Функция зависимости иногда
известна, иногда нет. Например, для параметра температуры функция зависимости
известна всегда (уравнение Аррениуса, уравнение Эйринга). Для параметра концен-
трации функция зависимости может быть исследована, а механизм реакции пред-
полагаться. В данном случае задача состоит в количественном исследовании ана-
логичных субстратов. Параметр концентрации может быть неизвестен при новых
исследованиях. В этих случаях уравнение (21) можно записать в виде:
,23>
Форма записи указывает на то, что основной необходимый расчет требуется для
оценки производной кинетического профиля. Отношение производной к А — А^,
т. е. к значению в точке профиля минус значение параметра в конце реакции, по-
зволяет получить значение коbs в каждой точке.
Далее будет представлено несколько примеров, имеющих отношение к фарма-
цевтическим системам.
7.5.4. Примеры кинетических экспериментов с переменными
параметрами
7.5.4.1. Кинетические эксперименты с переменным параметром
температуры
На рис. 7 представлен кинетический профиль, полученный в условиях УТЖ'-экспе-
римента, изучающего рацемизацию (—) адреналина в водном растворе, в кислой
среде [49].
Профиль получен поляриметрическим методом путем отслеживания параметра
оптического вращения а. Использовали модулирующую функцию Т= Тв + yt, где
Тй = 309,6 К, а у = 0,001694 К/с, надлежащую температуру обеспечивали с помощью
циркулирующей воды, поступающей из бани с устройством для программирования
температуры. Температуру считывали в поляриметрической ячейке с помощью пла-
тинового резистора. Результаты измерения температуры и поглощения автоматиче-
ски вводили в компьютер, соединенный с прибором. Типичная сигмовидная форма
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...
817
Рис. 7. Изменение оптического вращения (сплошная линия) в процессе рацемизации (-)
адреналина в водном растворе (1 М HCI), который протекает в условиях вариации параметра
температуры. М: Г(К) = 309,6 + 0,001694f (пунктирная линия)
профиля описывается уравнением г = kobs[ Д/)] С, т. е. скорость реакции определя-
ется произведением двух членов &о(и[ ДО], которое всегда возрастает с повышением
температуры, и величиной С, которая постоянно уменьшается в процессе реакции.
Поэтому реакция ускоряется в первой ее фазе, однако после точки перегиба сниже-
ние концентрации С становится преобладающим и реакция замедляется до нулевой
скорости, что означает ее окончание (рис. 8).
Прямая подгонка этого профиля к уравнению (24), где а0, ат, AS* и АН* —
параметры, которые следует оптимизировать, дала параметры активации
(AS* = (—27 ± 1) Дж/К моль, АН* = (95 ± 1) кДж/моль, /?2 = 0,99999). Их значения
идентичны значениям, полученным в ходе пяти традиционных VTK-экспериментов:
Рис. 8. Изменение скорости реакции в процессе рацемизации; профиль построен по методу
Савицкого-Голея [58] путем получения производных VTK-профиля, представленного на рис. 7
818
Часть 7. Стабильность лекарств
а = (а0-аво)ехр
Г к ГЛЛ’*Тгх^ ч
г 1ехр ДОо+г);
I *i L л Jo
exp
АН*
. Л+а„.
7?(r0 + yt)J J
(24)
На рис. 9 представлен профиль, полученный по результатам другого VTK-
эксперимента, выполняемого с помощью спектрофотометрического метода [45].
Изучали кинетику реакции гидролиза аспирина [12,13, 59, 60] при pH 4,50. Тем-
пературу в ячейке регулировали с помощью температурного программатора Пель-
тье и измеряли платиновым резистором, встроенным в фотометрическую ячейку.
Адекватное перемешивание обеспечивали при помощи магнитной мешалки. Мно-
жество точек данных было введено в компьютер, соединенный с аналитическим
прибором; проведена дифференциальная и интегральная обработка данных. Зна-
чения параметров активации согласовывались друг с другом и со значениями, по-
лученными в ходе сравнительных кинетических экспериментов с постоянным па-
раметром температуры, выполняемых в тех же условиях (AS* = —115 ± 1 Дж/К моль,
АН* = (69 ± 1) кДж/моль, R1 = 0,99999).
Сигмовидный профиль на рис. 10 описывает реакцию гидролиза аспирина, про-
текающую при pH 7,00 в условиях варьирования параметра температуры.
В этом случае в качестве аналитического инструмента использовали флуориметр,
облучающий салициловую кислоту на 310 нм и записывающий флуоресцентный
сигнал на 404 нм [45, 61]. Баня с программированием температуры обеспечивала
линейное повышение температуры (То = 323,05 К, а = 1,63110-3 К/с) в термоста-
тируемой флуориметрической ячейке. Температуру внутри ячейки контролирова-
ли и регистрировали и вводили в компьютер вместе со значениями интенсивности
флуоресценции.
Флуориметр является более чувствительным прибором в сравнении со спектро-
фотометром, что позволяет работать с очень малыми концентрациями субстрата.
Это может оказаться удобным на первых этапах фармацевтических испытаний.
Рис. 9. V7K-профиль (сплошная линия), полученная спектрофотометрическим методом
(X = 298,5 нм) для гидролиза аспирина в воде (pH=4,50). М: Т(К) = 304,36 + 3,647-10 4t (пунктирная линия)
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...
819
Рис. 10. Изменение интенсивности флуоресценции (сплошная линия; 404 нм) в процессе
гидролиза аспирина (pH 7,00) в условиях кинетического эксперимента с переменным параметром
температуры. М: Т(К) = 323,05 + 1,631-10^ (пунктирная линия)
К сожалению, зависимость интенсивности флуоресценции от температуры неиз-
бежна. Холостое сканирование в конце реакции позволяет получить независимый от
температуры сигнал [45], полезный при обработке результатов РТХ-эксперимента.
Результаты (AS* = (—109 ± 1) Дж/Кмоль, АН* = (71 ± 1) кДж/моль, 7?2 = 0,99999) со-
ответствовали результатам, полученным спектрофотометрическим методом в усло-
виях постоянной температуры и VTK
С помощью поляриметров, спектрофотометров и флуориметров, соединенных
с компьютером, в процессе ИРдХ-эксперимента можно получить сотни или, при не-
обходимости, тысячи информационных точек. Это увеличивает точность подгонки
экспериментальных данных к математической модели или оценки производной ки-
нетического профиля.
На рис. 11 представлен РТ/С-профиль, полученный с помощью ВЭЖХ исследо-
вания гидролиза аспирина при pH 7,00 при линейном повышении температуры, где
Го = 323,2 К и а = 1,631-10 3 К/с |50].
Эксперимент проводили в закрытом реакционном сосуде, погруженном в баню
с программируемым изменением температуры. Через определенные интервалы
времени отбирали пробы реакционной смеси и вводили их в колонку для жидкост-
ной хроматографии (ZQ. Число полученных точек не было велико из-за длитель-
ности хроматографического разделения Тем не менее, подгонка к соответствую-
щей математической модели позволила получить значения параметров активации,
близкие к параметрам, полученным в спектрофотометрическом эксперименте,
хотя и с большей статистической погрешностью (AS* = (—102 ± 8) Дж/К моль,
АН* = (73 ± 2) кДж/моль, Я2 = 0,9997).
7.Б.4.2. Кинетические эксперименты с переменным параметром
концентрации
Зависимость от концентраций веществ, присутствующих в реакционной среде, не-
сомненно, является важнейшим фактором с точки зрения изучения механизма ре-
820
Часть 7. Стабильность лекарств
t, с
Рис 11. УТК-профиль, построенный по значениям площадей хроматографических пиков
(РА) салициловой кислоты (круглые маркеры), образующейся при гидролизе аспирина при
pH = 7,00. М: Т(К) = 323,2 + 1,631 х 10-3t (пунктирная линия). Сплошная линия — соответствующая
теоретическая кривая
акции. Различные вещества могут влиять на реакцию, принимая в ней участие в ка-
честве реагента или катализатора, или же просто нарушая определенным образом
физико-химические характеристики реакционной среды, например, нуклеофил
при нуклеофильном замещении, ион металла в реакции, где он является катализа-
тором, или соль, изменяющая ионную силу, или молекулы растворителя, изменяю-
щие диэлектрическую постоянную. В первом случае «сердцевиной» закона, которо-
му подчиняется скорость реакции, является функция зависимости, обозначающая
вещества, участвующие в реакции на этапе, определяющем скорость, и помогающая
понять механизм их взаимодействия [1, 3,7,8]. Поэтому на поиск формы и числен-
ных значений членов уравнения следует обратить серьезное внимание.
Изучение нуклеофильного замещения в квадратно-плоскостном комплексоне
транс-[Pt(PEt3)2Cl2] — веществе, аналогичном цисплатину и другим соединениям,
широко используемым в качестве противоопухолевых агентов [53,62—65], проводи-
ли спектрофотометрическим методом при изменении концентрации нуклеофила со
временем. Реакционный сосуд помещали в водяной термостат, прибавление опре-
деленного количества концентрированного раствора нуклеофила (тиомочевины,
йодида, бромида, тиоцианида) проводилось с помощью автоматической бюретки;
сигнал поглощения считывали при помощи оптоволоконной ячейки; адекватное
перемешивание обеспечивалось при помощи погружной мешалки.
Были обеспечены условия протекания реакции первого порядка. В классиче-
ских экспериментах с постоянным параметром концентрации это означает, что
концентрация реагента достаточно высока в сравнении с концентрацией субстра-
та, и расходом его в процессе реакции можно пренебречь. В этом случае для обра-
ботки данных можно воспользоваться простым уравнением кинетики первого по-
рядка. В ИСА'-экспериментах эти условия формулируют по-другому: концентрация
веществ должна изменяться во времени так, чтобы отклонением от модулирующей
функции, вызванным расходом реагента, можно было пренебречь. Концентриро-
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...821
ванные растворы, в которых образуется необходимый избыток реагента в реакци-
онной среде, не трудно приготовить. В особых случаях, там, где это невозможно,
расход веществ может быть учтен в выражении для модулирующей функции [53].
На рис. 12 представлен кинетический профиль с переменным параметром кон-
центрации для реакции платинового комплекса с SCN'. Модулирующая функция
представляет собой выражение [SCN-] = а/, где а = 0,048 М/с.
Даже в этом случае наблюдается ускорение реакции в первой части кинетическо-
го профиля, однако это вызвано увеличением концентрации нуклеофила. Функция
зависимости для данной реакции описывается выражением
^ = ^+*y[Y], (25)
где ks представляет собой константу сольволиза; к^ — константу скорости реакции
при прямой атаке нуклеофила Y [62, 63]. Подгонка профиля к модели
ЯД
- = (fcs + ку аГ)(А - А.) (26)
at
дала оптимизированные значения к. и ку (соответственно, 0,92 КНс-1 и 0,351М_1с-1).
На рис. 13 представлена программа SCIENTIST, применяемая для подгонки ки-
нетического профиля, где KS, KY, АО и AI — параметры, которые необходимо опти-
мизировать.
Эксперимент, где реакцию того же субстрата в условиях переменной концентра-
ции тиомочевины контролировали кондуктометрическим методом, дал хорошие
результаты [16]. Реакция протекала в термостатируемом реакционном сосуде. Для
добавления в систему концентрированного раствора тиомочевины использовали
автоматическую бюретку. Данные по электропроводности считывали с помощью
кондуктометрической ячейки и вводили в компьютер.
Среди исследований зависимости кв1а от концентрации веществ, присутствую-
щих в реакционной среде, в фармацевтических исследованиях особое место уделя-
Рис. 12. VCK-профиль, полученный спектрофотометрическим методом (круги, выбранные точки)
и теоретическая кривая (сплошная линия) (280 нм) для реакции транс-[РЦРЕу/ЗЦ + 2SCN’ транс-
[Pt(PEt3)2(SCN)J + СГ в метаноле. М: [SCN-] = 0,048гМ (пунктирная линия); Т= 303,2 К
822
Часть 7. Стабильность лекарств
//VCK_SIM
IndVars: t
DepVars: A, V, K,Y
Params: KS, KY, AO. Al
a = g*M/VO
- > Y= a*t
- > К = KS + KY* Y
A’ = -K*(A-AI)
V = -A'
// Parameter values
KS = 0.0001
KY = 0.3
- > g = 0.59
- > M = 0.63
- > VO = 20000
AO = 0.3
Al = 0.7
// Initial conditions
t=0
A = A0
Рис. 13. Программа SCIENTIST, применяемая для обработки профиля VCK на рис. 11. Первые
три стрелки указывают соответственно на модулирующую функцию, функцию зависимости
и уравнение кинетики первого порядка. Три стрелки внизу указывают на экспериментальные
параметры д, Ми 1/0.
ется профилям pH — скорость реакции, то есть зависимости [Н+] и/или [ОН-]. Они
несут обширную информацию о механизмах реакций и способах решения проблем,
связанных со стабильностью [1, 3, 4]. Зачастую такие исследования требуют про-
ведения множества кинетических экспериментов, поскольку диапазон колебаний
концентрации иона водорода охватывает 14 порядков. Для построения профиля
pH — скорость реакции для аспирина, например, требуется 50 кинетических экс-
периментов. С учетом различных температур, сходных молекул и других различных
эффектов изучение гомологических серий может быть сильно затруднено.
В ходе КСАГ-экспериментов единственного экспериментального цикла может
быть достаточно для построения профиля pH — скорость реакции для реакцион-
ного субстрата.
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств... 823
t, с
Рис. 14. Изменения поглощения (сплошная линия) в ходе VpHKэксперимента по изучению реакции
гидролиза аспирина при Т = 342,5 К, >. = 298,5 нм. Пунктирная линия показывает колебания pH.
М: не известна по результатам анализа
На рис. 14 представлен кинетический профиль для переменного pH ( VpHK), по-
лученный спектрофометрическим методом для реакции гидролиза аспирина с pH,
варьирующимся в диапазоне 2—10, при Т= 342,5 К.
Условия переменной концентрации были реализованы путем добавления кон-
центрированного раствора NaOH (0,6 М) в термостатируемый реакционный сосуд,
содержащий водный раствор ацетилсалициловой кислоты и буфер, состоящий из
уксусной кислоты (0,01 М), фосфорной кислоты (0,01 М) и борной кислоты (0,01 М).
В этом случае подъем pH был почти линейным. Сигнал поглощения считывали при
помощи оптоволоконной ячейки и вводили в компьютер. Контроль pH проводили
при помощи pH сенсора, соединенного с компьютером.
Данный профиль более сложен по сравнению с ранее встречавшимися. Причи-
ной является то, что в уравнении скорости реакции, выраженном через произве-
дение kobs и концентрацию субстрата, £/н[(рН(/)] варьируется в зависимости от pH
необычным образом из-за специфической формы профиля pH-скорость реакции.
На рис. 15 представлена производная профиля pH-скорость реакции, полученная
методом Савицкого—Голея [58]. График показывает колебания скорости реакции
в ходе VpHKэксперимента. В первой части наблюдается ускорение, вызванное уве-
личением kobs с повышением pH, с последующим замедлением, связанным со стаби-
лизацией kobs и постоянным уменьшением концентрации субстрата. Затем скорость
реакции возрастает с увеличением значений k^vi вновь падает при приближении
концентрации к нулю.
Отношение производной профиля к А — ^позволяет получить, в соответствии
с уравнением (27), полный профиль pH-скорость реакции за один прогон (рис. 15).
~d{A~AJ = {*,JI //+1 «>]} 04 - A J. (27)
at
Для столь больших диапазонов pH функция зависимости может иметь весьма
сложный вид. Например, в случае аспирина при переменном параметре pH на-
блюдали четыре различных механизма реакции, а выражение для общей константы
824
Часть 7. Стабильность лекарств
Рис. 15. Изменение производной профиля VpHK, представленного на рис. 14 (сплошная линия)
и профиля /cobs[pH(f)J уравнения (27) (пунктирная линия)
скорости реакции включает несколько членов [3]. Тем не менее, после построения
профиля pH — скорость реакции и определения механизма реакции функцию за-
висимости можно ввести в уравнение (27) и провести полную подгонку для оценки
значений элементарных констант.
7.5.4.3. Кинетические эксперименты с переменным параметром ионной
силы
На рис. 16 представлен кинетический профиль для эксперимента с переменным
параметром ионной силы (КЖ) для реакции гидролиза индометацина [54, 66, 67]
при 0,005 MNaOH и Т= (299,0 ± 0,1) К при переменной ионной силе раствора
I = 1й + а/, где /0= 0,005 М по концентрации NaOH, а а = 1,77-10-5 М/с и рассчи-
тано по уравнению а = gM/V0. Экспериментальная аппаратура была аналогичной
применяемой в ходе Р^ТЖ-эксперимента. В этом случае концентрированный рас-
Рис. 16. Изменение параметра поглощения (круги, выбранные точки) входе VW-эксперимента
по изучению взаимодействия индометацина с NaOH 0,005 М и Т = (299,0 ± 0,1) К. Пунктирная
линия показывает увеличение ионной силы раствора, сплошная — соответствует теоретической
модели
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств... 825
твор LiCl (3 М) высвобождался в реакционный сосуд при помощи автоматической
бюретки. Были получены сотни экспериментальных точек по результатам анализа
поглощения. Подгонка экспериментальных данных к математической модели (28)
проводилась при помощи программы MicroMath SCIENTIST с параметрами Ло, Ат,
к0 и ZAZB, которые необходимо оптимизировать. Подгонка получилась отличной
(R2 = 0,99999) и результаты хорошо согласовывались с результатами, полученными
традиционным путем [Ро = (1,02+0,04)-10-4 с-1; ZKZB = 1,01 ± 0,04]:
НА
10 (28)
°'>S V 00' V z
Дифференциальный метод был также применен для обработки эксперименталь-
ных данных. Результаты хорошо согласуются.
7.5.5. Заключение
Стабильность является важнейшей характеристикой лекарственного препарата.
Быстрый скрининг новых лекарственных веществ и подробные ускоренные иссле-
дования отобранных веществ-кандидатов помогают избежать задержек и связанно-
го с ними удорожания первой части фармацевтических испытаний.
Метод VPaK представляет собой новый эффективный способ накопления ки-
нетических данных. Это новый взгляд на кинетические эксперименты. Вместо
получения единого значения наблюдаемой константы скорости в ходе одного экс-
перимента можно получить полную картину зависимости константы скорости от
физического параметра. Как правило, при изучении механизма реакции исследу-
ют характер взаимодействия реагентов на этапе, определяющем скорость реакции.
Природа реагентов и продукты реакции хорошо известны благодаря доступности
очень качественных аналитических приборов. Кинетические эксперименты пред-
ставляют собой рутинные операции; тем не менее, они имеют фундаментальное
значение, поскольку знание кинетики — единственный способ получить информа-
цию об этом аспекте реакционной способности основного вещества.
Без опасения потерять слишком много времени можно получить полную пано-
рамную картину химического поведения длинных рядов соединений-гомологов.
Прямые исследования механизмов реакций можно выполнять с помощью доступ-
ных приборов и программного обеспечения. Кроме того, кинетические данные
получают на одном образце, что позволяет избежать неоднородности, характери-
зующей традиционные СРаК-экспериментальные данные, потери времени на под-
готовку новых образцов и зачастую, что бывает немаловажно, использования мень-
шего количества соединения.
Литература
1. Carstensen J. Т. (2000), Solution Kinetics; Kinetic pH profiles; Oxidation in solution; Catalysis,
Complexation, and Photolysis, in Carstensen J. T., Rhodes С. T., Eds., Drug Stability, Principle and
Practice, 3rd ed., Marcel Dekker, New York, Chapters 2-5, pp. 19-143.
2. Connors K. A., Amidon G. L., Stella V. J. (1986), Chemical Stability of Pharmaceuticals: A Handbook
for Pharmacists, 2nd ed., Wiley-Interscience, New York.
826 Часть 7. Стабильность лекарств
3. Loudon G. М. (1991), Mechanistic interpretation ofpH-rate profiles, J. Chem. Ed., 68, 973-984.
4. Jenks W. P. (1969), Catalysis in Chemistry and Enzymology, McGraw Hill, New York.
5. Kerns E. H. (2001), High throughput physicochemical profiling for drug discovery, J. Pharm. Sci., 90,
1838-1858.
6. Avdeef A., Testa B. (2002), Physicochemical profiling in drug reserch: A brief survey of the state of
the art of experimental techniques, Cell. Mol. Life Sci., 59,1681—1689.
7. Moore J. W., Pearson R. G. (1981), Kinetics and Mechanism, Wiley, New York.
8. Wilkins R. G. (1991), Kinetics and Mechanism of Reactions of Transition Metal Complexes, VCH,
Weinheim.
9. Kresge A. J. (1987), Unusual reactivity of prostacyclin: Rational drug design through physical organic
chemistry, Acc. Chem. Res., 20, 364-370.
10. Page M. I., Webster P. (1990), The hydrolysis of azetidinyl amidinium salts. Part 1. The unimportance
of strain release in the four-membered ring, J. Chem. Soc. Perkin Trans., 2, 805-811.
11. Lajis N. H., Noor H. M., Khan M. N. (1995), Kinetic and mechanism of the alkaline hydrolysis of
securinine, J. Pharm. Sci., 84, 126-130.
12. Edwards L. J. (1950), The hydrolysis of aspirin. A determination of the thermodynamic dissociation
constant and a study of the reaction kinetics by ultraviolet spectrophotometry, Trans. Faraday Soc.,
46, 723-735.
13. Edwards L. J. (1952), The hydrolysis of aspirin. Part 2, Trans. Faraday Soc., 48, 696-699.
14. Romeo R., Alibrandi G. (1997), Structure-reactivity correlations for the dissociative uncatalyzed
isomerization of monoalkylbis(phosphine)platinum(II) solvento complexes, Inorg. Chem., 36, 4822—
4830.
15. Carstensen J. T. (1970), Kinetic salt effect in pharmaceutical investigation, J. Pharm. Sci., 59, 1140-
1143.
16. Koryta J., Dvorak J., Kavan L. (1993), Principles of Electrochemistry, Wiley,Chichester.
17. Alibrandi G. (1994), Variable-concentration kinetics, J. Chem. Soc. Chem. Commu., 23, 2709—2710.
18. Koch E. (1977), Non-Isothermal Reaction Analysis, Academic, London.
19. Brown M. E, Phillpotts C. A. R. (1978), Non-isothermal kinetics, J. Chem. E., 55, 556-560.
20. Kissinger H. E. (1957), Reaction kinetics in differential thermal analysis, Anal. Chem.,29, 1702—
1706.
21. Flynn J. H. (1969), in Schwenker R. V., Gam P. D., Eds., Thermal Analysis, Vol. 2, Academic, New
York, p. 1111.
22. Daniels T. (1973), Thermal Analysis, Kogan Page, London.
23. Dollimore D. (1996), Thermal analysis, Anal. Chem., 68, 63R—71R.
24. Ozawa T. (2000), Thermal analysis — Review and prospect, Thermoch. Acta, 335, 35—42.
25. Glass B. D., Vbvak Cs., Brown M. E. (2004), The thermal and photostability of solid pharmaceuticals.
A review, J. Therm. Anal. Calorim.,11, 1013—1036.
26. Rogers A. R. (1963), An accelerated stage test with programmed temperature rise, J. Pharm.
Pharmacol., 15, 101T-105T.
27. Ahlberg P., Wold S. (1970), Evaluation of activation parameters for a first order reaction from one ki-
netic experiment. Theory, numerical methods and computer program, Acta Chem. Scand., 24,618-632.
28. Ahlberg P. (1970), Determination of activation parameters in one kinetic experiment. Acta Chem.
Scand., 24, 1883-1893.
29. Madsen B. W., Anderson R. A. (1974), Integral approach to nonisothermal estimation of activation
energy, J. Pharm. Sci., 63, 777—781.
30. Edel B., Baltzer M. O. (1980), Nonisothermal kinetics with programmed temperature steps, J. Pharm.
Sci., 69, 287-290.
31. Tucker I. G., Owen W. R. (1982), High information kinetic studies: Non-isothermal programmed acid
concentration kinetics, Int. J. Pharm., 10, 323—337.
Глава 7.5. Альтернативные ускоренные методы изучения стабильности лекарств...827
32. Gonzalez J. L., Salvador F. (1982), Kinetics of reactions in solution: Method for the treatment of data
from non-isothermal chemical kinetic experiments, React. Kinet. Catal. Lett., 21 (1-2), 167—171.
33. Gonzalez J. L., Salvador F. (1984), Comparative studies of non-isothermal methods in linear and non-
linear temperature variation, React. Kinet. Catal. Lett., 25 (1—2), 125-130.
34. Mason T. J., Lorimer J. P. (1983), A method for the determination of the activation energy for a reac-
tion from a single kinetic run, Comp. Chem., 7 (4), 159-163.
35. Li Wan Po A., Elias A. N., Irwin W. J. (1983), Non-isothermal and non-isopH kinetics in formulation
studies, Acta Pharm. Suec., 20,277-286.
36. Ortiz Uribe M. L, Romero Salvador A., Irabien Gulias A. (1985), Kinetic analysis for liquid-phase re-
actions from programmed temperature data. I. Simple analysis of potential kinetic laws, Thermochem.
Acta, 94, 323-331.
37. Kipp J. E. (1985), Non-isothermal kinetics - Comparison of two methods of data treatment, Int. J.
Pharm., 26, 339-354.
38. Kipp J. E., Jensen M. M., Kronholm K., McHalsky M. (1986), Automated liquid chromatography for
non-isothermal kinetic studies, Int. J. Pharm., 34, 1—8.
39. Bunce N. J., Forber C. L., Mclnnes C. (1988), Single-step methods for calculating activation param-
eters from raw kinetic data, J. Chem. Soc. Perkin Trans., II, 363—368.
40. Schoenemann E., Hahn H., Bracht A. (1991), Determination of kinetic parameters from non-isother-
mal conductivity measurements by an integral method, Thermochim. Acta, 185 (1), 171-176.
41. Alibrandi G. (1994), Non-isothermal spectrophotometric kinetics applied to inorganic reactions,
Inorg. Chim. Acta, 221, 31—34.
42. Zhang S., Brown T. L. (1995), Application of non-isothermal approach to the kinetics of organome-
tallic reactions: The substitution of (h5-pentamethylcyclopentadienyl) dicarbonylrhodium(I), Inorg.
Chim. Acta, 240, 427^133.
43. Junnarkar G. H., Stavchansky S. (1995), Isothermal and nonisothermal decomposition of famotidine
in aqueous-solution, Pharm. Res., 12 (4), 599-604.
44. Lee M. L., Stavchansky S. (1995), Isothermal and nonisothermal decomposition of thymopentin and
its analogs in aqueous-solution, Pharm. Res., 15 (11), 599-604.
45. Alibrandi G., Micali M., Trusso S., Villari A. (1996), Hydrolysis of aspirin studied by spectrophoto-
metric and fl uorometric variable-temperature kinetics, J. Pharm. Sci., 85, 1105-1108.
46. Maeder M., Molloy K. J., Schumacher M. M. (1997), Analysis of non-isothermal kinetic measure-
ments, Anal. Chim. Acta, 337, 73—81.
47. Hodgson S. C., Ngeh L. N., Orbell J. D., Bigger S. W. (1998), Astudent experiment in non-isothermal
chemical kinetics, J. Chem. Ed., 75 (9), 1150-1153.
48. Ficarra R., Villari A., Micali N., Tommasini S., Calabn M. L., Di Bella M. R., Melardi, S., Agresta
M. F., Coppolino S., Stancanelli R. (1999), Stability study of piroxicam and cinnoxicam in solid phar-
maceuticals, J. Pharm. Biomed. Anal., 20, 283—288.
49. Alibrandi G., Coppolino S., D’Aliberti S., Ficarra P., Micali N., Villari A. (2002), Temperature-rate
profiles by polarimetric variable-temperature kinetic experiments to study racemization reactions,
J. Pharm. Biomed. Anal., 29, 1025—1029.
50. Alibrandi G., Coppolino S., D’Aliberti S., Ficarra R., Micali N., Villari A. (2003), Fast drug stabil-
ity determination by LC variable-arameter kinetic experiments, J. Pharm. Biomed. Anal., 32, 1073—
1079.
51. Mood A. R. H., Haghighi S., Gholami M. R. (2004), Fluorometric variable-temperature kinetic inves-
tigations of the transesterifi cation reaction of procaine with aliphatic alcohols, J. Pharm. Pharm. Sci.,
7 (1), 88-91.
52. Alibrandi G., Coppolino S., Micali N., Villari A. (2001), Variable-pH kinetics: An easy determination
of pH-rate profile, J. Pharm. Sci., 90, 270-274.
53. Alibrandi G., D’Aliberti S., Tresoldi G. (2003), Spectrophotometric variable-concentration kinetic
experiments applied to inorganic reactions, Int. J. Chem. Kinet., 35,497-502.
828 Часть 7. Стабильность лекарств
54. Alibrandi G., Coppolino S., D’Aliberti S., Ficarra P., Micali N., Villari A. (2003), Variable-ionic
strength kinetic experiments to study drug stability, J. Pharm. Sci., 92, 1730-1733.
55. Alibrandi G., D’Aliberti S., Pedicini R. (2001), Computer simulation of variable-parameter kinetic
experiments, Chem. Ed., 6, 185—191.
56. MicroMath Scientific Software, Salt Lake City, UT.
57. Press W. H., Flannery В. P., Teukolsky S. A., Vetterling W. T. (1986), Numerical Recipes, Cambridge
University Press, Cambridge.
58. Savitzky A., Golay M. J. E. (1964), Smoothing and differentiation of data by simplifi ed least squares
procedures, Anal. Chem., 36, 1627-1639.
59. Garrett E. R. (1957), The kinetics of solvolysis of acyl esters of salycilic acid, J. Am. Chem. Soc., 79,
3401-3408.
60. Fersht A. R., Kirby A. J. (1967), The hydrolysis of aspirin. Intramolecular general base catalysis of
ester hydrolysis, J. Am. Chem. Soc., 89, 4857—4863.
61. Miles C. 1., Schenk G. H. (1970), Fluorescence of acetylsalicylic acid in solution and its measurement
in presence of salicylic acid, Anal. Chem., 42, 656-659.
62. Tobe M. L. (1987), Substitution reactions, in Wilkinson G., Gillard R. D., McCleverty J. A., Eds.,
Comprehensive Coordination Chemistry, Vol. 1, Pergamon, Oxford, Chapter 7.1, pp 281—329.
63. Belluco U., Cattalini L., Basolo F., Pearson R. G., Turco A. (1965), Nucleophilic constants and sub-
strate discrimination factors for substitution reactions of platinum(II) complexes, J. Am. Chem. Soc.,
87, 241-246.
64. Sherman S. E., Lippard S. J. (1987), Structure aspect of platinum anticancer drug interactions with
DNA, Chem. Rev., 87, 1153-1181.
65. Nicolini M.,Ed.(1988), Platinum and Other Metal Coordination Compounds in Cancer Chemiotherapy,
Martinus Nijoff Publishing, Boston, MA.
66. Hajratwala B. R., Dawson J. E. (1977), Kinetics of indomethacin degradation I: Presence of alkali, J.
Pharm. Sci., 66, 27-29.
67. Cipiciani A., Ebert C., Linda P., Rubessa F., Savelli G. (1983), Kinetics and mechanism of the basic
hydrolysis of indomethacin and related compounds: A reevaluation, J. Pharm. Sci., 72, 1075-1076.
Часть 8
ВАЛИДАЦИЯ
Глава 8.1. Валидация аналитических методик:
принципы и подходы
Чунг Чжон Чан
Azopharma Contract Pharmaceutical Services (Мирамар, Флорида)
8.1.1. Введение
Валидация аналитической методики представляет собой процесс, в котором с по-
мощью лабораторных исследований устанавливается, что характеристики методики
соответствуют требованиям предполагаемого применения. Все аналитические ме-
тодики, предназначенные для анализа любых клинических образцов, должны быть
провалидированы. Валидация аналитических методик — важная, но требующая
больших затрат времени процедура для большинства аналитических испытательных
лабораторий. Поэтому важно детально понимать требования валидации методики
и возможные варианты оптимального использования аналитических ресурсов ис-
пытательной лаборатории.
8.1.2. Цели валидации аналитических методик
Существует множество причин, обосновывающих необходимость валидации ана-
литических методик. К ним относятся требования регуляторных органов, научного
подхода и контроля качества. В Своде федеральных нормативных актов США ( CFR,
Code of Federal Regulations') в пункте 311.165с четко указано: «Правильность, чувстви-
тельность, специфичность и воспроизводимость используемых фирмой испыта-
тельных методик должны быть определены и задокументированы». Конечно, как
ученые, мы хотели бы применить надлежащий научный подход для демонстрации
того, что используемая аналитическая методика обладает правильностью, чувстви-
тельностью, специфичностью и воспроизводимостью. Наконец, руководство отдела
контроля качества хотело бы гарантировать, что аналитические методики, исполь-
зуемые отделом для выпуска продукции, должным образом провалидированы для
предполагаемого использования с целью обеспечения безопасности продукта для
конечного потребителя — человека.
8.1.3. Действующие правила надлежащей производственной
практики в двадцать первом веке
Основой действующих правил надлежащей производственной практики {cGMP)
в двадцать первом веке и современных систем качества является принцип «качество
должно быть встроено в продукт, нельзя гарантировать качество продукта только
конечной проверкой». С аналитической точки зрения это означает, что аналитические
методики, используемые для проверки продукта, должны иметь встроенные в них
показатели качества. Чтобы показатели качества были встроены в аналитические
методики, требуется, чтобы основные показатели качества использовались спе-
832
Часть 8. Валидация
циалистами на производстве. Эта новая парадигма требует, чтобы научный работ-
ник на производстве обладал научным и техническим пониманием производства,
знанием продукта, процесса и/или имел навыки оценки рисков для надлежащего
выполнения функций качества при валидации аналитической методики. Таким об-
разом, требуется: 1) соответствующая подготовка отраслевого научного работника
для понимания принципов валидации методики и умение выполнять ее; 2) надле-
жащая документация, понимание и обработка данных; 3) понимание влияния этой
деятельности на продукт и конечного потребителя (пациента). На руководстве ле-
жит ответственность за проверку того, что полученные во время обучения навыки
применяются в ежедневной работе.
8.1.4. Цикл аналитической методики
Деятельность по валидации аналитической методики не является однократным ис-
следованием. Это наглядно отражено и обобщено в жизненном цикле аналитиче-
ской методики на рис. 1. Аналитическая методика разрабатывается и валидируется
для использования при анализе образцов на стадии ранней разработки фармацевти-
ческой субстанции или лекарственного препарата. Поскольку процесс разработки
лекарственного препарата начинается с 1-го этапа и заканчивается организацией
серийного производства, те же стадии проходит и аналитическая методика. Окон-
чательная методика валидируется на пригодность для предполагаемого использова-
ния на лекарственном препарате с окончательным составом и технологией (то есть
заявляемом к регистрации) и переносится в лабораторию отдела контроля качества
для запуска производства препарата. Однако если имеются какие-либо изменения
в процессе производства, способные изменить аналитический профиль фармацев-
тической субстанции или лекарственного препарата, то валидированный ранее ме-
тод необходимо ревалидировать (валидировать повторно), чтобы гарантировать, что
метод все еще пригоден для анализа фармацевтической субстанции или лекарствен-
ного препарата для предполагаемого применения.
Разработка методики
Лаборатория ОКК
Рис. 1. Жизненный цикл аналитической методики
Глава 8.1. Валидация аналитических методик: принципы и подходы 833
8.1.5. Валидационные характеристики аналитической
методики
Ниже приведены стандартные рабочие аналитические характеристики, которые
следует рассматривать при валидации методик, описанных в этой главе. Каждая
валидационная характеристика подробно описана для обеспечения однозначности
интерпретации и терминологии:
• правильность;
• прецизионность:
а) повторяемость;
б) промежуточная прецизионность;
• специфичность;
• предел обнаружения;
• предел количественного определения;
• линейность;
• диапазон;
• робастность.
8.1.5.1. Правильность
Международная конференция по гармонизации (ICH, International Conference on
Harmonization) определяет правильность аналитической методики по близости зна-
чений, принятых в качестве общепринятых верных или контрольных (эталонных),
и значений, полученных в испытании. Для фармацевтической субстанции правиль-
ность может определяться путем применения аналитической методики к образцу ве-
щества известной чистоты (например, к стандартному образцу). Для лекарственного
препарата правильность определяют путем проведения аналитического испытания
на модельных смесях компонентов препарата, к которым добавляют известные ко-
личества анализируемого вещества в пределах данной методики. В документации
ICHтакже рекомендуется оценивать результаты не менее девяти определений из не
менее трех концентраций, охватывающих определенный диапазон (например, три
концентрации/три повторности).
Правильность обычно выражается как процент обнаружения при количествен-
ном определении (с использованием предлагаемой методики) известного добав-
ленного количества анализируемого вещества в образце или как разница между
средним и принятым истинным значением с учетом доверительных интервалов.
Диапазон пределов правильности должен находиться в пределах диапазона линей-
ности методики. Обычно правильность (как % обнаружения) для методики количе-
ственного определения фармацевтической субстанции ожидается около 99—102%.
Значения данных правильности методики (как % обнаружения), выходящие за эти
рамки, необходимо дополнительно исследовать соответствующим образом.
8.1.5.2. Прецизионность методики
Прецизионность аналитической методики выражает близость результатов (степень
разброса) серий измерений, полученных на множестве проб одного однородного
834 Часть 8. Валидация
образца при заданных условиях. Обычно исследуются три уровня прецизионности:
повторяемость, промежуточная прецизионность и воспроизводимость. Для просто-
го состава важно, чтобы прецизионность определялась на аутентичных однородных
образцах. Если однородные образцы отсутствуют и используются модельные образ-
цы или испытуемые растворы, то требуется обоснование.
Повторяемость. Повторяемость — мера прецизионности при одинаковых услови-
ях эксплуатации в течение короткого промежутка времени, то есть при нормальных
условиях эксплуатации аналитической методики на одном и том же оборудовании.
Этот показатель иногда называется внутрианалитической прецизионностью.
По рекомендации ICH повторяемость следует оценивать, используя результаты
как минимум девяти определений, охватывающих установленный диапазон мето-
дики (например, три концентрации/три повторности, как в испытании на правиль-
ность), или как минимум шести определений при 100%-ной концентрации1 испы-
туемого раствора. Требуется представление вычисленного стандартного отклонения,
относительного стандартного отклонения (коэффициента вариации) и доверитель-
ного интервала. Результаты определений — это результаты независимых испытаний
на количественное определение, включавших полное выполнение аналитической
методики от приготовления пробы до получения окончательного результата изме-
рения. В табл. 1 представлен пример данных, полученных при определении повто-
ряемости.
Таблица 1
Данные повторяемости
Номер пробы Процентное содержание от заявленного
1 100,6
2 102,1
3 100,5
4 99,4
5 101,4
6 101,1
Среднее значение 100,9
Процент относительного стандартного отклонения 0,90
Промежуточная прецизионность. Промежуточная прецизионность — вариабель-
ность внутри одной лаборатории. Степень, с которой должна определяться проме-
жуточная прецизионность, зависит от условий, при которых будет использоваться
методика. Стандартными определяемыми параметрами при этом являются вариа-
бельности по дням, аналитикам и оборудованию. В зависимости от размера иссле-
дования рекомендуется использовать план эксперимента. Такой план снизит число
экспериментов, которые необходимо провести. Важно отметить, что ICHразрешает
не определять промежуточную прецизионность, если доказана воспроизводимость.
Предполагается, что промежуточная прецизионность должна показывать вариа-
1 Под 100%-ной концентрацией понимается 100% от количества, указанного на этикетке
или в спецификации. — Примеч. перев.
Глава 8.1. Валидация аналитических методик: принципы и подходы
835
бельность того же порядка или меньшую, чем вариабельность при воспроизводимо-
сти. ICH рекомендует включать в отчет значения стандартного отклонения, отно-
сительного стандартного отклонения (коэффициента вариации) и доверительного
интервала.
Воспроизводимость. Воспроизводимость измеряет межлабораторную прецизион-
ность. Этот параметр рассматривается при стандартизации аналитической методи-
ки (например, при включении методики в фармакопеи и переносе методики между
разными лабораториями).
Для валидации данной характеристики следует провести одинаковые исследо-
вания в разных лабораториях, используя одинаковые однородные испытуемые об-
разцы и одинаковый план эксперимента. В случае переноса методики между лабо-
раториями возможно применение различных подходов для успешности переноса.
Наиболее распространенный метод заключается в прямом переносе методики из
исходной лаборатории в «лабораторию-преемник». Исходная лаборатория — ла-
боратория, разработавшая и провалидировавшая аналитическую методику, или ла-
боратория, которая ранее была сертифицирована на проведение методики и будет
участвовать в исследованиях по переносу методики. Лаборатория-преемник — ла-
боратория, в которую переносится аналитическая методика и которая будет уча-
ствовать в исследованиях по переносу методики. При прямом переносе методики
рекомендуется вести протокол, фиксирующий подробности экспериментов и кри-
терии приемлемости (в случае расхождения между средними значениями двух лабо-
раторий) для успешного переноса методики. В табл. 2 представлены примеры дан-
ных, полученных при переносе методики между двумя лабораториями.
Таблица 2
Результаты переноса методики между двумя лабораториями
Число повторов Средний процент
Исходная лаборатория 12 100,7
Лаборатория-преемник 4 100,2
8.1.5.З. Специфичность
ICH определяет специфичность как способность определять анализируемое веще-
ство в присутствии компонентов, наличие которых в препарате наиболее вероятно.
Во многих публикациях термины селективность и специфичность часто использу-
ются как взаимозаменяемые. Однако возникают споры по поводу использования
специфичности или селективности, а некоторые регуляторные органы, например
Международный союз теоретической и прикладной химии (ИЮПАК, International
Union of Pure and Applied Chemistry), предпочитают термин селективность, используя
термин специфичность только для полностью избирательных методик. Для фарма-
цевтического применения будет использоваться приведенное выше определение
ICH.
При испытании на подлинность компоненты со схожей родственной структу-
рой, наличие которых наиболее вероятно, должны быть отделимы друг от друга. Это
должно быть подтверждено получением положительных результатов на образцах,
содержащих анализируемое вещество (путем сравнения с известным стандартным
836 Часть 8. Валидация
образцом), наряду с отрицательными результатами на образцах, не содержащих
анализируемое вещество. Кроме того, испытание на подлинность может быть про-
ведено на веществе с близкой структурой или родственном аналиту веществе для
подтверждения отсутствия положительного результата. Выбор подобных потенци-
ально мешающих определению веществ должен основываться на строго научном
подходе с рассмотрением возможных взаимных влияний.
Специфичность количественного определения и испытаний на примеси следует
рассматривать с двух позиций:
1. Образцы примесей доступны. Специфичность методики количественного
определения устанавливается путем сравнения результатов испытаний образца,
содержащего примеси, продукты разложения или компоненты плацебо, с резуль-
татами испытаний образца без примесей, продуктов разложения или компонен-
тов плацебо. Для методики количественного определения, используемой при ис-
следовании стабильности, необходимо получить разрешенные (раздельные) пики
продуктов разложения и действующего вещества. Однако разрешение таких пиков
примесей между собой не требуется. При оценке методики испытания на примеси
определение должно проводиться при добавлении в контрольный раствор (содер-
жащий плацебо) действующего вещества или лекарственного препарата, содержа-
щих соответствующее количество примесей. Должно быть показано, что методика
обеспечивает получение индивидуальных пиков примесей и/или их разделение
с другими компонентами состава. Следует использовать репрезентативные хрома-
тограммы.
2. Образцы примесей не доступны. Специфичность методики устанавливается
путем сравнения результатов испытания образца, содержащего примеси или про-
дукты разложения, с результатами испытания другой подробно описанной или
провалидированной аналитической методики (ортогональный метод). Должны ис-
пользоваться образцы, которые хранились в соответствующих «стрессовых» усло-
виях (воздействие света, температуры и влажности, кислотный/щелочной гидролиз
и окисление). Для методики количественного определения следует сравнивать два
результата; для методики испытания на примеси следует сравнивать профили при-
месей. Оценку спектральной однородности (неоднородности) пика следует прово-
дить, используя диодно-матричный или масс-спектрометрический детектор для де-
монстрации того, что хроматографический пик анализируемого вещества относится
только к одному компоненту. На рис. 2 изображена селективность методики по раз-
решению пиков известных продуктов разложения и основного пика.
8.1.5.4. Предел обнаружения
Предел обнаружения (ПО) является характеристикой только методик на предель-
ное содержание. Это минимальное количество аналита в пробе, которое может быть
обнаружено, но не обязательно определено в количественном отношении при за-
данных условиях эксперимента. Предел обнаружения выражается как концентра-
ция аналита в пробе, например, в процентах, частях на миллион (ррт) или частях на
миллиард (ppb).
Существует несколько подходов определения ПО. Визуальная оценка может ис-
пользоваться для неинструментальных (например, цвет раствора) и инструменталь-
Глава 8.1. Валидация аналитических методик: принципы и подходы
837
Время, с
Рис. 2. Наложение хроматограмм раствора с примесями и испытуемого раствора
ных методик. В этом случае ПО определяется путем проведения серии испытаний
образцов с известными концентрациями и установления минимального уровня, при
котором аналит может быть достоверно обнаружен. Представление сопутствующих
хроматограмм или другой сопутствующей информации является достаточным для
обоснования значения ПО.
При валидации инструментальных методик при наличии фонового шума обыч-
но сравнивают измеряемые сигналы от образцов с известными низкими концентра-
циями аналита с контрольными (холостыми) пробами. Минимальная концентра-
ция, при которой аналит может быть достоверно определен, устанавливается путем
использования приемлемого соотношения сигнал/шум 2:1 или 3:1. Представление
соответствующих хроматограмм достаточно для обоснования значения ПО.
Другой подход заключается в расчете значения ПО на основе стандартного от-
клонения отклика и наклона калибровочной кривой. Стандартное отклонение
определяется либо на основании стандартного отклонения результатов многократ-
ного определения контрольных (холостых) проб, либо на основании стандартного
отклонения величин отрезков, отсекаемых регрессионными кривыми на оси у в ди-
апазоне предполагаемого ПО. Подобная оценка требует последующей валидации
путем проведения отдельных определений подходящего числа образцов, содержа-
щих аналит в количестве, близком или равном ПО:
ПО = Зс/S,
где о — стандартное отклонение отклика; S — наклон калибровочной кривой.
8.1.5.5. Предел количественного определения
Предел количественного определения (ПКО) является характеристикой количе-
ственных методик определения веществ, присутствующих в образце в низких кон-
центрациях, таких как примеси в промышленно выпускаемых фармацевтических
838 Часть 8. Валидация
субстанциях и продукты разложения в готовых лекарственных препаратах. ПКО
определяется как концентрация родственных веществ в образце, при которой со-
отношение сигнал/шум составляет 10:1. На ПКО-методики влияют чувствитель-
ность детектора и точность пробоподготовки при низких концентрациях примесей.
На практике ПКО должен быть ниже, чем рекомендуемый ICH предел содержания
примеси, о присутствии которой необходимо указать в регистрационном досье.
ICHрекомендует три подхода для оценки ПКО. Первый — на основе визуальной
оценки. Этот подход можно использовать при неинструментальных и инструмен-
тальных методиках анализа. ПКО определяют исследованием образцов с известной
концентрацией аналита и нахождением минимального уровня, при котором может
быть установлено количественное содержание аналита с приемлемой правильно-
стью и прецизионностью.
Второй подход заключается в определении соотношения сигнал/шум путем
сравнения измеренных откликов при анализе образцов с известной низкой концен-
трацией аналита и контрольных образцов. ПКО — минимальная концентрация, при
которой аналит может быть достоверно количественно определен при соотношении
сигнал/шум 10:1.
При третьем подходе ПКО определяют по формуле
ПКО = 10ст/5.
Наклон кривой Сможет быть определен по калибровочной кривой аналита. Зна-
чение ст можно определить двумя способами: 1) вычисляя стандартное отклонение
откликов, полученных при измерении аналитического фонового шума подходяще-
го числа контрольных образцов; 2) вычисляя остаточное стандартное отклонение
(стандартное отклонение остатков) линии регрессии от калибровочной кривой
с использованием образцов, содержащих аналит в диапазоне ПКО.
Независимо от того, каким образом был установлен ПКО, полученные данные
необходимо затем валидировать путем выполнения испытаний подходящего числа
образцов, содержащих количество аналита в диапазоне ПКО, и определения пре-
цизионности и правильности при данном уровне.
8.1.5.6. Линейность
ICHопределяет линейность аналитической методики как способность (в рамках за-
данного диапазона) получать результаты испытаний в виде переменных (например,
величины поглощения и площади под кривой), прямо пропорциональных концен-
трации (количеству анализируемого вещества) пробы. Переменные, которые могут
использоваться для количественного определения анализируемого вещества, — это
площади пиков, высота пиков и отношение площадей (высот) пиков анализируе-
мого вещества к пику внутреннего стандарта. Количественное определение анали-
зируемого вещества зависит от выполнения закона Бера для спектроскопического
метода по диапазону концентраций. Поэтому концентрации рабочего испытуемого
раствора и растворов, используемых при оценке правильности методики, должны
находиться в линейном диапазоне.
Существует два основных подхода для определения линейности методики. При
первом непосредственно берутся различные навески стандартного образца для при-
Глава 8.1. Валидация аналитических методик: принципы и подходы 839
готовления растворов разной концентрации для определения линейности. Однако
данный метод непригоден при приготовлении растворов с очень низкой концентра-
цией из-за достаточно большой погрешности при взвешивании.
При другом подходе готовится исходный раствор высокой концентрации. Ли-
нейность определяется на растворах, полученных прямым разведением исходного
стандартного раствора. Этот метод наиболее распространен и часто рекомендуется.
Линейность лучше всего оценивать визуально по графику зависимости значений
откликов прибора от концентрации анализируемого вещества. Обычно переменные
используются для расчета регрессионной кривой с помощью метода наименьших
квадратов. Следует использовать результаты определений по крайней мере пяти
концентраций. При нормальных условиях линейность считается приемлемой при
коэффициенте детерминации (квадрате коэффициента корреляции) Р > 0,997. В со-
ответствии с требованиями ICH также должны быть рассчитаны наклон кривой,
остаточная сумма квадратов и величина отрезка, отсекаемого кривой на оси у.
Наклон регресионной кривой дает представление о чувствительности регрессии
и, следовательно, о чувствительности валидируемой методики. Величина отрезка,
отсекаемого на оси у, дает оценку вариабельности методики. Например, процент
отношения величин отрезков, отсекаемых кривой на оси у, для переменных, полу-
ченных при измерении номинальной концентрации, иногда используется для оцен-
ки вариабельности методики.
Для методик количественного содержания фармацевтической субстанции или
лекарственного препарата обычный диапазон линейности должен составлять ±20%
целевой или номинальной концентрации. Для методик определения однородности
дозирования она должна составлять ±30% целевой или номинальной концентрации.
На рис. 3 изображена линейность набора данных.
о
с:
16 000
14000
12000
юооо
8000
6000
4000
2000
50 100 150 200
Концентрация, мкг/мл
250
Рис. 3. Линейный график зависимости площади пика от концентрации
8.1.5.7. Диапазон
Диапазон аналитической методики — интервал между максимальной и минималь-
ной концентрацией анализируемого вещества в образце, для которого был показан
приемлемый уровень прецизионности, правильности и линейности аналитической
методики. Диапазон обычно выражается в тех же единицах (например, процентах,
частях на миллион), что и результаты испытания, полученные с помощью аналити-
ческой методики.
840
Часть 8. Валидация
Для методик количественного определения фармацевтической субстанции или
готового лекарственного препарата обычно рекомендуется, чтобы диапазон состав-
лял 80—120% номинальной концентрации.
Для методик определения однородности дозирования (или однородности по со-
держанию) стандартный диапазон должен составлять 70—130% номинальной кон-
центрации, пока не будет обосновано использование более широкого и более под-
ходящего диапазона (например, дозирующие ингаляторы).
Для методик, использующихся в испытании растворения, стандартный диапазон
должен составлять ±20% от заданного диапазона. Если критерий приемлемости для
препарата с контролируемым высвобождением выходит за диапазон 20% через 1 ч
и доходит до 90% через 24 ч, валидированный диапазон должен составлять 0—110%
заявленного значения. В данном случае минимальная концентрация анализируемо-
го вещества, которую можно определить, будет являться нижним пределом, так как
0% — недопустимое значение.
8.1.5.8. Робастность
Робастность (устойчивость) аналитической методики — способность методики
оставаться неизменной при небольших, но преднамеренных вариациях в параме-
трах методики; она представляет информацию о надежности при обычном исполь-
зовании.
Оценка робастности обычно проводится во время разработки и зависит от типа
изучаемой методики. План эксперимента (например, дробный факторный дизайн
эксперимента или дизайн Плакетга—Бурмана) — стандартное средство для удобно-
го исследования множества параметров одновременно. Результат позволяет опре-
делить критические параметры, которые будут влиять на рабочие характеристики
методики. Стандартные параметры метода, которые могут повлиять на аналитиче-
скую методику, следует рассматривать с учетом аналитической техники и свойств
испытуемых образцов:
1. Подготовка пробы:
а) время экстрагирования;
б) растворитель для приготовления испытуемого раствора (pH ± 0,05 единиц,
% содержания органического растворителя ± 2% (количества чистого рас-
творителя));
в) мембранные фильтры;
г) стабильность испытуемого и стандартного образцов.
2. Условия высокоэффективной жидкостной хроматографии (ВЭЖХ):
д) состав подвижной фазы (pH ± 0,05 единиц, % содержания органического
растворителя ± 2% (количества чистого растворителя));
е) используемая колонка (эквивалентные колонки, серии и/или поставщи-
ки, возраст колонки);
ж) температура;
з) скорость потока.
3. Условия газовой хроматографии (ГХ):
и) используемая колонка (серии и/или поставщики, возраст);
Глава 8.1. Валидация аналитических методик: принципы и подходы 841
к) температура;
л) скорость потока.
Если на результаты влияют некоторые критические параметры испытания,
в аналитическую методику следует включить предупредительные указания, чтобы
гарантировать, что данный параметр строго контролируется между испытаниями.
Например, если процент образования ионных пар подвижной фазы значительно вли-
яет на результаты, аналитическая методика должна включать подробное описание
предупредительных мер по подготовке водных компонентов, например 40%-ный
водный раствор 20 мМ октансульфоновой кислоты + 2% (количества безводного
вещества).
Робастность для подтверждения неизменности аналитической методики при ва-
лидации следует рассматривать и в следующих двух случаях.
1. Экспграция при подготовке пробы. Предпочтительно механическое встряхива-
ние, а не воздействие ультразвуком, так как на последний влияет ряд факто-
ров, например уровень воды в бане и расположение образца.
2. Разведение раствора и растворитель. Минимизируйте число разведений для
уменьшения вероятности внесения ошибки. Растворитель должен быть мак-
симально близок по свойствам к подвижной фазе.
8.1.6. Процесс валидации аналитической методики
Стандартный процесс валидации аналитической методики можно представить
в виде следующих этапов:
1. Планирование и выбор испытаний для валидации методики.
2. Написание и утверждение протокола валидации методики.
3. Выполнение протокола валидации методики.
4. Анализ данных, полученных при валидации методики.
5. Составление отчета по валидации аналитической методики.
6. Доработка аналитической методики.
Испытания при валидации методики должны быть хорошо спланированы и вы-
строены для эффективного использования времени и ресурсов во время непо-
средственного проведения валидации. Наилучшим способом обеспечения хорошо
спланированного валидационного исследования является написание протокола ва-
лидации методики, который будет оцениваться и подписываться соответствующим
лицом (например, руководством лаборатории или отделом обеспечения качества).
Валидационные параметры, которые будут определяться, зависят от типа вали-
дируемой методики. Аналитические методики, наиболее часто подвергающиеся ва-
лидации, можно разделить на три основные категории: испытания на подлинность,
испытания на чистоту и количественное определение. В табл. 3 представлены реко-
мендации ICH для каждой из этих методик.
Выполнение протокола валидации методики должно быть тщательно спланиро-
вано для оптимизации ресурсов и времени, требуемых для полного проведение всех
валидационных исследований. Например, при валидации методики количествен-
ного определения линейность и правильность могут быть оценены одновременно,
так как в обоих испытаниях могут использоваться те же стандартные растворы.
842
Часть 8. Валидация
Таблица 3
Валидационные характеристики аналитических методик
Характеристика Испытания на подлинность Испытания на чистоту Количественное определение — растворение (только измерение) — содержание/активность
Количественные методы Пределы содержания
Правильность - + - +
Прецизионность -
Повторяемость - + - +
Промежуточная прецизионность +а - +а
Специфичность6 + + + +
Предел обнаружения Предел - в ч- -
количественного — + — —
определения Линейность — + — +
Диапазон - + - +
Примечания. «-» — определение параметра не проводят; «+» — определение параметра
обязательно.а В случаях, когда проверялась воспроизводимость, промежуточная прецизион-
ность не требуется. 6 Недостаточная специфичность одной аналитической методики может
быть компенсирована другими вспомогательными аналитическими методиками. а Может
требоваться в некоторых случаях.
Стандартный валидационный протокол должен содержать как минимум следующие
пункты:
а) цель протокола;
б) валидационные параметры, подлежащие оценке;
в) критерии приемлемости для всех оцениваемых валидационных параметров;
г) подробное описание запланированных испытаний;
д) проект аналитической методики.
Данные валидации методики должны анализироваться по мере получения и об-
работки для обеспечения плавного потока информации. Если в ходе испытания об-
наруживаются ошибки эксперимента, их следует устранять как можно скорее для
уменьшения влияния, которое они могут оказать на дальнейшие испытания. Ана-
лиз данных включает визуальный контроль числовых показателей данных и хрома-
тограмм, наряду со статистической обработкой данных при необходимости.
По завершении всех испытаний данные компонуются в подробном отчете о ре-
зультатах валидации, где делается вывод об успешном выполнении или неудаче ва-
лидационного испытания. В зависимости от стратегии компании валидационные
данные могут быть изложены кратко. Успешное выполнение валидации ведет к по-
лучению окончательной аналитической методики, которая может использоваться
лабораторией в будущей аналитической работе с фармацевтический субстанцией
или препаратом.
Глава 8.1. Валидация аналитических методик: принципы и подходы 843
8.1.7. Необходимая для аналитической методики
информация
Окончательная аналитическая методика должна содержать как минимум следую-
щую информацию:
1. Обоснование аналитической методики и описание возможностей метода.
Пересмотр аналитической методики должен включать предлагаемые новым
пересмотром преимущества.
2. Предлагаемая аналитическая методика. Этот раздел должен включать пол-
ное описание аналитической методики, достаточно детальное, чтобы другой
аналитик мог воспроизвести ее. Подробное описание должно включать все
важные рабочие параметры и специфические инструкции, например приго-
товление реактивов, проверку пригодности системы, меры предосторожно-
сти и развернутые формулы для вычисления результатов испытаний.
3. Перечень допустимых примесей и их нормы содержания при количественном
определении примесей.
4. Валидационные данные. Прилагается либо подробное описание, либо крат-
кий обзор валидационных данных.
5. История пересмотров.
6. Подписи автора, рецензента, руководителя и представителя отдела обеспече-
ния качества.
8.1.8. Валидация методик на стадии разработки
Первоначальной целью cGMP было описание разработанных стандартов и действий
для обеспечения количественного содержания действующего вещества, подлинно-
сти, безопасности, чистоты и других показателей качества фармацевтических пре-
паратов, выпускаемых в обращение. Однако в правилах GMP не говорится ни слова
о стадиях разработки фармацевтических препаратов в ряде областей.
Регуляторные органы считают, что знания о лекарственном препарате и приме-
няемых аналитических методиках развиваются по мере разработки. Это четко ука-
зано в руководстве ICH Q7A. «При разработке возможны изменения, и каждое из-
менение, затрагивающее продукт, спецификации или методики испытаний, должно
быть задокументировано надлежащим образом». Поэтому можно с уверенностью
ожидать, что с увеличением знаний о структуре вещества будут вноситься измене-
ния в испытания, производство, упаковку и т. д. Однако даже при внесении изме-
нений нельзя пренебрегать необходимостью обеспечения безопасности субъектов
в клинических исследованиях.
Согласно руководству ICH целью валидации методики является демонстрация
ее «пригодности для предполагаемого применения». Поэтому методика должна
быть связана с клиническими исследованиями и фармацевтическим назначением
изучаемого продукта.
Цель ранней стадии разработки препарата заключается в доставке известной
дозы препарата, которую получит пациент при клинических исследованиях. По мере
продолжения разработки препарата особое внимание уделяется поиску стабильного
844
Часть 8. Валидация
и устойчивого состава лекарственной формы, что позволит производить многочис-
ленные серии, биоэквивалентные друг другу, а при финальном масштабировании
и переносе даст возможность наладить контролируемый промышленный выпуск
продукта.
Разработка и валидация аналитических методик должна идти аналогичным пу-
тем. Цель аналитических методик на ранних этапах разработки — проверка содер-
жания действующих веществ, понимание профиля примесей и продуктов разложе-
ния и помощь в понимании основных характеристик лекарственного препарата.
По мере дальнейшей разработки препарата методика должна исследовать стабиль-
ность и измерять влияние ключевых производственных параметров для обеспече-
ния постоянства качества фармацевтической субстанции и препарата.
Для аналитических методик, используемых для оценки чистоты и количествен-
ного содержания экспериментальных действующих веществ, на ранних стадиях
разработки необходима менее строгая валидация, чем требуется для методики, ис-
пользуемой лабораторией отдела контроля качества на производственной площад-
ке. Проект на ранней стадии может иметь ограниченное число серий для анализа,
и испытания могут проводиться только в одной лаборатории ограниченным числом
аналитиков. Возможность лаборатории «настраивать» методику и ее применение
относительно высока, особенно если очевидно лидерство лаборатории в выполне-
нии таких работ.
Условия, при которых используется методика, значительно меняются при пере-
носе методики в лабораторию отдела контроля качества на производственной пло-
щадке. Методика может воспроизводиться в нескольких лабораториях, выполняться
различными аналитиками и может быть одной из многих, ежедневно используемых
в лаборатории. Лаборатория, в которой производилась разработка, должна учесть
потребности лабораторий-преемников, например лаборатории отдела контроля ка-
чества, и ожидания регуляторных органов в отношении успешной валидации мето-
дики, используемой при подтверждении серийно выпускаемого продукта.
Пример минимальных требований для методик количественного определения
для фармацевтической субстанции и лекарственного препарата приведен в табл. 4.
Обратите внимание, что в соответствии с предыдущим обсуждением отсрочка оцен-
ки промежуточной прецизионности вызвана тем, что на начальной стадии аналити-
ческая методика используется в одной лаборатории и очень ограниченным числом
аналитиков. Объем работ по валидации методик и процессов на разных фазах раз-
работки в каждой отдельной компании будет отличаться, но принципы, на которых
они основаны, одинаковы. Размер и ожидания валидации методик на начальной
стадии ниже требований, предъявляемых на последующих стадиях разработки. Ра-
боты по валидации становятся более объемными и детализированными, собирается
больший объем данных для подтверждения надежности метода и пригодности для
использования на производственной промышленной площадке.
Однако определенные фундаментальные принципы cGMP должны соблюдаться
независимо от используемой стратегии пофазового подхода к валидации методик.
Примерами могут служить: 1) необходимая документация; 2) управление измене-
ниями; 3) отклонения; 4) квалификация оборудования и вспомогательных средств;
5) надлежащее обучение персонала.
Глава 8.1. Валидация аналитических методик: принципы и подходы
845
Таблица 4
Валидация методики количественного определения на начальной стадии разработки
фармацевтической субстанции и лекарственного препарата
Фармацевтическая субстанция Лекарственный препарат
Правильность Оценивается по прецизионности, Обнаружение при 100%-ной линейности и специфичности концентрации действующего вещества для каждой дозировки (группировка при большом количестве дозировок)
Повторяемость Три образца при 100%-ной концентрации
Промежуточная прецизионность Специфичность Проводится на последующих стадиях разработки Разделение наиболее вероятных Разделение примесей примесей и вспомогательных веществ
Предел количественного определения/предел обнаружения Линейность Не требуется Минимум три уровня от 80 до 120%
Диапазон Робастность Определяется при определении линейности Стабильность раствора
Детальный отчет по валидации методик может не потребоваться до момента по-
дачи регистрационного досье на готовый продукт. Тем не менее, краткие отчеты
должны быть доступны для облегчения и эффективного получения данных и пред-
ставления требуемой информации по запросу регуляторных органов.
8.1.9. Верификация методики
Федеральное управление США по контролю за пищевой продукцией и лекарствами
(FDA, Food and Drug Administration) в томе 21 CFR, в пункте 211.194(a)(2) указывает,
что при использовании аналитических методик, описанных в Фармакопее США
и Национальном формуляре (USP/NF, U. S. Pharmacopeia/National Formulary), вали-
дация по параметрам правильности и надежности этих методик не требуется, необ-
ходимо только провести подтверждение пригодности методик в реальных условиях
использования. Руководство по верификации описано в общей главе <1226> USP.
В данной главе содержится общая информация для лабораторий по верификации
фармакопейных методик, выполняемых впервые с целью получения приемлемых
результатов, с помощью персонала, оборудования и реактивов данной лаборатории.
Верификация состоит в оценке выбранных аналитических валидационных харак-
теристик, описанных ранее, с целью получения приемлемых релевантных данных,
а не в повторении процесса валидации для выпускаемого продукта. Информация из
этой общей главы применима к таким методикам, как титрование, хроматографиче-
846
Часть 8. Валидация
ские методики (родственные вещества, количественное определение и определение
пределов содержания) и спектрофотометрические испытания. Однако общие ис-
пытания (например, определение воды, тяжелых металлов, остатка при сжигании)
обычно не требуют верификации.
В табл. 5 приводится обобщенное сравнение валидационных и верификацион-
ных требований для методики количественного определения методом ВЭЖХ для
готовой лекарственной формы. ICH требует проведения валидации по характери-
стикам правильности, прецизионности, специфичности, линейности и диапазона.
Обычно при верификации требуется только валидация по характеристикам преци-
зионности и специфичности. Определение правильности зависит от специфики го-
товой лекарственной формы.
Таблица 5
Валидационные и верификационные требования для методики количественного определения
методом ВЭЖХ для готовых лекарственных форм
Характеристики Валидация Верификация
Правильность Да Возможно
Прецизионность Да Да
Специфичность Да Да
Предел обнаружения (ПО) Нет Нет
Предел количественного определения (ПКО) Нет Нет
Линейность Да Нет
Диапазон Да Нет
8.1.10. Ревалидация методики
Существует ряд обстоятельств, при которых необходимо проведение ревалидации
(повторной валидации) методики. Наиболее типичными являются следующие си-
туации.
1. При оптимизации процесса синтеза фармацевтической субстанции в процесс
были внесены значительные изменения. Для гарантии того, что аналитическая ме-
тодика позволит в будущем проводить анализ потенциально другого профиля при-
месей, может потребоваться ревалидация.
2. При обнаружении новой примеси, делающей методику недостаточно специ-
фичной, используемую методику необходимо изменить или повторно разработать
и провести ее ревалидацию, чтобы гарантировать ее пригодность для предполагае-
мого использования.
3. При изменении вспомогательных веществ может измениться профиль приме-
сей. Подобное изменение делает методику недостаточно специфичной для количе-
ственного определения или испытания на примеси, поэтому может потребоваться
повторная разработка методики и ее ревалидация.
4. Замена оборудования или поставщиков критических материалов для произ-
водства фармацевтической субстанции или готового лекарственного препарата спо-
собны изменить профиль продуктов разложения. В этом случае требуется повторная
разработка методики и ее ревалидация.
Глава 8.1. Валидация аналитических методик: принципы и подходы 847
8.1.11. Заключение
В этой главе кратко описаны валидационные характеристики, проверка которых не-
обходима в соответствии с требованиями ICH Q2(R1) [1]. Подробно описано изме-
нение парадигмы cGMP в двадцать первом веке, в соответствии с которой научные
работники на производстве должны обладать научным и техническим пониманием,
знанием продукта и процесса и/или навыками оценки рисков для надлежащего вы-
полнения обязанностей по контролю качества при валидации аналитической мето-
дики. Также рассмотрен процесс валидации методики и минимальные требования,
необходимые для включения в официальную, согласованную с регуляторным ор-
ганом методику. Представлены обзоры валидации методик на стадии разработки,
верификации методик и ревалидации методик в качестве рекомендаций, которых
следует придерживаться при возникновении схожих ситуаций.
Литература
1. International Conference on Harmonization (ICH) (2005, Nov.), Harmonised tripartite guideline
Q2(R1), Validation of analytical procedures: Text and methodology.
2. International Conference on Harmonization (ICH) (1999, Oct.), Harmonised tripartite guideline Q6A,
Specifi cations: Test procedures and acceptance criteria for new drug substances and new drug prod-
ucts: Chemical substances.
3. International Conference on Harmonization (ICH) (2000, Nov.), Harmonised tripartite guideline Q7A
GMP for active pharmaceutical ingredient.
4. International Conference on Harmonization (ICH) (2006, Sept.), Guidance for industry: Quality sys-
tems approach to pharmaceutical cGMP.
5. Code of Federal Regulations (CFR), Part 211, Current good manufacturing practice for finished phar-
maceuticals.
6. Chan, С. C., et al., (2004), Analytical Method Validation and Instrument Performance Verifi cation, J.
Wiley, Hoboken, NJ.
7. U.S. Pharmacopeia (USP, General Chapter (1225), Validation of compendial procedures, USP, Rock-
ville, MD.
8. U.S. Pharmacopeia (USP), General Chapter (1226), Verifi cation of compendial procedures, USP,
Rockville, MD.
Глава 8.2. Валидация аналитических методик
и обеспечение качества
Изабель Таверньерс, Эрик ван Бокстель, Марк де Лууз
Институт исследований в области сельского хозяйства
и рыбоводства (ILVO), Научно-исследовательский институт
Фламандского сообщества (Мерелбек, Бельгия)
8.2.1. Введение
Достоверность аналитических данных никогда не привлекала внимание обще-
ственности так, как в настоящее время. В основном внимание уделяется качеству
и надежности окончательных результатов, а не используемым методикам и мето-
дологии как таковой. Это вызвано стремлением соответствовать требованиям ре-
гуляторных органов, возросшей компетентностью потребителей — клиент хочет
знать степень надежности заявленных результатов, а также влиянием новых, более
строгих европейских и международных стандартов, таких как стандарт 17025 по ак-
кредитации испытательных лабораторий Международной организации по стандар-
тизации и Международной электротехнической комиссии (ИСО/7ЕС, International
Organization for Standardization / International Electrotechnical Commission). Лежащий
в основе ключевой принцип — сопоставимость результатов разных лабораторий
на более широкой международной основе. Для того чтобы результаты были сопо-
ставимы, они должны представляться с указанием неопределенности измерений
и прослеживаться до исходных первичных стандартов1. Методики необходимо ва-
лидировать, чтобы показать, что они измеряют именно то, что должны измерять, то
есть пригодны для предназначенного применения.
Так как валидация и обеспечение качества применяются к определенной ана-
литической методике, важно подходить к каждой методике индивидуально. Ана-
литическая методика — сложный многоэтапный процесс, начинающийся с отбора
образцов и заканчивающийся обработкой результатов. Хотя у каждой методики есть
своя цель, применение и аналитические требования, основные принципы валида-
ции и обеспечения качества одинаковы независимо от типа методики и области
применения. Информация в настоящей главе в основном относится к аналитиче-
ской химии, но также применима и к другим областям. Валидация аналитических
методик, установление прослеживаемости результатов (связи средств измерений
с первичными эталонами) и оценка неопределенности измерений должны осущест-
вляться единым согласованным образом в соответствии с международно приняты-
ми стандартами таких организаций, как Европейская ассоциация по аналитической
химии (JEurachem), Международный союз теоретической и прикладной химии (ИЮ-
ПАК) и ИСО.
1 Под первичными стандартами подразумеваются международные и национальные этало-
ны длины, массы и др. — Примеч. перев.
Глава 8.2. Валидация аналитических методик и обеспечение качества 849
Важно разработать общие подходы к валидации методик, прослеживаемости
и неопределенности измерений. В данной главе рассматриваются взаимосвязи
между валидацией методик, прослеживаемостью и неопределенностью измерений
при получении результатов. На основе руководств и стандартов была выбрана, со-
ставлена и обобщена наиболее важная информация. Большое значение придается
различным рабочим параметрам методик и их определению, способам выражения
и подходам к практической оценке. Роль валидации методик в обеспечении ка-
чества испытаний обсуждается наряду с темами по стандартизации, внутреннему
и внешнему контролю лабораторий (соответственно IQC и EQC, internal и external
quality control) и аккредитации, а также связи между этими разными понятиями.
В главе приводится полный и актуальный обзор вопросов качества аналитиче-
ских измерений в широком смысле слова. Информация будет полезна как для спе-
циалистов, не имеющих опыта в данной области, так и тем, кто непосредственно
связан с обсуждаемой проблематикой уже долгое время, кто заблудился в лабиринте
и ищет объяснения по конкретным вопросам или хочет достичь более глубокого по-
нимания проблем и расширить знания.
8.2.2. Прослеживаемость и неопределенность измерений
8.2.2.1. Введение: качество аналитических результатов
Бессчетное количество аналитических методик существует в аналитической и био-
аналитической химии, биохимии, биологии, клинической биологии, фармакологии
и связанных прикладных областях, таких как судебный, токсикологический, эколо-
гический, сельскохозяйственный и пищевой анализ. Независимо от типа методи-
ки, ее назначения и применения, лаборатории должны получать надежные данные
при проведении аналитических испытаний по заказу клиентов или регуляторных
органов. Наряду с быстрым развитием методологии аналитических исследований,
огромное значение в настоящее время придается понятию «качества» измеряемых
данных. Качество данных аналитического измерения заключается в двух важных
критериях: практичности и надежности (рис. 1) [1]. Практичность означает, что
аналитические результаты позволяют принять обоснованное решение. Ключевым
аспектом надежности, или валидности, результатов является их сопоставимость не-
зависимо от их происхождения. Сопоставимость результатов обеспечивается про-
слеживаемостью до соответствующих стандартов. Прослеживаемость до общих
контрольных стандартов лежит в основе возможности сравнения (т. е. разграниче-
ния) различных результатов. Если результаты необходимо сравнить в терминах ко-
личественных данных или уровней содержания анализируемых веществ, потребует-
ся дополнительная информация по аналитическим результатам — неопределенность
измерений. Погрешность результатов возникает при объединении всех погрешно-
стей1 значений стандартов (до которых результаты прослеживаются) и всех допол-
1 Современные зарубежные документы, регламентирующие порядок выражения результа-
тов в аналитической химии, сознательно не используют термин «погрешность», а применяют
термин «неопределенность», подразумевая под ним степень доверия к полученному резуль-
тату. В российских документах по метрологии изменение терминологии только началось.
Учитывая, что оба термина очень близки, для удобства читателя в данной книге оба термина
используются как взаимозаменяемые. — Примеч. ред.
850
Часть 8. Валидация
нительных погрешностей, связанных с методикой измерения. Неопределенность
измерения и прослеживаемость являются связанными понятиями, определяющими
качество аналитических данных (рис. 1) [2, 3].
Качество
Практичность
Т
Надежность
(валидность)
У1 = У2
Сопоставимость
1. Сравнимы ли два 1
результата, т. е.
отличимы ли друг I
от друга? I
Калибровка
2. Какой из двух результатов
содержит с достаточной
доверительной
вероятностью наибольшее
количество анализируемого
вещества?
У1 > У2 или У1 < У2?
Рис. 1. Связь качества, прослеживаемости и неопределенности измерения
Качество результатов отражает адекватность (или неадекватность) методики
с точки зрения степени, до которой методика соответствует требованиям или при-
годна для своей аналитической цели (см. ниже). Качество всегда является относи-
тельным понятием, ссылающимся на требования, установленные ранее на основе
национальных или международных регуляторных положений или нужд потребите-
лей [1, 4]. Важность обеспечения надежности аналитических данных подчеркива-
ется тем фактом, что результаты измерения могут стать основанием для принятия
решений. Ненадежные результаты приводят к высокому риску принятия непра-
вильных решений и могут привести к более высоким затратам, риску для здоровья,
незаконным методам и т. д. Представьте, например, последствия, если результаты
оказываются ложно-положительными или если погрешность значительно больше,
чем заявлено [1,5,6].
8.2.2.2. Роль валидации методики в прослеживаемости
и неопределенности измерений
Химический анализ — сложное многостадийное исследование свойств материалов,
определение подлинности и концентрации отдельного компонента в данном об-
разце материала [2, 7]. Ван Зунен (Van Zooneri) с соавт. [1] представили химический
анализ как циклический процесс, в котором конечной целью является получение
химической информации. Этот составной процесс начинается с определения основ-
ной аналитической проблемы (определения аналитических требований) и заканчи-
вается оценкой и представлением аналитических данных. В идеале на последнем
этапе дается ответ на поставленный вопрос, заданный клиентом или основанный
на нормативных требованиях.
Глава 8.2. Валидация аналитических методик и обеспечение качества
851
Рис. 2. Роль валидации методик в обеспечении качества аналитических
измерений. Валидация — процесс, показывающий соответствие
аналитической системы своему назначению [4, 8, 14,15]
Процесс поиска ответа на поставленную аналитическую задачу представлен на
рис. 2. Аналитическая система, представляющая «протокол определенной методи-
ки, применяемой для заданного типа испытуемого материала и при определенной
концентрации исследуемого вещества (аналита)», должна «соответствовать опре-
деленной аналитической цели» [4]. Эта аналитическая цель отражает достижение
аналитических результатов с приемлемой нормой правильности. Без указания нео-
пределенности (погрешности) результат не может быть интерпретирован и не имеет
ценности как таковой [8]. Результат должен выражаться с расширенной погрешно-
стью, которая в большинстве случаев составляет доверительные интервалы с веро-
ятностью 95% области значения результата. Вероятность того, что среднее значение
измерения находится с расширенной погрешностью, составляет 95% при условии,
что это значение без смещения (без значительной систематической ошибки) с воз-
можностью прослеживаемости до международно признанного эталона или стан-
дарта. В таком случае установление прослеживаемости и определение неопределен-
ности измерений связаны друг с другом. До оценки неопределенности измерений
необходимо показать, что результат прослеживается до эталона или стандарта, ко-
торый должен представлять истинное (опорное) значение [9,10].
Прослеживаемость и неопределенность измерений являются составными частя-
ми цели аналитической методики. Валидация играет важную роль в том смысле,
что она «подтверждает пригодность для назначенного использования определенной
аналитической методики» [4]. По определению ИСО валидация — это «подтверж-
дение на основе представления объективных свидетельств того, что требования,
установленные для конкретного применения, выполнены» [7]. Валидация являет-
ся инструментом, показывающим, что определенная аналитическая методика дей-
ствительно измеряет то, что должна измерять, и поэтому пригодна для предназна-
ченного применения [2,11].
В п. 8.2.3 описывается классический подход к валидации методики, основанный
на оценке ряда рабочих характеристик методики. В общих словах, основанный на
852
Часть 8. Валидация
ключевых параметрах процесс валидации состоит из оценки прецизионности и си-
стематической ошибки методики, проверки специфичности или селективности,
проверки линейности, исследовании робастности и, наконец, в зависимости от
практических требований методики, из оценки пределов обнаружения и/или ко-
личественного определения. Цель валидации состоит в проверке того, что условия
измерения и используемые расчетные формулы для вычисления окончательного
результата учитывают все влияния, способные воздействовать на этот результат. Ва-
лидация измеряет влияние различных факторов на результат внутри аналитической
системы и подтверждает, что остальными факторами можно пренебречь. Испыта-
ние на специфичность подтверждает, что методика регистрирует только определен-
ное анализируемое вещество без посторонних веществ и примесей. При проверке
линейности подтверждается, что предполагаемая зависимость величины сигнала от
количества единиц, используемых для измерения анализируемого вещества, дей-
ствительно существует и может использоваться. Оценка ошибки представляет со-
бой проверку правильности измерения сертифицированного стандартного образ-
ца и показывает, что методика не обладает значительным смещением результата.
Исследования прецизионности и робастности учитывают влияние вариабельности
условий, аналитиков, оборудования и времени проведения испытания.
Роль валидации методики в получении надежных результатов состоит: 1) в учете
всех возможных факторов, влияющих на конечный результат; 2) в прослеживаемо-
сти результатов до установленных стандартов (стандартные методики, стандартные
материалы или международная система единиц СИ); 3) в получении знаний о по-
грешностях, связанных с каждым таким фактором и со стандартами. Таким обра-
зом, валидация — это инструмент установления прослеживаемости до этих стан-
дартов [2,4]. В данном контексте важно видеть разницу между прослеживаемостью
и правильностью. Методика, обладающая правильностью, с точки зрения «истин-
ности» (т. е. близости к истинному значению) всегда прослеживается до принятого
истинного значения. Однако обратное утверждение не верно. Методика, которая
прослеживается до установленного стандарта, не обязательно обеспечивает получе-
ние истинных результатов (не обязательно обладает правильностью). В такой мето-
дике возможны ошибки в зависимости от стандарта [12].
Валидация аналитической методики образует первый уровень обеспечения ка-
чества в лаборатории. Обеспечение аналитического качества заключается в выпол-
нении лабораторией полного перечня мероприятий, гарантирующих постоянное
получение высококачественных данных. Кроме использования валидации и/или
стандартизованных методик к этим мероприятиям относятся эффективные проце-
дуры внутреннего контроля качества испытаний (использование стандартных мате-
риалов, контрольных карт и т. д.) наряду с участием в программах профессиональ-
ного тестирования (внешнего контроля качества лабораторий) и аккредитации на
соответствие международным стандартам, обычно ИСО//2ГС 17025 [4]. Валидация
методик и различные аспекты обеспечения качества рассматриваются в п. 8.2.3.
8.2.2.3. Руководства по прослеживаемости и погрешности результатов
В табл. 1 приведен обзор ведущих учреждений, разрабатывающих руководства
и стандарты по прослеживаемости, неопределенности измерений и смежным те-
Глава 8.2. Валидация аналитических методик и обеспечение качества 853
мам. В Европе лидирующую позицию занимает Eurachem — Европейская ассо-
циация по аналитической химии, организованная Лабораторией государствен-
ных химиков Великобритании {United Kingdom’s LGC, Laboratory of the Government’s
Chemists). Основными стандартами являются руководства CITACIEurachem по ка-
честву в аналитической химии [2] и прослеживаемости в химических измерениях
[3], а также руководство Eurachem по неопределенности измерений [13, 14]. Кроме
того, Eurachem публикует руководства по смежным темам, таким как стандартные
материалы [7] и валидация методик [15].
Таблица 1
Обзор европейских и международных организаций и регуляторных органов, издающих
руководства и стандарты по прослеживаемости, неопределенности измерений и смежным темам
Учреждение Полное название Тематика руководства Ссылки
Eurachem/СПАС Европейская ассоциация по аналитической химии/ Международная ассоциация по прослеживаемости в аналитической химии {Cooperation on International Traceability in Analytical Chemistry) Прослеживаемость, неопределенность измерений, стандартные материалы, валидация [2, 3, 7 13,14, 15]
ИЮПАК, ИСО, Международный союз теоретической Неопределенность [4, 8,
АОАС International и прикладной химии, Международная организация по стандартизации, Международная ассоциация химиков- аналитиков {Association of Official Analytical Chemists) измерений 16,17]
ФАО/ВОЗ: Codex/ ССМAS Продовольственная и сельскохозяйственная организация ООН/ Всемирная организация здравоохранения: Комитет по аналитическим методикам и отбору образцов {Committee on Methods of Analysis and Sampling) Комиссии по «Кодексу Алиментариус» Неопределенность измерений [18-21]
ЕА Ассоциация органов по аккредитации стран-членов ЕС {European Cooperation for Accreditation) Неопределенность измерений [22]
ИЛАК Международная ассоциация органов по аккредитации лабораторий {ILAC, International Laboratory Accreditation Cooperation) Неопределенность измерений [23]
На международном уровне соответствующие стандарты изданы ИЮПАК, ИСО,
АОАС International [4, 8, 16, 17] и CCMAS (Комитет по аналитическим методикам
и отбору образцов) Комиссии по «Кодексу Алиментариус» {Codex Alimentarius) [18—
21]. Другие информативные руководства опубликованы ЕА [22] и ИЛАК [23] (рас-
шифровку аббревиатур см. в табл. 1).
854 Часть 8. Валидация
8.2.2.4. Концепция прослеживаемости
Определения. Прослеживаемость — относительно новое понятие, привлекающее
все больше внимания в методологии аналитических измерений. Прослеживаемость
может относиться к различным аспектам, связанным с измерением, — рассматри-
вается прослеживаемость результатов, метода, методики, лаборатории, продукта,
сырья, оборудования и т. д. Также нет единственно верного определения прослежи-
ваемости. Прежде чем приступать к исследованию различных понятий прослежива-
емости, следует обратиться к более широкому, обобщенному значению. В соответ-
ствии с определением, которое предлагают Валькарсель и Риос (Valcarc I, Rios) [24],
основное значение термина «прослеживаемость» включает: 1) установление одной
или более связей с утвержденными эталонами и стандартами; 2) ведение докумен-
тированной «истории» продукта или системы. Эти две части основного значения
данного понятия могут быть вновь выделены, если определить прослеживаемость
как свойство или параметр различных аналитических аспектов. Различные понятия
термина «прослеживаемость» приведены на рис. 3.
Наиболее наглядно прослеживаемость можно определить как «свойство ре-
зультата измерения или значения стандарта, при помощи которого они могут быть
связаны с принятыми эталонами, обычно национальными или международными
стандартами, через непрерывную цепочку сопоставлений, имеющих измеренные
погрешности» [2]. Различные элементы и практическое использование этого опре-
деления будут рассмотрены в следующем разделе «Прослеживаемость на практике».
Прослеживаемость результата связана с прослеживаемостью методики, которая
в свою очередь связана с прослеживаемостью стандартов и прослеживаемостью
оборудования, используемого при выполнении аналитической методики (рис. 3).
Методика считается прослеживаемой, когда она обеспечивает получение результа-
тов (с определенной погрешностью), характеризующихся четкой прослеживаемо-
стью до утвержденных эталонов [24]. Уолш (Walsh) [25] определяет прослеживаемые
методики как «валидированные официальные или стандартные методики либо ва-
лидированные методики, содержащие заявленную погрешность, которые встроены
в систему качества и связаны с общей контрольной точкой».
Как видно из рис. 3, прослеживаемость стандартов является наиболее важной
основой прослеживаемости результатов [26]. Прослеживаемость оборудования
1.Связь(и)
с утвержденными
эталонами
и стандартами
2. Ведение доку-
ментированной
«истории»
продукта
или системы
Рис. 3. Различные понятия и обобщенное значение термина «прослеживаемость» [24]
Глава 8.2. Валидация аналитических методик и обеспечение качества
855
представляет «подробное, своевременное и отлаженное ведение записей по его
установке, неисправностям и ремонту, периодической калибровке и настройке
(если необходимо), времени использования, об измеренных с его помощью образ-
цах, использованных стандартах и т. д., при этом на все вопросы (что? как? кто?
и т. д.) должны быть даны подробные ответы в относящихся к делу документах». Ка-
либровка — ряд операций, используемых для установления связи между значения-
ми, показываемыми измерительным оборудованием, и значениями эталонов. При
осуществлении такой проверки результаты измерений становятся связанными со
значениями стандартов или эталонов и, следовательно, прослеживаемыми до них.
На практике калибровка проводится путем измерения образцов с известным со-
держанием анализируемого вещества, таких как сертифицированные стандартные
образцы, и контролем получаемых данных [2, 3]. Эти определения подтверждают
связь между прослеживаемостью оборудования, стандартов и результатов (рис. 3).
Прослеживаемость на практике. Практическое установление прослеживаемости
основано на поэтапном внедрении определения. Определение прослеживаемо-
сти по ИСО, основанное на метрологии (см. выше), может быть разделено на три
основных этапа [12, 24, 27]:
1) установление одной или более связей с принятыми эталонами;
2) непрерывная цепочка сопоставлений;
3) оценка погрешностей, связанных с такими сопоставлениями.
Такое определение согласуется с более применимым на практике определением,
предложенным Eurachem/СПАС [3]. Их методика определения прослеживаемости
состоит из следующих шагов [3]:
1) определить измеряемую величину, область измерения и требуемую погреш-
ность;
2) выбрать методику выполнения измерения;
3) провалидировать методику выполнения измерения;
4) определить или количественно выразить все возможные влияния на резуль-
тат;
5) выбрать подходящие стандарты;
6) оценить компоненты неопределенности, связанные с посторонним влиянием
и стандартами.
Основной принцип обоих подходов заключается в установлении связи с этало-
нами посредством непрерывной цепочки сопоставлений. На практике это означает,
что аналитическая методика сначала описывается как блок-схема или структурная
схема (этап 2 в определении ИСО; этапы 1 и 2 в определении Eurachem/СПАС). Тер-
мин непрерывная означает, что при рассмотрении различных этапов аналитической
методики, ведущих в итоге к получению результата измерений, потеря информа-
ции отсутствует. Каждый этап методики должен быть связан со стандартным мето-
дом, стандартным веществом или единицей СИ (этап 1 в определении ИСО; этап 5
в определении Eurachem/СПАС) [12,24, 27].
На рис. 4 изображены последовательные типы принятых стандартов (сырье или
методы) в так называемой цепи прослеживаемости. Установление прослеживаемо-
сти с помощью такой цепи вносит определенный уровень погрешности, называе-
856
Часть 8. Валидация
мый калибровочной погрешностью или погрешностью прослеживаемости (см. ниже)
[3, 7, 28]. Это подводит нас к третьему основному элементу определения прослежи-
ваемости: заявленным погрешностям. Каждый этап цепи прослеживаемости с эле-
ментами погрешности стандартов будет влиять на результат измерения и, следова-
тельно, на связанную с ним погрешность. Элементы, составляющие погрешность,
должны оцениваться на каждом этапе аналитического процесса (этап 3 в определе-
нии ИСО; этап 6 в определении Eurachem/CITAC). Как описано выше, валидация
является инструментом определения возможных эффектов или факторов внутри
аналитической методики, которые могут повлиять на конечный результат. По суще-
ству этапы 3 и 4 в определении Eurachem/CITAC являются дополнительными, они
могут быть очень полезны при определении прослеживаемости [3].
Пример того, как прослеживаемость определяется на практике, можно най-
ти в литературе. В 2004 г. журнал «Тенденции в аналитической химии» {Trends in
Analytical Chemistry) опубликовал специальный выпуск по проблемам обеспечения
прослеживаемости экологических измерений (№ 23, 2004). Данный выпуск со-
держит большое количество современной информации и практических примеров
в области экологического анализа. Многие авторы описывают наиболее важный
и сложный этап при установлении прослеживаемости: выбор стандартов и этало-
нов. Кевовиллер и Донард {Quevauviller, Donard) [ТТ\, Шарле и Маршаль {Charlet,
Marschal) [29], Сегура {Segura) с соавт. [30] приводят примеры и описания для раз-
личных этапов, указанных в цепи прослеживаемости на рис. 4. Другие авторы, на-
пример Пан {Pan) [28], Фёрстнер {Frstner) [31] и Теохаропулос {Theocharopoulos)
с соавт. [32], используют определение ИСО для установления прослеживаемости
в различных типах методов экологического анализа. Схожий подход, но на осно-
ве руководства Eurachem/CITAC по прослеживаемости [3], используют Сабэ и Рорэ
{Sab , Rauref) [33] и Дролк {Drolc) с соавт. [34]. В своих примерах все авторы учиты-
вают специфические этапы или факторы, влияющие на выполнение аналитической
Рис. 4. Цепь прослеживаемости и связь между прослеживаемостью и неопределенностью
измерений. Три способа установления прослеживаемости, показанные на рис. 5, выделены
жирным шрифтом [25, 28]
Глава 8.2. Валидация аналитических методик и обеспечение качества
857
методики, которые могут привести к разрыву цепи сопоставлений, такие как отбор
образцов, подготовка проб или подготовительные шаги. Некоторые авторы отмеча-
ют погрешности, связанные непосредственно с отбором образцов [35, 36].
8.2.2.5. Концепция неопределенности измерений
Неопределенность измерения — наиболее важный параметр как при валидации ме-
тодики, так и при внутреннем контроле качества испытаний. Ее определяют как «па-
раметр, связанный с результатами измерений и характеризующий степень разброса
значений, которые мотут относиться к измеряемой величине» [14]. Измеряемой ве-
личиной является определенное количество или концентрация анализируемого ве-
щества. Данный параметр может быть выражен стандартным отклонением или ши-
риной доверительного интервала [14,37]. Этот доверительный интервал представляет
интервал в масштабе измерений, внутри которого лежат истинные значения с задан-
ной вероятностью при условии, что учтены все источники ошибок [37]. Внутри дан-
ного интервала результат считается правильным, то есть точным и истинным [11].
Следует особо подчеркнуть, что неопределенность измерения отличается от
ошибки. Ошибка отдельного аналитического результата, разница между результа-
том и истинным значением измеряемой величины — всегда одиночное значение
[38]. Часть значения известной ошибки, систематическая ошибка, может использо-
ваться для коррекции результата. Это означает, что после коррекции результат ана-
лиза может быть очень близок к истинному значению. Однако неопределенность
измерения все еще может быть весьма значительной, так как имеются сомнения
или недостаточные знания о близости результата к значению. Неопределенность
выражается в виде диапазона и относится к конкретной аналитической методике
и конкретному типу образца, но к различным определениям и соответственно ре-
зультатам измерений. Величина неопределенности не может использоваться для
коррекции результата измерения.
Ошибка аналитического результата связана с (не)правильностью аналитической
методики и состоит из систематической и случайной составляющих [14]. Исследо-
вания прецизионности и систематической ошибки (смещения) образуют основу
оценки правильности аналитической методики [18]. Правильность результатов от-
носится только к пригодности аналитической системы для предназначенного ис-
пользования, что оценивается при валидации методики. Надежность результатов
касается не только валидации методики. Неопределенность измерений является
больше чем просто единичным выражением правильности. Она включает все ис-
точники ошибок, которые относятся ко всем концентрациям анализируемого ве-
щества. Неопределенность измерений — ключевой индикатор пригодности для на-
значенной цели и надежности результатов, связывающий вместе идеи пригодности
для предназначенного применения и контроль качества, тем самым охватывая всю
систему обеспечения качества [4, 37].
Таким образом, неопределенность измерений аналитической методики проис-
ходит из ошибки единичного аналитического результата, но отличается от него.
Отклонение результата измерения от истинного значения заключает в себе ряд си-
стематических и случайных ошибок, что показано на рис. 5. Каждая из этих оши-
бок добавляет собственную погрешность в общую погрешность аналитической
Результат = истинное значение +
Систематическая
ошибка методики
Ошибка
лаборатории
Ошибка
испытания
Погрешность
повторяемости
Результат = истинное значение +
। Погрешность
I прослеживаемости
I :
Погрешность
предполагаемого
отклонения
Ошибка
лаборатории
Систематическая ошибка
Ошибка Погрешность
испытания повторяемости
I I______________
Случайная
у ошибка
Случайная
ошибка
Погрешность =
Промежуточная прецизионность
Систематическая
ошибка оценки
находится
1)стандартной
методикой
2) методом добавок
Погрешность
стандарта = О
Погрешность
обнаружения =0
Разница между двумя
методиками
Разница между
двумя полученными
результатами
3) сертифицированным
стандартным
образцом
Погрешность
ССО = 0
Разница между
измеренным и
истинным значением
V
Случайная
ошибка
Внутрилабораторная
погрешность
Прецизионность воспроизводимости
Погрешность воспроизводимости
Промежуточная прецизионность
Погрешность с учетом систематической ошибки
Промежуточная прецизионность
Общая погрешность
Рис. 5. Погрешность аналитического результата во взаимосвязи с неопределенностью измерений. Различные рабочие определения
неопределенности измерения [10]. Внизу слева приведены три возможных способа определения прослеживаемости (также см. рис. 4)
858_____________________________________ Часть 8. Валидация
Глава 8.2. Валидация аналитических методик и обеспечение качества
859
методики. Поэтому различные источники ошибок называют источниками погреш-
ности. В зависимости от источника погрешности и условий измерений общая нео-
пределенность измерений будет меняться, и будет применяться другой способ на-
хождения неопределенности измерений. Это означает, что не существует единого
простого способа нахождения неопределенности измерений. Это, скорее, понятие,
интерпретация которого изменяется в зависимости от условий измерения и стан-
дартов, до которых прослеживается результат [10]. Различные определения неопре-
деленности измерений образуют предмет следующей темы.
8.2.2.6. Различные рабочие определения неопределенности измерения
Как показано на рис. 5, погрешность аналитического результата для определенной
концентрации анализируемого вещества состоит из ряда компонентов, совместно
образующих «лестницу погрешностей»:
• систематическая ошибка методики — систематическая ошибка, связанная
с методикой как таковой;
• ошибка лаборатории — либо систематическая ошибка, если лаборатория рас-
сматривается сама по себе, либо случайная ошибка, если лаборатория являет-
ся одной из группы, что происходит при межлабораторных исследованиях;
• ошибка испытания рассматривается как систематическая ошибка при одно-
кратном испытании и как случайная вариабельность при проведении лабора-
торией нескольких испытаний;
• погрешность повторяемости — случайная ошибка, вызванная повторными
измерениями, выполненными в одном и том же опыте [10].
Так как рассматриваемая ошибка относится только к определенной концентра-
ции анализируемого вещества, экстрагированного из образца или смеси компонен-
тов определенного типа, ошибки отбора образцов и эффекты вариабельности мате-
риала не рассматриваются в данном случае [4].
Более традиционным считается разделение ошибок на случайные ошибки и систе-
матические ошибки. При таком классическом подходе случайные ошибки обычно
называют «прецизионностью» (включает повторяемость, промежуточную преци-
зионность и воспроизводимость), в то время как систематические ошибки обычно
связывают с неопределенностью оценки смещения и погрешностью калибровки.
К этой классификации добавляются другие факторы погрешности, такие как про-
цедура отбора образцов, состав образца и погрешности, связанные с определенны-
ми допущениями, которые лежат в основе методики измерения и/или расчетной
формулы [2].
Как уже было сказано выше, каждая составляющая ошибки представляет потен-
циальный источник погрешности. В зависимости от условий, при которых прово-
дится анализ, различные источники погрешностей вносят вклад в общее значение
погрешности. Хуцц {Hund) с соавт. [10] предложили различные рабочие определе-
ния погрешности в соответствии с числом рассматриваемых источников погреш-
ности (рис. 5):
• внутрилабораторная погрешность происходит из промежуточной прецизион-
ности и включает только ошибку повторяемости и ошибку испытания;
860
Часть 8. Валидация
• погрешность воспроизводимости получается из прецизионности воспроизво-
димости (межлабораторные испытания) и вычисляется как ошибка воспро-
изводимости, испытаний и вариабельности лабораторий;
• погрешность с учетом систематической ошибки и общая погрешность допол-
нительно учитывают систематическую ошибку методики.
Систематическая ошибка методики — наиболее важный источник погрешности,
так как относится к эталону или стандарту, до которых методика должна просле-
живаться. Если рабочая методика не является первичной, то есть прослеживаемой
до единиц СИ (см. рис. 4), ее всегда сравнивают с другой стандартной методикой
или проводят испытания с использованием подходящих сертифицированных стан-
дартных образцов (материалов). Данные стандарты или эталоны учитываются при
вычислении погрешности, связанной с систематической ошибкой метода. В допол-
нение к погрешности, связанной с эталоном или стандартом, существует неопре-
деленность найденной систематической ошибки (смещения) (рис. 5). Различные
способы оценки систематической ошибки — и, как следствие, прослеживаемо-
сти — изображены на рис. 5. Если методику сравнивают со стандартной методикой,
погрешность, связанная с этой стандартной методикой, считается незначительной,
и систематическая ошибка находится как разница результатов, полученных двумя
методиками (случай на рис. 5). Если методика сравнения отсутствует, то системати-
ческую ошибку можно определить по степени обнаружения аналита, добавленного
в пробу. В данном случае погрешность при использовании метода добавок также бу-
дет близка к нулю (случай 2), и единственной систематической ошибкой методики
будет являться разница между измеренным образцом без добавки и образцом с до-
бавкой. Общая погрешность может быть найдена лишь в случае, когда используют-
ся сертифицированные стандартные образцы (материалы) (случай 3). Только в этом
случае можно гарантировать полную прослеживаемость до единиц СИ [10].
8.2.2.7. Подходы к нахождению неопределенности измерений
В общем случае для нахождения общей погрешности определенного результата не-
обходимо знать: 1) все погрешности, возникающие непосредственно при процедуре
измерения; 2) все погрешности, связанные с эталонами и стандартами, до которых
аналитические результаты прослеживаются [3]. Существуют различные подходы
к оценке общей неопределенности измерений. Обобщенные обзоры нескольких ав-
торов [9, 10, 39] на данную тему представлены в табл. 2.
Наиболее известный и традиционный подход основан на выявлении, измере-
нии и суммировании всех индивидуальных вкладов в неопределенность. При таком
«восходящем подходе» общая погрешность образуется из погрешностей отдельных
компонентов. Оценка неопределенности измерений пошагово (для каждого ком-
понента) изначально была разработана для физических измерений и адаптирова-
на Eurachem к химическим измерениям [13]. Однако из-за своей сложности данная
методология требует значительных затрат времени и сил, в связи с чем никогда не
находила широкого применения на практике.
Упрощенный подход к оценке неопределенности измерений — подход пригодно-
сти для назначенного использования, определяющий единственный параметр, назы-
Глава 8.2. Валидация аналитических методик и обеспечение качества
861
Подходы к нахождению неопределенности измерения
Таблица 2
Название подхода Основной принцип Преимущества Недостатки_________Ссылки_____
Восходящий Выявление, измере- Холистический [ Сложно, дорого, Eurachem [13],
подход, сум- марная по- грешность, распространение ошибки или по- шаговый подход ние и суммирование подход: все вкладов всех источ- важные источ- ников погрешности ники ошибки должны быть учтены долго ИСО [16]
Подход при- Нахождение функ- Простота, нео- Некоторые ис- «Кодекс
годности для ции пригодности пределенность точники погреш- Алиментариус»/
назначенного и —f{c) на основа- измерений ности можно ССЛ£45[18—21]
использования нии прецизионно- сти и исследований систематической ошибки оценивается для различных концентраций упустить
Нисходящий На основе данных Неопреде- Некоторые ис- Комитет по
подход межлабораторных исследований (пре- цизионности) ленность измерений оценивается для различных концентраций точники погреш- аналитическим ности можно методикам [37] упустить. Подход осуществим, только если до- ступны данные межлаборатор- ных исследова- ний
Подход на осно- На основе данных Расширение Некоторые ис- Eurachem
ве валидации межлабораторных и внутрилаборатор- ных валидацион- ных исследований (прецизионность, правильность, ро- бастность) валидацион- ных работ, не требуется проведения до- полнительных работ точникипогреш- [14], Барвик ности можно и Эллисон упустить {Barwick, Ellison) [47]
Подход на осно- На основе исследо- Простой,бы- Некоторые ис- Хунд с соавт. [39]
ве робастности ваний робастности как внутрилабора- торной симуляции межлабораторных исследований стро выполни- мый подход точники погреш- ности можно упустить. Метод должен обладать робастностью
ваемый функцией пригодности. Функция пригодности выражается в виде алгебра-
ической зависимости и = Дс) и описывает взаимосвязь между неопределенностью
измерений и концентрацией анализируемого вещества. Например, и = 0,05с озна-
чает, что неопределенность измерения составляет 5% концентрации. Вычисление
неопределенности измерения будет зависеть отданных, полученных при оценке ра-
862
Часть 8. Валидация
бочих характеристик отдельного метода, в основном по показателям повторяемости
и прецизионности воспроизводимости, а также, желательно, и по систематической
ошибке методики [21, 40, 41]. Данный подход можно рассматривать как упрощен-
ный вариант поэтапного протокола исследования неопределенности измерений,
описанного Eurachem [14].
Хотя неопределенность измерений включает не только систематические и слу-
чайные ошибки, ее можно определить по данным валидации методики. Данные
исследований рабочих характеристик методики могут предоставить всю или прак-
тически всю необходимую информацию для оценки погрешности [2, 4, 18, 37].
К подобной информации относятся данные внутреннего контроля качества лабо-
раторных работ и совместной валидации (обычно данные прецизионности), про-
граммы профессионального тестирования (внешнего контроля) (обычно данные,
содержащие систематическую погрешность) и любая связанная с погрешностью
информация, генерируемая системой обеспечения качества испытаний. Если по-
добная информация доступна и используется для определения погрешности, оценка
неопределенности измерений с использованием пошагового подхода не применя-
ется [18, 19]. В частности, валидационные исследования и данные контроля каче-
ства испытаний рассматриваются как важные источники информации для оценки
неопределенности измерений [42, 43].
В табл. 2 описаны три методологических подхода оценки неопределенности из-
мерений на основе валидационных данных. В нисходящем подходе Комитета по
аналитическим методикам [37] лаборатория рассматривается с более высокого уров-
ня, как член популяции групп. В результате систематические ошибки внутри одной
лаборатории становятся случайными, а вычисленная погрешность становится по-
грешностью воспроизводимости (см. рис. 5). Примеры исследований неопределен-
ности измерений с использованием данных межлабораторных испытаний приводят
Дехоук (Dehouck) с соавт. [44] и Марото (Maroto) с соавт. [45,46]. Два других подхода,
описанные в табл. 2 [14, 39,47], используют различные рабочие характеристики ме-
тодики. Все три методологии на основе валидационных исследований рассматрива-
ются как упрощенные, более быстрые в выполнении и менее дорогостоящие проце-
дуры, расширяющие процесс валидации. Однако необходимо отметить, что не все
источники погрешностей учитываются в рабочих данных методики. Некоторыми
источниками неопределенности, вклад которых может потребоваться учесть в до-
полнение к имеющимся данным, являются отбор образцов, предварительная об-
работка, систематическая ошибка методики, вариабельность условий и изменения
состава испытуемого раствора [14, 18,41].
Многие из этих принципов оценки неопределенности измерений применялись
в исследованиях Армишоу (Armishaw) [48] при определении содержания толуола
в грунтовой воде. Идея заключается в том, что для рутинного метода, ранее прошед-
шего валидацию и выполняемого в лаборатории с эффективной системой контро-
ля качества испытаний, возможно оценить неопределенность измерений в течение
одного рабочего дня. Ключевым моментом является получение всей необходимой
информации из уже имеющихся данных: валидационных исследований (данные по
систематической ошибке, проценту обнаружения и прецизионности), данных ка-
либровки оборудования (погрешность стандартных образцов (материалов)), данных
Глава 8.2. Валидация аналитических методик и обеспечение качества
863
периодически проводимых мероприятий по контролю качества испытаний (парал-
лельные анализы и контрольные образцы) и данных самой процедуры выполнения
методики (отбор образцов, однородность образцов и т. д.). После определения ком-
понентов, вносящих вклад в общую погрешность, и распределения данных из со-
ответствующих источников информации были вычислены стандартные погрешно-
сти, сумма которых составляет совокупную стандартную погрешность. Результаты
определения содержания толуола в воде выражались какх± U, где U — расширенная
погрешность, полученная умножением совокупной стандартной погрешности и, на
коэффициент охвата, равный 2.
В дополнение к восходящему методу вычисления неопределенности измерений
(подход 1) Армишоу определяет расширенную погрешность как функцию концен-
трации толуола (подход 2). В заключение авторы сравнивают экспериментально
найденную неопределенность измерений с ее значениями, вычисленными на осно-
ве: 1) внутрилабораторной оценки воспроизводимости; 2) данных схем профессио-
нального тестирования; 3) моделей Горвица (Horwitz) [49] и Томпсона и Лоусиана
(Thompson, Lowthian) [50]. Эти модели позволяют вычислить (в %) относительное
стандартное отклонение (RSB) как функцию концентрации анализируемого веще-
ства. Чтобы получить значения расширенной погрешности, Армишоу умножает
расчетное стандартное отклонение (SB) данных моделей на 2 [48]. Все вычисления
неопределенности измерений являются вариациями подходов, основанных на дан-
ных валидации (подходы 3 и 4, табл. 2).
8.2.2.8. Значение прослеживаемости и неопределенности измерения
Основной причиной, требующей установления прослеживаемости и неопреде-
ленности измерений, является необходимость принятия решений на основе по-
лученных аналитических результатов или необходимость соответствия регуля-
торным требованиям (для количественных определений) [51]. Неопределенность
измерений является неотъемлемым свойством аналитических результатов по ряду
причин. Во-первых, необходимо иметь представление о распределении результа-
тов и сравнимости результатов, полученных в разных лабораториях [43]. Любой
результат должен сопровождаться указанием неопределенности измерений, что-
бы конечный пользователь знал с каким уровнем достоверности получены данные
[52]. Понятия сравнимости и надежности результатов кратко обсуждались в п. 8.2.1
и представлены на рис. 1. Во-вторых, неопределенность измерений необходима,
так как она показывает прослеживаемость. Прослеживаемость до стандартов или
эталонов должна быть установлена еще до того, как неопределенность измерений
можно оценить. Мозер (Moser) с соавт. [43] утверждают, что прослеживаемость до-
казывается надлежащим использованием стандартных материалов или эталонов
и общей неопределенностью. Это дает нам третью причину необходимости оцен-
ки неопределенности измерений — необходимость знания методики, понимания
основных принципов и механизмов процедуры измерения. Недостаток глубоких
знаний методики как таковой приведет к добавлению неизвестной погрешности,
не принимаемой во внимание, и образованию пробелов в общей погрешности.
Неопределенность измерений можно определять, только если методика хорошо
понята [43, 53].
864 Часть 8. Валидация
Неопределенность измерений привлекает все больше внимания, в частности при
проведении аккредитаций. Новый стандарт по аккредитации ИСО//ЕС17025 [17],
вступивший в силу с декабря 2002 г., содержит четкие требования по оценке нео-
пределенности измерений и указания, когда и каким образом их следует включать
в отчет. Стандарт ИСО/ТЕС 17025 обязует сообщать о неопределенности измерений
по требованию клиента и в случаях использования и интерпретации результатов
измерений в рамках заданных спецификаций или пределов. Неопределенность из-
мерений должна быть легко доступна и должна указываться вместе с результатом
в виде Х± U, где U— расширенная погрешность [17,47, 51, 54]. Eurachem и Комитет
CCMAS Комиссии по «Кодексу Алиментариус» рассматривают неопределенность
измерений как отдельную тему [14, 18—20]. Некоторые даже утверждают, что не-
определенность измерений станет основным объединяющим принципом качества
аналитических данных [37].
8.2.2.Э. Заключение
В настоящее время внимание уделяется не столько используемым методам анали-
за и методологии, сколько качеству и надежности окончательных результатов. Это
происходит под влиянием более строгих регуляторных требований, большей ком-
петентности клиентов, желающих знать уровень достоверности заявленных резуль-
татов, и под влиянием новых веяний более жестких европейских и международных
стандартов, таких как ИСО//ЕС 17025 по аккредитации лабораторий. На основе
качества и надежности аналитических данных устанавливается межлабораторная
сравнимость результатов. Для того чтобы результаты были сравнимы, они должны
записываться с указанием неопределенности измерений и прослеживаться до обще-
принятых стандартов. Методики необходимо валидировать для демонстрации того,
что они действительно измеряют то, что должны, то есть пригодны для назначенно-
го использования.
Аналитическая методика представляет собой сложный многоэтапный процесс,
начинающийся с отбора проб и заканчивающийся обработкой результатов. Хотя
у каждой методики есть своя цель, область применения и аналитические требова-
ния, основные принципы валидации и обеспечения качества одинаковы независи-
мо от типа методики и ее применимости. Информация в настоящей главе в основ-
ном относится к аналитической химии, но также актуальна и в других областях.
Валидация аналитических методик, установление прослеживаемости результатов
и оценка неопределенности измерений должны осуществляться единым согласо-
ванным образом, в соответствии с международно принятыми стандартами таких
организаций, как Eurachem, ИЮПАК и ИСО.
Новый взгляд на аналитическое качество основывается на общем понимании
вопросов валидации методик, прослеживаемости и неопределенности измерений.
В данном разделе была рассмотрена взаимосвязь этих понятий. Была выбрана, об-
работана и обобщена наиболее полезная информация из большого числа руководств
и нормативных документов. Обсуждались различные подходы к оценке прослежи-
ваемости и неопределенности измерений аналитических методик в целом. Главное
место было отведено объяснению важности этих двух понятий и их связям с валида-
цией методик и с обеспечением аналитического качества.
Глава 8.2. Валидация аналитических методик и обеспечение качества
865
8.2.3. Валидация методики и обеспечение качества
8.2.3.1. Роль валидации методики в обеспечении аналитического качества
Термины «валидация» и «обеспечение качества» широко распространены. Однако
многие аналитики и лаборатории не знают их точного значения - ни разницы, ни
взаимосвязи между этими терминами. Валидация методики — проверка, достигну-
та ли аналитическая цель методики, то есть получение аналитических результатов
с приемлемым уровнем погрешности [4]. Валидация аналитической методики об-
разует первый уровень обеспечения качества в лаборатории (рис. 6). Обеспечение
аналитического качества — выполнение лабораторией полного перечня мер, гаран-
тирующих, что она способна постоянно получать данные высокого качества. Поми-
мо использования валидации и/или стандартизованных методик, к данным мерам
относятся эффективные процедуры внутреннего контроля качества (использова-
ние стандартных материалов (образцов), контрольных карт и т. д.), участие в про-
граммах профессионального тестирования (внешнего контроля) и аккредитация по
международным стандартам, обычно UCO/IEC17025 [2,4, 6].
Различные уровни, изображенные на рис. 6, представляют меры, которые долж-
на предпринять лаборатория для гарантии того, что она компетентна в проведении
аналитических измерений, соответствующих установленным требованиям. Лабо-
ратория должна представлять аналитические данные требуемого качества. «Уста-
новленные требования» аналитической методики и «требуемое -качество» анали-
тического результата относятся к пригодности методики для предназначенного
применения [4, 8, 15].
Рис. 6. Различные уровни обеспечения качества измерений для химико-аналитических
лабораторий и лабораторий по контролю пищевых продуктов [4, 8,15]
866 Часть 8. Валидация
Определение валидации по ИСО: «Подтверждение путем изучения и представ-
ления объективных свидетельств того, что определенные требования по указанно-
му предполагаемому предназначению выполнены» [15]. Валидация необходима для
«подтверждения пригодности определенной аналитической методики своему на-
значению», а это означает демонстрацию того, что «определенный протокол мето-
дики, применимый к определенному типу испытуемого материала и определенной
концентрации анализируемого вещества [они вместе называются аналитической
системой} пригоден для назначенной аналитической цели» [4]. Эта аналитическая
цель отражает достижение аналитических результатов с приемлемым уровнем пра-
вильности. Аналитический результат всегда должен сопровождаться указанием по-
грешности, определяющей интерпретацию результата (рис. 6). Другими словами,
интерпретация и использование любых измерений полностью зависят от связанной
с ними погрешности (при заявленном уровне достоверности) [8]. Таким образом,
валидация — это инструмент, показывающий, что определенная аналитическая ме-
тодика действительно измеряет то, что должна измерять, тем самым подтверждая
свою пригодность для предназначенного применения [11, 55, 56].
Валидация требуется, прежде всего, для любых новых методик. В соответствии
с определением валидация всегда касается определенной аналитической системы.
Это означает, что для определенного материала в указанном рабочем диапазоне
концентраций методика должна решать поставленную аналитическую задачу [4].
Как следствие, ревалидация необходима при изменении любого компонента анали-
тической системы или если появились признаки того, что используемая методика
больше не соответствует предъявляемым требованиям [15, 56, 57].
Валидация методики близко связана с разработкой методики. При разработке
новой методики некоторые параметры проверяются уже на этапе разработки, хотя
на самом деле эти действия составляют часть этапа валидации [15]. С другой сто-
роны, валидационные исследования могут указывать на необходимость изменения
протокола методики, что может потребовать ревалидации [58].
До начала валидации любой методики должна быть определена область примене-
ния валидации, заключающая в себе аналитическую систему и аналитические требо-
вания. Описание аналитической системы включает назначение и тип методики, тип
и диапазон концентраций анализируемого вещества или веществ, типы материалов
или сред, для которых применяется методика, и протокол методики. В основе каче-
ственного анализа лежит четкое указание аналитических требований. Этот анализ
должен отражать минимальный критерий пригодности для назначенного использо-
вания или различные рабочие параметры, которым должна соответствовать методи-
ка, чтобы решать конкретную задачу. Например, может потребоваться минимальная
прецизионность 5% (RSD, см. ниже) или предел обнаружения 0,1% (м./м.) [2,4, 15,
58]. Установленные критерии рабочих характеристик образуют основу для оконча-
тельной приемлемости аналитических данных и валидированной методики [58].
Валидация новой аналитической методики обычно проводится на двух уровнях.
Первый этап — предвалидация, направленная на установление области примене-
ния валидации. Второй этап — расширенная, или «полная», валидация, проводимая
в виде совместного исследования (двумя лабораториями) или межлабораторных
испытаний (несколькими лабораториями). Цель полной валидации, проводимой
Глава 8.2. Валидация аналитических методик и обеспечение качества 867
с участием минимального количества лабораторий, — демонстрация того, что мето-
дика обеспечивает такое же качество результатов, как и после предвалидации.
8.2.3.2. Руководства и рекомендации по обеспечению аналитического
качества
Как видно из рис. 6, использование валидированных методик — первый необходи-
мый этап обеспечения качества испытаний в рамках внутреннего контроля. Второй
этап необходим для участия в программах профессионального тестирования, кото-
рые являются необходимым условием для аккредитации [4].
Руководства и рекомендации для различных этапов обеспечения качества, пред-
ставленных на рис. 6, подробно описаны несколькими регуляторными органами,
агентствами по стандартизации, рабочими группами и комитетами. Что касается
прослеживаемости и неопределенности измерений (п. 8.2.2), то соответствующие
руководства перечислены в табл. 3. Руководства Eurachem касаются лабораторного
качества в целом [2], валидации методик [15] и программ профессионального тести-
рования [59]. Руководство объединенной группы Eurachem—Eurolab—ЕА [60] посвя-
щено профессиональному тестированию. На европейском уровне также существует
CEN, состоящий из различных технических комитетов и рабочих групп по стандар-
тизации аналитических методик для всех областей [61].
На международном уровне выделяются ИЮПАК, ИСО и АОАС International. Все
эти организации разрабатывают подходы к валидации и стандартизации в анали-
тической химии. Ассоциацией АОАС International была разработана программа со-
вместной проверки методик [62]. При совместном сотрудничестве ИЮПАК, ИСО
и АОАС International был разработан ряд согласованных друг с другом руководств
[4, 8, 63—67] в дополнение к стандартам ИСО [68—71]. Кроме того, FDA, USPuICH
разработали руководства специально для фармацевтических и биотехнологических
методик [55, 72—74].
Международная комиссия по «Кодексу Алиментариус», работающая в рамках
ФАО ООН и программы пищевых стандартов ВОЗ, создала Комитет по аналити-
ческим методикам и отбору образцов (CCMAS). Данный комитет разрабатывает
критерии оценки приемлемости методик анализа, а также руководства по внутри-
и межлабораторной валидации методик [75—78]. Для внутрилабораторной валида-
ции CCMAS придерживается согласованных руководств ИЮПАК [4]. На между-
народном уровне также имеются руководства по программе профессионального
тестирования (внешний контроль) [79] и аккредитации [80, 81] (табл. 3), разрабо-
танные ИЛАК.
8.2.3.3. Подходы к оценке приемлемых методов анализа
Целью аналитической методики является представление количественного и/или
качественного результата с приемлемым уровнем погрешности. Поэтому, теорети-
чески, «валидация» сводится к «измерению погрешности». На практике валидация
методик проводится путем оценки ряда рабочих характеристик методики, таких как
прецизионность, правильность, селективность или специфичность, линейность, ра-
бочий диапазон, процент обнаружения, предел обнаружения, предел количествен-
ного определения, чувствительность, устойчивость или робастность, пригодность.
868
Часть 8. Валидация
Таблица 3 Обзор европейских и международных организаций и регуляторных органов и разработанных ими руководств и стандартов по различным вопросам обеспечения аналитического качества
Организация Полное название Тематика руководств Ссылки
Eurachem/ Европейская ассоциация Валидация методик, [2,15,
CITACu ЕА по аналитической химии/ Международная ассоциации по прослеживаемости в аналитической химии и Ассоциация органов по аккредитации стран-членов ЕС проверка квалификации, обеспечение качества, аккредитация 59,60]
CEN Европейский комитет по стандартизации Стандартизация [61]
ИЮПАК, Международный союз Валидация методик, [4,8,
ИСО и АОАС International теоретической и прикладной химии, Международная организация по стандартизации, Международная ассоциация химиков-аналитиков стандартизация, внутренний контроль качества, профессиональное тестирование, аккредитация 62-71]
FDA, USP и ICH Федеральное управление США по контролю за пищевой продукцией и лекарствами, Фармакопея США, Международная конференция по гармонизации Валидация методик [55, 72-74]
ФАО/ВОЗ: Codex/CCMAS Продовольственная и сельскохозяйственная организация ООН/ Всемирная организация здравоохранения, Комитет по аналитическим методикам и отбору образцов Комиссии по «Кодексу Алиментариус» Валидация методик [21, 75-78]
ИЛАК (ILAQ Международная ассоциация органов по аккредитации лабораторий Профессиональное тестирование (внешний контроль), аккредитация [79-81]
Калибровка и прослеживаемость также упоминались как рабочие характеристики
методики [2, 4]. К ним можно добавить неопределенность измерений, хотя она яв-
ляется основным показателем как пригодности для предназначенного применения
(соответствия назначению) методики, так и стабильной надежности аналитических
результатов, получаемых в лаборатории (внутренний контроль качества). Неопре-
деленность измерений — всеобъемлющий параметр, включающий все источники
ошибок, и, таким образом, она выходит за рамки только валидации методики. На
практике данные, полученные при валидации методики и межлабораторных испы-
таниях, образуют основу, но не заменяют вычисления неопределенности измере-
ний. Таким образом, неопределенность измерений выходит за рамки определения
Глава 8.2. Валидация аналитических методик и обеспечение качества 869
«рабочий параметр методики», сформулированного в п. 8.2.2. На протяжении мно-
гих лет понятие неопределенности измерений привлекало все большее внимание во
всех аналитических областях, что привело к появлению двух различных подходов,
принимаемых и используемых в настоящее время для валидации аналитических ме-
тодик.
Традиционный подход оценки по критериям заключается в выявлении опреде-
ленных рабочих параметров и присвоении им числовых значений. Последние пред-
ставляют предельные или пороговые значения, которым должны соответствовать
параметры методики, чтобы она считалась приемлемой. Альтернативный подход
больше внимания уделяет соответствию назначению и погрешностям измерений.
При таком подходе, учитывающем соответствие назначению, общая неопределен-
ность измерений оценивается как функция концентрации анализируемого веще-
ства (см. п. 8.2.2).
В общем случае подход оценки по критериям используется для методик прямого
измерения, то есть методик, в которых результаты измерений могут быть получены
независимо от используемой методики. В противоположность рациональным мето-
дам существуют эмпирические методики (косвенный метод измерения), в которых
измеряемая величина зависит от используемой методики. Для эмпирических мето-
дик подход оценки по критериям неприменим. Вместо этого в качестве основы для
оценки неопределенности измерений и валидации обычно используются данные
прецизионности из межлабораторных исследований [75, 76].
Валидация необходима для того, чтобы показать, что аналитическая методика
соответствует установленным критериям различных рабочих характеристик [82].
Для аналитической методики эти характеристики оцениваются индивидуально: на
входе — очищенный или экстрагированный аналит, на выходе — аналитический
результат. Однако неопределенность измерений охватывает всю процедуру про-
ведения испытания, начиная с исходной серии, от которой отбираются образцы.
Оценка неопределенности измерений (см. и. 8.2.2) согласуется с так называемым
модульным подходом к валидации. Модульная валидация соответствует «модуль-
ности» (этапности) процедуры проведения испытания, разделенной на несколь-
ко последовательных этапов, необходимых для анализа материала. Сюда можно
отнести подготовку проб, экстракцию определяемых соединений и определение
анализируемого вещества (рис. 7). Каждый этап процедуры можно рассматривать
как аналитическую систему, валидацию которой можно провести отдельно, а затем
объединить с другими «модулями» в нужной последовательности. Модульная вали-
дация, таким образом, представляет собой пошаговую валидацию всей процедуры
испытания, учитывающую все возможные сложности или факторы неопределен-
ности на каждом ее этапе. Концепция модульной валидации берет начало в сфере
предиктивной микробиологии и в настоящее время предлагается для методов ана-
лиза генетически модифицированных организмов [83]. Взаимосвязь между тремя
валидационными подходами, описанными выше, представлена на рис. 7.
8.2.3.4. Рабочие характеристики методик и подход оценки по критериям
Объем работ по валидации зависит от типа валидируемой методики. С одной сторо-
ны, объем работ по валидации и выбор оцениваемых рабочих характеристик зависят
870
Часть 8. Валидация
— предел обнаружения и
предел количественного
определения;
— процент обнаружения;
— робастность или
устойчивость
Рис. 7. Схематическое представление аналитической методики в процессе анализа и различные
подходы к валидации; f(c) — функция концентрации
от статуса методики и опыта, имеющегося по ее выполнению. С другой стороны,
план валидации определяется аналитическими требованиями на основе потребно-
стей клиента или в соответствии с требованиями нормативных документов. Если
методика ранее была полностью провалидирована в соответствии с международным
протоколом [63, 68], то лаборатории не обязательно проводить обширные самосто-
ятельные валидационные исследования. Следует проверить только, что могут быть
достигнуты такие же рабочие характеристики, как и при межлабораторных иссле-
дованиях. По крайней мере следует провести определение прецизионности, систе-
матической ошибки, линейности и устойчивости. Схожая сокращенная валидация
требуется в случаях, когда рассматривается полностью валидированная методика,
выполняемая на новом материале, хорошо отработанная методика, но не прошед-
шая межлабораторных исследований, и методика, описанная в научной литературе,
с приведенными характеристиками. Более основательная валидация требуется для
методик, описанных в литературе без указания каких-либо характеристик, а также
для методик, разработанных внутри компании [84].
Какие рабочие параметры следует определять, зависит также от назначения ме-
тодики. Разные рекомендации ICH/USP специально установлены: 1) для методик
идентификации; 2) методик испытаний на примеси; 3) методик количественного
определения. Для методик идентификации подтверждают подлинность анализируе-
мого вещества в пробе путем сравнения с известным стандартным материалом (об-
разцом). Методики оценки примесей предназначены или для подтверждения нали-
чия примеси (испытание на предельное содержание) или для определения точного
содержания примеси, при этом под примесью подразумевается вещество, которое
обычно отсутствует в образце. Наконец, методика количественного определения
Глава 8.2. Валидация аналитических методик и обеспечение качества
871
предназначена для определения содержания значимого компонента (например, ан-
тимикробного консерванта) или действующего вещества в образце и для измерения
содержания действующего вещества в коммерческой фармацевтической субстан-
ции или действующего вещества в лекарственном препарате.
Для методик количественного определения, при которых значимый компонент
или действующее вещество должны присутствовать в больших количествах, следует
рассматривать параметры, отличные от используемых при валидации методик для
примесей. Этот принцип относится и к методикам количественного определения
в сравнении с методиками на подлинность и предельное содержание примесей
(табл. 4) [55, 56,72, 85].
Таблица 4
Критерии, определяемые для различных категорий аналитических методик
Рабочий параметр методики Испытание на подлинность Испытания на примеси Количественное определение
Предельное содержание Количественное содержание
Прецизионность а - + +
Правильность - а + +
Специфичность + + + +
Предел обнаружения (ПО) а + - -
Предел количественного а +
определения (ПКО)
Линейность а — + +
Диапазон применения _а а + +
Устойчивость + + + +
а Испытание может проводиться. Источник:. [56].
В литературе приводится большое число практических рекомендаций по опреде-
лению рабочих характеристик методик [58]. Помимо многообразия подходов, также
различны терминология и способы оформления результатов. Различия в основном
зависят от назначения и области применения методики, валидационные исследо-
вания могут усложниться с увеличением сложности анализов [86]. Далее приво-
дятся термины и формулы из принятой ИЮПАК номенклатуры для представления
результатов химических анализов [66]. Для каждого валидационного параметра
в табл. 5 приведены определения, способы выражения, руководства по определе-
нию и критерии приемлемости.
Точность. Прецизионность и систематическая ошибка, являющиеся частью
оценки неопределенности измерений, являются наиболее важными валидацион-
ными параметрами.
Определение прецизионности разделяют на: 1) определение прецизионности по-
вторяемости 5 или SD (sr или SD) и RSD (RSD); 2) внутрилабораторное определение
прецизионности воспроизводимости или определение промежуточной прецизион-
ности SD и RSD; 3) межлабораторное определение прецизионности воспроизводи-
мости s или SD (sR или SDr) и RSD (RSDr) [66].
872
Часть 8. Валидация
Обобщающая таблица
Параметр Определение
Точность Степень близости результата испытания принятому стандартному значению
1. Прецизионность Степень близости результатов независимых испытаний, полученных при заданных условиях
1.1. Прецизионность Прецизионность при условиях, когда получают результаты
повторяемости (внутриопытная прецизионность) независимых испытаний с помощью одной и той же методики на идентичных образцах в одной лаборатории, одним аналитиком, с использованием того же оборудования за короткий промежуток времени
1.2. Промежуточная прецизионность (межопытная прецизионность) Прецизионность при условиях, когда получают результаты независимых испытаний с помощью одной и той же методики на идентичных образцах в одной лаборатории, но разными аналитиками, с использованием разного оборудования в течение более длительного периода времени
1.3. Прецизионность Прецизионность при условиях, когда получают результаты испытаний
воспроизводимости (межлабораторная прецизионность) с помощью одной и той же методики на идентичных образцах в разных лабораториях, разными аналитиками, с использованием разного оборудования
2. Правильность Степень близости ожидаемого результата испытания (ожидаемое среднее значение) принятому стандартному значению (истинному значению)
Глава 8.2. Валидация аналитических методик и обеспечение качества
873
по рабочим параметрам методики
Таблица 5
Математическое выражение
Требования
Оценка на практике
• Стандартное отклонение s
или SD.
• Относительное стандартное
отклонение s , или RSD.
• Коэффициент вариации
%СКили%.
. RSD =100-50/х.
• Предел повторяемости
г= 2,83 50. и предел воспро-
изводимости R — 2,83 50л.
• Доверительный интервал
С1=х + С, где C = S t^
•Jn
• RSD= 0,5 при 0,6-кратном
теоретическом значении,
определенном функцией
Горвица 2ехр(1 — 0,51ogQ,
где С — концентрация
аналита в виде десятичной
дроби.
• RSDr = 0,5 при 2-кратном
теоретическом значении,
определенном функцией
Горвица.
• Коэффициент Хоррата —
RSDr (в опыте)/ RSDr (по
Горвицу) < 2.
• Значения RSDr и RSDr
в соответствии с програм-
мой АОАС по совместной
проверке методик
• Минимум на трех уровнях кон-
центрации, охватывающих весь
диапазон аналитической мето-
дики (при пределе обнаруже-
ния, среднем и максимальном).
• Минимум на трех повторно-
стях для каждого уровня кон-
центрации.
• Вычислить прецизионность
повторяемости SDr, RSDt,
r=2,8SDr, С, CI.
• Вычислить промежуточную
прецизионность 5D(n(, RSD.nl,
r=2,8SD „С, CI.
• Вычислить прецизионность
воспроизводимости SDR,
RSDR,r=2fiSDR, С, CI.
• Составить столбчатую диа-
грамму или контрольную карту
Если используется сертифици- Общие: сравнить результаты • Минимум на трех уровнях
рованный стандартный образец с результатами второй ва- концентрации, охватывающих
вещества: лидированной стандартной
систематическая ошибка = % методики.
ошибки: разница между по- Если используются стан-
лученной величиной и истин- дартные образцы:
ным (опорным) значением; Z-показатель < |2|.
Z-показатель: разница между используется метод до-
полученной величиной и зна- бавок:
чением сертифицированного
стандартного образца, %:
у — Y
g___ лопыт л стандарт
весь диапазон аналитической
методики (при пределе обна-
ружения, среднем и макси-
мальном).
Минимум три параллели по
10 повторностей для каждого
уровня концентрации.
процент обнаружения до- • Используются стандартные
бавленного аналита в соот-
ветствии с руководствами
USP/ICH (в зависимости от
уровня концентрации ана-
лита)
образцы: вычислить система-
тическую ошибку (% ошибки)
и/или Z-показатель.
Без стандартных образцов:
добавить известное количе-
ство анализируемого веще-
ства в контрольный раствор
(содержит все компоненты,
обычно присутствующие в ис-
Если сертифицированные стан-
дартные образцы не доступны:
процент обнаружения извест-
ного количества определяе-
мого вещества, добавленного
в контрольный раствор
пытуемом растворе, кроме
аналита) и раствор сравнения
(растворитель); вычислить %
обнаружения в виде 100 [кон-
трольный раствор с аналитом/
раствор сравнения с анали-
том]
874
Часть 8. Валидация
Параметр Определение
Процент обнаружения Известное количество аналита, добавляемое в пробу (аналит прибавляется или в контрольный раствор или в испытуемый раствор) до проведения анализа и измеряемое с помощью методики
Специфичность Способность методики однозначно и избирательно определять анализируемое вещество в присутствии других компонентов в образце (наличие которых в образце наиболее вероятно) при заданных условиях испытания (специфичность = 100% селективности)
Глава 8.2. Валидация аналитических методик и обеспечение качества
875
Таблица 5 (продолжение)
Математическое выражение
Требования
Процент обнаружения
известного количества
определяемого вещества
Процент обнаружения
в соответствии
с руководствами USP/ICH
(в зависимости от уровня
концентрации аналита)
Примечание: нельзя выразить
математически, необходимо
показать на опыте; зависит от
типа и назначения методики.
Для методик идентификации:
процент правильной
классификации:
• образцов, не содержащих
анализируемого вещества,
как отрицательных;
• образцов, содержащих
анализируемое вещество,
как положительных.
Способность различать
компоненты с родственной
структурой (отрицательные
результаты).
Для количественных методик:
процент обнаружения
в образцах, в которые
добавлены возможные
мешающие идентификации
вещества
Для методик
идентификации:
процент правильной
классификации ~ 100.
Для методик
количественного
определения:
процент обнаружения
в соответствии
с руководствами USP/ICH
(в зависимости от уровня
концентрации аналита)
______Оценка на практике_____
• Минимум шесть
повторностей контрольного
раствора или испытуемого
раствора без добавления
аналита и раствора
(растворов) с добавлением
аналита при разных
концентрациях.
• Вычислить % степени
обнаружения в виде 100
[контрольный раствор
с аналитом/ контрольный
раствор без аналита]
Методики идентификации
• Определить процент ложно-
положительных результатов
для минимального числа
растворов сравнения (не со-
держащих аналита).
• Определить процент ложно-
отрицательных результатов
для минимального числа об-
разцов, содержащих аналит.
• Испытать минимальное число
образцов, содержащих веще-
ства с похожей или родствен-
ной структурой, результаты
испытаний должны быть
отрицательными.
Методики количественного
определения и на содержание
примесей
• Ввести определенное количе-
ство возможных мешающих
испытанию веществ в разные
образцы и вычислить процент
обнаружения.
• Если возможные мешающие
испытанию вещества недо-
ступны, сравнить с результа-
тами второй методики
876
Часть 8. Валидация
Параметр Определение
Предел обнаружения Наименьшая концентрация или количество анализируемого
(ПО) вещества, которое: • может быть достоверно обнаружено (т. е. подтверждено его наличие); • может быть определено или измерено с приемлемой статистической значимостью
Предел количественного определения (ПКО) Наименьшая концентрация или количество анализируемого вещества, которое может быть определено количественно с приемлемым уровнем повторяемости, прецизионности и правильности
Глава 8.2. Валидация аналитических методик и обеспечение качества 877
Таблица 5 (продолжение)
Математическое выражение Требования Оценка на практике
• Примечание. ПО выражается как концентрация или количество анализируемого вещества, рассчитанное по величине измеренного сигнала. Наименьший сигнал: XL = ХЫ + ^SbV • Наименьшая концентрация/ количество: ПО = ?£(с£) = xL/S= 3stl/S. • Примечание'. SDblank трехкратного значения сигнала соответствует %RSD, равному 33% • Примечание-. ПКО выражается как концентрация или количество анализируемого вещества, рассчитанные на основании величины измеренного сигнала. • Наименьший сигнал: = хы + 10$ы. • Наименьшая концентрация/ количество: ПКО = ^(е£)=х£/5= 10sbl/S. • Примечание: SDblank • Измерить по крайней мере 10 параллелей раствора сравнения (растворителя) в одной повторности (Х£ = Х„+ЭМИЛИЮ параллелей раствора сравнения, содержащих аналит в минимально допустимой концентрации (Хг=35). • Выбрать уровни низких концентраций, находящихся около ожидаемого ПО. • Построить калибровочную кривую: сигнал х=А - концентрация + Б; таким образом, xL = А ПО. • Определить наклон кривой Л и вычислить ПО: ПО = х£/Л=[хы+35и]/Л • Измерить однократно не менее 10 параллелей раствора сравнения (xL = xbl + 10siZ). • Выбрать уровни низких концентраций, находящихся около вычисленного ПО. • Построить калибровочную кривую: сигнал х=А концентрация + В; таким образом, xL = А • ПКО. • Определить наклон кривой Л и вычислить ПКО: ПКО = х£/Л= [хы+lOsJ/Л
десятикратного значения
сигнала соответствует
%RSD, равному 10%
878 Часть 8. Валидация
Параметр Определение
Линейность. Линейный диапазон = рабочий диапазон = пределы линейности Способность методики получать результаты испытаний, пропорциональные концентрации анализируемого вещества (в рамках заданного диапазона). Диапазон концентрации или количества анализируемого вещества, при котором: • методика дает результаты, пропорциональные концентрации анализируемого вещества, или • может применяться линейная калибровочная модель с известным уровнем достоверности
Глава 8.2, Валидация аналитических методик и обеспечение качества 879
Таблица 5 (продолжение)
Математическое выражение Требования Оценка на практике
• Примечание: линейность Для методик количествен него нельзя выразить определения: математически, необходимо диапазон = 80-120% показать графически. уровня аналита. • Диапазон: диапазон Для методик на содержание концентраций между примеси: верхним и нижним диапазон = ПКО 50—150% пределами линейности или 0—150% в соответствии с руководствами USP/ICH и ИЮПАК; нижний предел линейности = ПКО • Первичная грубая оценка линейного диапазона: измерить раствор сравнения + отдельно приготовленные растворы не менее шести концентраций; построить калибровочную кривую и определить линейный диапазон визуально. Выбрать сертифицированные стандартные образцы или образцы с известной концентрацией (минимум шесть различных концентраций), входящие в установленный линейный диапазон. ’ Вычислить остаточные У-значения по калибровочной кривой и построить график их зависимости от концентрации. • При случайном распределении -> линейность; при систематическом тренде -> отсутствие линейности. • Вычислить относительные сигналы в виде сигнал/ концентрация; построить график функции их зависимости от концентрации. При горизонтальной линии —> линейность; если пределы линейности соответствуют 95 и 105% значений относительных сигналов —> диапазон
880
Часть 8. Валидация
_____Параметр_______________________________Определение________________________
Устойчивость Характеристика процедуры испытания (проверенная при
внутрилабораторных исследованиях) при внесении незначительных
изменений в рабочие и/или производственные условия
(общепринятый термин)
Робастность Мера способности аналитической методики сохранять неизменными свои характеристики несмотря на преднамеренное внесение незначительных изменений в различные условия выполнения испытания, что позволяет судить о надежности методики при нормальном использовании (термин используется только USP/ICH)
Чувствительность Отношение изменения ответа измерительного прибора к соответствующему изменению входного сигнала
Источник: [4, 15, 21, 55, 56, 72, 75,76].
Примечания. В таблице приведены определения, способы выражения, требования или
критерии приемлемости, руководства по практической оценке (подробнее см. текст). 1.1 —
критерий Стьюдента, соответствующий уровню достоверности 1 — а и v степеням свободы.
Символ р обозначает процентиль или процентный пункт распределения t. Для односторон-
них интервалов р = 1 — а, для двусторонних р = 1 — а/2. Значения t можно найти в номен-
клатуре ИЮПАК (Г = 2,776 для п = 5 и t = 3,182 для п = 4 при р = 0,95) [67]. 2. X— среднее
найденное значение; п — число измерений, для которых вычислялось SD. Если данные по SD
стандартных образцов недоступны, можно использовать границы доверительного интервала
на уровне 95% для оценки SD стандартных образцов (см. второй вид формулы определения
Z-показателя) [21]. 3. хы — среднее значение измерений растворов сравнения; зи — АОрезуль-
татов измерения растворов сравнения; S— чувствительность методики или наклон калибро-
вочной функции. Калибровочная функция — взаимосвязь между измеренным откликом xL
и концентрацией cL или количеством qL [56,72,95—96].
Глава 8.2. Валидация аналитических методик и обеспечение качества 881
Таблица 5 (окончание)
Математическое выражение Требования Оценка на практике
Устойчивость: мера вариабельности (воспроизводимость результатов, полученных при разных условиях); выражается как %RSD (межлабораторный) • Оценить по отдельности влияние различных незначительных изменений условий испытания (в днях, оборудовании, аналитиках, реактивах, материалах, количестве используемого для приготовления испытуемого образца и т. д.). • Вычислить данные по прецизионности
Наклон калибровочной
кривой (произвольно
выбранный)
882
Часть 8. Валидация
Помимо стандартного отклонения и коэффициентов вариации, значения и пре-
делы повторяемости или воспроизводимости (г, К) являются дополнительными
параметрами, имеющими высокое значение при оценке прецизионности (см. фор-
мулы в табл. 5). Эти параметры означают, что абсолютное отклонение между дву-
мя независимыми результатами — полученными в одной и той же лаборатории или
в разных лабораториях — может выходить за рамки значений г и R максимум в 5%
случаев [2]. Другой мерой прецизионности является доверительный интервал, при
котором все измерения попадают в определенный вероятностный или доверитель-
ный уровень 1 — а (а обычно 0,05 при вероятности 95%) [66].
Вычисленные значения повторяемости, промежуточной прецизионности и вос-
производимости можно сравнить со значениями существующих методик. Если
методики, с которыми можно сравнить параметры прецизионности, отсутствуют,
возможно найти стандартные отклонения теоретической относительной воспроиз-
водимости и повторяемости по уравнению Горвица (Horwitz) и коэффициенту Хор-
рата (Horrat) (табл. 5). Значения RSD по Горвицу представлены в табл. 6. Более высо-
кая вариабельность ожидается при приближении уровней анализируемого вещества
к пределу обнаружения (см. ниже). Наряду с уравнением Горвица программа АОАС
по совместной проверке методик предлагает собственные уровни приемлемости
%RSDb виде функции уровня концентрации аналита [56,72].
Таблица 6
Функция Горвица
Содержание анализируемого вещества в % (в образце) Доля аналита (в образце) Единица %RSD по Горвицу AOACPVM
100 1 100% 2 1,3
10 1-ю-1 10% 2,8 2,8
1 1-ю-2 1% 4 2,7
0,1 1-Ю-3 0,10% 5,7 3,7
0,01 110-’ 100 ррт 8 5,3
0,001 1-10-5 10 ррт п,з 7,3
0,0001 110-6 1 ррт 16 11
0,00001 1-Ю-7 100 ppb 22,6 15
0,000001 1-10-8 10 ppb 32 21
0,0000001 1-10-’ \ppb 45,3 30
Примечание. Эта функция дается как эмпирическая взаимосвязь между прецизионностью
аналитической методики и концентрацией анализируемого вещества независимо от природы
аналита, состава и используемого метода. Приемлемые значения RSDX и RSDr даются в соот-
ветствии с [75] и АОАС International [56, 62]; %RSD — процент относительного стандартного
отклонения; PVM — программа совместной проверки методик.
Данные прецизионности можно представить в виде столбчатых диаграмм или
контрольных карт, таких как контрольные карты Шухарта (Shewhart) (см. обсужде-
ние внутреннего контроля качества в п. 8.2.3.5). Столбчатые диаграммы показывают
значения %RSDc соответствующим доверительным интервалом. Контрольные кар-
ты отражают результаты отдельных измерений и средние значения ряда измерений
Глава 8.2. Валидация аналитических методик и обеспечение качества
883
с их доверительным уровнем (или с горизонтальными линиями, обозначающими
«пределы», см. ниже) как функцию числа измерений и числа опытов соответствен-
но [15, 55, 56, 58, 72, 85].
Прецизионность относится к случайной ошибке измерительной системы (рис. 8)
и является компонентом неопределенности измерений (см. также п. 8.2.2 и рису-
нок) [2].
Правильность выражается посредством систематической ошибки или процента
ошибок. Систематическая ошибка — разница между средним найденным значением
для анализируемого вещества и принятым истинным (опорным) значением или из-
вестным действительным содержанием [87]. Она представляет систематическое от-
клонение измеренных результатов от истинных значений. Правильность методики —
индикатор пригодности и применимости этого метода для реальных образцов [88].
Различные источники систематических ошибок вносят вклад в общую систе-
матическую ошибку (см. рис. 8). Томпсон и Вуд (Thompson, Wood) [8] описывают
постоянную систематическую ошибку как систематическую ошибку, влияющую на
все данные аналитической системы в течение длительных промежутков времени,
которая при этом является относительно малой, но постоянно присутствующей.
В общую систематическую ошибку вносят вклад различные компоненты, такие как
лабораторная систематическая ошибка, систематическая ошибка метода и влияние
компонентов образца. Наряду с постоянной систематической ошибкой наклады-
вается влияние испытания — систематическая ошибка аналитической системы при
проведении испытания [4, 8,15].
Как минимум один из этих компонентов систематической ошибки возникает при
проведении анализа стандартных материалов (стандартных образцов). Стандартные
материалы разделяют на сертифицированные (аттестованные общепризнанным ор-
ганом) стандартные материалы (CRM, certified reference material — это либо чистые
вещества и их растворы, либо многокомпонентые CRM) и лабораторные (несерти-
фицированные) стандартные материалы (LRM, laboratory reference material), также
называемые образцами для контроля качества [89]. Материалы CRM могут отно-
ситься ко всем типам систематической ошибки (метода, лаборатории и испытания);
они определяются с указанием неопределенности и прослеживаются до междуна-
родных стандартов. Поэтому CRMсчитаются удобными инструментами получения
прослеживаемости аналитических измерений, калибровки оборудования и методик
(в определенных случаях), контроля лабораторных работ и валидации методик и по-
зволяют сравнивать методики [4, 15, 30]. Однако использование CRMне обязатель-
но гарантирует правильность результатов. Лучшим способом оценки системати-
ческой ошибки на практике является многократное проведение анализа образцов
с известными концентрациями, таких как стандартные материалы (см. п. 8.2.2).
Идеальным стандартным материалом является многокомпонентный CRM, так как
он схож с анализируемыми образцами (такая схожесть называется совпадением по
компонентам). Правильный результат, полученный с помощью многокомпонент-
ного CRM, не гарантирует, что результат на неизвестном образце с другим составом
будет правильным [4, 89].
Пригодность CRM для валидации (в особенности при оценке правильности)
и определения прослеживаемости обсуждалась в течение многих лет. На данную
884
Часть 8. Валидация
Аналитический результат
Истинное значение
Ошибка
Ожидаемое значение
(предельное значение)
Разница между
аналитическим результатом
и истинным значением
| Погрешность |
Систематическая ошибка
Случайная ошибка
Систематическая
ошибка оценки
Разница между
ожидаемым и
истинным значением
Постоянная
систематическая
ошибка
Влияние
проведения
опыта
Разница между
аналитическим
результатом и
ожидаемым средним
значением
Случайная
погрешность
Воспроизводимость Промежуточная
[—Прецизионность^
Повторяемость
Межлабораторная
вариабельность,
исследуется в
межлабораторных
исследованиях
Вариации
внутри целой
аналитической
системы в течение
длительных
периодов
^Систематическая^
ошибка
। испытания
Вариации при
конкретном опыте
Внутрилаборатор-
ная вариабель-
ность, вызванная
случайными
воздействиями, —
вариабельность в
течение длитель-
ных периодов вре-
мени при разных
условиях
Прецизионность
испытаний —
вариабельность
в течение
коротких
интервалов
времени при
одинаковых
условиях
Влияние
изменений
состава
образца
Система-
тическая
ошибка
методики
Систематиче-
ская ошибка
лаборатории
Случайная систематическая ошибка
Показатель разницы
между ожидаемым и
истинным значениями
Истинность
Показатель разницы
между результатом и
ожидаемым значением
Прецизионность
Я
Анализ CRM +
статистический контроль
Анализ повторностей
Валидация внутри одной лаборатории
Минимально необходимо при валидации методики
Рис. 8. Формирование ошибки аналитического результата во взаимосвязи с правильностью
аналитической методики
Глава 8.2, Валидация аналитических методик и обеспечение качества
885
тему было опубликовано бессчетное число статей. В списке литературы мы приво-
дим лишь наиболее интересные [89—91]. Использование стандартного чистого веще-
ства, матричного сертифицированного стандарта или LRM описано в специальных
статьях журналов «Аккредитация и обеспечение качества» {Accreditation and Quality
Assurance', том 9, 2004) и «Аналитическая и биоаналитическая химия» {Analytical and
Bioanalytical Chemistry-, том 278, 2004) на примере биологических и экологических
стандартных материалов, а также в специальной статье журнала «Тенденции в ана-
литической химии» {Trends in Analytical Chemistry-, том 23, 2004) по вопросам дости-
жения прослеживаемости экологических измерений.
Если подобные (сертифицированные) стандартные материалы недоступны,
можно использовать контрольный раствор (раствор, содержащий все компоненты
испытуемого образца, за исключением аналита) и добавить в него известное коли-
чество чистого и стабильного вещества, приготовленного внутри компании. Такой
раствор называют раствором с добавкой или модельной смесью. Процент обнару-
жения вычисляется как процентное отношение найденного количества аналита,
добавленного в контрольный раствор, к найденному количеству аналита, добав-
ленного в раствор сравнения (растворитель), или известному количеству вещества,
добавленного к образцу. Чем меньше процент обнаружения, тем больше система-
тическая ошибка, влияющая на методику, и тем меньше правильность результатов
[4, 56, 72, 92, 93]. Показатель правильности также может быть получен сравнением
методики с другой хорошо известной стандартной методикой при условии, что из-
вестна прецизионность этой стандартной методики. Затем сравнивают результаты,
полученные двумя методиками на одинаковых образцах или сериях образцов. Об-
разцами могут быть CRM, внутренние (рабочие) стандартные образцы или просто
обычные образцы [15]. Сравнение трех способов нахождения систематической
ошибки описано в п. 8.2.2 по аналитическому качеству. Следует понимать, что ис-
пользование оценки процента обнаружения и сравнение методик являются альтер-
нативными путями, содержащими серьезные ограничения. Они могут дать общее
представление о сравнимости данных, но не гарантируют их правильность [89].
Правильность, или безошибочность, аналитического метода может быть пока-
зана на контрольной карте. На карте можно отразить либо разницу между средним
и истинным значением анализируемого RM{CRM) наряду с доверительными преде-
лами, либо процент обнаружения известного добавленного количества аналита [56,
62]. В данном случае опять же следует предпринять меры предосторожности в отно-
шении используемого стандарта. Контрольные карты могут быть полезны для оцен-
ки правильности, только если используется CRM, обладающий прослеживаемостью
до единиц СИ. Все остальные типы стандартных образцов позволяют лишь устано-
вить прослеживаемость до «согласованного» значения, которое не обязательно рав-
няется «истинному» значению [89]. Ожидаемые значения правильности или про-
цента обнаружения зависят от концентрации анализируемого вещества. Поэтому
правильность оценивают по крайней мере на трех различных концентрациях. Изме-
ренные значения процента обнаружения нужно сравнивать с принятыми критерия-
ми в соответствии с программой АОАС по совместной проверке методик (табл. 7)
[56, 62]. Помимо систематической ошибки и процента обнаружения, другой мерой
правильности является Z-показатель (табл. 5). Важно отметить, что значительная
886
Часть 8. Валидация
часть общей неопределенности измерений будет относиться к неопределенности
измерений систематической ошибки аналитической системы, включая погрешно-
сти стандартных материалов (образцов) (рис. 5 и 8) [2].
Таблица 7
Приемлемый процент обнаружения как функция концентрации анализируемого вещества
Процент содержания анализируемого вещества в образце Доля аналита Единица Среднее значение процента обнаружения, %
100 1 100% 98-102
10 1-10-1 10% 98-102
1 1-10-2 1% 97-103
од 110-’ 0,10% 95-105
0,01 1-Ю-4 100 ррт 90-107
0,001 1105 10 ррт 80-110
0,0001 140-6 1 ррт 80-110
0,00001 1-10-7 100 ppb 80-110
0,000001 1108 10 ppb 60-115
0,0000001 1-10-9 ippb 40-120
Процент (степень) обнаружения обычно выделяют как отдельный валидацион-
ный параметр (табл. 5). Аналитические методики предназначены для определения
истинного значения концентрации анализируемого вещества с погрешностью, при-
емлемой для предназначенного применения. Однако в подобных аналитических
методиках анализируемое вещество переносят из сложных многокомпонентых рас-
творов в более простые, при этом имеет место потеря аналита. Как следствие, изме-
ряемое значение будет меньше, чем истинная концентрация в исходном материале.
Поэтому оценка эффективности методики в определении полного содержания при-
сутствующего в образце аналита является частью процесса валидации. Eurachem,
ИЮПАК, ИСО и АОАС International указывают на необходимость определения про-
цента обнаружения в рамках валидации методики. Исследования процента обна-
ружения в простых или в модельных смесях должны проводиться для различных
типов составов смесей, нескольких вариантов каждого типа смесей и для каждого
типа смесей при различных уровнях концентрации аналита [2,4,15].
Специфичность и селективность. Специфичность и селективность дают пред-
ставление о надежности аналитической методики (определения см. в табл. 5). Одни
авторы дают различные определения для обоих терминов, в то время как другие
считают эти понятия идентичными. Термин специфичный, как правило, относится
к методикам, определяющим только одно анализируемое вещество, в то время как
селективные (избирательные) методики дают результаты для различных химических
молекул или аналитов, которые можно отличить друг от друга. Методика считается
селективной, если отклик на аналит отличим от всех остальных откликов. В этом
случае методика способна точно измерить аналит в присутствии других мешающих
определению веществ [56, 94]. По Eurachem специфичность и селективность отра-
жают одинаковую характеристику и близко связаны друг с другом таким образом,
Глава 8.2. Валидация аналитических методик и обеспечение качества
887
что специфичность означает 100% селективности. Другими словами, методика мо-
жет быть специфичной, только если она селективна на 100%. Другим близким по-
нятием является подтверждение подлинности, которое означает доказательство того,
что «измеряемый сигнал, принадлежащий аналиту, относится только к аналиту, а не
к присутствующим химически или физически сходным веществам, и не является
результатом совпадений» [15]. Методика должна показать высокую специфичность
прежде, чем будет проведен количественный анализ [87].
Не существует единой формы выражения специфичности. Скорее, эту характе-
ристику следует демонстрировать наглядно. То, каким образом это делается, зависит
от назначения и типа аналитической методики (см. ниже). Для испытаний подлин-
ности необходимо подтверждение подлинности аналита. Специфичность в данном
случае является способностью различать компоненты с родственной структурой,
которые могут присутствовать в пробе.
Для испытаний на примеси (испытание на предельное содержание примесей,
испытание на количественное содержание примесей) и испытаний на количествен-
ное определение акцент смещается на способность определения или различения
аналита в присутствии других веществ. Селективность может быть оценена путем
внесения в испытуемые образцы возможных мешающих определению веществ (на-
пример, продуктов разложения) [55, 56, 72].
Предел обнаружения. Нет других аналитических терминов или параметров, для
которых разнообразие терминологии и формулировок так же велико, как для преде-
лов обнаружения и количественного определения. Предел обнаружения или предел
чувствительности (детектирования) — наиболее широко используемый термин,
принятый Eurachem. ИСО использует термин минимально обнаружимая суммарная
концентрация, а ИЮПАК предпочитает минимально детектируемое (истинное) зна-
чение [15]. Тем не менее все официальные организации ссылаются на одно определе-
ние: наименьшее количество анализируемого вещества в пробе, которое может быть
обнаружено, но не обязательно определено количественно. В большинстве случаев
ПО выражается как концентрация cL или количество qL, полученное от наиболее
слабого сигнала xL, который может быть зафиксирован с достаточной достоверно-
стью для данной процедуры испытания. Наименьший сигнал xL — сигнал, который
в к раз больше SDblank — среднего значения отклика для раствора сравнения (раство-
рителя), где к — числовой коэффициент, выбираемый в соответствии с требуемым
уровнем достоверности [56, 72, 95—97].
Чем больше значение к, тем больше уровень достоверности. Eurachem и ИЮПАК
рекомендуют значение коэффициента к = 3, при котором вероятность того, что
сигнал величиной более 3s соответствует раствору сравнения (растворителю, под-
вижной фазе), составляет менее 1%. Предел обнаружения, таким образом, является
концентрацией или количеством, соответствующим уровню измерения (отклика,
сигнала), на три единицы sbl превышающему базовую линию (отклик при отсутствии
аналита в пробе) (табл. 5). При уровне концентрации или количества, вызывающем
сигнал, трехкратно превышающий sw, RSD или коэффициент вариации измеренно-
го сигнала составляет 33% (мера погрешности) [2, 4, 15, 75, 95, 98]. В соответствии
с USP/ICH предел обнаружения соответствует тому сигналу, в котором соотношение
сигнал/шум равно 2:1 или 3:1 [72, 85].
888
Часть 8. Валидация
Ошибочно считается, что обнаружение или количественное определение анали-
та невозможно ниже предела измерения, — просто на этих нижних уровнях погреш-
ность обнаружения или количественного определения выше, чем сама искомая ве-
личина [21]. В связи с этим Хубер (Huber) [56] определяет предел обнаружения как
точку, в которой измеряемая величина выше, чем связанная с ней погрешность.
Крулл и Шварц (Krull, Swartz) [88] рассматривают ПО как точку концентрации,
в которой возможно только качественное определение, но не точное и достоверное
количественное определение.
Для количественных методов ПО определяется как пороговая концентрация,
при которой испытание становится недостоверным. Серии растворов сравнения
с добавленными различными концентрациями анализируемого вещества исследуют
не менее чем в 10 повторностях. Пороговая, или предельная, концентрация опреде-
ляется визуально на основе кривой отклика, отмечающей процентное содержание
положительных результатов по отношению к концентрации. В связи с этим ПО так-
же определяют как концентрацию, при которой 95% испытаний дают однозначный
положительный сигнал [15].
ПО не следует путать с чувствительностью методики. Последняя является спо-
собностью методики определять незначительные различия в концентрации или
массе анализируемого вещества и равняется наклону калибровочной кривой (см.
ниже) [56].
Предел количественного определения. Для предела количественного определения или
предела измерения определения и формулы очень схожи с таковыми для ПО, только
для ПКО коэффициент к берется равным 5,6 или даже 10 [2,4,15, 56, 72, 96]. Значе-
ние коэффициента к, равное 10, означает, что %RSD при пределе количественного
определения равен 10%. Таким образом, ПКО соответствует такой концентрации
или количеству анализируемого вещества, которое возможно количественно изме-
рить при коэффициенте вариации не выше 10% [98]. ПКО всегда выше ПО и часто
принимается как фиксированный множитель (обычно равный 2) предела обнару-
жения [4]. Также предел измерения выражается как сигнал, в 10 раз превышающий
шум или фоновый сигнал, что отражается соотношением сигнал/шум 10:1 [72, 85].
На практике ПКО может быть вычислен аналогичным образом, как и ПО, что
показано в табл. 5. Альтернативный способ практической оценки ПО и ПКО за-
ключается в следующем. На первом этапе измеряют однократно 10 независимо при-
готовленных растворов сравнения (параллелей), вычисляют стандартное отклоне-
ние результатов измерения раствора сравнения sbP а затем вычисляют наименьшие
сигналы, соответствующие ПО и ПКО, по формулам хпо = хы + 3jw и хпко = хы + 10хы
соответственно. На втором этапе в растворы сравнения вносят различные количе-
ства анализируемого вещества (например, для получения шести концентраций),
близкие к ПО. Для каждой концентрации проводят измерение 10 независимых по-
вторностей и вычисляют стандартное отклонение измеренных сигналов. Эти стан-
дартные отклонения s (или %RSD) затем наносят на график функции от концен-
трации. Значениями ПО и ПКО будут те концентрации анализируемого вещества,
которые соответствуют значениям %RSD, равным 33 и 10% соответственно [15, 21].
Так же, как и для ПО, неверно полагать, что количественное определение невоз-
можно на уровне и ниже ПКО. Количественный анализ возможен, однако данные
Глава 8.2. Валидация аналитических методик и обеспечение качества
889
становятся недостоверными, так как погрешность, связанная с анализом на столь
малых уровнях, превышает саму измеряемую величину. Количественный анализ
становится достоверным, как только неопределенность измерений оказывается
меньше измеряемой величины [21].
Предел разрешения и возможность обнаружения: только для определенных обла-
стей. В контексте валидации аналитических методик часто используются термины
предел решения (ССа), возможность обнаружения (CC0), а также минимально необхо-
димые рабочие пределы (MRPL, minimum required performance limits), которые требуют
разъяснения. Эти термины применимы для измерения остаточных органических
веществ, контаминантов и химических элементов в живых животных и продуктах
животного происхождения, что регламентируется внутри Европейского союза ди-
рективами ЩЯ/ЕС [99], 2002/657/ЕС [82] и 2003/181/ГС [100]. Комиссия выделя-
ет вещества группы А, для которых максимальные допустимые пределы содержания
(максимальные уровни остатка MRL, maximum residue level) не установлены, и суб-
станции группы В с фиксированным допустимым пределом содержания.
Предел решения ССа — предел, при котором или выше которого можно сделать
вывод с вероятностью ошибки а, что образец не соответствует требованиям. Если
для субстанции был установлен допустимый предел содержания (PL, permitted limit)
(группа В, или регулируемые соединения), то образец считается не соответствую-
щим требованиям, когда предел решения превышен (ССа=xpL + 1,645^). Если для
вещества не был установлен допустимый предел (группа А), то предел решения явля-
ется наименьшим уровнем концентрации, при котором методика может подтвердить
со статистической достоверностью 1 — а присутствие определенного анализируемо-
го вещества (ССа = хы + 2,33^^). Возможность обнаружения CC0 — наименьшее
содержание вещества, которое может быть обнаружено, определено и/или количе-
ственно определено в образце с вероятностью ошибки 0 (CC0 = ССа + l,65.so6pa3ija).
Минимально необходимые рабочие пределы были определены для веществ,
для которых не были установлены допустимые пределы, и в особенности для ве-
ществ, использование которых не разрешено законодательно или запрещено в ЕС
(группа A). MRPL — минимальное содержание анализируемого вещества в образце,
которое должно быть по крайней мере обнаружено и подтверждено. В директиве
2003/18\/ЕС был опубликован ряд MRPL для остаточного содержания определен-
ных ветеринарных препаратов.
Для субстанций группы A (PL не установлен) ССа и CC0 сравнимы с ПО и ПКО
соответственно, так как их концентрации соответствуют измеренным сигналам,
в у раз превышающим сигнал раствора сравнения (растворителя). Для субстанций
с установленным PL (группа В) ССа и CC0 не связаны с ПО и ПКО, но выражаются
через PL. Важно отметить, что эти характеристики применяются специально для
контроля животных и свежего мяса на присутствие остаточного содержания вете-
ринарных препаратов и специфических контаминантов и поэтому отличны от ПО
и ПКО [82, 99-102].
Линейность и диапазон. Для оценки линейности аналитической методики не-
достаточно вычисления только линейной регрессии. Дополнительно следует рас-
считать значения остатков (табл. 5). Последние представляют разницу между дей-
ствительным значением у и значением у, определенным по кривой регрессии для
890
Часть 8. Валидация
каждого значения х. Если значения остатков, вычисленные простой линейной
регрессией, распределены в случайном порядке около линии регрессии, то линей-
ность считается подтвержденной, даже если систематические тренды указывают на
нелинейность. Если наблюдается подобный тренд или модель, то данные лучше об-
рабатывать с помощью взвешенной регрессии. Как для простой, так и для взвешен-
ной линейной регрессии линейность предполагает, что величина отрезка, отсекае-
мая на оси у, незначительно отличается от нуля [4,15, 21, 75].
Альтернативный подход к установлению линейности заключается в разделении
отклика по соответствующим концентрациям и построении графика полученных
«относительных откликов» в виде функции концентрации на логарифмической
шкале. Полученная линия должна быть горизонтальной на всем линейном диапазо-
не с положительным отклонением (в плюсовую сторону) для низких концентраций
и с отрицательным отклонением (в минусовую сторону) для высоких концентра-
ций. При проведении параллельных горизонтальных линий, соответствующих, на-
пример, 95 и 105% горизонтальной линии относительного отклика, можно получить
точки пересечения, в которых методика становится нелинейной [56].
Важно, что линейная кривая может быть воспроизведена в любой день. Одна-
ко линейные диапазоны для разных составов образца мотуг отличаться. Причиной
этого является возможный эффект взаимодействия, связанный с компонентами со-
става. Испытание на общие эффекты состава можно провести посредством «стан-
дартных добавок» или методом добавок аналита. Для серии образцов, полученных
добавлением различных концентраций анализируемого вещества в контрольный
раствор с определенным составом, наклон калибровочной кривой сравнивается
с наклоном обычной калибровочной функции. Отсутствие значимости (кривые па-
раллельны) означает, что влияние компонентов смеси отсутствует [21,75].
Устойчивость и робастность. Хотя термины устойчивость и робастность часто
употребляют как синонимы и взаимозаменяемые понятия, существуют отдельные
определения для каждого из них, что отражено в табл. 5.
Для понимания принципа устойчивости Eurachem рекомендует вносить наме-
ренные изменения (отклонения) в методику, такие как различные дни, аналитики,
оборудование и реактивы, а также отклонения в подготовке образцов или исполь-
зуемых образцах. Изменения следует вносить по отдельности, а влияние каждого
набора экспериментальных условий оценивать по прецизионности и истинности
[4, 15, 85]. Для анализа влияния различных факторов может использоваться метод
планирования факторного эксперимента, так называемый «факторный план», опи-
санный Хольстом (yon Holst) с соавт. [ 103]. Комбинируя изменение условий выполне-
ния методики и проводя серию экспериментов, можно определить, какие факторы
имеют значительное или даже критическое влияние на аналитические результаты.
В руководствах ICH/USP устойчивость не рассматривается как отдельное понятие,
а включается в прецизионность воспроизводимости: «степень воспроизводимости
результатов, полученных при различных условиях, выраженная как %RSD» [56].
Робастность — термин, введенный USP/ICH [88]. Хотя Eurachem включила ро-
бастность в официальный список определений, термин не используется другими
официальными организациями кроме USP/ICH. В соответствии с Eurachem оба па-
раметра представляют одно и то же и поэтому являются синонимами [15, 72, 85].
Глава 8.2. Валидация аналитических методик и обеспечение качества 891
Чувствительность. Чувствительность методики является градиентом кривой от-
клика. На практике чувствительность рассматривается как наклон калибровочной
кривой. Чувствительность часто используется совместно с пределами обнаружения
и количественного определения. В действительности наклон калибровочной кри-
вой применяют для вычисления пределов обнаружения и количественного опреде-
ления. Метод считается чувствительным, если небольшое изменение концентрации
или количества анализируемого вещества влечет значительное изменение величи-
ны измеряемого сигнала [4,15,21]. Чувствительность не всегда упоминается в офи-
циальных руководствах как валидационная характеристика. По мнению Томпсона
с соавт. [4], чувствительность не требуется при валидации, поскольку обычно она
является переменной величиной, зависящей от настройки оборудования. USP/ICH
вообще не упоминают чувствительность в своих руководствах.
8.2.3.5. Обеспечение аналитического качества
Обеспечение качества испытаний представляет собой комплексную организаци-
онную инфраструктуру, образующую основу для получения достоверных анали-
тических измерений [8]. Оно включает все запланированные и систематические
действия и мероприятия, проводимые в рамках системы качества [2, 104]. Система
качества имеет план качества, акцентирующий внимание на применении правил
надлежащей лабораторной практики (GLP, good laboratory practices). Правила GLP
сравнимы с правилами надлежащей производственной практики (GMP) и более об-
ширной методологией НАССР (Hazard Analysis and Critical Control Points — анализ
рисков и критические контрольные точки) систем качества при производстве пи-
щевых продуктов. Внимание уделяется всем аспектам менеджмента качества в ор-
ганизации работы лаборатории, включая обучение персонала, техническое обслу-
живание и калибровку всего используемого оборудования, лабораторные условия,
меры по обеспечению безопасности, систему идентификации образцов, ведение
учетной документации и хранение — последнее может быть упрощено применени-
ем системы управления лабораторной информацией, использованием валидиро-
ванных и стандартизованных методик, а также документированием этих методик
и всей информации, касающейся последующих процедур (СОПов — стандартных
операционных процедур).
Обеспечение качества включает контроль качества и оценку качества. Контроль
качества — механизм или практические действия, предпринимаемые для контроля
ошибок, в то время как оценка качества — механизм проверки того, что система
работает в рамках допустимых пределов. Оценка качества и контрольные меры обе-
спечивают стабильность и контролируемость процесса измерений [2, 8].
Использование валидированных методик: внутрилабораторная и межлабораторная
валидация. Всегда, когда это практически осуществимо, лаборатория должна ис-
пользовать методики, «полностью валидированные» посредством межлаборатор-
ных исследований, также называемых изучением эксплуатационных характеристик
методики. Валидация в межлабораторных исследованиях необходима для любых
новых аналитических методик, прежде чем они станут применяться в качестве стан-
дартных методик (см. ниже). Валидация внутри одной лаборатории также является
ценным источником информации, так как она демонстрирует соответствие ана-
892
Часть 8. Валидация
литической методики своему назначению. Внутрилабораторная валидация пред-
ставляет особый интерес в случаях, когда лаборатории неудобно или невозможно
присоединиться к межлабораторным исследованиям или самостоятельно их орга-
низовать [4, 5].
С одной стороны, даже если методика прошла внутрилабораторную валидацию
и показывает хорошие результаты и достоверную правильность, она не может сразу
применяться как стандартная методика. Методику, прошедшую внутрилаборатор-
ную валидацию, следует проверить в межлабораторных исследованиях, при этом
необходимо сравнить результаты как минимум восьми разных лабораторий. С дру-
гой стороны, межлабораторные исследования не следует проводить для неоптими-
зированной методики [58]. Такие исследования ограничиваются прецизионностью
и истинностью, в то время как другие важные рабочие характеристики, например
специфичность и ПО, не рассматриваются [105]. По этим причинам валидация вну-
три одной лаборатории и межлабораторная валидация не исключают друг друга,
а рассматриваются как две необходимые и взаимно дополняющие стадии процес-
са, представленного на рис. 9. Дополнительное значение валидации внутри одной
лаборатории заключается в том, что она упрощает следующий этап — межлабора-
торную валидацию — и, таким образом, сокращает разрыв между внутрипроизвод-
ственными (валидированными или нет) методиками и состоянием межлаборатор-
ной валидации. При оптимизации методики, в качестве подготовительной работы,
сначала внутри лишь одной лаборатории экономится колоссальное количество вре-
мени и денег, необходимых для межлабораторных исследований [58].
Важность проведения этапа предварительной валидации внутри одной лабора-
тории особенно подчеркивается международными организациями по стандартиза-
ции. ИЮПАК и АОАС International включили раздел по подготовительным работам
в свои руководства по межлабораторным исследованиям, указывая, что необходимы
данные исследований прецизионности, систематической ошибки, степени обнару-
жения и пределов применения, полученные при внутрилабораторных исследовани-
ях. Дополнительно для подготовительных работ необходимо четкое описание мето-
дики, включая указание назначения методики, типа и возможного использования
методики [62, 63]. Связь между предварительным этапом валидации внутри одной
лаборатории и межлабораторными исследованиями подчеркивается не только в со-
гласованных руководствах по межлабораторным исследованиям (см. ниже).
В последнее время ИЮПАК, ИСО и АОАС International опубликовали отдельные
руководства по проведению валидации методик анализа внутри одной лаборатории
[4]. Руководства ИЮПАК были рассмотрены и приняты CCMAS [77]. Кроме того,
специальные индивидуальные рабочие группы и некоторые ученые представляют
свои работы по предварительной валидации и валидации внутри одной лаборато-
рии методик анализа. Такие работы не относятся к официальным опубликованным
руководствам, но могут быть найдены в интернете [58, 85 и др.] или распространя-
ются через национальные или международные специальные рабочие группы. Цели
валидации внутри одной лаборатории и цели внутрипроизводственного процесса
валидации представлены на рис. 9. В зависимости от типа методики (табл. 4) мо-
гут быть получены данные для всех параметров, кроме прецизионности воспроиз-
водимости (межлабораторной). Однако именно межлабораторная вариабельность,
Глава 8.2. Валидация аналитических методик и обеспечение качества
893
Предвалидация
Оптимизация внутри
одной лаборатории
Валидация(полная) —> Стандартизация
Межлабораторные исследования: 1. ИСО 5725 [68] 2. ИЮПАК [63] Утверждение международным авторизованным органом по стандартизации
Описание аналитической
системы:
а) назначение методики;
б) тип аналита;
в) тип методики
4
Применимость или
назначенное применение
методики:
а) тип(ы) материалов или
состава(ов);
б)уровень концентрации
аналита
4
Написание стандартных
операционных процедур
(СОПов)
1
Установление
аналитических
требований
4
Оценка рабочих
характеристик методики
Минимум 5 материалов
г
Минимум 8 лабораторий
Данные по прецизионности
нужно привести в виде RSD
или CV (%)
1
Необходимо привести
данные по прецизионности
повторяемости
и воспроизводимости
1
Данные по прецизионности
необходимо привести
с выбросами
(резко выделяющимися
значениями) и без них
(тест Кохрана, тест Граббса)
Методика была
валидирована
в межлабораторных
исследованиях [63,68]
1
Оценка прецизионности
и других статистических
данных с помощью
утвержденной методики
статистического анализа
(Кохран, Граббс)
4
Прецизионность:
вычисленные значения RSD
должны соответствовать
значениям Горвица
(Хоррата)
4
Прецизионность: не более
1 из 5 наборов данных дают
более 20% статистически
отклоняющихся результатов
4
Обязательный стандарт
представления текстовых
данных и результатов
Рис. 9. Иерархическая связь между целями и требованиями предвалидации [106],
валидации [62, 63, 68] и стандартизации аналитических методик [62, 63, 67, 68, 75, 84]:
RSD — относительное стандартное отклонение; CV— коэффициент вариации
или воспроизводимость, является основным компонентом ошибки аналитического
измерения и, следовательно, главной причиной необходимости межлабораторной
валидации [106].
Межлабораторные валидационные исследования организуются лабораторией,
институтом или организацией. Предпочтительно они должны проводиться в соот-
ветствии с одним из следующих принятых протоколов: 1) ИСО 5725 по правильно-
сти (истинности и прецизионности) методов и результатов измерений [68]; 2) про-
токол ИЮПАК по планированию, проведению и интерпретации исследований
рабочих параметров методик [63]. Последние пересмотренные и согласованные
руководства были выпущены АОАС International в качестве рекомендаций для про-
граммы разработки официальных методов АОАС (АОАС Official Methods Program)
[62]. Основные требования к межлабораторным исследованиям, описанные в этих
руководствах, представлены на рис. 9.
Прецизионность играет главную роль в межлабораторных исследованиях. Вуд
[84] определяет межлабораторные исследования как «процедуру, посредством кото-
рой может быть оценена и количественно выражена прецизионность аналитической
методики». Именно определение прецизионности является целью межлаборатор-
894
Часть 8. Валидация
ных валидационных исследований, а не истинности или любого другого рабочего
параметра. Оценка приемлемости данных по прецизионности имеет важное значе-
ние при стандартизации методик (см. ниже).
Использование стандартизованных методик. Первым уровнем обеспечения анали-
тического качества является использование валидированных или стандартизован-
ных методик. Термины валидированный и стандартизованный в данном случае озна-
чают, что рабочие параметры методики были установлены и было подтверждено их
соответствие определенным требованиям. Как минимум, в таких методиках задо-
кументированы данные по прецизионности, что дает представление о погрешности
и, тем самым, об ошибке аналитического результата. В валидированных и стандар-
тизованных методиках рабочие параметры известны.
Валидированные методики могут разрабатываться самой лабораторией или
организацией по стандартизации после проведения межлабораторных исследова-
ний. Стандартизованные методики разрабатывают такие организации, как АОАС
International, ИСО, USP (см. табл. 3), Агентство по защите окружающей среды США
{ЕРА, U. S. Environmental Protection Agency), Американское общество по стандар-
тизации испытаний и материалов (ASTM, American Society for Testing and Materials)
и Ассоциация стандартов для пищевых продуктов (FSA, Food Standards Association)
[56]. Именно в этом и заключена разница между валидированными и стандартизо-
ванными методиками: аналитическая методика может быть стандартизована только
после валидации в межлабораторных исследованиях. Необходимым условием для
организаций по стандартизации является то, что методика была в достаточной мере
изучена, а ее прецизионность отвечает требуемым стандартам, как показано в об-
щем виде на рис. 9. Формат стандартной методики, описанный в руководстве ИСО
Guide 78/2 [58] приведен в табл. 8 [2]. Протокол ИЮПАК [67] подробно описывает,
как представлять данные по обеспечению аналитического качества, такие как рабо-
чие характеристики.
Эффективный внутренний контроль качества (IQC). В согласованных руковод-
ствах ИЮПАК по IQC Томпсон и Вуд [8] дают следующее определение IQC: «ряд
процедур, предпринимаемых лабораторным персоналом для постоянного монито-
ринга работы и результатов измерений с целью принятия решения о достаточной
надежности результатов для разрешения выпуска». IQC гарантирует пригодность
методик анализа для предназначенного применения, что означает постоянное по-
лучение аналитических результатов с требуемым уровнем точности. В действитель-
ности целью IQC является удлинение валидации методики для постоянной про-
верки правильности аналитических данных, получаемых ежедневно в лаборатории.
В этом отношении отслеживаются как систематические ошибки, приводящие к си-
стематической неопределенности измерений, так и случайные ошибки, приводя-
щие к случайной погрешности. Для того чтобы иметь возможность отследить эти
ошибки, они должны оставаться постоянными. Внутри лаборатории такие посто-
янные условия обычно достигаются в одном аналитическом опыте. Слово внутрен-
ний в IQC подразумевает, что достигнуты условия повторяемости. Таким образом,
отслеживание прецизионности как цели IQC затрагивает не воспроизводимость
или межлабораторную прецизионность, а только повторяемость или внутрилабо-
раторную прецизионность. Фактически, отслеживание точности аналитической
Глава 8.2. Валидация аналитических методик и обеспечение качества
895
Таблица 8
Обязательный текстовый формат для стандартизованных методик
1. Цель 2. Определения 3. Область применения 4. Основные положения 5. Оборудование Кратко указать, для чего предназначена методика Точное определение анализируемого вещества или определяемого с помощью методики параметра Тип материалов или составов, к которым применима методика Основные этапы методики Список необходимого оборудования для проведения определения
6. Реактивы Список необходимых реактивов аналитического класса чистоты для проведения определения
7. Отбор образцов Разделена на нумерованные пункты и подпункты. Включает
8. Методика этап приготовления исследуемых образцов и ссылку на процедуры обеспечения качества
9. Вычисления и Обозначение способов вычисления окончательных результатов
выражение результатов и единиц, в которых результаты необходимо выразить Дополнительная информация по методике; может быть
10. Примечания в форме примечаний, приведенных в данном разделе или размещенных по основному тексту Включают всю информацию по контролю аналитического качества, в частности условия прецизионности (данные
Приложения повторяемости и воспроизводимости) и таблицу статистических данных, описывающих правильность (истинность и прецизионность) методики Ссылки на опубликованные отчеты по межлабораторным
Ссылки исследованиям, которые проводились до стандартизации методики
Источник: по материалам руководства ИСО Guide 78/2 [2, 58].
методики при IQC может быть переведено в контроль (мониторинг) аналитической
системы [8].
Для IQC важны два аспекта: 1) анализ контрольных материалов (образцов), таких
как стандартные материалы или образцы с точно известной концентрацией анали-
та для контроля правильности; 2) проведение параллельных анализов для контроля
прецизионности. Большое значение в IQC имеют «холостые» и «слепые» образцы.
Оба аспекта IQC образуют часть статистического контроля — инструмента контро-
ля точности аналитической системы. На контрольной карте, например контрольной
карте Шухарта, измеренные значения повторных анализов стандартных материа-
лов отмечаются по отношению к номеру опыта. На основе данных в контрольной
карте методика признается аналитической системой, находящейся под контролем,
или неконтролируемой аналитической системой. Данная интерпретация возможна
путем изображения следующих горизонтальных линий на карте: х (среднее значе-
ние), х + х (SD) и х — s, х + 2s (верхняя предупредительная граница) и х — 2s (ниж-
няя предупредительная граница), х + 3s (верхний предел действия, или верхний
контрольный предел) и х — 3s (нижний предел действия, или нижний контрольный
896
Часть 8. Валидация
предел). Аналитическая система находится под контролем, если не более 5% изме-
ренных значений выходят за предупредительные границы [2, 6, 85].
Участие в программах профессионального тестирования (внешний контроль). Про-
фессиональное тестирование (РТ, proficiency testing) представляет периодическую
оценку компетентности или аналитической работы отдельных участвующих в про-
грамме лабораторий [23]. Независимый координатор распределяет индивидуальные
пробы однородного испытуемого материала. Участвующие лаборатории анализиру-
ют материалы выбранной ими методикой и возвращают результаты координатору.
Результаты, полученные различными лабораториями, последовательно сравнива-
ются друг с другом, и качество работы каждого участника оценивается на основе
единой шкалы компетентности [64, 107]. Существуют международные согласован-
ные протоколы по организации программ РТ[59, 60, 64, 69, 79].
Участие в программах РТ не является необходимым требованием или полной
заменой мер IQC, и наоборот. Однако участие в программах РТ бессмысленно без
хорошо развитой системы IQC, так как она лежит в основе этих программ. IQC
и участие в программах РТявляются важными составляющими обеспечения анали-
тического качества (рис. 6). Было показано, что лаборатории с наиболее строгими
процедурами контроля качества испытаний получили значительно более высокие
оценки в программах Р7’[8, 50]. Участие в программах РТ в определенной степени
может улучшить работу лаборатории; тем не менее, были отмечены неудовлетвори-
тельные показатели проведения программы (до 30% всех участников). Это означает,
что нет никакой связи между надлежащим выполнением аналитических требований
и участием в программах РТ [108]. Несмотря на это, РТ выполняет важную функ-
цию обучения, так как помогает лаборатории показать свою компетентность органу
по аккредитации или независимой третьей стороне [60].
Термины программы РТи межлабораторные исследования часто путают друг с дру-
гом, так как в обеих проверках обеспечения качества участвуют несколько различ-
ных лабораторий. Тем не менее, между этими терминами есть четкое различие. Эти
расхождения, в зависимости от целей и применений, результатов, используемых ме-
тодик, исследуемых материалов и участвующих лабораторий, обобщены в табл. 9.
Важно также отметить, что результаты, полученные при проведении программ РТ,
как и при межлабораторных исследованиях, могут быть использованы для оценки
неопределенности измерений (см. п. 8.2.2).
Внешний контроль качества и аккредитация. Участие в программах РТ является
объективным средством оценки надежности данных, полученных лабораторией.
Другим способом внешней оценки лабораторной работы является фактическая
инспекция лаборатории для гарантирования ее соответствия установленным внеш-
ним стандартам. Аккредитация лаборатории показывает применение ею требуемых
принципов обеспечения качества. «Золотой стандарт» ИСО//ЕС17025 [17], являю-
щийся пересмотренной версией руководства ИСО Guide 25 [70], описывает общие
требования к испытательным и калибровочным лабораториям. В Европе требования
к аккредитации изложены в Европейском стандарте E7V45001 [109]. Участие в про-
граммах РТ создает основу для аккредитации, так как РТ является эффективным
способом демонстрации возможностей лаборатории. Руководства по аккредитации
используют информацию, полученную посредством программ РТ[6, 17, 60, 64].
Глава 8.2. Валидация аналитических методик и обеспечение качества
897
Таблица 9 Разница между исследованиями характеристик методики и программами проверки квалификации
Характеристика Межлабораторные и внутрилабораторные исследования (характеристик методики) Программы профессионального тестирования
Основная цель Валидация новых методик Проверка уровня квалификации аналитических лабораторий
Применение Новые методики, требующие полной валидации и стандартизации; первое необходимое условие IQC и обеспечения качества Рутинные проверки (валидированные и/или стандартизованные методики), рекомендуемые в рамках IQC и обеспечения качества
Направленность Прецизионность: множественные Правильность: отдельный
результатов результаты, повторяемость и воспроизводимость; > %RSD сравнивается с теоретическими значениями Горвица и Хоррата результат по исследуемому материалу; > вычисление Z-показателя как меры систематической ошибки
Используемая Единственный описанный метод, для Множество методик;
методика которого строго должна соблюдаться участники имеют свободный
и протокол стандартная операционная процедура (СОП) выбор (валидированных и/или стандартизованных) методик
Испытуемые Минимум пять различных материалов Без установленного
материалы без ограничений по однородности и стабильности исследуемых проб минимума, на один цикл часто менее пяти исследуемых проб; однородность и стабильность материалов должна быть подтверждена
Участвующие Минимум восемь, предположительно Без установленного
лаборатории с равными возможностями минимума, допускается различие участников в рамках одной программы (разные циклы); не обязательно с равными возможностями (будут проверяться)
Источник: [8, 78,107].
Аккредитация — формальное признание того, что лаборатория обладает ком-
петенцией проводить определенные (по типам) калибровки или испытания [2].
Аккредитация по ИСО/7ЕС 17025 является четвертым основным принципом об-
щего обеспечения качества в лаборатории после использования валидированных
и стандартизованных методик, внедрения и использования методик IQC и участия
в программах РТ [4]. Руководства по внедрению ИСО//7ГС 17025, включая оценку
неопределенности измерений (см. п. 8.2.2), опубликованы в специальной литерату-
ре и официальными органами по аккредитации, такими как Eurachem, СПАС, ЕА,
Eurolab и ИЛАК (см. табл. 1) [2,60, 80, 81, ПО]. Стоит упомянуть, что аккредитация,
также как и участие в программах РТ, не обязательно означает надлежащую работу
лаборатории [108].
898
Часть 8. Валидация
8.2.4. Заключение
Наряду с быстрым развитием аналитических методологий, большое значение в на-
стоящее время придается качеству результатов измерений. Помимо необходимости
приведения результатов с указанием неопределенности измерений и прослеживае-
мости результатов до принятых стандартов или эталонов (п. 8.2.2), третьим критиче-
ским аспектом аналитических методик любого типа является статус их валидации.
Международно признано, что в аналитических лабораториях необходима валида-
ция. Однако меньше известно о том, что такое валидация и что следует валидиро-
вать, почему валидация так важна, когда и кем проводится валидация и, наконец,
как она проводится на практике. В настоящей главе авторы попытались ответить на
эти вопросы.
Подробно описана валидация методик и приведены различные подходы к оцен-
ке пригодности аналитических методик. Большое значение придается различным
рабочим характеристикам методик, их определению, способам выражения и под-
ходам к практической оценке. Валидация аналитических методологий помещена
в более широкий контекст обеспечения качества. Темы стандартизации, внутрен-
него и внешнего контроля качества, аккредитации рассматриваются как отдельно,
так и во взаимосвязи друг с другом. Так как валидация и обеспечение качества от-
носятся к определенной аналитической методике, важно подходить к каждой мето-
дике индивидуально. Аналитическая методика — сложный многоэтапный процесс,
начинающийся с отбора образцов и заканчивающийся обработкой результатов.
Хотя у каждой методики есть своя цель, применение и аналитические требования,
основные принципы валидации и обеспечения качества одинаковы независимо от
типа методики и области применения. Информация в настоящей главе в основном
относится к аналитической химии, но также применима и в других областях.
В п. 8.2.3, посвященном качеству в аналитической лаборатории, приводится
полный современный обзор важной информации по вопросам валидации и обе-
спечения качества испытаний. Данная информация будет полезна для не имеющих
достаточного опыта научных работников, а также для всех вовлеченных уже долгое
время в данную тематику специалистов, заблудившихся в лабиринтах путаных объ-
яснений и желающих найти разъяснения конкретных вопросов или углубить свои
знания.
Благодарность от авторов
Авторы хотят поблагодарить Саймона Кэя за предварительное обсуждение темы,
Жанну Пуумалайнен за замечания и комментирование ранних версий главы, Эндрю
Даманта за полезные предложения, Арне Хольст-Енсена за свежие идеи и Фридля
Ванхи за множество полезных обсуждений, замечаний и помощь в процессе напи-
сания.
Литература
1. van Zoonen, Р., Hoogerbrugge, R., Gort, S. M., Van de Wiel, H. J., and Van’t Klooster, H. A. (1999),
Some practical examples of method validation in the analytical laboratory, Trends Anal. Chem., 18,
584-593.
Глава 8.2. Валидация аналитических методик и обеспечение качества 899
2. CITAC/Eurachem Guide (2002), Guide to Quality in Analytical Chemistry—An Aid to Accreditation,
available: http://www.eurachem.org.
3. Eurachem/CITAC Guide (2003), Traceability in Chemical Measurement. A Guide to
Achieving Comparable Results in Chemical Measurement, Joint Eurachem/CITAC Work-
ing Group on Measurement Uncertainty and Traceability, available: http://www. eurachem.org.
4. Thompson, M., Ellison, S., and Wood, R. (2002), Harmonised guidelines for single-laboratory valida-
tion of methods of analysis, IUPAC Technical Report, Pure Appl. Chem., 74, 835-855.
5. Battaglia, R. (1996), Quality assurance in a food analytical laboratory — The introduction
of EN 45001 in the food analytical laboratories of a retail company, Accred. Qual. Assur., 1, 256-261.
6. Mesley, R. J., Pocklington, W. D., and Walker, R. F. (1991), Analytical quality assurance:
A review, Analyst, 116, 975-990.
7. Eurachem Guide EEE/RM/062rev3 (2002), The Selection and Use of Reference Materials. A Basic
Guide for Laboratories and Accreditation Bodies, available: http://www. eurachem.org.
8. Thompson, M., and Wood, R. (1995), Harmonised guidelines for internal quality control in analytical
chemistry laboratories, Pure Appl. Chem., 67, 649-656.
9. Maroto, A., Boqu ё, R., Riu, J., and Rius, F. X. (1999), Evaluating uncertainty in routine analysis,
Trends Anal. Chem., 18, 577—584.
10. Hund, E., Massar, D. L., and Smeyers-Verbeke, J. (2001), Operational deli nitions of uncertainty,
Trends Anal. Chem., 20, 394^406.
11. Fleming, J., Albus, H., Neidhart, B., and Wegschieder, W. (1996), Glossary of analytical terms (II),
Accred. Qual. Assur., 1, 87—88.
12. Quevauviller, Ph. (2004), Traceability of environmental chemical measurements, Trends Anal. Chem.,
23, 171-176.
13. Eurachem/CITAC Guide (1995), in Ellison, S. L. R., Rosslein, M., and Williams, A., Eds., Quantify-
ing Uncertainty in Analytical Measurement, 1st ed., available: http://www. eurachem.org.
14. Eurachem/CITAC Guide (2000), in Ellison, S. L. R., Rosslein, M., and Williams, A., Eds., Quantify-
ing Uncertainty in Analytical Measurement, 2nd ed., available: http://www. eurachem.org.
15. Eurachem Guide (1998), The Fitness for Purpose of Analytical Methods. A Laboratory Guide to
Method Validation and Related Topics, LGC, Teddington, available: http://www. eurachem.org.
16. International Organization for Standardization (ISO) GUM (1995), Guide to the Expression of
Uncertainty in Measurement, ISO, Geneva.
17. International Organization for Standardization (ISO)/IEC 17025 (1999), General Requirements for
the Competence of Calibration and Testing Laboratories, ISO, Geneva.
18. CX/MAS 01/8 (2001), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling (FAO/WHO), Measurement uncertainty. Relationship between the analytical result, the
measurement uncertainty and the specifi cation in Codex standards, agenda item 4a of the 23rd ses-
sion, Budapest, Hungary, Feb. 26-Mar. 2, 2001.
19. CX/MAS 02/6 (2002), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling (FAO/WHO), Proposed draft guidelines on measurement uncertainty, agenda item 5 of
the 24th session, Budapest, Hungary, Nov. 18—22, 2002.
20. CX/MAS 02/13 (2002), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling (FAO/WHO), The use of analytical results: Sampling, relationship between the ana-
lytical results, the measurement uncertainty, recovery factors and the provisions in Codex standards,
agenda item 9 of the 24th session, Budapest, Hungary, Nov. 18-22, 2002.
21. CX/MAS 02/4 (2002), Codex Alimentarius Commission, Codex Committee on Methods of
Analysis and Sampling (FAO/WHO), Proposed draft guidelines for evaluating acceptable meth-
ods of analysis, agenda item 4a of the 24th session, Budapest, Hungary, Nov. 18-22, 2002.
22. European Cooperation for Accreditation of Laboratories (EAL) (1996), The Expression of Uncer-
tainty in Quantitative Testing, EAL-G23, The Netherlands.
23. International Laboratory Accreditation Cooperation (ILAC) (2002), Introducing the Concept of
900
Часть 8.Валидация
Uncertainty ofMeasurement in Testing in Association with the Application of the Standard ISO/IEC
17025, ILAC-G17, ILAC Technical Accreditation Issues Committee, available: www.ilac.org.
24. Valcarc ё 1, M., and Rios, A. (1999), Traceability in chemical measurements for the end users, Trends
Anal. Chem., 18, 570-576.
25. Walsh, M. C. (1999), Moving from offi cial to traceable methods, Trends Anal. Chem., 18, 616-623.
26. Valcarc ё 1, M., and Rios, A. (1997), Is traceability an exclusive property of analytical results? An
extended approach to traceability in chemical measurement, Fresen. J. Anal. Chem., 359, 473—475.
27. Quevauviller, Ph., and Donard, O. F. X. (2001), Stated references for ensuring traceability of chemical
measurements for long-term environmental monitoring, Trends Anal. Chem., 20, 600-613.
28. Pan, X. R. (1996), The traceability scheme in chemical measurement, Accred. Qual. Assur., 1, 181-
185.
29. Charlet, P, and Marschal, A. (2004), Improvement in the traceability of environmental analysis by the
relevant use of certifi ed pure solutions and a matrix certifi ed reference material, Trends Anal. Chem.,
23, 178-184.
30. Segura, M., Camara, C., Madrid, C., Rebollo, C., Azcarate, J., Kramer, G. N., Gawlik, В. M., Lam-
berty, A., and Quevauviller, Ph. (2004), Certifi ed reference materials (CRMs) for quality control of
trace-element determinations in wastewaters, Trends Anal. Chem., 23, 194—202.
31. Forstner, U. (2004), Traceability of sediment analysis, Trends Anal. Chem., 23, 217-236.
32. Theocharopoulos, S. P., Mitsios, I. K., and Arvanitoyannis, J. (2004), Traceability of environmental
soil measurements, Trends Anal. Chem., 23,237-251.
33. Sabe, R., and Rauret, G. (2004), Challenges for achieving traceability of analytical measurements of
heavy metals in environmental samples by isotopic dilution mass spectrometry, Trends Anal. Chem.,
23,273-280.
34. Drolc, A., Ros, M., and Cotman, M. (2004), Establishment of traceability of ammonium nitrogen
determination in wastewater, Anal. Bioanal. Chem., 378, 1243-1250.
35. Thompson, M. (1998), Uncertainty of sampling in chemical analysis, Accred. Qual. Assur., 3, 117—
121.
36. Roy, S., and Fouillac, A.-M. (2004), Uncertainties related to sampling and their impact on the chemi-
cal analysis of groundwater, Trends Anal. Chem., 23, 185—193.
37. Analytical Methods Committee (1995), Uncertainty of measurement: Implications of its use in ana-
lytical science, Analyst, 120, 2303-2308.
38. Fleming, J., Albus, FL, Neidhart, B., and Wegschieder, W. (1997), Glossary of analytical terms (VIII),
Accred. Qual. Assur., 2, 160-161.
39. Hund, E., Massart, D. L., and Smeyers-Verbeke, J. (2003), Comparison of different approaches to
estimate the uncertainty of a liquid chromatographic assay, Anal. Chim. Acta., 480, 39-52.
40. Eurachem/EA Guide 04/10 (2002), Accreditation for Microbiological Laboratories, available: http://
www.eurachem.org.
41. Kiippers, S. (1998), Is the estimation of measurement uncertainty a viable alternative to validation?
Accred. Qual. Assur., 3, 412—415.
42. Ellison, S. L. R., and Barwick, V. J. (1998), Using validation data for ISO measurement uncertainty es-
timation. Part 1. Principles of an approach using cause and effect analysis, Analyst, 123, 1387-1392.
43. Moser, J., Wegscheider, W., and Sperka-Gottlieb, C. (2001), Quantifying the measurement uncer-
tainty of results from environmental analytical methods, Fresen. J. Anal. Chem., 370, 679-689.
44. Dehouck, P, Vander Heyden, Y., Smeyers - Verbeke, J., Massart, D. L., Crommen, P. H., Marini, R.
D., Smeets, O. S., Decristoforo, G., Van de Wauw, W., De Beer, J., Quaglia, M. G., Stella, C., Veuthey,
J.-L., Estevenon, O., Van Schepdael, A., Roets, E., and Hoog artens, J. (2003), Determination of un-
certainty in analytical measurements from collaborative study results on the analysis of a phenoxym-
ethylpenicillin sample, Anal. Chim. Acta, 481, 261-272.
45. Maroto, A., Riu, J., Boqu ё, R., and Rius, F. X. (1999). Estimating uncertainties of analytical results
using information from the validation process, Anal. Chim. Acta, 391, 173-185.
Глава 8.2. Валидация аналитических методик и обеспечение качества 901
46. Maroto, A., Boque, R., Riu, J., and Rius, F. X. (2001), Estimation of measurement uncertainty by us-
ing regression techniques and spiked samples, Anal. Chim. Acta, 446, 133-145.
47. Barwick, V. J., and Ellison, S. L. R. (2000, Jan.), VAM Project 3.2.1 : Development and Harmonisa-
tion of Measurement Uncertainty Principles. Part d. Protocol for Uncertainty Evaluation from Valida-
tion Data, LGC, UK, available: http://www.vam.org.uk.
48. Armishaw, P. (2003), Estimating measurement uncertainty in an afternoon. A case study in the practi-
cal application of measurement uncertainty, Accred. Qual. Assur., 8, 218-242.
49. Horwitz, W. (1982), Evaluation of analytical methods used for regulation of food and drugs, Anal.
Chem., 54, 67A-76A.
50. Thompson, M., and Lowthian, P. J. (1997), The Horwitz function revisited, J. AOACInt., 80, 676-
679.
51. King, B. (2001), Meeting the measurement uncertainty and traceability requirements of ISO/IEC
standard 17025 in chemical analysis, Fresen. J. Anal. Chem., 371, 714.
52. Mueller-Harvey, I. (2003), Do we need quality assurance and quality control of analytical measure-
ments in R & D laboratories? FoodAgric. Environ., 1, 9-11.
53. Rosslein, M. (2000), Evaluation of uncertainty utilising the component by component approach,
Accred. Qual. Assur., 5, 88—94.
54. Mueller, N. (2002), Introducing the concept of uncertainty of measurement in testing in as-
sociation with the application of the standard ISO/IEC 17025, Accred. Qual. Assur., 7, 79-80.
55. International Organization on Harmonization (1995), Guideline for industry: Text on validation of
analytical procedures, ICH - Q2A, available: http://www.fda.gov/cder/guidance/index.htm.
56. Huber, L. (1998), Validation of analytical methods. Part 9, in Huber, L., Ed., Validation and Qualifi
cation in Analytical Laboratories, Interpharm Press, Agilent Technologies, available: http://www.lab-
compliance.com.
57. Wells, R. J. (1998), Validation requirements for chemical methods in quantitative analysis — Horses
for courses? Accred. Qual. Assur., 3,189-193.
58. Green, M. (1996), Apractical guide to analytical method validation, Anal. Chem., 68, 305A—309A.
59. Eurachem Guide (2000), Selection, Use and Interpretation of Profi ciency Testing (PT) Schemes by
Laboratories, available: http://www.eurachem.org.
60. Eurachem/Eurolab/EA Guide EA - 03/04 (2001, Aug.), Use of Profi ciency Testing as a Tool for
Accreditation in Testing, available: http://www.eurachem.org.
61. European Committee for Normalisation (CEN), (2006), Standardization and related activi-
ties — General Vocabulary (from ISO/IEC Guide 2:2004), EN45020:2006, www.cenorm.be.
62. Association of Ofii cial Analytical Chemists (AOAC) International (2000), Method validation pro-
grams (OMA/PVM Department), including Appendix D: Guidelines for collaborative study proce-
dures to validate characteristics of a method of analysis, available: http://www.aoac.org/vmeth/devm-
ethno.htm.
63. Horwitz, W. (1995), Protocol for the design, conduct and interpretation of method performance stud-
ies, Pure Appl. Chem., 67, 331—343.
64. Thompson, M., and Wood, R. (1993), The international harmonized protocol for the profi ciency test-
ing of (chemical) analytical laboratories, Pure Appl. Chem., 65, 2123-2144 : also J. AOAC Int., 76,
26-940 (1993).
65. Currie, L. A. (1995), Nomenclature in evaluation of analytical methods including detection and quan-
tifi cation capabilities, Pure Appl. Chem., 67, 1699-1723.
66. Currie, L. A., and Svehla, G. (1994), Nomenclature for the presentation of results of chemical analy-
sis, International Union of Pure and Applied Chemistry (IUPAC), Pure Appl. Chem., 66, 595-608.
67. Pocklington, W. D. (1990), Harmonized protocols for the adoption of standardized analytical methods
and for the presentation of their performance characteristics, International Union of Pure and Applied
Chemistry (IUPAC), Pure Appl. Chem., 62,149-162.
68. International Organization for Standardization (ISO) Guide 5725 (1994), Accuracy (Trueness and
902
Часть 8. Валидация
Precision) of Measurement Methods and Results, ISO, Geneva. 69. International Organization for
Standardization (ISO) Guide 43 (1984), Development and Operation of Laboratory Proficiency
Testing, ISO, Geneva.
70. International Organization for Standardization (ISO) Guide 25 (1990), General Requirements for the
Competence of Calibration and Testing Laboratories, ISO, Geneva.
71. International Organization for Standardization (ISO)/IEC 17025 (1999), General Requirements for
the Competence of Calibration and Testing Laboratories, ISO, Geneva.
72. International Conference on Harmonization (1996), Guidance for Industry: Validation of Analytical
Procedures: Methodology, ICH-Q2B, available: http://www.fda.gov/cder/guidance/index.htm.
73. International Conference on Harmonization (1999), Harmonised Tripartite Guideline: Specifi cations:
Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, ICH-Q6B, avail-
able: http://www.fda.gov/cder/guidance/index.htm.
74. U.S. Food and Drug Administration (FDA)/CDER/CVM (2001), Guidance for industry — Bioana-
lytical method validation, available: http://www.fda.gov/cder/guidance/index.htm.
75. CX/MAS 01/4 (2001), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling, Criteria for evaluating acceptable methods of analysis for Codex purposes, agenda
item 4a of the 23rd session, Budapest, Hungary, Feb. 26-Mar. 2, 2001.
76. CX/MAS 02/5 (2002), Codex Alimentarius Commission, Codex Committee on Methods of
Analysis and Sampling (FAO/WHO), Criteria for evaluating acceptable methods of analysis
for Codex purposes, agenda item 4b of the 24th session, Budapest, Hungary, Nov. 18-22, 2002.
77. CX/MAS 02/11 (2002), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling (FAO/WHO), Requirements for single - laboratory validation for Codex purposes,
agenda item 8b of the 24th session, Budapest, Hungary, Nov. 18-22, 2002.
78. CX/MAS 02/12 (2002), Codex Alimentarius Commission, Codex Committee on Methods of Analysis
and Sampling (FAO/WHO), Validation of methods through the use of results from profi ciency testing
schemes, agenda item 8c of the 24th session, Budapest, Hungary, Nov. 18-22, 2002.
79. International Laboratory Accredidation Cooperation (ILAC) (2000), Guidelines for the requirements
for the competence of providers of profi ciency testing schemes, ILAC-G13 ILAC Technical Accredi-
tation Issues Committee, available: www.ilac.org.
80. International Laboratory Accredidation Cooperation (ILAC) (2001), Guidance for accreditation to
ISO/IEC 17025, ILAC-G15, ILAC Technical Accreditation Issues Committee, available: www.ilac.
org.
81. International Laboratory Accredidation Cooperation (ILAC) (2002), The scope of accreditation and
consideration of methods and criteria for the assessment of the scope in testing, ILAC-G18, ILAC
Technical Accreditation Issues Committee, available: www.ilac.org.
82. European Commission (EC) (2002), Commission decision 2002/657/EC implementing council direc-
tive 96/23/EC concerning the performance of analytical methods and the interpretation of results, Offi
J. Eur. Commun., L 221/8, 17.8.2002.
83. Holst-Jensen, A., and Berdal, K. G. (2004), The modular analytical procedure and validation approach
and the units of measurement for genetically modifi ed materials in foods and feeds, J. AOACInt., 87,
1-9.
84. Wood, R. (1999), Howto validate analytical methods, Trends Anal. Chem., 18, 624—632.
85. Waters Corporation (2004), Validation Guidelines: Terminology and Definitions, available: http://
www.waters.com/WatersDivision.
86. Hibbert, D. B. (1999), Method validation of modem analytical techniques, Accred. Qual. Assur., 4,
352-356.
87. Fleming, J., Neidhart, B., Albus, H., and Wegscheider, W. (1996), Glossary of analytical terms (Ш),
Accred. Qual. Assur., 1, 135.
88. Krull, I. S., and Swartz, M. (1999), Analytical method development and validation for the academic
researcher, Anal. Lett., 32, 1067-1080.
89. Quevauviller, Ph. (2004), Traceability of environmental chemical measurements, Trends Anal. Chem.,
Глава 8.2. Валидация аналитических методик и обеспечение качества 903
23, 171-176.
90. Holcombe, G., Lawn, R., and Sargent, M. (2004), Improvements in effi ciency of production and
traceability for certifi cation of reference materials, Accred. Qual. Assur., 9,198-204.
91. Lauwaars, M., and Anklam, E. (2004), Method validation and reference materials, Accred. Qual.
Assur., 9, 253—258.
92. Fleming, J., Albus, H., Neidhart, B., and Wegschieder, W. (1996), Glossary of analytical terms (TV),
Accred. Qual. Assur., 1, 191.
93. Thompson, M., Ellison, S. L. R., Fajgelj, A., Willets, R, and Wood, R. (1999), H armonised guidelines
for the use of recovery information in analytical measurement, PureAppl. Chem., 71, 337-348.
94. Vessman, J. (1996), Selectivity or specifi city? Validation of analytical methods from the perspective
of an analytical chemist in the pharmaceutical industry, J. Pharm. Biomed. Anal., 14, 867-869.
95. Analytical Methods Committee (1987), Recommendations for the defi nition, estimation and use of
the detection limit, Analyst, 112, 199-204.
96. Fleming, J., Albus, H., Neidhart, B., and Wegschieder, W. (1997) Glossary of analytical terms (VII),
Accred. Qual. Assur., 2, 51—52.
97. Huber, W. (2003), Basic calculations about the limit of detection and its optimal determination,
Accred. Qual. Assur., 8, 213—217.
98. Kuselman, I., and Sherman, F. (1999), Assessment of limits of detection and quantifi cation using cal-
culation of uncertainty in a new method for water determination, Accred. Qual. Assur., 4,124—128.
99. European Commission (EC), Council directive 96/23/EC of April, 29 1996, on measures to monitor
certain substances and residues thereof in live animals and animal products and repealing directives
85/258/EEC and 86/469/EEC and decisions 89/187/EEC and 91/664/EEC, Off. J. Eur. Commun.,
L125, 0010-0032, 23.05.1996.
100. European Commission (EC), Commission decision 2003/181/EC amending decision 2002/657/EC
as regards the setting of minimum required performance limits (MRPLs) for certain residues in food
of animal origin, Off. J. Eur. Commun.,СТ1/П, 0017-0018, 15.3.2003.
101. Antignac, J.-R, Le Bizec, B., Monteau, E, and Andre, F. (2003), V alidation of analytical methods
based on mass spectrometric detection according to the “2002/657/EC” European decision: Guide-
line and application, Anal. Chim. Acta, 483, 325-334.
102. De Wasch, K., De Brabander, H. F., Courtheyn, D., Van Hoof, N., Poelmans, S., and Noppe, H.
(2003), The commission decision 2002/657/EC: Disscussion on some new analytical aspects. EURO
FOOD CHEM XII, in Strategies on Safe Food, Proceedings, Vol. 1, Brugge, Belgium, Sept. 24—26,
2003, pp. 45-48.
103. von Holst, С., M ii Iler, A., Bjorklund, E., and Anklam, E. (2001), In-house validation of a simplifi
ed method for the determination of PCB’s in food and feedingstuffs, Eur. Food Res. Technol, 213,
154-160.
104. Prichard, E., Albus, H., Neidhart, B., and Wegscheider, W. (1997), Glossary of analytical terms (IX),
Accred. Qual. Assur., 2, 348—349.
105. van der Vbet, H., Van Rhijn, J. A., Van de Wiel, H. J. (1999), Inter-laboratory, time, and fitness-for-
purpose aspects of effective validation, Anal. Chim. Acta, 391, 159-171.
106. Horwitz, W, and Albert, R. (1996), Reliability of the determinations of polychlorinated contami-
nants (bifenyls, dioxins, furans), J. AOAC Int., 79, 589-621.
107. Thompson, M., and Lowthian, P. J. (1993), Effectiveness of analytical quality control is related to
the subsequent performance of laboratories in profi ciency tests, Analyst, 118,1495-1500.
108. King, B., Boley, N., Karman, G. (1999), The correlation of laboratory performance in profi ciency
testing with other QA characteristics, Accred. Qual. Assur., 4, 280-291. 109. The Joint European
Standard Institution (1989), General Criteria for the Operation of Testing Laboratories, EN45001,
CEN/CENELEC, Brussels.
110. Pritzkow, J. (2003), Practical experience of the laboratories in implementing the ISO/IEC 17025,
Accred. Qual. Assur., 8, 25—26.
Глава 8.3. Валидация лабораторного оборудования
Герман Лэм
Wild Crane Horizon, Inc., (Скарборо, Онтарио, Канада)
8.3.1. Введение
Надежность химических и физических измерений напрямую зависит от пригод-
ности и эксплуатационных параметров оборудования, на котором получены изме-
рения. Для любой лаборатории разработка практической программы по валидации
лабораторного оборудования различной сложности является трудной задачей. Тем
не менее, внедрение подобной программы имеет большое значение, поскольку обе-
спечивает соответствие оборудования эксплуатационным (рабочим) требованиям
и предназначенному применению. Подобное подтверждение требуется для соответ-
ствия правилам надлежащей производственной практики (GMP, good manufacturing
practices) и надлежащей лабораторной практики (GLP, good laboratory practices).
За последние годы многими организациями были приложены значительные
усилия по разработке рекомендаций по валидации самого разнообразного лабора-
торного оборудования различной сложности. В течение длительного времени на
конференциях и заседаниях обсуждались темы, связанные с валидацией оборудо-
вания. Доступно большое количество рекомендаций и справочной литературы по
этой теме [1—11]. Обобщенное представление ряда ключевых принципов и общая
структура валидации лабораторного оборудования выглядят следующим образом.
1. Основной целью валидации лабораторного оборудования является предостав-
ление гарантий того, что оборудование пригодно для предназначенного исполь-
зования. Гарантии обеспечиваются посредством документированного доказатель-
ства того, что система способна стабильно работать в рамках заданных параметров
(спецификаций) предназначенного применения.
2. Валидация лабораторного оборудования — обязанность организации-
пользователя, а не поставщика оборудования. Пользователи могут сотрудничать
с поставщиками для облегчения установки (монтажа) оборудования и валидацион-
ных испытаний.
3. Большая часть оборудования, используемого в лаборатории, является готовым
коммерческим оборудованием, доступным на рынке, поэтому конечные пользо-
ватели практически не влияют на стадию проектирования оборудования. Подход
разработки жизненного цикла системы в целом (SDLC, system development life-cycle)
[8], который используется для разработки сложных компьютеризированных си-
стем, таких как системы управления лабораторной информацией (LIMS, Laboratory
Information Management System), системы управления хроматографическими дан-
ными (CDS, Chromatographic Data System) или изготовленного под индивидуальный
заказ лабораторного оборудования, не подходит для коммерческого оборудования.
Некоторое лабораторное оборудование, например pH-метр или центрифуга, устро-
ено достаточно просто и поэтому не требует выполнения подхода SDLC.
Глава 8.3. Валидация лабораторного оборудования 905
4. Для обеспечения приемлемого уровня охвата лабораторного оборудования
различной сложности разрабатывается масштабируемая программа валидации в за-
висимости от предназначенного использования оборудования.
5. Лабораторное оборудование классифицируется по различным категориям
в соответствии с его сложностью, чтобы провести масштабируемую программу
валидации. Оформление отчетов по валидации и требования к документации для
каждого класса лабораторного оборудования можно описать в руководстве по вали-
дации для лаборатории.
6. Валидационные работы должны соответствовать сложности различных клас-
сов оборудования, что следует определить в руководстве по валидации, разработан-
ном для лаборатории.
8.3.2. Область применения
В настоящей главе приводится обзор валидации лабораторного оборудования для
правил GMP и GLP. общего обозначения GMP и GLP используется аббревиа-
тура GxP. В центре внимания находится валидация коммерческого лабораторного
оборудования с использованием масштабируемого подхода.
Валидация крупномасштабных компьютеризированных систем, таких как LIMS,
CDS и лабораторные системы по индивидуальным проектам (системы, сделанные
на заказ), рассматриваться не будут. Читателям настоятельно рекомендуется озна-
комиться с Руководством по валидации автоматизированных систем на фармацев-
тическом производстве GAMP (надлежащая автоматизированная производственная
практика, Good Automated Manufacturing Practice), версия 4, для валидации таких си-
стем [8].
8.3.3. Классификация лабораторного оборудования
Для обеспечения приемлемого уровня валидации широкого круга лабораторного
оборудования необходима разработка системы классификации этого оборудования
в зависимости от его функциональной сложности, назначения и риска сохранно-
сти данных. Для каждой категории или класса оборудования определяется набор
заранее заданных масштабируемых документов и испытаний. Эти параметры будут
использоваться для определения объема и типа испытаний, необходимых для вали-
дации различного оборудования. Подобная система классификации оборудования
и связанные с ней отчетные документы представляют простой и структурирован-
ный подход по решению валидационных задач, требующихся для большого числа
лабораторного оборудования: от простого pH-метра до полностью автоматизиро-
ванной системы для испытания «Растворение».
Система классификации, основанная на сложности оборудования, приведена
в табл. 1. Схожая классификация лабораторного оборудования приведена в «GAMP.
Руководство по надлежащей практике. Валидация лабораторных компьютеризиро-
ванных систем» [10] и в общей статье по квалификации аналитического оборудова-
ния Фармакопеи США (USP, U.S. Pharmacopeia) [11]. Так как возможно частичное
совпадение категорий оборудования, отдельным организациям рекомендуется при
необходимости вносить поправки в определения и типы оборудования в классифи-
906
Часть 8. Валидация
кационной системе, подстраивая ее под определенную операцию. После утверж-
дения классификации лабораторного оборудования определяются отчетные доку-
менты по валидации каждого класса оборудования для формирования структуры
процесса валидации. В табл. 2 перечисляется наиболее часто встречаемая в лабора-
ториях отчетная документация по лабораторному оборудованию.
Таблица 1
Классификация оборудования
Категория’ Определение Примеры6
А Оборудование и инструменты, используемые для пробоподготовки Электрические плитки, мешалки, шейкеры
В Простые программно-аппаратные средства для измерения фундаментальных физических параметров, таких как вес, размер, температура и pH Весы, pH-метры, цифровые термометры, центрифуги, ультразвуковые бани
С Сложные программно- аппаратные средства для спектрофотометрических, хроматографических методов, испытаний на растворение и т. д. Жидкостные хроматографы, газовые хроматографы, приборы для испытания «Растворение», титраторы по К. Фишеру
D Коммерческое лабораторное оборудование, контролируемое внешним компьютером. Гибридные системы, такие как автоматизированные лабораторные комплексы для испытания «Растворение», включающие интерфейс системы управления спектрофотометром или жидкостным хроматографом Жидкостные хроматографы, газовые хроматографы, спектрофотометры в УФ и видимой областях, инфракрасный спектрофотометр с Фурье-преобразованием (FTIR), спектрофотометр в ближней ИК- области, масс-спектрометры, атомно- абсорбционные спектрометры, термогравиметрические анализаторы, автоматизированные коммерческие лабораторные комплексные системы
а Категории могут отличаться. См. руководство предприятия по классификации лабора-
торного оборудования или справочную информацию в руководстве GAMP и общей главе
<1058> tZSP[ll, 12].
6 Примеры включают не все возможное оборудование.
8.3.4. Этапы валидации
По существу подход жизненного цикла к валидации лабораторного оборудования
можно разделить на четыре этапа, что показано на рис. 1.
8.3.4.1. Этап планирования и определения требований
Мероприятия этапа планирования начинаются после определения рабочих потреб-
ностей по применению оборудования в лаборатории. Данные мероприятия вклю-
чают краткое документальное изложение пользовательских и эксплуатационных
требований к оборудованию. После того как требования установлены, они исполь-
зуются для оценки подходящего оборудования, доступного у поставщиков. План
Глава 8.3. Валидация лабораторного оборудования
907
Таблица 2
Основная отчетная документация по валцдации оборудования
Отчетная документация По категориям
А В с D
Бизнес-требования2 V6 V6-' V V
Оценка соответствия регуляторным требованиям (оценка соответствия GxP) V6 V6-" V V
Валидационный план V6 V6 V" V
Оценка электронных записей и электронных подписей (ERES) X X X V
Пользовательские и функциональные требования (квалификация проектной документации) V6 Vя Vе V
Квалификация монтажа V6 V» Vе V
Квалификация функционирования V6 V" Vе V
Квалификация эксплуатации V6 V" Vе V
Отчет о валидации X V’ Vе V
Матрица прослеживаемости и описание конфигураций X X X V
Стандартные операционные процедуры V6 V V V
Проверка рабочих характеристик X V V V
Периодическая проверка X V6 V V
Вывод из эксплуатации V6 V V V
Примечание. v — обязательно; х — не обязательно.
Е Может потребоваться аудит поставщика для определенного оборудования категории D.
6 По желанию.
Е Можно использовать упрощенные стандартные формы.
валидации и отчетная документация, соответствующие классу используемого обо-
рудования, должны быть разработаны на этапе планирования.
План валидации. План валидации создается для описания мероприятий и пред-
ставляемой документации, требуемых для лабораторного оборудования. Степень
подробности плана валидации должна соответствовать сложности систем, подле-
жащих оценке, их надлежащему применению и влиянию на коммерческую деятель-
ность. Общий план валидации может использоваться для схожих типов лаборатор-
ного оборудования со схожим применением. Например, один план можно составить
для всех жидкостных хроматографов.
План валидации обычно включает следующие разделы:
• Цели и область применения проекта. Обоснование целесообразности исполь-
зования оборудования с указанием основных лабораторных задач, решаемых
с его помощью, и формулировкой допусков, исключений и ограничений по
проекту.
• Описание системы. Описание оборудования, программного обеспечения
и конфигурации системы (если система состоит из различных модулей или
компонентов).
908
Часть 8. Валидация
План
валидации
Требования
URS и FRS
DO и оценка
системы
Оценка рисков
и соответствия
Определить
потребности
Квалификация
Подготовка площадки Протокол квалификации Квалификация IQ, OQ, PQ Проверка и утверждение Краткий отчет
Функционирование
СОП Калибровка и проверка рабочих характеристик
Журналы
обслуживания
и эксплуатации
Периодический Контроль
осмотр изменений
Завершение
жизненного цикла
Вывод
из эксплуатации
Рис. 1. Этапы валидации. URS — спецификация требований пользователя (user requirement
specification)- FRS — спецификация функциональных требований (functional requirement
specification)-, DQ, IQ, OQ, PQ — соответственно квалификация проектной документации, монтажа,
функционирования и эксплуатации (design, installation, operational, performance qualifications);
СОП — стандартная операционная процедура
• Оценка соответствия. Указывается, отвечает ли используемое аналитическое
оборудование требованиям GxP и регуляторным требованиям. Оборудование,
попадающее под действие GxP, требует проведения валидации. Следует при-
вести обоснование принятого решения по оценке соответствия.
• Валидационный подход и документация. Указываются документы, квалифика-
ционные испытания и отчеты, которые необходимо включить в валидацион-
ный проект. Объем документации должен основываться на сложности и ри-
сках, связанных с предназначенным использованием системы. В документах
отражают критерии приемлемости для демонстрации того, что система отве-
чает требованиям для предназначенного использования.
• Обязанности и ответственность. Перечисляются группы или персонал, от-
ветственные за валидацию оборудования.
• График реализации проекта и основные этапы работ. Перечисляются планиру-
емые сроки выполнения основных работ по валидации. Данная информация
полезна при контроле выполнения проекта и подтверждении доступности
требуемых ресурсов на разных этапах валидации.
Глава 8.3. Валидация лабораторного оборудования
909
• Управление документацией. Описывается формат, нумерация, пересмотры,
контроль версий и хранение валидационной документации.
• Ссылки на необходимую информацию. Перечисляются внутренние и внешние
руководящие документы по валидации оборудования, фармакопейные стан-
дартные методики, квалификационные испытания и связанные стандартные
операционные процедуры по валидации.
Пользовательские требования. Пользовательские требования описывают анали-
тические задачи, для решения которых пользователь хочет эксплуатировать систе-
му аналитического оборудования, и основные функции, которыми оборудование
должно обладать. Требования должны быть четкими, недвусмысленными и поддаю-
щимися проверке или тестированию. Пользовательские требования устанавливают
необходимый объем работ по валидации. Успешное проведение валидации должно
показать, что оборудование отвечает пользовательским требованиям и может на-
дежно использоваться в назначенных аналитических целях. Очень важно ясно ука-
зать, что должно делать оборудование. Валидировать следует только обязательные
требования. Добавление дополнительных функций в пользовательские требования
(по типу «неплохо бы иметь») добавит ненужную работу при валидации.
Например, простая ВЭЖХ-система с изократической системой подачи подвиж-
ной фазы и многоволновым УФ-детекгором может быть достаточной для простой
повседневной проверки продукта для выпуска в обращение. Однако ВЭЖХ-система
для проведения количественного определения примесей или исследования скорее
всего потребует наличия градиентной системы подачи подвижной фазы и диодно-
матричного УФ-детектора. Если ВЭЖХ-система будет использоваться только для
выполнения методик с изократическим режимом, то валидация градиентой функ-
ции ВЭЖХ-системы не потребуется, даже когда система имеет такую функцию.
Другим примером является прибор для испытания «Растворение», который приме-
няется только для проведения методик растворения с использованием лопастной
мешалки. Хотя прибор позволяет проводить методики как с лопастной мешалкой,
так и «вращающейся корзинкой», необходима будет только валидация прибора для
выполнения методик растворения с лопастной мешалкой.
Функциональные требования. Требования компетентных пользователей образуют
основу функциональных спецификаций, которые устанавливают системные функ-
ции, режим работы и рабочую среду, необходимые для соответствия требованиям
пользователя. Опять же, требования должны поддаваться проверке или контролю,
чтобы они отвечали заданным параметрам во время квалификационных проверок.
Функциональные требования обычно включают рассмотрение следующих пара-
метров:
• Функции каждого компонента или модуля аппаратной системы: необходимо
определить ожидаемый эксплуатационный диапазон каждой функции и ее
рабочие параметры, такие как правильность, прецизионность и линейность,
необходимые для предназначенного применения.
• Требования, предъявляемые к рабочему месту, для обеспечения работы обо-
рудования: электроснабжение (электрическое напряжение и ток), вентиля-
ция, газоснабжение, водоснабжение и сливы.
910
Часть 8. Валидация
• Требования по охране труда и технике безопасности: механическая безопас-
ность автоматизированного движения, радиационная безопасность систем,
использующих радиоактивные источники, и лазерная безопасность для си-
стем, использующих высокомощные лазеры.
• Требования к компьютерной операционной системе и локальной сети.
• Тип данных (аналоговый и/или цифровой ввод/вывод) и объем (размер фай-
лов данных).
• Требования к электронным записям и электронным подписям [13—15]: без-
опасность системы, целостность данных, прослеживаемость, хранение и по-
иск данных.
• Взаимосвязь с другими системами и их контроль.
• Восстановление системы после серьезной неисправности или сбоя.
Типичные аппаратные функциональные требования для градиентной ВЭЖХ-
системы с детектором в УФ и видимой областях представлены в табл. 3 в качестве
примера.
Квалификация проектной документации. При приобретении аналитического ком-
мерческого оборудования пользователь влияет в малой степени или вообще не вли-
яет на проектную документацию. Обычно такая документация не представляется
пользователям. В подобном случае в квалификации проектной документации ука-
зываются пользовательские и функциональные требования, которые возможно удо-
влетворить рассматриваемым аналитическим оборудованием путем сравнения тре-
бований с техническими спецификациями поставщика. Для аналитических систем,
состоящих из множества коммерчески производимых компонентов (например,
система растворения с системой автоматического отбор проб для ВЭЖХ-анализа),
конфигурацию и совместимость компонентов различных поставщиков необходимо
указать в квалификации проектной документации.
Проверка системы и оценка поставщика. Как правило, существует большой вы-
бор поставщиков наиболее часто используемого лабораторного коммерческого обо-
рудования. Пользовательские и рабочие требования формируют основные крите-
рии выбора. Очевидно, что выбранное оборудование должно выполнять основные
требования в соответствии со своим предназначением. Следует учитывать и другие
факторы, касающиеся оборудования, в частности простоту использования и обслу-
живания, а также репутацию поставщика в плане качества, надежности и техниче-
ской поддержки. С практической точки зрения аудит поставщика может оказаться
невозможным или может не требоваться для обычно используемого лабораторного
коммерческого оборудования. Оценка поставщика иногда используется для про-
верки, имеется ли у поставщика надлежащая система качества, обеспечивающая
разработку и производство требуемого оборудования. Необходимость оценки по-
ставщика зависит от важности и сложности приобретаемой системы.
8.3.4.2. Этап квалификации (проверки)
Термины квалификация оборудования и валидация оборудования часто заменяют друг
друга. В данной главе термин квалификация относится к подготовке рабочего ме-
ста и проверке для демонстрации того, что оборудование должным образом уста-
Глава 8.3. Валидация лабораторного оборудования
911
Таблица 3
Типичные функциональные требования для ВЭЖХ-системы с детектором в УФ и видимой
областях
Модули Функциональные требования
Насос • Насос должен обеспечивать скорость потока от 0,50 до 5,00 мл/мин. • Насос должен быть градиентным, 4-канальным и иметь правильность смешения ±1,5% теоретических значений для четырех каналов. • Относительное стандартное отклонение (rsd) скорости потока должно составлять <2,0% для шести последовательных измерений откалиброванного расходомера
Автосамплер (устройство автоматического ввода пробы) • Автосамплер должен иметь инжектор, способный инжектировать пробы объемом от 1 до 100 мкл. • Прецизионность инжектора должна составлять <1,5% rsd. • Перенос остатка пробы инжектором должен составлять <0,5% площади пика. • Автосамплер должен выбирать соответствующий флакон. • Автосамплер должен использовать реле и/или контакты для связи с лабораторной системой управления хроматографическими данными (cds). • Термолабильные ячейки для проб должны поддерживать температуру пробы в диапазоне 4—15 °с (±3 °с)
Детектор в УФ и видимой областях • Детекторы должны работать в диапазоне длин волн от 200 до 800 нм. • Точность длины волны детектора должна составлять +2 нм. • Отклик детектора должен быть линейным с коэффициентом корреляции г2 не менее 0,999 на всем динамическом диапазоне. • Линейный диапазон должен доходить до 2,0 оптических единиц на всю шкалу
Колоночное отделение • Термостатируемое колоночное отделение должно поддерживать температурный диапазон на 5 °с выше внешней среды до 60 °сю • Термостат должен поддерживать температуру в пределах ±3 °с от установленной, а прецизионность температуры должна составлять <2,0%
новлено в надлежащих условиях, а рабочие параметры отвечают установленным
спецификациям для предназначенного использования. Квалификация является
частью жизненного цикла валидации. Валидация относится к процессам, удосто-
веряющим пригодность оборудования для предназначенного использования в те-
чение жизненного цикла оборудования. Квалификация монтажа (/Q), квалифика-
ция функционирования (OQ) и квалификация эксплуатации (PQ) необходимы для
представления доказательств, что спецификации требований пользователя (URS),
спецификации функциональных требований (FRS) и квалификация проектной до-
кументации (DQ) выполнены полностью. Последовательность подбора требований
и квалификационных событий, а также взаимосвязь между IQ, OQ, PQ и URS, FRS
912
Часть 8. Валидация
Рис. 2. V-диаграмма квалификации
и DQ в общем случае изображаются в виде V-диаграммы, приведенной на рис. 2.
Квалификация монтажа отображает выполнение этапа DQ. Аналогично, OQ ото-
бражает выполнение функциональных требований, a PQ отображает выполнение
пользовательских требований.
Подготовка рабочего места. До установки оборудования в лаборатории необхо-
димо завершить подготовительные работы, которые обеспечат его функционирова-
ние. Поставщик, как правило, предоставляет документацию по подготовке рабочего
места, в которой описаны условия, необходимые для функционирования оборудо-
вания. Документация по подготовке рабочего места обычно содержит следующую
информацию:
• физические размеры и вес оборудования;
• условия окружающей рабочей среды для надлежащего функционирования:
температуру, влажность и наличие виброизоляции;
• требования к инженерным системам: электроснабжению, водоснабжению,
газоснабжению, сливам, вентиляции, сетевому подключению;
• требования по охране труда и технике безопасности.
Частой ошибкой является недооценка затрат и времени, необходимых для под-
готовки рабочего места. Пользователи должны внимательно изучить руководство
по подготовке рабочего места, чтобы необходимые подготовительные работы были
наверняка завершены до установки оборудования. Данная подготовка, выполнен-
ная не в полном объеме, может стать причиной серьезных неудобств и длительных
задержек при монтаже оборудования. Подобная ситуация может стать тратой вре-
мени и денег, если инженер по эксплуатации не может работать в лаборатории из-за
незавершенной подготовки рабочего места.
Подходы к квалификации. Если аналитическое оборудование состоит из раз-
ных функционально отличающихся модулей, то к испытаниям по квалификации
Глава 8.3. Валидация лабораторного оборудования
913
функционирования возможно применить модульный подход, при котором провер-
ка функций конкретного модуля будет осуществляться по отдельности (например,
прецизионность скорости потока, проверка правильности ВЭЖХ-насоса и точно-
сти термостатирования колоночного отделения).
При проведении квалификации эксплуатации следует использовать целостный
(холистический) подход к проверке аналитической системы, при этом все необхо-
димые модули должны работать совместно для получения намеченного результата,
обозначенного в пользовательских требованиях (рис. 3). Надлежащее функциони-
рование каждого отдельного модуля аналитической системы не означает надлежа-
щего функционирования целой системы, состоящей из этих модулей.
Протоколы по квалификации. Для коммерческого оборудования поставщики
обычно предоставляют протоколы испытаний по квалификации монтажа и функ-
ционирования. Распространенной практикой является плата поставщику за оказа-
ние услуг по проведению квалификации. Поставщик должен обладать хорошими
знаниями устанавливаемого и квалифицируемого оборудования, поэтому исполь-
зование таких услуг может значительно сократить время и материальные затраты.
Несмотря на это, ответственность за проверку адекватности протоколов испыта-
ний, выбор надлежащих процедур и обоснованных критериев приемлемости ис-
пытаний, гарантирующих достижение поставленной цели, лежит на пользователе.
Процедуры и/или испытания поставщика должны быть совместимы с процедурами
и установленными порядками компании-пользователя. Слишком широкие кри-
терии приемлемости не обеспечивают достаточной гарантии работоспособности
оборудования. С другой стороны, слишком строгие критерии приемлемости могут
привести к нежелательным отказам, которые занимают много времени и усилий на
расследование и обоснования. Критерии приемлемости должны отражать как функ-
циональные, так и пользовательские требования. Протокол должен быть утвержден
до начала испытаний.
Квалификация монтажа
и функционирования
Квалификация эксплуатации
Проверка компонентов
модульное тестирование
Проверка пригодности системы
Рис. 3. Сравнение модульного и целостного подходов к квалификации
914 Часть 8. Валидация
Протокол квалификационных испытаний обычно содержит общее описание си-
стемы, ее конфигурацию и назначение. Сценарии тестирования в протоколах испы-
таний дают подробную информацию о процедурах испытаний. В каждом сценарии
тестирования следует отразить:
• описание тестируемого модуля;
• цель и назначение испытаний;
• область применения и ограничения;
• процедуру испытания;
• критерии приемлемости испытания;
• результаты испытания (в отдельных разделах);
• отклонения и исключения в ходе испытания (в отдельном разделе).
Квалификация монтажа. Квалификация монтажа обеспечивает документирован-
ное подтверждение того, что оборудование было получено и успешно установлено
в соответствии с утвержденными проектными требованиями и смонтировано над-
лежащим образом в рабочей среде, подходящей для его назначения. Надлежащая
установка (монтаж) является первым этапом обеспечения правильной работы обо-
рудования. Неправильно установленное оборудование с большой вероятностью вы-
зовет проблемы при квалификации функционирования и квалификации эксплуата-
ции. Далее приведено описание различных проверок на этапе IQ.
Проверки IQjio монтажа оборудования:
• проверить аппаратную часть и программное обеспечение по отгрузочной ве-
домости (накладной);
• проверить на наличие видимых повреждений;
• заполнить контрольный лист подготовки рабочего места;
• выполнить требования правил техники безопасности и охраны здоровья;
• внести оборудование в инвентарный перечень оборудования.
Проверки IQ во время монтажа:
• зарегистрировать местоположение оборудования;
• зарегистрировать все аппаратные компоненты, включая компьютер, принтер,
встроенное программное обеспечение аналитического оборудования, интер-
фейсы, сетевые подключения и т. д.;
• зарегистрировать все программные приложения, операционную систему
и расположение действующего программного пакета в системе;
• зарегистрировать структуру (конфигурацию) системы.
Проверки IQ после монтажа:
• проверить правильное подключение системы;
• проверить надлежащую инициализацию и начальное состояние;
• проверить надлежащую связь между модулями;
• откалибровать модули при необходимости;
• проверить версии программ;
• проверить надлежащий запуск программ и совместимость с аппаратурой;
• создать резервную копию важных данных системных настроек;
Глава 8.3. Валидация лабораторного оборудования
915
• создать журнал учета работы оборудования.
Должны быть доступны справочные документы, в частности:
• руководство по подготовке рабочего места;
• руководство пользователя системы;
• товарно-транспортные накладные;
• записи приемных испытаний производителя (если возможно);
• сертификат соответствия качества от поставщика.
Во время монтажа системы и квалификационных испытаний используйте, когда
возможно, функцию захвата изображения экрана монитора {Print screen, Prt Sc) для
печати информации в качестве объективного подтверждения выполнения провер-
ки, так как это более эффективный способ документирования результатов и наблю-
дений, чем простая запись информации на бумаге.
Квалификация функционирования. Квалификация функционирования обеспечи-
вает получение документированного подтверждения того, что оборудование будет
работать в соответствии с функциональными требованиями в рамках установленно-
го эксплуатационного диапазона в надлежащих рабочих условиях. OQ для простого
оборудования, такого как pH-метры, весы, мешалки, водяные бани и термометры,
можно завершить, проведя калибровку. Для относительно сложных систем при OQ
проверяется правильная работа аппаратной и программной частей оборудования.
Проверка аппаратной части должна включать проверку функциональных возмож-
ностей оборудования при нормальной работе. Например, проверка ВЭЖХ-системы
будет включать оценку функционирования насоса, инжектора и детектора [15].
Стандартные испытания О Q для модулей ВЭЖХ включают:
• для насоса: правильность скорости потока и градиента;
• для детектора: линейность отклика, шум, смещение и правильность длины
волны;
• для инжектора: прецизионность, линейность и точность переноса пробы;
• для термостатируемого колоночного отделения: правильность поддержания
температуры.
При стандартных испытаниях OQ для спектрофотометров в УФ и видимой об-
ластях проверяются [16]:
• правильность длины волны и воспроизводимость;
• рассеянное световое излучение;
• разрешение;
• фотометрическая правильность, воспроизводимость и линейность;
• шум;
• сглаженность и стабильность основной линии.
В дополнение к проверке системных компонентов проводится проверка функ-
ционирования программного комплекса, соответствия электронных записей и под-
писей (ERES) (в том числе проверка безопасности, целостности данных, резервной
копии данных и архива). Для проверки функционирования программного комплек-
са можно последовательно ввести определенный набор инструкций. Ответы систе-
мы сравниваются с ожидаемыми результатами; таким образом, выявляются любые
916 Часть 8. Валидация
проблемы при их выполнении. Некоторые поставщики представляют стандартный
набор данных, который обрабатывается системой для проверки работоспособно-
сти.
Рассмотрение электронных записей и электронных подписей (ERES). В компьюте-
ризированном оборудовании требуется проверка соответствия ERES для демонстра-
ции соблюдения функциональных требований в трех следующих областях [ 12—14].
1. Безопасность. Предупреждение несанкционированного доступа.
Контроль несанкционированного доступа к оборудованию осуществляется по-
средством физического, логического и процедурного управления. Физический
контроль обычно достигается контролем доступа на производственную площадку,
в здание, лабораторию и к оборудованию. Логическая безопасность реализуется
установкой различных уровней допуска пользователей, руководителей и систем-
ных администраторов, а также индивидуальными паролями для каждого санкци-
онированного пользователя. Двойной логический допуск обычно предусмотрен
программным обеспечением и операционной системой. Процедурный контроль,
использующий стандартные операционные процедуры, может быть применен для
назначения допусков пользователям.
2. Целостность данных. Демонстрация корректности и достоверности записей.
Запись представляет собой комбинацию исходных (первичных) данных и мета-
данных (параметров обработки и другой связанной информации, необходимой для
восстановления записей), к которым относятся:
• прослеживаемость — позволяет восстановить последовательность событий;
• использование журналов регистрации событий для отслеживания создания,
изменения или удаления записей; эта часть содержит сведения о том, кто что
делал, что писал, когда и зачем;
• встроенные отметки о дате и времени создания, изменения или удаления дан-
ных.
3. Сохранение данных. Копирование электронных данных на сменный или уда-
ленный накопитель и управление архивами данных для сохранения полного и точ-
ного набора записей.
Целостность данных не должна подвергаться риску во время процесса сохране-
ния. Проверяется:
• создание резервных копий и восстановление данных: активно используемые
данные периодически копируются с жесткого диска или персонального ком-
пьютера, контролирующего оборудование, на подходящий накопитель, на-
пример CD-ROM, DVD или отдельный контролируемый сервер;
• архивирование и получение данных: данные, более не используемые в по-
вседневной работе оборудования, архивируют так, чтобы их можно было лег-
ко восстановить.
Квалификация эксплуатации. Квалификация эксплуатации — процесс, представ-
ляющий документированное подтверждение того, что оборудование полностью
отвечает требованиям пользователя. Для PQ необходимо провести целостное рас-
смотрение системы, включающее все ее функциональные компоненты. Квалифи-
кацию эксплуатации можно выполнить, используя типичную методику, требующую
Глава 8.3. Валидация лабораторного оборудования 917
совместной работы всех модулей как единой системы, и получив ожидаемые резуль-
таты в соответствии с предназначенным применением.
Если возможно, полезным для PQ будет проведение высоконадежного испы-
тания, которое часто выполняется пользователями на определенном типе обору-
дования. Например, испытанием для PQ градиентной ВЭЖХ-системы будет такая
методика градиентной ВЭЖХ, с которой хорошо знаком пользователь. В данном
случае результаты испытания будут главным образом отражать рабочие параметры
оборудования и не будут подвержены влиянию погрешности используемой мето-
дики.
Исключения в испытаниях и обзор данных. Если результаты испытаний не соот-
ветствуют установленным критериям приемлемости, то необходимо провести рас-
следование для установления причины нарушения работы. Нарушения могут быть
вызваны ошибками в ходе выполнения процедуры испытания или проблемами,
связанными с оборудованием. На основе данных, полученных в ходе расследова-
ния, предпринимаются корректирующие действия для исправления проблемы.
После разрешения проблемы проводится повторная проверка для подтверждения
того, что работа оборудования соответствует требованиям. Расследование наруше-
ний работы, оценка воздействия, корректирующие действия и повторные испыта-
ния должны быть внесены в журнал особых ситуаций (исключений) по квалифика-
ционным испытаниям. После завершения квалификационных испытаний процесс
квалификации, собранная информация и результаты проверяются на правильность,
полноту и допустимые отклонения. Все основные проблемы, касающиеся соответ-
ствия требованиям, должны быть решены до разрешения оборудования к использо-
ванию в работе.
Итоговый отчет. После завершения мероприятий по квалификации составляется
итоговый отчет, в котором отражаются проведенные квалификационные исследо-
вания, полученные результаты и отклонения от плана валидации, а также дается
заключение о годности оборудования для предназначенного использования. Лю-
бые отклонения от запланированных мероприятий, включая нарушение выполне-
ния проверки и несоответствие системы заданным параметрам, необходимо указать
в отчете. Итоговый отчет содержит следующие разделы:
• введение;
• описание системы;
• ссылки на документы;
• валидационные мероприятия, испытания, критерии приемлемости и резуль-
таты;
• обзор отклонений, проблем и выводов для предупреждения рисков отказов;
• ограничения по системе (при наличии);
• соответствующая документация;
• заключение.
Для простого аналитического оборудования будет достаточно простой таблицы,
обобщающей квалификационные испытания, критерии приемлемости, результаты
и решение о соответствии или не соответствии результатов испытаний заданным
параметрам, поскольку требуется проведение небольшого числа испытаний, обыч-
918
Часть 8. Валидация
но относительно простых. Для сложных аналитических систем в итоговом отчете
составляется более сложная таблица, часто называемая матрицей прослеживаемо-
сти, в которой фиксируются требования, испытания, критерии приемлемости, ре-
зультаты испытаний, места хранения валидационных документов, данных испыта-
ний и другой сопутствующей документации.
После квалификационных испытаний и текущей проверки рабочих характери-
стик документы и связанные с ними данные испытаний — единственное доказа-
тельство того, что оборудование прошло подобные испытания, было надлежащим
образом установлено и проходило техническое обслуживание для поддержания
его в рабочем состоянии. Документы необходимо хранить в систематически упо-
рядоченных центральных хранилищах и поддерживать в хорошем состоянии.
Надлежащая система хранения документов может оказаться очень полезной при
подготовке к аудиту и позволит ускорить представление документов во время про-
верки.
8.3.4.3. Этап функционирования
После того как оборудование прошло квалификацию, его можно использовать в ра-
боте лаборатории. Мероприятия этапа функционирования включают ежедневное
использование и поддержание оборудования в валидированном состоянии.
Стандартная операционная процедура (СОП). СОП составляется для представле-
ния указаний по работе, техническому обслуживанию и калибровке нового обору-
дования. Как правило, СОП включает:
• общее описание системы;
• инструкции по эксплуатации;
• ответственность пользователей и администраторов системы;
• требования к калибровке или проверке требуемых рабочих параметров, кри-
терии приемлемости, частоту проверок и необходимых действий — в случае,
если оборудование не соответствует требуемым рабочим параметрам;
• требования по техническому обслуживанию;
• сервисное обслуживание, крупный и мелкий ремонт, замену изношенных ча-
стей, которые потребуют проведения повторной квалификации оборудова-
ния.
Например, замена УФ-лампы в УФ-детекторе не требует полной повторной ква-
лификации, в то время как замена электронных схем обязательно влечет ее про-
ведение.
Техническое обслуживание. Естественный износ, а также старение различных
компонентов могут подвергать риску функциональное состояние оборудования
и привести к нарушению его работы. Оборудование должно проходить техниче-
ское обслуживание для сохранения стабильной и надежной работы. Программа
профилактического обслуживания, при которой определяются и заменяются рас-
ходные материалы, позволит сэкономить деньги и время в долгосрочном плане.
Записи по использованию и обслуживанию хранятся совместно с оборудованием
для ведения истории эксплуатации системы. Эти записи могут явиться источни-
ком простого решения проблемы в случае неисправности оборудования и допол-
Глава 8.3. Валидация лабораторного оборудования 919
нительно помогают разработать осмысленную программу профилактического об-
служивания.
Проверка рабочих характеристик и калибровка. С целью поддержания оборудова-
ния в валидированном состоянии и демонстрации того, что оборудование выпол-
няет свои функции в соответствии с установленными пользовательскими требова-
ниями, необходима периодическая проверка рабочих характеристик и калибровка.
Действующие требования GMP (cGMP) также предписывают выполнение калибров-
ки оборудования в установленные промежутки времени в соответствии с принятой
письменной программой. Термины проверка рабочих характеристик и калибровка
часто используются как взаимозаменяемые. Калибровка заключается в измерении
и настройке отклика оборудования по известным стандартам. При проверке рабо-
чих характеристик эксплуатационные характеристики сравниваются с определен-
ным набором требований. Калибровку можно рассматривать как часть проверки
рабочих характеристик.
Периодичность проверки рабочих характеристик должна основываться на зна-
нии надежности функционирования вида оборудования и типе операций, поддер-
живаемых данным оборудованием. Начальная периодичность может быть установ-
лена по рекомендациям производителя-поставщика. Надлежащее ведение журнала
эксплуатации может в дальнейшем обосновать менее частое проведение проверок.
Потенциальный недостаток увеличения периода между проведением проверок —
увеличение времени воздействия посторонних факторов на получаемую информа-
цию с момента последней проверки в случае нарушения работы системы. Проверка
пригодности системы до анализа не заменяет периодическую калибровку оборудо-
вания. Такая проверка является специфичной для каждой методики, в то время как
калибровка системы проверяет общую работоспособность оборудования. Проверка
пригодности системы лишь показывает, что оборудование пригодно для определен-
ной методики во время анализа. Она не может показать крайние рабочие параметры
системы. Например, проверка пригодности системы количественного определения
методом ВЭЖХ с УФ-детектором скорее всего не обнаружит проблему точности
(правильности) длины волны, так как стандарты и образцы анализируются при оди-
наковой длине волны.
Контроль за изменениями. Контроль за изменениями представляет собой струк-
турированный механизм запросов, разрешений, оценок, проверок, внесения
и утверждения изменений в валидированных системах. Его следует проводить в со-
ответствии с утвержденными и задокументированными процедурами. Эти процеду-
ры должны включать следующие элементы:
до внесения изменений:
• описание предлагаемых изменений;
• оценку влияния предлагаемых изменений;
• согласование и утверждение перед выполнением;
• сообщение об изменениях пользователям системы;
• утверждение руководством проведения изменений;
после внесения изменений:
• осуществление изменения;
920
Часть 8. Валидация
• проверку после изменения;
• обучение и/или повторное обучение пользователей системы;
• утверждение руководством завершения процесса изменений.
Стандартные формы по контролю за изменениями приведены на рис. 4 и 5.
Тип и модель оборудования Идентификатор оборудования
Действующее программное обеспечение Новое программное обеспечение
Операционная система Ссылка на план валидации
Описание изменения
Причина изменения
Оказываемое влияние изменения
Ссылки на валвдационные документы
Документация, требующая обновления
Необходимость проведения повторной квалификации (приложить протоколы)
Инициатор запроса Дата
Утверждение руководства Дата
Утверждение ООК Дата
Рис. 4. Заполняемая форма до внесения изменений
Версия внутреннего программного обеспечения может быть непреднамеренно
обновлена инженером по эксплуатации при техническом осмотре или рутинном
техническом обслуживании без внесения изменений, известных владельцам систе-
мы. Изменение может не отразиться на функционировании оборудования и может
остаться незамеченным. Тем не менее, новая версия внутреннего программного
обеспечения будет в этом случае отличаться от версии, указанной в валидационных
документах. Поэтому владельцы системы должны работать совместно с инженера-
ми по эксплуатации, чтобы предотвратить возникновение возможных ошибок.
План обеспечения бесперебойной деятельности (аварийное восстановление). План
аварийного восстановления разрабатывается для обеспечения бесперебойной дея-
тельности лаборатории в случае возникновения нежелательных событий, связанных
с неисправностью оборудования и тем самым вызывающих прерывание процесса
производства. Нежелательные события, такие как неисправность критических ап-
паратных компонентов оборудования и неисправность программного обеспечения,
случаются в повседневной работе лаборатории. План аварийного восстановления
должен содержать необходимые шаги по восстановлению рабочего состояния си-
стемы. Основными этапами являются переустановка программного обеспечения
Глава 8.3. Валидация лабораторного оборудования 921
Тип и модель оборудования | Идентификатор оборудования
Общие результаты испытаний
Ссылка на повторную квалификацию Ссылка Дата
Согласованная документация Ссылка Дата
Журналы по оборудованию обновлены Ссылка Дата
Согласованные стандартные операционные процедуры Ссылка Дата
Системный журнал обновлен Ссылка Дата
Информация по программе валидации помещена в архив Ссылка Дата
Система готова к использованию Дата
Инициатор запроса Дата
Утверждение руководства Дата
Утверждение ООК Дата
Рис. 5. Заполняемая форма после внесения изменений
персонального компьютера, управляющего оборудованием, перенастройка обору-
дования и восстановление резервной копии данных.
Периодическая проверка. Работоспособность оборудования необходимо прове-
рять на регулярной основе, обычно один раз каждые два-три года, для обеспечения
его надежности и соответствия требованиям пользователей. Должны рассматри-
ваться следующие вопросы:
• Рабочая среда. Проверка изменений, таких как температура, влажность и ви-
брация, которые могут влиять на работу оборудования.
• Контроль за изменениями. Отслеживание изменений конфигурации системы,
аппаратной части, внутреннего программного обеспечения, изменений про-
граммного комплекса и контроль воздействия этих изменений.
• Отчеты. Эксплуатация, техническое обслуживание, ремонт, испытания по
проверке рабочих характеристик и местонахождение отчетов.
• Документация. Проверка того, что валидационные документы являются дей-
ствующими, а оборудование отвечает установленным требованиям. Проверка
рабочих инструкций, СОПов, плана аварийного восстановления и местона-
хождения документов.
• Обучение пользователей. Необходимое обучение по новой версии используе-
мой в работе программы.
Результатом проверки является определение, поддерживается ли оборудование
в валидированном состоянии. Если отчеты указывают, что оборудование подвер-
жено определенным типам неисправностей, желательно иметь программу профи-
лактического обслуживания для предотвращения неисправности системы во время
работы.
922
Часть 8. Валидация
8.3.4.4. Завершение жизненного цикла
Вывод из эксплуатации (списание) оборудования является последним этапом жиз-
ненного цикла валидации. Когда оборудование более не требуется в лаборатории,
оно подлежит снятию с эксплуатации. При этом необходимо выполнить следующие
мероприятия и оформить связанные с ними документы:
• обосновать в комплекте документации или журнале по оборудованию при-
чину списания и дату вступления в силу;
• поместить в архив все относящиеся к оборудованию отчеты, в частности до-
кументацию или журналы по оборудованию, программную часть и инструк-
ции по применению, более не требуемые в лаборатории;
• подтвердить, что все электронные отчеты помещены в архив в соответствии
с установленным порядком действий;
• отсоединить от оборудования все инженерные системы;
• составить распоряжение о демонтаже оборудования;
• изъять или исправить все затронутые СОПы;
• обновить программу калибровки и ведомость учета оборудования.
8.3.5. Заключение
Основной целью валидации лабораторного оборудования является представление
гарантий того, что данное оборудование пригодно для предназначенного примене-
ния. Валидационные работы, связанные с лабораторным оборудованием, должны
соответствовать сложности оборудования, его назначению и влиянию на получае-
мые данные. Подход к управлению жизненным циклом лабораторного оборудова-
ния, основанный на масштабируемой валидации, начиная с этапа планирования
и заканчивая выводом из эксплуатации, является эффективным способом обеспе-
чения бесперебойной работы и предотвращения неисправностей оборудования.
Систематический подход к валидации оборудования, основанный на строго науч-
ном обосновании баланса между потенциальными рисками и финансовыми воз-
можностями, позволит показать надежность выполняемых работ аудиторам во вре-
мя проверок лаборатории.
Литература
1. Freeman, М., Leng, М., Morrison, D., and Munden, R. (1995), Position paper on the qualification
of analytical equipment, Pharm. Technol. Eur., November 1995, 40.
2. Huber, L. (1995), Validation of Computerized Analytical Systems, Inteipharm Press. 3. Huber, L.
(1996), Quality assurance and instrumentation, Дсста/. Qual. Assur., 1,24.
4. Huber, L. (1999), Validation and Qualification in Analytical Laboratories, Interpharm Press.
5. International Organization for Standardization (ISO)/IEC 17025 (1999), General require- ments for
the compliance of testing and calibration laboratories, ISO, Geneva.
6. Miller, J. M., and Crowther, J. B. (2000), Analytical Chemistry in a GMP Environment—A Practical
Guide, Wiley Interscience, Hoboken, NJ.
7. International Conference on Harmonization (ICH) (2000), Quality guideline, Q7A, ICH, Geneva.
8. International Society for Pharmaceutical Engineering (ISPE) (2002), GAMP {Good Automated
Manufacturing Practice) Guidefor Validation of Automated System in Pharmaceutical Manufacturing,
Version 4, ISPE.
Глава 8.3. Валидация лабораторного оборудования
923
9. Lam, Н. (2004), Procurement qualification and calibration of laboratory instrument: An overview, in
Chan et al., Eds., Analytical Method Validation and Instrument Performance Verification, Wiley
Interscience, Hoboken, NJ, Chapter 9.
10. International Society for Pharmaceutical Engineering (ISPE) (2005), GAMP (Good Automated
Manufacturing Practice) Good Practice Guide: Validation of Laboratory Computerized Systems,
ISPE.
11. U.S. Pharmacopoeia (USP) (2006), general chapter (1058), Analytical instrument qualification, USP,
Rockville, MD.
12. U.S. Food and Drug Administration (FDA) (2005), Guidance for industry, 21 CFR Part 11: Elec-
tronic records and electronic signatures, FDA, Rockville, MD.
13. International Society for Pharmaceutical Engineering (ISPE) (2002), Good Practice and Compliance
for Electronic Records and Signatures, Part 1, Good Electronic Records Management, ISPE and
PDA.
14. International Society for Pharmaceutical Engineering (ISPE) (2005), GAMP (Good Automated
Manufacturing Practice) Good Practice Guide: A Risk-Based Approach to Compliant Electronic
Records and Signatures, ISPE.
15. Lam, H. (2004), Performance verifi cation of HPLC, in Chan et al., Eds., Analytical Method Validation
and Instrument Performance Verification, Wiley Interscience, Hoboken, NJ, Chapter 11.
16. Lam, H. (2004), Performance verification of UV — vis spectrophotometers, in Chan et al., Eds.,
Analytical Method Validation and Instrument Performance Verification, Wiley Interscience, Hobo-
ken, NJ, Chapter 10.
Глава 8.4. Принципы валидации фармацевтического
производства
Е. Б. Соуто1-3, Т Васконселос2-3, Д. С. Феррейра3, Б. Сараменто3
8.4.1. Введение
Фармацевтическая промышленность была пионером в разработке методов обеспе-
чения качества и безопасности, направленных на снижение риска в ее работе до
минимума. Внедрение в прошлом столетии правил надлежащей производственной
практики (GMP, good manufacturing practices) явилось маленьким шагом вперед в фар-
мацевтической работе, но гигантским шагом в уменьшении рисков для пациентов
и производственного персонала и в снижении финансовых потерь. Наиболее важ-
ным способом обеспечения требований GMP являются работы по валидации. Це-
лью валидации является подтверждение того, что качество обеспечивается по ходу
всего производства, а не контролируется лишь при завершении фармацевтических
процессов. Как правило, валидируется весь процесс целиком, при этом проверку
проходит каждый отдельный этап. Валидационные процедуры подтверждают, что
различные части, этапы и компоненты процесса подробно описаны и управляются
надлежащим образом, и тем самым гарантируют, что готовый продукт не изменится
в течение установленного срока годности.
В общем случае валидация представляет собой проверку соответствия какого-
либо процесса или объекта определенным критериям и получение точных доку-
ментированных доказательств того, что процесс или система (при работе в рамках
заданных параметров) могут эффективно и постоянно выполняться, отвечая при
этом установленным спецификациям и показателям качества. Таким образом, ва-
лидация — неотъемлемая часть обеспечения качества, касающаяся изучения си-
стем, производственных помещений и процессов, и направлена на определение,
выполняют ли они назначенные функции в соответствии с требованиями. После
проведения валидации процесс будет обеспечивать высокую степень гарантии
однородности серии, отвечающую требованиям спецификаций, и, следовательно,
регуляторным требованиям. Валидация не улучшает процессы, а подтверждает, что
они были надлежащим образом разработаны и находятся под контролем. Проведе-
ние валидации важно не только для выполнения требований регуляторных органов,
но оно также способствует улучшению фармацевтической промышленности как
таковой. Фармацевтическая промышленность получает пользу от валидационных
мероприятий за счет снижения риска возникновения проблем, поскольку обеспе-
чивается бесперебойное выполнение процессов. Помимо этого валидационные ме-
роприятия способствуют уменьшению затрат, возникающих вследствие брака про-
1 Свободный университет Берлина, Берлин, Германия.
2 Лаборатория фармацевтических разработок, BIAL, Сан-Мамеде-де-Коронаду, Португа-
лия.
3 Факультет фармацевтики, Университет Порто, Порто, Португалия.
Глава 8.4. Принципы валидации фармацевтического производства 925
дукции, снижению риска несоответствия регуляторным требованиям, повышению
надежности внутрипроизводственного контроля и проверки готового продукта для
выпуска в обращение.
Валидацию также можно определить как документирование того, что любая
процедура, процесс или действие постоянно приводят к ожидаемым результатам, то
же самое касается квалификации систем и оборудования. Валидируемый процесс
должен быть описан в сводном плане валидации (VMP — валидационный мастер-
план, validation master plan), который представляет собой утвержденный письмен-
ный план с перечнем целей и действий и с указаниями, как и когда компания до-
стигнет соблюдения требований GMP по валидации. Процедура валидации должна
быть четко описана в протоколе валидации (валидационном протоколе), который
содержит письменный план действий, определяющий способ проведения валида-
ции процесса, с указанием лиц, которые будут проводить различные испытания,
и параметров проверок. Валидационный протокол должен содержать план отбо-
ра образцов (проб), методики испытаний, спецификации характеристик продукта
и перечень используемого оборудования. Помимо этого в протоколе приводятся
критерии приемлемости и указываются лица, которые будут подписывать, утверж-
дать или отклонять заключения, сделанные по результатам этого научного исследо-
вания. Отчет о валидации (валидационный отчет) — итоговый документ процедуры
валидации, содержащий результаты и интерпретацию поставленных в валидацион-
ном протоколе задач.
Валидационные процедуры должны быть применимы к компьютерным систе-
мам, процессам очистки, производственным процессам, системам подогрева, вен-
тиляции и кондиционирования воздуха, системам водоподготовки, аналитическим
методикам и оборудованию.
Процесс валидации обеспечивает высокую степень гарантии того, что опреде-
ленный процесс постоянно приводит к получению продукта, отвечающего требова-
ниям спецификаций и имеющего установленные показатели качества. Валидация
представляет документированное подтверждение того, что процесс способен на-
дежно и неизменно производить продукт требуемого качества. Валидация систем
водо- и воздухоподготовки подтверждает, что системы находятся под контролем
в течение длительного периода времени. Аналитическая валидация подтверждает,
что аналитическая методика пригодна для назначенного использования. Валидация
оборудования применима к критическому оборудованию, работа которого может
повлиять на качество продукта, и гарантирует, что оно работает надлежащим об-
разом. Компьютерная валидация обеспечивает высокую степень гарантии того, что
компьютерные системы анализируют, контролируют и регистрируют данные пра-
вильно, а обработка данных соответствует установленным спецификациям. Валида-
ция очистки подтверждает, что процедуры очистки удаляют остаточные количества
материалов до приемлемого уровня, который зависит от таких факторов, как объем
серии, дозировка, токсичность и размеры оборудования.
Подводя итог, можно сказать, что валидационные процедуры представляют со-
бой инструмент разработки продукта с надлежащими и воспроизводимыми параме-
трами, обеспечивающий качество на протяжении жизненного цикла и повышаю-
щий безопасность пациентов и фармацевтической отрасли.
926
Часть 8. Валидация
8.4.2. Область применения процессов валидации
Область применения валидации весьма широко описывалась в литературе, и было
предпринято несколько попыток дать четкое определение этой области [1]. В на-
стоящее время понятно, что с точки зрения выполнения регуляторных требований
и для обеспечения соответствия им необходимо проведение валидации всех про-
цессов, включая производственные помещения, оборудование, аналитические ме-
тодики и компьютерные программы, используемые для аналитических испытаний
фармацевтических продуктов [2]. При проведении валидации необходимо с самого
начала особо сформулировать основные цели, которые она преследует, что означает
необходимость тщательной подготовки и планирования до начала валидации, а так-
же разработки определенной программы самих валидационных мероприятий.
Для своевременного проведения валидационных процедур необходима соот-
ветствующая и достаточная структурированная система, включающая организаци-
онную структуру и документацию, необходимый персонал и финансовые ресурсы.
К обсуждению необходимо привлекать руководство и персонал, ответственный за
обеспечение качества. Выполнение валидации должно проводиться опытным пер-
соналом из различных подразделений в зависимости от вида требуемых валидаци-
онных работ.
Валидацию с заданной периодичностью следует проводить для новых помеще-
ний и оборудования, новых инженерных сетей и целых систем, новых процессов
и методик, а также в случаях внесения серьезных изменений. Валидацию проводят
в соответствии с письменными протоколами. В зависимости от времени проведе-
ния различают перспективную, сопутствующую и ретроспективную валидацию.
Следует четко различать внутрипроизводственный контроль и валидацию. Вну-
трипроизводственные испытания проводятся во время выпуска каждой серии в со-
ответствии со спецификациями и методами, созданными на этапе разработки. Цель
таких испытаний — постоянный контроль за процессом, а не его валидация. Если
запускается производство препарата с новым составом или внедряется новый метод,
то необходимо до проведения валидации показать, что они пригодны для рутинной
работы. Определенный процесс с использованием заданных материалов и оборудо-
вания должен стабильно производить продукт требуемого качества.
Важная работа по валидации, таким образом, необходима для доказательства
того, что критические параметры процессов взяты под контроль. Для определения
объема работ по валидации следует использовать методологию оценки рисков.
8.4.3. Сводный план валидации ( VMP)
Валидация заключается в представлении документированного доказательства, име-
ющего высокую степень вероятности, что настроенный ранее процесс будет посто-
янно производить продукт, соответствующий заданным спецификациям.
По определению ВОЗ VMP — документ высокого уровня, содержащий всеобъ-
емлющий план валидации для целого проекта и обобщающий основную идеоло-
гию и подходы производителя, используемые для установления приемлемости его
деятельности. В плане приводится информация о программе валидационных работ,
включающая планируемые действия производителя и методы оценки адекватно-
Глава 8.4, Принципы валидации фармацевтического производства
927
сти работы оборудования, систем, процедур контроля и процессов, подлежащих
валидации. VMP — документ, подлежащий утверждению, содержащий подробное
описание и график работ по валидации, а также разграничение ответственностей,
связанных с планом. VMP можно определить как структурированный, детальный
план работ, содержащий информацию об основной идеологии производителя, пла-
нируемых действиях и подходах, используемых для установления соответствия вы-
полняемых работ установленным требованиям, и о контроле валидационных работ
по проекту.
VMP, бесспорно, должен быть наиболее важным документом любой програм-
мы по валидации. Стратегия проведения валидации на предприятии должна быть
четко изложена в данном документе с учетом организационной структуры всех ва-
лидационных мероприятий. В VMP даются основные указания по программе вали-
дации, определяется ответственность персонала, вовлеченного в валидационные
мероприятия, определяются все элементы, подлежащие валидации, и отражаются
характер и объем испытаний по каждому пункту [3]. Другие вопросы, которые не-
обходимо осветить в VMP, — это обзор существующих производственных участков,
систем и оборудования; перечень уже провалидированных процессов и тех, валида-
ция которых еще предстоит; планирование и составление временных графиков ва-
лидации; контроль изменений; ссылки на существующие документы, действующие
до разработки VMP. Таким образом, одновременно создается ретроспективный, со-
путствующий и перспективный план валидации. Все валидационные мероприятия,
касающиеся критических технических операций, связанных с контролем продуктов
и производственных процессов, должны быть включены в VMP. К этим мероприя-
тиям также относится квалификация критического производственного и контроль-
ного оборудования.
VMP является сводным документом, поэтому он должен быть кратким и четким.
В нем не следует повторять информацию, имеющуюся в других документах, но не-
обходимо привести ссылки на существующие документы, такие как регламенты,
стандартные операционные процедуры (СОПы), рабочие инструкции и валидаци-
онные протоколы и отчеты. Формат документов указывается в VMP.
VMP должен быть разделен на главы по различным аспектам. Во вступительной
части следует изложить стратегию производителя в отношении валидации и приве-
сти общее описание области применения указанных валидационных мероприятий,
их цели, задачи, места проведения и график работ. Затем необходимо описать все
валидационные мероприятия и их организационную структуру с учетом ответствен-
ности персонала за разработку VMP, валидационные протоколы, выполнение работ
по валидации, подготовку отчета и документов, утверждение аналогичных валида-
ционных протоколов, отчетность на всех этапах валидационных процессов, а также
необходимость в обучении, требуемом для проведения валидации. Другим требо-
ванием к VMP является наличие перекрестных ссылок на другие документы и на
определенные критические параметры процессов, влияющие на качество продук-
та. Затем все валидационные мероприятия, включенные в VMP, следует обобщить
и привести в табличной форме. Подобная таблица должна давать общее представле-
ние о валидации и содержать все включенные в VMP валидируемые объекты и опи-
сание требуемого объема валидационных работ. План должен включать валидацию
928
Часть 8. Валидация
аналитических методик, используемых при определении валидационного статуса
других процессов и систем, валидационные подходы, действия по ревалидации,
текущий статус и планирование на будущее. Наконец, в VMP необходимо в общих
чертах указать ключевые критерии приемлемости для всех валидационных меро-
приятий. Следует добавить раздел планирования с подробным описанием планов
подпроектов, состояния кадрового обеспечения, наличия необходимого оборудова-
ния, графика валидационных мероприятий и других специфичных требований для
успешного завершения валидации. Данный временной план-график можно вклю-
чить в упомянутую выше таблицу. VMP требует периодического обновления. Он
должен заканчиваться обязательством предприятия по управлению критическими
изменениями материалов, производственных помещений, оборудования и процес-
сов (включая аналитические методики), а также списком ссылок и терминов.
VMP должен содержать ссылки на СОПы (рабочие инструкции) и документы,
касающиеся любых аспектов валидационного процесса. Валидационные действия
должны выполняться в соответствии с письменными инструкциями, если произво-
дитель продукта хочет, чтобы были соблюдены правила 6Л/Рпри производстве про-
дукта (в ходе валидации). Перечень соответствующих СОПов должен быть включен
в VMP и должен определять, как эти операции валидировать. Путем планирования
и составления графика валидационных работ в VMP устанавливается периодич-
ность, необходимая для обеспечения приведенных в плане обязательств по вали-
дации. Валидация различных операций, помещений и систем проводится в заранее
определенных областях в соответствии с VMP. Кроме того, в ИМРуказываются лица,
ответственные за подготовку самого VMP, протоколов и СОПов (рабочих инструк-
ций), за выполнение валидационных работ, подготовку и контроль отчетной доку-
ментации, утверждение валидационных протоколов и отчетов, системы слежения
и определение потребностей в обучении.
VMP помогает не только производителю и рабочей группе, но также и аудиторам.
Он позволяет всем членам группы по валидации четко разделять задания и устанав-
ливает ответственность различных групп при валидации оборудования и систем
с учетом времени, людских ресурсов и денег. Он также помогает аудиторам понять
подход организации к валидации, структуру и организацию всех валидационных
действий. Более того, VMP — постоянно меняющийся документ, который обнов-
ляется и дорабатывается в ходе проекта. Это может быть обусловлено специфичны-
ми изменениями, требующими ревалидации, такими как изменения программного
обеспечения, производственной площадки и административной структуры, постав-
щиков исходных материалов, процесса, оборудования, производственных помеще-
ний и вспомогательных систем.
8.4.4. Валидационные протоколы и отчеты
8.4.4.1. Валидационные протоколы
Протокол является письменным набором инструкций, более широких по области
применения, чем стандартные операционные процедуры (СОПы). СОПы — под-
робные письменные инструкции для регулярно выполняемых процедур в рамках
любых действий, связанных с фармацевтическим производством. В протоколе опи-
сываются подробности тщательно спланированного исследования стабильности
Глава 8.4. Принципы валидации фармацевтического производства
929
работы новой системы, нового оборудования, новой процедуры или приемлемости
нового процесса перед его внедрением.
Протоколы содержат важную базовую информацию, обоснование работ и цели
исследования, в них приводится полное описание процедур, которые надо будет
выполнить. Протокол включает подробное описание: места проведения работы;
ответственности персонала; используемого оборудования; стандартов и критериев
соответствующих продуктов и процессов; вида валидации; отбора образцов (проб);
проведения испытания и контроля за соблюдением требований; порядка анализа
полученных результатов; заданных критериев приемлемости для однозначных вы-
водов. Для фармацевтических производителей валидация, исследования стабиль-
ности и клинические исследования являются примерами процедур, требующих на-
личия письменных протоколов.
Валидационный протокол — документ, в котором описывается объект, подле-
жащий проверке, испытания, с помощью которых будет производиться проверка,
а также ожидаемые результаты проверки. Он представляет собой комплект доку-
ментов, содержащий записи, результаты и оценку выполненной программы ва-
лидации. Также он может содержать предложения по улучшению процесса и/или
оборудования [1]. Валидационные протоколы играют важную роль в обеспечении
документального подтверждения стабильности оборудования, системы, процесса
или метода в рамках заданных пределов [4].
Валидационные протоколы необходимы для описания цели, методологии и кри-
териев приемлемости при проведении квалификации монтажа, функционирования
и эксплуатации. Они составляются для гарантии того, что методы проверки и кри-
терии приемлемости проверяются и утверждаются перед выполнением протоколов.
На практике существует несколько этапов составления протокола. Прежде всего
следует утвердить приемлемый формат протокола. Не существует универсальных
форматов протоколов, но в определенной степени стиль протокола будут опреде-
ляться типом оборудования, размером проекта и личными предпочтениями разра-
ботчика. Тем не менее, были выработаны определенные требования к протоколам
валидации. Как и другим контролируемым документам, протоколам присваивают
уникальные идентификационные номера и указывают номер версии. На каждой
странице приводится название протокола и номер страницы, отводится место для
подписей согласующих протокол лиц. Другими стандартными элементами прото-
колов являются краткое описание объекта, проходящего проверку, и четкое указа-
ние ответственностей.
Часто протокол включает заполняемые в ходе процедуры формы или разделы для
записи данных. Таким образом, после выполнения протокола в документе содер-
жатся записи с результатами и заключением [3]. В протоколе необходимо описать
действия, проводимые при валидации, а также критерии приемлемости для допуска
производственного процесса в целом или какой-либо его части для регулярного ис-
пользования [1].
Валидационный протокол требуется для определения специфических объектов
и действий, которые будут входить в любое валидационное исследование. Фирмам
рекомендуется составлять VMP, указывая общую стратегию валидации либо по груп-
пе продуктов, либо по типу оборудования или целой производственной площадке.
930
Часть 8. Валидация
Протокол должен быть составлен до начала исследования и должен включать ссыл-
ки на документацию, содержащую информацию об определенном процессе, его па-
раметрах, ответственности персонала и критериях приемлемости.
Следует заранее разработать и согласовать перечень объектов и методов, под-
лежащих проверке в соответствии с валидационным протоколом. Существует не-
сколько подходов к проведению валидационных испытаний. Следует понимать,
что выбираемый подход должен обеспечивать внутреннюю целостность валидации
и предупреждать исключения объектов из работ по невнимательности. Например,
возможно сгруппировать одинаковые объекты вместе в одном протоколе или для
каждого объекта создать отдельный протокол. Единичный или множественные про-
токолы можно составить для квалификации монтажа (IQ, installation qualification),
квалификации функционирования (OQ, operational qualification) и квалификации
проектной документации (DQ, design qualification). Для сложных объектов, таких как
крупные инженерные системы, допустимо создание отдельных протоколов для ге-
нерирующей и распределительной подсистем. Валидация компьютерных систем или
систем управления (контроля) технологическим оборудованием может быть оформ-
лена в виде подраздела протокола механической валидации или в виде отдельного
протокола.
Протокол необходимо утвердить до его использования. Кроме того, любые
изменения в протоколе должны утверждаться до практической реализации этих
изменений. После согласования подхода и формата можно начинать подготовку
протокола. Подготовка протокола требует наличия персонала, обученного из-
влекать информацию из различных источников и объединять ее в единое целое.
Обычно источниками информации являются данные об используемых материа-
лах, спецификации, технические данные, схемы трубопроводов и контрольно-
измерительных приборов, документация производителя и руководства по экс-
плуатации оборудования. Невозможно составить адекватные протоколы, если
количество источников информации недостаточно. Как только составлен первый
черновой вариант, протоколы необходимо проверить. Обычно рецензирование
проводит персонал из производственного отдела, инженерного отдела и отдела
обеспечения качества. Для исходного чернового варианта считается нормальным
внесение значительных изменений, поскольку заинтересованные стороны зна-
комятся с протоколом впервые. Вместо всего протокола предпочтительно отдать
специалистам на рецензирование только ту его часть, за которую они отвечают.
Например, отдел, ответственный за производство, должен проверить, что пред-
лагаемые критерии приемлемости соответствуют требованиям процесса; отдел,
ответственный за инженерные системы, должен подтвердить, что перечень обору-
дования, основные компоненты и требования к инженерным системам составле-
ны правильно; отдел обеспечения качества должен подтвердить, что нормативные
(например, фармакопейные) требования выполняются и протокол соответствует
ожидаемым нормам качества.
В большом проекте координация рецензирования протоколов, отслеживание
статуса версии, хранение и возвращение протоколов рецензентами — задачи, тре-
бующие строгого планирования. На разных этапах возможны ситуации, когда неко-
торые протоколы находятся на рецензировании, в некоторые вносятся изменения,
Глава 8.4, Принципы валидации фармацевтического производства 931
одновременно готовятся новые протоколы, а утвержденные протоколы отложены
на хранение до начала выполнения процедур. Следует использовать систему поис-
ка, позволяющую очень быстро найти нужный протокол с предоставлением инфор-
мации об актуальном статусе разрабатываемого протокола. Также следует заранее
согласовать процедуру рассмотрения протоколов и время, отводимое на рецензи-
рование. Способы организации рецензирования могут быть различными — от тра-
диционной передачи документа каждому специалисту до создания группы для со-
вместного рецензирования протокола в электронном виде и совместного внесения
изменений или комментариев. Выбор соответствующего способа должен основы-
ваться на имеющемся времени, ресурсах и технологии. В некоторых случаях рецен-
зирование протокола является ограничивающим фактором, который может оказать
критическое влияние на график выполнения работ [3].
8.4.4.2. Валидационные отчеты
Отчет о валидации — письменный документ, который содержит перекрестные ссып-
ки на валидационный протокол, обобщает полученные результаты, описывает все
наблюдавшиеся отклонения и представляет необходимые заключения, в том чис-
ле рекомендации по внесению изменений, требуемых для устранения недостатков,
обнаруженных при проведении квалификации и валидации [5]. В данном отчете
требуется представить как результаты и заключения, так и надлежащее утвержде-
ние исследования. Отчет должен содержать обзор процедур, использованных для
очистки, отбора образцов и проведения испытаний, а также результаты физических
и аналитических испытаний или ссылки на них. В отчет следует включить заключе-
ние о приемлемости результатов. Дополнительно приводится информация о статусе
валидируемых процедур, любые рекомендации по результатам испытаний и любая
сопутствующая информация, полученная во время исследования. К такой инфор-
мации относится проведение ревалидации (при необходимости), утвержденные за-
ключения и любые отклонения от протокола, которые могли произойти. В случаях,
когда дальнейшее серийное производство продукта в течение определенного перио-
да времени маловероятно, рекомендуется составлять временные отчеты по каждой
выпускаемой серии, пока не будет завершена валидация очистки [1].
Наконец, отделы, ответственные за проведение работ по квалификации и вали-
дации, должны согласовать заполненный отчет; в заключении отчета необходимо
указать, является ли проведенная квалификация и/или валидация успешной. Окон-
чательная проверка производится отделом обеспечения качества, который утверж-
дает отчет в соответствии с системой обеспечения качества предприятия [1].
8.4.5. Валидация производственных зданий
8.4.5.1. Общие положения
Валидация производственных помещений касается расположения, планировки
и конструкции производственных зданий и помещений, обеспечивающих надлежа-
щее выполнение процедур очистки, технического обслуживания и производствен-
ных операций, соответствующих типу и этапу процесса производства [6].
Все здания и помещения в фармацевтической промышленности должны быть
спроектированы и провалидированы с учетом минимизации рисков перекрестной
932
Часть 8. Валидация
контаминации. Валидация производственных помещений должна включать оцен-
ку материальных потоков и передвижений персонала, планировки помещений и их
уборки, систем воздухоподготовки и поддержания заданной влажности, а также во-
дораспределительных систем.
8.4. Б.2. Проектирование производственных зданий
В отношении планировки производственные здания и помещения всегда должны
разрабатываться с учетом самого простого пути материального потока и обеспече-
ния контроля перекрестной контаминации. С целью отделения выпускаемой про-
дукции от помещаемой в карантин и забракованной было описано несколько воз-
можных планировок площадок [7].
Одной из наиболее популярных планировок является центральное размещение
складских помещений или зон для исходных материалов, упаковочных материалов
и неупакованной (in bulk) продукции. По внешнему периметру этой зоны хранения
располагаются производственные и упаковочные зоны, что обеспечивает движение
исходных материалов и компонентов упаковки из зон приемки и карантина в зону
хранения разрешенных в производство материалов. После отвешивания необходи-
мого для серии количества материалов их передают в производственную зону. По
окончании процесса производства готовые продукты помещают в карантин, а затем
передают в зону хранения неупакованной продукции до разрешения на упаковку.
Процесс упаковки запускается в соответствии с установленным графиком, при этом
неупакованный продукт и упаковочные материалы доставляются из зон хранения
продукции и зон хранения материалов, разрешенных в производство. Преимуще-
ством подобной планировки является экономия пространства за счет близкого
размещения зон снабжения к снабжаемым зонам. Тем не менее, существенным не-
достатком является пересечение материальных потоков с возможным риском кон-
таминации или перепутывания.
Альтернативным проектом планировки является расположение зоны приемки,
зоны хранения разрешенных материалов и компонентов и зоны отпуска в произ-
водство с одной стороны, а производственной зоны, карантина, зоны хранения
неупакованной продукции и участка упаковки вдоль центрального коридора. Ма-
териальный поток из одной зоны в другую движется так же, как в предыдущей пла-
нировке. Однако в данном случае поток цикличен, тем самым исключена ббльшая
часть пересечений, описанных выше.
Для минимизации контаминации или перепутывания можно использовать тре-
тий вариант планировки — прямолинейный поток материалов, при котором пере-
мещение идет вдоль критического пути. В этом случае основное преимущество по
сравнению с вышеописанными планировками — минимальное пересечение мате-
риалов, в результате чего минимизируется возможный риск контаминации и пере-
путывания. Основным недостатком является то, что для создания подобной конфи-
гурации требуется дополнительная площадь.
Контроль перекрестной контаминации. При контроле перекрестной контамина-
ции наиболее эффективными являются система снабжения воздухом и пылеулав-
ливание.
Глава 8.4. Принципы валидации фармацевтического производства
933
В соответствии с требованиями правил GMP при проектировании системы воз-
духоснабжения предусматривается наружный забор воздуха, наличие соответству-
ющих систем фильтрации, требуемое положительное или отрицательное давление
воздуха, возможность рециркуляции или, наоборот, 100%-ный сброс использован-
ного воздуха, а также пылеулавливание и вытяжная вентиляция.
Системы фильтрации воздуха, включая фильтры предварительной очистки
и фильтры для удаления из воздуха частиц, должны быть установлены, если требует-
ся, при подаче воздуха в производственные зоны. Если воздух подвергается очистке
от производственной пыли в зонах, где происходит загрязнение воздуха во время
производства, то необходимо установить соответствующую вытяжную вентиляцию
или другие системы контроля загрязненности.
Стандартная конфигурация системы включает использование одного или более
фильтров средней очистки (пылеуловителей) либо фильтров со сменным фильтрую-
щим элементом, расположенных рядом с областью образования пыли. Эти устрой-
ства грубой фильтрации должны удалять 95% образуемой при фармацевтическом
производстве пыли [8]. Профильтрованный воздух затем смешивается с 10—15%
воздуха, взятого снаружи, и пропускается через высокоэффективный фильтр тон-
кой очистки (ЯЕРЛ-фильтр, high-efflciency particulate air), а затем вновь поступает
в помещения через подающую распределительную систему.
Наиболее нуждаются в системе улавливания пыли помещения для отбора образ-
цов и зона взвешивания (отпуска в производство). Эти зоны должны быть устроены
в виде замкнутых помещений с отдельными камерами или вытяжками, где возмож-
но отдельное взвешивание или отбор образцов. Данные помещения можно спро-
ектировать с использованием горизонтального ламинарного потока, подходящих
вытяжных систем или других пылеулавливающих устройств. Поступление воздуха
в эти системы будет происходить через НЕРЛ-фильтры, расположенные либо на
станциях забора воздуха, либо после пылеуловителя до возвращения в общую систе-
му или систему подачи воздуха в соответствующую зону.
Последней зоной, в которой проходит улавливание пыли, является зона упа-
ковки. Некоторые машины имеют встроенную вакуумную систему, возвращающую
воздух, профильтрованный через высокоэффективный фильтр абсолютной очист-
ки, обратно в зону упаковки. Для установок для вставки хлопкового наполнителя
следует предусмотреть особые требования к пылеулавливанию.
Контроль влажности и температуры. Системы контроля влажности и температуры
имеют важное значение, так как влияют на защиту продукции и комфорт рабочей
среды. Если не указано другое, 45%-ная влажность в помещениях и 21 °C — при-
емлемые рабочие условия в критических производственных зонах. Необходимо
обеспечить комфортные условия для всех выполняемых операций. При этом кон-
троль температуры должен гарантировать стабильные показания независимо от
температуры внешней (атмосферной) окружающей среды. Таким образом, созда-
ются комфортные условия для работы персонала и исключается негативное воздей-
ствие на характеристики полупродуктов. Складские операции должны проводиться
при адекватной вентиляции, особенно в зонах высокостеллажного хранения — или
с использованием стеллажей для паллет, или со складированием паллет. Вентиля-
ция может обеспечиваться большими крышными вентиляторами, обеспечивающи-
934
Часть 8. Валидация
ми циркуляцию воздуха. В зонах низких температур, таких как зоны погрузки или
приема товаров, можно использовать дополнительный нагрев воздуха с помощью
вентиляторных воздухонагревателей [7].
Контроль водоподготовки. Поступление питьевой воды в систему водоснабжения
производства не должно приводить к контаминации фармацевтической продукции.
Поэтому необходима эффективная система водоснабжения. В настоящее время воз-
можно использование нескольких способов получения воды высокого фармацевти-
ческого качества. К ним относятся ионообменная обработка, обратный осмос, дис-
тилляция, электродиализ и ультрафильтрация. Однако единственной оптимальной
системы для получения воды высокой степени чистоты не существует, выбор конеч-
ной системы зависит от качества исходной водопроводной воды, предполагаемого
использования, скорости подачи и стоимости. В фармацевтической промышлен-
ности обычно используют различные классы воды: артезианскую воду, водопрово-
дную воду, очищенную воду, а также разновидности специально очищенной воды,
такие как вода для инъекций (например, вода MilliQ).
Вода, получаемая непосредственно из скважины, называется артезианской. Она
может быть загрязнена химически или микробиологически, так как не проходила
обработку [7]. Поэтому использование артезианской воды должно быть ограниче-
но непроизводственными операциями и системами, такими как поливка газонов,
противопожарные и инженерные системы.
Водопроводная (питьевая) вода — это вода из общественного водопровода или
вода из скважины для частного пользования, которая подвергается определенной
микробиологической обработке, такой как добавление хлора [7]. Водопроводная
вода может применяться в производственных операциях для очистки и санитарной
обработки. Следует проводить периодический контроль точек водоотбора для обе-
спечения приемлемого остаточного уровня хлора и отсутствия микробной конта-
минации. Очищенная вода подвергается обработке для получения определенного
уровня химической чистоты и является тем классом воды, который используется
в большинстве производственных фармацевтических операций и финальной очист-
ке оборудования. Очищенная вода обычно получается деионизацией или дистил-
ляцией, хотя возможно применение систем обратного осмоса и ультрафильтрации,
если с их помощью может быть получена требуемая химическая чистота. В каче-
стве предварительной обработки для удаления ионов кальция и магния или хлора
и органических веществ часто используются умягчители или фильтрация с акти-
вированным углем. Ионный обмен и деминерализация посредством деионизации
являются широко распространенными методами получения очищенной воды для
фармацевтической промышленности.
Оборудование для проведения деионизации должно иметь надлежащий размер,
обеспечивающий возможность частой регенерации. Систему рециркуляции также
следует установить в блоке, соответствующем номинальной пропускной способно-
сти блока деионизации. Должны иметься письменные процедуры, гарантирующие,
что оборудование для водоподготовки эксплуатируется, контролируется, обслужи-
вается надлежащим образом и проходит регулярную санитарную обработку.
При проведении фильтрации необходима установка фильтров предварительной
очистки для предотвращения попадания частиц большого размера в систему и ми-
Глава 8.4. Принципы валидации фармацевтического производства 935
крофильтрация для удаления бактерий. Фильтры предварительной очистки обычно
представлены в виде сменных картриджей с размером пор до 25 мкм. Микрофиль-
трацию обычно дополняют высокоэффективным (абсолютным) фильтром 0,2 мкм,
удаляющим большинство бактерий.
После определения необходимого класса воды необходимо провести валидацию
системы водоснабжения для гарантии достаточного напора воды и ее чистоты (хи-
мической и микробиологической). После валидации следует выполнять периодиче-
ские проверки системы водоснабжения и точек забора воды. Такие проверки долж-
ны проводиться по тщательно разработанным СОПам.
Санитарную обработку лучше всего проводить несколькими методами. После
производственных этапов с малым использованием воды систему следует промы-
вать водой с достаточным содержанием хлора. Также рекомендуется периодическое
гиперхлорирование и микробиологическая обработка, которую можно проводить
путем поддержания температуры воды 80 °C. В качестве альтернативы можно ис-
пользовать ультрафиолетовое облучение.
Дератизация и дезинсекция. Все производственные участки, зоны обработки,
упаковки и хранения фармацевтической продукции должны содержаться в чистоте
с соблюдением санитарных требований. Это означает, что в данных зонах не должно
быть признаков присутствия грызунов, птиц, насекомых и других животных (кроме
лабораторных). Мусор и органические отходы следует хранить и своевременно вы-
брасывать в соответствии с санитарно-техническими требованиями.
Программа дератизации и дезинсекции должна быть разработана с целью обе-
спечения целостности и качества производимой продукции и соответствия суще-
ствующим требованиям. В ней должны быть обозначены общая цель и позиция
компании в этой области. Эффективность программы будет обеспечена назначе-
нием ответственного по предприятию с общей ответственностью по данной про-
грамме и указаниями, каким образом эта ответственность будет реализовываться.
Кроме того, группа по дератизации и дезинсекции — внутрипроизводственная или
нанятая по контракту — должна соответствовать четко установленным требовани-
ям к знаниям и опыту работы. Выполнению программы могут помочь и другие со-
трудники предприятия, указывая проблемные зоны, на которые следует обратить
внимание.
Данная программа должна быть доведена до сведения среднего и низшего ру-
ководящего производственного персонала (до начальников смен). В рамках про-
граммы дезинсекции необходимо периодически проверять перечень разрешенных
к применению пестицидов. К основной информации по пестицидам, которую не-
обходимо проверять, относятся: торговое название пестицида; классификация; тип
действия; химическое название и концентрация активного вещества; эффектив-
ность; практическое значение; область применения; способ и частота применения;
токсичность и любые специфические симптомы отравления; статус одобрения пра-
вительством; специфические ограничения и предосторожности при применении
[7]. Разработка перечней с подобной информацией служит сразу двум целям. Во-
первых, эти перечни должны быть согласованы отделом охраны труда предприятия,
чтобы подтвердить соответствие материалов требованиям Министерства охраны
труда США (OSHA, Occupational Safety and Health Administration) и других региональ-
936
Часть 8. Валидация
ных или местных регуляторных органов. Во-вторых, эти перечни способствуют
выполнению требований GMP. Письменные процедуры по использованию соот-
ветствующих родентицидов, инсектицидов, фунгицидов, фумигантов, чистящих
и дезинфицирующих средств необходимы для предотвращения контаминации обо-
рудования, комплектующих, емкостей для фармацевтических продуктов, укупороч-
ных, упаковочных, этикетировочных материалов или лекарственных средств.
Стены и пол во всех производственных зонах должны быть выполнены из мате-
риалов, не имеющих пор. Должно быть сведено к минимуму число выступающих
частей, таких как трубы и электрошкафы. Необходимо тщательным образом спла-
нировать размещение оборудования, чтобы предоставить достаточное пространство
для выполняемых операций. Освещение должно быть достаточным, а производ-
ственные зоны удалены от внешних выходов. Весь персонал должен пройти необ-
ходимое обучение для понимания требований GMP, в том числе и внешние под-
рядчики.
Проведение плановых инспекций и профилактических мероприятий должно до-
кументироваться с указанием возникающих проблем, а также специальных работ,
которые оказались эффективными в решении проблем.
Производственные помещения. Производственные помещения должны быть
спроектированы таким образом, чтобы гарантировать отвечающую требованиям
очистку и уменьшение перекрестной контаминации. При проектировании следует
избегать создания мест скопления пыли, таких как прямые углы (90°). Пылеулавли-
ватели или воздуховоды должны располагаться только на поверхности стен, а в по-
мещении должен быть только минимум необходимого оборудования.
Давление воздуха внутри производственных помещений должно быть положи-
тельным или отрицательным в зависимости от того, чем является продукт — жид-
костью или порошком (соответственно), с целью предотвращения перекрестной
контаминации. Давление должно контролироваться валидированными системами
контроля.
Контроль упаковки и маркировки. Устройство зоны упаковки имеет большое зна-
чение, поскольку в этой зоне очень велика вероятность перепутывания продукции
или этикеток. Элементы линий фасовки (первичной упаковки) должны иметь ми-
нимальное разделение друг от друга. В зависимости от используемого оборудования
может потребоваться больше места. Следует рассмотреть способ разделения линий
с помощью стенок. Стенки должны смыкаться с полом для предотвращения пере-
носа продукции и быть достаточно высокими, чтобы не допустить смешивания про-
дукции.
Начало операций упаковки может происходить на достаточно большой предва-
рительной площадке, вмещающей все необходимые компоненты для таких опера-
ций. Данная площадка должна быть отделенной от других, но прилегать к зоне на-
полнения или находиться перед ней. По возможности зона наполнения (фасовки)
должна быть оборудована системой воздушной фильтрации с высокоэффективны-
ми абсолютными фильтрами. В зоне наполнения должен располагаться пылеуло-
витель центральной системы пылеулавливания для удаления пыли из этой зоны.
Циркуляция воздуха в помещении должна начинаться с зоны наполнения, а затем
проходить в зоны, где производится вложение хлопкового наполнителя (влагопо-
Глава 8.4. Принципы валидации фармацевтического производства
937
глотителя), укупорка или маркировка. Так будет достигнута максимальная защита
продукции в зоне наполнения, где продукция наиболее уязвима, и станет возмож-
ным снижение уровня контроля в других зонах, поскольку там продукция уже на-
ходится в первичной упаковке.
Для всех операций упаковки необходимо разработать СОПы (письменные ин-
струкции). В них необходимо включить описание действий по настройке линии,
разрешению эксплуатации линии перед пуском в работу, периодической проверке
линии во время работы, прекращению работы линии, очистке линии по окончании
работы и учету (составлению материального баланса) продукции и компонентов.
Следует также обратить особое внимание на операции маркировки. В фармацев-
тической промышленности уже давно не применяются нарезные этикетки. Подсчет
и проверка электронных этикеток стали обычной практикой, все чаще использу-
ются штрих-коды, универсальные коды продуктов (UPC, universal product codes) или
штрих-коды системы здравоохранения (HIBC, health industry bar codes). Хранение
этикеток играет большую роль в вопросах безопасности и защиты от старения. В по-
следнее время популярной стала проверка этикеток на финальном этапе наклейки
этикеток, после того как Федеральное управление США по контролю за пищевой
продукцией и лекарствами (FDA, Food and Drug Administration) выявило основные
ошибки в этой области.
Необходимо разработать письменные инструкции для учета этикеток с установ-
ленным пределом отклонений, необходимо установить меры по надежному хране-
нию этикеток, например в запираемых шкафах на производственных линиях. Не-
обходимо вести листы учета и других компонентов, чтобы можно было составить
баланс использованных и полученных упаковок. По возможности следует преду-
смотреть зону для чистки фильтров и снимаемых частей оборудования, а также зону
для сборки и разборки фасовочных машин.
8.4.6. Валидация процесса производства
Цель валидации процесса производства лекарственного препарата — подтвержде-
ние того, что данный процесс полностью контролируется и обеспечивает стабиль-
ное получение продукции, соответствующей установленным параметрам качества.
Подобный процесс валидации охватывает все производственные операции от при-
готовления лекарственной формы до упаковки. Валидация процесса проводится
для обеспечения запуска в производство лекарственного продукта.
Также валидация представляет документированное подтверждение того, что
процесс производства, выполненный по утвержденному VMP и соответствующим
СОПам, позволяет стабильно получать лекарственное средство, отвечающее всем
критическим параметрам в соответствии с предназначенным применением. Вали-
дацию проводят для подтверждения того, что все требуемые условия выполнены,
и для технического анализа всех надлежащих производственных испытаний.
В настоящее время основное направление валидации производственного про-
цесса сосредоточено на фармацевтической разработке. Поэтому основной задача
экспериментального производства — обеспечение эффективности, экономичности
и постоянства производства продукции при последующем масштабировании. Так
как низкие производственные затраты обеспечивают конкурентные преимуще-
938
Часть 8.Валидация
ства, им следует уделить особое внимание. Каждый блок производственных опера-
ций поэтому должен быть оптимизирован. Для сокращения времени на выполне-
ние операций производственные инструкции, переносимые в отдел производства,
должны быть четко изложены, понятны и однозначны. Не следует переходить на
новое оборудование без экономического обоснования необходимости его покупки.
Однако если международные компании намерены производить продукцию на не-
скольких производственных площадках, может потребоваться альтернативное про-
изводственное оборудование и процедуры.
Физические свойства и спецификации производимых лекарственных препара-
тов должны соответствовать установленным ранее при разработке лекарственной
формы, проверенным на экспериментальном производстве и утвержденным отде-
лом контроля качества. Таким образом, производимый продукт при масштабирова-
нии должен обладать соответствующими показателями качества.
Дополнительной обязанностью персонала экспериментального производства
является проверка нового производственного оборудования с целью поиска при-
чин и нахождения решений проблем, которые могут возникнуть на производстве.
Так как отдел фармацевтических исследований и его отделение по разработке несут
ответственность за разработку состава лекарственных форм и процессов производ-
ства готовых препаратов, опыт, полученный при разработке этих процессов, будет
играть наиболее важную роль при проведении валидации. Валидация, выполняемая
на экспериментальном этапе, упрощает крупномасштабную валидацию.
Очевидно, что целью фармацевтических исследований является достижение
бездефектного производства с отсутствием забракованных серий, а это возможно
проверить с помощью валидации процесса. При этом следует помнить, что исчер-
пывающая окончательная проверка готовой продукции не может служить заменой
внутрипроизводственного контроля и валидации технологического процесса.
Процесс валидации позволит получать продукцию, напрямую связанную с лекар-
ственной формой, на которой определялась клиническая эффективность и безопас-
ность. Таким образом, целесообразно сопоставление производственного процесса,
используемого исходного сырья, внутрипроизводственного контроля и результатов
испытаний готового продукта. При масштабировании следует провести тщательное
изучение этих параметров на этапе разработки, чтобы обеспечить производство се-
рий надлежащего качества. Подбор совпадающего оборудования для эксперимен-
тального и промышленного производства позволит устранить возможные пробле-
мы при последующей валидации. Необходимо контролировать физико-химические
свойства используемых фармацевтических субстанций. Особое внимание следует
уделять препаратам с незначительным содержанием действующего вещества в еди-
нице лекарственной формы.
Физико-химические свойства исходных материалов имеют большое значение
для однородности дозирования и биодоступности. Поэтому биодоступность лекар-
ственного препарата необходимо подробно исследовать перед внесением значитель-
ных изменений. Поскольку физико-химические свойства используемых субстан-
ций (например, размер частиц исходных материалов) могут влиять на доступность
и фармакологический эффект препарата, необходимо проверять основные параме-
тры субстанции при проведении валидационной программы. Характеристики фар-
Глава 8.4. Принципы валидации фармацевтического производства
939
мацевтической субстанции мотуг отличаться от производителя к производителю,
а также от серии к серии у одного производителя. Контроль физико-химических
свойств вспомогательных веществ не менее важен и должен быть указан в специфи-
кациях.
Для разработки производственного процесса, обладающего воспроизводимо-
стью, необходимо уделять внимание особым инструкциям и процедурам проверки.
Например, во вспомогательных веществах не должно быть комков, а надлежащий
контроль способствует измельчению сырья. Дополнительно необходимо указывать
размер и форму емкостей для хранения и другого используемого оборудования.
Руководство и производственный персонал мозут вносить изменения в решения
по производственному процессу, касающиеся пригодности нового оборудования,
которое планируется использовать. Замена оборудования для некоторых продуктов
может потребовать дополнительного изучения растворения и/или однородности
дозирования. Объем и глубина исследований в значительной степени зависят от
конкретного продукта. Для некоторых продуктов с подробными данными за дли-
тельный период времени, полученными в воспроизводимом и контролируемом
процессе, определенные испытания можно исключить. Однако при разработке
сильнодействующих лекарственных препаратов такие испытания все же проводят-
ся (например, отбор проб из смеси при производстве таблеток) [9]. Если изучаются
продукты с недостаточным количеством архивных данных за определенный период
времени, то необходимы дополнительные испытания готового продукта. Например,
при производстве таблеток при испытаниях готового продукта проводят испытания
на растворение и однородность дозирования.
Серии, используемые приданном подходе к валидации производственного про-
цесса, должны изготавливаться в разных масштабах по мере разработки процесса
производства. Серии лабораторного масштаба очень невелики (например, в 100—
1000 раз меньше промышленных серий) и производятся на стадиях лабораторного
исследования и ранней разработки, когда идет работа над составом лекарственной
формы и упаковкой и ведутся доклинические и/или клинические исследования.
При разработке фармацевтических продуктов валидационные данные по сериям
лабораторного масштаба могут помочь при выборе подходящего производственно-
го процесса (развитие и определение критических рабочих параметров продукта).
Опытно-промышленные серии соответствуют по меньшей мере 10% промышлен-
ной серии или 100 000 таблеток (максимально) и используются на стадиях разработ-
ки и оптимизации для исследования стабильности. Опытно-промышленные серии
применяются для получения данных, прогнозирующих промышленные серии, и по-
этому являются связующим звеном между разработкой процесса и промышленным
производством фармацевтического продукта. Наконец, серии промышленного мас-
штаба производятся во время коммерческой реализации лекарственного препарата.
Как правило, надлежащее выполнение валидации процесса должно основывать-
ся на следующих принципах:
• Все исходные материалы, используемые для валидационных серий, должны
быть разрешены к применению отделом контроля качества.
• Лекарственные препараты валидируются одновременно. Образцы из трех
последовательно произведенных серий должны соответствовать всем уста-
940
Часть 8. Валидация
новленным спецификациям и не иметь необъяснимых несоответствий. Если
одна из трех серий не соответствует требованиям, валидацию проводят по-
вторно до трех раз, чтобы получить три приемлемых результата подряд. Если
результаты не соответствуют требованиям, испытания приостанавливаются
до пересмотра производственного процесса.
• Валидационные серии должны производиться на обычном производствен-
ном оборудовании и тем же персоналом.
• Должны быть определены допустимые пределы для критических внутрипро-
изводственных контрольных параметров и проведен комплексный монито-
ринг. Пределы, определенные как «худший случай», проверяются по крайней
мере дважды, другой предел проверяется по крайней мере один раз для завер-
шения комплексного мониторинга.
• Для критических контрольных параметров процесса, определяемых в кон-
трольных точках, величина должна проверяться в пределах допустимого уров-
ня, обычно ±1 единица.
• Все испытания должны проводиться обученным и опытным техническим
персоналом и документироваться строго научным образом с использованием
утвержденного формата протокола.
• Любая проверяемая функция, не соответствующая установленным параме-
трам в утвержденном протоколе, должна быть объяснена исчерпывающим
образом, а ее наличие должно быть согласовано; в противном случае квали-
фикация будет считаться невыполненной.
Образцы следует отбирать во время и/или после каждого критического этапа
производства. Все контрольные параметры производственного процесса должны
мониторироваться и записываться. Анализ образцов проводится в двух параллелях
с помощью валидированных или принятых фармакопейных методик. Результаты
испытаний образцов необходимы для подтверждения определенных в специфика-
циях параметров качества полупродуктов и готового продукта. Соответствие специ-
фикации подтверждает пригодность выбранных критических параметров, приме-
няемых при валидации процесса.
Валидационные данные должны создаваться для всех продуктов с целью демон-
страции соответствия производственного процесса заданным требованиям. Данные
по валидации процесса могут быть недоступны, если в нем используется нестан-
дартный метод производства. Поэтому данные, подтверждающие валидность ис-
пользуемого метода, необходимо включать в досье на регистрацию препарата.
8.4.7. Аналитические методики
Цель валидации аналитической методики — демонстрация того, что методика, ис-
пользуемая при анализе какого-либо продукта, например определение подлинно-
сти, испытание на примеси, количественное определение, растворение, определе-
ние размера частиц и воды или определение содержания остаточных растворителей,
оказывается удовлетворительной по наиболее важным характеристикам. Испыта-
ния на подлинность, определение содержания примесей, испытание на предельное
содержание примесей и количественное определение активного вещества в образ-
Глава 8.4. Принципы валидации фармацевтического производства 941
цах фармацевтического продукта — наиболее распространенные типы аналитиче-
ских методик, которые подвергаются валидации [1].
Однако для полноценного проведения валидации необходима проверка и других
аналитических методик, таких как тесты растворения лекарственной формы или
определение размера частиц фармацевтической субстанции. Ревалидация анали-
тической методики возможна при определенных обстоятельствах, например, если
требуется показать изменения в синтезе субстанции, составе готового препарата
или в самой аналитической методике. Тем не менее, ряд других изменений тоже мо-
жет потребовать проведения валидации.
Валидация методики должна подтвердить, что используемая аналитическая ме-
тодика пригодна для своего назначения. Валидация аналитической методики —
процесс, посредством которого в лабораторных исследованиях подтверждается, что
рабочие характеристики методики соответствуют требованиям предназначенного
применения. Это означает, что валидность методики можно продемонстрировать
только с помощью лабораторных исследований. Методики следует валидировать
или ревалидировать до их внедрения и регулярного использования при любых из-
менениях условий, в которых методика валидировалась ранее (например, оборудо-
вание с различными характеристиками), и при изменениях методики, выходящих за
рамки установленной области применения.
В зависимости от используемого метода количественного определения потребу-
ется установить различные параметры его валидации. Валидация методик количе-
ственного определения — процесс установления одного или более параметров (ха-
рактеристик), зависящих от вида количественного анализа. К этим видам относятся
правильность, прецизионность (повторяемость, промежуточная прецизионность),
линейность, диапазон, предел обнаружения, предел количественного определения,
специфичность и робастность [1]. Для физико-химических методов разработаны
определенные пределы параметров валидации.
Более подробно виды количественного определения могут быть описаны сле-
дующим образом.
1. Специфичность — способность однозначно определять анализируемое веще-
ство в присутствии компонентов, наличие которых ожидаемо, включая примеси,
продукты разложения, вспомогательные вещества и т. д.
2. Правильность — степень близости истинного или принятого (опорного) стан-
дартного значения найденной в ходе анализа величине.
3. Прецизионность — степень близости результатов серии измерений, получен-
ных при многократном отборе проб из одного однородного образца при заданных
условиях. Прецизионность можно разделить на три типа: повторяемость (прецизи-
онность при одинаковых рабочих условиях в течение короткого промежутка вре-
мени), промежуточную прецизионность (прецизионность внутрилабораторных
вариаций: разные дни, разные аналитики, разное оборудование и т. д.) и воспро-
изводимость (межлабораторную прецизионность при совместных исследованиях;
обычно устанавливается при стандартизации методологии).
4. Предел обнаружения — наименьшее количество анализируемого вещества
в пробе, которое может быть обнаружено, но не обязательно количественно опреде-
лено.
942
Часть 8. Валидация
5. Предел количественного определения — наименьшее количество анализи-
руемого вещества в пробе, которое может быть количественно определено с при-
емлемым уровнем прецизионности и правильности. Предел количественного опре-
деления устанавливается для компонентов, содержащихся в малых количествах,
и используется в основном для определения примесей и/или продуктов разложения.
6. Линейность — способность получать результаты испытаний в рамках заданно-
го диапазона, прямо пропорциональные концентрации анализируемого вещества
в пробе.
7. Диапазон — интервал между максимальной и минимальной концентрациями
анализируемого вещества в пробе, на котором показано, что аналитическая методи-
ка обладает приемлемым уровнем прецизионности, правильности и линейности.
8. Робастность — мера способности методики оставаться неизменной при вне-
сении малых, но преднамеренных изменений в ее параметры; показывает степень
надежности при нормальном использовании.
8.4.8. Комплексы оборудования и компьютерные системы
8.4.8.1. Комплексы оборудования
Общие положения. С начала промышленной революции в восемнадцатом веке
производственное оборудование играет важную роль в нашей жизни. Производ-
ственное оборудование является одним из важных составляющих нашего общества
в целом и фармацевтической промышленности в частности. В середине двадцатого
века многие действия на фармацевтическом производстве выполнялись вручную,
что требовало большого количества времени и человеческих ресурсов. Этап упаков-
ки являлся самым отстающим из всех; сотни людей, сидя за столами, упаковывали
фармацевтическую продукцию, и в то время это считалось нормальным. Начиная
со второй половины двадцатого века ручной труд постепенно был заменен на про-
изводственное оборудование. Блистерные машины, автоматическое наполнение
капсул, большая степень прессования и сушки стали нормой производства, суще-
ственно увеличив объем выпускаемой продукции и прибыль предприятий. С тех
пор оборудование, используемое для производства медикаментов, значительно
усовершенствовалось. В соответствии с правилами GMP все оборудование должно
располагаться, проектироваться, конструироваться, быть приспособленным и про-
ходить обслуживание таким образом, чтобы обеспечить выполнение назначенных
функций. Схема и конструкция оборудования должны свести к минимуму риск воз-
никновения ошибок, а также обеспечивать проведение эффективной очистки и вы-
полнение обслуживания для предотвращения перекрестной контаминации, образо-
вания пыли и грязи и вообще любых нежелательных влияний на качество продукта
[10]. Оборудование и помещения должны улучшать качество и безопасность про-
дукции и никогда не должны создавать риски для продукта. При создании нового
оборудования необходимо добиваться адекватной конструкции, которая позволит
улучшить эффективность и чистоту, а также уменьшит число ошибок и поломок.
Оборудование необходимо устанавливать в надлежащих рабочих условиях для
обеспечения точной работы и предотвращения риска возникновения ошибок или
контаминации. Окружающая среда должна представлять минимальный риск кон-
Глава 8.4. Принципы валидации фармацевтического производства
943
таминации материалов или продуктов при рассмотрении совместно с мерами по за-
щите производства [10].
Производственное оборудование должно проектироваться, располагаться и об-
служиваться таким образом, чтобы обеспечить выполнение назначенных функций.
Проведение ремонтных работ и технического обслуживания не должно представ-
лять угрозы качеству продукции [10]. Устройство оборудования должно позволять
проведение простой и в то же время тщательной очистки. Очистка должна про-
водиться по подробно описанным процедурам, а оборудование должно храниться
только в чистом и сухом состоянии. Следует выбирать и использовать оборудование
для мойки и чистки, которое не будет источником контаминации.
Производственное оборудование не должно быть источником опасности для
продукции. Части производственного оборудования, непосредственно контакти-
рующие с продукцией, не должны быть реакционно-способными, вносить примеси
или обладать абсорбционными свойствами в той степени, чтобы влиять на качество
продукта и тем самым быть источником опасности [10]. Оборудование не должно
содержать материалы, контаминирующие готовый продукт, поэтому производ-
ственное оборудование, как правило, выполняется из нержавеющей стали и по-
лимерных материалов, которые легко чистить. В соответствии с вышесказанным,
нельзя использовать натуральные материалы.
Весы и измерительное оборудование с соответствующим диапазоном и точно-
стью должны быть доступны для проведения производственных и контрольных
операций. Оборудование для измерения, взвешивания, записи и контроля должно
проходить калибровку и поверку с заданной периодичностью с помощью утверж-
денных процедур. Необходимо обеспечить надлежащее ведение записей подобных
испытаний [10]. Все оборудование, которое возможно калибровать, должно прохо-
дить периодическую калибровку. Периодичность калибровки должна основываться
на типе оборудования, оценке рисков и предыдущих результатах. Слишком частое
проведение калибровки достаточно дорого, а слишком редкое приводит к неудо-
влетворительным результатам проверок. Неудовлетворительность результатов озна-
чает, что они неверны, и поэтому все результаты, полученные с момента последней
калибровки, необходимо перепроверить. Таким образом, надлежащая оценка пе-
риодичности калибровки крайне важна.
Все оборудование должно четко обозначаться соответствующим названием и ко-
дом и должно иметь одну инструкцию по эксплуатации. Надлежащая инструкция по
эксплуатации содержит по крайней мере следующие пункты: 1) описание оборудова-
ния, в которое входит функция оборудования, название, код, серийный номер, мо-
дель, производитель, сопутствующие инструменты и приспособления, размеры, ис-
точник электроснабжения, соединительные интерфейсы и описание компонентов;
2) функционирование, включая описание процедуры эксплуатации оборудования
и всех необходимых этапов и параметров; 3) детальное описание методики калибров-
ки с указанием ее периодичности; 4) техническое обслуживание, при этом должны
быть предусмотрены простые и легкие в исполнении процедуры обслуживания;
5) подробное описание очистки оборудования; 6) порядок обеспечения безопасности.
Валидация комплекса оборудования. В GMP приведены характерные особенно-
сти валидации оборудования, в частности установлены этапы валидации, такие как
944
Часть 8.Валидация
квалификация проектной документации, квалификация монтажа, квалификация
функционирования и квалификация эксплуатации. Каждый этап описывается в от-
дельном документе, и поэтому на каждое оборудование предприятия должно быть
четыре подобных документа. Без любого из этих документов оборудование нельзя
считать соответствующим требованиям GMP. Хотя правила GMPwe, относятся к ва-
лидации оборудования, в них описаны эти четыре документа, и оборудование мож-
но считать провалидированным только после утверждения этих документов.
Валидация оборудования не обладает достаточной надежностью, поэтому требу-
ется рассмотрение ряда дополнительных параметров. Пригодность для целей GMP
проверяют с помощью нескольких процедур, после чего оборудование вводят в экс-
плуатацию. После начала эксплуатации необходимо проведение периодического
технического обслуживания и калибровки. Далее приводится характеристика эта-
пов валидации с описанием работ, которые должны быть проведены на новом обо-
рудовании перед пуском его в эксплуатацию.
Квалификация проектной документации (DQ). Когда на фармацевтическом про-
изводстве возникает необходимость покупки нового оборудования, в первую оче-
редь следует точно определить тип оборудования и его технические характеристи-
ки. Технические характеристики следует выбирать с учетом специфики требований
компании или производства. Все технические характеристики подлежат согласова-
нию. Этот этап называется квалификацией проектной документации. Требования
к оборудованию должны быть указаны в специальном документе до приобретения
оборудования фармацевтической компанией. Разработанный документ использует-
ся для обоснования выбора оборудования из большого числа аналогов.
Квалификация монтажа (IQ). После выбора оборудования необходимо подтвер-
дить, что оно правильно установлено. В документации по IQ описывается и про-
веряется установка оборудования. Таким образом, повышается уверенность в том,
что рабочее оборудование и вспомогательные системы могут согласованно работать
в рамках заданных пределов и допусков [ 10]. Производитель оборудования и фарма-
цевтическая компания должны согласовать и провести IQ, документацию по кото-
рой по завершении проверки утверждает фармацевтическое предприятие. Данный
документ подтверждает, что оборудование было установлено в соответствии с тре-
бованиями производителя и покупателя.
Квалификация функционирования (OQ). Документ OQ подтверждает, что оборудо-
вание работает так, как запланировано и определено производителем оборудования
и покупателем. Например, при покупке высокоскоростного лопастного смесите-
ля/гранулятора проверка ротации лопасти проводится откалиброванным тестером
скорости вращения, и если полученное значение соответствует заданным специфи-
кациям, вращение лопасти смесителя проходит проверку OQ. В противном случае
проводится повторная квалификация. Все результаты испытаний должны фикси-
роваться и проверяться в отчете по OQ, который утверждается фармацевтическим
предприятием. В документе по OQ описывается несколько испытаний оборудо-
вания, предназначенных для оценки его надлежащей работы, при этом проводи-
мые испытания должны быть описаны и утверждены производителем оборудова-
ния и покупателем. Таким образом, при проведении испытаний на оборудовании
для каждого из них должно быть приведено описание с подписями выполнившего
Глава 8.4. Принципы валидации фармацевтического производства
945
и проверившего. Обычно испытания проводятся производителем и проверяются
покупателем. В подобных испытаниях, как правило, проверяется надлежащая рабо-
та механических и электрических компонентов оборудования.
Квалификация эксплуатации (PQ, performance qualification). После проверки ра-
ботоспособности оборудования необходимо провести ряд испытаний в реальных
условиях, чтобы подтвердить соответствие результатов значениям, указанным
в спецификации. Этот этап называется квалификацией эксплуатации. С помощью
подходящих испытаний он повышает уверенность в том, что готовый продукт, по-
лученный в ходе определенного процесса, отвечает всем требованиям к эффектив-
ности и безопасности [11].
Как правило, при покупке таблеточного пресса для фармацевтического произ-
водства проводится PQ. Прежде всего компания должна выбрать ряд своих наиболее
изученных продуктов и провести их через все стадии производства непосредствен-
но до этапа таблетирования. Препарат должен пройти процесс таблетирования на
новом оборудовании при заданных условиях прессования, таких как сила сжатия
и скорость таблетирования. Полученные таблетки должны соответствовать всем
спецификациям продукта. Определяемыми параметрами могут быть внешний вид,
твердость на излом, толщина, диаметр, средняя масса и однородность массы, исти-
раемость и объем полученных таблеток. Если полученные таблетки соответствуют
спецификациям, то таблеточный пресс считается надежным и обладающим воспро-
изводимостью при получении продукта надлежащего качества, а оборудование —
прошедшим квалификацию эксплуатации.
На стадии PQ также создается документ с описанием проводимых испытаний
и связанных технических требований. Этот документ проверяется и утверждается до
проведения испытания. После испытаний результаты заносятся в документ, и фар-
мацевтическая компания утверждает окончательную версию.
Все документы по квалификации оборудования должны содержать назначение
оборудования, перечень необходимых испытаний и их технические требования,
перечень материалов, списки операторов (аппаратчиков), согласующих лиц и лиц,
ответственных за утверждение.
После одобрения результатов квалификации проектной документации, монта-
жа, функционирования и эксплуатации оборудование считается пригодным для це-
лей GMPvt может вводиться в эксплуатацию.
8.4.8.2. Компьютерные системы
Общие положения. Технический прогресс развивается с невероятной скоростью.
Контролируемые компьютерными системами процессы, которые казались фан-
тастическими еще несколько лет назад, в настоящее время просты в реализации
и широко распространены. Подобные достижения происходят практически каждый
день в различных областях. В фармацевтической промышленности, в особенности
за последнее десятилетие, было разработано разнообразное аналитическое обору-
дование, например для спектроскопии в ближней инфракрасной области, спектро-
скопии в инфракрасной области с Фурье-преобразованием {FTIR, Fourier transform
infrared) и рамановской спектроскопии, использующее сложное управляющее про-
граммное обеспечение, а также для высокоэффективной жидкостной хроматогра-
946
Часть 8. Валидация
фии (ВЭЖХ) и масс-спектрометрии с более простым и понятным программным
обеспечением. Подобное развитие аналитического оборудования позволяет про-
мышленности производить препараты лучшего качества и получать большую при-
быль. Революция вычислительных систем затронула не только аналитическое, но
также и производственное оборудование для фармацевтической промышленности.
В настоящее время в ней практически каждая операция выполняется при участии
компьютерных систем. Поэтому все компьютерные системы на фармацевтическом
производстве должны быть валидированы для подтверждения того, что результаты,
получаемые с их помощью, обладают правильностью и прецизионностью. Кроме
этого, при любой замене ручной операции на компьютерную автоматику не долж-
но снижаться качество продукта и не должны возникать проблемы в системе обе-
спечения качества. Особое внимание следует уделить риску утраты характеристик
предыдущей системы, что может быть вызвано уменьшением числа задействован-
ных операторов оборудования.
Компьютерная система состоит из программной и аппаратной частей, обору-
дования, процессора и пользователя. Обычно она используется для выполнения
конкретной процедуры. Независимо от того, была ли компьютерная система разра-
ботана компанией или подрядчиком, либо куплена готовой, разработка документи-
рованных требований конечного пользователя играет важную роль при проведении
валидации компьютерной системы. Без первоначального определения требований
конечного пользователя и предполагаемого использования практически невоз-
можно подтвердить, что система способна постоянно им соответствовать. После
установления требований следует получить доказательство того, что компьютерная
система выполняет эти требования правильно и что они прослеживаемы до требо-
ваний проекта системы и технических характеристик. Важно, чтобы в требованиях
конечного пользователя были учтены утвержденные правила [12].
Валидация компьютерной системы. Все компьютерные системы, используемые
в настоящее время на фармацевтическом производстве, необходимо подвергать
оценке рисков. Первой задачей валидации компьютерной системы является опре-
деление возможности влияния неполадок, возникающих в компьютерной систе-
ме, на безопасность, эффективность и качество продукта. Это относится ко всем
областям фармацевтической промышленности, связанным с надлежащей кли-
нической (GCP, good clinical practices) и дистрибьюторской практикой (GDP, good
distribution practices), надлежащей лабораторной практикой (GLP, good laboratory
practices) или GMP. Валидация компьютерной системы также служит доказатель-
ством того, что все компоненты системы работают слаженно, обеспечивая по-
лучение надлежащих результатов. Таким образом, необходимо валидировать все
компоненты системы, включая приложения, процессы, пользователей и произ-
водственные площадки.
Оценка рисков — первый критический этап при валидации компьютерной си-
стемы. После оценки риска необходимо создать протокол валидации, включающий
все этапы, указанные в VMP. Действия, которые будут выполняться в компьютер-
ной системе, необходимо описать, а также привести технические параметры и про-
водимые испытания. Такие испытания могут составлять IQ, OQ и PQ. После прове-
дения испытаний и оценки данных составляется отчет о валидации. Поэтому всегда
Глава 8.4. Принципы валидации фармацевтического производства 947
должны быть доступны подробно написанные и периодически пересматриваемые
(например, ежегодно) VMP, валидационные протоколы и отчеты о валидации.
Объем работ по валидации зависит от ряда факторов, в том числе от предна-
значенного применения системы, проведения перспективной или ретроспектив-
ной валидации и добавления новых элементов. Валидацию следует рассматривать
как часть полного жизненного цикла компьютерной системы. Этот цикл включает
этапы планирования, определения технических параметров, программирования,
тестирования, ввода в эксплуатацию, документирования, работы, контроля и вне-
сения изменений [13].
Ретроспективная валидация относится к используемым компьютерным систе-
мам с разработанным VMP. В этом случае для составления правильного плана ис-
пытаний следует обратить внимание на реальное использование системы.
Для проведения полноценной валидации потребуется тесное сотрудничество
между ведущими специалистами и персоналом, проводящим валидацию компью-
терной системы. Ответственный за компьютерные системы персонал должен прой-
ти обучение по управлению и использованию оборудования в рамках своих долж-
ностных обязанностей. Кроме того, должны быть доступны эксперты для оказания
консультаций по проекту, валидации, монтажу и эксплуатации компьютерных си-
стем [12].
После процедуры валидации и определения квалификации персонала необходи-
мо описать то, как обращаться с компьютерной системой, чтобы провалидировать
ее. Как было сказано ранее, компьютерная система используется для выполнения
конкретной процедуры с целью получения заданного результата. Поэтому внимание
следует уделить расположению оборудования в надлежащих условиях, при которых
посторонние факторы не будут влиять на систему [13]. Таким образом, физическое
расположение компьютерной системы должно быть описано в валидационном про-
токоле, а в случае перемещения оборудования потребуется составление плана по-
вторной валидации (ревалидации).
Необходимо составить подробное письменное описание системы (включая гра-
фики) и поддерживать его в актуальном состоянии. В нем должны быть приведены
основные положения, цели, меры по безопасности и область применения системы,
главные особенности использования компьютера и его взаимодействие с другими
системами и процедурами [13]. Компьютерная система, как и любое другое обору-
дование, требует наличия письменной инструкции, причем по возможности рядом
с оборудованием.
Программная часть, то есть програмное обеспечение, позволяющее компьютеру
выполнять определенное задание, — критический компонент компьютерной систе-
мы. Даже простой лист с вычислениями можно рассматривать как программу, по-
скольку он позволяет компьютеру выполнять определенную задачу. Пользователь
подобной программы должен предпринять все возможные шаги для гарантии того,
что программа была разработана в соответствии с системой обеспечения качества
ИЗ].
Система должна иметь встроенную проверку правильности ввода и обработки
данных. Для проверки данных по валидации в некоторые компьютерные системы
можно периодически вводить набор данных с заранее известными результатами,
948
Часть 8. Валидация
полученные значения необходимо сохранять. Если полученные результаты укла-
дываются в установленные нормы, то компьютерная система работает надлежащим
образом, а если результаты не соответствуют ожидаемым, то компьютерная система
работает неправильно и требуется техническое обслуживание. В подобной ситуации
все результаты, полученные с помощью компьютерной системы с момента предше-
ствующей валидации, считаются недостоверными и подлежат повторной проверке.
До ввода компьютерной системы в эксплуатацию ее необходимо тщательно про-
верить и подтвердить ее способность получать требуемые результаты. При замене
системы с ручным управлением обе системы какое-то время должны работать па-
раллельно для проведения проверок и выполнения валидации [13]. Это означает,
что каждый раз при внесении изменений в оборудование при техническом обслу-
живании, замене или настройке любой части потребуется ревалидация. Подобные
изменения при техническом обслуживании могут касаться как аппаратной, так
и программной частей.
Возможность вносить или изменять информацию должна быть только у уполно-
моченного персонала. Подходящими методами, препятствующими несанкциони-
рованному вводу данных, являются использование ключей, карт допуска, личных
кодов и ограниченного доступа к компьютерным терминалам. Должна быть разра-
ботана четкая процедура создания, отмены и изменения разрешений на ввод и ис-
правление данных, включая изменение личных паролей. Следует уделить внима-
ние системам, позволяющим вести запись попыток доступа неуполномоченными
для этого людьми [13]. Компьютерная система должна предусматривать различные
уровни доступа пользователей, а доступ к каждому приложению должен быть за-
щищен именем пользователя (логином) и паролем. Критические операции, такие
как изменения методов, калибровка и настройка, должны выполняться специаль-
но уполномоченным на это персоналом. Операторы должны иметь доступ лишь
к определенным процедурам, причем без возможности внесения изменений. Все
изменения должны автоматически записываться на оборудовании с указанием
имени сотрудника, времени и даты. Эти записи могут потребоваться при проверке
(аудите) системы обеспечения качества.
При вводе критических данных вручную (например, ввод массы и номера серии
компонента при отпуске в производство) следует проводить дополнительную про-
верку точности вносимых данных. Проверку может выполнять второй оператор или
валидированные электронные устройства [13]. Часто для выполнения определен-
ной задачи компьютерной системе требуются внешние данные. Ввод внешних дан-
ных является критическим этапом работы компьютерной системы, так как ошибка
при вводе данных может привести к ошибочному результату. Для предупреждения
ошибок возможно проводить двойную проверку с последующей автоматической
регистрацией данных или двойную проверку, при которой один оператор вводит
данные, а другой проверяет правильность введенных данных, после чего оба под-
писывают соответствующую регистрационную форму. Автоматическая регистрация
данных обеспечивает получение информации, введенной в другой компьютерной
системе. Например, для таблеток в испытании «Растворение» масса таблеток, вво-
димая в компьютер вручную, может быть получена с весов, связанных с системой
Глава 8.4. Принципы валидации фармацевтического производства 949
автоматической регистрации данных; подобная регистрация является подтвержде-
нием верности введенных данных.
Система должна вести запись данных операторов, вводящих или подтверждаю-
щих критическую информацию. Правом исправления введенных данных должен
обладать уполномоченный на это персонал. Любое изменение введенных крити-
ческих данных необходимо зарегистрировать и указать причину этого изменения.
Следует уделить внимание созданию полной записи всех произведенных исправле-
ний и дополнений («контрольный журнал регистрации событий») [13].
Как упоминалось выше, изменения в системе или компьютерной программе
должны осуществляться в соответствии с определенной процедурой и содержать
описание мероприятий по валидации, проверке, утверждению и внесению измене-
ний. Подобное изменение должно проводиться и фиксироваться по согласованию
с лицом, ответственным за рассматриваемую часть системы. Любое значительное
изменение должно проходить валидацию.
Для аудита качества необходима возможность получения четких печатных ко-
пий информации, хранимой в электронном виде. Данные должны быть защищены
физическими или электронными способами от преднамеренного или случайного
повреждения. Хранимую информацию следует проверять на открытость для досту-
па, устойчивость при хранении и точность. Если для компьютерного оборудования
или программ предлагается внесение изменений, описанные выше проверки следу-
ет проводить с частотой, соответствующей используемому носителю информации.
Данные следует защищать путем периодического резервного копирования. Резерв-
ная копия данных должна храниться в течение установленного срока в отдельном
безопасном месте [13].
Вся получаемая с помощью компьютерной системы информация должна быть
доступна, защищена от посторонних и храниться в надежном месте. Неуполномо-
ченные лица не должны иметь доступ к файлам данных. Перезапись данных должна
быть невозможной, а все перерасчеты должны создавать новый блок данных, не за-
меняя при этом предыдущий. Должны быть разработаны инструкции по обеспече-
нию безопасности данных. Резервное копирование данных следует периодически
проводить в соответствии с письменной инструкцией, а резервные копии должны
обозначаться и храниться в определенных безопасных местах. Все резервные копии
должны проверяться в процессе резервного копирования с целью подтверждения
надлежащего выполнения копирования. Данные необходимо периодически про-
верять на сохранность для гарантии того, что они хранятся надлежащим образом,
и потеря данных не происходит.
Следует разработать альтернативные меры для обеспечения работы систем
в случае возникновения неисправностей. Время, необходимое для введения в экс-
плуатацию альтернативных мер, должно быть связано с их важностью. Процедуры,
выполняемые при отказе или остановке системы, должны быть описаны и провали-
дированы. Любые неисправности и предпринимаемые корректирующие действия
необходимо регистрировать [13]. Особый случай неисправностей — неполадки
в системе электроснабжения. Для некоторых компьютерных систем требуется не-
прерывная работа в течение 24 часов после начала получения и накопления дан-
ных. Поэтому необходимо наличие внешних источников бесперебойного питания
950
Часть 8. Валидация
(ИБП), обеспечивающих работу подобных компьютерных систем при неисправно-
сти системы электроснабжения.
Необходимо разработать процедуру регистрации и анализа ошибок и предпри-
нимать корректирующие действия по их устранению. В каждой компьютерной си-
стеме могут возникать ошибки, и все они должны быть документально оформлены.
Каждый раз, когда происходит ошибка системы, ее необходимо проанализировать,
после чего, возможно, потребуется определенная перенастройка компьютерной си-
стемы. Все производимые изменения должны быть четко описаны и задокументи-
рованы; в некоторых случаях может потребоваться ревалидация системы.
Когда для проведения компьютерного технического обслуживания привлека-
ются внешние фирмы, необходимо составить соглашение, четко описывающее
ответственность фирмы-подрядчика [13]. При осуществлении выпуска серий в об-
ращение или поставки с использованием компьютерной системы выпуск должен
производиться только Уполномоченным лицом, а система должна однозначно
идентифицировать и зарегистрировать лицо, выпустившее серии. Это достигается
предоставлением Уполномоченному лицу операционного уровня доступа к ком-
пьютерной системе, позволяющего выпускать серии. Таким уровнем доступа не
должен обладать никто, кроме Уполномоченного лица.
Валидация программного обеспечения. Регуляторные органы требуют представле-
ния правил с описанием принципов валидации программного обеспечения, пред-
назначенного для проектирования, разработки и производства медицинского обо-
рудования или используемого в работе такого оборудования.
В руководствах рекомендуется совмещать управление жизненным циклом про-
граммного обеспечения и мероприятия по управлению рисками. Валидации про-
граммного обеспечения и мероприятия по проверке должны проводиться на про-
тяжении всего жизненного цикла программного обеспечения [12, 14]. Понятия
проверки (верификации) и валидации программного обеспечения часто путают.
Проверка программного обеспечения — процесс, представляющий объективные
доказательства того, что результат проектирования на определенном этапе жиз-
ненного цикла разработки программного обеспечения соответствует утвержден-
ным требованиям для данного этапа [14]. Верификация программного обеспечения
заключается в оценке надлежащего выполнения поставленных задач и получения
ожидаемых результатов. Верификация программного обеспечения — часть его ва-
лидации. Тестирование программного обеспечения является одним из многих ме-
роприятий, направленных на подтверждение соответствия результатов разработки
программного обеспечения исходным требованиям. Другие мероприятия при про-
ведении верификации — различные типы статических и динамических анализов,
проверка кода и документации, сквозной контроль и т. д. [ 14].
Хотя существующие руководства относятся лишь к программному обеспечению,
используемому как составная часть или вспомогательный элемент медицинского
оборудования (например, программное обеспечение станций переливания крови
(банков по заготовке донорской крови), программируемые логические контролле-
ры производственного оборудования, программное обеспечение для записи и веде-
ния отчета по истории работы устройства), подходы к валидации, представленные
в этих руководствах, применимы к валидации любого программного обеспечения.
Глава 8.4. Принципы валидации фармацевтического производства 951
Программное обеспечение не является физическим объектом, и, в отличие от
аппаратных неисправностей, проблемы с ним возникают без заблаговременного
предупреждения. Одним из наиболее частых дефектов программного обеспечения
является ветвление, то есть способность выполнять альтернативные серии команд
в зависимости от различных входных данных. Возможность ветвления программ-
ного обеспечения делает команды очень сложными, и валидация при возникнове-
нии ошибочных ответов на определенные входные данные будет затруднена до тех
пор, пока ошибочные входные данные не будут обнаружены. Входными данными
для программного обеспечения может быть любая информация, а так как в про-
грамму невозможно ввести все возможные данные, то валидация данных стано-
вится очень трудоемкой. Таким образом, результаты должны обладать достаточно
высокой достоверностью. Большинство проблем, связанных с программным обе-
спечением, — следствие ошибок при проектировании и разработке программы
и не связаны напрямую с поставщиком программного обеспечения. Весьма про-
стым является создание копии программы, которые будут так же отлично работать,
как и оригинал.
Валидация программного обеспечения не рассматривается отдельно в норма-
тивных документах по системам менеджмента качества. По определению FDA ва-
лидация программного обеспечения представляет «подтверждение посредством
проверки и представления объективного доказательства того, что технические ха-
рактеристики программного обеспечения соответствуют требованиям пользователя
и предназначенному применению и что определенные задачи с помощью данного
программного обеспечения могут быть выполнены» [14]. Валидация программно-
го обеспечения затрагивает установку, применение и использование программного
обеспечения. Такая валидация включает проведение нескольких испытаний, про-
верок и верификаций с целью подтверждения правильности установки и использо-
вания программного обеспечения, а также того, что решаемые на нем задачи соот-
ветствуют определенным спецификациям. Валидация программного обеспечения
должна проводиться в обычных рабочих условиях для данного оборудования. Это
особенно важно для медицинского оборудования, используемого в специальных
условиях, например в непосредственной близости к телу человека или внутри него.
В плане валидации программного обеспечения следует учитывать анализ рисков.
Критическое программное обеспечение должно иметь высокий уровень достовер-
ности и проходить доскональную валидационную проверку, в то время как некри-
тическое программное обеспечение может проходить упрощенные валидационные
испытания.
Незначительные, на первый взгляд, изменения в программном коде могут вы-
звать внезапные существенные проблемы в программном обеспечении. Процесс
разработки программного обеспечения должен быть достаточно хорошо спланиро-
ван, контролируем и задокументирован таким образом, чтобы можно было опреде-
лять и исправлять непредвиденные результаты изменения программного обеспече-
ния [14]. Обслуживание программного обеспечения необходимо проводить очень
осторожно, так как даже несколько маленьких изменений могут привести к нежела-
тельным результатам. Наличие точной и подробной документации — важная часть
валидации программного обеспечения.
952
Часть 8.Валидация
Валидация программного обеспечения — непростая задача, поэтому необходимо
заранее разработать соответствующий график и план действий по валидации и ве-
рификации программного обеспечения, чтобы в дальнейшем избежать ненужных
затрат времени и денег. Один день, потраченный на планирование без проведения
испытаний, лучше дня, потраченного на испытания без планирования.
Электронные документы. Были разработаны специальные регуляторные прави-
ла, описывающие использование разных видов электронных документов. К таким
документам относятся электронные записи и электронные подписи. Например,
FDA разработало нормативный документ, в котором указало критерии приемлемо-
сти (при наличии определенных условий) электронных записей, электронных под-
писей и рукописных подписей на электронных записях, используемых в качестве
эквивалента записям и подписям на бумаге. Эти инструкции, относящиеся ко всем
программам FDA, обеспечивают широкое применение электронных технологий, со-
вместимых с обязанностью FDA по охране общественного здоровья [12].
Некоторые указания данного руководства относятся практически к любой за-
писи, создаваемой на компьютере, а некоторые, достаточно простые, относятся
только к документам, непосредственно связанным с областью применения руко-
водства. Заполнение отчетов по строгим правилам, например, для подачи в FDA,
необходимо во всех случаях, когда электронный формат заменяет бумажные ко-
пии. С другой стороны, если для получения бумажных вариантов документов ис-
пользуется компьютер и полученные бумажные документы отвечают требованиям
установленных правил, FDA считает, что «использование электронных документов
вместо бумажных» не имеет места. То же самое относится к ситуациям, когда люди
имеют дело с бумажными документами в ежедневной работе. В подобных случаях
использование компьютерных систем для получения бумажных отчетов не требует
соответствия официальным требованиям [12]. Компьютерные системы, создающие
документы, которые впоследствии подписываются от руки в бумажном варианте,
в данном руководстве не рассматриваются.
Руководство описывает требования к документам, созданным в компьютерных
системах в виде файлов, подписываемых и изменяемых в электронном виде. Так как
документы не печатаются и не подписываются в бумажном варианте, применяется
ряд мер по обеспечению непротиворечивости данных, предотвращению внесения
нежелательных изменений, безопасности и персонификации электронной подписи.
Данное руководство относится только к тем документам, которые необходимо
обновлять. Все документы, созданные и подписанные электронным способом, но
не требующие обновления, не рассматриваются в данном руководстве. Решение
о том, какие документы попадают в область действия руководства, принимается
фармацевтическим производителем и должно быть задокументировано и обосно-
вано.
Электронные подписи предназначены для равноценной замены рукописных
подписей, инициалов и других способов идентификации, применяемых в офици-
альных документах. Это включает использование электронных подписей, напри-
мер, для документирования того, что некоторое событие или действие произошло
в соответствии с установленными правилами (например, утверждение, пересмотр
или проверка).
Глава 8.4. Принципы валидации фармацевтического производства 953
Валидационная группа. Для обеспечения надлежащего проведения валидацион-
ных испытаний необходимо создание вадидационной группы с четким разграниче-
нием ответственности внутри нее. Валидационная группа должна включать: ответ-
ственного за проведение валидации, руководителя группы, заведующего архивным
отделом, координатора испытаний, представителя отдела обеспечения качества,
технического специалиста (тестировщика) и наблюдателя. Лицо, ответственное за
проведение валидации, разрабатывает и утверждает VMP, протоколы и отчеты. Ру-
ководитель группы отвечает за валидацию и использование компьютерных систем.
Заведующий архивным отделом отвечает за систематизацию вадидационной доку-
ментации по всем компьютерным системам. Координатор испытаний отвечает за
проведение испытаний компьютерной системы и координирует разработку и про-
ведение оценки работы компьютерной системы. Представитель отдела обеспечения
качества должен периодически проверять и обучать персонал, а также проверять до-
кументацию по валидации. Тестировщик выполняет испытания в соответствии с ва-
лидационным протоколом. Наблюдатель контролирует действия тестировщика.
8.4.9. Валидация очистки
Валидация очистки — «процесс представления документированного доказатель-
ства того, что применяемые методы очистки на производственном участке стабиль-
но обеспечивают контроль возможного переноса продукта (включая полупродукт
и примеси), моющих средств и посторонних веществ в последующий продукт до
уровня, находящегося ниже заданных значений» [15].
Валидация очистки проводится для подтверждения безопасности и чистоты про-
дукта (требования потребителя), относится к обязательным требованиям при про-
изводстве субстанций и позволяет обеспечить надлежащее качество процесса с по-
зиций внутреннего контроля и соблюдения нормативных требований [15].
Очистка должна осуществляться относительно легко и с использованием стан-
дартных чистящих средств [7]. Вакуумные производственные системы должны быть
доступны для чистки, при этом загрязненные контактирующие с продуктом части
следует протирать и санировать с помощью дезинфицирующих средств. Оборудо-
вание следует мыть, сушить, накрывать чехлами и хранить в специальном помеще-
нии.
Подобно этапу производства, операции упаковки начинаются с составления
приказа (распоряжения) об упаковке. В нем должны быть указаны утвержденные
упаковочные материалы, номер серии подлежащего упаковке продукта и число
единиц продукта в каждой упаковке. Старший оператор проверяет правильность
и полноту распоряжения об упаковке, в том числе срок годности, используемую ли-
нию и любое другое оборудование, используемое для данной процедуры. Эти дей-
ствия выполняются до переноса материалов на упаковочную линию. Упаковочная
линия, включая оборудование, полностью проверяется на правильность разборки
и очистки от продуктов и материалов предыдущего процесса упаковки. После того
как ООК завершит проверку очистки рабочей зоны, все необходимые материалы
переносятся на упаковочную линию для механической установки. После заверше-
ния необходимой настройки старший оператор проводит предпусковые процедуры.
954
Часть 8. Валидация
К ним относятся: очистка продуктов, используемых во время установки; подсчет
этикеток для маркировки; проверка всех компонентов и номеров серий; проверка
числа флаконов, всех штампов и номеров серий; подписание распоряжения об упа-
ковке, подтверждающее готовность к началу процесса упаковки.
Выполнение ненадлежащих методик очистки приводит к получению серий не-
надлежащего качества из-за риска наличия в них ряда контаминантов, таких как
прекурсоры лекарственных веществ, продукты разложения, растворители и другие
вещества, используемые во время процесса производства, микроорганизмы, чистя-
щие вещества и смазочные материалы [15].
8.4.10. Заключение
С середины двадцатого века фармацевтическая промышленность заняла лидирую-
щие позиции по качеству и безопасности производства, соединив высокую эффек-
тивность производства с получением большей прибыли. Благодаря этому большое
значение приобрела валидация, представляющая собой подтверждение и доказа-
тельство того, что все производственные участки, оборудование и процессы работа-
ют как намечено, обеспечивая получение качественной продукции.
В настоящей главе рассматривалась система валидации в фармацевтической
промышленности. Для получения надлежащей валидационной системы сначала
требуется определить, какое оборудование, помещения и процессы будут валиди-
роваться, когда и кем будет проводиться эта работа. Такое определение основано на
методологии оценки риска и отражается в специальном документе, так называемом
сводном плане валидации (VMP). Для составления надлежащего отчета по валида-
ции необходимо описать все валидационные мероприятия в соответствующих про-
токолах, инструкциях и специальных методиках.
Валидация производственных зданий и помещений является критическим
процессом в фармацевтической промышленности, причем процедура ее выпол-
нения зависит от вида производимых лекарственных форм. Если на одном участ-
ке изготавливают различные лекарственные формы, необходимо разработать
специфические требования и различные критические параметры, основанные на
оценке рисков. Во всех производственных помещениях необходимо обеспечить
такое движение персонала, сырья, нерасфасованной и готовой продукции, чтобы
эти потоки не смешивались. Дополнительно для снижения риска перекрестной
контаминации необходимы наличие герметичных помещений, а также надлежа-
щие письменные инструкции (СОПов). Также требуется контроль температуры
и влажности воздуха, которые должны быть провалидированы для обеспечения
стабильности продукта.
VMP — ключевой документ, подтверждающий, что производство продукта соот-
ветствует установленным требованиям и обеспечивает стабильное получение про-
дукта заданного качества. Подробная информация об оборудовании, помещениях,
качестве сырья и параметрах процесса необходима для проведения валидации про-
изводственного процесса. Обеспечение постоянства этих параметров гарантирует
стабильность качества продукта после валидации. Изменения в оборудовании, сы-
рье или параметрах процесса требуют проведения ревалидации производственного
процесса.
Глава 8.4. Принципы валидации фармацевтического производства 955
Валидация аналитических методик является одним из наиболее строго контро-
лируемых мероприятий по валидации в фармацевтической отрасли. Валидация
аналитических методик необходима для подтверждения того, что используемые
методики являются наиболее подходящими для контроля конкретного продукта,
а получаемые с их помощью результаты обладают достоверностью и надежностью.
Все методики, применяемые для контроля сырья и готовой продукции, необходимо
валидировать.
Валидация оборудования состоит из четырех критических операций: квалифи-
кации проектной документации, квалификации монтажа, квалификации функ-
ционирования и квалификации эксплуатации. Эти операции подтверждают, что
оборудование соответствует указанным в документации техническим параметрам,
установлено надлежащим образом, правильно функционирует и производит про-
дукт с заданными свойствами. После этих процедур, независимо от места установки
оборудования, необходимо проведение периодической проверки и калибровки для
обеспечения надлежащей работы.
Компьютерные системы являются особым типом оборудования, которое также
необходимо валидировать. К этим системам предъявляются специальные требо-
вания, если они используются для сбора и обработки информации. С аппаратной
частью компьютерной системы связано программное обеспечение, проходящее
тщательную валидацию для подтверждения того, что получаемая с его помощью
информация правильна.
Другим вопросом, касающимся оборудования и обеспечивающим предотвра-
щение перекрестной контаминации, является процесс очистки, для которого не-
обходимо разработать соответствующие инструкции, обеспечивающие требуемую
чистоту. Валидация очистки должна проводиться с учетом методологии оценки ри-
ска и ситуаций «наихудшего случая».
Литература
1. World Health Organization (WHO) (2006), WHO expert committee on specifi cations for pharma-
ceutical preparations (fortieth report) — Supplementary guidelines on good manufacturing practices
(GMP): Validation, WHO, Geneva.
2. Thomas, E., G rochulski, A., Patel, R., George, S. M., and Zhang L. (2006, Sept.), The laboratory
control system: Fulfi Hing cGMP requirements, BioPharm Int., 26-32.
3. Slater, S. (1999), Biopharmaceutical Validation: An Overview in Biopharmaceuticals, an Industrial
Perspective, Springer-Verlag.
4. Chaloner-Larsson, G., Anderson, R., and Egan, A. A WHO Guide to Good Manufacturing Practice
(GMP) Requirements, 1997, World Health Organization, Geneva.
5. International Conference on Harmonization (ICH) (2000), Good manufacturing practice guide for
active pharmaceutical ingredients, ICH, Geneva.
6. European Medicines Evaluation Agency (EMEA) (2003), Good Manufacturing Practices, Chapter 4:
Building and facilities, EMEA.
7. Nash, R. A. (1990), The essential of process validation, in Pharmaceutical Dosage Forms, Tablets,
Marcel Dekker Inc, New York.
8. Hanna, S. A. (1990), Quality assurance, in Pharmaceutical Dosage Forms, Tablets, Marcel Dekker
Inc, New York.
9. Connolly, R. J., Berstler, F. A., and Coffin-Beach, D. (1990), Tablet production, in Pharmaceutical
Dosage Forms, Tablets, Marcel Dekker Inc, New York.
956 Часть 8. Валидация
10. European Medicines Evaluation Agency (ЕМЕА) (2003), Good Manufacturing Practices, Chapter 3:
Premise and equipment, EMEA.
11. U.S. Food and Drug Administration (FDA) (1987), Guideline on general principles of process valida-
. tion, FDA, Rockville, MD.
12. U.S. Food and Drug Administration (FDA) (2003), Guidance for industry, CFR Part 11, Electronic
records; electronic signatures, good manufacturing practices, FDA, Rockville, MD.
13. European Medicines Evaluation Agency (EMEA) (2003), Good Manufacturing Practices, Annex 11:
Computerised systems, EMEA.
14. U.S. Food and Drug Administration (FDA) (2002), general principles of software validation, good
manufacturing practices, Rockville, MD.
15. Active Pharmaceutical Ingredients Committee (APIC) (1999), Cleaning validation in active pharma-
ceutical ingredient manufacturing plants, APIC.
Список сокращений
ACPS
ANOVA
API
АРМА
ASTM
ATR
ВР
CAD
САРА
СВЕ
CBER
CCMAS
CDER
CDRH
CDS
CFD
CFR
cGMP
CIM
CIS
CNC
CORA
CTD
CUSUM-карга
DAB
DQ
EA
eCTD
EPA
EQC
ERES
Eurachem
EWG
ЕРЕМЛ-карта
FDA
FDCA
FMEA
FMECA
FMECA
FMS/FAS
FPA
FRS
FSA
GAMP
GCP
GDP
GLP
GMP
GTP
GXP
HACCP
HEPA
HIBC
Консультативная комиссия по фармацевтической науке
вариационный анализ
активный фармацевтический ингредиент
Ассоциация австралийских фармацевтических производителей
Американское общество (специалистов) по испытаниям и материалам
нарушенное полное внутреннее отражение
Британская фармакопея
автоматизированное проектирование
корректирующие и профилактические действия
внесенные изменения
Центр по экспертизе и исследованиям биологических препаратов FDA
Комитет по аналитическим методикам и отбору образцов
Центр оценки и исследований лекарств
Центр по изделиям и радиологической безопасности
система управления хроматографическими данными
вычислительная гидродинамика
Свод федеральных нормативных актов
текущая надлежащая производственная практика
система управления производством
система построения химического изображения
компьютерное числовое программное управление
компонентно-ориентированный анализ требований
общий технический документ
контрольная карта накопленных сумм
Немецкая фармакопея
квалификация проектной документации
Ассоциация органов по аккредитации стран-членов ЕС
электронный общий технический документ
Федеральное управление США по охране окружающей среды
внешний контроль качества
электронные записи и электронные подписи
Европейская ассоциация по аналитической химии
экспертная рабочая группа
контрольная карта экспоненциально взвешенного скользящего среднего
Федеральное управление США по контролю за пищевой продукцией и
лекарствами
Закон о пищевых продуктах, медикаментах и косметической продукции
анализ вида отказов и их последствий
анализ видов, последствий и критичности отказов
анализ видов и критичности отказов
гибкая фармацевтическая производственная и сборочно-упаковочная
система
фокально-плоскостная матрица
спецификация функциональных требований
Ассоциация стандартов для пищевых продуктов
надлежащая автоматизированная производственная практика
надлежащая клиническая практика
надлежащая дистрибьюторская практика
надлежащая лабораторная практика
надлежащая производственная практика
надлежащая практика обучения
международный стандарт качества в фармацевтической отрасли
анализ рисков и критические контрольные точки
высокоэффективный фильтр тонкой очистки
штрих-коды системы здравоохранения
958
Список сокращений
ICH IEC IFPAC IP IPEC IQC LCL LCTF EDA LGC LIMS LMS LNTL LRM MCR MCR-ALS Международная конференция по гармонизации Международная электротехническая комиссия Международный форум по процессно-аналитической химии Международная фармакопея Международный совет по фармацевтическим вспомогательным веществам внутренний контроль качества нижняя контрольная граница жидкокристаллический перестраиваемый фильтр линейный дискриминантный анализ Лаборатория государственных химиков Соединенного королевства система управления лабораторной информацией система управления обучения нижний естественный предельный допуск лабораторные стандартные материалы разрешение многомерной кривой разрешение многомерной кривой методом чередующихся наименьших квадратов
MIA MIR MIRS NF NPPI ОС OEE OLAP OPS OQ OSHA РАС PARAFAC PAS PAT PC PDA PFRA PHA PhRMA PIC/S PLC PLM PLS-DA PMI PQ QA QbR QC QM QOS QS QSR R&D RFID RMSEP ROI RSD RTR SD многомерный анализ изображений средний диапазон ИК-излучения спектроскопия в среднем диапазоне ИК-спекгра Национальный формуляр инновации для новых продуктов и процессов рабочие параметры общая эффективность оборудования аналитическая обработка в реальном времени Управление по фармацевтической науке квалификация функционирования Министерство охраны труда США процессно-аналитическая химия параллельный факторный анализ дополнение после утверждения процессно-аналитическая технология главные компоненты Ассоциация производителей парентеральных препаратов анализ рисков отказа процесса предварительный анализ опасностей Ассоциация фармацевтических производителей и разработчиков Организация по схеме взаимодействия фармацевтических инспекций программирование логических контроллеров управление жизненным циклом продукта РЬУ-дискриминационный анализ Институт управления проектами квалификация эксплуатации обеспечение качества обзор, основанный на вопросах контроль качества менеджмент качества общие аспекты качества система (менеджмента) качества правила систем качества научно-исследовательские разработки радиочастотная идентификация среднеквадратическая погрешность прогноза окупаемость инвестиций относительное стандартное отклонение выпуск в режиме реального времени стандартное отклонение
Список сокращений
959
SDLC разработка жизненного цикла системы в целом
SEC стандартная ошибка калибровки
SEP стандартная ошибка прогноза
SG&A продажные, общие и административные расходы
SIMCA метод формального независимого моделирования аналогий классов
SIMPLISMA простой интерактивный автомодельный анализ смесей
SNV стандартное нормальное отклонения
SRP потенциал риска объекта
TOC общий органический углерод
TPDM модель конструирования систем обучения
TQC тотальный контроль качества
TQM всеобщее управление качеством
UCL верхняя контрольная граница
UML универсальный язык моделирования
UNTL верхний естественный предельный допуск
URS спецификация требований пользователя
USL верхний допустимый предел для спецификации
USP Фармакопея США
VMP сводный план валидации
WEI вода для инъекций
WIP незавершенная продукция
АТФ аденозин-трифосфат
АФИ активный фармацевтический ингредиент
BO3 Всемирная организация здравоохранения
ВЭЖХ высокоэффективная жидкостная хроматография
ГХ газовая хроматография
ГХ-МС газовая хроматография с масс-спектрометрическим детектором
ГХ-МС/МС газовая хроматография с тандемным масс-спектрометром
ДНК дезоксирибонуклеиновая кислота
жх-мс жидкостная хроматография с масс-спектрометрическим детектором
ЖХ-МС/МС жидкостная хроматография с тандемным масс-спектрометром
ИЛАК Международная ассоциация органов по аккредитации лабораторий
ИСО Международная организация по стандартизации
ИСП-АЭС атомная эмиссионная спектроскопия с индуктивно-связанной плазмой
ИЮПАК Международный союз теоретической и прикладной химии
КОЕ колониеобразующая единица микроорганизмов
ПАУ полициклические ароматические углеводороды
ПВХ поливинилхлорид
ПНЖК полиненасыщенные жирные кислоты
ПП полипропилен
ПХБ полихлорированные бифенилы
ПЦР полимеразная цепная реакция
ПЭ полиэтилен
ПЭВА полиэтиленвинилацетат
ПЭНД, пэвп полиэтилен высокой плотности
ПЭТ полиэтилентерефталат
СМК система менеджмента качества
СОП стандартная операционная процедура
ЭДТА этилендиаминтетрауксусная кислота
Ш. К. Гэд (ред.)
ПРОИЗВОДСТВО ЛЕКАРСТВЕННЫХ СРЕДСТВ.
Контроль качества и регулирование
Практическое руководство
Перевод с английского
под редакцией Береговых В. В.
Ответственный редактор А. Стешко
ЦОП «Профессия»
Санкт-Петербург, 190005, а/я 25
Телефакс: (812) 251-46-76
URL: www.epcprof.ru,
e-mail: info@epcprof.ru
Интернет-магазин и онлайн-заказ книг издательства www.epcprof.ru
Подписано в печать 12.03.13. Формат 70x100/16. Усл. п.л. 60
Заказ № 31. Тираж 400
Отпечатано в типографии ООО «ИПК БИОНТ»
199026, Санкт-Петербург, Средний пр. дом 86
Тел.: (812)322-6843
Parallel
Мониторинг чистых помещений
Счетчики частиц в воздухе
М:1криииологические пробоотборники
Реакторные системы
для разработки и оптимизации
процесса производства
Контроль качества на всех этапах
Этапы создания фармацевтического производства /
Исследования и разработки
Строительство производства
Поляриметры
Химический
контроль
готовых
контроль
Фармацевтическое производство
Контроль качества продукции
Контроль качества
лекарственных форм
Тестеры твердых
лекарственных
форм
Счетчики частиц
в жидких
лекарственных
формах
Микробиологический
• Титраторы
• Хроматографы • Спектрометры
• Вискозиметры • Плотномеры
• Рефрактометры
Счетчики колоний
Гомогенизаторы
Средоварки
Автоклавы
• Инкубаторы
• Ламинарные шкафы
• Пластиковая посуда
ООО «Компания СокТрейд» г. Москва, Ленинский проспект 31, НОНК
Тел./факс: +7 (495) 604-44-44,926-38-40, infoyisoctrade.com, www.soctrade.com