/

























































































































































































































































































































































































































































































































































































































































































Author: Зеленин К.Н.
Tags: высшее образование университеты академическое обучение медицина химия медицинская химия
ISBN: 5-87685-109-4
Year: 1997




Text
К.Н. ЗЕЛЕНИН
ХИМИЯ
Учебник для медицинских вузов
Рекомендован экспертно-редакционным издательским
советом Военно-медицинской академии
Санкт-Петербург
"Специальная Литература”
1997
УДК 37»
61. 54
3 48
Рецензенты
проф А. И. Карпишенко (Военно-медицинская академия),
проф Б А Чакчир (Санкт-Петербургская химико-фармацевтическая академия)
Зеленин К. Н.
3 48 Химия: Учебник для медицинских вузов.— СПб: «Специальная Литера¬
тура*, 1997 —688 с.
В учебнике в ясной и доступной форме изложены основные понятия, законы
и методы современной химии в приложении к медико-бнологическим проблемам
Книга содержит множество примеров, заимствованных из фактического материала
биохимии, молекулярной биологии, биоиеорганической, биофизической и биоорга-
нической химии
Подход, лежащий в основе учебника, ориентирован на снятие традиционного
разделения курса на неорганическую, органическую, аналитическую, физическую и
коллоидную химию, и предназначен для формирования единого химического ми¬
ровоззрения, необходимого современному врачу. Задача учебника — внедрение фун¬
даментальных принципов химической науки в процесс обучения будущего врача
Его автор — заслуженный деятель науки РФ, академик РАЕН Кирилл Николаевич
Зеленин — на протяжении 25 лет заведует кафедрой химии Воеиио-медицииской
академии
Учебник соответствует программе, утвержденной Министерством здра¬
воохранения РФ, и предназначен для студентов медицинских вузов.
The basic concepts, laws and methods ot modem chemistry in the application to
medical and biological problems are stated in clear and accessible form in this textbook.
The book contains set of examples, borrowed from an actual material of biochemistry,
molecular biology, bioinoiganic, biophysical and biooiganic chemistry.
The approach, laying m the basis of the book, is orientated on the removal of borders
between unorganic, organic, physical, colloid and analitical chemistry. It is intended for
performance of common chemical outlook essential for modem physician. The aim of this
texbook is the establishment of common fundamental principles of chemical sciences in
the process of physician education. The author of this textbook, Honored scientist of
Russian Federation, member of Russian Academy of Natural Sciences Kirill Nikolae¬
vich Zelenin is a head of Department of Chemistry of Military Medical Academy during
25 years
The textbook corresponds to the program, authorized by Health Ministry of Russian
Federation and is intended for the students of medical universities.
Кни,п иаАяня »п.. поддержки науки и образования
чская книга»
^ Зеленин К Н. 1997
- © «Специальная Литература». 1997
КВЫ 5-87685-109-4 © Волошкнн О П, оформление обложки, 1997
Оглавление
Предисловие 12
Введение 14
Часть I. ФУНДАМЕНТАЛЬНЫЕ ЗАКОНЫ ХИМИИ
Глава 1. Строение атома и Периодический закон 17
1.1. Квантовомеханическое описание атома водорода 18
1.2. Строение многоэлектронных атомов 22
1.3. Периодическое сходство элементов 27
1.4. Периодическое изменение свойств элементов 30
Глава 2. Химическая связь 37
2.1. Ковалентная связь 38
2 1.1 Метод валентных связей 38
2.1.2. Гибридизация 43
2 1.3. Резонанс 48
2.1.4. Полярность 50
2.1.5 Метод молекулярных орбиталей 52
2.2. Ионная связь ... 61
2.3. Водородная связь 64
2.4. Межмолекулярные взаимодействия . . 66
2.4.1. Силы Ван-дер-Ваальса 66
2.4.2. Ионно-молекулярное взаимодействие 67
2 4.3 Гидрофобные взаимодействия .68
Глава 3. Комплексные соединения 71
3.1. Строение, классификация, номенклатура 71
3.2. Изомерия комплексных соединений . . 74
3.3. Химическая связь в комплексных соединениях .. 76
3.4. Комплексные соединения с полидентатными
лигандами ... 82
Глава 4. Общие сведения о биогенных элементах 85
Глава 5. Химическая кинетика 92
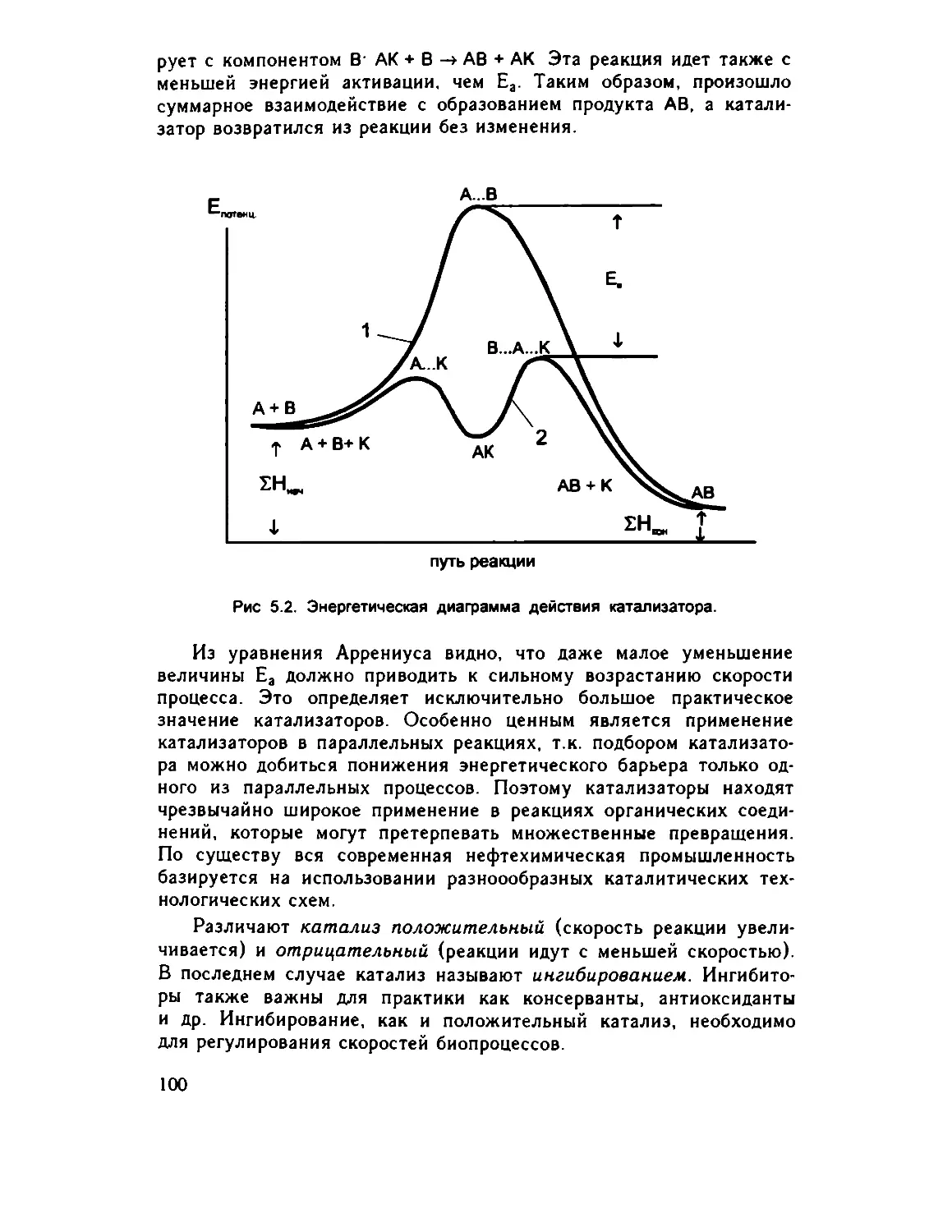
5 1. Скорость химической реакции 93
5 2 Уравнение Аррениуса Энергия активации 97
5 3. Инициация и катализ 99
5 4 Ферментативный катализ . ... 101
Глава 6. Понятие о химической термодинамике 107
6 1 Первый закон термодинамики Энтальпия . . 108
6.2. Термохимия. Закон Гесса . ... 110
3
6 3 Второй закон термодинамики. Энтропия
Свободная энергия Гиббса 113
Глава 7. Химическое равновесие ..121
7 1 Константа равновесия 121
7 2 Смещение химического равновесия 123
Часть II. РАВНОВЕСИЯ В БИОСРЕДАХ
Глава 8. Растворы 127
8 1. Вода и ее физико-химические свойства 127
8 2. Растворы неэлектролитов .. . 131
8 3 Осмос 138
8.4 Растворы электролитов 145
8.5. Водно-электролитный баланс 148
Глава 9. Кислотно-основное равновесие ... 152
9.1. Ионное произведение воды ^ 152
9.2. Диссоциация кислот и оснований 154
9.3. Вычисление pH растворов сильных кислот
и оснований 156
9.4. Вычисление pH растворов слабых кислот
и оснований 157
9^. Гидролиз солеи 159
9.6. Буферные растворы Г63"
9.6.1. Типы буферных растворов Механизм действия
и вычисление pH 164
9.6.2. Буферная емкость 167
9 6 3. Влияние разбавления на pH буферных растворов 169
9.7. Биологические буферные системы 170
9 7.1. Гидрокарбонатная (бнкарбонатная) буферная
система 170
9 7 2. Фосфатная буферная система 171
9.7.3 Белковые и аминокислотные буферные системы 172
9.7 4 Аминокислотные буферные растворы 173
9.7.5 Гемоглобиновый буферный раствор 174
9.8. Кислотно-основное состояние 176
Глава 10. Равновесие в комплексах 179
10.1. Константа нестойкости 179
10.2. Металло-лигандный гомеостаз и его нарушения . .. . 181
Глава 11. Электрохимическое равновесие. Ионометрия 184
11.1. Химическое равновесие в окислительно¬
восстановительных реакциях 184
11.2. Окислительно-восстановительные потенциалы 187
4
11.3. Классификация электродов 189
11.3.1. Металлический электрод 190
11.3.2. Водородный электрод 192
11.3.3. Каломельный электрод 193
11.3 4. Хлорсеребряный электрод 195
11.3.5. Сурьмяный электрод 196
11.3.6. Хингидронный электрод 197
11.3.7. pH-Зависимые электродные потенциалы 199
11.4. Диффузионный потенциал. Электролитический
мостик 201
11.5. Мембранный потенциал 202
11.6. Типы гальванических элементов 202
11.7. Окислительно-восстановительные реакции в
биологических процессах 206
117 1. Процессы с переносом электронов 207
11.7.2. Процессы с переносом ионов 210
11.8. Потенциометрия. Электрометрическое
измерение pH 212
11.9. Ионоселективные электроды 214
11 10. Типы ионоселективных электродов 218
11 10.1. Стеклянные электроды 218
11.10 2 Твердофазные электроды 220
11 10 3. Электроды с жидкой мембраной 221
11.10.4 Газовые, ферментные н бактериальные электроды 222
11.11. Ионометрия в медицине 223
Глава 12. Гетерогенное равновесие ... .... 226
12.1. Произведение растворимости ... .. 226
12.2. Влияние одноименного иона 229
12.3. Гетерогенные равновесия и организм 231
Часть III. МИКРОГЕТЕРОГЕННЫЕ СИСТЕМЫ
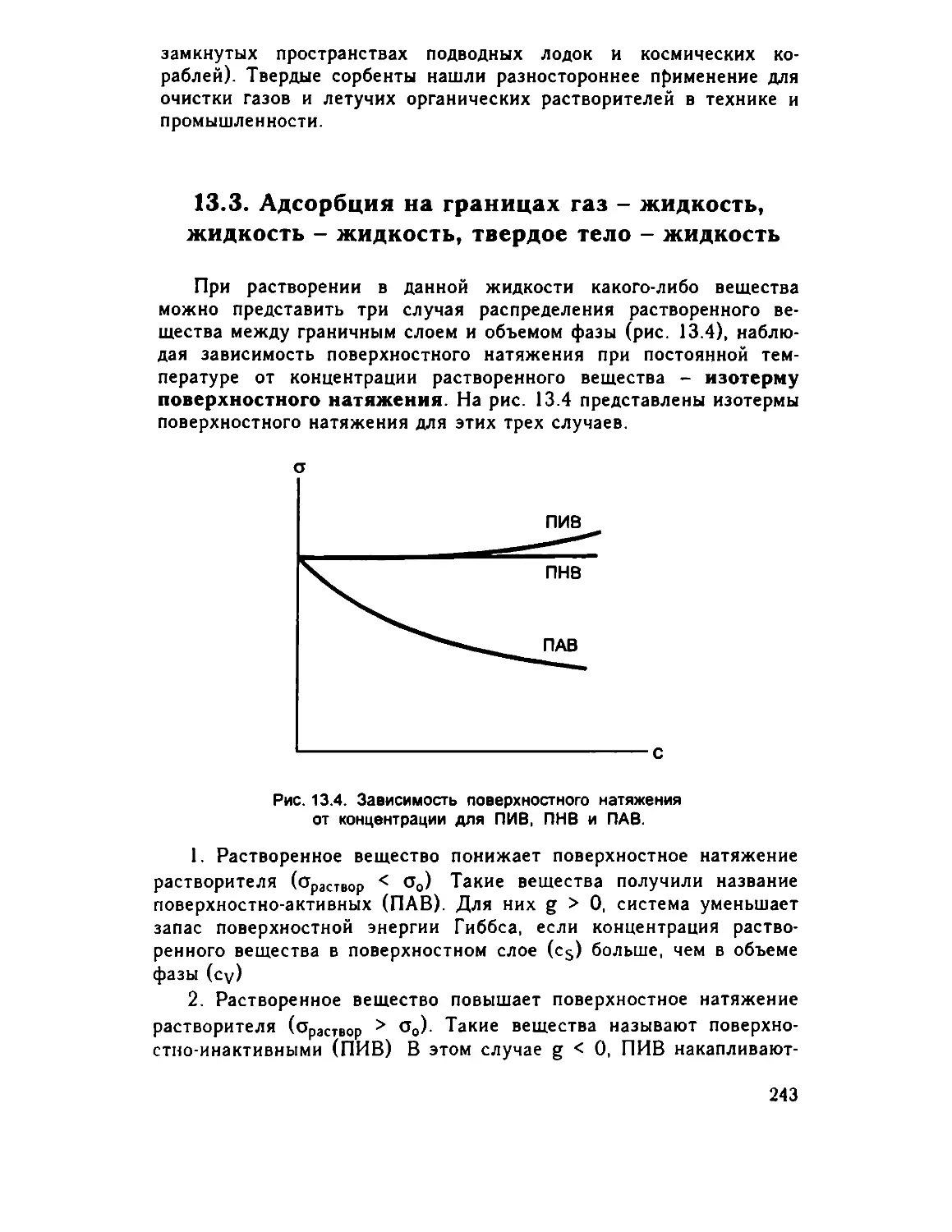
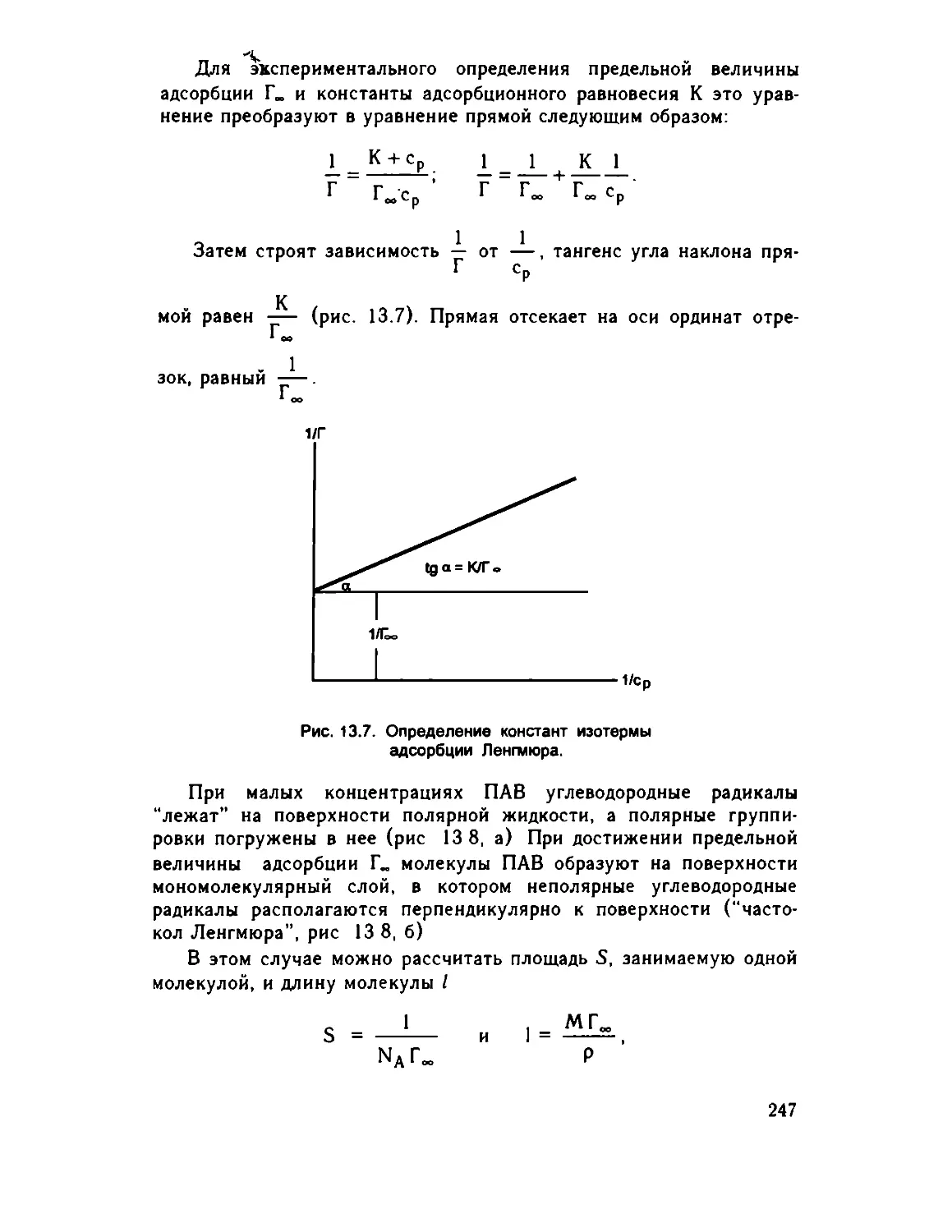
Глава 13. Физическая химия поверхностных явлений 235
13.1. Поверхностная энергия и поверхностное натяжение 235
13 2. Адсорбция на границе раздела твердое тело - газ . . 240
13 3 Адсорбция на границах газ - жидкость, жидкость -
жидкость, твердое тело - жидкость . . 243
13.4. Адсорбция на границе твердое тело - раствор . 249
13.5. Адсорбция сильных электролитов 251
13.6 Ионообменная адсорбция ... 253
5
Глава 14. Хроматографические методы 255
14 1 Тонкослойная хроматография (TCX) 256
14 2. Газо-жидкостная хроматография (ГЖХ) 259
14 2 1 Сущность метода 259
14 2 2 Техника газо-жидкостной хроматографии 260
14 2 3 Значение метода газо-жидкостной хроматографии 262
Глава 15. Химия дисперсных систем 265
15 1. Дисперсные системы 265
15 2 Способы получения коллоидных растворов .. 268
15.3. Очистка коллоидных растворов 270
15.4. Молекулярно-кинетические свойства коллоидных
растворов 272
15 4 1 Диффузия 272
15 4 2 Седиментация 274
15.5. Оптические свойства коллоидных растворов 275
15.6. Строение коллоидных частиц 276
15.7. Электрокинетические свойства коллоидных
растворов . 278
15.8 Электрокинетический потенциал 280
15 8.1 Влияние ^-потенциала на свойства коллоидных
растворов 280
15 8 2 Определение ^-потенциала методом свободного
электрофореза 281
15.9. Коагуляция 284
15.10. Коллоидная защита 287
15.11. Коллоидные растворы поверхностно-активных
веществ. Солюбилизация 288
Часть IV. ЭЛЕМЕНТЫ-ОРГАНОГЕНЫ
Глава 16. Водород 292
16.1 Разновидности водорода 293
16.2. Химические свойства 294
16.3. Катион водорода 297
16.4. Вода ... 301
16(5. Радиолиз воды ... . 303
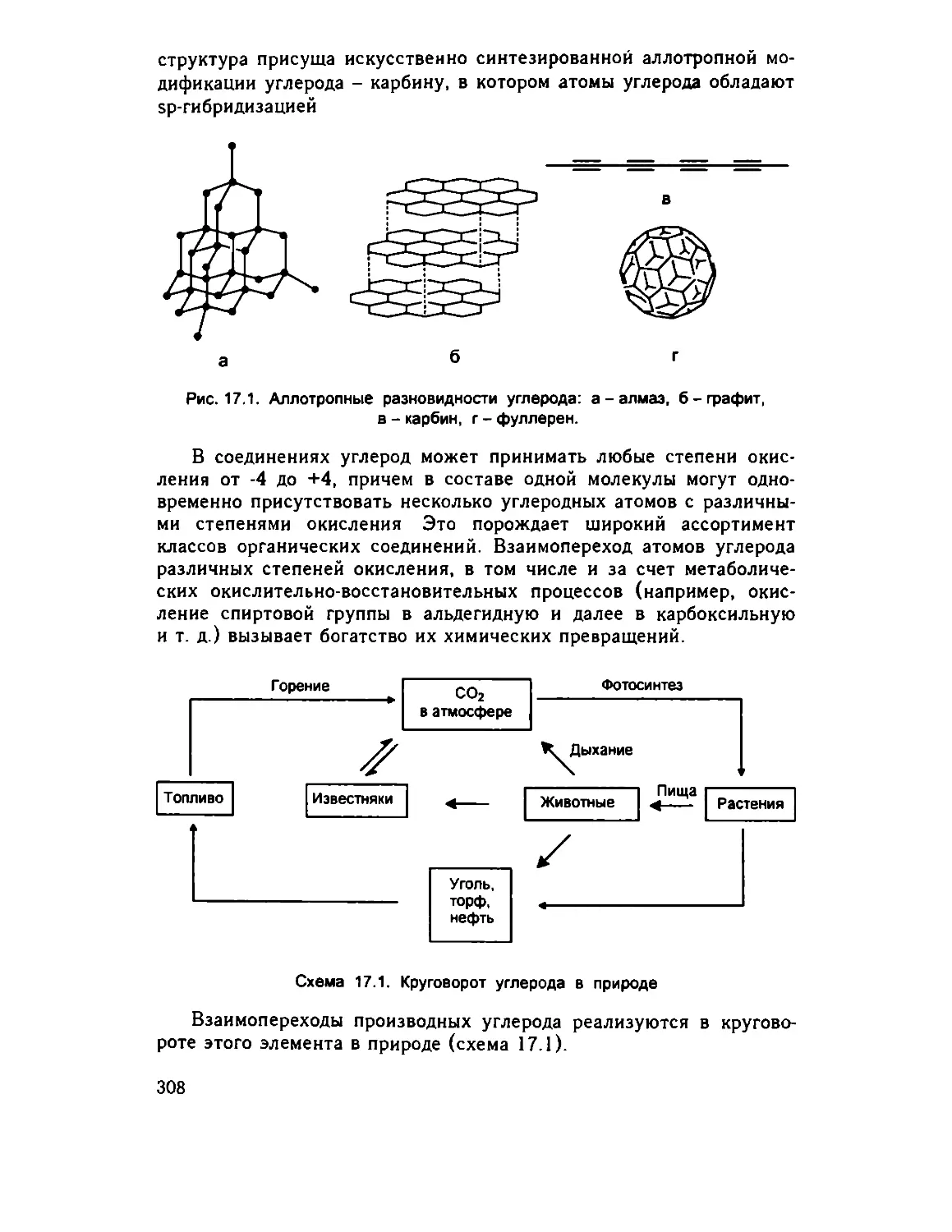
Глава 17. Углерод 306
17.1 Элемент, аллотропные модификации 306
17.2. Соединения углерода (+4) 309
17.3 Производные угольной кислоты 313
17.4. Соединения углерода (+2) 314
6
Глава 18. Углеводороды 319
18.1 Классификация 319
18.1.1. Алканы 319
18.1.2. Сведения о конформациях 320
18.1.3. Насыщенные циклические соединения 325
18.1.4. Алкены 329
18.1.5. Сопряженные алкаднены 330
18.1.6. Ароматические соединения 331
18.1.7. Алкины 334
18.2. Изомерия и номенклатура органических
соединений 334
18.2.1. Понятие о радикалах. Структурная изомерия 334
18.2.2. Оптическая изомерия 338
18.2.3 Геометрическая изомерия 342
18.2.4 Сведения о номенклатуре органических соединений 345
18.3. Свойства, получение и применение углеводородов . 347
18.3.1. Химические свойства алканов и циклоалканов . . . 347
18.3 2. Химические свойства алкенов 352
18.3.3. Химические свойства сопряженных алкадиенов 358
18.3.4 Химические свойства алкинов 361
18.3.5 Химические свойства аренов 364
18.3.6. Синтез углеводородов 378
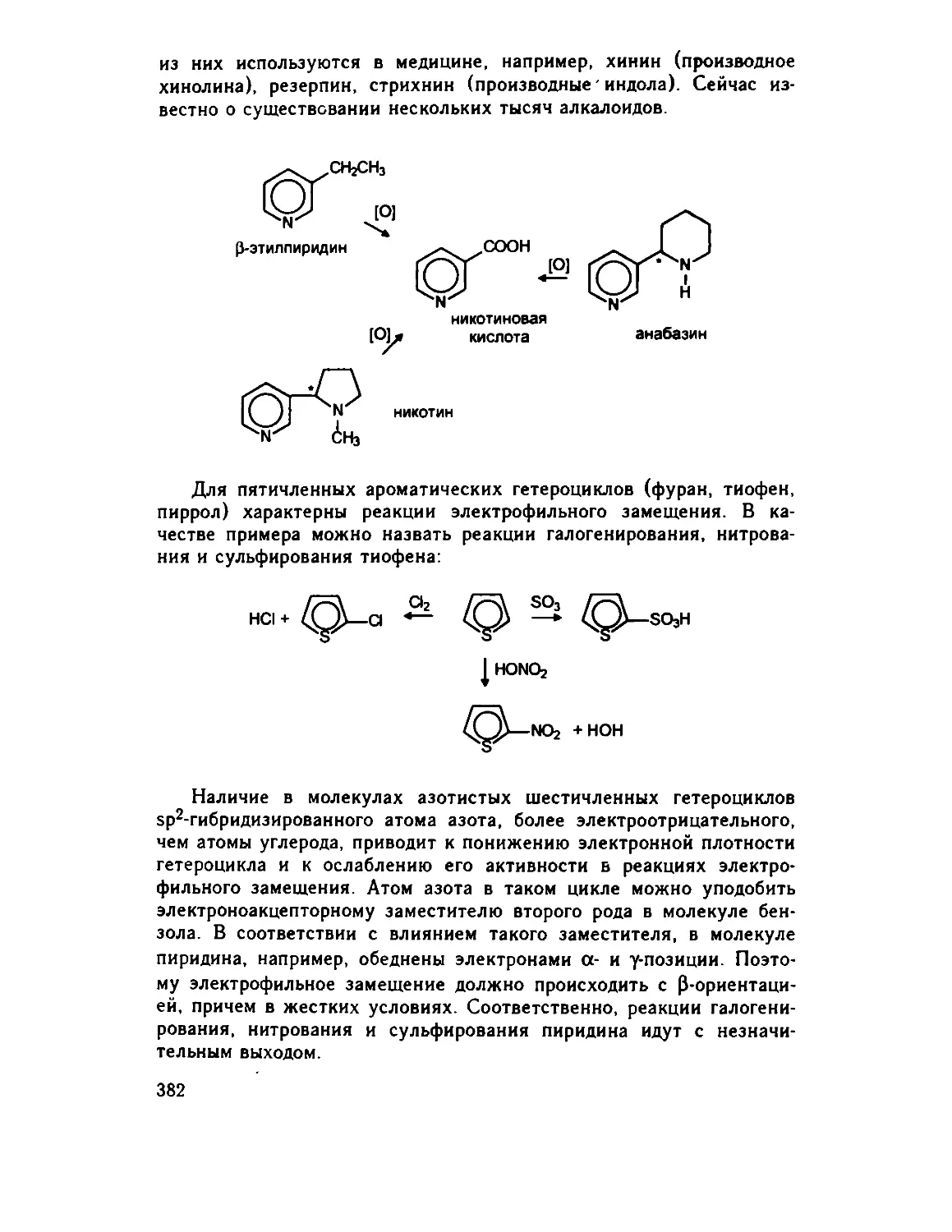
18.4. Химические свойства гетаренов 381
Глава 19. Галогены 384
19.1. Элементы, простые вещества 384
19.2. Галогенид-ионы 388
19.3. Галогенопроизводные углеводородов 392
19.4. Кислородные соединения галогенов 398
Глава 20. Кислород 403
20.1 Элемент, простое вещество 403
20 2 Супероксид-анион-радикал 407
20.3 Озон 409
20 4. Пероксиды . 411
20.5 Органические перекиси 413
20.6 Оксиды 415
20.7 Спирты, фенолы 416
20 7 1 Классификация и номенклатура 416
20 7.2. Способы получения спиртов ... .... 417
20 7 3 Свойства спиртов и фенолов 418
20 7 4 Важнейшие представители спиртов и фенолов,
их практическое значение .... 422
20.8. Простые эфиры ... 424
7
Глава 21. Сера • 428
21 1. Элемент, простое вещество 428
21 2. Соединения серы (-2) 430
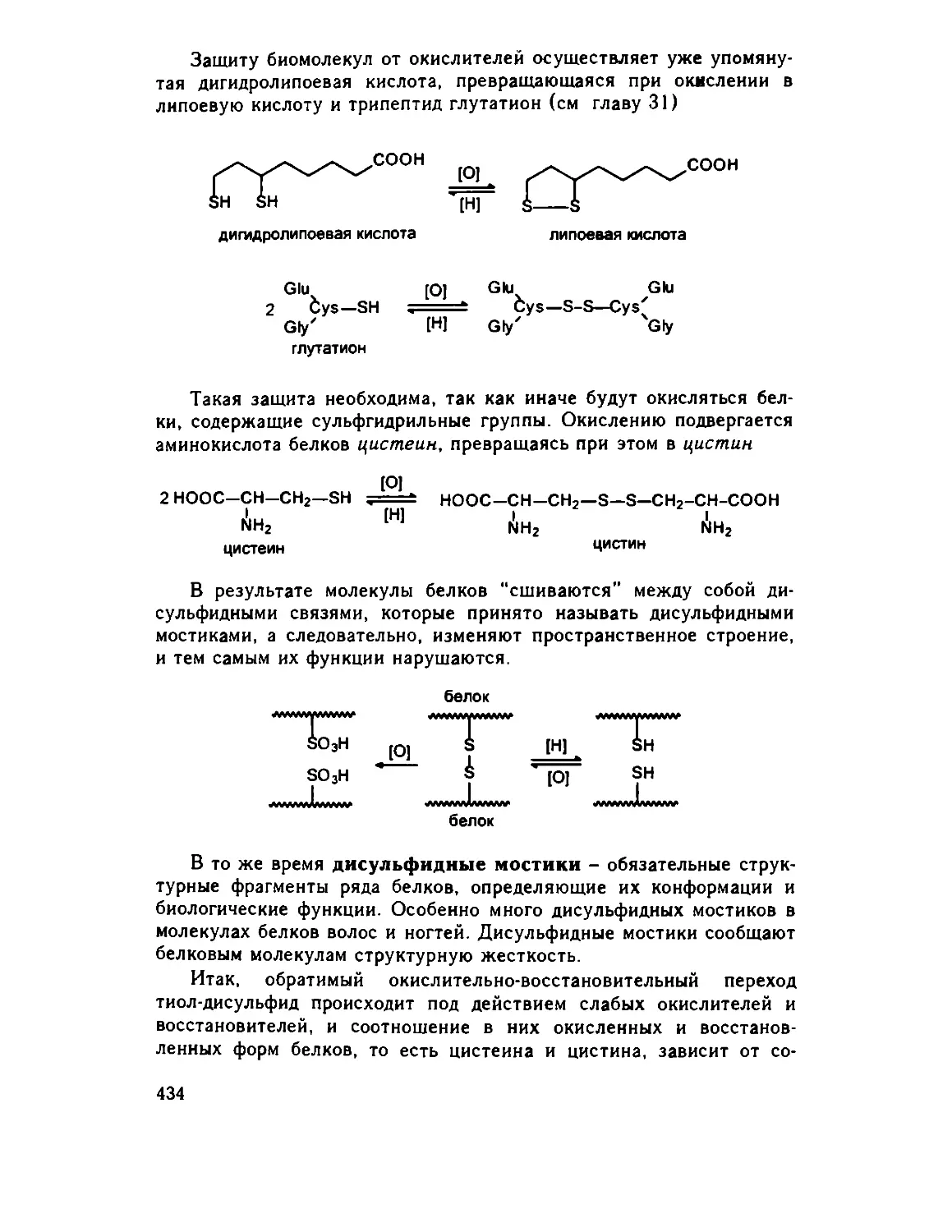
21.3 Тиолы, сульфиды 431
21 4. Соединения серы (+4) 437
21 5. Соединения серы (+6) 439
21.6. Тиосульфат-ион 442
21.7 Другие элементы 6 группы . . . 443
Глава 22. Азот • 445
22.1. Простое вещество, фиксация азота 445
22 2. Аммиак 448
22 3 Амины . .. ... . 449
22 3 1 Классификация и номенклатура 449
22 3.2. Амины как органические основания 451
22.3.3. Способы получения аминов 453
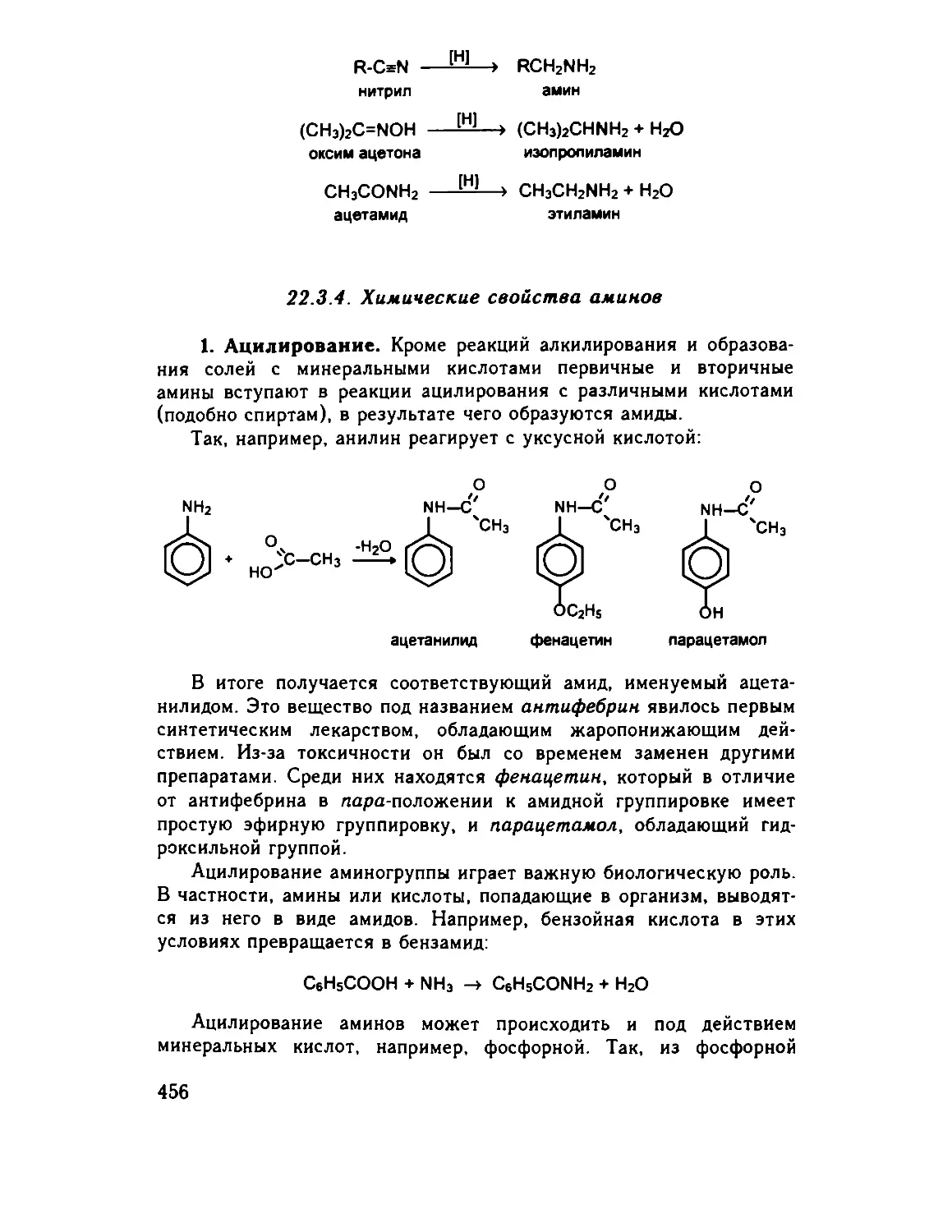
22.3.4 Химические свойства аминов 456
22.3 5. Важнейшие представители аминов и их медико¬
биологическое значение . 458
22.4. Гидразин и гидроксиламин .. 460
22.5. Нитрит-ион 461
22.6. Нитрат-ион 465
22.7 Оксиды азота 467
Глава 23. Фосфор . 469
23.1. Элемент, простое вещество 469
23.2. Фосфин, фосфиды 471
23.3. Кислородные соединения фосфора 471
23.4. Биологическая роль фосфатов .. 474
23.5. Органические производные фосфора 475
23 5 1 Получение и химические свойства 476
23 5 2 Биологическая роль органических производных
фосфорной кислоты ... 477
23 5 3 Биологическая активность алкилфосфонатов 480
23.6. Мышьяк, сурьма, висмут 483
23.6 1 Неорганические производные 483
23.6 2 Органические производные мышьяка . . . 485
Часть V. ОРГАНИЧЕСКИЕ БИОМОЛЕКУЛЫ
Глава 24. Оксосоединения
24.1. Классификация и номенклатура
24.2. Способы получения альдегидов и кетонов . .
487
487
489
8
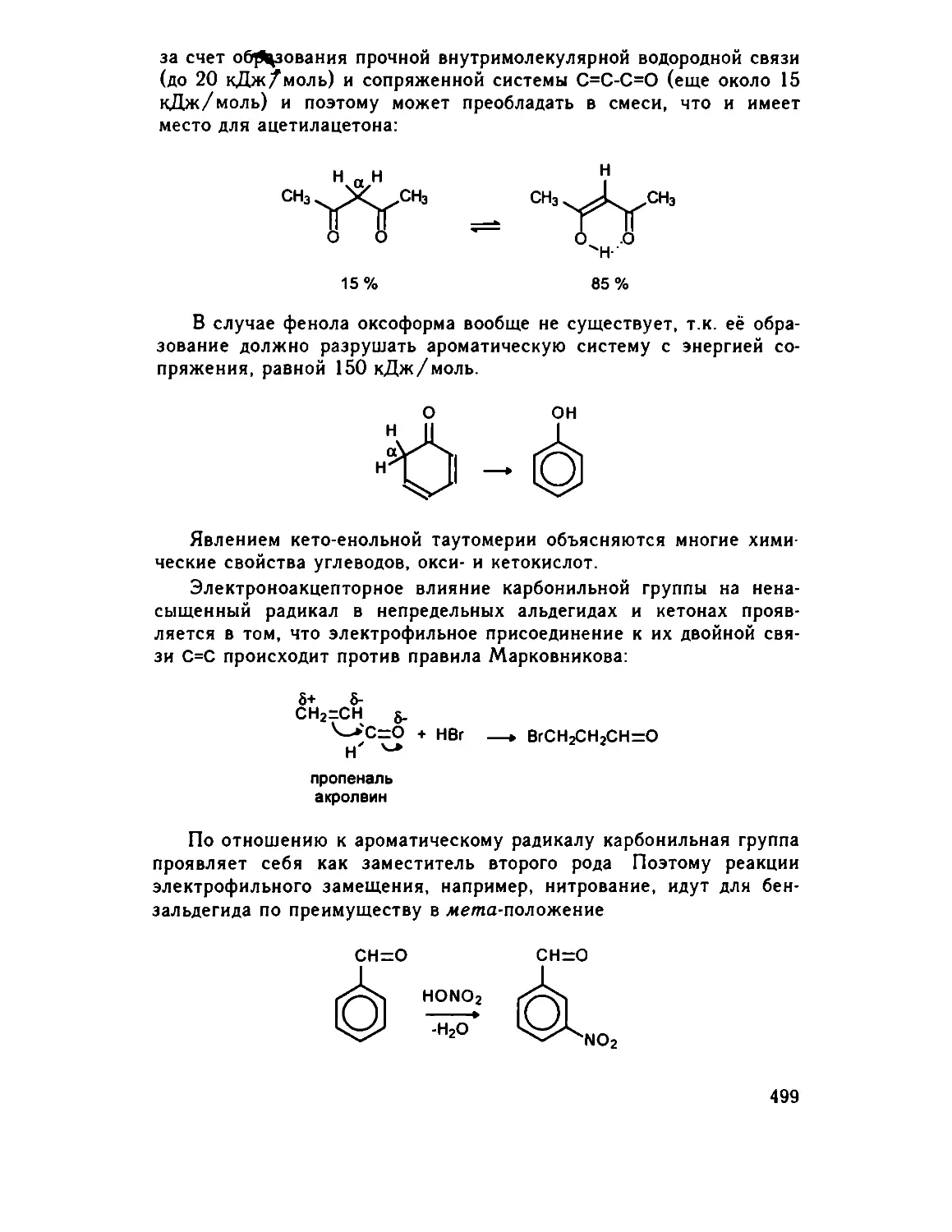
24.3. Химические свойства карбонильных соединений 491
24.3.1. Реакции присоединения карбонильной группы ... . 492
24.3.2. Реакции с участием заместителя при карбонильной
группе 498
24 3.3. Реакции полимеризации и конденсации альдегидов
и кетонов ... 500
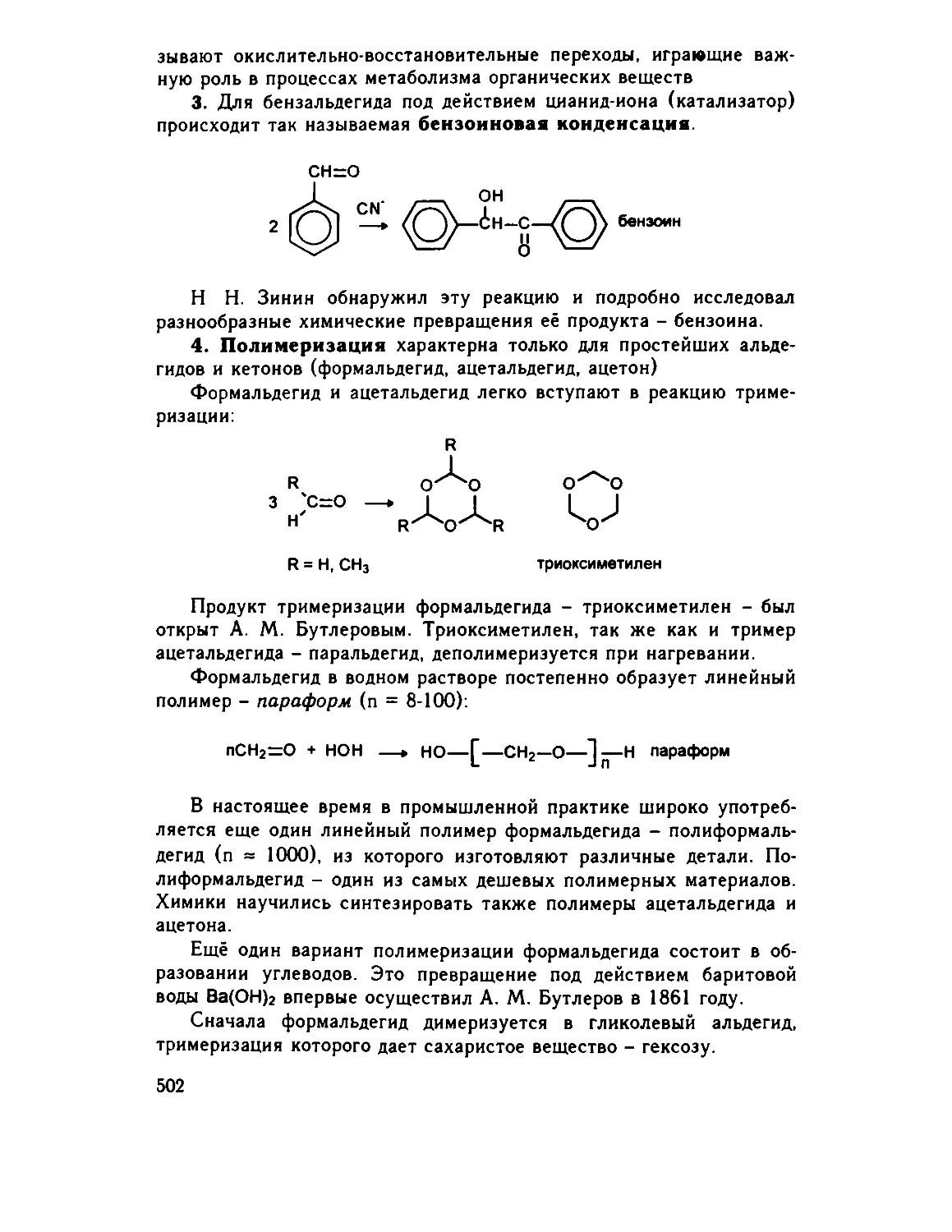
24.4. Отдельные представители карбонильных соединений 503
Глава 25. Углеводы 505
25.1. Введение 505
25.2. Моносахариды 506
25.2.1. Синтез Фишера-Килиани .... .... 507
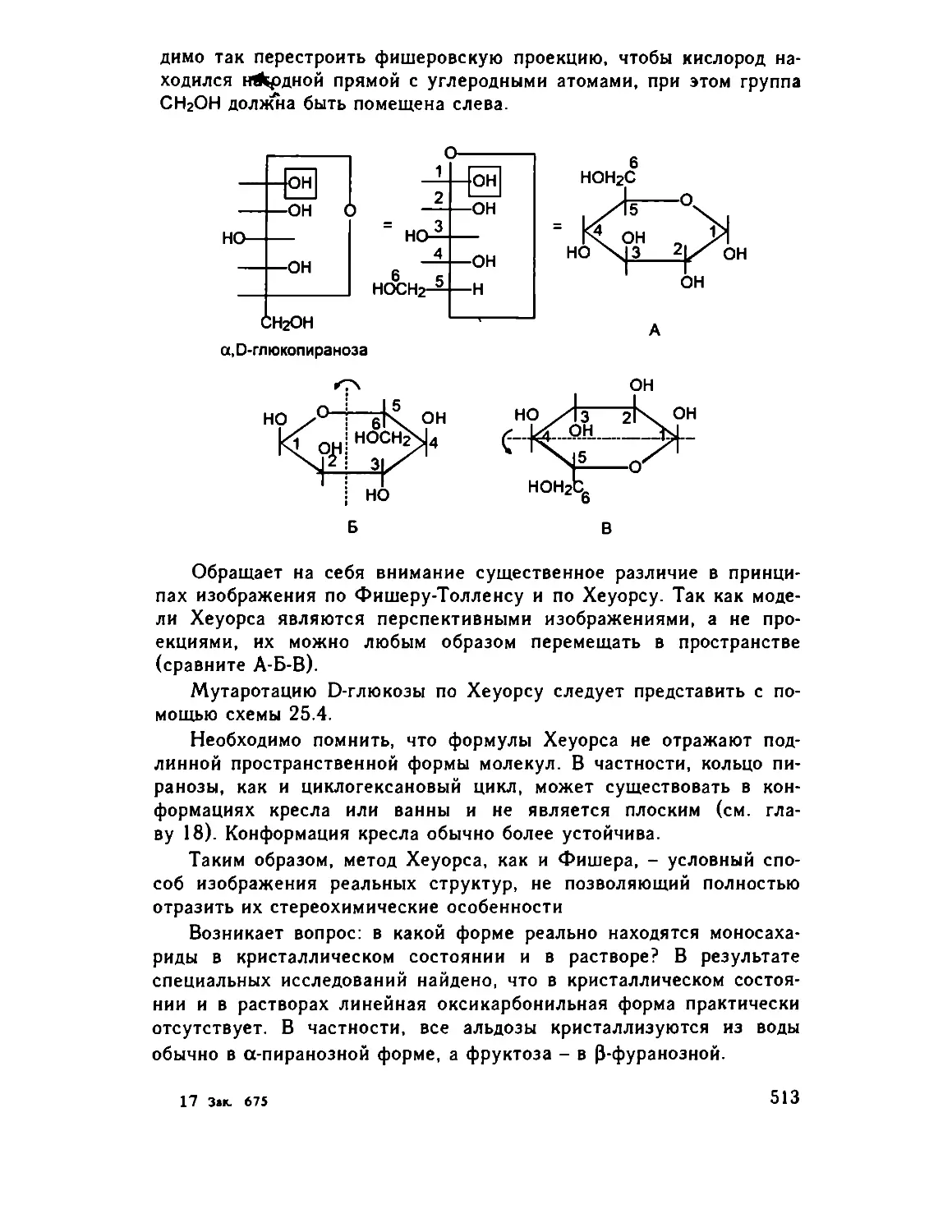
25.2.2. Свойства моносахаридов. Мутаротация .. 511
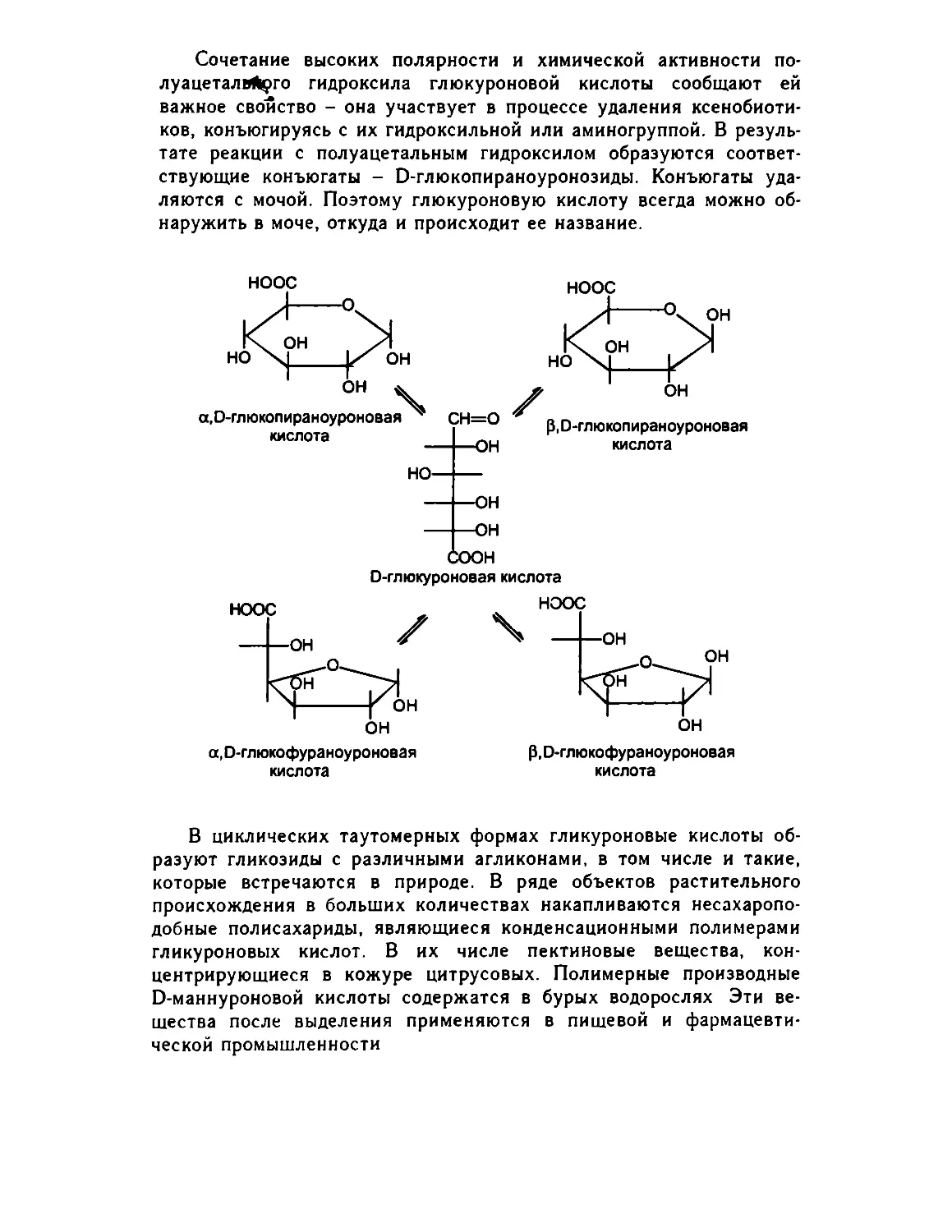
25.2.3. Реакции с участием гидроксильных групп .. 515
25 2.4 Реакции с участием карбонильной группы . . . 519
25.3. Сахароподобные полисахариды (дисахариды) 526
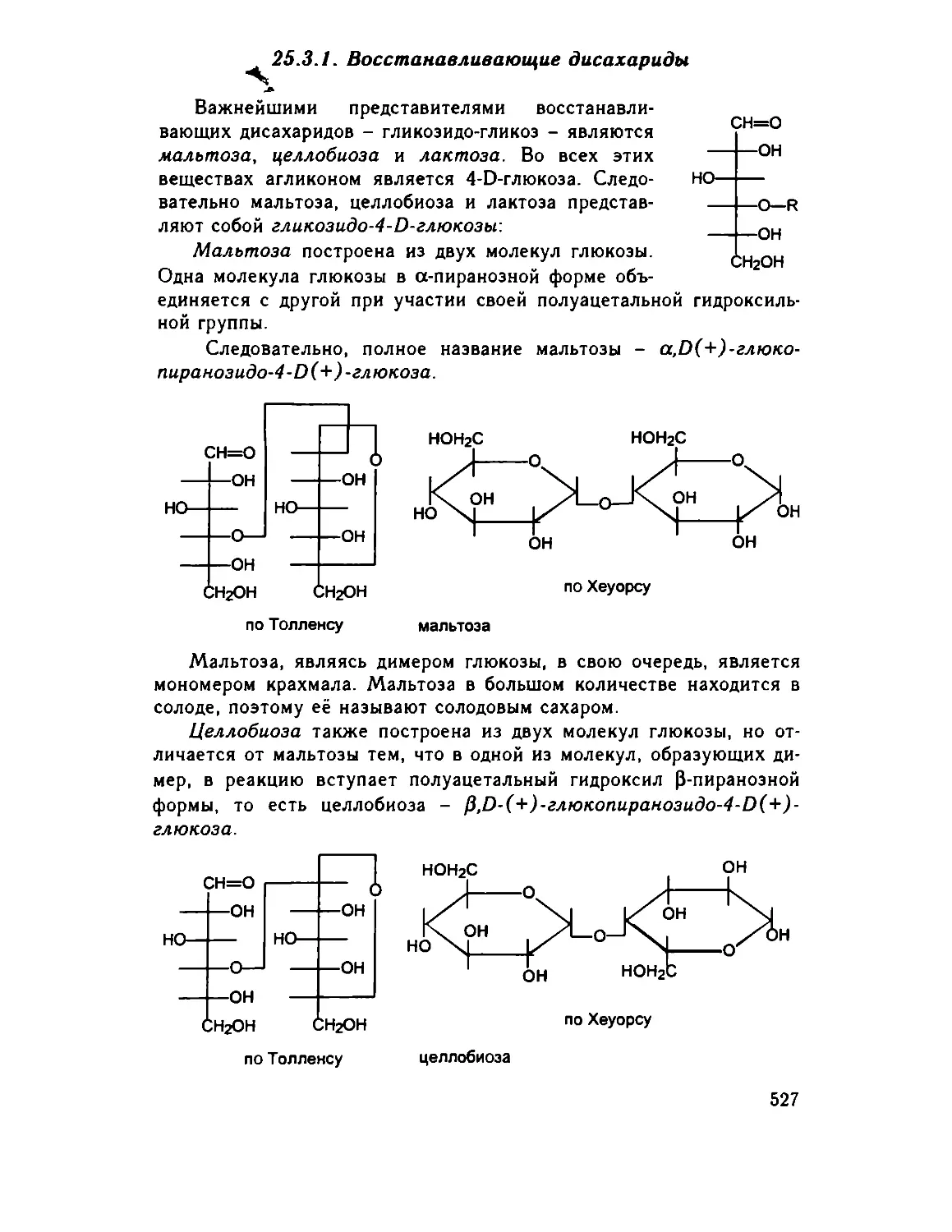
25.3.1. Восстанавливающие дисахариды 527
25.3.2. Химические свойства 528
25.3.3. Невосстанавливающие дисахариды (сахароза) . 530
25.4. Несахароподобные полисахариды 533
25.4.1. Гомополисахариды 533
25.4.2. Гетерополисахариды 538
25.5. Заключение 539
Глава 26. Карбоновые кислоты и их производные 541
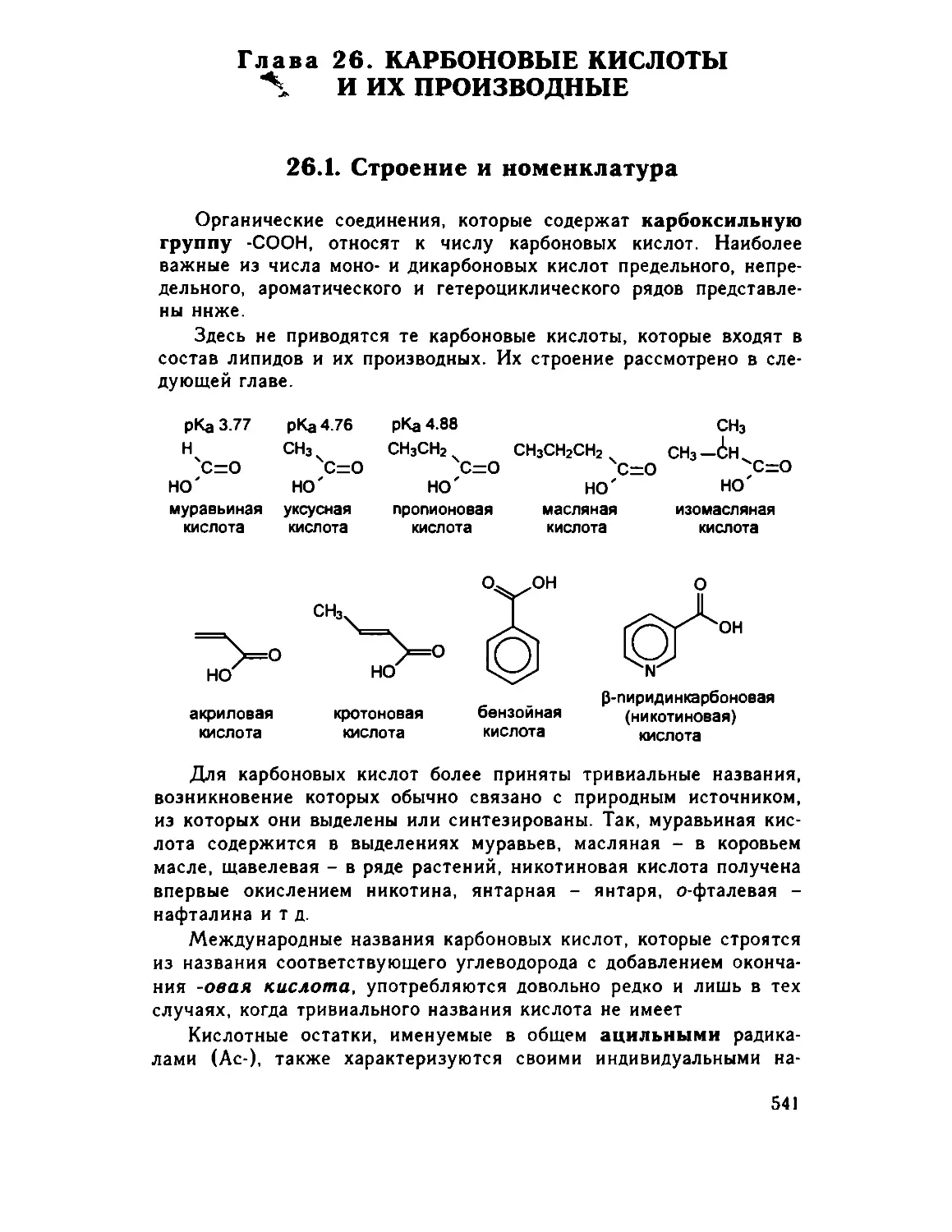
26.1. Строение и номенклатура . ... .541
26.2. Получение . 543
26.3. Свойства карбоновых кислот 545
26 3 1 Кислотные свойства . . ... 546
26.3 2. Окислительно-восстановительные превращения 547
26.3 3 Декарбоксилирование 548
26.3.4 Влияние карбоксильной группы на заместитель 548
26 4. Производные карбоновых кислот 550
26 4 1. Галогенангидриды 550
26 4.2 Ангидриды карбоновых кислот ... 551
26 4 3. Сложные эфиры 551
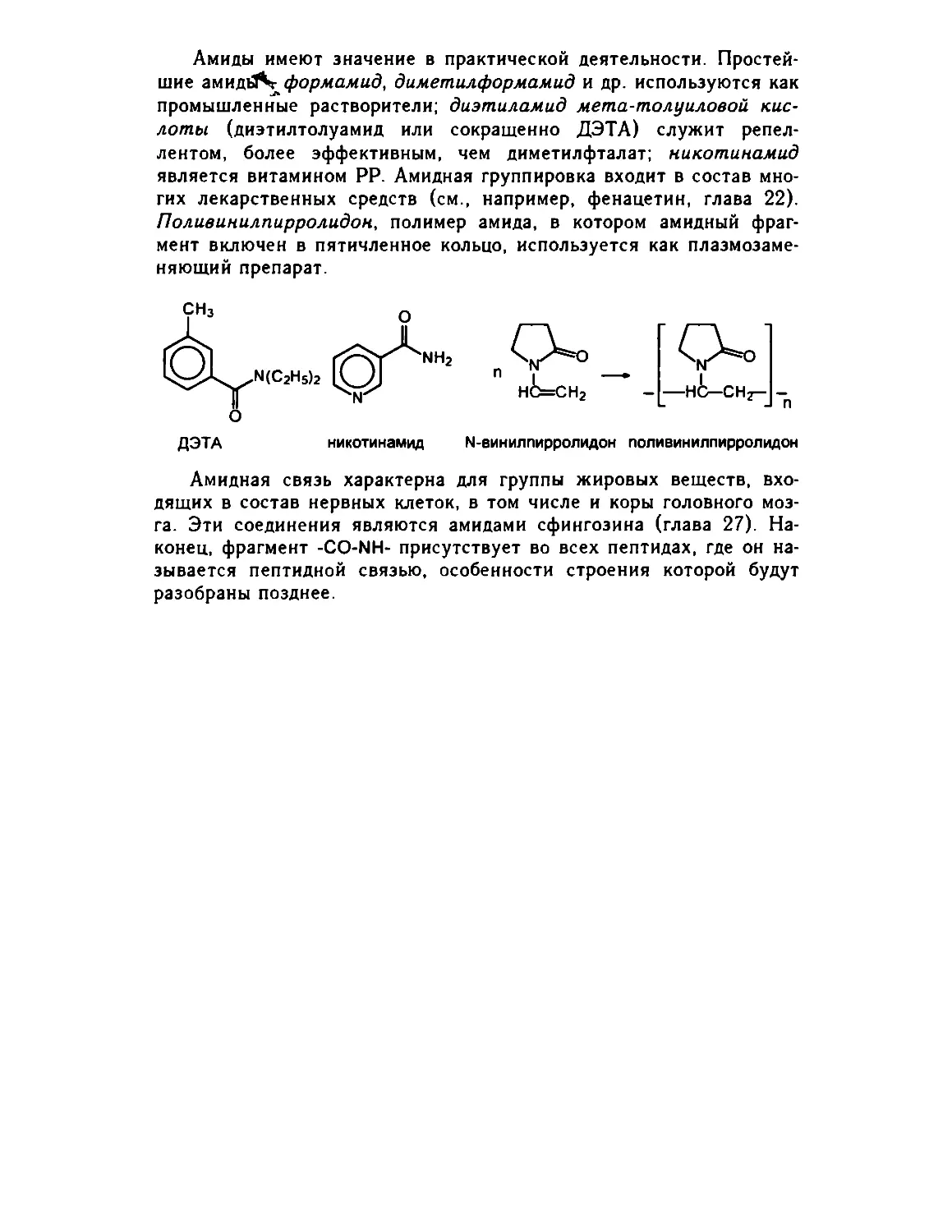
26 4 4 Амиды .. 553
Глава 27. Липиды 556
27 1. Строение и классификация 556
27 2 Стероиды 560
27.3. Простагландины . 562
27 4. Свойства липидов 563
9
Глава 28. Производные угольной кислоты 567
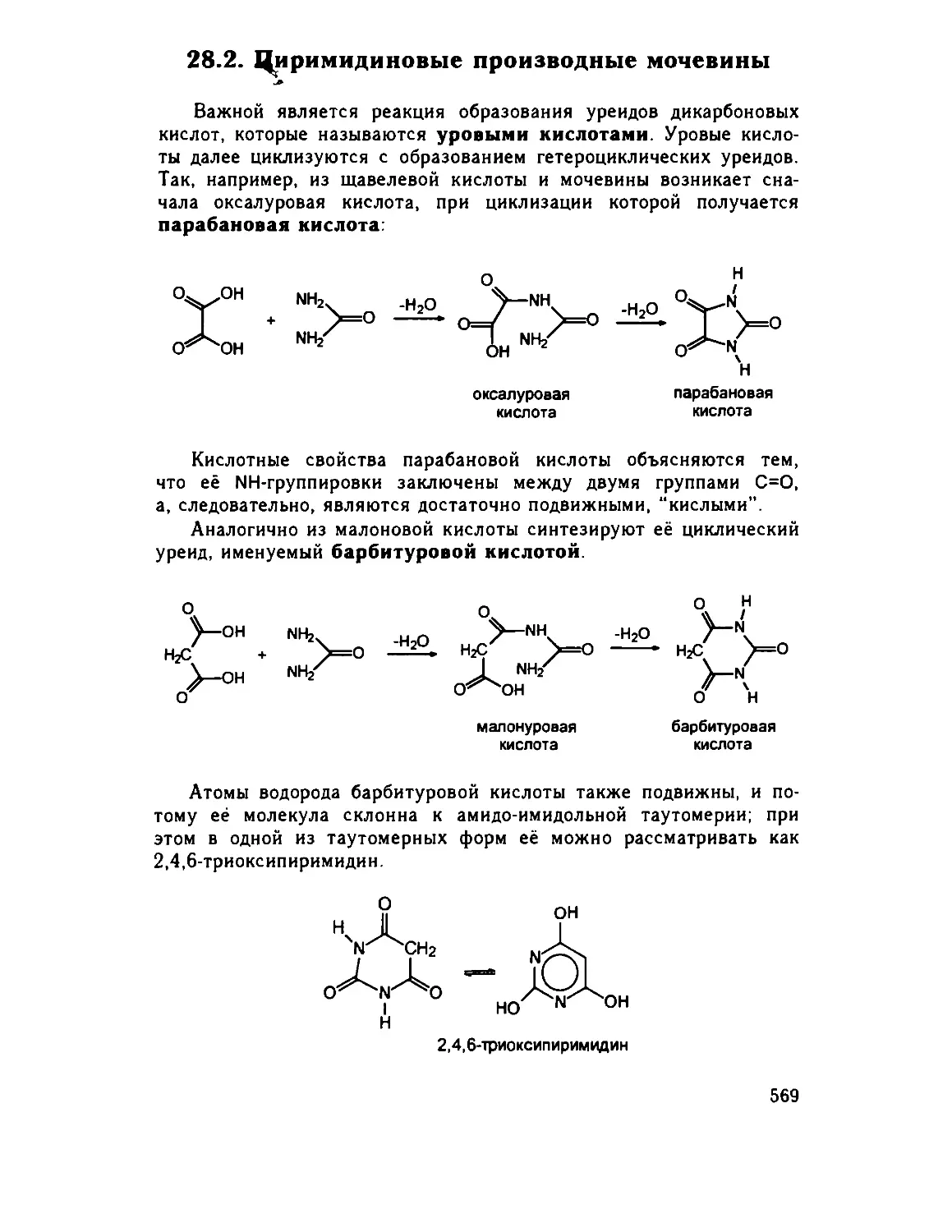
28 1 Мочевина и ее производные 568
28 2 Пиримидиновые производные мочевины 569
28 3. Пуриновые производные мочевины 571
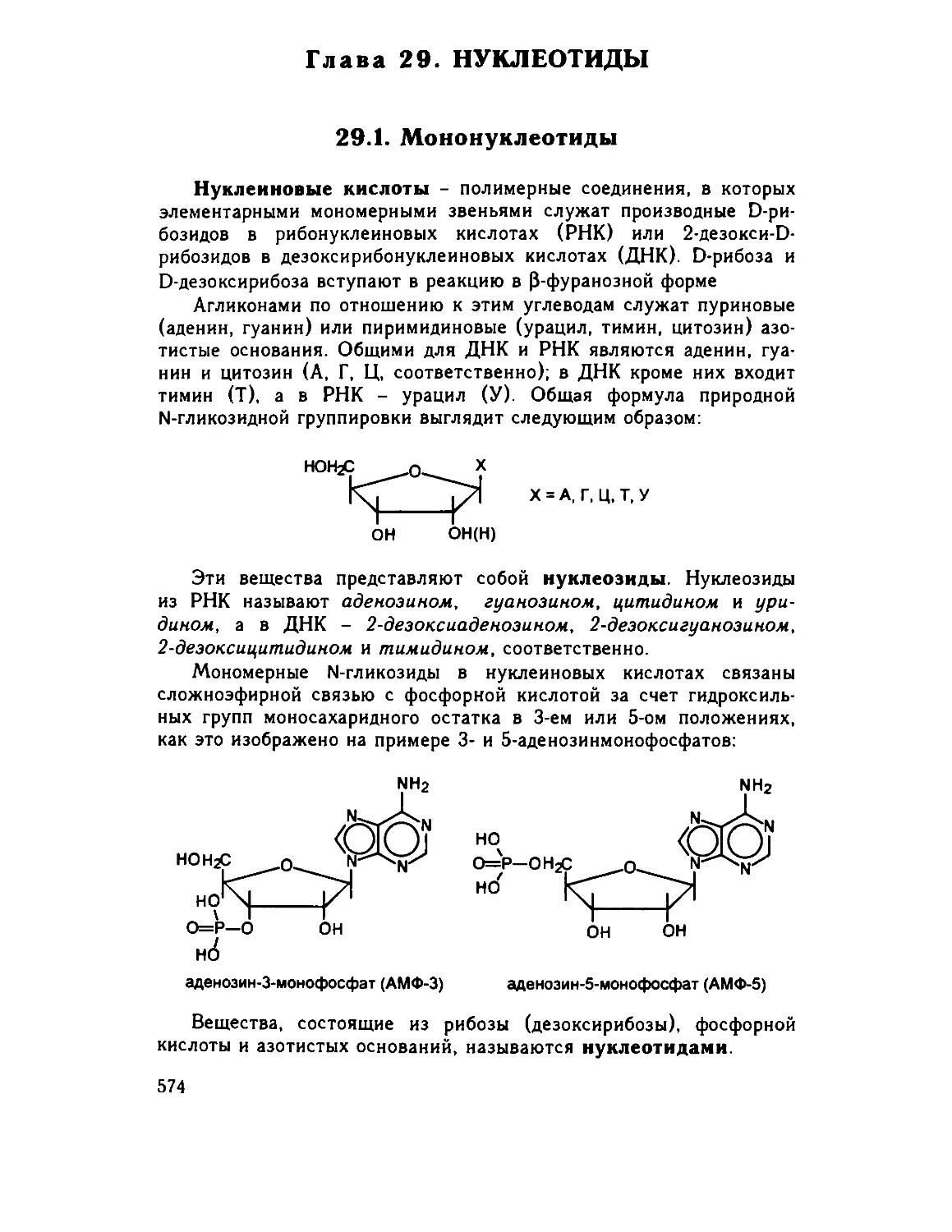
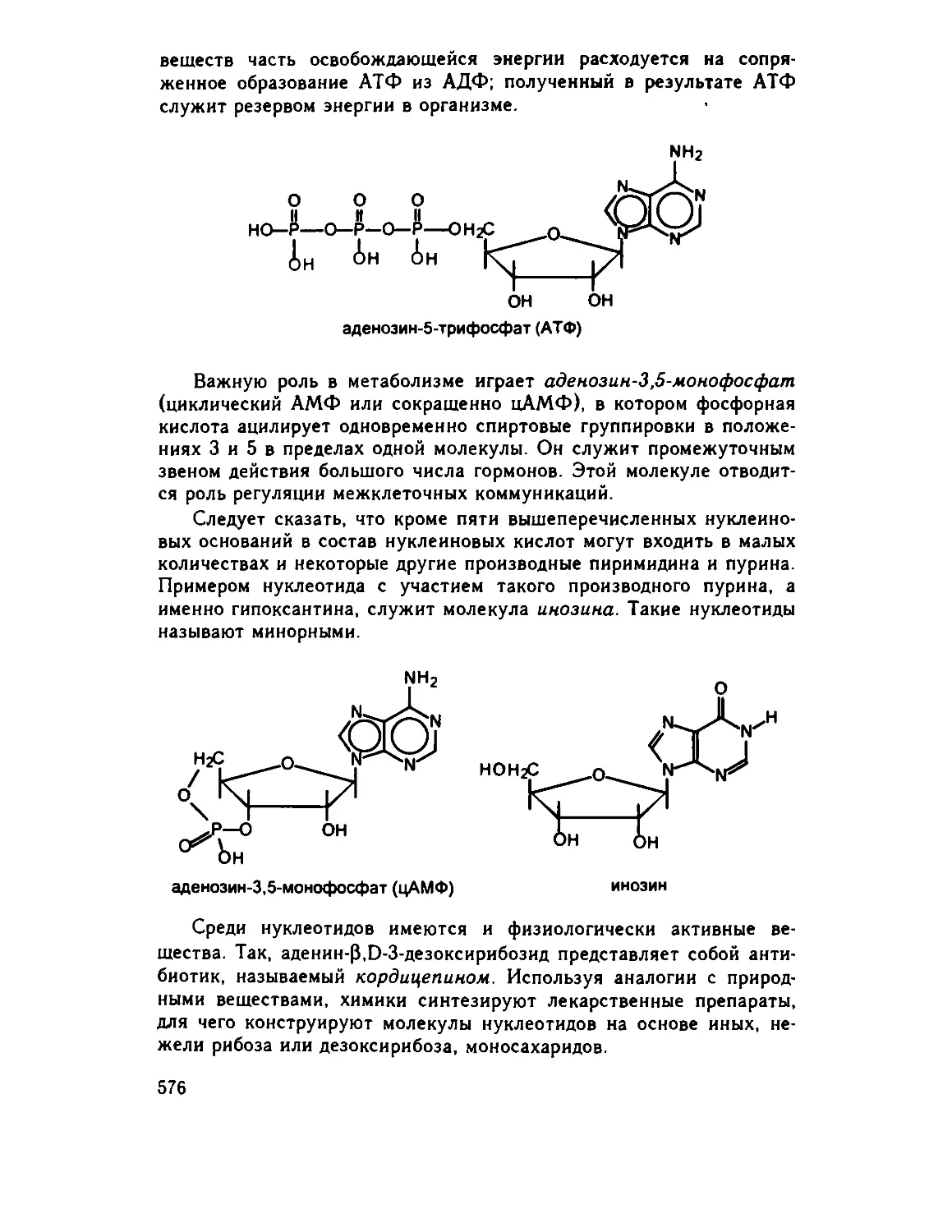
Глава 29. Нуклеотиды 574
29 1 Мононуклеотиды 574
29 2. Полинуклеотиды . .. 578
29.3 Физико-химические свойства полинуклеотидов 581
Глава 30. Окси- и оксокислоты .... 583
30.1. Оксикислоты 583
30 1 1 Классификация и номенклатура ... . 583
30 1 2 Стереоизомерия оксикислот 584
30 1 3 Способы получения 586
30 1 4. Химические свойства оксикислот 587
30 1 5. Значение оксикислот 589
30.2 Оксокислоты 590
30.2 1. Пировиноградная кислота .. 590
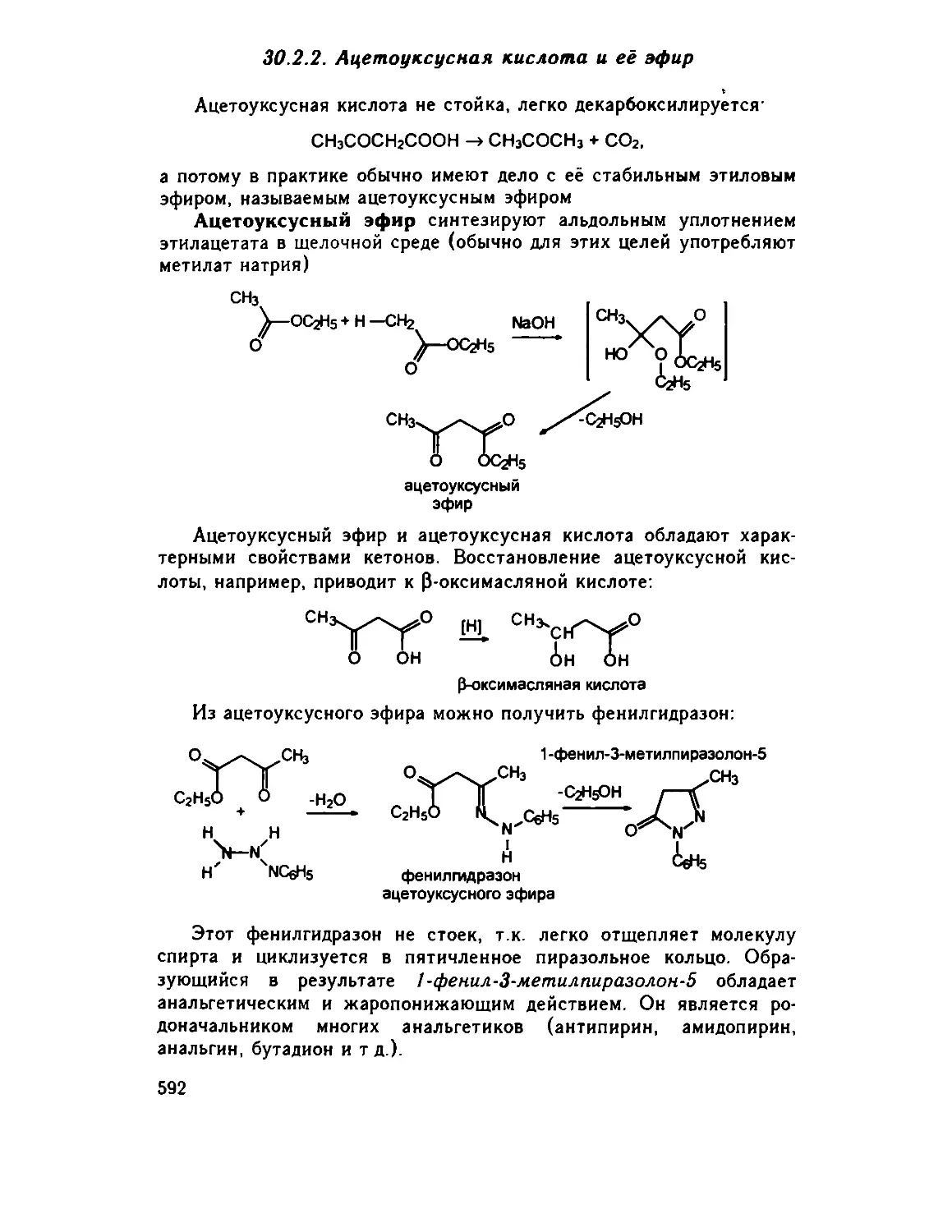
30 2 2 Ацетоуксусная кислота и ее эфир 592
Глава 31. Аминокислоты. Полипептиды 594
31.1. Классификация и получение 594
31 2. Свойства аминокислот 598
31 2.1. Реакции с участием аминогруппы 598
312 2 Реакции с участием карбоксильной группы 599
312 3. Реакции с участием групп 1МН2 и СООН 600
31.3. Полипептиды 601
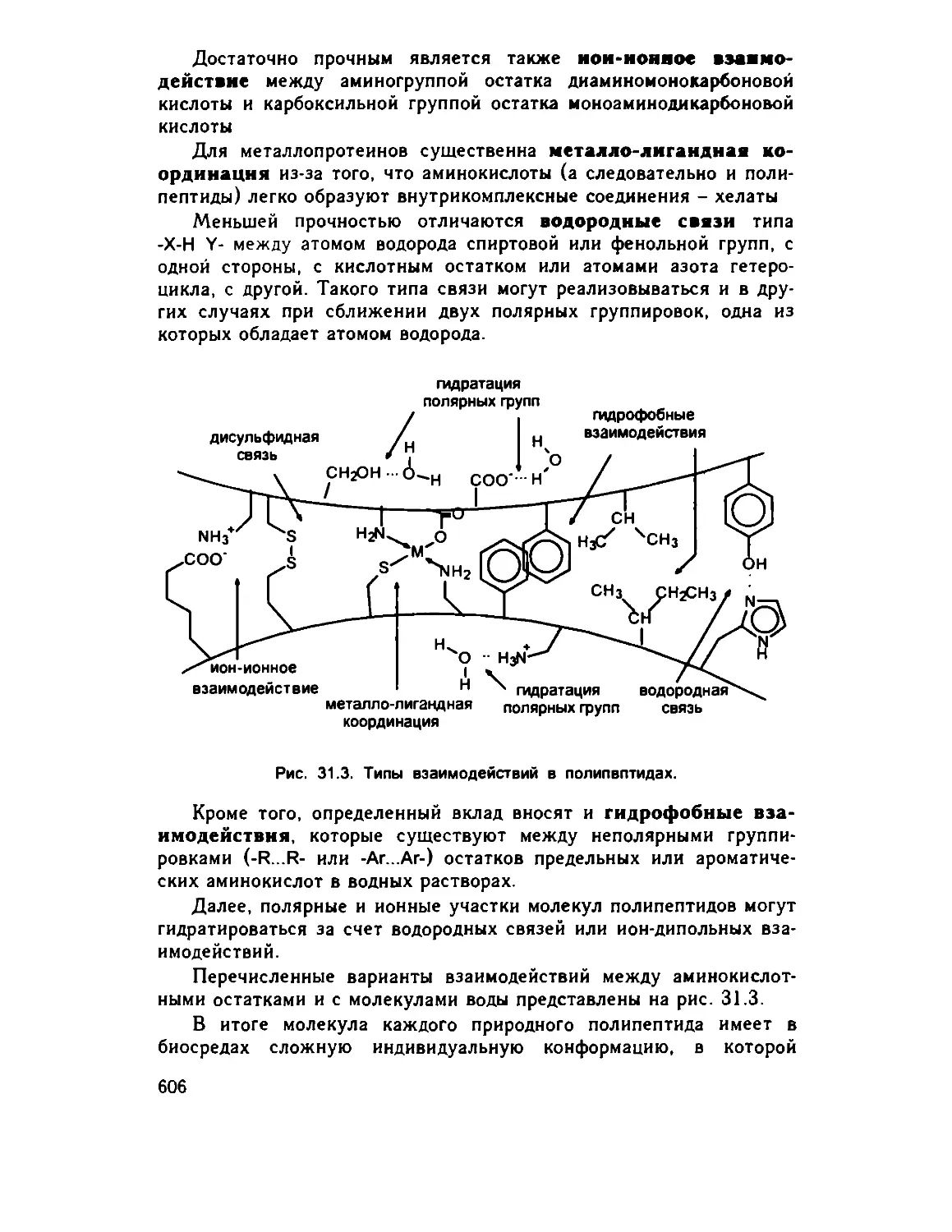
31.4. Пространственное строение полипептидов 603
31.5. Изоэлектрическая точка аминокислот и полипептидов 608
31.6 Полиамфолиты 609
Глава 32. Физико-химические свойства растворов
биополимеров 611
32.1. Основные понятия химии полимеров 611
32.2. Пространственное строение ВМС 612
32.3. Растворы ВМС 613
32 3.1. Коллигативные свойства растворов ВМС 614
32 3 2. Мембранное равновесие Доннана 615
32 3 3 Вязкость 618
32 3.4. Набухание и застудневание 620
32.3.5. Нарушение устойчивости растворов ВМС 622
32.3.6. Свойства студней 623
10
Часть VI. МЕТАЛЛЫ В ЖИВЫХ СИСТЕМАХ
Глава 33. Биометаллы и биолиганды 626
33.1. Общие сведения .. .. 627
33.2. Железосодержащие комплексы .. 629
33.2 1 Гемоглобин и миоглобин 630
33 2.2 Цитохромы 634
33 2 3. Каталаза и пероксидаза .... 635
33 2.4. Негемовые железосодержащие белки .... . 636
33 2 5 Железо-серусодержащие белки . . 639
33.3. Цинксодержащие комплексы . . .. 641
33.4. Медьсодержащие белки . 642
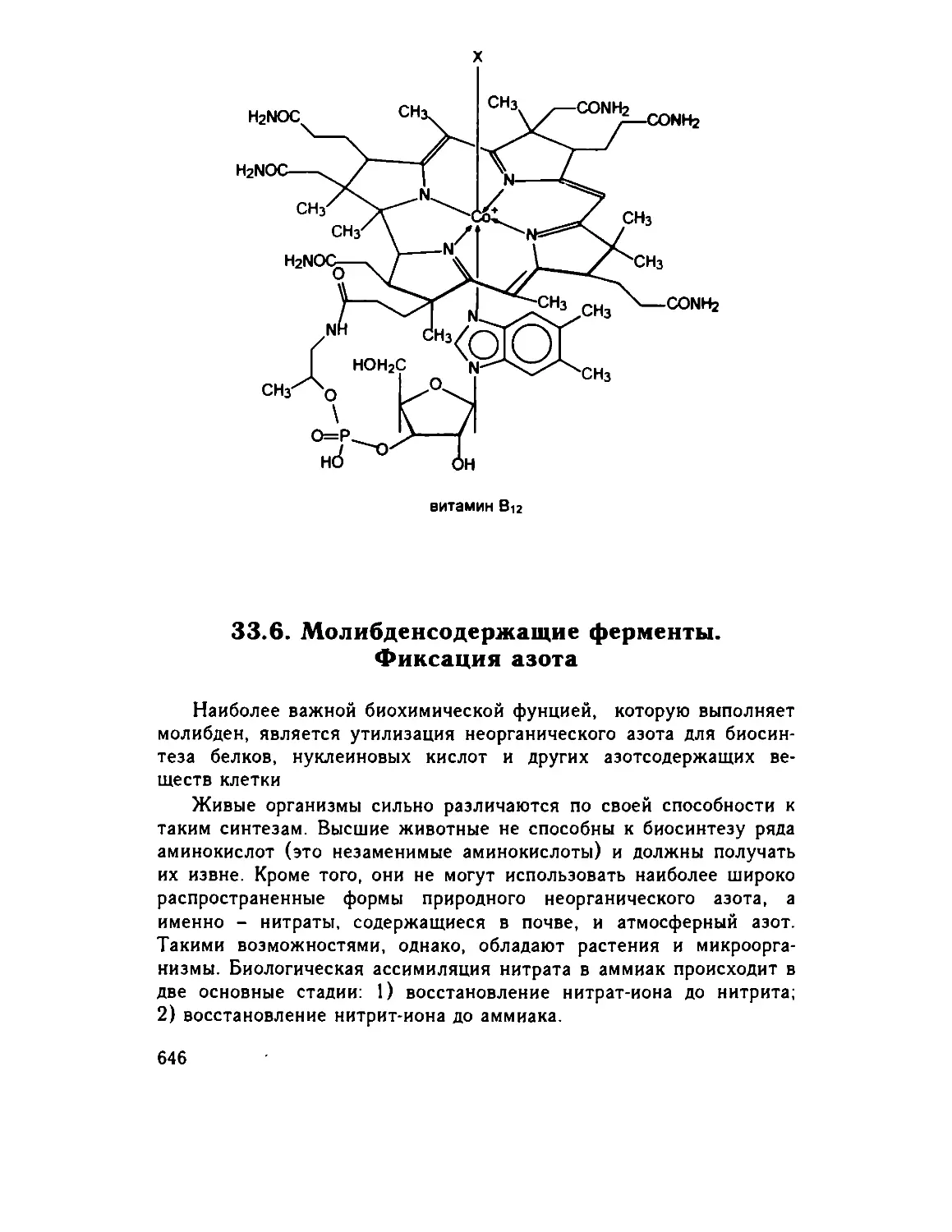
33.5. Корриноиды .. 645
33.6 Молибденсодержащие ферменты. Фиксация азота . 646
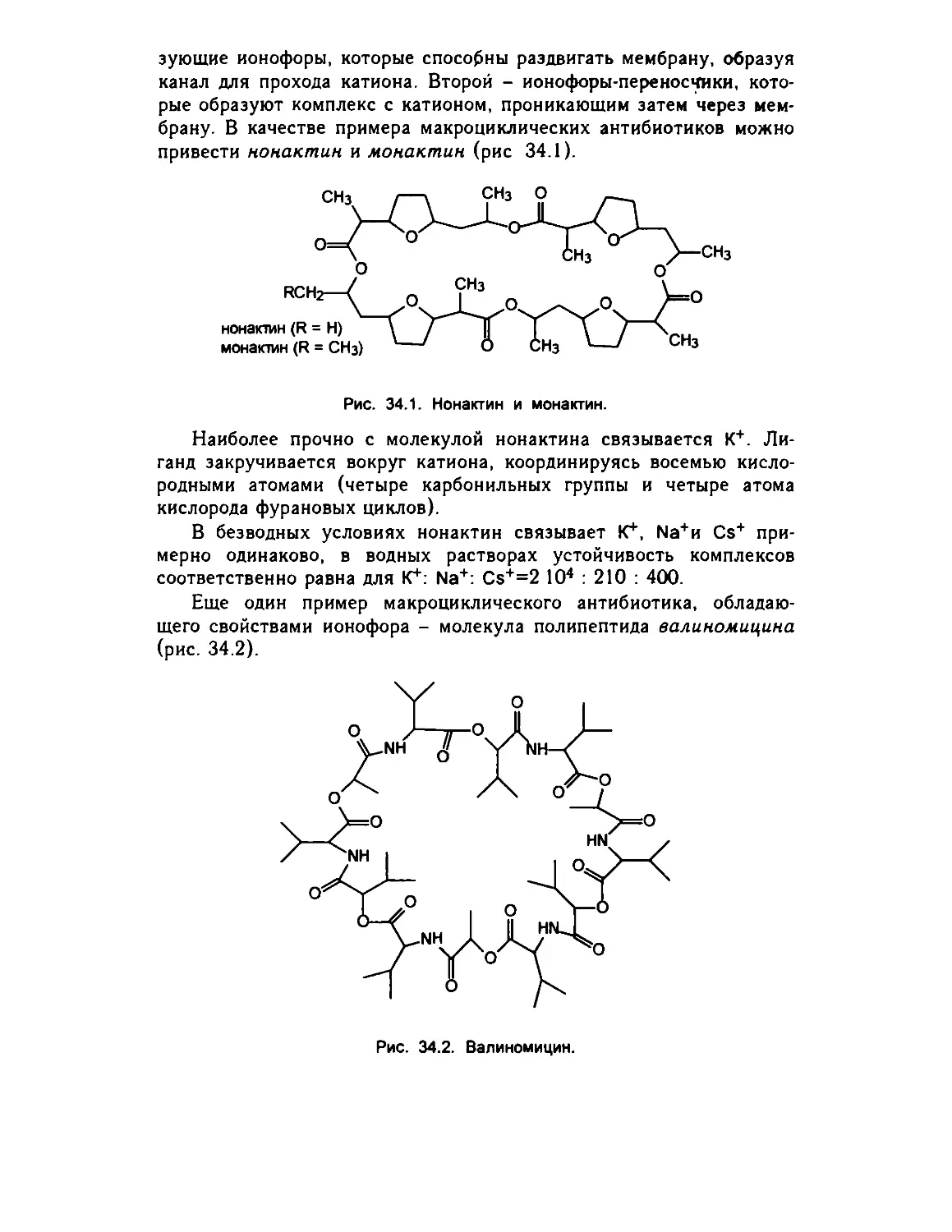
Глава 34. Катионы щелочных и щелочноземельных
металлов ... ... 650
34.1 Общие сведения 650
34.2. Комплексы катионов Б-металлов 652
34 3 Хлорофилл 656
Глава 35. Хелатотерапия 658
35 1. Комплексы как лекарственные средства 658
35 2 Комплексоны в медицине 659
Список рекомендуемой литературы 666
Предметный указатель 668
200-летию Военно-медицинской
академии посвящается
ПРЕДИСЛОВИЕ
Преподавание химии в медицинских вузах складывалось парал¬
лельно развитию химии в целом. Так как медицине обучали в основ¬
ном в университетах, то и курс химии для медиков представлял со¬
бой адаптированную к нуждам медицины модель университетского
химического курса. Медикам отдельно преподавали неорганиче¬
скую, физическую, коллоидную, аналитическую и органическую хи¬
мию в пределах первого курса обучения, в то время как химики
изучают эти дисциплины (наряду с другими) в течение всего пяти¬
летнего обучения.
Дробление курса, которое в ряде медицинских институтов в той
или иной форме сохранилось до настоящего времени, препятствует
выработке у будущих врачей целостного восприятия химии. Поло¬
жение усугубляется возникновением и бурным ростом таких биоло¬
гических (а не химических) дисциплин, как бионеорганическая, био¬
физическая и биоорганическая химия, которые зачастую стали под¬
менять собой химию как фундаментальную дисциплину.
Отражением ситуации является и положение с изданными в на¬
шей стране учебниками для медицинских специальностей. Нет еди¬
ного учебника, отвечающего государственной программе медицин¬
ских институтов. Не существует и профильных руководств, ориен¬
тированных на преподавание химии для клиницистов, фармацевтов,
токсикологов или гигиенистов.
В настоящее время в программах преподавания медицинских
институтов химия вновь рассматривается как единая естественно¬
научная дисциплина, как это и было во времена Н. Н. Зинина, зало¬
жившего основные принципы преподавания химии для будущих вра¬
чей. Эти принципы базируются на единстве курса как фундамен¬
тальной дисциплины, общие законы которой необходимо знать каж¬
дому образованному медику. Другой общий принцип состоит в ме-
дико-биологической направленности, профессиональной ориентиро¬
ванности курса. Наконец, необходимо соблюдение единства лекци¬
онного курса и химического практикума, который должен включать
в себя необходимый минимум химических методов, применяемых в
медицине безотносительно того, базируются ли эти методы на иде¬
ях аналитической или физической химии
В условиях все большего внедрения достижений естественных
наук в практическую медицину отчетливо прослеживается измене¬
ние приоритетов в преподавании химии в медицинских институтах,
12
требующего в настоящее время не столько ознакомления с фактоло¬
гической стороной изучаемой дисциплины, сколько формирования
целостного физико-химического подхода к изучению человеческого
организма. В этой связи традиционные принципы преподавания
химии оказываются малоэффективными.
Написание учебника в свете изложенного представляется непро¬
стой задачей. Настоящее издание и является первой попыткой в
этом направлении. Подход, положенный в его основу, ориентирован
на снятие традиционного разделения курса на неорганическую,
органическую и физическую химии. Такое расчленение учебного
материала, естественное и оправданное при подготовке профес¬
сиональных химиков, в медицинском вузе оказывается тормозом
на пути формирования единого физико-химического мировоззре¬
ния, крайне необходимого современному клиницисту. Остается на¬
деяться, что поставленную задачу автору удалось решить хотя бы
частично.
В основу учебника положен курс, много лет читаемый автором
на кафедре химии Военно-медицинской академии, которая почти
двести лет своего существования оставалась единой, и где, с одной
стороны, формировались общие принципы обучения (первый отече¬
ственный химический учебник создан А. Шерером в первой четвер¬
ти XIX века, химический практикум впервые в нашей стране вве¬
ден А. П. Бородиным в 60-х годах прошлого века, физическая и кол¬
лоидная химия - С. В. Лебедевым в 20-х годах нашего века), а
с другой стороны, утверждалась медико-биологическая ориентация
(первая в России кафедра биохимии отделилась от кафедры химии
в 90-х годах прошлого века).
Автор благодарит доцентов И. П. Бежан, 3. М Матвееву,
В. П. Сергутину и В. А. Хрусталева за участие в написании и об¬
суждении отдельных глав учебника, профессора В В. Алексеева
и доцента О. В. Солода за проделанные ими поиски по профори¬
ентации курса, кандидата химических наук О. Б. Кузнецову и
М. Ю. Смирнову за кропотливую работу по подготовке книги к из¬
данию.
ВВЕДЕНИЕ
Медик без довольного позна¬
ния химии совершенен быть не
может
М В Ломоносов
Соображая тесную связь,
существующую между знания¬
ми медицинскими и физико¬
химическими, делается понят¬
ным, что для каждого врача из¬
учение физики и химии должно
составлять один из предметов
первой важности в ряду его
занятий.
Н Н. Зинин
Химия как наука о веществах и их превращениях сложилась в
качестве самостоятельной дисциплины во второй половине XVIII
века благодаря усилиям таких великих исследователей, как
М. В. Ломоносов, А. Лавуазье и др., которые придали ей количест¬
венный характер. Однако связь химии с медициной началась еще до
ее количественного оформления. Возникшее в XVIII веке направле¬
ние в естествознании, известное под названием ятрохимии, отводи¬
ло главную роль в образовании болезней нарушению естественного
хода химических процессов в организме, а способом их лечения
считалось отыскание химических средств. Наивные представления
исследователей того времени получили научную основу уже в XIX
столетии, когда бурный прогресс науки и техники нашел, пожалуй,
свое самое яркое выражение в химии.
Развитие атомистических представлений привело к формулиров¬
ке двух фундаментальных обобщений в области химии, а имен¬
но - к Периодическому закону (Д. И. Менделеев, 1869 г.) и к от¬
крытию теории строения органической химии (А. М. Бутлеров,
1861 г.). Эти открытия, в свою очередь, породили блестящий этап в
развитии химии, который известен как “триумфальное шествие
органической химии” и который приходится на последнюю треть
XIX столетия. В этот период наряду с другими результатами были
достигнуты впечатляющие успехи в области химии природных со¬
единений - сахаров, пигментов и даже белков. Именно в это время
формируется такая область биологической науки, как биохимия,
которая изучает превращение химических веществ в живых орга¬
низмах. Стало очевидным, что “только физика и химия дают ключ
14
к разъяснению всех сложных и до бесконечности разнообразных
физиологических и патологических процессов, которые совершают¬
ся в организме” (Н. Н. Зинин). Однако подлинный триумф химия в
союзе с биологией и медициной празднует в настоящее время.
Используя современный арсенал физико-химических методов
синтеза, анализа и выделения веществ (хроматография, спектро¬
скопия, электронная микроскопия, рентгенография и др.), химия
успешно объясняет важнейшие медико-биологические закономерно¬
сти на молекулярном и субмолекулярном уровнях. Были интерпре¬
тированы химические механизмы таких фундаментальных явлений,
как фотосинтез, биологическое окисление, процессы метаболизма
и др. С помощью специальных экспериментов были вскрыты основ¬
ные закономерности химической эволюции, подводящие нас к ис¬
толкованию процесса возникновения жизни. С химических позиций
были объяснены явления наследственности и биосинтеза белков,
жиров, углеводов и прочих биомолекул. Благодаря исследованию
структуры и свойств ферментов, гормонов и витаминов стало воз¬
можным управлять процессами жизнедеятельности, решить пробле¬
мы многих патологических явлений и состояний, подойти вплотную
к химическому истолкованию возникновения таких сложных пато¬
логических процессов, как злокачественные опухолевые образова¬
ния, явления старения и др.
Химики в настоящее время успешно синтезируют чрезвычайно
сложные биомолекулы, относящиеся к семействам витаминов, гор¬
монов и ферментов, выясняют механизм их действия, разрабатыва¬
ют технологические способы их получения. Успехи химиотерапии
привели к получению физиологически активных и лекарственных
веществ самого широкого спектра действия (антибактериальные,
противовоспалительные, жаропонижающие, психофармакологиче¬
ские и др. средства).
Благодаря успехам химии стало возможным лечить некоторые
болезни обмена веществ (например, диабет), излечивать заболева¬
ния, ранее считавшиеся неизлечимыми Например, в результате
применения антибиотиков и химиотерапевтических средств смерт¬
ность от туберкулеза снизилась на 80 процентов. Принято считать,
что полезный эффект от лечения в развитых странах на 70 процен¬
тов зависит от применения лекарств Развитие химии инсектици¬
дов и репеллентов является важнейшим фактором в борьбе со мно¬
гими инфекционными заболеваниями (например, малярией, сонной
болезнью и др ) Прогресс в области синтетических материалов при¬
вел к разработке современных методов протезирования тканей и от¬
дельных органов (искусспвенная почка, искусственное сердце и т.д.)
Найдены новые эффективные кровезаменители
15
Медик должен быть ознакомлен со свойствами биоэлементов
(а их свыше пятидесяти). Содержание каждого из них поддержи¬
вается в узких и постоянных границах, и отступление от нормы
приводит к патологическим состояниям.
Медик должен ориентироваться в строении и свойствах химиче¬
ских веществ, а их насчитывается свыше пятнадцати миллионов.
Около 10 ООО из них входят в состав тканей человеческого организ¬
ма, многие тысячи насчитывают разнообразные лекарственные пре¬
параты и физиологически активные вещества растительного и жи¬
вотного происхождения. Многие и многие вещества оказывают
вредное (токсическое, канцерогенное и др.) действие на организм.
Достаточно сказать, что на каждого жителя Санкт-Петербурга в год
приходится до 100 килограммов потенциально опасных веществ, по¬
ступающих в окружающую среду в результате хозяйственной дея¬
тельности.
Медику необходимо ориентироваться в законах, управляющих
ходом химических реакций в живых системах. Ведь биохимические
процессы в целом определяются совокупностью нескольких тысяч
химических реакций, каждая из которых отличается своими специ¬
фическими особенностями.
Наконец, медик должен владеть арсеналом химических и физи-
ко-химических методов исследования, без которых немыслима дея¬
тельность современного врача.
Часть I
ФУНДАМЕНТАЛЬНЫЕ ЗАКОНЫ ХИМИИ
Химию в ее современном
состоянии... можно назвать
учением об элементах.
Д. И. Менделеев
Глава 1. СТРОЕНИЕ АТОМА
И ПЕРИОДИЧЕСКИЙ ЗАКОН
Свойства простых и слож¬
ных тел находятся в периоди¬
ческой зависимости от атом¬
ного веса элементов.
Д. И Менделеев
Конец XIX и начало XX века были ознаменованы фундаменталь¬
ными открытиями, во многом определившими пути развития совре¬
менной физики и химии. К ним следует отнести обнаружение ра¬
диоактивности (А. Беккерель, 1896 г.), рентгеновского излучения
(В. Рентген, 1895 г), идею квантования энергии атома (М. Планк,
1900 г.), открытие фотоэффекта (А. Эйнштейн, 1887 г.) и электро¬
на (Дж. Томсон, 1897 г.), установление линейчатого характера
спектров атомов и т.д.
Накопление экспериментального материала вызвало необходи¬
мость в создании такой теории строения атома, которая бы удовле¬
творительно объясняла имеющиеся экспериментальные данные и
находилась бы в согласии с Периодическим законом Д. И. Мен¬
делеева.
В свое время предлагались различные модели атома- “пудинго-
вая” модель (Дж. Томсон, 1904 г), планетарная (Э Резерфорд,
1907 г.), модель Бора (Н Бор, 1913 г ) Они представляют в настоя¬
щий момент лишь исторический интерес, т.к. внутренняя проти¬
воречивость и ограниченность заставили вскоре отказаться от этих
моделей.
В 20-е годы нынешнего столетия была создана квантово-меха¬
ническая модель, котораяла данном этапе развития науки дает наи¬
лучшее представление 9 ел^роеиии -«то^а
17
1.1. Квантово-механическое описание
атома водорода
Квантовая (или волновая) механика основывается на том, что
любые материальные частицы одновременно обладают и волновыми
свойствами. Впервые это было предсказано Л де Бройлем, кото¬
рый в 1924 г теоретически показал, что частица с массой т и ско¬
ростью V может быть ассоциирована с волновым движением, длина
волны которого X определяется выражением:
где Ь (постоянная Планка) = 6.6256-10'27 эрг е = 6.6256-10’34 Дж-с.
Вскоре это предположение было подтверждено явлениями дифрак¬
ции электронов и интерференции двух пучков электронов.
Двойственная природа элементарных частиц (корпускулярно¬
волновой дуализм) - частное проявление общего свойства материи,
однако ожидать его следует только для микрообъектов.
Это видно из следующего примера. Теннисный мяч массой 50 г,
летящий со скоростью 25 м/с, ассоциируется по расчету с волной,
имеющей ничтожно малую длину А. = 10'23 см. Поэтому волновыми
свойствами макрообъектов можно смело пренебречь. Таким обра¬
зом, корпускулярно-волновой дуализм не вносит изменений в опи¬
сание движения макроскопических объектов с помощью законов
классической механики. В то же время для микрочастицы электро¬
на, масса которого равна 9 • 10'28 г и в электронно-лучевой трубке
обычна скорость порядка 108 см/с, длина волны собственных коле¬
баний, вычисленная по вышеприведенному уравнению, составляет
= 10'8 см. Другими словами, пучок таких электронов ведет себя в
известном смысле как электромагнитное излучение той же длины
волны (это соответствует рентгеновскому излучению).
Волновые свойства микрочастиц выражаются в ограниченной
применимости к ним таких понятий, которыми характеризуется мак¬
рочастица в классической механике, как координата (х, у, г) и им¬
пульс (р = т • у). Если в макромире, задавая координаты и импульс
тела, можно однозначно предсказать, где оно будет находиться в
любой момент времени, то в микромире можно лишь вычислить
вероятность нахождения частицы в определенном объеме простран¬
ства. Другими словами, для микрочастиц всегда имеются неопреде¬
ленности в координате и импульсе, связанные соотношением Гей¬
зенберга:
где Дх - неопределенность координаты, а Дрх - неопределенность
импульса.
18
Согласно принципу неопределенности, движение микрочастицы
невозможно описать определенной траекторией и нельзя предста¬
вить движение электрона в атоме в виде движения по конкрет¬
ной круговой или эллиптической орбите, как это было принято в
модели Бора.
Описание движения электрона может быть дано при помощи
волн де Бройля. Волна, отвечающая микрочастице, описывается
волновой функцией у (х, у, г). Физический смысл имеет не сама
волновая функция, а только произведение квадрата ее модуля на
элементарный объем М2-^, равное вероятности нахождения элек¬
трона в элементарном объеме (IV = ёх ёу ёг.
Описать состояние частицы в квантовой механике означает оп¬
ределить вероятность нахождения этой частицы в любом элементе
пространства. Поэтому было необходимо найти уравнение, которое
описывало бы движение как свободной частицы, так и частицы в
силовом поле.
Таким уравнением явилось волновое уравнение Шредингера
(1926 г.), которое связывает волновую функцию с потенциальной
(и) и полной (Е) энергией электрона:
—^—2у + (Е - и)у = О,
8л т
с12у с12у
где 2у = —5-н 5- - сумма вторых производных по коорди-
Ах Ау Аг
натам; т - масса электрона; Ь - постоянная Планка.
Волновое уравнение Шредингера - это математическая модель
атома. Она отражает единство корпускулярных и волновых свойств
электрона. Не вдаваясь в анализ уравнения Шредингера, укажем,
что решить его - значит найти удовлетворяющую ему волновую
функцию у. Это дифференциальное уравнение имеет бесчисленное
множество решений, но интерес представляют лишь те, для кото¬
рых найденное значение |у|2 • Ау не противоречит физическим пред¬
ставлениям. Каждому приемлемому решению волнового уравне¬
ния (у], у2, Уз,.. уп) соответствует определенное значение энергии
(Е], Ег, Е3, . Еп), т.е. квантование энергии микросистемы непо¬
средственно подтверждается решением уравнения Шредингера.
Волновая функция, являющаяся решением уравнения, называет¬
ся орбиталью. Описать орбиталь, т.е. каждое состояние электрона
в атоме, можно с помощью набора трех квантовых чисел, значения
которых входят в модифицированное уравнение Шредингера и опре¬
деляют его приемлемые решения.
19
Главное квантовое число (п) принимает целые положитель¬
ные значения и определяет энергию, а, следовательно, и среднее
расстояние электрона от ядра Поэтому, когда речь идет о главном
квантовом числе, принято считать, что оно характеризует опреде¬
ленный энергетический уровень: п = 1,2, 3...
При п = 1 электрон находится на первом энергетическом уров¬
не и т.д.
Орбитальное (побочное) квантовое число (/) характеризует
“форму” орбитали - плотность вероятности нахождения электрона
у ядра и принимает целочисленные значения от нуля до (л-1).
Обычно квантовое число / обозначают буквами:
/ 0 12 3 4 5
Обозначение эре! Г е Ь
Магнитное квантовое число (т{) принимает значения: 0, ±1,
±2, ±3, ... ±/ и определяет ориентацию орбитали в пространстве.
Таким образом, все три квантовых числа п, I, т.1 связаны между
собой.
В табл. 1.1 приведены наборы квантовых чисел и символы
атомных орбиталей.
Таблица 1.1
Квантовые числа атомных орбиталей
«
1
т,
Символ орбитали
1
0
0
и
2
0
0
2б
1
-1 0 +1
2р
3
0
0
Зб
1
-1 0 +1
Зр
2
-2 -1 0 +1 +2
3(1
4
0
0
4э
1
-1 0 +1
4р
2
-2 -1 0 +1 +2
4(1
3
-3 -2 -1 0 +1 +2 +3
41
Из таблицы 1.1 видно, что для любого значения п имеется одна
орбиталь э-типа; начиная с п = 2, т.е. на втором энергетическом
уровне, появляется набор из трех р-орбиталей и т.д.
Любая э-орбиталь сферически симметрична, а по мере увеличе¬
ния главного квантового числа п максимум концентрации электрон¬
ной плотности располагается все дальше и дальше от ядра. Каждая
20
р-орбиталь состоит из положительной и отрицательной долей (знак
волновой функции в данной области пространства), расположен¬
ных вдоль положительного и отрицательного направления данной
координатной оси. В каждом наборе имеется три орбитали р-типа -
Рх» Ру. Рг-
Существует пять (1-орбиталей, которые можно представить раз¬
личными способами, чаще всего, как показано на рис. 1.1.
В каждом наборе, начиная с п = 4, имеется семь f-орбиталей.
Они слабо участвуют в образовании химических связей и здесь нет
необходимости их рассматривать.
Позднее для характеристики состояния электрона в атоме было
введено четвертое квантовое число - спиновое (ms), необходи¬
мость которого вытекала из теории относительности.
Смысл понятия спнн (от английского spin - веретено) в мо¬
дельном представлении означает, что электрон совершает вращение
вокруг собственной оси. При этом электрон обладает собственным
магнитным моментом, вектор которого параллелен вектору магнит¬
ного поля, но либо совпадает (параллелен) с ним по направлению,
либо противоположен (антипараллелен) ему. В соответствии с этим
спиновое квантовое число для электрона может иметь только два
значения ms = ±1 /2 Следует помнить, что спин - фундаментальное
свойство любой микрочастицы, которое не сводимо к какому-либо
другому более простому свойству.
Уравнение Шредингера можно строго решить только для атома
водорода - простейшего атома, состоящего из одного протона и од¬
ного электрона. В результате получается набор значений энергии,
сответствующих основному (энергия минимальна) и возбужденным
состояниям. Основным состоянием атома водорода будет ls-состоя-
Рис. 1.1. Форма и пространственная ориентация
электронных орбиталей в-, р- и (1- электронов.
21
ниє, а порядок последующих энергетических уровней будет опреде¬
ляться только значением главного квантового числа.
и < 2ь = 2р < Зэ = Зр = 3(1 < 4э = 4р = 4с1 = 41 < бэ. .
Поскольку квантовые числа I, т/, т$ не вносят ничего в энер¬
гию электронного состояния, все возможные состояния в пределах
данного уровня должны быть энергетически равны Это означает,
что в спектре испускания возбужденного атома водорода должны
наблюдаться только единичные линии (линейчатый спектр). Однако
из опыта хорошо известно, что в спектре водорода существует тон¬
кая структура. Такое несоответствие объясняется тем, что приве¬
денная выше последовательность энергетических уровней является
результатом расчетов, основанных на нерелятивистском (т.е. без
учета выводов теории относительности) подходе к атому водорода в
отсутствие внешнего магнитного или электрического поля. Исполь¬
зуя релятивистскую форму волнового уравнения, предложенного
Дираком, можно получить невырожденную последовательность
уровней, полностью соответствующую тонкой структуре спектра
атома водорода, при которой каждый тип орбиталей отличается от
других
1.2. Строение многоэлектронных атомов
Атомы всех элементов, кроме водорода, многоэлектронны. Рас¬
пределение электронной плотности в атоме, энергию связи электро¬
на с ядром и другие физические характеристики можно найти, ре¬
шив уравнение Шредингеиа. Однако для многоэлектронных атомов
точное решение этого уравнения пока не возможно, т.к. слишком
велики математические трудности, вызванные необходимостью уче¬
та взаимодействий электронов с ядром и друг с другом. В квантовой
механике можно найти лишь приближенное решение задачи для си¬
стемы многоэлектронных атомов. В настоящее время существуют
различные методы приближений, которые дают удовлетворительное
совпадение вычисленных и экспериментально найденных свойств
атомов
При определении реальной электронной конфигурации любого
атома следует руководствоваться принципом минимума энер¬
гии, который состоит в том, что электрон в первую очередь зани¬
мает наиболее энергетически выгодную орбиталь
Заполнение орбиталей электронами происходит в соответствии
с фундаментальным принципом исключения (принцип Паули,
1925 г.), который гласит: в одном атоме не может быть двух элек¬
тронов с одинаковым набором всех четырех квантовых чисел.
22
Важнейшее следствие из этого принципа состоит в том, что на
ка-ждой орбитали может быть не более двух электронов, имеющих
антипараллельные спины (различные знаки квантового числа т5).
Последовательность заполнения орбиталей электронами опреде¬
ляет электронную конфигурацию атома. Для атома водорода
это просто указание, на какой орбитали находится единственный
электрон. Так, 1$-орбиталь, имеющая наименьшую энергию, будет
занята электроном для атома водорода в невозбужденном состоя¬
нии, 2$, Зэ, 4э и т.д. для разных возбужденных состояний.
Для многоэлектронных атомов применяют такие же обозначе¬
ния. Количество электронов на орбиталях каждого типа указывают
верхним индексом. Так, для атома водорода электронная конфигура¬
ция записывается и1 , для гелия - 1э2 , для лития -1$22$1 и т.д.
Кроме приведенной буквенной записи электронной конфигура¬
ции, в которой находят отражения лишь главное и орбитальное
квантовые числа, используют графическую форму, отражающую все
четыре квантовых числа. Каждая орбиталь условно обозначается
квадратом, а электроны - стрелками, направление которых указыва¬
ет взаимное расположение векторов спина.
1Н и1 2Не и2
□ №
3Ы 1 в2 2з1 4Ве 1 в2 2%2 5В и2 2в2 2р*
При заполнении р- и ё-орбиталей пользуются правилом Хун-
даз наиболее устойчивой конфигурацией среди нескольких возмож¬
ных с одинаковой энергией является та, которая содержит наиболь¬
шее число неспаренных электронов.
Поэтому при последовательном заполнении р-орбиталей первые
три электрона располагаются по одному на разных орбиталях с
одинаковыми спинами (неспаренные электроны), и лишь затем про¬
исходит заполнение орбиталей электронами с антипараллельными
спинами. По этому правилу происходит заполнение электронных
уровней для атомов углерода, азота, кислорода, фтора и неона
У неона оказывается полностью заполненным второй уровень,
и при переходе от него к 11№ еще один электрон будет входить в
третью оболочку.
У следующих восьми элементов происходит постепенное запол¬
нение сначала Зэ, а затем Зр-орбиталей, которое заканчивается у
инертного газа аргона
По мере увеличения главного квантового числа происходит
уменьшение разницы энергий между уровнями: уровни начинают
взаимно проникать друг в друга и в результате оказывается, что 45-
орбиталь имеет более низкую энер¬
гию, чем 3(1-орбиталь. Поэтому у
калия и кальция заполняется 45-ор¬
биталь и лишь затем от скандия до
цинка происходит заполнение 3(1-
орбиталей. У последующих элемен¬
тов периода заполняются 4р-ор-
битали
На основании изучения линей¬
чатых спектров атомов была уста¬
новлена следующая последователь¬
ность по энергии атомных орбита-
лей (АО) для многоэлектронных
атомов:
1в < 2в < 2р < Зэ < Зр < 4э < 3(1 <
Рис. 1.2. Расположение
энергетических уровней 4р < 5э < 4(1 < 5р < 6э < 4Г < 5(1 <
в многоэлектронных атомах
(примерная схема). 6р < 7э и т.д.
24
Схема энергетической последовательности электронных уровней
приведена на рис. 1.2.
Для запоминания удобно пользоваться правилом Клейновско¬
го- атомные орбитали располагаются в последовательности возрас¬
тания суммы квантовых чисел (п + I), причем в группе с одинако¬
вым значением (п + I) первыми следуют уровни с меньшим значе¬
нием главного квантового числа п (табл. 1.2).
Таблица 1.2
Заполнение электронами энергетических уровней атомов
Сумма
п + 1
-
1
Состояние
Сумма
я + 1
-
1
1
1
0
15
6
4
2
4(1
6
5
1
5р
2
2
0
2ъ
6
6
0
бэ
3
2
1
2р
7
4
3
А\
3
3
0
Зб
7
5
2
5(1
7
6
1
6р
4
3
1
Зр
7
7
0
4
4
0
4э
8
5
3
Ъ\
5
3
2
3(і
8
6
2
6(1
5
4
1
4р
8
7
1
7р
5
5
0
5б
8
8
0
вБ
Небольшие отклонения от общей последовательности вызваны
тонкими эффектами, обусловленными полностью заполненными, за¬
полненными наполовину и свободными орбиталями одного уров¬
ня, которые имеют дополнительный выигрыш в энергии по сравне¬
нию с другими электронными конфигурациями Поэтому атом хро¬
ма имеет конфигурацию внешнего уровня 4$'3(15, (а не 4523с14), а
медь 4з13(110 (но не 4$23(19). Те же отклонения наблюдаются у ана¬
лога хрома молибдена, а также у элементов подгруппы меди - се¬
ребра и золота.
Взаимное проникновение электронных уровней обусловливает
дальнейший порядок их заполнения Для элементов с большими по¬
рядковыми номерами истинная электронная конфигурация устанав¬
ливается экспериментально (табл. 1 3).
Порядок заполнения электронных оболочек элементов и опре¬
деляет их расположение в Периодической системе Д. И. Менделее¬
ва, предложенной им еще в 1869 году (рис 1.3).
25
Таблица / 3
Электронные конфигурации атомов в их основном состоянии
Поряд¬
ковый
номер
Эле¬
мент
Электронная
конфигурация
Поряд¬
ковый
номер
Эле¬
мент
Электронная
конфигурация
1
Н
Ь1
53
1
4а105з25р5
2
Не
152
54
Хе
4с1105525р6
3
и
[Не^Б1
55
вэ
(Хе |6э1
4
Ве
252
56
Ва
бь2
5
В
2522р1
57
1_а
5(1'бэ2
6
С
2522р2
58
Се
7
N
2з22р3
59
Рг
4136з2
8
О
2з22р4
60
N(1
4Иб52
9
Р
2з22р5
61
Рт
4Г56з2
10
N6
2522р6
62
Бт
41«б52
11
N3
[Ме^Б1
63
Ей
4[7б52
12
Мд
352
64
вй
4Г75(11б52
13
А1
3523Р‘
65
ТЬ
4Рбз2
14
3
3з23р2
66
□у
4Г106з2
15
Р
3з23р3
67
Но
4Г"б52
16
Б
3523Р4
68
Ег
4Г12б52
17
С1
3з23р5
69
Тт
4113б52
18
Аг
3з23р6
70
УЬ
41146з2
19
К
[АгНб1
71
1.и
4Г,45(11б52
20
Са
4з2
72
Ж
4Г145(12 6б2
21
Бс
за^2
73
Та
4Г145(13б52
22
П
Зс12452
74
\Л/
4\н 5 ^бэ2
23
V
Зс13452
75
Ре
4Р4 5(15б52
24
Сг
Зс15451
76
Оэ
4Р4 5(16б52
25
Мп
3(15452
77
1г
4Р4 5(17652
26
Ре
Зс16452
78
Р1
4Р4 5(18б52
27
Со
79
Аи
651
28
N1
3(18452
80
нд
б52
29
Си
3(110451
81
Т1
б52
30
2п
3<110452
82
РЬ
6з26р2
31
вг
3(1104Б24р1
83
в.
б526р3
32
ве
Зс1104524 р2
84
Ро
6$26р4
33
Аэ
3с1104524р3
85
А1
6$26р5
34
Бе
3(110 4524р4
86
Рп
6э26р6
35
Вг
3(1104524р5
87
Рг
1Рп]75>
36
Кг
3(110 4&24р6
88
Ра
7б2
37
№
(Кг^Б1
89
Ас
1 Рп)б<^17 52
38
Бг
552 |
| 90
ТЬ
6(12752
26
Продолжение таблицы 1.3
Поряд¬
ковый
номер
Эле¬
мент
Электронная
конфигурация
I Поряд¬
ковый
номер
Эле¬
мент
Электронная
конфигурация
39
У
4(1‘552
91
Ра
5Рб(12752
40
гх
4р25з2
92
и
Ы36й17ъ2
41
№
4(14551
93
Мр
5\57%2
42
Мо
4<15551
94
Ри
5156ь2
43
Тс
4(15552
95
Ат
5177$2
44
Яи
4(17551
96
Ст
5Ш'7ь2
45
ЯЬ
4(18551
97
Вк
5Ш17%2
46
Рс1
4(110
или 51 97з2
47
Ад
4(110551
98
СГ
5Г107з2
48
Сс1
4(1,0552
99
Еэ
5111 7б2
49
1п
4с1105525р1
100
Рт
5Г12752
50
Бп
4(1105525р2
101
Мй
5Р3752
51
БЬ
4(1105525р3
102
N0
5Г147з2
52
Те
4(1105525р4
103
и
5Г146(117з2
1.3. Периодическое сходство элементов
Периодическая система элементов создана Д. И. Менделеевым
на основании наблюдаемого сходства химических и физических
свойств определенных групп элементов. Например, щелочные ме¬
таллы помещены в одну группу потому, что все они обладают уди¬
вительно похожими свойствами. Причина этого сходства была вы¬
яснена после создания удовлетворительной модели строения атома.
Согласно современной модели, щелочные металлы (и, N8, К, РЬ,
Се, Рг) имеют общую электронную конфигурацию лэ1 (п - главное
квантовое число). Это подобие и ведет к сходству химических и фи¬
зических свойств щелочных металлов. То же самое наблюдается и
в остальных группах Периодической системы (например, у инерт¬
ных газов или же у галогенов).
В дополнение к подобию по вертикали Периодической системы
для некоторых элементов существует сходство по горизонтали
Особенно ярко это выражено у лантаноидов и актиноидов ([-эле¬
менты), но довольно заметно оно и в случае элементов, у которых
происходит заполнение (1-подуровня ((1-элементы) Это объясняется
тем, что для Элементов происходит заполнение орбиталей, лежа¬
щих ниже валентных орбиталей, а, следовательно, не столь сильно
влияющих на свойства элементов, как в случае заполнения б- и р-
орбиталей (в- и р-элементы)
27
1
1
1
H
1 0
II
'руппь
l->
III
IV
V VI
р-элемепты
VII
VIII
2
He
40
2
3
Li
69
4
Вс
90
5
В
108
6
С
120
7
N
140
8
О
160
9
F
190
10
Nc
20 2
3
II
No
23
12
Mg
24 3
d-элсмснты
ІЗ
AI
27 0
14
Si
28 1
15
1»
31 0
16
S
32 1
17
CI
35 5
18
Ar
39 9
4
19
20
21
22
23
24
25
26
27
28
29
30
ЗІ
32
33
34
35
36
К
Ca
Sc
Ti
V
Cr
Mn
Fc
Co
Ni
Cu
Zn
Ga
Gc
As
Sc
Br
Kr
39 1
40 1
45 0
47 9
50 9
52.0
54 9
55 9
58 9
58 7
63 5
65 4
69 7
7 ,5
74 9
79 0
79 9
83 8
5
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
Rb
Sr
Y
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Ag
Cd
In
Sn
Sb
Tc
1
Xe
85 5
87 6
88 9
91 2
92 9
95.9
99
101 1
102 9
106 4
107 9
1124
1148
1187
121 8
127 6
126 9
131.3
6
55
56
57
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
Cs
Ba
La*
Hf
Ta
W
Rc
Os
Ir
Pl
Au
Hg
TI
Pb
Bi
Po
Al
Rn
132 9
137 3
138.9
178 5
181 0
183 9
186 2
190 2
102 2
195 5
197 0
200 6
204 4
207 2
209 0
210
210
222
7
пери¬
оды
87
Fr
223
88
Ra
226
89
Ac*
227
104
Ku
261
Лантаноиды* и актиноиды»
58
Сс
140 1
59
Рг
140 9
60
N<1
144 2
61
Рш
147
62
Бт
150 4
63
Ей
152.0
64
в!!
157 3
65
ТЬ
158 9
66
оу
162 5
67
Но
164 9
68
Ег
167 3
69
Тт
168 9
70
УЬ
173э0
71
1.и
175.0
90
91
92
93
94
95
96
97
98
99
100
101
102
103
1Ь
1>а
и
\р
Ри
Ат
Ст
Вк
СГ
Ел
Кт
Ш
N0
Ьг
232 0
231
238 1
237
242
243
247
245
251
254
253
256
254
257
Рис. 1.3. Периодическая таблица элементов.
Исходя из электронной конфигурации, можно различать четыре
класса элементов: инертные газы, типичные элементы, переходные
элементы и внутрирядные переходные элементы. Эта классифика¬
ция основывается на том, заполнены или нет те или иные элек¬
тронные орбитали.
Инертные газы. За исключением гелия, имеющего электрон¬
ную конфигурацию Ь2, все элементы этого класса имеют во внеш¬
нем уровне заполненные б- и р-орбитали. Это самый малочисленный
класс элементов-, состоящий всего из шести членов.
До недавнего времени все они считались химически неактивны¬
ми вследствие большой устойчивости полностью заполненной кон¬
фигурации П52пр6. Однако в 1962 году были получены первые сое¬
динения криптона, ксенона и радона. В настоящее время коли¬
чество соединений этих элементов достаточно велико, причем неко¬
торые из них уже нашли и практическое применение. Поэтому на¬
звание “инертные газы” сохранилось по традиции.
Типичные элементы. У этого класса элементов все уровни,
кроме внешнего, заполнены; они имеют электронную конфигура¬
цию от лб1 до ЛБ2лр5. В этом классе 46 членов, включая элемен¬
ты подгруппы меди и цинка, а также иттербий и 102-й элемент
(нобелий).
Химические свойства элементов этого класса в большей степе¬
ни определяются стремлением их атомов получить, отдать или
обобщить электроны таким образом, чтобы приобрести элек¬
тронную конфигурацию инертного газа с большим или меньшим по¬
рядковым номером (так называемую конфигурацию псевдоинерт-
ного газа (л-1Ь2р6с110 или Л52р6с110). К этому классу относятся мно¬
гие металлы и все неметаллы.
Переходные элементы. Эти элементы имеют два незаполнен¬
ных внешних уровня и, что особенно характерно, (л-1)с1-орбитали.
Все переходные элементы - металлы, по большей части очень проч¬
ные и хорошо проводящие тепло и электричество. Эти металлы
имеют переменную валентность и образуют огромное количество
окрашенных и парамагнитных комплексных соединений
Внутрирядные переходные элементы. В их атомах неза¬
полненными оказываются три уровня, в том числе (л-2)[-подуровни
Особенностью этих элементов, называемых лантаноидами и актино¬
идами, является большее сходство их химических свойств по срав¬
нению со сходством свойств элементов других классов
Еще одна классификация элементов основана на признаке за¬
полнения электронных подуровней и разделяет элементы на б-, р-,
с1- и [-элементы.
29
Существуют и иные принципы разделения элементов на классы
Каждая из классификаций имеет £вои достоинства и недостатки
К той или иной из них прибегают по соображениям удобства при
рассмотрении конкретных свойств элементов
1.4. Периодическое изменение свойств элементов
Закономерности изменения электронных конфигураций элемен¬
тов Периодической системы находят свое общее выражение в
Периодическом законе, впервые сформулированном Менделеевым.
В современной интерпретации Периодический закон гласит, что
свойства элементов находятся в периодической зависимости от за¬
ряда атомных ядер элементов
При этом под элементом Э подразумевают совокупность атомов
с одинаковым зарядом ядра, что не следует путать с простым ве¬
ществом Простые вещества записывают символом Э„, где п может
принимать различные целочисленные значения. Например, для эле¬
мента кислорода существуют такие разновидности простых ве¬
ществ, как молекулярный кислород Ог и озон Оз.
Собственно говоря, строение внешней, или, как принято гово¬
рить, валентной электронной оболочки уже представляет собой пе¬
риодическую функцию заряда ядра.
Радиус атома (гэ). Радиус атома во многом определяется ра¬
диусом внешних (валентных) орбиталей. С позиций квантовой ме¬
ханики изолированный атом не имеет строго определенного раз¬
мера, так как электронная плотность равна нулю лишь при беско¬
нечном удалении от ядра Однако электронная орбиталь становит¬
ся размытой уже на расстоянии нескольких ангстремов от ядра.
1 Ангстрем (А) равен 108 см.
Хотя абсолютные размеры атомов точно определить невозмож¬
но, достоверные суждения о геометрических параметрах атомов
можно получить, пользуясь представлением об эффективных ради¬
усах атомов. Их оценивают из экспериментальных данных по межъ-
ядерным расстояниям в молекулах и кристаллах. Тогда можно рас¬
сматривать атомы как несжимаемые шары, соприкасающиеся сво¬
ими поверхностями
Иными словами, эффективные радиусы атомов - это радиусы,
при которых начинается действие вандерваальсовых сил между
атомами. Радиусы некоторых атомов приведены в табл. 1.4.
Из данных таблицы 1 4 видно, что изменение эффективных ра¬
диусов атомов носит периодический характер. В периодах они
уменьшаются, что вызвано ростом сил электростатического взаимо¬
действия. С'увеличением номера периода радиусы растут, так как
30
увеличивается число электронных уровней. В подгруппах 6- и Г-
элементов радиусы атомов имеют тенденцию к плавному убыванию.
Эти явления называют (1- и Г-сжатием, соответственно.
Таблица 1.4
Радиусы некоторых атомов гэ, А
н
0.37
и
1.55
Ве
1.13
В
0.89
С
0.77
N
0.70
О
0.66
Р
0.64
№
1.89
Мд
1.60
АІ
1 43
1.17
Р
1.10
Б
1.04
СІ
0.99
К
2.36
Са
1.97
Вг
1.14
Энтальпия ионизации АН,0. Энтальпия ионизации (энергия
ионизации, потенциал ионизации) есть наименьшее количество
энергии, необходимое для удаления одного электрона от свободного
атома, находящегося в газовой фазе в низшем энергетическом со¬
стоянии.
Эг -> Эг+ + е + ДН,°
Полная картина изменения энтальпий ионизации в зависимости
от атомного номера элемента показана на рис. 1.4.
Рис 1.4 Зависимость энтальпий ионизации от порядкового
номера элементов
31
Следует отметить три основные тенденции изменения ДН,0.
Во-первых, максимумы наблюдаются для благородных газов,
минимумы - для щелочных металлов Это легко понять, т К 32
кнутые электронные оболочки инертных газов очень устойчивы и
разрыв требует больших затрат энергии Наоборот, у атомов и
лочных металлов имеется лишь одни валентный электрон, котор|
хорошо экранирован от ядра внутренними оболочками. Поэтому
относительно легко отрывается
Во-вторых, при движении от щелочного металла к инертно
газу наблюдается постепенное возрастание энтальпии ионизаш
Причина этого в том, что заряд ядра при движении по ряду в<
растает, а прибавляющийся на каждом этапе электрон лишь ч.
тично экранирует остальные электроны от растущего заряда ядра
В-третьих, возрастание энтальпии ионизации не является гл;
ким В каждом из периодов явно проявляются два скачка. Во вс
случаях энтальпия ионизации падает при переходе от коифигурац
б2 к Б2р* и от Б2р3 к б2р4. Как уже упоминалось, заполненные
полузаполненные слои обладают повышенной стабильностью. Э
“аномалии” в электронных конфигурациях и сказываются на ве;
чинах энтальпии ионизации.
Энтальпия присоединения электрона АН°еа*. Энтальп
присоединения электрона (или сродство к электрону) - ЭТО КОЛИ1
ство энергии, которое выделяется или поглощается при присое;
нении электрона к нейтральному атому в газовой фазе в его осн<
ном состоянии с образованием газообразного отрицательно за{
женного иона также в основном состоянии. Иначе говоря, энта,
пия присоединения электрона равна энергии процесса:
Эг + е -> Эг" ± ДН°еа
Прямое определение энтальпии присоединения электрона из
больших экспериментальных трудностей было сделано лишь для I
большого числа элементов, например, галогенов. Большинство 31
чений ДН°еа было получено расчетным путем (табл. 1.5).
Из данных табл 1.5 видно, что энтальпия присоединения эл<
трона к хлору наивысшая, т.е. он должен быть самым сильш
окислителем. Неожиданно низкое значение ДН°еа для фтора мож
приписать сильному взаимному отталкиванию пары электронов,
нимающих при приеме электрона компактную 2р-орбиталь. Эт
межэлектронным отталкиванием можно объяснить, по-видимол
’ От англ electron affinity - сродство к электрону.
32
и низкую энтальпию диссоциации молекулы фтора по сравнению с
хлором, а это, в свою очередь, вызывает более высокую хими¬
ческую активность фтора.
Таким образом, характер изменения энтальпии присоединения
электрона в зависимости от порядкового номера элемента будет на¬
поминать таковую для энтальпии ионизации.
И в этом случае налицо периодическая зависимость, однако
экстремальные точки смещены на один элемент влево по сравнению
с соответствующей зависимостью для ДН0,. В общем склонность к
образованию анионов возрастает слева направо в пределах периода
и убывает сверху вниз внутри группы.
Таблица 1.5
Энтальпия присоединения электрона (ДН°еа)
некоторых элементов в кДж/моль
н
-73
и
Ве
В
С
N
О
Р
-57
18
-29
-123
20
-142
-333
N3
Мд
А1
Р
Б
С1
-33
21
-48
-178
-71
-200
-350
К
-48
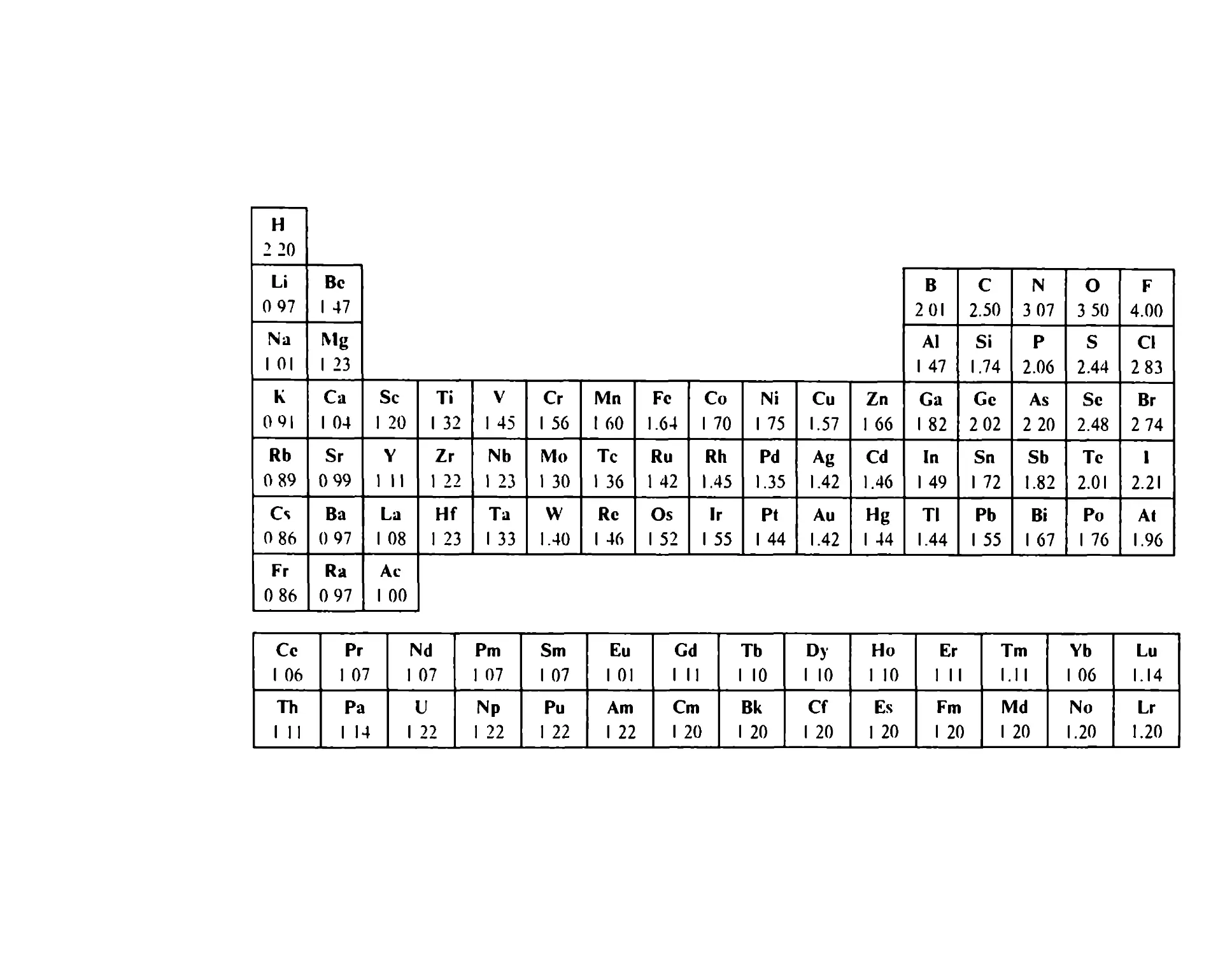
Электроотрицательность. Еще одной, причем очень важной,
характеристикой элементов является их электроотрицательность,
которая характеризует способность атома в молекуле притягивать к
себе электроны Не следует путать это понятие с энтальпией при¬
соединения электрона, т.к первое относится к атому в молекуле, а
второе - к изолированному атому.
Впервые понятие электроотрицательности ввел Л Полинг в
1932 году. Используя эмпирические данные и теоретические пред¬
ставления, произвольно приняв значение электроотрицательности
для водорода равным 2.2, он составил самую первую, широко из¬
вестную шкалу относительных электроотрицательностей ОЭО
(табл 1 6)
Электроотрицательность уменьшается сверху вниз по группе
для типичных элементов (например, от лития к францию) и увели¬
чивается слева направо в периоде Для переходных элементов она
несколько падает в пределах периода Таким образом, наименьшую
электроотрицательность среди всех элементов имеет франций, а
наибольшую - фтор.
2 Зек 675
33
Лучшее определение электроотрицательности было предложено
Малликеном, согласно которому она представляет собой полусумму
ДН° + ДН°
энтальпий ионизации и присоединения электрона .
Численные значения электроотрицательностей по Полингу и
Малликену различаются, однако характер изменений остается од¬
ним и тем же.
Строго говоря, электроотрицательность одного и того же атома
должна различаться в зависимости от партнера и от строения моле¬
кулы. Этим и объясняется наличие многих шкал электроотри¬
цательностей Следует поэтому помнить, что численные значения
электроотрицательностей не имеют смысла абсолютных параметров,
однако они чрезвычайно удобны для качественных оценок характера
и направления смешения электронов вдоль химических связей Так,
например, смешения электронов связей Ы-Н и Р-Н должны быть
противоположны, что и согласуется, как это показано, с четким
различием в химических свойствах этих гидридов.
1\ > Н Р < Н
0.97 Д(-1.23) 2.2 4.1 Д(+1.9) 2.2
В целом, в образовании, структуре и свойствах гидридов про¬
слеживаются периодические закономерности. Периодичность в из¬
менении свойств наблюдается и для других классов однотипных
соединений - оксидов, гидроксидов, галогенидов и др. Это позволя¬
ет говорить об аналогиях в свойствах элементов с однотипной элек¬
тронной конфигурацией.
Руководствуясь определением относительной электроотрица¬
тельности, можно разделить элементы на электроположительные и
электроотрицательные.
Наиболее электроположительные элементы, а следовательно
наиболее химически активные элементы, располагаются в левом
углу Периодической системы.
Наиболее электроотрицательные, а следовательно также наибо¬
лее реакционноспособные элементы располагаются в верхнем пра¬
вом углу таблицы.
Электроотрицательность непосредственно определяет тип хими¬
ческих реакций, в которые вступает данный элемент, а следова¬
тельно и тип соединений, образуемых этим элементом.
Величина электроотрицательности, в частности, разделяет эле¬
менты на металлы и неметаллы. Все б-, (1- и 1-элементы (кроме во¬
дорода и гелия) являются металлами. Первые из них наиболее
34
H
2 20
Li
0 97
Bc
1 47
В
201
С
2.50
N
3 07
О
3 50
F
4.00
Na
1 01
Mg
1 23
Al
1 47
Si
1.74
P
2.06
S
2.44
CI
2 83
к
091
Ca
1 04
Sc
1 20
Ti
1 32
V
1 45
Cr
1 56
Mn
! 60
Fc
1.64
Co
1 70
Ni
1 75
Cu
1.57
Zn
1 66
Ga
1 82
Gc
2 02
As
2 20
Sc
2.48
Br
2 74
Rb
0 89
Sr
0 99
Y
1 II
Zr
1 22
Nb
1 23
Mo
1 30
Tc
1 36
Ru
1 42
Rh
1.45
Pd
1.35
Ag
1.42
Cd
1.46
In
1 49
Sn
1 72
Sb
1.82
Tc
2.01
1
2.21
Cs
0 86
Ba
0 97
La
1 08
Hf
1 23
Ta
1 33
w
1.40
Rc
1 46
Os
1 52
Ir
1 55
Pt
1 44
Au
1.42
Hg
1 44
TI
1.44
Pb
1 55
Bi
1 67
Po
1 76
At
1.96
Fr
0 86
Ra
0 97
Ac
1 00
Cc
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
1 06
1 07
1 07
1 07
1 07
1 01
1 II
1 10
1 10
1 10
1 II
III
1 06
1.14
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
1 II
1 14
1 22
1 22
1 22
1 22
1 20
1 20
1 20
1 20
1 20
1 20
1.20
1.20
реакционноспособны, а потому их часто называют активными ме¬
таллами Находящиеся в левой нижней части таблицы р-элемеиты
также относят к числу металлов. Все неметаллы размещаются в
правой верхней части Периодической системы. На границе между
ними располагаются амфотерные элементы, сочетающие в себе
свойства и металлов, и неметаллов Таковыми являются бор, крем¬
ний, мышьяк, теллур и некоторые другие. Таким образом, величина
относительной электроотрицательности меньше 2.0 служит крите¬
рием отнесения элемента к числу металлов.
Следует обратить внимание на еще одну особенность в измене¬
нии свойств элементов в зависимости от их положения в Периоди¬
ческой таблице. Уменьшение электроотрицательности при переме¬
щении вниз по группе компенсируется перемещением вправо вдоль
периода. Это вызывает появление так называемых диагональных со¬
отношений между элементами. Это соотношение связывает между
собой пары элементов со сходными химическим свойствами. Тако¬
выми парами являются литий-магний, бериллий-алюминий, бор-
кремний, сера-бром и некоторые другие.
Глава 2. ХИМИЧЕСКАЯ СВЯЗЬ
Две атомные оболочки
взаимно проницаемы.
Д. Льюис
С момента создания теории химического строения и по сей день
в химии широко используется понятие валентности, которая ха¬
рактеризует способность атома давать химическую связь. Число
связей равно валентности. Доквантовая теория строения установи¬
ла целочисленность валентности, существование простых и кратных
(двойных и тройных) связей, постоянную и переменную валент¬
ность элементов. С развитием стереохимии’ представление о ва¬
лентности дополнилось учением о направленной валентности.
Качественные модели химической связи использовались хими¬
ками задолго до разработки квантовой теории. Так, в 1916 г. Кос-
сель предположил, что, вступая в соединение, атом какого-либо
элемента, теряя или присоединяя определенное число электронов,
приобретает наиболее устойчивую электронную оболочку ближай¬
шего инертного газа (правило октета). Эта гипотеза легла в основу
теории ионной химической связи. Тогда же Льюис выдвинул проти¬
воположный постулат о том, что химическая связь между двумя
атомами образуется двумя электронами, которые одновременно
принадлежат обоим атомам. С тех пор валентный штрих в хими¬
ческой формуле из обозначения связи вообще стал символом общей
пары электронов. Эта идея Льюиса - основа теории ковалентной
химической связи. Обе теории отражали известное химикам свой¬
ство особой устойчивости электронного октета, но они не могли
дать ответа на вопрос, как движутся электроны в молекуле.
Лишь с созданием квантовой теории атома стало ясно, что лю¬
бые статические модели не состоятельны. Квантовая механика раз¬
решила и принципиальный вопрос о природе химической связи
Описание химических связей в молекуле заключается в описа¬
нии распределения электронной плотности. Химическая связь -
это взаимодействие атомов, причем энергия этого взаимодействия
составляет от 40 до 1000 кДж/моль Столь широкий интервал
энергий может быть реализован различными взаимодействиями,
которые в настоящее время классифицируются как ковалентная,
ионная, водородная и металлическая связи В настоящей главе
мы рассмотрим указанные типы связей и их свойства
* Стереохимия - учение о пространственном строении молекул
37
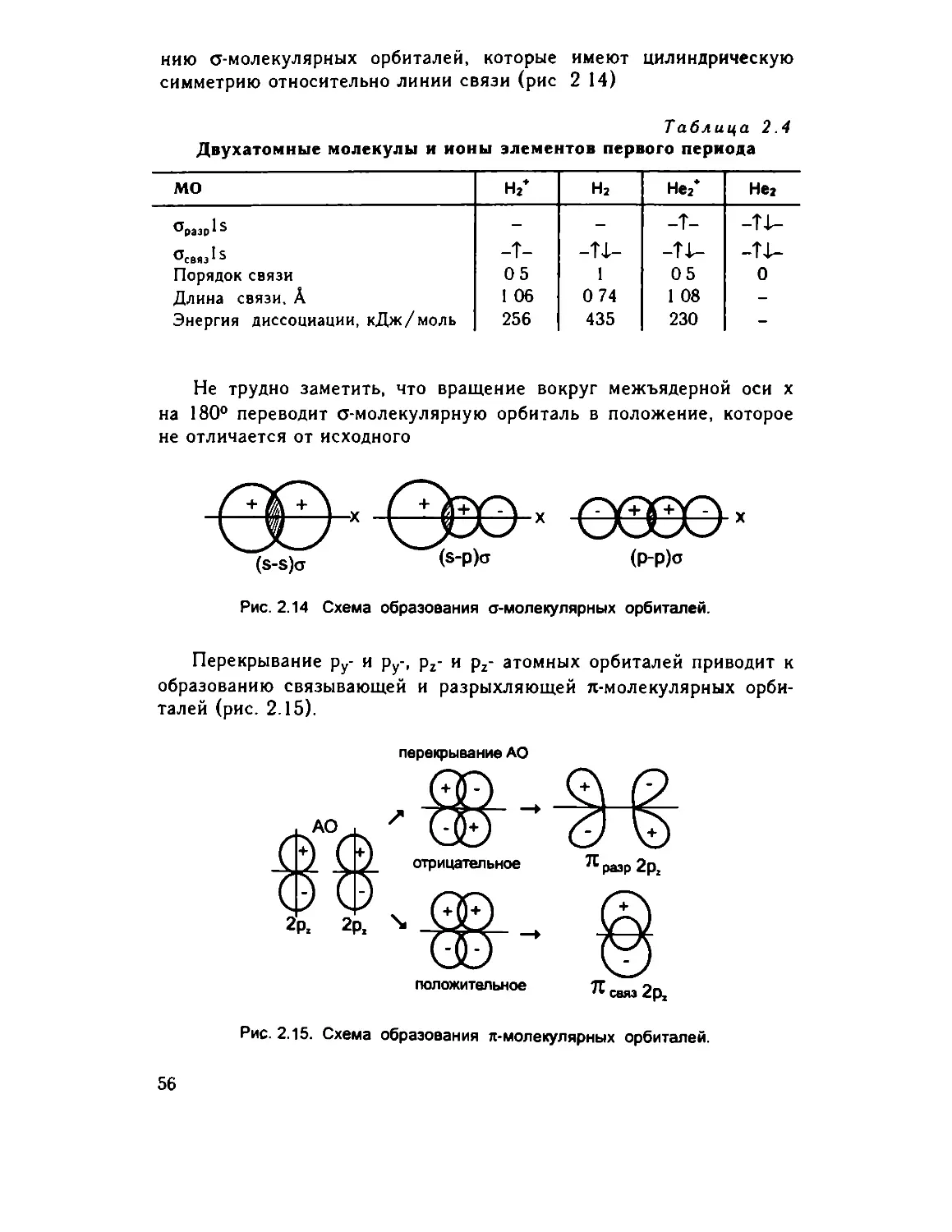
2.1. Ковалентная связь
При квантово-механическом подходе к исследованию строения
молекул необходимо составить и решить уравнение Шредингера для
системы из электронов и ядер, после чего дать физическую интер¬
претацию полученным решениям Решив уравнение Шредингера для
молекулы, мы получили бы геометрические параметры молекулы,
распределение электронной плотности, набор уровней энергии и все
связанные с этим характеристики молекулы Мы знаем, что точное
решение уравнения невозможно даже для многоэлектронного атома
Еще большие математические трудности возникают при расчете мо¬
лекул. Поэтому созданы различные приближенные методы расчета
Наиболее широкое распространение получили два подхода, метод
валентных связей (ВС) и теория молекулярных орбиталей
(МО), которые различаются методами подбора волновой функции
в уравнении Шредингера.
2.1.1. Метод валентных связей
В основе метода валентных связей лежит идея о спаривании
электронов. Предполагается, что каждая электронная пара может
связывать только два ядра, т.е. связь в методе ВС двухэлектронная
и двухцентровая.
При сближении двух атомов, имеющих неспаренные электроны,
становится возможным перекрывание одноэлектронных орбиталей,
в результате чего между атомами появляется область повышенной
электронной плотности.
Рассмотрим, например, процесс образования молекулы водоро¬
да. С уменьшением расстояния между атомами постепенно увели¬
чивается электростатическое притяжение между электронами и
ядрами разных атомов, но одновременно усиливается взаимное от¬
талкивание электронов По мере сближения атомов сила притяже¬
ния достигает максимума, соответствующего образованию молеку¬
лы водорода. При дальнейшем сближении резко возрастают силы
отталкивания.
Изменение энергии системы двух ядер можно представить в
виде расчетной кривой зависимости потенциальной энергии, изоб¬
раженной на рис. 2.1.
Стандартное состояние, для которого энергия равна нулю, соот¬
ветствует двум разделенным атомам (г = °°). В области притяжения
энергия отрицательна, а в области отталкивания - положительна,
следовательно, при образовании связи энергия выделяется.
38
Расстояние между атомами при минимуме потенциальной энер¬
гии называется равновесным расстоянием г°. Однако, поскольку
атомы в молекуле находятся в колебательном движении, в действи¬
тельности низшее состояние молекулы водорода соответствует го¬
ризонтальной линии, проходящей выше экстремальной точки. Пере¬
сечения этой линии с кривой потенциальной энергии дают два
крайних значения длины связи в процессе колебания.
Рис. 2.1. Зависимость потенциальной энергии системы
из двух атомов водорода от расстояния между ядрами.
Кривая потенциальной энергии для связывающего состояния со¬
ответствует процессу сближения двух атомов водорода с антипа-
раллельными спинами электронов. При сближении двух атомов во¬
дорода с паралельными спинами электронов кривая потенциальной
энергии не имеет минимума и отражает монотонное увеличение сил
отталкивания (разрыхляющее состояние).
Согласно этой модели, в молекуле водорода образуется одна ко¬
валентная СВЯЗЬ'
н и1 -Т- Н + Н -» НН
и
При сближении двух атомов гелия образование химической
связи не возможно, г к каждый электрон любого из атомов будет
иметь тот же спин, что и у одного из электронов другого атома
Другими словами, гелий не может образовать молекулу Нег
39
вследствие того, что оба электрона в атоме Не являются спаренны¬
ми. Распространяя проведенные рассуждения на другие системы,
можно сказать, что ковалентная связь возникает в тех случаях,
когда сближаются атомы, имеющие неспаренные электроны.
Так, например, в молекуле воды атом кислорода образует две
ковалентные связи с двумя атомами водорода за счет своих неспа¬
ренных электронов
_Т1_ -Т- -Т-
О 2з22р4 -Т4,- 2рх 2ру 2рг
2б
Приведенные примеры иллюстрируют так называемый обмен¬
ный механизм образования ковалентной связи, когда на образо¬
вание одной связи каждый атом дает по одному электрону.
Возможен и другой механизм образования двухэлектронной
двухцентровой связи: при образовании связи один партнер предо¬
ставляет пару электронов, а другой - свободную (вакантную) орби¬
таль. Примером такого взаимодействия является образование иона
аммония Атом азота имеет пять валентных электронов - три
неспаренных и неподеленную пару. За счет трех неспаренных элек¬
тронов азот образует три ковалентные связи с атомами водорода
в молекуле аммиака (обменный механизм). Далее, при взаимо¬
действии молекулы аммиака с протоном, образуется четвертая ко¬
валентная связь за счет неподеленной пары электронов атома азота
в аммиаке (донор) и валентной орбитали протона (акцептор):
□ н+ -
донор акцептор ион аммония
В молекуле ВЯз атом бора имеет вакантную 2р-орбиталь (элек¬
тронодефицитное соединение). При его взаимодействии с аммиаком
образуется твердое высокоплавкое и устойчивое соединение, в ко¬
тором валентность бора и азота возросли с трех до четырех.
Н
Н :Й :
Н
донор
Я
□ В:Р
Р
акцептор
Н Р
Н :Й : В: Р
Н Р
Это пример реакции между валентнонасыщенными молекулами,
происходящей за счет донорно-акцепторного взаимодействия.
40
Зная два механизма образования ковалентной связи, можно
объяснить ее насыщаемость, т.е. свойство атомов давать вполне
определенное количество ковалентных связей.
Среди элементов II периода для и, N. О, Я наблюдается полное
совпадение числа неспаренных электронов и валентности в про¬
стейших соединениях. Однако различие между валентностью и
числом неспаренных электронов для Ве, В и С заставило сделать
допущение, что в химическое взаимодействие эти атомы вступают
не в основном, а в возбужденом, так называемом валентном со¬
стоянии.
При возбуждении происходит распаривание электронной пары и
переход одного из электронов на вакантную орбиталь (табл. 2.1).
Принимается, что энергия, необходимая для возбуждения, компен¬
сируется энергией, выделяющейся при образовании связей.
Таблица 2.1
Основное и возбужденное состояния некоторых элементов
Переход
Э -» Э*
Энергия
возбуждения,
кДж/моль
Число иеспаренных
электронов
в возбужденном
состоянии
Валент¬
ность
Ве -» Ве’
1s22s2 ls*2sl2pl
264
2
2
В -» В*
ls22s22pl ls22s'2p2
356
3
3
С -» С*
ls22s22p2 ls22s‘2p3
406
4
4
Следует обратить внимание на большую величину энергии воз¬
буждения. Это подчеркивает гипотетический характер валентного
состояния, которое, однако, является полезным представлением,
необходимым для понимания особенностей химической связи
Переменная валентность атомов III периода объясняется суще¬
ствованием в третьем квантовом слое вакантных d-орбиталей По¬
этому валентность хлора равна единице, если он вступает во вза¬
имодействие в основном СОСТОЯНИИ'
CI _____ 3d H-CI
3s23P5 -Ti- -П- -T- 3P H_0_CI
HCl, НСЮ -TI- 3s
Эта возможность реализуется в молекулах HCI и НСЮ.
41
В первом возбужденном состоянии один из Зр электронов пере¬
ходит на Зс1-орбиталь, число неспаренных электронов возрастает до
трех. Трехвалентен хлор, например, в хлористой кислоте НСЮг
сГ -Т- — — — — за
3523р43с11 -Т1- -Т- -Т- Зр Н-0-С1=0
НСЮ2 -ТХ- Зэ
Во втором возбужденном состоянии происходит переход еще
одного Зр-электрона на Зс1-орбиталь, и, наконец, в третьем возбуж¬
денном состоянии - с Зэ-орбитали на Зс1-орбиталь. Число неспа¬
ренных электронов будет составлять, соответственно, пять и семь.
Пяти- и семивалентен хлор в кислородных кислотах НСЮ3 и НСЮ4,
соответственно.
Аналогичным образом можно интерпретировать переменную
валентность и для фосфора.
Для объяснения направленности валентностей в методе ва¬
лентных связей исходят из той же идеи о локализованных парах
электронов, осуществляющих связь между атомами.
Так, в молекуле воды две связи образованы двумя парами элек¬
тронов (обменный механизм). Оба валентных электрона кислорода
находятся в 2р-состоянии. Поскольку атомные р-орбитали взаимно
перпендикулярны в пространстве, именно в направлении осей этих
орбиталей должно происходить максимальное перекрывание с
э-орбиталями атомов водорода. Поэтому следовало ожидать, что
угол ZHOH должен быть равен 90°. Но наблюдаемый угол больше
прямого (104.5°).
Это можно объяснить как взаимным отталкиванием локализо¬
ванных пар электронов связей О-Н, так и отталкиванием частич¬
ных положительных зарядов на атомах водорода, возникающих
вследствие большей электроотрицательности атома кислорода. Так
же можно объяснить и строение аналогов молекулы воды (НгБ,
НгБе, Н2Те).
42
Пирамидальная структура молекулы аммиака объясняется вза¬
имной перпендикулярностью осей трех р-орбиталей, на которых на¬
ходится по одному неспаренному электрону. Перекрывание этих
орбиталей с э-орбиталями трех атомов водорода должно приводить
к трехгранной пирамиде с атомом азота в вершине и атомами водо¬
рода в основании. Предсказанный угол ZHNH равен 90°. Увеличе¬
ние угла для реальной молекулы до 107.3° объясняется так же, как
и для воды.
2.1.2. Гибридизация
Сложнее объяснить структуру молекулы метана. Так как угле¬
род четырехвалентен, то следует допустить, что в соединение с во¬
дородом он вступает в возбужденном состоянии С* (152252рх2ру
2р2). Однако только этого допущения недостаточно. Если следовать
прежним рассуждениям, то четырехвалентный углерод должен да¬
вать три связи под прямыми углами (за счет р-орбиталей) и одну
связь в произвольном направлении (за счет Б-орбитали). Но опыт¬
ные данные, согласно которым в молекуле метана все четыре связи
эквивалентны (т.е. молекула симметрична и имеет форму тетра¬
эдра), противоречат этому выводу.
Исходя из тетраэдрического строения, необходимо предполо¬
жить, что атом углерода образует связи за счет четырех равноцен¬
ных орбиталей, которые направлены под углом 109°28' друг к другу.
Такие орбитали могут быть описаны как линейные комбинации его
б- и р-орбиталей. Эти новые орбитали называются зр3-гибридными
орбиталями, т.к в каждой новой волновой функции, описываю¬
щей гибридную орбиталь, смешаны одна э- и три р-орбитали. Форма
зр3-гибридных орбиталей представлена на рис 2 2
Рис 2.2. Схема зр3-гибридизации
43
Коэффициенты смешения, т е. участия э- и р-состояний в гиб¬
ридной функции, подбираются так, чтобы оси гибридных орбиталей
были направлены в пространстве под углом 109°28', то есть к вер¬
шинам тетраэдра, и тогда перекрывание четырех гибридных орбита-
лей с Б-орбиталями четырех атомов водорода приводит к образова¬
нию четырех равноценных С-Н связей
Направление связей не предсказано в методе ВС. Исходя из
установленной экспериментально реальной конфигурации молекулы
метана, постулируют существование четырех равноценных орбита-
лей, направленных под углом 109°28', которые выражают через ли¬
нейные комбинации орбиталей свободного атома.
Гибридизация 5л3-типа_ характерна и для молекул аммиака, волы
и фтористого водорода лишь с той разницей, что в молекуле аммиа-
14а одна из орбиталей занята электронной парой; в молекуле воды
таких пар две; наконец, в молекуле НР три орбитали заняты непо-
деленными электронными парами.
Аналогично можно рассмотреть соединение типа ВНз. Известно,
что молекула ВНз плоская, с атомом бора в центре, а угол между
эквивалентными связями В-Н составляет 120°. Электронная конфи¬
гурация атома бора в возбужденном состоянии 152252рх2ру.
Для объяснения геометрии реальной молекулы допускается су¬
ществование у атома бора трех гибридных орбиталей, описываемых
смешением одной б- и двух р-орбиталей. Эти три зр2-гибридные ор¬
битали по форме подобны орбиталям, представленным на рис. 2.2,
но находятся в одной плоскости и направлены от центра под углом
120° (рис. 2.3).
Перекрывание трех Бр2-гибридных орбиталей с э-орбиталями
трех атомов водорода отвечает образованию плоской симметричной
молекулы ВНз* (рис. 2.4).
В действительности молекула ВН3 димерна и имеет состав В2Н6 Этот
вопрос обсужден в гл. 16
Рис. 2.3.
Схема зр2-гибридизации.
Рис. 2.4. Схема
образования молекулы ВНз.
44
Аналогично, атом Ве, имеющий в возбужденном состоянии
электронную конфигурацию 1522з12рх, образует две гибридные эр-
орбитали, расположенные под углом 180° (рис. 2.5).
- < - >
180°
э + р 2 ер
Рис. 2.5. Схема эр-гибридизации.
Перекрывание эр-гибридных орбиталей атома бериллия с Б-ор-
биталями двух атомов водорода приводит к образованию линейной
молекулы ВеНг.
Чтобы объяснить октаэдрическую структуру молекулы БРб, сле¬
дует принять, что сера участвует в образовании связи в возбужден¬
ном состоянии 3513р33<^2. В поле шести атомов фтора происходит
гибридизация шести орбиталей, на которых находится по одному
электрону (зр3с)2-гибридизация); коэффи¬
циенты в гибридной функции выбираются
таким образом, чтобы оси зр3с!2-гибридных
орбиталей были направлены к вершинам
октаэдра (рис. 2 6).
Гибридизация не исчерпывается рас¬
смотренными случаями. Существуют и
иные типы гибридных орбиталей.
Концепция гибридизации позволяет
дать описание стереохимии молекул, од¬
нако сомнения вызывает огромная вели¬
чина энергии возбуждения атома из рИс. 2.6. Молекуле БРв.
основного в валентное состояние соответ¬
ствующей гибридизации Так, для атома
углерода она составляет 406 кДж/моль (см. табл. 2 1). Избежать
этого позволяет динамическая теория гибридизации.
Ясно, что четыре атома водорода не могли одновременно при¬
соединиться к атому углерода в молекуле метана и занять тетра¬
эдрические позиции Следовательно, существует некоторая дина¬
мика образования гибридных орбиталей. В основном состоянии
атом углерода имеет два неспаренных электрона (С 2з22рх2ру). При
атаке первого атома Н сферически симметричное электронное об¬
лако атома С начнет искажаться Образуется молекула метилена
45
СН2 за счет неспаренных р-электронов. Два оставшихся э-электрона
атома углерода могут остаться спаренными, но будут находиться на
деформированной э-орбитали и пребывание на ней пары электронов
уже не так выгодно, как в свободном атоме
Энергетически предпочтительнее, чтобы э-электроны распари¬
лись и заняли две оставшиеся орбитали, которые также будут де¬
формированы под влиянием связей С-Н и кулоновского отталкива¬
ния электронов При этом молекула СН2 будет иметь валентный
угол намного больше 90° (опыт дает угол >120°).
Дальнейшее присоединение атомов водорода к молекуле СН2 с
образованием плоской частицы СНз и, наконец, тетраэдрической
молекулы СН4 может быть представлено аналогичным образом. При
таком динамическом описании стереохимии молекул, в котором
движущей силой гибридизации атомных орбиталей считаются куло-
новские взаимодействия, не надо вводить допущение о предвари¬
тельном возбуждении атома.
До сих пор мы обсуждали формы молекул, которые имеют толь¬
ко а-связи, то есть связи, образующиеся за счет перекрывания
орбиталей вдоль линии соединения атомов.
Одна из основных особенностей атомов углерода, азота и кис¬
лорода состоит в том, что они чаще, чем другие элементы, образуют
кратные - двойные и тройные связи.
Двойная связь, например, образуется между атомами углерода
в молекуле этилена Оба углеродных атома находятся в Бр2-гиб-
ридном состоянии, имея по три гибридных орбитали и одной
“чистой” р-орбитали Гибридные орбитали образуют три а-связи:
одну связь С-С и две связи С-Н, которые лежат в одной плоскости
под углом 120° друг к другу. Оставшиеся негибридизированными р-
орбитали атомов углерода перпендикулярны этой плоскости и их
перекрывание приводит к образованию л-связи (рис. 2.7). л-Связь
предполагает максимум перекрывания орбиталей вне линии, соеди¬
няющей центры атомов.
Перекрывание трех р-орбита-
лей двух атомов углерода в мо¬
лекуле ацетилена С2Н2 дает
тройную связь: рх-орбитали об¬
разуют а-связь, а ру и рг-орби-
тали - две л-связи.
Наряду с валентным углом
(углом между связями) наиболее
важными характеристиками хи-
Рис. 2.7. Схема образования мической связи являются ее
л-связи в молекуле этилена. длина и энергия.
46
Под длиной связи понимается равновесное расстояние между
ядрами двух связанных атомов. Эта величина определяется методом
дифракции электронов или рентгеновских лучей и находится обыч¬
но в интервале 1-2 А (1 ангстрем = 10'8 см). Энергия связи - это
количество энергии, необходимое для ее разрыва.
Как видно из табл. 2.2, в которой в основном представлены
важнейшие типы связей, встречающихся в биомолекулах, с повы¬
шением кратности связи уменьшается ее длина, но при этом растет
энергия диссоциации.
Таблица 2 2
Длины и энергии некоторых связей
Связь
Длина
связи,А
Энергия,
кДж/моль
Связь
Длина
связи,À
Энергия,
кДж/моль
с-с
1.54
348
H-N
1 03
393
с=с
1.35
620
Н-0
0.96
460
с=с
1.20
811
H-S
1.34
368
С-0
1 43
360
H-CI
1.27
431
с=о
1.17
724
N-N
1.45
160
C-N
1.47
276
N=N
1.10
418
C=N
1.34
615
NsN
1.01
947
C=N
1 16
761
N-O
1 46
176
C-S
1.81
255
0-0
1.45
146
C=S
1 55
477
S-S
2.05
226
H-C
1.09
374
P-О
1.62
502
Пространственное строение молекул принято представлять с
помощью шаростержневых моделей с учетом валентных углов и
длин связей, как это показано для метана на рис. 2.8.
Более совершенными являются полусферические модели. Ато-мы
при таком способе изображаются шарами, радиус которых отвечает
вандерваальсовым радиусам. Этот радиус отсекает сферу, внутрь
которой не может проникать другой атом, не имеющий связи
с данным атомом Чтобы получить правильное расстояние между
центрами атомов в молекуле (т е правильную длину связи),
надо отсечь часть сферы на расстоянии, именуемом ковалентным
радиусом.
Ковалентный радиус данного атома равен половине длины
химической связи между двумя одинаковыми атомами Например, в
молекуле водорода длина связи Н-Н составляет 0.74 А, следова¬
тельно, ковалентный радиус равен 0.37 А.
47
<ЗЕ) #>' Л
а б в
Рис. 2 8 Пространственное строение молекул: а - полусферическая модель
молекулы водорода, б - полусферическая модель молекулы метана,
в - шаростержневая модель молекулы метана.
Из таблицы 2.2 видно, что биомолекулы используют в своем со¬
ставе широкий ассортимент ковалентных связей
Углерод-углеродные простые и двойные связи составляют остов
органических молекул. Связями углерод-элемент на этом остове
крепятся функциональные группы - амино-, гидроксильная и др С
помощью связей О-Н и N-1-1 построены такие простые неорганиче¬
ские биомолекулы, как вода и аммиак.
Прочная связь Б-Б обеспечивает ковалентную сшивку молекул
белков. Менее прочная связь 0-0 присутствует в биомолекуле пе¬
роксида водорода, и эта невысокая прочность связи определяет
чрезвычайно легкое разрушение молекулы пероксида водорода. На¬
против, весьма богатая энергией связь Р-0 служит универсальным
источником энергии, обеспечивающим протекание всех метаболиче¬
ских процессов организма. Она приобрела название макроэрги-
ческой (то есть богатой энергией) связи.
2.1.3. Резонанс
Часто оказывается, что свойства молекулы не удается пол¬
ностью описать с помощью какой-либо структуры. Примером мо¬
жет служить карбонат-ион. Он имеет плоскую структуру с углом
Z0C0 120°. Такая геометрия легко объясняется, исходя из эр2-
гибридизации атома углерода.
Рис. 2.9. Строение карбонат-иона.
Экспериментально установлено, что все три связи С-0 имеют
одинаковую длину и энергию. Поэтому очевидно, что структурная
формула (а) на рис. 2.9 не дает истинного представления о струк¬
туре карбонат-иона.
Поскольку положение двойной связи С=0 совершенно произ¬
вольно, с равным основанием можно изобразить две другие струк¬
туры (рис. 2.9, б, в). Далее можно представить, что истинная
структура резонирует между этими структурами, и свойства иона
лучше всего объясняются всеми тремя резонансными структурами
одновременно.
Полезно обратить внимание, что символ резонанса между
структурами изображается обоюдоострой стрелкой.
Обычно для краткости пользуются одной формулой, в которой
делокализованные л-орбитали изображают пунктиром (рис. 2.9, г).
В методе ВС волновая функция карбонат-иона выражается в ви¬
де линейной комбинации
У = Са-Уа+Св-Ув+Сс-ус,
где уд, Ув> Ус. - волновые функции отдельных резонансных струк¬
тур, а СА, Св и Сс - коэффициенты, определяющие вклад каждой из
структур. В случае карбонат-иона, очевидно, все три структуры
эквивалентны и СА, Св и Сс - одинаковы.
О
НО—5—0
А
2-
0
НО—Р=0
А
2-
0
НО—Р—0
4
2-
0
1
а
6
в
Г
Рис. 2.10. Строение гидрофосфат-иона.
Еще один пример резонансной структуры - также биологически
значимый гидрофосфат-ион (рис. 2 10). Из рисунка следует, что
гидрофосфат-ион имеет тетраэдрическую конфигурацию с Бр3-гиб-
ридным атомом фосфора
Идея резонанса является умозрительной концепцией, а резо¬
нансные структуры не отражают какого-либо свойства реальной мо¬
лекулы и не имеют физического смысла Однако резонансные
структуры удобны для получения приближенного решения сложного
волнового уравнения Кроме того, они полезны тем, что позволяют
химику представить образование молекулы с помощью удобных ва¬
лентных схем, даже если реальное состояние молекулы не может
быть отражено одной традиционной структурной формулой
49
Рис 2 8. Пространственное строение молекул- а - полусферическая модель
молекулы водорода, б - полусферическая модель молекулы метана,
в - шаростержневая модель молекулы метана.
Из таблицы 2.2 видно, что биомолекулы используют в своем со¬
ставе широкий ассортимент ковалентных связей
Углерод-углеродные простые и двойные связи составляют остов
органических молекул. Связями углерод-элемент на этом остове
крепятся функциональные группы - амино-, гидроксильная и др С
помощью связей О-Н и N-1-1 построены такие простые неорганиче¬
ские биомолекулы, как вода и аммиак
Прочная связь Б-Б обеспечивает ковалентную сшивку молекул
белков. Менее прочная связь 0-0 присутствует в биомолекуле пе¬
роксида водорода, и эта невысокая прочность связи определяет
чрезвычайно легкое разрушение молекулы пероксида водорода. На¬
против, весьма богатая энергией связь Р-0 служит универсальным
источником энергии, обеспечивающим протекание всех метаболиче¬
ских процессов организма. Она приобрела название макроэрги-
ческой (то есть богатой энергией) связи.
2.1.3. Резонанс
Часто оказывается, что свойства молекулы не удается пол¬
ностью описать с помощью какой-либо структуры. Примером мо¬
жет служить карбонат-ион. Он имеет плоскую структуру с углом
ZOCO 120°. Такая геометрия легко объясняется, исходя из эр2-
гибридизации атома углерода.
48
Экспериментально установлено, что все три связи С-0 имеют
одинаковую длину и энергию. Поэтому очевидно, что структурная
формула (а) на рис. 2.9 не дает истинного представления о струк¬
туре карбонат-иона.
Поскольку положение двойной связи С=0 совершенно произ¬
вольно, с равным основанием можно изобразить две другие струк¬
туры (рис. 2.9, б, в). Далее можно представить, что истинная
структура резонирует между этими структурами, и свойства иона
лучше всего объясняются всеми тремя резонансными структурами
одновременно.
Полезно обратить внимание, что символ резонанса между
структурами изображается обоюдоострой стрелкой.
Обычно для краткости пользуются одной формулой, в которой
делокализованные л-орбитали изображают пунктиром (рис. 2.9, г).
В методе ВС волновая функция карбонат-иона выражается в ви¬
де линейной комбинации
V = СА • Vа + св' Ув + Сс • Ус.
где уд. Ув. Ус. - волновые функции отдельных резонансных струк¬
тур, а Сд, Св и Сс - коэффициенты, определяющие вклад каждой из
структур. В случае карбонат-иона, очевидно, все три структуры
эквивалентны и Сд, Св и Сс - одинаковы.
О
2-
О
2-
о
2-
О
II
НО—Р—о
, ,
но—р=о
,
НО—Р—о
1!
А
д
о
X О
о
о
О
а б в г
Рис. 2.10. Строение гидрофосфат-иона.
Еще один пример резонансной структуры - также биологически
значимый гидрофосфат-ион (рис. 2 10) Из рисунка следует, что
гидрофосфат-ион имеет тетраэдрическую конфигурацию с Бр3-гиб-
ридным атомом фосфора
Идея резонанса является умозрительной концепцией, а резо¬
нансные структуры не отражают какого-либо свойства реальной мо¬
лекулы и не имеют физического смысла Однако резонансные
структуры удобны для получения приближенного решения сложного
волнового уравнения Кроме того, они полезны тем, что позволяют
химику представить образование молекулы с помощью удобных ва¬
лентных схем, даже если реальное состояние молекулы не может
быть отражено одной традиционной структурной формулой
49
2.1.4. Полярность
Для химической связи между разными атомами характерно
смещение электронной плотности к более электроотрицательному
атому. Это приводит к возникновению частичных положительных и
отрицательных зарядов на атомах, т е связь обладает постоянным
электрическим дипольным моментом (ц) Он определяется как
произведение заряда (ц) на расстояние между ними (г)- ц = ц-г.
В системе СГС заряд электрона 4.8-1010 эл.ст.ед , и если сред¬
нее расстояние между атомами Ю'10 см (г), то дипольный момент
равен: ц = 4 8-10 |010 8 = 4.8 10 18 эл.ст.ед. = 4.80, где Ю =1018
эл.ст.ед.-см и называется дебаем (О) в честь ученого, который был
пионером в этой области.
Дипольный момент химической связи обычно мало изменяется
при переходе от одного соединения к другому, что дает возмож¬
ность рассчитывать дипольные моменты молекул как векторные
суммы дипольных моментов отдельных связей.
В табл. 2.3 приведены дипольные моменты некоторых связей.
Видно, что имеется приблизительная корреляция между величиной
дипольного момента связи и разностью электроотрицательностей
атомов (А ОЭО).
Таблица 2.3
Дипольные моменты связей
Связь
Дипольный
момент, О
Д ОЭО
1 Связь
Дипольный
момент, О
Д ОЭО
Н-0
1 5
1.30
с-м
02
0.57
н-м
1 3
0 87
С-Р
1.4
1 60
Н-Р
04
0 14
С-С1
1.5
0 33
с-н
04
0.30
С-Вг
1 4
0.24
С-0
07
1 00
С-1
1.2
0.01
Полярные молекулы (ц * 0) под действием внешнего электри¬
ческого поля ориентируются по направлению поля. Однако одно¬
временно происходит и изменение их дипольного момента за счет
смещения электронной плотности (отчасти и ядер).
Для не очень сильных полей величина наведенного (индуци¬
рованного) дипольного момента (циид) пропорциональна напряжен¬
ности поля (Н):
Циид = а • Н.
50
Коэффициент пропорциональности а называется поляризуе¬
мостью; он тем больше, чем легче смещается электронная плот¬
ность связей. Наведенный дипольный момент исчезает, как только
поле снимается.
Поляризуемость увеличивается по мере увеличения длины свя¬
зи. Неодинакова она для ст и л-связей. Электронное облако ст-связи
в значительно большей степени экранировано от действия внешнего
электрического поля, чем облако л-связи. Поэтому ст-связи поляри¬
зуются значительно слабее.
Поляризуемость во многом определяет свойства веществ. Так,
например, в ряду галогеноводородных кислот дипольный момент
связи падает от НР к Н1. Однако увеличение длины связи приводит
к возрастанию поляризуемости в этом ряду. Это объясняет значи¬
тельно большую степень диссоциации Н1 в водном растворе по срав¬
нению с НР.
Изложенный выше метод ВС обладает стройностью и нагляд¬
ностью. Он переводит на язык квантовых представлений привычные
структурные формулы, в которых под каждым валентным штрихом
понимается пара электронов. Однако метод ВС оказывается
недостаточным для объяснения некоторых свойств даже достаточно
простых молекул.
Так, в рамках метода ВС непонятен парамагнетизм кислорода.
Способность притягиваться магнитом свидетельствует о наличии в
молекуле неспаренных электронов. Действительно, молекула кис¬
лорода содержит два неспаренных электрона; изображение молеку¬
лы с двойной связью между атомами кислорода 0=0 не является
удовлетворительным описанием.
Это далеко не единственный случай, когда метод ВС не может
описать магнитных свойств веществ.
Если исходить из положения, что валентность элемента равна
числу неспаренных электронов на внешнем уровне, то инертные га¬
зы не должны вступать в химические реакции Между тем, в на¬
стоящее время известно более сотни соединений Хе, Кг, Рп с кис¬
лородом и галогенами.
Аналогичные затруднения возникают при описании металлоор¬
ганических соединений, молекул с “дефицитом электронов" (типа
диборана ВгНе) и др
Таким образом, несмотря на простоту и наглядность метода ва¬
лентных связей в качественной интерпретации, абсолютное пред¬
почтение в теоретической химии отдается методу молекулярных ор¬
биталей.
51
2.1.5. Метод молекулярных орбиталей
В методе молекулярных орбиталей (МО) к рассмотрению элек¬
тронной структуры молекулы подходят так же, как и к многоэлек¬
тронному атому. Логичность использования одной физической мо¬
дели вытекает из одинаковой природы электронов в атомах и моле¬
кулах, а также их взаимодействий с ядрами и между собой.
Основные принципы метода МО:
1 Молекула рассматривается как совокупность ядер и электро¬
нов, где каждый электрон движется в поле остальных электронов и
всех ядер.
2. Состояние электрона описывается волновой функцией у, ха¬
рактеризуемой определенным набором квантовых чисел. Эта функ¬
ция называется молекулярной орбиталью (МО). В отличие от
одноцентровой атомной орбитали (АО - электрон в поле одного
ядра) молекулярная орбиталь в общем случае многоцентровая. Как
и для электрона в атоме, квадрат модуля волновой функции I у I 2
определяет плотность вероятности нахождения электрона или
плотность электронного облака.
3. Каждой МО соответствует определенная энергия Е.
4. Совокупность МО молекулы, занятых электронами, называет¬
ся электронной конфигурацией молекулы. Она строится на основе
фундаментальных положений - принципа наименьшей энергии
(электрон занимает в молекуле свободную орбиталь с наименьшей
энергией), принципа Паули (на одной МО не может находиться бо¬
лее двух электронов, при этом спины их должны быть антипарал-
лельны) и правила Хунда. Следовательно, для описания электрон¬
ной конфигурации молекулы с 2п электронами требуется п молеку¬
лярных орбиталей.
Описать молекулу в методе МО - значит определить тип ее
орбиталей, их энергию и характер распределения электронов по
орбиталям.
Так как молекулярные орбитали являются многоцентровыми, то
в простейшем приближении их можно представить как линейную
комбинацию (сумма и разность) атомных орбиталей. В этом заклю¬
чается один из наиболее распространенных способов прибли¬
женного описания волновой функции электрона в молекуле (МО
ЛКАО - молекулярная орбиталь есть линейная комбинация атом¬
ных орбиталей).
Так, если атомные орбитали обозначить уд и Ув- то их линей¬
ная комбинация дает две молекулярные орбитали двух различных
типов:
52
у+ = С1 • уА + с2 • Ув
V- = С3 • Уд - С4' Ув-
Коэффициенты С], С2, С3, С4 указывают долю участия соответ¬
ствующих АО в формировании МО и вычисляются специальными
методами.
Рассмотрим форму и относительную энергию двухцентровых
МО, возникающих при линейной комбинации двух 1з-орбиталей
(например, в молекуле Н2). При данном межъядерном расстоянии
происходит перекрывание исходных атомных орбиталей.
Если знаки волновых функций атомных орбиталей в области
перекрывания одинаковы, то это отвечает положительному пере¬
крыванию (сложение АО). И наоборот, при разных знаках волно¬
вых функций АО имеет место отрицательное перекрывание
(вычитание АО).
При сложении АО образуется двухцентровая МО с повышенной
электронной плотностью между ядрами. Такая МО энергетически
более выгодна, чем исходные АО и называется связывающей МО
(обозначается стсвяз 1з).
При вычитании АО образуется двухцентровая МО с пони¬
женной электронной плотностью в межъядерной области. Эта орби¬
таль энергетически менее выгодна, чем исходные АО, и называется
разрыхляющей (обозначается как араЭр 1з). Процесс образования
МО показан на рис. 2.11.
Обычно образование МО из АО изображают в виде энергети¬
ческой диаграммы, где по вертикали откладывают значения энергии
(Е) орбиталей (рис 2 12)
При построении молекулярной орбитали по методу ЛКАО
должны соблюдаться следующие условия-
Рис. 2.11 Схема образования связывающих
и разрыхляющих молекулярных орбиталей
53
1 Комбинируемые АО должны
быть близки по энергии Это по¬
ложение очевидно, т к при боль¬
шой разнице в энергии АО элек¬
трон полностью перейдет к атому
с низшей энергией и МО не об¬
разуется.
2. АО, образующие МО,
должны перекрываться При этом
ядра располагаются так, чтобы
перекрывание было максимальным
Рис. 2.12. Диаграмма (принцип максимального перекры-
энергетических уровней АО и МО. вания). Чем полнее перекрывают¬
ся АО при образовании МО, тем
сильнее понижение энергии при
переходе электрона с атомной на молекулярную орбиталь, тем
прочнее химическая связь
3. АО, образующие МО, должны обладать одинаковыми
свойствами симметрии относительно межъядерной оси образую¬
щейся молекулы (рис. 2 13).
Это положение можно пояснить следующим образом Совмес¬
тим межъядерную ось с осью х системы координат При этом сим¬
метрия атомных орбиталей $ и рх относительно оси х будет одна и
та же: обе орбитали не изменяют знака при повороте на любой
угол вокруг оси х (рис. 2.13, а, б). В то
же время, атомная орбиталь ру отлича¬
ется от них по симметрии, она изменя¬
ет знаки своих долей на обратные при
повороте вокруг оси х на 180° (рис.
2 13, в)
То же самое относится к рг-орби-
тали. Поэтому могут комбинировать э
и э (а), в и рх (б), но не могут комби¬
нировать Б И Ру, Б и рг (в), т.к. при пе¬
рекрывании последних возникают две
области, равные по величине и проти¬
воположные по знаку, и суммарное пе¬
рекрывание оказывается равным нулю
Для описания электронной конфи¬
гурации гомоядерных двухатомных мо¬
лекул элементов первого периода мож¬
но воспользоваться энергетической
диаграммой, приведенной на рис. 2.12.
Рис. 2.13. Симметрия атомных
орбиталей.
54
Ион Нг+ состоит из двух протонов и одного электрона. Очевид¬
но, что единственный электрон молекулярного иона займет наибо¬
лее выгодную связывающую асвяз 1э орбиталь. Электронная конфи¬
гурация его запишется следующим образом:
Нг+ 1(асвяз и)1]
В молекуле Н2 присутствуют два электрона, которые по прин¬
ципу наименьшей энергии и принципу Паули займут связываю¬
щую СТсвяз.Ь орбиталь, при этом они будут иметь антипараллель-
ные спины.
Н21(асвяз 1з)2]
В молекулярном дигелий-ионе Нег+ три электрона, два из кото¬
рых занимают связывающую и один - разрыхляющую орбиталь.
Не2+[(стсвяз и)2 (стразр Ь)1]
В системе из двух атомов гелия Не2 четыре электрона - два на
связывающей и два на разрыхляющей орбитали.
Не2((асвяз Ы2(ара3р Ы2]
Распределение электронов по молекулярным орбиталям дает
возможность оценить энергию, длину и порядок (кратность) связи.
Мы уже выяснили, что нахождение электрона на связывающей ор¬
битали означает увеличение электронной плотности между ядрами,
уменьшение межъядерного расстояния и увеличение энергии диссо¬
циации молекулы. Если же электрон находится на разрыхляющей
орбитали, то наблюдается обратная картина
Согласно теории молекулярных орбиталей, порядок (кратность)
связи можно определить как полуразность числа связывающих и
разрыхляющих электронов-
Хпсвяз — £празр
Порядок связи =
Ниже приведены сведения о гомоядерных двухатомных молеку¬
лах и ионах элементов 1 периода (табл 2 4).
Элементы II периода имеют р-атомные орбитали, которые в от¬
личие от э-орбиталей не являются сферическими Поэтому при их
перекрывании возникают МО различной симметрии. Так, перекры¬
вание э- и рх-, рх- и рх- атомных орбиталей приводят к образова¬
55
нию а-молекулярных орбиталей, которые имеют цилиндрическую
симметрию относительно линии связи (рис 2 14)
Таблица 2.4
Двухатомные молекулы и ионы элементов первого периода
МО
Иг*
Н2
Не2*
Не2
Ораэр^
-
-
-Т-
-П-
Освяз1^
-Т-
-П-
-П-
-П-
Порядок связи
05
1
05
0
Длина связи, А
1 06
0 74
1 08
-
Энергия диссоциации, кДж/моль
256
435
230
-
Не трудно заметить, что вращение вокруг межъядерной оси х
на 180° переводит о-молекулярную орбиталь в положение, которое
не отличается от исходного
Рис. 2.15. Схема образования л-молекулярных орбиталей.
56
Рис. 2.14 Схема образования а-молекулярных орбиталей.
Перекрывание ру- и ру-, р2- и р2- атомных орбиталей приводит к
образованию связывающей и разрыхляющей л-молекулярных орби¬
талей (рис. 2.15).
Таким образом, поскольку у каждого атома имеется три р-
орбитали, то при взаимодействии атомов должно получаться три
МО - одна а- и две 71-орбитали. Отметим, что 2ру- и 2р2- АО одина¬
ковы по энергии и перекрываются одинаковым способом. Это опре¬
деляет одинаковую энергию и форму возникающих МО лсвяз2ру и
лсвяз2рг. лразр2ру И 71разр 2рг.
Энергия МО находится экспериментально по спектроскопиче¬
ским данным. Оказалось, что для двухатомных молекул II периода
порядок заполнения МО зависит от разницы энергий 2з- и 2р-
атомных орбиталей. При значительном различии, что характерно
для элементов конца периода (начиная с кислорода), молекулярные
орбитали располагаются по энергии в следующем порядке:
^связ^Б < Фразр^Б < <Усвяз2рх < Ясвяз^Ру — 71связ2р2 < Яра3р2ру —
— Яразр^Рг < ^разр 2рх-
При энергетической близости 2з- и 2р- атомных орбиталей от¬
талкивание электронов на с2в- и с2рх-орбиталях обусловливает
более низкое значение энергии лсвяз2ру и лсвяз2рг, чем (ТСвя32рх- В
этом случае порядок заполнения МО будет следующим:
^связ^Б < (Уразр^З < ясвяз2ру = 71связ2р2 < (Усвяз^Рх < яразр2ру =
= Лразр2рг < стразр2рх-
Энергетические диаграммы МО двухатомных молекул элементов
II периода даны на рис. 2.16.
В образовании химической связи принимают участие только
валентные электроны, т.е. находящиеся на незаполненном уровне
Поэтому для молекул, образованных элементами II периода, пере¬
крыванием 1в электронов можно пренебречь и рассматривать внут¬
ренние электроны как несвязывающие или принадлежащие отдель¬
ным атомам. В соответствии с полученными молекулярными диа¬
граммами приведем электронные конфигурации гомоядерных двух¬
атомных молекул элементов II периода (табл 2 5)
В рамках метода молекулярных орбиталей получает простое
объяснение наблюдающееся изменение в свойствах молекул- увели¬
чение порядка связи сопровождается уменьшением межъядерного
расстояния и возрастанием энергии диссоциации Так же просто
объясняются и магнитные свойства веществ- парамагнитные ве¬
щества (притягиваются магнитом) имеют неспаренные электроны
(молекулы бора и кислорода), а диамагнитные (слабо отталкиваются
магнитом) - их не имеют.
57
Рис. 2.16 Энергетическая диаграмма уровней двухатомных молекул
элементов II периода при значительном (а) и незначительном (6)
энергетическом различии 2э- и 2р-орбиталей.
58
Для описания гетероядерных двухатомных молекул метод МО
использует по существу такой же подход. Для приближенного
представления об их электронной структуре можно использовать
систему молекулярных орбиталей гомоядерных молекул, имеющих
такое же число электронов. Такие молекулы называются изоэлек-
тронными. Например, молекула СО и цианид-ион СМ' изоэлектрон-
ны молекуле N2. В каждой молекуле по 14 электронов и электрон¬
ная конфигурация такова:
(освяз 1 ьЖаразр 15)2(асвяз25)2(аразр25)2(71связ2ру)2(71связ2р2)2(асвяз2рх)2.
Отсюда следует, что порядок связи в СО и СМ" равен 3. Такое
представление согласуется, например, с высокой энергией диссо¬
циации молекулы СО (1076 кДж/моль) и малым межъядерным рас¬
стоянием (1.12 А). Обе эти молекулярные константы близки к та¬
ковым для молекулы N2.
Таблица 2.5
Электронные конфигурации и порядки связей
гомоядерных двухатомных молекул элементов II периода
Молекула
Электронная конфигурация
Порядок
связи
и2
(<Тсвяз25)2
1
Ве2
(<7связ25)2(аразр25)2
0
в2
(Освяз25)2(аразР25)2(лсвяэ2р)1(лсвяз2р2)1
1
с2
(асвяз25)2(араэр25)2(лсвяэ2ру)2(лсвяэ2рх)2
2
N2
(асвяз25)2(аразр25)2(лсвяэ2ру)2(лсвяз2рг)2(асвяз2рх)2
3
02
(асвяз25)2(а0аэ025)2(асвяз2р)2(лсвяз2ру)2(лсвяз2рг)2
(лразр2ру)1 (лразр2р2)1
2
(асвяз25) (аоазо25)2(аевяз2р)2(лсвяз2ру)2(лсвяз2рг)2
(лразр2Ру)2(лразр2р2)2
1
№2
(асвяз25)2(аоазо25)2(асвяз2рх)2(лсвяз2ру)2(лсвяз2рг)2
(лразр2ру)2(лразр2рг)2(аразр2рх)2
0
Однако предполагаемое электронное строение основано на
слишком грубом приближении и не может объяснить различия в
свойствах химически неактивной молекулы N2 и весьма реакцион¬
носпособной молекулы СО Для этого необходим детальный анализ
всех орбиталей молекулы
Рассмотрим электронное строение молекулы СО Электронные
конфигурации атомос: С 1 Б22522р:г и О 1522522р4. С увеличением
59
атомного номера энергии сходных атомных орбиталей понижаются
вследствие увеличения притяжения электронов к ядру. Поэтому
энергия 2э АО кислорода будет ниже, чем 2э АО углерода.
Так как в гетерояверных молекулах в одной МО участвуют раз¬
ные АО, например, 2в углерода и 2р кислорода, прежняя система
записи электронной конфигурации невозможна и орбитали просто
нумеруются по порядку Для молекулы СО она имеет вид:
(1 анесв)2(2анесв)2(3анесв)2(4асвяз)2( 1 лхсвяз)2( 1 яУСвяз)2(5анесв)2.
Схема образования МО молекулы СО приведена на рис. 2.17.
Рис. 2.17. Схема образования МО молекулы СО.
Из десяти МО молекулы СО семь заполнены электронами, а три
разрыхляющие орбитали свободны. Связь оказывается тройной за
счет шести электронов, находящихся на связывающих орбиталях:
4аСвяз* 1яхсвнз> ІлУсвяз- В отличие от молекулы N2 верхняя занятая
орбиталь 5а является несвязывающей. Электронная пара на такой
60
орбитали называется неподеленной парой углерода, что подчерки¬
вает ее атомный характер. Эта неподеленная пара углерода на
5аНесв орбитали и определяет высокую донорную способность моле¬
кулы СО. В этом причина образования более прочного комплекса
молекулы СО с гемоглобином по сравнению с молекулой Ог.
2.2. Ионная связь
В двухатомной молекуле, состоящей из разных атомов, элек¬
тронная плотность смещена к атому, обладающему большей элек¬
троотрицательностью. Такая связь является полярной и характери¬
зуется постоянным дипольным моментом. В предельном случае по¬
лярности, когда электронные облака взаимодействующих атомов
разделены настолько, что можно говорить об образовании противо¬
положно заряженных ионов, мы имеем дело с ионной связью.
Рис. 2.18. Взаимная ориентация ионов в кристаллах №С1 (а) и СэС! (6).
Классическим примером молекул с ионной связью являются га-
логениды щелочных металлов. Наиболее простая модель - модель
сферических ионов - объясняет эту связь следующим образом:
единственный внешний электрон атома щелочного металла нахо¬
дится над замкнутой сферической з2р6-оболочкой остова. Энтальпия
ионизации не велика, что обусловливает легкость отрыва валентно¬
го электрона и образование иона Ме*. Напротив, атом гало¬
гена имеет высокое значение энтальпии присоединения электрона и
характеризуется тенденцией к захвату электрона с образованием
иона X' с з2р6-оболочкой. При сближении атомов Ме и X возможен
переход электрона от металла к галогену и появление сферически
симметричных ионов Ме+ и X' Затрата энергии на такой переход
компенсируется энергией электростатического притяжения. На не¬
больших расстояниях начинают резко возрастать силы отталкива¬
ния между заполненными электронными оболочками. Равновесное
61
расстояние между атомами, отвечающее минимуму энергии систе¬
мы, определяется балансом сил притяжения и отталкивания. Таким
образом, кривые потенциальной энергии для двухатомных молекул с
ионной и ковалентной связями подобны (см. рис. 2.1).
Приведенная модель может использоваться для расчета межъ-
ядерных расстояний и энергии диссоциации в ионных молекулах.
Важнейшими особенностями ионной связи является ее ненасы-
щаемость и ненаправленность в пространстве. Эти свойства выте¬
кают из свойств сил электростатического взаимодействия. Поле,
создаваемое ионом, сферически симметрично, и все другие ионы,
находящиеся в этом поле, испытывают его действие.
Вследствие ненасыщаемости ионной связи ион в кристалле
может взаимодействовать одновременно с несколькими ионами
противоположного знака. Их число будет зависеть от протяжен¬
ности электронных облаков данного иона и окружающих его про-
тивоионов.
Та же картина наблюдается для любого иона в кристалле. Та¬
ким образом, кристаллические ионные соединения образуют трех¬
мерную бесконечную решетку, в узлах которой правильно череду¬
ются катионы и анионы. Отдельных молекул в решетках солей типа
КС1, №С1 нет.
Разность электроотрицательностей
Рис. 2.19. Зависимость степени ионности связи от ДОЭО.
Ненасыщаемость и ненаправленность связи приводит в ионных
кристаллах к структурам плотнейших упаковок. Важной характе¬
ристикой таких структур является координационное число (КЧ),
т.е. число противоионов, окружающих в кристалле данный ион.
Так, в решетке №С1 ион №+ окружен шестью нонами СГ и наобо¬
62
рот, следовательно, мы имеем гексагональную решетку (КЧ = 6).
В кристалле СэС1 координационное число равно восьми (рис. 2.18).
Координационные числа положительного и отрицательного ио¬
нов не обязательно должны совпадать. Они будут одинаковы в слу¬
чае бинарных структур с общей формулой АВ, где требование элек¬
тронейтральности означает равенство чисел противоионов. В струк¬
турах же типа АВ2, содержащих ионы А2* и В', для сохранения об¬
щей нейтральности число отрицательных ионов должно быть вдвое
больше числа положительных.
Запомним, что молекул с чисто ионной связью не существует.
Степень ионности связи будет определяться разностью электроот¬
рицательностей атомов (ДОЭО). Как видно из рис. 2.19, только для
фторидов щелочных и щелочноземельных металлов, для которых
ДОЭО = 2.8 3.3, связь можно считать близкой к ионной.
Ион-ионные взаимодействия, которые часто называют ионной
связью, достаточно широко представлены в биоструктурах. Водные
биосреды содержат многие виды катионов и анионов, что иллю¬
стрируется ионным составом плазмы крови и внутриклеточной
жидкости (рис. 2.20). Из этого рисунка следует, что для понимания
медицинских проблем необходимо знание свойств катионов натрия,
калия, магния, кальция и хлорид-, гидрофосфат-, гидрокарбонат- и
сульфат-анионов.
Труднорастворимые соли гидроксиапатит Са5(0Н)(Р04)з и фтора-
патит Са5р(Р04)з составляют основу костной ткани и зубной эмали,
соответственно.
Ион-ионное взаимодействие между аммонийным катионом и
анионом, образуемым карбоксильной группой, играет важную роль
в белковых структурах, имеет место в молекулах-передатчиках
нервных импульсов и др.
Металлическая связь, напротив, совершенно не характерна
для биоструктур, поэтому мы ограничимся самыми элементарными
представлениями о ней. Электронная структура атомов металлов
характеризуется наличием вакантных орбиталей, а также невысо¬
кими величинами энтальпий ионизации валентных электронов
Помня о том, что металлы образуют кристаллическую решетку
(которую в данном случае нельзя интерпретировать с помощью
ионной связи), можно объяснить существование металлической
связи тем, что валентные электроны легко перемещаются по всей
кристаллической решетке, представляя собой некоторый “элек¬
тронный газ” Этим определяются такие характерные свойства ме¬
таллов, как высокие электро- и теплопроводность Итак, металл
следует рассматривать как компактную структуру из катионов, ко¬
торые связаны друг с другом обобществленными электронами -
“электронным газом”.
63
Рис. 2.20. Ионный состав плазмы и внутриклеточной жидкости (в мг-экв/л).
2.3. Водородная связь
Если атом водорода связан ковалентной связью с каким-либо
электроотрицательным элементом, то он может одновременно при¬
тягиваться к другому атому, имеющему высокую электронную
плотность. Энергия такого притяжения составляет 4-40 кДж/моль,
т е. на порядок меньше энергии ковалентной связи. Связь, возни¬
кающая в итоге, получила название водородной (Н-связь).
64
Атом водорода имеет очень малый размер и частичный положи¬
тельный заряд из-за связи с электроотрицательным атомом. Водо¬
родная связь образуется вследствие.внедрения атома водорода в
электронную оболочку атома с высокой электроотрицательностью и
обычно имеющего неподеленную электронную пару. В качестве та¬
ких атомов могут выступать фтор, кислород, азот и, в меньшей сте¬
пени, хлор и сера.
Небольшая энергия водородных связей приводит к тому, что
они легко возникают и разрушаются. Длина водородной связи за¬
метно превосходит длину ковалентной связи. Так, например, если
длина ковалентной связи О-Н составляет 0.97 А, то водородная
связь О Н в воде имеет длину 1.75 А. Чтобы подчеркнуть отличие
водородной связи от ковалентной, ее обозначают тремя точками.
Х-Н У
Водородная связь широко распространена и играет важную роль
при ассоциации молекул, в процессах растворения и диссоциации,
образования кристаллогидратов и многих других
Образование водородной связи может существенно изменить
физические свойства вещества (теплоты плавления и испарения,
температуры кипения, вязкость, твердость и др.). Так, например,
аномально высокие точки кипения НР, МН3 и Н20 объясняются ас¬
социацией этих молекул в агрегаты за счет водородных связей.
Интересно, что наиболее прочная связь у НР (28 кДж/моль), у
воды энергия связи составляет 20-25 кДж/моль, а у аммиака -
18 кДж/моль В то же время самая высокая точка кипения у воды
(100 °С, +19.5 °С у НР и -33 4 °С у 1МНз). Причина этого заключает¬
ся в том, что на каждую молекулу воды приходится 4 водородных
связи (две - за счет атомов водорода и две - за счет двух неподе-
ленных пар атома кислорода), в то время как на молекулу
НР - всего две. Если фтористый водород образует линейные ассо-
циаты (НР)П, где п = 2-9, то вода в кристаллическом состоянии об¬
разует пространственную кристаллическую решетку типа алмаза.
Наряду с межмолекулярной водородной связью существует так¬
же и внутримолекулярная водородная связь
Образование последней возможно при
одновременном наличии в одной молекуле
электроноакцепторной группы Х-Н и элек-
тронодонорного атома У, причем расстоя¬
ние между атомами Н и У не должно
превышать обычной длины водородной
связи (1.6-2 0 А) Примером может слу¬
жить биомолекула салициловой кислоты
(рис. 2 21).
Рис. 2 21
Водородная связь
в салициловой кислоте.
3 Звк 675
65
Устойчивости такой связи благоприятствует образование шес¬
тичленного цикла, в котором нет деформации валентных углов.
Следует отметить, что только замыкание пяти- или шестичлеиных
циклов удовлетворяет стерическим требованиям возникновения во¬
дородной связи.
В молекуле полипептида, содержащей пептидные фрагменты
-ІЧН-СО-, образование водородной связи между аминокислотными
звеньями в положениях 1 и 5 приводит к известной а-спиральной
структуре Межмолекулярные водородные связи в двойной спи¬
ральной структуре образуются у дезоксирибонуклеиновой кислоты
(ДНК) - носителя генетической информации. Хотя энергия одной
водородной связи мала, множество таких связей в макромолекулах
могут определить устойчивость их пространственной структуры
(кооперативный эффект).
2.4. Межмолекулярные взаимодействия
2.4.1. Силы Ван-дер-Ваальса
Между молекулами, которые являются валентно насыщенными
в обычном представлении, существуют силы притяжения. Этими
силами объясняются отклонения газов от идеальности, сжижение
газов или их конденсация, аномалии теплот испарения жидкостей
и т.д. Вполне определенные величины плотностей твердых и жид¬
ких веществ указывают на существование и сил отталкивания меж¬
ду молекулами. При отсутствии последних плотность веществ воз¬
растала бы неограниченно. Таким образом, межмолекулярные вза¬
имодействия приводят к сближению молекул до расстояния, на ко¬
тором силы притяжения и отталкивания уравновешиваются.
Потенциальная кривая взаимодействия двух валентно насыщен¬
ных молекул отличается от таковой для двухатомной молекулы
(рис 2 1) только количественно: глубина потенциального минимума
значительно меньше (меньше энергия взаимодействия), а равновес¬
ное расстояние - больше. Силы притяжения между молекулами
являются дальнодействующими, довольно медленно спадают с рас¬
стоянием и называются силами Ван-дер-Ваальса.
Существование сил притяжения можно объяснить тремя при¬
чинами.
Электростатическое взаимодействие. Оно включает взаимо¬
действие электрически заряженных ионов или молекул с посто¬
66
янными дипольными моментами. Очевидно, что оно наиболее суще¬
ственно для ионных кристаллов и сильно полярных молекул.
Индукционное взаимодействие. Молекула, имеющая посто¬
янный дипольный момент, наводит в другой молекуле (полярной
или неполярной) индуцированный дипольный момент. Взаимодей¬
ствие постоянного и индуцированного диполей понижает энергию
системы из двух молекул и упрочняет ее. Такое взаимодействие на¬
блюдается при образовании гидратов инертных газов, в растворах
полярных веществ в неполярных и существенно только для молекул
со значительной поляризуемостью, к которым, в первую очередь,
относятся молекулы с сопряженными кратными связями.
Дисперсионное взаимодействие (силы Лондона). Если пер¬
вые две составляющие сил Ван-дер-Ваальса можно понять на основе
представлений электростатики, то дисперсионное взаимодействие
объяснимо только на основе квантовой механики. Грубое модельное
представление об этом взаимодействии можно составить, рассмат¬
ривая притяжение между двумя атомами инертного газа за счет
мгновенных диполей, возникающих при синхронном движении
электронов вокруг ядер. Направление диполя меняется с частотой
1015 циклов в секунду, вследствие чего атом инертного газа не об¬
ладает постоянным дипольным моментом. Однако при сближении
двух атомов мгновенные диполи ориентируются относительно друг
друга и их направление изменяется “в такт”.
Особенностью дисперсионного взаимодействия является его
всеобщность, т.к. во всех молекулах есть движущиеся электроны, а
для неполярных молекул это взаимодействие - главный источник
сил Ван-дер-Ваальса
Таковы три основных типа невалентных сил, ответственных за
притяжение между молекулами.
Энергия индукционного и дисперсионного взаимодействий зна¬
чительна для сильно поляризующихся молекул
2.4.2. Ионно-молекулярное взаимодействие
Промежуточным случаем между обычным химическим и ван-
дерваальсовым взаимодействием является взаимодействие иона с
молекулой Если из атома металла образуется катион, то остается
свободной низколежащая орбиталь, с которой удален один или два
электрона Возможен частичный перенос электронной плотности
молекулы на свободную орбиталь катиона с образованием связи ти¬
па донорно-акцепторного взаимодействия Квантово-химические
расчеты показывают, что такой перенос незначителен и поэтому
3*
67
можно считать, что в основе связи ион-молекула лежат силы Ван-
дер-Ваальса.
Ион-дипольное взаимодействие играет определяющую роль в
растворах электролитов в полярных растворителях (вода, спирт).
Особенно оно велико для ионов с заметной поляризуемостью
(например, Ад+), с высоким зарядом (например, Са2+, Мд +)
В растворах электролитов образуются довольно стабильные
(см. табл. 2 6) продукты взаимодействия иона с несколькими моле¬
кулами растворителя - сольваты (гидраты в водных растворах).
Так, катионы щелочных металлов 1_Г\ N8*, К+ присоединяют 6-8
молекул воды на ион. Анионы, как более крупные частицы, менее
сольватированы, так как заряд в них распределен по большему
объему.
Рис. 2.22. Гидратация катионов и анионов.
Полезно обратить внимание, что структуры гидратированных
катионов и анионов принципиально различаются (рис. 2.22).
2.4.3. Гидрофобные взаимодействия
Гидрофобными взаимодействиями (иногда неверно называе¬
мыми гидрофобными связями) объясняются взаимодействия между
неполярными молекулами (углеводороды) и неполярными группами
(в аминокислотах, липидах и др.) в водных растворах. Эти взаимо¬
действия связаны с изменением структуры воды при внесении в нее
таких веществ.
Как известно, вода имеет упорядоченную структуру за счет
межмолекулярных водородных связей. Внедрение неполярных моле¬
кул препятствует образованию таких связей, увеличивая беспоря¬
док в системе, т.е. сопровождается ростом энтропии’.
Следует помнить, что гидрофобные взаимодействия всегда эндо-
термичны (ДН > 0) в отличие от других типов взаимодействий.
* Чтобы роследующее объяснение стало понятным, необходимо прочи¬
тать главу 6
Свободная энергия системы понижается вследствие определяющего
вклада в ее изменение энтропийного фактора.
Таким образом, движущей силой гидрофобных взаимодействий и
их причиной является стремление воды воспрепятствовать измене¬
нию своей структуры, суммарное увеличение энтропии системы при
контакте неполярных веществ между собой.
Итак, вещества, построенные посредством ковалентных непо¬
лярных или мало полярных связей, не растворяются в воде, иными
словами, гидрофобны Гидрофобные взаимодействия между ними
способствуют их взаимной растворимости. Поскольку мало полярны
по своей природе липиды (жиры), такие вещества называют липо-
фильными. Напротив, взаимодействия с молекулами воды способ¬
ствуют растворению в ней сильно полярных и ионных соединений.
Эти вещества гидрофильны.
Термины гидрофильность и липофильность (обратны им
гидрофобность и липофобность) или, в общем, лиофильность
и лиофобность (понятия, характеризующие способность вещества
взаимодействовать с жидкой средой) чрезвычайно важны для пони¬
мания закономерностей распределения различных веществ в тканях
организма
В зависимости от природы вещества могут накапливаться либо
в липидных клеточных мембранах, либо в водных средах. Это долж¬
но учитываться в фармакологии при конструировании лекарствен¬
ных веществ, мишенью действия которых могут быть как клеточные
мембраны, так и водные биосреды, в токсикологии при выяснении
процессов накопления и выведения ядовитых веществ и во многих
других случаях.
Ниже приведена сводная таблица типов внутри- и межмолеку-
лярных взаимодействий (табл. 2 6)
Таблица 2 б
Типы внутри- и межмолекулярных взаимодействий
Тип взаимодействия
Энергия взаимодействия,
кДж/моль
Пример
Ковалентная связь
200 - 800
н-н
Ион-ионное
40 - 400
№+СГ
Ион-дипольное
4 - 40
Ма*(Н20)п
Ди пол ь-ди пол ьное
04-40
о2 о2
Диполь-индуцированный диполь
04-40
НС1 С6Нв
Дисперсионные силы
4 - 40
N6 N6
Водородная связь
4 - 40
Н20 Н20
69
Существует группа соединений, построенных без химических
связей Так, уже давно известны так называемые клатраты или
соединения включения Они образуются, когда молекулы одного
типа (“гости”) внедряются в полости других молекул ("хозяева”)
Естественно, что размеры и конформация (то есть пространст¬
венная форма) “гостей” должны строго соответствовать формам
и размерам полости “хозяев” Энергия взаимодействия “гость-
хозяин” (за счет сил Ван-дер-Ваальса) обычно не превосходит
40 кДж/моль, хотя иногда достигает и 125 кДж/моль В природе
такие соединения встречаются достаточно широко, например, сое¬
динения включения в организме образуют углеводы, а также неко¬
торые вещества жирового происхождения.
Другим способом сконструированы молекулы катенанов, ро-
токсанов и узлов (рис 2 23)
Катенаны построены по типу механической цепочки (а), ро-
токсаны собраны по принципу “колесо-ось” (б), а принцип узла
очевиден из рис. 2.23 (в). Такие соединения с механической связью,
по-видимому, играют определенную роль в химии полимерных
веществ, образующих живую материю (белки и нуклеиновые кис¬
лоты) Во всяком случае, в настоящее время известны катенановые
и ротоксановые дезоксирибонуклеиновые и узловые рибонуклеино¬
вые кислоты.
Рис. 2.23 Катвнаны, ротоксаны и узлы.
Глава 3. КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
При образовании сложной
аммиачной соли... наблюдается
изменение в функциях не одной
какой-либо составной части, ..
но и всех их одновременно.
Н. С. Курнаков
3.1. Строение, классификация, номенклатура
Любое взаимодействие между атомами, ионами, молекулами,
ионами и молекулами и т.д. сопровождается определенным про¬
странственным расположением частиц относительно друг друга или
координацией.
Простейшим примером является образование гидратной оболоч¬
ки у катионов металлов при растворении соли в воде (см. рис.
2.22). При взаимодействии СоСЬ с аммиаком образуются оранжевые
кристаллы, соответствующие составу СоС1э'6МНз. Это указывает
на образование прочной связи иона Со3* с молекулами аммиака.
В обоих случаях мы имеем дело с комплексообразованием
Комплексные соединения представляют собой наиболее обшир¬
ный и разнообразный класс химических соединений, а поскольку в
их состав могут входить как неорганические, так и органические
молекулы или ионы, то комплексные соединения связывают воедино
неорганическую и органическую химию.
Комплексные соединения нашли самое разнообразное практиче¬
ское применение. Особенно велика роль комплексов в биологиче¬
ских процессах. Достаточно сказать, что к комплексным соединени¬
ям относятся гемоглобин и хлорофилл, некоторые витамины и мно¬
гие ферменты.
Изучение комплексных соединений до конца XIX века носило
исключительно описательный характер и состояло из серии попы¬
ток объяснить существование и структуру гидратов, двойных солей
и аммиакатов Обобщением этих объяснений стала координаци¬
онная теория, предложенная швейцарским химиком А Вернером
(1893 г)
Эта теория, развитая и подкрепленная экспериментальными ис¬
следованиями, обеспечила быстрое развитие химии комплексных
соединений на рубеже двух столетий Однако широкое признание
теория Вернера получила лишь после создания электронной теории
валентности В развитии химии комплексных соединений важную
71
роль сыграли труды русских ученых Н. С. Курнакова, Л А. Чугаева,
И Н Черняева, А А Гринберга и др
В настоящее время химия координационных соединений из уз¬
кой и ограниченной области превратилась в наиболее интенсивно
развивающуюся область химии Ее сфера теперь настолько обшир¬
на, а число и разнообразие соединений настолько велики, что мы
ограничимся в данном разделе только основными понятиями
Химия комплексных (координационных) соединений изучает
ионы и молекулы, состоящие из центральной частицы и коорди¬
нированных вокруг нее лигандов. Центральная частица (ком-
плексообразователь) и непосредственно связанные с ней лиган¬
ды образуют внутреннюю (координационную) сферу ком¬
плексного соединения, а число этих лигандов называется коорди¬
национным числом Ионы, находящиеся за пределами координа¬
ционной сферы, образуют внешнюю сферу комплексного соедине¬
ния В формулах внутреннюю сферу комплексного соединения
заключают в квадратные скобки. Например. К2[Нд14], Кз[Ре(СМ)в],
[Сг(Н20)6]С1з и т.д
В подавляющем большинстве комплексных соединений в ка¬
честве комплексообразователя выступают ионы переходных метал¬
лов, хотя известны комплексные соединения практически для всех
элементов.
Наиболее важной характеристикой центрального атома является
его степень окисления, определяемая для комплексов как разность
между зарядом комплексной частицы и суммарным зарядом ли¬
гандов. Например, ион [Со(МНз)в]3+ относят к производным Со3+,
приписывая МНз нулевой заряд как нейтральной молекуле, а ион
[Со(ЗСМ)4]2" - к производным Со2+, приписывая группе ЭСМ" заряд
-1, как в роданидах щелочных металлов.
Обычными координационными числами в комплексах являются
4 и 6, однако для ряда комплексообразователей установлены числа
2, 3, 5, 7, 8 и более Существует статистическое правило, связы¬
вающее степень окисления комплексообразователя и координацион¬
ное число, отраженное в табл. 3.1.
Указанные в таблице 3.1 соотношения не имеют строгого ха¬
рактера и сохраняются главным образом в тех случаях, когда ли¬
ганды - нейтральные молекулы или однозарядные ионы.
В качестве лигандов выступают молекулы или ионы, содержа¬
щие донорные атомы, способные отдавать комплексообразователю
неподеленную электронную пару. Наиболее распространенными до-
норными атомами являются N. Р, О, галогены.
Число донорных атомов в лиганде может быть различным и оно
определяет его координационную емкость или дентатность.
72
Координационные числа некоторых комплексообразователей
Степень окисления
комплексообразователя
Координационное
число
Пример комплексного
соединения
+ 1
2
[Ад(МН3)2]С1 К[Ад(СМ)2]
+2
4
К2[НдЦ]
+3
6
[Сг(МНз)б]С1э
+4
6
Н2[&Р6]
+5
7
К2[ГаР7]
Монодентатные лиганды используют в качестве донорного один
атом и, следовательно, могут занимать только одно координацион¬
ное место у центрального атома. Монодентатными лигандами явля¬
ются ионы СГ, Р", ОН", молекулы N143, НгО, СО и др. К бидентатным
относятся многие органические молекулы, например, этилендиамин,
диметилглиоксим, дианион щавелевой кислоты и т.д
Кислородсодержащие анионы (С032_, Э042 и др ) могут быть как
монодентатными, так и бидентатными лигандами Например
МН3
+
МН3
МН3 I МНз
Со
”НЧ 1
Со БОз
I ^ОБОз
ш-/ | хо/
МН3
Мз
Названия комплексных соединении образуются аналогично на¬
званиям обычных солей с той лить разницей, что указываются
73
лиганды и степень окисления центрального атома. К названиям ли¬
гандов-анионов добавляют суффикс -о (сульфато-, хлоро-, бромо-,
циано-, и т д ) Наиболее важные лиганды-молекулы Н20 - аква,
МНз - аммин, СО - карбонил. Число лигандов каждого рода указы¬
вают греческими числительными- моно-, ди-, три-, тетра- и т д Если
комплексная частица является анионом, то ее название заканчи¬
вается суффиксом -ат
По характеру заряда комплексной частицы комплексы класси¬
фицируют как катионные, анионные и нейтральные Ниже приведен
ряд комплексных соединений и даны их названия
хлорид гвксаакваалюминия(Ш)
хлорид тетраамминцинка(Н)
тетрагидроксоалюминат(Ш) калия
гексацианоферрат(Н) калия
дихлородиамминплатина(М)
тетракарбонилникель(О)
Среди комплексных соединений различают кислоты, основания
и соли Есть и вещества, не диссоциирующие на ионы, т.е. неэлек¬
тролиты.
Например:
3.2. Изомерия комплексных соединений
Среди комплексных соединений широко распространено явле¬
ние изомерии, изучение которого и позволило установить их про¬
странственное строение. Рассмотрим основные типы изомерии
комплексов
Ионизационная изомерия связана с различным распределе¬
нием ионов между внутренней и внешней сферами комплекса, на¬
пример, [РЦЖзЬСЫВгг и [Р1(МНз)4Вг2]С12.
Координационная изомерия вызвана различным распределе¬
нием комплексообразователей или лигандов между комплексными
катионами и анионами'
74
Катионные:
Анионные:
Нейтральные:
Гидратная изомерия определяется характером связей моле¬
кул воды, входящих в состав комплекса. Так, для хлорида хрома(Ш)
известны три модификации:
Окраска кристаллов
сине-серая
светло-зеленая
темно-зеленая
Геометрическая изомерия характерна для комплексов, име¬
ющих плоское строение, либо форму пирамиды с центральным ато¬
мом в вершине. Так, например, в плоском квадратном комплексе с
двумя одинаковыми лигандами возни¬
кает возможность существования цис-
и транс-изомеров. \/
Так, например, дихлородиаммин- /\ /\
платина(Н) [Р1(МНз)2С12] существует в А с с А
виде двух геометрических изомеров. цис-изомер 7ранс-изомер
Интересно, что различие в строении
обусловливает различную физиологи¬
ческую активность этих соединений. Малотоксичный ^ис-изомер
обладает противоопухолевой активностью и нашел применение
в качестве лекарственного средства, а транс-изомер значительно
более токсичен и при этом не обладает противоопухолевым
действием.
Оптическая изомерия наблюдается у комплексов, структура
которых не имеет элементов симметрии (такие объекты называются
асимметричными и они не могут
быть совмещены со своими зер¬
кальными изображениями) В про¬
стейшем случае это тетраэдриче¬
ский комплекс с четырьмя разны¬
ми лигандами, для которого су¬
ществуют два изомера, относя¬
щиеся друг к другу как предмет
и его зеркальное изображение
(энантиомеры)
Такие изомеры называются оптическими антиподами Характер¬
ной их особенностью является способность вращать плоскость по¬
ляризации плоскополяризованного луча Однако, если один изомер
вращает плоскость поляризации влево, то другой - вправо на тот
же угол Подробнее вопросы оптической изомерии рассмотрены на
примере органических веществ.
зеркальная
плоскость
75
3.3. Химическая связь
в комплексных соединениях
При описании химической связи в комплексных соединениях,
объяснении их строения и свойств используют как электростатиче¬
ские представления, так и квантово-химический подход (методы
валентных связей и молекулярных орбиталей, теория кристалличе¬
ского поля, теория поля лигандов) Идеи и выводы каждого из ме¬
тодов с успехом применяются в характерных для них сферах химии
комплексных соединений, но приближенный характер методов огра¬
ничивает их использование.
Метод валентных связей в приложении к комплексным соеди¬
нениям базируется на тех же представлениях, что и в молекуляр¬
ных соединениях, т.е. ковалентная связь между лигандом и цент¬
ральным атомом двухэлектронная и двухцентровая, а образование
ее происходит по донорно-акцепторному механизму за счет неподе-
ленных электронных пар донорного атома лиганда и вакантных
орбиталей центрального атома. Орбитали комплекса рассматрива¬
ются как орбитали только центрального атома, гибридизация кото¬
рых приводит к образованию связывающих орбиталей.
Так, например, атом Со имеет электронную конфигурацию
4з2Зс17, а ион Со*3 - Зс16. Сильное взаимодействие Со*3 с молекулами
аммиака приводит к спариванию с1-электронов, и шесть электронов
занимают три из пяти 3-с1 орбиталей Остающиеся две вакантные 3с1
орбитали, а также одна 4э и три 4р-орбитали участвуют в гибриди¬
зации с образованием шести гибридных (с12зр3) орбиталей, направ¬
ленных к вершинам октаэдра.
76
При заполнении гибридных орбиталей парами электронов моле¬
кул аммиака образуется октаэдрический комплекс [Со(МН3)б]3*- По¬
скольку в образующемся комплексе нет неспаренных электронов,
он обладает диамагнитными свойствами (|о.= 0).
Неспаренные электроны, вращаясь вокруг собственной оси, соз¬
дают магнитный момент. Магнитный момент молекулы (ц) склады¬
вается из магнитных моментов неспаренных электронов. Если
молекула содержит неспаренные электроны, то ц * 0 и веще¬
ство парамагнитно, т.е. втягивается в магнитное поле. Если неспа-
ренных электронов нет и ц = 0, то вещество диамагнитно и слабо
выталкивается из магнитного поля.
Образующийся из атома мК4з2Зс18) двузарядный ион №2* имеет
электронную конфигурацию Зс18, где электроны располагаются по
орбиталям в соответствии с правилом Хунда.
Зd
М12+(Зс18) |И|И|И|1 III
При сравнительно слабом взаимодействии иона №2* с ионами
СГ электронные пары хлора занимают вакантные орбитали 4э и 4р,
которые участвуют в гибридизации с образованием четырех эквива¬
лентных зр3-гибридных орбиталей, направленных к вершинам тет¬
раэдра. Распределение электронов можно представить так:
3(1 4э 4р
1*«сц]2. ||||||||||| ||| И I -I-I-I
эрЗ-гибридизация
тетраэдрический комплекс (ц * 0)
Наличие двух неспаренных электронов у центрального атома
обусловливает парамагнетизм комплекса.
С молекулами аммиака ион №2* взаимодействует сильнее и об¬
разует комплекс [М|(МНз)б]2+, имеющий электронное строение:
3(1 4б 4р 4(1
икннлр* |11|11|11|114 В I -• I I -1 111
зрЗс12-гибридизация
октаэдрический комплекс ( ц * 0)
При еще более энергичном взаимодействии с цианид-ионами в
ионе №2* происходит спаривание двух с1-электронов В гибридизации
участвует одна вакантная 3(1-орбиталь, одна 4э и две 4р-орбитали
(с1$р2тибридизация) Образуется диамагнитный комплекс с плоским
квадратным строением
Зd 4э 4р
[ыксы)4]2- |И|11|11||!|»1 0 Г-1-1 1
зр2с1-гибридизация
плоский квадратный комплекс (ц = 0)
В таблице 3.2 приведено несколько примеров наиболее часто
встречающихся типов гибридизации
77
Таблица 3.2
Примеры часто встречающихся гибридизаций
с указанием распределения электронов
Комп-
лексо-
Конфигурация
кч
Геометри¬
ческая
форма
Распределение
электронов и
гибридизация
И
образо-
■атель
атома
иоиа
Со*3
Зсі74?2
Зсі6
6
октаэдр
3<1 45 4р
0
И11 и ЦТ 1 --1 -11-1 М--М
й2зрЗ-гибридизация
Мл*2
3<^54б2
за5
6
октаэдр
3(1 4з 4р
>0
1»|п1|_ш а н-н
<]2>рЭ-гибридиэация
ІЧі*2
Зс18452
Зсі8
4
плоский
3(1 45 4р
0
квадрат
1п|11|ц||||-:1 Г-1 1-1-1 1
4зр2-гибридизация
Си*2
3с1 104б 1
Зсі9
4
плоский
3(1 4* 4р
>0
квадрат
1и1и1и1иП Г-1 1-1-11 1
(1зр2-гибридизация
М*2
3<^84б2
Зсі8
4
тетраэдр
3(1 4з 4р
>0
ипшиишы 1-1-1"!
зрЗ-гибридиэация
ІЧІ*3
Зс18452
Зсі7
5
квадратная
пирамида
3(1 4з 4р
>0
ЩННПНМ Ы 1-1-1--1
йзрЗ-гибридизация
Из приведенных примеров видно, что гибридизация с участием
(1-орбиталей может быть двух видов- с использованием внешних
с1-орбиталей (4(1 в случае [МКМН3)б]2*) или с использованием внут¬
ренних (1-орбиталей (3(1 в случае [М1(СМ)4]2-).
Говорят о "внешней” и “внутренней” гибридизации или о
внешне- и внутриорбитальных комплексах. В рамках метода ва¬
лентных связей можно предсказывать и объяснять реакционную
способность комплексных соединений, которая во многом опреде¬
ляется скоростью обмена лигандов на другие молекулы или ионы в
растворе. Обмену лигандов благоприятствует. 1) “внешняя" гибри¬
дизация и 2) наличие у центрального атома свободных “внутрен¬
них” орбиталей. При “внешней” гибридизации связь лиганда с
комплексообразователем слабее, а при наличии у последнего ва¬
кантных с1-орбиталей становится возможным присоединение к ком¬
плексу дополнительной частицы с последующим отщеплением одно¬
го из лигандов. Согласно этому, из приведенных ниже аммиакатов
наименее реакционноспособным будет аммиакат кобальта.
78
Итак, наиболее распространенными пространственными форма¬
ми координационных соединений являются квадрат, тетраэдр и
октаэдр (рис 3.1)
а б в
Рис. 3.1. Координация лигандов (L) вокруг катиона-комплексообразователя М
в комплексных соединениях: а - квадрат, б - тетраэдр, в - октаэдр.
Эти же формы реализуются и в биокомплексах, однако следует
помнить, что из-за сложного лигандного окружения металла-
комплексообразователя конфигурации комплексов обычно выглядят
сильно искаженными. Тем не менее полезно запомнить, что для та¬
ких биометаллов, как железо и кобальт, обычно характерна окта¬
эдрическая конфигурация; катион цинка, как правило, образует
тетраэдрические комплексы Сложнее ситуация с медными биоком¬
плексами Для катиона Cu(I) чаще реализуется тетраэдрическая
конфигурация, катиону же Cu(II) свойственна геометрия квадрата,
однако в принципе пространственное строение медных комплексов
подвержено легко идущим изменениям
До сих пор предполагалось, что все лиганды имеют пару элек¬
тронов, которую они способны легко отдавать, образуя так назы¬
ваемую a-связь Однако это далеко от истинного положения вещей
79
Известно много лигандов (СО, РС1з, С2Н4, СвНв и др.), в которых
электронные пары обладают слабыми донорными свойствами, но
тем не менее образующих очень прочные комплексы с металлами.
Кроме того, сомнительно, чтобы на центральном атоме происходила
локализация высокого отрицательного заряда, который создавался
бы, если бы каждый лиганд отдавал свою пару электронов. Поэтому
считается, что наряду с а-связями образуются л-связи также по до-
норно-акцепторному механизму. При этом донором служит ион ме¬
талла, отдающий свои спаренные с1-электроны лиганду, имеющему
достаточно энергетически выгодные вакантные орбитали. Такие
связи называются дативными.
Дативные связи образуются.
а) за счет перекрывания вакантных р-орбиталей лиганда с с1-
орбиталью иона металла, на которой находятся электроны, не всту¬
пившие в а-связь (с1л-рл взаимодействие),
б) при перекрывании вакантных с1-орбиталей лиганда с запол¬
ненными с1-орбиталями металла (с1л-с1л взаимодействие, рис. 3.2).
Взаимодействие с1л-рл происходит, если лиганд координируется
через атомы элементов второго периода (С, N. О), а взаимодействие
с1л-с1л - через атомы третьего и других периодов (Р, Б).
Образование дативных л-связей сопровождается переносом
электронной плотности от металла к лиганду, что увеличивает по¬
ложительный заряд и акцепторные свойства металла, а, следова¬
тельно, упрочняет а-связь. И наоборот, чем прочнее а-связь, тем
выше электронная плотность на ионе металла, тем больше его до-
норные свойства, тем прочнее становится л-связь. Таким образом,
донорно-акцепторные а-связи и дативные связи взаимно усиливают
друг друга. -
Рис. 3.2. Схема образования дативных связей:
а - с1л-ря, б - с1л-с1л взаимодействие.
80
Так, например, ион №2+ (Зс18) может служить акцептором в
а-связях за счет вакантных орбиталей 4з, 4р, 46 (см. выше строение
[М1(МН3)б]2+), а также одной Зё-орбитали, если 3(1 электроны пред¬
варительно спарены (как в [М1(СМ)4]2-). Однако ион №2* может слу¬
жить донором, если атом лиганда имеет вакантные с1-орбитали.
Этим можно объяснить большее сродство никеля к серусодержа-
щим лигандам, чем к кислородсодержащим, т.к. в атоме кислорода,
в отличие от атома серы, нет энергетически выгодных вакантных
с1-орбиталей.
По той же причине, ион Ре2* в железосодержащих белках -
цитохромах, принимающих участие в переносе электронов в дыха¬
тельной цепи, связанный с атомом серы белковой молекулы, не
способен соединяться с кислородом.
Таким образом, метод валентных связей вполне удовлетвори¬
тельно описывает стереохимию комплексов и их свойства (особен¬
но магнитные). Однако он только качественно объясняет природу
комплекса и не позволяет объяснить или предсказать спектральные
свойства и устойчивость. Поэтому в настоящее время при количе¬
ственных расчетах предпочтение отдается другим методам.
Для объяснения склонности комплексообразователя к тем или
иным лигандам существует концепция “жестких” и “мягких" ре¬
акционных центров.
Можно разделить ионы металлов на два типа К “жестким” ме¬
таллам относят щелочные и щелочноземельные металлы, цинк и
первые представители с1-металлов четвертого периода (вплоть
до Сг). Они образуют более стабильные комплексы с лигандами,
имеющими донорные атомы из второго периода (14, О, Р), чем с
аналогичными лигандами, имеющими донорные атомы из третьего
периода (Р, Б, С1) Граница между классами в ряду с!-металлов при¬
ходится примерно на Мп, который проявляет промежуточные
свойства. Для “мягких” металлов наблюдается обратный порядок
стабильности комплексов В этом случае стабильность комплекса
будет больше, когда донорный атом относится к третьему периоду
Жесткие ионы металлов (или жесткие кислоты) подобны прото¬
ну, отличаются малыми размерами и с трудом поляризуются, тогда
как мягкие ионы металлов (или мягкие кислоты) имеют большие
размеры и легко поляризуются.
Лиганды с высокоэлектроотрицательными донорнымн атомами
являются жесткими основаниями, тогда как поляризуемые лиган¬
ды - мягкими основаниями. Серусодержащие доноры являются мяг¬
кими основаниями, тогда как азотсодержащие доноры - жесткими
По этим признакам можно расположить комплексообразователи и
лиганды в ряды:
81
Комплексообразоаатели:
увеличение жесткости комплексообраэователя
N8*. к\ Мд2+, Са2+, А*2*. Яе2*, Со2*, Ы2*, Си2*, 2п2*, СИ2*, РЬ2*, Нд2*
Б-металлы с1-металлы
биометаллы токсичные
металлы
Лиганды:
увеличение жесткости лиганда
Р, ОН’, СГ, Вг\ Г, РСОО, ЫН3, -БН, СЫ'
Общее правило гласит, что устойчивые комплексы - это ком¬
плексы, образующиеся между жесткой кислотой и жестким основа¬
нием или мягкой кислотой и мягким основанием.
Из этой схемы очевидно, что ионы металлов, имеющих
биологическое значение, являются в основном жесткими или
примыкают к жестким. Более того, важнейшие компоненты
клетки и те группы в них, которые выступают как потенциально
связывающие группы, относят к жестким элементам. Другими
словами, живая система является жесткой.
Напротив, токсичные тяжелые металлы, вызывающие отрав¬
ление и загрязнение окружающей среды, будут мягкими. Мягкие
металлы в основном токсичны; известно, например, что соединения
РЬ, Нд, Т1 и С<3 ядовиты. Принципы теории твердых и мягких кислот
и оснований позволяют более просто представить некоторые биоло¬
гические явления, такие, как отравление ионами металлов.
Рассматривая данные по ионам металлов, можно обнару¬
жить интересные параллели и различия между некоторыми био¬
логически важными ионами металлов. Имеется, к примеру, хороший
параллелизм между свойствами Мп2+ и Мд2+, и поэтому марганец
может замещать магний во многих биологических системах.
3.4. Комплексные соединения
с полидентатными лигандами
Полидентатные лиганды, занимая в координационной сфере два
и более места, образуют циклические комплексы, которые называют
также хелатными; это очень важный в биологическом отношении
класс комплексных соединений. К ним относится гемоглобин, хло¬
рофилл, витамин В12 и многие металлоферменты.
82
Образование циклического комплекса можно рассмотреть на
простейшем примере глицината меди. Известно, что осадок Си(ОН)2
легко растворяется в аминоуксусной кислоте - глицине.
ch2-nh2 H2N-CH2 2и20 ch2-nh2 н^-снг
+ Си + 6 * к ЧСи^
о' 'он но' 'он но' 'о о'' 'о-" ~^о' 'о
глицинат меди
При этом каждая из двух молекул глицина, участвуя в этом
процессе, использует обе функциональные группировки, а именно,
аминогруппа связывается с атомом меди по донорно-акцепторному
механизму, а карбоксильная - через кислород обычной ковалентной
связью. Центральный атом оказывается как бы втянутым внутрь ли¬
ганда и охвачен связями наподобие клешней рака. Отсюда и проис¬
ходит название - хелатные (от chelate - клешня)
Таблица 3.3
Полидентатные лиганды
Название
Формула
Дентатиость
Этилендиамин-
тетрауксусная
кислота (ЭДТА)
НООССН2^ СНзСООН
>1СН2СНг1<
НООССН2^ ^СНзСООН
4, 5, 6
Динатриевая соль
этилендиамин-
тетрауксусной кислоты
(трилон Б)
NaOOCCH2. .СНгСООЫа
”ГЫСН2СН2Г<
НООССНг^ ^СНгСООН
4. 5. 6
Диметилглиоксим
СНз—С-С-СНз
Л к
НО' 'ЧЭН
2
Порфирин
<й>
4
Дитизон
NH-NHC6H5
s=c'
N=NCeHs
2
Аммонийная соль
нитрозофенил-
гидроксиламина
(купферон)
N=°
\0/NvONHd
2
83
Высокая прочность хелатных комплексов обусловила широкое
применение пфлидентатных лигандов (или комплексонов) в ана¬
литической химии, токсикологии, гигиене и т.д. В таблице 3.3 при¬
ведены некоторые наиболее важные комплексоны.
Строение и устойчивость хелатных комплексов зависят от вели¬
чины и характера цикла Циклы, содержащие чередующиеся про¬
стые и двойные связи (сопряженные), обычно плоские, а несопря¬
женные связи дают неплоские структуры Наиболее стабильны
пяти- и шестичленные циклы (правило Чугаева)
Основную роль в хелатообразовании играют стереохимические
особенности лиганда, а не центрального атома. Разные типы гибри¬
дизации комплексообразователя сравнительно мало отличаются по
энергии. Поэтому может наблюдаться невыгодный способ гибриди¬
зации, если при этом возрастает устойчивость комплекса за счет
увеличения числа связей между центральным атомом и лигандом.
Так, например, в хлорофилле комплекс Мд2* имеет не тетраэдри¬
ческую, а плоскоквадратную конфигурацию, обусловленную пла¬
нарным строением порфиринового кольца.
Влияние иона металла на структуру комплекса увеличивается
по мере уменьшения его радиуса. Так, анион ЭДТА выступает как
пента- и гексадентатный лиганд при образовании комплексов с гп2*,
Нд2*, С<32+, Со2*, Си2* и т д., в то время как с монодентатными ли¬
гандами эти катионы имеют КЧ = 4. Однако в комплексах с Ве2*
анион ЭДТА имеет дентатность, равную четырем.
Известны примеры комплексов с четырех¬
членными циклами. Карбоксильная группа -
СООН или карбонат-анион С032', например, в
некоторых комплексах Со3* могут выступать
как бидентатные лиганды.
Образование четырехчленного цикла яв¬
ляется характерным для дитиокарбоксильной
группы, что еще раз указывает на чрезвычайно энергетически вы¬
годную связь атомов серы с ионами переходных металлов.
Отдельные примеры комплексов, наиболее важных в биологи¬
ческом отношении, будут рассмотрены ниже (часть 6).
Глава 4. ОБЩИЕ СВЕДЕНИЯ
О БИОГЕННЫХ ЭЛЕМЕНТАХ
Чем совершеннее техника
исследования состава организ¬
мов, тем большее число химиче¬
ских элементов находим мы
в них...
А Е. Ферсман
Физико-химические свойства элементов, а следовательно и их
физиологическая роль определяются положением этих элементов в
Периодической системе Д. И. Менделеева.
Элементы, необходимые для построения и жизнедеятельности
клеток и организмов, называют биогенными элементами.
В растительных и животных системах в значительных коли¬
чествах (в макроколичествах) содержатся легкие э- и р-элементы.
К жизненно необходимым макроэлементам относятся э-эле-
менты первого (водород), третьего (натрий, магний) и четвертого
(калий, кальций) периодов, а также р-элементы второго (углерод,
азот, кислород) и третьего (фосфор, сера, хлор) периодов. Большин¬
ство остальных э- и р-элементов первых трех периодов (А1, в!, Ве
и др.) физиологически активны. Расположенные в больших перио¬
дах э- и р-элементы редко выступают в качестве незаменимых.
Исключение составляют э-элементы - калий, кальций, р-элемент -
иод. К физиологически активным относят некоторые э- и р-элемен¬
ты четвертого и пятого периодов, например, мышьяк, селен, бром
Те биоэлементы, которые имеют крайние значения электроот¬
рицательностей, а именно сильно электроположительные и сильно
электроотрицательные элементы, в живых системах присутствуют в
виде ионных структур, а именно э-элементы в виде катионов, а
галогены (элементы седьмой группы) - в виде анионов
Хлорид-ион - основной анион плазмы крови и желудочного
сока. Кальций в виде труднорастворимых солей преимущественно
концентрируется в костной, а также в зубной ткани Натрий в
основном содержится во внеклеточных жидкостях, а калий и маг¬
ний - во внутриклеточных В виде фторидов натрий и калий входят
в состав костной и зубной ткани. Магний в виде фосфата присут¬
ствует в твердых тканях зубов
Основу всех живых систем составляют шесть элементов угле¬
род, водород, кислород, азот, фосфор, сера, получивших название
органогенов. Их содержание в организме достигает 97%.
85
Элементы-органогены имеют небольшие радиусы и промежу¬
точные значения электроотрицательностей, что благоприятствует
образованию прочных ковалентных связей
Отмеченные тенденции к образованию различных типов связей
представлены на схеме 4 1 для элементов II и III периодов, где на¬
ходятся многие биогенные элементы
Рис. 4.1. Закономерности в образовании соединений
элементами II и III периода.
Важнейшим органогеном, несомненно, является углерод. Он
способен к образованию прочных ковалентных связей.
Водород и кислород - макроэлементы. Они входят в состав во¬
ды, которой в организме взрослого человека в среднем содержится
около 65%. Кислород и водород в органических соединениях следу¬
ет рассматривать как носители окислительных и восстановительных
свойств. Соотношение кислорода и водорода в биомолекулах опре¬
деляет тенденцию этих соединений к окислительно-восстанови-
тельным переходам и взаимодействию их с водой - универсальной
биосредой.
Углерод, водород и кислород входят в состав углеводов, содер¬
жание которых в тканях животных невелико - примерно 2%. Эти
же элементы входят в состав лнпидов (жиров). Кроме них в состав
фосфолипидов входит фосфор в виде фосфатных групп В наи¬
большей степени липиды концентрируются в головном мозге (12%),
а затем в печени (5%), молоке (2%) и сыворотке крови (0.6%)
Однако основное количество фосфора (600 г) - содержится в кост¬
ной ткани Это составляет 85% от массы всего фосфора, находя¬
щегося в организме человека. Фосфор присутствует и в твердых
тканях зубов, в состав которых он входит вместе с кальцием,
хлором, фтором.
Органогены - углерод, водород, кислород, азот, сера, фосфор -
входят в состав белков, нуклеиновых кислот и других биологически
активных соединений организма. Содержание углерода в белках со¬
ставляет от 51 до 55%, кислорода - от 22 до 24, азота - от 15 до
18, водорода от 5 до 7, серы - от 0.3 до 2.5%.
Необходимо отметить, что элементы-органогены образуют и
многие важные для функционирования живых систем неорганиче¬
ские молекулы (например, углекислый газ, оксид азота(Н), являю¬
щийся важным биорегулятором, и др.) и анионы (карбонат-, фосфат-
, сульфат- и др. ионы).
Кроме шести основных макроэлементов-органогенов, из которых
состоят углеводы, жиры, белки и нуклеиновые кислоты, для нор¬
мального питания человека и животных необходимы неорганические
макроэлементы - кальций, хлор, магний, калий, натрий - и микро¬
элементы - медь, фтор, иод, железо, молибден, цинк, а также се¬
лен, мышьяк, хром, никель, кремний, олово, ванадий.
Среди с1-элементов жизненно необходимы в основном элементы
четвертого периода: марганец, железо, цинк, медь, кобальт. Уста¬
новлена физиологическая роль и некоторых других с1-элементов это¬
го периода: хрома, ванадия и др с1-Элементы с их невысокими зна¬
чениями электроотрицательностей склонны к существованию в виде
катионов, а наличие вакантных орбиталей в их электронных обо¬
лочках способствует легкому образованию для них комплексных со¬
единений. В виде комплексов с биолигандами они обычно и сущест¬
вуют в биосредах. Как правило, с!-биометаллы содержатся в орга¬
низме в виде комплексов с аминокислотами, белками, нуклеиновы¬
ми кислотами, гормонами, витаминами и т.д Так, ион Ре + в качест¬
ве комплексообразователя входит в состав гемоглобина,
Со2+ - в витамин В]2- Известно множество биокомплексов и других
элементов (Си, гп, Мо и др ), играющие важную биологическую
роль в организме.
с1-Элементы пятого и шестого периодов, за исключением молиб¬
дена, не проявляют выраженной положительной физиологической
активности. Молибден же входит в состав ряда окислительно¬
восстановительных ферментов (например, ксантиноксидазы, альде-
гидоксидазы), играет ключевую роль в протекании такого глобаль¬
ного биохимического процесса, как фиксация азота микроорганиз¬
мами почвы в составе их фермента нитрогеназы.
Некоторые Г-элементы, лантаноиды и актиноиды, в микроколи¬
чествах содержатся в организме человека, однако наличие боль¬
шинства из них не установлено Как правило, они высоко токсичны,
поскольку образуют устойчивые соединения с полидентатными
биолигандами Поэтому их попадание в организм сказывается на
течении многих биохимических реакций
87
Точно перечислить все биогенные элементы в настоящее время
еще не возможно из-за сложности определения очень низких кон¬
центраций микроэлементов и установления их биологических функ¬
ций. Биогенность установлена достоверно для 24 элементов.
Десять жизненно необходимых металлов - биометаллы Это -
кальций, калий, натрий, магний, железо, цинк, медь, марганец, мо¬
либден, кобальт. В организме человека массой 70 кг содержание
биометаллов составляет (в г): кальция - 1700, калия - 250, нат¬
рия - 70, магния - 42, железа - 5, цинка - 3, меди - 0.2, марганца,
молибдена и кобальта в сумме - менее 0 1
Исходя из значимости химических элементов для жизнедея¬
тельности, их подразделяют на три группы.
1. Незаменимые элементы. Они всегда содержатся в живом ор¬
ганизме, входя в состав его неорганических и органических соеди¬
нений - белков, нуклеотидов, липидов, ферментов, гормонов, биоре¬
гуляторов и витаминов- Н, О, Са, N. К, Р, N8, Б, Мд, С1, С, I, Мп, Си,
Со, Ре, гп, Мо, V Их дефицит приводит к нарушению жизнедеятель¬
ности.
2 Примесные элементы. Эти элементы также постоянно содер¬
жатся в организме животных и человека: ба, БЬ, Бг, Вг, Р, В, Ве, Ы,
81, Бп, Сэ, А1, Ва, ве, Аэ, РЬ, РЬ, Ра, В\, Сс1, Сг, N1, Т\, Ад, ТИ, Нд, и, Эе.
Однако их биологическая роль еще не всегда детально выяснена
или даже мало известна
3. Микропримесные элементы (Бс, Т1, 1п, 1_а, Рг, Бт, \Л/, Ре, ТЬ
и др.). Они обнаружены в организме человека и животных, но све¬
дения о содержании и биологическая функция не выяснены.
Однако эта классификация отражает только содержание эле¬
ментов в живых организмах, не указывая на биологическую роль и
физиологическое значение того или иного элемента.
Содержание в биосредах элементов-органогенов, ковалентно
связанных с органической частью биомолекул, уменьшается с рос¬
том размера атомов элементов в данной группе Периодической сис¬
темы. Например, содержание элементов шестой группы распределя¬
ется следующим образом: О > Б > Бе > Те. Это, вероятно, связано с
общей тенденцией по мере увеличения радиуса элементов к умень¬
шению прочности их ковалентых связей с углеродом, цепи которого
составляют структурную основу органического вещества.
Содержание элементов, находящихся в организме в виде ионов
(Б-элементы, р-элементы седьмой группы), с ростом радиуса атома
элемента в группе увеличивается до элемента с неким оптималь¬
ным ионным радиусом, а затем уменьшается. Так, например, во
второй группе при переходе от бериллия к кальцию содержание
элементов в организме увеличивается, а при дальнейшем переходе
от бария к радию снижается. Аналогично и в седьмой группе: при
переходе от фтора к хлору содержание элемента в организме уве¬
личивается, а затем уменьшается.
Как правило, токсичность элементов данной группы увеличива¬
ется (и соответственно уменьшается их содержание в организме)
при переходе к элементам с большими атомными и ионными радиу¬
сами, с высоким зарядом ядра. Это, вероятно, вызвано падением
растворимости соединений таких элементов, в силу чего они плохо
усваиваются живыми организмами. Во-всяком случае, среди
[-элементов нет биогенных. Если даже они и обнаруживаются в
ультрамикроскопических количествах в тканях, то вряд ли с их
присутствием связаны какие-либо биологические функции.
Близкие значения атомных и ионных радиусов, энтальпий иони¬
зации, координационных чисел, склонность к образованию связей с
одними и теми же элементами в молекулах биолигандов обусловли¬
вает эффекты замещения элементов в биологических системах. Та¬
кое замещение ионов может происходить как с усилением
(синергизм), так и с подавлением активности (антагонизм) за¬
мещаемого элемента.
Например, Б-элементы первой группы склонны к образованию
связен с атомом кислорода, все они находятся в растворе в виде
гидратированных ионов Э+(Н20)Х. Сходство лития с натрием обу¬
словливает их взаимозамещаемость, причем, как правило, они яв¬
ляются синергистами. Рубидий и цезий по физико-химическим ха¬
рактеристикам ближе к калию, поэтому в организме они также мо¬
гут замещать друг друга
Б-Элементы второй группы входят в состав биомолекул, связы¬
ваясь через атом кислорода с анионами фосфорной, угольной и кар¬
боновых кислот.
Магний в организме по преимуществу находится внутри клеток,
где образует соединения с белкамн и нуклеиновыми кислотами, со¬
держащие связи Мд-М и Мд-0 Сходство физико-химических харак¬
теристик ионов Ве2+ и Мд2+ обусловливает их взаимозамещаемость в
таких соединениях. Это объясняет, в частности, ингибирование
магнийсодержащих ферментов при попадании в организм бериллия,
следовательно, бериллий - антагонист магния Кальций, в основном
находящийся в составе костной ткани, по своим свойствам близок к
стронцию и барию, поэтому эти ионы могут замещать его в костях
При этом наблюдаются как случаи синергизма, так и антагонизма
р-Элементы третьей группы в микроколичествах входят в состав
биомолекул, связываясь с атомами кислорода или азота. Так, извес¬
тен антибиотик борамицин, в котором реализуется донорно-
акцепторное взаимодействие между атомами азота и бора Исклю¬
чение составляет таллий, для которого характерно образование свя¬
зи с атомами серы, чем объясняется его высокая токсичность.
р-Элементы четвертой группы входят в состав биомолекул, свя¬
зываясь с атомамн разных элементов Углерод в биомолекулах об¬
разует полимерные цепи углерод-углерод и прочно соединяется с
водородом, кислородом, азотом, серой, селеном и иодом. Прочие
элементы этой группы (кремний, германий, олово, свинец) образуют
предпочтительно связи с атомом кислорода, а свинец и с серой.
Различие в прочностях перечисленных выше связей элементов этой
группы обусловливает отсутствие аналогий в их физиологических
функциях Склонность свинца давать прочную связь с атомом серы
определяет его токсическое действие.
р-Элементы пятой группы также входят в состав биомолекул,
образуя связи с атомами многих элементов. Для азота в биомолеку¬
лах характерны связи с углеродом и водородом. Фосфор связывает¬
ся через кислород, мышьяк, сурьма и висмут - через кислород и се¬
ру. Это определяет малое сходство азота с фосфором, а также от¬
личие этих элементов от мышьяка, сурьмы и висмута. Наоборот,
склонность мышьяка, сурьмы и висмута к связыванию с серой бел¬
ков определяет их токсичность и в целом - синергизм в поведении
в живых системах.
р-Элементы шестой группы образуют в биомолекулах связи с
различными элементами. Однако сильноэлектроотрицательный ки¬
слород резко отличается по физико-химическим характеристикам от
серы и селена, в то время как они сходны по свойствам и выступа¬
ют в качестве синергистов
р-Элементы седьмой группы - бром и хлор обычно находятся в
организме в виде гидратированных галогенид-ионов, а фтор и иод -
в связанном состоянии Фтор связывается с металлами в трудно¬
растворимые соли (Са, Мд, Ре). По электроотрицательности и
склонности к координации с биогенными элементами фтор резко
отличается от других галогенов, поэтому он мало участвует в заме¬
щении ионов хлора, брома и иода. Три последних элемента близки
по свойствам и могут замещать друг друга в организме. Иод с его
невысокой электроотрицательностью в организме образует кова¬
лентные соединения со связью С-1.
Для с1-элементов более характерно горизонтальное сходство, чем
вертикальное. Ионы двухвалентных марганца, железа, кобальта,
никеля, меди, цинка имеют сходные физико-химические характери¬
стики: электронную конфигурацию, близкие радиусы ионов, од¬
нотипные координационные числа 4 и 6 (соответственно тетраэдри¬
ческое или квадратное и октаэдрическое окружение лигандов)
Сходство характеристик этих элементов обусловливает их хотя бы
90
частичную взаимозаменяемость и параллелизм в биологическом
действии. В виде определенных биокомплексов (в том числе, метал-
лоферментов) все они участвуют в регуляции обмена веществ, яв¬
ляясь катализаторами биосинтеза.
Большинство из них, за исключением цинка, стимулируют кро¬
ветворение. Их синергизм в этом процессе связан с участием их
ионов в различных этапах синтеза ферментных систем крови. Более
того, молекулы-переносчики кислорода для человека и большинства
высших животных содержат в своем составе железо, у червей эту
роль выполняют медьсодержащие белки, а у асцидий ту же функ¬
цию выполняют белки, в состав которых включен ванадий Ионы
никеля, марганца и железа замещают друг друга в живых организ¬
мах, участвуя в однотипных ферментативных превращениях.
Синергизм и антагонизм элементов изучен еще недостаточно.
Исследование этого вопроса важно, так как его решение дает воз¬
можность оценить биологическую роль элементов, создавать новые
лекарственные препараты.
Существующие частные зависимости между физико-химичес¬
кими характеристиками элементов и их биологическим действием
позволяют в определенной мере прогнозировать результат поступ¬
ления различных соединений этих элементов в организм человека.
Тем не менее простые и общие зависимости между физико-хими¬
ческими характеристиками элементов Периодической системы и их
функционированием в живых системах отсутствуют
Глава 5. ХИМИЧЕСКАЯ КИНЕТИКА
Для получения полного по¬
нятия о химических изменениях
тела, конечно, необходимо
обобщить все те реакции,
вследствие которых оно проис¬
ходит
Д И. Менделеев
В приложении к химическим
реакциям мы определяем ско¬
рость реакции как отношение
количества вещества, претер¬
певающего некоторое превра¬
щение, ко времени, в течение
которого это превращение со¬
вершилось.
И. А Меншуткнн
Химическая реакция - основной предмет изучения химии. Из¬
вестны реакции, которые идут чрезвычайно быстро. К их числу от¬
носятся взрывы, большинство ионных взаимодействий в растворах
(нейтрализация, образование осадка труднорастворимой соли и
другие). За тысячные доли секунды осуществляется передача нерв¬
ного импульса, в основе которого лежат химические процессы.
С другой стороны, известно много медленных химических превра¬
щений, например, коррозия. Таково и большинство биопроцессов -
биосинтез, в том числе фотосинтез, сбраживание и др. Так, белки
обновляются наполовину за 70 суток, а неорганическая основа
костных тканей полностью обменивается за 4-7 лет. Процессы об¬
мена веществ представляют собой несколько тысяч биохимических
реакций, идущих с согласованными между собой скоростями.
В зависимости от условий различным образом может осущест¬
вляться одна и та же химическая реакция. Например, не окисляе¬
мая прн обычных условиях глюкоза горит на воздухе при добавле¬
нии к ней каталитических микроколичеств солей некоторых метал¬
лов и взрывается в смеси с жидким кислородом Глюкоза медленно
“сгорает” в организме в процессе биологического окисления. Зна¬
чение проблемы регулирования скоростей химических реакций
очевидно. К примеру, в промышленности, как правило, выгодно
осуществлять быстрые процессы, а в биологических превращениях
часто более важным оказывается замедление - ингибирование.
Для этого необходимо знать, каков механизм данной химической
92
реакции. Изучение механизмов реакций и определение их скоро¬
стей и составляет предмет химической кинетики. Законы кинетики
универсальны, будь это явление оседания эритроцитов, процесс
усвоения лекарства, рост микробной популяции и т.д.
5.1. Скорость химической реакции
Скоростью химической реакции (V) называют число элементар¬
ных актов реакции, происходящих в единицу времени. Скорость ре¬
акции обычно измеряют изменением молярной концентрации исход¬
ных веществ илн продуктов реакции за секунду (моль/л с).
Если в момент времени Т] концентрация одного из веществ со¬
ставляет С], а в момент Тг - С2, тогда средняя скорость изменения
концентрации (Уср ) равна отношению:
ср ■
с2 ~ С1 _±Ас
т, - т2 Дт '
Изменение концентрации имеет положительный знак для про¬
дуктов и отрицательный - для реагентов. Скорость же реакции при¬
нято считать величиной положительной, поэтому убывание кон¬
центрации последних следует брать со знаком минус.
В ходе реакции ее скорость меняется непрерывно, поэтому пра¬
вильно пользоваться истинными, т.е. мгновенными значениями ско¬
ростей (V)- V = ± —, те изменением концентрации за бесконечно
с1т
малый промежуток времени - в виде производной концентрации
по времени Если скорость измеряют возрастанием концентрации
какого-либо вещества (образованием продукта), то производную
берут со знаком плюс
Опыт показывает, что скорость химических реакций обычно за¬
висит лишь от нескольких факторов, концентраций реагентов, их
физического состояния (газовая, жидкая или твердая фаза, природа
растворителя при взаимодействии в растворах), поверхности реаги¬
рующих веществ, если реакция идет в гетерогенной среде, давле¬
ния, температуры и катализатора
Уже давно известно, что увеличение концентрации (а также
давления в газовой фазе) реагирующих веществ увеличивает ско¬
рость химических превращений Первое истолкование связи скоро¬
сти химической реакции с концентрацией реагентов дал Н. Н Беке¬
тов (1865 г.). Обобщенно эту закономерность сформулировали
Гульдберг и Вааге (1867 г.)
93
Реакция может происходить только в результате контакта меж¬
ду молекулами - их столкновения между собой. Так как число
столкновений зависит от концентрации, то с ее ростом должна воз¬
растать и скорость химической реакции. Обычно измеряемая ско¬
рость реакции прямо пропорциональна концентрациям реагентов,
взятым в некоторой степени, что можно записать в кинетическом
уравнении для реакции аА + ЬВ + сС + . . ->...
у= 4^= к |а]“ [вр [с]' . .
с1т
где а, Р и у - показатели степени, определяемые опытным путем;
к - константа скорости реакции, не зависящая от концентрации
Константа скорости реакции (к) - фундаментальный кине¬
тический параметр, не зависящий от концентраций реагентов, а по¬
тому остающийся неизменным в течение реакции. Константа ско¬
рости химической реакции численно равна скорости химической
реакции при концентрациях всех реагирующих веществ, равных
1 моль/л. Константа скорости химической реакции зависит от при¬
роды реагирующих веществ, температуры и наличия в реакционной
среде катализатора.
Величины а, Р и у находятся специальными методами и назы¬
ваются порядками реакции по веществу А, В и С, соответствен¬
но. Сумма показателей степеней в кинетическом уравнении назы¬
вается общим порядком реакции п = а + Р + у. Порядок реак¬
ции определяет характер зависимости скорости от концентрации.
Наблюдаемые концентрационные кинетические зависимости тех
или иных реакций определяются их механизмом. Лишь немногие
химические превращения осуществляются в одну стадию. Большин¬
ство же процессов проходит через несколько элементарных стадий,
в которых могут принимать участие одна, две или три молекулы.
В соответствии с этим существуют моно-, би- или тримолекулярные
стадии. Число молекул, участвующих в отдельных стадиях, назы¬
вают молекулярностью.
Вероятность одновременного столкновения трех молекул, необ¬
ходимого для осуществления тримолекулярного процесса, в тысячи
раз меньше вероятности двойного соударения. Поэтому уже тримо¬
лекулярные процессы встречаются крайне редко и обычно элемен¬
тарные стадии любого химического превращения можно свести к
моно- или бимолекулярным взаимодействиям. Так, биологическое
окисление глюкозы до воды и углекислоты включает в себя 38 та¬
ких элементарных стадий. Лишь для очень простых реакций поря¬
док совпадает с молекулярностью. Интерпретация концентрацион¬
ных кинетических зависимостей чрезвычайно сложна, т.к. должна
94
исходить из реакционной схемы, учитывающей все элементарные
стадии и индивидуальные скорости этих стадий. Даже реакция син¬
теза иодистого водорода, для которой порядок равен молекулярно-
сти, имеет не бимолекулярный механизм. В большинстве случаев
скорость многостадийной реакции определяется скоростью ее самой
медленной стадии. В результате определяемый экспериментально
порядок реакции и отражает порядок этой стадии.
В зависимости от механизма реакции бывают простые (идут
в одну стадию) и сложные (многостадийные) Сложные реакции
могут быть последовательными, параллельными, сопряженными,
цепными и др.
Последовательные реакции (А -> В -> С -> О ->...) идут через
несколько различных промежуточных стадий, следующих одна за
другой. Примерами последовательных реакций могут служить фото¬
синтез, биологическое окисление глюкозы.
Параллельные реакции происходят одновременно в несколь¬
ких направлениях, т е. превращение вещества осуществляются че¬
рез различные промежуточные стадии:
В
Так, при нагревании бертолетовой соли КСЮз одновременно
идут два превращения:
KCI + ЗКСЮ4 <- 4КС10з -> 4KCI + 602.
Параллельно с крекингом углеводородов осуществляется их
изомеризация; замещение для производных бензола идет с образо¬
ванием смеси орто-, мета- и пара-изомеров Параллельно биологи¬
ческому окислению глюкозы могут происходить ее молочнокислое
или спиртовое брожение
Сопряженные реакции представляют собой совокупность раз¬
личных превращений, идущих через общую промежуточную стадию.
Осуществление одной из сопряженных реакций
(А -> В) обусловливает протекание другой (D -> Е), Ач - Е
причем вторая реакция невозможна без первой, ко-
торая индуцирует ее /
Явление химической индукции впервые исследо- D В
вал А Н Шилов (1905 г) Исключительна роль
сопряженных реакций в биологических процессах Например, кле¬
точное окисление углеводов или липидов вызывает синтез адено-
зинфосфорной кислоты, что в свою очередь индуцирует различные
95
превращения, в частности, биосинтез белков и нуклеиновых кислот.
Изучение механизма сопряженных реакций (особенно природы про¬
межуточного состояния С) является важнейшей задачей современ¬
ной физико-химической биологии.
Цепные реакции - процессы, в которых промежуточные ак¬
тивные соединения (как правило, содержащие неспаренные элек¬
троны) не исчезают в процессе образования конечных продуктов,
а стадии превращения исходных соединений в продукты многократ¬
но повторяются Простейшим примером цепной реакции является
синтез хлористого водорода по схеме:
С12 -» С1 + С1
СГ + Н2 -» НС1 + Н'
Н‘ + С12 -» НС1 + С1
СГ + СГ -» С12 (обрыв цепи)
Н' + Н' -» Н2 (обрыв цвпи)
Реакция начинается с диссоциации молекулы хлора (зарож¬
дение цепи). Встреча атома С1- с Н2 приводит к образованию НС1 и
атомарного водорода Н-, который в свою очередь атакует молекулу
С12. Таким образом происходит развитие цепи, включающее в себя
все большее число молекул реагентов Взаимодействие между собой
однотипных свободных атомов приводит к обрыву цепи. Цепные ре¬
акции играют важную роль в ряде патологических биопроцессов
(канцерогенез, лучевая болезнь и др ) В разработке теорий цепных
процессов большая заслуга принадлежит Н. Н. Семенову.
В зависимости от электронных изменений в ходе реакций раз¬
личают гомолнтические и гетеролитические реакции. В ходе
первых происходит разъединение пары электронов, образующих хи¬
мическую связь:
А:В -» А’ + В.
Гомолнтические процессы иначе называют радикальными, так
как в них образуются частицы, содержащие неспаренные электро¬
ны, которые именуют радикалами Приведенная реакция синтеза
хлористого водорода - типичный пример гомолитической реакции.
В гетеролитических процессах электронная пара остается у од¬
ного из партнеров связи:
А:В -» А:' + В+.
В живых системах имеют место оба вида реакций, причем около
двух третей происходящих в организме метаболических превраще¬
ний приходится на долю гетеролитических процессов.
96
5.2. Уравнение Аррениуса. Энергия активации
Известно, что с ростом температуры скорость реакции обычно
растет*. В 1879 году было сформулировано (Я Вант-Гофф) эмпири¬
ческое правило: с ростом температуры на 10° С скорость хими¬
ческой реакции возрастает в 2-4 раза
Объяснение роста скорости реакции с температурой заключает¬
ся в том, что не всякое столкновение приводит к химическому пре¬
вращению. Для осуществления реакции необходимо, чтобы молеку¬
лы обладали запасом энергии, достаточным для расшатывания тех
связей, которые перестраиваются в ходе реакции.
В качестве примера обратимся к рассмотренной выше цепной
реакции синтеза HCI. Зарождение цепи в ней возникает при разрыве
связи в молекуле Нг или СЬ Энергия связи молекулы Нг составляет
436 кДж/моль, а хлора - 240 кДж/моль, следовательно, мини¬
мальной необходимой для начала реакции энергией, явится энергия
разрыва связи CI-CI. Этой энергией обладает квант света с частотой
240
V = (h = 4* 10'13 кДж-с/моль ) или v = 6-1014 с1. Разделив
h
скорость света (V = 299.793-106 м/с) на найденное значение часто¬
ты, получим длину волны X = 510'5 см, что соответствует ближней
ультрафиолетовой части спектра. Именно такое излучение иниции¬
рует этот фотохимический процесс
Таким образом, для прохождения реакции нужно преодолеть не¬
который энергетический барьер, именуемый энергией активации
(Еа) Энергия активации - избыточная энергия, необходимая для
вступления реагирующих веществ в реакцию при их столкновении,
по сравнению со средней энергией, которой обладают молекулы.
Обычно значение Еа составляет от 40 до 200 кДж/моль
Математическая зависимость скорости химической реакции от
температуры выводится из теории соударений, считая реакцию би¬
молекулярной и идущей в газовой фазе
Прежде всего, молекулы должны встретиться, т е должно про¬
изойти их соударение Частота соударений достаточно велика и со¬
ставляет в 1 см3 газа при нормальных условиях -1028 в секунду
Однако число реагирующих частиц обычно оказывается всегда
значительно меньшим (в 1015 - Ю20 раз), т к не все сталкиваю¬
* В случае биологических превращении это справедливо лишь до опре¬
деленных температур, выше которых происходит деструкция биомолекул и
скорость реакций резко падает
4 Зак. 675
97
щиеся молекулы обладают энергией, большей чем энергия актива¬
ции (Еа) Доля таких молекул от их общего числа определяется
тепловым распределением Максвелла-Больцмана (рис 5 1)
Рис. 5.1. Распределение молекул по энергиям
при различных температурах.
Согласно уравнению Больцмана, доля частиц с большей, чем Еа
энергией (1^£а ), по отношению к общему их числу (Ы) составляет:
В соответствии с этим повышение температуры от Т) до Тг уве¬
личивает долю молекул с достаточной для реакции энергией. Сле¬
довательно, и зависимость скорости реакции от температуры долж¬
на носить экспоненциальный характер.
Эта зависимость (уравнение Аррениуса, 1889 г.) выглядит так:
Ез
к = А • е ~ КТ ,
где к - константа скорости реакции, А - предэкспоненциальный
множитель.
Предэкспоненциальный множитель А отражает долю эффек¬
тивных соударений в общем числе соударений. Очевидно, что его
значения должны находиться в интервале от 0 до 1 (при А = 1 все
соударения оказываются эффективными). Его истинное значение
обычно значительно меньше единицы, поскольку не всякое соударе¬
ние даже с энергией, большей, чем Еа, обязательно приведет к
реакции. Столкнувшиеся молекулы могут не взаимодействовать,
т.к. они не ориентированы должным образом в пространстве. Про¬
странственный фактор особенно проявляется в превращениях био¬
98
молекул, где трансформации должен подвергаться лишь реагирую¬
щий участок молекулы, небольшой по отношению ко всем ее разме¬
рам. Вероятность реакции может понижаться и за счет того, что в
некоторых случаях энергия перераспределяется внутри молекулы
без ее химических изменений.
5.3. Инициация и катализ
Многие реакции можно вызывать действием различных ини¬
циаторов - излучения, механического воздействия и др. Так, фото¬
химия занимается изучением реакций, идущих под действием света.
К числу фотохимических процессов относится выше приведенная
реакция водорода с хлором; под действием ультрафиолетового излу¬
чения идет синтез озона в верхних слоях атмосферы, процесс фото¬
графирования основан на фотохимическом разложении солей се¬
ребра. Самая масштабная из биореакций - фотосинтез - также яв¬
ляется фотохимической. Зрительная рецепция базируется на фото¬
химической изомеризации биоорганических соединений.
В последнее время бурно развивается лазерная химия, исполь¬
зуемая, к примеру, в синтезе алмазов. Радиационная химия иссле¬
дует процессы под действием радиоактивного излучения, в част¬
ности, реакции, вызывающие лучевую болезнь. Химия в электри¬
ческом разряде привела к разработке многих технологических про¬
цессов (электрокрекинг, синтез озона и др ). В атмосфере под дей¬
ствием электрических разрядов происходит образование N0, что
служит основой фиксации азота. Механохимия исследует реакции,
которые происходят в результате механического воздействия - тре¬
ния, удара. Последнее свойство предопределяет применение детона¬
торов. Интересные перспективы раскрываются при использовнии
ударного сжатия. Таким способом можно осуществить полимериза¬
цию, в частности, аминокислот в белковые молекулы
Важнейшими регуляторами химических превращений являются
катализаторы. Катализаторы - вещества, изменяющие скорость
химических реакций в результате их многократного участия в про¬
межуточных химических взаимодействиях с компонентами реакции,
сохраняющие при этом свой состав. Роль катализатора заключается
в понижении энергии активации данной реакции за счет того, что
он создает новый путь ее протекания.
Если реакция А + В -» АВ обладает энергией активации Еа (рис.
5 2), то катализатор (К) снижает ее на величину ДЕа за счет того,
что образует промежуточное соединение А + К -» АК, что требует
меньшей величины энергии активации Далее комплекс АК реаги¬
4'
99
рует с компонентом в- АК + В -» АВ + АК Эта реакция идет также с
меньшей энергией активации, чем Еа. Таким образом, произошло
суммарное взаимодействие с образованием продукта АВ, а катали¬
затор возвратился из реакции без изменения.
Рис 5.2. Энергетическая диаграмма действия катализатора.
Из уравнения Аррениуса видно, что даже малое уменьшение
величины Еа должно приводить к сильному возрастанию скорости
процесса. Это определяет исключительно большое практическое
значение катализаторов. Особенно ценным является применение
катализаторов в параллельных реакциях, т.к. подбором катализато¬
ра можно добиться понижения энергетического барьера только од¬
ного из параллельных процессов. Поэтому катализаторы находят
чрезвычайно широкое применение в реакциях органических соеди¬
нений, которые могут претерпевать множественные превращения.
По существу вся современная нефтехимическая промышленность
базируется на использовании разноообразных каталитических тех¬
нологических схем.
Различают катализ положительный (скорость реакции увели¬
чивается) и отрицательный (реакции идут с меньшей скоростью).
В последнем случае катализ называют ингибированием. Ингибито¬
ры также важны для практики как консерванты, антиоксиданты
и др. Ингибирование, как и положительный катализ, необходимо
для регулирования скоростей биопроцессов.
100
В том случае, когда продукты реакции влияют на скорость
взаимодействия исходных веществ, имеет место автокатализ.
Примером автокаталитической реакции является окисление под
действием КМпОд, скорость которого возрастает в присутствии ка¬
тионов Мп2+.
Катализ бывает гомогенный и гетерогенный. Гомогенный ката¬
лиз осуществляется катализатором, который образует однородную
(обычно жидкую или газовую) систему с реагирующими вещества¬
ми. Таков, к примеру, катализ окисления СО в СОг в присутствии
каталитических количеств водяного пара.
Гетерогенный катализ идет в присутствии катализатора, обра¬
зующего самостоятельную фазу, отделенную от реагирующей си¬
стемы границей раздела. В качестве гетерогенных катализаторов
выступают многие металлы, их оксиды и др. Теория гетерогенного
катализа сложна. Действие гетерогенных катализаторов связывают
с различными причинами (адсорбция реагентов на поверхности ка¬
тализатора, генерация цепных процессов, образование нескольких
связей между катализатором и молекулой реагента и т.д.).
Особое место занимает микрогетерогенный катализ, при кото¬
ром катализатор находится в коллоидном или высокомолекулярном
состоянии. Примером этого служат все биопроцессы, происходящие
под действием биокатализаторов - ферментов (энзимов).
5.4. Ферментативный катализ
Ферменты являются катализаторами химических реакций, про¬
исходящих в организме. В настоящее время известно около 10 ООО
биохимических реакций, каждая из которых катализируется фер¬
ментами.
Характерными особенностями ферментов являются чрезвычай¬
но высокая эффективность. Так, разложение пероксида водорода
без катализатора имеет энергию активации 75 кДж/моль, гетеро¬
генный катализ под действием платины снижает активационный
барьер до 48 кДж/моль, а в присутствии фермента этой реакции,
каталазы, энергия активации составляет лишь 23 кДж/моль Такое
изменение энергии активации приводит к увеличению скорости
реакции в 20 ООО и 3-1011 раз, соответственно
Гидролиз белков под действием HCI требует энергии активации
80 кДж/моль, а присутствующий в желудочном соке трипсин
понижает ее до 50 кДж/моль, благодаря чему и происходит усваи¬
вание белковой пищи желудком.
101
Один моль фермента может вызвать трансформацию 1000 -
100 ООО молей вещества (субстрата) в течение секунды, что экви¬
валентно увеличению скорости неферментативной реакции в Ю10-
1013 раз. Такая эффективность ферментов объясняется, во-первых,
концентрационным фактором -те активной сорбцией ферментом
субстрата, что эквивалентно увеличению его концентрации. Кон¬
центрационный фактор увеличивает скорость в тысячи раз.
Во-вторых, ферменты проявляют ориентационный эффект, кото¬
рый увеличивает скорость реакции еще примерно в тысячу раз.
Сущность ориентационного эффекта состоит в наличии стереоспе-
цифического контакта активного центра фермента с субстратом. Это
резко увеличивает вероятность эффективного столкновения, что
эквивалентно возрастанию предэкспоненциального множителя А
в уравнении Аррениуса.
В-третьих, ферменты обладают полифункциональным эффектом,
который имеет решающее значение. Этот эффект состоит в одно¬
временном действии на молекулу субстрата нескольких атакующих
групп фермента или в ряде последовательных воздействий на пре¬
вращающуюся связь.
Ферменты - это белки или их производные, которые благодаря
особой трехмерной структуре выступают в качестве высокоспеци¬
фичных биохимических катализаторов. В целом клетка нуждается в
разнообразных ферментах, каждый из которых катализирует строго
определенную реакцию Лишь ограниченное число ферментов ката¬
лизирует определенную разновидность реакций. Так, например, эс-
теразы являются катализаторами гидролиза сложных эфиров.
Процессы в живой природе, как правило, представляют собой
серию взаимосвязанных сложных реакций. Зачастую продукт одной
реакции служит исходным материалом (субстратом) для другой ре¬
акции. Реакции должны быть весьма специфичными, чтобы не воз¬
никало осложнений со стороны других процессов, протекающих од¬
новременно.
Необходимость для ферментативной реакции каких-либо допол¬
нительных низкомолекулярных компонентов можно проиллюстриро¬
вать таким опытом. Если раствор неочищенного фермента в целло¬
фановом мешочке поместить в дистиллированную воду, поры в цел¬
лофане позволяют низкомолекулярным компонентам диффундиро¬
вать в наружный водный раствор, в то же время молекулы белка
остаются внутри мешочка. Оказалось, что фермент становится не¬
активным до тех пор, пока отделенные таким способом низкомоле¬
кулярные компоненты не будут добавлены к нему вновь.
Активаторами ферментов могут быть ионы металлов или слож¬
ные органические молекулы, такие, как нуклеотиды или витамины.
Они называются коферментами.
102
Для описания механизма действия ферментов общепринято
сравнение с замком и ключом. Эта аналогия полезна, однако не от¬
ражает других происходящих взаимодействий, например, изменения
формы фермента при связывании его с субстратом.
Активный центр, т. е. та часть фермента, которая участвует в
непосредственном взаимодействии с субстратом, включает лишь
несколько аминокислотных остатков, причем эти остатки могут
принадлежать участкам белковой цепи, взаимно удаленным друг от
друга. Активный центр создается определенной конфигурацией бел¬
ковой молекулы, образующей щель, в которую встраивается акти¬
вируемый субстрат. Это объясняет специфичность ферментов.
Участок фермента, называемый активным центром, не только
обусловливает определенную стереоспецифичность реакции. Важна
природа боковых цепей, образующих этот центр, их гидрофильные и
гидрофобные свойства Так, наличие в активном центре неполярных
групп приводит к тому, что среда приобретает низкую диэлектри¬
ческую проницаемость по сравнению с водной фазой, в которой
находится фермент. Это приводит к изменению некоторых пара¬
метров иначе идут процессы электролитической диссоциации, ста¬
новятся более сильными ион-ионные взаимодействия. Все это важ¬
но для правильного функционирования фермента.
Кроме того, субстрат связывается с активным центром фермента
водородными связями и вандерваальсовыми взаимодействиями. На¬
чальное взаимодействие между субстратом и ферментом вызывает
последующие перестройки фермента, приводящие к еще более тес¬
ному сближению между субстратом и ферментом
Примером участия активного центра фермента в биохимической
реакции может служить реакция связывания углекислого газа водой
с помощью фермента карбоангидразы, в состав которого входит ка¬
тион цинка (рис. 5.3). Природа функциональных групп в активном
центре фермента, с которыми связан катион цинка, для упрощения
не указана.
активный
центр
Рис 5 3. Участие активного центра карбоангидразы
в гидратации углекислого газа.
103
Характерной чертой ферментативного катализа является то, что
скорость ферментативной реакции увеличивается до определенной
постоянной величины (Утах). Типичная кривая зависимости ско¬
рости ферментативной реакции от концентрации субстрата с5 пред¬
ставлена на рис. 5.4
Рис. 5.4. Зависимость скорости ферментативной реакции
от концентрации субстрата.
В 1913 году Михаэлисом и Ментен была предложена теория,
объясняющая эту зависимость. Ферментативный процесс можно
представить схемой:
^ Е8
к2
Е + Э
Е + Р.
в которой Е и Б - фермент и субстрат, ЕБ - промежуточный фер-
мент-субстратный комплекс, Р - продукт реакции, а кт-э - константы
скоростей соответствующих реакций.
Начальная скорость образования продукта реакции Р должна
быть пропорциональна концентрации промежуточного комплекса:
ат
С другой стороны, скорость реакции определяется соотношени¬
ем скоростей всех трех реакций:
V! = к, • [Е] • [Э] У2 = к2 • [ЕЭ] Уз = к3 • [ЕБ].
Концентрацию фермента через некоторый промежуток времени
после начала реакции можно представить, как [Е] = [Е]0 - [ЕЭ].
В обратимом процессе образования комплекса ЕЭ в состоянии
равновесия скорость его образования V), равна скорости его разло¬
жения У2+Уз-
104
V, - (У2 + Уз) = о = МеНэ] - к2[Е8] - к^ЕБ] =
= к,{ [Е]0 - [ЕЭ] Ив] - к^ЕЭ] - кз[ЕЭ].
Решая это уравнение относительно [ЕБ], находим:
Ме]0 (Э]
[еэ] =
к| [Э] + к2 н
В этом случае скорость ферментативной реакции будет описы¬
ваться уравнением
У, = ...кз к, Но N илиУо = ММ^.
[в] + 1<2 + кз
к2 + к3
где Кт = , которая называется константой Михаэлиса.
к,
В кинетических исследованиях используют величину макси¬
мальной скорости Утах, т.е. скорости, с которой реагирует фер¬
мент, полностью существующий в виде комплекса ЕБ. Тогда
[Е]0=[Е8], а Утах = к3 • [ЕЭ] = к3 • [Е]0.
Подставив эти выражения в предыдущее уравнение, получим:
V = V™, [э!
° К„+181
Если У0 = то Кт = [в], т е величина константы Михаэ¬
лиса численно равна концентрации субстрата, при которой скорость
реакции составляет половину максимальной (см рис 5.4).
Кинетическая константа к3 в уравнении Утах = к3[Е]0 назы¬
вается числом оборотов фермента, которое показывает количество
молекул субстрата, превращаемых в продукт реакции в условиях,
когда весь фермент находится в составе фермент-субстратного ком¬
плекса ЕБ. Одним из самых активных ферментов является карбо-
ангидраза, для которой число оборотов составляет 6-105 с1 Таким
образом, 10'6 моль/л раствор карбоангидразы способен катализи¬
ровать образование 0 6 моля Н2СО3 из СОг и НгО за секунду В этом
случае Утах = 6-105 с'ЧО'6 моль/л =06 моль/л с
Величина константы Михаэлиса зависит от pH, температуры,
природы субстрата и других факторов Ее значение, приводимое на¬
ряду с Утах и числом оборотов фермента, как количественный па¬
раметр ферментативной реакции, служит характеристикой конкрет¬
ной фермент-субстратной системы в определенных условиях
105
Ферментация уже давно используется в человеческой практике
Явления сбраживания, сквашивания, скисания, которые лежат в
основе процессов производства кисломолочных продуктов, сыра,
алкогольных напитков, обработки фруктов и овощей, являются
ферментативными процессами. Микробиологическая промышлен¬
ность производства антибиотиков и др. базируется на фермента¬
тивных реакциях В настоящее время перспективы промышленного
использования ферментативных реакций еще только раскрываются
В будущем биотехнологические ферментативные процессы явятся
основным источником промышленного производства аминокислот,
белков, углеводов, гормонов, лекарственных препаратов и других
био- и физиологически активных молекул
Уже в настоящее время в промышленности используется
несколько десятков технологических ферментативных схем (в том
числе, синтез некоторых окси- и аминокислот, гидролиз крахмала и
многие другие).
Интенсивно развивается медицинская энзимология, которая из¬
учает вопросы применения ферментов в качестве лекарственных
препаратов, а также разрабатывает методы диагностики с помощью
ферментов.
Еще одна важная область приложения закономерностей хими¬
ческой кинетики к медицинской науке - фармакокинетика и токси-
кокинетика, занимающиеся вопросами скоростей действия и выве¬
дения из организма лекарственных препаратов и ядовитых веществ,
соответственно. Фармакокинетика применяет методы математиче¬
ского моделирования Так, простейшей моделью организма можно
считать сосуд с объемом, равным объему его жидких сред, в кото¬
рый введено лекарство в определенном количестве т. Тогда ско¬
рость его выведения можно выразить формулой:
где ке - константа элиминирования, то есть выведения лекарствен¬
ного препарата из организма, которая при прочих постоянных усло¬
виях зависит от природы лекарственного препарата. С помощью из¬
вестных значений ке и последующего решения дифференциального
уравнения можно определить полупериод выведения лекарства из
организма. Это позволяет определить дозировку препарата и интер¬
валы его приема. Те же закономерности, базирующиеся на общих
законах химической кинетики, применимы и для токсикокинетиче-
ских расчетов.
106
Глава 6. ПОНЯТИЕ О ХИМИЧЕСКОЙ
ТЕРМОДИНАМИКЕ
Термодинамика - это един¬
ственная физическая теория,
относительно которой я уверен,
что... она никогда не будет
опровергнута.
А. Эйнштейн
...количество теплоты, вы¬
деляющееся при сгорании орга¬
нического вещества, будет
важным фактором, который
приведет нас к познанию
строения этого вещества.
Г. Гесс
Наука использует все возможные средства исследования для
понимания явлений природы. Одним из подходов к изучению хими¬
ческих процессов служит феноменологический подход, основанный
на изучении наиболее общих закономерностей, но не рассматри¬
вающий детальную природу явлений. Этот метод составляет суть
термодинамики.
Термодинамика исследует энергетику различных физических
и химических процессов. При этом она рассматривает только мак¬
роскопические объекты, т е. изучает коллективные свойства боль¬
шого числа молекул В основе термодинамики нет никаких модель¬
ных представлений, и она не зависит от изменений наших взглядов
на природу атомов и молекул или от создания новых физических
или химических теорий.
Знание энергетических изменений принципиально важно для
понимания важнейших биологических процессов. Так, существенны
сведения о количестве калорий, получаемых в процессе питания и
расходуемых при выполнении той или иной работы С точки зрения
биоэнергетики все живые существа делятся на аутотрофы, кото¬
рые накапливают энергию в организмах за счет биохимических
процессов (например, растения), и гетеротрофы, вырабатывающие
ее в результате окисления питательных веществ - жиров и углево¬
дов. Животные являются гетеротрофами Термодинамический под¬
ход плодотворно применяется в современной биологии
Детальное изложение химической термодинамики не служит
целью настоящего учебника. В данной главе мы сформулируем
107
лишь основные понятия и законы, представляющие по существу тот
язык, с использованием которых будут в дальнейшем описываться
различные химические явления.
6.1. Первый закон термодинамики. Энтальпия
Задачей химии является изучение химических реакций, т е
процессов превращения веществ В простейшем случае целесооб¬
разно рассматривать лишь начальное и конечное состояния взаимо¬
действующих тел, не принимая во внимание путь, по которому про¬
текает процесс, и время превращения В этом и заключается термо¬
динамический подход к изучению химических процессов
Для удобства изучения необходимо изолировать объект иссле¬
дования от окружающей среды. Такая совокупность тел, выделенная
из пространства, называется системой Если внутри системы воз¬
можен массо- и теплообмен, то система называется термодинами¬
ческой При отсутствии массо- и теплообмена с внешней средой
говорят об изолированной системе. Если это условие не соблю¬
дается, то система открытая. Термодинамические зависимости для
открытых систем имеют сложный характер.
Всякий живой организм и его отдельные части представляют
собой открытые системы, осуществляющие непрерывно как обмен
веществ, так и энергетический обмен. Для наших целей, т.е. для
истолкования основных термодинамических законов в их приложе¬
нии к химическим явлениям, мы будем оперировать изолированны¬
ми термодинамическими системами.
Состояние системы определяется совокупностью ее свойств и
характеризуется термодинамическими параметрами, к числу
которых относятся температура, давление, объем. Состояние си¬
стемы изменяется с изменением хотя бы одного из ее параметров.
Аналитически состояние системы можно представить в виде урав¬
нения состояния, которое связывает между собой все параметры.
Конкретный вид уравнения состояния известен лишь для небольшо¬
го числа простых объектов. Например, уравнение Клайперона-
Менделеева является уравнением состояния идеального газа. Для
большинства же систем при термодинамическом описании пользу¬
ются функциями состояния, которые могут быть однозначно оп¬
ределены через параметры Р, V, Т (давление, объем, температура).
Одной из основных функций состояния является полная
энергия Е, которая представляет собой сумму трех составляющих,
кинетической энергии К, потенциальной энергии П, обуслов¬
108
ленной действием внешних силовых полей (гравитационного, элек¬
тромагнитного и пр.), и внутренней энергии системы U:
Е= К + П + U.
При термодинамическом описании предполагают, что система
находится в относительном покое (К = 0) и действие внешних по¬
лей пренебрежимо мало (П = 0). В таком случае полная энергия
системы определяется запасом ее внутренней энергии U. Послед¬
няя складывается из кинетической энергии молекул, поступательно¬
го и колебательного движения отдельных атомов в молекулах,
энергии межмолекулярных, межъядерных, внутриядерных взаимо¬
действий и т.п.
Учет всех этих составляющих невозможен, но для термодина¬
мического анализа в этом нет необходимости, т.к. достаточно знать
лишь изменение внутренней энергии при переходе из одного со¬
стояния в другое, а не ее значение в этих состояниях.
Если отсутствует теплообмен системы с внешней средой, то
общий запас внутренней энергии системы остается постоянным.
По существу это - закон сохранения энергии, который и является
первым законом термодинамики.
Сообщенная системе теплота Q расходуется на рост внутренней
энергии AU и на совершение работы против внешних сил А:
Q = AU + А
Это уравнение представляет собой математическое выражение
первого закона термодинамики.
Для выяснения физического смысла работы внешних сил А
рассмотрим систему, представляющую собой газ в цилиндре, отде¬
ленный от внешней среды перемещающимся без трения поршнем
(рис 6.1).
Если поршень закрепить неподвижно (V = const), то сообщен¬
ная газу теплота полностью идет на увеличение запаса внутренней
энергии:
Qv = AU.
Если теперь дать поршню возможность j -у-
свободно перемещаться (р = const), то газ, 1 ^
перемещаясь, совершит работу р
А = fh = p-S-h,
где f - сила, действующая на поршень, h -
высота перемещения поршня, р - давление, Рис g 1 сжатие Порш-
S - площадь поршня нем газа в цилиндре.
109
Так как Б Ь = ДУ - изменение объема, то А = р-ДУ = р(У2 - У|)
и поэтому теплота при постоянном давлении (Зр = Д11 + р(У2 - VI),
а поскольку Д11 = 112 - и|. то <Зр = и2 - Ьг| + р(У2 - У|) =
(и2 + рУ2)- (и| + рУ 1)
Введем новую функцию СОСТОЯНИЯ-
Н = и + рУ,
которая больше внутренней энергии на величину работы расшире¬
ния. Эта функция состояния называется энтальпией. Таким обра¬
зом, при постоянном давлении (т е в изобарных условиях) теплота,
подводимая к системе, идет на увеличение ее энтальпии.
(Эр = Н2 - Н, = ДН
Другими словами, энтальпия эквивалентна внутренней энергии
системы при постоянном давлении
6.2. Термохимия. Закон Гесса
Химическое взаимодействие, как правило, сопровождается теп¬
ловым эффектом При этом тепло может выделяться (экзотерми¬
ческие реакции) или поглощаться (эндотермические реакции). Те¬
пловой эффект реакции необходимо характеризовать поэтому не
только абсолютной величиной, но и знаком. В термодинамике при¬
нято считать тепло, поглощенное системой (эндотермические реак¬
ции), положительным, а тепло, отданное системой в окружающую
среду (экзотермические реакции), - отрицательным. Обычно хими¬
ческие реакции протекают при постоянном давлении, поэтому теп¬
ловой эффект реакции в изобарных условиях определяется разни¬
цей энтальпий конечного и исходных состояний.
Рассмотрим следующую реакцию при 298 К (25 °С) и давлении
I атм ■
С (те ) + Ог( газ) — СОг (газ) АН = -393 5 кДж/моль
Эта запись означает, что при образовании 1 моля газообразного
С02 из графита и газообразного кислорода выделяется 393.5 кДж теп¬
лоты Величину АН можно представить в виде:
где Н - мольные энтальпии отдельных компонентов системы при дан¬
ной температуре и давлении
Если бы были известны энтальпии всех элементов и их соеди¬
нений, то можно было бы рассчитывать тепловые эффекту любых
реакций, не проводя никаких опытов. Однако не существует спосо¬
ба измерять абсолютные значения энтальпий.
Поэтому для расчетов в термодинамике вводится шкала моль¬
ных энтальпий, основанная на предположении, что каждый эле¬
мент в той форме, в которой он устойчив при 298 К (25° С) и дав¬
лении I атм. (стандартные условия), характеризуется нулевой эн¬
тальпией. (Для менее стойких аллотропных форм, а некоторые эле¬
менты существуют в нескольких аллотропных модификациях, на¬
пример, озон и алмаз - менее устойчивые формы кислорода и угле¬
рода, соответственно, имеется своя величина Н°, отвечающая теп¬
ловому эффекту аллотропного перехода).
Мольные энтальпии в стандартных условиях обозначаются Н°,
и в приведенной реакции Н£ = = 0. В таком случае энтальпия
реакции совпадает с энтальпией образования углекислого газа из
элементов: Н° = ДН° = -393.5 кДж/моль.
со2
Таблица 6 /
Стандартные энтальпии образования некоторых
неорганических и органических молекул
Соединение
(состояние) кДп,,„ил.
Соединение
(состояние)
АН®,
кДж/моль
НСІ (газ) 92 2 Метан (газ)
№01 (тв.) 4110 Этилен (газ)
02 (газ) 0 Ацетилен (газ)
03 (газ) -142 0 Бензол (жидк)
Н20 (газ) -241 8 Этанол (жидк)
Н20 (жидк ) -285 8 Глицерин (жидк.)
Б02 (газ) 296 9 Ацетальдегид (газ)
НгБ (газ) 20 4 Ацетон (жидк )
N1-13 (газ) -46 2 Уксусная кислота (жидк )
МНэ (водн ) -80 8 Масляная кислота (жидк )
HCN (газ) 132 0 Фумаровая кислота (тв )
Н3РО4 (тв ) -1281 1 Молочная кислота (водн )
С (графит) 0 Пировиноградная кислота (водн )
С (алмаз) +18 Глицин (тв )
СО (газ) -110 5 Мочевина(тв)
С02 (газ) -393 5 Мочевина (водн )
С02 (водн ) -699 6 Глюкоза (тв )
Н2 (газ) 0 Сахароза (тв )
52.3
226.7
49 0
-277.8
-670.7
-166 0
-246 8
-487.3
-524 3
-811 1
-694 0
-607 5
-524 7
-333 2
-319 2
-1274 4
-2222 0
-74.8
111
Стандартной энтальпией образования вещества называется
изменение энтальпии при получении одного моля этого вещества
из простых веществ, взятых в агрегатном состоянии, обычном для
них в стандартных условиях. Стандартные энтальпии ряда важных
в биологическом отношении веществ представлены в таблице 6 1
Необходимо обратить внимание, что все приведенные в таблице
природные органические молекулы имеют отрицательные значения
энтальпий образования.
Глубокое обобщение термохимических закономерностей дает
основной закон термохимии, сформулированный в 1836 г. рус¬
ским академиком Г. И. Гессом: тепловой эффект реакции не зависит
от числа промежуточных стадий, а определяется лишь начальным и
конечным состояниями системы.
Закон Гесса можно иллюстрировать схемой:
Образование соединения АВ представлено двумя путями: непо¬
средственным синтезом из компонентов (энтальпия образования
ЛН) или через стадию промежуточного соединения АС (энтальпия
образования ДН]), которое, реагируя с В (ДН2), дает тот же конеч¬
ный продукт. В соответствии с законом Гесса ДН = ДН| + ДНг-
Закон Гесса - прямое следствие первого закона термодинамики.
Его широкая применимость и значение определяется тем, что эн¬
тальпия является функцией состояния и, таким образом, ее измене¬
ние при переходе из одного состояния в другое всегда одинаково
независимо от числа стадий и пути перехода.
Из закона Гесса вытекает важное следствие: стандартная эн¬
тальпия реакции равна сумме стандартных энтальпий образования
продуктов реакции за вычетом суммы стандартных энтальпий обра¬
зования исходных веществ
Пример■ Вычислим значение АН0 гидролиза мочевины до С02 и N14у
Хотя в отсутствие фермента (уреазы) реакция практически не
идет, значение стандартной энтальпии этой реакции можно вычис¬
лить из термодинамических данных, если известны соответствующие
энтальпии образования всех веществ, участвующих в ней. Гидролиз
мочевины идет по уравнению
1ЧН2СОЫН2(еодн.,) +Н20(жидк) -» СОг(водн ) + 2ЫНэ(ео<?н)
А + В = АВ (ДН)
А
АВ
А + С = АС (ДН,)
АС + В = АВ + С (ДН2)
ДН°реакции — £ДН°Пр0Д — £ДН°ИСХ
112
Используя данные таблицы, по последнему уравнению находим-
^реакции = -699.6+2(-80 8)-1-319.2+(-285.8) 1=-256.2 кДж/моль
Приведенный пример показывает, каким образом можно узнать
тепловой эффект реакции, не прибегая к экспериментальным тер¬
мохимическим измерениям. Этот прием чрезвычайно полезен при
исследовании большинства химических реакций, происходящих в
живых клетках, которые часто недоступны для прямых измерений.
Если известны энтальпии образования для всех соединений, уча¬
ствующих в реакции, то можно определить теплоту реакции, мыс¬
ленно разложив все исходные вещества до элементов и соединив из
элементов продукты реакции.
Следует обратить внимание, что при термодинамических вычис¬
лениях необходимо учитывать агрегатное состояние реагирующих
веществ, т.к. только в этом случае расчеты могут иметь какую-либо
ценность.
6.3. Второй закон термодинамики. Энтропия.
Свободная энергия Гиббса
Первый закон термодинамики позволяет рассчитывать энергети¬
ческий баланс химического процесса Однако он ничего не говорит
о том, будет ли происходить то или иное превращение и в каком
направлении должна самопроизвольно (без действия внешних сил)
протекать химическая реакция
Попыткой воспользоваться первым законом термодинамики для
оценки направленности химических процессов явился принцип
Бертло-Томсона, согласно которому реакция самопроизвольно про¬
текает в сторону выделения теплоты, т е уменьшения энтальпии
(ДН < 0, экзотермическая реакция). Во многих случаях этот прин¬
цип действительно позволяет предвидеть направление химического
процесса, однако ограниченность его была показана большим чис¬
лом примеров самопроизвольно происходящих эндотермических яв¬
лений. Например, самопроизволен процесс растворения поваренной
соли в воде, происходящий с поглощением тепла.
Для прогнозирования возможности и направления процессов
необходимо ввести еще одну функцию, которая должна отвечать
ряду требований Во-первых, она должна быть функцией состояния,
т е не зависеть от пути реализации данного состояния. Во-вторых,
для всех самопроизвольных процессов изменение этой функции
должно иметь один и тот же знак
ИЗ
Физический смысл этой новой функции можно рассмотреть на
примере плавления индивидуального кристаллического вещества
Плавление происходит при постоянной температуре и сопровож¬
дается поглощением теплоты плавления ДНПЛ (энтальпия плавле¬
ния) Поглощение тепла должно, как казалось, способствовать уве¬
личению внутренней энергии системы, что выразилось бы в увели¬
чении температуры Однако этого не наблюдается. Следовательно,
в процессе плавления действует другой фактор, способствующий
сохранению постоянной температуры Аналитически это можно вы¬
разить в виде
ДНПЛ = Тпл АЬпл
где Д5ПЛ - величина, характеризующая процесс в системе, на кото¬
рый расходуется поглощаемое тепло при постоянной температуре.
Поскольку энтальпия плавления является функцией состояния, то и
величина Д5ПЛ должна быть функцией состояния При плавлении
кристалла происходит разрушение упорядоченной кристаллической
структуры и образование расплава с хаотическим расположением
частиц, т.е. увеличение беспорядка в системе.
Частным примером этой тенденции является переход тепловой
энергии от более нагретого тела к менее нагретому, но никак не
наоборот.
Другой пример - диффузия. Если сосуд разделить перегородкой
на две половины и поместить газ только в одну половину, то после
удаления перегородки газ распространится по всему сосуду, и в
обеих его половинах в среднем будет одинаковое количество моле¬
кул. Обратный процесс перехода молекул газа в одну половину со¬
суда не происходит.
В обоих приведенных примерах система вначале была упорядо¬
чена: температура двух соприкасающихся тел различалась, газ был
в одной половине сосуда. Конечное равновесное состояние - неупо¬
рядоченность: температура и числа молекул усреднялись.
Как показывает опыт, все самопроизвольные процессы в изоли¬
рованных системах протекают в сторону увеличения беспорядка.
Таким образом, критерием направленности процесса может служить
степень неупорядоченности системы. Мерой этой неупорядочен¬
ности является функция Б, называемая энтропией. Энтропия -
мера неупорядоченности. В наиболее неупорядоченном состоянии
энтропия будет максимальной.
Больцманом было высказано предположение, что энтропия Б
может быть выражена через термодинамическую вероятность
следующим образом:
Б = Ып"^ где к - константа Больцмана.
114
Поясним смысл величины следующим примером. Допустим,
что в сосуде, мысленно разделенном на две половины, находится
всего четыре одинаковых и невзаимодействующих молекулы. В
табл. 6.2 показаны все возможные способы их распределения по
двум сосудам. Распределения I и V наименее вероятны, т.к. каждое
из них реализуется лишь одним способом. Распределения II и IV в
4 раза более вероятны, т.к. каждое из них реализуется четырьмя
способами. Наконец, распределение III наиболее вероятно - оно
достигается шестью способами.
Таблица 6.2
Варианты распределения четырех молекул в двух сосудах
Макро-
состоянне
Способ распределения
микросостояний
Число микро¬
состояний,
Левая половина
Правая половина
I
12 3 4
• • • •
1
II
1 2 3
1
4
1 2 4
2
1 3 4
4
2 3 4
3
III
1 2
1 4
6
1 3
1 3
1 4
1 2
1 3
3 4
2 4
2 4
3 4
2 3
IV
1
1 2 4
4
2
1 2 3
3
2 3 4
4
1 3 4
V
12 3 4
....
1
Каждый конкретный способ распределения соответствует опре¬
деленному микросостоянию системы Поскольку молекулы не раз¬
личимы, то каждое макросостояние достигается определенным чис¬
лом микросостояний Число микросостояний, которым достигается
данное макросостояние, называется термодинамической вероят¬
ностью В приведенном примере термодинамическая вероятность
максимальна для распределения III (Ш = 6) Заметим, что в отличие
от математической вероятности, которая не может быть больше
единицы, термодинамическая вероятность выражается большими
числами.
115
При малых числах молекул максимум энтропии не резок - рас¬
пределение III лишь на 50% более вероятно, чем II и IV. Наоборот,
если число молекул велико, то вероятность равномерного распреде¬
ления очень велика, в то время как вероятность неравномерного
распределения (например, для тысячи молекул по двум сосудам в
отношении 333 667) окажется очень малой
Выяснив смысл новой функции состояния - энтропии, сформу¬
лируем второй закон термодинамики, согласно которому вся¬
кий самопроизвольный процесс в изолированной системе идет с
возрастанием энтропии Таким образом, если в результате процесса
ДБ > 0, процесс термодинамически возможен; если же ДБ < 0, то
его самопроизвольное протекание исключается.
В обратимом процессе изменения энтропии не происходит
(ДБ = 0). В рассмотренном выше примере плавления кристалличе¬
ского вещества происходит повышение энтропии при разрушении
кристаллической решетки, но это повышение компенсируется
уменьшением энтропии при охлаждении.
Возникает вопрос: можно ли обратить самопроизвольный про¬
цесс? Второй закон термодинамики отвечает, что это возможно, но
при определенных условиях. Обращение самопроизвольного процес¬
са может быть достигнуто только путем создания эквивалентной
или еще большей микроскопической неупорядоченности где-то
в другом месте.
Наглядным примером может служить биопроцесс фотосинтеза.
Двуокись углерода, вода и другие питательные вещества погло¬
щаются растениями, и за их счет синтезируются сложные высоко¬
организованные молекулы углеводов. Этот процесс сопровождается
понижением энтропии. Чтобы происходил процесс фотосинтеза,
растениям необходима солнечная энергия. Поэтому уменьшение эн¬
тропии при переходе от СОг и НгО к углеводам компенсируется
возрастанием энтропии на Солнце.
Многие другие фундаментальные биохимические процессы так¬
же осуществляются с уменьшением энтропии. Таков, например,
процесс образования биополимеров - белков и нуклеиновых кислот,
активный транспорт ионов через клеточные мембраны и др. Эти
примеры на первый взгляд противоречат термодинамическим зако¬
номерностям.
Живой организм есть открытая система, поэтому противоречие
между физической и химической эволюцией является кажущимся.
В открытой системе энтропия может возрастать, оставаться посто¬
янной или даже уменьшаться в зависимости от количества энтро¬
пии, производимого внутри системы, ее притока извне или оттока
во внешнюю среду.
116
Если считать Вселенную изолированной системой, то все собы¬
тия в ней должны приводить к возрастанию энтропии. В конечном
итоге должно быть достигнуто состояние с максимальной энтропи¬
ей, после чего уже невозможны никакие события. Это состояние
известно как “тепловая смерть Вселенной”. Однако оно никогда не
будет достигнуто, ибо Вселенная не статична и непрерывно эволю¬
ционирует.
В то время, как абсолютные значения энтальпии определить
невозможно, вычисление абсолютных значений энтропии - вполне
выполнимая задача.
При абсолютном нуле движение всех атомов прекращается, все
идеальные кристаллические вещества должны иметь нулевую эн¬
тропию. Одним из принципов термодинамики является недостижи¬
мость абсолютного нуля. Это становится понятным из того, что не
существует холодильника, имеющего еще более низкую температу¬
ру, к которому могли бы перейти последние очень малые доли теп¬
ловой энергии.
Этот принцип, который называют иногда третьим законом тер¬
модинамики, позволяет вычислять абсолютные значения энтро¬
пии Б0 (таблица 6 3).
Таблица 6 3
Абсолютные энтропии некоторых веществ при 298 К
Соединение
(состояние)
Б0,
Дж/моль К
Соединение
Б0,
Дж/мольК
НС1 (газ)
186 7
Метан (газ)
186 2
N301 (тв )
72 4
Этилен (газ)
2194
02 (газ)
205 0
Ацетилен (газ)
200 8
Оэ (газ)
238 8
Бензол (жидк )
173 2
Н20 (газ)
188 7
Этанол (жидк )
161 0
Н20 (жидк )
69 9
Глицерин (жидк )
204 5
Б02 (газ)
248 5
Ацетальдегид (газ)
264 2
НгЭ (газ)
205 6
Ацетон (жидк )
198 7
N143 (газ)
192 5
Уксусная кислота (жидк )
159 8
(газ)
201 7
Масляная кислота (жидк )
255 0
Н3Р04 (тв )
1105
Фумаровая кислота (тв )
166 1
С (графит)
5 7
Молочная кислота (водн )
221 7
С (алмаз)
24
Глицин (тв )
109 2
СО (газ)
197 9
Мочевина(тв )
104 6
С02 (газ)
2136
Мочевина (водн )
173 8
Н2 (газ)
130 6
Сахароза (тв )
360 3
117
Абсолютные значения энтропии используются для вычисления
стандартных энтропий образования веществ Энтропия образо¬
вания вещества (Д5°обра,) равна разности между суммами абсолют¬
ных энтропий продуктов реакции (5°пр) и абсолютных энтропий
реагирующих веществ (5°реаг)
Пример Пользуясь данными таблицы 6 3, определить энтропию
образования этанола
Напишем уравнение образования этанола из элементов
2С + ЗН2 + 1/202 -» С2Н5ОН
Подстановка значений Э0 из таблицы дает
^°образ = >64 0-(2 5 7+3 130 8+1/2 205 0) = -344 7 Дж/моль К.
В таблице 6.4 приведены значения Д5°образ для некоторых ве¬
ществ, представляющих биологический интерес.
Таблица 6 4
Стандартные энтропии образования некоторых веществ
при 298 К
Вещество
Д5°о6о«.
Дж/моль-К
Вещество
Д5°о6о»».
Дж/мольК
Аланин (тв )
-643 1
Масляная
Аспарагиновая
кислота (жидк.)
-523 8
кислота(тв )
-812.1
Уксусная
Ацетон (жидк )
-3109
кислота (жидк.)
-318.4
Глицерин (жидк )
-642 6
Янтарная кислота (тв.)
-648 9
Глюкоза (тв )
-1220 5
Этанол (жидк.)
-344 6
Глутаминовая
Фумаровая
кислота (тв )
-930 5
кислота (тв )
-528.0
Устойчивость любой системы определяется соотношением эн-
тальпийного и энтропийного факторов Энтальпийный фактор ха¬
рактеризует стремление системы к упорядочению, т.к. этот процесс
сопровождается уменьшением внутренней энергии. Второй фактор
отражает тенденцию к разупорядочению, поскольку такое состояние
наиболее вероятно.
Было целесообразно ввести такую функцию состояния, которая
учитывает совместное влияние обоих факторов Такая функция
представляет собой разность
Дй = ДН - ТДБ.
118
Эта функция состояния называется свободной энергией
Гиббса и является мерой устойчивости системы при постоянном
давлении. Стандартные свободные энергии Гиббса образования раз¬
личных веществ представлены в таблице 6.5. По данным этой таб¬
лицы можно рассчитать изменение свободной энергии при различ¬
ных реакциях.
Таблица 6.5
Стандартные энергии Гиббса образования некоторых веществ при
298 К
Соединение
(состояние)
ДО0,
кДж/моль
Соединение
(состояние)
ДС,
кДж/моль
НС1 (газ)
-95.3
Метан (газ)
-50 8
МаС1 (тв )
-384.1
Этилен (газ)
68 1
02 (газ)
0
Ацетилен (газ)
209.2
Оз (газ)
162.8
Бензол (жидк )
-124.4
НгО (газ)
-226 6
Этанол (жидк )
-174.1
НгО (жидк.)
-237 2
Глицерин (жидк )
-477 1
ЭОг (газ)
-300 2
Ацетальдегид (газ)
-1329
НгЭ (газ)
-33 5
Ацетон (жидк )
-155 4
МНз (газ)
-165
Уксусная кислота (жидк )
-389 4
НСМ (газ)
121 6
Масляная кислота (жидк )
-376 7
Н3Р04 (тв )
-11192
Фумаровая кислота (тв )
-653 6
С (графит)
0
Глицин (тв )
-366 8
С (алмаз)
2 8
Мочевина(тв )
-197 1
СО (газ)
-137 1
Глюкоза (тв )
-919 5
СОг (газ)
-393 4 1
Сахароза (тв )
-1544 7
По знаку функции свободной энергии судят о возможности са¬
мопроизвольного процесса: если Дв < 0, т е. процесс сопровождает¬
ся уменьшением свободной энергии, то процесс термодинамически
возможен Если Дв > 0, то самопроизвольный процесс невозможен
То есть все самопроизвольные процессы протекают в сторону
уменьшения свободной энергии Для системы, находящейся в рав¬
новесии, Дв = 0.
Из уравнения видно, что знак Дв зависит от относительных ве¬
личин ДН и ТДБ Если ДН отрицательна (экзотермическая реакция),
а ДБ положительна, то при любой температуре процесс будет идти
самопроизвольно Возможные соотношения между энтальпийной и
энтропийной составляющими и характеристика процесса приведены
в таблице 6.6
Изучение трансформации энергии в живых системах является
предметом биоэнергетики По мере выяснения молекулярных меха¬
119
низмов биохимических процессов ученые стараются применить тер¬
модинамические представления в исследованиях живых систем.
Однако классическая термодинамика имеет ряд черт, ограничи¬
вающих возможности ее применения для анализа процессов жизни.
Таблица 6.6
Факторы, определяющие ход процесса
дн
ДБ
Возможность самопроизвольного процесса
-
+
Процесс идет самопроизвольно при любой температуре
-
-
Процесс может идти самопроизвольно при низких температурах
+
+
Процесс может идти самопроизвольно при высоких температурах
+
Процесс не идет самопроизвольно ни при какой температуре
Во-первых, классическая термодинамика изучает изолированные
системы, а в живой природе таких систем нет. Во-вторых, она рас¬
сматривает равновесные состояния. Для живых организмов равно¬
весие - это смерть, и их состояние определяется как неравновесное
стационарное.
Стационарное состояние внешне похоже на равновесное тем,
что концентрация частиц в нем постоянна. Однако это постоянство
обеспечивается беспрерывным оттоком вещества из системы и при¬
током в нее. Поэтому для изучения живых систем необходима тер¬
модинамика необратимых процессов, которая в настоящее время
интенсивно развивается. По указанным причинам при термодина¬
мическом подходе к биохимическим системам следует соблюдать
осторожность в окончательных выводах. В то же время этот подход
чрезвычайно плодотворен в изучении химических процессов в не¬
живой природе.
Глава 7. ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Ведущей идеей... было рас¬
крытие роли энергии и энтро¬
пии в теории термодинамиче¬
ского равновесия.
Дж. Гиббс
7.1. Константа равновесия
Равновесие существует, когда все макроскопические свойства
системы (давление, объем, температура, концентрация) постоянны.
Для химических реакций равновесию отвечает равенство скоростей
прямого и обратного процессов.
Равновесие может существовать только для обратимых процес¬
сов. Лишь некоторые химические превращения носят практически
необратимый характер, например, взрывные явления, реакции с
выделением газа, выпадением осадка или с образованием недиссо¬
циирующих в растворе соединений, денатурация белков и т.д.
Большинство же превращений обратимо. Такова, например, реак¬
ция связывания кислорода гемоглобином, явление изменения кон¬
центраций катионов К* и №+ в нервных клетках при нервном им¬
пульсе и др.
Как видно из предыдущей главы, изменение свободной энергии
Гиббса Дв при постоянных температуре и давлении является про¬
стым критерием равновесия: в положении равновесия Дв = 0.
Поэтому для того, чтобы охарактеризовать любое химическое рав¬
новесие, необходимо осуществить детальный анализ свободной
энергии Гиббса вблизи состояния равновесия. Однако в химическом
процессе, как правило, участвуют по меньшей мере два различных
вещества (компонента). Следовательно, необходимо выяснить, как
изменяется свободная энергия системы в зависимости от соответ¬
ствующих свободных энергий каждого компонента.
Если обратиться к простому случаю, когда реакция происходит
в газовой фазе, причем реагируют идеальные газы, то из термоди¬
намических закономерностей выведено, что свободная энергия
1 моля идеального газа при его парциальном давлении р равна
(л = + ЯТ 1п р,
где в0 - свободная энергия идеального газа в стандартных условиях
(стандартная свободная энергия, р = 1 атм, Т = 298 К)
121
Рассмотрим такую химическую реакцию, в которой взаимодей¬
ствие между двумя идеальными газами А и В приводит к образова¬
нию двух других идеальных газов С и О согласно уравнению-
аА + вВ ^ сС + сЮ.
Так как свободная энергия является функцией состояния, ее
изменение в этой реакции определяется разницей свободных энер¬
гий продуктов и исходных веществ-
ДО = ейс + сЮв - айд - ЬОв-
Для компонентов
А: авд = аО°А + аИТ 1п рА,
В: Ьвв = ЬО°в + ЬИТ 1п рв,
С: евс = сО°с + сИТ 1п рс,
О: dG^) = dC0D + dRT 1п рр,
где рд, рв, Рс и Ро ~ парциальные (или частичные) давления компо¬
нентов А, В, С и О, соответственно.
Следовательно:
ДО = сО°с + dG0D - аО°д - ЬО°в +
+ сИТ 1п рс + dRT 1п ро - аИТ 1п рА - ЬИТ 1п рв,
но разница стандартных свободных энергий компонентов постоянна
и равна Дв0. Отсюда:
Дй =ЛО° + КТ 1п |РС|С|РВ|'
Ера1Мрв)ь
Так как при равновесии Дй = 0. то = -ЯТ |п
[РА1а[рВ]Ь
Смысл этого равенства состоит в том, что из постоянства левой
части (в том числе и независимости Лй0 от давления) следует и
постоянство правой части, которая также не зависит от давле¬
ния. Поэтому выражение под знаком логарифма есть величина по¬
стоянная и называется - константой химического равнове¬
сия Кравн :
к»“= !Рс1,?,Рр!ь °тсю1а ла°•-кт |п кр..»
Фа1 Фв1
122
Полученное выражение, как установлено, применимо практиче¬
ски к любым химическим реакциям, протекающим не только в газо¬
вой фазе для идеальных газов, но и в растворах.
В этом случае для вывода предыдущего уравнения при суммиро¬
вании свободных энергий компонентов пользуются выражением для
свободной энергии каждого компонента в растворе, аналогичным
выражению для свободной энергии идеального газа:
Оа = С°А + ЯТ 1п [А],
где (А] - равновесная концентрация компонента А в моль/л.
В общем виде эта зависимость известна как закон действую¬
щих масс (Гульдберг и Вааге, 1867 г.) для систем в положении
равновесия: частное от деления произведения равновесных кон¬
центраций продуктов реакции на произведение равновесных кон¬
центраций исходных веществ, взятых в степенях, равных их сте-
хиометрическим коэффициентам, является величиной постоянной,
именуемой константой равновесия Кравн .
Для реакции
аА + вВ + ... ^ сЮ + еЕ... + ...
в состоянии равновесия
|ЕеГ [р]“ I I
[А]а-|В]Ь | I '
где (А], [В], . - равновесные концентрации компонентов в моль/л;
а, Ь, (1, е - стехиометрические коэффициенты
Следует помнить, что константа равновесия при постоянной
температуре зависит только от величины стандартной свободной
энергии процесса, которая является термодинамической функцией
состояния. Следовательно и константа равновесия - термодинами¬
ческая функция состояния, зависящая лишь от природы реагирую¬
щих веществ, а следовательно - от величин их энтропий и энталь¬
пий Отсюда следует, что константу равновесия можно рассчитать,
пользуясь табличными термодинамическими данными
7.2. Смещение химического равновесия
Величина Кравн существенна для характеристики реакции Ее
значение дает возможность судить о направлении и глубине про¬
цесса в данных условиях, а также подбирать такие условия, когда
выход целевого продукта будет максимальным
123
Например, для обратимой реакции связывания кислорода гемог¬
лобином (НЬ) с образованием оксигемоглобина (НЬОг) величина
КраВн равна 1300 при 37 °С. Это значит, что при давлении кислоро¬
да, равном 1 атм , равновесная концентрация оксигемоглобина в
1300 раз превышает концентрацию гемоглобина. Другими словами,
равновесие реакции НЬ + Ог ^ НЬОг сильно смещено вправо.
Кр,.„ = 1нь°г) ° 1300
[НЬ| [02]
Постоянство Кравн и ее независимость от концентраций компо¬
нентов реакции и от давления позволяет смещать химическое рав¬
новесие, увеличивая или уменьшая концентрации исходных веществ
или продуктов.
Допустим, в реакции гемоглобина с кислородом мы увеличим
содержание кислорода. Это вызовет усиленное связывание его ге¬
моглобином, в результате чего выход оксигемоглобина возрастет.
Тем самым равновесие будет смещаться вправо. Процесс образова¬
ния оксигемоглобина будет идти до тех пор, пока не будет достиг¬
нуто равновесие с той же Кравн = 1300, т.е. с тем же соотношением
реагентов, хотя их концентрации уже будут отличны от предше¬
ствующих.
Рассмотрим другой вариант. Известно, что гемоглобин прочно
связывается с угарным газом (СО) с образованием карбоксигемо-
глобина (НЬ'СО). Поэтому введение СО в систему должно приводить
к уменьшению концентрации гемоглобина, однако его убыль вызо¬
вет разрушение оксигемоглобина и равновесие реакции гемоглобина
с кислородом сместится влево. Таким образом, в равновесной си¬
стеме изменение концентрации любого компонента влечет согласо¬
ванное изменение концентраций всех остальных.
Изменение давления влияет на положение равновесия в реак¬
циях, идущих с изменением объема. В самом деле, в процессе син¬
теза аммиака, происходящем с уменьшением объема:
N2 + ЗН2 ^ 2МНЭ,
увеличение давления увеличит концентрации исходных веществ в
большей степени, чем продуктов. По существу для газовых систем
изменение давления оказывается эквивалентным изменению кон¬
центраций. В итоге при синтезе аммиака увеличение давления
смещает равновесие в сторону увеличения его концентрации и на¬
оборот, чем ниже давление, тем в большей степени аммиак будет
распадаться на водород и азот.
124
Влияние температуры на константу равновесия видно из сле¬
дующих преобразований:
ДО0
1пКравн=—.
но Дв0 = ДН° - ТДБ0. Следовательно, 1п Кра
ДН° - ТДБ0
ЯТ
т , „ ДН° ДБ0 т , V дн° д5°
При Т,: 1п К] = а при Т2: 1п К2 = ——+ -г~-
К1| К К12 К
Поэтому 1пК,/К2 = -щй^гу.
Отсюда при ДН° < 0 (экзотермический процесс) и при Т] > Т2,
1п К1 /К2 < 0 и К] <К2, т.е. для экзотермического процесса при по¬
нижении температуры будет происходить увеличение Кравн или
увеличение концентрации конечных веществ в равновесной смеси
(равновесие смещается вправо).
Наоборот, при ДН° > 0 (эндотермический процесс) и при Т] >
Т2, 1п К,/К2 > 0 и К, > К2, т.е. понижение температуры для эндо¬
термического процесса приводит к уменьшению Кравн или умень¬
шению концентрации конечных веществ в равновесной смеси
(равновесие смещается влево).
Суммарно влияние условий (концентрации, давления, темпера¬
туры) на химическое равновесие можно выразить принципом Ле
Шателье: если на равновесную систему оказать какое-нибудь
внешнее воздействие, то в системе происходит процесс, ослабляю¬
щий это воздействие.
Применение принципа Ле Шателье позволяет управлять боль¬
шинством химических реакций, в том числе и такими, которые
осуществляются в промышленных масштабах. Примером такого
процесса является синтез аммиака, который, согласно приведенно¬
му выше химическому уравнению, следует производить при макси¬
мальном давлении, а поскольку он экзотермичен - при минимально
допустимой температуре.
Очевидно, что равновесие будет смещаться в одном направле¬
нии, если какой-либо из продуктов будет покидать сферу реакции.
Существует простое правило (именуемое правилом Бертло), что
химическое взаимодействие в растворах становится почти необра¬
тимым, если один из его продуктов является газообразным, выпа¬
дает в осадок или представляет собой мало диссоциированное со¬
единение
125
Это правило иллюстрируется простыми примерами.
МаНСОэ + НС1 -» №01 + Н20 + С02Т
ВаС12 + N82804 -> 2МаС1 + ВаБОд!
МаОН + НС1 -> №01 + Н20
Первая из реакций имеет место в желудке, препятствуя избы¬
точной кислотности его содержимого Второе превращение исполь¬
зуется в медицине для приготовления рентгеноконтрастного препа¬
рата, каким является сульфат бария. Третий пример не нуждается в
пояснениях. Таким образом, очевидно, что смещение химического
равновесия обычно для медико-биологической практики.
Ниже будут рассмотрены важнейшие значимые для биосистем
случаи химических равновесий в водных растворах с применением к
ним принципа Ле Шателье.
В целом следует помнить, что химические реакции, происходя¬
щие в биосредах, необходимо рассматривать как с термодинамиче¬
ских, так и с кинетических позиций Если термодинамика позволяет
определить принципиальную возможность прохождения той или
иной реакции и с помощью строгих расчетов подобрать оптималь¬
ные условия для ее осуществления (принцип Ле Шателье), то кине¬
тика дает возможность определить ее механизм и оценить скорость
протекания.
С этих позиций реакции в живой клетке можно разделить на
две группы реакции, регулируемые термодинамически, и реакции,
находящиеся под кинетическим контролем.
В качестве примера реакций первого типа можно привести
синтез дипептида из двух молекул аминокислоты. Дй0 этого
превращения составляет 17.2 кДж/моль, а рассчитанная константа
равновесия равна =10'3. Очевидно, сам по себе этот процесс в за¬
метной степени не пойдет. Однако он осуществим благодаря со¬
пряжению с гидролизом АТФ с участием определенного фермента.
Эта реакция - термодинамически контролируемое превращение: по¬
скольку сама по себе не является самопроизвольной, и для ее
осуществления требуется внешний источник свободной энергии.
Кинетически контролируемые реакции характеризуются отрица¬
тельным значением ДО0, то есть термодинамически выгодны.
К числу таких реакций относится, к примеру, экзотермическое
фосфорилирование углеводов. Однако в отсутствие ферментов ско¬
рость фосфорилирования ничтожно мала из-за его высокого актива¬
ционного барьера.
126
Часть II
РАВНОВЕСИЯ В БИОСРЕДАХ
Изменение любого факто¬
ра, могущего влиять на состо¬
яние химического равновесия
системы веществ, вызывает в
ней реакцию, стремящуюся про¬
тиводействовать производимо¬
му изменению.
А. Ле Шателье
Глава 8. РАСТВОРЫ
Раствор есть однородная
(гомогенная) жидкая система
непрочных диссоциирующих со¬
единений растворителя с рас¬
творенным веществом.
Д. И. Менделеев
8.1. Вода и ее физико-химические свойства
Растворы играли и играют важнейшую роль в возникновении и
развитии живой природы. В растворах формировались простейшие
белковые структуры - коацерваты, а в дальнейшем - водоросли, что
обусловило возникновение фотосинтеза
Общий объем воды Земли составляет 1.5-109 км3, из них
2.5-103 км3 содержится в составе живых организмов. Принято счи¬
тать, что основная масса воды биосферы (области Земли в интерва¬
ле от 25 км выше уровня моря до глубины океана 11 км) имеет
биогенное происхождение, то есть проходила через метаболические
превращения живых организмов.
Живые организмы получают пищу из водной среды, в воде осу¬
ществляется биосинтез, с водой из организмов выносятся отходы
В самом процессе биосинтеза также образуется вода - так назы¬
ваемая метаболическая вода В среднем человек потребляет с едой
и питьем около двух литров воды в сутки, из которых до
300 мл приходится на долю метаболической Вода выполняет в
организме и механическую роль смазки суставов, мышц, связок
127
Человек почти на 60% состоит из воды, кровь содержит ее 83%,
внутренние органы - сердце, почки - 80%, в костях 30% воды
Количество воды у людей индивидуально и зависит от выражен¬
ности жировой ткани, где ее содержание невелико и составляет
около 30%. Содержание воды в обезжиренном теле с учетом 10%
липидов, сохраняемых в любых случаях, равняется 72-73% Заме¬
тим, что количество воды в организме существенно зависит от
возраста. Наибольшее содержание воды наблюдается у новорож¬
денных, что объясняется большим внеклеточным пространством;
к первому году жизни количество воды заметно снижается, особен¬
но за счет внеклеточного пространства; далее к периоду полового
созревания ее содержание составляет у мужчин - 56%, а у жен¬
щин - 46%. Примерно половина всей воды организма находится в
мышечной ткани.
Внеклеточная вода - это жидкость кровеносной и лимфати¬
ческой сосудистых систем, межтканевая и межклеточная жидкости
Это посредник между внешним миром и клетками. Внеклеточная
жидкость омывает клетки, за счет чего организм точно регулирует
концентрацию внутриклеточного раствора.
Внутрисосудистая вода соответствует объему плазмы. Из объе¬
мов плазмы и эритроцитов складывается объем крови, который со¬
ставляет 6-7% массы тела в зависимости от пола и конституции.
Внутриклеточная вода занимает пространство, предназначенное
для метаболических процессов.
Межтканевая (интерстициальная) вода служит для приближе¬
ния тканей к "выводящей” крови. Содержание белков в ней очень
незначительно, а общий состав электролитов соответствует составу
плазмы. Она действует в качестве объемного буфера при кровопо-
тере и передозировке жидкости. Например, если общий объем вве¬
денной в организм жидкости в 5 раз превысит объем крови, то на
практике объем последней увеличивается лишь на 30%.
Межклеточная жидкость - это не гомогенная фаза. В таких
тканях, как кожа, кости и хрящи, обмен водой и растворенными в
ней веществами происходит наиболее медленно. Принято объеди¬
нять объем плазмы и небольшую часть жидкости плотных соедини¬
тельных тканей во внеклеточный физиологически активный объем
(обычно его доля в общем объеме жидких сред составляет 20%).
Трансцелюлярная вода охватывает ту часть жидкости, которая рас¬
пределена в полостях организма (желчные пути, пищеварительный
тракт). В норме она составляет 2.5% массы тела. Эти жидкости в
основном являются секретами желез, находятся в различных по¬
лостях и имеют четкие различия в химическом составе. За сутки
организмом выделяется около 2 л жидкости.
128
Важная роль воды в биологических процессах определяется ее
уникальными физико-химическими свойствами.
Во-первых, вода имеет необычайно высокую теплоемкость. Ог¬
ромные массы океанской воды поглощают или излучают колоссаль¬
ное количество тепла без заметных температурных изменений, что
играет определяющую роль в формировании погодных и климатиче¬
ских условий. Этот же фактор чрезвычайно важен в температурном
контроле растений и животных. Иными словами, вода выполняет
роль термостата для биомолекул, функционирование которых чрез¬
вычайно чувствительно к температурным изменениям.
Во-вторых, вода обладает большой энтальпией испарения.
Охлаждение за счет испарения с поверхности листьев растений и с
кожных покровов животных предохраняет их от перегрева. Кроме
того, высокая теплота испарения препятствует потере воды орга¬
низмом при повышении температуры
В-третьих, большая по сравнению с другими растворителями эн¬
тальпия плавления воды также выполняет термостатирующую
функцию и служит защитой от замерзания.
В-четвертых, вода по сравнению с другими гидридами (НЕ, ЫН3
и др.) имеет аномально высокие температуры плавления и кипения,
что и позволяет ей находиться в обычных земных условиях в жид¬
ком состоянии. Причина этого состоит в своеобразии структуры
воды за счет образования ею водородных связей. В воде число про¬
тонов, участвующих в водородных связях, равно числу неподелен-
ных электронных пар атомов кислорода, которые также необходимы
для водородной связи. В результате создается возможность по¬
строения трехмерной пространственной структуры, в отличие от
других гидридов, которые посредством водородных связей могут
давать лишь линейные или кольцевые ассоциаты.
На рис 8.1 представлена структура льда,’ в которой каждый
атом кислорода находится в центре тетраэдра, окруженный 4 ато¬
мами водорода. Два из них связаны с атомом кислорода ковалент¬
ными связями и находятся на расстоянии около 1 А, а два других -
водородными, имеющими длину 1 75 А.
Естественно, разрушение кристаллической структуры льда
требует значительной затраты энергии, что и объясняет его высо¬
кую энтальпию плавления и повышенную точку замерзания воды.
В жидком состоянии происходит частичное разрушение водород¬
ных связей, и жидкая вода построена из отдельных молекул, запол¬
няющих полости между структурными элементами с тетраэдриче-
’Известно 9 кристаллических модификаций твердой воды, существую¬
щих в различных интервалах температур и давлений Лед 1 - обычная
форма, существующая при нормальном давлении и ииже 0°С
5 Зек. 675
129
Рис. 8.1. Кристаллическая
структура льда.
ской конфигурацией. Принято
считать, что около 30% молекул
воды при 20° С находится в не¬
связанном состоянии.
Более плотная упаковка в
жидком состоянии, чем в кри¬
сталлическом, приводит к появ¬
лению еще одной аномалии
СВОЙСТВ ВОДЫ' плотность жидкой
воды больше плотности льда. Это
свойство также важно в биоло¬
гическом аспекте, т. к. лед, нахо¬
дясь на поверхности воды, вы¬
ступает в роли термоизолятора,
препятствуя ее охлаждению
Таким образом, вода представля¬
ет ассоциированную жидкость,
обладающую ажурной структу¬
рой, в которой часть ее молекул
находится в узлах каркаса, а
другие занимают пустоты.
Между обоими типами молекул существует динамическое рав¬
новесие, т. к. процесс разрыва и образования водородных связей
происходит непрерывно. Жидкую воду можно рассматривать как
трехмерную структуру с растянутыми, изогнутыми и частично ра¬
зорванными водородными связями. Считается, что время ЖИЗНИ
упорядоченной структуры составляет лишь Ю"10-!©"11 с. Однако
коллективное движение всех молекул жидкой воды приводит
к тенденции сохранения ее усредненной тетраэдрической конфи¬
гурации.
Присутствие элементов упорядоченной структуры жидкой воды,
а также значительная полярность ее молекул обусловливают боль¬
шую величину ее диэлектрической проницаемости (е при 25° С
равно 79.5). Следовательно, взаимодействие между зарядами в воде
примерно в 80 раз слабее, чем в пустоте. Благодаря этому ионные и
полярные соединения легко растворяются и диссоциируют в воде.
Вода является универсальным растворителем.
В грубом приближении морскую воду можно рассматривать
как 0.5 молярный раствор хлористого натрия (табл 8.1) Обра¬
щает внимание определенный параллелизм состава воды океана
и плазмы крови. В процессе эволюции происходило все большее
отклонение состава физиологических жидкостей от состава морской
воды (сравните состав крови медузы и человека). Сходство состава
130
физиологических сред и морской воды свидетельствует о глубокой
взаимосвязи внутренней и внешней жизненных жидких сред Земли
и, по-видимому, объясняется ее биологическим происхождением.
Таблица 8.1
Ионный состав некоторых водных сред
Компоненты,
мг-экв/л
Вода
океана
Кровь
медузы
Кровь
человека
На*
466
454
143
к+
10
10
4.5
Мд2+
53
51
20
Са2+
10
10
4.5
СГ
545
554
107
БОЛ
28
18
1
Итак, вода необходима в качестве среды и участника обмена
веществ и принимает участие в многообразных процессах в орга¬
низме, а именно:
- является структурной основой оптимального физиологически
активного обьема клетки;
- определяет пространственную структуру, а следовательно и
функции биомолекул;
- обеспечивает специфичность действия ферментов за счет ас¬
социации с полярными группами, особенно в их активных центрах;
- выступает в качестве субстрата в ряде ферментативных реак¬
ций, например, гидролиза белков, липидов и др ,
- формирует направленный поток веществ внутри клетки, между
клетками и между организмами и внешней средой,
-участвует в терморегуляции.
8.2. Растворы неэлектролитов
Для понимания многих биологических явлений необходимо
иметь представление об изменении ряда физических свойств водных
растворов в зависимости от их состава и концентрации
Прежде всего отметим, что растворами являются гомогенные
системы, состоящие не менее чем из двух компонентов
Образование растворов представляет собой сложное физико¬
химическое явление, при котором наблюдается взаимодействие
между молекулами растворенного вещества и воды (а в общем
5*
131
случае - растворителя). Так, ионы растворенной соли в воде гид¬
ратированы. На это указывал еще М. В. Ломоносов, который писал:
“. частицы соли отделяются от основной массы и, сцепляясь с вод¬
ными частицами, вместе с ними начинают двигаться поступатель¬
но". Впоследствии представления о растворах как соединениях пе¬
ременного состава (гидратах) были развиты Д. И. Менделеевым.
Гидратной оболочкой в растворах обладают не только ионы, но
и полярные частицы, к числу которых относятся важнейшие биомо¬
лекулы - углеводы, белки, нуклеиновые кислоты. По современным
воззрениям до 40% молекул воды клетки представляет собой
структурированую воду, т. е. входит в состав гидратных оболочек
биомолекул, что сказывается на их физико-химических свойствах.
Химические соединения с водой образуют и молекулы непо¬
лярных веществ при растворении в ней. Так, метан с водой образу¬
ет соединение состава СН^бНгО, а хлор - С12'8Н20. В данном случае
речь идет о соединениях включения, в которых молекулы раство¬
ренного вещества включены в тетраэдрические полости жидкой
воды, как это происходит и с самой водой. И жидкую воду также
можно рассматривать как соединение включения.
В первую очередь рассмотрим более простую ситуацию, когда
все межмолекулярные силы в растворах одинаковы независимо от
строения растворенных в воде молекул. Такие растворы называют
идеальными. К ним близки разбавленные растворы неэлектролитов.
К идеальным растворам применимы термодинамические закономер¬
ности, характерные для идеальных газов. В частности, свободная
энергия каждого компонента G; выразится уравнением:
G, = G°, + RT 1пр,
где р, - давление пара i-того компонента над раствором.
Полное давление пара (р) над раствором выражает закон
Дальтона
р = pi + р2 + рз + ... + Pi ,
где ppp, - парциальные давления компонентов раствора.
Если растворенное вещество обладает значительно большей ле¬
тучестью, чем вода (или в общем случае - растворитель), упру¬
гостью пара самой воды можно пренебречь, и тогда:
Р = P°2-N2,
где N2 - мольная доля растворенного вещества, р°2 - упругость пара
чистого растворенного вещества.
Мольная доля N есть отношение числа молей данного компо¬
нента к сумме чисел молей всех компонентов в растворе. Для рас-
132
твора, состоящего из растворителя и единственного растворенного
вещества (бинарный раствор), мольные доли растворителя N1 и
растворенного вещества N2 равны:
N1 =——— и N2=———, соответственно,
П1 + П2 Пх + п 2
где п, и П2 - числа молей растворителя и растворенного вещества.
Сумма мольных долей всех компонентов равна 1:
N! + N2 = 1 •
Для разбавленных растворов вместо р°2 применяют коэффициент
пропорциональности К:
р = КЫ2.
Это уравнение известно как закон Генри, а коэффициент про¬
порциональности К (выраженный в атм.) называют константой
Генри. Константа Генри зависит от природы растворителя и рас¬
творенного веществ, а также от температуры. Если заменить в
предыдущем уравнении мольную долю моляльностью, то закон Ген¬
ри представляется в виде:
р = Кш,
где К - постоянная Генри, выраженная в атм-моль'^кг воды.
Моляльная концентрация т соответствует числу молей рас¬
творенного вещества, содержащегося в 1000 г растворителя. Для
разбавленных водных растворов молярность и моляльность прибли¬
зительно одинаковы (с = т).
Отсюда видно, что растворимость газов в воде прямо пропор¬
циональна их парциальному давлению над раствором, что важно
для объяснения физиологии дыхания и газообмена. Так, зная кон¬
станту Генри К, можно легко определить растворимость данного
газа в воде.
С увеличением давления растворимость газов в воде значитель¬
но возрастает, поэтому на глубине, к примеру, 40 м под водой, ког¬
да парциальное давление кислорода достигает 6 атм, растворимость
кислорода возрастет почти в 27 раз по сравнению с нормальными
условиями (содержание кислорода в атмосфере = 21%).
Однако оптимальным физиологическим вариантом является со¬
держание кислорода в воде при давлении 1 атм, а следовательно
для дыхания на глубине 40 м следует использовать газовые смеси,
содержащие примерно в 27 раз меньше кислорода, чем в воздухе
атмосферы. Руководствуясь этим принципом, можно осуществить
погружение до глубин в несколько сотен метров.
133
Другим важным проявлением закона Генри служит кессонная
болезнь Ее причина - мгновенное выделение воздуха из плазмы
крови при быстром подъеме с больших глубин за счет резкого
уменьшения растворимости газов из-за падения давления. Возни¬
кающие при этом пузырьки газа в крови вызывают патологические
состояния, в худшем случае - гибель из-за закупорки кровеносных
сосудов.
Напротив, разреженная атмосфера и связанное с этим пониже¬
ние содержания кислорода в крови вызывает иное патологическое
состояние, именуемое горной болезнью и связанное с дефицитом
кислорода. Проблемы взаимоотношения организма с окружающей
газовой средой являются важнейшими вопросами физиологии под¬
водного плавания и авиационно-космической медицины.
Закон Генри соблюдается далеко не всегда, и отклонения от не¬
го вызваны химическими реакциями между газами и растворителем,
что принципиально важно для биологических процессов. К примеру,
аномально высокая растворимость углекислоты в воде объясняется
химической реакцией:
С02 + Н20 ^ Н2С03 ^ Н+ + НСОз".
Такова же причина высокой растворимости в воде аммиака, се¬
роводорода и хлористого водорода. Кислород мало растворим в воде,
однако прекрасно растворяется в крови, так как химически связы¬
вается с гемоглобином.
Растворимость газов в воде сильно понижается при растворении в
ней электролитов. И. М. Сеченов установил взаимосвязь между рас-
№
творимостью газа и концентрацией электролита. = > г&е № и
N - мольные доли газа в чистой воде и в растворе с концентрацией
соли с, соответственно; Л - эмпирическая константа, зависящая от
природы газа, электролита и температуры
И М Сеченов изучал и закономерности растворения кислорода и
углекислоты в крови. Они оказались достаточно сложными. В част¬
ности, Сеченов показал, что с увеличением количества кислорода
облегчается отдача кровью углекислоты, и, наоборот, с ростом давле¬
ния углекислоты растворимость кислорода в крови растет Эти зако¬
номерности, известные как эффект Бора, были объяснены значительно
позднее (см. главу 33).
Если в воде (или в другой среде) растворены нелетучие вещест¬
ва (а это - типичная для жидких биосред ситуация), давлением их
паров над раствором можно пренебречь. Следовательно, можно
учитывать только давление паров растворителя, то есть воды, по¬
этому можно записать:
134
Р = Р°н2оИн2о.
где р°Н2о иЫНг0 - соответственно давление пара над чистой водой
и мольная доля воды в растворе. Эта зависимость известна как
закон Рауля.
Тогда для идеальных растворов при постоянной температуре в
состоянии равновесия между раствором и газовой фазой над ним
давление насыщенного пара каждого компонента р, выражается
уравнением:
р; = р0,^,
где Ы, - мольная доля 1-того компонента, а р°, - давление пара
чистого 1-того компонента.
Так как концентрацию растворов удобнее выражать через кон¬
центрацию растворенного вещества, то для водных растворов:
Р = Р°н2о 'О -N2) или ~ = Ы2 >
Р
то есть относительное понижение давления пара растворителя рав¬
но мольной доле растворенного вещества.
Так как для разбавленных растворов в выражении N2 =———
П) + П2
числом П2 в знаменателе можно пренебречь (п 1 » П2), то N2 *
пг/пр
Для воды число молей в 1000 г п, = 1000/18, тогда
■Н £. = N2 = = 0.018 П2, но П2 - число молей растворенного
Р° Р°
вещества в 1000 г воды, а следовательно получим- = 0.018т
Р°
Так как для разбавленных растворов т = с, то:
Др/р° = 0.018с
Это уравнение представляет собой упрощенное выражение за¬
кона Рауля для разбавленных растворов нелетучих веществ, в кото¬
ром концентрация растворенного вещества выражена удобным для
расчета способом - в единицах молярности
Важными следствиями из закона Рауля для растворов нелету¬
чих веществ являются повышение температуры кипения рас¬
твора по сравнению с температурой кипения растворителя и
135
понижение температуры замерзания раствора по сравнению с
чистым растворителем Эти свойства растворов (наряду с пониже¬
нием упругости пара) обычно называют коллигативными (или
взаимосвязанными, то есть коллективными), так как они вызваны
общими причинами. В частности, количество частиц растворенного
вещества (а не их масса или размеры) определяют проявление этих
свойств Установление численного значения любого из коллига-
тивных свойств дает возможность расчета всех остальных.
Рис. 8.2. Фазовая диаграмма для чистой воды
и водных растворов с различной концентрацией.
Зависимость температур кипения и замерзания растворов от
концентрации, объясняемая понижением упругости пара, иллю¬
стрируется так называемой фазовой диаграммой. Фазовая диа¬
грамма связывает давление пара любого вещества в различных
агрегатных состояниях (пар, жидкость, твердое тело) с температу¬
рой. На рис. 8.2 представлена фазовая диаграмма для чистой воды
(с0) и для растворов с различными концентрациями (с! и с2) неле¬
тучего растворенного вещества.
Из рисунка видно, что с ростом температуры давление пара
увеличивается. Как показано выше, давление насыщенного пара над
раствором ниже, чем над чистой водой. Поэтому кривая давления
пара для раствора с концентрацией С1 располагается ниже, чем
кривая давления насыщенного пара над чистой водой, а кривая для
раствора с концентрацией с2 (с2 > с 1) — еще ниже.
136
Чистый растворитель застывает при более высокой температуре,
чем раствор.
Это явление используется в технической и медицинской практике,
для чего применяются специальные добавки - антифризы, понижающие
температуру замерзания водных растворов. На этом же основано
употребление солей для борьбы с гололедом. В лабораторной практике
применяют жидкие охладительные смеси, в частности насыщенный
раствор хлористого кальция остается жидким до -55 °С.
Понижение температуры замерзания' растворов - результат
понижения упругости пара при переходе от растворителя к рас¬
твору и при увеличении его концентрации. Кривая АС на фазовой
диаграмме (рис. 8.2) выражает зависимость давления насыщенного
пара льда от температуры. Точка С отвечает температуре плавле¬
ния льда Т°зам (темературе замерзания воды). Кривая СЬ выражает
зависимость давления пара чистой воды от температуры. Точки
пересечения кривых ВМ и АЫ с линией АС (Т^ам, Т2зам) являются
точками пересечения кривых сублимации чистого льда и пара рас¬
творов различной концентрации, то есть точками замерзания воды
из растворов при равновесном давлении. Из рис. 8.2 видно, что с
ростом концентрации раствора температура замерзания растворите¬
ля падает.
Разность температур замерзания АТзам = Т° - Т1 имеет линейную
зависимость от концентрации, что следует из подобия треугольни¬
ков ВСЭ и АСЕ (для идеальных растворов бесконечно малые
участки кривых АС, ВС, ВЭ и АЕ можно считать прямыми). Отсюда
следует, что:
АЕ/ВЭ = АС/ВС = АТ2зам/ДТ'зам = р° - р2/р° - р. =
Дрг/Др 1 = гп2/т.
Иными словами, наблюдаемая зависимость для понижения тем¬
пературы замерзания растворителя (воды) выражается уравнением
ДТзам = Кт = Кс,
где К - мольное понижение температуры замерзания или крио-
скопическая постоянная данного растворителя Ее значение для
воды составляет 1 86° С/моль
Температура кипения воды и растворов на рис 8 2 определя¬
ются как точки пересечения Ь, М и N кривых упругости пара
* Температура замерзания жидкости - та температура, при которой су¬
ществует равновесие между твердой и жидкой фазами данного вещества, в
нашем случае - воды
137
с прямой, отвечающей внешнему давлению (760 мм), так как жид¬
кость закипает, если давление ее паров оказывается равным внеш¬
нему давлению Из рисунка видно, что температура кипения рас¬
твора выше, чем температура кипения воды
Для разности температур кипения (повышения температуры ки¬
пения раствора) АТКИП = Т1 - Т° применимы те же рассуждения,
что и для понижения температуры замерзания. Следовательно.
ДТКИП = Ет = Ес,
где Е - коэффициент пропорциональности, называемый мольным
повышением температуры кипения растворителя или эбулиоско-
пической постоянной рас¬
творителя. Ее величина зави¬
сит только от природы раство¬
рителя и для воды имеет значе¬
ние 0.51° С/моль.
Значительно большая вели¬
чина криоскопической констан¬
ты по сравнению с эбулиоско-
пической постоянной объяс¬
няется тем, что удельная тепло¬
та плавления льда гораздо
меньше удельной теплоты испа¬
рения воды. Значения криоскопических и эбулиоскопических по¬
стоянных некоторых растворителей приведены в таблице 8.2.
8.3. Осмос
Еще одно коллигативное свойство растворов - осмотическое
давление, возникает вследствие явления осмоса. Осмос наблюдает¬
ся, например, в том случае, когда два раствора некоторого вещества
различной концентрации в одном растворителе разделены полу¬
проницаемой перегородкой, пропускающей молекулы воды (или
другого растворителя) и непреодолимой для молекул растворенного
вещества.
Такие перегородки или мембраны можно приготовить из неко¬
торых неорганических или органических материалов, однако осо¬
бенно широко они представлены в живой природе. Ведь именно
благодаря им и возможно существование жизни, начиная с клеточ¬
ного уровня. Оболочки клеток и органов, представляющие собой
мембраны, поддерживают постоянство состава их содержимого;
через биологические мембраны осуществляется транспорт необхо¬
Таблица 8.2
Эбулиоскопические
и криоскопические константы
Растворитель
Е
К
Вода
0.51
1 86
Бензол
2.53
5.12
Уксусная
2.93
3.90
кислота
138
димых для функционирования клетки веществ, через них же удаля¬
ются отходы ее жизнедеятельности. Таким образом, мембраны
являются необходимыми структурными элементами биосистем.
Существуют разнообразные типы мембран, одни из которых про¬
ницаемы для воды, другие - лишь для определенных видов ионов,
третьи - для некоторых малых молекул и т.д. Изучением биомем¬
бран занимается мембранология, представляющая одно из актуаль¬
ных направлений современной физико-химической биологии.
Явление осмоса, как неотъемлемое свойство биомембран, широ¬
ко распространено в живой природе. При разделении двух раство¬
ров различной концентрации мембраной молекулы воды мигрируют
через мембрану, но их число, перешедшее из раствора с большей
концентрацией, меньше, чем число молекул, перешедших из раство¬
ра с малой концентрацией В итоге имеет место перемещение воды
от менее концентрированного раствора к болеее концентрированно¬
му до наступления равновесия.
Процесс самопроизвольного проникновения растворителя через
полупроницаемую мембрану и представляет собой осмос.
Если бы мембрана оказалась подвижным поршнем, она бы сме¬
щалась в сторону более разбавленного раствора, однако можно
остановить движение поршня, приложив некоторое давление извне.
Это давление называют осмотическим. Осмотическое давление -
минимальное гидравлическое давление, которое нужно приложить к
раствору, чтобы осмос прекратился.
Можно вывести выражение для осмотического давления
Пусть в цилиндре объемом V (рис. 8.3) находится водный рас¬
твор вещества А. Мольные доли воды
и этого вещества соответственно П0ЛЭДр21«йвмыи
равны:
1ЧА =
П А
раствор
н2о —
'/НгО
пн2о+пд ’
-
N.
н2о
= 1 - ЫА =-
“н2о
Рис. 8.3. Осмотическое
давление
“Н20 + п А
У правой стенки цилиндра нахо¬
дится полупроницаемый поршень,
который мы переместим влево так, чтобы справа остался 1 моль
растворителя. Пусть объем его равен УНг0 (молярный объем рас¬
творителя). Поршень преодолевал осмотическое давление п, и рабо¬
та, затраченная на перемещение, равна
А =
н2о
139
Запишем выражение для свободной энергии чистого раствори¬
теля:
в = С0 + ЯТ 1п р°,
где р° - давление пара над чистым растворителем (водой) Свобод¬
ная энергия воды в растворе составит:
й = й0 + ЯТ 1п р,
где р - давление водяного пара над раствором.
По закону Рауля р = р° N^<3 = ^ Пн2° тогда
ПН2<Э + п А
& = С0 + ЯТ 1п Р _^°_= С0 + ЯТ 1 п р° + ЯТ 1п
ПН20 + п А пНгО + п А
Так как первые два слагаемых представляют собой свободную
энергию чистого растворителя, то
дС = С' - С = ЯТ 1 п ^22
ПН20 + п А
Изменение свободной энергии равно совершенной работе про¬
тив осмотического давления:
дС = А = -лУн2о = ЯТ 1 п Пн2° или
пНгО + п А
пун2о = ЯТ 1п (1 + пА/ Пн2о )
Известно, что 1п(1+а) = а, если а —» 0. Поэтому
ЯТ пА/ пНг0 , так как пА « пН2(Э .
По правилу аддитивности общий объем равен:
V = пн2о ^н2о + паУа,
гДе Ун2о и Уа - молярные объемы воды и растворенного вещества.
Отсюда
V = пН20 УНг0, а УНг0 = V/ пНг0 .
Подставив в предыдущее уравнение значение У^о. получим
.кУ/ пН2о = ЯТпА/ пН20 или лУ = ЯТ пА.
140
Если объем выражен в литрах, то я = ИТ пд/У = ЭТс, где с -
молярная концентрация растворенного вещества.
я = 1*Тс
Это выражение, связывающее величину осмотического давления
с концентрацией, известно как закон Вант-Гоффа.
Если осмотическое давление л выразить в н/м2, а концентра¬
цию в кмоль/л, то I* равна 8.3-103 Дж/К-кмоль и поэтому
я = 8.3-103Тс.
Если же осмотическое давление измерять в атмосферах, то при
I* = 0.082 л атм/К моль-
п = 0.082Тс.
Осмотическое давление раствора из нескольких растворенных
компонентов равно сумме давлений, создаваемых каждым из них в
отдельности:
Лобщ = 0.0821(0, + с2 + с3 + + с, + ...).
В биологических жидкостях (моча, плазма, слюна, молоко и др.)
в больших количествах содержатся неорганические и органические
вещества в виде молекул, ионов и коллоидных частиц. Их суммар¬
ная концентрация носит название осмотической концентрации
(осмолярности) и именно эта величина должна быть использова¬
на в уравнении Вант-Гоффа.
Растворы с одинаковым осмотическим давлением называют
изотоническими; раствор с большим осмотическим давлением
есть гипертонический раствор, в противоположном случае имеют
дело с гипотоническим раствором.
Следует отметить, что детальный механизм осмоса не вполне
понятен и в каждом конкретном случае может быть особым В
принципе термодинамический подход и не требует знания механиз¬
ма, полученное же удобное выражение для расчета осмотического
давления еще раз показывает эффективность термодинамического
подхода.
Роль осмотических явлений в различных физиологических про¬
цессах чрезвычайно велика Постоянство осмотического давления
(изоосмия) тех или иных физиологических сред (плазма, внутри¬
клеточная жидкость, моча и др.) представляет собой фундамен¬
тальное физико-химическое требование гомеостаза. Осмотическое
давление плазмы крови человека составляет около 8 атмосфер, у
рыб = 15, у отдельных растений может достигать 100, а у прорас¬
тающих семян - до 400 атмосфер Столь высокие значения осмоти¬
141
ческого давления достигаются за счет суммарного эффекта раство¬
ренных в биосредах низкомолекулярных веществ и ионов. В плаз¬
ме, в частности, наиболее значимым компонентом является хлорис¬
тый натрий
На долю высокомолекулярных компонентов - белков - прихо¬
дится незначительная часть от общего давления, называемая ои-
котическим давлением Величина онкотического давления кро¬
ви, вызываемого альбуминами и глобулинами, составляет только
0.03-0 04 атм Однако эта незначительная составляющая от общего
осмотического давления играет принципиальную роль в некоторых
физиологических явлениях, и изменение онкотического давления
может пагубно сказаться на их проявлениях
Стенка капилляров проницаема для воды и низкомолекулярных
веществ, но не для белков На артериальном конце капилляра соле¬
вой раствор вместе с питательными веществами переходит в меж¬
клеточное пространство. На венозном конце капилляра процесс
идет в обратном направлении, так как венозное давление ниже
онкотического давления. В результате в кровь переходят вещества,
отдаваемые клетками. При гнойных процессах происходящее при
этом разрушение белков сильно увеличивает величину онкотиче¬
ского давления. Оно играет важную роль в процессах всасывания
жидкости из тканей в кровеносные сосуды. При заболеваниях, со¬
провождающихся уменьшением концентрации белков крови, онко-
тическое давление падает. В результате происходит накопление
жидкости в межклеточном пространстве. Возникают так назы¬
ваемые онкотические отеки подкожной клетчатки.
На клеточное осмотическое давление влияет обмен веществ.
При распаде больших молекул осмолярность повышается; при син¬
тезе - снижается. Осмотические взаимоотношения ответственны за
распределение воды в жидкостных пространствах организма.
Так, в растительном мире транспирация (испарение воды
листьями в окружающую среду) поддерживается накоплением воды
через ствол и ветви за счет осмотического давления. Для того что¬
бы клетки не разбухали в результате поступления в них влаги
вследствие осмоса, они обладают жесткой оболочкой. Благодаря
этому в них развивается сильное внутреннее давление, которое
называют тургором. Когда тургорное давление равно осмотическо¬
му, клетка находится в состоянии водного баланса. Однако для
нормального функционирования тургорное давление должно быть
ниже осмотического, т.к. только в этих условиях наблюдается ак¬
тивный транспорт в клетку, что необходимо для ее существования.
Эти явления определяют объем и форму клеток (например, эри¬
троцитов). '
142
При нахождении клеток в сильно гипотоническом растворе они
набухают и разрываются. Такой распад клеток крови называют ге¬
молизом. Обратное явление, при котором клетка сжимается, нахо¬
дясь в гипертонической среде, представляет плазмолиз.
Основную задачу осморегуляции выполняют почки. Осмотиче¬
ское давление мочи в норме значительно выше, чем плазмы крови,
что и обеспечивает активный транспорт из крови в почку. Осморе¬
гуляция осуществляется под контролем ферментативных систем
Нарушение их деятельности приводит к патологическим процессам.
Так, при нефрите осмотическое давление у мочи может оказаться
ниже, чем у крови, что вызывает обратный транспорт веществ.
При внутривенных инъекциях, чтобы избежать нарушения ос¬
мотического баланса, следует использовать изотонические раство¬
ры. Изотоничен по отношению к крови физиологический раствор,
содержащий 0.9% хлористого натрия. Строго говоря, истинным
физиологическим раствором является раствор того же состава, что
и плазма крови (см. табл. 8.1), содержащий катионы натрия, калия,
магния, кальция и хлорид-, бикарбонат- и сульфат-анионы в опреде¬
ленном соотношении.
Явлением осмоса объясняют слабительное действие глауберовой
соли N32804-10420 и горькой соли МдЭО^НгО. Эти соли плохо
всасываются стенками желудка, а потому их высокая концентрация
в желудке и кишечнике вызывает интенсивный переход воды внуть
кишечника, способствуя послабляющему действию.
Обратное явление - переход жидкости из кишечника в брюш¬
ную полость - вызывают при перитониальном диализе созданием в
ней избыточной концентрации сильного электролита посредством
вскрытия брюшной полости и одновременным обильным введением
влаги в желудок и кишечник Это делают при острых отравлениях
проникающими через стенки желудка ядовитыми веществами Та¬
ким путем вызывают интенсивный переход воды из желудочно-
кишечного тракта в брюшную полость, увлекающий с собой токси¬
ческое вещество
В хирургии явлением осмоса пользуются, применяя гипертони¬
ческие марлевые повязки (марлю пропитывают 10%-ным раствором
хлорида натрия). При этом рана очищается от гноя и носителей
инфекции
Гипертонические растворы вводят внутривенно при глаукоме,
чтобы снизить внутриглазное давление из-за повышенного содержа¬
ния влаги в передней камере глаза
Таким образом, понимание и контроль осмотических процессов,
а также умение определять и рассчитывать осмотическое давление
имеют существенное значение для биологов и медиков
143
Экспериментально осмотическое давление можно определить с
помощью специальных приборов - осмометров, главной деталью
которых является полупроницаемая мембрана. Необходимость
иметь мембраны, через которые проходит только растворитель,
затрудняет использование осмометрии, однако в ней нет особой
нужды, т.к. измерение любого другого коллигативного свойства
раствора позволяет рассчитать и остальные, в том числе и осмоти¬
ческое давление
Измерение температуры кипения раствора обычно не применя¬
ют из-за разрушения биосред при нагревании, измерение упругости
пара раствора нецелесообразно из-за малой точности метода. По¬
этому самым распространенным способом является измерение тем¬
пературы замерзания - криоскопия, отличающаяся простотой и
необходимой точностью. Понижение температуры замерзания как
показатель гомеостаза - достаточно постоянная величина для раз¬
личных биологических жидкостей (табл. 8 3).
Таблица 8.3
Понижение температуры замерзания биосред
по сравнению с водой
Биосреда
дт„м.,°с
Кровь
0.56
Тканевой сок
06-08
Спинномозговая жидкость
0.56
Слюна
0.19 - 049
Желудочный сок
0 49 - 0.65
Пот
0 13-0.65
Моча
1.12 - 2.30
Молоко
0.55 - 0.57
Желчь
0 54 -0.61
Сок печени
0 81 - 0.96
Наиболее эффективным методом является термоэлектрическая
осмометрия, при которой с помощью термистеров определяют раз¬
ность температур кипения капель раствора и чистого растворителя.
Эти температуры различаются за счет неодинакового испарения с
поверхости этих капель, т.к. упругости паров различны. Величина
температурной разности пропорциональна концентрации раствора.
Определив перепад температур, можно рассчитать концентрацию, а
потом и осмотическое давление.
Измерение коллигативных свойств растворов используют не
только для оценки величины осмотического давления, но и для
определения молекулярной массы растворенного вещества. Ее лег¬
144
ко рассчитать, зная навеску вещества (^), растворенную в 1000 г
воды, т.к. ГЛ=ц/т.
В совокупности коллигативные свойства водных растворов
представлены в таблице 8.4.
Таблица 8.4
Физико-химические свойства водных растворов
Свойство
Метод измерения
Др/р° = 0 018с
тонометрия
ДТКИП = 0.51с
эбулиометрия
ДТэам = 1.86с
криометрия
к = 0 082Тс
осмометрия
8.4. Растворы электролитов
Полученные выше закономерности строго соблюдаются лишь
для идеальных разбавленных растворов, к числу которых нельзя
отнести такие биологически значимые объекты, как растворы элек¬
тролитов, которые в воде диссоциируют, а образовавшиеся в ре¬
зультате ионы энергично взаимодействуют с водой и между собой.
Еще до создания С. Аррениусом теории электролитической диссо¬
циации Вант-Гофф заметил, что для растворов кислот, оснований и
солей наблюдаемые величины Др/р°, ДТКИП , ДТзам и к превосходят
значения, полученные и рассчитанные для неэлектролитов той же
концентрации. Для того, чтобы достигнуть для электролитов соот¬
ветствия с теорией, Вант-Гофф ввел дополнительный поправочный
коэффициент I, именуемый изотоническим коэффициентом:
Др/р° = 0.018мсо, ДТзам=1.86чс0, ДТКИП = 0.5Нсо, п = 0.082 Т|со
где с0 - концентрация электролита без учета его диссоциации.
Физический смысл изотонического коэффициента состоит в том,
что в растворах электролитов общее число частиц (недис-
социированных молекул и образовавшихся ионов) больше, чем в
растворах неэлектролитов Реальная концентрация всех частиц в
растворе, называемая осмотической концентрацией (Сосм), и
будет превосходить заданную без учета диссоциации на величину
т е изотонический коэффициент показывает, во сколько раз осмо¬
тическая концентрация превосходит молекулярную-
1 “ СОСМ /С0
145
В общем случае величина изотонического коэффициента растет
с ростом степени диссоциации электролита
Для слабых электролитов вводится понятие степени диссо¬
циации (а) Степень диссоциации равна отношению концентрации
продиссоциировавших молекул (сдис) к начальной (аналитической)
концентрации электролита (с0 ), т е
а = сдис/со>
откуда
Сдис = с0а-
При концентрации электролита с0, распадающегося на v ионов,
их концентрация составит avc0, а число недиссоциированных моле¬
кул составит с0 - сдис = с0(1-а).
Следовательно, осмотическая концентрация (осмолярность),
представляющая собой сумму концентраций получившихся ионов
и недиссоциированных молекул, равна:
Сосм = c0(l-a) + avc0 = c0[l+a(v-l)], откуда
i = l+a(v-l).
Данное выражение справедливо лишь для растворов слабых
электролитов; сильные электролиты, которые в воде диссоциируют
полностью, не подчиняются этой зависимости, так как в их раство¬
рах имеет место сильное межионное взаимодействие. По этой при¬
чине электропроводность растворов сильных электролитов меньше,
чем следовало бы ожидать при полной диссоциации. В таких рас¬
творах каждый ион окружен противоположно заряженной ионной
атмосферой, взаимодействие с которой растет с увеличением кон¬
центрации. Эти взаимодействия изменяют свойства раствора таким
образом, как будто они вызваны уменьшением степени диссоциа¬
ции, т.е. приводят к уменьшению числа свободных “активных” ио¬
нов. Поэтому эффективные, то есть проявляющие себя в конкрет¬
ных физико-химических свойствах концентрации ионов и выражают
через их активности (а), которые отличны от аналитической кон¬
центрации электролита.
Активность пропорциональна концентрации, а коэффициент
пропорциональности (у) называют коэффициентом активности.
Отметим, что в любых истинных (а не идеальных) растворах эф¬
фективные концентрации не равны аналитическим, а следовательно
для любых растворов (а не только для случая сильных электроли¬
тов) следует говорить не о концентрациях, а об активностях. Итак,
а = су.
146
Активности разных ионов одного и того же электролита не рав¬
ны, а потому при практическом определении свойств его раствора
учитывают среднюю активность его ионов. В частности, для одно¬
валентного электролита (например, МаСі) средняя активность (а±)
выражается через активности его катиона (а+) и зниона (а.)
следующим образом:
Смысл введения понятий активности и коэффициента актив¬
ности состоит в том, что через них мы получаем возможность опре¬
делять свойства реальных растворов, пользуясь весьма простыми
закономерностями для идеальных растворов.
Оценка силы межионного взаимодействия с тем, чтобы предска¬
зать реальное физико-химическое свойство раствора сильного элек¬
тролита и, в частности, величину осмотического давления, пред¬
ставляет собой сложную задачу, которая была в определенной сте¬
пени преодолена в теории Дебая-Хюккеля. Согласно этой теории,
основы которой мы опускаем, можно рассчитать коэффициент ак¬
тивности ионов для водных растворов по формуле:
где г+ и г_ - величины зарядов ионов без учета знака, а I - так
называемая ионная сила растворов, численно равная:
где с, и г, - концентрация и заряд иона в электролите. Ионная сила
представляет собой удобную характеристику раствора, т.к свойства
раствора определяются суммарным вкладом всех ионов раствора, а
вклад, оценивающий взаимодействие, зависит от концентраций и
зарядов Это важно при изучении биологических систем, представ¬
ляющих собой растворы, содержащие в своем составе смеси раз¬
личных ионов.
Зная состав раствора сильного электролита, можно рассчитать
его ионную силу, далее - коэффициент активности и активность, а
затем предсказать его коллигативные свойства
В таблице 8.5 даны значения среднего коэффициента активно¬
сти ионов в зависимости от заряда иона и ионной силы раствора.
То же справедливо и для коэффициента активности-
У± = -0.51(г+- г_) 7Г ,
147
Таблица 8 5
Значения коэффициента активности иоиов
в зависимости от заряда и ионной силы раствора
I
У±
г = 1
г = 2
г = 3
2 = 4
10"4
0 99
0 95
090
0 89
10 3
0 96
0 86
0 73
0 56
ю2
0 89
0 73
0 39
0 19
ю1
0 78
0.63
0 08
0 01
2 101
0 70
0 24
004
0 003
Видно, что с уменьшением ионной силы раствора коэффициент
активности ионов стремится к единице, а при одном и том же зна¬
чении ионной силы раствора его величина заметно падает с ростом
заряда иона.
8.5. Водно-электролитный баланс
Величина ионной силы биологических сред существенна для
реализации разнообразных биохимических и физиологических про¬
цессов. Их оптимальные параметры достигаются лишь при посто¬
янном и вполне определенном значении ионной силы. Это иллю¬
стрируется хотя бы таким важным для живых систем обстоятель¬
ством, что диссоциация углекислоты заметным образом зависит от
изменения концентрации хлорида натрия в ее водном растворе
(а следовательно ее поведение в плазме отличается от свойств в
чистой воде).
Отсюда нетрудно сделать заключение, что постоянство концент¬
раций электролитов в водных биосредах (водно-электролитный
баланс) и определяемая этим постоянством величина ионной силы
биосред - одно из требований гомеостаза, т.е. координации проис¬
ходящих в организме физиологических и биохимических процессов
и поддержания постоянства внутренней среды.
Концентрация ионов - регулятор распределения воды между
внутриклеточным содержимым, межклеточным пространством и
мочей. К примеру, если при болезни или в результате применения
мочегонных препаратов происходит избыточное выделение мочи, то
вместе с влагой (восполняемой с питьем) организм теряет и соли.
Падение концентрации ионов в плазме крови приводит в итоге к
падению осмотического давления. Напротив, при жажде в резуль-
148
тате обезвоживания объем внутриклеточного пространства умень¬
шается из-за потери влаги. Тем самым увеличивается концентрация
электролитов в тканях, и в результате осмотическое давление по¬
вышается. Сложные концентрационные зависимости возникают
между содержанием влаги в тканях организма и в клетках болезне¬
творных бактерий, например, холерных вибрионов, которые при их
быстром размножении могут вызвать патологическое обезвожива¬
ние организма за счет перекачивания воды внутрь бактериальных
клеток и тем самым вызывать гибель человека.
Нарушения водно-электролитного баланса связаны с комплек¬
сом причин, приводящих к избытку или недостатку воды и (или)
электролитов. Нужно различать нарушения баланса (несоответ¬
ствие между поступлением и выведением) и нарушение распреде¬
ления между внеклеточным и внутриклеточным пространством.
В зависимости от содержания жидкости в организме и осмоти¬
ческого давления плазмы различают шесть различных состояний,
связанных с увеличением количества внеклеточной жидкости (ги¬
пергидратация) и ее уменьшением (дегидратация). Организм легче
переносит гипергидратацию, чем дегидратацию. Увеличение внекле¬
точного пространства вдвое и более совместимо с жизнью, а при
дегидратации быстрая потеря 20% жидкости смертельна.
Гипертоническая дегидратация характеризуется дефицитом
жидкости с повышением осмотического давления плазмы. При ги¬
пертонической дегидратации все жидкостные пространства умень¬
шаются. Она вызывается применением концентрированных пита¬
тельных смесей, потерей гипотонической жидкости при бронхитах,
пневмониях, лихорадках, сопровождаемых потоотделением, при
водянистом стуле, острой почечной недостаточности, диабете.
Изотоническая дегидратация характеризуется дефицитом
воды и растворенных веществ при нормальном осмотическом давле¬
нии плазмы При этом страдает прежде всего внеклеточное про¬
странство (объем плазмы и межклеточной жидкости). На первом
плане стоят нарушения кровообращения Рвота, кишечная непрохо¬
димость, потеря крови, применение мочегонных препаратов, пери¬
тонит, ожоги вызывают это явление, легко переходящее в шок
Гипотоническая дегидратация характеризуется дефицитом
воды и растворенных в ней веществ с падением осмотического
давления плазмы При гипотонической дегидратации внеклеточное
пространство уменьшено, а клетки пересыщены водой. Гипотонич-
ность плазмы способствует осмотическому перемещению воды из
внеклеточной жидкости в клетки В связи с этим внеклеточное
пространство дополнительно теряет воду Потеря солей при хрони¬
ческом пиелонефрите, слабительные, бедная натрием диета, вос¬
149
полнение простой водой потерянной жидкости, диурез при диабете
обычно приводят к гипотонической дегидратации.
Варианты обезвоживания представлены в табл 8.6.
Таблица 8 6
Обезвоживание организма
Явление
Гипертоническая дегидратация
Изотоническая дегидратация
Гипотоническая дегидратация
Причина
Применение концентрированных
питательных смесей, потоотделе¬
ние при пневмониях и лихорад¬
ках, водянистый стул, острая по¬
чечная недостаточность
Рвота, кишечная непроходимость,
потеря крови, применение моче¬
гонных препаратов, ожоги
Диурез, слабительные, потеря со¬
лей при восполнении водой поте¬
рянной жидкости, бедная натрием
диета, потеря солей при пиело¬
нефрите
Гипергидратация обычно связана с почечной недостаточностью.
Гипертоническая гипергидратация характеризуется избыт¬
ком воды и растворенных в ней веществ с повышением осмотиче¬
ского давления плазмы. Клетки при этом обезвоживаются. Внекле¬
точная гипергидратация и внутриклеточная дегидратация, наблю¬
дающиеся при этом, вызываются питьем морской воды, введением
гипертонических солевых растворов.
Изотоническая гипергидратация характеризуется избытком
воды и растворенных в ней веществ при нормальном осмотическом
давлении плазмы. При изотонической гипергидратации страдает в
основном внеклеточное пространство и увеличивается лишь коли¬
чество внеклеточной жидкости, что выражается в периферических
отеках. При наличии отеков организм хотя и пересыщен водой, но
непосредственно эта вода не используется. Явление характерно для
некоторых заболеваний сердца и цирроза печени, причем введение
физиологических растворов усугубляет их.
Гипотоническая гипергидратация характеризуется абсо¬
лютным или преимущественным перенасыщением водой с падением
осмотического давления плазмы. При гипотонической гипергидра¬
тации поражаются преимущественно клетки. Причиной является
чрезмерное введение воды или бессолевых растворов сахаров
150
Данные о случаях избыточного содержания воды (отеках)
стематизированы в табл. 8.7.
Таблица 8.7
Отеки при почечной недостаточности
Явление
Причина
Гипертоническая гипергидратация
Изотоническая гипергидратация
Гипотоническая гипергидратация
Питье морской воды, введение
гипертонических растворов
Цирроз печени, избыточное вве¬
дение физиологических растворов
Чрезмерное введение воды
Итак, организм обладает концентрационным гомеостазом, фи¬
зиологический механизм регуляции которого связан во многом с
функцией почек.
Электролиты выполняют в организме важную роль:
- отвечают за осмолярность и величину ионной силы биосред,
- образуют биоэлектрический потенциал,
- катализируют процессы обмена веществ,
- стабилизируют определенные ткани (костная),
- служат в качестве энергетических депо (фосфаты),
- участвуют в свертывающей системе крови.
Для практики полезно запомнить, что физиологическими рас¬
творами являются 1 /6 моль/л растворы солей, молекулы которых
полностью диссоциируют на 2 иона, и 1/3 моль/л растворы рас¬
творов неэлектролитов (например, 1/3 моль/л раствор глюкозы).
Физико-химические параметры гомеостаза таких растворов, а
следовательно и параметры гомеостаза плазмы - важнейшей био¬
среды человеческого организма составляют.
^плазмы = 7 6 — 8 1 атм, Лонкотическое — 0.03 - 0 04 атм,
АТзам ПЛазмы 0.56 °С, 1Внутриклет 0.35, Плазмы 0 15
Глава 9. КИСЛОТНО-ОСНОВНОЕ
РАВНОВЕСИЕ
Имеющую огромное значение
для химии чистую воду можно
рассматривать как слабую кис¬
лоту или слабое основание
С Аррениус
9.1. Ионное произведение воды
Вода, как полярное вещество, в малой степени ионизирована
по уравнению:
2Н20 ^ НзО+ + ОН- или упрощенно:
Н20 Ь Н* + ОН".
Константа равновесия этого процесса, представляющая в дан¬
ном случае константу диссоциации (Кдисс), в соответствии с за¬
коном действующих масс выразится уравнением:
= [Н+] [ОН")
[н2о] •
Ее величина может быть вычислена на основании данных изме¬
рения электропроводности чистой воды (что, кстати, представляло
собой сложную задачу, так как полностью очистить воду крайне
трудно). При 25° С константа Кдисс равна 1.8 10'16. Пренебрегая
незначительной степенью ионизации, концентрацию молекул воды
как в чистой воде, так и в разбавленных водных растворах можно
считать величиной постоянной и равной = 55.56 моль/л.
Тогда можно записать:
[Н+] [ОН1 = Кдисс-[Н20] = 1.8-10 16 • 55.56 = 1014.
Обозначим Кдисс [Н20] как К«,’. Тогда для 25° С получим:
К* = [Н+ПОН-] = 10-14
Это выражение известно как ионное произведение воды.
* От о>а/ег - вода.
152
Диссоциация воды - эндотермиче¬
ский процесс, а следовательно, ^ рас¬
тет с повышением температуры, что
видно из табл. 9.1.
Для наглядности удобно использо¬
вать величину = рКчу
В чистой воде при температуре
25° С [Н*] = [ОНИ = 7^ = 10-7 моль/л.
В соответствии с принципом Ле
Шателье при добавлении кислот или
оснований данное равновесие смеща¬
ется. В кислой среде [Н+] > 107, а
[ОН-] < 10'7, в щелочной среде наблю¬
дается обратная зависимость.
Для удобства в расчетах пользуются величинами водородного
показателя pH = ~^[Н+] и гидроксильного показателя
рон = -1йон“1.
Тогда после логарифмирования:
pH + рОН = 14
В кислой среде pH < 7, рОН > 7; в щелочной среде pH > 7,
рОН < 7. Так как эти показатели однозначно связаны между собой,
проще пользоваться только одним, а именно pH В нейтральной
среде водородный показатель pH = 7.
Таблица 9 2
Значения pH физиологических жидкостей
Среда
Вероятное
значение pH
Возможные
колебания
Желудочный сок
1 65
0 9-2 0
Желчь печеночная
7 35
6 2-8 5
Желчь пузырная
68
5 6-8 0
Кровь (плазма)
7.36
7.25-7.44
Моча
58
5 0-6 5
Пот
74
4 2-7 8
Слезная жидкость
7 7
7 6-7 8
Слюна
6 75
5 6-7 9
Спинномозговая жидкость
76
7 35-7 80
Сок верхнего отдела толстого кишечника
6 1
-
Сок поджелудочной железы
88
8 6-9 0
Сок тонкого кишечника
6 51
5 07-7 07
К№ при различных
температурах
^ °С
к*
рК*
0
1 2 1015
14 93
20
6.9 1015
14.96
25
1 0 1014
1400
37
2.5 1014
13 60
50
5.5 10 й
13 27
100
5 1 1013
13 29
153
Кислотность, определяемая концентрацией ионов гидроксония в
растворе, выражаемой величиной pH, является характеристикой
многих процессов жизнедеятельности От ее величины зависит по¬
ведение клеток, их биологическая активность Кровь, слюна, желу¬
дочный сок имеют определенную кислотность (см. табл 9 2), и от¬
клонение от нормы может быть причиной тяжелых заболеваний
Обменные процессы в организмах совершаются в водных сре¬
дах Вода и продукты ее диссоциации - ионы гидроксония и гидро¬
ксильные ионы - являются необходимыми факторами, определяю¬
щими структуру и биологические свойства белков, нуклеиновых
кислот и других клеточных компонентов.
9.2. Диссоциация кислот и оснований
Согласно определению кислоты - вещества, диссоциирующие
в растворе с образованием ионов Н+:
НА £+ Н* + А-
Константа равновесия этого процесса (Ка’) равна:
к = 1Н+][А~]
[НА]
Ее называют константой диссоциации кислоты. Чем силь¬
нее кислота, тем больше Ка. Для удобства часто применяют показа¬
тель константы рКа = -lgKa. Для сильных кислот величины рКа
отрицательны, для слабых - положительны, причем величина рКа
растет с ослаблением кислотности. Например, рКа для H2SO4 равна
-3, для HNO3 = -1.64. Это сильные кислоты. Для слабой уксусной
кислоты рКа = 4.75, а для еще более слабой хлорноватистой НСЮ
рКа = 7.3.
Сила бескислородных кислот определяется относительной
электроотрицательностью атома Н (2.1) и связанного с ним элемен¬
та, а также поляризуемостью связи Н-Э, которая возрастает с рос¬
том радиуса атома элемента Э. В соответствии с этим галогеново¬
дородные кислоты (HCl, HBr, HI) - сильные, причем их сила (т.е. ве¬
личина Ка) растет от HCI к Hl. H2S в водном растворе представляет
собой уже слабую кислоту, а NH3 в обычных условиях не проявляет
кислотных свойств.
* Индекс а от слова acid - кислота (англ ).
154
Сила кислородсодержащих кислот, общую формулу которых
можно представить как ЭОт(ОН)п, сильно зависит от т, но мало
чувствительна к изменению п. Это объясняется оттягиванием элек¬
тронов связями 3=0 от связей О-Н, что и вызывает более легкий
отрыв протона. Кислоты Э(ОН)п являются очень слабыми (рКа 7-10),
ЭО(ОН)п - слабыми (рКа 1.5-4), ЭОг(ОН)п - сильными, а ЭОэ(ОН)п -
очень сильными.
Отмеченная тенденция иллюстрируется примером кислородных
кислот хлора:
НСЮ НСЮ2 НСЮз НСЮ4
хлорноватистая хлористая хлорноватая хлорная
т = 0 т = 1 т = 2 т = 3
рКа = 7.3 рКа = 2 рКа = -1 рКа = -10
С уменьшением электроотрицательности элемента для соедине¬
ний формулы Э(ОН) в водных растворах становится возможной дис¬
социация по связи Э-О. В этом случае говорят об основаниях:
ЭОН ЬЭ* + ОН".
Константа равновесия этого процесса (Кь*) выразится так:
[Э+][ОН~]
Кь
[ЭОН]
Кь называют константой диссоциации основания.
Сила основания возрастает с ростом величины Кь и соответ¬
ственно убывает с увеличением рКь = -^Кь Щелочным металлам
отвечают сильные основания - щелочи (рКь для ИОН равна 0.17,
для МаОН рКь = -0.77), щелочноземельным металлам соответствуют
основания средней силы.
При близком значении относительных электроотрицательностей
элемента Э и атома Н в гидроксидах Э(ОН)п возможна одновремен¬
ная диссоциация по связям Э-О и О-Н, следовательно, такие гид¬
роксиды обладают одновременно и кислотными, и основными
свойствами. Они амфотерны. Амфотерными свойствами обладает,
например, НЮ (относительная электроотрицательность иода равна
2 2, водорода -21)
Г +ОН' ^ НЮ ^ Н+ + Ю'
рКь= 9 6 рКа =10 6
* От Ьа$е - основание (англ )
155
Амфотерными свойствами обладают Ве(ОН>2, А1(ОН)э, Сг(ОН)з, а
также ряд других гидроксидов типичных элементов средней части
периодической системы и переходных элементов.
В соответствии с принципом Ле Шателье кислотные свойства
амфотерных гидроксидов проявляются в их реакции с основаниями,
которые, связывая ионы ОН-, смещают равновесие вправо, и, наобо¬
рот, в кислой среде амфотерные гидроксиды ведут себя как основа¬
ния, т.к. протоны кислоты связывают гидроксильные ионы:
Э+ + ОН" ^ ЭОН ^ Н+ + ЭО‘
+ +
н2о <- Н+ ОН" -> н2о
9.3. Вычисление pH растворов
сильных кислот и оснований
Можно считать, что в разбавленных (10'2-10'5 моль/л) водных
растворах сильные одноосновные кислоты типа HCl, HNO3 ионизи¬
рованы нацело, следовательно равновесие
НА + Н20 U НзО+ + А+
полностью смещено вправо и концентрация ионов гидроксония
равна концентрации аниона:
[НзО+] = [А-].
Кроме того, ионы гидроксония образуются и в результате дис¬
социации воды:
н2о + н2о Ü НэО+ + ОН'.
Таким образом, их общая концентрация равна сумме концентра¬
ций ионов гидроксония, образующихся в процессах ионизации кис¬
лоты и воды. В то же время концентрация ионов НзО+, возник¬
ших при ионизации воды в соответствии с уравнением, равна кон¬
центрации ионов ОН', поэтому концентрация ионов гидроксония в
растворе составляет:
[НзО+] = [А-] + [ОН'].
Однако концентрацией ионов гидроксония, полученных при
диссоциации воды, можно пренебречь, т.к она на несколько поряд¬
ков ниже концентрации этих ионов, образовавшихся при ионизации
сильной кислоты.
156
[НзО+ ] = [А-] = сна
pH = —^СНА.
Следовательно, концентрация ионов гидроксония в растворах
одноосновных сильных кислот равна их исходной (т.е. аналити¬
ческой) концентрации.
Для очень разбавленных растворов сильных кислот
(сна < Ю'6 моль/л) этот способ вычисления концентрации ионов
водорода неприменим, т.к. концентрации Н+, образующихся в про¬
цессах ионизации кислоты и воды, становятся соизмеримыми, и для
нахождения точной концентрации ионов Н+ необходимо учитывать
оба процесса.
Из уравнения ионного произведения воды следует, что:
[ОН'] = КЧУ/[Н+]
тогда
[Н+] = [А'] + К*/[Н+]
или
[Н+]2 _ [А-][Н+] - Кж = 0.
Решением квадратного уравнения является выражение:
[А"] + л/[А ]2 +41^
[Н] = — 2 -•
Для однокислотных сильных оснований (щелочей) по аналогии
с растворами сильных кислот можно приближенно считать, что
[ОН*] = с0СН, где с0СН - концентрация основания. Отсюда
рОН = -1& [ОН-] = -1& с0Сн.
Для растворов сильных электролитов с высокой концентрацией
при расчетах pH и рОН следует использовать значения активности
ионов Н+ и ОН" (см. главу 8):
pH = ан+ = 7н+ сн* и рОН = а0н” = 7он' с0н"-
9.4. Вычисление pH растворов
слабых кислот и оснований
В водном растворе слабая кислота диссоциирует по уравнению:
НА ^ Н+ + А"
По закону действующих масс
к = [Н*][А~]
[НА]
157
В момент динамического равновесия
Сяис ~ [Ч ] — [АТ ~ С0 а
[НА] = с0 - Сдис = с0 - с0*а
Подставляя полученные значения равновесных концентраций в
выражение для константы диссоциации кислоты, получаем
2 2 2
_ cqQ с0а _ с0 a = c0ct
a c0 - c0a c0(l - a) 1 - a '
Для слабых кислот (при a —» 0) с достаточной для практики
точностью выражение (1-а) можно принять равным единице, тогда:
Ка = с0а2,
а = VKа / со
Это выражение называют законом разведения Оствальда
Подставляя значение а в выражение [Н*] = с0 а, получаем фор¬
мулу для расчета концентрации ионов гидроксония в разбавленных
водных растворах слабых кислот
[Н+] = VmT
Для выражения кислотности среды через водородный показа¬
тель (pH) полученное выражение логарифмируют:
lgIH*]=i|gKa+ ilgc„,
умножают обе части равенства на (-1):
-lg(H*]=-ilgKa-ilgc0
и получают выражение для расчета pH в водных разбавленных
растворах слабых кислот:
pH'^pKa-ilgc,,,
где рКа - отрицательный десятичный логарифм константы диссо¬
циации кислоты.
Аналогично можно вывести уравнение для расчета [ОН-] в рас¬
творах слабых оснований.
ВОН ^ В* + ОН' [ОН“] =
РОН = IpKb- i|gc0 РН= 14- ipKb+ ilgc0
158
Многоосновные кислоты диссоциируют ступенчато. Например,
трехосновная фосфорная кислота диссоциирует в три стадии.
1. НэР04 2= Н+ + Н2Р04‘
2 Н2Р04_ 25 Н*+ НР042-
з. нро42_ 2= н++ ро43-
Для каждой ступени ионизации можно отдельно записать кон¬
станту диссоциации. Известно, что константа диссоциации после¬
дующей ступени диссоциации на 4-6 порядков меньше предыдущей.
Это объясняется тем, что протон в каждой из последующих стадий
должен отрываться от все более отрицательно заряженного аниона,
что энергетически менее выгодно, чем отрыв от нейтральной моле¬
кулы кислоты на первой стадии диссоциации. В частности, для
фосфорной кислоты рКа^ = 2.15, рКа2 = 7.2 и рКаз = 12.3.
Так как Ка^ > Ка2 > Каз и концентрация ионов гидроксония, об¬
разовавшихся в результате первой ступени ионизации, значительно
превышает количество ионов гидроксония, которые появляются в
растворе за счет ионизации по второй и третьей ступеням и не ока¬
зывают существенного влияния на установление равновесия в си¬
стеме, то величину pH растворов многоосновных кислот обычно вы¬
числяют по Ка1. Иными словами, с точки зрения ионизации, много¬
основные кислоты можно рассматривать как одноосновные и учиты¬
вать лишь первую ступень их диссоциации. То же справедливо и
для многокислотных оснований.
9.5. Гидролиз солей
Нарушение равновесия ионизации воды может происходить не
только при растворении в ней кислот или оснований, но и некото¬
рых солей, причем изменение кислотности раствора обусловлива¬
ется характером ионов соли
Если соль К1А, в которой А" является анионом слабой кислоты,
растворить в воде, то А", взаимодействуя с водой, образует слабую
кислоту НА Ионы ОН' будут находиться в избытке и раствор станет
щелочным. Имеет место гидролиз по аниону
А + НОН ^ НА + ОН'
Если же растворить соль К1А, в которой Ю* является ионом
слабого основания, то образуя с гидроксильным ионом воды
159
слабое основание КЮН, способствует появлению избыточных нонов
гидроксония. Раствор в таком случае становится кислым - это гид¬
ролиз по катиону
+ НОН ^ КЮН + Н+.
Следовательно, гидролиз соли - это обратимый процесс вза¬
имодействия ионов слабого электролита, входящих в состав соли,
с водой, что приводит к образованию слабого электролита и сдвигу
ионного равновесия воды.
Интенсивность любого обратимого процесса характеризуется
величиной соответствующей константы равновесия, которая в слу¬
чае гидролиза солей носит название константы гидролиза (К^др).
При гидролизе солей, образованных слабыми кислотами и
сильными основаниями (гидролиз по аниону)
„ _ [НА][ОН"]
Лрави _
[А ][Н20]
Концентрацию воды можно считать практически постоянной ве¬
личиной, поэтому
у тп1-к _ [НАЦОН-]
Лравн — К.ГИДр - “ •
[А ]
Если концентрацию гидроксильных ионов выразить через ион¬
ное произведение воды, то
„ _ [НА]К*,
*'ТНДр _ 4. >
[А ][Н+]
[НА] 1
а отношение = —
[А"][Н+] Ка
Из этого следует, что константа гидролиза соли, образованной
слабой кислотой и сильным основанием (гидролиз по аниону), об¬
ратно пропорциональна константе диссоциации кислоты, образо¬
вавшейся в результате гидролиза
а произведение Кгидр на соответствующее значение Ка равно ионно¬
му произведению воды:
Кгидр-Ка = К*.
К аналогичному выводу можно прийти, рассмотрев гидролиз со¬
лей, образованных слабыми основаниями и сильными кислотами
(гидролиз'по катиону).
Из ионного произведения воды [Н+] = —. Подставив это
[ОН"]
значение в выражение для Кгидр, получим:
т е. в случае гидролиза соли, образованной слабым основанием и
сильной кислотой, константа гидролиза обратно пропорциональна
константе основности (Кь) этого основания, а произведение Кгндр на
Кь равно ионному произведению воды.
Таким образом, константа гидролиза равна отношению ион¬
ного произведения воды (К^ к константе ионизации (Ка или Кь)
слабого электролита, образовавшегося в результате гидролиза. По¬
этому сильнее гидролизуются соли, образованные более слабыми
электролитами.
Гидролиз усиливается при нагревании, поскольку при этом воз¬
растает значение Кж, и при разбавлении, так как при этом увеличи¬
вается концентрация воды.
По величине константы гидролиза можно вычислить pH раство¬
ра гидролизующейся соли.
При гидролизе соли, образованной слабой кислотой и сильным
основанием, [ОН*] = [НА], т е. [ОН-]2 = Кгидр [А*]. Но [А*] - это кон¬
центрация гидролизующейся соли сс. Отсюда
Логарифмируем это выражение и затем умножаем обе части ра¬
венства на (-1)
— и Кгидр- Кь КЧУ,
[041=^
гидрсс
Отсюда
1н*| = 10‘-> + 1|ВКа- ||есс
6 Ззк 675
161
-1в1н*| = -11* 10-»- ^к,+ 1]8сс
н = 7+
2
Для солей, образованных слабым основанием и сильной кисло¬
той (гидролиз по катиону),
+ НОН ^ КЮН + Н+
К„,
Кгидр '
рН = 7.РКь±1£^
Гидролиз кислых солей слабых многоосновных кислот осложнен
дополнительным процессом диссоциации аниона. Рассмотрим гидро¬
лиз гидрокарбоната-иона:
№НСОз -> Ма+ + НСОз”
НСОз'+НОН ^ Н2СО3 + ОН-
НСОз” 5 Н+ + СОз2-
Как видно из приведенных уравнений, гидрокарбонат-ион одно¬
временно участвует в двух реакциях. Гидролиз увеличивает кон¬
центрацию гидроксильных ионов, а диссоциация - ионов водорода.
Характер среды в растворах кислых солей зависит от соотношения
интенсивностей этих двух процессов, определяемых константами
равновесия, и практически не зависит от концентрации соли. Для
растворов солей двухосновных кислот кислотность можно опреде¬
лить, пользуясь следующими формулами:
[Н*1 = ч/Ка!Ка2 •
рк.,+Рк,г
Н 2
В более сложных случаях, например, для кислых солей фосфор¬
ной кислоты, в аналогичные формулы входит константа диссо¬
162
циации кислоты, определяющая константу гидролиза соли, и вто¬
рая - характеризующая диссоциацию аниона.
Растворы кислых солей могут иметь различный характер среды.
Так, в растворе гидрофосфата натрия создается щелочная среда, а в
растворе дигидрофосфата - кислая.
Раствор гидрокарбоната натрия - питьевой соды - имеет слабо¬
щелочную среду и используется в медицине (в частности, для пред¬
отвращения ацидозов) в отличие от технической соды - карбоната
натрия, раствор которого имеет сильно щелочной характер.
9.6. Буферные растворы
Биологические жидкости характеризуются определенной вели¬
чиной pH, отклонения от которой ограничены. Сохранение посто¬
янства кислотности жидких сред имеет для жизнедеятельности че¬
ловеческого организма первостепенное значение, потому что, во-
первых, ионы водорода оказывают каталитическое действие на мно¬
гие биохимические превращения; во-вторых, ферменты и гормоны
проявляют биологическую активность только в строго определенном
интервале значений pH; в-третьих, даже небольшие изменения кон¬
центрации ионов водорода в крови и межтканевых жидкостях ощу¬
тимо влияют на величину осмотического давления (см. главу 8) в
этих жидкостях.
Решающую роль в регулировании pH играют буферные системы
Еще точнее можно сказать, что буферные системы позволяют жи¬
вому организму, как открытой системе, реализовать принцип Ле
Шателье, противодействовать влиянию внешних факторов, направ¬
ленных как на снижение, так и на увеличение pH его жидких сред,
сохранять гомеостаз.
Необходимо рассмотреть механизм действия буферных систем
вообще и, в частности, в живом организме Кроме того, в медицин¬
ской практике часто возникает необходимость в приготовлении бу¬
ферных растворов, способных поддерживать постоянное значение
pH, например, для введения этих растворов в организм, для моде¬
лирования в лабораторных условиях биопроцессов, в клиническом
анализе и т.д
Кислотно-основными буферными растворами называются рас¬
творы, величина pH которых мало изменяется при добавлении к
ним сильных кислот или щелочей, а также при разбавлении.
6*
163
9.6.1. Типы буферных растворов.
Механизм действия и вычисление pH
По составу буферные растворы можно разделить на два типа.
Они обычно состоят из двух компонентов - слабой кислоты и ее
соли (кислотный буфер) или слабого основания и его соли
(основный буфер).
Следует отметить, что не только смеси, но и растворы некото¬
рых индивидуальных солей, например, тетрабората натрия, карбо¬
ната аммония и др., также обладают буферными свойствами, кото¬
рые объясняются сильным гидролизом этих солей и образованием
компонентов, необходимых для буферного действия.
Растворы, содержащие слабую кислоту и ее соль. Приме¬
ром кислотного буфера может служить ацетатный буферный
раствор, содержащий смесь уксусной кислоты и ацетата натрия
(СНзСООН + СНзСОО№). При добавлении к такому раствору кисло¬
ты она взаимодействует с солью и вытесняет эквивалентное коли¬
чество слабой кислоты:
СНзСОО№ + НС1 ^ СНзСООН + МаС1.
В растворе вместо сильной кислоты образуется слабая, и по¬
этому величина pH уменьшается незначительно.
Если к этому буферному раствору добавить щелочь, она нейтра¬
лизуется слабой кислотой, и в растворе образуется эквивалентное
количество соли:
СНзСООН + №ОН ^ СНзСООМа + Н20.
В результате pH почти не увеличивается.
Для расчета pH в буферном растворе на примере ацетатного
буфера рассмотрим процессы, в нем протекающие, и их влияние
друг на друга.
Ацетат натрия практически полностью диссоциирует на ионы,
ацетат-ион подвергается гидролизу, как ион слабой кислоты:
СНзСООМа -> №+ + СНэСОО‘
СНзСОО' + НОН ^ СНзСООН + ОН".
Уксусная кислота, также входящая в буфер, диссоциирует
лишь в незначительной степени:
СНзСООН ^ СНзСОО" + Н+
164
Слабая диссоциация СНэСООН еще более подавляется в при¬
сутствии СНзСООМа, поэтому концентрацию недиссоциированной
уксусной кислоты принимаем практически равной ее начальной
концентрации:
[СНэСООН] = ск.
С другой стороны, гидролиз соли также подавлен наличием в
растворе кислоты Поэтому можно считать, что концентрация аце-
тат-ионов в буферной смеси практически равна исходной концент¬
рации соли без учета концентрации ацетат-ионов, образующихся в
результате диссоциации кислоты:
[СНзСОСГ] = сс.
Согласно закону действующих масс, равновесие между продук¬
тами диссоциации уксусной кислоты и недиссоциированными моле¬
кулами подчиняется уравнению
к = [Н+][СН3СОО~]
а [СНэСООН]
Подставив общую концентрацию кислоты и соли в уравнение
константы диссоциации, получим:
[Н+] = Ка^-
Сс
pH = рКа + ^ ^
ск
Это уравнение называют уравнением буферного раствора
(уравнением Гендерсона-Гассельбаха). Его анализ для буферного
раствора, образованного слабой кислотой и ее солью, показывает,
что концентрация водородных ионов в буферном растворе опреде¬
ляется константой диссоциации слабой кислоты и соотношением
концентраций кислоты и соли
Следует подчеркнуть, что это уравнение является прибли¬
женным и его нельзя применять в следующих случаях
- если концентрации кислоты и соли несоизмеримы (отличают¬
ся более, чем в 100 раз),
- если кислота слишком сильная (рКл < 3), так как в этом слу¬
чае нельзя пренебречь ее диссоциацией,
- если кислота слишком слабая (рКа > 11), в этом случае нель¬
зя пренебречь гидролизом соли.
165
К кислотным буферам относятся гидрокарбонатный (бикарбо-
натный), карбонатный и фосфатные буферные растворы
Гидрокарбонатный буферный раствор представляет собой смесь
угольной кислоты и гидрокарбоната натрия (Н2СОз + МаНСОэ). Рав¬
новесие между кислотой и солью в этой системе отражается урав¬
нением-
Н2СОз 5 Н+ + НСОз”.
Поскольку уравнение соответствует первой ступени диссоциа¬
ции угольной кислоты, в формулу для вычисления pH этого раство-
pH = рКа +1е[МаНС°з' =6.35+ 1е1МЗНС°з'
к а1 [Н2СО,| 6 |Н2С03]
Другим примером кислотного буфера является карбонатный
буферный раствор - смесь гидрокарбоната и карбоната натрия
(№НСОз+ Ма2С03). Особенность этого буферного раствора состоит в
том, что оба его компонента являются солями. Роль “кислоты”
играет №НСОз, а роль "соли" - №2СОз. Равновесие между “кисло¬
той” и “солью” отражается уравнением, соответствующим второй
ступени диссоциации угольной кислоты:
НСОз- ^ Н+ + СОз2-
pH - рКа + 1в1^£а2°э1= ю 33 + | 1Ма2СОз2
г 32 [№НС03] [МаНСОз,
Таким образом, если кислота двухосновна, при нейтрализации
ее щелочью можно получить два вида буферных растворов. Соот¬
ветственно, при нейтрализации щелочью фосфорной кислоты можно
получить три фосфатных буферных раствора, из которых наиболь¬
шее практическое значение имеет смесь дигидрофосфата и гидро¬
фосфата натрия (или калия) - МаН2Р04, "кислота” + Ма2НР04,
“соль” Равновесие для такого буферного раствора отражается
уравнением, соответствующим второй ступени диссоциации фос¬
форной кислоты:
Н2Р04" 5 Н+ + нро42-
pH = рКа, ♦ |е!^нр°.' = 7.2 + , 1МагНР04]
1 ^ан2Р04| [МаН2Р04]
Растврры, содержащие слабое основание и его соль.
Примером может служить аммонийный буферный раствор, содер¬
166
жащий аммиак и хлорид аммония (МНз + МН4С1). При добавлении к
этой смеси сильной кислоты она будет нейтрализована присут¬
ствующим основанием:
N43 + НС1 15 МН4С1.
Если же к этому раствору добавить щелочь, то она взаимодей¬
ствует с солью и в результате вместо сильного основания в раство¬
ре образуется эквивалентное количество слабого основания:
(ЧЩС! + N304 1? N43 + Н20 + №С1.
В итоге величина pH в обоих случаях меняется незначительно.
Концентрация гидроксильных ионов и pH в растворах основных
буферов вычисляются по формулам, которые выводятся аналогично
формулам кислотного буфера:
[ОН"] = Кь С°сн
сс
рОН = рКь - -***-
сс
pH = 14 - рКь + С°сн .
Сс
Отсюда очевидно, что концентрация гидроксильных ионов бу¬
ферной смеси, образованной слабым основанием и его солью, опре¬
деляется константой основности слабого основания и отношением
концентраций основания и соли.
Так, для аммонийного буферного раствора:
pH = 14 - рКмн + Ь ■№?
к к мн3 ё[Мн4С1]
9.6.2. Буферная емкость
Количественной мерой устойчивости буферных систем является
буферная емкость Буферной емкостью (В) называется количество
сильной кислоты или сильного основания, которое нужно прибавить
к одному литру буферного раствора, чтобы изменить его pH на еди¬
ницу Она выражается в моль/л или чаще в ммоль/л и опреде¬
ляется по формуле:
ДрН Уб
167
где В - буферная емкость; с - концентрация сильной кислоты или
основания (моль/л); V - объем добавленного сильного электролита
(л), Уб - объем буферного раствора (л), ДрН - изменение pH
В, ммоль/л
Рис. 9.1. Изменение буферной емкости
в зависимости от величины отношения [соль]/[кислота].
Зависимость буферной емкости от соотношения концентраций
компонентов буферной смеси представлена на рис. 9.1.
Наибольшая величина буферной емкости буферной смеси дости¬
гается при равенстве концентраций обоих компонентов, когда
pH = рК. Поэтому применение любой буферной смеси ограничено
определенной областью pH (областью буферирования), а именно:
pH = рК ± 1
Следовательно, при использовании кислотных буферных рас¬
творов для обеспечения наибольшей буферной емкости надо вы¬
бирать такие кислоты, у которых значение рК наиболее близко
к заданному значению pH. При работе с основными буферными
растворами нужно сравнивать требуемое значение pH с величиной
“14 - РКЬ”.
Существуют универсальные буферные системы, которые обес¬
печивают высокую буферную емкость в широком интервале pH.
Они представляют собой смеси нескольких слабых кислот и их со¬
лей. Например, смесь фосфорной, уксусной и борной кислот, к ко¬
торой можно добавить переменное количество щелочи, обеспечи¬
вает высокую буферную емкость в пределах от 2 до 10 единиц pH.
168
Буферная емкость зависит не только от отношения концентра¬
ций компонентов буферного раствора, но и от общей концентрации
буферной смеси.
Пусть, например, даны два буферных раствора, один из которых
содержит по 100, а другой - по 10 миллимолей уксусной кислоты и
ацетата натрия. Сравним, как изменяются их pH при добавлении к
1 л каждого раствора 5 миллимолей соляной кислоты.
Добавляемая кислота вступит в реакцию с ацетатом натрия, и
это отношение в первом растворе станет равным 0.9, а во втором
0.33. В итоге у первого раствора отношение соль/кислота и, следо¬
вательно, величина pH изменились меньше. Отсюда видно, что пер¬
вый буферный раствор обладает большей буферной емкостью.
Если буферная система не обладает достаточной буферной ем¬
костью, ее можно повысить, увеличив концентрацию обоих компо¬
нентов в необходимое количество раз. Заданная величина pH при
этом почти не изменяется.
9.6.3. Влияние разбавления на pH буферных растворов
Нередко буферные растворы приходится разбавлять, особенно
при измерении pH малых объемов биологических сред (кровь, моча
и др.). В связи с этим возникает вопрос, как на pH буферных рас¬
творов влияет разбавление.
При разбавлении растворов концентрации обоих компонентов
смеси уменьшаются в одинаковое число раз. Следовательно, по
уравнению Гендерсона-Гассельбаха, величина pH буферных раство¬
ров при этом не должна изменяться. Однако опыт показывает, что
некоторое изменение pH, хотя и незначительное, все же происхо¬
дит (табл. 9 3).
Это расхождение объясняется тем, что все приведенные форму¬
лы для расчета pH буферных растворов не учитывают межионных
взаимодействий. При учете межион¬
ных взаимодействий в формулы должны
входить не молярные, а активные (см
главу 8) концентрации компонентов.
Нейтральная молекула кислоты долж¬
на слабее взаимодействовать с другими
частицами в растворе, чем ион Поэтому
для разбавленных растворов, когда ион¬
ная сила не превышает 0.1, коэффициент
активности молекул унд можно считать
равным 1
Таблица 9 3
Влияние разбавления
на pH ацетатного
буферного раствора
Концентрация
соль+кислота
(моль/л)
pH
0 1
4 62
001
4 67
0 001
4 74
169
pH = рКа + 1в^1= рКз + 1?
аНА СНА
Так как с разбавлением буферных растворов коэффициент ак¬
тивности ионов А' увеличивается, возрастает и pH раствора.
Хотя изменение величины pH вследствие разбавления незначи¬
тельно, при точных работах его следует учитывать Известно, что
изменение pH крови на 0.2-0.3 единицы pH приводит к серьезным
патологическим нарушениям. Для правильного истолкования значе¬
ний pH крови требуется воспроизводимость 0.003 единицы pH
В частности, ошибка в 0.02 единицы pH приводит к погрешности
более 4.5% в определении содержания углекислого газа в крови
9.7. Биологические буферные системы
Из буферных систем организма наибольшей емкостью характе¬
ризуются буферные системы крови, которые неравномерно распре¬
делены между эритроцитами и плазмой крови. И в плазме, и в эри¬
троцитах находятся гидрокарбонатная (бикарбонатная) буферная
система и буферные пары неорганических фосфатов. Только в плаз¬
ме локализуется буферная система плазменных белков (альбу¬
минов, глобулинов и др.). Гемоглобиновая буферная система и бу¬
ферные пары органических фосфатов находятся в эритроцитах.
Из кишечника и тканей в кровь при обмене веществ постоянно
поступают различные кислоты (угольная, молочная, масляная и др.)
и в меньшей степени основания (аммиак, креатин). В организме
человека в спокойном состоянии ежесуточно образуется количество
кислоты, эквивалентное -25л концентрированной соляной кисло¬
ты. Тем не менее, благодаря наличию вышеперечисленных буфер¬
ных систем, pH крови остается постоянным (7.4 ± 0.04)
9.7.1. Гидрокарбонатная (бикарбонатная)
буферная система
Величина pH крови зависит от концентраций свободной раство¬
ренной в крови Н2СОз и кислоты, связанной в гидрокарбонат-ион:
рН = рк, + !ї!£ї£2і! = рк, + ів
1Н2С03] [со5“6|
где [С02связ) - концентрация гидрокарбоната в пересчете на СОг в
объемных процентах; [С02сво6] - концентрация свободной угольной
кислоты в объемных процентах. В условиях плазмы крови (при
37°с>РкЦсо, = 61'
Концентрацию углекислоты, растворенной в крови, можно най¬
ти по формуле:
[С02сво6] = б- рСо2 ,
где рСо2 - парциальное давление углекислого газа в воздухе, нахо¬
дящемся в равновесии с кровью; в - коэффициент растворимости
углекислого газа в крови.
Для определения суммарной концентрации С02 в крови к ней
добавляют сильную кислоту и измеряют объем выделяющегося газа.
Таким образом, пользуясь газоаналитическим методом определения
гидрокарбонат-иона и С02, можно вычислить величину pH плазмы:
[СО^ум]-[СС>2Во6]
pH = 6 1 + 1г .
[СО|воб1
Из уравнения Гендерсона-Гассельбаха нетрудно рассчитать со¬
отношение гидрокарбонат-иона и угольной кислоты в крови при
pH = 7.4. Оно равно 20:1. Избыток гидрокарбоната обеспечивает
так называемый щелочной резерв крови. При поступлении в кровь
кислот гидрокарбонат нейтрализует их, а избыток С02 выводится
через легкие, вызывая увеличение легочной вентиляции. Таким об-
[№НС03]
разом, соотношение —, а следовательно, и величина pH
[Н2С03]
крови не изменяются.
У гидрокарбонатной буферной системы наибольшая взаимосвязь
со всеми буферными системами и вне-, и внутриклеточных жид¬
костей. Нарушение в любой буферной системе сказывается на кон¬
центрациях составляющих гидрокарбонатной буферной системы,
поэтому изменение ее параметров может достаточно точно характе¬
ризовать состояние дыхательных или метаболических нарушений,
то есть кислотно-основное состояние организма
9.7.2. Фосфатная буферная система
Величина рКгн ро в условиях плазмы крови (при 37° С) равна
6.8, поэтому уравнение Гендерсона-Гассельбаха принимает вид-
pH = 68 +
1Н2РО,‘]
171
Отношение ^НР^4—- в плазме крови (при pH = 7.4) равно 41
[Н2Р04-]
и не изменяется, так как при избыточном накоплении какого-либо
из компонентов он выделяется с мочой
Фосфатная буферная система крови характеризуется меньшей
буферной емкостью, чем гидрокарбонатная, из-за малой концентра¬
ции компонентов в крови. Однако эта система играет решающую
роль в других биологических средах - в клетке, в моче и соках
пищеварительных желез.
9.7.3. Белковые и аминокислотные буферные системы
Значительную долю буферной емкости крови обеспечивают бел¬
ковые буферные системы (гемоглобин, оксигемоглобин и, в меньшей
степени, белки плазмы). Клетки и ткани организма проявляют за¬
метное буферное действие благодаря белкам
Молекулы белков (Річії-Н) содержат остатки аминокислот
Н2М-СНВ-СООН, которые проявляют себя как амфотерные электро¬
литы. В них группы -СООН имеют слабые кислотные, а -МН2 -
слабоосновные свойства. Соответственно, белки противодействуют
как подкислению, так и подщелачиванию среды.
Следует подчеркнуть, что белковая (протеиновая) буферная си¬
стема работает совместно с гидрокарбонатной системой:
Равновесия (а) и (б) тесно связаны между собой. Рост кон¬
центрации С02 (за счет повышения продукции, например, при мы¬
шечной работе ьли за счет снижения скорости удаления при дыха¬
тельной недостаточности) сдвигает реакцию (а) вправо, а реакцию
(б) - влево
Следовательно, увеличение концентрации бикарбонат-иона со¬
ответствует снижению концентрации Prot'. Сумма концентраций
НСОз- и Prot" остается неизменной благодаря совместному дейст¬
вию этих буферных систем.
Если ионы водорода возникают из других источников, напри¬
мер, в связи с избыточным образованием молочной кислоты при ги¬
поксии или 3-гидроксимасляной кислоты при диабетическом кетозе,
то обе резкий сдвигаются влево, образуются формы Prot-H и СОг,
при этом избыток С02 удаляется через легкие.
172
Чтобы понять механизм действия буферных систем белков
плазмы, рассмотрим более простые аминокислотные системы с ана¬
логичным механизмом буферного действия.
9.7.4. Аминокислотные буферные растворы
В качестве примера возьмем простейшую аминокислоту - гли¬
цин (аминоуксусная кислота). В результате ионизации амино- и
карбоксильной групп глицин существует в водном растворе в виде
биполярного иона R*:
NH2—СНг—СООН (R) 5 NH3+—СН2—COO- (R1).
Концентрация биполярных ионов R* в водном растворе глицина
в 224 ООО раз больше концентрации нейтральных молекул R.
Если к водному раствору глицина добавить сильную кислоту, то
он присоединит протон по СОО--группе с образованием катиона
глицина R*. При добавлении к раствору глицина щелочи группа
NH3+ отдаст протон, и образуется анион глицина R-:
рКЭ1 = 2.6 рКа, = 9.8
+ Н+ + н+
мнз^-снг-соон — мнз^-снг-соо' — NH3—сн2—соо’
R+ (катион глицина) R* R' (анион глицина)
(биполярный ион глицина)
Из этой схемы видно, что катион глицина R* можно рассматри¬
вать как слабую двухосновную кислоту, которая характеризуется
двумя константами ионизации - К3| и Ка2 Следовательно, должны
существовать два вида глициновых буферных растворов
В водных растворах глицина все его три формы (R+, R*. R-) нахо¬
дятся в подвижном равновесии. Это равновесие при подкислении
должно сдвигаться в сторону увеличения концентрации R\ Следо¬
вательно, при добавлении к глицину определенного количества
сильной кислоты получается смесь двух форм - R+ и R*. которая
представляет собой глициновый кислотный буферный раствор
NH^-C^-COO" + Н + (недостаток) — NHj^C^-COOH
R- R+
В этом случае катион глицина играет роль кислоты, а глицнн -
соли Величина pH такого раствора вычисляется по формуле
173
При добавлении к глицину щелочи равновесие сдвигается в сто¬
рону увеличения концентрации Я” При этом можно получить смесь
форм Я* и Я". Такая смесь представляет собой глициновый щелоч¬
ной буферный раствор
МИз—СН2-СОО" + он- (недостаток) — мНг—СН2-СОО'
Я-
В этом случае роль кислоты играет биполярный ион глицина Я4,
а соли - анион глицина Я-. Величину pH такой буферной смеси
вычисляют по формуле:
рН=рКа, + 18 15^.
I
9.7.5. Гемоглобиновый буферный раствор
Гемоглобиновая буферная система является основной буферной
системой эритроцитов и обладает большой буферной емкостью.
Гемоглобиновый буфер является разновидностью белковой бу¬
ферной системы. Она состоит из двух форм гемоглобина - восста¬
новленного (ННЬ - гемоглобина) и окисленного (ННЬОг - оксиге-
моглобина). Условно гемоглобиновый буфер можно записать так:
а) Буферная система, образованная гемоглобином:
ННЬ и Н+ + НЬ'
кшв-> кГ+нь-
б) Буферная система, образованная оксигемоглобином:
ННЬ02 ^ Н+ + НЬОг”
КШЬОг -» Ю* + НЬОг-.
Уравнение Гендерсона-Гассельбаха для этих двух систем можно
записать следующим образом:
1ЧЬ ] [НЬ-]
pH = ркННЬ0! ♦ 1к£52Г1= 6.95 ♦
к к нньо2 6 [ННЬ02| 6
Так как гемоглобин ННЬ, присоединяя кислород, образует окси-
гемоглобин ННЬ02:
ННЬ + 02 ^ ННЬ02,
то системы гемоглобина и оксигемоглобина взаимосвязаны и су¬
ществуют как единое целое, причем гемоглобин является более
слабой кислотой (рКннь = 8.2, Кннь = 6.3-10'9), чем оксигемоглобин
(рКНньо2 = 6.95, КННЬо2= 1.12-10'7). Отсюда следует, что ион
НЬ~, являющийся анионом более слабой кислоты, способен активнее
связывать протон, чем ион НЬ02_.
Участие гемоглобина в регуляции pH крови связано с его ролью
в транспорте кислорода и углекислоты. Гемоглобиновые буферные
системы взаимодействуют с гидрокарбонатным буфером.
Углекислый газ, образующийся в значительных количествах в
периферических тканях, участвующих в процессе дыхания, поступа¬
ет в эритроциты, где эффективно превращается в угольную кислоту
под действием фермента карбоангидразы.
ферментативно
С02 + НгО ► Н2С03 5=—► Н++ НС03*
(в ткани)
При рНилетки -7.4 и рКд2н со = 6.1 более 90% образовавшей¬
ся угольной кислоты диссоциирует, поэтому связывание С02 приво¬
дит к повышению концентрации Н+ и грозит “закислить” кровь.
Для предотвращения опасного повышения кислотности крови в
действие вступают гемоглобиновые буферные системы Учитывая
отмеченную ранее различную кислотность гемоглобина и оксиге¬
моглобина, при pH = 7.4 равновесные концентрации каждой из со¬
пряженных пар (кислота-соль) будут различными-
ННЬ02 5=—- Н+ + НЬ02
преобладающая форма
ННЬ — Н++ НЬ'
преобладающая форма
Протоны, возникающие при диссоциации угольной кислоты, бу¬
дут взаимодействовать с преобладающей в растворе ионной формой
оксигемоглобина НЬОг”, образуя его молекулярную форму
Н+ + НЬ02" -> ННЬ02.
175
Известно, что при повышенной кислотности эффективность свя¬
зывания гемоглобином кислорода снижается. Это явление названо
эффектом Бора (см 33 2 1) Поэтому оксигемоглобин, освобождает
кислород (уходящий в ткани) и дает более слабую кислоту -
гемоглобин:
ННЬ02 -> ННЬ + 02.
Таким образом, в тканях повышение концентрации протонов,
вызванное диффузией СОг в клетку, в значительной мере нейтра¬
лизуется.
Однако вследствие указанных процессов нарушилось соотноше¬
ние соль/кислота в буферных системах: содержание НСОз' и ННЬ
увеличилось, а концентрация НЬОг" уменьшилась. Восстановление
этих соотношений происходит в легких. Когда венозная кровь до¬
стигает легких, кислород и гидрокарбонат-ион (из тканей) снова
проникает внутрь эритроцитов. При этом кислород связывается с
присутствующим в избытке гемоглобином:
02 + ННЬ -> ННЬ02.
Теперь в присутствии НСОз_-иона ННЬ02 выступает уже в роли
кислоты, образуя НЬОг” и угольную кислоту:
нньо2 + нсо3- -> ньо2-+н2со3.
И вновь возникающая угольная кислота под действием карбоан-
гидразы разлагается, уходя в легкие в виде углекислого газа:
Н2СО3 —> Н20 + С02 (ферментативно).
9.8. Кислотно-основное состояние
Кислотно-основное состояние - неотъемлемая составная часть
гомеостаза внутренней среды организма, который обеспечивает
оптимальные условия правильного течения обмена веществ.
Физиологические системы регуляции кислотно-основного со¬
стояния связаны с функциональной активностью легких и почек.
Процессы, происходящие в легких, связаны с тем, что образова¬
ние оксигемоглобина приводит к освобождению иона водорода из
гемоглобина. Ион водорода ассоциируется с бикарбонатом. Обра¬
зующаяся в результате угольная кислота распадается в легких под
действием фермента карбоангидразы, и удаление СОг в атмосферу
смещает равновесие этой реакции в сторону распада угольной
176
кислоты. Ион водорода при этом оказывается в составе воды, -
соединения, мало способного к диссоциации. В результате этих
процессов идет активное удаление гидрокарбонат-иона, потери ко¬
торого восполняются его ресинтезом в почках.
Действие почек заключается в удалении из организма ионов во¬
дорода и насыщении плазмы крови гидрокарбонат-ионом. При этом
принципиальную роль играет фермент карбоангидраза клеток ка¬
нальцев почек, имеющая ту особенность, что быстро образует
угольную кислоту и значительно более медленно ее разлагает, вне
зависимости от концентрации СОг и НгО.
Принципиальную роль играют биосинтез аммиака в почках и
фосфатная буферная система мочи. В результате ферментативной
реакции дезаминирования (то есть отщепления аммиака) глутами¬
новой кислоты образуется аммиак, который связывает протоны,
превращаясь в нон аммония. Процесс замены натрия на аммоний
в дигидрофосфате приводит к изменению соотношения гидрофос¬
фат/дигидрофосфат от 1:4 в крови до 1:50 в почках.
Способность почек выводить из организма ионы водорода на¬
столько велика, что в итоге соотношение между концентрациями
водородных ионов в моче и в крови может составить 800:1.
Рассмотренные буферные и физиологические механизмы в нор¬
ме обеспечивают стабильное значение pH. Дисбаланс между обра¬
зованием и (или) удалением ионов водорода, когда вышеуказанные
механизмы стабилизации их концентрации не справляются с на¬
грузкой, приводит к снижению или повышению pH. В первом слу¬
чае (при снижении pH) состояние называется ацидозом Во вто¬
ром случае (при повышении pH) - алкалозом.
В зависимости от механизма развития расстройств кислотно¬
основного состояния выделяют дыхательный и метаболический аци¬
дозы и алкалозы
Метаболический ацидоз характеризуется нарушением мета¬
болизма, которое приводит к нескомпенсированному или частично
компенсированому падению pH крови.
Метаболический ацидоз наступает вследствие.
а) избыточного введения или образования стойких кислот
(поступление кетокислот при голодании и диабете, повышенное
образование молочной кислоты при шоке, повышенное образование
серной кислоты при усиленном катаболизме, то есть в процессе
распада биомолекул, и др ),
б) неполного удаления кислот при почечной недостаточности,
в) избыточной потери гидрокарбонат-иона в результате поноса,
колита, язвы кишечника Процессы компенсации связаны с нейтра¬
лизацией ионов водорода гидрокарбонат-ионом и усилением легоч¬
ной вентиляции
177
Метаболический алкалоз характеризуется нарушением мета¬
болизма, которое приводит к нескомпенсированному или частично
компенсированому увеличению pH крови.
Метаболический алкалоз наступает вследствие-
а) потери водородных ионов (высокая кишечная непроходи¬
мость, рвота и др ),
б) увеличения концентрации бикарбоната (потеря воды, избы¬
точное введение бикарбонат-иона при метаболическом ацидозе, вве¬
дение солей органических кислот - молочной, уксусной, лимонной,
метаболизирующихся с поглощением ионов водорода и др.).
Компенсации этого явления достигают снижением легочной
вентиляции (соответственно, задержки С02), удалением гидрокар¬
бонат-иона почками.
Дыхательный ацидоз - это нескомпенсированное или час¬
тично компенсированное снижение pH в результате гиповентиляции
из-за:
а) заболевания легких или дыхательных путей (пневмония, отек
легких, инородные тела в верхних дыхательных путях и др.);
б) повреждения (заболевания) дыхательной мускулатуры;
в) угнетения дыхательного центра лекарственными средствами
или наркотиками - опиатами, барбитуратами и др.
Дыхательный алкалоз - это нескомпенсированное или час¬
тично компенсированное повышение pH в результате гипервентиля¬
ции из-за лихорадочного состояния или истерии. Процессы компен¬
сации осуществляются буферными системами, повышенным выведе¬
нием гидрокарбонат-иона почками.
Для коррекции кислотно-щелочного равновесия при ацидозах
обычно используют 4%-ный раствор гидрокарбоната натрия, кото¬
рый вводят внутривенно. Коррекция кислотно-щелочного равно¬
весия при алкалозах более сложна. В качестве одной из времен¬
ных мер целесообразно введение 5%-ного раствора аскорбиновой
кислоты.
Глава 10. РАВНОВЕСИЕ В КОМПЛЕКСАХ
10.1. Константа нестойкости
Комплексные соединения - электролиты - в разбавленных рас¬
творах ионизируются в две стадии. Сначала идет практически не¬
обратимый распад на внутреннюю и внешнюю сферы, например:
[Си(МН3)4]804 -» [Си(МНз)4]2+ + БОЛ
а затем комплексный ион в той или иной мере подвергается диссо¬
циации на комплексообразователь и лиганды:
[Си(МН3)4]2+ ±5 Си2+ + 4МН3.
Для второго процесса, который обратим, можно написать выра¬
жение для константы равновесия:
К - 1Си2*)[ИН314 _ ,3
лнест о 1и
[{Си(МН3)4}2+]
Эта константа ионизации внутренней сферы комплексного сое¬
динения называется константой нестойкости. Чем меньше ее
величина и чем соответственно больше величина рКнест = -1дКнест
(в данном случае рКнест = 12.1), тем более прочным является дан¬
ное комплексное соединение. В таблице 10.1 приведены типичные
значения констант нестойкости для нескольких примеров аммиач¬
ных комплексов и хелатов с участием
глицина (Гли) и двунатриевой соли
этилендиаминотетрауксусной кислоты
(трилон Б, Тр).
Обращает на себя внимание мень¬
шая прочность аммиачных комплексов
по сравнению с комплексами глицина,
которые в свою очередь уступают
хелатам с участием трилона Б.
Объяснение этому понятно из
следующих соображений Во-первых,
при разрушении аммичных комплек¬
сов для удаления из комплекса моле¬
кулы аммиака требуется разорвать
только одну связь металл-лиганд
Для разложения хелата с глицином необходимо разорвать уже
две связи металл-лиганд, что требует значительно больших энер¬
гетических затрат. Хелаты с трилоном Б разрушаются с раз¬
Таблица10 1
Константы нестойкости
комплексных соединений
Комплекс
К„„т
[Си(МН3)4]2*
8-Ю13
[Со(МН3)4Г
1-Ю5
Си(Гли)2
5-Ю16
Со(Гли)2
3 3-Ю9
СиТр
1 6-Ю19
СоТр
1-Ю16
179
рывом четырех связей комплексообразователя с этим лигандом. Та¬
ким образом, с ростом дентатности лиганда возрастает энтальпия
образования комплекса
Не менее важен вклад и энтропийного фактора Следует по¬
мнить, что в водных растворах катионы (1-металлов реально су¬
ществуют в виде аквокомплексов, а следовательно образование
любых комплексов есть результат обмена молекул воды на другие
лиганды с образованием более прочных, чем аквокомплексы, ком¬
плексных соединений В реакции с аммиаком число частиц не изме¬
няется, а поэтому не велико и изменение энтропии:
[Си(Н20)4]2* + 4МН3 —> [Си(МН3)4]2+ + 4Н20.
В реакции же с глицином число частиц - продуктов реакции (7)
больше, чем число исходных частиц (3), а следовательно в реакции
энтропия увеличивается:
[Си(Н20)4]2* + 2Гли -» Си(Гли)2 + 4Н20 + 2Н*.
В еще большей степени она увеличивается в реакции с трило-
ном Б, поскольку изменение числа частиц в этом случае еще боль¬
ше (семь вместо двух):
[Си(Н20)4]2* + Тр -» СиТр + 4Н20 + 2Н+.
Повышенная прочность комплексных соединений с полидентат-
ными лигандами называется хелатным эффектом.
Хелатный эффект дает ключ к пониманию состояния катионов
с1-металлов в биосредах. Все они оказываются прочно связанными
с теми или иными полидентатными биолигандами, как правило,
с полипептидами или циклическими полидентатными лигандами -
производными порфирина (например, в гемоглобине). В свободном
виде, точнее в виде аквокомплексов, эти катионы обнаруживаются
лишь в плазме, причем в ничтожных количествах.
Разрушение комплексного соединения состоит в смещении рав¬
новесия вправо, чего можно достигнуть уменьшением концентрации
лиганда или комплексообразователя, связыванием их в более проч¬
ное соединение или переведением в осадок. Этого можно добиться
осаждением катиона Си в виде сульфида:
[Си(МН3)4]2* + Б2’ -> СиБ4. + 4МН3
или переводом аммиака в ион аммония действием кислоты:
[Си(МН3)4]2* + 4Н* -» Си2* + 4МН4+.
Еще один способ разрушения комплексного соединения - свя¬
зывание комплексообразователя или лиганда переводом нх в более
прочное комплексное соединение, то есть имеющее меньшую кон¬
станту нестойкости.
Примером этого может служить действие трилона Б (Тр) на ам¬
миачный комплекс меди:
[Си^НэМ2* + Тр -> [СиТр]2+ + 4МН3 + 2Н*.
Случай обмена комплексообразователя иллюстрируется заменой
катиона кобальта на катион меди в аммиачном комплексе в соот¬
ветствии с величинами констант нестойкости соответствующих
комплексов (табл. 10.1):
[Со(МНз)4]2* + Си2*-» [Си(МН3)4]2* + Со2*.
Связывание гемоглобина молекулами СО является аналогичным
примером разрушения одного биокомплекса - оксигемоглобина за
счет образования более прочного карбоксигемоглобина:
НЬ02 + СО -» НЬСО + 02.
10.2. Металло-лигандный гомеостаз
и его нарушения
В организме непрерывно происходит образование н разрушение
биокомплексов из катионов биометаллов и биолигандов (порфири-
нов, аминокислот, полипептидов и др.). Обмен веществ с окружа¬
ющей средой поддерживает концентрации веществ, участвующих в
этом равновесии, на определенном
уровне, обеспечивая состояние ме-
талло-лигандного гомеостаза.
В принципе для каждого из
катионов биометаллов характерна
своя совокупность реакций металл-
лигандного равновесия Распреде¬
ление катиона металла между
биолигандами определяется как
величинами констант нестойкости
для комплексов этих лигандов, так
и концентрациями этих лигандов
Такое совокупное равновесие для каждого из катионов можно
выразить общей схемой, где М - катион металла, и, 1_2, 1о. - био-
лиганды
В кау«Стве примера суммарного равновесия можно привести
металлскпигандное равновесие для катиона железа Этот катион
находится в связанном виде в составе таких биокомплексов как
М-г1-з
М'УЧ^1-2 1г1-з м‘ти
XI. <г"Ц
М-хЦ^-1 м
М-П1-5
М-д!_б
181
переносчик кислорода гемоглобин, запасающий кислород в тканях
миоглобин, переносчики электронов - цитохромы (известно свыше
пятидесяти), ферменты - катал аза и пероксидаза В организме име¬
ются и железосодержащие белки - ферритины, в составе которых
содержатся резервные количества железа, высвобождающиеся от¬
туда по мере разрушения выше упомянутых биокомплексов
Кроме того, существуют и железосодержащие белки - транс-
феррины, выполняющие функцию транспорта железа из резервных
белков к месту биосинтеза указанных комплексов В частности,
транспорт катиона железа направлен в клетки ретикулоцитов, ко¬
торые выполняют биосинтез гемоглобина. Кроме трансферринов
транспортную функцию выполняют и железные комплексы с амино¬
кислотами, которые, будучи низкомолекулярными соединениями,
легче проникают через клеточные мембраны, чем трансферрины.
Нарушение этого суммарного равновесия приводит к ряду пато¬
логических явлений - железоизбыточных и железодефицитных со¬
стояний Такие же равновесия характерны и для каждого биокатио¬
на в биосредах. Их нарушение приводит к различным отклонениям
в метаболизме
Поступление, метаболизм, накопление и выделение катионов
металлов (а в целом - любых микроэлементов) регулируется специ¬
альной системой микроэлементозного гомеостаза. Глубокое изуче¬
ние этого вопроса, которое еще предстоит провести, позволит гово¬
рить о новом этапе молекулярной биологии - изучении жизненных
процессов на атомном уровне. Более или менее удовлетворительно
изучен лишь вопрос металло-лигандного гомеостаза железа. Между
тем в совокупности существуют тысячи патологических явлений -
микроэлементозов, связанных с теми или иными металлоизбыточ¬
ными или металлодефицитными состояниями.
В качестве примера можно привести неполный перечень забо¬
леваний, вызванных нарушением металло-лигандного баланса толь¬
ко лишь для одного иона - катиона меди. Дефицит этого элемента
в организме вызывает синдром Менкеса, синдром Морфана, бо¬
лезнь Вильсона-Коновалова, цирроз печени, эмфизему легких, аор-
то- и артериопатии, анемии. Избыточное поступление катиона меди
в организм может вызвать серию заболеваний самых различных
органов (ревматизм, бронхиальную астму, воспаление почек или
печени, инфаркт миокарда и др.), называемые гиперкупремиями.
Существует и профессиональный гиперкупреоз - медная лихорад¬
ка. Наконец, возможно и отравление различными медьсодержа¬
щими препаратами.
Нарушение металло-лигандного гомеостаза возможно по раз¬
ным причинам: из-за дефицита или избытка катионов биометаллов.
182
из-за поступления катионов токсичных металлов, из-за поступле¬
ния или образования посторонних лигандов.
В этих случаях в дополнение к естественным металло-ли-
гандным равновесиям прибавляется новое равновесие с образова¬
нием чужеродних комплексов, содержащих металлы-токсиканты или
лиганды-токсиканты, которые не выполняют необходимые биологи¬
ческие функции. Пример этого: поступление в плазму токсичного
лиганда - оксида углерода(Н), прочно связывающегося с гемоглоби¬
ном, который перестает переносить кислород.
В результате антропогенной деятельности в окружающую среду
поступают различные вещества - как катионы токсичных металлов,
так и токсичные лиганды. Особенно много ядовитых катионов ме¬
таллов поступает в окружающую среду в качестве отходов электро¬
химических производств (кадмий, медь), и с выхлопными газами
автотранспорта (свинец). Отходы металлургической и атомной про¬
мышленности поставляют широкий спектр разнообразных металлов-
токсикантов. Поступление токсичных лигандов связано с повсе¬
местным употреблением различных органических химикатов, на¬
пример, пестицидов, многие из которых могут быть лигандами.
Токсичность катионов с1-металлов во многих случаях связана с
устойчивостью образуемых ими биокомплексов. Если устойчивость
комплексного соединения с таким катионом выше, чем прочность
биокомплекса с каким-либо биокатионом в составе организма, при
его поступлении идет вытеснение последнего из биокомплекса. Этот
эффект выражен уравнением:
ма + М2 ^ М21. + М1
В этом уравнении - биокомплекс, а М2 - ион с1-металла. Ес¬
ли комплекс Мг1- более прочен, чем биокомплекс, происходит
вытеснение биокатиона чужеродным ионом н накопление чуждого
организму комплекса М21.. В этом и состоит суть токсикоза в дан¬
ном случае. К примеру, из данных таблицы 10.1 следует, что катио¬
ны меди прочнее связываются со многими лигандами, чем катион
кобальта. То же справедливо и в отношении катиона никеля Сле¬
довательно, эти катионы, вытесняя катион кобальта (и не только
его, но и катионы марганца, железа и др.) из относительно неста¬
бильных биокомплексов с их участием и тем самым подменяя био¬
комплексы своими хелатами, оказывают токсическое действие
Антидотная терапия при токсикозах, вызванных действием ка¬
тионов тяжелых металлов, основана также на образовании ими
прочных комплексов со специальными лигандами Этот метод - хе-
латотерапия - будет рассмотрен позже (см. главу 35)
183
Глава И. ЭЛЕКТРОХИМИЧЕСКОЕ РАВНОВЕСИЕ.
ИОНОМЕТРИЯ
обыкновенная форма, в
которой выделяется химичес¬
кая энергия - есть динами¬
ческое электричество
Н Н Бекетов
11.1. Химическое равновесие
в окислительно-восстановительных реакциях
С окислительно-восстановительными реакциями связаны дыха¬
ние и обмен веществ, гниение и брожение, фотосинтез и нервная
деятельность живых организмов. Окислительно-восстановительные
процессы лежат в основе горения топлива, коррозии металлов,
электролиза, металлургии и т.д.
Реакции, протекающие с изменением степени окисления атомов,
входящих в состав реагирующих молекул, называются окислитель¬
но-восстановительными. Процессы окисления и восстановления
протекают одновременно: если один элемент, участвующий в реак¬
ции, окисляется, то другой должен восстанавливаться.
Окислитель - это вещество, содержащее элемент, который
принимает электроны и понижает степень окисления. Окислитель в
результате реакции восстанавливается. Так, в реакции:
2Ре*3С1"з + 2К*Г -> 12° + 2Ре*2С12-+ 2К+СГ
окислителем является ион Ре*3.
Ре*3 + 1е ^ Ре*2
окисленная восстановленная
форма форма
Восстановитель - вещество, содержащее элемент, который
отдает электроны и повышает степень окисления. Восстановитель
в результате реакции окисляется. Восстановителем в предлагаемой
реакции является ион I":
21" - 2е ^ 12°
восстановленная окисленная
форма форма
Приведенные отдельно для окислителя и восстановителя урав¬
нения называются полуреакциями. Окисленная (ох) и восстанов¬
184
ленная ^recij формы, участвующие в полуреакции, составляют так
называемую редокс-пару (от английских слов oxydation - окисление
и réduction - восстановление).
Таким образом, окислительно-восстановительные реакции явля¬
ются примером единства двух противоположных процессов - окис¬
ления и восстановления.
Соединения, содержащие атом какого-либо элемента в низшей
возможной степени окисления, могут быть только восстановителя¬
ми, поскольку они способны лишь отдавать электроны и повышать
свою степень окисления. К ним относятся, например, металлы,
галогенид-ионы, сульфиды и т.д.
Соединения какого-либо элемента с высшей возможной степе¬
нью окисления, напротив, могут быть только окислителями, так как
способны принимать электроны и понижать свою степень окисле¬
ния [например, перманганат калия, оксид xpoMa(VI), азотная кисло¬
та, концентрированная серная кислота, оксид свинца(1У), висмутат
натрия и т.д.].
Соединения с промежуточными степенями окисления элемен¬
тов имеют двойственный окислительно-восстановительный харак¬
тер. Таковы оксид углерода(П), сульфиты, нитриты и т.д.
Окислительно-восстановительные реакции делят на три группы:
а) межмолекулярные, в которых изменяют степени окисления
атомы разных молекул, например:
2K4[Fe*2(CN)6] + Cl2° -> 2K3[Fe*3(CN)6] + 2KCI'1
6С*402+ 6Н2О-2 -» C60Hi206 + 60°2
(фотосинтез в зеленых растениях)
2H2S-2 + С*402 -» С°Н20 + 2S0 + Н20
(фотосинтез у некоторых бактерий)
б) внутримолекулярные, в которых изменяют степени окис¬
ления атомы, входящие в состав одной молекулы, например
(N^hU^C^O/ -» N2° + Сг2*303 + 4Н20
в) диспропорционирование, в котором атом одного и того
же элемента одновременно и повышает, и понижает первоначаль¬
ную степень окисления, например-
2Н202-1 -» 2W2 + 02°
(эндогенное разложение пероксида водорода)
20^° 5 + 2Н* -* 02° + Н202'1
(эндогенное разложение супероксид-анион-радикала)
c60Hi2Oe -> гс^ИйОн + 2С+4о2
(спиртовое брожение глюкозы)
185
Как известно, в окислительно-восстановительной реакции окис¬
литель (ох 1) принимает п электронов, превращаясь в восстановлен¬
ную форму (red |), а восстановитель (гес^), отдает п электронов и
окисляется в окисленную форму (0x2):
а 0x1 + b reÖ2 ^ а redi + b 0x2.
Это можно выразить двумя полуреакциями:
0x1 + Ь е ±5 redi
reÖ2 - а е 25 ох2
Общее количество электронов п = ab.
Суммарной реакции отвечает константа равновесия:
_ [red1]a[ox2]b
Равн г ,а, j ,Ь '
[ox1]a[red2]D
В принципе две полуреакции можно пространственно разделить,
а электроны, отдаваемые восстановителем, будут переходить к
окислителю по проводнику. Для замыкания электрической цепи со¬
суды с окислителем и восстановителем соединяют солевым (или
электролитическим) мостиком (стеклянная трубка с насыщенным
раствором KCl) и получают гальванический элемент т.е. устрой¬
ство для превращения энергии химической реакции в электриче¬
ский ток. Принцип действия гальванического элемента рассматри¬
вается в разделе 11.6. Гальванический элемент можно получить
комбинацией любой пары полуреакций.
Изменение свободной энергии (AG0) в окислительно-восстано-
вительной реакции реализуется через выполняемую в гальвани¬
ческом элементе электрическую работу:
AGo = -nFEo,
где п - число обмениваемых в реакции электронов (наименьшее
общее кратное а и b), F - число Фарадея (96 500 кулонов), Е° -
электродвижущая сила гальванического элемента (ЭДС, в вольтах),
измеряемая в стандартных условиях. Так как AG0 = -RTlnKpaBH, то
переходя к десятичным логарифмам, имеем
I аК - ПРЕ°
g равн 2.303RT
Следовательно, в окислительно-восстановительных реакциях из¬
мерение электродвижущей силы гальванического элемента в стан¬
дартных условиях дает возможность быстрого и точного определе¬
ния константы химического равновесия.
Например, известно, что для гальванического элемента Якоби
(медно-цинковая пара) ЭДС в стандартных условиях (т.е. при
25° С, 1 атм. и одномолярных концентрациях катионов Си2+ и 2гг*)
составляет 1.1 В. Отсюда легко вычислить Кравн :
1еКр„„= 11965002 = 37 и Кр„и = 103',
2.3031.98 298
Такая величина Кравн и определяет тот хорошо известный факт,
что реакция гп + Си2+ *5 2п2* + Си практически нацело идет слева
направо.
11.2. Окислительно-восстановительные
потенциалы
Изменение свободной энергии (AG) в окислительно-восстано¬
вительных реакциях есть разница между свободными энергиями ее
полуреакций, для каждой из которых можно записать (см. главу 7):
Gox/fed = GV/rea + 2.303RTIgM
Переходя к электрохимическому преобразованию, получим-
фох/red = Ф'ох/г«. + 2 303fjr'gj^Ji "ли "Р" Т = 298 К
^ 0059, [ох]
Фох/red = ф ox/red "*■ “ JredJ'
где [ох] и [red] - концентрации окисленной и восстановленной форм
вещества, участвующего в процессе окисления-восстановления,
П - ЧИСЛО обменивающихся электронов, Фох/red - потенциал редокс-
пары, участвующей в полуреакции, <p°ox/red ~ стандартный элек¬
тродный потенциал, то есть величина потенциала при
[ох] = [red] = 1 моль/л и при стандартных условиях
Последнее выражение, известное как уравнение Нернста, ха¬
рактеризует величину потенциала окислительно-восстановительной
полуреакции.
Любую окислительно-восстановительную реакцию можно пред¬
ставить как сумму двух гипотетических полуреакций, а разность
потенциалов (ДЕ°) как алгебраическую сумму потенциалов полуре¬
акций (ф| - Ф2). Зная величины стандартных потенциалов (а их
187
значения имеются в таблицах), можно рассчитать величину ДЕ°, а
из нее - Кравн и тем самым определить направление окислительно¬
восстановительного превращения.
Возникновение окислительно-восстановительного потенциала ф
можно рассмотреть на примере металлического электрода Если
опустить металлическую пластинку в воду, то металл в ней хотя
бы частично растворяется (даже если растворимость ничтожно
мала) Так, хорошо известно, что пропускание воды через серебря¬
ный фильтр дезинфицирует ее и поэтому является одной из стадий
очистки воды. Это объясняется бактерицидными свойствами ионов
серебра, переходящих в раствор Растворение характерно для всех
металлов и обусловлено возрастанием энтропии.
При растворении в воду переходят катионы металла, и металли¬
ческая пластина заряжается отрицательно. В результате возникает
двойной электрический слой. Система, в которой металл кон¬
тактирует с раствором электролита, характеризующаяся наличием
двойного электрического слоя на их границе с некоторым потенциа¬
лом ф, называется электродом (или полуэлементом).
Если погрузить металлическую пластину в раствор соли этого
же металла, то могут реализоваться два механизма образования
двойного электрического слоя. Химически активные металлы
(например, цинк, алюминий) при погружении в раствор своей соли
любой (практически достижимой) концентрации частично преходят
в раствор, и пластины заряжаются отрицательно. Для малоак¬
тивных металлов (медь, серебро, золото) наблюдается обратная
картина: при любой концентрации раствора происходит осаждение
ионов металла на пластине, которая при этом заряжается положи¬
тельно. Описанные выше процессы являются обратимыми и проте¬
кают до момента достижения динамического равновесия.
Мп+ + пе ^ М°
ох-форма гей-форма
Образование двойного электрического слоя сопровождается
возникновением электродного потенциала, т.е. разности потен¬
циалов на границе “металл-раствор”, которая определяется приро¬
дой металла, концентрацией раствора и температурой. Поэтому для
сравнения электродных потенциалов были выбраны стандартные
условия 0 = 25° С или Т = 298 К, активная концентрация равна
1 моль/л, р = 1 атм.).
Абсолютное значение электродного потенциала измерить невоз¬
можно, поэтому измеряют разность потенциалов между данным
электродом,и электродом сравнения, потенциал которого условно
принимают равным нулю.
В качестве электрода сравнения используют стандартный водо¬
родный электрод. В Нем газообразный водород находится в равнове¬
сии с ионами водорода, и механизм возникновения электродного
потенциала аналогичен таковому для металлического электрода.
2Н+ + 2е £; Н2°
ох-форма гей-форма
Электродные потенциалы, измеренные по отношению к стан¬
дартному водородному электроду, называются стандартными.
Принято все окислительно-восстановительные полуреакции за¬
писывать в виде реакций восстановления, и только в этом случае
можно сравнивать значения электродных потенциалов.
Чем выше положительная величина электродного потенциала,
тем более сильным окислителем является ох-форма редокс-пары.
Увеличению отрицательной величины потенциала соответствует
увеличение восстановительной способности гес!-формы.
Полезно уметь заранее оценить, будет ли протекать та или иная
окислительно-восстановительная реакция. Существует следующее
правило: окисленная форма одной редокс-пары будет взаимодей¬
ствовать с восстановленной формой другой редокс-пары, если ре-
докс-потенциал последней имеет меньшее значение.
Разность между стандартными окислительно-восстановитель¬
ными потенциалами окислительной и восстановительной редокс-пар
называют электродвижущей силой реакции (ДЕ или ЭДС). Ес¬
ли ЭДС, вычисленная путем вычитания из окислительного потен¬
циала редокс-пары, используемой в данной реакции в качестве
окислителя, потенциала редокс-пары, используемой в качестве вос¬
становителя, будет иметь положительное значение (ЭДС > 0), то
данная реакция осуществима. Если же вычисленная таким образом
ЭДС окажется отрицательной (ЭДС < 0), то предполагаемая реак¬
ция не пойдет; принципиально возможной будет обратная реакция,
для которой ЭДС будет иметь то же самое абсолютное значение, но
с обратным знаком.
11.3. Классификация электродов
Электроды можно разделить на 3 типа- 1) первого рода, обра¬
тимые только по отношению к катиону; 2) второго рода, обратимые
по отношению к аниону и катиону; 3) окислительно-восстано¬
вительные.
На металлических электродах, погруженных в раствор соли то¬
го же металла, идет процесс перехода катиона из металла в рас¬
189
твор или в противоположном направлении - в зависимости от знака
ЭДС цепи, в которую включен электрод. Эти электроды обратимы
относительно катиона и называются электродами первого рода,
к которым относится и водородный электрод.
К электродам второго рода относятся электроды, в которых
металл покрыт слоем его малорастворимой соли и находится в рас¬
творе, насыщенном этой солью и содержащем другую, легко рас¬
творимую соль с тем же анионом (например, Ag + AgCI + KCl).
Электроды этого типа обратимы относительно концентрации аниона
(СГ) и катиона (Ад+).
Существует группа окислительно-восстановительных электродов
(иначе редокс-электроды в узком смысле этого слова, так как
строго говоря, все электроды являются окислительно-восстано¬
вительными), т.е. электродов, потенциал которых возникает между
пластиной из благородного металла и раствором, содержащим окис¬
ленную и восстановленную формы какого-либо элемента Для них
присоединение или отдача электронов связаны с изменением степе¬
ней окисления веществ, находящихся в растворе. Сам электрод
(обычно это платиновая пластина) не принимает участия в элек¬
тродном процессе, а лишь служит переносчиком электронов. Так,
например, на платиновой пластине, опущенной в раствор, содер¬
жащий ионы Fe3+ (ох-форма) и Fe2+ (red-форма), возникает потен¬
циал, величина которого зависит от температуры и соотношения
активных концентраций ионов железа.
Смысл классификации электродов будет более понятным после
ознакомления с конкретными типами электродов.
11.3.1. Металлический электрод
Металлический электрод является электродом первого рода и
представляет собой металл, погруженный в раствор его соли. Урав¬
нение Нернста для металлического электрода приобретает следую¬
щий вид (при 25° С):
о 0.059, амп+
ф = ф „* + 1в—— (в).
мп+/м мп+/м п ам
Принято, что активность любого чистого кристаллического ве¬
щества равна единице (ам = 1), и, таким образом, она устраняется
из формулы. Это вполне согласуется с тем, что скорость любой хи¬
мической реакции не зависит от концентрации твердого вещества в
объеме фаз>1, а может зависеть лишь от площади поверхности
В данном случае, чем больше поверхность металла, тем больше
190
скорость ухода ионов в раствор и скорость их обратного перехода в
кристаллическую решетку металла. Таким образом,
о ^ 0.059, п+
+ ~''®а"'
В очень разбавленном растворе вместо активной концентрации
можно подставить молярную концентрацию:
п 0059
п* = <Р + ^ 1-
В этой формуле Ф°мп+/м - стандартный потенциал металличе¬
ского электрода при активности катионов, равной 1 моль/л. Он
зависит от температуры и природы металла.
Значения стандартных электродных потенциалов некоторых ме¬
таллов представлены в табл. 11.1. По мере роста величины потен¬
циала металлического электрода падает восстановительная актив¬
ность металла. Эта зависимость известна как ряд напряжений ме¬
таллов, впервые сформулированный Н. Н. Бекетовым.
Таблица 11.1
Ряд напряжений металлов
м/мп+
Ф°, В
M/Mn+
Ф°, В
Li/Li*
-3.00
Ni/Ni2*
-0 25
к/к+
-2.92
Sn/Sn2+
-0 14
Na/Na+
-2 71
Pb/Pb2+
-0 13
Са/Са2+
-2 37
Cu/Cu2+
+0.34
Mg/Mg2+
-2 36
Ag/Ag+
+0 80
Mn/Mn2*
-1 18
Hg/Hg2+
+0 85
Zn/Zn2+
-0 76
Pt/Pt2*
+ 1 20
Fe/Fe2+
-0 44
Au/Au*
+ 1 70
Потенциал металлического электрода зависит только от кон¬
центрации катионов металла и не зависит от концентрации анио¬
нов Следовательно, металлический электрод относится к электро¬
дам первого рода
191
11.3.2. Водородный электрод
К числу электродов первого рода принадлежит и водородный
электрод, стандартный потенциал которого условно принят за ноль
Водородный электрод представляет собой пластинку из инерт¬
ного металла (платины), погруженную в раствор, содержащий ионы
водорода, например, в раствор
НгБО« (рис. 11.1) Предварительно
электролизом раствора хлорида
платины на пластинку наносится
слой платиновой черни. Такая пла¬
тинированная пластинка обладает
большой активной поверхностью и
способна поглощать большое коли¬
чество водорода. Через раствор
пропускают ток чистого газообраз¬
ного водорода при постоянном
давлении (1 атм).
Газообразный водород адсорби¬
руется на платине и затем, распа¬
даясь на атомы, переходит в рас¬
твор в виде ионов. Таким образом,
между атомами водорода на плати¬
не и ионами водорода в растворе в
водородном электроде устанавли¬
вается равновесие:
Нг(газ) ^ 2Н ^ 2Н+ + 2е.
Схематически водородный электрод записывается так:
Н2{И} I н\
Очевидно, что он аналогичен металлическому электроду.
Применяя уравнение Нернста при 25° С для водородного элек¬
трода, получим:
| 0.059 аУ
2 & аН2 '
равна его давлению а = р , тогда при
Рис. 11.1. Водородный
электрод.
2Н+/Н2 '
атмосферном давлении (р„ = 1 атм.)
”2
0 059 , 2
и, = <р°
192
Но ’•'гн*/«, = °- тогда 2н*/н2 = 0059|еа„*
и так как -1§^ан+ = pH, то
<Р2н*,„г=-0 059рН<В)
Таким образом, потенциал водородного электрода зависит от ак¬
тивности ионов водорода и, следовательно, его можно применять
для измерения pH. При отсутствии посторонних окислителей и
восстановителей водородный электрод является самым точным
электродом для измерения кислотности среды в широком интерва¬
ле pH - от сильнокислой до сильнощелочной среды.
Посторонними окислителями могут быть многие органические
вещества, способные восстанавливаться водородом в присутствии
платины. Платина легко отравляется галогенами, соединениями се¬
ры, ртути и мышьяка, которые уменьшают ее способность погло¬
щать водород. Установка для измерения pH с водородным электро¬
дом включает электролизер для получения водорода и систему
поглотительных склянок для тщательной очистки водорода от дру¬
гих газов и потому является громоздкой. Кроме того, перед изме¬
рением pH требуется длительное насыщение раствора водородом.
Вместо водородного в качестве электродов сравнения использу¬
ют каломельный и хлорсеребряный электроды (см. ниже).
11.3.3. Каломельный электрод
К электродам второго рода, как уже отмечено, принадлежат
электроды, обратимые относительно катиона и аниона. В электро¬
дах второго рода металл погружен в раствор своей труднораство¬
римой соли и хорошо растворимого электролита с одноименным
анионом.
Так, каломельный электрод состоит из металлической ртути, ко¬
торая находится на дне сосуда, а сверху покрыта пастой из каломе¬
ли Нд2С12 (рис 11 2) Сосуд наполняется раствором КС1 определен¬
ной концентрации, насыщенным Нд2С12
Схема записи- НдЫдгОг, КС1.
На каломельном электроде устанавливаются равновесия
2Нд ^ Нд22* + 2е
Нд22* + 2СГ ^ Нд2С121
7 Зак 675
193
«|— Насыщенный
раствор КС1
Ж
— Нд + Нд2С12
Рис. 11.2. Каломельный
электрод.
Рассматривая этот электрод как
обратимый относительно катиона,
применим к нему уравнение Нернста
для металлического ртутного электро¬
да (при 25° С).
0059 ,
Ф 2* =Ф^ 2+ + \е а ,
Н®2 /2Нд Н®2 /2Нд 2 Н«| +
Активная концентрация ионов
Нд22+ в растворе, возникающая за счет
незначительной растворимости кало¬
мели, связана с активной концентра¬
цией ионов СГ:
ПРн,
Отсюда Знд2* =
Подстановка величины а н^+ в формулу приводит к выражению:
Фи- 2+ =Ф НП2+ ■*
Н02 /2Нд /2Нд
0 059 , Нд2С12
—
фНа2 + = Ф°на 2 +
"32 /2Нд /2Нд
Как видно нз формул, потенциал каломельного электрода дей¬
ствительно зависит и от концентрации катиона Ндг2+, и от концент¬
рации аниона СГ. Вообще, если металлический электрод находится
в насыщенном растворе своей соли, то его потенциал определяется
концентрацией катионов в растворе, но изменение концентра¬
ции анионов влияет на концентрацию катионов и в результате вы¬
зывает изменение потенциала электрода. Сумма постоянных вели-
0059 ,
чин <р° 2+ + 1£ПРНд С| является постоянной.
Нд2 /2Нд 2
Обозначая ее ф0«^, получим: ф^,*, = ф0клм - 0.059^ асГ (В)
* О произведении растворимости (ПР) см. главу 12.
194
Таблица 11.2
Потенциалы каломельного
электрода с различной
концентрацией КС1
Суммарная концентрация ионов
хлора в растворе практически равна
концентрации раствора KCI в
каломельном электроде. Потенциа¬
лы каломельного электрода с раз¬
личной концентрацией KCI приведе¬
ны в табл. 11.2.
Избыток KCI гарантирует, что
раствор остается насыщенным при
значительных изменениях темпера¬
туры. Наиболее удобен и устойчив,
а потому чаще применяется на практике насыщенный каломельный
электрод, содержащий насыщенный раствор хлорида калия.
CKCIf
Ф„м, В
моль/л
при 25 °С
0 1
0.336
1.0
0.282
Насыщ.
0.250
U.3.4. Хлорсеребряный электрод
\
/
Другим электродом второго рода является хлорсеребряный
электрод, простой в изготовлении и дающий хорошо воспроизводи¬
мую величину потенциала.
Он применяется как внутренний вспомогательный электрод в
стеклянных электродах (см. 11.10), а также в качестве внешнего
стандартного электрода сравнения при
измерении pH.
Хлорсеребряный электрод обычно
получают осаждением серебра на
платиновую проволоку с последующим
переведением поверхностного слоя
осажденного серебра в хлорид серебра
электролизом в растворе хлорида. Эта
проволока погружена в раствор хорошо
растворимого электролита, содержа¬
щего ионы СГ (например, раствор KCI,
насыщенный AgCl).
Схема электрода Ag | AgCl, KCI
На рис 11 3 приведена одна из кон¬
струкций хлорсеребряного электрода
Этот электрод состоит из стеклянного
корпуса 1 с впаянной асбестовой нитью
2, по которой просачивается раствор
хлорида калия (насыщ ), создавая элек¬
тролитический ключ Внутри электрода
Г
Рис 11.3 Хлорсеребряный
электрод.
7*
195
находятся серебряная проволока 5 и паста из хлорида серебра 4.
Корпус полностью заполняется насыщенным раствором хлорида ка¬
лия 3, контакт которого с парой Ад|АдС1 осуществляется при по¬
мощи асбестового фитиля 6 На электроде происходит реакция:
Ад ^ Ад+ + е
Ад+ + СГ и АдС11.
Потенциал хлорсеребряного электрода, выведенный из уравне¬
ния Нернста так же, как и для каломельного, имеет вид:
Фхс = Ф°хс - 0.0591£ асГ (В).
II.3.5. Сурьмяный электрод
Сурьмяный электрод является одним нз наиболее употребимых
металлоксидных электродов, которые обычно относятся к электро¬
дам второго рода. Формальное сходство сурьмяного электрода с
характерными электродами второго рода заключается в том, что
здесь металл (БЬ) погружен в концентрированный раствор трудно¬
растворимого оксида ёЬгОз, тогда как в рассмотренных выше элек¬
тродах второго рода - каломельном и хлорсеребряном - металл по¬
гружен в насыщенный раствор своей труднорастворимой соли.
Для бруска сурьмы, отлитого на воздухе, электрохимический
процесс протекает с участием сурьмы и тонкого слоя ее мало¬
растворимого оксида на поверхности металла:
БЬгОз + 6Н+ + бе ±5 2БЬ + ЗН20
Сурьма и ее оксид находятся в кристаллическом состоянии, и,
следовательно, их активности не изменяются. Активная концентра¬
ция воды в разбавленных растворах также приблизительно посто¬
янна. Поэтому потенциал электрода зависит только от активной
концентрации ионов водорода, и, следовательно, сурьмяный элек¬
трод можно использовать для измерения pH:
0059 л
Фсур = ф°сур + ^ 1еан+ = Ф°СУР + 0059’1баН+;
Фсур =ф°сур - 0.059рН.
Потенциал сурьмяного электрода зависит от состояния поверх¬
ности металла. Кроме того, на его величину влияют окислители и
восстановители. Поэтому с его помощью измеряют pH в тех случа¬
196
ях, когда не требуется высокая точность. Однако сурьмяный элек¬
трод имеет очень простое устройство, легко может быть выполнен в
виде электрода очень малых размеров, и потенциал его устанавли¬
вается очень быстро.
Сурьмяный электрод нашел, в частности, применение при изу¬
чении изменений pH в желудке в процессе пищеварения. Он пред¬
ставляет собой брусок сурьмы длиной 5 мм и диаметром 1 мм с
трехжильным медным проводом, который легко можно проглотить.
Вспомогательный каломельный электрод можно включать в цепь,
например, с помощью солевого раствора, в который помещены ноги
пациента.
11.3.6. Хингидронный электрод
Хингидронный электрод является представителем окислительно¬
восстановительных электродов.
Как известно, многие окислительно-восстановительные реакции
в водных растворах протекают с участием нонов водорода. В этом
случае потенциал редокс-электрода зависит от концентрации ионов
Н+ Такие электроды в принципе можно использовать для измере¬
ния pH. Из них наибольшее практическое применение нашел хин¬
гидронный электрод.
Для приготовления этого электрода исследуемый раствор взбал¬
тывают со щепоткой хингидрона (труднорастворимого темно¬
зеленого порошка) и таким образом получают его насыщенный рас¬
твор Хингидрон является молекулярным соединением хинона и
гидрохинона и при растворении в воде частично диссоциирует на
хинон и гидрохинон:
О • • • НО О но
«’-'М
хингидрон (ХГ) хинон (X) гидрохинон (ГХ)
В полученный раствор погружают платиновую пластинку Хи¬
нон и гидрохинон участвуют в окислительно-восстановительном
равновесии, от положения которого зависит потенциал платиновой
пластинки-
197
Здесь окисленной формой является хинон и ионы Н+, а восста¬
новленной формой - гидрохинон.
Схема электрода: 1 X, ГХ, Н+.
Применение формулы Нернста для этой окислительно-восстано¬
вительной реакции позволяет получить потенциал хингидронного
электрода:
п , 0.059, ах а2н+
Фхг = Ф°хг + ~1е
Концентрации хинона и гидрохинона в растворе равны, так как
они образуются в результате диссоциации хингидрона. Поэтому
Фхг = ф°хг + 0.059 ^ ан+,
где ф°хг - величина стандартного потенциала хингидронного элек¬
трода при ан+ = 1, равная 0.699 В. Отсюда
фхг = 0.699 - 0.059рН (В)
Хингидронный электрод удобен в работе и позволяет определять
pH кислых и слабощелочных растворов с достаточной точностью.
В щелочных растворах (при pH > 8) он не дает точных показаний
из-за диссоциации гидрохинона, в результате чего концентрация
гидрохинона меняется:
НО
1 - С Л*н*
Присутствие в растворах окислителей и восстановителей влияет
на потенциал хингидронного электрода. Кроме того, при измере¬
нии pH в исследуемый раствор приходится вносить постороннее
вещество -хингидрон.
Поэтому хингидронный электрод в отличие от более совершен¬
ного и универсального стеклянного электрода (см. 11.10) нельзя
применять для измерения pH многих биологических жидкостей.
Некоторые из звеньев окислительно-восстановительной дыха¬
тельной цепи (см. 11.7), например кофермент КоО, по механизму
своего действия близки к рассмотренной окислительно-восстанови-
тельной системе.
где Я - длинный разветвленный радикал.
Окисленная форма этого кофермента, присоединяя электроны,
превращается в соответствующий гидрохинон, а затем снова окис¬
ляется в хинон. Вообще хинонные и гидрохинонные структуры ши¬
роко распространены в живой клетке: их имеют, например, такие
важные для жизнедеятельности вещества, как витамины Е и К.
11.3.7. рН-Зависимые электродные потенциалы
Полуреакция, происходящая на хингидронном электроде, проте¬
кает с участием ионов гидроксония. Существует большая группа
полуреакций, в которых участвуют протоны или гидроксильные
ионы В таких случаях концентрация этих ионов входит в уравне¬
ние Нернста и pH раствора влияет на потенциал полуреакции.
Например, потенциал редокс-пары
Мп04~ + 81-Г + 5е 5 Мп2+ + 4Н20
будет вычисляться по уравнению:
, 0059, [Мп04 ][Н+]в
Ф + =<Р + + •б 51 •
Мп04~ + ВН+ Мп04~ +ВН 5 [Мп ]
Мп2 + Мп2+
Концентрация воды является величиной постоянной и потому ее
значение учитывается в постоянной величине стандартного потен¬
циала
В общем виде зависимость окислительно-восстановительного
потенциала от pH для уравнения
ох + тН+ + пе ^ гес!
можно записать так.
Фох/гес! = Ф°ох/гес1 + —1п
ИТ 1п [ох] [н+г
пР [гес1]
откуда
Из этого уравнения следует, что по мере подкисления раствора
окислительно-восстановительный потенциал будет увеличиваться.
Величина сдвига ф0Х/Гес1 ПРИ изменении концентрации ионов во¬
дорода зависит от числа электронов п и протонов т, участвующих в
реакции, т.е. от отношения т/п Такую разновидность окислитель¬
но-восстановительных полуреакций называют рН-зависимыми.
рН-Зависимые полуреакции (и не только они) нашлн широкое
применение в клинической диагностике, являясь основой аналити¬
ческих редокс-методов, прежде всего редоксиметрии - метода
объемного анализа, основанного на применении окислительно-вос¬
становительных реакций В зависимости от примененяемого титран-
та различают перманганатометрию, бихроматометрию, иодометрию,
цериметрию и др.
Перманганатометрия - метод, базирующийся на применении
в качестве титранта-окислителя перманганата калия в сильно кис¬
лой среде. В связи с высоким значением стандартного потенциала
менять для определения широкого набора веществ, способных
окисляться - сульфит-, сульфид-, нитрит-, арсенит-анионов, катио¬
на Ре2+, гидразина, ряда органических кислот (лимонной, винной,
яблочной, щавелевой, аскорбиновой и др.).
В гигиенической практике его используют для нахождения важ¬
ной характеристики воды - ее окисляемости, которая определяет¬
ся количеством миллиграммов перманганата калия, идущих на
окисление восстановителей, содержащихся в литре воды. Окисляе-
мость обусловлена присутствием гуминовых веществ, сероводоро¬
да, солей двухвалентного железа и др. примесей, в основном быто¬
вых или промышленных. Загрязненные воды имеют окисляемость
до 400 мг на литр воды, а незагрязненные - не более 4 мг на литр.
Наименее загрязнены артезианские воды (окисляемость менее 2 мг
на литр).
перманганата калия (ф°
Мп04' + 8Н+/Мп2+
= +1.51 В) его можно при-
200
Бихроматометрия основана на высокой величине редокс-
потенциала бихромат-иона в сильно кислой среде. Значение <р° для
полуреакции
Сг2072' + 14Н* + 6е ^ 2Сг3+ + 7Н20
составляет +1.33 В. Бихроматометрию используют для определения
многих восстановителей, а в гигиенической практике она нашла
применение для определения химического потребления кисло¬
рода - оценки содержания всех органических веществ, которые
растворены в воде. Они при кипячении в избытке бихромата калия
в присутствии серной кислоты окисляются до углекислоты.
11.4. Диффузионный потенциал.
Электролитический мостик
Чтобы гальванический элемент был замкнут, как уже упомина¬
лось, нужно соединить растворы электролитов электролитическим
мостиком. При выборе электролита для заполнения этого мостика
необходимо учитывать возможность возникновения диффузионного
потенциала.
В гальванических элементах имеет место соприкосновение рас¬
творов разных по составу или с разной концентрацией электроли¬
тов. На границе двух таких растворов обычно возникает разность
потенциалов.
Пусть, например, в сосуд с соляной кислотой осторожно, так,
чтобы растворы не перемешивались, приливают соляную кислоту
меньшей концентрации. В этом случае ионы Н+ и СГ должны диф¬
фундировать из нижнего раствора в верхний Однако эксперимен¬
тально установлено, что подвижность ионов Н+ почти в 5 раз
больше, чем ионов СГ; протоны малы по размерам и двигаются зна¬
чительно быстрее Поэтому в единицу времени в верхний раствор
будет переходить больше ионов Н+, чем ионов СГ В результате
верхний раствор приобретет положительный, а нижний - отрица¬
тельный заряд.
В общем, более разбавленный раствор всегда приобретает заряд
того иона, который движется с большей скоростью. Вследствие та¬
кого разделения зарядов на границе двух растворов возникает раз¬
ность потенциалов.
Разность потенциалов на границе соприкосновения двух раство¬
ров электролитов различной концентрации или различного состава,
обусловленная различными подвижностями ионов, называется
диффузионным потенциалом.
201
Хотя величина диффузионного потенциала не превышает не¬
скольких десятков милливольт, иногда он может заметно изменять
величину ЭДС гальванического элемента Для уменьшения ошибки
при измерении ЭДС элемента обычно стремятся уменьшить диффу¬
зионный потенциал Наиболее удобным способом является приме¬
нение солевого мостика из KCI, который соединяет растворы раз¬
личных электролитов Хлорид калия (обычно насыщенный раствор)
используется потому, что скорости движения ионов К+ и СГ почти
одинаковы. Обычно солевой мостик заполняют агар-агаром, насы¬
щенным KCI. Агар-агар - это студнеобразное вещество, предохра¬
няющее мостик от вытекания раствора электролита.
11.5. Мембранный потенциал
Если мембрана, проницаемая, например, только для ионов М+,
разделяет два раствора МХ различной концентрации, то часть ионов
М+ будет диффундировать через эту мембрану из раствора с боль¬
шей концентрацией (аг) в разбавленный раствор (ах). В результате
в первом растворе возникает некоторый избыток отрицательных
зарядов, а во втором - положительных. Таким образом, возникла
электрохимическая разность потенциалов между растворами, раз¬
деленными мембраной, которую называют мембранным потен¬
циалом (фмембр)- Его величина рассчитывается для идеальной
мембраны (селективной только по отношению к ионам М+) по фор¬
муле Нернста:
2.31ГГ, а2
Фмембр = =г!в—.
Vе а'
где - заряд диффундирующего иона в единицах заряда протона,
2.3ИТ
отношение называют угловым коэффициентом или крутиз-
V р
ной электродной функции (Б). При Т= 298 К и г = 1, Б = 0.059 В.
11.6. Типы гальванических элементов
В гальваническом элементе происходит окислительно-восстано-
вительная реакция, которая протекает раздельно на двух электро¬
дах: на одном - процесс окисления, в результате которого здесь
накапливаются электроны, а на другом - восстановления (элек¬
троны расходуются).
202
Примером гальванического элемента служит известный эле¬
мент Даниэля-Якоби (рис. 11.4).
Рис. 11.4. Схема гальванического элемента
(элемент Даниэля -Якоби).
Он состоит из цинкового и медного электродов. Цинковую плас¬
тинку погружают в раствор гпвО,», а медную - в раствор Си804.
Для каждого из электродов существует равновесие:
гп ±5 2п2* + 2е
Си ^ Си2+ + 2е
Однако цинк - более активный металл и легче, чем медь, посы¬
лает свои ионы в раствор Поэтому на цинковой пластинке накап¬
ливается больше избыточных электронов, чем на медной. Если обе
пластинки соединить проволокой, то электроны будут переходить по
ней с цинка на медь. В результате этого первое равновесие должно
сместиться вправо, то есть на цинковом электроде будет происхо¬
дить прямая реакция - переход ионов цинка в раствор-
гп->гп2+ +2е.
С другой стороны, переход электронов на медную пластинку
должен смещать второе равновесие влево Таким образом, на мед¬
ном электроде будет происходить только обратная реакция - пере¬
ход ионов меди из раствора на медную пластинку.
Си2* + 2е -» Си.
Казалось бы, такой гальванический элемент должен работать до
тех пор, пока вся цинковая пластинка не растворится или все
203
ионы меди из раствора не перейдут на медную пластинку Однако
работа элемента прекратится раньше. В самом деле, до соединения
двух пластинок проволокой оба раствора - ZnSC>4 и C11SO4 - были
электронейтральны, так как в них концентрация катионов была
равна концентрации анионов. В результате работы элемента соот¬
ношения изменятся [Zn2+] > (SO42'] и (Cu2+] < [SO42"], раствор у
цинкового электрода приобретает положительный заряд, а у медно¬
го электрода - отрицательный. Очевидно, это должно препятство¬
вать перетеканию электронов от цинка к меди. Поэтому оба раство¬
ра необходимо соединить электролитическим мостиком - раствором
KCI, по которому избыточные ионы SO42- будут переходить от мед¬
ного электрода к цинковому. В результате заряды растворов будут
выравниваться, и теперь элемент будет работать, пока не израсхо¬
дуются составляющие вещества. Схему гальванического элемента
представляют следующим образом
Zn | ZnS041 KCl | CuS041 Cu.
В такой схеме обычно сначала записывается электрод с более
отрицательным потенциалом, далее раствор, в который он погру¬
жен, электролитический мостик, затем состав второго раствора и,
наконец, электрод с более положительным потенциалом. Границы
раздела фаз обозначают вертикальными черточками. Электрод, на
котором происходит реакция окисления, называют анодом, а элек¬
трод, на котором происходит реакция зосстановления - катодом.
Так, цинковый электрод является анодом, а медный электрод -
катодом.
Количественной характеристикой электрохимического элемента
(или цепи элементов) является электродвижущая сила (ЭДС или
Е), которая равна разности потенциалов между двумя электродами.
При вычислении разности потенциалов вычитание производят из
потенциала более положительного электрода. Так, для элемента
Даниэля-Якоби:
Е = ф+Си - <p"zn.
При активных концентрациях ионов цинка и меди в растворах,
равных единице, ЭДС составляет 1.1 В. Электрическая работа,
которую можно получить от элемента при вытеснении цинком
1 моля атомов меди, равна А = nFE = 2-96500-1.1 = 212300 Дж.
Гальванические элементы можно разделить на три типа: хими¬
ческие, концентрационные и топливные. Химическими называются
такие гальванические элементы, у которых ЭДС возникает вследст¬
вие различней химической природы электродов. Примером может
служить описанный элемент Даниэля-Якоби.
204
Гальванические элементы могут быть составлены и из двух оди¬
наковых электродов, погруженных в раствор одного и того же элек¬
тролита с различной активной концентрацией ионов. Такие эле¬
менты называются концентрационными. Например, составим
элемент из двух медных пластинок, погруженных в растворы с раз¬
ной активностью ионов меди (а| и ag). Эти растворы соединены со¬
левым мостиком из KCI:
(_)Cu | C11SO41 KCl | CuS041 Cu<+>
aI а2
В данной цепи второй электрод погружен в раствор с более вы¬
сокой концентрацией ионов меди.
По уравнению Нернста потенциалы этих металлических элек¬
тродов равны:
. « 0.059,
V*'C.+—lg\u-'
, „ 0.059,
’о"** +~lga2cu- '
Так как а2Сиг+ > ai Си2+ • второй электрод более положителен.
Действительно, чем больше в растворе концентрация ионов
Си2+, тем больше вероятность их перехода в кристаллическую ре¬
шетку металла и тем положительнее должен быть потенциал ме¬
таллического электрода. Таким образом, между двумя медными
электродами возникает разность потенциалов
Е = ф 2 - ф 1
и, если соединить их проводником, а оба раствора - электролитиче¬
ским мостиком, то в цепи пойдет электрический ток Для каждого
электрода окислительно-восстановительная реакция выражается
уравнением- Си ^ Си2* + 2е Очевидно, что в результате перехода
электронов от первой медной пластинки ко второй, на первом
электроде будет происходить реакция растворения меди-
Си -> Си2+ + 2е, а на втором - осаждение меди- Си2++ 2е -» Си
Элемент будет работать до тех пор, пока первая пластинка не
растворится полностью или пока концентрации растворов не вырав¬
няются. Таким образом, источником энергии концентрационного
гальванического элемента являются диффузионные осмотические
силы, стремящиеся к выравниванию концентраций.
205
Топливными элементами называются такие гальванические
элементы, с помощью которых энергию, выделяющуюся при реак¬
ции окисления топлива (горючего), получают непосредственно в ви¬
де электрического тока. В топливных элементах те же реакции, что
и при сжигании топлива, идут гораздо медленнее, т.е. в значи¬
тельной степени обратимо В результате большее количество хими¬
ческой энергии превращается в работу.
Примером топливного элемента может служить пропан-кисло-
родный элемент Он состоит из раствора электролита, например,
серной кислоты или гидроксида натрия, и двух инертных электро¬
дов (например, платиновых). Пропан и кислород пропускают, соот¬
ветственно, через анодное и катодное пространства, где протекают
следующие реакции:
на аноде: СзНе + 6Н2О - 20е —» ЗСОг + 20Н+
на катоде: 50г + 20Н+ + 20е -» ЮНгО.
Суммарная реакция
СзНе + 502 -> ЗС02 + 4Н20
идентична реакции горения пропана в кислороде. Между электро¬
дами устанавливается разность потенциалов, а по проволоке, сое¬
диняющей электроды, течет ток от анода к катоду. Роль электродов
здесь двояка. Во-первых, анод служит источником электронов, а
катод - их ловушкой. Во-вторых, на поверхности электродов проис¬
ходит разложение молекул пропана и кислорода на атомы, т.е.
электроды здесь действуют как электрокатализатор.
При надлежащей конструкции удается достигнуть к.п.д. такого
элемента до 70%, что вдвое превышает к.п д. двигателя внутрен¬
него сгорания. Кроме того, топливные элементы в отличие от
обычных источников энергии не создают шума и вибрации, не обра¬
зуют выхлопных газов. Все перечисленные преимущества топ¬
ливных элементов создают широкие перспективы для внедрения их
в технику.
11.7. Окислительно-восстановительные реакции
в биологических процессах
Различные процессы жизнедеятельности сопровождаются воз¬
никновением в организме электрохимических процессов, играющих
существенную роль в обмене веществ. Электрохимические превра¬
щения в организме можно разделить на две основные группы:
206
1) процессы, связанные с переносом электронов и возникнове¬
нием окислительно-восстановительных потенциалов;
2) процессы, связанные с переносом ионов (без изменения их
зарядов) и с образованием биоэлектрических потенциалов (см. 11.4
и 11.5).
В результате этих процессов возникают разности потенциалов
между разными прослойками тканей, находящихся в различных
физиологических состояниях. Они связаны с различной интенсив¬
ностью окислительно-восстановительных биохимических процессов.
К ним относятся, например, потенциалы фотосинтеза, возникающие
между освещенными и неосвещенными участками листа, причем
освещенный участок оказывается положительно заряженным по от¬
ношению к неосвещенному.
Особенностью биологических окислительно-восстановительных
процессов первой группы является их многостадийность. Они про¬
ходят через ряд промежуточных стадий с образованием множества
кислородсодержащих продуктов (спирты, альдегиды, кетоны, карбо¬
новые кислоты), которые в конце концов окисляются до конечных
продуктов - СОг и НгО.
С помощью ступенчатого механизма организм предотвращает
нежелательные для него сильные метаболические отклонения, ко¬
торые происходят постепенно. Отдельные стадии биологического
окисления обратимы, что обеспечивает поддержание окислительно¬
восстановительного гомеостаза в организме.
11.7.1. Процессы с переносом электронов
Окислительно-восстановительные процессы первой группы в ор¬
ганизме можно разделить на три типа:
1. Непосредственный перенос электронов между веществами
без участия атомов кислорода и водорода, например, перенос элек¬
трона в цитохромах:
цитохром (Ре3+) + е ^ цитохром (Ре2+)
и перенос электрона в ферменте цитохромоксидазе:
цитохромоксидаза (Си2*) + е ^ цитохромоксидаза (Си1+).
2 Окислительный, связанный с участием атомов кислорода и
ферментов оксидаз, например, окисление альдегидной группы суб¬
страта в кислотную:
ЯСОН + О ^ ЯСООН.
207
3. рН-Зависимый, происходящий в присутствии ферментов
дегидрогеназ (Е) и коферментов (Ко), которые образуют активиро¬
ванный комплекс фермент-кофермент-субстрат (Е-Ко-Б), присоеди¬
няет электроны и катионы водорода от субстрата и вызывает его
окисление
Такими коферментами являются никотинамид-аденин-нуклеотид
(НАД+), который присоединяет два электрона и один протон.
Б-2Н - 2е + НАД* 5 Б + НАДН + Н*.
флавин-аденин-динуклеотид (ФАД), который присоединяет два элек¬
трона и два протона-
Б-2Н - 2е + ФАД 5 Б + ФАДН2,
и убихинон или кофермент О (КоО), который также присоединяет
два электрона и два протона:
Б-2Н - 2е + КоО ^ Б + КоОН2.
Все типы окислительно-восстановительных процессов происхо¬
дят при окислении субстратов в митохондриях, на внутренних мем¬
бранах которых размещаются ансамбли из ферментов-дегидрогеназ,
коферментов (НАД*, ФАД и КоО) и серии цитохромов Ь, С], с и а,
а также фермента - цитохромоксидазы. Они образуют систему кле¬
точной дыхательной цепи, с помощью которой происходит эстафет¬
ная передача электронов и катионов водорода от субстрата молеку¬
лам кислорода, доставленным гемоглобином к клетке.
Каждый компонент клеточной дыхательной цепи - участник
окислительно-восстановительного процесса - характеризуется оп¬
ределенным значением окислительно-восстановительного потенциа¬
ла (табл. 11.3).
Движение электронов в клеточной дыхательной цепи происхо¬
дит ступенчато от веществ с низким потенциалом (-0.324 В) к ве¬
ществам с более высоким потенциалом (+0.82 В).
В сответствии с этим биосубстрат (в первую очередь, глюкоза)
окисляется по следующей схеме с участием двух типов биологиче¬
ских окислительно-восстановительных процессов - с прямой пере¬
дачей электронов в дыхательной цепи цитохромов и посредством
рН-зависимых превращений с участием кофакторов.
Сначала происходит передача протона и пары электронов моле¬
куле НАД+, превращающейся в восстановленную форму НАДН За¬
тем приходит очередь превращения ФАД в ФАДНг- Далее происходит
восстановление КоО в КоСЖг-
Затем осуществляется процесс передачи электрона по цепи ци¬
тохромов в согласовании с величинами их потенциалов, представ¬
208
ленными в таблице 11.3. Последний из компонентов - цитохромок-
сидаза переносит электроны непосредственно молекуле кислорода.
1/202 + 2Н* + 2е 5 Н20.
Таблица 11.3
Стандартные редокс-потенциалы биомолекул
дыхательной цепи
Система
Полуреакция
Ф\ в*
над+/надн2
НАД* + Н*
+ 2е ^ НАДНг
-0.32
ФАД/ФАДНг
ФАД + 2Н*
+ 2е ^ ФАДН2
-0 30
КоО/КоОН2
КоО+ 2И*
+ 2е ±5 КоОН2
-0.04
цитохром Ь
+0 07
цитохром С!
Ре3н
^ 25 Ре2*
+0 23
цитохром с
+0 25
цитохромоксидаза
Си2*
ь е ±5 Си1*
+0 55
о2/н2о
02 + 4Н*
+ 4е ^ Н20
+0 82
V - величина потенциала в стандартных биологических условиях, то
есть при 37 °С и в растворах с pH = 7.4.
Энергия, выделяемая в процессах биологического окисления,
накапливается в клетках за счет синтеза молекул аденозинтрифос-
фата АТФ.
На каждую пару электронов, переданных по клеточной дыха¬
тельной цепи, синтезируются три молекулы АТФ. Таким обра¬
зом, окислительно-восстановительные процессы клеточной дыха¬
тельной цепи - источник энергии жизнедеятельности. За счет раз¬
личных окислительно-восстановительных процессов организм полу¬
чает 99 % энергии
Окислительно-восстановительные реакции необходимы для син¬
теза множества жизненно важных кислородных органических био¬
молекул (углеводы, жирные кислоты, гормоны и др.) Здесь наибо¬
лее значимы процессы окисления в митохондриях - органеллах кле¬
ток растений и животных
В их числе:
реакция С-гидроксилирования:
Р-Н + (О] Р-ОН,
окисление спиртовой группы в альдегидную
РСНг-ОН + [О] -> Р-СН=0 + Н20,
209
окисление последней в карбоксильную группу:
Р-СН=0 + [О] РСООН.
Эти процессы, служащие источником биосинтеза многих кисло¬
родсодержащих биомолекул, регулируются соответствующими фер¬
ментными системами с участием цитохрома Р450, алкогольдегидро-
геназы и альдегидоксидазы, соответственно. Чужеродные органиче¬
ские вещества (ксенобиотики) в тканях также подвергаются фер¬
ментативному окислению.
К числу общих биохимических окислительно-восстановитель¬
ных процессов относится явление перекисного окисления липидов.
Необходимым для метаболизма аминокислот является окисле¬
ние их аминогруппы под действием ферментов аминооксидаз.
Действие окислителей, содержание которых в условиях нашего
существования выше, чем восстановителей, направлено, в част¬
ности, на биомолекулы, содержащие группу -БН с образованием
дисульфидной группы -Б-Б-:
2-БН -Б-Б- + 21-Г + 2е.
Величина окислительно-восстановительного потенциала этой
полуреакции находится в пределах от 0 до -0.20 В (в зависимости
от природы биосубстрата), и поэтому в организме она идет легко и
обратимо. Это окислительно-восстановительное равновесие играет
важнейшую роль в регуляции биохимических механизмов.
С помощью окислительно-восстановительных реакций в орга¬
низме распадаются токсичные вещества, как образующиеся в ходе
метаболизма, так и попавшие в него извне. Так, в присутствии
ферментов каталазы и супероксиддисмутазы разрушаются сильные
окислители - промежуточные продукты биохимического восстанов¬
ления кислорода: пероксид водорода и супероксид-анион-радикал.
Действие на организм многих токсичных веществ (озон, галоге¬
ны, нитраты, оксиды азота) объясняется их сильными окислитель¬
ными свойствами, которые необратимо разрушают биосубстрат,
в первую очередь - ферменты.
Сильные окислители (перманганат калия, пероксид водорода,
раствор иода, хлорная известь и др ) используются в медицинской и
гигиенической практике в качестве дезинфицирующих средств.
11.7.2. Процессы с переносом ионов
В основе электрохимических процессов второй группы лежит
неравномерное распределение ионов по обе стороны какой-либо по¬
граничной поверхности, например, для большинства клеток ха¬
рактерна высокая внутренняя концентрация ионов калия и низкая
210
концентрация ионов натрия, напротив, во внешней среде этих кле¬
ток содержится большой избыток ионов натрия. Эти. процессы при¬
водят к возникновению биопотенциалов и биотоков. При возбужде¬
нии живой ткани и переходе ее в деятельное состояние возбужден¬
ный участок заряжается отрицательно по отношению к невозбуж¬
денному и возникает электрический ток.
Регистрация биотоков применяется в медицинской диагностике,
в частности, в электрокардиографии. Электрокардиографией на¬
зывают метод исследования физиологического состояния сердца пу¬
тем регистрации электрических потенциалов, возникающих при
работе сердечной мышцы, а электроэнцефалографией - метод
исследования деятельности головного мозга, основанный на реги¬
страции потенциалов, возникающих в нервных клетках.
Процессы, связанные с неравномерным распределением ионов в
биологических системах, вызывают появление трех типов потенциа¬
лов: диффузионных, мембранных и фазовых. Так, при повреждении
оболочек клеток электролиты диффундируют в клетку или из нее -
в зависимости от концентрации и подвижности ионов. Обычно по¬
врежденная ткань заряжается отрицательно по отношению к непо¬
врежденной, то есть возникает диффузионный потенциал поврежде¬
ния, величина которого составляет до 0.03-0 04 В.
Можно рассчитать соответствующую величину мембранного по¬
тенциала живой клетки, которая активно накапливает ионы калия.
Известно, что соотношение концентраций К+ внутри и снаружи
клетки составляет 20 : 1:
[К+В„утр„]/ 1К+С„аружи) = 20.
Е = 0.059^20 = 0.059-1.3 = 0 0767 В
Полученное значение близко к экспериментально определенно¬
му для живой клетки в состоянии покоя. Изменения мебранного
потенциала, сопровождающие передачу нервных импульсов или
мышечное сокращение, вызваны потоком катионов калия из клетки
и катионов натрия - внутрь ее Это приводит к падению потенциа¬
ла, который можно регистрировать с помощью микроэлектродов,
помещенных извне и внутри клетки.
Фазовые потенциалы возникают в системах, состоящих из
двух (или более) несмешивающихся друг с другом фаз Протоплаз¬
ма клеток и представляет собой такую многофазную систему Если
в одной из фаз более растворимы катионы, а в другой, наоборот,
анионы, то пограничная поверхность первой фазы приобретает по¬
ложительный заряд, а вторая заряжается отрицательно. Таким об¬
разом, на границе фаз возникает двойной электрический слой, об¬
условливающий появление фазового потенциала Фазовые потен¬
циалы особенно характерны для липидно-белковых систем клеток.
211
11.8. Потенциометрия.
Электрометрическое измерение pH
Потенциометрия - это ряд методов анализа и определения фи¬
зико-химических характеристик электролитов и химических реак¬
ций, основанных на измерении электродных потенциалов н электро¬
движущих сил гальванических элементов Потенциометрические
измерения являются надежными при изучении констант равновесия
электродных реакций, термодинамических характеристик реакций,
коэффициентов активности ионов, констант нестойкости комплекс¬
ных ионов, pH растворов.
Потенциометрические методы анализа имеют ряд преимуществ.
Они очень чувствительны; существуют модификации потенциомет¬
рического определения, позволяющие проводить анализ в пробах
объемом до десятых долей миллилитра, что важно для биологиче¬
ских исследований. Поскольку равновесное значение потенциала
устанавливается быстро, то потенциометрические измерения не
требуют значительных затрат времени. Их можно проводить в мут¬
ных и окрашенных растворах, вязких средах.
Потенциометрический метод анализа основан на измерении
электродного потенциала (<р) индикаторного электрода и нахожде¬
нии зависимости между его величиной и активностью потенциал-
определяющего иона в растворе.
Особенно широкое применение нашли потенциометрические
измерения при определении pH. Для этого используют электрохи¬
мическую цепь, составленную из индикаторного электрода и элек¬
трода сравнения.
Таблица П.4
Электроды сравнения и индикаторные электроды
Индикаторные электро¬
ды
Электроды сравнения
Водородный
Каломельный
Сурьмяный
Хлорсеребряный
Хингидронный
Стеклянный*
‘Стеклянный электрод относится к ионоселектив¬
ным электродам. Принцип его работы описан ниже.
Индикаторным электродом (или электродом определения)
называется электрод, который обратим относительно активности
212
анализируемого потенциалопределяющего иона. При измерении pH
потенциал индикаторного электрода должен зависеть от концентра¬
ции (активности) ионов водорода в растворе.
Электродом сравнения называют электрод с известной и по¬
стоянной величиной электродного потенциала.
Обычно используемые электроды приведены в табл. 11.4.
В принципе любую пару электродов можно использовать для
измерения pH. Для этого составляют гальваническую цепь из элек¬
трода определения, погруженного в исследуемый раствор, и элек¬
трода сравнения, и соединяют оба электродных раствора электроли¬
тическим мостиком. Электродвижущая сила гальванического эле¬
мента равна разности потенциалов двух электродов. Если более по¬
ложителен электрод сравнения*, то Е = <рСравн - Финд- Измерив ЭДС
и зная величину фСравн, можно найти финд, а отсюда по соответ¬
ствующей формуле вычислить величину pH. Например, если цепь
составлена из каломельного и водородного электродов,
Н2{И} I Нх* I КС11 Нд2С12, КС11 Нд,
Е = Ф+клм - Ф'вод = Фклм - (-0.059рНх) = фклм + 0.059рНх,
отсюда рНх = (Е - фклм)/0.059.
Описанный выше способ измерения pH основан на применении
химических гальванических элементов. Однако, для измерения pH
можно использовать и концентрационные элементы.
В этом случае в качестве электрода сравнения можно исполь¬
зовать тот же электрод, который взят в качестве индикаторного
электрода, но поместить этот электрод в раствор с известным зна¬
чением pH. Так, например, можно использовать цепь из двух водо¬
родных электродов, из которых один (индикаторный электрод) по¬
гружен в исследуемый раствор, а другой (электрод сравнения) -
в стандартный раствор с известным значением pH. Поскольку кон¬
центрация ионов водорода в обоих электродных растворах различна,
такой элемент является примером концентрационного гальваниче¬
ского элемента.
н2{и}1нх*|кс||н*стамд1{рпн2
Е —ф+вод сравн ‘ Ф вод опред — ‘0 059рНстанд + 0.059рНх
Этот случай более распространен В элементе, состоящем из кало¬
мельного и хингидронного электродов, положителен последний, т е инди¬
каторный электрод
213
11.9. Ионоселективные электроды
Одним из современных физико-химических методов анализа,
позволяющих контролировать состояние окружающей среды, сле¬
дить за изменением концентрации электролитов в биологических
жидкостях, является ионометрия - потенциометрический метод
исследования состава раствора с применением ионоселективных
электродов (ИСЭ)
В настоящее время выпускается около 30 ионоселективных
электродов, при помощи которых можно прямо или косвенно опре¬
делять концентрацию более 50 катионов, аннонов, а также молеку¬
лярных соединений Из них наибольшее применение нашли элек¬
троды, чувствительные к ионам Р-, СГ, СМ-, Б2-, N03", МЩ\ РЬ2+,
Си2+, М2*, Са2+, £(Са2*, Мд2+), а также ИСЭ для определения газов
(СОг, МНз, НС1, НгБ, НС1М, N0) и молекул (ацетилхолин, мочевина).
Ионометрия имеет принципиальные преимущества по сравне¬
нию с другими методами:
1. Она представляет собой идеальный метод анализа прежде
всего водных растворов и проб, хорошо растворимых в воде или
содержащих легкорастворимые в воде компоненты. Специфическая
особенность ионометрии состоит в том, что метод позволяет опре¬
делять активную концентрацию иона на фоне его общей концентра¬
ции, и в этом отношении ионометрия уникальна.
2. Измерения можно проводить в непрозрачных, мутных и
окрашенных средах, даже в вязких пастах. При этом исключаются
длительные, трудоемкие операции фильтрования, дистилляции и
экстрагирования.
3. Время установления равновесного потенциала ИСЭ обычно
невелико, что позволяет автоматизировать контроль за водной и
воздушной средами.
4 Ионометрия относится к группе неразрушающих методов
контроля, и анализируемый раствор может быть использован для
дальнейших исследований
5. Для ионометрии характерен широкий диапазон измерений,
что позволяет определять содержание как основных макрокомпо¬
нентов смеси, так и фиксировать микропримеси с достаточной
надежностью Интервал определения активности ионов в различных
природных и промышленных объектах находится в пределах
от 1 до 10б моль/л, но возможно определение и до 10-8 моль/л.
Погрешность определения при прямой потенциометрии (т.е. опреде¬
лении активности и концентрации по измеренным значениям элек¬
тродного потенциала и ЭДС) составляет 2-10%.
6. Унифицированность аппаратуры, ее сравнительная (в сопос¬
тавлении с другими физико-химическими методами анализа) деше¬
214
визна, возможность создания не только стационарных, но и пере¬
носных приборов также являются достоинствами ионометрии.
Перечисленные преимущества метода позволяют широко ис¬
пользовать его для медико-биологических целей.
Как и любому методу анализа, ионометрии присущ и ряд недо¬
статков. Так, селективность основной части электродов не так ве¬
лика, чтобы производить непосредственное измерение активности
интересующего иона в любой анализируемой среде. В некоторых
случаях состав раствора должен быть приблизительно известен,
чтобы оценить влияние мешающих ионов, на которые данный элек¬
трод дает отклик или предотвратить это влияние реакциями ком-
плексообразования, осаждения или ионообмена.
Возможность создания электродов, чувствительных к многоза¬
рядным ионам, ограничена точностью измерения ЭДС. Кроме того,
для всех электродов характерен дрейф стандартного потенциала,
что главным образом зависит от изменения температуры окружаю¬
щей среды. В лабораторных условиях электрод показывает дрейф
потенциала, составляющий - 2 мВ/сутки. Наличие дрейфа требует
периодической градуировки электрода.
В ионометрии в роли индикаторных электродов используют ио¬
носелективные электроды. Их аналитическими характеристиками -
селективностью, диапазоном линейности электродной функции,
временем отклика потенциала на изменение концентрации потен-
циалопределяющего иона - определяется точность и стабильность
ионометрии.
Основой ИСЭ является полупроницаемая мембрана, обла¬
дающая селективной ионной проводимостью
Возникновение мембранного потенциала (при возможности его
определения) и используют для измерения активности ионов
Селективная проницаемость большинства мембран объясняется
их ионообменными свойствами, способностью проявлять свойства
ионита (см. гл. 13). Основная особенность ионитов - способность к
ионному обмену между противоионами ионита и ионами контакти¬
рующего с ним раствора. Если катионит И-Х (то есть ионит с поло¬
жительно заряженными противоионами) приведен в контакт с рас¬
твором электролита, содержащим катион М+, то в этой гетероген¬
ной системе протекает процесс обмена ионами.
Х*(мембрана) + М*(раствор) X*(раствор) + М’(мембрана)
Процесс этот состоит из двух стадий а) проникновение иона
М+ в мембрану, б) его перезамещение внутри мембраны
Таким образом, за счет способности к ионообмену через тонкий
слой полупроницаемой мембраны ионита из одного раствора в
215
другой селективно перемещаются ионы и мембрана пробретает ион¬
ную проводимость Особенностью мембранного потенциала являет¬
ся то, что в сооответствующей ему электродной реакции не уча¬
ствуют электроны. Здесь имеет место обмен ионами между мембра¬
ной и раствором.
Разность потенциалов между двумя растворами, расположенны¬
ми с двух сторон ионоселективной мембраны, может быть измерена.
Для этого необходимо обеспечить электрический контакт между
внутренним раствором (стандартный раствор I с известной кон¬
центрацией электролита) и исследуемым раствором II, для чего ис¬
пользуют электроды сравнения 1 и 2 - хлорсеребряные или кало¬
мельные (рис. 11.5)
-(эдс)_
I
1 - внутренний электрод сравнения
2 - электрод сравнения
I - стандартный раствор
II - исследуемый раствор
- ионоселективная мембрана
Рис. 11.5. Схема работы ионоселективного электрода.
Следовательно, при потенциометрических измерениях с исполь¬
зованием ИСЭ собирают следующий гальванический элемент:
электрод I стандартный I I анализируемый I электрод
сравнения 1 | раствор I | мвмбрана | раствор II | сравнения 2
Собственно ИСЭ состоит из внутреннего электрода сравнения
1, внутреннего стандартного раствора I и ионоселективной мембра¬
ны. Этот полуэлемент погружают в анализируемый раствор II, куда
опущен электрод сравнения 2.
Электродвижущую силу (Е) собранного гальванического эле¬
мента можно измерить, применяя любые потенциометрические уст¬
ройства, сконструированные на основе токоусилительных систем.
Для этой цели служат иономеры и рН-метры.
Величина ЭДС (Е) системы связана с активностью определяе¬
мого иона ам следующим уравнением:
216
2 303' 1?Т
Е = ЕЧсопбО ± ——-—^ ам = Е°(сопз0 ± Б-^ам,
гг
где +Б - угловой коэффициент для катионов, -Б - угловой коэффи¬
циент для анионов.
Значение постоянной величины (Е°) зависит от выбора вспомо¬
гательных электродов сравнения (1 и 2) и их электродных потен¬
циалов, активности стандартного раствора I, а также небольшого
диффузионного потенциала.
Если мембрана не является идеально селективной и пропускает
также мешающие ионы X с активностью ах и зарядом гх, то вели¬
чину ЭДС цепи рассчитывают по уравнению Никольского:
Е = Е°(сопбО ± Б-^ам + К„.х ахг«/гх ),
где Км.х - коэффициент селективности электрода по отношению к
определяемому иону М на фоне мешающего иона X. Чем меньше
эта величина, тем более селективна мембрана. Для каждого элек¬
трода опытным путем определяют коэффициенты селективности по
отношению к основным мешающим ионам.
Прежде чем применить ИСЭ для аналитической цели, необхо¬
димо провести его электрохимическое изучение, которое предусма¬
тривает установление основных характеристик.
В частности, методом построения градуировочного графика
определяют область прямой концентрационной зависимости потен¬
циала электрода (линейный участок электродной функции).
Калибровка электрода заключает¬
ся в установлении зависимости
между ЭДС (Е) и активностью
или концентрацией изучаемых
ионов. Для этой цели строят эм¬
пирический градуировочный гра¬
фик (рис. 116) в координатах
Е - (-^а) или Е - (-^ См), приме¬
няя стандартные растворы анали¬
зируемого иона
Полученную для стандартных
растворов графическую зависи¬
мость в дальнейшем используют в
качестве рабочего графика при
определении активности или
концентрации в анализируемых
пробах
Рис. 11.6. Калибровочная кривая
ионоселективного электрода.
217
В настоящее время выпускают иономеры, в которых отсчет ак¬
тивности (точнее ее отрицательного логарифма, рХ) любого иона X
можно производить непосредственно по шкале (или цифровым по¬
казаниям) измерительного прибора, после его настройки по стан¬
дартным растворам При работе с такими приборами нет необходи¬
мости в построении калибровочного графика.
11.10. Типы ионоселективных электродов
Ионоселективные электроды классифицируют по агрегатному
состоянию электродноактивного материала.
11.10.1. Стеклянные электроды
Мембрана, изготовленная из натриевого (вЮг-МагО-СаО) или
литиевого (вЮг-ЫгО-СаО) стекла, обладает катионообменными
свойствами, так как в водном растворе ионы щелочного металла (N8
или и), гидратируясь, могут обмениваться только с ионами водоро¬
да внутреннего и внешнего раствора:
Н+(раствор) + №+(стекло) ^ Н+(стекло) + Ма+(раствор).
Таким образом, стеклянная мембрана приобретает свойства
ионного проводника и является проницаемой только для ионов во¬
дорода (рН-селективная мембрана).
Собственно стеклянный электрод представляет собой трубку
специального сорта стекла с выдутым на ее конце шариком с
очень тонкой стенкой (рис. 11.7, а). Внутрь электрода заливают
раствор электролита с известной величиной pH (чаще всего раствор
НС1, pH = 1) и помещают электрод сравнения - хлорсеребряный
электрод.
Схема записи стеклянного электрода:
Ад | АдС1, НС11 стекло | Н+х (раствор),
где Н+х - неизвестная концентрация ионов водорода в исследуемом
растворе
Для измерения кислотности (рис. 11.8) исследуемого раствора в
него погружают стеклянный электрод и соединяют со вторым элек¬
тродом сравнения (обычно используют хлорсеребряный или кало¬
мельный).
Измерив ЭДС (Е) полученной цепи и зная Е° в цепи стеклянно¬
го электрода, можно рассчитать pH исследуемого раствора:
218
’■'V Р - р°
pH = ^-(при 25°С).
0.059
Перед измерением стеклянный электрод калибруют - погружа¬
ют его в буферные растворы с точно известным pH.
Стеклянные рН-электроды - старейшие и наиболее распростра¬
ненные ИСЭ. Они являются в настоящее время лучшими рН-
электродами, поскольку они не чувствительны к окислительно¬
восстановительным системам и селективны в широком диапазоне
pH - от 1 до 12 5.
Рис. 11.7. Схема устройства ионселективных электродов,
а - стеклянный электрод, б - электрод с твердой мембраной, в - электрод с жидкостной
пластифицированной мембраной, г - ферментный электрод, д - электрод с твердой
мембраной с металлическим контактом
1 - ионселективная мембрана. 2 - внутренний стандартный раствор. 3 - внутренний
электрод сравнения. 4 - корпус для электрода (а, г - из стекле, б, в, д - из пластмассы),
5 - металлический контакт, 6 - слой ф
219
Исключительно высокая селективность стеклянных рН-электро-
дов (например, №+ 10 м) определяется тем, что радиус протона
на несколько порядков меньше радиуса любого катиона. Время от¬
клика электрода составляет 5-15 секунд. Электрод позволяет прово¬
дить измерения pH с точностью ±0 ОЗрН, причем возможна хими¬
ческая стерилизация электрода, что делает его особенно пригодным
для измерения кислотности биологических объектов Срок жизни
электрода измеряется годами.
Рис. 11.8. Схема измерения pH при помощи стеклянного электрода:
1 - стеклянная мембрана, 2 - стандартный раствор с постоянным значением pH,
3 - внутренний электрод сравнения, 4 - электрод сравнения, 5 - преобразователь.
Систематические исследования зависимости электродных
свойств стекла от его состава показали, что определенным его из¬
менением (введением оксидов бора, алюминия) можно получить
стекло, электроды из которого селективны по отношению к ионам
натрия, калия, аммония, серебра. Из них наиболее широкое приме¬
нение вследствие высокой селективности нашел р№-электрод.
Недавно стали применять стеклянные электроды и для измерения
окислительно-восстановительного потенциала раствора.
11.10.2. Твердофазные электроды
Мембрану этих электродов (рис. 11.7,6) создают из моно- или
поликристаллов труднорастворимых в воде солей. В этих мембранах
обычно один из двух составляющих соль ионов способен пере¬
мещаться в кристаллической решетке по ее дефектам. Поэтому
220
электроды "Ъ. твердой мембраной используются в качестве датчиков
для определения тех ионов, которые входят в состав мембраны, а
также тех ионов, которые способны взаимодействовать с активными
центрами материала мембраны.
Первый твердофазный высокоселективный мембранный электрод
был сконструирован с использованием монокристалла 1_аРз для
определения активности ионов фтора. Он нашел применение для
определения величины рГ в питьевой, морской и сточных водах, при
исследовании костей, зубов, слюны, зубных паст и т.д.
Электроды, чувствительные к ионам Ад+, в2-, Си2+, РЬ2+, Г, ВГ,
получены на основе Адгв (поскольку перечисленные ионы либо
замещают ион серебра, либо связываются с ним на поверхности
кристалла).
Монокристаллический АдС1 лежит в основе электрода, селек¬
тивного по отношению к ионам хлора. Этот электрод используется
при работе с цельной кровью и ее сывороткой, а также для опреде¬
ления концентрации хлорид-иона в воде и моче.
Существенным достоинством этих электродов является дли¬
тельный срок работы. Однако число твердых ионных кристалличе¬
ских соединений, обладающих ионной селективной проводимостью,
ограничено.
11.10.3. Электроды с жидкой мембраной
Эти электроды представляют собой диафрагму, поры которой
заполнены раствором электродноактивного вещества в органи¬
ческом растворителе (рис. II.7,в). В качестве электродноактивных
веществ используют ионообменные смолы (жидкие катиониты или
аниониты) или нейтральные молекулы - мембраноактивные ком-
плексоны, способные к образованию хелатов. Диафрагма разделяет
два водных раствора - раствор с известной концентрацией иона, по
отношению к которому селективна мембрана, и раствор с неизвест¬
ной концентрацией этого иона.
Селективность жидкостных мембран определяется избиратель¬
ностью комплексообразования или обмена ионов между мембраной
и раствором. Известны электронейтральные липофильные реагенты
со сравнительно небольшой молекулярной массой (например, анти¬
биотик валиномицин), которые образуют комплексные соединения с
некоторыми катионами. Такие лиганды способны экстрагировать ка¬
тионы из водных растворов в гидрофобную фазу мембраны и пере¬
носить их через мембрану из одного водного раствора в другой
Поэтому нейтральные переносчики ионов являются важными ком¬
221
понентами природных транспортных ионных систем. Их и вводят в
аналогичные искусственные системы, основным условием работы
которых является избирательность присутствующих в мембране пе¬
реносчиков к определенным ионам Электрод на основе валиноми-
цнна стал одним из наиболее важных ионоселективных электродов
благодаря его уникальной избирательности к ионам калия.
Ассортимент жидкостных ИСЭ постоянно увеличивается. Из¬
вестны электроды, селективные к катионам (Си2+, Мд2'*', Мп2+, МН4+
и др ), анионам (N03', СОз2-, БОд2' и др.), а также к ионогенным ор¬
ганическим соединениям. Среди них наиболее важными для реше¬
ния экологических проблем являются электроды, селективные к
поверхностно-активным веществам (определение загрязнений мою¬
щими средствами) и к ацетилхолину (определение загрязнений
фосфорорганическими ядохимикатами).
В свое время одной из больших трудностей в физиологических
исследованиях кальциевого метаболизма было отсутствие удобного
метода определения несвязанного катиона кальция. Теперь в меди¬
ко-биологических исследованиях применяют жидкостные Са-элек-
троды как на основе нейтральных лигандов, так и катионитов
(диалкил фосфатов).
Механическая непрочность пористых мембран, неизбежное
попадание органической фазы в анализируемый раствор затрудня¬
ют применение ИСЭ с жидкими мембранами в биомедицинских
исследованиях.
Этн недостатки частично устранены в пленочных (так назы¬
ваемых отвержденных) электродах, в которых электродноактивное
вещество и растворитель-пластификатор внедрены в полимерную
матрицу. Срок службы таких ИСЭ увеличивается до года.
11.10.4. Газовые, ферментные и бактериальные
электроды
Особое место в потенциометрических методах анализа зани¬
мают газовые (газочувствительные), ферментные (рис. 11.7, г), бак¬
териальные, иммуноэлектроды (называемые биологическими сен¬
сорами). Существенное отличие последних от обычных ионоселек¬
тивных - использование промежуточной реакции, в результате
которой из молекул определяемых веществ образуются ионы,
активность которых может быть определена одним из вышеперечис¬
ленных ИСЭ.
Газовые электроды позволяют определять активную концентра¬
цию следующих газов: С02, МН3, N02, Нгв, НХ (X = Р, С1, Вг, I).
222
В основе З^йствия газовых электродов лежит реакция с участием
воды, в результате которой изменяется характер среды, например:
С02 + Н20 ±5 Н+ + НСОз"
1МН3 + Н20 ^ ОН"+ ын/.
Индикаторными ИСЭ на выделяющиеся при этих реакциях ионы
(Н+, ОН") служат стеклянные рН-электроды.
Газочувствительные электроды находят все большее примене¬
ние, к примеру, при контроле содержания различных компонентов
в выхлопных газах автомобилей. Однако их широкому внедрению
препятствует сложность конструкции.
Позднее появились ферментные, бактериальные и иммуноэлек¬
троды, сочетающие селективность и чувствительность биохимиче¬
ских реакций со скоростью и простотой измерений потенциометри¬
ческого метода.
Так, ферментные электроды можно использовать для определе¬
ния концентрации не только продуктов ферментативной реакции,
но и любого участвующего в этой реакции вещества, что особенно
важно для многостадийных реакций, а также для определения ак¬
тивности фермента, концентрации его ингибиторов и активаторов.
11.11. Ионометрия в медицине
Применение биологических сенсоров существенно расширило
рамки ионометрии, позволив определять концентрацию органиче¬
ских соединений в водных растворах (глюкозы, мочевины, амино¬
кислот и др.), что перспективно для медицинской практики.
Дальнейший прогресс в развитии ионометрии связан, во-пер-
вых, с разработкой новых ИСЭ, с созданием (с помощью уже разра¬
ботанных или даже серийно выпускаемых электродов) аналитиче¬
ских методик определения ионов и низко- и высокомолекулярных
органических соединений, которые раньше методом ионометрии не
обнаруживались (например, белки, сахара и др ). Другое направле¬
ние - улучшение конструкции электродов, например, создание
электродов с твердым внутренним контактом между мембраной и
металлическим токоотводом Эти электроды не имеют внутреннего
жидкостного заполнения Твердый металлизированный контакт был
впервые применен к стеклянным электродам. Затем были разрабо¬
таны также электроды с металлическим внутренним контактом, со¬
держащие мембраны из галогенидов и сульфидов тяжелых метал¬
223
лов. Такие электроды считаются ионоселективными электродами
второго поколения (рис. 11 7, д).
В практике клинических лабораторий повсеместно применяется
как измерение потенциалов, связанных с переносом электронов,
с помощью окислительно-восстановительных (в широком смысле)
электродов, так и измерение потенциалов, обусловленных перено¬
сом ионов, с помощью ионоселективных электродов (см ниже).
Наряду с классическим методом исследования кислотности желу¬
дочного сока методом зондирования большое применение находит
электрометрический метод определения pH - введение в желудок
больного капсулы, содержащей миниатюрную пару электродов -
стеклянный и хлорсеребряный - для измерения pH желудочного
сока непосредственно в организме. Электрометрическое измерение
pH применяется также для непрерывного контроля кислотности
во время хирургической операции, при диагностике некоторых кож¬
ных заболеваний, при предварительном испытании новых лекар¬
ственных препаратов вне организма (in vitro) и во многих других
случаях.
Так же легко, как определяют pH, можно измерить активные
концентрации важнейших в медико-биологическом отношении ио¬
нов (например, Na+, К+, Са2+, NH4+, РЬ2+, СГ, В г. Г, N03" и др.),
используя соответствующие ионоселективные электроды Интен¬
сивное внедрение ионометрических методов в медико-биологичес¬
кие исследования обусловлено, во-первых, важностью контроля
водно-электролитного баланса и кислотно-щелочного состояния
организма и его отдельных органов и, во-вторых, необходимостью
определения состава лекарственных препаратов, ферментов, физио¬
логических растворов, продуктов питания, почвы, природных вод,
атмосферы и т.д.
Еще одна область применения метода ионометрии - нахождение
так называемого компромиссного (смешанного) потенциала тех или
иных тканей или органов с помощью микроэлектродов. Суммарная
совокупность смесей окислителей и восстановителей с учетом их
концентраций определяет величину этого потенциала. Оказалось,
что его величина специфична для тех или иных тканей. Например,
компромиссный потенциал кожи здорового человека находится в
интервале 220-280 мВ, а мышечной ткани - 170-220 мВ В част¬
ности, при ишемической болезни, когда суммарное окислительно¬
восстановительное равновесие в тканях смещено в сторону умень¬
шения содержания окислителей, величина этого потенциала в мыш¬
цах падает до 160 мВ, причем это отклонение в большей степени
затрагивает мышцы, чем кожу. Таким образом, величина компро¬
миссного потенциала становится диагностическим признаком.
224
Итак,’’окислительно-восстановительные процессы играют исклю¬
чительно важную роль в живых системах: снабжают их энергией,
необходимым “строительным материалом’’ и участвуют в механиз¬
мах биорегуляции. С помощью регулируемого обмена веществ
достигается постоянство содержания окислителей и восстановите¬
лей и продуктов их взаимодействия в организме, обеспечивая окис¬
лительно-восстановительный гомеостаз. Контроль окислительно¬
восстановительного гомеостаза, как и нахождение концентраций
практически любых ионов биосред, осуществляется с помощью
метода ионометрии, одного из универсальных физико-химических
методов для диагностических нужд.
Глава 12. ГЕТЕРОГЕННОЕ РАВНОВЕСИЕ
Растворение бывает двоя¬
кое целостное и частичное
Первое происходит, когда ра¬
створяющееся тело целиком
переходит в растворитель,
второе - когда какая-нибудь
составная часть выделяется
из растворяющегося тела си¬
лою растворителя и соединяет¬
ся с ним
М В Ломоносов
Рассмотренные выше случаи относятся к числу примеров гомо¬
генного равновесия, т.е. процессов, имеющих место в одной фазе, а
именно в растворе. Фазой называют любую часть системы, име¬
ющую одинаковый химический состав и обладающую равенством
термодинамических функций. Если компоненты химической реакции
находятся в разных фазах, разделенных границей - поверхностью
раздела, то речь идет о гетерогенных процессах. В случае гетеро¬
генных превращений в выражении для константы равновесия сле¬
дует считаться только с теми компонентами, концентрации которых
изменяются в ходе реакции.
Практически во всех реакциях в широком интервале температур
твердая фаза вносит постоянный вклад (const) в химическое равно¬
весие и может быть включена в константу равновесия.
12.1. Произведение растворимости
Случаем гетерогенного равновесия является и растворение
труднорастворимых веществ (в частности, солей) в любых раство¬
рителях, в том числе и в воде.
Энтальпия растворения солей (и любых других веществ) скла¬
дывается из энтальпии разрушения кристаллической решетки и
энтальпии гидратации, т.к. каждый ион в растворе окружен гид-
ратной оболочкой. Первая составляющая есть величина положи¬
тельная (эндотермический процесс), а вторая - отрицательная
(экзотермический процесс). В соответствии с этим их алгебраиче¬
ская сумма может принимать как положительные, так и отрица¬
тельные значения. Следовательно, одни соли растворяются с выде¬
лением тепла, а другие - с поглощением. Поэтому с увеличением
226
температзИры растворимость солей может либо увеличиваться, либо
уменьшаться. Например, растворимость МаС1 при нагревании увели¬
чивается, т.к. этот процесс эндотермический; напротив, раствори¬
мость Маг804 с ростом температуры падает, т.к. этот процесс экзо-
термичен. Однако, как уже отмечалось, растворение солей идет са¬
мопроизвольно, т.к. связано с резким увеличением энтропии, по¬
скольку происходит разрушение упорядоченной высокоорганизован¬
ной структуры кристаллической решетки.
В зависимости от содержания растворенного вещества и соот¬
ношения скоростей процессов растворения и кристаллизации раз¬
личают насыщенные, ненасыщенные и пересыщенные растворы.
Насыщенным называется раствор, находящийся в равновесии
с осадком растворяемого вещества, при этом скорость растворения
вещества равна скорости его кристаллизации. Насыщенный раствор
содержит максимально возможное при данных условиях количество
растворенного вещества.
Ненасыщенным называется раствор, концентрация которого
ниже концентрации насыщенного раствора, поэтому в таком рас¬
творе всегда можно растворить при тех же условиях дополнитель¬
ное количество растворяемого вещества.
Пересыщенным называется раствор, концентрация которого
выше концентрации насыщенного раствора. Пересыщенный раствор
обычно получают осторожным охлаждением насыщенного раствора,
и он является термодинамически неустойчивой, неравновесной си¬
стемой. Обычно при внесении в пересыщенный раствор кристаллов
или встряхивании начинается самопроизвольная быстрая кристал¬
лизация из раствора избытка растворенного вещества. Процесс кри¬
сталлизации продолжается до тех пор, пока раствор не станет на¬
сыщенным для данных условий.
Количественно растворимость различных веществ выражается
концентрацией насыщенных растворов. Растворимость (в) данного
вещества равна его молярной концентрации в насыщенном растворе
в моль/л Растворимость часто выражают и в граммах растворенно¬
го вещества на 100 г растворителя
Растворимость вещества зависит от природы растворяемого ве¬
щества, т е от его сродства к растворителю, температуры, концент¬
рации ионов в растворе При растворении большинства солей в во¬
де в раствор переходят не молекулы, а ионы Поэтому в водном на¬
сыщенном растворе малорастворимого сильного электролита между
твердой фазой (осадком) и ионами этого электролита в водной фазе
устанавливается динамическое гетерогенное равновесие
Рассмотрим гетерогенное равновесие между кристаллическим
осадком малорастворимой соли АдС1 и его водным раствором, со¬
держащим ионы Ад+ и СГ. При введении в воду соли в количестве,
227
большем, чем это необходимо для получения насыщенного раствора,
будет иметь место равновесие между твердой фазой и ионами соли
в растворе
(AgCl)re 5 Ад* + СГ,
а в общем виде для любого труднорастворимого электролита-
(АхВу)тв ^ хАу* + уВж'.
Учитывая постоянство концентрации твердого вещества
[А*Ву] = const, получим.
кравн const = [АУТ[Вж-]у = ПРд«ву
В частности, для AgCI:
ПРAgci = [Ад+][СГ]
В насыщенном растворе соли произведение концентраций ее
ионов’ в степенях, равных стехиометрическим коэффициентам,
есть величина постоянная при данной температуре, называемая
произведением растворимости (ПР).
Величина ПР характеризует растворимость электролита при
данной температуре. Она определяется природой электролита и
растворителя. Типичные величины ПР некоторых трудно раствори¬
мых солей даны в таблице 12.1. Величина ПР солей дается при
стандартных условиях, т.е. при 25° С, т.к. их растворимость, как
уже отмечалось, различным образом зависит от температуры.
Таблица 12 1
Произведения растворимости некоторых солей
Соль
ПР
Соль
ПР
АдСІ
1 8-Ю-10
HgS
1 610-52
АдВг
5 310-13
Мдз(Р04)2
1.010-13
Ад1
8.3-10"17
Са3(Р04)2
2.01 O'29
BaS04
1 1-10-10
СаНР04
2.7-10-7
PbS
2.5-1027
Са5(0Н)(Р04)3
1.6-1058
Иногда вместо ПР используют величины рПР. Например,
рПРАдс1 = 9.8, рПРддвг = 12.3, а рПРдд! =16.1. Это свидетельствует
об уменьшении растворимости в ряду АдС1 > АдВг> Ад1.
* Более правильно говорить об активностях ионов
228
Растворимость любого малорастворимого электролита состава
А,Ву (в, моль/л) можно рассчитать по формуле:
Смещение ионных гетерогенных равновесий происходит в соот¬
ветствии с принципом Ле Шателье, а именно, изменение концент¬
рации одноименных (то есть входящих в состав соли) ионов вызы¬
вает изменение растворимости электролита, поскольку произведе¬
ние растворимости - величина постоянная. Из этой закономерности
вытекают следствия.
а) Выпадение осадка. Осадок малорастворимого электролита
выпадает из пересыщенного раствора. Осадок образуется, если про¬
изведение реальных концентраций его ионов в растворе с учетом
коэффициентов в уравнении диссоциации данного электролита (ПИ)
больше произведения растворимости, т.е. ПИ > ПР, что имеет мес¬
то в пересыщенном растворе. Выпадение осадка продолжается до
тех пор, пока раствор не станет насыщенным.
б) Растворение осадка. Осадок малорастворимого электроли¬
та будет растворяться в том случае, если раствор над ним станет
ненасыщенным, то есть, когда в растворе над осадком этого мало¬
растворимого электролита стехиометрическое произведение кон¬
центраций ионов ПИ меньше его произведения растворимости
(ПИ < ПР, ненасыщенный раствор). Последнего можно достигнуть
разведением раствора, а также удалением или связыванием одного
из одноименных ионов в более прочное соединение.
Например, труднорастворимая соль ВаСОэ легко растворяется
в соляной кислоте из-за резкого понижения концентрации карбо¬
нат-иона, разрушающегося в углекислый газ, покидающий реакци¬
онную сферу:
Растворение малорастворимого гидроксида железа(Ш) в кислоте
происходит из-за более прочного связывания ионов ОН” в молекуле
Н20, чем они связаны в осадке
Ре(ОН)э + ЗНС1 -> РеСЬ + ЗН20
12.2. Влияние одноименного иона
ВаСОэ + 2НС1 -> ВаС12 + С02Т + Н20
229
Осадок АдС1 растворяется в водном растворе аммиака, посколь¬
ку взаимодействуя с аммиаком, образует растворимый комплекс,
устойчивый при избытке аммиака в растворе-
АдС1 + 2МН3 -»[Ад(МНэ)2]С1
Растворение осадков может происходить в результате окисли¬
тельно-восстановительной реакции
СиЭ + 4НМОэ -> Си(МОэ)2 + 2140 + ЭОг + 2Н20
Итак, растворение осадка является следствием конкуренции
между гетерогенным равновесием и химическими равновесиями, ко¬
торую выигрывает то равновесие, которое приводит к более проч¬
ному связыванию или полному удалению одноименных ионов, уча¬
ствующих в этих равновесиях.
в) Полнота осаждения ионов. На практике часто необходимо
удалять ионы из раствора переводом их в осадок. В частности,
таким образом можно удалить из раствора токсичный ион и тем
самым предотвратить отравление.
Для достижения полноты осаждения малорастворимого сильно¬
го электролита из его насыщенного раствора следует увеличить в
растворе концентрацию каких-либо ионов, входящих в состав этого
электролита.
В соответствии с принципом Ле Шателье при введении в рас¬
твор труднорастворимой соли хорошо растворимого электролита,
содержащего ионы Ау+ или Вх_ (одноименный ион), концентрация
противоиона должна уменьшаться за счет перехода его в осадок.
Другими словами, при добавлении одноименного иона раствори¬
мость соли уменьшается. Это значит, что растворимость АдС1, к
примеру, в чистой воде выше, чем в водном растворе МаС1.
г) Порядок осаждения ионов. Если к раствору, содержащему
смесь ионов, осаждаемых одним и тем же ионом, добавлять этот
ион, то образование осадков малорастворимых электролитов проис¬
ходит ступенчато: первым осаждается тот электролит, для дости¬
жения величины произведения растворимости которого требуется
меньшая концентрация добавляемого иона-осадителя.
Если к раствору, содержащему смесь хлорид-, бромид- и иодид-
ионов добавлять катион-осадитель - катион серебра, то в первую
очередь будет осаждаться йодид серебра, так как его произведение
растворимости минимально и будет достигаться прежде всего. Хло¬
рид-ион перейдет в осадок в последнюю очередь.
В биосредах следует учитывать, к примеру, что фосфат магния
растворим лучше фосфата кальция (см. таблицу 12.1), а это означа¬
ет, что из этих ионов, в близких количествах содержащихся в
230
плазме, в первую очередь в осадок будут переходить кальциевые
соли, которые и составляют основу костной ткани.
д) Солевой эффект. Растворимость солей зависит и от добав¬
ления электролитов, не имеющих общих с ними ионов. Обычно это
улучшает растворимость, причем она растет с увеличеием ионной
силы раствора. Причина этого явления, называемого солевым эф¬
фектом, вызвана понижением коэффициентов активности (а, следо¬
вательно, и активности) ионов, входящих в состав трудно раство¬
римой соли из-за роста сил межионного взаимодействия.
12.3. Гетерогенные равновесия и организм
В организме человека важнейшие гетерогенные процессы с
участием неорганических ионов связаны в первую очередь с обра¬
зованием и растворением минеральной основы костной ткани.
Ее основной компонент - гидроксиапатит, гидроксифосфат каль¬
ция Са5(0Н)(Р04)э. Его образование можно выразить общей схемой:
5Са2+ + ЗНР042‘ + НОН Са5(0Н)(Р04)3 + 4Н+
Уравнение показывает, что в кислой среде костная ткань раз¬
рушается.
Формирование костной ткани начинается с плазмы крови.
В плазме содержатся необходимые для этого катионы кальция, а
также дигидро- и гидрофосфат-ионы. Кроме этого, в ней же нахо¬
дятся и катионы, и анионы, обеспечивающие соответствующее кис¬
лотно-основное равновесие (см. главу 9). Концентрация катиона
кальция в плазме составляет 2.5-10'3 моль/л, однако лишь часть
его, а именно НО'3 моль/л, находится в ионизированном виде.
Концентрация гидрофосфат-иона в плазме - 2 9-10'4 моль/л.
Из данных табл. 12.1 видно, что этих концентраций достаточно
для образования осадка СаНР04 (ПР = 2.7107, ПИ = 2.910'7). Так
как раствор лишь слегка пересыщен, в плазме кристаллизация при¬
водит к образованию малых количеств микрокристалликов гидро¬
фосфата кальция
В омываемых кровью (а, следовательно, и микрокристалличе¬
ским осадком гидрофосфата кальция) клетках костной ткани, име¬
нуемых остеобластами, в результате ферментативного гидролиза со¬
держащихся в них биомолекул - сложных эфиров фосфорной кис¬
лоты - увеличивается концентрация фосфат-ионов Это создает
условия для еще большего пересыщения раствора фосфатов каль¬
ция, что способствует превращению гидрофосфата кальция в гид-
231
роксиапатит. Этому же благоприятствует и слабощелочная среда
плазмы. Таким образом, устанавливается динамическое равновесие,
состояние которого определяется совокупностью трех факторов, а
именно концентрациями фосфат-ионов и катионов кальция, а также
кислотностью среды. Следствием такого равновесия является еже¬
дневный обмен 700-800 мг кальция в составе костной ткани.
При увеличении концентраций свободных ионов кальция и гид¬
рофосфат-ионов в плазме происходит отложение гидроксиапатита в
костной ткани Их снижение приводит к растворению костей, что
наблюдается у детей при рахитах, у беременных, когда их костный
материал расходуется на формирование скелета плода, у космонав¬
тов из-за нарушения деятельности ферментов, ответственных за
кальциевый обмен в организме.
Повышение кислотности среды также приводит к растворению
гидроксиапатита. Особенно наглядно влияние кислотности среды
для случая разрушения зубной ткани, минеральную основу которой
также составляет гидроксиапатит. Анаэробные микроорганизмы
полости рта метаболируют с образованием органических кислот,
которые и растворяют гидроксиапатит зубов. В этом состоит причи¬
на кариеса.
Вместе с гидроксиапатитом в костной ткани могут осаждаться
и другие ионы. В первую очередь это относится к фторид-аниону.
Замена гидроксильной группы на фтор в гидроксиапатите приводит
к еще менее растворимому и более механически прочному фтор-
апатиту Са5р(Р04)э- Присутствие микроколичеств фторапатита
в костной ткани сообщает ей прочность. Особенно важен фторапа-
тит как прочное кислотоустойчивое покрытие зубов - зубная эмаль.
Очевидна необходимость добавок фторид-иона в зубные пасты.
Замещать кальций в составе костной ткани могут катионы дру¬
гих металлов второй группы Периодической системы: магния, бе¬
риллия и стронция. Если содержание первого в малых количествах
в костях естественно, причем это малое количество легко объясни¬
мо лучшей растворимостью фосфата магния по сравнению с каль¬
циевыми солями, то появление других крайне нежелательно. Ионы
стронция, замещая ионы кальция в костях, вызывают их ломкость
(стронциевый рахит). Особо опасен радиоактивный изотоп строн¬
ций-90, который при попадании в состав костной ткани облучает
костный мозг и нарушает кроветворные процессы. Даже небольшое
количество бериллия в составе костей приводит к заболеванию,
именуемому бериллозом, состоящем в размягчении костей. Это -
типичные примеры микроэлементозов (см. главу 10).
В некоторых органах человека могут откладываться и другие
малорастворимые соединения. Так, карбонат кальция, отлагаясь на
стенках кровеносных сосудов, вызывает кальциноз.
232
Поче*Й!о-каменная болезнь состоит в образовании камней - от¬
ложений солей различного состава: урата кальция - соли мочевой
кислоты (промежуточное вещество азотного обмена), малораство¬
римых фосфатов или оксалатов кальция. Их отложению способству¬
ет увеличение pH мочи, то есть щелочная среда. Кстати, одной из
причин гибели при отравлении этиленгликолем является закупорка
сосудов множественным отложением малорастворимого оксалата
кальция, который выпадает в осадок из-за того, что в плазме резко
возрастает концентрация щавелевой кислоты - продукта метаболи¬
ческого окисления этиленгликоля.
Печеночно-каменная болезнь связана с образованием карбоната
кальция, а также билирубината' кальция.
Химические приемы лечения этих патологических состояний
основаны на действии препаратов, растворяющих камни, для чего
кроме химиотерапии прибегают к специальным диетам и минераль¬
ным водам.
Отдельно следует сказать о малой растворимости сульфидов ря¬
да катионов <1-металлов (ртуть, кадмий, таллий и др.), а также ка¬
тионов свинца(Н) и мышьяка(Ш). Органические вещества, содер¬
жащие остаток сероводорода - сульфгидрильную группу, в первую
очередь, белки и ферменты, по этой же причине прочно и необра¬
тимо связываются с такими катионами В результате имеет место
денатурация белков и потеря ферментами их активности. Следова¬
тельно, эти ионы сильно токсичны Исходя из общих соображений,
простейшим противоядием при попадании ионов тяжелых металлов
в организм должны быть растворимые сульфиды, реагирующие с
такими катионами с образованием малорастворимых сульфидов, что
понижает концентрации токсичных катионов Для этой цели приме¬
няют так называемое щелочное сероводородное питье, а именно
сульфид натрия:
Нд2+ + Э2- -> НдвХ
Большую группу противоядий составляют органические вещест¬
ва, содержащие сульфгидрильные группы (см главу 21)
Следовательно, гетерогенное равновесие наряду с другими ви¬
дами равновесных биопроцессов вносит существенный вклад в об¬
щий гомеостаз организма, тканей, органов
Реакции осаждения широко применяют в клиническом, гигиени¬
ческом и фармацевтическом анализе С их помощью определяют со¬
держание хлорид-иона в плазме, моче и желудочном соке, анализи¬
руют токсичные ионы и др
* Билирубин - один из продуктов разложения гемоглобина
233
Итак, гомеостаз организма поддерживается за счет совокуп¬
ности взаимосвязанных равновесных систем
Водио-электролитный баланс обеспечивает постоянство осмоти¬
ческого давления, необходимого для поддержания постоянства кон¬
центраций множества компонентов биосред и для равномерного
транспорта питательных веществ в клетку и выведения продуктов
метаболизма из нее. Одновременно поддерживается постоянство
ионной силы внутриклеточных растворов и плазмы крови Ее вели¬
чина сильно сказывается на ферментативной активности
С ионной силой тесно связаны процессы диссоциации электро¬
литов биосред, в первую очередь, кислотно-основное равновесие
компонентов биологических буферных систем. С их помощью под¬
держивается кислотно-основное состояние отдельных тканей, био¬
сред и организма в целом. Тем самым достигается постоянство pH,
необходимое для успешного функционирования ферментов, чья ак¬
тивность чрезвычайно чувствительна к изменению кислотности.
В свою очередь, металло-лигандный баланс, являющийся важ¬
ным параметром гомеостаза, как и гетерогенное равновесие, регу¬
лирующее формирование твердых структур организма, зависят от
величины pH и тесно связаны с кислотно-основным состоянием.
Окислительно-восстановительные процессы, являющиеся по¬
ставщиком энергии биологических систем, представляют собой со¬
вокупность серии взаимосвязанных равновесных окислительно¬
восстановительных систем, причем часть из них также является
рН-зависимыми процессами. Таким образом, интенсивность проте¬
кания биологических окислительно-восстановительных превращений
тесно увязана с кислотно-щелочным состоянием организма.
Следовательно, понимание сути многочисленных патологических
состояний и болезней требует глубокого комплексного анализа хи¬
мических причин или последствий этих явлений с позиций наруше¬
ния функционирования равновесных систем, необходимых для под¬
держания гомеостаза. Это справедливо как для какой-либо разно¬
видности анемии, связанной с нарушением металло-лиганд-ного об¬
мена, так и для лучевого поражения, вызывающего в первую оче¬
редь нарушение кислотно-основного состояния плазмы крови, для
случаев отравления ионами тяжелых металлов, нарушающего гете¬
рогенное равновесие биосред, или для такой инфекционной болез¬
ни, как холера, самым важным последствием которой является
нарушение водно-электролитного баланса организма.
Часть III
МИКРОГЕТЕРОГЕННЫЕ СИСТЕМЫ
Вопросы, связанные с... кол¬
лоидальным состоянием ве¬
щества. относятся к числу
таких передовых, от решения
которых много подвинется
правильное понимание меха¬
низма множества химических
реакций
Д И Менделеев
Глава 13. ФИЗИЧЕСКАЯ ХИМИЯ
ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ
Общая площадь, поделенная
на количество молекул, равна
площади, занимаемой каждой
молекулой
И Ленгмюр
Биологические структуры являются гетерогенными системами,
состоящими не менее чем из двух фаз, разделяемых поверхностью
раздела. Граница раздела фаз, поверхность раздела, отличается по
термодинамическим параметрам от обеих фаз. Поэтому на поверх¬
ности раздела фаз имеют место поверхностные явления - по¬
верхностное натяжение, адсорбция и др. Так как многие физиологи¬
ческие процессы (дыхание, пищеварение, экскреция и др ) протека¬
ют на поверхности биомембран, для их понимания требуется знание
основных закономерностей поверхностных явлений
13.1. Поверхностная энергия
и поверхностное натяжение
Поверхностный слой, возникающий на границе различных фаз,
следует рассматривать как самостоятельную фазу толщиной в
несколько молекул В отдельных случаях поверхностный слой имеет
толщину, равную диаметру молекулы, и его называют мономо-
лекулярным. Поэтому поверхностный слой нужно рассматривать
как микрогетерогенную систему.
235
Поверхностный слой резко отличается по своим свойствам от
свойств фаз, которые он разделяет. Молекулы, атомы, ионы, нахо¬
дящиеся на границе раздела фаз, не равноценны тем же частицам,
находящимся в объеме фазы.
Силы, действующие на молекулы, находящиеся внутри жид¬
кости, одинаковы со всех сторон, и их равнодействующая Р равна
нулю. Силы, действующие на молекулы повехностного слоя грани¬
цы раздела жидкости с ее паром, не одинаковы со стороны раствора
(снизу и с боков) и газообразной фазы (сверху). Молекулярные
взаимодействия сверху отсутствуют, равнодействущая сил Ё не
равна нулю и направлена внутрь жидкой фазы (рис. 13.1). Для вы¬
хода молекул на поверхность требуется выполнить работу Ws про¬
тив этой силы. В итоге формируется поверхностный слой с избы¬
точной поверхностной энергией Гиббса 0$.
Все возможные поверхности раздела в зависимости от агрегат¬
ного состояния граничащих фаз делят на подвижные поверхности
раздела между жидкостью и газом (ж-г), двумя несмешивающимися
жидкостями (ж-ж) и неподвижные поверхности раздела между
твердым телом и газом (т-г), твердым телом и жидкостью (т-ж),
твердым телом и твердым телом (т-т).
Рис. 13.1. Взаимодействие между молекулами
в объеме жидкости и в поверхностном слое.
При измельчении какой-либо твердой или жидкой фазы суммар¬
ный объем и масса остаются теми же самыми, тогда как суммарная
площадь поверхности раздела возрастает.
Чтобы учесть влияние поверхности на свойства системы, вво¬
дят понятие удельной поверхности (Буд) фазы - величины, из¬
236
меряемой суммарной площадью граничной поверхности фазы (5сум),
отнесенной к ее объему (V):
5уд = 5сум/У.
Влияние поверхности раздела фаз на свойства системы воз¬
растает с увеличением удельной поверхности.
Энергия Гиббса й системы из двух фаз состоит из двух сла¬
гаемых - энергии Гиббса объемных фаз <3у и поверхностной энер¬
гии Гиббса
й = йу + Об-
Энергия Гиббса объемных фаз пропорциональна их массе, а,
следовательно, объему, занимаемому системой:
= кУ.
Поверхностная энергия Гиббса системы пропорциональна меж-
фазной поверхности:
йз = оБ,
где а - коэффициент пропорциональности, называемый поверх¬
ностным натяжением. Его величину измеряют в кДж/м2 (Н/метр)
или в Дж/см2.
Поверхностное натяжение а есть величина, измеряемая
энергией Гиббса, приходящейся на единицу площади поверхностно¬
го слоя. Оно численно равно работе, которую необходимо совер¬
шить в данной системе для образования в ней единицы поверхности
раздела фаз при постоянной температуре.
С учетом предыдущего получим уравнение:
й = кУ + сБ
0/У= к + с5уд.
Следовательно, энергия Гиббса, приходящаяся на единицу объ¬
ема системы, линейно увеличивается с увеличением ее удельной
поверхности. При малых значениях удельной поверхности 5уд по¬
верхностной энергией Гиббса можно пренебречь. При больших зна¬
чениях Буд с ней необходимо считаться.
Если поверхность кожи человека составляет 1.5 м2, то поверх¬
ность эритроцитов имеет величину уже 3 ООО м2. Трудно оценить
величину суммарной поверхности, разделяющей все клетки орга¬
низма, если помнить, что их общее число составляет около 1014, а
к ним еще следует добавить микроорганизмы кишечной флоры, ко¬
личество которых больше, чем общее количество клеток организма
Организм представляет собой совокупность систем с сильно раз¬
237
витыми поверхностями раздела (кожные покровы, стенки кровенос¬
ных сосудов, оболочки органов, клеточные мембраны, мембраны
органелл, то есть клеточных структур и т д ) В живых системах,
следовательно, величина поверхностной энергии должна иметь
большую величину
Поверхностное натяжение различных жидкостей (таблица 13 1)
на границе жидкость - газ возрастает с увеличением взаимодей¬
ствия между молекулами жидкости, являясь максимальным для во¬
ды из числа веществ, представленных в таблице 13 1.
Таблица 13 1
Поверхностное натяжение жидкостей на границе
с воздухом (298 К)
Жидкость
а, Н/м
Жидкость
а, Н/м
Вода
0.0728
Глицерин
0.0647
Плазма крови
0 0454
Этанол
0.0223
Уксусная кислота
0 0276
Оливковое масло
0.0330
Поверхностное натяжение жидкостей уменьшается с ростом
температуры и при критической температуре приобретает нулевое
значение, так как поверхность раздела фаз исчезает.
С ростом давления поверхностное натяжение на границе жид¬
кость - газ уменьшается, так как концентрация молекул в газовой
фазе возрастает, а, следовательно, увеличивается взаимодействие с
ними молекул поверхностного слоя, и сила Г уменьшается.
Способность растворенных веществ изменять поверхностное на¬
тяжение растворителя называется поверхностной активностью.
Растворенные вещества могут повышать, понижать или вообще не
влиять на поверхностное натяжение жидкостей.
Поверхностное натяжение биологических жидкостей использу¬
ют в диагностических целях. Так, поверхностное натяжение плазмы
крови варьируется в зависимости от заболевания (травма, шок, ра¬
на, рак и др.) С возрастом поверхностное натяжение сыворотки
крови уменьшается Поверхностное натяжение играет значительную
роль в таких явлениях, как деление клеток и движение бактерий,
фагоцитоз, влияет на проницаемость клеточных мембран и др.
Существует много несложных методов измерения поверхностного
натяжения, которые используют в медико-биологической практике.
Мерой поверхностной активности служит производная поверх¬
ностного натяжения по концентрации, взятая со знаком минус:
238
где de и'Но - бесконечно малые изменения концентрации и поверх¬
ностного натяжения.
В узких интервалах концентраций производная может быть за¬
менена отношением конечных изменений:
Да
g = - —•
Дс
Поверхностное натяжение на границе раздела жидкость - жид¬
кость зависит от природы жидкостей. Чем меньше силы молекуляр¬
ного взаимодействия между их молекулами, тем поверхностное на¬
тяжение больше. Температура и концентрации растворенных ве¬
ществ оказывают такое же влияние, как и в случае границы раздела
жидкость - газ. Давление на эту величину почти не влияет.
Любая система в соответствии со вторым началом термодина¬
мики стремится самопроизвольно перейти в такое состояние, в ко¬
тором она обладает минимальным запасом энергии Гиббса G. Сле¬
довательно она стремится к минимуму поверхностной энергии Гиб¬
бса (Gs —» min). Однокомпонентная жидкая система (а = const),
может понизить запас поверхностной энергии Гиббса только одним
путем - принять форму, при которой поверхность раздела мини¬
мальна (s -» min). Минимальной же поверхностью обладает сфера,
чем объясняется форма капель практически любой жидкости - во¬
ды, ртути, органических растворителей.
Раствор как двух- (или более) компонентная система может по¬
низить запас поверхностной энергии Гиббса и другим способом -
концентрированием на границе раздела фаз компонента с меньшим
поверхностным натяжением.
Самопроизвольное изменение концентрации растворенного ве¬
щества на границе раздела фаз называется адсорбцией. Поглоти¬
тель принято называть адсорбентом, а поглощаемое вещество -
адсорбтивом или адсорбатом. Величину адсорбции измеряют ко¬
личеством молей адсорбтива, приходящихся на единицу площади
поверхности адсорбента (Г, моль/см2).
Величину адсорбции в растворах можно определять по измене¬
нию поверхностного натяжения с концентрацией растворенного
вещества, используя уравнение Гиббса:
Г = °Р Aq
RT Дс ’
где Ср - равновесная концентрация вещества, моль/л или
кмоль/м3; Т - абсолютная температура; Да/Дс - поверхностная
активность (изменение поверхностного натяжения с концентрацией
при неизменной поверхности, Дж/м2 или Н/м); R - универсальная
газовая постоянная (8.31 103 Дж/кмоль град).
239
13.2. Адсорбция на границе раздела
твердое тело - газ
Адсорбцию газа на твердой поверхности измерять количеством
адсорбтива на единице поверхности затруднительно, поскольку
сложно оценить величину поверхности адсорбента Твердая поверх¬
ность является неровной Неровности - микродефекты - превышают
размеры молекул на два-три порядка (сотни ангстремов) Микроде¬
фекты увеличивают удельную поверхность системы и создают неко¬
торый избыток запаса поверхностной энергии Гиббса
Поэтому удельная адсорбция газа на твердом теле измеряется
его количеством, поглощенным единицей массы твердого тела
(моль/кг или моль/г) Количество поглощенного газа можно опре¬
делить по привесу адсорбента и по падению давления адсорбтива
Адсорбция обусловлена вандерваальсовыми силами Как и в
жидкости (см. рис. 13.1), силовое поле частиц адсорбента в его
объеме полностью компенсировано силовыми полями соседних час¬
тиц. Силовые поля частиц адсорбента, расположенных на его по¬
верхности, не уравновешены. Поэтому на поверхности адсорбента
действуют остаточные силы, способные притягивать молекулы ве¬
ществ, попавших на эту поверхность. Адсорбция в первую очередь
происходит на участках поверхности с максимальным локальным
значением поверхностной энергии Гиббса - на так называемых ад¬
сорбционных центрах.
В зависимости от характера действующих сил различают фи¬
зическую и химическую адсорбцию. Для физической адсорбции
характерна небольшая энтальпия, а потому процесс обычно обра¬
тим. Адсорбционное равновесие устанавливается очень быстро.
Если поглощение адсорбтива происходит вследствие его хими¬
ческого взаимодействия с адсорбентом, тогда имеет место хемо¬
сорбция Теплоты химической адсорбции находятся в пределах
от -40 до -400 кДж/моль, и потому процесс имеет тенденцию к
необратимости.
Адсорбция зависит от температуры, давления и природы ад¬
сорбтива, удельной поверхности и природы адсорбента.
На поверхности твердого тела при прочих равных условиях
лучше адсорбируются те газы, которые легче конденсируются в
жидкость. Так, активированный уголь хорошо адсорбирует аммиак
(Ткип = -33° С), но практически не адсорбирует оксид углерода(Н)
(ТКип = -190° С). Поэтому обычный противогаз не эффективен при
высоких концентрациях угарного газа.
Адсорбция зависит от давления адсорбтива, причем с ростом
давления адсорбция возрастает до некоторого предельного значе¬
240
ния а„ (рисгЛ3.2). Эта зависимость носит название изотермы ад¬
сорбции.
Так как адсорбция газов на твердых телах экзотермична, с рос¬
том температуры она уменьшается (рис. 13.2).
Рис. 13.2. Рассчитанные изотермы адсорбции газа
твердым телом при различных температурах.
Для изотермы адсорбции используют уравнение, выведенное
И. Ленгмюром, исходя из следующих положений:
1) адсорбция молекул адсорбата происходит только на вполне
определенных участках поверхности адсорбента, называемых ад¬
сорбционными центрами. При малых давлениях заполняется
лишь часть поверхности, соответствующая адсорбции,
2) адсорбционный центр удерживает только одну молекулу ад¬
сорбата. На поверхности адсорбционных центров образуется моно-
молекулярный слой, соответствующий предельной адсорбции Ины¬
ми словами, можно говорить о мономолекулярной адсорбции,
3) адсорбцию рассматривают как равновесный процесс, т е. при
равенстве скоростей адсорбции и десорбции.
В соответствии с законом действующих масс скорость адсорб¬
ции Уа пропорциональна давлению адсорбата р и числу свободных
центров а» - а/Ыд (^ - постоянная Авогадро)
Уа = кар(а„ - а)/ЫА,
где ка - константа скорости адсорбции.
Соответственно скорость десорбции Уд пропорциональна числу
занятых центров а
V, - к,а/нд.
где кд - константа скорости десорбции.
241
При равновесии скорости адсорбции и десорбции равны. Отсюда
получают уравнение адсорбции Ленгмюра
где а - адсорбция при данных условиях, а» - предельная адсорбция,
К = константа адсорбционного равновесия, К = кд/ка
На практике при больших концентрациях адсорбата на изотерме
после участка насыщения а», обычно наблюдается резкое увеличе¬
ние адсорбции (рис 13.3) Причина этого - переход от мономолеку-
лярной адсорбции к полимолекулярной, когда на адсорбционном
центре может находиться более одной молекулы адсорбата Проис¬
ходит такое явление вследствие капиллярной конденсации -
перехода поглощаемого газа или пара в жидкое состояние в узких
порах адсорбента. Она вызвана тем, что давление насыщенного па¬
ра над мениском жидкости в капилляре меньше давления его пара
над плоской поверхностью.
Рис. 13.3. Практическая изотерма адсорбции газа
твердым телом.
Кроме того, на процессы адсорбции накладывается и процесс
абсорбции - поглощения вещества не только поверхностью, но
и всей массой адсорбента (тогда речь идет об абсорбентах и абсорб-
тивах).
В реальных условиях разделить процессы адсорбции, капилляр¬
ной конденсации и абсорбции трудно. Поэтому для характеристики
взаимодействия сорбента с сорбтивом применяется более общий
термин - сорбция, сложный суммарный физико-химический про¬
цесс адсорбции, абсорбции и капиллярной конденсации.
Сорбция газов твердыми телами играет большую роль в процес¬
сах газообмена организма с окружающей средой. Сорбция газов и
паров на твердых поверхностях используется в системах автоном¬
ной очистки воздуха (противогаз, системы жизнеобеспечения в
242
замкнутых пространствах подводных лодок и космических ко¬
раблей). Твердые сорбенты нашли разностороннее применение для
очистки газов и летучих органических растворителей в технике и
промышленности.
13.3. Адсорбция на границах газ - жидкость,
жидкость - жидкость, твердое тело - жидкость
При растворении в данной жидкости какого-либо вещества
можно представить три случая распределения растворенного ве¬
щества между граничным слоем и объемом фазы (рис. 13.4), наблю¬
дая зависимость поверхностного натяжения при постоянной тем¬
пературе от концентрации растворенного вещества - изотерму
поверхностного натяжения. На рис. 13.4 представлены изотермы
поверхностного натяжения для этих трех случаев.
а
Рис. 13.4. Зависимость поверхностного натяжения
от концентрации для ПИВ, ПНВ и ПАВ.
1. Растворенное вещество понижает поверхностное натяжение
растворителя (сраСтвор < °о) Такие вещества получили название
поверхностно-активных (ПАВ). Для них 5 > 0, система уменьшает
запас поверхностной энергии Гиббса, если концентрация раство¬
ренного вещества в поверхностном слое (с5) больше, чем в объеме
фазы (су)
2. Растворенное вещество повышает поверхностное натяжение
растворителя (араствор > а0). Такие вещества называют поверхно-
стно-инактивными (ПИВ) В этом случае 5 < О, ПИВ накапливают¬
243
ся в объеме фазы (cs < cv), так как при этом запас поверхностной
энергии Гиббса уменьшается. По отношению к воде ПИВ - сильные
электролиты (кислоты, основания, соли) и сильно полярные орга¬
нические соединения (глицерин, аминокислоты и др)
3. Растворенное вещество не изменяет поверхностного натяже¬
ния растворителя (араствор = а0); поверхностно-неактивными (ПНВ)
по отношению к воде веществами является, к примеру, сахароза и
ряд других углеводов У веществ, не влияющих на поверхностное
натяжение растворителя, поверхностные и объемные концентрации
одинаковы (с$ = су)
Из-за биологической значимости подробнее рассмотрим ПАВ.
Поверхностно-активные вещества (ПАВ) - это вещества,
понижающие поверхностное натяжение, или, согласно уравнению
Гиббса, обладающие положительной адсорбцией. Их молекулы ди-
фильны, т.е содержат одновременно полярную группу (-ОН,
-СООН, -NH2, -SO3H и др.) и неполярную углеводородную цепь. Кон¬
центрируясь на границе раздела вода - воздух, дифильные молеку¬
лы ориентируются своими полярными группами в воду, а неполяр¬
ные углеводородные радикалы выталкиваются в воздух, тем самым
достигается уменьшение поверхностного натяжения.
ПАВ делятся на анионоактивные, катионоактивные,
неионогенные и амфотерные.
К анионоактивным ПАВ относятся:
а) соли высших карбоновых кислот RCOONa (Сц < R < Cie, на¬
пример, пальмитат натрия CisH3iCOO" Na+);
б) соли сульфокислот R-CeH4-S03" Na+ (R > С12, например, доде-
цилбензолсульфонат натрия Ci2H25-C6H4-S03' Na+);
в) соли алкилсерных кислот R0S03- Na+ (R > Ci2, например,
додецилсульфат натрия Ci2H2s-0-S03" Na+).
К катионоактивным ПАВ относятся:
а) соли и основания тетраалкиламмония [RR1R2R3N+]X-
(Се < R < С«), например, триметилцетиламмоний хлорид
[С1вНзз-М(СНз)з]+СГ;
б) соли алкилпиридиния (например, цетилпиридиний иодид)*.
Неионогенные ПАВ представлены соединениями следующей
общей формулы: R-X(CH2CH20)nH [R = Aik, X = О, N, S или
-СОО-, -CONH-, -СвН40-, п = 8-12; например, препарат ОП-Ю -
CeHi7-CeH4-0-(CH2CH20)ioHJ.
’ С формулой этого вещества так же, как и с формулами других ПАВ и
их названиями, можно ознакомиться в соответствующих главах данного
учебника.
244
Неионогенными ПАВ являются также высшие предельные
спирты, например, цетиловый спирт С^НззОН, длинноцепочечные
амины и др.
Амфотерные ПАВ имеют общую формулу: 0+-Р-К" (С9 < Р < С19,
0+ - основная группа, К" - кислотная группа, например, М-додецил-
р-аланин С^Нгз-МК^-СНг-СНг-СОСГ).
Существует правило Дюкло-Траубе: для низших членов го¬
мологического ряда жирных кислот, спиртов и аминов с увеличени¬
ем углеводородной цепи на группу -СНг- поверхностная активность
веществ возрастает в 3-3.5 раза при одинаковой молярной концент¬
рации. Это правило иллюстрируется рис. 13.5, где представлена
зависимость поверхностного натяжения от концентрации для гомо¬
логов карбоновых кислот.
Рис. 13.5. Семейство изотерм поверхностного натяжения
для гомологического ряда карбоновых кислот.
Моющее действие ПАВ заключается в следующем. По преиму¬
ществу частицы загрязняющих веществ (например, жира) гидро¬
фобии, а, следовательно, не смачиваются водой. Поэтому чистая
вода обладает слабым моющим действием. Если применить ПАВ,
его молекулы адсорбируются на частицах загрязнителя, ориентиру¬
ясь гидрофобными участками к его молекулам, а гидрофильны¬
ми - к молекулам воды (рис. 13.6).
Поскольку вода будет находиться в кон¬
такте только с полярными группами, которые
легко ассоциируются с ней, частицы загряз¬
няющих веществ окажутся растворенными
в воде и будут легко смываться ею В
результате молекулы ПАВ постепенно про¬
никают между очищаемой поверхностью и Рис. 13.6. Моющее
частицами загрязнителя Это явление назы- действие ПАВ.
245
вают расклинивающим эффектом. В итоге частицы грязи отделяют¬
ся от загрязненной поверхности
В живых системах ПАВ играют совершенно исключительную
роль, совмещая в своей структуре две системы - гидрофильную и
гидрофобную. Именно такие вещества формируют основу тех кле¬
точных мембан, которые обладают и гидрофильными, и гидрофоб¬
ными свойствами одновременно. Вещества этих клеточных мембран
могут в сильной мере отличаться химическим строением, однако их
общая особенность - наличие двух частей с принципиально разли¬
чающимися физико-химическими свойствами.
Несмотря на структурные различия, множество молекул при¬
родных ПАВ, например, содержащие остаток фосфорной кислоты
фосфолипиды и сфиголипиды, имеют весьма сходные размеры -
длину около 30 А и диаметр около 5 А.
Липофильная часть природных ПАВ состоит из углеводородного
радикала с достаточной для проявления липофильности величиной.
Этот радикал может быть линейного строения, и тогда его величина
должна превышать 12 углеродных атомов. Набор таких радикалов,
реализующихся в биоструктурах, превышает сотню разновидностей.
Другой тип радикалов - полициклические радикалы стероидного ря¬
да, как это, к примеру, имеет место в молекулах желчных кислот,
высокая поверхностная активность которых способствует дисперги¬
рованию пищи в мельчайшие частицы, необходимые для их после¬
дующего усвоения.
Гидрофильная часть природных ПАВ имеет в своем составе по¬
лярный остаток фосфорной, серной или карбоновой кислоты. Это
может быть также аммонийный ион (в белковых ПАВ) или сахарид-
ный остаток, например, в молекулах некоторых сфинголипидов.
Для однородной гладкой поверхности, каковыми являются гра¬
ницы раздела газ - жидкость и жидкость - жидкость, широко рас¬
пространена теория мономолекулярной адсорбции Ленгмюра. Со¬
гласно ей, адсорбция является равновесным процессом.
Уравнение изотермы адсорбции Ленгмюра на границе жид¬
кости имеет следующий вид:
где Г» - предельная величина адсорбции (предельная концентрация
вещества на 1 см2 поверхности жидкости, моль/см2); К - константа
адсорбционного равновесия, равная отношению констант скоростей
процессов десорбции и адсорбции, ср - равновесная концентрация
адсорбтива, моль/см2
246
тт
Для экспериментального определения предельной величины
адсорбции Г» и константы адсорбционного равновесия К это урав¬
нение преобразуют в уравнение прямой следующим образом:
1 _ К + Ср _1_ = _1_ _К__1_
Г Г^'Ср ' Г Гм + ГвСр'
Затем строят зависимость — от —, тангенс угла наклона пря-
г ср
мой равен (рис. 13.7). Прямая отсекает на оси ординат отре-
- 1
зок, равный —.
Рис. 13.7. Определение констант изотермы
адсорбции Ленпиюра.
При малых концентрациях ПАВ углеводородные радикалы
“лежат” на поверхности полярной жидкости, а полярные группи¬
ровки погружены в нее (рис 13 8, а) При достижении предельной
величины адсорбции Г.. молекулы ПАВ образуют на поверхности
мономолекулярный слой, в котором неполярные углеводородные
радикалы располагаются перпендикулярно к поверхности (“часто¬
кол Ленгмюра”, рис 13 8, б)
В этом случае можно рассчитать площадь 5, занимаемую одной
молекулой, и длину молекулы /
5 = 1 и , = МГ^
" Р
247
где Ыд - число Авогадро, Мир- соответственно молекулярная
масса и плотность адсорбтива
Следовательно, зная константу Г» из уравнения Ленгмюра,
можно определять размеры молекул
Рис. 13.8. Молекулы ПАВ на поверхности воды:
а - при малых концентрациях, б - в мономолекулярном слое.
При высоких концентрациях адсорбтива (когда имеет место бо¬
лее чем мономолекулярная адсорбция) справедливо уравнение
Фрейндлиха, в котором выражается эмпирическая зависимость
адсорбции от концентрации (или давления) в сравнительно широких
пределах концентраций:
где а - удельная адсорбция (количество молей адсорбтива, которое
может поглотить 1 г адсорбента); ср - равновесная молярная кон¬
центрация адсорбтива; Ь и п - постоянные, определяемые опыт¬
ным путем.
Для адсорбции из газовой фазы вместо концентрации с исполь¬
зуют р - равновесное давление адсорбируемого газа над поверх¬
ностью адсорбента.
Эмпирические постоянные Ь и п можно определять графически.
Логарифмируя уравнение Фрейндлиха, получаем уравнение прямой
линии:
1ц а = ^ Ь + п ^ ср.
Определив значение а при двух концентрациях и построив гра¬
фик в логарифмических координатах, определяют константы Ь и п
(см. рис. 13.9).
а
б
248
їдь
1 ід ср
Рис. 13.9. Определение констант в изотерме
адсорбции Фрейндлиха.
13.4. Адсорбция на границе
твердое тело - раствор
Адсорбция растворенных веществ твердыми адсорбентами яв¬
ляется более сложным процессом, чем выше описанные случаи, так
как она осложнена присутствием третьего компонента - раствори¬
теля, молекулы которого могут конкурировать с молекулами ад-
сорбтива за места на поверхности адсорбента, взаимодействовать с
адсорбтивом и с поверхностью адсорбента, если он является элек¬
тролитом.
Молекулярная адсорбция. Неэлектролиты и слабые электро¬
литы на поверхности адсорбента адсорбируются из растворов в виде
молекул. Такой процесс называют молекулярной адсорбцией.
В результате адсорбции концентрация растворенного вещества в
растворе уменьшается.
Л. А. Ребиндер сформулировал правило выравнивания полярно¬
стей: на полярных адсорбентах лучше адсорбируются полярные ад-
сорбтивы из малополярных растворителей; на неполярных адсор¬
бентах - неполярные адсорбтивы из полярных растворителей.
Для системы адсорбент - адсорбтив влияние природы раство¬
рителя на адсорбцию может быть сформулировано в виде другого
правила (правило Шилова)- чем лучше растворяется адсорбтив в
растворителе, тем он хуже адсорбируется поверхностью твердого
адсорбента, чем хуже растворяется - тем лучше адсорбируется
Эти правила можно объяснить тем, что процесс адсорбции из
растворов определяется суммарной энергией взаимодействия ак¬
тивных адсорбционных центров адсорбента между молекулами ад-
сорбата и молекулами растворителя.
249
Малополярные адсорбенты, например, активированный уголь,
лучше адсорбируют неполярные органические соединения, причем
тем больше, чем выше их молекулярная масса.
Полярные адсорбтивы лучше адсорбируются на поверхности
ионных кристаллов На поверхности адсорбентов, являющихся ок¬
сидами (силикагель, окись алюминия и др.), как правило, имеются
гидроксильные группы, поэтому они хорошо адсорбируют воду,
спирты, амины и другие полярные соединения.
Как известно, адсорбция экзотермична, а поэтому с повышением
температуры уменьшается Однако если растворимость адсорбтива
в данном растворителе падает с ростом температуры, то адсорбция
из раствора твердым адсорбентом может увеличиваться.
С ростом концентрации раствора адсорбция на границе раздела
твердое тело - раствор возрастает до предельного значения. Анализ
экспериментально наблюдаемой изотермы аналогичен анализу, при¬
веденному в предыдущем разделе. Для этого используются уравне¬
ния Фрейндлиха и Ленгмюра.
При адсорбции ПАВ на границе раздела твердое тело - рас¬
твор так же, как и на границе раствор - газ, имеет место простран¬
ственная ориентация молекул адсорбата.
активированный а (ЗЮ2)п ^
уголь
Рис. 13.10. Адсорбция ПАВ на границе раздела твердое тело - жидкость.
В системе полярный адсорбент - неполярный растворитель мо¬
лекулы адсорбтива-ПАВ полярной частью обращены к поверхности
адсорбента, а неполярная их часть погружена в растворитель (рис.
13.10, б). В случае системы неполярный адсорбент - полярный рас¬
творитель, наоборот, неполярная часть молекулы обращена к по¬
верхности адсорбента, а полярная часть погружена в растворитель
(рис. 13.10, а).
В качестве примера можно привести поведение дифильного ве¬
щества по отношению к неполярному адсорбенту - активированно¬
му углю в полярной среде - воде. В таких условиях молекулы
дифильной природы будут адсорбироваться на поверхности угля,
250
ориентируюсь по отношению к нему своими гидрофобными участка¬
ми. Напротив, те же молекулы в неполярном растворителе, бензоле,
адсорбируются на полярном адсорбенте (силикагеле БЮг), связы¬
ваясь с адсорбентом полярными участками молекул.
В системе полярный растворитель - малополярный адсорбент -
адсорбция ПАВ подчиняется правилу Дюкло-Траубе (см. выше).
При адсорбции ПАВ из неполярных растворителей полярными
адсорбентами выполняется обращенное правило Дюкло-Трау¬
бе: с ростом длины радикала адсорбция уменьшается. Обращение
правила объясняется тем, что с ростом углеводородной цепи рас¬
творимость ПАВ в неполярных растворителях увеличивается.
Закономерности распределения веществ между раствором и
твердой фазой важны для понимания процессов обмена веществ.
Законы распределения твердого вещества между неполярной фазой
липидного слоя биомембран и полярной фазой - внутри- и межкле¬
точной жидкостью - управляют поступлением питательных веществ
и удалением продуктов метаболизма. С явлением адсорбции на кле¬
точных мембранах из полярных биосред организма связано физио¬
логическое действие многих лекарств (барбитураты, анестезирую¬
щие средства и др.) и токсическое действие ОВ (например, иприта
и люизита). Существует специальный механизм окисления непо¬
лярных или малополярных чужеродных для организма веществ
(ксенобиотиков), в результате чего эти вещества приобретают по¬
лярные группировки и их способность адсобироваться на липидных
мембранах падает. В итоге они переходят в водную фазу, откуда
уже могут покинуть организм с выделениями
Молекулярная адсорбция твердыми адсорбентами из растворов
широко распространена в медицинской практике. Уже давно при
отравлениях растительными ядами, токсинами, малополярными ле¬
карствами (например, барбитуратами) применяют активированный
уголь для их удаления из пищеварительного тракта. В настоящее
время осуществляют сорбционную детоксикацию крови и лимфы
больного пропусканием их через активированный уголь. Гемо- и
лимфосорбция позволяют удалить из организма токсичные органи¬
ческие жидкости (например, дихлорэтан и другие галогенопроиз¬
водные), фосфорорганические соединения и др Адсорбцию исполь¬
зуют для очистки питьевой воды и сточных вод
13.5. Адсорбция сильных электролитов
В растворах сильных электролитов растворенное вещество на¬
ходится в полностью ионизированном состоянии, ионы адсорбиру¬
ются лишь на полярных и практически не адсорбируются на непо¬
лярных адсорбентах
251
Поэтому основным фактором, обусловливающим специфичность
адсорбции сильных электролитов, является знак заряда поверхности
полярного адсорбента - на положительно заряженных участках
поверхности адсорбируются из раствора анионы, на отрицательно
заряженных - катионы Величина и знак заряда иона, его радиус
и степень сольватации также влияют на процесс адсорбции
Например, чистый уголь является неполярным адсорбентом, на
котором электролиты почти не адсорбируются Однако поверхность
угля, окисленная кислородом при нагревании, приобретает поляр¬
ные группы. В водных растворах такая поверхность заряжается по¬
ложительно и начинает адсорбировать анионы. Если поверхность
угля обработать при нагревании водородом, она в водных растворах
заряжается отрицательно и адсорбирует катионы.
Адсорбционная способность ионов (особенно катионов) воз¬
растает с ростом их заряда. При одинаковых зарядах адсорбционная
способность больше у тех, радиус которых в сольватированном
состоянии меньше. В соответствии с этим правилом ионы по их
адсорбционной способности располагаются в ряды, именуемые
лиотропными рядами.
Лиотропный ряд адсорбции однозарядных катионов из воды
имеет вид:
Катион Сэ+ РЬ+ N4/ К* N3* Ы+
Радиус, А 1.69 1.48 1.43 1.33 0.95 0.60
Для однозарядных анионов лиотропный ряд выглядит так:
Анион N03" Г Вг" СГ ?'
Радиус, А 2.57 2.16 1.96 1.81 1.36
Различают три вида адсорбции сильных электролитов: экви¬
валентную, избирательную и обменную.
Эквивалентная адсорбция характеризуется тем, что и катио¬
ны, и анионы адсорбируются на поверхности адсорбента в эквива¬
лентных количествах. Встречается редко.
Избирательная адсорбция подчиняется правилу К. Фаянса:
на поверхности данного адсорбента преимущественно адсорбируют¬
ся те ионы, которые участвуют в достройке его кристаллической
решетки (или родственные им).
Рассмотрим образование осадка хлорида серебра по реакции:
КС1 + АдГЮз -►АдС14 + КГЮ3.
Если реагируют эквивалентные количества солей, поверхность
осадка не заряжена. При избытке КС1 поверхность осадка заряжена
252
отрицательно, поскольку на его поверхности адсорбируются хлорид-
ионы, а пр!Г избытке АдКЮз - положительно, так как Преимуще¬
ственно адсорбируются ионы серебра.
13.6. Ионообменная адсорбция
Известны адсорбенты, твердые природные или синтетические
вещества, нерастворимые в воде и органических растворителях и
имеющие общие формулы Р-Мх и Р-Ау, содержащие катионы М или
анионы А, которые способны в водных растворах к обмену с ка¬
тионом М1 и анионом Аь соответственно. Они получили название
ионитов.
Иониты обладают сетчатой структурой. С сеткой ковалентно
связаны группы атомов Р*, несущих положительный или отрица¬
тельный заряд (фиксированные ионы). Этот заряд компенсируется
противоионами (М+ или А"). Фиксированные ионы вместе с проти-
воионами (Р'М+ или Р+М‘) называют ионогенными или функцио¬
нальными группировками ионита.
По типу ионогенных групп иониты делят на катиониты Р~-М+
(кислотные иониты, обменивающие катион) и аниониты Р*-М~
(основные иониты, обменивающие анион).
По степени ионизации ионогенных групп различают сильно- и
слабокислотные катиониты и сильно- и слабоосновные аниониты.
Сильнокислотные катиониты содержат остатки серной,
фосфорной и других кислот, слабокислотные - карбоксильные,
сульфгидрильные и другие слабо диссоциирующие по кислотному
типу группы. Ионогенные группы сильноосновных анионитов
представляют собой обычно аммониевые основания, а слабооснов¬
ные - аминогруппы и остатки других органических оснований.
Элементарную ячейку катионита можно рассматривать как вы¬
сокомолекулярный поливалентный анион, отделенный поверх¬
ностью раздела от окружающей среды Такой поливалентный анион
пропитан раствором, содержащим большое число ионов водорода
(или других катионов), способных обмениваться на катионы, нахо¬
дящиеся в жидкости, окружающей зерно катионита Ячейку анио¬
нита можно рассматривать как высокомолекулярный поливалент¬
ный катион, противоионами которого являются гидроксильные ионы
(или другие анионы), способные обмениваться на анионы
Ионит, помещенный в водный раствор, поглощает значительное
количество воды (иногда до 50%), набухает Поглощенная вода
гидратирует ионогенные группировки и вызывает их ионизацию
253
Ионообменной адсорбцией называют процесс, в котором ио¬
нит и раствор обмениваются между собой в эквивалентных коли¬
чествах одноименно заряженными ионами. Ионообменные реакции
можно выразить следующими уравнениями:
для катионита - Р"-Н+ + ^ + Н\
для анионита - Р+-(ОН)" + СГ ^ Р+-СГ + ОН".
Активность ионитов - поглощающая способность, характери¬
зующаяся обменной емкостью, - количеством ионов (моль), свя¬
занных 1 г сухого ионита из раствора в равновесных условиях
Ионный обмен - обратимый процесс, что дает возможность ис¬
пользованные иониты регенерировать. Катиониты регенерируют
промыванием раствором кислоты, аниониты - раствором щелочи.
Иониты используют как катализаторы (в реакциях этерифика-
ции, гидратации и дегидратации), для обессоливания воды, очистки
сточных вод, в ионообменной хроматографии - для выделения и
очистки ионных соединений, в частности, аминокислот. В медицине
их применяют для консервирования крови, для беззондового опре¬
деления кислотности желудочного сока, для детоксикации при
отравлениях токсичными электролитами. Аниониты употребляют в
качестве антацидных (то есть понижающих желудочную кислот¬
ность) средств, для предотвращения ацидозов и др. В последнем
случае аниониты используются для обменной адсорбции ионов во¬
дорода, следовательно, в них ионы водорода предварительной обра¬
боткой были замещены катионами металла. Катиониты находят
применение для предупреждения и лечения отеков, связанных с де¬
компенсацией сердечной деятельности.
Свойствами ионитов обладают ткани растений и животных. Их
катионообменные свойства определяются присутствием карбоксиль¬
ных и фосфатных групп, а способность обмениваться с анионами -
аминогруппами белков.
Глава>4. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Следовало ожидать, ... что
всевозможные порошкообраз¬
ные вещества окажут воздей¬
ствие на хлорофилловые пиг¬
менты в лигроиновых раство¬
рах, и возникла надежда, что
систематическое изучение во¬
проса бросит некоторый свет
на сущность адсорбционных
явлений и позволит вырабо¬
тать на их основании новый
метод физического отделения
веществ
М. С. Цвет
Любую разновидность хроматографии можно определить как
физико-химический метод разделения смесей веществ, основанный
на их распределении между двумя несмешивающимися фазами,
одна из которых является неподвижной, а другая - подвижной.
Неподвижная фаза представляет собой поверхностно-активное
твердое тело или жидкость, закрепленную на поверхности инертно¬
го твердого носителя. Подвижная фаза - газ или жидкость, которые
проходят через слой неподвижной фазы.
Метод был открыт в 1903 году М. С. Цветом, который впервые
применил его для разделения пигментов листьев растений. Вот как
оценил один из английских химиков открытие Цвета “ ..был пред¬
ложен новый остроумный метод химического анализа, которому
предназначено оказать влияние на жизнь человечества и всего жи¬
вого мира”.
В адсорбционной хроматографии процесс разделения осно¬
ван на различии в относительном сродстве соединений к твердому
адсорбенту (неподвижная фаза), а подвижной фазой служит жид¬
кость (колоночная адсорбционная или тонкослойная хроматография)
или газ (газовая адсорбционная хроматография)
В распределительной хроматографии вещества распреде¬
ляются между двумя жидкими фазами (жидкостная распредели¬
тельная хроматография) или между неподвижной жидкой и газовой
фазами (газо-жидкостная хроматография) В жидкостной хромато¬
графии неподвижная фаза может представлять собой пленку или
слой (хроматография на бумаге или тонкослойная распределитель¬
ная хроматография) или может быть диспергирована на объемном
носителе (колоночная распределительная хроматография)
255
В основе ионообменной хроматография лежит обратимый
обмен ионов, содержащихся в растворе, на подвижные ионы иони¬
тов или ионообменников
При разделении веществ методом гель-хроматографии непо¬
движной фазой являются малоактивные материалы (гели) с задан¬
ной пористостью, способные удерживать молекулы вещества с
определенными размерами и формой, и разделять их по способности
проникать в поры геля, те по их способности к диффузии
Особняком в ряду других хроматографических методов стоит
аффинная или биоспецифическая хроматография В ее основе
лежит свойство высокомолекулярных и других биологически ак¬
тивных соединений “узнавать” в любой смеси “свои” строго опре¬
деленные вещества и взаимодействовать с ними Так, фермент
“узнает" свой субстрат, антиген “узнает” антитело, гормон - “свой”
рецептор и т.д.
Задачи хроматографического анализа
1. Идентификация нескольких компонентов в одном образце
2. Удаление соединений, мешающих анализу другим методом
3. Концентрирование компонента, присутствующего в виде сле¬
дов в сложной смеси с целью его дальнейшего анализа.
4. Решение специальных задач, например, вопросов химического
превращения компонентов (главным образом, токсических) в окру¬
жающей среде.
Методы тонкослойной (ТСХ) и газо-жидкостной (ГЖХ) хромато¬
графии широко используются как рутинные методы анализа органи¬
ческих соединений во многих областях науки, включая медицину, и
их значение постоянно возрастает. Эти методы и будут рассмотре¬
ны подробнее.
14.1. Тонкослойная хроматография (ТСХ)
Тонкослойная хроматография является одним из наиболее про¬
стых и эффективных экспресс-методов разделения и анализа ве¬
ществ в биосредах, пищевых продуктах и других сложных системах
и биоматериалах, не требующих сложного оборудования и доступ¬
ных для постановки в любых условиях. В то же время этот метод
обладает высокой избирательностью и чувствительностью.
В зависимости от природы неподвижной фазы ТСХ может быть
адсорбционной, распределительной и аффинной. Ниже рассмотрен
весьма широко применяемый адсорбционный вариант ТСХ.
Процесс разделения в этом варианте основан на различии в
относительном сродстве компонентов смеси к неподвижной фазе -
256
сорбенту й* осуществляется в результате перемещения подвижной
фазы - элюента под действием капиллярных сил по слою сорбен¬
та, нанесенного на хроматографическую пластинку из инертного
материала (стекло, алюминиевая фольга и др.).
Скорость перемещения вещества при хроматографировании в
стандартных условиях является постоянной величиной, характер¬
ной для данного соединения. Ее оценивают величиной
(рис. 14.1), которая представляет собой отношение расстояния от
стартовой линии хроматограммы до центра пятна этого вещества (/)
в любой момент времени к расстоянию, пройденному за то же вре¬
мя фронтом растворителя (/,).
Рис. 14.1. Типичная хроматограмма.
Ассортимент используемых сорбентов в настоящее время доста¬
точно велик. Это - универсальные сорбенты (силикагель, оксид
алюминия, целлюлоза), полиамиды, крахмал, кизельгур, гипс, агар-
агар, сульфат кальция, карбонат цинка и др. Главное требование к
сорбентам - отсутствие химического взаимодействия с анализируе¬
мым веществом.
Для приготовления подвижной фазы с высокой элюирующей
способностью используют смесь растворителей разной полярности.
В большинстве случаев пробы анализируемых веществ наносят
в виде растворов в подходящем низкокипящем растворителе. Нане¬
сение образца осуществляется при помощи калиброванных микро¬
пипеток или с помощью обычного стеклянного капилляра на рас¬
стоянии 1.5-2.0 см от нижнего края хроматографической пластинки
в виде серии пятен вдоль линии старта. Диаметр наносимого пятна
должен быть как можно меньше, так как по мере развития хромато¬
граммы он обычно увеличивается вследствие диффузии.
9 Зак. 675
257
В зависимости о.т того, в каком направлении поступает раство¬
ритель на пластинку, различают методы восходящей, нисходящей и
горизонтальной хроматографии Чаще всего используют восходящий
вариант- растворитель поднимается по пластинке вверх под дей¬
ствием капиллярных сил При этом в токе элюента перемещаются
также и исследуемые вещества со скоростью, зависящей от адсорб¬
ционных свойств. Когда фронт элюента достигнет верхней части
сорбента, хроматографическое разделение заканчивается
Окрашенные вещества не требуют специального обнаружения
Большинство же хроматографируемых соединений бесцветно, и по¬
этому существуют различные методы их обнаружения, а именно -
физические (УФ-облучение), химические (обработка хроматограмм
газами, например, аммиаком, парами иода, брома или опрыскивание
различными обнаруживающими реагентами, что вызывет появление
специфического окрашивания разделяемых компонентов в результа¬
те их реакции с действующим реагентом) и биологические (для об¬
наружения биологически активных соединений).
Идентификация хроматограмм осуществляется по стандартным
значениям величин Я* (литературные данные) или по методу “сви¬
детелей” (проведение параллельного хроматографирования смеси
неизвестного состава и индивидуальных соединений, присутствие
которых в исследуемой пробе предполагается, с последующим срав¬
нением величин Я*).
ТСХ перестает быть качественным методом анализа, когда с по¬
мощью какой-либо аппаратуры (спектрофотометров, фотоэлектроко¬
лориметров и др.) выполняют количественное определение кон¬
центраций веществ после разделения смесей.
Метод ТСХ позволяет медикам проводить определение токси¬
нов, осуществлять диагностику отравлений, исследовать продукты
метаболизма токсичных веществ в биологических средах (кровь,
моча, слюна).
Преимущества тонкослойной хроматографии, а именно: мини¬
мальное время, необходимое для подготовки пробы, высокая разре¬
шающая способность и чувствительность - позволяют врачу в крат¬
чайшие сроки провести не только дифференциальную диагностику
при острых химических отравлениях, но и следить при лечении
больного за детоксикацией организма, давая в руки клинициста на¬
дежный инструмент токсикокинетического контроля. Врач, получая
данные о результатах качественного определения анализируемых
компонентов в крови или другой биологической жидкости, имеет
возможность правильно оценить результаты лечения, эффектив¬
ность применяемых методов, установить необходимую длительность
проведения операций, перитонеального диализа и хирургических
методов детоксикации (гемодиализа, гемосорбции).
258
14.2. ^азо-жидкостная хроматография (ГЖХ)
14.2.1. Сущность метода
Газо-жидкостная хроматография - универсальный метод разде¬
ления смесей разнообразных веществ, испаряющихся без разложе¬
ния Следовательно ее возможности ограничены применимостью к
анализу сравнительно низкомолекулярных веществ (с молекулярной
массой не более нескольких тысяч).
сигнал, мВ
Рис. 14.2. Типичная хроматограмма.
А, Б - пики соответствующих компонентов, 1ЯА и 1яБ - времена удерживания соединений А и Б,
Ы2 - полувысота пика А, со - ширина пика А на его полувысоте
При этом компоненты разделяемой смеси перемещаются по
хроматографической колонке с потоком инертного газа (газа-
носителя) Разделяемая смесь распределяется между газом-носи¬
телем (подвижной фазой) и нелетучим растворителем (неподвиж¬
ной жидкой фазой), нанесенным на инертный материал (твердый
носитель), которым заполнена колонка. Компоненты смеси селек¬
тивно удерживаются неподвижной фазой, а затем выходят из ко¬
лонки и регистрируются детектором Сигналы детектора записы¬
ваются в виде хроматограммы автоматическим потенциометром
(самописцем). Каждому компоненту смеси на хроматограмме
(рис. 14.2) соответствует отдельный пик Положение пика опреде¬
ляется величиной времени удерживания Ои), которое представляет
собой время от момента ввода пробы до выхода максимума пика,
или величиной удерживаемого объема (Уд), который рассчитывает¬
ся по формуле
V,* = 1^,
где Р - объемная скорость газа-носителя.
9*
259
14.2.2. Техника газо-жидкостной хроматографии
Проведение анализа методом ГЖХ включает подготовительный
этап работы, проведение хроматографического разделения и обра¬
ботку результатов
В подготовительном этапе работы необходимо нанести непо¬
движную фазу в виде тонкой пленки на твердый носитель. Полу¬
ченным таким образом наполнителем заполняют колонку Системой
регулировки газов создают необходимый расход газа-носителя.
Разделение смесей веществ методом ГЖХ осуществляется на
приборах, называемых газовыми хроматографами (рис. 14 3).
Рис. 14.3. Принципиальная схема газового хроматографа.
1 - газ-носитель, 2 - испаритель, 3 - хроматографическая колонка, 4 - детектор,
5 - самопишущий регистратор, 6 - измеритель скорости потока, 7 - термостат.
Газ-носитель из баллона I (скорость подачи которого регулиру¬
ется измерителем 6) через редуктор и манометр поступает в
устройство для ввода пробы - испаритель 2. Захватив анализируе¬
мую пробу в виде пара или газа, газ-носитель направляется в хро¬
матографическую колонку 3, которая помещена в термостат 7.
В колонке анализируемая смесь разделяется на составные компо¬
ненты. Выходящие из колонки разделенные компоненты смеси в по¬
токе газа-носителя проходят через детектор 4, который подает сла¬
бый электрический сигнал на усилитель, а отсюда - на регистратор
(самописец) 5.
Устройство для ввода пробы - разной конструкции в зависи¬
мости от агрегатного состояния пробы и способа ввода в колонку.
Детектор - это устройство, которое преобразует в электриче¬
ский сигнал изменения физических или физико-химических
свойств газового потока, выходящего из колонки, по сравнению
с чистым газом-носителем.
Существует около 50 видов детекторов, однако широкое приме¬
нение находят только те из них, которые обладают высокой чувст¬
вительностью и универсальностью. Это - катарометр (детектор по
Проба
3
260
теплопроводности); детектор плотности (плотномер); пламенно¬
ионизационный детектор (ДИП), в котором водородное пламя слу¬
жит источником ионизации органического соединения; детектор
электронного захвата (ЭЗД); термоионный детектор (ТИД), который
обладает высокой селективностью к органическим веществам, со¬
держащим фосфор, азот и серу. Интерес к этому детектору заметно
возрос в связи с заменой хлорсодержащих пестицидов на фосфор¬
содержащие ядохимикаты.
Катарометр позволяет определять концентрации веществ в пре¬
делах 0.1-0.01%, ДИП - 10-3-10-5%, ЭЗД - Ю-б-1010%. Современ¬
ные детекторы позволяют определять даже пикограммы (10'12 г)
веществ.
Обработка результатов хроматографического разделения вклю¬
чает определение качественного и количественного состава анали¬
зируемой смеси (см рис. 14.2).
Определение качественного состава смеси проводится путем со¬
поставления времени удерживания данного компонента и эталона -
вещества известной структуры. При строгом воспроизведении всех
условий анализа время удерживания Ок) является такой же физи-
ко-химической характеристикой вещества, как его плотность, пока¬
затель преломления и т.д. Совпадение времени удерживания этало¬
на и определяемого компонента может указывать на их идентич¬
ность. Эталон чаще всего добавляется в исследуемую смесь (метод
метки). При этом число пиков на хроматограмме не должно изме¬
няться, а интенсивность пика одного из компонентов должна уве¬
личиваться.
При необходимости сопоставления данных об удерживании ис¬
следуемого вещества с литературными (например, при отсутствии
необходимого эталона), использование времен удерживания невоз¬
можно, т.к этот параметр сильно зависит от скорости газа-носи-
теля. В этом случае используется удерживаемый объем (Уд).
Определение количественного состава смеси основано на до¬
пущении того, что интенсивность пика каждого компонента пропор¬
циональна его содержанию в смеси. В качестве меры интенсивности
принимается обычно площадь пиков Существуют разные способы
измерения площадей пиков Наиболее простым методом является
умножение высоты пика И (см рис 14 2) на его ширину ш, изме¬
ренную на полувысоте пика
Б = И со
Результаты количественного анализа рассчитывают методами
абсолютной калибровки, внутреннего стандарта или простой нор¬
мировки. При анализе смеси веществ, близких по химическому
261
строению, хорошие результаты дает метод нормировки. Расчет про¬
центного содержания компонентов по этому методу производится
по формуле:
х,= ———100,
I в.
1=1
где х, - искомое процентное содержание 1-того компонента, Б, -
площадь пика 1-того компонента, I э,- сумма площадей пиков всех
1=1
компонентов
14.2.3. Значение метода
газо-жидкостной хроматографии
В исследовании липидов, а, в особенности, жирных кислот,
ГЖХ произвела настоящую революцию и до настоящего времени не
имеет альтернативы. Первым анализом, выполненным с помощью
ГЖХ, стало определение карбоновых кислот Джеймсом и Мартином
в 1952 году.
Приложением этого исследования в медицине стала микробио¬
логия В процессе своего метаболизма микробные клетки произво¬
дят низшие карбоновые кислоты, причем набор кислот является как
бы визитной карточкой того или иного микроорганизма.
Традиционные пути идентификации микроорганизмов - возбуди¬
телей инфекционных заболеваний или гнойно-воспалительных про¬
цессов включают в себя несколько этапов - посев биологического
материала на питательные среды, получение “чистых” (т.е. состоя¬
щих из одинаковых микробов) культур, выращивание их на средах
обогащения и лишь затем идентификация их по характеру деграда¬
ции тех или иных субстратов. Даже для микроорганизмов, обла¬
дающих способностью к быстрому росту, эти этапы исследования
занимают не менее двух суток. С помощью газовой хроматографии
можно проводить ускоренную (менее двух часов) идентификацию
микроорганизмов по спектру специфических компонентов их мем¬
бран или специфическим продуктам пиролиза.
Каждый двадцатый раненый в годы второй мировой войны
страдал от гнойно-септических осложнений, вызванных анаэроб¬
ными бациллами - возбудителями газовой гангрены. Успехи в ле¬
чении позволили снизить число осложнений, вызванных анаэроб¬
ными микроорганизмами, почти в 100 раз. Немалую роль в этом
сыграл тот факт, что определение карбоновых кислот непосредст¬
262
венно в рЬне сделало возможным верификацию диагноза анаэроб¬
ной инфекции в течение нескольких часов, тогда как традиционные
бактериологические методы дают ответ в лучшем случае через
4-5 суток.
Рис. 14.4. Хроматограмма гноя из плевральной полости при
анаэробном сепсисе: а - до лечения, б - после двухнедельного лечения
антибиотиком цефалоспорином;
1 - уксусная, 2 - пропионовая, 3 - масляная, 4 - изовалериановая кислоты.
В качестве примера на рис. 14.4а дана хроматограмма гнойных
выделений легких больного, пораженного анаэробной инфекцией, в
которых присутствует набор жирных кислот. Из рисунка 14.46 вид¬
но, как под действием антибиотика кислоты исчезают, исключая ук¬
сусную кислоту, являющуюся естественным метаболитом. Таким
образом, метод ГЖХ становится методом клинического контроля.
Исследования состава липидов крови привели к сегодняшнему
пониманию проблемы атеросклероза - болезни, ведущей к появле¬
нию ишемической болезни сердца, нарушений мозгового кровооб¬
ращения Для практической медицины сейчас разработаны простые
и эффективные средства диагностики нарушений метаболизма
липидов.
В своей повседневной практике врачи ежедневно назначают ис¬
следования холестерина, триглицеридов, липопротеидов При разра¬
ботке этих методик газовая хроматография использовалась как ре¬
ферентный метод, т.е метод-эталон Широкое внедрение во врачеб¬
ную практику исследований метаболизма липидов позволило разра¬
ботать патогенетические, направленные на устранение основы за¬
болевания, пути профилактики и лечения атеросклероза.
Возможность определения индивидуального состава жирных
кислот тех или иных липидов является чрезвычайно мощным инст¬
263
рументом в познании структуры и функции биологических мембран,
процессов внутриклеточного метаболизма.
Энергетическое обеспечение клетки осуществляется в так назы¬
ваемом цикле трикарбоновых кислот - цикле Кребса Газохромато¬
графическое определение карбоновых кислот цикла Кребса внесло
большой вклад в понимание интимных процессов внутриклеточного
метаболизма при различных патологических состояниях
Газовая хроматография дает возможность количественно оце¬
нить весь клинически значимый спектр стероидов. Были разработа¬
ны методы определения так называемых катехоламинов - адренали¬
на и норадреналина и родственных им соединений, гормонов щито¬
видной железы, альдостерона и кортизола
Метод газовой хроматографии нашел применение при гигиени¬
ческом анализе полимерных материалов, состава выхлопных газов,
анализа воздуха в производственных помещениях и операционных
палатах; хлор-, азот- и фосфорсодержащих пестицидов; определения
загрязнений в промышленных сливах (например, содержания фрео-
нов, различных кислот и их производных, ароматических соедине¬
ний, например, фенола, спиртов, нитрилов и т.д.); для оценки ка¬
чества пищевых продуктов; для концентрирования и выделения ор¬
ганических загрязнений стоков фармацевтических предприятий.
Технический прогресс сделал возможным получение так назы¬
ваемых метаболических профилей биосред - крови, мочи, слюны,
выдыхаемого воздуха. В одном образце анализируются несколько
сотен компонентов Метаболические профили так же индивидуаль¬
ны, как и отпечатки пальцев, но в отличие от папиллярных узоров
хроматограмма метаболитов человеческого организма несет в себе
массу медицинской информации - какие лекарства или продукты
получал человек в последнее время, каким микроорганизмом вызва¬
но его заболевание и многое другое.
Компьютерный анализ метаболических профилей является од¬
ним из мощнейших инструментов диагностики врожденных и при¬
обретенных нарушений метаболизма - таких заболеваний, как са¬
харный диабет, подагра и многие другие.
Быстро прогрессируют и другие инструментальные хроматогра¬
фические методы, среди которых особенно перспективна высокоэф¬
фективная жидкостная хроматография (ВЭЖХ), в которой элюен-
том служит жидкая фаза (специально подобранная смесь раствори¬
телей), под высоким (до 200 атмосфер) давлением подаваемая
в хроматографическую колонку. Этот метод позволяет анализиро¬
вать нелетучие биообразцы - белки, нуклеотиды, различные лекар¬
ственные средства и др.
ГлаЪ? 15. ХИМИЯ ДИСПЕРСНЫХ СИСТЕМ
Жизнь - это особая коллоид¬
ная система, это особое цар¬
ство природных вод.
В. И. Вернадский
15.1. Дисперсные системы
Коллоидная химия изучает физико-химические свойства гетеро¬
генных дисперсных систем, образованных из двух или более фаз с
сильно развитой поверхностью раздела. По крайней мере одна из
фаз распределена в виде мелких частиц и называется дисперсной
фазой, а другая, сплошная, фаза - дисперсионной средой
По агрегатному состоянию дисперсные системы можно разде¬
лить на суспензии, эмульсии, золи и т.д. (таблица І5.І).
Таблица 15.1
Классификация дисперсных систем по агрегатному состоянию
Дисперсионная
среда
Дисперсная фаза
газ
жидкость
твердое
тело
газ
-
туман
дым, пыль
жидкость
пена
эмульсия
суспензия
твердое тело
аэрозоль
жидкие включения
твердый
в твердом теле
золь
По величине (или по дисперсности) частиц дисперсной фазы
дисперсные системы можно разделить условно на три группы гру¬
бодисперсные, коллоидно-дисперсные и молекулярно(ионно)-дис-
персные:
< 10-9 М 10*7-10-9 М > 10-7 м
молекулярно(ионно)- коллоидно- тубодисперсные
дисперсные дисперсные
Коллоидно-дисперсные системы и относят к коллоидам (или
коллоидным растворам, если дисперсная среда - жидкость).
265
По интенсивности молекулярного взаимодействия фаз различа¬
ют лиофильные (в частности, гидрофильные) и лиофобные
коллоидные системы
В лиофильных системах молекулярное взаимодействие между
фазами велико, а поэтому поверхностное натяжение на границе
раздела между ними мало. Такие системы образуются самопроиз¬
вольно и имеют предельно высокую дисперсность. Лиофильные си¬
стемы термодинамически устойчивы и поэтому не разрушаются во
времени при сохранении условий их возникновения
В лиофобных системах взаимодействие между молекулами раз¬
личных фаз незначительно, межфазное поверхностное натяжение
велико, вследствие чего система имеет тенденцию к самопроизволь¬
ному укрупнению частиц дисперсной фазы. Вследствие избытка
свободной поверхностной энергии 0$ они термодинамически не¬
устойчивы, то есть имеют тенденцию к распаду. Агрегативная
устойчивость лиофобных коллоидов носит временный характер.
Коллоидный лиофобный раствор (золь) - это микрогетерогенная
система, состоящая как минимум из трех компонентов: дисперсной
фазы, дисперсионной среды и стабилизатора - вещества, которое
адсорбируется на поверхности раздела фаз и образует защитные
слои, препятствующие сближению частиц дисперсной фазы. С тер¬
модинамической точки зрения стабилизатор, адсорбируясь на меж-
фазной границе, понижает межфазное поверхностное натяжение и
приводит к образованию равновесных коллоидных систем.
Коллоиды широко распространены в природе. Почва, глина,
природные воды, воздух, облака, дым, пыль, многие минералы
(в том числе и драгоценные камни) - все это коллоидные системы.
Большое значение имеют коллоидные системы для биологии и
медицины. В состав любого живого организма входят твердые, жид¬
кие и газообразные вещества, находящиеся в сложном взаимо¬
действии с окружающей средой. Протоплазма клетки обладает
свойствами, характерными как для жидких, так и для твердых ве¬
ществ. С химической точки зрения организм в целом есть слож¬
нейшая совокупность многих коллоидных систем, включающих в
себя и жидкие коллоиды, и гели.
Такие биологические жидкости, как кровь, плазма, лимфа,
спинномозговая жидкость представляют собой коллоидные систе¬
мы, в которых ряд веществ, например, белки, холестерин, гликоген
и многие другие находятся в коллоидном состоянии. Вот почему
многие стороны явлений, происходящих в живом организме, могут
быть поняты лишь по мере познания природы коллоидного состоя¬
ния материи.
Важнейшие пищевые продукты - хлеб, молоко, масло - колло¬
идные системы. От величины капелек жира может зависеть ско¬
266
рость их1^асывания через стенки пищеварительных органов. Тонко
раздробленный жир в молоке и сливочном масле усваивается орга¬
низмом лучше, чем жир в сплошной массе, например, сало.
Сильно раздробленные вещества легче проникают через поры
кожи, эффективнее действуют на организм, поэтому в медицине
широко применяются лекарственные вещества в виде коллоидных
систем (суспензии, эмульсии, мази, кремы, пасты, аэрозоли). Тера¬
певтическое применение коллоидных препаратов металлов и других
сильнодействующих лекарств обусловлено, по-видимому, тем, что
они обеспечивают очень слабое, но продолжительное действие ма¬
лых доз вещества.
При лечении инфекционных заболеваний легких, а также при
заболеваниях дыхательных путей используются ингаляции аэрозо¬
лями различных антибиотиков.
Широко применяются в медицинской практике эмульсии.
Эмульсии классифицируют по двум типам: прямые, у которых в ка¬
честве дисперсной фазы выступает неполярная жидкость, а в ка¬
честве дисперсионной среды - полярная жидкость (их обозначают
“м/в” где “м” - масло или другая неполярная жидкость, “в” - вода
или другая полярная жидкость), и обратные (“в/м”), у которых
дисперсной фазой является полярная жидкость, а дисперсионной
средой - неполярная жидкость. Концентрированные эмульсии могут
существовать длительное время только благодаря введению специ¬
альных стабилизаторов, называемых эмульгаторами. В качестве
эмульгаторов могут применяться самые различные по природе по¬
верхностно-активные вещества (см. главу 13).
Жировые эмульсии применяются для энергетического обеспече¬
ния голодающего или ослабленного организма путем внутривенного
вливания. Жировые эмульсии должны иметь достаточно высокую
концентрацию и степень дисперсности, обладать устойчивостью и
не быть токсичными Обычно для получения эмульсии используют
хлопковое, соевое, оливковое и некоторые другие масла.
В санитарном деле очистка питьевой воды основана на процес¬
сах адсорбции и взаимной коагуляции; очистка воздуха от токсич¬
ных дымов и туманов построена на закономерностях, установлен¬
ных при исследовании аэрозолей.
Находят применение коллоиды и при образовании дымовых за¬
вес, распылении химикатов воздушно-капельным путем, приготов¬
лении дезинфицирующих растворов и т.д
Существенную роль играют коллоиды в промышленности, глав¬
ным образом, при добыче и переработке нефти, в металлургичес¬
кой промышленности, производстве строительных материалов, в
текстильной, лакокрасочной и пищевой промышленности. Поэтому
коллоидную химию называют химией реальных тел.
267
15.2. Способы получения коллоидных растворов
Для получения коллоидных растворов необходимы следующие
условия: достаточно малая растворимость дисперсной фазы в дис¬
персионной среде, определенная коллоидная степень дисперсности
(размеры частиц в пределах 10'7 - 1СГ9 м) и наличие в системе ста¬
билизатора (электролита или высокомолекулярного соединения),
препятствующего слипанию коллоидных частиц
Существуют два метода получения коллоидных систем: диспер¬
гирование - дробление крупных частиц до коллоидной степени дис¬
персности и конденсация путем создания условий, при которых
атомы, молекулы или ионы соединяются в агрегаты коллоидной
степени дисперсности.
Диспергирование можно проводить различными путями:
1. Механическое дробление осуществляется в шаровых и кол¬
лоидных мельницах, в которых диспергируемый материал вместе с
дисперсионной средой и стабилизатором дробится и истирается в
узком зазоре между трущимися поверхностями.
2. Электрическое распыление в вольтовой дуге. Для этого два
электрода из металла, золь которого хотят получить, погружают в
охлаждаемую жидкость, в которую добавлен электролит-стаби¬
лизатор, и пропускают электрический ток. Сближают электроды
для получения вольтовой дуги, металл при температуре вольтовой
дуги испаряется, а затем конденсируется в жидкости, образуя золь.
Этим методом получают золи золота, серебра, платины и других
металлов.
Одним из перспективных вариантов приготовления лекарствен¬
ных золей является использование аллотропа углерода, фуллерена
(см. 17.1). Его синтезируют в вольтовой дуге из графита в при¬
сутствии лекарственных веществ. В результате получают золь фул¬
лерена, полости в молекулах которого заполняют молекулы лекар¬
ственных препаратов.
3. Действием ультразвука. Ультразвуковые колебания - это
высокочастотные механические колебания (от 20 ООО до 1 ООО ООО
колебаний в секунду), которые получают с помощью специальных
генераторов. Разрывающие усилия возникают как вследствие че¬
редующихся локальных сжатий и расширений в системе, так и
вследствие кавитаций, т.е. образования и лопания пузырьков при
локальных растяжениях жидкости. При этом развиваются локаль¬
ные избыточные давления (порядка тысяч атмосфер) за ничтожно
малые промежутки времени, которые приводят к разрыву не только
жидкостей, но и твердых тел. Так получают гидрозоли серы, гипса,
графита, гидроксидов металлов и т.д.
268
В биоЛ^мбранологии ультразвуком обрабатывают водные сус¬
пензии липидов для получения сферических моделей биологических
мембран - липосом, т.е. липидных пузырьков со стенками, состоя¬
щими из бимолекулярного липидного слоя (см. ниже).
4. Методом пептизации, которая заключается в раздроблении
свежеприготовленных рыхлых осадков на отдельные коллоидные
частицы при добавлении небольшого количества электролита-
пептизатора. При этом степень дисперсности фактически не изме¬
няется, так как частицы рыхлого осадка уже имеют коллоидные
размеры. Различают адсорбционную и химическую пептизации.
В первом случае электролит-пептизатор адсорбируется на поверх¬
ности частиц осадка, сообщает им заряд и, таким образом, способ¬
ствует переходу их во взвешенное состояние. Так образуется золь
гидроксида железа(Ш) при добавлении к рыхлому осадку гидрокси¬
да железа(Ш) в качестве электролита-пептизатора хлорида желе-
за(Ш). Во втором случае пептизатор образуется в результате хими¬
ческой реакции добавляемого вещества с частицами на поверхности
осадка. Так можно получить золь гидроксида железа(Ш) при добав¬
лении небольшого количества соляной кислоты к свежеприго¬
товленному осадку гидроксида железа(Ш).
Пептизация имеет биологическое значение: рассасывание атеро¬
склеротических бляшек, почечных и печеночных камней; действие
антикоагулянтов при тромбофлебитах сводится, в сущности, к яв¬
лению пептизации.
Конденсационные методы образования коллоидных раство¬
ров разделяют на физические и химические. К физическим явлени¬
ям относится образование тумана, облаков, дыма, а также метод
замены растворителя, в котором вещество хорошо растворимо, на
растворитель, где данное вещество мало растворимо Так получают
гидрозоли серы, канифоли заменой органического растворителя
(например, спирта), в котором эти вещества хорошо растворимы, на
воду, в которой они малорастворимы.
В основе химических конденсационных методов лежат химиче¬
ские реакции (окисления, восстановления, гидролиза, обмена), при¬
водящие к образованию труднорастворимых веществ в присутствии
тех или иных стабилизаторов.
К примеру, реакцией гидролиза получают золи гидроксидов же¬
леза (III) и алюминия, которые применяют для очистки воды
РеС13 + ЗН20 -» Ре(ОН)3 + ЗНС1.
Стабилизатором этого золя является частично образующийся
при реакции оксихлорид железа
Ре(ОН)3 + НС1 -» РеОС1 + 2Н20.
269
Золь серы, который используется в медицине для лечения кож¬
ных заболеваний, получают реакцией окисления
гНгЭ + 02 -> 2Э + 2Н20.
Коллоидные растворы серебра и золота получают также окисли¬
тельно-восстановительными реакциями, например.
2АдШ3 + Н202 -> 2Ад + 02 + 2НГЮ3
2НАиСЦ + ЗН202 -> 2Аи + 8НС1 + 302
Ад20 + Н2 -> 2Ад + Н20
2КАиОг + ЗСН20 + К2СО3 -> 2Аи + ЗНСООК + КНС03 + Н20.
Бактерицидные свойства коллоидных растворов серебра широко
используются в медицине (лекарственные препараты колларгол и
протаргол), а золь радиоактивного золота применяют для лечения
злокачественных новообразований.
В формировании структуры почв играет большую роль золь ок¬
сида кремния, который образуется при гидролизе силикатов:
МагЭЮз + Н20 -> ЭЮг + 2№0Н.
Получение дымовых и туманных завес в военном деле осу¬
ществляется посредством гидролиза хлорида кремния:
ЭЮЦ + 2Н20 -> ЭЮг + 4НС1.
Золи сульфида сурьмы (III), йодида серебра, берлинской лазури
и многие другие золи получают реакцией ионного обмена:
28ЬС13 + ЗНгЭ -> ЭЬгЭз + 6НС1;
АдШз + К1 -> Ад1 + К1Ч0э.
Образование золя в каждом отдельном случае происходит в
присутствии того или иного стабилизатора.
15.3. Очистка коллоидных растворов
Коллоидные растворы всегда содержат примеси электролитов и
других низкомолекулярных веществ. Их можно отделить от колло¬
идных частиц путем диализа, электролиза или ультрафильтрации.
Диализ основан на применении мембран, задерживающих
крупные коллоидные частицы и пропускающих ионы и молекулы
низкомолекулярных веществ. Диализ проводится в приборе, кото¬
рый называется диализатором. Он представляет собой стеклянный
270
сосуд, нйУ^яя часть которого затянута полупроницаемой мембра¬
ной, сделанной из целлофана* или коллодия”. В диализатор нали¬
вают коллоидный раствор и погружают его в стакан с дистиллиро¬
ванной водой. Ионы и молекулы из золя через мембрану постепен¬
но переходят в наружный сосуд, воду в котором по мере загрязне¬
ния можно периодически менять. Коллоидные частицы ввиду малых
размеров пор мембраны не диффундируют через нее и будут оста¬
ваться внутри диализатора.
Процесс диализа длительный. Он может быть ускорен, если че¬
рез коллоидный раствор, подлежащий очистке от избытка электро¬
лита, пропустить постоянный электрический ток, тогда ионы низко¬
молекулярных примесей будут перемещаться к соответствующим
электродам. Такой процесс носит название электродиализа и про¬
водится в приборе - электродиализаторе, секции которого отделены
полупроницаемыми мембранами.
Ультрафильтрация - отделение дисперсной фазы от диспер¬
сионной среды - производится через специально приготовленные
плотные фильтры, непроницаемые для частиц дисперсной фазы.
Фильтрование обычно проводят под давлением, или используют
разрежение (вакуум). Применяя мембраны с определенной степе¬
нью пористости, можно в известной мере разделить коллоидные
частицы и приближенно определить их размеры. Этим методом
впервые были определены размеры ряда вирусов и бактериофагов.
Процесс ультрафильтрации лежит в основе функции почек.
При фильтрации через мембрану поток жидкости обеспечивается ее
гидростатическим давлением, создаваемым стенками капилляров.
Вещества с молекулярной массой до 10 ООО проходят через сито
мембраны свободно, а с молекулярной массой более 50 ООО - только
в ничтожных количествах.
Пример сочетания диализа и ультрафильтрации - аппарат
“искусственная почка”, предназначенный для временной замены
функции почек при острой почечной недостаточности Аппарат
оперативным путем подключают к системе кровообращения больно¬
го- кровь под давлением, создаваемым пульсирующим насосом
(“искусственное сердце”), протекает в узком зазоре между двумя
мембранами, омываемыми снаружи физиологическим раствором
‘Целлофан - тонкая прозрачная пленка из гидратированной целлюлозы,
получается при продавливании коллоидного раствора вискозы (ксанто-
гената целлюлозы) через узкую щель в кислотную ванну
"Коллодий - коллоидный раствор динитрата целлюлозы в смеси этило¬
вого спирта и эфира, после испарения растворителя остается плотная
пленка
271
Благодаря большой рабочей площади мембран (-15 ООО см2) из кро¬
ви достаточно быстро (3-4 часа) удаляются "шлаки” - продукты об¬
мена и распада тканей (мочевина, креатинин, ионы калия и др )
15.4. Молекулярно-кинетические свойства
коллоидных растворов
Свойства, связанные с тепловым движением частиц - броунов¬
ское движение, диффузия, осмос, - у коллоидных растворов
выражены гораздо слабее, чем у истинных растворов низкомолеку¬
лярных веществ, вследствие значительно больших размеров колло¬
идных частиц и меньшей их концентрации.
Коллоидные системы по молекулярно-кинетическим свойствам
принципиально не отличаются от обычных молекулярных растворов
Частицы золя подобно молекулам находятся в непрерывном движе¬
нии (броуновское движение). Вследствие хаотичности броуновского
движения частицы проходят сложный путь, который состоит из ог¬
ромного числа весьма малых отрезков прямых линий Учесть точно
эту траекторию нельзя, поэтому за величину, характеризующую ин¬
тенсивность броуновского движения, принимают величину среднего
смещения частицы за определенный промежуток времени.
Теория броуновского движения была разработана Эйнштейном
и Смолуховским. Было доказано, что квадрат среднего смещения
частицы пропорционален коэффициенту диффузии О-
х2 = 20т,
где О - коэффициент диффузии (м2/с), х - среднее смещение (м),
т - промежуток времени (с).
15.4.1. Диффузия
Для понимания особенностей транспорта растворенных молекул
через клеточные мембраны необходимы сведения о диффузии.
Диффузия - это процесс, который приводит к самопроизволь¬
ному уменьшению градиентов концентраций в растворе, пока не
установится однородное распределение частиц. Коэффициент
диффузии показывает количество вещества, которое диффундиру¬
ет через поперечное сечение площадью 1 м2 в течение 1 секунды
при градиенте концентрации, равном единице.
272
По формуле Стокса-Эйнштейна коэффициент диффузии обратно
пропорционален радиусу частицы:
N 6ят|г ’
где О - коэффициент диффузии (м2/с), Я - универсальная газо¬
вая постоянная, N - число Авогадро, т| - вязкость среды (н с/м),
г - радиус частицы (м). Коэффициент 6 указывает на сферическую
форму частицы.
Определение коэффициента диффузии золей является одним из
основных методов коллоидной химии при определении размеров
частиц дисперсной фазы, а также величины макромолекул.
Подставляя значение О в уравнение для величины среднего
смещения, получим:
Это уравнение показывает, что интенсивность броуновского
движения возрастает с уменьшением размеров частиц и с повыше¬
нием температуры.
В соответствии с размерами частиц дисперсной фазы скорость
диффузии в коллоидных растворах в сотни и тысячи раз меньше,
чем в истинных растворах низкомолекулярных веществ.
Аналогичное заключение можно сделать и об осмотическом
давлении коллоидных растворов. Так, 1%-ный золь золота имеет
осмотическое давление, равное 0.00045 атм., а раствор сахарозы
той же концентрации и в тех же условиях - 0.725 атм. Кроме того,
какая-то доля измеряемого осмотического давления в коллоидных
растворах обусловливается примесью электролитов.
Так как малые величины осмотического давления коллоидных
растворов точно измерить трудно, то для определения размера или
мицеллярного веса коллоидных частиц осмометрию применяют
очень редко
Понижение температуры замерзания и повышение температуры
кипения коллоидных растворов тоже очень малы (порядка 10"6 °С),
трудно измеримы, поскольку эти величины, подобно осмотическому
давлению, связаны с концентрацией частиц в единице объема
Криоскопические и эбулиоскопические методы неприменимы еще и
потому, что кипячение и замораживание коллоидных растворов мо¬
гут привести к их коагуляции. Поскольку концентрация коллоидных
растворов очень мала (порядка 1%), их вязкость также невелика;
она мало отличается от вязкости чистого растворителя
273
15.4.2. Седиментация
Под действием силы тяжести все коллоидные частицы оседают в
растворе. Этот процесс называется седиментацией Его скорость на¬
ходится в прямой зависимости от размера частиц Более крупные
частицы оседают быстрее, чем мелкие
Для шарообразных частиц скорость седиментации рассчиты¬
вается по уравнению
= 2г2(р-р0)&
9л
где и - скорость седиментации (м/сек), г - радиус частицы (м),
р - плотность дисперсной фазы (кг/м3), р0 - плотность дисперси¬
онной среды (кг/М3), Л - вязкость среды (н е/м), 5 - ускорение
силы тяжести ^ = 9.81 м/сек ).
Зная скорость седиментации, можно вычислить радиус осе¬
дающих частиц. Если частицы легче жидкости (например, в эмуль¬
сии масла в воде), то (р - р0) имеет обратный знак, и вместо оседа¬
ния происходит всплывание частиц по тому же закону
Однако седиментации в золях противодействует броуновское
движение, стремящееся равномерно распределить коллоидные час¬
тицы по всему объему раствора. В результате действия силы тя¬
жести и силы диффузии в золях устанавливается равновесное со¬
стояние - седиментационное равновесие, при котором концент¬
рация дисперсной фазы закономерно понижается от нижних слоев к
верхним и остается постоянной во времени. Распределение числа
частиц по высоте подчиняется закону Лапласа-Перрена:
П|_=Ш8НДр —р0( )
п2 ЯТ р 2 1
М^-^^М^р-роНЬг-Ь,),
П2 К1
где П] и пг - число частиц в единице объема на высоте ^ и Иг (м)
от дна сосуда; т - масса частицы (кг); Ыд - число Авогадро;
Я - универсальная газовая постоянная; р и р0 - соответственно
плотность дисперсной фазы и дисперсионной среды (кг/м3);
V - объем частицы (м3); д - ускорение силы тяжести.
Перрен доказал точность этого уравнения и вычислил с его по¬
мощью число Авогадро, величина которого оказалась близкой к по¬
лученной другими методами. Это еще раз подтверждает примени¬
мость молекулярно-кинетической теории к коллоидным растворам.
274
В настоящее время, когда Ыд известно с большой точностью,
метод подсчета частиц на двух уровнях используют для нахождения
массы частицы и, следовательно, ее радиуса.
Исследование седиментационного равновесия часто проводят в
центробежном поле в центрифугах с очень большой скоростью
вращения, так называемых ультрацентрифугах, которые позволяют
превышать ускорение силы тяжести в сотни тысяч раз. Ультрацент¬
рифуги широко используются в химии белков, нуклеиновых кислот,
вирусов и других клеточных структур.
15.5. Оптические свойства коллоидных растворов
При рассматривании коллоидного раствора в проходящем свете
он кажется совершенно прозрачным. Если луч света направлен на
коллоидный раствор сбоку, то его путь будет обнаруживаться на
темном фоне в виде светящегося конуса, получившего название
конуса Тиндаля.
В основе явления Тиндаля лежит рассеяние видимого света
коллоидными частицами, которое связано с размерами коллоидных
частиц и длиной волны падающего света. Частицы более крупные,
чем световые волны (10'3-10'6 м), отражают их; очень мелкие части¬
цы - молекулы и ионы низкомолекулярных веществ (Ю10 м) про¬
пускают свет; частицы, размеры которых соизмеримы с длиной по¬
луволны г = а именно такими являются коллоидные частицы,
рассеивают свет во все стороны: световые волны, наталкиваясь на
подобные частицы, огибают их, и луч отклоняется от прямой линии
(явление дифракции света).
По закону Рэлея, интенсивность рассеянного света (I) обратно
пропорциональна четвертой степени длины волны падающего света:
где I и 10 — интенсивности рассеянного и падающего света,
V - число частиц в 1 м3 золя (частичная концентрация), V - объем
отдельной частицы, X - длина волны падающего света, К - констан¬
та, зависящая от коэффициентов преломления дисперсной фазы и
дисперсионной среды.
Из этого уравнения следует, что короткие волны (синяя и фио¬
летовая часть спектра) рассеиваются сильнее, чем длинные волны
(желто-красная часть спектра). Следовательно, если исходный свет
275
белый, то рассеянный свет обогащается коротковолновыми компо¬
нентами и приобретает голубой оттенок, характерный для многих
коллоидных систем при боковом освещении. Этим объясняется го¬
лубая окраска неба. В проходящем свете остается больше длинно¬
волновых компонентов спектра, которые и придают ему красный
оттенок.
Яркая окраска многих золей (золи металлов, сульфида сурь-
мы(Ш), берлинской лазури) обусловлена не только рассеянием, но
и поглощением света, причем интенсивность окраски зависит от
степени дисперсности золя.
На явлении рассеяния света коллоидными частицами основаны
важнейшие методы исследования высокодисперсных систем - уль¬
трамикроскопия и поточная ультрамикроскопия, которые использу¬
ются для определения размеров коллоидных частнц.
15.6. Строение коллоидных частиц
Всякий золь состоит из мицелл и ннтермицеллярной жидкости.
Мицеллы составляют дисперсную фазу золя, а интермицеллярная
жидкость - дисперсионную среду, в состав которой входят раство¬
ритель и растворенные в нем электролиты и неэлектролиты.
Мицелла имеет сложное строение (рис. 15.1). Она состоит из
ядра, окруженного двойным электрическим слоем. Ядро составляет
основную массу мицеллы и представлет собой агрегат из атомов
или нейтральных молекул (обычно их число огромно - несколько
сотен или тысяч). Ядро имеет кристаллическое строение.
Двойной электрический слой (ДЭС) состоит из адсорбционного
и диффузного слоев. Образуется он двумя способами: адсорбцион¬
ным путем и путем поверхностной диссоциации.
При образовании ДЭС адсорбционным путем, согласно правилу
Панета-Фаянса, на ядре адсорбируются те ионы, которые входят в
состав кристаллической решетки ядра (или изоморфны с ней). Они
называются потенциалопределяющими ионами.
Они сообщают ядру заряд, вследствие чего вокруг ядра начи¬
нают группироваться противоионы, часть из которых располага¬
ется близко к ядру (на расстоянии ионного радиуса) и вместе с
потенциалопределяющими нонами образует адсорбционный слой.
Ядро и адсорбционный слой вместе составляют гранулу, заря¬
женную вследствие неполной компенсации заряда потенциалопре-
деляющих ионов.
276
Остальные противоионы испытывают электростатическое при¬
тяжение со стороны гранулы и, кроме того, находятся в состоянии
теплового движения, стремящегося равномерно распределить ионы
по всему объему раствора.
Рис. 15.1. Схема мицеллы:
1 - ядро, 2 - слой потенциалопределяющих ионов, 3 - адсорбционный слой,
4-диффузный слой, 5-слой противоионов, 6 - двойной электрический слой.
В результате действия этих двух противоположно направленных
сил противоионы распределяются в дисперсионной среде диффузно,
т.е. концентрация их по мере удаления от поверхности частицы
убывает.
Гранула вместе с диффузным слоем противоионов составляет
мицеллу. Мицелла электронейтральна
Электролит, один из ионов которого адсорбирован на ядре, на¬
зывается стабилизатором (обычно это тот электролит, который
взят в избытке), он сообщает устойчивость коллоидным частицам.
Рассмотрим строение мицелл золя Ад1 Его можно получить
взаимодействием разбавленных растворов АдМОз и К1 при избытке
одного из реагентов. В зависимости от того, какое из веществ взято
в большем количестве, заряд частиц золя будет разный
В первом случае (при избытке АдМОз) формулу мицеллы можно
записать следующим образом-
мицелла
гранула
{[т(АдІ)пАд*(п-х )ІЧ03~]х +х ІЧОз'}
ядро адсорбционный диффузный
слой слой
277
Во втором случае (при избытке К1) формула мицеллы приобре¬
тет вид:
мицелла
гранула
{[т(Ад1)пГ (п-х)К>- хК^}
ядро адсорбционный ''Чч диффузный
слой СЛОЙ
В этих формулах т означает количество молекул Ад1 в ядре,
п - число потенциалопределяющих ионов, адсорбированных на по¬
верхности ядра, (п-х) - число противоионов в адсорбционном слое,
х - их число в диффузном слое.
Из приведенных формул видно, что первая мицелла имеет по¬
ложительно заряженную гранулу вследствие преимущественной ад¬
сорбции на ядре катионов серебра (положительный золь), а гранула
второй мицеллы заряжена отрицательно за счет отрицательных
ионов иода (отрицательный золь).
В качестве примера образования ДЭС путем поверхностной
диссоциации можно рассмотреть строение мицеллы кремниевой
кислоты, при диссоциации которой образуются потенциалопреде-
ляющие ионы ЭЮз2-, фиксированные на поверхности, и противоио-
ны Н+, которые переходят в раствор:
{[т(8Ю2-уН20)п8Ю32- 2(п-х)Н+]2'" 2хН+}.
15.7. Электрокинетические свойства
коллоидных растворов
Электрокинетическими явлениями называются процессы, возни¬
кающие в гетерогенной системе при относительном перемещении
двух фаз с участием электрического тока. Открыты русским ученым
Ф. Ф. Рейссом в 1807 году.
Причина электрокинетических явлений - существование двой¬
ного электрического слоя и легкость смещения гранулы относи¬
тельно диффузного слоя. При действии электрического поля мицел¬
ла как бы разрывается на границе между адсорбционным и диффуз¬
ным слоями (эта граница называется поверхностью скольже¬
ния)- гранула движется к одному полюсу (электрофорез), а ноны
диффузного слоя движутся к другому полюсу, увлекая за собой
гидратные оболочки (электроосмос).
278
Электрофорез - перемещение заряженных частиц дисперсной
фазы относительно дисперсионной среды под действием внешнего
электрического поля.
Электроосмос - движение дисперсионной среды относительно
дисперсной фазы под действием внешнего электрического поля.
Если проталкивать жидкость под давлением через капиллярную
систему, на концах ее возникает разность потенциалов, получившая
название потенциала протекания (или течения)
При оседании коллоидных частиц в жидкой среде также появ¬
ляется разность потенциалов между верхним и нижним слоями
жидкости, называемая потенциалом оседания (седиментации).
Потенциал протекания можно рассматривать как явление, об¬
ратное электроосмосу, а потенциал оседания - электрофорезу.
По методике проведения электрофорез делится на электрофо¬
рез с движущимся пограничным слоем (свободный электрофорез
Тнзелиуса) и зонный электрофорез (с использованием носителей -
бумаги, гелей и т.д.).
Белки, бактерии, вирусы несут заряд и потому, находясь в бу¬
ферном растворе, способны двигаться под действием электрического
поля, причем скорость движения зависит от размера н заряда
частиц. Поэтому электрофорез чрезвычайно широко применяется
в медицине и биологии.
С помощью электрофореза проводят разделение и анализ смесей
макромолекул (например, белков сыворотки крови, спинномозговой
жидкости, мочн и др.). Этот метод применяется в медицине для ди¬
агноза и контроля за ходом болезни, так как в электрофореграммах
белков сыворотки крови прн различных патологических состояниях
наблюдаются резкие изменения, специфичные для каждого заболе¬
вания. Электрофорез лекарственных веществ как метод электроте¬
рапии давно и с успехом используется в лечении многих заболева¬
ний (например, ожоговых ран, атеросклероза, ревматизма, нервно-
психических заболеваний и др ) Введение через неповрежденную
кожу лекарственного вещества с созданием депо способствует бо¬
лее длительному действию его на организм больного
Электрофоретическим методом определяют изоэлектрическую
точку белков (изоэлектрическое фокусирование) Электрофорез
белков в присутствии ПАВ (например, додецилсульфата натрия) ис¬
пользуется для определения молекулярных масс белков
Явление электроосмоса довольно распространено в биологиче¬
ских системах. Введение лекарств через кожу облегчается еще и
потому, что при наложении разности потенциалов происходит
электроосмотический перенос жидкости через поры кожи - воздух
удаляется, проницаемость кожи увеличивается В медицине элек¬
троосмос применяется для очистки лечебных сывороток.
279
Потенциалы протекания и оседания представляют собой один
из механизмов возникновения биотоков при проталкивании крови
по сосудам; в частности, установлено, что один из пиков электро¬
кардиограммы (так называемый зубец 0) обусловлен течением кро¬
ви в коронарной системе.
Электрокинетические явления применяются в промышленности
и народном хозяйстве. Электрофорез используют для борьбы с то¬
почными дымами, при изготовлении посуды, резиновых изделий,
нанесении металлических покрытий на изделия сложных профилей
и т.д. Электроосмос применяется для интенсификации добычи неф¬
ти, для осушки и пропитки пористых материалов (например, осушка
торфа, пропитка древесины), для понижения уровня грунтовых вод
и т.д Изучение потенциалов протекания лежит в основе метода
геофизической разведки полезных ископаемых.
При транспортировке жидкого топлива возникают высокие по¬
тенциалы протекания и седиментации, которые могут быть причи¬
ной пожаров и взрывов.
15.8. Электрокинетический потенциал
15.8.1. Влияние потенциала на свойства
коллоидных растворов
Ионы, достраивающие кристаллическую решетку ядра, сооб¬
щают ему заряд, определяющий электротермодинамический
потенциал (Е, рис 15.2). Полное падение этого потенциала от его
значения на ядре до нулевого значения соответствует максимальной
разности потенциалов между твердой поверхностью и всем слоем
противоионов (порядка 1 вольта).
Рис. 15.2. Падение электрических потенциалов в мицеллв:
ОА - электродинамический потенциал Е, ВО - электрокинетический потенциал £.
280
"'V
Гранул» имеет электрический потенциал того же знака, что и
Е-потенциал, но величина его меньше (порядка 50 - 100 мВ) и за¬
висит от количества противононов в адсорбционном слое. Потенци¬
ал гранулы называется электрокинетическим или {^потен¬
циалом. Кинетическим его называют потому, что он может быть
обнаружен и измерен прн всех вндах перемещения дисперсной
фазы и дисперсионной среды относительно друг друга. Его можно
определить так же, как разность потенциалов между подвижной
(диффузной) и неподвижной (адсорбционной) частью двойного
электрического слоя.
Дзета-потенциал - мера интенсивности электрокинетических
явлений (чем больше заряд гранулы, тем больше ^-потенциал, и
тем больше, следовательно, электрофоретическая скорость кол¬
лоидных частиц).
Дзета-потенциал характеризует устойчивость коллоидных час¬
тиц (чем больше ^-потенциал, тем толще диффузный слой, который
предохраняет гранулы от слипания, т.е. коагуляции).
В живых организмах дзета-потенцнал оказывает существенное
влияние на размеры межклеточных пространств, противодействуя
силам притяжения Ван-дер-Ваальса.
15.8.2. Определение потенциала методом
свободного электрофореза
Коллоидые частицы, оказавшиеся в электрическом поле, уско¬
ряются в течение короткого времени, а затем достигается стацио¬
нарное состояние, поскольку электростатическая сила (Рэл - дви¬
жущая сила при электрофорезе) компенсируется силой трения со
стороны среды (Ртр - тормозящая сила при электрофорезе).
По законам электростатики:
Гэл = ЧН,
где я - заряд частиц, Н - напряженность электрического поля
По закону Стокса
Ртр = К ягт| V,
где К - коэффициент, зависящий от формы частиц (для сферических
частиц К = 6, для вытянутых К = 4), "П — вязкость среды,
г - радиус частиц, V - скорость движения частиц
281
Следовательно,
и Клпгу
ЯН = Кллгу или я = —-—
Если рассматривать двойной электрический слой как обкладки
плоского конденсатора, то в соответствии с законами электростати¬
ки получаем:
где С, - электрокннетическнй потенциал, ц - заряд частиц, г - ради¬
ус частиц, е - диэлектрическая проницаемость среды.
Отсюда:
К7СГ|У
* " еН
Если ввести электрофоретическую подвижность и, которая
определяется как скорость, отнесенная к единице напряженности
„ К7СП
и.
Если принять во внимание, что V =—, где I - путь золя, прой-
т
„ Е
денныи за время т, а Н =—, где Е - приложенная разность потен¬
циалов, а Ь - расстояние между электродами (путь тока), тогда
_ Ктгл 11_
е тЕ'
Для расчетов все величины следует брать в системе СИ, а
именно, / и Ь (м), т (сек), Е (В), Г| (Н-с/м), £(ВОда) = 9'Ю'9 Ф/м.
Электрокинетический потенциал можно рассчитывать, исходя
из данных электроосмоса по следующей формуле:
^ _ КяхуЛ
й
где х - удельная электрическая проводимость среды (Ом/м), V -
объемная скорость электроосмоса (м3/сек), Т] - вязкость среды
(Н е/м2 ), е - диэлектрическая проницаемость среды (Ф/м), 1 - си¬
ла тока (А).
282
Зная э^ектрокннетический потенциал, а также давление, под
которым проталкивается жидкость через капилляр (или диафрагму),
можно вычислить потенциал протекания по формуле:
Е =-&-
Ктспх ’
где Ет - потенциал течения (В), £ - дзета-потенциал (В), £ - диэ¬
лектрическая проницаемость среды (Ф/м), р - давление, приводя¬
щее жидкость в движение (Н2/м), Г) - вязкость среды (Н-с/м),
X - удельная электрическая проводимость среды (Ом/м).
Таблица 15.2
Дзета-потенциалы и скорость электрофореза
эритроцитов млекопитающих
Вид млекопитающих
Скорость
электрофореза,
vlO6 м/с
^-потенциал, В
Кролик
0 55
0 0070
Свинья
0.98
00125
Морская свинка
1.11
0.0142
Человек
1.31
00168
Обезьяна резус
1.33
0 0170
Кошка
1.39
0 0178
Крыса
1.45
0 0186
Собака
1.65
0.0211
Электрофорез и электроосмос наблюдаются и при прохождении
тока через ткани живых организмов В организме существование
заряженных групп на биологических мембранах приводит к образо¬
ванию двойного электрического слоя, в котором фиксированный от¬
рицательный заряд клеточной поверхности уравновешен положи¬
тельным зарядом, создаваемым ионами межклеточной среды.
Дзета-потенциал, измеренный у разных клеток, варьирует
от -10 до -30 мВ. Электрокннетический потенциал эритроцитов изу¬
чен достаточно хорошо. Было установлено, что величина
^-потенциала является характерной для данного вида животных и
варьирует в очень узких пределах (табл 15 2)
Выяснено, что ^-потенциал лейкоцитов у данного вида живот¬
ных ниже, чем эритроцитов, например, у лошади электрофоретиче¬
ская скорость эритроцитов равна 1 01*10'6 м/с, в то время как ско¬
рость электрофореза лейкоцитов равна 0 49 10'6 м/с
283
Существует предположение, что в механизме миграции лейко¬
цитов в воспаленный очаг определенную роль должен играть дзета-
потенциал лейкоцитов, в связи с тем, что между воспаленным н
здоровым участками ткани возникает градиент потенциала
Оказалось, что величина ^-потенциала бактериальных клеток
сильно зависит от наличия белков в среде и от возраста клеток
(у молодых клеток более высокий дзета-потенциал).
15.9. Коагуляция
Коллоидные растворы - это гетерогенные системы, обладающие
большой свободной энергией поверхности, то есть они термодина¬
мически неустойчивы. Различают кинетическую н агрегативную
устойчивость. Причиной кинетической устойчивости является бро¬
уновское движение, которое противодействует оседанию частиц под
действием силы тяжести. Причиной агрегативной устойчивости яв¬
ляется наличие у частиц одноименных зарядов и сольватных оболо¬
чек, которые мешают слипанию частиц.
Коагуляция - это процесс объединения коллоидных частиц в
более крупные агрегаты вследствие потери агрегативной устойчи¬
вости Коагуляция наступает тогда, когда силы притяжения между
частицами превышают силы отталкивания одноименно заряженных
гранул Процесс коагуляции могут вызвать различные факторы: из¬
менение температуры, увеличение концентрации, механическое воз¬
действие, облучение, добавление электролитов. Наиболее изучена и
имеет наибольшее практическое значение коагуляция коллоидов
электролитами. Электролиты, с одной стороны, необходимы для
стабилизации золей, а с другой стороны, их избыточное добавление
ведет к коагуляции золей.
Все сильные электролиты вызывают коагуляцию коллоидного
раствора при увеличении их концентрации в растворе до некоторого
значения, называемого порогом коагуляции.
Порог коагуляции (у) - это минимальное количество электро¬
лита (в молях), которое надо добавить к 1 л золя, чтобы вызвать
начало коагуляции за определенный промежуток времени. Порог
коагуляции рассчитывают по формуле:
где у - порог коагуляции (моль/л), с - концентрация электролита
(моль/л), V - объем раствора электролита (л), V/ - объем золя (л).
284
Коагулйрующее действие электролитов подчиняется правилу
Шульце-Гарди: коагуляцию вызывают ионы с зарядом, противопо¬
ложным заряду гранулы, и коагулирующая способность тем выше,
чем выше заряд коагулирующего иона.
Таким образом, лучшим коагулятором является тот электролит,
который имеет наименьший порог коагуляции для данного золя.
Дерягин и Ландау с помощью теоретических расчетов показали,
что значения порогов коагуляции для ионов различного заряда от¬
носятся как
или как 1 : 0.018 : 0.0013, то есть коагулирующее действие ионов
растет примерно пропорционально шестой степени заряда иона.
Правило Шульце-Гарди носит приближенный характер, так как
наряду с величиной заряда имеет значение и природа ионов: чем
выше гидратируемость иона, тем меньше его коагулирующее дей¬
ствие. По уменьшению степени гидратации ионы могут быть распо¬
ложены в следующие ряды (лиотропные ряды Гофмейстера):
С2042- > 3042- > СНэСОСГ > СГ > МОэ‘ > В Г > Г > СМЭ-
И* > На* > К+ > РЬ+ > Сэ* > Мд2+ > Са2+ > Эг2* > Ва2+.
Коагулирующее действие ионов электролитов связано, в основ¬
ном, с уменьшением ^-потенциала коллоидных частиц за счет сжа¬
тия диффузного слоя н за счет избирательной и ионообменной ад¬
сорбции на коллоидных частицах ионов электролита с зарядом, про¬
тивоположным заряду гранулы. Коагуляция, как правило, наблю¬
дается при уменьшении ^-потенциала до 25-30 мВ (критический по¬
тенциал). Прн падении ^-потенциала от максимального значения до
критического внешние изменения в золе не наблюдаются, поэтому
этот период коагуляции называется “скрытой” коагуляцией, а пери¬
од, сопровождающийся помутнением, - “явной” коагуляцией. Ско¬
рость коагуляции возрастает до максимальной в изоэлектрическом
состоянии (£ = 0).
Коагуляция происходит также при смешении двух золей с раз¬
личными знаками заряда (взаимная коагуляция) Такой тип коагу¬
ляции применяется в санитарно-гигиенической практике при очист¬
ке воды от взвешенных коллоидных частиц При добавлении к воде
солей алюминия (или трехвалентного железа) последние, гидроли-
зуясь, образуют положительно заряженные коллоидные частицы
гидроксидов, которые соединяются с отрицательно заряженными
коллоидными частицами, находящимися в воде.
285
Коагулирующая способность электролита зависит от способа его
прибавления к золю Если добавлять электролит малыми порци¬
ями, коагуляция наступает позже. Это явление носит название
"привыкания” золей Его следует учитывать при инъекциях. Иногда
наблюдается обратная картина (“отрицательное привыкание”)
Многозарядные ионы (например, ионы трех- и четырехвалент¬
ных металлов, органические ионы), а также ионы НэО+ и ОН- при
добавлении их к золям во все возрастающих количествах вызывают
явление “неправильных рядов” (чередование зон устойчивости и
коагуляции) 4
Сначала, по достижении порога коагуляции, они вызывают коа¬
гуляцию золя (первая зона коагуляции). Дальнейшее увеличение
концентрации ионов стабилизирует золь, и коагуляция отсутствует
(зона устойчивости). При еще больших концентрациях вновь насту¬
пает коагуляция золя (вторая зона коагуляции). Явление “не¬
правильных рядов” связано с перезарядкой коллоидных частиц. Пе¬
резарядка происходит в том случае, когда коагулирующий ион про¬
никает в адсорбционный слой гранулы в сверхэквивалентных коли¬
чествах, т.е. в количествах больших, чем надо для нейтрализации
потенциалопределяющих ионов.
Перезарядка золей объясняется тем, что многозарядные ионы
обладают большой адсорбционной способностью.
При коагуляции золя смесью электролитов можно наблюдать:
а) явление аддитивности - суммирование коагулирующего дей¬
ствия ионов;
б) явление антагонизма - ослабление коагулирующего действия
одного иона в присутствии другого;
в) явление синергизма - усиление коагулирующего действия
одного иона в присутствии другого.
Явление коагуляции играет существеную роль в живом орга¬
низме, так как коллоидные растворы клеток и биологических жид¬
костей находятся в соприкосновении с электролитами. Поэтому при
введении в организм какого-либо электролита надо учитывать не
только его концентрацию, но и заряд ионов. Так, физиологический
раствор хлорида натрия нельзя заменить изотоничным раствором
хлорида магния, поскольку в этой соли имеется двухзарядный ион
магния, обладающий высоким коагулирующим действием.
При введении смеси солей следует предварительно убедиться,
что эти соли не являются синергистами, чтобы избежать вредной
для организма коагуляции.
Решение многих проблем в медицине (например, протезирова¬
ние кровеносных сосудов, клапанов сердца и т.д.) связано с про¬
блемой свертывания крови. В хирургии во время операций в кровь
286
вводят антикоагулянты (гепарин), а после операций - для повыше¬
ния коагуляции - протамин-сульфат.
С явлением коагуляции эритроцитов вследствие уменьшения
их дзета-потенциала врачи постоянно имеют дело в клинических
лабораториях (метод определения СОЭ - скорости оседания
эритроцитов).
Это явление объясняется тем, что при патологии в крови уве¬
личивается содержание некоторых видов белков, место ионов
электролитов на поверхности эритроцитов занимают белки, заряд
которых ниже, чем у суммы замещенных ими ионов. Заряд эритро¬
цитов понижается, они быстрее объединяются и оседают.
В природе процессы коагуляции играют огромную роль в фор¬
мировании структуры почв. Образование плодородных дельт в
устьях рек легко объясняется тем, что электролиты морской воды
вызывают коагуляцию коллоидных частиц, содержащихся в речной
воде, а остановка течения способствует седиментации коагулиро¬
ванных агрегатов.
15.10. Коллоидная защита
При добавлении к золям некоторых высокомолекулярных ве¬
ществ устойчивость золей к действию электролитов значительно
повышается, что выражается в повышении порога коагуляции. Та¬
кое явление получило название коллоидной защиты. Защищен¬
ный золь поддается концентрированию и даже выпариванию досуха
и становится обратимым (то есть термодинамически устойчивым),
он как бы приобретает свойства раствора высокомолекулярного ве¬
щества Механизм защитного действия зависит от образования ад¬
сорбционного слоя введенного вещества вместе с его гидратными
оболочками на поверхности частиц гидрофобного золя. Защитными
веществами в водной среде могут служить белки, углеводы, пекти¬
ны (разновидность растительных полисахаридов).
Различные высокомолекулярные вещества защищают золи не
одинаково. Мерой защитного действия высокомолекулярных соеди¬
нений является так называемое золотое число - то минимальное
количество миллиграммов сухого высокомолекулярного соединения,
которое необходимо добавить к 10 мл стандартного (красного) золя
золота для того, чтобы предотвратить его коагуляцию (посинение)
при введении в систему 1 мл 10%-ного раствора хлорида натрия.
Аналогично можно оценить серебряное число, рубиновое число,
железное число и т д Более простое и легко доступное железное
287
число определяют как минимальное число миллиграммов защи¬
щающего высокомолекулярного соединения, способного защитить
10 мл золя гидроксида железа от коагулирующего действия 1 мл
0.005 моль/л раствора сульфата натрия
Сравнение золотых и железных чисел некоторых биполимеров
представлено в таблице 15 3.
Таблица 15 3
Золотые и железные числа некоторых веществ
Вещество
Золотое число, мг
Железное число,
мг
Желатин
0.008
50
Гемоглобин
0.25
-
Крахмал
25 0
20 0
Явление защиты играет большую роль в жизни организмов. Так,
белки крови защищают жир, холестерин, малорастворимые соли
кальция и мочевой кислоты от коагуляции и выделения на стенках
сосудов. При понижении защитной функции белков возникают за¬
болевания: атеросклероз, кальциноз, подагра, образование камней в
почках, печени и т.п Способность крови удерживать в растворен¬
ном состоянии большое количество газов (кислорода и углекислого
газа) также обусловлена защитным действием белков.
В медицине измерением золотого числа спинномозговой жид¬
кости пользуются для диагностики некоторых заболеваний, напри¬
мер, менингита.
В фармацевтической промышленности защитные свойства ве¬
ществ широко используются для получения концентрированных зо¬
лей серебра, ртути, золота и их радиоактивных изотопов (например,
лекарственный препарат колларгол - коллоидный раствор серебра, а
протаргол - коллоидный раствор оксида серебра, защищенные вы¬
сокомолекулярными соединениями).
15.11. Коллоидные растворы
поверхностно-активных веществ.
Солюбилизация
Мыла и некоторые красители, являющиеся ПАВ, при их малой
концентрации в воде (10'5-10'3 моль/л) образуют истинные раство¬
ры. Их более концентрированные растворы приобретают коллоид¬
ную структуру вследствие самопроизвольного образования в систе¬
ме коллоидных агрегатов (см. рис. 15.3).
288
ИСТИННЫЙ
раствор
<чи°о
°7\>
\ Л
о^|^о
Г ЗОЛЬ
(С2>
-О’ТТТт.^,
«а шаг»
дИТТ^
«'а а Г»
сз
■ » гель
(Сз > С2)
Рис. 15.3. Образование мицелл в коллоидных растворах ПАВ.
При этом липофильные части молекул ориентируются по на¬
правлению друг к другу с образованием гидрофобного ядра, кото¬
рое защищено от водной фазы оболочкой из гидрофильных частей
молекул.
При более высоких концентрациях сферические мицеллы пре¬
вращаются в более устойчивые пластинчатые мицеллы в виде
двойных слоев с полярными группами, направленными наружу, и
углеводородными цепями, ориентированными параллельно и на¬
правленными внутрь слоев.
Следует отметить важное значение мицеллообразования в био¬
логии, поскольку биологические мембраны представляют собой
сложные двойные слои с гидрофобным ядром и гидрофильным
окружением.
Образование сферических мицелл происходит при определенной
для каждого ПАВ концентрации, которая называется критической
концентрацией мицеллообразования (ККМ) При этом резко
изменяются физико-химические свойства раствора ПАВ' осмотиче¬
ское давление, электропроводность, мутность, поверхностное натя¬
жение (см. рис. 15.4)
ККМ определяют по изменению одного из этих свойств Метод
определения ККМ по изменению поверхностного натяжения а наи¬
более точен по сравнению с другими В отличие от асимметричных
единичных молекул ПАВ сферические мицеллы равномерно гидра¬
тированы по всей поверхности. Поэтому поверхностное натяжение
водных растворов ПАВ резко изменяется с ростом концентрации
10 Зек. 675
289
ПАВ вплоть до ККМ, после чего практически ие меняется с увели¬
чением концентрации. На кривых зависимости поверхностного на¬
тяжения от логарифма концентрации (см рис 15.4) обнаруживает¬
ся резкий излом, абсцисса которого соответствует ККМ
Рис. 15.4. Изменение поверхностного натяжения
коллоидного раствора ПАВ.
Свойством мицеллярных растворов, вытекающим из строения
мицелл ПАВ, является солюбилизация, т е. внедрение мало- или
практически нерастворимых в данном растворителе веществ в ми¬
целлы, приводящее к резкому увеличению растворимости этих ве¬
ществ в мицеллярных растворах. Например, бензол, гептан раство¬
ряются в водных растворах ПАВ при с > ККМ.
С процессом солюбилизации связано стабилизирующее действие
ПАВ, которые часто выступают в качестве эмульгаторов. Так, на¬
пример, процесс усвоения жиров
в организме начинается с солюби¬
лизации их солями желчных и
жирных кислот. Моющее действие
ПАВ также в известной степени
обусловлено способностью загряз¬
нений солюбилизироваться в рас¬
творах ПАВ.
Следует сказать, что многие
природные молекулы, обладающие
свойствами ПАВ, например, фос¬
фолипиды, могут в водных рас¬
творах образовывать бислойные
структуры, в которых их молеку-
Рис. 15.5. Строение липосомы. лы обращены друг к другу гидро¬
290
фобными 'Концами (гидрофобные взаимодействия), а полярными
группами - к молекулам воды (рис. 15.5). Такие структуры в прин¬
ципе могут быть многослойными, а не только бислойными.
Они могут включать в свою структуру воду, оказавшуюся в
промежутке между двумя бислойными структурами - внутренней и
внешней Такие образования называют липосомами Они удобны
как объекты изучения моделей клеточных мембран, с одной сторо¬
ны, а, с другой стороны, их используют для направленной доставки
лекарственных средств к тем или иным пораженным органам или
тканям В медицине в настоящее время широко применяются как
лекарственные средства протеолипосомы, т.е. липосомы, содержа¬
щие внутри молекулу белка.
Часть IV
ЭЛЕМЕНТЫ-ОРГАНОГЕНЫ
. между неорганическими ..
соединениями и органически¬
ми нет никакого особого раз¬
личия
А М Бутлеров
Глава 16. ВОДОРОД
Водород, как элемент, пред¬
ставляет составную часть,
наиболее подвижную в своих со¬
единениях.
Д. И. Менделеев
Символ Н, порядковый номер 1, относительная атомная масса
1.008. Впервые получен в 17 в. фламандским химиком И. Ван Гель-
монтом, тщательно изучен английским физиком и химиком Г. Ка¬
вендишем в конце 18 в. Название “Hydrogen” (от греческих слов
hydro - вода и genes - рождающий) ввел А. Лавуазье.
Согласно современным представлениям, этот элемент с элек¬
тронной конфигурацией Is1 нельзя отнести к какой-либо группе, а
следует считать элементом I периода или просто первым элементом
Периодической системы.
Чисто условно его можно поместить как в первую группу (так
как он обладает одним электроном), так и в седьмую (поскольку до
полного заполнения уровня ему не хватает одного электрона). Дей¬
ствительно, водород, с одной стороны, может при определенных
условиях проявлять металлические свойства, что роднит его со ще¬
лочными металлами, однако его энтальпия ионизации по сравнению
с ними аномально высока (ДН°; = 1310 кДж/моль, для лития 520,
а для натрия 500 кДж/моль). Это вызвано тем, что атом водорода
обладает лишь одной электронной оболочкой, а следовательно
единственный электрон не экранирован от действия заряда ядра
внутренними электронами. К тому же атом водорода мал, его ра¬
диус составляет 0.37 А.
С другой стороны, атом водорода способен присоединять элек¬
трон, поэтому указывают на его сходство с элементами VII группы.
Однако в отличие от них водород не является p-элементом. Это
различие проявляется в разнице энтальпий присоединения элек¬
трона галогенов и водорода (АН°еа = -73 кДж/моль для водорода,
292
-333 и -350 кДж/моль для фтора и хлора, соответственно). Таким
образом, водород уникален по свойствам и занимает особое место
в Периодической системе.
Уникальность водорода проявляется в таком важном явлении,
я связь, величина энергии которой обычно близка к
физико-химические свойства воды, димерное строение молекул фто¬
ристого водорода Н2р2, вторичную и третичную структуру белков,
устройство двойной спирали ДНК и др.
Еще одно фундаментальное явление с участием атомов водоро¬
да - трехцентровая связь для электронодефицитных молекул, при¬
мером которой служит строение молекулы диборана В2Н6. Именно в
таком виде существует соединение бора с водородом, что, если дать
простейшее объяснение, вызвано существованием двух водородных
связей в молекуле этого вещества.
По сути каждая из электронных пар двух центральных атомов
водорода принадлежит здесь одновременно трем атомам, что и по¬
зволяет говорить о новой разновидности химической связи.
Водород - самый распространенный элемент Вселенной. По ко¬
личеству в составе земной коры он стоит на девятом месте. Основ¬
ная часть его находится в виде воды, меньшее количество присут¬
ствует в нефти и в составе природного газа.
Существуют водородные бактерии, которые получают энергию,
утилизируя водород. В то же время имеются и микроорганизмы,
выделяющие водород при метаболизме. В частности, к их числу
относятся и некоторые бактерии желудочно-кишечного тракта. На¬
рушение определенных функций пищеварения связано с изменени¬
ем состава бактериальной флоры кишечника, а это, в свою очередь,
приводит к выделению водорода. Последнее обстоятельство исполь¬
зуется в диагностических целях. Например, лактазная недостаточ¬
ность, которая исключает вскармливание младенцев искусственны¬
ми питательными смесями, обнаруживается посредством определе¬
ния водорода в выдыхаемом воздухе
Существуют три изотопных разновидности водорода: протий
1iH, дейтерий D - iH и тритий Т - Э1Н, отличающиеся количеством
нейтронов Содержание дейтерия в природном водороде составляет
0 015 %, а трития в земной коре находится не более 2 кг.
Водородная связь определяет многие специфические
1-Г 'Н'' 'н
диборан
16.1. Разновидности водорода
293
Различия в химических свойствах изотопов малы, однако они
заметно различаются физическими свойствами Так называемая тя¬
желая вода D20 кипит при 101.3° С, плавится при 3.813° С, сверх-
тяжелая вода Т20 плавится при 4.5° С. В принципе изотопных раз¬
новидностей воды существует намного больше, если учесть суще¬
ствование устойчивых изотопов кислорода 160 и 180 (Н21вО, HD1 О и
т. д.). Тем не менее, химические различия в свойствах изотопов все
же имеются и проявляются они в несколько меньших скоростях
химических реакций молекул, содержащих дейтерий (или тритий),
а также в различных термодинамических параметрах реакций с
участием дейтеросодержащих молекул и без них. Это явление
носит название кинетического изотопного эффекта. Замена водоро¬
да на дейтерий несколько замедляет скорость метаболических про¬
цессов, а потому тяжелая вода токсична. Следовательно природное
содержание дейтерия в биосредах является оптимальным для под¬
держания гомеостаза.
В обычных условиях водород представляет собой двухатомную
молекулу Н2. Энергия ковалентной связи в этой молекуле составля¬
ет 436 кДж/моль, а следовательно образование атомарного водоро¬
да из молекулы происходит лишь при высоких температурах.
Атомарный водород, так называемый водород in situ, то есть в
момент выделения, чрезвычайно активен, как и большинство ради¬
кальных частиц, имеющих неспаренные электроны. Атомарный во¬
дород получают действием металлов на кислоты, а также растворе¬
нием водорода в некоторых металлах (платине, палладии и др.).
Малые размеры водородных молекул позволяют им легко диф¬
фундировать внутрь металла и при этом переходить в атомарное со¬
стояние, включаясь в межатомные полости металлической кристал¬
лической решетки. К примеру, в одном объеме палладия можно
растворить 850 объемов водорода. Высокая активность и большая
концентрация атомарного водорода в таких металлах делают по¬
следние эффективными катализаторами гидрогенизации, что широко
применяется при гидрировании непредельных органических соеди¬
нений, например, в случае жиров, что используется, в частности,
при получении маргарина.
16.2. Химические свойства
Относительная электроотрицательность водорода равна 2.2,
а это означает, что водород способен реагировать как с электропо¬
ложительными, так и электроотрицательными элементами. Актив¬
ность водорода высока, и он энергично взаимодействует со многи¬
294
ми элелЙмтами Периодической системы. В реакциях с более элек¬
троположительными элементами - щелочными и щелочноземельны¬
ми металлами - водород выступает в качестве окислителя, а по от¬
ношению к кислороду, галогенам, сере он проявляет свойства вос¬
становителя. В результате образуются гидриды, строение которых
определяется природой входящего в их состав элемента.
Так, с рядом переходных металлов водород дает нестехиометри-
ческие соединения с переменным количеством водорода за счет
встраивания атомов водорода в пространство между атомами ме¬
талла в кристаллической решетке. Для одних металлов количество
водорода не велико, например, в случае соединения \/Н0б. В некото¬
рых же случаях, как уже отмечалось, количество водорода по отно¬
шению к металлу может достигать многих сотен объемов. Следова¬
тельно, такие гидриды напоминают сплавы с их переменным соста¬
вом, а это в определенной мере роднит водород с металлами.
Сведения о строении гидридов можно получить из таблицы 16.1,
в которой представлены гидриды элементов II периода.
Таблица 16.1
Свойства гидридов элементов II периода
Гидрид
ин
ВеН2
В2Нв
сн4
ЫНз
Н20
№
ОЭО элемента
1.0
1.5
2.0
2.5
3.1
35
4.0
и.0
5 88
0
0
0
0.5
1 86
1 96
Как видно из величин дипольных моментов, в случае гидридов
щелочных металлов речь идет о ионных соединениях, в составе ко¬
торых водород находится в виде гидрид-аниона :Н_, то есть обладает
степенью окисления -1. То же характерно и для гидридов щелочно¬
земельных металлов, дипольный момент молекул которых равен ну¬
лю из-за симметричного линейного строения эр-гибридизованной
структуры. Однако связи металл-водород в них обладают высоким
дипольным моментом. Как и другие ионные соединения, эти гидри¬
ды - твердые кристаллические вещества.
Гидрид-анион по размеру больше атома водорода (его ради¬
ус - 1 5 А). Существование гидрид-аниона указывает на некоторую
аналогию водорода с галогенами, образующими галогенид-ионы, од¬
нако свойства тех и других сильно различаются Поведение гидрид-
иона характеризуется двумя особенностями
Во-первых, гидрид-ион - сильнейший восстановитель Стандарт¬
ный потенциал реакции
2: Н- - 2е 5 Н2
295
составляет -2 25 В. Поэтому солеобразные гидриды энергично реа¬
гируют с различными окислителями, восстанавливая их. В част¬
ности, они взаимодействуют с водой с выделением водорода, и эта
реакция служит удобным методом получения водорода наряду с
другими, хорошо известными:
СаН2-1 + 2Н+1ОН -> Са(ОН)2 + 2Н2°Т.
Во-вторых, этот ион обладает высокой донорной активностью,
что вызывает образование комплексных гидридов в реакциях с
электронодефицитными гидридами бора и алюминия:
Ы:Н + А1Нз -» И[А1Н4] - алюмогидрид лития
2Ма:Н + В2Н6 -» 2Ма[ВН41 - борогидрид натрия.
Эти гидриды как активные восстановители широко применяют в
органической химии для гидрирования многих классов веществ, на¬
пример, для восстановления спиртов в углеводороды:
Ы[А1Н4] + 4РОН -> ИОН + А1(ОН)3 + 4Р-Н.
Комплексные гидриды - потенциальные источники водорода при
взаимодействии с водой, при этом на одну молекулу комплексного
гидрида выделяется сразу же несколько молей водорода. Например,
при действии воды на 1 кг борогидрида алюминия А1[ВН4]з выделяет¬
ся около 3800 литров газообразного водорода.
При образовании гидридов с элементами правой части Периоди¬
ческой системы водород выступает в роли более электроположи¬
тельного партнера в образующейся ковалентной связи.
Возникает вопрос, почему с такими электроотрицательными
элементами, как фтор, кислород, хлор не образуются соединения
ионного характера. Причина этого заключается в малой величине
радиуса водородного атома, что приводит к достаточно сильному
взаимодействию электронной пары химической связи с протоном -
ядром атома водорода Поэтому все гидриды этой группы, а следо¬
вательно и молекулы, содержащие в своем составе связи атома во¬
дорода с элементами-органогенами, представляют собой ковалент¬
ные соединения.
В свою очередь, степень полярности связи элемент-водород в
ковалентных гидридах и ее способность к поляризации определяет
свойства этих соединений. В частности, неполярная связь углерод-
водород сообщает гидрофобный характер тем участкам органиче¬
ских молекул, в которых она находится. Таким образом, алкильные
группы биомолекул, построенные посредством неполярных связей
углерод-водород и углерод-углерод, ответственны за гидрофобные
296
взаимодействия в водных растворах биосред и формируют липо-
фильные участки биомолекул, а следовательно и сами липидные
структуры.
Полярные связи О-Н, ІЧ-Н и в-Н также широко представлены в
биомолекулах, причем различная величина положительного заряда
на атоме водорода в них сообщает спиртовой, амино- и сульфгид-
рильной группам различную кислотность. В принципе различают
С-Н, О-Н, ІЧ-Н и в-Н-кислоты. В этом ряду способность к диссоциа¬
ции по кислотному типу минимальна для С-Н кислот, невелика у
ІЧ-Н кислот, в то время как спиртовая и сульфгидрильная группы
обладают заметными кислотными свойствами. При этом связь в-Н
сильнее диссоциирует в водных средах с образованием протонов,
чем связь О-Н, так как она обладает большей поляризуемостью из-
за большего, чем у кислорода, радиуса атома серы.
Итак, связи элементов-органогенов с атомом водорода входят в
состав алкильных, спиртовых, амино- и сульфгидрильных групп
биомолекул. Биологически значима и более полярная молекула НСІ,
водный раствор которой - соляная кислота - входит в состав желу¬
дочного сока.
16.3. Катион водорода
Потеря электрона вызывает переход атома водорода в элемен¬
тарную частицу - протон. Так как радиус протона ничтожно мал
(105 А), плотность заряда на его поверхности очень велика. Это
приводит к высокой величине поляризующего действия протона, а
потому отрыв электрона от атома водорода требует больших энер¬
гетических затрат (ДН^ = 1310 кДж/моль). Поэтому ион водорода
образуется из молекул с полярной связью с участием атома водоро¬
да лишь в растворах, причем тогда, когда растворитель способен
эффективно сольватировать протон
Таким растворителем прежде всего является вода, которая
уменьшает свободную энергию протона на величину энергии гидра¬
тации, составляющую 1104 кДж/моль В итоге затраты на отрыв
электрона составляют 1310 - 1104 = 206 кДж/моль, то есть значи¬
тельно меньшую величину, чем в отсутствие растворителя
Процесс гидратации протона складывается из двух стадий. На
первой из них имеет место донорно-акцепторное взаимодействие, в
котором протон проявляет свойства типичного акцептора электрон¬
ной пары:
297
Н++ Н20: 5 Н30+ Крав„ =10100
Н
I
Н
%/Т
I
н .
I
н
катион гидроксония
гидратированный
катион гидроксония
В результате образуется катион гидроксония. Высокая величина
константы равновесия этого процесса означает, что все протоны в
водном растворе переходят в ион гидроксония. Далее катион гид¬
роксония гидратируется тремя молекулами воды за счет образова¬
ния трех водородных связей, и в итоге образуется катион Нэ04+,
в виде которого и существуют протоны в водном растворе.
Таким образом, диссоциацию воды с учетом гидратации не толь¬
ко катиона водорода, но и гидроксильного иона можно представить
схемой-
Гидратированный протон обладает следующими свойствами. Во-
первых, он является сильным акцептором электронной пары. Это
проявляется в его реакциях с аммиаком и аминами, а также с кис¬
лородсодержащими соединениями с образованием аммониевых и
оксониевых катионов, соответственно.
Во-вторых, протон является окислителем в соответствии с урав¬
нением:
Потенциал водородного электрода выражается уравнением:
а зависимость величины потенциала от кислотности среды можно
представить графиком, приведенным на рис. 16.1.
Так как pH плазмы равен 7.4, чему отвечает величина потен¬
циала, равная -0.42 В, это означает, что в биосредах значение по¬
тенциала меньшее, чем -0.42 В, будет вызывать восстановительные
процессы с выделением водорода. Следовательно, минимально до-
пН20 ^ Н(Н20)4+ + 0Н(Н20)п-5"
2Н+ + 2е ^ Н2.
Ф = -0.059рН,
298
пустимаЯч^ величина потен¬
циала биосред должна быть
не меньше этой величины.
Нельзя забывать, что
стандартный потенциал во¬
дородного электрода, приня¬
тый равным нулю, является
точкой отсчета при опреде¬
лении силы окислителей и
восстановителей. Именно по
отношению к водороду в
<р°, В
Рис. 16.1. Зависимость потенциала
водородного электрода от pH.
водных растворах построен предложенный Н. Н. Бекетовым элек¬
трохимический ряд напряжений металлов.
Протон играет исключительно важную роль в биопроцессах. Во-
первых, протон в растворах определяет их кислотные свойства.
Концентрация иона водорода является важным параметром гомео¬
стаза биосред. Многие вещества в составе биообъектов обладают
кислыми свойствами, будь это сильные и средние минеральные кис¬
лоты (соляная в желудочном соке, фосфорная в составе ее произ¬
водных - аденозинтрифосфорной и нуклеиновых кислот) или много¬
численные органические кислоты животных и растительных орга¬
низмов (уксусная, масляная, молочная, пировиноградная, аскорби¬
новая, никотиновая, лимонная, салициловая и др кислоты)
Во-вторых, с участием катиона водорода происходят многие
окислительно-восстановительные превращения, и в этих случаях
кислотность раствора сказывается на величине потенциала этих
рН-зависимых редокс-процессов. Типичным примером может слу¬
жить полуреакция, характеризующая окислительные свойства
бихромат-иона:
Сг2072' + 14Н+ + 6е 5 2Сг3* + 7Н20.
Редокс-потенциал этой полуреакции зависит от pH среды:
V 2 + = Ф 2
Сг?07 + 14Н Сг707 +14Н
0059
[сг2072-] [н+]14
2Сг +7Н20 2Сг +7Н20
откуда при [Сг2С>72_1 = [Сг3+] = 1 моль/л получаем
о 0.059 14
Сг3+1 [Н20]7
Сг207 +14Н
Сг?07 +14Н*
pH или
2Сг3 + +7Н20 2Сг3+ +7Н20
299
ф
усйо1
2Сг3 + +7Н20 2Сг3 + +7Н20
Из этого следует, что в сильно кислых средах бихромат-ион
представляет собой сильный окислитель (для однонормального рас¬
твора кислоты <р =<р° = 1.36 В), однако в нейтральной среде катнон
Сг3+ следует считать восстановителем (<р = <р° - 0.138-7 = 0.394 В.)
Многие метаболические редокс-процессы также происходят с
участием ионов водорода, а следовательно их направление опреде¬
ляется кислотностью среды. Среди них укажем такие важные пре¬
вращения, как восстановление кислорода
и фиксация азота:
N2 + 8Н+ + 6е 25 2МН4+ ( ф = ф° - 0.059рН )
М2+8Н+/2МН4+
Существует серия метаболических окислительно-восстанови-
тельных превращений биосубстратов (в), также зависящих от pH:
К их числу относятся окисление спиртовой группы в карбо¬
нильную, переход альдегидов в карбоновые кислоты, хинонов в гид-
рохиноны, восстановление кофермента - флавинадениндинук-
леотидфосфата (ФАД в сокращении) в восстановленную форму
ФАД-Нг и ряд других процессов, уравнение Нернста для которых
выражается общей формулой
Конкретное решение уравнения Нернста для каждой приведен¬
ной выше полуреакции при значении pH, равном 7 4, и с подста¬
новкой соответствующего значения стандартного потенциала опре¬
деляет истинное значение величины ее потенциала в биосредах.
Таким образом, концентрация катионов водорода является важ¬
нейшей характеристикой интенсивности прохождения в них окис¬
лительно-восстановительных превращений.
В-третьих, образование хелатов, а следовательно, связывание
катионов металлов в биокомплексы, происходит с участием ионов
водорода. Равновесие образования комплексов и металло-лиганд-
ный баланс в целом, таким образом, зависит от кислотности среды.
02 + 4Н+ + 4е 25 2Н20 (<р = <р°
02+4Н+/2Н20
- 0 059рН)
в + 2Н+ + 2е 2ц Б Н2.
Ф * = ф°
Э+2Н /Э Н2 8+2Н+/Э Н2
- 0.059рН.
300
В-че^ертых, во многих реакциях осаждения также принимают
участие ионы водорода. К их числу относится и процесс образова¬
ния минеральной основы костной ткани. Следовательно, кислот¬
ность среды является регулятором гетерогенных равновесий в
биосредах.
В-пятых, малые размеры катиона водорода определяют его вы¬
сокую каталитическую активность во многих, в том числе и биохи¬
мических, реакциях. Известно, что подвижность катиона водорода
примерно в десять раз выше скорости перемещения других ионов.
Возникает неясность, почему речь идет о малых размерах катиона
водорода, если он в действительности не мал из-за гидратации. Од¬
нако и здесь проявляются аномальные свойства водорода. Для иона
водорода имеет место так называемый туннельный эффект, состоя¬
щий в том, что его присоединение на одном конце ассоциирован¬
ной цепочки молекул воды сопровождается его одновременным от¬
щеплением на другом конце этой цепи.
Катализ ионами водорода важен, в частности при гидролити¬
ческом распаде липидов, пептидов и полисахаридов, вот почему вы¬
сока кислотность желудочного сока и процесс усвоения пищи в ки¬
шечнике требует участия желчных кислот. Следует помнить, что
пространственное строение ферментов зависит от кислотности сре¬
ды, а поэтому они проявляют свою каталитическую активность
лишь в узких интервалах значений pH.
16.4. Вода
Наиболее значимым для биологии и медицины гидридом явля¬
ется вода. Прежде всего, она важна как универсальный раство¬
ритель, необходимый для протекания биохимических реакций. Вода
прекрасно растворяет многие ионные и полярные ковалентные сое¬
динения.
Кроме того, сама вода способна выполнять каталитические
функции, на что уже давно обратили внимание Например, сухой
хлористый водород не реагирует с аммиаком и металлами Метал¬
лический натрий не горит в сухом хлоре, а алюминий не взаимо¬
действует с иодом, однако добавление воды вызывает бурную реак¬
цию между ними Таких примеров известно множество, и это по¬
зволяет считать воду самым универсальным катализатором.
Далее, вода, с точки зрения кислотно-основных свойств, пред¬
ставляет собой истинный амфолит, так как содержит равное коли¬
чество катионов водорода и гидроксильных анионов. Если о роли
301
протона в химических реакциях уже было сказано, то здесь необхо¬
димо обратить внимание на значение аниона ОН". Он выступает в
качестве катализатора многих реакций в водных растворах, и в та¬
ком случае говорят о щелочном катализе.
Вода вступает в химическое взаимодействие со многими про¬
стыми и сложными веществами. В частности, следует отметить ре¬
акции гидролиза, идущие без изменения степеней окисления.
Среди реакций гидролиза значимыми для биосистем являются
гидролитические процессы с солями, входящими в состав биосред,
а также гидролиз полипептидов, липидов, нуклеотидов. Обратимый
гидролиз молекулы аденозинтрифосфата (АТФ) является ключевой
реакцией биоэнергетики.
Нельзя забывать, что метаболическая вода, в свою очередь, об¬
разуется во многих биохимических реакциях - при нейтрализации,
в результате биологического окисления, как результат отщепления
от биомолекул, при конденсациях и т. д.
Еще одно важное свойство воды - ее донорная активность, вы¬
званная присутствием у атома кислорода неподеленных электрон¬
ных пар. Это приводит к тому, что все катионы переходных метал¬
лов, в том числе и биокатионы железа, меди, цинка и др., если они
не связаны с белками или другими органическими биомолекулами,
в биосредах представляют собой аквокомплексы.
Окислительно-восстановительные превращения воды определя¬
ются двумя полуреакциями:
2Н+ + 2е 25 Н2 и
2Н20 25 02 + 4Н+ + 4е.
О значении первой из них уже сказано, а вторая может проис¬
ходить в тканях, если окислители будут обладать большей величи¬
ной редокс-потенциала, чем его значение для второй полуреакции
при pH = 7.4. Зная, что ф° . = +1.24В, получаем, что это
02+4Н+/2Н20
должно происходить при
Ф + = 1.24 - 0.059 • 7.4 = 40.80В .
02+4Н /2Н20
Следовательно, границы редокс-потенциалов жизненных процессов
должны лежать в интервале -0.42 - +0.82 В. При потенциале мень¬
шем, чем -0.42 В, биосреды выделяют водород, иными словами, в
этих условиях идет необратимое восстановление биосубстрата.
Выше значения потенциала +0.82 В будет происходить выделе¬
ние кислорода, то есть необратимое окисление биомолекул. Редокс-
302
потенциалы всех вышеперечисленных окислительно-восстанови-
тельных превращений биосубстратов Б лежат внутри этого интер¬
вала (см. табл. 11.3).
Вода в биосредах находится частично в виде ассоциатов с неор¬
ганическими ионами и биомолекулами - белками, углеводами и др.
Иными словами, все биомолекулы окружены гидратными оболочка¬
ми. Такая вода называется связанной водой. Ее количество обычно
составляет около 40%. Остальная вода представляет собой ассо¬
циированную водородными связями подвижную структуру. При
этом идет непрерывный обмен между молекулами связанной и сво¬
бодной воды. При патологических процессах (воспалительных яв¬
лениях, образовании раковых клеток и др.) это соотношение ме¬
няется, а следовательно, должны меняться и физические свойства
биосред.
В частности, различна магнитная релаксация для молекул сво¬
бодной и связанной воды. Это явление может быть количествен¬
но измерено с помощью метода ядерного магнитного резонанса, что
и лежит в основе томографии ядерного магнитного резонанса,
успешно применяемой для диагностики различных поражений
тканей и органов, в том числе при обнаружении злокачественных
опухолей.
16.5. Радиолиз воды
Помимо гетеролитической диссоциации воды возможно и ее ге¬
молитическое разложение, которое не идет при обычных условиях,
так как стандартная энтальпия разложения воды на кислород и во¬
дород составляет +575 кДж/моль, то есть молекула воды обладает
высокой термодинамической стабильностью Однако воздействие
сильных источников энергии - ультрафиолетового и у-излучения
может вызывать распад воды при обычных температурах.
Распад воды под действием радиации лежит в основе химизма
лучевого поражения. Концентрация биосубстрата в тканях не
слишком высока (10-1 - 10"5 моль/л). Поэтому у-излучение по пре¬
имуществу будет оказывать воздействие на доминирующие молеку¬
лы воды.
При этом будут параллельно происходить два процесса: возбуж¬
дение молекул воды и выброс электрона
При возбуждении происходят следующие превращения.
303
1. Поглощение у-кванта молекулой воды и ее переход в возбуж¬
денное состояние:
нон НОН*
(Символ — применяется для обозначения химических реак¬
ций под действием радиации )
2. Далее возможен гомолитический распад возбужденной моле¬
кулы воды
НОН’ -» Н- + ОН
3. Образовавшиеся свободные радикалы могут претерпевать
следующие превращения:
2Н -»• Н2
2'ОН -> Н202
Н‘ + ОН НОН
Из их числа последняя реакция - рекомбинация с образованием
воды - не вызывает каких-либо нежелательных эффектов. Две же
первые приводят к появлению в плазме или тканях активного вос¬
становителя - водорода и сильного окислителя - перекиси водорода.
Они могут атаковать молекулы биосубстрата (ферментов, нуклеино¬
вых кислот, липидов клеточных мембран и др.) и приводить их к
необратимым трансформациям. Однако не следует забывать, что
атомарный водород и гидроксильный радикал - еще более активные
восстановитель и окислитель, соответственно.
Другое направление радиолиза воды заключается в том, что
при сильном возбуждении электрон покидает молекулу воды по
уравнению:
1. НОН НОН+- + ебыстр
Это направление преобладает, если речь идет о квантах высоких
энергий. В результате образуются так называемый быстрый элек¬
трон и ион-радикал воды. Основная энергия процесса приходится на
долю быстрого электрона, который может обладать энергией, доста¬
точной для последующей ионизации другой (или других) молекул
воды. Быстрый электрон уже через =10'11 секунды подвергается
гидратации, в результате чего образуется гидратированный элек¬
трон (егидр):
2- ббыстр + ПН20 —> бгидр
304
Ион-раЗЫкал воды реагирует с водой:
з. нон+-+нон-»н3о+ + -он,
а гидратированный электрон может взаимодействовать и с катионом
гидроксония по уравнению:
4. 6гидр +Н30+—» Н' + Н20
и с молекулой воды:
5. егидр +НОН Н' + ОН",
и с продуктами разрушения воды по механизму ее возбуждения.
В итоге в облученной воде накапливаются следующие частицы:
егидр, Н-, ОН, Н2, Н202 и НзО+.
Последняя из них - катион гидроксония - приводит к повыше¬
нию кислотности облученных водных растворов. -ОН и Н202 - силь¬
ные окислители, вгидр, Н- и Н2 - мощные восстановители. Таким об¬
разом, нарушаются кислотно-основной и окислительно-восстанови-
тельный баланс биосред
Надо помнить, что один у-квант может вызвать разложение бо¬
лее одной молекулы воды. В частности, при его энергии 100 эВ при
комнатной температуре распадается от шести до восьми молекул
воды При этом и соотношение продуктов радиолиза будет изме¬
няться, так как они возникают в результате серии превращений.
В дальнейшем молекулы биосубстрата подвергаются суммарной
атаке всей плеяды восстановителей и окислителей, образующихся
при радиолизе воды. Таким образом, гомолитический распад воды -
первичная химическая причина лучевого поражения.
Глава 17. УГЛЕРОД
Пропорционально количест¬
ву сгоревшего углерода, разви¬
вается в организме ряд сил, по¬
требных для разнообразных
движений, производимых жи¬
вотными
Д И Менделеев
17.1. Элемент, аллотропные модификации
В соответствии с положением атома углерода в Периодической
системе (IV-я группа II периода) и с его электронной конфигураци¬
ей (С = ...2s22p2) он обладает двумя валентными электронами, од¬
нако относительная близость по энергиям его s- и р-орбиталей до¬
пускает сравнительно легкий переход в возбужденное состояние
(С* = ...2s*2pxpypz), в котором углерод имеет четыре валентных
электрона. Этим он (как и кремний) отличается от ниже лежащих
элементов IV-й группы - олова и свинца, у которых энергии внеш¬
них s- и р-орбиталей уже существенно отличаются из-за их различ¬
ной чувствительности к эффектам экранирования поля ядра внут¬
ренними электронными оболочками. Последнее приводит к умень¬
шению участия s-электронов в образовании химических связей и
возрастанию металлических свойств.
Величина относительной электроотрицательности этого элемен¬
та (ОЭО = 2.4-2.5)’ указывает, что углерод способен к ковалентно¬
му связыванию как с электроположительными, так и электроотри¬
цательными элементами. Это иллюстрируется как способностью уг¬
лерода к диспропорционированию:
ЗС + СаО СаС2 + СО ,
так и экзотермическими реакциями с водородом:
С + 2Нг ^ СН< ДН° = -75 кДж/моль
и с кислородом:
2С + Ог^2СО ДН°=-140 кДж/моль.
Переменное значение ОЭО объясняется возможностью существова¬
ния атома углерода в различных гибридных состояниях, а следовательно, в
различии энергий взаимодействия электронов с ядром.
306
л
Таблица 17.1
Энергии некоторых связей с участием углерода
и связей “элемент-элемент”
Связь
Е, кДж/моль
Связь
Е, кДж/моль
С-0
360
С-1
180
с-н
415
1Ч-М
250
С-С
348
э-э
225
С-Р
452
0-0
146
С-С1
327
81-81
226
Высоки величины энергии связи С-С (табл. 17.1). В то же время
ближайшие соседи углерода по Периодической системе (и не только
они) характеризуются значительно меньшими значениями энергии
связи “элемент-элемент”. В результате этого углерод обладает уни¬
кальной склонностью к катенации - способности давать цепи угле¬
род-углерод любой длины и различной степени разветвления, а
также замыкать цепи в кольца различного размера. Галогены, обла¬
дающие прочной связью между собой, в принципе не могут давать
цепей, сера способна к катенации, но эти цепи не могут ветвиться и
не являются прочными из-за невысокой величины энергии связи
“сера-сера”. Кислород, азот, кремний и другие элементы с проме¬
жуточными значениями электроотрицательности также образуют
непрочные связи между собой, а потому дают лишь короткоцепоч¬
ные соединения.
На углеродном остове могут прочно "крепиться” самые разно¬
образные функциональные группировки, так как энергии связей
многих элементов с углеродом высоки. Наконец, сопоставимая ве¬
личина энергий э- и р-орбиталей углерода приводит к возможному
существованию разных типов гибридизации (эр-, эр2- и эр3-) с их
различной пространственной формой - линейной, плоскостной и
объемной. Такой набор свойств порождает неограниченное количе¬
ство структурных и пространственных комбинаций, реально прояв¬
ляющих себя в органических соединениях В этом состоит уникаль¬
ная роль углерода как основного элемента биосубстрата
Многообразие структур, порождаемых различными гибридными
состояниями углерода, иллюстрируется его аллотропными модифи¬
кациями (рис. 17 1) Связи зр3-5р3 реализуются в объемной конфи¬
гурации алмаза, зр2-гибридизированные атомы углерода характерны
для плоскостной (слоистой) структуры графита и сферической
конфигурации недавно синтезированной и интенсивно исследуемой
разновидности углерода - фуллерена Линейная (волокнистая)
307
структура присуща искусственно синтезированной аллотропной мо¬
дификации углерода - карбину, в котором атомы углерода обладают
эр-гибридизацией
Рис. 17.1. Аллотропные разновидности углерода: а-алмаз, б-графит,
в - карбин, г - фуллерен.
В соединениях углерод может принимать любые степени окис¬
ления от -4 до +4, причем в составе одной молекулы могут одно¬
временно присутствовать несколько углеродных атомов с различны¬
ми степенями окисления Это порождает широкий ассортимент
классов органических соединений. Взаимопереход атомов углерода
различных степеней окисления, в том числе и за счет метаболиче¬
ских окислительно-восстановительных процессов (например, окис¬
ление спиртовой группы в альдегидную и далее в карбоксильную
и т. д.) вызывает богатство их химических превращений.
Горение
С02
в атмосфере
Схема 17.1. Круговорот углерода в природе
Взаимопереходы производных углерода реализуются в кругово¬
роте этого элемента в природе (схема 17.1).
308
УглекйЬлый газ атмосферы в результате фотосинтеза накапли¬
вается в органическом веществе растений, которое, с одной сторо¬
ны, минерализируется в виде нефти, угля и других природных го¬
рючих ископаемых. Они сжигаются как топливо, вновь превращаясь
в углекислый газ. С другой стороны, растения как пища усваивают¬
ся животными и вновь превращаются частично в углекислоту, а па¬
раллельно минерализируются как в горючие ископаемые, так и в
негорючие карбонаты - известняки, мел, мрамор и др. Негорючие
минералы под действием влаги постепенно переходят в углекислый
газ. Таким образом, круговорот этого элемента включает соедине¬
ния углерода как с положительными, так и с отрицательными сте¬
пенями окисления, причем живая природа играет в этом процессе
важную роль.
Так как отрицательные степени окисления присущи в основном
углеводородам и их производным, то есть органическим соединени¬
ям, мы рассмотрим их позднее отдельно, а здесь остановимся на
так называемых неорганических производных углерода, то есть на
его соединениях с электроотрицательными элементами, причем не
имеющими углеродных цепочек. Следует помнить, что деление сое¬
динений углерода на органические и неорганические производные
является условным.
17.2. Соединения углерода (+4)
Окисление соединений углерода низших степеней окисления
под действием кислорода приводит к образованию воды и углекис¬
лого газа, степень окисления углерода в котором равна +4. Этот
процесс имеет место с биосубстратом тканей и клеток живых орга¬
низмов и служит энергетическим источником организма.
Подавляющую часть энергии организм черпает за счет так
называемого биологического окисления, то есть путем окисления
в клетках глюкозы, липидов и, в меньшей степени, - полипепти¬
дов (до 10%). Биологическое окисление глюкозы выражается урав¬
нением'
СвН12Ов + 602 —♦ 6С02 + 6Н20 Дй = -2880 кДж/ моль
Изменение свободной энергии этого процесса Дй (символ
употребляется при термодинамических расчетах в физиологических
условиях, т. е. при 310 К) и является основным источником энергии
тканей. Как уже отмечалось (глава 4), это превращение осу¬
ществляется через 38 элементарных стадий, каждая из этих стадий
сопряжена с образованием молекулы аденозинтрифосфата (АТФ),
309
которые в дальнейшем при гидролизе обеспечивают полезную рабо¬
ту клетки по биосинтезу, активному транспорту ионов и др. Так как
свободная энергия гидролиза АТФ AG/ равна -30 кДж/моль, то
коэффициент полезного действия (кпд) этой совокупности превра¬
щений составляет 30-38-100/2880 = 39%, то есть превосходит кпд
известных тепловых двигателей.
Биологическое окисление как окислительно-восстановительный
процесс можно представить совокупностью двух полуреакций:
ох Ог + 4Н+ + 4е ±5 2Н20 плазма
red CeHi2Oe + 6Н20 5 6СО2 + 24Н++ 24е клетка
Эти полуреакции составляют гальваническую пару. Восстано¬
вление кислорода происходит в плазме крови, а окисление глюко¬
зы - в клетке. Оба процесса разделены клеточной мембраной, сле¬
довательно должны существовать вещества - переносчики электро¬
нов из клетки в плазму. Эти соединения - цитохромы, содержат в
своем составе катионы железа, обратимое окисление-восстанов-
ление которых (Fe2+ ^ Fe3+) и выполняет функцию “мостика”, за¬
мыкающего цепь гальванического элемента.
Образующийся в результате “сгорания” глюкозы оксид углеро-
да(1У) представляет собой мало полярное вещество, а потому плохо
растворяется в воде. Его растворимость составляет 0.03 моль/л
при 298 К. Процесс растворения выглядит следующим образом:
сначала образуется гидрат Н2О СО2, который находится в окружении
молекул воды, а затем имеет место медленное превращение в моле¬
кулу Н2СОэ.
Образующаяся угольная кислота представляет собой (без учета
ее распада на ангидрид и воду) кислоту средней силы и диссоции¬
рует следующим образом:
Н2ОСО2 ±5 Н+ + НСОэ" *5 Н+ + СОэ2-
тдрокарбонат карбонат
Рк; = з.7 Рка" = ю.з
Диссоциация по второй стадии идет незначительно, а следова¬
тельно, необходимо считаться с появлением в растворе только гид-
рокарбонат-анионов. Однако концентрация углекислоты в растворах
не велика из-за того, что углекислота преимущественно распадается
до гидрата, а потому реально она проявляет слабые кислотные
свойства (ее истинный рКа' = 6.31 с учетом нестойкости).
Из-за этого, а также из-за присутствия в плазме гидрокарбо-
нат-иона в ней образуется гидрокарбонатная буферная система,
310
поддерживающая постоянство pH наряду с другими биологическими
буферными системами:
рн=б.31 + 1е1г№нс°3,).
[н2со3]
В этом состоит один из механизмов поддержания постоянства
pH в биосредах, в первую очередь, в плазме.
Внутрикле¬
точная
жидкость
Межкле-
точная
жидкость
Кровь
Плазма Эритроциты
Альвео-
лярный
воздух
С02
+
НОН ^
Н+ + нсоэ-
— со2
+
НОН ^
Н+ + нсо3-
карбоа>
— С02 ^ Я-ІЧН-СООИ —
♦ карбамин
НОН ^
Н+ + нсоэ-
«гидраза
-С02
Схема 17.2. Транспорт углекислоты из клетки в легкие.
Другой механизм состоит в удалении углекислоты из организма.
Образовавшийся в клетке углекислый газ частично диссоциирует во
внутриклеточной жидкости, однако его основная часть быстро диф¬
фундирует в межклеточное пространство (где тоже частично диссо¬
циирует) и далее - в плазму. В ней наряду с диссоциацией устана¬
вливается равновесие между углекислотой в растворе и той ее час¬
тью, которая проникает внутрь эритроцитов. В составе эритроцитов
она прочно удерживается в молекуле гемоглобина, обратимо связы¬
ваясь с его глобиновой (белковой) частью за счет образования не¬
стабильного карбамина:
Я-МНг + С02 5 Р-1ЧН-СООН (карбамин)
Эритроциты выносят связанную углекислоту к альвеолам лег¬
ких, где она мгновенно высвобождается под действием присут¬
ствующей здесь карбоангидразы. Изложенные выше процессы пред¬
ставлены на схеме 17.2.
Выделившийся углекислый газ удаляется с выдыхаемым возду¬
хом, в котором его концентрация достигает 4%. Концентрация уг¬
лекислого газа не должна быть здесь более высокой, так как С02
обладает токсическим действием.
Таким образом, два механизма поддержания постоянства pH
препятствуют возникновению ацидозов. Однако это достигается
311
лишь тогда, когда количества продуцируемой в результате биологи¬
ческого окисления и выдыхаемой углекислоты равны.
Угольная кислота образует кислые и средние соли - гидрокар¬
бонаты и карбонаты Растворимые в воде соли гидролизованы, при¬
чем карбонаты - в сильной степени Способность гидрокарбонатов к
гидролизу с созданием слабо щелочной среды реализуется в живых
организмах. В частности, гидрокарбонат иатрия продуцируется
стенками желудка, защищая их от разрушительного действия силь¬
но кислой среды, создаваемой в желудочном соке соляной кисло¬
той. Регуляция кислотно-щелочного баланса в желудке представля¬
ет собой непростую проблему. Может существовать по меньшей
мере два пути формирования повышенной кислотности, вызы¬
вающей язвенную болезнь, - перепроизводство соляной кислоты и
недостаточная секреция гидрокарбонат-иона. Естественно, что гид¬
рокарбонат натрия применяют в качестве антацида - средства про¬
тив повышенной кислотности:
МаНСОз + НС1 ->• С02Т + №01 + Н20.
В принципе антацидами могут служить любые слабо гидроли¬
зующиеся соли нетоксичных кислот или слабые основания.
Присутствие в воде гидрокарбонатов щелочноземельных метал¬
лов - причина временной (карбонатной) жесткости воды, которую
удаляют кипячением-
Са(НСОз)2 -» С02Т + СаСОз^ + НгО.
Содержание углекислоты в атмосфере составляет приблизи¬
тельно 0.03%. Она играет существенную роль в поддержании тем¬
пературного режима на земной поверхности. Молекулы СОг актив¬
но поглощают инфракрасное излучение и препятствуют уходу тепла
за пределы Земли.
СОг + Иу (инфракрасное излучение) ^ СОг*
Это явление, называемое парниковым эффектом, обеспечивает
дополнительный прогрев земной поверхности приблизительно на
20° С, что является мощным фактором поддержания жизни на зем¬
ле Подсчитано, что хозяйственная деятельность человека должна
уже через несколько лет вызвать увеличение концентрации угле¬
кислоты в воздухе до 0.04%, а это, как на первый взгляд кажется,
должно привести к глобальным климатическим изменениям. Однако
избыточная углекислота должна раствориться в объеме океанских
вод; концентрация углекислоты в них намного меньше равно-весной.
Этот пример показывает, что в экологических прогнозах следует
учитывать все возможные факторы.
312
17ГЗ. Производные угольной кислоты
Формальным приемом замены атомов кислорода в молекуле ок¬
сида углерода(1У) другими электроотрицательными атомами можно
произвести серию соединений углерода (+4), которые представляют
собой семейство производных угольной кислоты
Среди них находятся галогенопроизводные, например, четырех¬
хлористый углерод ССЦ, молекула которого неполярна, а следова¬
тельно гидрофобна и липофильна, что определяет использование
этого вещества в качестве органического растворителя, который яв¬
ляется токсичным из-за склонности вступать в биотрансформации с
участием молекул биосубстрата.
Этого свойства лишена молекула СР4 из-за высокой прочности
связи С-Р. Последнее определило широкое использование СР4 и его
аналогов общей формулы СпРхС1у, именуемых фреонами, в качестве
растворителей в различных аэрозолях, а также в роли хладоагентов
в холодильниках. К сожалению, химически инертные летучие фрео-
ны, накапливаясь в атмосфере, активно поглощают ультрафиоле¬
товое излучение, необходимое для образования озонового слоя
По этой причине фреоны постепенно изымаются из употребления.
Однако следует при этом учитывать тот факт, что фреоны наряду с
другими веществами в сопоставимых количествах образуются в ре¬
зультате вулканической деятельности.
Другим примером галогенопроизводных угольной кислоты яв¬
ляется фосген СОС12, молекула которого полярна, высоко реакцион¬
носпособна и крайне токсична.
Сернистый аналог углекислого газа СЭ2 - сероуглерод - нахо¬
дит применение в качестве органического растворителя и важного
промышленного реагента, однако он также высоко токсичен.
Наиболее важное производное угольной кислоты - мочевина,
являющаяся конечным продуктом азотного обмена млекопитающих.
Суммарно окисление белков в организме выражается уравнением.
СтНпОрМч +х02 -» аС02 + ЬН20 + ц/2 МН2СОМН2.
За сутки в организме человека продуцируется до 30 г мочевины
Ее содержание в моче является важным показателем азотного об¬
мена, а потому в биохимии широко определяют содержание моче¬
вины в моче, пользуясь для этого сконструированным А П Боро¬
диным прибором, в котором имеет место реакция
МН2СОМН2 + ЗМаВгО -» ЗМаВг + С02 + 2Н20 + 1Ч2
Исторически важен синтез мочевины Ф Велером в 1828 году,
когда она как вещество истинно биологического происхождения бы¬
ла получена из неорганической соли - циановокислого аммония
313
Мочевина широко используется в сельском хозяйстве, в част¬
ности, в качестве удобрения и пищевой добавки для скота, который
усваивает ее, обладая для этого соответствующими ферментами
Важна мочевина как полупродукт при получении многих биомоле¬
кул и лекарственных средств Поэтому ее производят в больших
количествах из углекислого газа или фосгена-
С02 + 2ЫНэ
ЫН4СМО —► МН2—С—МН2
циановокислый # ^ С0С1, + 2ЫН,
аммоний -2НС1
Органические производные мочевины будут подробно рассмот¬
рены в главе 28.
17.4. Соединения углерода (+2)
Типичным представителем этой группы веществ является оксид
углерода(П) - угарный газ.
Строение монооксида углерода можно объяснить с помощью
метода молекулярных орбиталей (глава 2). Связь между атомами
углерода и кислорода в его молекуле приближается к тройной, что
подтверждается величиной энергии связи в ней, которая равна
1076 кДж/моль, а длина связи составляет всего лишь 1.12 А.
Угарный газ образуется при сгорании органических веществ и
самого углерода при недостатке кислорода:
2С + 02 ->• 2СО,
а также при действии углерода на углекислый газ:
С02 + С -> 2СО.
Оксид углерода(П) формально является ангидридом муравьиной
кислоты. Нагревание последней в присутствии водоотнимающих
средств (концентрированная серная кислота) служит еще одним
способом получения этого оксида:
НСООН СО + Н20.
В соответствии с этим нагревание угарного газа со щелочью
приводит к образованию соли муравьиной кислоты - формиата:
СО + №ОН ->• НСООЫа.
314
Атом упіерода в молекуле муравьиной кислоты четырехвален¬
тен, что вызвано изомеризацией с миграцией протона от атома кис¬
лорода гидроксильной группы изомера с двухвалентным углеродным
атомом к атому углерода:
Еще один путь образования угарного газа - биосинтез из моле¬
кулы гемоглобина (НЬ) при его превращении в билирубин:
В сутки человеческий организм вырабатывает около 10 мл угар¬
ного газа, удаляемого из организма с выдыхаемым воздухом.
Оксид углерода(П) является активным восстановителем:
Важнейшим для физиологии свойством этого вещества является
способность к образованию комплексов, особенно с катионом Ре2+.
Высокая донорная активность оксида углерода объясняется с по¬
мощью теории МО (глава 2). Его сродство к катиону железа намно¬
го выше, чем у кислорода, а потому равновесие реакции:
смещено вправо уже при невысоких концентрациях угарного газа в
воздухе (Кравн составляет 310, в этом случае ее называют коэффи¬
циентом отравления). Так как в результате этого превращения весь
гемоглобин (или его большая часть) оказывается связанным с угар¬
ным газом в молекулы карбоксигемоглобина, то гемоглобин не мо¬
жет выполнять функцию переноса кислорода. Помня, что кислорода
в воздухе 21%, нетрудно подсчитать, что количество карбоксиге¬
моглобина окажется равным количеству оксигемоглобина (а это
уже вызывает удушье), если концентрация СО в воздухе достигнет
величины лишь 0.07%. В дополнение к этому оксид углерода свя¬
зывается и с другими железосодержащими биомолекулами, что усу¬
губляет его токсический эффект
НЬ 25 билирубин + СО.
2СО + 02 -> 2СОг СО + С12 -> СОС12
Ре20з + ЗСО -» ЗС02 + 2Ре.
Однако он может выступать и в качестве окислителя:
СО + Мд -» МдО + С.
нь о2 + со 5 нь со + о2
оксигемоглобин карбоксигемоглобин
315
Отравления окисью углерода происходят достаточно часто в
связи с возрастающим потреблением топлива, развитием химиче¬
ских производств с участием этого вещества, а также частыми по¬
жарами Для защиты от угарного газа существуют специальные
противогазы, в которых используется смесь МпОг (60%) и СиО
(40%), называемая гопкалитом. Эти оксиды обладают способностью
окислять угарный газ в СОг Особый аспект связан с необходи¬
мостью обеспечения нормальной воздушной среды для специалис¬
тов, работающих в системах автономного жизнеобеспечения, где
требуется очистка воздуха от эндогенной окиси углерода, чтобы из¬
бежать автоинтоксикации.
Управление химическим равновесием связывания гемоглобина с
окисью углерода - путь к отысканию средств детоксикации окиси
углерода. Простым антидотом (противоядием) при отравлении
окисью углерода является кислород, избыток которого создается с
помощью оксигенобаротерапии. Другое противоядие - комплексо-
образователь, имеющий повышенное сродство к окиси углерода.
Таковыми являются соли железа, которые и используют на практи¬
ке. Для этой цели можно применять и комплекс катиона кобальта
с трилоном Б (глава 10). Таким образом, современная медицина
располагает эффективными противоядиями при интоксикациях
окисью углерода.
Еще одно важное соединение углерода(Н) - цианистый водород
НСМ, водный раствор которого называют синильной кислотой. Это
вещество в связанном виде входит в состав некоторых природных
веществ растительного происхождения Особенно его много в кос¬
точках абрикосов и горького миндаля. В заметных количествах оно
образуется при сгорании современных азотсодержащих полимерных
материалов. В микроколичествах образуется и в организме челове¬
ка. Свойства этого соединения необходимо знать в связи с его вы¬
сокой токсичностью.
В водных растворах синильная кислота существует в виде двух
таутомерных форм:
Н-С=М С=М—Н
Таутомерия - явление обратимой изомерии, при котором два
или более изомера легко переходят друг в друга. В этом случае
изомеры называют таутомерами. Частный случай таутомерии - про-
тотропная таутомерия, при которой изомеризация осуществляется в
результате перехода протона. Таутомерия очень важна в процессах,
протекающих в живых организмах, что будет показано на многих
примерах в дальнейшем
316
ТаутомерГ' синильной - изосинильная кислота - присутствует в
равновесии в количестве около 1%. Электронное строение этого
таутомера аналогично структуре монооксида углерода, а следова¬
тельно он способен связываться с катионами железа биосубстрата,
чем и объясняется высокая токсичность НСГ'!.
Синильная кислота более ядовита, чем окись углерода, так как
обладает повышенным сродством к катионам железа(Ш) в составе
ферментов дыхательной цепи - цитохромов, содержание которых в
организме меньше, чем количество гемоглобина.
Синильная кислота - слабая кислота, диссоциирующая по урав¬
нению:
25 Н+ + С1^- рКа = 9.6.
Следовательно ее соли - цианиды легко гидролизуются, а так
как HCN - легколетучая жидкость, то хранить цианиды следует в
герметичной упаковке без доступа влаги.
Как уже сказано, синильная кислота и цианид-ионы являются
метаболитами. Они частично выделяются из организма с выдыха¬
емым воздухом, а частично с мочей, в виде нетоксичного роданид-
аниона, который образуется в результате связывания с серой, по¬
ставляемой для реакции серусодержащими белками:
С!1»]- + Б -» БСГ'Г (роданид).
На превращении цианид-иона в нетоксичный роданид основано
использование тиосульфата натрия в качестве противоядия при
отравлении цианидами:
+ N826203-» KSCN + N82803.
Другой антидот цианид-иона - глюкоза, реакция которой с HCN
будет рассмотрена позднее (глава 25)
Еще одно направление биотрансформации цианид-иона - коор¬
динация с катионом кобальта в составе витамина В|2 - цианкоба-
ламина, так как цианид-ион является активным лигандом Цианид-
ные комплексы переходных металлов широко применяются в про¬
мышленности, аналитической химии и других областях. Достаточно
привести примеры железосодержащих комплексов Кз[Ре(С^б] и
К4[Ре(С^б] (красная и желтая кровяная соли, применяются в фото¬
графии и для обнаружения катионов Ре+2 и Ре+3 в качественном
анализе).
Роданид-ион также активный лиганд Комплекс Маз[Со(8С^в],
на ярко-синей окраске которого основано качественное обнаруже¬
317
ние иона Со+3 в растворах - один из многих комплексов, образуе¬
мых с его участием.
В отличие от синильной кислоты роданистоводородная кислота
является сильной, что не удивительно, так как в ней протои при¬
соединен к более электроотрицательному атому серы, а не углерода,
как это имеет место в молекуле синильной кислоты:
Н-Б-С^ 15 Н+ + БСМ" рКа = 0.95.
В заключение отметим еще один аспект, связанный с синиль¬
ной кислотой, а именно - ее возможное участие в химической эво¬
люции - процессе синтеза биомолекул из простых молекул мине¬
рального происхождения. Известно, что ее можно превратить в
нуклеиновое основание - аденин (глава 28) по уравнению:
5НСМ -> СвНбМб - аденин (нуклеиновое основание).
Если учесть, что синильная кислота легко образуется из неко¬
торых простых неорганических молекул, то можно построить схему
образования нуклеиновых кислот из неорганических веществ.
^ Глава 18. УГЛЕВОДОРОДЫ
...ту часть нашей науки, кото¬
рую обычно называют органи¬
ческой химией, мы определяем как
химию углеводородов и их произ¬
водных.
К. Шорлеммер'
18.1. Классификация
Множество соединений углерода с отрицательными степенями
окисления принято считать органическими соединениями. Органи¬
ческая химия своим существованием как самостоятельная наука
обязана прежде всего неисчислимому многообразию таких соедине¬
ний углерода. В настоящий момент нам известно о существовании
около 15 ООО ООО углеродных соединений; цифра эта увеличивается
с каждым днем более чем на 1000. Систематическое изучение орга¬
нических соединений упрощается, если начать его с рассмотрения
углеводородов - веществ, содержащих лишь углерод и водород.
Принято рассматривать органическую химию как химию углеводо¬
родов и их производных.
18.1.1. Алканы
5р3-Гибридизированный атом углерода четырехвалентен и об¬
разует только а-связи. Простейшим представителем углеводородов,
основу которых составляют а-связи, является метан СН4. Молекула
этана СгНв содержит звено из двух атомов углерода - элементарную
часть углеродной цепочки.
сн4
метан
С6Ні4
гексан
С2н6
этан
С7Нів
гептан
СзНе
пропан
СеНіе
октан
С4Ню
бутан
С9Н20
нонан
С5Н12
пентан
С10Н22
декан
' К Шорлеммер (1834-1892) - немецкий химик, специалист в области
химии углеводородов, а также общественно-политический деятель
319
В принципе, посредством о-связей могут быть образованы цепи
из любого количества атомов углерода. Гомологический ряд пре¬
дельных (насыщенных) углеводородов, содержащих только простые
связи С-С и именуемых алканами, можно выразить общей фор¬
мулой СпНгп+2- Формулы десяти первых алканов вместе с их назва¬
ниями, которые следует запомнить, приводятся.
Наименования четырех начальных алканов сложились историче¬
ски; названия последующих гомологов (в том числе содержащих
более десяти атомов углерода) производят от греческих числитель¬
ных в сочетании с окончанием ан.
18.1.2. Сведения о конформациях
Для о-связей характерна наибольшая плотность электронного
облака на прямой, соединяющей ядра атомов. Поворот групп вза¬
имосвязанных атомов вокруг этой оси не изменит плотности элек¬
тронного облака на ней и, следовательно, не приведет к разрыву
данной связи. Таким образом, при вращении групп атомов вокруг
о-связи могут осуществляться различные варианты их взаимной
ориентации. Вследствие взаимных влияний атомов одной группы
на атомы другой число способов ориентации ограничено Каждый
из возможных вариантов называется конформацией.
Например, молекуле этана присущи две граничные конформа¬
ции - с наименьшим расстоянием между атомами водорода двух ме-
тильных групп (заслоненная конформация), и с наибольшим рас¬
стоянием между ними - заторможенная (рис. 18.1).
н н н н
■К ■ -И-
заторможенная заслоненная
4с . --у”
Рис. 18.1. Конформации этана.
Отдельные конформации можно изображать с помощью про¬
странственных формул (рис. 18.1, а), а также посредством проек¬
320
ционных куделей Ньюмена (рис. 18.1, б). При этом один из атомов
углерода молекулы этана (изображен кружком) помещается сзади
другого, который изображается точкой, а связи атомов углерода
с водородом (или с заместителями) проецируются на плоскость
рисунка.
Чем больше сила взаимного отталкивания атомов водорода, тем
выше энергия системы; поэтому заторможенной конформации будет
соответствовать минимум потенциальной энергии молекулы.
Энергия, необходимая для поворота метильных групп в мо¬
лекуле этана так, чтобы произошел переход от одной конформации
к другой, сравнительно невелика (11.7 кДж/моль). Поэтому уже
при комнатной температуре (25° С) молекула этана претерпевает
эти изменения с частотой около 1012 с-1. Таким образом, разделить
конформации этана при комнатной температуре невозможно.
Другой пример - конформации бутана (рис. 18.2). Конформация
с максимально удаленными друг от друга метильными группами
(анти), является энергетически более выгодной, чем какая-либо
другая из представленных на рисунке конформаций.
заслоненная скошенная (гош) заслоненная заторможенная (анти)
ф = Оо ф = 60° ф = 120о ф = 1800
Рис. 18.2. Конформации бутана.
В общем молекулы алканов стремятся пребывать в наиболее
“выгодной”, зигзагообразной конформации, предполагающей такое
же взаимное расположение соседних атомов углерода, как и в за¬
торможенной конформации бутана, а всей цепочки атомов углеро¬
да - в одной плоскости (рис. 18.3)
Рис. 18.3. Конформация цепи н-алкана.
11 Зак. 675
321
Жирными штрихами обозначено расположение заместителей
выше плоскости, в которой находится цепь полиалкана, пункти¬
ром - ниже этой плоскости, а атомы водорода для простоты
опущены
Такое пространственное строение цепи н-алкана знать необхо¬
димо, так как длинная неразветвленная насыщенная углеводород¬
ная цепь присуща большинству ПАВ и тем самым определяет физи-
ко-химические свойства этой липофильной части их молекул Еще
более важным является представление о конформации многозвен¬
ной углеводородной цепочки для понимания пространственного
строения липидов, а тем самым - структуры важного элемента
клетки, ее мембраны, которая по преимуществу состоит из липидов.
Помня, что жиры представляют собой эфиры глицерина и жирных
кислот, нетрудно представить, что три длинных углеводородных це¬
почки жирных кислот в пределах одной молекулы жира уклады¬
ваются параллельно, чтобы в минимальной степени препятствовать
друг другу.
Столь же легко сообразить, что молекулы жиров будут уклады¬
ваться на водной поверхности параллельно друг другу таким обра¬
зом, что полярные их части - остаток глицерина - будет обращен к
водной поверхности за счет диполь-дипольных взаимодействий и
водородных связей с молекулами воды. Таким образом, на поверх¬
ности клетки формируется мономолекулярный липидный слой.
Между этими слоями будет иметь место гидрофобное взаимодей¬
ствие, что приведет к образованию между клетками липидного бис¬
лоя, который и является основным структурным элементом клеточ¬
ной мембраны. В основе же ее строения лежит повторяющаяся за¬
торможенная бутановая конформация углеводородной цепочки жир¬
ных кислот. Изложенное представлено на рис. 18.4.
Преимущественное нахожде-
НгО
гидрофобные
взаимодействия
НгО
Рис. 18.4. Липидный бислой.
ние молекул природных и фи¬
зиологически активных веществ
в определенной конформации
означает не что иное, как
приобретение или утрату эти¬
ми веществами определенных
свойств. Это можно иллюстри¬
ровать примером конформаци-
онной однотипности группы
природных и синтетических сое¬
динений, воздействующих на
передачу нервного импульса.
Ацетилхолин является пере¬
датчиком нервных импульсов, и
322
эта его фикция осуществляется именно в той конформации, в ко¬
торой два заместителя в этановом фрагменте, как и метильные
группы в бутане, максимально удалены друг от друга. Синтетиче¬
ский аналог ацетилхолина карбохолин имеет то же пространствен¬
ное строение и свойства, а потому может временно выполнять
функцию ацетилхолина и употребим в качестве лекарства при де¬
фиците ацетилхолина в тех или иных клетках, что вызывает пара¬
лич. Природное вещество мускарин, содержащийся в мухоморе,
имеет более сложное строение, однако и он оказывает действие на
нервные ткани. Его молекулы, вытесняя молекулы ацетилхолина,
препятствуют нервной проводимости, а тем самым оказывают ток¬
сическое действие. Синтетические препараты фосфорилхолины
оказывают еще более сильное токсическое действие, являясь
мощными ядами нервно-паралитического действия. Таким образом,
здесь можно говорить о молекулярном соответствии указанных
веществ, вызванном их конформационной и структурной однотип¬
ностью.
СН3,
НО
№(СН3)3 X' Н Ы(СН3)3+ X”
ацетилхолин карбохолин
^3\ о ? Н РР(Х)00 И
\ . Н-М +
Н М(СН3)з X Н Ы(СНэ)з X'
N(043)3
фосфорилхолин
Рис. 18.5. Конформации ацетилхолина и его аналогов
Следует помнить, что, принимая различные конформации, моле¬
кулы того или иного вещества остаются химически однородными;
конформации не являются изомерами. Однако, при тесной упаков¬
ке атомов в молекулах некоторых веществ могут возникать пре¬
пятствия вращению групп атомов вокруг С-С связей, в результате
чего оказывается возможным разделить различные формы таких
веществ уже при обычной температуре.
При изучении методами рентгеноструктурного анализа про¬
странственного строения некоторых ферментов, витаминов, гемо¬
глобина и других соедимений было установлено, что даже незначи¬
11*
323
тельное изменение конформации этих веществ может привести к
потере их биологической активности.
Молекулы природных полимеров, к числу которых относятся, в
частности, белки и нуклеиновые кислоты, характеризуются вполне
определенным пространственным строением и вполне определен¬
ными конформационными изменениями, от которых зависят их
свойства
Структурные трансформации в биополимерах носят коопера¬
тивный характер - конформация каждого звена зависит от конфор¬
мации соседних звеньев и их изменение происходит согласованно.
В случае биополимеров часто говорят не о конформациях, а
используют термины “вторичная” или “третичная” структура, под¬
разумевая под первичной химическую структуру макромолекулы.
а-Связи могут возникать не только между углеродными и водо¬
родными атомами, но и между атомами других элементов. В состав
насыщенных (т. е. построенных посредством только а-связей) сое¬
динений наиболее часто входят атомы галогенов, кислорода, серы и
азота, которые принято называть гетероатомами. Соответственно,
выделяют различные классы насыщенных органических соединений:
галогенопроизводные, кислородсодержащие соединения - спирты и
простые эфиры, производные серы - меркаптаны, сульфиды, азотсо¬
держащие вещества - амины.
Энергетические и геометрические характеристики разных ©-свя¬
зей хотя и различаются между собой, варьируются не очень зна¬
чительно (табл. 17.1). Так, энергия самой слабой связи, угле¬
род-иод составляет 180 кДж/моль, а энергия самой прочной связи,
углерод-фтор - 452 кДж/моль. Длина связей тоже несколько раз¬
лична, но для большинства связей она не превышает 2.5 А. Еще в
меньшей степени разнятся валентные углы; их отклонения от тео¬
ретического значения 109°28’ не превышают, как правило, несколь¬
ких градусов.
Р-Х
Р-ОН
Р-О-Р
Р-БН
Р-Б-Р
галогенопроизводные
спирты
простые эфиры
меркаптаны
сульфиды
дисульфиды
Р-Б-Б-Р
Р-МН2, МНР1Р2
амины
324
*18.1.3. Насыщенные циклические соединения
Можно представить случай, когда зр3-гибридные атомы угле¬
рода на концах углеродной цепи связаны между собой о-связью и,
таким образом, замыкают цепь в кольцо. Для образования кольца
как минимум требуется три атома углерода. В общем случае речь
идет о циклической разновидности насыщенных углеводородов
СпНгп, которые называются циклоалканами Названия отдельных
представителей этого гомологического ряда образуют прибавлением
приставки "цикло" к названию соответствующего алкана.
Часто употребляемым способом изображения циклоалканов яв¬
ляется кольцо без указания атомов водорода и связей с ними.
н н
'с'
н н
Н-І-І-Н
нн Н Н
/ \ н. 'с' ,н
н-с-с-н н-с" ^с-н
н-с-с-н
н' н
Н—6—6—н
1 I
н н
“°\
о
о
У'х1
д
□
о
о
циклопропан циклобутан циклопентан циклогексан
Понятно, что замыкание цепочки в цикл приводит к уменьше¬
нию валентного угла: его величина вместо 109°28’ (нециклические
алканы) может стать 108° (циклопентан), 90° (циклобутан) и даже
60° (циклопропан).
Эта деформация связана с энергетическими затратами, в ре¬
зультате чего молекулы циклоалканов обладают энергией, большей,
чем соответствующие нециклические цепи. Этот избыток энергии,
который называют энергией напряжения, составляет, в част¬
ности, 116 кДж/моль для циклопропана, 110 кДж/моль для цик¬
лобутана и лишь 13.5 кДж/моль для циклопентана
Более правильным применительно к циклопропану и циклобута¬
ну было бы говорить не об искажении валентных углов, а о смеще¬
нии максимумов перекрытия зр3-орбиталей с прямых, соединяющих
атомы углерода, т е. об искривлении связей, которые из-за этого
становятся несколько слабее Действительно, определенная экспе¬
риментально длина С-С связи в циклопропане оказывается той же,
что и в алканах, а величина валентного угла - 106°, а не 60°, как
можно было бы полагать (рис 18 6)
Циклобутан и циклопропан обладают наибольшей энергией на¬
пряжения; в циклах большего размера она незначительна, посколь¬
ку молекулы этих веществ неплоские
325
Молекула циклопентана, например, пре¬
бывает обычно в конформации “конверта”,
а циклогексан может существовать в конфор¬
мациях “кресла" и “ванны”, причем первая
энергетически выгоднее (рис. 18 7) В этих
конформациях молекулы циклопентана и цик-
логексана не имеют искажений валентных
углов.
Молекулы пяти- и шестичленных цикло-
алканов и насыщенных гетероциклов непло¬
ские, но часто для удобства их изображают
плоскими, - так называемое изображение по Хеуорсу (рис. 18.7).
В этих изображениях атомы водорода для краткости опускаются.
Рис. 18.6.
Электронное строение
циклопропана
ксЬр] к}иЬ|
I ■ по Хеуорсу I I
Рис. 18.7. Конформации циклопентана и циклогексана
и плоскостное изображение по Хеуорсу
Если в составе насыщенных колец имеются гетероатомы (М, О, Б
и др ), то говорят о насыщенных гетероциклических соединениях.
Для насыщенных гетероциклов характерны те же закономерности,
что и для углеводородных аналогов.
Так, молекулы окиси этилена и этиленимина имеют простран¬
ственное строение, аналогичное строению молекулы циклопропана,
и, следовательно, обладают напряжением.
Молекулы же пятичленных гетероциклов - тетра-
гидрофурана и тетрагидропиррола - не имеют иска¬
жений валентных углов и существуют в неплоской
конформации "конверта".
Нет напряжения и в таких шестичленных гете¬
роциклах, как тетрагидропиран н пиперидин, кото¬
рые существуют преимущественно в конформа¬
ции “кресла”.
V
окись этилена
V
326
тетрагидрофуран
тетрагидропиррол
О кЦ»
тетрагидропиран
9 кд.
пиперидин
В число циклоалканов и их гетероциклических аналогов вклю¬
чают и производные не с одним, а с несколькими циклами.
Если циклы соединяются посредством атома углерода, получа¬
ются спирановые углеводороды. Спирановые структуры не ха¬
рактерны для природных объектов.
Если два цикла соединяются парой атомов углерода или парой
гетероатомов, то образуются конденсированные циклоалканы и
гетероциклы.
Примером такой разновидности углеводородов может служить
встречающийся в нефти декалин.
В целом же конденсированные кольца широко представлены в
природе. Среди них имеются и достаточно сложные по строению
молекулы. В частности, многочисленное семейство стероидных
гормонов обладает общей особенностью - четырьмя конденсирован¬
ными кольцами, одним циклопентановым и тремя циклогексановы-
ми Такое конденсированное кольцо носит название циклопентапер-
гидрофенантрен. Точнее, в состав стероидов входит структура гомо¬
лога последнего, именуемого стераном. В качестве примера природ¬
ного производного стерана приведена молекула холестерина.
327
сн3 ч^СН2СН2СН2СН<СНз)2
СНз[
стеран холестерин
Еще одной разновидностью циклических соединений являются
мостиковые углеводороды и гетероциклы, в которых углеводо¬
родные цепочки связаны более чем двумя атомами. Здесь изобра¬
жен углеводород, в котором таким образом сочленены пяти- и шес¬
тичленные кольца. Подобная структура
3\^ 3 присуща углеводороду камфану, который
N«4. лежит в основе ряда производных из
Г*\ ! ! числа терпенов, физиологически актив-
ных веществ растительного происхожде¬
ния. К числу производных камфана, в
частности, относится камфора, приме¬
няемая в медицине.
Наконец, следует назвать еще полиэдрические производные -
соединения с высокой степенью конденсированности.
СНз
камфан
кубан адамантан уротропин
Их молекулы могут иметь объемную структуру - с напряжени¬
ем, например, углеводород кубан или без напряжения, например,
углеводород адамантан и его гетероциклический аналог уротропин,
использующийся в медицине.
£Ш
Широкие возможности построения разнообразных полиэдриче¬
ских структур представлены выше.
328
18.1.4. Алкены
Комбинация двух р-орбиталей с в-орбиталью дает зр2-гибридные
орбитали, оси симметрии которых находятся в одной плоскости и
расположены под углом 120° друг к другу. Электростатическое от¬
талкивание атомов будет минимальным, если ось р-орбитали, не
участвующей в гибридизации, будет перпендикулярна к плоскости
гибридных орбиталей. В молекуле этилена две перекрывающиеся
зр2-гибридные орбитали атомов углерода образуют а-связь, а
р-орбитали - л-связь. л-Связь менее прочна, чем а-связь (её энергия
составляет 266 кДж/моль), - тем не менее вращение атомов вокруг
оси, соединяющей атомы углерода, исключается. Таким образом, в
молекуле этилена атомы углерода прочно соединены а- и л-связями
или, как принято говорить, двойной связью (длина 1.33 А), меньшей
по длине, чем простая С-С связь (1.54 А).
л-связь
Теоретически угол между атомами водорода в этилене должен
составлять 120°; это почти то же, что экспериментально устано¬
вленное значение - 117°.
По мере удлинения углеродной цепи путем присоединения к
молекуле этилена зр3-гибридных атомов углерода образуется гомо¬
логический ряд непредельных углеводородов, алкенов, который
определяется брутто-формулой СпНгп- Их названия производятся
от названий соответствующих алканов заменой в последних окон¬
чаний ан на ен.
л-Связь может существовать не только между атомами углеро¬
да; она может соединять также зр2-гибридные атомы кислорода,
серы, азота и других элементов. Соответственно, существуют
кратные связи- С=0 (альдегиды, кетоны, кислоты, амиды, сложные
эфиры), С=М (особенно часто встречается в гетероциклах) и неко¬
торые другие (например, связь N=1^, именуемая азосвязью)
Ниже приведены формулы и названия некоторых соединений со
связями С=0.
329
я
к
к
я
к
>=° >=° >=°
Г л/ НО
сложный
альдегид
кетон
кислота
амид
эфир
По геометрическим параметрам (длины связей и валентные уг¬
лы) эти кратные связи напоминают двойную связь углерод-углерод,
С=С. Близки эти связи к связи С=С и по энергетическим характе¬
ристикам (таблица 2 2).
Нетрудно представить углеродную цепь, содержащую две, три
или большее число двойных связей. В этих случаях говорят об
алкадиенах, алкатриенах и т. д., а в общем случае - о полиенах.
Алкадиенам соответствует формула СпНгп-г-
Обнаружено, что алкадиены, в которых двойные связи череду¬
ются с одной простой связью, более устойчивы, чем алкадиены с
изолированными (т. е. разделенными более, чем одной простой)
двойными связями. Алкадиены первого типа называются сопря¬
женными. Простейшим их представителем является бутадиен С4Н6,
для которого выигрыш в энергии (энергия сопряжения) составляет
14.7 кДж/моль.
Большая устойчивость сопряженных алкадиенов объясняется
следующим. Во-первых, а-связь между двумя зр2-гибридными ато¬
мами 2 и 3 (1.47 А) короче, чем а-связь между зр3-гибридными ато¬
мами в алканах (1.54 А), поскольку в зр2-гибридной орбитали доля
э-характера больше, чем в зр3-гибридной, а э-электроны, как извест¬
но, располагаются ближе к ядру, чем р-электроны. Во-вторых,
при такой укороченной а-связи, соединяющей две кратные связи,
р-орбитали перекрываются не только в пределах кратных связей,
но и частично между ними, при условии параллельности орбита-
лей. Вследствие этого вращение групп атомов вокруг зр2-гиб-
ридной а-связи затрудняется, что и определяет преимуществен¬
ное существование алкадиенов в конформации с параллельными
18.1.5. Сопряженные алкадиены
СН2=СН-СН=СН2
3 4 бутадиен-1,3
330
р-орбиталй»ли. Кроме того, перекрывание р-орбиталей приводит к
тому, что длина кратных связей в бутадиене (1.37 А) больше, чем
в несопряженных диенах (1.34 А).
Можно говорить, что четыре р-электрона двух сопряженных
кратных связей образуют общую молекулярную орбиталь, принад¬
лежащую всем четырем атомам углерода, - то есть говорить о со¬
пряженной системе кратных связей.
18.1.6. Ароматические соединения
Двойные связи могут быть включены в цикл. Так образуются
молекулы циклоалкенов и циклополиенов. Примером могут служить
бензол и его аналоги. Остов молекулы бензола образован шестью
зр2-гибридными атомами углерода и имеет форму плоского шести¬
угольника с валентными углами в 120°.
В результате сопряжения шести р-электронов (три двойные
связи) образуется молекулярная орбиталь (6л-орбиталь) - каждый
из р-электронов не связан с каким-либо одним из атомов углерода и
может перемещаться от одного атома к другому. Эффект сопряже¬
ния приводит к тому, что молекула бензола обладает значительной
энергией стабилизации - 150 кДж/моль.
бензол
Молекуле бензола присущ особый вид связи между атомами уг¬
лерода - так называемая ароматическая связь Длина этой связи
составляет 1 4 А - промежуточное значение между длиной простых
и двойных связей. Таким образом, в бензоле нет простых и двойных
связей. Поэтому молекулу бензола принято изображать не в виде
символа с чередующимися кратными и простыми связями, а в виде
символа, отображающего бл-электронную орбиталь (круг внутри
шестичленного цикла)
Бензол - родоначальник семейства ароматических углеводоро¬
дов, аренов, которые описываются брутто-формулой СпНгп-б Про¬
стейшим из гомологов бензола является толуол.
К числу аренов относят и конденсированные ароматиче¬
ские углеводороды, образующиеся путем сочленения циклов;
331
представителями конденсированных аренов являются нафталин,
антрацен, фенантрен, пирен, бензпирен
толуол нафталин антрацен фенантрен пирен бензпирен
Для образования ароматической системы необходимо, чтобы р-
электроны формально группировались в три, пять, семь двойных
связей. Математически это требование выражается правилом
Хюккеля, которое гласит, что соединение обладает ароматической
структурой, если в его молекуле содержатся плоские кольца, вклю¬
чающие замкнутую систему сопряженных двойных связей с коли¬
чеством л-электронов 4п+2, где п - натуральный ряд чисел
Согласно этому правилу, молекулы, содержащие шесть (бен¬
зол), десять (нафталин), четырнадцать и т.д. л-электронов, являют¬
ся ароматическими. В справедливости правила
Хюккеля можно убедиться на примере азулена -
V IV ароматического соединения, молекула которого
имеет в основе два сконденсированных ненасыщен-
азулен ных цикла - пятичленнный и семичленный; общее
число л-электронов в молекуле равно десяти.
В соответствии с правилом Хюккеля ароматическая связь долж¬
на существовать (и действительно существует) в ненасыщенных
гетероциклах - аналогах бензола, молекулы которых содержат
зр2-гибридные атомы азота, заменяющие атомы углерода.
Примерами таких соединений могут служить пиридин, пирида-
зин, пиримидин, пиразин, триазин.
Ненасыщенные ароматические гетероциклы именуются гетаре-
нами. Это весьма устойчивые соединения. В частности, энергия
стабилизации молекулы пиридина составляет 167 кДж/моль, то
есть больше, чем для молекулы бензола.
О О О О О
пиридин пиридазин пиримидин пиразин триазин
бл-Электронная конфигурация гетаренов может быть реализо¬
вана за счет двух р-электронов неподеленной электронной пары
гетероатома гетероцикла и р-электронов четырех зр2-гибридных
332
атомов углерода. Такую электронную конфигурацию имеют пяти¬
членные ароматические гетероциклы фуран, тиофен, пиррол и их
гетероаналоги (оксазол, пиразол, имидазол, тиазол и др.), образо¬
ванные заменой зр2-гибридных атомов углерода на атом азота.
ООО
I
н
фуран тиофен пиррол
О о о о
оксазол тиазол имидазол пиразол
Известно, что энергия стабилизации плоской молекулы фурана,
обусловленная ароматической связью, составляет 67 кДж/моль,
пиррола - 100 кДж/моль, тиофена - 115 кДж/моль.
индол хинопин акридин пурин птеридин
К гетаренам относятся также конденсированные ароматические
гетероциклы, образованные сочленением гетероцикла с одним или
несколькими бензольными кольцами (индол, хинолин, акридин),
либо с другим гетероциклом. Так, в молекуле пурина сконденсиро¬
ваны кольца имидазола и пиримидина, а гетарен птеридин образо¬
ван из пиримидинового и пиразинового колец.
В качестве еще одного примера, иллюстрирующего справедли¬
вость правила Хюккеля, можно назвать молекулу порфина, входя¬
щего в состав гема (играет важную роль в
процессе дыхательного обмена) и в состав
хлорофилла (обеспечивает фотосинтез).
Эта молекула имеет своей основой четыре
пиррольных кольца и содержит восем¬
надцать обобществленных л-электронов.
4п + 2, п = 4. Молекула порфина плоская
и обладает очень большой энергией стаби¬
лизации - свыше 1000 кДж/моль порфин
333
18.1.7. Алкины
Комбинация 2ь- и 2р-орбиталей дает две эр-гибридные орбитали,
противоположные одна другой, то есть расположенные по одной
оси. Негибридизированные 2ру- и 2р*-орбитали перпендикулярны
одна другой и перпендикулярны оси Бр-гибридных орбиталей При
сближении двух Бр-гибридных атомов углерода между ними возни¬
кает тройная связь одна а-связь и две л-связи После насыщения
двух оставшихся Бр-гибридных орбиталей атомами водорода образу¬
ется молекула ацетилена С2Н2, в которой все атомы лежат на одной
прямой. Наличие двух л-связей приводит к сокращению расстояния
между углеродными атомами до 1 2 А.
к-связь
Гомологический ряд ацетиленовых углеводородов - алкинов
описывается формулой СпН2п-2, а названия отдельных представите¬
лей этого ряда образуют заменой окончания ан в соответствующих
алканах на окончание ин.
Кроме алкинов, тройную связь включают в себя еще н нитри¬
лы - соединения, содержащие связанные между собой эр-гибрид-
ные атомы углерода и азота.
18.2. Изомерия и номенклатура
органических соединений
18.2.1. Понятие о радикалах. Структурная изомерия
Если в молекуле метана мысленно разрушить а-связь С-Н, то
образуется частица СН3, содержащая неспаренный электрон. Такие
частицы называются радикалами. Из метана можно получить ради¬
кал метил, из этана - этил и т. д. Радикалы можно создать и из
334
циклоалкаЬов, например, - циклопропил, циклогексил. В общем
случае алкильные и циклоалкильные радикалы обозначают
буквой Р.
Свободные радикалы - реальные частицы, промежуточный про¬
дукт тех или иных реакций. Однако они существуют очень короткое
время. Так, период полураспада метильного радикала составляет
6-10'3 секунды.
Химики, используя специальные экспериментальные приемы,
изучают свойства кратковременно живущих радикалов; варьируя
структуру заместителей, они научились синтезировать устойчивые
радикалы, достаточно долго существующие в обычных условиях.
Действие свободных радикалов связывают с механизмом канце¬
рогенеза, лучевой болезни и с другими патологическими явлениями.
Устойчивые радикалы вводят специально в состав биологически ак¬
тивных молекул, чтобы проследить их распространение и транс¬
формацию в живом организме. Свободные радикалы обладают пара¬
магнитными свойствами, которые легко регистрируются физически¬
ми методами.
Из метана и этана можно получить лишь по одному радикалу,
так как атомы водорода равноценны. Говоря о неравноценности уг¬
леродных атомов, их классифицируют по числу связанных с ними
углеродных же атомов на первичные, вторичные, третичные и чет¬
вертичные. Так, в молекуле пропана имеется два типа углеродных
атомов - первичные, по концам цепи, и вторичный - посредине.
перв. перв.
СН3—СН2—СН3 СН3—СН2—СН2— СН3—СН— СН3
втор. |
Пропану поэтому отвечает два радикала - пропил и изопропил. К
этому добавим, что последний входит в состав одной из двадцати
природных аминокислот, а именно - валина.
Молекулу того или иного гомолога можно произвести из соот¬
ветствующего алкана соединением алкильного и метильного ради¬
калов Осуществив такую процедуру с пропильными радикалами,
получают два изомерных бутана, в одном изомере углеродная цепь
линейна, это нормальный бутан (к-бутан) в другом изомере, изобу¬
тане, углеродный скелет разветвляется
СН3 -СН2 -СН2 -СН3 СН3 -СН -СН3
£н3
н-бутан изобутан
335
Изомерия бутана - простейший случай изомерии углеродного
скелета в ряду алканов Впервые изобутан синтезировал
А М. Бутлеров в 1866 году.
Из «-бутана в свою очередь можно произвести два радикала -
нормальный и вторичный бутилы. Изобутану также отвечают два
радикала - изобутил и третичный бутил. Два из них типичны для
природных структур, так как втор-бутил присутствует в аминокис¬
лоте изолейцине, а аминокислота лейцин содержит изобутильный
радикал.
СНЭ —СН2 — СНг —СН2 — СНЭ — СНг —СН —СН3
н-бутил втор-бутил
СНЭ —СН —СН2 СНЭ —СНЭ
СНэ <Ьнэ
изобутил грет-бутил
Из этих радикалов можно создать три изомера пентана:
СНЭ
СН3—СН2—СН2—СН2—СНЭ СНЭ—СН2—СН—СНЭ СНЭ— 6— СНЭ
<Ьн3 бнэ
Семейство пентильных радикалов С5Н11 насчитывает восемь
представителей, из которых можно образовать пять гексанов. С
увеличением длины цепи число изомеров в ряду алканов быстро
возрастает. Так, углеводороду СгоШг отвечает уже 366 319 изоме¬
ров, а углеводороду СэоН62 - 4 111 846 788 изомеров. Эти изомеры
отличаются порядком связей, образующих скелет молекулы, и их
называют изомерами углеродного скелета.
В случае циклоалканов изомерия скелета может быть обус¬
ловлена различным числом атомов углерода в кольце, примером че¬
му могут служить циклогексан и метилциклопентан.
Гомологические ряды изомерных алкенов и алканов строят ана¬
логичным образом: производят соответствующие непредельные ра¬
дикалы, которые соединяют с ме-
,—у тильным радикалом. Ниже приведе-
/ \—сн3 I \ ны формулы и названия часто встре-
чающихся простейших непредельных
метилциклопентан циклогексан радикалов.
336
сн2=&!*- сн2=сн-сн2— сн2=с-снэ —сн=сн-снэ
винил аллил изопропенил кротил
Из их числа винильный радикал входит в состав гемоглобина, в
его гемовую часть, а аллильный характерен для ряда растительных
веществ, обладающих бактерицидным действием, - фитонцидов,
продуцируемых луком, чесноком, горчицей и другими растениями.
Название аллил происходит от лат. allium - лук, чеснок.
Для ацетилена имеется радикал ацетиленил СН=С-
Для алкенов и алкинов, кроме изомерии скелета, характерна
изомерия положения кратной связи, что иллюстрируется изо¬
мерными бутенами (изомерия скелета и положения) и бутинами
(изомерия положения).
изомерные бутены
СН2=СН-СН2—СНЭ СНЭ—СН=СН—СНЭ СН2=С—СНЭ
изомеры положения изомеры скелета
изомерные бутины
НС=С-СН2—СН3 СН3—СЕС-СНз
В ряду аренов изомерия положения проявляется в том, что ал¬
кильные заместители боковых цепей могут занимать различные вза¬
имные положения в цикле.
Это становится понятным, если рассмотреть соответствующие
радикалы бензола, толуола и нафталина. Из их числа отдельно сле¬
дует отметить бензильный радикал, входящий в состав фенилалани¬
на - аминокислоты белков. Ароматические радикалы именуются
арильными и обозначаются Аг. Соответственно, арены в общем виде
выражаются формулой Аг-Н.
Произведенные из четырех радикалов толуола его гомологи
орто-, мета-, и лара-диметилбензолы (орто-, мета-, пара-
ксилолы, или проще - о-, м-, п-) являются изомерами положения,
по отношению к которым этилбензол является изомером скелета.
радикалы толуола
толуол бензил о-толил м-толил л-толил
337
гм.
РИ¬
ГИ,
СНз
этилбензол
о-, м- и л-ксилолы
а- и (З-Метилнафталины являются изомерами положения
Р
Р
а а
а-нафтил
,СН3
Р-нафтил а-метилнафталин р-метилнафталин
Изомерия скелета и изомерия положения - частные случаи
структурной изомерии Структурные изомеры существенно разли¬
чаются между собой физическими и химическими свойствами.
У органических соединений, содержащих гетероатомы, изомерия
положения обусловлена расположением гетероатома как в цепи, так
и в кольце В качестве простейших примеров изомеров положения
в ряду нециклических производных можно назвать этиловый спирт
СНэСНгОН и диметиловый эфир СНэОСНз, которые в данном случае
являются примерами межклассовой изомерии.
Примерами изомеров положения в ряду гетаренов являются
упомянутые выше пиразол и имидазол, а также пиридазин, пирими¬
дин и пиразин.
Радикалы гетаренов производятся по тому же принципу, что и
прочие. В качестве примера ниже изображены радикалы фурана и
пиридина.
Если изомеры различаются между собой расположением атомов
в пространстве, то говорят о стереоизомерии. Стереоизомерия де¬
лится на оптическую и геометрическую.
а-фурил р-фурил а-пиридил |3-пиридил у-пиридил
18.2.2. Оптическая изомерия
338
случаем стереоизомерии является оптическая изо-
мерия, характерная для Бр3-гибридных атомов, каким чаще всего
является углерод. Если такой атом связан с четырьмя различными
атомами или различными группами атомов, то возникают изомер¬
ные пары, в которых молекулы изомеров относятся по своей про¬
странственной организации одна к другой так же, как соотносятся
между собой предмет и его зеркальное изображение.
Атом углерода, имеющий четыре разных заместителя, называют
асимметрическим и обозначают С*. Молекулу, в целом не обла¬
дающую симметрией, называют хиральной - от слова хирос, что
означает по-гречески рука - образец несимметричной фигуры.
В качестве простейшего примера хиральной молекулы можно
назвать молекулу бутана, в которой при одном из вторичных атомов
углерода водород заменен на дейтерий. Приведем примеры еще
нескольких хиральных молекул - из алканов и циклоалканов.
Зеркальные изомеры - антиподы - принято называть оптиче¬
скими изомерами или энантиомерами.
Явление оптической изомерии, как свойство асимметрического
углеродного атома, было объяснено в 1874 г. Вант-Гоффом и
Ле-Белем. Возможность существования атома углерода в тетраэд¬
рической конфигурации до этого высказал А. М. Бутлеров.
В отличие от структурных изомеров энантиомеры идентичны
один другому в большинстве своих свойств, вследствие чего их
разделение оказывается затруднительным Однако, они отличают¬
ся по своему взаимодействию с плоскополяризованным светом,
а именно, вращают плоскость поляризации света в равной мере,
но в противоположных направлениях. Соответственно различают
правовращающий изомер или (+)-изомер и левовращающий или
(-)-изомер. Способность веществ изменять направление поляриза¬
ции света называют его оптической активностью Энантиомеры,
следовательно, оптически активны.
А'
энантиомерь
С2Н5
СН3—І-Н
I
С2Н5
СНз-І-Н СНз
339
Энантиомеры отличаются только теми свойствами, которые
проявляются у них под влиянием физических или химических воз¬
действий, асимметричных по своей природе. Этим объясняется
диаметрально противоположное взаимодействие энантиомеров с
поляризованным светом. Кристаллы энантиомеров, как правило,
являются зеркальными антиподами. Пользуясь различием физиче¬
ских свойств, а именно - асимметрией кристаллов энантиомеров,
Л Пастер впервые сумел разделить изомеры оптически активных
солей винной кислоты
Энантиомеры различаются некоторыми химическими свойства¬
ми - взаимодействием с веществами, которые в свою очередь явля¬
ются хиральными. Прежде всего следует отметить их взаимодей¬
ствие с ферментами, белковые молекулы которых тоже хиральны.
Немецкий химик Э. Фишер высказал в конце прошлого века ги¬
потезу, что специфичность биологического действия того или иного
соединения определяется его структурно-пространственным соот¬
ветствием ферменту, с ним взаимодействующему. Такую стереоспе¬
цифичность взаимодействия он уподобил соответствию между клю¬
чом и замком. Эта идея впоследствии развилась в концепцию ак¬
тивного центра фермента, объясняющую биологическое действие
стереоспецифическим контактом хиральных участков субстрата и
фермента. Как это поясняется графически ниже, в таком контакте
должны принимать участие по крайней мере три из четырех замес¬
тителей асимметрического атома углерода.
Ниже схематически представлен случай, когда конфигурация
энантиомера не соответствует конфигурации активного центра фер¬
мента и, следовательно, стереоспецифический контакт между ними
невозможен.
Этим и объясняется тот факт, что в живой природе хиральные
соединения представлены, как правило, лишь одной стереоизомер-
ной формой. На основании такого рода наблюдений Пастер еще в
1860 г. высказал предположение, что “асимметрия молекулярного
строения, быть может, образует единственную четкую границу,
разделяющую химию живой и неживой природы”. То, что сейчас
ведутся активные поиски оптически активных веществ вне Зем¬
энантиоме|
активный центр фермента
340
ли, - явление не случайное; их обнаружение послужит подтверж¬
дению гипотезы о существовании внеземной жизни.
Молекулы углеводородов, а также других органических веществ
могут содержать не один асимметрический атом углерода. Более
того, асимметричным может быть не только углеродный атом.
Асимметричным, в частности, может быть атом азота, имеющий три
неодинаковых заместителя - четвертым является неподеленная
электронная пара. В случае азота, однако, легко происходит инвер¬
сия, или выворачивание молекулы, поскольку энергия активации
этого процесса невысока. Инверсия атома азота приводит к тому,
что энантиомеры аминов легко переходят друг в друга.
Максимально возможное число оптических изомеров какого-
либо хирального вещества равно 2П, где п - число асимметрических
центров в его молекуле. К примеру, молекула холестерина
(см. 18.1.3) содержит 8 хиральных центров, а следовательно ей от¬
вечает 28, т. е. 256 оптических изомеров. Между тем природный
холестерин является единственным из всех возможных стерео¬
изомеров.
Следствием стереоселективности взаимодействия живого с оп¬
тическими изомерами является, в частности, различие в терапевти¬
ческой активности энантиомеров оптически активных лекарствен¬
ных веществ. Более физиологически активный из них принято на¬
зывать эвтомером, а другой - дистомером. При этом следует
учесть, что в настоящее время 40% лекарственных веществ явля¬
ются хиральными соединениями. Между тем, лишь ничтожная
часть из них представлена индивидуальными энантиомерами. Это
означает, что составляющий 50% препарата неактивный дистомер
может в лучшем случае представлять собой ненужный балласт.
Между тем, энантиомеры могут обладать в принципе разли¬
чающимся фармакологическим действием.
Так, анальгетический препарат дарвон - это эвтомер, а его дис¬
томер известен как лекарство против кашля Другой пример разли¬
чия в биологической активности - пестицид паклобутразол Один из
его стереоизомеров представляет собой фунгицид, а другой - силь¬
ный гербицид. Известны более сложные ситуации. Трагична исто¬
рия применения в медицине транквилизатора талидомида, исполь¬
зование которого беременными привело к тысячам случаев уродств
у новорожденных, так как дистомер, присутствовавший в равных
количествах в препаратах, обладает тератогенным (вызывающим
уродства) действием.
Существует проблема хиральной чистоты окружающей среды.
Производя множество хиральных веществ без их разделения
и очистки, промышленность постепенно приводит к загрязнению
341
среды обитания такими препаратами, последствия чего пока трудно
оценить
Более подробно вопросы оптической изомерии будут рассмотре¬
ны далее при обсуждении свойств биомолекул.
18.2.3. Геометрическая изомерия
Геометрическая изомерия характерна для замещенных циклоал-
канов и алкенов. Она обусловлена тем, что в молекулах этих ве¬
ществ свободное вращение атомов вокруг а-связей (у циклоалканов
и их гетероциклических аналогов) и относительно двойных связей
(у алкенов) оказывается невозможным.
гн гн гн н СНэ ЯНэ ^ /
сн, сн5 сн, н
н' н н сн3 / СНэ
цис- транс- цис- транс-
бутены-2 1,2-диметилциклопропаны
Эта изомерия имеет место при наличии у атомов углерода, об¬
разующих цикл или двойную связь, двух заместителей. Такие за¬
местители могут располагаться как по одну сторону плоскости цик¬
ла или кратной связи (цыс-изомеры), так и по разным сторонам
этой плоскости (транс-изомеры). Например, существуют цис- и
транс-изомеры бутена-2, а также цис- и транс-изомеры 1,2-диме-
тилциклопропана.
Вследствие того, что расстояния между одинаковыми атомами в
молекулах той или иной пары геометрических изомеров различны,
последние существенно различаются своими химическими и физи¬
ческими свойствами. Они могут быть разделены и существовать
индивидуально.
Должны быть различными и физиологические свойства цис- и
транс-изомеров. Например, известный фитотоксин (яд для расте¬
ний) - коричная кислота - оказывает свое физиологическое дей¬
ствие в цыс-изомерной форме. Транс-коричная кислота не является
фитотоксином. Все непредельные жирные кислоты липидов (приме¬
ры - олеиновая и линолевая кислоты) принадлежат к цис-изо-
мерам, независимо от длины углеводородной цепи и количества в
ней кратных связей.
342
л
СбН5 ■* соон
н>=<и
цис-коричная кислота
V
нсоон
транс-коричная кислота
СНЭ(СН2)7 ^(СН2)тСООН
н^н
олеиновая кислота
СНэ(СН2)э СНгч (СН^тСООН
н^н н>=<н
линолевая кислота
Примером биохимически значимого явления цис-транс-изоме¬
рии циклоалканов может служить молекула шестиатомного спирта
инозита, который имеет набор из 8 геометрических изомеров, од¬
нако в природе наиболее распространен
один из них, называемый миоинозитом.
Другой пример - явление стереоизо¬
мерии инсектицида гексахлорциклогекса-
на (сокращенно гексахлорана), который
также может иметь 8 изомеров. Они раз¬
личаются между собой токсичностью по миоинозит линдан
отношению к человеку и инсектицидным
действием. Один из них - линдан - наименее токсичен и обладает
максимальным инсектицидным эффектом.
Еще один пример. Молекула кокаина оказывает примерно в 85
раз более сильное наркотическое действие, чем молекула ее сте¬
реоизомера псевдокаина.
Барьер вращения вокруг кратной связи (связанный с необходи¬
мостью разрыва я-связи) должен быть значительно выше барьера
вращения вокруг одинарной связи в алканах Действительно, для
этого требуется около 80-100 кДж/моль Такая энергия активации
препятствует цис-транс-изомерному переходу при невысоких тем¬
пературах
Однако цыс-трамс-изомеризация в ряду алкенов - обычное яв¬
ление при нагревании или при воздействии света Фотоизомериза¬
ция цис-транс-алкенов имеет важное физиологическое значение. В
частности, световое превращение фумаровая кислота - малеиновая
343
малеиновая кислота
НООС СООН
.X
н соон
фумаровая кислота
кислота являётся метаболическим процессом
Его нарушение при дефиците ферментов вызы¬
вает трудно излечимое кожное заболевание
псориаз.
^/ис-транс-фотоизомеризация лежит в ос¬
нове фоторецепции. Одного кванта видимого
света достаточно, чтобы молекула транс-
ретиналя - вещества, находящегося в зритель¬
ном рецепторе, превратилась в цис-изомер
\^СНЭ Ч^АсНэ СНз-Л^^о
гранс-ретиналь
цис-ретиналь
Это вызывает каскад последующих биохимических превраще¬
ний, в результате чего и формируется соответствующий нервный
импульс. В дальнейшем происходит обратная изомеризация, и мо¬
лекула транс-ретиналя снова готова к световому воздействию.
Таким образом, стереоизомерия (оптическая и геометрическая)
играет важную биохимическую роль.
Следует помнить, что в действительности биомолекулы пред¬
ставляют собой достаточно сложные структуры, которые могут
иметь широкий набор хиральных центров, кратных связей и колец.
Это в какой-то мере можно продемонстрировать с помощью молеку¬
лы цинерина, продуцируемой некоторыми растениями и являю¬
щейся инсектицидом.
СНз,
СНэ'
6н3 с°9
I /
В молекуле цинерина может иметь место и цис-транс-изо¬
мерия относительно цикла и кратной связи, и оптическая изомерия
(хиральные центры отмечены звездочками). Однако природный ци-
нерин представлен единственной стереоизомерной формой.
344
18.2.4. бкедения о номенклатуре органических соединений
Способность атомов углерода и гетероатомов принимать раз¬
личную гибридизацию и образовывать многообразные комбинации,
неограниченная гомология углеводородов и гетероциклов, а также
их производных, различного рода изомерия, - все это приводит к
существованию множества типов органических веществ.
Изучающий органическую химию должен ориентироваться в
разнообразии органических соединений, для чего следует овладеть
основными принципами их номенклатурного упорядочения
В настоящее время в органической химии используются различ¬
ные, основанные на разных принципах, названия веществ. В боль¬
шинстве случаев химики до сих пор предпочитают использовать
условные “тривиальные” наименования веществ; происхождение
этих названий носит случайный характер. Некоторые соединения
названы по природному источнику, из которого их выделили или на
основе которого синтезировали, - например, муравьиная кислота,
лимонная кислота, мочевина, фруктоза. В этих названиях отра¬
жается физиологическое действие веществ или их физические
свойства: например, глюкоза названа так за сладкий вкус, азулен -
за голубую окраску, названия кубан и призман присвоены углево¬
дородам за форму их молекулы. Тривиальные названия не опреде¬
ляют структуру вещества, однако они употребляются как по тради¬
ции, так и ввиду того, что строго построенные систематические на¬
звания оказываются слишком громоздкими для частого практиче¬
ского употребления.
В связи с быстрым ростом числа органических веществ, став¬
ших известными науке, возникла необходимость в определении ло¬
гических принципов построения их наименований Как следствие
этого, уже в прошлом веке наметились принципы рациональной
номенклатуры органических соединений Эта номенклатура пред¬
усматривает сохранение в прежнем виде названий лишь неразвет-
вленных алканов (н-алканов); во всех остальных случаях за основу
наименования молекулы берется название её основного структурно¬
го узла с прибавлением названий радикалов, с ним связанных
В соответствии с этим, основным узлом разветвленного алкана счи¬
тается метановый атом углерода, а основным узлом алкенов и ал-
кинов - молекула этилена и молекула ацетилена В случае аренов
названия производятся от названия ключевого цикла - бензола,
нафталина и т.д По тому же принципу производятся и названия
гетаренов, а также названия производных углеводородов, содержа¬
щих кислород, серу, азот (спирты, эфиры, амины и др ).
При перечислении заместителей первыми называются про¬
стейшие радикалы. Греческими буквами а и (3 обозначается поло¬
345
жеНие заместителей при двух разных атомах углерода в этиленовом
фрагменте '
Положение заместителей в бензольном кольце определяется
приставками орто-, мета- или пара-, а в нафталиновом - так же,
как и в гетероциклическом ряду, буквами а и р.
В качестве примеров приведены рациональные названия изоме¬
ров гексана и бутенов.
СНзСНгСНгСНгСНгСНз СНзСНСНгСНгСНз
<Ьнз
н- гексан диметилпропилметан
СН3 СНз
СН3СН2СНСН2СН3 СН3СНСНСН3 СНз^СНгСНз
<Ьнэ ^Нэ (Ьнз
метилдиэтилметан диметилизопропилметан триметилэтилметан
цис- транс-
СН3СН2 Н СН3 СНз СНз Н СНз Н
чс=с' чс=с' С=С С=С
н' 'н н' 'н н' 'СНз СНз^ 'н
этилэтилен а, (3-диметилэтилен а,а-диметилэтилен
Рациональная номенклатура оказалась удобной для простейших
производных, и в этих случаях ею пользуются по-прежнему. Однако
для сильно разветвленных молекул с длинными углеводородными
цепями она неприменима, поскольку выбор основного узла стано¬
вится произвольным и подобрать названия примыкающих к нему
заместителей невозможно.
Этого недостатка лишена международная номенклатура.
Согласно ей, за основу названия алканов, алкенов, алкинов и их
нециклических производных берется название углеводорода, отве¬
чающее числу атомов углерода в главной цепи, а за основу назва¬
ния циклоалканов, аренов и гетаренов - название соответствующего
незамещенного циклического производного.
Главной цепью в ряду алканов считают цепь самую длинную и
самую разветвленную. Определяющим признаком главной цепи ал¬
кенов и алкинов является наличие в ней кратной связи, причем
двойная связь считается старше тройной.
Для построения названия необходимо пронумеровать атомы в
главной цепи. Выбор начала отсчета определяется при этом прави¬
лом наименьшей суммы номеров. Суть этого правила в том, что
сумма номеров, обозначающих положение заместителей в цепи,
должна быть минимальной. При нумерации следует помнить, что
заместители уступают по старшинству кратным связям.
346
Наконец, указывают местоположение и названия заместителей
и кратных связей; информацию о заместителях дают перед названи¬
ем главной цепи, а о кратных связях - после него.
В качестве примера приведем определенные по международной
номенклатуре названия углеводородов, которые выше были назва¬
ны рациональным способом: к-гексан, 2-метилпентан, 3-метилпен-
тан, 2,3-диметилбутан, 2,2-диметилбутан, бутен-1, цыс-бутен-2,
транс-бутен-2, 2-метилпропен.
18.3. Свойства, получение и применение
углеводородов
18.3.1. Химические свойства алканов и циклоалканов
В обычных условиях алканы, как правило, химически инертны,
так как о-связи С-Н и С-С в этих соединениях весьма прочны. В
связи с этим вещества этого класса называют еще парафинами -
от рагит - мало + а///ш'5 - сходство, родство (лат.). В мягких
условиях протекает лишь небольшое число реакций замещения ато¬
мов водорода на различные атомы и группы; обычно они осу¬
ществляются свободнорадикальным путем - это так называемые
гемолитические замещения.
Галогенированне. Энергия связи углерод-фтор составляет 452
кДж/моль, что намного превосходит тепловой эффект разрыва свя¬
зей С-Н. Поэтому фторирование алканов происходит чрезвычай¬
но энергично. Чтобы эта реакция подвергалась контролю, применя¬
ют специальные меры (разбавление фтора азотом, отвод тепла и
пр ), среди которых наиболее удачной мерой представляется фтори¬
рование алканов с помощью СоРз Вещество это при нагревании до
200° С медленно выделяет фтор.
СпН2п+2 ^(С°Рз) > Спр2п+2 + 2(П+1)НР
Полученные в результате фторирования фторуглеводороды не¬
горючи, химически инертны и термостабильны. Они используются
как смазочные масла, пригодные для использования в весьма жест¬
ких условиях, как нетоксичные хладоагенты (фреоны) и т. п.
Тепловой эффект хлорирования - 293 кДж/моль - значительно
меньше эффекта фторирования, поэтому реакция эта идет только
на свету, который играет роль инициатора Хлор последовательно
347
замещает атомы водорода в алканах; примером может служить его
взаимодействие с метаном:
1 СН4 + С12-> СНЭС1 + НС1 2. СН3С1 + С12-> СН2С12 + НС1
хлористый метил хлористый метилен
3. СН2С12 + С12-> СНСЬ + НС1 4. СНС1Э+С12-> ССЦ + НС1
хлороформ четыреххлористый углерод
Механизм хлорирования сходен с механизмом синтеза хлорис¬
того водорода и носит цепной характер. На первой стадии, под дей¬
ствием кванта света происходит гомолитическая диссоциация моле¬
кулы хлора-
С1 : С1 —► 2 СГ (инициирование цепи)
Радикалы СГ атакуют молекулу метана.
СН4 + СГ -» СНз‘ + НС1 (развитие цепи)
Метильный радикал активно реагирует с молекулой хлора:
СНэ' + С1: С1 -» СНзС! + СГ (развитие цепи)
В результате образуется радикал хлора и процесс начинается снова.
При столкновении двух свободных радикалов цепь обрывается.
СН3‘ + СГ -» СНзС! (обрыв цепи)
Полученные галогенпроизводные используются как хорошие
растворители.
Бромирование алканов происходит в очень жестких условиях;
реакция с иодом не идет.
Галогенирование циклоалканов. Аналогичным образом про¬
исходит галогенирование пяти- и шестичленных циклоалканов, по¬
строенных без напряжения. Иначе ведут себя напряженные цикло-
пропановые и циклобутановые углеводороды. Основой взаимодей¬
ствия циклопропана с галогенами является размыкание кольца, то
есть разрыв высокоэнергетических связей углерод-углерод, а не
связей углерод-водород. Затем происходит присоединение атомов
галогена к атомам углерода по освободившимся валентностям.
Хлорирование циклопропана частично сопровождается гомоли-
тическим замещением Циклобутан присоединяет галогены не
столь активно, как циклопропан, и доля продуктов замещения
возрастает.
Таким образом, трех- и четырехчленные циклоалканы вступают
в реакции присоединения с размыканием кольца, причем, как это
установлено, они присоединяют к себе не только галогены, но и га-
логеноводороды, а также водород. Так, циклопропан легко присое¬
диняет к себе Н1 и НВг и гидрируется в присутствии платинового,
палладиевого или никелевого катализаторов.
348
ВгСН2—СН2—СНгВг
СНз—сн2—сн3
ВгСН2—сн2—сн3
Циклобутановые углеводороды присоединяют водород при силь¬
ном нагревании. Циклопентан удается превратить в пентан лишь
в очень жестких условиях (нагревание, давление, катализатор),
циклогексан вовсе не гидрируется.
Итак, трех- и четырехчленные кольца термодинамически неста¬
бильны. По этой причине они обычно не встречаются в живой при¬
роде. В том же случае, когда это имеет место, такие производные
обладают обычно высокой биологической активностью вследствие
их реакционноспособности.
Так обстоит дело, например, для пиретроидов, содержащих в
своем составе циклопропановое кольцо. Пиретроиды - продуценты
некоторых растений из семейства ромашек (пиретрум - ромашка по
латыни), являющиеся инсектицидами. Вышеупомянутый цинерин -
типичный пиретроид.
Другой пример природных соединений с малыми циклами - ан¬
тибиотик пенициллин, содержащий четырехчленное кольцо. В при¬
роде также редки семи-, восьмичленные и больших размеров коль¬
ца, но здесь причина в другом, а именно, в энтропийном факторе -
малой вероятности замыкания колец из-за необходимости участия
в этом процессе достаточно удаленных друг от друга заместителей.
Таким образом, в природе обычно представлены соединения с
пяти- и шестичленными кольцами.
Нитрование. Эта реакция известна под названием реакции Ко¬
новалова, открывшего её в 1889 году. Она происходит под воздей¬
ствием разбавленной азотной кислоты при температуре 140° С и
повышенном давлении, приводя к замещению атома водорода на
нитрогруппу - N02, в результате чего образуются нитроалканы:
Р-Н + Н0М02 -> ^N02 + Н20
Нитроалканы, среди которых наиболее ценным является нитро¬
метан, используются как растворители, как добавки к топливам,
а также как полупродукт для синтеза взрывчатых веществ Они
сильно токсичны.
Сульфохлорирование. В эту реакцию алканы вступают на
свету при совместном действии на них хлора и сернистого газа, в
результате чего образуются алкилсульфохлориды Как и хлориро¬
вание, этот процесс имеет цепной свободнорадикальный механизм'
Вг2 / Ьу
Н2/ кат
349
1. С12 -* 2СГ
3. Р + Б02 -> рбо2
2. Р-Н + С1 -> Р + НС1
4. РБОг + С12 -> РБ02С1 + СГ
алкилсульфохлорид
Гидролиз алкилсульфохлоридов в присутствии шелочи приводит
к солям алкилсульфокислот - алкилсульфонатам, которые широко
используются в качестве синтетических моющих средств.
РБ02С1 + 2№ОН -> РБОгОМа + №С1 + Н20
алкил сульфонат
Лучше всего моющий эффект проявляется у алкилсульфонатов,
содержащих в цепи 10-18 атомов углерода В промышленности
сульфохлорированию обычно подвергается смесь углеводородов со¬
става С12-С10.
Обнаружено, что синтетические моющие средства, содержащие
цепочку с нечетным числом атомов углерода, не усваиваются мик¬
роорганизмами и загрязняют окружающую среду. В этой связи ал-
килсульфонаты стремятся заменять биологически более "мягкими"
веществами - то есть веществами, разлагающимися под влиянием
бактерий в природных условиях.
В реакциях галогенирования, нитрования и сульфохлорирования
разветвленных алканов и циклоалканов одновременному замещению
подвергаются атомы водорода при атомах углерода с различной
степенью замещения, что приводит к образованию сложной смеси
продуктов замещения. Установлено, что замещение происходит
преимущественно при третичном атоме углерода; первичный атом
углерода менее активен Объясняется это тем, что величина энер¬
гии связи атома водорода с первичным, вторичным и третичным
атомами углерода различна - она равна соответственно 418, 394 и
377 кДж/моль.
Крекинг. При нагревании алканов до 400-600° С их молекулам
сообщается энергия, достаточная для того, чтобы произошел гомо-
литический разрыв связей углерод-углерод. Образующиеся при этом
радикалы стабилизируются. Это происходит в основном за счет то¬
го, что один из них отдает другому атом водорода, в результате чего
образуются молекулы алкена и алкана. Например:
СНзСН2СН2СНз-> СН3СН2 + СН2СНз -> СН2=СН2 + СН3СН3,
а в общем виде:
Крекинг сопровождается частичной изомеризацией, при кото¬
рой из н-алканов образуются энергетически более устойчивые
СпН2п+2 —* СтН2т + СрН2р+2 , т+р = п
350
изоалканы^При крекинге частично разрушаются не только связи
углерод-углерод, но и более прочные связи углерод-водород, след¬
ствием чего является дегидрирование в алкены или циклоалканы и
ароматизация в арены.
Подбором условий (температура, катализатор) можно добиться
преимущественной активизации любого из названных здесь
процессов.
Окисление. В обычных условиях алканы и циклоалканы устой¬
чивы к действию кислорода и окислителей. Однако при под¬
жигании их на воздухе они сгорают до углекислого газа и воды,
причем сгорание происходит с выделением большого количества
тепла - почти 50 ОСЮ кДж на 1 кг топлива. Поэтому алканы и цик¬
лоалканы уже давно используются в качестве топлива.
Бензин - смесь жидких углеводородов состава Сб-Сю, кипящая
при температуре до 180° С. В состав бензина входит свыше ста
компонентов из числа изомерных алканов и циклоалканов. Бен¬
зин - основа карбюраторного топлива, используемого в автомобиль¬
ных двигателях.
Керосин. Кипит в пределах 180-230° С Содержит углеводороды
состава С11-С14. Керосин употребляется в больших количествах в
качестве топлива для реактивных двигателей и ракет.
Дизельное топливо. Имеет температуру кипения 230-400° С.
Состоит из смеси углеводородов состава выше С14.
Углеводороды ведут себя по-разному при сгорании в двигателях.
В частности, н-алканы обладают наиболее низкой температурой са¬
мовоспламенения. Поэтому они ценны как ракетное топливо На¬
против, использование их в автомобильных двигателях приводит из-
за их преждевременного воспламенения к детонации, снижающей
мощность двигателя и вызывающей его быстрый износ. В бензинах
высокого качества предпочтительно используются разветвленные
изоалканы.
За эталон горючего, обладающего максимальной склонностью к
детонации, принят к-гептан, а минимальной - 2,2,4-триметилпен-
тан, обычно именуемый изооктаном. Для сравнения различных сор¬
тов бензина с точки зрения их детонационных свойств введено по¬
нятие октанового числа. Для изооктана это число принято равным
100, для к-гептана - нулю Октановое число (например 92) показы¬
вает, что этот бензин обладает такими же детонационными
свойствами, как модельная смесь, содержащая 92% изооктана и
8% к-гептана. С целью повышения октанового числа (иногда до
130) к бензину добавляют различные антидетонаторы
Чаще всего для этого используют тетраэтилсвинец РЬ(С2Н5)4 -
ТЭС. Бензин с такой добавкой называют этилированным. Несмотря
351
на незначительное количество (меиее 5%) ТЭС, этилированный
бензин сильно токсичен По этой причине ТЭС постепенно заменя¬
ется на менее токсичные антидетонаторы - соединения марганца.
Окисление алканов и циклоалканов кислородом воздуха, проис¬
ходящее в более мягких условиях и в присутствии специальных ка¬
тализаторов, затрагивает не все их углеродные атомы и приводит к
образованию различных кислородосодержащих веществ - спиртов,
альдегидов, кетонов, кислот.
[°1 [О] [О] [О]
Я-СНз —» Р-СН2ОН Р-СН=0 —*- Я-ССЮН РСНгЯ —» РССЖ
алкан спирт альдегид кислота кетон
Многие из этих процессов имеют важное значение для про¬
мышленности - особенно переход алканов в жирные кислоты, кото¬
рые используются как поверхностно-активные вещества.
Окисление углеводородов до спиртов - так называемое С-гид-
роксилирование - осуществляется в живом организме фермента¬
тивным путем и является первой стадией выведения чужеродных
органических веществ, в том числе и углеводородов, из организма.
Чуждые организму органические вещества называют ксенобиотика¬
ми Эта же реакция, заключающаяся в замене связи С-Н связью
С-ОН, служит для введения гидроксильной группы в состав биомо¬
лекул и осуществляется в митохондриях.
18.3.2. Химические свойства алкенов
Электрофильное присоединение. Наличие в молекулах ал¬
кенов я-связи определяет их ненасыщенность, а, следовательно, -
способность вступать в реакции электрофильного присоединения.
Примером электрофильного присоединения может служить вза¬
имодействие алкенов с галогеноводородами. Реакция начинается с
присоединения по двойной связи электрофильной частицы - прото¬
на. В результате образуется положительно заряженный ион, карб-
катион, соединяющийся затем с галогенид-анионом.
СН2=СН2 + Н*-» СН3-СН2* карбкатион
СН3-СН2+ + Вг‘ -> СНз-СН2-Вг
При взаимодействии галогеноводорода с несимметричным алке¬
ном может произойти образование пары карбкатионов:
352
Л . в,-
СНЭ—СН—СН3 —► СН3—СН—СН3 2-Сромпропан
СНЭ—СН=СН2 —I
СНэ—СН2—СН2+-5* СН3—СН2—СН2—Вг 1-бромпропан
Эти карбкатионы энергетически неравноценны, более устойчи¬
вым является карбкатион с большей степенью разветвления, а по¬
тому именно он и образуется. Эту закономерность в присоединении
галогеноводородов к алкенам впервые обнаружил В. В. Марковни-
ков, ученик А. М. Бутлерова.
Согласно правилу Марковникова, присоединение галогеново¬
дородов к алкену происходит таким образом, что водород атакует
наиболее гидрогенизированный атом углерода.
Правило Марковникова применимо и в случае присоединения к
алкену серной кислоты.
НОЭОзН
СНз—СН2=СН2 1° г СНз-СН-СНз
* -НОЭОзН бэОзН
алкилсерная кислота спирт
Алкилсерные кислоты находят применение в органическом син¬
тезе, преобразующем алкены в другие классы веществ. Так, напри¬
мер, при их гидролизе образуются спирты, а при нагревании алкил-
серных кислот - снова алкены
Воду алкены присоединяют лишь в жестких условиях - при
сильном нагревании и при большом давлении, к тому же - в при¬
сутствии фосфорной кислоты и солей металлов. Реакция также
подчиняется правилу Марковникова.
НОН СН3—СН-СН3
сн3-сн=сн2 —— Ан
Гидрирование алкенов осуществляется лишь в условиях гетеро¬
генного катализа (в присутствии Рі, Р<1 N1) и имеет своим продук¬
том алканьг
СпН2п + Н2 (катализатор) -» СпН2п+2
Галогенирование алкенов также представляет собой электро-
фильное присоединение Молекула галогена, сблизившись с облас¬
тью двойной связи, поляризуется под действием я-электронов и
один из её атомов, приобретая положительный заряд, вступает во
взаимодействие с двойной связью В результате быстро протекаю¬
12 Зак. 675
353
щей реакции образуются вицииальные дигалогенпроизводные (лат
иісіпиз - соседний)
Р-СН=СН-Р
Окисление алкенов. Алкены, так же как и алкаиы, при под¬
жигании сгорают до углекислого газа и воды.
В отличие от алканов, алкены легко подвергаются действию
различных окислителей в сравнительно мягких условиях.
(01 . „Ліїи
«/ч нон Ан Ан
Так, например, окисление водным раствором перманганата ка¬
лия в щелочной среде приводит к образованию вицинальных двуха¬
томных спиртов - гликолей.
Эта реакция (её обнаружил русский химик Е. Е. Вагнер) проис¬
ходит быстро при обычной температуре. В результате реакции рас¬
твор перманганата обесцвечивается Поэтому реакция Вагнера ис¬
пользуется как проба на двойную связь. Образование гликолей из
непредельных соединений является важной биохимической реакци¬
ей, происходящей с последними в митохондриях. В частности, та¬
ким путем осуществляется метаболизм непредельных липидов.
Окисление алкенов кислородом происходит в присутствии ката¬
лизатора (мелко раздробленное серебро) и приводит к окисям алке¬
нов, в частности, из этилена получается окись этилена.
Это соединение, являясь насыщенным трехчленным, а следова¬
тельно, напряженным гетероциклом, вступает в реакции присоеди¬
нения еще легче, чем циклопропан, что и используется в синтети¬
ческих целях.
сн2=сн2 -5» Х7
(Ад) о
► НОСН2СН2ОН этиленгликоль
► НОСН2СН2МН2 коламин
► НОСН2СН2СІ (3-хлорэтиловый спирт
» СН?=СН—С—N акрилонитрил
► СН3СНО ацетальдегид
Путем гидрирования из окиси этилена получают этиловый
спирт; присоединение хлористого водорода приводит к важному
полупродукту - Р-хлорэтиловому спирту; реакция с водой дает
354
этиленгликйль, а с аммиаком - этаноламин (коламин), входящий в
состав клеток мозга и нервов. При действии на окись этилена си¬
нильной кислотой получается ценное сырье для синтеза полимер¬
ных материалов - акрилонитрил, а при нагревании окись этилена
изомеризуется в уксусный альдегид.
Следует представлять себе, что образование гликолей из непре¬
дельных соединений в биосредах идет через промежуточную стадию
окиси, которая далее присоединяет воду. Именно эти превращения,
как установлено, объясняют давно известный факт сильной канце¬
рогенной активности конденсированного углеводорода бензпирена,
который в высоких концентрациях присутствует в табачном дыме и
в каменноугольной смоле.
Одна из кратных связей молекулы бензпирена (положения 3
и 4) обладает повышенной активностью в реакциях электрофильно-
го присоединения. Эта кратная связь ферментативно трансформиру¬
ется в соответствующую окись, которая далее переходит в гликоль.
Кратная связь в молекуле последнего (в положениях 1 и 2) вновь
ферментативно присоединяет кислород. Получающаяся таким обра¬
зом окись обладает повышенным сродством к аминогруппе гуанина
(РМНг), входящего в состав ДНК. Под его воздействием окисное
кольцо раскрывается. В результате происходят необратимые изме¬
нения в молекуле ДНК, вызывающие последующее образование
раковых клеток.
Озонирование алкенов, разрушая двойную связь С=С, приводит
к малоустойчивым и взрывчатым озонидам, которые под действием
воды в свою очередь превращаются в карбонильные соединения -
альдегиды и кетоны.
Полимеризация алкенов. Впервые один из случаев этой ре¬
акции - удвоение 2-метилпропена - наблюдал А М Бутлеров, ко¬
торый еще в 1867 году утверждал, что "...уплотнение непредель¬
12*
355
ных углеводородов представляет, бесспорно, одну из самых замеча¬
тельных синтетических реакций, способных происходить под срав¬
нительно слабыми химическими влияниями". В настоящее время
полимеризация алкенов является основой синтеза многочисленных
полимерных материалов
Реакцию полимеризации алкенов можно выразить общим урав¬
нением
п СН2=СН — (— СН2—СН—)п
X X
мономер полимер
X = Н, СНз, ОН, С1, СЫ, С6н5 и т. д.
Полимеризация происходит обычно на основе гомолитнческого,
то есть радикального присоединения; для её осуществления требу¬
ется инициатор - вещество, легко распадающееся на свободные ра¬
дикалы. Возможно осуществление полимеризации и ионным путем.
Беря за исходный продукт соответствующие мономеры и подби¬
рая катализаторы, химики синтезируют полиэтилен (X = Н), поли¬
пропилен (X = СНз), поливиниловый спирт (X = ОН), поливинилхло¬
рид (X = С1), полиакрилонитрил (X = СМ), полистирол (X = СвНь) - по¬
лимерные материалы, широко используемые в народном хозяйстве,
в том числе и в медицине (табл. 18.1).
Еще один пример - полимеризация тетрафторэтилена, приводя¬
щая к образованию уникально устойчивого к воздействию темпера¬
туры и агрессивных сред материала - тефлона.
X СРг=СРг -> [-Ср2-Ср2-]* тефлон
С целью трансформации свойств полимерных материалов подбо¬
ром условий изменяют длину полимерной цепи, осуществляют
совместную полимеризацию (сополимеризацию) двух или более
мономеров или вводят в полимер разнообразные добавки-пласти-
фикаторы.
Физико-механические свойства полимеров (пластичность, тер¬
мостойкость, эластичность) в основном определяются простран¬
ственным строением их молекул. По форме молекулы полимеров
разделяются на линейные и трехмерные (глобулярные). Полимеры
с линейными молекулами обладают легкоплавкостью, прочностью,
хорошей растворимостью. Глобулярные полимеры неплавки, хрупки
и плохо растворимы. Синтез той или другой разновидности поли-
алкенов определяется условиями его осуществления (температурой,
давлением) и прежде всего - катализатором.
356
Таблица 18.1
Использование винильных полимеров в медицине
X
Название
Применение
н
полиэтилен
1. протезирование сосудов
2 замещение костных дефектов
3. сосуды для хранения крови
СНэ
полипропилен
в аппаратах для переливания крови
с6н5
полистирол
1. для замены дефектов черепа
2. изготовление искусственных органов
3. протезирование зубов
СІ
поливинилхлорид
1. в аппаратах для переливания крови
2. для устранения дефектов лица
он
поливиниловый
спирт
для получения полимеров
с бактерицидными свойствами
см
полиакрилонитрил
для склеивания кожи, сосудов и костей
ОС4Н9
поливинил-
бутнловый эфир
для очищения и заживления ран,
регенерации тканей
ОСОСНз
поливинилацетат
для получения полимеров
с бактерицидными свойствами
1
поливинил-
пиррол идон
заменитель плазмы крови
Линейный полиэтилен - (п>1000) имеет конформацию н-алка-
на, придающую достаточно длинным молекулам полимера тенден¬
цию скручиваться в спираль.
Для производных полиэтилена (X Ф Н) возможно образование
различных пространственных структур в зависимости от распо¬
ложения соседних заместителей по сторонам полимерной цепи,
что вызвано хиральностью каждого из звеньев, образующихся в
результате полимеризации. Помимо пространственно хаотичных
(атактических) структур возможно образование стереорегулярных
полимеров.
Стереорегулярные молекулы полипропилена (X = СН3), напри¬
мер, могут иметь изотактическое и синдиотактическое строение
В изотактическом полипропилене пространственное строение эле¬
ментарного звена полностью повторяется. В синдиотактическом
полипропилене идет регулярное чередование пространственной
конфигурации отдельных звеньев Полимеризация полипропилена с
357
применением катализаторов (ТіСЦ, триэтилалюминий и др ) носит
стереорегулярный характер, в результате чего получаются изотак-
тические или синдиотактические полимеры, обладающие высокой
прочностью.
Атактический полипропилен - продукт обычной полимеризации
пропилена; он не обладает прочностью и упругостью.
Таким образом, можно создавать полимеры с заранее заданными
и полезными для медицинских целей физическими свойствами -
прочностью, пластичностью, термоустойчивостью и многими
другими.
18.3.3. Химические свойства сопряженных алкадиенов
Реакция присоединения. Как уже было сказано, в алкадие-
нах двойные связи могут быть изолированными, а могут и образо¬
вывать сопряженную систему.
Существуют еще алкадиены с кумулированными кратными
связями У веществ такого рода двойные связи существуют при од¬
ном и том же атоме углерода. Простейшим представителем таких
углеводородов является аллен СН2=С=СНг. В этой молекуле первый
и третий атомы углерода находятся в состоянии зр2-гибридизации,
а центральный атом углерода - в ер-гибридизации. Такие углеводо¬
роды получить довольно трудно, в природе они не обнаружены, а
поэтому здесь не рассматриваются.
Алкадиены с изолированными двойными связями схожи по
своим свойствам с алкенами - каждая из двойных связей, незави¬
симо от других, вступает в обычную для неё реакцию электрофиль-
ного присоединения.
Иначе происходит электрофильное присоединение в случае дие¬
нов и их производных с сопряженной системой кратных связей, ти¬
пичными представителями которых являются бутадиен-1,3 (диви¬
нил), 2-метил бутадиен-1,3 (иэопрен), 2-хлорбутадиен-1,3 (хлоро-
прен) и циклопентадиен.
иэотактический полимер
синдиотактический полимер
Н
X
циклопентадиен
358
При ^яаимодействии бутадиена-1,3 с галогеноводородом прежде
всего происходит присоединение протона к одному из атомов угле¬
рода (он обозначен ниже индексом 1) в соответствии с правилом
Марковникова.
резонансный карбкатион
1 2 3 4 н+
СН2=СН-СН=СН2 —► СН3—СН+—СН=СН2 +-+ СН3—СН=СН-СН2+
[X
СН3—СН—СН=СН2 СН3—СН=СН—СНзХ
X
В образовавшемся карбкатионе положительный заряд сосед¬
ствует с двойной связью. 71-Электроны этой связи несколько сме¬
щаются в сторону заряда, в какой-то мере компенсируя его. В ре¬
зультате область атома углерода 4 приобретает положительный за¬
ряд. Другими словами, заряд распределяется между атомами угле¬
рода 2 и 4, или делокализуется. Таким образом, карбкатион можно
представить двумя резонансными формулами, следовательно, речь
идет о резонансном карбкатионе.
На следующей стадии реакции отрицательный галогенид-ион
присоединяется либо к атому углерода 2, либо к атому 4. В резуль¬
тате, в случае сопряженных алкадиенов может происходить как
1,2-, так и 1,4-присоединение.
Соотношение этих конкурирующих процессов зависит от приро¬
ды диена и электрофильного реагента, а также от условий, в кото¬
рых проходит реакция. Так, например, при гидрировании бутадиена
водородом in situ происходит исключительно 1,4-присоедине¬
ние; при каталитическом же гидрировании водород присоединяется
к атомам 1 и 2.
СНзСН2СН=СН2 сн2=сн-сн=сн2 Л* СН3СН=СНСН3
кат.
Хлор, взаимодействуя с бутадиеном, в одинаковой мере (50%)
вступает как в 1,2-, так и 1,4-присоединение:
СІ2 а2
аСНгСНаСН =СН2 СН2 =СН -СН =СНг —► ОСНгСН —CHCHjCI .
1,2-присоединение 1,4-присоединение
в то время как циклопентадиен с хлором, так же, как и с бромом,
образует продукты только 1,4-присоединения.
359
Тенденция сопряженных алкадиенов вступать в 1,4-присоеди-
нение особенно ярко проявляется в реакции диенового синтеза -
реакции их взаимодействия с алкенами.
с • I - ос
X
Частный случай диенового синтеза - димеризацию изопрена -
впервые наблюдал С В. Лебедев.
Продуктом реакции является лимонен - углеводород, встре¬
чающийся в эфирном масле лимона и других цитрусовых.
Диеновый синтез - это способ преобразования нециклических
углеводородов в производные, содержащие шестичленный цикл.
Эта реакция играет определенную роль в биосинтезе некоторых
природных шестичленных циклических структур (терпены, сте¬
роиды и др.).
У циклопентадиена склонность к диеновому синтезу выражена
настолько сильно, что этот углеводород даже в обычных условиях
быстро превращается в циклический димер, при нагревании распа¬
дающийся снова на две молекулы циклопентадиена:
0-0-00
димер циклопентадиена
Полимеризация алкадиенов. Каучуки. Склонность сопря¬
женных алкадиенов к реакциям 1,4-присоединения проявляется
также и в том, что они легко подвергаются полимеризации:
п СН2=С-СН=СН2 —► [—СН2—С=СН-СН2—] п
X X
х = Н, СН3, С1
Натуральный каучук - продукт млечного сока (латекса) расте¬
ний-каучуконосов - представляет собой линейный полимер изопре¬
на, точнее - смесь такого рода полимеров с молекулярным весом
360
от 5000% 300 ООО. Из дивинила получают бутадиеновый каучук,
а из хлоропрена - хлоропреновый.
Соседние звенья полимеров сопряженных 2-замещенных алка-
диенов могут приобретать различную пространственную структу¬
ру - цис- или транс-конфигурацию. Природный каучук имеет сте-
реорегулярное строение и все его звенья - цмс-конфигурацию.
Стереорегулярное строение имеет и другой природный полимер
изопрена - гутта, только, в отличие от каучука, звенья этого поли¬
мера имеют транс-конфигурацию. Гутта - компонент млечного со¬
ка некоторых тропических растений, из нее изготавливают гутта¬
перчу - материал, более жесткий, чем каучук.
—СН сн2—
X сн2—
м.Хм
Промышленное производство синтетического каучука из диви¬
нила впервые было организовано в России по методу, разработан¬
ному С. В. Лебедевым (1930 г.). В настоящее время синтетического
каучука производится больше, чем добывается природного. Наи¬
большее развитие получило производство стереорегулярного изо-
пренового каучука, выгодно отличающегося своими качествами.
Вообще же, существует много видов каучука, каждый со своими
особенностями.
Получение каучука с какими-то специальными свойствами до¬
стигается различными способами - как путем сополимеризации раз¬
личных мономеров, так и путем обработки полимеров. Одним из
них является вулканизация - термообработка в присутствии серы,
приводящая к сшиванию отдельных цепей полимера сульфидными
(-Б-) и дисульфидными (-Б-Б-) мостиками. Вулканизация придает
каучуку твердость. При оптимальном максимуме серы каучук в ре¬
зультате вулканизации превращается в эбонит - материал, вовсе
лишенный пластичности.
18.3.4. Химические свойства алкинов
Электрофильное присоединение. Алкины вступают в реак¬
ции электрофильного присоединения с водородом (в присутствии
катализатора), галогенами и галогеноводородами и последовательно
присоединяют к себе две молекулы этих веществ Подбором усло¬
вий реакцию можно остановить на стадии присоединения одной
361
молекулы. Присоединение галогеноводородов осуществляется по
правилу Марковникова
НХ \ / НХ II
Я_С=с-н —► )=( —► Я—I—|—н
х н 4 А
Алкины присоединяют к себе также некоторые другие вещества
кислого характера.
Характерным свойством алкинов является присоединение ими
воды в присутствии солей ртути - так называемая реакция Куче-
рова.
[СН2=СН 1
Ан ]
СН3—сн=о
виниловый ацетальдегид
Промежуточный продукт присоединения воды к ацетилену - ви¬
ниловый спирт - неустойчив; он изомеризуется в уксусный альде¬
гид - в соответствии с правилом Эльтекова, которое гласит, что
соединения с гидроксильной группой при двойной связи нестабиль¬
ны и преобразуются в карбонильные соединения. Правило Эльтеко¬
ва - проявление кето-енольной таутомерии (см. 24.3.2) для случая,
когда кетонная (альдегидная) таутомерная форма значительно
прочнее енольного таутомера.
Гомологи ацетилена присоединяют воду по правилу Марковни¬
кова, в результате чего образуются кетоны.
„ и НОН Г Я—С=СН21
Я—С=С—Н - ■—» —» Я—С—СН3 кетон
Ацетилен в присутствии СиС12 и МНз реагирует с синильной
кислотой; в результате реакции образуется акрилонитрил:
_ НСЫ
Н—С=С—Н —► СН2=СН—С—N акрилонитрил
В присутствии щелочи ацетилен может присоединять спирты:
НОС2Н5 этилвиниловый
Н-С=С-Н — СН2=СН-ОС2Н5 Эфир
362
Продеты такой реакции - виниловые эфиры - в отличие от
виниловых спиртов устойчивы. Этилвиниловый эфир применяют в
медицине для наркоза.
Присоединяются к ацетилену и карбоновые кислоты:
СН2=СН—ОССНз винилацетат
Например, присоединение уксусной кислоты к ацетилену (под
действием Н3РО4) дает винилацетат - мономер, используемый для
получения полимерных клеев (ПВА).
Полимеризация ацетилена. В присутствии водного раствора
хлорида меди и хлористого аммония ацетилен димеризуется в вини-
лацетилен.
сн2=сн-с=сн2 4— сн2=сн-с=сн «— пН—СЕС¬
ІЇ
винилацетилен
хлоропрен
[— СН=СН—] п
купрен
циклооктатетраен
Присоединение к тройной связи винилацетилена хлористого во¬
дорода приводит к хлоропрену, а водорода - к дивинилу. Оба алка-
диена используются в синтезе каучуков.
Нагревание ацетилена до 600° С в присутствии активированного
угля в качестве катализатора вызывает тримеризацию ацетилена с
образованием бензола.
Комплексные соли никеля инициируют тетрамеризацию ацети¬
лена. В результате этого процесса образуется циклооктатетраен.
При нагревании до 300° С в присутствии порошкообразной меди
ацетилен дает купрен - полимер, обладающий электроизоляцион¬
ными свойствами.
Реакции ацетиленового атома водорода. При переходе от
алканов к алкенам и к алкинам доля в-характера в гибридной орби¬
тали возрастает, длина связей С-Н уменьшается; в ацетилене они
соседствуют с атомами углерода теснее, чем в этилене и этане.
Увеличение доли в-орбитали в гибридизации приводит, таким обра¬
зом, к большей электроотрицательности атома углерода. Это обус¬
ловливает то, что ацетилен и его монозамещенные гомологи обла¬
дают свойствами слабых кислот
363
я—С=с—н
7-» Р-С=с' + н *
я—С=с—н
Р—С=С—№
♦ 1/2Н2
МН3
ацетиленид
Соли ацетилена и его гомологов - ацетилениды - легко гидроли¬
зуются и поэтому могут быть получены только в безводной среде,
например, в жидком аммиаке. Ацетилениды серебра и меди в воде
не растворимы, поэтому приведенный ниже процесс их образования
можно использовать как качественную реакцию на монозамещенные
ацетилены:
Ацетилениды широко используются в синтетических целях,
воздействуя на них галогенопроизводными, можно получать любые
гомологи ацетилена:
Ацетилениды тяжелых металлов легко взрываются и могут быть
использованы как детонаторы.
Электрофильное замещение. Простейший представитель
аренов, бензол, СвН6 - сильно ненасыщенное соединение, формально
его можно рассматривать как циклогексатриен.
Аналогично другим ненасыщенным соединениям - алкенам, ал-
кадиенам и алкинам - это вещество проявляет склонность к реак¬
циям с электрофильными реагентами. Однако, в отличие от пере¬
численных соединений, бензол вступает не в реакции присоедине¬
ния, а в реакции электрофильного замещения. Это объясняется
значительной энергией сопряжения в молекуле бензола, обуслов¬
ленной ароматической связью.
Характерным примером электрофильного замещения в ряду аре¬
нов может служить их галогенирование, катализаторами которого
обычно служат галогениды алюминия или железа. Роль катализато¬
ра в этой реакции заключается в образовании электрофильной час¬
тицы за счет поляризации молекулы галогена.
NN3
р_С=с—N3 +Х—Я, —► я_с=С-Р, + ЫаХ
18.3.5. Химические свойства аренов
364
СІ —СІ б- + АІ—СІ -
СІ—А|—СІ
СІ
Например, при галогенировании в присутствии хлористого алю¬
миния процесс состоит в обратимом донорно-акцепторном взаимо¬
действии атома алюминия (акцептор) с электронной парой одного
из атомов в молекуле хлора, в результате чего второй атом хлора
приобретает положительный заряд и оказывается способным к
электрофильному реагированию.
Электрофильная частица СГ далее взаимодействует с 671-орби¬
талью, что приводит к образованию на первой стадии процесса по¬
ложительно заряженного комплексного соединения - так назы¬
ваемого л-комплекса, в котором электрофильная частица координи¬
рована к электронной плотности всего кольца, а не к отдельным
атомам:
л-комплекс а-комплекс
Из шести электронов л-комплекса два образуют затем кова¬
лентную связь между электрофильной частицей С1+ и одним из
атомов углерода; остальные четыре электрона распределяются
между пятью атомами углерода. В итоге получается промежуточ¬
ный карбкатион - а-комплекс, который, теряя протон, переходит в
хлорбензол.
Суммарный процесс галогенирования ароматического соедине¬
ния можно отобразить так:
СІ
хлорбензол
Как видно из этого примера, электрофильное замещение в аре¬
нах принципиально отличается от электрофильного присоединения
к алкенам: происходит отщепление протона. В ароматическом ряду
электрофильное замещение не требует такой затраты энергии,
как присоединение, вызывающее разрушение стабильной бл-элек-
тронной системы.
365
Нитрование бензола также относится к реакции электрофильно-
го замещения. Оно осуществляется под действием так называемой
нитрующей смеси - смеси концентрированной азотной кислоты с
концентрированной серной кислотой В такой смеси идет реакция,
приводящая к образованию электрофильных частиц - катионов нит-
рония. Эти частицы атакуют молекулы бензола. Механизм реакции
аналогичен галогенированию и приведен ниже. Конечным продук¬
том реакции является нитробензол
НОГЮ2 ► N02+ + ОН-
нитроний-ион
л-комплекс о-комплекс
В общем виде реакцию можно записать так:
N02
О 6*1
нитробензол
Сульфирование аренов с образованием ароматических сульфо-
соединений происходит обычно при нагревании аренов совместно
с концентрированной серной кислотой.
©àSOзH Сульфирующим агентом служит серный
ангидрид БОз, в молекуле которого атом
серы обладает электрофильным характе¬
ром. Реакцию сульфирования можно запи-
сульфобензол сать общей схемой
Еще одним примером электрофильного
замещения в аренах является их алкилирование (реакция Фриделя-
Крафтса). Воздействуя на арены галогеноалкилами, можно получить
их гомологи.
Реакция происходит под действием галогенидов алюминия и со¬
стоит из нескольких последовательных фаз - возникновения карб-
катиона, образования я-комплекса, затем о-комплекса и отщепления
протона.
366
С2н5&<—С|5- + А1С13 —•> С2н5+[А1СЦГ
С2Н5
+ Н+
п-комплекс
о-комплекс этилбензол
Реакция алкенов с кислотами также дает карбкатионы. Поэтому
еще одним вариантом алкилирования аренов может быть их вза¬
имодействие с алкенами в присутствии кислых катализаторов.
Правила ориентации в электрофильном замещении. При
наличии в бензольном кольце хотя бы одного заместителя, молеку¬
ла соединения оказывается, с точки зрения электронного распреде¬
ления, несимметричной. Поэтому при электрофильном замещении
могут возникать три изомера - с орто-, пара- или мета-
расположением входящего заместителя относительно уже имеюще¬
гося. В связи с этим заместители разделяются на две группы.
Заместители первого рода обладают электронодонорным харак¬
тером (за исключением галогенов). Соединения, содержащие такие
заместители, в реакциях электрофильного замещения более актив¬
ны, чем бензол. Захватываемой электрофильной группе они прида¬
ют орто- или лара-ориентацию.
К числу таких заместителей относятся, в частности, алкильные
радикалы (-Р), гидроксильная группа (-ОН), алкоксильная
(-ОР), меркапто (-БН), аминогруппы (-МН2, -N4^ и галогены. Приме¬
ром проявления эффекта заместителей первого рода могут служить
продукты нитрования толуола - орто- и лара-нитротолуолы
X
X = Я, ОН, ОЯ, ГЖ2, МЯ, БН, На1
заместители первого рода
СНз
ЫОг
о- и л-нитротолуолы
367
Заместители второго рода обладают электроноакцепторными
свойствами. Соединения, их содержащие, менее активны, чем бен¬
зол. Наличие этих заместителей придает захватываемой электро-
фильной группе лета-ориентацию. К числу таких заместителей
относятся карбоксильная группа (-СООН), альдегидная группа (СНО),
нитро- (-N02), сульфо- (-БОзН) и циано-группа (-СМ).
СООН, СНО, N02, 503Н, СН,
заместители второго рода
Примером проявления эффекта
заместителя второго рода может
служить лета-нитросульфобензол,
образующийся при сульфировании
нитробензола.
Было бы ошибочно думать, что
все заместители действуют строго
избирательно. На самом деле это не так - их электронные эффекты
различны по силе. Поэтому в реакциях электрофильного замещения
возможно образование всех трех изомеров. Их соотношение будет
зависеть как от вида заместителя, так и от концентрации реагента,
от вида растворителя, катализатора, от температуры.
Например, при нитровании фенола образуются лишь орто- и
пара-изомеры (в соотношении 40% и 60%), а при нитровании
толуола - все три изомера: орто-, мета- и лара-нитротолуол
(56%, 4% и 40%, соответственно). При нитровании нитробензола
кроме лета-динитробензола (93%) образуется еще и орто-динит¬
робензол (7%).
Если в бензольном кольце заместителей больше, чем один, их
влияние может быть как согласованным, так и несогласованным, в
зависимости от их типа и расположения
Примером согласованной ориентации может служить нитрова¬
ние л-динитробензола. Пара заместителей второго рода в лета-
расположении придают захватываемой нитрогруппе также лета-
расположение, образуется 1,3,5-тринитробензол.
Примером несогласованной ориентации может служить нитро¬
вание орто-дихлорбензола, результатом которого является смесь
3-нитро- и 4-нитропроизводных.
Замещение в ароматическом ряду может быть не только элек-
трофильным; возможно и гомолитическое замещение, например,
у
м-нитросульфобензол
368
галогениромние. В процессах такого рода участвуют заместители
бензольного кольца. Например, под действием хлористого алюминия
можно осуществлять электрофильное хлорирование толуола с орто-
и лара-ориентацией в бензольном цикле, или под действием света
осуществлять хлорирование толуола в метильную группу.
Замещение в ароматическом ряду имеет важное практическое
значение. В синтетических целях оно осуществляется как в лабора¬
торных условиях, так и в промышленных масштабах. Примером
практического использования таких реакций служит синтез анти¬
микробного средства - стрептоцида.
ГШ2 ыНг
нозозС|. [<о>ґа
о- и п-сульфанилхлориды
При действии на анилин хлорсульфоновой кислотой по анало¬
гии с сульфированием имеет место сульфохлорирование, в резуль¬
тате чего получаются орто- и лара-сульфанилхлориды. Второй из
них после реакции с аммиаком дает пара-сульфаниламид, который
и является стрептоцидом. Его антимикробное действие основано на
молекулярном соответствии метаболиту микроорганизмов пара-
аминобензойной кислоте.
сульфамиды
Замещая последнее вещество в контакте с ферментом, стрепто¬
цид прерывает обмен веществ микроорганизма и тем самым убивает
его. Таким образом, стрептоцид является типичным антиметаболи¬
том. Антиметаболитами называют природные или синтетические
биологически активные соединения, близкие по строению метабо¬
литам и вступающие с ними в конкурентные отношения
369
Варьированием заместителей Я’-Я4 в молекуле стрептоцида син¬
тезируют его аналоги - сульфамиды, среди которых имеются сое¬
динения, обладающие некоторыми преимуществами перед стрепто¬
цидом, а потому нашедшие применение в медицинской практике.
К настоящему времени испытано несколько десятков тысяч сульфа¬
мидных препаратов, из числа которых около двадцати используют в
клинической практике.
Окисление гомологов бензола. В силу своей устойчивости
бензольный цикл в обычных условиях инертен даже по отношению
к таким сильным окислителям, как азотная кислота, перманганат
калия, перекись водорода. Поэтому обработка гомологов бензола
сильными окислителями "сжигает" лишь боковые цепи, то есть ал¬
кильные радикалы; от них остается атом углерода, непосредственно
связанный с бензольным кольцом и порождающий карбоксильную
группу -СООН.
При окислении гомологов бензола с одной боковой ветвью
образуется бензолкарбоновая или бензойная кислота.
При окислении гомологов бензола с двумя боковыми цепями об¬
разуются орто-, мета-, или лара-бензолдикарбоновые кислоты
(фталевые); при окислении гомологов с тремя цепями - трикарбо-
новые кислоты и т. д.
По числу и взаимной ориентации карбоксильных групп в про¬
дуктах окисления гомологов бензола можно судить о расположении
370
СН3
СООН
СНз,
ЧСН‘
-СН3
бензойная кислота
СНз
СООН
о- фталевая кислота
м-фталевая кислота
СНз
СООН
СН3' 'СНз
л-фталевая кислота
он
0 = 6
заместителей в исходном соединении; это обстоятельство может
быть использовано в аналитических целях.
При биологическом окислении арены ведут себя аналогично:
попадая в живой организм, гомологи бензола превращаются в соот¬
ветствующие карбоновые кислоты. Сам же бензол подвергается
медленному С-гидроксилированию с образо- он
ванием фенола, обладающего токсическим
действием. В этом - особо сильное токсичное
действие бензола по сравнению с другими
аренами. Бензол накапливается в организме,
т.е. является кумулятивным ядом.
Бензол в реакциях присоединения. Необратимое разруше¬
ние ароматической связи бензольного цикла происходит лишь при
взаимодействии с реагентами, богатыми энергией, или при высокой
температуре, когда бензол сгорает до углекислого газа и воды.
Кислород воздуха при температуре 450° С в присутствии ката¬
лизатора - оксида ванадия 0/2О5) - разрушает бензол по схеме:
° Ъ + 2С02 +2НгО
Продукт реакции - малеиновый ангидрид применяется для про¬
изводства некоторых пластмасс; реакция используется в промыш¬
ленности.
Хлорирование бензола идет при сильном освещении, вызываю¬
щем образование радикалов хлора. Продуктом реакции является
1,2,3,4,5,6-гексахлорциклогексан, часто называемый гексахлораном'
ояоа:ф:
циклогексан гексахлоран
Гексахлоран является эффективным и дешевым инсектицидом,
однако, он токсичен, подобно другим хлорсодержащим инсектици¬
дам (глава 19). Поэтому делаются попытки отыскать в замену ему
более совершенные препараты
Гидрирование бензола осуществляется лишь при нагревании
под давлением в присутствии никелевых или платиновых катализа¬
371
торов. Продукт реакции - циклогексан - служит сырьем дпя синтеза
капрона
Химические свойства нафталина. Особенности химического
поведения конденсированных аналогов бензола рассмотрим на
примере их простейшего представителя - нафталина.
8 1 а а
5 4 а а
В молекуле нафталина одна из пар я-электронов, создающих
ароматическую связь, является общей для того и другого кольца.
Поэтому энергия сопряжения у нафталина меньше, чем у бензола,
и химически он более активен.
Неодинаковое участие я-электронов углеродных атомов цикла в
образовании ароматической связи приводит к неравномерному рас¬
пределению электронной плотности в кольце Рентгенографический
анализ показывает, что и длины связей кольца неодинаковы. В
частности, длина а,Р-связи меньше длины бензольной связи, а дли¬
на Р,(3-связи больше.
Отличается также поведение атомов углерода в реакциях элек-
трофильного замещения: атомы 1, 4, 5 и 8 в реакциях такого типа
ведут себя активнее, чем атомы 2, 3, 6 и 7.
Электрофильное замещение в нафталиновом ряду - галогениро-
вание, нитрование, сульфирование, алкилирование - идет в сравни¬
тельно мягких условиях. Заместитель при этом взаимодействует
преимущественно с а-атомами углерода Так образуется, например,
а-нитронафталин:
а-нитронафталин
Правила ориентации заместителя в соединениях нафталина
несколько сложнее правил ориентации заместителей в производных
бензола.
Если молекула нафталинового соединения содержит замести¬
тель первого рода, то этот заместитель активирует цикл, к которому
принадлежит, и электрофильный реагент атакует один из а-атомов
углерода этого цикла.
372
Пример-такого случая - нитрование Р-метилнафталина:
N02
1-нитро-2-метил- 1-нитро- N02
нафталин 3-метилнафталин
,СН3
Присутствие заместителя второго рода дезактивирует цикл; ак¬
тивную роль в электрофильном замещении играют углеродные ато¬
мы смежного цикла, занимающие а-положение. В результате могут
образоваться два изомера. Так, из а-сульфонафталина при нитрова¬
нии образуются 1-сульфо-5-нитронафталин и 1-сульфо-8-нитро-
нафталин:
Различие в химической активности а- и Р-положений нафтали¬
нового цикла незначительно. Поэтому в жестких условиях реакции
замещения сопровождаются образованием заметного количества
Р-изомеров; Р-изомеры к тому же более стабильны, при нагревании
происходит переход а-изомеров в более устойчивые Р-изомеры.
Подбором условий можно синтезировать любой желаемый продукт
Гидрирование нафталина - частный случай реакции присоеди¬
нения Реакция состоит из нескольких стадий Сначала одно из ко¬
лец, выступая в роли сопряженного диена, присоединяет молекулу
водорода по 1,4-типу (в а-позиции) (при температуре 160° С и дав¬
лении 30 атм.), теряя тем самым ароматичность.
Образующийся при этом циклоалкен - 1,4-дигидронафталин -
легко присоединяет к себе по я-связи вторую молекулу водорода,
превращаясь в тетрагидронафталин (тетралин) При температуре
БОзН
ЭОзН N02 ЭОзН
+ НгО
N02
1,5- и 1,8-нитросульфонафталины
тетралин
декалин
373
250 - 300° С тетралин может присоединить к себе еще три молеку¬
лы водорода, превратившись в насыщенный декагидронафталин
(декалин)
Окисление нафталина происходит легче, чем окисление бензола
При этом затрагивается одно из колец, оно теряет ароматичность и
"сгорает" - как боковые цепи при окислении гомологов бензола
Продуктом окисления является орто-фталевая кислота, используе¬
мая в промышленности в синтетических целях.
о-фтапевая кислота
У замещенных нафталинов будет окисляться кольцо, содержа¬
щее заместитель первого рода. Заместители второго рода, напротив,
стабилизируют то кольцо, к которому присоединены.
Конденсированные арены, молекулы которых включают в себя
более двух колец, в общем обладают аналогичными химическими
тенденциями, а именно, склонны к электрофильному замещению и
плохо вступают в реакции присоединения (правила ориентации за¬
местителей в таких молекулах усложняются еще больше).
Таким образом, химические свойства углеводородов определя¬
ются особенностями строения их молекул. Для алканов, молекулы
которых построены на прочных о-связях, реакции с ионными или
сильно полярными реагентами не характерны. Они взаимодейству¬
ют с реагентами, богатыми энергией, вступая в реакции гомолити-
ческого замещения или подвергаясь окислению. Алкены, алкадиены
и алкины отличаются непрочными я-связями, легко поляризуемыми
р-электронами Поэтому их характерным свойством является элек-
трофильное присоединение. Ароматическая связь в аренах придает
им активность во взаимодействии с электрофильными реаген¬
тами, однако они больше предрасположены к реакциям замеще¬
ния, а присоединение происходит только в случае весьма активных
реагентов
Разнообразие химических свойств углеводородов объясняет
существование широкого спектра их производных, находящих все¬
возможное практическое применение. Свойства углеводородов об¬
общены в таблице 18.2.
Углеводороды в природе. Одним из естественных источников
углеводородов является природный газ, состоящий почти полностью
из метана.
374
Таблица 18.2
Свойства углеводородов
Тип, гибри¬
дизация
Механизм
реакций
Пример
Биологическое
значение
алкан, эр3
гомолитическое
галогенирование.
С-окисление
замещение
нитрование,
сульфирование
как путь
биосинтеза и
трансформации
ксенобиотиков
алкен,
электрофил ьное
гидрирование,
биосинтез
алкадиен,
присоединение
галогенирование.
гликолей.
алкин;
гидрогалогенирование,
канцерогенез
эр2, эр
полимеризация,
окисление
бензпирена
арен;
электрофильное
галогенирование.
С-окисление
зр2
замещение
нитрование,
сульфирование
как путь
биосинтеза и
трансформации
ксенобиотиков
Другим богатым источником углеводородов является нефть,
представляющая собой сложную смесь алканов, циклоалканов
(циклопентан, циклогексан и их гомологи), аренов (бензол, толуол,
диметилбензол и их гомологи), а также конденсированных углево¬
дородов (нафталин и его гомологи). В некоторых разновидностях
нефти содержатся еще и полиэдрические углеводороды, в част¬
ности, адамантан. Всего из нефти выделено около двухсот пятиде¬
сяти веществ, в том числе и неуглеводородного строения Как об¬
разуется нефть, науке пока не известно
Выделение из нефти отдельных компонентов основано на ис¬
пользовании особенностей физических свойств углеводородов По¬
скольку основой их молекул являются малополярные ковалентные
связи, они не проявляют склонности к межмолекулярным взаимо¬
действиям. Как следствие этого, им присущи относительно низкие
температуры кипения и плавления
Простейшие алканы, циклоалканы, алкены, алкадиены и алки-
ны, углеродная цепь которых содержит не более пяти атомов угле¬
рода, представляют собой при обычных условиях газы (исключая
бутин-2). Большинство компонентов, входящих в состав нефти, яв¬
ляются веществами жидкими.
Попутные газы нефти - это еще один источник углеводородов,
здесь присутствуют метан, этан, пропан и бутан
375
Простейшим способом пеработки нефти является её фракцион¬
ная перегонка Основными фракциями являются бензин, керосин н
дизельное топливо Остаток перегонки - мазут - применяют как ко¬
тельное топливо, а также для приготовления вазелина, смазочных
масел, гудрона; последний после соответствующей обработки ис¬
пользуется как асфальт.
По мере возрастания молекулярного веса углеводородов их тем¬
пература плавления повышается. Алканы, начиная с су¬
ществуют в твердом состоянии при обычной температуре, смесь та¬
ких веществ называют парафином Парафин применяют в медицине
в виде компрессов и ванн. Температура плавления углеводородов
зависит также от того, насколько плотно упакованы молекулы в
кристаллической решетке; поэтому углеводородам с более развет¬
вленной углеродной цепью соответствует более высокая температу¬
ра плавления Например, н-алкан, декан С10Н22 - вещество жидкое;
нафталин СюНв, с тем же числом углеродных атомов, становится
жидким при температуре 80°; адамантан СюН« плавится лишь при
нагревании до 269°.
Будучи слабо поляризованными веществами, углеводороды пло¬
хо растворимы в воде. Они, однако, липофильны. Поэтому углево¬
дороды широко применяются в качестве растворителей в процессах
синтеза и в процессах экстракции природных веществ - эфирных
масел, жиров и пр
Благодаря высокой липидной растворимости углеводороды, по¬
падая в живой организм, концентрируются в жировых тканях, а
также в оболочках нервных клеток, что приводит к снижению
нервной чувствительности. Этим объясняется наркотическое дей¬
ствие углеводородов (и вообще всех неэлектролитов). В отдельных
случаях, например, у циклопропана наркотический эффект весьма
значителен. Поэтому циклопропан иногда используют для общего
наркоза.
В организме углеводороды подвергаются С-гидроксилированию и
выводятся из клетки. Однако некоторые углеводороды, например,
бензол, окисляются очень медленно и оказываются, таким образом,
кумулятивными ядами С увеличением числа циклов в молекуле
арена возрастает не только его токсичность, но также и его канце¬
рогенная активность.
С физическими свойствами углеводородов связано существова¬
ние ряда экологических проблем. Важнейшей из них представляет¬
ся морская транспортировка нефти. Нефть, попадая по различным
причинам в воду, остается на её поверхности и, концентрируясь в
прибрежной зоне, богатой разнообразными живыми организмами,
вызывает необратимое нарушение экологического баланса.
376
К химическим способам обработки нефти относятся: крекинг,
дегидрирование, ароматизация и изомеризация. В сочетании с
перегонкой и гидрированием они позволяют получать те или иные
специфические продукты, используемые не только как топливо,
но и для целей органического синтеза. В развитие нефтеперера¬
ботки фундаментальный вклад внес выдающийся русский химик
В. Н. Ипатьев.
Нефть, таким образом, является важным комплексным сырьем,
запасы которого, к сожалению, ограничены.
В последнее время все большее внимание уделяется так назы¬
ваемой микробиологической обработке нефти, основанной на изби¬
рательном усваивании углеводородов некоторыми микроорганизма¬
ми, что позволяет получать из нефти белковое сырье.
Богатым источником углеводородов является каменноугольная
смола - побочный продукт коксования углей. Из неё в большом
количестве извлекают бензол, толуол, стирол, нафталин и его ана¬
логи - атрацен, фенантрен и др.
В большом количестве извлекают углеводороды и из раститель¬
ных источников. Так, добыча природного каучука из латекса со¬
ставляет в настоящее время около 3 млн тонн в год.
Многие растительные масла также содержат углеводороды, в
основном терпены. Примером смеси терпенов является скипи¬
дар - продукт перегонки смолы хвойных растений. Кроме углеводо¬
родов в состав таких масел входят терпеноиды - соединения, со¬
держащие спиртовые, альдегидные и кетонные группировки - в том
числе важные для медицины ментол и камфора. Характерным
представителем терпеновых углеводородов является лимонен С10Н1в.
Терпены широко используются для приготовления лаков и красок.
В соке моркови, томатов, шиповника содержатся углеводороды
ликопин и а,Р,у-каротины (С4оН5в).
Последние являются провитаминами А - веществами, которые
в организме человека превращаются в витамин А Молекулы кау¬
чука, терпеновых углеводородов, каротинов и ликопина построены
из чередующихся фрагментов, повторяющих скелет молекулы изо¬
прена. Поэтому их называют изопреноидами. К их числу относятся
также стероиды - основа гормонов
377
18.3.6. Синтез углеводородов
Алканы, циклоалканы. Для получения этих углеводородов
помимо нефтепереработки используются следующие процессы
а) Гидрогенизация ненасыщенных углеводородов - алкенов, ал-
кадиенов, алкинов, аренов - с сохранением длины углеродной цепи
исходного углеводорода
СпНгп-2 + Н2 —» СпН2п + Н2 —» СпН2п+2
алкин (алкадиен) алкен алкан
СбНе + ЗН2 -> СвН12
бензол циклогексан
б) Восстановление галогенопроизводных и кислородных соеди¬
нений - спиртов, альдегидов, кетонов, кислот - под действием йо¬
дистого водорода, гидридов металлов, гидразина и других восстано¬
вителей. Например:
Р-1 + Н-1 -> Р-Н + 12
4Р-ОН + УА1Н4 -> 4Р-Н + УОН + А1(ОН)3
в) Декарбоксилирование карбоновых кислот, осуществляемое в
лаборатории при сплавлении карбоновых кислот со щелочами:
Р-СООМа + МаОН -> Р-Н + Ма2СОэ
Эта же реакция происходит ферментативным путем в живом
организме.
Р-СООН -» Р-Н + С02
г) Углеродно-водородный синтез. Под воздействием вольтовой
дуги угольные электроды в атмосфере водорода превращаются в
метан:
С + 2Н2 -» СН4
В определенных условиях - давление, температура, катализа¬
тор - непосредственно из углерода и водорода можно получить
сложную смесь н-алканов, так называемый синтин - синтетический
бензин:
пС + (п +1)Н2 -» СпН2п+2
Синтетический бензин можно получить также гидрированием
окиси углерода при температуре 300° С в присутствии никелевого
катализатора. Этот процесс перспективен в странах, не имеющих
собственных запасов нефти.
378
д) Реакция Вюрца (1855 г.). Галогенопроизводные, при дейст¬
вии на них металлическим натрием дают соединения с "удвоенным"
углеводородным скелетом:
R-l + 2Na + l-R -> R-R + 2Nal.
При использовании смеси двух галогенопроизводных получают¬
ся три различных продукта:
3R-I + 6Na + 3I-R’ -> R-R + R’-R’ + R-R’ + 6Nal.
Разновидностью реакции Вюрца является взаимодействие цинка
с дигалогенопроизводными, в результате чего образуются цикло-
алканы:
1-(СН2)4-1 + Zn -> C4He + Znl2
циклобутан
Среди рассмотренных способов получения углеводородов про¬
мышленное значение имеют в основном гидрогенизация и крекинг.
В странах, не имеющих собственной нефти, практикуется производ¬
ство синтина.
Алкены, алкадиены. Основным промышленным способом по¬
лучения алкенов является крекинг нефти. Этим способом получают
ненасыщенные углеводороды с небольшой цепью - этилен, пропен,
бутены и пентены.
Вторым по важности является дегидрирование алканов, осуще¬
ствляемое при температуре 450° С в присутствии катализатора -
оксида хрома(Ш):
СпН2п+2 -» СпН2п + Н2
С увеличением температуры до 550 - 600° С алкены подверга¬
ются дальнейшему дегидрированию, образуя алкадиены. Именно так
производится основная масса дивинила (из бутана или бутена) и
изопрена (из 2-метилбутана) - сырья для получения синтетического
каучука
При лабораторном получении алкенов используются реакции
отщепления - дегидратация спиртов, осуществляемая в присутствии
серной (или фосфорной) кислоты или нагретой окиси алюминия, и
реакция дегидрогалогенирования галогеналканов под действием
спиртового раствора щелочи
Эти реакции удовлетворяют правилу Зайцева (1875 г), со¬
гласно которому водород уходит от того атома углерода, который
наиболее беден водородом
379
СН3-СН-СН2—СНз , сн3-сн=сн—сн3
Ан -н’°
КОН, спирт
СНз-СН—сн2—СНз ► сн3-сн=сн—СНз
Аг -НВг
Дегалогенирование вицинальных дигалогенопроизводных под
действием цинка - еще один способ получения алкенов:
рснх-снгх + гп -> Рсн=сн2 + гпХ2
Алкины. Основными промышленными способами получения
ацетилена является термический крекинг метана и окислительный
пиролиз метана - его сжигание при недостатке кислорода.
2СН4 (2000 °С) С2Н2+ ЗН2
4СН4 + 02 —» С2Н2 + 7Н2 + 2СО
Исторически более ранним промышленным способом получения
ацетилена является карбидный способ, основанный на разложении
карбида кальция водой. Однако этот метод дорогостоящий, поэтому
он утратил свое промышленное значение.
Гомологи ацетилена синтезируют взаимодействием ацетилени-
дов и галогеналканов.
Еще одним способом получения алкинов является дегидрогало-
генирование дигалогенопроизводных под действием спиртового рас¬
твора щелочи, согласно правилу Зайцева:
СН3—СВг2—сн2—сн3 ,КЯН-спирт, сн3—С =с —сн3
-2 НВг
Арены. Арены можно получать путем каталитической аромати¬
зации алканов и производных циклогексана - при температуре 450 -
500° С, под давлением и в присутствии оксида алюминия:
СпНгп+2 -» СпНгп-е + 4Н2
Бензол можно синтезировать путем тримеризации ацетилена.
Для синтеза гомологов бензола используется реакция Фриделя-
Крафтса.
Декарбоксилирование ароматических карбоновых кислот также
может служить способом получения бензола:
С6Н5-СООН СбНе + С02
бензойная кислота бензол
380
Таким образом, основными источниками углеводородов являют¬
ся природный газ (4лканы С1-С4) и нефть (алканы, циклоалканы,
арены), а также каменноугольная смола (арены).
18.4. Химические свойства гетаренов
Наличие ароматической связи в пяти- и шестичленных ненасы¬
щенных гетероциклах придает им свойства, подобные свойствам их
углеводородных аналогов - аренов. В частности, для них тоже не
характерны реакции присоединения; гидрирование этих соединений,
как и гидрирование бензола, осуществляется лишь в жестких усло¬
виях (температура, давление, катализатор). Например, фуран -
пятичленный гетероцикл - присоединяет к себе водород при повы¬
шенной температуре и давлении. Аналогичным образом в жестких
условиях присоединяет к себе водород пиридин - шестичленный
азотистый гетероцикл.
Устойчивость ароматического кольца гетероциклов проявляется
не только в их инертности по отношению к реакциям присоедине¬
ния, но и в поведении по отношению к окислителям. В реакциях
окисления гетероциклов, подобно окислению гомологов бензола,
разрушаются цепочки заместителей с образованием группы СООН,
ароматический же гетероцикл остается без изменения Примером
может служить окисление р-этилпиридина. Оно приводит к образо¬
ванию р-пиридинкарбоновой кислоты, известной под названием ни¬
котиновой кислоты.
Эта же кислота получается, как это можно ожидать, и при
окислении алкалоидов-изомеров никотина и анабазина. Полезно об¬
ратить внимание на то, что эти вещества имеют хиральный центр
(отмечен звездочкой).
Азотистые гетероциклические соединения растительного проис¬
хождения и обладающие физиологической активностью называют
алкалоидами Вещества эти в больших дозах весьма токсичны
для человека и для животных, но в небольших количествах многие
тетрагид рофуран
Н
пиперидин
381
из них используются в медицине, например, хинин (производное
хинолина), резерпин, стрихнин (производные' индола). Сейчас из¬
вестно о существовании нескольких тысяч алкалоидов.
Для пятичленных ароматических гетероциклов (фуран, тиофен,
пиррол) характерны реакции электрофильного замещения. В ка¬
честве примера можно назвать реакции галогенирования, нитрова¬
ния и сульфирования тиофена:
| НОЫОг
N02 + НОН
Наличие в молекулах азотистых шестичленных гетероциклов
зр2-гибридизированного атома азота, более электроотрицательного,
чем атомы углерода, приводит к понижению электронной плотности
гетероцикла и к ослаблению его активности в реакциях электро¬
фильного замещения. Атом азота в таком цикле можно уподобить
электроноакцепторному заместителю второго рода в молекуле бен¬
зола. В соответствии с влиянием такого заместителя, в молекуле
пиридина, например, обеднены электронами а- и упозиции. Поэто¬
му электрофильное замещение должно происходить с р-ориентаци-
ей, причем в жестких условиях. Соответственно, реакции галогени¬
рования, нитрования и сульфирования пиридина идут с незначи¬
тельным выходом.
382
| ножэ2
Шестичленные гетероциклы с двумя атомами азота - пирими¬
дин и пиразин - электрофильному замещению не подвержены
вообще.
Вследствие того, что в молекуле пиридина атомы углерода, за¬
нимающие места 2, 4 и 6, обеднены электронами, у них появляется
возможность участвовать в реакциях нуклеофильного замещения.
Еще сильнее проявляется это свойство у пиразина и пиримидина.
Примером нуклеофильного замещения может служить амини-
рование пиридина амидом натрия, именуемое также реакцией
Чичибабина.
Таким образом, комбинации атомов углерода различной гибри¬
дизации, вариация длины и разветвленности углеродных цепей и
колец и замены углеродных атомов на гетероатомы создают основу
для существования всего многообразия органических веществ.
N42
а- и у-аминопиридины
Глава 19. ГАЛОГЕНЫ
Нет других элементов со
столь развитыми кислотными
свойствами, как галоиды
Д И Менделеев
Галогены (gals - по гречески соль, genes - рождать) составляют
7-ю группу Периодической системы и имеют П52пр5-конфигурацию
Первым из них был открыт хлор (Шееле, Швеция, 1774 год).
В 1811 году француз Куртуа открыл иод, во Франции же были
открыты бром (Балар, 1826 год) и фтор (Муассан, 1886 год).
Астат - коротко живущий радиоактивный элемент, его известный
изотоп 2l0At распадается наполовину за 8.5 часов В земной коре
его содержится не более 30 г. Практического значения не имеет,
изучен мало.
19.1. Элементы, простые вещества
Галогены находятся в предпоследней группе соответствующих
периодов и являются наиболее типичными неметаллами, о чем сви¬
детельствуют величины их относительных электроотрицательностей
(табл. 19.1). Им присуща тенденция к присоединению электрона
с превращением в галогенид-анионы, что выражается полуреакцией
Х2 + 2е —» 2Х-, редокс-потенциал которой (табл. 19.1) указывает на
высокие окислительные способности галогенов. При этом величина
потенциала уменьшается сверху вниз параллельно уменьшению
величины ОЭО. В согласии с указанными особенностями галогены
в природе находятся в виде солей - фторидов, хлоридов, бромидов
и иодидов, что и определяет их общее название.
Общие сведения о галогенах Таблица 19.1
Элемент
ОЭО
Ф°, в
F
4.00
2.85
зубная эмаль
CI
2.90
1.36
СГ осмотический фактор,
HCI - желудочный сок
Вг
2.74
1.09
транквилизатор
1
2.21
0.53
щитовидная железа
384
Фтор^ своих соединениях как самый электроотрицательный
элемент всегда имеет степень окисления -1. В отличие от него дру¬
гие галогены с меньшей величиной электроотрицательности, с од¬
ной стороны, и d-подуровнями, с другой, могут приобретать наряду
со степенью окисления -1 и положительные степени окисления от
+ 1 до +7 (прежде всего в соединениях с кислородом).
Галогены играют важную роль в жизненных процессах. Фтор
следует отнести к микробиоэлементам - содержание этого элемента
в организме человека всего несколько миллиграммов. Однако инте¬
рес к этому элементу не ослабевает, прежде всего у стоматологов,
так как он концентрируется в первую очередь в зубах (в меньшей
степени в костях и ногтях). Основная часть фтора зубов входит в
состав зубной эмали в виде трудно растворимого фторапатита
CasF(P04)3. Избыток фтора в организме вызывает заболевание,
называемое флуорозом. При этом зубная эмаль и костная ткань
становятся хрупкими, а организм испытывает истощение.
В организме человека содержится около 100 г хлора, сущест¬
вующего в виде хлорид-иона, а именно хлористого натрия плазмы
крови и соляной кислоты желудочного сока. Хлористый натрий в
составе плазмы играет важнейшую роль в поддержании водно¬
электролитного баланса посредством создаваемого с его участием
осмотического давления. Суточная потребность организма в хлори¬
де натрия составляет до 10 г. Проблемы правильного питания, осо¬
бенно при сердечно-сосудистых заболеваниях, требуют контроля за
поступлением хлористого натрия в организм. Хлорид натрия необ¬
ходим для ферментативного синтеза соляной кислоты стенками
желудка по уравнению-
Н2СОз + СГ-» НСОз” + HCI.
Соляная кислота выполняет каталитическую роль в гидролизе
пептидных связей белков, а также имеет дезинфицирующее дей¬
ствие, так как высокая кислотность убивает микроорганизмы
Хлорсодержащие биомолекулы у высших организмов неиз¬
вестны, однако два атома хлора входят в состав антибиотика лево-
мицетина. Антибиотики - вещества биологического происхожде¬
ния, синтезируемые микроорганизмами и подавляющие рост бакте¬
рий и других микроорганизмов, а также вирусов и клеток.
Бром относится к микробиоэлементам (его количество в чело¬
веческом организме - несколько миллиграммов) Он концентриру¬
ется в железах внутренней секреции, в первую очередь в гипофизе
Роль брома в биопроцессах не ясна Однако хорошо изучено
действие бромид-иона на центральную нервную систему, которая
13 Зек. 675
385
высоко чувствительна к присутствию этого иона. Бромид-ион ока¬
зывает успокаивающее действие при повышенной возбудимости
Передозировка бромидных препаратов вызывает явление “бромиз-
ма” - хронического отравления этим ионом. Причина токсичности
бромид-иона заключается в его малой проницаемости через клеточ¬
ные мембраны из-за сравнительно большого по отношению к хло-
рид-иону размера, в результате чего скорость его выведения из ор¬
ганизма невысока. Для снижения токсического воздействия бромид-
иона на основе принципа Ле Шателье применяют введение избы¬
точных количеств хлорида натрия, что приводит к вытеснению бро-
мид-ионов из тканей и выведению их почками.
Больше известно о биологической функции иода, количество ко¬
торого в организме составляет около 25 миллиграммов. Более поло¬
вины находится в щитовидной железе в виде иодсодержащих гор¬
монов этой железы. Понижение ее активности вызывает заболева¬
ние гипотиреоз, а недостаток иода в пище и питьевой воде является
причиной другой болезни - эндемического зоба. Естественно, что
для лечения этих заболеваний применяют иодсодержащие препара¬
ты - иодиды натрия и калия. Остальная часть иода, содержащегося
в организме, по-видимому, необходима для биосинтеза некоторых
белков и липидов.
Таким образом, галогены являются незаменимыми для жизне¬
деятельности элементами. Следует обратить внимание на то об¬
стоятельство, что из-за сходства в свойствах галогены в биосредах
могут заменять друг друга, что может вызывать неожиданные эф¬
фекты как антагонизма в действии их ионов, так и явления усиле¬
ния действия (синергизма).
Поскольку галогены в природе существуют в виде галогенид-
ионов, получение свободных галогенов заключается в действии
окислителей на эти ионы, причем редокс-потенциал окислителей
должен превосходить потенциал самих галогенов (табл. 19.1).
В соответствии с этим не может существовать окислителя, способ¬
ного превратить фторид-ион в свободный фтор, так как фтор -
самый сильный окислитель. Следовательно, фтор синтезируют элек¬
тролизом фторидов в виде их расплавов (а не водных растворов,
чтобы избежать электролиза воды).
Проще обстоит дело с остальными галогенами, что видно из ре¬
акций получения хлора и брома:
2КМп04 + 16НС1 -» 5С12 + 2МпС12 + 2КС1 + 8Н20
Ф° =+1.51 В
Мп04 +8Н+
Мп2++4НгО
386
К2(3^р7 + 6КВг + 7Н2804 -» ЗВг2 + Сг2(804)э + 41^804 + 7Н20
ф° , =+1.36 В
Сг2072~+14Н+
2Сг3++7Н20
В этом отношении представляет интерес реакция СГ-иона с пе¬
роксидом водорода. Полуреакция пероксида водорода как окислите¬
ля в кислой среде выглядит следующим образом:
Н202+ 2Н++ 2е И 2Н20 ф° =+1.78 В
Н20?+2Н+
2Н20
Спрашивается, будет ли происходить образование хлора в плаз¬
ме крови, где присутствует в достаточных количествах хлорид-ион,
если известно, что пероксид водорода является метаболитом и не¬
прерывно образуется в плазме в процессе биотрансформации
кислорода.
Расчет по уравнению Нернста и сравнение полученного ре¬
зультата с данными таблицы 19 1, показывают, что в этих усло¬
виях потенциалы полуреакций практически равны (при усло¬
вии одномолярных концентраций всех веществ, за исключением
протонов).
ф = 1.78 - 0.059 pH (pH = 7.4 ф = 1.34 В)
Следовательно существует реальное равновесие, положение ко¬
торого в условиях организма определяется малой концентрацией
перекиси водорода, иными словами, образование хлора не происхо¬
дит. Когда среда становится более кислой, потенциал пероксида
возрастает.
Повышенную кислотность создают анаэробные бактерии, чей
метаболизм приводит к образованию ряда органических кислот. По¬
этому в ближайшем окружении таких бактерий создается кислая
среда. Это обстоятельство лежит в основе антибактериального дей¬
ствия лейкоцитов, которые атакуют анаэробные микробы при
их попаданиии в кровяное русло, а способствует этому то, что
стенки лейкоцитов содержат фермент пероксидазу, благоприят¬
ствующий продуцированию пероксида водорода. Таким образом,
в месте контакта лейкоцита с анаэробной бактерией произойдет
реакция:
2СГ + Н202 + 2Н+ 5 С12 + 2Н20,
в результате которой выделится хлор, убивающий микроорганизм
13* 387
Галогены широко применяются в промышлености. Достаточно
перечислить лишь некоторые области применения хлора Его ис¬
пользуют для получения соляной кислоты, некоторых хлоридов, на¬
пример, хлористого алюминия, солей хлорноватистой кислоты и
многочисленных дезинфицирующих и отбеливающих средств, хлор¬
новатой и хлорной кислот, брома, титана, германия, эмульгаторов
и детергентов, хлорированных углеводородов, применяемых в ка¬
честве растворителей, фосгена (из которого синтезируют полиуре¬
таны), хлоркаучука, различных пестицидов, монохлорукусной кис¬
лоты и многих других веществ.
Галогены в свободном виде чрезвычайно токсичны из-за их
сильного окислительного действия. Это определило применение
хлора в качестве боевого отравляющего вещества. Однако умень¬
шение окислительной активности при переходе к менее активному
иоду или использование хлора в малых концентрациях позволяет
употреблять их в качестве дезинфицирующих средств. Хлор приме¬
няют для дезинфекции воды, а йодную спиртовую настойку - для
обеззараживания ран.
19.2. Галогенид-ионы
Свойства соединений, в состав которых входят галогены со
степенью окисления -1, можно сопоставить на примере хлоридов
элементов III периода (табл. 19.2).
Хлориды элементов III периода
Таблица 19.2
Соединение
№С1
МдС12
А1С1Э
асц
РС1$
эось
ОЭО
элемента
0.9
1.2
1.5
1.8
2.1
2.5
Б-Элементы образуют соли в соответствии с их электроположи¬
тельными свойствами. Галогениды элементов 3-й группы занимают
промежуточное положение между ионными и ковалентными соеди¬
нениями. Начиная же с элементов 4-й группы, в галогенидах при¬
сутствует полярная ковалентная связь. Это наглядно видно по от¬
ношению галогенидов к гидролизу. Хлориды элементов 1-й группы,
как соли сильных оснований и сильной соляной кислоты, не гидро-
388
лизуютсЯ^<Хлорид магния как соль, образованная основанием сред¬
ней силы, подвергается слабому гидролизу при кипячении:
МдС12 + НОН 5 Мд(ОН)С1 + НС1.
Хлорид алюминия гидролизован в сильной степени уже на холоду:
А1С13 + НОН £* А1(ОН)С12 + НС1
А1(ОН)С12 + НОН £* А1(ОН)2С1 + НС1.
Хлориды элементов 4-6-й групп быстро и необратимо гидроли¬
зуются с образованием двух кислот: соляной и кислоты, соответ¬
ствующей элементу в составе хлорида.
ЭЮЦ + 2НОН -» ЭЮ2 + 4НС1
РС15 + 4НОН -» Н3Р04 + 5НС1
ЭОгСЬ + 2НОН -» Н2Э04 + 2НС1.
Иными словами, соединения галогенов с неметаллами ведут
себя по отношению к воде сходно с ангидридами, также образую¬
щими соответствующие кислоты. Это позволяет выделить все сое¬
динения, которые формально можно произвести заменой атомов
кислорода и (или) гидроксильных групп в кислотах на галогены,
в отдельную разновидность веществ, именуемую галогенангид-
ридами.
Тогда круг галогенангидридов для многих элементов должен
быть расширен. Так, для углерода кроме ССЦ галогенангидридом яв¬
ляется и фосген СОС12; для фосфора существует хлорокись фосфора
РОС13. Хлористый сульфурил ЭОгСЬ - не единственный хлорангид-
рид серной кислоты, существует и соединение 8С1б.
Все галогенангидриды объединены одним общим свойством -
гидролизом по связи Э-Х (X - галоген) по уравнению:
Э-Х + НОН -» Э-ОН + Н-Х.
Особый интерес представляют собой соединения галогенов с
водородом - галогеноводороды, некоторые свойства которых сведе¬
ны в таблицу 19.3.
Галогеноводороды можно получать несколькими способами.
Один из них - прямой синтез из элементов
Н2 + х2 ^ 2НХ
389
пригоден для получения фтористого и хлористого водорода, так как
эти реакции сильно экзотермичны, что видно из данных табл. 19.3:
Н2 + -» № Н2 + С12 —2НСІ
Реакция водорода со фтором происходит со взрывом уже в тем¬
ноте и на холоду Взаимодействие водорода с хлором также идет
энергично, но требует инициирования светом. Синтез бромистого
водорода осуществляется лишь при нагревании, а эндотермический
процесс образования иодистого водорода равновесен, и получать его
таким образом нецелесообразно:
Н2 + Вг2 !—> 2НВг Н2 + І2 і* 2НІ
Таблица 19.3
Свойства галогеноводородов
нх
Ц.О
г (X ), А
а, %
Енх. кДж/моль
АН0, кДж/моль
НР
1.98
1.30
10
568
-268
НСІ
1.04
1.81
92.5
436
-92.3
НВг
0 79
1 96
93.5
364
-36
НІ
0 38
2 20
95
298
+20
Другой путь получения галогеноводородов - вытеснение их из
солей действием сильных кислот в отсутствии воды (обычно ис¬
пользуют серную кислоту), также применим лишь к получению
фтористого и хлористого водорода:
СаР2 (тв.) + Н2Э04 (конц.) -» Н2Р2 + СаБС^
N301 (тв.) + Н2304 (конц.) -» НСІ + N314804.
По отношению к бромид- и иодид-ионам концентрирован¬
ная серная кислота выступает в качестве окислителя, в результате
чего вместо галогеноводородов выделяются свободные галогены по
уравнениям:
2NэBr(тв.) + 2Н2804 (конц.) -» Вг2 + Э02 + N8^04 + 2Н20
8Nal(тв.) + 5H2S04(кoнц.) -» 412 + Н2Э + 4Na2S04 + 4Н20.
390
ПоэТ%му для приготовления НВг и Н1 используют гидролиз соот¬
ветствующих бром- и иодангидридов:
2Р + ЗВг2 -» 2РВг3 РВг3+ ЗНОН -» НэРОэ+ ЗНВгТ
или соответствущие окислительно-восстановительные превращения
с участием галогенов:
Н2Э + 12 -» 2Н1 +
Галогеноводороды - газы, которые при растворении в воде обра¬
зуют кислоты за счет диссоциации сильнополярных (табл. 19.3)
связей:
НХ 5 Н+ + Х\
Сила галогеноводородных кислот растет с номером периода
(табл. 19.3), что объясняется параллельным ростом поляризуемос¬
ти, которая, в свою очередь, увеличивается по мере увеличения
радиуса галогенид-иона (табл. 19.3). Таким образом, иодистоводо-
родная кислота - самая сильная из галогеноводородных кислот
Аномально низкую диссоциацию плавиковой кислоты Н2Р2 объяс¬
няют ее димерным [и даже полимерным (Н2Р2)П) за счет водородных
связей строением, рассматривая ее как двухосновную и учитывая,
что второй ступенью диссоциации кислот такого типа можно пре¬
небречь.
Укажем на еще одну аномалию плавиковой кислоты - ее спо¬
собность реагировать со стеклом и растворять его. Эта способность
связана с тем, что фторид-ион может (как в отдельных случаях и
остальные галогенид-ионы) выступать в качестве лиганда. Процесс
растворения оксида кремния выразится реакциями:
2Н2Р2 + ЭЮ2 -> Э1р4 + 2Н20
Н2Р2 + Э1Р4 -» Нг^е].
Свойство же остальных галогенид-ионов быть лигандами пред¬
ставлено в уравнениях:
СиС12 + 2НС1 -» Н2[СиСЦ]
2К1 + Нд12 -» К2[Нд14].
Нахождение галогенид-ионов в биосредах требует умения опре¬
делять их присутствие, хотя бы на качественном уровне Для этого
существует групповая реакция на хлорид-, бромид- и иодид-анионы,
заключающаяся в образовании плохо растворимых галогенидов се¬
ребра при действии Ад1ЧОз:
391
X- + Ад* -» АдХІ.
Для качественного определения бромид-ионов используют вы¬
теснение брома из бромидов под действием хлора-
С12 + 2ВГ -> 2СГ + Вг2,
сопровождающееся окрашиванием раствора в желто-коричневый
цвет - окраску брома в водных растворах. На действии хлора осно¬
вано и обнаружение иодид-аниона Однако здесь действие избытка
хлора приводит к окислению иода до степени окисления +5. Снача¬
ла раствор, содержащий иодид-ионы, приобретает бурую окраску
выделившегося иода
С12 + 21" -» 2СГ + 12,
который затем обесцвечивается, переходя в Юз”-ион:
12 + 5С12 + 6Н20 -» 2Н10з + 10НС1.
Присутствие хлорид-иона определяют, основываясь на раство¬
рении осадка хлорида серебра в аммиаке, что объясняется тем, что
эта соль обладает большей растворимостью, чем бромид и иодид се¬
ребра (см. 12.1):
АдС11 + 2МНз -» [Ад(МНз)2]С1.
Фторид серебра неплохо растворим в воде, а потому определе¬
ние этого иона требует специальных процедур, основанных на спо¬
собности плавиковой кислоты разъедать стекло, о чем уже было
сказано. Для этого сначала действием серной кислоты вызывают
выделение фтористого водорода, который улетучивается и, попадая
на поверхность стекла, вызывает его помутнение.
19.3. Галогенопроизводные углеводородов
Получение органических галогенопроизводных основано на уже
известных реакциях углеводородов, а именно, на радикальном гало-
генировании алканов и циклоалканов:
Р-Н + Х2 -> Я-Х + Н-Х,
электрофильном присоединении галогенов и галогеноводородов к
алкенам:
392
и к алкинам (или алкадиенам):
CnH2n-2X2<- X2 + CnH2n-2+HX -» CnH2n-iX
а также на электрофильном замещении в ароматическом ряду:
Ar-H + Х2 -> Ar-X + Н-Х.
Комбинацией этих методов можно вводить заданное количество
атомов галогенов в необходимое положение цепи или цикла, что
видно из примеров хлорирования пропена и толуола, в зависимости
от условий приводящего к разным продуктам.
СН2=СН-СН3+С12 (hv) -» CH2=CH-CH2CI+CI2-> CH2CICHCICH2CI
пропен аллил хлористый 1,2,3-трихлорпропан
CHjQ СНэ СНз
6 € 6 ” и"
-на -на
хлористый толуол о-хлортолуол
бензил
Можно вводить атомы галогенов и замещением кислорода или
гидроксильной группы в кислородных органических соединениях,
действуя на них галогенангидридами, например, галогенангидридом
сернистой кислоты, называемым хлористым тионилом:
ROH + SOCI2 -» R-Cl + S02+ HCl
R2C=0 + SOCI2 -> R2CCI2+ S02.
Известны и другие способы введения галогена в состав органи¬
ческих молекул С некоторыми из них мы познакомимся позднее
Нуклеофильное замещение галогена - наиболее характерное
свойство органических галогенопроизводных. Объясняется это час¬
тичным положительным зарядом атома углерода, к которому при¬
соединен галоген, обладающий большой электроотрицательностью.
В общем виде это превращение выражено уравнением
R6+ х6- + у- R Y + ;Х-
нуклеофил
393
Способность к нуклеофильному замещению определяется при¬
родой галогена, в частности, величиной энергии связи углерод-
галоген, которая изменяется в той же последовательности, что и
для связи Х-Н. Связь углерод-фтор очень прочна (энергия связи
452 кДж/моль), что делает фтор малоподвижным в нуклеофильном
замещении, а фторалканы химически инертными веществами, о чем
уже говорилось выше
Значительно менее прочные связи углерода с остальными гало¬
генами делают их активными партнерами в нуклеофильном замеще¬
нии в соответствии с изменением энергии связи галоген-углерод,
величина которой составляет 252 кДж/моль для хлора, 239 и
180 кДж/моль в случае брома и иода, соответственно В согласии с
этим по химической активности галогенопроизводные можно распо¬
ложить в ряд: Р « С1 < Вг < I.
Другим фактором, сказывающимся на активности галогена
в нуклеофильном замещении, является природа атома углерода,
к которому этот галоген присоединен. В ряду ароматических и
особенно непредельных галогенопроизводных имеет место рд-сопря-
и хлорбензола, соответственно. Этот эффект приводит к полной по¬
тере подвижности галогена при кратной связи и к возможности
замещения галогена в аренах лишь в жестких условиях (темпе¬
ратура, давление).
Нуклеофильное замещение галогена в ряду производных алка-
нов и в меньшей степени для представителей аренов может проис¬
ходить под действием кислородных, сернистых, азотистых и других
нуклеофилов, что позволяет осуществлять на основе органических
галогенопроизводных синтез различных классов органических сое¬
динений. Многообразие таких превращений частично иллюстриру¬
ется схемой 19.1.
Различную подвижность атомов галогенов в предельных и аро¬
матических структурах можно использовать для направленного по¬
следовательного введения функциональных групп в состав органи¬
ческих молекул, что подтверждает приведенный ниже пример сни¬
жение между р-электронами атомов гало¬
генов И 71-СВЯЗЯМИ углеродного остова,
что приводит к упрочнению связи угле¬
род-галоген.
р,я-сопряжение
Об этом можно судить по уменьшению
длины связи, которая составляет 1.77 А
у хлоралканов (сопряжение отсутствует),
и 1.69 и 1.70 А в случае хлористого винила
394
спирт
Я-ОН
сульфид
Схема 19.1. Синтезы на основе галогенопроиэводных
теза /шра-гидроксибензилового спирта путем постепенной замены
атомов хлора на гидроксильные группы:
л-хлорбенэил- л-хлорбенэиловый л- гидроксибензиловый
хлорид спирт спирт
Другие возможности синтеза органических соединений из гало-
геналканов представляет реакция Вюрца:
ЗР'-Х + 6Ма + ЗХ-Р” -» Р’-Р” + Р’-Р’ + Р"-Р" + 6МаХ
и отщепление галогеноводородов (по правилу Зайцева, см 18 3 6)-
СпНгпоХ + МаОН (спирт) -» СпН2п + №Х + Н20,
позволяющие синтезировать алканы и алкены, соответственно.
Способность галогеналканов к нуклеофильному замещению
объясняет их известную высокую биологическую активность, преж¬
де всего, токсичность многих из них. Они реагируют в первую
очередь с аминогруппами белков и нуклеиновых кислот, вызывая
их необратимое поражение. На этом основано создание в ряду га¬
395
логенопроизводных (особенно среди хлорпроизводных) токсических
соединений направленного действия.
Среди них - боевые отравляющие вещества: сернистый ип¬
рит - Р,Р‘-дихлордиэтилсульфид (применявшийся в первую миро¬
вую войну), азотистый иприт - Р,Р‘,Р“-трихлортриэтиламин и
люизит - Р-хлорвинилдихлорарсин (также использованный в воен¬
ных операциях)
СН2СН2СІ £Н;СН;С1 (
Э' Ы—СН2СН2СІ С1СН=СН-Аз<(
'сНгСНгСІ Чн^ИгСІ <
сернистый азотистый люизит
иприт иприт
Другую группу хлорсодержащих токсичных соединений состав¬
ляют пестициды самого различного назначения. Пестициды -
химические средства борьбы с вредителями сельского хозяйства
и с переносчиками многих опасных заболеваний животных и
человека.
О гексахлоране и его действии уже сказано в разделе 18.2.3.
Отдельные представители хлорсодержащих пестицидов представле¬
ны ниже:
ОН ОСН2СООС«Нв
. . са3 ,
а—
ДДТ
полихлорированный эйджент орандж(2,4,5-Т) 1,1-ди(4-хлорфенил)-
фенол бутиловый эфир 2,4,5-трихлор- 2,2,2-трихлорэтан
феноксиуксусной кислоты
а он он
хлордан гексахлорофен диоксин
Среди них - гексахлорофен, являющийся бактерицидом. Его
применяют для дезинфекции ротовой полости в виде полосканий.
Хлордан, полихлорированный фенол и ДДТ представляют собой
типичные инсектициды. Эйджент орандж (2,4,5-Т), применявшийся
396
во время^ьетнамской войны, является дефолиантом - средством по
уничтожению листьев деревьев.
Будучи малополярными органическими веществами, хлорорга-
нические пестициды обладают кумулятивным токсическим эффек¬
том. Они накапливаются в жировых тканях и передаются с пищей
от одной особи к другой. К примеру, ДДТ обнаружен в жировых
тканях пингвинов Антарктиды, которые попали туда из рыб, явив¬
шихся резервуаром этого инсектицида. Поэтому имеется тенденция
к изъятию хлорсодержащих пестицидов из практики.
Всю опасность их применения демонстрирует “диоксиновая”
проблема. Обращает на себя внимание присутствие в ряде пестици¬
дов однотипного структурного фрагмента - полихлорированного
ароматического кольца. Оказалось, что два таких кольца входят в
состав диоксина - самого ядовитого из известных токсических сое¬
динений, синтезированных человеком.
Производство большинства хлорсодержащих пестицидов (и не
только их) сопровождается побочным образованием диоксина, точ¬
нее диоксинов, так как позднее выяснилось, что в зависимости от
размеров колец и числа галогенов в молекуле существует семейство
диоксинов, насчитывающее десятки представителей.
Диоксины, как оказалось, обладают не только кумулятивным
токсическим действием, но и пагубно воздействуют на наследствен¬
ный аппарат животных и человека, отражаясь на будущих поколе¬
ниях. Таким образом, необходимо полное уничтожение не только
таких производств, но и полная санитаризация почвы, влаги и
воздушной среды всех тех территорий, где когда-либо находились
или находятся производства с возможным образованием диоксина и
тех мест, где вещества, содержащие его в виде микропримесей,
применялись.
Однако это еще не делается повсеместно, несмотря на экологи¬
ческие катастрофы во Вьетнаме (где применялся эйджент орандж),
в Италии (Севезо, авария на производстве 2,4,5-трихлорфенола),
США (штат Миссури имеет около 100 зараженных диоксином уча¬
стков в результате производства эйджент орандж) и у нас в Уфе.
Как это ни прискорбно, применение хлорсодержащих инсектицидов
будет продолжаться в тропиках, так как это пока единственный
эффективный способ борьбы с переносчиками малярии, которая
по-прежнему уносит миллионы человеческих жизней
Нуклеофильное замещение хлора под действием воды в щелоч¬
ной среде с заменой хлора на гидроксильную группу составляет
397
основу метаболизма хлорсодержащих органических веществ. Ще¬
лочной гидролиз применяют для дегазации боевых хлорсодержащих
отравляющих веществ.
Некоторые галогенопроизводные применяются в медицине.
Хлористый этил, быстрое испарение которого вызывает охлаждение
смоченных им тканей и тем самым потерю болевой чувствитель¬
ности, употребляется для обезболивания. Для общего наркоза
применяется хлороформ (из-за токсичности сейчас употребляется
редко) и 1,1,1-трифтор-2-хлор-2-бромэтан (фторотан).
Способность производных фторалканов растворять в себе кис¬
лород, их химическая инертность и нетоксичность позволяют ис¬
пользовать их в качестве кровезаменителей.
В противоположность фторалканам, некоторые фторорганиче-
ские соединения, содержащие функциональные заместители, явля¬
ются сильными ядами. К ним относятся, например, фторуксусная
кислота, содержащаяся в ряде тропических растений (в частности,
в анчаре), и фторсодержащие боевые отравляющие вещества, такие,
как зарин и зоман (см. главу 23).
19.4. Кислородные соединения галогенов
Действие сильных окислителей на хлор и его аналоги может
служить способом получения соединений с положительными степе¬
нями окисления, которые в природе не встречаются. В большин¬
стве это кислородные соединения галогенов. В таблице 19.4 обоб¬
щены некоторые свойства кислородсодержащих кислот в ряду гало¬
генов. Среди них особое место представляет собой фторноватистая
кислота HOF и ее ангидрид, где фтор имеет степень окисления -1.
В действительности, число соединений с положительными степеня¬
ми окисления значительно больше, особенно среди оксидов, однако
они не представляют практического интереса.
Образование производных галогенов со степенью окисления +1
частично происходит уже при растворении галогенов в воде, одна¬
ко равновесие реакции самоокисления-самовосстановления (диспро-
порционирования)
Н20 + Х2 S НОХ + HX (X - галоген)
в большей степени смещено в обратную сторону, причем тем силь¬
нее, чем менее активен галоген.
398
Таблица 19.4
Кислородные кислоты галогенов
Степень
окислеыи
я
Г
а
Вг
1
Окончание
кислоты соли
+К-1)
0?2,
[НОР],
а>30%
нею,
рКа=
7 3
НВЮ,
рКа=
10.7
НЮ
рКа=
10.6
-ватис-
тая.
гипо-...
-ит
+3
"
[НСЮ2],
рКа= 20
-
-
-истая,
-ИТ
+5
-
НСЮз,
рКа= -1
НВЮз
НЮз
-оватая.
-ат
+7
-
НСЮ4,
рКа= -10
-
-
-ная,
пер- ..
-ат
Смещение указанного равновесия вправо достигается в щелоч¬
ной среде по уравнению:
Х2 + 2КОН ^ КОХ + КХ + Н20.
Таким образом, при растворении хлора в холодной щелочи про¬
исходит реакция:
С12 + 2КОН -> КСЮ + КС1 + Н20.
Если растворять хлор в кипящем щелочном растворе, диспро-
порционирование идет глубже, и в результате вместо гипохлорита
образуется хлорат калия:
ЗС12 + 6КОН -> 5КС1 + КСЮз + ЗНОН.
При более сильном нагревании, то есть исходя из расплава хло¬
рата калия, получают перхлорат калия-
4КС10з -> ЗКСЮ4 + КС1.
В этой серии превращений проявляется общая тенденция эле¬
ментов верхней части Периодической системы к существованию
в крайних степенях окисления как наиболее устойчивых Эта же
тенденция присуща азоту (наиболее стабильны степени окисления
-3 и +5) и сере (-2 и +6)
Однако по мере увеличения номера периода возрастают метал¬
лические свойства, растет разница в энергиях внешних б- и р-
399
орбиталей, а тем самым уменьшается вероятность их гибридизации,
и как следствие этих причин высшие степени окисления Становятся
менее прочными. Это наблюдается на примере галогенов, где иод и
бром не проявляют тенденции к образованию прочных соединений
со степенью окисления +7.
Итак, самоокисление-самовосстановление - основной путь к со¬
лям кислородных кислот галогенов, а от них и к самим кислотам,
на примере которых можно наблюдать некоторые закономерности в
изменении химических свойств.
Из данных таблицы 19.4, в частности, видно, что в ряду кислот
НХО сила кислот закономерно падает с уменьшением электроотри¬
цательности галогена Более того, иодноватистая кислота может
диссоциировать еще и по типу основания, то есть является амфоли-
том, демонстрируя тем самым появление у иода металлических
свойств-
HOI S Г + ОН' (рКь = 9.6)
Другую общую тенденцию проявляют кислородные кислоты
хлора, где по мере увеличения количества электроотрицательных
атомов кислорода растет сила кислот, и потому хлорная кислота
HCIO4 оказывается одной из самых сильных известных кислот
(табл. 19.4).
В соответствии со вскрытыми закономерностями соединения
хлора со степенью окисления хлора +1 являются, с одной стороны,
наиболее доступными среди хлорположительных соединений, а,
с другой, мало устойчивыми, проявляя свойства сильных окислите¬
лей. Величина редокс-потенциала этой степени окисления, менее
стойкой, чем +5 и +7, выше их потенциалов и выше, чем у самого
хлора. Эти обстоятельства и определяют область применения сое¬
динений со степенью окисления +1 - сильных и нестойких окисли¬
телей, которые принято называть соединениями, содержащими ак¬
тивный хлор (СГ).
Хлорактивные соединения широко применяются в качестве
дезинфицирующих средств, действие которых основывается на
окислении и замещении хлором атомов водорода в молекулах бел¬
ков микроорганизмов, что вызывает их гибель.
Простейший представитель дезинфицирующих средств среди
хлорактивных соединений - раствор хлора в гидроксиде калия, из¬
вестный под названием жавелевой воды. Другой всем известный
пример - хлорная известь, синтезируемая действием хлора на га¬
шеную известь.
Са(ОН)2 + С12
СаОС12 + Н20.
Хлорн^ известь представляет собой смешанную соль хлорид-
гипохлорит кальция и содержит один атом активного хлора. Можно
увеличить количество атомов активного хлора до двух, если выде¬
лить гипохлорит кальция Са(ОС1)2 в чистом виде.
Еще одна разновидность хлорактивных дезинфицирующих сое¬
динений - хлорамины, органические относительно стабильные про¬
изводные нестойкого хлорамина МН2С1. В их числе хорошо раство¬
римый в воде монохлорамин Б, хуже растворимый в воде, но зато
содержащий два активных хлора дихлорамин Б, и гексахлормеламин
с шестью атомами активного хлора.
сч,.
СІ
ОСІ
Са'
Са'
ОСІ
чОС1
хлорная
гипохлорит
известь
кальция
о02 $02
6 6
ск' СІ
монохлорамин Б дихлорамин Б гексахлормеламин
Активность хлорактивного соединения определяется количест¬
вом атомов активного хлора, приходящегося на молекулу хлорак¬
тивного соединения (ХАС). Эту активность измеряют количеством
молей свободного хлора, выделившегося при взаимодействии одного
моля хлорактивного соединения с соляной кислотой
При этом взаимодействии происходит окислительно-восстано-
вительная реакция, в результате которой два атома хлора с раз¬
личными степенями окисления (-1 и +1) превращаются в свобод¬
ный хлор. Имеет место разновидность окислительно-восстано-
вительных реакций, именуемая конмутацией Например, для широко
используемого монохлорамина Б реакция выглядит следующим
образом:
СбНбБОгМС^а + 2НСІ -> С6Н5302МН2 + №С1 + С12
Тогда содержание активного хлора в нем составит
С1‘(%) = ]^- = ^ = 333%
МХАС 2,35
Аналитически содержание активного хлора определяют с по¬
мощью метода редоксметрии, именуемого нодометрией Суть его
состоит в том, что всякий достаточно сильный окислитель (в дан¬
401
ном случае хлор) превращает иодид-ион в свободный иод, а послед¬
ний далее титруют тиосульфатом натрия, применяя в качестве
индикатора крахмал:
С12 + 2К1 -> 2КС1 + 12
12 + 2Ма2320з -» 2№1 + Ма234Ов.
Итак, соединения галогенов играют важную биологическую
роль, наибольшее же значение имеют производные хлора. Среди
них имеется несколько разновидностей физиологически активных
веществ - разнообразных пестицидов, отравляющих веществ и
дезинфицирующих средств.
Глава 20. КИСЛОРОД
В долгий период жизни земли
достигнуто то равновесие
между процессами, поглоща¬
ющими кислород и его разви¬
вающими, при котором сохра¬
няется в целой массе атмо¬
сферного кислорода определен¬
ное количество свободного кис¬
лорода.
Д И. Менделеев
20.1. Элемент, простое вещество
Кислород - элемент 6-й группы II периода. Впервые идентифи¬
цирован как самостоятельный элемент Шееле (1770 г., Швеция) и
Пристли (1774 г., Англия). Кислород является самым распростра¬
ненным элементом земной коры, где его содержание в виде силика¬
тов, карбонатов, воды и других оксидов и кислородных солей со¬
ставляет 55%. Много его и в атмосфере - 20.95% по объему. Его
масса в воздухе составляет около 1.1 1015т.
Весь свободный кислород - результат фотосинтеза, продуктив¬
ность которого составляет 2.5-1011 т в год. Практически весь этот
кислород потребляется животными и человеком для удовлетворения
его промышленных нужд. Первичная атмосфера Земли кислорода
почти не содержала и состояла из азота, водорода, метана и неко¬
торых других газов. Постепенное высвобождение кислорода в ре¬
зультате геохимических процессов, а также вследствие деятель¬
ности анаэробных микроорганизмов вызвало смену восстанови¬
тельной атмосферы на окислительную, а это привело к изменению
образа существования и к тому, что кислород стал использоваться
живыми организмами.
Кислород играет незаменимую физиологическую роль Его экзо¬
термическая реакция со многими биомолекулами, прежде всего с
глюкозой, является энергетическим источником организма. Кисло¬
род входит в состав многих неорганических биомолекул - воды,
фосфат-, карбонат-ионов и др. В качестве элемента-органогена кис¬
лород порождает множество кислородсодержащих органических
биомолекул и присутствует в кислородсодержащих функциональ¬
ных группировках - спиртовой, фенольной, эфирной, альдегидной,
кетонной, карбоксильной, сложноэфирной, амидной группах угле-
403
водов, липидов, пептидов, гормонов и др. Их взаимопревращения
служат основой большинства метаболических процессов, состав¬
ляющих сущность жизни. С участием кислорода или его соедине¬
ний связывают и некоторые частные жизненные проявления, на¬
пример, фагоцитарные, то есть защитные функции организма.
Высокая энтальпия ионизации (1313 кДж/моль), высокая эн¬
тальпия сродства к электрону (142 кДж/моль), а следовательно
сильная электроотрицательность (ОЭО = 3.5), уступающая лишь
фтору, а также малый радиус атома (0.75 А) сообщают кислороду
свойства сильного окислителя, с одной стороны, и полярный ха¬
рактер связей с его участием, с другой стороны, причем электроны
таких связей смещены к атому кислорода.
В соответствии с этим и с электронной конфигурацией кисло¬
род принимает степени окисления 0, -1 и -2. Исключение составля¬
ют его соединения со фтором, например, фторид кислорода Р20, где
он имеет степень окисления +2
В нулевой степени окисления элемент кислород может нахо¬
диться в двух аллотропных модификациях - в виде кислорода 02 и
озона Оз.
Молекула кислорода устроена необычно. Ее структура не может
быть описана с помощью метода валентных связей. Удовлетвори¬
тельное описание молекулы кислорода дает метод молекулярных
орбиталей (см. табл. 2.5). В соответствии с этим в молекуле кисло¬
рода имеется тройная связь, а сама молекула представляет собой
бирадикал с параллельными спинами неспаренных электронов, а
следовательно обладает парамагнетизмом, то есть притягивается
магнитом. Такая молекула кислорода носит название триплетного
кислорода (триплет 302).
|6:::0| +-£* ЦО:.:6 И* :6::0: Юг —*> Юг
триплет синглет I синглет II
Триплетный кислород имеет энергию диссоциации на атомы
496 кДж/моль. Эта высокая величина служит кинетическим фак¬
тором относительной химической инертности кислорода, что слу¬
жит одной из причин нахождения кислорода в свободном состоянии
в атмосфере. Нет нужды объяснять значение этого обстоятельства
для существования живых существ.
Молекулярный кислород применяется в медицине при гипокси-
ческих состояниях, сердечно-сосудистых заболеваниях, отравлениях
цианидами и угарным газом, в отдельных случаях в неврологи¬
ческой практике. Возможны три варианта употребления кислорода:
404
дыхание •Чцстым кислородом, оксигенобаротерапия, то есть помеще¬
ние пациента в атмосферу кислорода, иногда под давлением в
несколько атмосфер, и введение кислорода в желудок в виде так
называемого кислородного коктейля. При этом необходимо помнить,
что в избыточных дозах кислород токсичен. Не следует забывать
об опасности работы с чистым кислородом, в атмосфере которого
многие обычно негорючие материалы становятся легко воспламе¬
няемыми и даже взрывоопасными. Это нередко служит причиной
несчастных случаев в клинической практике.
При возбуждении триплетного кислорода под действием света
происходит электронная перестройка триплетной молекулы 3Ог, в
результате чего возникает молекула синглетного кислорода с парой
электронов, принадлежащей одному из атомов кислорода, так назы¬
ваемый синглет I. На это требуется энергия 92 кДж/моль. Такой
кислород нестабилен и быстро распадается. Период его полураспада
составляет 45 минут.
При дальнейшем возбуждении происходит еще одна трансфор¬
мация молекулы кислорода, и возникает молекула кислорода
синглет II. Это возбужденное состояние кислорода еще менее ста¬
бильно: его период полураспада составляет 10 ю секунды. На его
образование из синглета I требуется 63 кДж/моль. В целом же на
переход триплетного кислорода в синглет II необходимо затратить
155 кДж/моль. Это - именно та энергия, которой различается мо¬
лекула кислорода с тройной связью от молекулы кислорода, постро¬
енной посредством метода валентных связей, то есть с двойной
связью. Таким образом, под действием света происходит образова¬
ние активных синглетных форм кислорода 1Ог, причем их актив¬
ность тем больше, чем сильнее энергия облучения, и чем больше
время экспозиции.
С образованием синглетных форм кислорода на свету связано
важное физиологическое явление - фотодинамическое дейст¬
вие. Суть его состоит в следующем Существует группа веществ, в
том числе и в составе биосред (к ним, в частности, относятся пиг¬
менты крови), которые высоко чувствительны к действию света, и,
поглощая квант, переходят в возбужденное состояние Эти веще¬
ства обладают и другой особенностью - способностью передавать
энергию возбуждения молекулам кислорода, переводя их тем самым
в синглетное состояние Такие вещества называют сенсиби-
Юг
белок
белок-02
Т
405
лиэаторами (Б) Синглетный кислород, будучи химически актив¬
ным, реагирует с биомолекулами в биосредах, например, с белками
и, соединяясь с ними, вызывает их последующее разрушение.
Итак, фотодинамическое действие - это повреждение биострук¬
тур при поглощении света пигментами в присутствии кислорода
Простейший пример фотодинамического действия - солнечный
ожог С ним также связаны воспалительные процессы, интоксика¬
ция продуктами разрушения биоструктур, канцерогенез и другие
патологические явления. Для защиты организма в нем вырабаты¬
ваются специальные вещества - тушители (Т), которые перехваты¬
вают молекулы возбужденных сенсибилизаторов. К ним относят,
например, холестерин и витамин А. Следует сказать, что в отдель¬
ных случаях фотодинамическое действие используют в лечебных
целях, например, для поднятия общего тонуса, для чего порцию
крови пациента подвергают дозированному ультрафиолетовому об¬
лучению.
Химические свойства кислорода определяются, в частности,
наличием в его молекуле неподеленных электронных пар. Это опре¬
деляет его способность к координации с катионами металлов в ка¬
честве лиганда, а с биомолекулами - в роли биолиганда. Это чрез¬
вычайно важно для обеспечения процесса дыхания. Дело в том, что
растворимость кислорода в воде невелика - 3.1 объема кислорода в
100 объемах воды, чего совершенно недостаточно для обеспечения
процессов жизнедеятельности. К тому же поступление кислорода в
ткани за счет пассивного процесса диффузии не может снабдить их
кислородом в достаточной мере. Таким образом, необходимы биомо¬
лекулы-переносчики кислорода, которые, с одной стороны, должны
достаточно прочно связывать кислород, а с другой - легко высво¬
бождать его.
Такими свойствами обладают молекулы гемоглобина, содержа¬
щие катионы железа, и молекулы белков-гемоцианинов, которые
являются переносчиками кислорода у моллюсков, ракообразных и
пауков. Гемоцианины содержат катионы меди. Существуют синте¬
тические комплексы катиона кобальта с аминокислотами, которые
также могут связываться с кислородом. Эти комплексы служат мо¬
делями для изучения свойств кислорода как биолиганда.
Кислород, диффундирующий в кровь человека, связывается с
гемоглобином и в виде оксигемоглобина потоком крови переносится
в капилляры. Оксигемоглобии высвобождает в капиллярах кисло¬
род, который, диффундируя через их стенки, частично связывается
далее в комплексное соединение с железосодержащим веществом
тканей миоглобином (резерв кислорода в тканях). Основная же
часть кислорода в клетках расходуется на метаболические про¬
406
цессы. 0Д|1н литр крови одноразово переносит 250 мл кислорода.
Следует сказать, что лишь пятая часть вдыхаемого кислорода свя¬
зывается гемоглобином. Однако избыток кислорода обязателен, так
как обеспечивает необходимую для эффективной диффузии кисло¬
рода концентрацию.
Еще одна разновидность кислородных комплексов, играющих
существенную биохимическую роль, - комплекс кислорода с цито¬
хромом Р-450 (см. главу 33), выполняющим необходимую роль в
окислительных превращениях органических молекул в организме.
Важнейшее свойство кислорода - его окислительная способ¬
ность. Большинство элементов Периодической системы энергично
взаимодействуют с ним, превращаясь в оксиды. Большинство орга¬
нических веществ также окисляется кислородом. Хорошо известны
различные виды окисления - ржавление, коррозия, горение, гние¬
ние, скисание пищевых продуктов и т. д.
Восстановление кислорода в водных растворах, которое имеет
место в живых системах, описывается известным уравнением:
02 + 4Н+ + 4е ^ 2Н20 (ф = ф°о2+4н+/2н2о - 0.059рН ).
Как уже отмечалось, величина редокс-потенциала этой полуре-
акции в биосредах составляет +0 80 В. Эта относительно невысокая
величина является термодинамическим фактором, обеспечивающим
существование биологических систем.
Данная полуреакция распадается на ряд элементарных этапов,
подчиняющихся своим закономерностям, знание которых необходи¬
мо для понимания механизма связывания кислорода с био¬
субстратом:
02 + Н+ + е ^ Н02
Н02 + Н+ + е 5 Н202
Н202 + Н+ + е 5 Н20 + ОН
ОН + Н+ + е ^ Н20
20.2. Супероксид-анион-радикал
В свою очередь, первый этап начинается с еще более простой
стадии - присоединения электрона к молекуле кислорода
02 + е ^ 02 '
407
В результате возникает чрезвычайно активная частииа - супе-
роксид-анион-радикал кислорода 02'. Редокс-потенциал этой полу-
реакции составляет -0.3 В. Такая невысокая величина самой первой
стадии связывания кислорода служит еще одним термодинамиче¬
ским фактором, понижающим химическую активность кислорода.
Супероксид-анион-радикал кислорода играет существенную роль
в ряде биохимических процессов В малых количествах (не более
1-2%) он образуется в результате окислительно-восстановительных
процессов в молекуле оксигемоглобина:
НЬРе2*02 5 НЬТе3*02" ^ НЬТе3+ + 02'
В результате возникает метгемоглобин, содержащий ион желе¬
за 3+ и не способный переносить кислород. Супероксид-анион-
радикал далее вовлекается в окислительные превращения Так как
он чрезвычайно активен, существует специальный фермент, ответ¬
ственный за его разрушение - супероксид-дисмутаза (Е), содержа¬
щий катион Си2* (Е Си *). Супероксид-дисмутаза реагирует с ним по
следующей схеме:
ЕСи2* + 02 ’ ^5 Е'Си* + 02
ЕСи* + 02" + 2Н* ^ ЕСи2* + Н202
Таким образом, химически активный супероксид-анион-радикал
заменяется менее реакционноспособными молекулами перекиси во¬
дорода и кислорода. В тех случаях, когда концентрация супероксид-
анион-радикала оказывается аномально высокой, он вступает в ре¬
акции с биосубстратом, разрушая его.
Р-Н + 02" -> Р-0-0‘ + н-
02" + Н20 -> Н02 + ОН'
Н02 + Л-Н -> Р-0-0 +Н2
Именно такие превращения имеют место при лучевом пораже¬
нии, когда выбитый у-квантом из молекулы воды электрон захваты¬
вается молекулой кислорода плазмы крови. Это явление носит на¬
звание кислородного эффекта при радиолизе биосред. Оно зна¬
чительно усугубляет действие облучения.
Супероксид-анион-радикал кислорода образует соединения с
катионами щелочных металлов - супероксиды. Супероксиды ис¬
408
пользуют^ в системах автономного жизнеобеспечения в соот¬
ветствии с реакцией:
4К02 + 2С02 -> 2К2СОз + 302
20.3. Озон
Озон - другая аллотропная разновидность кислорода. Он об¬
разуется при облучении кислорода, а также при тихом электри¬
ческом разряде в кислороде. Его получе- 3о2 _> 1о2
ние можно выразить тремя последова- 302+102 -> Оз + О
тельными стадиями. 02 + О -> Оз
Строение молекулы озона можно
представить с учетом существования донорно-акцепторного взаимо¬
действия между молекулой кислорода (донор) и кислородным ато¬
мом (акцептор).
О 8Р2
, /'' % , 0=0—*о ► О— 0=0
ер3 о О 8Р
Поскольку молекула озона в таком виде может быть представ¬
лена двумя граничными структурами, то ее истинное строение есть
их суперпозиция, что приводит к заключению о нелинейном строе¬
нии молекулы озона с одинаковыми концевыми атомами кислорода.
Молекула озона нестабильна. Это выражается в положительной
величине энтальпии реакции его образования:
302 -> 20з АН0 = +284 кДж/моль
Поэтому характерным свойством озона является разрушение с
образованием молекулярного и атомарного кислорода. Если при ма¬
лых концентрациях озона этот процесс идет спокойно, то в газовых
смесях, сильно обогащенных озоном, распад озона может происхо¬
дить со взрывом
Другое свойство озона - высокая окислительная способность в
соответствии с уравнением:
Оз + 2Н* + 2е Ы 02 + Н20,
величина стандартного редокс-потенциала составляет +2 07 В Рас¬
чет показывает, что в биосредах при pH = 7 4 величина редокс-
потенциала этой реакции составит +1.63 В. Так как величины по¬
тенциалов биосред лежат ниже +0.82 В, их органическое вещество
будет немедленно подвергаться окислению при контакте с озоном.
409
По этой причине озон применяется для дезинфекции, и обработка
воды озоном для ее обеззараживания - одна из возможных стадий
водоподготовки Озон крайне токсичен (его летальная доза в воз¬
духе - 1%) и вызывает поражение дыхательных органов и кожи.
Примером окислительных свойств озона является реакция обна¬
ружения озона.
2К1 + 03 + Н20 -> 12 + 2КОН + 02.
Реакция озона с органическими веществами происходит различ¬
но для предельных и ненасыщенных соединений. Для насыщенных
молекул идет гомолитический процесс образования радикалов:
Р-Н + 03 -» Р02 + ОН
Это означает необратимую деструкцию таких молекул.
Алкены и другие соединения с кратными связями присоединяют
озон по двойной связи с образованием неустойчивых и взрывоопас¬
ных озонидов, представляющих собой пятичленные кислородные
гетероциклы. Озониды необратимо разрушаются водой, в результате
чего получаются карбонильные соединения, альдегиды или кетоны,
строение которых зависит от природы и числа заместителей при
двойной связи исходного алкена.
РЧ уНз 03 кЧ/0"°\укэ НгО Ъ
карбонильные м
алкен озонид соединения
Это означает, что озонирование может быть использовано для
нужд структурного анализа. Действительно, эта реакция явилась
ключевой для установления строения каучука. Озонирование по¬
следнего приводит к единственному продукту - кетоальдегиду, на¬
зываемому левулиновым альдегидом. Зная его строение, не состав¬
ляет труда реконструировать молекулу каучука.
г 1 о3. н2о
—СН2—С=СН-СН2—СН2—С=СН—СН2— — 2п СН3ССН2СН2СН
[ <1нз 4н, ]„ *** й «
каучук левулиновый
альдегид
Озон играет глобальную роль для поддержания жизненных
процессов на Земле, определяя характер поглощения солнечной
радиации в атмосфере. Хотя его содержание ничтожно мало
410
(10'5-10'®%^ по объему), причем эта часть преимущественно нахо¬
дится в верхних сЛоях атмосферы, именуемых озоновым слоем,
уникальная способность озона поглощать радиацию с длиной волны
менее 290 нм, то есть наиболее активную в биологическом отноше¬
нии, служит защитой биосферы от этого излучения. Вспомним, что
ключевой стадией получения озона является образование синглет-
ного кислорода. Следовательно тушители, то есть перехватчики из¬
лучения, должны препятствовать образованию озона. Присутствие
тушителей в атмосфере вызывает разрушение озонового слоя и
приводит к неблагоприятным экологическим последствиям
Именно это и происходит в результате накопления фреонов
(чаще всего, СР3С1 и СР2С12) в атмосфере. Стабильные фреоны ши¬
роко применяются в аэрозолях и в качестве хладоагентов, вследст¬
вие чего их концентрация в атмосфере растет. Поэтому в целях со¬
хранения озонового слоя фреоны начинают заменять другими ве¬
ществами.
20.4. Пероксиды
При неполном двухэлектронном восстановлении кислорода в
растворах происходит образование пероксида водорода - соедине¬
ния кислорода со степенью окисления -1:
02 + 21-Г + 2е 5 Н202
Химические свойства этого соединения определяются двумя об¬
стоятельствами. Во-первых, его молекула содержит связь О-О,
имеющую малую прочность ( Есвязи 146 кДж/моль), следовательно
перекись водорода легко разрушается. Во-вторых, промежуточное
значение степени окисления кислорода в ее молекуле сообщает ей
свойства и окислителя, и восстановителя в соответствии с уравне¬
ниями:
Н202 + 2е + 2Н+ 5 2Н20 (<р° = +1 78 В)
Н202 - 2е Ь 2Н+ + 02 (<р° = +0 68 В)
Так как восстановление кислорода в растворах до пероксида -
промежуточная стадия связывания кислорода в биосредах, и по¬
скольку перекись водорода является химически активным ве¬
ществом, в живых системах должны существовать способы утили¬
зации этого вещества
411
В соответствии с этим в организме существуют два фермента,
отвечающие за разложение перекиси водорода. В частности, дис-
пропорционирование перекиси водорода на кислород и воду проис¬
ходит с участием фермента каталазы.
каталаза
2 Н202 * 02 ♦ 2 НгО
Восстановление пероксида до воды регулируется посредством
фермента пероксидазы.
. пероксидаза окисленный
2 Н202 ♦ субстрат ► 2 Н20 ♦ субстрат
Следует отметить, что до 15% кислорода, участвующего в био¬
процессах, проходит через промежуточную стадию образования пе¬
рекиси водорода. Иными словами, перекись водорода - эндогенный
метаболит.
Перекись водорода в организме участвует в механизмах де¬
токсикации и интоксикации, транспорте глюкозы через мембраны и
других процессах.
В частности, перекись водорода участвует в детоксикации суль-
фит-иона, являющегося одним из промежуточных продуктов мета¬
болизма сернистых соединений, окисляя последний до нетоксичного
сульфат-иона по уравнению:
ЭОз2- + Н202 -> ЭОЛ + Н20
Как уже отмечалось, перекись водорода участвует в механизме
защитного бактерицидного действия лейкоцитов:
Н202 + 2СГ + 2Н+ -> С12 + 2Н20
Оказалось, что перекись водорода по аналогии с вышеприведен¬
ной реакцией вовлечена в процесс биосинтеза некоторых хлорсо¬
держащих антибиотиков, способствуя введению атома хлора в их
структуру:
Н202 + СГ + Р-Н + Н+ -> Р-С1 + 2Н20
Токсическое действие перекиси водорода заключается в ее ре¬
акции с гемоглобином, в результате чего происходит окисление ка¬
тиона Ре2+ в Ре3+-катион:
Н202 + Ре2+ -» Ре3+ + ОН-+ ОН
Образование в этой реакции гидроксильного радикала усугубля¬
ет ситуацию.
412
Перек1&ь водорода окисляет серусодержащие сульфгидрильные
и сульфидные группы ферментов:
ЗН202 + Я-ЭН -» Я-ЭОзН + ЗН20
2Н202 + Я-Э-Я' -» Я-802-Я' + 2Н20
Перекись водорода представляет собой жидкость, которая в
чистом виде разрушается, а потому используется в виде водных
растворов различной концентрации крепостью не выше 30%. Учи¬
тывая высокую химическую и физиологическую активность переки¬
си водорода, ее употребляют в качестве дезинфицирующего сред¬
ства, а также для отбеливания тканей, поскольку перекись водорода
окисляет и пигменты, обесцвечивая их. Как сильный окислитель пе¬
рекись водорода находит применение в качестве компонента ракет¬
ного топлива.
Перекись водорода представляет собой очень слабую кислоту,
однако замещение атомов водорода в ней возможно, что порождает
три разновидности пероксидов.
Замена атомов водорода катионами щелочных и щелочно-зе-
мельных металлов дает семейство солей - пероксидов металлов
(например, Ма202), которые используют в химической практике как
сильные окислители.
Замещение водородных атомов остатками кислородных кислот
дает класс пероксокислот (моно- при замене одного атома водорода
и ди- при замещении обоих).
О 0 0
НО—!»—О—О—н но- й-о-о-й -он
в в -в
Соли этих кислот, как и сама перекись, находят использование
в качестве отбеливателей.
20.5. Органические перекиси
Эти соединения можно рассматривать как производные переки¬
си водорода, в молекуле которой атомы водорода заменен^ органи¬
ческими радикалами. Соответственно, различают гидроперекиси
(РООН) и перекиси (РООР) Среди различных способов получения
этих соединений наиболее распространенным является окисление
органических веществ, зачастую происходящее на воздухе
413
У разветвленных алканов легче всего окисляются третичные
атомы углерода
СН3 СН3
СНз-А-Н .0, _ СНз-А-ООН
СНз СН3
В алкенах окислению подвергаются атомы углерода, соседние с
двойной связью. Именно эти атомы затрагиваются при перекисном
окислении липидов в организме
Р-СН2-СН=СН-Р' + о2 — Я—СН— СН=СН—Я'
6-о-н
Процесс образования гидроперекисей является гомолитическим
и, следовательно, инициируется ультрафиолетовым или гамма-из-
лучением. Он сопровождается образованием свободных радикалов,
так как связь кислород-кислород легко разрушается:
РООН -» ОД' + ОН (оксильные радикалы).
При этом образуются так называемые оксильные радикалы. Способ¬
ность перекисей и гидроперекисей к распаду на свободные радика¬
лы позволяет использовать некоторые из них как инициаторы в
процессах полимеризации.
В живом организме образование органических перекисей и гид¬
роперекисей интенсивно происходит из липидов биомембран.
Механизм этого явления по современным представлениям вы¬
глядит следующим образом. Под воздействием определенных фер¬
ментов образуются свободные радикалы липидов (PH):
PH -» № + -Н.
Далее активные формы кислорода (возникающие в тканях фер¬
ментативным способом) окисляют возникшие свободные радикалы:
я- + о2-»яо2- Н+02->Н02
Возникшие перекисные радикалы атакуют молекулы биосуб¬
страта по гомолитическому механизму с образованием гидропереки¬
сей, причем процесс будет продолжаться, так как сопровождается
появлением новых радикальных частиц:
Р02 + Р’Н -> РООН + Н02 + PH -> Р'ООН + Н.
414
Весьма^неустойчивые гидроперекиси легко разрушаются, об¬
разуя оксилъные радикалы (см. выше), также атакующие биосуб¬
страт. В результате таких процессов получается сложная гамма ве¬
ществ - активных радикалов и продуктов их последующих реакций
с биосубстратом. Свободные радикалы вызывают существенные из¬
менения в молекулах липидов, нуклеиновых кислот, ферментов,
других белков и прочих биомолекул.
Это явление, которое имеет место в тканях организма, пред¬
ставляет собой нормальный физиологический процесс и называется
перекисным окислением липидов. Превышение нормы перекис-
ного окисления липидов - показатель патологических состояний,
связанных с активацией гомолитических превращений под действи¬
ем таких факторов, как лучевое поражение, рана, травма и др. По¬
казано, что перекиси липидов являются промежуточными вещест¬
вами биосинтеза ряда гормонов и других физиологически активных
веществ и вызывают процесс обновления липидного состава био¬
мембран. Органические гидроперекиси обнаружены во многих орга¬
нах и тканях человека.
На важное биохимическое значение процессов образования пе¬
рекисей указывал еще в конце прошлого века А. Н. Бах. В настоя¬
щее время им придается значение при объяснении старения орга¬
низма, мутагенеза, канцерогенеза, лучевой болезни и др.
20.6. Оксиды
С большинством простых веществ кислород реагирует с образо¬
ванием оксидов - соединений кислорода со степенью окисле¬
ния -2. Высокий редокс-потенциал и определяет нахождение боль¬
шинства элементов Периодической системы в виде кислородных со¬
единений - силикатов, фосфатов, оксидов металлов и их солей с
кислородсодержащими кислотами.
Свойства оксидов в периодах и группах изменяются закономер¬
но в зависимости от структуры электронных оболочек элементов,
образующих соответствующие оксиды, что видно из данных
таблицы 20 1
По мере роста электроотрицательности в ряду натрий - хлор
меняется природа оксида от основных оксидов натрия и магния до
ангидридов кислот в случае оксидов кремния, фосфора, серы и хло¬
ра. Это легко прослеживается и на изменении свойств соответ¬
ствующих гидроксидов, где имеет место переход от очень сильного
основания через амфотерное соединение к сильной кислоте
415
Оксиды III периода
Оксид
Ыа20
МдО
АІ2О3
ЭЮг
Р2О5
ЭОз
СІ2О7
Гидрок
-сид
ЫаОН
Мд(ОН)2
А1(ОН)з
НгЭГОз
Н3РО4
НгЭОд
нсю4
Характе¬
ристика
гидрок¬
сида
щелочь
основание
средней
силы
амфолнт
слабая
кислота
средняя
кислота
сильная
кислота
очень
сильная
кислота
Итак, кислород со степенью окисления -2 формирует широкую
серию неорганических соединений - оксидов, оснований, кислород¬
содержащих кислот и их солей. Соединение кислорода в этой сте¬
пени окисления с органическими радикалами порождает спирты,
фенолы и простые эфиры.
20.7. Спирты, фенолы
20.7.1. Классификация и номенклатура
Спиртами называют органические вещества, представляющие
собой соединения алифатического радикала с одной или нескольки¬
ми гидроксильными группами (окси- или гидроксигруппами), а фе¬
нолами - соединения из гидроксигрупп и ароматического радикала.
По числоу оксигрупп в молекуле спирты и фенолы подразделяют на
одно-, двух-, трехатомные (или в общем случае многоатомные).
Примером двухатомного спирта является этиленгликоль; примером
двухатомного фенола - диоксибензол в трех своих изомерах -
орто-, мета- и пара- Глицерин - спирт трехатомный.
ОН ОН ОН
СНгОН
інон
бнгОН
этилен¬
гликоль орта- мета- пара- глицерин
диоксибензолы
ОдноЯ^емные спирты, в зависимости от радикала, с которым
связана группа ОН, подразделяются на первичные, вторичные и
третичные. Примеры: этиловый спирт - первичный, изопропиловый
спирт - вторичный, трет-бутиловый спирт - третичный.
СН3 СНз
СН3 —СН2 -ОН 'СН -ОН СН3 -С-ОН
СНз Ан,
этиловый изопропиловый грег-бутиловый
спирт спирт спирт
По международной номенклатуре, группе ОН соответствует
окончание -ол в названии соединения. Для некоторых спиртов ши¬
роко употребляются тривиальные названия:
СН3
СН3СН2СН2СН2ОН СН3СН2СНСН3 СН3СНСН2ОН СНз- бон
бн бнз £н3
тривиальные
н-бутиловый вгор-бутиловый иэобутиловый грег-бутиловый
спирт спирт спирт спирт
международные
_ , с- _ 2-метилпро- 2-метилпро-
бутанол-1 бутанол-2 пвнол-1 панол-2
20.7.2. Способы получения спиртов
Спирты можно синтезировать следующими способами-
- каталитическим окислением алканов (см. 18.3.1);
- гидратацией алкенов (см 18.3 2),
нон
РСН=СН2 н^о* ЯСНСНз
6н
- нуклеофильным замещением галогена в галогенопроизводных
под действием водного раствора щелочи (см 19 3),
- восстановлением карбонильных соединений - альдегидов и ке-
тонов; из альдегидов получаются спирты первичные, а из кетонов -
вторичные (глава 24)
14 Зак 675
417
20.7.3. Свойства спиртов и фенолов
Свойства спиртов и фенолов во многом определяются свойства¬
ми гидроксильной группы, входящей в состав их молекул
Если говорить о физических свойствах гидроксисоединений, то
способность гидроксильных групп образовывать водородные связи
объясняет тот факт, что простейшие спирты - метанол, этанол -
кипят при сравнительно высокой температуре (65 и 78° С, соответ¬
ственно), а фенол - даже не жидкость, а твердое вещество По той
же причине низшие гомологи спиртов и многоатомные спирты хо¬
рошо растворимы в воде Однако по мере усложнения углеводород¬
ного радикала растворимость спиртов в воде понижается, зато воз¬
растает липидная растворимость.
Высшие жирные спирты (С12-С20) обладают большой поверхност¬
ной активностью и поэтому широко используются в качестве мою¬
щих средств. Они обладают тем достоинством, что усваиваются
микроорганизмами, и, следовательно, хороши в экологическом от¬
ношении. В настоящее время этих веществ в мире производится
более 1 млн. тонн в год.
В водных растворах спирты и фенолы ведут себя как слабые
кислоты; они диссоциируют с образованием алкоголят-ионов или
фенолят-ионов, соответственно.
н-о • • • н-о. . • н-о ROH RO + H+ рКа 15-17
p!j ^ ^ алкоголят
водородные связи -°н рК.9.,1
Вследствие электронодонорных свойств алкильного заместителя
(R—ЮН) кислотные свойства у спиртов выражены слабее, чем у во¬
ды. Реакция нейтрализации для спиртов обратима, причем равнове¬
сие сильно смещено в сторону исходных веществ, однако отве¬
чающие им соли - алкоголяты - легко получить, воздействуя на
спирты активными металлами (например, натрием или калием). По¬
скольку ароматический цикл обладает электроноакцепторным дей¬
ствием (Аг<— ОН), кислотные свойства фенолов выражены достаточ-
он но сильно и фенолы легко нейтрали-
I зуются щелочами.
ОН кп Необходимо знать, что спиртовая
I тироэин группа входит в состав природных
серии сн2 аминокислот белков серина и трео-
»аллллялллллллллллллллал^л^л^ нина, а фенольная - тирозина, а
белок следовательно, сообщает тому уча-
стку пол^ептидной цепи, где находятся эти аминокислоты, соот¬
ветствующие химические свойства.
Подвижный атом водорода в спиртах и фенолах может заме¬
щаться не только металлом, но и кислотным остатком - ацилом (от
лат. acid - кислый). Ацилирование спиртов кислотами приводит к
образованию сложных эфиров. Процесс этот называют этерифи-
кацией.
ROH + НОАс U ROAc + НОН.
спирт кислота сложный эфир
Этерификация под действием серной кислоты приводит к обра¬
зованию алкилсерных кислот, например:
С2Н5ОН + H0S03H U C2H5OSO3H + НОН
этанол этилсерная кислота
Этерификация под действием азотной кислоты дает нитроэфиры.
В частности, из глицерина получают тринитрат глицерина, обычно
называемый нитроглицерином - взрывчатое вещество.
СНгОМОг сНгОН
ilH0N02 „ 1 бнОН
Ьн&Ы02 "ЗН2° (LH20H
Н3РО4
-н2о
тринитрат
глицерина
СНгОН
бнон О
£н20-^-0Н
6н
глицеринфос- глицеро-
форная кислота фосфат кальция
СНгОН
бнон О
^НгО-^-О
А-Аа
Подобно другим нитроэфирам, нитроглицерин обладает сосудо¬
расширяющим действием и поэтому служит лекарством.
Взаимодействуя с фосфорной кислотой, спирты образуют ал-
килфосфаты. В медицине находит применение глицеринфосфорная
кислота в виде кальциевой соли (глицерофосфат кальция - средство
от упадка сил и истощения нервной системы)
Реакция фосфорной кислоты со спиртами играет важную биоло¬
гическую роль, в частности, нуклеиновые кислоты - эфиры фосфор¬
ной кислоты и нуклеозидов, а фосфолипиды - производные глице-
ринфосфорной кислоты - входят в состав нервных клеток.
Сложные эфиры образуются также при взаимодействии спиртов
и фенолов с карбоновыми кислотами
СНз-С.
//
С*ДОН ^ CH3-cf0_C2Hs ♦ Н;0
этиловый эфир уксусной кислоты
(этилацетат)
14*
419
Ацилирование гидроксильных групп серииа, треонина и тирози¬
на белков кислотами - важная метаболическая реакция
Спирты легко подвергаются окислению, в результате чего пер¬
вичные спирты образуют альдегиды, а вторичные - кетоны
[О) (О)
СН3СН2ОН ^ СНзСН=0 СН3СНСН3 5=^ СН3ССН3
[Н] 6н и #
ацетальдегид ацетон
Эти процессы составляют промежуточную фазу окислительных
клеточных процессов, происходящих под воздействием ферментов.
Реакция обратима.
Третичные спирты в мягких условиях не окисляются, а в жест¬
ких претерпевают сложные превращения с разрывом углерод-угле-
родных связей.
При окислении первичной спиртовой группы глицерина образу¬
ется глицериновый альдегид, а при окислении вторичной - диокси-
ацетон:
сн=о сн=о
Н-С-ОН + НО-С-Н
^нгон ^нгон 'Н2°
глицериновый
альдегид
Окисление глицерина в диоксиацетон и глицериновый альдегид
является обратимым биохимическим превращением, связывающим
между собой липиды (глицерин - их составляющая) с углеводами
(по сути диоксиацетон и глицериновый альдегид являются простей¬
шими представителями природных углеводов).
Один из атомов углерода в молекуле глицеринового альдегида
асимметричен - он содержит четыре различных заместителя. По¬
этому глицериновый альдегид может существовать в виде двух
энантиомеров.
Стереоизомеры глицеринового альдегида можно отобразить с
помощью проекционных формул Фишера (рис. 20.1).
Для этого пространственное изображение хиральной молекулы
располагают так, чтобы вверху оказалась наиболее окисленная
группа, а ближе к наблюдателю - атом водорода и функциональный
заместитель с гетероатомом (ОН, МН2 и другие группы). Далее это
изображение проецируется на плоскость, как это показано на
рис. 20.1.
СН2ОН СН2ОН
(!;нон —!-!_* 6=о
(^НгОН 'Н2° ^НгОН
420
Проей^онные формулы глицеринового альдегида можно пред¬
ставить упрощенно, не изображая асимметрических атомов углеро¬
да (рис. 20.1, а), и совсем упрощенно - не изображая также атомов
водорода при асимметрических атомах углерода (рис. 20.1, б).
проекционные формулы
СН=0 ОНО СН=0
Н—I—ОН 1—ОН НО—I—
СН2ОН СН2ОН АнгОИ
Рис. 20.1. Стереоиэомерия глицеринового альдегида
По предложению М. А. Розанова фишеровское изображение
молекулы глицеринового альдегида с правосторонним расположе¬
нием группы ОН относительно асимметрического атома углерода
принято называть О-изомером, а его зеркальное отображение -
Ь-изомером. Согласно принятому обозначению направления
вращения плоскости поляризации света при прохождении его через
оптически активное вещество, первому изомеру приписывается знак
(+) [0(+)-глицериновый альдегид], а второму - знак (-)
[(Ц-)-глицериновый альдегид].
Все оптические изомеры природных молекул принято произво¬
дить от глицеринового альдегида. Согласно этому, стереоизомеры,
которые можно произвести без обращения конфигурации из глице¬
ринового альдегида с правым расположением ОН-группы, относят
к О-ряду. К нему принадлежат, в частности, почти все углеводы
природного происхождения. К Ь-ряду относятся аминокислоты бел¬
ков, так как все они имеют левое расположение аминогруппы в
проекционных формулах Фишера, то есть генетически связаны с
Ь-глицериновым альдегидом.
Фенолы - также легко окисляемые соединения. Среди них особо
следует отметить пара-дигидроксибензол - гидрохинон, окисление
которого приводит к хинону Данный процесс обратим Практиче¬
ское применение в хингидронном электроде находит эквимолеку¬
лярная смесь “хинон (X) - гидрохинон (ГХ)”, именуемая хингидро-
ном (ХГ) (см главу II) В организме подобные окислительно¬
восстановительные процессы реализуются для убихинонов, входя
щих в состав всех клеточных мембран.
421
гидрохинон
хинон
восстановленные
убихиноны п = 6-10
При взаимодействии спиртов с сухими галогеноводородами про¬
исходит замещение гидроксильной группы галогеном. Этот процесс
обратим, равновесие смещается вправо при связывании воды. Так
получают хлористый этил, применяемый в медицине:
С2Н5ОН + НС1 ^ С2Н5С1 + Н20.
Фенолы в эту реакцию не вступают. Галогенпроизводные могут
быть получены из фенолов (и из спиртов) действием пентагалоге-
нидов фосфора или хлорангидрида сернистой кислоты.
РВг5 БОСНз
к~он г-* Р-Вг Я-ОН — Р-С1
-РОВг3, -НВг -БОг, -НС1
20.7.4. Важнейшие представители спиртов и фенолов,
их практическое значение
Метанол. Широко используется как исходный продукт для по¬
лучения формальдегида (путем окисления). Используется также как
растворитель, как антифриз и как топливо. Обладает наркотическим
действием, однако сильно токсичен, - преобразуется в организме в
яды - формальдегид и муравьиную кислоту Это превращение мета¬
нола, происходящее под действием ферментов, служит причиной
его токсического действия. Явление, когда в результате метаболиз¬
ма из неядовитого вещества в организме образуются токсичные
продукты, называют летальным синтезом.
Существует несколько способов синтеза метанола; наиболее
эффективным среди них представляется синтез из СО и водорода в
присутствии катализатора:
СО + 2Н2 ->• СНзОН.
Этанол. Применяется для производства ацетальдегида, хлоро¬
форма, диэтилового эфира, этилена, уксусной кислоты, синтетиче¬
ского ка$цука. Используется в качестве моторного топлива, по¬
скольку обладает высоким октановым числом. В медицине ценен
как растворитель лекарственных средств. Употребляется при изго¬
товлении алкогольных напитков. Этанол - наркотик, возбуждающе
действующий на организм; его длительное и неумеренное употреб¬
ление приводит к алкоголизму.
Химизм возникновения алкоголизма чрезвычайно сложен и да¬
лек от окончательного выяснения. Однако ключевой стадией яв¬
ляется ферментативное окисление этанола в ацетальдегид, после¬
дующее взаимодействие которого с некоторыми биогенными амина¬
ми вызывает образование алкалоидоподобных веществ, блокирую¬
щих рецепторы, чувствительные к действию морфина и подобных
ему наркотиков. Следует знать, что этанол является продуктом ме¬
таболизма, а следовательно, это надо учитывать при судебно-
медицинских экспертизах по определению алкоголя в организме.
Синтезируют этиловый спирт путем гидратации этилена, а также
спиртовым брожением углеводов.
Цетиловый спирт (С16Н33ОН). Входит в состав восковых ве¬
ществ, представляющих собой сложные эфиры. Используется для
приготовления ПАВ.
Ментол (5-метил-2-изопропилциклогексанол). Относится к
терпенам. Извлекается из растений и входит в состав сердечно¬
сосудистых средств, зубных порошков
и паст.
Сорбит. Один из оптических изо¬
меров шестиатомных спиртов. Встре¬
чается в растительных источниках.
Им богаты фрукты и ягоды. Служит
заменителем сахара в питании боль¬
ных диабетом.
Инозит. Также относится к шес¬
тиатомным спиртам, но имеет цикли¬
ческое строение (см. 18.2.3). Содержится в мышцах, в печени, в
почках, в мозгу; обладает витаминным действием
Этиленгликоль. Синтезируется путем гидратации окиси этиле¬
на (см. 18.3 2). Применяется в качестве антифриза в смеси с водой
Сильный яд по той же причине, что и метанол - в результате ле¬
тального синтеза из него образуется токсичная щавелевая кислота
и другие не менее ядовитые соединения.
Глицерин Важнейший биогенный спирт Применяется в меди¬
цине как увлажнитель фармацевтических препаратов
Получают глицерин гидролизом липидов (см главу 27) или хло¬
рированием пропена с последующим действием щелочью.
ментол сорбит
423
сн3-сн=сн2 ———► СНгО -сн =СНг
^ і ІН 4
гс30"- СН2-СН-СН2
6н 6н 6н
Другой вариант промышленного синтеза глицерина - окисление
пропена до акролеина, с последующим присоединением пероксида
водорода и восстановлением
[О] Н202
Н2С=СН-СН3 ———♦ Н2С=СН-СН=0 СНг —сн —сн=о
акролеин Ди ^
—М— СНг —сн —СНг
<Ьн бн бн
Фенол. Извлекается из каменноугольной смолы - действием на
нее сначала щелочи, а затем, для нейтрализации, - кислотами.
В промышленности фенол чаще всего получают окислением кумола
(изопропилбензола), при этом синтезируется и другой ценный про¬
дукт - ацетон.
СН-». СН* ?°Н
СН3—СН-СНз
6 — 6—6•
СН3СОСН3
ацетон
фенол
Кроме того, фенол можно получить из бензолсульфокислоты
(см. 21.6) и гидролизом хлорбензола в жестких условиях.
Фенол и его гомологи - крезолы - используются в качестве ан¬
тисептиков. Из фенола получают лекарственные препараты, такие,
как салицилаты, салол и аспирин. Все фенолы сильно ядовиты.
20.8. Простые эфиры
К простым эфирам относятся соединения с общей формулой
Р’-О-Р2, где Р - алкильный, либо арильный радикалы. Названия
простых эфиров производятся от названий радикалов. Например,
424
С2Н5-О-С4Н5 - ДИЭТИЛОВЫЙ эфир, а СгН5-0-СН=СН2 - этилвиниловый
эфир. В простых эфирах атом кислорода может быть включен
в цикл.
I о Й1 0$ 0 о
этилена тетрагидрофуран тетрагидропиран диоксан фуран
Простейшие представители циклических эфиров - окись этиле¬
на, тетрагидрофуран, тетрагидропиран, диоксан и фуран.
Два из них представлены и формулами Хеуорса, широко ис¬
пользуемыми для изображения молекул углеводов, в которых эти
циклы и присутствуют.
Способы получения эфиров. Эфиры можно синтезировать
следующими способами:
- действием алкоголятов (или фенолятов) на галогенопроизвод¬
ные углеводородов:
С6Н5ОМа + С1СН3 -* СбН5-0-СНз + №01;
анизол
- действием спиртов на алкилсерные кислоты:
СгНзОБОзН + С2Н5ОН С2Н5-0-С2Н5+ НОЭОзН -
этим способом получают также тетрагидрофуран и диоксан
О
НОСН2СН2СН2СН2ОН Нг3°4» ГЛ 2 НОСНгСНгОН Н—4- Г ] ;
О ЧСГ
- электрофильным присоединением спиртов к ацетилену
НС=СН + НОС2Н5 -* Н2С=СН-0-С2Н5.
Свойства и применение эфиров. В отличие от спиртов про¬
стые эфиры не образуют водородных связей; по этой причине они
кипят при более низкой температуре Например, температура кипе¬
ния диэтилового эфира 36° С, в то время как этиловый спирт,
имеющий на два атома углерода меньше, кипит при 78° С По этой
же причине простые эфиры плохо растворимы в воде, зато облада¬
ют хорошей липидной растворимостью. Это объясняет анестези¬
рующее свойство некоторых эфиров (диэтиловый, этилвиниловый)
и их применение в медицине для общего наркоза
425
Диэтиловый эфир, тетрагидрофуран и диоксан широко исполь¬
зуются как растворители при экстракции эфирных масел, липидов и
других веществ.
Виниловые эфиры служат в качестве исходных мономеров при
получении полимерных материалов В частности, так называемый
бальзам Шостаковского, применяемый для заживления ран, - это
полимер винилбутилового эфира СН2=СН-0-С4Нв.
Особую группу циклических эфиров представляют собой так на¬
зываемые краун-эфиры (от англ crown - корона), молекулы
которых формой напоминают корону. Размер колец краун-эфиров
может меняться в любых пределах (в качестве примера приведены
две молекулы с разным количеством атомов в цикле), а это, как
оказалось, важно для их участия в качестве лигандов в комплексо-
образовании с катионами металлов, и, в частности, с катионами
щелочных металлов (глава 34).
Существует соответствие между
размерами колец краун-эфиров и ради¬
усами катионов, что обеспечивает из¬
бирательность взаимодействия. Послед¬
нее свойство применяется в химичес¬
кой практике для растворения солей
в органических растворителях, для
каталитических целей и, что более
важно, при интерпретации проблемы
транспорта ионов через клеточные мембраны.
По сравнению со спиртами простые эфиры химически менее ак¬
тивны. Характерным для простых эфиров является их взаимодей¬
ствие с сильными кислотами, приводящее к образованию оксоние-
вых солей:
гп Г"0
U(. J
I—I \ /
С2Н5-О-С2Н5 + HCI
С2Н5Чч(?ХС2Н5
оксониевая соль
Многие эфиры легко окисляются на воздухе под действием
света, причем окислению подвергаются атомы углерода, соседние с
эфирной связью:
СНЭ— СН2—О—СН2—СНЭ
02
—► СНЭ—СН2—О-СН—СНЭ
гидроперекись Іон
диэтилового эфира
426
В реЗ^пьтате окисления образуются неустойчивые и взрыво¬
опасные гидроперекиси. Из-за высокой воспламеняемости и лету¬
чести диэтилового эфира обращение с ним требует особых предо¬
сторожностей - его следует хранить в темноте, без доступа воздуха
и тепла.
Простая эфирная группировка присутствует во многих алкало¬
идах и лекарственных средствах, примером чего может слу¬
жить молекула папаверина, которая обладает четырьмя эфирными
группами.
Итак, кислород и множество его неорганических и органических
соединений необходимы для существования жизни. Имеется целая
гамма так называемых активных форм кислорода, которые включа¬
ются в те или иные жизненно важные процессы (синглетный кис¬
лород, супероксид-анион-радикал, перекись водорода, перекисные и
оксильные радикалы биомолекул) и которые играют определенную
роль в формировании и протекании разнообразных физиологических
явлений. Взаимоотношения живого организма с кислородом чрезвы¬
чайно сложны, включая различные виды химических превращений -
комплексообразование, окислительно-восстановительные реакции
и т. д. Множество биохимических процессов требует участия кис¬
лорода или его активных форм, будь это биологическое окисление,
перекисное окисление липидов, биосинтез гормонов и других биоре¬
гуляторов и т д. К ним примыкают и многие патологические явле¬
ния и заболевания, такие, как лучевое поражение, фотодинамиче-
ские явления, интоксикации и др
СН/'
СН30'
,осн3
чосн3
Глава 21. СЕРА
К этому классу веществ
принадлежат весьма важные
в физиологическом отношении
вещества, которые называются
протеинными, . . и составляют
главный материал, служащий
для постройки мелких частей
животных организмов; соедине¬
ния, сюда принадлежащие, за¬
ключают серу.
А М. Бутлеров
21.1. Элемент, простое вещество
Сера находится в шестой группе III периода Периодической си¬
стемы, а поэтому имеет 3з23р43(10-конфигурацию внешнего уровня.
Свойства серы определяются наличием свободного (1-подуровня,
что позволяет ей использовать в образовании химических связей
все шесть электронов внешней электронной оболочки. Таким обра¬
зом, сера имеет валентности 2, 4 и 6. Другая особенность этого
элемента - среднее по величине значение относительной электро-
отрицительности (ОЭО) - 2.5. Это означает, что сера может соеди¬
няться как с электроположительными, так и с электроотрицатель¬
ными элементами, принимая в результате как отрицательные, так и
положительные степени окисления. Действительно, самые харак¬
терные степени окисления серы: -2, О, +4 и +6.
Однако в таких реакциях химическая активность серы должна
быть умеренной. Следовательно сера относительно устойчива и в
свободном состоянии. В нем атомы серы способны к связыванию
между собой, но такая связь не может быть прочной, поскольку
атом серы обладает относительно большими размерами. Тем не ме¬
нее, свободная сера представляет собой в обычных условиях моле¬
кулу Бв, которая имеет циклическое строение. Однако известны и
другие ее модификации линейного строения с переменным числом
атомов в цепи - Бг, Бв.... Б«.
В природе сера встречается как в самородном состоянии, так и
в виде соединений со степенями окисления -2 (сульфиды, напри¬
мер, Рев) и +6 (гипс СаБ04 и др.). В составе живых организмов
сера присутствует в соединениях со степенью окисления -2 - в ос¬
новном в%^минокислотах белков цистеине, цистине и метионине
(некоторыб^белки содержат до 3% серы), в липидах, а также в не¬
которых витаминах и биорегуляторах. Присутствует в биосредах в
небольших количествах и сульфат-анион.
Свободная сера как умеренно активный реагент обладает неко¬
торым дезинфицирующим действием, а потому находит применение
как кожный противогрибковый препарат. В старину для тех же це¬
лей применялось окуривание серой, поскольку БОг, образующийся
при горении серы, также обладает бактерицидным действием.
Итак, сера представлена в природе широким набором соедине¬
ний, которые связаны между собой окислительно-восстановитель-
ными переходами. Если учесть еще одну особенность атома серы -
высокую поляризуемость, что объясняется экранированием валент¬
ных электронов от ядра электронами второго энергетического уров¬
ня, то можно сделать заключение, что и связи с участием серы
также легко поляризуемы, а потому легко разрываются, в особен¬
ности под действием нуклеофильных агентов. Иными словами, сера
обладает высокой нуклеофильностью.
Эти особенности отражаются на круговороте серы в природе
(схема 21.1). Сера и сульфиды легко окисляются кислородом воз¬
духа, превращаясь в диоксид серы и далее в сульфат-ион. Этот
процесс происходит также под действием тиобактерий и тионовых
бактерий.
Микробиологический процесс превращения сульфидов в серу
осуществляется под действием света и может быть выражен урав¬
нением-
окисление
тиобактерии тиобактерии
Э2-, Н2Э — —♦
окисление
окисление
десульфурирующие бактерии
тионовые бактерии
окисление
Я-БН, Я-5-5-Я
анаэробы биосубстрат
аэробы
Схема 21 1. Круговорот серы в природе.
6С02 + 12Н28 -» СвН12Ов + 6Н20 + 12Б
429
Это превращение - один из вариантов фотосинтеза, обеспечи¬
вающий серобактерии органическими веществами
Другая разновидность микроорганизмов - десульфурирующие
бактерии - вызывают переход сульфат-иона в сульфид Наконец,
аэробные процессы вызывают образование серусодержащих биомо¬
лекул, содержащих БН и Б-Б группировки, которые, в свою очередь,
под действием анаэробов трансформируются в сероводород.
21.2. Соединения серы (-2)
Важнейшее соединение серы со степенью окисления -2 - серо¬
водород. Сероводород - газ, хорошо растворимый в воде (в одном
объеме воды растворяется три объема сероводорода). Раствор се¬
роводорода в воде представляет собой слабую сероводородную
кислоту:
НгБ и Н+ + НБ- рКа! = 6.98
НБ- и Н' + Б2- рКа2 = 13
Это означает, что растворимые в воде соли сероводородной
кислоты, - сульфиды и гидросульфиды, гидролизованы согласно
уравнениям:
N328 + НОН ±5 №Н8 + №ОН
№Н8 + НОН ±5 Н28 + №ОН
Сульфиды ряда металлов (Рев, Мпв, гпБ) плохо растворимы в
воде, а некоторых - и в соляной кислоте (Си8, РЬБ, НдБ, ДэгЭз).
Различная растворимость и окраска сульфидов используется в каче¬
ственном анализе для разделения катионов и их обнаружения
Сульфид-ион - сильный восстановитель. Редокс-потенциал
сульфид-иона в щелочной среде составляет -0.48 В, а в кислой -
+0.14 В. Окислители превращают сульфиды в зависимости от усло¬
вий в соединения серы со степенями окисления 0, +4 и +6.
гНгБ + 302 -» 280г + 2НгО
бНгБ + 2КМп04 + ЗНгБО« -» 5Б + 2Мп804 + КгБО« + 8Н20
НгБ + 4Н202 -» НгБОд + 4Н20
Получ^от сероводород его вытеснением из сульфидов или пря¬
мым синтеЛм, который обратим:
N328 + 2НС1 -> НгБТ + 2NaCI
Н2 + Э и Н28
В природе он образуется при гниении в результате деятель¬
ности десульфурирующих и анаэробных серобактерий.
Сероводород сильно токсичен из-за способности прочно связы¬
ваться с катионами меди ферментов дыхательной цепи.
21.3. Тиолы, сульфиды
Замена атомов водорода органическими радикалами приводит к
двум классам соединений - тиоспиртам или тиолам R-SH и тиоэфи-
рам (сульфидам) R-S-R.
Тиолы содержат в своем составе группу SH, называемую
сульфгидрильной (меркапто-) группой. Сульфгидрильная группа
входит в состав белков и некоторых других биомолекул и выполня¬
ет важную биохимическую роль.
Будучи производными сероводорода, тиолы обладают слабо кис¬
лыми свойствами.
R-SH t? Н+ + RS' рКа = 10
Они образуют соли, называемые меркаптидами или тиолятами,
причем аналогия с сероводородом для них состоит в малой раство¬
римости солей тех же катионов, что и для сульфидов. Этому свой¬
ству тиолы обязаны своим тривиальным названием - меркаптаны
(от лат. corpus mercuria aptum - соединения, осаждающие ртуть).
Прочное связывание сульфгидрильной группы с катионами мно¬
гих металлов принципиально важно для жизненных процессов, по¬
скольку именно таким образом катионы многих биометаллов прочно
связываются с белками, образуя металлопротеины, среди которых
имеется множество металлоферментов
С другой стороны, связывание с сульфгидрильной группой пеп¬
тидов - причина высокой токсичности катионов свинца, ртути,
мышьяка, таллия, кадмия и ряда других тяжелых металлов. На
этом основано и создание некоторых боевых отравляющих
веществ, в качестве примера которых можно привести люизит
ф-хлорвинилдихлорарсин)
431
люизит дигидролипоевая кислота
&НСІ
Токсическое действие люизита состоит во взаимодействии с
сульфгидрильными группами молекулы дигидролипоевой (6,8-ди-
меркаптооктановой) кислоты, кофактора оксидазных ферментов
Кофакторы или коферменты - вещества, необходимые для
действия некоторых ферментов, которые потребляются и изменяют¬
ся в ходе биохимических реакций, но в последующих превращениях
восстанавливают свою структуру
Простейшим противоядием (антидотом) при отравлениях произ¬
водными тяжелых металлов может служить водный раствор суль¬
фида натрия, так называемое щелочное сероводородное питье. При
этом происходит уменьшение концентрации иона металла в резуль¬
тате образования его нерастворимого сульфида.
Еще более мощными антидотами здесь служат тиолы, содержа¬
щие более одной сульфгидрильной группы, что обеспечивает более
прочное связывание с ними токсичных катионов. Типичный пример
такого антидота - димеркаптопропанол, впервые примененный
в Англии, а потому именуемый британским антилюизитом
(сокращенно БАЛ). Для лучшей растворимости в воде гидроксиль¬
ную группу в молекуле БАЛ заменяют ионным остатком соли сер¬
ной кислоты, в результате чего получают более удобный для прак¬
тики антидот - унитиол.
О НЭ-СНг Э-СНг
асн=сн -а»( + I —► асн=сн -а</ І + гнсі
а нэ-бн э-сн
люизит БАЛ ІНгОН ІНгОН
сг~
а НЭ—СН2 Э—СНг
* нз-ін ^ Ч-ін *2НС'
ІНгБОзМа Ін^Оз^
унитиол
Сульфгидрильная группа обладает подвижным атомом водорода,
способным замещаться под действием кислородных кислот (АсОН),
то есть может вступать в реакцию тиоацилирования с образова¬
нием сложных тиоэфиров. Эта реакция обратима, а следовательно
432
сложные 1Цоэфиры легко гидролизуются, что объясняется сильной
поляризуемостью связи в их молекулах.
Р8Н + НОАс ^ Г^Ас + НОН
Столь же легко эта связь разрушается и под действием спиртов,
что приводит к превращению тиоэфиров в сложные эфиры.
Р8Ас+Р1ОН РБН + ^ОАс
Это свойство сульфгидрильной группы используется в биохими¬
ческих процессах для переноса ацетильной группы к кислородсо¬
держащим биосубстратам, с помощью молекулы кофермента А, со¬
держащего сульфгидрильную группу (КоА-БН). Так, в частности,
происходит превращение холина в ацетилхолин.
О
KoA-SH + СН3 —С' KoA-S—С-СНз + НгО
°Н 8
г СНз ,
Ко А—S—С—СН3 + НО -СН2 —СНг —N-CH3 ОН' .
8 I холин 6н3 J
СНз
СН3 —G—О—СНг —СНг —N-CH3 ОН'
8 ацетилхолин бнз J
С помощью кофермента А производится биосинтез жирных кис¬
лот, гормонов, а в некоторых микроорганизмах - антибиотиков
Как и сероводород, тиолы являются сильными восстановителя¬
ми. Обычно они претерпевают двухэлектронное окисление, величи¬
на редокс-потенциала которого лежит в интервале 0.2-0 3 В. Это
значение показывает, что действие окислителя in vivo, в первую
очередь, направлено на сульфгидрильную группу биомолекул Сле¬
довательно, такие молекулы выполняют защитную функцию, предо¬
храняя ткани от действия окислителей.
Окисление сульфгидрильной группы слабыми окислителями
происходит с образованием дисульфидов, причем реакция обратима,
а следовательно даже слабые восстановители биосред восстанавли¬
вают дисульфиды в тиолы.
2RSH + (О) S3 RS-SR + НОН
дисульфид
433
Защиту биомолекул от окислителей осуществляет уже упомяну¬
тая дигидролипоевая кислота, превращающаяся при окислении в
липоевую кислоту и трипептид глутатион (см главу 31)
^>^^С00И JO]^
SH éH "[H]” iI
дигидролипоевая кислота липоевая кислота
Glu [О] Glu Glu
2 Cys—SH . Cys— S-S—Cys'
Gly' [H] Gly' Gly
глутатион
Такая защита необходима, так как иначе будут окисляться бел¬
ки, содержащие сульфгидрильные группы. Окислению подвергается
аминокислота белков цистеин, превращаясь при этом в цистин
[О]
2 НООС—СН—СН2—SH i=± НООС—СН—СН2—S—S—СН2-СН-СООН
К " NHj АН;
цистеин цистин
В результате молекулы белков “сшиваются" между собой ди-
сульфидными связями, которые принято называть дисульфидными
мостиками, а следовательно, изменяют пространственное строение,
и тем самым их функции нарушаются.
белок
J0L~1
[О]
белок
В то же время дисульфидные мостики - обязательные струк¬
турные фрагменты ряда белков, определяющие их конформации и
биологические функции. Особенно много дисульфидных мостиков в
молекулах белков волос и ногтей. Дисульфидные мостики сообщают
белковым молекулам структурную жесткость.
Итак, обратимый окислительно-восстановительный переход
тиол-дисульфид происходит под действием слабых окислителей и
восстановителей, и соотношение в них окисленных и восстанов¬
ленных форм белков, то есть цистеина и цистина, зависит от со-
434
держанияЧдсислителей (восстановителей) в биосредах. Для поддер¬
жания гомеостаза это количество должно находиться в строго опре¬
деленных пределах.
Трагическими для белков являются трансформации, связанные
с действием сильных окислителей, которые, как уже сказано
(см. 21.2), должны переводить соединения серы в состояние со сте¬
пенью окисления +6. Такие окислители, например, эндогенная пе¬
рекись водорода, необратимо переводят и дисульфидные мостики, и
сульфгидрильные группы белков в сульфогруппы Р-БОзН, что озна¬
чает их денатурацию.
Особенно сильное нарушение окислительно-восстановительного
режима клеток происходит, как уже отмечалось, при лучевом пора¬
жении Не удивительно, что в качестве радиопротектора (то есть
препарата, предохраняющего организм от лучевого поражения)
применяется Р-меркаптоэтиламин (меркамин) МН2СН2СН28Н,
окисление которого активными формами кислорода - продуктами
кислородного эффекта при радиолизе воды приводит к образованию
цистамина.
[О],
2 ЫН2—СНгСН2—ЭН . ЫН2—СНгСН2—Э—Э—СНгСНг—ЫН2
[Н]
меркамин цистамин
Еще одна особенность сульфгидрильной группы - участие в го-
молитических процессах с образованием относительно мало реакци¬
онноспособных радикалов Р-Б'. Это свойство меркамина также слу¬
жит защитой от действия свободнорадикальных частиц - продуктов
радиолиза воды.
Итак, равновесие тиол-дисульфид связано с регуляцией актив¬
ности ферментов и гормонов, приспособлением тканей к действию
окислителей, восстановителей и радикальных частиц.
Тиолы обычно получают из галогенопроизводных углеводородов
реакцией нуклеофильного замещения или действием на спирты пя¬
тисернистым фосфором
Я-Х + МаНЗ Р-8Н + №Х
бЯ-ОН + Р285 -* 5Я-8Н + Р205
Простейшие предельные меркаптаны - газы, обладающие отвра¬
тительным запахом. Это свойство используется для обнаружения
утечки бытового газа, в который в микроконцентрациях добавлен
этилмеркаптан. Алкилмеркаптаны - основные компоненты отпуги¬
вающего секрета скунса.
435
Тиоэфиры R-S-R1 синтезируют нуклеофильным замещением га¬
логена действием меркаптидов на галогенопризводные:
R-X + NaS-R1 -> R-S-R1 + NaX
Неподеленные электронные пары атомов серы сообщают суль¬
фидам способность вступать в донорно-акцепторные взаимодей¬
ствия в качестве донора электронной пары, в результате чего с га-
логеналканами образуются сульфониевые соли. Сульфониевые соли
ввиду высокой поляризуемости связи С-Б охотно реагируют с
нуклеофилами, алкилируя их. Это свойство служит способом био¬
химического переноса метильной группы от одной молекулы био¬
субстрата к другой:
Конкретно в этой биохимической реакции принимает участие
молекула аминокислоты метионина (СНз-S-R, где R - аминокислот¬
ный остаток), которая при участии аденозина (А) образует соответ¬
ствующую метилсульфониевую соль. Последняя охотно передает
свою метильную группу азотистым нуклеофилам, например, моле¬
куле коламина, в результате чего in vivo получается холин:
3CH3-S*(R)A + HOCH2CH2NH2 -» 3R-S-A + HOCH2CH2N(CH3)3+ + 2И*
Другое общее свойство сульфидов - окисление, которое в зави¬
симости от силы и количества окислителя приводит к производным
серы в степени окисления +4 и +6 - сульфоксидам и сульфонам:
Среди сульфоксидов особого внимания заслуживает простей¬
ший представитель, диметилсульфоксид (СН3)280, полученный
впервые А. М. Зайцевым, учеником А. М. Бутлерова. Это веще¬
ство сочетает в своей молекуле как неполярные, так и сильно по¬
лярные связи, что сообщает ему уникальное свойство быть раство¬
рителем как мало полярных органических веществ, так и ионных
соединений Это позволяет молекулам диметилсульфоксида легко
проникать через клеточные мембраны. Поэтому под названием
R-S-R' +
+ HY —► R-S-R' + СНз-Y +НХ
_ метилированный
осубстрат биосубстрат
биосубстрат
коламин
холин
О
[О] [О] II
R-S-R —► R-S-R —► R-S-R
І і
сульфоксид сульфон
димексиЗ\р применяется в медицине как растворитель для накож¬
ного ведения некоторых лекарственных препаратов.
Среди сульфидов имеется сильно токсичное соединение -
Р,Р‘-дихлордиэтилсульфид, известное как боевое отравляющее ве¬
щество сернистый иприт. Его дегазация основана на окисле¬
нии, осуществляемом действием хлорной извести, и на щелочном
гидролизе.
^СНгСНгОН 2№0Н ^СНгСН^! [0] 0__8,СН2рНаР1 [0] О^СНгСНгС!
'СНгСНгОН *'2№С| 'СН^Н^! 'СН^С! ^ 'СН£НгС\
иприт
21.4. Соединения серы (+4)
Важнейший представитель - газообразный оксид серы(1У) или
сернистый ангидрид.
Сернистый ангидрид растворяется в воде с образованием сер¬
нистой кислоты средней силы, однако нестойкой:
Б02 + Н20 и Н2БОэ
Н2Б03 *5 Н+ + НБОз" КЯ| = 1.4 -10“2 рКа,=185
НБОз- и Н+ + БОз2' Ка, = 6.2 • 1(Г8 РК32 = 7.20
Ей отвечают два типа солей - сульфиты и гидросульфиты
Сульфиты слабо гидролизуются в растворах:
БОз2- + НОН 5 НБОз" + ОН-
10-14 10-'4 7
Кг = — = — — = 16 10“7 ,
' Ка2 б.2. Ю-8
при этом среда становится щелочной
Для гидросульфитов в растворах идут одновременно два конку¬
рирующих процесса - гидролиз и диссоциация
,п-14 1 л-14
НБОз" + НОН ^ Н2БОэ + ОН" к = — = — = 7 1 10'13
Гг К3| 14-10-*
НЭОэ- ЧН'^вОз2- К. =62 КГ*
437
Поскольку константа диссоциации много больше константы гид¬
ролиза, процесс диссоциации превалирует над гидролизом и среда в
растворе будет кислая.
Хлорангидрид сернистой кислоты - хлористый тионил SOCb Он
гидролизуется по уравнению:
SOCI2 + НОН ->• S02T + 2HCI
Его используют для введения хлора в молекулы органических
соединений, например, для синтеза галогенопроизводных из спиртов
(см. 20.7 3):
SOCI2 + ROH -> SO2T + R-Cl + HClT
Соединения cepbi(IV) активны в окислительно-восстановитель¬
ных реакциях, являясь как окислителями, так и восстановителями-
Na2S03 + 2Na2S + 3H2S04 -> 3Na2SO*+ 3S + 3H20
5Na2S03+2KMn04+3H2S04 -> 5Na2S04+2MnS04+K2S04+3H20
В этом - причина высокой токсичности оксида cepbi(IV) и его
производных. Поэтому существует биохимический механизм де¬
токсикации сульфит-иона с участием фермента сульфитоксидазы и
эндогенной перекиси водорода:
БОз2" + Н2Ог —» S042- + H20
Сернистый ангидрид накапливается в атмосфере в результате
вулканической деятельности, при сжигании содержащих серу низ¬
косортных каменных углей, в результате производства целлюлозы
сульфитным методом и вследствие промышленного получения окси¬
дов металлов из сульфидных руд. Это вызывает тяжелые экологиче¬
ские последствия. При высокой влажности воздуха формируется
туман, содержащий сернистую кислоту и серную кислоту (как ре¬
зультат окисления сернистого ангидрида), а также сажу и пыль.
Это явление чаще всего возникает в промышленных районах при
низких температурах и отсутствии ветра, что препятствует рассея¬
нию тумана. Оно называется токсическим смогом. Смог вызывает
поражения легких и в отдельных случаях - гибель людей.
Те же факторы, а также присутствие в атмосфере оксидов азота
и хлористого водорода (отхода ряда химических производств) слу¬
жат причиной кислотных дождей - еще одной формы загрязнения
окружающей среды. Кислотные дожди увеличивают кислотность
водоемов, а тем самым вызывают болезни и гибель их обита¬
телей, нарушают биохимические процессы в почвах, что приводит
к поражению растений. Закисление почвы вызывает растворение, а
438
затем вымй^ание из нее тяжелых металлов, что усугубляет токси¬
ческое действие. В атмосфере кислотных дождей разрушаются по¬
крытия зданий и сооружений, памятники и другие произведе¬
ния искусства, в частности, мраморные, корродируют металлокон¬
струкции и др.
21.5. Соединения серы (+6)
Этой степени окисления серы отвечает серный ангидрид 503.
Растворение его в воде приводит к сильной серной кислоте.
Н2804 -> Н+ + Н804_ рКа1 = -3
Н804" ±5 Н+ + Б042' РК32=2
Серной кислоте отвечают соли - сульфаты и гидросульфаты.
Ряд сульфатов находит применение в медицине. Так, сульфат нат¬
рия №2804'10Н20 - слабительное средство, Мд804'7Н20 применяют
как слабительный и желчегонный препарат, а также при гиперто¬
нии, медный и цинковый купорос Си804‘5Н20 и гп8047Н20 упот¬
ребляются в качестве антисептиков и рвотных препаратов. Гипс
Са804'2Н20 используют для изготовления гипсовых повязок, мало¬
растворимый Ва804 - рентгеноконтрастное вещество, применяемое
для рентгеноскопии желудочно-кишечного тракта.
Хлорангидрид серной кислоты - хлористый сульфурил ЭОгСЬ -
легко гидролизуется:
802С12 + 2Н20 -> Н2804 + 2НС1Т
Серному ангидриду отвечает еще одна кислота - пиросерная
Н282С>7, которую можно рассматривать как продукт присоединения
серного ангидрида к серной кислоте. Ее еще
называют олеумом. Олеум, как и безводная 0 0
НО
'-Ц-О-Ц-ОН
серная кислота, является сильным водоотни¬
мающим средством, используемым в практи- И И
ческих целях для выполнения реакций, иду- О О
щих с выделением воды и для осушения неко¬
торых газов
Концентрированная серная кислота - сильный окислитель, что
иллюстрируют реакции:
Си +2Н2804 (конц.) —> СиЭ04 + 802 + 2Н20
4Еп + 5Н2804 (конц.) —> 4Еп804 + Н28 + 4Н20
439
Соединяясь с перекисной группировкой, остатки сериой кисло¬
ты дают пероксокислоты - моно- и дипероксосерные КИСЛОТЫ, H2SO3
и H2S20e, сответственно (см. 20 4) Пероксокислоты и их соли, пер¬
сульфаты, - сильные окислители В частности, редокс-потенциал
персульфат-иона составляет +2 08 В
Это означает, что под действием персульфата возможно окис¬
ление таких веществ, которые в окисленном состоянии сами служат
сильными окислителями В качестве полезного примера можно при¬
вести превращение катиона марганца (2+) в перманганат-ион, что
используют как качественную реакцию для обнаружения солей мар¬
ганца в растворах:
5(NH4)2S2Oe + 2MnS04 + 8Н2О -» 5(NH4)2S04 + 2HMn04 + 7H2S04
Получают персульфаты электрохимическим путем по схеме:
2S042- - 2е -> 2S20e2' ф° = +2 08 В
Применяют персульфаты в качестве отбеливателей.
В органических соединениях остаток серной кислоты присут¬
ствует в составе сульфоэфиров (иначе называемых алкилсерными
кислотами) ROSO3H и сульфосоединений RSO3H.
Сульфоэфиры обычно синтезируют действием серной кислоты
на спирты:
ROH + HOSO3H -> ROSO3H + Н20
Сульфоэфиры выполняют определенную биохимическую роль.
Во-первых, присутствующий в малых количествах в плазме сульфат-
ион (а он присутствует в биообъектах как конечный продукт окис¬
ления серусодержащих биомолекул) реагирует с гидроксилсодер¬
жащими органическими веществами, попадающими в плазму, а об¬
разующиеся молекулы сульфоэфиров в виде ионных, а следователь¬
но хорошо растворимых в воде соединений - солей соответствую¬
щих сульфоэфиров - выводятся из организма с мочой.
Биохимическая реакция связывания спиртов сульфат-ионом -
частный случай связывания различных полярных групп (NH2i ОН
и др.) органических веществ в организме, который называется
конъюгацией.
Образование связи С-ОН вместо связи С-Н in vivo, как извест¬
но, называется С-гидроксилированием. Этот процесс происходит в
митохондриях и превращает неполярный или мало полярный ксено¬
биотик в более полярное, а следовательно лучше растворимое ве¬
щество, что позволяет ему попасть в плазму.
440
С-г^роксилирование конъюгация
СбНе + [О] -> С6Н5-ОН + НОБОзК -> С6Н5-0-803К + Н20
ксенобиотик
Суммарно С-окисление и конъюгация составляет суть так назы¬
ваемой сульфатной защиты организма от ксенобиотиков. К примеру,
токсичный бензол выделяется с мочой частично в виде калиевой
соли сульфоэфира фенола.
Тем же путем из кишечника выводятся вырабатываемые микро¬
организмами токсичные вещества - фенолы, произ¬
водные индола и другие соединения.
Во-вторых, сульфоэфирная группа входит в со¬
став сульфолипидов, которые составляют малую
часть от общего количества липидов.
Сульфосоединения (или сульфокислоты) полу¬
чают, как известно, гемолитическим сульфохлорированием алканов
(см. 18.3.1) и электрофильным сульфированием аренов и гетаренов
(см. 18.3.5).
Соли длинноцепочечных сульфоалканов - алкилсульфонаты
применяют в качестве моющих средств (ПАВ).
СНгОСОР1
бносстг
бигОБОзН
сульфолипиды
Ыа+
Ароматические сульфосоединения находят использование в ор¬
ганическом синтезе благодаря сильной поляризуемости связи С-Б в
их составе, что позволяет вводить их в реакции нуклеофильного
замещения, а тем самым - синтезировать из них фенолы, тиофено-
лы, нитрилы.
АгБН ♦ ЫаНвОз
Р | ыавн арилсульфохлорид
0 0 0 0
АЮН Аг—!—0№ <Гда0Н Аг—1—ОН 50С'2» Аг—1-С1 Аг— 1-ЫНг
->нво, - А -ион А -на 1 -т.* ь
арилсульфонат р|ксы арилсульфамид
АгСЫ ♦ КНвОз
Сульфосоединения - сильные кислоты, а потому со щелочами
дают соли арилсульфонаты. Подобно серной кислоте, сульфокисло¬
ты образуют галогенангидриды, в частности, хлорангидриды - суль-
фохлориды, действуя на которые аммиаком получают сульфамиды,
в том числе и сульфамидные антимикробные препараты
441
21.6. Тиосульфат-ион
При кипячении водных растворов сульфитов с серой образуются
тиосульфаты
МагБОз + Э -» №г8гОз тиосульфат натрия
Строение тиосульфатов можно представить двумя формулами,
включающими атомы серы в разных степенях окисления- -2 и +6
N3-0^ ^-N3 N3-0^ О-Ыа
✓\> ~ </Ч
Это сообщает молекуле тиосульфата способность к диспропор-
ционированию в кислой среде по схеме:
^ЭгОз + 2НС1 -> 2NaCI + Э + Б02 + Н20
Это означает, что тиосерная кислота не стабильна, существует
лишь в водных растворах и разрушается при хранении.
Свойства тиосульфат-иона определяются в первую очередь
атомом серы со степенью окисления -2, как более реакционноспо¬
собным. Следовательно, этому веществу присущи многие свойства
сульфид-иона. В частности, тиосульфат под действием окислителей
окисляется в свободную серу (при недостатке сильного окислителя
или в случае слабых окислителей) или в сульфат-ион (при избытке
сильного окислителя):
N828203 + С12 (недостаток) + Н2О -» 2№С1 + Э + Нг804
N328203 + 4С12 (избыток) + 5Н20 -> 8НС1 + 2NaHS04
С солями тяжелых металлов тиосульфат натрия дает плохо рас¬
творимые сульфиды, а следовательно его можно применять в ка¬
честве антидота при отравлениях тяжелыми металлами:
N828203+ НдС12 + Н20 -> N82804 + НдвХ + 2НС1
Тиосульфат-ион отдает атом серы и цианид-иону, тем самым
превращая его в нетоксичный роданид-ион:
N328203 + -> N82803 + ^N8
Под действием слабых окислителей, каким является, к примеру,
иод, происходит сшивка двух молекул тиосульфата, напоминающая
сшивку тиолов в дисульфид:
442
2 N5%! Э-Ма + 12 —> На—О-Ыа * 2Ча|
</%> </4 с/Ч>
тетратионат
В результате получается тетратионат натрия Ма2Б40б.
Эта реакция лежит в основе уже упомянутого (см. 19.4) метода
иодометрии, применяемого повсеместно, в том числе и в клинико¬
биохимической и гигиенической практике, для анализа как окисли¬
телей, так и восстановителей.
Следует сказать, что в биосредах наряду с конечным превраще¬
нием серусодержащего биосубстрата в сульфат-ион идет частичное
образование и тиосульфат-иона, а также тетратионат-иона, которые
в итоге также трансформируются в сульфат.
Итак, соединения серы в жизненных процессах выполняют не¬
заменимые функции: защиты тканей от окисления за счет восстано¬
вительных свойств соединений с низшими степенями окисления,
а посредством реакций нуклеофильного замещения - метилирования
и ацилирования - путей биохимического синтеза, а также конъюга¬
ции как защиты от токсичных веществ. Способность к образованию
связей между атомами серы является важным фактором формиро¬
вания пространственной организации белков, что необходимо для
их успешного функционирования.
21.7. Другие элементы 6 группы
В ряду О-Б-Бе-Те-Ро растут радиусы атомов и ионов, понижают¬
ся энтальпия ионизации и относительная электроотрицательность.
Ослабляется проявление неметаллических свойств, соответственно
накапливаются металлические свойства. Падает термодинамическая
стабильность гидридов Н2Э, зато растет сила их как кислот вследст¬
вие роста поляризуемости связи Э-Н В то же время увеличивается
восстановительная активность этих гидридов Кислоты состава
Н2ЭО3 нестойки, кислоты Н2ЭО4 стабильны, но являются сильными
окислителями
Селен является биоэлементом, но в отличие от кислорода и се¬
ры он содержится в биообъектах в микроколичествах (концен¬
трация соединений селена в плазме - не выше 410'3 ммоль/л)
Селен в избыточных количествах сильно токсичен
Будучи аналогом серы, селен замещает последнюю в составе
сульфгидрильных гр’'пп и дисульфидных мостиков В частности,
443
известно, что селен накапливается в волосах и ногтях. Являясь бо¬
лее сильным восстановителем в своих соединениях со степенью со¬
единения -2, селен выполняет защитную функцию, предохраняя
белки от действия эндогенных сильных окислителей, таких, как пе¬
рекись водорода Точно так же соединения селена предохраняют
ткани от токсического действия катионов тяжелых металлов - рту¬
ти и кадмия. Поскольку последний является канцерогеном, соеди¬
нения селена выполняют антиканцерогенную роль. Известны произ¬
водные селена, обладающие радиозащитным действием.
Прослежено несомненное участие селена в действии некоторых
ферментов, регулирующих окисление липидов и белков.
Теллур обнаружен в живых организмах, однако его роль остает¬
ся неясной, если не считать установленного факта, что он, подобно
селену, может замещать серу в серусодержащих пептидах. Это, ес¬
тественно, сказывается на активности ферментов. О биологическом
значении полония сведения отсутствуют.
Глава 22. АЗОТ
Ни растения, ни животные
прямо не поглощают азота
воздуха, а берут его из гото¬
вых азотистых соединений,
притом растения питаются
азотистыми веществами почвы
и воды, а животные азотис¬
тыми веществами, содержащи¬
мися в растениях или других
животных.
Д. И. Менделеев
22.1. Простое вещество, фиксация азота
Электронная конфигурация этого элемента 5 группы II периода
Периодической системы - 2з2р3. Химические свойства азота опре¬
деляются его высокой электроотрицательностью (ОЭО 3.1), а сле¬
довательно, способностью давать множество соединений, в первую
очередь, с электроположительными элементами, в частности, с во¬
дородом. Другая особенность - отсутствие с1-подуровня, что опреде¬
ляет существование у атома азота неподеленной электронной пары.
Это означает, что азот в своих соединениях выступает активным
донором в донорно-акцепторных взаимодействиях.
Хорошо известны соединения, отвечающие всем возможным
степеням окисления азота.
-3 -2
1ЧН3 1ЧН21ЧН2
аммиак гидразин
-1 О
1ЧН2ОН N2
гидроксиламин азот
+ 1 +2 +3 +4 +5
N20 N0 NN02 N02 NN03
закись азота, окись азота, азотистая двуокись азота, азотная
оксид азота(1) оксид азота(М) кислота оксид азота(1\/) кислота
Среди них наибольшей прочностью и значением обладают сое¬
динения со степенью азота -3 (аммиак и его органические произ¬
водные, в том числе, амины в составе биомолекул), +3 (нитрит-ион)
и +5 (нитрат-ион и его органические производные - нитросоедине¬
ния и нитроэфиры)
445
Химия молекулярного азота характеризуется необычайной ки¬
нетической и термодинамической стабильностью молекулы азота
Энергия, которая необходима для расщепления молекулы на атомы
по уравнению
:1^М: И 2Н,
составляет 947 кДж/моль Следовательно, любой процесс, требую¬
щий для своего осуществления предварительной диссоциации моле¬
кулы азота, должен иметь исключительно высокую энергию актива¬
ции, обычно отвечающую температурам порядка 1000° С или выше
Даже при 3000° С степень диссоциации молекулы азота составляет
лишь 0.1%. Восстановление до аммиака - экзотермическая реакция,
лежащая в основе как природного, так и промышленного процессов
связывания азота, - ускоряется, однако, действием катализаторов и
может поэтому протекать в значительно более мягких условиях.
Промышленный процесс синтеза аммиака происходит при 450° С и
давлении около 30 МПа.
N2+ ЗНг ^ 2МНз ДН° = -46 кДж/моль
Биологический процесс связывания азота реализуется при тем¬
пературах биосферы в соответствии с полуреакцией:
М2+6Н+ + 6е и 2МН3
Причина столь мягких условий биологической фиксации азота -
регуляция этого процесса ферментом нитрогеназой, содержащимся
в некоторых микроорганизмах почвы, которые выполняют эту реак¬
цию. Величина ее редокс-потенциала свидетельствует об умеренной
окислительной способности азота (Е° = +0.055 В).
Еще одна важная реакция - взаимодействие с кислородом, ко¬
торое в природных условиях происходит под действием электриче¬
ских разрядов гроз и выполняется в промышленности таким же
способом, хотя и в малых масштабах.
N2 + 02 ^ 21ЧО ДН° =+90 кДж/моль
Сочетание высокой химической активности азота в его соедине¬
ниях и прочности молекулы азота приводит к тому, что един¬
ственным природным ресурсом азота является молекулярный азот,
содержание которого в атмосфере приближается к 80%. Неболь¬
шие запасы селитры, которые разрабатываются в Чили, явля¬
ются минерализованными продуктами жизнедеятельности живых
организмов.
446
ГлобаЛной проблемой поэтому является вопрос фиксации
азота - перевода его в соединения. Общее количество связанного
азота Земли составляет 2.4-10® т Две трети этой массы - результат
биологической фиксации. Примерно 25% азота фиксируется в виде
аммиака, получаемого промышленным синтезом. Оставшиеся 10%
связываются путем образования оксида азота как в результате ат¬
мосферных явлений, так и промышленным способом.
Азот входит в состав молекул большинства классов органиче¬
ских биосоединений - белков, нуклеотидов, биогенных аминов, ге-
мов, витаминов, алкалоидов, мочевины и др.
Таким образом, круговорот азота в природе (схема 22.1) в
основном реализуется биогенным и антропогенным способами.
Микроорганизмы (Аго^ЬаМег) связывают его в аммиак. Другое се¬
мейство микроорганизмов, Nitrozomonas, переводят аммиак в нит¬
рит-ион, далее трансформирующийся под действием еще одной раз¬
новидности бактерий, й^гоЬаЫег, - в нитрат-ион. Совокупное мик¬
робиологическое превращение азота в нитрат называют нитрифи¬
кацией почвы. Кстати, простейший старинный способ получения
нитратов - помещение пищевых растительных отходов в почву на
длительное время с последующим вымыванием нитратов из продук¬
тов такой микробиологической обработки.
производство
ШгоготопаБ азотной
кислоты
I производство
—!— аммиака
ГЖз «
н
N0
N02
►
N03'
производство
азотной
кислоты
производство
азотной
кислоты
денитрификация
Схема 22.1. Круговорот азота в природе
Далее осуществляется процесс потери азота - денитрификация,
состоящая в том, что распад биосубстрата приводит к восста¬
новлению нитрат-иона в свободный азот. Таким образом, почва -
сложный комплекс микроорганизмов, нарушение режима суще¬
ствования которого приводит к серьезным экологическим послед¬
ствиям В частности, они вызываются избыточным введением
аммиачных удобрений, что приводит к перенаселенности почвы
447
Ыигоготопая и тем самым - к насыщению почвы и далее водоемов
токсичным нитрит-ионом.
Нитраты - основной источник азота для большинства зеленых
растений и грибов. Биологически процессы, в ходе которых почвен¬
ные нитраты превращаются в аммиак, называют восстановительной
ассимиляцией нитрата Нитраты могут также использоваться вместо
кислорода при анаэробной генерации энергии в некоторых бакте¬
риях. Продуцирование энергии этим способом называется нит¬
ратным дыханием или восстановительной диссимиляцией нитрат-
иона В некоторых типах бактерий могут реализовываться процессы
обоих типов.
22.2. Аммиак
Аммиак - газ, прекрасно растворимый в воде. В водных раство¬
рах аммиака создается щелочная среда, что видно из уравнения:
МНз+НОН 5 МН4+ + <Ж рКь = 4.75
Образующийся ион аммония ведет себя аналогично ионам ще¬
лочных металлов, образуя с кислотами соли, хорошо растворимые в
воде Соли аммония гидролизованы, имеют кислую реакцию. Вод¬
ный раствор смеси аммиака и соли аммония представляет собой
аммиачный буфер.
Хлорид аммония применяют в качестве мочегонного средства, а
потому изменение кислотности в плазме при его употреблении сле¬
дует учитывать. Его присутствие в биосредах вызывает некоторое
увеличение кислотности за счет гидролиза. Это может использо¬
ваться для предотвращения алкалоза.
Аммиак - активный лиганд и с большинством катионов
с1-металлов образует комплексы - аммиакаты.
Си2++ 4МН3 -> [Си(МН3)4]2+
Это обстоятельство, по-видимому, объясняет его токсичность,
поскольку при попадании в нервные ткани он координируется с ка¬
тионами биометаллов, нарушая их баланс.
Аммиак является восстановителем. Его окисление в зависимос¬
ти от условий и действующего окислителя может происходить по-
разному. Так, реакция с кислородом в отсутствии катализатора идет
до свободного азота:
4МН3 + 302 -> 2Н2 + 6Н20,
в присутй^ии катализаторов образуется оксид азота(Н), что ис¬
пользуется в промышленном способе получения азотной кислоты:
4МН3+ 502 -> 4140 +6Н20,
а ферментативный процесс окисления приводит к нитрит-иону:
2МН3 + 302 -> 2Н+ + 2М02- + 2Н20 (М(гоготопав)
22.3. Амины
22.3.1. Классификация и номенклатура
Аминами являются органические производные аммиака, в мо¬
лекуле которого один, два или три атома водорода заменены ради¬
калами. По этому признаку различают первичные (ШЧНг), вто¬
ричные Ш21ЧН) и третичные Шз1Ч) амины.
В зависимости от характера радикала амины могут быть пре¬
дельными или ароматическими, а также предельно-ароматическими
(метиламин, анилин и метиланилин, соответственно). С атомом
азота может быть связан и разветвленный радикал (например,
трет-бутиламин), и поликонденсированный, что демонстрируется
примером адамантиламина (аминоадамантана), обладающего биоло¬
гическим действием и применяемого в медицине.
СН3— Щ. СН3 —Ж СНз—гд—СН3 ИН2 ^
Ін3 Ін3 СНз —І—СН3
СНз
метиламин диметиламин триметиламин
первичный вторичный третичный грег-бутиламин
рКь = 3.37 рКь = 3.22 рКь = 4.20 аминоадамантан
&
Как можно догадаться, по принципам рациональной номенкла¬
туры название этого класса веществ складывается из названия
15 Эак 675
449
радикалов при атоме азота, именуемого амином. В названии пер¬
вичных аминов по международной номенклатуре аминному атому
азота присваивается название амино, употребляемое с указанием
его местоположения перед названием углеводородной цепи Впро¬
чем, многие амины сохранили свои тривиальные названия, напри¬
мер, анилин. (Анилин - одно из нескольких тривиальных наимено¬
ваний этого соединения, которое называли также кристаллином,
кианолом и бензидамом Термин анилин введен русским химиком
Фрицше в 1840 г )
Кроме аминогруппы в молекулах органических веществ могут
находиться и иные заместители, как это, к примеру, имеет место в
случае сульфаниловой кислоты. Аминный атом азота может быть
включен и в насыщенный цикл. К числу насыщенных гетероцикли¬
ческих аминов относится построенный с напряжением трехчленный
этиленимин, обладающий сильным мутагенным действием. Этилен-
иминовый цикл входит в состав молекул некоторых лекарств. Без
напряжения построены тетрагидропиррольный и пиперидиновый
циклы, присутствующие в молекулах ряда алкалоидов (в том числе
никотина и анабазина, см. 18.4). С их участием, как и с помощью
морфолинового кольца, построены молекулы многих лекарствен¬
ных средств.
Гетероциклическими ароматическими аминами являются, к при¬
меру, пиррол и пиридин. Наконец, аминогруппа может быть свя¬
зана и с гетероциклом, что иллюстрируется примером аденина
(6-аминопурина) - незаменимого фрагмента нуклеиновых кислот.
К числу производных аммиака относятся и органические веще¬
ства, которые можно построить из солей аммония или его гидрок¬
сида замещением всех четырех атомов водорода различными угле-
Н
Н
н
пиперидин
Н
морфолин
этиленимин тетрагидропиррол
Н
Н
пиррол
пиридин
аденин
450
водороднЛ(1и радикалами, как это видно на примере тетраметилам-
моний гидроксида:
СН3
СН3—Л—СН=СНг ОН
£н3
О
сГ
тетраметиламмоний
гидроксид
триметилвиниламмоний
гидроксид (нейрин)
Ы-алкилпиридиний
хлорид
Другим примером тетразамещенных аммонийных произ¬
водных - четвертичных аммониевых оснований или их солей -
служит нейрин, токсичное вещество, образующееся в процессе
гниения тканей животного происхождения.
Четвертичный атом азота может входить в состав гетероциклов,
например, соответствующей соли из ряда пиридина - Ы-алкил-
пиридиниевой соли. К таким четвертичным солям относятся неко¬
торые алкалоиды. Кроме того, четвертичный атом азота входит в
состав многих лекарственных веществ и некоторых биомолекул.
Выше приведенные примеры демонстрируют многообразие
аминосоединений и их большое медико-биологическое значение.
К этому необходимо добавить, что аминогруппа входит в состав та¬
ких классов биомолекул, как аминокислоты и белки, нуклеиновые
кислоты, присутствует в ряде природных производных углеводов,
именуемых аминосахарами. Аминогруппа является важнейшей
функциональной группой алкалоидов и многочисленных лекар¬
ственных препаратов самого различного назначения. Отдельные
примеры таких веществ будут приведены ниже.
Наличие свободной электронной пары азота сообщает аминам
свойства оснований. Поэтому характерной особенностью аминов
является реакция с кислотами с образованием соответствующих
аммониевых солей, что видно из реакции для первичного предель¬
ного амина:
Аналогично из анилина образуется анилиниевая соль, из пири¬
дина - пиридиниевая и т.д
22.3.2. Амины как органические основания
алкиламмоний
хлористый
Н
15*
451
Подобно аммиаку, амины в водных растворах создают щелочную
среду, согласно уравнению:
яын2 + н2о «=± го<Нэ+ + он-,
константа равновесия (Кь) которого характеризует силу данного
амина как основания (чаще используют величину рКь).
Электронодонорные заместители, к которым относятся алкиль¬
ные группы, должны увеличивать основность аминов, поскольку
увеличивают электронную плотность у атома азота. Так, метиламин
(рКь = 3.37) является более сильным основанием, чем аммиак
(рКь = 4.75), а диметиламин (рКь = 3.22) - более сильное основа¬
ние, чем метиламин. Однако при переходе к триметиламину, во¬
преки ожиданию, основность несколько падает (рКь = 4.20). Причи¬
на этого состоит в том, что с увеличением числа заместителей
у атома азота подход протона все более затрудняется Таким обра¬
зом, здесь речь идет не об электронном, а пространственном влия¬
нии заместителей. Это воздействие заместителей называют стери-
ческим фактором.
Ароматические амины - более слабые основания, чем предель¬
ные, из-за электроноакцепторного эффекта ароматического кольца.
Поэтому невысока основность и пиридина. Накопление фенильных
заместителей заметно подавляет активность электронной пары ато¬
ма азота. Так, рКь дифениламина составляет 13.12, а трифениламин
совсем не проявляет свойств основания.
Чрезвычайно низкая основность пиррола вызвана тем, что в его
молекуле электронная пара атома азота вовлечена в образование
бя-электронной ароматической связи. На её связывание с протоном
требуется значительная дополнительная затрата энергии. В резуль¬
тате образования пирролиевых солей ароматическая связь, а, следо¬
вательно, и стабильность молекулы исчезают. Этим объясняется то,
что пиррол в кислой среде быстро осмоляется.
Интересно отметить, что сильный электроноакцепторный эф¬
фект, оказываемый пиррольным циклом на атом азота, приводит к
ослаблению связи N-14, в силу чего пиррол способен проявлять
свойства слабой кислоты (рКа = 16.5). Под действием такого ак¬
тивного металла, как калий, может быть приготовлена его калиевая
соль - пиррол-калий.
Кислотные свойства связи N-14 пиррольного цикла объясняют, в
частности, способность порфина и его природных производных к
образованию солей с катионами металлов. Два пиррольных кольца
молекулы порфирина координируются с катионом за счет элек¬
тронных пар своих атомов азота, а два других - заменяя атомы
452
водорода^ак и молекула самого пиррола при образовании пиррол-
калия. Именно такими солями и являются хлорофилл и гемоглобин
(главы 33 и 34).
металлокомплекс
порфина
22.3.3. Способы получения аминов
1. Алкилирование аммиака и аминов галогеналканами
(синтез Гофмана). Эта реакция представляет собой нуклеофиль¬
ное замещение по поляризованной связи углерод-галоген с замеще¬
нием последнего атомом азота, обладающим нуклеофильной актив¬
ностью за счет электронной пары. На примере взаимодействия ам¬
миака с иодистым алкилом видно, что процесс приводит к посте¬
пенному замещению всех атомов водорода по схеме:
1. МНз + ^-Х5- -> [РМН3+]Х- + МНз 5 РМН2 + 1ЧН4Х
алкиламмоний алкиламин
2. РМН2 + ^-Х5- -> [Р2МН2+]Х- + МН3 ^ Р2МН + МЩХ
диалкиламмоний диалкиламин
3. + ^-Х5"-* [Р3МН+]Х- + МН3 5 РзМ + 1ЧН4Х
триапкиламмоний триалкиламин
4. Р3М + ^-Х5" -> [РХрС
тетраалкиламмоний
В результате образуется смесь первичного, вторичного, тре¬
тичного аминов и четвертичной аммониевой соли Чаще синтез
Гофмана применяют для синтеза третичных аминов и четвертичных
солей, поскольку на первых стадиях реакцию обычно остановить
не удается
453
Так, действие на триметиламин хлористым цетилом приводит в
одну стадию к четвертичной аммонийной соли - триметнлцетилам-
моний хлориду
Это вещество так же, как и его аналоги с достаточно длинной
углеводородной цепочкой (>С 12)> обладает моющим действием
СН3 СН3 СН3
СН3—N + С16НззС1 —► СН3—г!|-С16Нзз СҐ СН3—Л^С1вНзз Сі'
ІНз ІН3 ІН2
триметилцетил-
аммоний хлорид
диметилцетилбенэил-
аммоний хлорид
В отличие от обычного мыла такие соединения в поверхностно¬
активной органической части молекулы несут положительный за¬
ряд, а потому называются катионными или инвертными мылами.
Инвертные мыла используются в кислой среде. В структуру ин-
вертного мыла можно ввести заместитель, проявляющий антимик¬
робную активность. В этом случае синтезируют бактерицидные мы¬
ла, применяемые в хирургической практике. Примером бактерицид¬
ного мыла служит диметилбензилцетиламмоний хлорид. Инвертные
мыла можно синтезировать также алкилированием пиридина или
других циклических аминов. Так, из пиридина и хлористого цетила
получают хлорид цетилпиридиния.
СібНззСІ . . л.
і г м цетилпиридинии
^ хлорид
СІ
ІібНзз
2. Алкилирование аммиака и аминов спиртами. Реакция
аминов со спиртами, которая в промышленности осуществляется
при нагревании под давлением, идет, как правило, только по пер¬
вым двум стадиям, например:
Обрй¥чмая замена спиртовой группы на аминогруппу играет
важную роль в биологических процессах в живых тканях и проис¬
ходит ферментативным путем.
3. Декарбоксилирование аминокислот также является ха¬
рактерным биохимическим превращением. В лаборатории его можно
осуществить нагреванием в щелочной среде. Особенно легко оно
идет под действием микроорганизмов при гниении:
МН2СН(Р)СООН -> МН2СН2Р + С02
аминокислота амин
Резкий неприятный запах гниющих белков в основном опреде¬
ляется образованием аминов, а также сероводорода.
Белки состоят из 20 аминокислот, а это означает, что в биосре¬
дах присутствует не менее 20 биогенных аминов. На самом деле их
значительно больше, так как многие первично образующихся ами¬
нов претерпевают дальнейшие трансформации, порождая новые
разновидности биогенных аминов, многие из которых являются
биорегуляторами.
4. Восстановление азотсодержащих органических соеди¬
нений. Реакция Зинина (1842 г.) состоит в восстановлении нитро¬
соединений по схеме:
нитробензол нитрозобенэол фенилгидроксиламин анилин
Этой реакцией Зинин впервые синтезировал анилин, а также
выделил и исследовал некоторые промежуточные продукты Откры¬
тие Зинина играет важную практическую роль, т.к на основе ани¬
лина развились анилино-красочная промышленность и синтез цело¬
го ряда новых лекарственных соединений. Ценны и промежуточные
продукты этого восстановления - ароматические нитрозосоединения
и гидроксиламины. Поэтому, по словам Гофмана, имя Зинина за¬
служивает быть записанным золотыми буквами в историю химии
Можно получать амины восстановлением и других азотсодер¬
жащих органических соединений, в частности, нитрилов, оксимов и
амидов:
455
> РСН2ЫН2
нитрил амин
(СНз)2С=1ЧОН !* (СНэ)2СНМН2 + НгО
оксим ацетона иэопролиламин
CHзCONH2 > СН3СН2МН2 + Н20
ацетамид этиламин
22.3.4. Химические свойства аминов
I. Ацилирование. Кроме реакций алкилирования и образова¬
ния солей с минеральными кислотами первичные и вторичные
амины вступают в реакции ацилирования с различными кислотами
(подобно спиртам), в результате чего образуются амиды.
Так, например, анилин реагирует с уксусной кислотой:
ацетанилид фенацетин парацетамол
В итоге получается соответствующий амид, именуемый ацета-
нилидом. Это вещество под названием антифебрин явилось первым
синтетическим лекарством, обладающим жаропонижающим дей¬
ствием. Из-за токсичности он был со временем заменен другими
препаратами. Среди них находятся фенацетин, который в отличие
от антифебрина в лара-положении к амидной группировке имеет
простую эфирную группировку, и парацетамол, обладающий гид¬
роксильной группой.
Ацилирование аминогруппы играет важную биологическую роль.
В частности, амины или кислоты, попадающие в организм, выводят¬
ся из него в виде амидов. Например, бензойная кислота в этих
условиях превращается в бензамид:
С6Н5СООН + N43 -> СвН5СОМН2 + Н20
Ацилирование аминов может происходить и под действием
минеральных кислот, например, фосфорной. Так, из фосфорной
456
кислоты <41 диметиламина образуется СНз СНэ
гексаметиЛфосфотриамид (иначе гек- СНэ чм'
саметапол), нашедший применение 'N1—£=о Ьм—£=о
как эффективный растворитель. снз А N
Аналогичный амид, получаемый снз^ снэ ^
ацилированием этиленимина, под на- гексаметипфосфо- тэф
званием ТЭФ (триэтилениминфосфо- триамид
триамид) применяется как лекарственное средство для лечения не¬
которых злокачественных опухолей.
2. Реакции первичных аминов с альдегидами и кетонами
происходят, как и взаимодействие карбонильных соединений с ам¬
миаком (глава 24), и приводят к образованию иминов, например:
МН2 М=СНСНЭ
(^) ♦ С„зСН=0-Н-25 (^)
фенилимин ацетальдегида
3. Окисление происходит различным образом для первичных,
вторичных и третичных аминов.
Первичные амины при окислении дезаминируются, теряя моле¬
кулу аммиака и образуя карбонильное соединение.
[О] /Р
СН3-СН2-МН2 * СНз-С' + N43
У вторичных аминов при окислении наблюдается дезалкилиро-
вание, т.е. от молекулы амина отщепляется алкильная группа:
+ СН2=0 формальдегид
Г4-0кисление является характерным свойством третичных ами¬
нов, которое осуществляется по общей схеме.
Ю1
Я3м Я3Ы-*0 N-окись
457
Процессы дезаминирования, дезалкилирования и М-окисления
происходят при ферментативном окислении аминов и их надо учи¬
тывать, рассматривая процессы метаболизма аминопроизводных нз
числа лекарственных препаратов, алкалоидов, ОВ и т.д. при их по¬
падании в организм.
4. Замещение атомов водорода бензольного кольца аро¬
матических аминов. Как уже сказано ранее, аминогруппа яв¬
ляется заместителем первого рода Анилин и его аналоги легко
вступают в реакции галогенирования, нитрования и сульфирова¬
ния, причем входящий заместитель ориентирован в орто- и пара¬
положения.
5. Амины являются активными лигандами, образуя ком¬
плексы, как и сам аммиак-
Особенно активны ди-, три- и полиамины, так как выступают в
качестве полидентатных лигандов, образуя стабильные хелаты.
В качестве примера приводим реакцию катиона меди с бидентатным
этилендиамином-
Координация катионов биометаллов с содержащими атомы азота
группами белков, как и связывание их с сульфгидрильными группа¬
ми, и есть важнейшие виды взаимодействия между ними, опреде¬
ляющие образование и строение металлопротеинов, а также ток¬
сичность тяжелых металлов.
Метиламин, диметиламин, диэтиламин и другие простейшие
представители алифатических аминов находят применение в синте¬
зе лекарственных веществ, а некоторые из них (метиламин, диме¬
тиламин, триметиламин) содержатся в ряде биообъектов.
Анилин, метил- и диметиланилины, дифениламин используют
для получения многих медицинских препаратов (стрептоцид, и др.),
а также красителей, взрывчатых веществ и т.д.
М"++2пР1ЧН2-> (М^ЫНгЬГ
2+
22.3.5. Важнейшие представители аминов
и их медико-биологическое значение
458
Диамшш - путресцин и кадаверин - образуются в процессе
гниения белков при гнойных процессах и распаде трупов.
МН2(СН2)4МН2 МН2(СН2)5МН2 МН2(СН2)вМН2
путресцин
1,4-диаминобутан
кадаверин гексаметилендиамин
1,5-диаминопентан 1,6-диаминогексан
Их гомолог гексаметилендиамин вырабатывается для произ¬
водства полимерного материала найлона (глава 31).
Сильной биологической активностью обладают аминопроизвод¬
ные, имеющие функциональные заместители в (5-положении к
аминогруппе. Так, эмбихин, содержащий в своем составе два
Р-хлорэтильных заместителя, подавляет рост злокачественных опу¬
холей; заменой в его молекуле СНэ-группы на разнообразные замес¬
тители, синтезируют различные цитотоксические препараты, нахо¬
дящие применение в онкологической практике.
Замена метильного радикала еще одной Р-хлорэтильной груп¬
пой приводит к образованию сильного отравляющего вещества, об¬
ладающего кожно-нарывным действием - азотистого иприта.
Незаменимую физиологическую роль выполняют некоторые
Р-аминоспирты и их производные. Так, 2-аминоэтанол (или кола-
мин), и холин входят в состав липидов нервных тканей в виде
сложных эфиров с фосфорной кислотой - фосфорилколамина и
фосфорилхолина (глава 26).
сн3 снэ снэ
1+ |+ -н2о и
СН3-М-СН2СН2 ОН' СНэ-М-СН2СН2 ОН' =— СНЭ-М-СН=СН2 ОН
6нэ АсОСНз 6н3 Ан 6н3
ацетилхолин холин нейрин
При дегидратации холина образуется нейрин
Сложный эфир холина и уксусной кислоты - ацетилхолин вы¬
полняет функцию передачи нервных импульсов, то есть является
нейромедиатором
Сильное воздействие на центральную нервную систему оказ!>
вают симпатомиметические амины - эфедрин (1-фенил-2-метил-
аминопропанол-1), содержащийся в некоторых растениях, и адре¬
налин, входящий в состав ткани надпочечников Адреналин и ряд
СНЭМ(СН2СН2С1)2
метилди(Р-хлорэтил)амин
эмбихин
М(СН2СН2С1)3
триф-хлорэтил)амин
азотистый иприт
459
его аналогов (норадреналин, дофамин) объединяют общим названи¬
ем катехоламины. Катехоламины - гормоны надпочечников, яв-
дяющиеся медиаторами нервной системы
Адреналин принимает участие в передаче нервного возбужде¬
ния, оказывает влияние на сердечно-сосудистую систему и обмен
веществ Его используют при шоке, остановке сердца, падении кро¬
вяного давления, как и его аналог эфедрин. К числу симпатомиме-
тиков, то есть веществ, возбуждающих симпатическую нервную си¬
стему, относятся и некоторые синтетические препараты, например,
фенамин (2-амино-1-фенилпропан), который также применяют в
специальных целях.
Еще одним примером аминов-биорегуляторов служит трипта-
мин, 3-(Р-аминоэтил)индол, имеющий сильное симпатолитическое
действие (блокирующий нервную систему).
он
11
адреналин триптамин
Радиопротекторным, т.е. радиозащитным эффектом обладают
амины с ß-меркаптогруппой. Их простейшим представителем яв¬
ляется меркамин NH2CH2CH2SH (см. 21.3).
22.4. Гидразин и гидроксиламин
Степень окисления азота -2 имеет соединение, именуемое гид¬
разином. Его получают реакцией хлора с аммиаком с промежуточ¬
ным образованием хлорамина:
* NH3 + CI-CI -► NH2-CI + HCl
хлорамин
NH3 + NH2-CI -> NH2-NH2 + HCl
гидразин
460
Оба ац^ма азота в молекуле этого вещества способны к присое¬
динению пр&тонов:
Ш2-МН2 + НС1 -> [МН2^НЭ+1СГ+НС1 -» [МНэ+-МНз+1гСГ
гидразиний дигидразиний
Гидразин - сильнейший восстановитель, его горение в кислоро¬
де сильно экзотермично, а потому он используется в качестве ра¬
кетного топлива. 1,1-Диметилгидразин (СНз)2МН2 - также хорошее
топливо.
МН2-МН2 + 02 -> 1Ч2 + 2Н20 ДН° = -626 кДж/моль
Окислительные свойства гидразина выражены очень слабо, од¬
нако проявляются в реакции образования озазонов карбонильных
соединений (см. 24.3).
Многие органические производные гидразина находят примене¬
ние в медицине, т.к. обладают сильной биологической активностью.
Например, гидразид у-пиридинкарбоновой кислоты о=С—МНМН2
(изоникотиновой кислоты), получаемый ацилиро- 7
ванием гидразина по аналогии с ацилированием
аминов, под названием тубазид употребляется
при лечении туберкулеза N
Гидроксиламин - еще одно из соединений азо- тубазид
та. Степень окисления азота в нем -1, и следова¬
тельно, гидроксиламин обладает и окислительными, и восстанови¬
тельными свойствами.
Это вещество перспективно как источник антидотов ФОС (см.
главу 23).
При ацилировании гидроксиламина, идущему по атому азота,
получаются гидроксамовые кислоты, фрагмент которых присут¬
ствует в некоторых железосодержащих белках, сидерофорах (гла¬
ва 33) и входит в состав средств для лечения отравлений катионом
железа (глава 35).
♦ н*он - ^„нон "Т—"
22.5. Нитрит-ион
Степени окисления +3 отвечает азотистая кислота, которая яв¬
ляется нестойкой и разрушается в результате реакции диспропор-
ционирования. Поэтому ее следует считать кислотой слабой, хотя
461
по величине рКа ее следовало бы отнести к числу КИСЛОТ средней
силы.
ЗН1Ч02 -> 2140 + NN03 + н20
HN02 5 Н* + N0/ рКа = 3 4
Из-за неустойчивости азотистой кислоты обычно вместо нее
имеют дело с ее солями - нитритами. Нитрит-ион, как частица с
промежуточной степенью окисления азота, способна вступать в ре¬
акции как окисления, так и восстановления, что используется для
целей качественного анализа при обнаружении нитрит-иона, обла¬
дающего высокой токсичностью:
2КМп04+51™02+ЗН2804 -> 51^0з+2Мп804+2К2804+ЗН20
2К1 + 21^02 + 2Н2804 -> 2К2804 + 12 + 2N0 + 2Н20
Токсичность нитрит-иона, отчасти, объясняется его окисляющим
действием на катион Ре2+ в составе гемоглобина, в результате чего
тот превращается в катион Реэ+, а гемоглобин тем самым - в мет-
гемоглобин, который не связывает кислород, а потому возникает
кислородная недостаточность.
НЬ'Ре2+ + N0^ +2Н+ -> НЬ Ре3++ N0 + Н20
Важную роль играют реакции аминов с азотистой кислотой, ко¬
торые происходят различно для аминов разного типа.
Ключом к пониманию этой группы реакций может служить вза¬
имодействие с азотистой кислотой аммиака, представляющее собой
конмутацию - окислительно-восстановительное превращение, в ко¬
тором азот со степенями окисления -3 и +3 приобретает нулевую
степень окисления:
МНз+НМ02-> [МК^02] -» М2+2Н20
Сходно выглядит и реакция первичных алифатических аминов,
в которой одним из конечных продуктов также является азот, а ка¬
ким образом это происходит - ясно из схемы:
ямн2+ нсиую н 0» [ямн^д^о] — [я-м=м-он] — яон +
2 диазосоединвнив
В этом сложно идущем превращении в качестве промежуточно¬
го продукта возникает так называемое диазосоединение, которое
является нестойким и сразу разлагается.
462
Реакцию можно использовать в синтетических целях для пре¬
вращения Аминогруппы в спиртовую, а также и в анализе - для ко¬
личественного определения аминогрупп по объему выделившегося
азота. В частности, этот метод (метод Ван-Слайка) употребляется в
медико-биологической практике для определения первичных алифа¬
тических аминогрупп в биосредах.
Еще один аспект этой реакции - дезаминирование биомолекул,
содержащих аминогруппы, под действием нитрит-иона при его по¬
падании в организм. Это, в частности, приводит к повреждению
молекул ДНК, а следовательно, усугубляет токсическое действие
нитрит-иона.
Первичные ароматические амины под действием азотистой кис¬
лоты образуют соответствующие ароматические диазосоединения,
которые в отличие от предельных диазосоединений устойчивы в хо¬
лодных водных растворах из-за стабилизирующего действия арома¬
тического заместителя. Эти диазосоединения в солянокислом рас¬
творе обычно существуют в виде солей арилдиазония:
Аг— 1У1Н2 + НОЫО (№N02 + НС1) » [Аг— МН^Ээ] —
-Н20
[Аг—N=N-0^ -НС1 » [Аг—N=N1* СГ Н2° * А г—ОН + Ы2 + НС1
'н2° соль диазония 1
При кипячении растворов соли диазония превращаются в соот¬
ветствующие фенолы.
Реакции диазосоединений и солей диазония отличаются разно¬
образием, причем они могут происходить как с выделением азота,
так и без него. Превращения с выделением азота служат для осу¬
ществления реакций замещения диазогруппы на галоген, циано-,
меркаптогруппу или другие заместители:
АгВг
Си2Вг2|-М2
СиСЫ
[Аг—М=М+1а- —► АгСЫ
-М2
КЗН|-М2
АгЭН
К числу реакций без выделения азота относится восстановле¬
ние диазосоединений, которое является способом синтеза арилгид-
разинов:
НОН
АгОН 4—
-М2
463
Аг—N=N+01* АтЫНЫНуНС!
Поэтому ароматические диаэосоединения являются полезными
промежуточными веществами для превращения ароматических ами¬
нов в самые разнообразные продукты
Важное свойство ароматических диазосоединений представляет
собой реакция азосочетания, в результате которой диазогруппа Аг№
замещает атом водорода ароматического кольца, содержащего за¬
меститель первого рода X
@-н=м*сг * . НСІ
В этом превращении, которое является частным случаем элек-
трофильного замещения в ароматическом ряду, катион диазония
(Аг^+) играет роль электрофильной частицы. Источник катиона ди¬
азония называют диазосоставляющей; участвующее в реакции аро¬
матическое соединение, активированное заместителем первого рода,
названо азосоставляющей, а продукт - азосоединением.
Многие из полученных таким путем азосоединений интенсивно
окрашены, а потому составляют семейство азокрасителей. Азокра¬
сители применяются, в частности, в качестве кислотно-основных
индикаторов, например, метилоранж:
Для медицинских целей реакция образования азокрасителей
ценна как метод анализа фармпрепаратов с аминогруппами (напри¬
мер, стрептоцида) и способ их обнаружения в биосредах.
Вторичные амины, независимо от типа заместителей, с азотис¬
той кислотой образуют нитрозоамины.
РгМН + НОСЮ —> НгО + ЯгМ-^О диалкил(арил)нитрозоамин
Эти вещества являются сильно токсичными (проявляют канце¬
рогенную активность). Поскольку вторичные амины присутствуют
диазосоставляющая
азосоставляющая
метилоранж
азокраситель
464
в биосред%, становится понятной чрезвычайно высокая токсич¬
ность нитрит-иона, содержание которого в сточных водах не должно
превышать по гигиеническим требованиям 10 мг/л.
Третичные алифатические амины с азотистой кислотой практи¬
чески не реагируют, поскольку образующиеся соли слабой кислоты
неустойчивы.
Третичные ароматические амины в отличие от предельных всту¬
пают в реакцию с азотистой югслотой. Этот процесс представляет
собой электрофильное замещение, которое происходит под действи¬
ем нитрозоний-иона (N0*), образующегося из азотистой кислоты,
подобно нитроний-иону из азотной (см. 18 3 5). Реакция облегчена
тем, что ароматическое кольцо здесь активировано аминогруппой,
являющейся заместителем первого рода-
(Снэ)2М —^3^—н + нОМО —► (СНз)2М —М=° + Н0Н
л-нитрозодиметиланилин
‘22.6. Нитрат-ион
Степень окисления азота +5 представлена сильной азотной кис¬
лотой:
НМОэ -> Н++ N03" рКа = -1.6
Ее соли - нитраты, хорошо растворимы в воде Нитрат-ион яв¬
ляется сильным окислителем и, в зависимости от условий, может
восстанавливаться до различных продуктов Обычно он реагирует в
соответствии с полуреакциями:
N03" + 2Н* + 2е -» 1Ч02 + Н20
N03” + 4Н+ + Зе -> N0 + 2Н20
Одна из качественных реакций на нитрат-ион, в частности, про¬
исходит по второй схеме.
6РеЭ04 + 21^0Э + 4Н2504 -> ЗРе2(804)з + 2N0 + К2Э04 + 4Н20
Эта реакция имеет важное значение для регуляции сердечно¬
сосудистой деятельности Как выяснилось, в результате метаболи¬
ческого производства нитрат-иона (а он продуцируется в количест¬
вах до 400 мг в сутки) и его реакции с катионом Ре2+ в гемоглоби¬
465
не стенками сосудов продуцируется N0, что необходимо для их
расширения. Дефицит продукции нитрат-иона компенсируется ме¬
дикаментозным использованием препаратов общего типа RONO2, то
есть нитроэфиров. Последние, как всякие сложные эфиры, подвер¬
гаются in vivo гидролизу до нитрат-иона. Тринитроглицерин -
типичный, но не единственный препарат этой группы.
Несомненно, что в больших концентрациях нитрат-ион токси¬
чен. Однако вопрос о его опасности в малых дозах подвергается
сомнению и дискутируется в связи с проблемой использования
нитратов в сельском хозяйстве, а также в качестве пищевых кон¬
сервантов.
Окислительные свойства нитрат-иона зависят во многом от
природы действующего восстановителя. Подбором условий можно
реализовать переход азота практически в любую степень окисле¬
ния от +4 до -3. Примером этого может служить реакция нитрат-
иона с алюминием, которая также применяется для обнаружения
нитрат-иона:
Органические производные азотной кислоты содержат нитро¬
группу и их можно разделить на две группы: нитросоединения
Р-1Ч0г и нитроэфиры Р-0-№2.
Нитросоединения могут быть получены реакцией гомолитиче-
ского нитрования алканов и электрофильного замещения в ряду
аренов. Эти вещества не встречаются в природе, за исключением
антибиотика левомицетина (хлоромицетина).
Некоторые нитросоединения употребляются в производстве
пестицидов. В качестве примера можно упомянуть 2-метил-4,6-
динитрофенол.
Полинитросоединения представляют интерес как взрывчатые
вещества. Примером этого является наиболее важное бризантное
вещество 2,4,6-тринитротолуол, известный как тротил.
Значение нитросоединений в синтезе аминов обсуждено выше.
Нитроэфиры как лекарственные препараты уже упоминались.
Полинитроэфиры, такие, как тринитроглицерин и нитроклетчатка,
применяются в качестве взрывчатых веществ, как и полинитросое¬
динения.
8AI + 3KN03 + 5К0Н + 2Н0Н -» 8КАЮ2 + 3NH3T
левомицетин
466
22.7. Оксиды азота
Известны оксиды, отвечающие всем возможным положительным
степеням окисления азота: N20, N0, N203, N02, N205. Два из них -
азотистый и азотный ангидриды, N203 и N205, соответственно, прак¬
тического значения не имеют, а потому не рассматриваются. Окси¬
ды азота(1) и (II) - N20 и N0 - несолеобразующие оксиды, неспо¬
собные к реакциям с кислотами и основаниями.
Оксид азота(1), известный как закись азота, может быть полу¬
чен реакцией дисмутации нитрата аммония, происходящей при на¬
гревании:
N^N03 -> N20 + 2Н20
Закись азота применяется при наркозах (механизм действия не
известен) и при некоторых специальных криопроцедурах, например,
в офтальмологии.
N0 - оксид азота(Н) или окись азота, может быть получен как
действием восстановителей на нитрит- и нитрат-ионы, так и катали¬
тическим окислением аммиака или связыванием азота с кислородом
при высоких температурах или в электрическом разряде.
Оксид азота(П) имеет значение для медицины, так как выпол¬
няет роль биорегулятора кровяного давления.
Еще одно существенное в этом аспекте свойство - способность
окиси азота к координации с катионами с1-металлов, и в том числе,
с катионом ?е2+ в составе гемоглобина, что обусловливает токсич¬
ность окиси азота, вызывающей гипоксию, как и угарный газ.
Оксид азота(Н) легко окисляется уже на воздухе до диоксида
N02. Последний также токсичен, так как является сильным окисли¬
телем и оказывает разрушающее действие на живые ткани.
При работе двигателя внутреннего сгорания в нем в небольших
количествах имеет место связывание азота за счет окисления.
N2+ 02 -» 2N0 2N0 + 02 -» 2N02
Оксиды азота являются активными сенсибилизаторами (Б), вы¬
зывающими по уже обсужденному механизму (см. 20 1) образова¬
ние синглетного кислорода, озона и молекулярного кислорода-
Б + 1ти —» Э* Э* + 30г —» Э + 102
02 + 10г —> Оз + О
С другой стороны, при недостатке кислорода в двигателе внут¬
реннего сгорания может происходить не только полное сгорание
467
бензина до углекислого газа и воды, но и неполное окисление до
альдегидов-
Я-СНз + 02-> Р-СН=0 + Н20
альдегид
Присутствующие в выхлопных газах альдегиды реагируют с ак¬
тивными формами кислорода и диоксидом азота.
В результате в атмосфере накапливаются пероксиацилнитраты:
Эти вещества нестойки и как всякие вещества с перекисной
группировкой легко разрушаются по радикальному механизму при
взаимодействии с влагой. В результате образуются, с одной сторо¬
ны, агрессивная азотная кислота, а, с другой стороны, активные
свободные радикалы.
Это явление носит название фотохимического смога и тре¬
бует для возникновения интенсивного солнечного освещения,
активного транспортного движения и условий для появления за¬
стойной зоны воздуха в приземном слое атмосферы данной терри¬
тории. Фотохимический смог характерен для больших тропических
и субтропических городов. Впервые такой смог был отмечен в Лос-
Анджелесе в 1944 году.
Итак, N0, N02, Оз, Р-СН=0, Я-СОзГ^Ог - основные компоненты
фотохимического смога. Все эти вещества химически активны и
разрушают живые ткани, вызывая удушье, а в экстремальных слу¬
чаях и гибель людей. Агрессивные химические компоненты вызы¬
вают увядание растений, а также коррозию металлических кон¬
струкций, разрушение резины, красителей и других материалов и
сооружений.
В целом отметим, что азот незаменим в жизненных процессах.
При этом основная роль отводится соединениям азота со степенью
окисления -3, которые содержат в своем составе или аминогруппы,
или гетероциклические атомы азота (в кольцах имидазола, пирими¬
дина, индола, пурина, порфирина и др.). Атом азота сообщает тем
биомолекулам, в которых он присутствует, основные свойства,
высокую нуклеофильность и лигандную активность, проявляю¬
щуюся в комплексообразовании с катионами биометаллов из числа
с1-элементов.
.0 Оз. N02
"н -Н20
О—О—мо2
пероксиацилнитрат
Глава 23. ФОСФОР
Химия фосфорорганических со¬
единений. . является наглядным
классическим примером того, как
научные исследования чисто тео¬
ретического характера в извест¬
ный период своего развития начи¬
нают давать обильные плоды
практического значения.
Б. А. Арбузов
23.1. Элемент, простое вещество
Фосфор, как элемент V группы 3 периода Периодической сис¬
темы, имеет Зз2Зр3Зс10-конфигурацию внешней электронной оболоч¬
ки. Способность Зэ-электронной пары к распариванию с участием
вакантных d-орбиталей, с одной стороны, и сравнительно невысокое
значение относительной электроотрицательности (ОЭО = 2.2), с
другой, определяют особенности этого элемента.
Первая из них состоит в малой донорной активности неподе-
ленной электронной пары, а следовательно низкой основности сое¬
динений трехвалентного фосфора Другая особенность заключается
в малой устойчивости соединений трехвалентного фосфора, точнее,
любых других состояний фосфора, кроме пятивалентного. Таким об¬
разом, из наиболее типичных степеней окисления фосфора -3, +3 и
+5 самой устойчивой является последняя, в которой фосфор обычно
представлен кислородными соединениями Иными словами, для
фосфора типично существование в виде фосфат-иона.
В силу относительно большого радиуса (что является следстви¬
ем нахождения этого элемента в 3 периоде) не высока прочность
связи Р-Р. Эта связь присутствует в элементном фосфоре, который
представлен в целом одиннадцатью аллотропными модификациями,
из которых наиболее известны нетоксичные красный и черный фос¬
фор, и чрезвычайно ядовитый белый фосфор. К примеру, молекула
белого фосфора имеет состав Р< Еще один из немногих примеров
соединений со связью Р-Р - фосфорноватая кислота Н2О3Р-РО3Н2.
Не высока прочность связи фосфора и с такими элементами, как
Н (энергия связи Р-Н 322 кДж/моль) и С (272 кДж/моль) по
сравнению с прочностью связи Р-0 (502 кДж/моль) Последнее
469
обстоятельство является определяющим в химии фосфора Эта
связь сильно полярна и отличается сильной поляризуемо«тью из-за
относительно большого размера атома фосфора Следовательно
связь Р-0 активна в нуклеофильном замещении, что определяет
своеобразие химического поведения как неорганических, так и ор¬
ганических производных фосфатов К этому следует добавить пред¬
ставление о пространственном строении фосфат-иона, имеющего
тетраэдрическую конфигурацию (см главу 2)
В свободном состоянии фосфор стал известен, по-видимому, с
XII века, когда был описан арабским алхимиком Алхид Бехилом,
однако честь его открытия обычно приписывают Г Брандту (1667
год), получившему фосфор восстановлением фосфатов мочи дей¬
ствием угля.
В природе фосфор находится в виде малорастворимых фосфа¬
тов. В основном это минералы фосфорит Саз(Р04)2 и апатит
Са3(Р04)2Са(Р,С1)2. Его содержание в земной коре - 8-10*2%, что от¬
вечает общему количеству 15 млрд. тонн. Таким образом, ресурсы
фосфора ограничены, а следовательно, этим количеством ограниче¬
ны и масштабы земной жизни.
Фосфаты, прежде всего, кальциевые, широко потребляются зем¬
леделием в виде различных удобрений (суперфосфат, преципитат и
др.). Еще одна область, где расходуется значительное количество
фосфатов (до 50% от их общего количества) - производство мою¬
щих средств. Триполифосфат натрия - основной компонент сти¬
ральных порошков. Глицерофосфат кальция используется в медици¬
не. Органические производные фосфора необходимы сельскому хо¬
зяйству как пестициды.
В организме содержится 500-800 г фосфатов, из них 88% нахо¬
дится в скелете - остальная часть внутри клеток, небольшая часть
во внеклеточном пространстве. Концентрация неорганического фос¬
фата в плазме (сыворотке) крови - 1.12-1.45 ммоль/л. Фосфаты
различной степени замещения находятся в сыворотке преимуще¬
ственно в виде свободных ионов. Небольшая их часть (менее 15%)
связана с белком.
Биологическая роль фосфора в организме состоит в участии в
синтезе 2,3-дифосфоглицерата, определяющего кислородтранспор-
тную способность гемоглобина; в образовании фосфопротеинов,
нуклеиновых кислот, фосфолипидов клеточных мембран, кофермен-
тов; в фосфорилировании углеводов, что делает их доступными
для метаболических процессов; в образовании нерастворимого гид-
роксиапатита костной ткани; в формировании фосфатной буферной
системы крови и мочи.
470
Балашцфосфатов в плазме (сыворотке) крови складывается из
соответствующего равновесия между поступлением, депонировани¬
ем и выделением этих анионов. С обычной диетой за сутки в орга¬
низм поступает один грамм фосфора, 70% которого всасывается, а
остальная часть выводится.
Как уже отмечено, соединения фосфора в низшей степени окис¬
ления -3 непрочны. Их легче всего получить связыванием фосфора с
активными металлами при повышенной температуре:
ЗМд + 2Р -> Мд3Р2
фосфид магния
Гидролиз фосфидов приводит к получению летучего, токсичного
и горючего фосфина:
Мд3Р2 + 6Н20 -> ЗМд(ОН)2 + 2РН3
На воздухе он легко окисляется до фосфорной кислоты:
Основные свойства фосфина выражены слабо (рКь = 26), поэто¬
му соли фосфония можно получить лишь действием очень сильных
кислот (иодистоводородной, хлорной) на растворы фосфина в орга¬
нических растворителях.
Присутствие даже следов влаги полностью гидролизует эти со¬
ли с выделением фосфина
23.3. Кислородные соединения фосфора
Оксиды фосфора(Ш) и (V) получаются при сгорании фосфора
или каких-либо соединений фос¬
фора с низшими степенями 0
23.2. Фосфин, фосфиды
РНз + 202 -» Н3Р04
РНз + Н1 -»[РН4]+Г иодид фосфония
окисления в зависимости от ко¬
личества кислорода. Эти оксиды
имеют полициклическое строе¬
ние, причем в структурах реали¬
зуется тенденция фосфора к об¬
разованию прочных связей Р-0
Р406
Р4Ою
471
Существуют кислородные кислоты фосфора (и их соли), отве¬
чающие всем положительным степеням окисления фосфора от +1
до +5 (см. табл. 23.1). Особенности строения этих кислот опреде¬
ляются отмеченной тенденцией фосфора к нахождению в пятива¬
лентном состоянии. Проявлением этой тенденции служит явление
таутомерии фосфористой кислоты с переходом атома водорода от
кислорода (таутомерная форма А) к фосфору (таутомер Б). Послед¬
няя форма значительно более устойчива. Аналогичная таутомерия
имеет место для фосфорноватистой кислоты
ОН он
А ^ Н0-|Ь=0 Б
НО^ОН Д
Из данных таблицы 23.1 видно, что все кислородные кислоты
фосфора в первой ступени диссоциации близки между собой, пред¬
ставляя собой кислоты средней силы, что и не удивительно, так как
диссоциации во всех случаях подвергается связь Н-0 однотипного
фрагмента Н-О-Р.
Таблица 23.1
Кислоты фосфора
Степень
окисления
Формула
Структурная
формула
Название кислоты
и аниона
Константы
диссоциа¬
ции
+1
Н3РО2
Н
НО—^=0
н
фосфорноватистая,
гипофосфит
8.9-10-2
+3
НзРОз
н
НО-1^=0
6н
фосфористая,
фосфит
5.110-2
1.8107
+4
Н4Р2Ов
о
ц-ё
X | X
о-^-о
о
фосфорноватая,
гипофосфат
6.0-10-3
1.510-3
5.4-10-8
9.3-10"11
+5
Н3Р04
он
но-^о
6н
фосфорная,
фосфат
7.0-10-3
8.0-10-8
4.810-13
472
С друТ%Й стороны, видно, что диссоциация имеет место только
для атомов водорода, связанных с кислородом. Следовательно фос¬
фористая кислота двухосновна, а фосфорноватистая является одно¬
основной.
Типичные способы получения всех перечисленных выше кислот
или их солей изображены ниже:
Р4О10 + 6Н20 -» 4НэР04
2Р + 4КОН + 4Н202 -> К4Р206 + 6Н20
Р406 + 6Н20 —» 4Н3Р03
4Р + ЗКОН + ЗН20 -» РНз + ЗКН2Р02
Соединения РРб, РС15, РОС13, РС13, РВгз, Р13 - галогенангидриды
фосфорной и фосфористой кислот. Типичная их черта - необрати¬
мый гидролиз, в качестве примера которого изображен гидролиз пя¬
тихлористого фосфора:
РС15 + 4Н20 -» Н3Р04 + 5НС1
Замена галогена на кислород (или гидроксильную группу) воз¬
можна и под действием спиртов, что используют для превращения
последних в галогеналканы:
РОС13 + ЗРОН -» Н3Р04 + ЗЯ-С!
Степени окисления фосфора +5 отвечают еще две кислоты: пи-
рофосфорная Н4Р207 и метафосфорная (НР03)х.
ОН ОН
о=|!>-о-|!>=о
6н Ан
пирофосфорная
кислота
ОН
—1!»-0—
8
метафосфорная
кислота
В них проявляется характерная черта соединений фосфора -
легкое образование связей О-Р, что приводит к формированию как
циклических структур (оксиды фосфора), так и линейных с пере¬
менным числом атомов фосфора - двух (для пирофосфорной кисло¬
ты), трех (для молекулы аденозинтрифосфорной кислоты) (см. ни¬
же), или большего количества, как в полимерной метафосфорной
кислоте. Естественно, что и мета-, и пирофосфорная кислоты реаги¬
руют с водой, превращаясь в ортофосфорную кислоту.
473
23.4. Биологическая роль фосфатов
Соли фосфорной кислоты, фосфаты, играют наиболее значимую
роль, в том числе и в биологических системах
Растворимые фосфаты, а именно гидро- и дигидрофосфаты ка¬
лия, К2НРО4 и КН2РО4, формируют биологическую фосфатную бу¬
ферную систему, ответственную, вместе с белковыми буферными
системами, за постоянство pH внутриклеточной жидкости
Труднорастворимые кальциевые соли - гидроксиапатит
ЗСаз(Р04)2'Са(0Н)2 [иначе Са5(0Н)(Р04)з] и карбонатапатит
ЗСаз(Р04)2СаС0з НгО составляют минеральную основу костной тка¬
ни. Это - трудно растворимые соединения. Так, ПР для гидроксиа-
патита составляет 1.6-10'58. Как видно из структурной формулы,
гидроксиапатит представляет собой основную соль, а следователь¬
но, реагирует с кислотами по уравнению:
Са5(0Н)(Р04)з + Н+ 25 Са3(Р04)2 + 2Са2+ + РС^ + Н20
Это означает, что в организме имеет место постепенное раство¬
рение гидроксиапатита и существует равновесие между раствори¬
мыми и нерастворимыми фосфатами, в том числе, и органическими
Достаточно сказать, что в среднем каждые семь лет костная ткань
полностью обновляется. Подробнее процесс
образования костной ткани рассмотрен в
разделе 12.3.
Сходное явление имеет место и с зуб¬
ной тканью, которая также представляет
собой гидроксиапатит. Анаэробные микро¬
организмы полости рта метаболируют, соз¬
давая кислую среду, и тем самым служат
причиной кариеса. Этот процесс активиру¬
ется, если разрушена зубная эмаль. По¬
следняя содержит нерастворимый в кисло¬
тах и механически прочный фторапатит.
Для регенерации зубной эмали рекомендуется применять зубную
пасту, содержащую соли фтора, а потому способствующую образо¬
ванию фторапатита Другой вариант состоит в добавке в воду мик¬
роколичеств фторидов, однако он нежелателен, так как приводит
к хотя бы частичному образованию фторапатита в костной ткани,
что нарушает естественное равновесие между растворимыми и не¬
растворимыми фосфатами организма.
Труднорастворимые фосфаты цинка гп3(Р04)2 и алюминия А1Р04
входят в состав так называемых фосфат-цементов, пломбировочного
материала в стоматологии.
Са-0
Ч0-Р=0
СаС°'
'‘О
НО(Р)—Са-0-Р=0
О'
СаГ
'‘О
0-Р=0
Са-О'
гидрокси(фтор)-
апатит
474
Было (Анаружено, что смешанные соли метафосфорной кислоты
с катионами калия, кальция и магния и переменной длиной
цепи (х меняется от 3 до 1000) в больших количествах содержатся
в клетках микроорганизмов в виде гранул так называемого волюти-
на. У некоторых бактерий высокомолекулярные полифосфаты со¬
ставляют до трети весового количества их сухого вещества. Поли¬
фосфаты представляют собой резерв фосфора клеток. В условиях
дефицита фосфора происходит ферментативная реакция:
(ПолиФ)х + НгО -»(ПолиФ)х-1 + ортофосфат,
восполняющая этот дефицит. Одновременно эти полифосфаты пред¬
ставляют собой резервы катионов калия, магния и кальция. Кроме
того, их следует рассматривать как ионнообменники, регулирующие
уровень тех или иных ионов в клетках. В клетках высших живот¬
ных и человека полифосфаты присутствуют в клеточных ядрах и
мембранах, однако в малых количествах. В процессе биохимической
эволюции резервные фунции полифосфатов взяла на себя молекула
аденозинтрифосфорной кислоты. Роль полифосфатов в тканях чело¬
века заключается в другом, а именно - в участии в генетической и
структурной организации.
23.5. Органические производные фосфора
Как отмечено, наиболее распространены соединения пятива¬
лентного фосфора. Поэтому такие органические производные тре¬
хвалентного фосфора, как замещенные фосфины (ЯРН2, РгРН, РзР),
соли фосфония (Р4Р+Х"), триалкилфосфиты Р(СЖ)з, хотя и су¬
ществуют, однако не им принадлежит ведущая роль
Более важны производные фосфорной кислоты - её эфиры, ко¬
торые называют алкил- (или арил-) фосфатами, соединения пятива¬
лентного фосфора со связью Р-С, которые именуют алкилфосфоно-
выми кислотами или алкилфосфонатами, а также диалкилфосфино-
вые кислоты или, соответственно, диалкилфосфинаты
ОН
Я0-^=0
6н
алкилфосфорная
кислота,
алкилфосфат
ОН
Я—^—О
6н
алкилфосфоновая
кислота,
алкилфосфонат
И-^-О
бн
диалкилфосфиновая
кислота,
диалкилфосфинат
475
23.5.1. Получение и химические свойства
Получение. Алкилфосфаты синтезируют ацилированкем спир¬
тов фосфорной кислотой - их фосфорилированием (см 20 7.3) Сое¬
динения со связью Р-С получают с помощью метода, называемого
перегруппировкой Арбузова:
я-о в+ Г Я-О 1+
р—0\р к'5 х§ > р-О-Р-Р*' X" —55-* Я'—1>=0
р-с/ I р-</ ] •кх Ар
триалкилфосфит алкилфосфонат
В этой реакции реализуется тенденция фосфора переходить из
трех- в пятивалентное состояние. Она аналогична таутомерии фос¬
фористой кислоты. В настоящее время разработано множество и
других методов синтеза фосфорорганических соединений, например,
окисление фосфинов. Таким образом, фосфорорганические соедине¬
ния доступны и достаточно изучены.
н
[0]
0
1
'о
а
он
И-1>=0
Ан
чн
[ Ч0Н ]
[0]
[ % он 1 101
р
Р —(->=о
Ан
я'
1 ]
Свойства. Общим свойством акилфосфатов и алкилфосфонатов
является способность к нуклеофильному замещению ОН-групп. Это
явление называют фосфорилированием.
* галогенангидрид
р р—о алкилфосфоновой
Ан кислоты
НХ | -нон
СЖ1 ОН
р_*=о ^251 р-^о НМЯ’Я2. р-|1о
Ан -нон Ан -нон Ан
сложный эфир диалкиламидамид
алкилфосфоновой алкилфосфоновой
Можнв*£существить фосфорилирование различных соединений,
в том числе-Ъпиртов и аминов, в соответствующие эфиры и амиды.
Легко заменяется ОН-группа и на галоген с образованием галоге¬
на нгидридов.
В изображенных продуктах фосфорилирования остается ещё
одна ОН-группа, которая тоже может быть замещена. Тем самым в
пределах одной молекулы можно комбинировать между собой раз¬
личные функциональные группы (спиртовая, эфирная, амидная, га-
логенангидридная и др.), что в сочетании с варьированием замести¬
теля Р порождает многообразие органических соединений пятива¬
лентного фосфора.
В то же время хлорангидриды, эфиры и амиды фосфорной и
фосфоновой кислот также участвуют в реакциях нуклеофильного
замещения за счет своих функциональных группировок.
Так, они сравнительно легко реагируют с водой. Способность к
гидролизу, а, следовательно, потенциальная нестабильность являет¬
ся специфической особенностью алкилфосфатов и фосфонатов.
Необходимо упомянуть способность эфиров и хлорангидридов
фосфоновой и фосфорной кислоты участвовать в нуклеофильном
замещении с гидроксиламином и его производными (глава 22):
X 01^
Я-1>=0 + НОК!^ —► Р_|!>=0 + нх
I х I
У у
Эта реакция лежит в основе антидотного действия производных
гидроксиламина при отравлении токсичными фосфорорганическими
соединениями.
23.5.2. Биологическая роль органических производных
фосфорной кислоты
Производные алкилфосфатов, т.е. соединения с группировкой
Р-О-Р, широко представлены в живой природе Связывание фосфор¬
ной кислоты органическими молекулами биосубстрата с образова¬
нием их сложных эфиров или амидов называют биологическим
фосфорилированием. Остаток фосфорной кислоты входит в состав
многих производных углеводов, он обязателен для нуклеиновых
кислот и фосфолипидов
Для биоэнергетики важна молекула аденозинтрифосфорной
кислоты (АТФ) Ее образование в процессе биологического окис¬
ления глюкозы (и не только ее) называют окислительным фосфо¬
рилированием. Эффективность окислительного фосфорилирования
477
определяют коэффициентом Р/О - молярным отношением связан¬
ного фосфата, приходящегося на количество поглощенного кислоро¬
да Например, для окислительного фосфорилирования с участием
глюкозы Р/О составляет 38/12 = 3.17
Гидролиз АТФ до аденозиндифосфата (АДФ) и фосфорной кис¬
лоты, таким образом, сопряжен с другими биохимическими превра¬
щениями Накопление АТФ в клетках - аккумуляция ими энергии,
необходимой для функционирования (биосинтеза, активного тран¬
спорта питательных веществ, двигательной функции и др.).
Связь О-Р в молекуле АТФ является макроэргической Так
называются богатые энергией химические связи, разрушение кото¬
рых сопровождается выделением большого количества энергии
Обычно при разрушении связей высвобождается 8-10 кДж/моль,
а гидролиз АТФ вызывает выделение 30.5 кДж/моль.
ООО О О
А-^-О-Н-О-^-ОН + НОН «=* А-Н-О-^-ОН + Н* + Н2Р04'
Ан Ан Ан Ан Ан
аденозинтрифосфат аденозиндифосфат
АТФ АДФ
Макроэргической является и связь О-Р в молекуле АДФ В
сумме при гидролизе АТФ до аденозинмонофосфата (АМФ) и двух
молекул фосфорной кислоты выделяется около 50 кДж/моль.
0 0 о
А-^-О-^-О-Н + НОН А-^-О-Н + Н* + Н2Р04-
Ан Ан Ан
аденозиндифосфат аденозинмонофосфат
АДФ АМФ
Итак, последовательный разрыв макроэргических связей моле¬
кулы АТФ - основной поставщик энергии организма, предваритель¬
но выработанной им за счет биологического окисления. Разрыв пер¬
вой из них - источник обычных процессов жизнедеятельности. При
энергетическом дефиците отщепляется вторая фосфатная группи¬
ровка - это “второе дыхание” организма.
О
А—О—Р + НОН А + Н* + Н2Р04‘
Ан
аденозинмонофосфат
АМФ
Наконец, последняя из трех макроэргических связей - энерге¬
тический резерв организма, “неприкосновенный запас", расходова¬
ние которого приводит к гибели.
478
Молек)ШЫ АТФ, АДФ и АМФ, содержащие остатки фосфорной
кислоты, являются кислотами средней силы и в биосредах находят¬
ся в диссоциированном виде, причем в такой форме они связаны с
катионом Мд2+, и именно такие соединения представляют собой ак¬
тивные формы фосфорилирования различных биосубстратов. Иными
словами, катионы магния (а также и близкие им по свойствам ка¬
тионы марганца) являются катализаторами окислительного фосфо¬
рилирования.
Органический Неорганический
фосфор
фосфор
Органический Неорганический
водород водород
\
Органический Неорганический
азот „ азот
Органический Неорганический
углерод углерод
Органический Неорганический
кислород кислород
Органическая Неорганическая
сера сера
Схема 23.1. Сопряженные круговороты элементов-органогенов
Следует сказать, что макроэргическими связями обладают ди- и
трифосфаты других мононуклеозидов - гуанозина, уридина и тд, и
их гидролиз также реализуется в биохимических процессах для по¬
лучения энергии. Однако главенствующая роль в этом отношении
принадлежит молекуле АТФ, которую принято считать универсаль¬
ной “разменной монетой” биоэнергетики
Полезно знать, что макроэргическими могут быть не только
связи О-Р. Так, макроэргична связь С-Б ацилмеркаптанового фраг¬
мента кофермента А (Ас-Б-Р). Такими же свойствами обладает и
связь некоторых карбаминов (НООС^<) Эта связь образуется
479
в ряде биохимических процессов с участием витамина тиамина и
служит для введения карбоксильной группы в состав некоторых
биомолекул. Эти превращения также поставляют необходимую
энергию тем реакциям, с которыми они сопряжены. Однако их
масштабы несопоставимы с биоэнергетической ролью аденозинтри-
фосфорной кислоты.
Таким образом, обратимый гидролиз АТФ - универсальный ис¬
точник обмена веществ всех живых организмов. Следовательно,
биокруговорот всех элементов-органогенов (схема 23.1) является
следствием биокруговорота фосфора, реализуемого в реакции гид¬
ролиза АТФ.
Энергия этого процесса в конечном итоге - солнечное излуче¬
ние, используемое в фотосинтезе. Это позволило дать одно из опре¬
делений жизни как свойства материи, приводящего к сопряженной
циркуляции биоэлементов в водной среде, движимой энергией сол¬
нечного излучения по пути увеличения сложности.
Среди алкилфосфатов имеются и лекарственные препараты, на¬
пример, фосфакол, применяется при лечении глаукомы
Вопросы химии соединений с фосфатными группировками рас¬
смотрены далее в соответствующих разделах.
23.5.3. Биологическая активность алкилфосфонатов
Здесь приведены необходимые сведения об алкилфосфонатах,
т.е. веществах со связью С-Р. Эти соединения в природе встреча¬
ются редко. Исключение составляют немногие фосфонолипиды и
цилиатин МН2СН2СН2Р(0Н)2=0 ((3-аминоэтилфосфоновая кислота),
который является продуктом жизнедеятельности некоторых видов
морских анемонов.
Отличительной особенностью многих производных алкилфосфо-
новых кислот является высокая токсичность, причем одни из них
более ядовиты для теплокровных животных и человека (а потому
являются ОВ), другие - для насекомых (т.е. представляют собой
инсектициды).
К числу фосфорорганических ОВ относятся такие вещества,
как табун, зарин и зоман. Табун, зарин, зоман и родственные им
фосфакол
л-нитрофениловый эфир
диэтилфосфорной кислоты
480
соединенй%* являются ядами нервно-паралитического действия, т.е.
они поражают нервную систему.
Механизм действия фосфорорганических ОВ заключается в том,
что они являются ингибиторами фермента холинэстеразы. Они бло¬
кируют его активные центры, препятствуя тем самым взаимодей¬
ствию фермента с ацетилхолином - медиатором нервных импульсов.
Таким образом, токсичность фосфорорганических соединений -
результат нуклеофильного замещения с участием функциональных
группировок активного центра фермента.
Естественно, что фосфорорганические аналоги ацетилхолина -
фосфорилхолины обладают ещё большей токсичностью К числу
сильнейших ОВ относятся и тиоаналоги фосфорилхолинов - фос-
форилтиохолины.
Типичными представителями фосфорорганических инсектицидов
служат хлорофос и тиофос, изображенные ниже.
Полезно обратить внимание, что тиофос является эфиром не
фосфоновой, а тиофосфорной кислоты Кроме тиофоса существует
еще много и других органических тиофосфатов, которые используют
в качестве инсектицидов.
ОС2Н5
N=0-^=0
М(СНЭ)2
ОСН(СН3)2
сн3-|!>=о
*
СН3
о6нС(СНэ)з
сн3—|!>=о
табун,
диметиламид
этилового эфира
цианфосфоновой
кислоты
изопропиловый эфир
фторангидрида
метилфосфоновой
кислоты
зарин,
зоман,
пинаколиновый эфир
фторангидрида
метилфосфоновой
кислоты
ОСН2СН2М*(СНз)з
X—^=0
8СН2СН2М*(СН3)з
х—|!>=о
I
У )
фосфорилхолины
X = алкил, У = СЖ, ?
фосфорилтиохолины
хлорофос,
0,0-диметил(2,2,2,-трихлор-
1 -оксиэтил)фосфонат
тиофос,
0,0-диэтип-0-(4-нитрофенил)-
тиофосфат
16 Эак 675
481
Фосфорорганические инсектициды в отличие от хлорсодержа¬
щих инсектицидов сравнительно быстро разрушаются из-за уже от¬
меченной способности к гидролизу и не накапливаются в природе.
Однако они обладают сравнительно высокой токсичностью для че¬
ловека и поэтому с их нежелательным действием приходится стал¬
киваться в токсикологической практике
Принципы борьбы с отравлениями здесь такие же, как и в слу¬
чае фосфорорганических ОВ. В их числе важное место занимает
упомянутая выше реакция с производными гидрокснламина, кото¬
рые употребляются в качестве противоядий.
Гидролиз амидных, галогенангидридных и сложноэфирных
групп, входящих в состав ОВ, является способом нх дегазации.
Проблема ликвидации запасов боевых отравляющих веществ яв¬
ляется в настоящее время актуальной.
Среди производных алкилфосфонатов имеются и лекарственные
средства. Так, армии, близкий аналог фосфакола (см. 23.4.2), также
является препаратом для излечения глаукомы.
/—ч ОС2Н5 армии
^о2 __/())_ О—Р=0 л-нитрофениловый, этиловый эфир
у—У/ этилфосфоновой кислоты
Кроме производных фосфоновой кислоты существуют семейства
органических производных диалкилфосфинистой, диалкилфосфино-
вой и алкилфосфонистой кислот, среди которых имеется много
представителей физиологически активных веществ.
н
Г*-1>=0 я-Ь=о я.—}>=о
н Ан Ан
диалкилфосфинистая диалкилфосфиновая алкилфосфонистая
кислота кислота кислота
Следует упомянуть, что фосфорорганические соединения нахо¬
дят применение также и в качестве растворителей, поверхностно¬
активных веществ, катализаторов технологических процессов, ком¬
плексонов и т.д. Таким образом, органическая химия фосфора пред¬
ставляет собой перспективную и быстро развивающуюся область
химической науки. Существенный вклад в изучение фосфороргани¬
ческих соединений внесли отечественные ученые А. Е. Арбузов и
Б. А. Арбузов.
Таким образом, роль фосфора как элемента-органогена опреде¬
ляется его нахождением в биосредах в виде фосфат-иона, который
482
входит відстав неорганических компонентов живых организмов в
виде как мйЛо, так и хорошо растворимых фосфатов, с одной сторо¬
ны, и в состав органических биомолекул в ряду углеводов, нуклеи¬
новых кислот и липидов, с другой стороны. Нуклеофильное заме¬
щение при атоме фосфора в них составляет суть важнейшей реак¬
ции биологического фосфорилирования. Существует сложная си¬
стема равновесия между различными формами фосфатов в составе
биосубстрата, регулирующая распределение соединений фосфора в
различных тканях и средах организма.
23.6. Мышьяк, сурьма, висмут
23.6.1. Неорганические производные
С ростом радиуса атома от азота к висмуту падает величина
относительной электроотрицательности, уменьшается энтальпия
ионизации, все большую роль начинают играть в формировании хи¬
мических связей с!-орбитали. При движении вниз в пределах группы
усиливаются металлические свойства Если мышьяк является амфо-
терным элементом, то сурьма и висмут уже относятся к металлам.
Устойчивы для них степени окисления -3, +3 и +5. При этом
устойчивость соединений с крайними значениями степеней окисле¬
ния сверху вниз уменьшается в связи с нарастанием металлич-
ности. Поэтому гидриды этих элементов мало прочны. Не прочной
является и висмутовая кислота, как и ее соли, висмутаты, являю¬
щиеся сильными окислителями.
В частности, висмутаты способны окислять катион марганца
(2+) в перманганат-анион-
2Мп(ГЮэ)2 + 5№ВЮ3 + 16НГЮ3 -» 2НМп04 + бВКЫОзЬ + 5№ГЮ3 + 7Н20
Оксиды и гидроксиды трехвалентного мышьяка, сурьмы и вис¬
мута проявляют амфотерные свойства Однако самым значимым для
медицины свойством этих соединений является образование трудно
растворимых сульфидов, приводящее к их прочному связыванию с
сульфгидрильными группами белков, что и служит причиной их вы¬
сокой токсичности В первую очередь, этот эффект связывают с
блокировкой сульфгидрильных групп биорегулятора глутатиона, ко¬
торый восстанавливает в тканях токсичные пероксиды - продукты
перекисного окисления липидов (см. 21 3)
16*
483
Для судебио-медицииской практики важно обнаружение мышья¬
ковых препаратов, которое основано на восстановлении соединений
Ав*3 до соединения Ав-3 - арсина Последующее термическое раз¬
ложение арсина до свободного мышьяка с образованием так назы¬
ваемого мышьякового зеркала и служит для обнаружения этого
элемента при отравлениях Этот метод, чувствительность которого
7-10"7 г мышьяка, носит название пробы Марша:
АвгОз ♦ бгп + 12НС1 -» 2АзН3Т + 62пС12 + ЗН20
гАвНз -» 2АвХ + ЗН2 (нагревание)
Мышьяковые соединения накапливаются в ногтях и волосах за
счет связывания с сульфгидрильными группами белков. Последнее
обстоятельство используется в методе радиационного анализа при
определении хронических мышьяковых отравлений.
Столь же токсичны соединения сурьмы и висмута, но их эффект
выражен слабо, так как в желудке они гидролизуются с образова¬
нием малорастворимых соединений, содержащих катионы ЭО+ и
не всасывающихся через стенки желудочно-кишечного тракта:
эьсь + н2о -»эьоаХ + 2на
хлористый антимония
ВКГЮз)э + Н20 -» ВЮЫОэ4 + 2НГЮ3
азотнокислый висмутил
О биологической роли мышьяка, сурьмы и висмута, которые от¬
носятся к числу микробиоэлементов, известно немного. Мышьяк,
возможно, незаменимый элемент, во всяком случае, в микроколи¬
чествах он полезен. Относительно сурьмы и висмута ничего опре¬
деленного сказать нельзя. Известно лишь, что соединения этих
элементов угнетают действие некоторых ферментов.
Соединения мышьяка(Ш) в микродозах применяют в стоматоло¬
гической практике для омертвления мягких тканей зубов. Исполь¬
зуют мышьяковые препараты и для лечения некоторых кожных
заболеваний.
Еще одна область применения мышьяковых соединений в меди¬
цине - лечение нервных расстройств и малокровия с помощью
1%-ного раствора гидроарсената натрия №2НА804'71-120. Вероятно,
лечебное действие достигается за счет частичного блокирования
сульфгидрильных групп ферментов.
Сульфид сурьмы входит в состав медицинской резины.
Оксид висмута(Ш) - составная часть препарата ксероформа, кото¬
484
рый слу?И^т наружным подсушивающим и антисептическим сред¬
ством. Основной нитрат висмута ВЮГМОз употребляют в качестве
вяжущего и антисептического средства при желудочно-кишечных
заболеваниях.
23.6.2. Органические производные мышьяка
Существуют органические соединения трех- и пятивалентного
мышьяка. Среди них обнаружены как сильно ядовитые вещества,
так и лекарственные средства.
Взаимодействием АэСЬ с ацетиленом получают Р-хлорвинил-
дихлорарсин, который обладает кожно-нарывным действием и яв¬
ляется боевым ОВ, известным под названием люизит.
С1 н о Ю1 он
С1—СН=СН-Аз/ С1—СН=СН-Аз=0 —* С1—СН=СН-Аз=0
С1 -2НС1 I
люизит ОН
Так как в состав люизита входят два "галогенангидридных"
атома хлора, он легко гидролизуется, причем продукт гидролиза -
Р-хлорвиниларсеноксид тоже сильно токсичен.
Гидролиз люизита в присутствии окислителей (перекись водо¬
рода, хлорамин, хлорная известь и т.д.) превращает его в
Р-хлорвинилмышьяковую кислоту, которая значительно менее ядо¬
вита, как и все соединения пятивалентного мышьяка в целом.
В такой обработке и заключается дегазация люизита. Его токсиче¬
ский эффект состоит в действии на сульфгидрильные группировки
белков, а, следовательно, противоядиями являются БАЛ и унитиол.
Токсичны и другие арсины. Например, дифенилхлорарсин, ди-
фенилцианарсин и адамсит являются отравляющими веществами
раздражающего и общеядовитого действия
6\
дифенилхлорарсин дифенилцианарсин адамсит
Напротив, соединения пятивалентного мышьяка - производные
алкиларсоновой и диалкиларсиновой кислот - менее токсичны и ис¬
пользуются в качестве лекарственных препаратов
485
Натриевая соль диметиларсиновой кислоты применяется в дер¬
матологии. Для лечения сонной болезни ранее применялся препа¬
рат атоксил - натриевая соль арсаниловой кислоты. Однако
атоксил сильно токсичен и вызывает, в частности, слепоту.
Атоксил, как типичный аналог лара-аминобензойной кислоты, пред¬
ставляет собой антиметаболит.
Поиски лекарственных средств среди соединений мышьяка свя¬
заны с именем основателя химиотерапии П. Эрлиха. Эрлих получил
противосифилитический препарат "сальварсан", формула которого
была установлена позднее. Сальварсан является полимерным сое¬
динением со связями -As-As-. В настоящее время мышьяковые пре¬
параты вытесняются антибиотиками.
Часть V
ОРГАНИЧЕСКИЕ БИОМОЛЕКУЛЫ
Лаборатория живой клетки
с ее каталитическими процес¬
сами - вот идеал органической
химии.
А. Е Арбузов
Глава 24. ОКСОСОЕДИНЕНИЯ
24.1. Классификация и номенклатура
Соединения, содержащие карбонильную группу С=0, именуемые
оксосоединениями, представлены альдегидами ЯСН=0 - вещества¬
ми, у которых один из заместителей при карбонильной груп¬
пе - атом водорода, и кетонами Я1Я2С=0, в которых карбонильная
группа связана с двумя радикалами.
Отличие в строении этих классов оксосоединений обусловли¬
вает и принципиальное различие в их химических свойствах. Альде¬
гиды (но не кетоны) легко окисляются в соответствующие кислоты
уже такими слабыми окислителями, как аммиачный раствор оксида
серебра или свежеприготовленный гидроксид меди:
ЯСН=0 + Ад20 -> Я-СООН + 2Ад4-
карбоновая кислота
Генетическая связь альдегидов с карбоновыми кислотами отра¬
жается в том, что тривиальные названия многих альдегидов произ¬
водятся от названий тех кислот, в которые они окисляются.
Международные правила присваивают альдегидной группе
окончание -аль, причем эта группа определяет начало нумерации.
Часть альдегидов имеет свои тривиальные названия, как и многие
кетоны.
По правилам рациональной номенклатуры название кетонов на¬
чинается с названий радикалов, присоединенных к карбонильной
группе, которой соответствует окончание кетон По международ¬
ной номенклатуре кетонной карбонильной группе отвечает оконча¬
487
ние он, положение которой указано цифрои, причем нумерацию на¬
чинают с ближайшего к ней конца цепи.
Альдегиды
Н СН3 СНзСН, С1зС
с=о с=о с=о "с=о
н' н' и' н'
муравьиный уксусный пропионовый трихлорэтаналь
альдегид, альдегид, альдегид, хлораль
формальдегид ацетальдегид пропаналь
этаналь
СН3
СН3СН2СН2ч снз-бн^
С=0
иэомасляный акролеин
альдегид альдегид пропеналь
бутаналь 2-метилпропаналь
и' Н
СН=0
СНгОН
Н Н3С^ N
2-метил-З-окси-
а-фуранкарб- 5-оксиметилпиридин-
альдегид, 4-карбальдегид,
фурфурол пиридоксаль
Кетоны:
о О
СНз'
ацетон, метилэтилкетон, эцвтофвнон, циклогексанон
пропанон бутанон мвтилфвнилкетом
В зависимости от характера заместителей у карбонильной груп¬
пы оксосоединения могут быть насыщенными, ненасыщенными или
гетероциклическими.
Кроме карбонильной группы в их молекулах могут содержаться
и различные функциональные заместители, в том числе и другие
карбонильные группы. В последнем случае говорят о ди-, три- и т.д.
альдегидах и кетонах
СН3ч СН3СН2
с=о чс-
СН3' СН/
488
ДиаЛ^егиды и дикетоны:
нч /? СН\ /? И\^Н СН’-„^Снз
«М. о^сн, II ТТ
этандиаль,
глиоксаль
диметил-
глиоксаль,
бутандион
малондиальдегид, ацетилацетон,
пропандиаль пентандион-2,4
О
О
,СН3
О
л-хинон
О
• » АИПиП л .
циклогексадиен-2,5-дион-1,4 2-метилнафтохинон-1,4
Карбонильная группа присутствует во многих разновидностях
биомолекул. В частности, она характерна для углеводов (глюкоза,
например, обладает альдегидной карбонильной группой, а фруктоза
представляет собой кетон), присутствует в ряде стероидных гор¬
монов, а также терпенов. Ею обладают природные кетокислоты.
Пиридоксаль представляет собой кофактор многих ферментов.
Малондиальдегид - важнейший продукт перекисного окисления
липидов, а потому его определение в биосредах практикуется
в биохимическом анализе. О роли убихинонов уже говорилось
(см. 20.7 3) Нафтохинонная структура присутствует в витаминах
группы К. 2-Метил-/,4-нафтохинон является, к примеру, витами¬
ном К4 - антигеморрагическим фактором, отвечающим за сверты¬
ваемость крови.
Многие альдегиды и кетоны используют на практике, в том чис¬
ле в синтезе лекарств Например, фурфурол - сырье для производ¬
ства фурацилина и ряда других препаратов.
24.2. Способы получения альдегидов и кетонов
1. Оксосоединения можно получить ранее рассмотренной реак¬
цией окисления спиртов (глава 21), при этом первичные спирты
дают альдегиды, а вторичные - кетоны Окисление гидрохинона
служит методом синтеза л-хинона. Реакции окисления спиртовых
или фенольных гидроксильных групп, катализируемые ферментами,
например, алкогольдегидрогеназой, являются важнейшим путем
введения карбонильной группы в биомолекулы
489
2. Реакция Кучерова (глава 18) с ацетиленом приводит к
ацетальдегиду, а с гомологами ацетилена - к различным кетонам
3. Гидролиз геминальных дигалогеипроиэводиых приво¬
дит к образованию альдегидов и кетонов.
<Оьснс*Н£ї СИн=°
-СНСІ2
хлористый бенэилиден
4. Универсальным методом синтеза оксосоединений служит
реакция декарбоксилирования карбоновых кислот, осу¬
ществляемая при их сильном нагревании над оксидами металлов
(М) МпО или ТІ1О2 (или их гидроксидами). Из схемы нетрудно заме¬
тить, что использование одной кислоты (Я1 = ІЗ2), а не смеси из
двух различных кислот приводит к симметричному по строению ке¬
тону. Именно таким способом из уксусной кислоты и Са(ОН)г и был
получен впервые ацетон, откуда и происходит его название. Если
одна из кислот представляет собой муравьиную (Р1 = Н), в реакции
получается альдегид.
"-С
М(ОН)2
-нон
-МС03
Из дикарбоновых кислот образуются кетоны циклоалканового
ряда с числом атомов углерода в кольце меньше на единицу, чем в
исходной кислоте, например:
5. Широко применяется в промышленности метод оксосинте-
за, заключающийся в действии на алкены смесью оксида углеро-
да(Н) и водорода в присутствии кобальтового катализатора и приво¬
дящий к альдегидам:
490
сн2=сн2
10, Р
СН3СН2 —с
чн
СО + Н2 кат.
6. Альдегиды можно также в принципе синтезировать восста¬
новлением карбоновых кислот, которое удается осуществить в
определенных условиях:
24.3. Химические свойства карбонильных
соединений
Для двойной связи карбонильной группы, как и для всякой
другой кратной связи, характерны реакции присоединения. Оксосо-
единения значительно активнее алкенов, что обусловлено большей
электроотрицательностью атома кислорода. Это вызывает появление
значительного дипольного момента карбонильной группы (=2.5 Б),
причем положительный конец диполя находится у карбонильного
атома углерода, который обладает поэтому электронодефицитным
характером. Высокая полярность и поляризуемость связи С=0
приводят к тому, что присоединяющийся реагент будет направлять¬
ся своим отрицательным концом к атому углерода. Сказанное вы¬
ражается схемой:
Следовательно, для оксосоединений характерно нуклеофиль¬
ное присоединение
Донорные заместители (а в их числе находятся и алкильные
группы) должны снижать электрофильность карбонильного углеро¬
да и тем самым уменьшать реакционную способность Поэтому
в ряду формальдегид - ацетальдегид - ацетон последний обладает
минимальной реакционной способностью, а в целом альдегиды ак¬
тивнее кетонов Напротив, введение электроноакцепторных групп
облегчает присоединение Так, хлораль значительно активнее
ацетальдегида.
Несмотря на высокую полярность, карбонильные соединения
имеют меньшие температуры плавления и кипения, чем родствен¬
ные спирты. Это объясняется отсутствием нх ассоциации из-за не
Я-СООН + Н2 -> Я-СН=0 + Н20
/
491
возможности образования водородной связи. Простейшие альдегиды
и кетоны, однако, хорошо растворимы в воде, т.к с ее молекулами
такая связь у карбонильной группы образуется.
24.3.1. Реакции присоединения карбонильной группы
1. Присоединение водорода к карбонильной группе, как уже
отмечалось (глава 20), служит способом синтеза спиртовой группы.
Это явление носит обратимый характер и играет существенную
биохимическую роль.
Действие сильными восстановителями приводит к восстановле¬
нию оксосоединений до алканов:
2Р1ССЖ2 + ЫА1Н4 + 2Н20 -> 2Я1СН2Р2 + ЫОН + А1(ОН)3
2. Гидратация альдегидов и кетонов является обратимой
реакцией:
Для наиболее активного в реакциях присоединения формальде¬
гида (131 = Р*2 = Н) степень гидратации составляет 100%, ацетальде-
гид (К1 = СНз, Я2 = Н) гидратирован на 58%, а ацетон (Я1 = Я2= СНз)
практически не образует гидрата. Таким образом, большинство ок¬
сосоединений воду не присоединяют. Исключение составляют аль¬
дегиды и кетоны с электроноакцепторными заместителями. Напри¬
мер, продукт взаимодействия хлораля с водой - хлоральгидрат
СС1зСН(ОН)2 настолько устойчив, что его легко выделить в чистом
виде Хлоральгидрат обладает снотворным действием и применяется
в клинической практике.
3. Присоединение спиртов, которое легко осуществляется
для альдегидов, приводит первоначально к обратимому образованию
неустойчивых продуктов, именуемых полуацеталями.
хлоральгидрат
полуацетальный гидроксил
'С=0 + н-СЖ
Гон|
Р1_6_СЖ
полуацеталь
ацеталь
492
Гидрофильная группа полуацеталей, так называемый полуаце-
тальный гйдроксил, непрочно связан с углеродным атомом, т.к.
этот атом связан с еще одним электроотрицательным атомом - кис¬
лородом группы (Ж1. Поэтому полуацетальный гидроксил легко за¬
мещается под действием второй молекулы спирта, в результате чего
образуется ацеталь.
Интересен случай, когда карбонильная и спиртовая группы на¬
ходятся в одной молекуле. В таких веществах - оксикарбонильных
соединениях - имеется возможность внутримолекулярного взаимо¬
действия между этими группами, что должно приводить к образова¬
нию кольчатых полуацеталей. Естественно, что взаимодействие
может осуществляться тогда, когда замыкающийся цикл лишен на¬
пряжения, т е будет пяти- или шестичленным. Это условие выпол¬
няется для тех оксикарбонильных соединений, в которых реаги¬
рующие функциональные группы разделены тремя или четырьмя
атомами углерода. Рассмотрим это превращение на примере бута-
нол-4-аля (у-оксимасляного альдегида):
В образующемся циклическом полуацетале с тетрагидрофурано-
вым кольцом полуацетальный атом углерода (С1) является хираль-
ным центром, а, следовательно, получается смесь двух энантио-
меров (рацемат). Энантиомеры циклических полуацеталей именуют
а- и р-изомерами. При изображении по Хеуорсу (глава 18) принято
полуацетальную гидроксильную группу а-изомера помещать под
плоскостью кольца, а в Р-изомере - наоборот.
Аналогичный процесс с образованием шестичленного тетрагид-
ропиранового производного изображен и для пентанол-5-аля
3 2
у-оксимасляный
альдегид
кольчатый полуацеталь
у-оксимасляный
альдегид
а-форма
р-форма
а-форма
р-форма
493
Эти реакции идут достаточно быстро и обратимо, следовательно,
одно и то же вещество представлено одновременно тремя изо¬
мерными формами. В этом случае говорят о явлении таутомерии
(динамической изомерии). Рассматриваемые превращения являются
примерами кольчато-цепной таутомерии Кольчато-цепная изо¬
мерия представляет собой характерное свойство таких важных
в биологическом отношении веществ, как углеводы Наблюдается
она и у многих других природных веществ, содержащих одновре¬
менно и окси-, и оксогруппы, например, у некоторых природных
стероидных гормонов
4. Синильная кислота чрезвычайно легко присоединяется к
оксосоединениям, в результате чего получаются продукты, содер¬
жащие гидроксильную и нитрильную группы при одном и том же
атоме углерода (а-оксинитрилы) Для альдегидов эта реакция в об¬
щем виде выглядит следующим образом:
С=0 + Н-СЫ
н'
р_А10Н —£-°н но_£—
О- и (.-изомеры
Получающиеся а-оксинитрилы содержат асимметрический атом
углерода и следовательно представляют собой рацематы, т.е. смесь
Б- и Ь-энантиомеров, которые изображены с помощью проекцион¬
ных формул Фишера (глава 20).
Синтез а-оксинитрилов имеет большое практическое значение,
т.к. из них получают углеводы, окси-, кето- и аминокислоты, т.е.
разнообразные биомолекулы. Реакция синильной кислоты с карбо¬
нильной группой используется в борьбе с отравлениями синильной
кислотой, для чего обычно употребляют глюкозу (см. главу 25).
Дегазацию помещений, содержащих пары синильной кислоты,
осуществляют посредством формалина:
Н
N
;с=0 + н-сы —» Н-А-<
■он
5. Присоединение аммиака и аминов к альдегидам в
первой стадии происходит, как и другие рассмотренные реакции
нуклеофильного присоединения, а затем от первичного продукта
присоединения отщепляется вода за счет группы ОН и атома водо¬
рода аминогруппы. Образующееся вещество называется имином:
494
Образование иминов обратимо, при действии воды они распада¬
ются на исходные ^мин и оксосоединение.
Имины альдегидов обычно нестойки и претерпевают циклотри-
меризацию:
Реакция аминогруппы с образованием C=N связи является необ¬
ходимой стадией в процессе транс-аминирования - биохимиче¬
ского превращения, суть которого состоит в передаче аминогруппы
от одной молекулы к другой; транс-аминированием одни аминокис¬
лоты превращаются в другие in vivo.
В этом процессе принимает участие молекула пиридоксаля
Сначала он образует имин с молекулой аминокислоты (или амино¬
группой белка). Затем в образовавшемся имине имеет место тау¬
томерия за счет перемещения кратной связи С=М (имин-иминная
таутомерия), в результате получается имин с иным положением
связи С=М Этот имин ферментативно гидролизуется, давая кето¬
кислоту и пиридоксамин. Другая кетокислота взаимодействует с
R
Н
СН=0 ♦ NH2—6н—СООН CH=N—бн-соон
пиридоксапь
имин-иминная
імин-иминная <<
таутомерия |1
R
СН2МН2 ♦ О-С'
.R
пиридоксамин
пиридоксамином, а продукт этой реакции за счет таутомерии и по¬
следующего гидролиза (то есть реакции в обратном направлении)
превращается в аминокислоту и пиридоксаль Таким образом, сум¬
марное превращение служит путем замены карбонильной группы на
аминогруппу (и наоборот) и тем самым служит приемом передачи
аминогруппы от одной аминокислоты к другой, то есть представляет
собой транс-аминирование
Сложным образом происходит реакция аммиака с формальдеги¬
дом, в которой образуется уротропин Получение уротропина разо¬
брано ниже
6. Продукты взаимодействия оксосоединений с гидрокси-
ламином называют оксимами. Практическое значение имеет оксим
циклогексанона, получение которого служит промежуточной стади¬
ей синтеза полимера капрона.
Реакцией диметилглиоксаля с гидроксиламином получают ди-
метилглиоксим (реактив Чугаева), являющийся активным биден-
татным лигандом. Это его свойство используется для определения
ряда катионов переходных металлов в аналитической химии.
В биологических целях такие комплексы также подвергаются
исследованию, поскольку во многом имитируют свойства значи¬
тельно более сложно построенных биокомплексов (например, ви-
_.Н=МОН оксимы, особенно карбонильных соединений ге-
7. С гидразином и его ароматическим производным - фени-
лгидразином альдегиды и кетоны также образуют продукты, содер¬
жащие связь С=1М, которые называются гидразонами и фенилгид-
разонами, соответственно.
<0° * НгМОН -нг О-
:МОН циклогексанона
оксим
СНЧ
+ гНгШН
О СНэ
снэ ыон
диметилглиоксаль
диметилглиоксим
тамина В12).
В качестве противоядий при отравлении
фосфорорганическими ОВ применяют некоторые
Анэ
тероциклического ряда. В качестве примера
можно привести пралидоксим - оксим альдеги¬
да, содержащего пиридиновое кольцо с четвер¬
тичным атомом азота.
пралидоксим
Р1Р2С=0 + Нг^НР -> Р1Р2С=ММНР + Н20
Р = Н, гидразон; Р? = СбН5, фенилгидразон
Гидразоны под действием щелочи выделяют азот и превра¬
щаются в алканы. Эта реакция, открытая в начале XIX в. русским
химиком Н. М. Кижнером и носящая его имя, является эффек¬
тивным способом превращения различных оксосоединений в алка¬
ны, например:
СНзч снэ ч кпн
С=0 + ДО№г ——♦ СНзСНгСНгСНэ
ОгН/ -Н20 -М,
гидразон
метилэтилкетон метилэтилкетона ьутън
Своеобразно идет реакция фенилгидразина с а-оксикарбониль-
ными соединениями, например, с глицериновым альдегидом. В пер¬
вой стадии получается соответствующий фенилгидразон:
СН=0 + Н2ММНС6Н5
ОН
СНгОН
глицериновый
альдегид
Далее вторая молекула фенилгидразина окисляет вторичную
спиртовую группу в кетонную, которая снова взаимодействует с
фенилгидразином и в результате получается озазон
сн=ммнс6н5 сн=ммнс6н5 СН=ММНС6Н5
Тон н,шнс,н5| ^ Н;ММНС8Н5 |1ММНГ<И1
СНгОН -ННз.-С.Н^Н, -нг°
сн2он
оэаэон
Образование фенилгидразонов из альдегидов или кетонов и оза-
зонов из а-оксикарбонильных соединений используется в иденти¬
фикационных целях, в частности, в химии углеводов.
Реакция с фенилгидразином некоторых веществ, содержащих
карбонильную группу, нашла употребление в синтезе таких широко
известных антипиретиков и анальгетиков, как антипирин, амидопи¬
рин, анальгин и др (см. главу 30).
Оксосоединения могут вступать в реакции нуклеофильного
присоединения и с некоторыми другими веществами (металло-
7Г- СН=ММНС6Н5 фвнидг1дааз0н
2 —ОН гицеринового
сн2он альдегида
497
органические соединения, меркаптаны, ацетилен и др ) Эти реак¬
ции имеют значение в органическом синтезе, но менее интересны
в медико-биологическом аспекте
24.3.2. Реакции с участием заместителя
при карбонильной группе
Карбонильная группа обладает сильным электроноакцепторным
действием и потому оказывает воздействие на связанный с ней за¬
меститель В случае предельного радикала карбонильная группа
ослабляет связь С-Н при углеродном атоме в а-положении к карбо¬
нильной группе (а-водород).
Это выражается в повышенной склонности а-водорода к заме¬
щению, например, в реакциях галогенирования. Так, ацетальдегид
хлорируется уже в обычных условиях с последовательным замеще¬
нием всех трех а-водородных атомов и образованием хлорал я:
СНз\ +С1гасН2\ +С1г агСН +С( аэС
с=о —; с—о —* с=о 'с=о
и' -на н' -НС1 н' -НС1 и'
этанаяь хлораль
Малая прочность связи С-Н у а-водородного атома может при¬
водить к отрыву а-водорода в виде протона, который далее атакует
атом кислорода карбонильной группы. В таком случае можно ожи¬
дать частичную изомеризацию оксосоединения в непредельный
спирт (енол):
кетон О ;=Ь ^с=с/ енол
| ^ / ОН
Таким образом, возможна таутомерия кетон^енол, так назы¬
ваемая кето-енольная таутомерия.
Соотношение таутомеров определяется величинами свободных
энергий. У предельных альдегидов и кетонов свободная энергия ке-
тоформы ниже энергии енольной формы на 63 кДж/моль, поэтому
степень их енолизации ничтожно мала (=10’4%). Устойчивость ок-
соформы по сравнению с её таутомером - непредельным спиртом -
находит отражение в правиле Эльтекова (глава 18).
При стабилизации енольной формы действием каких-либо фак¬
торов её долю в таутомерном равновесии можно заметно повысить
или сделать эту форму единственно возможной. В частности, у
Р-дикарбонильных соединений енольный таутомер стабилизирован
498
за счет об^ования прочной внутримолекулярной водородной связи
(до 20 кДж/моль) и сопряженной системы С=С-С=0 (еще около 15
кДж/моль) и поэтому может преобладать в смеси, что и имеет
место для ацетилацетона:
В случае фенола оксоформа вообще не существует, т.к. её обра¬
зование должно разрушать ароматическую систему с энергией со¬
пряжения, равной 150 кДж/моль.
О ОН
-чб -6
Явлением кето-енольной таутомерии объясняются многие хими¬
ческие свойства углеводов, окси- и кетокислот.
Электроноакцепторное влияние карбонильной группы на нена¬
сыщенный радикал в непредельных альдегидах и кетонах прояв¬
ляется в том, что электрофильное присоединение к их двойной свя¬
зи С=С происходит против правила Марковникова:
8+ 5-
СН2=СН
'Ч->С=0 + НВг
Н' ^
пропеналь
акролеин
—» ВгСН2СН2СН=0
По отношению к ароматическому радикалу карбонильная группа
проявляет себя как заместитель второго рода Поэтому реакции
электрофильного замещения, например, нитрование, идут для бен-
зальдегида по преимуществу в лета-положение
499
24.3.3. Реакции полимеризации и конденсации
альдегидов и кетонов
1. Наличие подвижного а-водородного атома в молекуле пре¬
дельных альдегидов и кетонов сообщает им способность вступать
в реакцию нуклеофильного присоединения по карбонильной группе
другой молекулы. Эта обратимая реакция, катализатором которой
является щелочь, на примере ацетальдегида выглядит следующим
образом
СН3 а
0=0 + н-сн2ч кон
Н СпО —» СНЭ—СН—СН2—СН=0 альдоль
н' Ан
Её продукт содержит альдегидную и спиртовую группы и по¬
этому называется альдолем, а реакция представляет собой аль-
дольное уплотнение. Альдольное уплотнение впервые наблюдал
А. П. Бородин.
Альдоль нестоек и если в его молекуле имеется а-водородный
атом, при нагревании отщепляет воду, превращаясь в непредель¬
ный альдегид, бутен-2-аль, который имеет тривиальное название
кротоновый альдегид.
СНЭ Н
СН3 —СН -СНг -СН =0 —Х=<
Ан -Нг° н^>=°
кротоновый альдегид
Поэтому суммарное превращение ацетальдегида в две стадии
называется кротоновой конденсацией.
Эта реакция, в которую могут вступать различные оксосоедине-
ния, причем перекрестно (например, ацетон с ацетальдегидом),
имеет большое практическое значение, т.к. позволяет синтезиро¬
вать разнообразные (3-оксикарбонильные производные или ненасы¬
щенные альдегиды и кетоны, причем увеличивается длина углерод¬
ной цепи. Число способов увеличения атомов углерода в органиче¬
ских молекулах ограничено. Следует учитывать, что и продукты
альдольного уплотнения, в свою очередь, могут претерпевать новое
альдольное уплотнение и т.д.
Примером перекрестного альдольного уплотнения может слу¬
жить реакция между глицериновым альдегидом и диоксиацетоном,
которая приводит к фруктозе:
500
-ч
СНгОН
диоксиацетон
СНгОН
)Н
=0
НО
фруктоза
сн=о
ОН
СНгОН
глицериновый
альдегид
Этот процесс в живых организмах обратим и идет под дейст¬
вием специального фермента - альдолазы. В растительных клетках
так осуществляется биосинтез фруктозы, а в животных происходит
обратный процесс, представляющий собой одну из стадий углевод¬
ного обмена и биотрансформации молекул углеводов в липиды и
наоборот.
2. С альдегидами, не имеющими а-водородного атома (напри¬
мер, формальдегид, бензальдегид, фурфурол) и поэтому не способ¬
ными претерпевать альдольное уплотнение, под действием щелочи
происходит иное превращение - окислительно-восстановительное
диспропорционирование до соответствующих спирта и карбоновой
кислоты (в виде соли), например, в случае бензальдегида:
Реакцию впервые наблюдал Канниццаро в 1853 г. Для альдеги¬
дов, обладающих а-водородным атомом, аналогичный процесс уда¬
лось осуществить В. Е. Тищенко (1906 г.) под действием алкоголята
алюминия. В результате образуется не смесь спирта с кислотой, а
продукт их взаимодействия - сложный эфир, например.
СНзч А1(ОР)э ,СН3 этилоаый эфир
2 ^ —* СН3—СН2—О—С^ уксусной кислоты
Поэтому это превращение названо реакцией Канниццаро-Ти¬
щенко или сложноэфирной конденсацией Оно есть следствие
того, что альдегиды по степени окисления занимают промежуточ¬
ное положение между спиртами и кислотами, с которыми их свя¬
сн=о
соок
СНгОН
бензоат калия бензиловый спирт
501
зывают окислительно-восстановительные переходы, играющие важ¬
ную роль в процессах метаболизма органических веществ
3. Для бензальдегида под действием цианид-иона (катализатор)
происходит так называемая бензоиновая конденсация.
Н Н. Зинин обнаружил эту реакцию и подробно исследовал
разнообразные химические превращения её продукта - бензоина.
4. Полимеризация характерна только для простейших альде¬
гидов и кетонов (формальдегид, ацетальдегид, ацетон)
Формальдегид и ацетальдегид легко вступают в реакцию триме-
ризации:
Продукт тримеризации формальдегида - триоксиметилен - был
открыт А. М. Бутлеровым. Триоксиметилен, так же как и тример
ацетальдегида - паральдегид, деполимеризуется при нагревании.
Формальдегид в водном растворе постепенно образует линейный
полимер - параформ (п = 8-100):
пСН2=0 + НОН —» НО—[—СН2—О—]—Н параформ
В настоящее время в промышленной практике широко употреб¬
ляется еще один линейный полимер формальдегида - полиформаль¬
дегид (п = 1000), из которого изготовляют различные детали. По¬
лиформальдегид - один из самых дешевых полимерных материалов.
Химики научились синтезировать также полимеры ацетальдегида и
ацетона.
Ещё один вариант полимеризации формальдегида состоит в об¬
разовании углеводов. Это превращение под действием баритовой
воды Ва(ОН)2 впервые осуществил А. М. Бутлеров в 1861 году.
Сначала формальдегид димеризуется в гликолевый альдегид,
тримеризация которого дает сахаристое вещество - гексозу.
СН=0
Я
Р = Н, СН3
триоксиметилен
502
Эта реакция дает ключ к пониманию одного из вопросов хими¬
ческой эволюции - проблемы синтеза углеводов из простейших мо¬
лекул. Представляется, что их образование происходило в водной
среде под действием минеральных щелочных катализаторов из фор¬
мальдегида, присутствовавшего в земной атмосфере.
Формальдегид СНг=0 в больших количествах получают окисле¬
нием метанола, а также из смеси оксида углерода(П) и водорода.
Он важен для получения фенолформальдегидных смол, полифор¬
мальдегида, как полупродукт органического синтеза. В медицинской
практике его употребляют в виде 40%-ного водного раство¬
ра - формалина - для дезинфекции и консервирования препаратов.
Из формальдегида синтезируют уротропин, впервые получен¬
ный А. М. Бутлеровым (1860 г.). Реакция состоит в действии ам¬
миака на формальдегид и сначала дает, как обычно, тример.
24.4. Отдельные представители
карбонильных соединений
н
,н
ЗСН2=0 + ЗЫНз —► з[сн2=мн] —»
формальдимин
I
АнгОИ
уротропин
гексаметилентетрамин
Тример далее взаимодействует еще с тремя молекулами формаль¬
дегида, а промежуточный продукт - с молекулой аммиака, в
результате чего и образуется гексаметилентетрамин (тетрааэа-
адамантан), именуемый уротропином.
Молекула уротропина представляет собой поликонденсирован-
ный насыщенный гетероцикл, состоящий из четырех шестичленных
колец Уротропин применяют для синтеза полимеров, а также как
лекарство (мочегонное средство, составная часть антигриппозного
препарата кальцекса и др )
Ацетальдегид СН3СНО производят из ацетилена, а также непо¬
средственным каталическим окислением этилена кислородом возду¬
ха Он является полупродуктом для синтеза этилового спирта, эти-
лацетата, паральдегида, хлораля и др
Фурфурол получают термической обработкой отрубей, соломы,
кукурузных початков. Продукт нитрования фурфурола - 5-нитро-
фурфурол служит для производства фурацилина.
Пиридоксаль является важнейшей структурной единицей вита¬
мина Ве Он выполняет ряд незаменимых функций в животных
и растительных организмах, в частности, при его участии осу¬
ществляется процесс транс-аминирования.
Ацетон СНэСОСНз синтезируют в основном окислением кумола
(глава 18), а также изопропилового спирта, получают его из уксус¬
ной кислоты или микробиологическим путем - ацетон-бутиловым
брожением некоторых микроорганизмов. Ацетон незаменим как
растворитель, он необходим в синтезе хлороформа, его применяют
при получении изопрена и др При диабете наблюдается аномально
высокое содержание ацетона (а также и некоторых других предель¬
ных кетонов) в крови.
Камфора представляет собой терпеновый кетон, извлекаемый
из сока ряда растений. Из 4 возможных стереоизомеров камфоры
(молекула которой содержит 2 хиральных центра) в природе встре¬
чается только один изомер, обладающий правым
вращением поляризованного света. Камфора ис¬
пользуется в медицине как средство, возбуж¬
дающее сердечную деятельность. Ее употребляют
в производстве пластмасс, например, целлулоид,
первый известный синтетический пластический
материал, представляет собой смесь камфоры с
Раствор целлулоида в спиртоэфирной смеси из¬
вестен под названием коллодия, применяемого для закрепления по¬
вязок и покрытия ран.
камфора
Глава 25. УГЛЕВОДЫ
25.1. Введение
Синтез сахаров начинается с работы А. М. Бутлерова (1861 г.),
в которой он получил сахаристое вещество полимеризацией фор¬
мальдегида (глава 24).
Для сахаристых веществ был предложен термин углеводы
(К. Шмидтом в 1844 г.), так как состав многих представителей са¬
харов отвечает формуле Ст(Н20)„. Например, глюкоза и фруктоза
имеют состав Сб(НгО)б, тростниковый или свекловичный сахар
(сахароза) - С12(Н20)ц, а клетчатка - [Сб(Н20)5]х. На самом деле не
все сахара соответствуют предложенной формуле. Однако название
“углеводы” укоренилось и в настоящее время является обще¬
признанным для этих веществ.
Углеводы наряду с жирами (липидами), белками и нуклеиновы¬
ми кислотами являются важнейшими химическими соединениями
растительного и животного мира. Достаточно сказать, что около
12% сухого веса любых организм'ов приходится на долю сахаристых
веществ. Они присутствуют во всех частях человеческого организ¬
ма (кровь, клеточная ткань, мозг, печень и т.д.). Например, глико¬
ген - крахмал животного происхождения - может составлять до
20% от общего веса печени В одной бактериальной клетке присут¬
ствуют 4.2 млн. молекул глюкозы и фруктозы, 1.8 млн. молекул
рибозы и 3.9 млн. молекул дезоксирибозы
Согласно принятой классификации, углеводы подразделяют на
две основные группы: моносахариды и полисахариды.
Моносахариды, в свою очередь, по числу углеродных атомов
разделяются на триозы, тетрозы, пентозы, гексозы' и т.д., а в
зависимости от природы карбонильной группы, которая входит в их
состав, они делятся на альдозы и кетозы.
Полисахариды - это конденсационные полимеры моносахаридов
с отщеплением молекулы воды. Так, сахароза С12Н22ОМ - конденса¬
ционный димер глюкозы СбН^Об и фруктозы СбН12<Эб.
Полисахариды в свою очередь делятся на низкомолекулярные
(или сахароподобные) и высокомолекулярные (или несахаропо¬
добные)
Среди сахароподобных полисахаридов важны дисахариды, часто
встречающиеся в природе сахароза, мальтоза, целлобиоза, лактоза
Окончание оза - необходимое указание на принадлежность вещества
к классу углеводов
505
Дисахариды, как и моносахариды, легко растворяются в воде, из ко¬
торой выделяются в виде кристаллов Они сладки иа вкус
Сладость моно- и дисахаридов может различаться в довольно
широких пределах Так, самый сладкий углевод - фруктоза - в 1 5
раза слаще сахарозы, которую принимают за эталон Сахароза, в
свою очередь, в 2 раза слаще глюкозы и в 4-5 раз - лактозы, кото¬
рая уже почти безвкусна
Полимеры моносахаридов с большим молекулярным весом ли¬
шаются физических свойств сахаристых веществ, о которых сказано
выше Несахароподобные углеводы - высокомолекулярные полиме¬
ры - лишены сладкого вкуса. Высокомолекулярные полисахариды
(крахмал, клетчатка, агар-агар и др.), как правило, обладают малой
растворимостью. Они, подобно другим высокомолекулярным соеди¬
нениям, образуют коллоидные растворы. Для них характерны явле¬
ния набухания и застудневания, что широко используется в прак¬
тике (например, застудневание крахмала - в пищевой промышлен¬
ности, агар-агара - в микробиологии).
В схеме 25.1 отражена классификация углеводов:
глицериновый
альдегид, диоксиацетон, крахмал,
рибоза, фруктоза сахароза клетчатка
глюкоза
Схема 25.1. Классификация углеводов
25.2. Моносахариды
Под углеводами в настоящее время подразумевают производ¬
ные многоатомных спиртов, содержащие карбонильную группу
(альдегидную или кетонную).
Глицериновый альдегид и диоксиацетон (глава 20) можно
считать простейшими моносахаридами - триозами. В соответствии
506
с принятой классификацией глицериновый альдегид - альдотриоза,
а диоксиаЦЙ^он - кетотриоза. Эти вещества, хотя в индивидуаль¬
ном виде в природе обычно не встречаются, являются промежуточ¬
ными продуктами углеводного обмена.
25.2.1. Синтез Фишера-Килиани
При взаимодействии 0(+)-альдотриозы с синильной кислотой
образуется два стереоизомерных а-оксинитрила, а из её энантиоме-
ра - Ц-)-альдотриозы - еще два изомерных продукта.
Это объясняется тем, что после присоединения синильной кис¬
лоты к альдегидной группе 0(+)-глицерииового альдегида в молеку¬
ле возникает новый асимметрический атом углерода (бывший до
присоединения углеродом карбонильной группы). Напомним, что
общее число стереоизомеров любого вещества выражается форму¬
лой 2П, где п - число асимметрических атомов углерода. В молекуле
продукта присоединения НСМ к глицериновому альдегиду два асим¬
метрических атома углерода. Поэтому из О- и Ь-глицериновых аль¬
дегидов можно получить 4 изомерных а-оксинитрила.
й-глицериновый
альдегид
СН=0
ОН
:НгОН
сн=
“Ь
Ьглицериновый
альдегид
сн=о
НО—I
СН2ОН
|нсм
I нем
1
N
1
N
1
N
1
N
8
15
15
к
0 (°й] [но] [но]
О.а-оксинитрилы
1,а-оксинитрилы
Известно, что нитрильная группировка (-С=М) под действием
воды превращается в карбоксильную группу (СООН), которую в
определенных условиях можно восстановить до альдегидной груп¬
пировки При суммировании превращений (присоединение НСМ,
507
гидролиз СвЫ группы, восстановление СООН группы) мы переходим
от одной альдотриозы к двум стереоизомерным альдотетрозам.
1) нем
ДСН=0 — ► РСН(ОН)СМ
-NN3
► РСН(ОН)СООН
3) [Н]
-НтО
РСН(0Н)СН=0
Эти стереоизомерные альдотетрозы не являются энантиомерами
(как О- и Ь-альдотриозы) Разновидность оптических изомеров, ко¬
торые не являются энантиомерами, называется диастереомерами.
Диастереомеры отличаются физическими и химическими свойства¬
ми; следовательно, их легко отличить друг от друга.
СН=0
СНзОН
СНзОН
й-глицериновый альдегид
і 1)НСМ
2)Н20
3) [Н]
—0
СНзОН
й-эритроза
СН=0
НО—^
СНзОН
О-треоэа
, 1) НСМ
2) Н20
3) [Н]
О-альдотетроэы
1)НСМ
2) Н20
3) [Н]
СН^ОН
й-рибоза
СНаОН
й-арабиноза О-ксилоза
СНаОН
й-ликсоза
й-альдопентозы
Схема 25.2. Синтез Фишера-Килиани.
508
Такой переход от глицеринового альдегида к диастереомерным
альдотетроНм был осуществлен впервые Фишером и Килиани в
80-х годах прошлого столетия и носит название синтеза Фишера-
Килиани (схема 25.2).
При превращении триоз в тетрозы конфигурация нижнего
асимметрического атома углерода не изменяется, то есть продукты
сохраняют генетическую связь с тем глицериновым альдегидом
(О- или Ь-), из которого они получены.
Генетическая связь с исходной альдотриозой - основа дополни¬
тельной классификации углеводов. Существуют два ряда моносаха¬
ридов: О-ряд (при изображении по Фишеру гидроксильная группа у
нижнего асимметрического атома углерода находится справа) и
Ь-ряд (гидроксильная группа у того же атома углерода стоит слева).
Таким образом, возможны две диастереомерные О-альдотетрозы
и две энантиомерные им Ь-альдотетрозы. В природе встречается
О-эритроза как промежуточный продукт обмена веществ.
При обработке любой из четырех изомерных альдотетроз (две
О- и две Ь-) последовательно синильной кислотой, водой и водо¬
родом можно синтезировать все возможные 8 изомерных альдо-
пентоз, четыре из которых принадлежат к О-ряду, а четыре дру¬
гих изомера, являясь попарно энантиомерами соответствующих
О-стереоизомерных форм, относятся к числу Ь-альдопентоз. Все
О-альдопентозы широко распространены в живых системах.
Легко подсчитать, что должно существовать 16 альдогексоз, во¬
семь из которых являются О-моносахаридами, а восемь -
Ь-моносахаридами.
Сказанное можно представить в виде общей схемы 25.2, приве¬
денной для моносахаридов: альдотриоз, -тетроз и -пентоз О-ряда.
Ряд нетрудно распространить на синтез альдогексоз, просто такой
же ряд построить и для Ь-моносахаридов. Углеводы О-ряда выбраны
в связи с тем, что именно они находятся в природе.
Приведем структуры важнейших природных моносахари¬
дов, которые необходимо запомнить
В их числе находится 0(-)-кетогексоза - й-фруктоза. Она
служит примером левовращающего моносахарида, принадлежащего
к Ь-ряду Другое тривиальное название О-фруктозы - левулоза
Структурными изомерами О-фруктозы являются О-альдогек-
созы - й-глюкоза, й-манноза и О-галактоза, которые в свою оче¬
редь являются диастереомерами по отношению друг к другу
О-глюкоза и О-манноза отличаются конфигурацией только у
второго атома углерода. Разновидность оптических изомеров моно¬
сахаридов, которые отличаются конфигурацией атома углерода,
509
смежного с карбонильной группой (С2), относят к числу эпиме¬
ров. Следовательно, О-глюкоза и О-маииоза - эпимеры. Эпимерами
по отношению друг к другу являются также альдопеитозы -
О-рибоза и О-арабиноза.
С№=0
|Н Н(
Н1
ОН
I | | | и П "“ип п^/— ~и П
@ [он] [он] [он] [он]
СНзОН СНзОН |—1 1—1 1—1 1—1
СН=0 СНзОН
1Н
СН20Н СНзОН СНзОН СНзОН
О-рибоза О-дезоксирибоза О-глюкоза О-манноэа 0-галактоза 0-фруктоза
Полезно обратить внимание на одинаковую конфигурацию у
3-го, 4-го и 5-го атомов углерода для широко встречающихся в при¬
роде углеводов: О-глюкозы, Ь-маннозы и О-фруктозы, что позволяет
легко запомнить формулы этих моносахаридов.
Индивидуальные названия углеводов имеют тривиальное про¬
исхождение. Глюкозу называют именно так за сладкий вкус, фрук¬
тозу - по источнику, из которого извлекают, как впрочем, и манно-
зу. Манна - это сладковатые выделения ряда растений - ясеня,
некоторых лишайников и др. Галактоза входит в состав молочного
сахара (лактозы), а арабиноза обнаружена во многих растительных
камедях (вишневый клей, гуммиарабик и др.).
Все перечисленные моносахариды имеют общую формулу
Ст(НгО)п. Однако уже давно известны и найдены в живых объектах
и моносахариды, имеющие иную суммарную формулу. Важнейшей
из них является изображенная выше дезокси-О(-)-рибоза, форму¬
ла которой С5Н10О4. Она входит в состав дезоксирибонуклеиновых
кислот (ДНК). Приставка дезокси- указывает на отсутствие атома
кислорода. В данном случае он отсутствует в положении 2. Более
правильно этот углевод следует именовать 2-дезокси-0(-)-рибозой.
2-Дезокси-0(-)-рибоза относится к числу так называемых “не¬
правильных” моносахаридов. Известны и дезоксигексозы, напри¬
мер, 2-дезокси-0(+)-глюкоза, которая также обнаружена в природ¬
ных объектах. Изредка в растительных источниках встречаются
дезоксимоносахариды, в которых отсутствует атом кислорода у по¬
следнего атома углерода. Подобные моносахариды называются
метилозами.
Другая “неправильность” углеводов может заключаться в раз¬
ветвлении углеродной цепи, что в природе наблюдается редко.
510
К числу “неправильных” углеводов относятся и такие сахара, в
которых гидроксильная группа заменена другими функциональными
заместителями. Здесь важны аминосахара (одна или несколько гид¬
роксильных групп заменены МНг-группами). В частности, строи¬
тельный материал насекомых и ракообразных относится к числу не¬
сахароподобных аминополисахаридов. Он называется хитином.
Фрагменты аминосахаров входят в состав молекул ряда гормонов,
нервных клеток, компонентов крови (например, у-глобулина) и др.
25.2.2. Свойства моносахаридов. Мутаротация
Любой из индивидуальных моносахаридов, обладая рядом кон¬
кретных физических свойств (температура плавления, раствори¬
мость и т.д.), характеризуется и своей специфической величиной
удельного вращения.
ОН
бНзОН
(3,О-глюкофураноза
=51
СНгОН
а,0-глюкопираноза
»ЕЛ
СНгОН
|3,0-глюкопираноза
Схема 25.3. Мутаротация О-глюкозы по Толленсу.
Удельное вращение любого моносахарида после растворения в
воде меняется во времени, достигая в итоге нового, вполне опреде¬
ленного значения. Так, например, свежеприготовленный раствор
О-глюкозы имеет [а)о +112°, а после длительного стояния ее удель-
511
ное вращение становится равным +52 5° и далее ие изменяется.
Изменение удельного вращения растворов углеводов во времени на¬
зывается мутаротацией. Очевидно, это явление должно вызывать¬
ся изменением симметрии молекулы, а, следовательно, трансформа¬
цией ее структуры в растворе
Явление мутаротации объясняется кольчато-цепной таутомерией
(глава 24) углеводов за счет взаимодействия карбонильной группы с
гидроксильными группами в положениях 4 или 5 (в альдозах) и
в положениях 5 или б (у фруктозы)
Обе кольчатые полуацетальные формы (с пяти- или шес¬
тичленным кольцом) существуют в виде а- и |3-форм, каждая из ко¬
торых должна характеризоваться своей величиной удельного вра¬
щения [а)о, отличного от удельного вращения цепной (оксикар-
бонильной) формы.
При установлении равновесия между всевозможными формами
в растворе удельное вращение должно измениться от величины, от¬
вечающей одной из начальных таутомерных форм, в виде которой
моносахарид находится в твердом состоянии, до конечного значе¬
ния, отвечающего суммарному вкладу всех таутомерных форм, уча¬
ствующих в равновесии. На схеме 25 3 изображена мутаротация
глюкозы
Схема отвечает принципам изображения по Толленсу, при ко¬
тором для наглядности углеродный остов оксикарбонильной формы
сохраняется без изменения и в циклических изомерах, а кислород¬
ный мостик соединяет полуацетальный атом углерода с углеродом в
четвертом (пятичленное кольцо) или в пятом (шестичленное коль¬
цо) положениях.
При взаимодействии карбонильной группы О-глюкозы с четвер¬
той гидроксильной группой замыкается пятичленный тетрагидро-
фурановый гетероцикл, и поэтому пятичленные кольчато-полу-
ацетальные формы называют фуранозами. Реакция альдегидного
карбонила с пятым гидроксилом дает пирановое кольцо, и такие
таутомерные формы называют пиранозами.
Обобщая сказанное, можно дать полное название каждой тау-
томерной форме (см. схему 25.3).
При способе изображения по Хеуорсу (глава 18) заместители
помещают сверху и снизу от плоскости молекулы в зависимости от
конфигурации соответствующего углеродного атома, а именно:
группы, расположенные справа в фишеровской проекции, помещают
ниже плоскости кольца в формуле Хеуорса, а группы слева - выше
этой плоскости.
Рассмотрим переход от формул Фишера-Толленса к формулам
Хеуорса на примере а,0-глюкопиранозы. Предварительно необхо¬
512
димо так перестроить фишеровскую проекцию, чтобы кислород на¬
ходился нЛрдной прямой с углеродными атомами, при этом группа
СНгОН должна быть помещена слева.
СНгОН —' А
а,0-глюкопираноза
*7\ ОН
Б В
Обращает на себя внимание существенное различие в принци¬
пах изображения по Фишеру-Толленсу и по Хеуорсу. Так как моде¬
ли Хеуорса являются перспективными изображениями, а не про¬
екциями, их можно любым образом перемещать в пространстве
(сравните А-Б-В).
Мутаротацию О-глюкозы по Хеуорсу следует представить с по¬
мощью схемы 25.4.
Необходимо помнить, что формулы Хеуорса не отражают под¬
линной пространственной формы молекул. В частности, кольцо пи-
ранозы, как и циклогексановый цикл, может существовать в кон¬
формациях кресла или ванны и не является плоским (см. гла¬
ву 18). Конформация кресла обычно более устойчива.
Таким образом, метод Хеуорса, как и Фишера, - условный спо¬
соб изображения реальных структур, не позволяющий полностью
отразить их стереохимические особенности
Возникает вопрос: в какой форме реально находятся моносаха¬
риды в кристаллическом состоянии и в растворе? В результате
специальных исследований найдено, что в кристаллическом состоя¬
нии и в растворах линейная оксикарбонильная форма практически
отсутствует. В частности, все альдозы кристаллизуются из воды
обычно в а-пиранозной форме, а фруктоза - в |3-фуранозной.
17 Зак. 675
513
В растворе устанавливается равновесие между всеми кольчаты¬
ми формами с заметным преобладанием пираиоз Сказанное иллю¬
стрируется табл 25 1
а, й-глюкофураноза р.О-глюкофураноза
Схема 25.4. Мутаротация О-глюкозы по Хеуорсу.
Мутаротация - явление динамического химического равновесия,
и к ней полностью применимы все принципы смещения равновесия.
В зависимости от условий моносахарид может проявлять химиче¬
ские свойства любой из пяти возможных таутомерных форм и да¬
вать продукты взаимодействия в той таутомерной форме, реакция с
которой смещает равновесие в сторону её образования, даже в том
случае, когда содержание формы незначительно. Именно поэтому
химики, наблюдая образование продуктов присоединения по очень
активной карбонильной группе, ранее считали, что моносахариды
существуют в нециклической форме.
Таким образом, моносахариды следует рассматривать как ве¬
щества, обладающие свойствами:
а) многоатомных спиртов (в любой из таутомерных форм);
б) альдегидов или кетонов (в нециклической форме);
в) полуацеталей (в любой из четырех циклических форм).
514
Таблица 25.1
Мутаротация моносахаридов
[а]о
Равновесная смесь
в водном растворе,
Моносахарид
Кристалличес¬
кое состоя¬
Раствор
содержание
отдельных форм,%
ние, форма
Пираноза
Фураноза
а
Р
а
Р
0(-)-рибоза
-18 8°,
а-пираноза
-23.7°
18
54
12
16
0(+)-глюкоза
+ 112°,
а-пираноза
+52.5°
35
65
0(+)-манноза
+30°,
а-пираноза
+ 14.5°
67
33
-
0(+)-галактоза
+ 15°,
а-пираноза
+80°
25
75
0(-)-фруктоза
+ 132°,
(3-фураноза
-93°
76
4
20
25.2.3. Реакции с участием гидроксильных групп
Так как моносахариды в кристаллическом состоянии и в ра¬
створах существуют в кольчатых полуацетальных формах, целесо¬
образно прежде всего рассмотреть химические свойства, связанные
с присутствием именно этих форм, то есть обсудить реакции полуа-
цетального гидроксила.
Полуацетальный гидроксил непрочно связан с атомом угле¬
рода и может замещаться другими группировками в реакциях со
спиртами, кислотами, аминами (НХ) и т д. по общей схеме.
Вещество, которое действует на полуацетальный гидроксил мо¬
носахарида (гликозы или оэы), называется агликоном, а продукт
реакции - гликоэидом Гликозиды - вещества, как правило,
17*
515
нестойкие и под действием воды (особенно в присутствии кислот)
вновь распадаются на моносахарид и агликон
Гликозиды в отличие от самих исходных моносахаридов уже не
способны к мутаротации, так как подвижный атом водорода полуа-
цетальной группы в них отсутствует, поскольку и сам полуацеталь-
ный гидроксил исчез.
Если в качестве агликонов выступают спирты, фенолы или кар¬
боновые кислоты, то продукты реакции называют О-гликоэидами.
Например, при реакции метилового спирта (агликон) с глюкозой
(допустим, в а-пиранозной форме) образуется продукт алкилирова-
ния - метил-а,0-глюкопиранозид:
метил-а,0-глюкопиранозид ацетил-а.О-глюкопираноэид
Если же подействовать на а.Э-глюкопиранозу уксусной кисло¬
той, то можно получить ацетил-а,0-глюкопиранозид, являющийся
продуктом ацилирования.
К числу О-гликозидов относятся многие важные в физиологи¬
ческом отношении вещества, образующиеся в результате жизнедея¬
тельности. О-гликозидами являются ди- и полисахариды.
Сложно устроенные молекулы витаминов К, Р, антибиотика
стрептомицина и других природных соединений включают в себя
О-гликозидный фрагмент.
Большую группу природных растительных веществ составляют
так называемые сердечные гликозиды, извлекаемые из наперстян¬
ки, ландыша и т д. Они с давних пор широко используются при ле¬
чении сердечно-сосудистых и других заболеваний. Большинство из
этих веществ сложны по строению, однако среди них встречаются
простые соединения.
516
Так, например, гликозид толокнянки - арбутин - это гидрохи-
нон-а,0-гЛ1&копиранозид:
НОН2С
н^он
но
ОН -Н20 но
ОН
а.О-глюкопираноэа
арбутин
Если полуацетальный гидроксил углевода взаимодействует с
меркаптогруппой (-БН), продукт реакции называют Б-гликозидом.
Б-гликозиды очень редки. Они, в частности, найдены в горчице.
Более активные алкилирующие и ацилирующие агенты могут
реагировать кроме полуацетального также и со спиртовыми гид*
роксильными группами
Если вместо метилового спирта взять иодистый метил (СНэО в
присутствии щелочи и подействовать им на О-глюкозу, то в про¬
дукте реакции кроме гликозидной метильной группы образуется
еще четыре простых эфирных группировки.
НОН2С СН,ОН,С
Полученный продукт алкилирования глюкозы следует назвать:
метил-тетраметил-а.О-глюкопиранозид Одна из метильных групп в
названии выделена специально, чтобы показать, что она является
гликозидной, а, следовательно, отличается от остальных по своим
химическим свойствам Это легко заметить, если подействовать на
полученный продукт метилирования водой При действии воды гли-
козидный фрагмент разрушается на агликон и производное моноса¬
харида, в котором простые эфирные группировки, не склонные к
гидролизу, сохраняются без изменения
Н<
метил,тетраметил-а,0-глюкопираноэид
н2о)-сн3он
СН3ОН2С
ОСНз
517
Нужно указать, что продукт гидролиза в отличие от исходного
гликозида способен к мутаротации, но с участием только трех
таутомерных форм (а не пяти, как сама глюкоза), а именно, а- и
Р-пиранозных и нециклической 2,3,4,6-тетраметил-0-глюкозы.
Если в реакции с глюкозой вместо уксусной кислоты взять её
более активный ангидрид (см главу 26), в результате получится
продукт полного ацилирования а.О-глюкопиранозы. ацетил,тетра-
ацетил-а.О-глюкопиранозид В нем ацетильная группа при первом
атоме углерода (“полуацетальная”), как в предыдущем случае ме-
тильная, заметно отличается от других ацетильных (сложноэфир¬
ных) групп и легко удаляется гидролизом
Подбором условий можно получить продукты ацилирования од¬
ной или нескольких гидроксильных групп В живых организмах под
действием ферментов происходит избирательное ацилирование мо¬
носахаридов фосфорной кислотой по гидроксилу у последнего атома
углерода (пятый - у рибозы и дезоксирибозы, шестой - у глюкозы и
фруктозы).
Например, из Э-глюкозы образуется й-глюкозо-б-фосфат:
В процессе реакции, как и следует ожидать, сначала фосфори-
лируется более активный полуацетальный гидроксил молекулы глю¬
козы в ее таутомерной пиранозной форме, а затем остаток фосфор¬
ной кислоты с участием ферментов переносится из первого поло¬
жения в шестое. Эти реакции фосфорилирования углеводов
являются начальными стадиями углеводного обмена.
Широко распространены в живой природе, особенно в нуклеи¬
новых кислотах, М-гликозиды (см. главу 30), получаемые действи¬
ем аминов на гликозы с замещением полуацетального гидроксила
аминогруппой.
518
25.2.4. Реакции с участием карбонильной группы
Хотя линейная оксикарбонильная форма в сколько-нибудь зна¬
чительных количествах и не присутствует в кристаллических моно¬
сахаридах и их растворах, её участие в таутомерном равновесии
сообщает моносахаридам все свойства, присущие альдегидам
(в альдозах) или кетонам (в кетозах). Рассмотрим некоторые из
этих свойств, заключающиеся прежде всего в высокой активности
карбонильной группы в реакциях присоединения. С двумя реакция¬
ми мы уже познакомились - это синтез Фишера-Килиани
(присоединение синильной кислоты) и мутаротация (внутримолеку¬
лярное присоединение спирта).
Восстановление моносахаридов. Моносахариды легко гид¬
рируются по связи С=0 и при этом превращаются в многоатомные
спирты. Из Э-маннозы, в частности, получается многоатомный
спирт Х)-маннит, а из Э-глюкозы - Э-сорбит. Восстановление
Э-фруктозы приводит к эквимолекулярной смеси эпимеров -
О-маннита и Э-сорбита, так как в продукте её гидрирования вто¬
рой атом углерода становится асимметрическим.
СНзОН
Э-маннит
Получающиеся в результате восстановления шестиатомные
спирты (их общее название - гекситы) вступают во все реакции,
характерные для многоатомных спиртов В том числе они ацилиру-
ются, давая сложные эфиры, которые находят применение в прак¬
тике. Например, гексанитроэфир 6-маннита, как и нитроглицерин,
519
является взрывчатым веществом и, кроме того, применяется как со¬
судорасширяющее средство
Реакции с феиилгидразином. Известно (глава 24), что
а-оксиальдегиды и а-оксикетоны дают с феиилгидразином озазоны
Так же ведут себя и моносахариды. Эта реакция требует участия
трех молекул фенилгидразина на одну молекулу моносахарида, и её
продуктами являются озазон, аммиак и анилин.
Так как в результате реакции атом углерода, соседний со
связью С=М, теряет асимметричность, различие в строении озазонов
из двух эпимерных моносахаридов должно исчезать. Другими сло¬
вами, озазоны эпимеров идентичны. О-глюкоза и О-манноза дают
один и тот же продукт - О-глюкозазон. Тот же озазон получается и
из О-фруктозы, так как в ней при второй стадии дегидрируется пер¬
вичная спиртовая группа.
СН=ММНСвН5 НаММНСбНб
=ММНСвН5 .755
-МН2С^5
ІН -2НгО
ОН
ОН
бНзОН
І-Жз
-МН2СбН5
-2НгО
СНзОН
СН20Н
Э-фруктоза
Отметим, что из О-рибозы и О-арабинозы (а они эпимерны)
также образуется один и тот же озазон - О-рибозазон.
Озазоны - окрашенные в ярко-желтый цвет, высокоплав¬
кие, кристаллические вещества. Они плохо растворимы в воде и
поэтому используются в целях идентификации углеводов. То, что
О-глюкоза, О-манноза и О-фруктоза дают один и тот же озазон, яв¬
520
ляется строгим доказательством их одинаковой конфигурации с
третьего пятый атом углерода. Это было впервые установлено
Э. Фишером.
Эпимериэация моносахаридов. Карбонильная группа, как
известно, сообщает подвижность атому водорода у а-углеродного
атома, что приводит к явлению кето-енольной таутомерии альдеги¬
дов и кетонов. Применительно к О-глюкозе в её оксикарбонильной
форме явление кето-енольной таутомерии выразится следующим
уравнением:
В данном случае имеет место таутомерия - а-оксикарбонил ^
ендиол. Точно такой же ендиол возникает и из эпимера Э-глюко-
зы - Э-маннозы, так как при енолизации атом углерода С2 (а кон¬
фигурацией именно у этого атома углерода и отличаются эпимеры)
перестает быть асимметрическим. Этот же ендиол должен возник¬
нуть и при енолизации О-фруктозы с участием водорода при
Сгатоме углерода Следовательно, в процессе кето-енольной тауто¬
мерии моносахаридов должны принимать совместное участие три
моносахарида О-глюкоза, О-манноза и О-фруктоза В итоге должно
наблюдаться частичное взаимопревращение всех трех моносахари¬
дов через промежуточный ендиол до установления равновесия ме¬
жду ними. Из О-маннозы, О-глюкозы и О-фруктозы по отдельности
образуется одна и та же равновесная смесь трех моносахаридов
521
В водных растворах в отсутствие катализаторов такое равнове¬
сие устанавливается крайне медленно. Однако реакция резко ус¬
коряется под действием щелочи а также некоторых фермейтов. Ес¬
тественно, такой процесс, происходящий с обращением конфигура¬
ции у Сг-атома, сопровождается изменением удельного вращения.
Изменение удельного вращения моносахаридов до определенного
значения под действием щелочи, вызванное кето-енольной таутоме¬
рией, называется эпимериэацией
Эпимеризация глюкозы - одна из стадий метаболизма глюкозы,
причем в этом биохимическом процессе принимает участие фосфо-
рилированная форма глюкозы - й-глюкозо-б-фосфат-
СНгО—Р=0
Ьн
О-глюкоэо-б-фосфат
СН2ОН
Э-фруктозо-б-фосфат
ОН
СНгО—Р^)
|==0 он
СНгОН
монофосфат
диоксиацетона
сн=о
1—он ОН
СН2О—Р=0
сн=о
ОН он
СнгО—Р=о
он
монофосфат
глицеринового
Н3РО4 |-НгО
ОН
9Н20—рУ=о
ОН
НО-
ОН ОН
СНгО—Р=0
ОН
О-фруктоэо-1,6-дифосфат
Эпимеризация приводит к образованию й-фруктозо-б-фос¬
фата, который далее фосфорилируется в й-фруктозо-],6-дифос¬
фат. Ретроальдолизация последнего (см. главу 24) вызывает пре¬
вращение в 3-монофосфаты глицеринового альдегида и диоксиаце¬
тона. Эти фосфорные эфиры, в свою очередь, претерпевают вза-
имопереход за счет эпимеризации. Путем серии последующих фер-
522
ментативных превращений они превращаются далее в фосфат енола
пировиноГ^дной кислоты (см. главу 30).
Окисление моносахаридов. Под действием окислителей в
молекулах моносахаридов, в первую очередь, будет окисляться аль¬
дегидная группировка, которая наиболее активна в химических
реакциях. Для её превращения в карбоксильную группу достаточ¬
но использования даже таких слабых окислителей, как Си(ОН)2
или АдгО.
Продукты окисления альдегидной группы альдоэ называются
гликоновыми кислотами.
О-глюконовая
кислота
О-глюкаровая
кислота
Реакция “серебряного зеркала” с Э-глюкозой дает О-глюконо-
вую кислоту, с О-галактозой - Э-галактоновую кислоту и т д.
Реакцию “серебряного зеркала” дает и Э-фруктоза Дело в том,
что эта реакция происходит в щелочной среде, что вызывает эпиме-
ризацию Э-фруктозы в Э-глюкозу и Э-маннозу. Они и вступают в
реакцию с АдгО.
Э-глюконовую кислоту изготавливают в промышленности. Её
применяют в некоторых лекарственных препаратах, в частности, в
виде кальциевых солей (глюконат кальция).
Если обработать моносахарид более сильным окислителем
(например, НМОэ или К2СГ2О7), то окислению может подвергнуться
не только альдегидный, но и первичный спиртовой атом углерода
(5-й - у пентоз, 6-й - у гексоз) В результате возникают дикарбо-
новые кислоты, которые называют гликаровыми кислотами
Другое (тривиальное) название гликаровых кислот - сахарные
кислоты Из О-глюкозы, в частности, образуется О-глюкаровая
кислота.
Окисление в гликаровые кислоты используется в целях иденти¬
фикации моносахаридов.
Из Э-фруктозы после окисления сильным окислителем образу¬
ется О-фруктаровая кислота, которую можно получить также и
523
иным путем - окислением вторичнои гидроксильном группы
Э-глюкаровой кислоты.
СНзОН
О-фруктаровая
кислота
О-глюкаровая
кислота
При действии сильных окислителей не на сам моносахарид, а на
его гликозид (в нем нет карбонильной группы) окисляется первич¬
ная спиртовая группа. При последующем гидролизе получается
окисленное производное моносахарида, в котором альдегидная
группа осталась неокисленной Такие вещества называют глику-
роновыми кислотами. Из О-глюкозы, в частности, получается
О-глюкуроновая кислота
НОН2С ноос
I—г*
Гликуроновые кислоты обладают всеми характерными свойства¬
ми моносахаридов. Это - мутаротация, эпимеризация под действием
щелочей, реакция с синильной кислотой, фенилгидразином и т.д.
Они вступают в реакцию "серебряного зеркала" и образуют в ре¬
зультате этого гликаровые кислоты. Ниже приведена мутаротация
Э-глюкуроновой кислоты.
524
Сочетание высоких полярности и химической активности по-
луацеталвАдго гидроксила глюкуроновой кислоты сообщают ей
важное свойство - она участвует в процессе удаления ксенобиоти¬
ков, конъюгируясь с их гидроксильной или аминогруппой. В резуль¬
тате реакции с полуацетальным гидроксилом образуются соответ¬
ствующие конъюгаты - Э-глюкопираноуронозиды. Конъюгаты уда¬
ляются с мочой. Поэтому глюкуроновую кислоту всегда можно об¬
наружить в моче, откуда и происходит ее название.
й-глюкуроновая кислота
НООС
та.
ОН
а.О-глюкофураноуроновая
О ОН
З.О-глюкофураноуроновая
кислота
В циклических таутомерных формах гликуроновые кислоты об¬
разуют гликозиды с различными агликонами, в том числе и такие,
которые встречаются в природе. В ряде объектов растительного
происхождения в больших количествах накапливаются несахаропо¬
добные полисахариды, являющиеся конденсационными полимерами
гликуроновых кислот. В их числе пектиновые вещества, кон¬
центрирующиеся в кожуре цитрусовых. Полимерные производные
Э-маннуроновой кислоты содержатся в бурых водорослях Эти ве¬
щества после выделения применяются в пищевой и фармацевти¬
ческой промышленности
25.3. Сахароподобные полисахариды
(дисахариды)
В природе широко представлены полимерные производные мо¬
носахаридов - полисахариды с молекулярным весом, который может
очень сильно меняться (от нескольких сотен до десятков мил¬
лионов) Они построены из молекул моносахаридов, связанных
друг с другом в результате отщепления молекул воды, то есть поли¬
сахариды следует рассматривать как конденсационные полимеры
моносахаридов
Полисахариды относятся к О-гликозидам. Их образование про¬
исходит с участием в конденсации полуацетального гидроксила мо¬
носахарида в одной из четырех возможных циклических таутомер-
ных форм, которые возникают в результате мутаротации Следую¬
щая молекула моносахарида является агликоном и может реагиро¬
вать двояко: или также своим полуацетальным гидроксилом, или
одной из своих спиртовых групп. Обычно (но не обязательно) в
образовании полисахарида участвует гидроксильная группа в поло¬
жении 4.
Рассмотрим эти закономерности для конденсационных димеров
моносахаридов - дисахаридов. Их можно разделить на два типа.
Первому типу дисахаридов дано название гликозидо-гликозы,
второму типу - гликозидо-гликозиды.
В гликозидо-гликозах, где одна молекула связана полуацеталь¬
ным гидроксилом, а вторая - спиртовым, карбонильная группа со¬
храняется во второй части дисахарида, и поэтому для них проба с
Ад20 является положительной.
В гликозидо-гликозидах оба моносахарида реагируют своими по-
луацетальными гидроксильными группами. Карбонильная группа
в них отсутствует, поэтому они не обнаруживают ни одной из ре¬
акций, характерных для группы С=0, в том числе и реакцию
“серебряного зеркала”. Это позволяет легко отличить дисахариды
двух типов между собой. Гликозидо-гликозы называют восстана¬
вливающими дисахаридами, а гликозидо-гликозиды - невос¬
станавливающими дисахаридами.
Дисахариды обоих типов являются сахароподобными полисаха¬
ридами и имеют много общего в физических свойствах. Однако в их
химическом поведении кроме отношения к реагенту на карбониль¬
ную группу (АдгО) существуют и другие принципиальные различия,
которые требуют специального рассмотрения.
526
^25.3.1. Восстанавливающие дисахариды
Важнейшими представителями восстанавли¬
вающих дисахаридов - гликозидо-гликоз - являются
мальтоза, целлобиоза и лактоза. Во всех этих
веществах агликоном является 4-0-глюкоза. Следо- Н
вательно мальтоза, целлобиоза и лактоза представ¬
ляют собой гликозидо-4-О-глюкозы:
Мальтоза построена из двух молекул глюкозы.
Одна молекула глюкозы в а-пиранозной форме объ¬
единяется с другой при участии своей полуацетальной гидроксиль¬
ной группы.
Следовательно, полное название мальтозы - а,0(+)-глюко-
пиранозидо-4-0(+)-глюкоза.
СН2ОН
-ОН
СигОН
СНгОН
по Хеуорсу
по Толленсу
Мальтоза, являясь димером глюкозы, в свою очередь, является
мономером крахмала. Мальтоза в большом количестве находится в
солоде, поэтому её называют солодовым сахаром.
Целлобиоза также построена из двух молекул глюкозы, но от¬
личается от мальтозы тем, что в одной из молекул, образующих ди¬
мер, в реакцию вступает полуацетальный гидроксил Р-пиранозной
формы, то есть целлобиоза - Р,0-(+)-глюкопиранозидо-4-й(+)-
глюкоза.
целлобиоза
Целлобиоза в чистом виде в природе встречается редко, одиа-
ко она является мономером, образующим полимер - клетчатку, и в
связанном виде в этом соединении чрезвычайно распространена.
Лактоза также является восстанавливающим дисахаридом, по¬
строенным из Э-глюкозы, которая связывается своим 4-м гидрокси¬
лом с Э-галактозой. Э-галактоза реагирует своим полуацетальным
гидроксилом в Р-пиранозной форме Итак, полное название лакто¬
зы - р,0(+)-галактопиранозидо-4-й(+)-глюкоза.
Лактоза, или молочный сахар, содержится в молоке млекопи¬
тающих (до 4-6%). Этот дисахарид выделяют выпариванием сыво¬
ротки после отделения свертывающихся белков при скисании моло¬
ка. Из-за малой сладости лактозу применяют как наполнитель в
драже и таблетках в фармацевтической промышленности.
Химические свойства восстанавливающих дисахаридов опреде¬
ляются тем, что та из частей молекулы дисахарида, в которой не
затронут полуацетальный гидроксил, вследствие мутаротации может
существовать в растворах в различных таутомерных формах.
В отличие от моносахаридов в мутаротации дисахаридов прини¬
мают участие только три таутомерные формы: оксикарбонильная и
две пиранозные. Фуранозные формы здесь невозможны, так как в
дисахаридах нет свободной гидроксильной группы в 4-м положе¬
нии. Мутаротация для всех трех восстанавливающих дисахаридов
(мальтозы, целлобиозы и лактозы) в общем виде выглядит так:
ОН
СНзОН СНзОН
по Толленсу
по Хеуорсу
лактоза
25.3.2. Химические свойства
НОН2С
сн=о
он
НОН2С
он
Н(
го
он
он
СНгОН
он
528
В таутомерии, приводящей к мутаротации, участвует та часть
дисахаридаТ^оторая образована агликоном - 4-0-глюкозой.
Лактоза, которая в твердом виде находится в Р-пиранозной
форме и имеет удельное вращение (а]о +90°, после мутаротации
в водном растворе приобретает удельное вращение [а]о +55°. Цел-
лобиоза в кристаллическом состоянии находится в а-форме с
[а)о +24°, а после её мутаротации [а]о становится равным +35°.
По аналогии с моносахаридами для восстанавливающих дисаха¬
ридов характерны все те реакции, которые присущи как полуаце-
тальному гидроксилу, так и карбонильной группе.
Например, при действии метанола на мальтозу можно получить
а-метилмальтозид, что является результатом химической актив¬
ности её полуацетального гидроксила в таутомерной а-форме:
Мальтозу (как, впрочем, и лактозу, и целлобиозу) можно не
только алкилировать, но и ацилировать по полуацетальному гид¬
роксилу с образованием соответствующих О-гликозидов.
При взаимодействии с Ад20 альдегидная группа восстанавли¬
вающих дисахаридов окисляется до карбоксильной группы
СН—о
-—он
СООН
[О] ОН 1—ОН
— НО но
НО-
-О—1 он
-О—1 он
он
снгон снзон
СНзОН СНзОН
мальтоза
мальтобионовая кислота
529
Дисахарид вступает в реакцию в таутомерией оксикарбонильиой
форме и получаются гликоновые кислоты, которые в отличие
от гликоновых кислот из моносахаридов называют биоиоаыми
кислотами. Так, из лактозы получают лактобионовую кислоту,
а из мальтозы - мальтобионовую.
Для обсуждаемых дисахаридов характерны все остальные реак¬
ции по карбонильной группе: присоединение синильной кислоты,
взаимодействие с фенилгидразином с образованием озазонов, вос¬
становление в соответствующие многоатомные спирты, эпимериза-
ция в щелочных растворах и т.д.
Лактоза, мальтоза и целлобиоза представляют наглядные при¬
меры различия физиологического действия оптических изомеров в
зависимости от их конфигурации. Мальтоза полностью усваивается
человеческим организмом Лактоза, отличающаяся от нее конфигу¬
рацией двух хиральных центров, усваивается не всеми из-за так на¬
зываемой лактазной недостаточности, что важно в педиатрии.
Невозможно вскармливать детей, страдающих лактазной недоста¬
точностью, искусственными молочными смесями. Целлобиоза сов¬
сем не переваривается человеком, хотя легко гидролизуется в же¬
лудке травоядных животных.
Изредка в природных объектах происходит такое сочленение
моносахаридов, когда один из них реагирует не 4-й, а 6-й гидро¬
ксильной группой. Таким дисахаридом является, например, алло¬
лактоза, которая, как и лактоза, находится в молоке. Она тоже
образована из Э-глюкозы и Э-галактозы и представляет собой
р,0-галактопиранозидо-6-0-глюкозу. Полезно обратить внимание на
то, что в мутаротации аллолактозы в отличие от лактозы участвуют
пять таутомерных форм, так как здесь возможно образование не
только пираноз, но и фураноз.
Иногда, но крайне редко, в образовании природных дисахаридов
в качестве одного из двух моносахаридов участвуют пентозы, на¬
пример, арабиноза и др.
25.3.3. Невосстанавливающие дисахариды (сахароза)
Количество известных невосстанавливающих дисахаридов при¬
родного происхождения значительно меньше, чем восстанавливаю¬
щих. Однако один из числа их представителей чрезвычайно широко
распространен в природе и играет важную роль в жизнедеятельно¬
сти человека. Это - сахароза (тростниковый сахар), в построении
530
молекулы которой участвуют два моносахарида - Э-глюкоза и
О-фруктоза^глюкоза - в а-пиранозной форме, а фруктоза - в виде
Р-фуранозы.
Сахароза - это а,0-глюкопиранозидо-Р,0-фруктофуранозид, что
можно изобразить по Толленсу и по Хеуорсу:
по Толленсу сахароза
В молекуле сахарозы нет полуацетальных гидроксильных групп
и карбонильной группы, поэтому отсутствуют реакции, характерные
для этих функциональных группировок (мутаротация, образование
гликозидов, реакция “серебряного зеркала”, восстановление, вза¬
имодействие с фенилгидразином, синильной кислотой и т.д.).
Сахароза, как и всякий гликозид, может разрушаться на исход¬
ные компоненты при действии воды в присутствии кислот, высту¬
пающих в качестве катализатора этого превращения. Процесс гид¬
ролиза сахарозы сопровождается изменением удельного вращения
её растворов, так как в результате гидролиза возникают индивиду¬
альные моносахариды (Э-глюкоза и Э-фруктоза), каждый из кото¬
рых характеризуется своим удельным вращением, отличным от
удельного вращения сахарозы. Явление изменения знака удель¬
ного вращения при необратимом гидролизе сахарозы называется
инверсией сахарозы. При инверсии удельное вращение сахарозы
меняется от +66.5° до -40.5°. Изменение знака вращения вызвано
тем, что Э-фруктоза (левулоза) имеет сильное отрицательное вра¬
щение, которое составляет -93°.
Напомним, что явление гидролиза, как характерное свойство
всяких гликозидов, в той же мере присуще и восстанавливающим
дисахаридам - мальтозе, целлобиозе, лактозе.
Процесс инверсии необратим и при отщеплении воды от смеси
Э-глюкозы и Э-фруктозы сахароза вновь не образуется Дело в
том, что число возможных комбинаций сочленения моносахаридов
между собой, даже если учитывать только взаимодействие полуа¬
цетальных гидроксилов в кольчатых формах, составляет 16, а с
учетом возможного участия и спиртовых групп число таких комби¬
531
наций неизмеримо возрастает. Этот пример служит наглядной ил¬
люстрацией трудности решения проблемы синтеза дисахаридов за¬
данного строения из моносахаридов. Тем не меиее разработаны
специальные синтетические приемы, которые позволяют получать
отдельные индивидуальные дисахариды Родоначальником разра¬
ботки синтеза дисахаридов из моносахаридов явился А Колли
В природе синтез дисахарида определенного строения (например,
сахарозы) осуществляется за счет избирательного действия фер¬
ментативных систем Химиками выполнен ферментативный синтез
сахарозы
Для сахарозы практически можно наблюдать только реакции со
спиртовыми гидроксильными группами. Это, в частности, окисление
сильными окислителями, при котором три первичных спиртовых
гидроксила превращаются в карбоксильные группы и в результате
образуется сахартрикарбоновая кислота:
СНгОН
сахартрикарбоновая
кислота
Спиртовые гидроксильные группы сахарозы можно подвергать
также алкилированию и ацилированию, причем, как и для моноса¬
харидов - в жестких условиях. Например, при обработке сахарозы
йодистым метилом в присутствии щелочи получается продукт пол¬
ного метилирования сахарозы - октаметилсахароза:
октаметилсахароза
Еще одним важным свойством сахарозы, а также мальтозы яв¬
ляется участие в процессе брожения. Брожение происходит под
532
действием Ферментов и начинается с гидролиза дисахарида до мо¬
носахаридов которые далее и подвергаются собственно брожению.
Другими невосстанавливающими дисахаридами, также распро¬
страненными в природе, являются трегалозы. Эти вещества -
0-глкжопиранозидо-Е)-глкжопиранозиды, то есть продукты объеди¬
нения за счет полуацетальных гидроксилов двух молекул Э-глю-
козы в пиранозной форме. Возможны все варианты их сочетания
между собой - а,а-, Р,Р- и а,Р-, обнаруженные в тех или иных при¬
родных источниках (от пекарских дрожжей до лишайников и неко¬
торых видов грибов).
В процессе рафинирования (одной из стадий очистки тростни¬
кового сахара) может быть получена в чистом виде рафиноза, не¬
восстанавливающий трисахарид, который является 6-р,0(+)-гала-
ктопиранозидо-а,0(+)-глюкопиранозидо-Р,0(+)-фруктофуранозидом.
Рафиноза, кстати, самый распространенный после сахарозы полиса¬
харид из числа сахароподобных полисахаридов.
25.4. Несахароподобные полисахариды
При образовании из моносахаридов полимерной цепи несахаро-
доподобных (высокомолекулярных) полисахаридов одна молекула
моносахарида в одной из возможных кольчатых таутомерных форм
взаимодействует своим полуацетальным гидроксилом с каким-либо
спиртовым (обычно четвертым) гидроксилом второй молекулы мо¬
носахарида. Вторая молекула моносахарида также своим полуаце¬
тальным гидроксилом соединяется со спиртовым гидроксилом тре¬
тьей молекулы и т.д. Таким образом, высокомолекулярные полиса¬
хариды являются поли-О-гликозидами. В свою очередь, полисахари¬
ды делятся на гомо- и гетерополисахариды. Гомополисахариды со¬
стоят из одного моносахарида Гетерополисахариды составлены из
двух (или более) различных моносахаридов.
25.4./. Гомополисахариды
К гомополисахаридам, встречающимся в природных объектах,
относятся крахмал, гликоген и клетчатка.
Эти соединения отличают большие молекулярные веса порядка
105-107 - для крахмала, 106-107 - для гликогена, 107 - для клетчат¬
ки Общая молекулярная формула всех вышеназванных полисахари¬
дов (СвНю05)п-
533
Высокомолекулярные полисахариды по своим физическим
свойствам отличаются от низкомолекулярных полисахаридов и от
моносахаридов Они некристалличны. Так, крахмал и гликоген
представляют собой аморфные порошки Только гликоген хорошо
растворим в воде, крахмал растворяется в ней лишь при нагрева¬
нии, а клетчатка является не растворимым в воде полисахаридом.
Крахмал и гликоген образуют коллоидные растворы.
Несахароподобные полисахариды являются оптически активны¬
ми веществами Растворы крахмала и гликогена имеют сильное
правое вращение [а]о +195° Целлюлоза же обладает небольшой
оптической активностью, её удельное вращение [а)о -3°.
Крахмал представляет собой смесь двух различных полисаха¬
ридов - амилозы (до 20%) и амилопектина. Амилоза хорошо рас¬
творима в теплой воде и не дает клейстера. Амилопектин в горячей
воде растворим с трудом, и образует при этом вязкий клейстер.
Причина различий кроется в неодинаковом строении этих поли¬
сахаридов.
амилоза
Амилоза представляет собой регулярный а-1,4-глюкозидный по¬
лимер, то есть построена из молекул глюкопиранозы в а-форме,
связанных между собой с участием гидроксильной группы в поло¬
жении 4.
НОНгС
амилопектин
534
Амилопектин в отличие от амилозы имеет разветвленное
строение. Основная цепь построена, как и в амилозе, посредством
а-1,4-глюкозидной связи, однако она имеет ответвления за счет
образования а-1,6-глюкозидных связей.
Условно различие в строении амилозы и амилопектина может
быть изображено следующим образом:
ОссОеЛЬсооссоосьооооооо
охРса^>ссосаоорэо0оооо
амилоза амилопектин
Рис. 25.1. Амилоза и амилопектин.
Еще одно различие заключается в неодинаковой молекулярной
массе этих полисахаридов. Амилоза легче, ее цепь состоит пример¬
но из двух тысяч звеньев. Амилопектин содержит в своем составе
до сотен тысяч остатков глюкозы. Таким образом, крахмал - ассо-
циат амилозы и амилопектина, связанных между собой водород¬
ными связями.
При нагревании крахмала происходит расщепление его молекул
на полисахариды с меньшим молекулярным весом, которые назы¬
ваются декстринами Декстрины имеют ту же общую формулу
(СвНю05)х, что и крахмал, только х < п Под действием ферментов
(амилазы) крахмал подвергается гидролизу до дисахарида мальтозы;
при более глубоком гидролизе в присутствии минеральных кислот
расщепление идет до моносахарида - глюкозы:
Н20 Н20 Н20
крахмал » декстрины ► мальтоза ► глюкоза
Крахмал содержится в хлебе, картофеле, овощах и имеет важ¬
ное значение как источник питания Он используется также в ка¬
честве исходного материала при получении глюкозы. В фармацевти¬
ческой промышленности крахмал употребляют для приготовления
паст, таблеток и т.д.
Гликоген представляет собой аналог амилопектина Полимер¬
ная цепь гликогена также состоит из молекул глюкозы в их а-
пиранозной форме, связанных между собой а-1,4- и а-1,6-глико-
зидными связями.
Гликоген (или животный крахмал) является резервным углево¬
дом животных организмов Он содержится в печени (до 20%),
535
мышцах (-4%). В процессе жизнедеятельности, в первую очередь
при мышечной работе, происходит расщепление гликогена, который
отдает сосредоточенную в нем энергию. Гликоген еще более развет¬
влен, чем амилопектин, что имеет важное физиологическое значе¬
ние. Отщепление от гликогена молекул глюкозы, необходимых
для энергетического использования, осуществляется, в первую оче¬
редь, на концах цепей. Чем таких концов больше в молекуле, тем
быстрее компенсируются затраты глюкозы в процессе биологиче¬
ского окисления
Еще одним примером несахароподобного гомополисахарида, со¬
стоящего из молекул О-глюкозы, связанных а-1,6-гликозидными
связями, является декстран. Однако кроме 1,6-связей основной
цепи в молекуле декстрана имеются и ветвления посредством
а-1,4-, а-1,3- и а-1,2-связей.
Декстран синтезируют микробиологическим путем из сахарозы.
Декстран применяют при больших потерях крови, лечении ожо¬
гов и нефротических отеков. Частично гидролизованный декстран,
называемый “клиническим” декстраном, используется как замени¬
тель крови, поскольку по свойствам соответствует плазме. Сульфо-
эфиры декстрана подавляют свертываемость кровн и являются за¬
менителями гепарина (см. ниже).
Клетчатка (или целлюлоза) представляет собой линейный
полимер целлобиозы, то есть состоит нз глюкозных остатков
в их р-пиранозной форме. Таким образом, в целлюлозе имеются
р-1,4-гликозидные связи.
Клетчатка является основным веществом растительных клеток.
Она составляет 50-70% древесины. Хлопок, волокна льна и конопли
почти целиком состоят из клетчатки.
В результате частичного гидролиза клетчатки меняются
свойства целлюлозного материала: снижается прочность волокон,
увеличивается растворимость в щелочах. Полный кислотный (или
ферментативный) гидролиз клетчатки приводит к образованию
Ь-глюкозы.
536
целлюлоза
Человек, а также плотоядные животные не усваивают клетчат¬
ку, так ка!&в их организме нет ферментов, которые способствуют
её гидролизу. Переваривание клетчатки травоядными объясняется
присутствием в их пищеварительной системе микроорганизмов, ко¬
торые могут её гидролизовать.
Мономерный фрагмент молекулы клетчатки имеет три свобод¬
ные гидроксильные группы, по которым может идти её алкилирова-
нне и ацилирование. Образующиеся при этом сложные эфиры
имеют практическое значение. Так, при обработке остатков клет¬
чатки смесью концентрированных азотной и серной кислот образу¬
ется нитроклетчатка - нитроэфир клетчатки'.
В зависимости от условий нитрования можно получить моно-,
ди- и тринитроэфиры клетчатки. Смесь моно- и динитроклетчатки
называют коллоидной ватой или коллоксилином. Он используется в
медицине под названием коллодия. Из смесн коллоксилина, камфо¬
ры и спирта приготовляют целлулоид. Тринитроклетчатка пред¬
ставляет собой взрывчатое вещество, применяемое под названием
пироксилин.
Уксуснокислые эфиры илн ацетаты клетчатки получают при
действии на неё уксусного ангидрида в присутствии уксусной и
серной кислот:
‘Нитроклетчатка - общепринятое, но неверное название нитроэфира
клетчатки
нитроклетчатка
п
триацетилцеллюлоэа
537
Ди- и триацетаты клетчатки (ди- и триацетилцеллюлоза) ис¬
пользуется для приготовления искусственного шелка, лаков* и т.д.
Хитин построен из остатков О-глюкозы, связанных, как и в
целлюлозе, р-1,4-гликозидными связями Однако в его молекуле
гидроксильная группа в положении 2 отсутствует. Она заменена
ацетиламиногруппой Хитин нерастворим в воде, щелочи, разбав¬
ленных кислотах, органических растворителях
Хитин - основной компонент наружного скелета ракообразных,
насекомых, оболочек некоторых грибов Сульфоэфиры хитина обла¬
дают антикоагулирующим действием и могут поэтому применяться
в медицине.
Гиалуроновая кислота - полимер ацетилированного по амино¬
группе дисахарида р,0-глюкоуранозидо-3-(2-амино)-р,Р-глюкозы:
Гиалуроновая кислота входит в состав межклеточного вещества
тканей человека. Ей отводится множество жизненных функций. Она
выполняет роль межклеточной смазки, поэтому ее много в сухожи¬
лиях, коже, хрящах. Ее высокие вязкость и липкость определяют ее
роль в качестве барьера, препятствующего проникновению микроор¬
ганизмов внутрь соединительной ткани. В то же время в при¬
сутствии гиалуроновой кислоты возрастает упругость тканей, то
есть она служит неким цементирующим материалом. Ее содержание
высоко в стекловидном теле. Участвует гиалуроновая кислота и в
процессе распределения веществ в тканях. Известно, что ее ком¬
плекс с белком, именуемый протеогликином, способен связывать
ионы натрия, калия и кальция, то есть является ионнообменником,
и тем самым регулирует процесс обмена этих ионов.
Гепарин - гетерополнсахарид сложного строения. В его состав
входят Р-глюкозамин и Р-глюкуроновая кислота в а-пиранозной
форме. Соединены они чередующимися а-1,4- и а-1,3-гликозидными
25.4.2. Гетерополисахариды
ОН
р,0-глюкоураноэидо-3-(2-ацетиламино)- (З.О-глюкоэа
538
связями. К тому же аминогруппы полимера сульфатированы, то
есть представляют собой сульфамиды. Сульфатированы и гидро¬
ксильные группы в положении 4 (сульфоэфиры). В положениях 2
и 6 гидроксильные группы сульфатированы лишь частично. Сказан¬
ное представлено в формуле:
гепарин
Гепарин можно обнаружить в малых количествах в самых раз¬
личных тканях - в печени, крови, легких и селезенке. Вырабаты¬
вается гепарин особыми клетками, имеющими название тучных.
Главная функция гепарина - предотвращение свертывания крови.
В качестве антикоагулянта гепарин широко применяется в самых
различных областях медицины.
Другой представитель гетерополисахаридов - агар-агар, кото¬
рый получают из морских водорослей, находит применение в конди¬
терской промышленности и микробиологии из-за характерной осо¬
бенности давать плотные гели.
Агар-агар состоит из смеси полисахаридов сложного строения.
Основными (но не единственными) мономерными звеньями этих
полисахаридов являются глюкуроновая кислота и галактоза.
К настоящему времени из различных природных источников,
по преимуществу из микроорганизмов, выделены и исследованы
десятки полисахаридов самой разнообразной природы, причем не¬
которые из них обладают сильной биологической активностью
(противомикробное, противоопухолевое и др. действие). Ряд из них
уже производится промышленностью
25.5. Заключение
Приведенные сведения наглядно демонстрируют роль углеводов
в живой природе Суммируя данные, можно обратить внимание на
следующие моменты:
- углеводы и их производные выступают в роли источника энер¬
гии, выделяющейся в результате углеводного обмена К числу таких
углеводов относятся гликоген и глюкоза
- многие углеводы выполняют функцию резервных питательных
веществ, в определенных условиях превращающихся в Р-глюкозу
539
или другие питательные вещества. Резервными углеводами, в част¬
ности, являются сахароза, гликоген и крахмал.
- полисахариды и их производные являются конструктивными
материалами некоторых организмов. Строительным каркасом расте¬
ний служит клетчатка - главное сырье углеводных строительных
материалов в человеческой практике (древесина), основа целлюло¬
зообрабатывающей промышленности. Клетчатка выступает как цен¬
ное сырье и в химической промышленности (после гидролиза, аци-
лирования и т.д.). Полиаминосахарид хитин составляет остов орга¬
низмов насекомых и др.
- ряд сложных производных углеводов относится к числу биоло¬
гически активных веществ, применяемых в качестве лекарственных
препаратов (сердечные гликозиды и др.), витаминов (например,
аскорбиновая кислота), антибиотиков (стрептомицин и др.); поли¬
сахаридам, входящим в состав крови, присуща иммунологическая
активность.
- углеводы являются фрагментом нуклеиновых кислот (ДНК и
РНК), а также незаменимыми компонентами сложных белков
(гликопротеидов) и некоторых жировых веществ (гликолипидов).
Глава 26. КАРБОНОВЫЕ КИСЛОТЫ
*4 И ИХ ПРОИЗВОДНЫЕ
26.1. Строение и номенклатура
Органические соединения, которые содержат карбоксильную
группу -СООН, относят к числу карбоновых кислот. Наиболее
важные из числа моно- и дикарбоновых кислот предельного, непре¬
дельного, ароматического и гетероциклического рядов представле¬
ны ннже.
Здесь не приводятся те карбоновые кислоты, которые входят в
состав липидов и их производных. Их строение рассмотрено в сле¬
дующей главе.
рКа 3.77
рКа4.76
рКа 4.88
Н
СНз
СН3СН2 ^
СН3СН2СН2
О
II
О
О
II
О
О
II
О
О
II
О
но
но
но
но'
муравьиная
уксусная
пропионовая
масляная
кислота
кислота
кислота
кислота
СН3
сн3 —6н %
с=о
но"
о
N
р-пиридинкарбоновая
(никотиновая)
кислота
Для карбоновых кислот более приняты тривиальные названия,
возникновение которых обычно связано с природным источником,
из которых они выделены или синтезированы. Так, муравьиная кис¬
лота содержится в выделениях муравьев, масляная - в коровьем
масле, щавелевая - в ряде растений, никотиновая кислота получена
впервые окислением никотина, янтарная - янтаря, о-фталевая -
нафталина и т д.
Международные названия карбоновых кислот, которые строятся
из названия соответствующего углеводорода с добавлением оконча¬
ния -овая кислота, употребляются довольно редко и лишь в тех
случаях, когда тривиального названия кислота не имеет
Кислотные остатки, именуемые в общем ацильными радика¬
лами (Ас-), также характеризуются своими индивидуальными на¬
акриловая кротоновая бензойная
кислота кислота кислота
541
званиями. С их помощью удобно называть производные карбоновых
кислот - галогенангидриды, амиды, эфиры.
Н СН3 СН3СН2 СН3СН2СН2 Л.
;с=о /с=о /=о ;с=о 0
формил ацетил пропионил бутирил бензоил
Янтарная кислота - представитель предельных дикарбоновых
кислот, а изомерные фталевые кислоты принадлежат к аромати¬
ческому ряду.
Фумаровая и малеиновая кислоты представляют собой харак¬
терный пример цис- и транс-изомеров этиленового ряда, которые
сильно различаются между собой (см. ниже).
н соон н соон
СООН СООН .-СООН V V
боон СООН С00Н НООС Н Н СООН
щавелевая малоновая янтарная фумаровая малеиновая
СООН
г1^г'СООИ НООС. СООН
©С» 'От О
аэон
о-, м- и п-фталевыв кислоты
Кислоты имеют большое значение в практике. Муравьиная,
уксусная и бензойная кислоты в больших количествах вырабаты¬
ваются промышленностью, т.к. являются важными полупродукта¬
ми для синтеза разнообразных материалов - в том числе и лекар¬
ственных веществ. Соли длинноцепочечных карбоновых кислот с
щелочными металлами представляют собой мыла. Акриловую и
л-фталевую кислоты используют как мономеры в синтезе полимер¬
ных материалов, среди них и медицинского назначения.
Многие необходимые вещества, в том числе лекарства, получа¬
ют из малоновой, янтарной и о-фталевой кнслот.
Медико-биологическое значение карбоновых кислот состоит в
том, что многие из них являются метаболитами и присутствуют в
542
различных растительных и животных биосредах. Уксусная кислота
и ее проив^рдные, ацетаты, представляют собой структурную син¬
тетическую Единицу, из которой построены многие сложные биомо¬
лекулы, например, стероидные гормоны, липиды и др. Широкий на¬
бор предельных и ненасыщенных карбоновых кислот, именуемых
жирными кислотами, участвует в построении молекул лнпидов.
Карбоксильная группа входит в состав таких важных групп бно-
молекул, как окси-, кето- и аминокислоты. Никотиновая кислота
является провитамином витамина РР (никотинамида).
26.2. Получение
1. Карбоновые кислоты синтезируют окислением углеводоро¬
дов, первичных спиртов, альдегидов:
[О] [01
к—СНз —► я—соон •*—- я—СН2ОН
|[0]
я—сн=о
Окисление н-алканов осуществляется в промышленности дей¬
ствием на углеводороды нефти кислородом в присутствии катализа¬
тора - солей Мп под давлением и при высокой температуре. Осо¬
бенно характерен такой способ для синтеза ароматических и гете¬
роциклических кислот. Бензойную кислоту, как известно, легко по¬
лучать действием окислителей на монозамещенные гомологи бензо¬
ла, о-фталевую кислоту синтезируют из нафталина, а никотино¬
вую кислоту - из р-метилпиридина. Последняя образуется и при
окислении алкалоидов никотйна и анабазина (глава 18), т.к. у них
в р-положении пиридинового кольца находится насыщенное
(а, следовательно, разрушаемое окислением) кольцо.
Окисление спиртовой группировки используется в микробиоло¬
гическом способе синтеза уксуса, при котором происходит уксусно¬
кислое брожение спирта до уксусной кислоты
В целом, в живых организмах самые различные органические
вещества (углеводороды, спирты, альдегиды) обычно претерпевают
ферментативное окисление до соответствующих карбоновых кислот.
Иногда это имеет нежелательные последствия Так, токсичность
этиленгликоля объясняется его окислением при попадании в орга¬
низм до сильно ядовитой щавелевой кислоты
Метаболизм органических биомолекул до карбоновых кислот -
общая биохимическая реакция.
543
Один из биохимических путей образования карбоновых кислот:
явление перекисного окисления липидов (ПОЛ), при котором ак¬
тивные формы кислорода атакуют а-углеродные атомы кратных свя¬
зей непредельных липидов
таутомерия
Р1СНгСН=СНГС гаСН^СНСДО
Р1снсн=снге етсн^хснге
(^О-н гидроперекиси н-о5!>
РЮН=0 + |сН=СНРг| | Р1СН=СН | + 0=СНР2
он енолы но
Р1СН=0 + 0=НСХН2Р2 Р1СНгСН=0 + 0=НСГО
О.) 1р,
Р1С00Н + НООССН2Р2 РЮНгСООН + НООСР2
Процесс ПОЛ особенно активируется при лучевом поражении,
что и является основной причиной повреждения биомембран.
Уровень содержания некоторых карбоновых кислот в биосредах
является поэтому полезным биохимическим показателем, а обнару¬
жение в них низших карбоновых кислот служит дигностическим
признаком анаэробной инфекции, поскольку такие кислоты, как
пропионовая, масляная, изовалериановая, продуцируются анаэроб¬
ными бактериями.
2. Гидроксиформилирование алкенов также является про¬
мышленным способом, который состоит в действии на алкен смесью
СО + Н20 в присутствии металлических катализаторов:
^ Р
СНг=СН2 + СО + НзО кат » СНзСНгСООН
пропионовая
кислота
3. Гидролиз нитрилов позволяет синтезировать карбоновую
кислоту из галогенопроизводного углеводорода, при этом число
544
атомов углерода, как и в предыдущем методе, увеличивается на
единицу, Н^Ьример:
4. К карбоксильной группе приводит и гидролиз тригалоген-
производных, содержащих все три галогена при одном атоме
углерода:
5. Карбоновые кислоты образуются при гидролизе различных
производных карбоновых кислот (галогенангидриды, ангидриды,
сложные эфиры, амиды), в том числе и природного происхождения,
таких, как липиды. Об этом будет сказано подробно дальше.
Кроме перечисленных методов существует много и других спо¬
собов образования карбоксильной группы, которые, однако, носят
частный характер и потому здесь не рассматриваются.
Карбоновые кислоты относятся к числу высококипящих и высо¬
коплавких веществ по сравнению с другими органическими соеди¬
нениями с тем же числом атомов углерода. Это объясняется обра¬
зованием димерных ассоциатов, т е. объединением двух молекул по¬
средством водородных связей за счет взаимодействия гидроксиль¬
ных и карбонильных групп различных молекул Разрушение диме¬
ров происходит только при сильном нагревании. Поэтому даже в
газовой фазе большая часть молекул кислоты димеризована:
- СНг СНоон
26.3. Свойства карбоновых кислот
О—Н о
ассоциативный димер
18 Эак. 675
545
Низшие предельные кислоты хорошо растворимы в воде, но уже
начиная с С9-кислот, они почти не смешиваются с водой.
Химические свойства карбоновых кислот определяются особен¬
ностями строения карбоксильной группы, которая представляет со¬
бой не формальную комбинацию спиртовой и карбонильной групп, а
новую функциональную группировку Поэтому кислоты принципи¬
ально отличны по свойствам как от спиртов, так и от альдегидов
или кетонов С одной стороны, электроотрицательный атом кисло¬
рода ОН понижает полярность связи С=0, а поэтому для карбоно¬
вых кислот не характерно нуклеофильное присоединение по этой
связи, свойственное оксосоединениям. С другой стороны, поло¬
жительно заряженный карбонильный атом углерода притягивает
к себе электронные пары кислорода и тем самым ослабляют связь
О-Н, усиливая положительный заряд на атоме водорода Поэтому
карбоновые кислоты обладают более сильными кислыми свойства¬
ми, чем спирты.
26.3.1. Кислотные свойства
Карбоновые кислоты диссоциируют в водных растворах на кар-
боксилат-ион и протон:
карбоксилат-
ион
В карбоксилат-анионе, как было доказано, атомы кислорода рав¬
ноценны, т.е. происходит выравнивание длины двух связей С-0 и
равномерное распределение отрицательного заряда между атомами
кислорода. Такое выравнивание зарядов легко объяснимо с позиций
резонанса. Оно энергетически выгодно и потому является дополни¬
тельной причиной повышенной кислотности карбоксильной группы
по сравнению со спиртовой.
Сила карбоновых кислот определяется характером заместителя
при карбоксильной группе. Так, муравьиная кислота (см. выше)
несколько сильнее уксусной, т.к. алкильный заместитель является
донором электронов. Акцепторный характер непредельного и аро¬
матического заместителя по сравнению с СН3 группой вызывает не¬
которое уменьшение рКа при переходе от уксусной кислоты к
акриловой и бензойной. Влияние электроноакцепторного замести¬
теля на кислотность карбоксильной группы удобно проследить на
546
примере дикарбоновых кислот. По мере удаления электроноакцеп¬
торных карбоксильных групп друг от друга сила кислоты заметно
падает в ряду щавелевая - малоновая - янтарная кислоты. В об¬
ратном направлении меняется кислотность хлоруксусных кислот:
монохлоруксусная кислота (С1СН2-СООН) - рКа 2.86, дихлорук-
сусная (С12СН-СООН) - рКа 1.29, трихлоруксусная (С13С-СООН) -
рКа 0.65.
Соли карбоновых кислот, которые образуются при их нейтра¬
лизации, находят применение в практике. Так, натриевые и калие¬
вые соли непредельных и предельных кислот с длинным углеводо¬
родным радикалом представляют собой мыла. Основная свинцовая
соль уксусной кислоты НО-РЬ-ООС-СНз известна как свинцовая
примочка, применяемая в медицине. Нетоксичные соли бензойной
кислоты, бензоаты, обладают консервирующим действием и потому
используются как добавки ко многим пищевым продуктам. Некото¬
рые соли карбоновых кислот употребляются в органическом син¬
тезе, например, их пиролизом получают альдегиды и кетоны
(см. предыдущую главу).
Следует помнить, что соли карбоновых кислот подвержены за¬
метному гидролизу, а в смеси с самими кислотами они образуют
буферные растворы.
26.3.2. Окислительно-восстановительные превращения
Кислоты не имеют выраженной склонности к действию восста¬
новителей и окислителей Только сильные восстановители действу¬
ют на них. Так, с иА1Н4 они дают спирты:
4Р-СООН + ЫА1Н4-> 4Р-ОН + ИОН + А1(ОН)3,
а с Н1 - алканы:
Р-СООН + 6Н1 -» Р-СНз + 312 + 2Н20.
С окислителями реакции идут еще труднее Исключение состав¬
ляет муравьиная кислота, которая сохраняет в своем составе альде¬
гидную группу и потому дает реакцию “серебряного зеркала”
НСООН + Ад20 -» 2Ад + С02 ♦ Н20.
Легко окисляется и щавелевая кислота (или её соли), что и ис¬
пользуется в методе перманганатометрии
5Н2С204 + 2КМп04 + ЗН2804 -> ЮС02 + 2Мп804 + К2304 + 8Н20.
18*
547
В прочих предельных кислотах обычно окисляется а-угле-
родный атом с образованием а-окси- или а-кетокислот (с* ниже)
Ферментативным путем окислению подвергается и р-углеродиьж
атом до Р-оксикислот.
С перекисью водорода из кислот образуются пероксокислоты -
вещества, содержащие перекисную группировку
/Р //>
н-с' + н202 — + кио
"ОН ООН ^
пероксокислота
Пероксокислоты являются сильными окислителями.
26.3.3. Декарбоксилирование
При сильном нагревании карбоновых кислот наблюдается их де¬
карбоксилирование:
Я-СООН -> Я-Н + С02Т.
Способность к этому превращению определяется прочностью
связи карбоксильной группы с радикалом, которая понижается, ес¬
ли в нем содержатся электроноакцепторные заместители. Поэтому,
например, щавелевая кислота декарбоксилируется уже при слабом
нагревании:
НООС-СООН -» Н-СООН + С02Т,
малоновую кислоту для этого нужно нагреть значительно сильнее:
НООС-СН2-СООН -» СНз-СООН + С02Т,
а янтарную кислоту (как и уксусную или другие предельные кисло¬
ты) практически уже не удается декарбоксилировать нагреванием.
Однако декарбоксилирование любых кислот легко осуществляется
ферментативным путем и является одной из необходимых стадий
превращения органических веществ в организме.
26.3.4. Влияние карбоксильной группы на заместитель
Электроноакцепторный эффект карбоксильной группы заклю¬
чается в повышенной склонности к замещению атомов Н в
а-положении молекул предельных кислот. В результате можно вво¬
дить атомы галогена в это положение:
548
л
СНз-СООН + СІ2 -» СН2С1-СООН + неї
СН2С1-СООН + СІ2 -» СНСІ2-СООН + неї
СНСІ2-СООН + СІ2 -» ССІЗ-СООН + неї.
Еще легче осуществляется замещение с образованием из уксус¬
ной кислоты монофтор-, дифтор- или трифторуксусной кислот. Сле¬
дует отметить, что ядовитая монофторуксусная кислота содержится
в соке некоторых тропических растений. Её соли - монофторацета-
ты применяются как средства борьбы с грызунами.
Особенно сильно проявляется влияние на а-положение в моле¬
куле малоновой кислоты и её эфиров. Суммарный эффект двух кар¬
боксильных групп приводит к тому, что а-атом водорода может про¬
являть “кислый" характер и замещаться активными металлами. Это
используется в синтетических целях для получения из малоновой
кислоты и ее эфиров а-замещенных гомологов:
В непредельных кислотах эффект группы -СООН заключается
в том, что электрофильное присоединение идет вопреки правилу
Марковникова:
В ароматических кислотах карбоксильная группа выступает как
заместитель второго рода, ориентируя дальнейшее замещение в
мета-положение и затрудняя реакцию:
НОэЭ'
м-сульфобенэойная
кислота
549
26.4. Производные карбоновых кислот
Ещё одной общей особенностью карбоновых кислот является их
способность к нуклеофильному замещению ОН-группы, в результате
чего получаются производные карбоновых кислот: галогенангидри-
ды, ангидриды, сложные эфиры и амиды. Прежде чем обратиться к
отдельным представителям производных, необходимо обратить вни¬
мание на то, что они, с одной стороны, могут гидролизоваться в
карбоновые кислоты, а, с другой стороны, более активные из них
(хлорангидриды и ангидриды) способны к реакциям нуклеофильного
замещения с образованием менее реакционноспособных (сложных
эфиров и амидов).
Таким образом, перечисленные соединения связаны взаимными
переходами:
сложный
эфир
26.4.1. Галогенангидриды
Эти вещества (ІЗСО-Х) получают действием на кислоты галоге-
нангидридами минеральных кислот (ЭОСЬ, РСІ3, РОСІз, РСІ5, РВг3
и др.). Особенно удобно применять ЭОСЬ, т.к. кроме желаемого га-
логенангидрида остальные продукты представляют собой газы.
Известны фтор-*, хлор-, бром- и иодангидриды. Их названия
строят по типу ал кил галоген идов, исходя из соответствующего
‘Первый фторангидрид (бензойной кислоты) получил А П. Бородин в
1862 г.
550
ацильного радикала. Например, СН3-СО-С1 это хлористый ацетил,
а CeHs-CO-%- бромистый бензоил.
Наиболее важны ацилхлориды, которые чрезвычайно реакцион¬
носпособны и используются для ацилирования спиртов и аминов.
Причина их активности по отношению к нуклеофильным агентам -
высокий положительный заряд на атоме углерода, который поэтому
легко вступает в реакции присоединения с последующим отщепле¬
нием HCI. По той же причине все галогенангидриды легко гидро¬
лизуются.
26.4.2. Ангидриды карбоновых кислот
Ацилированием галогенангидридами солей карбоновых кислот
можно получить их ангидриды.
По химическим свойствам ангидриды похожи на галогенангид¬
риды: они легко гидролизуются и являются ацилирующими агента¬
ми по отношению к спиртам и аминам. Наибольший практический
интерес представляет уксусный ангидрид, который производится
для синтеза ацетатного волокна.
0 0 О
СНЛ л л
уксусный янтарный малеиновый
ангидрид ангидрид ангидрид
Особую группу составляют ангидриды дикарбоновых кислот,
которые получаются за счет внутримолекулярной потери молекулы
воды с образованием пяти- (или шестичленных) гетероциклических
колец - янтарный, малеиновый и фталевый ангидриды Они важны
для синтеза пластмасс, красителей, лекарственных средств.
26.4.3. Сложные эфиры
Обычным методом синтеза сложных эфиров являются ацили-
рование спиртов действием кислот, их галогенангидридов или ан¬
гидридов
Другой способ их получения - сложноэфирная конденсация
Сложные виниловые эфиры, как отмечено раньше, синтезируют
присоединением карбоновых кислот к ацетилену
551
Название сложных эфиров строится из названия радикала спир¬
та в сочетании с корнем латинского наименования кислоты, к кото¬
рому добавляется окончание -ат (или -оат).
Сложные эфиры широко распространены в живой природе.
Многие из них входят в состав плодов, цветов и определяют их
аромат. Сложные эфиры являются обязательными компонентами
эфирных масел - сложных смесей растительного происхождения,
в состав которых входят также терпены и кислородные гетероцик¬
лические соединения и др
Сложными эфирами глицерина, как известно, являются жиры
(липиды). Воскоподобное вещество кашалотов - спермацет - со¬
стоит, в основном, из цетилового эфира пальмитиновой кислоты
(СізНзіСООН). Пчелиный воск также представляет собой многоком¬
понентную смесь, в которой преобладает сложный эфир - мирици-
ловый эфир пальмитиновой кислоты.
Простейшие эфиры (например, этилацетат) употребляются как
растворители, экстрагенты, пластификаторы полимерных материа¬
лов и т.д. Ряд эфиров используется для приготовления фруктовых
эссенций в парфюмерии и пищевой промышленности.
Сі5НзіСООСі6Нзз
цетиловый эфир
пальмитиновой кислоты
спермацет
СізНзіСООСзіНбз
мирициловый эфир
пальмитиновой кислоты
воск
О
О
О
этилформиат
пентилацетат
этилбутират
О
этилбенэоат
диметилфталат
ІЧН?
СН3
СНз'^СНз
валидол
новокаин
552
Так, этилфомиат обладает запахом рома, изопентилацетат
входит в с&тав грушевой эссенции, бензилацетат пахнет жасми¬
ном, а этилбутират - абрикосами.
Соединения со сложноэфирной группировкой имеют широкий
спектр физиологического действия. Диметилфталат, например,
является репеллентом, отпугивающим кровососущих насекомых.
Сосудорасширяющий препарат валидол представляет собой менти-
ловый эфир изовалериановой кислоты. Сложноэфирные группиров¬
ки присутствуют в молекулах лекарственных средств аспирина,
салола (глава 30), новокаина и др.
26.4.4. Амиды
Названия амидов карбоновых кислот строятся из названий ис¬
ходных кислот. Так, НСО-ІЧНг это амид муравьиной кислоты или
формамид, а СвН5-СО-М(СН3)2 - это диметиламид бензойной кислоты
или диметилбензамид и т.д.
Амиды получают ацилированием аминов карбоновыми кислота¬
ми или лучше - хлорангидридами или ангидридами. Ещё один метод
их синтеза - гидролиз нитрилов:
Ацилированием аммиака действием циклических ангидридов
(янтарного, фталевого, малеинового и т.д ) получают циклические
диамиды, которые называются имидами. Например, из фталевого
ангидрида получают фталимид:
Имиды можно получить и с участием остатков различных кис¬
лот. В качестве примера можно привести сахарин - имид
о-сульфобензойной кислоты, который из-за сладкого вкуса и малой
токсичности употребляют как суррогат углеводов
553
Химические свойства амидов и имидов определяются тем, что
неподеленная электронная пара азота в них участвует в сопряже¬
нии с двойной связью С=0, а потому, с одной стороны, у амидов
подавлены свойства оснований, а, с другой стороны, атом водорода
1ЧНг-группы становится подвижным. Это означает, что у амидов
имеется тенденция к проявлению кислотных свойств В целом же
амиды следует рассматривать как соединения, у которых и основ¬
ные, и кислотные свойства выражены крайне слабо.
Подвижность атома водорода группы 1ЧН2 может проявляться
также и в явлении амид-имидольной таутомерии (аналогичной
кето-енольной).
По аналогии с альдегидами и кетонами в амидах “имидольный"
таутомер присутствует в незначительных количествах, зато доля
такого таутомера у циклических имидов (например, у фталимида),
как и доля енола у Р-дикарбонильных соединений, может заметно
возрастать.
фталимид
Увеличена в имидах и подвижность атома водорода группы
-ССМНСО-, который можно заместить на металл действием щелочи.
Это используется в органическом синтезе для получения первичных
аминов (метод Габриэля). Синтез Габриэля состоит в алкилирова-
нии фтал им ид-калия галогеналканом, а затем в последующей обра¬
ботке водой продукта, который гидролизуется, как и остальные
имиды (и амиды в целом), с образованием алкиламина.
фталимид фталимид-калий
Явление амид-имидольной таутомерии характерно для оксипро-
изводных азотистых гетероциклов и составляет важную отличи¬
тельную черту тех из них, что входят в состав нуклеиновых кислот
(см. следующую главу).
554
Амиды имеют значение в практической деятельности. Простей¬
шие амид1ЯЧ формамид, диметилформамид и др. используются как
промышленные растворители; диэтиламид мета-толуиловой кис¬
лоты (диэтилтолуамид или сокращенно ДЭТА) служит репел¬
лентом, более эффективным, чем диметилфталат; никотинамид
является витамином РР. Амидная группировка входит в состав мно¬
гих лекарственных средств (см., например, фенацетин, глава 22).
Поливинилпирролидон, полимер амида, в котором амидный фраг¬
мент включен в пятичленное кольцо, используется как плазмозаме¬
няющий препарат.
ДЭТА никотинамид М-винилпирролидон поливинилпирролидон
Амидная связь характерна для группы жировых веществ, вхо¬
дящих в состав нервных клеток, в том числе и коры головного моз¬
га. Эти соединения являются амидами сфингозина (глава 27). На¬
конец, фрагмент -СО-1ЧН- присутствует во всех пептидах, где он на¬
зывается пептидной связью, особенности строения которой будут
разобраны позднее.
Глава 27. ЛИПИДЫ
27.1. Строение и классификация
Липидами называют большую группу природных соединений, в
которую входят разнообразные по химическому строению вещества.
Общим свойством, позволившим объединить их в единую группу,
явилась растворимость в органических растворителях и нераство¬
римость в воде В соответствии с этим к липидам были отнесены
жирные кислоты, простые и сложные эфиры глицерина, ами¬
ды - производные аминоспирта сфингозина, холестерин и его эфи¬
ры, воск£ и даже каротин. По мере выяснения деталей структуры и
изучения функций этих соединений в клетке стало очевидным, что
такое определение липидов не отражает ни общности химического
строения, ни единства биологических функций и не может быть
признано удачным.
Более целесообразно к липидам относить природные биоло¬
гически активные производные высших жирных кислот и спиртов.
Обычно это простые и сложные эфиры, образующиеся в результате
взаимодействия глицерина и стероидных спиртов с высшими жир¬
ными кислотами. В молекуле липида непременно присутствует один
или несколько гидрофобных заместителей, обеспечивающих хоро¬
шую растворимость в неполярных растворителях. Многие липиды
содержат наряду с гидрофобными также и гидрофильные замести¬
тели, имеющие сродство к полярным растворителям. Наличие в ли¬
пидах такого типа полярных и гидрофобных заместителей определя¬
ет их участие в образовании структуры биологических мембран и
функциональную роль, связанную с переносом веществ и ионов че¬
рез мембраны, энергообеспечением клетки и защитными реакциями
организма. В последние годы широко обсуждается роль липидов как
биорегуляторов.
Разнообразие химического строения чрезвычайно осложняет
классификацию липидов, вследствие чего в настоящее время единая
система классификации отсутствует. В соответствии с химическими
свойствами липиды делят на две группы:
1) нейтральные липиды,
2) полярные липиды.
Каждая из групп включает несколько классов липидов. Обычно
классификация базируется на наиболее существенных особенностях
строения, но в сложных соединениях выбор определяющего призна¬
ка не всегда однозначен.
556
К группе нейтральных липидов относятся глицеролипиды,
сфинголиИмды, эфиры холестерина, воска. В каждый класс
входят соединения нескольких типов в зависимости от природы,
числа и положения заместителей.
Полярные липиды разделены на две группы в соответствии с
характером кислотного остатка: фосфолипиды и сульфолипиды.
Последние, содержащие сульфоэфирную группу (-О-БОг-ОН), содер¬
жатся в природных объектах в малых количествах.
Ниже приведены типичные карбоновые кислоты, именуемые
жирными кислотами, из которых построены молекулы липидов.
'х^'ЧчСООН масляная кислота С3Н7СООН
пальмитоолеиновая кислота С15Н29СООН
СООН
,СООН
Нетрудно заметить, что эти жирные кислоты имеют неразветв-
ленную цепочку с четным числом атомов углерода, что объясня¬
ется особенностями их биосинтеза, который происходит из остат¬
ков уксусной кислоты (Сг). В действительности ассортимент ки¬
олеиновая кислота С17Н33СООН
линолевая кислота С17Н31СООН
557
слот, обнаруженных в липидах, значительно шире (свыше 150),
причем в их числе обнаружены и кислоты с разветвленной цепью,
а также с кислородсодержащими функциональными группировками
(окси-, кетокислоты)
Другими типичными составляющими липидов разных групп яв¬
ляются глицерин, сфингозин и холестерин
СН2ОН
У"1 Анон
Н2°Н £н=СН(СН2)12СН3
глицерин сфингозин
[СН2)3—<
СНз
холестерин
Сложные эфиры жирных кислот и глицерина образуют группу
простых липидов или жиров Жиры составляют основную массу
природных липидов. Наиболее часто в жирах присутствуют остатки
пальмитиновой, стеариновой, олеиновой кислот, а в составе коро¬
вьего масла - и масляной кислоты.
Как правило, этерификации в жирах подвергнуты все три спир¬
товые группировки в молекуле глицерина (как первичные, обозна¬
ченные а-положениями, так и вторичные - в (3-положении). Полные
эфиры глицерина и жирных кислот являются простыми тригли*
церидами.
снгосот
гесоо IР
“снгосорз
простой триглицерид
СНгОСОт
№000—1 О
сн2о—ОН
Ан
фосфатидная кислота
Жиры - производные глицерина. Например, если глицерин был
ацилирован пальмитиновой, стеариновой и олеиновой кислотами
(Р1 - пальмитил, Р2 - стеарил, Р - олеил), то образуется простой
триглицерид - а-пальмитил-Р-стеарил-а‘-олеилглицерин. Молекулы
триглицеридов с различными а-кислотными остатками (Р1 * Рэ) хи-
ральны, т.к. Р-углеродный атом асимметричен. Поэтому боль¬
шинство молекул жиров оптически активно. Асимметричные при¬
родные триглицериды относятся к числу Ь-изомеров.
Продукты неполной этерификации глицерина жирными кисло¬
тами - моно- или диглицериды распространены намного реже и об-
558
наруживаются обычно в качестве промежуточных продуктов мета¬
болизма триглицеридов. Жиры относятся к нейтральным липидам.
При замене одного из а-остатков карбоновых кислот триглице¬
ридов фосфорной кислотой образуются фосфатидные кислоты,
из которых построены молекулы сложных липидов - фосфатидов,
принадлежащих к группе полярных липидов.
Фосфатиды характеризуются тем, что остаток фосфорной кис¬
лоты в них, в свою очередь, фосфорилирует молекулу биогенного
амина - коламина или холина*. Все природные фосфатиды имеют
а,Ь-структуру.
кефалины (фосфатидилколамины) лецитины (фосфатидилхолины)
Фосфатидилколамины (или кефалины) и фосфатидилхолины
(или лецитины) - незаменимые компоненты нервных клеток. Они
вместе с некоторыми белками дают белково-липидные комплексы -
липопротеиды, представляющие собой компоненты клеточных мем¬
бран. Фосфатидами богаты печень, сердечная мышца, эритроциты.
Фосфатиды присутствуют в таких продуктах, как яичный желток,
соевые бобы, икра Их недостаток порождает малокровие, заболева¬
ния нервной системы и т.д.
К числу сложных липидов относятся и сфинголипиды, кото¬
рые являются производными сфингозина.
Сфинголипиды представляют собой амиды сфингозина и некото¬
рых жирных КИСЛОТ, обычно лингоцериновой (С24). Из этих двух
компонентов образуется церамид.
Первичная спиртовая гидроксильная группа молекулы церамида,
в свою очередь, может быть связана с остатком моносахарида га¬
лактозы или фосфорилирована фосфорилхолином. В первом случае
образуется нейтральный липид, так называемый психозин, во вто¬
ром - полярный липид, сфингомиелин. Этерификация спиртовых
групп в галактозидной части психозина (в положении 3, например)
приводит к природным сульфолипидам, другой разновидности по¬
лярных липидов.
Еще одно из природных веществ, которое фосфорилируется фосфа-
тидными кислотами, - аминокислота серии (см главу 31) Такие производ¬
ные называют фосфатидилсеринами
559
°* — СН2СН2—14—сн3
снгО^^о' і Нз
Ін—ЫН—С—(СНг)22СНз
інон а
СН=СН(СН2)і 2СН3
СН2ОН
Ін—N4—С—(СН2)22СНз
інон і
ІН=СН(СН2)12СН3
церамид
сфингомиелин
НОН2С
Ін—ГЧН—С—(СН2)22СН3
інон і
ІН=СН(СН2)12СН3
психозин
Эти и родственные им вещества выполняют важнейшие функ¬
ции в нервных тканях. В частности, сфингомиелин входит в состав
миелиновой оболочки нервных волокон.
К нейтральным липидам относятся также и сложные эфиры
жирных кислот и холестерина. В организме эта разновидность ли¬
пидов особенно часто встречается в нервных тканях, печени, а так¬
же в плазме крови.
Холестерин содержит в своем составе насыщенную структуру
стерана (глава 18). Его молекула содержит 8 асимметрических ато¬
мов углерода и, следовательно, может иметь 256 стереоизомеров,
однако лишь один из них находится в природе.
Как известно, отложение холестерина на стенках кровеносных
сосудов приводит к атеросклерозу. Однако причиной этого следует
считать не присутствие холестерина в организме, а нарушение об¬
мена веществ.
Важной функцией холестерина является то, что из него в орга¬
низме осуществляется биосинтез большого семейства физиоло¬
гически активных веществ, именуемых стероидами. Все стероиды
объединяет наличие в их структуре стеранового цикла.
27.2. Стероиды
560
В печени из холестерина синтезируются необходимые для пи-
щеварений^келчные кислоты:
он н<
холевая кислота
ОН
таурохолевая кислота
СОМНСН2СООН
глицинхолевая кислота
Желчные кислоты - типичные ПАВ, которые выполняют ак¬
тивные функции в процессах пищеварения. Особенно они важны
для усвоения липидов.
Стероидные гормоны вырабатываются из холестерина желе¬
зами внутренней секреции и регулируют важнейшие жизненные
процессы. Половыми железами производятся гормоны, которые ока¬
зывают влияние на функции размножения, процессы роста и старе¬
ния организма. В качестве примера приведена формула мужского
полового гормона тестостерона.
НОНгС
Надпочечниками вырабатываются гормоны, регулирующие об¬
мен веществ, ионный баланс и т.д Например, кортизон, который
561
управляет углеводным обменом, используется в качестве лекарства
при ревматизме, астме, аллергии и др.
В настоящее время развивается промышленный синтез природ¬
ных стероидов и их синтетических аналогов (например, преднизо-
лона), которые находят разнообразное применение во многих об¬
ластях медицины
В этом отношении интересен пример с диэтилстильбэстролом
Это вещество, которое относительно несложно синтезировать,
представляет собой тетразамещенный этилен \транс-3,4-ди-(л-ок-
сифенил)гексен-3] Если мысленно соединить концевые атомы
этильных групп с бензольными циклами (на формуле это показано
пунктиром), молекула становится похожа на структуру с четырьмя
конденсированными кольцами, как и молекулы стероидов. Это ве¬
щество нашло применение для лечения эндокринных расстройств и
применяется как противоопухолевое средство.
Холестерин и некоторые родственные ему вещества относят к
зоостеринам, т.к. они встречаются у животных и человека. В расте¬
ниях их заменяют родственные стероиды, именуемые фитостерина-
ми. Из фитостеринов в растительных тканях осуществляется синтез
некоторых витаминов, алкалоидов, а также гликозидов (например,
сердечных гликозидов). В промышленном производстве стероидных
препаратов в качестве сырья используются некоторые виды расте¬
ний из числа пасленовых и лилейных.
В настоящее время общее число найденных в природе стероидов
составляет многие сотни соединений.
Простагландины были обнаружены в семенной жидкости барана
шведским исследователем У. фон Эйлером в тридцатых годах
XX века. Он же наблюдал, что простагландины вызывают пониже¬
ние кровяного давления и сокращение мышц. Оказалось, что про¬
стагландины - гормональные регуляторы многих биопроцессов.
диэтилстильбэстрол
27.3. Простагландины
562
Простагландини обладают широким спектром биологической актив¬
ности и встречаются в небольших количествах почти во всех тканях
организма.
В частности, они вызывают болевые ощущения. Анальгетики
(например, анальгин) уменьшают боль, так как подавляют биосин¬
тез простагландинов.
В организме простагландины образуются из арахидоновой ки¬
слоты. Молекулы простагландинов (как и арахидоновой кислоты)
построены из двадцати углеродных атомов.
арахидоновая кислота
Все простагландины имеют сходное строение. Их молекулы со¬
стоят из двух углеродных цепей, присоединенных в транс-поло¬
жении к циклопентановому кольцу. Разные функциональные груп¬
пы в составе молекулы, как и в случае стероидов, и порождают се¬
мейство простагландинов, которых в настоящее время известно
свыше 30. Ниже показаны формулы двух типичных простагланди¬
нов, для классификации которых приняты буквенные обозначения:
■СООН
ОН
простагландин Р 2
Концентрации простагландинов в тканях чрезвычайно низки,
они крайне нестойки. Поэтому эксперименты по их выделению, из¬
учению механизма действия представляют очень сложную задачу.
В настоящее время простагландины уже применяют в гинекологии,
для лечения сердечно-сосудистых заболеваний и астмы.
27.4. Свойства липидов
Основная часть липидов построена посредством неполярных
связей. Поэтому мало полярные в целом молекулы липидов гидро¬
фобии. Нерастворимые в воде клеточные оболочки и построены из
липидного материала, что обеспечивает функционирование водных
563
растворов содержимого клеток. Напротив, липиды хорошо смеши¬
ваются с неполярными или мало полярными веществами. Поэтому
жиры используются для растворения пахучих веществ в Парфюме¬
рии; в свою очередь, липиды экстрагируют с помощью таких рас¬
творителей, как углеводороды, эфир, хлороформ. Следует помнить о
высокой липидной растворимости многих отравляющих веществ,
молекулы которых построены посредством ковалентных малополяр¬
ных связей.
По принципу гидрофобности к группе липидов кроме перечис¬
ленных выше соединений относят также и другие не растворимые в
воде природные вещества - некоторые витамины (например, вита¬
мин А), убихиноны и т.д. Особенностью липидов является способ¬
ность к образованию в определенных условиях водных эмульсий,
что важно для питания организма. Примером такой эмульсии яв¬
ляется молоко.
Неполярная природа липидов служит причиной их низкой
электро- и теплопроводности. Поэтому липиды выступают в ка¬
честве защитных оболочек многих живых организмов от элек¬
трических или термических воздействий (как от охлаждения, так и
перегрева), а, кроме того, и от механических воздействий.
Невысокая плотность (0.91-0.97 г/см3) оказывается необходи¬
мой для сообщения многим организмам плавучести.
Липиды не имеют постоянной температуры плавления или за¬
стывания, т.к. представляют собой многокомпонентные смеси. Тем¬
пература застывания жиров (а их свыше 1300 разновидностей)
определяется относительным содержанием в них остатков непре¬
дельных кислот. Чем оно выше, тем при более низкой температуре
жир застывает. Высоко содержание непредельных кислот в жидких
растительных маслах и в жирах водоплавающих животных, липиды
наземных млекопитающих обогащены предельными жирными кисло¬
тами. Ниже других застывает ореховое масло (-27°), выше - баранье
сало (+55°).
Характерным химическим свойством липидов, как и всяких ор¬
ганических веществ, является окисление. Эта реакция сопровож¬
дается выделением 39 кДж энергии на 1 г жира, что более чем в
два раза превосходит тепловой эффект окисления углеводов или
белков. Причина этого - значительно более отрицательная суммар¬
ная степень окисления углерода в липидах, чем в белках и углево¬
дах. Поэтому жиры играют роль энергетических ресурсов, состав¬
ляя в норме до 20 % веса человеческого организма.
Другая важная особенность окисления липидов состоит в том,
что в его результате из 1 г жира образуется до 1.4 г НгО. Это су¬
щественный вклад в поддержание общего водного баланса организ¬
564
ма. Отдельные виды обитающих в пустынях животных эндогенной
водой полостью удовлетворяют свои потребности во влаге.
Биологическое окисление липидов - многостадийный процесс,
который начинается с их гидролиза до жирных кислот и глицерина
(в случае жиров) или других компонентов. Гидролиз жиров идет
ступенчато по схеме:
триглицерид -»диглицерид -» моноглицерид -»глицерин.
Лишь после гидролиза происходит ступенчатое окисление глицери¬
на и жирных кислот.
Как известно, первичная ступень окисления глицерина дает ди-
оксиацетон и глицериновый альдегид, альдолизация которых приво¬
дит к фруктозе. Этот процесс обратим и лежит в основе многосту¬
пенчатого взаимопревращения углеводов (гликогена) и липидов,
осуществляемого в организме.
Ферментативное окисление жирных кислот является основным
источником энергии при окислении липидов. Первая стадия про¬
цесса, который реализуется с участием кофермента А, состоит в
а,Р-дегидрировании жирной кислоты. Остальные этапы представле¬
ны в схеме 27.1:
-2Н Н,0 [01
СНг—СНг—СООН Р-СН=СН—СООН —=— СН—СНг—СООН !—!*
Ан
—► Р-С—СНг—СООН _ .. _
& &
Р-кетокислота
Схема 27.1. (3-Окисление жирных кислот.
В результате образуются Р-кетокислоты, поэтому процесс на¬
зывается Р-окислением. Далее кетокислоты вновь вовлекаются в
Р-окисление
Окислению могут подвергаться и остатки непредельных жирных
кислот по месту их кратных связей - происходит процесс перекис-
ного окисления липидов (см главу 26). Этот процесс может проис¬
ходить и на воздухе Он, а также частично идущий гидролиз явля¬
ются причинами прогоркания пищевых жиров
Промышленный гидролиз жиров или их омыление, осу¬
ществляемый в нескольких технологических вариантах, служит
способом получения глицерина и солей различных жирных кислот,
обладающих моющим действием (мыла).
СНзОСОт
РЮСО—I
снзосстэ
ЗМа0Н г £иОН + Р2СООЫа
(^НзОН РэСОО№
СНзОН РЮООЫа
Непредельные остатки липидов можно подвергнуть гидрогениза¬
ции и тем самым превратить жидкие растительные масла в твердые
жиры, что лежит в основе производства маргаринов.
Итак, к липидам причисляют многие классы веществ, объеди¬
няемых одним общим физическим свойством - гидрофобностью.
Среди них находятся вещества самого разного строения и выпол¬
няющие самые разнобразные функции - от оболочек клеток до гор¬
монов, биорегуляторов и витаминов.
Глава 28. ПРОИЗВОДНЫЕ
*4 УГОЛЬНОЙ КИСЛОТЫ
Для угольной кислоты, как и для всякой двухосновной кисло¬
ты, могут существовать два типа производных - монозамещенные
за счет одной гидроксильной группы и дизамещенные, образован¬
ные с участием двух ОН-групп. Монозаещенные производные
угольной кислоты - соответствующие галогенангидриды, эфиры,
амиды общей формулы Х-С(0Н)=0 (где X = С1, ОК МН2 и т.д.)
неустойчивы.
Продукты присоединения аммиака и первичных аминов к С02
(НО-СО-МНК) называются карбаминами. Образование непрочного
карбамина в реакции С02с аминогруппой глобина - белковой части
молекулы гемоглобина - лежит в основе транспорта углекислоты
из клеток к легким (см. главу 17).
Доступными и практически важными являются дизамещенные
производные:
Фосген СОС12, газообразное вещество со слабым запахом све¬
жего сена, легко синтезировать реакцией хлора с окисью углерода.
Фосген является боевым ОВ удушающего действия. Как и всякий
галогенангидрид, фосген вступает в нуклеофильное замещение с во¬
дой, аммиаком, спиртами (глава 17)-
Способность фосгена к нуклеофильному замещению, с одной
стороны, используется при его дегазации (щелочной гидролиз), а с
другой стороны, служит для синтетических целей С помощью фос¬
гена получают многие лекарства, красители, а также карбамид
В органическом синтезе применяют и диалкилкарбонаты
Важны для практики смешанные эфиры и амиды угольной кис¬
лоты - уретаны. В частности, этилуретан является снотворным
средством Некоторые другие уретаны также являются лекарст¬
венными препаратами О значении мочевины будет сказано далее
дихлорангидрид диалкил- алкил- карбамид
угольной кислоты карбонат уретан мочевина
іиомочевина гуанидин
алкиловый эфир
хлоругольной кислоть
567
Укажем здесь, что сернистый и азотистый аналоги мочевины -
тиомочевина и гуанидин, также находят широкое применение
Гуанидин входит в состав ряда важных природных соединений Так,
в мышечной и нервной тканях животных, а также в моче содержит¬
ся креатин (М-метилгуанидинуксусная кислота)
Гуанидиновый фрагмент присутствует и в молекуле аминокис¬
лоты белков аргинина (см. главу 31).
Мочевина или карбамид - кристаллическое вещество, хорошо
растворимое в воде. Её характерной особенностью является способ¬
ность к образованию соединений включения (см. главу 8) с
н-алканами и другими длинноцепочечными соединениями с нераз-
ветвленной углерод-углеродной цепью, по отношению к которым
она вступает в качестве “хозяина”.
В кристаллической решетке мочевины имеются шестигранные
полости, в которых и могут размещаться молекулы алканов. Изоал-
каны с разветвленными углеводородными цепями в полости не про¬
ходят. В этом заключается способ разделения нормальных и изоал-
канов, который важен для практики, в частности, для улучшения
качества топлива. При удалении н-алканов с помощью мочевины из
топлива повышается его октановое число и понижается температу¬
ра замерзания.
Мочевина способна к реакции ацилирования карбоновыми кис¬
лотами, их ангидридами или галогенангидридами. Эта реакция, от¬
крытая Н. Н. Зининым, приводит к уреидам, например:
креатин N1-1
ЧСН2—СООН
СН31ЧН
^=1ЧН
28.1. Мочевина и ее производные
ацетилмочевина илу
ацетуреид
568
28.2. Циримидиновые производные мочевины
Важной является реакция образования уреидов дикарбоновых
кислот, которые называются уровыми кислотами. Уровые кисло¬
ты далее циклизуются с образованием гетероциклических уреидов.
Так, например, из щавелевой кислоты и мочевины возникает сна¬
чала оксалуровая кислота, при циклизации которой получается
парабановая кислота:
Кислотные свойства парабановой кислоты объясняются тем,
что её МН-группировки заключены между двумя группами С=0,
а, следовательно, являются достаточно подвижными, “кислыми”.
Аналогично из малоновой кислоты синтезируют её циклический
уреид, именуемый барбитуровой кислотой.
Атомы водорода барбитуровой кислоты также подвижны, и по¬
тому её молекула склонна к амидо-имидольной таутомерии; при
этом в одной из таутомерных форм её можно рассматривать как
2,4,6-триоксипиримидин.
оксалуровая
кислота
Н
парабановая
кислота
малонуровая
кислота
барбитуровая
кислота
О
Н
2,4,6-триоксипиримидин
569
Некоторые производные барбитуровой кислоты, называемые
барбитуратами, являются эффективными снотворными препаратами
К их числу относятся веронал (диэтилбарбитуровая кислота) и
люминал (этилфенилбарбитуровая кислота).
диэтилбарбитуровая этилфенилбарбитуровая
кислота кислота
веронал люминал
Важно знать, что аналоги барбитуровой кислоты, содержащие
пиримидиновое кольцо, входят в состав нуклеиновых кислот. Таков
и 2,6-диоксипиримидин или урацил, обозначаемый в биохимии
буквой У-
5-фторурацил
урацил (У) тимин (Т)
Урацил склонен к амид-имидольной таутомерии. Более прочную
(обведенную рамкой) форму, в которой урацил находится в составе
нуклеиновых кислот, необходимо запомнить.
цитозин (Ц)
Кроме урацила в нуклеиновых кислотах находятся его 5-метиль-
ный гомолог тимин (Т) и 2-окси-6-аминопиримидин или цито¬
зин (Ц). Для них также в рамке представлены более прочные тау-
томерные формы.
570
Среди производных пиримидина существуют и лекарственные
препарать^Чз частности, обладающие противоопухолевым действи¬
ем. Примером может служить молекула 5-фторурацила, замести¬
тель которой (атом фтора) по своим размерам занимает промежу¬
точное положение между атомом водорода в молекуле урацила и
метильной группой в тимине. Следовательно он структурно подобен
этим веществам и может замещать их в составе молекул нуклеино¬
вых кислот в процессе их биосинтеза. Получающаяся при этом нук¬
леиновая кислота уже не может выполнять своих функций, а следо¬
вательно клетка гибнет. Таким образом, 5-фторурацил - типичный
антиметаболит.
28.3. Пуриновые производные мочевины
Осторожным окислением барбитуровой кислоты синтезируют
оксибарбитуровую кислоту (2,4,5,6-тетраоксипиримидин), кото¬
рая способна ацилировать мочевину с образованием циклического
уреида - мочевой кислоты. Молекула мочевой кислоты содержит
конденсированный пуриновый гетероцикл и представляет собой
2,6,8-триоксипурин
Мочевая кислота, как и мочевина, является продуктом азотис¬
того обмена в организме
У млекопитающих содержание мочевой кислоты в моче невели¬
ко и лишь при некоторых нарушениях обмена возрастает Отложе¬
ния мочевой кислоты в суставах служат причиной подагры Камни
мочевого пузыря и почек также состоят из мочевой кислоты или ее
солей - уратов У пресмыкающихся и птиц мочевая кислота, а не
ОН ОН он
2,4,5,6-тетраоксипиримидин мочевая кислота
?Н о
мочевая кислота
Н м
2,6,8-триоксипурин
571
мочевина, является главным продуктом азотистого обмена. Её со¬
держание в гуано, используемом как удобрение, достигает 26%.
Аналогами мочевой кислоты являются производные пурина, на¬
ходящиеся в нуклеиновых кислотах - аденин (А) и гуаиии (Г).
Аденин - это 6-аминопурин При его гидролизе происходит
замена аминогруппы на гидроксильную с образованием 6-окси-
пурина или гипоксантина, молекула которого способна к амид-
имидольной таутомерии-
———• гипоксантин
аденин (А)
Следует обратить внимание, что в имидазольном кольце произ¬
водных пурина в принципе возможна имин-иминная таутомерия
(см. 24.3.1), например, для гипоксантина:
гипоксантин
Действительно, такое равновесие имеет место.
Превращение аденина в гипоксантин легко осуществляется фер¬
ментативным способом, поэтому гипоксантин обнаружен во многих
тканях растений и животных.
Гуанин представляет собой 2-амиио-б-оксипурии, таутомер-
ные формы которого изображены ниже.
гуанин (Г) ксантин
Как и аденин, гуанин в организме дезаминируется с образова¬
нием 2,6-диоксипурина, именуемого ксантином.
572
Ксантин представляет собой важное промежуточное звено азо¬
тистого обст$на. Следует обратить внимание на то, что окисление
гипоксантина приводит к ксантину, а из него, в свою очередь, при
окислении возникает мочевая кислота.
Важными природными производными ксантина являются алка¬
лоиды теофиллии, теобромин и кофеин, обнаруженные в чае,
какао и кофе.
Они представляют собой метильные гомологи ксантина, а имен¬
но, теофиллин - это 1,3-диметилксантин, теобромин представляет
собой 3,7-диметилксантин, а кофеин - 1,3,7-триметилксантин.
О
СН3.
СН3
& да' ул
Анэ Ан3 Анэ
1,3-диметилксантин 3,7-диметилксантин 1,3,7-триметилксантин
теофиллин теобромин кофеин
Искусственным путем теобромин и кофеин, оказывающие сти¬
мулирующее действие на центральную нервную систему, можно по¬
лучить из ксантина.
Глава 29. НУКЛЕОТИДЫ
29.1. Мононуклеотиды
Нуклеиновые кислоты - полимерные соединения, в которых
элементарными мономерными звеньями служат производные О-ри-
бозидов в рибонуклеиновых кислотах (РНК) или 2-дезокси-0-
рибозидов в дезоксирибонуклеиновых кислотах (ДНК). О-рибоза и
О-дезоксирибоза вступают в реакцию в Р-фуранозной форме
Агликонами по отношению к этим углеводам служат пуриновые
(аденин, гуанин) или пиримидиновые (урацил, тимин, цитозин) азо¬
тистые основания. Общими для ДНК и РНК являются аденин, гуа¬
нин и цитозин (А, Г, Ц, соответственно); в ДНК кроме них входит
тимин (Т). а в РНК - урацил (У). Общая формула природной
М-гликозидной группировки выглядит следующим образом:
Эти вещества представляют собой нуклеозиды. Нуклеозиды
из РНК называют аденозином, гуанозином, цитидином и ури-
дином, а в ДНК - 2-дезоксиаденозином, 2-дезоксигуанозином,
2-дезоксицитидином и тимидином, соответственно.
Мономерные ІЧ-гликозидьі в нуклеиновых кислотах связаны
сложноэфирной связью с фосфорной кислотой за счет гидроксиль¬
ных групп моносахаридного остатка в 3-ем или 5-ом положениях,
как это изображено на примере 3- и 5-аденозинмонофосфатов:
Вещества, состоящие из рибозы (дезоксирибозы), фосфорной
кислоты и азотистых оснований, называются нуклеотидами.
х = А, г, ц, Т, У
ОН ОН(Н)
0=Р-0 он
н 6
он он
аденозин-3-монофосфат (АМФ-3)
аденозин-5-монофосфат (АМФ-5)
574
Нуклеотиду с фосфорной группой в положении 5 присваивают
окончание ^вая кислота. Например, аден.озин-5-мон.офосфат на¬
зывается адениловой кислотой. Изложенное выше представлено в
таблице 29.1.
Таблица 29.1
Классификация нуклеозидов и нуклеотидов
Азотистое
основание
Сокра¬
щение
Нуклеознд
Нуклеотид
Нуклеинова
я
кислота
Аденин
А
Аденозин
Адениловая
кислота
РНК
2-Дезокси-
аденозин
2-Дезоксиадени-
ловая кислота
ДНК
Гуанин
Г
Гуанозин
Гуаниловая
кислота
РНК
2-Дезокси-
гуанозин
2-Дезоксигуа-
ниловая кислота
ДНК
Цитознн
Ц
Цитидин
Цитидиловая
кислота
РНК
2-Дезокси-
цитидин
2-Дезоксицити-
диловая кислота
ДНК
Тимин
Т
Тимидин
Тимидиловая
кислота
ДНК
Урацил
У
Уридин
Уридиловая
кислота
РНК
Наряду с нуклеотидами, входящими в состав молекул нуклеино¬
вых кислот, некоторые мононуклеотиды и их производные встреча¬
ются в свободном состоянии. Так, изображенная выше адениловая
кислота присутствует в мышечной ткани АМФ-5 может присоеди¬
нять еще два остатка фосфорной кислоты, образуя в результате
аденозин-5-трифосфат (АТФ).
Аденозин-5-трифосфат является основным веществом, осу¬
ществляющим перенос энергии в биологических системах
(см. 23.5.2) Процессы, связанные с расходованием энергии, сопро¬
вождаются сопряженным расщеплением АТФ до аденозин-5-
дифосфата (АДФ) и Н3РО4 Совершение работы происходит за
счет выделения энергии при разрыве химической связи Р-0
(-20-30 кДж/моль) Напротив, в процессах распада питательных
575
веществ часть освобождающейся энергии расходуется на сопря¬
женное образование АТФ из АДФ; полученный в результате АТФ
служит резервом энергии в организме.
N42
ООО
НО-?—0-Р-0-?—О Н гС
ін Ан Ан К
он он
аденозин-5-трифосфат (АТФ)
Важную роль в метаболизме играет аденозин-3,5-монофосфат
(циклический АМФ или сокращенно цАМФ), в котором фосфорная
кислота ацилирует одновременно спиртовые группировки в положе¬
ниях 3 и 5 в пределах одной молекулы. Он служит промежуточным
звеном действия большого числа гормонов. Этой молекуле отводит¬
ся роль регуляции межклеточных коммуникаций.
Следует сказать, что кроме пяти вышеперечисленных нуклеино¬
вых оснований в состав нуклеиновых кислот могут входить в малых
количествах и некоторые другие производные пиримидина и пурина.
Примером нуклеотида с участием такого производного пурина, а
именно гипоксантина, служит молекула инозина. Такие нуклеотиды
называют минорными.
Среди нуклеотидов имеются и физиологически активные ве¬
щества. Так, аденин-Р,0-3-дезоксирибозид представляет собой анти¬
биотик, называемый кордицепином. Используя аналогии с природ¬
ными веществами, химики синтезируют лекарственные препараты,
для чего конструируют молекулы нуклеотидов на основе иных, не¬
жели рибоза или дезоксирибоза, моносахаридов.
аденозин-3,5-монофосфат (цАМФ)
инозин
576
Примерами такого рода могут служить аденинф,0-арабинозид,
являющий^ противовирусным препаратом, и цитозин-^.й-араби-
нозид, обладающий антилейкемическим действием.
Химики осуществляют и более глубокие трансформации нуклео¬
тидов, для чего в молекулы моносахаридов вводятся дополнитель¬
ные функциональные группы, как это имеет место, к примеру, с мо¬
лекулой аз идо т им ид и на (препарат против СПИДа), или вместо
фуранозного цикла используется нециклический радикал, напоми¬
нающий по строению моносахаридный остаток Таким веществом
является мощный антивирусный препарат - ацикловир.
NH2
ОН
аденин-р,0-3-дезоксирибозид
(кордицепин)
адени н-ß, D-араби нозид
цитози н-ß, D-арабинозид
О
О
N3
азидотимидин
ацикловир
’ Азидо - остаток азотистоводороднои кислоты HN3
19 Зак 675
577
Среди нуклеотидов имеются и кофакторы, необходимые для
функционирования некоторых ферментов, например, кофермент А.
пантотеновая кислота (остаток)
ОН СН3 О О
СН—А—СНг—0-1>—О—Р—ОНгС
о=А Ан3 Ан Ан
ЫН
—мнснгснгэн
ОН ОН
О меркамин (остаток) кофермент А
Кроме остатка аденозиндифосфорной кислоты в составе этого
вещества имеются фрагменты пантотеновой кислоты и меркамина
Кофермент А принимает участие в активации карбоновых кислот,
для чего служит сульфгидрильная группа меркаминового остатка,
вовлекающаяся в процесс тиоацилирования (см. 21.3).
29.2. Полинуклеотиды
Полимеризация нуклеотидов с образованием нуклеиновых кис¬
лот осуществляется посредством эфирной связи между фосфор¬
ной кислотой и спиртовыми гидроксильными группами в положе¬
ниях 3 и 5.
Ниже дан фрагмент молекулы нуклеиновой кислоты с остатками
А, Г и Ц (рис. 29.1).
Молекулы ДНК находятся в ядрах клеток. Для данного вида
животного или растительного организма количество ДНК в одной
клетке постоянно для всех его клеток. Постоянным является и раз¬
мер молекулы ДНК, молекулярная масса которой обычно велика и
лежит в пределах от 107 до Ю10. Еще одной отличительной особен¬
ностью молекулы ДНК является её состав - он постоянен для всех
клеток данного организма в любом возрасте и в любых физиологи¬
ческих условиях, хотя и заметно отличается для различных биоло¬
гических видов. Это означает, что для ДНК данного организма
последовательность азотистых оснований в цепи полинуклеотида
носит индивидуальный характер.
Специфической чертой ДНК является то, что число остатков
аденина в ней равно числу тиминовых остатков (А = Т) а количе¬
ство остатков гуанина равно количеству цитозиновых (Г = Ц).
578
Этот факт объясняется особенностью её вторичной структуры, ко¬
торая был^асшифрована в 1953 году Д. Уотсоном и Ф. Криком.
6
0=р—ОНг<
но7
9 он(н)
Рис. 29.1. Фрагмент молекулы нуклеиновой кислоты.
Оказалось, что молекула ДНК представляет собой двойную спи¬
раль, построенную из двух спирализованных полинуклеотидных
цепей. Связь между цепями осуществляется посредством водород¬
ных связей, которые существуют между парами аденин-тимин и
гуанин-цитозин. Первая пара ассоциирована с участием двух водо¬
родных связей, вторая объединяется за счет трех водородных свя¬
зей. Расстояние между связанными между собой водородными свя¬
зями атомами кислорода и (или) азота варьируется от 2 8 до 3.0 А.
Такое соответствие между парами азотистых оснований называ¬
ют их комплементарностью. В целом речь идет о комплементар-
но.сти двух цепей молекулы ДНК.
нчГ29М
ТИМИН
аденин
цитозин
гуанин
19*
579
Обычно длина витка двойной спирали ДНК составляет - 34 А,
причем на один её виток приходится 10 остатков азотистых основа¬
ний Однако длина витка и угол закручивания спирали мог^т варьи¬
роваться в зависимости от тонких деталей строения молекулы ДНК.
Поэтому известно более десятка разновидностей различных двуспи¬
ральных структур ДНК. Следует сказать, что кроме водородных
связей комплементарных пар оснований, действующих поперек спи¬
рали, некоторую роль играют и межмолекулярные взаимодействия
между соседними основаниями в цепи полимера, действующие
вдоль спирали Такие взаимодействия между нуклеиновыми основа¬
ниями, уложенными в стопку, называют стэкинг-взаимодействиями
(англ. stack - штабель). Они дополнительно скрепляют структуру
двойной спирали
Из принципа комплементарности следует, что последователь¬
ность нуклеотидов в одной цепи определяется их последователь¬
ностью в другой (рис. 29.2).
Постоянство состава и комплементар-
ность цепей молекулы ДНК в её спирали и
определяет её уникальную и фундамен¬
тальную биохимическую роль. ДНК яв¬
ляется хранителем всей совокупности ге¬
нетической информации в каждой клетке и
в целом организме.
Молекулы РНК в пределах одной клет¬
ки в отличие от ДНК не характеризуются
таким постоянством. Они различаются как
величиной и составом, так и местона¬
хождением в клетке. Это определяет и раз¬
нообразие их биологических функций.
Кроме того, в отличие от ДНК молекулы
рибонуклеиновых кислот не имеют такой пространственной упоря¬
доченности. Лишь в некоторых видах РНК обнаруживаются
участки, в которых водородные связи соединяют между собой ком¬
плементарные основания. Однако такое связывание имеет место в
пределах одной цепи, а не между двумя комплементарными цепями.
Молекулы РНК проявляют тенденцию к сворачиванию в хаотичные
клубки и т.д.
Так называемая рибосомная РНК, которая содержится в рибо¬
сомах и составляет около 80% РНК клеток, может иметь самые
различные размеры (М от 35 ООО до 1 ООО ООО). Транспортная РНК
невелика по размеру (М 23 ООО - 30 ООО), в то время как величина
информационной РНК варьируется в очень широких пределах
(М от 25 ООО до 1 500 000). Роль информационной РНК заключает¬
ся в том, что она выполняет функции матрицы, структура которой
определяет строение синтезируемых в клетке полипептидов.
Рис. 29.2.
Двойная спираль ДНК.
580
Хотя генетическая информация определяется последователь¬
ностью нуЙ&еиновых оснований в молекуле ДНК, сама эта молекула
в синтезе белка участия не принимает. Она передает всю необхо¬
димую информацию путем образования молекулы РНК. Этот про¬
цесс называется транскрипцией.
Синтез белков, основанный на информации, содержащейся в
РНК, называется трансляцией. Итак, РНК - посредник в синтезе
белков:
Транскрипция Трансляция
ДНК ► РНК ► Белок
В изучении строения полинуклеотидов, в исследовании синтеза
белка с их участием наука за последнее время достигла наиболее
впечатляющих достижений, которые открывают грандиозные пер¬
спективы выяснения механизмов интимнейших биологических про¬
цессов с целью управления ими.
29.3. Физико-химические свойства
полинуклеотидов
Каждое звено полимерной цепи полинуклеотида содержит оста¬
ток фосфорной кислоты, представляющую собой кислоту средней
силы (рКа = 2). Таким образом, нуклеиновые кислоты - это доволь¬
но сильные многоосновные кислоты, полностью ионизованные в
биосредах, и поэтому их поверхность несет отрицательный заряд.
Специфика пространственного строения ДНК определяет ту ее
особенность, что ионизированные остатки фосфорной кислоты со¬
ставляют наружную оболочку двойной спирали. Именно это обстоя¬
тельство объясняет большую склонность нуклеиновых кислот к ион-
ионному взаимодействию с веществами, содержащими аминогруп¬
пы. Особый интерес вызывает взаимодействие нуклеиновых кислот
с белками, которые содержат дополнительные аминогруппы (так на¬
зываемые слабоосновные белки) Такие белки иначе называют ги-
стонами. С ними ДНК образует комплексные ассоциаты -
нуклеопротеиды. В данном случае речь идет об образовании нуклео-
гистонов Полагают, что образование и диссоциация нуклеогистонов
регулируют синтез РНК, а тем самым - синтез белков.
Рибонуклеиновые кислоты также за счет ион-ионного взаимо¬
действия с белками дают нуклеопротеиды, рибосомы Рибосомы -
внутриклеточные частицы, осуществляющие биосинтез белка Они
присутствуют во всех клетках живых организмов бактерий, расте¬
ний и животных В каждой клетке содержатся тысячи или десят¬
ки тысяч рибосом Рибосомы имеют несколько разновидностей
581
Типичные рибосомы состоят из 50-65% рибосомной РНК и 35-50%
слабоосновных белков. Молекулярная масса одной рибосомной
субъединицы, имеющей сферическую форму, составляет около
3 I О6, а диаметр - 200-300 А. При увеличении pH (до 12) и ионной
силы среды (например, при растворении в 0 5-1.0 М растворах со¬
лей) происходит отделение белка от РНК в рибосомах.
Вирусы также представляют собой устойчивые комплексные ас-
социаты, содержащие до 30% нуклеиновых кислот (ДНК, возможно
присутствие и РНК) и большое число белковых молекул, уло¬
женных в определенном порядке и образующих специфическую для
данного вируса трехмерную структуру.
Как полидентатные лиганды, к тому же несущие остатки фос¬
форной кислоты, все нуклеотиды легко связываются с катионами
металлов, образуя соли или внутрикомплексные соединения (в по¬
следнем случае с участием донорных группировок и атомов азота
пиримидиновых и пуриновых основа¬
ний). Для молекул ДНК и РНК ком-
плексообразование вызывает измене¬
ние конформаций молекул, а следова¬
тельно и изменение биологических
свойств. Для мононуклеотидов связы¬
вание с катионами металлов необхо¬
димо для выполнения ими биологиче¬
ских функций. Так, АТФ функциони¬
рует в виде магниевого комплекса.
Молекула АТФ может связываться
in vivo и с катионами марганца или
кальция, что, естественно, сказывает¬
ся на участии АТФ в биохимических процессах.
Полинуклеотиды не содержат склонных к окислению группиро¬
вок, а потому в мягких условиях не подвергаются окислительно¬
восстановительным превращениям. При жестком окислении в вод¬
ной среде нуклеиновые кислоты превращаются, как и прочие орга¬
нические соединения, в углекислоту и воду. Азотистая часть их мо¬
лекул трансформируется в мочевину, мочевую кислоту или соли
аммония. Естественно, что при этом образуются и неорганические
фосфаты.
НО О
О—>-0
0=^Р—О-Mg.—NH2
¥ ^6"
он он
Глава ЗО. ОКСИ- И ОКСОКИСЛОТЫ
-ч
30.1. Оксикислоты
30.1.1. Классификация и номенклатура
Органические соединения, содержащие в своем составе одно¬
временно спиртовые (или фенольные) группы и карбоксильные
группировки, называют оксикислотами. При этом количество кар¬
боксильных групп определяет основность оксикислот, а количество
групп ОН (как спиртовых, так и кислотных) - их атомность.
Большинство оксикислот обладает тривиальными названиями.
Тривиальные латинские названия применяют и для солей этих
кислот
Простейшая одноосновная двухатомная оксикислота - глико¬
левая кислота, её гомологами являются молочная и (3-оксимас-
ляная кислоты
К числу одноосновных двухатомных кислот относится и про¬
стейшая из ароматических оксикислот - салициловая (или
Р
НО—СН2-СООН
гликолевая
кислота
СН3—СН—СООН
Ан
молочная
кислота,
лактаты
СН3 —СН—СН2 -СООН
Ан
(3-оксимасляная
кислота
СООН
у-оксимасляная
кислота,
у-оксибутираты
У
салициловая
кислота
СООН
яблочная кислота,
малаты
лимонная кислота,
цитраты
НООС—СН—СН—СООН
Ан Ан
винная кислота,
тартраты
583
о-оксибензойная) кислота. Яблочная кислота - представитель тре¬
хатомных дикарбоновых кислот, лимонная кислота содержит три
карбоксильных и одну ОН группу, а потому является четьфехатом-
ной трехосновной, четырехатомной двухосновной кислотой являет¬
ся винная Оксикислоты различают и по взаимному положению
функциональных заместителей, в зависимости от этого бывают
а-, (3-, у- и т д оксикислоты.
30.1.2. Стереоизомерия оксикислот
Многие оксикислоты (например, молочная, яблочная, винная)
обладают асимметрическими атомами углерода (в формулах отмече¬
ны звездочкой), а, следовательно, для них характерно явление оп¬
тической изомерии. Молочная кислота, имеющая один хиральный
центр, представлена двумя энантиомерами, изображенными ниже с
помощью проекционных формул Фишера-
Ь-глицрриновый Ц+)-мясомол очная
альдегид кислота
Тот из энантиомеров молочной кислоты, который имеет ту же
конфигурацию заместителей, что и О-глицериновый альдегид, при¬
нято считать принадлежащим к О-ряду. Его антипод служит пред¬
ставителем Ь-ряда.
Сопоставление конфигурации изомеров молочной кислоты и
глицеринового альдегида осуществляется путем таких химических
превращений, которые не изменяют конфигурации асимметрическо¬
го атома углерода. Окислением альдегидной группы О-глицери-
нового альдегида с последующим восстановлением первичной спир¬
товой группы и должна быть получена О-молочная кислота. При
этом совсем не обязательно, чтобы она обладала правым вращением
поляризованного света, т.к. вращение определяется тем, какие
функциональные заместители связаны с асимметрическим атомом
углерода, природой растворителя, концентрацией раствора и его
584
температурой. В частности, О-молочная кислота в водных раство¬
рах обладаем левым вращением и, следовательно, это - 0(-)-молоч-
ная кислота, а её антипод - Ц+)-мясомолочная кислота.
Таким образом, критерием определения конфигурации оптиче¬
ских изомеров и принадлежности к О-или Ь-ряду является их срав¬
нение с энантиомерами глицеринового альдегида.
Ц+)-Молочная кислота выделена Ю. Либихом в 1832 г. из
мышечной ткани и называется поэтому мясомолочной кислотой.
При сбраживании сахаров при помощи особых возбудителей
брожения образуется й(-)-молочная кислота. При закисании раз¬
личных продуктов, в том числе и молока, образуется смесь обоих
энантиомеров, т.е. их рацемат, не обладающий оптической актив¬
ностью. Рацемат носит название молочной кислоты брожения
Ф.Ь-молочной кислоты).
Молекула винной кислоты содержит 2 хиральных центра и,
следовательно, должна иметь 4 стереоизомера (2П, где п - число
хиральных центров). Эти стереоизомеры изображены ниже:
(+)-винная (-)-винная
кислота кислота
СООН
ОН
ОН
СООН
СООН
ОН
-—ОН
СООН
НО-
НО-
виноградная кислота
(рацемат)
Здесь должны существовать две пары антиподов - энантиоме¬
ров. Энантиомеры (I-II и III-IV) должны обладать попарно одинако¬
выми физико-химическими характеристиками, но различаться по
свойствам между парами, тку этих пар расстояния между отдель¬
ными заместителями различны, а, следовательно, различно и их по¬
ведение Следовательно они являются диастереомерами
Формулы III и IV тождественны, т к при повороте формулы IV
на 180° в плоскости рисунка она превращается в формулу III
Причина заключается в том, что эта молекула (в отличие от I и 11)
симметрична, т к обладает плоскостью симметрии, сечение которой
показано пунктиром
Действительно, оба хиральных центра форм III и IV облада¬
ют одинаковым набором заместителей с противоположной конфи-
585
гурациеи, а, следовательно, должны взаимно компенсировать вы¬
зываемое нми оптическое вращение Итак, III и IV - молекулы од¬
ного и того же вещества, которое не должно обладать оптической
активностью за счет внутримолекулярной компенсации. Такая раз¬
новидность стереоизомеров называется мезоформой
Таким образом, существует три стереоизомера винной кислоты:
два оптически активных энантиомера (+) и (-) и их диастереомер,
мезовинная кислота, который оптически не активен. Кроме мезо-
винной кислоты существует еще одна оптически недеятельная раз¬
новидность винных кислот, а именно рацемат - (±)-винная кислота,
называемая иначе виноградной кислотой’. Оптическую изомерию
винных кислот изучал Л. Пастер, который выделил из виноградной
кислоты оба энантиомера - (+)- и (-)-вннные кислоты.
30.1.3. Способы получения
Для получения оксикислот существует множество способов.
Большинство из них основано на окислительно-восстановительных
превращениях кислородсодержащих соединений. Так, гликолевую
кислоту можно в принципе получить окислением этиленгликоля
НО-СНзСНг—ОН -12!. НО—СНдСИ=0 -!2Ц НО—СНгС=0
■Нг0 Ан
гликолевый гликолевая
альдегид кислота
Окисление этиленглколя до гликолевого альдегида и далее -
гликолевой кислоты - промежуточные стадии окисления этиленгли¬
коля в организме до щавелевой кислоты. Эти вещества крайне ток¬
сичны и служат причиной отравлений этиленгликолем.
Молочную кислоту можно синтезировать обратным процес¬
сом - восстановлением соответствующей кетокислоты (пировино-
градной):
[Н]
СНз—С—СООН СНз—СН—СООН
4 го 4н
пировиноградная молочная
кислота кислота
* Отсюда и взято название рацемат, т к по латыни виноградная кисло¬
та это - асісіит гасетісит.
586
а-Оксикислоты удобно синтезировать гидролизом а-галоген-
кислот, а^&сже гидролизом а-оксинитрилов:
СН3—СНг—СООН —Д»сНз—СН—СООН СН3—СН—СООН ™2° СНз—сн—с=м-
•на А, ■НС| Ан мн> Ан
молочная
кислота
Полезно обратить внимание, что приведенные выше способы
получения молочной кислоты приводят к рацемической молочной
кислоте брожения.
30.1.4. Химические свойства оксикислот
Оксикислоты, обладающие полярными гидроксильными и кар¬
боксильными функциями, обычно представляют собой твердые вы¬
сокоплавкие вещества, легко растворимые в воде и других поляр¬
ных средах.
Химическое поведение этих соединений определяется способ¬
ностью к реакциям по спиртовой и карбоксильной группам, а также
свойствами, связанными с участием в реакциях обеих функций.
Реакции гидроксильной группы.
1. Предельные оксикислоты легко окисляются, например, мо¬
лочная кислота превращается в пировиноградную кислоту.
2. Как и прочие спирты, оксикислоты способны к образованию
сложных эфиров за счет реакций их ацилирования кислотами. На¬
пример, салициловая кислота ацетилируется с образованием аце¬
тилсалициловой кислоты, известной как жаропонижающее и
анальгетическое средство аспирин.
Реакции карбоксильной группы.
1. Оксикислоты относятся к числу слабых кислот (рКа - 4-5) и
образуют соли. Соли салициловой кислоты (салицилаты) приме¬
няют при лечении ревматизма.
2. Оксикислоты ацилируют спирты и фенолы с образованием
сложных эфиров. Так, из салициловой кислоты и метилового спирта
получают метилсалицилат, а с фенолом синтезируют фенилсали-
цилат
Метилсалицилат употребляют как наружное болеутоляющее
средство при ревматических воспалениях; фенилсалицилат (салол)
является дезинфицирующим препаратом при желудочно-кишечных
заболеваниях
587
Кроме сложных эфиров оксикислоты образуют другие производ¬
ные - галогенангидриды, ангидриды, амиды. К примеру, саышцила-
мид - жаропонижающий препарат
салицилат
натрия
салициловая
кислота
ацетилсалициловая
кислота
аспирин
,СНз
СН3ОН
°
О мм*
,ОН
,он
,он
метилсалицилат
фенилсалицилат
салициламид
Реакции с совместным участием спиртовой и кислотной
групп. Такие превращения обычно вызываются нагреванием окси¬
кислот и происходят различно для а-, (3- и у-оксикислот.
а-Оксикислоты дегидратируются межмолекулярно с образовани¬
ем двух сложноэфирных группировок в шестичленном кислородном
гетероциклическом производном, именуемом лактидом:
(З-Оксикислоты в этих условиях тоже теряют воду, поскольку
их а-водородный атом подвижен, и переходят в непредельные
кислоты:
лактид
СНз—СН—СНз—соон
Дн
р-оксимасляная
кислота
кротоновая
кислота
588
у-Оксикислоты, (а также и 5-) образуют внутримолекулярные
пятичлен1?Йке (или шестичленные) сложные эфиры - лактоны:
носн2сНгсн2соон ^_^!аао НОСН2СН2СН2СН2СООН -н^~
-у-оксимасляная 0 6-оксивалериановая
кислота у-бутиролактон кислота 5-валеролактон
30.1.5. Значение оксикислот
Оксикислоты в свободном виде или в составе некоторых произ¬
водных находятся в растительных и животных объектах и прини¬
мают важное участие в ряде биологических процессов. Яблочная,
лимонная, винные кислоты содержатся в соке многих растений или
продуктах их переработки (свекла, виноград, рябина, яблоки, пло¬
ды цитрусовых и т д.). Они безвредны для человека и находят
применение в кулинарии, а также при пищевом консервирова¬
нии. Салициловая кислота содежится в коре ивовых растений
(ЗаИсит), которая издавна применялась для лечения воспалитель¬
ных процессов.
Молочная кислота - продукт молочнокислого брожения углево¬
дов по суммарному уравнению:
С6Н1206 -> 2СэН60з.
Молочнокислое брожение лежит в основе процессов скисания
молока в кефир, простоквашу и подобные молочные продукты, а
также в основе квашения капусты, огурцов, помидоров и других
плодов. Силосование - процесс молочнокислого брожения расти¬
тельной зелени
Такие оксикислоты, как лимонная, яблочная и особенно молоч¬
ная кислота, принимают участие в биохимических явлениях кле¬
точного дыхания и обмена веществ (в частности, в цикле Кребса)
В процессе работы в мышцах накапливается мясомолочная кисло¬
та в результате расщепления углеводов по схеме, аналогичной
молочнокислому брожению Ее накопление является причиной ме¬
таболического ацидоза у-Оксимасляная кислота и ее производные,
у-оксибутираты, являются нейрорегуляторами.
589
30.2. Оксокислоты
Оксокислоты подразделяются на альдо- и кетокислоты В прак¬
тическом отношении и в медико-биологическом аспекте важны а- и
(3- кетокислоты, которые и рассматриваются здесь Среди кетокис-
лот главенствующая роль принадлежит пировиноградной и аце-
тоуксусной кислотам
X снтх°
пировиноградная ацетоуксусная
кислота кислота
Пировиноградная кислота - представитель а-кетокислот, ацето¬
уксусная кислота является (3-кетокислотой.
30.2.1. Пировиноградная кислота
Получение: 1) окислением молочной кислоты (см. выше),
2) действием КСМ на хлористый ацетил с последующим гидро¬
лизом:
3) пиролизом виноградной кислоты, откуда и возникло её на¬
звание:
ноосснснсоон р сн\ /?
нА Ан ° °н
Свойства. Пировиноградная кислота обладает всеми свойства¬
ми, присущими кетонной и карбоксильной группам. Являясь кисло¬
той, она дает соли, галогенангидриды, а также подвергается декар-
боксилированию Её соли и другие производные называют пирува-
тами.
В то же время пировиноградная кислота реагирует и своей ке¬
тонной группой. Так, её можно восстановить в молочную кислоту,
из неё легко синтезировать фенилгидразон.
590
Пировиноградная кислота проявляет склонность к кето-
енольной таутомерии, причем её енольная форма может, в свою
очередь, принимать участие в химических превращениях своим
спиртовым гидроксилом. В частности, при его ацилировании фос¬
форной кислотой образуется фосфат енола пировиноградной
кислоты, играющей определенную роль в процессах углеводород¬
ного обмена.
пируват натрия этилпируват пирувил хлорид
снзч .О СН3 О СН3 О
х^аОН
-Н20\
ОС2Н5
СгН-рн | -Н20 >^о2, -НС1
СН3,
СеН^Н —ы' 'ОН
фенилгидразон
СеНбМН-МНг
-Н
СИЗ 0 СНз
сЛ\)Н СОг о
пировиноградная
кислота ацетальдегид
| X
?снУч° — руч° с%ч°
но-р—О ОН [но он] НО ОН
Ан фосфат енола енол молочная кислота
пировиноградной кислоты
Медико-биологическое значение Пировиноградная кислота
является ключевым соединением многостадийного процесса угле¬
водного обмена При клеточном дыхании в присутствии кислорода
она в конечном итоге “сгорает”:
[О]
► С0г + СНзСН=0
С02+Н20
молочнокислое брожение
• С2Н5ОН спиртовое брожение
аэробное окисление
В отсутствии кислорода она под действием ферментов восста¬
навливается в молочную кислоту, которая и накапливается в мыш¬
цах при интенсивных физических нагрузках, связанных с большими
затратами кислорода То же явление происходит и при молоч¬
нокислом брожении Иной процесс, а именно ферментативное де-
карбоксилирование, идет при спиртовом брожении.
591
30.2.2. Ацетоуксусная кислота и её эфир
Ацетоуксусная кислота не стойка, легко декарбоксилируется-
СН3СОСН2СООН -» СН3СОСН3 + С02,
а потому в практике обычно имеют дело с её стабильным этиловым
эфиром, называемым ацетоуксусным эфиром
Ацетоуксусный эфир синтезируют альдольным уплотнением
этилацетата в щелочной среде (обычно для этих целей употребляют
метилат натрия)
Ацетоуксусный эфир и ацетоуксусная кислота обладают харак¬
терными свойствами кетонов. Восстановление ацетоуксусной кис¬
лоты, например, приводит к (3-оксимасляной кислоте:
Этот фенилгидразон не стоек, т.к. легко отщепляет молекулу
спирта и циклизуется в пятичленное пиразольное кольцо. Обра¬
зующийся в результате 1-фен.ил-З-метилпиразолон-5 обладает
анальгетическим и жаропонижающим действием. Он является ро¬
доначальником многих анальгетиков (антипирин, амидопирин,
анальгин, бутадион и тд ).
СН3
ОС2Н5+Н—СНг
О У~ОС2Н5
О
№ОН <*3
НО
<ЬгіН5
ацетоуксусный
эфир
СН
>0
р-оксимасляная кислота
Из ацетоуксусного эфира можно получить фенилгидразон:
1 -фенил-З-метилпиразолон-5
Н
Н
Н'
>-<
^СбН5
н
фенилгидразон
ацетоуксусного эфира
592
Характерной особенностью ацетоуксусного эфира является вы¬
раженная склонность к кето-енольной таутомерии за счет того, что
а-атом водорода испытывает сильное одновременное электроноак¬
цепторное действие карбоксильной и карбонильной групп.
Енол ацетоуксусного эфира дает характерные реакции спиртово¬
го гидроксила (в частности, алкоголят под действием №).
Н
Таким образом, ацетоуксусный эфир обладает разнообразной
реакционной способностью, как за счет функциональных группиро¬
вок двух таутомерных форм, так и за счет сложноэфирной группы.
Эти его свойства широко используются химиками в синтетических
целях. Синтезы с ацетоуксусным эфиром составляют важный раздел
препаративной органической химии.
Ацетоуксусная кислота представляет собой метаболит, который
накапливается в тканях при нарушениях углеводного обмена, вызы¬
ваемых диабетом, а также при углеводном голодании. Вместе с ней
в тканях всегда присутствуют и продукты её ферментативных пре¬
вращений, а именно (3-оксимасляная кислота, получаемая восста¬
новлением, и ацетон, возникающий при декарбоксилировании. Эти
соединения именуют ацетоновыми телами.
Глава 31. АМИНОКИСЛОТЫ. ПОЛИПЕПТИДЫ
31.1.Классификация и получение
Аминокислоты, т.е вещества с амино- и карбоксильными груп¬
пами, подразделяются, как и другие классы органических веществ,
на предельные и ароматические производные Среди ароматических
аминокислот важны п-аминобензойная кислота, служащая факто¬
ром роста микроорганизмов (ее антиметаболитами являются суль¬
фамидные препараты) и п-аминосалициловая кислота или ПАСК,
обладающая противотуберкулезным действием
л-аминобензойная л-аминосалициловая
кислота кислота (ПАСК)
Аминокислоты подразделяются в зависимости от расположения
групп МНг и СООН на а-, (3-, у- и т.д. аминокислоты. Так, в
е-аминокапроновой кислоте, из которой получают полимерный ма¬
териал капрон, эти группы разделены пятью группами СН2.
а р е
мнзснзсоон мнзснзснгсоон мнзСНгСНгСНзСНгСНгСООН
глицин р-аминопропионовая е-аминокапроновая
кислота кислота
В (3-аминопропионовой кислоте, служащей продуктом промежу¬
точного обмена аминокислот и входящей в состав ряда биологиче¬
ски активных веществ (например, одного из витаминов группы В -
пантотеновой кислоты), расстояние между МН2 и СООН группами
составляет два СН2-звена.
Белки, как известно, построены из а-аминокислот. Молекулы а-
аминокислот, за исключением простейшей (глицин, Я = Н, см. табл.
31.1), хиральны. Молекулы природных а-ами-
СООН нокислот принадлежат к Ь-ряду, что изображено с
ЫНг—— помощью проекции Фишера.
р Сведения о важнейших а,Ь-аминокислотах, кото¬
рые входят в состав белков, даны в табл. 31.1.
594
Таблица 31.1
Аминокислоты белков ЫНгСН(Р)СООН
Название
Формула
Сокращение
русское междунар.
рК,
Глицин
МНгСНгСООН
Гли
01у
2 60
9 80
Аланин
мн2снсоон
£н3
Ала
А1а
2 35
9 87
Валин
МН2СНСООН
А
сн/ снз
Вал
Уа1
2 29
9 40
Лейцин
МН2СНСООН
£н2
А
сн/ СНз
Лей
Ьеи
2 33
9.74
Изолейцин
мн2снс00н
сн^
сн3сн/ снз
Иле
Ией
2 32
9 76
Серии
МН2СНСООН
£н20н
Сер
Бег
2 19
9 21
Треонин
мнгснсоон
сн
СН3" 'ОН
Тре
ТИг
2 09
9 11
Цистеин
МН2СНСООН
^НгБН
Цис
Суз
1 92
8.35
10 46
Цистин
мнгснсоон
бНг-Б-Б-СНг
мнгбнсоон
ЦисБ-
БЦис
СуэБ-
БСуэ
1 90
10 40
Метионин
МН2СНСООН
^Нг-СНг-БСНз
Мет
Ме1
2 13
9 28
Аспарагиновая
кислота
Ж2СНСООН
£н2-соон
Асп
Аэр
1 99
3 90
9 90
Аспарагин
МНгСНСООН
£н2-сомн2
Асн
Абп
2 10
8 84
595
Продолжение таблицы 31.1
Название
Формула
Сокращение -
русское неждуиар.
рк.
Глутаминовая
кислота
ынгснсоон
<!:н2-сн2-соон
Глу
б1и
2 10
4 07
9 47
Глутамин
ынгснсоон
Анг-снг-соынг
Глн
Оп
2 17
9.13
Лизин
ГЖ2СНСООН
(^г-СНг-СНг-СНг-ыиг
Лиз
1ув
2 16
9.18
10 79
Аргинин
ын2снсоон ын
Аиг-СНг-СНг-ЫН-^
N42
Арг
Аге
1 82
8.99
2 48
Фенилаланин
Ш2СНСООН
Фен
РЬе
2 16
9 18
Тирозин
р
ын2снсоон
О
Тир
Туг
2 20
9 11
10.13
Триптофан
он
ынгснсоон
0^
Три
Тгр
2 43
9 44
Г истидин
ынгснсоон
н 1н2
&
Гис
Н15
1.80
6.04
9.35
Пролин
( У—соон
1
Про
Рго
1 95
10 64
Гидрокси-
пролин
н
но
( У—соон
н
НО-Про
НО-
Рго
1.99
10.66
596
Как видно из формул, в числе аминокислот белков есть пре¬
дельные алТЛ^окислоты (Р = Н или алкил). Кроме того, в состав ра¬
дикалов Р могут входить разнообразные функциональные группы:
спиртовая гидроксильная в серине и треонине или фенольная -
в тирозине, меркаптогруппа в цистеине, тиоэфирная - в метионине,
дополнительная карбоксильная группа в моноаминодикарбоновых
кислотах, аминная - в диаминомонокарбоновой кислоте - лизине
или гуанидиновая - в аргинине, амидная функция - в аспарагине и
глутамине.
Молекулы аминокислот могут также содержать бензольное
кольцо или азотистый гетероцикл - индольный, имидазольный или
насыщенный пиррольный (тетрагидропиррольный). Таким образом,
природа заместителя Р в аминокислотах белков варьируется в ши¬
роких пределах, что в итоге определяет разнообразие химического
поведения, а тем самым и биохимических функций белков.
Тривиальные названия аминокислот белков связаны с тем или
иным их свойством (например, Ьглицин обладает сладким вкусом)
или с природным источником, из которого они выделены (так, аспа¬
рагин извлечен впервые из спаржи).
Аминокислоты белков делятся на заменимые и незаменимые.
Незаменимые аминокислоты (валин, лейцин, изолейцин, треонин,
метионин, лизин, фенилаланин и триптофан) не синтезируются в
организме и должны быть получены с пищей. Заменимые аминокис¬
лоты образуются при их недостатке в результате биосинтеза из
других аминокислот или из небелковых компонентов.
Общее число а,Ь-аминокислот, извлеченных из различного рас¬
тительного или животного материала, намного больше приведен¬
ных в таблице и составляет в настоящее время несколько десят¬
ков. Примером такой довольно распространенной в биообъектах
аминокислоты может служить гомолог лизина - орнитин
МН2(СН2)зСН(МН2)СООН. В отдельных случаях в состав полипептидов
входят и О-аминокислоты.
Способы получения. а-Аминокислоты получают кислотным
гидролизом белков. Существует много разнообразных методов и\
синтеза, из числа которых укажем два важнейших-
1 из а-галогенкарбоновых кислот действием избытка аммиака,
например
<32 1ЧН3
СН3СН2СООН » СН3СНСООН 1* СН3СНСООН
-НС1 \ -нс1 П
6 1ЧНг а-аланин
2 гидролизом а*оксинитрилов с последующей обработкой из¬
бытком аммиака
597
сн^снсоон
мн2
Вариантом этого метода, предложенного Зелинским, является
действие МНиСМ, которое сразу же приводит к а-аминонитрилу, а
его дальнейший гидролиз - к аминокислоте
Принципиальное значение для выяснения вопроса о химиче¬
ской эволюции - формировании аминокислот из неорганических
веществ - имел эксперимент по получению аминокислот из смеси
аммиака, метана и углекислоты под действием электрического раз¬
ряда под давлением и при нагревании. В результате опыта в реак¬
ционной смеси были обнаружены почти все природные амино¬
кислоты
Аминокислоты как вещества, обладающие двумя функциональ¬
ными группами, дают реакции за счет обеих функциональных групп,
а также с их совместным участием.
Аминокислоты проявляют все свойства первичных предельных
аминов (см. главу 22), т.е. образуют соли с минеральными кислота¬
ми, подвергаются ацилированию под действием ангидридов и хлор-
ангидридов, образуют имины в реакциях с альдегидами и кетонами,
а с азотистой кислотой претерпевают дезаминирование - пре¬
вращаются в оксикислоты.
Взаимное превращение оксикислота - аминокислота происходит
в клетке под действием ферментов. Важно отметить, что этот про¬
цесс идет с обращением конфигурации хирального центра, уча¬
ствующего в реакции нуклеофильного замещения:
Механизм этого превращения доказан специальными экспери¬
ментами.
31.2. Свойства аминокислот
31.2.1. Реакции с участием аминогруппы
598
31.2.2. Реакции с участием карбоксильной группы
Аминокислоты, как и другие вещества с группой СООН, прояв¬
ляют слабые кислотные свойства. Как видно из данных табл.
31.1, величина рКа1, отвечающая диссоциации по кислотному типу,
колеблется в пределах 1.8-2.6.
Основность аминокислот характеризует величина рКа2 , кото¬
рая находится в интервале 8-11. Следует обратить внимание на то,
что ряд аминокислот, содержащих дополнительную функциональ¬
ную группировку, характеризуется величиной рКаз , значение кото¬
рой может оказаться в различных пределах в зависимости от при¬
роды этой группировки.
Так, у моноаминодикарбоновых кислот кислотность дополни¬
тельной группы СООН выражается величиной рКаз , близкой к 4, а
основность второй аминогруппы диаминомонокарбоновых кислот
определяется величиной рКаз 10.8-12.5.
Аминокислоты участвуют в ацилирова-
нии аминов и спиртов. В медицине применяет¬
ся для обезболивания продукт взаимодействия
этанола с лара-аминобензойной кислотой, из¬
вестный как препарат анестезин.
Еще большей эффективностью обладает но¬
вокаин - продукт ацилирования той же самой
кислотой (3-диэтиламиноэтилового спирта (см.
главу 22).
Аминокислоты могут претерпевать декар-
боксилирование. В организме этот процесс
происходит под действием ферментов декар¬
боксилаз.
Некоторые из аминов, получаемых в ре¬
зультате ферментативного декарбоксилирова-
ния, играют важную роль Так, например, обра¬
зующиеся из гистидина и триптофана гистамин
и триптамин (см главу 22) обладают физиоло¬
гической активностью. Гистамин, в частности, является сосудорас¬
ширяющим агентом, регулирует секрецию желудочного сока, влияет
на явление аллергии.
этиловый эфир
л-аминобензойной
кислоты
(анестезин)
СНзСНгМНг
I
н
гистамин
599
З!.2.3. Реакции с участием групп ЫН? и СООН
1. Как уже отмечалось, аминокислоты проявляют свойства и
кислот, и оснований. Такие соединения представляют собой амфо-
терные электролиты или амфолиты. Существенно, что при раство¬
рении аминокислот в воде (а они в ней хорошо растворимы за счет
полярных группировок 1ЧН2 и СООН) амино- и карбоксильная группа
реагируют между собой с образованием биполярного иона (или
амфиона) В таком виде аминокислоты и находятся в водном рас¬
творе Суммарные кислотно-основные превращения биполярного
иона обсуждены в главе 9
2. Бифункциональность аминокислот проявляется и в том, что
они представляют собой комплексоны (см главу 4), т.е. легко об¬
разуют внутрикомплексные соединения с катионами металлов.
3. Термические превращения аминокислот также связаны с
участием обеих функциональных групп и напоминают в этом отно¬
шении свойства оксикислот.
Так, а-аминокислоты при нагревании образуют внутренние ге¬
тероциклические (пиразиновые) амиды - дикетопиперазины:
Р-Аминокислоты отщепляют аммиак, так как в их молекулах
имеется а-водородный атом:
у-, 5- и е-Аминокислоты образуют внутримолекулярные цикличе¬
ские амиды (лактамы) с пяти-, шести- или семичленными кольца¬
ми соответственно, например:
Н
Н
дикетопиперазин
СН3—СН—СНг—СООН
ііїНг
р-аминомасляная
кислота
кротоновая
кислота
є-аминокапроновая
кислота
Н
е-капролактам
31.3. Полипептиды
Важнейшим свойством аминокислот, которое требует участия
как N142, так и СООН группы, является их линейная поликонденса¬
ция с отщеплением воды и образованием амидной связи. Продукты
такой реакции называют пептидами, а амидную связь в них - пеп¬
тидной связью. Из двух молекул аминокислот получаются дипепти¬
ды, из трех - трипептиды и т д. В целом о таких продуктах говорят
как о полипептидах. Синтетические способы образования пептид¬
ных связей включают некоторые специальные приемы и здесь под¬
робно не рассматриваются В животных и растительных клетках
синтез полипептидов осуществляется сложным образом с участием
молекул нуклеиновых кислот и ферментов.
В настоящее время в практике находят широкое применение
высокомолекулярные синтетические полипептиды капрон и найлон,
отличающиеся высокой механической прочностью.
Капрон синтезируют полимеризацией е-капролактама, а найлон
совместной поликонденсацией гексаметилендиамина с адипиновой
кислотой:
—СН2СН2СН2СН2СН2С- МН— 1
II п
О 2
е-капролактам капрон
пМН2(СН2)бМН2 + пНООС(СН2)4СООН 2^° | —МН(СН2)6МН—С—(СН2)4С— 1
I & & \п
гексаметипендиамин адипиновая кислота найпон
Однако оба эти полимера по своим качествам заметно уступа¬
ют некоторым природным полипептидам, например, кератину волос,
паутине или шелку. Причина этого состоит в многообразных струк¬
турных возможностях, которые реализуются в природных полипеп¬
тидах, что позволяет им эффективно выполнять те или иные специ¬
альные функции.
Во-первых, в природных полипептидах может широко варьиро¬
ваться длина цепи Так, активатор ферментов глутатион представ¬
ляет собой трипептид, антибиотик грамицидин Б состоит из 10
аминокислотных остатков, гормон инсулин построен из 51 амино¬
кислоты, а фермент рибонуклеаза содержит 124 аминокислотных
остатка. Рибонуклеазу уже по существу можно отнести к белкам,
т.к высокомолекулярные полипептиды (М - 5 ООО), выполняющие
601
какую-либо специальную биологическую функцию, и принято счи¬
тать белками Некоторые белки имеют молекулярный вес свыше
1 ООО ООО, а следовательно, состоят из многих тысяч аминокислот¬
ных остатков.
Во-вторых, в природных полипептидах широко комбинируется
состав аминокислотных звеньев. В общем виде структуру любого
полипептида можно представить формулой.
в которой Р’-Р5 и т.д. - остатки, отвечающие любой из природных
аминокислот.
Например, трипептид у-глутатион, являющийся антиоксидан¬
том и кофактором, построен из глутаминовой кислоты, цистеина и
глицина:
МНг-СН—(СНг)2—с—ЫН-СН-С—ЫН-СНг-СООН тч-лутатион
При написании формулы полипептида слева помещают ту кон¬
цевую аминокислоту, которая реагирует своей карбоксильной груп¬
пой (С-концом), а при составлении названия аминокислотным
остаткам с прореагировавшей карбоксильной группой придают
окончание -ил. Название молекулы, реагирующей своим 1М-концом,
сохраняется без изменения. В соответствии с этим глутатион пред¬
ставляет собой у-глутамил-цистеил-глицин. В сокращенной записи
по-русски это выглядит Н-у-Глу-Цис-Гли-ОН, а в международной
форме - Н-у-01и-Су5-01у-0Н.
Нетрудно посчитать, что глутатиону может отвечать еще пять
структурных изомеров с иной последовательностью аминокислотных
звеньев. С возрастанием цепи число всевозможных полипептидных
структур чрезвычайно быстро растет. Так, для полипептидной цепи
из 50 аминокислотных остатков, в построении которой могут уча¬
ствовать 20 аминокислот, количество возможных вариантов состав-
В-третьих, разнообразие структуры и свойств белков достигает¬
ся тем, что в состав заместителей аминокислот входят функцио¬
нальные группы (алкил, арил, гетарил, спиртовый или фенольный
гидроксил, сульфгидрильная, тиоэфирная, аминная, амидная, гуа¬
£оон О £н2 О
Ін
ляет 2050.
602
нидиновая или карбоксильная группы, см. табл. 31.1), что позволя¬
ет им проявлять полифункциональные химические свойства. В за¬
висимости от состава полипептид (или его отдельный участок) мо¬
жет обладать кислотными или основными свойствами, проявлять
склонность к образованию водородных, ионных или ковалентных
связей, комплексных соединений, вступать в окислительно-восста¬
новительные превращения и т.д.
31.4. Пространственное строение полипептидов
Последовательность аминокислот в молекуле полипептида об¬
разует его первичную структуру. Из неё нельзя получить пред¬
ставления о пространственном строении белков Между тем,
функции белков в значительной степени определяются особенно¬
стями их стереохимии. Сведения об этом дает знание вторичной,
третичной и четвертичной структуры полипептида
Они, в первую очередь, определяются конформацией пептидной
связи. Как известно (глава 26), связь С-М в амидах (а следователь¬
но, и в пептидах) занимает промежуточное положение между про¬
стой и двойной связями за счет сопряжения между 71-СВЯЗЬЮ С=0 и
электронной парой атома азота. Это приводит к стабилизации
плоской конформации фрагмента -СО-МН-, причем аминокислотные
остатки, как установлено, находятся в трансоидном положении:
В результате вся полипептидная цепь выглядит следующим об¬
разом-
ОРіНОРзНОРб
603
Нужно отметить, что в преде¬
лах цепи находятся кзк гибкие,
т.е. способные к быстрым кон-
формационным переходам, обыч¬
ные с-связи, так и относительно
малоподвижные пептидные связи,
которые, однако, не представляют
собой истинно двойных связей, а
следовательно, тоже способны к
конформационным переходам, хотя
и к более медленным. Сочетание
в цепи полипептида жестких и
подвижных участков делает её
эластичной и позволяет ей гибко
перестраивать структуру в зави¬
симости от условий.
Особенностью пептидных свя¬
зей является способность к обра¬
зованию водородных связей между
амино- и карбонильными группами
(-С=0 Н-М-). Если такие связи
возникают внутри полипептид-
ной цепи, это приводит к её скру¬
чиванию в спираль, называемую
а-спиралыо (рис. 31.1).
Длина одного шага спирали
полипептида равна 5.4 А, причем
один её виток составляет 3.6 ами¬
нокислотных остатка.
При наличии межмолекулярных водородных связей возникает
плоская параллельная конформация, состоящая из нескольких по-
липептидных цепей, так называемая ^-складчатая конформация
полипептидов (рис. 31.2).
Нахождение молекулы белка в а-спиральной или Р-складчатой
конформации и определяет вторичную структуру полипептида.
Малая прочность водородных связей обусловливает относитель¬
но легкую трансформацию вторичной структуры под воздействием
внешних условий (изменение pH, температуры, механическое воз¬
действие). К примеру, в грубом приближении кератин волоса имеет
конформацию а-спирали, а при натяжении переходит в Р-конформа-
цию, в результате чего его длина может увеличиваться обратимо
без разрыва почти в два раза.
Рис. 31.1. Строение а-спирали.
Тонкими линиями показаны
водородные связи.
604
Третичная структура полипептидов выражается в том, что ни
один белое не имеет-правильной а- или Р-конформации. Более вы¬
сокий порядок пространственной организации определяется наличи¬
ем изгибов полипептидной цепи.
Рис. 31.2. Строение р-складчатой структуры полипептидов.
Одна из причин таких изгибов - отсутствие водородных связей
между отдельными звеньями. Это, в частности, обязательно проис¬
ходит в присутствии остатков пролина и гидроксипролина, в кото¬
рых аминный атом азота является вторичным, а следовательно, пеп¬
тидная связь с его участием не имеет атома водорода при атоме
азота. Таким образом, это звено в принципе не может участвовать в
образовании водородной связи:
Другая причина - взаимодействие между собой функциональных
заместителей аминокислотных остатков.
Здесь важную роль играют ковалентные связи - дисульфид-
ные мостики между остатками цистеина. Такие дисульфидные мос¬
тики могут связывать между собой участки одной полипептидной
цепи, например, в молекуле рибонуклеазы имеется 4 внутримолеку¬
лярные дисульфидные связи. Часто белок образован несколькими
цепями, соединенными между собой дисульфидными мостиками.
Так, молекулы некоторых белковых антител устроены из 4 полипеп-
тидных цепей, связанных дисульфидными мостиками.
605
Достаточно прочным является также иои-иояиое взаимо¬
действие между аминогруппой остатка диаминомонокарбоновой
кислоты и карбоксильной группой остатка моноаминодикарбоновой
кислоты
Для металлопротеинов существенна металло-лигаидиая ко¬
ординация из-за того, что аминокислоты (а следовательно и поли¬
пептиды) легко образуют внутрикомплексные соединения - хелаты
Меньшей прочностью отличаются водородные связи типа
-Х-Н У- между атомом водорода спиртовой или фенольной групп, с
одной стороны, с кислотным остатком или атомами азота гетеро¬
цикла, с другой. Такого типа связи могут реализовываться и в дру¬
гих случаях при сближении двух полярных группировок, одна из
которых обладает атомом водорода.
гидратация
Рис. 31.3. Типы взаимодействий в полипептидах.
Кроме того, определенный вклад вносят и гидрофобные вза¬
имодействия, которые существуют между неполярными группи¬
ровками (-Р...Р- или -Аг...Аг-) остатков предельных или ароматиче¬
ских аминокислот в водных растворах.
Далее, полярные и ионные участки молекул полипептидов могут
гидратироваться за счет водородных связей или ион-дипольных вза¬
имодействий.
Перечисленные варианты взаимодействий между аминокислот¬
ными остатками и с молекулами воды представлены на рис. 31.3.
В итоге молекула каждого природного полипептида имеет в
биосредах сложную индивидуальную конформацию, в которой
606
отдельные участки регулярной структуры а-спирали и Р-склад-
чатого Л^Ьта сменяются сложными изгибами, петлями и т.д.
В качестве примера можно привести изображение весьма про¬
стого природного полипептида грамицидина Б*. обладающего анти¬
микробной активностью (рис. 31.4). Первичная структура этого
циклического полипептида выражается формулой:
І— \/аІ-С)т-І_еи-0-РІіе-Рго-\/аІ-От-І_еи-0-РІіе-Рго J
Рис. 31.4. Конформационная модель грамицидина Б.
В этой молекуле можно увидеть как реализацию Р-складчатой
структуры с четырьмя водородными связями, так и неучастие
остатков пролина в образовании водородных связей.
Единство химического состава и пространственной конформации
молекулы каждого полипептида и определяет его ярко выраженную
биохимическую индивидуальность. Особенно наглядно это иллю¬
стрируется специфичностью действия такой разновидности поли¬
пептидов, как ферменты. Структура ряда из них расшифрована,
а для некоторых имеются и представления о механизме действия.
В реакции принимает участие не весь полипептид, а лишь неболь¬
шой его участок - активный центр. При этом далеко не всегда ами¬
нокислотные остатки активного центра расположены в цепи по со¬
седству. Они могут быть сближены пространственно при формиро¬
вании третичной структуры. Конфигурация активного центра опре¬
деляет специфичность действия фермента
Конформации полипептидов чрезвычайно чувствительны к воз¬
действию внешних условий. Так, действие слабых восстановителей
* Выбранный пример интересен и другими моментами- во-первых, в
этом полипептиде присутствуют остатки б-аминокислот (Э-фенилалании),
что случается редко, и, во-вторых, в его состав входит аминокислота ории-
тин МН2(СН2)зСН(МН2)СООН, обычно не участвующая в построении природ¬
ных полипептидов
607
или окислителей может разрушить или, наоборот, создать дисуль-
фидные связи, изменение pH среды столь же сильно сказывается
на существовании ион-ионных взаимодействий и водородных свя¬
зей. Изменение ионной силы раствора также сильно влияет на все
виды ионных и полярных взаимодействий. Смена растворителя
влияет на степень проявления водородных связей и гидрофобных
взаимодействий
Не удивительно, что белки легко разрушаются и при повышении
температуры В результате таких воздействий происходит денату¬
рация белков, т.е необратимые конформационные (или химичес¬
кие) изменения, вследствие которых активный центр фермента раз¬
рушается.
Четвертичная структура полипептидов определяет объедине¬
ние их в белковые комплексы, обычно (но необязательно) за счет
слабых, т.е. водородных связей и гидрофобных взаимодействий Так,
в гемоглобине объединены четыре попарно одинаковые субъедини¬
цы, а белковая оболочка вируса табачной мозаики состоит из 2 130
полипептидных молекул. Кроме того, многие природные полипепти¬
ды связаны с остатками фосфорной кислоты (фосфопротеиды), с
липидами (липопротеиды), углеводами (гликопротеиды), катионами
металлов (металлопротеиды), нуклеиновыми кислотами (нуклео-
протеиды). Вопросы структуры сложно построенных белков рас¬
сматриваются в курсе биохимии.
31.5. Изоэлектрическая точка
аминокислот и полипептидов
Как известно, аминокислоты обладают свойствами амфолитов
(подробнее см. главу 9). Их диссоциация, взятая на примере глици¬
на, выражается суммарным уравнением:
рКа^г.6 + рКа2 = 9.8
+ н + н
мнз^-снг-соон — МНЗ^СН2-С00' — МН2-СН2—СОО'
Я+ (катион глицма) д+- (анлон глмрна)
(биполярный ион глицииа)
В случае биполярного иона аминокислоты, например, глицина,
МНз+-СН2СОО”, молекула электронейтральна. Такое состояние назы¬
вается изоэлектрическим состоянием. То значение pH, при котором
достигается изоэлектрическое состояние и при котором молекула не
перемещается под воздействием внешнего электрического поля, на¬
зывается изоэлектрической точкой (р1).
608
Изоэлектрическая точка зависит от величин рК аминокислот и
может б!к|ь рассчитана на примере глицина следующим образом.
„ _ [Н+][МН3+СН2СОСГ] „ [Н+][МН2СН2СОСГ]
К1 : и к2 = ; •
[МН3 СН2СООН] [МН3 СН2СОО_]
[Н+]2[МН2СН2СОО-]
Следовательно,К1К2 = £—* , а в изоэлектричес-
[МН3 + СН2СООН]
кой точке [МН2СН2СОО-] = [МН3+СН2СООН], поэтому К,К2 = [Н+]2.
Отсюда [Н+] = л/к^Г. После логарифмирования получаем:
pH = р1 =рК| *рКа.
2
Отсюда для глицина р1 = (2.6 + 9.8)/2 = 6.2.
Картина выглядит более сложной для тех аминокислот, которые
содержат дополнительную группу, способную к ионизации
(например, для аспарагиновой кислоты). Ее диссоциация может
быть представлена следующим образом:
СООН _н+ +?00 -Н+ соо‘ _н+ СОО*
МНз-С-Н — Мз-С-Н — Ж3-С-Н — МН2-С-Н
СН2СООН СН2СООН СН2СОО' СН2СОО'
Для этапов диссоциации А5Б,Б58иВ5Г величины рКа
составляют 1.99, 3.90 и 9.90, соответственно. Только форма Б нахо¬
дится в изоэлектрическом состоянии (с равным количеством поло¬
жительных и отрицательных зарядов). При расчете здесь изоэлек-
трической точки следует учитывать значения лишь двух первых ве-
„ т , 1.99 + 3.90 опс
личин рКа. Тогда р1 = = 2 95
31.6. Полиамфолиты
Звенья полипептидной цепи, содержащие дополнительные ами-
но- или карбоксильные, то есть ионогенные группы (например,
лизин, гистидин, аспарагиновая или глутаминовая кислоты), сооб¬
щают полимеру свойства полиэлектролитов Полиэлектролиты дис¬
социируют в растворах на макроионы (высокомолекулярные заря¬
20 Зак. 675
609
женные частицы) и малые ионы (низкомолекулярные заряженные
частицы). Они растворимы в полярных растворителях, электропро-
водны, и на их свойствах сильно отражается электростатическое
взаимодействие. Таким образом, полиэлектролиты находятся в та¬
ком же отношениии к полимерам-неэлектролитам, как низкомоле¬
кулярные электролиты - к неэлектролитам
Более широко полипептиды следует рассматривать как поли-
амфолиты - полимеры, несущие как положительные, так и отрица¬
тельные заряды
В кислои среде молекулы полиамфолитов заряжены положи¬
тельно. С ростом pH протоны нейтрализуются, слабокислые группы
заряжаются отрицательно, что приводит к падению общего заряда
макромолекулы. Дальнейшее повышение pH вызывает отрыв прото¬
нов, что ведет к тому, что в сильнощелочных средах молекулы по¬
лиамфолитов приобретают суммарный отрицательный заряд.
При некотором промежуточном зна¬
чении pH суммарный заряд макромоле¬
кулы белка становится равным нулю, то
есть достигается изоэлектрическая точ¬
ка. Число катионных и анионных групп
молекул белка совпадает (рис. 31.5).
Величина р1 белков зависит от приро¬
ды буферной системы и ионной силы
раствора.
Изоэлектрическая точка белков может быть измерена с по¬
мощью электрофореза, так как в этой точке подвижность полипеп¬
тида равна нулю. При этом используется прием изолектрического
фокусирования. Суть его состоит в том, что между электродами
устанавливают градиент pH, так что разные белки образуют ста¬
ционарные зоны в тех местах, где их р1 совпадает с pH. Если от¬
дельные зоны затем отделить друг от друга, тогда возникает метод
разделения природной смеси белков. Для определения изоэлектри-
ческой точки могут быть использованы также данные по набуха¬
нию и коагуляции полиамфолитов в растворах с различными значе¬
ниями pH (см. главу 32).
Ж;
N143-
Шз
Рис. 31.5. Полиамфолит
в изоэлектрической точке.
Глава 32. ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
*4 РАСТВОРОВ БИОПОЛИМЕРОВ
Растворы составляют еще
нерешенную задачу естество¬
знания, потому что, будучи
химическими соединениями, они
не подчиняются законам ато¬
мизма или кратных отно¬
шений. .
Д. И. Менделеев
32.1. Основные понятия химии полимеров
Структурной химической основой организмов являются природ¬
ные высокомолекулярные соединения (ВМС), биополимеры: белки,
нуклеиновые кислоты и полисахариды, а также смешанные биопо¬
лимеры - липопротеиды, гликопротеиды и липополисахариды.
Необходимо знание свойств ряда промышленных полимеров,
применяемых в медицинской практике - винильных полимеров, кау-
чуков, тефлона, капрона, найлона и др.
Сюда отнесем и неорганические полимеры - полисиликаты, в
частности, кремниевую кислоту. Наконец, полезно знать и об эле¬
ментоорганических ВМС. В их числе, к примеру, находятся силок-
саны, содержащие связи кремния с углеродом и кислородом.
[ °н I ( ? 1
[х°_]п [_г-]п
кремниевая
кислота силоксан
ВМС - это вещества, молекулы которых состоят из большого
числа (не менее 1000) химически связанных атомов, а потому их
молярные массы находятся в пределах 104-107
Полимеры представляют собой такую разновидность ВМС,
молекулы которых состоят из одинаковых групп атомов, мономер
ных звеньев - п раз повторяющихся структурных единиц X, !-Х-)г
Представители полимеров - полиэтилен, винильные полимеры, кау
чуки. К их числу можно отнести некоторые полиамиды, например
капрон, если он был получен полимеризацией капролактама.
Прочие ВМС получают поликонденсацей К этому классу при
надлежат полипептиды (если они получены из аминокислот), поли
сахариды.
20*
611
Линейные полимеры представляют собой химически не свя¬
занные одиночные цепи мономерных звеньев.
-х-х-х-х-
К ним относятся каучук, целлюлоза, найлон
Разветвленные полимеры выглядят следующим образом
I
-Х-Х-Х-
I
-Х-Х-Х-Х-
I
Разветвленную структуру имеет полисахарид амилопектин
Имеются и лестничные полимеры:
-Х-Х-Х-Х-
1111
-х-х-х-х-
К ним относятся некоторые искусственные волокна.
Сетчатые полимеры - это трехмерные полимеры, звенья ко¬
торых образуют единую химически связанную пространственную
сетку. В качестве примера сетчатых полимеров можно привести по¬
лисахарид гликоген.
В зависимости от элементного состава цепи различают гомо-
цепные и гетероцепные полимеры Пример гомоцепных полиме¬
ров - полиэтилен [-СН2-СН2-)п, гетероцепных - полиамиды
[-МН-СО-(СН2),-]п.
Полимеры, содержащие в одной макромолекуле различные типы
мономерных звеньев -Х1-Х2-Х3-, называются сополимерами. Регуляр¬
ные и нерегулярные сополимеры различают порядком чередования
мономерных звеньев Так, найлон с его однотипными звеньями, по¬
строенными из адипиновой кислоты и гексаметилендиамина, пред¬
ставляет собой регулярный сополимер. Природные полипептиды
являются полимерами различных аминокислот, т.е. нерегулярными
сополимерами как и полинуклеотиды, построенные из разных зве¬
ньев мононуклеотидов
32.2. Пространственное строение ВМС
Химические связи между мономерными звеньями прочны, как и
любые ковалентные связи (их энергия около 400 кДж/моль).
Кроме них между полимерными цепями или разными участками од¬
ной цепи возникают межмолекулярные взаимодействия - так
называемые молекулярные связи. Обычно их величина - не более
612
20 кДЬк/моль. Примером такого рода связей могут быть водородные
связи ЛЬ. полипептидах, закручивающие их в спираль или уклады¬
вающие в складчатую структуру, и сворачивающие молекулы поли¬
нуклеотидов в двойные спирали. Молекулярные связи делают моле¬
кулы ВМС нелетучими.
Характер и число молекулярных связей (а оно в многозвенном
полимере велико) определяет конформацию молекулы ВМС.
Подсчитано, что при 25° С средняя кинетическая энергия теп¬
лового движения одного звена составляет около 5 кДж/моль, что
меньше величины энергии молекулярных связей. При малых вели¬
чинах активационного барьера между различными конформациями
(а он обычно невелик, так как звенья полимера соединены между
собой а-связями с их свободным вращением) молекула ВМС пред¬
ставляет собой свободно-сочлененную цепь Отдельные звенья по¬
лимерной цепи поэтому находятся в состоянии хаотического тепло¬
вого броуновского движения, в результате чего молекула ВМС при¬
обретает форму клубка. Такая форма молекулы ВМС называется
глобулой.
Для величины среднего радиуса глобулы существует связь со
значением молекулярной массы ВМС (М):
ге = К>/М ,
где гг - средний радиус глобулы, К - коэффициент пропорцио¬
нальности.
Отношение эффективного радиуса макромолекулы г, определен¬
ного экспериментально, к рассчитанному по этой формуле значе¬
нию гг, характеризует жесткость полимерной цепи Оно варьирует¬
ся в широких пределах, составляя, к примеру, величину 1 7 для
гибкой цепи каучука, и 5.0 для жестких цепей нитроцеллюлозы
32.3. Растворы ВМС
Молекулы биополимеров дифильны Они состоят из неполярных
и полярных группировок. У неполярных радикалов, например, угле¬
водородных цепей, отсутствует сродство к полярному раствори¬
телю - воде, у полярных групп оно достаточно велико
Полисахариды построены из неполярных связей С-С и С-Н, с
одной стороны, и сильно полярных связей С-0 и ОН, с другой
Молекулы полинуклеотидов обладают высокой степенью полярно¬
сти их поверхности, на которой размещаются остатки фосфорной
613
кислоты, в то время как внутренняя часть этих молекул менее по-
лярна. Полипептиды содержат в своем составе наборы аминокис¬
лот с алифатическими и ароматическими радикалами, имеющими
гидрофобный характер, и с полярными группами (ОН, вН, ЫНг,
СООН и др ), обладающими высокой гидрофильностью Вдоль поли-
пептидной цепи располагаются фрагменты с принципиально разли¬
чающимся соотношением гидрофобность - гидрофильность.
Вследствие этого биополимеры обладают поверхностной актив¬
ностью и в водных растворах выступают в качестве стабилизаторов
различных гидрофобных микрогетерогенных систем.
Их поэтому относят к коллоидным ПАВ, которые с одним и тем
же растворителем в зависимости от условий образуют истинный
или коллоидный растворы.
Для таких систем характерно существование динамического
равновесия между истинным и коллоидным растворами.
Коллоидные растворы ВМС получаются самопроизвольно из ис¬
тинных растворов за счет ассоциации молекул. Процесс образова¬
ния мицелл в растворах коллоидных ПАВ термодинамически выго¬
ден. Ядро образовавшихся мицелл составляют неполярные радика¬
лы, а внешнюю обкладку - полярные группы, что обеспечивает
наименьший контакт гидрофобных групп с водой.
32.3.1. Колигативные свойства растворов ВМС
Осмотическое давление растворов ВМС существенно отличает¬
ся от рассчитываемого согласно уравнению Вант-Гоффа я = сЯТ,
причем экспериментально полученная кривая лежит выше теорети¬
ческой прямой (рис. 32.1).
Это объясняется уже отмеченной
относительной независимостью тепло¬
вого движения отдельных участков по¬
лимерной молекулы. Иначе говоря,
каждая макромолекула ведет себя как
совокупность нескольких молекул
меньшего размера, что и проявляется в
увеличении осмотического давления.
Очевидно, что для однотипно построен¬
ных молекул ВМС, количество та¬
ких автономных участков будет тем
большим, чем больше молекулярная
масса М.
Рис. 32.1. Зависимость
осмотического давления
от концентрации ВМС.
614
Длд расчета осмотического давления растворов ВМС предло¬
жено уравнение Галлера:
КТ о 2
* = — с+Рс,
в котором с - концентрация раствора ВМС, г/л; М - его молярная
масса, г/моль; Р - коэффициент, учитывающий гибкость и форму
молекулы ВМС в растворе.
Число подвижных единиц в растворе учитывается дополнитель¬
ным слагаемым рс2. При небольших концентрациях полимера зна¬
чение этого слагаемого невелико, и уравнение Галлера совпадает с
уравнением Вант-Гоффа.
Из всех коллигативных
свойств растворов ВМС осмомет-
рический метод наиболее чувстви¬
телен при определении молеку¬
лярной массы полимеров. Измеряя
осмотическое давление для рас¬
творов с разными концентрация¬
ми, получают графическую зави¬
симость величины л/с от с, из
которой и находят значение моле¬
кулярной массы полимера М и ко¬
эффициента Р (рис. 32.2).
Рис. 32.2. График зависимости
л/с от с.
32.3.2. Мембранное равновесие Доннана
Осмотические свойства растворов полимерных электролитов
(полиэлектролитов), а таковы растворы белков и полинуклеотидов,
проявляют ряд особенностей.
Известно, что малые и высокомолекулярные ионы распределя¬
ются неравномерно по обе стороны мембран, если они непрони¬
цаемы для ВМС.
Рассмотрим типичную для биологических систем ситуацию
По разные стороны мембраны находятся соль высокомолекулярного
соединения, белка (допустим, МаЯ - слева) и низкомолекулярный
электролит (N301, справа). Пусть в начальный момент их концент¬
рации составляют С] и С2, соответственно.
При установлении равновесия в такой системе малые анионы
СГ перемещаются преимущественно из правой части в левую
Макроанионы Я” через мембрану проникать не могут. Поэтому для
615
сохранения электронейтральности вместе с СГ-анионами справа
налево перемещаются и катионы натрия. В результате в момент
равновесия, когда через мембрану переместилось х моль/л хлорис¬
того натрия, концентрации ионов в обеих частях сосуда принимают
значения:
мембрана
слева
(Ма+) = сі + х
(С Г] = х
ЇР"] = с,
справа
[Ма'*'] = С2 - х
[СГ] = с2 - х,
где [Ма+1, [СГ], (Р-] - равновесные концентрации ионов; х - измене¬
ние концентраций ионов натрия и хлора в результате перехода час¬
ти хлористого натрия через мембрану справа налево.
В равновесии в соответствии с законами термодинамики сво¬
бодные энергии частиц слева (й ) и справа (й ) от мембраны
должны быть равны.
Для соли МаС1 это равенство имеет вид:
сЫаС1 = сЫаС1
0 <_°
Так как ОмаС1 = ОмаС1, а О = С5 + ИТ 1па±, где а± - актив¬
ность хлористого натрия (а± = а± = ^а Ма+- а С(- , см. главу 8).
Следовательно, ИТ1п ^а Ма+- а С(- = НТ1п д/а Ма+- а С(- , отсюда:
Это равенство называется мембранным равновесием Дон-
нана. Если коэффициенты активности слева и справа равны
(у, = у, , где у, - коэффициент активности 1-того компонента), полу¬
чают соотношение Доннана для концентраций ионов №01 по разные
стороны мембраны:
С С=°2 ■
Для конкретно обсуждаемого случая это уравнение выглядит:
(С! + х)х = (с2 - х)2.
616
а изменение концентрации NaCI равно:
Со
X = ,
С! + 2с2
откуда следует, что низкомолекулярный электролит распределяется
по обе стороны мембраны неравномерно. Данное явление названо
эффектом Гиббса-Доннана.
При этом, если концентрация низкомолекулярного электролита
намного больше концентрации полимера (с2 >> с ! ), то х = с2/2,
т. е. малые ионы равномерно распределяются по обе стороны мем¬
браны. При обратном соотношении концентраций (с2 << Cj) изме¬
нение х = 0, а следовательно низкомолекулярный электролит через
мембрану практически не перемещается.
В первом случае, таким образом, эффект Гиббса-Доннана будет
проявляться в сильной степени, а именно этот случай и типичен
для биосистем. Осмотическое давление такого раствора будет скла¬
дываться из осмотического давления, вызванного присутствием
ВМС (онкотическое давление), и осмотического давления низкомо¬
лекулярного соединения.
Эффект Гиббса-Доннана важен для жизнедеятельности организ¬
мов, ведь биомембраны проницаемы для неорганических солей и
воды и непроницаемы для биополимеров Этот эффект - в числе
причин неравномерного распределения ионов вне и внутри клетки.
В частности, влияние доннановского равновесия заставляет би¬
карбонат-ионы, образующиеся в эритроцитах, диффундировать пре¬
имущественно в плазму. В эритроцитах концентрации НЬ" и НЬОг”
(они по преимуществу находятся в анионной форме, см. главу 9)
очень высоки. Поэтому проникающие из эритроцита в плазму анио¬
ны распределяются между эритроцитами и плазмой неравномерно.
В результате установления доннановского равновесия концентрации
НСОз", СГ и ОН"-ионов в плазме оказываются выше, чем в эритро¬
цитах Далее, должно выполняться такое соотношение-
[нсо3-]э = [СГ]Э = [он-]э
[нсо3-]п [СГ]П [ОН"]/
где индексы “э” и "п” обозначают эритроциты и плазму, соответ¬
ственно.
В капиллярных сосудах СОг диффундирует в эритроциты и пре¬
вращается в Н2СОз (см главу 17). Угольная кислота реагирует с
НЬ" с образованием бикарбонат-иона, что приводит к увеличению
617
отношения концентраций ^^3—[э_ Бикарбонат-ионы диффунди-
[НС03-]п
руют в плазму, а хлорид- и гидроксил-ионы проникают в клетки,
поддерживая электронейтральность, пока не восстановятся приве¬
денные выше равенства.
Описанное выше явление называют бикарбонатно-хлоридным
сдвигом
В легких процесс обращается. Здесь бикарбонат-ионы реаги-
|нсо3-],
руют с НЬОг, что приводит к снижению отношения 22—-
[нсо3-]п
Бикарбонат-ионы диффундируют из плазмы в эритроциты, а СГ и
ОН'-ионы двигаются в обратном направлении, пока отношения кон¬
центраций снова не выравняются.
32.3.3. Вязкость
Вязкостью или внутренним трением называют меру сопротивле¬
ния среды движению.
Измерение вязкости - самый простой и доступный способ из¬
учения свойств макромолекул. Он ценен тем, что позволяет опреде¬
лять молекулярную массу биополимеров (М).
И. Ньютон для ламинарного (послойного) течения жидкости об¬
наружил зависимость:
Р=л^1$1
г2-г1
где Г - тангенциальная касательная сила, вызывающая сдвиг слоев
жидкости относительно друг друга; Т| - коэффициент динамической
вязкости; ——— - градиент скорости течения (быстрота изменения
г2 -гх
скорости течения у2 - VI от слоя жидкости г2 к слою г\\ Б - пло¬
щадь слоя, по которому происходит сдвиг.
Коэффициент динамической вязкости или абсолютной вязкости
т| количественно характеризует сопротивление жидкости смещению
ее слоев. Измеряют Т1 в пуазах, г/см-с. Величина, обратная Т|,
Ф = 1/т|, называется текучестью. Зависимость Т| от действующей
силы Г носит сложный характер и может быть описана лишь при-
618
ближеныо, а экспериментальное измерение затруднительно, поэто¬
му удобнЬ пользоваться относительными вязкостями растворов.
Относительная вязкость (т|отн) выражается формулой:
где Т| и Т|0 - вязкости раствора и стандарта, в качестве которого
обычно используют чистый растворитель, а t и t0 - соответствую¬
щие времена истечения. Нахождение этой величины совсем не
сложно. Для этого надо сравнить время истечения чистого раство¬
рителя и раствора биополимера из специально подобранного для
этой цели капилляра.
Поскольку наличие молекул растворенного вещества, как пра¬
вило, нарушает ламинарное течение жидкостей, приводя к возрас¬
танию вязкости, относительная вязкость Т|отн обычно больше едини¬
цы. Вязкость раствора выражают несколькими величинами.
1) удельная вязкость: Т|уд = Т|отн - 1,
2) приведенная вязкость: Лпрнвед = — .
3) характеристическая вязкость: Т|хао = lim —,
с—»0 С
где с выражается в г/мл или г/100 мл.
Относительная вязкость является мерой изменения вязкости
раствора по сравнению с вязкостью чистого растворителя.
Удельная вязкость отражает возрастание относительной вязко¬
сти по сравнению с единицей. Чтобы учесть влияние концентрации
раствора, т.е. оценить, насколько велика удельная вязкость, отне¬
сенная к единице концентрации растворенного вещества, т|уд делят
на с. При этом получается Лпривед-
Кроме того, Лпрнвед сама зависит
от концентрации, так что прихо¬
дится вводить еще одну величи¬
ну - Т|хар (собственную или харак¬
теристическую вязкость), которую
получают путем экстраполяции,
показанной на рис 32.3.
Ни одна из обсужденных ве¬
личин не имеет размерности вяз¬
кости, так что следует соблюдать
известную осторожность при рас¬
четах с ними.
Рис. 32.3. Приведенная
и характеристическая вязкость.
619
Штаудингером было предложено соотношение между молеку¬
лярной массой и характеристической вязкостью несфериЧескнх мо¬
лекул В модифицированном позднее виде оно выглядит так.
Лхар = КМ“,
где К - постоянная, экспериментально определяемая для данных
макромолекул и растворителя
Показатель степени а зависит от формы макромолекул Для бел¬
ков компактной структуры, так называемых глобулярных белков,
форма которых близка к шарообразной, показатель а составляет
около 0.5. Таковым, к примеру, является миоглобин. Для молекул,
находящихся в конформации беспорядочного клубка, показатель
степени возрастает до 2. Примером таких белков может служить
актин, белок мышечных волокон, когда он находится в фибрилляр¬
ной форме, т.е. в виде волокон.
32.3.4. Набухание и застудневание
Набухание - явление проникновения растворителя в полимер¬
ное вещество, сопровождаемое увеличением его объема и массы.
Мерой набухания служит степень набухания (0^):
где т0 - начальная масса полимера; т - масса после поглощения
растворителя.
Можно определять степень набухания и сопоставлением объе¬
мов биополимера до и после набухания. Тогда мера набухания
выразится:
V-V0
°^=—г-®-.
где У0 - начальный объем; V - объем после поглощения раство¬
рителя.
Набухание - самопроизвольный процесс, а следовательно при¬
ращение энергии Гиббса здесь отрицательно.
Степень набухания полимера зависит от его природы и природы
растворителя.
Закономерности набухания определяются жесткостью полимер¬
ных цепей. Так, степень набухания у сетчатых и лестничных поли¬
620
меров с большим числом сшивок между цепями невелика. Таковы,
наприлф», эбониты - вулканизированные резины.
Желатин в холодной воде характеризуется ограниченным набу¬
ханием, однако в горячей воде набухает неограниченно. Ограничен¬
ное набухание приводит к образованию студней (см. ниже).
Полимер лучше набухает в растворителе, который сильнее вза¬
имодействует с его молекуламии. Поэтому полярные полимеры
(белки) набухают в полярных жидкостях (вода), неполярные
(каучуки) - в неполярных средах (бензол).
На набухание ВМС влияют также значение pH и присутствие
электролитов.
Влияние ионов на набухание ВМС связано с их способностью к
дегидратации молекул ВМС. Известно, что жесткие основания и
кислоты (см. главу 3) гидратируются сильнее, чем мягкие. Типич¬
ные жесткие основание и кислота - это фторид-анион и катион ли¬
тия. Иодид-ион и катион цезия - характерные примеры сравнитель¬
но мягких однозарядных ионов.
В соответствии с этим степень набухания падает под влиянием
ионов в соответствии с лиотропным рядом (ср. главу 15) :
Сэ+ < ЯЬ+ < К+ < < и+
СМБ" < Г < Вг" < N03" < С Г < СНзСОО" < Г < БО.,2"
Анионы, которые находятся правее в таком ряду, подавляют на¬
бухание, мешая образованию гидратной оболочки молекул ВМС.
Анионы же, находящиеся левее, гидратируются слабее. Они ослаб¬
ляют слабые взаимодействия между молекулами ВМС, а следова¬
тельно, способствуют набуханию с последующим растворением.
Те же ионы образуют ряды с обратным влиянием на способ¬
ность растворов ВМС к коагуляции (ср. главу 15).
Набуханию благоприятствуют также адсорбционные способ¬
ности ионов. Так, предварительная адсорбция желатином иодид- и
роданид-ионов приводит к легкому набуханию и далее - к растворе¬
нию белка.
Степень набухания белка а минимальна в изоэлектрической
точке, что может быть использовано для ее определения Если
добавлять к раствору ВМС органический растворитель, вызыва¬
ющий десольватацию молекул ВМС и тем самым - коагуляцию
(см главу 15), то последняя в большей степени идет при значениях
pH, близких к изоэлектрической точке Это свойство также можно
применять для нахождения р1. Зависимость степени набухания и
порога коагуляции у от р! представлена на рис. 32.4
621
Рис. 32.4. Влияние pH на набухание (1) и коагуляцию
желатина (2). Р1*елатииа 4.8.
32.3.5. Нарушение устойчивости растворов ВМС
Высаливание ВМС. Высаливание ВМС - выделение ВМС
из раствора при введении ионов или неэлектролитов. Лиотропные
ряды по влиянию ионов на набухание ВМС имеют обратную после¬
довательность по высаливающему эффекту. Наименьший выса¬
ливающий эффект проявляют мягкие основания-катионы, кото¬
рые слабо гидратируются и хорошо адсорбируются на молекулах
полимеров
Стабильность растворов ВМС падает с уменьшением лиофиль-
ности полимера. Лиофильность понижается при добавлении к вод¬
ному раствору ВМС растворителя, в котором полимер хуже раство¬
рим. Например, этанол высаливает желатин из воды.
Коацервация. Кроме высаливания при нарушении устойчи¬
вости раствора белка или полисахарида возможно образование коа-
цервата - новой жидкой фазы, обогащенной биополимером. Коацер-
ват может образовать сплошной слой (расслаивание) или выделить¬
ся в виде капель
Коацервация связана с понижением взаимной растворимости
компонентов раствора, что можно вызвать изменением как темпера¬
туры, так и состава раствора. С образованием коацерватов связы¬
вают процесс зарождения жизни (А. И. Опарин). В экспериментах
их используют как модели клетки и ее отдельных структур.
Коацервацию используют в фармацевтической практике. Лекар¬
ство измельчают в растворе полимера, а затем вызывают образова¬
ние мелких капель коацервата. Для этого охлаждают или изменяют
622
кислотность, частично испаряют растворитель или вводят высали-
ватель^Сапли оседают на поверхности капсулируемых частиц.
Этот прием называют микрокапсулированием. Микрокапсули-
рование способствует устойчивости и увеличению длительности
(пролонгации) действия лекарств.
32.3.6. Свойства студней
При ограниченном набухании ВМС или частичном испарении
растворителя из раствора ВМС образуются студни.
Студни - структурированные гомогенные системы, состоящие из
ВМС и низкомолекулярных жидкостей. Студни упруги, нетекучи,
способны сохранять форму. Это обусловлено существованием в
них пронизывающей весь объем пространственной сетки макромо¬
лекул, соединенных в отдельных “узлах” силами межмолекулярного
взаимодействия.
При старении студни теряют гомогенность. Это явление назы¬
вают синерезисом. Он сопровождается уплотнением простран¬
ственной структурной сетки и уменьшением объема студня за счет
выделения жидкой фазы. Примеры синерезиса - свертывание крови,
отделение сыворотки, явление свертывания скисшего молока и др.
Таким образом, студни отличает от гелей неспособность восстана¬
вливать свою структуру со временем после ее механического раз¬
рушения, то есть отсутствие тиксотропии.
Скорость диффузии ионов и молекул в студнях зависит от кон¬
центрации студня, что проявляется в своеобразии химических реак¬
ций, имеющих место в студнях
Р. Лизеганг, исследуя образование осадка бихромата серебра
действием АдМОз в студне желатина, пропитанного раствором
бихромата калия, наблюдал образование чередующихся колец
(называемых кольцами Лизеганга)' окрашенных - содержащих
бихромат серебра, и бесцветных, не содержащих этой соли. По ме¬
ре удаления от места введения реагента растет ширина неокрашен¬
ных колец, а интенсивность окраски падает
Сущность этих явлений заключается в том, что раствор нитрата
серебра диффундирует внутрь геля, где и образует осадок при вза¬
имодействии с К2СГ2О7 по уравнению:
К2СГ2О7 + 2АдМ03 —» АдгСггО/ + 2КМОэ
В зону выпадения осадка диффундирует К2СГ2О7 из нижележа¬
щего слоя, поэтому при дальнейшем движении АдМОз попадает в
зону с недостаточной концентрацией бихромат-иона и осадка не
623
образуется. Ниже К2СГ2О7 содержится уже в достаточном коли¬
честве, и там появляется вторая полоса осадка.
Имеется и другое объяснение происходящего образующийся
осадок движется вместе с диффундирующим веществом в виде кол¬
лоидного раствора Постепенно диффундирующий электролит на¬
капливается в среде до уровня порога коагуляции, после чего про¬
исходит коагуляция коллоидного раствора с образованием хорошо
заметного слоя осадка. Затем весь процесс повторяется.
Такие реакции называют периодическими Это явление имело
место при образовании слоистых минералов, таких, как агат и яш¬
ма. Подобным образом формируются и камни в почках. Периодиче¬
ские реакции наблюдаются и для других биологических явлений,
например, передачи нервных импульсов и генерации биоритмов.
Таким образом, растворы ВМС обладают набором специфических
свойств, которые позволяют рассматривать их как промежуточное
звено между истинными растворами и микрогетерогенными систе¬
мами (коллоидными растворами). Эта специфика позволяет опреде¬
лять ряд важнейших параметров биополимеров - их молекулярную
массу, изоэлектрическую точку, коллигативные свойства, а тем са¬
мым - понимать особенности функционирования биополимеров в
живых системах.
Специфика свойств таких систем во многом определяется свой¬
ствами поверхности раздела между дисперсной средой (мицеллой) и
дисперсионной средой (водой и растворенными в ней низкомолеку¬
лярными компонентами - электролитами, метаболитами и др. реа¬
гентами). Ведь биохимические реакции происходят на этой поверх¬
ности, величина свободной энергии которой (0$) не может не ока¬
зывать существенное воздействие на их ход. Количество адсорби¬
руемого и далее проникающего внутрь мицеллы биосубстрата, кото¬
рый метаболирует на активном центре фермента в составе мицеллы,
возрастает с ростом его поверхностной активности и уменьшается с
уменьшением величины поверхности мицеллы.
Поэтому поверхность раздела играет важную роль в средах,
являющихся лиофильными коллоидными системами, а адсорбцион¬
ная способность субстрата является существенным фактором,
влияющим на скорость биохимических реакций. Это находит свое
выражение в однотипной математической зависимости уравнения
адсорбции по Ленгмюру (глава 13) и уравнения ферментативной
кинетики Михаэлиса-Ментен (глава 5).
Свойства и величина поверхности раздела фаз зависят от приро¬
ды молекул биологических ПАВ, их количества, а также от формы
и размера мицелл.
624
Менсп,у тем, два последних фактора чрезвычайно чувствительны
к различным внешним воздействиям. Эти воздействия - изменения
pH, природы электролитов, растворенных в дисперсной водной сре¬
де, температуры и др Данные факторы зависят и от характера и
типа межмолекулярных взаимодействий между молекулами ВМС
в составе мицеллы и субстратом дисперсной фазьг ион-ионных,
ион-дипольных, гидрофобных, водородных связей и др. Это, в свою
очередь, в случае ферментов определяется набором функциональ¬
ных групп в их активных центрах и в их окружении. Заместители
должны быть так ориентированы в пространстве, чтобы происхо¬
дило узнавание определенного субстрата, который метаболирует на
данном ферменте.
Эти же факторы зависят от того, в каком состоянии находится
система - в виде истинного раствора или мицеллы гидрофильного
коллоидного раствора. Между тем, лиофильные коллоидные раство¬
ры - весьма лабильные системы. Величина активационного барьера
перехода истинный раствор - коллоидный раствор не превосходит
нескольких кДж/моль, что меньше, чем большинство перечислен¬
ных выше разновидностей межмолекулярных взаимодействий. Сле¬
довательно, даже при наличии слабых воздействий может осущест¬
вляться легкий переход системы из одного состояния в другое, что
должно принципиально сказываться на скорости и характере тех
или иных метаболических процессов
Часть VI
МЕТАЛЛЫ В ЖИВЫХ СИСТЕМАХ
. .прибавленные к углеродис¬
тым соединениям минеральные
вещества понижают темпера¬
туру реагирования и вообще об¬
легчают химические реакции и
тем содействуют превращению
простейших питательных ве¬
ществ в сложные составные
части организма
Д И Менделеев
В живых организмах обнаружено свыше 60 элементов Периоди¬
ческой системы. Большая часть из них относится к металлам и
в клетках встречается в виде разнообразных комплексных соедине¬
ний с биолигандами. Установлено, что даже те металлы, содержа¬
ние которых невелико (так называемые микроэлементы), не явля¬
ются случайными примесями, а выполняют определенную биологи¬
ческую роль.
Известно множество патологических нарушений, связанных с
недостаточностью в клетке железа, меди, цинка, марганца, молиб¬
дена, кобальта, не говоря уже о более распространенных в живых
тканях катионах кальция, магния, натрия и калия (последние четы¬
ре элемента составляют около 99% от общего содержания биоме¬
таллов).
Исследования биологических процессов, в которых участвуют
ионы различных металлов, представляют сравнительно новую, бы¬
стро развивающуюся область науки, называемую бионеоргани-
ческой химией.
Находясь на стыке двух наук - биохимии и неорганической хи¬
мии, бионеорганическая химия использует методы исследова¬
ния, заимствованные, с одной стороны, из арсенала энзимологии, а
с другой, - из химии координационных соединений. Другими слова¬
ми, бионеорганическую химию можно определить как приложение
принципов координационной химии металлов к биологическим
проблемам.
В живых организмах большинство катионов металлов связано с
белками, и именно в изучении металлзависимых ферментов
(комплекс металл-белок) в последнее время достигнут наибольший
прогресс.
626
Гл^ва 33. БИОМЕТАЛЛЫ И БИОЛИГАНДЫ
33.1. Общие сведения
Как известно, ферменты, благодаря их особой трехмерной
структуре, могут работать как высокоспецифические катализаторы с
огромным каталитическим эффектом. Они увеличивают константы
скорости биопроцессов по сравнению с модельными более чем в
1012 раз. Обычно фермент - это комплекс иона металла с белком.
Ионы металлов в белках и ферментах выполняют ряд каталити¬
ческих и структурных функций. О структурной роли ионов метал¬
лов в биологических системах свидетельствует существование фер¬
ментов, в которых ионы металла непосредственно не участвуют в
каталитическом акте, но являются необходимыми для выполнения
этими ферментами их функции. Таким образом, ионы металлов в
белках и ферментах можно разделить на два класса - “химические”
и “структурные" металлы.
“Химические” металлы принимают непосредственное участие в
реакции, как, например, ион железа в цитохромах или гемоглобине.
“Структурные” металлы стабилизируют пространственную форму
фермента, необходимую для выполнения его функции, как, напри¬
мер, кальций в термолизине (таблица 33.1).
Другая классификация металлоферментов основана на проч¬
ности связи между ионом металла и макромолекулой белка. Все
системы металл-белок можно условно разделить на две группы:
- металлопротеины, в которых ион металла настолько прочно
связан с молекулой белка, что его можно рассматривать как со¬
ставную часть белковой структуры. Извлечь такой ион металла
можно лишь сильным химическим воздействием, причем тогда ме¬
таллопротеины теряют свою активность. Потеря активности проис¬
ходит также при замене исходного металла на другой.
- белки, активизируемые металлами В эту группу входят
те соединения, в которых ион металла связан с белком обратимо
Связь металл-белок в таких системах гораздо слабее, чем в систе¬
мах первого типа, однако именно она определяет механизм реакций,
идущих с их участием
В табл. 33.1 приведены некоторые биометаллы, выполняющие
важные биохимические функции Видно, что в природе с белками
связано относительно небольшое число металлов Если с приведен¬
ными в таблице рассматривать также ферменты, активируемые ме¬
таллами, то к этому списку элементов следует добавить лишь ка¬
лий, натрий и магний
21*
627
Таблица 33 I
Ионы металлов в металлоферментах
Металл
Белок
Функции
Ре
Гемэритрин
Гемоглобин
Миоглобин
Клетки крови
морских червей
Эритроциты
млекопитающих
Мышцы
млекопитающих
Запасание и (или)
перенос кислорода
Цитохром
Ткани животных
и растений
Перенос электрона
при дыхании
митохондрий
Каталаза
Печень,
эритроциты
Диспропорционирование
Н2О2 до НгО И Ог
Мо
Нитрогеназа
Аго1оЬас1ег
Фиксация азота
Мо, Ре
Ксантин-
оксидаза
Молоко
Метаболизм
пуриновых оснований
в мочевую кислоту
Ре, Си
Цитохром-
оксидаза
Ткани
животных
Перенос электрона
при восстановлении
кислорода
Си
Гемоцианин
Кровь моллюсков
Перенос кислорода
Карбокси-
пептидаза
Поджелудочная
железа
Гидролиз белков
Алкоголь-
дегидрогеназа
Печень
Метаболизм и
окисление этанола
Инсулин
Р-Клетки подже¬
лудочной железы
Метаболизм глюкозы
Термолизин
В. Игегторго-
1ео1у1'1СЫ5
гп - гидролиз белков,
Са - термостойкость
1п. Са
а-Амилаза
Слюна
гп -связь в димерной
макромолекуле фермента,
Са - гидролиз углеводов
Мп
Пируват-
карбоксилаза
Печень
Образование
пировиноградой кислоты
Со
Метионин-
синтетаза
Микроорганизмы
Биосинтез метионина,
перенос метильной группы
628
Принято считать, что ионы металлов в металлопротеинах несут
двойнукЕ^функцию. Первая из них - матричный (или ориентацион¬
ный) эффект, вторая - концентрирование биосубстрата на участке
протекания реакции.
Биологическая роль иона металла характеризуется высокой спе¬
цифичностью, однако в зависимости от типа белка один и тот же
ион осуществляет разные функции. Из этого можно сделать есте¬
ственный вывод о том, что биологическая специфичность функции
металла имеет, по-видимому, стереохимическую природу, т.е. про¬
странственное строение фермента (третичная структура) определя¬
ет его активность.
Кроме белков к биолигандам следует также отнести молекулы
воды, кислорода, азота, порфирина, коррина и нуклеиновых кислот,
цианид- и фторид-ионы.
Взяв за основу атом металла, имеющийся в соединении, коротко
рассмотрим строение и функции некоторых биокомплексов.
33.2. Железосодержащие комплексы
Представители этого класса биокомплексов ответственны за
транспорт и хранение кислорода (гемоглобин и миоглобин), элек¬
тронный перенос в цитохромах, катализ окисления кислородом и
пероксидом водорода (оксидазы и пероксидазы), катализ разложе¬
ния пероксида водорода (каталазы). Широкий спектр функций, ко¬
торый могут выполнять железосодержащие комплексы, еще раз
подчеркивает, насколько влияние белка может разнообразить
свойства явно близких по строению соединений
Интересно, что во всех указанных ферментах атом железа в
степени окисления +2 находится в центре плоской порфириновой
системы, образуя гем. Порфирины - высокосопряженные молекулы,
содержащие четыре пиррольных кольца и удовлетворяющие усло¬
вию ароматичности (см. правило Хюккеля, глава 18). Все восемь
атомов углерода пиррольных колец являются замещенными. Замес¬
тители в порфириновом кольце оказывают большое влияние на
свойства порфириновой системы
В молекуле порфирина два протона, связанные с азотом, могут
быть легко заменены на атом металла Еще две связи двух остав¬
шихся атомов азота с атомом железа образуются по донорно-
акцепторному механизму Пример гема с типичным набором замес¬
тителей приведен ниже
Гемопротеины образуют октаэдрические комплексы (координа¬
ционное число 6). Пятое положение в координационной сфере за¬
629
нято атомом азота аминокислоты (гистидил) в полипептидной цепи
Шестое положение в гемоглобине и миоглобине (дезоксиформы*)
занято молекулой воды. Гем, полипептидная цепь и шестой лиганд
образуют так называемую субъединицу Важные физиологические и
химические свойства гемопротеинов связаны с быстрым и обрати¬
мым присоединением соответствующих лигандов по шестому коор¬
динационному месту.
33.2.1. Гемоглобин и миоглобин
Гемоглобин - это белок с молекулярной массой 64 450. Он со¬
стоит из двух идентичных пар субъединиц, расположенных прибли¬
зительно в форме тетраэдра. Миоглобин является более простой
молекулой с молекулярной массой 17 500, напоминающей мономер¬
ную единицу гемоглобина.
Гемоглобин выполняет две биологические функции:
1 Он связывает молекулы кислорода своими атомами железа и
переносит из легких к мышцам, где кислород передается молекулам
миоглобина.
2. С помощью концевых аминогрупп связывает несколько мета¬
болических молекул углекислого газа и переносит их в легкие.
Интересно сравнить сродство к кислороду гемоглобина (НЬ) и
миоглобина (МЬ). Для миоглобина имеется простое равновесие:
МЬ + 02 5 МЬ02.
Константа равновесия имеет вид:
к= [МЬ 021
[МЬ][02]
* Дезоксиформы не содержат кислорода в качестве лиганда
630
Вд^дем величину I - степень насыщения:
{= [мьо2]
[МЬ 02] + [МЬ] ’
г.1 [мь о2]
из константы равновесия [МЬ1 = —и, следовательно,
К[02]
, _ [021 К102|
102]+1 1 + кг°2]
Так как активная концентрация газообразного кислорода равна
его парциальному давлению (р), то
Это - уравнение гиперболы, которое превосходно описывает
связывание кислорода миоглобином (рис. 33.1).
Гемоглобин с его четырьмя субъединицами ведет себя значи¬
тельно сложнее, что приблизительно можно описать уравнением
Хилла:
У- К-р"
1 + Кр" ’
где п в нормальных физиологических условиях составляет примерно
2.8 и называется константой Хилла.
Кривая насыщения кислородом гемоглобина имеет Б-образную
форму, а тот факт, что константа Хилла больше единицы, с хими¬
ческой точки зрения означает следующее: присоединение первой
молекулы кислорода к одному из четырех гемов увеличивает кон¬
станту связывания для следующей молекулы кислорода, что, в
свою очередь, приводит к повышению константы для следующей и
т.д. Это явление называется кооперативным эффектом и связа¬
но, очевидно, с конформационными изменениями гемоглобина при
последовательном присоединении молекул кислорода
Анализ кривых насыщения, представленных на рис 33.1, пока¬
зывает, что если при высоких давлениях кислорода гемоглобин
связывает его так же хорошо, как и миоглобин, то при низкнх дав¬
лениях сродство гемоглобина к кислороду существенно меньше.
Поэтому гемоглобин передает кислород миоглобину в мышцах Бо¬
лее того, метаболическая СОг в мышечной ткани уменьшает вели-
631
чину pH, что еще больше увеличивает способность гемоглобина от¬
давать кислород (эффект Бора, правая кривая на рис 33ч])
парциальное давление
в мышцах в легких
Насыщение, % Т т
МЬ,
/нь /
/рН=7.бХ
Унь
>^Н=6.8
40
100
Парциальное давление кислорода, мм рт. ст.
Рис 33.1. Кривые насыщения кислородом миоглобина (МЬ)
и гемоглобина (НЬ).
Механизм кооперативного связывания кислорода и эффекта
Бора сложен и до конца не установлен Экспериментами было
установлено, что координация кислорода с атомом железа сопро¬
вождается изменением структуры гема.
а б
Рис. 33.2. Структура гема в дезоксигемоглобине (а) и
в оксигемоглобине (б).
Если в оксигемоглобине железо находится в плоскости порфи-
ринового кольца, то в дезоксигемоглобине оно выходит из плос¬
кости на 0.7-0.8 А, образуя конфигурацию квадратной пирамиды
(рис. 33.2).
632
Следует указать на важную роль белковой части молекулы. До-
статоИ^ сказать, что гем, содержащий Ре(Н) и не связанный с по-
липептидной цепью, не способен обратимо связываться с кислоро¬
дом и вместо этого окисляется до Ре(Ш)-содержащего комплекса,
который уже не взаимодействует с кислородом. Некоторые вещест¬
ва, например, нитриты, нитросоединения, способствуют переходу
Ре(Н) —» Ре(Ш) в гемоглобине, который при этом теряет способ¬
ность к переносу кислорода (метгемоглобин).
Помимо воды и кислорода, шестое координационное место в ге¬
моглобине и миоглобине могут занимать и другие лиганды, напри¬
мер, молекула СО (карбокси-формы), цианид-ион Высокая токсич¬
ность оксида углерода (И) обусловлена его большим сродством к
атому железа по сравнению с молекулой кислорода. Это объясняет¬
ся повышением степени двоесвязанности между железом и лиган¬
дом за счет дативной связи:
/М х-ч N N
НЫум-Де-^О — нкОм-Уе=С=0
НЬ + 02^НЬ02 (V,) НЬ + СО ^ НЬСО (У2)
оксигемоглобин карбоксигемоглобин
Так как К?*??» 3500К”*??, а У2 = 250УЬ то вдыхание
воздуха, содержащего угарный газ, приводит к его накоплению
в крови. Так, при содержании последнего в количестве 0.07%
половина имеющегося гемоглобина переходит в карбокси-форму
([НЬ'02] крови = [НЬСО] крови). Другими словами, равновесие
НЬ02 + СО 5 НЬСО + 02
[НЬ'С0][0 2 ]
сдвинуто вправо, а константа равновесия Кр = , назы-
[НЬ02][С0]
ваемая коэффициентом отравления, для человека лежит в ин¬
тервале 200+280 Отсюда понятны методы антидотной терапии при
отравлении угарным газом вдыхание чист го кислорода (смещение
равновесия влево) и внутривенные инъекции сахарата двухвалент¬
ного железа’ (конкурентное связывание молекул СО)
Соль глюкаровой кислоты
633
33.2.2. Цитохромы
Цитохромы - широко распространенная группа внутриклеточ¬
ных окислительно-восстановительных катализаторов Все клетки
содержат по крайней мере три близких по строению белка, назван¬
ных цитохромами а, в, и с Цитохромами называются гемсодер-
жащие белки, принцип действия которых состоит в переносе элек¬
тронов в результате обратимого изменения валентности атома же¬
леза в геме
Наиболее изучены цитохромы с, у которых гем связан с белком
ковалентно через боковую цепь порфирина Все цитохромы с мле¬
копитающих содержат одну группу гема и полипептидную цепь Гем
является железным комплексом порфирина, а пятое и шестое коор¬
динационные места заняты гистидиновым и метиониновым остатка¬
ми полипептидной цепи:
Прочная связь катиона железа с серой метионина предотвра¬
щает связывание кислорода гемом. Цитохром с из сердца лошади с
молекулярной массой около 12 400 содержит в белковой цепи 104
аминокислотных остатка. Пептидная цепь цитохрома с так “обмо¬
тана" вокруг гема, что образуется гемовая полость (карман). Гем
окружен гидрофобными боковыми цепями, в то время как полярные
боковые цепи находятся снаружи молекулы. Особенностью строе¬
ния цитохрома с является наличие “двух каналов”, которые ведут с
поверхности молекулы внутрь, к гемогруппе и пространственно
приспособлены для входа и выхода электронов.
Перенос электрона цитохромом с сопровождается изменением
степени окисления железа:
Интересно, что при переходах железа между состояниями Ре(Н)
и Ре(Ш) не наблюдается каких-либо конформационных изменений
в биокомплексе, что исключает изменение характеристик связи
металл-лиганд в ходе реакции.
белок
белок-Ре2+ + - Ог - е ^ белок-Ре3+ + - Ог~
2 2
и далее - 022~ + 2Н+ -* Н20.
2
634
Было замечено, что при отравлениях цианидами венозная кровь
приобретает алый цвет, т.е. цвет артериальной крови, насыщенной
кислородом. Значит, под влиянием цианид-иона организм теряет
способность усваивать кислород. Механизм токсического действия
в данном случае объясняется образованием прочного комплекса
иона СМ' с Ре3+ в окисленной форме цитохрома. Модификация
фермента делает его неактивным, становится невозможным пере¬
нос электрона на кислород, нарушается процесс клеточного дыха¬
ния, и быстро формируется патологическое состояние тканевой
гипоксии.
Интересно, что одним из методов антидотной терапии является
применение метгемоглобинобразователей (нитритов, нитросоедине¬
ний). Последние переводят часть гемоглобина крови в метгемогло-
бин, который конкурирует с цитохромом за цианид-ион.
НЬ(Ре2+) ———2—» МШЬ(Ре3+)
белок(Ре3+)СМ + МШЬ(Ре3+) 5 МШЬ(Ре3+)СМ + белок(Ре3+)
ингибированный активный
цитохром цитохром
Таким образом, искусственно создавая кровяную гипоксию, бо¬
рются с тканевой гипоксией. Это уникальный случай, когда ядови¬
тые нитриты выступают как противоядие.
В последние годы интенсивно изучается группа так называемых
цитохромов Р450- Это железогемсодержащие белки с молекуляр¬
ной массой 45 ООО - 55 ООО, выделенные из печени человека и жи¬
вотных. Кроме того, они входят в состав клеток многих бактерий,
дрожжей, грибов и некоторых высших растений. Специфической
особенностью этой группы ферментов является активация молеку¬
лярного кислорода и включение одного из его атомов в органиче¬
ские соединения. Это чрезвычайно важный процесс, который опре¬
деляет не только биосинтез и метаболизм гормонов, но и инактиви¬
рует чужеродные химические агенты (ксенобиотики).
33.2.3. Каталаза и пероксидаза
Перекись водорода и супероксид-анион-радикал Ог-" - химически
активные и токсичные вещества, которые могут образовываться при
окислении многих органических соединений в водной среде in VIVO
Образование атмосферы, содержащей кислород, и развитие орга¬
низмов с аэробным типом обмена потребовало возникновения
635
ферментов, которые могли бы возможно эффективнее удалять пере¬
кись водорода и 02'.
Возможны два пути удаления перекиси водорода, а именно: ре¬
акции диспропорционирования 2Н202 -» 2Н20 + Ог и восстановления
Н202 + red -» 2Н20, причем восстановителем может быть чисто орга¬
ническое соединение, иодид-ионы и пр.
Существует несколько типов ферментов, которые катализируют
преимущественно или диспропорционирование (каталазы), или вос¬
становление (пероксидазы) Они широко распространены в живот¬
ных и растительных микроорганизмах. Точная физиологическая
роль их не установлена, и кроме удаления перекиси водорода, они
выполняют, по-видимому, и какую-то другую, дополнительную
функцию.
Пероксидаза представлена одной субъединицей с молекулярной
массой около 40 ООО. Кроме белка и гем-группы с атомом Fe(III) в
состав пероксидазы входит также углевод, который, вероятно,
представляет собой уроновую кислоту
Шестое координационное место могут занимать различные ли¬
ганды- Н20, CN" и т.д По-видимому, обмен лигандов в этом шестом
положении имеет существенное значение для функционирования
пероксидазы.
Каталаза имеет молекулярную массу около 240 ООО и сформи¬
рована из четырех одинаковых субъединиц, каждая нз которых со¬
держит один гем с железом(Ш) в качестве центрального атома.
Пятым лигандом является аминокислотный остаток полипеп-
тидной цепи, а шестым - вода.
33.2.4. Негемовые железосодержащие белки
Существуют железосодержащие белки, которые не содержат
порфирина В них атомы железа связаны обычно с атомами серы
полипептидной цепи.
Гемэритрин был обнаружен в эритроцитах некоторых червей,
он выполняет роль запасания кислорода. С этой его ролью согла¬
суется небольшой кооперативный эффект связывания кислорода
(константа Хилла равна 1.2-1.4). Гемэритрин имеет молекулярную
массу около 107 ООО и состоит из восьми одинаковых полипеп-
тидных цепей, каждая из которых содержит два атома железа.
Обратимое связывание молекулярного кислорода гемэритрином
является его физиологической функцией, а главное отличие этого
636
соединения от гемоглобина заключается в стехиометрии реакции.
Для 1%го чтобы связать одну молекулу кислорода в гемэритрине,
требуются два атома железа, а гемоглобину достаточно одного.
Первичная структура гемэритрина известна. Полипептидная
цепь состоит из 113 аминокислот и может быть связана с атомами
Ре3+ остатками тирозина, метионина, глутаминовой или аспараги¬
новой кислот. Различные состояния гемэритрина представлены в
таблице.
Таблица 33.2
Различные состояния гемэритрина
Комплекс
Лиганд
Степень окислення железа
Оксигемэритрин
02
+3
Метгемэритрин
БСМ-, СҐ, Н20
+3
Дезоксигемэритрин
Н20
+2
При длительном хранении оксигемэритрин медленно переходит
в метгемэритрин, координирующий самые разнообразные лиганды и
теряющий способность связывать кислород.
Накопление и транспорт железа. В организме взрослого че¬
ловека около 65% всего железа содержится в гемоглобине и
миоглобине, большая часть оставшегося запасается в специальных
белках (ферритине и гемосидерине), и только очень небольшая
часть находится в различных ферментах и системах транспорта.
Трансферрины - класс железосвязывающих молекул, в кото¬
рый входят лактоферрин (из молока), кональбумин или овотранс-
феррин (из яичного белка) и сывороточный трансферрин Это тран¬
спортные белки, переносящие железо из обломков гемоглобина
клеток селезенки и печени в костный мозг, где на специальных его
участках вновь синтезируется гемоглобин
Весь сывороточный трансферрин человека, единовременно свя¬
зывая лишь приблизительно 4 мг железа, ежедневно переносит в
костный мозг около 40 мг железа Больные с генетическими нару¬
шениями синтеза трансферрина страдают одновременно железоде¬
фицитной анемией и интоксикацией от избытка железа.
Трансферрин - белок с молекулярной массой 80 ООО, состоит из
одной полипептидной цепи, свернутой так, что она образует два
участка, каждый из которых способен связать один ион Ре(Ш)
Высокая устойчивость комплекса железа с трансферрином де¬
лает его отличным переносчиком, но зато возникает проблема вы¬
637
свобождения железа из комплекса. Механизм этого процесса не
установлен.
Ферритии и гемосидерин - белки, служащие для Запасания
железа. Гемосидерин является малоизученным и, возможно, пред¬
ставляет собой продукт распада ферритина. Последний состоит из
двадцати четырех полипептидиых цепей, образующих пустотелую
сферическую оболочку с внешним диаметром -125 А и диаметром
полости -70 А. Во внутренней полости находится мицеллярное ядро
из неорганического комплексного оксогидроксофосфата железа(Ш)
состава (РеООН)8 (РеООРОзНг). Содержание железа в мицелле
-57% (до 4 500 атомов железа), если ферритин полностью насы¬
щен Белковую оболочку пронизывают шесть каналов, служащих
для приема и отдачи железа Прием железа происходит при катали¬
тическом окислении Ре(Н) -> Ре(Ш), а высвобождение - при вос¬
становлении до Ре(П)
Большинство микроорганизмов имеют весьма эффективные
транспортные системы для принятия железа в нужных количествах
Железо вне организма существует в виде нерастворимых при фи¬
зиологических величинах pH соединений Ре(Ш).
В ответ на нехватку железа в клетке организм синтезирует и
выделяет специальные лиганды, высокоспецифичные для Ре(Ш). Это
сидерофоры или железонесущие лигаиды. Все они используют
для связывания железа донорные атомы кислорода и дают октаэд¬
рический комплекс. Сидерофоры разделяются на две группы: гид-
роксаматные и фенолятные, в зависимости от того, содержат ли они
в своем составе фрагменты гидроксамовой кислоты (а) или пиро¬
катехина (б).
а б
гидроксамовые производные
кислоты пирокатехина
У большинства природных гидроксамовых кислот на одну моле¬
кулу приходится три кислотных группировки, которые дают с Ре(Ш)
нейтральные комплексы. Феррихромы представляют собой цикли¬
ческие пептиды, а ферриоксамины могут иметь либо линейную,
либо циклическую структуру. Пирокатехиновые сидерофоры обычно
находят в бактериях, а гидроксаматные - в более высокоорганизо¬
ванных организмах.
638
Отдача железа сидерофором происходит вследствие его восста¬
новлен^ до Ре(И) физиологическими восстановителями, причем
молекула лиганда либо расщепляется, либо остается неповрежден¬
ной и может быть использована вновь.
33.2.5. Железо-серусодержащие белки
К этому классу относятся такие белки, в которых железо не
связано с порфириновым кольцом и в состав которых входят два
типа серусодержащих комплексообразующих групп: сульфгидриль-
ная группа аминокислоты цистеина (-SH) и так называемая
“лабильная сера”, природа которой не установлена.
Количество обнаруженных железо-серусодержащих белков
стремительно растет, а интерес исследователей к ним определяется
большим разнообразием выполняемых биологических функций. Они
участвуют в окислительно-восстановительных процессах во всех из¬
вестных нам формах жизни, осуществляют перенос электрона при
фотосинтезе, фиксации азота, метаболических превращениях.
Железо-серусодержащие белки обозначают n-Fe-S белками (где п -
количество атомов железа в составе белка). Лабильную серу обо¬
значают звездочкой.
Рубредоксин, выделенный из бактерии Clostridium paste-
uranium, относится к 1-Fe-S типу. Его физиологическая функция не
ясна, однако известно, что он служит переносчиком электронов
Белки l-Fe-S-типа содержат от 51 до 60 аминокислотных остатков,
что соответствует молекулярной массе около 6000-7000.
остатки
639
Во всех этих белках имеет¬
ся четыре остатка йистеина,
которые и связывают атомы
железа, образуя тетраэдриче¬
ский комплекс (рис 33 3) При
подкислении из каждого моля
белка можно извлечь один моль
железа, причем сера не выде¬
ляется, что свидетельствует об
отсутствии лабильной серы
Ферредоксин относится к
белкам типа в-Ре-Б’ и обес¬
печивает передачу электронов
между окислительной и восстановительной цепочками метаболизма
в ряде анаэробных и фотосинтезирующих бактерий. Его биохимиче¬
ские функции многообразны, среди них участие в фиксации азота,
окислении уксусного альдегида до ацетата, формиата до углекислого
газа, восстановлении протонов до водорода, гидроксиламина до
аммиака и пр.
Таблица 33 3
Характеристики некоторых железо-серусодержащих белков
Тип
белка
Название
Происхож¬
дение
М
Функции
1-Ре-Б
Рубредоксин
Анаэробные
бактерии
6 380
Не известны, но может
заменять ферредоксины
Ферредоксин
хлоропластов
Растения
10 650
Перенос электронов
при фотосинтезе
г-Ре-э
Адрено-
доксин
Кора надпо¬
чечников
13 090
Перенос электронов
при гидоксилировании
стероидов
Ферредоксин
Микрококки
6 200
Восстановитель
при фиксации азота,
окислитель при
расщеплении фосфатов
в-Ре-Б
Ксантин-
оксидаза
Молоко
250 000
Окисление ксантина и
др пуринов, а также
альдегидов кислородом
Альдегид-
оксидаза
Печень
280 000
Окисление ароматиче¬
ских и алифатических
альдегидов
Рис. 33.4. “Клетка" в структуре
Ре-Б белков.
640
Ст|>уктурной особенностью большинства Ре-Б белков явля¬
ется своеобразная группировка атомов серы и железа в виде
“клетки”, связанной с цистеиновыми остатками полипептидной це¬
пи (рис. 33.4). Некоторые примеры железо-серусодержащих белков
приведены в таблице 33.3.
33.3. Цинксодержащие комплексы
Известно свыше шестидесяти цинксодержащих биологически
активных веществ, среди которых наиболее изученными являются
широко распространенные в живых организмах ферменты карбоан-
гидраза и карбоксипептидаза.
Карбоангидраза - металлофермент с молекулярной массой
30 ООО, содержащий один ион Тп2+ на молекулу. Он широко рас¬
пространен в растениях, животных и бактериях, а физиологическая
роль его заключается в быстрой гидратации метаболической С02,
образующейся в тканях, дегидратации Н2СО3 в легких, а также пе¬
реносе и накоплении Н+ и НСОз" в органах секреции.
а) Н20 + С02 ^ Н2СОз ^ Н+ + НСОз'
б) ОН" + С02 ^ НСОз'
Реакция связывания двуокиси углерода может быть записана с
водой (а) или с гидроксил-ионом (б) в качестве активных частиц.
Окончательно этот вопрос пока не решен, но имеются вполне убе¬
дительные данные в пользу второго (б) механизма.
Известны две формы этого фермента - карбоангидраза В (КАВ)
и карбоангидраза С человека (КАС), различающиеся по аминокис¬
лотному составу и активности. Карбоангидраза С существенно бо¬
лее эффективна, и число оборотов этого фермента прн гидратации
С02 составляет б 105 сек1, что позволяет считать эту реакцию са¬
мым быстрым из всех известных фермента¬
тивных процессов
Данные рентгеноструктурного анализа
показывают, что тетраэдрический ион гп2+
находится примерно в центре скрученной
молекулы Три координационных места за¬
няты атомами азота гистидиновых остатков
полипептидной цепи, а четвертым лигандом
является гидроксильная группа
о=с=о
;
"О;
641
Механизм обратимой гидратации СОг до конца не ясен, а пред¬
ложенные гипотезы обладают определенными недостатками.
Карбоксипептидаза (молекулярная масса 34 300) представля¬
ет собой фермент, содержащий 307 аминокислот в единственной
полипептидной цепи, которая прочно связывает один ион 2п2+ Два
координационных места у атома цинка заняты гистидиновыми ато¬
мами азота, третье - кислородом карбоксильной группы глутамино¬
вой кислоты в белковой молекуле, а четвертым лигандом является
молекула воды.
Этот фермент поджелудочной железы млекопитающих катали¬
зирует гидролиз пептидной связи на карбоксильном конце белко¬
вой цепи.
Другим примером участия цинксодержащих металлоферментов
в гидролизе пептидов является термолизин, выделенный из
В. 1Иегторго1ео1уИсиз. Он, как и карбоксипептидаза, имеет молеку¬
лярную массу 34 600 и содержит один ион 2п2*, лигандами у кото¬
рого являются остаток аминокислоты глутамина, два остатка гисти¬
дина и молекула воды. Интересно, что эти бактерии могут функ¬
ционировать короткое время при высокой температуре, что обус¬
ловлено присутствием в комплексе четырех ионов кальция, предот¬
вращающие раскручивание, а, следовательно, денатурацию макро¬
молекулы белка.
К цинксодержащим биокомплексам относятся также фруктозо-
1,6-дифосфатаза, р-лактамаза, геморрагический токсин из яда гре¬
мучей змеи и др. (см.таблицу 33.1).
33.4. Медьсодержащие белки
Медь входит в качестве необходимого элемента в различные
ферменты, участвующие в биологических процессах от переноса
электронов до окисления различных субстратов. Физиологические
функции некоторых из них до сих пор не известны.
Некоторые представители медьсодержащих белков представлены
в таблице 33.4
Получены данные, свидетельствующие о влиянии содержания
меди в организме на развитие нормальных вкусовых ощущений, на
рост костных и соединительных тканей.
Физиологические последствия дефицита меди проявляются
только у животных; для человека такие случаи не известны, так как
в пище человека медь содержится в большом избытке.
642
-ч
Медьсодержащие белки
Таблица 33.4
Белок
М
Число молей Си
на моль белка
Функция
Пластоцианин
10 500
1
Перенос
электронов
Лакказа
60 000-
4
Восстановление
120 000
кислорода до воды
Галактозооксидаза
68 000
1
Окисление
галактозы
Цитохром-с-
100 000
2+2Ре
Перенос
оксидаза
электронов
Супероксид¬
32 000
2+22п
Диспропорцио-
дисмутаза
нирование 02"
Гемоцианин
25 000-
2
Перенос
(моллюски и
35 000
электронов
членистоногие)
Несмотря на поступления большого количества меди в организм
человека с пищей, концентрация этого металла в тканях здорового
человека поддерживается строго постоянной. Следовательно, в ор¬
ганизме должны существовать какие-то системы, препятствующие
непрерывному накоплению меди в тканях путем ее адсорбции или
стимуляции ее выведения при физиологических условиях. Меха¬
низм этого процесса пока не известен. Однако известно, что по¬
ступление катиона меди в ткани осуществляется с участием медно¬
го комплекса с трипептидом глицил-гистидил-лизином, являющимся
универсальным фактором клеточного роста, который по-видимому,
образуется в плазме.
Широкий спектр физиологического действия медьсодержащих
ферментов можно продемонстрировать на примере супероксиддис-
мутазы и гемоцианина.
Супероксиддисмутаза - фермент, выполняющий важную за¬
щитную функцию, а именно, катализирующий диспропорционирова-
ние токсичного супероксид-анион радикала на кислород и пероксид
водорода-
202 - + 2Н+->Н202+02
Образовавшийся при этом пероксид водорода разлагается затем
ферментом каталазой. Таким образом, эти два фермента защищают
клетки от токсичных побочных продуктов кислородного дыхания
643
Супероксиддисмутаза выделена в очищенной форме из широкого
круга микроорганизмов, растений и животных, причем она содер¬
жит медь и цинк. В некоторых случаях она может содержать желе¬
зо и цинк или марганец и цинк.
Наиболее изученной является супероксиддисмутаза из эритро¬
цитов быка. Она имеет молекулярную массу 31 400 и состоит из
двух одинаковых субъединиц, каждая из которых содержит 151
аминокислотный остаток, один атом меди и один - цинка. Геомет¬
рия окружения меди - искаженная плоскоквадратная с четырьмя
гистидиновыми лигандами. Ион цинка находится в тетраэдрическом
окружении с тремя азотными донорами гистидина и одним кисло¬
родным донором - аспарагином Интересно, что одно гистидиновое
звено является общим для обоих металлов лигандом.
Ион 1п2+ выполняет роль по организации и стабилизации ак¬
тивного центра в ферментах, поэтому супероксид-анион-радикал,
по-видимому, взаимодействует с катионом меди, претерпевая сле¬
дующие превращения (Е - фермент):
ЕСи(И) + 02-->ЕСи(1) + 02
Е Си(1) + 02 - + 2Н+ -> Е Си(11) + Н202
Гемоцианин (что в переводе с греческого означает “синяя
кровь”) - медьсодержащий белок, выполняющий функцию тран¬
спортировки кислорода у моллюсков и членистоногих. В зависи¬
мости от источника молекулярная масса гемоцианина составляет от
25 ООО до 37 ООО, а содержание меди 0.25% и 0.17%, соответ¬
ственно.
Было установлено, что активные центры гемоцианина, связы¬
вающие кислород, содержат по два эквивалентных и независимых
друг от друга атома меди. В дезоксиформе гемоцианина медь нахо¬
дится в степени окисления +1, а в оксигенированной форме пере¬
ходит в Си(П).
Для гемоцианинов так же, как и для гемоглобинов, характерен
кооперативный эффект связывания кислорода, причем величина п в
уравнении Хилла достигает значений 3.5-4.0.
644
^ 33.5. Корриноиды
Биологическая активность кобальта в большей степени ограни¬
чивается его ролью в действии коферментов ряда витамина В12.
Структура этого витамина включает частично сопряженное корри-
новое кольцо, структурно родственное порфириновому, за исключе¬
нием того, что содержит на одну метиновую (=СН-) группу меньше.
коррин
Корриновый лиганд, как и порфириновый, поставляет атому
Со(Ш) четыре донорных атома азота, при этом отщепляется един¬
ственный МН-протон. Координационное число равно шести. Поэтому
Со(Ш) в плоскоквадратном корриновом комплексе присоединяет
еще два аксиальных лиганда, образуя октаэдрическую структуру.
Пятым лигандом является атом азота нуклеотидного фрагмента в
боковой цепи корриновой системы (кобаламины), а природа шестого
может быть различной.
Если лигандом выступает цианид-ион (X = СМ"), то мы имеем
цианкобаламин или витамин В12- Отметим, что цианид-ион входит в
состав выделяющего из организма комплекса, и его нет ни в одной
из активных форм витамина. В биологических системах роль шесто¬
го лиганда обычно играет молекула воды (X = НгО).
Был выделен также 5-дезоксиаденозилкобаламин, в котором
атом углерода С-5 аденозильной группы связан с кобальтом. Это
был первый пример устойчивого природного металлоорганического
соединения со связью кобальт-углерод.
Витамины В12 действуют в сочетании с большим числом фер¬
ментов и участвуют в переносе метильной группы между биомоле¬
кулами с участием молекулы метионина (см. главу 21), перегруппи¬
ровках углеродного скелета биомолекул и в различных реакциях
восстановления.
Перенос метильной группы с участием микроорганизмов вызы¬
вает неблагоприятные экологические последствия. Это связано с
переходом относительно безопасной элементарной ртути в чрезвы¬
чайно токсичный и растворимый катион НдСНз+, что приводит к
рассеянию ртути в окружающей среде.
645
X
33.6. Молибденсодержащие ферменты.
Фиксация азота
Наиболее важной биохимической фунцией, которую выполняет
молибден, является утилизация неорганического азота для биосин¬
теза белков, нуклеиновых кислот и других азотсодержащих ве¬
ществ клетки
Живые организмы сильно различаются по своей способности к
таким синтезам. Высшие животные не способны к биосинтезу ряда
аминокислот (это незаменимые аминокислоты) и должны получать
их извне. Кроме того, они не могут использовать наиболее широко
распространенные формы природного неорганического азота, а
именно - нитраты, содержащиеся в почве, и атмосферный азот.
Такими возможностями, однако, обладают растения и микроорга¬
низмы. Биологическая ассимиляция нитрата в аммиак происходит в
две основные стадии: 1) восстановление нитрат-иона до нитрита;
2) восстановление нитрит-иона до аммиака.
Первая стадия катализируется молибденсодержащим ферментом
нитратрт^уктазой. Аммиак может образовываться как из нитрит-
иона, так и из атмосферного азота, а процессы эти требуют другого
молибденсодержащего фермента - нитрогеназы. Этот фермент со¬
держится в некоторых почвенных бактериях, живущих в клубеньках
корней ряда овощных культур.
Растения ежегодно связывают около Ю10 тонн азота. Уже по
одной этой причине молибден следует считать одним из важнейших
биологически активных металлов.
В восстановительной ассимиляции нитрат-иона (см. главу 22)
участвует фермент - нитратредуктаза, которая представляет со¬
бой многомерный белок с молекулярной массой 230 ООО, состоящий
из цитохрома, связанного с молибденом и с органической молеку¬
лой флавина (ФАД, см. главу 11).
Роль ФАД в биохимических окислительно-восстановитель-
ных системах сводится к двум основным аспектам: активации
С-Н-связей и распариванию электронов. Двухэлектронный перенос
при восстановлении нитрат-иона в нитрит связан с переходами
ФАД2Н ^ ФАД и молибдена(У+) в молибден(+У1). Тонкие детали
механизма этого процесса пока не известны.
Нитрогеназа - фермент, ответственный за фиксацию азота
(образование аммиака) бактериями и сине-зелеными водорослями.
Процесс происходит в водной среде при атмосферном давлении и
обычных температурах. В противоположность этому промышленный
синтез аммиака (процесс Габера-Боша) основан на применении вы¬
соких температур и давлений
Способность микроорганизмов восстанавливать в мягких усло¬
виях свободный азот, который по традиции рассматривается как в
высшей степени инертное вещество, объясняет повышенный инте¬
рес химиков к нитрогеназным ферментам Интерес этот основы¬
вается не только на желании понять механизм важнейшего биохи¬
мического процесса, но и на стремлении создать синтетический ка¬
тализатор по типу нитрогеназы для получения соединений азота.
Азотфиксирующие бактерии подразделяются на симбиотические
и асимбиотические, т е свободнообитающие
Симбиотические бактерии могут связывать азот только после
того, как они поселятся в корнях некоторых видов овощных куль¬
тур. Эти бактерии снабжают растения азотом в обмен на потреб¬
ляемые углеводы
Асимбиотические бактерии связывают азот непосредственно из
Почвы Количество такого азота существенно меньше, чем связы¬
ваемого симбиотическими бактериями Все бактерии могут фикси¬
ровать азот только при полном отсутствии кислорода.
647
Нитрогеназа из \zotobacter
была первым ферментом, для кото¬
рого было показано существен¬
ное значение молибдена. Для свя¬
зывания азота необходимы также
железо и магний. Нитрогеназа со¬
стоит из двух белков, а именно,
Мо-Ре-белка с молекулярным ве¬
сом 270 ООО - 300 ООО, содержа¬
щего молибден, железо, цистеин и
лабильную серу в соотношении
1 20 : 20 : 15, а также
Ре-белка с массой 40 000.
Активный центр молибденсодержащего белка имеет структуру
“клетки” (рис. 33.5), устроенную аналогично “клетке” ферредок-
сина (рис. 33.4).
Каждый белок по отдельности не проявляет нитрогеназной ак¬
тивности. Восстановление азота до аммиака происходит только в
случае сопряжения этой реакции с гидролизом аденозинтрифосфата
(АТФ) в аденозиндифосфат (АДФ), для протекания которой, в свою
очередь, необходим ион Мд2+.
По современным представлениям, фиксация азота может быть
выражена с помощью приведенной ниже схемы 33.1, в которой Ре¬
белок и Мо-Ре-белок обозначены как (Ре) и (Мо-Ре), а Ф - неоргани¬
ческий фосфат
(Ре) -1* (Ре)”
изменение конфигурации I Мпдтф
и потенциала ' ► МдАТФ(Ре)'
перенос
(Ре) МдАДФ Ф (МоРе)~(М2). электРона. (Ре)- МдАТФ(МоРе) (N2)
I гидролиз АТФ
(Ре) + МдАДФ + Ф + (МоРе) + 2ЫН3
Схема 33.1. Каталитическое действие нитрогеназы.
Из этой схемы видно, как нитрогеназа [(МоРе) + (Ре)] возвра¬
щается неизменной и может вновь включаться в каталитический
цикл, который требует для участия комплекса катиона магния с
АТФ (см. главу 29).
(МоРе)
N2
Рис. 33.5. "Клетка" молекулы
нитрогеназы
Вс^-реакции, катализируемые нитрогеназой, имеют восстанови-
тельныижарактер. Следовательно, для проявления ферментам ката¬
литической активности необходим физиологический источник элек¬
тронов, а также соответствующий переносчик, который бы вза¬
имодействовал с нитрогеназой, но не являлся ее частью. К таким
природным агентам относятся электротранспортные железо-серу-
содержащие белки - ферредоксины, а физиологическим источником
электронов является пируват - анион пировиноградной кислоты.
Нитрогеназные реакции в стандартных условиях являются тер¬
модинамически выгодными (Дй < 0), однако для протекания их не¬
обходима энергия в специфической форме АТФ.
Зависимое от восстановителя потребление АТФ нитрогеназой
является фундаментальным процессом для всех реакций нитрогена¬
зы, однако его механизм относится к числу наименее понятных во¬
просов биологической фиксации азота.
Рассмотренные примеры лишь частично иллюстрируют роль ка¬
тионов переходных биоэлементов в виде комплексов - металлопро-
теинов в биохимических трансформациях, из которых, однако, вид¬
на их уникальная роль в качестве координаторов сложнейших про¬
цессов - от транспорта кислорода и углекислого газа до разнооб¬
разнейших метаболических реакций.
Глава 34. КАТИОНЫ ЩЕЛОЧНЫХ
И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
34.1. Общие сведения
Химия э-элементов определяется их стремлением принять
электронную конфигурацию инертного газа с образованием ионов
М+ и М2+ Вследствие этого она является в основном “ионной” хи¬
мией Исключение составляют лишь бериллий и литий, небольшой
радиус ионов которых (табл. 34 1) делает их сильно поляризую¬
щимися, поэтому связи лития, и особенно бериллия, имеют кова¬
лентный характер Сами эти металлы не представляют особого био¬
логического интереса, однако литий во многих отношениях сходен
с магнием (диагональное сходство). Соли лития применяются при
терапии маниакальной депрессии. Механизм действия лития неиз¬
вестен, но его способность легко проникать через все мембраны
позволяет предполагать конкуренцию между Ы+ и Мд2+ за центры
связывания Мд2+.
Таблица 34 1
Сведения о гидратированных ионах
некоторых $-элементов I и II групп
Ион
Ионный
радиус, А
ДН° гидратации,
кДж/моль
Радиус гидра¬
тированного
иона
Число
гидратации
(среднее)
Ы+
0 60
-519
3 40
25.3
N3*
0 95
-406
2.76
16.6
К+
1 33
-322
2.32
10.5
Ве2+
0.31
-2512
4.08
13.5
Мд2+
0 65
-1936
3 46
120
Са2+
0 98
-1595
309
10.0
Величины ионных радиусов и зарядов играют большую роль,
т.к. именно они определяют поведение катионов I и II групп в рас¬
творах - теплоту гидратации, радиус гидратированного иона и число
молекул воды, входящих в гидратную оболочку (число гидрата¬
ции) По-видимому, этим и объясняется тот факт, что основными
биометаллами являются натрий и магний, калий и кальций, в то
время, как литий, бериллий, стронций и барий - токсичны Радиу¬
650
сы дв)гх последних (1.10 и 1.29 А) превосходят радиусы катионов
магния и^кальция.
Кроме активации множества ферментов, катионы щелочных ме¬
таллов играют важную роль в осмотическом балансе, действуют как
переносчики заряда при передаче нервного импульса, стабилизиру¬
ют структуру нуклеиновых кислот и т. д.
Ионы Са2+ инициируют некоторые физиологические процессы,
такие, как сокращение мышц, секреция гормонов, свертывание
крови и др
Концентрации неорганических ионов внутри и вне клетки нахо¬
дятся в динамическом равновесии. От внешней среды клетка отде¬
лена мембраной, через которую в одном направлении проходят пи¬
тательные вещества, а в другом - продукты жизнедеятельности.
Избирательность мембраны очень велика и распределение ионов
внутри и снаружи клетки невозможно сводить только к диффузии.
Так, внутри клетки содержится большое количество К+, но относи¬
тельно мало Ма+, концентрации Мд2+ и фосфат-иона внутри выше,
чем снаружи. Для ионов Ма+, Са2+ и СГ существует обратное соот¬
ношение (см. рис. 2 19 главы 2)
Чтобы поддерживать повышенную концентрацию К+ внутри
клетки, необходимо постоянно транспортировать его из внеклеточ¬
ной жидкости во внутриклеточную, т.е. против градиента концент¬
рации. Такой процесс сопровождается понижением энтропии
(ДБ < 0), следовательно, термодинамически не выгоден (Дй > 0) и
не может идти самопроизвольно. Объяснение состоит в том, что ак¬
тивный транспорт ионов сопряжен с гидролизом АТФ и суммарное
изменение свободной энергии оказывается меньше нуля. Стацио¬
нарное состояние достигается при равенстве потоков ионов К+
внутрь клетки (активный транспорт) и из клетки за счет диффузии.
Обратное явление наблюдается при транспорте ионов Ма+
Существование калиево-натриевого градиента концентраций по¬
перек мембраны имеет важные физиологические последствия
Разница концентраций приводит к возникновению мембранного по¬
тенциала, величина которого около 80 мВ Благодаря ему нервные
волокна способны передавать импульсы, а мышцы - сокращаться.
Нарушение ионного баланса может иметь трагические последствия
Так, например, увеличение концентрации К+ в межклеточном про¬
странстве в два раза ведет к сбою сердечного ритма и смерти, хотя
это количество К+ ниже нормального содержания внутри клетки
Тот факт, что кальций не используется в клетке, связан с его
применением в организме в качестве строительного материала
(в костях, зубах). Концентрация кальция контролируется двумя
гормонами: кальцитонином и паратироидным гормоном. Основным
651
хранилищем кальция является скелет. Кальцитонин ингибирует вы¬
свобождение ионов кальция, а паратнроидный гормон способствует
их извлечению из костей. Совместное действие гормонов сохраняет
и поддерживает структуру костей. Накопление кальция в нетипич¬
ных участках приводит к образованию камней, полиартриту, остео¬
хондрозу, катарактам и артериальным нарушениям
34.2. Комплексы катионов в-металлов
Удивительна способность некоторых биолигандов селективно
связывать Ма+ или К+, Мд2+ или Са2+. Многие из них “различают"
катионы непосредственно по их размерам, предоставляя “дырку"
или полость определенного размера, подходящую для одного катио¬
на в большей степени, чем для другого. Для одинаковых по ради¬
усам катионов I и II групп причиной селективного связывания в
комплекс является различие в зарядах, обусловливающих поляри¬
зацию и гидратацию. Лиганды с гидрофобной внешней частью высо¬
ко селективны по отношению к однозарядным катионам, и наобо¬
рот, отрицательно заряженные лиганды гораздо более избирательно
связывают двухзарядные катионы.
В последние годы большой интерес привлекли к себе некоторые
группы макроциклических лигандов, способность которых к селек¬
тивному связыванию катионов очень велика. Это циклические эфи¬
ры, криптанды и некоторые природные ионофоры (вещества, кото¬
рые обладают свойством селективно связывать ионы металлов и
осуществлять их перенос через мембраны), чаще всего антибиотики
нескольких типов структур и их синтетические аналоги.
Циклические эфиры, или краун-эфиры (crown - корона по ан¬
глийски), содержат от 3 до 12 атомов кислорода и образуют ста¬
бильные комплексы с рядом катионов, обычно в соотношении 1:1.
дибензо-12-краун-4
18-краун-б дициклогексил-18-краун-б
652
Эти циклические эфирные комплексы имеют две стороны -
гидрофильную внутренюю и гидрофобную наружную, что позволяет
им растворяться в неводных растворителях.
Константы нестойкости комплексов с участием краун-эфиров
значительно изменяются от катиона к катиону, что определяется
относительными размерами катиона и полости краун-эфира. Для
катионов щелочных металлов существует простая зависимость
между ионным радиусом и числом атомов кислорода в краун-эфире,
а именно: и+ (4), Ма+ (5), К+ (6) и Сэ+ (8).
Следует различать селективность краун-эфира по катиону и
прочность связывания с ним. Так, например, наиболее селективным
по отношению к Ма+ по сравнению с другими катионами является
дициклогексил-16-краун-5, в то же время другие краун-5-эфиры свя¬
зывают Ма+ более прочно.
Во время комплексообразования происходит постепенное заме¬
щение сольватной оболочки катиона кислородными атомами краун-
эфира, которому предшествуют конформационные изменения в ли¬
ганде. Очевидно, что взаимодействия лиганд-катион будут чувстви¬
тельны к природе растворителя.
Гидрофобность наружной стороны краун-эфира позволяет его
комплексам растворяться в гидрофобных растворителях. Так, на¬
пример, КМп04 может быть растворен в бензоле при добавлении
подходящего краун-эфира, что используется при количественном
окислении ряда органических соединений.
Некоторые из больших краун-эфиров могут связывать сразу по
два катиона.
Криптанды - это макробициклические лиганды, которые свя¬
зывают катионы еще более специфично, чем краун-эфиры В назва¬
ние криптанда входит обозначение числа
атомов кислорода в каждой из трех
цепей, связывающих головные мостико-
вые атомы азота. Размер полости криптан¬
да задается по трем направлениям, а
не в плоскости, как это было в случае
краун-эфира. Это обусловливает тот факт,
что комплекс К+ с 2,2,3-криптандом в
104 раз стабильнее комплекса К+ с ионофорным антибиотиком ва-
линомицином и в 105 раз - с аналогичным краун-эфиром При до¬
бавлении 2,2,3-криптанда КМп04 растворяется в бензоле, а сульфат
бария в воде (вплоть до 50 г/л)
Ионофорные антибиотики встречаются в природе и оказывают
существенное влияние на проницаемость мембран для катионов
Ионофоры делятся на два класса Один представляет каналообра¬
2,2,3-криптанд
653
зующие ионофоры, которые способны раздвигать мембрану, образуя
канал для прохода катиона. Второй - ионофоры-переносчики, кото¬
рые образуют комплекс с катионом, проникающим затем через мем¬
брану. В качестве примера макроциклических антибиотиков можно
привести нонактин и монактин (рис 34.1).
Рис. 34.1. Нонактин и монактин.
Наиболее прочно с молекулой нонактина связывается К+. Ли¬
ганд закручивается вокруг катиона, координируясь восемью кисло¬
родными атомами (четыре карбонильных группы и четыре атома
кислорода фурановых циклов).
В безводных условиях нонактин связывает К*, Ма+и Св+ при¬
мерно одинаково, в водных растворах устойчивость комплексов
соответственно равна для К4-: Ма+: Сз+=2 104 : 210 : 400.
Еще один пример макроциклического антибиотика, обладаю¬
щего свойствами ионофора - молекула полипептида валиномицина
(рис. 34.2).
Рис. 34.2. Валиномицин.
Координация катиона калия с ним осуществляется за счет шес¬
ти слсй&ноэфирных групп, обращенных, как это и изображено,
внутрь цикла.
Внутриклеточное и внеклеточное пространство разделено кле¬
точной мембраной, липидная структура которой обеспечивает гид¬
рофобный барьер на пути прямого проникновения гидратированных
катионов щелочных и щелочноземельных металлов Это - причина
поддержания постоянного мембранного потенциала. Ионофоры,
перенося катионы калия через мембрану, как это показано на
рис 34 3 на примере валиномицина, уменьшают мембранный по¬
тенциал и тем самым осуществляют разобщение жизненно необхо¬
димых процессов клеточного дыхания. В результате валиномицин и
обладает свойствами антибиотика.
Рис. 34.3. Транспорт катиона калия через биомембрану
с участием валиномицина.
Постоянство концентраций ионов щелочных и щелочноземель¬
ных металлов внутри и вне клетки обусловлено специальным меха¬
низмом транспорта через мембрану Перенос катионов осущест¬
вляется двумя способами:
1) активным транспортом против градиента концентраций, за
счет энергии гидролиза АТФ,
2) диффузией по градиенту концентраций, которая зависит от
наличия ионофоров
Гидролиз АТФ происходит в присутствии фермента - аденозин-
трифосфатазы, которому для выполнения своей функции необходи¬
мы катионы К+, Ма+ и Мд2+ Этот же фермент выполняет и транс¬
портные функции при переносе катионов через мембрану Он имеет
димерную структуру, состоящую из двух пар больших и малень¬
ких полипептидных цепей. Конформационные изменения, происхо¬
дящие в ферменте, приводят к тому, что катионы принимаются по
655
одну сторону мембраны, а освобождаются - по другую, а именно
Ыа+ выводится из клетки, а К+ транспортируется внуц>ь. Таким
образом, одновременно с гидролизом АТФ идет перемещение
катионов.
34.3. Хлорофилл
Особого разговора заслуживают катионы магния Они играют
совершенно исключительную роль в поддержании жизни на Земле
Катионы магния входят в состав биокомплекса хлорофилла, кото¬
рый является звеном между энергией Солнца и жизнью на Земле
Молекула хлорофилла играет ключевую роль в процессе фотосинте¬
за, который является самой масштабной химической реацией на
Земле Хотя утилизируется не более 1% световой энергии, па¬
дающей на лист растения, продукция органического вещества фото¬
синтеза (1011 тонн в год) в сотни раз превосходит продукцию всего
нефтехимического производства, а запасаемая в результате энер¬
гия во столько же раз превышает энергию сжигаемого челове¬
чеством топлива.
хлорофилл
Молекула хлорофилла напоминает гем с той разницей, что за¬
местители в кольце несколько различаются, причем в числе этих
заместителей - радикал фитил, остаток высокомолекулярного спир¬
та стероидного ряда фитола. Другое отличие состоит в том, что в
состав хлорофилла входит еще одно конденсированное циклопента¬
новое кольцо.
Хлорофилл представляет собой лишь часть сложной биохими¬
ческой машины по преобразованию солнечной энергии в процесс
фотосинтеза.
656
Его роль вкратце можно свести к тому, что его молекула раз¬
мещается ме%ду молекулами - донорами и акцепторами электронов.
Свет возбуждает хлорофилл, переводя его электроны на более вы¬
сокие энергетические уровни Один из возбужденных электронов
хлорофилла передается молекуле-акцептору электрона, а хлорофилл
синхронно возвращает себе электрон от молекулы-донора электро¬
на Такие процессы происходят очень быстро- за секунду их проис¬
ходит около 1500.
Между донором и акцептором электронов тем самым возникает
поток электронов - круговой ток, который, совершая свой путь,
выполняет работу, часть которой расходуется на биосинтез АТФ
и коферментов
Итак, катионы щелочных и щелочноземельных металлов вы¬
полняют ряд важных биохимических функций. В полной мере
оценить роль катионов щелочных и щелочноземельных металлов
в процессах жизнедеятельности организма не представляется
возможным.
Глава 35. ХЕЛАТОТЕРАПИЯ
Концентрации ионов металлов в живых организмах являются
величинами постоянными (гомеостаз), колеблются в очень узких
пределах и сложным образом контролируются, как это было час¬
тично прослежено на примере железа (глава 33), определенными
белками и гормонами.
Расстройства, связанные с нарушением ионного баланса, могут
проявляться как в результате повышенного, так и пониженного по
сравнению с нормой содержания ионов металлов Поэтому каче¬
ственное и количественное исследование жидкостей и тканей орга¬
низма на содержание ионов металлов является важным диагности¬
ческим тестом
Долгое время при болезненных состояниях изучался только ба¬
ланс щелочных и щелочноземельных металлов внутри и на поверх¬
ности клетки. Оказалось, однако, что большое число болезней свя¬
зано с изменением концентраций микроэлементов
Такие элементы, как железо, медь, цинк, кобальт, марганец и
другие, необходимы для ферментных систем, но их повышенная
концентрация оказывает токсическое действие. Так, например, за¬
болевание мозга у детей (синдром Менкеса) связано с дефицитом
меди, так как в мозговой ткани не хватает цитохромоксидазы. На¬
против, при болезни Вильсона содержание меди во многих тканях
увеличено в сотни раз по сравнению с нормой.
Поэтому в медицине существует проблема поиска эффективных
хелатирующих агентов для использования в лекарственных целях.
35.1. Комплексы как лекарственные средства
Токсический эффект некоторых комплексов используется для
создания противораковых и противомикробных препаратов. К при¬
меру, издавна известно бактерицидное действие серебра, которое
объясняется тем, что ничтожных концентраций иона серебра, кото¬
рые появляются в воде при ее помещении в серебряную посуду, до¬
статочно для угнетения активности тиолсодержащих ферментов
микроорганизмов.
К 2500 году до н. э. восходит история применения в медицине
золота, которое использовали в Китае для лечения проказы. В на¬
стоящее время соединения золота употребляют в основном для ле¬
чения ревматоидного артрита. Среди них имеются неорганические
соединения, например, хризолан Маз1Аи(820з)2], а также ряд органи¬
ческих производных (рис. 35.1).
658
СНзОН
о
Аи—БСНСООЫа
Ан^соон
Аи —N
4
миокриэин
солганол
тетрасукцин-
имидоэолото (III)
Рис. 35.1. Лекарственные производные золота.
Механизм действия золота при лечении артрита детально не
установлен. Предполагают, что эти соединения ингибируют гидро¬
литические ферменты, разрушающие суставы.
Найдены активные соединения золота против бактериальных
инфекций, в частности, туберкулеза, обнаружено и их противоопу¬
холевое действие.
Среди соединений платины наибольшее значение имеет ней¬
тральный комплекс - дихлородиамминплатина(П) [Р^ЫНэЭгСЬ].
//ыс-изомер этого плоскоквадратного комплекса (цис-ДДП) обладает
противоопухолевой активностью. Установлено, что цис-ДДП связы¬
вается в опухолевой клетке с молекулой ДНК и ингибирует ее
синтез (репликацию).
Известны лекарственные средства и среди комплексов других
металлов (например, соединения цинка уже давно применяются в
дерматологии как противомикробные средства)
Растет интерес медиков к использованию комплексонов для
поддержания металло-лигандного гомеостаза и выведения из орга¬
низма ионов токсичных металлов.
Увеличение уровня промышленного производства и связанное с
ним загрязнение окружающей среды привело к резкому росту числа
отравлений ионами тяжелых металлов - мышьяка, кадмия, ртути,
свинца, стронция, бериллия, таллия.
Для того, чтобы выполнять функцию антидотов при отравле¬
нии тяжелыми металлами, комплексоны должны отвечать ряду тре¬
бований.
Во-первых, они должны быть нетоксичными.
Во-вторых, комплексоны не должны подвергаться разложению
или какому-либо изменению в биологической среде
35.2. Комплексоны в медицине
22*
659
Далее, их антидотное действие зависит от прочности образую¬
щегося металлокомплекса, что, в свою очередь, определяется вели¬
чиной константы нестойкости соответствующих комплексов'. Исходя
из этой величины, можно установить степень химического сродства
отдельных катионов к тем или иным комплексонам, а значит, пред¬
видеть возможность избирательного связывания
Одновременно необходимо учитывать, что эффективность ком¬
плексонов в отношении токсичных металлов зависит не только от
стабильности образуемого комплекса металл-хелат, но и от проч¬
ности связи извлекаемого металла с биокомплексами организма
С учетом этих требований среди комплексонов наибольшее
распространение в качестве антидотов получили различные соли
этилендиаминтетрауксусной кислоты (ЭДТА), среди которых
наиболее доступной является динатриевая соль, известная
как трилон Б.
Его применение показано, к примеру, при отравлении соедине¬
ниями кальция - СаО (негашеная известь), Са(ОН)г (гашеная из¬
весть), СаСг (карбид кальция). При этом трилон Б, связывая ионы
кальция, превращается в тетацин.
По возрастающей степени устойчивости комплекса металл-
ЭДТА металлы располагаются в таком порядке: Бг, Мд, Са, Ре2+,
Мп, Со, гп, Сс1, РЬ, Си, Нд, №. Отсюда следует, к примеру, что тета¬
цин является эффективным антидотом при отравлении свинцом и
кадмием, так как катионы этих металлов вытесняют из комплек-
трилон Б
тетацин
660
сона ион кальция, образующий менее прочный комплекс с ЭДТА.
По тЗ^же причине выведение из организма стронция и магния не
будет осуществляться кальциевой солью ЭДТА, а марганца и желе¬
за - ее кобальтовой солью.
Получающийся в реакции тетацина с катионом свинца комплекс
СаРЬЭДТА хорошо растворим в воде и легко удаляется из организ¬
ма через почки.
В соответствии с приведенным рядом прочности хелатов тета-
цин обменивает ион кальция на ионы ртути, кобальта, кадмия, ба¬
рия. Он, таким образом, достаточно универсален.
Его можно вводить в организм в виде 5-10%-ного раствора,
основой которого является физиологический раствор (хлорида
натрия или глюкозы). Другой вариант использования - промывание
тетацином желудка для связывания яда, еще не всосавшегося в
кровь. Наконец, эффективно применение тетацина посредством
ингаляции, при которой антидот быстро всасывается и долго цирку¬
лирует в крови.
Помимо тетацина и трилона Б практическое значение в ка¬
честве противоядий имеют и некоторые другие соли этилендиамин-
тетрауксусной кислоты.
Перспективен еще один комплексон - производное диэтилен-
триаминпентауксусной кислоты - СаЫазДТПА (пентацин). Его
особенно успешно применяют при отравлениях радиоактивными
элементами.
пентацин
Обращает внимание структурное сходство реакций комплексо¬
нов с реакциями связывания яда молекулами дитиолов, например,
БАЛ: в обоих случаях образуется замкнутая структура хелатного
типа. Вот почему хотя бы частично оправдано причисление БАЛ,
унитиола и других дитиолов к комплексообразующим антидотам.
В отличие от соединения яд-ЭДТА соединение яд-дитиол мало или
почти нерастворимо в воде, что уменьшает скорость его выведения
из организма.
661
Данные последних лет свидетельствуют о высокой антидотиой
эффективности при свинцовых отравлениях еще одного кояплексо-
образующего вещества - О-пеиицилламина Ф-ГТАМ), который
представляет собою диметилцистеин, т е аминокислоту следующего
строения-
Защитное действие Э-ПАМ обусловливается наличием трех
групп (сульфгидрильной, аминной, карбоксильной). Оказалось, что
Ь-ПАМ особенно хорошо проявляет себя при хронических формах
отравлений, когда необходим длительный прием препарата.
Еще одна перспективная для медицины группа комплексонов
принадлежит к семейству полициклических хелатирующих реаген¬
тов - криптандов (см. главу 34), с которыми катионы метал¬
лов координируются таким образом, что ион оказывается “спрятан¬
ным” в циклической полости лиганда. Изображенный выше пред¬
ставитель криптандов высоко селективен по отношению к катиону
стронция.
Специфичным для катиона железа является комплексон дефе-
роксамин, применяемый для удаления железа при некоторых
железоизбыточных состояниях. Это вещество содержит фрагмен¬
ты гидроксамовой кислоты, которая присутствует в сидерофорах
(см. главу 33)
О-пеницилламин
криптанд
СОО'ЫН/ СООЫН/
О,
О
ОНО
НО,
>0
-мн—(СН2)5-
-сн3
мн
он
—(СНг)2—р—М—(СН2)5МН2
о
дефероксамин
алюминон
ОН
'СОО' ЫН/
662
Для связывания токсичного катиона бериллия применяется
алю1к^нон, получивший такое название из-за способности коорди¬
нироваться с катионом алюминия. Его эффективность по отноше¬
нию к бериллию - проявление диагонального сходства пары берил¬
лий-алюминий.
Высокой степенью комплексообразования отличается также
фитин - сложный органический препарат, представляющий собой
смесь кальциевых и магниевых солей инозитфосфорных кислот.
Его получают из конопляных жмыхов. Фитин полностью защищает
животных, отравленных смертельными дозами свинца. При этом он
в отличие от солей ЭДТА выводит яд преимущественно через желу¬
дочно-кишечный тракт, а не через почки.
Таблица 35.1
Комплексоны в медицине
Болезнь
Избыточный
Применяемый
нон металла
комплексен
Гемохроматоз, гемосидероз,
Ре
Дефероксамин,
интоксикация железом
пеницилламин
Катаракта,
Са
Трилон Б,
атеросклероз
пеницилламин
Болезнь Вильсона
Си
Смесь пеницилламин
+ тетацин
Болезнь
С<1
Криптанд,
“итаи-итаи-био”
тетацин, БАЛ
Болезнь Минимата
Нд
Тетацин,
пеницилламин
Интоксикация плутонием
Ри
Пентацин
Свинцовая интоксикация
РЬ
БАЛ, тетацин
Бериллоз,
Ве
Алюминон
бериллиевый рахит
Фитин - совершенно безвредный лечебный препарат, он может
быть использован и при отравлении ионами других металлов
Имеется еще ряд перспективных комплексонов, среди которых при¬
сутствуют и вещества растительного происхождения.
Хотя разброс в структуре применяемых в клинической практи¬
ке комплексонов очевиден, можно обратить внимание на тенден¬
цию, связанную с представлением о жестких и мягких кислотах
и основаниях Наиболее распространенные хелатирующие агенты.
663
а именно, БАЛ, О-пеницилламин, тетацин и алюминон можно рас¬
положить в порядке уменьшения их мягкости БАЛ координируется
с участием атомов серы, О-пеницилламин является О.М.Э-дЪнором,
тетацин координируется с участием атомов кислорода и азота, а
алюминон является кислородным донором
Комплексоны и их комплексы применяют при лечении различ¬
ных металлоизбыточных и металлодефицитных состояний, связан¬
ных с заболеваниями, которые вызываются нарушениями обмена
кальция, железа, меди и др. (рахит, психические заболевания, про¬
филактика радиационных поражений и т д). Типичные примеры
использования хелатотерапии даны в таблице 35 1.
Более подробное представление о современном состоянии хела¬
тотерапии можно получить из схемы 35.1, в которой приведен пе¬
речень важнейших применяемых комплексонов, причем указано, с
какими металлами они координируются
Схема 35.1. Комплексоны в хелатотерапии.
Иногда длительное поступление в организм малых количеств ядови¬
тых металлов приводит к их накоплению в различных внутрен¬
них органах и тканях, вследствие чего их концентрация в крови и
моче существенно не повышена. В таких случаях коплексоны мож¬
но использовать в целях диагностики. Введение комплексонов уве¬
личивает выведение яда с мочой и тем самым указывает на его
присутствие в организме. Иными словами, процесс комплексообра-
зования приводит к нарушению установившегося равновесия меж¬
ду ионизированным металлом плазмы и металлом, содержащимся,
например, в жировых тканях, а также в эритроцитах, печени, кост¬
ной 1^ани и т.д.
Например, тетацин используется при диагностике хронических
свинцовых отравлений. Диагностическим показателем здесь служит
выведение металла с мочой в результате однократной инъекции
комплексона. Надо, однако, отметить, что при этом возможно и
усиление интоксикации, по-видимому, из-за увеличения обратного
всасывания связанного с тетацином свинца из пищеварительного
тракта, куда он переходит из плазмы через стенку кишечника.
Еще один, на первый взгляд, неожиданный пример использова¬
ния хелатотерапии - защита от газовой гангрены. Оказалось, что
введение раствора тетацина вызывает в данном случае связывание
ионов цинка и кобальта, выполняющих функцию активаторов дей¬
ствия фермента лецитиназы, который и является токсином газовой
гангрены. Поэтому, связывая эти ионы, удается резко снизить дей¬
ствие токсина.
Таким образом, в настоящее время можно говорить о несом¬
ненных успехах и широких перспективах хелатотерапии в
изыскании и применении лекарственных средств. Практическое
использование этих средств оказалось особенно результативным
при профессиональных хронических интоксикациях соединениями
мышьяка, свинца, ртути
Список литературы
Абрамзон А А , Зайченко Л П, Файнгольд С И
Поверхностно-активные вещества- Синтез, анализ, свойства и
применение. - Л • Химия, 1988 - 200 с
Ахметов Н С Общая и неорганическая химия Учебник для
вузов - М • Высш шк , 1981 - 679 с
Вайзман Ф. Л Основы органической химии. Учебное пособие
для вузов. Пер с англ - СПб Химия, 1995. - 464 с.
Глинка Н Л. Общая химия. Учебное пособие для вузов. -
25-е изд., исправленное. - Л.: Химия, 1986. - 704 с.
Дюга Г., Пенни К. Биоорганическая химия: Пер. с англ. - М.:
Мир, 1983. - 512 с.
Ершов Ю. А., Попков В. А., Берлянд А. С., Книжник А. 3., Михайли-
ченко Н. И Биофизическая химия. Химия биогенных элементов.
Общая химия. - М.: Высшая школа, 1992. - 560 с.
Захарченко В. Н. Коллоидная химия. 2-е изд., перераб. и
доп. - М : Высш. шк., 1989. - 238 с.
Кирхнер Ю. Тонкослойная хроматография: В 2 т.: Пер. с
англ. - М.: Мир, 1981. - Т 1. - 616 с.; Т. 2. - 523 с.
Крю Ж. Биохимия. Медицинские и биологические процессы:
Пер. с франц. - М.: Мир, 1979. - 510 с.
Ленский А. С. Введение в бионеорганическую и биофизическую
химию. - М.: Высш. шк., 1989. - 256 с.
Марри Р., Греннер Д., Мейес П., Родуэлл В. Биохимия
человека: В 2 т: Пер. с англ. - М.: Мир, 1993. - Т. 1. - 384 с.;
Т. 2. - 415 с.
Маршелл Э. Биофизическая химия: Принципы, техника и при¬
ложение: В 2 т.: Пер. с англ. - М.: Мир, 1981. - Т. 1,2,- 820 с.
Митрука Б. М. Применение газовой хроматографии в
микробиологии и медицине: Пер. с англ. - М.: Медицина, 1978. -
608 с.
Мусил Я., Новакова О., Куни, К. Современная биохимия в
схемах- Пер. с англ. - М.: Мир, 1981. - 215 с.
Некрасов Б В. Учебник общей химии. - 4-е изд., перераб. -
М.: Химия, 1981. - 560 с
Неорганическая биохимия / Под ред. Г. Эйхгорна: В 2 т.: Пер.
с англ. - М.: Мир, 1978. -Т. 1.-711 с.; Т. 2. - 736 с.
Потапов В. М., Татаринчик С. Н. Органическая химия. - М.:
Химия, 1980 - 464 с.
Равич-Щербо М. И., Новиков В. В. Физическая и коллоидная
химия. - 3-е изд., испр. и доп. - М.: Высшая школа, 1975. - 255 с.
666
Райлс А , Смит К., Уорд Р. Основы органической химии для
студе1^врв биологических и медицинских специальностей- Пер. с
англ - М.. Мир, 1983 - 352 с.
Рут Г. Кислотно-щелочное состояние и электролитный баланс:
Пер. с англ. - М.: Медицина, 1978. - 118 с.
Рэмсден Э. Н. Начала современной химии: Справ, изд.: Пер с
англ. - Л. Химия, 1989 - 784 с.
Терней А. Современная органическая химия: В 2 т.: Пер. с
англ. - М . Мир, 1981. - Т. 1. - 678 с ; Т. 2. - 651 с.
Тюкавкина Н. А , Бауков Ю. И Биоорганическая химия. -
М.: Медицина, 1991. - 480 с.
У гай Я А. Общая химия. - М.- Высш. шк., 1984. - 440 с.
Фримантл М. Химия в действии: В 2 т.: Пер с англ. - М.:
Мир, 1991. - Т. 1.- 528 с.; Т. 2. - 620 с.
Хаваш Е. Ионо- и молекулярно-селективные электроды в
биологических системах: Пер. с англ. - М.: Мир, 1988 - 221 с.
Хмельницкий Р. А. Физическая и коллоидная химия. М :
Высш шк., 1988. - 400 с.
Хьюз М. Неорганическая химия биологических процессов: Пер.
с англ - М.: Мир, 1983. - 416 с.
Хьюи Дж. Неорганическая химия. Строение вещества и
реакционная способность: Пер. с англ. - М.: Химия, 1987. - 696 с.
Чанг Р Физическая химия с приложениями к биологическим
системам- Пер. с англ. - М.: Мир, 1980. - 662 с.
Химия: Справ изд./Шретер В., Лаутеншлегер К.-Х.,
Бибрак X. и др.- Пер. с нем. - М.: Химия, 1989. - 648 с.
Эппликвист Д., Де Пюи Ч., Райнхарт К. Введение в
органическую химию: Пер. с англ - М.: Мир, 1985. - 384 с.
Предметный указатель
Абсорбция 242
Агар-агар 506, 539
Агликоны 515, 574
Адамантан 328
Адамсит 487
Аддитивность 286
Адениловая кислота 575
Аденин 318, 450, 572, 574, 586
Аденозин 575
Аденозин-3,5-монофосфат 577
Аденозиндифосфат (АДФ) 484,
575, 648
Аденозинмонофосфат (АМФ)
484, 576
Аденозинтрифосфат (АТФ) 209,
302, 483, 575, 582, 648, 656
Адипиновая кислота 496, 601
Адреналин 465
Адсорбент 244
Адсорбтив (адсорбат) 239
Адсорбционный слой 276
Адсорбция 239
- избирательная 252
- ионообменная 253, 254
- удельная 240
- эквивалентная 252
Азидотимидин 577
Азокрасители 464
Азосочетание 465
Азот 59, 445
Азотистая кислота 461
Азотная кислота 465
Азулен 332
Акридин 333
Акриловая кислота 541
Акролеин 424, 488, 499
Активированный уголь 254, 256
Активность 146
Активный центр 102
Акцептор 40, 368, 491, 548
Аланин 595
Алкадиены 330, 359, 379, 392
Алкалоз 177, 178
Алкалоиды 381
Алканы 319, 347, 379, 392
Алкены 329, 352, 392
Алкилирование 366, 453, 517,
532
Акрилонитрил 354, 362
Алкилсерные кислоты 353, 419,
441
Алкилсульфокислоты 350
Алкилсульфонаты 350, 441
Алкилфосфаты 475
Алкилфосфонаты 475
Алкилфосфонистая кислота 482
Алкины 334, 380, 393
Алкогольдегидрогеназа 489, 628
Алкоголяты 418
Аллен 358
Аллил хлористый 393
Аллолактоза 529
Аллотропия 307, 469
Алмаз 307
Альдегидоксидаза 640
Альдегиды 330, 355, 468, 487
Альдозы 501
Альдоль 500
Альдольное уплотнение 500
Алюминон 662
Амидопирин 592
Амиды 330, 456, 553, 603
Амилоза 534
Амилопектин 534, 612
Аминокапроновая кислота 594,
600
Аминокислоты 455, 594
Аминомасляная кислота 600
Аминопиридин 383
Аминопропионовая кислота 594
668
Аминосахара 511
Амин*, 324, 449, 456, 458
Аммиак 43, 446, 448, 458, 646
Амфолит 301, 400, 600, 608
Амфотерность 155, 156
Анабазин 381, 543
Анальгин 592
Ангидриды 545, 550
Анестезин 599
Анилин 450, 455
Аниониты 253
Анод 204
Антагонизм 89, 286, 386
Антибиотики 385
Антидот на
- окись углерода 316
- тяжелые металлы 233, 432,
442, 659
- ФОС 461, 477
- цианиды 317, 442, 494, 635
Антикоагулянты 287
Антиметаболиты 369, 571
Антипирин 497, 592
Антрацен 332
Апатит 470
Арабиноза 520, 577
Арахидоновая кислота 563
Арбутин 517
Аргинин 596
Арены 364, 380
Арилгидразины 463
Арилсульфонаты 441
Ароматизация 351, 377, 380
Арсин 484
Арсиновые кислоты 485
Арсоновые кислоты 485
Асимметрический атом 339, 494,
584
Аскорбиновая кислота 178, 609
Аспарагин 595
Аспарагиновая кислота 595, 610
Атоксил 485
Атом водорода 16, 21
Атомы углерода
- вторичные 335, 350
- первичные 335, 350
- третичные 335, 350
Ауротерапия 658
Ацеталь 492
Ацетальдегид 362, 488, 491, 504,
504, 591
Ацетилацетон 489, 499
Ацетилен 47, 363
Ацетилениды 363, 380
Ацетилсалициловая кислота 587
Ацетилхолин 322, 433, 459, 481
Ацетилцеллюлоза 538
Ацетон 488, 491, 504, 592
Ацетоновые тела 592
Ацетоуксусная кислота 590, 592
Ацетоуксусный эфир 592
Ацетофенон 488
Ацидоз 177, 178, 589
Ацикловир 577
Ацилирование 419, 456, 461, 516,
529, 532, 552, 698,
Барбитуровая кислота 569, 579
Барий 650
Белки 48, 602
Бензальдегид 488, 499, 500
Бензамид 553
Бензил хлористый 393
Бензилацетат 553
Бензин 351, 376
Бензоин 502
Бензойная кислота 370, 541
Бензол 331, 363, 366, 369
Бензпирен 332, 355
Бериллий 89, 650
Бикарбонатно-хлоридный сдвиг
618
Биогенные элементы 85
Биолиганды 629
Биометаллы 88
Бионовые кислоты 529
Биопотенциал 211
Бихроматометрия 201
Британский антилюизит (БАЛ)
432, 485, 661
Бром 385
Бутадиен 330, 368
Бутадион 592
Бутан 319, 321
Бутандион 489
Бутиролактон 589
Буферная емкость 167, 169, 172
Буферный раствор 163, 165
- аминокислотный 172, 173
- аммонийный 166
- ацетатный 164
- белковый 172
- биологический 170
- гемоглобиновый 174
- гидрокарбонатный 166, 170,
175, 176, 310
- глициновый 174
-универсальный 168
-фосфатный 166, 171, 474
Валентное состояние 41
Валентность 37
Валентные электроны 57
Валентный угол 47, 324
Валеролактон 589
Валидол 552
Валин 335, 595
Валиномицин 654
Веронал 570
Взаимодействие
- электростатическое 67
- гидрофобное 68, 296, 606
- дисперсионное 67
- индукционное 67
- ион-дипольное 68
- ион-ионное 63, 606
- ионно-молекулярное 67
- межионное 146, 147
- межмолекулярное 66
Винилацетат 363
Винилацетилен 363
Виниловый спирт 362
Виниловый эфир 362
Винная кислота 583, 585, 588
Виноградная кислота 585, 590
Висмутаты 483
Витамин А 377, 406
Витамин В12 87, 317, 645
Витамин Вб 504
Витамин Е 199
Витамин К 199, 489
Внешне- и внутриорбитальные
комплексы 78
Внешняя сфера 72
Внутренняя сфера 72
Вода 40, 42, 127, 129, 301, 303
Водно-электролитный баланс 149
Водород 39, 55, 86, 292
Водородная связь 65, 103, 293,
492, 579, 604
Водородный показатель (pH) 153,
299
- определение 212, 218, 222
- расчет 156, 157, 162, 165
Волновая функция 19, 52
Волновые свойства 18
Волны де Бройля 19
Восстановитель 184
Восстановление органических
соединений 378, 417, 455,
491, 519, 547, 586, 591
Время удерживания 261
Вулканизация 361
Высаливание 622
670
Высокодолекулярные соединения
(BMfc) 611
Вязкость 618
Галактоза 510
Галогенангидриды 389, 473, 550,
Галогенирование 347, 353, 364,
371, 382, 392
Галогеноводороды 51, 389, 391
Галогенопроизводные 324, 378,
392, 422, 490
Галогены 90, 384, 393
Гальванический элемент 186, 202
- концентрационный 205
- топливный 206
- химический 204
Гексаметапол 457
Гексаметилендиамин 458, 601
Гексан 319
Гексахлоран 343, 371
Гексахлормеламин 401
Гексахлорофен 396
Гексозы 505
Гелий 39, 55
Гем 630, 634, 656
Гемоглобин 87, 123, 175, 182,
315, 406, 462, 628, 630
Гемолиз 143
Гемопротеины 629
Гемосидерин 637, 638
Гемосорбция 251
Гемоцианин 406, 628, 643
Гемэритрин 628, 636
Геометрия комплексов 79
Гепарин 287, 538
Гептан 319
Гетарены 332, 380
Гетеротрофы 107
Гиалуроновая кислота 538
Гибридизация 43, 45, 77, 78
- типы 43, 329, 334
Гидразин 445, 460, 496
Гидразоны 497
Гидратация 353, 417, 492
Гидраты 68, 132
Гидриды 34, 295
Гидрирование 353, 371, 373,
378, 566
Гидрокарбонат 163, 178, 312
Гидроксамовые кислоты 461, 638
Гидроксиапатит 63, 231, 474
Гидроксиламин 445, 461, 477,
482, 496
Гидроксилирование 209, 352,
371, 376, 440
Гидроксильный показатель 153
Гидроксимасляная кислота 172
Гидроксипролин 596, 605
Гидроксиформилирование 544
Гидроксоний 152, 298
Гидролиз 160, 302, 312, 388, 398,
482, 490, 544, 553, 587, 597
Гидролиз солей 160, 437
- по аниону 159, 161
- по катиону 160, 162
- кислых 162, 437
Гидроперекиси 413, 426, 544
Гидрофильность 69, 614
Гидрофобность 69, 556, 614
Гидрофосфат-ион 49
Гидрохинон 197, 422
Гипергидратация 149
Гипоксантин 572
Гипохлориты 399, 400
Гистамин 599
Гистидин 596, 610
Глауберова соль 143
Гликаровые кислоты 523
Гликоген 533, 535, 612
Гликозиды 515, 518, 526
Гликозидо-гликозиды 526
Гликозидо-гликозы 526
Гликолевая кислота 583, 586
Гликолевый альдегид 503, 586
Гликоль 354
671
Гликуроновые кислоты 521, 524
Глиоксаль 489
Глицерин 322, 416, 420, 423, 558
Глицериновый альдегид 420, 497,
501, 506, 507, 509, 584
Глицеринфосфорная кислота 419
Глицеролипиды 557
Глицин 83, 173, 179, 594, 595,
601, 608
Глицинхолевая кислота 561
Глобин 567
Глобула 613
Глутамин 596
Глутаминовая кислота 596,
602, 611
Глутатион 434, 601
Глюкаровая кислота 524
Глюкоза 94, 309, 489, 494, 503,
505, 510, 519, 521
Глюкозо-6-фосфат 518, 522
Глюконат кальция 523
Глюконовая кислота 523
Глюкопирануронозид 525
Глюкуроновая кислота 519, 539
Гомеостаз 148, 176, 181, 207,
234, 658
Гопкалит 316
Градуировочный график 217
Грамицидин 601, 607
Гранула 276
Графит 307
Гуанидин 567
Гуаниловая кислота 575
Гуанин 355, 572, 574, 579
Гуанозин 574
Гутта 361
Дативные связи 80
Двойной электрический слой
188, 278
ДДТ 396
Дегалогенирование 379, 380
Дегидратация 149, 379, 588
Дегидрирование 351, 377, 380
Дегидрогалогенирование, 379
Дезаминирование 463, 598
Дезинфицирующие средства 388,
400, 309, 429
Дезоксиадениловая кислота 575
Дезоксиаденозин 574
Дезокс игуан иловая кислота 575
Дезоксигуанозин 574
Дезоксирибоза 510, 576
Дезоксирибонуклеиновая кисло¬
та (ДНК) 66, 574, 575, 578
Дезоксицитидиловая кислота
575
Дезоксицитидин 574
Декалин 327, 373
Декан 319
Декарбоксилирование 378, 380,
455, 490, 548, 592, 599
Декстран 536
Декстрины 535
Дентатность 72
Детектор 260
Дефероксамин 662
Диазосоединения 462
Диализ 270
- перитонеальный 143
- электро- 270
Диалкилкарбонат 567
Диалкилфосфинистая кислота
482
Диалкилфосфиновая кислота 482
Диамагнетизм 57, 76
Диастереомеры 508
Диборан 293
Дивинил 379
Дигидролипоевая кислота 432,
434
Диеновый синтез 359
Дизельное топливо 351, 376
Дикарбоновые кислоты 490
Дикетопиперазин 600
Димеркаптопропанол (БАЛ) 432
672
Диметш1глиоксаль 489, 496
ДиметиЛглиоксим 73, 496
Диметилсульфоксид 436
Диметилфталат 552
Диоксан 425
Диоксиацетон 421, 501, 506
Диоксин 396
Дипольный момент 50, 491
Дисахариды 506, 526
- восстанавливающие 526
- невосстанавливающие 526, 530
Диспергирование 268
Дисперсионная среда 265
Дисперсная фаза 265
Дисперсные системы 265
Диспропорционирование
(дисмутация) 185, 306, 398,
442, 461, 467, 501, 636
Дистомер 341
Дисульфидная группа 210, 361,
434, 606
Дисульфиды 324, 433, 434
Дифенилхлорарсин 485
Дифенилцианарсин 485
Диффузия 114, 272
Дихлорамин Б 401
Дихлородиамминплатина 659
Диэтиловый эфир 425
Диэтилстильбэстрол 562
Длина связи 39, 47, 55, 324,
325, 394
Донор 40, 367, 491, 656
Ендиол 521
Енол 498, 544, 591
Желатин 621
Железное число 287
Железо 626
Желчные кислоты 56!
Жесткие кислоты и основания
81, 621, 664
Жесткость воды 312
Жиры 322, 558
Закон
- Вант-Гоффа 141
- Генри 133
- Гесса 112
- Дальтона 132
- действующих масс 123
- Лапласа-Перрена 274
- разведения Освальда 158
- Рауля 135
- Сеченова 134
Заместители второго рода 367
Заместители первого рода 367
Зарин 398, 480
Золото 658
Золотое число 287
Золь 266
Зоман 398, 480
Идеальные растворы 132
Идеальный газ 108
Изобутан 335
Изолейцин 336, 695, 597
Изомасляная кислота 541
Изомеризация 350, 377
Изомерия
- ионизационная 74
- геометрическая 75, 342
- гидратная 75
- координационная 74
- межклассовая 338
- оптическая 75, 338
- положения 337
- структурная 338
- углеродного скелета 335, 336
Изопрен 358, 360
Изопреноилы 377
673
Изосинильная кислота 316, 317
Изотерма
- адсорбции 241
- поверхностного натяжения 243
Изотонический коэффициент 145
Изотопы 293
Изоэлектрическая точка 608, 609
Имидазол 333
Имины 457, 494, 598
Инверсия 341, 531
Инвертные мыла 454
Ингибирование 92, 100
Индол 333
Инертные газы 29, 51
Инициаторы 99
Инозин 576
Инозит 343, 423
Инсулин 601, 628
Иод 386
Йодноватая кислота 400
Иодометрия 200, 401, 443
Ион аммония 40
Иониты 215, 253
Ионная сила 147
Ионное произведение воды 152
Ионный состав плазмы 64
Иономеры 216, 218
Ионометрия 214
Ионофоры 652
Иприт азотистый 396, 459
Иприт сернистый 396, 437
Кадаверин 458
Кадмий 660
Калий 650, 651
Кальций 233, 628, 650, 651, 660
Камфан 328
Камфора 328, 377, 504
Капиллярная конденсация 242
Капролактам 600, 601, 611
Капрон 601, 611
Карбамид 567
Карбамин 311, 479, 567
Карбид кальция 380
Карбии 308
Карбкатион 352, 359, 365
Карбоангидраза 103, 175,
311, 641
Карбоксигемоглобин 124, 181,
315, 633
Карбоксильная группа 541,
546, 583
Карбоксипептидаза 628, 641
Карбонаты 48, 312
Карбонатапатит 474
Карбонильная группа 487, 491
Карбоновые кислоты 487, 541,
553
Карбохолин 323
Каротин 377, 556
Каталаза 182, 210, 412,
628, 636
Катализ 100
Катализатор 99, 365, 367
Катенаны 70
Катенация 307
Катехоламин 460
Катиониты 253
Катод 204
Каучук 360, 410, 612
Квантовомеханическая модель 17
Квантовые числа 19, 20
Керосин 351, 376
Кетозы 505
Кетокислоты 495, 565
Кетоны 330, 355, 362, 487
Кинетика 92
Кинетический контроль 126
Кислород 51, 86, 403, 404
- синглетный 405, 411
- триплетный 404
Кислородный эффект 408
Кислотность 154
Кислотность органических
молекул 297, 418, 452, 599
674
Кислотные дожди 438
Кислот^ 55
- многоосновные 159
- сильные 156
- слабые 157, 452
Клатраты 70
Клетчатка (целлюлоза) 506,
533, 536
Коагуляция 284, 610, 621
Коацервация 622
Кокаин 343
Коламин 355, 436
Коллигативные свойства 136
Коллоид 265
Коллоидная защита 287
Коллоидные растворы 506, 614
Комплексные соединения 71, 179
- разрушение 180
Комплексоны 84, 600, 659
Комплексообразователь 71, 180
Комплементарность 579
Конденсация 269, 500
- бензоиновая 502
- кротоновая 500
- сложноэфирная 501
Конденсированные углеводороды
327, 331
Конмутация 401, 462
Константа
- гидролиза 160
-диссоциации 152, 154
- нестойкости 179
- равновесия 122, 123
- скорости 94
- Хилла 631, 636, 645
Конус Тиндаля 275
Конформация 603, 604
- ванна 326, 513
- заслоненная 320
- заторможенная 320
- конверт 326
- кресло 326, 513
- скошенная (гош-) 321
Конъюгация 440
Кооперативный эффект 66, 631
Координационная емкость 72
Координационная теория 71
Координационное число 62, 72
Кордицепин 576, 577
Коричная кислота 343
Корпускулярно-волновой дуализм
18
Коррин 645
Корриноиды 645
Кортизон 561
Кофеин 573
Кофермент (3 199, 208
Кофермент А 433, 479, 578
Коферменты (кофакторы)
102, 432
Коэффициент активности 146,
Коэффициент отравления 633
Краун-эфиры 426, 652
Крахмал 506, 533
Креатин 568
Крезолы 424
Крекинг 350, 377, 379
Кремниевая кислота 611
Криоскопия 144
Криоскопическая постоянная 137
Криптанды 653, 662
Кристаллическая решетка 63
Кротоновая кислота 541,
589, 600
Кротоновый альдегид 500
Ксантин 572
Ксантиноксидаза 628, 641
Ксенобиотики 251, 352
Ксилол 344
Кубан 328
Кумол 424
Купрен 363
Лактам 600
Лактаты 583
675
Лактид 588
Лактоза 505, 506, 527, 528
Латекс 360
Левомицетин 466
Левулиновый альдегид 410
Левулоэа 531
Лейцин 336, 595, 597
Летальный синтез 422
Лиганды 72, 74, 458, 652
Лиганды полидентатные 84, 180
Лизин 596, 609
Ликопин 377
Лимонен 360, 377
Лимонная кислота 583, 588
Лингоцериновая кислота 557
Линдан 343
Линолевая кислота 343, 557
Лиотропный ряд 252, 285, 621
Лиофильность 69, 266, 622
Лиофобность 69, 266
Липиды 210, 322, 556, 563
Липоевая кислота 343, 434
Липопротеиды 608
Липосомы 269, 291
Липофильность 69, 376
Липофобность 69
Литий 650
Люизит 396, 431, 485
Люминал 570
Магний 88, 582, 648, 656
Макросостояние 115
Макроэлементы 85
Малаты 583
Малеиновая кислота 542
Малеиновый ангидрид 371, 551
Малондиальдегид 489
Малоновая кислота 542, 547,
548, 549, 696
Мальтобионовая кислота 529
Мальтоза 506, 527, 528
Маннит 519
Манноза 510, 519, 521
Марганец 628
Масляная кислота 541, 557
Медь 203, 205, 628, 642
Мезовинная кислота 585
Мезоформа 586
Мембрана 139, 201, 215, 322,
615
Мембранное равновесие Доинана
615
Ментол 377, 423
Меркамин 435, 460, 578
Меркаптаны 324, 431, 435
Меркаптогруппа 210, 233
Метаболизм 177, 178, 182, 207,
251, 258, 262, 264, 398, 404,
422, 543, 641
Метаболиты 263, 369, 412, 593
Метаболический профиль 264
Металлопротеины 606, 608, 627
Металлы 34, 626
Метан 43, 48, 319
Метанол 422, 423, 517
Метафосфорная кислота 473
Метгемоглобин 408, 462, 633
Метиламин 449
Метилоранж 464
Метилсалицилат 588
Метилэтилкетон 488, 497
Метионин 436, 595, 628, 645
Метод валентных связей 38
Метод молекулярных орбиталей
38, 52
Механизм
- донорно-акцепторный 40, 76
- обменный 40
- реакции 93, 348, 352, 364
Микросостояние 115
Миоглобин 182, 628, 630
Миоинозит 343
Миристиновая кислота 557
Мицелла 276, 289, 614
Молекулярная орбиталь 38, 51
Молекулярность реакции 94
676
Молекулярные связи 613
Молишгеы 628, 646
Молочная кислота 170, 583, 584,
585, 586, 588, 589, 591
Мольная доля 132
Моляльная концентрация 133
Монактин 654
Мономер 356
Мононуклеотиды 574
Моносахариды 505, 523
Монофторуксусная кислота 549
Монохлорамин Б 401
Морфолин 450
Мостиковые углеводороды 334
Мочевая кислота 237, 578
Мочевина 319, 574
Муравьиная кислота 541
Мускарин 323
Мутаротация 511, 524, 528
Мышьяк 483, 484
Мягкие кислоты и основания
81, 621, 663
Мясомолочная кислота 583, 584
Набухание 612, 620
Найлон 458, 601, 612
Натрий 650
Нафталин 332, 372, 543
Нейрин 451, 459
Неметаллы 34, 36
Необратимые процессы 121, 125
Несахароподобные углеводы
505, 533
Нефть 375, 377
Никотин 381, 543
Никотинамид 554
Никотинамидадениннуклеотид
(НАД+) 208, 209
Никотиновая кислота 382, 541
Нитратредуктаза 647
Нитраты 448, 465, 647
Нитрилы 334, 455
Нитриты 462, 635, 647
Нитробензол 366, 455
Нитрование 349, 366, 368, 382
Нитрогеназа 446, 628, 647
Нитроглицерин 419, 465
Нитрозамины 464
Нитрозобензол 455
Нитроклетчатка 537
Нитросоединения 466
Нитроэфиры 466, 467
Новокаин 552
Номенклатура
- международная 346, 488
- рациональная 345
- тривиальная 345
Нонактин 654
Нуклеиновые кислоты 574
Нуклеозиды 574
Нуклеопротеиды 581, 608
Нуклеотиды 574
Обменная емкость ионита 254
Обратимые процессы 121
Озазоны 461, 497, 520, 529
Озон 404, 409
Озониды 355, 410
Озонирование алкенов 355, 410
Озоновый слой 411
Окисление органических
соединений 351, 354, 370,
381, 417, 420, 457, 483, 489,
523, 543, 584, 587
Окислитель 184
Окисляемость воды 200
Окись этилена 326, 354, 425
Оксазол 333
Оксалуровая кислота 569
Оксибарбитуровая кислота 571
Оксибутираты 583
Оксивалериановая кислота 589
Оксигемоглобин 123. 175, 181,
315, 406, 632
677
Оксигенобаротерапия 405
Оксид углерода(И), угарный газ
59, 314
Оксидазы 207, 629
Оксиды 415, 467
Оксиды азота 445, 465, 466
Оксиды фосфора 471
Оксикислоты 583, 584, 598
Оксильные радикалы 414
Оксимасляная кислота 583, 588,
592, 594
Оксимасляный альдегид 493
Оксимы 455, 496
Оксинитрилы 494, 507
Оксокислоты 590
Оксониевые соли 426
Оксосинтез 490
Оксосоединения 487, 489
Октан 319
Октановое число 351
Олеиновая кислота 343, 557
Олеум 439
Омыление липидов 565
Онкотические отеки 142
Онкотическое давление 142
Оптическая активность 339
Оптическая изомерия 584
Орбиталь 19, 20, 21
- атомная 19, 52
- двухцентровая 53
- молекулярная 52
- а-молекулярная 56
- тс-молекулярная 56
- разрыхляющая 53
- связывающая 53
Орнитин 597, 607
Осаждение 229
Осмолярность 141
Осмометрия 144
Осморегуляция 143
Осмос 139
Осмотическая концентрация
141, 145, 146
Осмотическое давление 138, 139,
273, 614
Основность органических
соединений 452, 583, 599
Пальмитиновая кислота 552, 557
Пальмитоолеиновая кислота 557
Пантотеновая кислота 578
Папаверин 427
Пара-аминобензойная кислота
369, 594
Пара-аминосалициловая кислота
594
Парабановая кислота 569
Паральдегид 502
Парамагнетизм 51, 57, 76, 77
Параметры состояния 108
Парафин 383
Параформ 502
Парацетамол 456
Парниковый эффект 312
Пеницилламин 662
Пентан 319
Пентацин 661
Пентозы 505
Пептидная связь 601
Пептиды 600, 603
Пептизация 269
Перегруппировка Арбузова 476
Перекиси 413, 468
Перекисное окисление липидов
210, 414, 415, 544, 565
Периодическая система 27
Периодический закон 30
Периодичность 34
Перманганат калия 210
Перманганатометрия 200
Пероксиацилнитрат 468
Пероксид (перекись) водорода
48, 210, 211, 387, 407, 635
Пероксидаза 182, 412, 629, 635,
636
678
Пероксида 411
Пероксоктелоты 413, 440, 548
Пероксомоносерная кислота
413, 440
Пероксодисерная кислота 413,
440
Персульфаты 440
Перхлорат калия 399
Пестициды 396
Пиперидин 326, 450
Пиразин 332, 383
Пиразол 333
Пиранозы 512
Пирен 332
Пиретроиды 349
Пиридаэин 332
Пиридин 332, 381, 450, 454
Пиридинкарбоновая кислота
541
Пиридоксаль 488, 495, 504
Пиридоксамин 495
Пиримидин 332, 333, 383
Пировиноградная кислота 586,
587, 590
Пирокатехин 638
Пироксилин 537
Пиросерная кислота 440
Пирофосфорная кислота 473
Пиррол 333, 382, 450, 452
Пируваты 590, 659
Плавиковая кислота 391
Плавление 114
Плазмолиз 143
Поверхностная активность 238
Поверхностно-активные вещества
(ПАВ) 244, 250, 288, 322, 441,
613
Поверхностное натяжение 237
Поверхностные явления 235
Поверхность
- раздела 226, 235
- скольжения 278
- удельная 236
Полиакрилонитрил 357
Полиамиды 611
Полиамфолиты 608, 610
Поливинилацетат 357
Поливинилбутиловый эфир 357
Поливиниловый спирт 357
Поливинилпирролидон 357, 555
Поливинилхлорид 357
Поликонденсация 611
Полимер 356, 611
Полимеризация 355, 360, 502,
611
Полимеры 358, 611
Полинуклеотиды 614
Полипептиды 66, 601, 602, 611
Полипропилен 357
Полисахариды 505, 516, 526, 611,
613
Полистирол 357
Полиформальдегид 502
Полифосфаты 475
Полихлорфенол 396
Полиэтилен 357, 611
Полуацетали 492, 512, 515
Полуацетальный гидроксил 493
Поляризуемость 51, 491
Полярность 50
Порог коагуляции 284
Порфин 333
Порфирины 452, 629
Порядок реакции 94
Порядок связи 55, 57
Потенциал
- биоэлектрический 207
- диффузионный 201, 211
- зависимость от pH 207, 299
- компромиссный 224
- мембранный 202, 211, 215, 651,
655
- окислительно-восстановитель-
ный 207, 386, 409, 411, 430
- оседания 279
- протекания 279
679
- фазовый 211
-электродный 188, 197
- электрокинетический 281
- электротермодинамический
280
Потенциалопределяющий ион
212, 276
Потенциометрия 212
Правила ориентации 374, 379
Правило
- Вант-Гоффа 97
- Дюкло-Траубе 245, 251
- Зайцева 379, 395
- Клечковского 25
- Марковникова 353, 359, 362,
499, 549
- октета 37
- Панета-Фаянса 252, 276
- Ребиндера 249
- Хунда 23, 52
- Хюккеля 332, 333
- Чугаева 84
- Шилова 249
- Шульце-Гарди 285
- Эльтекова 362, 498
Пралидоксим 496
Принцип
- Бертло-Томсона 113
- Ле Шателье 125
- минимума энергии 22, 52
- неопределенности Гейзенберга
18
- Паули 22, 52
Природный газ 375
Проба Марша 484
Проекции Ньюмена 321
Произведение растворимости
(ПР) 228
Пролин 596, 605
Пропан 206, 319
Пропаналь 488
Пропионовая кислота 541
Простагландины 562
Пространственный фактор 99
Противоионы 276
Протон 297
Псевдокаин 343
Психозин 559
Птеридин 333
Пурин 333
Путресцин 458
Равновесие 121, 124, 226
Радикалы 96, 334
Радиолиз воды 303
Радиопротекторы 435, 460
Радиус
- атома 30
- вандерваальсов 47
- ковалентный 47
- иона 650
Разрыхляющая орбиталь 53
Разрыхляющее состояние 39
Растворимость 227
- газов 134
Растворы 127, 131
- гипертонические 141, 143
- гипотонические 141
- изотонические 141
- коллоидные 265
- насыщенные 227
-физиологические 151
- электролитов 145
Рафиноза 533
Рацемат 493, 494, 578
Реакции
- гетеролитические 96
- гомолитические 96
- замещения 347, 364, 372, 382,
383, 393, 394, 395, 453, 598
- окислительно-восстановитель¬
ные 185
- параллельные 95, 100
- периодические 624
- последовательные 95
680
- присоединения 358, 361, 371,
392, «1
- простые 95
- сложные 95
- сопряженные 95, 96
- цепные 96
Реакция
- Вагнера 354
- Вюрца 379, 395
- Габриэля 554
- Гофмана 453
- Зинина 455
- Канниццаро-Тищенко 501
- Кучерова 362, 490
- серебряного зеркала 523, 524,
526, 547
- Фриделя-Крафтса 366, 380
- Чичибабина 383
Редоксиметрия 200
Резонансные структуры 48, 359
Ретиналь 344
Рибоза 510, 520, 576
Рибонуклеаза 601, 605
Рибонуклеиновая кислота (РНК)
574, 575, 580, 581
Рибосомы 581
Роданид-ион 317, 318, 442
Рубредоксин 639, 640
Ряд напряжений металлов 191
Салициламид 588
Салициловая кислота 65, 583,
588
Сальварсан 486
Сахарин 553
Сахароза 530
Сахароподобные углеводы 505,
525
Сахартрикарбоновая кислота
532
Свинец 660
Свободная энергия Гиббса 119,
186
Свободные радикалы 335
Связывающая орбиталь 53
Связывающее состояние 39
Связь
- ароматическая 331
- а- 46, 320, 324
- тс- 46, 329, 334
- кратная 46, 329
- макроэргическая 48, 478
Седиментация 274
Селен 443
Сенсибилизаторы 405, 467
Сера 428
Серин 418, 595, 597
Серная кислота 390, 439
Сернистая кислота 437
Сернистый ангидрид 437, 439
Серный ангидрид 439
Сероводород 233, 430
Сероуглерод 313
Сидерофоры 638
Сила кислоты 154, 391, 400,
546, 581
Сила основания 155
Силоксан 611
Силы Ван-дер-Ваальса 66, 103
Синергизм 89, 286, 386
Синерезис 623
Синильная кислота 316, 494,
507, 524, 529
Синтез Килиани-Фишера 507,
509
Система
- изолированная 108
- открытая 108, 117
Скипидар 377
Скорость реакции 93
Соединения включения 132
Солевой эффект 231
Сольваты 68
Солюбилизация 288, 290
681
Соляная кислота 385
Сополимеры 612
Сопряжение 330, 394, 603
Сорбент 242, 257
Сорбит 423, 519
Спин 21
Спирановые углеводороды 327
Спирты 324, 416
Сродство к электрону 32, 404
Стабилизатор 266, 277
Стационарное состояние 120
Стеариновая кислота 557
Степень диссоциации 146
Стеран 327, 560
Стереоспецифичность 340
Стероиды 327, 560
Стрептоцид 369
Стронций 232, 650
Студни 623
Субстрат 102
Сульфамиды 369
Сульфаты 439
Сульфгидрильная группа 210,
233, 431, 578, 639
Сульфиды 324, 430, 436
Сульфирование 366, 382
Сульфиты 437
Сульфобенэойная кислота 553
Сульфоксиды 436
Сульфолипиды 441, 557
Сульфониевые соли 436
Сульфоны 436
Сульфосоединения 440
Сульфохлорирование 349
Сульфоэфиры 440
Сульфурил хлористый 445
Супероксид-анион-радикал 210,
407, 635
Супероксид-дисмутаза 210, 408,
643
Супероксиды 408
Сфингозин 556, 558
682
Сфииголипиды 557, 559
Сфиигомиелин 559
Табун 480
Тартраты 583
Таурохолевая кислота 561
Таутомерия 316, 472, 494, 495,
544, 572
- амид-имидольная 554, 570
- кето-енольная 362, 498, 521
- кольчато-цепная 494, 512
Температура
- замерзания 136
- кипения 136
Теобромин 573
Теория строения атома 17
Теофиллин 573
Тепловой эффект 110
Термодинамика 107
- второй закон 116
- первый закон 109
Термодинамическая вероятность
114
Термодинамический контроль
126
Термолизин 628, 642
Терпеноиды 377
Терпены 377
Тестостерон 561
Тетацин 660
Тетрагидропиран 326, 425, 493
Тетрагидропиррол 326, 450
Тетрагидротиофен 326
Тетрагидрофуран 326, 425, 493
Тетралин 373
Тетраэтилсвинец 351
Тетрозы 505
Тефлон 356
Тиазол 333
Тиксотропия 623
Тимидиловая кислота 575
Тимидин 574
Тимин 570, 574
Тиомоч^^ина 567
Тионил хлористый 393, 438
Тиосерная кислота 442
Тиоспирты 431
Тиосульфат натрия 317, 402, 442
Тиофен 333, 382
Тиофос 481
Тиоэфиры 431, 436
Тирозин 418, 596, 597
Токсический смог 438
Токсичность
- галогеналканов 395
- галогенов 388
- люизита 432, 485
- мышьяка 90, 483
- нитритов 462
- окиси углерода 181, 315, 633
- оксида серы(1У) 438
- оксидов азота 467
- сероводорода 431
- синильной кислоты и цианидов
317, 635
- тяжелых металлов 90, 183, 431
- фосфорорганических
соединений 480, 481
- этиленгликоля 543
Толуол 331, 370, 393
Транс-аминирование 495
Транс-изомер 342, 342
Транспирация 142
Трансферрины 182, 637
Трегалозы 533
Треонин 595, 597
Триазин 332
Трилон Б 179, 660
Тринитротолуол 466
Триозы 505
Триоксиметилен 502
Триптамин 466
Триптофан 596, 597
Триэтилениминфосфотриамид
(ТЭФ) 457
Тубазид 461
Тургор 142
Тушители 406, 411
Убихиноны 208, 422
Угарный газ, оксид углерода(И)
59, 314
Углеводороды 319
Углеводы 505
Углекислый газ 171, 175, 309
Углерод 86, 306
Угольная кислота 170, 310, 567
Уксусная кислота 518, 541
Уксусный альдегид 355
Уксусный ангидрид 518, 551
Ультрафильтрация 271
Унитиол 432, 485, 661
Уравнение
- Аррениуса 98, 100
- Вант-Гоффа 614
- Галлера 614
- Гендерсона-Гассельбаха 165,
171
- Гиббса 239
- кинетическое 94
- Клайперона-Менделеева 108
- Ленгмюра 242, 246
- Михаэлиса-Ментен 104
- Нернста 187, 387
- Никольского 217
- Фрейндлиха 248
- Хилла 631
- Шредингера 19, 21, 38
Ураты 571
Урацил 570, 574, 579
Уреиды 568, 569
Уретаны 567
Уридиловая кислота 575
Уридин 574
Уровые кислоты 569
Уротропин 328, 496, 503
683
Фаза 226, 255
Фазовая диаграмма 136
Фенамин 460
Фенантрен 332
Фенацетин 456
Фенилаланин 337, 596
Фенилгидразин 496, 520, 524
Фенилгидразон 497, 590, 592
Фенилгидроксиламин 455
1 -Фенил-З-метилпиразолон-5
592
Фенилсалицилат 588
Фенол 371, 416, 422, 424, 499
Ферменты 101, 106, 340,
607, 627
Ферредоксин 640, 649
Ферриоксамины 638
Ферритины 182, 637, 638
Феррихром 638
Фиксация азота 447, 646
Фитин 663
Флавинадениндинуклеотид
(ФАД+) 208, 209, 300, 647
Формалин 494, 503
Формальдегид 488, 491, 492, 503
Формамид 553
Формиат 314
Формула
- Дебая-Хюккеля 147
- Стокса-Эйнштейна 273
Формулы
- Толленса 512, 527, 528, 531
- Фишера 420, 494, 584, 594
- Хеуорса 326, 425, 493, 512,
527, 528, 531
Фосген 313, 567
Фосфакол 480
Фосфат енола пировиноградной
кислоты 591
Фосфатидилколамины
(кефалины) 559
Фосфатидилхолины (лецитины)
559
Фосфатидные кислоты 558
Фосфиды 471
Фосфин 471, 475
Фосфолипиды 557
Фосфопротеиды 608
Фосфор 86, 469
Фосфорилирование 476, 518
Фосфорилколамины 459
Фосфорилтиохолины 481
Фосфорилхолины 323, 459, 481
Фосфористая кислота 472
Фосфорит 470
Фосфорная кислота 471, 472
Фосфорноватая кислота 469, 472
Фосфорноватистая кислота 472
Фотодинамическое действие 405
Фотоизомеризация 343
Фотосинтез 116, 403
Фотохимическая реакция 99
Фотохимический смог 468
Фреоны 313, 411
Фруктаровая кислота 523, 524
Фруктоза 489, 501, 505, 510,
513, 519, 521, 522, 523, 531
Фруктозо-1,6-дифосфат 522
Фруктозо-6-фосфат 522
Фталевая кислота 370, 374, 542
Фталевый ангидрид 551, 553
Фталимид 553, 554
Фтор 385
Фторалканы 398
Фторапатит 63, 232, 385, 474
Фторид бора 40
Фторид-ион 391, 474
Фторноватистая кислота 398
Фторотан 398
Фторуксусная кислота 398
5-Фторурацил 570
Фуллерен 307
Фумаровая кислота 344, 542
Функции состояния 108
Фуран 333, 381, 382, 425
684
Фуранозы 512
ФурфуроЦ.488, 501, 504
Хелатные комплексы 82, 83
Хелатный эффект 180
Хелатотерапия 658, 664
Хелаты 614
Хемосорбция 240
Химическая связь 37
- водородная 64, 103, 298, 492,
579, 604
-дативная 80
- ионная 37, 61
- ковалентная 37, 38
- металлическая 63
Химическое потребление
кислорода 201
Хингидрон 197
Хинолин 333
Хинон 197, 422, 489
Хиральные молекулы 339
Хитин 538
Хлор 385, 388
- активный 400
Хлорактивные соединения 400
Хлораль 488, 491, 498
Хлоральгидрат 492
Хлорамины 401, 460
Хлорат калия 399
Хлорид аммония 448
Хлорид натрия 385
Хлориды 388
Хлористая кислота 42, 399
Хлористый ацетил 551, 590
Хлорная известь 210, 401
Хлорная кислота 42, 399
Хлорноватая кислота 42, 399
Хлорноватистая кислота 41, 399
Хлороводород 41, 96
Хлоропрен 358
Хлорофилл 84, 656
Хлороформ 398
Хлорофос 481
Хлоруксусные кислоты 547
Холевая кислота 561
Холестерин 328, 406, 556, 558,
560
Холин 433, 436, 459
Хроматограф газовый 260
Хроматографическая
подвижность 257, 259
Хроматография 255
- адсорбционная 255
- аффинная 256
- бумажная 256
- газо-жидкостная 255, 259, 262
- ионообменная 256
- колоночная 255
- распределительная 255
- тонкослойная 255, 256
Целлобиоза 506, 527, 528
Целлулоид 537
Целлюлоза 536, 612
Церамид 559
Цетиловый спирт 423
Цианид-ион 59, 317, 442
Цианкобаламин 645
Циклоалканы 325, 348, 351, 392
Циклобутан 325, 348
Циклогексан 326, 336
Циклогексанон 488
Циклооктатетраен 363
Циклопентадиен 358, 360
Циклопентан 325
Циклопентанон 490
Циклопропан 326, 348, 376
Цинерин 344, 349
Цинк 203, 628, 641
Цис-изомер 342, 344
Цистамин 435
Цистеин 434, 595, 605, 639
Цистин 434, 595
Цитидиловая кислота 575
685
Цитидин 574
Цитозин 570, 574, 579
Цитохромоксидаза 207, 208, 628
Цитохромы 182, 207, 208, 209,
310, 317, 407, 628, 634
Цитраты 583
Четыреххлористый углерод 313
Число гидратации 650
Щавелевая кислота 73, 233, 541,
542, 543, 547, 548, 569, 586
Эбулиоскопическая постоянная
138
Эвтомер 341
Экзотермические реакции 110
Электрод 188
- водородный 189, 192, 298
- второго рода 189, 190, 193
- индикаторный 212
- каломельный 193
-металлический 189, 190
- окислительно-восстановитель¬
ный 189, 190
- первого рода 189, 190
- сравнения 195, 213
- сурьмяный 196
- хингидронный 197
- хлорсеребряный 195
Электроды ионселективные 214
- газовые 222
- жидкостные 221
- стеклянные 218
- твердофазные 220
- ферментные 222
Электродвижущая сила 189, 204
Электродный потенциал 187, 196
Электрокинетические явления
278
Электролитический мостик 186,
201
Электролиты 146
Электронная конфигурация 22,
23, 26, 30, 52
Электроосмос 279
Электроотрицательно<;ть 33, 34,
35, 36
Электрофил 365
Электрофорез 279, 281, 610
Элемент Даниэля-Якоби 203
Элементы 30
- биогенные 85
- внутрирядные переходные 29
- микропримесные 88
- незаменимые 88
- органогены 85
- переходные 29
- примесные 88
- типичные 29
Эмбихин 459
Эмульгатор 267
Эмульсия 267
Энантиомеры 75, 339, 493, 584
Эндотермические реакции 110
Энергетическая диаграмма 57
Энергетический уровень 20, 24,
25. 54
Энергия
- активации 97
- внутренняя 109
- возбуждения 41
- Гиббса 118, 119, 237, 243
- диссоциации 57
- кинетическая 108
- напряжения 325
- полная 108
- потенциальная 38, 108
- связи 47, 55, 324, 350, 393, 469
Энтальпия 110
- ионизации 31, 292, 404
- мольная 111
- присоединения электрона 32
- растворения 226
- стан^ртная образования 111
ЭнтропйЬ 114, 180, 651
- абсолютная 117
- стандартная образования 117
Эпимеризация 521, 522, 530
Эпимеры 510
Эритроза 509
Этан 319, 320
Этанол 422
Этаноламин 355
Этерификация 419
Этилбензол 338
Этилбутират 553
Этиленгликоль 233, 416, 423,
543, 586
Этилендиамин 73, 458
Этилендиаминтетрауксусная
кислота (ЭДТА) 179, 181,
660
Этиленимин 326, 450
Этилформиат 552
Эфедрин 460
Эфиры простые 324, 424, 517
Эфиры сложные 419, 501, 550
Эффект Бора 134, 176, 632
Эффект Гиббса-Доннана 617
Яблочная кислота 583, 588
Янтарная кислота 541, 547, 548
Янтарный ангидрид 551
Учебное издание
Кирилл Николаевич ЗЕЛЕНИН
ХИМИЯ
Учебник для медицинских вузов
Ответственный за выпуск Бровко А. В.
Оформление обложки Волошкин О. П.
Технический редактор Иванова О. £
Корректоры Макеева Л. А., Селезнева Л. М.
Компьютерная верстка Кузнецова О. Б.
Лицензия № 071099 от 01.11.94. Подписано к печати 29.10.97.
Формат 60x88 /їв. Бумага офсетная. Печать офсетная.
Гарнитура Таймс. Уел. печ. л. 43. Тираж 5000 экз. Заказ 675.
Издательство «Специальная Литература*
при участии издательства «Санкт-Петербург оркестр».
198052, Санкт-Петербург, Измайловский пр., 29.
Отпечатано с готовых диапозитивов
в ордена Трудового Красного Знамени ГП «Техническая книга»
Комитета Российской Федерации по печати.
198052, Санкт-Петербург, Измайловский пр., 29.