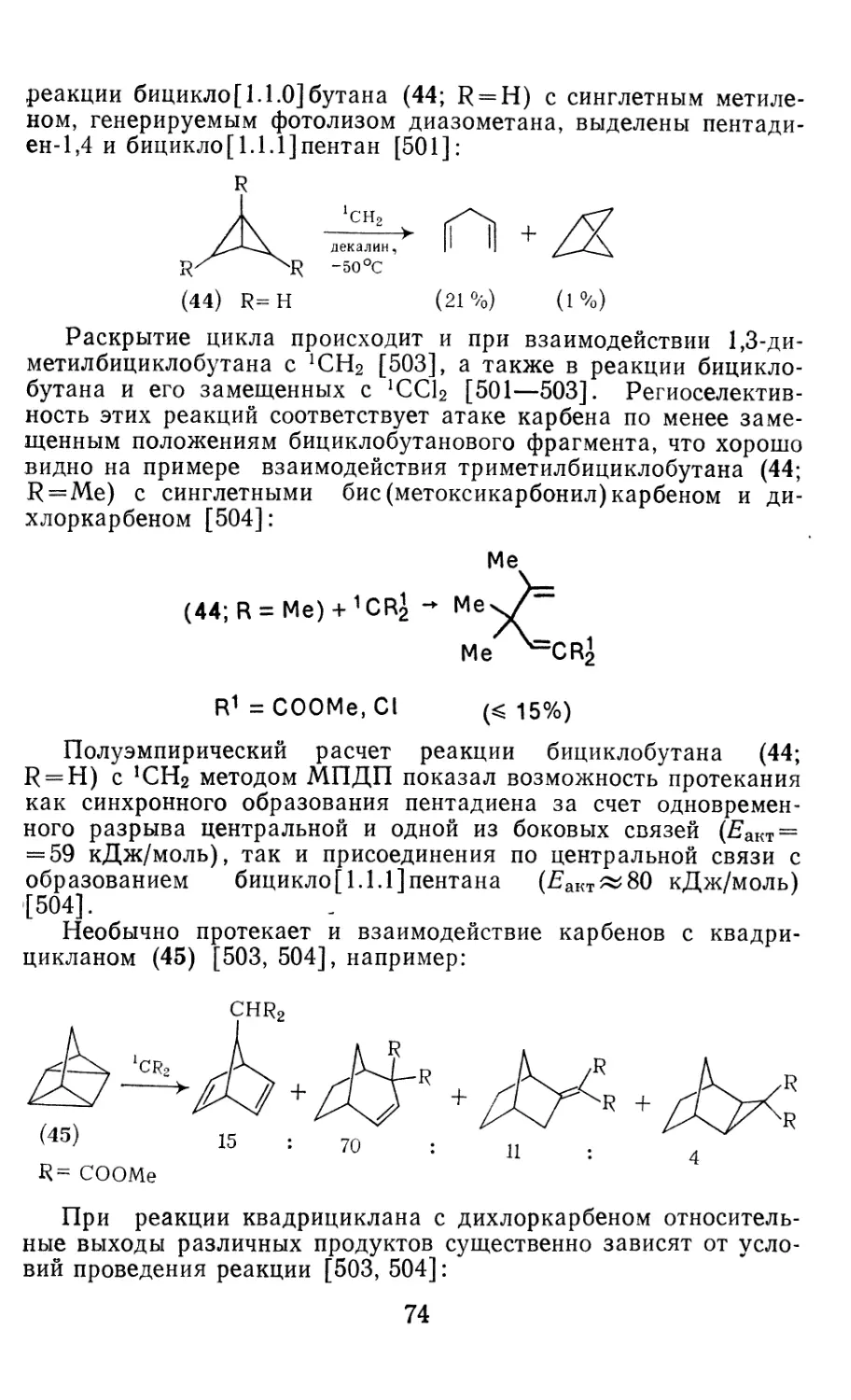
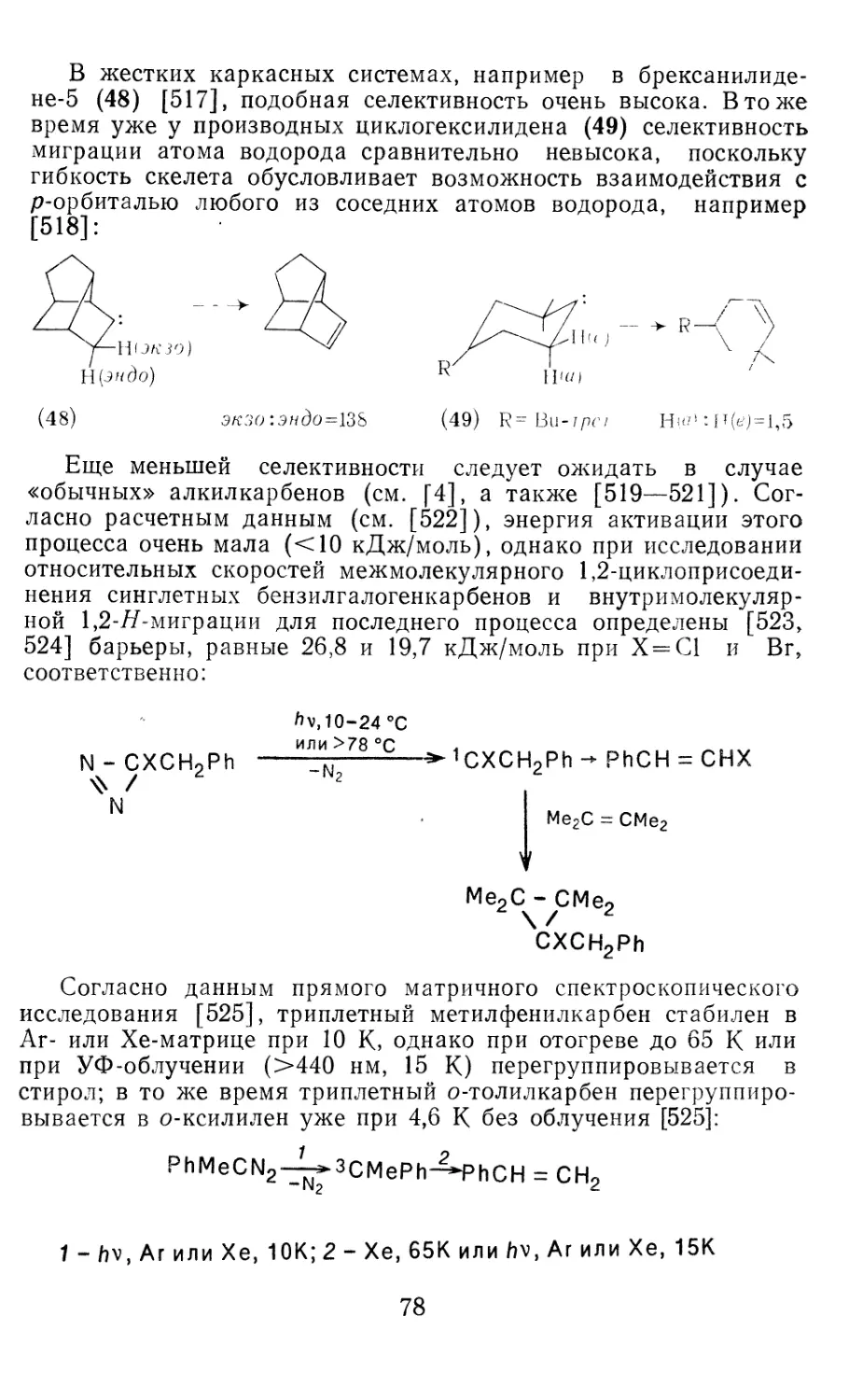
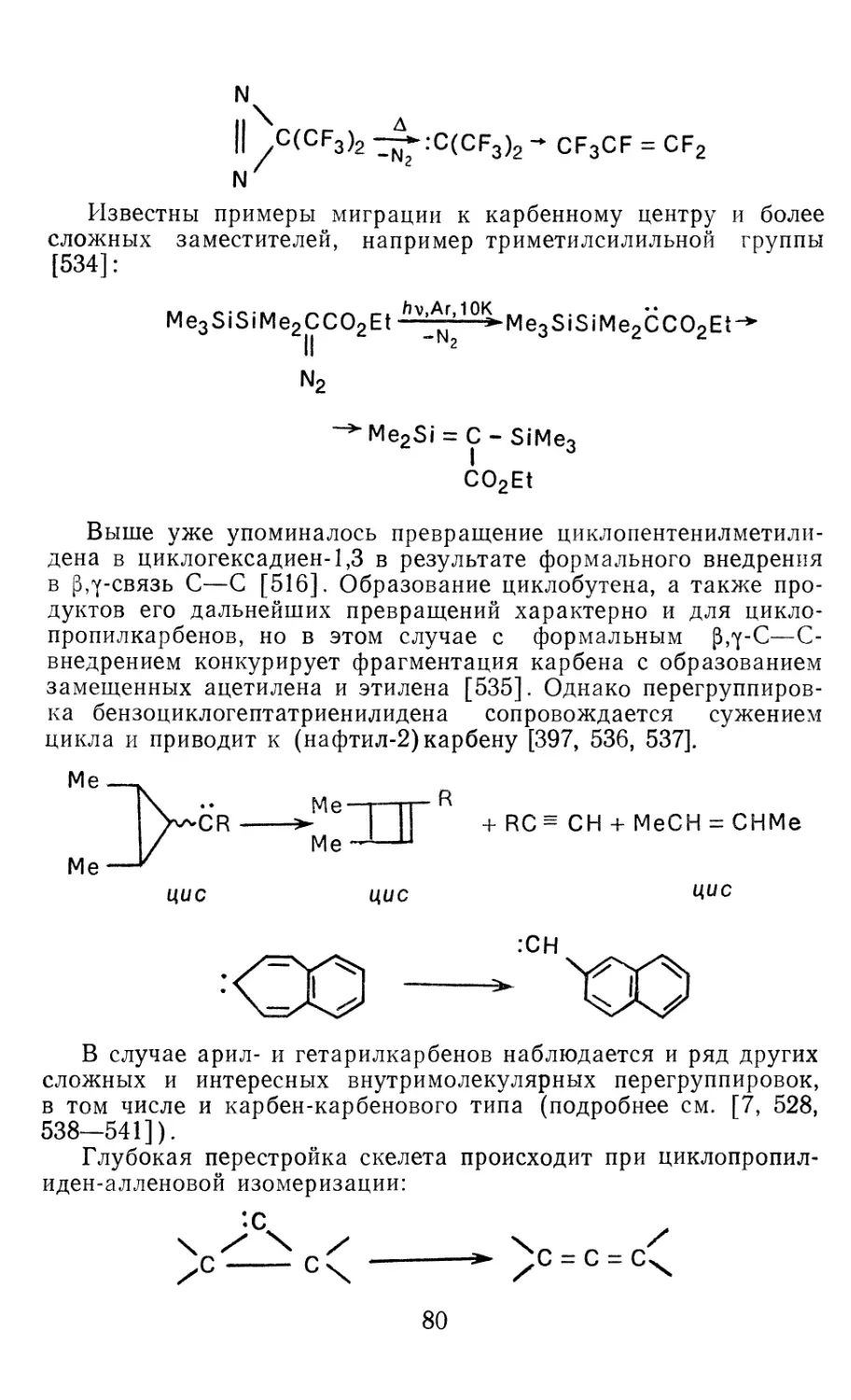
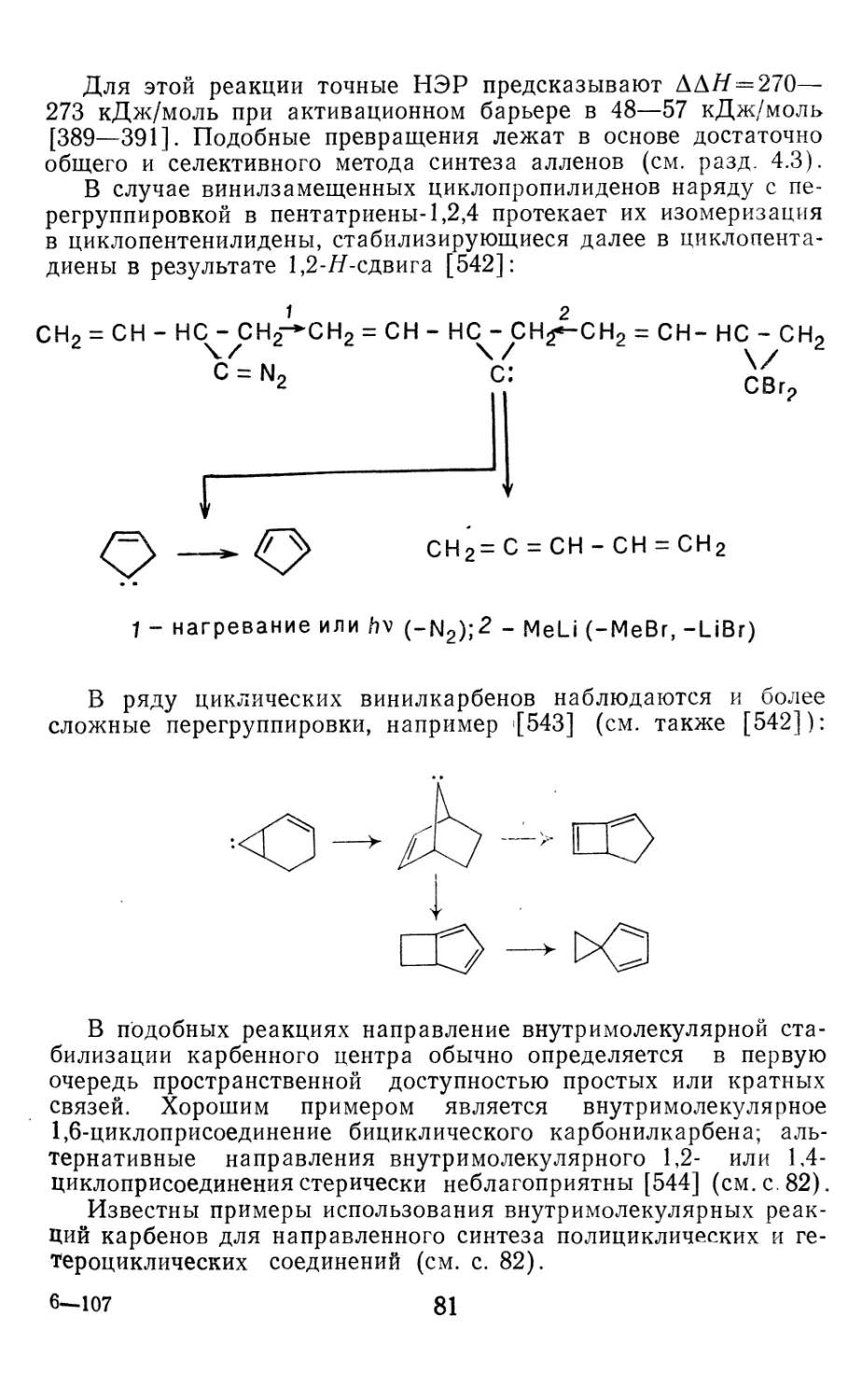
/





































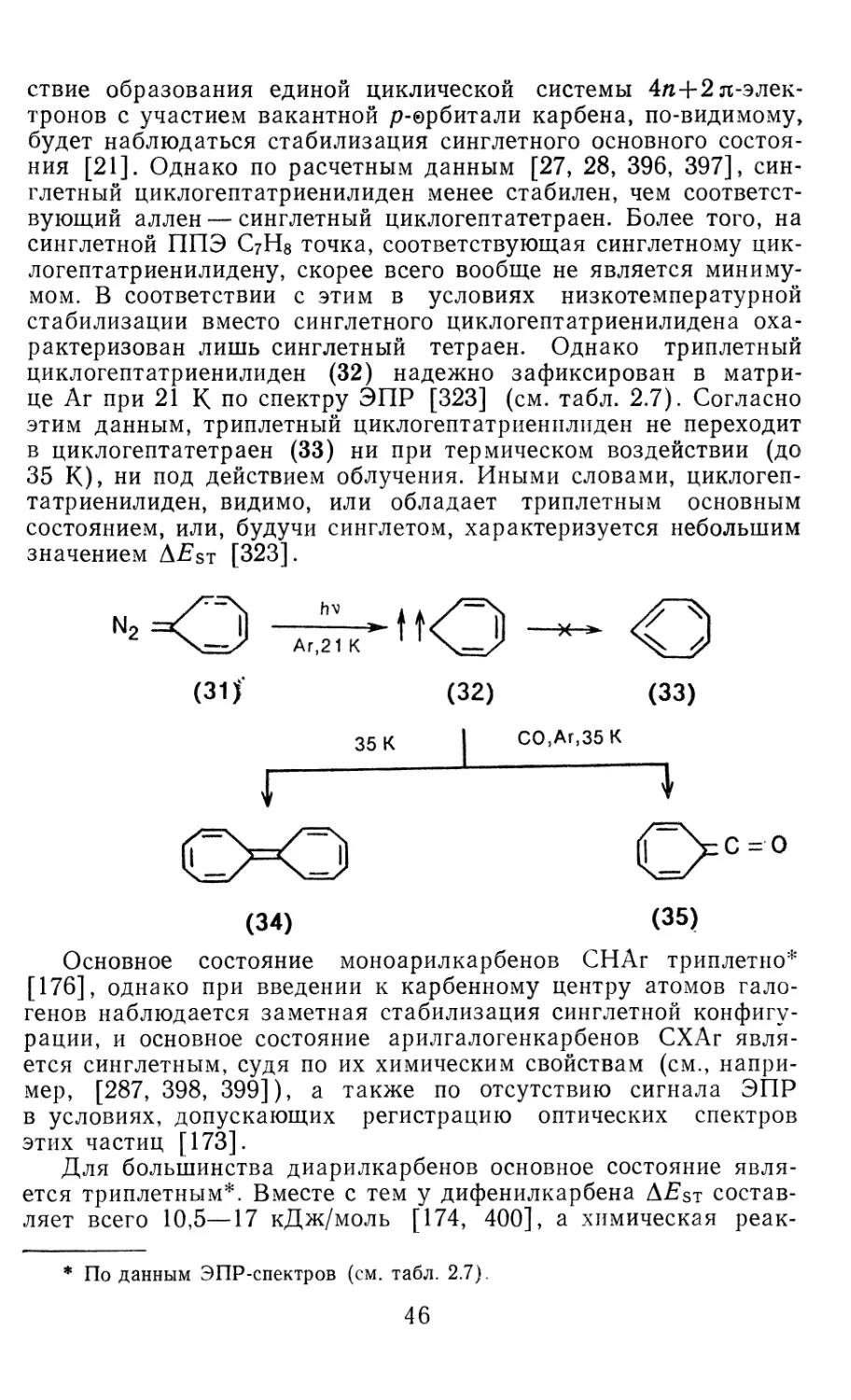


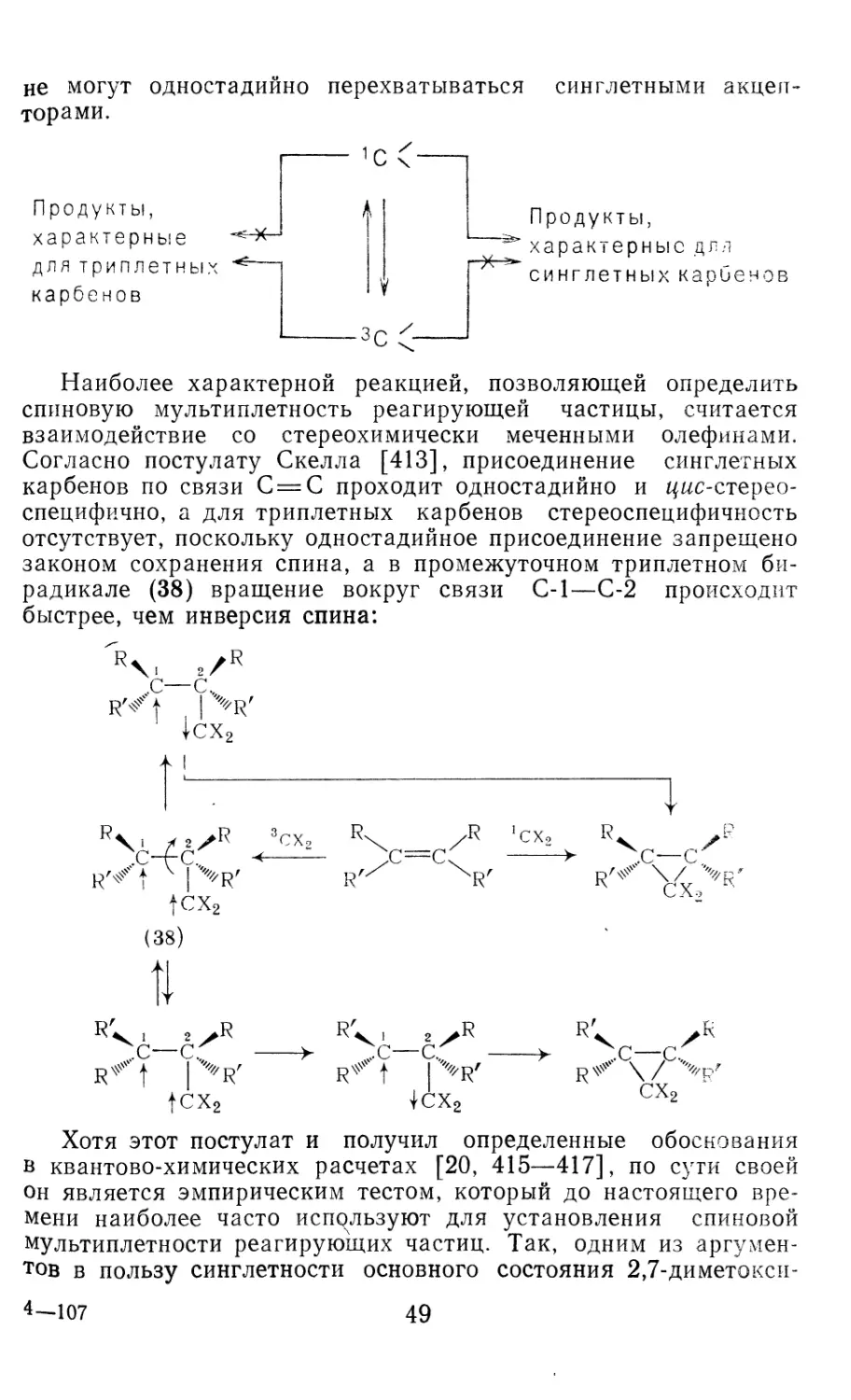
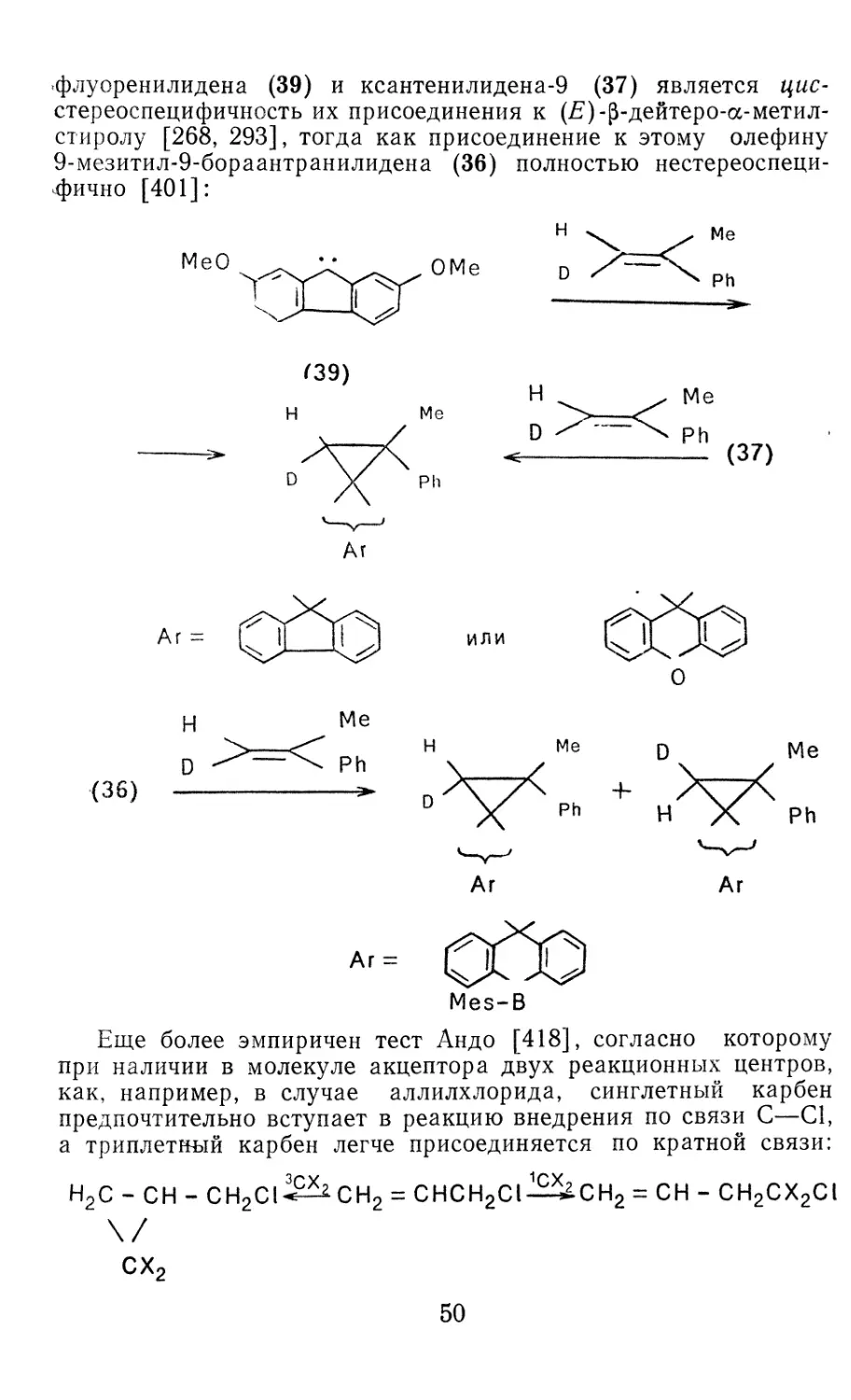
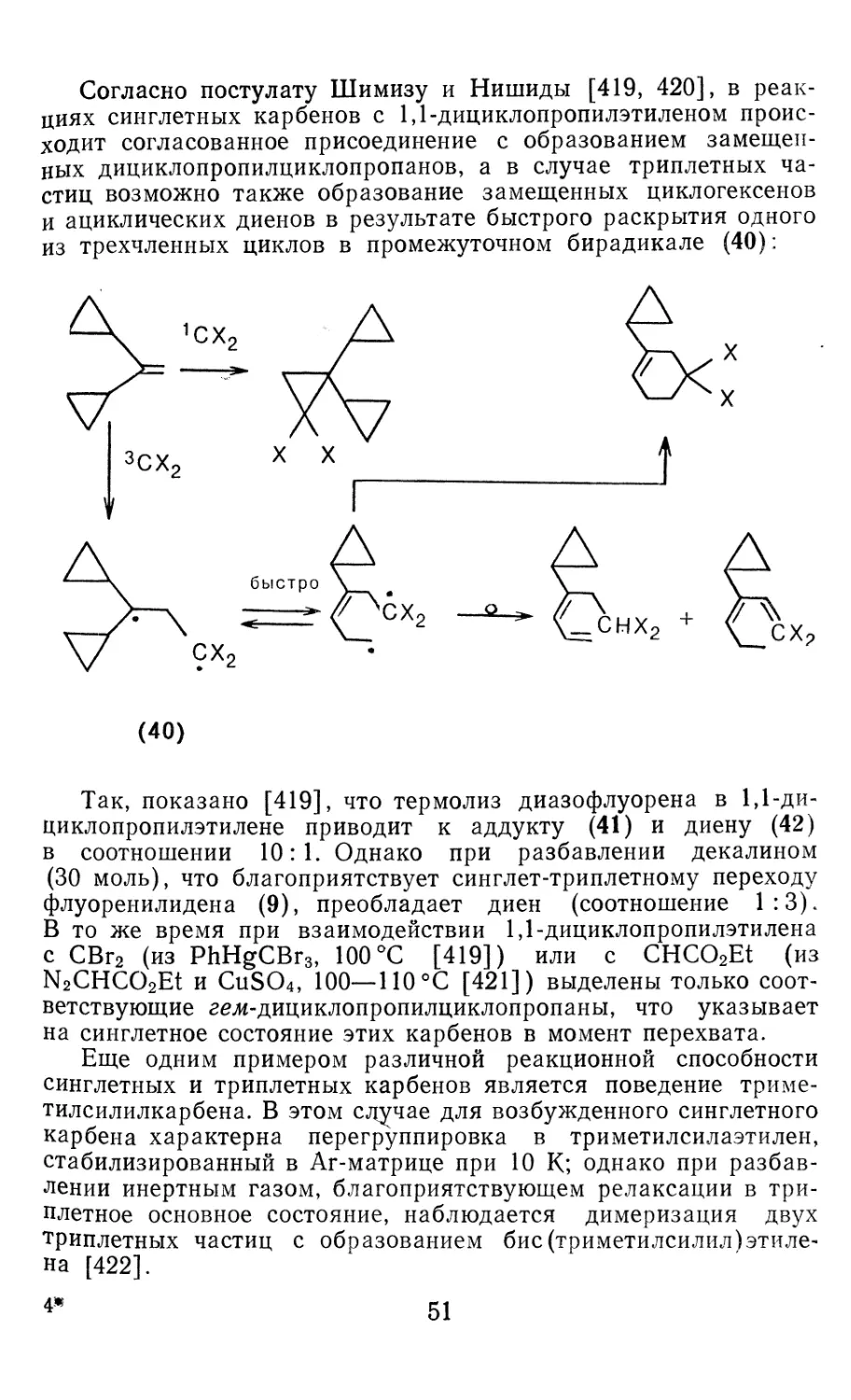








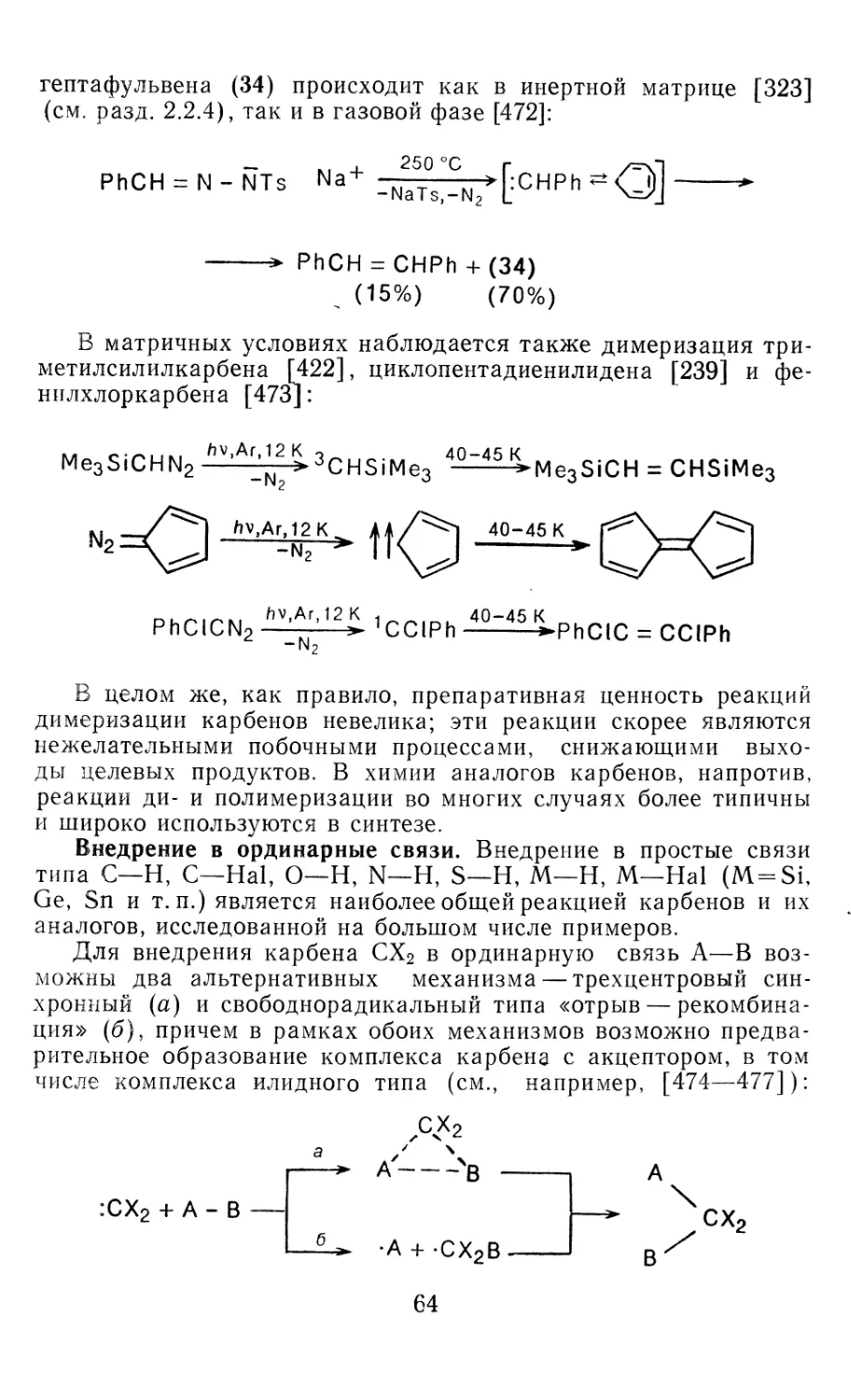



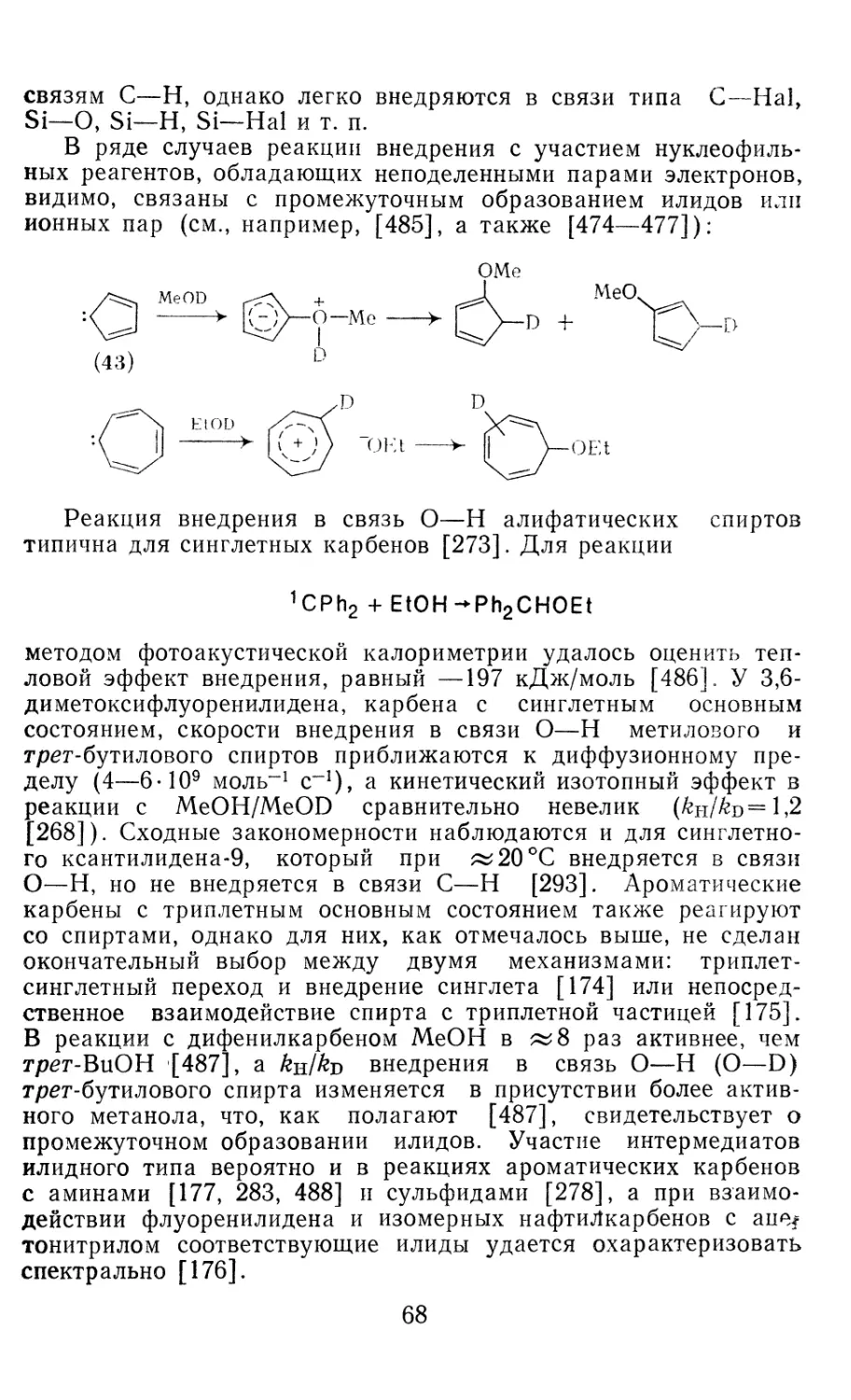


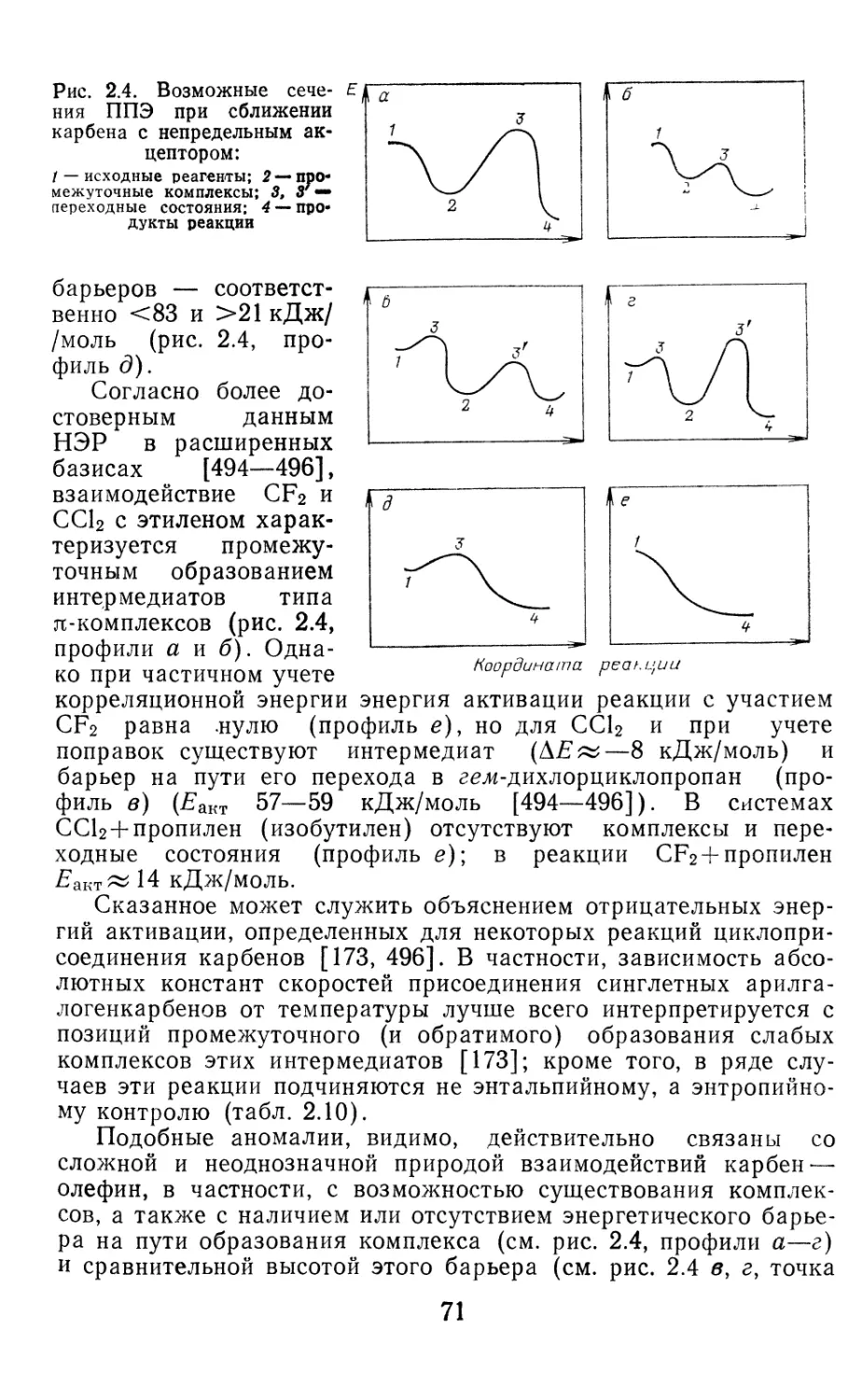
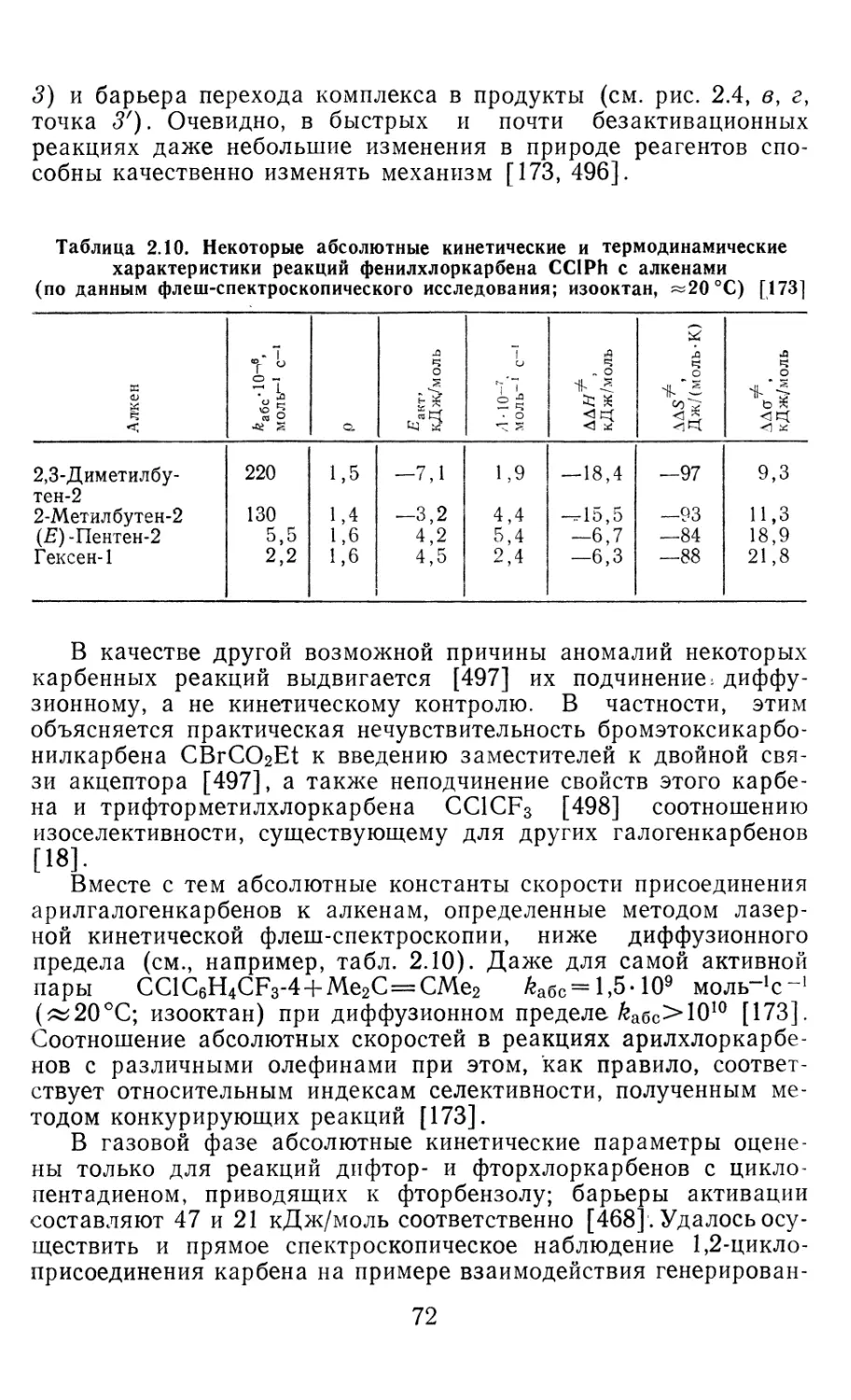

















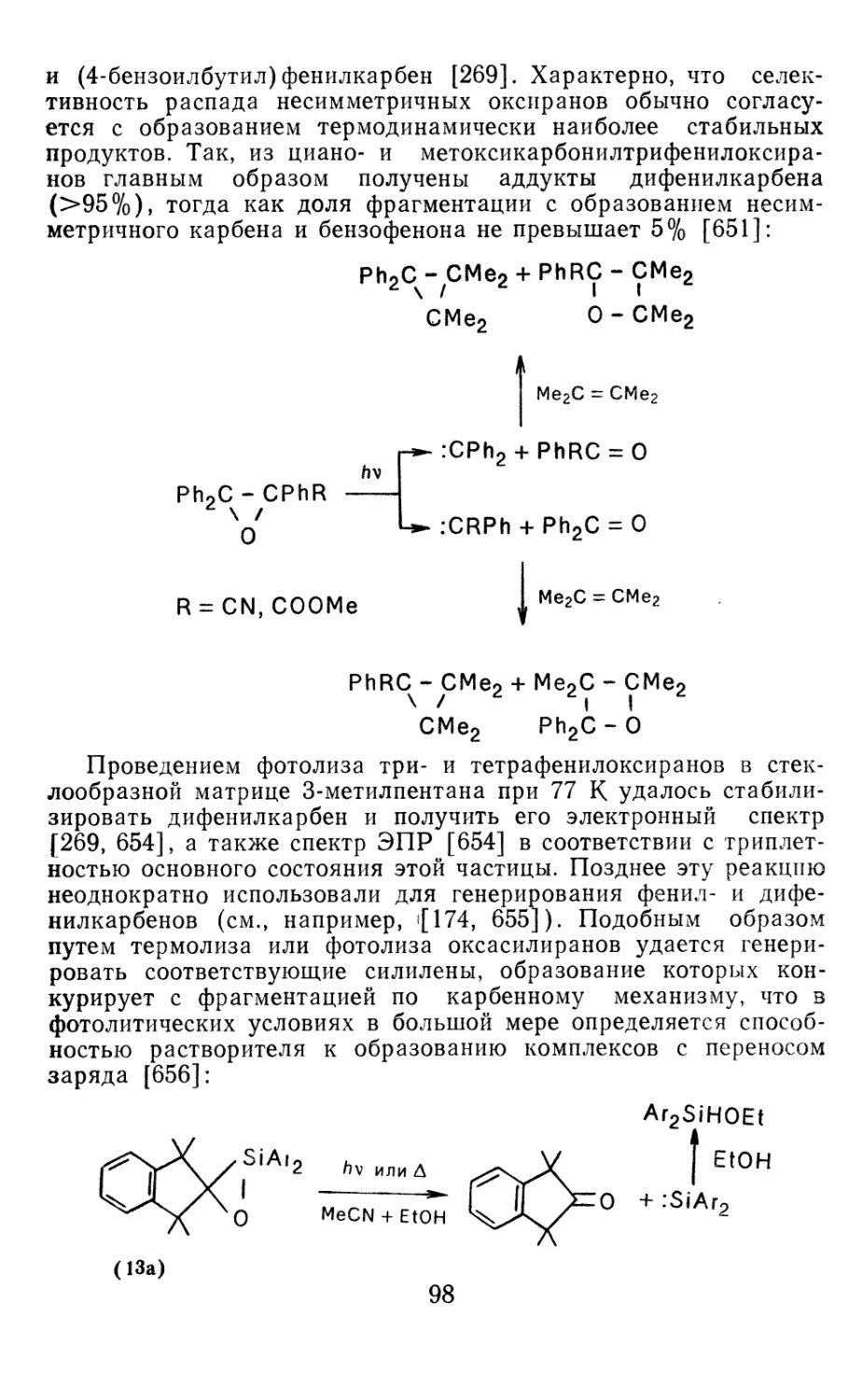


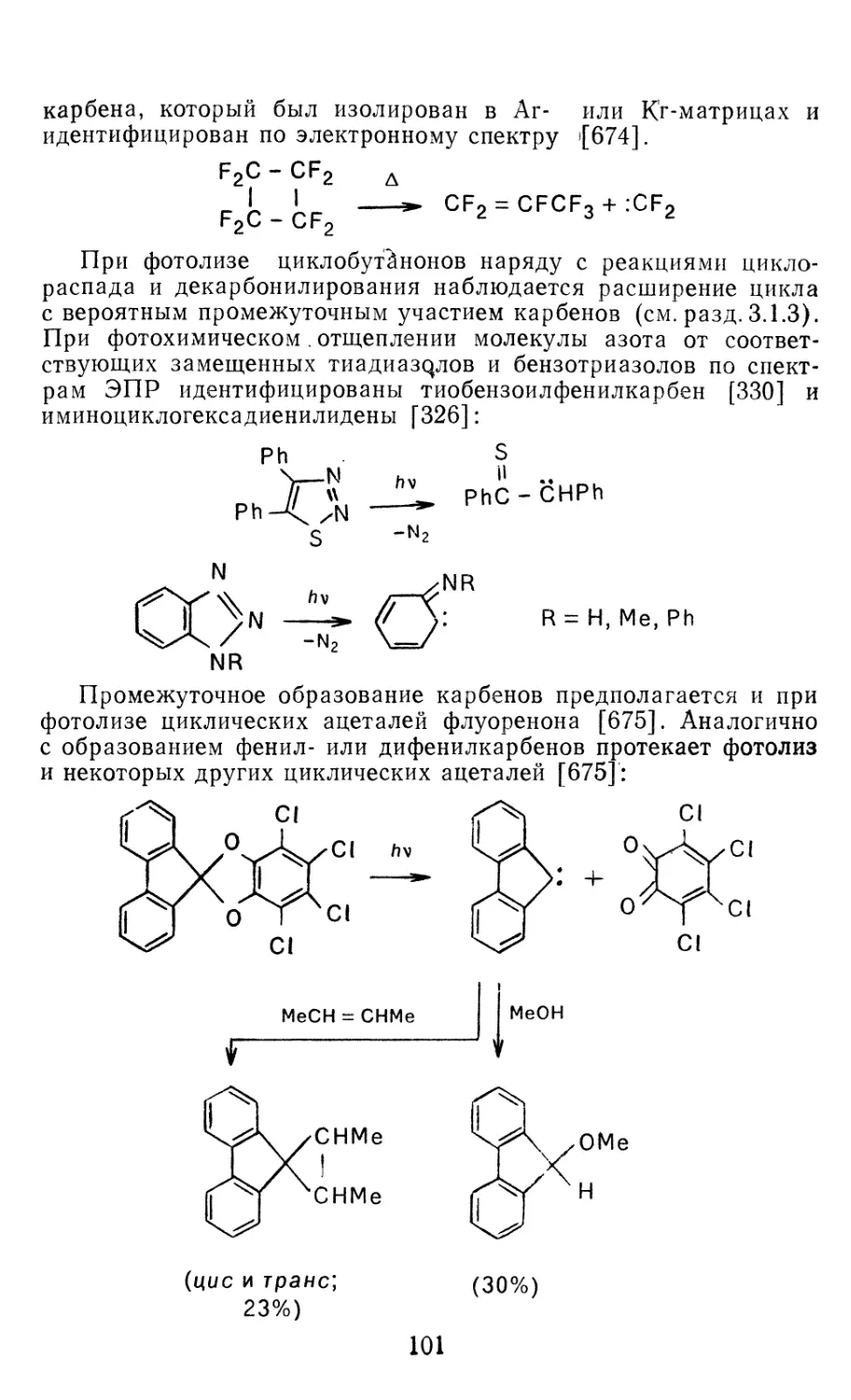
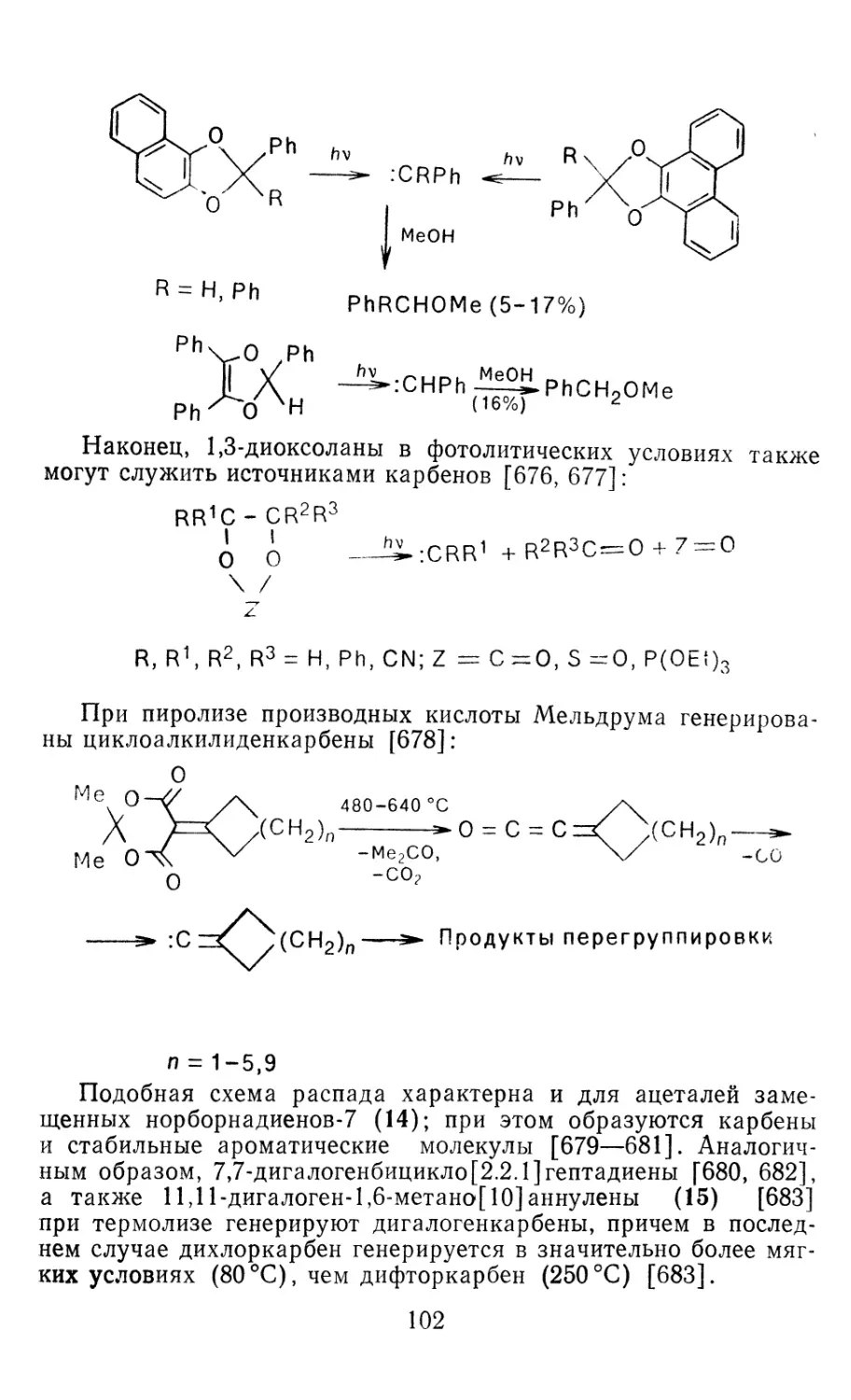
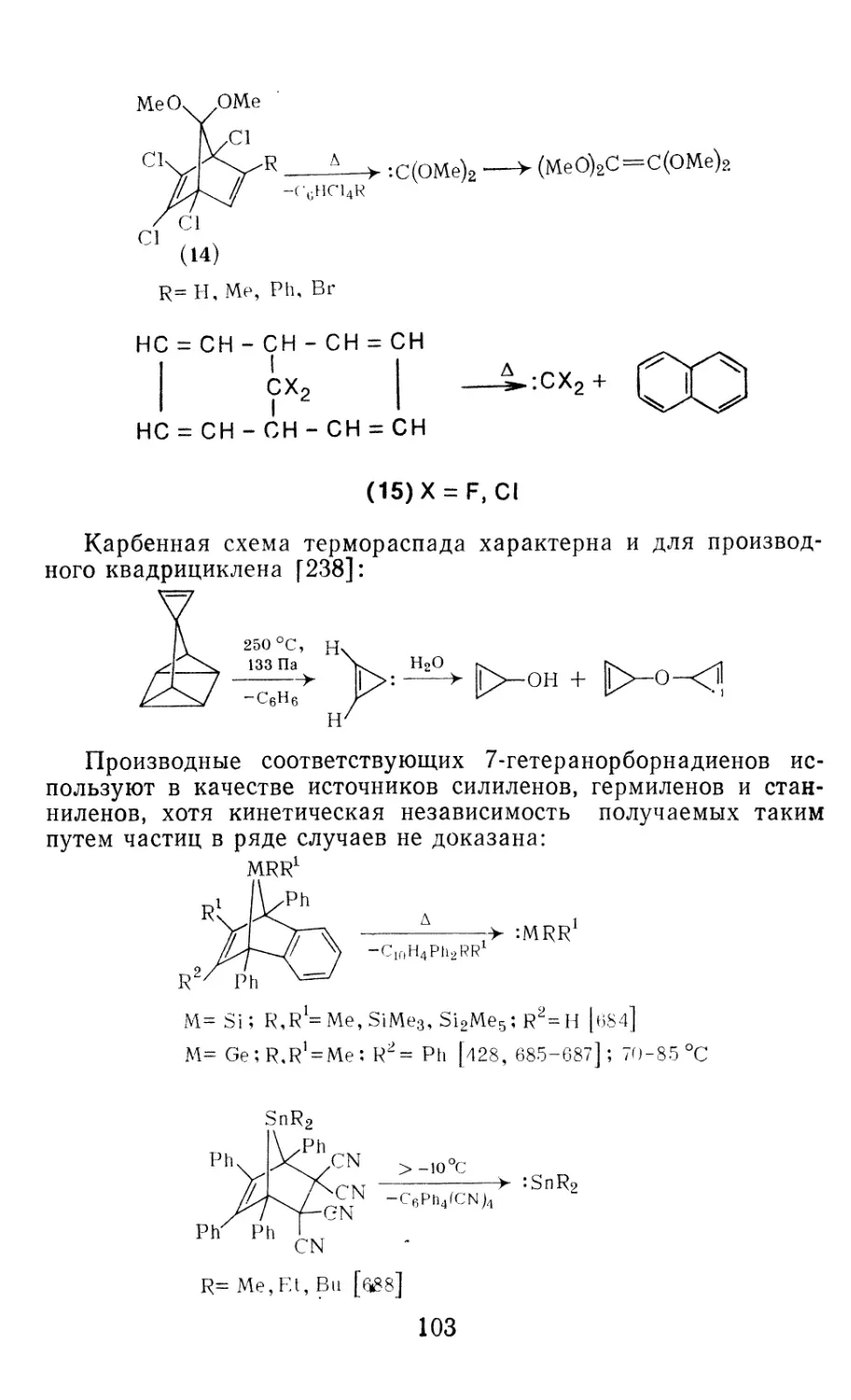










































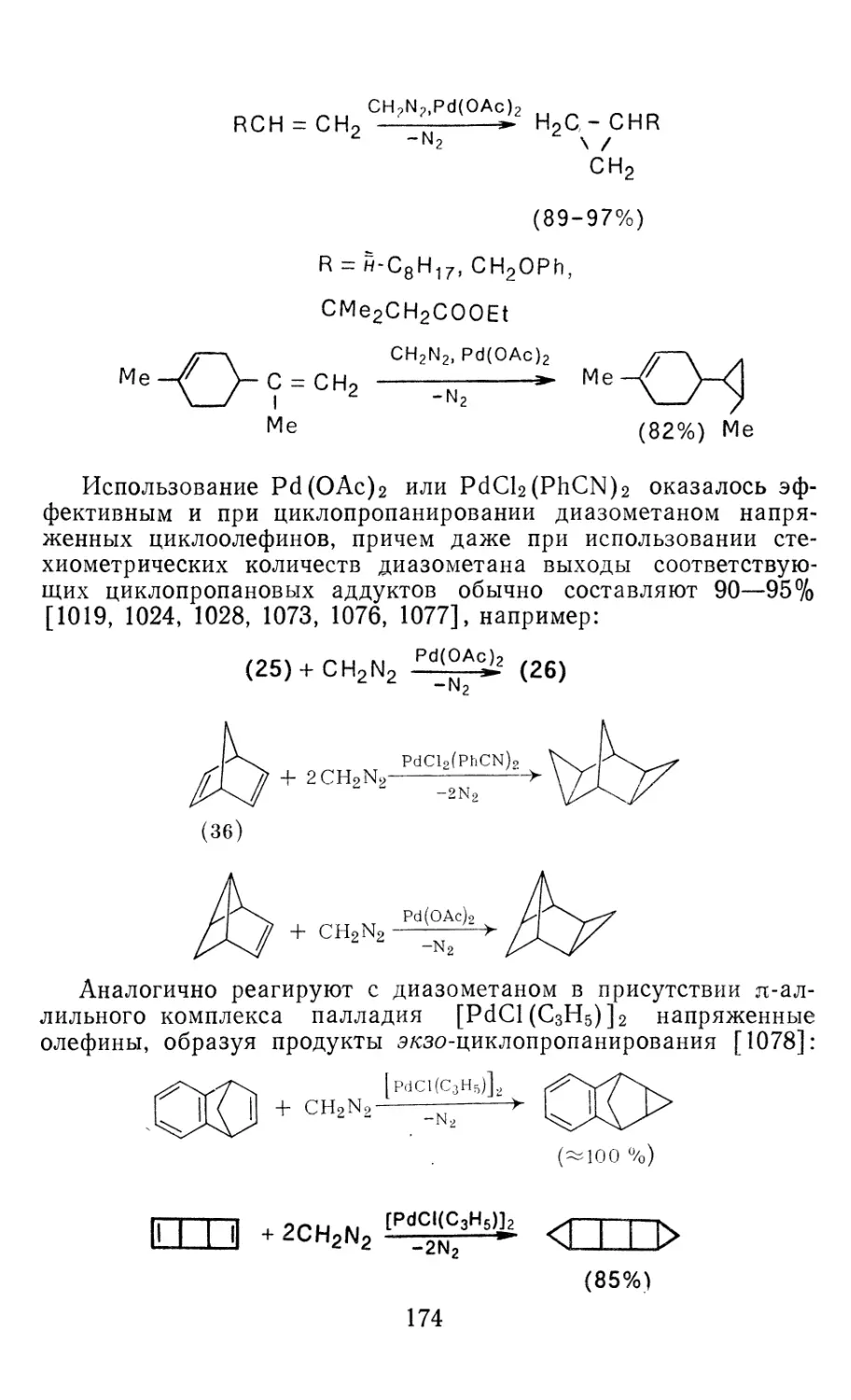
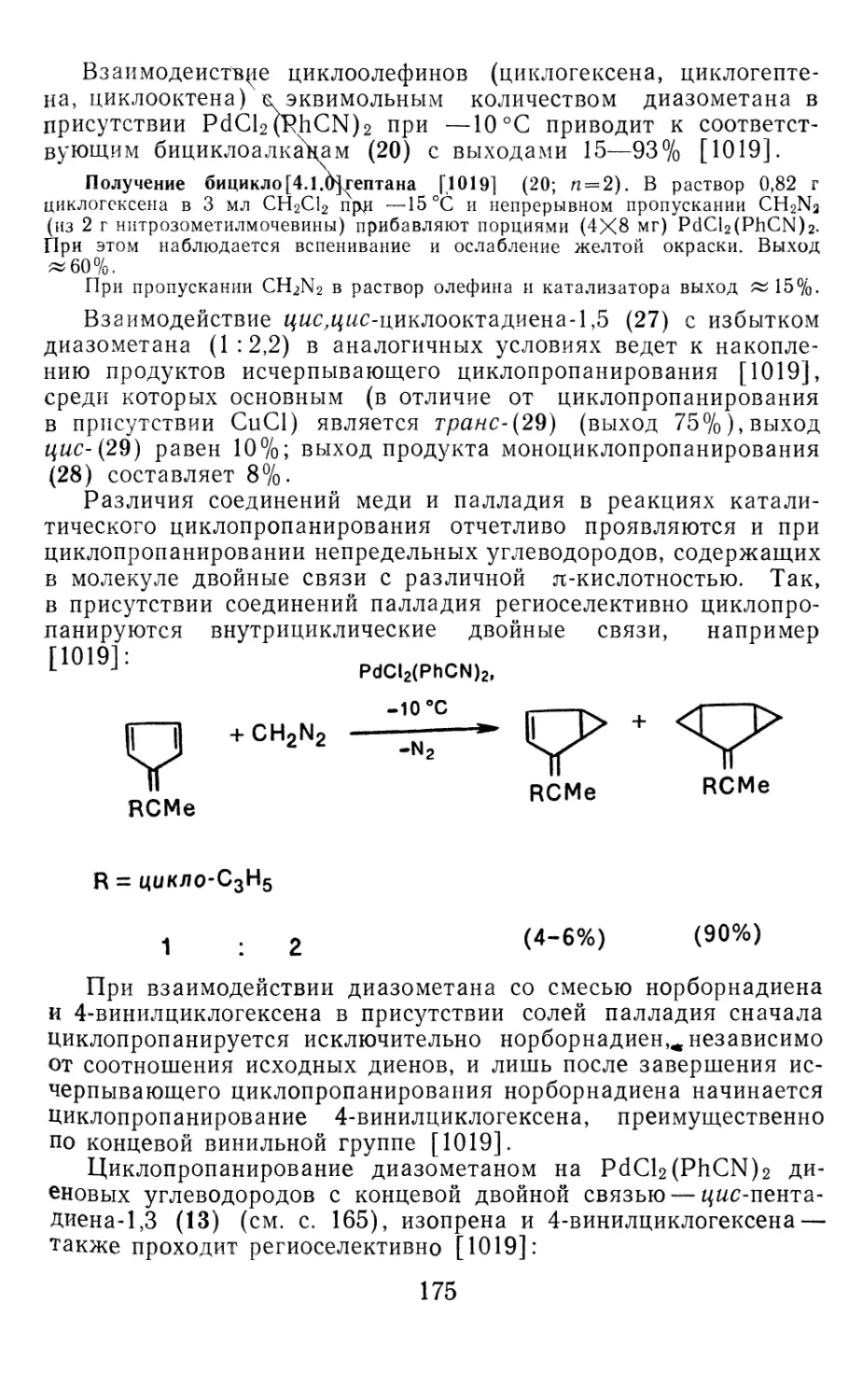



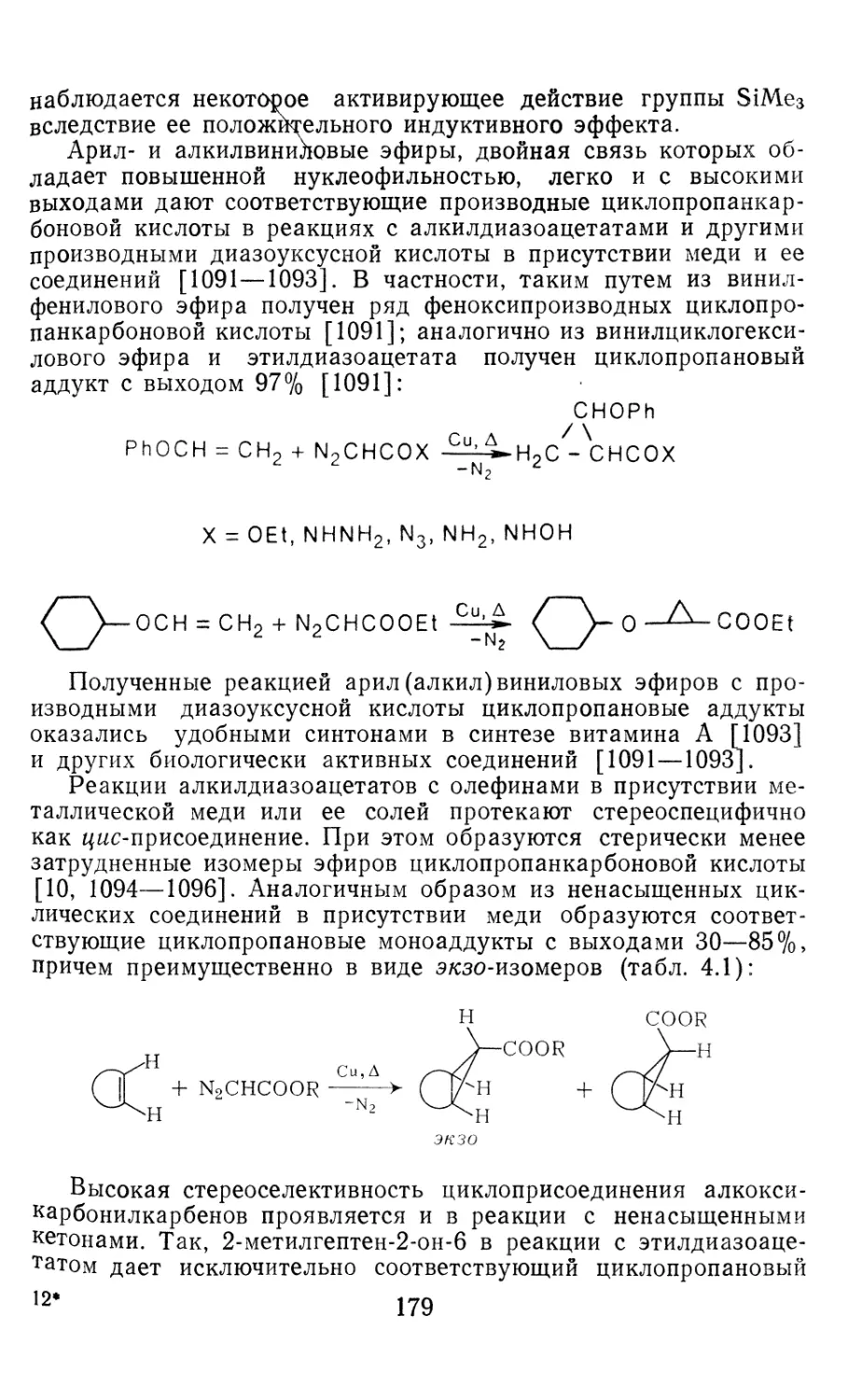


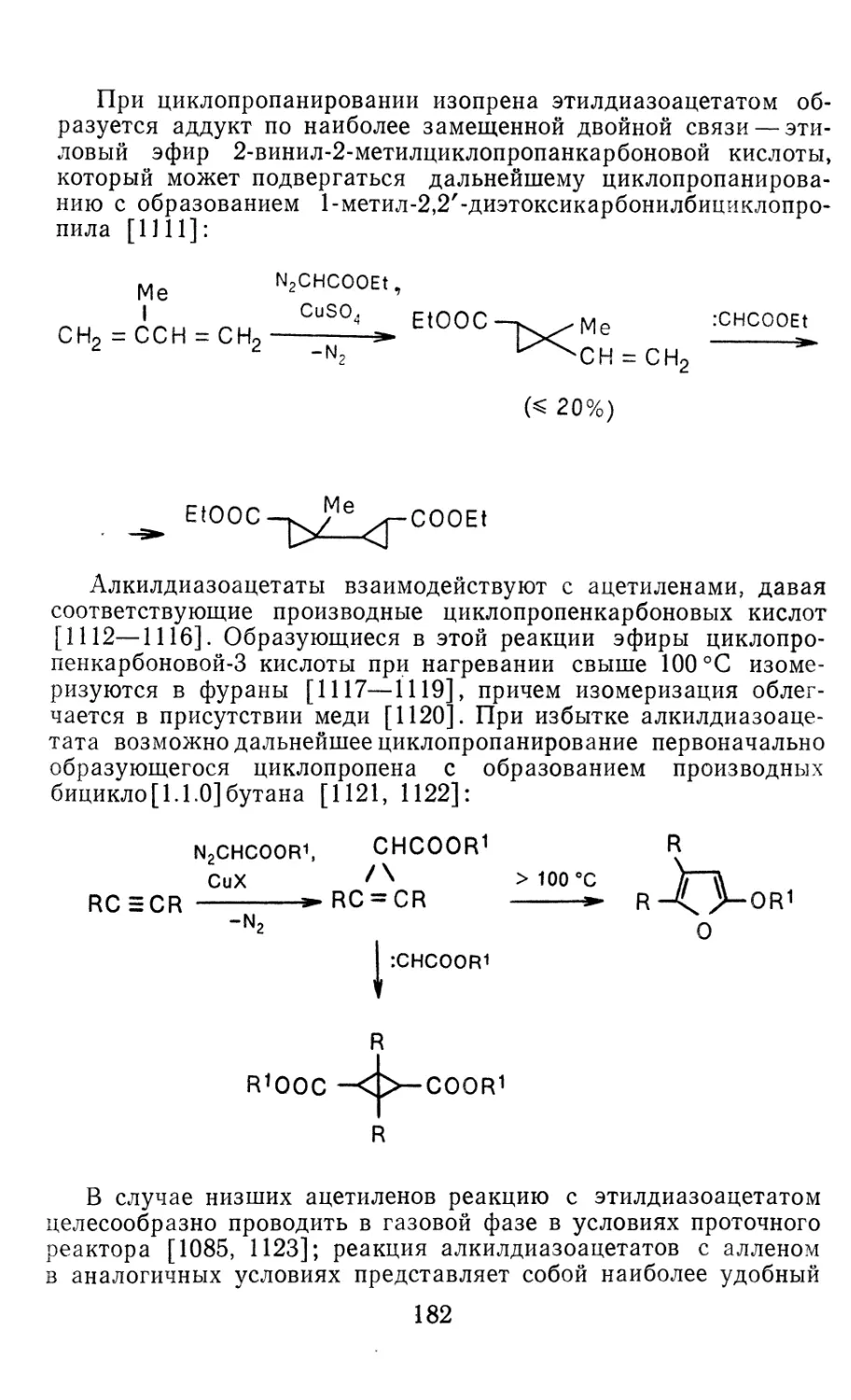




















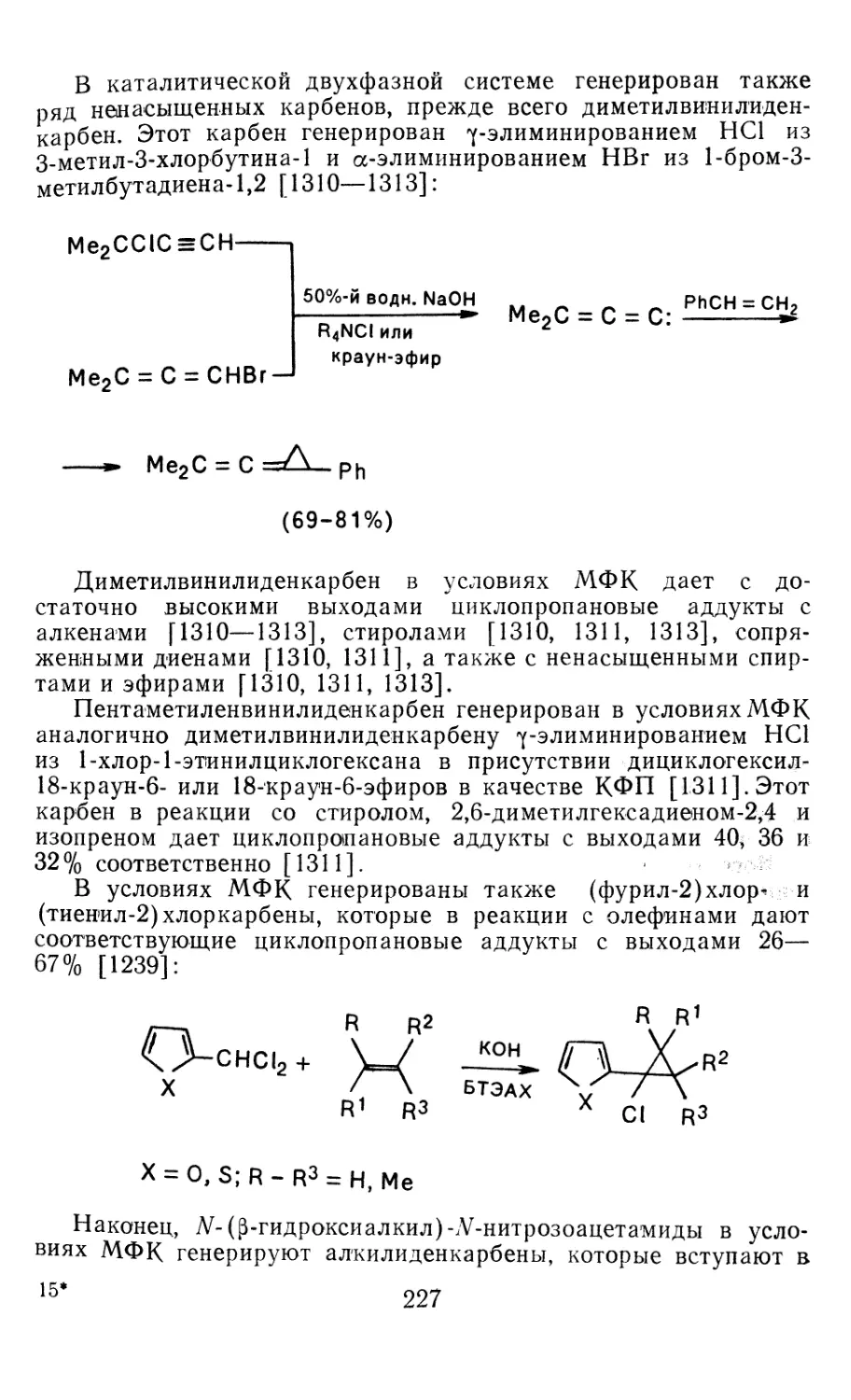

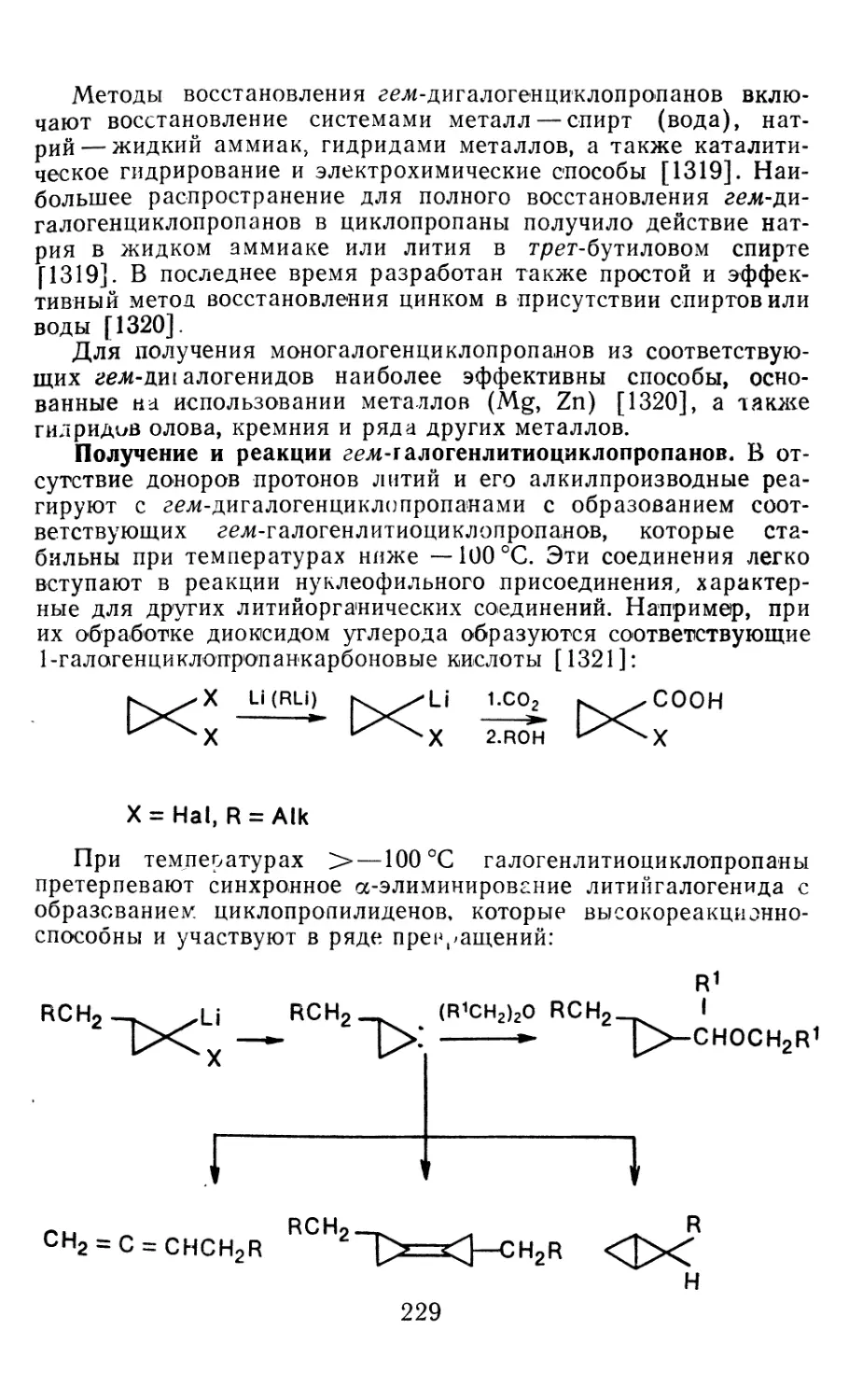

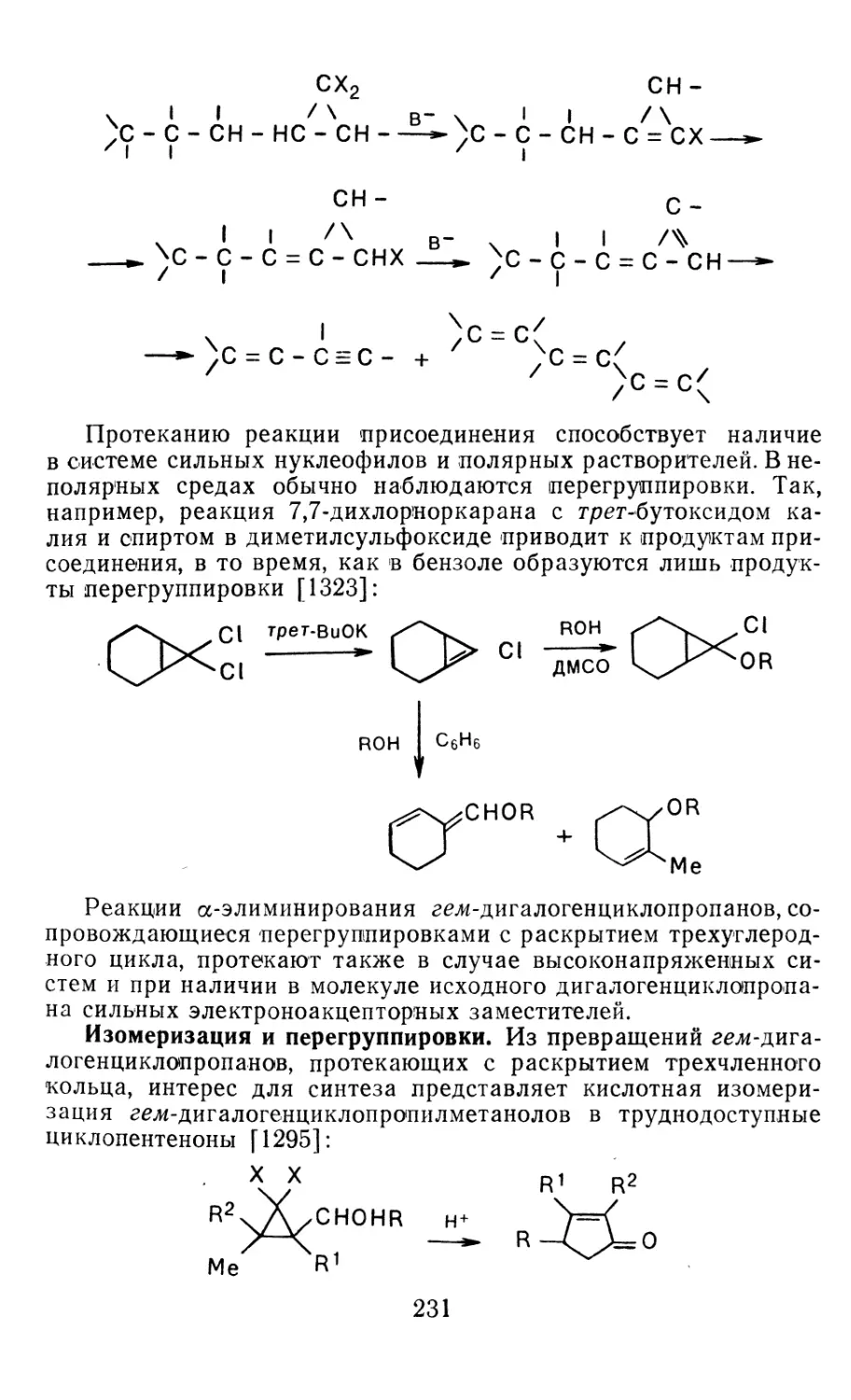

















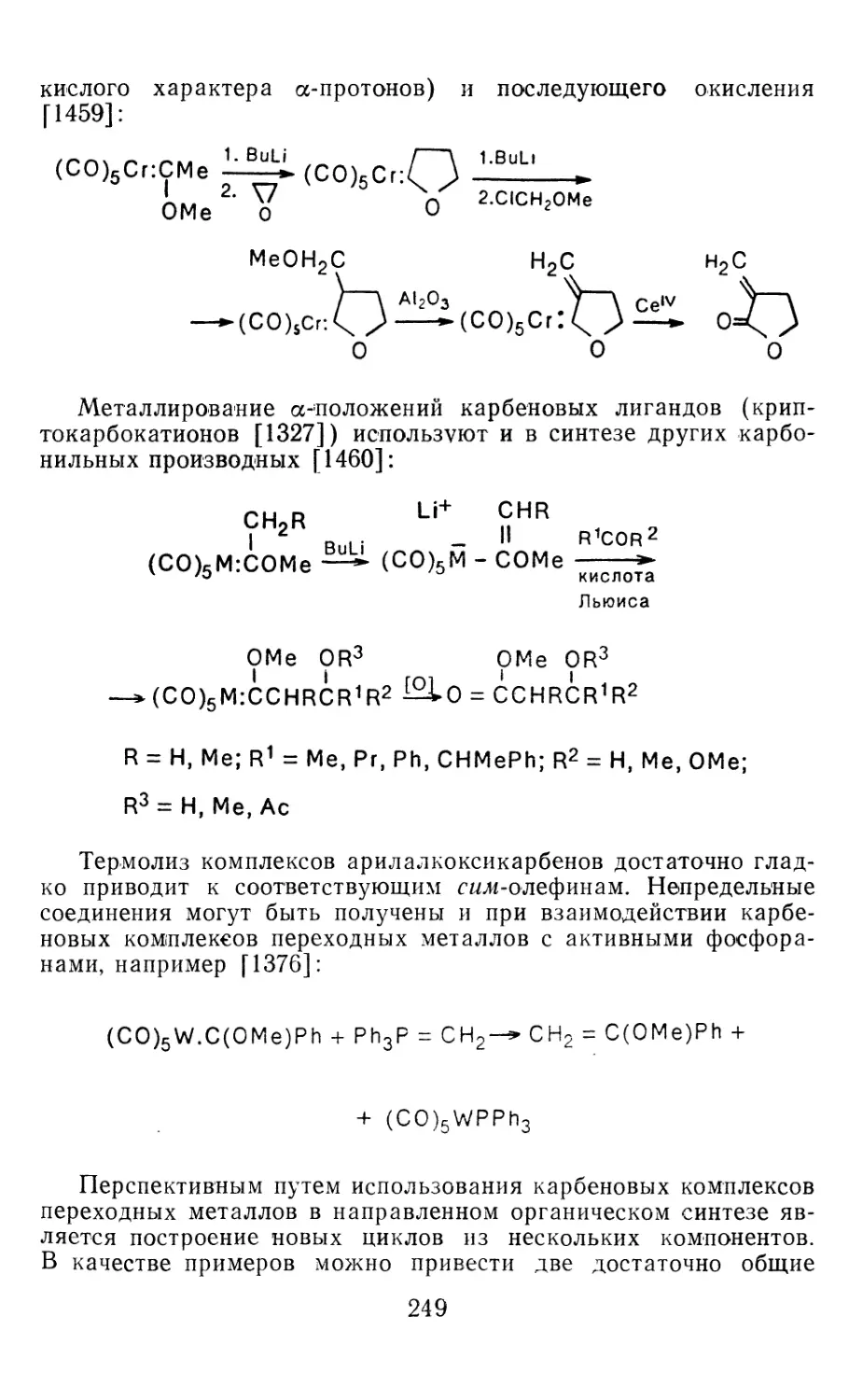





















































Author: Иоффе А.И. Нефедов О.М. Менчиков Л.Г.
Tags: органическая химия химия химические элементы химические соединения
ISBN: 5-7245-0568
Year: 1990



Text
ХИМИЯ
КАРБЕНОВ
О.М.НЕФЕДОВ,А.И.ИОФФЕ,Л.Г.МЕНЧИКОВ
ХИМИЯ
КАРБЕНОВ
lllllllllllllliillllllllllllllllllllllll
МОСКВА ХИМИЯ' 1990
ОГЛАВЛЕНИЕ
Введение °
Глава Г Номенклатура карбенов 12
Глава 2. Строение и реакционная способность карбенов и их аналогов 15
2.1 Теоретический и квантовохимический анализ строения карбе-
нов 16
2.2 Исследование карбенов инструментальными методами 20
2.2.1. Импульсные методы получения карбенов и регистрация
их спектров поглощения 23
2.2.2. Использование метода матричной изоляции для получе-
ния ИК-, УФ- и ЭПР-спектров карбенов 28
2 2.3. Исследование карбенов в газовой фазе 39
2 2.4 Энергетическое расщепление уровней и ЙЛ у карбе-
нов и их аналогов 42
2.2.5. Анализ спиновой мультиплетности основного и возбуж-
денных состояний карбенов по их химическим превращениям.
Метод ХПЯ 48
2.3 Закономерности электронного и геометрического строения кар-
бенов и их аналогов 56
2.4 . Основные типы карбенных реакций 61
2.4.1. Межмолекулярные реакции карбенов и их аналогов 61
2.4.2. Внутримолекулярные реакции карбенов и их аналогов 77
Глава 3. Методы генерирования карбенов и их аналогов 83
3.1. Генерирование карбенов из энергетически активированных
предшественников 84
3.1 1. Реакции а-элиминирования 84
3 1.2. Другие реакции элиминирования 120
3.1.3. Внутримолекулярные перегруппировки 131
3.2. Сольволитические методы генерирования карбенов 138
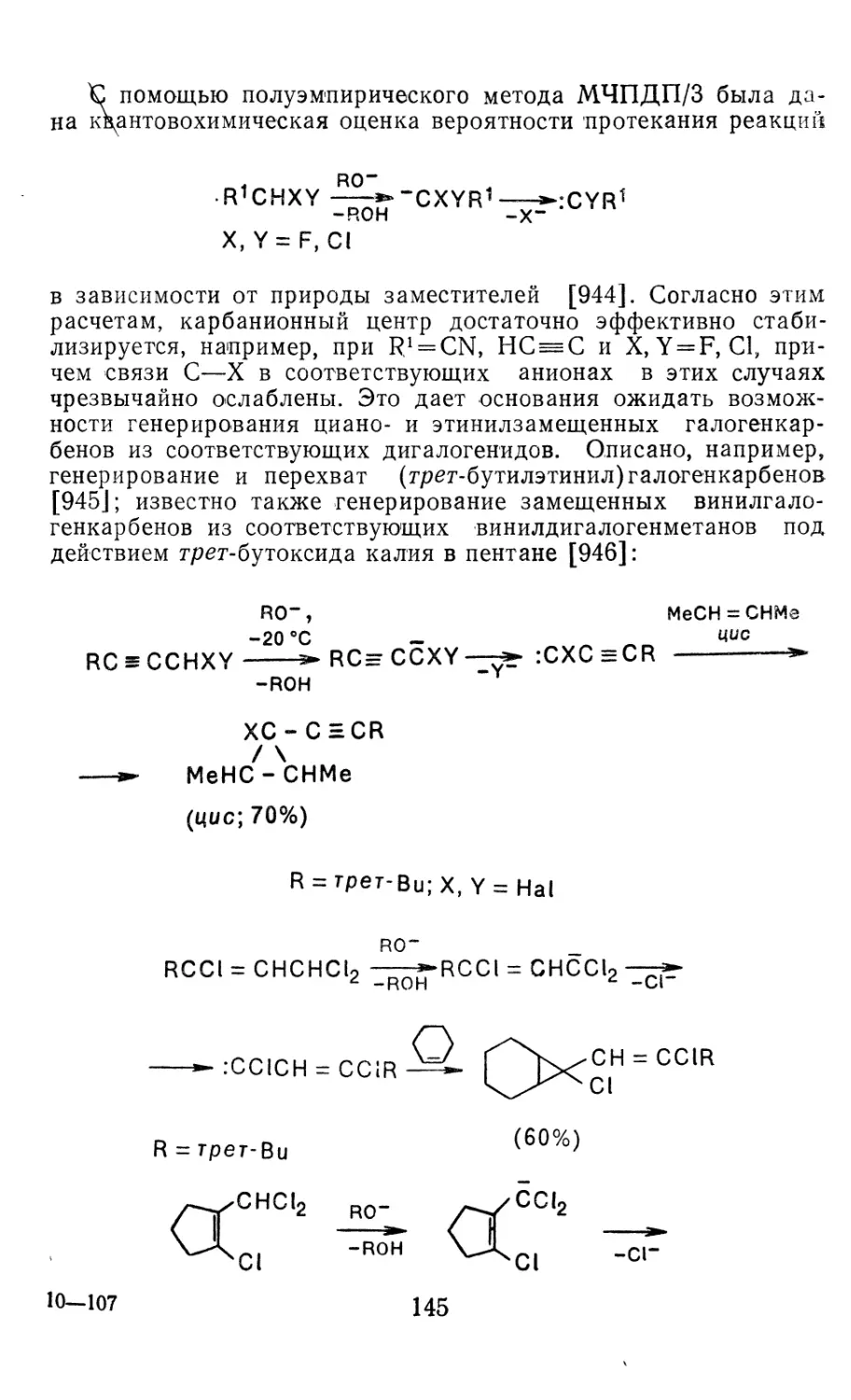
3 2 1. Генерирование дигалогенкарбенов 139
3 2.2. Генерирование моногалогенкарбенов из дигалогенмета-
нов и их производных 144
3 2.3 Дегидрогалогенирование органилгалогенидов 146
3.3. Генерирование карбенов и их аналогов из атомарных элсмен-
юв подгруппы IVB 149
3.4. Сравнительная реакционная способность карбенов, генериро-
ванных различными методами 154
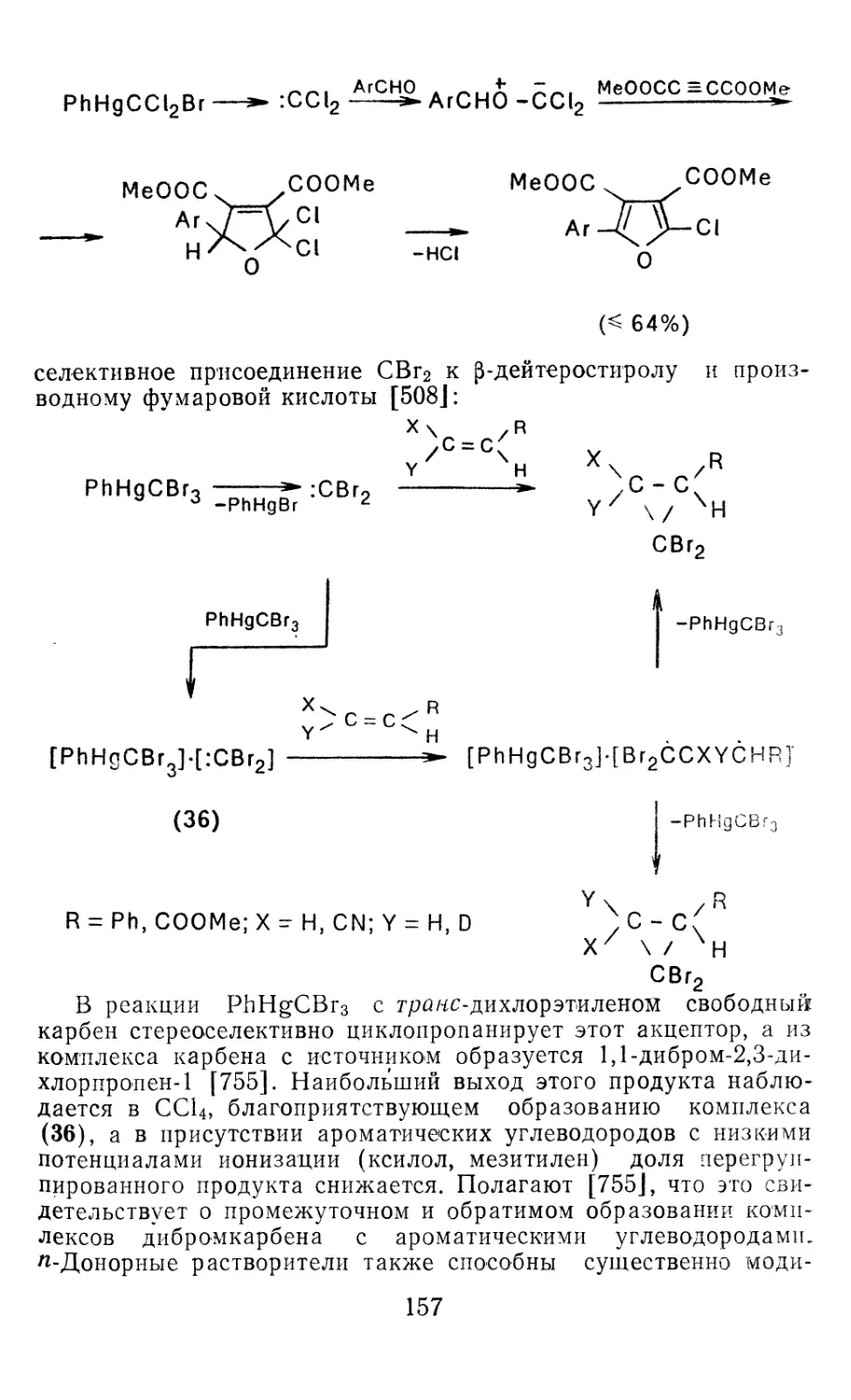
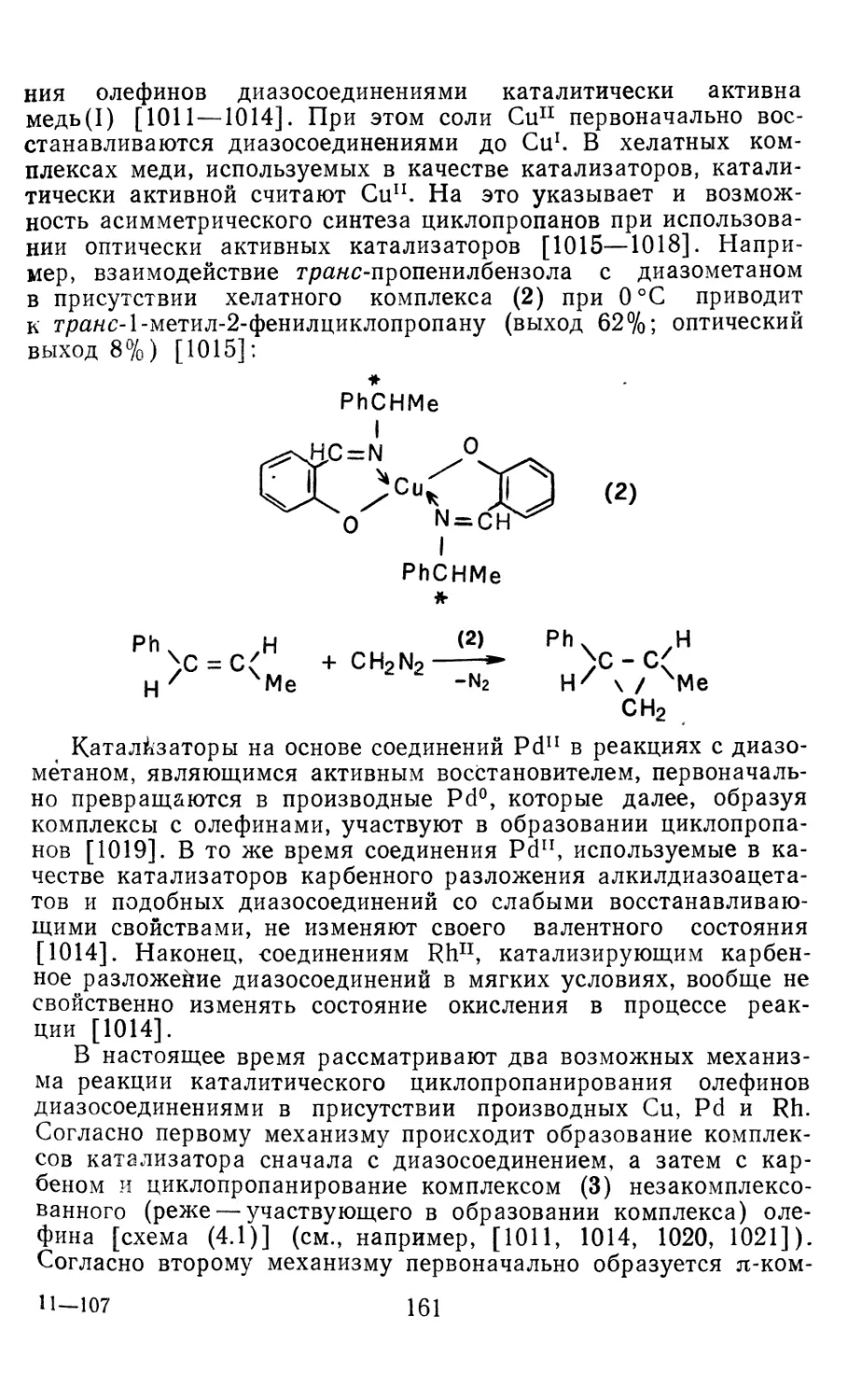
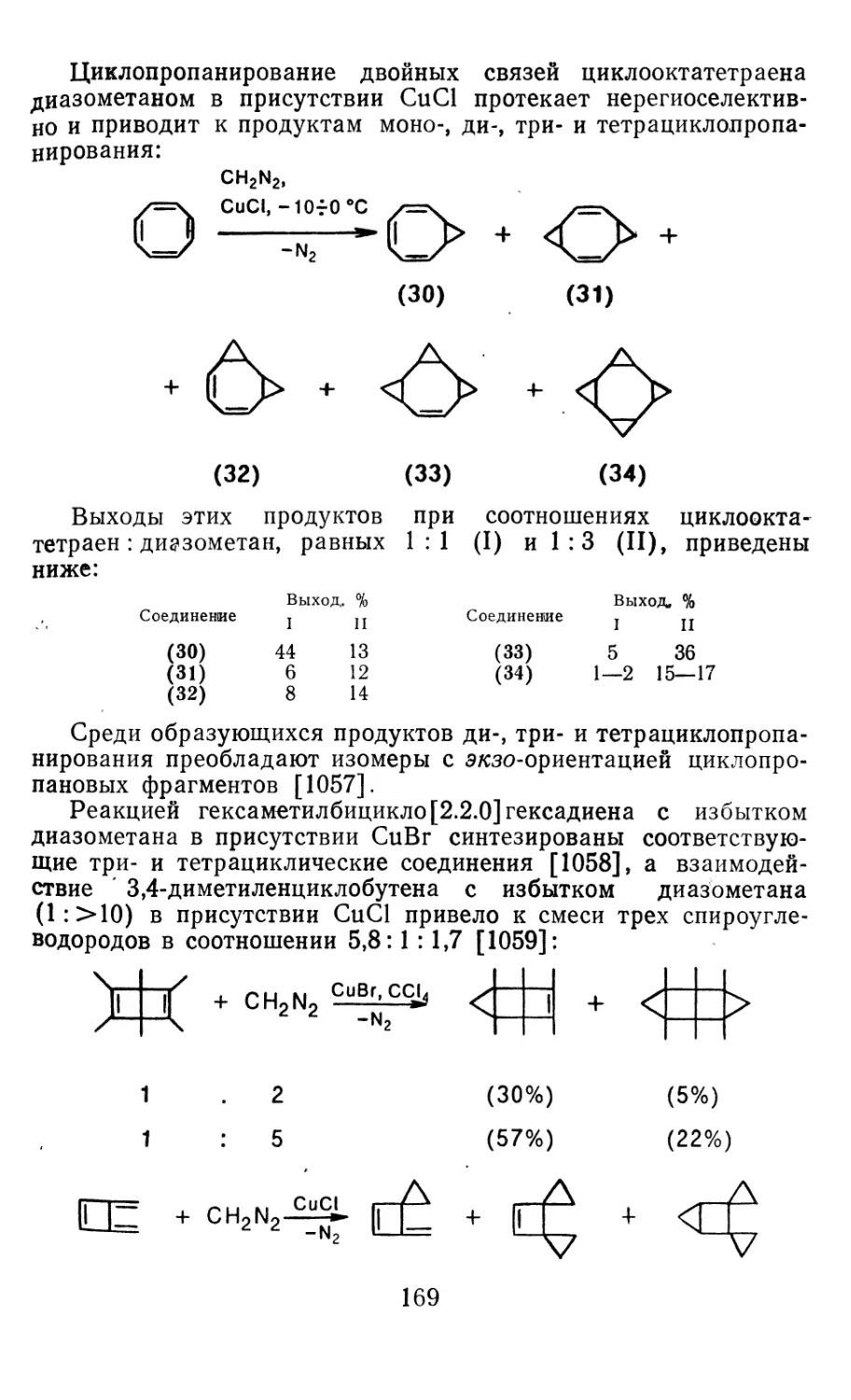
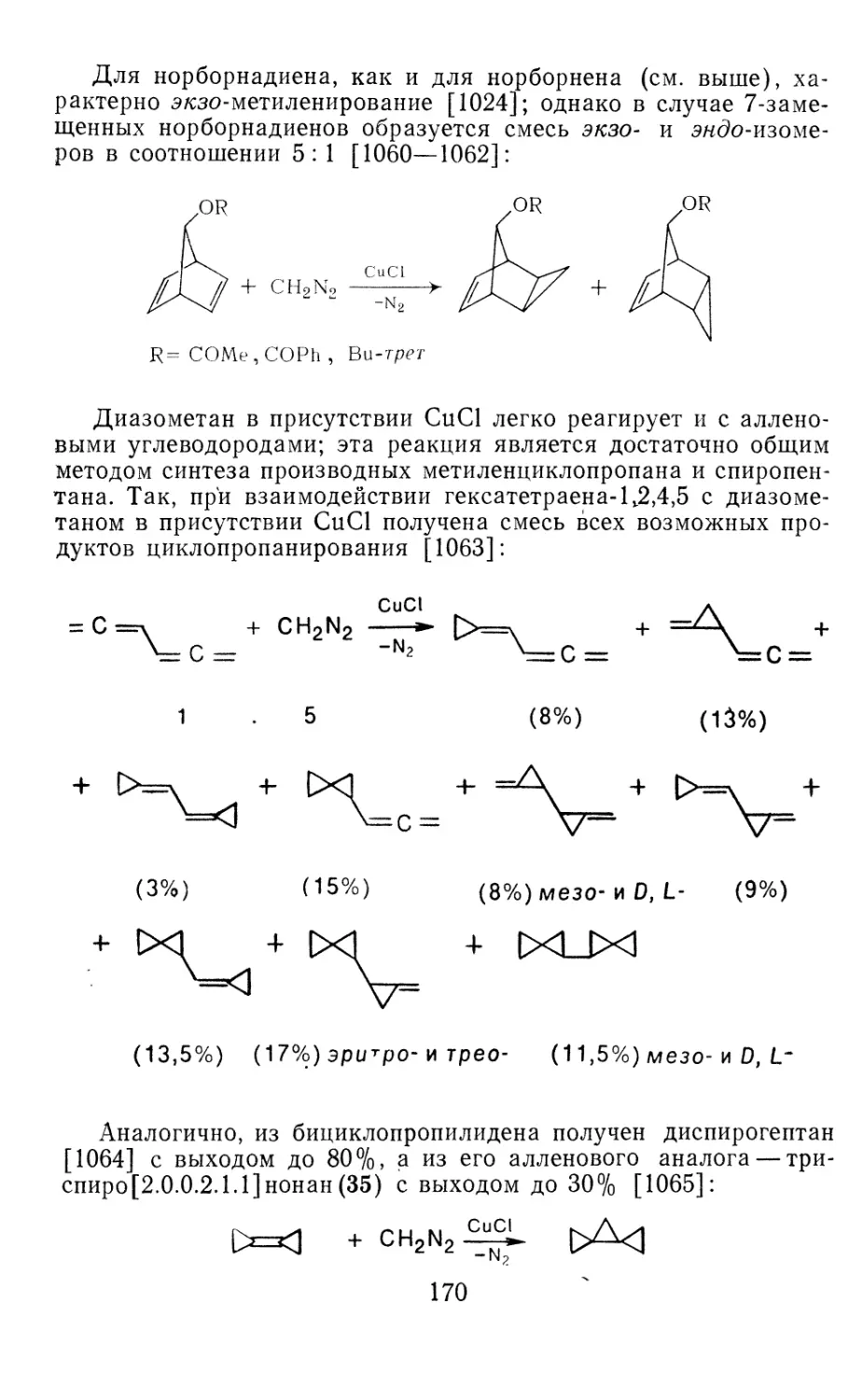
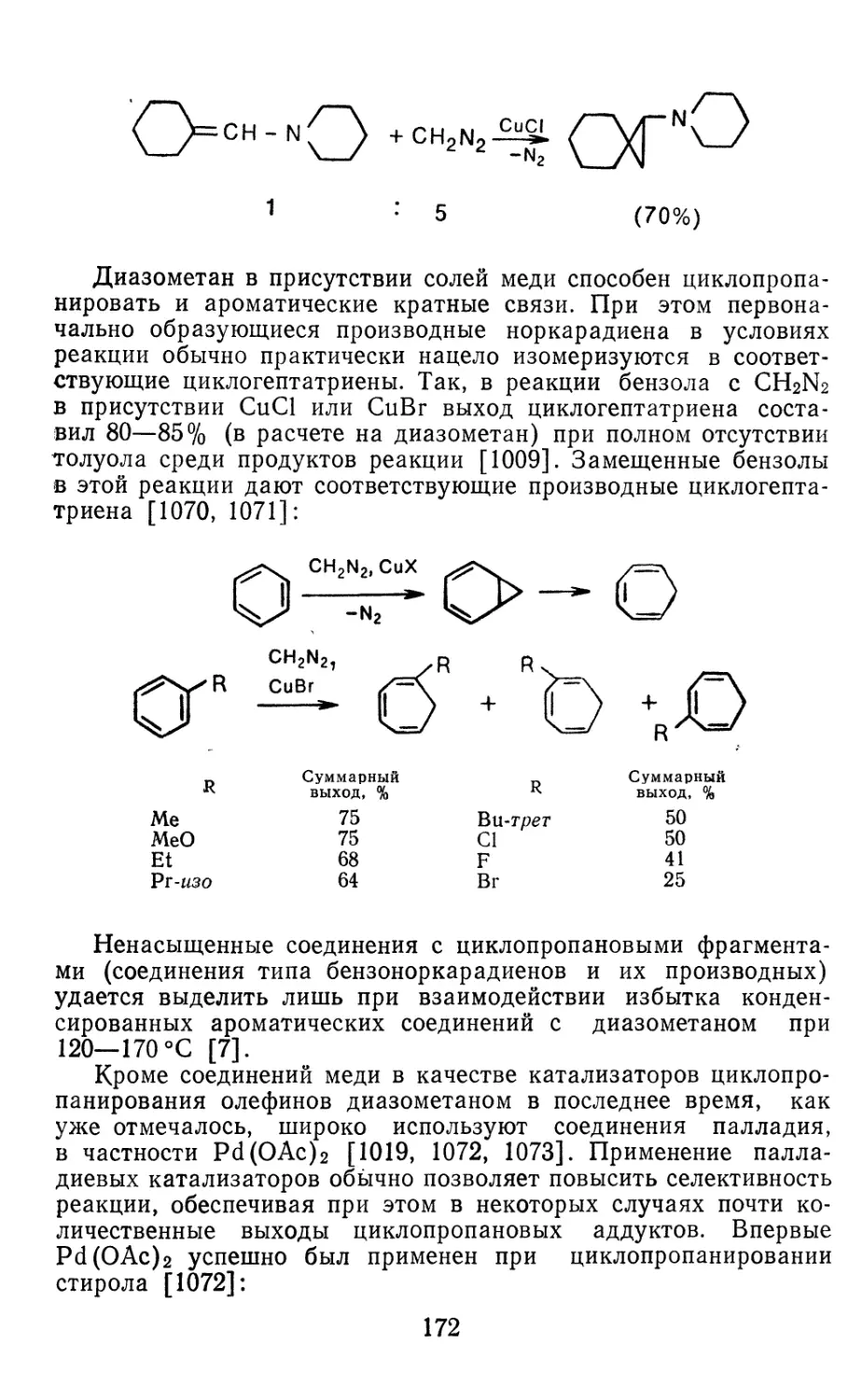
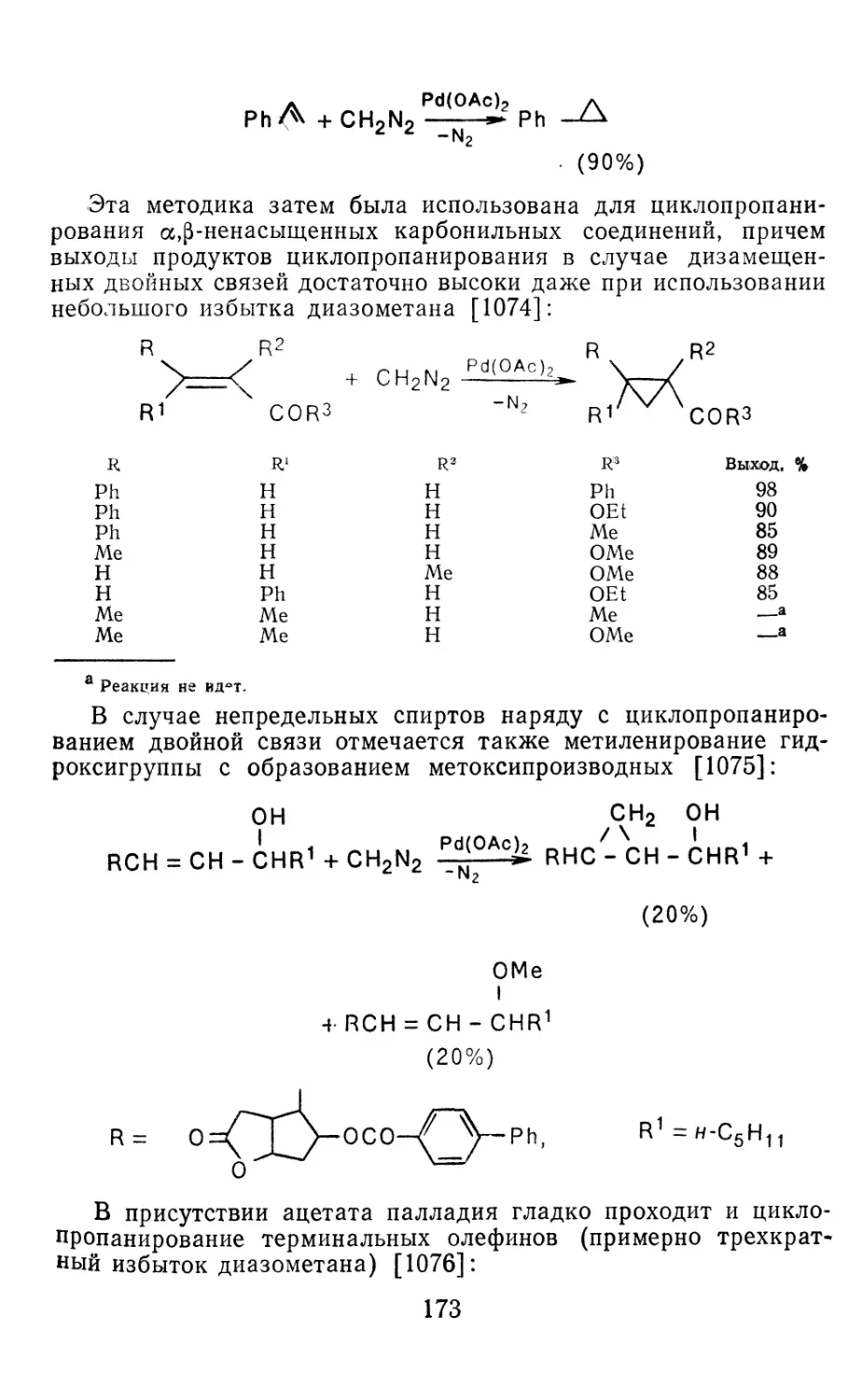
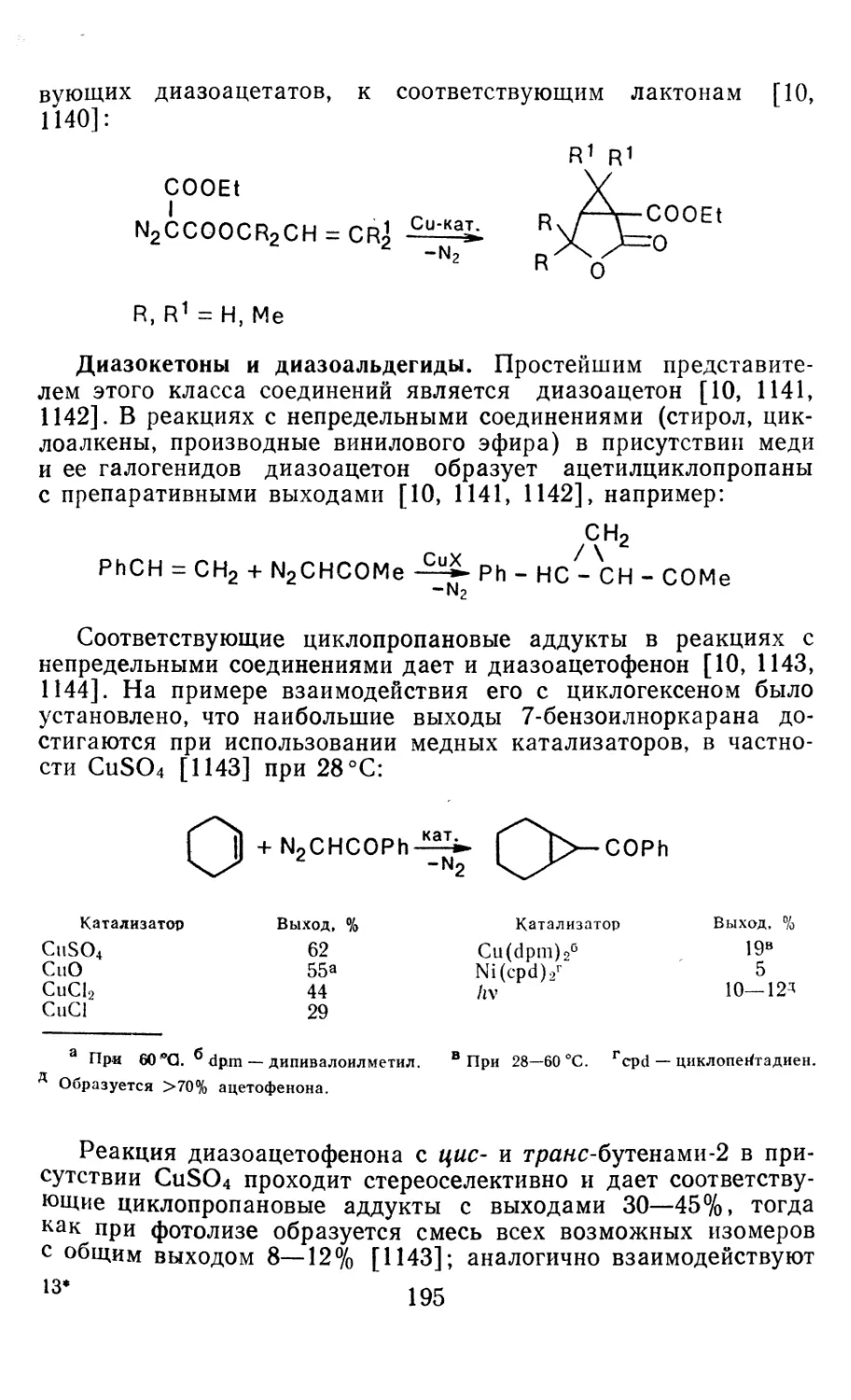
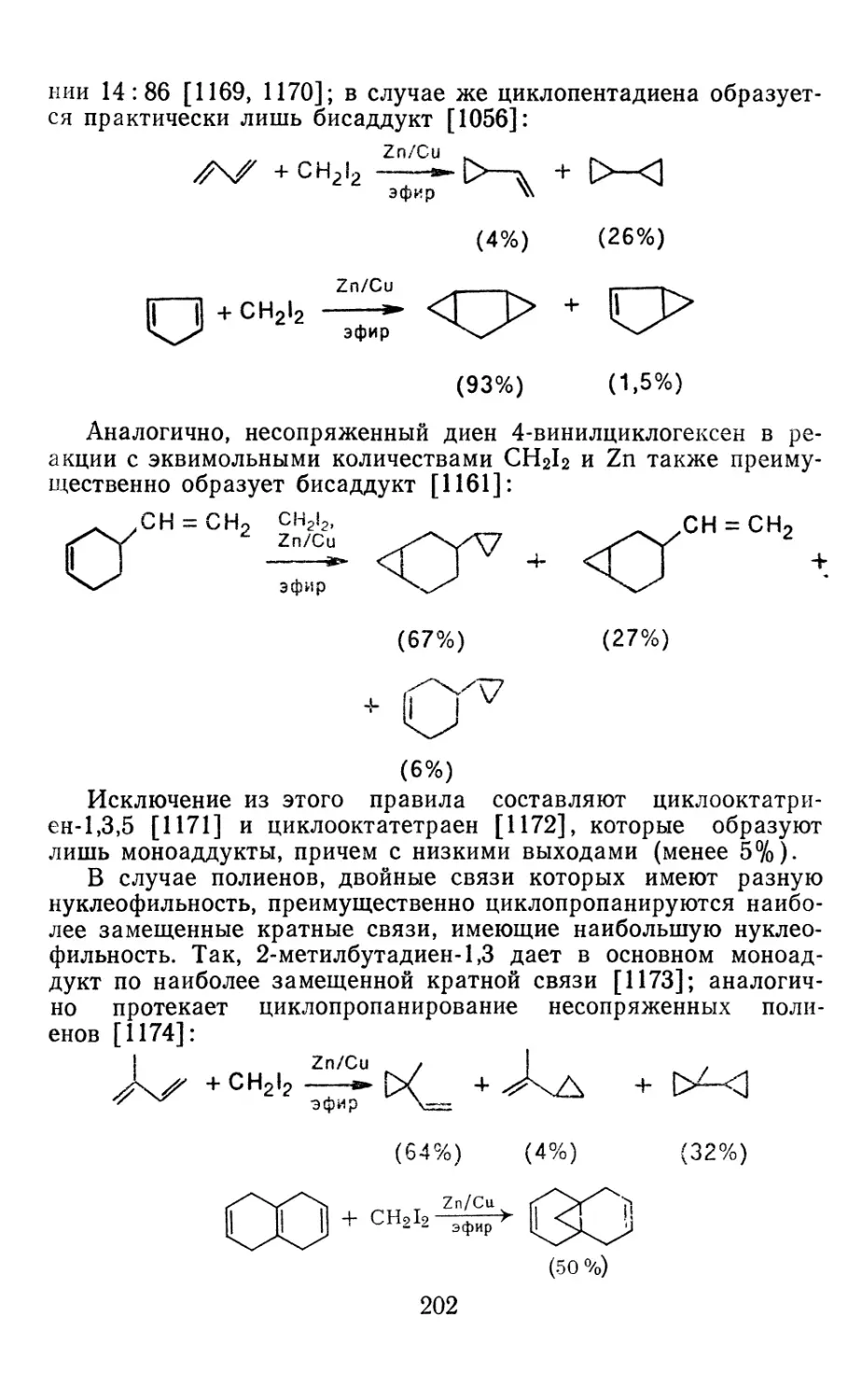
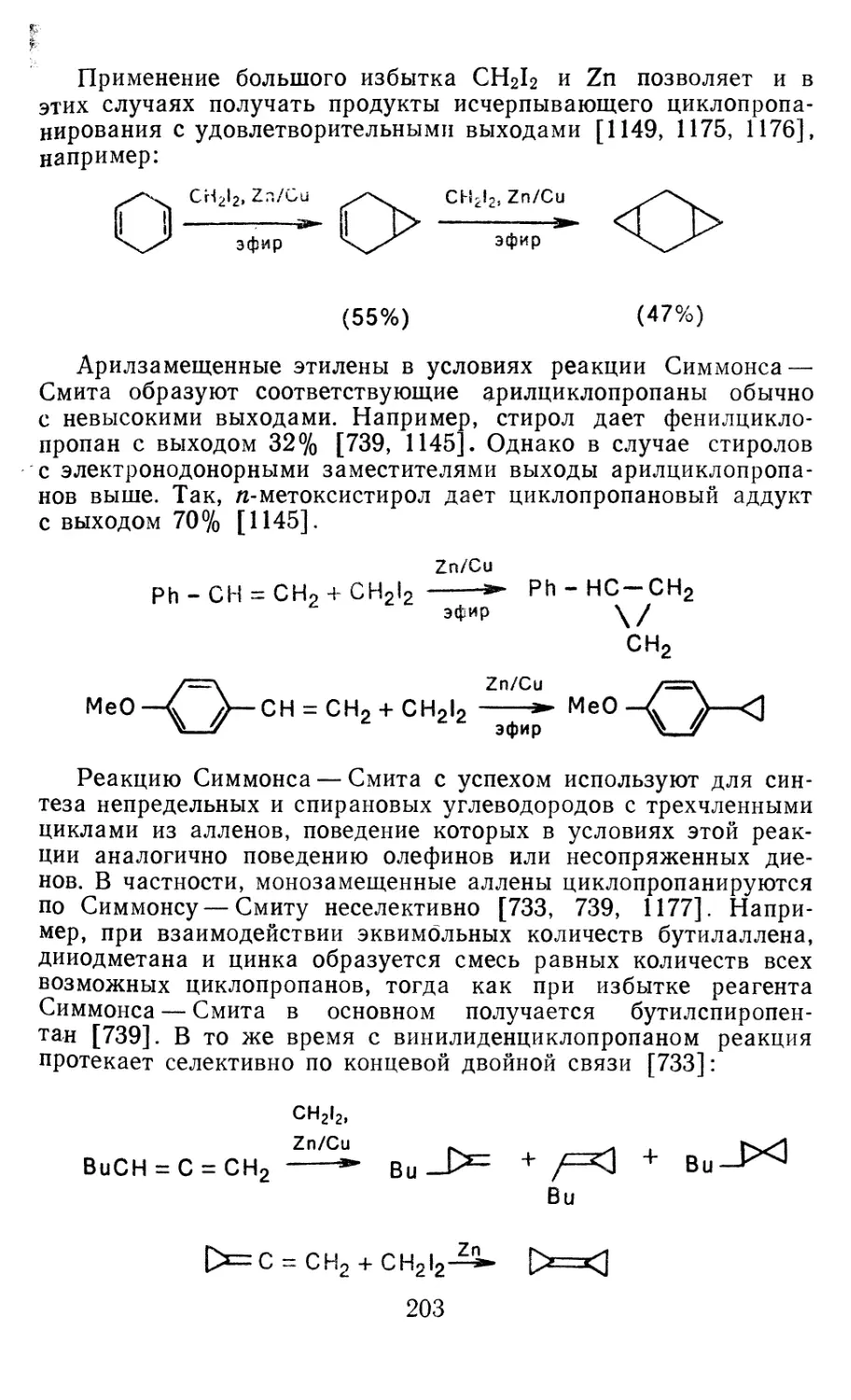
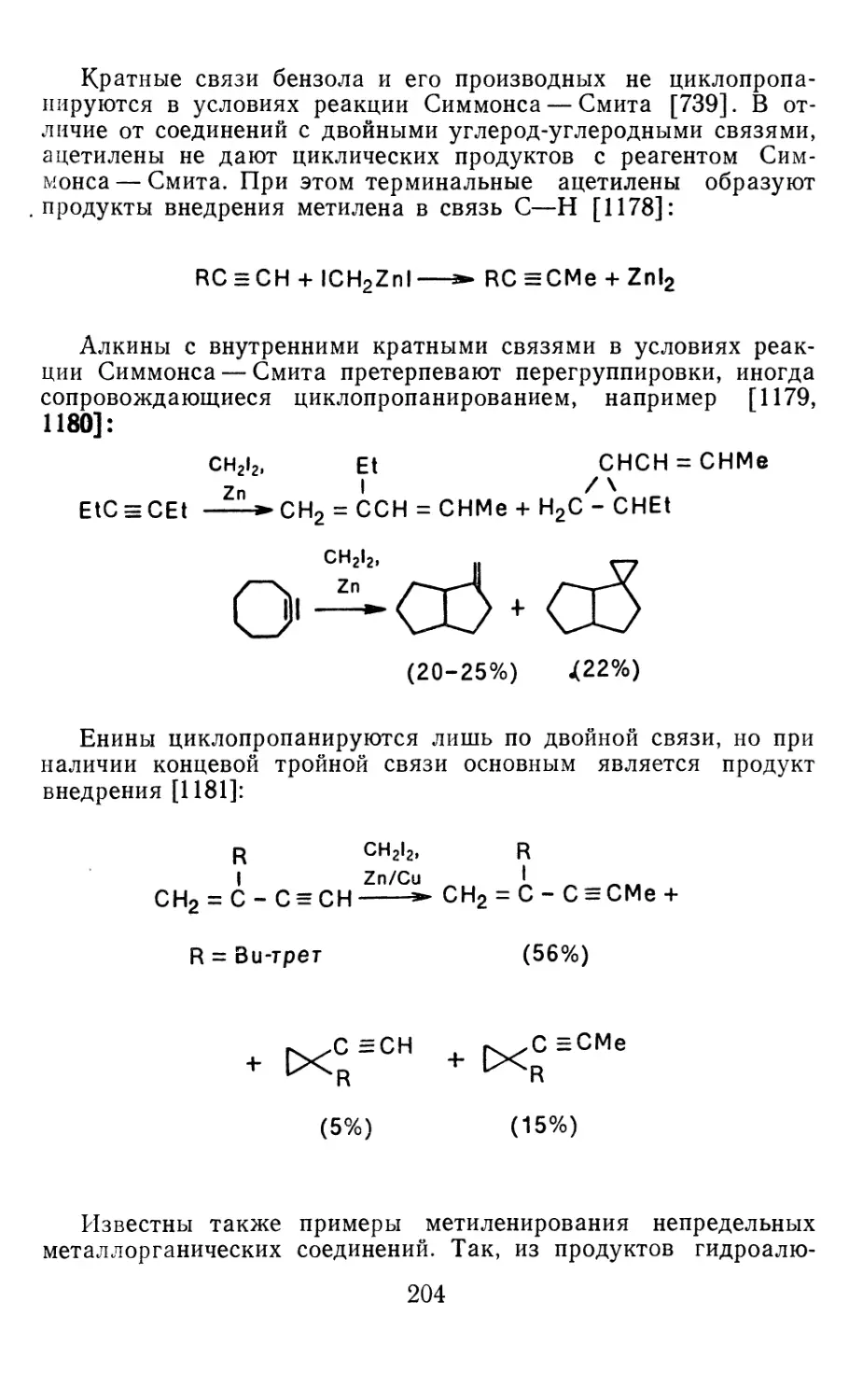
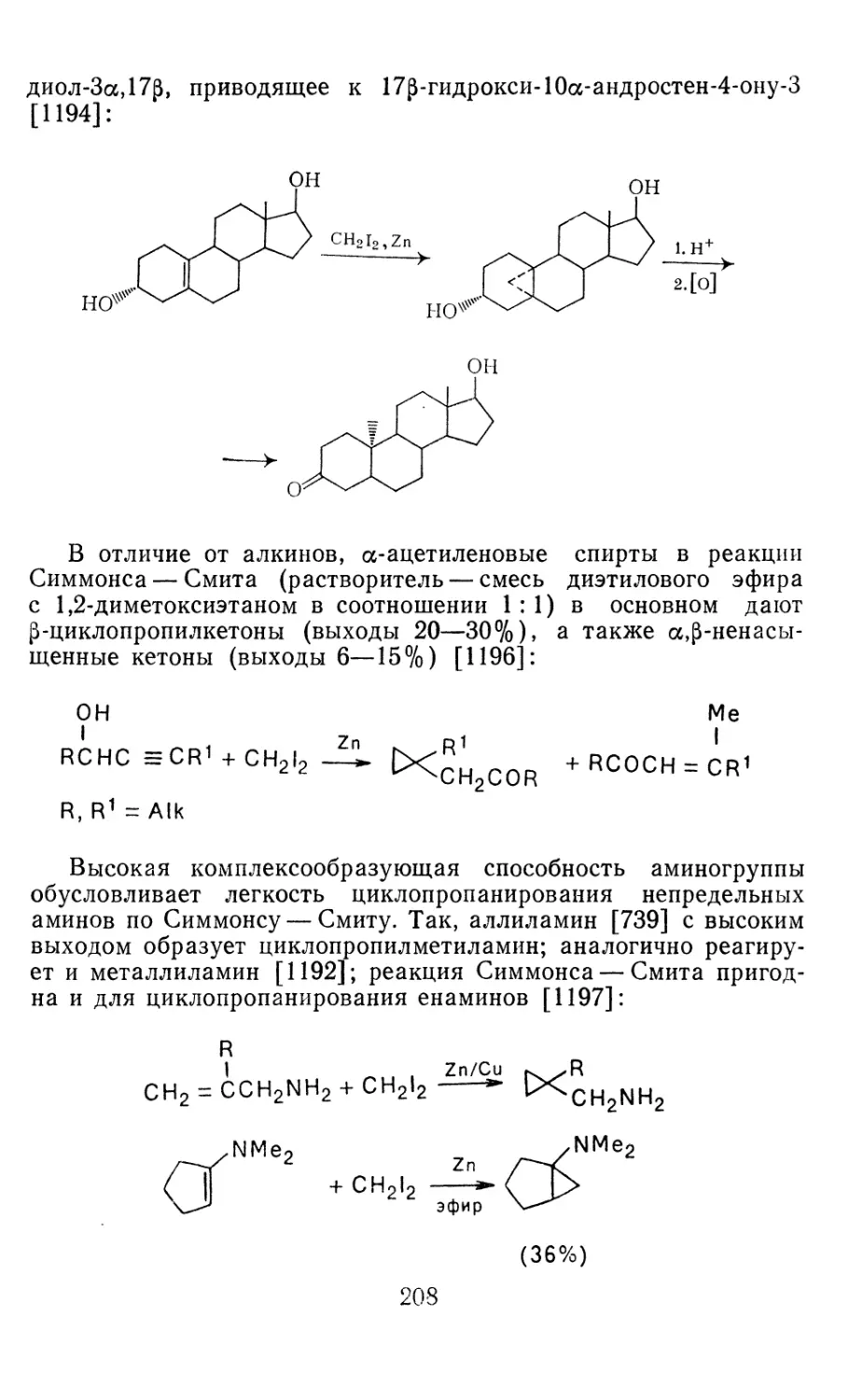
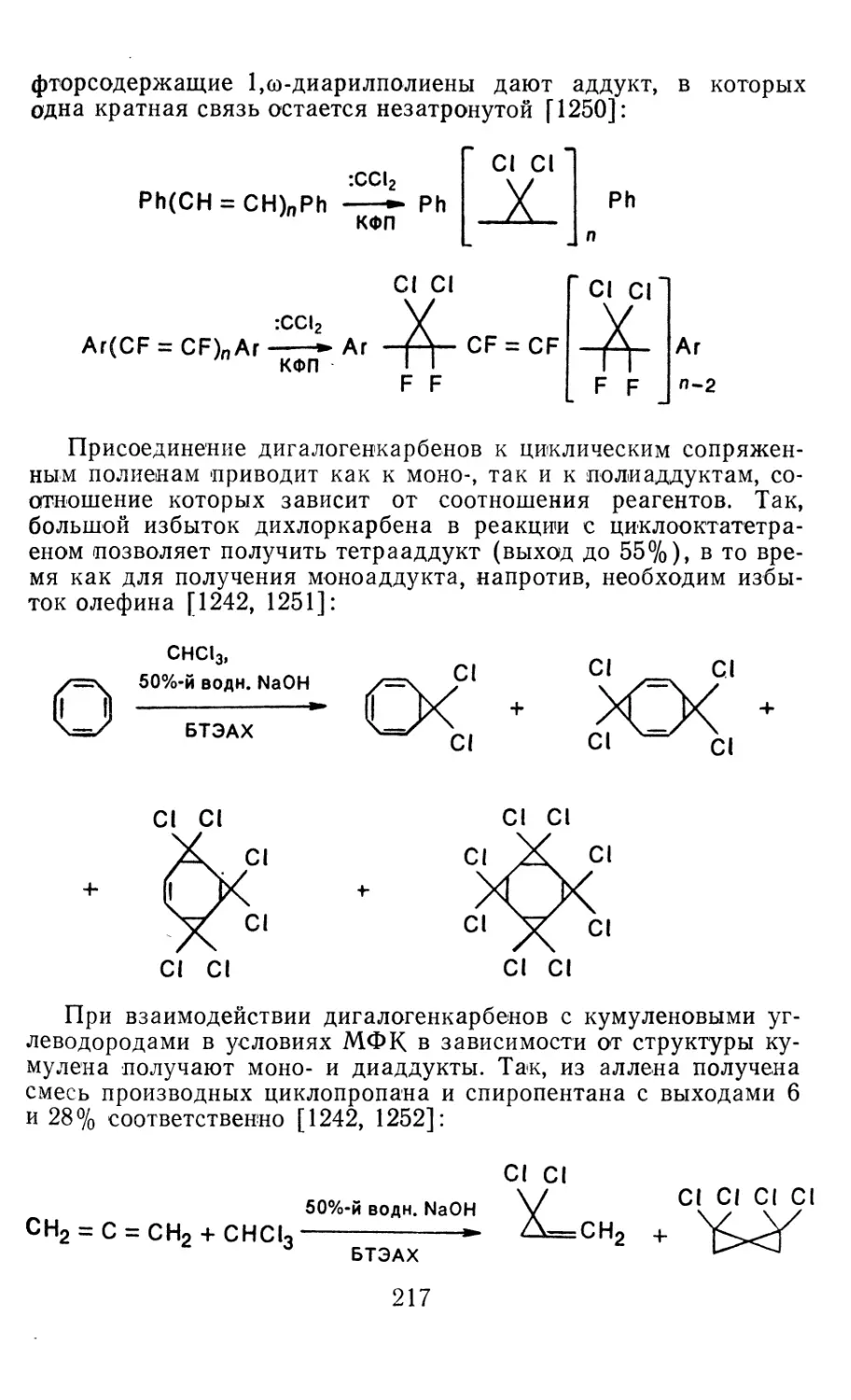
Глава 4. Циклопропанирование непредельных соединений 158
4.1 Генерирование карбенов из диазосоединений. Синтез цикло-
пропанов и их функциональных производных 158
3
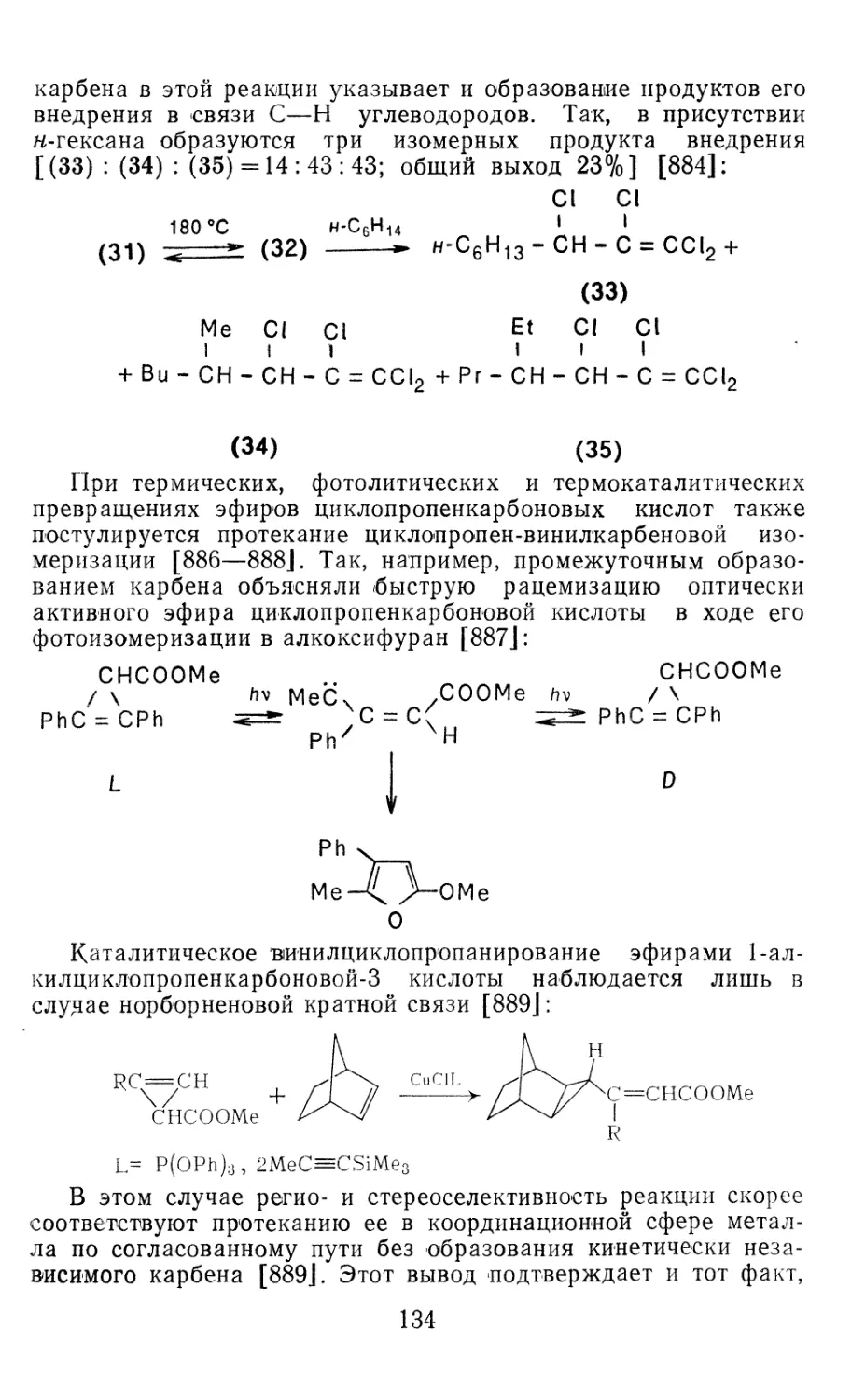
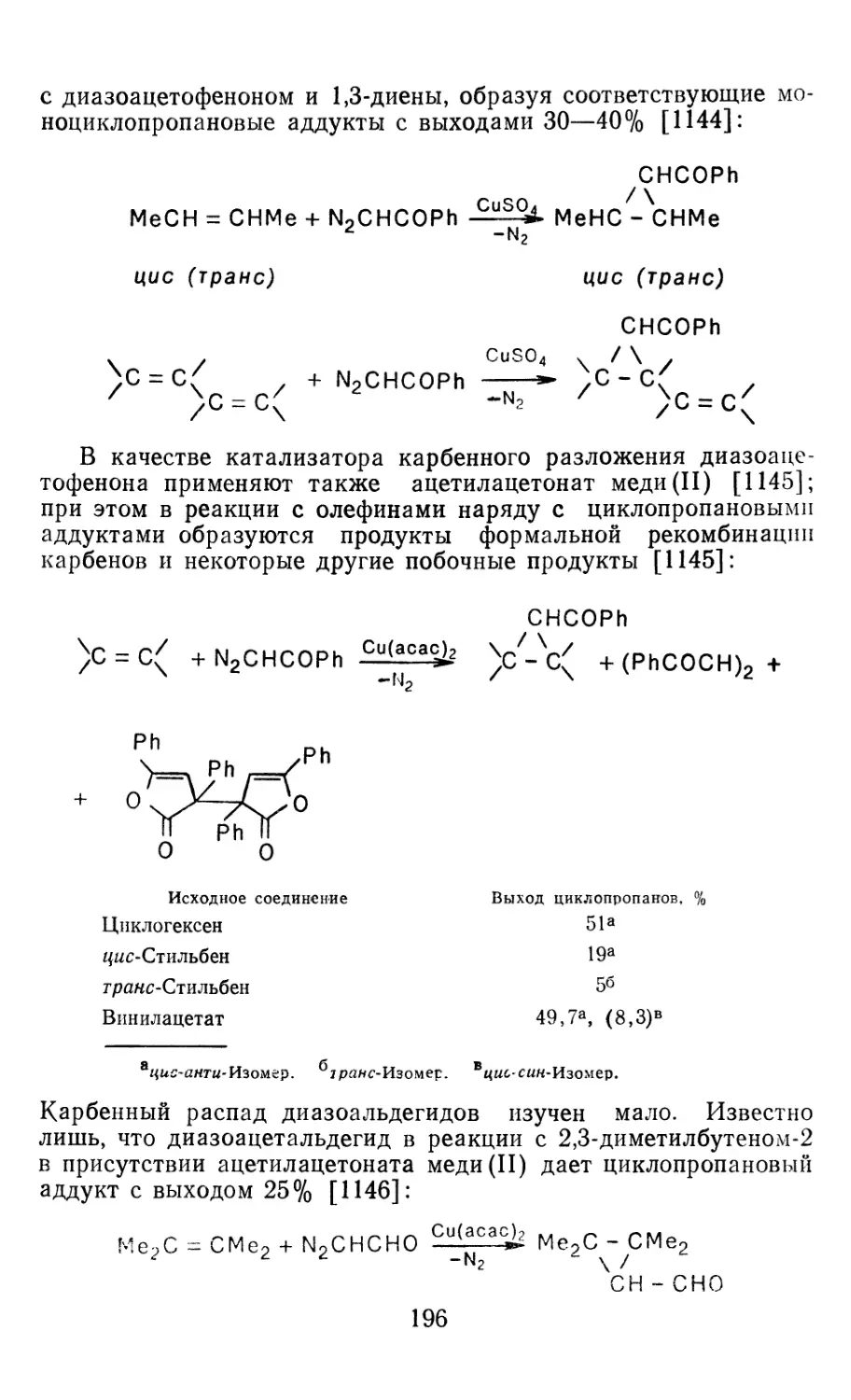
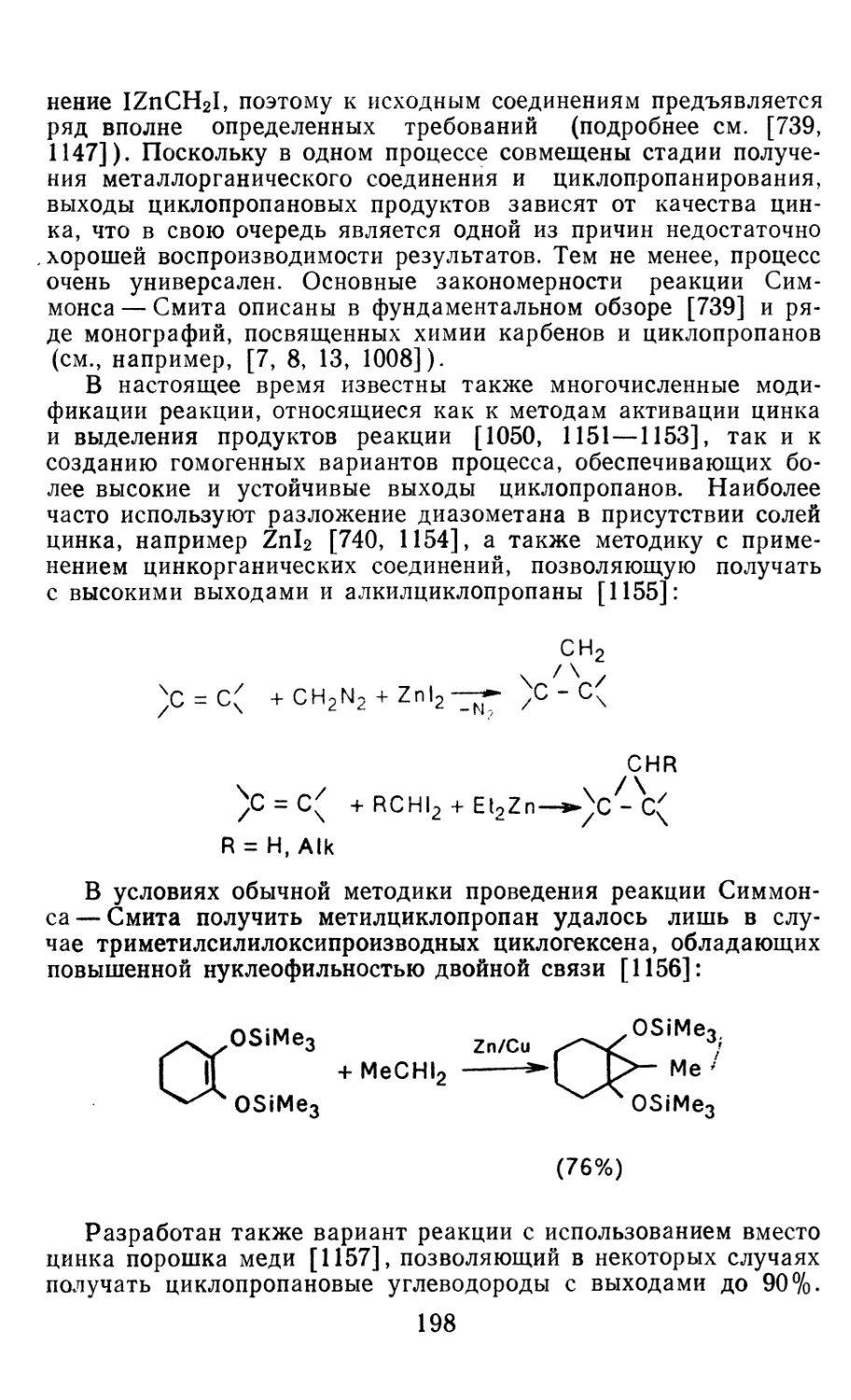
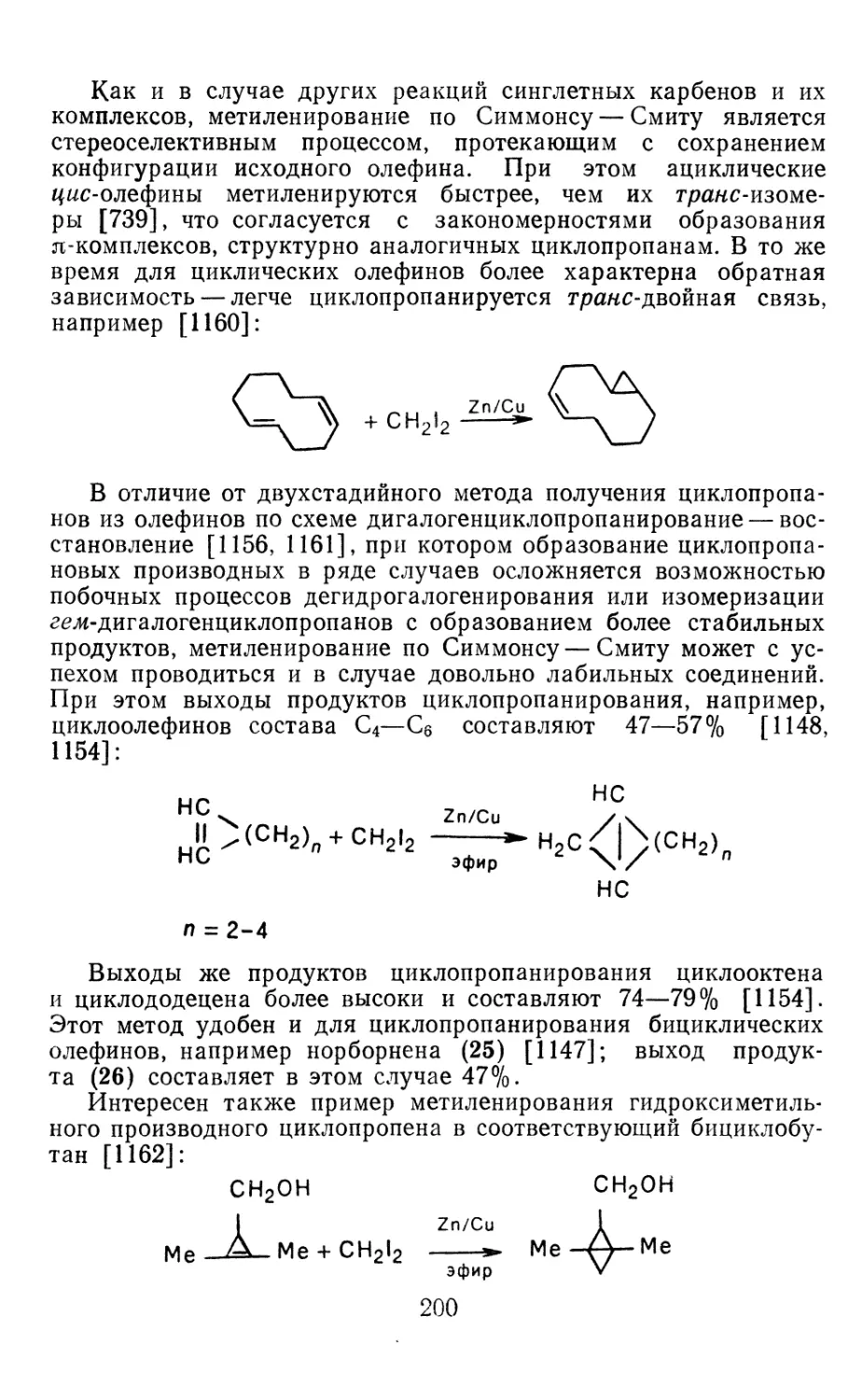
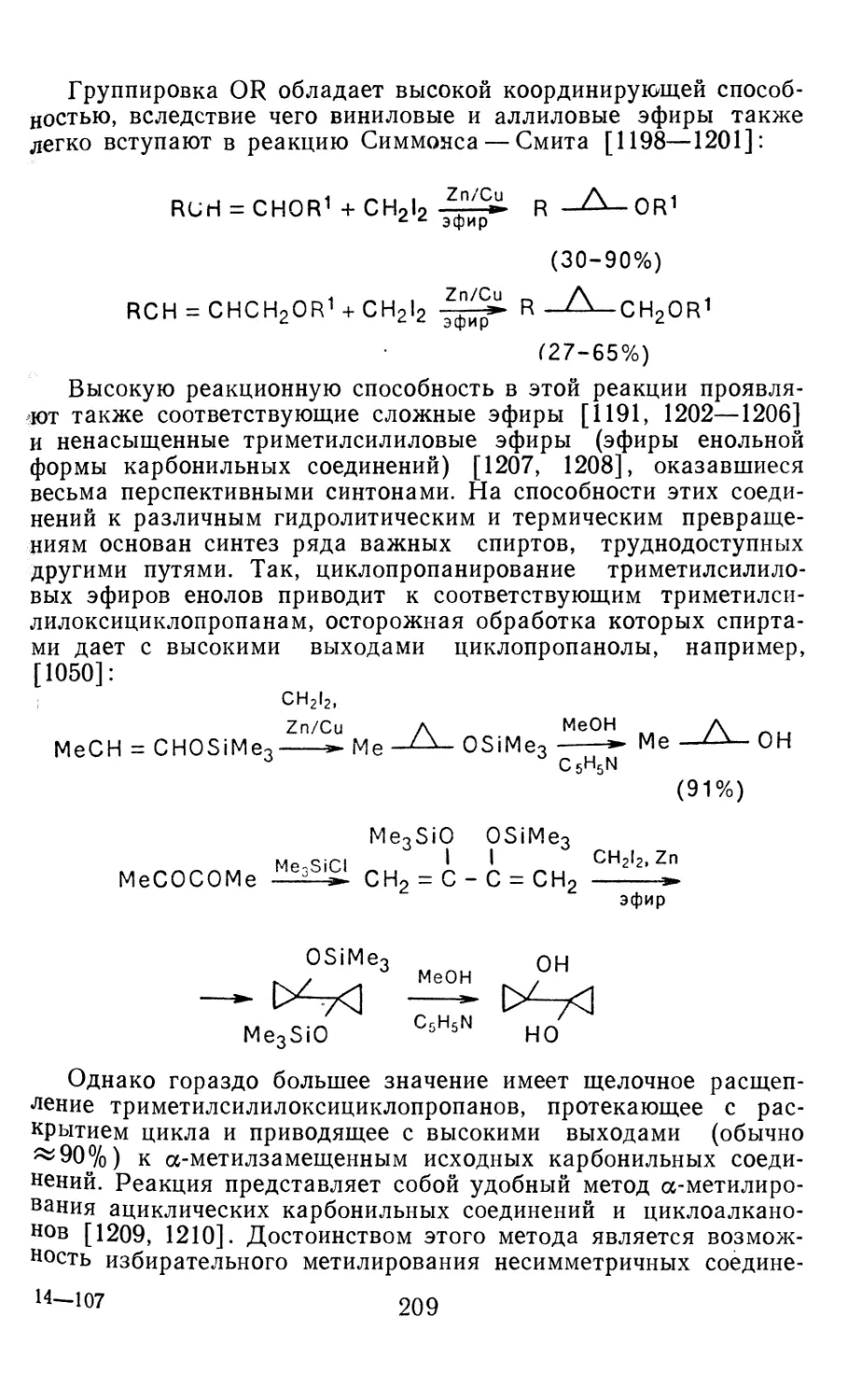
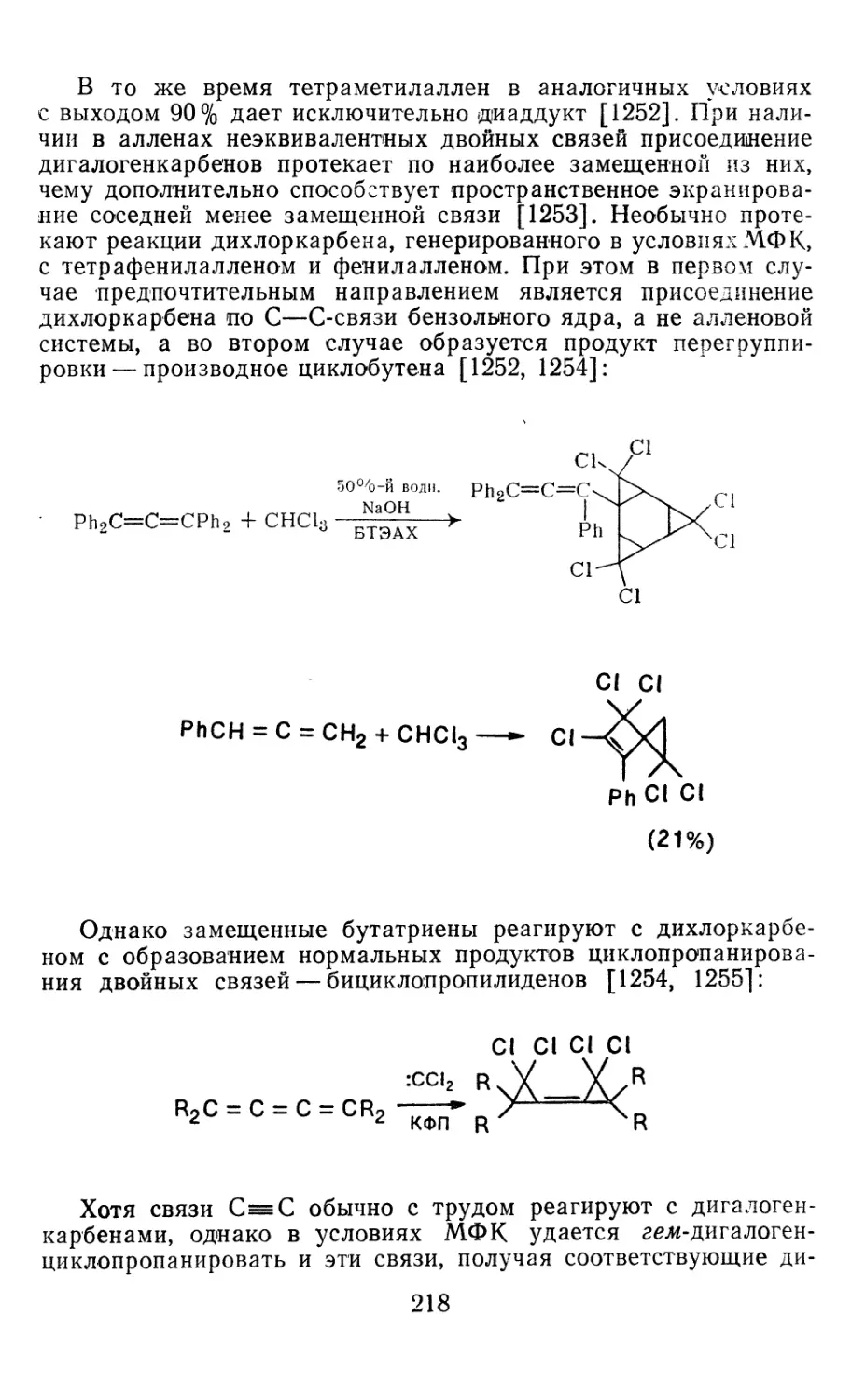
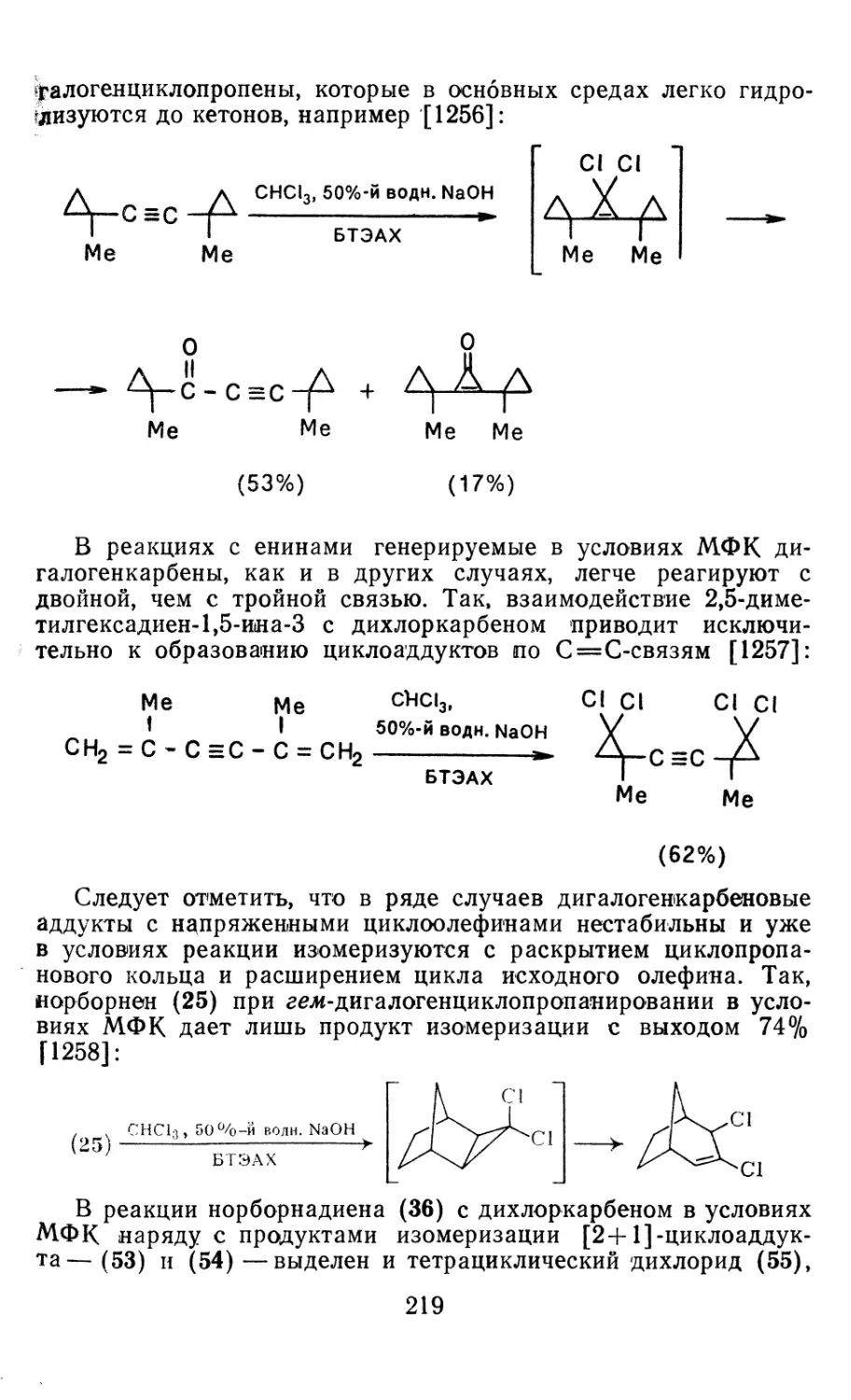
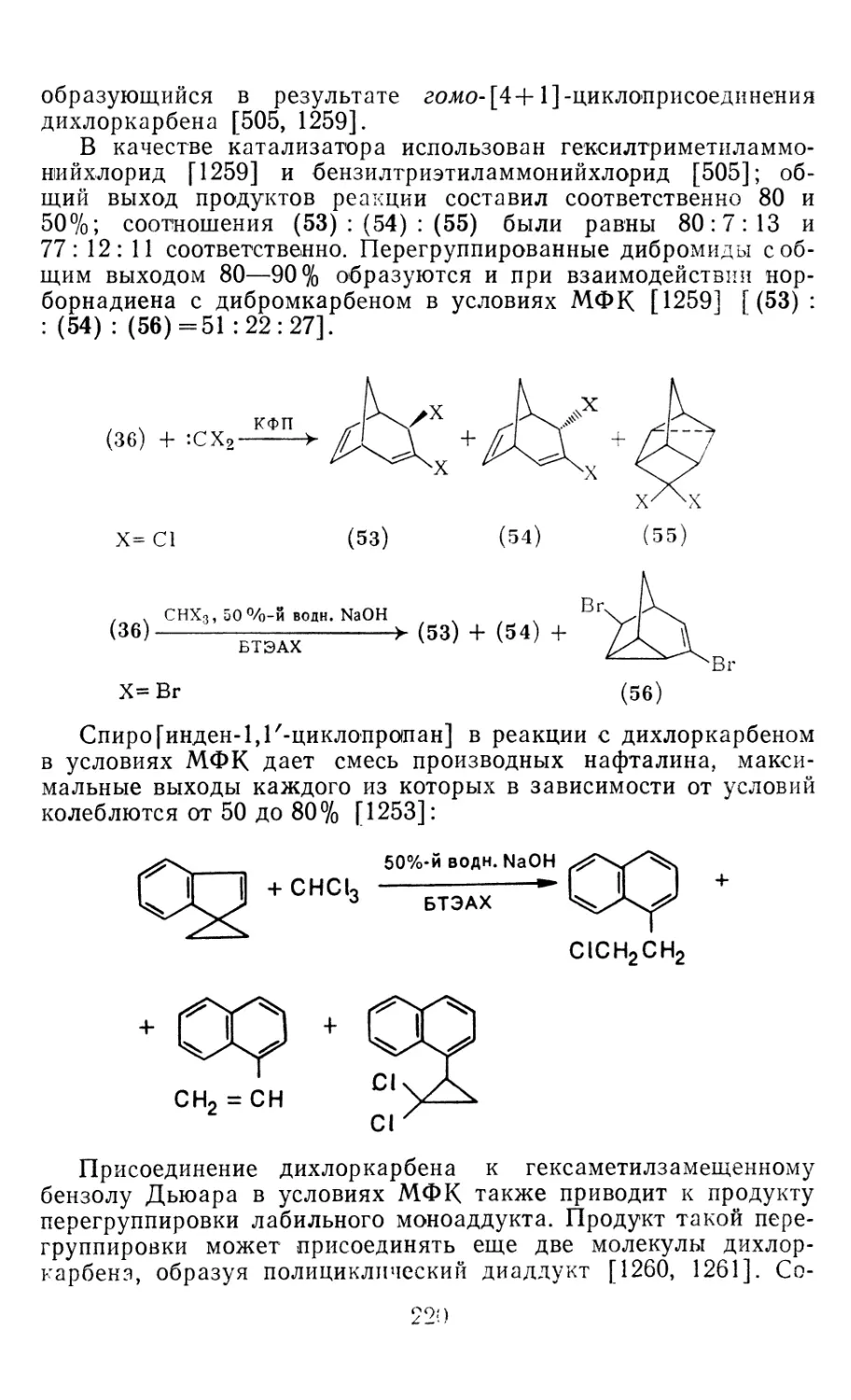
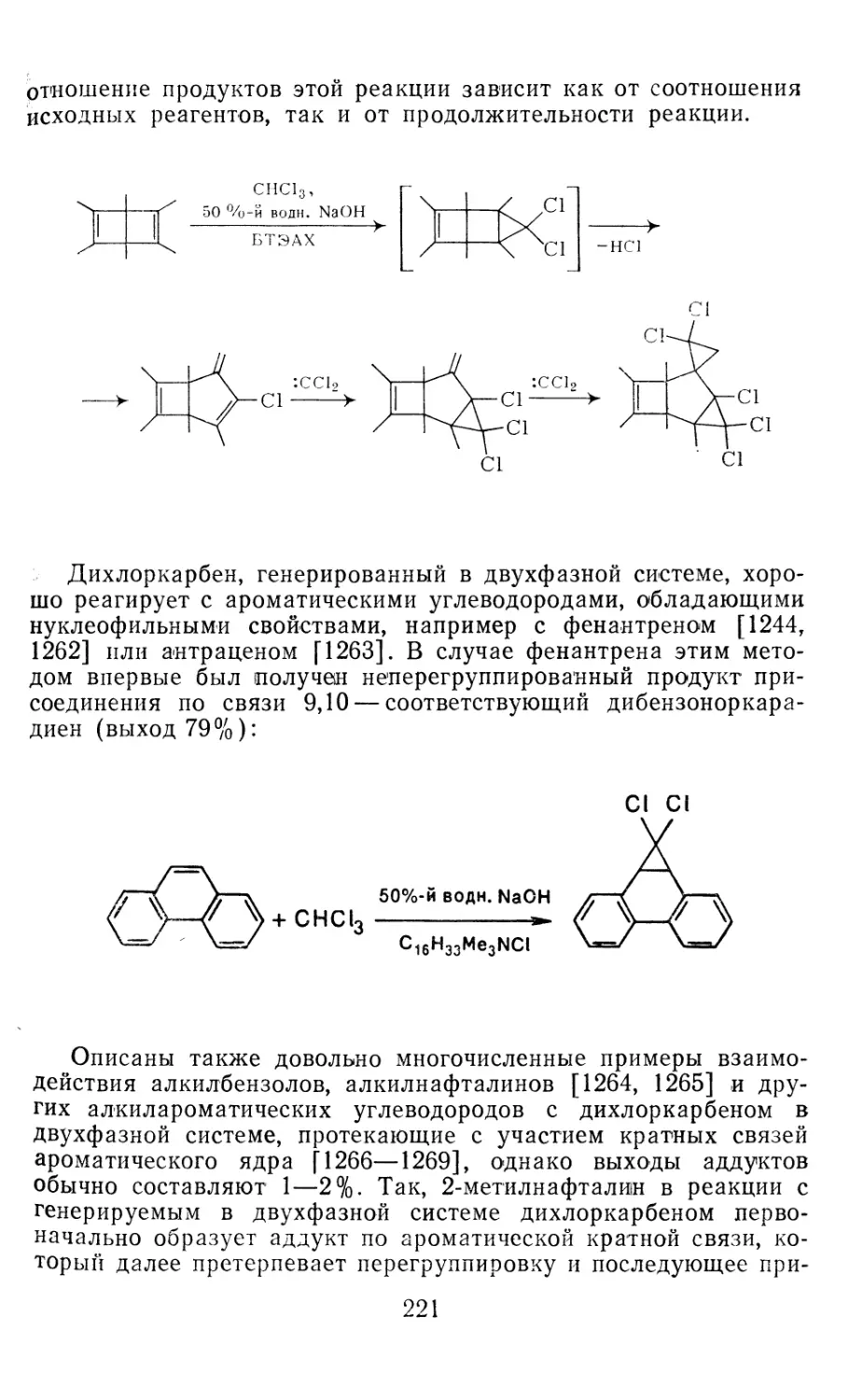
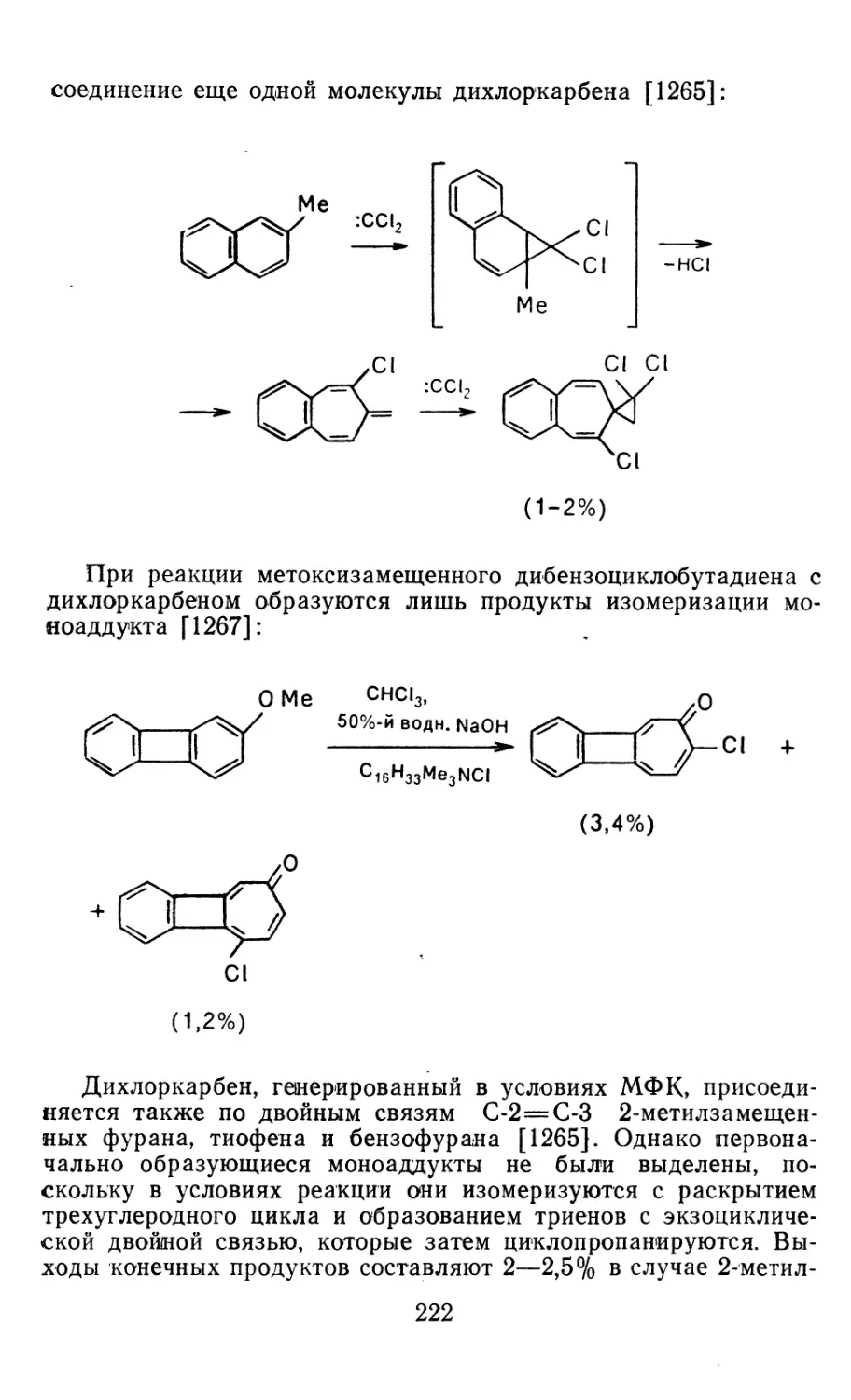
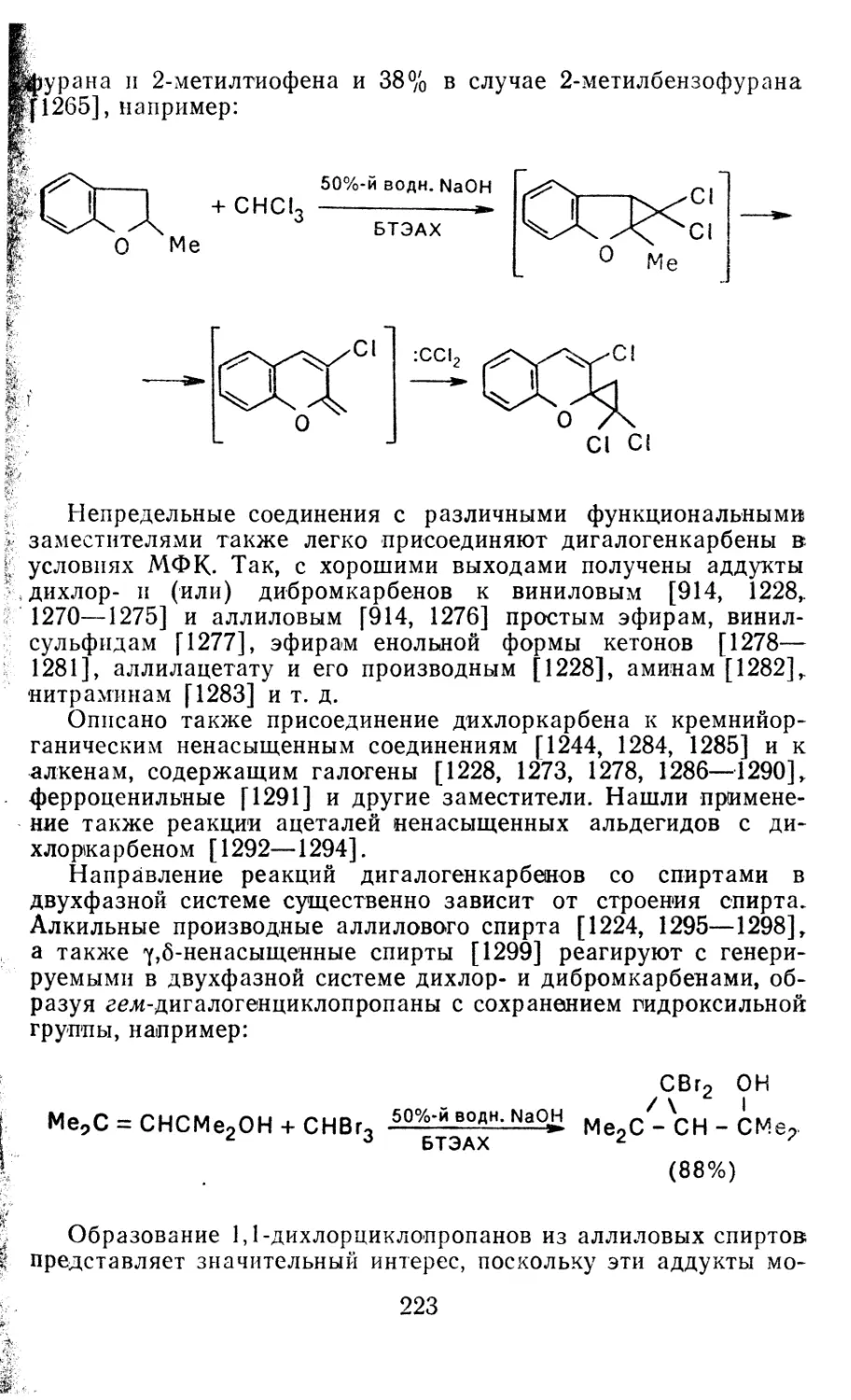
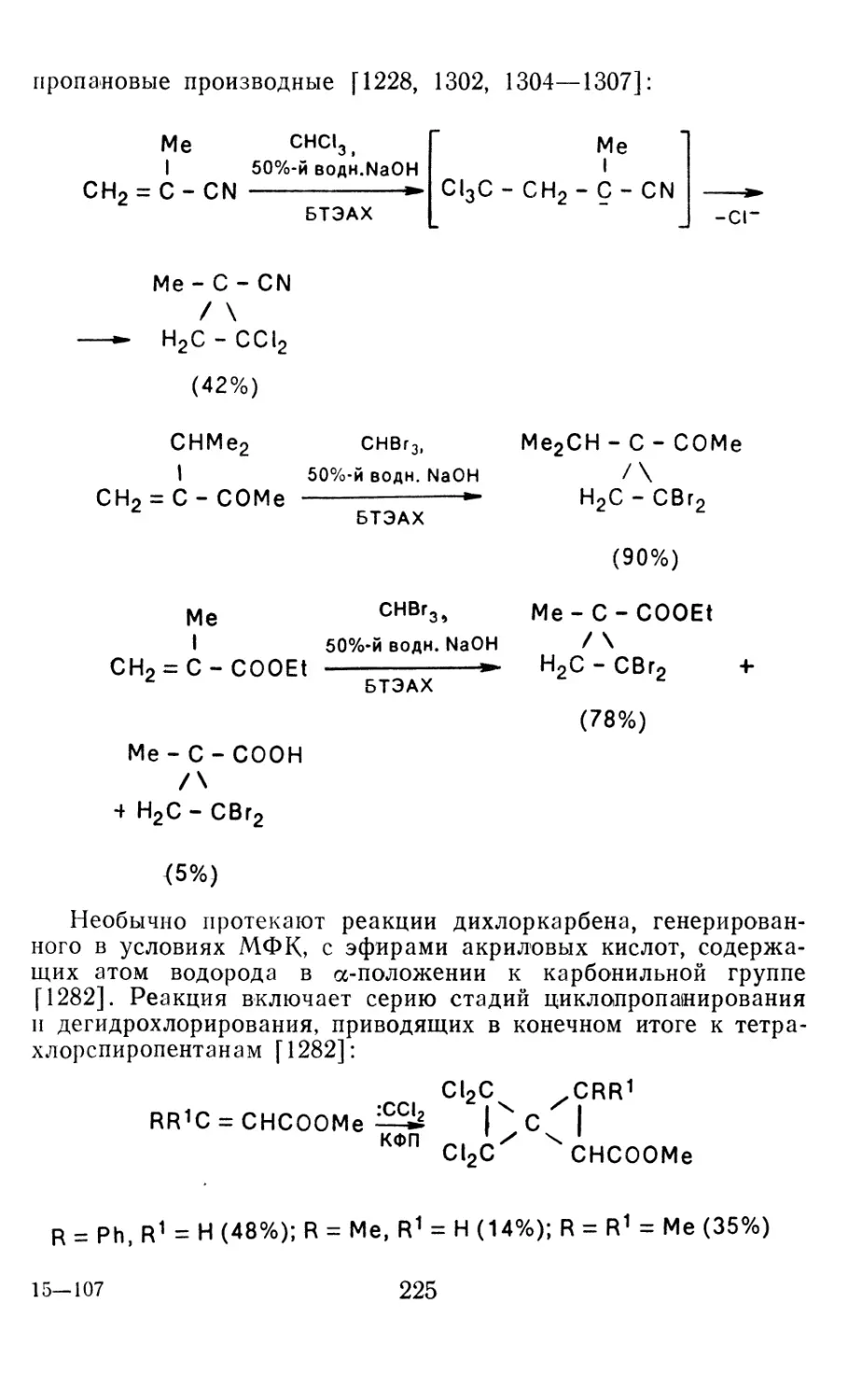
4.1.1. Диазометан 4.1.2. Алкилдиазоацетаты 4.1.3. Другие диазокарбонильные соединения 4.2. Метиленирование непредельных соединений по Симмонсу Смиту 4.3. Генерирование и реакции карбенов в условиях межфазного катализа. Химические превращения дигалогенциклопропанов 4.3.1. Механизм генерирования карбенов в условиях МФК 4.3.2. Реакции дигалогенкарбенов 163 177 192 197 212 214 215
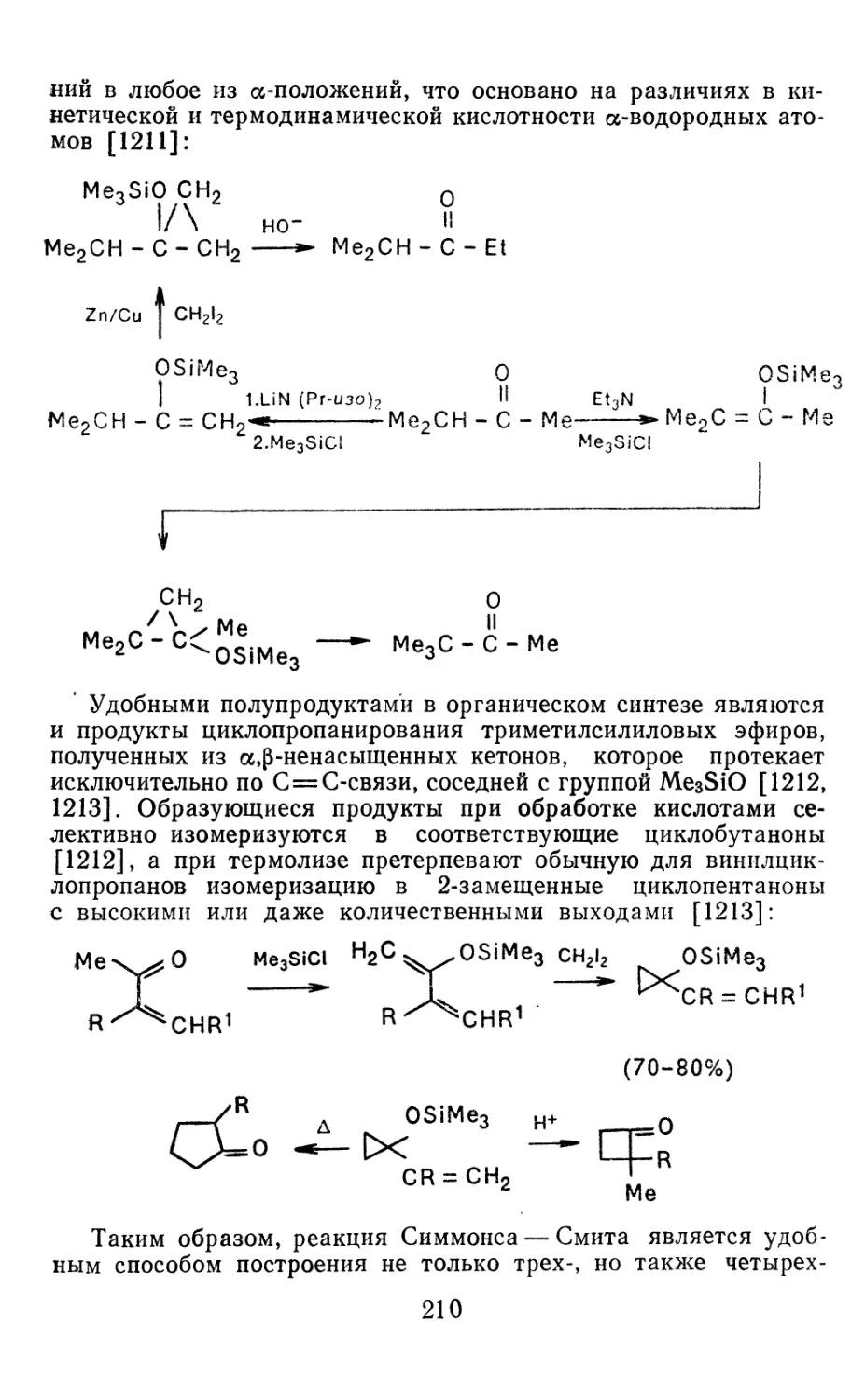
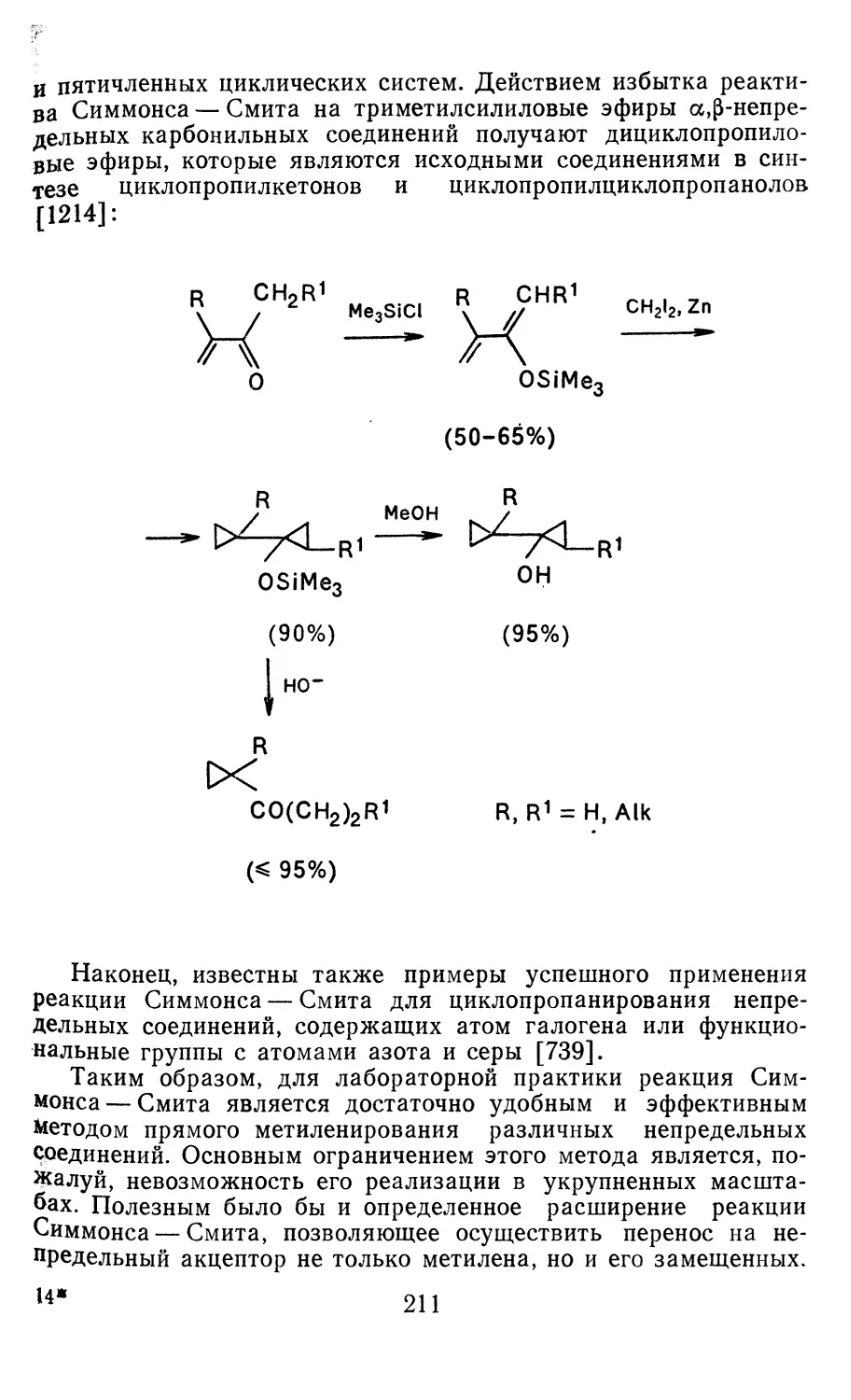
4.3.3. Реакции других карбенов, генерируемых в условиях МФК 4.3.4. Химические превращения г^и-дигалогенциклопропанов 226 228
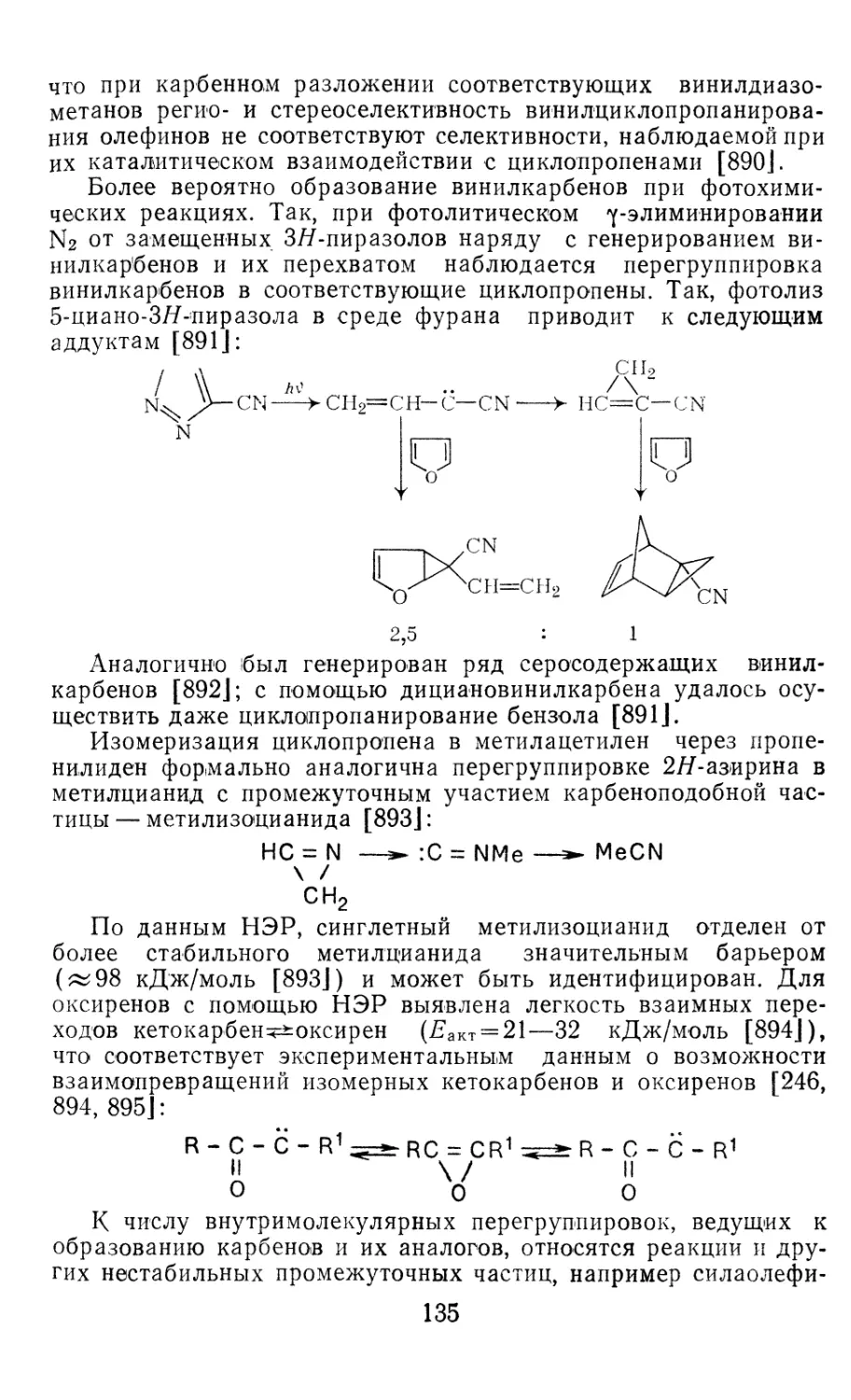
Глава 5. Карбеновые комплексы переходных металлов и их практиче-
ское использование . 233
5.1. Строение карбеновых комплексов переходных металлов 233
5.2. Каталитическое диспропорционирование олефинов 236
5.3. Полимеризация циклоолефинов с раскрытием цикла 242
5.4. Другие каталитические реакции карбеновых комплексов 245
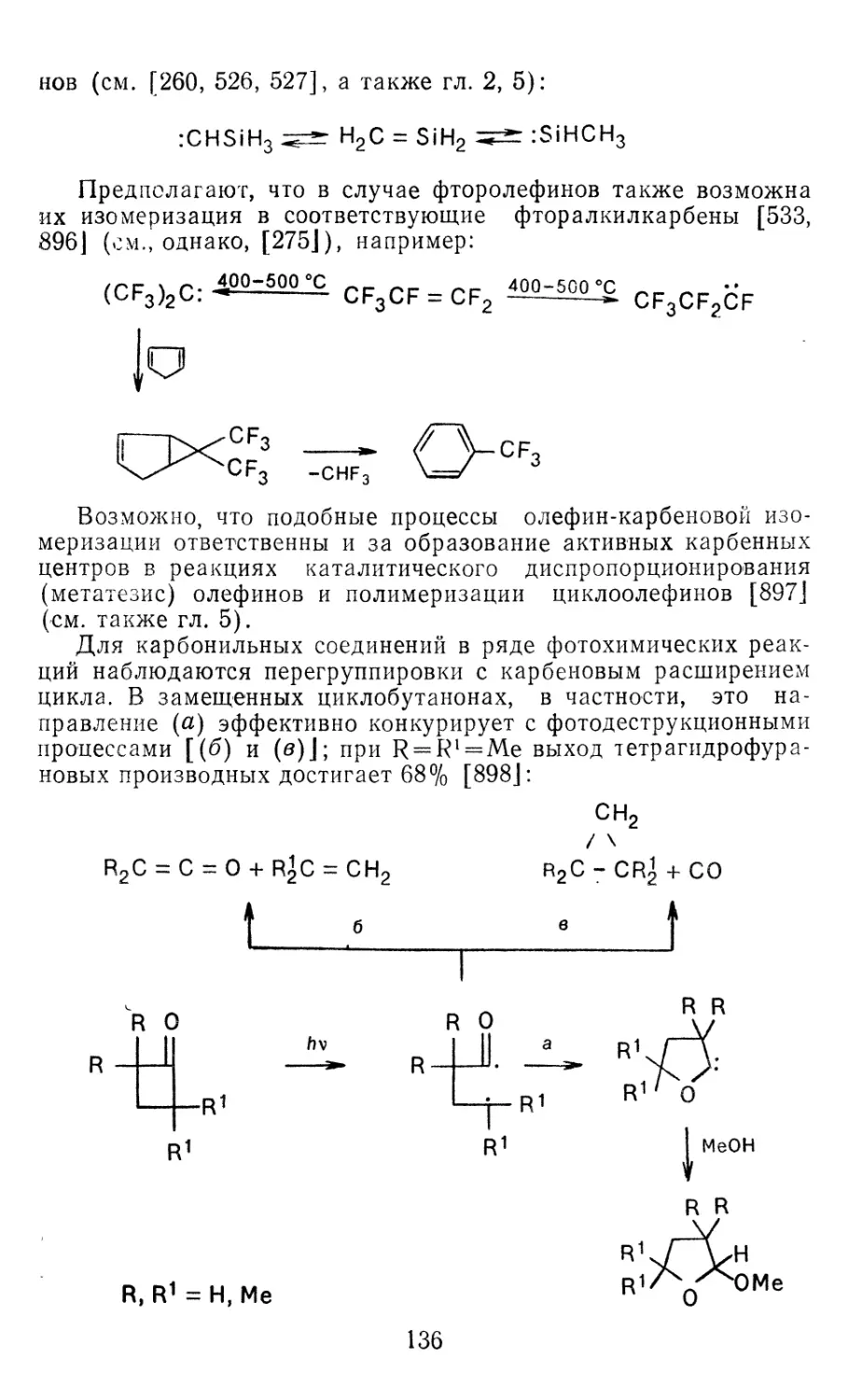
Глава 6. Некоторые перспективы применения карбенных реакций в про-
мышленном органическом синтезе 251
6.1. Термические превращения углеводородов 251
6.2. Производство фторсодержащих мономеров 254
6.3. Получение фторароматических соединений 255
6.4. Получение циклопропановых производных 258
6.5. Процессы с участием карбеновых комплексов переходных ме-
таллов 261
Библиографический список 263
ВВЕДЕНИЕ
Карбены — нестабильные соединения двухвалентного углерода — являются в
органической химии наряду с ионами и свободными радикалами одним из
основных классов высокореакционных промежуточных частиц (интермедиа-
тов), которые определяют направление'Протекания химических реакций и ха-
рактер образующихся продуктов.
Карбены и их аналоги участвуют во многих химических реакциях и
практически важных процессах, причем круг таких реакций по мере совер-
шенствования методов синтеза и исследования механизмов реакций постоян-
но расширяется. С участием карбенов связаны многие пиролитические про-
цессы (например, получение тетрафторэтилена и гексафторпропилена, пиро-
лиз углеводородов); по мере повышения температуры пиролиза роль карбе-
ноподобных интермедиатов в этих процессах возрастает. Кремниевые и гер-
маниевые аналоги карбенов участвуют в таких важных промышленных про-
цессах, как «прямой синтез» кремнийорганических соединений, получение
высокочистых кремния и германия. Широкие перспективы открывают кар-
беновые методы в тонком органическом синтезе, в том числе в промышлен-
ном.
В качестве примеров можно упомянуть карбеновые методы синтеза пи-
ретроидов, моно- и дифторароматических соединений, ряда лекарственных
препаратов и феромонов. Все возрастающую роль играют карбеновые комп-
лексы переходных металлов, с участием которых связано большинство ката-
литических методов генерирования карбенов. Комплексообразование пред-
ставляет собой наиболее действенный путь управления реакционной способ-
ностью и селективностью карбенов. С участием карбеновых комплексов
переходных металлов протекают такие важные процессы, как реакция Фи-
шера — Тропша, метатезис олефинов, полимеризация циклоолефинов с рас-
крытием цикла, синтез хиральных циклопропановых и ряда других органи-
ческих соединений. Наконец, нестабильные молекулы карбенового типа,
и прежде всего дикарбены общего состава Сп (например, :С = С:, :С = С = С:),
обнаруженные, в частности, в хвостах комет, играют важную роль в космохи-
мии. Карбеноподобные молекулы участвуют также в процессах горения и
плазмохимии.
В изданном Международным союзом теоретической и прикладной хи-
мии (IUPAC) «Глоссарии терминов, используемых в физической органиче-
ской химии» дано следующее определение карбенов: «родовое название час-
тиц :СН2 и их замещенных производных, содержащих электрически нейт-
ральный двухвалентный атом углерода с двумя несвязывающими электро-
нами» [1].
Согласно определению М. Е. Вольпина, Д. Н. Курсанова и О. М. Нефе-
дова [2], для них характерны следующие структурные (электронные) и хи-
мические признаки: наличие у центрального атома не менее одной пары не-
связывающих 5- или р-электронов и не менее одной вакантной р-орбитали
(или двух электронов на двух р-орбиталях); отсутствие свободных J-уров-
5
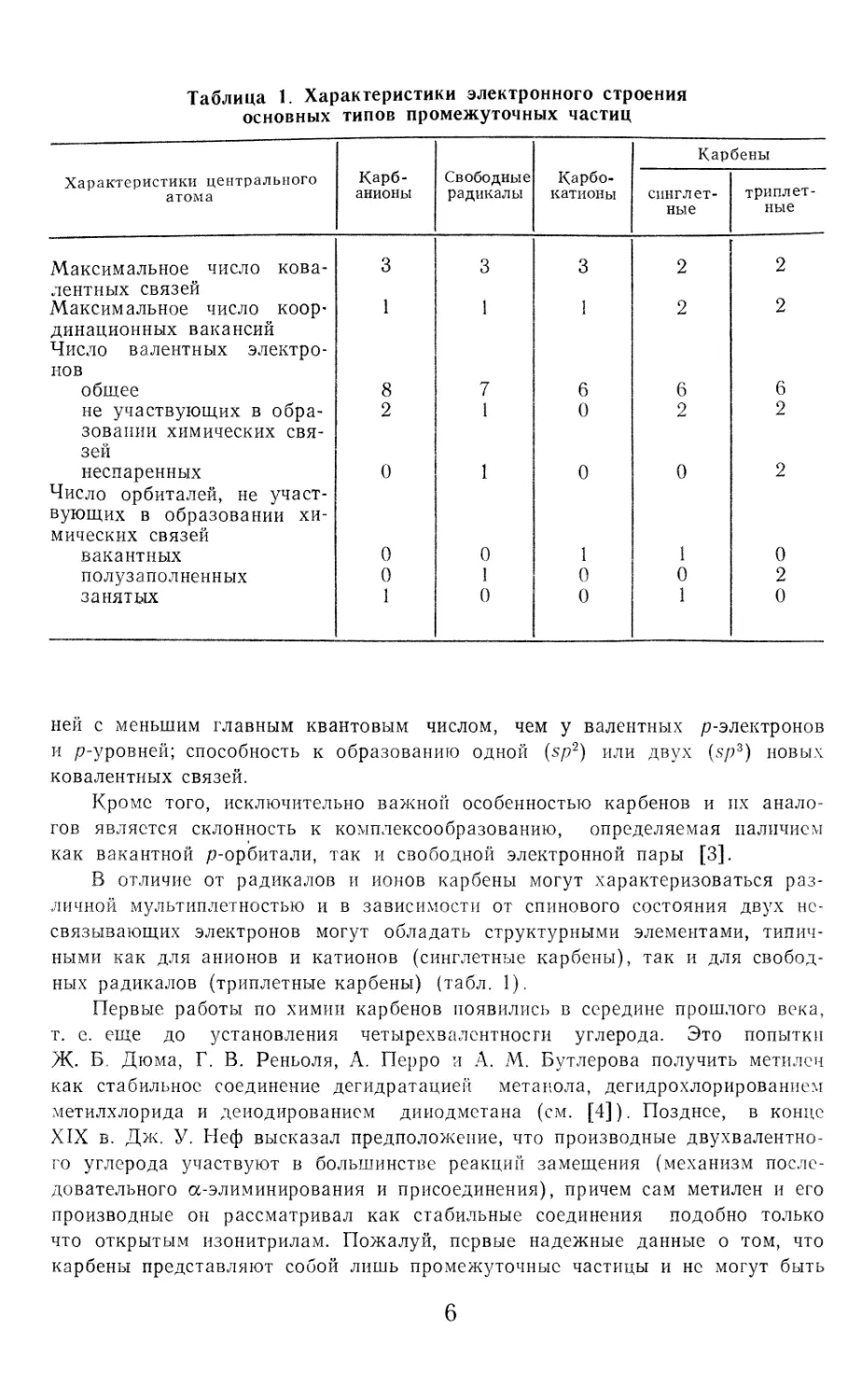
Таблица 1. Характеристики электронного строения
основных типов промежуточных частиц
Характеристики центрального атома Карб- анионы Свободные радикалы Карбо- катионы Карбены
синглет- ные триплет- ные
Максимальное число кова- лентных связей 3 3 3 2 2
Максимальное число коор’ динационных вакансий Число валентных электро- нов 1 1 1 2 2
общее 8 7 6 6 6
не участвующих в обра- зовании химических свя- зей 2 1 0 2 2
неспаренных Число орбиталей, не участ- вующих в образовании хи- мических связей 0 1 0 0 2
вакантных 0 0 1 1 0
полузаполненных 0 1 0 0 2
занятых 1 0 0 1 0
ней с меньшим главным квантовым числом, чем у валентных р-электронов
и р-уровней; способность к образованию одной (sp2) или двух (sp3) новых
ковалентных связей.
Кроме того, исключительно важной особенностью карбенов и их анало-
гов является склонность к комплексообразованию, определяемая наличием
как вакантной р-орбитали, так и свободной электронной пары [3].
В отличие от радикалов и ионов карбены могут характеризоваться раз-
личной мультиплетностью и в зависимости от спинового состояния двух нс-
связывающих электронов могут обладать структурными элементами, типич-
ными как для анионов и катионов (синглетные карбены), так и для свобод-
ных радикалов (триплетные карбены) (табл. 1).
Первые работы по химии карбенов появились в середине прошлого века,
т. е. еще до установления четырехвалснтносги углерода. Это попытки
Ж. Б. Дюма, Г. В. Реньоля, А. Перро и А. М. Бутлерова получить метилен
как стабильное соединение дегидратацией метанола, дегидрохлорированием
метилхлорида и деиодированием дииодметана (см. [4]). Позднее, в конце
XIX в. Дж. У. Неф высказал предположение, что производные двухвалентно-
го углерода участвуют в большинстве реакций замещения (механизм после-
довательного а-элиминирования и присоединения), причем сам метилен и его
производные он рассматривал как стабильные соединения подобно только
что открытым изонитрилам. Пожалуй, первые надежные данные о том, что
карбены представляют собой лишь промежуточные частицы и не могут быть
6
идентифицированы традиционными химическими методами, были получены
в начале нашего века Г. Штаудингером и сотр. при исследовании распада
диазосоединений и кетенов. Более поздние работы действительно подтверди-
ли образование карбенов при распаде этих соединений, так что по существу
именно с работ Штаудингера и следует рассматривать начало развития хи-
мии карбенов (см. [4]).
На первых порах развитие это не было бурным, и лишь в 50-е годы
после установления карбенного механизма щелочного гидролиза галофор-
мов [5] и обнаружения препаративной значимости данной реакции интерес к
химии карбенов заметно возрос. Последующие годы характеризовались рас-
ширением именно препаративных аспектов химии карбенов. Здесь в первую
очередь следует отметить разработку удобных карбенных методов синтеза
циклопропановых углеводородов и их производных из олефинов — получе-
ние циклопропанов по Симмонсу — Смиту, галогенциклопропанирование оле-
финов с помощью галогензамещенных карбенов (прежде всего дигалогенкар-
бенов), синтез производных циклопропанкарбоновой кислоты термокаталити-
ческими реакциями этилдиазоацетата, а также широкое исследование хими-
ческих превращений циклопропанов, получаемых с участием карбенов. Таким
образом, в 50—60-е годы карбенныс методы прочно вошли в синтетическую
практику органической химии, хотя строгого обоснования и доказательств ре-
ального участия карбенов в большинстве случаев не было. Тем не менее уже
в эти годы с целью оценки возможности участия в исследуемых реакциях
карбенов и их мультиплетности были предложены и широко использовались
качественные и полуколичественные тесты и подходы типа постулатов Скел-
ла и метода конкурирующих реакций.
Физико-химические аспекты химии карбенов стали интенсивно развивать-
ся с середины 60-х годов, прежде всего в связи с успехами инструменталь-
ных методов прямой спектроскопической идентификации промежуточных час-
тиц.
В первую очередь необходимо отметить успешное применение для ста-
билизации и прямого спектроскопического изучения карбенов и их аналогов
метода матричной изоляции, заключающегося в замораживании исследуемых
нестабильных молекул в органические стекла или матрицы инертных газов.
Этот метод, несмотря на необходимость использования при работе с инерт-
ными газами довольно сложной и дорогостоящей криогенной аппаратуры,
позволил в значительной мере снять ограничения по быстродействию спект-
ральных приборов, связанные с малым временем жизни (обычно от 10~3 до
10~6 с) и низкими текущими концентрациями карбенов в химических реак-
циях.
Усовершенствование спектральных приборов, прежде всего повышение
их чувствительности и быстродействия, позволило разработать и широко ис-
пользовать импульсные методы генерирования и регистрации карбенов в ус-
ловиях газовой фазы. В конце 60-х и начале 70-х годов для изучения жид-
кофазных карбенных реакций стали применять разработанный в это время
метод химической поляризации ядер (ХПЯ).
Эти же годы характеризуются и большим прогрессом в области теоре-
7
тических и квантовохимических методов, благодаря которым расчетная ин-
формация о строении и механизмах реакций карбенов стала достаточно до-
стоверной не только в сравнительном, но и в абсолютном плане. Тогда же
и сформировались современные представления о месте карбенов среди про-
межуточных частиц органических реакций и о единстве химии карбенов и их
аналогов, в первую очередь силиленов, гермиленов и других соединений эле-
ментов подгруппы IVB.
В середине 60-х годов Е. О. Фишером были синтезированы и охаракте-
ризованы первые стабильные карбеновые комплексы переходных металлов,
а в нашей лаборатории — комплексы дихлоргермилена GeCl2 с п-донорными
лигандами (1,4-диоксаном и др.). Впоследствии были получены и охаракте-
ризованы карбеновые комплексы переходных металлов «шроковского» типа,
а работами Б. А. Долгоплоска и сотр. вскрыта роль карбеновых комплексов
в процессах метатезиса олефинов и полимеризации циклоолефинов с раскры-
тием цикла и в ряде других каталитических процессов. Эти исследования,
которые вначале развивались изолированно от собственно химии карбенов,
позволили впоследствии с единых позиций рассмотреть общую проблему
комплексообразования в химии карбенов.
В этот период существенно прогрессировала и синтетическая химия кар-
бенов Усовершенствовались методы генерирования карбенов, открывались и
выявлялись новые превращения с участием карбенов и их аналогов, разра-
батывались карбенные методы синтеза различных практически полезных ве-
ществ и синтонов. В первую очередь необходимо отметить появление и раз-
работку метода межфазного катализа, позволившего в химии карбенов, а по-
том и в других областях органической химии, избавиться от использования
абсолютированных растворителей и «неудобных» реагентов типа алкоголятов
щелочных металлов и литийорганических соединений, расширить область
синтетического применения карбенных методов, а в ряде случаев и поднять
выходы целевых продуктов.
И хотя во многих конкретных (экспериментальных) исследованиях син-
тетические и физико-химические аспекты химии карбенов рассматривались
раздельно, все больше начало вырисовываться объединение синтетических,
кинетических, инструментальных и расчетных аспектов химии карбенов и их
аналогов в единое научное направление. Этому в большей мере способство-
вало повышение качества и расширение границ применения расчетных мето-
дов и появление новых экспериментальных возможностей регистрации кар-
бенов и подобных им интермедиатов с помощью пико- и наносекундной ла-
зерной спектроскопии. Это позволило, в частности, следить за генерировани-
ем и исчезновением карбенных центров в реальных многокомпонентных жид-
кофазных системах, определять спектральные, кинетические и термодинами-
ческие параметры карбенов и реакций с их участием. Появлялись и совер-
шенствовались другие инструментальные методы прямого наблюдения и изу-
чения карбеноподобных интермедиатов (пиролитическая масс-спектрометрия,
газовая электронография в сочетании с масс-спектрометрией, фотоэлектрон-
ная спектроскопия). Например, недавно в нашей лаборатории метод низко-
температурной матричной ИК-спектроскопии впервые удалось использовать
8
для прямого наблюдения и изучения реакции (1 + 2)-циклоприсоединения
карбена к связи С = С. В свою очередь разработка новых и усовершенство-
вание известных методов инициирования химических реакций, таких, как
электрохимия, фотолиз, адиабатическое сжатие, механохимия, воздействие
ультразвука и высоких давлений со сдвигом, не только содействует расши-
рению применения карбенов и их аналогов в синтезе, но и открывает новые
возможности для изучения кинетических и структурных аспектов реакций с
участием этих частиц.
Все это позволяет высоко оценить современное состояние химии карбе-
нов и их аналогов и ожидать по-прежнему интенсивного развития этой об-
ласти химии в ближайшем будущем.
Следует отметить, что несмотря на публикацию нескольких монографии
и большого числа обзоров как общего, так и частного характера моногра-
фическая [4—7] и обзорная литература явно не поспевает за быстрым раз-
витием данной области. Достаточно сказать, что с момента выхода в свет
монографии [7] прошло свыше 19 лет. Еще хуже обстоит дело с монографи-
ческой литературой по химии карбенов на русском языке. Единственная вы-
шедшая еще в 1966 г. небольшим тиражом книга В. Кирмсе [4] существен-
но устарела, поскольку характеризует состояние этой области на начало 60-х
годов.
Весьма устаревшие к настоящему времени монографии Дж. Хайна
[5] и В. Кирмсе [6, 7] на русский язык вообще не были переведены. Раз-
личным аспектам химии карбенов посвящен ряд обзоров [8—10], но эти ма-
териалы, во-первых, достаточно фрагментарны и узки, а во-вторых, прак-
тически недоступны советскрму читателю. Существенно более доступны тема-
тические выпуски таких журналов, как Журн. ВХО им. Д. И. Менделеева
(1979. т. 24, вып. 5), Tetrahedron (1984; т. 41, вып. 8) и Успехи химии (1989,
т. 58, вып. 7, 8), но и они, безусловно, не могут дать полного представления
о современном состоянии химии карбенов и перспективах ее развития, по-
скольку посвящены достаточно узким вопросам химии карбенов. Набор от-
дельных обзоров представляет собой и сборник [И], а монография Н. С. Зе-
фирова и сотр. [12] посвящена лишь одному, хотя и важному аспекту хи-
мии карбенов — циклоприсоединению дихлоркарбена к олефинам. Практиче-
ски только «циклопропановый» аспект химии карбенов рассмотрен и в очень
подробном обзоре Д. Вендиша [13]. Наконец, монография В. Львовски [14]
и сборник [15] посвящены химии нитренов — частиц, во многом напомина-
ющих карбены, но существенно от них и отличающихся. Детальное рассмот-
рение карбенов и их аналогов с общих позиций проведено лишь в обзорах
[16, 17] (см. также гл. 2).
Предлагаемая читателю монография представляет собой попытку своего
рода «моментальной фотографии» химии карбенов. В ней детально рассмот-
рены инструментальные и расчетные методы установления строения и реак-
ционной способности карбенов и их аналогов, современное состояние этих
методов и перспективы их развития. Приведенные данные дают, на наш
Взгляд, достаточно полное представление о возможностях физико-химиче-
ских исследований самих карбенов и реакций с их участием. Рассмотрены
9
также методы генерирования карбенов и их аналогов, дана исчерпывающая
классификация методов генерирования карбенов, впервые предложенная в
работах [16, 17], и показаны проблемы, связанные с генерированием опре-
деленных типов карбенов и доказательством их реального участия в этих
процессах.
При описании возможностей препаративного использования наиболее
важной карбенной реакции — циклопропанирования непредельных соедине-
ний — основное внимание уделено трем наиболее известным методам получе-
ния циклопропанов (использование диазосоединений, циклопропанирование
по Симмонсу — Смиту и использование карбенов, генерируемых в условиях
межфазного катализа); сопоставлены возможности, преимущества и недо-
статки этих методов.
Небольшая глава посвящена молодой, но бурно развивающейся и весь-
ма перспективной области химии карбенов—карбеновым комплексам
переходных металлов, играющим ключевую роль во многих процессах с уча-
стием металлорганических соединений и катализаторов. В последней главе
рассмотрено использование карбенных реакций в промышленном органиче-
ском синтезе, в том числе для получения мономеров, лекарственных препара-
тов, пестицидов, красителей и т. д.
Авторы отдают себе отчет в том, что ограниченный объем монографии
не позволил им детально проанализировать многие интересные аспекты хи-
мии карбенов. В частности, отдельного рассмотрения заслуживают аналоги
карбенов на основе элементов не только группы IVB, но и других групп
Периодической системы, в первую очередь нитрены. Сказанное относится и к
реакциям карбенов с ароматическими и гетероциклическими акцепторами,
а также к разнообразным химическим превращениям галогенциклопропа-
нов— продуктов циклоприсоединения галогенкарбенов к олефинам. Вместе
с тем, в книге отражены именно основные аспекты химии карбенов, определя-
ющие ее дальнейшее развитие, продемонстрирована необходимость общего
подхода к рассмотрению данной области, единства ее физико-химического и
синтетического аспектов.
Наша страна традиционно занимает одно из ведущих мест в мире по
исследованиям в области химии карбенов, их комплексов и аналогов. По-
мимо уже упомянутых исследований А. М. Бутлерова по генерированию ме-
тилена следует отметить работы И. А. Дьяконова и сотр. в области химии
алифатических диазосоединений [17а], наши и М. Е. Вольпина и Д. Н. Кир-
санова исследования в области кремниевых и германиевых аналогов карбе-
нов, работы Б. А. Долгоплоска и других советских химиков в области кар-
беновых комплексов переходных металлов и процессов с их участием, пер-
вый серьезный обзор по химии карбенов И. Л. Кнунянца и сотр. [17 б],
первое рассмотрение с общих структурно-химических позиций карбенов и их
аналогов [16].
Начиная с 1972 г. в нашей стране регулярно проводятся всесоюзные
конференции по химии карбенов и их аналогов. Работы по данному направ-
лению координируются Научным советом АН СССР по химической кинети-
ке и строению. Хочется надеяться, что настоящая монография даст объек-
тивное и достаточно полное представление о современном состоянии и пер-
10
спективах этой интересной и важной области современной химии, будет со-
действовать росту интереса к ней, способствовать ее развитию в нашей
стране.
В заключение авторы считают своим приятным долгом выразить благо-
дарность проф. Л. В. Гурвичу (Термоцентр АН СССР) за предоставление
последней версии сводки термодинамических характеристик карбенов и их
аналогов из банка данных ИВТАНТЕРМО, |А. К. Мальцеву[, чье участие в
создании главы 2 данной книги трудно переоценить, а также Т. А. Бункиной,
Лэ А. Егоровой, Т. Л. Кирсановой, Н. Н. Курковой, М. Б. Липкинд,
Н. М. Цветковой и другим сотрудникам лаборатории химии карбенов и дру-
гих нестабильных молекул Института органической химии им. Н. Д. Зелин-
ского АН СССР за большую помощь в подготовке рукописи.
Глава 1
НОМЕНКЛАТУРА КАРБЕНОВ
К настоящему времени наиболее распространенной номенкла-
турой соединений двухвалентного углерода является карбеновая
номенклатура. Термин «карбен» был впервые предложен
в 1951 г. Дёрингом.
Согласно карбеновой номенклатуре, соединения этого типа
рассматривают как производные простейшего их представите-
ля—метилена, для которого сохраняется это «некарбеновое»
название. В рамках данной системы названия производных ме-
тилена (карбена) строят по обычным принципам «заместитель-
ной» номенклатуры, например дихлоркарбен СС12, этоксикарбо-
нилкарбен CHCOEt, метилхлоркарбен CCIMe, винилиденкарбен
С = С = СН2 (табл. 1.1). Для циклических карбенов с карбено-
вым атомом в цикле используют приставку «карбена», напри-
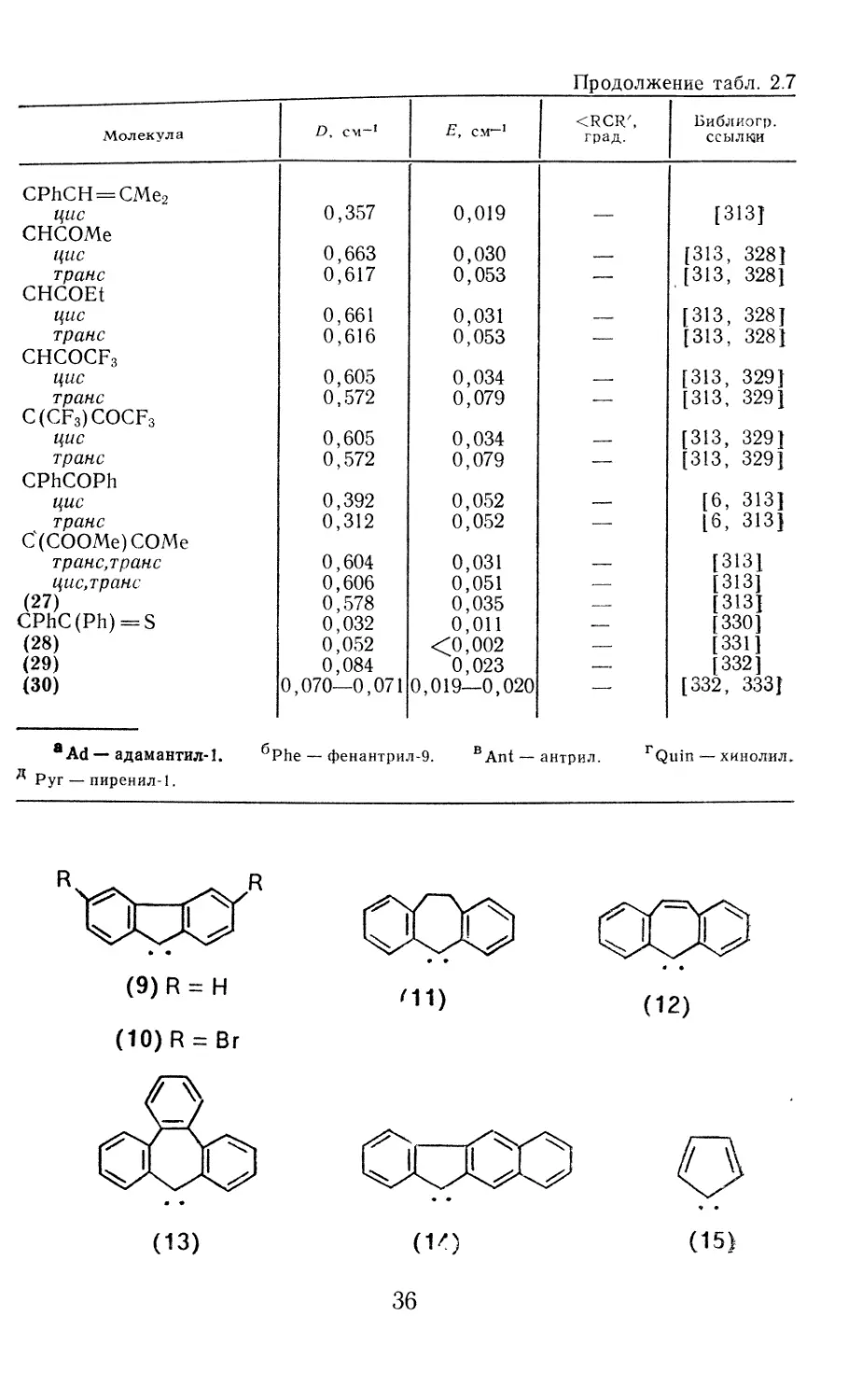
мер, карбенациклопентан (1), 1 -карбенациклогептатриен-2,4,6
(2), 1-карбенациклопентадиен-2,4 (3) и т. п.
(1) (2) (3)
Существенно реже используют «метиленовую» номенклатуру,
согласно которой, например, СС12 называют дихлорметиленом,
CHPh — фенилметиленом и т. п. С учетом триплетности основно-
го состояния метилена предлагали использовать метиленовую
номенклатуру для всех триплетных частиц, а карбеновую сохра-
нить только для синглетных интермедиатов. Однако это предло-
жение, как и любое другое, направленное на учет мультиплетно-
сти в названии частицы, представляется совершенно нереаль-
ным из-за трудности однозначного определения мультиплетности
карбеновой частицы в конкретных условиях химического экспе-
римента, а также легкости протекания синглет-триплетных пе-
реходов для многих из этих интермедиатов.
В тех случаях, когда рассматривается спиновое состояние
карбенов, его следует обозначать словом «синглетный» или
«триплетный», или надстрочным цифровым индексом перед кар-
беновым атомом углерода, или с помощью двух параллельных
или антипараллельных стрелок, например:
синглет: 1СН2 или UCH2; триплет. 3cHPh или HCHPh
12
Таблица 1.1. Названия некоторых карбенов по карбеновой
и илиденовой номенклатурам
Карбен Названия по карбеновой номенклатуре Название по илиденовой номенклатуре
сн2 СС12 CHCOOEt CHEt CHPh CClAr снсн=сн2 с=с==сн2 (1) (3) Метилен Дихлоркарбен Этоксикарбонилкарбен Этилкарбен Фенилкарбен Арилхлоркарбен Винилкарбен Винилиденкарбен 1 -Карбенациклопентан 1-Карбенациклопента- диен-2,4 Метилиден Дихлорметилиден Этоксикарбонилметилиден Пропилиден-1 Фенилметилиден Арилхлорметилиден Пропен-2-илиден-1 Пропадиен-1,2-илиден-1 Циклопентилиден Циклопентадйен-2,4-или- ден-!
Менее удачна дискриминация синглетного и триплетного сос-
тояний карбенов с помощью «спаренных» или «расспаренных»
точек в соответствии с обычным обозначением свободных элек-
тронов. например :СН2— синглетный метилен, »СН2— триплет-
ный.
В последние годы в связи с разработкой ряда проектов по номенклатуре
химических соединений предложен ряд новых вариантов и для названия ос-
новных классов интермедиатов в органической химии, в том числе и для
карбенов. В частности, Комиссией по номенклатуре в органической химии
IUPAC была разработана так называемая Х-конвенция, призванная, по мне-
нию ее авторов, решить общие проблемы номенклатуры молекул (интермедиа-
тов), содержащих тот или иной атом в необычном валентном состоянии [17в].
Согласно ее основным положениям, атом в необычном валентном состоянии,
например карбеновый центр, обозначают символом X", где п — максимальное
число связей у данного атома (для карбенов п = 2, для свободных радикалов
п — 3, для карбен-радикалов — карбинов — /2=1). Этому символу предшеству-
ет цифровой локант в соответствии с местонахождением данного атома. На-
пример, дихлоркарбен СС12 называют дихлор-Х2-метаном, диэтилкарбен (4) —
ЗХ2-пентаном и т. д. Естественно, такая система выглядит весьма надуманной
и вряд ли получит распространение.
СН3 - СН2 - С - СН2 - СН3 (4)
1 2 3 4 5
Позднее той же комиссией предложена унифицированная номенклатура
свободных радикалов и карбенов ГД1- Согласно этой номенклатуре, карбены
и их аналоги рассматривают как «бивалентные радикальные центры, получае-
мые путем удаления двух атомов водорода от моноядерных гидридов СН4,
NH3 и т. д.». В соответствии с этим для названия карбеновой молекулы (не-
зависимо от ее мультиплетности) предлагают использовать окончание диил,
которому предшествует цифра (локант), обозначающая местоположение «би-
радикального» центра в молекуле, например: дихлорметандиил для дихлор-
карбена СС12, 2-метилпентан-3,3-диил для этил (изопропил) карбена (5), проп-
2-ен-1.1-диил для винилкарбена (6) и т. д. Однако такой формализованный
подход к названию производных двухвалентного углерода находится в явном
13
противоречии с их химической природой, так как искусственно объединяются
в общий класс интермедиатов свободные радикалы и карбены, в том числе
синглетные, методы генерирования и свойства которых практически не имеют
ничего общего с таковыми для свободных радикалов.
СН3 У---V
сн3 - сн - с - сн2 - сн3 :сн-сн = сн2 •/ \
1 2 3 4 5 1 <б\
(5) (6) (7)
Более логичной и универсальной является «илиденовая» но-
менклатура, согласно которой карбеновый центр обозначается
окончанием илиден с соответствующим цифровым локантом
(см. табл. 1.1). Согласно этой номенклатуре, которая, кстати,
довольно давно используется для названия интермедиатов с кар-
беновым центром в цикле, рекомендуется, например, СС12 назы-
вать дихлорметилиденом, винилкарбен (6) —пропен-2-илиде-
пом-1, карбен (7) —циклогексилиденом, (3) —циклопентади-
ен-2,4-илиденом-1 (циклопентадиенилиденом) и т. д. (в порядке
исключения допускается использование названий метилен для
СН2, силилен для SiH2 и т.п.).
Таким образом, в соответствии с рекомендациями IUPAC
термин «карбены» следует использовать в качестве общего (ро-
дового) названия для всего класса этих интермедиатов или не-
скольких представителей их, тогда как в конкретных случаях
предпочтительна илиденовая номенклатура. Такая номенклату-
ра, безусловно, оптимальна для циклических карбенов типа
циклоалкилиденов. В то же время, учитывая сложившуюся
к настоящему времени в химической литературе практику, сле-
дует полагать, что для большинства сравнительно простых аци-
клических соединений карбенового типа карбеновая номенкла-
тура будет по-прежнему использоваться и для названия кон-
кретных интермедиатов данного типа.
Рассматривая номенклатуру карбенов, нельзя не остановить-
ся и на довольно широко используемом в химической литерату-
ре термине «карбеноид». Первоначально [2, 16, 17] этот термин
был предложен для названия электронных и химических
аналогов карбенов типа SiRR', GeRR' и т.п., причем между
терминами карбен и карбеноид подразумевалась такая же
связь, как между понятиями металл и металлоид, лантан и лан-
таноид.
Однако впоследствии карбеноидами довольно часто стали
называть различные комплексы карбенов и их лабильные пред-
шественники типа сс-галогенлитийорганических соединений, лег-
ко претерпевающих сс-элиминирование молекулы галогенида
лития с образованием слабого солевого комплекса карбена.
Так, согласно подготавливаемому Комиссией по номенклатуре
14
в органической химии IUPAC «Глоссарию наименований клас-
сов соединений в органической химии» под карбеноидами пред-
полагается понимать «молекулы, которые непосредственно реа-
гируют как карбены или проявляют себя источниками карбе-
нов». Естественно, что такая широкая и нечеткая формулировка
не позволяет установить отличия карбенов не только от их ком-
плексов, но и даже от их предшественников (источников). Бо-
лее того, учитывая высокую склонность синглетных карбенов
и их аналогов к комплексообразованию (см. разд. 2.4.4)., вся
химия карбенов в жидкой фазе в большой мере представляет
собой химию тех или иных комплексов карбенов, обычно не
идентифицируемых и специально не рассматриваемых, посколь-
ку солевые и подобные им слабые карбеновые .комплексы по
химическому поведению принципиально не отличаются от кине-
тически свободных карбенов. В связи с этим во избежание пу-
таницы и неоднозначного толкования лучше всего не употреб-
лять термин карбеноид, используя для обозначения подобных
карбенам соединений других элементов термин аналоги карбе-
нов, а для их комплексов — термин комплексы карбенов (напри-
мер, солевые комплексы карбенов, карбеновые комплексы пе-
реходных металлов).
Что касается номенклатуры аналогов карбенов, то с учетом
рекомендаций IUPAC и сложившейся в химической литературе
практики соединения азота (I) NR следует называть нитренами,
кремния(П) SiRR' — силиленами, германия(П) GeRR' — гер-
миленами, олова(Н) SnRRz— станниленами, свинца (II)
PbRR'— плюмбиленами и т. д.
Глава 2
СТРОЕНИЕ И РЕАКЦИОННАЯ СПОСОБНОСТЬ КАРБЕНОВ
И ИХ АНАЛОГОВ
Посвящается памяти А. К. Мальцева — одного из
основоположников физической химии карбенов
2Л ТЕОРЕТИЧЕСКИЙ И КВАНТОВОХИМИЧЕСКИЙ АНАЛИЗ
СТРОЕНИЯ карбенов
Карбены и их ближайшие аналоги по подгруппе IVB, образуя
только две связи с заместителями, имеют две несвязывающие
орбитали, заселенные двумя электронами. Простая, на первый
взгляд, задача о распределении двух электронов по двум МО не
15
р
Рис. 2.1. Строение граничных МО карбенов и их низшие электронные конфи-
гурации
имеет однозначного решения, и своеобразие строения и реакци-
онной способности карбенов в большой мере основывается
именно на различных вариантах заселения.
Традиционно в химии карбенов несвязывающую орбиталь,
симметричную относительно плоскости, которая проходит через
центральный атом и два его заместителя, называют о-орби-
талью, а антисимметричную МО — р-орбиталью (рис. 2.1); вы-
бирая оси координат, как показано на рисунке, можно сказать,
что в о-орбитали участвуют s- и рх-АО центрального атома, а в
р-орбитали — только р^-АО.
В одноэлектронном приближении карбен может иметь одну
из трех электронных конфигураций: о2, ор или р2. Конфигура-
ция ор может быть синглетной или триплетной (!ор и Зор), пол-
носимметричные* конфигурации о2 и р2 синглетны. При этом
о- и р-орбитали, будучи формально несвязывающими, сравни-
тельно близки по энергии и в то же время отделены значитель-
ной энергетической щелью от занятых и вакантных скелетных
МО. Поэтому при конфигурационном взаимодействии наибо-
лее существенно смешивание конфигураций ]о2 и 1р2, приводя-
щее к двум новым состояниям — низшему с энергией 1Е~ и воз-
бужденному с энергией 1£+. Если учесть, что вследствие спин-
орбитального взаимодействия конфигурация Зор всегда предпо-
чтительнее соответствующего синглета ^р, то энергии четырех
низших электронных состояний карбена, согласно [4], могут
располагаться в соответствии с одной из трех последователь-
ностей**:
1Е^3Еор^Еор^Е+ (2.1)
* Справедливо для изогнутой молекулы, где точечная группа симметрии
не выше C2v (нет вырожденных неприводимых представлений). В случае ли-
нейной молекулы понятия п- и р-орбиталей теряют смысл, так как для них
выделенная па рис. 2.1 плоскость ху отсутствует.
** Неравенства (2.1) —(2.3) справедливы только для фиксированного рас-
положения ядер. Поскольку для каждой конфигурации оптимальное положение
ядер, вообще говоря, неидентично, то реальные последовательности электрон-
ных состояний могут отличаться от (2.1) — (2.3). Впрочем, электронные пере-
ходы тоже рассматриваются в адиабатическом приближении, т. е. для них
соблюдаются зависимости (2.1) — (2.3).
16
3Eqp 1^OP (2.2)
3Еор^Еор^Е^<Е+ (2 3)
Существенно, что для карбенов, описываемых последова-
тельностью (2.1), основное состояние будет синглетным, а пер-
вое возбужденное — триплетным, в то время как при реализа-
ции последовательностей (2.2) или (2.3) основное состояние
триплетно, а все ближайшие возбужденные — синглетны.
Очевидно, триплетные карбены по свойствам должны быть
близки к свободным радикалам, хотя из-за наличия двух полу-
заполненных МО вместо одной они более реакционноспособны.
В то же время реакционная способность синглетных карбенов
двойственна: неглубокая по энергии дважды занятая МО при-
дает им нуклеофильные, анионоподобные свойства, а вакантная
МО невысокой энергии — свойства электрофильные, катионо-
подобные. Поскольку карбен в целом электронейтрален, то энер-
гия его верхней занятой МО (ВЗМО) должна быть ниже, чем
у соответствующих анионов; аналогично, энергия нижней сво-
бодной МО (НСМО) должна быть выше, чем у соответствую-
щих катионов. Это предопределяет, с одной стороны, умерен-
ную (по сравнению с катионами и анионами в газовой фазе)
реакционную способность карбенов, а с другой стороны, — зна-
чительную чувствительность энергии и электронной плотности
на обеих граничных МО к влиянию заместителей, иными слова-
ми, большее разнообразие всех свойств карбенов.
В первом приближении карбены, для которых доминирует
взаимодействие НСМО карбена с занятыми МО второго реаген-
та, можно считать электрофильными. Если же ведущую роль
играют взаимодействия ВЗМО карбена с вакантными МО реа-
гента, то такой карбен является нуклеофильным. В отсутствие
выраженной электрофильности или нуклеофильности карбен
считается амбифильным. Разумеется, «фильность» карбена — ха-
рактеристика не абсолютная; она' зависит от второго реагента.
Наиболее разработана классификация карбенов по их «филь-
ности» [18] для реакций присоединения по кратной связи (под-
робнее см. гл. 3 и 4), для которых кроме количественной оценки
фильности карбенов предложены и качественные тесты. Приме-
ром является реакция с 6,6-диметилфульвеном [19], в котором
эндоциклические связи — акцепторы нуклеофильных карбенов,
а экзоциклическая связь — акцептор электрофильных карбенов.
Расчет строения карбенов. На ранних этапах развития химии
карбенов такие характеристики этих частиц, как мультиплет-
ность основного состояния, положение граничных орбиталей
и т.п., анализировали в рамках различных полуэмпирических
2—107
17
Spliner
схем господствовавших в теоретической opi анической химии.
Та к,’в классической работе Р. Хоффмана и сотр. [20] предпо-
лагалось, что стабилизация синглетного основного состояния
дигалогенкарбенов может быть связана с делокализацией непо-
деленных электронных пар галогенов на вакантную р-орбиталь
карбена. Для ряда циклоалкилиденов было предсказано [21]
понижение энергии синглетного состояния по мере уменьшения
размера цикла и показано, что для непредельных циклических
карбенов с числом л-электронов 4п + 2 должно стабилизировать-
ся синглетное состояние, тогда как в случае 4п л-электронов
в ненасыщенном фрагменте карбена в сопряжение дополнитель-
но вовлекается один из электронов карбенного центра, т. е. ста-
билизируется триплетное состояние. Примером первого случая
является циклопропенилиден (ц = 0), а второго — циклопента-
диенилиден (и=1). Подобные предсказания основаны на свой-
ствах симметрии МО, и их качественная справедливость бес-
спорна. Однако в настоящее время ясно, что получаемые из
полуэмпирических расчетов структурные характеристики следу-
ет рассматривать лишь как оценочные или систематизирующие,
поскольку они более или менее корректно передают не абсолют-
ные значения, а лишь тенденции, и то в определенных рядах.
В то же время сейчас для сравнительно несложных карбе-
нов, содержащих 5—6 тяжелых атомов, существуют высокока-
чественные неэмпирические расчеты (НЭР), позволяющие адек-
ватно и полностью описать всю молекулу в целом* и, в частно-
сти, воспроизвести такие доступные эксперименту характеристи-
ки, как геометрическое строение, электронный спектр, потенци-
ал ионизации, мультиплетность основного состояния и т. д.
Это позволяет для труднодоступных экспериментально карбенов
рассматривать расчетную информацию как заслуживающую
высокой степени доверия. Разумеется, речь идет только о НЭР
в достаточно полном базисе и (что особенно существенно для
карбенов — молекул с открытой оболочкой) с учетом конфигу-
рационного взаимодействия; при этом дальнейшее расширение
базиса не должно существенно менять рассчитываемые харак-
теристики. Расчеты же в минимальном базисе ОСТ-ЗГФ не име-
ют преимуществ перед современными полуэмпирическими схе-
мами типа МЧПДП/З и МПДП.
Надежные НЭР в настоящее время возможны для систем
с 10—15 атомами (из них до 5—6 тяжелых), которые представ-
ляют собой также основные объекты экспериментальной физи-
ческой химии карбенов, поскольку дают сравнительно легко
интерпретируемые спектры. Так, экспериментально наиболее
изученные трехатомные карбены и их аналоги, в первую оче-
редь метилен, являются излюбленным объектом для оценки точ-
* О точности НЭР в описании строение органических молекул см. [22].
18
ности любого нового расчетного подхода. Не имея возможности
остановиться здесь на многочисленных (несколько сотен) рас-
четах, мы ограничимся ссылкой на обзоры [23, 24].
Конкретные примеры высококачественных неэмпирических
расчетов карбенов и их аналогов использованы ниже при об-
суждении строения отдельных молекул. Здесь же отметим лишь
несколько моментов принципиального характера. Так, да-нные
НЭР, по мнению Харрисона и сотр. [25], ставят под сомнение
сделанный Р. Хоффманом в 1968 г. вывод о доминирующем
влиянии л-донорной способности заместителя на строение кар-
бена [20]. По-видимому, ведущую роль здесь играет электроот-
рицательность заместителей, причем донорные группы стабили-
зируют триплетное, а акцепторные — синглетное состояние; роль
же л-эффектов вторична. Не укладываются в рамки концепции
«л-сопряжения» и результаты расчета циклопентадиенилидена
в двухэкспоненциальном базисе сгруппированных гауссовых
функций [26].
Неожиданные результаты принесли недавние МИДИ-расче-
ты циклогептатриенилидена (1) [27, 28]. Оказалось, что на по-
верхности потенциальной энергии (ППЭ) системы С7Н6 струк-
тура (1) является не минимумом, а седловой точкой* и соответ-
ствует переходному состоянию энантиомеризации циклогептате-
траена (2)—напряженного циклического аллена с £акт, равной
96 кДж/моль [27]:
(2а) (1) (26)
Было отмечено, что все известные реакции, приписываемые
промежуточному участию синглетного циклогептатриенилиде-
на (1), с тем же успехом можно рассматривать как реакции,
протекающие с участием аллена (2) (см. также [29] и
разд. 2.2.4). Подобным образом с помощью метода МПДП,
правда без анализа всей ППЭ, показано [30], что в соответст-
вии с данными о химическом поведении циклический аллен (3)
стабильнее, чем карбен (4) или цвиттер-ион (5).
MeN ц
MeN
(4) •
R— Н,Ме
* Матрица вторых производных энергии по внутренним степеням свободы
Для структуры (1) имеет одно отрицательное собственное значение. Можно от-
метить, что геометрия и энергетика экстремумов на ППЭ довольно сильно за-
висят от метода расчета, однако топология ППЭ сравнительно устойчива.
2‘ 19
Аналогично для системы С2Н4 по данным неэмпирического
расчета в расширенном базисе с поляризационными функция-
ми и учетом электронной корреляции вплоть до четвертого по-
рядка теории возмущений структура, отвечающая синглетному
метилкарбену, не является локальным минимумом на ППЭ
и безактивационно переходит в этилен [31]. Эти данные ставят
под сомнение существование синглетного метилкарбена в каче-
стве кинетически независимой частицы (ср. [13]).
Приведенные примеры показывают, что промежуточное уча-
стие даже, казалось бы, привычных карбенов необходимо дока-
зывать физическими методами. Вследствие этого данные теоре-
тического расчета следует считать достоверными либо для
структуры, зафиксированной инструментальными методами,
либо в случае идентификации ее на ППЭ в виде минимума, т. е.
при расчете не только первых, но и вторых производных*. Есте-
ственно, подобная трудоемкая процедура возможна лишь для
небольших молекул. То же относится и к расчету карбенных
реакций, где также необходимо определение профиля ППЭ.
Б этой связи неудивительно практически полное отсутствие
высококачественных НЭР карбенных реакций, за исключением
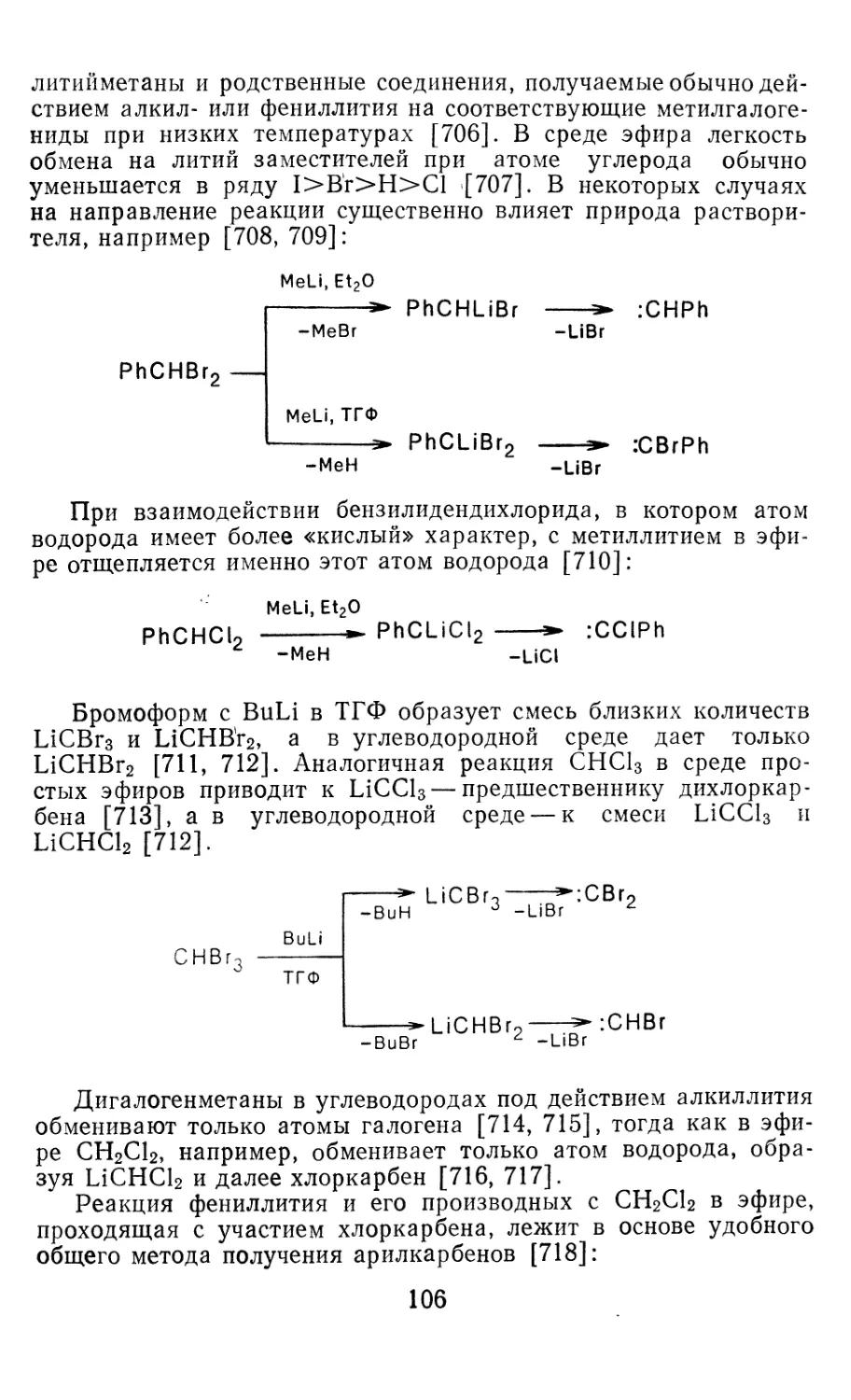
систем СН2 + Н2 и СН2+СН4 (см. ниже). Данные полуэмпири-
ческих расчетов и НЭР в небольших базисах для всех типов
карбенных реакций рассмотрены в разд. 2.4 и использованы при
обсуждении механизмов соответствующих реакций.
2.2. ИССЛЕДОВАНИЕ КАРБЕНОВ
ИНСТРУМЕНТАЛЬНЫМИ МЕТОДАМИ
В настоящее время выполнено большое число исследований
карбенов и их аналогов физическими методами, которые дают
возможность обнаружения и прямого изучения этих реакцион-
носпособных частиц [32, 33]. Применение инструментальных
методов к этим объектам затруднено из-за высокой реакцион-
ной способности карбенов, определяющей их малое время жиз-
ни (обычно 10~34-10~6 с) и низкую текущую концентрацию
в условиях обычных химических реакций. Поэтому для достиже-
ния достаточных для обнаружения концентраций карбенов
обычно применяют импульсные методы или методы, связанные
с предотвращением быстрого исчезновения частиц в результате
вторичных реакций.
Импульсные методы — импульсный фотолиз, метод ударной
волны, импульсный электрический разряд — создают высокую
* Методы оптимизации геометрии, как правило, основаны на сгонке к ну-
лю градиента полной энергии, что возможно не только в минимумах (все соб-
ственные значения матрицы вторых производных положительны), но и в дру-
гих экстремумах, в частности, в седловых точках (одно отрицательное собст-
венное значение).
20
Таблица 2.1. Структурные параметры карбенов
и их аналогов MRR' в основном состоянии
Молекула Терм основ- ного состоя- ния <RMR/, град. Библиогр. ссылки
сн2 “В, 0,1075—0,1085 133,9—136 [36—40]
cf2 ‘At 0,1300—0,1304 104,78 [34, 41—45]
CHF ‘A' 0,1120(0,1314) 101,8 [36, 46—48]
CC12 ‘At 0,170—0,1758 108 [32, 34, 49—53]
СНС1 0,1119—0,1120 (0,1689—0,1696) 101 — 103 [46, 52, 54, 55]
CFC1 ‘A' 0,130(0,170) 105 [56—58]
CBr2 0,187—0,1907 109—110 [34, 59—61]
CI2 Mi 0,212a 110* [62, 63]
SiH2 Mt 0,1516 92,1 [64—67]
SiF2 ‘Ai 0,1590—0,1594 100,88 [34, 68—81]
SiHF 0,152a (0,160)a 100* [62, 82]
SiCl2 0,2044—0,2083 102,8—105 [34, 35, 83—85]
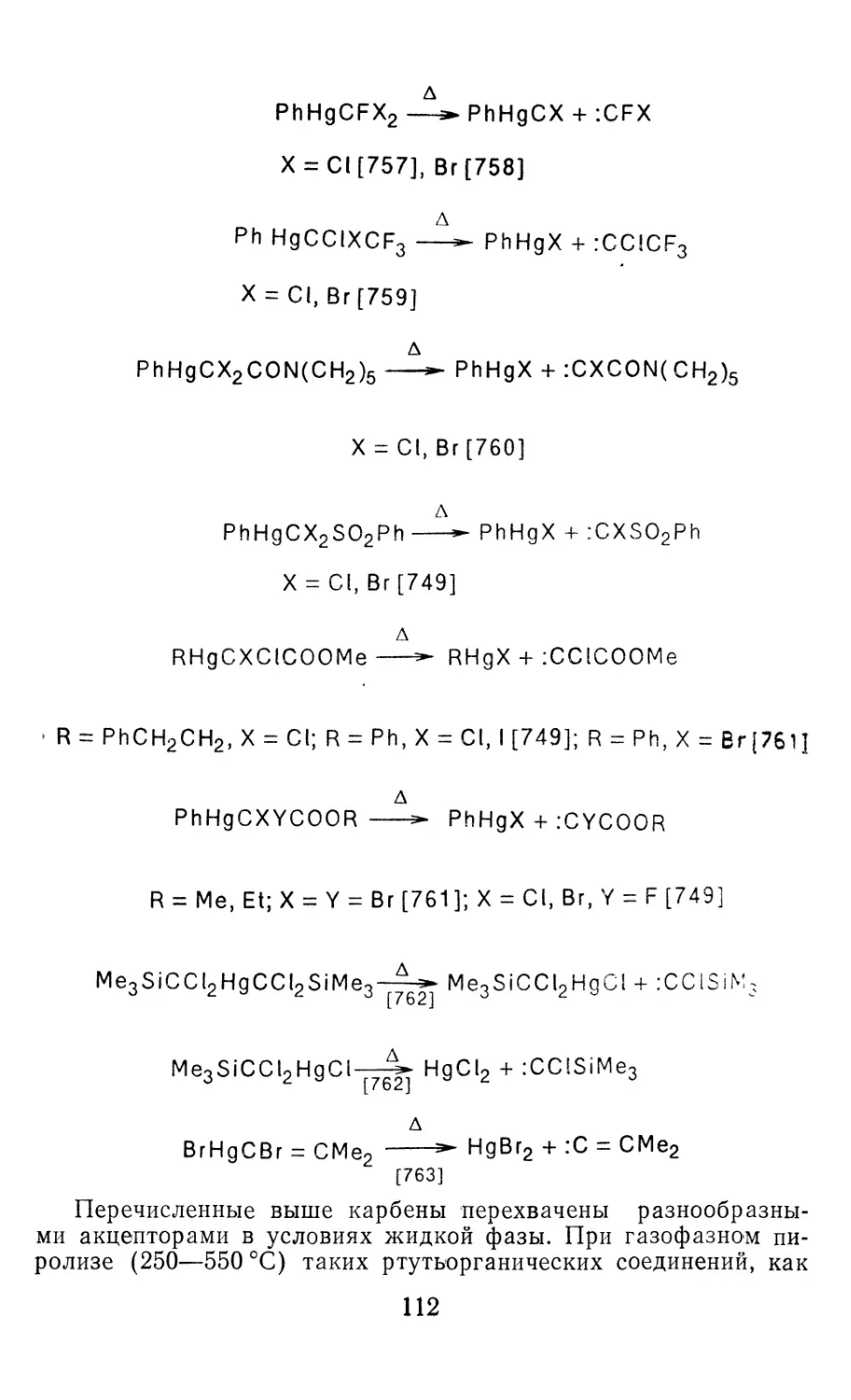
SiHCl ‘A' 0,1561(0,2064) 102,8 1 186]
SiFCl ‘A' 0,160a (O,2O5)a 100* [62]
SiBr2 ‘At 0,2194—0,2243 102,7—105 [34 35]
SiHBr lA' 0,156(0,2231) 102,9 [87]
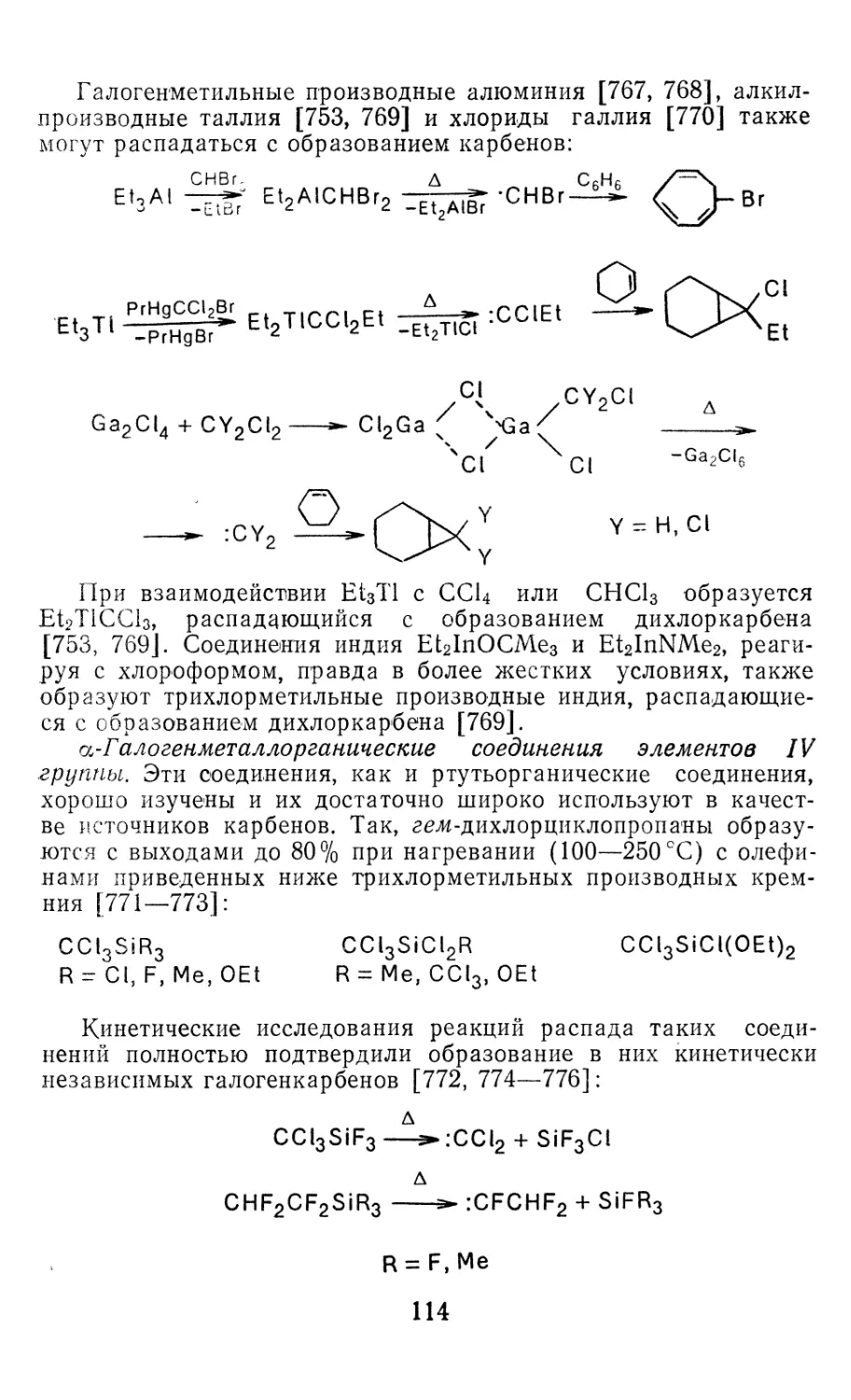
Sil2 Ai 0,244a 110a [62]
SiHI lA' 0,156a(0,2451) 102,7 [82]
SiHOH ‘A' — 113—114,5 [88]
GeH2 ‘At 0,160* 90* [89]
GeF2 ‘Ai 0,1732 97,2 [90—94]
GeCl2 ‘Ai 0,2186 100,4 [95, 96]
GeBr2 ‘A-, 0,2337 101,2 [34, 97]
GeHBr 'A' 0,153(0,230) 100 [98]
SnF2 ’Ai 0,1893—0.196 94—96 [34, 99]
SnCl2 ‘A, 0,2342—0,2346 98,5—99 [34, 100]
SnBr2 ‘Ai 0,2497—0,264 100,5 [34, 101]
Snl2 ‘At 0,273* 95* [102, 103]
(6)6 ‘At 0,209 96 [Ю4]
PbF2 ’A. 0,1989—0,203 90—95 [34, 99, 105]
PbCl2 ‘Ai 0,2444—0,2445 98,3—98,5 [34, 99, 102, 106]
PbBr2 ‘Ai 0,2594 100 [34]
Pbl2 ‘At 0,279* 95* [99, 102]
„ Л I I
а Оценочное значение. о8п[КСМе2(СН2)зСМе2]2 (6).
мгновенную концентрацию нестабильных частиц путем кратко-
временного мощного энергетического воздействия на молекулы
их предшественников, что позволяет регистрировать эти частицы
с помощью быстродействующих (^10~5 с) приборов (спектро-
графы, ИК-спектрометры быстрого сканирования) до того, как
они исчезнут в результате вторичных реакций.
Другой подход основан на предотвращении исчезновения
карбенов по вторичным реакциям путем уменьшения числа их
активных столкновений. Это достигается генерированием и ана-
лизом нестабильных частиц в высоком вакууме или разбавле-
21
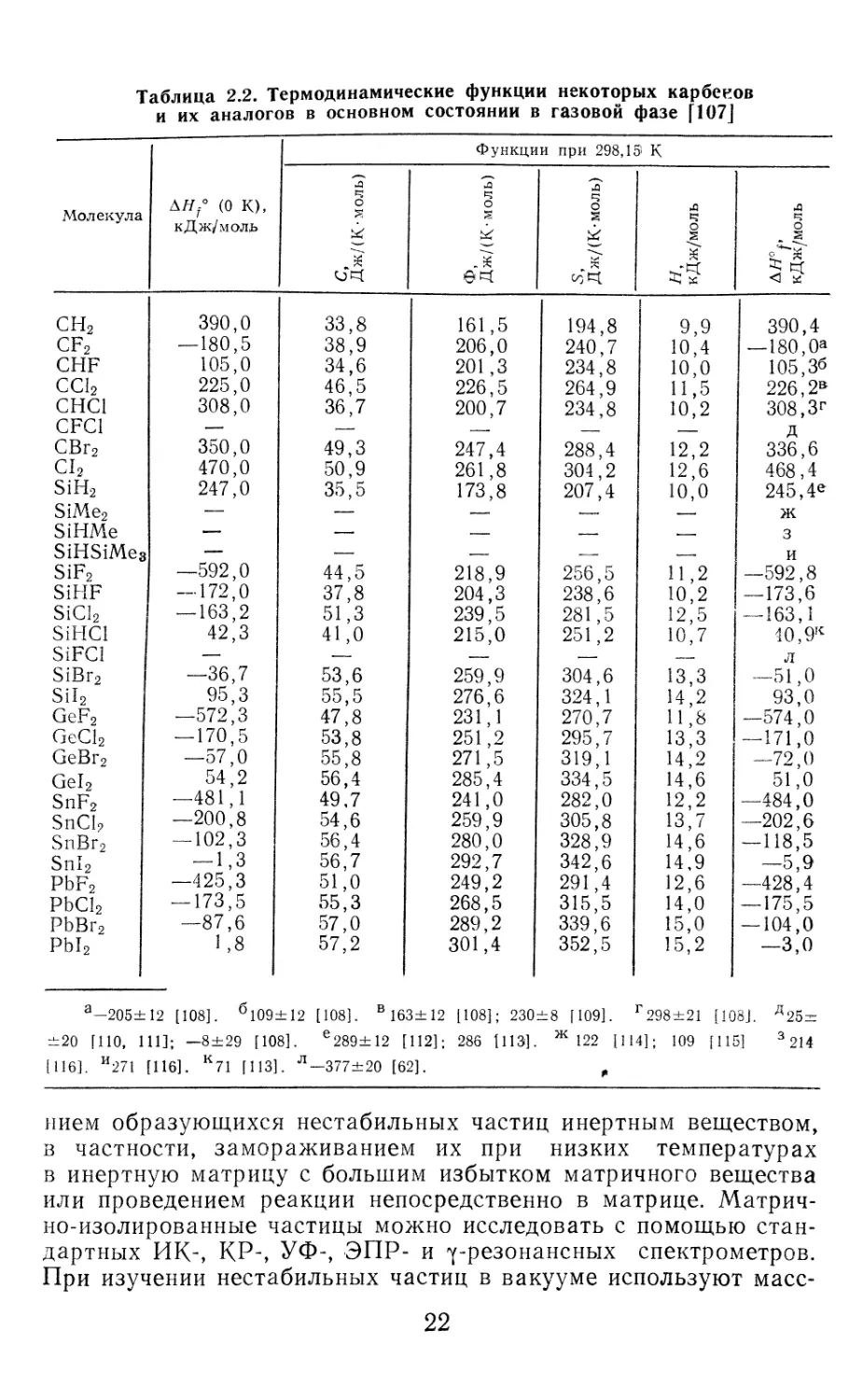
Таблица 2.2. Термодинамические функции некоторых карбенов
и их аналогов в основном состоянии в газовой фазе [107J
Молекула (о K), кДж/мОЛЬ Функции при 298,15' К
л ч о £ 1 ф, Дж/(К-моль) л О н, кДж/моль кДж/моль
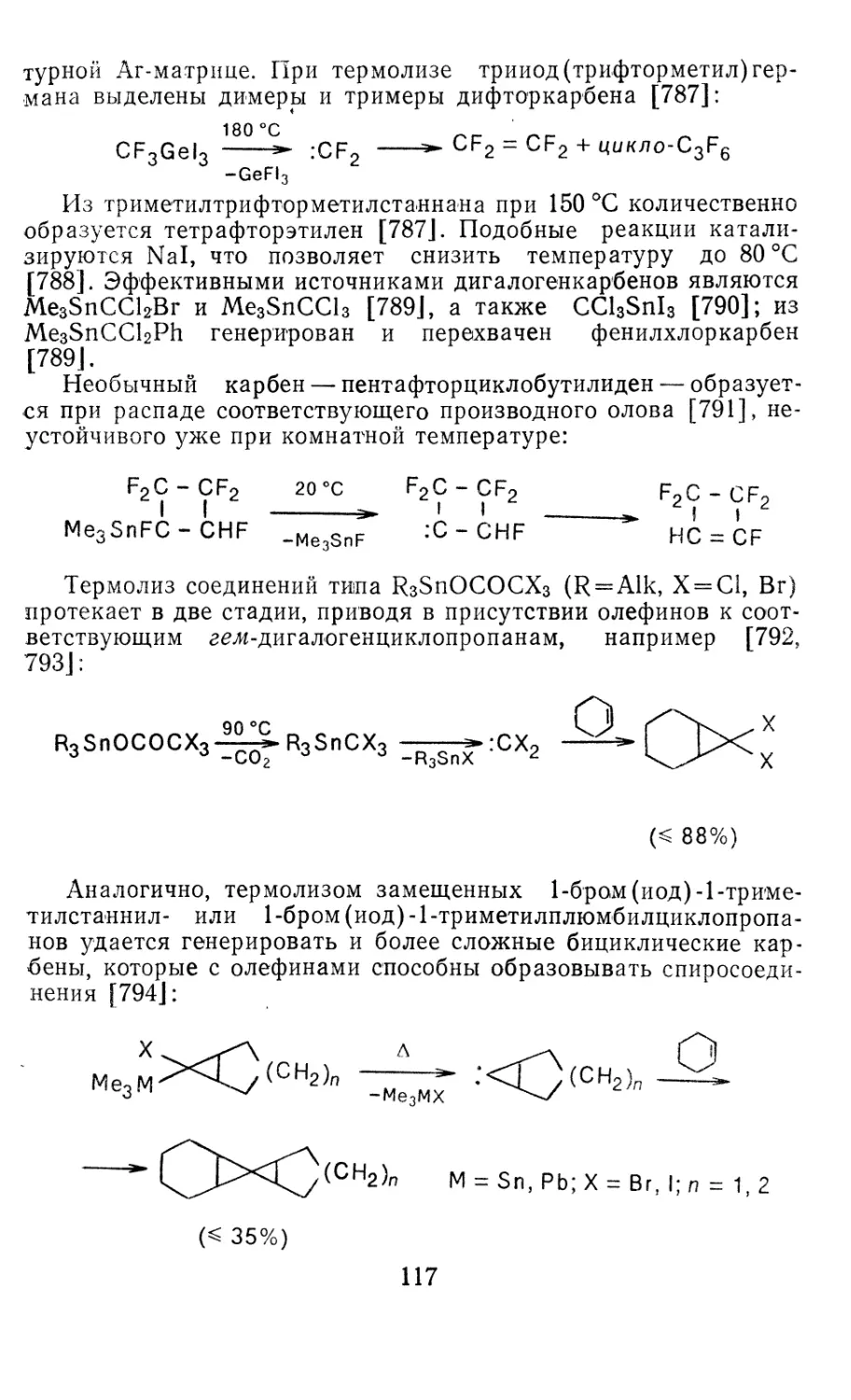
сн2 390,0 33,8 161,5 194,8 9,9 390,4
cf2 — 180,5 38,9 206,0 240,7 10,4 — 180,0*
CHF 105,0 34,6 201,3 234,8 10,0 105,36
СС12 225,0 46,5 226,5 264,9 11,5 226,2В
СНС1 308,0 36,7 200,7 234,8 10,2 308,Зг
CFC1 — — — Д
СВг2 350,0 49,3 247,4 288,4 12,2 336,6
С12 470,0 50,9 261,8 304,2 12,6 468,4
SiH2 247,0 35,5 173,8 207,4 10,0 245,4е
SiMe2 — —. —. — —. ж
SiHMe — — — — , 3
SiHSiMeg — — — —. и
SiF2 —592,0 44,5 218,9 256,5 И,2 —592,8
SiHF — 172,0 37,8 204,3 238,6 10,2 — 173,6
SiCI2 — 163,2 51,3 239,5 281,5 12,5 — 163,1
SiHCl 42,3 41,0 215,0 251,2 10,7 40,9К
SiFCl — —- — — — л
SiBr2 —36,7 53,6 259,9 304,6 13,3 —51,0
Sil2 95,3 55,5 276,6 324,1 14,2 93,0
GeF2 —572,3 47,8 231,1 270,7 И, 8 —574,0
GeCl2 — 170,5 53,8 251,2 295,7 13,3 — 171,0
GeBr2 —57,0 55,8 271,5 319,1 14,2 —72,0
Gel2 54,2 56,4 285,4 334,5 14,6 51,0
SnF2 —481,1 49,7 241,0 282,0 12,2 —484,0
SnCl? —200,8 54,6 259,9 305,8 13,7 —202,6
SnBr2 — 102,3 56,4 280,0 328,9 14,6 — 118,5
Snl2 — 1,3 56,7 292,7 342,6 14,9 —5,9
PbF2 —425,3 51,0 249,2 291,4 12,6 —428,4
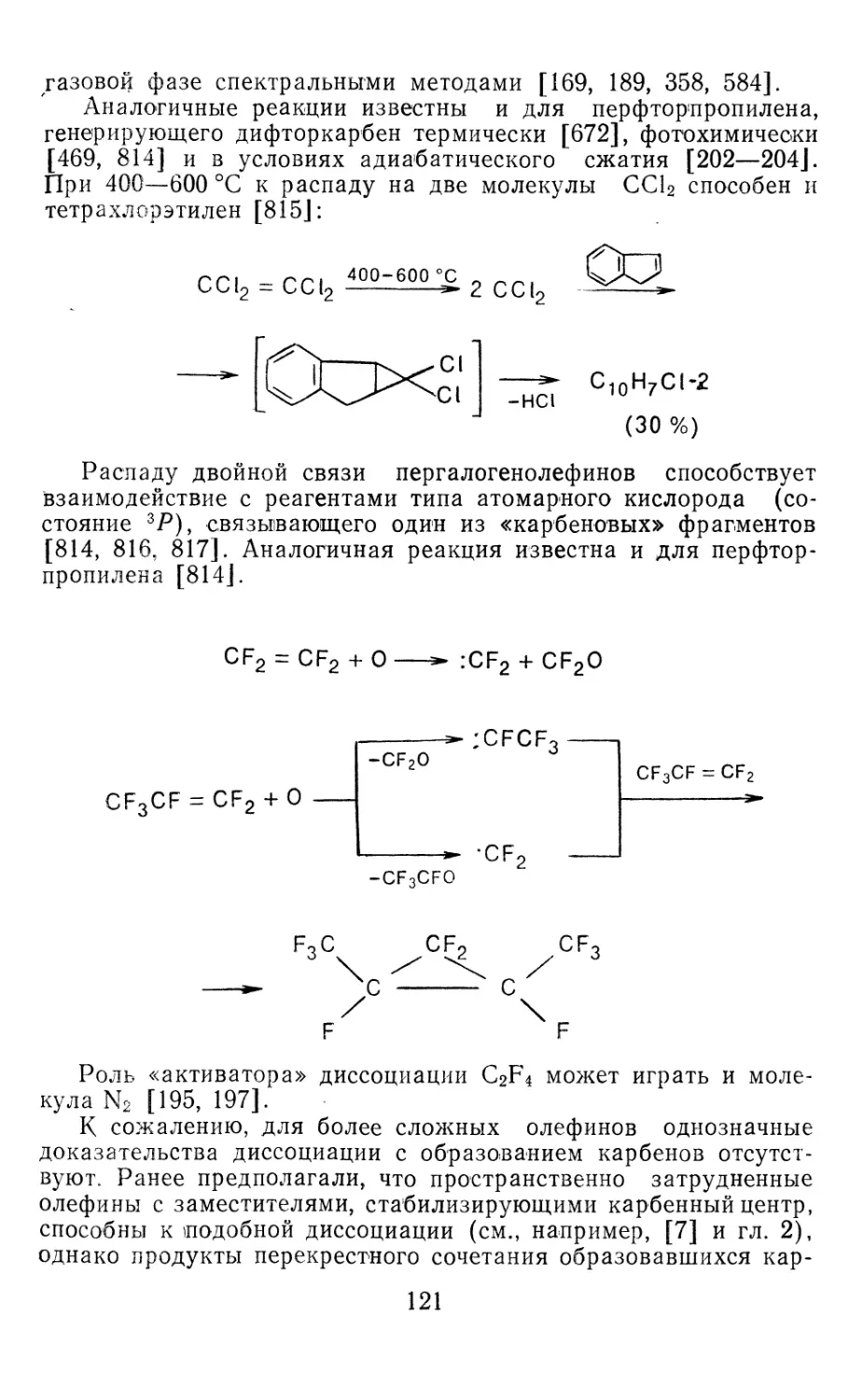
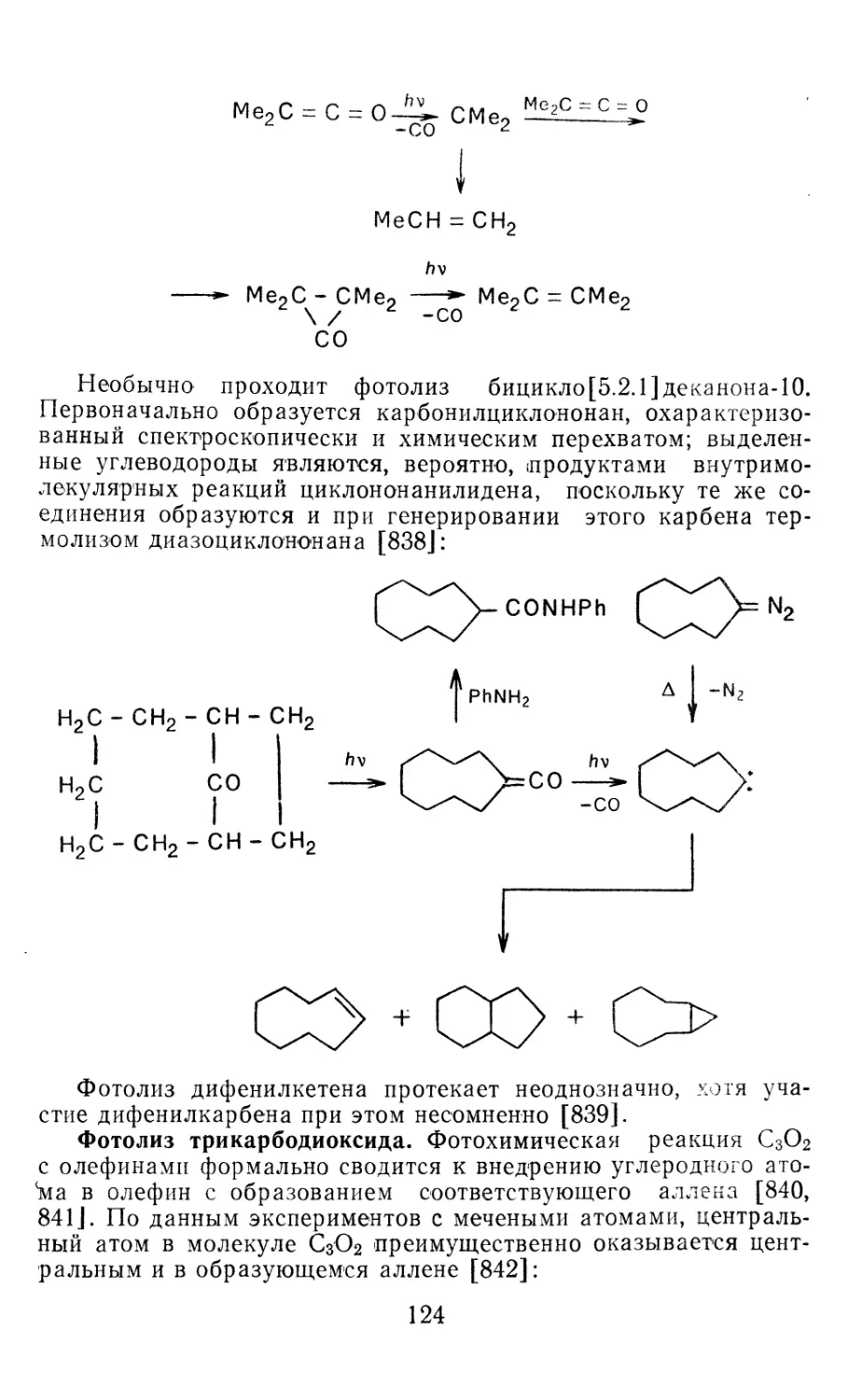
PbCl2 — 173,5 55,3 268,5 315,5 14,0 — 175,5
PbBr<? —87,6 57,0 289,2 339,6 15,0 — 104,0
Pbl2 1 ,8 57,2 301,4 352,5 15,2 —3,0
а—205±12 [108]. б109±12 [108]. в 163± 12 [108]; 230±8 [109]. г298±21 [108]. д25т=
±20 [ПО, 111]; —8±29 [108]. е289±12 [112]; 286 [113]. Ж 122 [114]; 109 [115] 3 214
[116]. И271 [116]. К71 [ИЗ]. Л—377±20 [62]. ,
нием образующихся нестабильных частиц инертным веществом,
в частности, замораживанием их при низких температурах
в инертную матрицу с большим избытком матричного вещества
или проведением реакции непосредственно в матрице. Матрич-
но-изолированные частицы можно исследовать с помощью стан-
дартных ИК-, КР-, УФ-, ЭПР- и ^-резонансных спектрометров.
При изучении нестабильных частиц в вакууме используют масс-
22
Таблица 2.3. Первые потенциалы ионизации некоторых карбенов
и их аналогов в основном состоянии
Молекула ПИ, эВ Библиюгр. ссылки Молекула ПИ, эВ Библиогр. ССЫЛКИ
сн2 10,40 [36, 117— 126] GeCI2 10,2—10,4 [95, 139, 140]
cf2 11,8 [НО, 120— 122] GeBr2 SnF2 9,5 11,5 [139] [140]
CFC1 10,8 [НО, 127] SnCl2 10,2 [141]
СС12 9,10—9,76 [109, 128] SnBr2 10,0 [141]
СВг2 10,0±0,2 [62, 129, 130] PbFo PbCl2 11,6 10,3—11.2 [140] [142, 143]
СНВг 11,7±0,4 [130, 131] PbBr2 10,2 [143]
SiF2 11,0—11,3 [132—135] (7p 6,90 [104]
SiCl2 GeF2 10,0—10,6 11,6—11,8 [136, 137] [94, 132, 138] (6) 6,80 [104]
а Ge[NCMe2(CH2)3CMe2]2 (7).
спектрометрию или фотоэлектронную и флуоресцентную оптиче-
скую спектроскопию.
Указанные методы позволяют не только регистрировать при-
сутствие карбенов или подобных им нестабильных частиц, но
и определять такие важные характеристики их, как энергии
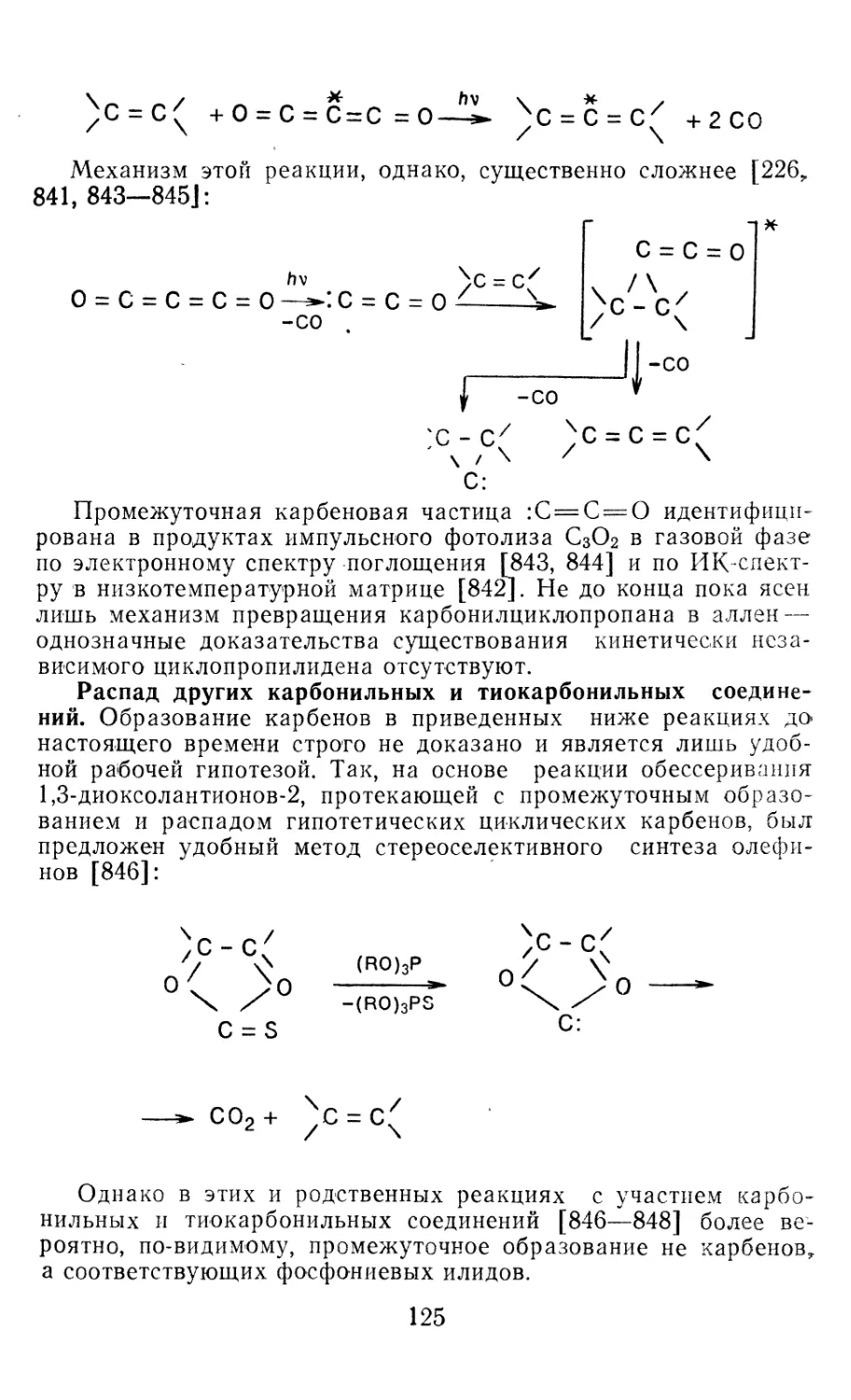
связей и электронных переходов, частоты колебаний, геометри-
ческие параметры, стандартные энтальпии образования, потен-
циалы ионизации (ПИ), а также кинетические константы реак-
ций образования и исчезновения карбенов. Правильное сочета-
ние объектов исследования с оптимальными условиями генери-
рования позволяет применять для структурных исследований
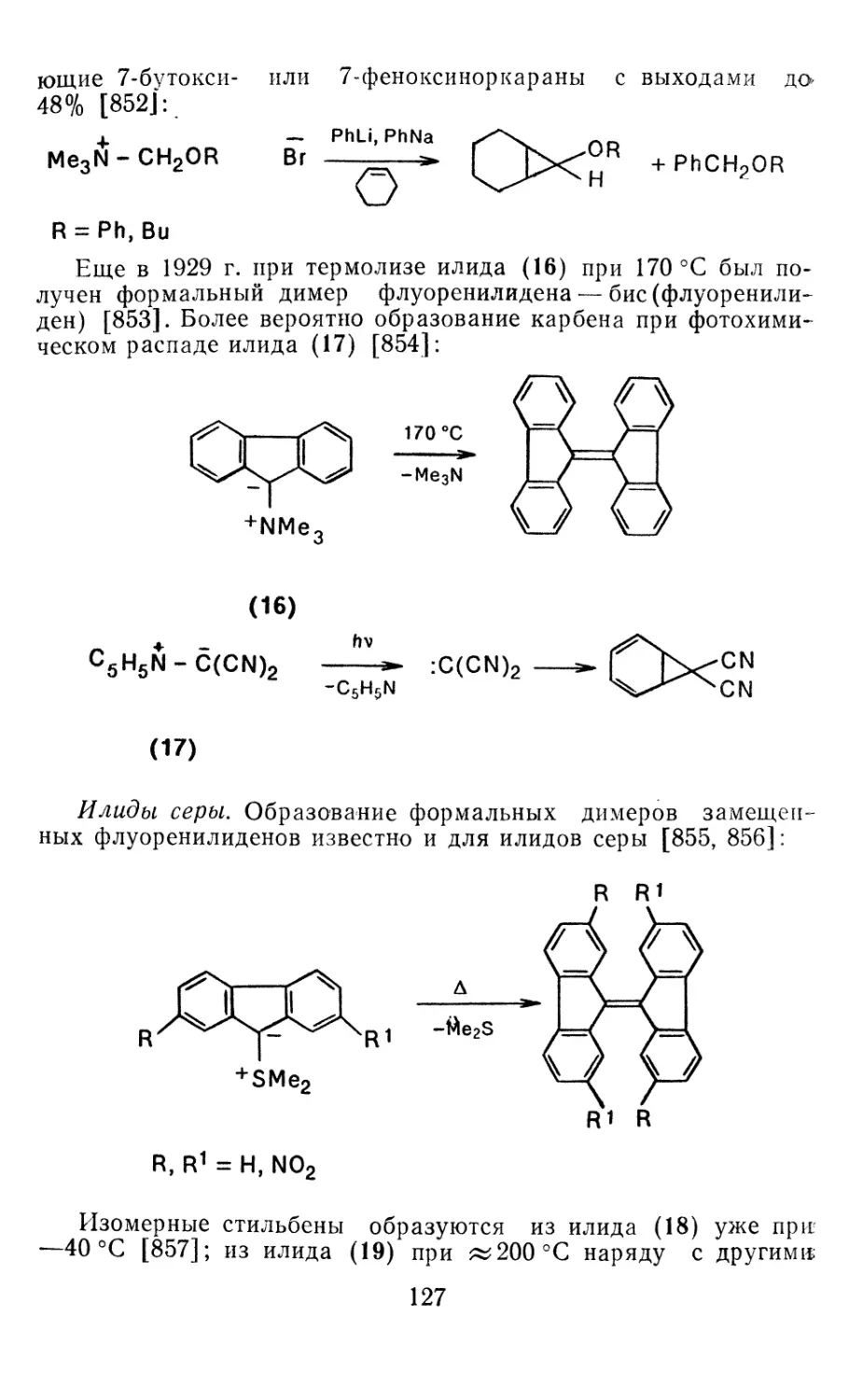
карбеноподобных молекул и других нестабильных частиц даже
газовую электронографию (в сочетании с квадрупольной масс-
спектрометрией) [34, 35].
Информация, полученная из анализа экспериментальных
данных по исследованию карбенов и их аналогов физическими
методами, суммирована в табл. 2.1—2.6 в виде сводки рекомен-
дуемых значений.
Ниже кратко рассмотрены основные методы генерирования
карбенов и их аналогов с целью изучения их строения различ-
ными физическими методами.
2.2.1. Импульсные методы получения карбенов
и регистрация их спектров поглощения
Импульсные методы позволяют создать высокую мгновенную
концентрацию нестабильных высокореакционноспособных ча-
стиц путем кратковременного мощного энергетического воздей-
23
Таблица 2.4. Энергия возбужденных состояний
некоторых трехатомных карбенов и их аналогов
Молекула Терм iBO3- бужДейнюго состояния Энергия возбужде- ния из основного состояния, эВ Библиогр. ссылки
СНо Mi 0,39 [144]
1,31 [36, 67]
cf2 зв. 1,86 [62]
4,62—5,0 [41, 53, 145]
CHF 0±0,6 [146]
*4" 2,14 [46]
СС12 8в, 1,49 [1.62]
’В, 2,16—2,5 [1, 147, 148}
СНС1 М" 0,99 [62]
1Д/7 1,52 [62]
CFC1 М" 1,61 [62]
1Д/7 3,11 [56—58]
CBr2 8в, 1,24 [62]
в, 1,85 [61]
С12 зв, 1,7* [62]
'В, 1,7* [62]
SiH. ЗВ, 0,2—0,4 [149, 150]
>В, 1,93 [64—66]
SB2 3,0* [149
SiHF гЛ" 0,62 [62
’4" >2.5 [62
SiHCl 'Л" 2,57 [86
SiHBr 'А" 2,47 [86
SiHI М" 2,26 [15Г
SiF2 ®В, 3,26 [71. 152]
!В, 5,47 [64, 73, 74, 153]
ЗВ2 7,71 [62]
SiCl2 8В, 2,84 [101, 51]
‘В, 3,61—3,94 [51, 154, 155]
SiFCl 8Л" >2,5 [62]
SiBr2 8В, и ‘В, >2,5 [62]
Sil2 8В, и «В, >2,5 [62]
GeF2 8В, 3,72 [156]
’В, 5,44 [157]
8В2 8,43 [72, 134]
GeCl2 3В, 2,77 [51, 158]
>В, 3,84 [51, 158}
триплет 2,17 [159]
SnF2 8В, 3,47 [62]
,В1 5,05 [160]
SnCI2 8В, 2,76 [154, 161]
‘В, 3,85—4,00 [51, 155}
PbF2 ЗВ, 3,47а [62]
в. 5,03 [160]
PbCl2 3В, 2,72а [62]
’В, 3,84 [51, 155]
а Оценочное значение.
24
Таблица 2.5. Структурные параметры некоторых трехатомных карбенов
и их аналогов MRR' в возбужденных состояниях
Молекула Терм возбужден- ного состояния ГМ.-R (rM-r' )’ нм <RMRZ, град. Библиогр. ссылки
сн2 Mi 0,111 102—104 [36, 38, 67, 162]
‘В, 0,106 140 [36, 67]
CHF Ч" 0,112 102,2 [140]
(0,1537)
3AZ/ 0,1077 120,4а [146, 148]
(0,1321)
cf2 0,1305 134,3 [95, 139]
СНС1 M" 0,1075 123,3 [148]
(0,1735)
ссь 3B; 0,1730 125,5 [148]
СВК зв{ 0,1746 1506 [60]
SiH2 зв{ 0,155в 123,5В [149, 150]
1В{ 0,149 122—123 [64—66]
SiHCl 0,148 116,1 [86]
(0,205)
SiHBr Ч" 0,150 116,6 [80]
(0,221)
SiHI М" 0,150 116,2 [151]
(0,243)
SiF2 3В1 0,171в 114,9В [71, 152]
хв, 0,160 115,9 [64, 73, 74, 153[
GeF£ 3Bi — 111,6 [156]
аДанные НЭР. ^Данные ненадежны (см. [33]). вОценочное значение.
ствия (фотолиз, электрический разряд, ударная волна и т.п.) на
соответствующие стабильные предшественники, пригодные, как
правило, для генерирования карбенов и в «нормальных» усло-
виях (диазосоединения, диазирины, органогалогениды и т.п.;
см. гл. 3). Выбранный источник должен генерировать карбено-
вую частицу достаточно селективно, поскольку ее спектроскопи-
ческая идентификация и без того сопряжена со значительными
экспериментальными трудностями, обусловленными малым вре-
менем жизни активных интермедиатов. В качестве регистрирую-
щих приборов используют быстродействующие (<10-5 с)
спектрографы, ИК-спектрометры быстрого сканирования и т.п.
До последнего времени главными объектами физических
исследований служили трехатомные карбены, в первую очередь
сам метилен, генерируемые и исследуемые в основном в газо-
вой фазе. Как раз с целью обнаружения метилена в 1947 г. был
предложен [163] широко распространенный в настоящее время
метод импульсного фотолиза, хотя только в 1959г.
удалось зафиксировать спектр метилена в УФ-области, а пра-
вильная интерпретация этого спектра была дана лишь в 1971г.
25
Таблица 2.6. Полосы поглощения некоторых ароматических карбенов
в жидкой фазе при ~20 С [,170—177]
Карбен, Растворитель -макс- нм
3CPh2 Ацетонитрил 295, 266а
3Флуоренилиден То же 470
3СНС10Н7-2 «-Пентан 362
!СС1С6Н5 Изооктан 310
1CClC6H4CF3-4 То же 298
1СС1С6Н4С1-4 » 320
!СС1С6Н4Ме-4 » 317
!СС1С6Н4ОМе-4 » 340
’CBrPh » 330
а Спектр лазерно-индуцированной флуоресценции.
[118]. Получение спектра метилена при газофазном импульсном
фотолизе диазометана [164, 165] явилось первым эксперимен-
тальным подтверждением возможности прямого обнаружения
и исследования этой частицы. Оно дало возможность опреде-
лить геометрические параметры СН2 в основном (3Bi) и воз-
бужденном (Mi) состояниях (см. табл. 2.1 и 2.5).
При действии фотолитического импульса температура систе-
мы обычно изменяется незначительно, поэтому благодаря малой
заселенности возбужденных колебательно-вращательных со-
стояний и, следовательно, относительно небольшой диффузности
полос оказывается доступной тонкая вращательная структура
электронного перехода, что обеспечивает большую информатив-
ность регистрируемых электронно-колебательно-вращательных
спектров высокого разрешения. Получаемые таким путем из
спектров значения энергий диссоциации и потенциалов иониза-
ции карбенов обычно наиболее надежны. Анализ спектров прос-
тейших трехатомных карбенов позволяет определить их основ-
ное и низшие возбужденные состояния, энергии перехода меж-
ду ними, межатомные расстояния и валентный угол, получить
сведения о некоторых частотах колебаний (см. [62]).
Импульсный фотолиз был успешно применен для генериро-
вания и регистрации электронных спектров поглощения в газо-
вой фазе следующих карбенов и силиленов: СНС1 и CHF [46],
CF2 [166—168], CFC1 [169], SiH2 [65], SiHCl и SiHBr [86],
SiHI [151] (см. табл. 2.1, 2.4, 2.5). Однако получение информа-
ции о структуре более сложных карбенов затруднено чрезвы-
чайным усложнением колебательно-вращательной структуры
линейчатых спектров [33]. Для ряда многоатомных молекул
из-за диффузности линий и их слияния в широкие полосы элект-
ронные спектры являются неспецифичными. Тем не менее метод
26
импульсного фотолиза в жидкой фазе позволил получить спект-
ральные полосы карбенов, содержащих ароматические циклы
(см. табл. 2.6).
Применение метода импульсного фотолиза для изучения
жидкофазных реакций карбенов основано на том факте, что
время образования нестабильной частицы существенно меньше
времени ее жизни и при соответствующем подборе условий за
время импульса наносекундного лазера (4—20 нс) удается ге-
нерировать до 1-Ю”5 моль карбена [173]. Далее через некото-
рое время задержки (50 нс—1 мкс) с применением быстродей-
ствующих регистрирующих приборов идентифицируются оптиче-
ские спектры поглощения карбена [173—175, 177] и (или)
продуктов его реакций [176, 177]. Таким способом удается уве-
ренно идентифицировать карбены со временем жизни ^50 нс,
хотя в ряде случаев исследованы и более лабильные частицы,
например триплетный флуоренилиден в растворе MeCN при
^20 °C, имеющий в этих условиях т./2^ 17 нс [177]. Для более
активных частиц используют также пикосекундные лазеры
в двухимпульсном режиме; в этом случае после первого им-
пульса (25—30 пс) и задержки 1 нс регистрируются лазерно-
индуцированные спектры флуоресценции [174, 178].
Однако даже эти, достаточно уникальные методы позволя-
ют исследовать лишь частицы со сравнительно большим време-
нем жизни и интенсивным поглощением (е>1000). При этом
необходимо также хотя бы неполное перекрывание полос погло-
щения источника карбена, самого карбена и продуктов его ре-
акции. Полосы же эти обычно довольно близки: например,
у (нафтил-2) диазометана 2-Ci0H7CHN2 ХМакс = 355 нм,
у 2-СюН7СН: ХМакс = 362 нм, а у 2-СюН7СН2’—продукта его ра-
дикальных реакций — Хмакс = 378 нм [176].
Метод импульсного фотолиза применяли пока лишь в случае
ароматических карбенов; для этих частиц достаточно глубоко
изучена абсолютная кинетика их реакций с различными акцеп-
торами в обычных жидкофазных условиях (см. разд. 2.4). Есте-
ственно, в избытке акцептора кинетика исчезновения карбена
подчиняется псевдопервому порядку.
Применение сходных методов в принципе возможно и в га-
зовой фазе; так, методом лазерной резонансно-абсорбционной
импульсно-кинетической спектроскопии исследована кинетика
реакций SiH2 + H2 и SiH2+D2 в газовой фазе [179].
Кроме электронных спектров применение методики импульс-
ного фотолиза позволило с помощью специальных ИК-спектро-
метров быстрого сканирования получить колебательные спект-
ры ряда карбенов в газовой фазе. Так, в частности, был полу-
чен ИК-спектр дифторкарбена [180—182].
Другой разновидностью импульсных методов является им-
пульсный электрический разряд. С его помощью
27
удалось получить интенсивный электронно-колебательный
спектр CF2 [183]. Однако для наблюдения этого и других дига-
логенкарбенов с относительно большим временем жизни (полу-
период исчезновения 10~3—10"4 с) могут быть использованы
и непрерывный электрический [184, 185] или высокочастотный
(ВЧ) [41, 55, 186—189] разряды. В частности, ВЧ-разряд позво-
лил зарегистрировать с помощью импульсного лазера спектры
флуоресценции фторхлор- и дихлоркарбенов в газовой фа-
зе [190].
Закономерно, что для более стабильных кремниевых и гер-
маниевых аналогов дигалогенкарбенов разнообразные спект-
ральные данные в газовой фазе часто могут быть получены
с помощью обычных спектрометров без использования импульс-
ных методик [51, 71—73, 90, 157, 191—194]. Результаты иссле-
дований карбенов и их аналогов методом электрического разря-
да (в том числе импульсного) приведены в табл. 2.1, 2.4, 2.5.
Для изучения кинетики и равновесия газофазных реакций
термической диссоциации при высоких температурах (вплоть до
12000 К) применяют метод ударной волны. При темпера-
турах 1000—3000 К в газовой фазе протекают реакции диссо-
циации исходных молекул с образованием карбенов, а также
диссоциация карбенов на более мелкие фрагменты.
Известно, что измерение констант равновесия является од-
ним из самых точных способов определения теплот реакций, и
именно этим методом получены наиболее надежные значения
стандартной энтальпии образования CF2, а также констант ско-
рости, констант равновесия и теплот реакций с участием этой
частицы [195—200]. Теплоты образования некоторых карбенов
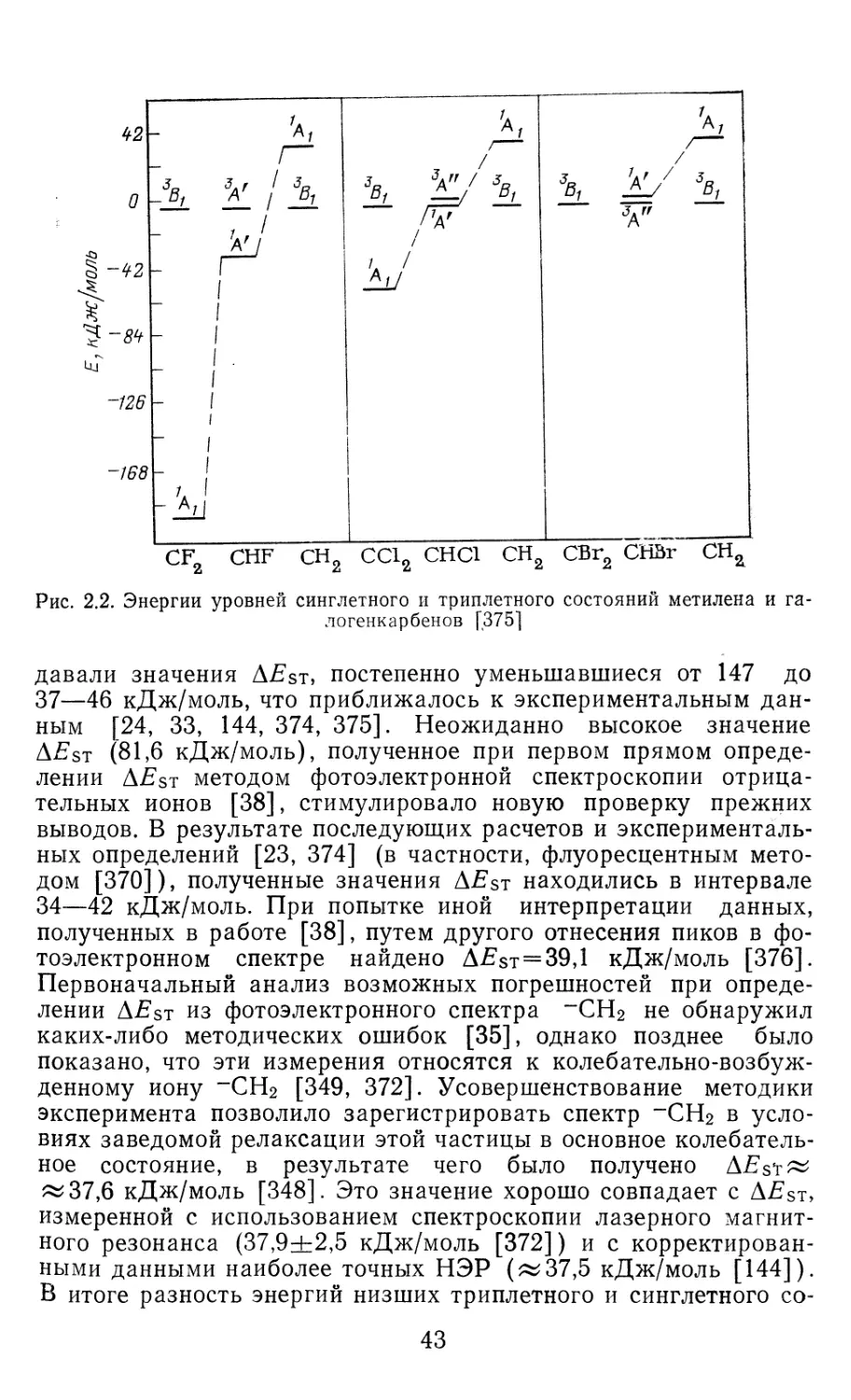
и их аналогов, полученных с помощью метода ударной волны,
приведены в табл. 2.2.
Своеобразной разновидностью этого метода является метод
адиабатического сжатия, позволяющий исследовать
кинетику газофазных реакций в строго гомогенных условиях
[201]. При этом кинетические эффекты проявляются в виде не-
симметричности кривой зависимости оптического поглощения
карбена от времени, а также сдвига максимума этой кривой от-
носительно максимума давления (точки остановки поршня).
С помощью специальных расчетных методик из этих характери-
стик удается определять кинетические параметры реакций об-
разования и рекомбинации карбенов, в частности CF2 [202] „
СС12 [203], CFCF3 и C(CF3)2 [204].
2.2.2. Использование метода матричной изоляции
для получения ИК-, УФ- и ЭПР-спектров карбенов
При получении карбенов без использования импульсных мето-
дов удается создать лишь ничтожную текущую их концентра-
цию. Однако исчезновение карбенов по вторичным реакциям
28
можно затормозить, проведя их генерирование, например, в ва-
кууме и применяя разбавление первичных продуктов реакции
большим избытком инертного газа.
В качестве матричного вещества используют чаще всего ар-
гон, образующий при 10—18 К достаточно жесткую и прозрач-
ную матрицу, а также Ne, Кг, Хе, N2, SFe и другие инертные
вещества, не обладающие собственным колебательным спектром
в исследуемой области. Температуру матриц обычно поддержи-
вают в пределах 4,2 (жидкий гелий) —20 К (жидкий водород),
а необходимое разбавление матричным веществом нестабиль-
ных частиц обычно составляет не менее 1 :1000 (подробнее
см. [32, 33, 205, 206]).
Если замораживать разбавленную инертным газом реакци-
онную смесь при низких температурах, то образующаяся твер-
дая матрица может быть получена в объеме, достаточном для
обнаружения изолированных в ней нестабильных частиц обыч-
ными спектроскопическими методами. В инертной матрице мо-
гут быть изолированы и стабильные предшественники карбена,
способные в результате последующего облучения или матрич-
ной реакции образовывать искомую частицу.
Метод матричной изоляции [32, 33, 205, 206] позволил обна-
ружить и спектроскопически изучить без ограничений во вре-
мени ряд карбенов. Для их идентификации используют спектро-
скопию в ИК-, УФ- и видимой областях спектра, а в случае кар-
бенов с триплетным основным состоянием также и спектроско-
пию ЭПР. Подробный анализ матричных исследований карбе-
нов проведен в обзорах [32, 206].
Матричная ИК-спектроскопия. Достоинства этого метода за-
ключаются не только в возможности обнаружения с помощью
стандартных спектрометров высокореакционных карбенов и ус-
тановления их частот колебаний, но и в большой информатив-
ности матричных спектров, а именно в малой полуширине спек-
тральных полос (до нескольких десятых долей см -1) из-за поч-
ти полного отсутствия вращения и межмолекулярных взаимо-
действий в низкотемпературной матрице. Это позволяет иденти-
фицировать все компоненты сложных смесей и даже различать
близко расположенные полосы частиц с различным изотопным
составом.
Наибольшие успехи матричной ИК-спектроскопии достигну-
ты в изучении галоген- и дигалогенкарбенов, которые облада-
ют синглетным основным состоянием и поэтому не могут быть
обнаружены методом ЭПР. Изучены матричные ИК-спектры
СНС1 [52, 54], CHF [47], CF2 [148, 207, 208], СС12 [50. 53, 147,
209—215], СВг2 [59, 216—218], С12 [63], CFC1, CFBr и CFI [56,
57, 219], а также CCIBr [59, 210]. Изотопное расщепление
ИК-полос СС12 и CCIBr было использовано для расчета сило-
вых полей этих частиц [59, 210—212]. Матричный ИК-спектр
29
СВг2 изучен менее детально [59, 216], однако частоты деформа-
ционных колебаний СВг2 и CCIBr позднее были определены из
спектров флуоресценции [61].
В то же время ИК-спектр наиболее активного карбена —
метилена — до последнего времени не был получен несмотря
на многочисленные попытки на протяжении последних 20 лет,
хотя среди продуктов фотолиза CH2N2 в Аг- или Хе-матрицах
методом ИК-спектроскопии предположительно детектирован
триплетный метилен [220]. Причина этого, очевидно, заключа-
ется в достаточной диффузии и высокой реакционной способ-
ности СН2 даже в матрицах при 4,2 К. В 1970—71 гг. эту части-
цу удалось стабилизировать в матрицах в концентрациях, до-
статочных для обнаружения более чувствительным методом
ЭПР [221—225] (см. ниже).
ИК-спектры SiH2 и GeH2, а также менее реакционноспособ-
ных кремниевых и германиевых аналогов галогенкарбенов
(SiF2, GeF2, SiCl2, GeCl2, SiBr2 и GeBr2) получены в матри-
цах Ar, Ne, Кг и N2, а в случае SiF2 и GeF2 — и в газовой фазе
(подробнее см. [32]). Методом ИК-спектроскопии исследованы
некоторые матричные реакции атомарного кремния (Аг-матри-
ца, 15 К), ведущие к образованию силиленов:
НрО HF(DF)
SiH(OH)-*— Si —*>SiHF (SiDF)
[88] [82] '
Матричными реакциями атомарного углерода с СО и HCN
получены два необычных по строению карбена — С = СО [226,
227] и CHCN [228]—и выполнено их ИК-спектроскопическое
исследование. Углерод-углеродная связь в карбонилкарбене по
значению силовой постоянной является промежуточной между
ординарной и двойной, причем этот карбен, вероятно, обладает
линейной структурой [226]. По результатам исследования
ПК-спектров цианокарбена CHCN и расчета его силового поля
[228] (см. также газофазное исследование [229]), эта частица
обладает линейным строением алленового типа (1,3-бирадика-
ла) (8), что согласуется с ранней интерпретацией его спектров
ЭПР [230—232], однако может быть ошибочным в свете недав-
них экспериментальных и расчетных работ (см. разд. 2.2.3).
Известны матричные ИК-спектры дицианокарбена C(CN)2
[233], а также молекулы Si = NH, формально являющейся кар-
беноподобной частицей [234], и ее углеродного аналога C = NH
[235]. Последняя молекула исследована методом колебательной
спектроскопии и в газовой фазе [236, 237].
Н - С = С = N (8)
Метод матричной ИК-спектроскопии использован и для иден-
тификации более сложных карбенов и их аналогов, в частности,
30
циклопропенилидена [238], циклопентадиенилидена [239, 240].
фенилкарбена [241], фенилхлоркарбена [242], (пиридил-3) кар-
бена [243], метоксихлоркарбена [244, 245], трех перфторкето-
карбенов [246], диметилсилилена [247] и диметилстаннилена
[248]. Вместе с тем, для подобных сложных молекул характер-
но комплексное использование матричной спектроскопии в види-
мой, УФ- и ИК-областях спектра, а также спектроскопии ЭПР,
что дает более полную и надежную структурную информацию.
В последние годы начались исследования матрично-изолирован-
ных частиц и с помощью лазерной спектроскопии КР [249—
251].
Электронные спектры карбенов. Поскольку в твердой инерт-
ной матрице карбены находятся в основном электронном со-
стоянии, то сопоставление их электронных спектров поглощения,
полученных в газовой фазе (например, при использовании им-
пульсного фотолиза) и в матрице, сразу вносит ясность в отне-
сение нижних состояний в наблюдавшихся переходах. В частно-
сти, таким путем установлено, что нижнее синглетное состояние
у карбенов CHF [47] и СНС1 [52] является основным. Элект-
ронные полосы поглощения в матрице были обнаружены у дига-
логенкарбенов CF2 [148, 207, 252], ССЬ [52, 53] и CFC1 [56],
а также у дикарбена С3 (:С = С = С:) [253].
Значительные достижения в изучении электронных перехо-
дов в матрицах .получены благодаря применению флуорес-
центных методов. В большинстве случаев дигалогенкарбе-
ны были получены непосредственно в матрице фотолизом мат-
рично-изолированных предшественников (СН2С12, CH2F2,
CH2CIF, CHCI3), бомбардировкой тетрагалогенметанов атома-
ми щелочных металлов или реакциями атомарного углерода
с галогенами или галогеноводородами. Спектры флуоресценции
позволили выявить тонкую колебательную структуру электрон-
ных полос. Таким путем получены частоты колебаний основно-
го состояния, в том числе деформационных колебаний V2, кото-
рые обычно малоинтенсивны и с трудом обнаруживаются
в ИК-спектрах карбенов, а также важная информация о часто-
тах колебаний, времени жизни и релаксации возбужденных со-
стояний.
Для хорошо изученного дифторкарбена спектры флуоресцен-
ции в матрице [44, 254] хорошо согласуются с известными элек-
тронными спектрами высокого разрешения в газовой фазе [41].
Применение перестраиваемых лазеров открыло большие
перспективы для получения флуоресцентных спектров путем се-
лективного возбуждения отдельных колебательных уровней
электронного перехода [57]. Такая методика была применена
к СС12 [147, 211], СВг2 и CCIBr [61, 255], а также к CFC1
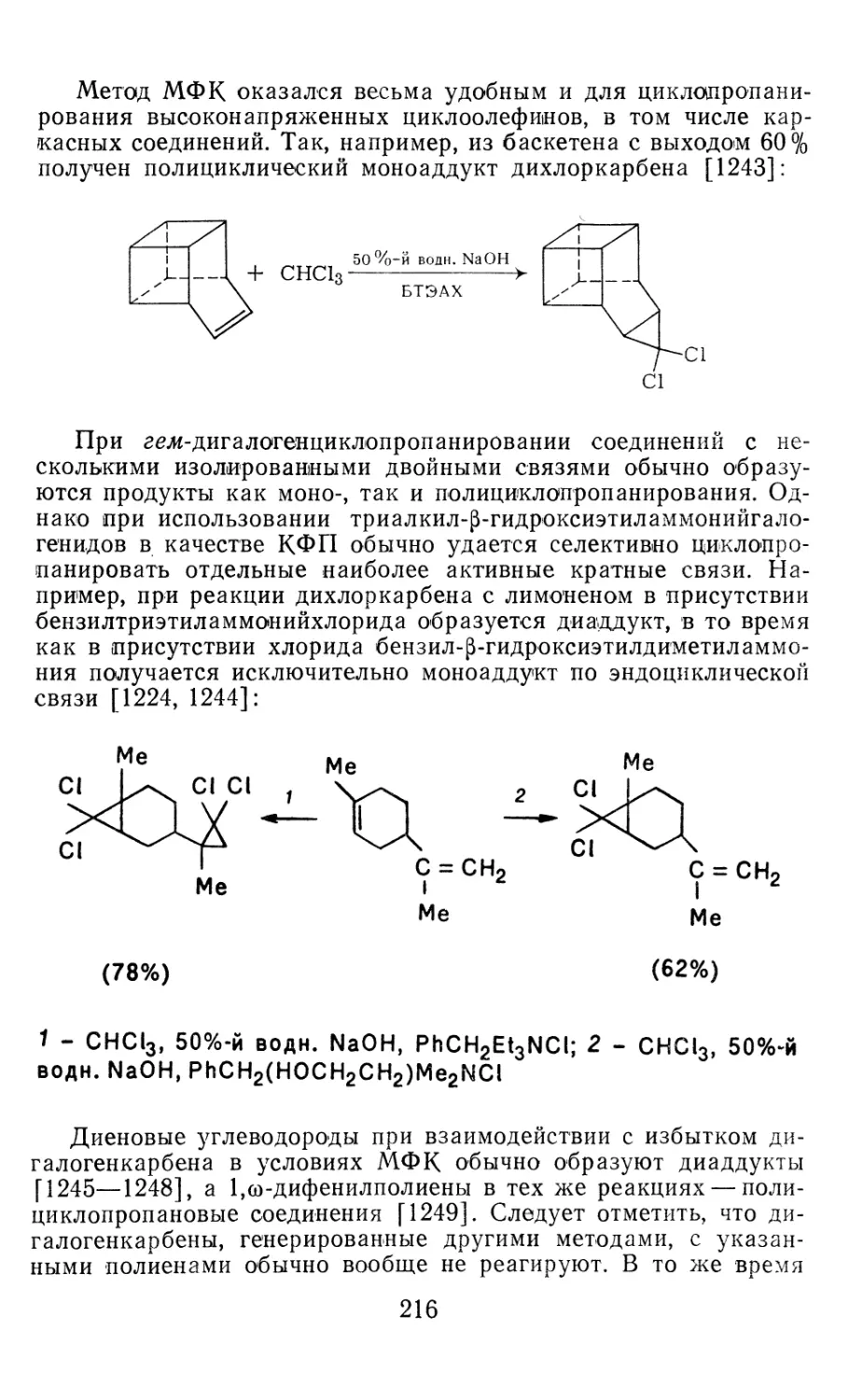
[56—58] и CFBr [256]. В ряде случаев матричные коле-
Нательные и оптические спектры получены для молекул карбе-
31
и их аналогов, содержащих органические заместители, на-
пример для диметилсилилена, генерированного из различных
источников:
Me3SiSiMe2OMe —
MeOSiMe2SiMe2OMe —_ , , S i M p
(Me3Sn)2SiMe2 2 PhMe2SiSiMe2SiMe2Ph —- ~ • и i г 1 c 2
2
----(Me2Si)6
4
----СН2 = SiHMe
— Me2Si(N3)2
1 — флеш-пиролиз с улавливанием в матрицу [257]; 2 — hv, углеводород-
ные стекла, 77 К или hv, Аг, 28—30 К [247, 258]; 3 — hv — углеводородное
стекло, 77 К [259]; 4 — hv, Аг, 10 К [257]; 5 — hv, Аг, 10—25 К [259, 260].
Зарегистрирован также лазерно-индуцированный электрон-
ный спектр диметилгермилена, генерированного фотолизом
Me2Ge(SePh)2 [261]. Подробно исследованы дифенилкарбен
и его замещенные, а также полициклические ароматические
карбены, генерированные в инертных матрицах или органиче-
ских стеклах при фотолизе соответствующих диазометанов
и диазиринов [173, 262—268]. Описан оптический спектр фенил-
карбена [269], приведены данные ИК- и УФ-спектров метокси-
хлоркарбена и феноксихлоркарбена в Аг- или Ы2-матрицах при
10 К, из которых, в частности, следует наличие у этих карбенов
/{^^тра^с-изомерии [245]. Вместе с тем спектры сложных
карбенов в матрицах, а тем более в стеклах, малоинформативны,
их используют обычно лишь для идентификации промежуточ-
ных частиц и исследования кинетики реакций их исчезновения.
Значительное число работ посвящено спектроскопическому ис-
следованию матричных реакций, в основном для арилкарбенов
(см., например, [242, 267, 270—276]).
Использование нано- и пикосекундных лазеров позволяет де-
тектировать некоторые сравнительно стабильные карбены и их
аналоги в условиях их фотолитического генерирования в раство-
рах при обычных температурах, а также определять абсолютную
кинетику их реакций с различными субстратами. С помощью
этих лазеров были исследованы реакции многих карбенов с не-
предельными акцепторами, кислород-, азот- и серосодержащи-
ми соединениями: реакции дифенилкарбена и его замещенных
[172, 174, 175, 277—283], нафтилкарбенов [176, 284], бензил-
хлоркарбена [285], арилгалогенкарбенов [173, 286—289], флуо-
ренилидена и его замещенных [177, 268, 278, 290—292], ксантил-
идена-9 [293]. Та же методика применена и для детектирова-
ния метилфенилсилилена — продукта лазерного фотолиза ме-
тилбис (триметилсилил)фенилсилана, а также для определения
32
констант скоростей реакций этого силилена и его димера с кис-
лородом, этанолом и триэтилсиланом [294].
Возвращаясь к арилкарбенам, необходимо отметить, что для
исследований строения и реакционной способности этих частиц
в целом характерно сопоставление результатов импульсных и
матричных исследований в ИК-, УФ- и видимой областях спект-
ра с данными спектроскопии ЭПР, поскольку большинство этих
карбенов обладает триплетным основным состоянием.
ЭПР-Спектроскопия. Спин-гамильтониан, описывающий
энергетические уровни и спектр ЭПР триплетной системы с сум-
марным спином S=l, в магнитном поле с напряженностью Я о
имеет вид [32]:
н =g₽SH0 + D(S2- 1/3 S2) + E(S2 - S2).
Первый член этого уравнения — энергия магнитного момента
электрона в поле HQ (р — магнетон Бора, g‘ = 2,0023 для свобод-
ного электрона, S— оператор спина), а второй и третий члены
характеризуют дипольное взаимодействие спинов с компонен-
тами Sx, Sy и Sz вдоль соответствующих осей координат. Коэф-
фициенты D и Е (так называемые параметры расщепления ну-
левого поля) соответствуют расщеплению уровней в отсутствие
внешнего магнитного поля; их определение и является одной
из главных целей эксперимента. Известно, что параметр D об-
ратно пропорционален кубу эффективного расстояния между
неспаренными электронами, и при их локализации на орбиталях
соседних атомов (как, например, в возбужденной в триплетное
состояние молекуле нафталина) значение D близко к 0,10 см-1.
При сокращении расстояния, в частности в случае расположе-
ния неспаренных электронов на двух орбиталях одного атома
(что имеет место у карбенов), значения D должны быть сущест-
венно выше. Так, у 3СН2 D=;0,76 см~].
По параметру Е можно судить об отклонении системы от
цилиндрической симметрии, т.е. о различии магнитных свойств
вдоль осей хну, например, о степени изогнутости карбена.
Для линейного карбена Е должно быть равно нулю. Повыше-
ние отклонения отношения Е/D от нулевого значения связано
с увеличением s-характера о-орбитали — одной из двух орбита-
лей карбена, не занятых связывающими парами электронов
(см. рис. 2.1), т.е. с ростом степени изогнутости карбена. Спект-
роскопия ЭПР является удобным методом изучения карбенов,
имеющих триплетное основное состояние, что, как оказалось,
характерно почти для всех карбенов, у которых центральный
атом связан только с атомами С и Н. При наличии же у кар-
бенного центра даже одного атома галогена основным является
синглетное состояние.
3 — 107
33
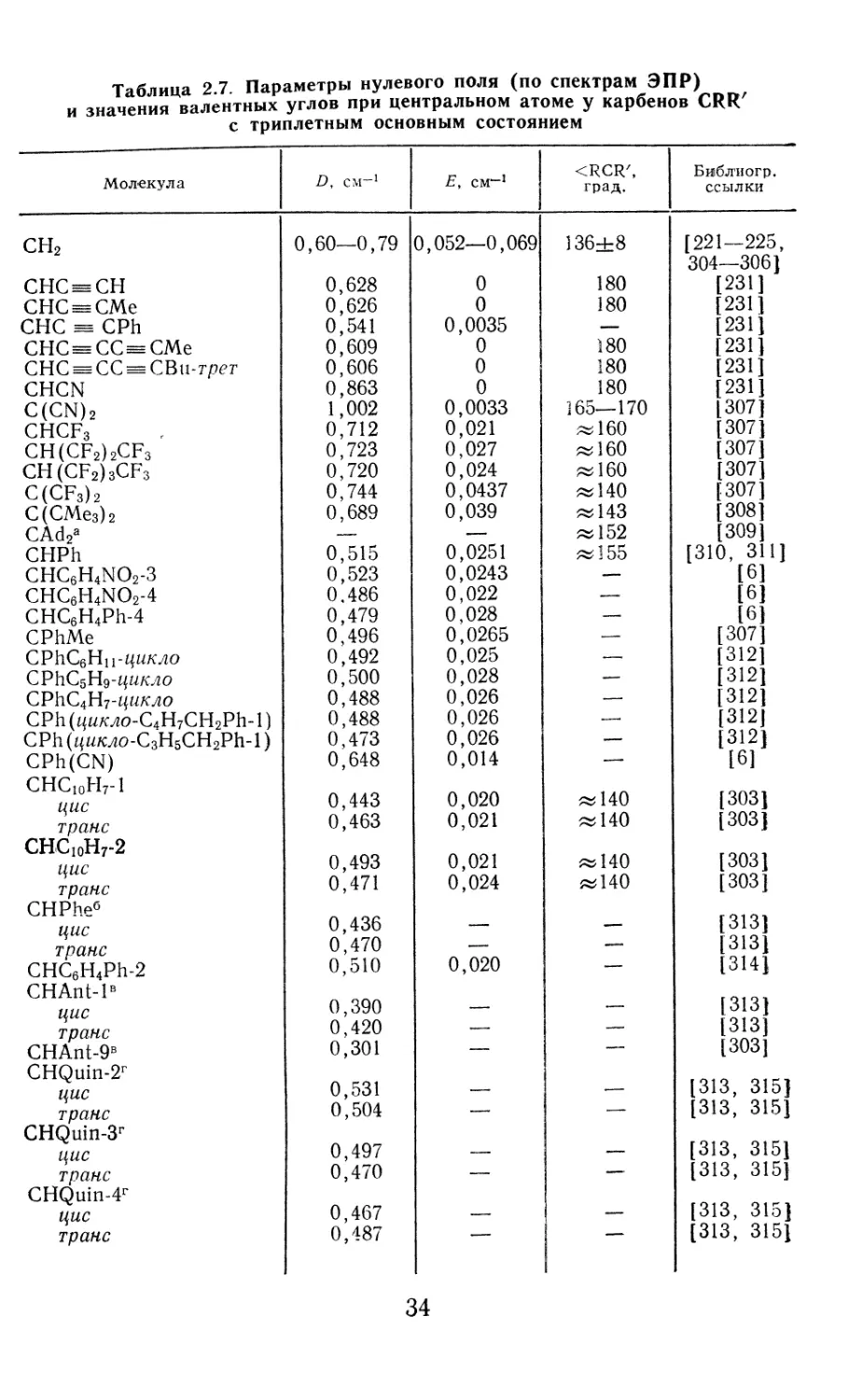
Таблица 2.7. Параметры нулевого поля (по спектрам ЭПР)
и значения валентных углов при центральном атоме у карбенов CRRZ
с триплетным основным состоянием
Молекула D, см-1 Е, см*-1 <RCR', град. Библиогр. ссылки
сн2 0,60—0,79 0,052—0,069 136±8 [221—225,
304—306 [
СНС — СН 0,628 0 180 [231]
СПС^СМе 0,626 0 180 [231]
СНС CPh 0,541 0,0035 — [231]
СНС^СС^СМе 0,609 0 180 [231]
СНС = СС == СВи-трет 0,606 0 180 [231]
CHCN 0,863 0 180 [231]
C(CN)2 1,002 0,0033 165—170 [307]
CHCF3 0,712 0,021 « 160 [307]
CH(CF2)2CF3 0,723 0,027 «160 [307]
CH(CF2)3CF3 0,720 0,024 «160 [307]
C(CF3)2 0,744 0,0437 «140 [307]
С(СМе3)2 0,689 0,039 «143 [308]
CAd2a — — «152 [309]
CHPh 0,515 0,0251 «155 [310, 311]
CHC6H4NO2-3 0,523 0,0243 — [6]
CHC6H4NO2-4 0,486 0,022 — [6]
CHC6H4Ph-4 0,479 0,028 —, [6]
CPhMe 0,496 0,0265 — [307]
СРИСбНц-цикло 0,492 0,025 — [312]
СРЬС5Н9-цикло 0,500 0,028 —, [312]
СР11С4Н7-ф/л'ло 0,488 0,026 — [312]
CPh (ц/7Л'ло-С4Н7СН2РЬ- 1) 0,488 0,026 .—. [312]
CPh(/{Z7oo-C3H5CH2Ph-l) 0,473 0,026 — [312]
CPh(CN) 0,648 0,014 — [6]
CHCI0H7-l
цис 0,443 0,020 «140 [303]
транс 0,463 0,021 «140 [303]
CHC10H7-2
цис 0,493 0,021 «140 [303]
транс 0,471 0,024 «140 [303]
CHPhe6
цис 0,436 — .— [313]
транс 0,470 — — [313]
CHC6H4Ph-2 0,510 0,020 — [314]
CHAnt-lB
цис 0,390 — .— [313]
транс 0,420 — — [313]
CHAnt-9B 0,301 — — [303]
CHQuin-2r
цис 0,531 — — [313, 315}
транс 0,504 — — [313, 315]
CHQuin-3r
цис 0,497 — — [313, 315]
транс 0,470 — — [313, 315]
CHQuin-4r
цис 0,467 — — [313, 315]
транс 0,487 — — [313, 315]
34
Продолжение табл. 2,7
Молекула D, см-1 Е, см-1 <RCRZ, град. Бпблиогр. ссылки
CHQuin-8r
цис 0,433 — — [313, 315]
транс 0,464 — [313, 315]
СНРугд 0,40 — — [316]
CPhCH2Ph 0,493 0,029 — 16]
CPh2 0,408 0,021 147,6±1,8 [296, 300,
СРНСбН«Ме-4 0,407 0,019 .— UUU J [6] 1 1
CPhC6H4OMe-4 0,404 0,019 — [6] 1
CPhC6H4Cl-4 0,403 0,019 — [6]
CPhC6H4Br-4 0,392 0,019 — [6] 1
CPhC6H4NO2-4 0,377 0,018 — [6] 1
C(C6H4OMe-4)2 0,407 0,019 — 16]
CPhe2 0,301 0 180 [6] 1
(9) 0,408 0,028 135 [297, 298,
317—3 19]
(10) 0,399 0,028 — [320]
(11) 0,393 0,017 150 [321
(12) 0,379 0,016 — [321
(13) 0,422 0,019 — [32Г
(14) 0,39 0,030 — [322]
Инденилиден-1 0,378 0,016 — [317
(15) 0,409 0,012 — [317
(2) 0,317 0,009 — [3231
(16) 0,318 0,006 — [324]
(17) 0,328 0,009 — [3241
(18) 0,347 0,001 — [324]
(19) 0,314 0,005 — [3241
(20) 0,333 0,011 — [324
(21) 0,364 0,018 — [325^
(22) 0,38 0,03 — [6]
(23) ' 0,170 0,003 —. [326]
(24) 0,146 — — [3261
(25) 0,105 0,003 — [326]
(26) 0,13 0,004 — [326
CHCH==CH2
цис 0,458 0,020 -— [313, 327]
транс 0,408 0,023 — [313, 327]
CHCH==CHMe
цис 0,457 0,021 — 1313]
транс 0,409 0,024 — [313]
СНСН = СМе2
цис 0,454 0,020 — 1313]
транс 0,412 0,024 — 1313]
СНСМе=СН2
цис 0,431 0,025 — [313]
транс 0,401 0,025 — 13131
СМеСН=СН2
цис 0,453 0,020 — [313]
транс СМеСН = СМе2 0,399 0,022 — [313]
цис 0,442 0,023 —. [313]
транс 0,400 0,022 — [313]
35
Продолжение табл. 2.7
Молекула D, см-1 Е, см—1 <RCRZ, град. Библиогр. ссылки
CPhCH = CMe2
цис СНСОМе 0,357 0,019 — [313J
цис 0,663 0,030 — [313, 3281
транс CHCOEt 0,617 0,053 — [313, 328J
цис 0,661 0,031 —, [313, 328J
транс CHCOCF3 0,616 0,053 — [313, 3281
цис 0,605 0,034 — [313, 329J
транс C(CF3)COCF3 0,572 0,079 — [313, 329]
цис 0,605 0,034 — [313, 329]
транс CPhCOPh 0,572 0,079 — [313, 329]
цис 0,392 0,052 — [6, 313]
транс С(СООМе)СОМе 0,312 0,052 — [6, 313}
транс,транс 0,604 0,031 — [3131
цис,транс 0,606 0,051 — [313]
(27) 0,578 0,035 — [313]
CPhC(Ph) = S 0,032 0,011 — [330]
(28) 0,052 <0,002 — [331]
(29) 0,084 0,023 — [332]
(30) 0,070—0,071 0,019—0,020 —. [332, 333}
а Ad — адамантил-1.
Д Руг — пиренил-1.
6phe — фенантрил-9. в Ant — антрил.
rQuin — хинолил.
(9) R = Н
(10) R = Вг
'11) (12)
(13)
(1/) (15)
36
о
(27)
(23) R = Н- (24) R = Me;
(25)R = Ph (26) R = C10H2r«
PhC —Z~V-CPh <—PhC==/=y=CPh
(28)
(29)R = H
(30) R = Ph
Впервые методом ЭПР было зафиксировано триплетное ос-
новное состояние у дифенилкарбена [295, 296]. Значение £> =
= 0,41 (см. табл. 2.7) указывает на локализацию неспаренных
электронов преимущественно у карбенового атома углерода,
а отличное от нуля значение £ = 0,02 см~' свидетельствует об
изогнутости 3CPh2. В результате неоднократных исследований
£295__303] выяснено распределение плотности неспаренных
электронов, а также геометрия дифенилкарбена в различных
матрицах.
Триплетное основное состояние и значения параметров нуле-
вого поля D и Е установлены для ряда алкил- и арилзамещен-
ных карбенов, что позволило в большинстве случаев опреде-
лить и геометрическое строение этих частиц, в первую очередь
валентный угол при центральном атоме (табл. 2.7).
У большинства арилкарбенов валентный угол, оцениваемый
из соотношения £/£, составляет 140—150° [297, 303, 310, 317,
318].
Опубликованный в 1970 г. [221] первый спектр ЭПР метиле-
на доказал триплетность его основного состояния, а параметры
D и Е (0,69 и 0,003 см-1 соответственно), по мнению авторов
[221], соответствовали слегка изогнутой (почти линейной)
структуре метилена. Однако в более поздних работах [39, 222—
225, 304, 328] низкое значение Е было объяснено вращением
метилена в матрице Хе при существенном отклонении от линей-
ности. Валентный угол НСН впервые был определен на основа-
нии спектров ЭПР CD2 и CHD [223], это значение (136+8°)
в дальнейшем было уточнено [224, 225, 304] измерениями изо-
тропного и анизотропного сверхтонких взаимодействий с ядром
13С в 13CD2. Спектры ЭПР метилена получены также в матри-
цах Кг, SF6 и перфторциклобутана [305], смесях Кг и Хе [304],
Хе и Аг [306]. Позднее параметры нулевого поля молекулы ме-
тилена были определены также и из спектров лазерного маг-
нитного резонанса в газовой фазе (£> = 0,7784, £ = 0,03991 см"1
[39]).
После того как на примере метилена было установлено, что
близкие к нулю значения £ могут быть связаны с вращением
частицы в матрице, а не с ее линейным строением, были выска-
заны подобные предположения об изогнутости цианокарбена
[222, 334] и этинилкарбена [335], у которых £^0 (см.
табл. 2.7). Получены также спектры ЭПР других карбенов с од-
нокоординироваиным карбенным атомом — карбонилкарбена
+ —
:С = С=О и диазокарбена :C = N=N [307, 336], а также их
+ —
кремниевых аналогов :Si = C = O и :Si = N —N [337].
Методом ЭПР удается регистрировать и поликарбеновые ча-
стицы, например (28) — (30) (см. табл. 2.7). Триплетные фенпл-
бензоилкарбен, (4-метоксифенил) фенилкарбен, бис (4-метоксифе-
нил)карбен и антранилиден (9,10-дигидроантраценилиден-9)
в каждом случае объединяются в радикальную пару с соответ-
ствующим квинтетным состоянием, характерным для дикарбе-
38
на [324, 338, 339]. Зарегистрирован [340] спектр ЭПР тетра-
карбена— частицы нонетной мультиплетности, представляющей
собой модель молекулярного суперпарамагнетика:
Вместе с тем основной задачей метода ЭПР, несомненно,
является установление спиновой мультиплетности того или ино-
го карбена, релаксирующего при регистрации спектра в низко-
температурных матрице или стекле в основное состояние.
2.2.3. Исследование карбенов в газовой фазе
Обнаружению и прямому изучению карбенов физическими ме-
тодами в газовой фазе благоприятствует использование вакуу-
ма, что уменьшает число межмолекулярных столкновений.
При поддержании в системе сравнительно высокого давления
(~1,3 кПа) некоторые .карбены и их аналоги удается исследо-
вать традиционными структурными методами. Прежде всего
к ним относится метод газовой электронографии (ГЭ),
позволяющий получать значения межатомных расстояний, ва-
лентных углов и амплитуд колебаний; однако необходимость
введения в вакуумную камеру электронографа достаточно плот-
ного молекулярного пучка ограничивает применимость данного
метода. Тем не менее была сделана попытка определить этим
методом структуру дибромкарбена, генерируемого пиролизом
СВг4 [60]. Однако одновременное присутствие в анализируемой
смеси других продуктов пиролиза СВг4 не позволяет считать по-
лученные результаты надежными.
Значительно более успешным оказалось использование газо-
вой электронографии для исследования строения более стабиль-
ных аналогов карбенов — SiCl2 и SiBr2 [35], GeCl2 и GeBr2 [34,
95]. Полученные таким путем геометрические характеристики
этих частиц хорошо согласуются со значениями, определенными
другими методами (см. табл. 2.1). С помощью газовой электро-
нографии (100 °C, 103 Па) исследован также станнилен (6)
[104] (см. табл. 2.1).
Метод микроволновой спектроскопии (МВ)
применяют при исследовании строения сравнительно малореак-
ционноспособных карбенов и их аналогов. Известны МВ-спект-
ры SiF2 [70, 71, 75] и GeF2 [90, 91], позволившие определить
39
дипольные моменты этих частиц (1,23 и 2,61Д соответственно),
а также рассчитать их геометрическое строение (см. табл. 2.1).
Значительно более универсальным является масс-спект-
рометрический метод исследования, целью которого
обычно является измерение потенциалов ионизации (ПИ) и рас-
чет теплот образования карбенов. Так, этим методом путем
измерения ПИ было впервые непосредственно доказано образо-
вание метилена при пиролизе кетена [119]; полученное значе-
ние ПИ хорошо согласуется со значением, получаемым из спект-
роскопических данных [117] (см. табл. 2.3). В масс-спектромет-
рических исследованиях кроме термических методов генерирова-
ния карбенов и их аналогов [34, 119, 341] используют и фотоли-
тические методы, например многофотонный ИК-фотолиз полига-
логенидов в ячейке Кнудсена, присоединенной к масс-спектро-
метру [342]:
C3F7I ——-*--C3F7 + I -C3F7^:CF2 + -C2F5
•C2F5-^:CF2 + -CF3
Потенциалы ионизации трехатомных карбенов и их аналогов
приведены в табл. 2.3.
Значительный прогресс в изучении карбенов достигнут в по-
следние годы благодаря применению принципиально новых ме-
тодов изучения нестабильных частиц при низких давлениях. Так,
разработан метод лазерной фотоэлектронной спек-
трометрии отрицательных ионов, с помощью которо-
го можно получать энергетический спектр электронов, отщепля-
ющихся от отрицательных ионов под действием лазерного излу-
чения, и таким образом, изучать расположение электронных
уровней как в этих ионах, так и в соответствующих им ней-
тральных молекулах, включая карбены [343—346]. Данный ме-
тод основан на том, что пучок отрицательных ионов, образован-
ных при электрическом разряде низкого давления (^1,3 гПа)
в исходном веществе, вытягивают из разряда, ускоряют и фоку-
сируют; затем выделяют с помощью масс-фильтра отрицатель-
ные ионы определенной массы, которые подвергают действию
поляризованного лазерного излучения. Электроны, возникающие
в результате фотоотщепления от отрицательных ионов, анализи-
руют по энергиям в электростатическом монохроматоре. Из
спектров электронов получают значения энергий переходов меж-
ду уровнями данного отрицательного иона и образующейся из
него нейтральной частицы, а следовательно, и расщепление меж-
ду различными энергетическими уровнями в последней.
При использовании СН4 и CH2N2 в качестве источника ио-
нов ~СН2 этим методом удалось впервые непосредственно из-
мерить [38, 347] энергию синглет-триплетного расщепления
49
уровней 3Bi и Mi в метилене; A£st = 81,6 кДж/моль. Однако до-
стоверность этого неожиданно высокого значения позднее была
поставлена под сомнение другими экспериментальными и рас-
четными данными, и после улучшения методики эксперимента
(см. ниже) было получено значение ^37,6 кДж/моль [348].
Сродство к электрону молекулы 3СН2 по данным различных
спектроскопических методов составляет 0,65+0,02 эВ [144, 349,
350].
Метод спектрометрии электронов, фотоотщепляющихся от
отрицательных ионов, позволил изучить расположение электрон-
ных уровней в молекулах—аналогах метилена. Для силилена
таким путем выявлены переходы [351]:
:SiH2(3B1)-^r~SiH2(2S1) —^:SiH2(1A1 J
При этом установлено, что основное состояние силилена син-
глетно, однако расщепление уровней A£St(3^i—Mi), как и в
СН2, относительно невелико, во всяком случае не превышает
63—67 кДж/моль [351]. Этот результат согласуется и с данны-
ми НЭР [149, 150]. Сродство же к электрону частицы SiH2 со-
ставляет 1,124+0,020 эВ [351], а сродство к протону, опреде-
ленное методом ионно-циклотронного резонанса, равно 844+
+7 кДж/моль [П2].
Определенные достижения в исследованиях карбенов физи-
ческими методами связаны с усовершенствованием флуорес-
центного метода в газовой фазе, давно используемо-
го для получения спектров испускания как стабильных соедине-
ний, так и свободных радикалов (см. выше). Этот метод успешно
применен (см., например, [352, 353]) и при низких давлени-
ях— до 0,1 гПа. Использование метода индуцированной лазер-
ной флуоресценции в исследовании карбенов CFC1 и CFBr, по-
лученных в разрядно-проточной установке [354, 355] (атомы О
получены СВЧ-разрядом в смеси Аг—О2)
F2C = CFX + О - :CFX + F2CO X = Cl, Br
позволило исследовать кинетику их исчезновения и эксперимен-
тально определить энергии диссоциации химических связей
С—F, С—С1 и С—Вг в этих карбенах, которые оказались рав-
ными 490, 340 и 300 кДж/моль соответственно [354]. Характер-
но, что связь С—F в CFC1 и CF2 (486 кДж/моль [92, 356, 357])
заметно ослаблена по сравнению со связью С—F в CF4
(544 кДж/моль [92, 356, 357]) (подробнее эта закономерность
рассмотрена в разд. 2.3).
Спектры лазерно-индуцированной флуоресценции получены
и для CFC1, генерированного мультифотонным ИК-фотолизом
C2F3C1 или его реакцией с возбужденными атомами Не [358],
а также для СНС1 [55] и CHF [359], генерированных по реак-
ции СН3Х (X = F, Cl) с продуктами MB-разряда в CF4. Спект-
ры лазерно-индуцированной флуоресценции получены также
и для некоторых аналогов карбенов — S1H2 (вакуумный фото-
лиз PhSiH3 в присутствии Не [360]; см. также [353]) и SiHF,
генерированного по реакции SiH4 + F2 [361]. Получены спектры
возбуждения и флуоресценции карбонилкарбена С=СО [362];
в ряде случаев зарегистрированы также спектры хемилюмине-
сценции карбенов и их аналогов, генерируемых в возбужденных
состояниях химическими реакциями в газовой фазе. Так, среди
продуктов горения СН4 во F2 идентифицирован CHF [363],
в продуктах реакции SiH4+F2 зафиксирован SiHF [361].
При взаимодействии SiH4 или Si2H6 с СЬ, F2 или C1F, судя по
спектрам, образуются SiHCl и SiCb [364], а в продуктах реак-
ции GeH4 с СЬ идентифицированы GeHCl и, возможно,
GeCl2 [365].
При УФ-облучении (Х== 104,8-5-123,6 нм) метана обнаружены
полосы, принадлежащие метилену, который в этих условиях
образуется в возбужденном состоянии и, флуоресцируя, пе-
реходит в нижнее синглетное состояние Mi [365, 366]. Возбуж-
денные молекулы СН2 и CD2 получены также при разряде в сме-
сях Не—CF4—СН2=СО (CD2=CO) [367], CF4—СН4 и
CF4 — СН2—СО [368], Не—CD4—F2 [369]. Дальнейшее усовер-
шенствование данного метода связано с применением импульс-
ных лазеров для получения спектров возбуждения. Так, хотя
в электронных спектрах СС12 и CFC1 наблюдали полосы прак-
тически без тонкой структуры [190], сочетание лазерного фото-
лиза в вакууме и лазерного возбуждения отдельных спектраль-
ных переходов позволило зафиксировать вращательную струк-
туру спектра СН2 и CD2 [39, 367—372] и непосредственно
определить значения A£st(35i—Mi).
2.2.4. Энергетическое расщепление уровней 3Bi и Mi
у карбенов и их аналогов
Определение значения A£’st(3^i—Mi) для молекулы метилена
и других карбенов является предметом многочисленных экспе-
риментальных и теоретических исследований, так как карбены
могут участвовать в реакциях как в основном, так и в возбуж-
денном состояниях. В ранних работах по экспериментальному
определению A£st(CH2) при анализе различных термохимиче-
ских и кинетических характеристик реакций с участием метиле-
на было определено, что A£’st:=29—42 кДж/моль [4, 32, 33,
144]. При измерении кинетической энергии отдачи продуктов
фотолиза различных источников метилена оценочное значение
AFst составило 35,5+2,1 кДж/моль [373]. Квантовохимические
расчеты метилена по мере их усложнения и совершенствования
42
давали значения A£st, постепенно уменьшавшиеся от 147 до
37—46 кДж/моль, что приближалось к экспериментальным дан-
ным [24, 33, 144, 374, 375]. Неожиданно высокое значение
A£st (81,6 кДж/моль), полученное при первом прямом опреде-
лении A£st методом фотоэлектронной спектроскопии отрица-
тельных ионов [38], стимулировало новую проверку прежних
выводов. В результате последующих расчетов и эксперименталь-
ных определений [23, 374] (в частности, флуоресцентным мето-
дом [370]), полученные значения A£st находились в интервале
34—42 кДж/моль. При попытке иной интерпретации данных,
полученных в работе [38], путем другого отнесения пиков в фо-
тоэлектронном спектре найдено A£’st = 39,1 кДж/моль [376].
Первоначальный анализ возможных погрешностей при опреде-
лении A£st из фотоэлектронного спектра "“СН2 не обнаружил
каких-либо методических ошибок [35], однако позднее было
показано, что эти измерения относятся к колебательно-возбуж-
денному иону "“СН2 [349, 372]. Усовершенствование методики
эксперимента позволило зарегистрировать спектр “СН2 в усло-
виях заведомой релаксации этой частицы в основное колебатель-
ное состояние, в результате чего было получено AEst~
^37,6 кДж/моль [348]. Это значение хорошо совпадает с AZTst,
измеренной с использованием спектроскопии лазерного магнит-
ного резонанса (37,9+2,5 кДж/моль [372]) и с корректирован-
ными данными наиболее точных НЭР (^37,5 кДж/моль [144]).
В итоге разность энергий низших триплетного и синглетного со-
43
cf2
сн(сн3)
СН(ОН) СС12 СНГ СНС1 СНВг сн2с(ск^си2снд
б
SiH2
• синглетное основное состояние
SiHti
триплетное основное состояние
40 80 120
-200 -160 -120 -80 -40 О
АЕ^кДж^моль
Рис. 2.3. Синглет-триплетное расщепление некоторых карбенов и силиленов
{3751:
а — синглетное основное состояние; б — триплетное основное состояние
стояний молекулы метилена можно считать надежно установ-
ленной в пределах 37—38 кДж/моль.
Неэмпирические расчеты геометрии и энергетического рас-
щепления между основным и первым возбужденным состояния-
ми проведены и для ряда других карбенов и их аналогов (см.
[375], а также рис. 2.2 и 2.3). По данным этих расчетов, в пол-
ном соответствии с экспериментальными данными, видно, что
при наличии в сс-положении к карбеновому центру атомов F, С1,
N или О основным состоянием является синглет, а при наличии
только атомов С или Н — триплет, что соответствует результа-
там исследования методом ЭПР.
Обнаружено [377], что вычисленные неэмпирически значе-
ния AEst синглет-триплетного расщепления ряда карбенов СНХ
дают хорошую линейную корреляцию с л-донорной способно-
стью заместителей X, определяемой по изменению полной
л-электронной плотности при переходе от СбНб к CeHgX. По-
скольку л-донорная способность коррелирует с эмпирическими
константами заместителей X o°R, то рассчитанные значения
AEst также хорошо коррелируют с o°R. В соответствии с этим
для карбенов CXY предложено [377] простое соотношение,
предсказывающее значение AESt (в кДж/моль):
AfST = 20,2(0° + о®) + 3J
Значения AESt для карбеноподобных молекул МХУ связаны
с изменением прочностей связей в аналогичных им насыщенных
соединениях МХУН2 и ненасыщенных молекулах XYM=MXY
[378].
Для алкилзамещенных карбенов НЭР уверенно выявляют
триплетное основное состояние [20, 379—381]. При этом увели-
чение объема заместителей, сопровождающееся увеличением
валентного угла при карбенном атоме углерода, в соответствии
44
с теоретическими соображениями [20, 21, 382] усиливает отно-
сительную стабилизацию триплетной частицы. Например, у
бис(трет-бутил)карбена низшее синглетное состояние, согласно
НЭР [383], дестабилизировано по сравнению с основным трип-
летным уже на »105 кДж/моль. За влиянием введения к кар-
бенному центру гетероатомов с неподеленными парами электро-
нов (см., например, [20, 21, 24, 25, 377, 384, 385]) можно просле-
дить на примере НЭР ряда фторзамещенных карбенов [386]
(A£st>0, если основное состояние триплетно):
Карбен A£ST, кДж/моль Карбен AEST, кДж/моль
сн2 37—38 CHCF3 «54
CHF «—26 CFCF3 «—38
cf2 «—190 C(CF3)2 «74
Из приведенных данных видно, что атом фтора стабилизи-
рует синглетное состояние карбена (A£st<0), тогда как триф-
торметильная группа, напротив, стабилизирует триплетное со-
стояние частиц CHCF3 и C(CF3)2- По данным НЭР [387], для
хлор- и бромкарбенов также характерно синглетное основное
состояние, однако абсолютное значение AFst невелико (—17-4-
—23 кДж/моль).
Подробно исследовано электронное строение винилкарбенов.
Для этих частиц в соответствии с данными ЭПР (см. выше,
а также [388]) НЭР определяют триплетное основное состоя-
ние, а относительную дестабилизацию низшего синглета
(AFst) оценивают в 42—51 кДж/моль [389, 390]. Изомер винил-
карбена — циклопропилиден, согласно данным НЭР, имеет син-
глетное основное состояние Мр, относительная энергия триплет-
ной частицы (3Bi) составляет 37—160 кДж/моль [389—391],
т.е. ABst<0.
Первым представителем циклических ненасыщенных карбе-
нов является циклопропенилиден. Для него в соответствии
с моделью Р. Хоффмана (см. разд. 2.2.1) по данным НЭР уста-
новлена [392, 393] синглетность основного состояния (MJ.
Этот карбен должен характеризоваться высокой нуклеофиль-
ностью, что согласуется с поведением его 2,3-дифенилзамещен-
ного [393, 394]. Триплетный циклопропенилиден дестабилизи-
рован на ^290 кДж/моль, причем энергии состояний 3Bi и 3Л2
близки [392]. Известен и НЭР силациклопропенилидена-1, кото-
рый оказался самым стабильным изомером состава C2SiH2
[395]. Молекула циклопентадиенилидена, также в согласии с
моделью Р. Хоффмана, обладает триплетным основным состоя-
нием (см. параметры спектра ЭПР в табл. 2.7); относительная
дестабилизация низшего синглета составляет около 88 кДж/моль
[26, 28].
Наиболее интересной в этом ряду оказалась молекула цик-
логептатриенилидена. Напомним, что для этой частицы вслед-
45
ствие образования единой циклической системы 4л+2 л-элек-
тронов с участием вакантной р-орбитали карбена, по-видимому,
будет наблюдаться стабилизация синглетного основного состоя-
ния [21]. Однако по расчетным данным [27, 28, 396, 397], син-
глетный циклогептатриенилиден менее стабилен, чем соответст-
вующий аллен — синглетный циклогептатетраен. Более того, на
синглетной ППЭ С7Н8 точка, соответствующая синглетному цик-
логептатриенилидену, скорее всего вообще не является миниму-
мом. В соответствии с этим в условиях низкотемпературной
стабилизации вместо синглетного циклогептатриенилидена оха-
рактеризован лишь синглетный тетраен. Однако триплетный
циклогептатриенилиден (32) надежно зафиксирован в матри-
це Аг при 21 К по спектру ЭПР [323] (см. табл. 2.7). Согласно
этим данным, триплетный циклогептатриенилиден не переходит
в циклогептатетраен (33) ни при термическом воздействии (до
35 К), ни под действием облучения. Иными словами, циклогеп-
татриенилиден, видимо, или обладает триплетным основным
состоянием, или, будучи синглетом, характеризуется небольшим
значением A2?st [323].
(34) (35)
Основное состояние моноарилкарбенов СНАг триплетно*
[176], однако при введении к карбенному центру атомов гало-
генов наблюдается заметная стабилизация синглетной конфигу-
рации, и основное состояние арилгалогенкарбенов СХАг явля-
ется синглетным, судя по их химическим свойствам (см., напри-
мер, [287, 398, 399]), а также по отсутствию сигнала ЭПР
в условиях, допускающих регистрацию оптических спектров
этих частиц [173].
Для большинства диарилкарбенов основное состояние явля-
ется триплетным*. Вместе с тем у дифенилкарбена A£St состав-
ляет всего 10,5—17 кДж/моль [174, 400], а химическая реак-
* Поданным ЭПР-спектров (см. табл. 2.7).
46
ционная способность этой частицы отвечает существованию
в растворах быстрого равновесия 1CPh2+=3CPh2. Значение /Сравн
составляет (5,4+1)-103 [277], а для AG этой реакции в ацето-
нитриле и изооктане получены значения —13,2+1,3 и —19,3+
+ 1,3 кДж/моль соответственно [174]. При введении в аромати-
ческое ядро алкильных заместителей валентный угол при кар-
бенном атоме углерода, а вместе с ним и AEst увеличиваются
[282] в ряду:
CPh2 < CPhMes < С(С6Н4Ме-2) Mes < CMes2
(Mes = 2,4,6-Me3C6H2)
У бис(мезитил)карбена, частицы с почти линейной геомет-
рией при карбенном атоме [175], AESt на 8 кДж/моль больше,
чем у дифенилкарбена [282], и химическое поведение этой ча-
стицы не соответствует равновесию с участием ^Меэг [175].
Другим примером ароматического карбена с высоколежащим
синглетным состоянием является 9,10-дигидро-9-мезитил-9-бора-
антраценилиден (36) (AESt>22 кДж/моль [401]).
(36) (37)
Напротив, в молекулах флуоренилидена [400, 402] и его
2,3-бензопроизводного [322] AESt<8 кДж/моль, и в жидкофаз-
ных реакциях с различными акцепторами участвуют как син-
глетные, так и триплетные частицы [174, 175, 273]. В то же
время у 3,6-диметоксифлуоренилидена [268] и ксантенилидена-9
[293] основное состояние синглетно, причем для ксантенилиде-
на (37) энергия дестабилизации триплета составляет
~17 кДж/моль [293]. Относительная стабильность синглетных
и триплетных состояний карбенов (36) и (37) полностью соответ-
ствует теоретическим представлениям [21] об участии вакант-
ной АО атома бора или неподеленной пары электронов атома
кислорода в' циклическом сопряжении.
Экспериментальные и расчетные оценки энергии синглет-
триплетного расщепления проведены и для ряда аналогов кар-
бенов. Так, для силилена SiH2 основное состояние является
синглетным; по данным лазерной фотоэлектронной спектроско-
пии энергия возбуждения в триплетное состояние оценивается в
47
^58,6 кДж/моль [351]. В то же время на основании лучших
НЭР дестабилизация 3S1H2 составляет 63—78 кДж/моль [150,
403—405]. При введении к силиленовому центру электроот-
рицательных заместителей с неподеленными парами электронов,
как и в случае карбенов, наблюдается стабилизация синглетно-
го состояния (см., например, [88, 406—408]); в частности, для
SiF2 по расчетным [403] и экспериментальным [71] данным
оценочное значение энергии дестабилизации триплета составля-
ет 306—314 кДж/моль, а для SiHF по расчетным данным оно
равно 158,2 кДж/моль [409]. В то же время гипотетическая
частица SiHLi, согласно НЭР [410], должна иметь триплетное
основное состояние при значении A£St~29 кДж/моль; триплет -
+ —
ны в основном состоянии и силилидены Si = C = O и Si = N = N
[337]. Для гермиленов и станниленов по расчетным и экспери-
ментальным данным A£st<0, т.е. основное состояние синглет-
но [248, 411, 412]; энергии возбуждения в нижний триплет для
GeH2, GeF2 и GeMe2 составляют соответственно 37—42, 268—
310 и 58 кДж/моль [411, 412], а для молекулы SnH2 AEst~
—92 кДж/моль [248].
2.2.5. Анализ спиновой мультиплетности основного
и возбужденных состояний карбенов
по их химическим превращениям. Метод ХПЯ
Вопрос о спиновой мультиплетности реагирующего карбена яв-
ляется одним из центральных вопросов химии этих частиц. Ос-
новную информацию о спиновой мультиплетности (см. табл. 2.3,
2.4) дают физико-химические исследования изолированных ча-
стиц. Помимо расчетного пути важную роль играют исследова-
ния матричных спектров ЭПР, которые непосредственно позво-
ляют детектировать триплетное основное состояние. Для простых
трехатомных карбенов используют значительно более широкий
набор физико-химических методов.
Разработан ряд эмпирических тестов на спиновую мульти-
плетность реагирующей частицы. В первую очередь необходимо
отметить постулаты Скелла [413, 414], согласно которым спи-
новое состояние карбена предопределяет его реакционную спо-
собность. Так, при реакции синглетных частиц с синглетными
акцепторами нет запретов, обусловленных необходимостью
сохранения спина, и возможен одностадийный процесс, напри-
мер стереоспецифическое присоединение к олефинам или одно-
стадийное внедрение в ординарные связи. В то же время синг-
летные карбены не должны взаимодействовать с триплетными
частицами типа О2, которые, однако, должны эффективно реа-
гировать с триплетными карбенами. Последние в свою очередь
48
не могут одностадийно перехватываться синглетными акцегь
торами.
1С
Продукты,
характерные
для триплетных
карбенов
А
V
Продукты,
характерные для
синглетных карбенов
Наиболее характерной реакцией, позволяющей определить
спиновую мультиплетность реагирующей частицы, считается
взаимодействие со стереохимически меченными олефинами.
Согласно постулату Скелла [413], присоединение синглетных
карбенов по связи С = С проходит одностадийно и ^цс-стерео-
специфично, а для триплетных карбенов стереоспецифичность
отсутствует, поскольку одностадийное присоединение запрещено
законом сохранения спина, а в промежуточном триплетном би-
радикале (38) вращение вокруг связи С-1—С-2 происходит
быстрее, чем инверсия спина:
2/R
.с—с.
R'X+ |
lcx2
А I
I - ;
R\1 X2Zr 3сх2 R\ ZR 'сх2 RK z:
c-f-c *-------- /C=cC -------------► ;c—-c
R'Z' f v | >R' R'X \r' r'z \ /
fcx2 CXi
(38)
R'L ! 2 >R 1 г
у _у
f |_____________________________t I """R' \/
fCX2 |CX2 CX2
Хотя этот постулат и получил определенные обоснования
в квантово-химических расчетах [20, 415—417], по сути своей
он является эмпирическим тестом, который до настоящего вре-
мени наиболее часто используют для установления спиновой
мультиплетности реагирующих частиц. Так, одним из аргумен-
тов в пользу синглетности основного состояния 2,7-диметокси-
4—107
49
флуоренилидена (39) и ксантенилидена-9 (37) является цис-
стереоспецифичность их присоединения к (£)ф-дейтеро-а-метил-
стиролу [268, 293], тогда как присоединение к этому олефину
9-мезитил-9-бораантранилидена (36) полностью нестереоспеци-
фично [401]:
Еще более эмпиричен тест Андо [418], согласно которому
при наличии в молекуле акцептора двух реакционных центров,
как, например, в случае аллилхлорида, синглетный карбен
предпочтительно вступает в реакцию внедрения по связи С—С1,
а триплетный карбен легче присоединяется по кратной связи:
н2с - сн - ch2ci«£^ch2 = chch2ci^Uch2 = СН - СН2СХ2С1
сх2
50
Согласно постулату Шимизу и Нишиды [419, 420], в реак-
циях синглетных карбенов с 1,1-дициклопропилэтиленом проис-
ходит согласованное присоединение с образованием замещен-
ных дициклопропилциклопропанов, а в случае триплетных ча-
стиц возможно также образование замещенных циклогексенов
и ациклических диенов в результате быстрого раскрытия одного
из трехчленных циклов в промежуточном бирадикале (40):
(40)
Так, показано [419], что термолиз диазофлуорена в 1,1-ди-
циклопропилэтилене приводит к аддукту (41) и диену (42)
в соотношении 10:1. Однако при разбавлении декалином
(30 моль), что благоприятствует синглет-триплетному переходу
флуоренилидена (9), преобладает диен (соотношение 1:3).
В то же время при взаимодействии 1,1-дициклопропилэтилена
с СВг2 (из PhHgCBr3, 100 °C [419]) или с CHCO2Et (из
N2CHCO2Et и CuSO4, 100—НО °C [421]) выделены только соот-
ветствующие гел-дициклопропилциклопропаны, что указывает
на синглетное состояние этих карбенов в момент перехвата.
Еще одним примером различной реакционной способности
синглетных и триплетных карбенов является поведение триме-
тилсилилкарбена. В этом случае для возбужденного синглетного
карбена характерна перегруппировка в триметилсилаэтилен,
стабилизированный в Ar-матрице при 10 К; однако при разбав-
лении инертным газом, благоприятствующем релаксации в три-
плетное основное состояние, наблюдается димеризация двух
триплетных частиц с образованием бис(триметилсилил)этиле-
на [422].
4*
51
R = цикло-С3Н5
Me3SiCHN2-^*1CHSiMeo
-n2 j
MeCH = SiMe2
3CHSiMe3------------^Me3SiCH = CHSiMe3
Для синглетных карбенов характерно также внедрение в
связи О—Н спиртов, а для триплетных — гомолитический раз-
рыв менее прочных связей С—Н [273]. Примеры использования
таких тестов для установления спиновой мультиплетности реа-
гирующих карбенов весьма многочисленны (см., например,
[4—11, 175, 273]). В последние годы определение абсолютных
кинетических характеристик карбенных реакций, проводимых
при низких температурах и в растворах, позволило несколько
уточнить подобные постулаты и придать им определенное теоре-
тическое обоснование. Поскольку скорости карбенных реакций
обычно очень велики, а зачастую даже достигают диффузионно-
го предела [174—177, 273, 286, 287], то в неравновесных усло-
виях удается эффективно перехватывать карбен в момент его
образования, т. е. до релаксации этой частицы в основное со-
стояние. В этом случае постулат Скелла оказывается работо-
способным; примером этому может служить генерирование и пе-
рехват синглетных арилкарбенов — частиц с триплетным основ-
ным состоянием [423, 424]:
52
hv
ArCHN2 _____
1CHAr
hv
-n2
-n2
CHAr
R1
N = N
R
H^C = C
H
R R1
H \ / H
CHAr
R H
+ ^.C—
R1
CHAr
>20 : 1
Вместе с тем для многоатомных карбенов взаимный переход
синглетной и триплетной форм также является быстрым про-
цессом (й=109—1010 с-1) [175, 277, 400]), и в равновесных
условиях постулату Скелла, позволяющему различать синглет-
ную и триплетную реакционную способность, будут подчинять-
ся только карбены с большой энергетической щелью между
синглетным и триплетным состояниями (согласно [175] | AEst| >
>10 kT). Для карбенов же с малой энергетической щелью, на-
пример для дифенилкарбена и флуоренилидена, имеющих три-
плетное основное состояние, даже в этих условиях возможно
образование продуктов, типичных для синглетных частиц.
При этом использование пико- и наносекундного лазерного
флеш-фотолиза и лазерно-индуцированной флуоресценции по-
зволяет определить скорости не только взаимопревращения
1iCPh2 и 3СР112, но и скорости реакций их исчезновения в раство-
ре при 20 °C [174] (см. схему на с. 54).
Образование типичных для синглетного состояния продуктов
возможно и путем безызлучательного перехода при взаимодейст-
вии триплетного карбена с акцептором, т. е. в реакциях типа
3CPho + MeOH—>MeOCHPh2 [175]. Однако для практических
целей этот вопрос является в известной мере схоластическим,
поскольку в любом случае для триплетной частицы характерна
синглетная реакционная способность, т. е. в равновесных усло-
виях постулаты Скелла для карбенов с малой энергетической
щелью между основным и возбужденным состоянием непри-
менимы.
Еще одна возможность исследования спинового состояния
карбенов, реагирующих в жидкой фазе, т. е. в условиях обычно-
53
MeOCHPh2
Ph2CN2
Ph2C - CPh2
\/
0
k, MeOH
—* 1CPh2
3CPh2
--------Ph?CN2
сенсибили- *
затор
k3 CH2 = CHCH = CH2
I
Me
I
Ph?C - сб
\ Z 4
CH2
Me
CH = CH2+
Ph2C -/CH - C = CH2
CH2 Me
k, = 7.77-109, k3 = 1,36-Ю6 моль"1 с"1;
kST = 3,23-Ю9, kTS = 6,29-Ю6 с"1
го химического процесса, появилась благодаря созданию мето-
да химически индуцированной динамической
поляризации ядер (ХПЯ) [32,33,425,426].
Наблюдение эффекта ХПЯ в спектрах ЯМР позволяет уста-
навливать электронно-спиновое состояние промежуточно обра-
зующихся карбенов и выявлять дальнейшие их химические
превращения. Однако непосредственным объектом исследования
при этом служат стабильные конечные продукты, образовавшие-
ся из тесной радикальной пары, возникшей в свою очередь в ре-
зультате реакций карбенов:
m[X2C:] + R - Y "* m[X2YC- -R] - X2YC - R
Если возникновение радикальной пары явилось следствием
отрыва карбеном атома Y от соседней молекулы R—Y, то по
знаку эффекта ХПЯ у продуктов реакции можно судить о муль-
типлетности т этого карбена [425, 426]. В частности, таким
путем была установлена мультиплетность генерированных раз-
личными путями метилена, дифенил-, циано- и алкоксикарбо-
нилкарбенов, а также некоторых циклогексадиеноновых карбе-
нов (подробнее см. [425, 426]).
Помимо установления мультиплетности частицы метод ХПЯ
одновременно позволяет определить и селективность их реак-
ций. Так, в реакции с дейтерохлороформом синглетный метилен,
генерированный прямым фотолизом диазирина, отрывает атом
хлора, и в спектре продукта — дейтерированного 1,1,2-трихлор-
54
этана — наблюдается значительная эмиссия метиленовых прото-
нов. В то же время триплетный метилен, полученный при сенси-
билизированном фотолизе, отрывает атом дейтерия, и рекомби-
нация триплетной радикальной пары приводит к дейтеро-1,1,1-
трихлорэтану, в спектре ЯМР которого усилен сигнал метилено-
вых протонов [427]:
, hv
’СН2 *---------- сн2
CDCI3
v
1 [CI2DC CH2Cl]
CI2DC - CH2Cl
hv
Ph2CO
3CH2
CDCI3
3[H2DC Cci3]
H2DC - CCI3
Аналогичным образом с помощью ХПЯ была подтверждена
синглетность диметилрермилена и исследован механизм его
внедрения в связь С—Вг молекулы PhCH2Br [428].
Таким образом, метод ХПЯ, не регистрируя сами карбены,
дает возможность в ряде случаев устанавливать их спиновое
состояние и дальнейшие превращения в реакциях, карбенный
механизм которых доказан другими способами. Вместе с тем
продуманное использование метода ХПЯ позволяет решать мно-
гие актуальные вопросы химии карбенов, в частности, оценить
реакционную способность синглетных и триплетных состояний,
возможность их взаимных переходов, выявить ряд тонких дета-
лей механизма внедрения карбенов в ординарные связи. Подоб-
ную информацию трудно, а порой и невозможно получить дру-
гими методами. Скорее всего, возможности метода ХПЯ далеко
не исчерпаны, и его использование в дальнейшем позволит глуб-
же понять природу карбенов и их аналогов. К числу недостат-
ков метода ХПЯ следует отнести, пожалуй, лишь невозможность
количественной оценки доли продукта, образующегося по сво-
боднорадикальному механизму, в том числе и с участием кар-
бенов.
Применение метода ХПЯ в исследовании реакционной спо-
собности карбенов подробнее рассмотрено в обзорах [32, 33,
425, 426].
55
2.3. ЗАКОНОМЕРНОСТИ ЭЛЕКТРОННОГО И ГЕОМЕТРИЧЕСКОГО
СТРОЕНИЯ карбенов и их аналогов
Определение геометрического строения карбенов представляет
собой сложную задачу. Непосредственно дифрактограммы по-
лучены лишь для некоторых неорганических аналогов карбе-
нов— SiCl2, SiBr2, GeCl2 и GeBr2 [34, 35, 95], а для нестабиль-
ных органических молекул лишь недавно удалось впервые осу-
ществить электронографическое исследование аллильного
радикала [429]. В большинстве структурных исследований кар-
бенов и их аналогов геометрия подбирается из условия наилуч-
шего соответствия экспериментально наблюдаемым характери-
стикам типа колебательно-вращательной структуры электронно-
го спектра, частот и изотопных сдвигов в колебательных
спектрах, значений параметров D и Е, сигналов ЭПР и т.д.
В этой связи особое значение приобретает использование для
определения геометрического строения нескольких независимых
методов. Для большинства простейших карбенов и их аналогов
эта задача успешно решена (см. табл. 2.1—2.4), причем данные
различных методов не только хорошо согласуются между со-
бой, но п позволяют построить общую феноменологическую мо-
дель электронного и геометрического строения карбенов и их
аналогов.
В первую очередь следует отметить, что по многочисленным
экспериментальным данным в молекулах трехатомных карбенов*
и их аналогов МХ2 связи М—X являются менее прочными, чем
в соответствующих валентнонасыщенных соединениях МХ4.
Это видно при сопоставлении не только длин связей М—X, но
и силовых постоянных соответствующих связей (табл. 2.8).
Изменение энергий диссоциаций (Едис) связей М—F в моле-
кулах MF2 и MF4 также соответствует этой закономерности;
энергии диссоциации таких связей [115, 449] приведены ниже
(в кДж/моль):
р
дис
F—CF
F—SIF
F—GeF
486—516
645—650
486
•£дис
F—CF3 544
F-SiF3 671-712
F— GeF3 519
Такая тенденция полностью согласуется с анализом строе-
ния молекул МХ2 и МХ4 методом МО [450]. В синглетных моле-
кулах МХ2 часть занятых орбиталей образована в результате
взаимодействия неподеленных пар электронов атомов М и X, не
участвующих в образовании валентных связей. Эти орбитали
карбенов МХ2 не имеют аналогий среди МО молекул МХ4 и яв-
ляются разрыхляющими в области связей М—X, как, напри-
мер, орбиталь 2&2 в молекулах МХ2 [449, 450]:
56
Таблица 2.8. Длины связей и силовые постоянные молекул МХ2 и МХ4
Молекула МХ2 Молекула МХ4
м X £ ^М-Х’ 10-2 Н/м библиогр. ссылки й я % к. ^М-Х’ 10-2 Н/м библиогр. ссылки
с н 0,108 5,2 [37, 62] 0,1093 5,4 [62, 433]
F 0,130 6,0—6,19 [34, 41, 42, 56, 207] 0,132 5,6—7,0 [32, 34, 433]
С1 0,170— 0,176 3,0 [212] 0,177 3,1—3,6 [34, 212, 433]
Вг 0,187— 0,191 2,38 [59] 0,1942 2,4 [62, 433]
Si н 0,152 2,4—2,8 [64] 0,148 2,8 [434—436]
F 0,159 5,0—5,4 [191] 0,155 6,2—6,6 [34, 437, 438]
С1 0,204— 0,208 2,2—2,4 [83, 84, 430] 0,202 2,8—3,4 [34, 99, 439—441]
<де н 0,160 2,1 [89] 0,153 2,6 [436]
F 0,173 4,1 [90, 92] 0,167 5,6 [34, 440— 442]
С1 0,219 2,1 [430— 432] 0,211 2,4—2,9 [34, 206, 430, 439]
Sn Вг 0,234— 0,235 1,6 [97] 0,227— 0,234 1,9—2,4 [34, 443, 444]
С1 0,234 1,9—2,1 [34, 102, 103, 430] 0,228— 0,230 2,5—2,6 [34, 430, 440, 441, 445—447]
Pb С1 0,245 1,7 [34, 102, 103, 430] 0,243 2,1 [430, 445— 448]
Это и обусловливает некоторое ослабление связей в карбе-
нах. Тем самым, и экспериментальные, и теоретические данные
позволяют решительно отвергнуть как некорректную раннюю
гипотезу [451, 452] о «частичной двоесвязанности» атомов М
и X в галогенкарбенах вследствие возможного вклада биполяр-
ных структур, возникающих в результате взаимодействия ва-
кантной р-орбитали центрального атома М и свободных элект-
ронных пар атомов галогенов X:
+ “ • ♦ — 4-
X = М-Х^Х- М -Х^Х - М = X
57
Другой важной структурной характеристикой карбенов, оп-
ределяющей их электронное строение, является валентный угол
в основном и возбужденных состояниях (см. табл. 2.1, 2.5). На-
помним, что низшее синглетное состояние является основным
для таких аналогов карбенов, как силилены, гермилены, станни-
лены и плюмбилены. Для самих карбенов, как было показано
выше, основное состояние синглетно только при наличии у цент-
рального атома электроноакцепторных заместителей, в частно-
сти, это справедливо для галоген- и дигалогенкарбенов.
Из данных, приведенных в табл. 2.1 и 2.5, нетрудно видеть,
что молекулы всех трехатомных карбенов изогнуты и в основ-
ном, и в возбужденном состояниях, причем для синглетных ча-
стиц значения валентного угла значительно меньше. Эту зако-
номерность интерпретируют [449] на основе электростатической
теоремы Гельмана — Фейнмана, согласно которой равновесная
конфигурация молекулы определяется сбалансированностью
для каждого атома сил притяжения его ядра с центрами элект-
ронной плотности неподеленных пар и валентных связей, а так-
же менее существенного кулоновского взаимодействия несвязан-
ных атомов. В случае синглетного карбена наличие двух
электронов на нецентросимметричной (7-орбитали* и предопреде-
ляет [449, 450] меньшее значение валентного угла в сравнении
с триплетной частицей, где один электрон находится на центро-
симметричной p-АО (см. рис. 2.1).
В ряду трехатомных карбенов и их аналогов с увеличением
порядкового номера центрального атома уменьшается валент-
ный угол в основном состоянии молекулы, а увеличение поряд-
кового номера заместителя сопровождается увеличением валент-
ного угла (табл. 2.9). Эти закономерности тоже могут быть ин-
терпретированы на основе теоремы Гельмана — Фейнмана (см.
[449, 450]), причем наиболее существенно, что такое изменение
валентного угла отражается на электронном строении карбенов
и их аналогов. Так, уменьшение валентного угла снижает энер-
гию c-орбитали вследствие стабилизирующего взаимодействия
сближающихся АО концевых атомов и увеличения вклада
5-АО центрального атома, однако р-орбиталь карбена практи-
чески нечувствительна к изменению валентного угла.
Энергии а-орбиталей карбенов могут быть определены из их
потенциалов ионизации (ПИ) (см. табл. 2.3 и 2.9). Характерно,
что по значениям ПИ синглетные карбены и их аналоги, содер-
жащие несвязывающие, но спаренные электроны с относительно
невысокой энергией, занимают промежуточное положение между
* Здесь и далее предполагается, что в низшее синглетное состояние карбе-
на с энергией 1£_ (см. разд. 2.1) основной вклад вносит низшая по энергии
конфигурация Щ2, что вполне согласуется с данными НЭР при учете конфигу-
рационного взаимодействия (см., например, [23, 24]).
58
Таблица 2.9. Некоторые характеристики геометрического
и электронного строения ряда карбенов
и их аналогов МХ2 [449]
Молекула <XMX в состоянии Mi, град.а ПИ, эВ Энергия перехода эВ
сн2 102 10,40 1,7
SiHo 92 — 1,9
GeH2 90 — —
cf2 105 11,8 5,0
SiF2 101 11,0—11,3 5,5
GeF2 97 11,6—11,8 5,4
SnF2 95 11,5 5,1
PbF2 95 11,6 5,0
CC12 108 9,10—9,76 2,5
SiCl2 105 10,0—10,6 3,7
GeCl2 100 10,4 3,8
SnCl2 99 10,2 3,9
PbCl2 98 10,3 3,9
CBr2 110 10,0 1,9
SiBr2 Ю5 — >2,5
GeBr2 lol 9,5 •—
SnBr2 ioo 10,0 _—.
PbBr2 10° 10,2 1—
а Оценочное значение. Рекомендуемые значения см. в табл. 2.5.
соответствующими радикалами *МХ и *МХ3 с неспаренным
электроном и валентнонасыщенными молекулами МХ4 (подроб-
нее см. [449, 450]). При этом для дигалогенидов элементов
подгруппы IVB увеличение порядкового номера центрального
атома практически не сказывается на потенциале ионизации,
а увеличение порядкового номера атома X систематически сни-
жает его (см. табл. 2.9).
Поскольку непосредственное определение энергии р-орбита-
ли карбенов невозможно, удается проследить лишь за влия-
нием природы атомов М и X на разность энергий о- и р-орбита-
лей, оцениваемую из энергий переходов Mi—^Bi (см. табл. 2.9).
Оказывается, что при переходе от карбенов к их аналогам по
подгруппе IVB энергия р-орбитали, как правило, существенно
возрастает, хотя электронное строение силиленов, гермиленов,
станниленов и плюмбиленов весьма сходно. Особенно характер-
но это для гермиленов, станниленов и плюмбиленов, где (при
одних и тех же заместителях X) достаточно единообразно не
только электронное, но и геометрическое строение (см. табл. 2.9).
В то же время при переходе от дигидридов элементов подгруп-
пы IVB к соответствующим дигалогенидам, в частности от ме-
тилена к дигалогенкарбенам, энергия р-орбитали заметно воз-
растает [449, 450]. Напомним, что положение о- и р-орбиталей
59
карбена в первую очередь и определяет его электрофильную
(р-орбиталь) и нуклеофильную (а-орбиталь) реакционную спо-
собность.
К сожалению, результаты физико-химических исследований
строения более сложных карбенов существенно менее полны,
что связано с трудностью получения и неоднозначностью интер-
претации соответствующих экспериментальных данных. Вместе
с тем, основываясь на достаточно полной и непротиворечивой
картине строения трехатомных карбенов, можно с определенной
уверенностью описать предполагаемое электронное строение не-
которых карбенов со сложными органическими заместителями.
Так, диалкильные и диарильные производные, не содержащие
в a-положении электроотрицательных заместителей с неподе-
ленными парами электронов, должны напоминать метилен, по-
скольку влияние подобных заместителей на энергию и форму
о- и р-орбиталей невелико. Иными словами, основное состояние
таких карбенов будет триплетным, что соответствует данным
ЭПР и неэмпирических расчетов (см., например, [25, 453]).
Карбены с заместителями, содержащими в а-положении
электроотрицательные атомы с неподеленными парами электро-
нов*, по свойствам должны быть близки к галоген- и дигало-
генкарбенам. В частности, основное состояние амино- и алк-
оксикарбенов должно быть синглетным с доминирующим вкла-
дом конфигурации 1о2.
Описанные выше правила определения электронного строе-
ния карбенов, в частности их мультиплетности, разумеется, яв-
ляются достаточно приближенными и не в состоянии описать
весь спектр реакционной способности карбенов, основанный на
тонких различиях в их электронном строении из-за влияния за-
местителей. Подобные детали будут по мере необходимости
обсуждаться при рассмотрении конкретных классов карбенов
и типов их реакций.
Приведенные в этом разделе примеры показывают, как ис-
пользование различных физико-химических инструментальных
и расчетных методов позволяет построить цельную и непротиво-
речивую картину строения карбенов и их аналогов. Знание фи-
зико-химических характеристик карбенов в большинстве слу-
чаев позволяет корректно планировать и, тем более, интерпре-
тировать химический эксперимент. Вместе с тем для практиче-
ских целей более важно исследование механизмов карбенных
* Ранее доминирующим считалось влияние неподеленных пар а-атомов
[20], однако недавние НЭР [25] на первый план выдвинули индуктивную
электроотрицательность. Для практики этот вопрос является в известной мере
схоластическим, так как большинство индуктивно-акцепторных заместителей
одновременно обладает и неподеленной парой электронов (атомы N. О, Hal).
60
реакций, а не самих этих частиц. К систематической классифи-
кации карбенных реакций и рассмотрению общих закономерно-
стей их механизмов мы и переходим.
2.4. ОСНОВНЫЕ ТИПЫ КАРБЕННЫХ РЕАКЦИЙ
Высокая активность карбенных центров предопределяет их бы-
строе исчезновение в реакционных смесях, причем типы «моле-
кул-перехватчиков» достаточно разнообразны. Вместе с тем
карбенные реакции удается достаточно четко классифициро-
вать. Поскольку большинство реакций характерно для всех ти-
пов карбенов, в основе такой классификации лежит тип реак-
ционного центра, перехватывающего карбен, — центральный
атом другой молекулы карбена (реакции ди- и полимеризации),
двойная или тройная связь (реакции присоединения), ординар-
ная связь (реакции внедрения), гетероатом с неподеленной па-
рой электронов или атом переходного металла (реакции комп-
лексообразования). Большинство таких реакций возможны и во>
внутримолекулярном варианте, однако ряд внутримолекулярных
реакций карбенов обладает определенной спецификой и заслу-
живает отдельного рассмотрения. В настоящем разделе для
большинства типов карбенных реакций приведены только общие
сведения об их протекании и наиболее вероятном механизме;
возможность использования реакций того или иного типа рас-
смотрена отдельно (см. гл. 4—6).
2.4.1. Межмолекулярные реакции карбенов и их аналогов
Димеризация и полимеризация. Простейшим случаем меж-
молекулярного взаимодействия карбенов и их аналогов являет-
ся рекомбинация с образованием димеров и (или) полимеров.
Многочисленные экспериментальные данные показывают, что
в отсутствие подходящих акцепторов молекулы карбенов реком-
бинируют с образованием димеров, тогда как аналоги карбенов
образуют высшие циклические и линейные олигомеры и по-
лимеры.
Ряд квантово-химических расчетов различного уровня стро-
гости показывает, что димеризация синглетного метилена про-
ходит не по пути «наименьшего движения», а по достаточно
сложной траектории [405, 454—462] (см. схему на с. 62).
На больших расстояниях (г>0,3 нм) взаимная ориентация
карбенов соответствует оптимальному взаимодействию вакант-
ной р-орбитали одной молекулы с занятой о-орбиталью дру-
гой (а). При г«0,2 нм начинает осуществляться переход на
траекторию наименьшего движения (б), окончательно форми-
рующуюся при г<0,2 нм (в) (подробнее см. [463]). Барьер ак-
61
тивации этой реакции невысок (^33 кДж/моль [458]) или да-
же отсутствует [461].
6
Путь наименьшего движения не является оптимальным
маршрутом и для сближения синглетных карбена и силилена,
а также двух молекул образование лабильных димеров
H2C = SiH2 и H2Si = SiH2, соответственно, не требует барьера
активации [405].
Для взаимодействия синглетного и триплетного метиленов
расчет предсказывает оптимальность пути наименьшего движе-
ния; Еакт образования триплетного этилена высока и составляет
^577 кДж/моль (г = 0,4 нм) [461]. Рекомбинация двух триплет-
ных частиц в зависимости от взаимной ориентации спинов моле-
кул может приводить к этилену в основном или синглетном воз-
бужденном состояниях [461]. По расчетным данным [461, 464,
465], для первого процесса барьер активации отсутствует, что
согласуется с экспериментально определенным высоким значе-
нием константы скорости димеризации 3СН2 [5,3-108 л/(молеку-
ла-с)] при 298 К [466]; однако высокий тепловой эффект
(^730 кДж/моль*) делает достаточно вероятным распад «го-
рячего» этилена. Именно это и наблюдается в эксперименте:
димеризация триплетного метилена приводит к ацетилену и во-
дороду [466]:
23СН2~* [СН2 = СН2]**-> СН = СН + Н2
Согласно расчетным данным, описанные выше закономерно-
сти сохраняются и для фтор- и дифторкарбенов [461], хотя
введение к карбенному атому заместителей вносит определен-
ную специфику.
Димеризация синглетного дифторкарбена в газовой фазе, ле-
жащая в основе промышленного метода получения тетрафтор-
* Рассчитано по (3СНз) (см. табл. 2.2) и A£/f° (СН2=СН2) [467].
62
этилена [468], изучена достаточно подробно; энергия активации
этой реакции составляет 7—21 кДж/моль.
21CF2- C2F4
к = 106’87Т1/2ехр (-5,0/ЯТ) [469]
к = 106’4Т1/2ехр (-1J/RT) [470]
При димеризации более активного фторхлоркарбена в газо-
вой фазе энергия активации равна нулю [471]:
21CCIF- FCIC = CCIF к = 109>7ехр (-0/ЯТ)
Склонность сравнительно стабильных карбенов к димериза-
ции может быть использована, например, для получения трудно-
доступных другими методами фторсодержащих мономеров,,
прежде всего тетрафторэтилена и других фторолефинов, в част-
ности 1,1-дифторэтилена и трифторхлорэтилена [468]. Реакция
рекомбинации двух разных карбенов лежит в основе синтеза
афф-трифторстирола и ряда фторакрилонитрилов (подробнее
см. [468]):
:CF2 + :CFPh- F2C = CFPh 2 :CFCN- NCFC = CFCN
Вместе с тем в ряде случаев, как уже отмечалось выше, об-
разование продуктов формальной рекомбинации двух карбенов
обусловлено не их прямым взаимодействием, а атакой карбена
стабильной молекулы (предшественник карбена) [4]. В част-
ности, такой процесс достаточно характерен для диазосоедиие-
ний (см. разд. 3.1.2); известна и матричная реакция метилена
с диазометаном при 6—10 К, сопровождающаяся хемилюмине-
сценцией возбужденного этилена [220]:
3СН2 + CH2N2 [Н2С = СН2Г - н2с = сн2
— N2
Однако для ряда карбенов, как уже отмечалось, димериза-
ция является ярко выраженным процессом. Так, димеризация^
Например, циклогептатриенилидена, генерируемого фотохимиче-
ски или термически из диазосоединения (31), с образованием
63
гептафульвена (34) происходит как в инертной матрице [323]
(см. разд. 2.2.4), так и в газовой фазе [472]:
., . 250 °C г 7=^1
PhCH = N - NTs Na+ [:CHPh Q]
PhCH = CHPh 4- (34)
(15%) (70%)
В матричных условиях наблюдается также димеризация три-
метилсилилкарбена [422], циклопентадиенилидена [239] и фе-
нилхлоркарбена [473]:
.я bv,Ar,12K о 40-45 К
Me3SiCHN2----—>3CHSiMe3 —^-^Me^iCH = CHSiMe3
PhCICN2 1CCIPh -4-°~45K>PhCIC = CCIPh
-n2
В целом же, как правило, препаративная ценность реакций
димеризации карбенов невелика; эти реакции скорее являются
нежелательными побочными процессами, снижающими выхо-
ды целевых продуктов. В химии аналогов карбенов, напротив,
реакции ди- и полимеризации во многих случаях более типичны
и широко используются в синтезе.
Внедрение в ординарные связи. Внедрение в простые связи
типа С—Н, С—Hal, О—Н, N—Н, S—Н, М—Н, М—Hal (M=Si,
Ge, Sn и т. п.) является наиболее общей реакцией карбенов и их
аналогов, исследованной на большом числе примеров.
Для внедрения карбена СХ2 в ординарную связь А—В воз-
можны два альтернативных механизма — трехцентровый син-
хронный (ц) и свободнорадикальный типа «отрыв — рекомбина-
ция» (б), причем в рамках обоих механизмов возможно предва-
рительное образование комплекса карбена с акцептором, в том
числе комплекса илидного типа (см., например, [474—477]):
:СХ2 + А - В
zCX2
А-----'в
•А -г -СХ2В
64
Для реакций триплетных карбенов одностадийное образова-
ние синглетных продуктов внедрения запрещено законом сохра-
нения спина, что предопределяет реализацию механизма (б) с
образованием триплетной радикальной пары. Эта пара может
рекомбинировать с образованием продукта внедрения, однако не
исключен и выход радикалов из клетки в объем. Для синглет-
ного карбена разрешен и прямой одностадийный путь внедрения
(«)•
В большинстве расчетных работ исследованы простейшие
реакции внедрения метилена в связь Н—Н молекулы водорода
или в связь С—Н молекулы метана [417, 456, 463, 478—481].
Для реакции 3СН2 + Н2 расчеты выявили невыгодность образо-
вания триплетного метана, причем энергетически наиболее вы-
годно (£акт = 67—109 кДж/моль) образование пары радикалов
• СНз+-Н [463]. Для реакции ^Нг + Нг барьер активации не
превышает 25 кДж/моль, а путь реакции не соответствует пути
наименьшего движения, включая на первой стадии электрофиль-
ную атаку р-орбитали карбена на связь Н—Н. В соответствии
с этими закономерностями находится известный эксперимен-
тальный факт: скорость реакции 3СН2+Н2 на два-три порядка
меньше скорости реакции ^Нз + Нг ([463].
То же соотношение скоростей характерно и для реакции
метилена с метаном [463]. Согласно расчетным данным, в слу-
чае синглетного метилена [схема (2.4)] [417, 478—482] первая
стадия (а) (г>0,3 нм) соответствует электрофильной атаке
р-орбитали карбена (М = С, Х = Н) на связь С—Н [связь А—В
в схеме (2.4)], а затем проходит безактивационный (или почти
безактивационный) перенос атома водорода к карбенному цент-
ру, соответствующий нуклеофильному взаимодействию о-орби-
тали с мигрирующим атомом (б) и приводящий к тесной ради-
кальной паре (в) и далее к конечному продукту (г):
а б в г
По результатам очень точного неэмпирического расчета
в базисе ОСТ-6-31 * ГФ с введением поправок третьего порядка
теории возмущений [480] для реакции 1СН2 + Н2->СН4 барьер
активации равен нулю. С помощью НЭР для реакции ^Нг+Нг
найдено, что барьер активации равен 8 кДж/моль; при введении
к карбенному центру атомов фтора наблюдается последователь-
ное увеличение барьера: 64 кДж/моль для реакции ^НР + Нг
5—107
65
и 197 кДж/моль для реакции ^Рг + Нг [481].
По данным полуэмпирического расчета методом МЧПДП/3,
при введении к карбенному атому или атакуемой связи алкиль-
ных заместителей, экранирующих реакционный центр, также
наблюдается некоторое увеличение барьера активации [482].
Характерно, что тепловые эффекты реакций внедрения синглет-
ного метилена в связи С—Н зависят от типа связи. Так, внедре-
ние метилена во вторичную связь С—Н на 6—8 кДж/моль
более экзотермично, чем внедрение в первичную связь С—Н, а
переход к третичной связи С—Н повышает тепловой эффект еще
на ж 4 кДж/моль*. Эта тенденция полностью соответствует
известной предпочтительности внедрения !СН2 в более замещен-
ные связи С—Н. Определенные основания ожидать снижения
теплового эффекта внедрения есть и при переходе от метилена
к алкилзамещенным карбенам [482]1.
При взаимодействии со связями, образованными атомами с
неподеленными парами электронов (связи С—Cl, N—Н, О—Н,
F—Н, Р—Н, S—Н, Н—С1 [480, 483]), изменение механизма
* Найденная закономерность не является артефактом расчета. Для иллю-
страции можно сопоставить реакции, в которых стандартные энтальпии обра-
зования алканов определены экспериментально [467]:
(СН3)2СНСН3ХсН3СН2СН3 + 1сн2 ^*сн3сн2сн2сн3
ААН(а-б) = 8 кДж/моль
а_>_(СН3СН3)2СНСН3
(СН3)2СНСН2СН3 + 1сн24
[б_^(СН3)2СНСН2СН2СН3
(СН3)2СНСН(СН3)2 (СН3)3ССН2СН3
ДА Н(а - б) = 2,7 кДж/моль; ДДН(а - в) = 7,3 кДж/моль;
ДД Н(а - г) = 14,3 кДж/моль
66
внедрения метилена не наблюдается — по-прежнему центром
электрофильной атаки остается именно эта связь (связь А—В)
[см. схему (2.4)], а не атом с неподеленной парой электронов.
Переход от карбенов к их аналогам, как правило, снижает теп-
ловой эффект реакции внедрения, одновременно увеличивая
барьер активации, Так, согласно уже упоминавшемуся НЭР
[480], £акт для реакции 1SiH2 + CH4->CH3SiH3 равна 115 кДж/
/моль при тепловом эффекте —209 кДж/моль, тогда как для
реакции ]СН2 + СН4->С2Нб £^ = 0 и ДДЯ = —428 кДж/моль.
Специфика карбеноподобной молекулы МХ2 и атакуемой
связи А—В во многом определяют и судьбу радикальной пары.
Так, расстояние переноса концевого атома В к карбенному
центру М [см. схему (2.4)] определяется по уравнению:
° га + гв + гв + гм
В итоге в тесной радикальной паре (в) расстояние между
центрами М и А может либо соответствовать, либо не соответст-
вовать существованию достаточно прочной связи М—А. Степень
растяжения связи М—А характеризуется параметром 7л
X = D/(rM + гА) = 1 + 2гв/(гм + Гд)
Следовательно, увеличение порядкового номера (ковалент-
ного радиуса гв) атакуемого атома увеличивает расстояние
переноса и вероятность расхождения радикалов в объем без их
непосредственной рекомбинации. Напротив, увеличение поряд-
кового номера центрального атома М или атома В реагирующей
связи А—В повышает вероятность непосредственной рекомбина-
ции радикалов [449]'. В соответствии с этим в реакциях внедре-
ния синглетных карбенов, например, по связям С—С1 (%^2,3)
детектированы свободные радикалы, тогда как при внедрении
в связи С—Н (X ~1,4) эффекты ХПЯ отсутствуют [426].
Обсуждаемый механизм внедрения синглетных карбенов
позволяет понять и их селективность по отношению к связям
различных типов. Действительно, в ходе внедрения доминирую-
щим на первой стадии является взаимодействие р-орбитали
карбена с занятой о-МО атакуемой связи. Поэтому связи с бо-
лее высоколежащими ВЗМО (М—М, М—О, М—И, М—Hal;
M=Si, Ge, Sn и т. п.) легче акцептируют карбены. При этом,
если подобное различие не столь заметно для наиболее неселек-
тивного из карбенов — метилена, который, например, в реакции
с алкилфторсиланами образует продукты внедрения в связи всех
типов 1[484], то более селективные галоген- и дигалогенкарбены,
а также аналоги карбенов, как правило, полностью инертны к
5‘ 67
связям С—Н, однако легко внедряются в связи типа С—Hal,
Si—О, Si—Н, Si—Hal и т. п.
В ряде случаев реакции внедрения с участием нуклеофиль-
ных реагентов, обладающих неподеленными парами электронов,
видимо, связаны с промежуточным образованием илидов или
ионных пар (см., например, [485], а также [474—477]):
Реакция внедрения в связь О—Н алифатических спиртов
типична для синглетных карбенов [273]. Для реакции
1CPh2 + EtOH -*Ph2CHOEt
методом фотоакустической калориметрии удалось оценить теп-
ловой эффект внедрения, равный —197 кДж/моль [486]. У 3,6-
диметоксифлуоренилидена, карбена с синглетным основным
состоянием, скорости внедрения в связи О—Н метилового и
трет-бутилового спиртов приближаются к диффузионному пре-
делу (4—6-Ю9 моль"1 с-1), а кинетический изотопный эффект в
реакции с MeOH/MeOD сравнительно невелик (&h/&d=1,2
[268]). Сходные закономерности наблюдаются и для синглетно-
го ксантилидена-9, который при ^20 °C внедряется в связи
О—Н, но не внедряется в связи С—Н [293]. Ароматические
карбены с триплетным основным состоянием также реагируют
со спиртами, однако для них, как отмечалось выше, не сделан
окончательный выбор между двумя механизмами: триплет-
синглетный переход и внедрение синглета [174] или непосред-
ственное взаимодействие спирта с триплетной частицей [175].
В реакции с дифенилкарбеном МеОН в ^8 раз активнее, чем
трет-ВиОН [487], a &h/&d внедрения в связь О—Н (О—D)
трет-бутилового спирта изменяется в присутствии более актив-
ного метанола, что, как полагают [487], свидетельствует о
промежуточном образовании илидов. Участие интермедиатов
илидного типа вероятно и в реакциях ароматических карбенов
с аминами [177, 283, 488] и сульфидами [278], а при взаимо-
действии флуоренилидена и изомерных нафтиЛкарбенов с ане^
тонитрилом соответствующие илиды удается охарактеризовать
спектрально [176].
68
Наиболее характерной реакцией триплетных ароматических
карбенов является отрыв атома водорода; за ходом этой реак-
ции удается наблюдать с помощью современной лазерной спект-
роскопической пико- и наносекундной техники одновременно по
убыли поглощения карбена и накоплению соответствующего
радикала (см., например, [176, 280]). Скорости этих реакций
существенно ниже диффузионного предела (4—6-Ю9 мольбе1);
например, в реакции нафтил-2-карбена с пентаном, циклогекса-
ном и 2,2,3-триметилпентаном k = 2—6-Ю6 молы^с-1 [176], а
перенос атома водорода, видимо, идет по туннельному механиз-
му (см. также [270]). В реакциях менее реакционноспособного
sCPh2 с циклогексаном и толуолом скорость отрыва атома водо-
рода составляет 4—5-Ю5 моль-1 с-1, однако при взаимодействии
с тетрагидрофураном и триэтиламином скорость исчезновения
3СР112 возрастает до 2—3-106 моль-1 с-1, что также может сви-
детельствовать в пользу образования интермедиатов илидного
типа [280]. Согласно данным [489], для реакций триплетных
дифенилкарбена, флуоренилидена, дибензоциклогептатриенили-
дена и его дигидропроизводного с метилхлоридом характерна
предпочтительность отрыва атома водорода по сравнению с ато-
мом хлора; подобный же пример различной селективности син-
глетного и триплетного метиленов приведен при обсуждении
эффектов ХПЯ в аналогичных реакциях (см. разд. 2.2.5).
В заключение необходимо отметить, что реальный механизм
внедрения, видимо, несколько сложнее описанного выше. Подоб-
ные реакции с малым или нулевым барьером активации не обя-
зательно будут следовать по оптимальной траектории, а, скорее,
всего, пойдут по альтернативным путям, определяемым только
исходной (и случайной) взаимной ориентацией реагентов [490,
491]. Ясно, что такие реакции не могут обладать высокой се-
лективностью, поскольку активный карбен с высокой вероятно-
стью будет реагировать с той связью, на которую он ориентиро-
ван, не перестраиваясь для атаки по наиболее выгодному на-
правлению. При реакциях в жидкой фазе существенное влияние
оказывают также эффекты сольватации — десольватации, а так-
же возможность образования карбенами и их аналогами слабых
молекулярных комплексов с растворителем. Именно из-за низ-
кой селективности реакции межмолекулярного внедрения не
нашли широкого использования в препаративной химии карбенов.
Присоединение по кратным связям. Взаимодействие с непре-
дельными соединениями, приводящее к образованию циклопро-
панов, циклопропенов или продуктов их вторичных реакций,
является самой распространенной и изученной реакцией карбе-
нов, которая наиболее широко применяется в синтезе. Подроб-
но присоединение карбенов по связям С = С, С = С, C = Nht. п.
рассмотрено в гл. 4, здесь же мы ограничимся общими сведе-
ниями о механизме этих реакций.
69
Согласно одному из постулатов Скелла [413] (см. разд.
2.2.5), присоединение синглетных карбенов по связи С —С про-
ходит строго ч^с-стереоспецифично, а присоединение триплетных
частиц — нестереоспецифично. Данные квантовохимических рас-
четов модельных реакций полностью соответствуют этому
эмпирическому наблюдению. Так, для триплетного метилена
о-подход к этилену [путь наименьшего движения; а на схеме
(2.5)] разрешен по симметрии, однако на промежуточной стадии
образуется триплетный триметиленовый бирадикал [415]; барь-
ер активации этой реакции ^20 кДж/моль, а тепловой эффект
равен 209 кДж/моль. Для замыкания же связи С-1—С-3 необ-
ходима инверсия спина, проходящая медленнее, чем почти
свободное вращение метиленовых групп, что и приводит к поте-
ре стереоспецифичности.
(2.5)
В то же время для синглетных карбенов о-подход запрещен
по симметрии, и на больших расстояниях (г>0,25 нм) молекула
карбена ориентируется своей вакантной р-орбиталью на один
из атомов углерода кратной связи [так называемый л-подход;
б на схеме (2.5)] [20, 415, 417, 492]. На этой стадии на карбе-
новом атоме углерода накапливается избыточный отрицатель-
ный заряд (до 0,65 е), соответствующий электрофильной атаке
р-орбитали карбена на л-ВЗМО олефина 1[20, 415, 456, 492].
Характерно, что при расчете реакций с участием дифторкарбена
[456, 492] перенос электронной плотности на карбен меньше,
чем в случае !СН2 [20, 415, 417, 492]. При этом в системе изо-
бутилен— оптимальной оказывается антиперипланарный
подход карбена к наименее замещенному атому углерода олефи-
на [492] {в на схеме (2.5)], а три альтернативных варианта
сближения менее выгодны.
Согласно расчетным данным [20, 415, 417, 492], присоедине-
ние синглетного метилена не имеет барьера активации (рис. 2.4,
профиль е), и на более близких расстояниях (г^0,2 нм) про-
исходит просто переориентация карбена на о-тип подхода с
последующим коллапсом в циклопропан. В то же время для
присоединения к этилену синглетных дифторкарбена ^[492] и
циклопропилидена [493] полуэмпирические расчеты обнаружи-
вают существование сравнительно невысоких активационных
70
Рис. 2.4. Возможные сече-
ния ППЭ при сближении
карбена с непредельным ак-
цептором:
/ — исходные реагенты; 2 —про-
межуточные комплексы; 3, 3' •—
переходные состояния; 4 — про-
дукты реакции
барьеров — соответст-
венно <83 и >21 кДж/
/моль (рис. 2.4, про-
филь <9).
Согласно более до-
стоверным данным
НЭР в расширенных
базисах [494—496],
взаимодействие CF2 и
СС12 с этиленом харак-
теризуется промежу-
точным образованием
интермедиатов типа
л-комплексов (рис. 2.4,
профили а и б). Одна-
ко при частичном учете
корреляционной энергии энергия активации реакции с участием
CF2 равна .нулю (профиль е), но для СС12 и при учете
поправок существуют интермедиат (А£^—8 кДж/моль) и
барьер на пути его перехода в геж-дихлорциклопропан (про-
филь в) (£акт 57—59 кДж/моль [494—496]). В системах
СС12 +пропилен (изобутилен) отсутствуют комплексы и пере-
ходные состояния (профиль е); в реакции CF2 +пропилен
£акт~ 14 кДж/моль.
Сказанное может служить объяснением отрицательных энер-
гий активации, определенных для некоторых реакций циклопри-
соединения карбенов [173, 496]. В частности, зависимость абсо-
лютных констант скоростей присоединения синглетных арилга-
логенкарбенов от температуры лучше всего интерпретируется с
позиций промежуточного (и обратимого) образования слабых
комплексов этих интермедиатов [173]; кроме того, в ряде слу-
чаев эти реакции подчиняются не энтальпийному, а энтропийно-
му контролю (табл. 2.10).
Подобные аномалии, видимо, действительно связаны со
сложной и неоднозначной природой взаимодействий карбен —
олефин, в частности, с возможностью существования комплек-
сов, а также с наличием или отсутствием энергетического барье-
ра на пути образования комплекса (см. рис. 2.4, профили а—г)
и сравнительной высотой этого барьера (см. рис. 2.4 в, г, точка
71
3) и барьера перехода комплекса в продукты (см. рис. 2.4, в, г,
точка 3'). Очевидно, в быстрых и почти безактивационных
реакциях даже небольшие изменения в природе реагентов спо-
собны качественно изменять механизм [173, 496].
Таблица 2.10. Некоторые абсолютные кинетические и термодинамические
характеристики реакций фенилхлоркарбена CCIPh с алкенами
(по данным флеш-спектроскопического исследования; изооктан, —20 °C) [173]
Алкен *а60-ЮЛ МОЛЬ1-1 с-1 о. Е акт» кДж/моль /ЬЮ-7. моль-1 с~! кДж/моль AAS , Дж/(моль-К) ААсг^, кДж/моль
2,3-Диметилбу- тен-2 220 1,5 —7,1 1,9 — 18,4 —97 9,3
2-Метилбутен-2 130 1,4 —3,2 4,4 —И 5,5 —93 11,3
(Е) -Пентен-2 5,5 1,6 4,2 6,4 —6,7 —84 18,9
Гексен-1 2,2 1,6 4,5 2,4 —6,3 —88 21,8
В качестве другой возможной причины аномалий некоторых
карбенных реакций выдвигается [497] их подчинение диффу-
зионному, а не кинетическому контролю. В частности, этим
объясняется практическая нечувствительность бромэтоксикарбо-
нилкарбена CBrCC^Et к введению заместителей к двойной свя-
зи акцептора [497], а также неподчинение свойств этого карбе-
на и трифторметилхлоркарбена CC1CF3 [498] соотношению
изоселективности, существующему для других галогенкарбенов
[18].
Вместе с тем абсолютные константы скорости присоединения
арилгалогенкарбенов к алкенам, определенные методом лазер-
ной кинетической флеш-спектроскопии, ниже диффузионного
предела (см., например, табл. 2.10). Даже для самой активной
пары CClC6H4CF3-4 + Me2C=CMe2 &абС= 1,5-109 мольбе"1
(^20°C; изооктан) при диффузионном пределе £абс>Ю10 [173].
Соотношение абсолютных скоростей в реакциях арилхлоркарбе-
нов с различными олефинами при этом, как правило, соответ-
ствует относительным индексам селективности, полученным ме-
тодом конкурирующих реакций [173].
В газовой фазе абсолютные кинетические параметры оцене-
ны только для реакций дифтор- и фторхлоркарбенов с цикло-
пентадиеном, приводящих к фторбензолу; барьеры активации
составляют 47 и 21 кДж/моль соответственно [468] . Удалось осу-
ществить и прямое спектроскопическое наблюдение 1,2-цикло-
присоединения карбена на примере взаимодействия генерирован-
72
ного в матрице циклопентадиенилидена (43) с этиленом [239]:
bv,Ar,12K
-n2
(43)
С2Н4
Аг.40-45 К
Глубокое изучение механизма присоединения карбенов по
кратным связям только начинается.
Реакцию (1+ 2)-циклоприсоединения с образованием трех-
членных гетероциклов ранее считали нехарактерной для анало-
гов карбенов — силиленов и гермиленов. Однако появившиеся
сравнительно недавно многочисленные примеры успешного син-
теза сила- и гермациклопропанов и -циклопропенов, в том числе
реакциями с участием аналогов карбенов [409], позволяют
считать реакцию (1+2)-циклоприсоединения достаточно общей
и для аналогов карбенов.
Целесообразно кратко рассмотреть сравнительную селектив-
ность синглетных и триплетных карбенов в реакциях присоеди-
нения и внедрения. Напомним, что одним из эмпирических те-
стов на спиновую мультиплетность является тест Андо, согласно
которому в реакции с аллилхлоридом синглетные частицы пред-
почтительно внедряются в связь С—С1, а триплетные — присое-
диняются по кратной связи [418] (см. разд. 2.2.5). Данные по
абсолютной кинетике карбенных реакций также показывают,
что для синглетных карбенов внедрение в связи с участием
гетероатомов, прежде всего в связи О—Н, предпочтительнее
присоединения по связям С = С. Так, для 2,6-диметоксифлуоре-
нилидена, частицы с синглетным основным состоянием, констан-
ты скорости реакций со стиролом и метанолом (бензол, ^20 °C)
равны 4,3 -107 и 5,6 -109 мольбе-1 соответственно [268], а для
триплетного (нафтил-2)карбена (2,2,4-триметилпентан, ^20 °C)
составляют 4,3 • 107 и 7,3 ДО6 молы^с-1 [176].
Для арилкарбенов в возбужденном синглетном состоянии
соотношение продуктов присоединения по кратной связи алке-
нов и внедрения в связи О—Н спиртов существенно зависит от
природы заместителей в ароматическом ядре: электронодонор-
ные заместители благоприятствуют реакции с более активным
субстратом [424]; значительные изменения селективности при-
соединения— внедрения наблюдаются и при переходе от раство-
ров к различным органическим матрицам (стеклам) [273, 500].
Иными словами, экстраполяция эмпирических тестов типа теста
Андо должна проводиться с известной осторожностью. Сказан-
ное не относится, пожалуй, лишь к постулату Скелла о цис-
стереоспецифичности присоединения синглетных карбенов при
цестереоспецифичности реакции триплетных частиц.
В заключение кратко рассмотрим сложные для классифика-
ции в терминах «присоединение — внедрение» реакции карбенов
с напряженными насыщенными молекулами. Так, из продуктов
73
реакции бицикло[1.1.0] бутана (44; R = H) с синглетным метиле-
ном, генерируемым фотолизом диазометана, выделены пентади-
ен-1,4 и бицикло [1.1.1] пентан [501]:
R
‘сн2
декалин,
-50 °C
(21%) (1%)
(44) R=H
Раскрытие цикла происходит и при взаимодействии 1,3-ди-
метилбициклобутана с [503], а также в реакции бицикло-
бутана и его замещенных с [501—503]. Региоселектив-
ность этих реакций соответствует атаке карбена по менее заме-
щенным положениям бициклобутанового фрагмента, что хорошо
видно на примере взаимодействия триметилбициклобутана (44;
Р = Ме) с синглетными бис (метоксикарбонил) карбеном и ди-
хлоркарбеном [504]:
(44; R = Ме) + 1CR2
Me
R1 = СООМе, Cl (< 15%)
Полуэмпирический расчет реакции бициклобутана (44;
R = H) с 'СНг методом МПДП показал возможность протекания
как синхронного образования пентадиена за счет одновремен-
ного разрыва центральной и одной из боковых связей (£'акт =
= 59 кДж/моль), так и присоединения по центральной связи с
образованием бицикло[1.1.1]пентана (£акт»80 кДж/моль)
[504].
Необычно протекает и взаимодействие карбенов с квадри-
цикланом (45) [503, 504], например:
R= СООМе
При реакции квадрициклана с дихлоркарбеном относитель-
ные выходы различных продуктов существенно зависят от усло-
вий проведения реакции [503, 504]:
74
Cl Cl
(46)
Сходный набор продуктов образуется и при взаимодействии
дихлоркарбена с норборнадиеном (см., например, [505, 506]);
особенно интересно здесь образование тетрациклического сое-
динения (46) — продукта гомо-(4+ 2)-циклоприсоединения кар-
бена.
Каждая из этих реакций является по-своему уникальной, и
можно предположить, что вовлечение в карбенные реакции
других напряженных молекул приведет к открытию новых нео-
бычных путей их превращений.
Комплексообразование. Вряд ли будет особым преувеличени-
ем сказать, что химия реакционноспособных промежуточных
частиц в растворах во многом определяется способностью их к
комплексообразованию. В этом отношении карбены и их анало-
ги не только не являются исключением, а наоборот, идеально
адаптированы для взаимодействия с любым комплексообразо-
вателем. Действительно, у синглетных частиц вакантная р-ор-
биталь склонна к взаимодействию с нуклеофильными центрами,
а о-орбиталь, несущая два электрона, — с электрофильными.
Следовательно, подходящими партнерами для комплексообразо-
вания синглетных карбенов и их аналогов могут быть как до-
норные, так и акцепторные молекулы (кислоты или основания
Льюиса), а также молекулы, объединяющие обе функции, на-
пример, соединения переходных металлов. Естественно, наи-
больший интерес представляют слабые молекулярные комплек-
сы, способные легко высвобождать координированный карбен.
Образование таких комплексов является одним из путей измене-
ния селективности и управления реакционной способностью
карбенов (подробнее см. [507]).
Комплексы карбенов и их аналогов с основаниями Льюиса
наиболее' распространены и изучены {507, 508]. Хотя возмож-
ность координации неподеленной пары электронов атомов типа
О, S, N, Р и т. п. на вакантную р-орбиталь синглетного карбена
не вызывает сомнений, энергии такого взаимодействия оказыва-
ется недостаточно для связывания в стабильные комплексы вы-
сокореакционных карбенов [508]. В то же время для аналогов
карбенов — силиленов, гермиленов, станниленов и плюмбиле-
нов — известны достаточно стабильные молекулярные комплек-
сы с n-донорными лигандами. В частности, комплекс дихлоргер-
милена с диоксаном является наиболее удобным источником
75
GeCl2 в различных реакциях [509, 510]. Рентгеноструктурный
анализ комплексов С4Н8О2-МС12 (M = Ge [511], Sn [100]) вы-
явил для них цепочечную структуру, в которой атомы Ge и Sn
связаны с двумя атомами кислорода различных молекул диокса-
на. Геометрия фрагмента МС12 практически не изменяется по
сравнению с геометрией свободной частицы, а тригонально-би-
пирамидальная конфигурация при атоме М с углом О~М—О,
равным 180°, однозначно указывает на координацию неподелен-
ных пар атомов кислорода на вакантную р-орбиталь аналога
карбена [100, 507, 511]. Для комплекса дихлоргермилена с
1,4-диоксаном оценочное значение, прочности координационной
связи в растворах, полученное различными методами, состав-
ляет И—30 кДж/моль [507].
Подобное строение комплексов найдено и при полуэмпири-
ческом расчете модельной системы ^Нг + ЬГО [512], где мини-
мум с энергией стабилизации 23,4 кДж/моль отвечает комплексу
состава ^Нг-НгО с координацией электронной пары атома кис-
лорода на р-орбиталь метилена. Сходное строение, по данным
НЭР в расширенном базисе [496], характерно и для модельных
комплексов ^Рг-НгО и ^СЬ-НгО. Комплекс дифторкарбена с
Н2О имеет энергию стабилизации ^41 кДж/моль и довольно
«рыхл» (гс-о = 0,223 нм), однако комплекс дихлоркарбена отве-
чает более тесному сближению молекул (гс-о = 0,168 нм) и го-
раздо более стабилен (Д£=109 кДж/моль). Столь высокие
значения £Стаб, полученные при расчете достаточного уровня
строгости (базис 3-21 ГФ с поправками МП/2 [496]), показы-
вают, что в реальных условиях жидкофазных реакций комплек-
сообразование карбенов с основаниями Льюиса (эфиры, галоге-
ниды, амины и т. п.) достаточно существенно (см. [474—477,
508]); во всяком случае, его следует учитывать при анализе де-
тального механизма карбенных реакций. Напомним, что .проме-
жуточное образование комплексов арилкарбенов с гетероатом-
ными нуклеофилами установлено при исследовании абсолютной
кинетики [176, 177], хотя структура подобных интермедиатов
не совсем ясна; во всяком случае предположение об их илидном,
а не n-донорном строении в настоящее время декларативно.
Характерно, что и модельный расчет [512], и эксперимен-
тальное исследование комплексов GeCl2 с трифенилфосфинОхМ
[513] и бензотиазолом [514] не обнаруживают характерного
для илидов плоского расположения фрагмента С1—Ge—Cl и
атома фосфора или атома азота. Полагают [507], что эти ком-
плексы можно рассматривать как илиды необычного строения —
с сохранением присущей двухвалентному состоянию геометрии
аналога карбена. Известны и другие комплексы аналогов кар-
бенов с n-донорными лигандами (подробнее см. [507].
Комплексы аналогов карбенов с кислотами Льюиса доста-
точно немногочисленны, в основном это комплексы малоактив-
76
ных циклопентадиенилзамещенных станниленов, координирован^
ных с соединениями бора(III) [507]. Структура этих комплек-
сов отвечает координации нуклеофильной о-орбитали на вакант-
ную орбиталь кислоты Льюиса, причем прочность координацион-
ной связи невелика [515].
Комплексы карбенов и их аналогов с соединениями переход-
ных металлов хорошо изучены и играют ключевую роль во мно-
гих типах химических реакций (подробнее см. гл. 5). Большая
прочность таких комплексных соединений может быть связана
с одновременным участием о- и р-орбиталей карбена во взаимо-
действии с орбиталями переходного металла.
2.4.2. Внутримолекулярные реакции карбенов и их аналогов
Описанные выше типы реакций карбенов, прежде всего присое-
динение по кратным связям и внедрение в ординарные связи,
возможны и во внутримолекулярном варианте, если молекула
сложного карбена содержит подходящие реакционные центры.
Такие процессы могут эффективно конкурировать не только
с межмолекулярным перехватом карбенов, но в ряде случаев и
между собой. Так, в случае карбена (47) возможны следующие
реакции: внедрение в у-связь С—Н (/), присоединение по связи
С—Н (2), карбен-олефиновая изомеризация (внедрение в р-
связь С—Н; <?), внедрение в р,у-связь С—С (4) [516]:
Из приведенного примера видно, что среди внутримолекуляр-
ных реакций карбенов есть ряд чисто специфических перегруп-
пировок. В первую очередь это карбен-олефиновая изомеризация
(<?), формально отвечающая внедрению в p-связь С—Н. Данная
реакция исследована на большом числе примеров. Согласно
расчетным данным [20, 379—381], к карбенному центру преи-
мущественно мигрирует атом, копланарный с осью вакантной
р-орбитали, т. е. путь 1 предпочтительней, чем путь 2:
77
В жестких каркасных системах, например в брексанилиде-
не-5 (48) [517], подобная селективность очень высока. В то же
время уже у производных циклогексилидена (49) селективность
миграции атома водорода сравнительно невысока, поскольку
гибкость скелета обусловливает возможность взаимодействия с
р-орбиталью любого из соседних атомов водорода, например
[518]:
~ ~► мА
у— Н( эк 30) А
H(sMo)
(48) экзо :эндо=138 (49) МВи-/рг/ Нк-’СПМД
Еще меньшей селективности следует ожидать в случае
«обычных» алкилкарбенов (см. [4], а также [519—521]). Сог-
ласно расчетным данным (см. [522]), энергия активации этого
процесса очень мала (<10 кДж/моль), однако при исследовании
относительных скоростей межмолекулярного 1,2-циклоприсоеди-
нения синглетных бензилгалогенкарбенов и внутримолекуляр-
ной 1,2-Я-миграции для последнего процесса определены [523,
524] барьеры, равные 26,8 и 19,7 кДж/моль при Х = С1 и Вг,
соответственно:
bv, 10-24 °с
N - CXCH9Ph ----“ли>~—~ »iCXCH2Ph - PhCH = CHX
/ 2 N2
N Me2C = CMe2
V
Me2C - CMe,
CXCH2Ph
Согласно данным прямого матричного спектроскопического
исследования [525], триплетный метилфенилкарбен стабилен в
Аг- или Хе-матрице при 10 К, однако при отогреве до 65 К или
при УФ-облучении (>440 нм, 15 К) перегруппировывается в
стирол; в то же время триплетный о-толилкарбен перегруппиро-
вывается в о-ксилилен уже при 4,6 К без облучения [525]:
PhMeCN2—^3CMePh-^PhCH = СН9
** IM 2
1 - h», Аг или Хе, ЮК; 2 - Хе, 65К или hv, Аг или Хе, 15К
78
bv, Ar, 4,6K
-------э*
4,6K
1,2-Сдвиг атома водорода к карбенному центру характерен
и для аналогов карбенов, содержащих в ^-положении атомы
водорода. В частности, изолированный в Аг- или Ыг-матрице
(10—20 К) диметилсилилен при облучении способен переходить
в 1-метилсилаэтилен (см., например, [247, 260]). Описана [257,
259] также обратная реакция в условиях матричного облуче-
ния и исследована {526] кинетика термической перегруппировки
1-метилсилаэтилена в диметилсилилен в газовой фазе:
(CH3)2Si:5==^CH3SiH = сн2
В условиях матричной изоляции силен-силиленовая перегруп-
пировка наблюдается и для других силаолефинов [527]:
Аг- или N^-маIрица,
10-12 К
I AV I
XHSi=CH2 4==>:SiXMe
Х= Н,Ме,С1
Карбен-олефиновая изомеризация является также общим
методом синтеза разнообразных антибредтовских олефинов с
двойной связью в голове моста [528].
В ходе карбен-олефиновой изомеризации возможна 1,2-миг-
рация не только атома водорода, но и органического заместите-
ля или атома галогена. Так, для перегруппировки диметилвини-
лиденкарбена в диметилацетилен оценочное значение ЕакТ~
^50 кДж/моль [529] (см. также [389, 530]). Для кетокарбенов
характерна перегруппировка Вольфа, которая формально пред-
ставляет собой 1,2-сдвиг заместителя R к карбенному центру с
образованием соответствующих кетенов [531, 532] (см. также
разд. 3.1.2):
RCOCR1-* RR1C = C = O
Описана [533] изомеризация фтор (трифторметил) карбена в
тетрафторэтилен с миграцией атома фтора к карбенному цент-
ру, а также изомеризация бис (трифторметил) карбена в пер-
фторпропилен [275]:
600 °C
cf3cf2cooh _C0-;_-h^cfcf3 -* f2c = cf2
79
II /C(CF3)2~:C(CF3)2- CF3CF = CF2
n'
Известны примеры миграции к карбенному центру и более
сложных заместителей, например триметилсилильной группы
[534]:
Me3SiSiMe2CCO2Et ^^^*"Me3SiSiMe2CCO2Et_*’
N2
~>Me2Si = С - SiMe3
CO2Et
Выше уже упоминалось превращение циклопентенилметили-
дена в циклогексадиен-1,3 в результате формального внедрения
в [3,у-связь С—С [516]. Образование циклобутена, а также про-
дуктов его дальнейших превращений характерно и для цикло-
пропилкарбенов, но в этом случае с формальным р,у-С—С-
внедрением конкурирует фрагментация карбена с образованием
замещенных ацетилена и этилена [535]. Однако перегруппиров-
ка бензоциклогептатриенилидена сопровождается сужением
цикла и приводит к (нафтил-2) карбену [397, 536, 537].
В случае арил- и гетарилкарбенов наблюдается и ряд других
сложных и интересных внутримолекулярных перегруппировок,
в том числе и карбен-карбенового типа (подробнее см. [7, 528,
538—541]).
Глубокая перестройка скелета происходит при циклопропил-
иден-алленовой изомеризации:
С = С = С
80
Для этой реакции точные НЭР предсказывают ДДЯ = 270—
273 кДж/моль при активационном барьере в 48—57 кДж/моль
[389—391]. Подобные превращения лежат в основе достаточно
общего и селективного метода синтеза алленов (см. разд. 4.3).
В случае винилзамещенных циклопропилиденов наряду с пе-
регруппировкой в пентатриены-1,2,4 протекает их изомеризация
в циклопентенилидены, стабилизирующиеся далее в циклопента-
диены в результате 1,2-Я-сдвига [542]:
1 2
сн2 = сн - нс - сн2-*сн2 = сн - нс - сн^-сн2 = сн- нс - сн2
У-
СН2= С = сн - сн = сн2
1 - нагревание или hv (-N2);2 - MeLi (-МеВг, -LiBr)
В ряду циклических винилкарбенов наблюдаются и более
сложные перегруппировки, например [543] (см. также [542]):
В подобных реакциях направление внутримолекулярной ста-
билизации карбенного центра обычно определяется в первую
очередь пространственной доступностью простых или кратных
связей. Хорошим примером является внутримолекулярное
1,6-циклоприсоединение бициклического карбонилкарбена; аль-
тернативные направления внутримолекулярного 1,2- или 1,4-
циклоприсоединения стерически неблагоприятны [544] (см. с. 82).
Известны примеры использования внутримолекулярных реак-
ций карбенов для направленного синтеза полициклических и ге-
тероциклических соединений (см. с. 82).
6—107 81
CN
COCH=N2 RbfOAr).,
CH;
CN
COCH
r1 Br МеМ.эфир,
I • 20-35 °C
R2 C — С C H2 C — S i M 62 >
gr -MeBr,-LiBr
CR2
----* R1 - C - C - SiMe->
\/ 3
CH2
(<53%) [545]
R1 *
I
R2C = CCH2C — SiMs2 *
R, R1 = H, Me
(«71%) [546, 547]
.CH-CH HC-CH
( /~'SiHMe2——;£HCf ^CH
О XOSiHMe2 X) - SiMe2
1 - вакуумный флеш-пиролиз 05/6) [548]
Таким образом, приведенные в данной главе сведения дей-
ствительно убеждают в своеобразии строения карбеноподобных
молекул и многообразии их реакционной способности. В резуль-
тате комплексного использования различных физических мето-
дов и химических тестов установлены четкие критерии для от-
несения того или иного соединения к классу карбенов. Вместе
с тем даже краткое рассмотрение различных типов карбенных
реакций показывает полную справедливость сделанного в
1966 г. дополнения этих критериев «химическим признаком»,
82
определяющим тенденцию карбенов к образованию одной sp2 или
двух sp3 новых ковалентных связей [16]. Проведенный анализ
позволяет перейти к систематическому рассмотрению методов
генерирования карбенов и их аналогов, а также путей их исполь-
зования.
Глава 3
МЕТОДЫ ГЕНЕРИРОВАНИЯ КАРБЕНОВ
И ИХ АНАЛОГОВ
В настоящее время известно большое число методов генерирова-
ния карбенов и их аналогов, и перед исследователем, нуждаю-
щимся в той или иной нестабильной частице для физико-хими-
ческих исследований или решения синтетических задач, встает
проблема выбора наиболее удобного и (или) наиболее эконо-
мичного метода. Источники для генерирования новых карбенов
обычно можно подобрать по аналогии с уже описанными.
Мы не ставили перед собой задачу дать исчерпывающий об-
зор всех известных примеров генерирования карбенов (образо-
вание которых часто, кстати, строго не доказано): в данном
разделе в основном дана классификация этих методов, позво-
ляющая ориентироваться в большом числе разноплановых
экспериментальных исследований. Наиболее важные в практи-
ческом отношении методы — генерирование карбенов из диазо-
соединений, реакция Симмонса — Смита и получение дигалоген-
циклопропанов в условиях межфазного катализа детально рас-
смотрены в отдельных разделах гл. 4.
Возвращаясь к общим вопросам генерирования карбенов и
их аналогов, можно в первую очередь отметить разнообразие
попыток классификации соответствующих методов как по типам
генерируемых частиц (см., например, [4]), так и по используе-
мым источникам [7]. Методически более оправданно разделение
методов генерирования карбенов на две большие группы. В пер-
вую группу попадают все «энергетические» методы, при исполь-
зовании которых карбеноподобная молекула образуется из
стабильного в нормальных условиях предшественника в резуль-
тате подвода дополнительной энергии в виде тепла, фотолиза,
радиолиза, электрического разряда и т. п. Реакции этого типа
формально мономолекулярны, хотя не исключается, например,
последовательный разрыв двух о-связей у центрального атома,
а также участие катализатора или материала поверхности реак-
тора, что позволяет проводить реакцию в более мягких усло-
виях.
6* 83
Во вторую группу попадают «химические» методы генериро-
вания карбенов, основанные на взаимодействии двух (или бо-
лее) стабильных веществ, возможно также в присутствии ката-
лизатора и (или) подвода энергии. Такое разделение, естествен-
но, не является абсолютным, поскольку реакции первой группы,
протекающие с участием катализатора или стенки реактора,
строго говоря, не являются мономолекулярными, а большая
часть реакций второй группы обычно сводится к получению
нестабильного предшественника, распадающегося с образова-
нием карбена. Вместе с тем подобная классификация позволяет
четко разграничить «энергетические» и «химические» способы
активации стабильных молекул-предшественников.
Следует подчеркнуть, что далеко не во всех случаях образо-
вание карбенов строго доказано, поскольку участие карбенов
часто постулируется на основании анализа стабильных конеч-
ных продуктов реакции без исследования кинетики процесса и,
тем более, без фиксации и прямого изучения самих частиц.
3.1. ГЕНЕРИРОВАНИЕ КАРБЕНОВ ИЗ ЭНЕРГЕТИЧЕСКИ
АКТИВИРОВАННЫХ ПРЕДШЕСТВЕННИКОВ
Реакции этого типа достаточно универсальны и широко распро-
странены, хотя находят сравнительно ограниченное применение.
Среди них можно выделить процессы а-элиминирования с раз-
рывом (не обязательно одновременным) двух геминальных
о-связей и процессы диссоциации различных непредельных
соединений, а также полимеров, олигомеров, илидов и т. д.
К процессам диссоциации относится также разложение диазо-
соединений и диазиринов с образованием карбенов (подробнее
см. гл. 4).
Особое место занимают процессы, в которых карбенный
центр образуется не в результате реакции отщепления, а фор-
мально вследствие внутримолекулярной перегруппировки пред-
шественника.
3.1.1. Реакции а-элиминирования
К таким реакциям по чисто формальному признаку разрыва
двух геминальных связей при одном атоме углерода относятся
процессы синхронного cc-элиминирования (а) и реакции, идущие
в несколько стадий (б):
84
Детальный механизм подобных процессов зачастую неизве-
стен, и с той или иной степенью уверенности можно констатиро-
вать лишь сам факт образования карбена из стабильного пред-
шественника реакций формального а-элиминирования.
а-Дегидрирование. Реакции с отщеплением молекулы водо-
рода, которому обычно способствуют катализаторы и раствори-
тели основного характера, наиболее характерны для ди- и поли-
гидридов кремния, германия и олова:
SiH4 40?°-»:SiH2 + Н2 [549-552]
1408-1563 °C (ударная труба)
Me2SiH2 1163_1273оС(проточнь|0реактор5 Me2Si: + Н2 [553]
Me2SnH2-^M^Me2Sn: + Н2 [248]
Дегидрирование диалкилстаннанов облегчается в присутст-
вии оснований (C5H5N, ДМФА, Et2NH и т. п.) [554—556].
Термическое дегидрирование, правда в более жестких усло-
виях, наблюдается и при пиролизе метана, в том числе в избыт-
ке водорода [557—560], причем для этой реакции определены
кинетические параметры [468]. Фотолиз метана или его смесей
с водой в твердой или газовой фазе также приводит к метилену
[561—563]; аналогично проходит ^-радиолиз метана [562, 564],
облучение его электронами [565], а также пропускание через
него электрического разряда (см., например, [4]). Аналогично
протекает и фотолиз силана [66, 549].
hv
СН4-:СН2 + Н2 SiH4-^:SiH2 + Н2
Образование ряда галоген- и дигалогенкарбенов путем
«-дегидрирования при УФ-облучении замороженных в инертных
матрицах моно- и дигалогенметанов удалось зафиксировать по
матричным УФ- п ИК-спектрам этих карбенов (Аг- или ^-мат-
рицы, 14 К):
hv
СН2Х2 *:СХ2 + Н2 X - F, С1 [52], Вг [255]
hv
СН2ВгХ-*:СХВг + Н2 X = F [256], CI [255]
hv
СН3Х-*:СНХ + Н2 X = F [47], CI [52]
CH2CIF—*:CCIF + Н2 [56]
85
Подобные реакции известны и в химии аналогов карбенов,,
например [98]:
GeH3X-^:GeHX + Н2 X = CI, Вг
Для высших алканов также известны реакции а-дегидриро-
вания с образованием карбенов. Так, при крекинге алканов до-
минирует карбенный распад, а доля цепных реакций не превы-
шает 5% [566]. Образование карбенов в реакциях такого типа
подтверждено результатами кинетических исследований (см.
[468, 566]).
RR1CH2-^-k“h> RR1C: + н2 R, R1 = Н, Aik
Однако наряду с а-дегидрированием при крекинге углеводо-
родов могут протекать и другие реакции, также приводящие к
образованию карбенов [468, 566], например:
RCHR1R2 - R1R2C: + RH
R = Me, Et и т.п.; R1, R2 = H, Me, Et, Pr и т.д.
Диапазон энергий активации крекинга алканов (264—
300 кДж/моль) перекрывается энергиями образования карбено-
вых частиц по реакциям а-дегидрирования [468]. Квантовохи-
мический расчет методом МЧПДП/3 показал, что синхронное*
а-дегидрирование алканов характеризуется энергией активации
350—400 кДж/моль, причем образуются наиболее замещенные
карбены [326]. В термических процессах генерирования карбе-
нов с участием алканов могут одновременно достаточно эффек-
тивно протекать процессы образования карбенных центров,
^-дегидрирования, карбен-олефиновой и скелетной изомеризации
[567]. С помощью меченых атомов подтверждено образование
алкилкарбенов при фотолизе этана [561, 568, 569]. пропана
[570—572] и изобутана [573].
а-Дегидрогалогенирование. Эта реакция имеет значительно
более общий характер, чем а-дегидрирование, особенно в усло-
виях термических превращений. Так, галоформы, в том числе
и смешанные, при термолизе достаточно селективно генерируют
соответствующие дигалогенкарбены по синхронному механизму:
СНХ3 - :СХ2 + НХ X — СI [574-579], F [580]
CHFClo------------:CFCl + НС1
2 [471,581,582]
86
CHF2X - :CF2 + HX X = Cl [471,579, 581-584], Br [585]
Аналогично из CHF2CN удается термически генерировать
фторцианокарбен [586]:
CHF2CnA;CFCN + HF
Для большинства этих реакций исследована формальная ки-
нетика. Полученные результаты подтверждают их протекание
по синхронному (одностадийному) механизму а-элиминирования
молекулы НХ {471, 575, 576, 580—582, 585, 586].
Реакции а-дегидрогалогенирования хорошо известны также
для тригалогенсиланов и тригалогенгерманов [16, 98, 549, 587].
Данные по фотохимическому распаду галоформов менее од-
нозначны. Так, фотолиз хлороформа приводит к дихлоркарбену
в результате двухстадийного дегидрохлорирования [588—591]:
А? у
СНС13-^ГСНС12^Д5Г:СС12
В то же время импульсный фотолиз CHF2Br при Z>200 нм
проходит, по-видимому, путем молекулярного элиминирования
НВг с образованием дифторкарбена, поскольку в тех же услови-
ях CF3I и CF3Br, распадающиеся по радикальному механизму,
не образуют дифторкарбена [167]. Дифторкарбен иден-
тифицирован спектральными методами в газовой фазе и в
продуктах импульсного фотолиза CHF2C1 [469], а также при
действии ударной волны на фтороформ [195]. При УФ-облуче-
нии (Х<120 нм) замороженного при 20 К в Ar-матрице СНВг3
зарегистрирован спектр дибромкарбена [216]; аналогично, из
CHFC12 при 4 К в Ar-матрице генерирован фторхлоркарбен
[57]. Хлороформ генерирует дихлоркарбен при у-радиолизе или
пропускании тихого электрического разряда [592], а также при
электролизе [593, 594].
В ряду дигалогенметанов реакция термического а-дегидро-
галогенирования также носит достаточно общий характер, хотя
протекает при более высоких по сравнению с соответствующи-
ми галоформами температурах:
СН2Х2-^:СНХ + НХ X = CI [468, 595, 596], F [580]
CH2FCl~*-:CHF + HCI [581]
Хлоркарбен идентифицирован по его матричному ИК-спект-
ру и среди продуктов УФ-фотолиза замороженного в Аг- или
М2-матрице СН2С12 (см. [52]).
87
В случае моногалогенметанов протекание термического
а-дегидрогалогенирования с образованием метилена надежно
установлено лишь на примере CH3F [597, 598]. В то же время
при УФ-фотолизе замороженного в Аг- или ^-матрице СН3С1
не удалось идентифицировать метилен [52], возможно, из-за его
высокой реакционной способности даже при 4,1 К (см. гл. 2).
«-Дегалогенирование. Этот процесс также является доста-
точно общим, однако харакФерен главным образом для тетрага-
логенметанов. В отличие от синхронной реакции термического
а-дегидрогалогенирования галоформов термическое а-дегалоге-
нирование, по-видимому, протекает в две стадии. Так, СС14 в
вакууме при 1500 °C ступенчато диссоциирует с образованием
дихлоркарбена [599]; присутствие катализаторов (Fe, Си, Zn)
позволяет значительно снизить температуру этого процесса
д
СС14—.СС13—:СС12
Соответствующие дигалогенкарбены СХ2 зафиксированы
масс-спектрометрически при пропускании паров СХ4 (X = F, Cl,
Вг) над раскаленной W-проволокой l[129, 601]. По данным
масс-спектрометрии двухстадийным является и процесс генери-
рования дифторкарбена термолизом CF4 и CF3I [199]. Образо-
вание CF2 из CF4 и CF3I происходит также под действием удар-
ной волны [195, 197], МВ-разряда [184, 185], радиочастотного
разряда [42, 188] или при пиролизе i[208], причем образующий-
ся дифторкарбен зарегистрирован в газовой фазе по его УФ-
спектрам поглощения или испускания [184, 185, 195, 197], МВ-
спектру [42], колебательному спектру [188] или в инертной
матрице по ПК-спектру [208]. а-Дегалогенирование с образова-
нием дифторкарбена происходит также при импульсном элект-
рическом разряде в CF3Br (CF2 идентифицирован по УФ^спект-
ру) [183]' и при пропускании электрического разряда через
CF2C12 [602]. Образование дибромкарбена при термолизе СВг4
показано с помощью метода газовой электронографии [60].
Импульсный фотолиз тетрагалогенметанов типов CF2X2 и
CF3X (Х = С1 или Вг) проходит мономолекулярно по синхронно-
му механизму [166, 167]. Однако при фотолизе CF2BrCl и
CF2Br2 реализуются как синхронный, так и ступенчатый меха-
низмы [168, 603]:
hv hv
CF2x2T^:CF2-^-CF3X
hv hv
•CF2x^-CF2BrX-BT:CF2
X = Br, Cl
88
Импульсный фотолиз CHFBr2 и СНС1Вг2 сопровождается
дебромированием и образованием соответственно фтор- и хлор-
карбенов, которые идентифицированы по их электронно-колеба-
тельным спектрам [46].
Описано электрохимическое генерирование дихлоркарбена
при гальваностатическом восстановлении СС14 или СНС13 на
Pb-катоде в СНС13—Bu4NBr или СН2С12—Bu4NBr при —5 °C
[594].
К образованию соответствующих карбенов приводит и фото-
химическое а-деиодирование геж-дииодидов. Так, при фотолизе
СН212 метилен был перехвачен различными реагентами, хотя
вопрос об участии в этой реакции кинетически независимого
карбена до конца не ясен [604, 605]. Аналогичным образом при
фотолизе СН13 или СНСП2 в присутствии олефинов получены
циклопропановые аддукты иод- и хлоркарбенов соответственно
[606]. При фотохимическом распаде 1,1-дииодпропана и 1,1-
дииод-2,2-диметилпропана (геж-дииоднеопентан) в присутствии
олефинов циклопропановые аддукты соответствующих карбенов
получены только при фотолизе в газовой фазе с использованием
коротковолнового света (2i<240 нм) [607].
Следует отметить, что термолиз и фотолиз пергалогеналка-
нов могут протекать не только путем а-дегалогенирования с
образованием карбенов, но и путем гомолиза связей С—С или
С—На! с образованием свободных радикалов, при дальнейшей
фрагментации которых также возможно образование карбенов.
Так, термолиз гексафторэтана в ударной трубе приводит, по
данным УФ-спектроскопии, к дифторкарбену, образующемуся,
как полагают [199], .из первичного трифторметильного ради-
кала:
C2F6-A-2'CF3-^-2:CF2
Дифторкарбен в газовой фазе образуется также при элект-
рическом разряде в h-C5Fi2 [183] и при MB-разряде в C2F6
[189]; стадийные гомолитические реакции, ведущие к дифтор-
карбену, происходят и при мультифотонном лазерном ИК-ф°Т0’
лизе C3F7I и C2F5I [342]:
Чт?1 Tj7^C3F7-----*-:CF2 + -C2F5
с2f5 1 ^^-с2?5 ——*:CF2 + -CF3
Завершая обзор рассмотренных реакций а-элиминирования,
наиболее характерных для галогенметанов, можно отметить, что
в условиях термолиза, как правило, преимущественно протекает
89
реакция синхронного а-дегидрогалогенирования, а при ее невоз-
можности— ступенчатое отщепление тяжелых атомов галоге-
нов. В условиях фотолиза обычно расщепляется наименее проч-
ная связь С—I или С—В'г.
Здесь же можно отметить и результаты исследования распа-
да возбужденных галогенметанов, полученных по реакции заме-
щения с участием «горячих» атомов. Так, возбужденная молеку-
ла СНТХ2 селективно распадается с образованием :СТХ (про-
дукты-перехвата его немеченого аналога :СНХ не могли быть
детектированы применявшимися радиоизотопными методами)
[596, 608]:
СН2Х2-~^[СНТХ2]* - :СТХ + НХ
X = F, CI
Подобным образом при взаимодействии атомов 18F с CF4
получен CF18F, а с CH2F2 — меченные 18F фтор- и дифторкар-
бены [609]:
18F- + CH2F2
[CH^8FF]* :CH18F + HF
^r*[CHF£8F]*-:CF18F + HF
Аналогично, бомбардировкой «горячими» атомами фтора,
образующимися при MB-разряде в CF4, из СН4 и CD4 генери-
рованы :СН2 и :CD2 [367—369], а из СН3Х (X = F или С1) —
соответствующие галогенкарбены :CHF ,[359] и :СНС1 [55].
В заключение следует отметить, что в большинстве работ
спиновая мультиплетность генерируемого карбена специально
не рассматривалась и предопределялась либо способом генери-
рования, либо условиями стабилизации карбена. Рассмотрен-
ные выше методы осуществляли преимущественно в газовой
фазе, что, в частности, для термолитических реакций определя-
ется довольно жесткими требованиями к температуре процесса
(обычно >500°C). Наконец, большинство рассмотренных мето-
дов имеют ограниченное применение в синтезе; они были исполь-
зованы при проведении различных физико-химических исследо-
ваний. Исключение составляют газофазные пиролитические
методы генерирования и реакции фторсодержащих карбенов
(см., например, [468]), а также силиленов [549, 610].
Рассматриваемые ниже реакции а-элиминирования карбенов
из оксиранов, диазиринов и других карбо- и гетероциклических
соединений, а тем более из галогенметаллорганических пред-
шественников обычно протекают в значительно более мягких
условиях, что делает данную группу методов генерирования
90
карбенов интересной не только для физико-химических иссле-
дований, но и для широкого использования в синтезах.
а-Элиминирование карбенов из циклопропанов и трехчлен-
ных гетероциклов. Циклопропан и его гетероаналоги — оксиран
и азиридин — являются достаточно напряженными молекулами
с энергиями напряжения «113 кДж/моль [611]. Это в сущест-
венной мере обусловливает повышенную реакционную способ-
ность трехчленных циклов, в том числе и в реакциях внутримо-
лекулярной экструзии карбенов. Существенно, что энергии
активации таких процессов на 84—105 кДж/моль ниже, чем
энергии активации реакций а-дегидрогалогенирования галоген-
метанов [468]. Так, при диссоциации перфторциклопропана на
тетрафторэтилен и дифторкарбен £акт=163—180 кДж/моль
[612, 613], а для подобного распада перфтораллилциклопропана
£акт=180 кДж/моль [614]:
р Г 253-276 °C
2---------»CF2 = CF2 + :CF2
cf2
cf2
CF2 = CFCF2 - FC - CF2 200~250 °£ (CF2 = CF)2CF2 + :CF2
Источником дифторкарбена является и дифторметилперфтор-
циклопропан [615]:
CF2
f2cZ- cf - chf2 C22Xf2c = CF - CHF2 + :CF2
Еще более «мягкими» источниками CF2 являются смешанные
фторхлорциклопропаны, сравнительно легко получаемые присое-
динением дихлоркарбена к фторолефинам [616]. Распад этих
соединений проходит при ^300 °C и отличается высокой селек-
тивностью. Так, из тетрафтор-1,4-дихлорциклопропана, трифтор-
1 Л,2-трихлорциклопропана и 1,1-дифтортетрахлорциклопропана
главным образом генерируется CF2, а не CFC1 или СС12 [616]:
F2C-CCI2 -* :CF2 + CCI2 = СХХ1
СХХ1
I__________________.
V |
:СС12 + CF2 = СХХ1
:СХХ1 + CF2 = CCl2
X, X1 = F, Cl
91
Напротив, тетра- и пентахлорциклопропаны при 500—540 °C
в основном перегруппировываются в соответствующие пропиле-
ны, а не элиминируют карбены, например ([617]. В то же время
гексаметоксициклопропан легко и селективно диссоциирует на
диметоксикарбен и тетраметоксиэтилен [618].
С12с - CHCI —~540О£ RCCl2CH = СС12
R-C-C1 (90-93%)
R = Н, CI
200 °C
(МеО)2С-/С(ОМе)2 —
С(ОМе)2
:С(ОМе)2 + (МеО)2С = С(ОМе)2
Фотолитическое расщепление трехуглеродного цикла с обра-
зованием карбенов наиболее характерно для арилциклопропа-
нов. Так, предполагают, что при фотолизе 1,1,2,2- и 1,1,2,3-тетра-
фенилциклопропанов, а также 1,1,2-трифенилциклопропана
образуется дифенилкарбен [619]:
Ph2CCRPh —Л :CPh2 -^Ph2CHOMe
CHR1 (< 50%)
R, R1 = Н,Ph
При фотолизе 1,1-диметил-3- (2-метилпропенил)2,2-дифенил-
циклопропана также образуется дифенилкарбен, идентифици-
рованный по продукту внедрения в связь О—Н трет-бутилового
спирта [620]; судя по образованию диметилдпфенилэтилена, в
этой реакции генерируется также и изобутен-1-илкарбен, иден-
тифицировать который, однако, не удалось [620]:
Ме2С - CPh?__________-CPh R0H г,,. „
г\/ 2 -Ме2С = СНСН = СМе2 •C/Ph2 *Ph^HOR
СН = СНСМе2 (19%)
/7 V
(45%)
:СНСН — СМе2 + ^^2^ ~ СМе2 R — Ви-трет
Фотолиз транс- 1,2-дифенилциклопропана [619], а также
1,2,3-трифенилциклопропана [621, 622] дает фенилкарбен; в
92
первом случае этот карбен удалось перехватить соответствую-
щими акцепторами [619]:
\ /
сн2
транс
MeOCH9Ph PhHC— СНМе
СМе2
цис и транс
Фотолиз 2-фенил-1,1 -дихлорциклопропана в среде олефинов
приводит к соответствующим циклоаддуктам дихлоркарбена,
причем присоединение дихлоркарбена к цис- и транс-бутену-2
проходит, как и следовало ожидать для синглетного карбена,,
стереоспецифично, например [591]:
МеСН = СНМе
Н9С-СС1о-----~----> :СС12--—>МеНС - СНМе
2\/ 2 -PhCH-CH г \/
CHPh СС12
цис
Алкилиденкарбены образуются при фотолизе арилзамещен-
ных алкилиденциклопропанов [623, 624]; карбены идентифици-
рованы в виде циклоаддуктов с непредельными акцепторами и
продуктов внедрения в связи С—Н (однако выходы всех продук-
тов невысоки), например:
Ph2C~CH2
С = СМе2
hv
-Ph2C ~ СН2
(< 1%)
Предполагают ф625—629], что при фотолизе фенилциклопро-
пана и метиленциклопропана образуется метилен.
93
а-Элиминирование с образованием соответствующих карбе-
нов характерно и для полициклических ароматических производ-
ных циклопропана; движущей силой большинства таких про-
цессов является образование (наряду с карбеном) термодинами-
чески выгодного ароматического соединения. Так, при термолизе
полициклического спирана (1) получены аценафтилен и флуоре-
нилиден (2), идентифицированный в виде димера (3) [630].
Фотолизом спиропроизводного норкарадиена (4) генерирован
гелг-диметилциклогексадиенилиден (5) (и бензол) [631]. Фото-
лиз дигидроциклопропафенантрена (6) приводит к метилену
и фенантрену [625].
Предполагают, что метилен генерируется при термолизе [632,
633] или фотолизе [625] тропилидена, находящегося в равнове-
сии с норкарадиеном. Термолизом или фотолизом 7-диметилами-
ноциклогептатриена (7) ’[629], 7,7-дицианоциклогептатриена
(8) [634] и 2,5,7-трифенилноркарадиена (9) [635] генериро-
94
ваны соответствующие карбены, стабилизированные заместите-
лями:
NMe2
(7)
70-110 °C (7)
------Э» :CHNMe2 ----»
2
(5-8%)
(8)
hv
:C(CN)2
-С6Н6
(9)
Аналогично ведут себя при фотолйзе замещенные бензонор-
карадиены (10) [634, 636]. О возможности образования метиле-
на при фотолизе незамещенного бензоноркарадиена (циклопро-
па[а]нафталина) (11) свидетельствует образование дицикло-
пропа[а,с]тетрагидронафталина (12), а в присутствии циклогек-
сена— образование норкарана (13) [637].
(Ю) R = R1 = CN [634]
R = СООМе, R1 = Н [636]
(11) R = R1 = н [637]
95
(11) н > ;СН2
(и)
(12)
(1з-
В сравнительно мягких условиях как переносчики силиленов
ведут себя соответствующие силациклопропаны (см., например,
[499, 638]), хотя образование кинетически независимых сили-
ленов в этих реакциях вызывает сомнения:
Me Me
60-80 °с Ме>^\
Ме2С-СМе2 —-----------:SiMe2 ----------|| SiMe,
\/ -Ме2С = СМе2
SiMe2
Силилены образуются также при фотолизе замещенных ди-
сила- и трисилациклопропанов. Образующиеся в этих реакциях
силилены, а также дисилены и трисилены (на схеме не показа-
ны) стабилизированы за счет объемистых заместителей. Анало-
гичная реакция описана [644] и для тригермациклопропанов.
Me Me
t f Me к Me
4, с; с-л с-а СН, = С-С = СН, ГЛ
“/SlA'2 :SlA,2 —----------> 4 >
CH2 S;Ar2
ROH
Ar2RSiOH
Ar = 2,6-Me2C6H3 [639]
RR1Si - SiRR1—:SiRR1 + RR1Si = SiRR1
SiRR1
R = R1 = CHEt2[640], CHCMe3[641], CMe3[642];
R = трет-Ви, R1 = 2,4,6-Me3C6H2 [643]
hv 1
Ar2 \~/GeAr2 *~:GeAr2 + Ar2Ge = GeAr2 Ar = 2,6-Et2C6H3|
GeAr2 r
96
Производные оксирана ведут себя в условиях термолиза и
фотолиза аналогично замещенным циклопропанам. Так, сам
оксиран в газовой фазе при 930 °C распадается на формальде-
гид и метилен с £акт —230 кДж/моль {645], а карбенный распад
тетрафтороксирана и трифторметилтрифтороксирана проходит
значительно легче (£акт=134—151 кДж/моль) [612, 646]:
RFC - CF2 1°°-140 °£. ;CF2 + RFC = О
О
R = CF3, F
Трифторметилтрифтороксиран был использован в качестве
источника дифторкарбена в препаративных опытах по дифтор-
циклопропанированию галогензамещенных олефинов (выходы до
85%), проводимых в автоклаве при 170—200 °C и повышенном
давлении акцептора дифторкарбена [647].
Арилзамещенные оксираны, подобно арилциклопропанам,
также склонны к фотофрагментации с образованием замещен-
ных карбенов и соответствующих карбонильных соединений.
Так, при фотолизе 2,3-дифенилоксирана в присутствии олефи-
нов, используемых в качестве акцепторов образующихся карбе-
нов, выходы соответствующих циклопропанов достигают 75%,
однако наряду с ними образуются и соответствующие оксета-
ны — продукты [2 + 2] -фотоциклоприсоединения карбонильных
соединений к тем же олефинам, например {648, 649]:
PhHC - CHPh-^:CHPh + PhCH = О Ме2° ~ см£2
V
---9- PhHC - СМе2 + <Ме2С - CHPh
\ ' 1 ’
СМе2 Ме2С - О
Необходимо подчеркнуть, что фенилкарбен, генерируемый
фотолизом транс-2,3-дифенилоксирана, проявляет ту же реак-
ционную способность, что и фенилкарбен из других источников.
Это свидетельствует о кинетической независимости этой частицы
[649].
Аналогично, при фотолизе соответствующих фенилоксиранов
генерированы дифенилкарбен [649], метилфенилкарбен [650],
фенилцианокарбен [651, 652], метоксикарбонилфенилкарбен
[651], метоксифенилкарбен [653], циклопропилфенилкарбен [8]’
7-Ю7 97
и (4-бензоилбутил) фенилкарбен [269]. Характерно, что селек-
тивность распада несимметричных оксиранов обычно согласу-
ется с образованием термодинамически наиболее стабильных
продуктов. Так, из циано- и метоксикарбонилтрифенилоксира-
нов главным образом получены аддукты дифенилкарбена
(>95%), тогда как доля фрагментации с образованием несим-
метричного карбена и бензофенона не превышает 5% [651]:
PhnC - СМе2 + PhRC - СМе2
СМе2 О - СМе2
Ме2С - СМе2
Ph?C - CPhR
* \ /
О
г- :CPh? + PhRC = О
hv
;CRPh + Ph2C = О
R = CN, СООМе
МегС = СМе?
PhRC - CMe2 + Me2C - СМе2
СМе2 Ph2C - О
Проведением фотолиза три- и тетрафенилоксиранов в стек-
лообразной матрице 3-метилпентана при 77 К удалось стабили-
зировать дифенилкарбен и получить его электронный спектр
[269, 654], а также спектр ЭПР [654] в соответствии с триплет-
ностью основного состояния этой частицы. Позднее эту реакцию
неоднократно использовали для генерирования фенил- и дифе-
нилкарбенов (см., например, {174, 655]). Подобным образом
путем термолиза или фотолиза оксасилиранов удается генери-
ровать соответствующие силилены, образование которых кон-
курирует с фрагментацией по карбенному механизму, что в
фотолитических условиях в большой мере определяется способ-
ностью растворителя к образованию комплексов с переносом
заряда [656]:
fiv или А
MeCN + EtOH
Ar2SiHOEt
(13а)
98
(13а)
hv,
CH2CI2,
(NC)2C = C(CN)2
+ Ar2Si = 0 Ar2Si(OH)OEt
Ar = 2,4,6-Me3C6H2
В отличие от циклопропенов и оксиранов реакции карбенной
фрагментации азиридинов изучены мало, тем не менее можно
отметить факт перехвата фенилкарбена в реакции фотолиза
1,2,3-трифенилазиридина [657]:
РПЦ
PhHC-CHPh -----------:CHPh ^i> PhCHnOR
х / -PhCH = NPh 2
NPh
Ненасыщенные азотсодержащие гетероциклы — диазирины,
представляющие собой циклические изомеры диазосоединений,
генерируют карбены легко и селективно как в термических,
так и, особенно, в фотохимических условиях, представляя собой
один из наиболее распространенных и универсальных источни-
ков разнообразных карбенов в виде кинетически независимых
частиц. Наиболее вероятная побочная реакция при этом — изо-
меризация диазиринов в соответствующие диазосоединения, что,
в частности, наблюдается при фотолизе дизамещенных диазири-
нов в Аг-матрицах при 10—15 К [239, 242, 275]. Однако образо-
вание диазометана при фотолизе незамещенного диазирина,
замороженного в И2-матрице, является результатом не изомери-
зации диазирина, а взаимодействия образовавшегося из него
метилена с молекулами азота матрицы [658].
Термолиз диазиринов в газовой фазе проходит гомогенно и
является реакцией первого порядка [659—661]. Диазометан
стабильнее, чем диазирин, на 126 кДж/моль [662]. Однако
даже будучи первичным продуктом превращения диазирина,
диазометан в условиях газовой фазы деазотируется быстрее, чем
передает избыток энергии другим молекулам или стенке реак-
тора. В то же время в растворе или в низкотемпературной
матрице такая изомеризация более реальна [241, 243, 663],
особенно для замещенных диазиринов, где возможно эффектив-
ное внутримолекулярное перераспределение избыточной энергии
в «горячем» диазосоединении. Именно при фотолизе или пиро-
лизе замещенных диазиринов наряду с продуктами карбенных
7‘ 99
реакций часто получают и соответствующие азины и иные
продукты вторичных превращений с участием диазосоединений,
но не азиринов [663, 664]; образование азинов отмечено даже
при газофазном пиролизе бис (трифторметил)диазирина при
300 °C [664].
Наиболее широко диазирины в качестве источников карбе-
нов используют в физико-химических исследованиях. Так, имен-
но фотолизом диазирина в Аг-, Ne-, Кг + Хе- или SFe-матрицах
при 10—20 К впервые удалось осуществить низкотемпературную
стабилизацию триплетного метилена и, зарегистрировав его
спектр ЭПР, установить нелинейную структуру этого простей-
шего карбена [223, 304]. Соответствующие замещенные диази-
рины использовали в качестве источников карбенов при регист-
рации их спектров ЭПР в низкотемпературных матрицах или
стеклах (см. [8]), при исследовании абсолютной кинетики кар-
бенных реакций в растворах методами пико- и наносекундной
лазерной спектроскопии [173], а также при исследовании мат-
ричных реакций карбенов [239, 242, 275], (подробнее см. гл. 2).
В частности, именно фотолизом соответствующих диазиринов
были генерированы метилкарбен [665], диметилкарбен '[666],
метилэтилкарбен [667, 668], диэтилкарбен [688], изопропилкар-
бен [668], метилизопропилкарбен [668], трет-бутилкарбен [665,
668], трет-бутилметилкарбен [668], пентилкарбен [389], цикло-
пентилиден [663], циклогексилиден '[665], фенилхлоркарбен и
его замещенные [173, 242, 399, 669], бромфенилкарбен [670],
бис (трифторметил) карбен [275], метокси- и феноксихлоркарбе-
ны [244, 245], бензилхлоркарбен [523] и бензилбромкарбен
[524]. Прямой и сенсибилизированный фотолиз диазирина в
дейтерохлороформе при ^20 °C использовали для исследования
методом ХПЯ реакций внедрения заведомо синглетного и трип-
летного метиленов [427]:
ci2dcch2ci-£^1ch2^—n = n —----------------
сн2
—^зСн2£2Цз ci3cch2d
Импульсным газофазным фотолизом дифтордиазирина гене-
рирован дифторкарбен и получен его ИК-спектр '[181, 671]:
CF2 „
N = N ---*- :CF2 + N2
а-Элиминирование карбенов из других карбо- и гетероцик-
лических соединений. Карбенный распад циклобутанового ске-
лета надежно установлен лишь в случае перфторциклобутана.
Для этой реакции получена кинетическая схема [672, 673].
Позднее эту реакцию использовали для генерирования дифтор-
100
карбена, который был изолирован в Аг- или Kr-матрицах и
идентифицирован по электронному спектру {674].
f2c - cf2 д
* ' ---» CF2 = CFCF3 + :CF2
«2^ Ьг2
При фотолизе циклобут^нонов наряду с реакциями цикло-
распада и декарбонилирования наблюдается расширение цикла
с вероятным промежуточным участием карбенов (см. разд. 3.1.3).
При фотохимическом, отщеплении молекулы азота от соответ-
ствующих замещенных тиадиазцлов и бензотриазолов по спект-
рам ЭПР идентифицированы тиобензоилфенилкарбен [330] и
иминоциклогексадиенилидены [326]:
Ph S
phC-CHPh
Ph-</N ------
S ~n2
N
NR
R = H, Me, Ph
Промежуточное образование карбенов предполагается и при
фотолизе циклических ацеталей флуоренона [675]. Аналогично
с образованием фенил- или дифенилкарбенов протекает фотолиз
и некоторых других циклических ацеталей [675]:
МеСН = СНМе
МеОН
V
(цис и транс; (30%)
23%)
101
PhV° /ph
ДЛ ^««Нгои.
Наконец, 1,3-диоксоланы в фотолитических условиях также
могут служить источниками карбенов [676, 677]:
RR1C - CR2R3
О о —^:CRR1 + R2R3C — О + 7 = 0
\ /
R, R1, R2, R3 = Н, Ph, CN; Z = 0=0, S =0, P(OEt)3
При пиролизе производных кислоты Мельдрума генерирова-
ны циклоалкилиденкарбены [678]:
О
Me о—/\
X _/=ОСН2)п
Me оЧ
О
480-640 °C
-СО
-Ме2СО,
-СО?
О = С = С
п = 1-5,9
Подобная схема распада характерна и для ацеталей заме-
щенных норборнадиенов-7 (14); при этом образуются карбены
и стабильные ароматические молекулы [679—681]. Аналогич-
ным образом, 7,7-дигалогенбицикло [2.2.1] гептадиены [680, 682],
а также 11,11-дигалоген-1,6-метано[10]аннулены (15) [683]
при термолизе генерируют дигалогенкарбены, причем в послед-
нем случае дихлоркарбен генерируется в значительно более мяг-
ких условиях (80°C), чем дифторкарбен (250°C) [683].
102
R= Н, Me, Ph, Вг
нс = сн - сн - сн = сн
:С(ОМе)2---> (МеО)2С —С(ОМе)2
сх2
НС = сн - сн - сн = сн
(15)X = F, Cl
__^:СХ2 +
Карбенная схема термораспада характерна и для производ-
ного квадрициклена [238]:
-с6н6
250 °C, Н-
133 Па
он + Г>-о
Н
Производные соответствующих 7-гетеранорборнадиенов ис-
пользуют в качестве источников силиленов, гермиленов и стан-
ниленов, хотя кинетическая независимость получаемых таким
путем частиц в ряде случаев не доказана:
MRR1
R1 IVPh
У :MRR
Ч -СщНдРПгРР1
R2/Ph \=/
M=Si; R,R1= Me, SiMeg, Si2Me5; R2= Н [б84]
M= Ge;R,R' = Me: R2= Ph [428, 685-687]; 70-85 °C
R= Me,Et, Bu [®8]
> -io c
-----------> :SnR2
-CePh4(CN)4
103
Удобными источниками силиленов являются не только рас-
смотренные выше силациклопропаны с одним — тремя атомами
кремния в цикле, но и циклополисиланы большого размера, а
также их ациклические аналоги — полисиланы. Пиролиз или
фотолиз линейных или циклических полисиланов представляет
собой один из общих методов генерирования силиленов, содержа-
щих органические заместители (см. обзор [689]); изучен [690]
термолиз большого числа алкил- или арилзамещенных ди- и
трисиланов. Несколько примеров генерирования силиленов по-
добным путем приведены ниже:
/м o x hv [247, 691, 692]
(Me2Si)6—----------:SiMe2
hv
(H-C6H13MeSi)n—»- :SiMeC6H13-H
n = 5-7 [693]
Si Ro
h,
(R2Si)4 —=»-:SiR2 + R2Si - SiR2 R -CHMe? [694]
RR1 Si(SiMe3)2 Аили^ :SiRR1 + Me6Si2
R = Me, R1 = Ph [294]; R = Ph, R1 = SiMe3 [695, 696];
R = R1 = SMe [697, 698]; R = SMe, R1 = Bu-rper [643];
R = R1 = SiMe3 [699]
Согласованный механизм отщепления силиленов из таких
соединений подтвержден [700] сохранением в продукте реакции
стереохимической конфигурации исходного трисилациклогепта-
на, что указывает на координацию a-заместителей в процессе
отщепления силилена:
С SiMePh hyj SiMePh
^SiMe2 ----: SiMe2 + ^SiMePh
SiMePh '
цис
цис
Аналогичные реакции используют и для термического или
фотохимического генерирования диорганилгермиленов и -стан-
ниленов из соответствующих полигерманов и полистаннанов:
104
:GeMe2 +
Me2Ge - GeMe2
д
(Me2Sn)6 ------» :SnMe2
[248]
Термическое и фотохимическое генерирование карбенов и
их аналогов из циклических предшественников подробно рас-
смотрено в обзорах [702, 703].
Г енерирование карбенов из галогенметаллорганических
предшественников. Реакции этого типа имеют достаточно общий
характер и находят широкое применение (см. обзор [704]).
Как и во всех остальных случаях а-элиминирования, распад
а-галогенметаллорганического соединения (а-ГМОС) может
происходить по ступенчатому (а или а') или по синхронному
(б) механизму*:
ЧС:
/
\ -
—хс; 1
-мх /
м
С:
б
Согласно литературным данным (см., например, [704, 705]),
наиболее вероятен синхронный механизм (б), движущей силой
которого является внутримолекулярная координация атомов га-
логена и металла. Именно эта схема позволяет корректно про-
анализировать зависимость стабильности а-ГМОС от теплового
эффекта реакции распада, разности энергий координирующихся
орбиталей и расстояния между отщепляющимися атомами М и
X. Конечно, такая упрощенная схема является в известной сте-
пени прокрустовым ложем для реальных, в большинстве своем
сольволитических процессов, связанных с образованием слож-
ных ассоциатов, однако возможность объединения в рамках
единого механизма достаточно разнородных реакций, на наш
взгляд, вполне оправдывает принятый подход.
Галогеналкилъные производные щелочных металлов. К наи-
более изученным соединениям этого типа относятся галоген-
* При ступенчатом механизме возможно как гомолитическое, так и гете-
роциклическое (различные варианты) расщепление связей С—М и С—X.
105
литийметаны и родственные соединения, получаемые обычно дей-
ствием алкил- или фениллития на соответствующие метилгалоге-
ниды при низких температурах [706]. В среде эфира легкость
обмена на литий заместителей при атоме углерода обычно
уменьшается в ряду 1>Вг>Н>С1 {707]. В некоторых случаях
на направление реакции существенно влияет природа раствори-
теля, например [708, 709]:
MeLi, Et2O
PhCHBr2 — * PnCHLiBr * -MeBr -LiBr MeLi, ТГФ =► PhCLiBr2 -MeH -LiBr
:CHPh
:CBrPh
При взаимодействии бензилидендихлорида, в котором атом
водорода имеет более «кислый» характер, с метиллитием в эфи-
ре отщепляется именно этот атом водорода [710]:
MeLi, Et2O
PhCHClo ------------- PhCLiCI2------Э- :CClPh
-MeH -LiCI
Бромоформ c BuLi в ТГФ образует смесь близких количеств
ЫСВгз и LiCHB'r2, а в углеводородной среде дает только
LiCHBr2 [711, 712]. Аналогичная реакция СНС13 в среде про-
стых эфиров приводит к LiCCl3— предшественнику дихлоркар-
бена [713], а в углеводородной среде — к смеси LiCCl3 и
ЫСНС12 [712].
---* LiCBr->—-*-;CBr2
-BuH J -LiBr
СНВг3
BuLi
ТГФ
-BuBr
LiCHBr2
---* :CHBr
-LiBr
Дигалогенметаны в углеводородах под действием алкиллития
обменивают только атомы галогена [714, 715], тогда как в эфи-
ре СН2С12, например, обменивает только атом водорода, обра-
зуя ЫСНС12 и далее хлоркарбен [716, 717].
Реакция фениллития и его производных с СН2С1г в эфире,
проходящая с участием хлоркарбена, лежит в основе удобного
общего метода получения арилкарбенов [718]:
106
A r Li,
Et2O
CH2CI2--* LiCHCl2—*
2 -ArH "L|C1
:CHCI-^U ArCHLiCl----* :CHAr
-LiCl
Металлирование ди-, три- и тетрагалогенметанов, приводя-
щее к последующему образованию соответствующих галогенкар-
бенов, может быть осуществлено не только литийорганическими
соединениями, но и непосредственно металлическим литием или
натрием в ТГФ при —404-4-40°C [719]. В этих условиях в та-
ких соединениях, как СН2С12, СНС13, PhCHCl2 и СНВгз заме-
щается только атом водорода, а в случае СН2Вг2—атом брома
[720].
Реакции этого типа использованы для генерирования карбе-
нов не только в обычных химических системах, но и в низко-
температурных инертных матрицах с целью последующей ста-
билизации карбенов. Так, при взаимодействии атомов лития,
натрия или калия с замороженными в Ar-матрицах при 15 К
молекулами СС14 [209, 211], СВ’г4 или СС1Вг3 [59, 61] и СС12Вг2
[59] были детектированы с помощью ИК-спектроскопии дихлор-
карбен, дибромкарбен и смесь дибром- и бромхлоркарбенов
соответственно. Напротив, из СН2С1Х и СН2ВгХ (X = Br, Cl, F)
в тех же условиях удалось детектировать лишь радикалы
•СН2С1 и *СН2Вг, а также продукты вторичных реакций — С2Н4,
• СН3 и СН4, образование которых можно объяснить участием
метилена [721].
Низкая стабильность галогенлитийметанов в инертных
матрицах даже при 4 К при одновременной относительной
устойчивости их в эфирных растворах (обычно стабильны до
—70°C) объясняется возможностью образования в последнем
случае сложных ассоциатов с растворителем (подробнее см.
[704, 722, 723]). Подобная ассоциация (образование эфиратов)
существенно влияет как на стабильность, так и на реакционную
способность а-ГМОС, которые в растворах до температуры их
распада (обычно —704—90°C) ведут себя как обычные литий-
органические соединения, например [704]:
BuLi, эфир или
ТГФ,<-90’С >-80 °C
CHCI,------------* LiCClo ---------»:СС12
-BuH -LiCl
ROD
CDCl3
<-90 °C
1.CO2
I
СС13СООН
107
В связи с этим следует еще раз подчеркнуть необоснован-
ность довольно широко используемого для этого класса а-ГМОС
термина «карбеноиды». Тем не менее образующиеся в этих ус-
ловиях из галогенлитийметанов карбены связаны в слабые со-
левые комплексы типа LiX-CRR1, которые, однако, по реакцион-
ной способности практически не отличаются от одноименных
кинетически независимых карбенов. В частности, для дихлор-
карбена, генерированного в широком интервале температур
(—1964-1100°C), в том числе и из LiCCl3 в эфире при —70°C,
относительная реакционная способность (относительные кон-
станты скорости присоединения к олефинам) подчиняется урав-
нению [724, 725]:
AAG = АДН- ТДДЗ.
а-ГМОС щелочных металлов используют и для генерирова-
ния ряда аналогов карбенов. В частности, удобными источника-
ми соответствующих дигалогенидов германия и олова являются
трифтор (цезио) герман [726], трихлор (цезио)герман [727, 728],
а также трихлор (цезио)станнан [729]. При взаимодействии
дигалогенидов диорганилзамещенных элементов подгруппы IVB
со щелочными металлами в различных органических раствори-
телях образуются продукты формальной олигомеризации анало-
гов карбенов, хотя, скорее всего, без участия этих частиц в
кинетически независимом состоянии. Гораздо более вероятно
участие свободных аналогов карбенов при проведении этих
Реакций в газовой фазе, например, с калий-натриевыми парами
730—732]:
RRiMCl2-^lRR1M:
M = Si,Ge;R,Ri =Alk
Для генерирования простейших карбенов в настоящее время
разработаны более удобные методы, в первую очередь методы,
основанные на использовании принципов межфазного катализа
(см. ниже). Однако для генерирования сложных карбенов, в
первую очередь замещенных циклопропилиденов — удобных
предшественников алленов, до сих пор широко применяют имен-
но методы литийорганической химии, например:
Br Br Br Li
к/ PhLi, Et2o
-PhBr
-LiBr
108
N=C = CH2[733]
(СН2)П
£3(СНг)"
п 3-7 [734]
В последнем случае использование хирального амина NRS
позволяет получать оптически активные циклические аллены.
Описано [735] успешное препаративное использование диме-
тилвинилидена, генерируемого из соответствующего а-ГМОС, в
синтезе замещенного [3]радиалена:
Ме2С = СВг2 Ме2С = С В rl_i —Ме2С = С:—
z “BuBr “Libr
С = СМе2
—/ \
Ме2С = С - С = СМе2
а-Галогенметильные производные магния. Эти соединения,
подобно их литиевым аналогам, при нормальных условиях про-
являют склонность к карбенному распаду с а-элиминированием
дигалогенида магния. Исходные а-ГМОС получают обменной
реакцией соответствующих полигалогенметанов с реактивами
Гриньяра в эфирных растворах при низких температурах, при-
чем даже при наличии у исходных галогенметанов достаточно
кислых атомов водорода, замещается исключительно атом гало-
гена [736]. Эффективен в этих реакциях (особенно в случае ди-,
три- и тетрабромметанов) и «суперактивный» магний, получае-
мый взаимодействием соответствующих солей магния с калием
в ТГФ [737]. В то же время при взаимодействии паров магния
и других металлов (Al, Сг, Ti, Fe, Си) с дихлор- или дибром-
метаном, или с 1,1-дибромциклопропаном в условиях «мат-
ричного синтеза» (испарение металла в замороженный органо-
галогенид) происходит радикальный отрыв атома галогена
металлом без промежуточного образования ГМОС, в том числе
и в присутствии сольватирующего растворителя (эфир, ТГФ)
[738].
Цинк- и кадмийорганические соединения. Их широко исполь-
зуют в качестве метиленирующих агентов, причем наибольшее
препаративное значение имеет реакция Симмонса — Смита, ос-
нованная на использовании дигалогенметанов (обычно CH2I2)
и активированного порошка Zn [739, 740] (подробнее эта реак-
ция рассмотрена в разд. 4.2; см. также [704]).
109
CH2I2
Zn
Znl?
ch2n2
x c =
_^ICH2Znl -------
~Znl2
-N2
zc“cC
z \ / 4
CH2
а-Галогенртутъорганические соединения. Эти соединения до
сих пор сохранили свое значение в химии карбенов, поскольку
с их помощью возможно генерирование карбенов в нейтральных
средах при невысоких температурах в отсутствие часто конку-
рирующих с карбенами галогенметильных анионов. В связи с
этим данные реагенты широко применяют для циклопропаниро-
вания соединений, отличающихся повышенной чувствительно-
стью к основаниям и (или) протонным средам, а также к гало-
генметильным анионам.
Синтез а-галогенртутьорганических источников карбенов
обычно основан на реакции генерированного иным методом кар-
бена или предшествующего ему аниона с дигалогенидом или
органогалогенидом ртути [741]:
RHgX
~СХХ1Х2 Х“+ :СХ1Х2 -------------RHgCXX1X2
-х~
Карбенный распад полученных соединений происходит при
умеренном нагревании в нейтральных растворителях (диметок-
сиэтан, бензол, тетрагидрофуран, дихлорметан) и в присутствии
олефина идет в две стадии [741]. Обратимость первой стадии
карбенного распада, в результате которой имеется возможность
регенерации исходного а-ГМОС, позволяет с высокой степенью
эффективности использовать эти источники карбенов и цикло-
пропанировать таким путем даже малоактивные олефины или
другие акцепторы.
RHgCXX1X2 ^-д{!енн3° RHgX + :СХ1Х2
СХ1Х2
:СХ1Х2+ )С = С( S )С-ЧС/
Закономерности распада а-галогенртутьорганических соеди-
нений установлены довольно надежно: их стабильность зависит
от природы галогена X и, например, в ряду PhHgCX3 увеличи-
вается в ряду: F»Cl>Br>I. Тот же ряд относительной ак-
тивности галогенов определяет и направление распада смешан-
ных производных вида PhHgCXX^ и PhHgCXX^2 [741]. Так,
ПО
в случае PhHgCCl2Br в присутствии Nal дихлор- и бромхлор-
карбены образуются в соотношении «6:1 [716, 742, 743]:
:CCI2^-_ PhHgCCl2Br -------:—»:СС1Вг
z -PhHgBr 2 -PhHgCI
В соответствии с принятым для а-ГМОС синхронным меха-
низмом «-элиминирования карбенов, уменьшение электроотри-
цательности R в RHgCX3 дестабилизирует молекулу, облегчая
ее карбенный распад (подробнее см. [704, 741]). Карбены, ге-
нерированные из различных а-галогенртутьорганических соеди-
нений, приведены ниже:
д
RHgCH2X —>- RHgX +:СН2
R = CH2Br, X = Вг [744]; R = СH2I, X = I [745]; R = X = I [745];
R = PhCH2, X = I [746]
д
PhHgCHXY ----»- PhHgX + :CHY
X = Y = Cl [747]; X = Br, Y = Cl [748]; X = Y = Br [748]; X = Cl,
Y = СООЕЦ749]
Д
RHgCCl2X —* RHgX + :CCl2
R = CCl3, X = Cl [749]; R = X = Cl [750]; R = Ph, X = Cl, Br [751],
I [752]; R = H-C3H7, X = Cl, Br [753]; R = цикло-CgHi j, X = Cl,
Br 17551
д
RHgCBr2X------ RHgX + :CBr2
R = CBr3, X = Br [754]; R = Ph, X = Br [508, 751, 755];
R = KUOo-C6H11,X = Br[752]
Д
RHgCCIBrX----* RHgX + :CClBr
R = Ph, X = Br [751 ], I [756]; R = цикло-С6Н11, X = Br [752]
111
A
PhHgCFX2 —» PhHgCX + :CFX
X = Cl[757], Br [758]
A
Ph HgCCIXCF3-----PhHgX + :CC!CF3
X = Cl, Br [759]
A
PhHgCX2CON(CH2)5----* PhHgX + :CXCON(CH2)5
X = Cl, Br [760]
A
PhHgCX2SO2Ph------ PhHgX + :CXSO2Ph
X = Cl, Br [749]
A
RHgCXCICOOMe-----* RHgX + :CClCOOMe
R = PhCH2CH2, X = Cl; R = Ph, X = Cl, I [749]; R = Ph, X = Br[761 ]
A
PhHgCXYCOOR -----* PhHgX + :CYCOOR
R = Me, Et; X = Y = Br [761]; X = Cl, Br, Y = F [749]
Me3SiCCI2HgCCl2SiMe3-p^-* Me3SiCCI2HgCl + :CCISiM?
Me3SiCCI2HgCI-^|U- HgCl2 + :CC!SiMe3
A
BrHgCBr = CMe2 --* HgBr2 + :C = CMe2
[763]
Перечисленные выше карбены перехвачены разнообразны-
ми акцепторами в условиях жидкой фазы. При газофазном пи-
ролизе (250—550 °C) таких ртутьорганических соединений, как
112
PhHgCClBr2, PhHgCCl2Br, PhHgCCl3, Hg(CCl3)2 и ClHgCCl3,
также образуются соответствующие дигалогенкарбены; они бы-
ли изолированы из газовой фазы в низкотемпературные инерт-
ные матрицы и идентифицированы методом ИК-спектроскопии
[49, 50, 210]. Однако в этих условиях, особенно при повышен-
ных температурах, подобные ртутьорганические соединения, по-
видимому, из-за ослабления внутримолекулярной координации
атомов галогена и ртути диссоциируют и по гомолитическому
механизму с образованием тригалогенметильных радикалов
•СХ3, полученных и при фотолизе PhHgCCl3 и Hg(CCl3)2 в
твердой фазе при —196-4—20 °C, а также в газовой фазе при
80 °C и в растворе диметоксиэтана при 18 °C [764]. Тем не ме-
нее в некоторых случаях фотолиз а-галогенртутьорганических
соединений приводит к соответствующим карбенам, например
[765]:
PhHg\
Cl
/7 V
2 -PhHgcT
:С = СС12
С = CCI
Галогенметильные соединения элементов III группы. Эти со-
единения в качестве потенциальных источников карбенов иссле-
дованы сравнительно мало. Можно отметить имеющую общий
характер реакцию получения внутренних олефинов при карбен-
олефиновой изомеризации частиц, полученных из борорганиче-
ских предшественников:
(RCH2CH2)3B^^ (RCH2CH2)2BCCI2CH2CH2R -----Э-
R = Me,Et [766]
80 °C
*RCH,CH2BCICCI(CH2CH2R)2 гн > :C(CH2CH2R)2
— r\L>rl2^'rl2D<-'<2
rch2ch - chch2ch2r
Описано также генерирование фенилхлоркарбена из а-хлор-
борорганических соединений, полученных внедрением дихлор-
карбена (из ртутьорганического источника) в связь В—С [766]:
’СС1 А
PhBR2 —± PhCCI2BR2 -A-sj. ;CCIPh
— r\2DUl
(21%)
R = Ph, OMe
8—107
113
Галогенметильные производные алюминия [767, 768], алкил-
производные таллия [753, 769] и хлориды галлия [770] также
могут распадаться с образованием карбенов:
СНВг. д С6Н6 Z=\
Е'зА1--Г Et2AlCHBr2 -СНВг-А^ / J-Br
PrHgCCI^Br Е сс :CClEt
tzl3 1 -РгНдВг 2 £ -Et2 • Кя
При взаимодействии Et3Tl с ССЦ или СНС13 образуется
Et2TlCCl3, распадающийся с образованием дихлоркарбена
[753, 769]. Соединения индия Et2InOCMe3 и Et2InNMe2, реаги-
руя с хлороформом, правда в более жестких условиях, также
образуют трихлорметильные производные индия, распадающие-
ся с образованием дихлоркарбена [769].
а-Галогенметаллорганические соединения элементов IV
группы. Эти соединения, как и ртутьорганические соединения,
хорошо изучены и их достаточно широко используют в качест-
ве источников карбенов. Так, гем-дихлорциклопропаны образу-
ются с выходами до 80% при нагревании (100—250 °C) с олефи-
нами приведенных ниже трихлорметильных производных крем-
ния [771—773]:
СС l3SiR3 CCl3SiCl2R
R Cl, F, Me, OEt R = Me, CCl3, OEt
CCl3SiCl(OEt)2
Кинетические исследования реакций распада таких соеди-
нений полностью подтвердили образование в них кинетически
независимых галогенкарбенов [772, 774—776]:
а
CCl3SiF3 —:СС12 + S i F3 СI
А
CHF2CF2SiR3-----:CFCHF2 + SiFR3
R = F, Me
114
Для термолиза CCl3SiCl3 также надежно установлено пре-
имущественное протекание карбенной фрагментации; доля го-
молитического распада здесь невелика, а альтернативное на-
правление с отщеплением молекулы дихлорсилилена вообще не
реализуется [774]:
• С С + • S i СI3
:SiCl2 + CCl4JX-CCI3SiCI3 --
к 2
—Э- :СС12 + SiCl4
= 1016exp[-184/(RT)]; к2 = 1010exp[-124/(RT)]; fc3 = О
Характерно, что для подобных соединений карбеновый путь
распада является и наиболее экзотермичным, т. е. легкость
а-элиминирования определяется тепловым эффектом и возмож-
ностью внутримолекулярной координации геминальных замес-
тителей с донорными и акцепторными свойствами [449]. В со-
ответствии с этим фторметильные производные кремния распада-
ются при значительно более низких температурах, чем их хлор-
содержащие аналоги, что, видимо, связано с уникальной проч-
ностью образующейся связи Si—F (700—800 кДж/моль [777]).
Так, CF3SiF3 гладко отщепляет молекулу дифторкарбена уже
при 100 °C [778]. При наличии в алкильном заместителе сила-
на одновременно атомов фтора и хлора термолиз протекает
только с миграцией атома фтора [771], например:
2CFCt2CF2SiCl3 -^5^. ;CFCFCl2 + С С12 = CF2 + 2S i FC l3
Гексагалогенсиланы являются эффективными и достаточно
селективными источниками соответствующих дигалогенсилиле-
нов; в частности, пиролиз Si2Cl6 широко используют для гене-
рирования SiCl2 [549, 610]:
д
Si2x6---* :SiX2 + SiX4 X = Cl, Br
Очень удобным источником диметилсилилена оказался тет-
раметил-1,2-диметоксисилан, легкость термораспада которого
во многом определяется высокой прочностью образующейся свя-
зи Si—О (500—600 кДж/моль) [549, 777]:
Me2(MeO)Si - Si(OMe)Me2 :SiMe2 + Me2Si(OMe)2
8*
115
Аналогичные реакции широко используют для генерирования
различных силиленов, например [779—781]:
:Sl<SiMe3>2 (Me3Si)3SiOMe ; S i< О Me) Si М е3
МеО
)Si
Me3Si
-MeOSiMe3
д
(MeO)3SiSi(OMe)3 —^QMe> :Si(OMe)2
Аналогичным образом, высокая энергия связи Si—О пред-
определяет малую стабильность ацеталей силилзамещенных
альдегидов и кетонов [782, 783]—перспективных предшествен-
ников метоксикарбена; монометоксиметилсиланы более ста-
бильны, но также способны разлагаться по карбенному меха-
низму [782]:
125 °C /С=С\ ч /
(MeO)3SiCH(OMe)2--------э- :СНОМе ------)с - С;
-Si(OMe)4 ' \ /
СНОМе
Me2PhSiC(OMe)Ph2 ^XMe2PhSiOMe + :CPh2
Для метоксигидридов германия характерна реакция отщеп-
ления метанола. Эта реакция является достаточно общим ме-
тодом генерирования органилгермиленов, например [784]:
MeOGeHClPh 20 °с> :GeCIPh
-МеОН
GeCiPh
(< 70%)
Галогенметильные соединения Ge, Sn, Pb претерпевают кар-
бенный распад при несколько более низких температурах, чем
аналогичные производные кремния (за исключением фторпро-
изводных). Так, из (дифенилхлорметил)трихлоргермана при
150—220 °C образуется, по-видимому, дифенилкарбен (иденти-
фицирован только его димер — тетрафенилэтилен) [785]; пи-
ролиз трихлор (трихлорметил) германа при 220 °C дает GeCl4 и
дихлоркарбен, который идентифицирован в виде аддуктов с
олефинами [785, 786], а также по ИК-спектру в низкотемпера-
116
турной Ar-матрице. При термолизе трииод (трифторметил)гер-
мана выделены димеры и тримеры дифторкарбена [787]:
180°с гс гс п с
CF3Gel3 ----:CF2 ------CF2 = CF2 + цикло-С3Е6
~GeFI3
Из триметилтрифторметилстаннана при 150 °C количественно
образуется тетрафторэтилен [787]. Подобные реакции катали-
зируются Nal, что позволяет снизить температуру до 80 °C
[788]. Эффективными источниками дигалогенкарбенов являются
Me3SnCCl2Br и Me3SnCCl3 [789], а также CCl3SnI3 [790]; из
Me3SnCCl2Ph генерирован и перехвачен фенилхлоркарбен
[789].
Необычный карбен — пентафторциклобутилиден — образует-
ся при распаде соответствующего производного олова [791], не-
устойчивого уже при комнатной температуре:
F2C-CF2 20 °C F2C-CF2 f2c - cf2
Me3SnFC-CHF _Me3SnF -C - CHF ~ * HC = CF
Термолиз соединений типа R3SnOCOCX3 (R = Alk, X = C1, Br)
протекает в две стадии, приводя в присутствии олефинов к соот-
ветствующим гем-дигалогенциклопропанам, например [792,
793]:
90 °C
R3SnOCOCX3—* R3SnCX3 ——-j*:CX2
““L/V2 ТлЗОПА
88%)
Аналогично, термолизом замещенных 1-бром(иод)-1-триме-
тилстаннил- или 1-бром (иод)-1-триметилплюмбилциклопропа-
нов удается генерировать и более сложные бициклические кар-
бены, которые с олефинами способны образовывать спиросоеди-
нения [794J:
л
-Ме3МХ
Ме3М(сн2)п
(=S 35%)
М = Sn, Pb; X = Br, I; п = 1,2
117
При разложении полигерманов, содержащих заместители с
неподеленными парами электронов, образуются соответствую-
щие гермилены [795—798]. Аналогичная реакция достаточно
характерна и для ди- и полистаннанов [555, 556].
R3Ge-GePhClY 't?! :GePhY -^7—- PhY2Ge - GeY2Ph
J -R3GeCI -PhGeY3 z z
Y = OMe, SMe, NMe2, PEt2
A
XR2Sn-SnR2Y------R2SnXY + :SnR2
R = Me, Et, Bu; X, Y = HCl
Движущей силой этих процессов является внутримолекуляр-
ная координация заместителя, имеющего неподеленную элект-
ронную пару, с соседним атомом Ge или Sn и образование в
результате распада наряду с карбеноподобными молекулами
соединений четырехкоординированного германия или олова с
термодинамически прочными связями типа М—Cl, М—F, М—О,
М—N и т. п. Следует также отметить, что реакции этого типа
ускоряются в присутствии пиридина, а кислоты Льюиса [799],
ТГФ либо Со2(СО)8 [800] катализируют даже станниленовую
фрагментацию гексаметилдистаннана (но не его гексаэтильного
аналога [556]).
Соединения элементов V группы. В этом ряду источниками
карбенов чаще всего оказываются соответствующие илиды.
Примеров карбенного распада галогенорганических соединений
элементов V группы известно немного. Так, термолиз трис(три-
фторметил) сурьмы при 180—220 °C и дифенилтрихлорметил-
сурьмы при 120 °C, а также распад нестабильной в обычныхусло-
виях трис(трихлорметил)сурьмы, видимо, протекают с проме-
жуточным образованием соответствующих дигалогенкарбенов
[801—803]. При термолизе перфторметилфосфоранов [804] об-
разуется дифторкарбен:
(CF3)nPF5_n --- А > CF2 —-
-(СЕз)п_1 rrg_n
cf2
/ \
—CF2 = CF2 + F2C - CF2 + (CF2)m n = 1-3,m>2
Органические соединения переходных металлов в качестве
источников карбенов рассмотрены в гл. 5.
а-Элиминирование с участием производных серы и азота.
Эти реакции не носят общего характера, однако можно приве-
118
сти ряд примеров, где легкость сс-элиминирования с генерирова-
нием карбенов определяется образованием радикалов бензиль-
ного типа, например [805]:
RCH(SO2CH2Ph)2----:CHR + 2SO2 + 2-CH2Ph
Аналогично протекает термолиз S-алкилтиосульфонатов
[806]:
PhRCHSO2SR1 --*- :CRPh + SO2 + HSR1
При термическом распаде азосоединений а-элиминирование
с образованием карбенов происходит ступенчато [807, 808]; это
установлено масс-спектрометрически на примере перфторазоэта-
на, в ходе газофазного пиролиза которого перфторэтильные ра-
дикалы образуются при 800 °C, а их распад с образованием ди-
фторкарбена— только при 950 °C [120].
CF3N = NCF3 —-------2-CF3 —э» CF2 + -F
PhCH2N = NCH2Ph 20°n°£-2-CH2Ph —* :CHPh + PhCH3
с с M _ МГ c 800 °c о rc rc 950 -ntr , ,rc
^2 ’ 5 N — N C2 F&-2 • C F2 C F3 « 2 * 3
—N?
Эффективными переносчиками диметилкарбена являются
соединения типа MCMe2NO2 (M = Li, К) [809].
Образование карбенов предполагают и при пиролизе амино-
производных малоновой кислоты [810], а также тригалогенук-
сусных [811] и перфторпропионовой кислот [533]:
Me2NCH(CN)COOR ——* :C(CN)NMe2
— Me2NH
СХ3СООН
300-400 °C 600 "''.
:СХ2 CF3CF2COOH :CFCF3
X = F, Cl
Еще одним перспективным методом генерирования дифтор-
карбена является и газофазное разложение тригалогенуксус-
ных кислот в присутствии водяного пара при 300—500 °C, про-
текающее, по-видимому, в одну стадию [812J:
119
н2о
СХ3СООН----->
—> -СХ2 - со2+ нх
3.1.2. Другие реакции элиминирования
Общим для реакций этого типа является отщепление карбена
от стабильного предшественника, активированного тем или
иным энергетическим воздействием, причем разрыву подверга-
ются не две геминальные о-связи, как в реакциях сс-элиминиро-
вания (см. разд. 3.1.1), а одна связь повышенной кратности
(двойная, илидная и т. п.). В этом разделе не рассматривается
генерирование карбенов из диазосоединений и тозилгидразонов
(реакция Бамфорда — Стивенса), поскольку в этих чрезвычай-
но важных для синтетической органической химии реакциях
практически невозможно провести грань между энергетической,
химической и каталитической активацией предшественника.
Карбенная диссоциация олефинов. В принципе, любой оле-
фин можно рассматривать как продукт рекомбинации двух оди-
наковых или разных карбенов. Однако обратная реакция—тер-
мическая или фотохимическая карбенная диссоциация олефи-
нов— возможна далеко не всегда, поскольку в этих условиях \
процесс часто проходит с разрывом не двойной, а другой, менее
прочной о-связи. Тем не менее карбенная диссоциация олефинов,
особенно при наличии в них слабой л- и сильных о-связей, идет
во многих случаях, что надежно установлено различными хими-
ческими и физико-химическими методами.
Таким путем протекает распад тетрафторэтилена. Известны
надежные кинетические данные о равновесии и скоростях пря-
мой и обратной реакций в различных условиях [582, 583J. Эту
реакцию неоднократно исследовали методами ударной волны
[122, 195—197] и адиабатического сжатия [202—204], причем
анализ продуктов проводили с помощью масс-спектрометрии
и газофазной УФ-спектроскопии; при пиролизе при низком дав-
лении среди образовавшихся продуктов, замороженных в инерт-
ной матрице и охарактеризованных по ИК-спектрам, основным
был именно дифторкарбен [208]:
д
CF2 = CF2 2 CF2
Фотолиз тетрафторэтилена, как прямой [813], так и сенси-
билизированный ртутью [814], а также импульсный фотолиз
[469] тоже приводят к дифторкарбену.
1,1 -Дифтор-2,2-дихлорэтилен и трифторхлорэтилен под дей-
ствием импульсного фотолиза или MB-разряда образуют ди-
фтор- и дихлор- или фторхлоркарбены, зарегистрированные в
120
газовой фазе спектральными методами [169, 189, 358, 584].
Аналогичные реакции известны и для перфторпропилена,
генерирующего дифторкарбен термически [672], фотохимически
[469, 814] и в условиях адиабатического сжатия [202—204].
При 400—600 °C к распаду на две молекулы СС12 способен и
тетрахлорэтилен [8151:
СС1г = СС12^2«гсс,2
-НС1
С10Н7С1-2
(30 %)
Распаду двойной связи пергалогенолефинов способствует
взаимодействие с реагентами типа атомарного кислорода (со-
стояние 3Р), связывающего один из «карбеновых» фрагментов
[814, 816, 817]. Аналогичная реакция известна и для перфтор-
пропилена [814].
CF2 = CF2 + О —:CF2 + CF2O
Роль «активатора» диссоциации C2F4 может играть и моле-
кула N2 [195, 197].
К сожалению, для более сложных олефинов однозначные
доказательства диссоциации с образованием карбенов отсутст-
вуют. Ранее предполагали, что пространственно затрудненные
олефины с заместителями, стабилизирующими карбенный центр,
способны к подобной диссоциации (см., например, [7] и гл. 2),
однако продукты перекрестного сочетания образовавшихся кар-
121
беновых фрагментов удается выделить не всегда; например,
в приведенных ниже реакциях только при R = Alk [818]:
RN NR NR
Образование продуктов взаимодействия электрофила с про-
дуктами карбенной диссоциации пространственно затрудненного
олефина типа 1,1,2,2-тетракис (а-нафтил) этилена [819, 820], ско-
рее всего не связано с участием кинетически свободных кар-
бенов.
Кремниевые и германиевые аналоги олефинов обычно чрез-
вычайно лабильны, однако введение объемистых заместителей
к кратной связи позволяет стабилизировать их, делая карбено-
подобную диссоциацию этих соединений термодинамически вы-
годным процессом. Так, описана фотохимическая диссоциация
тетраарилдигермена с образованием соответствующего герми-
лена [644]:
Ar2Ge = GeAr22:GeAr2 Ar = 2,6-Et2C6H3
Диссоциация кетенов. Механизм газофазного фотолиза кете-
на был предметом длительного и скрупулезного исследования.
Зависимость выходов продуктов от длины волны, парциальных
давлений кетена и других вводимых газов удалось рационали-
зировать в рамках следующей схемы [821—824]:
hv
сн2 = С = О “*[СН2 = С = О]* —- -сн2 + со
:СН2 + СН2 = С = О-^ СН2 = СН2 2:СН2
Позднее эта схема была уточнена за счет разделения кана-
лов, приводящих к синглетному и триплетному метиленам
[825]:
СН2 = С = О СН2 = С = О (S) —* 1сн2 + со
X
I--------СН2 = С = О (Г) —» Зсн2 + со
При определении доли синглетного и триплетного метиле-
нов по продуктам реакций с различными акцепторами на при-
веденную и без того непростую схему накладывается возмож-
ность, безызлучательной релаксации синглетного метилена в
122
триплетное основное состояние при простых столкновениях с
другими частицами. Неудивительно поэтому, что точной оценки
соотношения синглетного и триплетного метиленов при фотоли-
зе кетена нет до сих пор. Полагают, что доля триплетного ме-
тилена возрастает от 25—30 [826] до 60—75% [822, 827, 828]
при увеличении длины волны облучения от 213,9 до 366 нм.
В то же время при сенсибилизированном ртутью фотолизе ке-
тена преимущественно образуется триплетный метилен [829—
832].
По данным спектроскопических исследований [466], им-
пульсный фотолиз кетена в газовой фазе приводит в основном
к синглетному метилену, причем с увеличением парциального
давления инертного газа в газовой смеси доля триплетного ме-
тилена, как и следовало ожидать, возрастает; те же закономер-
ности отмечены и при фотолизе диазометана в газовой фазе.
Термическое разложение кетена и его производных также
приводит к соответствующим карбенам (см., например, [4]);
однако эти реакции детально не исследованы, за исключением
реакции термолиза дихлоркетена [833], получаемого дехлори-
рованием трихлорацетилхлорида цинковой стружкой. В этом
случае была осуществлена низкотемпературная матричная ста-
билизация (Аг, 12 К) и ИК-спектроскопическая идентификация
образующегося дихлоркарбена:
Zn, 420 °C, 10 Па ~ п 420 °C
CCLCOCI -------—“—С С Ip — С — О .СС12
—ZnC^ ~СО
При фотолизе алкилкетенов в газовой фазе образуются ал-
килкарбены, из которых наряду с олефинами «димерного со-
става» получаются и «мономерные» олефины — продукты хо-
рошо известной для алкилкарбенов карбен-олефиновой изоме-
ризации, например [834, 835]:
hv
СН3СН = С = О—*• :СНСН3 —* [СН2 = СН2]‘ —* СН2 = СН2
-СО
сн3сн = с = о -со -н2
СН3СН=СНСН3 НС=СН
Аналогично, из диметилкетена образуются 2,3-диметилбу-
тен-2 и пропилен [836]. При этом при фотолизе в жидкой фазе
наряду с этими олефинами удается обнаружить тетраметилцик-
лопропанон, распадающийся далее с образованием 2,3-диме-
тилбутена-2 [837]:
123
Ме2С = С = 0-^- СМе2 —;С ? С =>°
МеСН = СН2
hv
Ме2С - СМе9 —► Ме9С = СМе?
\ / г -со г
СО
Необычно проходит фотолиз бицикло [5.2.1 ] деканона-10.
Первоначально образуется карбонилциклононан, охарактеризо-
ванный спектроскопически и химическим перехватом; выделен-
ные углеводороды являются, вероятно, продуктами внутримо-
лекулярных реакций циклононанилидена, поскольку те же со-
единения образуются и при генерировании этого карбена тер-
молизом диазоцикло'нонана [838J:
Н2С - сн2 - сн - сн2
н2с со
н2с - сн2 - сн - сн2
Фотолиз дифенилкетена протекает неоднозначно, хотя уча-
стие дифенилкарбена при этом несомненно [839].
Фотолиз трикарбодиоксида. Фотохимическая реакция С3О2
с олефинами формально сводится к внедрению углеродного ато-
<ма в олефин с образованием соответствующего аллена [840,
841]. По данным экспериментов с мечеными атомами, централь-
ный атом в молекуле С3О2 преимущественно оказывается цент-
ральным и в образующемся аллене [842]:
124
л л * hv \ * у
+ о = с = с=с=о—=► ;c = c = cf +2 со
Механизм этой реакции, однако, существенно сложнее [226.
841, 843—845]:
с = с = о
hv
о=с=с=с=о —с = с = о
-со .
с = с,
-со
-со
с = с = с
С:
Промежуточная карбеновая частица :С = С = О идентифици-
рована в продуктах импульсного фотолиза С3О2 в газовой фазе
по электронному спектру поглощения [843, 844] и по ИК-спект-
ру в низкотемпературной матрице [842]. Не до конца пока ясен
лишь механизм превращения карбонилциклопропана в аллен —
однозначные доказательства существования кинетически неза-
висимого циклопропилидена отсутствуют.
Распад других карбонильных и тиокарбонильных соедине-
ний. Образование карбенов в приведенных ниже реакциях до*
настоящего времени строго не доказано и является лишь удоб-
ной рабочей гипотезой. Так, на основе реакции обессеривания
1,3-диоксолантионов-2, протекающей с промежуточным образо-
ванием и распадом гипотетических циклических карбенов, был
предложен удобный метод стереоселективного синтеза олефи-
нов [846]:
ф ’ %
о( >0
(RO)3P
с - с
-(RO)3PS
С = s
X °
С:
с = с
Однако в этих и родственных реакциях с участием карбо-
нильных и тиокарбонильных соединений [846—848] более ве-
роятно, по-видимому, промежуточное образование не карбенов,
а соответствующих фосфониевых илидов.
125
Участие алкоксикарбенов предполагается и при фотолизе
эфиров тиокарбоновых кислот [849]:
hv
СН3 - CS - OR----сн3 - С - OR + S
Генерирование карбенов из илидов и бетаинов. Предполага-
ют, что интермедиаты илидного типа образуются во многих жид-
кофазных реакциях карбенов при наличии у растворителя или
субстрата соответствующего гетероатбма (см., например, [176,
177, 474—477, 850, 851]). Вместе с тем многие интермедиаты та-
кого типа мало отличаются от слабых лабильных молекулярных
комплексов карбенов (см. разд. 2.4.1), они не могут быть выде-
лены в индивидуальном виде, и их наличие лишь несколько мо-
дифицирует реакционную способность карбенов (см. ниже).
В данном разделе в качестве источников карбенов рассмотре-
ны лишь стабильные илиды и бетаины.
Для реакций этого типа проблема доказательства участия
кинетически независимых карбенов (путь а) особенно актуаль-
на, поскольку в данном случае в конденсированной фазе наблю-
дается взаимодействие заряженных частиц. Строго говоря,
с уверенностью предполагать образование карбенов можно
только в случае фотодиссоциации илидов, а в термических про-
цессах перенос «карбенного» фрагмента на его акцептор, оче-
видно, происходит при непосредственном участии положитель-
но заряженного центра (путь б), хотя, возможно, и без обра-
зования промежуточного аддукта с илидом (путь в):
в -С-С-
Однако эта проблема носит значительно более общий ха-
рактер и относится, пожалуй, ко всем жидкофазным, особенно
гионным, методам генерирования карбенов.
Илиды азота. При взаимодействии /i-бутоксиметил- или
феноксиметилтриметиламмонийбромида с литийорганическими
реагентами в присутствии циклогексена получены соответству-
126
ющие 7-бутокси- или 7-феноксиноркараны с выходами до
48% [852J:
4. — PhLi, PhNa
Me3N - CH2OR Br —— - » + PhCH20R
R = Ph, Bu
Еще в 1929 г. при термолизе илида (16) при 170 °C был по-
лучен формальный димер флуоренилидена — бис(флуоренили-
ден) [853]. Более вероятно образование карбена при фотохими-
ческом распаде илида (17) [854]:
(16)
C5H5N-C(CN)2
hv
-----* :C(CN)2
-c5h6n
(17)
Илиды серы. Образование формальных димеров замещен-
ных флуоренилиденов известно и для илидов серы [855, 856]:
R, R1 = Н, NO2
Изомерные стильбены образуются из илида (18) уже при
—40 °C [857]; из илида (19) при «200 °C наряду с другими
127
продуктами образуется тетрацианоэтилен — формальный димер
дицианокарбена [858].
Ph2S - CHPh Me2S-C(CN)2
(18) (19)
При разложении илида (20) выделены изобутен («27%) и
метилциклопропан («9%), рассматриваемые как продукты
внутримолекулярных реакций изопропилкарбена [859]. Анало-
гично, при фотолизе илида (21) выделен циклопропен — про-
дукт внутримолекулярной реакции винилкарбена [860].
СНМе
+ _ / \
Ph2S - СНСНМе2 :СНСНМе2—* Ме2С = СН2 + Н2С - СН2
(20)
PhzS - СНСН = СН2 (21)
Продукты циклопропанирования получены в реакции аце-
нафтилена с алкил- и арилсульфониевыми илидами [861], од-
нако этот результат, особенно в случае илида (22; R = Pr), ско-
рее свидетельствует против участия свободных карбенов, по-
скольку продукт карбен-олефиновой изомеризации пропилкар-
бена — бутен — не обнаружен.
(22)
-Ph2S
R = Ph, Рг
Илид (23) в присутствии медного катализатора является
циклогаропанирующим агентом [862]. Реакция с олефинами
проходит стереоспецифично, давая соответствующие циклопро-
паны с выходами от 35% (циклогексен) до 48% (транс-окт-
ена-2) [862]:
+ - \ , СЩасас)?
Ph2S - СН2 + )С = С( ------------*
' ' ТГФ, 25 °C
(23)
сн2
+ Ph2 S
Участие карбенов вполне вероятно при фотолизе илида (24);
в этом случае об участии в реакции бензоилкарбена свидетель-
128
ствует образование дибензоилэтилена и транс-трибензоилцик-
лопропана, а также его перехват циклогексеном [863]:
I _ /tv
Me,S-CHCOPh -----► :CHCOPh —=»- PhCOCH = CHCOPh
(24) -Me2S :CHCOPh PhCO4z~<| J PhCOHC - CHCOPh \ / CHCOPh
транс
Стабильные илиды (26) выделены при каталитическом раз-
ложении диметилдиазомалоната [Rh(OAc)2, 20 °C] в присутст-
вии 2-пропенил- или 2-изобутенилтиофена [864,865]. Эти илиды
способны к внутри- и межмолекулярному переносу бис (меток-
сикарбонил) карбенового фрагмента с образованием соответст-
вующих продуктов циклопропанирования двойной связи или
продуктов внедрения в связь С—Н, что полностью оправдывает
неоднократно высказывавшиеся предположения о промежуточ-
ном участии илидов в реакциях замещенных фуранов и тиофе-
нов с карбенами [864, 866J.
СН = СНМе
S
N2C(COOMe)2,
Rh(OAc)2,
20 °C
S - С(СООМе)2
(25) (26)
СН = СНМе
S
(27) (28)
(25) ^С(С?°^ЬСиС1-80;с (27) + (28) ]°П(26)
Илиды фосфора. Хотя при разложении илида (29) и выде-
лен 1,2-ди-н-бутоксиэтилен — формальный димер бутоксикарбе-
на, попытки перехватить этот карбен олефинами к успеху не
привели [867]. Напротив, при фотолизе илида (30) в присутст-
вию? 129
вии 1,1-дифенилэтилена с выходом 73% получен 1,1,2,2-тетра-
фенилциклопропан [868J:
Ph3P - CHOBu (29)
Ph3P - CPh2 :CPb2 Ph2C-CPh2
-rn3r \
(30) CH2
Бетаины. Промежуточное участие алкилкарбенов предпола-
гается и при разложении биполярных диазонийкарбоксилатов
[869], однако выход продуктов перехвата этих карбенов невы-
сок:
RCHCOCT
+ NH3
изопентил-
нитрит
---------
63 °C
R — Me, Et, Pt-изо
-n2,-co2 I
RCHCOCT—RCHCOCT —*:CHR
I -n2 -co2
+n2 h
~co2
rCHN2 —-------------------
Полагают, что 2,3-дифенилциклопропенилиден является про-
межуточной частицей в реакциях дифенилциклопропенилий-
карбоксилата [870J:
Ph3C С1О7
PhC=CPh —----------у PhC — CPh ----->
\ / Li2CO3 \+/ -СОо
СНСООН с—СОО“
PhoC С1О4
---> PhC=CPh --------> PhC—CPh
\ / ‘ W
С: С—CPh3 СЮ;
От гетероциклических бетаинов пиридинового, 2-хинолиново-
го и 1-изохинолинового рядов при 60—80 °C отщепляется СО2;
предполагают, что при этом в качестве промежуточных частиц
образуются нуклеофильные гетероциклические карбены, пере-
хваченные разнообразными электрофилами, например [871]:
MeN+
-go2
60-80 °C
MeN+ MeN
130
Характерно, что в этих реакциях скорость декарбоксилиро-
вания не зависит от природы электрофила, что согласуется с
предположением об участии кинетически независимых карбен-
ных частиц.
3.1.3. Внутримолекулярные перегруппировки
Общим для реакций этого типа является возможность образо-
вания карбена в результате формально внутримолекулярной пе-
регруппировки энергетически активированного предшественни-
ка. Среди реакций такого рода наиболее интересной и изучен-
ной является циклопропен-винилкарбеновая изомеризация:
С = С-
\
С
hv, А
или катализатор
:С-С =
Согласно данным высококачественных НЭР [389], на ППЭ
системы С3Н4 имеются локальные минимумы, соответствующие
синглетным винилкарбену и пропенилидену (рис. 3.1). Вместе
с тем невысокий энергетический барьер на пути перехода про-
пенилидена в метилацетилен (^3 кДж/моль [389]) не позво-
ляет ожидать выделения продуктов перехвата этого карбена
даже в низкотемпературных условиях. В то же время винилкар-
бен, для которого барьеры значительно выше (^45 кДж/моль;
см. рис. 3.1) может быть идентифицирован или перехвачен при
проведении реакции при температурах <700—750 °C. Действи-
тельно, при изомеризации 3,3-диметилциклопропена в 3-метил-
бутин-1 и изопрен при ^900 °C в условиях адиабатического
сжатия промежуточные карбены не удалось перехватить таки-
ми акцепторами, как этилен, бутадиен и циклопентадиен [872]:
г*:С = СНСНМе2—^СН = ССНМе2
873 °C
нс ~ сн-------
\ /
СМе2
L*:CHCH = СМе2—*СН2 = СМеСН = CHg
В то же время возможность изомеризации циклопропенов,
содержащих углеводородные заместители, в соответствующие
винилкарбены в условиях фотолиза, термолиза или в присутст-
вии катализаторов показана на большом числе примеров
[873—882]. Так, например, термолиз З-метил-З-фенилцикло-
Рис. 3.1. Сечения ППЭ системы С3Н4
по данным НЭР [389]:
1 — СН2=С=СН2, £=3 кДж/моль; 2, 4»
6, « — переходные состояния, £=290, 263,
269 и 186 кДж/моль соответственно; <3 ~
:СНСН = СН2, £==244—253 кДж/моль; 5 —
НС=СН, £=95 кДж/моль; 7 — :С=СНМе,
\ /
сн2
£=183 кДж/моль; 9 — СН—СМе; £=0
Координата реакции
пропена в среде 2,3-диметилбутена-2 приводит к винилцикло-
пропановому аддукту и небольшому количеству индена:
MeCPh
/ \ 1яп°с —.. _, Мере = СМе2
нс - сн 1?9-£-;СНСН = CMePh —-------i-
—» PhMeC = СН - НС - СМе2
\ f
СМе2
Me
Для проведения циклопропен-винилкарбеновой изомериза-
ции в присутствии катализаторов до недавнего времени обычно
использовали комплексы Ni° с циклооктадиеном-1,5 (cod). С их
помощью удалось осуществить винилциклопропанирование
3,3-диалкил(арил)замещенными циклопропенами ряда эфиров
непредельных карбоновых кислот, обладающих пониженной
нуклеофильностью двойной связи (акрилаты, фумараты и т.п.)
с выходами аддуктов до 85% [874, 883J:
нс =сн \ / CRR1 Ni(cod)2 + R2CH = CHCOOR3 - U 40-45 °C транс
* R2HC—CHCOOR3
\/
СНСН = CRR1
R, R1 = Me, Ph; R2 = Н, Me, COOR3; R3 = Aik
Применение катализаторов на основе комплексов меди поз-
волило осуществить винилциклопропанирование и других оле-
финов; выходы циклоаддуктов составляют от 5—10% для мо-
нозамещенных этиленов (гексен-1, гептен-1) до 55—70% в слу-
132
чае других непредельных углеводородов [880, 882]:
НС = СН 9“c|P<OPh)3>CHzOg rRR1C-CH_ HC:CuC|p(OPh).
у / — oO т —20 G I
CRR1
RR’C = CHCH = CHCH = CRR1
CHCH = CRR1
R = Me, цикло-С3Н5; R1 = цикло-С3Н5
В ряде случаев наблюдали внутримолекулярные реакции
генерируемых таким путем винилкарбенов. В частности, при
термолизе полиарилциклопропенов получены следующие поли-
циклические продукты [881]:
PhC=CPh Д
\ /
МеС(СН2)2СН = СН2
сн2сн = сн2 д
с - СМе
Phx ' ,f
rn CPh
Термическая изомеризация в соответствующий винилкарбен
хорошо известна для перхлорциклопропена (31) [878, 884,
885]:
С1С = СС1 ^=^:СС1СС1 = СС12 )с-С(
\ / 2 z \/ 4
СС12 С1ССС1 = СС12
(31) (32)
Таким путем осуществляют также винилциклопропанирова-
ние непредельных карбоновых кислот и олефинов; выходы цик-
лоаддуктов достигают 80% [884, 885]. Помимо выделения ви-
нилциклопропанов на промежуточное участие перхлорвинил-
133
карбена в этой реакции указывает и образование продуктов его
внедрения в связи С—Н углеводородов. Так, в присутствии
н-гексана образуются три изомерных продукта внедрения
[(33) : (34) : (35) = 14: 43 : 43; общий выход 23% ] [884]:
Cl CI
180 °C н-С6Н14 ' ।
(31) < (32) -----» н-С6Н13 - СН - С - СС12+
(33)
Me Cl Cl Cl Cl
ill ill
+ Bu - CH - CH - C = CCI2 + Pr - CH - CH - C = CCI2
(34) (35)
При термических, фотолитических и термокаталитических
превращениях эфиров циклопропенкарбоновых кислот также
постулируется протекание циклопропен-винилкарбеновой изо-
меризации [886—888]. Так, например, промежуточным образо-
ванием карбена объясняли быструю рацемизацию оптически
активного эфира циклопропенкарбоновой кислоты в ходе его
фотоизомеризации в алкоксифуран [887]:
СНСООМе .. СНСООМе
/ \ hv МеСч .СООМе hv / \
PhC = CPh ;С = С( PhC = CPh
Phz H
L О
V
Ph x__
Me-( Д-ОМе
О
Каталитическое В1инилциклопропанирование эфирами 1-ал-
килциклопропенкарбоновой-3 кислоты наблюдается лишь в
случае норборненовой кратной связи [889J:
RC=/CH + /Г> ^Me-СНСООМе
СНСООМе I
R
L= Р(ОРЬ)з, 2MeC=CSiMe3
В этом случае регио- и стереоселективность реакции скорее
соответствуют протеканию ее в координационной сфере метал-
ла по согласованному пути без образования кинетически неза-
висимого карбена [889]. Этот вывод подтверждает и тот факт,
134
что при карбенном разложении соответствующих винилдиазо-
метанов регио- и стереоселективность винилциклопропанирова-
ния олефинов не соответствуют селективности, наблюдаемой при
их каталитическом взаимодействии с циклопропенами [890].
Более вероятно образование винилкарбенов при фотохими-
ческих реакциях. Так, при фотолитическом 7-элиминировании
N2 от замещенных ЗЯ-пиразолов наряду с генерированиехМ ви-
нилкарбенов и их перехватом наблюдается перегруппировка
винилкарбенов в соответствующие циклопропены. Так, фотолиз
5-циано-ЗЯ-пиразола в среде фурана приводит к следующим
аддуктам [891]:
, А сн,
/ \\ AV .. /\“
CN >СН2=СН-С—CN У НС=С~CN'
N ___
CN
2,5 : 1
Аналогично был генерирован ряд серосодержащих винил-
карбенов [892J; с помощью дициановинилкарбена удалось осу-
ществить даже цикло1пропанирование бензола [891].
Изомеризация циклопропена в метилацетилен через пропе-
нилиден формально аналогична перегруппировке 2Я-азирина в
метилцианид с промежуточным участием карбеноподобной час-
тицы— метилизоцианида [893]:
НС = N —э- :С = NMe —* MeCN
\ /
СН2
По данным НЭР, синглетный метилизоцианид отделен от
более стабильного метилцианида значительным барьером
(^98 кДж/моль [893]) и может быть идентифицирован. Для
оксиренов с помощью НЭР выявлена легкость взаимных пере-
ходов кетокарбен^оксирен (Яакт = 21—32 кДж/моль [894]),
что соответствует экспериментальным данным о возможности
взаимопревращений изомерных кетокарбенов и оксиренов [246,
894, 895]:
R - С - С - R1^*-rc = CR1
И \ /
^s=*R-C-C-R1
II
О
К числу внутримолекулярных перегруппировок, ведущих к
образованию карбенов и их аналогов, относятся реакции и дру-
гих нестабильных промежуточных частиц, например силаолефи-
135
нов (см. [260, 526, 527], а также гл. 2, 5):
:CHSiH3 Н2С = SiН2 :SiHCH3
Предполагают, что в случае фторолефинов также возможна
их изомеризация в соответствующие фторалкилкарбены [533,
896] (см., однако, [275]), например:
(СЕз)2С:^0-500°с CF3CF = CF2 4^00^ CF3CF2CF
-CHF3
Возможно, что подобные процессы олефин-карбеновой изо-
меризации ответственны и за образование активных карбенных
центров в реакциях каталитического диспропорционирования
(метатезис) олефинов и полимеризации циклоолефинов [897]
(см. также гл. 5).
Для карбонильных соединений в ряде фотохимических реак-
ций наблюдаются перегруппировки с карбеновым расширением
цикла. В замещенных циклобутанонах, в частности, это на-
правление (л) эффективно конкурирует с фотодеструкционными
процессами [(б) и (в)J; при R = R1 = Me выход тетрагидрофура-
новых производных достигает 68% [898]:
СН2
/ \
R2C = С = О + R2C = СН2 R2c - CR2 + СО
I_______ ! e I
RO R
hv
---R--------
R1
R1
R
R, R1 = H, Me
136
При облучении 7,7-диметилбицикло[3.2.0]гептен-2-она-Ь
вклад подобной перегруппировки с участием циклического кар-
бена достигает 50% [899]. Аналогичная реакция описана и для
циклопроизводного камфанона, где карбен удалось перехватить
не только спиртом, но и циклогексеном [900].
СН2СН = С = О СН2СН2СООМе
I—CH = СНСН = СМе2^^СН = СНСН = СМе2
Промежуточное участие бициклического алкоксикарбена при
облучении камфоры в этаноле или гексане подтверждено при
исследовании распределения D-метки в образовавшемся 3-эток-
си-2-оксабицикло [3.2.1J октане [901J:
h\) EtOH
137
При фотолизе бензоциклобутендиона первоначально образу-
ется соответствующий дикетен, идентифицированный в виде
аддукта с малеиновым ангидридом; однако наряду с этим на-
блюдается и перегруппировка дикетена в ацилоксикарбен,
идентифицированный в виде циклоаддуктов с непредельными
акцепторами (выходы до 65%) [902]:
В ряду спиротиагексанонов также известен пример конку-
ренции фотохимической десульфуризации и перегруппировки с
образованием циклического карбена, причем последнее направ-
ление становится основным при увеличении длины волны света,
используемого для фотолиза [903]:
МеОН
Генерирование замещенных винилиденов в результате внут-
римолекулярных перегруппировок замещенных ацетиленов
[547] и а-силилированных эфиров енолов [548] в условиях ва-
куумного флеш-пиролиза описано в разд. 2.4.2.
3.2. СОЛЬВОЛИТИЧЕСКИЕ МЕТОДЫ ГЕНЕРИРОВАНИЯ КАРБЕНОВ
Основу сольволитических методов генерирования карбенов со-
ставляют реакции а-элиминирования, отличающиеся от рас-
смотренных в разд. 3.1 термических и фотохимических процес-
сов в первую очередь ионным характером промежуточных час-
тиц. Как следствие, реакции сольволмтического а-элиминиро-
138
вания проводят обычно в хорошо сольватирующих растворите-
лях и под действием органических или неорганических осно-
ваний.
3.2.1. Генерирование дигалогенкарбенов
Щелочной гидролиз галоформов. Эта реакция (на примере
хлороформа) была первой, где участие карбена доказано кине-
тическими методами [904], и сохранила препаративную цен-
ность до сих пор.
Многочисленными исследованиями кинетики сольволиза
(гидролиза) галоформов и дейтеро-водородного обмена в них
было надежно установлено [904, 905], что щелочной гидролиз
галоформов, не содержащих атома фтора, в водной среде опи-
сывается схемой:
СНХ3 + ~СХ3 + Н2О (3.1)
X = Cl, Br, I
Участие трихлорметильного аниона в качестве первичного
продукта гидролиза хлороформа подтверждается возможностью
его химического перехвата в этих условиях. Наилучшими «ло-
вушками» являются карбонильные соединения, превращаю-
щиеся при этом в соответствующие трихлорметилметанолы (см.,
например, [4, 906], а также гл. 4):
СС13 ОН
С13С" + RR1C = О —Ч>. RR1C - O’-^RRlQ - СС13
С последовательностью реакций (3.1) и (3.2), в частности с
быстрым и обратимым характером стадии (3.1), согласуются
значения скоростей гидролиза и дейтеро-водородного обмена
различных галоформов: для галоформов, не содержащих атома
фтора, этот обмен проходит на один —три порядка быстрее,
чем гидролиз, а для монофторгалоформов скорости обмена и
гидролиза сопоставимы; ряд активности галоформов при дей-
терообмене приведен ниже [907]:
CDI3 «= CDBr3 > CDBr2CI > CDBrCl2 =» CDCI2I > CDFI2 > CDBr2F >
> CDCI3 > CDBrCIF > CDCI2F
Стадия (3.2) диссоциации тригалогенметильных анионов с
образованием дигалогенкарбенов также была исследована ме-
139
тодами формальной кинетики [905, 907J. Образование «свобод-
ного» дихлоркарбена согласуется и с положительным значени-
ем свободного объема активации реакции (3.2): ДУ* =
= 16 см3/моль [908]. Склонность к отщеплению различных га-
логенид-анионов от смешанных тригалогенметильных анионов
уменьшается в ряду [909]: Вг>1>С1.
В то же время для дифторгалогенметанов на основании ре-
зультатов кинетических исследований был предложен одно-
стадийный механизм генерирования дифторкарбена в условиях
щелочного гидролиза [907, 908]:
6- б-
HCF2X + НО” —♦НО - - - Н - - - CF2 - - - Х~ —♦HgO + :CF2 + X”
Реакции гидролиза галоформов подробно исследованы в
протонных средах, где препаративный перехват дигалогенкар-
бенов, например, олефинами конкурирует с их реакцией с во-
дой, приводящей к монооксиду углерода и (после гидролиза) к
формиат-аниону:
но-
:CCt2 + Н2О ---СО + НСОО~
н2о
В препаративных целях сольволиз галоформов проводят в
апротонных средах, где можно рассчитывать на более селектив-
ный перехват дигалогенкарбенов целевыми субстратами. Имен-
но по этой схеме Е. Дёринг и А. К. Хоффман впервые в 1954 г.
осуществили ге-и-дигалогенциклопропанирование олефинов, ге-
нерировав дихлор- и дибромкарбены из соответствующих гало-
формов и трет-бутоксила калия в углеводородной среде [910].
Позднее в этой реакции использовали также метоксид натрия,
изопропокоид калия и даже безводный NaOH [4], а также наф-
талид натрия [911] и анионы типа О2“ [912].
Таким путем были генерированы СС12, CBr2, CBrCl и другие
дигалогенкарбены, в том числе дииодкарбен С12 (из йодофор-
ма и трет-бутоксида калия [4]). Полезной модификацией этого
метода является использование в качестве основания оксирана,
что позволяет генерировать дигалогенкарбены (в том числе
CF2 из Тазообразного CHF2C1) в нейтральной среде в присутст-
вии катализатора типа галогенидов тетраэтиламмония при по-
вышенных температурах и давлении [913J:
нсх3 +
Et4NBr
Примененные в этой
соли аммония—позднее
сх2
-сх3 -----:СХ2 )с - С<
-х- /
реакции катализаторы — четвертичные
совершили подлинную революцию в
140
препаративной химии карбенов, в первую очередь дигалоген-
каро^нов. Как было обнаружено [914—916], в двухфазной си-
стеме, состоящей из 50 %-й водной щелочи, олефина, хлорофор-
ма и каталитических количеств хлорида триэтилбензиламмо-
ния, удается осуществить ге-и-дихлорциклопропанирование оле-
финов с выходами до 70%. По современным представлениям,
в этих условиях катализатор обеспечивает транспорт трихлор-
метильного аниона с поверхности раздела фаз (ПРФ) в орга-
ническую фазу (ОФ), где и протекает циклопропанирование
олефина [917] (ВФ — водная фаза):
СНС13(ОФ) + МаОН(ВФ) С13С~ Ыа+(ПРФ)
С13С~ Ма+(ПРФ) + ^N+X_(00)=j2±rCI3C_+N<-(00) + ЫаХ(ВФ)
CI3G-+fjJ< (ОФ)^±- :СС12(ОФ) + ->Ы+СГ(ОФ)
:СС12 (ОФ) + )С = С( (ОФ)— )С (ОФ)
СС12
Естественно, возможность проведения реакций дигалоген-
карбенов без применения абсолютированных растворителей,
алкоголятов щелочных металлов и автоклавного оборудования
вызвала большой интерес. К настоящему времени этот метод,
включающий разнообразные катализаторы и каталитические
системы межфазного переноса, распространен на большое чис-
ло галоформов и их аналогов типа RR^HX с «кислым» водо-
родом и в значительной мере вытеснил описанные выше методы
генерирования дигалогенкарбенов и ряда других карбенов. Для
циклопропанирования чувствительных к щелочам и протонсо-
держащим растворителям субстратов применяют, как правило,
ртутьорганические источники карбенов (см. разд. 3.1.1). По-
дробнее механизм генерирования карбенов с использованием
межфазного катализа и возможности использования этой мето-
дики в синтезе рассмотрены в разд. 4.3.
Альтернативные источники тригалометильных анионов и ди-
галогенкарбенов. Возможность генерирования тригалогенме-
тильных анионов и далее соответствующих дигалогенкарбенов
из других, помимо галоформов, источников хорошо известна
(см., например, [4]). Однако и эти методы практически вытес-
нены методом генерирования дигалогенкарбенов в условиях
141
межфазного катализа, что позволяет нам ограничиться лишь
кратким их перечислением.
Декарбоксилирование солей тригалогенуксусных кислот
представляет прежде всего интерес как метод генерирования
тригалогенметильных анионов и далее дигалогенкарбенов в ап-
ротонной среде, т. е. в нейтральных условиях [918, 919]. Опти-
мальным растворителем является 1,2-диметоксиэтан (80—
90 °C), причем скорости распада солей уменьшаются в ряду:
СС13СООК > CCI3COONa > CCi3COOLi
Обычно используют натриевую соль как наиболее дешевую
[919]:
ХпССООМ 55==-Х3СС00" —*- Х3С"—<-:СХ?
° 6 -со? 6 -х~ z
X = Ci, Br; М = К, Na, LJ
Следует отметить, что эту реакцию можно осуществлять
в условиях межфазного катализа в среде CHCI3, что позволяет
несколько снизить температуру распада (до ^70 °C) [920,
921J.
Определенный препаративный интерес представляет и воз-
можность генерирования дифторкарбена из дифторхлорацетата
натрия [922, 923], поскольку получить этот карбен из соответ-
ствующих галоформов в условиях межфазного катализа не уда-
ется [917].
Щелочное расщепление тригалогенкарбонильных соедине-
ний также позволяет генерировать дигалогенкарбены и являет-
ся достаточно общей реакцией. Так, алкилтригалогенацетаты
под действием трет-бутоксида калия, этоксида или метоксида
натрия успешно геж-дигалоциклопропанируют олефины с выхо-
дами аддуктов до 80% [924—926]:
CIXYCCOOR —:CXY У - с(
-ROCOOR1, -СГ 7 \ / 4
XCY
X = Y = Cl; X = Ci, Y = F
Однако получить таким путем дифторкарбен из метилхлор-
дифторацетата не удалось [925]. Аналогично протекает щелоч-
ное расщепление тригалогенкетонов и тригалогенальдегидов
[925, 927]. В случае симметричных бис (тригалогенметил) кето-
нов образующийся эфир тригалогенуксусной кислоты может
142
служить источником и второй молекулы дигалогенкарбена
[925X928-931]:
RO-
| -ROCOORI
(XCl2C)2CO-^£ xci2ccoor + ХС12С-
1 -сг
X = Cl, F :СХС|
И в этом случае генерировать дифторкарбен из (C1F2C)2CO
не удается [925].
С промежуточным участием трихлорметильного аниона и
далее дихлоркарбена происходит также щелочное разложение
трихлорметилсульфината [932], трихлорметилсульфонилхлори-
да [932] и диэтилтрихлорметилфосфоната [933]:
Cl3CSO2Me
Cl3CSO2CI
Cl3CPO(OEt)2
RO”
-ROSO2Me
RO”
-(RO)2SO2
RO”
-ROPO(OEt)2
cci3 TBT:CCl2
Описанные в настоящем разделе сольволитические, а в
разд. 3.1.1 термические и фотохимические методы генерирова-
ния дигалогенкарбенов позволяют получать эти частицы в ши-
роком диапазоне температур и условий. При этом, как было по-
казано на примере реакции циклоприсоединения к различным
олефинам, дихлоркарбен, полученный разнообразными метода-
ми при температурах от —260 до 1500 °C, обладает практиче-
ски идентичной активностью [724, 725]. Более того, даже не-
которое изменение относительной скорости присоединения того
или иного карбена при генерировании его разными методами и
в различных условиях, в частности в результате образования
солевых комплексов, практически не сказывается на препара-
тивных результатах. Таким образом, можно уверенно считать,
что проблемы простого и эффективного генерирования дигало-
генкарбенов для синтетических и даже промышленных целей
успешно разрешены: для жидкофазных условий это метод меж-
фазного катализа или (для лабильных субстратов) применение
тригалогенметильных соединений ртути и некоторых других
металлорганических предшественников или тригалогенацетатов
Щелочных металлов, а для реакций в газовой фазе — преимуще-
143
ственно пиролиз соответствующих галоформов или перфтороле-
финов [468]. Пожалуй, единственным «белым пятном» остается
отсутствие универсального, простого и эффективного метода
генерирования дифторкарбена в жидкой фазе в интервале тем-
ператур 0—100 °C.
3.2.2. Генерирование моногалогенкарбенов
из дигалогенметанов и их производных
Кинетические исследования механизма щелочного гидролиза ди-
галогенметанов привели к выводу о невозможности или малой
вероятности протекания реакции а-элиминирования с образова-
нием соответствующих галогенкарбенов [934]. Так, при взаи-
модействии СН2Х2 с трет-бутоксидом калия в присутствии бен-
зола выход продукта расширения цикла, обусловленного уча-
стием галогенкарбенов, составляет не более 1,4% [935]:
RO _ rGHG
СН2Х2 СНХ2 :СНХ ----------->-
Однако при наличии заместителей, способных стабилизиро-
вать промежуточный дигалогенметильный анион, образование
замещенных галогенкарбенов происходит достаточно эффек-
тивно. Так, в реакциях бензилидендигалогенидов с трет-буток-
сидом калия в присутствии олефинов выходы соответствующих
геж-галогенфенилциклопропановых аддуктов достигают 90%
[708, 936—939]:
XCPh
PhCHXY~^"CXYPh-^ :CXPh = & )с - с(
X = F, Cl, Br; Y = С I, Br; R = трет-С4Н9
Аналогично, из PhSCHCl2 и трет-бутоксида калия генериро-
ван (фенилтио)хлоркарбен [940], а из 2- и 3-дихлорметилфура-
нов и 2-дихлорметилтиофена и трет-бутоксида калия при —20 °C
образуются соответствующие гетерилхлоркарбены [941, 942]:
CICAr
ArCHCl2 ^g^-CC^Ar -^:СС1Аг
Аг = фурил-2, фурил-3, тиенил-2
Описано генерирование монохлоркарбена при щелочном рас-
щеплении а,а-дихлорацетофенона [943].
144
X) помощью полуэмпирического метода МЧПДП/3 была да-
на кдантовохимическая оценка вероятности протекания реакций
• R1CHXY —*“CXYR1—*:CYR1
“ROH “X
X, Y = F, Cl
в зависимости от природы заместителей [944]. Согласно этим
расчетам, карбанионный центр достаточно эффективно стаби-
лизируется, например, при Р? = СЫ, НС = С и X, Y = F, Cl, при-
чем связи С—X в соответствующих анионах в этих случаях
чрезвычайно ослаблены. Это дает основания ожидать возмож-
ности генерирования циано- и этинилзамещенных галогенкар-
бенов из соответствующих дигалогенидов. Описано, например,
генерирование и перехват (трет-бутилэтинил^алогенкарбенов
[945]; известно также генерирование замещенных винилгало-
генкарбенов из соответствующих винилдигалогенметанов под
действием трет-бутоксила калия в пентане [946]:
RO-, МеСН = СНМе
“20 °C — Uuc
RCBCCHXY-----5- RC= CCXY—:CXC=CR -----------*•
-ROH
XC-CBCR
/ \
--MeHC-CHMe
(цис; 70%)
R = трет-Ви; X,Y = Hal
RCCl = CHCHCl2 —; “ HOl — :CCICH = CCiR - R = трет-Ви ^yCHCI2 RQ; \-ACi -roh *RCCl = GHCCl2--*- n “Cl 0 KXV<CH = CCIR (60%) /--/CC(2 \-XC| -Cl-
10—Ю7
145
Ме2С = СН2
Cl
С/ Cl
Vc-CMe2
сн2
R~rper-Bu (67%)
Судя по расчетным данным [944], возможно, по-видимому, и
сольволитическое генерирование хлор (циклопропил) карбенов
из дихлорметилциклопропана. Однако при включении карбани-
онного центра непосредственно в состав напряженного фраг-
мента циклопропана, бицикло[1.1.0] бутана, спиропентана либо
[1.1.1]прО1пеллана стабилизация недостаточно эффективна, так
что образование соответствующих карбенов в этом случае ма-
ловероятно [944].
Приведенные данные показывают, что генерирование орга-
ны лзамещенных моногалогенкарбенов сольволитическим сту-
пенчатым элиминированием галогеноводородов является вполне
реальной задачей и может позволить синтезировать разнообраз-
ные моногалогенциклопропаны, в первую очередь геж-арилга-
логен- и гем-(арилтио)галогенциклопропаны. Вместе с тем в
связи с неудачами при генерировании незамещенных моногало-
генкарбенов из дигалогенметанов в щелочной среде наиболее
общими методами генерирования этих частиц остаются разло-
жение дигалогенметиллития (см. разд. 3.1.1), а также реакции
дигалогенметанов с сильными основаниями типа бис(триметил-
силил) амида натрия (см., например, [947—949]). Для получе-
ния моногалогенпроизводных циклопропанового ряда целесо-
образнее двухстадийный путь: дигалогенметиленирование не-
предельных соединений и последующее моновосстановление ди-
галогенциклопропанов. Обе стадии этого превращения могут
быть проведены достаточно селективно и с высокой конверсией.
3.2.3. Дегидрогалогенирование органилгалогенидов
В ряду бензилгалогенидов типа дифенилметилхлорида и 9-га-
логенфлуоренов хорошо известна реакция дегидрогалогениро-
вания в основной среде с образованием соответствующих «ди-
мерных» олефинов:
2 ^СНХ + 2В“—)С = С( + 2ВН + 2Х"
Для этой реакции нетрудно представить себе промежуточное
участие карбеновых частиц, однако неудачные попытки их пе-
рехвата, а также данные кинетических исследований позволи-
146
ли Ьделать вывод об исключительном протекании реакции че-
рез соответствующие анионы, а не карбены (см., например,
[4]); тр же относится и к образованию стильбенов из замещен-
ных бензилгалогенидов [950, 951J. Напротив, из хлорметокси-
бензола, его тио- и селеноаналогов под действием тр^т-буток-
сида калия [940, 952] или оксирана [953] удается генерировать
соответствующие карбены:
ro-
PhZCH2CI ----~CHClZPh-------:CHZPh
г -ROH -СГ
Z = О, S, Se
сс-Замещенные фенилтиокарбены CRSPh (R = Me, Et, Pr,
Ph) генерированы из соответствующих аеж-фенилтиоалкилга-
логенидов под действием литийорганических реагентов в ТГФ
по сольволитическому механизму с промежуточным участием
анионов “CRCISPh [954].
Фурил-2- и (тиенил-2)карбены генерированы из соответст-
вующих галогенидов под' действием трет-BuOK в присутствии
дибензо-18-краун-6-эфира, а также в условиях межфазного ка-
тализа [941].
Алкилгалогениды обычно инертны к сольволитическому
а-элиминированию. В то же время из алифатических винилга-
логенидов, обладающих более «кислым водородом» при крат-
ной связи, реакциями а-элиминирования удается генерировать
алкилиденкарбены, например [955, 956]:
Ме2С = CHCI
Ме2С = CBrSiMe3
трет-BuOK
Me4N+ F“
:С = СМе2
Пожалуй, наибольшее значение, в том числе и в синтезе,
имеют реакции ступенчатого а- и у-дегидрогалогенирования,
позволяющие генерировать винилиденкарбены в сольволитиче-
ских условиях под действием оснований (0—10 °C, непредель-
ный акцептор) из алленил- и (пропин-2-ил)галогенидов соот-
ветственно [957—960].
При проведении этой реакции в среде олефинов с хороши-
ми выходами (до 55%) образуются алкенилиденциклопропаны,
причем относительные скорости алкенилиденметиленирования
при использовании как алленил-, так и (пропин-2-ил)галогени-
Дбв практически одинаковы и зависят лишь от активности суб-
страта [957—960]. В отсутствие олефинов в этой реакции об-
разуются соответствующие гексапентаены — формальные ди-
меры алкенилиденкарбенов [961, 962].
10!
147
R2G'= С = С - СГ
\ / 4
- С -
(« 55%)
С =С,
R2C = С = СНХ-^*- R2C = С = СХ —* R2C = С = С:
—ВН —ХС
D— — 4- —
R2XC - С = СН -^*R2XC - С = С —*• R2C - С =С:
—вн —х
RgC — С — С — С — С — CR2
R - Me, Et, Bu-трет; X = Cl; Вг (=S 30%)
Описано генерирование пентадиен-1,3-илиденкарбена фор-
мально по реакции е-элиминирования [963]:
сн3снхсн = снс =сн сн3сн-снсн = с = С: 2е
р/
—сн3сн = снсн = с = с р
X = С1, Вг, ОАс
43%)
Достаточна общим препаративным методом генерирования
алкилиденкарбенов является также взаимодействие замещен-
ных винилтрифторметансульфонатов (трифлатов) с трет-буток-
сидом калия [964—968]. Отщепление трифторметансульфокис-
лоты обычно протекает под действием сильных оснований, од-
нако карбеновое расщепление триметилсилильных производных
винилтрифторметансульфонатов гладко идет и в более мягких
условиях [968J.
148
трет-BuOK,
-20-0 °C
RR1C = CHOSO2CF3---------* RR1C = C:
z 4 -CF3SO2OH
rper-BuOH
RR1C = C-C<
\ /
-C-
RR1C = СНОВи-трет
R, R1 = Alk, Ph
0 SO2 C Fg
Me2C - C - SiMe3
PhN = NPh
r4nf, -20 °c,
G2H4(OMe)2
—-----------:C = CMe9
-CF3SO2OSiMe3 2
CHMe2
(43%)
В целом же, за исключением винилиден- и алкилиденкар-
бенов, сольволитическое дегидрогалогенирование органилгало-
генидов нельзя считать столь же общим методом генерирования
разнообразных функционально замещенных органилкарбенов,
каким является превращение замещенных дигалогенметанов в
органилгалогенкарбены (см. разд. 3.2.2).
3.3. ГЕНЕРИРОВАНИЕ КАРБЕНОВ И ИХ АНАЛОГОВ
ИЗ АТОМАРНЫХ ЭЛЕМЕНТОВ ПОДГРУППЫ IVB
Атомарный углерод и его аналоги по подгруппе IVB, как и лю-
бые атомы, являются термодинамически стабильными частица-
ми, однако их кинетическая стабильность, особенно углерода,
невысока и они проявляют чрезвычайно высокую реакционную
способность, стремясь перейти в стабильное четырехвалентное
состояние. Атом углерода формально является дикарбеном,
и для него можно ожидать протекания (наряду с' другими) двух
последовательных карбенных реакций, каждая из которых в
149
принципе может быть заменена двумя гомолитическими стадия-
ми, например:
V
RH ,ruD RH ’
С---------------:снк-------------------»• KgbHj
Наиболее удобными и достаточно селективными источника-
ми атомарного углерода могут быть ядерные реакции [969,
970]; фотолиз цианазида [47, 53, 54, 207, 226], а также фото-
лиз трикарбодиоксида [227] (см. разд. 3.1.2):
12С-^-Пс т1/2 = 20,4 мин
N3ChA:NCN-!X:C: С3О2
4 -n2 -n2 -2СО 15 z
Эти методы селективно приводят именно к атомарному уг-
лероду, в отличие от методов угольной дуги в высоком вакууме
[971] или лазерного испарения графита [972], где наряду с
«истинно атомарным» углеродом образуются дикарбеновые мо-
лекулы С2 и Сз- Эти частицы также высокореакционноспособ-
ны, и образующиеся из них продукты, естественно, усложняют
анализ реакционных смесей.
Попытки провести реакцию :С:4-Н2->:СН2 в Ar-матрице при
14 К не привели к успеху; ИК-спектроскопически был обнару-
жен лишь метан, образовавшийся, видимо, в результате мат-
ричных реакций с участием метилена [972]. В то же время в
низкотемпературных инертных матрицах из элементов удалось
синтезировать дифтор- и дихлоркарбены [53, 207], что, в част-
ности, было показано с помощью матричного колебательного
спектра. Аналогичным образом из атомарного углерода и HF
или НС1 в матрице при 14 К были получены фтор- и хлоркар-
бены соответственно; зафиксированы их электронные и колеба-
тельные спектры [47, 54].
Реакции атомарного углерода в газовой фазе проходят
сложнее, поскольку их не удается остановить на стадии образо-
вания карбена, а релаксация возбужденных продуктов по срав-
нению с низкотемпературными матрицами затруднена (подроб-
нее см., например, [973]). Так, при газофазной реакции «лег-
ких» углеродных атомов ПС с н-алканами состава С3—Cs обра-
зуются этилен и ацетилен, а также продукты неселективного
внедрения метилена по связям С—Н; в жидкой и твердой фа-
зах, где возможность релаксации выше, выход продуктов дест-
рукции— этилена и ацетилена — ниже [974, 975].
150
Аналогичным образом в реакции атомов ПС с этиленом в
газовой фазе первоначально образующийся «горячий» цикло-
пропилиден преимущественно распадается на ацетилен и мети-
лен, тогда как в конденсированной фазе он перегруппировыва-
ется в аллен [976]:
С:
СН2
= С = сн2
:С: + Н2С = СН2 —
Н2С - СН2—
^:СН2 + СН = СН
При взаимодействии «горячих» атомов НС с циклопентадие-
ном образуются бензол и фульвен, причем в качестве проме-
жуточных частиц здесь также рассматриваются карбены, воз-
никающие в результате присоединения атома углерода по крат-
ной связи или его внедрения в связь С—Н [977]:
Аналогичным образом из НС и бензола в бензольной мат-
рице при —196 °C образуются толуол, тропилиден, фенилацетп-
лен, бифенил и дифенилметан, источниками которых, по дан-
ным изотопного анализа, являются метилен, фенилкарбен и
циклогептатриенилиден [978].
Была исследована реакционная способность атомов углеро-
да в основном (3Р) и возбужденных (lD и электронных со-
стояниях [971]. При генерировании атомов углерода в глубо-
ком вакууме в угольной дуге в присутствии олефинов среди
продуктов обнаружены аллены и спиропентаны. Если же гене-
рированные атомы имели возможность «остывать» в инертной
матрице, т. е. релаксировали в основное состояние и лишь
затем в матрицу вводили олефины, то единственными продук-
тами оказывались спиропентаны [971]:
:С:(13)+
:С:(3Р)+
151
При внедрении C^S) и С(3Р) в связи С—Н алканов также
образуются соответственно синглетные и триплетные алкилкар-
бены [979]. Внедрение атомарного углерода в связи С—С1 при-
водит к соответствующим алкилхлоркарбенам [980], которые
во внутримолекулярных реакциях активнее, чем частицы, гене-
рированные сольволитическими методами:
Ме2С = CMeCt
:С: (1S) + Ме3СС1 —:СС1СМе3
Ме?С - CHCI
\ /
сн2
Описано и взаимодействие атомарного углерода с водой
[981].
Если в реакциях внедрения в ординарные связи и присоеди-
нения по кратным связям образовавшийся карбен как бы со-
храняет два несвязанных электрона исходного атома углерода,
то в реакциях атомарного углерода с карбонильными соедине-
ниями новый карбеновый центр образуется из карбонильного
соединения в результате отщепления от него атома кислорода
карбонильной группы, а исходный атом углерода образует ста-
бильную молекулу СО. Однако эта интересная реакция изуче-
на явно недостаточно. Взаимодействие атомарного углерода с
карбонильными соединениями при —196 °C приводит к моноок-
сиду углерода и соответствующим карбенам, которые далее
изомеризуются в олефины; это позволило приписать способ-
ность к отрыву карбонильного кислорода исключительно воз-
бужденным атомам углерода (JS или !D) [982]:
RCOC^R1 + :С:—-» :CRCH2R1 ---* RCH = CHR1
По аналогичной реакции из СОС12 генерирован дихлоркар-
бен, а из НСООМе — метоксикарбен [983].
Образующиеся одновременно с атомарным углеродом при
испарении в дуге [971] или лазерным лучом [972] молекулы
С3 также проявляют свойства дикарбенов. В частности, с оле-
финами они образуют соответствующие бис(этано) аллены, оче-
видно, с промежуточным участием этаноалленилиденов [984]:
Описанные в данном разделе реакции атомарного углерода
и молекул Сз в настоящее время не представляют препаратив-
ной ценности; однако реакции атомарного углерода, по-види-
152
мому, протекают в плазмохимических процессах и при глубо-
кой переработке угле- и нефтехимического сырья [468]; деталь-
ные сведения по этому вопросу пока отсутствуют.
Сходные реакции известны и для атомарного кремния. От-
метим, в частности, матричные реакции [66, 82, 88]:
| н2о
:SiH(OH)
Известен также и ядерно-химическпй метод генерирования
силиленов с участием атомарного кремния [985—989]:
РХп 31.gj. РХз» 3bcjY
X = Н, F
Необходимо упомянуть также хорошо известные реакции эле-
ментных кремния и германия с различными органическими и
неорганическими галогенидами («прямой синтез»). Эти реак-
ции лежат в основе промышленного получения практически
всех галогенидов кремния и германия (см., например, [587,
990—992])—исходных веществ для синтеза большинства эле-
менторганических соединений. Механизм этих реакций доста-
точно хорошо изучен; в частности, с применением различных
физических методов показано, что в этих процессах участвуют
силилены и гермилены MR2, MXR и МХ2 [16, 549, 587, 610].
RX + М---RnMX4_n
М = Si, Ge; X = Hal
Хорошо известна и реакция высокотемпературного диспро-
порционирования галогенидов кремния и германия, которую, в
частности, неоднократно использовали для генерирования час-
тиц МХ2 с целью их последующего исследования методами
Масс-спектрометрии, низкотемпературной матричной спектро-
скопии, газовой электронографии, а также в препаративных це-
лях (подробнее см., например, [32—35, 991]). Менее характер-
ны, хотя и известны, подобные реакции для олова и свинца
[993—995].
М + МХ4 2 :МХ2
М = Si, Ge; X = F, Cl, Br
153
3.4. СРАВНИТЕЛЬНАЯ РЕАКЦИОННАЯ СПОСОБНОСТЬ КАРБЕНОВ,
ГЕНЕРИРОВАННЫХ РАЗЛИЧНЫМИ МЕТОДАМИ
Рассмотренные методы генерирования карбенов достаточно
разнообразны как по набору молекул — источников, так и по
условиям проведения реакции — от матричных реакций, прово-
димых при низких, вплоть до 4 К, температурах (фотолиз ди-
азосоединений или диазиринов, реакции атомов щелочных ме-
таллов, а также атомарных С и Sic галогенидами), до высо-
котемпературных пиролитических процессов (распад полига-
логенметанов, гексагалогенидов, реакции «прямого» синтеза).
В этой связи возникает вопрос о сравнительной реакционной
способности карбенов, генерированных различными методами,
имеющий, по сути, два аспекта.
Первый из них относится к избыточной энергии карбена,
в частности, к взаимосвязи спиновой мультиплетности этой ча-
стицы в основном состоянии, в условиях ее генерирования и при
участии в реакции.
Основные характеристики реакционной способности синг-
летных и триплетных карбенов, установленные как в виде ряда
эмпирических правил, так и при современных импульсно-кине-
тических исследованиях были рассмотрены в гл. 2. Так, для
синглетных карбенов характерны стереоспецифическое /^-при-
соединение по кратным связям, преимущественное одностадий-
ное внедрение, в частности, в связь О—Н спиртов и в связь
С—С1 аллилхлоридов в условиях внутримолекулярной конку-
ренции. У триплетных карбенов реакции внедрения проходят
по механизму «отрыв — рекомбинация» с возможным выходом
образующейся радикальной пары из клетки; характерно также
нестереоспецифичное присоединение по кратным связям, пре-
имущественное присоединение по связи С = С аллилхлорида
(а не внедрение по связи С—С1) и взаимодействие с триплет-
ными партнерами, в частности с молекулой О2 (см. разд. 2,2—
2.4).
Не все из этих признаков в равной мере универсальны, од-
нако в целом они, в совокупности с прямыми физическими ис-
следованиями, позволяют четко определить спиновую мульти-
плетность реагирующего карбена. В то же время не во всех
случаях карбен вступает в реакцию в том же состоянии, что и
образуется. Так, большинство методов генерирования исходит
из синглетных предшественников и как в «мономолекулярном»,
так и в сольволитическом варианте приводит к синглетной час-
тице. Если для данного карбена синглетное состояние является
основным, например для карбенов с гетероатомами в сополо-
жении, то анализ реакционной способности не представляет
особых проблем. Если же основное состояние триплетно (мети-
лен, арил- и диарилкарбены и т. п.), то состав продуктов ре-
154
акции будет определяться достаточно тонким балансом скоро-
стей внутримолекулярного (или индуцированного другой части-
цей) безызлучательного перехода в основное, триплетное со-
стояние и перехвата возбужденного синглета, т. е. результат бу-
дет существенно зависеть от условий эксперимента. Так, для
сравнительно малоактивных карбенов снижение температуры
среды (переход от раствора к стеклам и матрицам) благопри-
ятствует релаксации их из возбужденного в основное триплет-
ное состояние [273]. В то же время при высокой концентрации
перехватчика большая доля карбена (особенно активного) мо-
жет успеть прореагировать в возбужденном синглетном состоя-
нии.
Для генерирования заведомо триплетных карбенов практи-
чески единственным методом является сенсибилизированный
фотолиз соответствующих диазосоединений или диазиринов.
Вместе с тем современные импульсно-кинетические исследова-
ния показали, что и в этом случае существенную роль играет
равновесие синглетных и триплетных карбенов, поскольку ре-
акционная способность триплетных частиц обычно ниже.
Таким образом, помимо условий реакции (температура,
концентрация перехватчика, метод генерирования), важную
роль играет и энергия синглет-триплетного расщепления (AEst,
см. гл. 2). Полагают [175], что карбен с триплетным основным
состоянием в равновесных условиях будет реагировать только в
этой форме, если A£st>10/?T; если же A£st<5/?E, то вклад
реакций более активной синглетной частицы может оказаться
доминирующим.
В то же время в неравновесных условиях большое значение
имеет избыточная энергия карбенов и продуктов их реакций,
поскольку в большинстве методов генерируются «горячие» час-
тицы, а реакции исчезновения карбенов, как правило, сильно
экзотермичны (несколько сотен кДж/моль). 'Хемилюминесцен-
ция продуктов карбенных реакций наблюдалась даже в инерт-
ных матрицах при низких температурах (см., например, [220,
272]), т. е. в условиях, благоприятствующих релаксации. Еще
больше проблем вызывает избыточная энергия карбенов в га-
зофазных реакциях; достаточно вспомнить неверное определе-
ние AEst у метилена, обусловленное «горячими» полосами в
фотоэлектронном спектре иона ~СН2 (см. гл. 2).
Другим важным аспектом сравнительной реакционной спо-
собности карбенов является вопрос об их кинетической незави-
симости. Многие формально карбенные реакции .могут быть ин-
терпретированы как взаимодействие источника с акцептором
(А), не включающее стадию образования свободного карбена
(яркий пример — реакция Симмонса — Смита, см. гл. 4). Одна-
ко до необратимого исчезновения карбен может образовать
слабый интермедиат координационного типа, способный обра-
155
тимо регенерировать карбен или «переносить» его к акцепто-
ру; в роли «координационного акцептора» (L) может высту-
пать растворитель, источник карбена или даже его акцептор:
А
Источник -------——---------------------Продукт
IL I
l-cr2-----------------1
-L
Веским аргументом в пользу кинетической независимости
карбенов, генерированных различными методами, является
идентичность их реакционной способности. Наиболее известным
примером является дихлоркарбен [724, 725, 742, 996, 997]. Для
этой частицы, генерированной из различных источников в ин-
тервале температур —196 -т-1100 °C, относительная реакцион-
ная способность подчиняется уравнению
AAG = ДДН- ТДД5
что свидетельствует о практической идентичности реагирующей
частицы, т. е. о практически полной кинетической независимо-
сти дихлоркарбена во всех рассмотренных случаях [724]. Для
ряда других галогенкарбенов (CF2, CFC1, CCIBr, CBr2, CCIPh)
также исследована температурная зависимость их селективно-
сти, правда, в менее широком диапазоне температур, и эти час-
тицы также оказались кинетически независимыми [18, 173,
998—1000]. Присоединение арилгалогенкарбенов к непредель-
ным соединениям проходит с промежуточным образованием
комплексов карбен — олефин [173] (подробнее см. разд. 2.4.1).
В реакции арилкарбенов со спиртами ход температурной
зависимости распределения продуктов свидетельствует об об-
разовании промежуточных комплексов [850, 1001]. Промежу-
точное образование подобных интермедиатов, предположитель-
но илидного строения, установлено спектральными или кинети-
ческими методами при взаимодействии карбенов с карбониль-
ными соединениями, спиртами, нитрилами, аминами, галогени-
дами, сульфидами и селенидами [176, 177, 474—477, 851, 1002—
1004]. В ряде случаев нестабильные илиды перехвачены хими-
ческими методами; см., например, схему на с. 157 [1005].
По данным кинетических исследований [508, 755], ртутьор-
ганические источники карбенов способны координировать кар-
бены; образование таких комплексов ответственно за нестерео-
156
nuu D _____-rri ArCHP A,run Fri MeOOCC=CCOOMe
PhHgCCI2Br—* .CCl2---* ArCHO-CCI2 ---------
MeOOC COOMe
Ar'Vycl
hXz^CI
-HCl
селективное присоединение <
водному фумаровой кислоты
PhH3CB'3^?:CB'2
СВг2 к ji-дейтеростиролу и произ-
[ [508]:
х \ zr
/с = с\
Y Н
Х\ R
;с-с(
YZ \/ ЧН
С Вг2
PhHgCBr3
-PhHgCBr
X
[PhHgCBr3H:CBr2]
(36)
R
H
[PhHgCBr3]-[Br2CCXYCHRJ
-PhHgCBr3
R = Ph, COOMe; X - H, CN; Y = H, D
/R
c - c;
Xх \ / XH
CBr2
В реакции PhHgCBr3 с т/щ/тс-дихлорэтиленом свободный
карбен стереоселективно циклопропанирует этот акцептор, а из
комплекса карбена с источником образуется 1,1-дибром-2,3-ди-
хлорпропен-1 [755]. Наибольший выход этого продукта наблю-
дается в СС14, благоприятствующем образованию комплекса
(36), а в присутствии ароматических углеводородов с низкими
потенциалами ионизации (ксилол, мезитилен) доля перегруп-
пированного продукта снижается. Полагают [755], что это сви-
детельствует о промежуточном и обратимом образовании комп-
лексов дибромкарбена с ароматическими углеводородами.
я-Донорные растворители также способны существенно моди-
157
фицировать реакционную способность синглетных карбенов и
их аналогов. Так, в конкурентной реакции внедрения диметил-
силилена по связям О—Н этилового и трет-бутилового спиртов
в различных растворителях селективность при переходе от цик-
логексана к эфиру и тетрагидрофурану, используемым в каче-
стве растворителя, возрастает в 2,6 и 5,3 раза соответственно
[1006]; аналогично, в присутствии 1,4-диоксана селективность
О—Н-внедрения синглетного фенилкарбена (пара МеОН —
трет-BuOH) повышается в 33 раза [1007]. Несколько меньшие
эффекты наблюдаются для метоксикарбонилфенилкарбена, ди-
фенилкарбена и флуоренилидена [1007]. Напомним, что имен-
но п-донорные молекулы типа ТГФ и 1,4-диоксана являются
наиболее подходящими партнерами для координации синглет-
ных карбенов и их аналогов; такая координация должна сни-
жать активность этих частиц, тем самым повышая их селектив-
ность.
Подобные изменения сравнительной реакционной способно-
сти карбенов, как правило, не сказываются на конечных ре-
зультатах, и интересный и важный для практики вопрос о воз-
можности регулирования реакционной способности карбенов в
растворах путем подбора растворителей и добавок до настоя-
щего времени практически не исследован.
Глава 4
ЦИКЛОПРОПАНИРОВАНИЕ
НЕПРЕДЕЛЬНЫХ СОЕДИНЕНИЙ
4.1. ГЕНЕРИРОВАНИЕ КАРБЕНОВ ИЗ ДИАЗОСОЕДИНЕНИЙ.
СИНТЕЗ ЦИКЛОПРОПАНОВ И ИХ ФУНКЦИОНАЛЬНЫХ
ПРОИЗВОДНЫХ
Генерирование карбенов из диазосоединений в настоящее время
стало одним из наиболее универсальных и широко используе-
мых общих методов получения этих частиц, нашедшим и разно-
образное применение в синтезе, в первую очередь в синтезе
циклопропановых углеводородов и их функциональных произ-
водных:
RR1CN2^^^U RR1C;
-n2 7 \ / х
CRR1
R, R1 = H, COAlk, COOAIk, Hal, CN, Ar и др.
158
Несомненным достоинством этого метода являются доступ-
ность исходных диазосоединений, простота проведения реакции,
возможность изменения реакционной способности и даже муль-
типлетности получаемых карбенов в результате использования
различных условий их генерирования. В последние годы появи-
лись примеры использования этих реакций в промышленности,
например в производстве пиретроидов, лекарственных препара-
тов и других продуктов тонкого органического синтеза [468,
1008] (см. также гл. 6).
Карбены могут быть генерированы из диазосоединений тре-
мя основными способами — термически, фотохимически или ка-
талитически (термокаталитически).
Термический метод долгое время достаточно широко
применяли для генерирования карбенов из диазосоединений,
поскольку традиционно использовавшиеся при термокаталити-
ческом генерировании медьсодержащие катализаторы, также
эффективны при довольно высоких температурах, лишь нена-
много (на 20—30 °C) уступающих температурам чисто термиче-
ских реакций (120—150°C). Однако появление в последние годы
ряда высокоактивных катализаторов карбенного разложения
диазосоединений, таких, как трифлат меди [Cu(OTf)2], соли
родия и палладия, которые позволяют проводить эти реакции
при комнатных и в отдельных случаях даже при минусовых
температурах, резко снизило препаративное значение термиче-
ских методов и интерес к ним. Единственное исключение состав-
ляют реакции диазосоединений, получаемых щелочным расщеп-
лением тозилгидразонов алифатических (алициклических) кето-
нов и альдегидов (реакция Бэмфорда — Стивенса). Эта реакция
идет при таких температурах (130—150°C), при которых обра-
зующиеся диазосоединения обычно разлагаются, что не позво-
ляет выделить их в свободном виде.
RR’C = 0 RR,C = N . ЙТз 130-1И-С
-Н2О -Ts-
- RR’cn^RR'C: R<CH = CHR2
>С - с'
\/
CRR1
(D
159
При фотохимическом генерировании карбенов из ди-
азосоединений используемое УФ-излучение* (обычно полный
спектр излучения ртутной лампы, в некоторых случаях более
узкие области этого спектра или даже отдельные линии) спо-
собно вызывать нежелательные побочные превращения субстра-
та и (или) продуктов реакции, например, их полимеризацию,
осмоление, изомеризацию. Карбены и их аддукты, образовав-
шиеся в результате фотохимических реакций, часто обладают
избытком энергии, что повышает вероятность побочных процес-
сов и значительно снижает селективность основных превраще-
ний. Кроме того, проведение фотохимических реакций требует
специального экспериментального оборудования. Все это суще-
ственно ограничивает область применения фотохимических ре-
акций, которые в отличие от термических или термокаталитиче-
ских сравнительно редко используют в синтетических целях.
В то же время фотохимический метод позволяет генерировать
карбены практически при любой температуре в различных аг-
регатных состояниях реакционной смеси. Более того, именно фо-
толиз, обычно сенсибилизированный ртутью, бензофеноном или
другими сенсибилизаторами, позволяет получать и исследовать
карбены в триплетном основном состоянии. В связи с этим фо-
тохимический метод генерирования карбенов из диазосоедине-
ний широко используют в физико-химических исследованиях
карбенов и их реакций. В ряде случаев путем использования
низкотемпературного фотолиза удается получить и стабилизиро-
вать в инертной матрице сами карбены и другие лабильные
соединения, которые не удается получить другими методами ив
иных условиях (подробнее см. гл. 2).
Каталитический (термокаталитический) ме-
тод генерирования карбенов из диазосоединений, как уже от-
мечалось, является основным в синтезах с использованием
диазосоединений. В качестве катализаторов деазотирования
диазосоединений обычно используют соединения переходных
металлов. Карбенное разложение диазосоединений способны ка-
тализировать многие металлы и их соединения (Си, Ag, Zn, Al,
Со, Pt) [8, 1009], однако к настоящему времени в синтетиче-
скую практику химии карбенов прочно вошли лишь катализато-
ры на основе Си, Rh и Pd. Несмотря на почти столетнюю исто-
рию использования медных катализаторов и попытки изучения
механизма их каталитического действия в этих реакциях [8,1010].
этот механизм полностью не выяснен, не говоря уже о механиз-
мах реакций с использованием катализаторов на основе других
металлов. Обычно считают, что в реакциях циклопропанирова-
* Было показано влияние характеристик используемого УФ-света на на-
правление реакции и состав образующихся продуктов [4, 8], однако система-
тические исследования в этой области не проводились.
160
ния олефинов диазосоединениями каталитически активна
медь(1) [1011—1014]. При этом соли Си11 первоначально вос-
станавливаются диазосоединениями до Си1. В хелатных ком-
плексах меди, используемых в качестве катализаторов, катали-
тически активной считают Си11. На это указывает и возмож-
ность асимметрического синтеза циклопропанов при использова-
нии оптически активных катализаторов [1015—1018]. Напри-
мер, взаимодействие тра«с-пропенилбензола с диазометаном
в присутствии хелатного комплекса (2) при 0°С приводит
к транс- 1-метил-2-фенилциклопропану (выход 62%; оптический
выход 8%) [1015]:
♦
PhCHMe
(2)
PhCHMe
*
Ph Н
/с = с<
Н z чМе
(2)
+ ch2n2-------•
-n2
Hz \ / чМе
СН2
Катализаторы на основе соединений Pd11 в реакциях с диазо-
метаном, являющимся активным восстановителем, первоначаль-
но превращаются в производные Pd°, которые далее, образуя
комплексы с олефинами, участвуют в образовании циклопропа-
нов [1019]. В то же время соединения Pd11, используемые в ка-
честве катализаторов карбенного разложения алкилдиазоацета-
тов и подобных диазосоединений со слабыми восстанавливаю-
щими свойствами, не изменяют своего валентного состояния
[1014]. Наконец, соединениям Rh11, катализирующим карбен-
ное разложейие диазосоединений в мягких условиях, вообще не
свойственно изменять состояние окисления в процессе реак-
ции [1014].
В настоящее время рассматривают два возможных механиз-
ма реакции каталитического циклопропанирования олефинов
диазосоединениями в присутствии производных Си, Pd и Rh.
Согласно первому механизму происходит образование комплек-
сов катализатора сначала с диазосоединением, а затем с кар-
беном и циклопропанирование комплексом (3) незакомплексо-
ванного (реже — участвующего в образовании комплекса) оле-
фина [схема (4.1)] (см., например, [1011, 1014, 1020, 1021]).
Согласно второму механизму первоначально образуется л-ком-
11—107
161
плекс (4) олефина с катализатором, а затем происходит атака
этого комплекса диазосоединением [схема (4.2)1 [1011, 1014,
1022—1024]:
N2 n2
М + и —М-»И -----*
CRR1 CRR1 -N2
м + п > = С(----)с = с( )nM N.?-CRS1
(4)
( Ч/С = С( )nMCRRi—Н )С = С( )nMCRRl
' II -n2
N2
(1) + ( )С = С( )П_!М
(4.1)
(4.2)
М - активный центр катализатора
При этом реальный механизм действия медных катализато-
ров в значительной мере зависит от природы лигандов и проти-
воионов, обусловливающих различную способность катализа-
торов координировать олефины. Так, соединения меди, которые
с трудом дают л-комплексы с олефинами [прежде всего соеди-
нения меди(II), для которой образование подобных комплексов
вообще не характерно], очевидно, катализируют циклопропани-
рование олефинов диазосоединениями через комплекс карбена
с катализатором [(3); см. схему (4-1)]. В случае же соедине-
ний меди, легко дающих л-олефиновые комплексы (прежде
всего трифлат меди), предпочтительно первоначальное образо-
вание л-комплекса (4), который далее реагирует с диазосоеди-
нением [см. схему (4.2)] [10, 1014].
Соединения родия катализируют циклопропанирование оле-
финов диазосоединениями через комплекс (3) по схеме (4.1),
а соединения палладия — по схеме (4.2) с предварительным
образованием комплекса (4) [1014, 1025].
Для реакций, протекающих по схеме (4.1), степень и регио-
селективность циклопропанирования определяются совокупно-
стью стерических и электронных факторов координационного
комплекса карбена и незакомплексованного олефина и, как пра-
вило, одинаковы внутри каждого класса непредельных соеди-
нений (см., например, [1014, 1025—1027]).
В то же время при реализации схемы (4.2) определяющее
влияние на циклопропанирование оказывает комплексообразо-
вание олефина с катализатором, причем легче циклопропаниру-
ются двойные связи, в большей степени склонные к образова-
нию л-олефиновых комплексов (см., например, [1014, 1019,
162
1024, 1027, 1028]). Следует отметить, что легче образуют ком-
плексы с переходными металлами олефины с более нуклеофиль-
ными двойными связями, тогда как само комплексообразование
с переходными металлами понижает л-электронную плотность
олефина и делает возможным его реакцию с нуклеофильными
реагентами [1029]. Это особенно характерно для л-олефиновых
комплексов палладия. Прочность координационной связи зави-
сит также от напряжения в циклических олефинах. Так, соли
меди обычно образуют более устойчивые комплексы с эндоцик-
ли^ескими олефинами, чем с олефинами с открытой цепью
[1030—1032]. По этой причине наибольшей устойчивостью ха-
рактеризуются л-комплексы CuCl с такими олефинами, как нор-
борнен, норборнадиен, цг/с,цщ>циклооктадиен-1,5; причем в по-
следнем случае комплекс с CuCl может быть выделен [1024].
Именно эти непредельные углеводороды при каталитическом
взаимодействии с диазосоединениями дают циклопропановые
аддукты с максимальными выходами. В частности, реакции
диазометана непосредственно с л-олефиновыми комплексами
CuCl, например с комплексами норборнадиена и циклооктадие-
на-1,5 [1033, 1034], приводят к соответствующим циклопропано-
вым углеводородам с выходами до 90%.
Региоселективность циклопропанирования различных крат-
ных связей также определяется преимущественным комплексо-
образованием и, как следствие, предпочтительным циклопропа-
нированием стерически наиболее доступных двойных связей
[1011, 1030]. В полном соответствии со сказанным выше опти-
мальным является проведение этих реакций в отсутствие раст-
ворителя или в растворителях с низкой полярностью, не препят-
ствующих образованию л-олефиновых комплексов [1035]. Сни-
жение нуклеофильности аниона в используемом медном
катализаторе (например, переход от С1~ к BF4~ или “OSO2CF3)
повышает склонность иона меди к избирательному комплексо-
образованию с кратными связями олефинов, а следовательно,
и степень их циклопропанирования [1011, 1030]. Напротив, та-
кие соли, как CuCN и Cui, для которых не характерно образо-
вание л-олефиновых комплексов [1036], проявляют низкую ка-
талитическую активность в этих реакциях.
Что же касается диазосоединений, используемых в реакциях
каталитического метиленирования кратных связей, то наиболее
широко для получения циклопропановых производных применя-
ются диазометан и алкилдиазоацетаты.
4.1.1. Диазометан
Несмотря на высокую токсичность и взрывоопасность, диазоме-
тан является одним из основных источников простейшего кар-
бена— метилена, что объясняется, с одной стороны, его доступ-
11*
163
ностью, а с другой — сравнительной экспериментальной просто-
той методик работы с ним.
В обычных условиях диазометан представляет собой газ
(т. кип. —27 °C) и может служить источником метилена как
в газовой, так и в жидкой фазах.
Получение диазометана [1024]. В снабженную трубками для ввода и вы-
вода газа круглодонную колбу без шлифов, центральное отверстие которой
соединено со шнековым устройством (стекло и тефлон), помещают 100 мл
45—50 %-го раствора КОН и 25 мл додекана, продувая инертный газ (Аг или
N2) со скоростью «80 мл/мин. С помощью шнекового устройства в колбу
подают N-метил-ЛДнитрозомочевину со скоростью 6—7 г/ч. Образующийся
CH2N2 потоком газа транспортируют через осушительную трубку с твердым
кон в реактор, снабженный обратным холодильником (температура охлаж-
дения до —70 °C). Выход CH2N2 60—65%.
Следует отметить, что в синтетических целях применяют
исключительно жидкофазные реакции диазометана, используя
в качестве катализаторов соединения меди (обычно галогени-
ды) и палладия (1—2% на вводимый диазометан). При этом
диазометан берется в избытке по отношению к циклопропани-.
руемому олефину, поскольку целевая реакция циклопропаниро-
вания в жидкой фазе практически всегда сопровождается по-
бочными реакциями димеризации, олигомеризации и полимери-
зации метилена, а также его взаимодействием с растворителем,
что снижает выход целевого продукта. Вместе с тем образую-
щиеся в результате этих побочных реакций полиметилен, а так-
же этилен и другие газообразные продукты (этан, циклопропан,
пропилен) обычно не затрудняют выделение и очистку цикло-
пропановых продуктов.
Первая удобная методика циклопропанирования диазомета-
ном заключалась в пропускании разбавленного азотом газооб-
разного диазометана в олефин, содержащий CuCl, при 0°С
[1037]. Реакция протекает стереоспецифично [1029, 1038].
Так, метиленирование ^^с-1,4-дихлорбутена-2 приводит исклю-
чительно к ц«с-1,2-ди (хлорметил) циклопропану [1039]:
С1Н2СХ
HZ
/СНгС!
ХН
CuCl
+ С Но No----•
-N2
С1Н2СХ
Hz
с-с
\ /
СН2С1
н
сн2
с = с
Однако региоселективность подобных реакций, как правило,
невысока. Например, из ^с-гексатриена-1,3,5 (5). были получе-
ны все возможные продукты циклометиленирования.
Выходы этих продуктов зависят от мольного соотношения
гексатриена и диазометана [1037] и даже избыток диазомета-
на не приводит к полной конверсии гексатриена; выходы при
164
ch2n2,
CuCl
*-N2
ch2n2,
CuCl
соотношениях гексатриен: диазометан, равных 1:2 (I) и
1:5 (II), приведены ниже:
Выход, % Выход, %
Соединение (5) I 40 II 6 Соединение (10) I 6 11 3
(в) 31 8 (11) 2 5
(7) 8 12 (12)-цис 0,5 3
(8) 8 12 (12) -транс 0,5 3
(9) . 4 48
При циклопропанировании цис- и трй/^с-пентадиенов-1,3 ди-
азометаном в присутствии CuCl также образуется смесь возмож-
ных продуктов циклопропанирования, причем, несмотря на
предполагаемый электрофильный характер карбенного интерме-
диата, несколько лучше циклопропанируется менее замещенная
двойная связь [1040]:
(13)
1
+ ch2n2 2S W=/ + +
(14) (22%) (15) (19%) (16) (14%)
1
165
1 : 0,6
1 : 1,2
ch2n2
CuCI
-n2
(21%)
(29%)
(14%) (7%)
(18%) (22%)
Интересные результаты по относительной реакционной спо-
собности олефинов получены при использовании в качестве ка-
тализаторов трифлата и ацетилацетоната меди [1011]. Так, при
пропускании диазометана в смесь 2,3-диметилбутена-2 и гексе-
на-1 (в соотношении 1:1), содержащую каталитическое коли-
чество ацетилацетоната меди, выделена смесь, состоящая из
тетраметилциклопропана и бутилциклюпропана (87:13). Одна-
ко соотношение образующихся продуктов резко меняется при
замене ацетилацетоната меди на трифлат меди. В этом случае
циклопропанирование протекает преимущественно по менее
замещенной кратной связи гексена-1, и отношение тетраметил-
циклопропана к бутилциклопропану составляет 1 : 4.. [1011].
+ АЛ/ + CH2N2
См(асас)2
vw
Подобная зависимость направления реакции циклопропани-
рования от степени замещения кратной связи и используемой
соли меди (природы аниона) четко проявляется и при цикло-
пропанировании 2-метилоктадиена-2,7. При использовании аце-
тилацетоната меди преимущественно циклопропанируется более
замещенная двойная связь. При конверсии 10% в реакционной
смеси содержалось 86% соединения (17) и 14% соедине-
ния (18). При повышении конверсии до 90% в значительном
количестве образовывался продукт двойного циклопропанирова-
ния (19) и соотношение (17) : (18) : (19) составляло 1 :0,1 :0,6
[1011]. В присутствии трифлата меди соотношение (17) : (18)
при низких степенях конверсии исходного диена равно 1 :2.
При дальнейшем пропускании диазометана до ^50%-й конвер-
сии 2-метилоктадиена-2,7 соотношение продуктов (17) : (18) :
166
Взаимодействие циклоолефинов с диазометаном в присутст-
вии CuCl гладко приводит к соответствующим бицикло[п. 1.0]-
алканам (20) [1024], выходы которых увеличиваются по мере
повышения устойчивости циклоолефиновых комплексов хлори-
да меди(1) (ср. [1041]):
н
Т)(СН2)П + ch2n2 CuC>;~ °t°4£
н 1:1
<О(СНг)"
(20)
п = 2 (выход 20-25%), 3(50-55%), 4(75-80%)
Каталитическое разложение диазометана на медных катали-
заторах успешно используют для синтеза малостабильных или
труднодоступных соединений. Так, циклопропанированием
3-метилциклобутена была получена смесь цис- и трйнс-2-метил-
бицикло [2.1.0] пентанов [1042]:
Взаимодействием диазометана с бицикло[3.2.0] гептеном-1
(21) получен с выходом до 65% трицикло [3.2.1.01’5] октан (22),
который легко полимеризуется уже при комнатной температу-
ре [1043]. Реакцией трицикло [4.2.1.025] нонена-2 (23) с избыт-
ком диазометана (соотношение 1:2) в присутствии каталити-
ческих количеств OuBr синтезирован с выходом 87% еще один
пропеллановый углеводород (24) [1044]. Аналогичным образом
был получен ряд других полициклических углеводородов
[1045—1050].
В случае норборнена (25) п ,
Ю52] наблюдается селективное эязо-присоединение
По двойной связи:
и его производных । [1024, 1051,
‘ метилена
167
К) : 1
Получение экзо-трицикло[3.2.1.02*4]октана (26) [1024]. К раствору 0,95 г
норборнена в 3 мл СН2С1г прибавляют 0,02 г CuCl, охлаждают до —10-=-0 °C
и пропускают CH2N2, генерированный из 2,0 г JV-нитрозометилмочевины. Про-
дукт перегоняют. Выход 88%; т. кип. 134—136 °C.
Полинепредельные циклоолефины образуют с диазометаном
сложные смеси продуктов [1040, 1053—1055], в которых преоб-
ладают моноциклопропановые аддукты. При избытке диазоме-
тана доминирует образование продуктов исчерпывающего цик-
лопропанирования. Так, в реакции циклопентадиена с диазо-
метаном (соотношение 1:3) образуется смесь углеводородов,
содержащая исходный диен, моноаддукт, транс- и 1рс-диаддук-
ты в соотношении 0,2 : 3,4 : 3,9 : 1 [1056]. ^сдес-Циклоокта-
диен-1,5 /27) с диазометаном (1:2) при —15 — 0°С в присут-
ствии CuCl дает также продукты моно- и дициклопропанирова-
ния с общим выходом до 90% [1024]:
+
цис и транс
(28) (35%) (29) (51%)
(27)
168
Циклопропанирование двойных связей циклооктатетраена
диазометаном в присутствии CuCl протекает нерегиоселектив-
но и приводит к продуктам моно-, ди-, три- и тетрациклопропа-
нирования:
Выходы этих продуктов при соотношениях циклоокта-
тетраен : диазометан, равных 1:1 (I) и 1:3 (II), приведены
ниже:
Выход, %
Соединение j п
(30) 44 13
(31) 6 12
(32) 8 14
Соединение
(33)
(34)
Выход, %
I II
5 36
1—2 15—17
Среди образующихся продуктов ди-, три- и тетрациклопропа-
нирования преобладают изомеры с экзо-ориентацией циклопро-
пановых фрагментов [1057].
Реакцией гексаметилбицикло [2.2.0] гексадиена с избытком
диазометана в присутствии CuBr синтезированы соответствую-
щие три- и тетрациклические соединения [1058], а взаимодей-
ствие ' 3,4-диметиленциклобутена с избытком диазометана
(1: >10) в присутствии CuCl привело к смеси трех спироугле-
водородов в соотношении 5,8:1:1,7 [1059]:
169
Для норборнадиена, как и для норборнена (см. выше), ха-
рактерно э/сзо-метиленирование [1024]; однако в случае 7-заме-
щенных норборнадиенов образуется смесь экзо- и з//(?о-изоме-
ров в соотношении 5: 1 [1060—1062]:
R=COMe,COPh, Ви-трет
Диазометан в присутствии CuCl легко реагирует и с аллено-
выми углеводородами; эта реакция является достаточно общим
методом синтеза производных метиленциклопропана и спиропен-
тана. Так, при взаимодействии гексатетраена-1^2,4,5 с диазоме-
таном в присутствии CuCl получена смесь всех возможных про-
дуктов циклопропанирования [1063]:
(13,5%) (17%) эригро- и трео- (11,5%) мезо- и D, L~
Аналогично, из бициклопропилидена получен диспирогептан
[1064] с выходом до 80%, а из его алленового аналога — три-
спиро[2.0.0.2.1.1]нонан(35) с выходом до 30% [1065]:
z-ч | । к I CuCl
+ ch2n2
-n2
170
. CuCl
>=с=<| + ch2n2 —->{Х]_Л
(5°%) (35)30о/о)
Циклопропанирование диазометаном некоторых производных
метиленциклопропана положено в основу синтеза изомерных
этоксикарбонилспиропентанов [1066, 1067], например:
+ ch2n2
1 : 15
Me COOEt
(29 %)
Наиболее подробно изученной реакцией диазометана с не-
предельными акцепторами является взаимодействие с аллилга-
логенидами, приводящее к смеси продуктов циклопропанирова-
ния и формального внедрения в связь С—X [1039]:
сн2 = CHRCH2X \Хсн2Х + сн2 = crch2ch2x
X = Hal, OR1, NR2; R = Н, Aik
Взаимодействие диазометана (5:1) с винил галогенидами
в присутствии меди и ее соединений при —ЗОН—10 °C обычно
дает соответствующие циклопропаны с низкими выходами
[1039, 1068]:
^Br + CH2N2^^ Д-Вг
”2 (13%)
R1 CI ж
vy +CH2N2^^ W-R1
Г~ -n2 r '
R = цикло-С3Н5; R1 = H (выход 35%), CHMe или
цнкло-С3Н5 (24%), CH2CH2Cl (19%)
В то же время диазометан оказался удобным циклопропа-
нирующим агентом для виниламинов [1069], например:
171
1 •’ 5
(70%)
Диазометан в присутствии солей меди способен циклопропа-
нировать и ароматические кратные связи. При этом первона-
чально образующиеся производные норкарадиена в условиях
реакции обычно практически нацело изомеризуются в соответ-
ствующие циклогептатриены. Так, в реакции бензола с CH2N2
в присутствии CuCl или СиВг выход циклогептатриена соста-
вил 80—85% (в расчете на диазометан) при полном отсутствии
толуола среди продуктов реакции [1009]. Замещенные бензолы
в этой реакции дают соответствующие производные циклогепта-
триена [1070, 1071]:
CH2N2) CuX
-n2
ch2n2,
CuBr
Суммарный
выход, %
Суммарный
выход, %
Me 75
MeO 75
Et 68
Pr-r/зо 64
Bu-трет 50
Cl 50
F 41
Br 25
Ненасыщенные соединения с циклопропановыми фрагмента-
ми (соединения типа бензоноркарадиенов и их производных)
удается выделить лишь при взаимодействии избытка конден-
сированных ароматических соединений с диазометаном при
120—170 °C [7].
Кроме соединений меди в качестве катализаторов циклопро-
панирования олефинов диазометаном в последнее время, как
уже отмечалось, широко используют соединения палладия,
в частности Pd(OAc)2 [1019, 1072, 1073]. Применение палла-
диевых катализаторов обычно позволяет повысить селективность
реакции, обеспечивая при этом в некоторых случаях почти ко-
личественные выходы циклопропановых аддуктов. Впервые
Pd(OAc)2 успешно был применен при циклопропанировании
стирола [Ю72]:
172
Ph A + ch2n2
Pd(OAc)2
-N2 *
Ph
• (90%)
Эта методика затем была использована для циклопропани-
рования а$-ненасыщенных карбонильных соединений, причем
выходы продуктов циклопропанирования в случае дизамещен-
ных двойных связей достаточно высоки даже при использовании
небольшого избытка диазометана [1074]:
R R2 R R2
+ CH2N2
R1? COR3 -n2 RV V XCOR3
К R1 R3 R3 Выход, %
Ph H H Ph 98
Ph H H OEt 90
Ph H H Me 85
Me H H OMe 89
H H Me OMe 88
H Ph H OEt 85
Me Me H Me a
Me Me H OMe a
а Реакция не ид°т.
В случае непредельных спиртов наряду с циклопропаниро-
ванием двойной связи отмечается также метиленирование гид-
роксигруппы с образованием метоксипроизводных [1075]:
ОН СН2 ОН
RCH = СН - CHR1 + CH2N2 Pd(0.^. RHC - СН - CHR1 +
- Nj
(20%)
R =
OMe
l
4 RCH = CH - CHR1
(20%)
О
Ph,
R’^H-CgHu
В присутствии ацетата палладия гладко проходит и цикло-
пропанирование терминальных олефинов (примерно трехкрат-
ный избыток диазометана) [1076]:
173
RCH = CH2
CH?N2,Pd(OAc)2
-n2 *
H2C, - CHR
CH2
(89-97%)
R = h-C8H17, CH2OPh,
CMe2CH2COOEt
C = CH2
CH2n2, Pd(OAc)2
-n2
Me
(82%) Me
Использование Pd(OAc)2 или PdC12(PhCN)2 оказалось эф-
фективным и при циклопропанировании диазометаном напря-
женных циклоолефинов, причем даже при использовании сте-
хиометрических количеств диазометана выходы соответствую-
щих циклопропановых аддуктов обычно составляют 90—95%
[1019, 1024, 1028, 1073, 1076, 1077], например:
(25) + CH2N2 ^-°АД2 (26)
-n2
+ ch2n2
Аналогично реагируют с диазометаном в присутствии л-ал-
лильного комплекса палладия [PdCl (С3Н5)]2 напряженные
олефины, образуя продукты эязо-циклопропанирования [1078]:
[ Р(1С1(С3НД]2
+ CH2N2--------—-------
(^100 %)
пл * 2ch2n2 <rm>
(85%)
174
Взаимодействие циклоолефинов (циклогексена, циклогепте-
на, циклооктена) еэквимольным количеством диазометана в
присутствии PdCl2(PhCN)2 при —10 °C приводит к соответст-
вующим бициклоалкаЦам (20) с выходами 15—93% [1019].
Получение бицикло[4.1 .бргептана [1019] (20; п = 2). В раствор 0,82 г
циклогексена в 3 мл СН2С12 при —15 °C и непрерывном пропускании CH2N3
(из 2 г нптрозометилмочевины) прибавляют порциями (4X8 мг) PdCl2(PhCN)2.
При этом наблюдается вспенивание и ослабление желтой окраски. Выход
»60%.
При пропускании CH2N2 в раствор олефина и катализатора выход ^15%.
Взаимодействие ццс?^с-циклооктадиена-1,5 (27) с избытком
диазометана (1 :2,2) в аналогичных условиях ведет к накопле-
нию продуктов исчерпывающего циклопропанирования [1019],
среди которых основным (в отличие от циклопропанирования
в присутствии CuCl) является транс-(29) (выход 75%), выход
цис-(29) равен 10%; выход продукта моноциклопропанирования
(28) составляет 8%.
Различия соединений меди и палладия в реакциях катали-
тического циклопропанирования отчетливо проявляются и при
циклопропанировании непредельных углеводородов, содержащих
в молекуле двойные связи с различной л-кислотностью. Так,
в присутствии соединений палладия региоселективно циклопро-
панируются внутрициклические двойные связи, например
f1019-! ’ PdCI2(PhCN)2,
___ -10 °C
+ ch2n2 —
RCMe
RCMe
RCMe
R = цикло-С3Н5
1 . 2 (4-6%) (90%)
При взаимодействии диазометана со смесью норборнадиена
и 4-винилциклогексена в присутствии солей палладия сначала
циклопропанируется исключительно норборнадиен,^ независимо
от соотношения исходных диенов, и лишь после завершения ис-
черпывающего циклопропанирования норборнадиена начинается
Циклопропанирование 4-винилциклогексена, преимущественно
по концевой винильной группе [1019].
Циклопропанирование диазометаном на PdCl2(PhCN)2 ди-
еновых углеводородов с концевой двойной связью — г{цс-пента-
Диена-1,3 (13) (см. с. 165), изопрена и 4-винилциклогексена —
также проходит региоселективно [1019]:
175
При реакции 4-винилциклогексена с диазометаном в присут-
ствии солей меди выходы соединений (37), (38) и (39) состав-
ляют соответственно 10, 13 и 3% [1024].
При использовании солей меди высокие препаративные вы-
ходы циклопропановых аддуктов зачастую достигаются приме-
нением большого избытка диазометана, тогда как в случае
палладиевых катализаторов увеличение избытка диазометана
сверх указанных соотношений [1 : (1—3)] обычно практически
не сказывается на выходах продуктов. Диазометан при этом
разлагается с образованием легколетучих соединений, в основ-
ном циклопропана, а реакционная смесь постепенно чернеет
вследствие выделения металлического палладия.
Ограничением в применении палладиевых катализаторов яв-
ляется их высокая чувствительность к стерическому экраниро-
ванию циклопропанируемой двойной связи, что, как уже отмеча-
лось выше, связано с первоначальным образованием л-олефино-
вых комплексов палладия, которые далее реагируют с
диазометаном. Именно поэтому Pd-катализаторы наиболее перс-
пективны в реакции циклопропанирования таких олефинов,
двойные связи которых способны к комплексообразованию
с металлами, в первую очередь терминальных и напряженных
циклических олефинов.
Большинство алкил- и алкенилзамещенных диазометанов
в фотохимических или термических (термокаталитических) ус-
ловиях также претерпевают карбенный распад. Вместе с тем
образующиеся при этом алкил- и алкенилкарбены более склон-
ны к внутримолекулярным реакциям С—Н-внедрения и карбен-
олефиновой изомеризации, чем к межмолекулярному циклопро-
панированию непредельных акцепторов [8]. В связи с этим при-
менение таких диазосоединений обычно ограничивается генери-
рованием из них соответствующих карбенов для физико-химиче-
ских исследований (см. гл. 2), а также синтезом различных вы-
соконапряженных структур, включая циклические аллены и ан-
тибредтовские олефины (полициклические соединения с двойной
связью в голове моста) [528]. Внутримолекулярное С—Н-внед-
рение винилкарбенов представляет собой один из общих методов
176
синтеза циклопропенов [1079]:
\ или hv
RCH = CHCR1 —V- » RCH = CHCR1
n2
R, R’ = Alk
Для синтеза алкил- и алкенилциклопропанов из соответст-
вующих олефинов эти диазосоединения практически не исполь-
зуют; пожалуй, единственным исключением является примене-
ние диазоэтана для синтеза метилциклопропановых производ-
ных на Pd-катализаторах [1080]. Определенное значение в син-
тезе имеют также арилдиазометаны [8], производные диазоцик-
лопентадиена [7, 8, 240] и других диазоциклоалкадиенов, в пер-
вую очередь замещенные диазоциклогексадиеноны [1081, 1082].
Наиболее перспективны для синтеза карбонилзамещенные
диазометаны, в первую очередь соответствующие диазоацетаты.
4.1.2. Алкилдиазоацетаты
Алкилдиазоацетаты легко деазотируются с образованием алкок-
сикарбонилкарбенов, которые, присоединяясь к двойным связям
непредельных соединений, дают производные циклопропанкар-
боновой кислоты обычно с достаточно высокими выходами:
„ ,А ч CHCOOR
N2CHCOOR :CHCOOR х/ V
-|\12 ~
Обычно используют каталитическое (термокаталитическое)
разложение алкилдиазоацетатов, причем в качестве катализа-
торов наиболее широко применяют соединения меди и, в послед-
нее время, соединения родия (II). При использовании в качестве
катализаторов солей меди, в частности C11SO4, реакцию прово-
дят при 90—ПО °C в избытке олефина (мольное соотношение
алкен: диазоацетат обычно »8: 1) [1083, 1084]. Дальнейшее
увеличение избытка олефина практически не сказывается на
выходе эфира циклопропанкарбоновой кислоты, затрудняя лишь
его выделение [1083]. Напротив, при уменьшении избытка непре-
дельного акцептора выход циклопропановых продуктов снижа-
ется из-за заметного образования в этих условиях продуктов
формальной рекомбинации алкоксикарбонилкарбенов — эфиров
Фумаровой и малеиновой кислот [1083]:
2N?CHCOOR-----^ROOCCH = CHCOOR
2 -2N2
12—107
177
Эфиры фумаровой и малеиновой кислот в/ значительных ко-
личествах образуются и при понижении температуры реакции
до 60—65 °C [1084]. При температурах выше 110 °C в жидко-
фазных условиях увеличивается осмоление продуктов реак-
ции [1084], однако при проведении реакции с непредельными
акцепторами в газовой фазе (проточный реактор, насадка из
СиО на пемзе или кварце) оптимальной является температура
150—200 °C [1085].
Алкоксикарбонилкарбены отчетливо проявляют электро-
фильные свойства [421], что, например, наглядно видно в реак-
циях с эфирами сорбиновой кислоты, где образуется исключи-
тельно продукт присоединения карбена по наиболее нуклео-
фильной двойной связи, удаленной от сложноэфирной группы
[1086—1989]:
Me
МеСН = СНСН = ССООМе + N2CHCOOEt
-n2
—*МеНС - СНСН = ССООМе
\ / I
EtCOOCH Me
(< 70%)
При реакции этилдиазоацетата с силилзамещенными олефи-
нами в присутствии CuSO4 при температуре кипения олефина
[1090] основными продуктами являются трамс-изомеры соответ-
ствующих эфиров циклопропанкарбоновых кислот:
CHR
А
RCH = СН2 + N2CHCOOEt--»-Н2С - CHCOOEt
R = SiMe, CH2SiMe3, (CH2)2SiMe3
Общий
выход, %
SiMe3 >2
CH2SiMe3 66,5
(CH2)2SiMe3 54,9
С5Нц-н 50
транс : цис
69,3:30,7
61,0:39,0
65,5:34,5
58,9:41,1
В случае винилтриметилсилана (R = SiMe3) дезактивирую-
щее влияние силильной группы связано с dn—ря-сопряжением,
типичным для соединений кремния. При отделении силильной
группы от реакционного центра (двойной связи) группами СН2
178
наблюдается некоторое активирующее действие группы SiM.es
вследствие ее положительного индуктивного эффекта.
Арил- и алкилвиниловые эфиры, двойная связь которых об-
ладает повышенной нуклеофильностью, легко и с высокими
выходами дают соответствующие производные циклопропанкар-
боновой кислоты в реакциях с алкилдиазоацетатами и другими
производными диазоуксусной кислоты в присутствии меди и ее
соединений [1091 —1093]. В частности, таким путем из винил-
фенилового эфира получен ряд феноксипроизводных циклопро-
панкарбоновой кислоты [1091]; аналогично из винилциклогекси-
лового эфира и этилдиазоацетата получен циклопропановый
аддукт с выходом 97% [1091]:
CHOPh
PhOCH = СН2 + N2CHCOX -£JL^h?C-'cHCOX
-N,
X = OEt, nhnh2, n3, nh2, nhoh
OCH = CH2 + N2CHCOOEt
Си, Д
-n2
^-COOEt
Полученные реакцией арил (алкил) виниловых эфиров с про-
изводными диазоуксусной кислоты циклопропановые аддукты
оказались удобными синтонами в синтезе витамина А [1093]
и других биологически активных соединений [1091 —1093].
Реакции алкилдиазоацетатов с олефинами в присутствии ме-
таллической меди или ее солей протекают стереоспецифично
как ^^-присоединение. При этом образуются стерически менее
затрудненные изомеры эфиров циклопропанкарбоновой кислоты
[10, 1094—1096]. Аналогичным образом из ненасыщенных цик-
лических соединений в присутствии меди образуются соответ-
ствующие циклопропановые моноаддукты с выходами 30—85%,
причем преимущественно в виде экзо-изомеров (табл. 4.1):
4- N2CHCOOF>
СООР
Высокая стереоселективность циклоприсоединения алкокси-
карбонилкарбенов проявляется и в реакции с ненасыщенными
кетонами. Так, 2-метилгептен-2-он-6 в реакции с этилдиазоаце-
татом дает исключительно соответствующий циклопропановый
12‘ 179
Таблица 4.1, Циклопропанирование некоторых
циклоолефинов этилдиазоацетатом в присутствии меди
Исходное соединение Выход цикло- пропановых моноаддуктов, % экзо : эндо Библиогр. ссылки
Циклопентен 30 6:1 [162, 163,
1097, 1098]
Циклогексен 66 16:1 [1099]
Циклогептен 67 5:1 [1098]
Циклопентадиен — 5:1 [1100]
Циклогексадиен-1,3 — 5:1 [1Ю1]
Циклогексадиен-1,4 85 7:1 [1101]
60:1 [1102]
Циклогептатриен — 5:1 [1098, 1103]
Норборнадиен — 2:1 [1104]
аддукт трамс-строения (выход 35%) [1105]; аналогично реаги-
рует винилметилкетон, образуя с выходом 34% этиловый эфир
тра«с-2-ацетилциклопропанкарбоновой кислоты [1106]:
Ме2С = СН(СН2)2СОМе + N2CHCOOEt—
СМе2
Н (СН2)2СОМе
с-с'
EtOOCZ ЧН
СН2 = СНСОМе + N2CHCOOEt
CUj
Н\ /С\Н2 С0Ме
/С'<
EtOOC Н
Циклопропановые аддукты этоксикарбонилкарбена доста-
точно легко образуются и из разветвленных олефинов [1107].
Так, например, 2,3,3-триметилбутен-1 в реакции с этилдиазоаце-
татом при 100 °C в присутствии медной бронзы или сульфата
меди дает этиловый эфир 2-трет-бутил-2-метилциклопропанкар-
•боновой кислоты с выходом 63% [1107]:
Me
1 Си или CuSO4, 100 °C
МезСС = СН2 + N2CHCOOEt ------------4-----*•
Me - С - СМе3
Н2С —CHCOOEt
180
Аналогично реагирует 4-пропилгептен-З, образуя этиловый
эфир 2,2-дипропил-^\этилциклопропанкарбоновой кислоты (вы-
ход 56%) [1107]. В то же время этиловый эфир 2,2-диизопро-
пил-3,3-диметилциклопропанкарбоновой кислоты образуется из
соответствующего тетразамещенного этилена с выходом лишь
14% [1107]:
МегС = С(Рг-изо)2 + N2CHCOOEt —-^00
-n2
----МегС—-С (Рг-изо)г
CHCOOEt
При взаимодействии алкилди'азоацетатов с диенами реакцию
можно остановить на стадии моноприсоединения. Так, бута-
диен- 1,3 реагирует с этилдиазоацетатом при 100 °C в отсутствие
катализатора или при 20 °C в присутствии CuSC>4 с образова-
нием этилового эфира 2-винилциклопропанкарбоновой кислоты
с выходом 20—40% [1108]. При этом в случае термической
реакции соотношение цис- и трамс-изомеров составляет 45:55
[1054, 1108, 1109], а в термокаталитическом варианте оно равно
40:60 [1108]. Проводя эту реакцию с 6-кратным мольным из-
бытком бутадиена в автоклаве при 100—НО °C, удается повы-
сить выход продукта до 80% (в расчете на этилдиазоацетат)
[1054, 1109]:
СН2 = СНСН = СН2 + N2CHCOOEt а
СН2
/\
---* СН2 = СН - НС - СН - COOEt
Алкилдиазоацетаты и их функциональные замещенные обра-
зуют циклопропановые моноаддукты и в реакции с 2,5-диметил-
гексадиеном-2,4; при этом соотношение цис- и транс-изомеров
близко к 1:1 [1026, 1110]:
Ме2С = СНСН = СМе2 + N2CHCOOCH2CH2X Cu+^uS°4. Ю0.^С
снсоосн2сн2х
---* Ме2С - СНСН = СМе2
(=? 50%)
х = Н, Cl, Br, I, NR2, SR
181
При циклопропанировании изопрена этилдиазоацетатом об-
разуется аддукт по наиболее замещенной двойной связи — эти-
ловый эфир 2-винил-2-метилциклопропанкарбоновой кислоты,
который может подвергаться дальнейшему циклопропанирова-
нию с образованием 1-метил-2,2'-диэтоксикарбонилбициклопро-
пила [1111]:
Ме N2CHCOOEt,
I CuSO4
сн, = ссн = сн,-----------
2 2 -n2
EtOOC
COOEt
Е t О О С М е
*^><S'CH = СН2
:CHCOOEt
(< 20%)
Алкилдиазоацетаты взаимодействуют с ацетиленами, давая
соответствующие производные циклопропенкарбоновых кислот
[1112—1116]. Образующиеся в этой реакции эфиры циклопро-
пенкарбоновой-3 кислоты при нагревании свыше 100 °C изоме-
ризуются в фураны [1117—1119], причем изомеризация облег-
чается в присутствии меди [1120]. При избытке алкилдиазоаце-
тата возможно дальнейшее циклопропанирование первоначально
образующегося циклопропена с образованием производных
бицикло[1.1.0]бутана [1121, 1122]:
N2CHCOOR1, CHCOOR1
Сих / \ >100 °с
RC = CR ----------* RC = CR ------------»
-n2
I tCHCOOR1
R
R’OOC -O-COORl
R
В случае низших ацетиленов реакцию с этилдиазоацетатом
целесообразно проводить в газовой фазе в условиях проточного
реактора [1085, 1123]; реакция алкилдиазоацетатов с алленом
в аналогичных условиях представляет собой наиболее удобный
182
метод синтеза эфиров метиленциклопропанкарбоновой кислоты
[1085, 1124]: CuO/пемза, COOEt
МеС = CH + N2CHCOOEt —~17° £ Me-А . н
-N,
(35%)
СиО/пемза,
150-170 °C
СН2 = С = сн2 4- N2CHCOOR------НрС^А—COOR
-n2
R = Me,Et (45-50%)
При взаимодействии алкилдиазоацетатов с непредельными
соединениями, содержащими одновременно двойные и тройные
углерод-углеродные связи, преимущественному алкоксикарбо-
нилметиленированию подвергаются двойные связи [1125]. Так,
при реакции гексен-1-ина-4 с этилдиазоацетатом в присутствии
медной бронзы наряду с этиловыми эфирами малеиновой и фу-
маровой кислот (суммарный выход 25%) образуются продукты
циклопропанирования по двойной связи (выход 19%, соотноше-
ние цис : транс = 2^ : 76) и незначительное количество диаддук-
та [1125]. Однако в аналогичной реакции 2-метилгексен-1-ина-4
образуется смесь продуктов моноциклопропанирования как по
двойной, так и по тройной связям [1126]:
МеС = ССН.СН = CH, + N„CHCOOEt Си~бронза.^
2 2 Z _м
EtOOC - CH = CH - COOEt + МеС= ССН2
НС - CH - COOEt +
\ /
сн2
] + Me - С - С - СНО - НС - CH - COOEt
\ / 2 \ /
ЕЮОС-СН СН2
МеС = ССН„С =СН2 + N„CHCOOEt
a z -N2
Me CHOOEt CHCOOEt Me
l/\ /\ 1
—»- MeC= CCH2 - C - CH2 + MeC = C— CH2C = CH2
(34%) (11%)
При сравнении каталитической активности различных соеди-
нений меди в реакциях карбенного разложения этилдиазоацета-
та [1027] было найдено, что селективность циклопропанирова-
183
Таблица 4.2. Моноциклопропанирование некоторых непредельных
соединений этилдиазоацетатом на медных
катализаторах при 25 °C [1027]
Исходное соединение Выход продуктов, % транс : цис
CuClP(OPr-wso)3
Стирол 88 2,8
Винилэтиловый эфир 61 1,9
3,3-Диметилбутен-1 23 7,3
Циклогексен 28 6,8
1 -Метоксициклогексен 54 4,2
Изопрен 53« 1,3»
286 3,46
2-Фенилбута диен-1,3 21» 1,1»
76 3,56
2,5-Диметилгексадиен-2,4 55 2,7
CuClP(OPh)3
Стирол 84 2,5
Винилэтиловый эфир 67 2,2
Циклогексен 25 7,0
1 -Метоксициклогексен 35 4,5
Изопрен 48» 1,2»
256 3,66
2,5-Диметилгексадиен-2,4 66 2,7
Cl! (OSO2CF3) 2
Стирол 97® 1,9®
Винилэтиловый эфир 55г 2,4г
3,3-Диметилбутен-1 54 5,5
Циклогексен 80 6,8
Изопрен 38® 1,0®
19« 1,86
2-Фенилбутадиен-1,3 34“ 1,0®
14б 1,86
2,5-Диметилгексадиен-2,4 93 2,3
Медная бронза
Стирол 53в 1,9В
3,3-Диметилбутен-1 5 8,1
Циклогексен 23 7,5
Изопрен 13» 1,2»
76 4,06
2 - Фени л бута диен-1,3 22» 1,2»
86 3,26
2,5-Диметилгексадиен-2,4 34 2,7
Си(асас)2
Стирол 71® 2,6В
Винилэтиловый эфир 15 1,6
3,3-Диметилбутен-1 20в 10,4В
Циклогексен 18в 6,5В
184
Продолжение табл. 4.2
Исходное соединение | |Выход продуктов, % | транс : цис
1-Метоксициклогексен 44 4,8
Изопрен 37а 1 ,За
186 3,3б
2-Фенилбутадиен-1,3 31а 1,0а
цб 3,16
2,5-Диметилгексадиен-2,4 76в 1,8В
аПо связи 1,2. бПо связи 3,4. вПри 60 °C. г При 0 °C.
ния незначительно зависит от валентного состояния металла
и его окружения (табл. 4.2). Среди использованных катализа-
торов наивысшую каталитическую активность проявил трифлат
меди(II) (Tf = CFsSO2). Однако этот катализатор не является
универсальным; в частности, он неприменим для циклопропани-
рования виниловых эфиров [1027], которые при этом полимери-
зуются, а также дают другие побочные продукты. Ацетилацето-
нат меди (II) и медная бронза являются наименее активными
катализаторами в реакции циклопропанирования этилдиазоаце-
татом. Препаративные выходы циклопропановых аддуктов в
этих случаях достигаются лишь при проведении реакции при
60 °C [1027] (см. табл. 4.2).
Использование в качестве катализаторов оптически актив-
ных хелатных комплексов меди, например (40) — (42), позволи-
ло получить соответствующие эфиры циклопропанкарбоновых
кислот с высокой оптической чистотой [1016—1018] (табл. 4.3).
Таблица 4.3. Циклопропанирование некоторых алкенов диазоацетатами
в присутствии комплекса (42) [1%(мол.), С1(СН2)2С1, 23 °C] [1016]
ch2=chr + n2chcoor1 -->
н
COOR1
(43) (44)
R RJ Общий выход цик- лопропанов, % транс : цис Оптический выход, %
для I (43) 1 1 для 1 (44)
Ph Et 65 78:22 85 68
Ph Bu-трет 60 84:16 93 92
Ph (—) -Ментил 65—75 85:15 91 90
Ph (4-) -Ментил 60—70 82:18 97 95
сн=сн2 То же 60 63:37 97 95
СзНц-и » 25—30 82:18 92 92
185
При этом энантиоселективность катализаторов возрастает в ря-
ду (40) <(41) <(42).
(40) R=COOMe
(41) R= CH2OSiMe2Bu-Tper
(42) R= CMe2OH
Среди соединений других переходных металлов, катализи-
рующих циклопропанирование олефинов алкилдиазоацетатами,
значительный интерес представляют соединения палладия и осо-
бенно родия, позволяющие проводить реакцию при комнатных
температурах. Сравнительное исследование катализаторов кар-
бенного разложения алкилдиазоацетатов, проведенное на при-
мере реакции этилдиазоацетата со стиролом (количество ката-
лизатора <1%; 22°C) [1014], показало высокую эффектив-
ность соединений палладия (II) и родия (II), в частности их аце-
татов, в этих превращениях:
Катализатор Выход циклопропанового аддукта, % транс : цис
PdCl2 70 —
Pd(OAc)2 98 2,0
PdCl2(PhCN)2 65 2,3
Pd(PPh3)4 57 2,2
Pd/C 0 —
Cu(acac)2 RhCl3-3H2O 65 7 2,1
RhCl(PPh3)3 12 —
Rh(OAc)2 92 1,5
Rh(OOCBu-Tper)2 60 1,5
Rh(OOCBu)2 95 1,3
Rh(OOCCF3)2 66 0,9
Mo2(OAc)4 5 —
Ru2(OAc)4C1 38 1,8
В то же время, как и в случае диазометана (см. разд. 4.1.1),
соединения палладия (II) эффективно катализируют циклопро-
панирование диазоацетатами лишь напряженных олефинов, ко-
торые легко образуют комплексы^ соединениями палладия(II),
и мало активны в реакциях с ациклическими олефинами [1014]
(табл. 4.4). Соединения родия(II) одинаково эффективны
в реакциях алкилдиазоацетатов как с ациклическими, так и с
циклическими олефинами [1014].
186
Таблица 4.4. Циклопропанирование некоторых ненасыщенных соединений
этилдиазоацетатами на различных катализаторах при 22 °C [1014]
Исходное соединение Выход циклопропанового моноаддукта (в %) на катализаторе
Pd(OAc)3 Cu(OTfh Rh(OAch
ццс-Бутен-2 24а — 54а
тргшс-Бутен-2 21а — —
Гексен-1 30а 36а 86а
ф/с-Октен-2 5а 40а 65а
траяс-Октен-2 2а 14а 24а
цис-Октен-4 12а 8а 7а
1,2-Диметилбутен-2 5 30 70
Циклопентен 60 60 95
Циклогексен 21 54 88
Стирол 98 80 90
Инден 20 55 71
Норборнен 87 95 95
Винилацетат 5 22 77
Винилэтиловый эфир 42 Об 85
Норборнадиен 95 47 88
аРеакция с N2CHCOOMe.
б Происходит полимеризация
исходных реагентов.
При циклопропанировании алкилдиазоацетатами в присутст-
вии соединений родия (II), палладия (II) и меди(1) как ацикли-
ческих, так и циклических непредельных соединений с электро-
нодонорными заместителями при кратных связях наблюдаются
аналогичные закономерности [1026] (табл. 4.5). Из использо-
Таблица 4.5. Моноциклопроланирование некоторых непредельных
соединений на различных катализаторах при 25 °C [1026]
Исходное соединение Выход циклопропанового моноаддукта^ (в %) на катализаторе
Rhe (СО) не* PdCl2(PhCN)2 cud- Р(ОРг-изо)и
Винилэтиловый эфир 62 43 61
Бутилвиниловый эфир 69 34 51
2-Метоксипропен 72 66 67
Дигидропиран 82 41 18
3,4-Дигидро-2-этокси-2Я-пиран 70 33 42
1 -Метоксициклогексен 59 39 54
Циклогексен 88 31 28
Стирол 87 52 80
2>5-Диметилгексадиен-2,4 87 20а 55а
При использовании Pd(OAc)2 и Си(асас)2 выход аддукта снижается.
187
Таблица 4.6. Моноциклопропанирование непредельных соединении
этилдиазоацетатом при 25 °C на различных катализаторах [1027]
Исходное соединение Выход про- дукта3, % транс : циа
Rh(OAc),
Аллилбромид 55 1,1
Аллилхлорид 90 1,2
Стирол 93 1,6
Винилэтиловый эфир 88 1 Л
Бутилвиниловый эфир 84 1,7
Винилфениловый эфир 92 1,4
Винилацетат 59 1,6
Гексен-1 95 1,5
З-Метилбутен-1 58 2,0
3,3-Диметилбутен-1 87 4,2
2-Фенилбутадиен-1,3 25б 1,5
56в 1,0
2-Хлорбутадиен-1,3 226 1,4
54в 1,1
Изопрен 366 1,8
57в 1,1
2-трет-Бутилбутадиен-1,3 456 3,7
25в 1,3
1 -Метоксибутадиен-1,3 806 1,4
10в 0,73
транс-1 -Хлорбутадиен-1,3 73(73) 1,6
цнс-1-Хлорбутадиен-1,3 49(49) 1,6
транс-1 -Фенилбутадиен-1,3 886 1,6
2В 1,7
ф/с-1-Фенилбутадиен-1,3 556 1,6
7В 1,7
траноПентадиен-1,3 54(61) 1,6
транс-5,5-Диметилгексадиен-1,3 54(54) 1,6
2-Метоксипропен 78 1,0
3,3-Диметил-2-метоксибутен-1 70 0,71
2-Метоксибутадиен-1,3 88(88) 0,80
1-Метоксистирол 88 0,96
2,З-Д иметоксибутадиен-1,3 76 0,85
транс-1 -Бромпропен 25 2,2
Циклопентен 96 2,1
Циклогексен 90 3,8
З-Оксациклогексен 91 6,5
цас-2-Метоксистирол 60 2,0
2-Метилбутен-2 97 1,5
2,5-Диметилгексадиен-2,4 81 1,8
1 -Метилциклогексен 80 2,7
1 -Метоксициклогексен 78 2,5
Rh6 (СО) 16
Аллилбромид 30 1,1
Аллилхлорид 24 1,2
Стирол 86 1,7
Винилэтиловый эфир 62 1,7
Бутилвиниловый эфир 69 1,8
3,3-Днметилбутен-1 42 4,5
188
Продолжение табл. 4.&
W— ’ Исходное соединение Выход про- дукта3, % транс : цис
2-Хлорбутадиен-1,3 24б 1,76
56® 1,1®
2-Фенилбутадиен-1,3 20(71) 1,7
Изопрен 276 1,7б
43в 1,1в
2-трет-Бутилбутадиен-1,3 5б 3,86
4® 1,4В
1 -Метоксибутадиен-1,3 58(63) 1,3
1 -Хлорбутадиен-1,3 21(21) 2,0 1 л
2-Метоксипропен 72 1,1) А 70
3,3-Диметил-2-метоксибутен-1 83 0, /Z
2-Метоксибута диен-1,3 63(63) 0,82
Циклогексен 88 3,9
З-Оксациклогексен 82 6,8
2,5-Диметилгексадиен-2,4 87 1,9
1-Метоксициклогексен СиС1Р(ОРг-«зо)з 59 2,6
Аллилбромид 7 1,8
Аллилхлорид 50 1 ,з
Стирол 88 2,8
Винилэтиловый эфир 61 1,9
Бутилвиниловый эфир 51 2,0
3,3-Диметилбутен-1 23 7,3
2-Хлорбутадиен-1,3 5б 28в 2,1б 1,3В
2-Фенилбутадиен-1,3 Изопрен 53в 2,5 3,46 1,3«
2-трег-Бутилбута диен-1,3 8б 12в 6,4б 1,3В
1 -Метоксибутадиен-1,3 48(57) 1,5
1-Хлорбутадиен-1,3 17(17) 2,6
2-Метоксипропен 67 1,1
3,3-Диметил-2-метоксибутен-1 7 1,2
2-Метоксибутадиен-1,3 38(38) 1,0
Циклогексен 28 6,8
З-Оксациклогексен 18 6,3
2,5-Диметилгексадиен-2,4 55 2,7
1 -Метоксициклогексен PdCl2(PhCN)2 54 4,2
Стирол 52 1,6
Винилэтиловый эфир 43 1,5
Бутилвиниловый эфир 34 1,6
3,3-Диметилбутен-1 34 2,5
2-Хлорбутадиен-1,3 <8 —
2(4)» 0,93»
2-Фенилбутадиен-1,3 5(Ю) 1,4
189
Продолжение табл. 4.6
Исходное соединение Выход про- дукта3, % транс : цис
Изопрен 166 1,06
8В 0,9в
2-трет- Бутилбута диен-1,3 46 2,26
2В 0,7®
1 -Метоксибутадиен-1,3 16(22) 1,2
1-Хлорбутадиен-1,3 <8 —
2-Метоксипропен 66 0,93
3,3-Диметил-2-метоксибутен-1 28 0,61
2-Метоксибута диен-1,3 12(12) 0,71
Циклогексен 31 2,2
З-Оксациклогексен 41 3,8
2,5-Диметиллгексадиен-2,4 20 2,3
1 -Метоксициклогексен 39 1,5
аВ скобках приведен общий выход циклопропановых продуктов. ^По связи 1,2.
в По связи 3,4.
ванных катализаторов лишь кластерное соединение родия(П) —
гексадекакарбонилгексародий — во всех случаях проявляет вы-
сокую активность: выходы циклопропановых аддуктов высоки
и практически не зависят от строения исходного непредельного
соединения [1026].
Соединения родия успешно применены также для циклопро-
панирования аллилгалогенидов этилдиазоацетатом (общий вы-
ход продуктов реакции 76—95%) [477]:
N2CHCOOEt, COOEt СНСН2Х
СН„ = СНСН-Х Rh<0AC--j> СН? = СНСН2СНХ + Н2С - CHCOOEt
2 2 _N 2 г.
(45) (46)
X = Br, CI
(45): (46) = 28:72 (X = Вг) и 5:95 (X = CI)
Изучение стереоселективности циклопропанирования олефи-
нов этилдиазоацетатом в присутствии соединений Rh, Си и Pd
(табл. 4.6) показало, что Cu-катализаторы в общем проявляют
несколько большую транс (анти)-селективность, чем соединения
родия(II) и палладия(II) [1025, 1027]. Тем не менее соедине-
ния родия(II), в частности его ацетат, являются исключительно
эффективными катализаторами реакций карбенного распада
диазосоединений с переносом карбенного фрагмента на крат-
190
ную связь [1025, 1127—1129]. Именно использование таких ка-
тализаторов в реакциях алкилдиазоацетатов с олефинами, про-
водимых при комнатной температуре, позволило получить раз-
личные эфиры циклопропанкарбоновой кислоты [1027] (см.
табл. 4.6). При этом ацетат родия (II) не восстанавливается
диазосоединениями, не образует л-комплексов с олефинами и не
обменивает карбоксилат-ион в обычно принятых условиях ката-
лиза; даже при мольном отношении катализатора к этилдиазо-
ацетату, равном всего 0,0005, выход продукта циклопропаниро-
вания 1-метоксициклогексена составляет 80% [ИЗО]:
аОМе ОМе
+ IXLCHCnOFt рЬ(ОАс)2(5-10-5моль), Et?O
2 —COOEt
(0,1 моль) (0,1 моль)
Таким образом, хотя ранее в реакциях с алкилдиазоацетата-
ми использовали [420, 1131—1133] гетерогенные катализаторы
на основе медной бронзы или C11SO4, а также гомогенные ката-
лизаторы— хелаты меди [1134] или растворимые комплексы
типа CuClP(OR)3 [1023], в настоящее время применяют и дру-
гие соединения переходных металлов, прежде всего Rh(OAc)2
[1025], Cu(OTf)2 [1011], Pd(OAc)2 [1072] и Rh6(CO)i6 [1026].
Следует отметить, что определенное значение в синтезе имеют
и функционально замещенные диазоацетаты, позволяющие в ря-
де случаев направленно получать соответствующие производные
эфиров циклопропанкарбоновых кислот. Взаимодействие заме-
щенных диазоацетатов с диенами может быть использовано для
синтеза производных пиретроидных кислот (см. гл. 6); в ряде
случаев определенный интерес представляют и внутримолеку-
лярные реакции функционально замещенных арилоксикарбонил-
и алкоксикарбонилкарбенов. Так, аллилдиазоацетат в отсутст-
вие непредельного акцептора образует продукт внутримолеку-
лярного циклопропанирования — бициклический лактон, а также-
димер соответствующего карбена [1135]:
Си-кат,
N,CHCOOCH„CH = СН ---------э-’.СНСООСН2СН = сн2—
2 z г -N2
—* сн2 = снсн2ооссн = снсоосн2сн = сн2 +
(20-60%)
(40-50%)
191
Аналогичным образом, высшие алкилдиазоацетаты в услови-
ях карбенного разложения дают соответствующие лактоны по
реакции внутримолекулярного С—Н-внедрения [1136]:
N„CHCOOCH,CHRR1 кваРц- 200^ :CHCOOCH„CHRR1 —
2 2
R, Rl = Aik
4.1.3. Другие диазокарбонильные соединения
Диазомалоновые эфиры. Диазомалоновые эфиры обычно терми-
чески стабильны; в присутствии катализаторов они разлагаются
с умеренной скоростью, образуя в реакциях с олефинами как
циклопропановые аддукты, так и продукты внедрения в орди-
нарные связи исходного непредельного акцептора. Так, при
взаимодействии алкилдиазомалонатов с алкенами основными
продуктами являются циклопропановые производные, продукты
внедрения в аллильные связи С—Н и формальный димер карбе-
на — тетракис (алкоксикарбонил)этилен. Кроме того, обычно
образуется также незначительное количество соответствующего
тетракис(алкоксикарбонил)этана [10, 1137]:
I I
)с = С - СН - + N2C(COOR)2 кат" а3
-n2
C(COO.R)2
/ \
)с-с-сн- +
I I
+ )с = С - С - CH(COOR)2 + (ROOC)2C = C(COOR)2 +
+ (ROOC)2CH - CH(COOR)2
Следует отметить, что в реакциях алкилдиазомалонатов
с олефинами в присутствии соединений меди выход циклопро-
пановых продуктов существенно зависит от количества катали-
затора и характера применяемого растворителя [10]: макси-
мальные выходы циклопропанов достигаются при использова-
нии 0,1—0,2 ммоль СиХР(ОМе)3 на 1 моль диазомалоната
и проведении реакции без растворителя или в гексафторбензо-
ле [10]. В реакции диметилдиазомалоната с цис- и т/?а«с-гексе-
нами-2 стереоселективно образуются соответствующие цикло-
192
пропановые аддукты с выходами 81—95 и 41—76% соответст-
венно [1138]:
С(СООМе)2
МеСН = CHPr N*C(COOMe)2,CuXP(OMe)3 менс'-'сНРг +
-n2
(47) цис
(81-95%)
СН(СООМе)2 СН(СООМе)2
+ МеСН = С-Рг + МеС = СНРг + (МеООС)2С = С(СООМе)2
^48) (49)у (50) (4-6%)
(0,5-12%)
N2C(COOMe)2,
МеСН = СНРг — — —-MJ^rpaHc-(47) + (48) + (49) + (50)
-N2 *--'
транс (41-76%) (6-35%) (9-23%)
X = Ct, Br
Циклоалкены в реакции с диметил- и ди-трет-бутилдиазома-
лонатами также дают соответствующие циклопропановые про-
дукты с выходами до 80% [10, 1138]:
[ /сн2)п + N2C(COOR)2
Cu®[Cu, CuCl,
CuCIP(OMe)3]
-N2
ROOC
ROOC
\ A-CH(COOR)2 + (ROOC)2C = C(COOR')2
(CH2)n
n = 2-4; R = Me, Bu-rper
Аналогично реагируют с диалкилдиазомалонатами и алкил-
замещенные циклогексены [10]. Циклические диены — норбор-
надиен (36) и циклооктадиен-1,3 в реакциях с диметил- и ди-
трет-бутилдиазомалонатами образуют соответствующие моно-
13-107 193
аддукты, причем в первом случае в виде смеси экзо- и эндо-изо-
меров [10]:
( +N2C(COOR)2 с.иХр(°Ме)з / V^-COOR
\_/ -N2 \_^<\COOR
R = Me, Bu-rperJX = Hal
В то же время реакция диалкилдиазомалонатов с циклогек-
садиенами-1,3 и -1,4 приводит в основном к бензолу; в незначи-
тельном количестве образуются циклопропановые аддукты [10].
Побочные реакции окисления протекают также при использо-
вании в качестве катализатора медной бронзы [10]; однако
в реакции диметилдиазомалоната с 1-метилциклогекса дие-
ном-1,4 с хорошими выходами образуются циклопропа-
ны [1102]:
+ N2C(COOMe)2
Си-кат.
-N2
Me
МеООС
МеООС
(основной продукт)
СООМе
СООМе
Подобно алкилдиазоацетатам диметилдиазомалонат легко
реагирует с виниловыми эфирами, образуя с высокими выхода-
ми соответствующие циклопропаны [1139], например:
CHOEt + N2C(COOMe)2
Си-кат.
-N2*"
/—\ CHOEt
\ Xj
7 С(СООМе)2
(85%)
Аллилдиазомалонаты претерпевают внутримолекулярное
циклопропанирование, приводящее, как и в случае соответст-
194
вуклцих диазоацетатов, к соответствующим лактонам [10,
1140]:
COOEt
N2CCOOCR2CH = CPU -Ctl'Ka^
2 -n2
R1 R1
R, R1 = H, Me
Диазокетоны и диазоальдегиды. Простейшим представите-
лем этого класса соединений является диазоацетон [10, 1141,
1142]. В реакциях с непредельными соединениями (стирол, цик-
лоалкены, производные винилового эфира) в присутствии меди
и ее галогенидов диазоацетон образует ацетилциклопропаны
с препаративными выходами [10, 1141, 1142], например:
СН2
PhCH = СН2 + N2CHCOMe Ph - НС -СН - СОМе
-n2
Соответствующие циклопропановые аддукты в реакциях с
непредельными соединениями дает и диазоацетофенон [10, 1143,
1144]. На примере взаимодействия его с циклогексеном было
установлено, что наибольшие выходы 7-бензоилноркарана до-
стигаются при использовании медных катализаторов, в частно-
сти CuSO4 [1143] при 28 °C:
О+ N2CHC0Ph-!^
2 -n2
COPh
Катализатор
Cu(dpm)26
Ni(cpd)2r
hv
Катализатор Выход, %
CtlSO4 62
CuO 55а
CuCl2 44
CuCl 29
Выход, %
19B
5
10—123
a При 60 °G. $ dpm— дипивалоилметил.
д Образуется >70% ацетофенона.
в При 28—60 °C. rcpd — циклопе^тадиен.
Реакция диазоацетофенона с цис- и тракс-бутенами-2 в при-
сутствии C11SO4 проходит стереоселективно и дает соответству-
ющие циклопропановые аддукты с выходами 30—45%, тогда
как при фотолизе образуется смесь всех возможных изомеров
с общим выходом 8—12% [1143]; аналогично взаимодействуют
с диазоацетофеноном и 1,3-диены, образуя соответствующие мо-
ноциклопропановые аддукты с выходами 30—40% [1144]:
CHCOPh
МеСН = СНМе + N2CHCOPh --uS°4 МеНС - СНМе
-N,
цис (транс)
. CuSO4
С = С( + NnCHCOPh -----*
/С = с( “N2
цис (транс)
CHCOPh
В качестве катализатора карбенного разложения диазоаце-
тофенона применяют также ацетилацетонат меди(II) [1145];
при этом в реакции с олефинами наряду с циклопропановымп
аддуктами образуются продукты формальной рекомбинации
карбенов и некоторые другие побочные продукты [1145]:
CHCOPh
)с = с( +N2CHCOPh 2g.ca.c.k )cZ-Xc( +(PhCOCH)2 +
Исходное соединение
Циклогексен
^wc-Стильбен
гракс-Стильбен
Винилацетат
Выход циклопропанов, %
51а
19а
56
49,7а, (8,3)в
аци5-анты-Изомер. ^ураяс-Изомер. вцмс-син-Изомер.
Карбенный распад диазоальдегидов изучен мало. Известно
лишь, что диазоацетальдегид в реакции с 2,3-диметилбутеном-2
в присутствии ацетилацетоната меди (II) дает циклопропановый
аддукт с выходом 25% [1146]:
Ме?С = СМе? + N2CHCHO Cu(acac^ Ме2С - СМе2
z -М2 \ /
сн ~ сно
196
Из приведенных в настоящем разделе данных видно, что ряд
диазосоединений (диазометан, алкилдиазоацетаты, диалкилдиа-
зомалонаты и некоторые другие диазокарбонильные соединения)
являются не только достаточно общими источниками соответ-
ствующих карбенов, но и весьма эффективными циклопропани-
рующими реагентами. Использование палладиевых и родиевых
катализаторов позволяет проводить циклопропанирование в мяг-
ких условиях, при низких температурах, хотя возможности уп-
равления стерео- и региоселективностью этих реакций, видимо,
далеко не исчерпаны. Применение комплексных катализаторов
с хиральными лигандами позволяет получать оптически актив-
ные циклопропаны. Дальнейших успехов следует ожидать и в
расширении круга синтетических полезных диазосоединений,
в первую очередь функционально замещенных.
4.2. МЕТИЛЕНИРОВАНИЕ НЕПРЕДЕЛЬНЫХ СОЕДИНЕНИЙ
ПО СИММОНСУ—СМИТУ
Одним из наиболее распространенных лабораторных методов
прямого циклопропанирования олефинов и их производных яв-
ляется реакция Симмонса — Смита [1147]—взаимодействие
непредельных соединений с дииодметаном и активированной
цинковой пылью (чаще всего с цинк-медной парой):
СН2
CH2l2-*[IZnCH2l] )с"с(
Хотя скорость взаимодействия CH2I2 с цинком существенно
зависит от растворителя, уменьшаясь в ряду: диметоксиэтан>
>тетрагидрофуран>эфир [739], тем не менее последний обес-
печивает наибольшую селективность процесса и является наи-
лучшим растворителем. С большим успехом здесь также мож-
но использовать и диметоксиэтан, в то время как тетрагидро-
фуран дает гораздо худшие результаты. Реакцию обычно
проводят кипячением смеси компонентов в эфире, при этом оп-
тимальным является соотношение (мольное) олефин : CH2I2: Zn :
эфир, равное 1 : 1 : 1 :3 [1148—1150]. В этих условиях выходы
соответствующих циклопропанов в зависимости от природы ис-
ходного олефина могут изменяться в широких пределах: от не-
скольких процентов до почти 100%. Если исходное непредельное
соединение нестабильно в присутствии Z11I2 как кислоты Льюи-
са, то в реакцию вводят 1 экв диметоксиэтана, связывающего
Znl2 в нерастворимый комплекс. Для проведения реакции необ-
ходимы абсолютированные растворители и реагенты. Промежу-
точным продуктом реакции является металлорганическое соеди-
197
нение IZnCH2I, поэтому к исходным соединениям предъявляется
ряд вполне определенных требований (подробнее см. [739,
1147]). Поскольку в одном процессе совмещены стадии получе-
ния металлорганического соединения и циклопропанирования,
выходы циклопропановых продуктов зависят от качества цин-
ка, что в свою очередь является одной из причин недостаточно
хорошей воспроизводимости результатов. Тем не менее, процесс
очень универсален. Основные закономерности реакции Сим-
монса — Смита описаны в фундаментальном обзоре [739] и ря-
де монографий, посвященных химии карбенов и циклопропанов
(см., например, [7, 8, 13, 1008]).
В настоящее время известны также многочисленные моди-
фикации реакции, относящиеся как к методам активации цинка
и выделения продуктов реакции [1050, 1151—1153], так и к
созданию гомогенных вариантов процесса, обеспечивающих бо-
лее высокие и устойчивые выходы циклопропанов. Наиболее
часто используют разложение диазометана в присутствии солей
цинка, например Znl2 [740, 1154], а также методику с приме-
нением цинкорганических соединений, позволяющую получать
с высокими выходами и алкилциклопропаны [1155]:
СН2
)с = с( + CH2N2 + Znl2T^ )с - с(
CHR
)с = с( + RCHI2 + Et2Zn—>^C -Хс(
R = Н, Aik
В условиях обычной методики проведения реакции Симмон-
са— Смита получить метилциклопропан удалось лишь в слу-
чае триметилсилилоксипроизводных циклогексена, обладающих
повышенной нуклеофильностью двойной связи [1156]:
^x.OSiMejj
OSiMe3
+ МеСН12
Zn/Cu
OSiMe3,
>— Me
OSiMe3
(76%)
Разработан также вариант реакции с использованием вместо
цинка порошка меди [1157], позволяющий в некоторых случаях
получать циклопропановые углеводороды с выходами до 90%.
198
При этом вместо дииодметана можно с успехом использовать
бромиод- и иодхлорметаны [1157]. Использование порошка ме-
ди позволяет получать также алкил- [1158], алкоксикарбонил-
[1157] и бензоилзамещенные [1159] циклопропаны:
СНМе
МеСН12,
Си
75 °C
ROOCCHBr2,
Си
---------
65-120 °C
CHCOOR
(11-32%)
(20-50%)
100 °C
PhCOCHBr2,
Си
CHCOPh
(2-12%)
Однако большинство предложенных модификаций реакции
Симмонса — Смита связано с существенным усложнением мето-
дики, что оправданно лишь в исключительных случаях. По этой
Причине до настоящего времени в основном используют ее пер-
воначальный вариант с возможными небольшими изменениями,
относящимися к получению цинк-медной пары и выделению
продуктов.
Как и в большинстве других карбенных процессов, в реакции
Симмонса — Смита в качестве непредельных соединений чаще
всего используют непредельные углеводороды. В сопоставимых
условиях выходы циклопропанов в заметной степени зависят от
природы олефина, возрастая в ряду [739]:
СН2 = CH2<RCH = CH2<RCH = CHR<R2C = CR2<R2C =CHR<
<R2C = CH2
R = Me, Et и т.п.
Такая зависимость свидетельствует об электрофильной при-
роде метиленирующего агента. При этом метиленирование оле-
финов состава С5 и более приводит к соответствующим цикло-
пропанам с выходами в среднем 50—80% • Низшие олефины,
в частности этилен [739], можно метиленировать под давле-
нием, однако выходы продуктов при этом невелики (например,
выход циклопропана из этилена составляет 29% [1147]).
199
Как и в случае других реакций синглетных карбенов и их
комплексов, метиленирование по Симмонсу — Смиту является
стереоселективным процессом, протекающим с сохранением
конфигурации исходного олефина. При этом ациклические
цас-олефины метиленируются быстрее, чем их транс-изоме-
ры [739], что согласуется с закономерностями образования
л-комплексов, структурно аналогичных циклопропанам. В то же
время для циклических олефинов более характерна обратная
зависимость — легче циклопропанируется транс-двойная связь,
например [1160]:
В отличие от двухстадийного метода получения циклопропа-
нов из олефинов по схеме дигалогенциклопропанирование — вос-
становление [1156, 1161], при котором образование циклопропа-
новых производных в ряде случаев осложняется возможностью
побочных процессов дегидрогалогенирования или изомеризации
гсж-дигалогенциклопропанов с образованием более стабильных
продуктов, метиленирование по Симмонсу — Смиту может с ус-
пехом проводиться и в случае довольно лабильных соединений.
При этом выходы продуктов циклопропанирования, например,
циклоолефинов состава С4—Сб составляют 47—57% [1148,
1154]:
НС НС
Zn/cu /.к
J ^(СН2)п + СН212 ------*-н2с< >(СН2)
ЧС Эфир V/ п
НС
п = 2-4
Выходы же продуктов циклопропанирования циклооктена
и циклододецена более высоки и составляют 74—79% [1154].
Этот метод удобен и для циклопропанирования бициклических
олефинов, например норборнена (25) [1147]; выход продук-
та (26) составляет в этом случае 47%.
Интересен также пример метиленирования гидроксиметиль-
ного производного циклопропена в соответствующий бициклобу-
тан [1162]:
СН2ОН СН2ОН
AZn/Cu 1
_ Ме + СН212 -»- Me-А-Me
эфир V
200
Реакцию циклопропанирования по Симмонсу — Смиту широ-
ко используют в синтезе полициклопропановых соединений
[1163, 1164]:
Zn/Cu
эфир
Zn/Cu ДАЛ
>—СН = СН —<] + СН212-------*-
эфир
Таким путем был получен и ряд высоконапряженных спира-
новых углеводородов [1165—1167]:
АЛ- + сн212
Zn/Cu
эфир
Zn/Ag
+ СН212 ------э
эфир
СП +СН212
Zn/Cu
эфир
(20-25%)
Высокая эффективность реакции Симмонса — Смита при
циклопропанировании 1,1 -дизамещенных этиленов позволяет
совмещать эту реакцию с метиленированием кетонов по Витти-
ту, что дает общий метод получения (обычно с высокими выхо-
дами) циклопропанов из карбонильных соединений [739]:
СН2
= )с = снг )cZ-CH2
Аналогичным путем были синтезированы высоконапряжен-
ные углеводороды — ротаны [ 1168].
Поведение диенов и других полиенов (как сопряженных, так
и несопряженных) в реакции Симмонса — Смита аналогично
поведению олефинов: эти соединения легко циклопропанируют-
ся, причем реакция протекает ступенчато [739]. Поскольку при
этом в случае полиенов, нуклеофильности двойных связей кото-
рых одинаковы или близки, скорость образования бисаддукта
обычно превышает скорость образования моноаддукта, в полу-
чаемых продуктах часто преобладают дициклопропановые соеди-
?лоИЯ' Так’ бутадиен-1,3 в этой реакции образует с выходом
/о смесь винилциклопропана и бициклопропила в соотноше-
201
нии 14:86 [1169, 1170]; в случае же циклопентадиена образует-
ся практически лишь бисаддукт [1056]:
Zn/Cu
+ сн212 —** D>——<П
эфир V
(4%) (26%)
Zn/Cu
эфир
(93%) (1,5%)
Аналогично, несопряженный диен 4-винилциклогексен в ре-
акции с эквимольными количествами CH2I2 и Zn также преиму-
щественно образует бисаддукт [1161]:
СН212,
Zn/Cu
" -ЗЕ»-
эфир
(67%) (27%)
(6%)
правила составляют циклооктатри-
Исключение из этого
ей-1,3,5 [1171] и циклооктатетраен [1172], которые образуют
лишь моноаддукты, причем с низкими выходами (менее 5%).
В случае полиенов, двойные связи которых имеют разную
нуклеофильность, преимущественно циклопропанируются наибо-
лее замещенные кратные связи, имеющие наибольшую нуклео-
фильность. Так, 2-метилбутадиен-1,3 дает в основном моноад-
дукт по наиболее замещенной кратной связи [1173]; аналогич-
но протекает циклопропанирование несопряженных поли-
енов [П74]:
(32%)
202
Применение большого избытка CH2I2 и Zn позволяет и в
этих случаях получать продукты исчерпывающего циклопропа-
нирования с удовлетворительными выходами [1149, 1175, 1176],
например:
CH2I2, Zn/Cli
------------£»•
эфир
CH2f2s Zn/Cu
------------з».
эфир
(55%)
(47%)
Ари л замещенные этилены в условиях реакции Симмонса —
Смита образуют соответствующие арилциклопропаны обычно
с невысокими выходами. Например, стирол дает фенилцикло-
пропан с выходом 32% [739, 1145]. Однако в случае стиролов
с электронодонорными заместителями выходы арилциклопропа-
нов выше. Так, п-метоксистирол дает циклопропановый аддукт
с выходом 70% [1145].
Zn/Cu
Ph - СН = СН2 + СН212
эфир
Ph - НС-СН2
МеО
СН = СН2 + СН212
сн2
Zn/Cu
-----ы МеО
эфир
Реакцию Симмонса — Смита с успехом используют для син-
теза непредельных и спирановых углеводородов с трехчленными
циклами из алленов, поведение которых в условиях этой реак-
ции аналогично поведению олефинов или несопряженных дие-
нов. В частности, монозамещенные аллены циклопропанируются
по Симмонсу — Смиту неселективно [733, 739, 1177]. Напри-
мер, при взаимодействии эквимбльных количеств бутилаллена,
дииодметана и цинка образуется смесь равных количеств всех
возможных циклопропанов, тогда как при избытке реагента
Симмонса — Смита в основном получается бутилспиропен-
тан [739]. В то же время с винилиденциклопропаном реакция
протекает селективно по концевой двойной связи [733]:
сн212,
Zn/Cu
BuCH = C = CH2
= СН2 + СН212-^
203
Кратные связи бензола и его производных не циклопропа-
нируются в условиях реакции Симмонса — Смита [739]. В от-
личие от соединений с двойными углерод-углеродными связями,
ацетилены не дают циклических продуктов с реагентом Сим-
монса — Смита. При этом терминальные ацетилены образуют
продукты внедрения метилена в связь С—Н [1178]:
RC = CH + ICH2Znl—* RC=CMe + Znl2
Алкины с внутренними кратными связями в условиях реак-
ции Симмонса — Смита претерпевают перегруппировки, иногда
сопровождающиеся циклопропанированием, например [1179,
1180]:
CH2i21 Е1 СНСН = СНМе
Zn ' /
EtC = CEt ----*СН2 = ССН = СНМе + Н2С - CHEt
(20-25%) 422%)
Енины циклопропанируются лишь по двойной связи, но при
наличии концевой тройной связи основным является продукт
внедрения [1181]:
R CH2I2, R
I Zn/Cu •
СН2 = С-С = СН------► СН2 = С - С = СМе+
R = Ви-трет
(56%)
+ IX
с =сн
R
С =СМе
R
(5%) (15%)
Известны также примеры метиленирования непредельных
металлорганических соединений. Так, из продуктов гидроалю-
204
минирования терминальных алкинов по реакции Симмонса —
Смита получены замещенные циклопропаны [1182]:
сн2вг2, сн
R2AIH Zn/Cu / X
RC = CH------»-R2AICH = CHR---------*rJaI- НС - CHR
Et2O
CH2 CH2
/\ / \
h2c-chr x-hc-chr
(35-62%) (47-58%)
R = Bu, Bu-rper, CgH^-цикло; R1 = Bu-изо; X = Br, I
Аналогичным образом из дивинилртути получена труднодо-
ступная другими методами дициклопропилртуть [1183]:
(СН2 = СН)2Нд + СН212-|~ >-Нд-<|
Реактив Симмонса — Смита проявляет в реакции циклопро-
панирования непредельных соединений свойства слабого элект-
рофила, поэтому введение в молекулу субстрата элект-
роотрицательных заместителей, к которым относится большин-
ство функциональных групп, тормозит реакцию. Вследствие этого
функционально замещенные непредельные соединения обычно
менее активны в этой реакции, чем соответствующие углеводо-
роды. Поэтому непредельные соединения с электроотрицатель-
ными функциональными группами циклопропанируются тем
легче, чем дальше от двойной связи находится функциональ-
ная группа. Например, этилолеат, у которого влияние функцио-
нальной группы на двойную связь практически отсутствует,
превращается в соответствующий циклопропан с выходом
51% [1147]:
н-С8Н17СН = CH(CH2)7COOEt + CH2I2
—** H"CqHi7 ——(CH2)7COOEt
Однако известны случаи, когда тормозящее влияние элект-
роноакцепторного заместителя не сказывается и на значитель-
205
но меньших расстояниях. Так, этиловый эфир 2-метиленцикло-
пропанкарбоновой кислоты превращается в производное спиро-
пентана с выходом 80% [1184] :
Н2С -A—COOEt + CH2I2 [XL-COOEt
В то же время эфиры аф-ненасыщенных кислот образуют
продукты циклопропанирования связи С = С обычно с низкими
выходами. Так, диметилфумарат и этиловый эфир циклобутен-
карбоновой-1 кислоты образуют соответствующие циклопропа-
новые аддукты с выходами, не превышающими 5% [1185, 1186].
В случае метилкротоната в обычных условиях реакции выход
циклопропанового аддукта составляет 10% и лишь в модифици-
рованном варианте процесса его удается повысить до 25%
[1187].
Подобные закономерности характерны и для непредельных
кетонов, хотя кетогруппа проявляет обычно гораздо меньшее
тормозящее влияние. Так, несопряженные алкенилкетоны [1185,
1188—1190] дают продукты метиленирования с выходами 70—
90%, тогда как винилметилкетон и его алкилзамещенные — с
выходами 20—50% [1188, 1191]. В то же время циклические
еноны — циклопентенон-2 и циклогексенон-2 — образуют соответ-
ствующие циклопропановые аддукты с выходами 80—90%
[И88]:
Интересно отметить, что обе последние реакции полностью
подавляются при введении метильной группы в положение
3: З-метилциклогексенон-2 и 3,4,4-триметилциклопентенон-2
в этих условиях вообще не образуют циклопропановых продук-
тов [1188]. Подобное поведение — полное торможение реакции
из-за весьма незначительных пространственных затруднений —
характерно для нестабильных промежуточных частиц, имеющих
несколько равновероятных каналов необратимого превращения.
В отличие от других непредельных карбонильных соедине-
ний, ненасыщенные альдегиды в реакции Симмонса — Смита
лишь полимеризуются.
Циклопропанирование ненасыщенных спиртов, эфиров и ами-
нов проходит легко и с хорошими выходами, что связано со
способностью таких функциональных групп образовывать коор-
206
динационные связи с цинкорганическим реагентом. Так, заме-
щенные аллиловые и у.б-непредельные спирты циклопропаниру-
ются более эффективно, чем 0,7-производные [1191—1193]:
ОН ОН
1 Zn/Cu л I
МеСН = СНСНМе + СН212-----*- Me—сА_СНМе
(60%)
СН2 = СН(СН2)2ОН + CH2I2 Zn/-C> А—(СН2)2ОН
(25%)
МеСН = СН(СН2)2СНМе + СН212 Me-Д-(СН2)2СНМе
(65%)
Аллиловый спирт в условиях реакции Симмонса — Смита
дает не циклопропилметанол, а соответствующий ацеталь (вы-
ход 90%), что также указывает на первоначальную координа-
цию реагента по гидроксильной группе [1193]; повышение
электронной плотности двойной связи при переходе от аллило-
вого спирта к металлиловому увеличивает скорость циклопропа-
нирования, и главным продуктом реакции становится 1-гидрок-
симетил-1-метилциклопропан [1193]:
СН2 = СНСН2ОН + СН212
СН2ОСН2ОСН2
Me сн2'г. 1 Zn/Cu СН2 = ССНоОН Me С Н2 0 Н X + О^СНгО Me сн2 2
(70%) (10%)
Способность гидроксильной группы к координации с реаген-
том Симмонса — Смита, обусловливающая региоселективность
циклопропанирования, широко используют в химии стероидов
для стереоспецифичного введения метильных групп. Реакцию
обычно проводят в смеси тетрагидрофурана с эфиром, причем
выходы циклопропановых аддуктов составляют в среднем 40—
50% [1194, 1195]. Таким путем, например, осуществлено сте-
реоспецифичное введение 19а-метильной группы в эстрен-5(10)-
207
диол-3а,17р, приводящее к 17£-гидрокси-10а-андростен-4-ону-3
[И94]:
В отличие от алкинов, а-ацетиленовые спирты в реакции
Симмонса — Смита (растворитель — смесь диэтилового эфира
с 1,2-диметоксиэтаном в соотношении 1:1) в основном дают
p-циклопропилкетоны (выходы 20—30%), а также а,p-ненасы-
щенные кетоны (выходы 6—15%) [1196]:
ОН Me
I Zn 1 ।
RCHC =CR1 + CH2I2—1- CxJL PnD + RCOCH = CR1
Ь п2си H
R, R1 = Aik
Высокая комплексообразующая способность аминогруппы
обусловливает легкость циклопропанирования непредельных
аминов по Симмонсу — Смиту. Так, аллиламин [739] с высоким
выходом образует циклопропилметиламин; аналогично реагиру-
ет и металлиламин [1192]; реакция Симмонса — Смита пригод-
на и для циклопропанирования енаминов [1197]:
R
I о.. . Zn/Cu
сн2 = cch2nh2 + с Н212 —
R
CH2NH2
Zn
+ CH2I2 ----*
эфир
(36%)
208
Группировка OR обладает высокой координирующей способ-
ностью, вследствие чего виниловые и аллиловые эфиры также
легко вступают в реакцию Симмонса — Смита [1198—1201]:
RUH = CHOR1 + CH2I2 Z.n-^ R Л—OR1
эфир
(30-90%)
RCH = CHCH2OR1 + CH2I2 R —CH2OR1
(27-65%)
Высокую реакционную способность в этой реакции проявля-
ют также соответствующие сложные эфиры [1191, 1202—1206]
и ненасыщенные триметилсилиловые эфиры (эфиры енольной
формы карбонильных соединений) [1207, 1208], оказавшиеся
весьма перспективными синтонами. На способности этих соеди-
нений к различным гидролитическим и термическим превраще-
ниям основан синтез ряда важных спиртов, труднодоступных
другими путями. Так, циклопропанирование триметилсилило-
вых эфиров енолов приводит к соответствующим триметилси-
лилоксициклопропанам, осторожная обработка которых спирта-
ми дает с высокими выходами циклопропанолы, например,
[1050]:
СН212,
Zn/Cu А МеОН А ли
МеСН = CHOSiMen------* Me -Z-^- OSiMe3----* Me z..a- OH
° C5H5N
(91%)
Me3SiO OSiMe3
Me-SiCI I I CH2I2» Zn
MeCOCOMe CH2 = C - C = CH2 ----
эфир
OSiMe3
Me3SiO
MeOH
C5H5N
Однако гораздо большее значение имеет щелочное расщеп-
ление триметилсилилоксициклопропанов, протекающее с рас-
крытием цикла и приводящее с высокими выходами (обычно
«90%) к а-метилзамещенным исходных карбонильных соеди-
нений. Реакция представляет собой удобный метод а-метилиро-
вания ациклических карбонильных соединений и циклоалкано-
нов [1209, 1210]. Достоинством этого метода является возмож-
ность избирательного метилирования несимметричных соедине-
14—107 2 09
ний в любое из a-положений, что основано на различиях в ки-
нетической и термодинамической кислотности а-водородных ато-
мов [1211]:
Me3SiO СН2 q
1/\ НО" 11
Ме2СН - С - СН2-----*• Ме2СН - С - Et
Zn/Cu | CH2I2
OSiMe3 О OSiMe3
I I.LiN (Pr-изо), II Et3N I
Me2CH - c = CH2-«-----------Me2CH - C - Me------*- Me2C = C - Me
2.Me3SiCl Me3SiCI
CH?
/\
Me2C-C<
Me
OSiMe3
О
II
Me3C - C - Me
Удобными полупродуктами в органическом синтезе являются
и продукты циклопропанирования триметилсилиловых эфиров,
полученных из аф-ненасыщенных кетонов, которое протекает
исключительно по С=С-связи, соседней с группой МезЭЮ [1212,
1213]. Образующиеся продукты при обработке кислотами се-
лективно изомеризуются в соответствующие циклобутаноны
[1212], а при термолизе претерпевают обычную для винилцик-
лопропанов изомеризацию в 2-замещенные циклопентаноны
с высокими или даже количественными выходами [1213]:
Мех . О Me3SiCI H2C^x,OSiMe3 сн212 ^_OSiMe3 T '* ^CR = CHR1
Rx ^CHR1 R^^CHR1 (70-80%) r^o ? °SIMe3 Н». 0 CR = CH2 !“r * Me
Таким образом, реакция Симмонса — Смита является удоб-
ным способом построения не только трех-, но также четырех-
210
и пятичленных циклических систем. Действием избытка реакти-
ва Симмонса — Смита на триметилсилиловые эфиры аф-непре-
дельных карбонильных соединений получают дициклопропило-
вые эфиры, которые являются исходными соединениями в син-
тезе циклопропилкетонов и циклопропилциклопропанолов
[1214]:
r CH2R1
Me3SiCl
OSiMe3
CH2I2, Zn
(50-65%)
(95%)
(90%)
НО-
CO(CH2)2R1
R, R1 = H, Aik
(« 95%)
Наконец, известны также примеры успешного применения
реакции Симмонса — Смита для циклопропанирования непре-
дельных соединений, содержащих атом галогена или функцио-
нальные группы с атомами азота и серы [739].
Таким образом, для лабораторной практики реакция Сим-
монса— Смита является достаточно удобным и эффективным
Методом прямого метиленирования различных непредельных
соединений. Основным ограничением этого метода является, по-
балуй, невозможность его реализации в укрупненных масшта-
бах. Полезным было бы и определенное расширение реакции
Симмонса — Смита, позволяющее осуществить перенос на не-
предельный акцептор не только метилена, но и его замещенных.
,4“ 211
4.3. ГЕНЕРИРОВАНИЕ И РЕАКЦИИ КАРБЕНОВ
В УСЛОВИЯХ МЕЖФАЗНОГО КАТАЛИЗА.
ХИМИЧЕСКИЕ ПРЕВРАЩЕНИЯ ДИГАЛОГЕНЦИКЛОПРОПАНОВ
Метод межфазного катализа (МФК) [1215—1219], основанный
на применении катализаторов фазового переноса (КФП), позво-
ляет осуществить перенос одного из реагентов из твердой или
водной фазы в органическую, тем самым снимая ограничения,
связанные с нерастворимостью неорганических соединений и
плохой растворимостью некоторых органических солей в орга-
нических растворителях. В качестве катализаторов обычно ис-
пользуют краун-эфиры и криптаты, образующие комплексы
с катионами щелочных металлов, или соли с липофильным
катионом (например, триэтилбензиламмонийхлорид или другие
соли четвертичного аммония). Метод межфазного катализа
дает возможность использовать дешевые и легкодоступные
растворители, а также облегчает выделение продуктов реакции.
Особый интерес представляет применение этого метода для по-
лучения карбенов. Именно возможность генерирования дигало-
генкарбенов из галоформов под действием 50 %-го водно-
го NaOH в присутствии хлоридов бензилтриэтиламмония
(БТЭАХ)* [914] и метилтриоктиламмония [915] легла в основу
метода МФК.
СХ2
)с = с( ♦ снх3 W. )с'- с(
(51)
Простота и эффективность генерирования карбенов этим
методом существенно упростила синтез гел/-дигалогенциклопро-
панов (51), сделав их дешевыми и доступными полупродуктами.
Более того, это методика не требует абсолютирования реагентов,
а также повышенных (или пониженных) давления и темпера-
туры. Наконец, успешное применение метода МФК для синтеза
геж-дигалогенциклопропанов привело к широкому использова-
нию его и для осуществления других реакций, в том числе для
получения функционально замещенных циклопропанов.
Общая методика получения дихлорциклопропанов в условиях МФК Г1220].
Ненасыщенное соединение смешивают с СНС13 и 50%-м водным NaOH (взя-
тыми в 4-кратном мольном избытке по отношению к олефину) и «1% (мол.)
КФП при 0—5 °C и интенсивном перемешивании (>800 об/мин). Получившую-
ся эмульсию перемешивают 1—2 ч при комнатной температуре и затем еще
2—4 ч при 50 °C. Затем смесь разбавляют примерно 2-кратным количеством
* Чаще для этого соединения используют сокращенное название ТЭБАХ.
212
еоды, экстрагируют эфиром, экстракт высушивают. Растворитель упаривают,
остаток подвергают дальнейшей очистке (обычно перегоняют).
При получении геж-дибромциклопропанов в реакционную смесь необходи-
мо добавлять 1—2 мл этанола на 1 моль бромоформа.
В качестве катализаторов при синтезе ге;и-дигалогенцикло-
пропанов в условиях МФК нашли применение четвертичные
соли аммония, соли фосфония и арсония, триалкиламины, кра-
ун-эфиры. В последнее время стали применять также катализа-
торы, иммобилизованные на полимерных носителях (трехфаз-
ный катализ) [1221]. Кроме того, реакция может протекать и в
отсутствие катализатора, но при воздействии на реакционную
смесь ультразвука [1222]. В качестве КФП наиболее часто ис-
пользуют бензилтриэтиламмонийхлорид, хотя в этой реакции
эффективны и другие хлориды четвертичного аммония, включая
выпускаемую промышленностью техническую смесь высших
алкилбензилдиметиламмонийхлоридов (катамин АБ) [1223].
Помимо галогенидов возможно применение и гидроксидов.
Так, например, применение гидроксида триалкил-^-гидрокси-
этиламмония позволяет селективно дигалогенциклопропаниро-
вать наиболее активные двойные связи в полиенах [1224].
Следует отметить, что активность соли тетраалкиламмония
обычно повышается с увеличением длины цепи алкильного ра-
дикала, увеличением «рыхлости» ионной пары и повышением
эффективного положительного заряда на атоме азота [1225].
Для разбавления ненасыщенных соединений, применяемых в ка-
честве акцепторов карбенов, применяют апротонные раствори-
тели, обычно дихлорметан. В качестве второй (неорганической)
фазы возможно использование не только водных, но и безвод-
ных щелочей [1226, 1227], что позволяет, в частности, проводить
реакцию даже при температурах ниже О °C и осуществлять цик-
лопропанирование олефинов, газообразных при обычных усло-
виях [1226]. Катализаторы фазового переноса катализируют не
только щелочной гидролиз галоформов, но и карбенный распад
Na-солей тригалогенуксусных кислот в нейтральной среде при
20 "С [920].
С помощью данного метода удается генерировать и ввести
в реакцию [1+2]-циклоприсоединения практически все дигало-
генкарбены; дихлор- [914, 915], дибром- [1228, 1229], дииод-
[1229, 1230], фторхлор- [1229—1232], бромфтор- [1229, 1232],
иодфтор- [1233], бромхлор- [1229, 1234], иодхлор- [1230, 1234]
и бромиодкарбены [1235]. Труднее всего генерировать в этих
условиях дифторкарбен, дающий низкие выходы геж-дифторцик-
лопропанов [1217, 1232]. Указанный метод пригоден также для
получения ряда моногалогенкарбенов: фенилхлор- [1236], ме-
тилтиохлор- [1237], фенилтиохлоркарбенов [1238], а также
некоторых других [1215—1219], в том числе с гетероциклически-
ми заместителями [1239].
213
4.3.1. Механизм генерирования карбенов в условиях МФК
Механизм генерирования карбенов в условиях МФК подроб-
но изучен на примере дигалогенкарбенов [1215—1219, 1238].
При этом в двухфазной системе [галоформ — акцептор карбе-
на— концентрированный водный раствор NaOH — четвертичная
аммониевая соль (в каталитических количествах)] первона-
чально из галоформа и щелочи образуется адсорбированный на
поверхности раздела фаз тригалогенметильный анион, который
с солью аммония образует соответствующую аммониевую
соль. Эта соль переходит в органическую фазу и постепенно
превращается в исходную соль и дигалогенкарбен. Последний
далее реагирует с олефином, образуя гел^-дигалогенцикло-
пропан:
NaOH (ВФ) + СНХ3 (ОФ) Ыа+-СХз(ПРФ) + Н2О
Na+“CX3 (ПРФ) + Q+X- (ОФ) Q+-CX3 (ОФ) + Na+X- (ВФ)
Q+ ~СХ3 (ОФ) :СХ2 (ОФ) + Q+ X"
:СХ2+ )С = С< -*(51)
где ВФ — водная фаза; ПРФ — поверхность раздела фаз; ОФ — органическая
фаза; QX — соль тетраорганиламмония.
Следует отметить, что в отсутствие КФП из циклогексена
и хлороформа под действием 50%-ного водного КОН 7,7-ди-
хлорноркаран образуется с выходом менее 0,5%, при этом хло-
роформ в основном гидролизуется [910]. Поскольку гидролиз
хлороформа под действием водной щелочи в любом случае
протекает через дихлоркарбен, то считают [1215—1219, 1240],
что в двухфазной системе эта частица образуется и в отсутст-
вие катализатора, но лишь на поверхности раздела фаз. В этих
условиях карбен не способен реагировать с алкенами. Это под-
тверждается тем, что реакции дихлоркарбена с алкенами в
двухфазной системе эффективно катализируются не только
четвертичными аммониевыми солями, но и триалкиламинами
[1215—1219]. При катализе третичными аминами первоначаль-
но на поверхности раздела фаз в результате взаимодействия
триалкиламина с дигалогенкарбеном образуется илид (52), ко-
214
торый затем мигрирует в органическую фазу, где действует как
основание [1240]:
СНХ3 (ОФ) + NaOH (ВФ) Na+-CX3 (ПРФ) + Н2О
- NaX + :СХ2(ПРФ) + Н2О
:СХ2 (ПРФ) + NR3 (ОФ) - - R3N - СХ? (ОФ)
(52)
(52) + СНХ3 (ОФ) * R3NCHX2 "СХ3 (ОФ) -
+ ' чс = с7
-*R3NCHX2 Х~ (ОФ) + :СХ2 (ОФ) ---*(51) + R3N 4- СНХ3
Специальными опытами показано, что образование илида —
необратимый процесс [1240].
Активность краун-эфиров в образовании дигалогенкарбенов
и дигалогенциклопропанировании олефинов в условиях МФК
определяется их способностью образовывать комплексы с иона-
ми щелочных металлов и тем самым переводить щелочь в орга-
ническую фазу.
4.3.2. Реакции дигалогенкарбенов
Почти все ненасыщенные углеводороды присоединяют дигало-
генкарбены, генерированные в условиях МФК, причем выходы
гельди галогенциклопропанов обычно значительно выше, чем при
генерировании тех же карбенов другими методами [12, 1215—
1219]. Легкость циклопропанирования алкилзамещенных эти-
ленов в условиях МФК, как и при других методах генерирова-
ния дигалогенкарбенов, возрастает по мере увеличения степени
замещения кратной связи [12]. Особым достоинством метода
МФК является достаточно высокая эффективность циклопропа-
нирования кратных связей, обладающих низкой активностью
вследствие дезактивирующего влияния электроноакцепторных
заместителей, сопряжения или пространственных факторов. На-
пример, аддукты дихлоркарбена с биадамантилиденом [1241]
н трифенилэтиленом [1242] получены в этих условиях с выхо-
дами 70—80%:
215
Метод МФК оказался весьма удобным и для циклопропани-
рования высоконапряженных циклоолефинов, в том числе кар-
касных соединений. Так, например, из баскетена с выходом 60%
получен полициклический моноаддукт дихлоркарбена [1243]:
50%-й водн. NaOH
--------------у
БТЭАХ
При геж-дигалогенциклопропанировании соединений с не-
сколькими изолированными двойными связями обычно образу-
ются продукты как моно-, так и полициклопропанирования. Од-
нако при использовании триалкил-^гидроксиэтиламмонийгало-
генидов в качестве КФП обычно удается селективно циклопро-
панировать отдельные наиболее активные кратные связи. На-
пример, при реакции дихлоркарбена с лимоненом в присутствии
бензилтриэтиламмонийхлорида образуется диаддукт, в то время
как в присутствии хлорида бензил-р-гидроксиэтилдиметиламмо-
ния получается исключительно моноаддукт по эндоциклической
связи [1224, 1244]:
(78%) (62%)
t - СНС13, 50%-й водн. NaOH, PhCH2Et3NCl; 2 - СНС13> 50%-й
водн. NaOH, PhCH2(HOCH2CH2)Me2NCI
Диеновые углеводороды при взаимодействии с избытком ди-
галогенкарбена в условиях МФК обычно образуют диаддукты
[1245—1248], а 1,со-дифенилполиены в тех же реакциях — поли-
циклопропановые соединения [1249]. Следует отметить, что ди-
галогенкарбены, генерированные другими методами, с указан-
ными полиенами обычно вообще не реагируют. В то же время
216
фторсодержащие 1,со-диарилполиены дают аддукт, в которых
одна кратная связь остается незатронутой [1250]:
:СС12
Ph(CH = CH)nPh ------Ph
КФП
Cl Cl
:CCI2
Ar(CF = CF)nAr-— Ar
КФП
Присоединение дигалогенкарбенов к циклическим сопряжен-
ным полиенам приводит как к моно-, так и к полиаддуктам, со-
отношение которых зависит от соотношения реагентов. Так,
большой избыток дихлоркарбена в реакции с циклооктатетра-
еном позволяет получить тетрааддукт (выход до 55%), в то вре-
мя как для получения моноаддукта, напротив, необходим избы-
ток олефина [1242, 1251]:
СНС13,
50%-й водн. NaOH
БТЭАХ
При взаимодействии дигалогенкарбенов с кумуленовыми уг-
леводородами в условиях МФК в зависимости от структуры ку-
мулена получают моно- и ди аддукты. Так, из аллена получена
смесь производных циклопропана и спиропентана с выходами 6
и 28% соответственно [1242, 1252]:
Cl CI
50%-й водн. NaOH у
сн2 = с = СН2 + СНС1,---------------- Д=сн2
БТЭАХ
Cl Cl Cl CI
217
В то же время тетраметилаллен в аналогичных условиях
с выходом 90 % дает исключительно диаддукт [1252]. При нали-
чии в алленах неэквивалентных двойных связей присоединение
дигалогенкарбенов протекает по наиболее замещенной из них,
чему дополнительно способствует пространственное экранирова-
ние соседней менее замещенной связи [1253]. Необычно проте-
кают реакции дихлоркарбена, генерированного в условиях МФК,
с тетрафенилалленом и фенилалленом. При этом в первом слу-
чае предпочтительным направлением является присоединение
дихлоркарбена по С—С-связи бензольного ядра, а не алленовой
системы, а во втором случае образуется продукт перегруппи-
ровки— производное циклобутена [1252, 1254]:
Ph2C==C==CPh2 + СНС13
PhCH = С = СН2 + CHCI, —-
* W
Cl CI
Ph Cl Cl
(21%)
Однако замещенные бутатриены реагируют с дихлоркарбе-
ном с образованием нормальных продуктов циклопропанирова-
ния двойных связей — бициклопропилиденов [1254, 1255]:
R2 С — С — с — с r2
:СС12
КФП
Cl Cl Cl CI
Хотя связи CsC обычно с трудом реагируют с дигалоген-
карбенами, однако в условиях МФК удается гаи-дигалоген-
циклопропанировать и эти связи, получая соответствующие ди-
218
уалогенциклопропены, которые в основных средах легко гидро-
лизуются до кетонов, например [1256]:
Me Me
СНС13, 50%-й водн. NaOH
БТЭАХ
(53%)
Me Me
(17%)
В реакциях с енинами генерируемые в условиях МФК ди-
галогенкарбены, как и в других случаях, легче реагируют с
двойной, чем с тройной связью. Так, взаимодействие 2,5-диме-
тилгексадиен-1,5-ина-3 с дихлоркарбеном приводит исключи-
тельно к образованию циклоаддуктов по С=С-связям [1257]:
Me Me сУюц,
1 I 50%-й водн. NaOH
СН2 = С - С =С - С = СН2--------------
БТЭАХ
Cl Cl CI С1
Me Me
(62%)
Следует отметить, что в ряде случаев дигалогенкарбеновые
аддукты с напряженными циклоолефинами нестабильны и уже
в условиях реакции изомеризуются с раскрытием циклопропа-
нового кольца и расширением цикла исходного олефина. Так,
норборнен (25) при геж-дигалогенциклопропанировании в усло-
виях МФК дает лишь продукт изомеризации с выходом 74%
[1258]:
В реакции норборнадиена (36) с дихлоркарбеном в условиях
МФК наряду с продуктами изомеризации [2+1]-циклоаддук-
та— (53) и (54) —выделен и тетрациклический дихлорид (55),
219
образующийся в результате гомо-[4 +1]-циклоприсоединения
дихлоркарбена [505, 1259].
В качестве катализатора использован гексилтриметиламмо-
нийхлорид [1259] и бензилтриэтиламмонийхлорид [505]; об-
щий выход продуктов реакции составил соответственно 80 и
50%; соотношения (53) : (54) : (55) были равны 80:7:13 и
77: 12: 11 соответственно. Перегруппированные дибромиды с об-
щим выходом 80—90 % образуются и при взаимодействии нор-
борнадиена с дибромкарбеном в условиях МФК [1259] [(53) :
: (54) : (56) =51 : 22 : 27].
Х=С1 (53) (54) (55)
, СНХ3, 50%-й водн. NaOH
(36)
Вг
> (53) + (54) +
БТЭАХ
В г
Х= Вг (56)
Спиро [инден-1,Г-циклопропан] в реакции с дихлоркарбеном
в условиях МФК дает смесь производных нафталина, макси-
мальные выходы каждого из которых в зависимости от условий
колеблются от 50 до 80% [1253]:
+ CHCI3
50%-й водн. NaOH
БТЭАХ
Присоединение дихлоркарбена к гексаметилзамещенному
бензолу Дьюара в условиях МФК также приводит к продукту
перегруппировки лабильного моноаддукта. Продукт такой пере-
группировки может присоединять еще две молекулы дихлор-
карбенэ, образуя полициклический диаддукт [1260, 1261]. Со-
229
отношение продуктов этой реакции зависит как от соотношения
исходных реагентов, так и от продолжительности реакции.
СНС13,
50 %-й водн. NaOH
-----------------у
БТЭАХ
Дихлоркарбен, генерированный в двухфазной системе, хоро-
шо реагирует с ароматическими углеводородами, обладающими
нуклеофильными свойствами, например с фенантреном [1244,
1262] пли антраценом [1263]. В случае фенантрена этим мето-
дом впервые был получен неперегруппированный продукт при-
соединения по связи 9,10 — соответствующий дибензоноркара-
диен (выход 79%):
Описаны также довольно многочисленные примеры взаимо-
действия алкилбензолов, алкилнафталинов [1264, 1265] и дру-
гих алкилароматических углеводородов с дихлоркарбеном в
двухфазной системе, протекающие с участием кратных связей
ароматического ядра [1266—1269], однако выходы аддуктов
обычно составляют 1—2%. Так, 2-метилнафталин в реакции с
генерируемым в двухфазной системе дихлоркарбеном перво-
начально образует аддукт по ароматической кратной связи, ко-
торый далее претерпевает перегруппировку и последующее при-
221
соединение еще одной молекулы дихлоркарбена [1265]:
При реакции метоксизамещенного дибензоциклобутадиена с
дихлоркарбеном образуются лишь продукты изомеризации мо-
ноаддукта [1267]:
Дихлоркарбен, генерированный в условиях МФК, присоеди-
няется также по двойным связям С-2=С-3 2-метилзамещен-
ных фурана, тиофена и бензофурана [1265]. Однако первона-
чально образующиеся моноаддукты не были выделены, по-
скольку в условиях реакции они изомеризуются с раскрытием
трехуглеродного цикла и образованием триенов с экзоцикличе-
ской двойной связью, которые затем циклопропанируются. Вы-
ходы конечных продуктов составляют 2—2,5% в случае 2-метил-
222
фурана ii 2-метилтиофена и 38% в случае 2-метилбензофурана
1265], например:
CI
С1
Me
50%-й водн. NaOH
+ CHCL --------------
я БТЭАХ
Me
Непредельные соединения с различными функциональными
< заместителями также легко присоединяют дигалогенкарбены в
условиях МФК. Так, с хорошими выходами получены аддукты
. дихлор- и (или) дибромкарбенов к виниловым [914, 1228,.
1270—1275] и аллиловым [914, 1276] простым эфирам, винил-
сульфидам [1277], эфирам енольной формы кетонов [1278—
1281], аллилацетату и его производным [1228], аминам [1282],
нитраминам [1283] и т. д.
Описано также присоединение дихлоркарбена к кремнийор-
ганическим ненасыщенным соединениям [1244, 1284, 1285] и к
алкенам, содержащим галогены [1228, 1273, 1278, 1286—1290],
ферроценильные [1291] и другие заместители. Нашли примене-
ние также реакции ацеталей ненасыщенных альдегидов с ди-
хлоркарбеном [1292—1294].
Направление реакций дигалогенкарбенов со спиртами в
двухфазной системе существенно зависит от строения спирта.
Алкильные производные аллилового спирта [1224, 1295—1298],
а также ^б-ненасыщенные спирты [1299] реагируют с генери-
руемыми в двухфазной системе дихлор- и дибромкарбенами, об-
разуя гел«-дигалогенциклопропаны с сохранением гидроксильной
группы, например:
СВг2 ОН
Me,С = СНСМе,ОН + СНВг, 50%-й водн- NaOjj Ме,С-СН - СМе?
i 3 БТЭАХ 2 7
(88%)
Образование 1,1-дихлорцикло-пропанов из аллиловых спиртов
представляет значительный интерес, поскольку эти аддукты мо-
223
гут перегруппировываться в кислых средах,
выходами циклопентеноны [1300]:
давая с хорошими
СНСН.ОН
Ме2С = СНСН2ОН + СНС!3
£-0% * годя. NaOH
C16H=3Me3NC!
/\ н-
Ме2С - CCI, —*
(74%)
При наличии в молекуле спирта нескольких кратных связей
при использовании в качестве КФП бензил-2-гидроксиэтилди-
метиламмонийгидроксида удается селективно пиклопропаниро-
вать двойную связь, соседнюю с гидроксиметильной группой
[1300], например:
Me Me СС12
I СНСН \ / \
МеСН = СНСН2С = СНСН2ОН-------МеСН = СНСН, - С - СНСН ,ОН
1
(57%)
1 - 50%-й водн. NaOH, PhCH2(HOCH2CH2)Me2NOH
Как указывалось выше, при генерировании дигалогенкарбе-
нов в условиях МФК первоначально образуется тригалогенме-
тильный анион, который затем распадается до дигалогенкарбе-
на. Однако в реакциях с электрофильными олефинами, содер-
жащими сильные электроноакцепторные группы, возможно при-
соединение по кратной связи недиссоциированного тригалоген-
метильного аниона (по Михаэлю), что приводит к соответствую-
щим тригалогенметильным производным, например [1301]:
СН2 = CHCN + CHCl3 50%_й^-^ cci3c:i2ch2cn
(72%)
Аналогичные результаты получены с аф-ненасыщенными со-
единениями, нитрилами и сульфонами. Если же используемый
непредельный акцептор имеет алкильный заместитель в а-поло-
жении, то основным продуктом реакции становится геж-дигало-
генциклопропан [1302, 1303]. По аналогичной схеме реагируют
также незамещенные и замещенные в ^-положении эфиры ме-
такриловой кислоты, образуя соответствующие дибромцикло-
224
пропановые производные [ 1228, 1302, 1304—1307]:
Me chci3> Me
I 50%-й водн.NaOH I
CH2 = С - CN --------------— CI3C - СН2 - С - CN
БТЭАХ
Me - С - CN
/ \
—- Н2С - СС12
(42%)
СНМе2 снвг3,
I 50%-й водн. NaOH
СН2 = С - СОМе ---------------
БТЭАХ
Me
I
CH2 = C-COOEt
СНВГ3,
50%-й водн. NaOH
БТЭАХ
Ме2СН - С - СОМе
/\
Н2С СВг2
(90%)
Me-C-COOEt
/ \
Н2С — СВг2 +
(78%)
Ме-С-СООН
/\
4 Н2С - СВг2
<5%)
Необычно протекают реакции дихлоркарбена, генерирован-
ного в условиях МФК, с эфирами акриловых кислот, содержа-
щих атом водорода в a-положении к карбонильной группе
[1282]. Реакция включает серию стадий цикло1пропанирования
и дегидрохлорирования, приводящих в конечном итоге к тетра-
хлорспиропентанам [1282]:
CI2C XCRR1
RR1C = СНСООМе |Х С I
кфп ' / х 1
С12С СНСООМе
R = Ph, R1 = Н (48%); R = Me, R1 = Н (14%); R = R1 = Me (35%)
15—107
225
В реакциях генерируемого в условиях МФК дихлоркарбена
с алкадиенами-1,3 [1300] и алкадиенами-1,2 [1302], содержа-
щими электроноакцепторные заместители, гелг-дихлорциклопро-
паны образуются с выходами 20—70%. При этом дихлоркарбен
обычно присоединяется по двойной связи, наиболее удаленной
от электроноакцепторной группы:
СНС13,
50%-й водн. NaOH
МеСН = СНСН = СНСООМе -------т———
БТЭАХ
СС12
/ \
—» МеНС - СНСН = СНСООМе
(22%)
снс13> СС12
50%-й водн. NaOH / \
Ме2С = С = СНСООМе ------------” Ме2С " с = СНСООМе
* БТЭАХ
(69%)
4.3.3. Реакции других карбенов,
генерируемых в условиях МФК
Метод МФК успешно применен и для генерирования серосодер-
жащих карбенов. При обработке фенил (хлорметил)сульфида
или фенил (дихлорметпл)сульфида 50%-м водным NaOH в при-
сутствии олефина и КФП (обычно БТЭАХ) образуются фенил-
тиоциклопропаны и фенилтиохлорциклопропаны с выходами
44—79% [1308, 1309]. В качестве непредельных акцепторов
применяли алкены, циклогексен, стирол и простые виниловые
эфиры [1308, 1309]:
Y-C-SPh
,С = с<
+ PhSCHClY
50%-й водн. NaOH
БТЭАХ
/\
)с - с'
Y = Н, С1
В аналогичных условиях генерирован метилтиохлоркарбен
из метилдихлорметилсульфида. В реакции с 2,3-диметилбуте-
ном-2 этот карбен образует соответствующий циклопропаниро-
ванный аддукт с выходом 43% наряду с 1,2-бис (метилтио)-1,2-
дихлорэтиленом (выход 24%) [1237].
226
В каталитической двухфазной системе генерирован также
ряд ненасыщенных карбенов, прежде всего диметилвинилиден-
карбен. Этот карбен генерирован 7-элиминированием НС1 из
З-метил-З-хлорбутина-1 и а-элиминированием НВг из 1-бром-З-
метилбутадиена-1,2 [1310—1313]:
Ме2СС1С==СН
Ме2С = С = СНВг—1
50%-й водн. NaOH
---------------*>
R4NCI или
краун-эфир
Ме2С = С = С:
---Ме2С = С
(69-81%)
Диметилвинилиденкарбен в условиях МФК дает с до-
статочно высокими выходами циклопропановые аддукты с
алкенами [1310—1313], стиролами [1310, 1311, 1313], сопря-
женными диенами [1310, 1311], а также с ненасыщенными спир-
тами и эфирами [1310, 1311, 1313].
Пентаметиленвинилиденкарбен генерирован в условиях МФК
аналогично диметилвинилиденкарбену 7-элиминированием НС1
из 1-хлор-1-этинилциклогексана в присутствии дициклогексил-
18-краун-6- или 18-краун-6-эфиров в качестве КФП [1311]. Этот
карбен в реакции со стиролом, 2,6-диметилгексадиеном-2,4 и
изопреном дает циклопропановые аддукты с выходами 40, 36 и
32% соответственно [1311].
В условиях МФК генерированы также (фурил-2)хлор* и
(тиенил-2) хлоркарбены, которые в реакции с олефинами дают
соответствующие циклопропановые аддукты с выходами 26—
67% [1239]:
R1 R3
R R1
О Х^
БТЭАХ у*** / \
Х Cl R3
X = О, S; R - R3 = н, Me
Наконец, А^-(р-гидроксиалкил)-1У-нитрозоацетамиды в усло-
виях МФК генерируют алкилиденкарбены, которые вступают в
15* 007
характерные для карбенов реакции [1314—1317]:
ОН NO
R2C - CH2NCOMe 5.°°/о^ R2C = С: —
2 2 (C8H17)3NMeCI
R = цикло-С3Н5 (64%)
* ♦
Таким образом, наиболее распространенными препаративны-
ми методами получения циклопропановых соединений являются
диазометод, реакция Симмонса — Смита и дигалогенциклопро-
панирование в условиях МФК. Диазометод используют в основ-
ном для синтеза циклопропановых углеводородов, эфиров цик-
допропанкарбоновых кислот, других функциональных производ-
ных с трехуглеродным циклом, а реакцию Симмонса — Смита —
исключительно для метиленирования непредельных соединений,
в основном олефинов. Пожалуй, наиболее широкое применение
в синтезе имеет рассмотренная выше реакция дигалогенцикло-
пропанирования в условиях МФК, прежде всего из-за большого
разнообразия химических превращений галоген- и гел«-дигало-
генциклопропанов (см. обзор [1318]).
4.3.4. Химические превращения
гел/-дигалогенциклопропанов
В настоящее время область использования гелг-дигалогенцикло-
пропанов в синтезе существенно расширилась. Ниже кратко
рассмотрены наиболее важные в препаративном плане превра-
щения этих соединений, которые могут протекать с сохранени-
ем или с раскрытием трехчленного цикла.
Восстановление. К важнейшим превращениям геж-дигало-
генциклопропанов, протекающим с сохранением трехуглеродно-
го цикла, относится восстановительное дегалогенирование, по-
зволяющее получать как моногалогенциклопропаны — важные
полупродукты в синтезе циклопропенов и других соединений
циклопропанового ряда, так и циклопропановые углеводороды.
Последнее превращение в сочетании с дигалогенметиленирова-
нием в условиях МФК представляет собой общий двухстадий-
ный метод синтеза циклопропанов из олефинов, во многих слу-
чаях более удобный и эффективный, чем известные одностадий-
ные варианты циклопропанирования олефинов (например, по
Симмонсу — Смиту или каталитическим разложением диазоме-
тана)' сх2 сн2
:СХ2
[Н]
228
Методы восстановления гел/-дигалогенциклопроланов вклю-
чают восстановление системами металл — спирт (вода), нат-
рий— жидкий аммиак, гидридами металлов, а также каталити-
ческое гидрирование и электрохимические способы [1319]. Наи-
большее распространение для полного восстановления гем-ди-
галогенциклопропанов в циклопропаны получило действие нат-
рия в жидком аммиаке или лития в трет-бутиловом спирте
[1319]. В последнее время разработан также простой и эффек-
тивный метод восстановления цинком в присутствии спиртов или
воды [1320].
Для получения моногалогенциклопропанов из соответствую-
щих аельдш алогенидов наиболее эффективны способы, осно-
ванные на использовании металлов (Mg, Zn) [1320], а также
гидридов олова, кремния и ряда других металлов.
Получение и реакции гт-галогенлитиоциклопропанов. В от-
сутствие доноров протонов литий и его алкилпроизводные реа-
гируют с гел/-дигалогенциклопропанами с образованием соот-
ветствующих гел/-галогенлитиоциклопропанов, которые ста-
бильны при температурах ниже —100 °C. Эти соединения легко
вступают в реакции нуклеофильного присоединения, характер-
ные для других литийорганических соединений. Например, при
их обработке диоксидом углерода образуются соответствующие
1-галогенциклопропанкарбоновые кислоты [1321]:
ix^xLi
Li (RLi)
1.CO2
2.ROH
СООН
X
Х = Hal, R = Aik
При температурах >—100 °C галогенлитиоциклопропаны
претерпевают синхронное а-элиминирование литийгалогенида с
образованием циклопропилиденов, которые высокореакциэнно-
способны и участвуют в ряде превращений:
R1
(r1ch2)2o RCH2
229
Наибольший интерес представляет циклопропилиден-аллено-
вая перегруппировка, которая широко изучена на примере
геж-дибромциклопропанов и является одним из наиболее про-
стых общих методов получения алленов [7, 13, 1318].
Реакции элиминирования — присоединения. При взаимодей-
ствии гети-дигалогенциклопропанов с нуклеофильными агента-
ми, являющимися сильными основаниями, обычно протекают
процессы ^-элиминирования. Эти реакции представляют боль-
шой интерес в качестве простых и эффективных путей синтеза
циклопропенов, других ненасыщенных соединений с трехчлен-
ным циклом, а также диенов и триенов, для многих из которых
нет альтернативных способов получения. Обычно для проведе-
ния таких реакций в качестве реагента используют трст-буток-
сид калия в диметилсульфоксиде или других полярных раство-
рителях; в последнее время разработан более простой вариант
проведения этого превращения, основанный на использовании
КОН в присутствии катализаторов фазового переноса [1322].
В этих превращениях первоначально образуются высокореак-
ционноспособные циклопропены, которые в условиях реакции
нестабильны и претерпевают дальнейшие превращения: экзо-
изомеризацию циклопропеновой двойной связи, присоединение
нуклеофилов, перегруппировки:
сх2 —с- —с-
. f \ В~ / NuH I
\с - сн—- - с = сх—-НС - с
1.В-
2.NuH
—с —
/ \
-HC-CNu
СХ2
/\
^СН-НС-С/
\ /ч
)сн-с = сх
с = с-снх
I
сх, нс - сн - сн(
к /\ / X /Ч
;сн - нс - с - сн - сн(—* ;сн - с = сх —*
I I
нс - сн - сн( с-сн-сн(
/ \ в_ А\
—»-)с = с-снх —► )с = с-сн —»•
- с - с = с(
—► )с = с - сн2
230
сх2 сн-
\ I I /X В- \ I I /\
;с - с - сн - нс - сн-- ;с - с - сн - с = сх_
z । । ' ।
СН- с_
. 1 1 /х В- ч I I А
\с-с-с = с-снх—\с - с - с = с - сн—*
—*)с = с-с = с- + /С С>с = с(
)с = с(
Протеканию реакции присоединения способствует наличие
в системе сильных нуклеофилов и полярных растворителей. В не-
полярных средах обычно наблюдаются перегруппировки. Так,
например, реакция 7,7-дихлорноркарана с трет-бутоксидом ка-
лия и спиртом в диметилсульфоксиде приводит к продуктам при-
соединения, в то время, как в бензоле образуются лишь продук-
ты перегруппировки [1323]:
трет-ВиОК
ROH
ДМСО
ROH С6Н6
Реакции а-элиминирования ееж-дигалогенциклопропанов, со-
провождающиеся перегруппировками с раскрытием трехуглерод-
ного цикла, протекают также в случае высоконапряженных си-
стем и при наличии в молекуле исходного дигалогенциклопропа-
на сильных электроноакцепторных заместителей.
Изомеризация и перегруппировки. Из превращений гел/-дига-
логенциклопропанов, протекающих с раскрытием трехчленного
кольца, интерес для синтеза представляет кислотная изомери-
зация гел/-дигалогенциклопропилметанолов в труднодоступные
циклопентеноны [1295]:
CHOHR
R1
231
Широко используют в синтезе и термическую перегруппиров-
ку винил-геж-дигалогенциклопропанов в циклопентены:
СХ2
сх2
В частности, эта реакция лежит в основе промышленного ме-
тода получения дифторбензолов из 1,3-диенов и галогенфторкар-
бенов [1324].
Другим широко изученным превращением геж-дигалогенцик-
лопропанов является циклопропил-аллильная перегруппировка,
которая в сочетании с последующим восстановительным дегало-
генированием представляет собой удобный путь гомологизации
олефинов:
сх2 х X
RCH = CHR1 RHC - CHR1—♦RCHC = CHR1-^
—*► RCH2CH = CHR’
Повышенная напряженность трехуглеродного цикла в поли-
циклических соединениях или возможность образования арома-
тической системы в результате перегруппировки способствуют
ее протеканию уже в условиях реакции дигалогенциклопропани-
рования. Однако в ряде случаев эта перегруппировка идет лишь
при нагревании, иногда длительном, при высоких температурах
[1324], например:
Циклопропил-аллильная перегруппировка в сочетании с де-
гидрогалогенированием в присутствии оснований или при тер-
молизе позволяет синтезировать циклические полиолефины, на-
пример, [1318, 1325, 1326]: ’
PhNR2
или А
(<80%)
X = Cl, Вг; R = Me, Et
232
Таким образом, химические превращения геж-дигалогенцик-
лопропанов открывают широкие перспективы для их использо-
вания в органическом синтезе. Дальнейшее изучение химиче-
ских свойств этих соединений, прежде всего в реакциях с нук-
леофильными агентами, откроет новые интересные возможности
их использования.
Глава 5
КАРБЕНОВЫЕ КОМПЛЕКСЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
И ИХ ПРАКТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ
5.1. СТРОЕНИЕ КАРБЕНОВЫХ КОМПЛЕКСОВ
ПЕРЕХОДНЫХ МЕТАЛЛОВ
Как уже отмечалось в гл. 2, многие свойства карбенов и их
аналогов удается интерпретировать при рассмотрении гранич-
ных МО. В частности, ярко выраженная склонность синглетных
карбенов и их аналогов к комплексообразованию с координа-
ционно-ненасыщенными соединениями переходных металлов
объясняется возможностью образования о-связи М—Ск путем
передачи пары несвязывающих электронов с нуклеофильной
о-орбитали карбенного углерода Ск на атом металла М. Одно-
временно возможно образование и л-связи в результате взаи-
модействия подходящих по симметрии d-AO металла с вакант-
ной электрофильной р-орбиталью карбена [507].
Как правило, карбеновый лиганд является эффективным
о-донором и сравнительно слабым л-акцептором, что предопре-
деляет поляризацию большинства карбеновых комплексов пере-
ходных металлов по типу:
6-6 +
LnM CKXY
Это соответствует криптокарбокатионному характеру карбе-
новых лигандов [1327]. Вместе с тем известен и ряд карбеновых
комплексов переходных металлов с обратной поляризацией свя-
зи М—Ск; в первую очередь это алкилиденовые комплексы тал-
лия, в которых карбеновый лиганд проявляет нуклеофильные
свойства [1328, 1329].
Отдельные представители карбеновых комплексов переход-
ных металлов-, видимо, были синтезированы еще в начале
XX века [1330], однако первый полностью охарактеризованный
представитель этого ряда — метоксифенилкарбеновый комплекс
пентакарбонилвольфрама (1)—был получен Э. О. Фишером в
233
1964 г. [1331]. К настоящему времени известно около 500 комп-
лексов карбенов и их аналогов практически для всех групп пе-
реходных металлов, которые в этих комплексах обычно нахо-
дятся в нульвалентном состоянии или в низших состояниях окис-
ления. Синтезированные комплексы обычно электрически ней-
тральны, хотя известны и ионные (в основном — катионные)
формы [507]. Наиболее изучен класс так называемых фишеров-
ских комплексов (2), у которых карбеновый лиганд в а-поло-
жении к карбеновому атому углерода содержит гетероатом
[1332—1340]:
(CO)5W:C(OMe)Ph LnM:C(ZR)R1
(1) (2) Z = О, N, S
Хорошо известны и комплексы без стабилизирующего а-ге-
тероатома, в частности комплексы 2,3-дифенилциклопропилиде-
на [1341], дифенилкарбеновые комплексы вольфрама [1342],
хрома [1337], молибдена [1343], марганца [1344]. Описан ряд
.алкилиденовых и фенилкарбеновых комплексов вольфрама
[1345, 1346], тантала и ниобия [1328, 1347—1353], а также
комплексы метилена с производными тантала [1347], циркония
[1354], рения [1355], железа [1356, 1357], марганца [[1358], ти-
тана [1359] и других металлов. Известен и ряд комплексов
соединений переходных металлов с аналогами карбенов — сили-
.ленами, гермиленами, станниленами и плюмбиленами. Сходст-
во и отличие таких комплексов от собственно карбеновых
комплексов переходных металлов рассмотрены в обзоре [507].
Общим закономерностям синтеза, строения и реакционной спо-
собности карбеновых комплексов переходных металлов, а так-
же их отдельным классам посвящено большое число обзоров и
монографий [507, 1332—1340, 1360—1365]; ниже мы кратко
суммируем лишь основные выводы, касающиеся строения этих
молекул.
В большинстве карбеновых комплексов переходных метал-
лов карбеновый атом углерода имеет гибридизацию, близкую к
sp2, а группировка МСХУ обычно планарна. В неопентилидено-
вых комплексах тантала (так называемые шроковские комплек-
сы) наблюдается довольно необычная ориентация карбенового
лиганда: ZM—Ск—С достигает 170°, a ZM—Ск—Н равен 80°
[1366, 1367]. Это приводит к ослаблению связи Ск—Н и доволь-
но эффективному взаимодействию М---Н, хотя для этих комп-
лексов такая деформация может быть и следствием стерической
перегруженности первого окружения атома Ср. Более того, по
данным [1368], тенденция к внутримолекулярной координации
М---Н характерна и для других комплексовэлектронодефицит-
234
ных переходных металлов с карбеновыми лигандами, имеющими
а-атом водорода.
<* р .
L^//CK - С<
*н
В карбеновых комплексах переходных металлов связи
М—Ск, как правило, на 0,01 нм длиннее связей М—СО в род-
ственных металлкарбонильных комплексах, однако на — 0,015 нм
короче, чем ординарные связи М—С. Сопоставить длины раз-
личных связей М—С удобно- на примере комплекса, содержа-
щего одновременно алкильный, карбеновый и карбиновый ли-
ганды [1369]:
Ме0РСН9СНРМе? 1 ~ 0,226 нм; 2 - 0,194 нм;
I
’ 2 3 - 0,179 нм
трет-ВиС Н9—W =СНВи-трет
з III
СВи-трет
Подобное же соотношение длин связей металл—углерод с
участием атомов углерода различных типов лигандов характер-
но и для комплексов других металлов. Эти закономерности чет-
ко свидетельствуют в пользу заметной двоесвязанности атомов
М и Ск в карбеновых комплексах, что, как отмечалось выше,
обусловлено возможностью взаимодействия d-kO металла с ва-
кантной р-орбиталью карбенового лиганда (см., например,
[1370, 1371]). Вместе с тем барьер вращения вокруг связи
М—Ск в комплексах фишеровского типа обычно не превышает
25—30 кДж/моль, однако в комплексах типа (3) он выше
85 кДж/моль [1348, 1372, 1373]. Экспериментально определенные
значения прочности связей М—Ск в фишеровских комплексах от-
сутствуют, однако высокая термостабильность таких комплексов,
в частности способность возгоняться без разложения [1337],
свидетельствуют о прочности связей М—Ск. Для метиленовых
комплексов переходных металлов в настоящее время экспери-
ментально определены прочности связей М—Ск [1358]: >380
(Fe:CH2), 402±17 (Мп:СН2), 322±21 кДж/моль [(СО)5Мп:
:СН2]. По расчетным данным [1374] для Сг:СН2 и Ni:CH2 эти
значения равны 210 и 272 кДж/моль соответственно.
(ср)2МеТа:СН2 (3)
В этой связи неудивительно, что карбеновые комплексы пере-
ходных металлов, как правило, не являются источниками сво-
бодных карбенов и что разнообразные превращения карбеновых
лигандов, в частности образование олефинов — формальных ди-
меров карбенов, а также карбонильных соединений, продуктов
35
CR2R3
CR6R7 и т.д.
циклопропанирования и т. п.» обычно протекает в координацион-
ной сфере переходного металла (см., например, [1375—1378],
а также раздел 5.3).
Роль карбеновых комплексов переходных металлов в реак-
циях распада диазосоединений и метиленирования реагентами
Симмонса — Смита и их аналогами уже рассматривалась ра-
нее; здесь мы кратко рассмотрим промежуточное участие кар-
беновых комплексов в ряде важных каталитических процессов,
а также возможности практического использования подобных
комплексов.
5.2. КАТАЛИТИЧЕСКОЕ ДИСПРОПОРЦИОНИРОВАНИЕ ОЛЕФИНОВ
Реакция каталитического диспропорционирования (метате-
зис) олефинов—наиболее важная из катализируемых металла-
ми органических реакций, открытых за последние 30 лет [1327].
Этот процесс открыт независимо несколькими группами ученых
[1379—1381] и представляет собой равновесную каталитическую
реакцию, при которой общее число о- и л-связей в исходных и
конечных продуктах не изменяется [1382]:
RR1C = CR2R3 CRR1
+ —* II +
r4r5c = cr6r7 cr4r5
В большинстве случаев реакция является практически изо-
термической и энергия активации ее невысока ( — 25—
30 кДж/моль). В этой реакции могут быть использованы как
гомогенные, так и гетерогенные катализаторы, содержащие
соединения W, Mo, Re и реже Та, Ti, Nb, 1г и Ru. Для гомоген-
ных систем циглеровского типа в качестве сокатализатора ши-
роко используют алюминийорганические соединения RnAlCU-n
(п = 0—3), а также органические производные кремния, олова,
магния и кислоты Льюиса; гетерогенные катализаторы — это
обычно оксиды или карбонилы W, Mo, Re на АЬОз/БЮг [1383].
Ранние представления о механизме инициирования, роста и
обрыва цепи реакции каталитического диспропорционирования
олефинов рассмотрены в обзорах [1382—1387].
В настоящее время общепринято, что в этой реакции ключе-
вую роль играет образование и превращение карбеновых комп-
лексов переходных металлов. Необходимо подчеркнуть, что та-
кая схема впервые была предложена еще в 1972 г. Б. А. Долго-
плоском и сотр. [1388]. Решающим доказательством ключевой
роли карбеновых комплексов переходных металлов в протека-
нии данной реакции является ее инициирование путем заведо-
мого генерирования карбенов при разложении их традиционных
источников — диазосоединений — в присутствии производных пе-
реходных металлов [1383]. В реальных условиях наиболее ве-
236
роятным путем возникновения активных карбеновых центров
считают распад органических производных переходных метал-
лов, которые образуются при алкилировании их солей
сокатализаторами — органическими соединениями Al, Mg, Li,
Sn, Si и т. п. Распад алкильных производных переходных ме-
таллов возможен лэ различным механизмам, в том числе и пу-
тем внутримолекулярного отщепления атома водорода от
а-атома углерода алкильной группы:
XnMCH2R- XnHM:CHR
2XnMCH2R - XnM:CHR + MeR + MXn
Xm M(CH2R)2 -XmM:CHR + MeR
XnMCH2Z- Xn_1M:CH2 + ZX
Подобные реакции хорошо известны в химии соединений
переходных металлов [1327] и исследованы на большом числе
модельных объектов — производных Mo, W, Re, Та, Nb, Ti
и т. д. (подробнее см. [1383, 1389, 1390]). По данным радиоизо-
топных исследований в системе WC16—SnMe4 лишь « 1 % WC13
образует карбеновые центры, активные в реакции каталитиче-
ского диспропорционирования олефинов; именно такое коли-
чество и в состоянии обеспечить высокую каталитическую ак-
тивность этой и подобных систем [1391].
Еще одним возможным путем инициирования метатезиса яв-
ляется образование карбеновых комплексов переходных метал-
лов в результате олефин-карбеновой изомеризации в сфере пе-
реходного металла [893, 1388, 1389, 1392]:
R1R2C
RHrML„-L„M:C<CHRlR2
Стадия роста цепи реакции каталитического диспропорцио-
нирования олефинов может быть описана схемой:
L^M-.CRR1
R\ ,R4 r\ I R4 £Rr1
Um-.crr’ + V/ )cr-r:;4 ^
R3/ 'R R3/ \r5 \Z .
(4) CR Rf'
(5)
LnM = CR4R5
RR1C==CR2R3 L„M-.CR4R5 + RR*C=CR2R3
(6)
237
Этот механизм («металлациклобутановый»), совпадающий
с предложенным ранее Б. А. Долгоплоском и сотр. [1388],
включает образование олефин-карбенового комплекса (4), его
переход в металлациклобутановую структуру (5) и обратный
процесс, приводящий через комплекс (6) к новым олефину и
карбеновому комплексу. Такая схема подтверждается рядом
фактов. Так, относительные скорости метатезиса уменьшаются
в ряду: вырожденный обмен метиленовых звеньев между конце-
выми олефинами > перекрестная реакция концевых и внутрен-
них олефинов > диспропорционирование внутренних олефи-
нов > образование этилена и внутренних олефинов из конце-
вых [1393] [реакции (5.1) — (5.4) соответственно]:
RCH = CH2 + R1CH = CD2^ RCH = CD2 + R1CH = СН2 (5.1)
RCH = CH2 + R’CH = CHR1 * RCH = CHR1 + R1CH = CH2 (5.2)
RCH = CHR + R’CH = CHR1 2RCH = CHR1
2RCH = CH2 5= RCH = CHR + CH2 = CH2
(5.3)
(5.4)
Такой ряд полностью соответствует относительной стабиль-
ности промежуточных карбеновых комплексов. Действительно,
сравним, например, стадии роста цепи для реакций типа (5.1)
и (5.4):
б а
Х„М:СН2 + RCH = CHR1 * XnM:CHR + R’CH = СН2 * XnM:CHR1 +
+ RCH = СН2
По направлению а образуется сравнительно стабильный
комплекс алкилкарбена, а образование внутреннего олефина
(путь б) требует участия менее стабильного метиленового комп-
лекса и будет реализовываться со значительно меньшей вероят-
ностью. Подобные же закономерности выявлены и при исследо-
вании модельных процессов — стехиометрических реакций кар-
беновых комплексов с олефинами [893, 1394, 1395], например:
Аг2С = СН2 + (CO)5W:CMe2
(CO)5W:CAr2 + Ме2С = СН2---
Аг2С = СМе2 + (CO)5W:CH2
ka:k6> 1200:1
238
—► Ar2C = CH2 + (CO)5W:CHPr
(CO)5W:CAr2 + PrCH = CH2--
Ar2C = CHPr + (CO)5W:CH2
Ar = C6H4Me-n ka:k6 = 600:1
На этих примерах хорошо видна предпочтительность образо-
вания комплексов алкил- и, особенно, диалкилкарбенов по срав-
нению с метиленовыми комплексами.
Невысокая, но отчетливая стереоселективность реакции ка-
талитического диспропорционирования олефинов также вполне
согласуется с металлациклобутановым механизмом. В этой ре-
акции обычно наблюдается тенденция к сохранению в продуктах
исходной (Z)- или (£)-конфигурации [1396]. Предполагают, что
за такую стереоселективность ответственно неплоекое строение
металлациклобутанового интермедиата типа (5) [1393, 1397].
Этому выводу соответствуют и результаты изучения химических
превращений металлациклобутанов, генерированных независи-
мыми путями (см., например, [1398]). При распаде металлацик-
лобутанов (7), получаемых реакцией [М:СН2]+ с циклопропа-
ном в газовой фазе, доминирующим процессом является обра-
зование карбен-олефинового комплекса [1399]:
МСН3--->
Последней стадией реакции каталитического диспропорцио-
нирования олефинов является обрыв цепи. Этот процесс естест-
венно связать с дезактивацией активного карбенового центра
в результате, например, карбен-олефиновой изомеризации или
превращения карбена в радикал [1383]:
239
XnM:CHCH2R- RCH = CH2 + MXn
XnM:CHR- RCHX + MXn.1
Еще одной возможностью обрыва цепи является образова-
ние значительно более стабильного (и нереакционноспособно-
го!) карбенового комплекса. Иллюстрацией может служить
прекращение реакции при введении, например, добавок винил-
триметилсилана — предшественника комплекса триметилсилил-
карбена, намного более стабильного, чем комплексы алкилкар-
бенов [1400]:
Хл M:CHR + СН2 = CHSiMe3 - RCH = СН2 + XnM:CHSiMe3
С образованием стабильного комплекса, видимо, связана и
низкая активность в реакциях метатезиса диенов [1384] —
предшественников винилкарбеновых 'комплексов; в модельных
реакциях с карбеновыми комплексами вольфрама диены деме-
тиленируются. Образование винилкарбеновых комплексов в этом
случае предполагается на основе выделения продуктов изоме-
ризации этих карбеновых лигандов [893]. Аналогичным обра-
зом, метатезис хорошо инициируется при разложении в присут-
ствии WC16 фенилдиазометана и этилдиазоацетата, дающих ак-
тивные комплексы фенил- и этоксикарбонилкарбенов; более ста-
бильные комплексы дифенил- и триметилсилилкарбенов менее
эффективны, а в присутствии комплексов еще менее активного
фенилбензоилкарбена реакция не идет вообще [1383].
Обрыв цепи метатезиса возможен также в результате обра-
зования циклопропановых аддуктов; формально это является
одним из возможных путей распада металлациклобутана (5).
Действительно, при пропускании этилена над Мо-катализато-
ром выходы циклопропана и метилциклопропана достигают со-
ответственно 8 и 12% (на превращенный этилен) [1381], одна-
ко такие высокие выходы продуктов циклопропанирования в це-
лом для этих процессов нетипичны. Так, в системе цис-бу-
TeH-2+PhWCl3—А1С1з при введении эквимольного (к бутену)
количества этилакрилата выход циклопропанового аддукта со-
ставил всего 0,04% [1401]:
МеСН = СНМе PhWCIj-AICJJ ClnW:CHMe <="^<="”<4!
цис
СН2
/\
- Me - НС - CHCOOEt
транс
240
Однако именно такие количества и должны быть характер-
ны для распада активных центров, концентрация которых в ка-
талитических системах невысока. Здесь уместно вспомнить, что
циклопропанирование непредельных соединений без участия
свободных карбенов, т. е. в координационной сфере переходного
металла, достаточно характерно для многих карбеновых комп-
лексов (см. разд. 4.1 и 5.4).
Отметим также, что известна реакция кросс-метатезиса цик-
лопропанов с олефинами («переметиленирование») [1401] [ре-
акция (5.5)], а также каталитическое диспропорционирование
ацетиленовых соединений в присутствии катализаторов, содер-
жащих W(CO)6 и фенол [1402].
Z^R + СН2 = CHCOOEt ^^А'С^ил^А'С^)
-CH2 = CHR + Д-cOOEt (5,5)
R = Aik
Реакция каталитического диспропорционирования олефинов,
видимо, уже в ближайшие годы займет важное место в нефте-
химических процессах, поскольку позволяет легко и эффектив-
но получать различные мономеры (этилен, бутен, изобутилен).
Наибольшие технические перспективы, пожалуй, имеет равно-
весная реакция бутена-2 с этиленом, которая может быть сме-
щена как в одну, так и в другую сторону [1403, 1404]:
2МеСН = СН2?± МеСН = СНМе + СН2 = СН2
Не менее важна возможность вовлечения в метатезис олефи-
нов, содержащих функциональные группы. Исследования в этом
направлении развиваются достаточно интенсивно. Для ряда
функционально замещенных олефинов достаточно эффективны-
ми оказались гомогенные системы WC16—SnAlk4 [1405] и
WC16— 1,1,3,3-тетраметил-1,3-дисилациклобутан [1406], а так-
же гетерогенные системы на основе Re2O7, промотированного
оловоорганическими соединениями [1407]; в этих условиях
удается достаточно эффективно проводить метатезис функцио-
нально замещенных олефинов и их сометатезис с алкенами, на-
пример [1406, 1408]:
RezOy/AlaQa
+ С2Н4
70%)
16—107
241
сн2 = CH(CH2)2COOEt »ЕЮОС(СН2)2СН = CH(CH2)2COOEt
(8) (47%)
WCIeL RCH = CH2
u
RCH = CH(CH2)2COOEt + RCH = CHR + (8) + C2H4
(42%) (25%) (4%)
L = /SiQjSi^ ;R = C5H11-h
5.3. ПОЛИМЕРИЗАЦИЯ ЦИКЛООЛЕФИНОВ С РАСКРЫТИЕМ ЦИКЛА
Этот процесс во многом аналогичен каталитическому диспропор-
ционированию олефинов и сводится к образованию полимеров,
полностью сохраняющих ненасыщенность исходного циклооле-
фина, в качестве которого могут быть использованы циклобу-
тен, циклопентен, циклогептен, циклооктен, циклооктадиен, цик-
лодецен, циклододецен и их замещенные:
НС = СН катали--^ ~ СН = СН(СН2)П ~
(СН2)П
Реакция проходит практически в тех же условиях, что и
диспропорционирование олефинов; в частности, обычно исполь-
зуют катализаторы тех же типов [1383], хотя, естественно, в ос-
новном гомогенные. Полагают, что механизмы этих реакций
близки, однако ряд особенностей полимеризации циклоолефинов
позволил внести существенный вклад в понимание ее механиз-
ма. Так, исследование полимеризации циклоолефинов позволяет
значительно проще и надежнее установить кинетические зако-
номерности на основе молекулярно-массового распределения об-
разующихся полимеров. Именно этим путем был доказан не
ступенчатый, а цепной механизм процесса — уже на ранних ста-
диях (конверсия циклоолефина <10%) в присутствии различ-
ных каталитических систем образуются полимеры с высокой мо-
лекулярной массой, а не низшие олигомеры, которые обычно
образуются в результате деструкции высокомолекулярных по-
лимеров, также протекающей по схеме метатезиса [1388, 1409].
Важным для понимания механизма данной группы реакций яви-
лось и проведение полимеризации ряда циклоолефинов при
инициировании комплексом [ (CO)5W:CPh2] [1410—1412]. В об-
242
разевавшихся полимерах на каждую молекулу в среднем оказа-
лась одна группа CPh2, что свидетельствует в пользу цепного
механизма, при котором карбеновый комплекс переходного ме-
талла ведет много актов полимеризации, вплоть до момента его
дезактивации.
Стадия роста цепи реакции полимеризации циклоолефинов
также имеет ряд особенностей. В соответствии с общим меха-
низмом (см. разд. 5.2) каждый акт этой стадии можно изобра-
зить схемой [1383]:
г^НС: МХЛ
НС.
11>СН2)т
иг*'
не:мх.п
(CH2)m
нс—мх„
нс—сн
Ьн2)т
-^НС нс. p[Q
Ц*-мх„ Х„м—>r>CH2)m
НС /СН --------> — сн=сн(сн2)тсн с
(сн2)т
~CH=CH(CH2)mCH=CH(CH2)mCH:MX„ и т.д.
При этом взаимодействовать с активным центром способны
двойные связи не только мономера (что приводит к увеличению
молекулярной массы полимера), но и ранее образовавшейся по-
лимерной цепи. При взаимодействии активного центра с другой
полимерной молекулой изменяется лишь молекулярно-массовое
распределение, тогда как взаимодействие с двойной связью
собственной цепи приводит к деструкции полимера:
** СН = СН НС
Х„М:СН(СН2,”в'СН:МХ"+ нР(СНг)т
Именно поэтому молекулярные массы образовавшихся поли-
меров максимальны при низких степенях конверсии мономера,
когда скорость полимеризации превышает скорость деструкции.
При глубокой конверсии роль циклодеструкции возрастает, и мо-
лекулярная масса полимера снижается [1383, 1390].
Возможность подобной циклодеструкции под действием ка-
тализаторов диспропорционирования показана и для других по-
линепредельных полимеров, в частности для полибутадиена
[1413—1417]. Именно в результате процесса деструкции при
полимеризации циклоолефинов наблюдается равновесие поли-
мер^мономер [1418, 1419], причем известная неспособность
циклогексена полимеризоваться по схеме с раскрытием цикла
16* 243
связана со смещением этого равновесия в сторону мономера
[1383].
Полимеризация циклоолефинов с раскрытием цикла позво-
ляет синтезировать принципиально новые полимерные структу-
ры, недоступные другими методами, с использованием нестан-
дартных мономеров. Так, полимеризация циклоолефинов позво-
ляет получать полимерные цепи с повторяющимися звеньями из
5—12 атомов углерода, в отличие от полимеризации диенов, где
повторяющееся звено максимально содержит четыре атома уг-
лерода [1390]. При этом полимеры на основе циклоолефинов
характеризуются меньшей долей двойных связей, т. е. более
устойчивы. Значительный интерес представляет сополимериза-
ция циклоолефинов с ациклическими непредельными соедине-
ниями, протекающая по той же схеме и позволяющая получать
линейные олигомерные продукты с регулируемой степенью нена-
сыщенности.
Еще одной интересной особенностью полимеризации цикло-
олефинов является образование небольших количеств катена-
нов. В частности, такие продукты выделены при полимеризации
циклододецена по схеме с раскрытием цикла в присутствии
WC16—EtAlCl2—EtOH [1420, 1421]:
НС=СН
НС—
II (СН2)10
нс—
WC16—EtAICIo
------------->
EtOH
-НС—СН
'т
Целесообразно здесь же кратко рассмотреть поведение на-
пряженных циклических молекул в присутствии катализаторов
того же типа. Ранее уже упоминалась реакция переметилениро-
вания (кросс-метатезис олефинов и циклопропанов; см.
разд. 5.2). Производные циклопропана под влиянием хлоридов
вольфрама изомеризуются в олефины, реагирующие далее по
обычной схеме метатезиса [1422]. Вместе с тем в присутствии
каталитической системы PhWCl3—А1С13 наблюдается деметиле-
нирование циклопропана и его алкилзамещенных [1401]. Ана-
логично, деметиленирование циклопропанов происходит при
взаимодействии комплекса [ (CO)sW:CPli2] с производными цик-
лопропана [1423], а катализаторы на основе WC16 или M0CI5
вызывают обессеривание тииранов [1424].
H2C-CHR
СН2
PhWCI3 - AICI3
~ RCH = СнТ
LnW:CH2- C2H4
R = H, Aik
244
Н2С - CHR + (CO)5W:CPh2 - Ph2C = CH2
R, R1=Alk CHR1
H2C - СНМе -C‘6 и™ Мо<^5 CH2 = СНМе
\ / “sx
S
При наличии в циклической молекуле одновременно цикло-
пропанового кольца и двойной связи на каталитической систе-
ме (1)—TiCl4 наблюдается полимеризация с раскрытием цикла,
причем в полученном полимере полностью сохраняются как
двойные связи, так и циклопропановые группировки [1425]:
(1)-TiCl4
= СНСН2СН2 - НС - сн - СН2СН2СН = ]п
Эта же каталитическая система активна и при полимериза-
ции бицикло[5.1.0]октена-2, в котором двойная связь сопряже-
на с трехчленным циклом [1425].
5.4. ДРУГИЕ КАТАЛИТИЧЕСКИЕ РЕАКЦИИ
КАРБЕНОВЫХ КОМПЛЕКСОВ
Реакции каталитического диспропорционирования олефинов и
полимеризации циклоолефинов отнюдь не исчерпывают список
процессов, в которых участвуют карбеновые комплексы пере-
ходных металлов. Еще одним фундаментальным процессом это-
го рода является реакция Фишера — Тропша — синтез углево-
дородов из СО и Н2 (см., например, [1426]). Этот процесс, от-
крытый в начале века, активно использовали в годы второй ми-
ровой войны в Германии для получения искусственного жидкого
топлива (740 тыс. т в 1942 г. [1426]), а сейчас он вновь привле-
кает к себе внимание в связи с перспективами каталитической
переработки бурых углей. Катализаторами служат соединения
Ni, Со, Fe, промотированные AI2O3, ThO2, SiO2, ZnO, оксиды
редкоземельных элементов; промышленными катализаторами
являются Со—ZrO2 (или ThO2)—MgO на кизельгуре, Fe3O4—
А12Оз—К2СО3—SiO2, Fe—Си—Na2SiO3 [1426].
Детальный механизм этого процесса неоднозначен и зависит
от используемого катализатора. Вместе с тем ясно, что сущест-
венную роль в реакции Фишера — Тропша играют молекулы
метилена и, возможно, высших алкилкарбенов, координирован-
ные с металлом [1427]. Характерно, что такой механизм по сути
был предложен еще в начале 40-х годов Н. Д. Зелинским и
Я. Т. Эйдусом, однако справедливость его доказана лишь срав-
245
нительно недавно [1427]. В пользу карбенного механизма реак-
ции Фишера — Тропша свидетельствует и образование углево-
дородов на металлах VIII группы из смеси CH2N2+H2 [1428,
1429]—заведомого источника карбеновых комплексов переход-
ных металлов («поверхностных частиц СН2»). Аналогичным об-
разом, оказалось, что на катализаторах Фишера — Тропша и их
аналогах (Fe/SiO2, Ru/SiO2, Os/SiO2, Rh/SiO2) механизм гомо-
логизации олефинов совпадает с механизмом реакции Фише-
ра— Тропша, т. е. связан с промежуточным участием карбено-
вых частиц [1430].
Промежуточное участие карбеновых комплексов предполага-
ется и во многих других процессах, например при гидрировании
ряда ненасыщенных соединений гидридами Ni, Со, Cr, Pd и дру-
гих металлов [1389, 1390]:
RCH = СН2 + Н - М - H-^(RCH2CH2)2M->RCHMe + M:CHCH2R
R = Aik, SiMe3, COR1 и др.
Предполагают [1389] также, что карбеновые комплексы пе-
реходных металлов участвуют в реакциях дегидрирования угле-
водородов и в процессах их скелетной изомеризации на катали-
заторах, содержащих А1—Pt, Pt, Mo, W, Re и т. п. В частности,
промежуточное участие циклопропановых производных в реак-
циях скелетной изомеризации может быть связано с внутримо-
лекулярными реакциями внедрения координированных на пере-
ходном металле карбенов [1389], а не только с реакциями
типа у-элиминирования. Наконец, в процессах олигомеризации и
полимеризации олефинов и диенов образование карбеновых
комплексов переходных металлов является побочной реакцией,
вероятность которой увеличивается при наличии у одного атома
переходного металла двух или более углеводородных остатков
(полигидриды металлов, комплексы типа jt-R3Cr, jt-R2Cr,
л-Р2СгС1, tt-R2CrOR, R4Ti, R3Ti и т. п.). В частности, образова-
нием карбеновых комплексов объясняют [1389, 1390] понижение
эффективности катализатора с увеличением глубины превра-
щения.
Возможно участие карбеновых комплексов в полимеризации
по Циглеру — Натта [1431] и в реакциях крекинга углеводоро-
дов [1432]. В последнем случае полагают, что образуются ме-
таллациклобутановые интермедиаты типа (2) (см. разд. 5.2),
характерные, видимо, и для процессов скелетной изомеризации
углеводородов [1432—1434], эпоксидирования и деэпоксидиро-
вания [1435, 1436]. Участие карбеновых интермедиатов неодно-
кратно обсуждалось при исследовании механизма С—Н-акти-
вации на различных переходных металлах (см., например,
[1437—1441]); вместе с тем показано [1442], что на Pt-катали-
246
заторах роль карбеновых комплексов не является опреде-
ляющей.
Следует подчеркнуть, что во всех этих процессах, за исклю-
чением, пожалуй, реакции Фишера — Тропша, промежуточное
участие карбеновых комплексов нельзя считать строго доказан-
ным. Это относится и ко многим частным реакциям, где проме-
жуточное участие карбеновых комплексов переходных металлов
зачастую постулируется без специальных исследований. Отме-
тим, например, реакции изомеризации напряженных углеводо-
родов в присутствии производных Rh и Ag [1443, 1444] и при-
соединение нуклеофилов к изоцианидам в присутствии солей Си
и Pd [1445, 1446].
Каталитические свойства карбеновых комплексов переход-
ных металлов используют и в тонком органическом синтезе.
Так, метоксифенилкарбеновый комплекс пентакарбонилвольфра-
ма катализирует реакцию, являющуюся своеобразным гибри-
дом метатезиса и полимеризации ацетиленов и приводящую к
производным поликонденсированных ароматических углеводоро-
дов, например [1447]:
С = СН
СН = СНМе
цис
(CO)5W:CPh(OMe)
Карбеновые комплексы переходных металлов являются так-
же эффективными катализаторами реакции переалкилирования
третичных аминов — общего метода синтеза несимметричных
третичных аминов [1448, 1449].
Однако применение карбеновых комплексов в органическом
синтезе не ограничивается использованием их каталитических
свойств. Известен ряд реакций, в которых карбеновые комплек-
сы переходного металла участвуют в качестве стехиометриче-
ских реагентов. В первую очередь это относится к реакции цик-
лопропанирОвания непредельных соединений. Так, комплексы
фишеровского типа достаточно эффективно циклопропанируют
электронодефицитные эфиры а,[3-ненасыщенных кислот [1377],
например:
СООМе Ph
+ (СО)5М:СОМе—*
Me
Ph СООМе
Me
247
МеО СООМе
+ рМ
Me
При использовании хиральных комплексов таким путем уда-
ется получить оптически активные производные циклопропа-
на [1327].
Эффективными метиленирующими агентами являются комп-
лексы (9; R = R1 = H); (10; М = Мо, W) и т. п.; при этом выходы
продуктов циклопропанирования достигают 98% [1356, 1450—
1452]. Аналогичным образом источником метилкарбена служит
комплекс (9; R = H, R!=Me) [1453, 1454], а комплексы (11)
[1455] и (9; R=H, R! = Ph) [1456] являются эффективными
переносчиками фенилкарбена. Следует подчеркнуть, что реак-
ции циклопропанирования подобными карбеновыми комплекса-
ми отличаются от традиционных металлорганических или диазо-
методов высокой цис(син)-селективностью [1453—1457]. Нако-
нец, недавно как достаточно эффективные источники соответ-
ствующих карбенов охарактеризованы также комплексы (9;
R = Ri=Me и R = H, R1 = СН = СМе2) [1458].
[ср (CO)2Fe:CRR1]+“BF4 [ср (PPh3)(CO)2M:CH2]+
(9) (10)
[(CO)5W:CHPh]+ (11)
СНСН = СМе2
м. г ГЦ [ср (СО)гРе:СНСН = СМе;]*~BF4 м /_\Н
Ме2С = СН2 ------------—---------► те2и им2
(56%)
Карбеновые лиганды в комплексах переходных металлов
могут быть использованы и в других реакциях. В частности,
под действием окислителей (CeIV, О2, пиридин-М-оксид и т. п.)
они превращаются в карбонильные соединения, например
[1378]:
О-окислитель
(CO)5W:C(OMe)R -----------RCOOMe
Еще одним примером подобных превращений является доста-
точно общий метод синтеза а-метиленлактонов, основанный на
последовательной модификации карбенового лиганда (за счет
248
кислого характера а-протонов) и последующего окисления
[1459]:
п 1-BuLi / \ I.BuLi
(СО)5Сг:СМе —* (СО^Сг:^ ) ----------------►
' 2- v X 2.С1СНг0Ме
ОМе о и
МеОН2С Н2С
(СО)5Сг:^3-^-(СО)5Сг:1Э
О О
Ce'v
Металлирование а-положений карбеновых лигандов (крип-
токарбокатионов [1327]) используют и в синтезе других карбо-
нильных производных [1460]:
CH2R
I BuLi
(СО)5М:СОМе —*
Li+ CHR
_ И
(СО)5М - СОМе
R1COR2
------
кислота
Льюиса
ОМе OR3 ОМе OR3
—» (CO)5M:CCHRCR1R2 Яо = CCHRCR’R2
R = Н, Me; R1 = Me, Pr, Ph, CHMePh; R2 = H, Me, OMe;
R3 = H, Me, Ac
Термолиз комплексов арилалкоксикарбенов достаточно глад-
ко приводит к соответствующим с/иьолефинам. Непредельные
соединения могут быть получены и при взаимодействии карбе-
новых комплексов переходных металлов с активными фосфора-
нами, например [1376]:
(CO)5W.C(OMe)Ph + Ph3P = СН2-^ СН2 = C(OMe)Ph +
+ (CO)5WPPh3
Перспективным путем использования карбеновых комплексов
переходных металлов в направленном органическом синтезе яв-
ляется построение новых циклов из нескольких компонентов.
В качестве примеров можно привести две достаточно общие
249
схемы построения шестичленного цикла циклогексадиенонов с
участием комплексов метоксикарбенов [1461 —1463]:
I
ОМе
МеО. ..
> С" чс
Карбеновые комплексы переходных металлов используют в
направленном синтезе природных соединений, например в син-
тезе фурохромона хеллина [1464]. Использование хромового
комплекса метоксифурилкарбена на ключевой стадии (построе-
ние бензофуранового ядра) позволяет полностью обеспечить
нужную региоселективность [ 1464] -
OMe OSiMe2Bu-rper ai2o3,
I /Г“А I NEt3
(СО)5Сг:С-<ч/^ + MeCHC = COEt ---------~
О
(43%)
В заключение необходимо подчеркнуть, что применение кар-
беновых комплексов переходных металлов в тонком органиче-
ском синтезе только начинается, но в этой области в ближай-
шем будущем следует ожидать определенных достижений. По-
мимо возможностей реализации этим путем принципиально но-
вых синтетических схем такой подход открывает и перспективы
значительного повышения селективности карбенных реакций в
результате снижения реакционной способности карбенов при их
координации с соединениями переходных металлов.
250
Глава 6
НЕКОТОРЫЕ ПЕРСПЕКТИВЫ ПРИМЕНЕНИЯ
КАРБЕННЫХ РЕАКЦИЙ В ПРОМЫШЛЕННОМ
ОРГАНИЧЕСКОМ СИНТЕЗЕ
Несмотря на сравнительно короткий срок самостоятельного раз-
вития химии карбенов, на основе карбенных реакций уже разра-
ботаны способы получения ряда важных мономеров, пестицидов,
лекарственных препаратов, красителей и других важных техни-
ческих продуктов. Некоторые из этих методов внедрены в про-
мышленное производство или находятся на стадии внедрения.
Кроме того, детальное исследование механизма таких извест-
ных промышленных процессов, как пиролиз, электрокрекинг,
окислительный пиролиз углеводородов, метатезис олефинов по-
зволило выявить существенный вклад карбенных реакций в эти
процессы.
Для установления механизма карбенных реакций наряду с
надежными и точными физико-химическими методами прямого
изучения этих реакций используют также изучение кинетиче-
ских закономерностей. Однако кинетических данных, относя-
щихся к реакциям образования и превращения карбенов, край-
не мало [468], причем в отличие от радикальных реакций, на-
дежность их определения в ряде случаев невысока, что в боль-
шой степени связано с высокой активностью карбенов и труд-
ностью выделения в карбенных процессах отдельных химических
стадий [468]. Этим в значительной мере обусловлена немного-
численность внедренных в промышленность процессов. Эти про-
цессы рассмотрены ниже.
6.1. ТЕРМИЧЕСКИЕ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ
Основным промышленным методом получения этилена, пропи-
лена, ацетилена, бутенов и сопутствующих им аллена, цикло-
пентадиена и других углеводородов является пиролиз природ-
ных углеводородов [1465], на что расходуется около 5% добы-
ваемой нефти, причем предполагается расширение использова-
ния этого сырьевого источника [1465]. Пиролиз углеводородов,
протекающий, как отмечалось, с участием карбеновых интер-
медиатов, в настоящее время проводят при 900—1000 °C; более
того, есть тенденция к повышению температуры проведения та-
ких процессов до 1800—2000 °C, что позволяет получать, напри-
мер, этцлен из сырой нефти [1466] и деасфальтированного
сырья [1467] с хорошим выходом при малых временах контак-
та. Сочетание неполного горения метана с пиролизом, а именно
251
окислительный пиролиз метана, является одним из основных
методов производства ацетилена [1468].
Термическое разложение метана включает в качестве первич-
ной стадии образование метилена (£аКт = 382,2 кДж/моль) [558,
560, 1469, 1470]. Аналогично протекает и пиролиз метана в из-
бытке водорода [560], а также гетерогенное разложение метана
с образованием пироуглерода [1470].
Весьма вероятно образование карбеновых промежуточных
частиц и при термических превращениях высших углеводородов.
Показано [566], что доля цепного протекания крекинга алканов
в ряде случаев незначительна (не более 5%); диапазон энергий
активации их распада (265—300 кДж/моль) перекрывается теп-
лотами образования карбеновых частиц [566, 1471, 1472], на-
пример:
RCH3 -* :CHR + Н2 АН = 330,5 кДж/моль
R1RCH2- :CRR1 + Н2 АН = 308:7 кДж/моль
С2Н6 “*:СН2 + сн4 А Н = 287,3 кДж/моль
(СН3)3СН :С(СН3)2 + СН4 АН = 265,4 кДж/моль
Поскольку механизм процесса пиролиза углеводородов вы-
яснен недостаточно, в каждом отдельном случае кинетическая
модель составляется исходя из схемы, включающей, как пра-
вило, ряд радикальных реакций; число реакций и их параметры
варьируются до получения удовлетворительного согласия расче-
та и эксперимента. Выяснение роли карбеновых промежуточных
частиц в процессах термических превращений углеводородов
позволит полнее использовать возможности производства. Так,
учет реакции образования метилена из метильного радикала в
процессе окислительного пиролиза метана с образованием аце-
тилена позволил достичь качественного совпадения данных рас-
чета и эксперимента [1473]:
•СН3- :СН2 + Н
СН3 + СН2 - С2Н2 + Н2 + Н Д = 65,9 кДж/моль
252
Без учета этих реакций данные расчета и эксперимента не
согласуются. Образующийся метилен, очевидно, вступает и в
другие реакции, например [1474]:
н2 + С02 *- :СН2 + о2 —-
н2 + со + о
н- + со + но-
При повышении температуры направление процесса окисли-
тельного пиролиза метана смещается в сторону образования
ацетилена (через метилен) [1473]. Повышение температуры пи-
ролиза должно также увеличивать вклад реакций таких карбе-
новых частиц, как дикарбены :С: и :С = С:, а также вклад кар-
бина :СН. В частности, с реакцией карбина [1475] связано уве-
личение концентрации ионов в диффузионном пламени пропана
при 1400—1500 °C:
:СН + О-*СНО + е
При получении ненасыщенных соединений электрокрекингом
углеводородов образуются такие же промежуточные частицы,
как и при высокотемпературном пиролизе. Так, об участии ме-
тилена в качестве интермедиата в этом процессе свидетельству-
ют данные исследования распада СН4 в тлеющем разряде
[1476] и в плазме дугового разряда переменного тока [1477].
При этом наряду с метиленом здесь участвуют также частицы
С, С2 и СН.
Поскольку пиролиз нефтяных фракций остается главным ис-
точником сырья для нефтехимической промышленности и общей
тенденцией его развития является повышение температуры
[1478], концентрации дикарбеновых частиц С и С2 в реакцион-
ной зоне при высоких температурах могут стать настолько вы-
сокими, что реакции этих частиц на холодной поверхности или
при закалке холодными реагентами смогут приобрести практи-
ческое значение [982, 983, 1479—1482].
В производстве топлив из нефти большие перспективы име-
ет характерная для карбенов реакция циклопропанирования
олефинов, которая может заменить или дополнить широко ис-
пользуемую в этом производстве стадию гидрирования стадией
циклопропанирования, что позволит получать высококачествен-
ное топливо со «сверхоктановым» числом [1483]. Для промыш-
ленного получения циклопропановых углеводородов может быть
использовано диспропорционирование олефинов по схеме мета-
тезиса с участием карбеновых активных центров [1401]. Так,
например, продукты диспропорционирования этилена на катали-
заторе Мо(СО)б/А12Оз содержат до 8% циклопропана и 12%
метилциклопропана [1381].
253
6.2. ПРОИЗВОДСТВО ФТОРСОДЕРЖАЩИХ МОНОМЕРОВ
Реакции галогенкарбенов (CF2, CFCI, CFCN, CCICN, CFPh и
др.) лежат в основе многих способов получения фторолефинов
[1484]. Наибольшее применение в промышленном фтороргани-
ческом синтезе имеет димеризация дифторкарбена в тетрафтор-
этилен— наиболее многотоннажный фторсодержащий мономер
[468, 1485, I486]:
CHCIF2* :CF2 + HCI 2:CF2^C2F4
Для получения тетрафторэтилена, свободного от микропри-
месей хлорорганических соединений, используют бесхлорный
источник дифторкарбена, в частности фтороформ; для разделе-
ния продуктов пиролиза применяют специальные методы, в том
числе экстракционную дистилляцию [1487, 1488].
Тетрафторэтилен применяют для получения полимеров,
в частности политетрафторэтилена [1489], а также как полу-
продукт в синтезе ряда других важных фторорганических соеди-
нений [1490—1493]:
f2c - cf2*2-c2f4
о
ch2 = chcn
F2f - CF2
f2c - cf2
CF2 = CHCN + CH2 = CF2
Промышленное производство второго важного фторсодержа-
щего мономера — гексафторпропилена — также основано на ре-
акциях дифторкарбена [1494, 1495]:
CF2 = CF2 + :CF2 цикло-С3Р6 - CF3CF = CF2
В качестве сырья для производства перфторпропилена мо-
жет применяться также тетрафторэтилен [1494, 1496], хотя ис-
пользование для данного процесса фтороформа более экономич-
но [468, 1497, 14-98]. Следует отметить, что графитовая стенка
реактора катализкрует дальнейшие превращения перфторпро-
пилена, также протекающие с участием карбенов [1484]:
CF3CF = CF^* :C(CF3)2^CF2 = C(CF3)2
254
Перфторпропилен исйрльзуют также для синтеза перфтор-
(метилоксирана) [1490]: 4
CF3CF = CF2-^^^F3 -FC-^F2
О
На основе карбенных реакций могут быть получены и другие
фтормономеры ряда этилена, например 1,1-дифтор- и трифтор-
хлорэтилены [1499, 1500], и ряда стирола [1501, 1502].
Фторсодержащие производные акрилонитрила также получа-
ют реакциями с участием карбеновых частиц [468, 1503—1505],
например:
CHFXCN —^»;CFCN - NCCF = CFCN
—НХ
Х = Hal
F F
NCCF = CFCN + C2H4ANC4d""CN 2CH2 = CFCN
Разработаны и одностадийные методы получения фторакри-
лонитрилов [586].
Большое значение для защиты окружающей среды и повы-
шения экономичности основных производств имеет переработка
в полезные продукты отходов различных фторорганических про-
изводств. Наиболее универсальным и перспективным путем пе-
реработки являются высокотемпературные пиролитические и
плазмохимические превращения самых разнообразных смесей.
При этом, как и в случае пиролиза углеводородов, важную роль
должны играть реакции промежуточных карбеновых частиц С,
С = С и CF2- Примером таких процессов является получение
тетрафторэтилена и других ценных продуктов из смесей, содер-
жащих CF4, его гомологи, SiF4 и другие фториды, во вращаю-
щейся-углеродной дуге, причем содержание СгР4 в продуктах
реакции достигает 64% [1506].
6.3. ПОЛУЧЕНИЕ ФТОРАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Фторароматические производные представляют большой прак-
тический интерес как полупродукты в синтезе лекарственных
препаратов, пестицидов, красителей, полимерных материалов
и т. п. Обычно их получают из соответствующих ароматических
аминов по методу Шимана — Шерера или замещением хлора на
фтор в ароматических полихлоридах [1484].
Дигалогенкарбены, присоединяясь по кратной связи цикло-
пентадиенов-1,3, образуют 6,6-дигалогенбицикло [3.1.0] гексе-
255
ны-3, которые под действием основании или термически претер-
певают легкое дегидрогалогенирование с расширением пяти-
членного цикла и образованием термодинамически выгодной
ароматической системы:
Эти реакции могут быть объединены со стадией термическо-
го генерирования дигалогенкарбена в один пиролитический про-
цесс, что позволило разработать новый общий промышленный
метод получения фторбензола и его производных с выходами до
90—100% [815,911, 1324, 1507—1511]:
F
CHXYF+
+ НХ + HY
X, Y = Hal
Аналогично, исходя из индена и его производных, получают
2-фторнафталины (выходы 50—8О°/о) [815, 1510, 1512]:
X = Hal
Следует отметить, что при синтезе этих соединений по Ши-
ману — Шереру [1513] необходимо использование канцероген-
ных 2-аминонафталинов. Для получения 2-фторнафталинов кар-
беновым методом вместо инденов могут быть использованы ал-
лилбензол и его гомологи [1514], термически легко превращаю-
щиеся в индены [1510].
Используя легкость термической изомеризации винилцикло-
пропанов, в том числе 1-винил-2,2-дигалогенциклопропанов,
в соответствующие циклопентены [1318] и легкость термическо-
го дегидрогалогенирования геж-дигалогенциклопентенов в гало-
генциклопентадиены, удалось на основе сопиролиза источника
галогенфторкарбена с ациклическими 1,3-диенами предложить
простой одностадийный способ получения изомерных дифтор-
256
бензолов (выходы 30—\0%) [911, 1507, 1509, 1511]. Аналогич-
но, термическим взаимодействием галогенфторкарбенов со сти-
ролом и его замещенными удалось получить неизвестные ранее
2,3-дифторнафталины с выходами 30—60% [911, 1324, 1507„
1512, 1515]:
СН2 = СНСН = СН2 CHF*^ YFC - СНСН = СН2-А^
-нх \ /
СН2
X, Y = Hal
X = Hal
По аналогичной схеме получается 2,3-дифторфенантрен из
2-винилнафталина [911].
Технологически карбеновый метод получения ароматических
фторидов оформляется в виде газофазного одностадийного не-
прерывного процесса [1516]. Процесс прост в аппаратурном ис-
полнении и наиболее экономичен среди известных методов полу-
чения фторароматических соединений. Проведение процесса в
присутствии паров воды, промотирующих карбеновый распад
дифторхлор- и фтордихлорметанов, позволяет значительно по-
высить селективность образования фторбензола [468]. Исполь-
зование в качестве источника галогенфторкарбена фтордихлор-
метана, который является промежуточным продуктом при про-
изводстве дифторхлорметана [1486], повышает экономичность
процесса, поскольку при этом не теряется связанный фтор и
17—107
257
упрощается система выделения и очистки продуктов реакции
[468]. По приблизительной оценке [1517] карбеновый метод
синтеза фторбензола при мощности установки 10 т/год более
чем в 20 раз экономичнее метода Шимана — Шерера [1513].
6.4. ПОЛУЧЕНИЕ ЦИКЛОПРОПАНОВЫХ ПРОИЗВОДНЫХ
Для карбенов наиболее характерно присоединение по кратным
связям непредельных соединений с образованием производных
циклопропана и циклопропена:
CRR1 CRR1
/ \ -CSC- >с = с< ч / \
- С = С - ------:CRR’ -------- )С—С<
Как уже отмечалось, для дигалогенциклопропанирования
олефинов особый интерес с точки зрения перспектив промыш-
ленной реализации представляет генерирование дигалогенкар-
бенов щелочным гидролизом галоформов в условиях межфазно-
го катализа [914]. Этот метод не требует применения сильных
оснований и абсолютированных растворителей (см. разд. 4.3).
Использование катализаторов фазового переноса, иммобилизо-
ванных на твердой инертной матрице, позволяет значительно
упростить технологическое оформление процесса в непрерывном
режиме.
Одним из примеров возможной промышленной реализации
двухфазного метода является получение биологически активных
соединений, например 2,2-диметил-3,3-дихлорциклопропанкарбо-
новой кислоты и ее производных [1518]:
Ме2С = CHCONHCMe3 + СНС13
50%-й водн. NaOH
КФП
СС12
Ме2С—-CHCONHCMe3
Доступность геж-дигалогенциклопропанов позволяет по-но-
вому оценить и перспективы практического использования раз-
нообразных химических превращений соединений этого класса,
делающих доступными моногалогенциклопропаны, циклопропа-
новые и циклопропеновые углеводороды, аллены, циклогепта-
триен и его гомологи, галогеналлиловые спирты, индены и дру-
гие соединения (см. разд. 4.3). Например, легко получаемый из
изобутилена и галоформов через 1,1-дигалоген-2,2-диметилцик-
лопропаны 3,3-диметилциклопропен [1322] может быть исполь-
258
зован для синтеза хризантемовой кислоты и ее производных
[874, 1519-1523]: СМе2 СМе2
1 2 А з
СН2 = СМе2—*н2с— сх2—»н2с— СНХ—*
СМе2 СНСН = СМе2 СНСН = СМе9
/\ 4 5 /\
—нс = СН--*Ме2С~CHMgBr—*Ме2С — СНСООН
I 6 цис
7 СНСН = СМе9
1 8 /\
:СНСН = СМе2—*Ме2С—CHCOOR
1 - СНХ3, водн. NaOH, КФП; 2- восстановление; 3- КОН, КФП
или трет-ВиОК; 4- Ме2С = CHMgBr; 5- СО2; 6- Н3О+;
I7 - Ni(cod)2; 8- Ме2С = CHCOOR
Другим важным и широко применяемым источником карбе-
нов являются диазосоединения. В частности, каталитическое
разложение диазосоединений широко используют в промышлен-
ности тонкого органического синтеза для получения биологиче-
ски активных веществ циклопропанового ряда. Наибольшую
практическую значимость имеют синтезы с участием алкилди-
азоацетатов, являющихся доступными и достаточно стабильны-
ми реагентами. Так, например, каталитическое разложение ал-
килдиазоацетатов в присутствии диенов лежит в основе ряда
промышленных методов получения пиретроидов — производных
перметриновой (R = C1) и хризантемовой (R = Me) кислот [1519,
1520, 1524—1526]:
Ме2С = СНСН = CR2
n2chcoor1 ,
катализатор
-N2
СНСН = cr2
.. х Н3О+
Ме2С - CHCOOR1 ---
CHCH = CR2
R2R3CHOH
Ме2С - СНСООН --------’
СНСН = CR?
/\ 2
Ме2С - CHCOOCHR2R3
R = Cl, Br, Me; R1 = Aik; R2 = H, CN; R3 = C6H4OPh-3, Ph,
2-бензилфурил-4, 2-феноксипиридил-6, 1,3-диоксо-4 5 6 7-тет
рагидроизоиндолинил-2 ’ ’ ’
17’
259
Реализованы и другие процессы метатезиса, например, про-
цесс «SHOP» (Shell High Olefin Process) [1543]. Процесс на-
правлен на получение высших олефинов, необходимых для про-
изводства поверхностно-активных веществ, путем последова-
тельной олигомеризации в а-олефины и их метатезиса [54].
Другой промышленный процесс фирмы «Shell» связан с получе-
нием а,со-алкадиенов сометатезисом этилена с соответствующи-
ми циклоолефинами. Так, гексадиен-1,5 получен из этилена и
циклооктадиена-1,5, а декадиен-1,9 — из этилена и циклоокте-
на [1544]:
Re2O7/А12О3
С2Н4 + П Н--------► СН2 = СН(СН2)2СН = сн2
Re2o7/Al2o3
ГЧ I I II I J
С2Н4 + III-----------* сн2 = СН(СН2)6СН = сн2
Сометатезис этилена и 2,4,4-триметилпентена-2 используется
для промышленного производства душистых веществ [1545,
15461:
С2н4 + трет-BuCH = СМе2 У°з5Ю2-МдО трег.ВиСН = +
+ СН2 = СМе2
Реакции метДтезиса используют и в других малотоннажных
процессах тонкого органического синтеза, в частности, для по-
лучения различных феромонов (трикозена-9, гептакозена-13.
нонакозенов-13 и -14 и т. п.) [1547—1551].
Нашли применение в промышленности и реакции полимери-
зации циклоолефинов с раскрытием цикла (см. разд. 5.3)
[1552—1554]. Ряд примеров практического использования реак-
ции метатезиса олефинов и полимеризации циклоолефинов рас-
смотрен в обзоре [1555] и в монографии [1556].
Широкие перспективы получения большого числа крупнотон-
нажных химических продуктов открывают новые реакции окис-
ления в условиях метатезиса [1557], например:
Интенсивные исследования карбеновых комплексов переход-
ных металлов, особенно в реакциях метатезиса и полимериза-
ции циклоолефинов, и выявление новых возможностей их прак-
тического использования приведут, очевидно, к тому, что химия
комплексных соединений карбенов, подобно оксосинтезу, в бли-
жайшее время выделится в большую самостоятельную область
химии.
262
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Номенклатурные правила ИЮПАК по химии. В 6 т. Т. 5. Физическая ор-
ганическая химия: Пер. с англ. М.: ВИНИТИ. 1985. С. 63, 219.
2. Вольпин М. Е., Кирсанов Д. Н.ЦЖ. общ. хим. 1962. Т. 32. С. 1142; Nefe-
dow О. М., Manakow М. N.f/Amgew. Chem. 1966. В. 78. S. 1966.
3. Колесников С. П.Ц7К. ВХО им. Д. И. Менделеева. 1979. Т. 24. С. 505—512.
4. Кирмсе В. Химия карбенов: Пер. с англ./Под ред. Д. Н. Курсанова. М.:
Мир, 1966. 302 с.
5. Hine J. Divalent Carbon. N. Y.: Ronald Press, 1964. 196 p.
6. Kirmse W. Carbenes, Carbenoids und Carbene-Analoge. Weinheim: Verlag
Chemie, 1969. 260 p.
7. Kirmse W. Carbene Chemistry. 2-nd Ed. N. Y.: Acad. Press, 1971. 615 p.
8. Carbenes/Eds. R. A. Moss, M. Jones. In 2 V. N. Y.: Wilev, V. 1. 1973.
356 p.; V. 2. 1975. 374 p.
9. «Reactive Intermediates/Eds. R. A Moss, M. Jones. N. Y.: Wiley. In 3 V.
V. 1. 1978. 350 p.; V. 2. 1981. 396 p.; V. 3. 1985. 436 p.
10. Reactive Intermediates/Ed. R. A. Abramovitch N. Y.: Plenum Press. In 2 V.
V. 1. 1980. 450 p; V. 2. 1982. 522 p.
11. Chemistry of Carbenes and Small-Cized Cyclic Compounds./Ed. О. M. Ne-
fedov. M.: Mir. 1989. 310 p.
12. Зефиров H. С., Казимирчик И. В., Лукин К. А. Циклоприсоединение ди-
хлоркарбена к олефинам. М.: Наука, 1985. 202 с.
13. Wendisch D. //Houben-Weyl Methoden der organische Chemie. Stuttgart:
Verlag. Bd. IV/III. 1971. 880 p.
14. Lwowski W. Nitrenes. N. Y.: Wiley-Interscience. 1970. 316 p.
15. Azides and Nitrenes/Ed. E. F. V. Scriven. Orlando: Acad Press. 1984. 210 p.
16. Nefedov О. M., Manakow M. N.l/Amgew. Chem. 1966. B. 78. S. 1039.
17. Нефедов О. Af./Дис ... д-ра хим. наук: 02.00.03. М., 1967. 356 с.
17а. Дьяконов И. А. Алифатические диазосоединения. Л.: Изд. ЛГУ, 1956,
138 с.
176. Кнунянц И. Л. и др.//Усп. химии. 1958. Т. 27. С. 1361.
17в. Pure a. Appl. Chem. 1984, V. 56. Р. 769.
18. Moss R. А. //Асе. Chem. Res. 1980. V. 13. Р. 58—64.
19. Moss R. A., Young С. М., Perez L. A., Krogh-Jespersen K-//L Amer. Chem.
Soc. 1981. V. 103. P. 2413—2415.
20. Hoffmann R., Zeiss G. D., Van Dine G. W.HL Amer. Chem. Soc. 1968.
V. 90. P. 1485—1499.
21. Gleiter R., Hoffman R.//J. Amer. Chem. Soc. 1968. V. 90. P. 5457—5460.
22. DeFrees A. J., Raghavachari K, Schlegel H. B., Pople J. A.//Ibid. 1982.
V. 104. P. 5576—5580.
23. Borden W. T., Davidson E. R. //Ann. Rev. Phys. Chem. 1979. V. 30.
P. 125.
24. Harrison J. F./fNcc. Chem. Res. 1974. V. 7. P. 378—384.
25. Harrison J. F., Liedtke R. C., Lieb man J. F.//J. Amer. Chem. Soc. 1979.
V. 101. P. 7162—7168.
26. Shepard R., Simous /.//Int. J. Quantum Chem. 1980. Quant. Chem. Symp.
№ 14. P. 349.
27. Waali E. E.//J. Amer. Chem. Soc. 1981. V. 103. P. 3604—3606.
28. Kassaee M. Z., Nimlos M. R., Downie K> E. e. a.//Tetrahedron. 1985. V. 41.
P. 1579.
29. Saito K, Ishihara //.//Bull. Chem. Soc. Jap. 1986. V. 59. P. 1095—1098.
30. Cuthbertson A. F., Glidewell C., Lloyd D.//L Chem. Res. Part S. 1982.
P. 80.
31. Raghavachari K-, Frisch M. J., Pople J. A. e. a.//Chem. Phys. Lett 1982.
V. 85. P. 145.
263
32. Мальцев А. /(.//Новое в химии карбенов. М.: Наука, 1973. С. 10.
33. Мальцев А. К.ЦЖ. ВХО им. Д. И. Менделеева. 1979. Т. 24. С. 445.
34. Shultz G., Tremmel J., Hargittai I. e. a. //J. Mol. Struct. 1979. V. 55.
P. 207—214; 1982. V. 82. P. 107—113.
35. Hargittai I., Schultz G., Tremmel J. e. a.//J. Amer. Chem. Soc. 1983. V. 105.
V. 77. P. 5348—5362.
36. Herzberg G.//Angew. Chem. 1972. B. 84. S. 1126—1142.
P. 2895—2896.
37. Herzberg G., Johus J. W. C.//J. Chem. Phys. 1971. V. 54. P. 2276.
38. Zittel P. F., Ellison G. B., ONeil S. V. e. а.Ц]. Amer. Chem. Soc. 1976.
V 98 P 3731________3732
39. Sears T. Y., Bunker P. R., Me Ke liar A. R. W. e. aJ/J. Chem. Phys. 1982.
40. Yensen P., Bunker P. R., Hoy A. RJ/J. Chem. Phys. 1982. V. 77. P. 5370.
41. Mathews C. W.//J. Chem. Phys. 1966. V. 45. P. 1068; Can. J. Phys. 1967.
V. 45. P. 2355.
42. Powell F. X., Lide D. R.//J. Chem. Phys. 1966. V. 45. P. 1067.
43. Kirchhoff W. H., Lide D. R., Powell F. Х.Ц1. Mol. Spectrosc. 1973. V. 47.
P. 491.
44. Bondybey V. E.//J. Mol. Spectrosc. 1976. V. 63. P. 164.
45. Ishiguro T., Hamada Y., Tsuboi A4.//Bull. Chem. Soc. Jap. 1981. V. 54.
P. 367—374.
46. Merer A. J., Travis D. A.//Can. J. Phys. 1966. V. 44. P. 525, 1541.
47. Jacox M. E.t Milligan D. E.//J. Chem. Phys. 1969. V. 50. P. 3252.
48. Samsonov Yu. N., Petrov A. K.//Chem. Phys. Lett. 1981. V. 84. P. 183.
49. Мальцев А. К., Микаэлян P. Г., Нефедов О. Л4.//Докл. АН СССР. 1971.
Т. 201. С. 901; Изв. АН СССР. Сер. хим. 1971. С. 199.
50. Maltsev А. К., Mikaelian R. G., Nefedov О. Л4.//Ргос. Nat. Acad. Sci. USA.
1971. V. 68. P. 3238.
51. Hastie J. W., Hauge R. H., Margrave J. L.//L Mol. Spectrosc. 1969. V. 29.
P. 152.
52. Jacox M. E., Milligan D. E.ffJ. Chem. Phys. 1970. V. 53. P. 2688.
53. Milligan D. E., Jacox M. Е.Ц1. Chem. Phys. 1967. V. 47. P. 703.
54. Jacox M. E., Milligan D. E.ffJ. Chem. Phys. 1967. V. 47. P. 1626.
55. Kakimoto M., Saito S., Hirota Е.Ц}. Mol. Spectrosc. 1983. V. 97. P. 194—
203.
56. Smith С. E., Milligan D. E., Jacox M. Е.Ц1. Chem. Phys. 1971. V. 54.
P. 2780.
57. Bondybey V. E., English J. H.//J. Mol. Spectrosc. 1977. V. 68. P. 89.
58. Tevault D. E., Andrews L.//J. Mol. Spectrosc. 1975. V. 54. P. 54.
59. Andrews L., Carver T. G.UJ. Chem. Phys. 1968. V. 49. P. 896.
60. Ivey R. C.//J. Chem. Phys. 1974. V. 60. P. 3174.
61. Tevault D. E., Andrews L.HJ. Amer. Chem. Soc. 1975. V. 97. P. 1707—
1710.
62. Термодинамические свойства индивидуальных веществ. В 4 т. Т. 2. М.:
Наука, 1979. Книга 1. 440 с., книга 2. 344 с.; Термические константы ве-
ществ/Под ред. В. П. Глушко. Вып. 4. М.: Наука. 1971. 320 с.
63. Smith D. W., Andrews L.HJ. Phys. Chem. 1972. V. 76. P. 2718.
64. Dubois L, Herzberg G., Verma R. Z).//Ibid. 1967. V. 47. P. 4262.
65. Dubois /.//Can. J. Phys. 1968. V. 46. P. 2485.
66. Milligan D. E., Jacox M. Е.Ц1. Chem. Phys. 1970. V. 52. P. 2594.
67. Герцберг Г. Спектры и строение простых свободных радикалов: Пер. с
англ./Под ред. В. М. Татевского. М.: Мир, 1974. 208 с.
68. Hastie J. W., Hauge R. H., Margrave J. L.//J. Amer. Chem. Soc. 1969. V. 91.
P. 2536—2538.
69. Dixon R. H., Halle М.Ц1. Mol. Spectrosc. 1970. V. 36. P. 192.
70. Shoji H., Tanaka T., Hirota Е.Ц1. Mol. Spectrosc. 1973. V. 47. P. 268.
71. Rao D. 7?.//Ibid. 1970. V. 34. P. 284; J. Chem. Phys. 1965. V. 43. P. 2775.
72. Gole J. L.//J. Mol. Spectrosc. 1972. V. 43. P. 441.
264
73. Johns J. W. C., Chantry G. W., Barrow R. F.//Trans. Faraday Soc. 1958.
V. 54. P. 589.
74. Milligan D, E., Jacox M. Е.Ц1. Chem. Phys. 1968. V. 49. P. 4269.
75. Rao D. R., Venkateswarlu Р.Ц1. Mol. Spectrosc. 1961. V. 7. P. 287.
76. Sankaranarayanam S.//Proc. Nat. Sci. India. 1962. V. A28. P. 311.
77. Bassler J. M., Timms R. L., Margrave J. £.//Inorg. Chem. 1966. V. 5.
P. 729.
78. Rao U. M., Curl R. F.//Trans. Amer.^Crystallogr. Assoc. 1966. V. 2. P. P.
183, 187.
79. Wilson E. ^.//Science. 1968. V. 162. P. 59.
80. Чаркин О. И., Дяткина М. B.HJR. структ. хим. 1964. Т. 5. С. 924.
81. Searcy A. U7.//J. Chem. Phys. 1958. V. 28. Р. 1237.
82. Ismail Z. R., Fredin L., Hauge R. H. e. a.//J. Chem. Phys. 1982. V. 77.
P. 1626.
83. Milligan D. E., Jacox M. E.//J. Chem? Phys. 1968. V. 49. P. 1938.
84. Maas G., Hauge R. H., Margrave J.L.//Z. anorg. Chem. 1972. B. 392. S. 295.
85. Нефедов О. M., Мальцев A. R., Святкин В. А.//Изв. АН СССР. Сер.
хим. 1974. С. 958.
86. Herzberg G., Verma R. D.//Can. J. Phys. 1964. V. 42. P. 395.
87. Молекулярные постоянные неорганических соединений/Под ред.
К. С. Краснова. Л.: Химия, 1979. 446 с.
88. Ismail Z. R., Hauge R. Н, Fredin L. e. a.//J. Chem. Phys. 1982. V. 77.
P. 1617. *
89. Smith G. R., Guillory W. А.ЦЗ. Chem. Phys. 1972. V. 56. P. 1423; J. Appl.
Spectrosc. 1973. V. 27. P. 137.
90. Takeo H, Curl R. F., Wilson P. W.//J. Mol. Spectrosc. 1971. V. 38. P. 464.
91. Takeo H//J. Nat. Chem. Lab. Ind. 1973. V. 68. P. 340.
92. Hastie J, W., Hauge R., Margrave J. L.//J. Phys. Chem. 1968. V. 72.
P. 4492.
93. Takeo H, Curl R. F.//J. Mol. Spectrosc. 1972. V. 43. P. 21.
94. Huber //.//Can. J. Chem. 1974. V. 52. P. 95.
95. Vaida E./fJ. Organomet. Chem. 1976. V. 105. P. 33.
96. Мальцев A. R., Святкин В. А., Нефедов О. M. //Док л. АН СССР. 1976.
Т 227 С 1151
97. Izabel R.' J.//L Chem. Phys. 1973. V. 58. Р. 818.
98. Izabel R. J., Guillory W. A.//L Chem. Phys. 1971. V. 55. P. 1197; 1972.
V. 57. P. 1116.
99. Hastie J. W.t Hauge R. H., Margrave J. L.//Ann. Rev. Phys. Chem. 1970.
V. 21. P. 475; J. Mol. Spectrosc. 1973. V. 45. P. 420.
100. Hough E., Nicholson D. G.//J. Chem. Soc. Dalton Trans. 1976. P. 1782.
101. Andrews R. H., Donaldson J. D./JAsAa crystallog. 1977. V. 1333. P. 307.
102. Brever L.t Somayajulu G. R., Brackett £.//Chem. Rev. 1963. V. 63. P. 111.
103. Lister M. W., Sutton L. E,//Trans. Faraday Soc. 1941. V. 37. P. 406.
104. Lappert M. F., Power P. P., Slade M. J. e. a.HL Chem. Soc. Chem. Comm.
1979. P. 369—370.
105. Акишин И. А., Спиридонов В. П., Ходченков А. И.//JR. физ. хим. 1958.
Т. 32 С. 1679.
106. Hargittai /.//J. Mol. Struct. 1977. V. 42. Р. 147.
107. Банк данных ИВТАНТЕРМО, версия 1989 г.
108. Lias S. G., Rar pas Z., Lieb man J. F./IL Amer. Chem. Soc. 1985. V. 107.
P. 6089—6096.
109. Rademan R., Yochims H.-W., Baumgartel H. e. a.//J. Phys. Chem. 1985.
V. 89. P. 3459.
110. Сырватка Б. Г., Гильбурд M. М.//Новое в химии карбенов. М.: Наука,
1973. С. 134.
111. Сырватка Б. Г., Бельферман А. Л.» Гильбурд М. М., Моин Ф. Б.!/Ж. орг.
хим. 1971. Т. 7. С. 9.
112. Shin S. R., Beauchamp J. L.//L Phys. Chem. 1986. V. 90. P. 1507.
265
113. Но Р., Coltrin М. Е., Binkley J. S. е. a.HJ. Phys. Chem. 1985. V. 89..
Р. 4647.
114. Davidson I. М. Т., Matthews J. 1.1 /J. Chem. Soc. Faraday Trans. Part I.
1976. V. 71. P. 1403—1409.
115. Walsh R.f/Acc. Chem. Res. 1981. V. 14. P. 246—252.
116. Vanderwielen A. J., Ring M. A., O’Nieal H. E.//J, Amer. Chem. Soc. 1975.
V 97 P 993_________998
117. Herzberg G.//Can. J. Phys. 1961. V. 39. P. 1511.
118. Энергии разрыва химических связей. Потенциалы ионизации и сродство
к электрону. М.: Наука, 1974. 351 с.
119. Reineke W., Strein /С//Вег. Bunsenges. phys. Chem. 1976. B. 80. S. 343.
120. Fisher I. P., Homer J. B., Lossing F. Р.ЦЗ, Amer. Chem. Soc. 1965. V. 87.
P. 957—960.
121. Pottie R. F.//J. Chem. Phys. 1965. V. 42. P. 2607.
122. Zmbov K. F., Uy О. M., Margrave J. L.//J. Amer. Chem. Soc. 1968. V. 90.
P. 5090_______5092
123. Ehlert T. C.//J. Phys. Chem. 1969. V. 73. P. 949.
124. Hildebrand D. L.//Chem. Phys. Lett. 1975. V. 30. P. 32.
125. Biordi J. C., Lazzara С. P., Papp J. F.//J. Phys. Chem. 1976. V. 80.
P. 1042—1048.
126. Dyke J. M., Golob L., Jonathan N. e. a.//J. Chem. Soc. Faraday Trans. Part
II. 1974. V. 70. P. 1828—1836.
127. Curran R. A.//J. Chem. Phys. 1961. V. 34. P. 2006.
128. Shapiro J. S., Lossing F. P.HJ. Phys. Chem. 1963. V. 72. P. 1552.
129. Reed R. L, Sneeden 1F.//Trans. Faraday Soc. 1958. V. 54. P. 301.
130. Kaposi О.ЦКсХъ chim. hung. 1976. V. 89. P. 229.
131. Kaposi O., Riedel M., Deutsch 7.//Act a chim. hung. 1974. V. 81. P. 43.
132. Ehlert T. C., Margrave J. L.HJ. Chem. Phys. 1964. V. 41. P. 1066.
133. McDonald J. D., Williams С. H., Thompson J, C. e. a.//Adv. Chem. Ser.
1968. V. 72. P. 261.
134. Fehlner T. P., Turner D. IF.//Inorg. Chem. 1974. V. 13. P. 754.
135. Westwood N. P. C.//Chem. Phys. Lett. 1974. V. 25. P. 558.
136. Андрианов К. А.//Докл. АН СССР. 1970. Т. 194. С. 1077.
137. Hastie J. W., Margrave J. L.//J. Phys. Chem. 1969. V. 73. P. 1105.
138. Zmbov K. F., Hastie J. W., Hauge R. e. a.//Inorg. Chem. 1968. V. 7.
P. 608.
139. Uy О. M., Muenow D. W., Margrave J. L.//Trans. Faraday Soc. 1969. V. 65.
P. 1296.
140. Zmbov K-, Hastie J. W., Margrave J. L.//Trans. Faraday Soc. 1968. V. 64.
P. 861.
141. Knowless D. J., Nicholson A. J., Swingler D. L.HJ. Phys. Chem. 1970. V. 74.
P. 3642.
142. Bloom H., Hastie J. IF.//Austral. J. Chem. 1966. V. 19. P. 1003.
143. Hastie J. W., Bloom FL, Morrison J. D.//J. Chem. Phys. 1967. V. 47.
P. 1580.
144. Shavitt /.//Tetrahedron. 1985. V. 41. P. 1531.
145. Основные свойства неорганических фторидов/Под ред. Н. П. Галкина.
М.: Наука. 1976. 400 с.
146. Goldfield D., Simons J. //J. Phys. Chem. 1981. V. 85. P. 569.
147. Bondybey V. E.HJ. Mol. Spectrosc. 1977. V. 64. P. 180.
148. Milligan D. E.//J. Chem. Phys. 1964. V. 41. P. 1199.
149. Wirsam B.//Chem. Phys. Lett. 1972. V. 14. P. 214.
150. Meadows J. H., Schaefer H. F.//J. Amer. Chem. Soc. 1976. V. 98. P. 4383—
4386.
151. Billingsleg /.//Can. J. Phys. 1972. V. 50. P. 531.
152. Wersam B.//Chem. Phys. Lett. 1973. V. 22. P. 360.
153. Hopkins H. P., Thompson J. C., Margrave J. L.HJ. Amer. Chem. Soc. 1968.
V. 90. P. 901—902.
266
154. Asundi R. К, Karim M. S., Samuel R.//Proc. Phys. Soc. 1938. V. 50.
P. 581.
155. Wieland H., Heise Af.//Angew. Chem. 1951. B. 63. S. 438.
156. Martin R. W., Merer A. /.//Can. J. Phys. 1973. V. 51. P. 727.
157. Hauge R. H., Khanna V. M., Margrave J. L.//3. Mol. Spectrosc 1968. V. 27.
P. 143.
158. Tewarson A., Palmer H. BJ/J. Mol. Spectrosc. 1966. V. 22. P. 117.
159. Pathak С. M., Palmer H. B.//Chem. Phys. L$tt. 1969. V. 31. P. 170.
160. Hauge R. H., Hastie J. W., Margrave J. L.//5. Phys. Chem. 1968. V. 72.
P. 3510.
161. Bauschlicher C. W., Schaefer H. F., Bag us P. SJ/J. Amer. Chem. Soc. 1977.
V. 99. P. 7106—7110.
162. Herzberg G„ Johns J. W. C.//Proc. Chem. Soc. 1966. V. A295. P. 107.
163. Портер Дж.//Усп. химии. 1970. Т. 39. С. 319.
164. Herzberg G., Shoosmith /.//Nature. 1961. V. 183. Р. 1801.
165. Herzberg GJ/Proc. Roy. Soc. London. 1961. V. A262. P. 291.
166. Mann D. E., Thrush В. A. I /J. Chem. Phys. 1960. V. 33. P. 1732.
167. Simons J. P., Jarwood A. /.//Nature. 1961. V. 192. P. 943.
168. Brannon J. HJ/J. Phys. Chem. 1986. V. 90. P. 1784.
169. Tgerman W. J. RJ/L Chem. Soc. Chem. Comm. 1968. P. 392—393.
170. Moritani I., Murahashi S.-I., Ashitaka H. e. aJ/J. Amer. Chem. Soc. 1968.
V 90 P 5918__________5919
171. Yamamoto /.//Tetrahedron. 1970. V. 26. P. 251.
172. Closs G. L., Rabinow В. EJ/J. Amer. Chem. Soc. 1976. V. 98. P. 8190—
8198.
173. Gould I. R., Turro N. J., Butcher J. e. «.//Tetrahedron. 1985. V. 41.
P. 1587.
174. Eisenthal К В., Turro N. J., Sitzmann E. V. e. «.//Tetrahedron. 1985. V. 41.
P. 1543.
175. Griller D., Nazran A. S., Scaiano J. C.//Tetrahedron. 1985. V. 41. P. 1525.
176. Horn K. A., Chateauneuf J. ^.//Tetrahedron. 1985. V. 41. P. 1465.
177. Zupancic J. J., Grasse P. B., Lapin S. C. e. «.//Tetrahedron. 1985. V. 41.
P. 1471.
178. Dupny C., Korenowski G., McAuliffe M. e. «.//Chem. Phys. Lett. 1981.
V. 77. P. 272.
179. Jasinski J. MJ/J. Phys. Chem. 1986. V. 90. P. 555.
180. Pimentel G. C., Herr K. C.//J. Chem. Phys. 1964. V. 41. P. 1509.
181. Herr K. C„ Pimentel G. C.//Appl. Opt. 1965. V. 4. P. 25.
182. Herr K. C.} Carlson G. A., Pimentel G. С.//Кагаку-по рёики. 1967. T. 21.
С. 11.
183. Thrush В. A., Zwolenik J. /.//Trans. Faraday Soc. 1963. V. 59. P. 582.
184. Venkateswarlu P.//Phys. Rev. 1950. V. 77. P. 676.
185. Laird R. K, Andrews E. B., Barrow R. £.//Trans. Faraday Soc. 1950.
V. 46. P. 803.
186. Gilberg R., Theoret AJ/J. Phys. Chem. 1976. V. 80. P. 1017—1022.
187. Quach-Tat-Trung J/Chem. Phys. Lett. 1977. V. 47. P. 404.
188. Davies P. B., Hamilton P. A., Elliott J. M. e. a.//J. Mol. Spectrosc. 1983.
V. 102. P. 193—203.
189. Davies P. B., Lewis-Bevan W., Russell D. KJ/J. Chem. Phys. 1981. V. 75.
P. 5603.
190. Huie R. E., Long N. J. T., Thrush B. A.//Chem. Phys. Lett. 1977. V. 51.
P. 197.
191. Rao V. M., Curl R. F., Timms P. L. e. a.//J. Chem. Phys. 1967. V. 47.
P. 2557.
192. Rao V. M., Curl R. F.//J. Chem. Phys. 1966. V. 45. P. 2032.
193. Caldow G. L., Deeley С. M., Turner P. H. e. «.//Chem. Phys. Lett. 1981.
V. 82. P. 434—438.
267
194. Jasinski J. M., Whittaker E. A., Bjorklund G. C. e. a.//Appl. Phys. Lett. 1984.
V. 44. P. 1155.
195. Modica A. P„ La Graff J. EJ/J. Chem. Phys. 1965. V. 43. P. 3383; 1966.
V. 44. P. 3375.
196. Carlson G. AJ/J. Chem. Phys. 1971. V. 75. P. 1625.
197. Modica A. P., La Graff J. EJ/J. Chem. Phys. 1966. V. 45. P. 4729.
198. Keating E. L., Matula R. AJ/J. Chem. Phys. 1977. V. 66. P. 1237.
199. Modica A. P., Sillers S. JJ/J. Chem. Phys. 1968. V. 48. P. 3282.
200. Modica A. P.//J. Chem. Phys. 1966. V. 44. P. 1585.
201. Колбановский Ю. А., Щипачев В. С., Черняк Н. Я- и др. Импульсное
сжатие газов в химии и технологии. М.: Наука, 1982. С. 240.
202. Колбановский Ю. А., Мамиконян Е. Р., Матвеева Л. Н., Щипачев В. CJ/
Труды IV Всесоюзной конференции по химии карбенов. М.: Наука, 1987.
С. 117—118.
203. Колбановский Ю. А., Чернышева А. С., Щипачев В. С. Там же С. 119—
120.
204. Буравцев Н. Н., Григорьев А. С., Колбановский Ю. А. Там же. С. 118—
119.
205. Крейдок С., Хинчклиф А. Матричная изоляция. Пер. с англ./Под ред.
Нефедова О. М. М.: Мир, 1978. 173 с.
206. Мальцев А. К-, Королев В. А., Нефедов О. Л4.//Физическая химия. Со-
временные проблемы. М.: Химия, 1982. Т. 6. Гл. 7.
207. Milligan D. Е., Jacox М. EJ/J. Chem. Phys. 1968. V. 48. Р. 2265.
208. Snelson A.//High Temp. Sci. 1970. V. 2. P. 70.
209. Andrews /..//Tetrahedron Lett. 1968. P. 1423; J. Chem. Phys. 1968. V. 48.
P. 972 979.
210. Maltsev A. KJ/J. Phys. Chem. 1971. V. 75. P. 3984.
211. Tevault D. E., Andrews LJ/J. Mol. Spectrosc. 1975. V. 54. P. 110.
212. Святкин В. А., Мальцев А. К., Нефедов О. MJ/Изв. АН СССР. Сер. хим.
1977. С. 2236—2242.
213. Нефедов О. М., Мальцев А. К., Святкин В. А.//Изв. АН СССР. Сер. хим.
1976. С. 1907.
214. Мальцев А. К., Хабашеску В. Н., Нефедов О. М. //Докл. АН СССР. 1977.
Т. 233. С. 421.
215. Shirk J. SJ/J. Chem. Phys. 1971. V. 55. Р. 3608.
216. Rogers Е. EJ/J. Chem. Phys. 1970. V. 52. P. 2198.
217. Prochaska E. T., Andrews LJ/J. Chem. Phys. 1977. V. 67. P. 1091.
218. Ault B. S., Andrews LJ/J. Chem. Phys. 1975. V. 63. P. 1411.
219. Prochaska E. T., Andrews LJ/J. Chem. Phys. 1980. V. 73. P. 2651.
220. Lee J.-P., Pimentel G. CJ/J. Chem. Phys. 1981. V. 75. P. 4241.
221. Bernheim R. AJ/J. Chem. Phys. 1970. V. 53. P. 1280.
222. Wasserman E., Jager W. A., Kuck V. J.//Chem. Phys. Lett. 1970. V. 7.
P. 409.
223. Wasserman E., Kuck V. J., Hutton R. S., Yager W. AJ/J. Amer. Chem. Soc.
1970. V. 92. P. 7491—7493.
224. Bernheim R. AJ/J. Chem. Phys. 1971. V. 54. P. 3223.
225. Wasserman EJ/J, Chem. Phys. 1971. V. 54. P. 4120.
226. Milligan D. EJ/J. Chem. Phys. 1965. V. 43. P. 3734.
227. Moll N. G., Thompson W. EJ/J. Chem. Phys. 1966. V. 44. P. 2684.
228. Dendranis A., Leroi G. EJ/J. Chem. Phys. 1977. V. 66. P. 4334.
229. Merer A. J.//Can. J. Phys. 1967. V. 45. P. 4103.
230. Bernheim R. AJ/J. Chem. Phys. 1964. V. 41. P. 1156.
231. Bernheim R. A., Kempf R. J., Gramas J. V. e. aJ/J. Chem. Phys. 1965.
V. 43. P. 196.
232. Bernheim R. A., Kempf R. J., Reichenbecker E. FJ/J. Magn. Reson. 1970.
V. 3. P. 5.
233. Smith W. H., Leroi G. fV/Spectrochim. acta. 1969. V. 25A. P. 1917.
268
234. Ogilvie J. E., Cradock S. //J. Chem. Soc. Chem. Commun. 1966. P. 364—
365.
235. Milligan D. E., Jacox M. E.//J. Chem. Phys. 1963. V. 39. P. 712.
236. Naki A. G., Sams R. L.//J. Chem. Phys. 1981. V. 75. P. 4178.
237. Pearson E. F.HZ. Naturforsch. 1976. B. 31a. S. 221.
238. Reisenauer H. P., Maier G.t Riemann A. e. a.//Angew. makromol. Chem.
1984. B. 96. S. 596.
239. Мальцев А. К., Зуев П. С.г Томилов Ю. В., Нефедов О. Л4.//Изв. АН СССР.
Сер. хим. 1987. № 10. С. 2202.
240. Baird М. S., Dunkin I, R., Hacker N. е. а.//J. Amer. Chem. Soc. 1981.
V. 103. P. 5190—5195.
241. West P. R., Chapman O. L., LeRoux J.-P.//J. Amer. Chem. Soc. 1981.
V. 103. P. 5190—5195.
241. West P. R., Chapman O. L., LeRoux J.-P.//J. Amer. Chem. Soc. 1982.
V. 104. P. 1779—1782.
242. Мальцев А. K-, Зуев П. С., Нефедов О. Л1.//Изв. АН СССР. Сер. хим.
1987. № 2. С. 463.
243. Chapman О. L., Sheridan М. S.//J- Amer. Chem. Soc. 1979. V. 101. Р. 3690—
3692.
244. Cheridan R. S.t Kesselmayer M. A.HJ. Amer. Chem. Soc. 1984. V. 106.
P. 436—437.
245. Kesselmayer M. A., Sheridan R. S.//J. Amer. Chem. Soc. 1986. V. 108.
P. P. 99—107, 844—845.
246. Torres M., Bourdelande J. L., Clement A.f Strausz О. Р.ЦЗ. Amer. Chem..
Soc. 1983. V. 105. P. 1698—1700.
247. Arrington C. A., Klingensmith K. A., West R., Michl J.HJ. Amer. Chem. Soc.
1984. V. 106. P. 525—530.
248. Bleckmann P., Maly Н.» Minkwitz ^.//Tetrahedron Lett. 1982. V. 23..
P. 4655.
249. Andrews L.//Appl. Spectrosc. Revs. 1976. V. 11. P. 125.
250. Barnes A. L, Bignail J. C., Purnell C. J.//J. Raman. Spectrosc. 1975. V. 4..
P. 159.
251. Ozin G. A., Van der Voet A.//J. Chem. Phys. 1972. V. 56. P. 4768.
252. Bass A. M., Mann D. E.//J. Chem. Phys. 1968. V. 36. P. 3501.
253. Chang K. W., Graham W. R. M.//J. Chem. Phys. 1982. V. 77. P. 4300.
254. Smith С. E., Jacox M. E., Milligan D. E.//J. Mol. Spectrosc. 1976. V. 60.
P. 381.
255. Bondybey V. E., English J. H.HJ. Mol. Spectrosc. 1980. V. 79. P. 416—
423.
256. Miller J. C., Andrews L.HJ. Phys. Chem. 1980. V. 84. P. 401—403.
257. Maier G., Mihm G., Reisenauer H. P. e. a.//Chem. Ber. 1984. B. 117.
3 2369_______2381.
258. Drahnak T. J., Michl J.t West R./JJ. Amer. Chem. Soc. 1979. V. 101.
P. 5427. 1981. V. 103. P. 1845—1846.
259. Vancik H., Raabe G., Michalczyk M. J. e. a./ /J. Amer. Chem. Soc. 1985.
V. 107. P. 4097—4098.
260. Raabe G., Vancik H., West R. e. a.HJ. Amer. Chem. Soc. 1986. V. 108.
P. 671—677.
261. Shimoda M.t Tomoda S., Takeuchi Y. e. a.//Eight IUPAC Conference on
Physical Organic Chemistry. Abstracts. Tokyo. 1986. P. 296.
262. Closs G., Hutchison C. A., Kohler B. E.HJ. Chem. Phys. 1966. V. 44..
P. 413.
263. Gibbons W. A., Trozzolo A. M.//J. Amer. Chem. Soc. 1966. V. 88. P. 172—
173.
264. Moritani A.//Tetrahedron Lett. 1966. P. 373.
265. Trozzolo A. M.t Gibbons W. A.//J. Amer. Chem. Soc. 1967. V. 89. P. 239—
243.
269
266. Ware W. R., Sullivan P. J.//J. Chem. Phys. 1968. V. 49. P. 1445.
267. Dunkin I. R.t Bell G. 4.//Tetrahedron. 1985. V. 41. P. 339.
268. Chuang C., Lapin S. C., Schrock A. R. e. a.UL Amer. Chem. Soc. 1985.
V. 107. P. 4238—4243.
269. Becker R. S., Bost R. O., Role J. e. aJ/L Amer. Chem. Soc. 1970. V. 92.
P. 1302—1311.
270. Savino T. G., Soundararajan N., Platz M. S.//J. Phys. Chem. 1986. V. 90.
P. 919.
271. Ganzer G. A., Sheridan R. S., Lin M. T. HjUJ. Amer. Chem. Soc. 1986.
V. 108. P. 1517—1520.
272. Sander 1F.//Angew. Chem. 1986. B98. S. 255—256.
273. Wright B. B.//Tetrahedron. 1985. V. 41. P. 1517.
274. Bell C. A., Dunkin I. R., Shields C. /.//Spectrochim. acta. 1985. V. 41 A.
P. 1221.
275. Мальцев A. R.f Зуев П. С., Нефедов О. Л1.//Изв. АН СССР. Сер. хим.
1985. С. 957.
276. Chapman О. L., Hess Т. С.ЦЗ. Amer. Chem. Soc. 1984. V. 106. Р. 1842—
1843.
277. Eisenthal R. B.t Turro N. J., Aikawa M. e. a.J/J. Amer. Chem. Soc. 1980.
V. 102. P. 6563—6565.
278. Alberti A., Griller D., Nazran A. S., Pedulli G. F.//J. Amer. Chem. Soc.
1986. V. 108. P. 3024—3028.
279. Sitzman E. V., Wang Y.t Eisenthal R. В.ЦЗ. Phys. Chem. 1983. V. 87.
P. 2283.
280. Hadel L. M., Platz M. S., Scaiano J. С.ЦЗ. Amer. Chem. Soc. 1984. V. 106.
P. 283—287.
281. Griller D., Nazran A. S., Scaiano J. C.//Accounts Chem. Res. 1984. V. 17.
P. 283—289.
282. Gilbert B. S., Griller D.t Nazran A. S.jlL Org. Chem. 1985. V. 50. P. 4738—
4742.
283. Nazran A. S., Griller D.HL Amer. Chem. Soc. 1985. V. 107. P. 4613—
4615.
284. Hadel L. M., Platz M. S., Scaiano J. C.//Chem. Phys. Lett. 1983. V. 97.
P. 446.
285. Tomioka H., Hayashi N., Izawa Y., Liu M. T. Н.ЦТ. Amer. Chem. Soc. 1984.
V. 106. P. 454—456.
286. Griller D., Liu M. T. H., Scaiano J, C.//J. Amer. Chem. Soc. 1982. V. 104.
P 5549_______5551
287. Griller D., Liu M. T. H., Montgomery С. P, e. а.ЦЪ. Org. Chem. 1983. V. 48.
P. 1359—1360.
288. Moss R. A., Perez L. A., Turro N. J. e, ^.//Tetrahedron Lett. 1983. V. 24.
P. 685.
289. Cox D. P., Gould I. R., Hacker N. P. e. ^.//Tetrahedron Lett. 1983. V. 24.
P 5313
290. Zupancic J. J., Schuster G. B.//L Amer. Chem. Soc. 1980. V. 102. P. 5958—
5960.
291. Wong P. C., Griller D., Scaiano C.//J. Amer. Chem. Soc. 1981. V. 103.
P. 944; Chem. Phys. Lett 1981. V. 83. P. 69.
292. Griller D., Montgomery C. R., Scaiano J. C. e. a.f/L Amer. Chem. Soc.
1982. V. 104. P. 6813—6814.
293. Lapin S. C., Schuster G. В.ЦЗ. Amer. Chem. Soc. 1985. V. 107. P. 4243—
4248.
294. Gaspar P. P., Roo В. H., Chari S. e. a.//Chem. Phys. Lett. 1984. V. 105.
P. 153—157.
295. Murray R. W., Trozzolo A. M., Wasserman Е.» Yager W. А.Ц5. Amer. Chem.
Soc. 1962. V. 84. P. 3213—3214.
270
296. Brandon R. W., Closs G. L., Hutchison C. AJ/J. Chem. PhyS. 1968. V. 37.
P. 1878.
297. Wasserman EJ/J. Chem. Phys. 1964. V. 40. P. 2408.
298. Brandon R. WJ/J. Chem. Phys. 1965. V. 43. P. 2006.
299. Wasserman E., Yager W. AJ/J. Phys. Chem. 1967. V. 71. P. 205.
300. Hutchison C. A., Kohler B. EJ/J. Chem. Phys. 1969. V. 51. P. 3327.
301. Cheng T., Tien-Sung L.> Sloop D. J.//Chem. Phys. Lett. 1976. V. 44.
P. 576.
302. Humphreys R. W. R., Arnold D. RJ/Can. J. Chem. 1977. V. 55. P. 2286.
303. Trozzolo A. M., Wasserman Е.» Yager W. A.//J. Amer. Chem. Soc. 1965.
V. 87. P. 129—130.
304. Bernheim R. AJ/J. Chem. Phys. 1976. V. 64. P. 2747.
305. Wasserman EJ/J. Chem. Phys. 1971. V. 55. P. 2539.
306. Bicknell B. R., Graham W. R. M., Weliner WJ/J. Chem. Phys. 1976. V. 64.
P. 3319.
307. Wasserman E., Barash L., Yager W. AJU. Amer. Chem. Soc. 1965. V. 87.
P. P. 2075—2076, 4974—4975.
308. Gano J. E., Wettach R. H., Platz M. S. e. a. J. Amer. Chem. Soc. 1982.
V. 104. P. 2326—2327.
309. Myers D. R., Senthilnathan V. P., Platz M. S., Jones M. Jr J/J. Amer. Chem.
Soc. 1986. V. 108. P. 4232—4233.
310. Trozzolo A. M., Murray R. W.f Wasserman EJ/J. Amer. Chem. Soc. 1962.
V. 84. P. 4990—4991.
311. Higuchi J. 11 J. Chem. Phys. 1963. V. 39. P. 1339.
312. Barcus R. L., Palik E. С.» Platz M. S.//Tetrahedron Lett. 1982. V. 23.
P. 1323.
313. Roth H. D., Hutton R. S.//Tetrahedron. 1985. V. 41. P. 1567.
314. Palik E. C., Platz M. SJ/J. Org. Chem. 1983. V. 48. P. 963—969.
315. Hutton R. S., Roth H. D., Schilling M. L. M., Suggs J. WJ/J. Amer. Chem.
Soc. 1981. V. 103. P. 5147—5151.
316. Senthilnathan V. P., Platz M. SJ/J. Amer. Chem. Soc. 1981. V. 103.
P. 5503—5511.
317. Wasserman E„ Barash L., Trozzolo A. M. e. aJ/J. Amer. Chem. Soc. 1964.
V. 86. P. 2304—2305.
318. Hutchison C. A., Pearson G. AJ/J. Chem. Phys. 1967. V. 47. P. 520.
319. Hutchison C. AJ/J. Phys. Chem. 1967. V. 71. P. 209.
320. Murahashi S., Moritani I., Nagai TJ/B»\iW. Chem. Soc. Jap. 1967. V. 40.
P. 1655—1660.
321. Moritani I., Murahashi S.-L, Nishino MJ/J. Amer. Chem. Soc. 1967. V. 89.
P. 1259—1260.
322. Grasse P. B., Zupancic J. J., Lapin S. CJJJ. Org. Chem. 1985. V. 50.
P 2352_______2356
323. McMahon R. J., Chapman O. LJ/J. Amer. Chem. Soc. 1986. V. 108. P. 1713—
1714.
324. Wasserman E., Murray R. WJ/J. Amer. Chem. Soc. 1964. V. 86. P. 4203—
4204.
325. Murai H., Torres M., Strausz O. PJ/J. Amer. Chem. Soc. 1980. V. 102.
P 7390_______7391.
326. Murai H., Torres M., Strausz O. PJ/J. Amer. Chem. Soc. 1980. V. 102.
P. 1421—1422.
327. Hutton R. S., Manion M. L., Roth H. DJ/J. Amer. Chem. Soc. 1974. V. 96.
P. 4680—4682.
328. Hutton R. S., Roth H. DJ/J. Amer. Chem. Soc. 1978. V. 100. P. 4324—
4325.
329. Murai H., Ribo J., Torres M., Strausz O. PJ/J. Amer. Chem. Soc. 1981.
V. 103. P. 6422—6426.
330. Murai H., Torres M., Strausz O. PJ/J. Amer. Chem. Soc. 1979. V. 10L
P. 3976—3979.
271
331. Trozzolo A. MJ/J. Amer. Chem. Soc. 1963. V. 83. P. 2526.
332. Wasserman E., Murray R. W., Yager W. А.ЦЗ. Amer. Chem. Soc. 1967.
V. 89. P. 5076—5078.
333. Itoh K,//Chem. Phys. Lett. 1967. V. 1. P. 235.
334. Zandler M. E., Goddard J. D., Shaefer H. FJ/J. Amer. Chem. Soc. 1979.
V. 101. P. 1072—1076.
335. Hehre W. J., Pople J. A., Lathan W. AJIL Amer. Chem. Soc. 1976. V. 98.
P. 4378—4383.
336. Smith G. R., Weltner WJ/L Chem. Phys. 1975. V. 62. P. 4592.
337. Lembke R. R., Ferrante R. F., Weltner WJIL Amer. Chem. Soc. 1977. V. 99.
P. 416—423.
338. Mural Н.» Torres M., Strausz O. PJ/L Amer. Chem. Soc. 1980. V. 102.
P. 5104—5106.
339. Sugawara T., Tukada H., Izuoka AJIL Amer. Chem. Soc. 1986. V. 108.
P. 4272—4278.
340. Sugawara T., Bandow S., Kimura KJ IL Amer. Chem. Soc. 1986. V. 108.
P. 368—371.
341. Каграманов H. Д., Мальцев А. К., Дубинский M. Ю. и dpJ/Изв. АН СССР.
Сер. хим. 1983. C. 537.
342. Rossi M. J., Barker J. R., Golden D. MJ/J. Chem. Phys. 1982. V. 76.
P. 406.
343. Lineberger W. C.t Woodward B. №.//Phys. Rev. Lett. 1970. V. 25. P. 424.
344. Siegel M. №.//Phys. Rev. 1972. V. 6A. P. 607.
345. Celotta R. /.//Phys. Rev. 1972. V. 6A. P. 631.
346. Hotop H., Patterson T. A., Lineberger W. C.//Phys. Rev. 1973. V. 8. P. 762.
347. Engelking P. CJ/L Chem. Phys. 1981. V. 74. P. 5460.
348. Leopold D. G., Murray K. K*> Lineberger W. CJ/L Chem. Phys. 1984. V. 81.
P. 1048.
349. Okumura М.» Yeh L. L-С., Normand Z).//Tetrahedron. 1985. V. 41. P. 1423.
350. Sears T. J., Bunker P. RJ/L Chem. Phys. 1983. V. 79. P. 5265.
351. Kasdan A., Herbst E., Lineberger W CJIL Chem. Phys. 1975. V. 62.
P. 541.
352. Davis D. D., Okabe HJJL Chem. Phys. 1968. V. 49. P. 5526.
353. Hirota EJIL Phys. Chem. 1983. V. 87. P. 3375.
354. Purdy J. R., Thrush B. A.,//Chem. Phys. Lett. 1981. V. 80. P. 11.
355. Meunier H., Purdy J. R., Thrush В. A JI'J. Chem. Soc. Faraday Trans. Part II.
1980. V. 76. P. 1304—1313.
356. Wang J. L.-F., Margrave J. L., Franklin J. LJ/L Chem. Phys. 1974. V. 60.
P. 2158.
357. Hildebrand D. LJ/Chem. Phys. Lett. 1975. V. 32. P. 533.
358. Bialkowski S. Е.» King D. S., Stephenson J. CJjL Chem. Phys. 1979. V. 71.
P. 4010.
359. Hakuta KJ/L Mol. Spectrosc. 1984. V. 106. P. 56—63.
360. Inoue G., Suzuki Af.//Chem. Phys. Lett. 1984. V. 105. P. 641—644.
361. Lee H. U., Deneufville J. P.//Chem. Phys. Lett. 1983. V. 99. P. 394—398.
362. Pitts W. M., Donneley V. M., Baronavski A. R.//Chem. Phys. 1981. V. 61.
P. 451—464.
363. Patel R. L, Stewart G. W., Casleton KJICh&n. Phys. 1980. V. 52. P. 461 —
468.
364. Conner С. P., Stewart G. W., Lindsay D. M. e. aJ/L Amer. Chem. Soc. 1977.
V. 99. P. 2540—2544.
365. Welsh A. R., Yudge D. LJ/L Chem. Phys. 1972. V. 57. P. 286.
366. Masanet J., Vermeil CJ/L chim. phys. et phys.-chim. biol. 1975. V. 82.
P. 820.
367. McKellar A. R. W., Yamada C., Hirota E. e. aJ/J. Chem. Phys. 1983. V. 79.
P. 1220.
368. Sears T. J., Bunker P, R., McKellar A. R. Wj/L Chem. Phys. 1982. V. 77.
P. 5363.
272
369. Bunker P. R., Sears T. J., McKellar A. R. W. e. aJ/J. Chem. Pnys. 1963.
V. 73. P. 1211 — 1219.
370. Lengel R. K-, Zare R. N.H J. Amer. Chem. Soc. 1978. V. 100. P. 7495—
7499.
371. Danon /.//Chem. Phys. 1978. V. 29. P. 345.
372. McKellar A. R. W., Bunker P. R., Sears T. J. e. a.//J. Chem. Phys. 1983.
V. 79. P. 5251.
373. Hayden С. C.. Neumark D. M., Shobatake K. e. a.//J. Chem. Phvs. 1982.
V. 76. P. 3607.
374. Lengel R. K-, Zare R. NJ/J. Amer. Chem. Soc. 1971. V. 93. P. 4112—4119.
375. Harrison J. F., Liedtke R. C., Lieb man J. FJ/J. Amer. Chem. Soc. 1979.
V. 101. P. 7162—7168.
376. Harding L. B., Goddard W. AJ/J. Chem. Phys. 1977. V. 67. P. 2777.
377. Mueller P. H., Rondan N. G., Houk K. N. e. a.//J. Amer. Chem. Soc. 1981.
V. 103. P. 5049—5052.
378. Carter E. A., Goddard W. A J/J. Phys. Chem. 1986. V. 90. P. 998.
379. Altmann J. A., Csizmadia I. G., Yates KJ/J. Amer. Chem. Soc. 1974. V. 96.
P. 4196—4201; 1975. V. 97. P. 5217—5222.
380. Bodor N., Dewar M. Y. SJ/J. Amer. Chem. Soc. 1972. V. 94. P. 9103—
9106.
381. Kyba E. PJ/J. Amer. Chem. Soc. 1977. V. 99. P. 8330—8332.
382. Metcalfe J., Halevi E. A.//J. Chem. Soc. Perkin Trans. Part II. 1977. P. 634—
639.
383. Mueller P. H., Rondan N. G., Houk K. N. e. ^.//Tetrahedron Lett. 1983.
V. 24. P. 485.
384. Feller D., Borden W. T., Davidson E. RJ/J. Chem. Phys. 1978. V. 71.
P. 4987.
385. Feller D., Borden W. T., Davidson E. 7?.//Chem. Phys. Lett. 1980. V. 71.
P. 22.
386. Dixon D. AJ/J. Phys. Chem. 1986. V. 90. P. 54.
387. Scuseria С. E., Duran M., Maclagan R. G. A. R., Schaefer H. FJ/J. Amer.
Chem. Soc. 1986. V. 108. P. 3248—3253.
388. Feller D., Borden W. T., Davidson E. RJ/J. Phys. Chem. 1983. V. 87.
P. 4833.
389. Honjou N., Pacansky J., Yoshimine MJ/J. Amer. Chem. Soc. 1984. V. 106.
P. 5361—5363; 1985. V. 107. P. 5332—5341.
390. Rauk A., Bouma W. J., Radom LJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 3780—3786.
391. Valtazanos P., Elbert S. T., Ruedenberg KJ/J. Amer. Chem. Soc. 1986.
V. 108. P. 3147—3149.
392. Lee T. J., Bunge A., Schaefer H. FJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 137—142.
393. Jones W. M., Stowe M. E., Wells E. E. e. aJ/J. Amer. Chem. Soc. 1968.
V. 90. P. 1849—1859.
394. Mitsuhashi T., Jones W. MJ/J. Chem. Soc. Chem. Common. 1974. P. 103—
104.
395. Frenking G., Remington R. B., Schaefer H. F. IIIJ'/'J. Amer. Chem. Soc.
1986. V. 108. P. 2169—2173.
396. Radom L., Schaefer H. F., Vincent M. A.//Nouv. j. chim. 1980. V. 4. P. 411.
397. Balci M., Winchester W. R., Jones W. MJ/J. Org. Chem. 1982. V. 47.
P. 5180—5186.
398. Turro N. J., Lehr G. F., Butcher J. A. e. aJ/J. Amer. Chem. Soc. 1982.
V. 104. P. 1982.
399. Turro N. J., Butcher J. A., Moss R. A. e. aJ/J. Amer. Chem. Soc. 1980.
V. 102. P. 7576—7578.
400. Grasse P. B., Brauer В. E., Zupancic J. J. e. aJ/J. Amer. Chem. Soc. 1983.
V. 105. P. 6833—6845.
18—107
273
401. Lapin S. C., Brauer В. E., Schuster G. B.//J. Amer. Chem. Soc. 1984. V. 106.
P. 2092—2100.
402. Griller D., Hadel L., Nazran A. S. e. а.//J. Amer. Chem. Soc. 1984. V. 106.
P 2227_______2235
403. Colvin M. E., Grev R. S., Schaefer H. F. e. a.//Chem. Phys. Lett. 1983. V. 99.
P. 399.
404. Rice J. E., Handy N. C.//Chem. Phys. Lett. 1984. V. 107. P. 365.
405. Ohta K, Davidson E. R., Morokuma K//J. Amer. Chem. Soc. V. 107.
P. 3466—3471.
406. Krogh-Jespersen KUJ. Amer. Chem. Soc. 1985. V. 107. P. 537—543.
407. Apeloig Y., Kami M.//J. Chem. Soc. Chem. Comm. 1985. P. 1048—1049.
408. Truong T. N., Gordon M. S.//J. Amer. Chem. Soc. 1986. V. 108. P. 1775—
1778.
409. Luke В. T., Pople J. A., Krogh-Jespersen M.-B. e. a.//J. Amer. Chem. Soc.
1986. V. 108. P. 270—284.
410. Colvin M. E., Breulet J., Schaefer H. F.//Tetrahedron. 1985. V. 41. P. 1429.
411. Barthelat J.-C., Roch B. S., Tringuier G., Satge J.//J. Amer. Chem. Soc.
1980. V. 102. P. 4080—4085.
412. Olbrich 6.//Chem. Phys. Lett. 1980. V. 73. P. 110.
413. Skell P. S., Woodworth R. C.//J. Amer. Chem. Soc. 1956. V. 78. P. 4496—
4497.
414. Skell P. ^.//Tetrahedron. 1985. V. 41. P. 1427.
415. Fujimoto H, Yamabe S., Fukui /(.//Bull. Chem. Soc. Jap. 1972. V. 45.
P. 2424—2429.
416. McIver J. W.//J. Amer. Chem. Soc. 1972. V. 94. P. 4782—4783.
417. Bodor N., Dewar M. J. S., Wasson Y. S.//J. Amer. Chem. Soc. 1972. V. 94.
P. 9095—9102.
418. Ando W., Kondo S., Migita T.//J. Amer. Chem. Soc. 1969. V. 91. P. 6516—
6517.
419. Shimizu N., Nishida S.//J. Chem. Soc. Chem. Comm. 1972. P. 389.
420. Marchaud A. P., MacBrockway A.//Chem. Rev. 1974. V. 74. P. 431.
421. Нефедов О. M., Долгий И. Е., Шведова И. Б. и др.//Изв. АН СССР. Сер.
хим. 1972. С. 1885.
422. Мальцев А. К-, Королев В. А., Хабашеску В. Н., Нефедов О. /И.//Докл.
АН СССР. 1980. Т. 251. С. 1166.
423. Closs G. L., Moss R. A.//J. Amer. Chem. Soc. 1964. V. 86. P. 4042—
4053.
424. Tomioka H., Tabayashi K-, Ozaki J. e. «.//Tetrahedron. 1985. V. 41. P. 1435.
425. Roth H. £>.//Acc. Chem. Res. 1977. V. 10. P. 85—91.
426. Савин В. И.//УК. ВХО им. Д. И. Менделеева. 1979. Т. 24. С. 454—466.
427. Roth Н. D.//J. Amer. Chem. Soc. 1971. V. 93. Р. 4935—4936.
428. Kocher J., Lehnig M.//Organometallics. 1984. V. 3. P. 937.
429. Vaida E., Tremmel J., Rozsondai B. e. a.//J. Amer. Chem. Soc. 1986. V. 108.
P. 4352—4353.
430. Andrews L., Frederick D. L./Д. Amer. Chem. Soc. 1970. V. 92. P. 775—
778.
431. Guillory W. A., Smith G. R.//J. Chem. Phys. 1970. V. 53. P. 1661.
432. Brunvoll Y., Hargittai I.//Acta Chem. Scand. 1976. V. АЗО. P. 230.
433. Вилков Л. В., Мастрюков В. С., Садова Н. И. Определение геометриче-
ского строения свободных молекул. Л.: Химия, 1978. 224 с.
434. Thyagarajan G., Herraks J., Cleveland F. F.//J. Mol. Spectrosc. 1961. V. 7.
P. 154.
435. Willets D. V., Jones W. J., Robiette A. G.//J. Mol. Spectrosc. 1975. V. 55.
P. 200.
436. Lindeman L. P., Wilson M. KJ/Z. phys. Chem. (BRD). 1956. B. 9. S. 29.
437. Duncan J. L., Mills I. M//Spcctrochirn. acta. 1964. V. 20. P. 1089.
438. McDowell R. S., Reisfeld M. J., Patterson C. W. e. a.//J. Chem. Phys. 1982.
V. 77. P. 4337.
274
439. Freeman D. E., Wilson M. KJ/Z. phys. Chem. (BRD). 1962. B. 35. S. 335.
440. Clark R. J. H., Mitchell P. DJ/J. Chem. Soc. Faraday Trans. Part II. 1975.
V. 71 P 515________524.
441. Koniger F., Muller A., Orville-Thomas W. J.I/J. Mol. Struct. 1977. V. 37.
P. 199.
442. Caunt A. D., Mackie H., Sutton L. £.//Trans. Faraday Soc. 1951. V. 47.
P. 943.
443. Long D. A.//Proc. Roy. Soc. London. 1957. V. A240. P. 499.
444. Sousa G. G., Wieser J. DJ/J. Mol. Struct. 1975. V. 25. P. 442.
445. Bruger EL, Ruoff A.//Spectrochim. acta. 1968. V. 24A. P. 1863.
446. Fujii H., Kimura M.//Bull. Chem. Soc. Jap. 1970. V. 43. P. 1933—1939.
447. Koniger F., Muller AJ/J. Mol. Spectrosc. 1975. V. 56. P. 200.
448. Neu J. T., Gwinn W. DJ/L Amer. Chem. Soc. 1948. V. 70. P. 3463—
3465.
449. Иоффе А. И., Нефедов О. М//Ж. ВХО им. Д. И. Менделеева. 1979. Т. 24.
С. 475—484.
450. Nefedov О. М., Kolesnikov S. Р., Ioffe A. /.//Organometal. Chem. Rev.
Library. 1977. V. 5. P. 180.
451. Hine Y., Ehrenson S. J J/J. Amer. Chem. Soc. 1958. V. 80. P. 824—830.
452. Simons J. PJ/J. Chem. Soc. 1965. P. 5406—5413.
453. Baird N. C., Taylor K. FJ/J. Amer. Chem. Soc. 1978. V. 100. P. 1333—1338.
454. Sustman R., Binsch GJ/Moi. Phys. 1971. V. 20. P. 9.
455. Фукуи /(.//Реакционная способность и пути реакций. Пер. с англ. М.:
Мир, 1977. С. 57; Fujimoto Н., YamabeS., Fukui KJ/Bull. Chem Soc. Jap.
1972. V. 45. P. 1566.
456. Nagase S., Fueno T,//Theor. chim. acta. 1976. V. 41. P. 59.
457. Hoffmann R., Gleiter R., Mallory F. BJ/J. Amer. Chem. Soc. 1970. V. 92.
P. 1460—1466.
458. Kollmar //.//Tetrahedron Lett. 1970. P. 3337.
459. Basch HJ/J. Chem. Phys. 1971. V. 55. P. 1700.
460. Tantardini G. F., Simonetta MJ/\sv&e\ J. Chem. 1972. V. 10. P. 581.
461. Cremaschi P., Simonetta MJ/J. Chem. Soc. Faraday Trans. Part II. 1974.
V. 70. P. 1801 — 1809.
462. Feller D„ Davidson E. R.//J. Phys. Chem. 1983. V. 87. P. 2721.
463. Костиков P. P., Хлебников А. Ф., Беспалов В. flJ/JK. ВХО им. Д. И. Мен-
делеева. 1979. T. 24. С. 438—445.
464. Cheung L. М., Sundberg К. R., Ruedenberg KJAM. J. Quantum Chem.
1979. V. 16. P. 1103.
465. Ruedenberg K-> Schmidt M. W., Gilbert M. M. e. a.//L Chem. Phys. 1982.
V. 71. P. 41.
466. Brown W., Bass A. M., Pilling MJ/J. Chem. Phys. 1970. V. 52. P. 5131.
467. Pedley J., Rylance J. Computer-analysed Thermochemical Data: Organic
and Organometallic Compounds. Univ, of Sussex. 1983. 310 p.
468. Политанский С. Ф.ЦУК. ВХО им. Д. И. Менделеева. 1979. Т. 24. С. 529—
539
469. Dalby F. WJ/J. Chem. Phys. 1964. V. 41. Р. 2297.
470. Tyerman W. J. R.//Trans. Faraday Soc. 1969. V. 65. P. 1188.
471. Кутина И. Д., Бельферман А. Л., Шевчук В. У.//Кинет. и катализ. 1972.
Т. 13. С. 843.
472. Joines R. С., Turner А. В., Jones W. MJ/J. Amer. Chem. Soc. 1969. V. 91.
P. 7754—7755.
473. Мальцев А. К., Зуев П. С., Нефедов О. MJ/Изв. АН СССР. Сер. хим.
1985. С. 2159.
474. de March Р., Huisgen RJ/J. Amer. Chem. Soc. 1982. V. 104. P. 4952.
475. 'Wong P. C., Griller D., Scaiano J. CJ/J. Amer. Chem. Soc. 1982. V. 104.
P. 6631—6635.
18'
275
476. Rende A. S., Hebeisen P., Sanfilippo P. J., Toder В. H.//J. Amer. Chem.
Soc. 1982. V. 104. P. 4244—4245.
477. Doyle M. P., Tamblyn W. H., Bagheri V.//J. Org. Chem. 1981. V. 46.
P. 5094—5102.
478. Dobson R. C., Hayes D. M., Hoffmann R.//J. Amer. Chem. Soc. 1971. V. 93.
P. 6188—6192.
479. Bauschlicher C. W., Bender C. F.t Schaefer H. F.i/J. Amer. Chem. Soc. 1976.
V. 98. P. 3072—3074.
480. Gordon M. S., Cano D. R.//J. Amer. Chem. Soc. 1984. V. 106. P. 5421 —
5425; Raghavachari R., Chandrasekhar J., Gordon M. S., Dykema R. J.-/
J. Amer. Chem. Soc. P. 5853—5859.
481. Sosa C., Schlegel H. B.//J. Amer. Chem. Soc. 1984. V. 106. P. 5847—5852.
482. Нефедов О. M., Иоффе А. И.//Изв. АН СССР. Сер. хим. 1987. С. 801.
483. RoAecHtiKoe С. П., Иоффе А. И., Нефедов О. М.ЦИзв. АН СССР. Сер.
хим. 1973. С. 2622.
484. Bell Т. N., Sherwood A. G., Soto-Garrido G.//J. Phys. Chem. 1986. V. 90.
Р. 1184.
485. Rirmse W., Loosen R., Slutna H. D.//J. Amer. Chem. Soc. 1981. V. 103.
P 5935_______5937
486. Simon J. D., Peters R. S.//J. Amer. Chem. Soc. 1983. V. 105. P. 5156—
5158.
487. Bethell D., N ewall A. R„ Whittaker D.//J. Chem. Soc. Part B. 1971. P. 23—
31.
488. Bethell D., Hayes J., Newall A. R.//J. Chem. Soc. Perkin Trans. Part II.
1974. P. 1307—1312.
489. Wright В. B., Ranakarajan R., Platz M. S.//J. Phys. Chem. 1985. V. 89.
P. 3574.
490. Bauschlicher C. W., Schaefer H. F., Bender C. F.//J. Amer. Chem. Soc. 1976.
V. 98. P. 1653—1658.
491. Wang I. S. Y., Rar plus M.//J. Amer. Chem. Soc. 1973. V. 95. P. 8160—
8164.
492. Hoffmann R., Hayes D. M., Skell P. S.//J. Phys. Chem. 1972. V. 76. P. 664.
493. Bodor N., Dewar M. J. S., Maksic Z. B.//J. Amer. Chem. Soc. 1973. V. 95.
P 5245_______5249
494. Rondan N. G., Houk R. ^//Tetrahedron Lett. 1984. V. 25. P. 5965.
495. Houk R. N., Rondan N. G., Mareda J.//J. Amer. Chem. Soc. 1984. V. 106.
P. 4291—4293; Houk R. N., Rondan N. G.//Ibid. 1984. V. 106. P. 4293—
4294.
496. Houk R. N., Rondan N. G., Mareda /.//Tetrahedron. 1985. V. 41. P. 1555.
497. Giese B., Mehl W. H„ Lee W. ^.//Tetrahedron. 1985. V. 41. P. 1565.
498. Moss R. A.t Guo W., Denney D. Z. e. a.//J. Amer. Chem. Soc. 1981. V. 103.
P. 6164—6169.
499. Seyferth D.//J. Organomet. Chem. 1975. V. 100. P. 237.
500. Savino T. G., Ranakarajan R., Platz M. S.//J. Org. Chem. 1986. V. 51.
p 1305_______1309.
501. Wiberg R. B., Lampman G. M., Cinla R. P. e. «.//Tetrahedron. 1965. V. 21.
P. 2749.
502. Von Doering W. E., Coburn J. F.//Tetrahedron Lett. 1965. P. 991.
503. Jef ford C. W., Gehret J. С. E., de los Heros V.//Bull. Soc. chim. Belg. 1979.
V. 88. P. 973.
504. Jackson J. E., Mock G. B., Tetef M. L. e. «.//Tetrahedron. 1985. V. 41.
P. 1453.
505. Rwantes P. M., Rlumpp G. W.//Tetrahedron Lett. 1976. P. 707.
506. Jef ford C. W„ Bernardinelli G. J., Rossien J. C. e. «.//Helv. chim. аса.
1987. V. 65. P. 1467.
507. RoAecHUKoe C. H./IJR. ВХО им. Д. И. Менделеева. 1979. T. 24. С. 505—
512.
276
508. Lambert J. В., Larson E. G., Bosch R. /.//Tetrahedron Lett. 1983. V. 24.
P 3799
509. Колесников C. LL, Ширяев В. И., Нефедов О. Л4.//Изв. АН СССР. Сер.
хим. 1966. С. 584.
510. Колесников С. П., Рогожин И. С., Нефедов О. MJ/Изв. АН СССР. Сер.
хим. 1974. С. 2379.
511. Кулишов В. И.ЦУК. структур, химии. 1970f Т. 11. С. 71.
512. Колесников С. П., Иоффе А. И.., Перльмуттер Б. Л., Нефедов О. М.,1/
Изв. АН СССР. Сер. хим. 1975. С. 755—759.'
513. Бокий Н. Г., Колесников С. П., Стручков Ю. Т. и др.//Изв. АН СССР.
Сер. хим. 1975. С. 812—815.
514. Brauer D. J., Kruger С.//Тезисы Второго Европейского конгресса по кри-
сталлографии. Кестхей (Венгрия). 1974. С. 435.
515. Harrison Р. G., Zucherman J. JJ/3. Amer. Chem. Soc. 1970. V. 92. P. 2577—
2578
516. Larrabee R. B.//Tetrahedron Lett. 1965. P. 287; Hill E. A.//J. Org. Chem.
1972. V. 37. P. 4008—4012.
517. Nickon A., Huang F., Weglein R. e. a.//J. Amer. Chem. Soc. 1974. V. 96.
P. 5264—5265.
518. Kyba E. P., John A. AL//J. Amer. Chem. Soc. 1977. V. 99. P. 8329—8330.
519. Dellacoletta B. A., Schechter //.//Tetrahedron Lett. 1979. P. 4817.
520. Chang K-Т., Schechter H./JJ. Amer. Chem. Soc. 1979. V. 101. P. 5082—
5084.
521. Fukushima M., Jones M., Brinker U. //.//Tetrahedron Lett. V. 23. P. 3211.
522. Schaefer H. FJ/Acc. Chem. Res. 1979. V. 12. P. 288—296.
523. Liu M. T. HJ/J. Chem. Soc. Chem. Comm. 1985. P. 982—984.
524. Liu M. T. H., Subramanjan R.//J. Phys. Chem. 1986. V. 90. P. 75.
525. McMahon R. J., Chapman O. LJ/J. Amer. Chem. Soc. 1987. V. 109. P. 683—
692.
526. Conlin R. T., Kwak Y.-WJRL Amer. Chem. Soc. 1986. V. 108. P. 834—835.
527. Reisenauer H. P., Mihm G., Maier G.//Angew. Chem. 1982. B. 94. S. 864—
865.
528. Jones MJ/Acc. Chem. Res. 1974. V. 7. P. 415—421.
529. Burnett S. M., Stevens A. E., Feigerle C. S. e. aj/Chem. Phys. Lett. 1983.
V. 100. P. 124.
530. Osamura Y., Schaefer H. F., Gray S. K-, Miller Ж HJjJ. Amer. Chem. Soc.
1981. V. 103. P. 1904—1907.
531. Родина Л. Л., Коробицына И. KJ/Усп. химии. 1967. T. 36. С. 611.
532. Fenwick J., Frater G., Ogi K-, Strauss O. PJ/J. Amer. Chem. Soc. 1973.
V. 95. P. 124—132.
533. Политанский С. Ф., Липкинд M. Б., Ивашенко А. А., Нефедов О. M. /
Изв. АН СССР. Сер. хим. 1980. С. 734.
534. Sekiguchi A., Ando Ж./ZTetrahedron Lett. 1985. V. 26. Р. 2337.
535. Шарп Дж. Т.//Общая органическая химия. В 12 т. Т. 1: Пер. с англ./Под
ред. Н. К- Кочеткова. М.: Химия, 1981. С. 601—617.
536. Krajca К Е., Jones W. ^.//Tetrahedron Lett. 1975. Р. 3807.
537. Becker J., Wentrup CJ/J. Chem. Soc. Chem. Comm. 1980. P. 190—191.
538. Wentrup C.//Fortschr. Chem. Forsch. 1976. V. 62. P. 173.
539. Crow W. D., Paddon-Row M. NJ/J. Amer. Chem. Soc. 1972. V. 94. P. 4746—
4747; Jones W. MJ /Ibid. 1973. V. 95. P. 826.
540. Gaspar P. P., Hsu J.-P., Chari S. e. ^.//Tetrahedron. 1985. V. 41. P. 1479.
541. Wentrup C., Mayor C., Becker J. e. ^.//Tetrahedron. 1985. V. 41. P. 1601.
542. Kirmse W., Chiem P. V., Henning P.-G.//Tetrahedron. 1985. V. 41. P. 1441.
543. Brinker U. H., Ritzer J J/J. Amer. Chem. Soc. 1981. V. 103. P. 2116—
2119.
544. Birkhahn M., Dehmlow E. V., Bogge //.//Angew. Chem. 1987. B. 99. S. 80—
81.
277
545. Baird M. S., Buxton S. R., Mitra MJ/Tetrahedron Lett. 1982. V. 23.
P. 2701.
546. Bloch R., Orvane ^.//Tetrahedron Lett. 1981. V. 22. P. 3597.
547. Barton T. J., Groh B. LJfJ. Org. Chem. 1985. V. 50. P. 158—166.
548. Barton T. J., Groh B. LJfT. Amer. Chem. Soc. 1985. V. 107. P. 8297—
8299.
549. Этвелл У. X., Вейенберг Д. Р.ЦУ&1. химии. 1970. Т. 39. С. 1244; At-
well J. Н., Weienberg D. R.//Intra-Science Chem. Repts. 1973. V. 7. P. 139.
550. Gordon M. S., Cano D. R., Binkley J. S., Frisch M. J.//J. Amer. Chem. Soc.
1986. V. 108. P. 2191—2195.
551. Noor Batcha I., Raff L. M., Thompson D. L. e. a.JT. Chem. Phys. 1984.
V. 81. P. 3715; V. 82. P. 5762; 1985. V. 82. P. 1543; 1986. V. 84. P. 4341.
552. Viswanathan R.f Thompson D. L., Raff L. M.//J. Chem. Phys. 1984. V. 80.
P. 4230; V. 81. P. 828, 3363.
553. Rickborn S. F., Rogers D. S., Ring M. A. e. а.ЦТ. Phys. Chem. 1986. V. 90.
P. 408.
554. Neumann W. P. The Organic Chemistry of Tin. L.: Wiley. 1970. 190 p.
555. Bos K- D. Organic and Organometallic Chemistry of Divalent Tin. Ut-
recht. 1976. 210 p.
556. Ширяев В. И., Миронов В. Ф., Кочергин В. П. Соединения двухвалент-
ного олова в синтезе оловоорганических соединений. М.: НИИТЭХИМ,
1977. 202 с.
557. Murgulescu I. G., Schneider I. А.ЦТС phys. Chem. (DDR). 1961. B. 218.
S. 338.
558. Kozlov G. L, Knorre V. G.//Combust, a. Flame. 1962. V. 6. P. 253.
559. Козлов Г. И., Кнорре В. FJ/Ж- физ. хим. 1963. Т. 37. С. 2082.
560. Левуш С. С. и др.//Кинет, и катализ. 1973. Т. 14. С. 277.
561. Okabe Н., McNesby J. RJ/J. Chem. Phys. 1961. V. 34. P. 668.
562. Ausloos P., Rebbert R. E., Lias S. GJfT. Chem. Phys. 1965. V. 42. P. 540.
563. Stief L. J., DeCarlo V. L, Hillman J. J J /J. Chem. Phys. 1965. V. 43.
P. 2490.
564. Gevantman L. H., Williams R. RJ/J. Phys. Chem. 1952. V. 56. P. 569.
565. Hearne J. А.//РЖХим. 1966. 13Б 859.
566. Витвицкий А. И.ЦУК- орг. хим. 1969. T. 5. C. 1718.
567. Иоффе А. И., Фаустов В. И.ЦИзъ. АН СССР. Сер. хим. 1987. С. 1793.
568. Hamson R. F. е. а.ЦТ. Chem. Phys. 1964. V. 40. Р. 1099.
569. Roquitte В. С.ЦТ. Phys. Chem. 1970. V. 74. Р. 1204.
570. Okabe Н., McNesby J. RJfT. Chem. Phys. 1962. V. 36. P. 3505.
571. Ausloos P., Lias S. GJfT. Chem. Phys. 1966. V. 44. P. 521.
572. Rebbert R. E., Ausloos Р.ЦТ. Chem. Phys. 1967. V. 46. P. 4333.
573. Tschuikow-Roux E., McNesby R.//Trans. Faraday Soc. 1966. V. 62. P. 2158.
574. Semeluk G. P., Bernstein R. ВДТ. Amer. Chem. Soc. 1957. V. 79. P. 46—
49.
575. Шилов A. E., Сабирова P. ДЛЖ- физ. хим. 1960. T. 34. С. 860.
576. V acherot М., Par ant NJ/С. г. 1963. V. 257. Р. 3938.
577. Kung F. Е., Bissinger W. Е.ЦТ. Org. Chem. 1964. V. 29. P. 2739—2742.
578. Миронов В. Ф., Максимова Н. Г.//Изв. АН СССР. Сер. хим. 1965. С. 2193.
579. Gozzo F., Patrick С. /.//Tetrahedron. 1966. V. 22. Р. 3329.
580. Политанский С. Ф., Шевчук В. У.//Кинет. и катализ. 1968. Т. 9. С. 496.
581. КушинаИ. Д., Бельферман А. Л./ДАзъ. АН СССР. Сер. хим. 1977. С. 1484.
582. Ку шина И. Д., Бельферман А. Л., Шевчук В. У.//Изв. АН СССР. Сер.
хим. 1974. С. 946.
583. Edwards J. W., Small Р. A.//Nature. 1964. V. 202. Р. 1329; Edwards J. W.,
Small P. A.//Ind. Eng. Chem. 1965. V. 4. P. 396.
584. Simons J. ^//Nature. 1965. V. 205. P. 1308.
585. Cox R. A., Simmons R. FJfT. Chem. Soc. Part B. 1971. P. 1625—1631.
586. Иванык Г. Д., Политанский С. Ф.//Новое в химии карбенов. М.: Наука,
1973. С. 150.
278
587. Лебр М., Мазероль П., Сатже Ж. Органические соединения германия:
Пер. с англ./Под ред. Г. А. Разуваева. М.: Мир, 1974. 232 с.
588. Разуваев Г. А., Василейская Н. С.//Изв. АН СССР. Сер. хим. 1965.
С. 1285.
589. Dolphin D., Girigg R., Johnson A. W., Leng J.ffL Chem. Soc. 1965.
P. 1460—1467.
590. Unger I., Semelic G. P.//Can. J. Chem. 1966. V. 44. P. 1427.
591. Jones AL, Sachs W. H., Kulczycki A., Waller F. J.//J. Amer. Chem. Soc.
1966. V. 88. P. 3167—3168.
592. Werner H. R., Firestone R. F.RL Phys. Cherti. 1965. V. 69. P. 840.
593. Wawzonek S., Duty R.//L Electrochem. Soc. 1961. V. 108. P. 1135.
594. Fritz H. P„ Kornrumpf WJ/Ann. 1978. S. 1416.
595. Нефедов О. M., Иващенко А. А.//Изв. АН СССР. Сер. хим. 1965. С. 1716.
596. Fang Y.-N., Rowland F. S.//L Amer. Chem. Soc. 1965. V. 87. P. 1625—
1626.
597. Политанский С. Ф., Шевчук В. У.//Кинет. и катализ. 1967. Т. 8. С. 12.
598. Schung R. Р., Wagner Н. G.HZ. phys. Chem. (BRD). 1973. В. 86. S. 59.
599. Wescott H. D., Skell P. S.//J. Amer. Chem. Soc. 1965. V. 87. P. 1721 —
1724.
600. Stefanescu P.//Rev. roum. chim. 1959. V. 10. P. 524.
601. Blanchard L. P., Le Goff P.//Can. J. Chem. 1957. V. 35. P. 89.
602. Fielding W., Pritchard H. O.HL Phys. Chem. 1960. V. 64. P. 278.
603. Taylor P. D., Tuckerman R. T., Whittle E.HL Photochem. 1982. V. 19.
P. 277.
604. Blomstrom D. C., Herbig K-, Simmons H. E.//L Org. Chem. 1965. V. 30.
P. 959—964.
605. Kaplan L.//J. Amer. Chem. Soc. 1967. V. 89. P. 4566; Kaplan Ь.ЦА. Chem.
Soc. Chem. Comm. 1969. P. 106—107.
606. Yang N. C., Marolewski T. A.RL Amer. Chem. Soc. 1968. V. 90. P. 5644—
5646.
607. Newman R. C., Wolcott R. G.//Tetrahedron Lett. 1966. P. 6267.
608. Tang Y.-N., Rowland F. S.//L Amer. Chem. Soc. 1966. V. 88. P. 626—
627; 1967. V. 89. P. 6420—6427; 1968. V. 90. P. 574—582.
609. Tang Y.-N., Smail T., Rowland F. S.//L Amer. Chem. Soc. 1969. V. 91.
P. 2130—2131.
610. Чернышев E. А., Комаленкова H. Г., Башкирова С. Д.//Усп. химии. 1976.
T. 45. С. 1782.
611. Иоффе А. И., Святкин В. А., Нефедов О. М.//Строение производных ци-
клопропана. М.: Наука, 1986. 160 с.
612. Kennedy R. С., Levy J. B.//L Fluor. Chem. 1976. V. 7. Р. 101.
613. Atkinson В., McKeagan D.HL Chem. Soc. Chem. Comm. 1966. P. 189—
190.
614. Mitsh R. A., Neuvar E. W./fL Phys. Chem. 1966. V. 70. P. 546.
615. Haszeldine R. N., Speight J. G.HL Chem. Soc. Chem. Comm. 1967. P. 995—
996.
616. Birchall J. M., Hazseldine R. N., Roberts D.W.fjL Chem. Soc. Chem. Comm.
P. 287—288.
617. Fields R., Haszeldine R. N., Peter D.HL Chem. Soc. Chem. Comm. P. 1081 —
1983; Fields R., Haszeldine R. N., Peter D.HL Chem. Soc. Part C. 1969.
P. 165—172.
618. Moss R. A., Cox D. ^.//Tetrahedron Lett. 1985. V. 26. P. 1931.
619. Kristinsson H. e. a.//Chem. a. Ind. 1966. P. 1562.
620. Zimmerman H. E., Pratt A. C.//L Amer. Chem. Soc. 1970. V. 92. P. 6259—
6267.
621. Dietrich H., Griffin G. W., Petterson P. C.//Tetrahedron Lett. 1968. P. 153.
622. Valyocsik E. W., Sigal P.//L Org. Chem. 1971. V. 36. P. 66—72.
623. Gilbert J. C., Butler J. R.ffL Amer. Chem. Soc. 1970. V. 92. P. 7493—
7494.
279
624. Kende A. S., Goldschmidt Z., Smith R, F.HL Amer. Chem. Soc. 1970. V. 92.
P. 7606—7607.
625. Huttner G., Schelle S., Mills O. S.//Angew. Chem. 1969. B. 81. S. 536.
626. Leermakers P. A., Ross M. E.HL Org. Chem. 1966. V. 31. P. 301—304.
627. Brunton R. K.//J. Phys. Chem. 1968. V. 72. P. 321.
628. Richardson D. B., Durrett L. R., Martin J. M. e. a.//J. Amer. Chem. Soc.
1965. V. 87. P. 2763—2765.
629. ter Borg A. P., Razenberg E., Kloosterziel HJ/Rec. trav. chim. 1966. V. 85.
P. 774.
630. Schonberg A., Mustafa A., Latif N.//J. Amer. Chem. Soc. 1953. V. 75.
P. 2267.
631. Levin R. H., Jones ^.//Tetrahedron. 1971. V. 27. P. 2031.
632. Woods W. G./IL Org. Chem. 1958. V. 23. P. 110—112.
633. Нефедов О. M.t Новицкая H. H., Исаев И. С.//Докл. АН СССР. 1966.
Т. 168. С. 106.
634. Ciganek Е.ЦУ Amer. Chem. Soc. 1967. V. 89. Р. 1458 — 1468.
635. Toda Т., Nitta М., Mukai T.//Tetrahedron Lett. 1969. P. 4401.
636. Swenton J. S., Krubsack A. J.//J. Amer. Chem. Soc. 1969. V. 91. P. 786—
787.
637. Pomerantz M., Gruber G. W./J. Amer. Chem. Soc. 1967. V. 89. P. 6798—
6799.
638. Seyferth D., Annarelli D. C., Duncan D. P.//Organometallics. 1982. V. 1.
P. 1288.
639. Masamune S., Murakami S., Tobita H, Williams D. J.HL Amer. Chem.
Soc. 1983. V. 105. P. 7776—7778.
640. Masamune S., Tobita H., Murakami S.//L Amer. Chem. Soc. P. 6524—6525.
641. Watanabe H., Okawa T., Kato M., Nagai Y.HL Chem. Soc. Chem. Comm.
1983. P. 781—782.
642. Schafer A., Weidenbruch M.HJ. Organomet. Chem. 1985. V. 282. P. 305.
643. Murakami S., Collins S., Masamune S.//Tetrahedron Lett. 1984. V. 25.
2131.
644. Collins S., Murakami S., Snow J. T. e. a./ Tetrahedron Lett. 1985. V. 26.
P. 1281.
645. Crocco L., Classman L, Smith I. Е.Ц5. Chem. Phys. 1959. V. 31. P. 506.
646. Lenzi M., Mele A.//J. Chem. Phys. 1965. V. 43. P. 1974.
647. Sargeant P. B.//J. Org. Chem. 1970. V. 35. P. 678—682.
648. Bayless J. H., Friedman L., Cook F. B., Shechter H.HL Amer. Chem. Soc.
1968. V. 90. P. 531—533.
649. Kristinsson H., Griffin G. IF.//Angew. Chem. 1965. B. 77. S. 859; J. Amer.
Chem. Soc. 1966. V. 88. P. 1579—1580.
650. Kristinsson //.//Tetrahedron Lett. 1966. P. 2343.
651. Petrellis P. G., Dietrich H., Meyer E., Griffin G. W.//L Amer. Chem. Soc.
1967. V. 89. P. 1967—1969; Petrellis P. G., Griffin G. W.//L Chem. Soc.
Chem. Comm. 1967. P. 691—692.
652. Темникова T. И., Степанов И. П., Семенова Л. О.//Ж. орг. хим. 1967.
Т. 3. С. 1708.
653. Темникова Т. И., Степанов И. П., Доценко Л. А.//Ж. орг. хим. 1967.
Т 3 С 2203
654. Trozzolo А. М., Yager W. A., Griffin G. W. е. a.//J. Amer. Chem. Soc. 1967.
V. 89. Р. 3357—3358.
655. Tomioka Н., Griffin G. W., Nishiyama KJfL Amer. Chem. Soc. 1979. V. 101.
P. 6009—6012.
656. Ando W., Hamada Y., Sekiguchi A.//Tetrahedron Lett. 1984. V. 25. P. 5057.
657. Nozaki H., Fujita S., Noyori /^.//Tetrahedron. 1968. V. 24. P. 2193.
658. Moore С. B., Pimentel G. C.ffL Chem. Phys. 1964. V. 41. P. 3504.
659. Frey H. M., Stevens I. D. R.//J. Chem. Soc. 1962. P. 3865—3867.
660. Frey H. M.; Wscaplehorn A.HL Chem. Soc. Part A. 1966. P. 968.
661. Neuvar E. W., Mitsch R. A.//L Phys. Chem. 1967. V. 71. P. 1229.
280
662. Paulet G. S., Ettlinger R.//J. Chem. Phys. 1963. V. 39. P. 825. P. 3534;
1964. V. 41. P. 2557.
663. Frey H. M.//Adv. Photochem. 1966. V. 4. P. 225.
664. Gale D. M., Middleton W. J., Krespan C. GJ/J. Amer. Chem. Soc. 1966.
V. 88. P. 3617—3623.
665. Frey H. M., Stevens I. D. R.//J. Chem. Soc. 1963. P. 3514—3519; 1964.
p. 4700—4706; 1965. P. 1700—1706. P. 3101—3108; J. Amer. Chem. Soc.
1962. V. 84. P. 2647.
666. Bredereck H., Effenberger F., Bredereck H. /.//Angew. Chem. 1966. B. 78.
S. 984.
667. Frey H. M., Stevens I. D. RJ/J. Amer. Chem. Soc. 1962. V. 84. P. 2647.
668. Mansoor A. M., Stevens I. D. R.//Tetrahedron Lett. 1966. P. 1733
669. Tomioka H., Ozari Y., Koyabi Y. e. a.h Chem. Lett. 1982. P. 843.
670. Moss R. A.//Tetrahedron Lett. 1967. P. 4905.
671. Pimentel G. C., Herr K. C»//J. Chem. Phys. 1964. V. 41. P. 1509.
672. Atkinson B., Atkinson V. A.//J. Chem. Soc. 1957. P. 2087.
673. Butler J. AL//J. Amer. Chem. Soc. 1962. V. 84. P. 1393—1398.
674. Bondybey V. E., Pimentel G. C.//J. Chem. Phys. 1972. V. 56. P. 3832.
675. Nair R. M. G., Meyer E., Griffin G. IF.//Angew. Chem. 1968. B. 80. S. 442—
443.
676. Smith R. L., Manmade A., Griffin G. W.//J. Heterocvcl. Chem. 1969. V. 6.
P. 443; Tetrahedron Lett. 1970. P. 663.
677. Petrellis P., Griffin G. W.//J. Chem. Soc. Chem. Comm. 1968. P. 1099—
1100.
678. Baxter G. J., Brown R. F. C.//Austral. J. Chem. 1978. V. 31. P. 327.
679. Hoffmann R. W., Wilnsche C.//Tetrahedron Lett. 1964. P. 197; Tetrahedron.
1965. V. 21. P. 891; Chem. Ber. 1967. B. 100. S. 943—949.
680. Lemal D. M., Gosselink E. P., McGregor S. D./;Tetrahedron Lett. 1964.
P. 579; J. Amer. Chem. Soc. 1966. V. 88. P. 582—600.
681. Mackenzil /C//J. Chem. Soc. 1964. P. 5710.
682. McBee E. T., Smith D. K., Ungnade H. Ej.J. Amer. Chem. Soc. 1955. V. 77.
P. 387—389.
683. Rautenschtrauch V., Scholl H.-J., Vogel £.//Angew. Chem. 1968. B. 80—
81. S. 278.
684. Sakurai H., Sakaba H., Nakadaira Y.f/J. Amer. Chem. Soc. 1982. V. 104.
P. 6156—6158.
685. Kocher J., Neumann W. P.//Organometallics. 1985. V. 4. P. 400.
686. Schriewer M., Neumann W. P.//J. Amer. Chem. Soc. 1983. V. 105. P. 897—
901.
687. Kocher J., Neumann IF. P.//J. Amer. Chem. Soc. 1984. V. 106. P. 3861 —
3862.
688. Gross L.-W., Moser R., Neumann IF. P. e. a.//Tetrahedron Lett. 1982. V. 23.
P. 635.
689. Kumada AL//Intra-Science Chem. Repts. 1973. V. 7. P. 121.
690. Fritz G., Grunert B.//Z. anorg. Chem. 1981. B. 473. S. 59.
691. Tortorelli V. J., Jones MJ /J. Amer. Chem. Soc. 1980. V. 102. P. 1425—
1426.
692. Nakao R., Oka K-, Dohmaru T. e. a.HJ. Chem. Soc. Chem. Comm. 1985.
P. 706.
693. Trefonaz Р.» West R., Miller R. D.HJ. Amer. Chem. Soc. 1985. V. 107.
P. 2737—2742.
694. Watanabe H., Kongo Y., Nagai Y.//J. Chem. Soc. Chem. Comm. 1984.
P. 66—67.
695. Ishikawa M., Nakagawa K-, Katayama S., Kumada M.//J. Amer. Chem. Soc.
1981. V. 103. P. 4170—4174.
696. Ishikawa M., Nakagawa K.-E, Kumada M. e. aJlJ. Organomet. Chem. 1981.
V. 214. P. 277.
281
697. Ando W., Hamada Y., Sekiguchi A.//Tetrahedron Lett 1982. V. 23. P. 5323;
1983. V. 24. P. 4033.
698. Ando W., Ikeno M., Hamada YJfS. Chem. Soc. Chem. Comm. 1981. P. 621 —
622.
699. Chen Y.-S., Gaspar P. P.//Organometallics. 1982. V. 1. P. 1410.
700. Sakurai H., Kobayashi Y., Nakadaira YJ/E Amer. Chem. Soc. 1974. V. 96.
P. 2656—2657.
701. Sakurai H., Sakamoto K., Kira Af.//Chem. Lett. 1984. P. 1379—1382.
702. Hoffmann R. IF.//Angew. Chem. 1971. B. 83. S. 595—603.
703. Griffin G. IF.//Angew. Chem. 1971. B. 83. S. 604—613.
704. Нефедов О. M., Дьяченко А. И., Прокофьев А. Kj/Усп. химии. 1977. Т. 46.
С. 1787.
705. Дьяченко А. И., Иоффе А. И., Нефедов О. MJ/Изв. АН СССР. Сер. хим.
1979. С. 366.
706. Miller W. Т., Kim С. S. YJ/J. Amer. Chem. Soc. 1959. V. 81. Р. 5008—
5009.
707. Wittig G, Pockets UJ/Ber. 1939. В. 72. S. 884—886; Wittig G„ Witt Н.Ц
Ber. 1941. B. 74. S. 1474—1491.
708. Closs G. L., Coyle J. J.//J. Org. Chem. 1966. V. 31. P. 2759—2765.
709. Kobrich G., Fischer R. //.//Chem. Ber. 1968. B. 101. S. 3208—3218.
710. Нефедов О. M., Ширяев В. И.//УК. общ. хим. 1967. Т. 37. С. 1223.
711. Fischer R. Н., Kobrich G.//Chem. Ber. 1968. В. 101. S. 3230—3233.
712. Нефедов О. М., Дьяченко А. И.//Изв. АН СССР. Сер. хим. 1972. С. 487.
713. Kobrich К, Flory К> Fischer R. //.//Chem. Ber. 1966. В. 99. S. 1793—
1804.
714. Huisgen R., Burger (/.//Tetrahedron Lett. 1970. P. 3049. P. 3053.
715. Friedman L., Honour R. J., Berger J. GJ/F Amer. Chem. Soc. 1970. V. 92.
p. 4640—4645.
716. Closs G. L., Closs L. EJ/J. Amer. Chem. Soc. 1960. V. 82. P. 5723—5728.
717. Hoeg D. F., Lusk D. L, Crumbliss A. LJ/L Amer. Chem. Soc. 1965. V. 87.
P. 4247.
718. Нефедов О. M., Ширяев В. И., Петров А. Д.//УК- общ. хим. 1962. Т. 32.
С. 662.
719. Нефедов О. MJ/Изв. АН СССР. ОХН. 1962. С. 367.
720. Нефедов О. М., Ивашенко А. А.//Изв. АН СССР. Сер. хим. 1965. С. 2209;
Докл. АН СССР. 1964. Т. 156. С. 884.
721. Andrews L., Smith D. WJ/J. Chem. Phys. 1970. V. 53. P. 2956; 1971. V. 55.
P 5295
722. Vincent M. A., Schaefer H. FJ/J. Chem. Phys. 1982. V. 77. P. 6103.
723. Harada T., Nozaki Y., Yamaura Y. e. aJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 2189—2190.
724. Skell P. S., Cholod M. S.//J. Amer. Chem. Soc. 1969. V. 91. P. 7131—7137.
725. Нефедов О. M.t Шафран P. //.//Изв. АН СССР. Сер. хим. 1965. С. 538.
726. Muetterties Е. £.//Inorg. Chem. 1962. V. 1. Р. 342.
727. Lesbre MJ /С. г. 1937. V. 204. Р. 1822.
728. Poskozin Р. SJ/J. Organomet. Chem. 1968. V. 12. Р. 115.
729. Tchakirian A., Lesbre М., Lewinsohn KJ/С. г. 1936. V. 202. Р. 138.
730. Гусельников Л. Е., Кервина 3. А., Поляков Ю. П. и др.//Изв. АН СССР.
Сер. хим. 1982. С. 219.
731. Gusel’nikov L. Е., Polyakov Yu. Р., Volnina E. A. e. aJ/J. Organomet.
Chem. 1985. V. 292. P. 189.
732. Нефедов О. M., Скелл П. С.ЦДокл. АН СССР. 1981. Т. 259. С. 377.
733. Le Parchee Р., Conia J. AL//Tetrahedron Lett. 1970. P. 1587.
734. Nozaki //.//Tetrahedron Lett. 1971. P. 905.
735. Kobrich G., Heineman H., Zirndorf W.//Tetrahedron. 1967. V. 23. P. 565.
736. Villieras JJ/J. Organomet. Chem. 1972. V. 34. P. 209.
737. Ческис Б. А., Дьяченко А. И., Моисеенков A. M., Нефедов О. MJ/Изв.
? И СССР. Сер. хим. 1981. С. 692.
282
738. Колесников С. П., Поваров С. Л., Цветков С. Ю. и др.ЦИзъ. АН СССР.
. Сер. хим. 1984. С. 861.
у 39. Simmons Н. Е., Seyferth D./fOrg. Reactions. N. Y.: Wiley. 1973. V. 20.
* P. 1.
740. Wittig G., Schwarzenbach /(.//Ann. 1962. B. 650. S. 1.
741. Seyferth D.HKce. Chem. Res. 1972. V. 5. P. 65—74.
742. Seyferth D., Burlitch J. AL//J. Amer. Chem. Soc. 1964. V. 86. P. 2730—
2731.
743. Seyferth D., Gordon M. E., Mui J. Y.-P., Burlich J. М.ЦЕ Amer. Chem. Soc.
1967. V. 89. P. 959—966.
744. Seyferth D., Eisert M. А.ЦЕ Amer. Chem. Soc. 1964. V. 86. P. 121 —122.
745. Seyferth D., Haas C. KJ IE Organomet. Chem. 1972. V. 39. P. 41.
746. Sheffold R., Michel UJ/Angew. Chem. 1972. B. 84. S. 160—161.
747. Seyferth D., Simmons H. D„ Shih H. MJ IE Organomet. Chem. 1969. V. 29.
P. 359.
748. Seyferth D., Andrews S. B., Simmons H. DJ/E Organomet. Chem. 1969.
V. 17. P. 9.
749. Seyferth D., Woodruff R. А.ЦЕ Org. Chem. 1973. V. 38. P. 4031—4039;
J. Organomet. Chem. 1974. V. 71. P. 335.
750. Logan T. J.ЦЕ Ovg. Chem. 1963. V. 28. P. 1129—1131.
751. Seyferth D., Burlilch J. M., Minasz R. J J IE Amer. Chem. Soc. 1965. V. 87.
P. 4259—4270.
752. Seyferth D., Haas С. КЦЕ Organomet. Chem. 1972. V. 46. P. C33; 1972.
V. 30. P. C38.
753. Щербаков В. И.//Ж- общ. хим. 1971. Т. 41. С. 1095.
754. Robson R., Dickson I. Е.ЦЕ Organomet. Chem. 1968. V. 15. P. 7.
755. Lambert J. B., Bosch R. J., Larson E. G.//E Org. Chem. 1985. V. 50.
P. 3054—3059.
756. Seyferth D., Haas С. K-, Hopper S. Р.ЦЕ Organomet. Chem. 1977. V. 33.
P. Cl.
757. Seyferth D., Darragh K. VJ/E Org. Chem. 1970. V. 35. P. 1297—1302.
758. Seyferth D., Hopper S. Р.ЦЕ Organomet. Chem. 1973. V. 51. P. 77.
759. Seyferth D., Mueller D. С.ЦЕ Organomet. Chem. 1970. V. 25. P. 293; J.
Amer. Chem. Soc. 1971. V. 93. P. 3724.
760. Johansson N. G.//Acta Chem. Scand. 1971. V. 25. P. 1927; 1973. V. 27.
P. 1417.
761. Seyferth DJIE Organomet. Chem. 1972. V. 43. P. 55.
762. Seyferth D., Hanson E. MJ/E Amer. Chem. Soc. 1968. V. 90. P. 2438—
2440.
763. Seyferth D., Dagani D.HE Organomet. Chem. 1976. V. 104. P. 145.
764. Мальцев A. K-, Каграманов H. Д., Нефедов О. М.Ц\Азв. АН СССР. Сер.
хим. 1974. С. 1993. С. 2146; Докл. АН СССР. 1975. Т. 224. С. 630.
765. Sakakibara Т., Odaira Y., Tsutsumi S.//Tetrahedron Lett. 1968. P. 503.
766. Seyferth D„ Prokai В.ЦЕ Amer. Chem. Soc. 1966. V. 88. P. 1834—1835.
767. Miller D. ^.//Tetrahedron Lett. 1964. P. 989.
768. Colette J. W.//E Org. Chem. 1963. V. 28. P. 2489—2490.
769. Щербаков В. И., Жильцов С. Ф.//Новое в химии карбенов. М.: Наука,
1973. С. 162; Ж. общ. хим. 1970. Т. 40. С. 2046.
770. Lind W., Worrant I. /.//Inorg. NucL Chem. Lett. 1971. V. 7. P. 1153.
771. Bewan W. J., Haszeldine R. N„ Young J. C.//Chem. a. Ind. 1961. P. 789.
772. Lee E., Roberts D. WJIE Chem. Soc. Perkin Trans. Part II. 1973. P. 437—
444.
773. Cunico R. F., Chou В. В.ЦЕ Organomet. Chem. 1978. V. 154. P. C45.
774. Seibt D., Heydtman H. UY. phys. Chem. (BRD). 1973. B. 83. S. 256.
775. Anderson F., Birchall J. M., Haszeldine R. N., Tyler В. J J IE Chem. Soc.
Perkin Trans. Part II. 1975. P. 1051 —1054.
776. Haszeldine R. N., Parkinson C., Robinson P. J.ЦЕ Chem. Soc. Perkin Trans.
Part II. 1973. P. 1018—1020.
283
777. Флеминг //.//Общая органическая химия. В 12 т. Т. 6: Пер. с англ./Под
ред. Н. К. Кочеткова и Ю. Н. Бубнова. М.: Химия. 1986. Т. 6. С. 66—232/
778. Sharp К. G., Koyle Т. D.//J. Fluor. Chem. 1971/1972. V. 1. Р. 249. /
779. Gaspar Р. Р., Chen Y.-S., Helfer А. Р. е. a.f/J. Amer. Chem. Soc. 1981.
V. 103. P. 7344—7345.
780. Barton T. J., Burns S. A., Gaspar P. P. e. «.//Synth. a. Reactiv. Inorg. a.
Metallorg. Chem. 1983. V. 13. P. 881—885.
781. Maier G., Schotter K-, Reisenauer H. ^.//Tetrahedron Lett. 1985. V. 26.
P. 4079.
782. Brook A. G., Dillon P. /.//Can. J. Chem. 1969. V. 47. P. 4347.
783. Atwell W. H., Weyenberg D. R., Uhtmann J. G.ffJ. Amer. Chem. Soc. 1969.
V. 91. P. 2025—2028.
784. Massol M., Satge J., Riviere P., Barrau J.HJ. Organomet. Chem. 1970.
V. 22. P. 599.
785. Колесников С. П., Перльмуттер Б. А., Нефедов О. М.ЦИзв. АН СССР.
Сер. хим. 1973. С. 1418.
786. Massol М., Barrou J., Satge /.//Inorg. Nucl. Chem. Lett. 1971. V. 7. P. 895.
787. Clark H. C.f Willis C. J.//J. Amer. Chem. Soc. 1960. V. 82. P. 1888—1891;
1962. V. 84. P. 898—900.
788. Seyferth D.f Dertouzos H., Suzuki R., Mui J. Y.-P.//J. Org. Chem. 1967.
V. 32. P. 2980; Seyferth D., Mui J. Y.-P., Gordon M. E., Burlitch J. MJf
J. Amer. Chem. Soc. 1965. V. 87. P. 680—681.
789. Seyferth D., Ambrecht F. M.HJ. Organomet. Chem. 1966. V. 6. P. 573; J.
Amer. Chem. Soc. 1969. V. 91. P. 2616—2623.
790. Hani R., Geanagel R. A.//Inorg. chim. Acta. 1985. V. 96. P. 225.
791. Cullen W. R., Styan G. E.HJ. Organomet. Chem. 1966. V. 6. P. 633.
792. Seyferth D., Prokai B., Cross R. J.//J. Organomet. Chem. 1968. V. 13.
P. 169.
793. Ambrecht F. M., Tronich W., Seyferth D.HJ. Amer. Chem. Soc. 1969. V. 91.
p 3218_______3222
794. Seyferth D., Lambert R. L.f/J. Organomet. Chem. 1975. V. 91. P. 31.
795. Riviere P., Satge J., Soula D.HJ. Organomet. Chem. 1974. V. 72. P. 329.
796. Riviere P., Satge J., Dousse G. e. a.HJ. Organomet. Chem. 1974. V. 72.
P 339.
797. Riviere P., Castel A., Satge J.//J. Organomet. Chem. 1981. V. 212. P. 351.
798. Castel A., Escudie J., Riviere P. e. a.//J. Organomet. Chem. 1981. V. 210.
P. 37.
799. Kettle S. F. A.HJ. Chem. Soc. 1959. P. 2936.
800. McWilliam D. C., Wells P. R.//J. Organomet. Chem. 1975. V. 85. P. 165.
801. Ayscough P. P., Emeleus H. J.//J. Chem. Soc. 1954. P. 3381.
802. Muller R., Reichel S., Dathe C./fJ. pract. Chem. 1969. V. 311. P. 930.
803. Несмеянов A. H., Борисов A. E., Кизим H. Г.//Изв. АН СССР. Сер. хим.
1974. C. 1672.
804. Mahler W.HJ. Amer. Chem. Soc. 1962. V. 84. P. 4600—4601; Inorg. Chem.
1963. V. 2. P. 230.
805. Leonard E. C.//J. Org. Chem. 1965. V. 30. P. 3258—3260.
806. Kice J. L., Engebrecht R. H., Pawlowski N. E.HJ. Amer. Chem. Soc. 1965.
V. 87. P. 4131—4137.
807. Гинсбург В. А.//Докл. АН СССР. 1962. Т. 142. С. 88.
808. Bickel A. F.l/J. Chem. Soc. 1963. Р. 1958.
809. Krief A., Hevesi L., Chaboteaux G. e. a.HJ. Chem. Soc. Chem. Comm. 1985.
P. 1693—1695.
810. De Vries L./fJ. Amer. Chem. Soc. 1978. V. 100. P. 926—933.
811. Политанский С. Ф., Липкинд M. Б., Ивашенко A. А., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1979. С. 1654.
812. Ивашенко А. А., Политанский С. Ф.//Втор. Всес. сов. по химии карбенов
и и< аналогов. Тезисы докладов. М.: Наука, 1977. С. 15.
284
813. Caglioti V., Site D., Lenzi M., Mele AJ/J. Chem. Soc. 1964. P. 5430—
5433.
^14. Heicklen J., Saunders D./fJ. Chem. Phys. 1965. V. 42. P. 221; 1965. V. 43.
P. 871; 1967. V. 47. P. 4203; J. Amer. Chem. Soc. 1965. V. 87. P. 2088—
2092.
Й15. Нефедов О. M., Ивашенко А. А.//Изв. АН СССР. Сер. хим. 1968. С. 446.
816. Johnston Т., Heicklen JJ/J. Chem. Phys. 1967. V. 47. Р. 475.
817. Mitchell R. C., Simons J. PJ/J, Chem. Soc. Part B. 1968. P. 1005—1007.
818. Vorsanger J. /.//Bull. Soc. chim. France. 1966. P. 1772.
819. Franzen V., Joschek H. /.//Ann. B. 633. S. 7.
820. Wanzlick Н.-WJ/Angew. Chem. 1962. B. 74. S. 129; Angew. Chem. 1963.
B. 75. S. 1024; Chem. Ber. 1963. B. 96. S. 3024—3027; Chem. Ber. 1964.
B. 97. S. 3513—3516; Ann. 1967. B. 708. S. 155.
821. Noyes W. A., Unger /.//Pure a. Appl. Chem. 1964. V. 9. P. 461.
822. Ho S., Unger E, Noyes W. AJ/J. Amer. Chem. Soc. 1965. V. 87. P. 2297—
2298.
823. Traylor G. A., Porter G. В J/J. Chem. Phys. 1962. V. 36. P. 1353.
824. Kistiakowsky G. B., Sauer A.//J. Amer. Chem. Soc. 1956. V. 78. P. 5699—
5700; 1958. V. 80. P. 1066—1071.
825. Strachan A. N„ Thornton D. EJ/J. Phys. Chem. 1966. V. 70. P. 952.
826. Kistiakowsky G. B., Walter T. A.//J. Phys. Chem. 1968. V. 72. P. 3952.
827. Ho S.-Y., Noyes W. AJ/J. Amer. Chem. Soc. 1967. V. 89. P. 5091—5098.
828. Dorer F. H., Rabinovitch B. SJ/J. Phys. Chem. 1965. V. 69. P. 1952.
829. Duncan F. J., Cvetanovic R. JJ/J. Amer. Chem. Soc. 1962. V. 84. P. 3593—
3594.
830. Avery H.-E., Cvetanovich R. JJ/J. Chem. Phys. 1968. V. 48. P. 380.
831. Montague D. C., Rowland F. SJ/J. Phys. Chem. 1968. V. 72. P. 3705.
832. Frey H. M.f Walsh RJ/J. Chem. Soc. Chem. Comm. 1969. P. 158; J. Chem.
Soc. Part A. 1970. P. 2115.
833. Хабашеску В. H., Мальцев А. К., Нефедов О. Л4.//Докл. АН СССР. 1987.
С. 2187.
834. Kistiakowsky G. В., Mahan В. HJ/J. Amer. Chem. Soc. 1957. V. 79.
р 2412_______2419.
835. Chong D. P„ Kistiakowsky G. В J/J. Phys. Chem. 1966. V. 70. P. 126.
836. Holroyd R. A., Blacet F. EJ/J. Amer. Chem. Soc. 1957. V. 79. P. 4830—
4834.
837. Haller I., Srinivasan RJ/J. Amer. Chem. Soc. 1965. V. 87. P. 1144—1145.
838. Gutsche C. D., Baum J. 1F.//Tetrahedron Lett. 1965. P. 2301; J. Amer. Chem.
Soc. 1968. V. 90. P. 5862—5867.
839. Nozaki H., Nakano M., Kondo /(.//Tetrahedron. 1966. V. 22. P. 477.
840. Bayes К DJ/J. Amer. Chem. Soc. 1962. V. 84. P. 4077—4080; 1963. V. 85.
P. 1730—1733.
841. Willis C., Bayes К DJ/J. Phys. Chem. 1967. V. 71. P. 3367; J. Amer. Chem.
Soc. 1966. V. 88. P. 3203—3208.
842. Mullen R. T., Wolf A. PJ/J. Amer. Chem. Soc. 1962. V. 84. P. 3712.
843. Devillers CJ/C. r. 1966. V. C262. P. 1485.
844. Morrow T., McGrath W. D.//Trans. Faraday Soc. 1966. V. 62. P. 3142.
845. Stief L. J., DeCarlo V. JJ/J. Amer. Chem. Soc. 1969. V. 91. P. 839—843.
846 Corey E. J., Winter R. A. FJ/J. Amer. Chem. Soc. 1963. V. 85. P. 2677—
2678; 1965. V. 87. P. 934—935; Tetrahedron Lett. 1967. P. 3201.
847. Ramires F., Yamanaka H., Basedow O. HJ/J. Amer. Chem. Soc. 1961. V. 83.
P. 173—178.
848. Bird C. W., Wong D. YJ/J. Chem. Soc. Chem. Comm. 1969. P. 932.
849. Schmidt UKabitzke К HJ/Angew. Chem. 1964. B. 76. S. 687.
850. Turro N. J., Cha Y., Gould I. /^.//Tetrahedron Lett. 1985. V. 26. P. 5951.
851. Ibaia T., Kashiuchi Af.//Bull. Chem. Soc. Jap. 1986. V. 59. P. 929—930.
852. Wittig G., Krauss DJ/Ann. 1964. B. 679. S. 34.
853. Ingold С. K., Jessop J. AJ/J. Chem. Soc. 1929. P. 2357.
285
854. Streith J., Cassul J. М.ЦС. r. 1967. V. C264. P. 1307.
855. Hughes E. D., Kuriyan К. 1.11 J. Chem. Soc. 1935. P. 1609.
856. Johnson A. W., La Count R. B.HJ. Amer. Chem. Soc. 1961. V. 83. P. 417
423. ।
857. Johnson A. W., Hruby V. J., Williams J. L.HJ. Amer. Chem. Soc. 196n.
V. 86. P. 918—922. I
858. Middleton W. J., Buhle E. L., McNally J. G., Zanger M.//J. Org. Chem. 1905.
V. 30. P. 2384—2386.
859. Franzen V., Schmidt H.-J., Mertz C.//Chem. Ber. 1961. B. 94. S. 2942—
2950.
860. Trost B. M.f La Rochelle R. W.//J. Amer. Chem. Soc. 1970. V. 92. P. 5804—
5806.
861. Nozaki H., Kondo K-, Takaku 7W.//Tetrahedron Lett. 1965. P. 251; Tetrahed-
ron. 1966. V. 22. P. 2145.
862. Cohen T., Herman G., Chapman T. M. e. a.HJ. Amer. Chem. Soc. 1974. V. 96.
P. 5627—5628.
863. Trost B. M.HJ. Amer. Chem. Soc. 1966. V. 88. P. 1587—1588; 1967. V. 89.
P. 138—142.
864. Шостаковский В. M., Златкина В. Л., Васильвицкий А. Е., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1982. С. 2126.
865. Юфит Д. С., Стручков Ю. Т., Шостаковский В. М. и др.//Изв. АН СССР.
Сер. хим. 1985. С. 594.
866. Шостаковский В. М., Васильвицкий А. Е., Златкина В. Л., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1980. С. 2180.
867. Wittig G., Boll №.//Chem. Ber. 1962. В. 95. S. 2526—2534.
868. Ritter A., Kim B.//Tetrahedron Lett. 1968. P. 3449.
869. Neuman R. C.f/J. Org. Chem. 1964. V. 29. P. 2096—2097.
870. McGregor S. D., Jones W. M.HJ. Amer. Chem. Soc. 1968. V. 90. P. 123—
126.
871. Quast H., Frankenfeld E.//Angew. Chem. 1965. B. 77. S. 680; Ann. 1970.
B. 732. S. 43.
872. Колбановский Ю. А., Шапиро E. А., Купцова T. С. и др.//Изв. АН СССР.
Сер. хим. 1987. С. 2143.
873. Stechl tf.-tf.//Chem. Ber. 1964. В. 97. S. 2681—2688.
874. Binger P., McMeeking /.//Angew. Chem. 1974. B. 86. S. 518—519.
875. Davis J. H., Goddard W. A., Bergman R. G.//J. Amer. Chem. Soc. 1976.
V. 98. P. 4015—4017.
876. Elliot N. ^.//Thermal Electrocycl. React. 1980. P. 83.
877. Padwa A.HCng. Photochem. 1979. V. 4. P. 261.
878. Liese T., Splettstaber G., de Maijere A.//Tetrahedron Lett. 1982. V. 23.
P. 3341.
879. Домнин И. H., Костиков P. P., де Майере А.ЦЖ. орг. хим. 1983. T. 19.
C. 2206.
880. Томилов Ю. В., Бордаков В. Г., Цветкова Н. М. и др.ЦИзв. АН СССР.
Сер. хим. 1983. С. 336.
881. Padwa A., Rieker W. F., Rosenthal R. J.HJ. Org. Chem. 1984. V. 49.
P. 1353—1360.
882. Томилов Ю. В., Бордаков В. Г., Цветкова Н. М. и др.//Изв. АН СССР.
Сер. хим. 1982. С. 2413.
883. Binger Р., Brinkmann А.ЦСХмп. Вег. 1978. В. 111. S. 2689—2695.
884. Weber W., de Meijere A.//Chem. Ber. 1985. В. 118. S. 2450—2471.
885. Kostikov R., de Meijere A.HJ. Chem. Soc. Chem. Comm. 1984. P. 1528—
1529.
886. Долгий И. E., Томилов Ю. В., Штейншнейдер А. Я., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1983. С. 700.
887. Pincock J. A., Moutsokapas A. A.//Can. J. Chem. 1977. V. 55. Р. 979.
888. Протопопова М. Н.ЦДвс. ... канд. хим. наук. 02.00.03. М., 1984. 161 с.
286
889. Томилов Ю. В., Шапиро Е. А., Протопопова М. И. и др.//Изв. АН СССР.
Сер. хим. 1985. С. 631.
890. Томилов Ю. В.//Труды IV Всесоюзн. конференции по химии карбенов.
М.: Наука, 1987. С. 61.
$91. Frank-Neumann М., Buchecker C.//Angew. Chem. Int. Ed. 1970. V. 9.
. P. 526.
Д92. Frank-Neumann M., Lohmann J. /.//Tetrahedron Lett. 1978. P. 3729; An-
gew. Chem. 1977. B. 89. S. 331—332.
8^3. Lohr L. L., Hanamura M., Morokuma /C//J. Amer. Chem. Soc. 1983. V. 105.
P. 5541—5547.
894. Bouma W. J., Nobes R. H., Radom L., Woodward С. Е.ЦЕ Org. Chem. 1982.
V. 47. P. 1869—1875.
895. Maier G., Reisenauer H. P., Sayraq T.//Chem. Ber. 1982. B. 115. S. 2192—
2201' S. 2202_____2213
896. Martin K. V.//E Chem. Soc. 1964. P. 2944—2947.
897. Колесников С. П., Охрименко H. И., Нефедов О. Л1.//Докл. АН СССР.
1978. Т. 243. С. 1193.
898. Turro N. L, Southam R. M.//Tetrahedron Lett. 1967. P. 545; Morton D. R.,
Lee-Ruff E., Southam R. M.//J. Amer. Chem. Soc. 1970. V. 92. P. 4349.
8QQ Hostettler H. (/.//Tetrahedron Lett. 1965. P. 687.
Q00 Yates P., KUmurry L.//Tetrahedron Lett. 1964. P. 1739; J. Amer. Chem.
Soc. 1966. V. 88. P. 1563—1564.
901. Agosta W. C., Herron D. К-ЦЕ Amer. Chem. Soc. 1968. V. 90. P. 7025—
7030.
902. Staab H. A., Ipaktschi /.//Tetrahedron Lett. 1966. P. 583; Staab H. A.//
Chem. Ber. 1968. B. 101. S. 1457—1472.
903. Pacifici J. G., Diebert С.ЦЕ Amer. Chem. Soc. 1969. V. 91. P. 4594—4596.
904. Hine J.//E Amer. Chem. Soc. 1950. V. 72. P. 2438—2445.
905. Hine J., Thomas С. H., Ehrenson S. J.HL Amer. Chem. Soc. 1955. V. 77.
P. 3886—3889; Hine J., Dowelt A. M., Singlev J. E.//Ibid. 1956. V. 78.
P. 479—482.
906. Merz A., Tomahogh R.//Chem. Ber. 1977. B. 110. S. 96—106.
907. Hine J., Peek R. C., Oakes B. D.HE Amer. Chem. Soc. 1954. V. 76. P. 827—
829; Hine J., Burske N. W.//Ibid. 1956. V. 78. P. 3337—3339; Hine J., Bur-
ske N. W„ Hine M., Langford P. B.//Ibid. 1957. V. 79. P. 1406—1412;
Hine J., Butterworth R., Langford P. B.//Ibid. 1958. V. 80. P. 819—824.
908. Le Noble W. J.f/E Amer. Chem. Soc. 1965. N. 87. P. 2434—2438; Le Nob-
le W. J., Duffy M//Ibid. 1964. V. 86. P. 4512.
909. Hine J., Prosser F. Р.ЦЕ Amer. Chem. Soc. 1958. V. 80. P. 4282—4285.
910. Doering W. E., Hoffman A. K//E Amer. Chem. Soc. 1954. V. 76. P. 6162—
6165.
911. Ивашенко А. А.//Дис. ... канд. хим. наук. 02.00.03. М., 1972. 190 с.
912. Purringion S. Т., Kenion G. В.ЦЕ Chem. Soc. Chem. Comm. 1982. P. 731 —
732.
913. Weyerstahl P., Klamann D., Finger C. e. tz.//Chem. Ber. 1967. B. 100.
S. 1858—1869; Ann. 1968. B. 710. S. 17.
914. Makosza M., Wawrzynievich AL//Tetrahedron Lett. 1969. P. 4659.
915. Starks С. М.ЦЕ Amer. Chem. Soc. 1971. V. 93. P. 195—199.
916. Пат. 1573167. Франц. МДИ2 С 07с. Catalytic reaction.
917. Макоша М., Федорыньски М.ЦУК. ВХО им. Д. И. Менделеева. 1979.
Т. 24. С. 466—474.
918. Wagner W. Л1.//Ргос. Chem. Soc. 1959. Р. 229.
919. Wagner W. М. е. a.//Rec. trav. chim. 1961. V. 80. P. 740; 1962. V. 81.
P 925 P 933
920. Dehmlow E. ^.//Tetrahedron Lett. 1976. P. 91.
921. Dehmlow E. V., Remmler Т.ЦЕ Chem. Res. Part S. 1977. P. 72.
922. Birchall J. M~. Cross G. W., Haszeldine R. V.//Proc. Chem. Soc. 1960
P. 81.
287
854. Streith J., Cassul J. M.//C. r. 1967. V. C264. P. 1307.
855. Hughes E. D., Kuriyan К. I.//J. Chem. Soc. 1935. P. 1609.
856. Johnson Л. W., La Count R. B.//L Amer. Chem. Soc. 1961. V. 83. P. 417-/-
423.
857. Johnson A. W., Hruby V. J., Williams J. L.//J. Amer. Chem. Soc. 196/4.
V. 86. P. 918—922.
858. Middleton W. J., Buhle E. L., McNally J. G., Zanger M.//J. Org. Chem. 1965.
V. 30. P. 2384—2386.
859. Franzen V., Schmidt H.-J., Mertz C.//Chem. Ber. 1961. B. 94. S. 2942—
2950.
860. Trost B. M., La Rochelle R. W.//J. Amer. Chem. Soc. 1970. V. 92. P. 5804—
5806.
861. Nozaki H., Kondo K-, Takaku ^.//Tetrahedron Lett. 1965. P. 251; Tetrahed-
ron. 1966. V. 22. P. 2145.
862. Cohen T., Herman G., Chapman T. M. e. a.//J. Amer. Chem. Soc. 1974. V. 96.
P. 5627—5628.
863. Trost B. M.//L Amer. Chem. Soc. 1966. V. 88. P. 1587—1588; 1967. V. 89.
P. 138—142.
864. Шостаковский В. M„ Златкина В. Л., Васильвицкий А. Е., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1982. С. 2126.
865. Юфит Д. С., Стручков Ю. Т., Шостаковский В. М. и др.//Изв. АН СССР.
Сер. хим. 1985. С. 594.
866. Шостаковский В. М., Васильвицкий А. Е., Златкина В. Л., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1980. С. 2180.
867. Wittig G., Boll №.//Chem. Ber. 1962. В. 95. S. 2526—2534.
868. Ritter A., Kim B.//Tetrahedron Lett. 1968. P. 3449.
869. Neuman R. C.//J. Org. Chem. 1964. V. 29. P. 2096—2097.
870. McGregor S. b., Jones W. MJ/J. Amer. Chem. Soc. 1968. V. 90. P. 123—
126.
871. Quast H., Franken/eld E.//Angew. Chem. 1965. B. 77. S. 680; Ann. 1970.
B. 732. S. 43.
872. Колбановский Ю. А., Шапиро E. А., Купцова T. С. и др.//Изв. АН СССР.
Сер. хим. 1987. C. 2143.
873. Stechl H.-HJ/Chem. Ber. 1964. B. 97. S. 2681—2688.
874. Binger P., McMeeking /.//Angew. Chem. 1974. B. 86. S. 518—519.
875. Davis J. H., Goddard W. A., Bergman R. G.f/J. Amer. Chem. Soc. 1976.
V. 98. P. 4015—4017.
876. Elliot N. M.//Thermal Electrocycl. React. 1980. P. 83.
877. Padwa A.//Org. Photochem. 1979. V. 4. P. 261.
878. Liese T., Splettstaber G., de Maijere A.//Tetrahedron Lett. 1982. V. 23.
P. 3341.
879. Домнин И. Н.г Костиков Р. Р., де Майере A.//JK. орг. хим. 1983. Т. 19.
С. 2206.
880. Томилов Ю. В., Бордаков В. Г., Цветкова Н. М. и др.ЦИзв. АН СССР.
Сер. хим. 1983. С. 336.
881. Padwa A., Rieker W. F., Rosenthal R. JJ/J. Org. Chem. 1984. V. 49.
P. 1353—1360.
882. Томилов Ю. В., Бордаков В. Г., Цветкова Н. М. и др.//Изв. АН СССР.
Сер. хим. 1982. С. 2413.
883. Binger Р., Brinkmann A.//Chem. Вег. 1978. В. 111. S. 2689—2695.
884. Weber W., de Meijere A.//Chem. Ber. 1985. В. 118. S. 2450—2471.
885. Kostikov R., de Meijere A.//L Chem. Soc. Chem. Comm. 1984. P. 1528—
1529.
886. Долгий И. E., Томилов Ю. В., Штейншнейдер А. Я., Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1983. С. 700.
887. Pincock J. A., Moutsokapas A. A.//Can. J. Chem. 1977. V. 55. Р. 979.
888. Протопопова М. Н.ЦДвс. ... канд. хим. наук. 02.00.03. М., 1984. 161 с.
286
889. Томилов Ю. В., Шапиро Е. А., Протопопова М. И. и др.//Изв. АН СССР.
Сер. хим. 1985. С. 631.
890. Томилов Ю. В.//Труды IV Всесоюзн. конференции по химии карбенов.
М.: Наука, 1987. С. 61.
S91. Frank-Neumann М., Buchecker C.//Ange\v. Chem. Int. Ed. 1970. V. 9.
' P. 526.
892. Frank-Neumann M., Lohmann J. /.//Tetrahedron Lett. 1978. P. 3729; An-
gew. Chem. 1977. B. 89. S. 331—332.
8(p3. Lohr L. L., Hanamura Nl., Morokuma K.//J. Amer. Chem. Soc. 1983. V. 105.
P. 5541—5547.
894. Bouma W. J., Nobes R. FL, Radom L., Woodward С. Е.Ц5. Org. Chem. 1982.
V. 47. P. 1869—1875.
895. Maier G., Reisenauer H. P., Sayraq T.//Chem. Ber. 1982. B. 115. S. 2192—
2201' S. 2202______2213
896. Martin K. V.//J. Chem. Soc. 1964. P. 2944—2947.
897. Колесников С. П., Охрименко H. И., Нефедов О. Л1.//Докл. АН СССР.
1978 Т 243 С 1193
898. Turro N. J., Southam R. М.//Tetrahedron Lett. 1967. Р. 545; Morton D. R.,
Lee-Ruff E., Southam R. М.Ц5. Amer. Chem. Soc. 1970. V. 92. P. 4349.
8Q9 Hostettler H. (/.//Tetrahedron Lett. 1965. P. 687.
Q00 Yates P., KHmurry /„//Tetrahedron Lett. 1964. P. 1739; J. Amer. Chem.
Soc. 1966. V. 88. P. 1563—1564.
901. Agosta W. C., Herron D. K-lfL Amer. Chem. Soc. 1968. V. 90. P. 7025—
7030.
902. Staab H. A., Ipaktschi /.//Tetrahedron Lett. 1966. P. 583; Staab H. A.//
Chem. Ber. 1968. B. 101. S. 1457—1472.
903. Pacifici J. G., Diebert С.Ц5. Amer. Chem. Soc. 1969. V. 91. P. 4594—4596.
904. Hine /.//J. Amer. Chem. Soc. 1950. V. 72. P. 2438—2445.
905. Hine J., Thomas С. H., Ehrenson S. J.ЦЕ Amer. Chem. Soc. 1955. V. 77.
P. 3886—3889; Hine J., Dowell A. M., Singlev J. E.//Ibid. 1956. V. 78.
P. 479—482.
906. Merz A., Tomahogh R.//Chem. Ber. 1977. B. HO. S. 96—106.
907. Hine J., Peek R. C., Oakes B. D.HL Amer. Chem. Soc. 1954. V. 76. P. 827—
829; Hine J., Burske N. IE.//Ibid. 1956. V/78. P. 3337—3339; Hine J., Bur-
ske N. W., Hine M., Langford P. B.//Ibid. 1957. V. 79. P. 1406—1412;
Hine J., Butterworth R., Langford P. B.//Ibid. 1958. V. 80. P. 819—824.
908. Le Noble W. J.//L Amer. Chem. Soc. 1965. V. 87. P. 2434—2438; Le Nob-
le W. J., Duffy M//Ibid. 1964. V. 86. P. 4512.
909. Hine J., Prosser F. Р.ЦЗ. Amer. Chem. Soc. 1958. V. 80. P. 4282—4285.
910. Doering W. E., Hoffman A. /C//J. Amer. Chem. Soc. 1954. V. 76. P. 6162—
6165.
911. Ивашенко A. A.//JLnc. ... канд. хим. наук. 02.00.03. M., 1972. 190 с.
912. Purrington S. Т., Kenion G. B.//J. Chem. Soc. Chem. Comm. 1982. P. 731 —
732.
913. Weyerstahl P., Klamann D., Finger C. e. a.//Chem. Ber. 1967. B. 100.
S. 1858—1869; Ann. 1968. B. 710. S. 17.
914. Makosza M., Wawrzynievich ^.//Tetrahedron Lett. 1969. P. 4659.
915. Starks С. М.ЦУ Amer. Chem. Soc. 1971. V. 93. P. 195—199.
916. Пат. 1573167. Франц. МДИ2 С 07с. Catalytic reaction.
917. Макоша М., Федорыньски М.Ц7К. ВХО им. Д. И. Менделеева. 1979.
Т. 24. С. 466—474.
918. Wagner W. M//Proc. Chem. Soc. 1959. Р. 229.
919. Wagner W. М. е. a.//Rec. trav. chim. 1961. V. 80. Р. 740; 1962. V. 81.
Р 925 Р 933
920. Dehmlow Е. ^.//Tetrahedron Lett. 1976. Р. 91.
921. Dehmlow Е. V., Remmler Т.Ц5. Chem. Res. Part S. 1977. P. 72.
922. Birchall J. M- Cross G. W., Haszeldine R. V.//Proc. Chem. Soc. 1960.
P. 81.
287
923. Fuqua S. A., Duncan W. G., Siversfein R. ^.//Tetrahedron Lett. 1965.
P. 521; J. Org. Chem. 1965. V. 30. P. 1027—1029; P. 2543—2545.
924. Parham W. E., Loew F. С.ЦЗ. Org. Chem. 1958. V. 23. P. 1705—1708; Pan
ham W. E., Schweizer E. E.//Ibid. 1958. V. 24. P. 1733—1735.
925. Moore R. A., Levine R.//J. Org. Chem. 1964. V. 29. P. 1883—1887.
926. Ando /.//Tetrahedron Lett. 1967. P. 1123.
927. Nerdel F., Dahl H., Weyerstahl ^.//Tetrahedron Lett. 1969. P. 809. .
928. Kodaba P. K., Edwards J. О //J. Org. Chem. 1960. V. 25. P. 1431 —1433.
929. Grant F. W., Cassie W. B.//L Org. Chem. 1960. V. 25. P. 1433—1434. I
930. Farah B., Horensky S.//J. Org. Chem. 1963. V. 28. P. 2494—2495.
931. Oliver J. P., Rao V. V., Emerson M. /.//Tetrahedron Lett. 1964. P. 3419.
932. Schollkopf U., Hilbert P.//Angew. Chem. 1962. B. 74. S. 431.
933. Berry J. P., Arnold J. R.} Isbell А. Р.Ц5. Org. Chem. 1968. V. 33. P. 1664—
1665.
934. Hine J., Duke R. B., Glod E. F.HL Amer. Chem. Soc. 1969. V. 91. P. 2316—
2324.
935. Вольпин M. E., Дулова В. Г., Курсивов Д. //.//Докл. АН СССР. 1959.
Т. 128. С. 951; Tetrahedron. 1960. V. 8. Р. 33.
936. McElvain S. М., Weyna Р. L.//J. Amer. Chem. Soc. 1959. V. 81. Р. 2579—
2588.
937. Hodgkins J. E., Woodyard J. D., Stephenson D. Е.ЦУ Amer. Chem. Soc.
1964. V. 86. P. 4080—4085.
938. Moss R. A.//Tetrahedron Lett. 1965. P. 3445: 1968. P. 1961; 1966. V. 22.
P. 2637; 1969. V. 25. P. 647.
939. Ando T. e. a.//Tetrahedron Lett. 1968. P. 2479.
940. Schollkopf LL, Woerner F. P., Wiskott E.//1962. P. 165; Chem. Ber. 1966.
B. 99. S. 806—811.
941. Долгий И. E., Шаврин К. И., Крылова И. В., Нефедов О. М.//Изв. АН
СССР. Сер. хим. 1987. С. 135.
942. Лукевиц Э., Игнатович Л. М., Рубина К. И. и др.//Труды IV Всесою н.
конференции по химии карбенов. М.: Наука, 1987. С. 5—6.
943. Saxena М. К-, Bokadia М. М.//Chem. a. Ind. 1966. Р. 666.
944. Иоффе А. И., Нефедов С. М.Ц}Азв. АН СССР. Сер. хим. 1987. С. 2329
945. Шаврин К. Н., Нефедов О. М.ЦИзь. АН СССР. Сер. хим. 1987. С. 1196.
946. Шаврин К. Н., Крылова И. В., Долгий И. Е.//Труды IV Всесоюзн. кон-
ференции по химии карбенов. М.: Наука, 1987. С. 169.
947. Martel В., Hiriart J. M.//Angew. Chem. Int. Ed. 1972. V. 11. P. 326; Syn-
thesis. 1972. P. 201.
948. Martel B., Aly Е.ЦЕ Organomet. Chem. 1971. V. 29. P. 61.
949. Arora S., Binger P.//Synthesis. 1974. P. 801.
950. Doleib D. M., Iskander У.ЦЕ Chem. Soc. Part B. 1967. P. 1154—1158.
951. Rothberg L, Thornton E. R.//J. Amer. Chem. Soc. 1963. V. 85. P. 1704—
1705; 1964. V. 86. P. 3296—3301; P. 3302—3306.
952. Schollkopf U., Lerch A., Paust /.//Chem. Ber. 1963. B. 96. S. 2266—2280;
1964. B. 97. S. 1527—1541; Tetrahedron Lett. 1963. P. 105.
Q53. Klamann D., Weyerstahl P.f Nerdel FJ/Nnn. 1968. B. 710. S. 59.
954. Harada T., Karasawa A., Oku А.ЦЕ Org. Chem. 1986. V. 51. P. 842—
846.
955. Tanabe M., Walsh R. A.//J. Amer. Chem. Soc. 1963. V. 85. P. 3522—3523.
956. Curiico R. F.. Han Y. K//J. Organomet. Chem. 1976. V. 105. P. C29; 1978.
V. 162. P. 1.
957. Harlzler H. D.//J. Amer. Chem. Soc. 1959. V. 81. P. 2024—2025; 1961. V. 83.
P. 4990. P. 4997—4999; J. Org. Chem. 1964. V. 29. P. 1311 — 1312.
958. Bramwell A. F., Crombie L., Knight M. //.//Chem. a. Ind. 1965. P. 1265.
959. Landor S. /?., Whiter P. FJ/L Chem. Soc. 1965. P. 5625—5629.
960. Hennion G. F., Maloney D. Е.Ц5. Amer. Chem. Soc. 1951. V. 73. P. 4735—
4737.
961. Cadiol P.//Ann. chim. (France). 1956. В. 1. S. 214.
288
9^2. Hartzler H. DJ/J- Amer. Chem. Soc. 1966. V. 88. P. 3155—3156.
96B. Gore J., Douthean ^.//Tetrahedron Lett. 1973. P. 253.
96|. Stang P. J., Mangum M. G., Fox D P e aJ/J. Amer. Chem. Soc. 1974
V. 96. P. 4562—4569.
963. Stang P. J., Davis J., Fox D, P.//J. Chem. Soc. Chem. Comm. 1975. P. 17.
9661 Stang P. J., Mangum M. G.i/y Amer. Chem. Soc. 1975. V. 97. P. 1459—
! 1464. P. 3854—3856. P. 6478—6481.
9671 Stang P. J., Fox D. PCollins C. J. e. a.//J. Org Chem. 1978. V. 43.
’ P. 364—365.
968. Krageloh K-> Anderson G. H., Stang P. JJ/J. Amer. Chem. Soc. 1984.
V. 106. P. 6015—6021.
969. Wolfgang R.//Progr. React. Kinet. 1965. V. 3. P. 99.
970. Mackay C., Wolfgang ^.//Science. 1965. V. 148. P. 899.
971. Skell P. S., Engel R. RJ/J. Amer. Chem. Soc. 1965. V. 87. P. 1135. P. 2493—
2494; 1966. V. 88. P. 3749—3758; 1967. V. 89. P. 2912—2915.
972. Schaeffer R., Pearson R. K.//J. Amer. Chem. Soc. 1969. V. 91. P. 2153—
2154.
973. McKee M. L., Shelvin P. BJ/J. Amer. Chem. Soc. 1985. V. 107. P. 5191 —
5198.
974. Stocklin G., Wolf A. PJ/J. Amer. Chem. Soc. 1963. V. 85. P. 229—230.
975. Wolf A. P./ZAnn. Rev. Nucl. Chem. 1960. V. 10. P. 259.
976. Nicholas J., Mackay C.f Wolfgang RJ/J. Amer. Chem. Soc. 1966. V. 88.
P. 1610—1616; Tetrahedron. 1966. V. 22. P. 2967.
977. Rose T. LJ/J. Chem. Soc. Chem. Commun. 1969. P. 95—96; Rose T. L.,
Mackay C., Wolfgang RJ/J. Amer. Chem. Soc. 1966. V. 88. P. 1064—1065.
978. Pohlit H. M., Lin T.-FL, Lemmon R. MJ/J. Amer. Chem. Soc. 1969. V. 91,
P. 5425—5429.
979. Engel R. R., Skell P. SJ/J. Amer. Chem. Soc. 1965. V. 87. P. 4663—4664;
1966. V. 88. P. 4883—4890; Skell P. S., Plonka J. H., Wood L. SJ/J. Chem.
Soc. Chem. Comm. 1970. P. 710—711.
980. Skell P. S., Harris R. FJ/J. Amer. Chem. Soc. 1965. V. 87. P. 5807—5808.
981. Ahmed S. N., McKee M. L., Shevlin P. BJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 1320—1324.
982. Skell P. S., Plonka J. H., Engel R. EJ/J. Amer. Chem. Soc. 1967. V. 89.
P. 1748; 1970. V. 92. P. 836—839.
983. Skell P. S., Plonka J. HJ/J. Amer. Chem. Soc. 1970. V. 92. P. 2160—2161.
984. Skell P. S., Wescott L. D., Goldstein J.-P., Engel R. R.//J. Amer. Chem,
Soc. 1965. V. 87. P. 2829—2835; Skell P. S.f Harris R. F.//Ibid. 1969.
V. 91. P. 699—701.
985. Gaspar P. P., Konieczny S., Mo S. HJ/J. Amer. Chem. Soc. 1984. V. 106.
P. 424—425.
986. Gaspar P. P., Hwang R.-L, Eckelman W. CJ/J. Chem. Soc. Chem. Comm.
1974. P. 242—243.
987. Hwang R. J., Gaspar P. PJ/J. Amer. Chem. Soc. 1978. V. 100. P. 6626—
6635.
988. Sieferth E. E., Loh K-L., Ferrieri R. A., Tang Y.-N.//J. Amer. Chem. Soc.
1980. V. 102. P. 2285—2289.
989. Sieferth E. E., Witt S. D.f Tang Y.-N.//J. Chem. Soc. Chem. Comm. 1981.
P 217—218.
990. Bazant V., Joklik J., Rathousky /.//Angew. Chem. Int. Ed. 1968. V. 7.
P. 112.
991. Muller KJ/Z. Chem. 1985. B. 25. S. 309.
992. Fritz G., Wdsching AJ/Z. anorg. Chem. 1984. B. 512. S. 131.
993. Поллер P. К //Общая органическая химия. В 12 т. Т. 7: Пер. с англ./Под
ред. Н. К- Кочеткова и Ф. М. Стояновича. М.: Химия. 1984. С. 155—
221.
994. Nicholson J. W., Douek J. A., Collins J. DJ/J. Organomet. Chem. 1982.
V. 233. P. 169—173.
1 9—107
289
995. Kizlink /.//Chem. listy. 1984. V. 78. P. 134.
996. Doering W. E., Henderson W. A.//J. Amer. Chem. Soc. 1958. V. 80. P. 527(1-
5277. 1
997. Starks С. М.ЦЕ Amer. Chem. Soc. 1971. V. 93. P. 195—199. ,
998. Giese B., Meister /.//Angew. Chem. Int. Ed. 1978. V. 17. P. 595; Ann. 1980.
S 725
999. Giese B., Lee W.-В./1Angew. Chem. Int. Ed. 1980. V. 19. P. 835; Chem.
Ber. 1981. B. 114. S. 3306—3312. '
1000. Giese B., Lee W.-B., Neumann C.//Angew. Chem. Int. Ed. 1982. Vi 21.
P. 310.
1001. Oikawa S., Tsuda M.//J. Amer. Chem. Soc. 1985. V. 107. P. 1940—1946.
1002. Tomioka H.f Hayashi M., Izawa Y. e. «.//Tetrahedron Lett. 1984. V. 25.
P. 4413.
1003. Doyle M. P., Terpstra J. W., Winter C. //.//Tetrahedron Lett. 1984. V. 25.
P. 901.
1004. Bekhazi M., Warkentin 1.ЦЗ. Amer. Chem. Soc. 1983. V. 105. P. 1289—
1292.
1005. Gill H. S., Landgrebe J. A.//Tetrahedron Lett. 1982. V. 23. P. 5099.
1006. Steele К. P., Weber W. P.//J. Amer. Chem. Soc. 1980. V. 102. P. 6095—
6097.
1007. Tomioka H., Ozaki Y., Izawa Y.//Tetrahedron. 1985. V. 41. P. 4987.
1008. Яновская Л. А., Домбровский В. А., Хусид A. X. Циклопропаны с функ-
циональными группами. M.: Наука, 1980. 223 с.
1009. Muller Е., Fricke //.//Ann. 1963. В. 661. S. 38.
1010. DePuy С. H., Mahoney L. R., Eilers K. L.//3. Org. Chem. 1961. V. 26.
P. 3616—3617.
1011. Salomon R. G., Kochi J. K-//S. Amer. Chem. Soc. 1973. V. 95. P. 3300—
3310.
1012. Дьяконов И. А., Витенберг А. Г.ЦХК- орг. хим. 1967. T. 3. C. 1153.
1013. Saegusa T., Ito Y., Shimizu T., Kobayashi S.//Bull. Chem. Soc. Jap. 1969.
V. 42. P. 3535—3538.
1014. Anciaux A. L, Huber A. J., Noels A. F. e. а.ЦЕ Org. Chem. 1980. V. 45.
P. 695—702.
1015. Nozaki //.//Tetrahedron Lett. 1966. P. 5239.
1016. Fritschi H., Leutenegger UPfaltz A.jlNng^x. Chem. 1986. B. 98. S. 1028—
1029.
1017. Nozaki H., Takaya H.f Moriuti S., Noyori R.//Tetrahedron. 1968. V. 24.
P. 3655.
1018. Nakamura A., Kinishi A., Tatsuno Y., Otsuka S.//J. Amer. Chem. Soc. 1978.
V. 100. P. 3443—3448.
1019. Томилов Ю. В., Бордаков В. Г., Долгий И. Е., Нефедов О. М.//Изв. АН
СССР. Сер. хим. 1984. № 3. С. 582—8.
1020. Muller Е., Kessler Н., Zech B.//Fortschr. Chem. Forsch. 1966. В. 7. S. 128.
1021. Nakamura A., Yoshida T., Cowie M. e. a.//J. Amer. Chem. Soc. 1977. V. 99.
P. 2108—2117.
1022. Moser W. R.//J. Amer. Chem. Soc. 1969. V. 91. P. 1135—1140.
1023. Moser W. R.//J. Amer. Chem. Soc. 1969. V. 91. P. 1141 — 1146.
1024. Нефедов О. M., Долгий И. Е., Томилов Ю. В., Бордаков В. Г.//Изв. АН
СССР. Сер. хим. 1984. С. 119.
1025. Doyle М. РДХсс. Chem. Res. 1986. V. 19. Р. 348.
1026. Doyle М. Р., Tambtyn W. Н., Buhro W. Е., Dorow R. L.//Tetrahedron Lett.
1981. V. 22. Р. 1783.
1027. Doyle М. Р., Dorow R L., Buhro W. E. e. «.//Organometallics. 1984. V. 3.
P. 44—52.
1028. Томилов Ю. В., Бордаков В. Г., Долгий И. Е.//Тезисы докл. XII Менде-
леевского съезда. М.: Химия, 1981. Т. 2. С. 36.
1029. Dewar М. J. S.//Bull. Soc. chim. France. 1951. V. 18. P. C79.
290
lOBO. Олейникова А. Л., Темкин О. Н., Богданов М. И., Флид Р. Л4.//Ж. физ.
хим. 1970. № 44. С. 2418.
10^1. Harvilchuck J. М.г Aikens D. A., Murray R. C.//Inorg. Chem. 1969. V. 8.
P. 539.
103fe. Cvetanovic R. J., Duncan F. J., Falconer W. E., Irwin R. S.f/3. Amer. Chem.
Soc. 1965. V. 87. P. 1827—1832.
103^. Hende J. H., Baird W. С.ЦЕ Amer. Chem. Soc. 1963. V. 85. P. 1009—
< 1010.
1034. Cook B. W., Miller R. G. J., Todd P. F.HJ. Organomet. Chem. 1969. V. 19.
P. 421.
1035. Kirmse W., Kapps M., Hager R. B.//Chem. Ber. 1966. B. 99. S. 2855—2868.
1036. Schrauzer G. N., Fichler S.//Chem. Ber. 1962. B. 95. S. 260—267.
1037. Doering W. E., Roth Г.//Tetrahedron. 1963. V. 19. P. 715.
1038. Mango F. D., Dvoretzky I.//3. Amer. Chem. Soc. 1966. V. 88. P. 1654—
1657.
1039. Kirmse W., Kapps M., Hager R. B.//Chem. Ber. 1966. B. 99. S. 2855—
2868.
1040. Roth W. R:> Konig /.//Ann. 1965. B. 688. S. 28.
1041. Херберхольд M. л-Комплексы металлов: Пер. с англ./Под ред. И. И. Мо-
исеева. М.: Мир, 1975. С. 205.
1042. Chesick J.//J. Amer. Chem. Soc. 1962. V. 84. P. 3250—3253.
1043. Gassman P. G., Topp A., Keller J. W.//Tetrahedron Lett. 1969. P. 1093.
1044. Aue D. H., Reynolds R. N.//E Org. Chem. 1974. V. 39. P. 2315—2316.
1046. De Meijere A., Weitemeyer C., Schallner O.//Chem. Ber. 1977. B. 110.
1045. De Meijere A., Weitemeyer C.//Angew. Chem. 1970. B. 82. S. 359.
1046. De Meijere A., Weitemeyer C., Schallner O.//Chem. Ber. 1977. B. 110.
S. 1504—1522.
1047. Hemetsberger H., Brauer W., Tartier D.//Chem. Ber. 1977. B. 110. S. 1586—
1593.
1048. Bosse D., De Meijere A.//Tetrahedron Lett. 1978. P. 965; 1975. P. 871.
1049. Prinzbach H., Rucher C.//Tetrahedron Lett. 1977. P. 4195.
1050. Denis J. M.f Girard C., Conia J. ^.//Synthesis, 1972. P. 549.
1051. Pincock R. E., Wells J. I.//E Org. Chem. 1964. V. 29. P. 965—967.
1052. Battiste M. A., Brennan M. E.//Tetrahedron Lett. 1966. P. 5857.
1053. Roth IF.//Ann. 1964. B. 671. S. 10.
1054. Vogel E.//Angew. Chem. 1962. B. 74. S. 829.
1055. Vogel E., Wiedemann W., Kiefer H., Harrison W. F.//Tetrahedron Lett.
1963. P. 673.
1056. Kirmse W., Pohlmann /(.//Chem. Ber. 1967. B. 100. S. 3564—3577.
1057. Томилов Ю. В., Митенина T. Л., Луценко А. И. и др.//Изв. АН СССР.
Сер. хим. 1986. С. 77—84.
1058. Muller Е., Kessler //.//Tetrahedron Lett. 1968. Р. 3037.
1059. Blickle Р., Hopf Н., Bloch М., Jones Т. B.//Chem. Ber. 1979. В. 112.
3 3691_________3702
1060. Haywood-Farmer J., Pincook R. E., Wells J. /.//Tetrahedron. 1966. V. 22.
P. 2007.
1061. Klumpp G. W., Veefkind A. H., de Graaf W. L., Bickelhaupt F.//Ann. 1967.
B. 706. S. 47.
1062. Halton B., Battiste M. A., Rehberg R. e. a.//J. Amer. Chem. Soc. 1967.
V. 89. P. 5964—5965.
1063. Heinrich F., Liittke IF.//Angew. Chem. 1972. B. 84. S. 263—265.
1064. Fitjer L., Conia J. M//Angew. Chem. 1973. B. 85. S. 349—350.
1065. Fitjer L., Conia J. M//Angew. Chem. 1973. B. 85. S. 832—833.
1066. Gajewski J. J., Burka L. T.//E Org. Chem. 1970. V. 35. P. 2190—2196.
1067. Gajewski J. J., Burka L. Т.ЦЕ Amer. Chem. Soc. 1972. V. 94. P. 8860—
8864.
1068. Долгий И. E., Шаврин К. H., Нефедов О. М.ЦМзь. АН СССР. Сер. хим.
1987. С. 135.
19'
291
1069. Wenkert E., Mueller R. A., Reardon E. J. e. aJ/J. Amer. Chem. Soc. 1970.
V. 92. P. 7428. I
1070. Muller E., Fricke H., Kessler //.//Tetrahedron Lett. 1963. P. 1501.
1071. Muller E., Fricke H., Kessler HJjAnn. 1964. В. 675. S. 63.
1072. Paulissen R., Hubert A. J., Teyssie P.//Tetrahedron Lett. 1972. P. 146$.
1073. Kottwitz J., Vorbruggen //.//Synthesis. 1975. P. 636.
1074. Mende U., Raduchel B., Skubalta W.t Vorbrilggen //.//Tetrahedron Lett.
1975. P. 629.
1075. Raduchel B., Mende U., Cleve G., Hoger G. A. e. a.//Tetrahedron Lett.
1975. P. 633.
1076. Suda Л1.//Synthesis. 1981. P. 714.
1077. Казимирчик И. В., Лукин К. А., Зефиров Н. С.ЦУК. орг. хим. 1986.
С. 1554.
1078. Dinulescu G., Enescu L. N., Chencialescu A., Avram MJ/J. Chem. Res.
Part S. 1978. P. 456.
1079. Closs G. L.//Alicycl. Chem. 1966. V. 1. P. 53-127.
1080. Томилов Ю. В., Бордаков В. Г., Луценко А. И. и др.//Изв. АН СССР.
Сер. хим. 1987. С. 1338.
1081. Никифоров Г. А., Ершов В. В.//Ж. ВХО им. Д. И. Менделеева. 1979.
№ 24. С. 523.
1082. Никифоров Г. А., Свиридов Б. Д., Ершов В. В J/Изв. АН СССР. Сер.
хим. 1974. С. 373.
1083. Мещеряков А. Н., Долгий И. Е.//Изв. АН СССР. Сер. хим. 1980. С. 931.
1084. Мещеряков А. П., Долгий И. Е.//Изв. АН СССР. Сер. хим. 1980. С. 1874.
1085. Шапиро Е. А.//Дис. ... канд. хим. наук: 02.00.03. М., 1976. 105 с.
1086. Shimizu N., Nishida S.//J. Amer. Chem. Soc. 1974. V. 96. P. 6451—6456.
1087. Alonso M. E., Gomes ^.//Tetrahedron Lett. 1979. P. 2763.
1088. Carlsson D. J., Ingold K. UJ/J. Amer. Chem. Soc. 1968. V. 90. P. 747.
1089. Gajewski J. J., Weber R. J., Braun R. e. a J/J. Amer. Chem. Soc. 1977. V. 99.
P. 816—821.
1090. Дьяконов И. А., Голодников Г. В., Репинская И. Б.//УК. общ. хим. 1965.
Т. 35. С. 2181.
1091. Finkelstein R. е. a./fJ. med. Chem. 1966. V. 9. Р. 319. 440; 1965. V. 8.
Р. 432.
1092. Julia М., Thullier G. L.//Bull. Soc. chim. France. 1966. P. 717.
1093. Julia M., Baillarge AL//Bull. Soc. chim. France. 1966. P. 734—743.
1094. Blatchford J. K-, Orchin MJ /J. Org. Chem. 1964. V. 29. P. 839—843.
1095. Дьяконов И. А., Костиков P. PJ/УК. общ. хим. 1964. T. 34. P. 3843.
1096. Wharton P. S., Bair T. IJ/J. Org. Chem. 1965. V. 30. P. 1681—1684.
1097. La Londe R. T., Tobias M. AJ/J. Amer. Chem. Soc. 1964. V. 86. P. 4068—
4073.
1098. Kirmse W.t Pook K-H J/Chem. Ber. 1965. B. 98. S. 4022—4026.
1099. Skell P. S., Etter R. MJ /Proc. chem. Soc. 1961. P. 443.
1100. Warkintin J.//Can. J. Chem. 1965. V. 43. P. 3456.
1101. Berson J. A., Hand E. SJ/J. Amer. Chem. Soc. 1964. V. 86. P. 1978—1988.
1102. Musso H., Beithan (A//Chem. Ber. 1964. B. 97. S. 2282—2288.
1103. Korte FJ/Ann. 1964. B. 664. S. 114.
1104. Sauers R. R., Sonnet P. E.//Chem. a. Ind. 1963. P. 786.
1105. Owen J., Simonsen J. LJ/J. Chem. Soc. 1932. P. 1424; 1933. P. 1225.
1106. Пат. 3010971 США, МКИ2 С 07. Cyclopropylamine derivs.
1107. Мещеряков А. //.//Изв. АН СССР. Сер. хим. 1966. С. 1235.
1108. Дьяконов И. AJ/УК. орг. хим. 1965. Т. 1. С. 1189.
1109. Vogel Е., Erb RJ/Angew. Chem. 1962. В. 74. S. 76.
1110. Шапиро Е. А., Романова Т. Н., Долгий И. Е., Нефедов О. М.//Изв. АН
СССР. Сер. хим. 1984. С. 2529, 2535.
1111. Дьяконов И. А., Мызникова В. Ф.//С6. статей общей химии. Л.: АН
СССР, 1953. Т. 1. С. 489.
292
11\12. Дьяконов И. А., Комендантов М. Иф/Ж. общ. хим. 1961. Т. 31. С. 3881;
\ Дьяконов И. А., Комендантов М. И.//Вестник ЛГУ. Т. 11. Сер. физ. и
) хим. 1956. № 4. С. 166.
1113. Breslow R., Battiste AL//Chem. a. Ind. 1958. Р. 1143.
1114. Breslow R., Winter R., Battiste М.ЦЗ. Org. Chem. 1959. V. 24. P. 415—
416.
1115. Breslow R., Chipman Z).//Chem. a. Ind. i960. P. 1105.
1116. Vidal ^.//Tetrahedron Lett. 1967. P. 1073.
1117. Дьяконов И. А., Комендантов M. И., Коршунов С. П.) Ж- общ. хим. 1982.
Т 32 С 923.
1118. Дьяконов И. А., Комендантов М. И.ЦУК. общ. хим. 1963. Т. 33. С. 2448.
1119. Комендантов М. И., Дьяконов И. А., Гохманова И. и др.//Ж. орг. хим.
1965. Т. 1. С. 209.
1120. Комендантов М. И., Дьяконов И. А., Смирнова Т. С./Ж. орг. хим. 1966.
Т. 2. С. 559.
1121. Дьяконов И. А., Комендантов М. И., Разин В. В./ Ж общ. хим 1963.
Т. 33. С. 2420.
1122. Dyakonov I. A., Komendantov М. I., Razin V. V.//Tetrahedron Lett. 1966.
Р. 1127, 1135.
1123. Шапиро Е. А., Долгий И. Е., Нефедов О. М.//Изв. АН СССР. Сер. хим.
1980. С. 2096.
1124. Долгий И. Е., Шапиро Е. А., Нефедов О. Л1.//Изв. АН СССР. Сер. хим.
1983. С. 2405.
1125. Дьяконов И. А., Данилкина Л. П.ЦУК. общ. хим. 1964. Т. 34. С. 738.
С. 3129. Ж- орг. хим. 1966. Т. 2. С. 3.
1126. Дьяконов И. А., Гмызина Р. Н., Данилкина Л. П.ЦУК. орг. хим. 1966.
Т. 2. С. 2079.
1127. Paulissen R., Reimlonger Н., Hayez Е. е. ^.//Tetrahedron Lett. 1973.
Р. 2233.
1128. Hubert A. J., Noels A. F., Anciaux A. J., Teyssie P.//Synthesis. 1976. Р. 600.
1129. Anciaux A. J., Demonceau A., Hubert A. J. e. a.//J. Chem. Soc. Chem. Comm.
1980. P. 765—766.
1130. Doyle NL P., van Leusen D., Tamblyn W. //.//Synthesis. 1981. P. 787.
1131. Dave V., Warnhoff EJ/Org. Reactions. N. Y.: Wiley, 1970. V. 18. P. 217.
1132. Burke S. D., Grieco P. A.//Ibid. 1979. V. 26. P. 361.
1133. Wenkert Е.ЦАсс. Chem. Res. 1980. V. 13. P. 27—31.
1134. Nozaki H., Moriuti S., Yamade M., Noyori R.//Tetrahedron Lett. 1966.
P 59
1135. Kirmse W., Dietrich //.//Chem. Ber. 1965. B. 98. S. 4027—4032.
1136. Долгий И. E., Шапиро E. А., Лунькова Г. В., Нефедов О. Л1.//Изв. АН
СССР. Сер. хим. 1979. С. 1652.
1137. Peace В. W., Wulf man D. S.//Synthesis. 1973. P. 137.
1138. Wulf man D. S., McGibboney B. G., Steffen E. K- e. ^.//Tetrahedron. 1976.
V. 32. P. 1257.
1139. Wenkert E., Alonso M. E., Buckwaiter B. L., Chou R. J.HJ. Amer. Chem.
Soc. 1977. V. 99. P. 4778—4782.
1140. Ledon H., Linstrumelle G., Julia S.//Bull. Soc. chim. France. 1973. P. 2071.
1141. Novak J. e. a.//Coll. Czech. Chem. Comm. 1957. V. 22. P. 1836.
1142. Novak J., Ratusky J., Sneberg V., Sorm FJ/Chem. listy. 1957. V7. 51.
P. 479.
1143. Cowan D. O., Couch M. M., Kopecky K. R-> Hammond G. S.//J. Org. Chem.
1964. V. 29. P. 1922—1925.
1144. Петров А. А., Калинина В. А., Херузе Ю. И./[УК- орг. хим. 1967. Т. 3.
С. 637.
1145. Takebayashi М., Ibata Т., Kohara Н., Ueda /(.//Bull. Chem. Soc. Jap. 1969.
V. 42. Р. 2938—2944.
1146. Arnold Z.HJ. Chem. Soc. Chem. Comm. 1967. P. 299—300.
293
1147. Simmons H. E.f Smith R. D.HE Amer. Chem. Soc. 1958. V. 80. P. 5323—
5324; 1959. V. 81. P. 4256—4264. 1
1148. Smith R. D.f Simmons H. E.//Org. Synthesis. 1961. V. 41. P. 72.
1149. Simmons H. E., Blanchard E. P., Smith R. D.HE Amer. Chem. Soc. 1964.
V. 86. P. 1347—1356.
1150. Blanchard E. P., Simmons H. Е.ЦЕ Amer. Chem. Soc. 1964. V. 86. P. 1337.
1151. Neumann R. (/.//Tetrahedron Lett. 1964. P. 2541.
1152. Le Goff E.//E Org. Chem. 1964. V. 29. P. 2048—2050.
1153. Rawson R. J., Harrisson I. T.//E Org. Chem. 1970. V. 35. P. 2057—2058.
1154. Wittig G., Wingler F.//Chem. Ber. 1964. B. 97. S. 2146—2164.
1155. Furukawa J., Rawalata N., Nishimura /.//Tetrahedron Lett. 1966. P. 3353;
1968. P. 3495.
1156. Lewicka S., OkamurA W. //.//Synth. Comm. 1980. V. 10. P. 415.
1157. Rawabata N., Ramemura L, Naka М.ЦЕ Amer. Chem. Soc. 1979. V. 101.
P. 2139—2145.
1158. Rawabata N., Yamagishi N., Yamashita S.//Bull. Chem. Soc. Jap. 1977.
V. 50. P. 466—468.
1159. Rawabata N., Rujii T., Naka M., Yamashita S.//Bull. Chem. Soc. Jap.
1977. V. 50. P. 1005—1006.
1160. Nazaki H., Roto S., Noyori 7?.//Can. J. Chem. 1966. V. 44. P. 1021.
1161. Roch S. D., Rliss R. M., Lopiekes D. V., Wineman R. J.HL Org. Chem.
1961. V. 26. P. 3122_3125
1162. Breslow R., Bozino H., Wolf P.//Tetrahedron Lett. 1970. P. 2395.
1163. Effenberger F., Podszun W.llAngw. Chem. 1969. B. 81. S. 1046.
1164. Harker A.//Angew. Chem. 1967. B. 79. S. 103.
1165. Dolbier W. R., Akiba R, Riemann J. M. e. a.//E Amer. Chem. Soc. 1971.
V. 93. P. 3933—3940.
1166. Applequist D. E., Landgrebe J. А.ЦЕ Amer. Chem. Soc. 1964. V. 86.
P. 1543—1549.
1167. Erden /.//Synth. Comm. 1986. V. 16. P. 117.
1168. Conia J. M., Denis J. Л4.//Tetrahedron Lett. 1969. P. 3545.
1169. Sheldon R. A., Rochi J. R.HE Amer. Chem. Soc. 1970. V. 92. P. 4395—
4404.
1170. Overberger C. G., Halek G. W.//E Org. Chem. 1963. V. 28. P. 867—868.
1171. Radlick P., Winstein S.//E Amer. Chem. Soc. 1961. V. 85. P. 344—345.
1172. Vogel E.HAngew. Chem. 1961. B. 73. S. 548.
1173. Нагапетян Л. А., Сафонова И. Л., Еазанский Б. А.//Изв. АН СССР. Сер.
хим. 1962. С. 902.
1174. Nelson Р. Н., Untch R. (/.//Tetrahedron Lett. 1969. Р. 4475.
1175. Boikess R. S., Winstein SJ/E Amer. Chem. Soc. 1963. V. 85. P. 343—344.
1176. Nozaki H., Rawanishi M., Noyory R.HE Org. Chem. 1965. V. 30. P. 2216—
2217.
1177. Fitjer L., Wehle //.//Angew. Chem. 1979. B. 91. S. 927—928.
1178. Vo-Quang L., Cadiot P.t Willemart А.ЦС. r. 1960. V. 255. P. 950.
1179. Andrews S. D., Smith J. C.//Chem. a. Ind. 1966. P. 1636.
1180. Wittig G., Hutchinson J. /.//Ann. 1970. B. 741. S. 70.
1181. Vo-Quang L., Cadiot Р.ЦВМХ. Soc. chim. France. 1965. P. 1525.
1182. Zweifel G., Clark G. M., Whitney С. C.//E Amer. Chem. Soc. 1971. V. 93.
P. 1305—1307.
1183. Toller E., Foster D. J.HZ. Naturforsch. 1962. B. 17. S. 135.
1184. Ronzelman L. M., Conley R. T./fE Org. Chem. 1968. V. 33. P. 3828—3838.
1185. McCarty J. F.//E Med. Chem. 1964. V. 7. P. 72.
1186. Gassman P. G., Mansfield R. Т.ЦЕ Org. Chem. 1967. V. 32. P. 915—920.
1187. Sawada S., Oda J., Inouye Y.//E Org. Chem. 1968. V. 33. P. 2141—2143.
1188 Limasset J. C., Amice P., Conia J. M.//Bull. Soc. chim. France. 1969.
P. 3981.
1189. Wilcox C. F., Jesaitis R. G.HE Org. Chem. 1968. V. 33. P. 2154—2156.
1190. Felix D., Stoll M., Schenmoser A.//Chimia. 1969. V. 18. P. 174.
294
1191. Armand Y., Perraud R., Pierre J. L., Arnaud P.//Bull. Soc chim France
1965. P. 1893.
1192. Perraud R., Armand /.//Bull. Soc. chim. France. 1968. P. 154Q.
1193. Majerski Z., Schleyer P. RJ/J. Org. Chem. 1969. V. 34. P. 3215—3216.
1194. Ginsing R., Cross A. DJ/J. Amer. Chem. Soc. 1965. V. 87. P. 4629 4635.
1195. Tanabe M., Crowe D. /.//Tetrahedron. 1967. V. 23. P. 2115.
1196. Vidal M., Dumont C., Arnand /.//Tetrahedron Lett. 1966. P. 5081.
1197. Blanchard E. P., Simmonth H. E., Taylor J. SJ/J. Org. Chem 1965 V 30
P. 4321—4322.
1198. Sawada S., Inouye YJ/BwW. Chem. Soc. Jap. 1969. V. 42. P. 2669___2672.
1199. Nishimura J., Furukawa J., Rawabata N., Ritayama M.//Tetrahedron 1970
V. 26. P. 243.
1200. Ретинский А. А., Шостаковский C. MJ/Изв. АН СССР. Сер. хим 1967
C. 413.
1201. Шостаковский C. M.f Львов A. M., Диммельфелъд Я- MJ/Изв. АН СССР.
Сер. хим. 1966. C. 1754.
1202. De Puy С. H., Mahoney L. R.//J. Amer. Chem. Soc. 1964. V. 86. P 9653—
2657.
1203. De Puy С. H., Mahoney L. R., Eilers R. LJ/J. Org. Chem. 1961. V 26
P. 3616.
1204. De Puy С. H., Dappen G. M., Eilers R. L., Rlein R. AJ/J. Org. Chem. 1964.
V 29 P 2813
1205. Wassermann E. H., Clagett D. C.//Tetrahedron Lett. 1964. P. 341.
1206. Goh S. H., Closs L. S., Closs G. L. //J. Org. Chem. 1969. V. 34. P. 25—31.
1207. Le Goaller R-, Pierre J. L.//Bull. Soc. chim. France. 1973. P. 1531.
1208. Rubottom G. M.f Lopez M. IJ/J. Org. Chem. 1973. V. 38. P. 2097—2099.
1209. Conia J. M., Robson M. /.//Angew. Chem. 1975. B. 87. S. 505—516.
1210. Conia J. M.//Pure a. Appl. Chem. 1975. V. 43. P. 317.
1211. Amice R., Blanco L., Conia J. ^.//Synthesis. 1976.. P. 196.
1212. Ginard C., Amice P., Barnier J. P., Conia J. ^.//Tetrahedron Lett. 1974.
P 3329
1213. Trost В. M', Bogdanowicz M. JJ/J. Amer. Chem. Soc. 1973. V. 95. P. 5321 —
5334.
1214. Girard C., Conia J. AL//Tetrahedron Lett. 1974. P. 3333.
1215. Вебер В., Гокель Г. Межфазный катализ в органическом синтезе: Пер. с
англ./Под ред. И. П. Белецкой. М.: Мир, 1980. 327 с.
1216. Яновская Л. А., Юфит С. С. Органический синтез в двухфазных систе-
мах. М.: Химия, 1982. 184 с.
1217. Dehmlow Е. S., Dehmlow S. S. Phase Transfer Catalysis: Monographs
in Modern Chemistry. V. 11. Weinheim. (F. R. G.): Verlag Chemie. 1983.
316 p.
1218. Starks C., Liotta C. Phase Transfer Catalysis: Principles and Technique.
N. Y. — San Francisko— London: Academic Press, 1978, 110 p.
1219. Ж. BXO им. Д. И. Менделеева. 1986. T. 31. № 2.
1220. Dehmlow Е. V., Lissel MJ/J. Chem. Res. Part S. 1978. P. 310.
1221. Гольдберг Ю. ILL, Шиманская M. В J/YR. BXO им. Д. И. Менделеева.
1986. T. 31. С. 149—157.
1222. Regen S. L., Singh AJ/J. Org. Chem. 1982. V. 47. P. 1587—1588.
1223. Агавелян Э. С., Нефедов О. MJ/Изв. АН СССР. Сер. хим. 1987. С. 681.
1224. Hiyama T.f Sawada Н., Tsukanaka М., Nozaki //.//Tetrahedron Lett. 1975.
Р. 3013.
1225. Herriott A. W., Picker DJ/J. Amer. Chem. Soc. 1975. V. 97. P. 2345—
2349.
1226. Дьяченко А. И., Савилова С. Ф., Абрамова H. M. и др.ЦИзв. АН СССР.
Сер. хим. 1980. С. 1688—1690.
1227. Julia S.t Ginebrela A.//Synthesis. 1977. Р. 682.
1228. Makosza М., Fedorynski Af.//Synth. Comm. 1973. V. 3. P. 305.
295
1229. Новокрещенных В. Д„ Мочалов С. С., Шабаров Ю. С. ПУК. орг. хим. 1979.
Т. 15. С. 485.
1230. Mathias R., Weyerstahl Р.//Angew. Chem. Int. Ed. 1974. V. 13. P. 132.
1231. Mathias R., Weyerstahl P.//Angew. Chem. 1974. B. 86. S. 42—43.
1232. Weyerstahl P., Blume G., Muller S. C.//Tetrahedron Lett. 1971. P. 3869.
1233. Weyerstahl P., Mathias R-, Blume G.//Tetrahedron Lett. 1973. P. 611.
1234. Fedorynski M//Synthesis. 1977. P. 923.
1235. Dehmlow E. V., Broda IF.//Chem. Ber. 1982. B. 115. S. 3894—3897.
1236. Moss R. A., Pilkiewicz F. G.//J. Amer. Chem. Soc. 1974. V. 96. P. 5632—
5633.
1237. Moss R. A., Pilkiewicz F. (/.//Synthesis. 1973. P. 209.
1238. Makosza T4.//Pure a. Appl. Chem. 1975. V. 43. P. 439.
1239. Долгий И. E., Шаврин К. H., Крылова И. В., Нефедов О. ЛТ/уИзв. АН
СССР. Сер. хим. 1986. С. 2380.
1240. Makosza М., Kacprowicz A., Fedorynski М.//Tetrahedron Lett. 1975. Р. 2119.
1241. Костиков Р. Р., Гришина Е. Н., Слободин Я. М.//УК. орг. хим. 1979. Т. 15.
С 331
1242. Dehmlow Е. V., Schonefeld /.//Ann. 1971. В. 744. S. 42.
1243. Sasaki Т., Kanematsu К., Okamura N.HJ. Org. Chem. 1975. V. 40. P. 3322—
3325
1244. Joshi G. C., Singh N., Pande L. Af.//Tetrahedron Lett. 1972. P. 1461.
1245. Kaspar F., Beier Т.Ц2. Chem. 1976. B. 16. S. 435.
1246. Слободин Я. M., Ашкинази Л. А., Климчук Г. HJ/jK. орг. хим. 1984. Т. 20.
С. 1001.
1247. Красуцкий П. Л., Фокин А. А., Чекменева Л. А. и др.//Ж. орг. хим. 1984.
Т. 20. С. 1807.
1248. Костиков Р. Р.> Гришина Е. Н., Слободин Я. М.ЦУК. орг. хим. 1984. Т. 20.
С. 1426.
1249. Костиков Р. Р., Дрыгайлова Е. А., Оглоблин К. А.//Ж. орг. хим. 1978.
Т. 14. С. 2008.
1250. Кремлев М. М., Фиалков Ю. А., Ягупольский Л. Л4.//Ж. орг. хим. 1981.
Т 17 С 332
1251. Dehmlow V., Klabuhn Н., Hass Е. С.//Ann. 1973. S. 1063.
1252. Greibrokk Г.//Acta Chem. Scand. 1973. V. 27. P. 3207.
1253. Костиков P. P., Воробьева И. С., Молчанов А. П.//УК. орг. хим. 1983.
Т 19 С 256_________262
1254. Dehmlow Е. ^.//Tetrahedron Lett. 1975. Р. 203.
1255. Костиков Р. Р., Васильева И. А., Слободин Я. Н.//УК. орг. хим. 1974.
Т 10 С 2325
1256. Dehmlow Е. V., Dehmlow S. S., Marschner F.//Chem. Ber. 1977. В. ПО.
S. 154—164.
1257. Dehmlow S. S., Dehmlow E. V.//Ann. 1973. S. 1753.
1258. Makosza M., Gajos /.//Roszn. Chem. 1974. V. 48. P. 1883—1893.
1259. Jef ford C. W., de los Heros V., Burger (/.//Tetrahedron Lett. 1976. P. 703.
1260. Dehmlow E. V.//Tetrahedron Lett. 1972. P. 175.
1261. Hart H., Nitta M//Tetrahedron Lett. 1974. P. 2109.
1262. Joshi G. C., Singh N., Pande L. M.//Synthesis. 1972. P. 317.
1263. Blume G., Neumann T., Weyerstahl P.//Ann. 1975. S. 201.
1264. Blume G., Weyerstahl P.//Tetrahedron Lett. 1970. P. 3669.
1265. Weyerstahl P„ Blume (/.//Tetrahedron. 1972. V. 28. P. 5281.
1266. Tsunetsugu J., Sato M., Ebine S.//J. Chem. Soc. Chem. Comm. 1972. P. 363—
364.
1267. Sato M., Ebine S., Tsunetsugu J.//J. Chem. Soc. Chem. Comm. 1974.
P. 846—847;//J. Chem. Soc. Perkin Trans. Part I. 1977. P. 1282—1287.
1268. Sato M., Tsunetsugu J., Ebine S.//Bull. Soc. Chem. Jap. 1976. V. 49.
P 2230________2235
1269. Sato M., Uchida A., Tsunetsugu J. e. a.//Tetrahedron Lett. 1977. P. 2151.
1270. Арбузов Б. А.//Изв. АН СССР. Сер. хим. 1975. C. 331.
296
1271. Шостаковский С. М., Ретинский А. А., Бобров А. BJ/VLsn. АН СССР.
Сер. хим. 1974. С. 1818.
1272. Аксенов В. С., Мин Р. С.//Изв. СО АН СССР. Сер. хим. 1974. С. 139.
1273. Фешин В. П., Воронков М. Г., Шостаковский С. М. и др. /Ж. орг. хим.
1976. Т. 12. С 2302.
1274. Braverman S., Duar Y., Segev ^.//Tetrahedron Lett. 1976. P. 3181.
1275. Бредихин А. А., Племенков В. В.//Ж. орг. хим. 1976. Т. 12. С. 1001..
1276. Ranken D. F.//Synth. Comm. 1973. V. 3. Р. 311.
1277. Voronkov М. G.//Topics in Organic Sulfur Chemistry. Bangor, 1974. P. 8.
1278. Jef ford C. W., Burger U., Delay F.//Helv. chim. acta. 1973. V. 56. P. 1083.
1279. Klein G., Kraus ^.//Tetrahedron. 1977. V. 33. P. 3121.
1280. Hiyarna T., Mishima T., Kitatani К e. ^.//Tetrahedron Lett. 1974. P. 3297.
1281. Fedorynski M., Gorzkowska I., Makosza MJ/Synthesis. 1977. P. 120.
1282. Graefe J., Adler M., Muhlstadt MJ/Z. Chem. 1975. B. 15. S. 14.
1283. Ившин В. П., Кемешен М. С., Белик Н. П.'/УК. орг. хим. 1980. Т. 16.
С. 980.
1284. Chan Т. Н., Massuda DJ/Тetrahedron Lett. 1975. Р. 3383.
1285. Miller R. В.//Synth. Comm. 1974. V. 4. P. 341.
1286. Костиков P. P., Молчанов А. П.ЦУК. орг. хим. 1975. T. 11. C. 1861.
1287. Makosza M., Fedorynski MJ/Synthesis. 1974. P. 274.
1288. Klein G., Paquette L. A.//Tetrahedron Lett. 1976. P. 2419.
1289. Boswell R. F„ Bass R. GJ/J. Org. Chem. 1977. V. 42. P. 2342—2344.
1290. Boswell R. F., Bass R. GJ/J. Org. Chem. 1975. V. 40. P. 2419—2420.
1291. Gokel G. W., Shepherd J. P., Weber W. P. e. aJ/J. Org. Chem. 1973. V. 38.
P. 1913—1918.
1292. Хусид А. Х.//Изв. АН СССР. Сер. хим. 1977. C. 2135.
1293. Крылиталь F. В., Яновская А. А.//Изв. АН СССР. Сер. хим. 1976. C. 424.
1294. Khusid A. K., Kryshtal G. V., Kucherov V. F. e. a.//Tetrahedron Lett. 1977.
P. 907.
1295. Hiyama T., Tsukanaka H., Nozaki HJ/J. Amer. Chem. Soc. 1974. V. 96.
P. 3713—3714.
1296. Damiano J. C., Luche J. L., Grable ^.//Tetrahedron Lett. 1974. P. 779.
1297. Luche J. L., Damiano J. C., Grable PJ/J. Chem. Res. Part S. 1977. P. 33.
1298. Kleveland K„ Skattebl L., Sydnes L. KJ/Ncta Chem. Scand. 1977. V. B31.
P. 463.
1299. Vogel E., Ippen /.//Angew. Chem. 1974. B. 86. S. 778—779.
1300. Dehmlow E. V., Hofle GJ/Chem. Ber. 1976. B. 107. S. 2760—2767.
1301. Makosza M., Ludwikov MJ/Angew. Chem. Int. Ed. 1974. B. 13. S. 665.
1302. Dehmlow E. V.//Ann. 1972. B. 758. S. 148.
1303. Bar let M. RJ/C. r. 1974. V. 278C. P. 621.
1304. Sydnes L. A.//Acta Chem. Scand. 1977. V. B31. P. 823.
1305. Makosza M., Gajos /.//Bull. Pol. Acad. sci. Chem. 1972. V. 20. P. 33.
1306. Пат. 2201514 ФРГ, МКИ2 C 07ic. Alkyl 2,2-dihalocyclopropanecarboxylates.
1307. Sydnes L. /<.//Helv. chim. acta. 1975. V. 58. P. 2061.
1308. Makosza M.t Biolecka ^.//Tetrahedron Lett. 1971. P. 4517.
1309. Boche G., Schneider D. ^.//Tetrahedron Lett. 1975. P. 4247.
1310. Sasaki T., Eguchi S., Ogawa T.//J. Org. Chem. 1974. V. 39. P. 1927—1930.
1311. Sasaki T„ Eguchi S., Ohno M., Nakata FJ/J. Org. Chem. 1976. V. 41.
P. 2408—2411.
1312. Patrick /.//Tetrahedron Lett. 1974. P. 1407.
1313. Julia S., Michelot D., Linstrumelle GJ/C. r. 1974. V. 278C. P. 1523.
1314. Newman M. S., Gromelski S. JJ/J. Org. Chem. 1972. V. 37. P. 3220—3224.
1315. Newman M. S.t Din ZJ/J. Org. Chem. 1973. V. 38. P. 547—549.
1316. Newman M. S., Liang W. CJ/J. Org. Chem. 1973. V. 38. P. 2438—2441.
1317. Newman M. S.t Zwan M. CJ/J. Org. Chem. 1974. V. 39. P. 761—763, 1186—
1189.
1318. Barlet R., Vo-Quang У.//Ви11. Soc. chim. France. 1969. P. 3729—3760.
1319. Pinder A. ^.//Synthesis. 1980. P. 425—452.
297
1320. Корнева О. С.//Дис. ... канд. хим. наук: 02.00.03. М., 1985. 124 с.
1321. Norden W., Sander V., Weyerstahl P.//Chem. Ber. 1983. В. 116. S. 3097—
3111.
1322. Нефедов О. M., Мечников Л. Г., Дьяченко А. И., Агрэ С. А.ЦУК. ВХО
им. Д. И. Менделеева. 1986. Т. 31. С. 182.
1323. Arct J., Migay В.I/Tetrahedron. 1981. V. 37. Р. 953.
1324. Нефедов О. М., Ивашенко А. Л.//Новое в химии карбенов. М.: Наука,
1973. С. 145.
1325. Нефедов О. М., Новицкая Н. Н.//Изв. АН СССР. Сер. хим. 1965. С. 395—
396; Nefedov О. M.t Novitskaya N. N., Ivaschenko A. A.//Ann. 1967. В. 707.
S. 217—226.
1326. Нефедов О. М., Агавелян Э. С.//Изв. АН СССР. Сер. хим. 1971. С. 2608—
2610; 1972. С. 481—482.
1327. Блек Д. С. К-, Джексон У. Р., Свен Дж. М.//Общая органическая химия.
В 12 т. Т. 7.: Пер. с англ./Под ред. Н. К. Кочеткова и Ф. М. Стояновича.
М.: Химия, 1984. С. 240—461.
1328. McLain S. J., Wood С. D., Schrock R. R.//J. Amer. Chem. Soc. 1977. V. 99.
p 3519________3520
1329. Schrock R. R., Sharp P. R.//J. Amer. Chem. Soc. 1978. V. 100. P. 2389—
2399.
1330. Чугаев Л. А., Сканави-Григорьева M. С.//Ж. русск. физ.-хим. общ., 1915.
Т. 47. С. 776.
1331. Fischer Е. О., Massbol A.//Angew. Chem. Int. Ed. 1964. V. 3. P. 580.
1332. Cardin D. J., Cetinkaya B., Lappert M. F.//Chem. Rev. 1972. V. 72. P. 545.
1333. Fischer E. O.//Pure a. Appl. Chem. 1972. V. 30. P. 353.
1334. Фишер Э. О.//Усп. химии. 1972. Т. 41. С. 1161.
1335. Cardin D. J., Cetinkaya В., Doyle М. L, Lappert М. F.//Chem. Soc. Rev.
1973 V 2 P 99
1336. Cotton F. A., Lukehart C. M.//Progr. Inorg. Chem. 1972. V. 16. P. 487.
1337. Fischer E. O.//Angew. Chem. 1974. B. 86. S. 651—663.
1338. Connor J. A.//Organomet. Chem. Synth. 1975. V. 4. P. 235.
1339. Fischer E. O.l/Mv. Organomet. Chem. 1976. V. 14. P. 1.
1340. Brown F. /.//Progr. Inorg. Chem. 1980. V. 27. P. 1.
1341. Ofele A.//Angew. Chem. 1968. B. 80. S. 1032—1033//J. Organomet Chem.
1970. V. 22. P. C9.
1342. Casey С. P., Burkhardt T. J.//I. Amer. Chem. Soc. 1973. V. 95. P. 5833—
5834.
1343. Beatty R. P„ Maher J. M., Cooper N. J.//J. Amer. Chem. Soc. 1981. V. 103.
p 238_________239.
1344. Herrmann W. A.//Chem. Ber. 1975. B. 108. S. 486—499.
1345. Clark D. N., Schrock R. R.//J. Amer. Chem. Soc. 1978. V. 100. P. 6774—
6776.
1346. Wengrovius J. H., Schrock R. R., Churchill M. R. e. а.Ц1. Amer. Chem. Soc.
1980. V. 102. P. 4515—4516.
1347. Schrock R. R.//J. Amer. Chem. Soc. 1974. V. 96. P. 6796—6797; 1975. V. 97.
P. 6577—6578.
1348. Guggenberger L. L, Schrock R. R.//J. Amer. Chem. Soc. 1975. V. 97.
P. 6578.
1349. Schrock R. R., Fellmann J. D.//J. Amer. Chem. Soc. 1978. V. 100. P. 3359—
3370.
1350. Schrock R. R., Messerle L. W., Wood C. D., Guggenberger L. /.//Ibid.
P. 3793—3800.
1351. Sharp P. R., Schrock R. R.//J. Organomet. Chem. 1979. V. 171. P. 43.
1352. Messerle L. W.t Jennische P., Schrock R. R.f Stucky G.//I. Amer. Chem.
Soc. 1980. V. 102. P. 6744—6752.
1353. Churchill M. R., Youngs W. /.//Inorg. Chem. 1979. V. 18. P. 1930.
1354. Schwartz J., Gell К. 1.ЦУ Organomet. Chem. 1980. V. 184. P. Cl.
298
1355. Wong W.-R., Tan W., Gladysz J. A.//J. Amer. Chem. Soc. 1979. V 101.
P. 5440—5442.
1356. Brookhart M., Tucker J. R., Flood T. C., Jensen J.HJ. Amer. Chem. Soc.
1980. V. 102. P. 1203—1205.
1357. Chang S.-С., Rafafi Z. H., Hauge R. H. e. a.//J. Amer. Chem. Soc. 1985.
V. 107. P. 1447—1448.
1358. Stevens A. E., Beauchamp J. L.HJ. Amer. Chem. Soc. 1978. V. 100. P. 2584—
2585; 1979. V. 101. P. 6449—6450.
1359. Buchwald S. L., Anslyn E. W., Grubbs R. H.HJ. Amer. Chem. Soc., 1985.
V. 107. P. 1766—1768.
1360. Schrock R. RJ/Acc. Chem. Res. 1979. V. 12. P. 98—104.
1361. Calderon N., Lawrence J. P., Of stead E. A.//Adv. Organomet. Chem. 1979.
V. 17. P. 449.
1362. Schubert (/.//Coord. Chem. Revs. 1984. V. 55. P. 261.
1363. Dotz R. //.//Pure a. Appl. Chem. 1983. V. 55. P. 1689.
1364. Dotz R. H., Fischer H., Hoffmann P. e. a. Transition Metal Carbene Com-
plexes. Verl. Chemie, 1984. 150 c.
1365. Henrici-Olive G., Olive S. The Chemistry of the Catalyzed Hydrogenation
of Carbon Monooxide. Berlin: Springer—Verlag, 1984. P. 231.
1366. Schultz A. J., Williams J. M., Schrock R. R. e. a.HJ. Amer. Chem. Soc. 1979.
V. 101. P. 1593—1595.
1367. Schultz A. J., Brown R. R., Williams J. M., Schrock R. R.//J. Amer. Chem.
Soc. 1981. V. 103. P. 169—176.
1368. Goddard R. J., Hoffmann R., Jemmis E. D.HJ. Amer. Chem. Soc. 1980.
V. 102. P. 7667—7676.
1369. Churchill M. R.t Youngs W. /.//Inorg. Chem. 1979. V. 18. P. 2454.
1370. Marynick D. S., Rirkpatrick C. M.HJ. Amer. Chem. Soc. 1985. V. 107.
P. 1993—1994.
1371. Zeigler T., Versluis L., Tschinke V.//J- Amer. Chem.. Soc. 1986. V. 108.
P. 612—617.
1372. Lauher J. W., Hoffmann R.//J. Amer. Chem. Soc. 1976. V. 98. P. 1729—
1742.
1373. Casey С. P., Tuinstra H. E.HJ. Amer. Chem. Soc. 1978. V. 100. P. 2270—
2272.
1374. Rappe A. R., Goddard W. A.//J. Amer. Chem. Soc. 1977. V. 99. P. 3966—
3968; 1980. V. 102. P. 5114—5115.
1375. Casey С. P., Anderson R. L.[[J. Chem. Soc. Chem. Comm. 1975. P. 895—
896.
1376. Casey С. P., Burkhardt T. J.HJ. Amer. Chem. Soc. 1972. V. 94. P. 6543—
6544.
1377. Dotz R. H., Fischer E. O.//Chem. Ber. 1972. B. 105. S. 1356—1367.
1378. Casey С. P., Boggs R. A., Anderson R. L.HJ. Amer. Chem. Soc. 1972.
V. 94. P. 8947—8949.
1379. Truett W. L., Johnson D. R., Robinson I. M., Montague B. A.//Ibid. 1960.
V. 82. P. 2337—2340.
1380. Dall’Asta G.//Makromol. Chem. 1962. V. 56. P. 224.
1381. Banks R. L., Bailey G. C.//Ind. a. Eng. Chem. Prod. Res. a. Development.
1964. V. 3. P. 170.
1382. Mango F. D.//Coord. Chem. Rev. 1975. V. 15. P. 109.
1383. Маковецкий R. Л., Долгоплоск Б. A., RoptuaK Ю. В. и dp.HJR. ВХО им.
Д. И. Менделеева. 1974. T. 24. С. 513.
1384. Hains R. J., Leigh G. /.//Chem. Soc. Rev. 1975. V. 4. P. 155.
1385. Mol J. C., Moulijn J. A.//Adv. Catal. a. Relat. Subj. 1975. V. 24. P. 131.
1386. Hughes W. B.//Organomet. Chem. Synth. 1972. V. 1. P. 341.
1387. Calderon M//Acc. Chem. Res. 1972. V. 5. P. 127—132.
1388. Долгоплоск Б. А., Маковецкий R. Л., Тинякова E. И.ЦДокл. АН СССР.
1972. T. 202. C. 871.
1389. Долгоплоск Б. A.HRmti. и катализ. 1981. T. 22. С. 807.
299
1390. Долгоплоск Б. А., Тинякова Е. И. Металлорганический катализ в процес-
сах полимеризации. М.: Наука, 1985. 510 с.
1391. Исагулянц Г. В., Климов А. FL, Коваленко Л. И., Коган В. Л/.//Изв. АН
СССР. Сер. хим. 1986. С. 2805.
1392. Lappert М. F.//Adv. Chem. Ser. 1976. V. 150. Р. 256.
1393. Casey С. P.t Albin L. D., Burkhardt T. J.//J. Amer. Chem. Soc. 1977. V. 99..
P 2533________2539
1394. Casey С. P., Burkhardt T. J.'/J. Amer. Chem. Soc. 1974. V. 96. P. 7808—
7809.
1395. Casey С. P., Tuinstra H. E., Saeman M. С.ЦЗ, Amer. Chem. Soc. 1976.
V. 98. P. 608—609.
1396. Wang J.-L., Menapace H. R.//J, Org. Chem. 1968. V. 33. P. 3784.
1397. Katz T. J., McGinnis J.HL Amer. Chem. Soc. 1975. V. 97. P. 585.
1398. Puddephatt R. J., Quyser M. A., Tipper C. F. Н.ЦЗ. Chem. Soc. Chem. Comm.
1976. P. 626—627.
1399. Jacobson D. B., Freiser B. S.//J. Amer. Chem. Soc. 1985. V. 107. P. 2605—
2612; P. 4373—4378.
1400. Смирнов С. А.//Докл. АН СССР. 1978. T. 239. C. 392.
1401. Gassman P. G., Johnson T. Н.ЦЗ. Amer. Chem. Soc. 1976. V. 98. P. 6055—
6059.
1402. Mortreux A., Blanchard M.//J. Chem. Soc. Chem. Comm. 1974. P. 786—787.
1403. Теддер Дж., Нехватал А., Джубб А.//Промышленная органическая хи-
мия: Пер. с англ. М.: Мир, 1977. С. 121.
1404. Жоров Ю. М. //Термодинамика химических процессов. М.: Химия, 1985.
С 219
1405. Levisalles J., Willemin Z).//Tetrahedron. 1980. V. 26. Р. 3181.
1406. Вдовин В. М., Беспалова Н. Б., Бовина М. А.//Изв. АН СССР. Сер. хим.
1986. С. 2618.
1407. Ellison A., Coverdale А. К., Dearing Р. F.HL Mol. Catal. 1985. V. 28..
Р. 141.
1408. Финкельштейн Б. Ш., Стрельчик Б. С., Заикин В. Г., Вдовин В. М.Ц
Нефтехимия. 1975. Т. 15. С. 667—672.
1409. Коршак Ю. В., Варданян Л. М., Долгоплоск Б. А.//Докл. АН СССР. 1973.
Т. 208. С. 1138.
1410. Katz Т. J., McGinnis J., Altus C.//J. Amer. Chem. Soc. 1976. V. 98. P. 606—
608.
1411. Lee S. J., McGinnis J., Katz T. J.//J. Amer. Chem. Soc. 1976. V. 98.
P. 7818—7819.
1412. Katz T. J., Lee S. J., Aston A.//Tetrahedron Lett. 1976. P. 4247.
1413. Кропачева E. Я.//Докл. АН СССР. 1970. T. 195. C. 1388.
1414. Tленкопачев M. А., Копьева И. А., Бычкова H. А., Коршак Ю. В. и dp.ff
Докл. АН СССР. 1976. Т. 277. С. 889.
1415. Коршак Ю. В., Маковецкий К. Л., Долгоплоск Б. А. и др.//Докл. АН
СССР. 1976 Т. 224. С. 1344.
1416. Кропачева Е. Я.//Докл. АН СССР. 1972. Т. 206. С. 872.
1417. Pampas G., Witte J., Hoffmann M.//Rev. gen. caoutch. et plast. 1970.
V. 47. P. 1343.
1418. Маковецкий К. А., Редькина Л. И.ЦДокл. АН СССР. 1976. Т. 231. С. 131.
1419. Of stead Е. A., Calderon A.//Macromol. Chem. 1972. V. 154. Р. 21.
1420. Wolovsky R./IL Amer. Chem. Soc. 1970. V. 92. P. 2132—2133.
1421. Ben-Efraim D. A., Batich C., Wasserman Е.ЦЗ. Amer. Chem. Soc. 1970.
V. 92. P. 2133—2135.
1422. Выдрина T. К.//Изв. АН СССР. Сер. хим. 1977. C. 244.
1423. Иоффе А. И., Охрименко H. И., Колесников С. П., Нефедов О. М.//Изв.
АН СССР. Сер. хим. 1977. С. 1936.
1424. Коротнева Л. А., Белоновская Г. П., Долгоплоск Б. А.ЦДокл. АН СССР.
1972. Т. 207. С. 899.
300
1425. Колесников С. П., Поварова Н. И., Штейншнейдер А. Я-, Нефедов О. М.Ц
Изв. АН СССР. Сер. хим. 1979. С. 2398.
1426. Иванский В. И. Катализ в органической химии. Л.: Изд. ЛГУ, 1985.
С. 104.
1427. Muetterties Е. L., Stein /.//Chem. Rev. 1979. V. 79. Р. 479.
1428. Brady R. C.f Pettit R.//J. Amer. Chem. Soc. 1980. V. 102. P. 6181—6182;
1981. V. 103. P. 1287—1289.
1429. Osterloh W. T., Cornell M. E., Pettit RJ/J. Amer. Chem. Soc. 1982. V. 104.
P. 3759—3761.
1430. Leconte M., Theolier A., Rojas D., Basset J. MJ/J. Amer. Chem. Soc. 1984.
V. 106. P. 1141 — 1142.
1431. Green M. L. //.//Pure a. Appl. Chem. 1978. V. 50. P. 27.
1432. Foger K-, Anderson J. PJ/J. Catal. 1978. V. 54. P. 318.
1433. Dartigues J.-M., Chambellan A., Gault F. GJ/J. Amer. Chem. Soc. 1976.
V. 98. P. 856—857.
1434. Irvin K. J-, Rooney J. J., Stewart C. D. e. aJ/J. Chem. Soc. Chem. Comm.
1978. P. 604—606.
1435. Sharpies K. B.t Teranishi A. Y., Backvall J.-EJ/J. Amer. Chem. Soc. 1977.
V. 99. P. 3120—3128.
1436. Sharpless К. B., Umbriet M. A., Nieh M. T., Flood T. CJ/J. Amer. Chem.
Soc. 1972. V. 94. P. 6538—6540.
1437. Mintsa-Eya V., Hilaire L., Choplin A. e. aJ/J. Catal. 1983. V. 82. P. 267.
1438. Zaera F., Somorjai G. AJ/J. Amer. Chem. Soc. 1984. V. 106. P. 2288—
2293.
1439. Demuth J. EJ/Swrt. Sci. 1980. V. 93. P. L82.
1440. Salmeron M., Somorjai G. A//J. Phys. Chem. 1982. V. 86. P. 341.
1441. Shilov A. E., Shteinman A. A.//C,Oord. Chem. Rev. 1977. V. 24. P. 97.
1442. Lebrilla C. B., Maier W. FJ/J. Amer. Chem. Soc. 1986. V. 108. P. 1606—
1616.
1443. Paquette L. A.//Acc. Chem. Res. 1971. V. 4. P. 280—287.
1444. Greenberg A., Liebman J. F. Strained Organic Molecules. N.-Y.: Acad.
Press, 1978. 220 p.
1445. Saegusa T., Ho /.//Synthesis. 1975. P. 291.
1446. Ito Y., Hirao T., Saegusa TJ/J. Organomet. Chem. 1974. V. 82. P. C47.
1447. Katz T. J., Sivavec T. MJ/J. Amer. Chem. Soc. 1985. V. 107. P. 737—738.
1448. Wilson R. B., Laine R. MJ/J. Amer. Chem. Soc. 1985. V. 107. P. 361 —
369.
1449. Adams R. D., Kim H.-S., Wang SJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 6107—6108.
1450. Jolly P. W., Pettit RJ/J. Amer. Chem. Soc. 1966. V. 88. P. 5044—5045.
1451. Brandt S., Helquist PJ/J. Amer. Chem. Soc. 1979. V. 101. P. 6473—6475.
1452. Kegley S. E., Brookhart M., Husk G. R.//Organometallics. 1982. V. 1.
P. 760.
1453. Brookhart M., Tucker J. R., Husk G. RJ/J. Amer. Chem. Soc. 1983. V. 105.
P. 258—264.
1454. Kremer К. A. M., Helquist P., Kerber R. CJ/J. Amer. Chem. Soc. 1981.
V. 103. P. 1862—1864.
1455. Casey С. P., Polochnowski S. W., Shusterman A. J., Jones C. RJ/J. Amer.
Chem. Soc. 1979. V. 101. P. 7282—7292.
1456. Brookhart M., Humphrey M. B., Kratzer H. J., Nelson G. OJ/J. Amer.
Chem. Soc. 1980. V. 102. P. 7802—7803.
1457. Kuo G. H., Helquist P., Kerber R. C.//Organometallics. 1984. V. 3. P. 806.
1458. Casey С. P., Miles W. H., Tukada HJ/J. Amer. Chem. Soc. 1985. V. 107.
P. 2924—2931.
1459. Casey С. P., Brunsvold HJ/J. Organomet. Chem. 1975. V. 102. P. 175.
1460. Wulff W. D., Gilbertson S. RJ/J. Amer. Chem. Soc. 1985. V. 107. P. 503—
505.
301
1461. Tang P.-С., Wulff W. D.//J. Amer. Chem. Soc. 1984. V. 106. P. 1132—1133;
Wulff W. D., Yang D. C.//Ibid. P. 7565—7567.
1462. Dotz К. H., Kuhn W.flAngew. Chem. Int. Ed. 1983. V. 22. P. 732.
1463 Wulff W. D., Kaesler R. W., Peterson G. A., Tang Р.-С.ЦУ Amer. Chem.
Soc. 1985. V. 107. P. 1060—1062.
1464. Yamashita A.//J. Amer. Chem. Soc. 1985. V. 107. P. 5823—5824.
1465. Тихонова P. А., Кислякова H. А.//Хим. пром, за рубежом. 1973. С. 54.
1466. Chem. Econ. a. Eng. Rev. 1977. V. 9. N 5. P. 37.
1467. Prescott T. //.//Chem. Eng. 1977. V. 28. N 3. P. 63.
1468. Романюк И. М.//Хим. пром. 1977. С. 254.
1469. Федосеева Т. В.ЦХтш.. высок, энерг. 1970. Т. 4. С. 268.
1470. Рогайлин М. И.ЦХт. тверд, топлива. 1977. № 4. С. 64.
1471. Витвицкий А. И.ЦДркл. АН СССР. 1968. Т. 183. С. 856.
1472. Витвицкий А. И.ЦУсп.. химии. 1971. Т. 40. С. 1879.
1473. Гутор И. ЛЕ//Докл. АН УССР. Б. 1977. С. 910.
1474. Russell R. L., Rowland F. S.//J. Amer. Chem. Soc. 1968. V. 90. P. 1671 —
1673.
1475. Gonzaler Fl.-N., Diamy A.-M., Ben-Aim R. ///J. chim. phys. et phys.-chim.
biol. 1977. V. 74. P. 253.
1476. Alcock W. G. e. a./ICan. J. Chem. 1972. V. 50. P. 3813.
1477. Heinrich G.HBzr. Kernforschungsanlage Tillich. 1966. B. 1338. S. 121.
1478. Мухина T. H., Меньшиков В. А., Барабанов H. Л.//Ж. ВХО им. Д. И. Мен-
делеева. 1977. Т. 22. С. 8.
1479. Plonka J. Н., Skell Р. S.//Tetrahedron Lett. 1970. Р. 4557.
1480. Skell Р. S., Plonka J. //.//Tetrahedron. 1972. V. 28. P. 3571.
1481. Skell P. S., Fagone F. A., Klabunde K. ЕЦУ Amer. Chem. Soc. 1972. V. 94.
P. 7862—7866.
1482. Skell P. S., Villaume J. E., Fagone F. A.//J. Amer. Chem. Soc. 1972. V. 94.
P. 7866—7867.
1483. Пат. 2407717 США, МКИ2 С 07. Aviation fuel.
1484. Шепард У., Шартс Ш. Органическая химия фтора: Пер. с англ./Под ред.
И. Л. Кнунянца. М.: Мир, 1972. С. 206.
1485. Гудлицкий М.//Химия органических соединений фтора. М.: Госхимиздат,
1961. С. 238, 297.
1486. РЖХим. 1966. 11Н22П.
1487. С. А. 1965. V. 62. 2707.
1488. С. А. 1967. V. 64. 12547.
1489. Осипова Л. В., Новикова Ю. А.//Хим. пром, за рубежом. 1972. С. 3.
1490. Таррант Н., Стамп Б.//Ж- ВХО им. Д. И. Менделеева. 1970. Т. 15. С. 34.
1491. Dronnan G. A., Matula Р. А.ЦУ Phys. Chem. 1968. V. 72. Р. 3462.
1492. Пат. 2773089 США, МКИ2 С 07с. Fluorinated olefinic nitriles.
1493. Platonov V. E., Yakobson G. (/.//Synthesis. 1976. P. 374.
1494. Политанский С. Ф.//Кинет, и катализ. 1969. T. 10. С. 500.
1495. Dasey J. R., LiAler J. (/.//Can. J. Chem. 1969. V. 47. P. 3871.
1496. Ловлейс А., Роуч Д., Постельнек У. Алифатические фторсодержащие со-
единения: Пер. с англ./Под ред. И. Л. Кнунянца. М.: Издатинлит. 1961.
С. 111.
1497. Пат. 3009966 США, МКИ2 С 07с. Pyrolysis of fluoroform.
1498. Pass GJ/У Chem. Soc. 1965. P. 824—826.
1499. Пат. 3073870 США, МКИ2 С 07с; 12о. Pyrolysis of CHC1F2 and CH3C1
to prepare CH2 : CF2.
1500. A. c. 170962 СССР, МКИ2 С 07c; 12o, 19o2- Способ получения хлортри-
фторэтилена и тетрафторэтилена.
1501. РЖХим. 1967. 23Н35П.
1502. Пат. 3489807. США, МКИ2 С 07с. Phenyltrifluoroethylene preparation.
1503. РЖХим. 1965. 17Н8П.
1504. РЖХим. 1966. 5Н86П.
302
1505. А. с. 455951 СССР, МКИ2 С 07с 121/30. Способ получения р, ^-дифтор-
хлоракрилонитрила.
1506. РЖХим. 1962. 10Л42П.
1507. Нефедов О. М., Ивашенко А. А.//Изв. АН СССР. Сер. хим. 1968. С. 446.
1508. А. с. 277759 СССР, МКИ2 12о 2/01. Способ получения фторароматиче-
ских соединений.//Открытия. Изобретения. 1970. № 25.
1509. Нефедов О. М., Ивашенко А. А.//Изв. АН СССР. Сер. хим. 1968. С. 1417.
1510. А. с. 279608 СССР, МКИ2 12о 2/01. Способ получения ароматических
фторидов.//Открытия. Изобретения. 1970. № 27.
1511. А. с. 214530 СССР, МКИ2 12о, 2/01. Способ получения ароматических
фторидов.//Открытия. Изобретения. 1968. № 12.
1512. А. с. 339535 СССР, МКИ2 С 07с 19/08. Способ получения 2-фторнафтали-
на или его производных.//Открытия. Изобретения. 1972. № 17.
1513. Сушницкий //.//Успехи химии фтора. Л.: Химия, 1970. Т. 3—4. С. 334
1514. А. с. 406820 СССР, МКИ2 С 07с 15/22. Способ получения индена или его
производных.//Открытия. Изобретения. 1973. № 46.
1515. А. с. 290900 СССР, МКИ2 С 07с 25/18. Способ получения 2,3-дифторнаф-
талина или его производных.//Открытия. Изобретения. 1971. № 3.
1516. А. с. 572446 СССР, МКИ2 С 07с 25/04. Способ получения ароматических
фторуглеводородов.//Открытия. Изобретения. 1977. № 34.
1517. Нефедов О. М., Ивашенко А. А., Политанский С. Ф.Н'Хежяя докл. III
Всес. конф, по химии фторорганич. соединений. Одесса, 1978. С. 35.
1518. Пат. 2324390 ФРГ. МКИ2 С 07с, A 01n. Cyclopropane derivatives.
1519. Arlt D., Jantelat M., Lantzsch /?.//Angew. Chem. Int. Ed. 1981. V. 20. P. 703.
P. 818.
1520. Arlt Von D., Jantelat M., Lantzsch 7?.//Angew. Chem. 1981. B. 93. S. 719.
1521. 74/02879 Нидерл., МКИ2 C 07c, A Oln. Substituted cyclopropane carbo-
xylic.
1522. Lehmktihl H„ Mehler K.//Ann. 1978. S. 1841.
1523. Дмитриева E. Ф., Домнин И. Я.//Современ. проблемы орг. химии. Л.:
Изд. ЛГУ, 1986. С. 43—75.
1524. Нефедов О. М., Шапиро Е. А., Цветков С. Ю. и др.//Химия и технология
синтетических пиретроидов и их применение в сельском хозяйстве. Тези-
сы докладов Всес. совещ. М.: ВДНХ СССР, 1984. С. 6.
1525. Справочник по пестицидам/Мелбников Н. Н., Новожилов К. В., Бе-
лан С. Р., Пылова Т. Н. М.: Химия, 1985. 351 с.
1526. Мельников Н. Н., Либман Б. Я., Дечер Р. М.//Ж. ВХО им. Д. И. Менде-
леева. 1985. Т. 30. С. 268—283.
1527. А. с. 406543 СССР, МКИ2 А61к 27/00. Лекарственное средство.//Откры-
тия. Изобретения. 1973. № 46.
1528. А. с. 477998 СССР, МКИ2 С 07с 69/74; С 07с 61/04. Способ получения
сложных эфиров циклопропанкарбоновых кислот.//Открытия. Изобрете-
ния. 1975. № 27.
1529. Пат. 3897478 США, МКИ2 С 07с 67/00; С 07с 64/174. Continuous process
for the manufacture of alkylchrysanthemate esters.
1530. Пат. 1533381 Великобрит., МКИ2 С 07с 69/74. Preparation of esters of
cyclopropanecarboxylic acids.
1531. Пат. 1459289 Великобрит., МКИ2 С 07с. Preparation of esters of cyclopro-
panecarboxylic acids.
1532. A. c. 576313 СССР, МКИ2 С 07c 61/38. Способ получения 2-винилцикло-
пропанкарбоновой кислоты или ее эфиров.//Открытия. Изобретения. 1977.
№ 38.
1533. А. с. 523078 СССР, МКИ2 С 07с 69/52. Способ получения эфиров бутин-3-
овой кислоты//Открытия. Изобретения. 1978. № 28.
1534. Hocks L.//Bull. Soc. chim. France. 1975. P. 1893.
1535. Calderon N., Ofstead E. A., Judy W. A.//Angew. Chem. 1976. B. 88.
S. 433—442.
1536. Streck 7?.//Chem. Ztg. 1975. B. 99. S. 397.
303
1537. Долгоплоск Б. А.//Усп. химии. 1977. Т. 46. С. 2027.
1538. Calderon N.//J. Macromol. Sci. 1972. V. С7. Р. 105.
1539. Janowski F.//Chem. Techn. (BRD). 1977. V. 29. P. 313.
1540. Чаманов E. M.//Катализаторы, содержащие нанесенные комплексы. Но-
восибирск: Наука, 1977. С. 231.
1541. Hydrocarbon Process. 1967. V. 46. Р. 232.
1542. Stern Н. №.//Erd61 u. Kohle-Erdgas-Petrochem. 1975. В. 26. S. И.
1543. Freitas E. R.f Gum C. R.//Chem. Eng. Progr. 1979. V. 75. P. 73.
1544. Appl. Catal. 1986. V. 28. P. 324.
1545. Banks R. L., Regier R. B.//Int. Eng. Chem. Prod. Res. Dev. 1971. V. 10.
P. 46.
1546. Banks R. L.//J. Molec. Catal. 1980. V. 8. P. 269—276.
1547. Rossi R.//Chim. et Ind. (Milan). 1975. V. 57. P. 242.
1548. Kupper F. W., Streck R.//Chem. Ztg. 1975. B. 57. S. 464.
1549. Kupper F. W., Streck R.HZ. Naturforsch. 1976. B. 31B. S. 1256.
1550. C. A. 1977. V. 87. 22390.
1551. C. A. 1977. V. 86. 155197.
1552. Le Delliou P.//Rev. Gen. Caout. Plast. 1977. V. 54. P. 77.
1553. Draxler A.//Kautsch. Gummi. Kunstst. 1980. B. 81. S. 185.
1554. Mateika L., Houtman C., Macosko C. W.HJ. Appl. Polym. Sci. 1985. V. 30.
P. 2787—2790.
1555. Фелъдблюм В. Ш.1ГЖ- BXO им. Д. И. Менделеева. 1977. Т. 22. С. 23.
1556. Dragutan V., Balaban А. Т., Dimonie М. Olefin Metathesis and Ring-Ope-
ning Polymerisation of Cycloolefines. Editura Academici Bucuresti—J. Wi-
ley, 1985. 544 p.
1557. Kim L., Raley J. H., Bell C. S.//Proc. Int. Symp. on Metathesis. 1977. V. 96.
P. Ml36.
Научное издание
Нефедов Олег Матвеевич,
Иоффе Александр Исаакович,
Менчиков Леонид Геннадьевич
химия карбенов
Редактор М. Н. Пастушенко
Художник Е. В. Бекетов
Художественный редактор К- К. Федоров
Технический редактор Е. Н. Крумштейн
Корректор М. В. Черниховская
ИБ № 2227
Сдано в набор 15.02.90. Подписано в печать 25.07.90. Т-10183.
Формат бОХЭО’Лб. Бумага тип. № 1. Гарнитура Литератур-
ная. Печать высокая. Усл. печ. л. 19,0. Усл. кр.-отт. 19,0.
Уч.-изд. л. 21,54. Тираж 4370 экз. Заказ 107. Цена 4 р. 60 к.
Ордена «Знак Почета» издательство «Химия», 107076, Моск-
ва, Стромынка, 21, корп. 2.
Московская типография № И Государственного комитета
СССР по печати. 113105, Москва, Нагатинская ул., д. 1.
Рецензент: д-р хим. наук Р. Р. Костиков
УДК 547.0
Химия карбенов/О. ЛК Нефедов, А. И. Иоффе, Л. Г. Мен-
чиков. — М.: Химия, 1990. — 304 с. — ISBN 5—7245—0568.
Первая отечественная монография по химии карбенов — соединений
двухвалентного углерода, являющихся высокореакционноспособными
промежуточными частицами. Рассмотрены строение и реакционная спо-
собность карбенов и их ближайших аналогов, использование карбенов
и продуктов их реакций в тонком и промышленном органическом син-
тезе. Описаны типовые методики.
Для физикохимиков и инженеров-технологов, работающих в области
органического синтеза и смежных областях. Может быть полезна пре
подавателям, аспирантам и студентам вузов.
Табл. 17, ил. 5. Библиогр : 1557 назв.
„ 1705000000—001
X-------------1—90
050(01)—90
ISBN 5—7245—0568
© О. М. Нефедов, А. И. Иоффе,
Л. Г. Менчиков, 1990