/





















































































































































































































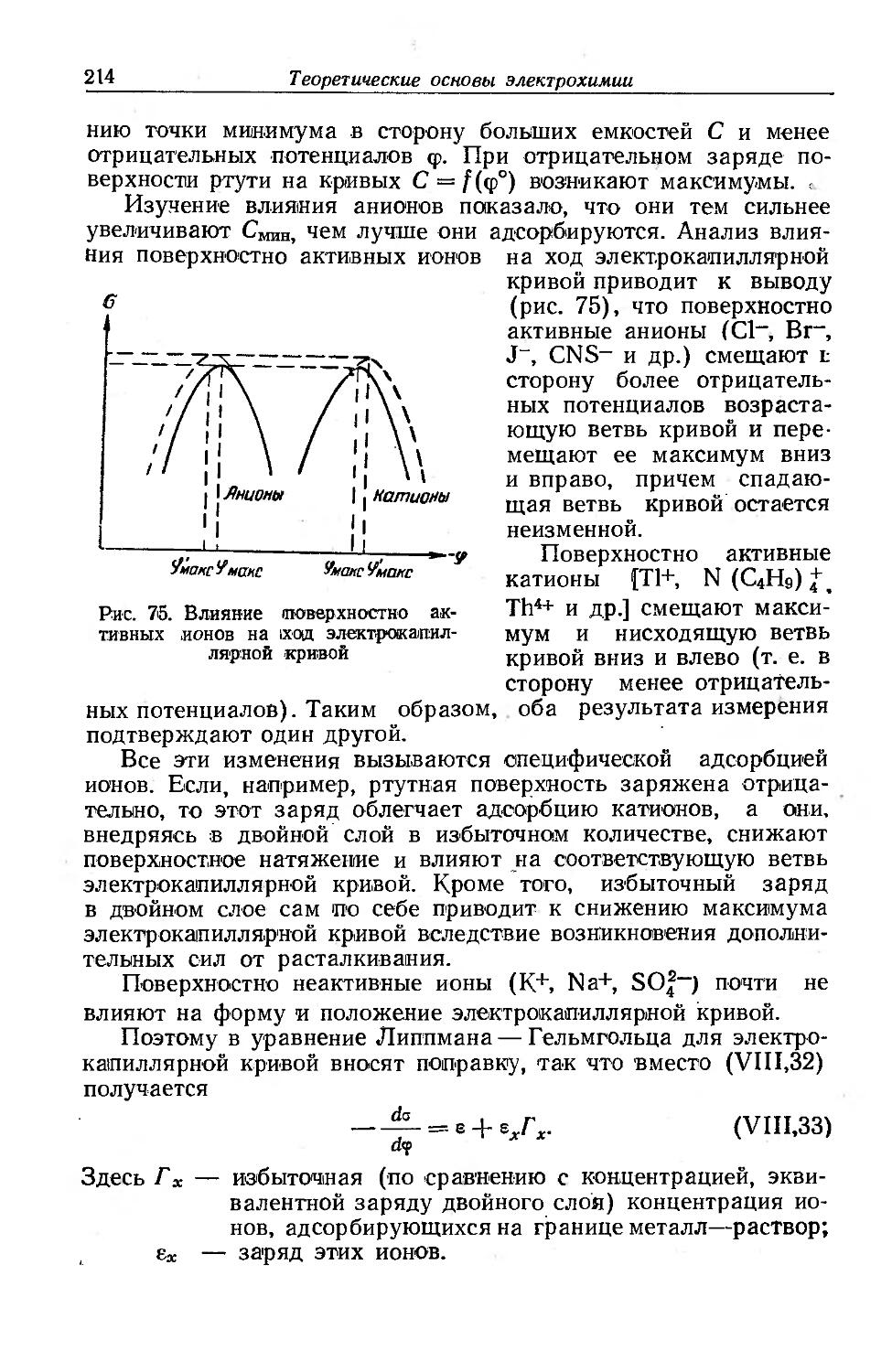































































































































































































































Text
А. И. ЛЕВИН
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
ЭЛЕКТРОХИМИИ
допущено
МИНИСТЕРСТВОМ ВЫСШЕГО И СРЕДНЕГО СПЕЦИАЛЬНОГО ОБРАЗОВАНИЯ СССР В КАЧЕСТВЕ УЧЕБНОГО ПОСОБИЯ ДЛЯ СТУДЕНТОВ ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ВУЗОВ И ФАКУЛЬТЕТОВ
ГОСУДАРСТВЕННОЕ
НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО ЛИТЕРАТУРЫ ПО ЧЕРНОЙ И ЦВЕТНОЙ МЕТАЛЛУРГИИ
Москва 1963
УДК 541.13.001.1(075.8)
АННОТАЦИЯ
В книге изложены современные теории прохождения тока через растворы электролитов; приведены основные положения теории слабых и сильных электролитов; рассматриваются электродвижущие силы гальванических элементов и скачки потенциалов, возникающие на границе фаз; описываются концентрационные элементы и условия их применения; анализируется строение, свойства и теория двойного электрического слоя; даны сведения об электрокапиллярных и электроки-негических явлениях; приводится анализ природы и особенностей электродной поляризации; рассматриваются современная теория и закономерности электроосаждения металлов из растворов их простых и комплексных солей; представлены новейшие данные по коррозии металлов и явлению пассивности.
Книга предназначена для студентов старших курсов химико-технологических вузов и факультетов, а также для инженерно-технических работников -в области электрометаллургии, технической электрохимии, коррозии металлов, гальванотехники. Пособие представляет интерес для аспирантов и научных работников соответствующих научно-исследовательских институтов и лабораторий. Книга будет также полезной работникам смежных областей при организации и осуществлении физико-химических методов анализа и в других специальных случаях.
ПРЕДИСЛОВИЕ
Программа, принятая XXII съездом КПСС, поставила перед советской наукой ответственные задачи, связанные с созданием в нашей стране материально-технической базы коммунистического общества. В этом историческом документе, в частности, определено преимущественное развитие большой химии, энергетики, металлургии.
Решение проблемы дальнейшего совершенствования и интенсификации производства цветных, редких, благородных и рассеянных металлов возможно лишь на основе широкого применения новых, высокоэффективных технологических процессов, базирующегося на глубоком понимании механизма этих процессов. Здесь особое значение приобретает теоретическая электрохимия — наука, обобщающая законы статики и кинетики электродных реакций.
Советским электрохимикам удалось создать тонкую экспериментальную методику исследования электродных процессов: построение поляризационных кривых в стационарных и нестационарных условиях, метод с использованием переменных токов, осциллографический метод, позволяющий установить временную зависимость потенциала электрода при пропускании тока постоянной силы, метод меченых атомов и др. Новые инструментальные методы раскрыли перед исследователями более широкие горизонты. Так, было показано, что основным фактором, определяющим возникновение скачка потенциала на границе между, металлом и раствором, является двойной электрический слой из зарядов металла и ионов раствора. Было найдено, что на условия появления и величину скачка потенциала между металлом и раствором большое влияние оказывает адсорбция и ориентация дипольных молекул. Сопоставление данных, полученных при изучении электрокапиллярных явлений, пролило яркий свет на роль поверхностно активных и коллоидных веществ, адсорбирующихся на поверхности электродов.
Однако до настоящего времени все еще имеется некоторое несоответствие между теоретическим обоснованием закономерностей электролиза и соответствующими реальными процессами электрокристаллизации, прочно вошедшими в современные технологические схемы цветной металлургии.
4 П редисловие
Установленное жизнью единство задач и целей, стоящих перед электрометаллургией и технической электрохимией, убедительно свидетельствует о важности электрохимической науки, о ее исключительной практической направленности и широте.
Поэтому целесообразность и своевременность издания новых учебников, учебных пособий по теоретической электрохимии, рассчитанных на подготовку квалифицированных кадров в области цветной металлургии и технической электрохимии, не вызывает сомнений.
Автор отдает себе отчет в том, что далеко не все проблемы теоретической электрохимии изложены в книге достаточно полно, он ставил перед собой цель осветить проблемы, наиболее существенные для студентов технологических вузов.
ВВЕДЕНИЕ
В настоящее время в важнейших отраслях промышленности многие технологические операции осуществляют с применением электрохимического метода. Получение тяжелых цветных, благородных, легких и редких металлов высокой чистоты, осуществление гальванических покрытий, обладающих особыми механическими и антикоррозионными свойствами, изыскание новых и совершенствование имеющихся химических источников тока, производство разнообразных продуктов окисления и восстановления, гальванопластика — вот далеко не полный перечень производств, использующих электрохимический метод.
Интенсификация электрохимических процессов и создание новых производств невозможны в настоящее время без глубокого изучения теоретических проблем электрохимии.
Характерная особенность теоретических исследований, выполненных за последние десятилетия, — их тесная связь с практикой.
Применение ряда теоретических обобщений, касающихся, например, строения двойного электрического слоя, и использование оригинальных прецизионных методов исследования позволили обобщить громадный опытный материал и осветить многие экспериментальные наблюдения, накопленные в области перенапряжения при разряде ионов водорода и ряда металлов.
При современных масштабах производства особый интерес проявляется к вопросам интенсификации и совершенствования электролитического извлечения и рафинирования различных цветных металлов. Следует иметь в виду, что состояние технической электрохимии и гидрометаллургии в данное время таково, что поиски новых возможностей в технологии без глубокого и всестороннего теоретического анализа процессов могут привести лишь к случайным результатам и обобщениям.
Чтобы создавать наиболее рациональные процессы и сознательно совершенствовать электрохимическую технологию, необходимо глубоко разбираться в механизме электродных реакций, знать законы и особенности, сопровождающие взаимные превращения электрической энергии в химическую и обратно. Теоретическая электрохимия перекликается со многими обла
16
Введение
стями науки и более всего с физической и коллоидной химией. .Для электрохимии характерно, что она имеет дело с управляемыми и самопроизвольно .протекающими процессами, проходящими в электролите и главным образом на границе фаз электрод — электролит.
В сферу изучения этой области знаний входят преимущественно гетерогенные системы, состоящие из двух или более гомогенных областей. Это существенно отличает электрохимию от ряда других разделов химической науки и технологии.
Электрохимические реакции, протекающие на границе раздела двух фаз, совершаются при наличии двойного электрического слоя из зарядов, находящихся в металле, и ионов другого знака в растворе. Подобные ионные двойные слои, возникающие на границе соприкосновения фаз, приводят к глубоким изменениям физико-химических свойств поверхностных слоев. Процесс ионного обмена протекает таким образом, что значение электродного потенциала отвечает термодинамическому равновесию между металлом и электролитом.
Однако и в случае отсутствия двойного ионного слоя па поверхности раздела металл — раствор здесь 'может возникнуть скачок потенциала. Избыток пли недостаток вещества в поверхностном слое по сравнению с содержанием вещества в объеме раствора приводит к адсорбции растворенных дипольных молекул и даже нейтральных атомов или ориентации их относительно поверхности.
В последнее время особое внимание проявляется к проблемам электрохимической кинетики, которые являются центральными при осуществлении любых электродных реакций. Теоретические обобщения и выводы электрохимической кинетики все более широко применяются не только при изучении теоретических закономерностей электрохимии, но и в электрохимической технологии. Результаты теоретических исследований позволяют, в частности, глубже осознать связь между явлениями, которые протекают при возникновении двойного электрического слоя на границе фаз, и теми глубокими изменениями- вещества, которые наблюдаются на электродах при пропускании тока.
До недавнего времени при исследовании электродных процессов усилия электрохимиков были сосредоточены главным образом на выборочном, иногда случайном изучении некоторых промежуточных стадий суммарных реакций, протекающих на электродах. Эти промежуточные стадии часто рассматривались как важнейшие, определяющие скорость электрохимического процесса в целом.
В настоящее время в значительной степени благодаря фундаментальным исследованиям советских электрохимиков все большее внимание уделяется выяснению взаимосвязи между
Введение
7
адсорбционными и электрохимическими «явлениями, наиболее широкому и систематическому обобщению механизма разряда ионов и кинетики электродных процессов, при этом в качестве объектов исследования привлекаются различные газообразные и металлические вещества, способные восстанавливаться или окисляться на электродах.
Так, в последние годы были особенно подробно изучены закономерности разряда водородных и частично металлических ионов, исследовалась кинетика обмена между амальгамами и ионами металлов, реакции взаимодействия простых и сложных комплексных ионов на катоде с .перезарядкой или восстановлением их до металла и ряд других существенных проблем.
Весьма большое значение для развития электрохимической кинетики приобрело применение новых экспериментальных методов изучения электрохимических процессов и установления достаточно объективных теоретических предпосылок для определения физической сущности изучаемых технологических процессов.
Благодаря развитию теоретической электрохимии и большому числу экспериментальных исследований в СССР созданы крупнейшие электрохимические предприятия и отдельные производства.
Техническая электрохимия в применении к цветной металлургии в Советском Союзе развивалась самостоятельными путями с использованием достижений передовой советской науки.
Опыт работы многих заводов по производству и электрорафинированию цветных металлов свидетельствует о том, что основные технологические показатели у нас в стране в большинстве случаев превосходят зарубежные. Это преимущество обусловлено коренными отличиями социалистической системы хозяйства от капиталистической, позволившими устранить различные препятствия на пути технического прогресса.
По решению Коммунистической партии и Советского правительства у нас сооружаются новые сверхмощные гидро- и теплоэлектростанции. Осуществление сплошной электрификации страны и создание единой энергосистемы сильно снижают стоимость электроэнергии, позволяют еще больше использовать дешевую электроэнергию в электрохимической и электрометаллургической промышленности.
Задача исследователей и технологов, таким образом, состоит в том, чтобы всемерно совершенствовать методы электролитических производств на основе всестороннего и глубокого теоретического изучения и обоснования электрохимических процессов.
В течение последних десятилетий курсы теоретической электрохимии в достаточно большом объеме преподаются не только
8
Введение
в химико-технологических институтах, но и в институтах цветных металлов, горнометаллургичеоких, на технологических и физико-технических факультетах ряда высших учебных заведений.
Чтобы общие закономерности электрохимии были поняты и достаточно хорошо усвоены, в (пособии дается приближенное к действительности рассмотрение их прикладного значения.
В учебном пособии специально рассматриваются физико-химические методы анализа (кулонометрический, кондуктометрический, полярографический, амперометрический и др.), где электрохимические закономерности используются достаточно широко. Не меньше используются теоретические основы электрохимии при рассмотрении особенностей электроосаждения металлов в компактной и порошкообразной форме (в гидрометаллургии и гальванотехнике) и т. д.
Работа студентов в области электрохимической теории не заканчивается непосредственным усвоением теоретических основ электрохимии. Наоборот, эта работа вступает далее, при изучении прикладной электрохимии и специальных курсов, в фазу последующей конкретизации, углубления и дальнейшего совершенствования.
Круг вопросов, рассматриваемых в книге, ограничен областью водных растворов электролитов, так как другие области и, в частности, электрохимия расплавленных сред уже давно выделились в самостоятельные дисциплины.
Глава I
ПРЕДМЕТ И СОДЕРЖАНИЕ ЭЛЕКТРОХИМИИ
§ 1. Организация электрохимического процесса
Химическая реакция обычно сопровождается выделением или поглощением энергии. Представим себе одну из таких реакций в общем виде:
Vl^l + V2^2 + v3^3 + • • • + V2^2 + ',Д±^
Если это не гетерогенный процесс, то условием протекания подобной реакции является взаимодействие реагирующих частиц (молекул, атомов, ионов), которое происходит в любой точке раствора. При этом электроны проходят путь, длина которого не превышает радиуса атома или молекулы. Место встречи и направление электронных переходов ориентированы в пространстве любым образом. Из сказанного следует, что такой процесс идет беспорядочно, неорганизованно в гомогенной системе, свойства которой во всех частях либо одинаковы, либо непрерывно меняются от одной точки раствора к другой. Такая система, помимо отсутствия поверхности раздела фаз твердое тело — раствор, характеризуется тем, что энергетические изменения в ней чаще всего сопровождаются выделением или поглощением тепла (тепловой эффект реакции). Примером подобного процесса может служить экзотермическая реакция восстановления трехвалентного железа при введении в раствор йодистого калия:
Fe2 (SO4)3 + 2KJ — 2FeSO4 + K2SO4 + J2 + Q. (1,2}
Так как реагирующие вещества в растворе находятся в виде ионов, то уравнение (I, 2) может быть переписано следующим образом:
2Fe3+ + 2J- -> 2Fe2+ + J2 + Q.
Задача электрохимического метода — так организовать протекание химической реакции, чтобы следствием процесса явилось выделение или поглощение электрической энергии. Для этого:
90 Теоретические основы электрохимии
1) не допускают непосредственного взаимодействия реагирующих веществ;
2) расчленяют единый процесс на два электродных акта: а) с отдачей электронов ионами Х~ и б) с получением электронов ионами Л4+;
3) создают пространственную направленность электронных переходов;
4) увеличивают пути электронных переходов до макроскопических размеров.
Таким образом, в отличие от протекания химических реакций, где основным условием является наиболее тесное соприкосновение реагирующих веществ в одной фазе (в растворе, расплаве и т. п.), при проведении электрохимического процесса необходимо разделение реагентов и образование гетерогенной системы, в которой переход электронов от одной группы атомов к другой осуществлялся бы через особые каналы (металлические проводники — электроды). Развитая поверхность электродов как бы увеличивает вероятность взаимодействия электронов с определенными группами атомов или ионов.
При осуществлении электрохимического процесса понятие системы включает не одну, а, по крайней мере, две или несколько соприкасающихся между собой фаз, которые разделены четкими поверхностями, при этом реагирующий компонент под влиянием тока существенно меняет свои свойства (например, при электроосаждении или ионизации металлов) либо образует совершенно новую фазу, отличную от исходных (выделение газообразных веществ на металлических электродах).
Процесс восстановления или окисления веществ на электродах, сопровождаемый приобретением, или потерей электронов частицами вещества в результате электрохимической реакции, называется электролизом. Для электролиза необходима система, которая состоит из следующих элементов:
1) раствора электролита — проводника второго рода, в котором реагирующие вещества диссоциированы на ионы, напри-мер Н+ и С1~;
2) двух проводников первого рода, погруженных в раствор электролита, так называемых электродов. В этом случае при соответствующем направлении тока на границах фаз электрод — электролит будут идти процессы:
Н+ + е_-1-Н2; С1~ — е —»С12, (1,3)
что приведет к образованию новых химических веществ — газообразного водорода и хлора:
Н++ С1~ —»Н2-)--i-С12; (1,4)
Предмет и содержание электрохимии И
3) проводника первого рода, 'соединяющего электроды между собой либо с внешним источником тока.
Электрод, па котором идет процесс приобретения электронов частицами вещества, носит название катода. Электрод, с которого электроны переходят во внешнюю цепь, называется анодом.
По предложению М. Фарадея (1833 г.), положительно заряженные атомы (Н+, Cu2+, Fe3+ и др.) получили название катионов, отрицательно заряженные (С1~, ОН-, SO4~n т. п.) —анионов. При пропускании постоянного электрического тока частицы, несущие заряд, направляются к соответствующим электродам: анионы к аноду, катионы к катоду. Здесь вследствие взаимодействия ионов с электронами (с отдачей или приобретением электронов) вещество либо выделяется в виде нейтральных атомов или молекул, либо перезаряжается.
В основе всякого электролиза, следовательно, лежит разделение единого процесса на два элементарных электрохимических электродных акта, представляющих собой реакции электрона с ионом или нейтральной молекулой. Осуществление такого течения реакции, при котором система и среда обмениваются электрической энергией, мы и будем называть организацией электрохимического процесса. При этом, если суммарная реакция протекает с выделением электрической энергии, система носит название гальванического элемента; если же процесс сопровождается поглощением энергии, систему называют электролитной ванной или электролизером (эти же названия сохраняются и для ячеек, в которых осуществляется соответствующий электрохимический процесс).
Примерами электрохимических систем могут служить следующие:
1. Раствор ZnSO4 с цинковым электродом и отделенный от него пористой диафрагмой раствор CuSO4 с медным электродом:
Zn | ZnSO41 CuSO41 Cu.
В этом случае на медном электроде (положительный полюс) протекает процесс
Сп2+ + 2е -> Си, а на цинковом (отрицательный полюс): Zn — 2е —♦ Zn2+.
В данном примере суммарная реакция идет с выделением электроэнергии, т. е. мы имеем дело с гальваническим элементом.
2. Раствор нитрата серебра с погруженными в него двумя серебряными электродами (катод — чистое серебро, анод — серебро, содержащее примеси):
Ag+|AgNO3[-Ag.
12 Теоретические основы электрохимии
В этом случае на катоде идет .процесс выделения чистого серебра:
Ag+ 4- е Ag, а на аноде, кроме ионизации серебра , :
Ag — е -> Ag+, образуются примеси, накапливающиеся в электролите либо попадающие в шлам.
Приведенная система представляет собой электрохимическую ячейку, работающую с поглощением электроэнергии. В данном примере мы имеем дело с электролитной ванной для рафинирования (очистки) серебра.
3. Электрохимически при наличии диафрагмы может быть проведено также восстановление либо окисление вещества. В этом случае электродами служат инертные, например платиновые электроды, не образующие в данных условиях ионов, а электролитом — раствор, содержащий ионы высшей и низшей степени окисления. Так, например, в системе
Pt+1 Fe2 (SO4)s, FeSOJ-Pt
на катоде пойдет реакция
Fe3+ + е -> Fe2+, на аноде - 1
Fe2+ — е -> Fe3+.
В итоге в католите будут накапливаться продукты восстановления, а в анолите — окисленные вещества. В общем случае процесс потери электронов — окисление — протекает на аноде; процесс приобретения электронов — восстановление — на катоде.
Таким образом, отличительным признаком всякого электрохимического процесса, протекающего на границе фаз электрод — электролит в гальванических элементах или электролитных ваннах, является непременное участие электрона. Электрохимия— отрасль химической науки, изучающая наиболее общие законы превращения веществ в электролитах и на границе фаз электрод — электролит при поглощении либо отдаче молекулами, атомами или ионами электронов. Именно электронный переход и реакция между ионами и электронами на границе металл—раствор определяют наблюдаемые при электролизе превращения электрической энергии в новые химические вещества в электролитных ваннах либо глубокие качественные превращения вещества на полюсах элементов с возникновением электрического тока. Нетрудно заметить, что механизм электрохимических процессов существенно отличается от обычной картины химического превращения материи.
Предмет и содержание электрохимии 13
В электрохимических системах имеют дело с проводниками первого рода, в которых электрический ток переносится электронами, и с проводниками второго рода, в которых наблюдается исключительно ионный перенос электрического тока. К проводникам первого рода, или электронным проводникам, относят все металлы и сплавы, графит, уголь, а также некоторые твердые «кислы, карбиды и сульфиды металлов. Металлические проводники состоят из положительно заряженных ионов и отрицательно заряженного «газа», образованного коллективизированными электронами. Этот «газ» равномерно заполняет все пространство между ионами.
Согласно электронной теории, протекание тока в проводниках первого рода сопровождается различными физическими явлениями (нагревание, намагничивание и т. д.), но не приводит к .химическим изменениям. Электропроводность таких проводников относительно очень высока.
Ниже приведена удельная электропроводность некоторых материалов при 18 °C.
Удельная .
электропроводность —I —1 ом -см
Проводки к и первого рода
Серебро................................. 615000
Алюминий ............................. 360 000
Платина ................................ 86 200
Ртуть .................................. 10 400
Уголь ..................................... 200
Проводники второго рода
NaF (расплавленный при 1000 °C) , . . 4,01
NaCl ( » » 750 °C) . . . 3,40
AgNOs ( » » 209 °C) . . - 0,65
H2SO4 (30%-ный раствор)..........0,740
КС1 (1-н. раствор) -............. 0,09784
Этиловый спирт................... 0,000003
Вода............................. 0,000000043
Диэлектрики
Сера................................... 2-10~16
Кварц............................ 5-10—17
Парафин................................... Ю-18
Как показывает опыт, удельная электропроводность проводников первого рода с ростом температуры уменьшается:
= [! — «(/—18)]. (1,5)
Здесь х/, Xie — удельные электропроводности при температурах t и 18 °C;
а — температурный коэффициент.
12 Теоретические основы электрохимии
В этом случае на катоде идет .процесс выделения чистого серебра:
Ag+ + e-»Ag, а на аноде, кроме ионизации серебра .
Ag — е -> Ag+, образуются примеси, накапливающиеся в электролите либо попадающие в шлам.
Приведенная система представляет собой электрохимическую ячейку, работающую с поглощением электроэнергии. В данном примере мы имеем дело с электролитной ванной для рафинирования (очистки) серебра.
3. Электрохимически при наличии диафрагмы может быть проведено также восстановление либо окисление вещества. В этом случае электродами служат инертные, например платиновые электроды, не образующие в данных условиях ионов, а электролитом — раствор, содержащий ионы высшей и низшей степени окисления. Так, -например, в системе
Pt+1 Fe2 (SO4)3, FeSO4|~Pt
на катоде пойдет реакция
Fe3+ 4- е -> Fe2+,
на аноде J
Fe2+ — е -+ Fe3+.
В итоге в католите будут накапливаться продукты восстановления, а в анолите — окисленные вещества. В общем случае процесс потери электронов — окисление — протекает на аноде; процесс приобретения электронов — восстановление — на катоде.
Таким образом, отличительным признаком всякого электрохимического процесса, протекающего на границе фаз электрод — электролит в гальванических элементах или электролитных ваннах, является непременное участие электрона. Электрохимия— отрасль химической науки, изучающая наиболее общие законы превращения веществ в электролитах и на границе фаз электрод — электролит при поглощении либо отдаче молекулами, атомами или ионами электронов. Именно электронный переход и реакция между ионами и электронами на границе металл — раствор определяют наблюдаемые при электролизе превращения электрической энергии в новые химические вещества в электролитных ваннах либо глубокие качественные превращения вещества на полюсах элементов с возникновением электрического тока. Нетрудно заметить, что механизм электрохимических процессов существенно отличается от обычной картины химического превращения материи.
Предмет и содержание электрохимии
13
В электрохимических системах имеют дело с проводниками первого рода, в которых электрический ток переносится электронами, и с проводниками второго рода, в которых наблюдается исключительно ионный перенос электрического тока. К проводникам первого рода, или электронным проводникам, относят все металлы и сплавы, графит, уголь, а также некоторые твердые окисли, карбиды и сульфиды металлов. Металлические проводники состоят из положительно заряженных ионов и отрицательно заряженного «газа», образованного коллективизированными электронами. Этот «газ» равномерно заполняет все пространство между ионами.
Согласно электронной теории, протекание тока в проводниках первого рода сопровождается различными физическими явлениями (нагревание, намагничивание и т. д.), но не приводит к .химическим изменениям. Электропроводность таких проводников относительно очень высока.
Ниже приведена удельная электропроводность некоторых материалов при 18 °C.
Удельная -электропроводность —1 —1 ом -см
Проводники первого рода
Серебро................................. 615000
Алюминий ............................. 360 000
Платина ................................ 86 200
Ртуть .................................. 10 400
Уголь ..................................... 200
Проводники второго рода
NaF (расплавленный при 1000 °C) , . . 4,01
NaCl ( » » 750 °C) . . . 3,40
AgNO3 ( » » 209 °C) . . . 0,65
H2SO4 (30%-ный раствор).........0,740
КС1 (1-н. раствор) -............ 0,09784
Этиловый спирт.................. 0,000003
Вода............................ 0,000000043
Диэлектрики Сера................................ 2-10~16
Кварц................................... 510~17
Парафин......................... 10“18
Как показывает опыт, удельная электропроводность проводников первого рода с ростом температуры уменьшается:
х, = х1в[1-а(/-18)]. (1,5)
Здесь xt, Xie — удельные электропроводности при температурах t и 18 °C;
а — температурный коэффициент.
14______________Теоретические основы электрохимии____________
Примеси также уменьшают электропроводность проводников, первого рода.
К электронным проводникам полностью применим закон Ома:
/ = (1,6>
где Е — падение напряжения на концах проводника, в;
R — сопротивление проводника, ом;
I — ток, а,
т. е. проводимость проводников первого рода при данной температуре не зависит от падения напряжения в цепи.
Системы, проводящие ток электролитически, т. е. с участием ионов — материальных частиц, несущих положительные ил» отрицательные заряды, называют электролитами, или проводниками второго рода1. В соответствии с приведенным определением весьма часто и растворенное вещество, способное диссоциировать на ионы, называют электролитом.
Электролитические проводники делят на две группы: а) вещества, обладающие электролитической проводимостью в чистом состоянии (расплавленные соли, твердые галогениды некоторых металлов, вода, спирты и многие подобные органические растворители), и б) растворы одного или нескольких веществ в воде и других полярных растворителях.
Проводники второго рода обладают значительно меньшей проводимостью, чем проводники первого рода. Электропроводность проводников второго рода возрастает с ростом температуры:
^ = ^[1 + 4/ -IS)]- (1.7>
Промежуточное положение занимают смешанные проводники, в которых ток переносится с участием электронов и ионов. Примерами смешанных проводников служат некоторые твердые соединения металлов (AggS, ZnO, Cu2O и др.), а также концентрированные растворы щелочных и щелочноземельных металлов в жидком аммиаке. Рассмотрение такого рода проводников: выходит за рамки данного курса.
Так как течение электрохимических реакций связано с переходом электричества, то для любой электрохимической системы можно написать выражение для работы электрического тока:
W — QE,
где Q — фактор емкости;
Е — фактор интенсивности.
1 .В отличие от проводников первого рода inpox-ождение электрического-тока через проводники второго рода сопровождается выделением иа электродах веществ — продуктов электрохимической реакции, т. е. продуктов взаимодействия ионов с электронами. (Ред.)
Предмет и содержание электрохимии 15»
Следовательно, здесь имеются две группы закономерностей. Первая группа связана с прохождением некоторого количества электричества через границы катод — электролит (QK)^ анод — электролит (Qa) и собственно электролит (Q3). Нетрудно заметить, что
Q = Q =0 = 0
чк ^-внешн. цепи.
Кроме того,
Q = h,
где I — сила тока, о;
т— время, сек, мин, ч.
Электрические единицы включены в Международную систему единиц СИ1. В этой системе единиц сила измеряется в ньютонах (н), заряд (количество электричества) в кулонах (к),, длина — в метрах (л<), масса в килограммах (кг), время в секундах (сек). Сила в 1 н сообщает покоящемуся телу массой в 1 кг ускорение, равное 1 м в секунду за секунду: 1 н = 1 кг-• 1 м{сек2 *. Сила в 1 н на пути в 1 м совершает работу в джоуль: 1 дж =1 н • 1 м.
Выражая работу электрического тока через количественный' фактор Q и фактор интенсивности Е, ;можно определить электрические единицы следующим образом.
Единица силы тока — ампер (а) есть сила неизменяюшегося тока, который, будучи поддерживаем в двух параллельных прямолинейных проводниках бесконечной длины ничтожно малого круглого сечения, расположенных на расстоянии 1 м один от другого в пустоте, вызывает между этими проводниками силу, равную 2» 10~7 единиц силы Международной системы на каждый-метр длины.
Единица работы — джоуль (дж) представляет работу, производимую силой в 1 н на пути в 1 м .при совпадении направлений силы и перемещения точки приложения силы.
Единица мощности — ватт (вт) —это мощность, при которой в течение одной секунды равномерно производится работа, равная 1 дж.
Единица количества электричества — кулон (к), или ампер-секунда (а-сек), есть количество электричества, протекающее-через поперечное сечение проводника в течение 1 сек при неиз-меняющемся токе в 1 а. Количество электричества и электрический заряд имеют одинаковую размерность.
Вторая группа закономерностей относится к фактору интенсивности Е и связана со скачками потенциалов на границе ка-
1 Все недостающие производные и внесистемные единицы, допускаемые к.
применению, следует брать из государственных стандартов на единицы по от-
дельным видам измерения.
1.8
Теоретические основы электрохимии
Количество граммов вещества (элемента), выделившегося при прохождении единицы количества электричества, носит название электрохимического эквивалента. На практике его чаще всего относят не к постоянной Фарадея F, а к одному ампер-часу. Согласно закону Фарадея, электрохимические эквиваленты пропорциональны химическим эквивалентам. В таком случае элект-
А
рохимическии эквивалент выразится как —-. Некоторые
значения этих величин приведены в табл. 1.
Таблица 1. Атомные массы и электрохимические эквиваленты
Л ГО 3 о и л ГО
« © го ©“ JS ® ж-р го
Элемент го £ Ь ? * i Элемент £ н ж *
<0 К <и О ж К Ф
ч го CQ О <5 ч g гс и со ч го £Q с •58 ч 3^° ©8 go аз Kfco
Алюминий . . 3 26,9815 0,3356 Олово . . 2 118,69 2,2154
Барий .... 2 137,34 2,5636 » 4 1,1077
Бром 1 79,09 2,9832 Палладий 2 106,4 1,9914
Висмут .... 3 208,980 2,6005 > . . 4 0,9957
Водород 1 1,00797 0,037636, Платина 2 195,09 3,6437
Железо . . . 2 55,847 1,0424 » . . . 4 1,8219
В . . . 3 —- 0,6949 Ртуть . . . 1 200,59 7,4882
Золото . . . 1 197,00 7,3610 » 2 3,7441
» . . . 3 — 2,4537 Свинец 2 207,19 3,8673
Йод 1 126,9044 4,7376 Селен 2 78,96 1,4337
Кадмий . . 2 112,40 2,0980 Сера .... 2 32,064 0,5983
Калий .... 1 39,102 1,4595 Серебро .... 1 107,870 4,0269
Кальций . . . 2 40,08 0,7480 Стронций . . 2 87,62 1,6355
Кислород . . . 2 15,9994 0,2986 Сурьма .... 3 121,75 1,5150
Кобальт . . 2 58,332 1,1000 Таллий 1 204,37 7,6293
Магний . . . 2 24,312 0,4539 Теллур . . . 2 127,60 2,3817
Марганец . . . 2 54,9381 1,0252 Фтор 1 18,9984 0,7092
Мышьяк . . 3 74,9216 0,9321 Хлор 1 35,453 1,3236
Медь 1 63,54 2,3729 Хром 3 51,996 0,6471
» 2 —- 1,1864 6 .— 0,3236
Натрий .... 1 22,9898 0,8585 Цинк . 2 65,37 1,2202
Никель .... 2 58,71 1,0954
Хотя закон Фарадея и не имеет исключений в применении к границе между проводником первого рода и проводником второго рода (к границам, в которых участвуют смешанные проводники с ионно-электронной проводимостью, он, очевидно, неприменим), на практике приходится встречаться с кажущимися отклонениями от этого закона.
Приведем некоторые причины таких отклонений.
Предмет и содержание электрохимии 19
Совместный разряд ионов
Представим себе электролитную ванну, содержащую раствор сернокислого цинка и серной кислоты:
Pt+|ZnSO4, H2SO4, HaO|-Pt.
Здесь в электролите присутствуют ионы Н+, Zn2+ и SOf-. На катоде желательно выделение цинка:
Zn2+ + 2е -> Zn.
Однако при пропускании 1 а-ч электричества выделяется не 1,22 г цинка, как следовало бы по закону Фарадея, а меньшее количество. Это обусловлено тем, что одновременно с разрядом Zn2+ на катоде идет разряд Н+-ионов:
Н+ + е->-^-Н2.
Суммарное же количество выделившихся на катоде цинка и водорода точно соответствует закону Фарадея.
Наличие в системе ионов разной валентности
Рассмотрим ванну для электрорафинирования золота:
Аи+1 AuCl3, AuCl, НС1, Н2О |~Au.
В электролите присутствуют ионы Au3+, Au+, Н+ и С1_. На катоде идут процессы:
Аи3+ + Зе -> Au; (а)
Аи+ + е -> Au. (б)
Если допустить возможность совместного разряда ионов Аи3+ и Аи+, то может оказаться, что золота выделяется больше, чем требуется по реакции (а). Это кажущееся отклонение легко объясняется, если учесть реакцию (б).
Перезарядка
В той же системе, очевидно, возможен еще и процесс частичного восстановления Аи^-ионов, т. е.
Аи3+ 4- 2е -> Аи+,
что также должно повести к кажущимся отклонениям от закона Фарадея.
2*
20 Теоретические основы электрохимии
Взаимодействие продуктов электродных реакций между собой или с электролитом (вторичные реакции)
Рассмотрим ванну для электролитического получения хлора:
C+|NaCl, H20|~Fe.
Здесь на аноде выделяется хлор:
2С1~ — 2е-> С12.
Но хлор может частично реагировать с раствором едкого натра, проникающим из катодного пространства с образованием хлората и гипрохлорита. Это приведет к отклонению от закона Фарадея.
При получении магния электролизом расплавленных солей в ванне:
С+1 расплав MgCla |~Fe
катодный процесс сводится к выделению магния:
Mg2+ + 2е -* Mg,
анодный — к образованию хлора:
2С1- —2е->С12.
Если допустить проникновение хлора в катодное пространство, то возможен обратный процесс окисления:
• Mg + С12 MgCl2,
что приведет к снижению количества магния, выделившегося на катоде.
Катодное восстановление анодных продуктов
При техническом электролизе с получением перманганата на аноде идет реакция
МпО2~ — е -> МпО— 4 4
Если допустить проникновение МпОТ-ионов к катоду, то на катоде возможен обратный процесс, ведущий к увеличению расхода электрического тока.
Причинами кажущихся отклонений от закона Фарадея могут быть также неустойчивость конечных продуктов, диспергирование выделяющегося металла при использовании расплавленных солей (образование «металлического тумана»), утечки тока, короткие замыкания и т. п. затруднения.
Предмет и содержание электрохимии £1
Отклонения от закона Фарадея влияют на выход по току /1, который характеризует полезное использование тока:
А = —-100%, (1,10)
(7т
где Цср — количество граммов основного (полезного) продукта, выделившегося при пропускании определенного количества электричества;
9т — количество граммов основного продукта, которое должно было выделиться по закону Фарадея при пропускании того же количества электричества.
Величина А показывает, насколько правильно и целесообразно организован процесс электролиза.
Выход по току можно представить также в виде отношения
(1,11) <2пР
где QT — количество электричества, которое должно было быть затрачено на проведение данного процесса по закону Фарадея;
Сцр — действительные затраты электричества на единицу вещества.
Таким образом, в общем случае А 100%.
Следует различать катодный (Лк) и анодный (Ла) выходы по току, так как они почти всегда не равны один другому. Особенно большие отклонения от 100%-ного выхода по току наблюдаются при электролизе расплавленных солей. В последнем случае выход /по току зависит от /большого числа факторов: /высокой температуры и относительно большой скорости диффузии, плотности тока, примесей, расстояния между электродами и пр.
На использовании закона Фарадея основан способ измерения количества электричества — кулонометрия. Приборы, применяемые для этого, называются кулонометрами. Существуют три группы кулонометров: весовые, объемные и титрационные.
Весовые кулонометры основаны на определении количества электричества, прошедшего через систему по привесу металла катода. К этому типу относится серебряный кулонометр (рис. 1). В простейшем виде он состоит из платинового тигля 4, служащего катодом, и серебряного анода 2, который подвешивается на стеклянный крючок. Между электродами на стеклянном кольце 1 находится пористый сосуд 3—-диафрагма, препятствующая возникновению побочных реакций. Электролитом служит нейтральный или слегка подкисленный раствор AgNO3. Катодная плотность тока 0,02 а/см2, анодная — не более 0,2 а]см2. Точность серебряного кулонометра достигает 0,005%.
32
Теоретические основы электрохимии
Менее точным является медный кулонометр (рис. 2), состоящий из двух медных анодов 1 и катода 2, опущенных в электролит состава, г/л: 50 H2SO4 (уд. вес 1,84); 150 CuSO4-5H2O;
50 спирта (или сахара).
Для удержания анодного шлама пластины, служащие анодом, обертывают пергаментной бумагой. На. отогнутых отростках анодных пластин укрепляют разрезные клеммы 3, соединенные проволокой 4, охватывающей сосуд. Катод закрепляется на
Рис. il. Серебряный кулонометр
Рис. 2. Медный кулонометр
штативе 5. Катодную плотность тока берут в пределах от 0,002 до 0,02 а/см2. Спирт (или сахар) добавляют в электролит для предотвращения реакции
2Си+ Сп2+ + Си.
Благодаря своей простоте медный кулонометр получил наибольшее распространение в лабораторной практике.
К объемным кулонометрам относятся газовый и ртутные кулонометры. В газовом (рис. 3) осуществляется электролиз 15— 20%-ного раствора едкой щелочи. Электродами служат пластинки из никелевой жести с приваренными к ним молибденовыми проволоками, впаянными в стекло. Электролизер соединен с уравнительным сосудом и газовой бюреткой, позволяющей по объему выделившегося гремучего газа судить о количестве пропущенного тока.
Предмет и содержание электрохимии
23
Ртутный кулонометр (рис. 4) состоит из стеклянного сосуда 1 с круговым желобом, в который залита ртуть, служащая анодом. Желоб сообщается с ртутным резервуаром 3. Кольцо 2 предохраняет нижний резервуар 5 от попадания в него ртути из желоба. Электролитом здесь служит раствор: 225 г/л HgJ2 + + 750 г/л KJ. Ртуть выделяется на угольном катоде 4 и стекает
Р,ис. 3. Газовый кулонометр:
1 — кулонометр:
2 — компенсирующий сосуд
Рис. 4. Ртутный кулонометр
Рис. S. Титрационный кулонометр (йодный): 1 — анод; 2 — катод;
3 — кран
в резервуар 5, проградуированный на ампер-часы. Точность прибора небольшая, всего 1%, но зато он может работать при относительно больших силах тока.
В титрационных кулонометрах изменение количества вещества, реагирующего на катоде, определяется титрованием. Наиболее распространены йодные кулонометры (рис. 5):
Pt/Ir+1 НС1разб! КЛконц, НС1, Н2О |-Pt/Ir.
на аноде которых выделяется и затем оттитровывается йод, и кулонометр Кистяковского:
Ag+1KNO3 (15—20 % -ный) | HNOS (0,5 % -ная) Pt/1г,
на аноде которого растворяется серебро и раствор титруется на содержание Ag+-HOHOB. Точность этих кулонометров достигает 0,001%-
В простейшем случае при отсутствии более точных способов количество пропущенного электричества можно определить, зная
24
Теоретические основы электрохимии
силу тока и время, в течение которого ток пропускался, так как Q = /r, (1,12)
если I выражено в амперах, а т в часах.
Точность показаний большинства кулонометров весьма сильно зависит от плотности тока:
1 , 2
i — — а/сж,
где I — сила тока;
S — площадь электрода, так как качество осадка (например, меди в медном кулонометре) находится в прямой зависимости от этой величины i.
Следует различать катодную и анодную плотность тока, которые в соответствии с размерами электродов могут быть различными. Плотность тока определяет скорость электродных реакций и их направление, а также существенно влияет на выход по току продуктов электрохимической реакции и в конечном счете обусловливает рентабельность процесса, снижая или повышая расход электрической энергии на единицу готового продукта.
§ 3. Электровесовой метод анализа — кулонометрия
На применении закона Фарадея основан электровесовой анализ, принцип которого достаточно прост: анализируемое вещество отлагается на инертном, например платиновом электроде, и таким образом по привесу определяется исходное количество вещества. Обычно при этом образуются металлические осадки, но имеются случаи использования электровесового метода и с неметаллическими осадками. Так, например, свинец можно определить анодно, выделив его в виде двуокиси свинца.
При электролизе можно поддерживать постоянным либо напряжение, наложенное на ячейку, либо наложенный извне ток. Чаще всего на электровесовых методах задают вполне определенную плотность тока, которая в большинстве случаев подбирается для создания оптимальных условий электролиза эмпирически.
Установка для кулонометрического титрования при постоянной силе тока показана на рис. 6.
Электролизер 1 присоединяется к источнику тока 2, который поддерживает постоянную силу тока при электролизе независимо от процессов, проходящих на электродах. Замкнув ключ 3, который одновременно запускает электрический счетчик времени 4, присоединенный к сети переменного тока, начинают электролиз.
Рабочий электрод погружен непосредственно в электролизер; вспомогательный же электрод помещается в стеклянной трубке,
Предмет и содержание электрохимии 25
заполненной хорошо проводящим электролитом (типа H2SO4, KNO3 и т. п.) и не содержащим общих ионов с анализируемым раствором. Трубка, содержащая вспомогательный электрод, имеет впаянный стеклянный фильтр; этим устраняется возможность смешения продуктов электродных реакций. Испытуемый раствор в электролизере перемешивается с помощью мешалки.
Рис. 6. Схема прибора для кулонометрического титрования при постоянной силе тока
Кулонометрическим методом анализа можно пользоваться для непосредственного определения ряда элементов, а также для их количественного разделения. Такое электрическое разделение можно проводить не только при постоянной силе тока, но и при заданном потенциале. Последним способом пользуются в тех случаях, когда на электроде возможно одновременное выделение нескольких элементов. Пользуясь законом Фарадея, определяют количество прореагировавшего у электрода вещества (Р) по равенству:
р AQ
96510г‘
Здесь А — атомный или молекулярный вес восстановленного или окисленного на электроде вещества; Q — число кулонов, затраченных на электролиз; z — количество электронов, участвующих в электродном процессе.
ЛИТЕРАТУРА
Б а й д а Л. И., Д о б ротворсюий Н. С., Оршанский Д. Л. и др. Электрические измерения. М.— Л., Госэнергоиздат, 1950.
Биргер Н. И. Задачи по электрохимии, ГОНТИ, 1939.
Биллитер Ж. Основы гальванотехники. Металлургиздат, 1949.
Бродский А. Н. Физическая химия, т. II. Госхимиздат, 1948.
26 Теоретические основы электрохимии
Глесстои С. Введение в электрохимию, ИЛ, 1951.
Деляхей П. Новые приборы и методы в электрохимии, ИЛ, 1957.
Дол М. Основы теоретической и экспериментальной электрохимии (пер. под ред. А. Н. Фрумкина). ОНТИ, 1937.
Кар апетья нц М. X. Химическая термодинамика. Госхимиздат, 1949.
Левин А. И., Пом осов А. В, Укше Е. А. Руководство к лабораторным занятиям по теоретической электрохимии, Изд. УПИ им. С. М. Кирова, 1957, стр. 26.
Мансуров Н. Н., Попов, В. С. Теоретическая электротехника. Госэнер-гоиздат, 1958.
О к а т о в А. Н. Химические источники тока. М.— Л., Госхимиздат, 1948.
Р о т и н я и А. Л., Хейфец В. Л. Теоретические основы гидрометаллургии. Основы металлургии, т. I, Металлургиздат, 1960.
Сочеванов В. Г. Гальванические элементы. М.— Л., Госэнергоиздат, 1951.
Эйкен А. Курс химической физики, вып. II. Химиздат, 1938.
Глава 11
ДВИЖЕНИЕ ИОНОВ
ПОД ДЕЙСТВИЕМ ЭЛЕКТРИЧЕСКОГО ПОЛЯ
§ 1. Подвижность и злектропроводность ионов
Электричество в растворах электролитов при прохождении тока переносится двумя видами ионов: катионами и анионами (например, Н+ и SOf--ионами в растворах серной кислоты, Na+ и ОН~-ионами в растворах едкого натра и т. д.). Катионы переносят электричество к катоду, анионы — к аноду. На границе электрод — электролит ток не прерывается благодаря протеканию электродных реакций, идущих с приобретением или отдачей электронов.
Например:
Си2+ + 2е Си (на катоде), 1 „
Си — 2е->Си2+ (на аноде). J
При этом хотя в переносе тока участвуют все ионы, присутствующие в растворе, однако далеко не все они являются участниками электродных процессов. Так, например, в ванне для электроосаждения меди
Cu+|CuSO4, H2SO4, H2O|-Cu
в переносе тока участвуют ионы Н+, Cu2+, SOi-, а электродные реакции (II, 1) идут с участием только Си2+-ионов.
Электрохимическая ячейка представляет собой последовательно соединенный проводник, поэтому к обоим электродам всегда доставляется одинаковое количество зарядов. Это позволило Фарадею высказать предположение, что скорости движения всех ионов одинакового заряда в растворе одни и те же и что •они зависят только от заряда иона и величины тока, протекающего через электролит. На самом деле это предположение оказалось неверным, и в 1853 г. Гитторф обнаружил, что при прохождении тока через электрохимическую систему в слоях электролита, прилегающих к катоду (католита) и к аноду (анолита), ппоисходят различные изменения концентрации растворенного
28
Теоретические основы электрохимии
вещества. Эти изменения были объяснены различием в скоростях движения ионов.
Рассмотрим концентрационные изменения, происходящие в приэлектродных слоях в электрохимической ячейке с нерастворимым анодом при прохождении через нее электрического тока (рис. 7).
Рис. 7. Схема движения ионов и изменения концентрации /в приэлектродиых слоях при прохождении тока
Для простоты предположим, что в электролите присутствуют одновалентные катионы ( + ) и анионы (—) и что при отсутствии тока ионы в растворе неподвижны (схема а). Пропустим два фарадея электричества. В этом случае на катоде и на аноде разрядится по 2 г-экв каждого сорта ионов (схема б). Если скорости катиона и аниона равны:
го каждый сорт ионов перенесет в растворе по одному фарадею электричества и концентрационные изменения у катода и анода будут одинаковы.
Картина будёт иной, если скорость катиона не равна скорости аниона, например
VK ~ б^а-
Тогда при пропускании шести фарадеев электричества получим следующую картину (схема в).
У катода разрядилось 6 г-экв-, перенесено катионами 5F, у анода разрядилось 6 г-экв-, перенесено анионами IF. Следова-
Движение ионов под действием электрического поля
29
тельно, концентрационные изменения в анолите будут более значительными, чем в католите.
Приведенная модель электрохимической системы весьма схематична (рис. 7), однако она показывает, каким образом Гитторф пришел к выводу о неравенстве скоростей различных ионов.
Если обозначить изменения концентрации в католите и анолите соответственно через ДСК и ДСа, тогда:
&СК Со — Ск уа g.
&Са Со —Са vK
где Со — начальная концентрация раствора.
Таким образом, изменения концентрации в приэлектродных слоях обратно пропорциональны скоростям движения отдельных ионов (правило Гитторфа).
Чтобы выяснить факторы, влияющие на скорость движения ионов, представим себе работающую электролитную ванну. Пусть падение напряжения между электродами составляет Де в, а расстояние между ними I см. В этом случае электрическая сила, действующая на ион заряда Ztle, где Zt — валентность, а е — элементарный заряд, равна
7е = у^. (П.З)
Если пренебречь взаимодействием ионов между собой, то при движении иона под действием силы fe ему придется преодолевать только сопротивление среды, т. е. силу внутреннего трения жидкости, которая пропорциональна скорости движения ионов:
I = (П.4)
Здесь ki — коэффициент пропорциональности;
Vi — скорость 1-того иона.
Результирующая сила, действующая на ион, таким образом, равна:
7= Ге — kPi- и1-5)
Хотя величины f, fe, fs, v, be — векторы, но мы для простоты можем рассматривать движение ионов по прямой, а указанные величины считать скалярными. Для стационарного процесса, когда f= О
v = ' (Ц.6)
1 kt I
30 Теоретические основы электрохимии
В полученной таким образом формуле имеется произведение двух величин, из которых Первая зависит только от свойств иона и растворителя, вторая — только от внешнего поля. Если поле одинаково, то скорости ионов все же остаются различными.
Скорость иона при градиенте потенциала, равном единице, принято называть абсолютной подвижностью:
Ц = (II,7>
Абсолютная подвижность иона £/< — скорость иона, см)сек, когда градиент потенциала равен 1 в/см. Так как eNo — F (где No — число Авогадро), то
• (П, 8>
1 Nokt
В электрохимии чаще пользуются понятием относительной подвижности иона. Она равна абсолютной подвижности, умноженной на постоянную Фарадея F:
I. = FU. = . (II,9>
Эта величина называется ионной электропроводностью. Электропроводности ионов /к и /а зависят от концентрации раствора, температуры и природы растворителя. Зависимость электропроводности ионов от концентрации иллюстрируется табл. 2.
Таблица 2. Электропроводность ионов при 18 °C
Концентрация моль)л Электропроводность ионов, ом 1-см,/г-экв
Н+ Li+ Na+ К+ ОН С1 F
0 314,5 33,3 43,5 64,5 172,0 64,9 46,2
0,001 314,8 37,3 42,3 63,1 171,0 65,0 45,5
0,01 307,0 30,1 40,0 60,4 167,0 61,5 43,2
0,1 294,4 27,5 35,4 55,4 157,0 55,8 38,0
Из таблицы следует, что:
1) с ростом концентрации электропроводность всех ионов уменьшается. Это можно объяснить, если учесть, что ионы несут электрические заряды и потому должны взаимодействовать между собой. Такое взаимодействие ведет к замедлению движения ионов, и очевидно, что межионное взаимодействие будет возрастать при увеличении их концентрации;
2) электропроводность «быстрых» ионов с ростом концентра
Движение ионов под действием электрического поля 31
ции убывает, медленнее, чем электропроводность «медленных» ионов *;
3) электропроводность ионов возрастает с увеличением их радиуса. На первый взгляд это представляется парадоксальным. Действительно, по закону Стокса (который может быть применен к движению ионов с некоторыми оговорками),
ki - 6тт/;, (11,10)
где 1] — вязкость раствора.
По формуле (II, 9) для двух одинаково заряженных ионов будем иметь:
й .. ki_ _ г2 ,ц . ..
I? ki гг
т. е. с уменьшением размеров ионов скорость его должна расти. Однако если учесть, что rt — радиус гидратированного иона и что толщина гидратной оболочки должна возрастать с уменьшением истинного радиуса иона, так как она определяется электрическим потенциалом иона — (где at
at — истинный радиус иона), то. смысл разбираемого явления становится вполне определенным. В самом деле, если взять ионы щелочных металлов: Cs+, Rb+, К+, Na+, Li+, у которых Zte одинаково, и рассчитать на основании электропроводности их радиусы, то получим величины, приведенные в табл. 3.
С другой стороны, известно, что методом рентгенограмм также можно определить радиусы ионов, находящихся в твердой кристаллической решетке. В этом случае, однако, получаются другие их значения.
Из табл. 3 следует, что: 1) значения радиусов, найденные по электропроводности га, всегда больше их значений, определенных по рентгенограммам гр; 2) величины га растут от Cs к Li, а гр уменьшаются в том же порядке. Чем объясняется такое несовпадение? Строго говоря, нельзя применять закон Стокса к движению ионов под влиянием силы тока внешнего поля, но эта причина не главная. Главная причина расхождений заключает-
Таблица 3. Радиусы ионов, рассчитанные по электропроводности (гэ) и рентгенографически (гр)
Ионы О Радиусы ионов, А
гэ ’’р
Cs+ 5,05 2,57
Rb+ 5,09 2,21
к+ 5,32 2,2
Na+ 7,9 1,76
Li+ 10 1,42
1 Объяснение этого факта, как и того, что Н+ и ОНионы обладают аномально высокой скоростью, будет дано ниже.
32
Теоретические основы электрохимии
ся в том, что в растворе ионы взаимодействуют с молекулами растворителя, и поэтому радиусы их больше радиусов ионов, находящихся в твердой кристаллической решетке.
При этом силы взаимодействия ионов одного заряда с молекулами воды обратно пропорциональны радиусам ионов. Таким образом, сила связи ионов Li+ с водой больше, чем ионов Cs+, т. е. ионы с меньшим радиусом более прочно связаны с молекулами воды и радиус гидрооболочки у таких ионов, естественно, больше.
Пользуясь изложенными соображениями, иногда по измеренной подвижности рассчитывают степень гидратации ионов. Полученные значения, хотя и не дают количественных представлений, однако могут служить качественной характеристикой, описывающей относительную гидратацию различных ионов.
С ростом температуры электропроводность ионов растет. Это связано, во-первых, с понижением вязкости раствора при повышении температуры и, во-вторых, с уменьшением радиусов ионов вследствие дегидратации.
Зависимость электропроводности ионов от природы растворителя изучали Л. В. Писаржевский и П. Вальден. Они пришли к следующей закономерности:
zco40 = const, (11,12)
где /оо — электропроводность иона при бесконечном разведении; т)о — вязкость растворителя (табл. 4).
Таблица 4. Электропроводности ионов в различных растворителях при 25 °C
Растворитель Вязкость растворителя при 20 °C относительные единицы Электропроводность ионов при бесконечном разбавлении, ом см* ! г-экв
н+ к+ С1 N(C2Ho)J
Вода 1,000 350 74 76
Метиловый спирт 0,593 143 54 52 53
Этиловый спирт 1,188 62 25 21 27
Ацетон 0,328 — 70 105 93
Хлористый этилен 0,830 — — 32 28
Эта закономерность вытекает из уравнений (11,9) и (II, 10), так как
l^- ZiF = В. 6тгА0г(-7]0 6itN0r,-
(И, 13)
Движение ионов под действием электрического поля
33
Если Гг не зависит от температуры и природы растворителя, то В = const. Последнее близко к истине лишь для крупных ионов (например, N'('C2Hs)t) в неводных растворах, для которых правило Писаржевского — Вальдена соблюдается достаточно хорошо.
§ 2. Числа переноса и методика их определения
, Зная число прошедших фарадеев электричества и убыль концентрации в католите и анолите, можно определить лишь соотношение скоростей движения ионов, но не их скорости в отдельности. Чтобы охарактеризовать участие данного типа ионов в переносе тока, вводим понятие о числах переноса ионов.
Числом переноса t-того иона называется доля тока, переносимая этим ионом в растворе:
(ПД4)
Если в растворе присутствуют два сорта ионов, то
Здесь пк и п& — числа переноса катиона и аниона. Числа переноса тесно связаны с электропроводностями ионов. Чтобы уяснить себе эту связь, мысленно проведем через электрохимическую систему плоскость, параллельную электродам, и будем подсчитывать число ионов, проходящих через эту границу за единицу времени (см. рис. 7) ,в ту и другую стороны. Число катионов и анионов, прошедших через плоскость в течение 1 сек., будет пропорционально их скоростям, площади сечения S и концентрации ионов в растворе:
хк = CKvKS- ха = CavaS. (II, 16)-
С другой стороны, произведение xtFzi есть не что иное, как величина тока, обусловленная движением t-того иона.
тельно, по (11,6) и (11,7)
7K = I7KFCKS^zK; Ia = UaFCaS z&.
Отсюда и _ IkCkZk . „ ____________________ leCaZa
» *— • * J / - ---—-------- ,
4<QkZk -J- ^a^a^a ^a^a^a
Следова-
(11,17)
(11,18)
3 Заказ 1338
34
Теоретические основы электрохимии
Если электролит состоит из ионов одинакового заряда, т. е. Ск = Са, Zk = 2а, ТО
Очевидно, что
Пк + «а = 1 (П,20)
* и в общем случае
Snz = 1.
Далее, согласно правилу Гитторфа (для систем с нерастворимыми анодами),
= (П,21)
Пк /к АСа
т. е. числа переноса характеризуют концентрационные изменения в приэлектродных слоях и, кроме того, показывают, какую часть тока берет на себя в переносе каждый ион данного электролита.
Числа переноса зависят от состава электролита. Так, например, в растворах КС1 и NaCl числа переноса С1~-иона различны и равны:
в КС1 иС1_ =1 — «к+;
в NaCl n~,_ =1 — п +
C1 Na^'
Числа переноса зависят от концентрации раствора (табл. 5).
Таблица 5. Числа переноса некоторых ионов при температуре 18 °C
Электролит Ион Числа переноса при концентрации раствора, молъ1л
10 0,1 0,01 0
НС1 н+ 0,8410 0,8314 0,8251 0,8209
С1- 0,1590 0,1686' 0,1749 0,1791
КС1 к+ 0,4882 0,4898 0,4902 0,4906
С1~ 0,5118 0,5102 0,5098 0,5094
CH3COONa Na+ — 0,3828 0,3848 0,3863
СН3СОО“ — 0,6172 0,6152 0,6137 '
AgNO3 Ag+ NO~ 0,4710 0,5290 0,4740 0,5260 0,4740 0,5260 —
Движение ионов под действием электрического поля
35
Эта зависимость может быть выражена математически. Так, для растворов КС1 найдено:
п^п^-А^С, (11,22)
где А — постоянная;
tikm — число переноса катиона при бесконечном разбавлении.
Таким образом, в большинстве случаев числа переноса по мере разведения сближаются. Зависимость чисел переноса от
концентрации в некоторых случаях столь велика, что они формально становятся отрицательными. Так, например, число пе
реноса Сб2+-ионов в 0,02-51. растворе CdJs равно 0,296, а в 2-н. растворе равно —0,15. Объясняется это явление образованием комплексных ионов [CdJJ1 2-, число которых возрастает с увеличением концентрации. Ионы, содержащие кадмий, будут в этом случае приближаться к аноду, а не удаляться от него. Повышение температуры также приводит к сближению чисел переноса различных ионов (рис. 8).
Причина этого явления сводится к росту подвижностей ионов вследствие деги-
Рис. 8. Зависимость чисел переноса от температуры
дратации, а также уменьшения вязкости растворителя. Та же причина приводит, по-видимому, к изменению чисел переноса в зависимости от концентрации раствора (табл. 5).
Основное положение теории Гитторфа:
__ йС;|
па ДСк
дает возможность экспериментально определять числа переноса по изменению концентрации в приэлектродных слоях электролита *.
В сосуд, изображенный на рис. 9, наливают испытуемый электролит, а затем в течение некоторого времени пропускают ток (количество пропущенного электричества определяется с помощью кулонометра). После отключения тока электролит вылива-
1 В общем случае это не совсем верно, так как ДСа и ДСК зависят от
объемов анодного и катодного пространства н от характера электродных процессов.
3*
36
Теоретические основы электрохимии
ют из отдельных частей электролизера и каждую порцию раствора титруют. Таким образом определяют величины концентрационных изменений в приэлектродных слоях ДСК, АСа. Так как
= 1, ТО
Пк = —АЬ_ . п = АС« . (11,23)
ДСк-+ДСа ДСК-(ДСЙ
Рис. 9. Прибор для определения чи-сел переноса:
/ и 2 — анод и катод; 3 — среднее пространство; 4 — источник тока; 5 — кулонометр; 6 и 7 — краны
Рис. ilO. Прибор для определения чисел переноса то методу движущейся границы
Другой метод определения чисел переноса — метод движущейся границы — сводится к следующему. Два раствора, различающиеся по цвету или коэффициенту преломления, наливаются в прибор, снабженный электродами (1 и 2 на рис. 10). Если растворы различаются по плотности, то в средней калиброванной части сосуда 3 можно наблюдать четкую границу (а — Oi) между ними. При пропускании тока граница между растворами начнет перемещаться. Если концентрация растворов С г-экв^м?, то при прохождении F кулонов электричества граница раздела лройдет объем V' где пк — число переноса ка-
тионов. При пропускании q кулонов будет пройден объем V = ---— Следовательно,
ст7’
VFC пк =-----
Ч
(11,24)
Движение ионов под действием электрического поля 3?
регули-
(П,25)
должна опреде-
Величина q измеряется кулонометром, соединенным последовательно с прибором.
Важное условие образования резкой границы между двумя растворами — то, что исследуемые катионы kz+ и kz2+, находящиеся по обе стороны границы, имеют одинаковые скорости. Для достижения равенства этих скоростей соблюдается рующее соотношение Кольрауша: г я
пк _ «к С С" ’
т. е. эквивалентная концентрация каждого электролита быть пропорциональна числу переноса его катиона. При
лении чисел переноса по методу движущейся границы объемную концентрацию индикаторного раствора выбирают таким образом, чтобы она соответствовала уравнению (11,25). Причем для вычисления С используют приближенные значения чисел переноса. Затем опыт повторяют, меняя концентрацию индикаторного электролита до тех пор, пока не будут получены постоянные значения чисел переноса.
Числа переноса в твердых солях также можно определять экспериментально, для этого соли прессуют в форме цилиндриков, которые в дальнейшем используют в исследованиях значений пк и па (цилиндрики зажимают между соответствующими электродами).
Таким образом, по изменению веса катодного и анодного цилиндрика, содержащего твердую соль, было установлено,, что в случае AgJ ток переносится ионами серебра. Перенос тока только катионами Na+ наблюдается и при электролизе стекла, в то время как ионы остаются неподвижными.
Однако в галоидных солях свинца наблюдается обратное явление: носителями тока :в этом случае являются анионы. В NaCl при низкой температуре ток переносится одними ионами Na+. но при повышении температуры ионы С1- также обнаруживают подвижность, так что в этом случае, как и для водных и расплавленных электролитов, наблюдаемые числа переноса лежат между 0 и 1.
В теории Гитторфа имеется важное упущение. Дело в том, что ионы в растворе при перемещении несут с собой некоторое количество воды.
Перенос растворителя (А) может быть учтен, и вычисленные таким образом числа переноса обычно называют истинными (Wi) в отличие от гитторфских (п ). Они связаны между'собой следующими уравнениями:
Wk Пк ± Wa na i (П,26)
38 Теоретические основы электрохимии
В разбавленных растворах Wt и щ почти совпадают, но с ростом концентрации разница между ними становится ощутительной.
Нернст предложил для определения истинных чисел переноса измерять величину иг одним из описанных выше методов, а затем повторить измерение, добавив к исследуемому раствору какой-либо индифферентный раствор неэлектролита, например сахар. Предполагалось, что концентрация сахара в приэлектрод-ных слоях будет неодинаковой вследствие изменения содержания воды. Подобные расчеты основывались только на представлениях о связывании ионами молекул воды вследствие перманентной гидратации. Однако в настоящее время имеется достаточно юснований полагать, что гидратацию -ионов в растворе нельзя объяснить простым связыванием ионами того или иного числа молекул воды раствора. Этот результат весьма сложного действия ионов на трансляционное движение ближайших молекул воды. Размеры ионов -в общем случае отличаются от размеров молекул растворителя, поэтому трансляционное движение молекул воды вблизи ионов происходит с другой частотой, чем в чистой воде. В таком случае средняя плотность расположения молекул воды вокруг ионов становится отличной от плотности расположения молекул воды в воде. Как предполагают, именно по этой причине ряд ионов способен уменьшить подвижность ближайших к ним молекул растворителя, в то время как около других ионов молекулы воды становятся более подвижными, чем в чистой воде. Описанное явление получило название «отрицательной гидратации». Из катионов щелочных металлов отрицательная гидратация свойственна К+, Rb+ и Cs+.
Отказ от представлений о перманентной гидратации ионов в растворах электролитов не противоречит результатам опытов по переносу ионами воды при электролизе, а следовательно, и изменению чисел переноса. Эти изменения сводятся главным образом к переносу объемов большей или меньшей плотности. Так, например, в случае водных растворов хлоридов щелочных и щелочноземельных металлов движение гидратирующихся катионов к катоду сопровождается весьма эффективным переносом воды в том же направлении, в то время как движение негидратирую-щихся анионов к аноду связано с переносом воды в обратном направлении. Непосредственное экспериментальное определение переноса воды ионами при электролизе в большинстве случаев осложняется тем, что поток воды, переносимый ионами в одном направлении, непременно приводит к возникновению оттока воды в обратном направлении, так как объем раствора в катодном и анодном пространствах (в случае стационарного процесса электролиза) не меняется. Это не учитывалось при попытке определения истинных чисел переноса по методу Нернста.
Движение ионов под действием электрического поля
39
§ 3. Аномальная подвижность Н+ и ОН_-ионов
Опыт показывает, что подвижности, а следовательно, скорости и 'числа переноса Н+ и ОН--монов аномально велики (табл. 6). Эта аномалия не была объяснена Гитторфом и не может быть приписана малому радиусу Н+ и ОН--ионов, так как радиус ОН~-ионов 1,40 А соизмерим с радиусами других ионов (см. табл. 3), а протон Н+, хотя и обладает очень малым радиусом, но в водных растворах он существует лишь в виде иона гидроксония Н3О+, радиус которого также сравним с радиусами многих ионов. Для объяснения аномальной подвижности Н+ и ОН~-ионов были привлечены представления Гроттгуса, который предполагал, что механизм прохождения тока через электролит сводится к вполне определенной ориентации молекул раствора, состоящих из положительных и отрицательных частей, относительно электродов с последующим разрушением ближайших к противоположным полюсам молекул.
В последующей стадии оставшиеся после разряда на электродах положительные и отрицательные части молекул присоединяются к соответствующим частям смежных не разрушенных молекул раствора, причем этот процесс повторяется вдоль всей неподвижной цепи молекул, находящихся между электродами.
Таблица 6. Абсолютные подвижности различных ионов при 18 °C
А е г, 1‘
при — = = 1 в/см, рассчитанные по уравнению
Катион vK, см 1 сек Аннон иа, см/сек
н+ 0,003263 сг 0,000679
Li+ 0,000346 NO- 0,000639
Na+ 0,000450 С107 0,000570
к+ 0,000669 ОН- 0,001803
В противоположность Гитторфу, где, как это следует из рис. 7, ионы двигаются цепочками, механизм Гроттгуса уподобляется удару твердых шаров (где вся цепочка молекул остается в покое и отлетают только крайние частицы). Это и должно было объяснить аномальную подвижность ионов воды.
Однако взгляды Гроттгуса не могут быть приняты во внимание, так как он не предполагал возможности диссоциации молекул и существования свободных ионов в растворе до прохождения тока.
В настоящее время хорошо известно, что диссоциация молекул на ионы происходит при отсутствии тока и что избыточные
40
Теоретческие основы электрохимии
протоны, имеющиеся в водных растворах кислот, не закреплены за определенными молекулами воды, с которыми они образуют ионы Н3О+, а постоянно перемещаются от одной молекулы к другой. Этот процесс перемещения протона был впервые подробно рассмотрен Берналом и Фаулером, которые исходили из следующих предпосылок. Представим себе растворение газа HCI в воде:
НС1 + Н2О -> Н3О+ + С1-.
Ионы гидроксония в растворе будут окружены молекулами воды. Ток переносится скачкообразным переходом протона Н+ от иона Н3О+ к соседней молекуле воды (рис. 11). При этом про-
нго н,о'
Рис. 1'1. Сясема (протонного перехода от иона Н3О+ к гмолекуле воды
тон каждый раз попадает внутрь одной из двух незанятых протонами электронных орбит молекулы воды — в один из двух ее отрицательных полюсов. На основании имеющихся данных о структуре молекулы воды было подсчитано, что от иона Н3О+ к молекуле HgO протон должен пройти расстояние 0,86- 10-8cju, а положительный заряд в результате такого перехода перемещается на 3,1 • 10~8 см (в результате дальнейшей перестройки комплекса). В таком случае электропроводность будет значительно больше, чем это соответствует обычным представлениям Гитторфа.
Поскольку избыточные протоны перемещаются по всем молекулам воды раствора, они сообщают этим молекулам некоторый положительный заряд е. Поэтому, как полагает О. Я. Самойлов,
каждая молекула
ляет собой ион
воды в растворе кислоты в среднем представ-е 14~
Н2 О2 , где е — заряд электро-
на. Очевидно, что е растет с ростом концентрации кислоты. В разбавленных растворах кислоты е е. Величина е пред-
ставляет собой средний заряд молекулы воды за достаточно большой промежуток времени, только небольшую часть которого избыточный протон пребывает в каждом из молекул (эта часть растет с ростом концентрации кислоты).
Движение ионов под действием электрического поля
41
Аналогичным переходом протона от молекулы Н2О к ОН~-иону объясняется кажущееся движение гидроксильных ионов в. обратном направлении. Но так как отрыв протона от молекулы воды происходит с большими трудностями, чем переход протона от иона Н3О+, то подвижность гидроксила меньше, чем подвижность Н+ и Н3О+.
Правомерность изложенных взглядов подтверждается тем фактом, что некоторые другие растворители, которые способны давать водородную связь, также обладают аномальной подвижностью ионов водорода (например, в метиловом и этиловом спиртах). Таким образом, ион водорода, образующий в спирте ион ROHs", способен переходить от иона ROH’t к молекуле спирта ROH в полной аналогии!С1механизмом, описанным для воды. Так как этот переход связан с преодолением энергетического барьера, то эффект аномальной электропроводности уменьшается с увеличением длины цепи молекул спирта.
§ 4. Числа переноса и электродные балансы (электрохимическая стехиометрия)
Хотя количество переносимого электричества остается постоянным вдоль всей цепи, внутри электролита в зависимости от состава компонентов токи разветвляются на параллельные цепи — две, три и т. д. Рассмотрим систему:
Pt+|KNO3, NaCl, H2O|~Pt.
При пропускании через нее тока I он будет переноситься ионами К+ и NO?, Na+ и С1~. Поэтому мы может представить себе, что ток на границе металл — раствор разветвляется и течет по двум параллельным проводникам — K'N'O3 и NaCl (рис. 12). При ЭТОМ I = /кыоэ+ /NaCl-
По закону Кирхгофа,
^KNQ3"^KNO3 = AsiaCl'^NaCl
ИЛИ
ZKNOa __ ^NaCl (И 27)
/NaCl RKN03
Так как далее
где х—'удельная электропроводность проводника, ом 1-см г;
I — длина его;
S — площадь сечения, то /
' /kno.^ xKN03> (П>28>
/NaCl zNaCl
42
Теоретические основы электрохимии
Если обозначить
_ ^NaCl . _ 7KNOs
XNaCl — j ’ XKNO3 j ’
TO
I v ______ '''NaCl . _ *KNOa ztt pox
XNaCl — ’ XKNOa ~ _ * V11»^
'-NaCl + xKNOa xNaCl + xKNOa
Причем
XNaCl = П№+ + nCl—’ XKNOa = Пк+ nNO—3‘ (H,30)
Рис. 12. Разветвление тока в электролите, содержащем две соли (<KNOa и NaCl)
Величины х характеризуют долю участия данной соли в переносе электричества в растворе электролита, содержащем несколько компонентов (солей).
Как отмечалось, изменения концентрации раствора и возникновение новых продуктов электролиза происходят в определенных частях электрохимической системы у поверхности электродов. В связи с этим рассмотрим количественные изменения, происходящие в приэлектродных слоях (католите и анолите) в процессе электролиза.
Следует заметить, что при составлении электродных балансов допускается ряд существенных упрощений, которые искажают картину действительных изменений, происходящих в приэлектродных слоях электролитной ванны. Здесь учитывается только миграция ионов, но не принимается в расчет влияние диффузии и конвекции электролита, которые выравнивают концентрацию раствора: пренебрегают также барботирующим действием газов, выделяющихся при электролизе на электродах. Полученные итоговые балансы справедливы только до тех пор, пока концентрационные изменения, вызванные перемещением ионов, малы *.
1 Балансы составлены для данного .начального момента времени для бес-коненно малого количества прошедшего электричества.
Движение ионов под действием электрического поля
43
Тем не менее даже приближенная картина показывает, что ‘Имеющиеся изменения в составе и содержании веществ в при-электродных слоях (с учетом только миграции и разряда ионов) не всегда сводятся к простой убыли концентрации, но имеют гораздо более сложный характер. Подведение балансов дает основание судить об особенностях и направлении электродных реакций. Можно различить, по крайней мере, четыре возможных /случая связи чисел переноса с изменениями концентрации вблизи электродов.
Электродные балансы в случае уменьшения количества растворенного вещества в электролите
1. Уменьшение концентрации обусловлено уходом из раствора разряжающихся ионов.
Рассмотрим систему
Pt+|HC1, H2O|-Pt.
Так как диссоциация воды ничтожно мала, то следует считать, что в переносе тока участвуют только ионы Н+ и С1~ из соляной кислоты; переносом же тока ионами воды можно пренебречь (последнее относится не только к данному, но и ко всем (Последующим балансам).
При пропускании через систему одного фарадея электричества электродные балансы будут иметь следующий вид:
Катодный баланс Анодный баланс
Разряд: Н+ -f- F -> Н2 С1~ — F -> С12
Перенос: п +Н+— (1 —/I +)С1~ —п +Н+4-(1 — и +)С1~
н н н н
«Сумма: — (1 — п +)НС1 + _п НС1 _1_С1 р , - 2
+ 4-H.-F
Или так как число переноса Н+-иона в НС1 примерно 0,83, то сумма будет соответственно равна: —0,17 НС1+—,Н2—F; — 0,83 HCI + -|-С12 + F.
Суммарный процесс в ванне сводится к реакции НС1^—На + —С12.
2 2
В приэлектродных слоях раствора концентрация кислоты понижается.
2. Уменьшение концентрации происходит вследствие пере-:ход.а ® другие ионные комбинации.
44
Теоретические основы электрохимии
Рассмотрим систему
Pt+|Na2SO4, H2O|-Pt, в которой катодное и анодное пространства разделены диафрагмой.
Так как в реальных условиях ни Na+, ни ЗО^-ионы не могут разряжаться на электродах, то при пропускании тока вода фактически будет электролитически разлагаться. В таком случае получим:
Катодный баланс Анодный баланс
Диссоциация: Н2О -> Н+ -(-ОН- Разряд: Н+ 4. F -> — Н2 2 2 Перенос: 4-0,4Na+ — О.бЗО?- 2 4 Н2О -> Н+ + он-он- — F -» — Н2О 4- -А- о2 — 0,4Na+ 4- — 0.6SO2- 2 4
Сумма: NaOH — 0,6Na„SO. 4- 2 Z 4 4-Ah2-H2O-F vH^.-yO®W + + фо,-фн,о + т
т. е. в католите растет концентрация щелочи и выделяется водород, в анолите повышается концентрация кислоты и образуется кислород *. Защелачивание католита и подкисление анолита — обычные явления, сопровождающие электролиз водных растворов солей металлов. Суммарный процесс в системе выражается уравнением
— Na2SO4 + — Н2О -> NaOH + — H2SO4 4- — Н2 + — О2.
2 2 2'2 4
Электродные балансы при постоянном содержании растворенного вещества в электролите
1. Уменьшение концентрации вследствие разряда ионов компенсируется за счет молекул растворителя.
Рассмотрим систему
Ni+|NaOH, H2O|~Ni, которая представляет собой модель ванны для электролиза воды. Так как электролиты подобных ванн состоят из достаточно концентрированных щелочей (pH раствора больше 14 единиц),
1 Образующиеся в процессе электролиза новые ионы в дальнейшем также ^начинают принимать участие в переносе тока, а доля тока, переносимого «первичными ионами», соответственно падает.
Движение ионов под действием электрического поля
45
то естественно предположить, что из-за весьма малой концентрации ионов Н+ и отсутствия других катионов, которые могут принимать участие в катодном процессе, единственно возможной катодной реакцией будет разряд молекул воды.
Таким образом, будем иметь:
Катодный баланс Анодный баланс
Разряд: Н2О+ F -> — Н2 + ОН- ОН- — F -> — + Н2О + — О2
2 2 4
Перенос: 0,2Na+ — 0,8ОН~ — 0,2Na+ Ц- 0,8ОН—
Сумма: 0,2NaOH—Н2О+^-Н2—F —0,2NaOH+ H2O+-^-O24-F
Суммарный процесс в системе
— Н2О-> —н2 + — О2, 2 * 2 * 2 2 4 2
т. е. разлагается вода, между тем как общая концентрация едкого натра при электролизе не меняется.
2. Уменьшение концентрации вещества в электролите к о м-пенсируется растворением металлического инода.
Рассмотрим систему
Cu+|CuSO4, Н2О|-Сн.
В электролите имеются ионы Cu2+, SO2-, Н+ и ОН-. Пропуская один фарадей электричества, получим;
Катодный баланс Анодный баланс
Разряд: J- Сц 2+ + F — — Си — Си — F— Си2+ 2 2 2 2
Перенос: J- 0,ЗСи2+— - 0,7SO2~ — — 0,ЗСи2+ + — 0,7SO2~
2 2 4 2 2 4
Сумма:----L 0,7CuSO4 + -Си—F + — 0,7CuSO4----------- Си + F
2 2 2 2
Суммарной химической реакции в этом случае не будет, так
как катодный и анодный процессы проходят таким образом, что восполняют один другой. Смысл этого процесса, имеющего большое практическое значение, состоит в растворении чернового металла на аноде и осаждении того же металла уже без примесей на катоде. Балансы свидетельствуют, что в результате прохождения тока и в таком случае концентрация веществ в приэлект-родных слоях изменяется. Такие ванны применяются для электрорафинирования (очистки) тяжелых цветных и благородных
46
Теоретические основы электрохимии
металлов (меди, серебра, сурьмы и др.). В качестве анодов-здесь используют металлы, содержащие ряд примесей, от которых освобождаются при электролизе.
Для первых двух случаев электродных балансов, когда общее количество молекул вещества в электролите уменьшается^ изменения концентрации основного электролита подчиняются уравнению
причем при пропускании одного фарадея электричества убыль концентрации в грамм-эквивалентах пропорциональна соответственно:
Дб?к Па> ^^а
Для третьей и четвертой групп балансой, когда общее число молекул электролита в растворе не уменьшается, справедливо уравнение:
___ Изменение количества вещества у катода а Число пропущенных фарадеев электричества
ИЛИ
(11.32) mF
Рассмотрим примеры составления электродных балансов для реальных ванн, когда составы электролитов сложнее рассмотренных выше.
Электродные балансы ванн со сложным составом электролита 1
Ванна для электроизвлечения цинка. В упрощенном виде такую ванну можно представить схемой:
TWe+|ZnSO4, Н2О, H2SO4|-Ме2.
В этом случае на катоде будет проходить разряд ионов Zn2+ и Н+. Если обозначить выход по току цинка А, то выход по току водорода выразится как (1—А). В переносе тока участвуют как ионы Zn2+ и SOf- (соли), так и ионы Н+ и SO4~ (кислоты). Если обозначить долю тока, переносимую ионами соли, через X, то
х _ *ZnSO4
xZnSO4+ XH2SO4
1 Имеется в виду начальный состав электролита.
Движение ионов под действием электрического поля 4Т
а доля тока, переносимая кислотой, 1-х=- — .
*ZnSO4+ ''H2SO4
где х — удельная электропроводность электролита.
При пропускании двух фарадеев электричества получим: Катодный баланс:
Разряд: AZn2+ + 2AF -> AZn
2(1 — А)Н+ + 2(1 — A)F ->(1 — А)Н2
Сумма: AZn2+ + 2(1 — А)Н+ + 2F -> AZn + (1 — А)Н2
Перенос: ХП 2 Zn2+ — х (1 — П 2 ,) SO*-
Zn Zn
+ (1 - х) п + 2Н+ - (1 - х) (1 - п +) SO2-м п
Сумма: хп Zn2+-}-(l—х)п ,2Н+—[х(1—п 2,) + (1—*)(1—п ,)]SO
zn "* н"г Zn ~ i-r 4
Суммарный процесс на катоде:
+ Лгп + (1 — А)Н2 — (А — хп 2+)ZnSO4 —
— (1 — А — п , + хп ,)H2SO, — 2F. н-г нт
Анодный баланс (в случае нерастворимого анода в кислой среде):
Разряд: Н2О — 2F -> 2Н+ + -у- О2
Перенос:—хп 2 Zn2+ + X (1 — П ) SO2-
Zn 2+ Zn2+ 4
— 2(1 — х)п ,Н+ + (1 — х)(1 — п JSO2-н'1' н"1”
Сумма: —’Xn ZnSO. — Н2О 4- [(1 — П , ) ф- ХП , ]
’ Zn + н +
H2SO4+^-O2 + 2F
Суммарный процесс, идущий в системе, может быть описан уравнением
AZnSO4 + Н2О -> AZn + (1 — А) Н2 + AH2SO4 + -у О2.
Таким образом, катодный и анодный балансы в цинковой ванне, снятые в начальный момент, без учета диффузии и конвекции показывают, что по мере осаждения металлического цинка на катоде в электролите уменьшается концентрация ZnSO4 и
48 Теоретические основы электрохимии
возрастает концентрация H2SO4. Этот вывод полностью согласуется с опытными данными.
Ванны, содержащие комплексные соли металлов. Рассмотрим картину концентрационных изменений в католите в предположении, что преобладающей формой комплекса при сравнительно небольшом избытке комплексообразо-вателя (КХ) будет соединение типа КМеХ2.
В таком случае ванну для электроосаждения металла можно представить схемой:
/Ие+|КЛ1еХ2; КХ, H2O|~Me.
В электролите возможны ионы: К+. Н+, 7ИеХ7, Х~ и ОН-. Обозначим через у долю тока, переносимого в электролите ионами комплексного соединения КА4еХ2 и через (1 — у) — долю тока, переносимого ионами комплексообразователя КХ. Пусть далее ni и п2 выражают числа переноса анионов в этих солях, а Л и (1 —А) — выходы по току Me и Н2.
Тогда, исходя из возможности совместного разряда на катоде ионов МеХТ и Н+, получим следующий катодный баланс:
Разряд: АМеХ~'+ AF -> АМе + 2АХ~~
(1—Л)Н2О-^(1—Л)Н+ + (1—Л)ОН-
(1— Л)Н+ ->(1- Л)К-(1 — Л)Н2
Перенос: у (1 — п() К+ — уп{МеХ~ + (I — у)(1 — П2) К+ +
+ (I — у)п2Х~
Сумма: — (Л + уп^ КМеХ2 + (2Л — п2 ц2у) КХ — (1 -—
— Л)Н2О + (1 — Л)КОН Н-ЛМе+ ~ (1—Л)Н2 — F.
Таким образом, у катода концентрация КМеХ2 убывает, в то время как содержание комплексообразователя (КХ) и щелочи (КОН) непрерывно увеличивается.
Прикладное значение чисел переноса не исчерпывается составлением электродных балансов. Очень часто числа переноса определяют выход по току в реальных электрохимических системах.
Например, в ванне для электролитического получения хлора:
Fe-|NaCl, Н2О|+С выход по току хлора определяется уравнением
I 1 . G1 I
Движение ионов под действием электрического поля 49
где лон~ — число переноса ионов ОН~;
а — постоянная;
С\ и С2— концентрация NaCl и щелочи в ванне (щелочь накапливается в процессе работы).
Знание чисел переноса во всех случаях углубляет наши представления о действительных процессах, происходящих в электрохимических системах, и позволяет решать ряд важных для технологии задач. С помощью чисел переноса можно судить о строении некоторых молекул. Так, например, было показано, что в смесях двух солей типа KCN и Fe(CN)2 при пропускании тока ионы, содержащие железо, концентрируются у анода. Последнее указывает., на комплексный характер этой соли: K4Fe(CN)6->4K+ + Fe(CN)t"
То же было установлено для 2NaCl • PtCl4(Na2PtCl6). Для CuBr2 была показана непрерывность перехода от простой соли к комплексной (по мере роста концентрации).
ЛИТЕРАТУРА
Bernal J. D., Fowler R. J. Chem. Phys. I, 515, 1933; Успехи физ. наук, 14, 586, 1934.
Гаев А. И. и Есин О. А. Электролиз цинка. Металлургиздат, 1937.
Грубе Г. Основы электрохимии. Госхимиздат, 1932.
Крейтон Ж. Основы электрохимии. ОНТИ, 1937.
Левин А. И., П о м о с о в А. В., У к ш е Е. А. Руководство к лабораторным занятиям по теоретической электрохимии. Изд. УПИ им. С. М. Кирова, 1957, гл. II.
Л е в и н А. И. Труды УПИ, Ns 43, Машгиз, 1953; № 27, Металлургиздат, 1947. Стендер В. В. Прикладная электрохимия. Изд. Харьковского университета, 1961.
Семенченко В. К. ЖНХ, 1, 1956, 1131.
Соколов Н. Д. Успехи физ. наук, 37, 1955, 205.
Строение и физические свойства вещества в жидком состоянии, Материалы совещаний, Киев, Изд. КГУ, 1954.
Яцимирский К. В. Термохимия комплексных соединений. Госхимиздат, 1951.
4 Заказ 1338
Глава III
ПАДЕНИЕ НАПРЯЖЕНИЯ В ЭЛЕКТРОЛИТЕ. КОНДУКТОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА
§ 1. Электропроводность электролитов
Падение напряжения в системе при прохождении электрического тока есть результат того, что система оказывает току некоторое сопротивление и тем большее, чем больший ток проходит через единицу сечения проводника в единицу времени.
В соответствии с законом Ома падение напряжения в элек-ролите
еэ = /7? (II 1,1)
определяется при заданном токе его сопротивлением 7<. Сопротивление проводника зависит от его природы и геометрических размеров — длины I и поперечного сечения 5:
= (III,2)
О
Формула эта справедлива для постоянного значения S.
Из формулы (III, 2) следует, что удельное сопротивление электролита равно сопротивлению объема раствора, имеющего форму кубика с длиной ребра 1 см. В электрохимии чаше пользуются величиной, обратной удельному сопротивлению, носящей название удельной электропроводности:
х=—. (III,3)
Р
Единицей электропроводности является «обратный ом» (ом~х) — электропроводность проводника с сопротивлением в один ом. Удельная электропроводность * х измеряется в ом~х • см~1.
1 По Международной системе СИ удельное электрическое сопротивление измеряется в омметрах (ом-м), а удельная электропроводность в сименс на метр (сим/м).
Падение напряжения в электролите
51
Пользуясь данным ранее определением плотности тока i из уравнений (III, 1), (III, 2) и (III, 3), легко получить
еэ = —. (Ш,4)
Следовательно, падение напряжения в электролите определяется плотностью тока t, расстоянием между электродами I и ущельной электропроводностью я. Первые две величины .мы можем изменять по своему желанию. Последняя является свойством изучаемых систем; она зависит от природы электролита и растворителя, их состава и температуры. В некоторых случаях при весьма высоких напряжениях и частотах тока величина я для растворов электролитов начинает зависеть и от напряженности внешнего поля.
Ток, переносимый i-тым сортом ионов, связан с подвижностью и концентрацией этих ионов в растворе уравнением
/. = СЛДеу-. (П1.5)
Если для простоты ограничиться раствором, содержащим только два сорта ионов (бинарный электролит), то общий ток, проходящий через систему, выразится как сумма токов, переносимых катионами и анионами:
/ = /к + /а (П 1,6)
и, следовательно,
1 = + (III,7)
так как
CK=Ca = Ci.
Но Де есть не что иное, как падение напряжения в электролите и, следовательно, подставляя его значение из формулы (Ш, 1) в выражение (IIL7), получим
откуда
Сравнивая это уравнение с уравнением (III, 2) и учитывая, что I, = FUi (где F — константа Фарадея, С7,- — подвижность ионов), можем написать:
р = -----1-----= -----J—- (III,9)
С; (/к + la) FiF (UK -рUa)
И
(Ш,10)
* — (1К + /а).
4»
52
Теоретические Основы электрохимии
Рассмотрение полученного равенства приводит к выводу, что связь удельной электропроводности с концентрацией и природой растворенных ионов, а также с температурой и природой растворителя определяется влиянием этих параметров на подвижность ионов.
Зависимость удельной электропроводности от температуры часто выражается эмпирическими формулами типа
х/ = х18[1+а(/-18)+Р(г-18)г+ (111,11)
причем температурные коэффициенты для растворов солей и щелочей лежат в интервале 0,02 — 0,025, а для растворов кислот— в интервале 0,01—0,016.
Электропроводность проводников второго рода увеличивается с ростом температуры. Однако зависимость удельной электропроводности от температуры часто отклоняется от прямолинейной и при некоторых температурах наблюдается максимум электропроводности. Эта зависимость является следствием ряда причин:
а) снижение вязкости растворов с повышением температуры ведет к росту подвижностей ионов, а следовательно, и к увеличению электропроводности;
б) уменьшение радиусов ионов вследствие частичной дегидратации с ростом температуры также приводит к росту электропроводности и подвижностей;
в) уменьшение диэлектрической постоянной раствора с ростом температуры связано с увеличением взаимодействия ионов и, таким образом, со снижением подвижности и электропроводности.
Если бы концентрация раствора не влияла на подвижность ионов, то удельная электропроводность изменялась бы прямо пропорционально концентрации. Однако на самом деле между этими двумя 1вел.ичина(М'И 1существует гораздо более сложная зависимость, так что кривые х = f(C) имеют максимум (рис. 13).
Максимальная удельная. электропроводность растворов NaOH наблюдается при 4 г-экв/л (20 °C), NaCl — при 5 г-экв]л, КОН — при 7 г-экв]л и т. д.
Для малорастворимых электролитов максимум может лежать за пределами растворимости (пунктир на рис. 13). Такой вид кривых объясняется тем, что рост концентрации ионов в растворе соответствует числу частиц, переносящих ток. Но, с другой стороны, при этом увеличивается тормозящее взаимодействие ионов.
Ввиду сложности зависимости х = f(C) в электрохимии для характеристики состояния электролитов часто пользуются понятиями эквивалентной и молекулярной электропроводностей. Эти понятия были введены Ленцем в 1887 г.
Падение напряжения в электролите
53
Эквивалентная электропроводность есть электропроводность грамм-эквивалента электролита, заключенного между электродами соответствующей поверхности и имеющего толщину слоя I см (рис. 14). Аналогично определяется молекулярная электропроводность Лм. Очевидно:
= . (HI.12)
С*
где V = -----разведение, л!г-экв. Если концентрация выражена
в молярности, то V = и л = 100(1 —. Так как х имеет размерность ом-1 • см-1, то размерность X равна ом~~! • см2.
о 2 4 6 Я 10 12 14 пан цент рация Z-экв/л
Рис. ,13. Зависимость удельной электропроводности от концентрации
Рис. 14. Связь менаду эквивалентной и удельной электропроводностями
Зависимость эквивалентной электропроводности от разведения электролита иллюстрируется рис. 15. Эта зависимость определяется уравнениями (III, 10) и (III, 12), из которых вытекает
+ (П1ДЗ)
и в общем случае
Однако полученное уравнение относится к случаю, когда все молекулы электролита продиссоциировали на ионы, т. е. когда Ск = Са = С. В противном случае
Ас = «0к + О- (111,14)
Как будет показано в дальнейшем, а — 1 при бесконечном разбавлении раствора * 1 и поэтому уравнение (III,13) определя-
1 Практически раствор считают бесконечно разбавленным при содержании
1 моль вещества в 10 000 л.
54
Теоретические основы электрохимии
«ет электропроводность при бесконечном разбавлении (Лоо), а «формула (III, 14) —электропроводность при некоторой концентрации С(Хс). Из этих уравнений следует:
а=—. (Ш,15)
Лоо
Как видно из рис. 15,'зависимость Л от С такова, что Л стремится к некоторому пределу, отвечающему Лоо- Эта зависимость
g/50
>§§./50 з м&го
лп
1000
Разведение =V, nji-эиЁ
Рис. 15. Зависимость эквивалентной электро-1 \ проводности (X) от разведения (V = ~ 1
электролита
приближенно описывается эмпирическим уравнением Коль-рауша:
(Ш,16)
где А — постоянная.
Последнее уравнение дает хорошие результаты только в применении к достаточно разбавленным растворам. Для более концентрированных растворов (С = 0,001 —0,1 г-экв/л) многих одновалентных солей типа КС1, NaCl справедливо уравнение Гоша: f
^с = ^-А/С, (111,17)
где величина Aj близка к 400.
Часто применяется уравнение к растворам, концентрации которых не превышают 0,5 г-экв/л-.
С (Ле — Лк,)
= Д,+Д2(С1хсг,
(111,18)
где А1, Аг, m — эмпирические постоянные.
Так как в растворах электролитов электрический ток переносится ионами, то для одинаковых концентраций и валентностей
Падение напряжения в электролите 55
ионов лучшим проводником будет тот раствор, ионы которого движутся быстрее. Кольрауш экспериментально нашел, что разница в значениях электропроводности при бесконечном разведении для растворов солей, содержащих один общий ион, постоянна: Л сод^ —^coKsz = const. В самом деле, используя правило (III, 13), можно написать для уксусной кислоты:
Хсн,соон ZH+ + Zch3coo~
и, так как
I + -|-1 — = I 4. ® Ч-1 4- “Ь ——I + — —»
Н СН3СОО H^ Cl Na^ CN„COO NaT Cl
TO
\oCH3COOH ~ \oHCl ~Ь \oCOsCOONa \»NaCl
ИЛИ
\oCH3COOH ^ooHCl ~ ^coCH3COONa ^coNaCH
В общем случае
X „ . == X „ . 4- X „ . — X „ . . (111,19)
ooK,4t coKtA2 1 coKg/l, ооКгЛ2- ' ’ '
Различия между величинами Ас и у сильных электролитов в водных растворах не могут быть объяснены с помощью представлений о неполной диссоциации. Было сделано предположение, что количество ионов в растворе остается неизменным, а электропроводность падает в связи с падением подвижности ионов в растворах. В действительности по отношению к концентрированным растворам сильных электролитов это предположение также неприменимо, так как электропроводность в таких растворах зависит .и от изменения подвижности ионов и от перемены величины степени ассоциации. Что касается разбавленных растворов сильных электролитов, в которых допускается полная диссоциация молекул на ионы, электропроводность меняется с концентрацией также вследствие изменения подвижности ионов. Подвижность ионов в зависимости от концентрации изменяется вследствие изменения плотности ионной атмосферы, возникающей вокруг ионов.
§ 2. Методы экспериментального измерения электропроводности
Электропроводность — величина, обратная сопротивлению, поэтому ее измерение сводится к определению сопротивления электролитов. В связи с тем что при пропускании через электролит постоянного тока на электродах происходят процессы, резко увеличивающие сопротивление системы (поляризация), сопротивление электролитов обычно измеряют с помощью переменно
56
Теоретические основы электрохимии
го тока высокой частоты и с применением достаточно больших электродов (с этой целью платиновые электроды платинируют). Исследуемый раствор помещают в1 ячейки различной формы (в зависимости от электропроводности испытуемых растворов) с впаянными в стекло электродами (рис. 16). Ячейки для определения электропроводности готовят из специального стекла с ми-
Рис. 16. Ячейки для измерения электропроводности:
а — для растворов с большой электропроводностью; б, е, г — для растворов с малой электропроводностью
нимальным коэффициентом теплового расширения. Это делается для предотвращения возможного изменения расстояния между электродами.
Для измерения сопротивления электролитов пользуются мостами переменного тока (рис. 17). Натянутая калиброванная проволока ab имеет три контакта — неподвижные а и b и подвижный d. К неподвижным контактам присоединены: исследуемое сопротивление Rx и магазин сопротивления /?м. Скользящий контакт соединяется с телефоном Т или другим нуль-инстру-ментом, например с катодным осциллографом или ламповым вольтметром. Второй провод от телефона соединяется с точкой с, лежащей между Rx и /?м. Кроме того, к контактам а и b присоединяется генератор переменного тока звуковой частоты ГЗ.
Измерение электропроводности (или сопротивления) с помощью моста состоит в том, что перемещением движка d по проволоке ab добиваются минимума звука в телефоне, что указывает
Падение напряжения в электролите
57
на отсутствие тока в цепи cTd. В этом случае, согласно закону Кирхгофа (рис. 17):
IaRu =- Wad, iaRx = Wdb, (111.20>
откуда условие равновесия моста
_ (III,21)
RM Roa
Если поменять местами генератор и телефон, равновесие моста не нарушится.
Рис. 17. Мост переменного тока для измерения сопротивления электролитов
Так как измерительная проволока ab однородна по сечению и сопротивление любого ее отрезка пропорционально его длине, то
= (1П.22)
lad lad
где lab, lad и 1аъ — длины соответствующих участков струны, изменяющиеся при передвижении скользящего контакта. Перепишем уравнение (III, 22) в виде:
А
и продифференцируем его по X
R'x(X) = -R±-. (111,23)
58 Теоретические основы электрохимии
Относительная погрешность минимальна, когда R’XWV 2Х-1
RAX)I (1-XfX* U’
что отвечает условию X = ~. Иными словами, погрешность измерения по методу моста будет минимальной в том случае, когда компенсация происходит вблизи середины струны ab. Следовательно, наилучшие условия компенсации достигаются при
RX = RU. (111,25)
Отсутствие звука в телефоне и, следовательно, отсутствие тока в цепи возможно при равенстве потенциалов в точках с и d. Но полностью достигнуть этого в мостике переменного тока нельзя в связи с тем, что к омическому сопротивлению цепи добавляется реактивное сопротивление, обусловленное самоиндукцией L и емкостью с.
Чтобы уточнить результаты измерений, нужно устранить реактивные сопротивления в отдельных ветвях измерительного контура. Для этого соединительные провода следует брать короткими с большим сечением, избегать их пересечения и закручивания спиралью. Контакты нужно тщательно зачищать и припаивать. Кроме того, ветви мостика экранируют, а экран заземляют.
Однако все эти меры не устраняют емкостного сопротивления ячейки, вследствие чего в момент компенсации сила тока в цепи не равна нулю и в телефоне слышится минимум звука. При ответственных измерениях емкость измерительной ячейки нужно специально скомпенсировать. Для этого пользуются тем, что емкостное сопротивление отстает по фазе от сопротивления самоиндукции. В цепь ячейки последовательно включают переменную индуктивность (магазин индуктивности) и подбирают Ам таким образом, чтобы выполнялось условие:
<^=-1-. (II 1.26)
0>С
В этом случае точке компенсации будет соответствовать полное отсутствие тока в телефоне и точность измерений повысится.
Для многих целей, однако, точность, достигаемая при работе на обычной схеме (до 0,1%), вполне достаточна.
§ 3. Кондуктометрический метод анализа
На измерении электропроводности раствора основан метод установления эквивалентной точки — кондуктометрический анализ. Аппаратура для кондуктометрического титрования несколь-
Падение напряжения в электролите
59
ко отличается от той, которая применяется при измерении электропроводности. При титровании весьма неудобно пользоваться телефоном, поэтому его в схемах для кондуктометрического анализа обычно заменяют гальванометром с выпрямителем (кенотрон), как это показано на рис. 18.
Так как величина электропроводности концентрации и от подвижности отдельных ионов, то кондуктометрическое титрование может быть применено к изучению процессов, которые сопровождаются изменением и числа и характера ионов в растворе.
Чтобы яснее представить себе принцип кондуктометрического анализа, рассмотрим изменение электропроводности какого-либо сильного электролита, например AgNO3, при добавлении к нему реагента NaCl, дающего с одним из ионов (Ag+) нерастворимое соединение:
раствора зависит от LubuHuuuJ.
=. ,у= ТР
О
Рис. 18. Схема кондуктометрического моста с выпрямителем:
Тр, Tpi — трансформаторы; Э — электролизер;
В — выпрямитель; Г — гальванометр; R реостат; А—Б — реохорд
Ag+ + Na+ + NO- +
4- Cl- - AgCl + Na+ + NO-. Электропроводность
участвующих
в реакции ионов: При "
25 °C
62
50,5
72
При
18 °C
53,8
43,3
61,6
65,2
добавления NaCl ввиду
76
мере
ZAg+...........
ZNa +..........
ZNO-............
zci—...........‘
Так как/Ag+ >1 Na+, то no
замещения Ag+-HOHOB ионами Na+ электропроводность раствора будет 'уменьшаться до тех пор, пока все ионы Ag+ не будут связаны в AgCl. Дальнейшее добавление NaCl приведет к росту электропроводности.
Таким образом, откладывая на графике электропроводность раствора против количества добавленного реагента (NaCl), мы можем определить точку эквивалентности по минимуму электропроводности. Для кондуктометрического титрования совсем не обязательно вычисление электропроводности, так как ни сопро-
60
Теоретические основы электрохимии
тивление R, ни электролитическая емкость сосуда с при титровании в большинстве случаев не изменяются. Электропроводность х, таким образом, пропорциональна отношению отрезков реохорда (см. рис. 17).
Поэтому на практике при титровании строят график х = = f(m), где m — число миллилитров прибавляемого реактива. Практически титрование обычно ведут в закрытом (рис. 19, а) или открытом (рис. 19, б) сосуде, в
который опущены два спаянных стеклянной каплей платинированных элек-трода (рис. 19, в). Во время измерений необходимо сохранять постоянство температуры, которая сильно влияет на точность измерений.
При кондуктометрическом анализе титрованный раствор подбирают таким образом, 'чтобы после достижения эквивалентной точки и появления в растворе избытка реагента ход изменения электропроводности раствора менялся по возможности наиболее резко.
Точность кондуктометрического титрования вообще невелика и не превышает 0,5—4,0%. Более точные результаты можно получить, если учесть изменение объема при титровании. Преимущество кондуктометрического метода анализа — возможность приме-
Рис. .19. Сосуды для кондуктометрического титрования; а — закрытый; б — открытый; в —электроды, применяющиеся при кондуктометрическом титровании
нения его для технически загрязненных и окрашенных жидкостей. Здесь отсутствует необходимость точного определения эквивалентной точки, так как его находит графически.
Рассмотрим основные случаи применения кондуктометрического титрования.
Титрование сильной кислоты сильным основанием или силь-
ного основания сильной кислотой (рис. 20):
НС1 + NaOH -> NaCl + Н2О;
Н+ + Cl- + Na+ -Ь ОН- -> Na+ + Cl- + Н2О.
Таким образом, реакция сводится к образованию нейтральной молекулы воды из Н+ и ОН-. Ионы Na+ и С1“ не принимают участия в реакции. Так как ион водорода обладает наибольшей подвижностью (/ + = 350 при 25°C), то прибавление щелочи, т. е. замена водорода на Иа+-ион, .ведет к сильному уменьшению электропроводности.
Падение напряжения в электролите-
61
После достижения точки эквивалентности избыток Опционов вызывает резкий подъем кривой, так как в раствор попадают обладающие большой подвижностью ионы ОН~(/он_ = 196 при 25 °C). Минимум выражен очень резко и при аккуратной ра-
Количество No ОН, см3
Рис. 20. Титрование сильной кислоты сильным основанием-
боте почти точно отвечает pH = 7.
Титрование слабой кислоты сильным основанием или слабого основания сильной кислотой (рис. 21). В этом случае форма кривой нейтрализации зависит от концентрации и степени диссоциации слабой кислоты (или основания). Например, для уксусной КИСЛОТЫ;
СН3СООН + NaOH-> CH3COONa + Н2О
Здесь уксусная кислота диссоциирована лишь частично, а уксуснокислый натрий — полностью. Поэтому при добавлении сильной щелочи, с одной стороны, электропроводность будет падать вследствие нейтрализации диссоциированной части кислоты, а с другой — возра-" - - — -------------------------- диссоциирующей соли.
хорошо
стать вследствие образования
Рис. 21. Титрование слабой кислоты сильным основанием:
а и с — -конец титрования; Ь — касательная в этих точках
Рис. 22. Титрование борной кислоты. Точка перелома кривой указывает на конец титрования
Точка эквивалентности в таких случаях выражена очень слабо. Добавление избытка NaOH вызовет резкий подъем кривой. Точку эквивалентности находят обычно, продолжая солевую линию acb (рис. 21).
В случае, если кислота очень слабая (например 0,1-н.. Н3ВО3), электропроводность непрерывно повышается вследствие образования соли (рис. 22). Но в точке эквивалентности происходит перегиб (избыток ОН-ионов).
62
Теоретические основы электрохимии
Результаты измерений вблизи точки эквивалентности сильно искажаются вследствие влияния одновременно идущего процесса гидролиза, поэтому при построении кривой титрования таких осложнений следует избегать.
Нейтрализация слабых кислот слабыми основаниями
В случае нейтрализации уксусной кислоты 4рис. 23):
аммиаком
СН3СООН + NH4OH NH4CH3COO + Н2О
[одержание ИН^0Нгсм3
Рис. 23. Титрование 'слабых кислот слабыми основаниями
в начале титрования возможно небольшое снижение электропроводности ввиду нейтрализации Н+-ио-нов (участок аЪ), затем образование хорошо диссоциирующей соли CH3COONH4 приводит к возрастанию электропроводности до точки эквивалентности (fee). Дальнейшее добавление аммиака, который является слабым основанием, почти не изменяет электропроводности (cd). И в этом случае результаты могут быть сильно искажены вследствие влияния процесса гидролиза.
Титрование смеси сумной и слабой кислот сильным основанием
При титровании смеси уксусной и соляной кислот едким натром (рис. 24) вначале идет титрование сильной кислоты НС1 с точкой эквивалентности Ь. Затем добавление щелочи вызывает появление «солевой» линии fee, а во второй точке эквивалентности имеется перегиб с и дальше наблюдается резкое увеличение электропроводности вследствие появления избытка гидроксильных ионов.
Титрование сильным основанием смеси сильной кислоты и ее соли с образованием плохо растворимой гидроокиси металла
Примером может служить анализ смеси CuSO4 + H2SO4. При титровании щелочью сначала оттитровывают кислоту, а затем соль меди по реакции
CuSO4 + 2NaOH Си (ОН)2 + Na2SO4.
Избыток щелочи обусловливает повышение электропроводности. Таким образом, в этом случае кривая титрования также
Падение напряжения в электролите
63
имеет три ветви. Возможность титрования смеси двух компонентов — одно из важнейших преимуществ кондуктометрического метода анализа.
Очень важен правильный подбор реагентов для титрования. Так, если в рассмотренном выше примере с AgNO3 (см. стр. 59}
вместо NaCl использовать для титрования хлористый барий, то ввиду того что электропроводность Ва2+-иона (/Ва2+ = = 64)почти не отличается от электропроводности Ag+-HOHa, точка эквивалентности будет очень неявно выражена. Наоборот, при применении LiCl (ZLi = 39) можно получить наилучшие результаты. Опыт показывает, что при определении катионов реактив должен быть таким, чтобы его катион был менее подвижен, чем определяемый.
Определение растворимости труднорастворимых солей кондуктометрическим методом
/ П , Содержание НаОН,см^
Рис. 24. Титрование смеси сильной и слабой кис-
лот.
Участок ab титрование сильной кислоты (Ь — точка эквивалентности Z). Ъс — добавление щелочи (солевая линия); cd — появление избытка гидроксильных ионов (с — вторая точка эквивалентности II)
Концентрация растворов труднорастворимых солей обычно столь мала, что можно пренебречь силами междуионно-го взаимодействия и считать, что эквивалентная электропроводность таких рас
творов равна электропроводности при бесконечном разбавлении, т. е. что коэффициент электропроводности (см. стр. 96) их равен единице:
7-ос
С другой стороны, электропроводность таких растворов сама по себе весьма незначительна и при ее определении приходится учитывать электропроводность воды (хв). Таким образом, измеренная электропроводность насыщенного раствора труднорастворимой соли связана с электропроводностью этой соли (хс) равенством:
X = X -г X .
р-ра с 1 в
Эквивалентная электропроводность соли равна:
(1.11,27)
2^-----Б_.1000,
С
€4
Теоретические основы электрохимии
откуда концентрация соли
С = ХР-ра~>_. 1000, хс
а так как = /к + /а. то
с = .->Р.а~Хв . 1 000. (III ,28)
1к + ia
Измерив удельную электропроводность раствора и чистой воды и воспользовавшись табличными данными для ионных электропроводностей, можно определить растворимость труднорастворимой соли.
Исследование комплексных соединений кондуктометрическим методом
Измерения электропроводности позволяют решить ряд задач в химии комплексных соединений. В частности, этим методом можно определить состав и константу нестойкости комплексных ионов в растворе.
Представим себе, что мы имеет два раствора солей CuSQj и СбН5О7Иаз. Соли эти практически нацело диссоциированы:
CuSO4 ? Cu2+ + SO2-; C6H6O7Na3 3Na+ + С6Н «.Of-. ,
При сливании растворов образуется комплексный цитрат меди:
Си2+ + С6Н5()з- 3 [Си (С6Н5О7)]-. (а)
В результате электропроводность смеси оказывается гораздо меньшей, чем она была бы, если бы реакция комплексообразования не происходила.
Так как в реакции комплексообразования участвуют только ионы Си2+ и СбНбОт-, то разность между предполагаемой электропроводностью смеси (хс) и суммарной электропроводностью (хЕ ) можно подсчитать по уравнению
1000 Az -= 1000 (xs - zc) = Ckz+ l+ + 2Cz_ Г - Crzk Ik,
где Z , ZK — ионные электропроводности соответственно, Cu2+, CeHsO? hCu (C6H5O7)3 g
z ,'z_, zK —заряды соответствующих ионов;
Ск — концентрация Си (С6Н5О7)-, г-ион!л.
В общем случае:
mMez+ + /?QZ“ = МетС1гпк (zK = z+ — z_), (б)
где MemQ*K — комплекс,
Падение напряжения в электролите
65
можно написать:
1 000 Дх = mC z. I, + пС z I — Cz I (111,29)
Если известны электропроводности и заряды соответствующих ионов, а также состав комйлекса, то, определив экспериментально величину Дх, можно подсчитать по уравнению (111,29) значение Ск. Известно, что константа нестойкости комплексного иона типа MemQ** определяется равенством
Наиболее точным является графический метод исследования состава и прочности комплексных ионов или метод непрерывных изменений.
Величина Дх при смешивании растворов, содержащих ионы Mez+ и Qz~ в разных пропорциях, будет тем больше, чем больше концентрация образующегося комплекса MemQ^K.
Если приготовить растворы соли, содержащей ион металла Mez+ концентрации Со и анион Qz~ концентрации Со = рСо и слить их в такой пропорции, чтобы на единицу объема (1 л) приходилось (1 —х) л первого и х л второго раствора, то после установления равновесия (б) концентрации соответствующих ионов будут определяться равенствами:
= (Ш,30)
C+=C0(l-x)-mCK, (III,31)
С_ = Сорх — пСк. (III ,32)
Изменяя х, т. е. смешивая растворы в различных пропорциях, получим ряд концентраций комплекса, так как Ск зависит от х. Максимум концентрации ионов MemQ*K при изменении х-можно найти из условия:
= 0. (III,33)
dx
Продифференцировав уравнения (111,30) — лучим: dCc „ dC mC —i + пС' = 0, dx "* dx (111,32) no x, no- UH,34)
dC+- с dx (111,35)
dC- , r h рСо, dx (111,36)
5 Заказ 1338
66 Теоретические основы электрохимии
что в совокупности с предыдущими четырьмя уравнениями позволяет найти:
= Срп^гП *РП 1 [(pm 4- п) Хо — n]m+n (Ju
mn— 1 пт— 1 — ^rn-J-n—I п)х0]
где х0 — значение х при = 0.
dx
Построив кривую зависимости Az от jc и определив х0 как значение х, соответствующее максимальному значению Ах, рассчитывают константу нестойкости комплекса.
Если же исходные растворы эквимолекулярны (Со = Со или р = 1), тогда определить значение константы нестойкости не удается, но зато значение х, отвечающее максимальной величине Ск (или Ах), будет определяться соотношением
х0 = —— . (111,38)
т 4- п
Таким образом оказывается возможным определить стехиометрический состав комплексного иона.
Метод непрерывных изменений оказывается весьма точным: недостатком его является трудность определения состава ионов, если в растворе образуется несколько комплексов.
ЛИТЕРАТУРА
Биргер Н. И. Задачи по электрохимии. ГОНГИ, 1939, гл. IV.
Вальден П. Электролиты и растворители. Госхимиздат, 1934.
Воробьев Н. К., Гольдшмидт В. А., Карапетьянц М. X. Практикум по физической химии. Госхимиздат, 1950.
Глессон С. Введение в электрохимию. ИЛ, 1951.
Горен бей н Е. Я. Труды II конференции по теоретической и прикладной электрохимии. Изд. АН УССР, 1949.
Деляхей П. Новые приборы и методы в электрохимии. ИЛ, 1959, стр. 40. Кольтгоф И. М., Л а й т и н е н Г. А. Определение концентрации водородных ионов и электротитрование. ИЛ, 1947.
Левин А. И., П о м о с о в А. В., У к ш е Е. А. Руководство к лабораторным занятиям по теоретической электрохимии. Изд. УПИ им. С. М. Кирова, гл. II, 1957.
Ляликов Ю. С. Физико-химические методы анализа. Металлургиздат, 1951. С'тро.мберг А. Г., Захаров М. С., Аткмин Г. Ф. и др. Сборник примеров и задач по электрохимии, гл. III, изд. Томского университета, 1962.
Хейфец В. Л., Авдеев Д. К., Р е й ш а х р и т Л. С. Практикум по теоретической электрохимии. Изд. ЛГУ, 1954.
Глава IV
СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
§ 1. Слабые и сильные электролиты
Электролиты отличаются от так называемых идеальных растворов 1 рядом специфических свойств. В частности, осмотическое давление, понижение точки замерзания и повышение точки кипения электролитов гораздо больше зависят от концентрации, чем этого следовало ожидать исходя из теории идеальных растворов по законам Рауля — Вант-Гоффа.
Отношение экспериментальных значений для указанных величин к вычисленным по теории идеальных растворов носит название изотонического коэффициента I. Он, как показывает опыт, не является постоянной величиной, что связано с изменением общего числа частиц в растворе из-за диссоциации части молекул на ионы. Теория Гроттгуса, изложенная ранее, не объяснила этих явлений. Они получили объяснение только после того, как в 1887 г. Сванте Аррениус высказал гипотезу об электролитической диссоциации.
Основные положения теории электролитической диссоциации сводятся к следующему.
1. Молекулы сложного вещества в растворах диссоциируют на заряженные атомы или группы атомов —- ионы. При этом ионы представляются частицами, подобными частицам идеального таза (или раствора). Их взаимодействием между собой и с молекулами растворителя Аррениус пренебрегает.
2. Все изменения в растворах электролитов, связанные с диссоциацией и вызывающие отклонения от законов идеальных растворов, характеризуются степенью диссоциации:
* = (IV3)
т. е. отношением числа молекул, распавшихся на ионы (п), к первоначально взятому их числу в данном объеме раствора (N).
* Идеальным называется раствор, образование которого из- компонентов не сопровождается тепловым эффектом и изменением объема, а измеиёние энтропии такое же, как и при смешении идеальных газов.
5*
68
Теоретические основы электрохимии
Степень диссоциации связана с изотоническим коэффициентом простым соотношением. В самом деле, если каждая молекула, диссоциирующая в растворе, распадается на v ионов, то общее число ионов и молекул равно
N (1 —а) a.vN.
Осмотическое -давление растворов пропорционально концентрации. Поэтому изотонический коэффициент равен
. _ N (1 —а) + cr.N I «
N
т. е. отношению числа частиц, действительно присутствующих в разбавленном растворе, к числу частиц, которые были бы в растворе той же аналитической концентрации при отсутствии диссоциации. Таким образом,
i=l+(v—1)а. (IV,2)
Все электролиты могут быть условно разделены на две группы.
1. Сильные электролиты, т. е. такие вещества, которые даже в относительно концентрированных растворах (0,1-н.) полностью диссоциированы на ионы. К сильным электролитам относится большинство растворимых солей неорганических кислот: серная, соляная, азотная кислоты и основания типа NaOH, КОН и др.
2. Слабые электролиты, т. е. вещества, имеющие в 0,1-н. растворах степень диссоциации менее 3%. К ним относятся некоторые неорганические кислоты (H2S, HCN, Н2СОз, Н3ВО3 и др.), большинство органических кислот и соли (HgCl2, Fe(CNS)3, Hg(CH2, FeF3 и др.) ’. По числу ионов, на которые распадаются электролиты, они называются бинарными (КС1), тернарными (ВаС12), кватернарными (FeCK) и т. д. Однако разделение электролитов на сильные и слабые не имеет общего значения; в зависимости от среды электролит может быть и сильным и слабым. В неводных растворителях некоторые сильные электролиты становятся слабыми, что связано с величиной диэлектрической постоянной растворителя и с взаимодействием молекул растворителя с ионами электролита. В водных раство рах имеются электролиты средней силы, занимающие промежуточное положение между сильными и слабыми электролитами: в 0,1-н. растворах они диссоциируют в пределах от 30 до 3%. Отметим некоторые характерные свойства растворов электролитов.
--------- л
1 Все слабые электролиты характеризуются тем, что они диссоциируют в результате химических реакций (обмена заряженными частицами), приводящих к образованию ионов.
Свойства растворов электролитов 69
Поскольку ни растворы электролитов в целом, ни отдельные молекулы, не продиссоциировавшие на ноны, свободного электрического заряда не имеют, то очевидно, что суммы имеющихся в растворе положительных и отрицательных зарядов равны во всем растворе и в любой его части. Математически условие электронейтральности выражается равенством
= 0. (IV.3)
Ионы различных типов химически отличаются от аналогичных им атомов или незаряженных радикалов. Электрохимические представления» связали атом вещества с электрическим зарядом и показали, что свойства материи зависят от этого заряда. Так, натрий вытесняет водород из воды; при этом получается ион натрия, который уже более не в состоянии реагировать с водой. Ион Сп2+ обладает химическими свойствами, весьма отличными от свойств атома меди; ион Н+ ведет себя иначе, чем атом водорода, и т. д.
При электролитической диссоциации многие вещества образуют в растворе ионы, которые реагируют между собой. Ионные реакции протекают с большой скоростью. Достаточно, например, к раствору AgNO3 прилить раствор NaJ, как мгновенно выпадает осадок AgJ. Именно ионные реакции особенно широко используются в аналитической химии.
Взгляды, развитые Аррениусом, оказали благотворное влияние на развитие теории образования комплексных соединений *. По поведению веществ в водных растворах можно судить, является данное вещество двойной солью или комплексным соединением.
Таким образом, теория электролитической диссоциации, выдвинутая Аррениусом, объяснила ряд опытных фактов, позволила выяснить многие новые электрохимические закономерности и вскоре получила широкое распространение. Взгляды, очень близкие к теории Аррениуса, высказал в 1881 г. Н. Н. Каяндер, впервые указавший на диссоциацию как на процесс, объясняющий химические особенности растворов электролитов.
Помимо указанных выше фактов, теория электролитической диссоциации объяснила другие закономерности. Укажем некоторые из них. ,
1. Скорость растворения металлического магния в кислотах обратно пропорциональна вязкости раствора и пропорциональна электропроводности кислоты. Эти свойства легко объяснить, предполагая ионный механизм реакции.
1 Комплексным 'соединением называют 'определенные молекулярные соединения, которые при диссоциации распадаются :на 'Сложные ‘положительно или отрицательно заряженные ионы. В частном случае заряд сложного иоиа может быть нулевым.
70
Теоретические основы электрохимии
2. Постоянство' теплоты нейтрализации сильных кислот и оснований, равной теплоте образования воды из ионов Н+ и ОН* (13 650 кал), прямо указывает на ионный характер реакции нейтрализации.
3. Увеличение электропроводности при разбавлении объясняется ростом числа ионов вследствие увеличения степени диссоциации.
Аррениус предполагал, что диссоциация соединений является химической реакцией, ведущей к установлению равновесия между ионами и непродиссоциированными молекулами. Увеличение разведения ведет к возрастанию степени диссоциации а, которая достигает единицы при бесконечном разбавлении, т. е. тогда, когда наступает полная диссоциация.
Эти положения были облечены в математическую форму в работах В. Оствальда, который рассматривал диссоциацию бинарного электролита, например кислоты СНзСООН, на ионы:
СН3СООН Н+ + СН3СОО-. (IV,4)
Обозначим концентрацию Н+-ионов в растворе через Сн+ = — концентрацию СН3СОО_-ионов через Сх_ и концентрацию молекул СНзСООН через Со = — - . Константа равновесия реакция (IV,4), по закону действующих масс, равна
IH+nCHgOOT “ 5
[СНзСООН] 1—а (1—а)У
где V = ----общий объем электролита.
Уравнение (IV,5) называют законом разведения Оствальда, который связывает константу диссоциации со степенью диссоциации электролита. Из уравнения (IV,5) следует, что с увеличением объема электролита V степень диссоциации а уксусной кислоты должна возрасти. Определить ее можно различными путями. Наиболее часто степень диссоциации устанавливают измерением эквивалентной электропроводности при разбавлении. В таком случае, пользуясь формулой
(IV,6)
можно написать
------С. (IV,7)
У» ~ М
Свойства растворов электролитов^71
Последнее уравнение дает возможность из измерений электропроводности вычислять константу равновесия диссоциации.
Теория электролитической диссоциации с успехом была применена к разработке ряда .вопросов, имеющих практическое и теоретические значение. Рассмотрим некоторые из них.
§ 2. Ступенчатая диссоциация
Закон разведения разъясняет явление ступенчатой диссоциации многоосновных кислот и многокислотных оснований; они имеют несколько ступеней диссоциации и соответственно несколько констант диссоциации ’.
В ряде случаев практически сказывается только первая ступень диссоциации, так как при ступенчатой диссоциации электролита константа диссоциации последующей ступени всегда значительно меньше константы диссоциации предыдущей ступени. Так, например, фосфорная кислота Н3РО4 имеет три ступени диссоциации:
Н3РО4^Н+ + Н2РО-;
H2PO^ ? Н+ + HPQ2-;
НРО2-5±Н+ + РО43-.
Константы диссоциации этих промежуточных стадий (ступеней) приведены в табл. 7. Как видно из приведенных данных, по мере увеличения номера стадии (ступени) диссоциации уменьшается величина Кь Электролиты, имеющие несколько ступеней диссоциации, диссоциируя по первой ступени, в большинстве случаев являются сильными электролитами, а по второй и особенно третьей — слабыми. Для органических кислот значение константы тем меньше, чем длиннее алифатическая цепочка. Объясняется это явление тем, что оторвать Н+-ион от аниона гораздо труднее, чем от незаряженного атома. В случае же длинной цепи в органической кислоте кулоновские силы взаимодействия между Н+-ионом и отрицательно заряженным концом молекулы уменьшаются. Н. А. Измайлов показал, что при переходе от растворителя к растворителю константы диссоциации кислот изменяются в миллионы раз, а соотношения в силе кислот в тысячи раз.
1 Диссоциация любых электролитов происходит в несколько последовательных стадий. Весьма часто вещество типа КД, взаимодействуя с растворителем, образует продукт присоединения КА + пМ^КАМп, который в результате дальнейшей сольватации диссоциирует на ионы КАМп + тМ ^к++д7.
72- Теоретические основы электрохимии
Таблица 7. Константы диссоциации многоосновных кислот при 18 °C
Кислота Ступень * Кислота Ступень к
н2со3 I II 4,3-10—7 4,7-10—11 С —ООН С —ООН I II 3,8-10-2 4,9-КГ"5
Н3Р04 I II III 8,3-10—3 6,3-io-8 2,2-10—13 С —ООН (cih С —ООН I II 6,6-10“5 2,7-10-6
§ 3. Ионное произведение воды и гидролиз
Чистая вода является слабым электролитом и диссоциирует по уравнению
Н2О^Н+ + ОН“. (IV,8)
Так как константа равновесия этой реакции очень мала, второй ступени диссоциации гидроксильного иона воды по схеме
ОН-^О2“ + Н+ (IV,9)
практически не обнаруживается.
Поскольку концентрация воды Н2О значительно больше концентрации Н+ и ОН--ионов, то ее можно считать постоянной. Обычно для воды рассматривают не величину /<ь а так называемое ионное произведение воды:
Kw - [Н2О] = [Н+] [ОН-]- (IV, 10)
В чистой воде имеет место равенство
VX; = [H+] = [OH-]. (IV, 11)
Величины Kw для чистой воды, найденные из измерений электропроводности при разных температурах, имеют следующие значения:
Температура Ионное произведение воды Kw
16 18 22 25 34 50 0,63 ю-14 0,74 • 10~14 1,0 - 10~14 1,2 - 10~14 2,6 10~14 5,4 • 10~14
Из приведенных данных следует, что в нейтральном растворе при комнатной температуре
[Н+] = [OH_j = 10~7 г-ион/л. (IV, 12)
Свойства растворов электролитов
73
Величина (—1g Сн+) обычно обозначается pH и называется водородным показателем, т. е.
pH = -lgC. (IV, 13)
м
Поскольку Kw сохраняется постоянной независимо от концентрации и состава раствора, величина pH является полной характеристикой кислотности или щелочности для любых растворов в воде. В табл. 8 приведены значения pH для различных растворов.
Таблица 8. Водородный показатель (pH) для различных растворов
Растворы [Н+] pH [ОН ] Растворы [Н+] pH [ОН ]
Сильные кислоты 10° 10“’ ю-2 0 1 2 Ю-14 10 13 10“12 Нейтральный раствор io-7 7 10-7
Слабые основания 10“8 10~9 10-ю 10_11 8 9 10 11 10-6 io-5 10“4 10~3
Слабые кислоты 10- 3 Ю~4 кг5 10-6 3 4 5 6 10-" 10-1 о 10~9 10—8
Сильные основания 10-12 10-'3 10-14 12 13 14 10_2 10-' 10°
Диссоциацию воды следует учитывать при подсчете концентрации водородных ионов или ионов ОН- в растворах слабых кислот и оснований. Возьмем, например, раствор слабой кислоты НА, константа диссоциации которой
К(IV, 14) [НЛ]
Очевидно, вследствие диссоциации воды
[Н+] = [Д-] + [ОН-]
и, следовательно,
или
1Н+1 = 1/ЛДНЛЦ- /<w.
Аналогично для слабых оснований Л1еОН [ОН“] = ]Л^[7ИЮН] + 2(Ш,
74
Теоретические основы электрохимии
где Ль — константа диссоциации основания;
К, = Mte+llOH-1 ]V 15
b [ТИеОН] '
Для сильных кислот и щелочей величиной Kw можно пренебречь по сравнению с произведением константы диссоциации кислоты (Ка) или основания (Кь) на концентрацию электролита.
Диссоциация воды является причиной гидролиза солей. Так. известно, что растворы слабых кислот и сильных оснований обладают щелочной реакцией, а растворы солей сильных кислот и слабых оснований имеют кислую реакцию. Для объяснения этого явления примем, что между солью и водой идет обратимая реакция:
МеА + Н,0 £ МеОН + НА, (IV, 16)
константа равновесия которой
„ = [Me ОН] [НД]
~ [МеА] [Н2О] ’
а так как концентрация воды практически постоянна, то
__ [МеОН] [НД] {jy I?)
h [МеА]
Величина Kh называется константой гидролиза.
Пусть в испытуемом растворе, содержащем моль соли МеА в V л, гидролитически распалась часть х, тогда соли осталось (1—х) и в соответствии с уравнением (IV,16) образовалось по х молей основания (МеОН) и кислоты (НА). Следовательно, концентрации
[МеА]; [МеОН] = [НА] - А (IV, 18)
И
v2
Kh =—------ (IV,19)
* V(l— х)
Рассмотрим соль слабой кислоты и сильного основания. Если НА — слабая кислота, то
НАХН+ + А-.
Константа диссоциации кислоты:
К = JH+] [ДП _ (IV,20)
'° [НД] 7
Свойства растворов электролитов
75
Концентрация [НД]=-^ (по формуле (IV,18). Концентрация анионов определяется концентрацией самой соли, т. е. [Л-] = = ———, а [ОН-] — концентрацией основания [Л1еОН], т. е. [ОН-] =^.
Так как МеОН — сильное основание, то оно полностью диссоциировано на ноны. В таком случае к [Н+Щ-х]
X 9
откуда
[Н+] = (IV,21)
Константа диссоциации воды К» = [Н+] [ОН-]; следовательно, из уравнений (IV,16) и (IV,21) найдем
к* Л/ * откуда
= (IV,22)
Ад
Нетрудно показать, пользуясь аналогичными рассуждениями, что для соли сильной кислоты и слабого основания
= (IV,23)
Л*
Здесь Кь — константа диссоциации слабого основания:
МеОН Ме+ + ОН-.
Значительный интерес представляет случай гидролиза в растворе, содержащем соль слабого основания и слабой кислоты например CH3COONH4. В таком растворе имеют место равновесия:
CH3COONH4 Z NH4 4- СН3СОО~, (а)
Н2О Z Н+ 4- ОН-, . (б)
но так как кислота СН3СООН и основание NH4OH диссоциированы в малой степени, будет иметь место тенденция ионов Н+ и СН3СОО-, а также NH+ и ОН- соединяться с образованием недиссоциированных молекул вплоть до установления равнове
76 Теоретические основы электрохимии
сий. Таким образом, происходит взаимодействие между ионами растворенного электролита и ионами воды:
СН3СОО~ + Н +-> СНзСООН, (в>
NH++ ОН~ —> NH4OH. (г)
Так как соль диссоциирует полностью, в итоге получим
NHt + СН3СОО~ + Н2О Z NH4OH + СН3СООН. (IV,24)
ti
CH3COONH4
Очевидно, если Ка « Кь, то раствор остается нейтральным, но если К.а > Къ, он делается слабо кислым, в противном случае, когда К.а< Къ — слабо щелочным.
Каждому из трех равновесий—(б), (в), (г) отвечает своя константа Kw, Ка и К.ъ', константа гидролиза реакции (IV,24) равна
„ [СНзСООН] [NH4OH] Ал — т •
[НН+] [СН3СОО~] [Н2О]
Так как [Н2О] = const, то
„ _ [СН3С00Н] [nh4oh] _ ,IV 25)
h [NH+][CHSCOO-] А«Ай *
Следовательно, гидролиз соли сводится к образованию в ее растворе слабой кислоты или основания либо того и другого вместе.
§ 4. Теория кислот и оснований
В теории электролитической диссоциации были даны определения понятиям «кислота» и «основание», в основу которых было положено то, что кислоты характеризуются избытком Н+-ионов, а основания—.избытком ОН--ионов ,в растворе. Однако эта простая и в течение многих лет общепринятая точка зрения на кислоты и основания оказалась слишком ограниченной, так как и для водных растворов она не совсем точна. Диссоциация воды в действительности протекает по более сложному уравнению
2H2OJ Н3О+ + ОН~.
Следовательно, в водных растворах кислота характеризуется ионами гидроксония [Н3О+].
В абсолютной уксусной кислоте наблюдается равновесие
2СН3СООН J CH3COOHt + СН3СОО~
Свойства растворов электролитов
77
причем роль гидроксония выполняет ион СН3СООН2, а роль гидроксила — ион СН3СОО_. Таким образом, CH3COONa в воде — соль, а в уксусной кислоте — щелочь. Стало быть, ион водорода не является характерной особенностью кислоты, так же как ион гидроксила не характеризует, основных свойств. В различных растворителях самые разнообразные ионы выступают как представители кислот и оснований.
Современная теория кислот и оснований, развитая Бренсте-дом, Лаури и Бьеррумом, объединила физические и химические взгляды на диссоциацию кислот и оснований и носит количественный характер. Бренстед исходил из того, что в образовании кислотных группировок всегда принимает участие протон, подобно тому как в окислительно-восстановительных процессах участвует электрон.
Как известно, окислителем называют вещество, присоединяющее электроны:
Fe3+ + е -> Fe2+,
г восстановителем — отдающее электроны:
2
Суммирование этих уравнений приводит к уравнению окислительно-восстановительной реакции:
Fe3+ +j"->Fe2+ + -J2.
По аналогии Бренстед предложил называть кислотой вещество, способное отдавать протоны, а основанием — вещество, способное присоединять их. Реакции, сводящиеся к протонным переходам, были названы протолитическими.
С каждой кислотой А сопряжено основание В, в которое она переходит, отщепляя протон:
Д = В“ + Н+. (IV,26)
Ори чтении справа налево это же уравнение изображает присоединение протона к основанию, которое переходит в сопряженную с ним кислоту. Однако протоны не существуют в растворах в свободном виде, поэтому реакция (IV,26) не может протекать изолированно. Необходимо присутствие одновременно другого основания, которое, присоединяя протон, превращается в кислоту.
Обратная реакция перехода кислоты в основание также тре бует непременного присутствия другой кислоты, доставляющей протон с переходом в сопряженное основание. В растворе, таким
78 Теоретические основы электрохимии
образом, всегда протекают два процесса, например:
НС1 — н+ч-сг
кислота основание.
Н2О + Н+-Н3О+.
основание кислота
что в сумме дает
на + н2о н3о+ + СГ.
Следовательно, кислотно-основная реакция протекает между соответствующими кислотами и основаниями. Она сопровождается перебазированием протона по аналогии с окислительно-восстановительной реакцией, при которой электрон передается от восстановителя к окислителю.
Ниже приводятся примеры сопряженных кислот и оснований, связанных между собой уравнением типа (IV,26):
кислота = Н+ + основание;
Н2О =--Н+4-ОН“;
Н3О+ = Н+ + Н2О;
NHt = Н+ + NH3;
НС1 = Н+ + СГ;
СН'зСООН = Н+ Ф СН3СОСГ
Заряд, как это следует из сказанного, не является существенным признаком кислоты или 'основания; необходимо лишь чтобы при превращении кислоты в сопряженное с ней основание заряд уменьшался на одну положительную единицу.
Что касается воды, аммиака и ряда других соединений, то они могут являться либо кислотой, либо основанием, т. е. способны и отщеплять и присоединять протон:
Н2О 4- Н+ — Н3О+ NH3 + Н+ - NHt
Н2О — Н+- ОН~ NH3 — H+->NH2
2Н2О = Н3О+ + ОН- 2NH3NHt 4- NH?
Подобные соединения являются электролитами, имеющими и кислотные и основные свойства. Теория Бренстеда наиболее полно расшифровывает понятия «кислота» и «основание». Если в качестве растворителя взять воду, то точка зрения Бренстеда совпадает с представлениями Аррениуса, являющимися частным случаем теории Бренстеда. Но теорией Бренстеда не кончается рассмотрение проблемы кислот и оснований. Лыоис, например, в понятие кислоты и основания положил электронный механизм взаимодействия между кислотами и основаниями. Согласно
Свойства растворов электролитов 79 этой теории, основание представляет собой донор электронов, кислота — акцептор электронов и кислотно-основное равновесие — акцепторно-донорную реакцию.
Рядом исследований, однако, было показано, что те взаимодействия, которые Льюис приводил для подтверждения предложенного им механизма, в действительности протекают по другому механизму.
М. И. Усанович полагает ошибочным представление о кислотах и основаниях как о классах соединений. Он представляет кислотность и основность как химические функции, присущие всем веществам, независимо от того, к какому классу соединений эти вещества принадлежат.
Так, например, в реакции
2Na + С12Х 2Na+ + 2СГ, по Усановичу, хлор является 'кислотой, потому что может присоединить анион-электрон, а натрий — основанием, так как он этот анион-электрон отдает.
Критика теорий Льюиса и Усановича привела А. И. Шатен-штейна к представлению о том, что наряду с водородными кислотами имеются вещества, которые сходны со свойствами водородных кислот. А. И. Шатенштейн считает, что, придерживаясь определения кислоты как вещества, способного выделить протон, нет оснований игнорировать сходство с кислотами ряда веществ, йе содержащих водород.
Позднее другие исследователи пришли к выводу о необходимости сохранения бренстедовско'го определения кислот и о целесообразности выделения группы веществ, подобных кислотам.
В последнее время Н. А. Измайлов предложил количественную теорию диссоциации кислот и оснований, в которой учитывается многообразие химических и физических процессов в растворах и объясняется дифференцирующее действие растворителей на силу кислот. Особенности кислотно-основного взаимодействия как электрохимического процесса являются следствием особых свойств протона как элементарной заряженной частицы. Кислотой называется вещество, содержащее водород и участвующее в кислотно-основном взаимодействии в качестве донора протона. Основанием называется вещество, участвующее в кислотно-основном взаимодействии в качестве акцептора протона. В завершенном кислотно-основном процессе протон передается от кислоты к основанию, в результате чего образуется катион и анион кислоты.
Обычно полагают, что сила кислот и оснований в растворах определяется их способностью образовывать свободные ионы. Мерой этой силы является константа диссоциации кислоты на ионы.
80
Теоретические основы электрохимии
§ 5. Амфотерные электролиты (амфолиты)
Единство кислот и оснований подтверждается существованием целого класса соединений, обладающих и кислотными и основными свойствами, амфотерных электролитов, или амфолитов. К амфолитам, помимо воды и аммиака, относятся многие гидроокиси металлов — Zn(OH)2, А1(ОН)3 и др. Так, для Л4е(ОН)ж возможны равновесия: в кислой среде
Me (ОНД X Мег+ ф- хОН“ в щелочной среде
Me (ОН)Х Z МеОг~ + хН+.
Весьма часто амфолиты встречаются среди органических соединений, особенно белков, состоящих из аминокислот КНгДСООН. Общая форма подобных амфолитов НХОН. В кислой среде идет процесс:
НХОН Д НХ+ 4- ОНГ. (IV,27)
В щелочной среде амфолит ведет себя как кислота:
НОХН Д н+ 4- XOIГ. (IV,28)
Следовательно, для любого амфолита существуют две константы диссоциации: кислотная Ка и основная Кь-
[н+цхон~] . _ [нх+цон-] nv2q.
= [НХОН] ’ Кь~ [НХОН]' • (1 v’ }
Преимущество может иметь либо одна, либо другая константа, что зависит от прочности связей Н+ и ОН_-иопов с остальной частью молекулы. Это связано также со структурой и природой вещества.
В зависимости от соотношения величин констант диссоциации растворы амфолитов могут иметь нейтральную (Ка ~ Кь), щелочную (Ка < Кь) или кислую (Ка > Кь) реакцию. Таким образом, кислоты и основания это предельные случаи ряда амфолитов.
Для любого амфолита можно подобрать такую среду, чтобы он диссоциировал в обе стороны одинаково. Точка, соответствующая этому значению pH, называется изоэлектрической точкой. Для нее
[ХОН“] = [ХН+].
При диссоциации органических амфолитов в этой точке происходит максимальное образование так называемых диполярных
Свойства растворов электролитов SI
ионов, имеющих вид ~Х+:
НХОН J Н+ + ~Х+ 4- ОН“.
В изоэлектрической точке
[Н+] = [ОН"] = [~Х+]. (IV,30)
Эта точка характеризуется минимумом диссоциации и раст-воримости.
Примером диполярных ионов могут служить внутренние соли аминокислот, образующиеся в результате реакции
nh2j?cooh х+ nh3£coo~.
Так как длина цепи в аминокислотах часто очень велика, соответственно и дипольные моменты этих молекул весьма значительны. Диполярный ион нельзя считать в подлинном смысле слова ионом, так как здесь нет избыточного заряда. Это по существу нейтральная молекула, которая обладает очень большим дипольным моментом. Поэтому присутствие таких ионов сильно повышает диэлектрическую постоянную раствора. Кроме того, они отличаются большой поверхностной активностью. Та кое образование молекул с очень большим дипольным моментом наблюдается не только в растворах, но и в парах, что и было показано экспериментально.
§ 6. Нейтрализация и буферное действие
При постепенном прибавлении кислоты к основанию (или наоборот), т. е. при титровании, происходит реакция нейтрализации с образованием воды и соли:
НЛ + МеОН->МеА г Н2О.
Точка, в которой количество прибавленного реагента эквивалентно количеству титруемого вещества, называется эквивалентной точкой, а кривые pH — количество добавленного реагента называются кривыми титрования. Такие кривые подробно рассматриваются в курсах объемного анализа; здесь же мы рассмотрим лишь общие принципы процессов титрования
В качестве примера возьмем титрование 100 см3 0,1-н. раствора соляной кислоты 0,1-н. раствором едкого натра (табл. 9)'.
Оба электролита полностью диссоциированы. Для простоты принимаем, что объем раствора во время титрования не меняется (хотя это, строго говоря, неверно). Очевидно, начальный раствор характеризуется pH = 1. После приливания 100 см3 NaOH достигается точка pH = 7. Изменение pH от 4 до 10 происходит при приливании всего 0,2 см3 щелочи (вблизи эквивалентной точки pH растет очень быстро).
6 Заказ 1338
82
Теоретические основы электрохимии
Таблица 9. Изменение pH кислоты с увеличением концентрации щелочи
Добавка NaOH, см3 Количество НС1, не подвергшейся нейтрализации, % Н+, г-экв!л pH
0 100 1(Г’ 1
50,0 50 0,5-10-1 1,3
90,0 10 10~2 2
99,0 1,0 10~3 3
99,9 0,1 10~4 4
100,0 0,0 кг-7 7
100,1 Избыток щелочи 10-ю 10
101,0 » » 10-" 11
110,0 » » 10-12 12
Гораздо медленнее растет pH при титровании 10~4-н. кислоты щелочью такой же концентрации (рис. 25, кривая II). Отсюда следует, что титрование разбавленных растворов дает более
Рис. 25. Кривые титрования сильных и слабых кислот:
I - 0,1-н НС1; II - 10-М. НС1; III — СНзСООН; IV - фенол
точные результаты.
В случае титрования слабых кислот или оснований необходимо учитывать, что образующаяся при титровании соль понижает диссоциацию титруемого реагента и сама подвергается гидролизу. Как видно из рис. 25 (кривая III), в этом случае положение точки эквивалентности несколько искажено: кривая идет гораздо выше, чем для НС1, и более круто. Интервал pH, отвечающий нейтрализации, сужен до 7—10. Очень слабые кислоты, например фенол (рис. 25, кривая IV), вовсе не имеют резкого перехода вблизи точки эквивалент-
ности (pH — 11).
Знание особенностей хода кривых титрования позволяет правильно подходить к выбору индикаторов титрования. А это — одна из важнейших задач в аналитической химии.
Существование слабых кислот и оснований дает возможность получать так называемые буферные смеси — раствор соли вместе с ее кислотой или основанием. Под буферным действием понимают способность буферной смеси противостоять изменению pH при разбавлении, концентрировании или при до
Свойства растворов электролитов
83
бавлении кислоты или щелочи в количествах, не превышающих некоторого предела. Примером буферной смеси может служить смесь 0,1-н. СН3СООН и 0,1-н. CH3COONa. Вычислить pH этой буферной смеси можно следующим образом. Из уравнения диссоциации
СНзСООН Z Н+ + сн8соо~
получим
[Н+] [СН3СОО~] (СНзСООН]
откуда
[Н+] = Ка
[СН3СООН]
[СН3СОО~]
В присутствии хорошо диссоциированной натриевой соли уксусной кислоты равновесие диссоциации сместится в сторону образования недиссоциированной СНзСООН, поэтому концентрацию СНзСООН можно приравнять к той, которая получилась бы от диссоциации одной соли (уксуснокислого натрия).
В таком случае
[Н+] - Ка
[СН3СООН]
[CH3COONa] — а
(IV,31)
где а — степень диссоциации уксуснокислого натрия. Соотношение (IV.31) называют формулой буферной смеси. Для 0,1-н. CH3COONa а = 0,79 и, следовательно, при Ка =
= 1,86 • IO-5 [Н+] = 1,86 • IO-5 °-1 = 2,355 • КГ5; pH = 4,628. 0,1-0,79 .
При разбавлении раствора в 10 раз а = 0,87, pH = 4,670. При еще большем разбавлении, например для 0,001-н. CH3COONa, а = 1 и тогда
[Н ь] = 1,86 • КГ5 • ; pH = 4,73.
0,001 г
Таким образом, pH при разбавлении почти не меняется. Так как приведенная выше буферная смесь имеет вполне определенное pH, ее используют для приготовления водородного (ацетатного) электрода сравнения, который служит эталоном при измерениях электродвижущих сил (э. д. с.).
Если к подобной буферной смеси добавить сильную кислоту,, например НС1, то благодаря образованию недиссоциированной СНзСООН из СНзСОО- и Н+ ионы водорода будут связываться и pH будет оставаться постоянным.
В самом деле, если добавить НС1 в таком количестве, что содержание ее будет соответствовать 0,01-н. раствору в чистой
84 Теоретические основы электрохимии
воде, pH достигнет почти двух единиц. В присутствии же буферной смеси:
н+ _ к 1сн£оон) + |на1 _ о.н ...
{[CH3COONa] — [НС1]} а 0,09-0,79
откуда pH = 4,6.
Прибавленная сильная кислота вытесняет из буферной смеси слабую кислоту; образовавшаяся же соль NaCl не влияет на pH и в смеси устанавливается концентрация [Н+], соответствующая константе диссоциации слабой кислоты.
Таким же образом буферное действие сказывается и при добавлении раствора сильной щелочи, например едкого натра. В таком случае концентрация ионов водорода практически не будет меняться из-за того, что по мере связывания Н+-ионов в воде будут образовываться новые Н+-ионы вследствие диссоциации слабого электролита—уксусной кислоты.
Буферные смеси имеют большое практическое применение в аналитической химии, в технологии электрохимических производств при электроосаждении металлов, т. е. везде, где почему-либо возникает необходимость сохранять постоянной концентрацию водородных ионов. Особо ценными буферными смесями являются смеси бикарбоната и . карбоната натрия, а также NaHgPOs и Na2HPO4, играющие большую роль в регулировании процессов, протекающих в живых организмах.
§ 7. Индикаторы
Аналитическое определение эквивалентной точки при нейтрализации обычно производится с помощью специальных реагентов, большей частью органического происхождения, меняющих свой цвет при изменении pH. Такие вещества принято называть индикаторами (т. е. указателями).
Впервые теория индикаторов была разработана В. Оствальдом на основе теории Аррениуса. Она базировалась на двух основных положениях.
1. Индикаторы — это слабые органические кислоты или основания.
2. В недиссоциированной форме такие соединения имеют одну окраску, а в диссоциированной — другую. Например,
Н1пХН+ + 1п“.
Применяя закон действующих масс, можем написать:
[Н+1 = Ка в- = Кд . (IV,32)
[In ] а
Свойства растворов электролитов 85
где [Н1п] — концентрация недиссоциированной формы, имеющей одну окраску;
{In-] — концентрация диссоциированной формы, имеющей другую окраску;
Кп — константа.
Прибавление кислоты повышает концентрацию Н+-ионов, что ведет К'сдвигу равновесия в сторону образования Н1п. Прибавление щелочи оказывает обратное действие.
Так как индикаторы характеризуются весьма малыми константами диссоциации (Кд), то и отношение также очень Щ+]
мало, т. е. самое незначительное изменение pH вызовет перемену окраски раствора.
Область изменения цвета индикатора обычно называется областью перехода. Она зависит от свойств человеческого глаза и величины Кд. Глаз наблюдателя замечает перемену цвета, если к начальной окраске добавлено не менее 9% окраски другого цвета. Стало быть, мы замечаем начало перехода при а = 9% и конец при а = 91 %, т. е. начало перехода:
[Н+] = Кд • = ЮК,
д 0,09
конец перехода:
[Н+] = Кд • — = 0,1К. д 0,91
Следовательно, область перехода равна примерно
ДрН = рН1-рН2 = -1бК+ 1-1- lgK+ 1~2.
Значения pH, при которых начинается и кончается переход, определяются величиной Кд. Так, при Кд = 10-s началу перехода отвечает точка pH = 4, концу — pH = 6; при Кд = Ю-7 начало перехода при pH = 6, конец — при pH = 8 и т. д.
В основу теории Оствальда, однако, принято положение о том, что молекула и ион слабого электролита имеют в растворе различную окраску. Поэтому некоторые исследователи указывали на несостоятельность этой теории.
Вместо теории Оствальда были выдвинуты различные представления, исходившие из более оправданных гипотез. Наиболее приемлемы из них — представления о том, что изменения окраски индикатора являются следствием внутримолекулярных, таутомерных превращений.
Таутомерная теория допускает, что индикатор может существовать в двух таутомерных формах, каждая из которых различ-
86
Теоретические основы электрохимии
но окрашена. Одна из таких форм может носить щелочной, а другая — кислый характер, поэтому в кислом растворе превалирует одна из этих форм, в щелочном — другая. Например,
Н1п, ^Н1пиХН++ In”.
Добавление Н+-ионов вызывает сдвиг равновесия влево и наоборот. Примером такого превращения может служить превращение фенолфталеина:
ОН*
Равновесие
С-О StJccouuouuu (
он*
гпаутометрии Бесцветная
форма
Форма .окрашенная 6 розовый цвет
Можно написать:
[Н+] = /<д
[HInil
С другой стороны:
[Н1пц]
IHInj]
где Лт — константа таутомерного равновесия и, следовательно,
[Н+1 == КЛКТ
[HIn,] llnRl
(IV,33)
Полученное уравнение принципиально не отличается от уравнения (IV,32), но роль константы здесь играет произведение КпКч и начало и конец области перехода определяются величиной константы диссоциации, а также величиной константы таутомерного превращения. Например, при /Ся = 1(Н и Кт = К)-2 индикатор применим для области перехода, где в эквивалентной точке pH = 7.
Свойства растворов электролитов 87
§ 8. Развитие взглядов на свойства растворов электролитов
Аррениус распространял свою теорию не только на разбавленные растворы слабых электролитов, но также и на концентрированные растворы и на растворы сильных электролитов. Но очень скоро были установлены опытные факты, противоречащие взглядам Аррениуса.
Противоречия теории Аррениуса опыту можно проиллюстрировать следующими примерами:
1. Степень диссоциации а может быть найдена различными способами, а именно:
а) из измерений осмотического давления растворов, по величине изотонического коэффициента
= (IV,34)
б) из измерений электропроводности растворов
(iv,35) со
в) из определений электродных потенциалов 1
<Р = <f>° + In афС (IV,36)
(здесь а., = f(<p) характеризует электрическую работу, совершаемую ионами).
Если теория Аррениуса применима к сильным электролитам, то степени диссоциации, вычисленные из осмотического давления, электропроводности и электрического действия ионов в растворе, должны быть одинаковыми. Однако довольно точные опыты дают расхождение для а, и от 2,2 до 41,5%, а между а и ах часто вообще не наблюдается никакого соответствия.
Сказанное иллюстрируется рис. 26, на котором приведены опытные данные, показывающие изменения ах и a.f с изменением концентрации растворов сильной кислоты (НС1). Как видно а оказывается больше единицы, что противоречит физическому смыслу понятия о степени диссоциации.
2. Из закона разведения Оствальда, являющегося прямым следствием теории Аррениуса, следует, что величина
гх «2С
1 См. гл. VI.
88
Теоретические основы электрохимии
должна оставаться постоянной при заданных условиях и не
должна зависеть от концентрации раствора.
Этому требованию хорошо удовлетворяют растворы слабых электролитов. Однако в случае сильных электролитов, когда а близка к единице, К оказывается функцией концентрации раствора и, таким образом, закон разведения Оствальда здесь не-
применим.
Рассмотрим опытные факты, указывающие на полную (или почти полную) диссоциацию молекул сильного электролита на
Р.ис. 26. Опытные данные по определению и % в сильном электролите (НО)
ионы, вследствие чего теория Аррениуса в данном случае оказалась недостаточной.
Большинство сильных электролитов имеет в твердом состоянии ионную кристаллическую решетку, об этом свидетельствуют рентгенографическое и электронографическое (изучение по-
добных веществ.
Процесс
диссоциации
сильных электролитов, .вообще говоря, нельзя изобра-
жать схемой, принятой для слабых электролитов. Возьмем для примера хлористый натрий. Уже твердый кристалл NaCl состоит из ионов Na+ и С1~. В присутствии растворителя, например воды, ионы реагируют с ней (гидратируются) и переходят в
раствор:
[Na+Cl-]TB + Н2О = Nato + С1н2о- (IV,37)
Таким образом, молекулярная структура растворителя имеет весьма большое значение для свойств растворов электролитов.
Присутствие заметных количеств недиссоциированных молекул в растворе можно было бы обнаружить оптическим путем — по спектрам поглощения и комбинационного рассеяния. Однако оптические методы показывают полное отсутствие молекул в растворах сильных электролитов.
Поглощение света окрашенными растворами сильных электролитов пропорционально концентрации электролита в растворе. Если бы сильный электролит не полностью диссоциировал на ионы, то степень диссоциации менялась бы с концентрацией и пропорциональность между поглощением света и концентрацией электролита непременно нарушилась.
Ряд свойств растворов обусловлен существованием ионов и меняется пропорционально их концентрациям. Так, например, некоторые реакции ускоряются водородными и гидроксильными
Свойства растворов электролитов
89
ионами; от концентрации ионов Cu2+, Ni2+, MnOf- и др. зависит величина поглощения света при его прохождении через раствор и другие физические свойства.
Как показали исследования таких свойств для растворов сильных электролитов, они меняются пропорционально изменению общей концентрации раствора. Это указывает на то, что степень диссоциации здесь во всех случаях не зависит от концентрации.
По этой же причине константа диссоциации, вычисленная по формуле
с использованием кажущихся степеней диссоциации, вычисленных из изотонического коэффициента или из измерений электропроводности, лишена для сильных электролитов какого-либо физического смысла. Кроме того, если по этим кажущимся значениям степени диссоциации вычислить константы равновесия при разных концентрациях сильного электролита, то никакого постоянства эта константа равновесия не покажет. Так, например, для КС1 в интервале концентраций от 0,01 до 0,10 м значе ние константы равновесия меняется.
По теории Аррениуса произведение растворимости малорастворимых солей типа AgCl, Т1С1 и др.
C+-C_=Lp
должно быть величиной постоянной. Однако при добавлении к такому раствору электролита, имеющего общий ион с труднорастворимой солью, например КС1, величина Lp оказывается переменной.
Совершенно необъяснимо с точки зрения теории Аррениуса также заметное влияние одних электролитов на растворимость других, не имеющих с ними общих ионов. Например, растворимость Т1С1 увеличивается при прибавлении к раствору KNO3 и т. п.
Теории, объясняющие поведение сильных электролитов, можно разделить на две большие группы — физические и химические теории растворов.
В физической теории, целиком опирающейся на гипотезу полной диссоциации молекул в растворах на ионы, учитывали главным образом физический характер взаимодействия заряженных частиц между собой. Эту теорию для сильных электролитов развивали Бьеррум, Гош, Дебай, Гюккель и др. Интересное толкование эта теория нашла в работах В. К. Семенченко, которые привели ее к сближению с химической теорией.
’90 Теоретические основы электрохимии
Химическая теория растворов, предполагающая химическое взаимодействие ионов электролита между собой, основывается на исследованиях В. А. Плотникова, И. А. Каблукова, Д. П, Коновалова, А. Н. Саханова, М. И. Усановича и др.
Обе теории — физическая и химическая — полностью исходят из представлений Аррениуса — Каяндера о возможности распада молекул в растворе на ионы.
§ 9. Причины электролитической диссоциации
Известно, что при растворении многих кислот, солей и оснований в воде выделяется или поглощается тепло. Это свидетельствует о происходящих при растворении энергетических -изменениях. Большинство солей, подобно NaCl, уже в твердом состоянии построено из ионов, связываемых между собой электростатическими силами.
Повышая температуру, можно добиться полного распада диссоциации) и других, ранее не диссоциированных солей (расплавленные электролиты). Что касается кислот и оснований, то их молекулы характеризуются полярной связью, которая легко переходит в ионную. Рассмотрим условия, необходимые для ослабления взаимного влияния ионов.
Электростатические силы взаимодействия описываются законом Кулона:
f =---—, (IV,38)
Dr2
где /—сила взаимодействия двух зарядов;
г — расстояние между зарядами;
D — диэлектрическая постоянная среды.
Нет оснований полагать, что между ионами в каком-либо растворе кулоновские силы отсутствуют. Но поскольку для воздуха D 1, а для воды D ~ 80, то при перенесении молекулы электролита из воздуха в воду сила взаимодействия между ионами, ее составляющими, падает в 80 раз. С точки зрения природы происходящих процессов это означает, что химическое сродство ионов между собой меньше, чем между ними и молекулами растворителя.
Энергия разрыва ионной связи равна работе, которую нужно произвести, чтобы увеличить расстояние между ионами до такой величины, когда силы взаимодействия между ними будут практически равны нулю (т. е. г ~ оо).
Очевидно,
СО
v = _f_?Ldr=-±. (IV,39)
I Dr2 Dr
Свойства растворов электролитов 91
Для одной молекулы типа КС1 подсчет по этой формуле е2
дает при D = 1 (воздух) v = — = 7 • 10-12 эрг.
При растворении КС1 в воде, диэлектрическая постоянная которой D ~ 80, энергия разрыва ионной связи, подсчитанная по формуле (IV,39), становится равной 6,8- 10~14 эрг, а эта величина будет того же порядка, что и кинетическая энергия молекул воды при комнатной температуре.
Таким образом, чем больше диэлектрическая постоянная среды D, тем В большей степени ионы изолированы один от другого. Растворитель с большой постоянной D ослабляет стремление ионов соединиться в молекулы.
Эти рассуждения согласуются с эмпирическим правилом Томсона: степень диссоциации электролита при заданных условиях пропорциональна диэлектрической постоянной растворителя.
Однако нельзя сводить причины диссоциации только к диэлектрической постоянной растворителя. Наоборот, имеется большое число данных, указывающих на химическое взаимодействие между растворителем и растворенным веществом. Впервые это с особой ясностью подчеркнул Д. И. Менделеев, исследовавший зависимость плотности растворов электролитов от их состава и обнаруживший появление максимумов и минимумов на кривых плотность — состав.
Это подтверждается еще и следующими экспериментальными фактами.
1. Установлено, что взаимодействие между растворителем и электролитом облегчает переход электролита в раствор.
2. Растворение в воде электролитов большей частью уменьшает растворимость в ней нейтральных молекул (эффект высаливания). Например, растворимость хлора (СЬ) в воде при 20 °C и нормальном давлении составляет 2—3 л в 1000 г воды, а в 26 %-ном растворе NaCl растворимость хлора падает до 300 см3 в 1000 г раствора.
3. Найдено, что объем смеси электролита и растворителя меньше суммы их объемов до смешения (эффект электрострикции). Это понижение объема уменьшается с разбавлением электролита и исчезает при бесконечном разбавлении.
4. На взаимодействие растворителя с растворенным веществом указывает также существование кристаллогидратов и кристаллосольватов.
Д. И. Менделеев на основании глубокого и всестороннего анализа опытных данных пришел к выводу, что неопределенные химические соединения — растворы нельзя резко отделить от определенных, или так называемых истинных, химических соединений.
92
Теоретические основы электрохимии
Таким образом, в растворе существуют химические соединения растворенного вещества с растворителем. Эти соединения могут иметь различный состав и находятся в равновесии с растворителем.
Идеи Д. И. Менделеева развивались в работах И. А. Каблукова, открывшего явление аномальной электропроводности, показывающее, что растворитель ни в коем случае не может являться индифферентной средой и что нужно принять во внимание некоторое химическое взаимодействие между растворенным телом и растворителем.
Признавая правильность гипотезы Аррениуса об электролитической диссоциации, И. А. Каблуков отмечает, что вода, разлагая молекулы растворенного тела, входит с ионами в непрочное соединение, находящееся в состоянии диссоциации. По мнению же Аррениуса, ионы свободно двигаются, подобно тем отдельным атомам, которые образуются при диссоциации молекул галоидов при высокой температуре.
Изучая зависимость электропроводности от состава раствора, Д. П. Коновалов подтвердил теорию Д. И. Менделеева. Эта теория, дополненная и развитая И. А. Каблуковым, сохраняет свое значение до настоящего времени и подтверждает несомненное химическое взаимодействие электролита и растворителя. Такое взаимодействие способствует диссоциации сложного вещества на ионы и в общем виде может быть описано уравнением:
А+ + иН2О А (Н2О)+ 4- Q. (IV, 40)
Таким образом, взаимодействие растворителя с ионами сопровождается энергетическими изменениями Q, носящими название энергий или теплот гидратации (сольватации).
Нахождению величины энергии гидратации ионов было посвящено много работ. Экспериментальным путем было найдено, что:
а) с увеличением радиусов ионов энергия гидратации уменьшается;
б) разности энергий гидратации соединений с одним одинаковым ионом не зависят от природы этого иона. Иными словами, энергия гидратации электролита слагается из энергий гидратации ионов, его составляющих;
в) рост диэлектрической постоянной растворителя приводит к росту энергии сольватации.
Из установленных зависимостей следует, что константа диссоциации показывает, как сильно диссоциирует электролит в данном растворителе. Для этого необходимо знать, насколько молекула электролита подготовлена к диссоциации. Чем больше дипольный момент молекулы, тем в большей степени она
Свойства растворов электролитов 93
подготовлена к диссоциации, тем, следовательно, больше а. Молекулы какого-либо соединения можно рассматривать состоящими из нескольких наружных электронов, связанных с атомными остатками.
Распределение наружных электронов по отношению к атомным остаткам может быть различным. Если средний электрический центр тяжести наружных электронов лежит не симметрично по отношению к центрам тяжести атомных остатков, то молекула имеет полярный характер. В случае полной симметричности и равномерности в распределении наружных электронов молекула будет иметь строго неполярный характер (Н2, N?. О2)-
Полярные молекулы в свою очередь можно подразделить на: а) совершенно полярные, когда наружные электроны полностью смещены в сторону одного из атомных остатков; в этом случае атомные остатки становятся ионами (Na+ + С1_; Са2+ + + 2F-);
б) слабо полярные, в которых смещение электронов к одному из остатков не настолько заметно, чтобы говорить об образовании действительных ионов (Н2О).
Само строение молекулы указывает на наличие или отсутствие в ней ионов. Так, полярные молекулы целиком составлены из ионов. Слабо полярные содержат как бы потенциально возможные ионы. В неполярных молекулах ионы отсутствуют.
Возьмем слабый электролит — уксусную кислоту, структурную формулу которой можно представить в следующем виде:
Н О
\ S
И—С—С нУ Хо-н — 4-
Здесь у водорода электрон сдвинут в сторону кислорода. Электроны трех других водородов сдвинуты несколько к углероду. Следствием этих причин является возникновение очень малого дипольного момента и диссоциация на СН3СОО~ и Н+-ионы очень небольшая. Вводя в молекулу вместо Н+-ионов другие группы, мы будем менять природу электролита вследствие изменения дипольного момента. Различные заместители влияют различно. Например, NO2, Cl влияют в одну сторону, СН3 — в другую. Одни группы (электронофильные) притягивают к себе электронное облако, другие (электронофобные) — отталкивают. Таким образом, если вместо атомов Н в СН3СООН ввести атомы С1, то центр тяжести молекулы переместится и молекула будет обладать большим дипольным моментом и большей кон
94
Теоретические основы электрохимии
стантой диссоциации. Наоборот, если вместо атомов С1 ввести в СН3СООН группу СН3
С1 О СН8 О
С1—с—с сн8—с—с
C1Z Хон+ СН8 Хон+
то центр тяжести отрицательных зарядов еще больше приблизится к положительному полюсу молекулы; следовательно, дипольный момент будет еще меньше.
Если взять молекулу NH4OH и заменить в ней водород на группу CH3[N(CH3)4OH], то здесь эффект будет обратный, т. е. дипольный момент молекулы заметно возрастет.
Таким образом, «заместители» могут либо притягивать, либо отталкивать электронное облако и вследствие изменения дипольного момента менять константу диссоциации электролита.
ЛИТЕРАТУРА
Бродский А. И. Физическая химия, т. II, Госхимиздат, 1948.
Биргер Н. И. Задачи по электрохимии. Госхимиздат, 1939, гл. V.
Grunwald Е., Loewenstein A., Meibrom S. J. Chem. Phys., 27, 1957, 630, 641.
Измайлов Н. А. Сборник работ по физической химии. Изд. АН СССР, 1947, стр. 310.
Карякин Ю. В. Кислотно-основные индикаторы. Госхимиздат, 1951.
К и.с т >я к о в с к и й В. А. Теория электролитической диссоциации Аррениуса и эволюция современной химии. Госхимиздат, >1929.
Кричевский Н. Р. Фазовые равновесия в растворах при высоких давлениях. Госхимиздат, 1952.
Людер В., Цуффанти С. Электронная теория кислот и основании. ИЛ, 1950.
Менделеев Д. И. Сочинения, т. 4, ОНТИ, 1937, стр. 1, 227, 281.
Писаржевский Л. В. Свободная энергия химической реакции и растворитель. Москва, 1912.
Соловьев Ю. И. История учения о растворах. Изд. АН СССР, 1959.
Усанов ич М. И. Что такое кислоты и основания. Изд. АН Каз.ССР, 1953.
Шатенштейн А. И. Теория кислот и оснований. Госхимиздат, 1940; Успехи химии, 24, 1955, 377.
Шахпоронов М. И. Введение в молекулярную теорию растворов. Тех-теоретиздат, 1956.
Яцимирский к. В. Термохимия комплексных соединений. Госхимиздат, 1951.
Глава V
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
§ 1. Термодинамика растворов электролитов
Как отмечалось, сильные электролиты не подчиняются закону разбавления Оствальда. Степени диссоциации сильных электролитов, вычисленные из данных криоскопии, электропроводности и электродвижущих сил, заметно расходятся между собой даже в относительно разведенных растворах. Рентгенографическое изучение показало, что электролиты в твердом кристаллическом состоянии имеют ионную решетку. При растворении благодаря высокой диэлектрической постоянной воды и других растворителей с полярными молекулами электростатические силы между ионами уменьшаются. Этому же способст--вует и большая энергия гидратации (сольватации) ионов. Сильные электролиты практически полностью диссоциированы на ионы. Наблюдавшееся на опыте отклонение свойств растворов сильных электролитов от идеальных вызвано действием электрических межионных сил в растворе.
Для характеристики электролитов Бьеррум предложил ввести термодинамические коэффициенты, показывающие отклонения свойств реальных растворов от идеальных.
Коэффициент активности
Коэффициент активности обычно выражается следующей формулой:
(V.1) О
где а — активность;
С — концентрация раствора.
Величина а есть та эффективная концентрация, которой должна была бы обладать идеальная система, чтобы производить такое же действие при химических реакциях, как и изучаемая реальная система. Таким образом, активность — это неко
«6
Теоретические основы электрохимии
торая функция концентрации, которую нужно подставить в выражение для химического потенциала данного компонента в идеальном растворе, чтобы получить .наблюдаемые на опыте значения химического потенциала этого компонента в неидеальном растворе.
Все термодинамические законы идеальных растворов, например закон действующих масс, применимы к реальным растворам, если вместо концентрации использовать активность
д - Cfa. (V.2)
Поправочный коэффициент fa учитывает влияние сил взаимодействия, отклоняющих систему от простейшего в термодинамическом отношении состояния.
Осмотический коэффициент
В некоторых случаях для характеристики отклонения свойств растворов от идеального состояния целесообразно пользоваться не коэффициентом активности, а осмотическим коэффициентом:
(V,3)
где Р и Ро — осмотическое давление реального и идеального растворов.
Осмотический коэффициент f0 равен отношению действительно найденного осмотического давления Р к давлению Ро, которое было бы при полном отсутствии электростатического взаимодействия между ионами. Имея в виду, что
PV = imRT; POV = vmRT, (V,4)
где tn — молярная концентрация, получим для разбавленных растворов
fo = -- (V.5)
Так как v — число ионов, на которое распадается молекула, есть величина постоянная, то осмотический коэффициент fo меняется с изменением изотонического коэффициента I, который зависит от межионных электрических сил.
Коэффициент электропроводности
h=--~ (V,6)
оо
определяет изменение эквивалентной электропроводности ионов в результате взаимодействия их зарядов с изменением концен-
Основные положения теории растворов электролитов 97
трации. —- выражает отношение электропроводности раствора при данной концентрации к электропроводности при бесконечном разведении.
Чтобы установить связь между осмотическим коэффициентом и коэффициентом активности, запишем уравнения осмотического давления для идеального и реального растворов:
Po = --^lnn*; P = -^-ln£n*. (V.7)
где fa, ti* — коэффициент активности и молярная доля растворителя;
V — объем растворителя.
Следовательно,
, __ Р_ In п* — In fa
Ро^ In п*
или
lnfXfo-l)lnn*.
Пользуясь уравнением Гиббса — Дюгема:
(V.8)
(V.9)
дп дп
где n= 1—п* — молярная доля растворенного вещества, получим
fo+n—^ 1 + п^Ь. (V,10)
дп дп
Физический смысл коэффициента активности fa может быть установлен следующим образом. Представим себе два граничащих между собой раствора одного и того же вещества в одном и том же растворителе, причем один из этих растворов идеальный, а другой — реальный. Поскольку в идеальном растворе отсутствует взаимодействие между частицами растворенного вещества, а в реальном оно имеется, то переход из одного раствора в другой требует некоторой затраты работы W.
Согласно законам термодинамики, эта работа при постоянных давлении и температуре равна разности термодинамических потенциалов вещества в реальном (Ф) и в идеальном (Ф^ состоянии, т. е.
Ц7 = Ф —(V,H)
Дифференцируя это уравнение по числу молей i-того компонента при постоянных давлении Р, температуре Т и числе молей 7 Заказ 1338
98
Теоретические основы электрохимии
остальных компонентов раствора п', получим:
» = (».) _(«.) , (V.12)
\ dtli ]р. т.п’ \ дщ )р, т.п’ \ дщ ]р, т, п’
причем величины, приведенные в правой части уравнения, представляют собой химические потенциалы i-того компонента в реальном (щ) и в идеальном (цг) растворах. Следовательно,
= . (V.13))
\ дщ /Р, т, п’
Но, как известно,
pz = р.« + RT In С; = [i° 4- RT In ait (V, 14)
где р° — стандартный химический потенциал.
Активность и химический потенциал — это тесно связанные понятия, что и следует из уравнения (V, 14). Формально говоря, можно было бы вместо активности вычислить химический потенциал данного компонента в растворе по одному из свойств раствора при данной концентрации его, а затеям по найденному из опыта значению химического потенциала вычислить по соответствующим уравнениям другие равновесные свойства раствора.
Однако на практике все же удобнее пользоваться коэффициентом активности.
Так как az = fiCit то
Н = + RT In С£ + RT In fa. = + /?Т1п fa.. (V, 15)
Очевидно, lnffl£ = ^—(V.16) i\l
Кроме того, In 1 • (V,17) RT \ дщ p, т.п’
Таким образом, коэффициент активности характеризует отклонение свойств рассматриваемой реальной системы от свойств идеальных систем или, точнее, от свойств, которыми обладала бы система, состоящая из данных компонентов, если бы она принадлежала к идеальным системам. Однако непосредственно из термодинамики вычислить коэффициент активности (и активность) не представляется возможным.
Коэффициенты активности могут быть найдены сравнением с экспериментальным значением какой-либо термодинамической величины, вычисленной из термодинамических уравнений для идеальных растворов. С этой целью можно воспользоваться рас
Основные положения теории растворов электролитов
99
творимостью плохо растворимых солей, давлением пара растворителя, электродвижущими силами или результатами криоскопических и эбулиоскопических измерений.
В зависимости от того, в каких величинах представлена концентрация раствора, различают несколько выражений для коэффициентов активности. Наиболее часто применяют так называемые практические коэффициенты активности, при использовании которых концентрация выражается в молях на литр раствора (молярность); в этом случае концентрация обычно обозначается буквой С, а коэффициент активности fc. Если же измерять концентрацию в молях на 1000 г растворителя (молярность), соответствующие обозначения в этом случае будут m и у. Числовые значения концентраций, выраженные через m и С, практически не различаются при концентрациях раствора меньше 0,1-м. В таком случае безразлично, будем ли мы пользоваться коэффициентами активности f или у. Существует также понятие рационального коэффициента активности fn, в этом случае концентрация выражается в молярных долях. Это различие следует учитывать при пользовании справочниками, а также при расчетах. Практический коэффициент активности fc и рациональный fn связаны соотношением:
In =- In (1 + O.OOlvMj/n) + In fc, (V, 18)
где Mi — молекулярный вес растворенного вещества;
m — молярность растворенного электролита;
v — число ионов, получающихся из одной молекулы электролита при диссоциации.
Кроме того,
1П fn ~~ In fc + In P + + , (V.19)
Po
где TU2 — молекулярный вес растворителя;
p, Ро—плотность раствора и растворителя.
Следует учитывать, что коэффициенты активности отдельных ионов не могут быть найдены экспериментально. Опыт дает возможность найти лишь средний коэффициент активности:
(V,20)
где faK — коэффициент активности катиона;
faa —коэффициент активности аниона.
В общем случае, когда электролит диссоциирует на k катионов и а анионов:
„ k In fa, — с In
In fa± =--- - • (V.21)
7*
100 Теоретические основы электрохимии
Пользуясь условием электронейтральности, можно также написать:
Inf . = .-ln-gK-+-alnIg^, (у, 22)
° гк 4- za
где zK, za — зарядности катиона и аниона.
В табл. 10 приведены средние коэффициенты активности для ряда электролитов. Во всех случаях средний коэффициент активности электролита равен среднему геометрическому из коэффициентов активности ионов, на которые этот электролит диссоциирует в растворе.
Можно, вообще говоря, получить некоторые условные численные значения для коэффициентов активности отдельных ионов, если сделать произвольное предположение об относительной величине коэффициентов активности какого-либо катиона и какого-либо аниона в соответствующей молекуле электролита. Например, иногда в таблицах помещают числовые значения коэффициентов активности отдельных ионов, вычисленные в предположении (совершенно произвольном), что коэффициенты активности ионов калия и хлора в растворе хлористого калия равны между собой при всех концентрациях хлористого калия в растворе. Поскольку при вычислении равновесных термодинамических свойств растворов в расчетах всегда участвует средний коэффициент активности ионов f±, то на результаты термодинамических расчетов в растворах электролитов это дополнительное произвольное предположение не влияет. В практике термодинамических расчетов в растворах сильных электролитов использование коэффициентов активности отдельных ионов пока не находит широкого применения.
В полной аналогии с f± средняя активность ионов электролита а± равна среднему геометрическому из активностей катиона и аниона электролита, и средняя концентрация ионов электролита С± равна среднему геометрическому из концентрации катиона и аниона. Средняя концентрация ионов электролита (С±) отличается от концентрации электролита (С2) на множитель v±, который равен единице только в одно-одновалентном электролите (где v+ = v_ = 1). Для несимметричных электролитов, у которых v_, он отличается от единицы. Например, в двух-трехвалентном электролите он равен (22-33)1/s = (108)1/ь-
Как отмечалось, числовое значение средней активности ионов электролита и среднего коэффициента активности электролита зависят от способа выражения концентрации.
Используя коэффициенты fa, f0, f\, можно производить необходимые расчеты для сильных электролитов с помощью обычных термодинамических уравнений. Однако необходимость экспериментально определять величины этих коэффициентов делает такой метод неудобным для практического применения.
Таблица 10. Средние коэффициенты активности для ряда сильных электролитов в зависимости от концентрации при температуре при 25 °C
Средние коэффициенты активности электролитов при концентрациях, моль/1000 г растворителя
Электролит 0, 001 0,005 0,01 0,05 o, 1 o, 2 0, 4 0,6 0,8 l.o 1.2 1.4 1,8 2, 0 2,5 3,0 4.0
НС1 0,966 0,928 0,904 0,830 0,796 0,767 0,755 0,763 0,783 0,809 0,876 0,960 1,009 1,147 1,316 1,762
HNO3 0,965 0,927 0,902 0,823 0,791 0,754 0,725 0,717 0,718 0,724 — 0,745 0,775 0,793 0,846 0,909 0,982
HaSO4 0,830 0,639 0,541 0,340 0,265 0,209 —, — — 0,130 — — — 0,124 0,141 0,170
NaOH —. — 0,82 0,766 0,727 0,697 0,685 0,679 0,678 — 0,686 0,700 0,709 0,743 0,784 0,903
КОН 0,92 0,90 -0,82 0,798 0,760 0,734 0,733 0,742 0,756 — 0,800 0,856 0,888 0,974 1,081 1,352
NaCl 0,966 0,929 0,904 0,823 0,778 0,735 0,693 0,673 0,662 0,657 — 0,655 0,662 0,668 0,688 0,714 0,783
NaBr 0,966 0,934 0,914 0,844 0,782 0,741 0,704 0,692 0,687 0,687 — 0,699 0,718 0,731 0,768 0,812 0,929
NaJ 0,97 0,94 0,91 0,86 0,787 0,751 0,727 0,723 0,727 0,736 — 0,763 0,799 0,820 0,883 0,963 —
KC1 0,965 0,927 0,901 0,815 0,769 0,719 — — —. 0,606 — — —— 0,576 0,571 0,579
KBr 0,965 0,927 0,903 0,822 0,777 0,728 — — 0,625 — — —— 0,602 0,603 0,622
KJ 0,965 0,927 0,905 0,84 0,80 0,76 —— — — 0,68 — — — 0,69 0,72 0,75
KNOa —. — — 0,739i 0,663 0,576 0,519 0,476 0,443 — 0,390 0,350 0,333 0,297 0,269 ——
NaNO3 0,966 0,93 0,90 0,82 0,77 0,70 —— — —. 0,55 — —. —— 0,48 — 0,44 0,41
Na2SO4 0,887 0,778 0,714 0,53 0,45 0,36 — —> — 0,20 — — — —- — — —.
CuSO4 0,74 0,53 0,41 0,21 0,150 0,104 0,071 0,057 0,048 0,043 0,039 0,037 — — — —
ZnSO4 0,70 0,48 0,39 — 0,150 0,104 0,071 0,057 0,048 0,043 0,040 0,038 — 0,035 0,037 0,041 —
CdSO4 0,73 0,50 0,40 0,21 0,150 0,102 0,069 0,055 0,046 0,043 0,038 0,035 — 0,032 0,032 0,033 —
NiS04 , — 0,150 0,105 0,071 0,05 0,047 0,042 0,039 0,036 — 0,034 0,035 —. —
A1C13‘ , — 0,337 0,305 0,313 0,356 0,429 0,539 0,701 0,936 — — —— — —
CrCl3 —. — — 0,331 0,298 0,300 0,375 0,397 0,481 0,584 —. — — — — —.
ZnCl2 0,88 0,77 0,71 0,56 0,50 0,45 — — — 0,33 — — — — — — —
Pbda 0,86 0,70 0,61
CuCla 0,89 0,78 0,72 0,58 0,52 0,47 —. —. — 0,43 — — — 0,51 — 0,59 —
Al (NOs)3 — — — 0,20 0,16 — — — 0,19 — — — 0,45 — 1,0 1,2
AgNO3 — 0,90 0,90 0,79 0,72 0,64 —. — — 0,40 — — — 0,28 — —• —
FeCl2 0,89 0,80 0,75 0,62 0,58 0,55 — — — 0,67 — — — — —- — —
K4Fe (CN)„ — — —. 0,19 0,14 0,11
(S’H4)2SO4 0,874 0,726 0,67 0,48 0,40 0,32 — — — 0,16 — — — — —, — —
102
Теоретические основы электрохимии
§ 2. Электростатическая теория растворов
Количественная теория сильных электролитов разработана Дебаем и Гюккелем. Она исходит из положения о полной дис
социации сильных электролитов на ионы и учитывает совокупность взаимодействия каждого данного (центрального) иона со всеми окружающими его ионами. Необходимо иметь в виду, что ионы взаимодействуют и с молекулами растворителя.
Выберем в достаточной
Рис. 27. Исследование элементарного объема электролита
близости (г) от центрального иона некоторый элементарный объем раствора dV = dxdydz и будем наблюдать за ионами, проходящими через этот объем (рис. 27). В течение некоторого отрезка времени в объеме d.V побывает большое число положительных и отрицательных и с шов. Но так как центральный положительный ион будет отталкивать от себя положительные и притягивать отрицательные ионы, то за любой, достаточно большой отрезок времени т число отрицатепьных ионов, побывавших в исследуемом объеме, будет больше соответству-
ющего числа положительных ионов. Получится, что объем dV в среднем по времени обладает некоторым избыточным отрицательным электрическим зарядом. Мы выбрали наш объем в пространстве вокруг иона произвольно, и поэтому такая же картина будет наблюдаться в любой точке пространства, достаточно близкой к центральному .иону, так, чтобы .силы электростатического взаимодействия F =-^-
не стали исчезающе малыми. Следовательно, вокруг |централь-Н'Ого иона существует как бы 'облако противоположного заряда, которое так и навивается ионным облаком, или ионной атмосферой.
Поскольку в качестве центрального может быть выбран любой ион в растворе, то очевидно, что каждый из ионов, составляющих ионную атмосферу, будет окружен своей ионной атмосферой, несущей противоположный заряд и взаимодействующей с данным ионом.
Так как даже в очень разведенном растворе находится гро-
Основные положения теории растворов электролитов
103
:. 28. Зависимость плотности заряда Р от расстояния г
мадное число ионов *, то каждая точка раствора испытывает суммарное действие всех этих ионов.
В связи с указанным элементарный объем ионной атмосферы может быть охарактеризован некоторой плотностью заряда у (количество заряда, приходящееся на единицу объема) и некоторым потенциалом ip. Очевидно, что чем больше расстояние г от центрального иона до выбранного элементарного объема dV, тем меньше будет в этом объеме плотность заряда, так । как сила электростатического взаимодействия убывает обратно пропорционально квад-рату расстояния (закон Кул о-на). Зависимость р = f(r) гра- | фически представлена на рис. 28. |
Существование потенции- § ла ф ионной атмосферы озна- В чает, что перемещение поло- с: жительного или отрицательного иона из бесконечности в объем dV требует затраты некоторой работы против элект- { ростатических сил, равной + ezip или —егф (где ez — заряд иона).
По закону Больцмана, число ионов, обладающих энергией, достаточной для совершения такой работы в растворе при постоянной температуре Т, равно: для катионов
егк , рт Ф Ск=С°е dV;
для анионов , ега ,
*4- ру Ф
Са = С°е -dV, (V.23)
где С°, С° — общее число катионов и анионов в единице объема раствора;
Ск, Са —число катионов и анионов, находящихся в объеме dV.
1 Например, 1 моль содержит 6,0235 • 1023 молекул или 1024 ионов; при со-
1024
держании 1 моль в 1000 л, т. е. в 106 мл, в каждом кубике будет = 10” 10°
ионов.
104
Теоретические основы электрохимии
Плотность заряда в объеме dV для бинарного электролита равна
Р — (^к^к ^•а^-'а) е — е >> Г) я о * 1 ЗУ । -G- М Г) и о л + ini m , •* - • (V.24)
В общем же случае
i р- zi \ztC\e (V.25)
1
Кроме того потенциал и плотность заряда ионной атмосферы должны удовлетворять известному в теории электричества уравнению Пуассона, которое устанавливает связь между средним значением и плотностью заряда р :
У2Ф = -~Р- (V.26)
Здесь ф — потенциал в данной точке;
D — диэлектрическая постоянная среды;
V2 — оператор Лапласа,
так что
(V.27)
Применяя уравнение Пуассона, Дебай уподобляет раствор сильного электролита электричеству, заполняющему пространство с различной плотностью.
Таким образом, по уравнениям (V,25) и (V,26)
X ze .
= kT . (V,28)
i
Это уравнение является исходным в теории Дебая — Гюкке-ля. Однако при его выводе были сделаны следующие допущения. Считалось, что: а) к ионам применим статический закон распределения Больцмана, что позволило сложное взаимодействие многих ионов заменить более простым взаимодействием их ионных атмосфер; б) растворение не изменяет диэлектрической постоянной, т. е. диэлектрические постоянные раствора и растворителя равны.
Эти допущения ограничивают применимость уравнения (V,28). Поэтому авторы теории прибегли к двум дополнительным упрощениям.
1. Ионы характеризуются только своим зарядом, размерами их пренебрегают.
Основные положения теории растворов электролитов 105
2. Для интегрирования уравнения (V,28) ' экспоненциальный член разлагается в ряд:
два первых члена разложения сохраняются, а остальные отбрасываются.
По условию электронейтральности,
ECz-zz=-0 (V.30)
и, следовательно, первый член разложения (V,29) под знаком суммы обращается в нуль. Для симметричных электролитов, т. е. таких, у которых zK = za, уравнение (V,28) еще упрощается, так как в этом случае сокращаются все члены в четной степени. Таким образом,
v2* = ±^ECzz-<p. (V.31)
v ‘ DkT т
Чтобы решить это уравнение, обозначим
Е C°zf = х. (V,32)
DkT
В полярных координатах:
Г2Ф = 1 ГА 1Г2 *Н1 = 1 AL
’ г2 L dr \ dr /J dr2 rdr ’
d2^ 2d£ = у2ф (V 33)
dr2 rdr ‘
Если далее обозначить трг == у, получим ^. = ГА+ф dr dr
и
d2y dfy i dty d2<p „ d2<p , ---- --- -+~--------H Г --- =2 Г----------
dr2 dr dr dr2 dr2
(V,34)
2^. dr
_ d2y
dr2 ‘
Решение такого уравнения имеет вид:
у = А^ + А^г. Следовательно,
Ф = + (V,35)
106
Теоретические основы электрохимии
Чтобы определить постоянные At и Д2, заметим, что при г—>оо величина чр стремится к нулю. Следовательно, Д2 = 0.
Предполагая далее, что ионы не имеют размеров, получим, что при г->0 потенциал чр должен стремиться к потенциалу точечного заряда ez<, который в диэлектрике равен eZilDr, т. е.
(V.36)
откуда
4 = -^- (V.37)
Таким образом, потенциал в точке, отстоящей на расстоянии г от центрального иона, определяется равенством
(V,38)
Но этот потенциал создается как зарядом центрального иона, так и ионной атмосферой. Потенциал, создаваемый центральным ионом, равен чр* = и потенциал, создаваемый ионной Dr
атмосферой, определяется равенством
ф =; ф' — ф* = -gL (e-zr _ i). (V,39)
Потенциал ионной атмосферы в точке, занимаемой центральным ионом г = 0, устанавливается по правилу Лопиталя:
ф° = Игл Ф, п =--— У;
т D
откуда потенциальная энергия центрального иона
₽2~2
V = еггф° = — X. (V,40)
Рассматривая это уравнение, легко заметить, что величина 1
— аналогична радиусу ионнои атмосферы и имеет размерность длины (см).
1 __ / DkT х 1/ 4яе2ХС2- ??
Таким образом, характеристическая длина — зависит от ди-
X
электрической постоянной, концентрации и заряда иона. С ростом концентрации С, радиус ионной атмосферы уменьшается, т. е. атмосфера сжимается. Рост диэлектрической постоянной и
Основные положения теории растворов электролитов 107
температуры, наоборот, вызывает расширение атмосферы. В водных растворах при 18 °C
4?3 •,19~8_ (V.42)
х ]/ С^,г?
где С — концентрация раствора, моль!л\
v—число ионов, на которое распадается одна молекула.
Пользуясь значением потенциальной энергии ионов, мы можем подсчитать работу заряжения W. Для этого введем некоторый множитель К при заряде .ezz так, чтобы удовлетворялось условие
0<Х<1. (V.43)
Если %= 1, то заряд иона Q, = Xez, = ezit что отвечает реальному раствору. Для идеального раствора 1 = 0 и Qi = 0. Т1ри переходе от идеального раствора к реальному % будет расти •от 0 до 1, a Qi — от 0 до ez,-. Если такой переход совершать при постоянных температуре и давлении, то для любого промежуточного состояния можно написать:
/4ге2Л2 2 , .
DkT 1
Qi. = «А;]
dQ} eZidk. Работа заряжения одного иона от до г-того составит
а для заряжения всего раствора, содержащего Сг- ионов г-того типа:
> = 1
х С
I m.
D J о
Следовательно,
Г = —(V44> 30
Пользуясь же равенством . г _____________ 1 ды
n'ai — ЙС7 ’
108
Теоретические основы электрохимии
получим для среднего коэффициента активности inf, =—^L-1/E!!d
• ± 2DkT у DRT
(У,45)
или
lg f± = — hzjVp.,
где
h = .9-4343g2 = const*, (V.46)
2DkT у DRT
p.=:lECzz?, (V,47)
где p— ионная сила раствора, равная полусумме произведений из концентраций всех ионов в растворе на квадрат их заряда.
Таким образом, средние коэффициенты активности ионов данного электролита зависят только от концентрации и электрических зарядов всех ионов раствора. Так, например:
для одно-одновалентных электролитов (К'С1)
ц = С;
для одно-двухвалентных электролитов (Na2SO4)
ц = ЗС;
для двух-одновалентных электролитов (MgCl2)
ц = ЗС;
для двух-двухвалентных электролитов (CoS04)
ц = 4С;
для трех-одновалентных электролитов (А1С13)
ц = 6С
И т. д.
Понятие об ионной силе имеет большое значение при изучении растворов смесей электролитов. Средний коэффициент активности данного иона в растворе зависит только от ионной силы, и при неизменном значении ионной силы он остается постоянным и не зависит от остальных электролитов, присутствующих в растворе (закон ионной силы).
Отсюда следует, что в разбавленных растворах с одинаковой ионной силой средний коэффициент активности любого электролита данного типа (одно-одновалентные, двух-трехвалентные
* Числовые значения величины h для различных температур приведены ниже.
Основные положения теории растворов электролитов
109
и т. д.) одинаков независимо от природы самой соли и от природы прочих электролитов, присутствующих в растворе.
Например, средний коэффициент активности КС1 будет приблизительно одинаков в следующих растворах: 0,01 -м. КС1; 0,004-м. КС1 + 0,002-м. Са(1ЧОз)2; 0,001-м. КС1 + 0,006-м. NaCI + + 0,001-м. IJ2SO4 и т. д„ так как ионная сила всех этих растворов одинакова и равна 0,01.
Уравнения (V.45) и (V,46) обычно называются уравнениями первого приближения теории Дебая и Гюккеля, или предельным законом Дебая. Опытная проверка этих уравнений показала, что они оправдываются в очень разбавленных растворах, если величина ионной силы ц не превышает 0,02. Результаты такой проверки иллюстрируются данными табл. 11.
Таблица 11. Результаты измерения коэффициентов активности солей (f±)
Раствор NaCl Раствор ZnSO<
концентрация С г-экв!л значение концентрация С г-экв/л значение
опытное теоретическое опытное теоретическое
0,0000 0,980 0,979 0,0005 0,780 0,863
0,0003 0,975 0,971 0,0010 0,700 0,812
0,0010 0,965 0,959 0,0020 0,608 0,745
0,0020 0,952 0,946 0,0050 0,477 0,627
0,0100 0,906 0,890 0,0100 0,378 0,517
0,0200 0,867 0,848 —• — —
Кроме того, было найдено, что рост диэлектрической постоянной растворителя и уменьшение валентности ионов способствуют лучшему совпадению опытных данных с теоретическими.
Применимость закона ионной силы к разбавленным растворам сильных электролитов объясняет опытные данные по влиянию посторонних электролитов на растворимость малорастворимых солей. Как отмечалось, произведение растворимости малорастворимой соли Kv+ А_ определяется выражением
Ар = а~,
гдеа+ и а_—активности катиона и аниона малорастворимой соли в растворе;
Lp — произведение растворимости соли, которое является постоянной величиной при данной температуре и, следовательно, не зависит от присутствия в растворе других электролитов.
110
Теоретические основы электрохимии
Таким образом,
Нужно различать два случая влияния постороннего электролита на растворимость малорастворимой соли. В первом случае растворимый электролит не имеет одноименных ионов с малорастворимым электролитом и влияет на его средний коэффициент активности. Учитывая, что ’ (С^+ Cv_y)I/v = s± = v±s, получим
где s— растворимость малорастворимой соли.
В связи с тем что средний коэффициент активности обычно-уменьшается с увеличением общей концентрации электролитов, в растворе, т. е. с увеличением ионной силы раствора, то растворимость соли (s) и средняя концентрация ионов электролита (С±) с повышением концентрации постороннего электролита без-одноименного иона (например, Т1С1 в присутствии KNO3) обычно увеличивается.
Во втором случае растворимый электролит имеет один общий ион с малорастворимым электролитом (например, анион). При этом растворимость соли определяется по концентрации иона малорастворимой соли, который не имеет одноименного иона в растворимом электролите (например, по концентрации катиона С+). А средняя концентрация ионов малорастворимого электролита определяется из выражения
гдеС+/у+= C_/v_ =С—концентрация малорастворимого электролита;
C+/v+ = С_/э_ —С' —концентрация растворимого электролита с общим анионом. Отсюда следует:
В этом случае растворимость соли (концентрация катиона С+) уменьшается с увеличением содержания растворимого электролита (например, Т1С1 в присутствии КС1), так как концентрация аниона (С_ + С’_ ) возрастает быстрее, чем уменьшается /±. Средняя концентрация ионов малорастворимого электролита (С±) в этом случае возрастает, как и в первом случае, из-за уменьшения /±.
Основные положения теории растворов электролитов
111
Опытные данные по влиянию различных электролитов на растворимость Т1С1 показывают, что с увеличением ионной силы раствора средняя концентрация ионов электролита (С±) увеличивается (---уменьшается). При значении ионной силы менее
с±
0,02 независимо от природы постороннего электролита средняя концентрация ионов, а значит, и средний коэффициент активности в соответствии с правилом ионной силы одинаковы.
Недостаточное совпадение результатов расчета по уравнениям (V,45) и (V,46) с опытом побудило Дебая и Гюккеля попытаться улучшить свою теорию. Прежде всего необходимо было освободиться от неверного по существу допущения об отсутствии собственных размеров у ионов. На самом деле ионы обладают конечными размерами. Если представить себе ионы в виде шариков радиуса г, несущих на своей поверхности заряд zte, то можно написать:
ф = Ф* т г~а т г=а
(V,48)
где ф — потенциал вне шара радиуса а; ф*—потенциал внутри шара радиуса а. Кроме того,
Ф = — — + const. ‘ ГЛ., 1
(V,49)
Используя эти трудно получить уравнения, по аналогии с предыдущим не-е2г? 7. Vi = -С. . (V.50) D 1+ах \ » /
Соответственно этому для среднего коэффициента активности имеем
е2г? у
Inf =—z , ± 2DkT 1 + ах
(V.51)
Это уравнение обычно называется вторым приближением теории Дебая — Гюккеля и записывается в форме
, /гг? К и
1п/+= —------. (V,52)
- l+ag-KH V '
Подстановка числовых значений для одно-двухвалентных электролитов дает
Igf 0,505/С
- ' 1 + 0,328 108аУС
(V.53J
112
Теоретические основы электрохимии
Значения величины g, входящей в равной:
уравнение (V,52)
и
1 Г 8ке2 ё ~ V ~DkT'
приводятся ниже:
Температура, °C . . h .................
g .................
О 18 25 100
0,486 0,499 0,505 0,568
0,324-108 0,327-Ю8 0,328-108 0,324-108
Экспериментальная проверка второго приближения теории
Дебая — Гюккеля затруднена тем, что величина а точно не из-
вестна.
Рис. 29. Минимальное расстояние, на которое могут приблизиться ионы
Величина а представляет собой то предельное расстояние, на которое могут сблизиться разнозаряженные ионы в растворе и которое, очевидно, должно быть равно сумме их радиусов (рис. 29). Чтобы проверить совпадение теории с опытом, можно, измерив коэффициенты активности, подсчитать а. Результата™ таких измерений приведены ниже:
о о
Электролит а, А Электролит а, А
НС1.................5,3 СиС12...........5,2
NaCl ...............4,4 MgSO4 .... 3,4
КС1.................4,1 K2SO4...........3,0
CsNO3...............3,0 CuSO4 . . . . 3,1
La2(SO4)3 .... 3,0 KNO3.............0,43
Второе приближение дает хорошие результаты для значений ионной силы р. до 0,1. Гюккель сделал попытку улучшить тео-
Основные положения теории растворов электролитов
113;
рию сильных электролитов. Он заметил, что в очень концентрированных растворах коэффициент активности становится больше единицы. Это указывает на то, что в таких растворах преобладает отталкивание ионов из-за их взаимодействия с молекулами растворителя. Это взаимодействие могло быть принято во внимание, если учесть изменение диэлектрической постоянной при растворении и ее зависимость от концентрации раствора. Расположение молекул растворителя под влиянием ионных полей упорядочивается, молекулы ориентируются. Такая ориентация
Рис. 30. Влияние ионов на расположение дипольных молекул:
а — полная ориентация (диэлектрическое насыщение); б — частичная ориентация; в — беспорядочное расположение диполей
дипольных молекул воды или другого растворителя с полярными молекулами должна привести к понижению диэлектрической постоянной раствора. Влияние ионов на расположение дипольных молекул растворителя схематично представлено на рис. 30.
Так как с ростом концентрации ионов растет доля ориентированных молекул, то диэлектрическая постоянная раствора падает. Из этих соображений Гюккель пришел к необходимости ввести в уравнение (V,52) дополнительное слагаемое так, что
1п4=- 1/-^--+^- <v’54>
- 1 + ag р и
Однако исходные положения, принимавшиеся Гюккелем при выводе его формулы, не соответствуют опыту, так как диэлектрическая постоянная растворов начиная с некоторых концентраций не уменьшается, а возрастает.
§ 3. Осмотический коэффициент
Отклонение величины осмотического давления в реальном растворе (Р) от значения осмотического давления в идеальной системе (Ро) обычно объясняется действием ионных зарядов,
R Заказ 1338
• Л
114
Теоретические основы электрохимии
На основании кинетической теории можно предположить, что
осмотическое давление полностью диссоциированного электролита равно сумме импульсов на 1 см2!сек, т. е. числу ударов, умноженному .на ——•. Пока ионы находятся в глубине раство
ра, они расположены более или менее симметрично. Но движение каждого иона тормозится действием ионной атмосферы, которое, по Дебаю, сводится к действию одного иона с зарядом противоположного знака. Поэтому с возрастанием концентра-
ции число ударов движущихся в растворе ионов о стенку сосуда будет уменьшаться. Это объясняется тем, что ионы, находящиеся у стенки, окружены только полусферой, вследствие чего движение иона тормозится силами, направленными в глубину раствора.
Такая асимметрия может быть выражена уравнением
р — р ___р
1 го гвз-
Таким образом, наблюдаемое осмотическое давление уменьшается по сравнению с его теоретическим значением на величину ДВз, которая зависит от межионного взаимодействия.
Разделив обе части уравнения на Ро, получим
— = 1 2лз или f
Ро Ро ° Ро
Теория Дебая и Гюккеля дает следующую зависимость для осмотического коэффициента:
(V.55)
здесь v — число ионов, на которое распадается молекула; Яцг2 —сумма произведений числа катионов и анионов на квадраты их валентностей.
Например:
для одно-одновалентных электролитов (КС1)
Ev.?2 = 2;
для двух-одновалентных электролитов (BaCh)
EvjZ? = 6;
для двух-двухвалентных электролитов (ZnSO4)
== 8;
для трех-двухвалентных электролитов [Fez/SO^s]
= 30
и т. д.
Основные положения теории растворов электролитов 115
Подставляя числовые значения в уравнение (V.55) для водных растворов при О °C, получим
/Sv.z?\V.
/о = 1 — 0,263 —— YvC. (V.56)
\ V /
Так, например, для Fe2(SO4)3 (трех-двухвалентный электролит) будем иметь
fo = 1 — 0,263 • 6 Уб". V5C.
Результаты проверки уравнения (V,56) показали, что и в этом случае теория Дебая и Гюккеля применима только для больших разбавлений.
В некоторых случаях для характеристики отклонения свойств растворов от идеальных целесообразно пользоваться не коэффициентом активности, а осмотическим коэффициентом, имея в виду, что
с __ Apt
'° — Ди,
“•*1, ид
Следовательно, осмотический коэффициент равен отношению изменения химического потенциала растворителя (Api = = ц.1 — ц ° ) при образовании неидеального раствора сильного электролита данного состава к изменению химического потенциала растворителя при образовании идеального раствора с такой же концентрацией сильного электролита. Или:
Др.1 = fij — рО = fo = foRT In Сх.
Из данной формулы осмотический коэффициент можно определить так же, как коэффициент, на который можно умножить натуральный логарифм концентрации растворителя для химического потенциала растворителя в идеальном растворе, чтобы получить химический потенциал растворителя в неидеальном растворе.
С учетом этого обстоятельства получаем выражения, связывающие активность и коэффициент активности растворителя с осмотическим коэффициентом:
In — fo In С/, ==(?{";
Jnf±=(fo-l)lnCi; =
т. e. зная осмотический коэффициент, можно вычислить активность и коэффициент активности растворителя. По данным для осмотического коэффициента можно вычислить также и средний коэффициент активности электролита (растворенного вещества).
8*
116
Теоретические основы электрохимии
§ 4. Эквивалентная электропроводность сильных электролитов
Несмотря на полную диссоциацию сильных электролитов, их электропроводность с ростом концентрации раствора уменьшается. Теоретическое обсуждение этого явления, предпринятое Дебаем, Гюккелем и Онзагером, исходит из того факта, что при наложении внешнего электрического поля, помимо поступательного движения ионов к электродам, осложняемого их тепловым движением, необходимо учесть силы трения сольватированных ионов о растворитель и тормозящий эффект, возникающий вследствие электростатического взаимодействия ионов между собой.
Если рассматривать движение одного (центрального) иона, то, как отмечалось, вблизи его будут преимущественно концентрироваться ионы противоположного знака. Это приводит к возникновению тормозящих сил, которые делятся на катафоретические и релаксационные.
Возникновение катафоретических сил объясняется тем, что ионная атмосфера под воздействием внешнего поля стремится двигаться в направлении, обратном направлению движения центрального иона, а это ведет к появлению сил трения, вызванных увеличением скорости движения центрального иона относительно прилегающих к нему слоев растворителя, увлекаемых ионной атмосферой.
Действие релаксационных сил связано с тем, что ионы во время движения создают впереди себя новую ионную атмосферу, в то время как старая ионная атмосфера позади иона исчезает. Но эти изменения не могут происходить мгновенно. Представим себе картину исчезновения или формирования ионной атмосферы в том случае, когда центральный ион тем или иным способом внезапно извлечен из раствора. Рассасывание, перестройка в расположении ионов от ориентированного к беспорядочному, хаотическому будет проходить не мгновенно, но в течение некоторого времени, точно так же, как и при внесении иона в раствор, создание вокруг него ионной атмосферы требует некоторого времени т. Это время называется временем релаксации. Оно составляет 10-8—10-1 сек и зависит от температуры, диэлектрической постоянной, концентрации раствора и других факторов.
Ионная атмосфера рассасывается вследствие диффузии ионов, и поэтому величина т зависит также от коэффициента диффузии. Для бинарного электролита время релаксации приближенно определяется уравнением
(V.57)
т _ Kt __ At RTv. С ’
Основные положения теории растворов электролитов 117
где Л, — постоянная для данного раствора величина при Т — const;
С — концентрация электролита.
Следует иметь в виду, что ионное облако позади движущегося иона всегда толще, чем впереди его. Поэтому несимметричное расположение ионов противоположного знака вокруг центрального иона оказывает тормозящее действие на перенос тока.
Если бы катафоретические и релаксационные силы были равны нулю, то раствор в этом случае имел бы ту же эквивалентную электропроводность, что и при бесконечном разведении (Аоо). Поскольку в реальном растворе данной концентрации релаксационная и катафоретическая силы противодействуют движению ионов под действием внешнего поля, они приводят к уменьшению эквивалентной электропроводности раствора, так что для эквивалентной электропроводности при конечной концентрации можно написать:
Х„ = Х — X—X (V.58)
где X], Хп—уменьшение эквивалентной электропроводности вследствие релаксационного и катафоретического эффектов соответственно.
Так как обе величины Xi и Ли пропорциональны j/C, то это же соотношение можно представить в виде уже известной формулы Кольрауша:
хг = хм-лус. (V,59)
Постоянная А была вычислена Дебаем и Онзагером. Конечным результатом их расчетов явилось следующее выражение: А = 9,838_- 10^ _ 2qz^( }*/. _ 28,95 ( ./ (у б0)
(D7)a/= 1+V<7 (РТ) /гц ’
где
ZkZb 1к — 1а
2к + Za Za/к + Zk/э
/к, 4 —электропроводности катиона и аниона;
т]— вязкость раствора.
Для водных растворов при 18 °C это приводит к следующим выражениям.
Для одно-одновалентного электролита
- (0,159Хет + 35,7) ]/2С.
Для двух-двухвалентного электролита
V = - (0,664Хсо - 84,46) V4С
и т. д.
118
Теоретические основы электрохимии
Здесь С — концентрация, г-экв!л.
В общем случае
= (V.61)
где а и р — коэффициенты.
В достаточно разбавленных растворах (С = 0,003 г-экв/л) уравнения Дебая — Онзагера хорошо согласуются с опытом. Однако это относится к одно-двухвалентным электролитам, когда растворителем является вода. С падением диэлектрической постоянной растворителя и ростом концентрации и заряд-ности ио,нов уравнения Дебая — Онзагера становятся неприменимыми.
Для концентрированных водных растворов часто оказывается возможным применять эмпирическое уравнение Шидловско-го, которое по внешнему виду отличается от уравнения Дебая — Онзагера введением слагаемого АХС + A2ClgC + А3С2. Таким образом, _
хоо = АС - А2с 1g С + А3С2. (V.62)
1 — а у С
Однако это уравнение не улучшает результаты, так как и в таком случае имеются отклонения от опытных значений. Можно предполагать, что здесь имеется неполная диссоциация электролита. В таком случае
* = 8 - («L + ₽) /бС], (V.63)
где 6 играет роль степени диссоциации, но она не идентична аррениусовской степени диссоциации а, так как для сильных электролитов степень диссоциации при всех концентрациях остается близкой к единице, а отклонение равновесных свойств и электропроводности раствора от теоретического значения при полной диссоциации электролита является характеристикой сил межионного взаимодействия. Внешне же создается впечатление, что с повышением концентрации число ионов в растворе сильного электролита уменьшается, и поэтому б называют «кажущейся степенью диссоциации».
Зависимость Xv от температуры в уравнениях Дебая — Онзагера (V,60), (V.61) на первый взгляд очень сложна, но можно ожидать, что должна существовать такая температура 4, при которой электропроводность данного раствора максимальна. В самом деле, слагаемое в уравнении (V,61) с ростом, температуры возрастает, так как уменьшается вязкость раствора.
Такая сложная функция, очевидно, должна иметь экстремум, что и подтверждается опытом, причем величина возрастает с увеличением зарядности ионов и падает с уменьшением диэлектрической постоянной растворителя.
Основные положения теории растворов электролитов
119
Важным экспериментальным доказательством правильности теории Дебая — Онзагера является рост электропроводности с увеличением частоты поля (эффект Дебая — Фалькенгагена) и его напряженности (эффект Вина).
Эффект Дебая — Фалькенгагена, или дисперсия электропроводности, сводится к тому, что электропроводность электролитов возрастает с ростом частоты переменного тока. Это явление легко объяснить на основании теории Дебая — Онзагера.
При движении иона в результате существования остатков ионной атмосферы возникает тормозящая сила (релаксационный эффект), являющаяся следствием асимметрии в распределении зарядов вокруг иона. Если направление поля меняется за промежуток времени, меньший, чем время релаксации, то ионная атмосфера не будет успевать разрушаться, что приведет к уменьшению асимметрии. При достаточно большой частоте релаксационный эффект сведется к нулю и сохранится только влияние катафоретического эффекта. Следовательно, электропроводность возрастет. Поясним сказанное примером. Пусть скорость ионов равна Ю-3 см/сек. Тогда при частоте 50 пер!сек за один период ионы пройдут, расстояние
z — -О =2-10 5 см,
что составляет величину, в 2000 раз большую, чем радиус самого иона (~10~8 см).
Но если увеличить, частоту до 6 гц, тогда путь иона, пройденный за один период:
10-3 — Щ-9 гм у ’ lv СМ.
10е
что составляет лишь 0,1 радиуса иона. Таким образом, ион будет колебаться около одной точки, сохраняя ионную атмосферу неизменной.
Так как время релаксации обратно пропорционально концентрации [см. уравнение (V,57)], например для КС1 при 18°C
0,553-Ю-10 VRA
то дисперсия уменьшается при увеличении концентрации раствора.
Рост подвижности ионов, очевидно, должен уменьшать дисперсию, что и наблюдается на самом деле, и дисперсия при прочих равных условиях для растворов НС1 меньше, чем для КС1.
Уменьшение валентности ионов и увеличение диэлектрической постоянной раствора благоприятствуют дисперсии, так как при этом уменьшаются силы, действующие между ионами и при
120 Теоретические основы электрохимии
водящие к образованию ионной атмосферы. Увеличение температуры ослабляет дисперсию, так как при этом возрастает подвижность ионов и уменьшается диэлектрическая постоянная.
Эффект Вина состоит в том, что при увеличении напряжения на электродах электропроводность электролитов возрастет, стремясь к величине Аоо. Вин опытным путем показал, что при сильных полях, порядка 100 000 в 1см, скорости ионов измеряются в метрах в секунду. В таком случае за время релаксации ион проходит расстояние, в несколько раз превышающее толщину ионного облака.
Это явление легко объяснить с точки зрения теории Дебая. Действительно, скорости, приобретаемые ионами под влиянием больших электрических полей, могут стать столь значительными, что фактическое время взаимодействия ионов станет меньше времени, необходимого для образования ионной атмосферы. В связи с этим ионное облако не сможет образоваться, и ионы начнут двигаться так быстро, как если бы они испытывали только сопротивление, вызванное вязкостью растворителя.
Экспериментальное изучение эффекта Вина представляет большие технические трудности, так как опыты необходимо проводить при падениях напряжения в сотни киловольт на сантиметр.
Исследования, проведенные Вином, показали, что:
1) интенсивность повышения электропроводности с ростом напряжения зависит от природы электролита. Так, например, электропроводности растворов КС1 и KFe(GN)6 в обычных условиях близки, но при градиентах потенциала 10 кв) см электропроводность раствора KFe(CN)6 становится на 35% больше, чем электропроводность аналогичного раствора КС1;
2) для слабых электролитов интенсивность увеличения электропроводности в 5—10 раз выше, чем для сильных, что можно объяснить дополнительной диссоциацией их под напряжением.
Чтобы более подробно выяснить влияние поля на электропроводность, вспомним уравнение скорости ионов, приводившееся ранее:
v 21 е Де ‘ kt ' I
За время релаксации т ион проходит расстояние
г;С Де
X = ШТ = Т ------.
Т kt I
Отношение пути, пройденного ионом за время т, к толщине ионной атмосферы равно
Основные положения теории растворов электролитов 121
Для 0,01-н. растворов КО три 18 °C это дает
х и = — : 4 • 104. т I
Ниже (приведено время релаксации т для 0,001-и. растворов при 18 °C:
KCl HCl MgCl, CdS04 LaCIs
т, сек 0,553.10’ 0,189-10’ 0,324-10’ 0,315-10’ 0,162-10’
Очевидно, ионная атмосфера успеет сформироваться и (затормозить движение тока только (при условии, если 1. Следовательно, закон Ома для 0,01-н. растворов КС1 будет справедлив до значений у = 4 • ГО4 в!см. Увеличение градиента потенциала выше 4 • 104 в/см перестает удовлетворять закону Ома, наступает эффект Вина.
§5. Ионные ассоциации в растворах сильных электролитов
Выводы электростатической теории сильных электролитов Дебая и Еюккеля соответствуют опытным данным в области разбавленных растворов. Однако уже при относительно низких концентрациях становится заметным отклонение от теоретических предельных законов. В частности, теория Дебая—Гкжкеля не объясняет аномальный ход кривых электропроводности в растворителях с малыми диэлектрическими постоянными.
Теория сильных электролитов признает, что взаимное притяжение ионов усиливается с повышением концентрации раствора вследствие уменьшения расстояния .между ионами. Усиление взаимного притяжения ионов приводит к изменению свойств растворов в том же направлении, что и частичное соединение ионов в молекулы, т. е. в направлении уменьшения кажущейся степени диссоциации. Однако теория Дебая и Гкжкеля использует чисто физические методы и не учитывает специфичности химического взаимодействия, которое несомненно имеет .место.
«Теория ионных ассоциаций», выдвинутая В. К. Семенченко, была опубликована (в 1922 г., т. е. за год до появления теории Дебая и Гкжкеля. Смысл теории Семенченко сводится к тому, что при сближении на достаточно малые расстояния ионы под .влиянием сил взаимодействия будут двигаться один относительно другого по замкнутым орбитам. Взаимной деформации ионов при этом не происходит и ионы удерживают гидратацион-ную воду. Таким образом, ионная ассоциация является соединением гидратированных или сольватированных ионов.
Такие ассоциированные ионы носят название квазимолекул. Они, очевидно, подобно недиссоциированным молекулам, не
122
Теоретические основы электрохимии
должны участвовать в переносе электрического тока через электролит.
Чтобы найти то минимальное расстояние г0, при сближении на которое ионы образуют квазимолекулу, напомним, что кинетическая энергия движения ионов определяется по закону Больц-з
мана как — kT. Если энергия взаимодействия ионов не меньше этой величины, т. е.
(V.69)
то ионы ассоциируют и величина го, определяемая этим выражением, и есть максимальный радиус ассоциированной группы:
2 e2zKza
г0 = — "* 3 DkT
(V.70)
Таким образом, г0 зависит от валентности ионов zK и za, а также от температуры и диэлектрической постоянной среды. Отсюда следует, например, что для одно-одновалентного электролита, растворенного в воде при 18 °C:
2
'’о- —
(4,774)2 1О~20
81,7 • 1,67 10-16 • 291
= 4,67 • 1(Г8 СМ. .
В этих условиях квазимолекулу могут образовать лишь такие два иона, сумма радиусов которых меньше чем 4,67-• 10-8 см. Следовательно, все ионы, расположенные внутри сферы с радиусом г0 =*4,67 • 10-8, образуют ассоциированные ионные пары, в то время как ионы, расположенные вне этой сферы, можно считать свободными.
Чем больше г0, тем больше объем вокруг данного иона, в котором можно найти противоположно заряженные ионы, и тем больше вероятность возникновения ионных пар. Позднее эту же величину г0 вычислил и Бьеррум, при этом оказалось, что она
равна
2DkT
__g
= 3,5 • 10 см.
По Бьерруму, например, вводных растворах CsCNSnpn 18°C о о
(rCs+ -- 1,65 A, rCNS_ — 1,95 А) квазимолекулы образоваться не могут. С другой стороны, такие ионы, как Си2+ и SO2- (rCu2+ = о О.
= 0,82 A, rso2—= 2,3 А) вполне могут образовывать квазимо-
лекулы, так как для них г0 = 1,4 • 10-7 см.
При постоянной температуре
r0D = = const. (V,71)
Основные положения теории растворов электролитов
123
При .18 °C эта величина равна 2,8 • 1.0-6 для одно-одновалент-ных электролитов. Следовательно, для ацетона (£) = 21) г0 = = 13,3* 10“8 см, для хлоральбутила (D = 10) г0 = 28-10~8 см, ,т. е. ассоциация происходит тем легче, чем меньше диэлектрическая постоянная среды.
Среднее расстояние между ионами в растворе одно-однова-лентного электролита при разведении V л/г-экв можно полагать равным
У V _ 3/~ 1000 _ 10-7 _ 0,938 10~7
Z 2N0 ~ V 2N^C “ 1;0663<с “ Ус
(V.72)
где \No = 6,0235 1023. В частности, при С = 0,037 г-экв!л получим
гср^28 • 10 8 = г0,
т. е. три такой .концентрации в хлора льбутиловых растворах все ионы практически будут участвовать в ассоциации. Между тем в водном растворе полная ассоциация была бы возможна лишь при 'громадной концентрации— 27 г-экв/л.
Эти очень (приближенные расчеты 'показывают, что в растворителях с малыми диэлектрическими постоянными ассоциация ионов должна играть исключительную роль.
Существование ионных ассоциаций дает возможность говорить о степени ассоциации q, показывающей отношение числа ассоциированных ионов к первоначальному их числу. Бьеррум произвел подсчеты степени ассоциации в зависимости от концентрации электролита и суммы радиусов катиона и аниона а = гк + га.
Результаты этих расчетов для одно-одновалентных электролитов при 18 °C в водном растворе приведены в табл. 12.
' Таблица 12. Степени ассоциации ионов в водном растворе при 18 °C
Сумма ’радиусов см Го а Степени ассоциации ионов при концентрациях, моль [л
о о о о 0, 0002 0,0005 0,001 0,002 0,005 О о 0,02 0,05 О сч о
2,82 • 108 2,5 0,002 0,002 0,005 0,008 0,017 0,029 0,048
2,35 • 10» 3 0,001 0,002 0,004 0,008 0,013 0,028 0,048 0,079
1,76 • 108 4 _ — —- 0,001 0,003 0,007 0,012 0,022 0,046 0,072 0,121
1,01 - 10» 7 — — 0,002 0,004 0,007 0,016 0,030 0,053 0,0105 0,163 0,240
0,70 • 108 10 0,001 0,002 0,006 0,011 0,021 0,048 0,083 0,137 0,240 0,336 0,437
0,47 • 10» 15 0,027 0,049 0,106 0,177 0,274 0,418 0,529 0,632 0,741 0,804 0,854
124
Теоретические основы электрохимии
Как (видно из табл. 12, степень ассоциации возрастает с уменьшением суммы гк + га, диэлектрической постоянной растворителя и температуры. В концентрированных растворах квази-молекулы (могут образовываться также из трех, четырех и т. д. ионов, так как пары ионов могут взаимодействовать между собой и с отдельными ионами. Ассоциированная пара ионов представляет собой диполь с моментом т. Потенциальная энергия взаимодействия такого диполя с ионом заряда ге определяется уравнением
t/^_^|cos0, (V,73>
где 0 — угол между осью диполя и линией наименьшего расстояния х между ионом и одним 'ИЗ концов диполя.
В лучшем случае, когда 0=0, минимальное расстояние для образования ионного тройника
2тге . (V,74>
3DkT
Таким образом, может быть подсчитана степень образования тройных ассоциаций.
Теория Семенченко предполагает существование при определенных условиях следующих равновесий:
МеХМе+ t-MeX + Ме+;
ХМеХ~ t МеХ + Х~, (V,75>
характеризующихся константами равновесия Ki и /<2 и, кроме того, равновесия образования ионных пар:
ХМе^Ме++Х~
с константой равновесия Ко-
Величины Х\ и К2 Для ионных равновесий должны быть близки между собой, так как вероятность образования ассоциаций МеХМе+ и ХМеХ~ примерно одинакова. Поэтому
К, Kl= к, = _ IWtHX-1 . (v>76)
[МеХМе+] [ХМеХ~]
Следовательно,
[Ме+] _ [Л~] [МеХМе] [ХМеХ~]
ИЛИ
[flfe+] _ [МеХМе+]
[Х“] ~ [ХМеХ-1
Основные положения теории растворов электролитов 125
Отношение концентраций ионных тройников равно отношению концентраций соответствующих '.простых ионов. Для бинарного электролита
[МеХМе+] = [zYAleX-] (V.77)
ионные тройники должны участвовать в переносе тока. Поэтому эквивалентная электропроводность электролита может быть представлена в виде суммы:
^ = (1-<7)^+?з3^, (V.78)
где q3 —• константа ассоциации для тройников;
Лео и зЛоо — электропроводности при бесконечном разведении (в отсутствие взаимодействия ионов), обеспечиваемые соответственно простыми ионами (первое слагаемое) в уравнении (V, 78) и тройниками (второе слагаемое) в том же уравнении, или (в зависимости от концентрации раствора
Ч—Vx+₽VC.
(V.79)
С ростом концентрации первое слагаемое уменьшается, а второе растет и поэтому функция Av = | (С) должна пройти через минимум.
Уравнение (V, 79) весьма приближенное, но оно правильно отражает найденные на опыте аномалии электропроводности. На самом деле, в растворителях с малыми диэлектрическими постоянными, в которых возможно образование ионных тройников, первоначальное увеличение концентраций должно приводить к уменьшению электропроводности вследствие образования парных ассоциаций, не переносящих ток. Однако начиная с некоторого момента рост концентрации приводит к увеличению электропроводности ввиду образования тройников, переносящих ток. Этим и объясняется появление минимумов на кривых Av = f(C), впервые обнаруженное И. А. 'Каблуковым.
в t/i/ p'DkT V
Величина Л1 = Л2= -----—— пропорциональна (------------
Яз \zKzal2 ).
Если же мы попытаемся найти концентрацию Смин, при которой электропроводность минимальна, и для этого приравняем к нулю дифференциал по уравнению (V, 79), то получим
dC
г а- тг ^оо , / DkT \3 Лу
Смин = — = Ai-------- — COnst -------I —— ,
(V.80)
откуда следует правило Вальдена:
---- const.
О3
126 Теоретические основы электрохимии
Таким' образом, теория Семенченко объясняет явления аномальной электропроводности. Однако, как указывает сам автор, математически она является приближенной. Кроме того, она сохраняет и следующие недостатки теории Дебая — Гюккеля:
1) взаимодействие ионов с молекулами растворителя описывается с помощью его диэлектрической, что неправильно;
2) по-прежнему остается недоказанной допустимость применения к ионам в растворе статистического закона Максвелла — Больцмана.
§ 6. Химическая теория концентрированных растворов сильных электролитов
В отличие от (представителей «физического» направления в теории сильных электролитов А. Н. Саханов объясняет аномальное поведение растворов в растворителях с малой диэлектрической постоянной и концентрированных растворов сильных электролитов химическим взаимодействием ионов, ведущим к образованию «автокомплексов», т. е. комплексно-проводящих ионов типа Ag2NO3+, Ag(NO3)2~ и полимерных молекул типа (AgNO3)2. Как нетрудно видеть, эта точка зрения близка к теории В. К. Семенченко, так как автокомплексы ничем не отличаются от ионных тройников. Разница состоит в том, что представители «химической» теории считают причиной образования автокомплексов не электростатическое, а химическое взаимодействие ионов.
Впервые идея об образовании токопроводящих автокомплексов была высказана В. А. Плотниковым в 1902 г.
В 1913 г. А. Н. Саханов развил теорию В. А. Плотникова. Он исходил из следующих положений.
1. Растворенное вещество в данном растворителе может образовывать комплексы, состоящие из молекул растворенного вещества и растворителя или только ’из молекул растворенного вещества.
2. Образовавшийся комплекс в растворе диссоциирует как на сложные ионы, так и на обычные молекулы и ионы.
Так, наряду с равновесием
AgNO3?Ag+ + NOr (а)
в концентрированных растворах простые молекулы полимеризуются:
2AgNO8->(AgNO3)2 (б)
и образуют сложные анионы и катионы:
(AgNO3)2 ?Ag+ 4- Ag (NO3)7; (в)
(AgNOs)2 X Ag2NOt -I- NO?. (r)
Основные положения теории растворов электролитов
127
В концентрированных растворах [схема (а)] равновесие сдвинуто влево, здесь простых ионов Ag+ и МОз~ почти нет. В рас-
творе в этом случае находятся молекулы, которые полимеризуются по схеме (б) и дают комплексные ионы. Так как этих ионов
достаточно много, то электропроводность раствора хорошая (точка 1 на рис. 31). При разбавлении часть комплексных ионов
распадается и равновесия [схемы (в) и (г)] сдвигаются влево с образованием молекул. Вследствие уменьшения количества сложных ионов и возрастания числа недиссоци-ированных молекул электропроводность уменьшается (точка 2 на рис. 31). При дальнейшем разведении диссоциация будет проходить по схеме (а), что приведет к увеличению количества простых ионов в растворе, и электропроводность возрастет до предельного значения (точка 3 на рис. 31) при бесконечном разведении. Исследуя зависимость лого серебра от разведения,
Рис. 31. Зависимость электропровод-иости от разведения электролита
электропроводности азотнокис-А. Н. Саханов обобщил свои
выводы, придя к следующему экспериментальному закону:
к = В 4- АСп,
(V,81)
частным случаем которого является правило Кольрауша. При отсутствии простых ионов, очевидно, В = О.
§ 7. Современное состояние и развитие теории растворов электролитов
Чисто физическая теория ассоциации ионов Семенченко — Бьеррума, дополненная Фуоссом и (Краусом, объяснила аномальную проводимость за счет кулоновского взаимодействия, но она не стала общей теорией, так как не учитывала, что ассоциация ионов связана не только с кулоновским, но и с химическим взаимодействием между ионами и молекулами растворителя. Для создания единой теории сильных и слабых электролитов необходимо рассматривать поведение ионов и молекул в растворах электролитов во взаимодействии, учитывая реальные условия и среду.
В настоящее время возникла возможность плодотворного объединения физической точки зрения Вант-Гоффа—-Аррениу-
128 Теоретические основы электрохимии
са с химической теорией растворов, изложенной Д. И. Менделеевым.
В развитии современных представлений нашла свое место и координационная теория комплексных соединений А. Вернера, позволившая рассматривать растворы как системы комплексов растворенного вещества и растворителя.
Существенное значение приобрела работа Бернала и Фауле-< ра о квазикристаллической структуре воды и водных растворов. Она позволила выяснить самый механизм образования координационных структур ион-дипольного характера и сделала возможным замену статических представлений динамическими. При этом весьма важную роль сыграли исследования Я. И. Френкеля, обнаружившие изменения координационного числа при плавлении тела. Согласно имеющимся представлениям, тепловое движение атомов жидкости состоит из колебаний атамов около временных состояний равновесия и скачкообразных перемещений из одного положения равновесия в другое. Второй вид движения соответствует самодиффузии частиц жидкости и, как уже упоминалось, известен под названием «трансляционного движения». Трансляционное движение ионов и молекул воды в растворах представляет собой активированные скачкообразные перемещения.
В настоящее время внимание исследователей обращено на систематическое изучение термодинамических свойств растворов электролитов <в свете успехов, достигнутых в структурном анализе подобных систем. Изучению природы водных растворов электролитов и характеристике состояний отдельных ионов в растворе уделено особое внимание в работах А. Ф. Капустинского и его сотрудников. Введение кристаллохимических характеристик ионов позволило обобщить обширный фактический материал по энтропиям, теплоемкостям и парциальным объемам ионов, а также представить картину гидратации в виде своеобразного, замещения ионами молекул воды в ее подвижной квазикристалличе-ской структуре. Еще Д. И. Менделеев обратил внимание на то, что вода имеет различную степень химического родства с растворенным веществом, т. е. часть воды имеет большую связь с растворенным веществом по сравнению с остальной массой растворителя. Действительно, как показали многолетние работы О. Я. Самойлова, предложенный им и нашедший широкое применение термохимический метод определения координационных чисел ионов в водных растворах позволил установить, что гидратация ионов в растворах может быть разделена на две части — ближнюю и дальнюю. Первая связана со взаимодействием иона с молекулами воды, составляющими в растворе непосредственное окружение иона, вторая — со взаимодействием с более удаленными объемами воды. Что касается дальней гидратации,
Основные положения теории растворов электролитов
129
то она состоит в ’поляризации под действием поля иона окружающих молекул воды. Дальняя гидратация всегда сопровождается выделением довольно больших количеств энергии при переходе ионов в раствор.
Ближнюю гидратацию, согласно этим представлениям, следует рассматривать как действие ионов на тепловое и прежде всего на трансляционное движение ближайших молекул воды раствора.
Именно с ближней гидратацией связаны так называемые кинематические свойства растворов и механизм протекания в растворах ряда процессов. Таким образом, напрашивается вывод, что гидратация ионов в водных растворах состоит не в связывании ионом большей или меньшей оболочки из молекул воды. Ближняя гидратация сводится лишь к более или менее сильному взаимодействию иона с ближайшими молекулами воды, число которых в разбавленных растворах определяется квази-кристаллической структурой воды.
В случае разбавленных растворов электролитов под координационным числом ионов понимается среднее число постоянно сменяющихся молекул воды, составляющих в растворе непосредственное окружение ионов. Определение этого числа показало, что координационное число ряда одноатомных ионов близко к среднему координационному числу самих молекул воды в воде.
О 'физическом и химическом взаимодействии между ионами и молекулами растворителя свидетельствуют прежде всего данные Вальдена. Вальден показал, что одинаково диссоциированные в воде соли по-разному ведут себя в неводных растворителях с одинаковой диэлектрической проницаемостью. Для объяснения особенностей влияния различных растворителей на электропроводность Фуосс и Краус также признали существование химического взаимодействия в растворах.
В исследованиях последних лет, особенно в работах Н. А. Измайлова, было показано, что под влиянием неводных растворителей изменяются свойства любых электролитов: кислот, оснований, солей. В зависимости от свойств и структуры растворителя одно и то же вещество может быть неэлектролитом, сильным или слабым электролитом, кислотой или основанием или же вовсе не проявлять кислотно-основных свойств. Подобная зависимость и изменение свойств вещества под влиянием растворителей широко используются в данное время для решения ряда аналитических задач при электрометрическом титровании, полярографическом, амперометрическом и других методах физико-химического анализа для: а) повышения либо понижения растворимости вещества; б) усиления либо ослабления силы кислот, оснований и солей; в) изменения соотношения между ионным 9 Заказ 1338
130
Теоретические основы электрохимии
произведением среды и константы диссо'циаци1и вещества, а также в других целях.
Расширение применения неводных растворителей стало возможным благодаря возникновению единой точки зрения на растворы сильных и слабых электролитов как на вещества, молекулы и ионы которых подвержены физическим и химическим превращениям.
Все научные направления в этой области, как нетрудно заметить, исходят из теории образования непостоянных соединений между растворенным веществом и растворителем, изложенной впервые Д. И. Менделеевым.
ЛИТЕРАТУРА
Бродский А. И. Современная теория электролитов. ОНТИ, 1934.
Бродский А. И. Физическая химия, т. II. Госхимиздат, 1948.
Горенбейн Е. Я. Теория аномальной молекулярной электропроводности.
Труды II Всесоюзной конференции по теоретической и прикладной электрохимии. Изд. АН УССР, 1949.
Давтян О. К. ЖФХ, 1946, т. 20, вып. 9.
Дол М. Основы теоретической и экспериментальной электрохимии. ОНТИ, 1937.
Измайлов Н. А. Электрохимия растворов. Изд. Харьковского университета, 1959.
Краткий справочник физико-химических величин, под ред. К. П. Мищенко. Госхимиздат, 1959.
Мелвин-Хьюз Э. А. Физическая химия, кн. 1-2. ИЛ, 1962.
Самойлов О. Я. Структура водных растворов электролитов и гидратация ионов. Изд. АН СССР, 1957.
Семенченко В. К. Физическая теория растворов. Госхимиздат, 1941; ЖНХ, 1, 1956, 1131.
Стромберг А. Г. Термодинамика растворов электролитов. Томск, Изд.
Томского политехнического института, 1958.
Фалькенгаген Г. Электролиты. ОНТИ,' 1935.
Фаулер Р., Г у гг е н г е й м Э. Статистическая термодинамика. ИЛ, 4949. Френкель Я. И. Кинематическая теория жидкостей. Изд. АН СССР, 1945. Харнед Г., Оуэн Б. Физическая химия растворов электролитов. ИЛ, 1952.
Глава VI
ЭЛЕКТРОДВИЖУЩИЕ СИЛЫ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
§ 1. Обратимые электрохимические системы
Электрическое напряжение в общем случае выражается ли-
2
нейным интегралом U = \EsdS, взятым вдоль пути, по которо-1
му перемещается заряд Es (слагающая напряженности электрического поля) вдоль элемента пути dS.
Электрическое напряжение гальванического элемента или напряжение, наложенное извне на клеммы электролитной ванны и измеряемое в обоих случаях во внешней цепи (проводнике первого рода), может быть представлено в виде алгебраической суммы:
^ = ±[(<Ра-Тк)+е9л]. (VI, 1)
где U — электрическое напряжение электрохимиче-
ской системы;
Фа—'потенциал анода;
фк—'Потенциал катода;
Бэл — ‘К. = — —падение напряжения в электролите.
X
Знак плюс относится к электролитной ванне, а минус — к'гальваническому элементу.
В тальваническом элементе катодом является положительный полюс, а анодом — отрицательный. При применении уравнения (VI, 1) к аккумулятору следует иметь в виду, что при зарядке он ведет себя как электролитная ванна, а при разряде как гальванический элемент. Таким образом, во время зарядки аккумулятора катодом является отрицательный полюс, а анодом — положительный; при разряде положительный полюс является катодом, а отрицательный — анодом.
Из уравнения (VI, 1) следует, что измеряемое электрическое напряжение элемента всегда меньше разности потенциалов электродов фк — фа на величину падения напряжения в электро-fl*
132 Теоретические основы электрохимии
лите (е0л). Напряжение, измеряемое 'на клеммах электролитной ванны, наоборот, всегда больше <ра — фк на ту же самую величину (еэл)- Разность электродных потенциалов (гальванического элемента при отсутствии тока (ф°— <?°) равна его полной электродвижущей силе; именно исходя из этого принципа в электрохимии и определяют электродвижущую 'силу любого элемента компенсационным методом.
Электродвижущая сила (э. д. с.) в общем случае определяется как та часть работы переноса единичного положительного заряда из одной точки электрического поля в другую, которая совершается «сторонним» электрическим полем. Наиболее распространенный случай возникновения Естор или э. д. с. ,в электрохимических системах наблюдается в результате взаимодействия между зарядами, которые обусловлены физико-химической неоднородностью системы (гальванические элементы, аккумуляторы) .
С энергетической точки зрения э. д. с. в источниках тока появляется в результате преобразования неэлектрических видов энергии (например, химической) в электрическую, т. е. за счет работы.
Взаимный переход различных видов энергии делает принципиально возможным такое протекание электрохимического процесса, при котором осуществляется обратимое превращение химической энергии в электрическую либо, наоборот, обратимое превращение электрической энергии в химическую.
Электрическая энергия, затрачиваемая в электролитной ванне или передаваемая во внешнюю цепь от работающего элемента, может быть выражена равенством
• dA = UdQ.
При пересчете на 1 г-ион прореагировавшего у электрода вещества получим работу:
А zFU (VI, 2)
или с учетом (VI, 1)
А = + гР [(<ра — ?к) + еэл]. (VI .3)
Здесь часть общей энергии гВеЭл расходуется на преодоление омического сопротивления (внутри системы), а другая часть zF(<pa — фк) —на осуществление электрохимических реакций на электродах. Так, например, в электролитной ванне Pti+jHCl, H2O|2-Pt электрическая энергия Т(фа—фк) расходуется на осуществление химической реакции НС1-> l/2H2 + V2CI2, представляющей собой сумму двух электродных процессов: анодного — с выделением газообразного хлора и катодного — с выделением водорода.
Электродвижущие рилы и электродные потенциалы
133
В элементе типа Р1(С12)ф| НС1, Н2О I7 (H2)Pt, наоборот, электрическая энергия F(<ра — фк) .получается во внешней цепи вследствие процессов ионизации на электродах хлора V2CI2 + е—(положительный полюс) и водорода ‘/гНг— е—>Н+ (отрицательный полюс).
Таким образом, энергия источника тока — элемента появляется в результате протекания токообразующей химической реакции:
УаНг-’-НСЕ
Пользуясь уравнением (VI, 3) можно написать: для гальванического элемента
Л.Э К<ркг—’?а)г.э — еэл] ZF, (VI, 4а)
для электролитной ванны
Л = К?а — ?к)в + еэ J zF (VI, 46)
но для обратимых систем при условии, когда 1->0 и, следовательно, Ав = Аг.э, получим
(VI. 5)
Если i > 0, то на электродах любой электрохимической системы возникает электродная поляризация Аф, тем большая, чем больший ток проходит через цепь, т. е. <ра — фк = (ф° —Ф°) +' + Аф, или, учитывая (VI, 3):
А = + zF ((ср., — <рк) + Д<р + еэл]*. (VI, 6)
Электродная поляризация Аф это часть напряжения U, которое мы теряем безвозвратно в связи с необратимостью процесса, протекающего при вполне определенной скорости электродных актов; Аф, очевидно, пропорционально заданной плотности тока.
В общем случае протекание тока через электрохимическую систему сопровождается глубокими качественными изменениями, которые претерпевают частицы вещества (ионы, атомы и молекулы) на границе раздела фаз электрод — электролит. При этом электрическая энергия, расходуемая или получаемая в результате осуществления электродных реакций, зависит прежде всего от интенсивности процесса, т. е. в данном случае от силы или плотности тока.
Поскольку разность потенциалов, затрачиваемая на суммарный процесс, протекающий на обоих электродах в ванне, и на
* Знак (+) относится к электролитной ванне, а (—) к гальваническому элементу.
134
Теоретические основы электрохимии
пряжение, получаемое от источника тока, зависят от силы проходящего через 'систему электрического тока, то сравнивать между собой можно лишь обратимые (термодинамические) электрохимические системы. Обратимыми электрохимическими реакциями называют реакции, идущие одновременно в прямом и обратном направлении и предусматривающие возможность возвращения системы в первоначальное состояние, т. е. без того, чтобы в’ окружающей среде появились и остались какие-либо изменения.
Электрохимическое равновесие является равновесием динамическим: оно обусловлено не тем, что дойдя до него процесс полностью прекращается, а тем, что .взаимно противоположные электродные реакции при бесконечно малом токе протекают с одинаковыми скоростями. Например, на катоде (отрицательном полюсе) обратимой системы: Pt('C12) + | НО, H2O|~(H2)Pt все время идет и образование атомов водорода и их ионизация, но число образующихся за единицу времени атомов водорода равно числу распавшихся (перешедших в раствор в ионную форму). Поэтому создается впечатление, что никаких видимых изменений в системе не происходит.
Подобная же картина наблюдается на аноде (положительном полюсе) указанной обратимой электрохимической системы при осуществлении реакций ’/гСЬ^СГ". ‘
Если на равновесную электрохимическую систему производить внешнее воздействие, то равновесие будет смещаться в сторону, указываемую этим воздействием, и до тех пор, пока нарастающее в системе противодействие не станет равным внешнему действию (принцип смещения равновесий Ле-Шателье). Наоборот, при бесконечно медленном процессе (z->0) состояние электрохимической системы в каждый данный момент времени бесконечно мало отличается от равновесного. Полностью обратимый электрохимический процесс, характеризуется, кроме z->0, тем, что вся совокупность элементов (веществ), принимающих в нем участие, проходит в точности через те же состояния, что и при прямом процессе, но в обратной последовательности. Если же это условие не удовлетворяется, то имеет место необратимость. Примером необратимой системы в указанном смысле может служить элемент Zn-| H2SO4 |+Си, тде даже при разомкнутой внешней цепи (Z = 0) протекает самопроизвольная химическая реакция взаимодействия между цинком и серной кислотой.
Таким образом, электрохимическая система является полностью обратимой при двух условиях: во-первых, если проходящий через нее ток (при Р, Т и С = const) стремится к .нулю и, во-вторых, если в системе не происходит каких-либо самопроизвольных химических процессов. Примером обратимой системы, очень часто применяемой в практике электрохимических изме
Электродвижущие силы и электродные потенциалы
135
рений, является нормальный элемент (рис. 32), который может быть изображен схемой
Hg+|Hg2SO4(TB.HaCbnu.p.p), 3CdSO4 8Н2О|—12%Cd в Hg.
Здесь положительным полюсом является ртуть, покрытая пастой Hg2SO4, отрицательным—кадмиевая амальгама. Электролитом служит насыщенный раствор 'CdSO4, причем для поддержания насыщения часть сосуда заполнена кристаллами CdSO4. Токообразующий процесс в элементе протекает по следующей схеме:
Cd + Hg2SO44-%H2O->
vcdso4.%H2o+;2Hg,
Этот элемент является международным эталоном вольта, так как принято, что международный вольт равен 1/1,018300 электродвижущей силы нормального элемента при 20 °C. Вообще же э. д. с. равна:
En = 1,018300 — 4,06.
• 10-5(f—'20) — 0,95. I0“56(Z—20)2+
+ 1 • 10~8(f — 20)3.
Рис. 32. Нормальный элемент:
1 — насыщенный раствор CdSO4;
2 — кристаллы CdSO4 • 8/з Н2О; 3 — амальгама Cd; 4 — ртуть; 5 — паста Hg2SO4
Следует иметь в виду, что для обратимой работы нормального г элемента от него можно отбирать лишь очень малые токи. Поэтому при измерениях с этим прибором ток включают лишь на несколько
секунд, и после каждого включения дают ему отстояться без тока для выравнивания концентрационных изменений, происшедших в приэлектродных слоях.
Опыт показывает, что в нормальном элементе при размыкании внешней цепи ни один из электродов не подвергается каким-либо химическим изменениям (в противоположность рассмотренному выше случаю для цинка в H2SO4).
Хотя процессы могут быть полностью обратимыми только в предельном случае, т. е. при бесконечно медленном их протекании, а все реальные процессы, протекающие с конечной скоростью (плотностью тока), необратимы, ,в некоторых случаях, схематизируя явление, неправильно считают обратимыми процессы, идущие с конечной скоростью. Обратимым, например, иногда представляют процесс зарядки и разрядки аккумулятора,
136
Теоретические основы электрохимии
однако при этом пренебрегают неизбежными потерями на преодоление омических сопротивлений (еэл) и поляризации (Дфп)-Обратимые системы и процессы играют весьма существенную роль в электрохимии.
Обращаясь к выяснению условий, описывающих состояние равновесия, необходимо рассмотреть закономерности термодинамики обратимых электрохимических систем.
§ 2. Изменение свободной энергии обратимых элементов и электродов
В самом общем виде реакцию, обратимо протекающую в гальваническом элементе, можно описать уравнением
Vl«l + v2«2 + ....? V . . . , (VI, 7)
где vi, V2, v/, V2Z и т. д. — стехиометрические коэффициенты реакции;
аь а2, а/, и т. д. т— активности химических веществ, участвующих в реакции.
Максимальная работа такого элемента:
,”1 , ”2
А = — ДФ -=ЯПп/С — RT In v, (VI, 8)
fil а2
Самопроизвольное протекание физико-химических процессов сопровождается уменьшением свободной энергии ДФ. Для электрохимического процесса эта величина может быть приравнена к электрической работе zFE. Поэтому э. д. с. обратимого элемента
Е
(VI, 9)
где Ео — э. д. с. элемента, когда
«X2
В том случае, когда при реакциях, происходящих в системах, через границу системы переносится вещество, совершается работа против сил химического взаимодействия, поэтому величина Е может быть выражена и через химические потенциалы:
Р t = р° --J- RT 1г q
Электродвижущие силы и электродные потенциалы
137
Здесь р,— химический потенциал или изменение изобарного потенциала системы данного состава при обратимом введении .в нее одного моля z-того компонента при условии постоянства температуры и давления и при неизменном содержании всех других компонентов системы.
р9 — постоянная, не зависящая от состава, но зависящая от температуры.
Для рассматриваемого выше элемента Pt(H2)+|HCl. Н.2О| - Pt (С12) в общем виде имеем
— ДФ = zFE = рс12 + рНг — zpHCP (VI, 10)
откуда при z = 2 получим
2
с RT . „ RT . «НС! р 2RT . ZVT
In/С--“Ь-—— = £о--------------^lnaHC1. (VI, И)
2F 2F Ра,Рн2 F
Рассмотрим далее элемент, составленный из металлического и водородного электрода. Суммарная реакция при работе такого элемента будет описываться уравнением.
Мег+ + -j- Н2 -> Me + zH+.
Если принять ан+ = 1 и рн2 = К то э. д. с. такого элемента, согласно уравнению (VI, 9), будет равна:
E = -^lnX-^In-J-. zF zF z+
aMe
Таким образом, Е здесь выражает потенциал полуэлемента —• металлического электрода, рассчитанный по водородному электроду, принятому за условный нуль шкалы потенциалов, т. е.
? z+. .. . -^-Ina ,+. (VI,12)
гМе /Ме тMez4Me^ гр Мег^
Сказанное в равной мере относится не только к металлическим электродам, но и к газовым электродам, обратимым по отношению к анионам:
Хп = nX2 — nze,
где п — число атомов в молекуле;
е — заряд электрона.
Учитывая, что в таком случае в токообразующем процессе принимают участие отрицательно заряженные ионы Xz—, при раскрытии значения Е (по водородной шкале) получим
<?х/хг-==^---RIV’13)
138 Теоретические основы электрохимии
Таким образом, обратимые элементы состоят из обратимых электродов. Потенциал электрода, обратимого относительно катионов, с ростом активности потенциалопределяющих ионов изменяется в положительную сторону, а потенциал электрода, обратимого относительно анионов, -— в отрицательную сторону. Приведя эти уравнения к виду, удобному для вычисления, получим
?+ 0,0002 — 1g а., (VI, 14)
так как R = 8,313 дж)град • моль и F = 96 500 к/г-экв, а модуль перехода от натуральных логарифмов к десятичным равен 2,3026; следовательно, в формуле Нернста (VI, 14) для катионообразующих веществ перед логарифмом следует писать знак ( + ), для анионообразующих — знак (—). Очевидно, что <р, = = <р{° при условии, когда активности реагирующих веществ или отношение активностей реагирующих веществ равны единице.
Таким образом, <p,-° есть стандартный или нормальный электродный потенциал, который зависит только от природы электрода и температуры.
§ 3. Уравнение Томсона и Гиббса—Гельмгольца
Приближенное к действительности осуществление условия обратимости системы (£—>0) на практике может быть достигнуто включением во внешнюю цепь настолько большого сопротивления, чтобы внутреннее сопротивление системы было по сравнению с ним исчезающе мало.
Рассмотрим элемент Якоби—Даниэля при условии включения такого сопротивления:
Zn | ZnSO4| CuSO41 Си.
Здесь осуществляется реакция
Zn 4- CuSO4 = Си + ZnSO,. (VI, 15)
Если поместить подобный элемент в калориметр, то количество тепла Qp, выделившееся при его работе,'по закону Гесса, будет равно тепловому эффекту реакции (VI, 15).
Томсон, не учитывавший второго закона термодинамики, предположил, что тепловой эффект химической реакции эквивалентен электрической работе элемента, т. е.
4,19Qp = zfE,
где 4,19 — переводной множитель для перехода от калорий к вольт-ампер-секундам.
Электродвижущие силы и электродные потенциалы
139
Тогда
Величина QP для реакции (VI, 15) составляет 50,13 ккал!моль, откуда
Е = 4,19-50,13 . 1000 = 1 083,
2 • 96 500
что почти точно соответствует действительной э. д. с. системы.
Однако очень скоро выяснилось, что правило Томсона не согласуется с опытом ’. На самом деле законным является равенство:
Е = — —19АФ-. (VI, 17)
zF
Последняя величина связывается с теплотой реакции уравнением Гиббса — Гельмгольца:
-ДФ = (2р-т(^-)р. (VI, 18)
Смысл приведенного уравнения и его отличие от правила Томсона состоят в том, что тепловой эффект реакции не равен ее максимальной работе А. Пользуясь равенствами (VI, 17) и (VI, 18), можно получить
Е —т— = 4’19<3р . (VI,19)
dT zF
. Уравнение (VI, 1'9) дает хорошее совпадение с опытными данными, но на практике оно почти не применяется ввиду отсутствия значений температурных коэффициентов.
§ 4. Проблема топливных элементов
Этим термином определяются гальванические элементы, в которых осуществляется реакция окисления газообразного, жидкого или твердого «топлива» и которые дают возможность получить энергию, .выделяющуюся в этой реакции, непосредственно в виде электрического тока. Нахождение технически приемлемых форм топливного элемента позволило' бы значительно повысить к. п. д. процесса по сравнению с обычно принятыми методами использования горючего для паровых котлов, турбин и т. п.
1 Один из примеров неточности правила Томсона .приводится А. И. Бродским. Так, если в гальваничеоком элементе протекает реакция: 2Ag + HgiC12= = 2AgCls + 2Hg, то э. д. с. элемента равна +0,0456 в. Между тем тепловой эффект этого процесса Qp = —2546 кал!моль. Таким образом, здесь Qp и Е имеют разные знаки.
140 Теоретические основы электрохимии
Как известно, до сих пор основным источником энергии является химическая энергия различных видов топлива, в первую очередь угля и нефти. Но на пути к потребителю она проходит через целую цепь превращений. 'Сначала она превращается в тепло при сгорании топлива, затем — в механическую работу двигателя и лишь после этого — в электрический ток. На каждом таком этапе неизбежно теряется значительная часть энергии. Сократить число этих промежуточных стадий — значит намного уменьшить потери. Топливный элемент примечателен тем, что химическая энергия «горючего» здесь сразу превращается в электрическую энергию.
В топливном элементе запас электрохимического горючего непрерывно пополняется извне. В качестве такого горючего в принципе можно использовать достаточно доступные вещества: некоторые природные топлива или продукты их переработки.
Теоретически .в топливном элементе можно полностью использовать свободную энергию горючего, которая не очень отличается от его теплотворной способности. На самом деле это не совсем так, поскольку и здесь потери энергии неизбежны. Однако можно получить коэффициент полезного действия, равный 65—70%, т. е. значительно выше, чем у самых лучших тепловых двигателей.
В низкотемпературных топливных элементах электролитом служит водный раствор. Главная трудность создания таких элементов — это подбор электродов, на поверхности которых реакция образования ионов из электрохимического горючего протекала бы с достаточной скоростью. Для этого электроды делают пористыми, получая большую поверхность соприкосновения газа и жидкости, используют материалы, увеличивающие реакционную способность горючего.
Есть и 'другой тип — высокотемпературные элементы. В них вместо водного раствора электролитом служит расплавленный или твердый проводник, .в котором ток переносят не электроны, а заряженные атомы или группы атомов. Подобные элементы рассчитаны на работу при 600—900 °C. При таких относительно высоких температурах электрохимические реакции идут быстрее и подобрать материал электродов довольно просто. У высокотемпературных’ элементов есть важное преимущество. В них можно использовать более широкий круг горючих, в том числе особенно перспективное горючее — окись углерода в виде генераторного газа. Он будет окисляться на отрицательном электроде в углекислоту, которую можно затем использовать для газификации твердого топлива и получения из него новых порций генераторного газа. Топливо при этом подогревается избыточным теплом, выделяющимся при работе элемента. Такой круговой процесс позволяет использовать в топливном элементе.
Электродвижущие силы и электродные потенциалы 141
хотя и косвенным путем, химическую энергию угля. В отдельных элементах этого типа, работающих при 750 °C, уже достигнута большая продолжительность непрерывной работы. К сожалению, эти элементы допускают лишь незначительную нагрузку током, так как в твердом электролите продукт реакции (двуокись углерода) обладает малой скоростью диффузии « медленно удаляется из реакционной зоны.
Бесшумность топливных элементов, отсутствие движущихся частей, относительно высокая величина мощности на единицу объема и веса, отсутствие выделения каких-либо газов или излучений делают их особенно ценными :в экеплуатации.
По запасу энергии на единицу веса и объема топливные элементы могут превзойти большинство известных в настоящее время типов элементов и аккумуляторов. Можно полагать, что топливные элементы в недалеком будущем явятся весьма удобным источником энергии на электростанциях. Здесь не будет ни котлов, ни вращающихся частей динамомашин, ни сложных сооружений для защиты от излучений, которые неизбежны на атомных станциях.
§ 5. Скачки потенциалов на границе фаз и механизм их возникновения
Подобно обратимым элементам можно представить себе и обратимые электроды. Уравнения (VI, 1-8) и (VI, 19) справедливы и для отдельного обратимого электрода. В этом случае Q будет теплотой, выделяющейся или поглощающейся при превращении 1 г-экв металла (в случае металлического электрода) в ионы. Эта величина называется теплотой ионизации.
Таким образом, любое состояние обратимого (равновесного) электродного потенциала также определяется температурой, давлением и -составом электролита. Электролит можно нагреть, охладить, сконцентрировать, разбавить, изменить давление, но всегда представляется возможным заранее предсказать для определенных температур, давления и соотношения компонентов значения равновесного потенциала. Изменяются условия, изменяется и потенциал. Но как только система возвратится в первоначальные условия, электрод будет обладать первоначальными свойствами.
Таким образом, в истинно обратимом электроде все процессы полностью обратимы.
В термодинамике показывается, что если имеется система, состоящая из одной или нескольких фаз, находящихся в равновесии, то каждый компонент такой системы описывается с помощью химического потенциала этого компонента щ-.
Если система включает в -себя ряд1 компонентов, состоящих
142 Теоретические основы электрохимии
из заряженных частиц, то каждый такой компонент обладает некоторым электрохимическим потенциалом:
V = И» + ez?z, (VI,20)
где е, — заряд i-того иона;
<pj — электрический потенциал.
Свободная энергия перехода какого-либо компонента, несущего заряд, из одной фазы в другую будет равняться разности электрохимических потенциалов, которые могут быть определены из термодинамического условия равновесия двух фаз
* гг
Т, = Т-, I l’ откуда
д<р. (VI, 21)
Ч
При наличии границы фаз всегда возникает некоторое перераспределение электрического заряда, образуется двойной электрический слой и возникает скачок потенциала.
Таким образом, мы приходим к неизбежности межфазного скачка потенциала на границе двух фаз, содержащих заряженные компоненты. Подобными фазами могут быть не только металл и раствор электролита. Скачки потенциалов на границе фаз можно .наблюдать в различных случаях, например:
а) «а границе металл — раствор электролита (электродный потенциал);
б) на границе двух различных металлов (контактный потенциал) ;
в) на границе металл — газ (контактный потенциал II рода);
г) на границе двух растворов в разных растворителях (фазовый жидкостный потенциал);
д) на границе раствор — газ (фазовый контактный потенциал);
е) на границе соприкосновения двух растворов в одном растворителе, различающихся или по виду растворенного вещества, или по его концентрации (диффузионный потенциал);
ж) если такие растворы разделены полупроницаемой перегородкой (мембраной), то возникают мембранные потенциалы;
з) скачок потенциала, возникающий при перемещении двух фаз одна относительно другой, отличается по величине от скачка потенциала на границе двух неподвижных фаз. Такой скачок играет важную роль в электрокинетических явлениях; обычно он называется электрокинетическим или ^-потенциалом.
Определение абсолютных значений перечисленных скачков потенциалов на границе двух фаз практически затруднено, но
Электродвижущие силы и электродные потенциалы
143
тем не менее нет никаких сомнений в реальности их существования, так как появление скачков потенциалов является следствием стремления всякой системы к термодинамическому равновесию.
Вернемся еще раз к вопросу о потенциале отдельного электрода, так как при изучении электрохимических систем и реакций решающую роль играет именно скачок потенциала на границе раздела фаз электрод — электролит, так называемый электродный потенциал. В связи с этим приведенное выше рассмотрение значений э. д. с. любого элемента хотя и является термодинамически правильным, но не учитывает процессы, протекающие на границе фаз электрод — раствор.
Рассмотрим систему, которую в электрохимии принято называть «электрод в растворе своих ионов». Для этого опустим в раствор сульфата меди медный электрод и постараемся представить себе те явления, которые произойдут после этого.
И в металле, и в растворе имеются одинаковые ионы — ионы меди. Только .в металле эти ионы находятся в узлах кристаллической решетки, а в растворе они связаны с молекулами воды — гидратированы. Чтобы вывести ион меди из раствора, необходимо затратить работу, равную энергии гидратации, т. е. энергии связи этого иона с молекулами воды £7Р. Чтобы вывести ион из кристаллической решетки, тоже нужно затратить энергию UM, которую можно назвать работой выхода иона из металла (по аналогии с работой выхода электрона).
При опускании металлической пластинки в раствор своих ионов металл и раствор взаимодействуют и становится возможным переход ионов из металла в раствор и обратно. В первый момент направление этих переходов связано соотношением величины UK и Up. Если UM>UP, т. е. если энергия связи ионов в кристаллической решетке металла больше, чем энергия гидратации этих ионов, как в меди, то после погружения металла в раствор ионы металла будут переходить из раствора в кристаллическую решетку.
Но металл и раствор — проводники, поэтому электричество здесь, как во всех проводниках, растекается по поверхности. Следовательно, электроны будут тесно прижаты к граничной поверхности металла. Аналогично этому, положительные ионы Mez+ будут продвигаться к граничной поверхности раствора. Из .всех поверхностей, которые имеются у металла и раствора, лишь одна является граничной для обеих фаз, а именно—-поверхность раздела металл — раствор. Следовательно, все избыточные заряды в металле (электроны) и катионы Mez+ в растворе собираются на границе раздела фаз, образуется двойной ионный электрический слой и возникает скачок потенциала, который стремится вернуть ионы Mez+ в металлическую решетку
144
Теоретические основы электрохимии
(рис. 33). Так как ионы Си2+ несут положительные заряды, то это ведет к заряжению медного электрода положительным электричеством, а раствора, в котором ионов меди недостает, — отрицательным. Это затрудняет дальнейшее осаждение Си2+-ионов, и в конце концов в системе устанавливается равновесие:
Си2+ 2 Си2+ 9 связанные ионы в металле свободные ионы в растворе
имеющее динамический характер, т. е. переход ионов Си2+ из раствора в металл продолжается, но одновременно и с той же скоростью происходит обратный переход Си2+-ионо'в из твердой
Рис. 33. Схема возникновения двойного слоя: а — на электроде — избыток отрицательных зарядов; б ~ двойной слой отсутствует; в — на электроде — избыток положительных зарядов
фазы в раствор. Таким образом, медный электрод обычно заряжен положительно по отношению к раствору своих ионов. Если вместо системы Cu|CuSO4 рассмотреть систему Zn|ZnSO4 (цинк, погруженный в раствор сульфата цинка), то окажется, что в этом случае металл заряжается отрицательно, а раствор— положительно. Таким образом, ионы цинка в первый момент после погружения переходят из металла в раствор, а двойной электрический слой образуется избыточными электронами на металле и ионами Zn2+ в растворе. Очевидно, для цинка £7м<;£7р.
Равновесия и в таком случае достигаются выравниванием скоростей перехода вещества из металла в раствор и обратно. В обычных случаях при установившемся равновесии мы не наблюдаем описанного перехода ионов, так как поверхность соприкосновения металла с раствором очень мала, и поэтому то количество ионов, которое переходит из одной фазы в другую, практически не изменяет концентрации этих ионов в рас
Электродвижущие силы и электродные потенциалы 145
творе. Чтобы заметить эти изменения, надо увеличить поверк-ность металла по сравнению с его весом и уменьшить объем раствора, в который погружен металл, либо применить способ меченых атомов, позволяющий убедиться в существовании таких процессов, которые -нельзя обнаружить обычными аналитическими методами.
Характерная черта ионных двойных слоев — то, что составляющие их заряды расположены в обеих фазах.
Очевидно, что для металлического и анионообразующего газового (например, хлорного) электродов, при одном и том же соотношении UM и Up -знаки зарядов двойных ионных слоев будут противоположными, что должно быть учтено в уравнении Нерн-ста для электродного потенциала. Но во -всех случаях, если заряд de, переходя из одной фазы в другую, пересекает двойной слой, он на очень коротком пути (толщина двойного слоя составляет Ю-8 см) подвергается действию интенсивного электрического поля и производит определенную работу. Эта работа равна:
dA = cpde,
где <р есть разность потенциалов точки в растворе, лежащей вблизи границы электрод—-электролит, и точки в металле вблизи той же границы. Эта разность потенциалов ф, возникающая между металлом и раствором, называется электродным скачком потенциала или абсолютным электродным потенциалом. Интенсивность возникающих при этом полей достигает нескольких десятков миллионов вольт на 1 см (при разности потенциалов 1 в и радиусе ионов 1,5 • КУ'8 см)'. Естественно, что такие большие поля должны влиять на свойства поверхности электрода, а также на скорость процессов, протекающих на (границе фаз электрод — электролит. В настоящее время мы не можем измерить абсолютные скачки потенциалов точек, расположенных в различных фазах: ib металле и растворе, так как переход заряженной частицы через границу фаз сопровождается работой, равной разности химических потенциалов веществ в двух фазах. Экспериментально удается измерить лишь разности потенциалов для точек, лежащих в пределах одной фазы, так как при этом разность химических потенциалов равна нулю.
Практическое измерение электродных потенциалов сводится к изучению электродвижущих сил гальванических элементов. Однако не всякая пара электродов пригодна для этой цели. Например, при измерении э. д. с. элемента Якоби — Даниэля:
Cu+|CuSO4, И2о|н2О, ZnS04|-Zn.
Она слагается из электродных скачков потенциалов медного Cu|CuSO4 и цинкового Zn|ZnSO4 электродов, а также из скач-
Ю Заказ 1338
146
Теоретические основы электрохимии
Рис. 34. Схема элемента Якоби — Даниеля и распределение отдельных слагаемых э. д. с.
fZn/ZnSO4 и <PCu/CuSO4 - электродные потенциалы; Vzn/Cu ~ контактный потенциал: f>g — диффузионный по-
тенциал; Г — гальванометр; R — сопротивление
ка 'потенциала, возникающего .на границе двух растворов CuSO4/ZnSO4 (диффузионного потенциала <рд). Помимо этого, вследствие разного уровня энергии электронов в двух металлах (Cu/Zn) при соприкосновении .их электроны переходят из одного металла в другой, что сразу же нарушает электрическую нейтральность обоих металлов и приводит к возникновению контактного скачка потенциалов VZn/Cu (рис. 34), который может быть выражен через работы выхода электрона1 из соответствующих металлов (f7fn и ). Таким образом, VZn/cu = ^n-t4Cu. (VI,22) А. Н. Фрумкин показал, что контактная разность потенциалов определяется разностью потенциалов между точками нулевых зарядов этих металлов. Следовательно, в самом общем виде абсолютная величина э. д. с. элемента.
Е = Zn/Cu “Ь ^Cu/CuSO. +
+ Тд-?аяюо,. (VI,23)
При обсуждении относительной величины э. д. с. элемента мы можем не учитывать контактного потенциала на границе двух проводников первого рода. Если контактная разность потенциалов близка к разности потенциа
лов между нулевыми точками различных металлов, то очевидно, что эта последняя величина не должна зависеть от природы электролита. Таким образом, можно считать, что VZn/Cll = = const и не влияет на относительную величину э. д. с. Диффузионным потенциалом в известных условиях можно пренебречь либо устранить его искусственным путем.
В таком случае для э. д. с. становится справедливым равенство
Е ~ <Pcu/CuSO. ?Zn/ZnS04’ (VI,24)
составленное из двух известных слагаемых.
1 Чтобы вырваться из металла, электрон должен совершить работу против сил электрического поли внутри металла и двойного электрического слоя, а также против сил зеркального электрического отображения, т. е. на расстоянии, примерно равном 10-® см. Эта работа называется работой выхода. Работа выхода обычно измеряется в электрон-вольтах (эв).
Электродвижущие силы и электродные потенциалы 147
Характерно, что при всех подобных изменениях определяются не абсолютные .величины электродных потенциалов, а лишь их относительные значения. Поэтому необходимо было условиться о том, относительно какого электрода измерять все потенциалы. Как уже отмечалось, в качестве стандартной шкалы потенциалов была принята водородная шкала, за нуль которой условно принимается потенциал нормального водородного электрода, т. е.
И.Р1(На)]|Н+(Сн+=1).
Потенциал нормального водородного электрода считают равным нулю при всех значениях температуры. Так, например, потенциалом медного электрода <PCu2+/Cu называют э. д. с. элемента:
I. (Н2) Pt (рн> = 1)| Н+ (ан+ = 1), Н2О | Си2+, Н2О | Си, а потенциалом цинкового электрода ?zn2+/zn э- д- с- эле-мента:
II. (H2)Pt(pHf= 1)|Н+(Он+ = 1), H2O|Zn2+, H2O|Zn.
Таким образом, электродным потенциалом любого неизвестного электрода, опущенного в раствор, содержащий его ионы, принято называть электродвижущую силу элемента, составленного из исследуемого электрода и водородного электрода, находящегося в нормальных условиях. Если все вещества, участвующие в электрохимическом процессе, протекающем в обратимом элементе, .находятся в нормальных условиях, т. е. их активности или отношение их активностей равны единице, э. д. с. такого элемента равна своему нормальному (стандартному) значению. В соответствии с этим нормальным (стандартным) электродным потенциалом называют потенциал любого электрода, опущенного в раствор, содержащий его ионы, при условии, если активность или отношение активностей ионов, относительно которых электрод является обратимым, равны единице.
Исходя из правила знаков для элементов и цепей, согласно которому электродвижущая сила любого элемента положительна, для нормальных электродных потенциалов принимают тот знак, который он имеет в паре с водородным нормальным электродом, если водородный электрод записан слева в цепи. Это значит, что в цепи I — Cu2+/Cu положительный полюс, а в цепи II — Zn2+/Zn отрицательный полюс. В соответствии с этим при ан+ — аСи2+ — 1 ион+ = °zn2+ = l нормальный потенциал медного электрода <PCu2+/cu = +'0,'34 в, а цинкового электрода <Pzr2+/zn =— °>76 в-
10*
148
Теоретические основы электрохимии
В сводной таблице (табл. 13) указан знак нормальных потенциалов электродов по водородной шкале. Здесь следует обратить внимание, в каком направлении записана электродная
Таблица 13. Нормальные потенциалы электродов при 25 °C
Электрод Электродный процесс Потенциал в Электрод Электродный процесс Потенциал в
F2 F2 + 2e->2F“ 4-2,85 H2 2Н+4-2е->Н2 0,00
Au Au+ + le-> Au + 1,50 Fe Fe3+ + 3e->Fe —0,04
Au Au3+ 4- 3e -> Au + 1,29 Pb Pb2+4-2e->Pb —0,13
Pt Pt2+ + 2e->Pt + 1,20 Sn Sn2"F + 2e->-Sn —0,14
Hg Hgl+ + 2e - 2Hg 4-0,80 Ni Ni2+ + 2e->Ni —0,23
Hg Hg2+4-2f->Hg +0,86 Co Co2+ 4- 2e T1 —0,27
Pd Pd2+ + 2e -» Pd +0,82 T1 T1+ + le -> T1 —0,34
Pb Pb4+ + 4e Pb +0,80, Cd Cd2+4-2e^Cd —0,40
Ag Ag+ + le -> Ag +0,80 Fe Fe2+ 4- 2e Fe —0,44
T1 Tl3+ + 3e->Tl +0,72 Cr Cr3+ + 3e-»Cr —0,51
J2 J2 + 2e > 2J— +0,53 Zn Zn2+ + 2e-»Zn —0,76
Cu Cu+ + le -> Cu +0,52 Mn Mn2+ 2e - Mn —1,10
Co Co3+ + 3e->Co +0,43 Al Al3+ + 3e -> Al —1,30
o2 4H+ + O2 + 4- 4e _> 2H2O + 1,23 Mg Na Mg2+ 4- 2e -> Mg Na+ 4- le-> Na —1,55 —2,71
Br2 Br2 + 2e -> 2Br~ + 1,07 Ca Ca2+ + 2e->Ca —2,76
Cl2 Cl2 + 2e > 2C1~ + 1,36 К K++ le->K —2,92
Cu Sn Cu2+ + 2e -> Cu Sn4+ + 4e -> Sn +0,34 +0,01 Li Li+ 4- le - Li —2,96
реакция. Если бы для меди реакция была записана в обратном порядке, т. е. Сц/Сц2+, то для данного случая потенциал электрода следует принять равным —0,34 в.
Таким образом, знак отдельного электрода (полуэлемепта) зависит не только от того, каков заряд электрода (полуэлемента) в цепи с водородным электродом, но и от порядка записи электродной реакции. Однако .во всех случаях процесс ионного обмена протекает таким образом, что значение электродного потенциала отвечает термодинамическому равновесию между электродом и раствором электролита.
Электродвижущие силы и электродные потенциалы
149
§ 6. Квантовомеханическая теория электродных потенциалов
Ряд нерешенных вопросов сделал неизбежными дальнейшие попытки создания такой теории электродных потенциалов, которая дала бы возможность более точно рассчитывать стандартные или нормальные потенциалы. Кроме того, необходимо было получить ответ на вопрос о происхождении скачков потенциалов, составляющих э. д. с. гальванической цепи.
Попытка построения квантомеха-V нической теории электродных потен-
циалов была предпринята Р. Герни.
Рис. 35. Кривая потенциальной энергии .положительного нона в зависимости от расстояния г
Рис. 36. Потенциальный' барьер (уровни энергии Z и W), который необходимо преодолеть иону для перехода его в раствор и обратно
Представим себе металлический электрод, помещенный в вакууме. Чтобы вывести ион металла из кристаллической решетки, нам придется совершить работу для преодоления энергии связи этого иона в кристаллической решетке. Эта энергия быстро уменьшается с расстоянием, так что кривая потенциальной энергии положительного иона будет иметь вид, .изображен- . ный на рис. 35, где горизонтальная линия г отвечает уровню иона металла на поверхности. С другой стороны, если ион находится в растворе, то он также находится в «потенциальной яме» (рис. 36), т. е. располагается рядом с молекулами растворителя так, чтобы его потенциальная энергия была минимальной. Таким образом, на поверхности металла существует некоторый потенциальный барьер, который необходимо преодолеть иону, чтобы перейти в раствор или обратно.
Однако с точки зрения квантовой механики такие переходы возможны, причем вероятность их зависит от соотношения глубин «потенциальных ям». Если энергетический уровень иона в растворе ниже, чем в металле, то наиболее вероятными становятся переходы ионов из металла в раствор. В противном случае, наоборот, наиболее вероятны переходы ионов из раствора в металл.
150
Теоретические основы электрохимии
Возможные соотношения «потенциальных ям» иона в ме-
талле и растворе представлены на рис. 37. В случае б, когда уровни ям одинаковы, переходы ионов в обоих направлениях
равновероятны и совершаются обратимо. В случае а возможны лишь переходы ионов из металла в раствор. Подобные перехо-
ды необратимы, так как ионы не способны к самопроизвольному
переходу с низшего уровня энергии к более высокому уровню ее.
Рис. 37. Возможные соотношения «потенциальных ям»
Когда катионы металла переходят в раствор, металл заряжается отрицательно, вследствие чего уровни энергии смещаются в таком направлении, что устанавливается положение равновесия ионов (рис. 37, а) и возникает скачок потенциала на границе металл— раствор.
В положении а ионы будут разряжаться на металле, что вызовет передвижение уровней и создание, противоположного потенциала. В этом случае металл зарядится положительно.
Когда два металла, погруженные в раствор своих ионов, приводятся в соприкосновение через проводник первого рода (как, например, в элементе Якоби — Даниэля), то электроны пе
реходят с более высокого уровня энергии в металлическом цинке на более
низкий уровень в меди. При этом энергетический уровень электронов в цинке будет понижаться, а в меди повышаться. Но понижение уровня электронов в цинке означает повышение уровня положительного иона в этом металле. Это нарушает условие равновесия, указанное на рис. 37, б, и создает положение, изображенное на рис. 37, а, только при этом разность в условиях потенциальной энергии становится значительной. В меди наблюдается обратная картина: уровни элект-
ронов повышаются, уровни ионов понижаются и на поверхности металлов создается положение, изображенное на рис. 37, в.
Когда элемент работает, т. е. проходит ток, электроны у контакта медь — цинк совершают переход между одинаковыми энергетическими уровнями; таким образом, здесь энергия ни те-ряется, ни приобретается. На поверхности же металлов ионы переходят с высокого уровня на более низкий, поэтому энергия элемента берется от химической реакции у электродов, хотя разность в уровнях положительного иона на границе металл—раствор обусловлена прежде всего разностью в работе выхода электрона для обоих металлов.
Электродвижущие силы и электродные потенциалы 151
V
Рис. 38. Схема, изображающая вероятность перехода ионов металла в раствор
Нернст .полагал, что в ряду напряжений металлы располагаются по своей способности посылать положительные ионы в раствор; из квантовой механики следует, что электроны у контакта металл — металл определяют порядок металлов в этом ряду.
Перейдем к выводу уравнения для электронного потенциала. Обозначим (рис. 38) через Z— уровень энергии, необходимый для вырывания положительного иона металла из кристаллической решетки в вакуум, W— энергию сольватации иона, Ум — низший колебательный уровень иона в металле, — низший колебательный уровень иона в растворе. Затрата энергии при переходе ионов из металла в раствор будет равна
A = -(Z-W) = Vu-Vp.
Количество ионов в растворе и в металле (пр и пм), обладающих запасом кинетической энергии, не меньшим, чем W = V— Vp или Z => = V—VM, равно, по закону Больцмана:
v-vp lzp-v
пр m k[e RT ь= k\e RT ,
v~vu Vv~v
nM~k# RT =K2eRT . (VI,25)
Экспоненциальные члены представляют собой вероятности перехода ионов из раствора в металл и обратно. Однако при ближайшем рассмотрении оказывается, что скорости перехода определяются не только «глубиной» потенциальных ям, но также поверхностью соприкосновения металла с раствором. На поверхности металла в растворе могут находиться дополнительные молекулы растворителя, «вытягивающие» ионы металла в раствор (рис. 39). Поэтому вероятность переходов ионов из металла в раствор пропорциональна поверхностной концентрации молекул растворителя Мр, т. е.
(VI,26)
152
Теоретические основы электрохимии
Вероятность же перехода ионов из раствора в металл, очевидно, пропорциональна поверхностной концентрации ионов металла, т. е.
k^NKe RT ,
(VI,27)
Пусть вероятность прямого процесса, т. е. перехода ионов из металла в раствор, больше, чем вероятность обратного перехо-
Рис. 39. Ориентация дипольных молекул расгвэрителя
да. Тогда и скорость прямых переходов будет больше скорости обратных, так как скорость процесса пропорциональна его вероятности. Следовательно,
VP-V
k^e RT >k2NMe ™ , (VI,28)
где fej, k2 — коэффициенты пропорциональности.
В этом случае металл получит относительно раствора отрицательный заряд, вследствие чего уровень энергии в левой потенциальной яме понизится на такую величину, что скорости прямого и обратного переходов будут равны.
С другой стороны, понижение уровня потенциальной энергии будет примерно равно zFq>, где <р — скачок потенциала на границе металл — раствор. Следовательно,
ум_у+фгг vp-v kxN^ RT = k2NMe .
Отсюда следует
exp /,Vm~Vp + zF? \ = ____N*
Р \ RT ) kx Np
(VI,29)
Нетрудно заметить, что выражение, стоящее в правой части, пропорционально концентрации ионов или их активности:
А_.-^_=йсм/м. (VI,зо)
kx Np
Тогда
kCMfM = exp
Ум ~ VP + zFV
RT
Ур-Ум
T = ---7--
zF
zF zF
Электродвижущие силы и электродные потенциалы
153
а так как Vp— VM = Z— W, то
т = + —- In k + In С JM, (Vl,31) zF zF zF
где o = Z-W _RT jn k (VI,32) T zF zF
И, наконец, <P = <P° + -^lnCMfM. (VI,33) zF
Таким образом, квантовомеханическая теория указывает на зависимость нормальных потенциалов от энергии сольватации W.
§ 7. Измерение электродвижущих сил
Для измерения электродвижущих сил пользуются компенсационным методом, преимущество которого состоит в том, что он позволяет измерять э. д. с. элемента, когда в цепи отсутствует ток (г = 0). На рис. 40 приведена принципиальная схема компенсационной установки.
1
Рис. 40. Схема установки для определения э. д. с.:
1 — аккумулятор; 2 — исследуемый гальванический элемент; 3 — гальванометр; 4 — нормальный элемент; 5 — телеграфный ключ; 6 — переключатели; 7 — подвижный контакт; ВС — реохорд
Принцип работы ее состоит в том, что при перемещении скользящего контакта по реохорду можно добиться такого положения контакта 7, когда падение напряжения на отрезке ВК будет равно измеряемой э. д. с. Ех. Тогда ток в цепи станет равным нулю, что и будет зафиксировано показанием гальванометра 3. При этом изучаемый элемент должен быть включен
t54 Теоретические основы электрохимии
против аккумулятора 1. В точке компенсации будет соблюдаться равенство
£, == Екк.
Но величина Евк, согласно закону Ома, равна IRbk, где Rbk — сопротивление проволоки на участке В К; I — текущий через нее ток. Так как проволока калибрована, то сопротивление ее будет пропорционально длине, и при прочих равных условиях
Ех = К • BR.
Величина К есть не что иное, как цена деления реохорда в -вольтах. Для ее определения достаточно найти точку компенсации с каким-либо элементом, э. д. с. которого известна. Обычно для определения цены деления реохорда используют нормальный элемент, э. д. с. которого при 20 °C равна 1,0183 в. Очевидно,
1,0183— вк
Вторично компенсацию проводят с изучением элементов х и тогда
Ех = 1,0183-^-. вк
При точных измерениях метровая проволока со скользящим контактом (реохорд) оказывается уже недостаточной и ее заменяют системой магазинов сопротивления, так называемым потенциометром, в котором цена деления реохорда откалибрована в милливольтах. Схема лабораторного потенциометра П-4 приведена «а рис. 41.
В качестве нуль-инструмента при потенциометрических измерениях обычно пользуются стрелочными гальванометрами, чувствительность которых составляет обычно 10~6—10~7 а на одно деление шкалы.
Часто при электрохимических измерениях применяют также зеркальные гальванометры (рис. 42). В зеркальных гальванометрах при прохождении тока вращается зеркальце, отбрасывающее световой зайчик на шкалу с делениями. Зеркальные гальванометры обладают большой чувствительностью (до 10-12 а на одно деление шкалы, что позволяет использовать их при достаточно точных работах.
При измерениях очень малых э. д. с., когда возможна заметная ошибка, часто пользуются способом компенсации с 'Применением дополнительного нормального элемента. Для этого в цепь последовательно с измеряемым элементом и против а'кку-
Электродвижущие силы и электродные потенциалы
155
мулятора включают второй нормальный элемент и замеряют суммарную э. д. с. Затем переключают полюса у излучаемого
элемента и вновь производят замер.
Таким образом, получают два значения
Ei ~ Еш + Ех, Е% — Еш — Ех, откуда следует, что р_________________Е\ — Ег
э. д. с., причем
Рис. 42. Зеркальный гальванометр:
1 — зеркальный гальванометр; 2 — призма; 3 — шкала; 4 — зеркало; Б — пампа для освещения зеркала в гальванометре
Рис. 41. Схема лабораторного потенциометра:
1 — аккумулятор; 2 — нормальный элемент;
8 — искомая э. д. с.; 4 и 5 — реостаты грубой и плавной настройки; 6 — контакт
При этом ни в коем случае не следует переключать полюса дополнительного нормального элемента, так как его э. д. с. гораздо больше, чем Еж, и при таком переключении будет еще труднее добиться компенсации.
В случае, если необходимо измерить отдельные электродные потенциалы, составляют элемент из изучаемого электрода и какого-либо электрода сравнения с известным потенциалом, например водородного (ацетатного), каломельного, хлорсеребряного, ртутноокисного и т. п.
Величину искомого потенциала вычисляют по уравнению
Е = ?а — <рк, причем неизвестный электрод может оказаться либо катодом, либо анодом. В первом случае его потенциал
<Рк = 'Pep — Et во втором
=?ср + £'-
Здесь фср — потенциал электрода сравнения.
156
Теоретические основы электрохимии
§ 8. Полуэлементы (электроды сравнения)
Важное место в практике электрохимических измерений занимают электроды, применяемые в качестве электродов сравнения при определении электродных потенциалов методом измерения э. д. с.
Для измерения электродных потенциалов необходимо иметь электрод сравнения, обладающий постоянным и известным потенциалом. В качестве стан
Р.ис. 43. Водородный электрод:
1 — платиновая пластинка; 2 — стек-лянйая трубка; 3 — ртуть; 4 — стеклянный сосуд; 5 — сифон; 6 н 7 — жидкостные затворы
да точно равна единице, создать довольно трудно, для
дартного электрода сравнения наиболее часто применяют водородный электрод.
Конструкции водородного электрода весьма разнообразны. Наиболее простая из них приведена на рис. 43. Он состоит из платинированной платиновой пластинки, опущенной в раствор, содержащий ионы водорода, и омываемой весьма чистым (электролитическим) водородом. При этом платиновый электрод приобретает свойства водородного, так как потенциал его обратимо изменяется по отношению к изменению концентрации ионов Н+ в растворе.
Так как электрод, в котором активность ионов водоро-измере-
ния практического характера обычно пользуются ацетатным буферным раствором, который, как было показано выше, отличается весьма устойчивым значением pH. Для ацетатного водородного электрода готовят смесь из 50 мл 1-н. NaOH и 100 мл СНзСООН в 500 мл раствора. Таким образом, получают буферную смесь 0,1-н. СНзСООН + 0,1-н. CHsCOONa, pH которой при 25 °C равен 4,627. Аналогично могут быть приготовлены 0,01-н. и 0,001-н. ацетатные растворы, pH которых при 25 °C соответственно равны 4,67 и 4,78.
Как легко показать, потенциал водородного электрода зависит только от pH раствора, если давление водорода постоянно:
? = <р°0,00027 pH.
(VI,34)
Электродвижущие силы и электродные потенциалы
157
Водородный электрод является типичным газовым электродом, так как платинированная платина, насыщенная водородом, проявляет себя как электрод, обратимый относительно Н+-ио-нов.
Помимо классификации относительно водородного электрода и классификации по знаку заряда потенциал определяющего иона, электроды 'подразделяются на электроды первого и второго рода.
Электроды первого рода — это металлические или газовые электроды, погруженные в раствор своих ионов и обратимые по отношению к этим ионам (металла, водорода, хлора ,и т. п.). Примером электродов первого рода могут служить: медный Cu/Cu2+, водородный Pt(H2)/H+, хлорный Pt (С12)/С1_ и др.
В тазовых электродах потенциалопределяющий материал (водород, хлор и др.) не является электронным проводником. Поэтому электрический контакт здесь осуществляется с помощью инертного металла типа платины, иридия, золота, которые служат передатчиками электронов от газа к ионам в растворе или наоборот.
Электроды второго рода — это металлические электроды, покрытые малорастворимой солью этого металла и опущенные в раствор хорошо растворимой соли, имеющей общий анион с малорастворимой солью.
В некоторых случаях металлический электрод может быть и не покрыт труднорастворимой солью, а просто опущен в насыщенный раствор ее в присутствии другой хорошо растворимой соли с общим анионом. Потенциал электрода второго рода, например хлорсеребряного, определяется уравнением
ф = </> (VI,35)
‘Ag YAg+/Ag ' р Ag+’ 7
но активность Ag+-HOHOB определяется из условий постоянства произведения активностей труднорастворимой соли через:
а , • а — const = L. Ag+ ci- р
Поэтому
= ?° ,, + —InA — -—Ina
YAg YAg+/Ag 1 F P F Cl-
ИЛИ
<₽. —^-Ina . (VI,36)
TAg ‘ Ag/AgCl F Cl- ' 7
RT
Подставляя числовые значения для —— и переведя In в 1g,
Г получим
^Ag = C/Agci - 0.0002Т 1g aCI_ (VI,37)
158
Теоретические основы электрохимии
Потенциал хлорсеребряного электрода определяется, таким образом, активностью С1_-ионов. Величина нормального потенциала хлорсеребряного электрода: TAg/Agci ='0,2224 в при
25 °C.
Для определения электродных потенциалов <ра и <рк и падения напряжения в
Рис. 44. Хлорсеребряный электрод:
/ — ртутный контакт; 2 — капиллярная трубка; 3 — электрод, покрытый AgCl
электролите еЭл применяют специальные электролитические ключи с впаянным хлорсеребряным электродом. Для приготовления хлорсеребряного электрода, прокаленную и хорошо очищенную с поверхности серебряную проволочку длиной до 50 мм и диаметром 0,5—2,0 мм свертывают в спираль и . обрабатывают анодно при плотности тока 10—20 Maj см2 в 0,1-н. растворе соляной кислоты в течение 1—2 ч. Затем проволочку тщательно промывают и впаивают в место сочленения колена со стеклянной трубкой (рис. 44). Вместо серебряной спирали можно взять платиновую и покрыть серебром в цианистом электролите при плотности тока 0,008—0,01 а!см2 в течение 8—10 ч. Состав раствора, г/л: 25— 35 AgCN, 25—38 KCN, 30—40 К2СО3. Анодом является чистая серебряная пластинка. Полученный осадок тщательно отмывают и затем анодно хлорируют в 0,75-н. растворе HCI или в плотности тока 0,005 а)см2 в течение
1-н. растворе NaCl при 5 ч.
Потенциал хлорсеребряного электрода определяется активностью ионов хлора, поэтому трубку 2 заливают раствором КС1. Если концентрация хлор-ионов в исследуемом растворе строго постоянна, то в трубку втягивают испытуемый раствор и определяют потенциал хлорсеребряного электрода 3, сравнивая его с каломельным или водородным.
Такой метод использования хлорсеребряного электрода возможен в том случае, если в изучаемом растворе нет ионов Вг~, J-, NH+, CN-, NO- ит. п.
Потенциал хлорсеребряного электрода по отношению к нормальному водородному электроду равен:
'PAg/ABci= 0,2224 - 6,4 - 1СГ4 (/-25)-3,2 . 10~6(/-25)2-
— [0,591 + 2 • 1(Г4 (t — 25)] 1g а в.
Электродвижущие силы и электродные потенциалы
159
Рис. 46. Схема каломельного электрода: 1 — ртуть; 2 —• платиновая проволока; 3 — стеклянная трубка с внешним вводом; 4 —паста из каломели; 5 — ртуть; 6 — пробка; 7 — боковая трубка; 8 — сифон; 9 — раствор хлористого калия
В нерабочем состоянии хлорсеребряный электрод хранят в дистиллированной воде.
Составление электрода сравнения оказалось наилучшим методом оценки работы промышленных ванн, в особенности при выяснении влияния различных факторов на величину и характер изменения катодного и анодного потенциалов и омических потерь в электролите в отдельности. Именно таким путем были определены все известные балансы напряжения ванн (цинковых, кадмиевых, никелевых, медных и др.).
Наиболее распространенный тип электродов второго рода — каломельные электроды:
Hg|Hg2Cl2, КС1, н2о.
Потенциал каломельного электрода (рис. 45), подобно хлорсеребряному, может быть выражен уравнением
?кал = ?кОал -0,00027 lg аа_. (VI,38)
В практике применяют каломельные электроды с различными концентрациями КС1. Наиболее распространены из них следующие: децинормальный (0,1-н. раствор КС1):
<Рк°ал =-- 0,3365 — 6,0 • 10“5(/ — 25 °C);
нормальный (1-н. раствор КС1):
Ткал = 0,2828 — 2,4 • 10~4 (f — 25 °C);
насыщенный (насыщенный раствор КС1):
Ткал = °»2438 — 0,00065 (t — 25 °C).
Более точным из перечисленных электродов является децинормальный каломельный электрод, обладающий наименьшим температурным коэффициентом. Однако каломельный электрод с насыщенным раствором КС1 нашел наибольшее применение в связи с простотой его приготовления и устойчивостью на воздухе, хотя температурный коэффициент самый значительный.
Для измерений в сернокислых растворах удобно пользоваться свинцовым и ртутным сернокислыми электродами второго рода:
РЬ | PbSO41SO4-; Hg I Hg2SO4 I SO4“.
160 Теоретические основы электрохимии
Нормальные потенциалы этих электродов при 25 °C соответственно равны:
<Р°Ь = — 0,3505 в; <pog = -f- 0,6141 в.
Концентрация же применяемых растворов SO^--ионов может быть различной.
Большое распространение нашел также сурьмяной электрод Sb | Sb (ОН)31 КОН; <р°ь = + 0,64 в и др.
Электроды второго рода особенно незаменимы при определении концентрации анионов Cl-, SO|— и др., так как использование с этой целью, например, газового (хлорного) электрода связано с большими осложнениями, а в случае определения концентрации SO^- составить анионный газовый электрод вообще невозможно. Электроды второго рода нашли наибольшее практическое применение как электроды сравнения при любых электрохимических измерениях.
Водородный ацетатный электрод и перечисленные выше электроды второго рода (электроды сравнения) часто называют полуэлементами, так как они дают возможность при сочетании их с неизвестным электродом составить гальванический элемент, в котором потенциал электрода сравнения известен. Таким образом, измерив э. д. с. составленного элемента, можно определить потенциал неизвестного, интересующего нас электрода.
§ 9. Окислительно-восстановительные процессы и их потенциалы
Окислительно-восстановительные процессы чрезвычайно распространены и проявляются обычно в самых разнообразных формах.
Окислением первоначально называли процессы присоединения кислорода, либо реакции, сопровождающиеся отдачей водорода, например превращение этана в этилен:
сн3 сн2 I - II
СН3 СН2 + Н2
или реакция превращения этилового спирта в ацетальдегид:
СН3СН2ОН + -^-О2 -> СН3СОН + Н2О.
Однако известны окислительные процессы, протекающие без участия кислорода или водорода, например, окисление меди трехвалентным железом:
Cu+ + Fe3+ -> Cu2+ + Fe2+.
Электродвижущие силы и электродные потенциалы 161
В общем случае приобретение положительного заряда или потеря отрицательного заряда молекулами, атомами илй ионами вещества есть процесс окисления. Наоборот, приобретение отрицательного или потеря положительного заряда частицами вещества есть процесс восстановления. Таким образом, во всякой электрохимической системе процессы на электродах могут быть рассматриваемы как окислительно-восстановительные, если они сопровождаются перемещением электронов и перезарядкой молекул, атомов или ионов.
Известны разные типы окислительно-восстановительных систем. Простейшие системы состоят из ионов одного и того же металла в двух степенях валентности, например:
Fe3+ + е ? Fe2+.
Другой тип систем состоит из анионов, несущих разные заряды:
МпОГ + е. МпО2-.
В некоторых случаях процесс протекает между анионами и катионами, содержащими один и тот же металл:
МпО? + 8Н+ + 5е г Мп2+ + 4Н2О.
Известны также окислительно-восстановительные системы, где участвует одно или несколько твердых веществ:
РЬО2 + 4Н+ + SO2~ +2е^ PbSO4 -4-- 2Н2О.
Наконец, большое число обратимых окислительно-восстановительных систем можно встретить при рассмотрении органических соединений. Такие равновесия могут быть выражены реакцией
R + 2Н+ + 2е £ Н2Д.
В данном разделе мы рассмотрим обратимые окислительно-восстановительные процессы, идущие в электролите без образования новой фазы.
Если металлический индифферентный электрод опустить в раствор, где присутствуют ионы какого-либо вещества в окисленной и восстановленной формах, то потенциал такого электрода будет целиком определяться соотношением концентраций и природой веществ, находящихся в испытуемом растворе. Соответствующие потенциалы называются окислительно-восстановительными. Они, очевидно, отвечают равновесному состоянию каждой из систем, участвующих в окислительно-восстановительной реакции.
Металлический индифферентный электрод в данном случае выполняет роль передатчика электронов. При увеличении кон-11 Заказ 1338
162 Теоретические основы электрохимии
центрации окисленной формы металл приобретает (некоторый излишек электронов и заряжается отрицательно относительно раствора и наоборот.
Очевидно:
RT < ,, , RT , [Окисленная форма]
<р . = ----In Д’ 4-----In —1-----------——-—
0/в гР р zF [Восстановленная форма] или
То;.-^. + тг1п”- <VI'39>
ивосст
Величина <рв/в является нормальным или стандартным потенциалом. При равенстве активностей окисленной и восстановленной форм окислительно-восстановительный потенциал ср будет равен нормальному потенциалу <р°. Результаты измерения нормальных потенциалов некоторых окислительно-восстановительных систем приведены в табл. 14.
Таблица 14. Нормальные электродные потенциалы <р окислительно-восстановительных систем при 25 °C
Система 0 «’c/b’ « Система 0 ^O/B’ *
Co3+/Co2+ + 1,82 Fe3+/Fe2+ +0,75
Pb4+/Pb2+ + 1,75 MnO^/MnOf- +0,66
Mn4+/Mn3+ +1,64 Fe(CN)|-/Fe(CN)4- +0,49
Mn4+/Mn2+ + 1,58 Ti4+/Ti2+ +0,37
Ce4+/Ce3+ + 1,55 Cu2+/Cu+ +0,16
MnC>7 + 8Н+/Мп2+ + 1,48 Ti4+/Ti3+ +0,06
T13+/T1+ + 1,21 V4+/v3+ —0,20
JO^ + 6H+/i/2 J2 + 1,20 Sn4+/Sn2+ —0,50
Hg2+/Hg2+ +0,90 Cr3+/Cr2+ —0,41
Mo (CNJ^/Mo (CN)g— +0,82
Однако эти данные ненадежны, так как точно не известны величины активностей соответствующих растворов.
Таким образом, при ферри-ферропроцессе потенциал его выразится уравнением
fo/B = <Ро/в
RT In °Fe3+
* °Fe2+
(VI,40)
Электродвижущие силы и электродные потенциалы
163
Для процесса, включающего анионы и катионы,
'Ро/в =
RT 5F
а • а° , МпОТ н+
In-----
°Мп2+ ' ОН,О
(VI,41)
и т. д.
Как следует из выражения (VI, 41), концентрация Н+-ионов играет большую роль в некоторых окислительно-восстановительных процессах.
От изменения pH зависят нормальные электродные потенциалы <р° органических окислительно-восстановительных систем. Поэтому некоторые из них широко применяются при потенциометрическом определении pH. В качестве примера рассмотрим поведение хингидронной окислительно-'восстановительной системы или хингидронного электрода, который нашел большое распространение при определении pH.
Хингидронным электродом называют гладкий платиновый электрод, погруженный в исследуемый раствор, насыщенный хингидроном. Поскольку потенциал такой системы устанавливается даже при минимальной диссоциации молекул хингидрона на ионы, предполагают, что гидрохинон отдает электроду атом водорода, т. е. протон с электроном, который отрывается от одной из групп ОН- — 'ближайшей к поверхности электрода.
Таким образом, вблизи платинового электрода идет уже не просто электронный, но протонно-электронный обмен, приводящий к установлению равновесия:
О ОН
4-2(H+ + <3^
Хинон
Гидрохинон
Гидрохинон — это двухатомный фенол, а хинон — отвечающий ему дикетон. Формулу хингидрона можно представить в виде С6Н4О2 • СбН4('ОН)2. Он в очень малой степени растворим в воде. Если в раствор внести такое количество хингидрона, чтобы образовался насыщенный раствор, то в растворе создаются эквимолекулярные концентрации хинона и гидрохинона. Последний является слабой двухосновной кислотой с константами диссоциации Ki = 10_1° и К2 = 3- 10~12. Диссоциация гидрохинона по уравнению
С6Н4 (ОН)2^± с6н4о!~ + 2Н+
11*
164
Теоретические основы электрохимии
приводит к образованию аниона, одинакового по составу с хиноном. При окислении протекает реакция
СбН4О|“ — 2е->С6Н4О2.
Потенциал хингидронного электрода зависит от концентрации водородных ионов и выражается уравнением
0 , RT , °хин°н+
? = ?хин 4--— 1П--------
zF #гидр
ИЛИ
<Р = ?“>,» + 0,000\Т 1g ---0,00027 pH.
°гндр
При измерениях в испытуемый раствор вносят такое количество хингидрона, чтобы часть его оставалась в осадке (практически одну щепотку). В таком случае вследствие образования эквимолекулярной смеси хинона и гидрохинона потенциал хингидронного электрода зависит только от pH раствора и поэтому его можно выразить так:
<Р - ®°ив — 0,00027 pH. (VI,42)
Здесь <p°HH — нормальный потенциал хингидронного электрода, который в интервале от 0 до 37 °C определяется равенством:
'Рх.ш = 0,7177 — 0,00074/.
Хингидронный электрод очень часто используется при измерениях pH. Простота в работе и хорошая воспроизводимость результатов — основные преимущества этого электрода. Но он может быть использован при измерениях только в кислых растворах. При pH > 7 хингидрон медленно окисляется воздухом и разлагается.
Для определения стандартных окислительно-восстановительных потенциалов можно воспользоваться уравнением типа (VI, 39), если раствор, содержащий окислительно-восстановительную систему, достаточно разбавлен. Поэтому измеряют окислительно-восстановительный потенциал при убывающих равновесных значениях концентраций окисленной и восстановленной форм вещества и затем экстраполируют до нулевых «он-центраций. Для этого строят график, на оси абсцисс которого откладывают концентрации, а на оси ординат — вычисленные по уравнению (VI, 39) значения <р °/в. Отрезок на оси ординат, отсекаемый при экстраполяции полученной кривой до нулевой концентрации, и представляет собой искомую величину
Однако в реальных условиях часто приходится иметь дело с растворами, содержащими не только исследуемые вещества,
Электродвижущие силы и электродные потенциалы
165
но и другие ионы, не участвующие непосредственно в окислительно-восстановительной реакции, но влияющие на величину окислительно-восстановительного потенциала. Такой реальный потенциал окислительно-восстановительного электрода в нормальных условиях называют формальным потенциалом.
Величина формального потенциала в большинстве случаев не поддается расчету и может быть определена лишь опытным путем. Прежде всего эта величина зависит от активности ионов водорода в растворе, т. е. от его pH.
Зависимость потенциала окислительно-восстановительного электрода от pH не означает, что его можно уподобить газообразному водородному электроду. Например, потенциал электрода Pt |Fe2+, Fe3+, 0,1-н. НС1 равен 0,7 в. Если же рассматривать его как газовый электрод, то получим, по уравнению Нернста: <рн = 0,0596 lgCn+ — 0,0298 lg pHi; РНг = 10~27, что лишено физического смысла.
Для определения истинного значения формального потенциала в растворах с различным значением pFI на опыте приходится экстраполировать эту зависимость до pH = 7 (до нулевой концентрации кислоты).
В случае простейших окислительно-восстановительных систем, состоящих из двух ионов (например, Fe2+ и Fe3+, Fe(CN) 36~ и Fe(CN) и др.), потенциал электрода определяется по уравнению
Фо/в = 'Ро/в + — In In ,
о/в <,/Б zF бвосст zF f BOCCT
где f —соответствующие коэффициенты активности.
Если растворы достаточно разбавлены, то, используя известное уравнение Дебая — Гкжкеля при 25 °C
получаем
1g
/восст
0,509 (2ок ^восст) 1^5
следовательно.
ToZB = ^ + ^6ig^-г восст
^^(Zok-Zbocct) Ур. (VI,43)
Последнее слагаемое даже в разбавленных растворах может быть весьма велико. Так, например, при ц = 0,010 для системы Fe(CN)3- ^Fe(CN)^- величина последнего слагаемого достигает 0,030(16—9) ]/0,01 = 0,021 в.
16б Теоретические основы электрохимии
Пользуясь уравнением (VI,43) в простейших случаях, можно приближенно определить <р °/в, измерив значения потенциала при нескольких значениях Сок/СВосст- Зная <р°/в , можно по уравнению <р°/в =- In Лр подсчитать значение константы равновесия реакции.
§ 10. Правило Лютера и окислительно-восстановительные равновесия
Если металл способен образовывать ионы различной валентности, то стандартные потенциалы различных реакций прямого или ступенчатого образования этих ионов по схеме
Me Me' Me*
| ^i/o гге I (VI.44)
’2/0
связаны правилом Лютера:
'Pl/O2! + Т2/1 (г2 21) = ^2/ог2 (VI,45)
(знак * относится к стандартным 'потенциалам).
Правило Лютера является прямым следствием закона сохранения энергии: изменение свободной энергии при любом процессе не зависит от пути процесса и определяется только начальным и конечным состоянием системы.
Когда металл и раствор пришли к равновесию, равновесные потенциалы всех возможных процессов:
рт
Ф1/0 = ?i/o + In [Me ],
71 г
<Р2/О = 'Р2/о + “-7-1П1Л1^ ]
И
2 4-
, . RT . [Me 2
92/1 ?2/1 4 I 4
должны быть между собой равны. Из этого, в частности, следует:
Фцо + ] = Ф;/о + 1п \МеЛ ]
ИЛИ
РУ z'f' 2~j"
-J--[z2ln[Me 1 ] — z1ln[Me 2 1} = — ф7/0.
Электродвижущие силы и электродные потенциалы
167
откуда +
In .4 (VI,46)
Уравнение (VI, 46) свидетельствует, что после установления электрохимического равновесия между металлом и раствором его катионов в растворе всегда должны преобладать те катионы, потенциал образования которых менее положителен. Равновесная концентрация катионов другой валентности уже при разности потенциалов (<р1/о — <р2/о) порядка нескольких десятых вольта становится, как легко подсчитать, исчезающе малой.
Небезынтересно отметить, что хотя формально и в уравнении Лютера, и в приведенном только что выводе, и во всех таблицах стандартных потенциалов потенциалы <р,/о и <р2/о для различных реакций вполне равноправны, однако на практике более положительный из них всегда оказывается фиктивной величиной и не отвечает реальному протеканию соответствующего процесса. Он не поддается непосредственному экспериментальному измерению и находится расчетом по правилу Лютера, исходя из реально замеренного потенциала образования ионов, преобладающей валентности и реально же замеренного потенциала окислительно-восстановительной реакции <р2/1 .
Окислительно-восстановительным равновесием в системе 2+ — (г2—Z,) е ,+
Me ' <- ~ Me 2 (VI,47)
и определяется, как правило, действительное соотношение концентрации ионов обеих валентностей. Объясняется это тем, что в момент образования катионы находятся в очень плотном контакте с поверхностью электрода и легко обмениваются с ней электронами, что создает идеальные условия для установления указанного окислительно-восстановительного равновесия.
Знание нормальных окислительно-восстановительных потенциалов чрезвычайно важно при вычислении изменения свободной энергии и констант равновесия химических реакций. Имея окислительно-восстановительные потенциалы данных систем, можно заранее сказать, в каком направлении пойдет тот или иной процесс в таких системах при заданных условиях (температуре и концентрации).
Известно, что при смешении двух обратимых окислительно-восстановительных систем устанавливается определенное равновесие, которое в значительной степени связано с нормальными потенциалами обеих систем. Например, для реакции между системами ферри и ферро и двух-четырехвалентным оловом равновесие, устанавливающееся при работе цепи
Pt|Fe2+, F3+|Sn2+, Sn4+| Pt,
168
Теоретические основы электрохимии
определяется уравнением
Е о ~ 1п К ~ Ш f
zF ZF \ °Sn2+OFe3+/
и выражается в виде разности соответствующих нормальных потенциалов
РЧ) —— ег- 0 — zqO
‘ Fe3+/Fe2+ ™Sn4+/Sn2+‘
Таким образом, константа равновесия зависит от разности нормальных потенциалов взаимодействующих систем.
Для ряда неорганических окислительно-восстановительных систем нахождение нормального потенциала связано с большими затруднениями. Сложность экспериментального определения нормальных потенциалов объясняется тем, что в приведенном примере с железом и оловом и в ряде других случаев и окисленная и восстановленная формы представляют собой ионы с высокой валентностью. Поэтому, например, при потенциометрическом способе определения отдельных окислительно-восстановительных потенциалов при титровании активности будут сильно изменяться вследствие изменения ионной силы раствора.
В связи с указанным обстоятельством данные для нормальных электродных потенциалов, приведенные в табл. 14, не вполне надежны. Правило Лютера позволяет установить значение некоторых окислительно-восстановительных потенциалов помимо их экспериментального определения.
§ 11. Влияние растворителя на обратимые электродные потенциалы
Зависимость нормальных электродных потенциалов от растворителя представляет собой вполне реальное явление; изменение электродного потенциала при замене растворителя определяется разностью химических энергий сольватации потен-циалопределяющего иона. Хотя в настоящее время эта разность не может быть строго определена на опыте или вычислена, приближенное решение этой задачи вполне возможно при условии применения таких электродов сравнения, которые бы сами только в незначительной степени меняли свой потенциал с переменой растворителя.
Водородный электрод с этой точки зрения оказался непригодным, поэтому измерения потенциалов в неводных растворителях, по предложению В. А. Плескова, проводятся относительно цезиевого или рубидиевого электродов, так как ионы этих металлов обладают большим радиусом и сольватация их незначительна. Кроме того, работа выхода этих ионов из раствора
Электродвижущие силы и электродные потенциалы
169
в разных электролитах отличается очень мало. С помощью таких электродов возможно приближенное сравнение рядов напряжений в разных растворителях.
В табл. 15 приведены значения потенциалов различных металлов в метиловом и этиловом спиртах, а также в жидком аммиаке, гидразине, безводной муравьиной кислоте, ацетонитриле и воде.
Из табл. 15 следует, что растворитель сильно влияет на ве-
Таблица 15. Потенциалы электродов в различных растворителях при 25 °C
Растворитель Потенциалы электродов, в
3 Z Z С N 04 С К +х п О 04 3 О Ьй Д 04 ъд Д ьа ьл 6 6
СН3ОН * С2Н5ОН * NH3** N2HZ* СНООН** CH3CN** Н2О —3,09 —3,04 —0,31 —0,19 —0,03 —0,06 —0,09 —2,73 —2,66 —0,08 —0,18 —0,03 —0,30 —0,22 —0,74 —0,64 1,40 1,60 2,4 2,43 2,17 0,00 0,00 1,93 2,01 3,45 3,17 2,93 0,34 0,21 2,36 3,31 2,89 3,28 0,74 0,76 2,68 3,42 3,79 0,76 0,75 2,76 2,78 3,62 3,40 3,74 1,12 1,05 3,96
Примечание. Значками * и ** помечены растворители, потенциалы электродов в которых вычислены соответственно по водородной или по рубидиевой шкале.
личину электродных потенциалов. Потенциалы в метаноле и этаноле такие же, как в воде. Это можно объяснить близостью свойств воды и спиртов. В аммиаке и гидразине, обладающих сильнощелочными свойствами, потенциалы галоидов и щелочных металлов меняются сравнительно слабо. Наоборот, потенциалы Н2, Си, Ag, Zn, Cd резко сдвинуты в отрицательную сторону относительно их значений в водных растворах. В муравьиной кислоте, типично кислом растворе, потенциалы перечисленных металлов, наоборот, сдвинуты в положительную сторону.
Ацетонитрил является растворителем с нерезко выраженным основным характером и значительной склонностью к комплексообразованию с ионами тяжелых металлов. Этим объясняется сдвиг потенциалов серебра и меди в этом растворителе в отрицательную сторону; по отношению к другим металлам он ведет себя как растворитель с кислыми свойствами. Например, электродные потенциалы щелочных металлов в ацетонитриле, как и в других растворителях (жидком аммиаке, гидразине, муравьиной кислоте), по сравнению с водой практически неизменны.
170
Теоретические основы электрохимии
Чтобы выяснить характер влияния растворителя на электродный потенциал, рассмотрим металл, находящийся в равновесии с растворам, содержащим его ионы (рис. 46). Условием равновесия будет равенство
=
(VI,48)
гДе i<?e—скачок потенциала на границе металл — раствор; р‘”, p'J — химические потенциалы ионов в растворе и в ме-
талле.
Рис. 46. Зависимость электродного потенциала от энергии сольватации
Таким образом,
Последние же величины связаны с энергиями сольватации
ионов. Чтобы . выяснить эту связь, представим себе, что над раствором имеется вакуум и одновалентный ион переносится из находящегося в вакууме металла в раствор.
Работа такого перехода, с одной стороны, равна сумме работ выхода иона из металла и энергии сольватации иона W + Нр и, с другой стороны, может быть представлена в виде e,V = е,<р —+ е®°, 1 е 1“е “1 1 ч’
где <р ° — скачок потенциала на границе металл — вакуум;
<р° — скачок потенциала на границе раствор — вакуум.
Величина Яр + е<рО обычно называется «химической» энергией сольватации в отличие от «реальной» энергии сольватации Яр. Стало быть,
о , W Н: Л = ?? + ------------;
(VI,49)
Здесь ясно видно, что электродный потенциал зависит от энергии сольватации W, а значит от природы растворителя. От природы растворителя, очевидно, должны зависеть также и электродвижущие силы.
Чтобы найти эту зависимость, рассмотрим э. д. с. цепи, состоящей из двух пластинок одного и того же металла, опущенных в раствор одной и той же соли одинаковой активности, но
Электродвижущие силы и электродные потенциалы 171
в разных растворителях. Очевидно, что при этом
£ = <₽? — = (VI,50)
Y| '2 ZF Кг
Здесь К\ w Кг— константы . равновесия электродных реакций в различных растворителях.
Эти величины, как показал Л. В. Писаржевский, не будут •одинаковы в различных растворителях и поэтому разность — <р£ будет отличаться от нуля.
Ёсли электродная реакция может быть описана уравнением А<*В,
то
&=- —, (VI,51)
аА
где а — соответствующие активности.
Приведем в соприкосновение оба раствора. Тогда вещества Л и В распределятся между ними таким образом, что
Сд S» Сп Sp
= = (VI,52)
аАг SAs аВг SB2
тле S растворимость вещества А и В в обоих растворителях. По уравнениям (VI, 51) и (VI, 52)
(VI,53)
sa. $Вг
«откуда следует, что
?0_-?0 = -^1п zF SB, SB 5д, ’ (VI,54)
Это соотношение проверялось А. И. Бродским и А. С. Афа-
насьевым для цепей
Hg | Hg2Cl2 КВг | KC1, Hg2Cl21 Hg, Ag | AgBr, KBr | KC1, AgCl | Ag в спиртовых растворах.
Тогда уравнение (VI, 49) примет вид:
?о _ ?о _BL. 1п «ксц . ^квг^ . (VI,55)
^КСЦ SKBr,
Опыт подтверждает справедливость этого соотношения. Однако можно сказать, что оно применимо не всегда, а лишь в тех случаях, когда с изменением растворителя не меняется сама природа электродных процессов.
172
Теоретические основы электрохимии
ЛИТЕРАТУРА
Б а г о ц к и й В. С. Достижения в области химических источников тока. Химическая наука и Промышленность, 1958, № 4, стр. 448.
Бродский А. И. Физическая химия, т. 2. Госхимиздат, 1948.
Герни Р. Ионы в растворах. ГОНТИ, 1938.
Гринберг А. А. Введение в химию комплексных соединений. Госхимиздат, 1951.
Давтян О. К. Проблема непосредственного превращения химической анергии топлива в электрическую. Госхимиздат, 1947.
Карашетьянц М. X. Химическая термодинамика. Госхимиздат, 1949.
Карпачев С. В. Проблемы электродвижущей силы гальванического элемента в работах советских электрохимиков. Труды Уральского филиала АН СССР, вып. 4, 7, Свердловск, 1951.
Латимер В. М. Окислительные состояния элементов и их потенциалы в водных растворах. ИЛ, 1954.
Левин А. И., Помосов А. В., Укше Е. А. Руководство к лабораторным занятиям по теоретической электрохимии, ч. I. Изд. УПИ им. С. М. Кирова, 1957.
Ляликов Ю. С. Физико-химические методы анализа. Госхимиздат, 1960.
Михаэлис Л. Окислительно-восстановительные потенциалы и их физиологическое значение. ИЛ, 1938.
Некоторые проблемы современной электрохимии (под ред. Дж. Бокриса), гл. III. ИЛ, 1958.
Плесков В. А. Труды совещания по электрохимии. Изд. АН СССР, 1953.
Рейтер В. Л. и Юза В. А. Сборник «XX лет Института физической химии им. Л. В. Писаржевского». Изд. АН СССР, 1950.
Свен Т. Электрохимические методы получения органических соединений. ИЛ, 1951.
Стромберг А. .Г., Захаров М. С., Аткни.н .Г. Ф. и др. Сборник примеров и задач по электрохимии, гл. IV, изд. Томского университета, 1962.
Темкин М. И. Изв. АН СССР, серия химич., 1946, 235.
Фрумкин А. Н. Успехи химии, 1946, № 15, стр. 385.
Харнед Г., Оуэн Б. Физическая химия растворов электролитов. ИЛ, 1952.
Царев Б. М. Контактная разность потенциалов, Техтеоретиздат, 1955.
Эршлер Б. В. Труды совещания по электрохимии. Изд. АН СССР, 1953.
Глава VII
КОНЦЕНТРАЦИОННЫЕ ЭЛЕМЕНТЫ.
ПОТЕНЦИОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА
§ 1. Концентрационные элементы с переносом
Механизм возникновения и действия концентрационных элементов (цепей) давно привлекает к себе внимание ввиду их большого прикладного и теоретического значения. Типичными являются два вида концентрационных элементов: а) цепи с переносом ионов (с жидкой границей) и б) цепи без переноса (при отсутствии жидкой границы).
Примером элементов с переносом могут служить цепи с двумя одинаковыми электродами, погруженными в растворы одного и того же электролита, но с разной концентрацией.
В таких элементах источником возникновения электродвижущей силы является изменение концентрации ионов в различных зонах раствора. Здесь в соответствии со вторым законом термодинамики должно произойти самопроизвольное выравнивание концентраций за счет диффузии вещества из более концентрированного раствора в менее концентрированный.
Так, в системе
Cu+1 CuSO41 CiiSO, Cu; а± > а2-
SPi tZi <РД a2 <?2
Медный электрод, погруженный в разбавленный раствор ионов Си2+ («2), приобретает при этом потенциал, менее положительный по отношению к раствору, чем электрод, погруженный в более концентрированный раствор (<Zi).
Потенциалы электродов в этом случае соответственно равны:
„ , 0,00027" , 0 0,00027" <
+----2---' gG'; ?2 = +----2--- g 2’
Так как постоянные <Рси равны между собой, то э. д. с. такой цепи (без учета диффузионного потенциала) будет определяться уравнением
р 0,00027" ci ,,,тт ,,
£ = —?2=-----------lg—- (VII,1)
Z
174
Теоретические основы электрохимии
Положительным полюсом составленного таким образом концентрационного элемента с переносом будет электрод, погруженный в более концентрированный раствор.
Этот элемент будет работать до тех пор, пока не выравняются концентрации (активности) обоих растворов.
Концентрации будут выравниваться вследствие диффузии ионов через границу раздела между растворами, а также в результате электрохимических процессов, происходящих на электродах, в результате которых электрод, погруженный в разбавленный раствор, будет растворяться, а на электроде, погруз женном в концентрированный раствор, будет выделяться медь из раствора.
Цепи с переносом применяются при решении ряда теоретических и практических задач, например в аналитической химии (потенциометрический анализ), при определении заурядностей ионов, измерении произведений растворимости, изучении реакций комплексообразования и т. и. Частично возможные области применения таких цепей будут рассмотрены далее.
, Кроме того, равенством (VII, 1) можно пользоваться для определения неизвестной концентрации (или активности) ионов а2, если известны а\ и Е (э. д. с. цепи). Таким путем можно определить растворимость труднорастворимых солей типа BaSO4> AgCl и т. п.
Так, в элементе
Ag| 0,1-н. AgNO3|AgCl, <асыщ |Ag,
в котором водная суспензия AgCl соединена с 0,1-н. AgNO3 в концентрационную цепь, концентрацию С2 насыщенного раствора хлористого серебра можно считать равной активности ионов серебра, так как в очень разбавленных растворах f± = 1. Таким образом, измерив э. д. с. приведенной цепи, можно определить растворимость AgCl.
При определении э. д. с. концентрационных цепей представляется возможным вычислить концентрацию ионов металла, которые имеются в растворах комплексных солей. Известно, например, что AgCN дает с KCN растворимую в воде комплексную соль KAg(CN)2, которая диссоциирует по схеме:
KAg (CN)2 К+ + Ag (CN)7, Ag(CN)r-Ag+ + 2CN-
Бодлендер и Эберлейн нашли, что обменные процессы с. растворителем и комплексообразователем, приводящие к возникновению некоторого количества простых ионов, могут быть обнаружены из э. д. с. концентрационной цепи:
Agjl-н. Ag+|0,05-H. KAg(CN)2 в 0,95-н. KCN|Ag.
Концентрационные элементы. Потенциометрический метод анализа 175
Оказалось, что при 18 °C величина Е исследуемой цепи равна 1,327 в, поэтому концентрация не связанных в комплекс ионов серебра представляется исчезающе малой величиной, равной 1 • 10-23 г-ион]л. В таком случае время существования свободных ионов серебра не превышает 10-20 сек.
Однако э. д. с. концентрационных элементов не всегда практически равна Е = <р( — <рг, так как в большинстве случаев здесь добавляется еще скачок потенциала, возникающий на границе между двумя растворами (диффузионный потенциал <рд).
§ 2. Диффузионный потенциал
Этим термином определяют скачки потенциала, возникающие на границе двух растворов. Они зависят от разности концентраций растворов и от подвижности ионов. Чтобы уяснить эту зависимость, рассмотрим систему
(I)HC11НС1 (II); сц > а2.
а1 ?Д а2
Вследствие разности концентраций ионы Н+ и С1~ будут перемещаться в этом случае от первого раствора ко второму. Но ионы Н+ и С1~ двигаются с различными скоростями (подвижность ионов Н+ примерно в 5 раз больше подвижности ионов С1_). Поэтому за единицу времени через границу соприкосновения растворов пройдет больше Н+-ионо>в, чем С1_-ионов. Это ведет к заряжению одного раствора относительно другого, т. е. к возникновению двойного электрического слоя на жидкостной границе и появлению скачка потенциала.
Пропустим один фарадей электричества через такую границу, для чего придется проделать работу FcpR. Эта работа будет осуществлена за счет перехода пк грамм-ионов Н+-ионов из рас-.твора с большей концентрацией (I) в раствор с меньшей концентрацией (II), что при обратимом процессе выразится через
А = пк/?Г1п-^-. (VII,2)
а2
Ионы хлора в, количестве (1 — /гк) грамм-ионов остаются в более слабом растворе, что требует затраты работы
Лп = —(1—пк)/?Пп-^. (VII,3)
а2
Таким образом, общая работа
Л = F<?a nKRT In (1 — nK) RT In = (2/гк — 1) RT In .
Cbi ^2 ^2
176
Теоретические основы электрохимии
Так как
пк =
/к
/к 4* 4
то очевидно, что
/а ах
1к la F а2
(VII,4)
где /к и /а — электропроводности катиона и аниона.
В става
I
I
I
I
случае, когда электролит содержит новы различного со-
и валентности,
выражение для диффузионного потенциала получается более сложным.
В общем случае для получения | уравнения диффузионного потенци-
। ала рассмотрим границу между
। двумя растворами, содержащими
I , ионы различных сортов с активно-СТЯМИ itZi, 1<Z2, 1<2з, \С1п И 2<71, 2^2, 2а3> । 2Оп (рис. 47). Вдоль границы между
। соприкасающимися растворами об-
। разуется некоторый переходный
I слой, в котором происходит измене-
' ние активностей каждого иона от
Рис. 47. Схематическое изображение границы между двумя растворами
i<Zj до 2^г- Представим себе в этом пограничном слое бесконечно тонкий слой, в котором активность /-того иона меняется на dait и пропу-
стим через систему один фарадей электричества. Очевидно, что при этом через тончайший слой пройдут t-тые ионы в количестве
г/
Здесь щ — число переноса;
Zi — зарядность г-того- иона.
Такой переход ионов обязательно приведет к изменению термодинамического потенциала:
i i
<*Ф = S = S
• I I
где dfii — изменение химического потенциала ионов i-того сорта, равное RTd In Of.
Таким образом,
Концентрационные элементы. Потенциометрический метод анализа 177
но дифференциал термодинамического потенциала равен дифференциалу максимальной работы с противоположным знаком (при постоянных температуре и давлении), т. е.
dA = — RTV^-dlna,. zi
1
(VII 5)
Кроме того,
dA — Fd^>a
и тогда
d<fi
F г.
или
2 i
1 1
(VII,6)
Это уравнение является основным в теории диффузионного потенциала. Оно может быть переписано в виде:
2
RT
(VI 1,7)
i
В ряде частных случаев последнее уравнение упрощается. Так, если рассматривать границу между растворами, имеющими общий катион: MeAtf МеА2, at = а2, тогда
RT Щ ®_ =-----ш-------,
* Д р t •• 1
F 1к + 1а что может быть выражено уравнением RT In ?д F Х2 ’ где Х| и 7.2 — эквивалентные электропроводности.
Последнее уравнение для диффузионного потенциала было предложено Льюисом и Серджентом и подтверждено экспериментально.
В частном случае (бинарный электролит) получается уравнением типа (VII, 4), т. е.
RT
Фд с-
" zF
(VI 1,8)
1к — la С1
1к + 1а а2
(VII,9)
12 Заказ 1338
178
Теоретические основы электрохимии
По абсолютной своей величине диффузионные потенциалы, вообще говоря, относительно невелики. Рассмотрим, например, цепь
Ag|AgNO3|AgNO3|Ag.
0,1-н. 0,01-н.
По уравнению (VII,4) диффузионный потенциал такой цепи равен
RT
’Рд р
Рис. 48. Электролитический ключ
in О.РЬ 0,938 = 0>003б в 54,0 4-61,6 0,1-0,815
Здесь f ^gNOj = 0,938;
f" " = 0,815; I. . = 54,0; l_ = 61,1.
Опытное значение <рд = 0,0035 в, тогда как общая э. д. с. цепи оказалась равной 0,0590 в. Таким образом, диффузионный потенциал составляет лишь 5,9% от общей э. д. с. Однако из уравнения (VII.4) следует, что диффузионный потенциал становится тем большим, чем значительнее разница концентраций двух растворов и чем больше отличаются одна от другой подвижности ионов. Если /к = 1а, ТО, очевидно, <рд = 0. Наоборот, на границах растворов кислот и щелочей диффузионный потен
циал достигает весьма заметной величины благодаря аномальной подвижности иоиов Н+ и ОН".
При точных измерениях диффузионный потенциал должен быть выражен количественно и учтен в выражении для э. д. с. гальванических цепей или же максимально уменьшен (элиминирован). Для устранения диффузионного потенциала применяют промежуточные сосуды и солевые мостики (электролитические ключи), заполненные раствором электролита, состоящим ИЗ ИОНОВ С близкими ПОДВИЖНОСТЯМИ (КС1, KNO3, NH4NO3), которые соединяют оба полуэлемента (рис. 48).
Чтобы воспрепятствовать возможной диффузии испытуемого раствора в раствор полуэлемента (и обратно), растворы, заполняющие солевые мостики и промежуточные сосуды, берут насыщенными.
Рассмотрим пример, иллюстрирующий действие такого солевого мостика. Составим цепь
Hg| 1-н. LiCl
0,1-Н. LiCl |Hg насыщен Hg2Cl2 насыщен Hg2Cl2
Здесь X— промежуточный насыщенный солевой раствор.
Концентрационные элементы. Потенциометрический метод анализа 179
Электродвижущая сила этой цепи составляет 53 мв, однако непосредственное измерение разности потенциалов при различных X дает следующие результаты:
Промежуточный солевой раствор
NH4NO3 _
NH4NO3
KNO3
КС1
Э. д. с., мв
48,0
54,3
50,2
51,5
Как видно, этот прием значительно снижает величину диффузионного потенциала в цепях с Переносом, однако не устраняет его полностью.
§ 3. Концентрационные элементы без переноса ионов
В отличие от концентрационных цепей с переносом в концентрационных элементах без переноса концентрации веществ раз-
личаются не в растворах, а в электродах.
К таким цепям относятся, в частности, эле менты, составленные из двух металлов или сплавов, опущенных в один и тот же раствор и состоя
Cf fy
Рис. 49. Схема концентрационного элемента без лереноса ионов
щих из одного и того же электродного материала, но различающихся по концентрации в них активного вещества (амаль-
гамные и газовые электроды). Могут быть отличия и по другим физическим свойствам электродов.
Рассмотрим два амальгамных цинковых электрода в растворе одной и той же соли (рис. 49), например ZnSO4:
Zn(Hg) [ ZnSO4 I Zn(Hg)
G . C2 ’
Ci > Cz-
Чем больше концентрация цинка в амальгаме, тем легче он будет растворяться и посылать освобождающиеся электроны во внешнюю цепь. Таким образом, отрицательным полюсом такого элемента является электрод с большей концентрацией активного вещества — цинка, положительным — с меньшей концентрацией цинка.
12*
180
Теоретические основы электрохимии
Э. д. с. такой цепи, следовательно, зависит от G и С2 и в рассмотренном случае будет выражаться уравнением
Е = (VII,10)
2F С2 ' '
Концентрационный элемент без переноса ионов (рис. 50) может быть составлен из двух одинаковых газовых электродов:
Рис. 50. Концентрационная цепь, составленная из !В1одородных электродов
различающихся парциальным давлением газа (например, водорода). В этом случае, если, например, pa>Pi, то возникает электродвижущаяся сила:
Е = <{=!-— <р2 =---------— In----------—
г Р
( RT In
\ F Р /
которая в конечном итоге может быть выражена равенством
2Д Р1
В том случае, когда можно применить электроды, обратимые как относительно катиона, так и относительно аниона данного электролита, возможно специальное изготовление химических
Концентрационные элементы. Потенциометрический метод анализа 181
элементов без переноса ионов. Составим цепь Pt(H2)|HCl, AgCl„JAg, где концентрация НС1 раина m молей на 1000 г раствора.
Э. д. с. этой цепи
Е = ?н+/н2 — ?Ag/AgCl + 1п С1Л> где fi — средний коэффициент активности НС1.
Соединив два таких элемента с разными концентрациями, получим цепь
Ag(AgCl)| НС1 | (H2)Pt | НС1 I AgCl | Ag.
C2
Если Ci < C2, то в левой части будет протекать реакция 1/2Н2 + Ag+ + СГ -> Ht + С1Г + Ag+, ведущая к росту концентрации НС1. В правой части, согласно схеме
Hit + С1Й + Ag+ - i/2H2 + Ag+ + СГ, будет наблюдаться убыль концентрации НО. В конечном итоге процесс сводится к перемещению НО справа налево.
Э. д. с. всей цепи
Аобщ = — Е2
или
Д' 1 гч2г2 1 /-'З с2
Еобщ — —— ’пС]/]-----— 1пС2/2
F г
или, наконец,
Описанная только что цепь также называется концентрационной цепью без переноса ионов, так как ее э. д. с., хотя и зависит от отношения концентраций обоих растворов, по работа цепи не сопровождается непосредственным переносом ионов из одного раствора в другой. Перенос здесь осуществляется косвенным путем в результате химических реакций.
С помощью такого рода цепей производятся наиболее точные измерения стандартных (нормальных) потенциалов. Отсутствие переноса ионов в химических элементах без жидкостного соединения позволяет применять их также для нахождения чисел переноса ионов и для определения диффузионных потенциалов. Так, например, для экспериментального опреде
182
Теоретические основы электрохимии
ления диффузионного потенциала на границе двух растворов НС1 разных концентраций измеряют э. д. с. следующих двух элементов:
Pt(H2) ’ НС1 || НС1 I (H2)Pt
Сх С2
Hg | Hg2Cl2,'HCl II НС1, Hg2Cl2 I Hg.
Ci c2
Э. д. с. первого из этих элементов может быть выражена как
= In.(VI 1,11)
Г С>2 а э. д. с. второго как
£"=-^1п-^- + <рд. (VII,12)
Диффузионный потенциал в случае соприкосновения двух одинаковых растворов различной концентрации
? in . (VII,13)
А /к + la F Сг
Кроме того,
(VII,14)
Для измерения чисел переноса, помимо Е' и Е", определяют э. д. с. элемента Pt (Н2) | НС11 Hg2Cl2| Hg, не содержащего жидкостных соединений.
Из уравнений (VII,11), (VII,12) и (VII,13) следует, что
= 2/а
1к + /а
___ 2/к
1к + 1а
In — = 2naIn—
F С2 F С2
поэтому в сочетании с уравнением (VII,13) будем иметь:
Е"
Е ’
(VII,15)
Полученные значения чисел переноса представляют собой средние для интервала концентраций от Ci до С2.
Концентрационные элементы без переноса незаменимы во всех случаях, когда в потенциометрических измерениях необходимо устранить ошибки, вносимые в измерение э. д. с. диффузионным потенциалом. Большое применение такие элементы нашли также и в технике. Главная область использования элементов без переноса ионов — производство химических источ-
Концентрационные элементы. Потенциометрический метод анализа 183
ликов тока. Для этой цели преимущественно используют щелочные и свинцовые аккумуляторы, а также цинк-двуокисномар-ганцевые, окиономедные, цинк-угольные, магний-серебряные и другие гальванические элементы, которые работают с одним раствором электролита, т. е. при отсутствии диффузионных потенциалов.
Очень широко распространенный процесс самопроизвольного растворения металлов в растворах электролитов (коррозия металлов) также связан с действием концентрационных цепей без переноса.
§ 4. Потенциометрический анализ
Измерение э. д. с. концентрационных цепей используется в потенциометрическом методе анализа.
И обычное, и потенциометрическое титрование основано на том, что вблизи так называемой эквивалентной точки (когда количество прибавленного реагента почти эквивалентно количеству титруемого вещества) концентрация титруемого вещества резко уменьшается, а логарифм концентрации прибавляемого реагента быстро растет.
Недостатком обычного метода титрования является то, что для некоторых реакций не удается найти подходящего индикатора. Кроме того, в окрашенных и загрязненных растворах применение индикатора становится затруднительным.
Основная идея потенциометрического метода титрования заключается в том, что изменение цвета индикатора здесь заменено изменением потенциала какого-либо электрода, обратимого относительно ионов титруемого вещества. Такой электрод называют индикаторным электродом. Потенциал индикаторного электрода вблизи эквивалентной точки, как и в течение всего времени пребывания его в растворе/ является логарифмической функцией активности, и резкое изменение потенциала наблюдается потому, что логарифм активности действующих в реакции ионов в точке эквивалентности резко изменяется от одной-двух капель приливаемого реагента. Методы потенциометрического титрования применимы в случаях реакций осаждения, комплексообразования и окислительно-восстановительных реакций.
Для использования той или иной реакции при потенциометрическом анализе необходимо, чтобы:
а) реакция 'протекала с достаточно большой скоростью и равновесие устанавливалось быстро;
б) отсутствовали побочные процессы;
в) реакция протекала стехиометрически;
г) была возможность подобрать соответствующий индикаторный электрод.
184
Теоретические основы электрохимии
Преимущества потенциометрического метода титрования по сравнению с химическим сводятся к возможности титрования окрашенных и содержащих осадок растворов и к возможности одновременного определения нескольких компонентов.
Невозможность полного устранения диффузионного потенциала фд и отсутствие точных данных о средних коэффициентах активности ионов f± не позволяют абсолютно точно определить концентрацию ионов в растворе по изменению э. д. с.
Рис. 51. Схема установки для /потенциюмет.р'И’ческого титрования:
1 — каломельный электрод; 2 — соляной мостик; 3 — водородный электрод; 4 — мешалка; 5 — бюретка
цепей с жидкостными границами. Но при аналитических определениях нет необходимости знать абсолютные значения концентрации ионов в титруемом растворе в процессе анализа.
В простейшей форме экспериментальное определение концентрации электролитов с помощью потенциометрического титрования сводится к следующему (рис. 51).
Известный объем испытуемого раствора, содержащего определенные ионы, приводят в соприкосновение с индикаторным (например, водородным) электродом и с помощью солевого мостика соединяют с электродом сравнения (каломельным, хлор-серебряным, водородным, ацетатным и т. п.). При титровании резко изменяется потенциал индикаторного электрода и соответствующее ему количество добавленного реагента. Пусть, на
Концентрационные элементы. Потенциометрический метод анализа 185
пример, титруется NaCl раствором азотнокислого серебра (рис. 52). Индикатором служит серебряный электрод. Как видно из рисунка, потенциал серебряного электрода изменяется вначале очень медленно, но вблизи эквивалентной точки х происходит резкий скачок.
Значение эквивалентной точки находят по максимуму величины Д<р и ДИ—изменение потенциала и разность объ
емов реагента, добавленных к титруемому раствору.
Максимум величины может быть определен непосредственно без графических построений, если воспользоваться методом дифференциального титрования.
Этот метод состоит в том, что часть раствора помещают в капиллярную трубку объемом около 0,03 см3. Она погружена в испытуемый раствор, так что жидкость в капилляре соприкасается с платиновым электродом, служащим
Рис. 52. Кривая 1потенциометрического титрования
во время титрования элект-
родом сравнения. Другой электрод (капиллярная трубка, обмо-
танная платиновой проволокой) служит индикаторным электродом. Так как диффузия в капилляре происходит медленно, состав раствора внутри трубки практически не меняется во время титрования. Таким образом, электрод, изолированный от основ-
ного раствора с помощью стеклянного капилляра, сохраняет свой потенциал неизменным. После каждого измерения капиллярный электрод поднимают и раствор перемешивают до исчезновения разности потенциалов. Затем добавляют новую порцию реагента. В точке эквивалентности разность потенциалов между электродами достигает максимального значения. Прибор для дифференциального титрования изображен на рис. 53.
Метод .дифференциального титрования характеризуется быстротой выполнения и простотой аппаратуры. При этом нет необходимости в изготовлении специального электрода сравнения (полуэлемента). Точность дифференциального метода достигает 0,002%.
Возможность отказаться от применения специальных электродов сравнения дает также метод титрования с биметалличе-
186
Теоретические основы электрохимии
сними электродами. Сущность этого метода состоит в том, что один из применяемых электродов не реагирует на изменение состава раствора в эквивалентной точке и, следовательно, служит своеобразным электродом сравнения — это так называемый инертный электрод. Второй электрод — индикаторный, потенциал которого сильно меняется в зависимости от состава электролита. Схема установки для некомпенсационного титро-
Р.ис. 54. Схема установки для енекоштенсацион-н ого ти тр о в а ни я:
1 — аккумулятор: 2 — гальванометр: 3 — платиновый электрод; 4 — вольфрамовый электрод; 5 — мешалка;
6 — бюретка; 7 — ключ
p.ис. 53. Схема прибора для дифференциального титрования:
1 — стеклянная трубка; 2 — электрод сравнения; 3 — платиновая проволока — индикаторный электрод
вания приведена на рис. 54. Сухой элемент или аккумулятор 1 включают последовательно с высокоомным (JR = 20—80 тыс. ом) сопротивлением для того, чтобы увеличить отклонение стрелки гальванометра, происходящее вследствие резкого изменения потенциала индикаторного электрода, в то время как потенциал инертного электрода почти не меняется. Скачок стрелки гальванометра отвечает эквивалентной точке титрования.
Недостаток некомпенсационного метода — трудность подбора подходящих электродов, а также отсутствие явных указаний на приближение к эквивалентной точке -в процессе титрования, за тем исключением, что в начале титрования стрелка гальванометра остается неподвижной, тогда как вблизи эквивалентной точки она несколько отклоняется от нуля.
Метод нёкомпенсационного титрования вследствие затруднений, связанных с выбором инертного электрода, применяется лишь в ограниченном числе случаев.
Концентрационные элементы. Потенциометрический метод анализа 187
§ 5. Индикаторные электроды
При обычном и при видоизмененных способах потенциометрического титрования большое значение имеет правильный подбор индикаторных электродов. Если индикаторный электрод обратим по отношению к ионам титруемого или титрующего вещества, то изменение потенциала такого электрода однозначно указывает на изменение концентрации ионов в испытуемом растворе.
В качестве индикаторных могут применяться самые различные электроды. Так, для реакций осаждения и комплексообразования наилучшими являются, металлические электроды первого и второго рода; для окислительно-восстановительных процессов— инертные электроды (платиновые, вольфрамовые, графитовые); при потенциометрическом титровании галогенидов азотнокислым серебром пользуются серебряным индикаторным электродом и т. д.
При потенциометрическом титровании кислот и оснований в качестве индикаторного электрода может быть взят водородный электрод особой формы. Устройство такого электрода видно из рис. 50. Во время работы следят за тем, чтобы платиновая пластинка примерно наполовину была погружена в раствор и чтобы внутрь стеклянного колокола электрода непрерывно поступал водород. В качестве второго электрода сравнения обычно пользуются насыщенным каломельным электродом. Таким образом, составляется цепь
Н2 (Pt • Pt) | испытуемый раствор | КС1 I Hg2Cl2, КС1 | Hg, насыщ. насыщ.
Э. Д. С. которой Е = фкал —<рн+/н2.
Водородный индикаторный электрод применяется как эталон в лабораторных исследованиях pH от 0 до 14. Однако показания водородного электрода неверны в присутствии окислителей (солей азотной, хромовой, марганцевой, хлорноватой кислот, солей окисного железа и т. п.), а также восстановителей (сернистого ангидрида, сульфидов и др.). Кроме того, водородный электрод отравляется, когда раствор содержит поверхностно активные органические вещества. Применение водородного индикаторного электрода сопряжено с неудобствами из-за необходимости насыщения его чистым сухим водородом. Потенциал водородного электрода устанавливается по времени не сразу. Поэтому большое применение нашли металлоокисные электроды.
Если окись металла мало растворима в воде, то такой раствор насыщен относительно гидроокиси, и так как
/Ие(ОН)2^/Ие2+ + 2ОН“
188
Теоретические основы электрохимии
[Ме^] [ОН"]2-Lp, то
[М/+] - —i [н+]2 = К, [Н+]2. (VII,16)
[он-12
Потенциал такого электрода
? = ?° + 0,029 1g аМе 2+
или, по уравнению (VII, 16),
<Р = <?° + 0,029 lg КгС2.
ГТ и
+0,0581gC+. (VII, 17)
1 н
Таким образом, окисный электрод «обратим» относительно Н+-ИОНОВ.
Из таких электродов нашли применение окиснортутный и окисносеребряный, которые могут употребляться для измерения pH в сильнощелочных растворах (pH = 9,0). Однако их употребление ограничено из-за возможного протекания процессов комплексообразования, например:
HgO + 4СГ + Н2О HgCl!- 4- 2ОН~.
Кроме того, даже при малой концентрации ионов водорода ионы двухвалентной ртути взаимодействуют с металлической ртутью с образованием черной закиси ртути. Более распространен сурьмяноокисный индикаторный электрод.
Сурьмяноокисный электрод представляет собой электрод второго рода. Так как окись сурьмы плохо растворима, то в растворе устанавливается равновесие типа
Sb2O3 + 6H+^2Sb3+ + ЗН2О.
Константа равновесия
гак как активности Sb2O3 и Н2О постоянны.
Из сказанного потенциал сурьмяноокисного электрода tP = 'Psbto. + ^lnGsb3+ и, следовательно,
Т “ W, + I" К + °н+ = 14 - pH. (VII, 18)
Концентрационные элементы. Потенциометрический метод анализа 189
Таким образом, потенциал сурьмяноокисного электрода определяется активностью Н+-ионов.
Сурьмяноокисный электрод не дает солевых и коллоидных ошибок, т. е. его потенциал не искажается в присутствии коллоидов или в концентрированных растворах солей. Им можно измерять pH в мутных растворах, желеобразных или кашеобразных массах.
Сурьмяноокисный электрод позволяет определять pH в интервале от 2 до 12 включительно, однако он недостаточно точен. Обычная точность его показаний 0,1—0,2 единицы pH. Сурьмяный электрод нельзя употреблять, если в .растворе имеются соли металлов, более благородных, чем сурьма, например Си, Bi, Rb, Sn, Ag и др., так как эти металлы могут контактно выделяться на поверхности сурьмы. Соли сернистой кислоты, сероводород Н2О2, СгО3 и другие окислители и восстановители влияют на показания сурьмяноокисного электрода. Так же влияют некоторые органические вещества (например, лимонная кислота).
При 25 °C потенциал сурьмяноокисного электрода определяется уравнением
? = 0,254 — 0,0536 pH. (V11,19)
Как видно, здесь коэффициент перед pH несколько меньше теоретического.
При изменении температуры на кривой <p = f(pH) для сурьмяноокисного электрода наблюдается излом (около pH = 4—• 5). Поэтому в интервале 20—50 °C pH рассчитывают по уравнениям:
для pH = 2—4
£ —<р" 4-0,254 — 0,0013 (/ —25) рН=--------------ад ;
для pH = 5—12
н _ £ — у" 4-0,254 — 0,0022(/— 25)
Р “ 0,0536
где Е — э. д. с. элемента;
<р" — потенциал каломельного электрода.
Сурьмяный электрод необходимо непрерывно очищать, так как малейшее загрязнение его поверхности в процессе работы делает потенциал неравновесным.
Изготовляют такой электрод из сурьмяного стержня (рис. 55), который укрепляется (с помощью пицеиновой замазки) «в футляре 'из плексигласа. Перед работой электрод шлифуют очень мелкой наждачной бумагой и промывают дистиллированной водой.
190
Теоретические основы электрохимии
Весьма большое распространение получил описанный выше хингидронный электрод, потенциал которого также зависит ,от pH раствора. Однако, как уже указывалось, хингидронный электрод неприменим при pH > 7 вследствие кислотной диссоциации
Рис. 55. Сурьмяные электроды:
/ — сурьмяный электрод; 2 — стеклянная трубка; 3 — пицеин;
4 — медная проволока
гидрохинона, а также окисления его кислоро-дом воздуха. Хингидронный электрод можно использовать для измерения pH в неводных растворах — в спиртах, бензоле, ацетоне, муравьиной кислоте и др.
§ 6. Стеклянный электрод; области его применения и теория
Большим достижением в методике определения концентрации водородных ионов являлось изобретение стеклянного электрода. Еще в 1906 г. Кремер заметил, что тонкая стеклянная мембрана, разделяющая два раствора, обнаруживает скачок потенциала, зависящий от концентрации Н+-ионов. Более детальное исследование показало, что потенциал такой мембраны зависит также от концентрации других ионов (Na+, К+, Rb+ и Cs+), от состава и толщины стекла и температуры.
Устройство стеклянного электрода видно из рис. 56. Из спе-
циального, хорошо проводящего ток стекла готовят тонкостенный шарик 1 (толщина стенок менее 0,001 мм) и заполняют его электролитом, например раствором КО. Шарик помещают в сосуд 2, в которой через воронку 3 наливают испытуемый раствор либо воду для промывки электрода, а также раствор кислоты для его хранения в промежутках между «измерениями. Эти растворы сливаются через кран 4. Электрод включают в систему с помощью платиновой проволоки 5, пропущенной через пробку 6 и слой парафина 7. Электродом сравнения служит обычно каломельный полуэлемент 8. Таким образом, составляется цепь (1):
Hg
Hg2Cl2, КС1насыщ.
буферный раствор 1, pHi
стекло
испытуемый раствор 2, рН2
KCl,Hg2Cl2Hg.
(1)
Такой элемент обладает некоторой э. д. с., причина появления которой объясняется тем, что на границе фаз стекло —
Концентрационные элементы. Потенциометрический метод анализа 191
раствор возникает скачок потенциала, величина которого зависит от pH раствора по уравнению
? = tp°T_ 0.0002Т pH. (VII,20)
Механизм возникновения этого потенциала до некоторой степени аналогичен механизму появления скачков потенциалов на (границе фаз металл—^раствор.
В описанном выше элементе возникают два скачка потенциала: один на границе раствор (1)—стекло (дц), другой на границе стекло—-раствор (2) (ф2) • Поэтому э. д. с. элемента
Е = <Р1 — или
£ = 0,00027 (рНх —рН2).
' Очевидно, если в первый сосуд залить буферный раствор, pH которого известен, то можно определить pH любого испытуемого раствора, измеряя э. д. с. элемента (1), при этом
рНх = рН0 +---------
0.0002Т
(VII,21)
Рис. 56. Стеклянный электрод
= а
При рНж = рН0 э. д. с. элемента (1) равна нулю. Значение pH при £=0, т. е. величину а, называют электрическим нулем стеклянно-
го электрода. Э. д. с. элемента (1) подчиняется уравнению (VII. 21) в интервале от pH = 1 до pH = 9,5.
Буферными смесями при калибровании служат, обычно, растворы с различными pH (от 1 до 8). Индикаторный стеклянный электрод считается пригодным для последующих измерений, если калибровочный график представляет собой прямую линию.
Поскольку внутреннее сопротивление стеклянного электрода в большинстве случаев весьма значительно, а ток, протекающий через систему, ничтожен, то прибор, используемый в качестве нуль-^инструмента, не должен практически потреблять ток от измеряемой ячейки. Этому условию удовлетворяет электрическая пепь, включающая ламповые усилители. Эти приборы основаны на том, что ток от измеряемой цепи подается на сетку электронной лампы. Незначительные изменения силы тока в цепи сетки
192
Теоретические основы электрохимии
вызывают большие изменения в силе тока в анодной цепи. Наиболее часто измерения осуществляют с помощью потенциометра (компенсационная схема). Здесь электронная лампа вместе с усилителем служит чувствительным гальванометром (нуль-ин-стру ментом).
Известно, что стекло содержит в простейшем случае ионы Na+ и силикатные анионы, которые располагаются в определенном порядке (подобие кристаллической решетки). При погружении стекла в раствор электролита ионы Na+ могут обмениваться с находящимися в растворе нонами водорода, которые с силикатными анионами стекла в образуют слабо диссоциирован-
I ные силикатные кислоты. Этот
j । обмен идет до установления рав-
I Стекло Растбор новесия и приводит к появлению
। скачка потенциала, причем вели-
I чина последнего определяется
] концентрацией ионов водорода.
। ца --------» При этом стекло только вполне
I определенного состава обладает
] —---------н* обратимостью по отношению к
водородным ионам и то лишь после длительного вымачивания его в растворах кислоты. Аномалия, наблюдаемая в поведении стеклянного электрода в сильнокислой и сильнощелочной средах, возникновение так называемого потенциала асимметрии наиболее полно были объяснены в теории Б. П. Никольского и квантовоме-сходной с теорией Р. Герни. Эти
теории в настоящее время общепризнаны, поэтому ниже дается их краткое содержание.
При вырывании из решетки стекла Иа+-иона его место может быть занято другим катионом. При погружении стекла в кислый раствор часть ионов Na+ из стекла переходит в раствор, а их места занимают Н+-ионы, которые вместе с силикатными ионами стекла образуют более или менее диссоциированные силикатные или алюмосиликатные кислоты. Таким образом, изменяется состав поверхностного слоя стекла ДА' — В В' (рис. 57).
Обмен Na+ и Н+-ионов, вероятно, идет до установления некоторого равновесия между стеклом и раствором. Положение равновесия зависит от концентрации этих ионов в стекле и в растворе и от .прочности их связи в стекле и в растворе.
J' д'
Рис. 57. Ионный обмен ..на границе стекло — р аствор:
АА' — измененное стекло; ВВ' — неизмененное стекло
ханической теории М. Дола,
Концентрационные элементы. Потенциометрический метод анализа 193
Граница В В' измененного и неизмененного отекла может передвигаться в глубь стеклянной фазы, но поверхностный слой АА' — ВВ' будет оставаться в равновесии с раствором. Если раствор кислый, то равновесие сдвинуто в сторону почти полного вытеснения Иа+-ионов Н+-ионами и потенциал растет с ростом pH. В щелочных растворах водородных ионов в стекле почти нет, преобладает переход Na+-ионов из стекла в раствор и стекло заряжается отрицательно относительно электролита.
Прямолинейная функция <р = f (pH) стеклянного электрода проходит через максимум. Это связано с тем, что в нейтральных и слабокислых областях зависимость потенциала от pH имеет один знак, а в щелочной области-—другой.
Погрузим стеклянный электрод в раствор известной кислотности. Условием термодинамического равновесия поверхностного слоя стекла с раствором будет равенство электрохимических потенциалов Н+ и Ы>а+-ионов в двух фазах, т. е.
^+^+^4--^+^ + ^+’
где ф, ф — потенциалы, стекла и раствора^
_ pi, gi — соответствующие химические потенциалы;
Ф—ф=ф — скачок потенциала на границе стекло — раствор.
Имея в виду, что
и так как
+ ВТ In аг, то
М “ ~RT Рн+ - |'н+ + %+ -S.+] = |п К-
Н ' Na
Отсюда
а ! а +
= К. (VII,22)
“н+ “Na+
Величина К называется константой обмена. В равновесном слое стекла
ар . 4- ар , —- an = const, так как число свободных мест, которые могут быть заняты катионами, примерно постоянно. Отсюда следует
flNa~t~ _ fl° OH~t~ _ ^'o^Na't'^Na+_д- °Na+ (уц 23)
°н+ “н+ ' “н+ ^н+ “н+
13 Заказ 1338
194
Теоретические основы электрохимии
ИЛИ
„ д_ ( ^я+
VL K'v+lV/g"+
сн+ “(/на+
(VII,24)
где fн+ и fNa+ — коэффициенты активности ионов в стекле, которые для данного образца стекла можно считать постоянными и не зависящими от величин Сн+ и Cnh+, так как они являются
функциями расстояний Полагая
между ионами, которые не изменяются.
^н+ ' ^Na+ 1 ’
получим
"н+ ан+ С другой стороны,
да 4. + а ।
Na+ 1 Н+
й0
(VII,25)
или
_ ----а + In (пн+ + №N1+), (V11,26)
г г г
откуда следует уравнение Никольского для потенциала стеклянного электрода:
<р ---= <р° + Т 1g (ан+ + AaNa+), (VII ,27)
где
у = 2,3—. ' F
В кислом растворе, когда он+^>А<^-а+. уравнение (VII, 27) принимает вид:
<р = <р° — у pH. (VII,28)
При больших pH, когда ац+<^ fiNa+A-
<р = + Т 1£ А- + у 1g aNa+- (VII,29)
Так как
„ _ с Кв
^Na+—-ОН"" с ’
н+
Концентрационные элементы. Потенциометрический метод анализа 195
то
? = ?’ + TpH. (VII,30)
Константа обмена К в уравнениях Никольского характеризует разницу в прочности связи Н+ и Ыа+-ионов в стекле и в растворе. Она может быть определена по положению максимума кривой <р = /(pH).
Исследования показали, что для обычных электродных стенок этот максимум лежит при pH около 12.
М. Дол, пользуясь квантовомеханическим методом, разработал свою теорию стеклянного электрода. Дол полагает, что в с(тличие от водородного электрода, через стеклянную мембрану проникает ион водорода вместе с гидратной оболочной. Конечные уравнения Дола полностью совпадают с уравнением Никольского (VII, 28). Отклонение потенциала электрода от водородной функции в кислой среде, по Долу определяется выражением
лт=-^|п<.„,о> (VII,31)
где ано —активность воды в испытуемом растворе.
Из уравнения (VII, 31) следует, что для неводных сред, когда активность воды исчезающе мала, ошибка стеклянного электрода достигает таких размеров, что он не может быть применим.
В концентрированных растворах сильных кислот при pH < 0, в весьма концентрированных нейтральных растворах солей, а также в неводных растворах потенциал стеклянного электрода становится более отрицательным, чем можно ожидать.
Это объясняется тем, что при прохождении тока через мембрану и при большом различии активностей воды, находящейся по разные стороны от мембраны, изменяется .свободная энергия воды .и, следовательно, величина потенциала.
Б. П. Никольский и Т. А. Толмачева изучили влияние состава стекла на свойства стеклянного электрода. Эти работы показали, что присутствие в стекле окислов бора и алюминия вызывает сильные отклонения потенциала от водородной функции (VII, 28), так как эти добавки упрочняют связь №+-ионов в стекле и ослабляют связь ионов водорода.
В щелочных растворах, где количество ионов водорода весьма незначительно (10-9 г-ион)л и меньше), начинают принимать участие в переносе электричества через мембрану другие положительные ионы, концентрация которых во много раз больше концентрации ионов водорода. В таких растворах стеклянный электрод перестает быть строго обратимым относительно' водородных ионов и становится одновременно обратимым относительно других ионов, например натрия, лития, калия или сереб-13*
196 Теоретические основы электрохимии
ра, присутствующих в исследуемом растворе (солевая ошибка). Потенциал его становится более положительным, чем должен быть, согласно теоретическому уравнению для стеклянного электрода. Таким образом, электрод перестает быть строго обратимым по отношению к водородным ионам. Из сказанного следует, что стеклянный электрод является достаточно точным при определении pH между 0 и 9. При малом или умеренном содержании положительных одновалентных ионов металлов, особенно натрия и лития, стеклянный электрод пригоден при pH = 11—12.
В качестве материала для изготовления стеклянного электрода нельзя применять обычные стекла вследствие их малых электропроводностей. Составы стекол, употребляющихся разными авторами для изготовления стеклянного электрода, приведены в табл. 16.
Большое электросопротивление стекла является существенным недостатком стеклянного электрода при потенциометрическом анализе, так как затрудняет измерение разности потенциалов. Поэтому при измерениях со стеклянным электродом, как уже отмечалось, приходится использовать сложные измерительные приборы — ламповые .потенциометры, электрометры, электромагнитные и электростатические системы.
Таблица 16. Химический состав стекла для изготовления стеклянных электродов
Сорт стекла Химический состав, % (вес.)
SiO« Na,0 CaO в,о, А1,О, MgO
Юза . 72,0 20,0 8,0 — — —
Мак-Иннеса 72,0 22,00 6,00 — —-
Ботвинкина 72,0 22,00 6,00 -—. — —
Никольского 64,6 20,95 5,65 8,8 — .—
Толмачевой 66,1 21,40 5,80 — 6,7 —
Пасынского 59,0 30,0 — — — 11,0
ЛИТЕРАТУРА
Белозерский С. С., Флоренский П. А., Денисов С. С., Андерс В. Р. Методы и приборы для измерения pH в нефтяной промышленности. Гостоптехиздат, 1953.
Бриттон X. Водородные ионы. Химтеоретиздат, 1936.
Виноградова Е. Н. Методы определения концентрации водородных ионов. Изд. МГУ, 1950.
Воскресенский П. И. Основы техники лабораторных работ. Госхимиздат, 1956.
Деляхей П. Новые приборы и методы в электрохимии. ИЛ, 1957.
Кольтгоф И. и Лайтинен Г. Определение концентрации водородных ионов и электротитрование. ИЛ, 1947.
Концентрационные элементы. Потенциометрический метод анализа 197
Краткий справочник физико-химических величин, под ред. К- П. Мищенко. Госхимиздат, Л., 1959.
Комаидин А. В. Электродвижущие силы. Изд. МГУ, 1951.
Латимер В. Окислительные состояния элементов и их потенциалы в водных растворах. ИЛ, 1954.
Л е в и н А. И., П о м о с о в А. В., Ушке Е. А. Руководство к лабораторным занятиям по теоретической электрохимии. Изд. УПИ им. С. М. Кирова, 1957.
Лопатин Б. А. Ламповые гальванометры постоянного типа. Госэнергоиз-дат, 1952.
Никольский Б. П. ЖФХ, 27, 1953, стр. 724.
Никольский Б. П. и М а т eip о в а Е. М. ЖФХ, 25, 1951, стр. 335-
Стромберг А. Г., Захаров М. С., А т к н и и Г. Ф. и др. Сборник примеров и задач по электрохимии, гл. V, изд. Томского университета, 1962.
Хейфец В. Л., Авдеев Д. К., Р е й ш а х ip и т Л. С. Практикум по теоретической электрохимии. Изд. ЛГУ, 1954.
Глава VIII
СТРОЕНИЕ ДВОЙНОГО ЭЛЕКТРИЧЕСКОГО СЛОЯ, электрокапиллярные И ЭЛЕКТРОКИНЕТИЧЕСКИЕ ЯВЛЕНИЯ
§ 1. Возникновение двойного электрического слоя на границе фаз
Понятие о двойном электрическом слое (который мы в дальнейшем будем для краткости называть двойным слоем) приводится в учении об электричестве. Образование двойных
слоев на границе раздела фаз представляет собой общее явление.
Представим себе две весьма близкие и параллельные одна другой поверхности S_ и S+, имеющие противоположные заряды (рис. 58). Пусть при этом плотности зарядов противолежащих поверхностей Е- И Е+ равны по
Рис. 58. Схема двойного электрического слоя (по Гюи—(Гельмгольцу)
абсолютной величине и противоположны по знаку, т. е.
причем е+ > 0 (и, следовательно, е-<0). Тогда, если расстояние б между поверхностями S. и 5+ исчезающе мало по сравнению с расстоянием от этих поверхностей до рассматриваемых точек пространства (например, точка Р), совокупность этих поверхностей и определяет характер двойного электрического слоя.
В двойном слое имеется скачок потенциалов, равный
Д<р = 4леб, (VIII,!)
т. е. чем больше плотность зарядов на обкладках двойного слоя и расстояние между ними, тем больше скачок потенциала. Скачки электродных потенциалов на границе фаз непосредственно связаны с образованием пограничных двойных слоев.
Строение двойного электрического слоя
199
Рассмотрим фазовую границу воздух — электролит (рис. 59). Чтобы измерить скачок потенциала на этой границе, нужно сделать слой воздуха между поверхностью раствора 1 и металлическим электродом 2 электропроводящим. С этой целью можно покрыть электрод слоем радиоактивного вещества, например полония, излучающего только а-частицы. Благодаря испусканию «-частиц воздух ионизируется, т. е. становится проводником тока. В жидкость погружают каломельный электрод 3. Воздушный
Рис. 59. Фазовая граница воздух — электролит:
Е — э. д. с. между электродами 2 и 1
Рис. <60. Возникновение двойного слоя на 'поверхности жидкости
электрод присоединяют к квадрантному электрометру, кало-мельный электрод — к потенциометру. Некоторые анионы адсорбируются на границе раствор — воздух сильнее катионов. В результате на поверхности раствора образуется двойной слой с отрицательной обкладкой снаружи и положительной обкладкой внутри жидкой фазы (рис. 60). Этот двойной слой можно получить с использованием раствора НС1 + KJ, причем J^-ионы будут адсорбироваться на границе раствор — воздух, а вторая обкладка двойного слоя будет составлена из катионов Н+ и К+. Такой двойной слой в соответствии с механизмом его возникновения называется адсорбционным.
Другой возможный механизм образования двойного слоя состоит в том, что адсорбированные на фазовой границе дипольные молекулы, например молекулы воды, ориентируются определенным образом. Такой двойной слой обычно называется лио-электрическим (рис. 61).
Наконец, можно себе представить третий механизм образования двойного слоя —так называемый ионогенный (рис. 62). Такой слой образуется путем перехода ионов из металла в раствор до установления термодинамического равновесия. При этом в металле оказывается избыточное количество эл ктронов. Они и составляют внутреннюю обкладку двойного слоя. Внешней же обкладкой служат гидратированные катионы металла.
200
Теоретические основы электрохимии
Такой слой создается на границе металл — раствор в том случае, когда энергия сольватации катионов металла больше, чем энергия связи между электронами и ионами в металле. В противном случае двойной слой образуется в результате избирательной адсорбции ионов какого-либо одного знака, например катионов, составляющих его внутреннюю обкладку (рис. 63). Внешняя же обкладка образуется избыточными анионами раствора. Такой двойной слой является по существу также адсорбционным.
Рис. 61. Лиоэлектри-ческий двойной слой
Рис. 62. Ионогенный двойной слой:
1 — электроны в металле; 2 — одновалентные катионы
Рис. 63. Адсорбционный двойной слой
Все приведенные здесь схемы гораздо проще имеющихся в действительности реальных двойных электрических слоев. Но именно такого рода слои определяют возникновение скачков потенциалов на границах фаз. В первом приближении можно считать, что реальные двойные слои полностью совпадают с тем, который был рассмотрен вначале (см. рис. 58). Такой двойной слой можно назвать простейшим. Его отличительные черты — параллельность обкладок и наличие на них равномерно и непрерывно распределенного разноименного электричества. Тогда для скачка потенциала в двойном слое Д<р (рис. 64) должно оправдываться уравнение (VIII,1).
Изложенные представления о строении и свойствах реального двойного слоя были высказаны в 1878 г. Р. А. Колли, который уподобил двойной слой на границе фаз плоскому конденсатору и экспериментально определил его емкость в 80— 120 мкф!см2.
Сгроение двойного электрического слоя
201
Теорию двойного электрического слоя иногда называют «теорией плоского конденсатора» и часто ошибочно связывают с именем Гельмгольца.
Первым практическим затруднением, с которым столкнулась, эта теория, была невозможность объяснить с ее позиций существование электрокинетического потенциала £, который всегда
меньше, чем ф, и иногда имеет противоположный знак. Поэтому
Гуи и Чепмэн сделали попытку создать новую теорию, так называемую диффузионную теорию двойного слоя. Эта теория исходит из непрерывного теплового движения ионов в растворе, которое должно привести к отклонению обкладок двойного слоя от параллельного расположения их у поверхности раздела фаз.
Весьма часто двойной слой имеет более сложное строение, чем схематически изображенное на рис. 58. Ионы двойного слоя вырываются тепловым движением из плоскости закрепленных обкладок конденсатора и уносятся в глубину раствора. Притягиваемым за-
Расстояние от поверхности металла
Рис. 64. Скачок потенциала в двойном слое:
8 = х' — толщина двойного слоя
рядом поверхности они возвращаются обратно, но под влиянием броунского движения и интерионного взаимодействия
снова вырываются. В результате часть избыточных ионов опре-
деленного знака, компенсирующих заряд поверхности металла,, оказывается расположенной не в закрепленной обкладке раствора, а в толще электролита. Концентрация избыточных ионов по мере удаления от поверхности металла, естественно, будет уменьшаться. Согласно взглядам Гуи, внешняя обкладка двойного слоя представляет собой ионную атмосферу (см. теорию Дебая — Гюккеля в гл. V). Толщина двойного слоя б опреде-
ляется уравнением
(VIII,2>
Здесь
р — ионная сила раствора;
Сь г, —концентрация и зарядность i-того иона; В — величина, не зависящая от ц;
— —радиус ионной атмосферы. .
202
Теоретические основы электрохимии
При очень больших концентрациях двойной слой по своему строению приближается к плоскому конденсатору; при малых концентрациях он, наоборот, носит резко выраженный диффузионный характер (рис. 65). По закону распределения Больцмана, концентрации одновалентных ионов на расстоянии х от поверхности электрода (+СХ и -Сх) равны соответственно г __ с ехп (— Гу* ) . +Сх со ехР rt )’
Рис. 65. Двойной электрический слой по Гуи:
-4- е — плотность заряда на поверхности металлического электрода; — е — плотность заряда диффузного слоя
_Сх = Сю’ехр(+-^-), (VI 11,3)
где <рж — потенциал на расстоянии х от металла;
Ссо— общая концен-
трация раствора на бесконечном удалении от металла;
+СХ\ _jCx— концентрации аниона и катио
на соответственно.
При значениях:
х = оо
%С = 'Р‘со = 0
= ?о — Д?-
С другой стороны, пространственная плотность диффузной части на расстоянии х
Следовательно,
ex = F(+Cx-_Cj.
zFcp zFcp "
----- -----------
RT ____e RT
По закону Пуассона,
Л DRTC
|/ 2nz
гТД?* гРЬц>х
2RT ____e 2RT
(VIII,4)
заряда e в
(VIII.5)
(VIII,6)
(VIII, 7)
(VI11,8)
d2<p _ 4тг D
Строение двойного электрического слоя
203
Таким образом, представление о диффузионном строении жидкостной обкладки слоя учитывает взаимодействие ионов в •растворе. Представление о диффузном слое помогло объяснить ряд электрокапиллярных явлений, например снижение и смещение максимума электрокапиллярной кривой в зависимости от концентрации и состава капиллярноактивных ионов в электролите.
Однако теория Гун не в состоянии была объяснить перезарядку в коллоидных системах, т. е. обращение знака потенциала по мере прибавления электролита. Эта теория не учитывает специфической адсорбции ионов, т. е. взаимодействия ионов с поверхностью металла, а также не принимает во внимание дискретного строения внутренней (обращенной к металлу) обкладки двойного слоя.
Все это привело к несовпадению теории Гуи с опытом. Она, например, дает для емкости двойного слоя значения, близкие к 250 мкф!см\ т. е. величины, примерно в 10 раз большие, чем опытные. Слишком большие значения для емкости двойного слоя ведут к физически невероятному представлению, что заряды, связанные с ионами, находятся на расстоянии от поверхности металла, меньшем чем 1 • 10~8 см (величина среднего ионного радиуса). Кроме того, теория Гуи, как и теория Колли, не учитывает существования потенциалов диффузного слоя.
Дальнейшее развитие теория двойного слоя получила после работ О. Штерна, который предположил, что часть ионов внешней обкладки прочно адсорбирована на металле и образует двойной слой Колли, т. е. плоский конденсатор с потенциалом ф. Остальные ионы, составляющие жидкостную обкладку, распределены в диффузной части слоя, имеющей потенциал ф1 (рис. 66).
Таким образом, строение двойного слоя зависит от состава и концентрации раствора электролита и от потенциала электрода. .Двойной слой, следовательно, можно мысленно разделить на две части, одна из которых («закрепленная» часть) находится между поверхностью электрода и плоскостью, отстоящей от нее на ионный радиус, т. е. не превышает нескольких ангстрем, другая (диффузная часть) простирается от этой плоскости в глубь раствора и достигает относительно больших величин (в чистой коде эта толщина а может превысить 1 А).
Не весь диффузный слой может свободно передвигаться. Если бы при электрокинетических процессах жидкость смещалась как раз на поверхности раздела фаз, то разность потенциалов между движущейся жидкостью и неподвижным электродом была бы равна термодинамическому потенциалу <р. Но так как в действительности приэлектродная часть слоя жидкости
204
Теоретические основы электрохимии
неподвижно связана с поверхностью твердой фазы, то видимое
смещение слоев жидкости происходит на некотором расстоянии от поверхности раздела, т. е. в толще диффузного слоя. Скачок потенциала между неподвижной и движущейся фазой, таким об-
разом, меньше термодинамического потенциала <р и равен фр
Соотношение между термодинамическим потенциалом <р и по-
тенциалом ф] графически представлено на рис. 66. Здесь 6 — толщина закрепленной
Рис. 66. Строение двойного слоя по
части диффузного слоя, ордината <р соответствует полному скачку потенциала. Электрокинетичес-кий потенциал выражен ординатой фр Как видно' из графического построения, разность (ф — ф1) составляет величину падения потенциала в закрепленной части двойного слоя) На потенциал ф1 влияет только толщина диффузного слоя. С уменьшением ее понижается потенциал фр В случае, когда толщина слоя уменьшается до величины 6 (молекулярные размеры), т. е. двойной слой приобретает простейшее строение, ф1 становится
1П равным нулю, в то время теРнУ как величина не изменяется. Даже в том слу-
чае, когда потенциал фд меняет свой знак на обратный (см. рис. 66), термодинамический потенциал <р остается неизменным. Изменение знака потенциала ф] можно объяснить избиратель-
ной адсорбцией некоторых ионов. Например, ионы тория адсорбируются в обращенной к жидкости обкладке двойного слоя и
делают потенциал металла более положительным.
Если бы диффузная часть двойного слоя проходила абсолютно точно в плоскости, отстоящей от электрода на расстоянии среднего ионного радиуса, то потенциал ф1 был бы тождественным электрокинетическому потенциалу £. Однако в действительности имеется различие между ф] и t-потенциалами, особенно при больших концентрациях растворов (например, кислот),. С разбавлением ф1 и ^-потенциалы сближаются.
Строение двойного электрического слоя
205
При рассмотрении количественной стороны вопроса становится очевидным (рис. 66), что
Je0| = leil + l^l, (VIII,9)
где ео — абсолютное значение плотности заряда на границе фаз металл — раствор;
Bi — значение плотности заряда в адсорбционной части слоя;
В2 — значение плотности заряда в диффузной части слоя. Кроме того, заряд на границе фаз металл — раствор может быть рассчитан из уравнения
Z (VIII,10)
4тго
По теории Гуи [см. уравнение (VIII,8)], плотность заряда диффузной части двойного слоя определяется соотношением:
~е 2RT
(VIII.1I)
S2
= zFl\
где ipi — потенциал, создаваемый частью двойного слоя, находящейся на расстоянии одного ионного радиуса от поверхности электрода.
Плотность заряда плоской части двойного слоя в случае отсутствия специфической адсорбции определяется уравнением
1_________________I
_ -L гГ^г
1 RT , , 1 "Г RT
— e 1 4- — e
C C
количество ионов на единице поверхности, г-экв, концентрация электролита, моль/см?.
, (VIII,12)
С —
Когда С достаточно мало, единицей в знаменателе можно пренебречь. В таком случае
(гГф, гГф,
ейт-+е--«т-
откуда следует уравнение Штерна:
(zF^i ____zFtyi \
е RT — е 2,<1 ) +
4Пб ’
--------гГф, гГф, DCRT 2RT 2RT
в t'
2тгг
(VI 11,13)
(VI 11,14)
Анализ этого уравнения позволяет сделать выводы, согласующиеся с опытом. С понижением концентрации электролита плотность заряда в адсорбционной части слоя (ej) уменьшается
206
Теоретические основы электрохимии
по сравнению с плотностью заряда в диффузной части (е2)> т. е. диффузная часть обкладки растет. В таком случае ф| будет Приближаться К (р и |е0] = |е2|.
Изменение плотности заряда поверхности |е0| при изменении^ потенциала на единицу равняется емкости конденсатора
_____q ду
(VII 1,15>
Поэтому, учитывая уравнения чнм выражение для эффективной
(VIII,11) и (VIII,15), полутолщины диффузного слоя:
6 =
Vdrt
in------
d<p
(zFg> _____ zFtp
(VIII,I6>
D
откуда следует, что увеличению диффузности двойного слоя способствуют повышение температуры и диэлектрической постоянной среды, а также уменьшение концентрации электролита, валентности ионов и потенциала <р.
Теория Штерна объяснила влияние концентраций и природы • ионов на строение двойного слоя. Она оказалась также полезной при объяснении ряда фактов из области электрокапилляр-ных и электрокинетических явлений. Поэтому основные представления о строении двойного слоя были использованы в современной теории перенапряжения водорода.
Вместе с тем количественное выражение теории Штерна наряду с громоздкостью характеризуется рядом недостатков, из= которых укажем следующие:
1. В теории Штерна не учитывается взаимодействие частиц,, образующих двойной слой.
2. Величина диэлектрической постоянной D, входящей в уравнение Штерна, не равна диэлектрической постоянной растворителя и точно не известна, но для водных растворов она меньше 10. Кроме того, самый факт оперирования величиной D при; рассмотрении микроявлений неправомерен.
А. Н. Фрумкин с сотрудниками предпринял серию исследований по выяснению влияния многовалентных катионов и анионов на емкость ртутного электрода в растворах электролитов. Эти исследования также подтвердили недостаточность теории1 Штерна. Указанные недостатки побудили предпринять разработку более совершенной теории двойного слоя на границе металл— раствор. О. А. Есин рассматривает двойной слой, образующийся на поверхности металла вследствие адсорбции ионов какого-либо одного знака, например катионов (рис. 67). При этом ионы противоположного знака образуют вторую обкладку двойного слоя. В результате получается такая картина, как:
Строение двойного электрического слоя
207
будто бы на поверхности металла адсорбированы диполи, взаимодействующие между собой и потому располагающиеся на поверхности в определенном порядке. Б. В, Эршлер несколько, видоизменил картину, представленную О. А. Есиным, указав.
Рис. 67 Двоимой слой по О. А. Есину: 1 — раствор; 2 — металл
что ионы внешней обкладки не будут прочно связаны с адсорбированными ионами внутренней обкладки (рис. 68).
Но по аналогии с теорией Дебая — Гюккеля можно предположить, что внешняя обкладка будет состоять из ионного об-
Рис. 68. Двойной слой по Б. В. Эршлеру
лака. Максимальная толщина этого облака (рис. 68), очевидно, равна радиусу ионной атмосферы, т. е.
(VIII,17)-
Адсорбированные ионы, взаимодействуя между собой, стремятся так распределиться по поверхности, чтобы их потенциальная энергия была минимальна.
Полный скачок потенциала <р° между поверхностью металла и раствором в этом случае можно разбить на два слагаемых: скачок <ра между поверхностью металла и плоскостью АА', в ко
208
Теоретические основы электрохимии
торой находятся центры адсорбированных ионов, и скачок <р*, описывающий поле, лежащее за плоскостью АА'.
Сделав ряд вспомогательных допущений, Б. В. Эршлер подсчитал величину фа:
<pa = 0,5<p°p + 0,37-^-j
(VIII,18)
где г — расстояние между центрами адсорбированных ионов; б — расстояние между адсорбированными ионами и плоскостью 00', ограничивающей нижний край ионного облака (см. рис. 67).
Общий скачок потенциала
<Р° = 0,5<р° /1 + 0,37 <р*.
(VIII,19)
Если, адсорбция незначительна, то число ионов, адсорбированных на единице поверхности металла, может быть подсчитано по уравнению Больцмана
q>F
С\ =
где К' — постоянная;
а±—активность ионов в растворе.
Тогда общий скачок потенциала
epF
„ 4п 4тг ts, RT
<р° —------------С т = — ------------тК а+е
r D D *
где т — момент «диполя» — ион-ионное облако. Следовательно,
In (—<р°) = In а± + ? + const;
RT dy° _ F
ain0± —_0 ,5(1+0,56
F<p° \
(VI11,20)
(VI11,21)
(V 111,22)
Величина 6, по Эршлеру, примерно равна 0,75- IO-8 см. Значение г можно вычислить, задавшись каким-либо определенным порядком в расположении ионов на поверхности. Так, если ионы расположены в узлах гексагональной,плоской решетки:
________2_________ /3 • 6,06 • \0мС
(VI 11,23)
Строение двойного электрического слоя 209
Для проверки теории Б. В. Эршлер построил график функции (рис. 69) от общего скачка потенциала:
F
RT
(VII 1,24)
Как видно из рис. 69 теоретические данные Есина — Эршлера (кривая 1) достаточно хорошо совпадают с опытными (кривая 2).
§ 2. Общая теория электрокапиллярных явлений
Совокупность ряда методов исследования, в том числе изучение электрокапиллярных явлений, позволяют развить представления о строении двойного электрического
д In а±
Рис. 69. Проверка теории Есина — Эршлера
слоя на поверхности металла, погруженного в раствор.
Еще в начале прошлого века было замечено, что форма поверхности ртутной капли, находящейся в растворе, зависит от сообщенного ей заряда. Если с поверхности ртути периодически снимать заряд, что достигается, например, с помощью укрепленной иглы, то капля ртути начинает совершать сложные движения («ртутное сердце»). Это явление можно объяснить, если предположить, что поверхностное натяжение ртути зависит от возникновения двойного электрического слоя на металле и, следовательно, от скачка потенциала на границе ртуть — раствор. Наблюдать такую зависимость очень удобно с помощью капиллярного электрометра (рис. 70), который состоит из двух ртутных электродов, сообщающихся при помощи разбавленного раствора серной кислоты. Один из электродов — анод (ртуть в каломельном полуэлементе 4) обладает большой поверхностью и при прохождении тока практически не поляризуется; другой же электрод, поляризуемый током,—катод — находится в тонкой стеклянной трубке 2, заканчивающейся капилляром. Вследствие весьма ограниченной поверхности катода (капля ртути) потенциал его может быть изменен в широких пределах в за
висимости от величины приложенного заряда.
14 Заказ 1338
210
/
Теоретические основы электрохимии
Работа обратимого изотермического образования единицы поверхности твердого или жидкого тела называется коэффициентом поверхностного натяжения, или удельной свободной поверхностной энергией. Поверхностное натяжение по системе СИ измеряется в н/м:
°общ = aS’
(VI 11,25)
где о — поверхностное натяжение;
S — величина поверхности.
При этом Ообщ стремится к минимуму.
Вполне определенным значением поверхностного натяжения обладают не только жидкости, но и твердые тела. Однако изме
Рис. 70. Капиллярный электрометр:
1 — аккумулятор; 2 — стеклянный
рение поверхностного натяжения твердых тел затруднительно из-за условности оценки их истинной поверхности. Кроме того, на разных гранях кристалла поверхностное натяжение имеет различные значения.
Изменение поверхностной энергии жидких металлов, например ртути, выражается в изменении поверхностного натяжения ртути в капилляре, которое можно определить по высоте ртутного столба в трубке 2 (рис. 70).
Совокупность значения поверхностного натяжения при разных потенциалах образует так называе-
мую электрокапиллярную кривую. Известно, что при отсутствии поляризации поверхность ртути относи-
капилляр; з - гальванометр; 4 - тельно раствора заряжается поло-каломельный лолуэлемент; 5 — г г г
ключ; 6 и 7 — подвижные контакты ЖИТСЛЬНО, ТЭК КЭК рабОТЭ ВЫХОДЯ ионов из металлической ртути UM
больше работы дегидратации Up.' Положительные заряды Hg2+
на поверхности ртути, отталкиваясь один от другого, стремятся увеличить поверхность, способствуя уменьшению поверхностного натяжения. Если ртуть катодно поляризовать, т. е. сообщить ей отрицательный заряд, то положительные ионы Hg2+ будут нейтрализованы и поверхностное натяжение на границе ртуть — раствор начинает возрастать. Однако поверхностное натяжение будет увеличиваться до тех пор, пока не будут нейтрализованы все ионы Hg2+, т. е. пока приложенный извне потенциал не станет равным скачку потенциала на границ^ фаз ртуть — раствор. Дальнейшее сообщение отрицательного заряда приводит к пе
Строение двойного электрического слоя
211,
реходу от схемы а к схеме б (рис. 71) и к снижению поверхностного натяжения из-за отталкивания на поверхности частиц, получивших избыточный отрицательный заряд.
Количество катионов металла, исчезающих из раствора или появляющихся в нем при увеличении поверхности раздела фаз металл—электролит, пропорционально величине образующейся поверхности. Изменение поверхности электрода на единицу ведет к изменению плотности заряда — количества ионов в двойном слое. Эта связь выражается известным термодинамическим уравнением Гиббса
do = —(VI 11,26)
Здесь — поверхностная концентрация катионов /-того сорта в грамм-ионах на единицу поверхности;
|Л< — химический потенциал /-того компонента раствора;
2 — знак суммы всех компонентов поверхностной фазы, кроме катионов металла.
Для ртути уравнение (VIII,26)
Рис. 71. Перезарядка |ртут,и:
1 — стеклянная трубка; 2 — ртуть;
3 — электролит
можно переписать в виде
- Г^+ • %62+ - (VI11,27)
Если е — заряд единицы поверхности металла (плотность заряда), a Hgj+ является единственным потенциал образующим ионом, то
(VIII’28>
Следов ател ьно,
~ %g2+ - (VI11,29)
Известно, что равновесный потенциал также связан с активностью ионов
d<f> = — In at zF
или
zFd^^RTXnai, (VI11,30)
14*
212
Теоретические основы электрохимии
а так как
d^t = RTd In а„ (VI11,31)
то, учитывая уравнения (VIII,29) и (VIII,31), получим
da = — ed<p — Гtd In а,-.
Если раствор имеет неизмененные состав и концентрацию, т. е. d\n = 0, то do = — ₽с?<р, откуда da е =---------------------------------
d<p
Уравнение (VIII,32) Липпмана— Гельмгольца определяет зависимость поверхностного
(VII 1,32)
Электрокаоиллярная кривая в лреимущественной физической
Рис. 72. области адсорбции катионов, анионов и молекулярных веществ
натяжения о от потенциала <р и плотности заряда е при Т = = const.
Зависимость поверхностного натяжения ртути от величины потенциала, т. е. о = f (<р), графически представлена на рис. 72. Элект-рокапиллярная кривая в первом приближении имеет параболическую форму. Максимум ее отвечает максимуму поверхностного натяжения и соответствует такому потенциалу, при котором заряд ртутной поверхности равен нулю. В точке нулевого заряда при <р = фк на границе фаз нет двойно
го ионного слоя. Поверхностная энергия ртути у вершины параболы достигает предела в случае, если она не снижена адсорбцией ионов. В таком случае поверхностное натяжение максимально. Было определено, что для ртути нулевая точка соответствует потенциалу tpN = —0,21 в (по водородной шкале).
В области более электроположительных потенциалов, т. е. слева от нулевой точки, вследствие того что поверхность ртути имеет положительный заряд, должна происходить преимущественная адсорбция анионов из раствора. Их поверхностная концентрация (плотность) растет по мере удаления от максимума электрокапиллярной кривой. Наоборот, в области, лежащей справа от нулевой точки, где поверхность ртути имеет отрицательный заряд, будет происходить адсорбция катионов и тем в большей степени, чем больше плотность отрицательного заряда на ртути.
Строение двойного электрического слоя
213
Вблизи точки нулевого заряда, где поверхность металла практически не заряжена, наиболее вероятна преимущественная адсорбция молекул *.
Теоретические соображения показывают, что наклон касательной в каждой точке электрокапиллярной кривой пропорционален электрическому заряду, расположенному на единицу поверхности ртути, соприкасающейся с раствором. У вершины капиллярной кривой наклон касательной равен нулю. Это подтверждает, что при потенциале максимума <pw поверхность не
-Е
Рис. 73. Электрокапилляр-ная кривая плотность заряда двойного слоя (е) на поверхности ртути
Рис. 74. Зависимость потенциала нулевого заряда от поливалентных катионов
имеет электрического заряда. С помощью электрокапиллярной кривой на основании уравнения Липпмана — Гельмгольца можно вычислить изменение плотности заряда е с изменением ср. Характер этой зависимости показан на рис. 73.
Как показали исследования, потенциал нулевого заряда в значительной степени зависит от присутствия в растворе поверхностно активных ионов или молекул, которые сильно искажают электрокапиллярные кривые. Подробное исследование этой зависимости было произведено А. Н. Фрумкиным и его сотрудниками, которые изучали положение потенциала нулевого заряда для ртутного электрода в 0,001-н. растворе НС1 с добавкой различных солей. Оказалось (рис. 74), что добавление поливалентных катионов ThCU, ЬаС13, ВаС12 приводит к смеще-
1 Изложенная точка зрения верна для физической адсорбции; что касается 'хемосорбции, то природа ее имеет более сложный специфический характер и часто не зависит от заряда поверхности.
214
Теоретические основы электрохимии
Ния поверхностно активных ионов
6
Рис. 75. Влияние поверхностно активных .ионов на 1ход электрокапиллярной кривой
ных потенциалов). Таким образом, подтверждают один другой.
Все эти изменения вызываются
нию точки минимума в сторону больших емкостей С и менее отрицательных потенциалов <р. При отрицательном заряде поверхности ртути на кривых С = f(<p°) возникают максимумы. -
Изучение влияния анионов показало, что они тем сильнее увеличивают Смин, чем лучше они адсорбируются. Анализ влия-на ход электрокапиллярной кривой приводит к выводу (рис. 75), что поверхностно активные анионы (С1~, В г-, J“, CNS- и др.) смещают t сторону более отрицательных потенциалов возрастающую ветвь кривой и перемещают ее максимум вниз и вправо, причем спадающая ветвь кривой остается неизменной.
Поверхностно активные катионы {Т1+, N(C4H9)4> Th4+ и др.] смещают максимум и нисходящую ветвь кривой вниз и влево (т. е. в сторону менее отрицатель-оба результата измерения специфической адсорбцией
ионов. Если, например, ртутная поверхность заряжена отрицательно, то этот заряд облегчает адсорбцию катионов, а они, внедряясь в двойной слой в избыточном количестве, снижают поверхностное натяжение и влияют на соответствующую ветвь электрокапиллярной кривой. Кроме того, избыточный заряд в двойном слое сам по себе приводит к снижению максимума электрокапиллярной кривой вследствие возникновения дополнительных сил от расталкивания.
Поверхностно неактивные ионы (К+, Na+, SO|—) почти не влияют на форму и положение электрокапиллярной кривой.
Поэтому в уравнение Липпмана — Гельмгольца для электро-капиллярной кривой вносят поправку, так что вместо (VIII,32) получается
---= е -I- ехГх. (VIII,33)
Здесь Гх — избыточная (по сравнению с концентрацией, эквивалентной заряду двойного слоя) концентрация ионов, адсорбирующихся на границе металл—раствор; ех — заряд этих ионов.
Строение двойного электрического слоя
215
В максимуме электрокапиллярной кривой
&Х^X — S-
(VI 11,34)
Снижение максимума электрокапиллярной кривой наблюдается также при добавлении в раствор поверхностно активных органических соединений (неэлектролитов). На рис.. 76 приведены электрокапиллярные кривые для 1-н. раствора NaCl с добав-
кой различных количеств третичного амилового спирта.
Исследования А. Н. Фрумкина показали, что:
а) понижение концентрации электролита и органиче ского соединения приводит к сглаживанию перехода от нормальной электрокапиллярной кривой к пониженной;
б) поверхностное натяжение при введении в раствор поверхностно активных веще-
Р.ис. 76. Электрокапиллярные кривые (1—6) в зависимости от добавки различных .количеств амилового спирта
ств почти не понижается при больших потенциалах, соответствующих максимумам кривых, полученных без добавок.
Адсорбция органических молекул и ионов на границе фаз ме-
талл— раствор играет существенную роль в электрохимических процессах. Она влияет на поверхностное натяжение и сма
чивание.
В ряде случаев адсорбционные процессы определяют структуру и свойства металлических покрытий в гальванотехнике и гидрометаллургии, оказывают решающее влияние на течение коррозионных процессов (ингибирующее действие).
Длительное время считалось, что электрокапиллярные явления могут быть изучены только на ртути, галлии и амальгамах жидких металлов в водных растворах при комнатной температуре. Однако С. В. Карпачеву и А. Г. Стромбергу удалось изучить электрокапиллярные явления на легкоплавких расплавленных металлах при высоких температурах (400—1000 °C), а О. А. Есину, Ю. П. Никитину, С. И. Попелю — в оксидных расплавах при 1300—1500 °C.
В 1945 г. П. А. Ребиндер и Е. К. Венстрем разработали оригинальный метод, позволяющий снимать электрокапиллярные кривые на твердых металлах. При этом изучали зависимость твердости металла (77). от потенциала, которая совершенно идентична зависимости о = />(ф).
216
7 еоретические основы электрохимии
Принцип метода П. А. Ребиндера весьма прост (рис. 77). На исследуемый металл наносят каплю раствора электролита, в которую опускают капилляр каломельного электрода сравнения.
В процессе поляризации твердость металла определяют по амплитуде царапающего его поверхность маятника с алмазной иглой (под каплей раствора).
Рнс. 77. Принцип снятия электрика-пиллярных кривых на твердых металлах:
1 — маятник; 2 — раствор; 3 — металл
жидкость — газ, твердое тело —
Рис. 78. Краевой угол смачивания твердого электрода жидкостью:
1 — газ; 2 — жидкость; 3 — металл
§ 3. Поверхностное натяжение и смачивание металлов
электролитами
Изучение смачиваемости позволяет проследить за изменением поверхностной энергии твердого тела с поляризацией.
На рис. 78 изображена капля жидкости на поверхно-
сти твердого электрода. Равновесие капли, очевидно, определяется величинами поверхностных натяжений на трех поверхностях раздела (твердое тело — газ, жидкость). Условие равновесия, согласно рис. 78, заключается в том, что
°т-г = °т_ж + %_г • cosfi
(V 111,35) или
cos е .= —Ст~ж, (Vi 11,36) —г
где 6 — угол, образованный касательной к границе раздела фаз жидкость — газ и границей раздела твердое тело — жидкость.
Угол 6, отсчитываемый в сторону жидкой фазы, называется краевым углом смачивания.
Жидкость легче смачивает электрод в том случае, когда поверхностное натяжение на границе жидкости с твердым телом меньше поверхностного натяжения твердого тела на границе с собственным насыщенным паром. Однако общее условие смачивания электрода жидкостью зависит и от поверхностного натяжения самой жидкости.
Строение двойного электрического слоя 217
С поверхностным натяжением жидкостей тесно связаны две производные характеристики, называемые адгезией и когезией.
Чтобы разорвать две несмешивающиеся жидкости по границе их соприкосновения, надо затратить работу, равную сумме поверхностных натяжений каждой из жидкостей за вычетом поверхностного натяжения на границе жидкость — жидкость. Эта результирующая работа называется работой адгезии. Для однородной жидкости вводится понятие работы когезии.
Понятия адгезии и когезии применимы и к твердым телам. Как для границы жидкость — жидкость, так и для границы твердое тело — жидкость работа адгезии тем больше, чем меньше межфазное натяжение.
Работа адгезии на границе твердое тело — жидкость может быть вычислена через краевой угол смачивания. Действительно:
Аадг ~~ ст—г + сж—т °т—ж- (V 111,37)
Складывая почленно соотношения (VIII,35) и (VIII,37), получим
Аадг = аж-г (1 + cos 6). (VI 11,38)
На основании определения краевого угла смачивания, Б. Н. Кабанов показал, что значения угла 6 меняются с изменением потенциала <р по кривой, близкой к электрокапиллярной кривой, причем 0 = 6 макс при <р = <pjv. Это указывает на то, что около точки нулевого заряда поверхность твердого электрода смачивается хуже, чем при значительной поляризации электрода. В точке нулевого заряда жидкость не смачивает поверхность электрода.
Исследования А. Н. Фрумкина и П. А. Ребиндера в области флотационных процессов и электро-капиллярных явлений вскрыли роль и значение адсорбции поверхностно активных веществ в процессах смачивания.
Смачивание весьма заметно изменяется при адсорбции капиллярно активных веществ на твердой поверхности. Адсорбируясь на поверхности твердого тела, полярно-асимметричные молекулы ориентируются в адсорбционном слое таким образом, что к поверхности обращены их полярные группы, а углеводородные радикалы направлены в окружающую среду. Подобного типа адсорбция может вызвать инверсию смачивания вследствие образования гидрофобной пленки из поверхностно активных веществ. Способность поверхностно активных веществ менять величину и знак смачивания, т. е. вызывать инверсию, изменяется всегда параллельно адсорбируемости и резко растет в гомологическом ряду с удлинением углеводородного радикала органических веществ.
218
Теоретические основы электрохимии
Исследование влияния поверхностно активных веществ на смачиваемость катодного цинка электролитом показало, что добавки, адсорбируясь на катоде, весьма существенно меняют величину краевого угла, при этом полярно асимметричные гидрофобные органические вещества увеличивают краевой угол, гид-рофилльные уменьшают его.
Если краевой угол мал, что указывает на хорошую смачиваемость поверхности металла электролитом, то пузырек газа легко отрывается от металла, не успев вырасти. Наоборот, с ростом краевого угла периметр прикрепления пузырька к поверхности катода увеличивается, благодаря чему его размеры становятся значительными. Следствием этого является образование ячеистых осадков металла, при этом глубина и размеры ячеек зависят от степени прилипания пузырьков таза к поверхности цинка.
Электрокапиллярные явления на твердых металлах позволили объяснить закономерности, сопровождающие процессы флотации, установить причины разрушения угольных анодов, разъяснить существо электролитического обезжиривания металлов, вскрыть механизм ряда других важных технологических процессов.
§ 4. Потенциалы нулевого заряда и проблема электродвижущей силы гальванического элемента
Изучение потенциалов нулевого заряда, проведенное главным образом советскими электрохимиками, позволило наметить пути разрешения проблемы электродвижущей силы гальванического элемента. Этот вопрос имеет принципиальное значение и заслуживает специального рассмотрения.
Больше 100 лет тому назад было установлено, что если два металла поместить в вакуум и замкнуть их, то между металлами появляется электрическое поле, т. е. возникает разность потенциалов, которая впоследствии получила название контактной разности потенциалов.
Это открытие в то время считали очень важным с точки зрения понимания электродвижущей силы гальванических элементов. В связи с тем что электроды гальванического элемента обычно являются различными металлами, предполагали, что электродвижущая сила гальванического элемента обусловлена контактной разностью потенциалов и равна ей. Таким образом, возникла так называемая контактная теория электродвижущей силы гальванического элемента.
Если расположить металлы в ряд, например по возрастанию их нормального электрода потенциала, то в этом ряду металлы следуют один за другим примерно так же, как если бы мы их
Строение двойного электрического слоя 219
расположили, руководствуясь не их нормальными потенциалами, а соответствующими величинами контактных разностей потенциалов. Это обстоятельство приводилось как доказательство справедливости контактной теории.
Однако указанное соответствие не является полным. В дальнейшем было обнаружено, что действие гальванического элемента связано с определенными химическими реакциями внутри него. Это стали расценивать как факт, опровергающий контактную теорию, по которой электролит не играет существенной роли с точки зрения величины электродвижущей силы гальванического элемента.
Открытие зависимости величины электродного потенциала от концентрации соответствующих ионов в растворе окончательно поколебало позиции контактной теории.
А. Н. Фрумкин впервые совершенно конкретно сформулировал роль ионного обмена в образовании электродного потенциала. Ионный обмен участвует в создании электродного потенциала наряду с контактной разностью потенциалов, причем процесс ионного обмена протекает таким образом, что значение электродного потенциала отвечает термодинамическому равновесию между металлом и электролитом.
С этой точки зрения представляет интерес установление зависимости потенциала электрокапиллярного максимума от природы металлической фазы.
В классической электрохимии предполагалось, что после удаления ионного двойного слоя с границы металл — раствор разность потенциалов сведется к абсолютному нулю. Так, например, считалось, что абсолютным нулем потенциалов является потенциал (tpjv), соответствующий максимуму электрокапиллярной кривой для ртути. Однако из предыдущего должно быть ясным, что если с поверхности раздела фаз металл — раствор удалить все заряды и затруднить адсорбцию посторонних поверхностно активных веществ, то и в этом случае нельзя исключить адсорбцию молекул растворителя, которые, обладая достаточным дипольным моментом, могут создать заметный скачок потенциала на границе фаз.
Кроме того, с помощью электрокапиллярных исследований и другими методами А. Н. Фрумкин показал недостаточность представления Нернста о том, что скачок потенциала целиком локализован на границе фаз электрод — электролит.
Нельзя забывать о контактной разности потенциалов, которая обусловлена различным уровнем электронов в металлах. Для разных металлов точки нулевого заряда различны. Так, например, разность между <pw для ртути и для жидкого таллия составляет около 0,4 в. Растворение металлов в ртути сдвигает положение q>N. Разность между <pw для чистой ртути и для 12%-
220 Теоретические основы электрохимии
ной амальгамы таллия составляет 0,33 в. Таким образом, потенциал на границе фаз — величина сложная. В нее входят значение величины скачка, обусловленного существованием электронов в металле и ионов в растворе, значение величины скачка, вызванного адсорбцией поверхностно активных ионов и молекул из раствора, и, наконец, контактная разность потенциалов.
Исследования абсолютных скачков потенциалов показали, что для каждого металла имеются свои точки нулевого заряда, положение которых характерно для данного электрода.' Этот важный вывод положен в основу специальных исследований электродвижущей силы и природы ее возникновения. Положение нулевых точек для ряда металлов было определено количественно (табл. 17).
Таблица 17. Потенциалы нулевого заряда <р д’
Электрод Состав раствора Метод измерения
Кадмий Cd 10-3-н. КС1 —0,9 II
Таллий Т1 10-3-н. КС1 —0,8 II
Свинец РЬ 10~3-н. КС1 —0,69 II, IV
Таллий, насыщенная амальгама Tl(Hg) 1-и. Na2SO4 —0,65 I
Цинк Zn 1-н. Na2SO4 —0,63 VI
Галлий Ga 1-н. КС1—0,1-н. НС1 —0,6 I
Ртуть Hg Разбавленные неактивные электролиты —0,48 I,II,III
Графит С 0,05-н. NaCl —0,07 VI
Серебро Ag 0,1-н. KNO3 —0,05 V
Медь Си —0,04 V
Уголь активированный С 1-н. Na2SO4-H-H. H2SO4 0,0—0,2 V
Платина в атмосфере J-н. Na2S04+0,01-H. H2SO4 ( 0,11 V
водорода Pt(H2) 1.0,27 IV
Теллур Те Гн. H2SO4 0,61 VI
Платина окисленная Pt(O2) 1-н. Na2S04-|-0,01H. H2SO4 0,4—1,0 V
Основываясь на указанных фактах, А. Н. Фрумкин высказал предположение, согласно которому разность потенциалов между электрокапиллярными максимумами различных металлов обусловлена контактной разностью потенциалов между этими металлами. Это наблюдение весьма существенно с точки зрения теории электродвижущей силы гальванического элемента. Очевидно, электродвижущая сила любого гальванического элемента будет равна разности потенциалов нулевых точек металлов (образующих электроды гальванического элемента) плюс некоторый концентрационный член, который возникает вследствие
Строение двойного электрического слоя
221
разности концентраций соответствующих солей в растворах около электродов, отличающихся от концентраций и растворов, отвечающих точкам нулевого заряда.
Из изложенного следует, что приведенные соображения Фрумкина способствуют решению проблемы о роли контактной разности потенциалов в образовании электродвижущей силы гальванического элемента. Опыты подтвердили эту точку зрения.
Если контактная разность потенциалов близка к разности потенциалов между нулевыми точками различных металлов, то очевидно, что эта последняя величина не должна зависеть от природы электролита.
С. В. Карпачев и А. Г. Стромберг исследовали электрокапил-лярные явления на ряде металлов. Чтобы увеличить число металлов, доступных исследованию, измерения производили при высоких температурах, причем в качестве электролита применяли расплавленную смесь хлоридов калия и лития.
В табл. 18 приведены значения потенциалов, отвечающих
Таблица 18. Потенциалы нулевого заряда <р fj, соответствующие максимуму электрокапиллярных кривых, для некоторых расплавленных и твердых металлов
Металл Потенциал точки нулевого заряда
по С. В. Карпачеву и А. Г. Стромбергу по П. А. Ребиндеру и Е. К- Венстрем
Ртуть' 0,00 0,00
Т еллур . +0,70 +0,70
Свинец —0,37 —0,37
Цинк . —0,45 —0,42
Галлий —0,57 —0,49
максимуму электрокапиллярной кривой для металлов в расплаве КС1 + LiCl при 420—450 °C, по данным С. В. Карпачева и А. Г. Стромберга, и для твердых металлов в 1-н. растворе H2SO4, по данным П. А. Ребиндера и Е. К. Венстрем.
Как видно, в пределах точности измерений такое глубокое изменение природы электролита, как переход от водного раствора к расплавленной соли, не меняет разности потенциалов между электрокапиллярными максимумами различных металлов.
Для однозначного подтверждения теории необходимо было сравнить разность потенциалов между нулевыми точками двух металлов с величиной контактной разности потенциалов между этими металлами.
Для решения этой задачи С. В. Карпачев и А. Г. Стромберг измерили контактную разность потенциалов между ртутью и 12%-ной амальгамой таллия, М. В. Смирнов — контактную раз-
222
Теоретические основы электрохимии
ность потенциалов между ртутью и 4%-ной амальгамой кадмия. Результаты этих измерений удовлетворительно совпадали с разностями потенциалов между нулевыми точками ртути и соответствующей амальгамы.
М. В. Смирнов измерил контактные разности потенциалов: между следующими парами жидких металлов: олово — свинец, висмут — таллий, олово — таллий.
Приводим результаты этих измерений с указанием разностей потенциалов между электрокапиллярными максимумами этих металлов.
Sn — Pb Bi — T1 Sn — T1
Разность потенциалов между электрокапиллярными максимумами .... 0,24
Контактная разность потенциалов . . 0,28
0,35 0,42
0,36 0,46
Как видно, совпадение чисел первой и второй строки вполне удовлетворительно, т. е. контактная разность потенциалов между металлами близка к разности потенциалов между точками нулевых зарядов этих металлов.
Следует, конечно, иметь в виду, как это указывал А. Н. Фрумкин, что контактная разность потенциалов совпадает с разностью потенциалов между нулевыми точками металлов только в том случае, если адсорбционные эффекты на обоих металлах либо отсутствуют, либо, что вероятнее, примерно одинаковы, в силу чего и компенсируют один другой.
Потенциал нулевого заряда данного металла можно выразить не только из максимума электрокапиллярной кривой, но и как потенциал электрода в «нулевом растворе», концентрация которого такова, что мета-лл, погруженный в него, не будет отдавать своих ионов и ионы из раствора в свою очередь не будут разряжаться на металле. В самом деле, если опустить в раствор AgNO3 с концентрацией С серебряную пластинку и если в этом растворе, кроме основного электролита, присутствует соль с общим анионом, например KNO3, то при С > Со (где Со — «нулевая концентрация») ионы серебра будут разряжаться на пластинке. При этом пластинка зарядится положительно и притянет эквивалентное количество NO3_-hohob.
Таким образом, вследствие потенциалопределяющей адсорбции соли в растворе как бы исчезнет некоторое количество AgNO3. Если С < Со, то, наоборот, серебро будет растворяться, выделяя Ag+-HOHbi и заряжаясь электроотрицательно. Этот заряд будет компенсироваться ионами К+ и таким образом в растворе возрастет концентрация AgNO3. При С = Со концентрация AgNO3 не будет изменяться. Определив Со экспериментально, можно было бы по уравнению Нернста вычислить потенциал нулевого заряда. Однако неопределенная зависимость
Строение двойного электрического слоя 223
потенциала электрода от адсорбции ионов на его поверхности не позволяет сделать это определение со всей строгостью.
Мы видели, что нулевая точка, соответствующая максимуму электрокапиллярной кривой, как бы разграничивает области потенциалов, соответствующих положительно и отрицательно заряженным поверхностям металла, а следовательно, представляет собой границу раздела с преимущественной адсорбцией катионов или анионов из раствора, в котором этот металл находится. Это обстоятельство оказывает большое влияние на кинетику неравновесных электродных процессов, определяя их характер и направление.
Для определения потенциалов нулевого заряда были применены следующие методы (см. табл. 17).
I. Метод максимума электрокапиллярной кривой. В точке максимума = 0; следовательно, плотность заряда е = 0 и потенциал <р, измеренный против нормального водородного электрода, соответствует точке нулевого заряда (см. § 2).
II. По емкости двойного электрического слоя. Вблизи точки нулевого заряда двойной слой обладает наибольшей диффузно-стью (см. § 5).
III. По кривым заряжения. Зависимость потенциала от количества электричества, пропущенного через электрод, позволяет установить емкость электрода, а следовательно, и потенциал нулевого заряда.
IV. По изменению краевого угла пузырька на поверхности металла в зависимости от потенциала поляризации (см. § 3).
V. По исследованию адсорбции потенциалопределяющих ионов. В нулевом растворе не будет наблюдаться ни положительная, ни отрицательная адсорбция. Потенциал, который будет устанавливаться на металле в нулевом растворе, соответствует потенциалу нулевого заряда.
VI. По исследованию твердости металлов с использованием методики П. А. Ребиндера (см. § 2).
Во всех случаях значения <pw отнесены к нормальному потенциалу водородного электрода.
§ 5. Емкость двойного слоя и ее измерение
Скачок потенциала в двойном слое определяется уравнением (VIII, 1).
Если же учесть, что диэлектрическая постоянная между обкладками двойного слоя в растворе отлична от единицы, то
224 Теоретические основы электрохимии
Емкость такого двойного слоя С, т. е. соотношение между плотностью заряда на единицу поверхности е и разностью потенциалов между его обкладками Дер:
С= ——. (VI 11,39)
4тгВ
Величина С, как правило, выражается в микрофарадах на квадратный сантиметр и поэтому, если б выражена в сантиметрах, то
С =------—----мкф/см?-. (VI 11,40)
4 г 89 • 10s
Очевидно, что величина этой емкости в первую очередь зависит от толщины двойного слоя, т. е. от того, на какое расстояние сближаются между собой противоположные заряды по обе стороны границы раздела.
В зависимости от того, какие ионы находятся в жидкостной обкладке двойного слоя, расстояние между обкладками б будет различным, а следовательно, будет меняться и емкость. Если предположить, что б — величина одного порядка с радиусом ионов, т. е. 10~8 см, а диэлектрическая постоянная воды D = 81, то для случая катионов в двойном слое С = 16—20 мкф!см2. При положительном заряде металла, т. е. когда в двойном жидкостном слое находятся анионы, емкость обычно в два раза больше. Это объясняется более легкой деформируемостью гидратной оболочки анионов и поляризацией самих ионов, что позволяет центрам зарядов ионов подойти ближе к металлической обкладке и таким образом увеличивает емкость.
Адсорбция молекулярных соединений приводит к уменьшению емкости двойного слоя, что зависит от увеличения толщины двойного слоя б вследствие внедрения в него молекул, адсорбировавшихся на электроде, и некоторого уменьшения диэлектрической постоянной D.
Толщина б двойного слоя зависит не только от природы ионов, его образующих, но и от величины электродного потенциала <р. Чем больше разность потенциалов между обкладками двойного слоя, тем большая электрическая деформация ионов возможна в двойном слое. При достаточно большом положительном заряде двойной слой построен из сильно деформированных отрицательных ионов. Затем поверхность металла перезаряжается, что приводит к росту толщины двойного слоя и уменьшению емкости. Вблизи от точки нулевого заряда электродный потенциал <р мал, деформация ионов ничтожна и в значительном промежутке остается практически постоянной. При переходе через qjjv анионный двойной слой исчезает и заменяется слоем катионов.
Строение двойного электрического слоя
225
Наибольшей диффузностью двойной слой обладает вблизи точки нулевого заряда. Метод измерения емкости двойного слоя позволяет исследовать изменения, происходящие в двойном электрическом слое, в частности кинетику адсорбции поверхностно активных веществ, деформацию ионов под влиянием электрического поля, изменение толщины двойного слоя при адсорбции атомов и молекул. Сравнительное изучение поведения ряда металлов в водных растворах показало, что строение ионного двойного слоя относительно мало зависит от природы металла. Вместе с тем определение значения емкости двойного слоя помогает судить о строении и истинной поверхности металлического электрода. Измерения емкости в разбавленных растворах позволили, например, непосредственно проверить на опыте теорию диффузионного строения двойного слоя и определить величину потенциала фь создаваемого частью двойного слоя, находящейся на расстоянии одного ионного радиуса от поверхности электрода.
Сопоставление потенциалов фь полученных из опытных данных в разбавленных растворах и рассчитанных по уравнению Штерна, показывает, что теория Штерна достаточно хорошо согласуется с опытом:
Электродный потенциал <р, в
—0,1
—0,2
—0,3
Опытные
41, «
—0,076
—0,123
—0,141
Расчетные ф, (из уравнения Штерна), в
—0,085
—0,147
—0,177
Емкости двойного слоя на границе металл — раствор измеряют переменным или постоянным током в условиях, когда исключена возможность протекания электрохимических реакций на поверхности металла.
Дело в том, что при пропускании тока часть его затрачивается на электрохимический процесс («фарадеевский» ток), часть же — на изменение заряда двойного слоя, если потенциал электрода меняется (нефарадеевский ток).
В малополяризующемся электроде преобладает первый процесс, в сильнополяризующемся — второй процесс. Емкость двойного слоя определяется так же, как емкость обычного конденсатора, имеющего утечку. Емкости конденсатора соответствует емкость двойного слоя, а сопротивлению утечки — величина, обратная скорости электродной реакции в процессе электролиза. Емкость удобно измерять в таких условиях, когда практически в конденсаторе нет утечки, т. е. когда- сила фарадеевского тока незначительна по сравнению с нефарадеевским.
Измерение емкости впервые было применено Боуденом и Райдилом для определения истинной поверхности никеля, серебра и платины. А. Шлыгин и А. Н. Фрумкин для ликвидации уте-15 Заказ 1338
226 Теоретические основы электрохимии
чек в конденсаторе измеряли емкости платины при пропускании слабого тока, что значительно упростило эксперимент и повысило точность результатов.
В методе Боудена и Райдила ликвидация утечки достигается тем, что заряжение производят быстро, при большой плотности тока.
Шлыгин и Фрумкин применяют электрод с большой поверхностью и из раствора удаляются неиндифферентные газы. Б. Эрш-лер использует чрезвычайно малое количество электролитов (несколько кубических миллиметров), а следовательно, и ничтожное количество деполяризатора.
Измерение емкости двойного слоя постоянным током с целью определения истинной поверхности электрода удачно использовали Б. Кабанов и Р. Юдкевич. Авторам удалось определить величину истинной поверхности свинцовых электродов. Сущность метода заключается в том, что снимают кривые заряжения свинцового электрода и по количеству протекшего электричества судят о величине емкости двойного слоя электрода.
Для определения истинной поверхности электрода емкость двойного слоя измеряют методам переменных токов. Этот метод используется в том случае, когда хотят определить истинную плотность тока при электроосаждении металлов. Однако здесь могут быть ошибки, заключающиеся в том, что из рассчитанной емкости получается общая величина поверхности, состоящая из растущих и нерастущих участков, поэтому рассчитанная плотность тока не соответствует истинной.
Для определения поверхности по измеренной емкости требуется, чтобы осадок металла был равномерным и мелкокристаллическим.
Метод измерения емкости с переменным током требует несколько более сложной аппаратуры, но имеет то преимущество, что не нуждается в длительной подготовке электрода, так как допускает более сильный «фарадеевский» ток. Применение обычного синусоидального переменного тока позволяет производить вычисления по простым закономерностям переменного тока. Особенно простое устройство можно применять в том случае, когда измеряется «чистая» емкость, т. е. когда сдвиг фазы потенциала от фазы силы тока в измеряемой цепи близок к 90 °C.
В основе применения метода лежит тот факт, что межфазовая граница электрод — раствор, на которой протекает электрохимическая реакция, может быть с известным приближением уподоблена плоскому конденсатору с утечкой. Величина утечки определяется скоростью электродной реакции. В общем случае граница электрод — раствор эквивалентна электрической схеме, изображенной на рис. 79, а. Величина Z в этой схеме представляет собой комплексное сопротивление реакции, включающее в
• Строение двойного электрического слоя 227
себя активную и реактивную составляющие. В том случае, когда скорость электродной реакции определяется замедленностью стадии разряда (ионизации), величина Z может быть заменена чисто активным сопротивлением (рис. 79, б).
Величины С и R (рис. 79, б) могут быть определены экспериментально с помощью мостовых или иных схем, применяющихся в электротехнике для такого рода измерений. Существенно, что при таких измерениях в раствор всегда вводится вспомогательный электрод, также представляющий собой конденсатор
Рис. 79. Электрические схемы, эквивалентные электрохимической ячейке.
В схеме д:
Сх — емкость испытуемого электрода; Rx — сопротивление утечки испытуемого электрода; Г — гальванометр; Со — известная емкость; Ro, Ri, jR2 — известные сопротивления
с утечкой и характеризуемый, следовательно, своими определен-ными значениями емкости (С') и сопротивления (R'). Между рабочим и вспомогательным электродами находится электролит, обладающий активным сопротивлением г. Полная схема ячейки показана на рис. 79, в.
С экспериментальной точки зрения важно, что имеется возможность выбрать вспомогательный электрод с достаточно малыми значениями емкостного и активного сопротивлений. В этом случае соответствующими величинами можно пренебречь и считать ячейку эквивалентной схеме, изображенной на рис. 79* г. Практически для этого достаточно, чтобы площадь вспомогательного электрода была значительно (например, в Г00 раз) больше площади рабочего электрода.
Величину г можно до известного предела уменьшить, сокращая расстояние между рабочим и вспомогательным электродами.
15*
228 Теоретические основы электрохимии
На практике обычно так и поступают, после чего либо вовсе пренебрегают сопротивлением г, т. е. считают ячейку эквивалентной схеме (рис. 79, б), либо вводят поправку на это сопротивление, предварительно определив его величину экспериментальным или расчетным путем. При этом в обоих случаях возникают некоторые погрешности в определении Си/?, ограничивающие возможности измерений, особенно на высоких частотах.
Примерная схема измерений приведена на рис. 79, д. На такой схеме можно измерить раздельно емкость Сх и сопротивление Rx утечки испытуемого электрода. По достижении равновесия моста в точке компенсации наблюдаются следующие равенства:
2?°-= 2L = . (VIII,41)
Их Иъ Со
Необходимо следить за тем, чтобы на испытуемом электроде не могли протекать быстрые реакции, т. е. чтобы ток, проходящий через ячейку, затрачивался только на заряжение двойного слоя.
§ 6. Электрокинетические явления
Во всякой системе, содержащей несколько противоположно заряженных фаз, при наложении внешнего электрического поля эти фазы смещаются в противоположных направлениях; в этом и кроется существо так называемых электрокинетических явлений. Скорость такого рода смещений зависит от ряда причин — величины наложенного поля, формы и размеров фаз, их природы, заряда частиц и т. п.
Все электрокинетические явления по своим внешим признакам могут быть разделены на две основные группы:
1. Электрофорез или катафорез — перемещение дисперсных частиц твердой фазы относительно жидкости или газа при градиенте потенциала, не равном нулю.
2. Электроосмос — перемещение дисперсной среды относительно твердой фазы (стенки капилляра), при градиенте потенциала, не равном нулю.
Взвешенные частицы, подвергающиеся электрофорезу, могут быть твердыми, жидкими, газообразными. Наиболее часто мы встречаемся с электрокинетическими явлениями при исследовании коллоидных систем. Жидкости, подвергающиеся электроосмосу, также могут быть самыми разнообразными (вода, спирт, керосин, ацетон, толуол и т. n.J^
В зависимости от природы жидкости, материала твердой фазы и других условий электрофорез (или электроосмос) может идти в направлении катода и в направлении анода. Направление
Строение двойного электрического слоя
229
движения определяется знаком заряда фаз и примесями (в жидкостях). Особенно сильное влияние оказывают в этом отношении электролиты. В некоторых случаях под воздействием введенных в испытуемые растворы электролитов направление движения может стать обратным по отношению к начальному движению в чистой жидкости.
При объяснении явлений электроосмоса и электрофореза исходят из того, что при соприкосновении двух различных по природе веществ на границе их раздела возникает заряд. Например, на границе фаз жидкость — твердое тело жидкость приобретает положительный заряд, а твердое тело — отрицательный.
Общим для определения заряда фазы является следующее правило.
При соприкосновении двух диэлектриков, находящихся в различном агрегатном состоянии, электроположительным становится тот из них, который имеет большую диэлектрическую постоянную. Возникающая разность потенциалов пропорциональна разности диэлектрических постоянных соприкасающихся веществ. Так, например, вода, обладающая большой диэлектрической постоянной (D ~ 80), соприкасаясь с различными веществами, приобретает положительный заряд и, следовательно, при электроосмосе движется к катоду. Однако это правило соблюдается только при употреблении чистых жидкостей и может утратить свое значение в присутствии посторонних электролитов.
Из закономерностей электрофореза вытекает, важный практический вывод, что скорость движения взвешенных в жидкости частиц не зависит от их размера и в изученных случаях находится в пределах от 10 до 40-10-5 см)сек. Эта величина близка к подвижности простых неорганических ионов (кроме ионов Н+ и ОН-). Скорость не зависит также от заряда частиц. Хевеши объясняет это тем, что отношение — (где е — плотность заряда, г — радиус частицы) для всех заряженных частиц жидкости одинаково. В жидкой среде каждая малая частица (ион или коллоидная частица) заряжается до некоторого постоянного потенциала, который для воды равен 0,07 в. При допущении таких весьма приближенных расчетов, действительно, все частицы должны двигаться с одинаковой скоростью.
Можно было ожидать, что если движущиеся частицы имеют, например, металлический характер, то величина £ диффузной части двойного электрического слоя должна равняться скачку потенциала на границе металл — раствор, т. е. термодинамическому потенциалу <р. Однако на самом деле это не так. Опыт показывает, что значение £ отличается от соответствующих электродных потенциалов.
В процессе определения электрокинетического потенциала на
230
Теоретические основы электрохимии
границе раздела фаз стекло — раствор при движении жидкости вдоль стенок стеклянной трубки выяснились, например, существенные различия между и ф-потенциалами.
Как следует из данных табл. 19, значения £ отличаются от соответствующих электродных потенциалов не только по величине, но и по знаку.
Таблица 19. Значение Z потенциала и скачка потенциала <р на границе раздела фаз стекло — растворы BaCh и Th('NOs)4
Раствор Концентрация моль[л Потенциалы, лее
с | «р
ВяС1г 10~8 —16 + 140
ВаСЦ 10~4 — 4 + 92
Th(NO3)4 10~8 — 5 + 137
Th(NO8)4 10“4 + 5 + 50
Известно далее, что потенциал ф на границе стекло — раствор резко меняется в зависимости от концентрации ионов Н+ в растворе, что закономерно, если вспомнить, что стеклянный электрод ведет себя подобно водородному, эти же водородные ионы почти не влияют на величину электрокинетического потенциала. Наоборот, наличие ряда других ионов, почти не изменяющих величину термодинамического потенциала ф, чрезвычайно резко влияет на ^-потенциал. Главную роль при этом играют зарядность и знак посторонних ионов, а также их электро-рапиллярные свойства.
Эти явления объясняются теорией двойного электрического слоя.
Из всех электрокинетических явлений наиболее изучен электрофорез, играющий особенно важную роль при изучении коллоидных растворов. Здесь мы можем непосредственно следить за изменением электрических свойств самих коллоидных частиц. Даже чисто качественные наблюдения за движением взвешенных частиц позволяют установить знак ^-потенциала. Непосредственное наблюдение позволяет уловить (что особенно важно) тот момент, когда этот потенциал становится равным нулю, что соответствует изоэлектрической точке, в которой оказывается коллоидная система.
Электроосмотические явления, относительно менее распространенные по сравнению с электрофорезом, также были изучены рядом авторов. При этом было показано следующее.
1. Если жидкость не содержит каких-либо ионов, а твердая фаза их не выделяет, то между жидкостью и твердой фазой возникает контактная разность потенциалов, причем положитель
Строение двойного электрического слоя
231
ный заряд получает та фаза, диэлектрическая постоянная которой больше.
2. Если твердая фаза способна выделять ионы, а раствор содержит их (электролит), то твердая фаза получает заряд со
знаком, противоположным заряду выделяемых ионов.
А. Н. Фрумкин и В. Г. Левич подробно изучили особенности
движения ртутных капель в жидкости при наложении внешне
го поля. Движение такого рода частиц отличается от обычных электрофоретических передвижений тем, что в данном случае как бы совмещаются электрокинетический и элект-рокапиллярный эффекты. В результате увеличения и уменьшения поверхностного натяжения в различных час-। тях ртутной капли возникают добавочные электрокапиллярные течения в жидкости, кото
Рис. 80. «Реактивное» движение в ртутной капле
рые в свою очередь сообщают
капле дополнительную («реактивную») скорость при ее движении в направлении поля (рис. 80).
Величина этой скорости движения, добавляющаяся к общей скорости электрокинетического движения, была теоретически рассчитана. В общем случае она равна
(21+3^) 1+ — +--------
к 2рх к. /
где р — сопротивление на границе металл — раствор;
т), т]1 — вязкость жидкости и ртути соответственно.
Если считать ртутную частицу идеально поляризуемой, т. е.
° л полагать -------- = О, то
2рх
V = -------—------— . (VI 11,43)
j е2
+ х (2>; + Зъ)
Явления электрофореза и электроосмоса приобрели большое прикладное значение и применяются при решении важных практических задач, которые не удавалось осуществить иными методами (электролитическое обезвоживание торфа и очистка воды и растворов от взвешенных примесей, обезвоживание коллоидных веществ и кашеобразных смесей, с трудом поддающихся фильтрованию, очистка коллоидов от посторонних примесей, раз
232 Теоретические основы электрохимии
деление водномасляных эмульсий, пропитка тканей, дубление кожи и т. п.).
Для технического осуществления электрофореза или электроосмоса необходимо, чтобы обрабатываемое вещество находилось в таком состоянии, когда скачок потенциала на границе фаз по возможности велик и поэтому электроосмотическое действие резко выражено. Этого можно добиться добавлением к жидкой фазе соответствующих электролитов в определенных концентрациях. Назначение электролитов — увеличить заряд частиц, т. е. стойкость коллоида, или увеличить скорость электроосмотического протекания жидкости через диафрагму.
Следует, однако, заметить, что промышленное использование электрокинетических явлений ограничивается растворами с весьма малой удельной электропроводностью. Это может быть объяснено тем, что в жидкостях, содержащих большое количество примесей, т. е. обладающих достаточной электропроводностью, наряду с электроосмосом или электрофорезом, будет происходить электролиз, что приведет к осложнениям.
Не меньшее значение приобретают в последнее время элект-рокинетические явления и для развития физико-химической и коллоидной науки. Так, упомянутая выше теория движения ртутных капель оказалась полезной в полярографии и других физико-химических методах анализа с применением капельного ртутного электрода.
Значительную роль сыграли исследования электрокинетических явлений в построении современной теории скачка потенциала на границе фаз. Опыты с частицами угля и платины позволили выяснить, в какой мере электролитические явления коллоидных систем связаны с величиной общего скачка потенциала на границе фаз. Так, например, старые представления Нернста, Гельмгольца и других не могли дать ответа на вопрос о том, почему при возникновении двойного электрического, слоя на границе фаз, кроме термодинамического, потенциала <р, появляется электрокинетический потенциал £. Более точное количественное изучение коллоидных систем и строения двойного слоя позволило не только обнаружить, но и вычислить величину ^-потенциала.
ЛИТЕРАТУРА
Адам Н. К. Физика и химия поверхностей. ИЛ, 1947. Антропов Л. И. ЖФХ, т. 25, в. 12, 1951, стр. 1494.
Бор и'с о в а Т. Н., Э р ш л е р Б. В. ЖФХ, т. 24, 1950, стр. 337.
Есин О. А., Никитин Ю. П., П one ль С. И. ДАН СССР т. 87, 1952, стр. 813.
Кабанов Б. Н. Электродные процессы и строение двойного слоя (приложение к книге С. Гиесстона «Введение в электрохимию»), ИЛ, 1951.
Строение двойного электрического слоя 233
К а р in а ч е в С. В., С тр о м б е р г А. Г. Труды Института химии УФАН СССР. Изд. УФАН СССР, 1952, № 4, стр. 3.
Кройт Г. Р. Наука о коллоидах. ИЛ, 1955.
К у и и н Л. Л. Поверхностные явления в металлах. Металлургиздат, 1955.
Левин А. И., Колеватова В. С., Мокрушин С. Г. Коллоидный журнал, т. 15, 1953, стр. 252.
Левич В. Г. Физико-химическая термодинамика. Изд. АН СССР, 1952.
Липатов С. М. Физико-химия коллоидов. Госхимиздат, 1948; Некоторые проблемы современной электрохимии (под ред. Дж. Бокриса). ИЛ, 1958.
Плесков В. А. Электродные (потенциалы и (Энергия гидратации. Успехи химии, 16, 1947, 254.
Ре бинде'р П. А., Венстрем Е. К. ЖФХ, т. 49, 1945, № 1.
Т.емкЦ.н И. М. Изв. АН СССР, т. 3, .4947, стр. 235.
Фрумкин А. Н. Изв. АН СССР, т. 1, 1945, стр. 230.
Хейфец В. Л., Авдеев Д. К., Рейшахрит Л. С. Практикум по теоретической электрохимии. Изд. ЛГУ, 1954.
Ч мутов К. В. Техника физико-химического исследования. Госхимиздат, 1954. Эршлер Б. В. Успехи химии, т. 21, 1952, стр. 237.
Глава IX
ПРИЧИНЫ ВОЗНИКНОВЕНИЯ ЭЛЕКТРОДНОЙ ПОЛЯРИЗАЦИИ
И МЕТОДЫ ЕЕ ЭКСПЕРИМЕНТАЛЬНОГО ИЗУЧЕНИЯ
§ 1. Ток обмена. Напряжение разложения водных растворов электролитов. Остаточный ток
Если металлический электрод опустить в раствор, содержащий его ионы, то при отсутствии тока между металлом и раствором устанавливается равновесие. Однако это равновесие we означает наличия полного покоя, так как некоторое количество ионов 'из металла может переходить в раствор, оставляя свободные электроны. Одновременно с этим такое же количество ионов из раствора будет разряжаться на металле, захватывая электроны.
Таким образом, при равновесии, когда измеряемый внешний ток равен нулю, между металлом и раствором в обоих направ-лениях непрерывно протекает некоторой ток i и г, причем
Г-?=г0, (IX, 1)
где io — ток обмена.
Обмен при фр может быть обнаружен методом радиоактивных индикаторов. Так, если взять металл, содержащий некоторое количество радиоактивного изотопа, и погрузить в раствор своей соли, то через некоторое время в растворе, ранее не содержащем меченых атомов, можно обнаружить радиоактивные ионы.
К такому же выводу приводит обратный опыт с металлом, не содержащим меченых атомов, при погружении его в раствор собственной соли, где имеются радиоактивные ионы металла. В таком случае в поверхностном слое металла появляется радиоактивность. Такие измерения были выполнены В. А. Плесковым и Н. Б. Миллером. Величина тока обмена—важная электрохимическая характеристика электрода. Понятие о токе обмена в принципе можно распространить на скорости реакций при лю-
Причины возникновения электродной поляризации 235
бом 'подвижном равновесии, в том числе и при обратимых окислительно-восстановительных реакциях.
Во всех случаях величина тока обмена выражает количество электричества, участвующее в единицу времени в итоге обмена, а влияние природы электрода проявляется в значении равновесного потенциала фР'
Рассмотрим обратимую электрохимическую систему, состоящую из двух одинаковых медных электродов, погруженных в один и тот же водный раствор сульфата меди:
Cu|CuS04, H2O|Cu.
Несмотря на отсутствие внешнего тока, здесь на электродах появятся токи обмена, соответствующие скорости разряда и ионизации ионов меди. При установившемся равновесии Cu^Cu2+ + 2e на границе фаз возникнут вполне определенные, равные скачки потенциалов. Таким образом, в рассматриваемой системе суммарные токи, проходящие через всю систему, при разомкнутой цепи и в случае замыкания электродов равны нулю. Если же на эту систему приложить некоторое напряжение извне, то вследствие обратимости таких электродов электролиз должен начаться при предельно малом внешнем напряжении. Это означает, что в данном случае достаточен минимальный сдвиг потенциалов от их равновесного значения, чтобы на катоде начался процесс преимущественного разряда ионов меди, а медный анод начал растворяться.
Потенциал, при котором медь будет переходить в раствор при некотором токе (ток ионизации), называется потенциалом растворения металла. На катоде медь будет выделяться при потенциале разряда иона (Си2+).
Так как величина приложенного извне напряжения в рассматриваемом случае из-за бесконечно малой силы тока весьма близка к величине обратимой э. д. с. элемента (при разомкнутой цепи), это напряжение имеет вполне определенную величину и может быть рассчитано термодинамическим путем через равновесные потенциалы по формуле Нернста либо по уравнению Гиббса — Гельмгольца. На аноде медь будет переходить ;в раствор при некотором токе i„ (ток ионизации), а на катоде процесс разряда катионов меди протекает при токе разряда ip. Величина поляризующего тока на катоде будет равняться разности плотностей токов разряда и ионизации, т. е.
iK^Tp—X,. (IX,2)
Ток анодной поляризации будет в свою очередь равен разности скоростей ионизации и разряда, пропорциональных их плотностям:
(IX,3)
236
Теоретические основы электрохимии
Таким образом, при поляризации электродов равновесие нарушается и число образовавшихся в единицу времени ионов уже не равняется количеству исчезнувших. В том же случае, когда: iK = ia = 0, нетрудно заметить, что ip = iH = i0.
Ток обмена (io), возникающий в результате реакций образования и разряда некоторых металлических ионов (типа меди), часто достигает большой величины. По этой причине для изучения такого рода реакций применяют обычно методы переменноточных измерений, так как в случае использования постоянного' тока возникают серьезные затруднения, связанные с необходимостью учета концентрационных изменений и омических потерь в приэлектродных слоях.
Есть, однако, инертные электроды (Pt, Pd, Au и некоторые другие), которые не посылают своих ионов в раствор и имеют вследствие этого исчезающе малый ток обмена. Различное поведение электродов и неодинаковая их поляризуемость связаны с величиной тока обмена. Опыт показывает, что чем меньше ток обмена, тем больше поляризация электрода.
Поляризация электродов приводит к нарушению равновесия,, вследствие чего число и характер образующихся в единицу времени ионов будет заметно отличаться от количества ионов, исчезнувших в тот же промежуток времени. На инертных электродах, погруженных в растворы H2SO4 и NaOH, электролиз может быть осуществлен только в том случае, если приложенное, извне напряжение достигает некоторой величины:
^.p-=?a-<PR. (IX,4 >
Здесь t/H.p — напряжение разложения электролита;
Фа и фк — анодный и катодный потенциалы.
Величину напряжения в системе с применением инертных электродов в момент, когда на них (или на одном из них) появляются газообразные продукты электролиза, называют обычно напряжением разложения (Пн.р).
Напряжение разложения определяется экспериментально путем снятия кривой величина тока — напряжение. Первоначально через ячейку проходит весьма малый остаточный ток, при этом на электродах не наблюдается видимого выделения продуктов электролиза — газообразных водорода и кислорода. Далее, по достижении соответствующей величины напряжения разложения ток в ячейке быстро возрастает и на электродах наблюдается газовыделение. Таким образом, заметный ток появится в системе только в том случае, когда приложенное извне напряжение достигнет разности потенциалов выделения продуктов электродных реакций (IX,4).
Принято считать, что напряжение разложения есть то извне приложенное минимальное напряжение, которое необходимо для
Причины возникновения электродной поляризации
237
того, чтобы через данную электрохимическую систему начал лроходить ток значительной силы (рис. 81, точка D).
Величина напряжения разложения всегда больше э. д. с. Явление напряжения разложения было впервые замечено Гельмгольцем и описано подробно Лебланом, который и ввел термин «напряжение разложения». По Леблану, до достижения вели
чины напряжения разложения рис. 81) будет расположена вблизи оси абсцисс, что указывает на прохождение через систему весьма малых «остаточных токов». Остаточные токи в системе вызываются различными причинами, например присутствием таких примесей в растворе, которые могут реагировать на электродах при относительно меньших потенциалах, чем потенциалы выделения кислорода и водорода. Предполагается также, что остаточный ток может быть вызван работой газового
кривая i — U (до точки D на
Рис. 81. Кривая напряжения разложения '(то Леблану)
элемента.
Вследствие протекания электродных реакций и выделения газообразных продуктов электролиза платиновые катод и анод превращаются в водородный и кислородный электроды. Следует иметь в виду, что положение точки D (рис. 81) не остается постоянным и дает лишь некоторую ориентировку для определения величины минимального напряжения, после которого может быть осуществлен электролиз с пропусканием тока значительной плотности.
В настоящее время можно считать, что понятие «напряжение разложения» не имеет определенного физического смысла. Величина t7H.p не может быть рассчитана термодинамическим путем. Действительно э. д. с. кислородно-водородного обратимого элемента составляет 1,23 в, в то время как наблюдаемое при разложении воды t7H.p =1,7 в. Разность в 0,43 в складывается из значений перенапряжений при выделении кислорода (rio) и водорода (т;Нг) на гладких платиновых электродах (аноде и катоде). Если в качестве инертных электродов взять другие, например палладиевые или золотые, то величина напряжения разложения (В'нр) изменится в соответствии с другими значениями и ^н, на таких электродах. Что касается э. д. с. (Ер), то она по-прежнему останется без изменения (1,23 в).
Напряжение разложения растворов кислородсодержащих со
238
Теоретические основы электрохимии
лей щелочных и щелочноземельных металлов на платиновых электродах больше, чем у соответствующих кислот и щелочей, и. составляет ~2,1 в. Последнее обстоятельство объясняется тем, что в процессе электролиза раствор вблизи катода подщелачивается, а анолит подкисляется; следовательно, разряд ионов-водорода происходит из более щелочного, а гидроксильных ионов — из более кислого раствора по сравнению с начальным составом электролита.
Таким образом, если э. д. с. элементов, составленных из обратимых газовых электродов (например, продуктов разложения воды), не зависит от состава раствора и электродов (она равна э. д. с. обратимого кислородно-водородного элемента), то напряжение разложения, напротив, весьма заметно зависит и от материала инертных электродов и от состава принятого электролита.
Величина <ра и <рк необратимых газовых электродов, помимо состава и активности ионов в растворе, зависит от давления газов, насыщающих инертные электроды; потенциал водородного электрода становится отрицательнее, а кислородного — положительнее. Подобное изменение величины потенциалов газовых электродов наблюдается до тех пор, пока давление насыщающего электрод газа не достигнет некоторой постоянной величины, максимальной в данных условиях. Очевидно, что при нормальных условиях это давление рГаз = 1 атм (101325 н1м1').-
В процессе электролиза при каждом новом повышении внешнего напряжения на ячейке величина обратной э. д. с., возникающей в итоге работы кислородно-водородного элемента, также будет возрастать, препятствуя электролизу. Действительно с ростом плотности тока количество насыщающих электроды газов увеличивается, но они не могут собраться в пузырек (ргаз < < I атм) и удалиться в атмосферу, а растворимость их в электролите ограничена. Если бы давление газов на электродах оставалось неизменным, то обратная э. д. с. образовавшегося газового элемента полностью воспрепятствовала бы прохожденик> тока извне. Однако вследствие частичного удаления газов с поверхности электродов из-за диффузии и растворения их в электролите через ячейку проходит весьма небольшой, «остаточный» ток, достаточный для того, чтобы выделенное им количество Нг и О2 компенсировало потери газов. Только в случае, когда давление образующихся на электродах газов сравняется с атмосферным, дальнейший рост величины рк* и рОг станет невозможным, так как продукты электродных реакций — газы будут выделяться в атмосферу. Хотя обратная э. д. с. кислородно-водородного элемента достигает при этом предельной величины, работа газового элемента уже не может препятствовать протеканию электролиза вследствие достижения величины напряже
Причины возникновения электродной поляризации
239
ния разложения. Следует подчеркнуть, что значительное перенапряжение у инертных электродов, применяемых при электролизе, способствует более заметному проявлению остаточного тока, так как для достижения атмосферного давления приходится затрачивать большее напряжение; следовательно, область остаточного тока (до точки D) на i—tZ-кривой (см. рис. 81) зависит и от материала электродов и от продуктов электродной реакции. Так, например, при электролизе водных растворов, содержащих хлориды, остаточный ток слабее, чем в случае кислородсодержащих растворов электролитов. Это связано с величиной т]ск, которая на платине невелика, а растворимость хлора в водном электролите значительно больше растворимости кислорода. Таким образом, значение С7н-р на платиновых электродах при использовании хлорсодержащих электролитов близко к теоретически подсчитанной э. д. с.
Можно, следовательно, сделать вывод, что прохождение постоянного тока через различные электролиты возможно при любом приложенном извне напряжении и непременно связано со смещением потенциалов электродов относительно их равновесных значений, т. е. с электродной поляризацией. Явление поляризации электродов было открыто еще в первой половине XIX столетия исследованиями Э. X. Ленца и А. С. Савельева. Уже в то время было замечено, что в первые моменты электролиза газы не выделяются, хотя ток проходит и напряжение растет.
Таким образом, между обычным и «остаточным» током принципиальной разницы нет. Во всех случаях пропускания через ячейку тока наблюдается торможение электродных реакций, связанное с возникновением или перестройкой двойного электрического слоя на границе между электродом и раствором.
Потенциал электрода при электролизе непрерывно изменяется в зависимости от количества пропущенного электричества или (при поляризации током постоянной силы) от времени. Кривые зависимости потенциала от количества электричества, сообщенного электроду, называются кривыми заряжения. По кривым заряжения можно определить емкость электрода. Так, Р. А. Колли впервые использовал измерения сдвига потенциала электрода при пропускании тока в течение определенного промежутка времени и установил емкость платинового электрода. По данным Колли, она близка к 150 мкф/см2. Правильное толкование эти наблюдения получили только в последние десятилетия.
§ 2. Природа и особенности электродной поляризации
Кинетика электродных реакций — это учение о скоростях и механизме электрохимических процессов, протекающих на границе фаз металл — раствор при прохождении через эту гра
240 Теоретические основы электрохимии
ницу электрической энергии. Механизм процессов, контролируемых активацией, занимает особое место в современных представлениях теоретической электрохимии и имеет большое практическое значение. Изучение кинетики электродных реакций способствует сознательному регулированию, совершенствованию и интенсификации разнообразнейших электродных процессов в техническом электролизе (в электролитных ваннах) и при работе различных источников тока (элементов и аккумуляторов).
Скорости химических реакций выражаются уравнением Аррениуса:
z.'-A'expJ— -^j. (IX,5)
Скорости электродных процессов рассматриваются обычно с применением тех же приемов, что и скорость химических реакций. Но при этом, однако, нужно иметь в виду сложность протекания большинства электрохимических превращений по сравнению с химическими, а также то, что решающая роль здесь принадлежит плотности тока L Процесс разряда ионов, как известно, происходит на фазовой границе электрод — электролит. Таким образом, электродные реакции являются гетерогенными процессами, кинетика которых определяется многими специфическими затруднениями. Помимо собственно разряда, т. е. перехода ионов из одной фазы (раствора) в другую (газ, металл), процесс обычно включает в себя миграцию, диффузию и конвекцию частиц, совместный разряд ионов примесей, некоторое растворение (коррозию) уже осажденного ранее металла и другие, сопутствующие процессу разряда явления, которые осложняют суммарный эффект. Реальная электрохимическая система не может быть правильно истолкована без учета всех явлений, предшествующих элементарному., акту разряда и сопровождающих его. Электродная реакция может быть представлена рядом последовательных стадий, через которые она проходит. Такими стадиями являются:
1) доставка ионов или молекул, участвующих в реакции, к поверхности раздела фаз металл — раствор (или отвод частив от электрода);
2) собственно электрохимическая реакция между ионом и электроном, т. е. элементарный акт разряда или ионизации, который в свою очередь может быть разбит на несколько промежуточных стадий.
В общем случае причиной поляризации является возникновение различия в скоростях передачи электрических зарядов на границе фаз электрод — электролит. Действительно, если в металлическом проводнике ток протекает без существенных торможений, мгновенно, то от металла к электролиту он передается
Причины возникновения электродной поляризации 241
в результате химической реакции, которая осуществляется с конечной и относительно небольшой скоростью. Наблюдаемое при этом изменение величины заряда электродов приводит к смещению равновесных потенциалов, устанавливающихся при отсутствии тока. С ростом плотности тока все большее количество зарядов не успевает нейтрализоваться, т. е. вступить в химическую реакцию, вследствие чего возрастает электродная поляризация.
Скорость стадии, протекающей наиболее замедленно, определяет собой скорость всего процесса и, следовательно, в той или иной степени влияет на характер и величину поляризации. Можно представить себе три случая связи между природой и абсолютной величиной поляризации и скоростью той или иной стадии электродного акта. Рассмотрим эти случаи на примере катодных реакций.
1. Пусть скорость доставки ионов из объема раствора к границе катод — электролит очень мала, в то время как процесс разряда ионов осуществляется с достаточной быстротой. Тогда очевидно, что для обеспечения нужной скорости суммарного процесса (необходимой плотности тока iK) придется затратить некоторую добавочную энергию на преодоление обратной концентрационной э. д. с., которая возникает вследствие замедленного передвижения ионов из глубины раствора к! электроду.
2. Возьмем случай, когда доставка ионов к катоду идет быстро, но скорость разряда невелика; очевидно, что при этом поляризация будет вызвана в основном тем, что металл или другой проводник первого рода будет замедленно передавать реагирующим частицам (молекулам, атомам или ионам) электроны.
3. В общем случае при обеспечении заданной плотности тока следует ожидать, что обе рассмотренные выше стадии протекания процесса имеют ограниченную скорость. В таком случае необходимо затрачивать энергию на доставку разряжающихся частиц к электроду, и на обеспечение достаточной скорости их разряда. Если скорости обеих стадий соизмеримы, то нужно учитывать оба тормозящих процесса. Они и будут совместно определять суммарную электродную поляризацию. Исходя из изложенного, катодную и анодную поляризацию в зависимости от природы их возникновения подразделяют на:
а) химическую поляризацию (или перенапряжение), сосредоточенную на границе раздела или на поверхности электрода и вызываемую замедленностью собственно электродного акта разряда или одной из его промежуточных стадий, и б) концентрационную поляризацию, которая обычно сосредоточена в электролите и является следствием замедленной миграции, диффузии и конвекции ионов, участвующих в электрохимической реакции, к поверхности электрода. (Если электродный процесс
16 Заказ 1338
242
Теоретические основы электрохимии
сводится к растворению металла, то концентрационная поляризация определяется скоростью отвода продуктов реакции от поверхности анода.)
В том случае, когда в реакции участвуют не ионы, а молекулы вещества, у электродов концентрация также изменяется. При этом молекулы не переносятся током и поступают к электроду не под действием электрического поля, а под влиянием осмотических сил, возникающих при разности концентраций т. е. за счет диффузии. В связи с указанным обстоятельством концентрационную поляризацию называют иногда диффузионной.
Экспериментальные исследования показали, что перенапряжение, помимо плотности тока, зависит от природы ионов, участвующих в электродной реакции, от материала электродов, от состава и концентрации посторонних ионов и поверхностно активных веществ, находящихся в растворе электролита. Оно зависит также от многих других факторов электролиза и отличается от концентрационной поляризации большим разнообразием и сложностью наблюдаемых явлений.
Этот вид торможения электродных реакций изучен еще недостаточно всесторонне. Наиболее полно механизм перенапряжения исследован лишь на примере восстановления водородных ионов. Это объясняется тем, что электродная реакция восстановления ионов водорода имеет большое практическое распространение и существенный теоретический интерес. Что касается концентрационной поляризации, то в этом случае изменения у поверхности электрода не зависят от материала электрода и вида ионов и сводятся к замедленной доставке ионов из толщи растцора в приэлектродные слои. Этот вид торможения электродной реакции зависит главным образом от диффузии, закономерности которой в настоящее время достаточно хорошо' изучены.
Во многих практических случаях электролиза поляризация заметно осложняет течение желаемых электродных процессов. Поляризация возрастает в зависимости от плотности тока, поэтому на преодоление торможения электродной реакции тратится значительное количество электроэнергии. Например, в случае электрорафинирования меди при среднем напряжении на клеммах 0,28 в около 21% этой величины приходится на поляризацию. При этом электроосаждение таких металлов, как медь, цинк, кадмий, серебро и ртуть, из растворов их простых солей сопровождается относительно небольшой, главным образом концентрационной поляризацией. Значительно труднее протекают процессы разряда и ионизации металлов группы железа. Особенно большой поляризацией сопровождаются разряд ионов водорода, а также окислительно-восстановительные реакции, протекающие на инертных электродах в электролитных ваннах.
Причины возникновения электродной поляризации 243
76в; ’н+/н,= -0-42 '
Не менее существенное влияние оказывает поляризация и на работу гальванических элементов. Здесь при отборе тока электроды также заметно поляризуются, вследствие чего напряжение на полюсах работающего элемента становится значительно меньше, чем его э. д. с. в равновесном состоянии (т. е. при разомкнутой цепи).
Во многих случаях поляризационные явления оказываются полезными и даже необходимыми. Рассмотрим катодный процесс в цинковой ванне, которую в простейшем случае можно представить схемой:
Zn+1 ZnSO4, Н2О |~ Zn. (IX,6)
Равновесные потенциалы ионов Н+ и Zn2+ при 18 °C определяются следующими уравнениями:
<PZn2+/Zn = “ 076 + С’029 lg OZn2+; Ж7)
?н + /нЛ'°’05818ан+- (IX>8)
Пусть aZn2+ — 1 г-ион/л-, концентрация водородных ионов в нейтральном растворе составляет 10~7 г-ион/л. Тогда при = 1 ^гп2+/гп — 9
Из двух возможных электродных актов на катоде происходит тот, которому отвечает более положительный потенциал. Следовательно, в нашем случае на катоде должен разряжаться водород. Найдем, при каких условиях станет возможным совместный разряд ионов водорода и цинка. Условием совместного разряда двух ионов является равенство соответствующих электродных потенциалов, т. е. в нашем примере при отсутствии электродной поляризации получим
—0,76 + 0,0291gа —0,42.
1 ь Zn2+
При = 1 это равенство отвечает следующей концентрации Zn2+-HOHOB:
0,76 — 0,42
CZn2+ = 10 °-029 X. 1012 г-ион/л.
Таким образом, при отсутствии затруднений в разряде ио--нов водорода цинк может выделяться лишь при колоссальной концентрации катионов Zn2+ (1012 г-ион!л), что соответствует содержанию около полутора миллионов тонн ZnSO4 в литре раствора. Практически достижение такой концентрации абсолютно немыслимо. Однако известно, что при определенных плотностях тока цинк осаждается на катоде в виде компактного металлического осадка с достаточно высоким выходом по току. 16*
244 Теоретические основы электрохимии
В этом последнем случае потенциалы разряда металлических и водородных ионов составляются из равновесных их значений и величины электродной поляризации тр сопровождающей разряд соответствующих ионов, для принятой плотности тока на катоде, т. е.
= Чме + ---~аМе2+ ~ ^Ме’ (1Х.9)
о ., 0,00027 . /IV
Тн = Тн + —;-----аи — riut- (IX,10)
Сопоставление экспериментально полученных значений т)мв и т]н при плотностях тока, равных реальным и близких к ним, показывает, что величина перенапряжения для разряда ионов водорода на катоде t]Hs при прочих равных условиях во много раз превышает величину т}Л/е. Это характеризуется поляризацией при разряде ионов Н+ и Zn2+ на цинковом катоде при t = = 20 °C:
i^MaJcM^ . . 0,5 1,0 4,0 5,0 7,5 10 20 30 40
ПН1, в..... 1,015 1,075 1,115 1,165 1,18 1,195 1,24 1,26 1,285
TqZn, в — — — 0,064 — 0,065 0,07 0,0720 0,075
Из приведенных данных видно, что разряд ионов водорода сопровождается очень большим перенапряжением.
В противоположность водороду разряд металлических ионов Zn2+ сопровождается незначительной поляризацией. Таким образом, можно, подбирая соответствующие условия (плотность тока, температуру, состав электролита, поверхностно активных веществ), осаждать цинк не только из нейтральных, но и из весьма кислых растворов простых солей с достаточно высоким выходом по току.
В большинстве случаев электродная поляризация существенно влияет на структуру, чистоту и характер осадка, а также на равномерность распределения металла по поверхности катода. Не меньшее влияние оказывает поляризация и на анодные процессы. Подобно тому, как разряд ионов металла на катоде ускоряется при переходе к более отрицательным потенциалам, анодные процессы ионизации ускоряются при сдвиге потенциала в положительную сторону.
Рассмотрение анодной поляризации имеет большое практическое значение, так как она является наиболее существенным фактором, обусловливающим устойчивость ряда металлов против коррозии. Даже на однородной поверхности могут с большей или меньшей скоростью происходить одновременно анодный процесс растворения металла и катодные процессы выделения водорода или ионизации кислорода. Особенность растворения
Причины возникновения электродной поляризации 245
или коррозии металлов можно заранее определить, изучая скорости катодных и анодных процессов на поверхности металла. Приведенные примеры иллюстрируют исключительную роль электродной поляризации при осуществлении и регулировании реальных электрохимических процессов. Весьма незначительное изменение поляризации в желаемом направлении может привести к заметному технологическому и экономическому эффекту. Поэтому изучение кинетики электродных реакций представляет наиболее важный раздел теоретической электрохимии и должно осуществляться с достаточной объективностью и строгостью.
§ 3. Методы экспериментального исследования кинетики электродных процессов
Метод снятия поляризационных кривых
В случае торможения электродной реакции, для того, чтобы сделать возможным прохождение тока определенной плотности, необходимо, очевидно, заметно изменить величину электродного потенциала по сравнению с равновесным его значением. Как было указано выше, смещение потенциала электрода от его равновесного значения при заданной плотности тока носит название электродной поляризации. При этом различают катодную Д<рк и анодную Д<ра поляризации, определяемые следующим образом:
Д<Рк = <р° —- <рк; Д<ра’= <ра — ср0,
где <р° — равновесное значение потенциалов электродов, а;
<рк и <ра — потенциалы поляризованных электродов *.
Для суждения о величине и характере электродной поляризации снимают поляризационные кривые, графически выражающие связь между плотностью тока и электродным потенциалом. По оси абсцисс обычно откладывают потенциал относительно какого-либо условного нуля, например потенциал относительно нормального водородного электрода, по оси ординат — значение плотности тока на исследуемом электроде.
Измерения начинают с равновесного потенциала, а затем постепенно увеличивают поляризующий ток и определяют величину потенциала электрода для каждой данной плотности тока. С увеличением нагрузки электрода током потенциал электрода все более отклоняется от его равновесного значения в данном растворе. Типичные кривые для анодной и катодной поляризации изображены на рис. 82.
Анодная I и катодная II, II' кривые могут иметь различные наклоны по отношению к оси абсцисс. Это обусловлено тем, что
Дф = Дф + Дфк [см. (VI, 6)].
246
Теоретические основы электрохимии
анодная и катодная поляризации различаются не только по абсолютной величине, но и по характеру описываемых процессов. Величина электродной поляризации Аф может быть непосредственно определена из графического построения. Очевидно, что чем больше торможение электродной реакции, тем положе ход i — ф-кривой.
Определяя зависимость силы тока от потенциала, в стацио-
нарных условиях установили основные закономерности течения
Гис. 82. Кривые, выражающие зависимость потенциала электрола <р от плотности тока i
ряда электрохимических реакций. Но опыт показал, что результаты, полученные при анализе поляризационных кривых, представляют значительный интерес только в том случае, если скорость самой электрохимической реакции заметно меньше скорости других сопутствующих стадий процесса.
Для измерения электродных потенциалов под
током существуют различные способы. Простейший — компенсационный метод снятия
поляризационных кривых.
Принцип компенсации состоит в уравновешивании определяемой э. д. с. равным по величине и противоположным по направлению падением напряжения.
Компенсирующее падение напряжения обычно создается с помощью свинцовых аккумуляторов.
Электрическая схема такой установки приведена на рис. 83. Измерения проводят в электролитической ячейке (электролизере) 3, имеющей два электрода, один из которых анод, а второй — исследуемый катод 1. Электроды поляризуют постоянным током от аккумулятора 4 через делитель напряжения (реостат) 5, причем силу тока измеряют точным миллиамперметром 7. Изучаемый электрод 1 соединен при помощи электролитического ключа и промежуточного сосуда с электродом сравнения 2. Электродвижущую силу системы измеряют с помощью обычной потенциометрической схемы, т. е. реохорда 9 с нормальным элементом 10 и гальванометром 11. В качестве электрода сравнения чаще всего применяют каломельный, хлорсеребряный или ртутноокисный полуэлементы. Промежуточный сосуд и электролитический ключ заполняют для снижения диффузионного по
Причины возникновения электродной поляризации
247
тенциала раствором КС1 или NH4NO3 в зависимости от условий опыта. Таким образом измеряют э. д. с. цепи:
Раствор соли I Раствор соли к--. I КС1, ме металла | металла + КС1 насыи* | Hg2Cl2
Если при измерениях контакт поляризуемого электрода с воздухом не желателен, то исследуемый электрод может быть помещен в атмосферу инертного газа (водород, азот). Так как
ь
Рис. 83. Схема установки для измерения электродных потенциалов (под током:
I — исследуемый электрод; 2 — каломельный электрод; 3 — электролизер; 4 — аккумулятор; 5 — реостат; 6 — вольтметр;
7 — миллиамперметр; 8 — шунт; 9 — реохорд; 10 — нормальный элемент; 11 — гальванометр
раствор электролита представляет собой заметное электрическое сопротивление, то при прохождении тока падение напряжения в электролите между электродом 1 и концом электролитического ключа входит в измеряемую э. д. с. и мешает точному определению электродной поляризации. Для уменьшения омических потерь применяют в качестве ключа стеклянную трубку — сифон с оттянутым в капилляр кончиком, который прижимают к поверхности исследуемого электрода. Однако при этом поверхность электрода частично экранируется стеклом капилляра, что приводит к перераспределению силовых линий электрического поля и к некоторому уменьшению плотности тока, а следовательно, и потенциала изучаемой точки поверхности.
Чтобы уменьшить экранирование, капилляр делают по возможности более тонким и оставляют незначительный зазор между концом капилляра и поверхностью электрода. Омическое сопротивление раствора в этом приэлектродном слое стремятся снизить, повышая электропроводность электролита. Правильным подбором капилляра и его расположения можно добиться того, что обе ошибки, т. е. омическое сопротивление раствора и экранирование электрода, станут исчезающе малыми.
248
Теоретические основы электрохимии.
Эти источники ошибок были изучены Пионтелли.' Автор исследовал поведение электродов с различными типами капилляров Луггина, изображенных на рис. 84. Капилляры типа а и г показали наилучшие результаты, так как они не вносили заметных отклонений в распределение тока.
Электролитический ключ г давал наиболее правильные экспериментальные данные для величины потенциала при заданной плотности тока. Готовили этот капилляр следующим образом. Платиновую проволоку впаивали в конец стеклянной трубки,
при этом угол между проволокой и стеклянной трубкой составлял примерно 45°. Конец стеклянной трубки сошлифовывали на плоскость, а платину растворяли в смеси соляной и азотной кислот.
Конец капилляра д был расположен на обратной стороне электрода, так что использовалась лишь одна сторона электрода (на рисунке — правая).
Компенсационным методом снятия поляризационных кривых широко пользуются при экспериментальных исследованиях. Объясняется это тем, что он особенно удобен и доступен при изучении реальных электрохимических систем.
Для определения величины электродной поляризации и снятия поляризационных кривых применяют электролизеры самых различных конструкций. На рис. 85 приведена схема установки, применявшейся для изучения поляризации при выделении водорода на ртутном катоде. Прибор состоит из трех основных частей: электролитической ячейки 6, трубки 5 для ампулы с раствором и трубки 4 для ампулы с ртутью, спаянных в одно целое. Нижняя расширенная часть электролитической ячейки предназначена для ртути, служащей катодом. Анодом служит платинированная платина. Анод вставляется на шлифе в часть /, которая отделена от катодного пространства краном 3, препятствующим диффузии продуктов электролиза из анодного пространства.
Электродом сравнения служит свежеплатинированная платина; электрод вставляется на шлифе в часть, отделенную от кат годного пространства двумя стеклянными фильтрами. Таким образом, измеряемые электродные потенциалы представляют собой непосредственно перенапряжение. Электролизер заполни-
Причины возникновения электродной поляризации 249
ется ртутью и электролитом из ампул, которые разбиваются специальными электромагнитными ударниками 7.
Такое устройство прибора гарантирует от попадания в электролизер случайных примесей, влияющих на поляризацию. При конструировании ячеек во всех случаях необходимо уделить большое внимание исключению корковых и резиновых пробок и соединений, удалению газов из раствора и обеспечению инертной атмосферы внутри аппарата (азот, водород, углекислый газ).
Рис. 85. Установка для .изучения /перенапряжения водорода на ртутном катоде:
1 “ анод; 2 — электрод сравнения; 3 — край, разделяющий анодное и катодное пространства; 4 — трубка для ампулы с ртутью; 5 — трубка для ампулы с раствором; 6 — электролитическая ячейка; 7 — электромагнитные ударники
На рис. 86 приведена схема электролизера для изучения катодной поляризации при осаждении металлов. Здесь катод 1 и аноды 2 изготовлены из платины, покрытой металлом, поляризация которого изучается, 3 мешалка, 4 капилляр, соединяющий катод с электродом сравнения. Электролизер имеет трубки для пропускания водорода или другого инертного газа.
Электролизер и электрод сравнения должны быть помещены в термостат с автоматической регулировкой температуры. Так как потенциал электрода зависит не только от силы тока, но также и от величины электродной поверхности, то при изучении поляризации для обеспечения возможности сравнения различных результатов необходимо фиксировать плотность тока i (в -ма/см2 или а!дм2).
Однако при определении истинной поверхности электрода часто возникают серьезные затруднения, так как видимая (гео-
•250
Теоретические основы электрохимии
Рис. 86. Электролизер с твердым катодом: 1 — катод; 2 — аноды; 3 — мешалка; 4 — капилляр; 5 — электрод сравнения; 6 — промежуточный сосуд
метрическая) величина поверхности только в редких случаях совпадает с истинной поверхностью. Такое совпадение наблюдается в случае применения жидких электродов (ртуть, галлий). У твердых же металлических электродов истинная поверхность обычно отличается от видимой, особенно при использовании шероховатой поверхности (платинированная платина и т. п.). Эта неопределенность в оценке значений истинной поверхности электрода еще более возрастает при электроосаждении или анодном растворении металлов, так как при этом поверхность электрода непрерывно меняется. В связи с этим некоторые исследователи считают компенсационный метод снятия поляризационных кривых непригодным при изучении поляризации, сопровождающей электроосаждение металлов. Однако есть основания
предполагать, что изменения поверхности в процессе электролиза довольно часто оказываются пренебрежимо малыми. Это относится, например, к электроосаждению металлов из комплексных электролитов, когда образуются особенно плотные и гладкие осадки, почти не отличающиеся по структуре. Для получения воспроизводимых результатов в этом случае необходимо лишь принять ряд специальных мер, а именно: проводить измерения достаточно быстро и в некоторых случаях переходя от больших плотностей тока к малым.
Об изменении истинной поверхности электрода можно судить по результатам определения емкости двойного слоя. С помощью таких измерений было показано, что изменение истинной поверхности электрода в процессе снятия поляризационных кривых при приведенных выше условиях настолько мало, что в первом приближении им можно пренебречь.
Чтобы освободиться от неопределенности в суждении о поверхности электрода и гарантировать себя от ошибок при определении величины этой поверхности, полезно исследовать поляризацию в электролизерах с капельным или струйчатым ртутным электродом (рис, 87). Такие работы проводились многими
Причины возникновения электродной поляризации
251
исследователями, причем в некоторых случаях результаты измерений на твердом и ртутном струйчатом электродах оказывались достаточно близкими.
Струйчатый ртутный электрод (рис. 88) представляет собой тонкую струйку ртути 1 толщиной не более 0,2 мм и высотой
Рис. 87. Электролизер с Рис. '88. Струй-жидким ртутным като- чатый ртутный дом: катод
1 — диоды; 2 — чашечка сифона; 3 — капилляры
2 см, вытекающую из капилляра и падающую в чашечку сифона 2, отводящего ртуть из электролизера. Истинная поверхность струйки ртути составляет около 0,15 см?. С двух сторон непосредственно к поверхности ртути подводятся оттянутые в капилляры концы стеклянной трубки 3, соединяющей ртутный электрод с электродом сравнения.
Капельный ртутный электрод рассмотрен при описании полярографического метода анализа.
Капельный и струйчатый ртутные электроды применяются главным образом для аналитических целей и для изучения диффузии электролита у границы раздела металл — электролит. Такие электроды
часто применяют для установления связи между структурой органических молекул и их восстановлением на электроде, при определении состава комплексных ионов и т. п.
Применение капельного или струйчатого ртутных электродов для исследования кинетики электродных реакций имеет то преимущество, что благодаря непрерывному обновлению поверхности электрода устраняется возможность изменения ее активности со временем и вследствие этого не предъявляются особенно строгие требования к предварительной очистке раствора. Сопоставление кинетики разряда ионов металла в условиях, когда металл выделяется на твердом и на жидком катоде (например, на ртути), способствует выявлению особенностей, связанных с природой фазовой поляризации.
552 Теоретические основы электрохимии
Вместе с тем самостоятельные измерения на ртутном катоде не могут дать представлений о течении процессов электрокристаллизации. В тех случаях, когда устанавливается важная для технологии связь кристаллической структуры с определенными условиями электролиза, такой электрод не пригоден. В ряде случаев необходимо также учитывать возможность загрязнений ртути и образования амальгамы (при катодном осаждении металлов), а также ионизацию ртути при использовании ее в качестве анода.
С применением ртутного капельного электрода изолирование продуктов электродной реакции полностью невозможно, что затрудняет определение характера и скорости течения реакции. Наличие емкостного (не фарадеевского) тока, потребляемого' для заряжения каждой капли, делает затруднительным применение такого электрода для изучения кинетики при малых плотностях тока, т. е. в условиях, когда могут быть получены важные характеристики процесса.
Этот метод не может быть также применен при исследовании условий осаждения таких металлов, как цинк, кадмий, никель и др., которое обычно осложняется совместным разрядом ионов-водорода. Следует иметь в виду, что величина перенапряжения водорода на ртути и на твердых электродах различна. Таким образом, для суждения о кинетике разряда ионов металла целесообразно электродные процессы изучать на твердых электродах и в условиях, максимально приближенных к реальным.
За последнее время в электрохимических исследованиях все возрастающее применение находят так называемые «быстрые» методы, особенностью которых в отличие от «ручных» измерений является автоматическая запись потенциала ср исследуемого электрода, либо плотности тока i, протекающего через электроды, либо функциональной зависимости <р — I. Используемые для этих целей установки по наименованию лежащих в их основе методов можно разделить на две группы — потенциостатические и гальваностатические. Под первыми подразумеваются схемы, при помощи которых автоматически регистрируется сила тока при постоянном (или изменяющемся по заданному закону) потенциале, под вторыми — схемы для записи потенциала исследуемого электрода при постоянной (или задаваемой) плотности тока.
В тех случаях, когда собственно электрохимический процесс протекает с достаточно большой скоростью и его особенности выявляются лишь при высоких плотностях тока, при которых доставка реагирующего вещества к электроду может ограничивать скорость процесса в целом, особое место в электрохимической кинетике занимают методы исследования, позволяющие в той .или иной степени снять диффузионные ограничения.
Причины возникновения электродной поляризации
253
Потенциостатический метод изучения электродных процессов
Наблюдение за изменением силы тока во времени при наложении на электролитическую ячейку постоянной разности потенциалов позволяет судить о характере процессов, протекающих на электродах.
Особенно это полезно в том случае, когда скорость изменения электрохимического процесса очень велика в начальный мо
Рис. 89. Катодные устройства, позволяющие определить выход иго току:
а — взвешиванием; б — улавливанием водорода
мент электролиза, а затем, по мере течения тока, скорость электродной реакции замедляется.
В таком случае наиболее полную картину кинетики электродного процесса дают i — т-кривые, полученные при постоянном потенциале. Преимущество этого метода особенно проявляется при электроосаждении металлов группы железа, хрома и в ряде других случаев, когда выделение металла на катоде сопровождается совместным разрядом водорода.
Для определения выхода по току металла и водорода удобно пользоваться специальными катодными устройствами (рис. 89).-Собирают измерительную схему, приведенную на рис. 90. В нее входят: электролизер 1, имеющий катод 2 и два анода 3, 4. Катод электролитическим ключом соединен с электродом сравнения 5. От катода и электрода сравнения разность потенциалов подается на электронный усилитель 9, соединенный с магазином сопротивления 7 и гальванометром 8. Все эти приборы вместе
254
Теоретические основы электрохимии
составляют катодный вольтметр, позволяющий непрерывно следить за потенциалом поляризуемого электрода.
В настоящее время разработано большое число ламповых схем, пригодных для измерений в самых различных условиях. Конструирование таких схем и их изготовление требуют специальных знаний по радиотехнике и освещены достаточно подробно в специальной литературе. Работа катодного вольтметра основана на том, что при включении измеряемого напряжения нарушается равновесие, моста и через гальванометр протекает ток.
Рис. 90. Схема измерения .поляризации .с помощью катодного вольтметра (при постоянном потенциале):
1 — электролитическая ячейка; 2 — катод; 3, 4 — аноды;
5 — каломельный полу элемент; 6 — низкоомное сопротивление;
7 — магазин сопротивления иа 1000 ом; 8 — гальванометр:
6 — усилитель; 10 — переключатель; 11 — однополюсный
ключ; 12 — аккумулятор
пропорциональный величине приложенного напряжения. Проградуировав шкалу гальванометра в вольтах, можно применять схему для измерения э. д. с. любых гальванических элементов.
Большое преимущество катодных (ламповых) вольтметров — возможность непрерывного измерения э. д. с., изменяющихся во времени. Если включить вместо гальванометра шлейф осциллографа, то можно непосредственно записать соответствующие кривые Е = /(т) или Е = /(/), если сила поляризующего тока изменяется во времени по какому-либо известному закону.
Постоянное напряжение на электродах можно поддерживать при помощи специальных электронных приборов, а также при помощи простой электрической схемы.
Необходимое условие постоянства величины подаваемого на электроды напряжения заключается в том, чтобы ток, идущий через делитель напряжения, был во много раз больше тока, идущего через электролитическую ячейку, т. е. если Ц >> 12, то ДЕ ~ const. Это условие осуществляется, когда сопротивление реостата (делителя напряжения) меньше сопротивления в- цепи
Причины возникновения электродной поляризации
255-
электролитической ячейки. Кроме того, необходимо, чтобы омическое сопротивление в цепи ячейки было также во много раз меньше, чем сопротивление самой ячейки. Такая схема применяется обычно в полярографах.
Электролизер 1 в схеме рис. 90 питается постоянным током от аккумуляторной батареи 12, включенной потенциометрически на низкоомный реостат 6. Особенностью схемы является необходимость применения низкоомного реостата, не превышающего 20 ом. Еще лучше использовать двойную схему включения реостатов, при которой регулировка напряжения, подаваемого на ячейку, значительно облегчается.
Ход исследования сводится к следующему: прежде всего градуируют катодный вольтметр. Для этого к входным клеммам усилителя подключают нормальный элемент, э. д. с. которого равна 1,0183 в. Затем, меняя сопротивление 7, при включенном усилителе устанавливают стрелку гальванометра на желаемое деление, которое отвечает напряжению 1,0183 в. После этого определяют цену деления шкалы гальванометра. Затем электролизер и кулонометр заливают электролитами и начинают пропускать ток. Амперметр в цепи 'служит для ориентировочного определения режима электролизера, при котором на катоде образуются качественные осадки. В определенном таким образом интервале плотностей тока и ведут электролиз. При этом устанавливают вполне точное значение выхода по току металла и водорода и величину катодного потенциала, которую и поддерживают постоянной с помощью реостата 6.
Потенциостатический метод снятия ' поляризационных кривых, существо которого заключается в том, что при каждом заданном значении потенциала электрод выдерживается до установления стационарной плотности тока, был применен А. И. Левиным с сотрудниками для определения характера электродной поляризации при осаждении цинка, железа, хрома и меди (в последнем случае из комплексного пирофосфатного электролита) с совместным выделением водорода. С помощью потенцио-статических измерений Деляхею удалось определить зависимость силы тока от потенциала для отдельных электрохимических процессов при одновременном протекании нескольких электродных реакций.
Метод может быть применен не только для исследования катодной поляризации, но и для изучения пассивирующих слоев на металлическом электроде. Он дает возможность полностью определить истинную поляризационную кривую на тех участках, где она при других методах нередко маскируется самопроизвольными скачками потенциала при одновременном противоположном скачке тока (например, во время пассивирования поверхности электрода).
256
Теоретические основы электрохимии
Коммутаторный метод измерения электродной поляризации
В случае работы с растворами, плохо проводящими электрический ток, величина омических потерь между капилляром электролитического ключа и поверхностью электрода достигает заметных размеров и должна быть особо учтена или устранена иным способом.
В связи с этим возникла идея на момент измерения электродной поляризации выключать поляризующий ток, что и достигается применением специального устройства — коммутатора, позволяющего изменением соединений в электрических цепях снизить промежуток времени между выключением внешнего тока и измерением потенциала электрода до 10~4—10-5 сек. В этом случае в измеренную величину сдвига потенциала не включается величина омического падения напряжения, так как /Д = 0. Если перерывы тока будут очень кратковременными, то поляризация в момент выключения будет достаточно близкой к значению ее при заданной плотности тока.
Для снятия поляризационных кривых в простейшем случае применяют вращающийся коммутатор, предназначенный для замены и переключения элементов электрических цепей. Действие коммутатора состоит в попеременном, с большой частотой, переключении исследуемого электрода с поляризующей на измерительную цепь. Таким образом, потенциал электрода определяется в момент, когда в системе отсутствует ток.
Однако рассматриваемый метод имеет некоторые недостатки. При большой скорости вращения возможно замыкание контактных пластин коллектора коммутатора через искровые разряды в момент разрыва поляризующей цепи. Кроме того, в момент выключения тока возникают экстратоки (токи Фуко), появление которых искажает истинную величйну потенциала поляризованного электрода. Применение этого метода связано с трудностями, которые обусловлены тем, что после поляризации электрода током большой плотности (десятые доли ампера на квадратный сантиметр) поляризация после выключения тока падает настолько быстро, что даже через 1О~5 сек потенциал электрода сильно отличается от его значения при прохождении тока.
В таких случаях приходится снимать кривую зависимости потенциала электрода от времени, прошедшего с момента выключения тока, и экстраполировать полученные значения поляризации на момент выключения тока. Такая экстраполяция часто связана со значительными ошибками.
В потенциометрических измерениях применяют и другие, более сложные коммутаторы с автоматическим управлением. Так, например, ламповые коммутаторы позволяют включать из-
Причины возникновения электродной поляризации 257
мерения через 10~5 сек. В литературе описаны многие приборы такого рода, но почти все они являются сложными и уникальными устройствами, изготовление которых связано с трудностями. В Тартуском государственном университете в последнее время разработан простой электронный коммутатор.
Принципиальная схема цепей коммутатора приведена на рис. 91. В качестве коммутаторной лампы применяют лучевой
Рис. 91. Принципиальная схема коммутатора (конструкция Тар туского университета)
выходной тетрод 6П7С(ЛХ), имеющий в режиме пентода малое внутреннее сопротивление. Запускающий задержанный импульс ЭНО-1 формируется при помощи цепи дифференцирования и диодного ограничителя (Л4) в более короткий однополярный импульс, который управляет схемой пересчета через диодный распределитель (Л3). Лампы и детали коммутатора выбраны из расчета получения наименьшего времени коммутации. С этой же целью применяют сетевой трансформатор особой конструкции с малой емкостью относительно земли. Для крепления всех деталей, присоединенных к катоду Л1, применяют изоляционную пластину. Для нормального режима работы Л1 не следует уменьшать ее анодное напряжение ниже 50 в. В случае необходимости работы с малыми токами питания ячейки в цепь коммутатора можно ввести дополнительное последовательное сопротивление.
17 заказ 1338
258 Теоретические основы электрохимии
Осциллографический метод исследования электродных процессов
Для непосредственного изучения хода различных электрод ных реакций нужны измерительные схемы и приборы высокой степени совершенства; Основные требования к приборам а) безынерционность регистрации измеряемых электрических величин; б) высокая чувствительность (прибор должен давать показания при очень слабой реакции на измеряемый электрический процесс). В настоящее время электроизмерительная техника позволяет проследить изменение электрических величин во времени с точностью, превышающей 10-8 сек. Измеряемые токи могут составлять всего несколько электронов в секунду.
В основу измерительной аппаратуры осциллографического метода положены вакуумные приборы, обеспечивающие фото-запись электронным лучом (электронный осциллограф).
Изучение кинетики электродных реакций связано с необходимостью записи различных переменных электрических величин и прежде всего силы тока и напряжения. Первые попытки таких измерений были осуществлены Ленцем в 1849 г. Он предложил способ измерения мгновенных значений этих величин. Идея Ленца вскоре была воплощена в конструкции так называемой шайбы Жубера. В 1891 г. была разработана первая конструкция шлейфового осциллографа. Этот прибор непрерывно совершенствовался, и в настоящее время, .пользуясь им, можно измерять переменные токи с частотой до 20 кгц.
В 1894 г. была произведена первая запись электронным лучом, управляемым переменным магнитным полем. В 1898 г. Браун разработал конструкцию электронно-лучевой трубки — прообраза современного электронного осциллографа. В настоящее время электронный осциллограф является техническим электроизмерительным прибором, широко используемым для изучения весьма многих явлений.
Современный низковольтный электронный осциллограф имеет следующее устройство (рис. 92): в одном конце стеклянной, откачанной до высокого вакуума трубки помещен источник электронов — горячий катод, в другом (широком) конце трубки — флуоресцирующий экран. Испускаемые катодом электроны под воздействием ускоряющих напряжений пролетают ряд диафрагм и в виде узкого луча бомбардируют экран, свечение которого можно наблюдать снаружи.
Во время полета от катода к флуоресцирующему экрану поток электронов может быть подвержен различным воздействиям электростатических или магнитных полей. Специальная электронная оптика позволяет получить концентрированный электронный луч с очень малой площадью поперечного сечения, от
Причины возникновения электродной поляризации
259
Рис. 92.
НИТЬ
Схема электронно-лучевого осциллографа:
накала; 2 — катод; 3 — управляющий электрод; 4 первый анод; 5 — второй аиод; 6 — отклоняющие пластины (гори->
зонтальные); 7 — отклоняющие пластины (вертикальные); 8 — экранирующий слой; 9 электронный луч; 10 — экран
дельные частицы которого имеют одинаковую скорость. Это достигается с помощью регулирования яркости (изменение отрицательного потенциала на специальном управляющем электроде —- цилиндре Венельта) и изменением напряжения на первом ускоряющем электроде.
Электронный луч отклоняется магнитным и электростатическим полями, чаще всего одновременно в двух взаимно перпендикулярных направлениях.
В одном (обычно верти- 8
кальном) направлении про- 3 5 в ] Д
изводится отклонение, свя- 1 х \ V-’O
занное с изменением изучаемого электрического тока или напряжения. В другом направлении (горизонтальном) отклонение луча производится по известному временному закону. Таким образом, луч вычерчивает на экране трубки осциллограмму в обычной системе координат.
В большинстве случаев
осциллограф применяют для записи процессов, не требующих точности записи по координате времени, большей чем 10-2 — 10-3 сек. Как правило, осциллографические установки при изучении кинетики электрохимических процессов применяют для записи периодических явлений, вследствие чего осциллограммы на экране получаются неподвижными.
Электронно-лучевой осциллограф был впервые введен в электрохимическую практику Лебланом. Для определения временной зависимости потенциала электрода при пропускании тока постоянной величины метод успешно использовали В. А. Рейтер, В. А. Юза и Е. С. Полуян. Они определяли токи обмена между твердыми металлическими электродами и электролитом. К этой же группе относится и осциллографическая полярография, основанная на измерении временной зависимости тока при изменении электродного потенциала.
Техника осциллографических измерений сводится к следующему (рис. 93). Первоначально заряжают конденсатор 2 от источника постоянного напряжения 1 через сопротивление 6. Напряжение от конденсатора подается на усилитель 3. После усиления напряжение подается на ячейку, соединенную последов вательно с калиброванным переменным сопротивлением 7.
Напряжение с усилителя 4 и передается на пластины, отклоняющие зайчик катодного осциллографа в вертикальном напра-17*
260
Теоретические основы электрохимии
влении. Таким же образом напряжение, накладываемое на ячейку, повышается в усилителе 5 и подается на горизонтально отклоняющие пластины осциллографа.
Кривые потенциал — время записываются с помощью схемы, приведенной на рис. 94. В данном случае ячейка последовательно соединяется с регулируемыми сопротивлениями 4 и 5 и источником постоянного тока 1 с выходным напряжением 200—300 в. Поскольку напряжение на клеммах ячейки небольшое (1—2 в), омическое падение в 4 и 5 составляет более 90% от напряжения
Рис. 93. Схема 'прибора для записи кривых 'сила тока—потенциал при большой 'скорости изменения шотен-щиала:
1 — источник постоянного тока; 2 — конденсатор; 3—5 — усилители; 6, 7 — сопротивления; 8 — КЛЮЧ
Р,ис. 94, Схема прибора для снятия кривых потенциал—‘’время при постоянном токе:
1 — источник постоянного тока; 2, 3 — потенциометры; 4, 5 — сопротивления; 6 — ячейка; 7 — ключ; 8—10 — электроды;
// — самописец
источника тока. В таком случае через ячейку идет ток практически постоянной силы, не зависящей от процессов на электродах. Силу тока, регулируемую с помошью сопротивления 4, определяют, измеряя с помощью потенциометра 2 омическое падение напряжения на калиброванном регулируемом сопротивлении 5. При измерении силы тока ячейку шунтируют для устранения поляризации электродов. Следует иметь в виду, что шунтирование вносит некоторую ошибку в измерение силы тока (порядка нескольких десятых процента).
Разность потенциалов между поляризуемым электродом 8 и электродом 9 подают на самописец, перо которого может пройти всю шкалу за одну секунду и лента которого имеет достаточную скорость движения. Если вместо самописца пользоваться катодным осциллографом, то компенсирующий потенциометр 5 не нужен, поскольку луч осциллографа можно установить в любом положении, изменяя напряжение в выходном контуре усилителя, управляющего горизонтальным смещением.
С помощью осциллографа удается количественно исследовать медленные химические реакции, предшествующие собственно электрохимической стадии. Метод осциллографической полярографии был успешно применен Я- Гейровским для изучения степени необратимости электродных процессов.
Причины возникновения электродной поляризации
261
Быстрый метод определения i — ^-кривых
Для выяснения механизма электролитического выделения металлов А. Т. Баграмян разработал метод, позволяющий пренебречь изменениями поверхности электрода во время электролиза.
Схема установки для исследования поляризации приведена на рис. 95. Ток для электролиза подается от аккумулятора 1 к
Рис. 95. Схема ирибора А. Т. (Баграмяна:
1 — аккумулятор; 2 — электролизер; 3 — реостат; 4 — стержень с контактом; 5 — электромотор; 6 — коробка распределения скоростей; 7 — вращающийся барабан с фотопленкой; 8 — зеркальный гальванометр; 9 — щель для «зайчика» гальванометра; 10 — электромагнит для открывания щели; 11 ~ трансформатор для питания электромагнита; 12 — выключатель; 13 — реостат к лампочке гальванометра; 14 — лампочка гальванометра; 15 — катодный вольтметр; 16 — исследуемый электрод; 17 — электрод сравнения
ячейке 2 через реостат-потенциометр 3, который состоит из кольцевого реостата и равномерно скользящего по нему латунного контакта 4. При вращении контакта от точки d1 сила тока в ячейке растет от нуля до максимальной величины, затем снова падает до нуля (d), после чего ток меняет направление и вновь растет до максимума и падает до нуля (di).
Поляризационная кривая записывается на вращающемся барабане 7 на фотопленку. При этом для синхронизации барабан 7 и контакт 4 вращаются на одной оси. Потенциал измеряют специально проградуированным зеркальным гальванометром.
262 Теоретические основы электрохимии
зайчик которого попадает на фотопленку через щель 9, открываемую автоматически посредством электромагнита, питающегося от трансформатора 11. Тот же трансформатор через реостат 13 питает лампочку гальванометра 14.
Прибор сконструирован для изучения процессов, протекающих при электроосаждении и растворении металлов, и предназначен для автоматической регистрации с разной скоростью зависимости катодной и анодной поляризации от плотности тока. Преимущества метода состоят в возможности пренебречь изменениями поверхности электрода в процессе электролиза благодаря короткому времени измерения. Метод позволяет применять высокие плотности тока, что дает возможность покрывать электрод слоями свежеосажденного металла; осаждение идет практически по всей поверхности катода равномерно.
Однако в методе А. Т. Баграмяна не соблюден принцип компенсации, измерения ведутся при силах тока, отличных от нуля.
Метод измерения с применением переменного тока
Для решения задачи о снятии диффузионных ограничений особое значение имеют переменноточные измерения при высоких частотах.
Впервые переменный ток для исследования электродных процессов был применен А. Н. Соколовым в 1887 г. Затем аналогичная методика была использована Лебланом и Шиком при исследовании закономерностей электроосаждения металлов. Детальная разработка метода была осуществлена А. Н. Фрумкиным, Б. В. Эршлером и П. И. Долиным, которые показали, что при наложении переменного тока в течение коротких отрезков времени фронт диффузии не успевает отойти от поверхности электрода на значительное расстояние. Это позволяет пропустить через ячейку гораздо большие токи по сравнению со стационарными условиями при обычных режимах размешивания.
Снятие диффузионных ограничений делает возможным выявление стадий, определяющих кинетику электродного процесса. Так, например, была изучена кинетика разряда ионов водорода на платине (по проводимости электрода по отношению к переменным токам различной частоты).
При поляризации электрода переменным током можно отдельно определить концентрационную и химическую составляющие. Преимущество метода — то, что он позволяет измерять химическую поляризацию, даже когда она относительно мала. В измерениях с помощью постоянного тока такую малую химическую поляризацию обычно обнаружить не удается и создается впечатление, что поляризация электрода имеет только концентрационный характер. Преимущество переменноточных из
Причины возникновения электродной поляризации 263
мерений связано с тем, что доля химической поляризации электрода возрастает с частотой тока.
Рассматриваемый метод позволяет исследовать и другие процессы, обладающие скоростью, в 104 — 105 раз большей, чем скорость процессов, изучаемых обычным методом поляризационных кривых.
При прохождении тока переменного напряжения через электрод сдвигаются фазы между током и напряжением; следовательно, полное сопротивление (импеданс) электрода можно рас-
Рис. 96. Эквивалентная схема .системы электрод— раствор электролита
сматривать как состоящее из омических и емкостных составляющих. Величина этих составляющих, а также соотношение между ними определяются свойствами электрода и процессами, которые с различной скоростью протекают на границе фаз при прохождении тока. Измеряя раздельно омическое и емкостное сопротивление электрода и исследуя их зависимость от различных факторов, можно сделать выводы о кинетике обусловливающих их реакций.
Необходимо, однако, заметить, что достаточно хорошие результаты здесь могут быть получены в случае применения электродов с совершенно гладкой поверхностью (ртуть и т. п.), так как в противном случае наблюдается некоторая замедленность в образовании двойного слоя на поверхности электрода, что затрудняет истолкование результатов измерений при высоких частотах.
Смысл метода измерения с применением переменных токов сводится к тому, что всякая электрохимическая система формально может быть представлена в виде электрически эквивалентной схемы (рис. 96), сочетающей в себе емкость двойного слоя (Сд.с) и реактивное сопротивление электрода (Сл, /?л).
Принципиальная схема для измерения емкости достаточно проста и аналогична мосту для измерения сопротивления. Одна из применяемых в электрохимии схем была приведена ранее (см. рис. 79). В ней были применены постоянные безреактивиые сопротивления (см. стр. 223—228).
264
Теоретические основы электрохимии
В качестве нуль-инструмента обычно используют электронный осциллограф или ламповый вольтметр. Условия равновесия такого моста:
2) RX = -^RO.
Практически мост составляют из изучаемой ячейки, в которой один из электродов имеет поверхность, гораздо большую, чем другой, так что его емкостным сопротивлением можно пренебречь.
Рис. 97. Принципиальна?! схема установки для переменноточ-иых измерений:
/ — исследуемый электрод; 2 — цилиндрический электрод для переменного тока; 3 — электрод сравнения; 4 — вспомогательный электрод для поляризации постоянным током; 5 — стеклянные фильтры;
6 — сосуд с раствором, нз которого предварительно удален газ;
7 — осциллограф в качестве нуль-ннструмента; 8 — прецизионный магазин сопротивления; 10 — два сопротивления, являющиеся плечами измерительного моста
Общая электрическая схема установки приведена на рис. 97.
Для изучения кинетики процессов наиболее интересным элементом является Rx (см. рис. 79), так как он зависит от электрохимической реакции. Для обозначения этого элемента эквивалентной цепи введен термин «фарадеевский импеданс». Rx по своей природе обусловливается скоростью акта разряда. Эту часть сопротивления Геришер называет «сопротивлением прохождения». С другой стороны, величина Rx определяется так на
Причины возникновения электродной поляризации 265
зываемой поляризацией пополнения. Это название объединяет явления поляризации, которые обусловлены недостаточным пополнением реагирующих ионов и ведут к изменению концентрации веществ, определяющих разряд на границе фаз.
Поляризация пополнения возникает в результате замедленной доставки ионов (диффузия, миграция, конвекция), а также вследствие затруднения реакций, обеспечивающих необходимую концентрацию потенциалопределяющих компонентов (диссоциация, десольватация, образование комплекса и др.).
Если один из электродов ячейки не поляризуется, то свойства ячейки по существу определяются исследуемым поляризующимся электродом.
Б. В. Эршлер рассмотрел зависимости величин полного сопротивления поляризации от кинетики электродных реакций и других факторов в случае растворения металлов, окислительно-восстановительных процессов, образования амальгам и адсорбционных слоев. Автору удалось показать, что независимо от механизма обмена ионами'между электродом и раствором скорость этого процесса в равновесных условиях равна производной от поляризующего тока по потенциалу, найденной для равновес-ного потенциала, умноженной на величину q = —— (q — число ионов, участвующих в обмене). Эту производную находят из сопротивления электрода, измеренного с помощью переменного тока.
Метод наложения переменного тока на постоянный позволяет, не прибегая к величинам перенапряжения, исследовать скорости отдельных стадий электродного процесса, которые недоступны при использовании только одного постоянного тока.
Другие методы исследования электродных процессов
Для дальнейшего развития наших знаний в области кинетики электродных процессов особое значение приобретает комбинирование методов исследования, позволяющее сочетать изучение скорости элементарного акта разряда ионов или ионизации атомов металла с такими, например, явлениями, как адсорбция. Опыт последних лет показывает, что введение радиоактивных индикаторов в одну из соприкасающихся фаз (в электродный металл или раствор электролита) дает исследователю в руки весьма совершенное средство для непосредственного изучения двух, а иногда и нескольких сопряженных процессов.
Метод меченых атомов позволяет измерять скорость осаждения металла при заданной плотности тока при обратном процессе коррозии, протекающем с достаточной скоростью. Таким образом, А. И. Левину и В. И. Фаличевой удалось выяснить
266
Теоретические основы электрохимии
механизм торможения коррозии цинка наложенным извне током. Скорость коррозии в данном случае характеризовалась количеством «меченого» цинка, перешедшего в раствор, т. е. радиоактивностью пробы испытуемого раствора. Так было установлено, что коррозия цинка оказалась наибольшей при отсутствии тока. С ростом катодной плотности тока (поляризации) коррозия металла заметно снижалась и при <рк —0,894 в (i > 840 а/м2) радиоактивность раствора становилась равной нулю, что свидетельствовало о прекращении коррозионного процесса. О торможении процесса коррозии катодного цинка свидетельствовал тот факт, что поляризующий ток при достижении защитного потенциала оставался постоянным или даже незначительно уменьшался к концу экспозиции. Для цинка, загрязненного примесями кобальта, обратное растворение металла приостанавливалось при <рк> —0,937 в 1500 а/м2). Для цинка, загрязненного примесями сурьмы и никеля, в исследуемом интервале плотностей тока (до 2500 а/м2) полной катодной защиты достигнуть не удалось. Экспериментальные данные показали, что защитное действие наложенного извне тока связано с созданием некоторого барьера, удерживающего ионы цинка в двойном электрическом слое; при этом процесс саморастворения металла затухает. Был сделан вывод, что выделение водорода на цинковом катоде при достижении определенного значения <рк не может быть обусловлено коррозией уже осевшего на катоде металла. Газовыделение является первичной электродной реакцией, т. е. зависит только от скорости взаимодействия ионов водорода с электронами.
С помощью меченых атомов В. В. Лосев снял полные поляризационные диаграммы в достаточно широком интервале потенциалов и тем самым установил соотношение между постоянными а и р в уравнении электрохимической кинетики, характеризующем катодный и анодный процессы.
Не меныйие возможности вскрывает методика определения зависимости скорости электродной реакции от сдвигов в ионных равновесиях и изменения концентрации реагирующих веществ в приэлектродных слоях.
Применение методов определения pH в католите и анолите позволяет решить вопрос об условиях торможения электродной реакции, а также о порядке реакции. Это весьма существенно для правильных представлений и выводов о механизме электродных процессов. Например, результаты исследования зависимости перенапряжения водорода на различных катодах от pH раствора позволили В. С. Багоцкому получить ценные данные о механизме этого процесса и природе реагирующих частиц при исследовании кинетики выделения водорода на ртути из растворов, содержащих НС1 и KCL
2 Причины возникновения электродной поляризации 267
Найденная в работе 3. А. Иофа и Э. А. Мазниченко полная независимость скорости выделения водорода на ртути из рас* гвора, содержащего катионы тетраметиламмония, от pH раствора (в интервале pH от 10 до 13) явилась доказательством того, что выделение водорода на ртути в щелочных растворах определяется кинетикой разряда не ионов водорода, а молекул воды.
Методы измерения pH в приэлектродных слоях были широко использованы при электроосаждении хрома, железа и других металлов для определения характера электродных реакций. Таким образом были установлены причины пассивирования растущих кристаллов гидратными пленками.
Метод измерения емкости двойного электрического слоя в сочетании с измерением заряжения поверхности позволяет судить о характере адсорбционных пленок та металлах. Определение емкости электрода переменным током заключается в том, что поверхности металлического образца и раствору сообщают малые количества электричества и измеряют изменение потенциала электрода.
Для нахождения величины заряда поверхности определяют количество электричества, которое необходимо сообщить границе раздела металл — раствор в момент ее образования. При этом предполагается, что все количество электричества тратится на заряжение поверхности, т. е. электрод обладает свойствами идеальной поляризуемости. Кривыми заряжения можно охарактеризовать зависимость потенциала электрода от сообщенного ему количества электричества. Кроме того, по этим кривым можно определить емкость электрода, а следовательно, и истинную поверхность его, что крайне необходимо при исследовании характера электродной поляризации и свойств пассивных пленок.
Весьма интересные выводы позволяет сделать исследование зависимости тока обмена от концентрации реагирующих веществ. X. Геришер на основании прямых измерений плотности тока обмена i0 цинкового и кадмиевого электродов при* равновесии пришел к выводу, что в большинстве случаев в разряде участвуют комплексы с низким координационным числом и наименьшим отрицательным зарядом.
Описанные в данной главе методы исследования электрохимических процессов не претендуют на исчерпывающую полноту, но нужно подчеркнуть, что многие из них весьма существенно влияли на развитие электрохимической кинетики в установлении современных теоретических представлений, позволяющих объяснить опытные данные, накопившиеся в технической электрохимии.
268
Теоретические основы электрохимии
ЛИТЕРАТУРА
Алексеев Н. Г., Прохоров В. А., Чмутов К. В. Электронные приборы и схемы в физико-химическом исследовании, Госхимиздат, 1961.
Байда Л. И., Добро тво р ский Н. С., Оршанский Д. Л. и др. Электрические изме|рения. Госэнергоиздат, 1950.
Баймаков Ю. В., Журин А. И. Электролиз в гидрометаллургии. Метал-лургиздат, 1963.
Багоцкий В. С., Яблокова И. Е. ЖФХ, 23, 1949, 413.
Баграмян А. Т. и Соловьева 3. А. Методы исследования электроосаждения металлов. Изд. АН СССР, 1960.
Ge,ri scher Н. Z. phys. chem., 1953, 302.
Делахей П. Новые приборы и методы в электрохимии. ИЛ, 1957.
Долин П. И., Эршлер Б. и Фрумкин А. ЖФХ, 14, 1940, 886.
Иофа 3. А., Мазниченко Э. А. Труды четвертого совещания по электрохимии. Изд. АН СССР, 1959.
Левин А. И., Сотникова В. И. Емкость двойного слоя и определение истинной поверхности электрода в процессе снятия поляризационных кривых. ЖОХ, 13, 1943, 661.
Левин А. И. и Фаличева В. И. Научные доклады высшей школы, Химия и химическая технология, 1958, № I.
Левин А. И., П о м о с о в А. В., У к ш е Е. А. Руководство к лабораторным занятиям по теоретической электрохимии. Изд. УПИ им. С. М. Кирова, 1957.
Левитин Е. А. Электронные лампы. Госэнергоиздат, 1960.
Лопатин Б. А. Ламповые гальванометры постоянного тока. Гостехиздат, М.—Л., 1952.
Лосев В. В. ЖФХ, 30, 1956, 1402.
Труды совещания по действию поверхностно активных веществ на процессы электроосаждения. Изд. АН Литовской ССР, 1957.
Плесков В. А. иМиллер Н. Б. Труды совещания по электрохимии, Изд, АН СССР 1953 165
Пальм У. В.’ Паст В. Э„ Резбеп В. А. ЖФХ, 35, № 5, 1961, 1136, Рой тер В. А., Юза В. А., Полуян Е. С. ЖФХ, 13, 1939, 605.
Скор че летти В. В. Теоретическая электрохимия. Госхимиздат, 1959.
Федотьев’ Н. П., Алабышев А. Ф., Ротинян А. Л., .Вячеславов П. М., Жив отинский П. Б., ГальнбекА. А. Прикладная электрохимия. Госхимиздат, 1962.
Фрумкин А. Н., Луковцев П. Д. Химическая наука и промышленность, III, № 4, 1958, 410.
Фрумкин А. Н. Некоторые итоги развития работ по механизму электрохимических реакций. Труды четвертого совещания по электрохимии. Изд. ДМ СССР 1QKQ
Эршлер Б. В. ЖФХ, 22, 1948, 683.
Глава X
' ДИФФУЗИОННАЯ кинетика и теория КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИИ
§ 1. Диффузионные ограничения как причина торможения электродных реакций
В процессе электролиза в приэлектродных слоях электролита наблюдаются заметные изменения концентраций реагирующих веществ (молекул, ионов), вследствие чего возникает перемещение частиц в направлении убывания их концентрации. Если скорость доставки ионов более медленная, чем скорость электрохимического -акта разряда или ионизации, то кинетика электродной реакции в целом определяется концентрационной поляризацией.
Смещение потенциала электрода под влиянием изменения концентрации электролита при электролизе, вызванное замедленностью доставки ионов к поверхности электрода или соответственно замедленностью отвода их в глубь раствора, принято называть концентрационной поляризацией.
В результате изменения концентраций в приэлектродных слоях возникает диффузия, которая способствует некоторому выравниванию концентраций вещества. Кроме того, процесс электролиза сопровождается самопроизвольным движением электролита под влиянием джоулева тепла и газовыделения на электродах. Возникающие при этом конвекционные токи жидкости направлены обычно вниз у анода и вверх у катодной поверхности. В еще большей степени процесс конвективной диффузии проявляется! при циркуляции или перемешивании электролита. Следовательно, для точного фиксирования концентрированных изменений, возникающих у электродов при электролизе, необходимо учитывать особенности конвективной диффузии.
Однако теория конвекции была разработана сравнительно-недавно и то для простейшего случая. Поэтому закономерности концентрационной поляризации устанавливают обычно, исходя из представлений Нернста, т. е. не принимая во внимание движения жидкости вблизи поверхности электрода.
270
Теоретические основы электрохимии
Количественная теория диффузии была развита впервые швейцарским физиком А. Фиком (1855 г.) Причина диффузии кроется в наличии градиента концентрации С по направлении* X, т. е. Первый закон Фика для массы вещества dm, диф-dX
фундирующей за время dr через поверхность S, при условии, если градиент концентрации перпендикулярен 5, выразится равенством
dm — — DLS dx.
dx
(Х,1>
dC
Минус указывает на то, что dm > 0, если —— < 0, т. е. если С dX
падает с возрастанием X. Здесь D, — коэффициент диффузии ионов, измеряемый количеством вещества, диффундирующим
, с , dC
за время dx = 1 через поверхность о = I при градиенте —— = dX
= 1; размерность Di-—cm1!сек. Если принять, что И,- не зависит от С, то из выражения (Х,1) получается (второй закон Фика *:
(Х,2>
di ‘ dX2
Это означает, что изменение С со временем в данной точке электрохимической системы может происходить только в случае раз-„ dC
личии градиентов---- с обеих сторон этого элемента, т. е. ког-
dX
d2C dC d2C
да не равно нулю. Когда же = const, то = 0
и по (Х,2) — = 0. di
Остановимся на примере, когда с изменением плотности тока будет меняться концентрация молекул или ионов, участвующих в электродной реакции.
Возьмем простейшую электрохимическую ячейку, типа Me+\MeA,H2Oj~Me,
где один и тот же вид ионов участвует в катодном и анодном процессах. В этом случае на катоде будет протекать реакция
/Иег+ + ге —» Me,
на аноде
Me— ze-+Mez+.
1 Для случая, когда градиент концентрации dC в каждой точке меняет-
ся вО' времени.
Диффузионная кинетика и теория концентрационной поляризации 271
Зададимся определенной плотностью тока и выясним, как изменится при этом потенциал катода по сравнению с равновесным, предполагая, что оба этих процесса идут без химической поляризации.
При отсутствии' электрического тока (/ = 0) концентрация ионов металла во всех точках раствора одинакова и равна Со-Если же через ячейку пропускать электрический ток, то на катоде разрядятся ионы Л4ег+. Естественно при этом полагать, что в результате диффузии некоторое количество ионов подойдет к катоду из глубины раствора, но так как ионы могут перемещаться только вследствие некоторого градиента концентрации, то у поверхности катода концентрация Мег+ понизится до некоторой величины Ск, где Ск < Со-
Наоборот, в прианодном слое вследствие замедленного отвода ионов их концентрация возрастет до некоторой величины Са. Таким образом, при прохождении тока возникает разность концентраций разряжающихся ионов в приэлектродных слоях, по сравнению с объемом раствора, тем большая, чем больший ток пропускается через ячейку (рис. 98).
Если не принять специальных мер к выравниванию возникшего градиента концентрации, то вследствие того что диффузия и миграция ионов идут с конечной скоростью, некоторая разница в концентрации будет сохраняться в течение всего процесса электролиза, так что со временем возникнет торможение реакции, выражаемое некоторой величиной Лфконц- Электрохимическая реакция вступит в область диффузионной кинетики, обусловленной диффузионными ограничениями.
Концентрационная поляризация может быть легко обнаружена, так как она уменьшается под действием любого фактора, который способен увеличить скорость диффузии.
Выясним зависимость концентрационной поляризации от плотности тока для катодного процесса. Пусть 5К — площадь катода, т — время, необходимое для прохождения одного фа-радея электричества (Е), z— зарядность ионов, участвующих в катодной реакции, iK — катодная плотность тока.
Так как
«kSkt = F, то 1 г-экв вещества выделится за время F
(Х,3)
(Х,4)
т =-------
Если пк — число переноса катионов при отсутствии посторонних проводящих ток солей, то на 1Е электричества общая убыль концентрации ионов под действием тока составит (1 — пк) г-экв.
272
Теоретические основы электрохимии
Таким образом, в начальный момент пропускания тока скорость убыли концентрации в прикатодном слое будет равна:
Ряс. 98. Схема концентрационных .изменений в приэлектрод-ных слоях при различных плотностях тока
(Х,5)
(Х,6)
Как показывает опыт, убыль концентрации на границе фаз гетерогенной реакции непременно будет пополняться за счет диффузии, скорость которой можно выразить, исходя из первого закона Фика:
v2 = + = DZSK . (Х,7)
ах о
С __С
Здесь —--------------градиент концентрации; б — толщина
6
диффузного слоя, т. е. расстояние между поверхностью катода, где концентрация разряжающихся ионов Ск, и ближайшей к ней точкой раствора, характеризующейся неизменной концентрацией (Со).
С уменьшением Ск скорость диффузии будет расти. Однако при постоянной плотности тока Ск будет уменьшаться до тех пор, пока убыль концентрации катионов за счет разряда не сравняется со скоростью их поступления за счет диффузии.
Диффузионная кинетика и теория концентрационной поляризации 273
В таком случае процесс достигнет стационарного состояния, при котором концентрация ионов в каждой точке раствора уже больше не будет меняться во времени. Так как анионы А*~ на поверхности катода не разряжаются, то постоянство их концентрации означает, что они в католите не движутся ни к катоду, ни к аноду, т. е. что сумма сил *, действующих на ионы Аг~, равна нулю 1 2.
Так как в стационарном состоянии электрическая и осмотическая силы, действующие на анион Аг~, взаимно компенсируются и уничтожаются, на катион Мег+, наоборот, будет влиять общая сила, равная суммарной осмотической силе. Таким образом, скорость движения катионов будет в ----• раза больше
1 —пк
той скорости, которая получилась бы при действии одной осмотической силы. На этом основании в случае, когда концентрационные изменения в диффузном слое достигли стационарного состояния, в переносе тока в диффузном слое начинают участвовать только разряжающиеся ионы (Мег+ ), а числа переноса неразряжающихся ионов (Л-), (Л+) падают до нуля. Вот почему далее в уравнение диффузионной кинетики множитель (1 —Пк) [см. уравнения (Х,5 и Х,6)] уже не входит. Следовательно, для установившегося процесса, когда концентрационные изменения в приэлектродном слое достигли стационарного состояния, можно, несколько упрощая действительную картину, написать
(Х.8)
zF В
t
Из условия (Х,8) выражаем концентрацию разряжающихся на катоде ионов:
ск = со----iK = Со - KiK, (Х.9)
zrL>i
т. е. концентрация ионов у поверхности катода отличается от концентрации их в глубине раствора на величину, пропорциональную катодной плотности тока iK.
Изменение концентрации вещества вблизи катода должно привести к изменению потенциала электрода. В таком случае
<Р1к = <Ро + ^1пСк. ' (Х.10)
1 Первая из этих сил — сила электрического поля, вторая — сила, вызы-зающая диффузию ионов.
2 См. А. Н. Фрумкин, В. С. Б а г о ц к и й, 3. А. И о ф а, Б. Н. К а б а-: н о в. Кинетика электродных реакций. Изд. МГУ, 1952, стр. 72.
18 Заказ 1338
274
Теоретические основы электрохимии
Имея в виду, что при отсутствии тока
?г-0 = In Со, (Х,11)
I—и zp
получим, что величина концентрационной поляризации
5-|П<- <Х’12>
пользуясь формулой (Х,9), получим
----^<«1- <х-13>
zF L zFDiC0 J
Вернемся к уравнению (X, 9) и определим из него:
iK = zFDi(co~c^ . (X, 14)
Из уравнения (X, 14) следует, что плотность тока на катоде не может быть произвольно большой, так как при предельных ее значениях Ск становится равной нулю и тогда величина АфКОнп-рассчитанная по уравнению (X, 12), обращается в бесконечность. Отвечающая этому значению плотность тока может быть найдена из условия:
5 • 1
zFDfio 4пр
откуда
zFDiC0 гпр- £
(Х.15)
(Х,16)
Подставляя (X, 16) в выражение (X, 13), получим уравнение концентрационной поляризации для катодного процесса:
ДтКонц=--^г1п(1--М. (Х,17)
zF \ 1Пр )
Из равенства (X, 16) вытекает очень важное следствие, что при прочих постоянных условиях предельная плотность тока пропорциональна начальной концентрации .(точнее активности) разряжающихся ионов. Экспериментально найденная зависимость (iK ф) для разряда катионов графически представлена на рис. 99. Из графика видно, что после достижения предельной плотности тока (znp) кривая iK — ф не остается параллельной оси потенциалов, а при постепенном сдвиге потенциала в отрицательную область вновь начинает подниматься (ветвь CD).
Большинство электролитов содержит несколько сортов ионов, или молекул, способных восстанавливаться на катоде, поэтому при плотности тока выше предельных значений потенциал возрастает настолько, что становится возможным разряд других.
Диффузионная кинетика и теория концентрационной поляризации 275
более электроотрицательных веществ, присутствующих в электролите. Когда на катоде способен разряжаться только один сорт ионов металла и электродная реакция не может протекать в несколько последовательных стадий восстановления, такими веществами являются молекулы растворителя (воды) или ионы водорода, которые и начинают восстанавливаться с возрастающей скоростью в известной области потенциалов.
Рис. 99. Кривая i — <р, выражающая совместный разряд металла и водорода:
ABCD — суммарная поляризационная кривая; — доля плотности тока, идущей на разряд металлических ионов; <н2 — доля плотности тока, расходуемой на выделение водорода; /fJp — плотность предельного тока
При плотностях тока выше предельных, когда происходит совместный разряд катионов металла и водорода, ток делится между обоими разряжающимися ионами так, что выход по току металла определяется соотношением
ЛМе = ........ • Ю0%- (X, 18>
1Ме + 'н2
Таким образом, ветвь I (рис. 99) для разряда ионов металла начинается, в точке А, отвечающей равновесному потенциалу этого иона (<р'). Далее она с ростом плотности тока проходит через точки В и С, асимптотически приближаясь к предельной для металла плотности тока (inp). Ветвь II для разряда ионов водорода начинается в точке А', отвечающей равновесному потенциалу этих ионов (ф"), и пересекается с кривой I в точке С при потенциале фк и плотности тока /пр. До точки С выход по току металла составляет 100%, но начиная с этой точки он непрерывно уменьшается, согласно уравнению (X, 18), так как 18*
276
Теоретические основы электрохимии
iMe остается неизменным и равным «пр, а «н> продолжает быстро расти.
Экспериментально предельная катодная плотность тока может быть определена из условия (X, 17). Построив по точкам кривую <рг = можно найти «пр по положению «скачка» потенциала ВС (рис. 99).
Такой метод был применен О. А. Есиным и А. И. Левиным при изучении катодной поляризации меди и цинка, при электроосаждении их из раствора простых солей.
Иногда делаются также попытки теоретически рассчитать «пР по уравнению (X, 16). При этом zFCp задаются условиями опыта, число переноса может быть рассчитано по равенству:
1к пк =----~--»
4с + 1а
Приближенно коэффициент диффузии ионов £)г- рассчитывают по уравнению
D,= - RT . (Х,19)
6^rN
т. е. он обратно пропорционален коэффициенту вязкости среды и радиусу диффундирующих ионов г.
Рассматривая возможность возникновения концентрационной поляризации на аноде, по аналогии с предыдущим можно написать для растворимого электрода:
Са = Со+-1-«а, (Х,20)
, zFD,
т. е. концентрация катионов будет увеличиваться. Выражение для концентрационной поляризации на аноде может быть записано следующим образом:
ДТа= KLln(l + А-). (Х,21)
zF \ «пр ]
Наиболее часто предельный или максимальный анодный ток будет возникать из-за достижения предела растворимости соли металла, концентрирующейся в непосредственной близости к аноду (в таком случае прохождение тока через границу раздела электрод — электролит тормозится солевой пленкой, образующейся на аноде). В случае электролиза раствора сернокислой соли металла эта пленка состоит из кристаллов AfeSO4.
Иногда при работе растворимого анода образуются тончайшие, адсорбционные пленки, пассивирующие электрод. В таком случае металл перестает растворяться и посылать свои ионы в раствор. Вместо ионизации атомов металла доминирующим здесь становится другой процесс, обычно окисление анионов
Диффузионная кинетика и теория концентрационной поляризации 277
I I
кислоты или разряд гидроксильных ионов. Не вдаваясь подробно в рассмотрение процесса анодной пассивности (см. по этому вопросу гл. XV), укажем лишь, что на нерастворимом аноде течение анодной реакции окисления анионов также ограничивается при достижении предельного тока диффузии. Так, вследствие разряда анионов кислоты концентрация их в толще электролита и у анода будет отличаться. В таком случае
Са = С0----(Х.22)
zFDi
откуда
za = ..гГ£>г^0-~Са1. (Х,23)
Здесь Со — концентрация анионов кислоты в глубине раствора электролита; Са — концентрация анионов кислоты у анода.
Из уравнения (X, 22) следует, что в полной аналогии с катодным процессом плотность тока на аноде может достичь предельного значения из-за замедленной доставки анионов, участвующих в электродной реакции, к аноду.
§ 2. Развитие теории концентрационной поляризации
Изложенная теория концентрационной поляризации Нернста достаточно проста и наглядна. Однако она имеет тот существенный недостаток, что не принимает во внимание конвекции электролита, не раскрывает физического смысла и значений диффузного слоя.
Теория концентрационной поляризации, основанная на представлениях Нернста, исходит из следующих положений: здесь постулируется существование вблизи электрода сравнительно толстого слоя жидкости, названного «диффузным» слоем, в котором происходит основное изменение концентрации. В остальной части раствора концентрация считается неизменной от точки к точке.
В этих условиях концентрационное и омическое (связанное с сопротивлением раствора) торможение электродной реакции определяется свойствами ионов и толщиной пограничного слоя. Однако толщина этого слоя 6 из теории Нернста не может быть рассчитана и в каждом отдельном случае находится из опыта. Помимо этого, в теории Нернста не приведены достаточно убедительные аргументы в пользу самого существования диффузного слоя. Поэтому многие исследователи указывали на несостоятельность этой теории, так как она не дает истинных представлений о гидродинамических явлениях, происходящих вблизи поверхности электрода при движении электролита. Необхо
278 Теоретические основы электрохимии
димость в каждом случае специально измерять ^величину 6 чрезвычайно сильно снижает возможность практического использования нернстовской теории концентрационной поляризации.
В 1938 г. А. Г. Самарцев предложил экспериментальный способ для определения толщины диффузного слоя 6, основанный на измерении коэффициента преломления раствора с помощью поляризационного интерферометра Лебедева. Измерения, проведенные А. Г. Самарцевым, показали, что для растворов сернокислой меди значение 6 заметно меняется в зависимости от скорости движения электролита. Таким образом, затруднения возникли при истолковании условий доставки реагирующих веществ к электродной поверхности при размешивании электролита. Это привело к необходимости разработать другую точку зрения на рассматриваемые процессы, свободную от произвольных представлений о диффузном слое.
Это и было сделано В. Г. Левичем и А. Н. Фрумкиным. На основе принципов гидродинамики они дали общее решение вопроса переноса вещества к электроду в размешиваемых растворах.
Известно, что скорость доставки ионов к электроду определяется тремя факторами — скоростью диффузии, скоростью миграции (т. е. переноса ионов под действием электрического поля) и скоростью движения или перемешивания раствора электролита, т. е. скоростью конвективного переноса. Таким образом,
~ = ^днф -И ^мнгр + ^ОНВ. (X.24)
Задача, следовательно, сводится к определению всех составляющих скорости движения ионов к электроду и особенно влияния перемешивания. Практически электролит перемешивается в результате естественной и искусственной конвекции. Естественная конвекция обусловлена главным образом неравномерным распределением температуры в растворе и газовыделением у электродов. Такая конвекция влияет на перенос ионов и на распределение плотности тока на электродах, но трудности ее расчета делают теоретические выводы в этом направлении весьма сомнительными. Поэтому с точки зрения теории и практики электрохимических исследований важно рассмотреть закономерности искусственной конвекции. При помощи искусственного перемешивания можно значительно увеличить скорость доставки реагирующих веществ к поверхности электродов и тем самым намного повысить предельную плотность тока, что необходимо при практическом осуществлении ряда технологических процессов.
Наиболее просто поставленная задача решается в случае поляризации электрода, представляющего собой вращающийся
Диффузионная кинетика и теория концентрационной поляризации 279
плоский диск с достаточной площадью. Диск вращается вокруг оси, перпендикулярной к плоскости электрода, с постоянной угловой скоростью со. Скорость вращения такова, что движение раствора остается ламинарным, характеризуясь определенным значением числа Рейнольдса ’.
Исследование гидродинамики движения жидкости в этих условиях, осуществленное В. Г. Левичем, приводит к следующей картине: вдали от вращающегося диска (диаметр которого до
статочно велик) жидкость движется нормально в направлении к электроду (рис. 100) со скоростью v0, т. е. с исходной скоростью струи. В тонком слое, непосредственно примыкающем к поверхности вращающегося диска, жидкость приобретает вращательное движение (рис. 101,а) с угловой скоростью, возрастающей по мере приближения к поверхности диска, до значения со (угловая скорость вращения диска). Наконец, ввиду наличия центробежных сил в «области увлечения» жидкость приобретает так-
ского (6) пограничных слоев
же и радиальную скорость.
Пользуясь приведенными представлениями о движении жид
кости, В. Г. Левич решил задачу для случая, когда катионы восстанавливаются именно на таком диске — катоде. При этом
оказалось, что весь граничный слой раствора по концентрации может быть разбит на две области. В первой, простирающейся от катода в глубь раствора на расстояние б, концентрация меняется от Ск на электроде до Ср. Как видно, этот слой (диффузный) значительно меньше толщины слоя Ьо, в котором меняется
скорость движения жидкости относительно электрода, а концентрация не меняется и равна Со. Такое распределение концентраций в растворе очевидно, но В. Г. Левич его не постули
рует, а приходит к нему на основании анализа законов гидродинамики и диффузии. Конечным выводом его теории явилось
следующее уравнение для предельного тока:
Ка + zk) DjCpF 0,896га
(Х,25)
1 Числом Рейнольдса называют безразмерное отношение Re — — , где V
I — характерные размеры твердого тела, .например плоского электрода, погруженного в струю жидкости, двигающуюся со скоростью Vo относительно электрода, v—кинематическая вязкость среды. Критическое значение числа Рейнольдса в пограничном слое на пластинке Re >. 10 s.
280 Теоретические основы электрохимии
где zK и 2а — валентности катиона и аниона;
Di — коэффициент диффузии разряжающихся ионов.
Полный ток, текущий через раствор, пропорционален площади электрода и равен |i — 2№inp, где фактор г введен из-за учета обеих сторон диска.
Уравнение (Х,25) показывает, что к вращающемуся катоду течет ток, интенсивность которого не может превышать 1Пр-В случае прохождения тока, превышающего это предельное значение, возрастает отклонение раствора от нейтральности, появляются очень высокие сопротивления и ток вновь падает до /пр-
Рис. 101. Распределение линии тока жидкости вблизи вращающегося диска (а) ,и неподвижного электрода (б)
Из уравнения (Х,25), кроме того, следует, что значения inp отвечают такому распределению ионов в растворе, что концентрация Со линейно падает до нуля вблизи вращающегося электрода. Это падение происходит внутри слоя, имеющего толщину
£)*/а^‘/в
8=1,62—^—. (Х,26)
ш '2
Существование такого диффузного слоя, как мы видели, постулировала и теория Нернста, но она не давала способа расчета его толщины. Таким образом, точная теория согласуется с качественной теорией Нернста. Вместе с тем в рассматриваемом случае величина 6, фигурирующая в уравнении (Х,25), зависит не только от скорости движения жидкости, но, как это следует из (Х,26), и от коэффициента диффузии D{ . Отсюда следует, что 6 зависит от природы участвующих в электродной реакции веществ (катионов и анионов) и таким образом приобретает определенный физический смысл.
Диффузионная кинетика и теория концентрационной поляризации 28J
Очевидно, что между толщиной диффузного слоя 6 и гидродинамическим слоем жидкости до (слой Прандтля), увлекаемой' вращающимся диском, существует некоторое соотношение. Математический расчет показывает, что это соотношение может быть выражено следующим образом:
d^(-^-Y/s80. (Х,27>
\ V /
Для водных растворов Дит имеют значения порядка 10-5 и 10-2 см2-сек~х. Следовательно, толщина диффузного слоя д составляет примерно одну десятую от толщины гидродинамического слоя до- Дальнейший анализ показывает, что соотношение (Х,27) имеет весьма общий характер и оправдывается во всех случаях, когда движение жидкости характеризуется числом1 Рейнольдса, сильно превышающем единицу. В общем случае, когда жидкость движется вдоль неподвижного электрода (рис. 101, б), скорость ее течения, начиная с некоторого расстояния до (толщина граничного слоя Прандтля1). уменьшается, доходя до минимума на поверхности электрода. Тогда толщина пограничного слоя до может 1быть определена из соотношения
(Х,28>
У v0
где Со — скорость движения струи относительно твердого тела; х— расстояние от точки набегания струи до’рассматриваемой точки.
Уравнения (Х,27) и (Х,28) показывают, что толщина пограничного слоя в случае раствора, текущего вдоль электрода, зависит, вообще говоря, не только от скорости движения струи относительно твердого тела и кинематической вязкости жидкости v, но и от положения выбранной точки на поверхности электрода. Этот вывод интересен, так как позволяет, например, предвидеть изменение толщины гальванического покрытия в разных точках катода.
Очевидно также, что и предельная плотность тока в различных точках электрода различна, так как она зависит от х. Для плоской пластины, достаточно большой, чтобы можно было пренебречь влиянием краев, расчет дает:
/пр = 0,67zFC0O;S1n/2v-,/“/if/% (Х,29>
где I и h — геометрические размеры пластины.
Из уравнения (Х,29) вытекает следствие: предельная плотность тока пропорциональна коэффициенту диффузии не в пер
1 Слой |Празддтля—гидродинамический граничный слой, внутри которого-нарушается равномерное движение струи.
282
Теоретические основы электрохимии
вой степени, как в теории Нернста, а в степени 2/з. Это хотя и не имеет практического значения, но показывает, что в теории .Левича величина 6 не задается произвольным распределением скоростей в жидкости, но определяется протеканием самого процесса диффузии. В случае, если движение раствора носит турбулентный характер (число Рейнольдса очень велико), предельная плотность тока зависит также и от состояния поверхности электрода. Неоднородность поверхности приводит к снижению величины предельного тока. Количественной теории для этого случая построить пока не удалось.
Полученное В. Г. Левичем уравнение для предельного тока диффузии на вращающемся дисковом электроде не содержит произвольных констант и поэтому может служить основой для расчетов на твердом электроде в такой же мере, как формула 'Илькевича 1 применима для капельного ртутного электрода.
Вращающийся твердый дисковый электрод имеет то преимущество перед ртутным капельным электродом, что позволяет изучать также анодные процессы, идущие при значительных плотностях тока. Все же применимость теории Левича ограничивается следующими условиями:
1) режим движения жидкости должен быть таков, чтобы число Рейнольдса было гораздо больше единицы 1);
2) теория не учитывает зависимости коэффициентов диффузии от концентрации;
3) теория не приложима к случаю естественной конвекции;
, 4) вычисление предельного тока было проведено лишь в двух случаях: в случае бинарного электролита и когда концентрация ионов, разряжающихся на катоде, очень мала по сравнению с концентрациями остальных ионов.
Опытная проверка теории Левича оказалась весьма плодотворной; в случае бинарного электролита предельный ток, по (Х,25), меняется прямо пропорционально коэффициенту диффузии разряжающихся ионов Di и обратно пропорционально корню .кубическому из эффективного коэффициента диффузии D. Приближенно можно считать, что Dj^Dn тогда
Такое соотношение подтверждено для случая растворения металла опытами Эйкена и других исследователей, причем показатель степени был найден равным 0,7—0.83.
Что касается зависимости /Пр от вязкости раствора, то она .может быть охарактеризована выражением
гп₽ ТТЛ •
1 См. стр 287.
Диффузионная кинетика и теория концентрационной поляризации 283
С другой стороны, коэффициент диффузии £>г также является функцией вязкости, причем
и, следовательно,
_ Л гпр ~ v 6
Это соотношение подтверждается опытом. Нйконец, наибольший интерес представляет зависимость предельного тока от скорости перемешивания. В. Г. Левич проверил свою теорию по данным, имеющимся в работе Эйкена. Результаты этой проверки показывают хорошее согласование теории и опыта:
Угловая скорость <и. рад . . . 88,3 19,3
«пр- теор» а . . . 1,33 -10~4 1,15 -10-4
«пр. опытн, а................ 0,671-10-4 0,685-10—4
Ю. Г. Сивер и Б. Н. Кабанов в целях проверки теории Левина изучали поляризацию при разряде ионов водорода в растворах 0,002-н. H2SO4 +0,1-н. K2SO4 и 0,002-н. НС1 +0,1-н. КС1 и др. на вращающемся со скоростью 1—40 об!сек дисковом катоде. Оказалось, что предельный ток прямо пропорционален корню квадратному из числа оборотов катода п. Это согласуется с теорией Левича, так как угловая скорость связана с числом оборотов соотношением
ш = 2ют.
Из изложенного следует, что подбором соответствующих условий, а именно: а) увеличением начальной концентрации раствора; б) повышением катодной -плотности тока; в) увеличением температуры; г) повышением интенсивности перемешивания концентрационная поляризация может быть доведена до минимума. Однако каким бы сильным ни было перемешивание и как бы ни были благоприятны остальные условия, полностью исключить концентрационную поляризацию затруднительно.
Как общее правило, при плотностях тока, не очень близких к предельной, величина концентрационной поляризации относительно невелика. Так, при -у— = 0,7
*пр
Дшк = — 0,0596 1g о, 1 = — 0,0596 в. 2 2
Увеличение плотности тока до 0,99 «пр приводит также к незначительному росту концентрационной поляризации, и она достигает величины 0,1192 в.
284
Теоретические основы электрохимии
При плотностях тока удовлетворяющих неравенству I < 0,5 inp концентрационная поляризация не превышает — 0,018 в.
§ 3. Полярографический метод анализа
Полярографический метод анализа был предложен в 1922 г, чешским ученым Ярославом Гейровским, который установил, что концентрация и природа восстанавливающихся или окисляющихся на ртутном капельном электроде веществ могут быть определены по кривым потенциал — плотность тока, т, е. по полярограммам, полученным с помощью двух электродов, один из которых очень малого размера (ртутная капля) и подвержен весьма сильной поляризации.
Сущность этого метода заключается в следующем: предположим, что в растворе имеется несколько сортов ионов; в таком: случае электродная реакция может протекать в несколько последовательных стадий. Повышая приложенное к полярографической ячейке напряжение с помощью делителя напряжения, мы первоначально получим кривую, характерную для восстановления того иона, потенциал которого имеет наиболее положительное значение. Во время катодного восстановления этого иона ток, который тратится на совместный разряд других ионов, еще очень мал и им можно пренебречь.
Так как предельная плотность тока /пр пропорциональна концентрации разряжающихся на электроде ионов, т. е. 1щ> = КСОт то площадка предельного тока соответствует концентрации восстанавливающихся ионов. При дальнейшем сдвиге потенциала в отрицательную сторону и приближении его к потенциалу полуволны другого иона, присутствующего в растворе, скорость второго процесса возрастает и он накладывается на первый процесс. Таким образом, на полярограмме появляется вторая волна и соответствующая ей площадка предельного тока inP, высота которой равна сумме предельных токов для восстановления первого и второго сортов ионов. Дальнейшее увеличение приложенного извне напряжения приводит к образованию новых ветвей на поляризационной кривой, характеризующих разряд соответствующих ионов (рис. 102).
При достаточном различии потенциалов каждый из интересующих нас ионов может быть расшифрован в отдельности. Как показывает опыт, в электродной реакции могут участвовать не только ионы, но и нейтральные молекулы. Так, например, растворенный в электролите кислород может восстанавливаться на платиновом катоде по реакции
О2 + 4е + 2Н2О-4ОН“.
Диффузионная кинетика и теория концентрационной поляризации 285
•ионов на ртутном
0тносигпельная насыщенность злектрода <f-B
Рис. 102. Потенциалы выделения различных электроде
течения органических
реакций и да-веществ. По-
Таким образом, полярографический метод пригоден для анализа и молекулярных веществ.
Область применения полярографии в настоящее время чрезвычайно расширилась. Этот метод может быть применен не только для быстрого определения природы и концентрации большинства катионов и ряда анионов, содержащихся в растворах в самых ничтожных количествах, но и в случае определения разнообразнейших органических соединений, способных электрохимически восстанавливаться или окисляться на электроде. В анализе многих органических соединений полярографический метод вообще незаменим. В •связи с этим можно предполагать, что в будущем эта область окажется наиболее важной для применения полярографии. Достаточно указать, что с помощью 'Полярографии •представляется возможным определять механизм
же некоторые особенности строения органических лярографический метод позволяет, кроме того, исследовать состав и прочность комплексных соединений, растворимость различных труднорастворимых веществ, каталитическое действие разнообразных катализаторов.
Теоретический фундамент полярографии в настоящее время настолько окреп, что с помощью имеющихся теоретических соотношений можно на основании опытных данных (по полярограмме) делать выводы: а) о числе электронов, принимающих участие в электродном процессе; б) об «обратимости» процесса, т. е. о том, определяется ли электродный процесс только замедленностью диффузии или наряду с процессом диффузии происходят также другие замедленные процессы; в) о величине коэффи-фицента диффузии вещества, которое восстанавливается на электроде; г) о величине окислительно-восстановительного потенциала.
Особенно широко применяется полярография в цветной металлургии. Здесь в большинстве случаев удается путем простых операций и за короткое время получить результаты, по точности обычно даже превосходящие другие методы анализа (в особенности при малых содержаниях вещества). Это относится в пер
286 Теоретические основы электрохимии
вую очередь, к следующим металлам (или их рудам, технологическим полуфабрикатам и растворам, а также к их солям):, меди, кадмию, свинцу, олову, цинку, кобальту, алюминию, магнию.
В настоящее время разработан ряд методик по определению примесей в цветных металлах и их соединениях. На многих заводах цветной металлургии полярографические методы успешно применяются в контроле производства.
Точность полярографического метода достигает 2%, а чувствительность его лежит в пределах концентраций 10-4—10-5. Качественные результаты могут 'быть получены при еще меньших концентрациях веществ.
Все эти свойства полярографического метода сделали его практически незаменимым при точнейших аналитических исследованиях в научных целях, а также в промышленных лабораториях.
Наибольшее .распространение получил полярографический анализ с ртутным капельным электродом, хотя в последнее время начинает развиваться полярография с твердым и особенно амальгамным электродом.
Капельный ртутный электрод может быть использован в качестве катода и в качестве анода. Однако при этом необходимо полностью исключить ионизацию самой ртути.
Ртутный капельный электрод имеет ряд преимуществ: 1) легкость достижения предельной плотности тока из-за малого диаметра капли — катода (0,05 мм); 2) непрерывное обновление электрода, обеспечивающее постоянство условий и воспроизводимость результатов анализа; 3) достаточную устойчивость металлической ртути в кислотах и щелочах; 4) высокое перенапряжение водорода на ртути, что обеспечивает возможность определения ряда электроотрицательных катионов.
Ртутный капельный электрод в простейшем виде состоит из стеклянного капилляра, из которого ртуть по каплям вытекает и падает на дно сосуда с испытуемым раствором. Диаметр капилляра обычно составляет 0,03—0,05 мм, так что период капания ртути составляет 3—5 сек. Для сохранения постоянства периода капания (т. е. времени образования и отрыва одной капли) необходимо поддерживать постоянным уровень ртути в воронке, для чего ее соединяют с капилляром с помощью гибкого резинового шланга. Чаще применяются электролизеры с разделенным катодным и анодным пространством. При этом в качестве второго электрода Ханода) служит каломельный или ртутно-окисный электрод второго рода, потенциал которого остается практически неизменным из-за относительно большой поверхности ртути на дне сосуда по сравнению с ртутной каплей (катодом).
Диффузионная кинетика и теория концентрационной поляризации 287'
В 1924 г. Гейровский и Шиката создали весьма совершенный
фоторегистрирующий прибор, который и до настоящего времени подвергался лишь небольшим изменениям.
Общая электрическая схема полярографа с автоматической записью приведена на рис. 103.
Однако при внедрении полярографического метода в производство выяснилось, что фотопроцесс сильно усложняет полярографические анализы. Тогда вместо фотополярографа стали создавать визуальные поляро-графы (или полярометры).
Принципиальная схема визуального полярографа представлена на рис. 104.
Ю
Рис. ЛОЗ. Схема полярографической •установки:
1 — аккумулятор; 2 — вольтметр; 3 — реостат; 4 — зеркальный гальванометр;
5 — электромотор; 6 — контакт; 7 — осветитель; 8 — барабанный мостик; 9 — электролизер; 10 — шунт гальванометра;
11 — барабан со светочувствительной бумагой для записи полярограмм
Рис. >104. Принципиальная схема визуального (полярографа:
1 — источник тока; 2 — делитель напряжения (потенциометр); 3 — электролизер;
4 — гальванометр
Преимущество визуальных полярографов — возможность быстро производить съемку полярограммы, так как в противоположность регистрирующим приборам-самописцам отсчеты в визуальных полярографах производятся по отклонению зайчика зеркального гальванометра.
Л. Илькович вывел уравнение для диффузионного тока на ртутном капельном электроде:
inp = 0,732г/7 (Х,30)
где 0,732 — числовой множитель, вытекающий из геометрической характеристики капельного электрода;
288
Теоретические основы электрохимии
zF— число кулонов, необходимое для того, чтобы на границе ртуть — раствор прореагировал один моль вещества;
С — концентрация разряжающегося вещества, моль/см?; Di — коэффициент диффузии ионов, см21сек\
m — масса ртути, вытекающая из капилляра за одну секунду, г/сек-,
т — период капания, сек.
Произведение зависит от свойств капилляра и на-
зывается постоянной капилляра. Д. Илькович при этом исходил из того положения, что скорость образования капли и ее отрыва зависит только от трения ртути в капилляре и остается неизменной во времени при постоянной высоте столба ртути, под давлением которого капля вытекает. Уравнение Ильковича оправдывается на опыте в том случае, если период капания т превышает 4 сек.
Поскольку величина т2/»т*/в зависит не только от свойств капилляра, но и от поверхностного натяжения ртути, которое определяется зарядом ее поверхности и потенциалом, то значения практически постоянны лишь в интервале потен-
циалов от 0 до —0,8 в (относительно насыщенного каломельного электрода). При точных измерениях всегда необходимо учитывать возможность снижения величины trf1*'?'* с дальнейшим увеличением потенциала.
Чтобы вычислить диффузионный ток по уравнению Ильковича, необходимо знать коэффициенты диффузии отдельных ионов, участвующих в электродной реакции:
10 5см2/сек D. . 10 5см2/сек Dt • 10 ЬсмР/сек
н+ . . . . 9,34 Zn2+ . . . 0,72 BrO^ . . . . 1,44
Na+ . . . . 1,35 Cu2+ . . . 0,72 Fe(CN)g— . . 0,89
к+ . . . . 1,98 Ni2+ . . . 0,69 CuCl2- . . 0,733
Т1+ . . . . 2,00 он- . . . 5,23
РЬ2+ . . . 0,98 Cl~ . . . . 2,03 Cu(NH3)|+ 0,885
Cd2+ . . . 0,72 JO? • . . 1,09 Cd(NH3)2+ 0,923
В разбавленных растворах эти коэффициенты могут быть подсчитаны по уравнению
д. = -gL^' . , (Х,31)
где I)г — подвижность иона; »
— его валентность.
При 25 °C это уравнение имеет вид:
Di = 2,67 • 10-7 смг/сек. (Х,32)
Zi
Диффузионная кинетика и теория концентрационной поляризации 289
Следует учитывать, что при комплексообразовании коэффициенты диффузии ионов изменяются.
Уравнение Ильковича выведено для случая, когда концентрация исследуемого вещества в растворе очень мала. При таких именно условиях практически и применяют установки для полярографического анализа. При этом для повышения электропроводности в раствор добавляют в избытке какой-либо индифферентный электролит, например КО, K2SO4, KNO3 и т. п., потенциал выделения которого значительно больше, чем у определяемого иона. В этих условиях исследуемые ионы практически не переносят никакого тока и потому предельный их ток равен диффузионному (id).
Если бы мы подвергали электролизу чистый раствор исследуемого вещества без индифферентного электролита (или, как его часто называют, фона), то предельный ток мог бы быть представлен в виде суммы:
lnp = h + *м + г'з» (Х,33)
где /м — ток миграции;
/з — ток заряжения.
Ток заряжения (i3) протекает при поляризации капельного ртутного электрода независимо от того, происходит ли на электроде какой-либо электрохимический акт или нет. Он обусловлен тем, что на свежеобразующейся поверхности ртути создается двойной слой. На ртутном электроде, обладающем постоянной поверхностью, после образования двойного электрического слоя устанавливается вполне определенный потенциал по отношению к потенциалу в глубине раствора или к потенциалу нулевого заряда. В таком случае ток заряжения становится постоянным.
Ток миграции (гм) возникает под действием электрического поля. К электроду мигрируют все ионы, имеющие противоположный знак, но дальнейшее их поведение здесь различно — часть ионов разряжается (уводится из электролита), другие же ионы, не способные разрядиться при данном потенциале, остаются у поверхности катода в количестве, соответствующем заряду поверхности. Электрическое поле электрода экранируется этими ионами, и в таком случае непрерывного подтягивания ионов из глубины раствора не происходит.
В полярографическом анализе создаются условия, при которых электрическое поле экранируется ионами постоянного индифферентного электролита. Для этого необходимо, чтобы концентрация постороннего электролита была по крайней мере в 100 раз больше испытуемого.
При электролйзё очень разбавленного раствора с применением фона iM становится пренебрежимо малым и плотность тока |9 Заказ 1338
290
Теоретические основы электрохимии
в любой момент, очевидно, равна
4 = К(С0-Ск), ' (Х,34)
где К — коэффициент пропорциональности;
Со, Ск — концентрация вещества в глубине раствора и на поверхности капли ртути (катода).
При наступлении предельного тока Ск = 0 и, следовательно.
/Пр = = К.С0. (Х,35)
Таким образом, по формулам (X, 34) и (X, 35)
ск = . (Х,36)
Сила тока в любой точке полярографической волны определяется кинетикой электрохимической реакции и скоростью транспортировки вещества, участвующего в электродном процессе.
Если электрохимическая реакция протекает настолько быстро, что на электроде достигается электрохимическое равновесие, то сила тока обусловливается только скоростью доставки вещества, восстанавливаемого или окисляемого на электроде. Такие волны принято называть обратимыми.
При обратимом выделении металлических ионов на ртути
Мег~ 4- ze + Hg Ate(Hg) потенциал электрода выражается формулой
«ад
zr ьам
где Сам — концентрация атомов металла в амальгаме;
Фам — стандартный потенциал амальгамы.
Концентрация амальгамы, по Гейровскому и Ильковичу, пропорциональна плотности тока, т. е.
Сам = №
поэтому
+ (Х.38)
Значение потенциала в точке i = в полярографии принято называть потенциалом полуволны <p,/s. При этом
ln^z£ = 0
i
И
(Х,39>
Диффузионная кинетика и теория концентрационной поляризации 291
Таким образом, получаем уравнение для концентрационной поляризации на капельном ртутном электроде:
RT jn М *' zF i
(X,40)
Последнее уравнение называется обычно уравнением обратимой полярографической волны. Как следует из уравнения (X, 39) потенциал полуволны при разряде ионов металла с образованием амальгамы зависит от нормального потенциала
-0,5
-1,0
-1,5
2(1
1
Fe^b^P^S^^r^Ni^fe^CrW^rAl^Ba^NH^
Л
ш
П
-С.5
-1,0
-15
Cd^Pb^Fe3*
~^05 чЦТ
1 Г i I i 1 ...
CtF Si3*- Pb! + №г*
-0,5 -10 -15
-2,0
ВгОд О4 50д
NOg
Ряс. 105. ‘Полярографический спектр (потенциалы итолуволн -в различных средах):
I — нейтральная среда; II — щелочная среда; III — цианистая н виннокислая среда; IV — анионы
амальгамного электрода. Величина потенциала полуволны является вполне определенной характеристикой данной реакции и целиком характеризует природу разряжающихся ионов. С помощью специальных таблиц (полярографических спектров, рис. 105), можно по опытным данным определить состав испытуемого раствора. Можно также решить вопрос о том, в каком растворе вести полярографический анализ сплава. Так, например, рассмотрение полярографического спектра показывает, что примесь сурьмы в свинце следует определить в солянокислом растворе, так как в нем потенциал сурьмы более положительный, и, наоборот, примесь свинца в сурьме нужно определять в щелочном растворе. Таким образом, рациональный выбор фона (комплексообразователя) позволит избежать необходимости в разделении (осаждении, фильтровании и т. п.) элементов.
19*
292
Теоретические основы, электрохимии
Если подобными методами не удается достигнуть положительных результатов, то полярографическому определению должны предшествовать специальные химические манипуляции.
Таблицы потенциалов полуволн ряда неорганических и органических веществ приводятся в соответствующих источниках.
Потенциал полуволны (рис. 106) не зависит от концентрации восстанавливаемого иона, а только от его природы. Величина <p,Zi служит, таким образом.
Рис. il<06. Сдвиг потенциалов восстановления и тостоянство потенциалов полуволны:
Hi, Нг, Из — высота предельного тока
для качественной характеристики ионов, имеющихся в испытуемом растворе.
Таким образом, из уравнения (X, 40) можно вычислить число электронов, принимающих участие в электрохимической реакции. Чем больше число электронов (г), тем более круто поднимается полярографическая волна.
Потенциал полуволны может быть определен графически. Для этого строят по экспериментальным данным прямую в координатах In =/(ф) и определяют значение <р, отве-
чающее нулю на оси ординат. При обратимом электролизе тан-гене угла наклона равен (0,0596 при 25°С, если z — 1).
Уравнение (X,40) верно только для случая обратимого электролиза, или, иными словами, когда химическая поляризация при электролизе отсутствует. В противном случае формула Гейровского — Ильковича становится непригодной.
Практически полярограмма [кривая i = f(<p)] имеет вид, показанный 1на рис. 102. Зигзагообразность ее объясняется природой капельного электрода, т. е. изменением плотности тока за время жизни одной капли. Природа присутствующих ионов определяется по потенциалам полуволн, а по предельным токам (/пр) находят их концентрацию.
При полярографировании могут мешать сопутствующие ионы, имеющие близкие потенциалы выделения, а также растворенный в электролите кислород, который удаляют из нейтральных или кислых растворов продуванием инертного газа (водорода, азота), а из щелочных и аммиачных растворов—химическим путем. Влияние ионов примесей также может быть устранено химическим путем (осаждением или переводом в комплексы) или применением компенсации, при которой поля
Диффузионная кинетика и теория концентрационной поляризации 293
рографическая волна подавляется электродвижущей силой противоположного направления.
Иногда вследствие увеличения предельного тока на полярограммах появляются «максимумы» и «пики», сильно искажающие форму нормальной кривой. Явление возникновения максимумов состоит в том, что при отсутствии в растворе поверхностно активных веществ на полярограмме получается резкий скачок в силе тока (полярографический максимум) и только при дальнейшем увеличении потенциала катода высота волны падает до нормальной величины. Следует отметить, что Гейровский дал неправильную теорию максимумов. Только после опубликования работы А. Н. Фрумкина (.1934 г.); в которой была высказана новая теория максимумов и были проведены чрезвычайно изящные и наглядные опыты, подтверждающие эту теорию, этот раздел полярографии получил прочную теоретическую основу и с тех пор продолжает развиваться силами почти исключительно советских ученых. Было показано, что причиной увеличения предельного тока является движение ртутной капли, вызывающее размешивание раствора и поэтому уменьшающее толщины диффузного слоя. В результате возрастает диффузия разряжающихся ионов к капельному электроду. Как указывает Б. Н. Кабанов, движение поверхности ртути мо.жет вызываться двумя причинами: во-первых, образованием капли при вытекании струи ртути из капилляра, во-вторых, неравномерной поляризацией капли, приводящей к тому, что в разных точках капли получается различное поверхностное натяжение. Изменение поверхностного натяжения связано со взаимным отталкиванием ионов двойного слоя, растущим с увеличением заряда двойного слоя. Максимумы могут подавляться добавкой веществ, адсорбирующихся на поверхности электрода (желатина, агар-агара, метилового красного и др.).
§ 4. Амперометрическое титрование
В последние годы широкое развитие получил метод амперометрического титрования, основанный на определенной зависимости концентрации восстанавливающегося (или окисляющегося) на микрбэлектроде иона от величины предельного тока.
Этот метод состоит в том, что по мере прибавления реагента к раствору испытуемого вещества замеряют диффузионный ток. т., е. высоту полярографической волны. Минимальное значение диффузионного тока указывает на эквивалентную точку. Конечная точка титрования определяется обычно графически по пересечению прямых, показывающих изменение величины предельного тока до и после достижения эквивалентной точки.
Амперометрическое титрование позволяет получить результа
294
Теоретические основы электрохимии
ты, не зависящие от температуры, характеристики капилляра и даже состава раствора. Кроме того, при многих методах амперометрического титрования можно не удалять кислород.
Одно из важных преимуществ амперометрического метода — возможность определять вещества, которые сами не восстанавливаются (либо не окисляются), но могут быть оттитрованы восстанавливающимся (или окисляющимся) реагентом.
В зависимости от того, что восстанавливается или окисляется при заданном микроэлектроду потенциале график амперометрического титрования будет иметь различный вид. Так, в случае восстановления определяемого иона по мере добавления реагента значение inp будет уменьшаться вплоть до достижения эквивалентной точки, В эквивалентной точке величина i будет равна остаточному току, так как добавляемый реагент индифферентен (рис. 107, а).
В случае, когда определяемый ион индифферентен, а на микрокатоде восстанавливается или окисляется добавляемый реагент, ход титрования регистрируется графиком, приведенным на рис. 107, б.
При одновременном восстановлении (или окислении) и определяемого иона, и реагента при заданном потенциале кривая титрования имеет вид, представленный на рис. 107, в.
В том случае, когда определяемое вещество окисляется, а реагент восстанавливается при заданном микроэлектроду потенциале, график амперометрического титрования будет иметь вид, представленный на рис. 107, г.
Таким образом, ртутный капельный электрод является индикатором при объемном титровании. В последнее время вместо ртутного капельного электрода при амперометрическом титровании с успехом применяют вращающийся платиновый микроэлектрод и другие твердые электроды.
Для амперометрического титрования можно применять любые визуальные или регистрирующие полярографы. В качестве электрода сравнения при титровании с ртутным капельным или твердым электродом употребляют каломельные, ртутно-сульфат-ные, серебряно-хлоридные, меркур-йодидные и другие электроды. Для контакта исследуемого раствора с электродом сравнения применяют агар-агаровые мостики, наполненные насыщенным раствором хлористого калия. Выбор сосуда зависит от объема титруемого раствора, его природы и т. п.
Титрование лучше проводить из микробюретки реагентом, в 10—15 раз более концентрированным, чем испытуемый рас-
1 В ходе титрования потенциал микро1электрода сохраняется постоянным при значении, отвечающем предельному таку, и после добавления очередной порции реагента регистрируется значение /Пр-
Диффузионная кинетика и теория концентрационной поляризации 295
твор. При этом необходимо заранее знать поведение титруемого вещества и реагента на данном электроде. Метод амперометрического титрования характеризуется следующими положительными особенностями:
а) изменения температуры и величины диффузионного тока не влияют на результаты титрования амперометрическим мето
6
С)
Рис. 107. Кривые амперометрического титрования
ОЬъем ¥ пл
г
дом; следовательно, этот метод требует более простой аппаратуры, а точность его иногда даже выше, чем полярографического;
б) амперометрическим методом можно определять вещества, не дающие полярографической волны, если полярографическую волну дает реагент;
в) амперометрическое титрование можно проводить со значительно более разбавленными растворами (0,001 моль!л), чем при обычном объемном титровании;
г) значительно расширяется число объемно-аналитических определений, так как нет необходимости подбирать индикатор, меняющий цвет в эквивалентной точке.
296 ,Теоретические основы электрохимии
В последнее время выполнен ряд интересных работ по амперометрическому титрованию. Разрабатывается методика с применением в полярографии осциллографа, что дает возможность изучать быстропрртекающие процессы и исследовать кинетику химических и электрохимических реакций, а также значительно повысить точность и чувствительность метода.
ЛИТЕРАТУРА
Г^йр о в еки й Я. Техника полярографического исследования. Госхимиздат, 1951.
Глесстон С. Введение в электрохимию. ИЛ, 1951.
Делахей П. Новые првборы и методы в электрохимии. ИЛ, 1957.
Есин О. А. Совместный разряд ионов водорода с ионами металла. Труды Уральского индустриального института им. С. М. Кирова. Сб. 27. Метал-лургиздат, 1947.
Кольтгоф И. М., Лайтинен Г. А. Определение концентрации водородных ионов и электротитрование. ИЛ, 1947.
Крюкова Т. А., Синякова С. И., Арефьева Т. В. Полярографический анализ. Госхимиздат, 1959.
Кольтгоф И. М. и Дж. Лингейн. Полярография. Госхимиздат, 1948. Кольтгоф И. М. и Дж. Лингейн. Успехи химии, 8, 11, 1939, 1652. Левин В. Г. Физико-химическая гидродинамика, изд. 2-е. Физматгиз, 1959. Новые методы физико-химических исследований, сб. под ред. Б. В. Дерягина.
Изд. АН СССР. 1957.
Р о т и н я н А. Л., Хейфец В. Л. Теоретические основы гидрометаллургии, в книге «Основы металлургии», т. I. Металлургиздат, 1960.
Сонгина О. А.,Амперометрическое титрование в анализе минерального сырья. Госгеолтехиздат, 1957.
Сивер Ю. Г., Кабан о в Б. Н. ЖФХ, ,22, 1948, 53; 23, 1949, 428.
Стромберг А. Г. Труды Уральского филиала АН СССР, № 4, УФАН СССР, 1951.
Фрумкин А. Н., Б а г о ц к и й В. С., И о ф а 3. А., Кабанов Б. Н. Кинетика электродных процессов, гл. I и II. Изд. МГУ, 1952.
Фрумкин А. Н. и Л е в и ч В. Г. ЖФХ, 21, 1947, ,1335; 21, 1947, 1163.
Глава XI
ПЕРЕНАПРЯЖЕНИЕ ПРИ РАЗРЯДЕ ИОНОВ ВОДОРОДА
§ 1. Влияние различных факторов на величину перенапряжения водорода
В данный момент реакция разряда (и ионизации) водорода — наиболее изученная из всех известных электродных реакций.
Первоначально под термином «перенапряжение водорода» понимали разность между равновесным потенциалом и значением потенциала, необходимым для разряда с выделением пузырьков газа, <р °, т. е.
”%ин <РК ?к(мин)'
Здесь <рК(мин)—'потенциал, при котором начинается видимое образование пузырьков газа.
Но минимальное перенапряжение Т]МИн, необходимое для выделения водорода на катодах из различных металлов, сильно' различается по данным разных авторов и, таким образом, не представляет собой строго постоянной величины, характеризующей физико-химические свойства электродов. Причина этих отклонений связана с ошибками визуальных наблюдений. Кроме того, момент начала выделения пузырьков меняется от условий электролиза и в зависимости от состояния поверхности электрода.
В настоящее время под термином «перенапряжение» подразумевается разность между равновесным потенциалом электрода <р° и потенциалом поляризованного электрода <рк- Таким образом, перенапряжение имеет положительный знак при катодной поляризации *:
7гнг = 'р°—V (ХМ>
Данные для перенапряжения водорода в интервале плотностей тока от 1 до 200 ма1см? на различных технически чистых ме-
1 См. А. Н. Ф р у м к и н, В. С. Б а г о ц к и й, 3. А. И о ф а, Б. Н. Кабанов. Кинетика электродных процессов. Изд. МГУ, 1952, стр. 1120.
298
Теоретические основы электрохимии
таллах приведены в табл. 20. Из таблицы видно, что перенапряжение водорода зависит от природы Металла.
Таблица 20. Перенапряжение водорода при выделении его
«а технически чистых металлах из 2-н. растворов серной кислоты при 25 °C, по данным А. Г. Печерской и В. В. Стендера
Потенциалы выделения водорода (—а), ма/см', при
Металлы 1 5 10 20 40 60 100 250
РЬ 1,05 1,11 1,14 1,17 1,20 1,22 1,24 1,26
Zn 0,83 0,95 1,01 1,05 1,10 1,13 1,17 1,22
Sb 0,63 0,70 0,72 0,76 0,79 0,81 0,83 0,86
Al 0,58 0,64 0,67 0,69 0,72 0,81 0,83 0,79
Sn 0,63 0,66 0,70 0,74 0,78 0,80 0,83 0,86
Cd 0,51 0,65 0,71 0,77 0,84 0,87 0,93 1,00
Cu 0,48 0,56 0,60 0,66 0,68 0,71 0,74 0,78
Ta 0,46 0,51 0,55 0,56 0,58 0,59 0,61 —
Ge 0,39 0,54 0,62 0,68 0,76 0,78 0,85 0,92
Fe 0,36 0,44 0,47 0,51 0,55 0,57 0,60 0,64
Mo 0,35 0,42 0,45 0,48 0,52 0,55 0,56 0,59
Ag 0,34 0,40 0,42 0,44 0,47 0,49 0,50 0,53
Co 0,32 0,39 0,42 0,45 0,48 0,50 0,52 0,56
Ni 0,30 0,37 0,40 0,42 0,44 0,46 0,49 0,51
W 0,26 0,33 0,37 0,39 0,42 0,43 0,46 0,49
Re — 0,09 — — —- 0,23 0,27 0,34
Pt Сплав 0,15 — 0,28 — — -— 0,40 0,45
(W—Ni) 0,13 — 0,21 — — — 0,30 0,37
Перенапряжение при разряде ионов водорода вообще весьма гвелико по сравнению с поляризацией, сопровождающей большинство других электродных реакций. Так, например, на ртутном катоде при плотности тока iK = 10-4 а)см2 (электролит — 1-н. раствор НС1) перенапряжение достигает 0,94 в.
Сравнивая величину перенапряжения на различных твердых катодах, нужно иметь в виду, что т] н зависит от плотности тока .iK, которую обычно находят как частное от деления наблюдаемой при электролизе силы тока на измеренную поверхность электрода. Но поверхность твердых тел не бывает совершенно гладкой и непосредственно измеренная величина ее не соответствует истинной поверхности. Для большинства твердых металлов поверхность, на которой протекает электродная реакция, в несколько раз больше, чем измеренная, т. е. действительная плотность тока в соответствующее число раз меньше. Эту особенность нужно иметь в виду при оценке величины водородного перенапряжения. Фактическое перенапряжение на твердых электродах больше, чем измеренное. Как было установлено в 1905 г. “Гафелем, зависимость перенапряжения от плотности тока при до
Перенапряжение при разряде ионов водорода
299
статочно больших плотностях тока укладывается в эмпирическое уравнение
%, = ° + b <Х1>2)
Здесь а и b — коэффициенты, не зависящие от плотности тока.
Это уравнение для большинства металлов согласуется с опытом в широком диапазоне плотностей тока (от 10~9 до 10 а!см2) при условии достаточной чистоты и однородности поверхности катода. Однако оно не является исчерпывающим, так как при по уравнению (XI, 2) т)На стремится к отрицательной бесконечности, между тем как по самому смыслу перенапряжения оно при гк = 0 должно обращаться в нуль. Фактически в области малых поляризаций имеется линейная зависимость между перенапряжением и плотностью тока, т. е.
сад
Уравнение (XI, 3) обычно оправдывается при достаточно малых плотностях тока.
Постоянная а в уравнении Тафеля, т. е. величина перенапряжения при плотности тока, равной единице (iK = 1 а/см2), зависит от природы металла электрода, состояния его поверхности, состава электролита и температуры. Эта постоянная характеризует собой степень необратимости процесса на электроде: чем больше а, тем больше при данной iK, тем больше отклонение от обратимого состояния.
В табл. 21 приведены значения коэффициента а по данным различных авторов. Из таблицы следует, что эти значения лежат в пределах от 0,3 до 1,7 в. Как нетрудно заметить, высокое значение а типично для определенной группы металлов (свинец, ртуть, цинк, кадмий, олово).
Таблица 21. Постоянные а и Ь из формулы Тафеля по данным различных авторов
Металл а, в (Лоренц) a. e (Стендер и Печорская) b (Стендер и Печорская) Металл a, в (Лоренц) a, в (Стендер и Печорская) ь (Стендер н Печорская)
РЬ 1,47 1,34 0,118 Au 0,75 .
Hg 1,43 — 0,119 Fe 0,72 0,72 0,12
Zn 1,30 1,34 0,17 Ag 0,95 0,58 0,08
Cd 1,25 1,14 / 0,21 Mo — 0,66 0,105
Bi 1,27 Re — 0,41 0,14
Ge .— 1,08 4 0,23 Co — 0,62 0,10
Sb — 0,93 0,10 Ni 0,59 0,585 0,095
Sn 1,24 0,93 0,10 w — 0,56 0,10
Al Cu 0,79 0,85 0,87 0,09 0,13 Pt 0,35 — 0,125
300
Теоретические основы электрохимии
Константа b (табл. 21) в отличие от постоянной а мало зависит от природы металла или состава раствора. Эта константа в широком интервале плотностей тока может быть выражена со-
отношением
b = -RT—
0,4343Fa
(XI,4)
где значение а для большинства металлов близко к 0,5, так что при 25 °C b ~ 0,12.
Заметные отклонения от этой величины наВлкщаются лишь для цинка, кадмия и германия. Эти отклонения, однако, определяются некоторыми побочными явлениями, например неоднородной структурой или окислением поверхности, приводящими к увеличению значения Ь.
Наиболее подробно температурная зависимость перенапряжения исследована 3. А. Иофа, К. П. Микулиным и В. А. Степановой. Ими было найдено, что в интервале температур от 10 до 80 °C с ростом плотности тока температурный коэффициент ( —) для ртути в кислых растворах падает, достигая нуля при-\dt J мерно при iK = 51 а/с-м2. Кроме того, температурный коэффициент зависит от концентрации и состава раствора.
В области средних плотностей тока изменение перенапряжения с температурой составляет около 2—4 мв на один градус. Исследование показало, что по крайней мере в пределах от 1 до 200 ат перенапряжение от давления не зависит.
Перенапряжение водорода зависит от природы растворителя. Так, С. Левина и М. Зильберфарб нашли, что при переходе от водных растворов к спиртовым перенапряжение при прочих равных условиях уменьшается, а коэффициент b возрастает.
О влиянии растворителя на коэффициент b на ртутном катоде при t = 20 °C были получены следующие данные:
Р астворитель . . .
Коэффициент Ь . . .
вода
0,119
метиловый метиловый спирт эфир 0,120 0,125
Однако это явление исследовано совершенно недостаточно.
Зависимость перенапряжения водорода от концентрации Н+-ионов (pH) подробно изучали С. Д. Левина, В. А. Заринский и В. С. Багоцкий для ртутного катода. В чистых растворах НСК не содержащих посторонних солей, было найдено, что в интервале от 0,001 до 1-н. с изменением pH перенапряжение не меняется. Однако дальнейшие исследования, в особенности проводившиеся В. С. Багоцким и И. Е. Яблоковой, измерявшими перенапряжения на ртутном катоде в буферных растворах, показали^ что в этом случае при увеличении pH на единицу в интервале pH от 2 до 7 перенапряжение возрастает на 0,055—0,058 в (при
Перенапряжение при разряде ионов водорода 301
22°C). Таким образом, характерной особенностью процесса выделения водорода является его сильная зависимость от pH электролита. Перенапряжение вообще зависит от состава и раствора и присутствия в растворе посторонних ионов, влияющих на величину электрокинетического потенциала и строение двойного слоя. Это явление было изучено С. Д. Левиной и В. А. Заринским, которые нашли, что добавление к раствору НС1 соли ЬаС1з, дающей поверхностно активные поливалентные катионы La, повышает перенапряжение на ртути. Аналогичные закономерности наблюдаются и на других металлах, например на никеле.
Присутствие в растворе поверхностно активных анионов (С1“, Вг~. J~), наоборот, при не очень больших плотностях тока приводит к снижению перенапряжения водорода. При больших плотностях тока эффект снижения перенапряжения посторонними анионами исчезает, что связано с десорбцией анионов с поверхности электродов при достаточно отрицательном заряде поверхности.
Заметно перенапряжение водорода возрастает при адсорбции на поверхности металлических электродов положительно заряженных органических ионов, например иона тетрабутиламмония. В отличие от поверхностно активных анионов подобные вещества адсорбируются на отрицательно заряженной поверхности электрода и десорбируются при более положительных потенциалах по сравнению с точкой нулевого заряда. На перенапряжение заметно влияют добавки в электролит некоторых других высокомолекулярных соединений, коллоидов и слабых органических кислот.
Действие такого рода добавок изучалось многими исследователями. Так, было найдено, что перенапряжение водорода на цинковом катоде изменяется при добавлении небольших количеств желатины, сначала увеличиваясь, и затем, с дальнейшим повышений концентрации желатины, уменьшаясь.
М. А. Лошкарев исследовал влияние различных поверхностно активных веществ в весьма малых концентрациях на перенапряжение водорода при выделении его на ртути. Результаты этих исследований приведены на рис. 108. Как видно из графика, действие этих добавок весьма различно, причем некоторые из них увеличивают перенапряжение, другие — уменьшают. Б. Н. Кабанов изучал зависимость перенапряжения водорода при выделении его па свинце в концентрированной кислоте в присутствии различных добавок — слабых органических кислот и оснований и молекулярных соединений. Все подобные добавки увеличивают перенапряжение. Действие этих добавок также, вероятно, объясняется тем, что они, адсорбируясь, препятствуют прохождению электродной реакции.
302
Теоретические основы электрохимии
Перенапряжение водорода на одном и том же электроде в одинаковых условиях изменяется в процессе электролиза. Первоначально при нестационарном режиме, длящемся доли секунды,, перенапряжение линейно растет, затем медленно изменяется в течение большого промежутка времени (несколько часов).
Зависимость перенапряжения от времени представлена на рис. 109. Из графика видно, что на участке АВ скорость роста перенапряжения практически постоянна, т. е. изменение ^[Г
Перенапряжение
Рис. ilO8. Влияние поверхностно активных веществ на перенапряжение водорода при выделении его на ртутном катоде (по М. А. Лошкареву). Электролит—1-и H2SO4:
1 — ацетофенон; 2 — & -нафтол, тимол и дифениламин; 3 — дифениламин; 4 — 3 -нафтол; 5 — тимол; 6 — камфора;
7 — трибензоил амин; 8 — чистый раствор
прямо пропорционально времени и, следовательно,, количеству протекшего* через катод электриче-ства. Это объясняется тем, что большая часть пропускаемого в первые мгновения тока расходуется на заряжение двой-
Рис. 109. Зависимость перенапряжения водорода от времени,
кого слоя, т. е. на увеличение потенциала, и лишь очень незначительная часть идет на выделение водорода.
В некоторых случаях обнаруживалось, что перенапряжение, достигнув максимума, начинает весьма медленно падать. Это явление наблюдалось на медном, железном и кобальтовом катодах и может быть объяснено тем, что в процессе электролиза поверхность металла становится более шероховатой вследствие восстановления поверхностной окисной пленки. Так как на шерехо-ватой поверхности истинная плотность тока гораздо меньше, чем на гладкой, то перенапряжение уменьшается. Этот вывод подтверждается тем, что на катодах из платины, серебра, ртути, такое явление не наблюдается.
Перенапряжение при разряде ионов водорода 303
Наконец, было найдено, что перенапряжение водорода уменьшается при наложении переменного тока на постоянный. Подобное влияние, вероятно, обусловлено деполяризацией катода кислородом. Степень деполяризации при этом зависит от материала катода, плотности тока и других факторов.
Объяснению явления перенапряжения водорода было посвящено большое число исследований — и экспериментальных и теоретических. Многие из этих теорий в настоящее время представляют лишь исторический интерес и мы на них остановимся весьма кратко. Другие, 1нашротив, получили весьма широкое распространение.
§ 2. Некоторые ранние гипотезы водородного перенапряжения
Изучение кинетики электрохимических реакций показывает,, что реальные процессы на электродах состоят из ряда диффузионных. химических и электрохимических стадий. Так, процесс разряда водородных ионов условно можно представить в виде следующей схемы:
Н8о+->Н+ + НаО + е Надс -> 1/2Н2адс -> 1/2Н2гаэ. (XI,5} Как известно, скорость многостадийного процесса в основном лимитируется скоростью наиболее медленной стадии. Не располагая возможностью непосредственно определять скорость течения промежуточных стадий процесса катодного выделения водорода, некоторые исследователи сосредоточили свои усилия на теоретическом изучении различных стадий.
Так, М. Леблан (1910 г.) предполагал, что скорость разряда ионов на катоде затруднена из-за связывания их в комплексные соединения с молекулами растворителя или комплексооб-разователя. Разряд комплексного ^ли сольватированного соединения происходит не сразу, а с некоторой кинетической задержкой и таким образом определяет собой течение всего электродного процесса. Однако эта точка зрения не была подтверждена какими-либо экспериментальными данными. Кроме того, ближайшее рассмотрение этой гипотезы показывает ее несостоятельность, поскольку, например, процесс дегидратации протона водорода, вряд ли. вообще возможен как самостоятельная стадия, так как энергия связи протона с молекулой воды очень велика (около 282 кал на 1 г-ион), а константа диссоциации гидроксо-ния
[Н+ПН8О]
[Н3О+]
по реакции
н3о+->н+4-н2о
304
Теоретические основы электрохимии
чрезвычайно мала и равна примерно 10-100, т. е. на 101СО ионов гидроксония приходится один протон. Поэтому наиболее вероятно предположение, что дегидратация и собственно заряд (нейтрализация) Н+-иона происходят одновременно, как одна стадия, т. е. что разряжаются не протоны, а ионы гидроксония:
Краевой угол р,град
Рис. ildO. Связь между 7,МИп и р, mo X. Мёллеру
Н3О+ + е -> Н + H2Q.
Гипотеза Леблана в настоящее время почти никем не разделяется.
X. Мёллер (1909 г.) исходил из неоднократно наблюдавшейся на опыте связи между величиной перенапряжения и поверхностным натяжением на границе металл — водород (рис. ПО). Эта связь подтверждается эмпирической формулой
— 1
^м„Н= , (XI,6)
о
где т]мин — минимальное напряжение водорода, в;
Р — краевой угол, образуемый пузырьком водорода на различных металлах при потенциале, отвечающем началу заметного выделения водорода, град\
k и b — постоянные.
Для начала выделения газа необходимо, чтобы на электроде создавалась определенная концентрация молекул водорода. X. Мёллер допускает, что у поверхности электрода существует капиллярный слой водорода толщиной около 10~7 см. Давление водорода в этом слое должно возрастать с ростом поверхностного натяжения на границе газ — жидкость и уменьшением поверхностного натяжения на границе металл — жидкость.
Работами А. Н. Фрумкина, Б. Н. Кабанова, А. В. Городецкой была показана полная несостоятельность гипотезы Мёллера. Однако, как замечает О. А', Есин, параллелизм между перенапряжением водорода и краевым углом р все же действительно имеется и может быть объяснен тем, что величина краевого угла зависит от скачка потенциала электрод — электролит, а не наоборот, как думал Мёллер. Таким образом, в гипотезе Мёллера перепутаны причина и следствие. В целом же следует, что процесс образования пузырьков газа имеет второстепенное значение в электродной реакции и не влияет на величину перенапряжения.
Перенапряжение при разряде ионов водорода
305
Таким образом, если отбросить возможность замедленной дегидратации иона водорода (Леблан) и замедленного удаления пузырьков водорода (Мёллер) и не прибегать далее'к предположению о существовании таких гипотетических ионов, как Н_ (Гейровский), то можно представить себе, что процесс разряда ионов водорода складывается из следующих промежуточных стадий.
В кислых растворах
1. Разряд иона гидроксония с образованием адсорбированного атома водорода:
/Ие(Н3О+) 4- е^ТИе(Н) + Н2О. (Х1,7)
2. Десорбция атомарного водорода, которая может протекать либо как простой химический процесс воссоединения водородных атомов (рекомбинация):
Me (Н) + Me (Н) -> 2Ме + Н2, (X 1,8)
либо как электрохимический процесс разряда иона гидроксония Vc/участием адсорбированного атома водорода (электрохимическая десорбция):
ТИе (Н) + Н3О++ е/Ие + Н2О + Н2. (XI,9)
В щелочных растворах
Так как концентрация Н3О+ здесь исчезающе мала, то в данном случае электрон переходит на молекулу воды с образованием адсорбированного водорода и гидроксила:
УИе(Н2О) + е — Me (Н) + ОН”. (XI, 10)
Вторая стадия процесса здесь аналогична процессам в кислых средах, т. е. протекает либо как простой акт рекомбинации:
ТИе (Н) + Ale (Н)27Ие + Н2, (XI,II)
либо по механизму электрохимической десорбции:
Же(Н) + Н2О + е —Ме + Н2 + ОН-. (XI.12)
Таким образом, если не учитывать возможность одновременного протекания процесса по всем вероятным направлениям с соизмеримыми скоростями (по П. Д. Луковцеву), то наиболее вероятны два крайних случая:
1) скорость процесса определяется замедленной рекомбина цией. В этом случае стадии разряда реакций (XI,7) или (XI,10) протекают быстро, а удельный вес процесса (XI,9) или (XI, 12) весьма ничтожен; ’
20 Заказ 1338
306
Теоретические основы электрохимии
2) скорость процесса определяется замедленностью одной из электрохимических стадий (XI,7), (XI,9), (XI,10), (XI, 12). а процесс химической рекомбинации протекает быстрее.
В соответствии с приведенными здесь крайними случаями возникли две основные теории перенапряжения при катодном выделении водорода: теория замедленной рекомбинации и теория замедленного разряда. Эти теории мы рассмотрим в их исторической последовательности.
§ 3. Теория замедленной рекомбинации атомарного водорода
Первая теория замедленной молизации ((рекомбинации) была предложена Тафелем исторически раньше других теорий (1905 г.). Она в своем первоначальном виде давала хорошее объяснение известным в то время опытным фактам.
Основой теории Тафеля служит положение, что наиболее медленной является стадия образования молекул водорода из адсорбированных на поверхности электрода атомов. При этом образование молекул сопровождается одновременной десорбцией водорода, так как опыт показывает, что молекулярный водород не адсорбируется. Таким образом, замедленным является процесс удаления водорода вследствие реакции
2Налс—»Н2 . аде ^газ
Скорость такой реакции по законам химической кинетики может быть выражена формулой
= (XI, 13)
С другой стороны, скорость образования атомов водорода, очевидно, пропорциональна плотности тока: dC,. ] + —У-= — i, dr К"
(XI,14)
1 .гл.
где —— коэффициент пропорциональности. К
Если пренебречь скоростью обратного процесса, т. е. диссоциацией молекул водорода на атомы, то при установившемся процессе число образующихся на катоде атомов водорода должно быть равно числу рекомбинирующихся. Отсюда, приравнивая выражения (XI,13) и (XI,14), мы получим зависимость между концентрацией атомов водорода на поверхности электрода и
плотностью тока:
= _L и
Перенапряжение при разряде ионов водорода
307
ИЛИ
сн = /^- (XI.15)
Равновесный потенциал водородного электрода определяется из выражения
0 = — 1п-^. (XI, 16)
,=t> F КС°Н .
Очевидно, при прохождении тока
^=-^-1п/<+^1пСн+_^-1пСн
или
Т. = - ^ 1П К + -^ Inс„+ + In W —In(XI, 17)
Вычитая из <рр значения потенциала <рг-, получим
^н. = Фр —Р,- = - ~ In К'К" (C°Hr + In i (XI, 18)
и, следовательно,
= а -f- b In i. (XI,19)
Здесь
а = -^-1пК'К"(С°)2, 2F н/ ’
6 = +—lge== + 0,029 (при 18°C).
2F
Отсюда следует, что коэффициент а в уравнении Тафеля зависит от природы металла через константу К'. Константа скорости реакции рекомбинации, очевидно, пропорциональна экспоненциальной функции энергии активации процесса, т. е.
А
К’ Г<т А — qe ,
где q — коэффициент пропорциональности.
Следовательно,
“ = > (in F + In К’) - Ш - g (- ± + S) -
—1пС;,=^{й-А_2|пГн1. (XI.20)
где В = In Fq.
20*
308 Теоретические основы электрохимии
Отсюда следует, что с ростом энергии активации реакции молизации возрастает величина а. Иными словами, чем хуже металл катализирует процесс образования молекулы водорода, тем выше перенапряжение. Проверка этого положения теории замедленной рекомбинации была осуществлена Бонгоффером (1927 г.), который изучал каталитическое действие металлов на скорость молизации атомов водорода.
С этой целью шарик термометра покрывали слоем исследуемого металла и на него подавали поток активированного (атомарного) водорода. Реакция рекомбинации экзотермична, и по скорости повышения температуры можно судить о степени каталитической активности различных металлов. Опыты Бонгоффера показали, что каталитическая активность металла меняется параллельно с величиной перенапряжения водорода:
Возрастание каталитической активности
*Pt, Pd, W, Ni, Fe, Ag, Cu, Zn, Sn, Pb
-------------------------------->
Увеличение перенапряжения
Это означает, что металлы с большим перенапряжением катализируют процесс рекомбинации слабее, чем металлы с меньшим перенапряжением. Эти выводы были подтверждены рядом исследований.
Несомненно, что скорость процесса рекомбинации атомарного водорода в молекулярный может сказаться на величине перенапряжения. Все же попытка объяснить причину перенапряжения только замедленностью этой стадии встречает ряд возражений.
1. Если причина водородного перенапряжения заключается в замедленности стадии молизации, то металлы, поглощающие водород (Pt, Pd, Fe, Ni, Co, Та и др.), должны обладать наименьшим перенапряжением. Это справедливо, если сопоставить металлы железной группы, легко поглощающие водород, со ртутью или цинком, на которых перенапряжение значительно выше; однако это не оправдывается для тантала. Тантал поглощает водород в значительно больших количествах, чем металлы железной группы, в то же время перенапряжение для разряда ионов водорода на нем очень велико.
2. Экспериментальные данные показывают, что Г)нг зависит от pH раствора, присутствия посторонних ионов, диффузности двойного слоя, наличия в электролите поверхностно активных веществ. Влияние, всех этих факторов сказывается на константе а, изменяя ее величину. Однако рекомбинационная теория не объясняет этих явлений.
3. Перенапряжение при разряде металлических ионов, а также в случае выделения кислорода при окислительно-восстанови
Перенапряжение при разряде ионов водорода 309
тельных реакциях также, как правило, подчиняется уравнению Тафеля. Это свидетельствует о том, что причины перенапряжения всех упомянутых реакций аналогичны. Реакция же рекомбинации в большинстве перечисленных процессов отсутствует.
4. Перенапряжение водорода имеет место при таких плотностях тока, когда нет видимого выделения Нг (в области остаточных токов). Специальное изучение кинетика заряжения электрода показало, что его потенциал в области малых токов меняется линейно с количеством пропущенного электричества. Последний опытный факт связывают с накоплением зарядов (ионов и электронов) в двойном электрическом слое, что противоречит взглядам Тафеля.
5. Рекомбинационная теория дает неправильное значение ко-, , г. КГ
эффициента Ь, равное ---, в то время как опыт показывает, что
л 2F 2/?Г А
для большинства металлов эта величина равна—т. е. в 4 раза больше.
Теория замедленной рекомбинации и десорбции водорода в течение многих лет разрабатывалась Н. И. Кобозевым с сотрудниками, которые полагают в отличие от Тафеля, что водород может десорбироваться не только в виде молекул, но и в виде атомов («эмиссия атомарного водорода»).
Для металлов с низким перенапряжением Н. И. Кобозев предполагает возможность непосредственной десорбции адсорбированных атомов с поверхности электрода с переходом их в глубину раствора вне сферы действия поверхностных сил. При чисто эмиссионном механизме десорбции перенапряжение при заданной плотности тока не должно зависеть от энергии адсорбции и, следовательно, от природы металла, а коэффициент b не
RT должен превышать .
Точка зрения Кобозева оказалась спорной и была подвергнута критике рядом исследователей. В частности, расчеты показывают, что если подобная эмиссия и имеется, то она столь мала, что не может играть заметной роли в удалении водорода с поверхности электрода.
§ 4. Теория замедленного разряда
На возможность замедленной нейтрализации иона на электроде впервые было указано еще в конце прошлого века Р. А. Колли.
В то время эта идея не получила широкого развития, но начиная с 1930 г. ряд авторов (Эрдей-Груз и Фольмер и др.) про
310
Теоретические основы электрохимии
вел исследования, показывающие, что именно медленность самого процесса разряда (нейтрализации) и является причиной перенапряжения при выделении водорода. Здесь же следует заметить, что полное развитие теория замедленного разряда получила благодаря работам советских электрохимиков, которые впервые отказались от обычных химических представлений, лежащих в основе рекомбинационной и других теорий перенапряжения, и сосредоточили свое внимание на электрохимической природе взаимодействия между ионом и электроном.
Представления о замедленности электрохимического акта разряда особенно перспективны, так как могут быть распространены на более общие проблемы электрохимической кинетики— не только на случай разряда ионов водорода, но и на электроосаждение металлов (естественно, с соответствующими дополнениями, вытекающими из особенностей процесса электрокристаллизации).
Эрдей-Груз и Фольмер, обсуждая зависимость между количеством пропущенного электричества и изменением потенциала г. процессе заряда электрода, исходили из прямолинейного изменения поляризации со временем (см. рис. 109). Для объяснения этой зависимости рассмотрим процессы рекомбинации
Me—Н + Н — Me- > 2Ме 4- Н2
и разряда
Н30+ + е (Me) — Me — Н -J- Н2О.
Допустим, что в начальный момент после включения тока происходит разряд, но вследствие медленности процесса рекомбинации водород не выделяется. Тогда, очевидно, весь ток будет расходоваться на образование атомов водорода, адсорбированных поверхностью металла.
Концентрация атомов водорода на поверхности при пропускании тока плотностью i а!см2 в момент времени после включе-ir
ння т изменяется на величину —, так что
Сн = 4+Сн, (XI,22)
г
где — концентрация водорода на поверхности до включения тока. Из уравнения процесса рекомбинации следует, что перенапряжение должно изменяться со временем по логарифмическому
закону
RT . сн RT .
=--------1П------ =---------1П
‘Hl F г0 P
(XI,23)
Перенапряжение при разряде ионов водорода
311
Рис. 111. Заряжение двойного слоя (по Эр-дей-Грузу и Фольмеру)
На самом же деле перенапряжение ц н> изменяется прямо пропорционально времени 1 (прямая АВ на рис. 109).
Если предположить, что замедленной стадией является разряд, то в первые моменты времени после включения тока атомы водорода не образуются, а происходит заряжение двойного слоя — процесс, аналогичный заряжению конденсатора, т. е. к границе .металл — раствор .из внешней цепи проникают электроны, а из раствора — ионы НзО+ (рис. 111) и плотность заряда двойного слоя возрастает.
Пусть ео — плотность заряда в двойном слое до включения тока, <р0 — равновесный потенциал электрода, с — емкость двойного слоя. Тогда до включения тока
?о = — • (XI,24)
С
После включения тока плотность заряда будет увеличиваться со временем, так что
ет = е0 + ti и, очевидно, что при этом возрастет потенциал до <рг- = —. Следовательно, С
(XI,25)
Таким образом, прирост потенциала пропорционален времени, протекшему с момента включения тока.
Скорость разряда водородных ионов vp при катодном потенциале ф, по Эрдей-Грузу и Фольмеру, может быть определена из учета поверхностной концентрации ионов Сн+ и энергии активации.
Таким образом, при прохождении тока не все электроны, притекшие к электроду, потребляются на процесс разряда ионоз НзО+; часть из них остается не нейтрализованной и образует одну из обкладок конденсатора. Вторую обкладку двойного слоя составляют ионы, притянутые из глубины раствора электростатическим полем электрода.
Чем больше заданная плотность тока, тем больше заряд пластин конденсатора и, следовательно, потенциал на границе металл — раствор. Это объясняет рост перенапряжения водорода с ростом плотности тока.
1 В начальный момент электролиза при нестационарном режиме.
312
Теоретические основы электрохимии
Из учения о скоростях химических реакций известно, что если при достаточно больших концентрациях реагирующих веществ химическая реакция протекает с конечной скоростью, то это означает, что в реакцию вступают не все молекулы или ионы вещества, находящиеся в сфере протекания реакции, а только часть их, обладающая известным избытком энергии и0 (энергией активации). Константа скорости реакции (К) при этом оказывается экспоненциальной функцией энергии активации (ио), т. е.
_ н»
К=Кое RT . (XI,26)
Стадия приобретения или отдачи электрона в электродной реакции протекает с конечной скоростью, поэтому Эрдей-Груз и Фольмер применяют к константе реакции перехода электрона из металла на ион водорода, находящейся в двойном слое, уравнение химической кинетики с тем исключением, что энергия активации здесь будет меняться на некоторую долю а с изменением потенциала <р, т. е.
и == «о + а?Д (XI, 27)
В этом заключается особенность и отличие электрохимической кинетики от обычной химической.
Тогда
dC । _ ~Ьaq>F _ач>р
ир = -^- = К'Сп+е *т . (XI,28}
Скорость обратного процесса ионизации водородных атомов по аналогии может быть представлена следующим образом:
(wa — bvF) РфГ
цн=К"Сне RT =К2Сне r<T , (XI,29)
где twc — энергия активации для ионизации атома водорода, а Р — некоторая доля от <р. При равновесном потенциале ир = Ун> т. е. обе скорости равны. В таком случае
Рфр/7 а'₽р/?
откуда
(а + ₽) <ppF
Сн = ^зСн+е (ХЬЗО)
Далее авторы произвольно полагают, что концентрация адсорбированных на поверхности водородных ионов Сн+ тождественна с их объемной концентрацией [Н+], фигурирующей в тер
Перенапряжение при разряде ионов водорода
313
модинамической формуле Нернста для равновесного потенциала Фр, т. е.
сн+ = /с4[н+]Г’^г. (XI.31)
Сопоставляя два последних уравнения, получим а + ₽ = I. При этом Эрдей-Груз и Фольмер предполагают, что вершина потенциального барьера прямого и обратного процесса расположена как раз посредине двойного слоя, что обусловливает одинаковое действие потенциала на процесс разряда и ионизации. В таком случае а = ₽ = 0,5 и при пропускании тока плотностью I при неравновесном потенциале, получим
Ч>{Е q^F
i^KlCli+e~^r-K2Ciie+~^. (XI,32)
Так как потенциал электрода фг равен разности
следовательно:
ер -F1 ер Р
Р tjF Р
х = л,сн+е 2ет е 2RT -К2Сне№Т е 2RT
или
; _ 17 С 2RT V- Г п 2RT
1 ~~ д6Сне
(XI,33)
Уже при потенциалах, незначительно больших обратимого 2.Р.Т
(ц > ~ ), вторым слагаемым уравнения (XI,33) можно rape-
Г
небречь.
Тогда
i^K5CH+e 2RT . (XI,34)
Отсюда при Сн+ = const получим формулу Тафеля:
= const 4- In i = а + b 1g i, (XI,35)
причем при 18 °C
Однако, как нетрудно заметить, это значение для коэффициента b Эрдей-Груз и Фольмер получили, пользуясь не вполне обоснованным доказательством того положения, что а = 0,5 и
314
Теоретические основы электрохимии
что поверхностная концентрация Сн+ тождественна объемной [Н+]. Кроме того, здесь нет упоминания о природе раствора и ее влиянии на т]Н1.
В теории Фольмера двойной электрический слой представляется в виде плоского конденсатора. В таком случае учитываются только электрические силы, действующие между заряженной поверхностью металла и ионами раствора. При этом количество ионов, находящихся во внешней обкладке двойного слоя, точно соответствует числу противоположных по знаку зарядов на электроде; эти ионы расположены вдоль поверхности, вплотную к ней.
Описанное представление о строении двойного слоя, как уже отмечалось, справедливо только в отдельных случаях, а именно при больших концентрациях электролита и больших плотностях заряда.
§ 5. Замедленный разряд, по А. Н. Фрумкину
В связи с изложенным появилась необходимость разработать более строгую теорию замедленного разряда, что и было осуществлено А. Н. Фрумкиным и сотрудниками. Представление о ме
ханизме процесса разряда
Рис. 112. Потенциальные кривые для вона Н3О+ и адсорбированного атома водорода
сти. Размер адсорбированного
можно получить из рассмотрения потенциальных кривых иона Н3О+ и адсорбированного атома водорода (рис. 112).
Ионы НзО+, участвующие в разряде, располагаются в слое, прилегающем к поверхности электрода и имеющем толщину, примерно равную радиусу ионов (6). Процесс состоит в том, что ион из положения А переходит в положение В, соединяясь с электроном и превращаясь .в атом водорода, адсорбированный на поверхно-гома меньше размера гидрати-
рованного иона, и точка В, в которой находится в равновесии центр атома, лежит ближе к поверхности. Кривая аа характеризует собой изменение энергии системы Н+ • Н2О при удалении протона из устойчивого равновесного состояния А. Кривая ЬЬ представляет собой изменение энергии при смещении адсорбированного атома водорода от своего равновесного положения В. Точка С, в которой потенциальные кривые пересекаются, выра-
жает энергию, при которой одна форма связи водорода перехо-
Перенапряжение при разряде ионов водорода 315
дит .в другую: ион водорода превращается в атом, адсорбированный на поверхности металла.
При разряде ион Н3О+ должен получить энергию, достаточную для преодоления энергетического барьера, высота которого и равна энергии активации процесса разряда. Аналогично и обратный процесс ионизации требует некоторой энергии активации w. Величины и и w, согласно законам химической кинетики, определяют собой скорости процессов нейтрализации и ионизации водорода:
и
^P = V’3o+e
W
Плотность тока, протекающего через катод, очевидно, будет равна
i = К(ор—ои) или
i — ip im
где ip = Fvp; iH = Fvn — токи обмена.
Таким образом, при поляризации электрода потенциальная кривая ЬЬ для атома водорода остается без изменения (поскольку атом не заряжен), а кривая, характеризующая положительный ион Н3О+, располагается на более высоком уровне а'а' (рис. 113). Это связано со сдвигом потенциала электрода в отрицательную сторону, что равносильно сдвигу потенциала раствора в положительную сторону. По Штерну, скачок потенциала <р на границе металл — раствор складывается из двух слагаемых:
Ф = Ф + Фь
где ф — падение потенциала между поверхностью металла и границей плотной части двойного электрического слоя на расстоянии примерно одного радиуса Н3О+-ионов;
Ф1 — падение потенциала в остальной части двойного слоя.
Как отмечалось, адсорбционный потенциал ф( не тождествен электрокинетическому потенциалу поскольку при движении жидкости толщина неподвижного слоя больше, чем толщина од кого ионного радиуса (б), но <в разбавленных растворах эти величины близки между собой. Электродные реакции протекают в пространстве между поверхностью электрода и первым ионным слоем в растворе. Таким образом, на энергию активации электродной реакции будет влиять не весь потенциал, создаваемый ионным двойным слоем, а лишь та часть его ф, которая падает в
316
Теоретические основы электрохимии
плоской части двойного электрического слоя. Чтобы пояснить это, обратимся вновь к потенциальной кривой (см. рис. 113), которая начерчена с учетом скачка потенциала на границе металл — раствор. Появление между плоскостью металла и плоскостью АА', в которой расположены центры ионов Н3О+, скачка потенциала ip, приведет к относительному смещению потенциальных кривых по вертикали (плоскость Б Б' характеризует расстояние, на которое может подойти адсорбированный атом). При этом сместится, очевидно, и точка пересечения этих кривых С'.
Л С Г' Л T
Рис. 113. Смещение потенциальной кривой иона Н3О+ три изменении потенциала электрода
положение которой определяет собой величины энергий активации и и w, которые изменятся на некоторую величину, пропорциональную ф/7, причем коэффициенты пропорциональности определяются наклоном обеих кривых. Если обозначить энергию активации разряда при отсутствии скачка потенциала «о, то, очевидно,
и = и0 + a^bF (XI,36)
и аналогично
w — w0 — а2фЕ, (X1,37)
так что теперь
_ (»о + «iW (w„ —
(XI,38)
Часть скачка потенциала ф определяет собой изменение концентрации Н3О+-ионоБ в слое толщиной б.
Катионы или анионы раствора, подходя к поверхности электрода, встречают на своем пути электрическое поле одного с ними или противоположного знака. Притяжение или отталкивание ионов сказывается на их поверхностной концентрации (в слое непосредственно у электрода), поэтому А. Н. Фрумкин подчеркивает, что поверхностная концентрация не равна объемной.
Поверхностная концентрация ионов определяется в общем случае уравнением изотермы адсорбции
^НаОадс
г(аСн,о+
I + “СНаО -
(XI,39)
где Сн,о+ — объемная концентрация Н3О+-ионов;
Перенапряжение при разряде ионов водорода
317
со — энергетический фактор адсорбции, который в случае небольших специфических адсорбционных сил
(Х1,40) так что
гё~ КТС С ._______________I
Н.о^с“
(XI,41)
Здесь z — поверхностная концентрация при насыщении или геометрический фактор адсорбции. Так как степень заполнения поверхности электрода ионами гидроксония мала (около 9%), то ехр(^г~) Снао+«1
и
Ch.o+„=z<W ”
Действие ф] — потенциала заключается в первую очередь в том, что он отталкивает одноименно заряженные и притягивает разноименные ионы, при этом изменяется поверхностная концен трация реагирующего вещества.
Таким образом,
ZK = К0Сн,о+е~ e~ (X 1,43)
где Ko = zK\e RT.
При отсутствии тока (i = 0), очевидно,
<?i=0 = Фо + фо (XI.44)
и
г/ F 0 wo
^СНв0+е е кт - К£'не е ™ .
К'2 и разделим обе части уравнения
Обозначим /С2ехр^~~^ = на е RT .
318
Теоретические основы электрохимии
Тогда
ф ' F
_2_ <а« + I/' Г
е RT е RT Л2 . СН
Ко сн,о+ или
(»> + «0fc+H—(XI,45) г До bHjO+
Сравнивая уравнения (XI, 44) и (XI, 45), нетрудно получить ai + a2=l. (Х1,46)
При малых значениях перенапряжения т]н из уравнений (XI,43) и (XI,46) следует
*1н, = —4. (XI,47)
Г 10
Более подробно вопрос о величине коэффициентов <Х| и аг рассматривался в работах О. А. Есина и В. А. Кожеурова и Н. Д. Соколова, в которых теоретически показано, что эти коэффициенты зависят от энергии связи водорода в комплексе Н3О+ и на металле (Me — Н) и что ai ~ аг ~ 0,5 (по Соколову, ai = 0,48). Последнее подтверждается также экспериментальными данными.
При значительных скоростях выделения водорода (i > > 0), скорость ионизации становится пренебрежимо малой по сравнению со скоростью разряда и i — Fvv, а так как а( == 0,5 и ф = = <р — фь
(ч>—ф,)Г
‘=*оСн,о+е 2RT ; (XI,48)
Ini — ln/G 4-InC —Jhf---+
'° H,o+ RT 2RT 2RT
lni = lnK0-J-lnCH1O+
Разделив обе части уравнения на
фг/;
2RT 2RT ‘
F ----, получим 2RT
2RT. .
----Int F
Учитывая, что
2RT । 2RT
— ln Ko + F
F <?. (XI,49)
4н. = <Pz=o — Tz
R>R 1т, >
?z=o — —In CH,o+, г
Перенапряжение при разряде ионов водорода
319
получим:
<Р = + —f’ in CHjO+ - фх In I + const; (XI,50) Чн. = lnCHjO+ + Фх + Ini + const +^1пСн,о+(Х1,51)
или
Чн. - фх = - In CHjO+ + In i + const.. (XI,52)
Таким образом величина фгпотенциала может весьма существенно влиять на величину перенапряжения и вообще на явления, протекающие у электрода. В самом общем случае следует иметь в виду, что фгпотенциал различен в растворах разных электролитов и меняется в зависимости от концетрации раствора.
RT
В разбавленных растворах кислот величина ----—1пСнао+»
F
как было показано Штерном, постоянна и тогда
Чн, = + in i + const, (XI,53)
F
т. е. имеем уравнение Тафеля.
Уравнение Фрумкина (XI,52) позволило объяснить ряд опытных явлений, возникающих при выделении водорода. Так, очевидно, при i — const
»!н, — Фх = —in C ++const (X 1,54)
и поэтому в разбавленных растворах кислот
Чн, = const, (Х1,55)
т. е. перенапряжение не зависит от pH раствора. При добавлении в разбавленный раствор кислоты избытка нейтральных солей общая концентрация ионов в растворе сильно возрастает, вследствие чего двойной слой становится плотным, a уменьшается и становится постоянной величиной, не зависящей от pH. Тогда при i = const
Чн, = In Сн 0+ + const, (Х1,56)
F 8
т. е. перенапряжение становится функцией pH.
Уравнения (XI,54) и (XI,56) позволяют приближенно вычислять потенциал фх из измерений поляризации в чистом растворе кислоты и в растворе с избытком нейтральной соли. Так как во
320
Теоретические основы электрохимии
втором растворе диффузный слой сильно уменьшается и фгпо-тенциал стремится к нулю, получим
'll —Т]2 = Дт]Н! = + const. (XI,57)
Уравнение (XI,57) проверялось С. Д. Левиной и В. А. Заринским, а также В. С. Багоцким, который определял изменение перенапряжения при добавлении к раствору НС1 соли КС1 и сравнивал полученные значения с рассчитанными по уравнению (XI,57) или (XI,54). Значения ф>1 при этом вычислялись по уравнению диффузного двойного слоя,-
-------I _ ДА. д.ДА\ 2RT_ + 2^)t (Х158)
где
с = [НСП + [КС1].
Результаты измерений Багоцкого приведены в табл. 22, из которой видно, что теория достаточно хорошо совпадает с опытом.
Таблица 22. Изменение перенапряжения Дт]к при добавлении КС1 к раствору НС1 (по В. С. Багоцкому) при i = 10-4 а! СМ1
[НС1] г-зкв!л [НС1] + [КС1] г-экв/л
0,001 0,001 0,215
0,001 0,01 0,163 0,052
0,001 0,1 0,112 0,103
0,001 0,33 0,085 0,130
0,001 1,0 0,062 0,153
0,01 0 01 0,156 —
0,01 0,1 0,106 0,050
0,01 1,0 0,057 0,099
Теория Фрумкина позволила также установить зависимость перенапряжения от присутствия поверхностно активных ионов, изменяющих £ (или ф1)-потенциал. Такая зависимость с очевидностью следует из уравнения (XI,54).
Уравнение замедленного разряда Фрумкина объясняет, почему, например, адсорбция анионов (галоидов) на ртути ускоряет реакцию разряда иона водорода. Снижение водородного перенапряжения наблюдалось и при адсорбции анионов на кадмии и таллии.
Наоборот, при наличии в двойном слое многозарядных катионов лантана перенапряжение на ртути повышается. Эти эффекты могут быть объяснены адсорбцией анионов и катионов и из-
Перенапряжение при разряде ионов водорода 321
менением ipi-потенциала. Влияние поля электрода на движение ионов в растворе возможно только при дуффузном слое (фгпо-тенциал не равен нулю). При плотном двойном слое (^-потенциал равен 0) поле электрода полностью экранировано, вследствие чего уменьшается скорость электрохимической реакции и растет перенапряжение водорода. В присутствии адсорбированных нейтральных органических веществ, не влияющих на величину фгпотенциала, перенапряжение может меняться только в том случае, если концентрация ионов водорода в поверхностном слое понижена (ориентировочно при pH > 4).
В сильнощелочных растворах, в которых ионы Н3О+ практически отсутствуют, водород выделяется по реакции
Мс(Н2О) + е Me (Н) + ОН“.
Теория Фрумкина и для кислых и для щелочных растворов хорошо подтверждается на опыте. Так, в случае щелочного раствора уравнение замедленного разряда имеет вид:
<р — <рх —-1 + const. (XI.59)
Так как
1= const Л aRT,
то поверхностная концентрация воды на электроде не зависит от концентрации и от величины фгпотенциала. В щелочных растворах удобнее выражать потенциал равновесного электрода в зависимости от концентрации ионов гидроксила, т. е.
<?₽ = <?<>-In Сон-- (XI,60)
Тогда из уравнения (XI,59) и (XI,60) получаем
71 + ‘1 = + ~аЕ 1Ш’-^rlnCOH- + const- (XI-61)
Следовательно, перенапряжение водорода при i = const должно повышаться с разбавлением чистых растворов щелочей. Так, например, скорость разложения амальгам щелочных металлов, определяемая перенапряжением водорода, изменяется в зависимости от концентрации щелочи в соответствии с уравнением (Х1,61).
Согласно теории двойного слоя Штерна, в этих растворах
, RT . ,
41 =----1пС„„—Pconst.
* * р ип *
21 Заказ 1338
322 Теоретические основы электрохимии
В присутствии избытка соли ф1 = const и по уравнению (XI,61) получим уравнение Тафеля:
т] = + In i + const. (XI,62)
aF
Необходимо, однако, отметить, что для некоторых электродов, например платинового, в щелочных растворах перенапряжение в зависимости от концентрации щелочи не подчиняется уравнению замедленного разряда. Поэтому возникла необходимость в экспериментальной проверке скорости процесса разряда, что и было осуществлено Б. В. Эршлером, П. И. Долиным и А. Н. Фрумкиным, которые показали, что в некоторых случаях удается подобрать такие условия, когда при измерении скорости суммарной электрохимической реакции можно непосредственно измерять скорость одного этапа реакции, например разряда иона с переходом его в адсорбированный атом. Для этого платиновый электрод в определенном интервале потенциалов покрывают адсорбированными атомами водорода: количество этих атомов на единице поверхности платинового электрода зависит от потенциала электрода. По мере увеличения анодной поляризации количество их убывает. При потенциале на одну десятую вольта положительнее, чем потенциал обратимого водородного электрода, выделение молекулярного водорода практически прекращается; таким образом, можно полагать, что по сравнению с другими процессами оно не играет существенной роли. Если теперь такому электроду сообщить через раствор некоторое количество электричества, то единственно возможной электродной реакцией становится реакция разряда ионов водорода с переходом их в адсорбированные атомы. Дальнейшие стадии — образование молекул водорода — здесь не могут протекать. Для определения скорости процесса разряда удобнее применять переменный ток различной частоты. В самом деле, если электрод включить в цепь переменного тока, то он будет вести себя подобно конденсатору, т. е. электроду будет эквивалентна электрическая схема, в котором емкость с и омическое сопротивление R включены параллельно.
Пользуясь уже описанными ранее схемами мостов переменного тока, можно измерить величину емкости и сопротивления электрода. Сопротивление электрода при увеличении частоты стремится к некоторой постоянной величине, которая определяет собой скорость процесса разряда.
Опыты проводили в интервале потенциалов от О до 1 ев анодную сторону от обратимого водородного потенциала в испытуемом растворе при атмосферном давлении. В этих условиях все стадии суммарного процесса, кроме стадии разряда и ионизации водорода, исключались. Таким образом, удалось непосредствен-
Перенапряжение при разряде ионов водорода
323
но измерить абсолютную скорость стадии разряда и ионизации водорода. При этом оказалось, что омический компонент прово-
димости электрода, измеренный током достаточной частоты,
обратен скорости разряда Н+-ионов и ионизации адсорбирован
ного водорода, умноженной на
величину Эти измерения
впервые позволили непосредственно определить скорость отдельно взятой стадии:
H3O+(Pt) + e->H(Pt) + H2O
и показать, что этот процесс протекает с конечной скоростью. Далее сравнивали скорость стадии разряда и ионизации со скоростью суммарного процесса 2Н+^Й2, и было показано, что скорость стадии разряда больше скорости суммарного процесса на платине при обратимом водородном потенциале в НС1 в 27 раз и в NaOH в 11 раз. В соответствии с этим было принято, что перенапряжение водорода на платине определяется скоростью двух реакций — разряда на свободных и заполненных участках поверхности электрода с образованием соответственно адсорбированного атома водорода и молекулы воды, а также скоростью обратного процесса.
По М. И. Темкину, замедленный разряд можно рассматривать как замедленную активированную адсорбцию, считая, что объединение электрона с адсорбированным гидратированным ионом водорода есть мгновенный акт. Уравнения, полученные при этом предположении, не отличаются от уравнений соответствующих теорий замедленного разряда.
Рассмотрим физическую сущность зависимости tjh = = /(Сн>о+). Из хода адсорбционной кривой (рис. 114) следует, что когда концентрация НзО+-ионов мала, адсорбция примерно пропорциональна этой концентрации (участок АВ кривой). Если значение СНа0+ велико, то CHj04- достигает адсорбционного насыщения (точка С), свободных для адсорбции мест на поверхности нет и перенапряжение сводится к борьбе ионов за место на поверхности электрода. Так как число таких мест зависит от других поверхностно активных ионов в растворе (La3+, С1~ и др.), то они также влияют на перенапряжение.
Опытным путем, однако, не удалось с достоверностью обнаружить существование предельного тока, обусловленного насыщением поверхности адсорбированным водородом. Во рсяком случае, как показали Б. Н. Кабанов и Дж. Бокрпс, он не наблюдается на ртутном электроде. Представление о рекомбинационном механизме удаления адсорбированного водорода встречает возражения и тогда, когда концентрация водорода на поверхности электрода очень мала. Приведенные соображения застав-21*
324
Теоретические основы электрохимии
ляют наряду с рекомбинацией рассмотреть и электрохимический механизм удаления адсорбированного водорода, при котором
атомы водорода переходят в молекулы не в процессе соединения с другими атомами [схемы (XI,8) и (XI.11)], а вследствие реакции с ионом гидроксония (НзО+) или молекулой воды [схемы (XI,9) и (XI,12)] Другими словами, предполагается, что разряд водородных ионов возможен не только на свободных от
Рис. 144. Адсорбционная кривая, выражающая зависимость ^н,= /(Сн,о+)
адсорбции участках поверхности, но и на тех местах, где уже имеется атом водорода.
Механизм электрохимической десорбции очень вероятен и, таким образом, удаление адсорбированных атомов водорода с поверхности электрода может осуществиться двумя путями:
а) обычной (так называемой каталитической) десорбцией, скорость которой, по подсчетам М. И. Темкина, равна
ок 5» 5 • 101ое12 а/см2, (Х1,63)
где
б) электрохимической десорбцией, скорость которой, как показал А. Н. Фрумкин, примерно равна:
v3 == 2 • 1О40 а/смК
Очевидно, если степень заполнения поверхности велика, основную роль играет каталитическая десорбция, которая соответствует, таким образом, большим перенапряжениям. Наоборот, при малых перенапряжениях, когда в мала, главную роль играет электрохимический механизм десорбции.
Естественно, что в общем случае могут быть осуществлены оба механизма разряда.
Если замедленными являются стадии нейтрализации и электрохимической адсорбции, то при малых поляризациях будет иметь:
i = 2FACHCHaO+ 4- ё~^ГбНЦ (XI,65)
откуда для перенапряжения получается;
1 См. стр. 305.
Перенапряжение при разряде ионов водорода 325
7) = const 4- -In CHso+ + In ГГ (XI,66)
причем const = /(Сн).
Это уравнение было выведено А. Н. Фрумкиным для объяснения поляризации на платиновом катоде.
Таким образом, опыт показывает, что как замедленный разряд, так и замедленная рекомбинация (и десорбция) водорода могут оказаться причиной перенапряжения. При этом рекомбинация (и десорбция) могут идти двумя путями — каталитическим и электрохимическим. Процесс разряда Н3О+ -> Н — Me, очевидно, сводится к дегидратации Н+-иона и адсорбции атома водорода. Поэтому при разряде затрачивается энергия активации и выигрывается энергия адсорбции водорода.
Если металл хорошо адсорбирует водород (платина, палладий), скорость разряда велика, а скорость рекомбинации мала и лимитирующей стадией является рекомбинация. Наоборот, если металл плохо адсорбирует водород (свинец, ртуть), т. е. 0Н мало, то скорость рекомбинации велика и перенапряжение определяется стадией разряда; для металлов со средними величинами энергии адсорбции могут существовать различные механизмы перенапряжения, что и было экспериментально показано П. Д. Луковцевым и С. И. Левиной при изучении выделения водорода на никеле.
ЛИТЕРАТУРА
Б а г о ц к и й В. С., Я б л оков а И. Е. ЖФХ, 23, 1949, 413. Bonhoeffer. Z. phys. Chem., 113, 1924, 199.
Ванюкова Л., Кабанов Б. ЖФХ, 14, 1940, 1620.
Глесстон С. Введение в электрохимию (примечание Б. Н. Кабанова). ИЛ, 1951, стр. 713.
Долин П. И., Эршлер Б. В. и Фрумкин А. Н. ЖФХ, 14, 7, 1940, 905—916.
Есин О. А., Кожеуров В. ЖФХ, 17, 4, 1943.
Заринский В. А. Перенапряжение водорода на ртутном катоде и g-потен-циал. ГОНТИ, 1938.
Иофа 3. А., Степанова В. ЖФХ, 19, 1945, 125.
Л евин А. И., Ф аличева В. И. ЖФХ, 33, 1959, 930.
Лошкарев М. А., Есин О. А. ЖФХ, 11, 1938, 410.
Лошкарев М. А., Крюкова А. А. ЖФХ, 23, 1949, 1457.
Луковцев П. Д., Левина С. И. ЖФХ, 21, 1947, 592.
Проблемы физической химии. Труды Физико-химического института им. Л. Я. Карпова. Госхимиздат, 1958.
Печерская А. Г., С т ен д е р В. В. ЖФХ, 24, 1950, 865.
Соколов Н. Д. ДАН СССР, 1961, 1948, 87.
Темки.нМ. И. ЖФХ, 15, 1941, 336; 22, 1948, 1081.
Фрумкин А. Н. ЖФХ, 24, 1950, 224.
Фрумкин А. Н., Луковцев П. Д. Химическая наука и промышленность. III, 4, 1958, 410.
Фрумкин А. Н., Иофа, 3. А., Б а го цки й В. С. ЖФХ, 25, 1951, 1117.
Глава XII
ПЕРЕНАПРЯЖЕНИЕ ПРИ РАЗРЯДЕ МЕТАЛЛИЧЕСКИХ ИОНОВ
§ 1. Общие сведения
Имеющиеся экспериментальные данные по катодному осаждению металлов недостаточны для теоретических обобщений. Особенность реакций разряда на катоде металлических ионов — возникновение новой твердой фазы. Поэтому здесь наряду с явлением переноса вещества и нейтрализации ионов необходимо учитывать трудности, связанные с построением кристаллической решетки.
Затруднения, вызванные переходом металлических ионов межфазной границы, обычно определяются термином «перенапряжение металлов».
В общем виде процессы разряда катионов металла можно выразить следующим образом:
[Ме(Н2О)л.]р^твор 4- ze МекрИст + хН2О. (XII, 1)
Многими исследователями было показано, что эти процессы протекают замедленно, причем скорость их зависит от природы металла, состояния его поверхности, температуры, состава электролита и других факторов, сопровождающих электролиз.
Процесс (XII, 1) условно можно разбить на ряд последовательных стадий:
[Me (Н2О)х]раствор [Me (Н2О)А]двойной слой (з);
[Me (Н2О)х]дХй1юй слой ? Mez+ + хН2О (б);
Мег+ + ге -> Меатом (в);
Меатом —» Мекриет. зародыш (г);
Мекриет. зародыш * МекрИСт. решетка (fl)-
Из всех этих стадий наиболее изучена лишь стадия (а), касающаяся диффузионных ограничений, т. е. доставки ионов из раствора к поверхности электрода. Хотя суммарная скорость
Перенапряжение при разряде металлических ионов 327
диффузии, миграции и конвекции ионов в общей сложности довольно большая, при значительных плотностях тока эта стадия может оказаться лимитирующей, вызывая заметную концентрационную поляризацию.
Однако повышая концентрацию и температуру электролита, применяя интенсивное перемешивание и другие меры, устраняющие транспортировку вещества, можно в значительной мере элиминировать влияние диффузионных ограничений, возникающих у электродов.
В данной главе рассматриваются процессы, связанные с переходом ионами фазовой границы1; их можно выразить уравнением
[Л1б (Н2О)х]деойной слой h -Л^бкрист Ч~ хН2О. (XII,2)
Анализ подобных электрохимических процессов весьма затруднителен из-за непрерывного изменения поверхности электрода вследствие отложения либо ионизации металлов.
Полагают также, что наряду с изменением величины истинной поверхности электрода не остается постоянной и ее «активность». Указанные и некоторые другие затруднения, наблюдаемые при изучении riMe, привели к заметной невоспроизводимое™ кривых потенциал — плотность тока, полученных различными исследователями. Можно привести примеры, когда даже одни и те же авторы прибегают к различным истолкованиям экспериментальных данных для объяснения природы явления перенапряжения на одинаковых металлических электродах.
Вместе с тем, если отбросить дегидратацию металлического иона как самостоятельную стадию по причинам, рассмотренным ранее, и не учитывать возможность одновременного протекания процесса по всем приведенным выше промежуточным стадиям с соизмеримыми скоростями, то станет ясным, что действительные затруднения при разряде металлических ионов можно свести к следующим двум причинам:
1) скорость процесса определяется замедленной кристаллизацией, т. е. стадией (г) или (д). В этом случае реакция (в) протекает быстро, а уд. вес процессов (а) и (б) настолько ничтожен, что ими можно пренебречь;
2) скорость процесса определяется стадией (в) —замедленного разряда, а все остальные промежуточные стадии не являются лимитирующими.
В соответствии с рассмотренными двумя крайними случаями к настоящему времени определились две основные теории пере
1 Поляризация в таком случае исключает стадию (а), связанную с диффузионными ограничениями.
328 Теоретические основы электрохимии ъ
напряжения металлов: теория замедленной кристаллизации и теория замедленного разряда. Эти теории и рассматриваются ниже в их исторической последовательности.
§ 2. Теория замедленной кристаллизации
Проблема возникновения новой фазы имеет общее значение. Если, например, в переохлажденной жидкости возникает . кристаллический зародыш, то при этом выделяется скрытая теплота плавления. Подобное выделение энергии может привести к нагреванию вещества выше температуры плавления возникшего кристалла. Это будет означать, что зародыш расплавится или вообще не возникнет. Поэтому появление кристаллического зародыша возможно только в том случае, если имеется флуктуаль-но формирующаяся группировка молекул с пониженным запасом энергии по сравнению со средним уровнем для молекул данной жидкости.
Современное учение о возникновении новой кристаллической фазы связано с работами М. Фольмера с сотрудниками, который показал, что при образовании металлического зародыша *, подобно зародышу в среде пересыщенного пара, необходимо затратить работу, величину которой можно определить из подсчета свободной энергии некоторого кругового процесса.
Рассматривая путь иона из гидратированного состояния в растворе до кристаллического состояния на катоде, Фольмер указывает, что получившиеся после разряда атомы должны принять в металле ориентированное положение. И даже в том случае, если разряд ионов совершается беспрепятственно на любых участках электрода, то стадия образования и роста кристаллов может оказаться замедленной. Исходя из предпосылки, что процесс электрокристаллизации является частным случаем фазовых превращений при образовании кристаллического зародыша внутри газообразной фазы, или расплава, Фольмер полагает, что плотность тока здесь играет такую же роль, как пересыщение при кристаллизации из раствора или величина температурного градиента при кристаллизации из расплава. При фазовых превращениях одна фаза может перейти в другую или путем возникновения зародышей новой фазы внутри прежней, или, если эти процессы не связаны с образованием зародышей, в результате удаления поверхностных атомов твердого тела.
1 Кристаллический 13арощыш, или центр кристаллизации, —- это уплотнение, вокруг которого начинается рост кристалла. Центры кристаллизации возникают благодаря тому, что вхождение атомов в кристаллическую решетку соответствует наиболее устойчивому состоянию системы. Зародыш — это наименьший кристалл, который может существовать при данных условиях (t, р, С и др.), и поэтому он не может исчезнуть самопроизвольно, но дает начало росту кристаллов.
Перенапряжение при разряде металлических ионов 329
Для возникновения зародышей новой фазы внутри прежней необходима затрата энергии на преодоление энергетического барьера, связанного с созданием границы зародыш — фаза.
1. Пусть W2— работа, необходимая для образования устойчивого двухмерного зародыша (характеристической капли для пересыщенных паров); тогда выражение для скорости образования, а следовательно, для пропорциональной ей плотности тока, будет иметь вид:
(ХП>3)
I = ,
где
W72=-^-. (XII,4)
RT In —
Роэ
Здесь р — реберное натяжение;
О — молярная поверхность;
рк — давление паров над поверхностью кристаллического зародыша;
Роо—давление паров над жидкостью.
Необходимое для образования характеристического зерна (зародыша) пересыщение связано с перенапряжением, т. е.
' ОО (XII,5)
откуда 1Г/2 = ^-zF^Me (XI 1,6)
и, следовательно, тгр2О
ДТгГэ].. i = Ke Me • (XII.7)
Если в первом приближении пренебречь изменением р с изменением потенциала, то можно все величины считать постоянными, т. е.
т.р2О т-р2О
тогда 1
1 Перенапряжение (чд[е) имеет положительный знак при катодной поляризации.
330 Теоретические основы электрохимии
Таким образом, если скорость образования двухмерного зародыша определяет собой величину перенапряжения, то из формулы (XII,8) получим
—b---a + &lni. (XII,9)
''‘Me
2. Аналогично предыдущему связь перенапряжения с замедленностью образования трехмерного зародыша устанавливается из следующих трех уравнений:
тег
/ = <ХП’10)
= / ?2МЗ Х2~ (ХП,П)
d2 / RT In ]
\ Р°° /
= (хп, 12)
Роо
Здесь W3 — работа образования устойчивого трехмерного зародыша;
М — молекулярный вес;
d — плотность осаждающегося металла.
Совместное решение этих уравнений приводит к зависимости -b- = a + 61ni. (XII, 13)
"ЧМе
3. Рассматривая состояние ионов металла, которые приносятся током ж электроду, но не могут в силу торможения войти в кристаллическую решетку, Брандес обсуждает следующую возможность. Ионы, не разряжаясь, входят в двойной слой, емкостно связывая протекшее количество электричества. Изменение потенциала по времени в таком случае зависит от емкости двойного слоя и увеличения заряда. Заряд составляется из разности двух величин: плотности поляризующего тока и скорости перехода иона металла из двойного слоя в кристаллическую решетку (т. е. плотности деполяризующего тока). Таким образом:
—О’-h). (ХИ, 14)
ат с
Здесь т] — катодная поляризация;
т — время;
с — емкость двойного слоя;
i — плотность поляризующего тока;
t'i — плотность деполяризующего тока.
Перенапряжение при разряде металлических ионов 331
Если теперь допустить, что ионы, приносимые к электроду током, разряжаются в двойном слое не на любом месте, а лишь в некоторых, энергетически подходящих точках электрода, то благодаря тому, что ионам приходится странствовать вдоль двойного слоя в поисках пункта, удобного для перехода в кристаллическую решетку, и преодолевать сопротивление среды, окружающей активные участки, необходимо добавочное напряжение. Поляризация и плотность тока, определяемая потоком ионов, будут связаны в таком случае уравнением, аналогичным закону Ома, т. е..
riMe=^Ki. (XII,15)
Таким образом, в теории замедленной кристаллизации Фоль-мера и Эрдей-Груза в процессе катодного выделения металла рассматриваются следующие варианты:
1. Разряд металлических ионов обладает достаточной скоростью, а кристаллизация 'металлов при электролизе является наиболее замедленной стадией. При этом необходимо различать возможности:
а) скорость образования двухмерного зародыша кристалла является наиболее замедленным процессом. В таком случае, исходя из работы образования зародыша, получим
-J— = а + b 1g /к;
ЧМе
б) скорость образования трехмерного зародыша определяет величину перенапряжения. Этот случай характеризуется зависимостью
—= a + 61g/K.
'ТМе
2. Если причиной перенапряжения металлов является замедленность доставки материала к месту формирования кристалла, что ведет к возникновению дополнительного омического сопротивления в электролите, окружающем активные центры, то
Фольмер, обобщая эти положения, делит металлы на две группы: 1) металлы с малой поляризацией и 2) металлы с большой поляризацией. К первой группе он относит: Hg, Си, Zn, Cd, Ag, Bi. Ко второй группе относятся металлы железной группы, промежуточное положение занимает РЬ.
Если у Hg имеется только концентрационная поляризация, то у металлов Си, Zn, Cd, Ag, Bi к ней добавляется еще «структурная» поляризация, обусловленная своеобразным слоевым
332 Теоретические основы электрохимии
ростом кристаллов. При этом Фольмер отмечает, что электролиз протекает лишь на активных участках, «ступенях», которые образуют эти слои, т. е. другими словами, фактическая плотность тока на растущих участках значительно больше той величины, которая получается вычислением для поверхности всего электрода. Так как число стационарных активных точек поверхности зависит от величины электродного потенциала, это вызывает довольно сложную зависимость перенапряжения от плотности тока.
Фольмер допускает также, что у металлов второй группы, имеющих особенно большую поляризацию и дающих обычно мелкокристаллические осадки, перенапряжение вызвано замедленностью процесса разряда, т. е. в этом случае зависимость T)Me = а + 61g i описывается уравнением Тафеля.
§ 3. Развитие теории перенапряжения металлов
Приведенное выше деление металлов на группы с большой и малой поляризацией верно лишь в первом приближении, так как характер поляризации зависит не только от природы металла, но и от свойств и природы электролита (валентность, комплексность ионов ит. п.) и условий, сопровождающих процесс элек-трокрпсталлизации. Весьма сильно на течение электрохимических реакций влияет адсорбция поверхностно активных веществ, приводящая к изменению значения ipi-потенциала.
Сам факт разделения единого катодного процесса на ряд гипотетических промежуточных стадий представляется недостаточно обоснованным и, по-видимому, противоречит действительности. Безусловно верным является лишь разделение поляризации на концентрационную и химическую, ибо эти виды торможений обусловлены по существу разными процессами: процессом диффузии и миграции или вообще замедленной доставкой реакционно-способного компонента к месту протекания реакции и самим электрохимическим актом, т. е. образованием атомов металла из гидратированных или комплексных ионов того же металла в электролите.
Богатый опыт, накопившийся при исследовании водородного перенапряжения, позволяет перекинуть мост к решению проблемы перенапряжения металлов. В данное время имеются многие экспериментальные факты, свидетельствующие о достаточной применимости основных положений теории замедленного разряда к процессам электроосаждения и ионизации металлов. Здесь, как и в случае истолкования закономерностей разряда водородных ионов, должны быть учтены в полной мере специфические свойства и строение двойного электрического слоя на границе фаз металл—-электролит.
Перенапряжение при разряде металлических ионов
333
Изучаемые закономерности перехода ионами фазовой границы в известных условиях могут быть условно приравнены к обратимым процессам, так как из рассмотрения кинетики электродных реакций следует, что различные условия электролиза, такие, например, как температура, перемешивание, изменение концентрации ионов, участвующих в, реакции (в приэлектрод-ных слоях), и др., влияют симметрично и на катодную, и на анодную (в случае растворимых электродов) поляризацию. Поэтому для процессов, осуществляемых на металлических электродах при заданной плотности тока [см. уравнение (XII,2)], можно использовать известное уравнение теории замедленного разряда А. Н. Фрумкина, связывающее плотность тока с перенапряжением и величиной равновесного тока обмена:
iK = Г—7 = КхСМе— К2СМее^
I п,Г(Ч>—<Ppi asF(4>—фр) \ , a^F atr,F \
= 4U — ё~кг ) =-- 4 U нт — е м /. (XII, 16)
Легко заметить, что это соотношение справедливо не только для любого электродного акта, протекающего в прямом (электроосаждение), и в обратном (ионизация) направлениях. Отсюда напрашивается вывод, что грань кристалла, характеризуемая наибольшим равновесным током обмена, должна (при прочих равных условиях) и расти, и растворяться быстрее всех остальных граней. Основное уравнение теории замедленного разряда, оправдавшееся полностью для водородного перенапряжения, может быть применено и для процессов, связанных с ростом или разрушением кристаллической решетки металлов. Величина энергетического барьера (или энергии активации) при электроосаждении металлов представляет собой сумму:
А — иМе + Up -|- Uх,
(XII, 17)
где UMe—химическое сродство разряжающегося иона к металлу;
Up — сродство того же иона к раствору;
Ux —некоторый дополнительный фактор, обусловленный внешним электрическим полем и полями молекул, ионов и атомов, присутствующих в двойном слое. • Величина UMe определяется энергией связи иона в металле, а значит, и тем, в какой точке кристалла (ребро, грань, угол) разряжается ион. Следовательно, особенностью процессов электрокристаллизации как раз и является то, что кристаллизационные факторы определяют скорость (или энергию активации) самого акта разряда.
334
Теоретические основы электрохимии
Имеющиеся представления о величине и природе энергии активации чрезвычайно недостаточны; можно лишь предполагать, ЧТО I ; ! |
Д = а + Р?/, (XII,18)
где аир — некоторые постоянные.
Таким образом, величины UMe, Up связаны линейно с электродным потенциалом <р. Скорость процесса в таком случае выражается зависимостью:
• "На+Рч>;) +Рч>;
« = 1 —/ССре *, (XII, 19)
что по форме и содержанию совпадает с уравнением теории замедленного разряда.
Так как
?£ = ?р + W ТО
, v +Р71 V Г +РТ‘
или
Цме = а + b 1g i. (X 11,20)
Если учесть еще конечную скорость доставки ионов к электродной поверхности, то
или
Ъме = a-\-b\g (--1— у (XII,21)
I 1—-А- /
' *пр '
Последнее уравнение описывает концентрационную и химическую поляризацию в предположении, что причиной торможения электродного процесса является замедленный разряд. В таком виде это уравнение справедливо для любого процесса, протекающего с диффузионными и химическими ограничениями на границе электрод — электролит.
Таким образом, рассматривая основные положения теории замедленного разряда, нетрудно убедиться, что эта теория в достаточной мере учитывает специфику процессов, связанных с ростом или разрушением кристаллической решетки.
Мы. видели, что учет условий роста кристалла [равенство (XII, 19)] является одной из основных предпосылок теории замедленного разряда. Обычно предполагают, что электрокристаллизация осуществляется только на «свободных» местах в кристаллической решетке против разряжающегося иона. В данном случае разряд иона на «идеальной» поверхности грани кристалла
Перенапряжение при разряде металлических ионов 335
потребовал бы значительно большей энергии активации, приближающейся к величине работы выхода электрона из металла. В действительности процесс осуществляется на особо активных участках поверхности, где для разряжающегося иона находится место в кристаллической решетке. Для процессов кристаллизации в условиях, близких к равновесным, такими активными местами могут служить углы и ребра кристалла, а также и любые дефектные места кристаллической решетки (микротрещины, дыры и т. п.). Так как каждому акту разряда, ведущему к заполнению этих дефектов, отвечает акт ионизации, вновь освобождающий место к решетке, число дефектных мест остается постоянным длительное время, что обеспечивает определенную плотность токов обмена (равенство (XII, 16)].
В случае пропускания тока при катодном осаждении металла количество разряжающихся ионов превалирует над числом ионов, переходящих в раствор. Для этих избыточных актов разряда случайные дефектные места в кристалле не могут уже являться постоянной базой, так как они вскоре оказались бы заполненными. Стационарное отложение металла при небольшой поляризации может, очевидно, происходить лишь на краю кристалла, начинаясь на ребрах и углах и распространяясь далее фронтом в виде нового слоя, покрывающего прежние грани кристаллов. Именно такую картину наблюдали К. М. Горбунова, П. Д. Данков и другие исследователи на растущих кристаллических осадках.
Естественно, что такой тип отложения должен приводить к воспроизведению правильной формы кристаллов. Таким образом, теория замедленного разряда наиболее правильно объясняет особенности роста и разрушения кристаллических решеток, т. е. явлений, не происходящих при газовыделении.
Работы последних лет полностью подтверждают сделанный вывод о единстве механизма элементарных электрохимических реакций на газовых (например, водородных) и металлических электродах и о распространении на оба случая теории замедленного разряда.
Однако многолетний опыт электролитического извлечения металлов показывает, что приведенные выше достаточно простые кинетические соотношения, описывающие затруднения, возникающие при электроосаждении металлов и вызывающиеся диффузионными (замедленная транспортировка ионов), электрохимическими (замедленный разряд ионов) и структурными (замедленная кристаллизация) ограничениями, могут быть еще осложнены образованием на поверхности электрода адсорбционных или фазовых пленок вследствие сдвига в ионных равновесиях. Эти специфические затруднения заслуживают специального рассмотрения.
336 Теоретические основы электрохимии
§ 4. Природа ионов, участвующих в электродной реакции
Известно, что в растворе ионы металла окружены сольватной оболочкой либо связаны с молекулами растворителя в устойчивом комплексном соединении. Поэтому любой ион, прежде чем перейти в кристаллическую решетку, должен освободиться от связывающей его оболочки. Природа растворителя или комплексообразователя оказывает при этом весьма значительное влияние, так как обусловливает величину затрат энергии на десольватацию. Особое значение в гидрометаллургии и гальванотехнике приобрели комплексные соли, которые нацело диссоциируют на ионы по схеме
Na„ [MeXm] -> nNa+ + \MeXm] n~. (XI 1,22)
Сложные комплексные катионы и анионы способны частично диссоциировать на простые (гидратированные) ионы, например:
\MeXm\n~ +аН2О ? Me (Н2О)*+ + тх (Н2О)*“. (X11,23)
Последнее равновесие в отличие от обычных ионных принято называть сольватационным. Если пренебречь участием в рассматриваемых процессах молекул растворителя, то диссоциация комплексного иона может быть записана:
[МеХт}п~ Mez+ + тХу~. (X11,24)
Такие и подобные им равновесия характеризуются величиной константы нестойкости комплексного иона:
к [Мег+][Х?~]т
[МеХт]
Значения их весьма малы и, например для
Ale(CN)TT, Me(CN)2—, лежат в пределах 10~16—10~28.
Легко заметить, что концентрация [Мег+ ], т. е. простых ионов металла, в растворах комплексных солей составляет исчезающе малую величину. Время существования свободных катионов в таком случае также столь ничтожно, что всякая возможность их участия в разряде представляется невероятной (обычно величина т не превышает 10~16—10-20 сек).
В противоположность ионным и кислотно-основным равновесиям, которые устанавливаются мгновенно, сольватационные равновесия типа (XII,23) протекают во времени. Комплексы, для которых сольватационное равновесие устанавливается достаточно быстро, называют лабильными; комплексы, которые образуются медленно, называются инертными.
(XII,25)
ионов типа
Перенапряжение при разряде металлических ионов 337
Инертность или лабильность вовсе не определяются прочностью комплекса. Так, ионы типа Ni(CN)2—, Pd(CN)2-, Hg(CN)2~ лабильны, а соединения типа Co(CN)|—, Fe(CN)|—, Mo(CN)4-инертны. Комплекс Мп (CN)|—занимает промежуточное положение. Опыт показывает, что равновесие в растворе, содержащем Ni(CN)®~, устанавливается примерно в 104 раз быстрее, чем в растворе Co(CN)g—, хотя ионы никеля и кобальта очень сходны по их физико-химическим свойствам. Комплексы меди, одновалентные и двухвалентные, в сочетании с Cl~, Br~, SO2- и NH3 являются лабильными.
Скорости установления сольватационных равновесий существенно влияют на кинетику электродных процессов. Так, например, ход поляризационных кривых в ряде случаев зависит от времени, прошедшего с момента приготовления комплексного электролита.
Другой важной характеристикой комплексных растворов электролитов являются величины констант нестойкости сложных ионов, которые выражают собой прочность комплекса и связаны с изменением стандартной свободной энергии АФ равенством:
Дф = —ЯТЧп/С. (XI 1,26)
Прочность комплексного иона определяется электростатическими и ковалентными силами, которые в зависимости от свойств комплексного иона могут играть преобладающую роль.
Таким образом, существующее мнение, что комплексный ион, прежде чем восстановиться на электроде, диссоциирует на простой ион и только в виде простого катиона восстанавливается на катоде, ошибочно. В ряде исследований показано, что комплексы восстанавливаются на катоде как таковые и обладают обычными для любых ионов предельными токами диффузии. Однако восстановление комплексных ионов во всех случаях сопровождается более заметной катодной поляризацией. О. А. Есин высказал предположение, что реакции распада комплексов могут лимитировать весь электродный процесс. Такая возможность в последнее время несколько раз обсуждалась в литературе.
При образовании какого-либо комплексного соединения металла, например меди, при избытке цианистого калия в растворе может существовать ряд комплексных форм; CuCN, Cu(CN)7, Си(СЫ)з—, Cu(CN)3— и т. п., концентрация которых определяется их константами нестойкости и сольватационными равновесиями.
При создавшихся в процессе электролиза условиях комплексный ион определенного состава и зарядности может доминиро-22 Заказ 1338
338
Теоретические основы электрохимии
вать в растворе, однако существование других возможных комбинаций его в электролите вовсе не исключается.
Стабильность комплекса ионного типа определяется зарядом и радиусом центрального иона. Стабильность же комплекса ковалентного типа характеризуется ионизационным потенциалом центрального иона.
Так как комплексы в растворе образуются ступенчато, то и стабильность каждой формы характеризуется соответствующей промежуточной константой нестойкости. В общем случае
= [xv ] = ] 2, 3 ...). (XI 1,27)
m [MeXm]
Деление на простые и комплексные электролиты все же в известной степени условно, так как невозможно провести абсолютно четкую границу между комплексными и «простыми» соединениями.
Комплексхые соединения являются в большинстве своем продуктом сочетания отдельных, способных к самостоятельному существованию «простых» соединений. При пропускании тока через электролит изменение состава комплексных или сольватированных ионов, участвующих в катодных реакциях, обусловливается не только их константами нестойкости и сольватационными равновесиями, но и условиями, которые устанавливаются при заданной плотности тока.
X. Геришер замечает, что при использовании цинковых солей непосредственными участниками электродной реакции являются комплексы типа: Zn(OH)2, Zn(C2H4), Zn(CN)2, Zn(CN)~ и т. п., т. е. соединения с низшим координационным числом и наименьшим отрицательным зарядом, чем это соответствует составу преобладающих компонентов раствора в равновесных условиях, т. е. при отсутствии тока.
К аналогичному выводу приходит А. Г. Стромберг, который полярографически определял состав непосредственно участвующих в электродном процессе и преобладающих в растворе компонентов. Это исследование основано на изучении зависимости разности анодного и катодного потенциалов полуволн необратимой анодно-катодной полярографической волны от концентрации комплексообразователя. Было показано, что состав непосредственно участвующих в электродном процессе комплексов цинка отличается от состава преобладающих в растворе комплексов, что согласуется с опытными данными, полученными Геришером методом переменного тока.
Позднее А. Г. Стромберг применил измененный метод расчета состава преобладающих в растворе и разряжающихся
Перенапряжение при разряде металлических ионов 339
комплексов, основанный на предположении, что электродный процесс при перезарядке комплексов полностью необратим.
Таким образом, было показано, что преобладающие в растворе комплексы Ti (четырехвалентного) и Ti (трехвалентного) имеют состав Ti(OH)2Cl2 и Ti3+, тогда как непосредственно в электродном процессе участвуют комплексы ТЮНС12+ и TiOHCl.
А. И. Левин и Е. А. Укше показали, что в тех случаях, когда в растворе присутствуют два комплексообразователя при относительно малых плотностях тока, в процессе разряда участвует наименее прочный комплекс. Так, например, при прибавлении к пирофосфатному электролиту определенного количества лимоннокислого натрия отмечалось, что реакция катодного восстановления протекает по схеме-
Си(СвН5О7Г + 2е - Си + СвН6ОГ, несмотря на присутствие в растворе в более значительной концентрации ионов Си(Р2О?)2- или Си(Р2О7)2~.
Можно, следовательно, заключить, что состав ионов, доминирующих в приэлектродном слое и непосредственно участвующих в электродной реакции, весьма часто отличается от состава комплексов или сольватированных ионов, преобладающих в объеме испытуемого раствора (или в равновесных условиях). Это связано с весьма заметными концентрационными изменениями, происходящими в приэлектродных слоях при электролизе. Опыт показывает, что в катодной реакции с изменением плотности тока могут непосредственно участвовать различные комплексные формы одного и того же металла, в том числе и сложные анионы, содержащие атомы металла.
Катодное восстановление анионов сопровождается иногда специфическими для этого процесса затруднениями — «спадами тока». На рис. 115 приведена i — ф-кривая для медного электрода в разбавленном пирофосфатном комплексном растворе. Как видно из рисунка, при потенциале, несколько более отрицательном, чем +0,05 в, резко уменьшается ток и тормозится процесс восстановления анионов СиР2О2~. Такой эффект может быть объяснен переходом i — ф-кривой через точку нулевого заряда и вызывается появлением сил отталкивания, которые возникают между отрицательно заряженной поверхностью металла (катода) и анионами. Подобное влияние отрицательного заряда поверхности на анионы может проявиться различным образом.
Подробно это явление было изучено в работах А. Н. Фрумкина с сотрудниками. Экспериментально установлено, что для ряда анионов, восстановление которых начинается при потенциалах, соответствующих положительным, зарядам поверхности электрода, поляризационная кривая особенно в случае разбав-22*
340
Теоретические основы электрохимии
ленных растворов имеет аномальную форму. К числу таких анионов, помимо СиР^О?-, относятся SoO^-, PtCl?-, PtCI?-, Hg(CN)*~, S40|- Fe(CN)6— и некоторые другие. В области по-
тенциала нулевого заряда скорость восстановления анионов резко уменьшается из-за электростатического отталкивания их от отрицательно заряженной поверхности электрода.
Полуколичественно это явление может быть объяснено на основании приближенного уравнения теории замедленного разряда: ;
« «гкг
(XI 1,28)
из которого следует, что поверхностная концентрация восстанавливающихся анионов зависит от всех тех факторов, которые влияют на ipi-inoTeH-циал.
+DJ10 +0,15 +ЦЮ +Щ№ О -OflS -0,15 Потенциал
Рис. 115. Поляризация медного катода в пирофосфатных электролитах вблизи точки нулевого заряда:
1 — 0,0115-м. CuSO4 4- 0,016-м. Na4P20z:
2 — 0,0098-м. CuSO4 + 0,08-м. Na4P2O7:
S - 0,02-м. CuSO4 + 0,03-м. Na4P2O7;
4 — 0,03-м. CuSO4 + 0,05-м. Na4P2O,
Приближенно величина ф1 -потенциала в этом случае может быть представлена выражением
<]>! = const -f- In (± <ра)-In С. (XI 1,29)
Здесь С—концентрация электролита;
za—валентность комплексного аниона;
<ра— потенциал ионной части двойного слоя, т. е. потенциал электрода, отнесенный к потенциалу нулевого
заряда.
Знак плюс перед <ра ставят, если ф1 > О, знак минус — если ф1 < 0.
Величина фь следовательно, зависит от потенциала электрода, концентрации электролита, валентности ионов, присутствия посторонних добавочных агентов и т. п.
Возможность разряда анионов не только на положительно заряженной поверхности электрода, но и после спада тока, когда катодная поверхность приобретает большой отрицательный заряд (правая ветвь электрокапиллярной кривой), объясняется по-разному. Ж. Биллитер, например, допускал, что в поле като
Перенапряжение при разряде металлических ионов 341
да комплексные анионы ведут себя как диполи, поворачиваясь к электроду положительным полюсом, т, е. катионом металла. Вследствие «растягивания» сложного комплексного иона из-за электростатических сил отрыв металла облегчается.
Т. А. Крюкова объясняет возможность разряда анионов на катоде экранирующим действием находящихся в растворе посторонних положительных ионов, которые то тем или иным причинам сами разряжаться не могут.
Поверхностно активные катионы (например, катионы тетра-бутиламмония) уменьшают отрицательный заряд катода и тем самым облегчают восстановление сложных анионов на катоде.
Этот факт •свидетельствует о том, что действие катионоактив-ных добавок является следствием их адсорбции и изменения структуры двойного слоя.
А. Н. Фрумкин замечает, что реакция восстановления анионов на отрицательно заряженных поверхностях может проходить по туннельному механизму1 без непосредственного соприкосновения разряжающихся ионов с металлом электрода.
Так, было показано, что скорость катодного восстановления анионов на отрицательно заряженной поверхности ртути в присутствии неорганических катионов, сдвигающих ^-потенциал в положительную сторону, возрастает в соответствии с уравнением (XII,28), причем увеличение скорости тем больше, чем выше заряд неорганического аниона. Восстановление анионов ускоряется и при увеличении радиуса катионов в ряду: Li+ < Na+ < < К+ < Rb+ < Cs+, так как с ростом радиуса катионов увеличивается их адсорбируемость на поверхности ртути.
В’ последнее время было установлено, что катион Н3О+, не обладающий специфической адсорбцией на ртутном электроде, влияет на скорость электровосстановления S2O|— примерно так же, как и катион Li+. Это указывает на облегчение подхода аниона к поверхности электрода благодаря образованию катионных мостиков, что приводит к снижению абсолютной величины отрицательного ф1-потенциала. Образование этих мостиков, как и сама адсорбция катиона в двойном слое, происходит, по-ви-димому, с нарушением гидратной оболочки катиона. В таком случае многозарядные анионы можно рассматривать «включенными последовательно» с ионами двойным слоем на границе раздела фаз (рис. 116).
С некоторым приближением характер электростатического влияния неорганических катионов на кинетику электровосстановления анионов может быть распространен на органические
1 Туннельный механизм (или туннельный эффект) характеризует прохождение частицы сквозь потенциальный барьер, т. е. сквозь такую область пространства, где потенциальная энергия частицы больше ее йолной энергии.
342
Теоретические основы электрохимии
катионы. Так, были исследованы электростатические эффекты органических катионов тетраалкиламмония, трибензиламина и двузарядного катиона N, N'-триметилен- (триэтил)-аммония на скорость восстановления S2O|~, Fe(CN)|—, PtCl^~, PtClg- и PtBr*— на капельном ртутном электроде. Было показано, что
Рис. 116. Схема образования катионного мостика
органические катионы ускоряют реакцию восстановления анионов при потенциалах адсорбции органических катионов на ртутном электроде. Эффективность действия катионов возрастает С повышением концентрации добавок, заряда органического катиона и с увеличением его адсорбируемости. Эффективность действия катионов зависит также от геометрического строения восстанавливаемого аниона и величины органического катиона.
Таким образом, с изменением плотно
сти тока в процессе электролиза в катодной реакции могут принимать участие различные комплексные формы одного и того же комплексообразователя. При
этом с ростом плотности тока на поляризационных кривых появляется площадка предельного тока для первой формы комплексного соединения, вслед за которой становится возможным разряд более сложного (имеющего больший отрицательный заряд) иона. В общем случае существование нескольких перегибов на Z —$р-кривых всегда связано с разрядом различных ионов, содержащихся или образующихся в процессе электролиза в электролите (точнее, в приэлектродных слоях электролизера).
§ 5. Явление адсорбции и кинетика электрохимических реакций
Адсорбция поверхностно активных веществ, меняя величину и знак ф1-потенциала, может существенно влиять на скорость электрохимической реакции. Торможение поверхностно активными веществами реакции восстановления различных ионов начали изучать только в последние годы. Систематические исследования в этой области были сделаны А. Н. Фрумкиным, О. А. Есиным, М. А. Лошкаревым, Т. А. Крюковой и другими исследователями в нашей стране >и за рубежом.
Если на какой-либо поверхности, например на грани кристалла, возрастает концентрация каких-либо молекул или ионов, то такое уплотнение вещества на поверхности раздела называется адсорбцией; происходит оно под действием поверхностных сил, называемых адсорбционными. Поглотитель называется адсорбентом, поглощаемое вещество— адсорбатом.
Перенапряжение при разряде металлических ионов 343
Наиболее полно процессы адсорбции различных веществ изучены на жидкой ртути. Результаты этих исследований показывают, что максимальное количество адсорбированного вещества удерживается на поверхности ртутного катода в интервале потенциалов, близких к точке нулевого заряда ртути. В случае поляризации такого электрода в анодную или катодную сторону происходит десорбция поверхностно активных молекул с поверхности ртути; потенциал десорбции при этом зависит от природы адсорбированного вещества и концентрации его в электролите.
В случае адсорбции поверхностно активных ионов наблюдаются несколько иные закономерности. Так, при разноименном заряде поверхности ртути и поверхностно активных ионов они обычно сильнее притягиваются к электроду и десорбция не происходит. При одноименном заряде ртути и поверхностно активных ионов молекулы воды вытесняют поверхностно активные ионы и ртутный электрод в таком случае полностью освобождается от адсорбированного вещества.
Увеличение концентрации поверхностно активных веществ приводит к монотонному возрастанию величины адсорбции вплоть до некоторого предельного значения ее, отвечающего созданию плотного адсорбционного монослоя на поверхности ртути (адсорбционное насыщение).
Имеющиеся в литературе данные по адсорбции поверхностно активных веществ на твердых электродах гораздо менее систематизированы. Хотя силы взаимодействия между частицами данного вещества в твердом и жидком состоянии не имеют существенных различий, все же опыт показывает, что совершенно однородна только поверхность чистого металла в жидком состоянии. Кристаллическое твердое тело не однородно, так как даже в спектроскопически чистом металле различные грани, ребра и углы кристаллов обладают неодинаковым запасом энергии. Твердые тела, кроме того, обычно шероховаты. В общем случае на неполированной поверхности поверхностное натяжение в разных точках твердого тела имеет различную величину.
Появление на поверхности твердого катода адсорбционного слоя, построенного из частиц, обладающих дипольными свойствами, или из ионов, вызывает и здесь глубокие изменения, влияя на ту часть скачка потенциала, которая зависит, от свободных зарядов поверхности металла и ионов двойного слоя.
В результате появления пассивирующих слоев и пленок скорость электродной реакции меняется, что качественно проявляется в изменении поляризации. О заметном действии адсорбированного слоя говорится в таких случаях, когда вызванное им торможение достаточно велико, чтобы быть обнаруженным на опыте наряду с другими видами поляризации (концентрационной, химической, структурной).
344
Теоретические основы электрохимии
Вместе с данными,' из которых вытекает возможность возникновения полислойной адсорбции, имеются многие исследования, приводившие к выводу о возможности возникновения на поверхности твердого электрода монослоя. В таком случае адсорбционные процессы связаны с более глубоким взаимодействием между адсорбированным веществом и металлом, приводящим к образованию новых химических соединений на поверхности электрода (хемосорбция).
Общим для всех рассматриваемых случаев является то, что пассивирующие электрод, адсорбционные поли- или монослои выполняют роль своеобразного регулятора роста кристаллов, что связано с преодолением заметных торможений в осуществлении электродной реакции.
Для большинства органических поверхностно активных веществ характерно линейное строение молекул, длина которых значительно превышает поперечные их размеры. При этом одна часть молекул состоит из групп, родственных по своим свойствам молекулам растворителя, а другая — из радикалов, по свойствам резко отличным от него.
В соответствии с этим один конец молекулы представляет собой углеводородный радикал, характеризующийся слабыми побочными валентными связями, а другой — состоит из гидрофильных полярных групп, обладающих резко выраженными валентными силами.
Чем больше молекулярный вес поверхностно активного вещества (больше групп СН2, входящих в углеводородный радикал), тем большей активностью обладает добавка. Как уже отмечалось, действие растворимых поверхностно активных веществ обусловлено возникновением на металле адсорбционного слоя, образующегося вследствие миграции молекул или ионов из объема раствора и ориентации их на поверхности раздела *. Поэтому существенное значение при применении поверхностно активных веществ имеет их растворимость. Однако многие вещества, будучи практически не растворимыми в воде, также обладают способностью образовывать на поверхности раздела адсорбционные слои, которые могут быть обнаружены в результате исследования изменений величин, характеризующих процесс адсорбции (изменение поверхностной энергии, скачка потенциа.-ла на этой поверхности, емкости двойного электрического слоя и т. п.).
Из уравнения Гиббса, связывающего значения концентрации вещества дС с вызываемым им изменением поверхностного
1 Молекулы, адсорбированные на поверхности твердого тела, сохраняют способность тангенциального перемещения. Многочисленные опыты подтверждают существование такого перемещения — миграции.
Перенапряжение при разряде металлических ионов
345
натяжения <5ст, следует:
да RTT дС С ’
(XI 1,30)
Здесь Г—- поверхностная концентрация вещества (влияющего на поверхностное натяжение), приходящаяся на единицу поверхности;
С — концентрация адсорирующегося вещества в растворе.
Рис. 1'17. Графический метод построения изотермы адсорбции
Зависимость адсорбции от концентрации может быть установлена на основании опытных данных при исследовании изменения поверхностного натяжения от концентрации. Соответствующую задачу решают обычно графическим путем. На графике (рис. 117) кривая АВ представляет собой o=f(C). Здесь выбирают ряд точек М, N, Р и т. д., в которых желательно определить величину Г. Легко заметить, что tg <р= —.
В треугольнике ТИоЛ^ТИ сторона Л4ОМ1—Л4ОМ tg<p. Так как М0М = См, a tgq>M= , то. M0Mi = Cm. Из уравнения Гиб-
\ дС/м
бса (XII, 30) величина С-^- пропорциональна Г. Принимая дС
последовательно для абсциссы См ординату, равную отрезку M0Mi, для абсциссы Ск ординату N0Ni, для абсциссы СР ординату PqPi, можно в определенном масштабе получить ряд точек, для кривой, описывающей P=f(C).
346 Теоретические основы электрохимии
Как видно из рис. 117, накопление поверхностно активных .веществ на границе фаз электрод—электролит стремится к некоторому пределу, который обозначается Гm и соответствует образованию на поверхности мономолекулярного слоя поверхностно активного вещества.
Таким образом, уже при очень малых добавках вся поверхность электрода может быть заполнена и дальнейшее увеличение концентрации поверхностно активного вещества не будет оказывать влияния.
Непосредственная зависимость адсорбции от концентрации раствора для однородной поверхности выражается уравнением Лангмюра:
6= КС - (XI 1,31)
1 + КС ’
тде 0 — степень заполнения поверхности;
К — константа адсорбции.
Часть поверхности, занятую адсорбированным веществом, можно считать пропорциональной ---, тогда
Г = КС (XI 1,32)
1 + КС
Если величину, вычисленную по изотерме адсорбции Лангмюра (XII, 31), подставить в уравнение Гиббса (XII, 30), то получим
da = R7T — R7T™ J«^C------------. (XI 1,33)
С (1+KQC
Интегрируя уравнение (XII, 33) в пределах от 0 до С, будем иметь
С = СО— RTrmln(KC + l). (XI 1,34)
Полагая далее. /?7'ГСО=Р, получим уравнение Шишковского:
°о — о == К In (1 аС0). (X11,35)
Здесь К и а — постоянные.
Из уравнения (XII, 35) так же, как и из графического построения, следует, что наиболее заметно поверхностно активные вещества влияют на поверхностное натяжение при малых значениях Со. При дальнейшем увеличении концентрации добавочного реагента потенциал электрода должен меняться сравнительно мало. Представленная на рис. 118 зависимость изменения стационарного потенциала цинкового электрода от концентрации поверхностно активного вещества напоминает по форме адсорбционную изотерму. Зависимость <p=f(C) свиде-
Перенапряжение при разряде металлических ионов
347
Рис. Iil8. Зависимость стационарного потенциала цинка от концентрации добавки. Электролит 2-н. Z11SO4, t = = 30 °C.
Добавь-т: 1 — тетрабутиламмоннй;
2 — октанол; 3 — камфара
тельствует о том, что процесс образования пассивирующих сло-ев на металле связан с явлением насыщения. Плотность вещества. в адсорбционном поле меняется по экспоненциальному закону вплоть до полного блокирования поверхности электрода. Поскольку токи обмена при этом ослаблены, равновесие устанавливается не сразу. Чем большее сопротивление оказывает адсорбционная пленка, тем длительнее по времени устанавливается стационарный потенциал. Так, в присутствии камфары потенциал цинка устанавливается через 2 ч, в присутствии же тетрабутил аммония — через 3 ч и т. д.
В процессе электрокристал-.лизации сильно адсорбирующиеся вещества взаимодействуют прежде всего с поверхностью свежеобравующихся кристаллов. Это значительно изменяет скорость роста различных граней кристалла, так как, по закону Г. В. Вульфа, скорость роста грани кристалла пропорциональна ее поверхностному натяжению о. Таким •образом, поверхностно активные вещества, понижающие поверхностное натяжение, должны
тер ограничения и, следовательно, на скорость роста кристаллов.
Однако причину влияния добавочных реагентов на электрокристаллизацию нельзя усматривать только в изменении поверхностной энергии. Необходимо учитывать также глубокое химическое взаимодействие молекул, атомов и ионов добавки с ионами и атомами металла, вследствие чего образующееся новое поверхностное соединение может не только изменить форму кристалла, но в некоторых случаях полностью прекратить его развитие (пассивировать). На возможность образования поверхностных химических (или хемосорбционных) соединений указала Л. К- Лепинь, отметившая, что прочность подобных соединений должна возрастать с уменьшением радиуса атомов металла и увеличением его плотности.
Отличие химической адсорбции от физической1 заключается в том, что в случае хемосорбции поверхность металла (электрода) будет стремиться образовывать химические связи с вещест
1 Физическая адсорбция .протекает <под действием ван-дер-ваальсовых сил, -тогда как химическая (хемосорбция) связана с поверхностными химическими реакциями.
весьма сильно влиять на
348 Теоретические основы электрохимии
вом соприкасающейся фазы. При этом электроны переносятся от адсорбента к адсорбату и обратно.
Хемосорбция отличается от физической адсорбции также тем, что не все поверхности, даже если они чисты, активны для хемосорбции. Физическая адсорбция, наоборот, происходит на всех поверхностях.
Химически насыщенная поверхность всегда может быть насыщена в результате хемосорбции только одного атомного или молекулярного слоя; поэтому, если известно, что величина адсорбции превышает монослой, то по крайней мере второй и последующие слои адсорбируются физически.
Касаясь механизма хемосорбции, следует заметить, что при физической адсорбции никакие химические связи не разрываются и не создаются вновь и поэтому химическая природа адсорбируемого вещества не меняется. При хемосорбции адсорбированное вещество претерпевает химические или электрохимические изменения и обычно распадается на независимые части.
Радикалы и атомы, несущие заряд, являются первичными частицами, из которых создаются хемосорбционные слои. При хемосорбции может образоваться любой из трех главных'типов химической связи — ионный, ковалентный и координационный.
Среди многих свойств, определяющих адсорбционные слои, особое место занимает величина дипольного момента ц, который возникает при адсорбции. Величина дипольного момента указывает на тип связи (большие значения ц— ионные связи, малые значения ц— ковалентные); кроме того, работа выхода электрона из металла, необходимая для удаления электрона в вакуум с наиболее высокого занятого уровня в твердом теле, изменяется вследствие возникновения при адсорбции поверхностных диполей. Последнее свойство связано с изменением потенциального барьера, через который проходят электроны при удалении с поверхности.
В последнее время был получен обширный экспериментальный материал по электрохимическим и химическим свойствам хемосорбционных слоев на металлах. При этом были использованы измерения адсорбционных потенциалов, применены радиоактивные индикаторы и другие методы, позволяющие определить влияние адсорбционных слоев на кинетику электродных процессов. Так, например, было установлено, что адсорбция йода на платине сопровождается значительным проникновением его в глубь металла. Поскольку связь между металлом и адсорбированными атомами имеет дипольный характер, образование атомных слоев приводит к нарушению строения двойного электрического слоя вплоть до изменения знака потенциала. Характерно также заметное снижение емкости двойного слоя, вызванное созданием адсорбционных слоев.
Перенапряжение при разряде металлических ионов
349
Для понижения адсорбционной способности металлических порошков по отношению к влаге А. И. Левин и А. В. Помосов исследовали возможность создания на поверхности частиц металла тончайших адсорбционных слоев (пленок), образованных высокомолекулярными органическими соединениями жирного и ароматического рядов. При этом было установлено, что полярный конец гетерополярной молекулы образует типичную химическую связь с атомами металла, что приводит к необратимости таких процессов, в то время как углеводородный радикал высокомолекулярных соединений придает явно выраженные гидрофобные свойства наружной поверхности пленки, образующейся на частицах металла. Чем длиннее углеводородная цепь стабилизатора, тем более гидрофобной должна быть адсорбционная пленка на поверхности частиц.
В качестве добавок, обладающих стабилизирующим действием, испытывали такие органические соединения, которые, помимо гидрофобных свойств, способны давать достаточно тонко диспергированные водные растворы. Были изучены: бензойная, антраниловая, оксибензойная и алеиновая кислоты, тиокрезол, а также различные мыла и среди них простое натровое мыло.
В последнем случае при стабилизации медных порошков коррозионная стойкость металла повышалась в 50—70 раз по сравнению с нестабилизированной порошковой медью. Было показано, что для гидрофобизации поверхности частиц металла требуется вполне определенная концентрация мыла, при достижении которой стабилизирующее действие пленки проявляется наиболее полно. Опыт показал, что расход стабилизатора весьма незначителен, во всех случаях он не превышал 0,01%.
При использовании мыла надежная стабилизация поверхности частиц металла наступает при условии, когда все адсорбционные центры замещены, что, по-видимому, соответствует мономолекулярному адсорбционному насыщению поверхности.
Важное условие надежности защиты частиц металла от коррозии — способность стабилизирующей пленки к восстановлению своей целостности. Молекулы под влиянием двухмерного давления слоя адсорбированного стабилизатора стремятся занять все активные центры поверхности. Последнее свойство обеспечивает цельность и эластичность стабилизирующих металл пленок, а следовательно, и высокую коррозионную стойкость металлического порошка.
Экспериментальные данные свидетельствуют о том, что гид-рофобизация поверхности металлических частиц мыльной пленкой и некоторыми другими поверхностно активными веществами является не только средством защиты порошкообразных металлов от окисления, но способствует прессованию изделий, действуя в качестве активной смазки. ...
350
Теоретические основы электрохимии
Весьма часто поверхностно активные вещества, входящие в состав хемосорбированных слоев, непосредственно участвуют в электродном процессе. В ряде случаев получение осадков с определенными свойствами, например блестящих, особенно мелкокристаллических, становится возможным из растворов простых солей серебра, меди и других металлов.
При этом в полной аналогии с ростом единичного кристалла наблюдается пассивация граней поликристаллического осадка— те грани кристаллов, на которых вещество сильно адсорбируется, не растут, в то время как грани с относительно чистой поверхностью продолжают расти беспрепятственно *.
К катодной пассивности близко примыкает явление адсорбционной поляризации, приводящее к снижению тока в присутствии некоторых поверхностно активных веществ, обнаруженное М. А. Лошкаревым. При этом поверхностно активные вещества образуют на катоде сплошной слой, почти не проницаемый для разряжающихся ионов, что ведет к резкому торможению или полному прекращению роста кристаллов. Возникающая при этом химическая поляризация может достигнуть необычайно больших значений, далеко превосходя все известные ее величины для катодного выделения металлов из чистых растворов их простых солей. Как показали исследования, явление адсорбционной поляризации охватывает многие электродные процессы. Образование адсорбционных слоев вблизи точки нулевого заряда обычно сопровождается явлением низкого адсорбционного тока, который определяется скоростью проникновения реагирующих частиц из объема раствора в двойной электрический слой.. М. А. Лошкарев с сотрудниками описал многочисленные случаи, когда предельно низкие значения тока в очень широкой области потенциалов не зависят от потенциала.
Механизм блокирующего действия адсорбционных поверхностных слоев, составленных из органических соединений, объясняют по-разному. Так, А. Г. Стромберг считает, что действие это определяется изменением чН-потенциала, вызванным зарядами ионов или диполями адсорбирующихся молекул. По М. А. Лошкареву, основное значение имеет заполнение поверхности адсорбированными частицами. Снижение предельного тока объясняется не диффузионными ограничениями, но чисто активационным торможением катодной реакции. А. И. Фрумкин считает, что действие адсорбированных слоев при высоком заполнении поверхности не может быть сведено к изменению фгпотенциала. Он полагает неправильным также совершенно не учитывать электростатические факторы в случае слоев со значительным
1 Адсорбция ловеркностно активных веществ гранями кристалла, вызывающая вамедление его роста, получила название катодной пассивности.
Перенапряжение при разряде металлических ионов 351
заполнением. Многим исследователям удалось экспериментально показать существенное изменение характера катодных осадков и возможность получения весьма совершенных высокодисперсных отложений металлов при использовании поверхностна активных веществ.
Не менее интересно торможение электродных реакций при возникновении адсорбционных слоев при отсутствии специальных посторонних добавок. Как было показано А. И. Левиным с сотрудниками, при достаточной энергии адсорбции значительная часть активных участков катодной поверхности может быть заполнена малорастворимыми соединениями, возникающими в приэлектродных слоях вследствие концентрационных изменений и сдвигов в ионных равновесиях, наблюдаемых в католите при электролизе. Энергия адсорбции промежуточных соединений, концентрирующихся у электродной поверхности, так же как и в случае действия посторонних поверхностно активных веществ, зависит от соотношения между потенциалом нулевого заряда, характерным для данного электродного металла, и рабочей областью потенциалов, при которых протекает процесс электроосаждения металла.
Картина появления и роста адсорбционных пленок, составленных из гидроокисей, может быть представлена следующим образом: первоначально образуются островки, цепочки, которые не связаны между собой и свободно перемещаются. Со временем концентрация золя увеличивается, что приводит к ускорению его коагуляции. Постепенно образуется сетчатая структура и пленка становится мало подвижной. Наконец, ячейки сетки заполняются вновь образовавшимися частицами гидроокиси и пленка превращается в сплошную, неподвижную. С дальнейшим сдвигом равновесия гидролиза и образованием гидроокисей и основных солей частицы коллоидной взвеси больших размеров-могут наслаиваться, вследствие чего адсорбционные слои приобретают характер полимолекулярных пленок незавершенной структуры.
Очевидно, что для правильного истолкования кинетики электрокристаллизации необходимо наряду с диффузионными затруднениями, замедленным разрядом и кристаллизацией учитывать адсорбционную поляризацию, сильно влияющую на характер электродных реакций при электроосаждении металлов. Приведенная на рис. 119 диаграмма распределения ряда металлов, составленная Л. И. Антроповым в результате сопоставления рабочих потенциалов, сопровождающих катодное выделение металлов, с величинами равновесных потенциалов и точек нулевого заряда, указывает на большое значение потенциалов нулевых зарядов. Точка нулевого заряда не только характеризует металл, но разграничивает области, отвечающие положительно
352
Теоретические основы электрохимии
и отрицательно заряженной поверхности металла, а следовательно, и области преимущественной физической адсорбции катионоактивных, анионоактивных и молекулярных поверхностно активных веществ. Однако наряду с физической адсорбцией необходимо учитывать специфическую адсорбцию.
Рис. 119. Диаграмма распределения ряда металлов по величинам их потенциалов (отнесенным к соответствующим потенциалам нулевых зарядов) при равновесных условиях (•) и при катодном осаждении (<Д) металлов (по Л. И. Антропову)
§ 6. Соотношение кинетики адсорбции и скорости электродных реакций при заданной плотности тока в реальных условиях электролиза
Изучению действия поверхностно активных веществ в реальных условиях электролиза, т. е. при заданной плотности тока, посвящено большое число исследований. Это связано с тем, что в гидрометаллургии и гальвайотехнике некоторые добавочные реагенты существенно влияют на структуру и свойства катодных отложений. Вместе с тем в литературе указывается на ряд затруднений, встречающихся при попытке установить какую-либо определенную зависимость между составом и свойствами «добавки» и ее действием на процесс электрокрнсталлизации.
Первое из затруднений связано с тем, что восстановление ионов данного металла на катоде часто сопровождается совместным разрядом других металлических и водородных иОнов. Поэтому для исчерпывающего суждения о характере влияния добавочных реагентов необходимо иметь полную картину дей-
Перенапряжение при разряде металлических ионов 353
ствительного распределения тока при электролизе между разряжающимися ионами.
Второе существенное затруднение при анализе закономерностей действия поверхностно активных веществ — то, что сильно адсорбирующиеся вещества искажают и смещают положение максимума электрокапиллярных кривых и сдвигают положение потенциала нулевого заряда ’. Причиной подобных изменений является специфическая адсорбция. Нетрудно заметить, что вследствие возникновения изменений в положении потенциала нулевого заряда меняются области <р — о-кривых, характеризующие положительно и отрицательно заряженную поверхность металла, а следовательно, и соотношение потенциалов, при которых осуществляется разряд ионов и преимущественное концентрирование на поверхности электрода различных по характеру поверхностно активных веществ.
Наблюдающиеся относительные изменения в распределении точек нулевого заряда при неизменных исходных условиях электролиза и возникновении специфической адсорбции оказывают в дальнейшем решающее влияние на условия разряда и ионизации металлов, а также на величину энергии активации, которая требуется, чтобы электродный процесс протекал в желаемом направлении.
Третье серьезное затруднение в истолковании действия поверхностно активных веществ — использование в практике электролиза комбинированных и сложных добавок, представляющих собой не вполне изученные смеси различных высокомолекулярных органических соединений.
Компоненты добавки, оказывающие наибольшее влияние при электролизе, в большинстве случаев остаются невыясненными. Можно, однако, предполагать, что содержание «действующего компонента» в комбинированной добавке пропорционально концентрации всей добавки и что наиболее заметным поверхностно активным действием здесь будут обладать компоненты, имеющие наибольший молекулярный вес и линейное строение молекул (длина молекул превышает поперечные их размеры).
Анализ действия отдельных компонентов поверхностно активных веществ, кроме того, затрудняется появлением в католите непроизвольных образований, возникающих вследствие сдвигов в ионных равновесиях при электролизе. Как отмечалось, такие вещества выполняют роль добавочных регуляторов роста кристаллов. При этом возникает затруднение в количественном определении непроизвольной добавки, так как обычно ни концентрация, ни состав возникающего в католите малорастворимого соединения не поддаются точному анализу и учету.
1 Положение зависит, кроме того, от кристаллографических особенностей поверхности металла.
23 Заказ 1338
354
Теоретические основы электрохимии
Перечисленные и некоторые другие затруднения свидетельствуют о том, что при заданной плотности тока в реальных условиях электролиза действие поверхностно активных веществ усложняется и требует дополнительных обоснований.
Изучение кинетики адсорбции поверхностно активных веществ при достаточной скорости электродной реакции показывает, что концентрирование различных веществ на электродах, а значит, и состояние адсорбционного слоя очень сильно зависят от конкретных условий электролиза, свойств металла и раствора и, следовательно, от строения двойного электрического слоя (потенциала ионного слоя).
В самом общем случае при электролизе поверхностно активные вещества могут вносить следующие существенные изменения в строение межфазной границы поляризуемый электрод — электролит:
1) менять структуру двойного электрического слоя вследствие изменения фрпотенциала;
2) вносить пространственные ограничения, вплоть до полного блокирования поверхности электрода фазовыми пленками;
3) менять физико-химическую природу поверхности металла, ее заряд, смачиваемость и другие свойства вследствие хемосорбции.
Поэтому одностороннее (т. е. только с учетом физической адсорбции) рассмотрение влияния поверхностно активных веществ совершенно недостаточно для объяснения многообразия процессов, которые можно наблюдать при электролизе. В этом случае только знание динамического фактора, т. е. кинетики (физической и специфической) адсорбции поверхностно активных веществ на поверхности металла в сочетании с рассмотрением кинетики электродных процессов может представить интерес для истолкования наблюдаемых явлений.
Исходя из адсорбционной теории влияния поверхностно активных веществ на электродные процессы можно различать следующие возможные случаи действия адсорбционного слоя на катодный процесс.
1. Если скорость электродного процесса на чистой, свободной от адсорбционных слоев и пленок поверхности катода превалирует над скоростью адсорбции, то поверхностно активные вещества экранируют его лишь частично, что приводит к снижению-скорости разряда и увеличению поляризации катода (вследствие изменения фгпотенциала либо увеличения расстояния между обкладками двойного слоя).
2. В том случае, когда скорость адсорбции явно превалирует над скоростью разряда ионов, адсорбционный слой может быть настолько прочным, что для его преодоления необходимо затратить большую энергию активации. При этом скорость процесса
Перенапряжение при разряде металлических ионое 355
почти не зависит от потенциала (в определенном интервале) и на поляризационной кривой появляется горизонтальный участок предельного тока адсорбционной поляризации, характеризующий торможение процесса.
В практике электролиза наиболее частым является случай, когда на поверхности имеются участки активные, а также покрытые поверхностно активным веществом. В зависимости от характера адсорбированных веществ разряд может протекать или только по активным участкам поверхности, или по активным и малоактивным одновременно. В последнем случае скорости разряда на активных и малоактивных участках могут сильно различаться, причем разница в Аг = гаКт— г’неакт может быть очень велика и иметь положительный или отрицательный знак в зависимости от природы поверхности электрода, природы поверхностно активного вещества и условий электролиза.
При таком соотношении скоростей адсорбции и разряда ионов можно представить следующие варианты процесса.
1. Если на катоде разряжаются простые (гидратированные) катионы металла, а катодный потенциал расположен отрицательнее точки нулевого заряда, то фгпотенциал отрицателен и наболее вероятна адсорбция катионов. В таком случае скорость процесса, пропорциональная плотности тока, определяется уравнением
azF(cp—tpj)
г=КСре (XI 1,36)
где Ср — поверхностная концентрация разряжающихся ионов, определяемая в свою очередь равенством
zF
Ср — Се~нт\ (XI 1,37)
откуда следует, что при i]?i < 0 уменьшение отрицательной величины ^-потенциала, происходящее при адсорбции катионов, должно привести к торможению процесса (уменьшению г), что и было установлено при изучении влияния тетрабутиламмония на разряд водорода 3. Иофа, Б. Кабановым, Е. Кучинским и Ф. Чистяковым. Такой же случай наблюдается при разряде цинковых ионов из растворов ZnSO4.
Как показано в работе А. И. Левина и В. С. Колеватовой, поверхностно активные анионы, такие, например, как сульфонафталиновая кислота, не влияют на величину и характер катодной поляризации цинка, а следовательно, и на свойства катодного осадка.
Наоборот, высокомолекулярные органические вещества, диссоциирующие с образованием поверхностно активного катиона (алкалоиды и органические производные аммония), адсорбируются на катоде и вызывают заметную поляризацию, что приво-23*
356
Теоретические основы электрохимии
дит к торможению катодного процесса. Например, добавка тет-рабутил аммония в концентр ации до 25 мг/л при обычно принятой плотности тока (iK = 500 afw2) позволяла осуществлять длительное катодное осаждение цинка с выходом по току, достигающим 95,2%. При этом образуются плотные, исключительно мелкокристаллические, матовые осадки металла.
2. Если разряд катионов на катоде происходит при потенциалах более положительных, чем потенциал нулевого заряда, то ipi <0. Тогда наиболее вероятным процессом становится адсорбция анионов, сдвигающая фгпотенциал в сторону более отрицательных значений и тем самым увеличивающая скорость процесса (уменьшающая поляризацию).
Этот эффект также наблюдался 3. Иофа и его сотрудниками при изучении влияния С1—, Вт- и 3”-ионов на водородное перенапряжение. Подобный же случай наблюдается при осаждении меди из сернокислых растворов. Экспериментально было установлено, что разряд Си2+-ионов на катоде тормозится поверхностно активными анионами (сульфокислоты) и молекулами (клей, тиомочевина, амиловый спирт) и не тормозится поверхностно активными катионами. При этом скорость процесса определяется уравнением
а(Ф—ф')гГ гф'А
i = KiCcu^ +f(De (X11,38)
где ф1' — измененное значение адсорбционного потенциала; /(Г) —функция адсорбции Г, причем f(0) = 1.
Разряд сложных анионов, содержащих атомы металла, например СиР2О72- и Си (РгСМг6-, происходит при потенциалах, столь отрицательных, что фт <0. В этом случае адсорбция катионов должна способствовать уменьшению поляризации. Этот эффект и был обнаружен А. И. Левиным и Е. А. Укше в условиях, полностью совпадающих с теоретическими данными. Несколько упрощая действительную картину, можно сказать, что положительно заряженные ионы, адсорбируясь на катоде, уменьшают отрицательный заряд его поверхности и тем самым облегчают разряд комплексных анионов. Уравнение замедленного разряда при этом имеет вид:
? = =-^-1пС + (XII,39)
azKF azKF azKF а
где zK — заряд катиона меди;
za — заряд комплексного аниона.
Такая трактовка явления предполагает, что в логарифмических координатах поляризационные кривые должны иметь вид параллельных прямых, что и было подтверждено опытными данными.
Перенапряжение при разряде металлических ионов 357
Как отмечалось, практика применения добавочных реагентов характеризуется тем, что во многих случаях в промышленные электролиты вводят комбинированные добавки в различных соотношениях и концентрациях.
Особенно часто применяют смеси коллоидов с истинными растворами. Введение в электролит комбинированных добавок при удачном их сочетании заметно усиливает влияние отдельных реагентов. Специфическая адсорбция способствует образованию более плотных мелкокристаллических осадков Об этом, в частности, свидетельствует опыт применения комбинированной добавки клея, р-нафтола и сурьмы при электроосаждении цинка. Характер действия комбинированной добавки, содержащей сурьму, занимает в данном случае особое место. В последнее время было установлено, что введение растворимых соединений сурьмы в весьма малых концентрациях облегчает процесс снятия катодного цинка с алюминиевых матриц. В связи с отмеченным свойством такой добавки сурьму в виде раствора «рвотного камня» специально вводят в электролит для создания разделительного слоя и предотвращения явления «трудной сдирки». Кроме того, оказалось, что сурьма в составе комбинированной добавки с клеем и р-нафтолом увеличивает катодную поляризацию и снижает скорость коррозии цинка, что обеспечивает получение компактных осадков цинка с высокими выходами по току. Благоприятное влияние следующего компонента комбинированной добавки — клея можно объяснить тем, что мицеллы его, адсорбируясь, претерпевают денатурацию, приводящую к повышению вязкости пленки. Вместе с тем мицеллы клея адсобиру-ются и коллоидными частицами гидроокиси сурьмы, вследствие чего комбинированная система сурьма + клей на поверхности цинка приобретает гидрофильные свойства. Если иметь в виду, что по своей молекулярной структуре металлы обладают гидрофобными свойствами, то легко заметить, что адсорбционная пленка приводит к весьма существенному изменению и величины и знака смачиваемости катода раствором, что соответствует глубоким изменениям химического состояния его поверхности.
Следующий благоприятный фактор, влияющий на свойства катодного осадка цинка, — способность клея уменьшать краевой угол. Это приводит к сокращению времени прилипания газовых водородных пузырьков к поверхности катода. В таком случае цинк осаждается в виде гладкого, плотного мелкокристаллического осадка со слабо обозначенными следами выделения водорода.
1 Этот вывод |цакод.пт подтверждение в работах А. Н. Фрумкина и сотрудников в области специфической адсорбции катионов на положительно ва-ряженной поверхности.
358 Теоретические основы электрохимии
Что касается присутствия р-нафтола, то он не влияет на катодную поляризацию цинка.
Положительная роль р-нафтола в комбинированной добавке сводится, по-видимому, к тому, что он связывает в сложный комплекс ионы кобальта и таким образом уводит их из реакционной зоны раствора.
§ 7. Влияние поверхностно активных веществ на смачиваемость поляризуемого катода электролитом 1
Исследования А. Н. Фрумкина, А. В. Городецкой, Б. Н. Кабанова и Н. Н. Некрасова показали, что смачиваемость металлов в значительной степени определяется электрическим зарядом поверхности.
В зависимости от потенциала поверхность металла, например ртути, может быть гидрофильной и гидрофобной в различных точках, электрокапиллярной кривой. Так, незаряженная поверхность ртути предельно гидрофобна; при наложении же поляризации, особенно катодной, она становится гидрофильной (гидрофилизирующее действие заряда).
Поверхность цинка при потенциалах, соответствующих техническому электролизу, имеет положительный заряд и, согласно изложенному положению А. Н. Фрумкина, хорошо смачивается электролитом в силу своей гидрофильности. Поэтому образующиеся пузырьки водорода задерживаются здесь сравнительно недолго, но все же оставляют на поверхности катодного цинка достаточно глубокие следы.
Однако, как показали исследования А. И. Левина и В. С. Колеватовой, при введении в цинковый электролит добавок краевой угол весьма заметно меняется, причем в присутствии камфары и октанола он становится больше, чем в растворе без добавок, т. е. смачиваемость ухудшается. Вместе с этим удлиняется «время жизни» пузырька водорода — пузырек удерживается на поверхности катода, например, в присутствии камфары в три и более раз дольше, чем в растворе без добавок. Полученный в этом случае осадок цинка отличается весьма ярко выраженной ячеистостью.
В противоположность камфаре и октанолу клей и тетрабу-тиламмоний, будучи гидрофобными веществами, адсорбируются на поверхности цинка и улучшают ее смачиваемость электролитом, т. е. уменьшают краевой угол. Вследствие этого флотационные силы ослабляются. Время прилипания пузырька водорода, его размеры становятся значительно меньшими, чем в растворе без добавок. Поэтому на осадке наблюдаются лишь отдельные
1 См. гл. VIII, § 3 (сТр. 216).
Перенапряжение при разряде металлических ионов 359
следы выделения водорода. Ячеистость осадка практически отсутствует.
Сопоставление опытных данных для меди показывает, что при большинстве испытанных добавок выделение газовых пузырьков начиналось при меньшей силе тока, чем в чистом растворе, в то время как величина катодного потенциала в момент отрыва пузырьков для них была значительно выше, чем в чистом растворе и в электролите, содержащем добавку клея. В последнем случае плотность тока, соответствующая началу отрыва водородных пузырьков, оказалась даже большей, чем в чистом растворе.
Установленные и в этом случае факты подтверждают вывод, что изменение смачиваемости поляризуемого катода связано с изменением заряда двойного электрического слоя на его поверхности.
Из экспериментальных данных следует, что зависимость поверхностного натяжения от электродного потенциала определяется главным образом 01,2 (металл — раствор), которое изменяется по закону, идентичному с изменением электрокапиллярной кривой, т. е. уменьшается по мере удаления потенциалов разряда катионов Си2+ от потенциала нулевого заряда поверхности.
Потенциал, замеренный в чистом электролите, содержащем 120 г/л CuSO4 и 100 г/л H2SO4, в момент отрыва газового пузырька оказался равным +0,11 в, т. е. находился в непосредственной близости к точке нулевого заряда меди, которая лежит в пределах от —0,04 до +0,1 в, в связи с чем величина краевого угла 0 достигает здесь наибольшего значения. В растворах же, содержащих добавочные реагенты молекулярного типа (тиомо чевина), либо анионоактивные (сульфитцеллюлозные щелоки), О’!,2 заметно уменьшается. Следовательно, с ростом электродной поляризации и сдвигом потенциалов, устанавливающихся на катоде при выделении меди, в электроотрицательную сторону величина краевого угла 6 закономерно уменьшается. Одновременно с этим улучшается смачиваемость металла раствором, а размеры пузырьков, срывающихся с электродной поверхности, становятся меньшими, соответственно сокращается и время жизни пузырьков.
Таким образом, возможность получения гладких, компактных отложений металлов связана с требованием относительно хорошей смачиваемости поверхности поляризуемого электрода электролитом. На этом основании можно подбирать добавочные реагенты в электролиты. В самом общем случае в качестве добавок могут быть рекомендованы те вещества, которые либо не изменяют смачиваемости, либо заметно увеличивают ее.
360 Теоретические основы электрохимии
ЛИТЕРАТУРА
Антропов Л. И. Успехи химии, 25, № 8, 1956, 1043.
Горбунова К. М., Данков П. Д. Успехи химии, 17, 1948, 710.
Е с и н О. А. Успехи химии, 2, 1933, 493. '
Ильин Б. В. Природа адсорбционных сил. Госхимиздат, 1952
Иоффе В. С. Успехи химии, 13, 1944, 48.
Кузнецов В. Л. Кристаллы и кристаллизация. Техтеоретиздат, 1953.
Л и х т м а н В. И., Щукин Е. Д., Р е б и н д е р П. А. Физико-химическая механика металлов. Изд. АН СССР, 1962.
Левин А. И. ЖФХ, 31, вып. 12, 1957.
Левин А. И., Колеватова В. С., Мокрушин С. Г. Коллоидный журнал, 15, вып. 4, 1953, 252.
Лошкарев М. А., Крюкова А.. А. ЖФХ, 26, 1952, 731.
Новые (проблемы современной электрохимии (под ред. Дж. Боириса). ИЛ, 1962.
Плесков В. А., Миллер Н. Б. Труды совещания по электрохимии. Изд. АН СССР, 1953.
Ройт ер В. А. Украинский химический журнал, 16, 3, 1950, 225.
Т реп не л Б. М. Хемосорбция. ИЛ, 1960.
Фольмер М. ЖФХ, 5, 1934, 319.
Фрумкин А. Н., Б а г о ц к и й В. С.. Иофа 3. А., Кабанов Б. Н. Кинетика электродных процессов. Изд. МГУ, 1952.
Фрумкин А. Н. Вестник МГУ, 9, 1952, 37.
Фрумкин А. Н. Адсорбция и окислительные процессы. Изд. АН СССР, 1951.
Фрумкин А. Н. Вестник МГУ, 12, 1955, 7.
Шварц А. и Дж. Перри. Поверхностно активные вещества. ИЛ, 1953.
Глава XIII
ЭЛЕКТРООСАЖДЕНИЕ металлов
Электрохимический метод получил особенно широкое распространение в технологии извлечения, разделения и рафинирования цветных металлов из водных растворов их простых и комплексных солей. Этот метод используется в разных областях техники для нанесения защитных и декоративных металлических покрытий.
Электролитическим путем получают металлические порошки, а также сплавы и специальные покрытия, обладающие определенными антифрикционными, механическими, оптическими, магнитными и антикоррозионными свойствами.
За последние десятилетия были получены достаточно убедительные экспериментальные и теоретические данные, вскрывающие особенности электродных реакций при осаждении металлов, позволяющие сделать некоторые обобщения и выводы о влиянии различных факторов электролиза на электрокристаллизацию.
§ 1. Закономерности образования единичных кристаллов
Электрокристаллизация металлов существенно отличается от обычного возникновения и роста кристаллов в жидкости, в расплаве или газе. Это объясняется прежде всего тем, что на силы кристаллической решетки накладываются силы внешнего электрического поля. Эти силы особенно велики на субмикроскопических остриях и ребрах. Поэтому на таких участках, как указывает В. Д. Кузнецов, происходит наиболее интенсивный рост кристаллов, что приводит к образованию иглообразных и нитеобразных единичных кристаллов. Кристаллическая нить, как это наблюдал А. Г. Самарцев, часто ограничивается правильными гранями. Иногда она дает резкие пространственные изломы по плоскостям двойниковых срастаний, но остается монокристаллической. Микрофотография таких нитей приводится на рис. 120. Направление роста нитей не находится в прямой связи с направлением линий тока в электролите. Нить разбивается нормально даже в том случае, если активная поверхность посте-
362
Теоретические основы электрохимии
пенно удаляется от анода. В дальнейшем регулярный рост нарушается. На некоторых участках поверхности кристалла появляются неровности и вскоре эти участки пассивируются, т. е. на них прекращается отложение металла. Пассивирование, распространяясь от углов и ребер, захватывает полностью некоторые грани. Через известный промежуток времени металл откла-
Рис. 120. Микрофотография митеобразного кристалла серебра (то А. Г. Самарцеву)
дывается только на одной грани. Плотность тока достигает при этом определенного уровня и в дальнейшем устанавливается и поддерживается на развивающемся кристалле как бы автоматически.
Рис. >121. Изменение сечения нитеобразного (единичного) кристалла серебра, вызванное изменением силы тока (по А. Г. Самарцеву)
Если по достижении некоторой предельной плотности ток# силу тока в цепи внезапно увеличить, то активная поверхность начинает быстро разрастаться (рис. -121). Далее развивается нить большего поперечного сечения, однако линейная скорость роста при этом не изменяется и плотность тока на активной поверхности остается более или менее постоянной. Если, наоборот, силу тока уменьшить, то величина активной поверхности соответственно сократится.
Электроосаждение металлов
363
Эту особенность электрохимического роста нитевидного кристалла можно объяснить следующим образом. Свежеобразованная поверхность кристалла частично пассивируется (отравляется) адсорбирующимся из раствора веществом. Если скорость возникновения новой поверхности меньше скорости пассивирования, то дальнейшее выделение металла на этой поверхности становится затруднительным. Поэтому в том случае, когда сила тока ограничивается, на краях растущей грани' усиливается пассивирование, фронт роста сужается и линейная скорость роста нити возвращается к прежнему значению. Если, наоборот, увеличить силу поляризующего тока, то на краях грани пассивация не успевает за разрядом и грань будет расширяться, пока плотность тока и соответствующая ей линейная скорость роста нити опять не станут прежними. Таким образом, в некоторых случаях плотность тока при электролитическом росте единичного кристалла остается неизменной.
§ 2. Закономерности образования поликристаллических осадков
При образовании поликристаллических осадков, обычно наблюдаемых в результате электролитического осаждения металлов, мы встречаемся с рядом новых явлений, связанных с концентрационной поляризацией, играющей здесь значительно большую роль, чем при выделении водорода, а также со специфическим торможением процесса, вызываемым образованием на поверхности катода адсорбционной пассивирующей пленки.
Схематическая картина роста поликристаллического осадка может быть представлена следующим образом. В каждой точке катода чередуются процессы возникновения и роста кристаллов и их пассивирования. Вместе с образованием новых зародышей часть тока будет расходоваться на их дальнейший рост, в то время как ранее развивающиеся кристаллы будут терять свою активность и их рост прекратится1.
Можно полагать, что процесс электрокристаллизации отличается от обычной кристаллизации в растворах тем, что пересыщение, необходимое для возникновения зародыша, здесь создается нарушением равновесия, вызываемым прохождением электрического тока. В процессе электролиза каждый ион должен быть доставлен к поверхности раздела фаз металл —раствор, адсорбироваться на этой поверхности, вступить в реакцию взаимодействия с электронами и в конце концов занять соответствующее место в кристаллической решетке. Из всех возмож-
1 Такая картина .наблюдается не всегда: иногда первоначальные зародыши продолжают расти в форме столбиков во все время осаждения, при этом наиболее быстро растущие грани кристаллов вырождаются в ребра, или верпггины.
364
Теоретические основы электрохимии
них стадий только процесс адсорбции протекает практически мгновенно, тогда как транспортировка ионов и собственно электродный акт нуждаются в дополнительной энергии активации для преодоления торможения. С ростом плотности тока все большее количество ионов не успевает вступить в реакцию с электронами, вследствие чего потенциал электрода все в большей степени смещается от равновесного значения. Фазовый
Рис. .1Й2. Три энергетических положения «строительного элемента» на поверхности кристалла (по К. М. Горбуновой и П. Д. Данкову)
Рис. 123. Схема изменения поля-
ризации электрода
переход является, следовательно, вынужденным, навязанным извне, поэтому элементарный акт разряда ионов, образование и разрастание зародышей кристаллов протекают с видимым торможением.
Как замечают К. М. Горбунова и П. Д. Данков, элементарный акт перехода иона из раствора в кристалл происходит в соответствии с величиной энергии каждого «строительного элемента». В случае простой кубической решетки (возможны при способа присоединения строительного элемента (рис. 122). Здесь наиболее активное место грани.показано стрелкой.
Итоговый акт перехода иона в кристалл определяется разностью вероятностей перехода монов в кристалл и обратного процесса ионизации атомов. Эта величина (т. е. разность вероятностей) пропорциональна плотности тока.
Процесс разряда металлических ионов (распадается на ряд промежуточных стадий (рис. 123).
1. В первый момент после наложения внешнего напряжения накапливаются электроны на поверхности металла и ионы в прилегающем к электроду слое раствора. При этом потенциал повышается почти линейно до момента, при котором становится возможным разряд ионов и образование двухмерных зародышей (участок ab на рис. 123).
2. Так как двухмерные зародыши образуются при некотором перенапряжении г]2, которое вызвано частично пассивированной
Электроосаждение металлов
365
поверхностью, то часть тока продолжает расходоваться на дальнейшее заряжение двойного слоя.
Таким образом, |ipa3p меньше плотности тока 1внеш, поступающего от внешнего источника. В таком случае перенапряжение со временем, меняется уже не линейно и связь между плотностью тока и перенапряжением описывается уравнением
_К2
(XIII,1)
г2 — К2е г> .
Наряду с возникновением двухмерных зародышей весьма быстро становится возможным их рост. Скорость этого процесса определяется из соотношения
ti = К^е ’i .
Плотность общего тока разряда складывается, таким образом, из суммы плотностей частных токов, т. е.
к’1 к;
(XI 11,2)
ipaap — l'l + С— Куб -Т K^c . (XI11,3)
Так как новый слой зародышей распространяется по пассивному металлу, а 'верхняя, свежеобразованная поверхность кристаллов остается активной, то рост слоя и возникновение на нем новых зародышей, при некотором перенапряжении, превышающем т}2, протекают одновременно (участок Ьс на рис. 123). В итоге образуется «пакет» двухмерных зародышей, распространяющихся в виде толстого слоя до краев грани.
Вследствие продвижения фронта роста плотность тока возрастает и в конце данной стадии кристаллизации плотность тока разряда становится равной плотности внешнего тока в цепи ячейки:
Ipasp — £внешн« (XIII,4)
3. Перенапряжение при /разр = 1'внеш становится максимальным (точка с на рис. 123). Развитие зародышей и их рост протекают при этом настолько бурно, что ток в цепи пополняется за счет извлечения из двойного слоя электронов, накопленных за предыдущие этапы, и /разр > 1‘внеш. Извлечение электронов (приводит к снижению потенциала (участок cd на рис. 123), т. е. кривая т] — т, пройдя через максимум, начинает падать и по достижении значения т]2 образование зародышей прекращается. Ток разряда при этом, также уменьшается из-за частичного уменьшения запаса электронов в двойном слое.
Скорость протекания третьего этапа будет тем выше, чем больше концентрационная поляризация, нарастание которой приводит к замедлению процесса образования зародышей.
366
Теоретические основы электрохимии
4. Когда перенапряжение снизится до величины, 'несколько меньшей т]2, процесс из-за невозможности образования новых зародышей сведется к продвижению фронта роста грани, но с постепенно убывающей скоростью за счет jpasp > бжет. Перенапряжение продолжает снижаться (участок de на рис. 123). В-та-ком случае связь между плотностью тока и перенапряжением выражается уравнением
(ХШ,5>
Л ii - ч .
5. Когда плотность тока достигнет iBHem, перенапряжение будет иметь значение цо, начинается пятый этап (участок ef на рис. 123) процесса, самый длительный, при котором фронт роста продвигается с постоянной скоростью и при неизменном перенапряжении.
Первый цикл изменения потенциала заканчивается, когда «фронт роста» достигает противоположного края грани. В момент завершения слоя концентрация у исходного края восстанавливается, вследствие чего начинается новая стадия, представляющая собой повторение предыдущей, но с тем отличием, что колебания потенциала во времени оказываются менее заметными, так как новый слой осаждается на, только что образовавшей-' ся, почти не пассивированной поверхности металла. Концентрация у ребер кристаллов восстанавливается быстрее, чем на середине грани. Поэтому новый слой должен возникать, как правило, у исходного края грани, что и наблюдается на опыте. Учет концентрационных изменений у поверхности катода приводит к выводу о том, что сопровождающая процесс кристаллизации поляризация представляет собой сложную величину, составленную из диффузионных ограничений и перенапряжения. Доля этих слагаемых по различным участкам электрода неодинакова. У места, где только что возник зародыш, концентрационная поляризация минимальна, а перенапряжение максимально. У фронта роста соотношения обратны.
Положения, развиваемые К. М. Горбуновой и П. .Д. Данковым, отражают результаты микрофотографических исследований, представляющих процесс роста кристаллов посредством послойного образований двухмерных зародышей. Подробные исследования роста кристаллов были предприняты Р. Каишев’ым на примере серебра. Наблюдения .показали, что кубические грани серебряных монокристаллов чаще всего растут спиральными фронтами. Условия роста спиралей можно менять, изменяя силу поляризующего тока. Было показано, что хотя истинная плотность тока остается практически постоянной при различных перенапряжениях, кажущаяся плотность тока возрастает с ростом перенапряжения за счет возрастания числа витков спирали.
Электроосаждение металлов
367
§ 3. Характеристика катодных отложений и требования к осадкам
В гидрометаллургии и особенно в гальванотехнике стремятся получить мелкокристаллические, компактные, беспористые, иногда блестящие отложения металлов. Размеры кристаллов,, образующих осадки, зависят от многих факторов, и прежде всего, от природы электролита, и поляризации, сопровождающей выделение металла. Электролитический осадок металла, как всякое поликристаллическое тело, характеризуется размером и формой (огранением) кристаллов, а также взаимной ориентированностью кристаллов — их текстурой. Текстурой поликристал-лического тела называется совокупность всех имеющихся ориентаций отдельных кристаллов. В зависимости от того, преобладает ли какая-либо определенная ориентация кристаллов или же кристаллы не ориентированы и расположены беспорядочно, судят о преобладании той или иной кристаллографической оси текстуры. Иногда под термином «текстура» понимают преобладание определенной ориентации кристаллов. Текстура определяет многие физико-механические свойства поликристаллического тела.
Величина кристаллов, образовавшихся из насыщенного раствора или расплава, зависит от соотношения скорости зарождения центров кристаллизации и линейной скорости роста кристаллов. Чем больше скорость образования центров кристаллизации и чем соответственно меньше линейная скорость кристаллизации, тем меньше размер кристаллов поликристаллического твердого тела. Эти закономерности полностью относятся и к процессу электрокристаллизации металлов. Многочисленными исследованиями установлено, что вое факторы, способствующие увеличению катодной поляризации, ведут к росту скорости зарождения центров кристаллизации. Такая связь между поляризацией и скоростью образования зародышей объясняется тем, что энергия активации, необходимая для образования зародыша, значительно больше энергии, затрачиваемой на рост уже имеющихся кристаллов. В связи с тем, что при электролизе изменяются и число и характер образующихся кристаллов, истинная плотность тока весьма заметно отличается от плотности тока, рассчитанной по геометрической поверхности электрода.
Скорость осаждения металла различна для разных граней; и поэтому изменение плотности тока вызывает появление той или иной оси текстуры. Степень ориентации осадков также зависит от плотности тока — она обычно до известного предела увеличивается с ростом плотности тока. Появление определенных осей текстуры связано с поляризацией электрода, поскольку различные грани не являются эквипотенциальными.
Преобладание процесса возникновения новых зародышей кри
368
Теоретические основы электрохимии
Сталлов над скоростью роста уже имеющихся кристаллов особенно характерно для комплексных электролитов, а также для растворов простых солей, содержащих поверхностно активные добавки. В последнем случае из-за образования адсорбированных слоев на растущих гранях линейная скорость роста кристаллов уменьшается и осадки получаются высокодисперсными.
Весьма часто они не имеют даже четко выраженной кристаллической структуры *.
При обратном соотношении скоростей, когда линейная скорость роста кристаллов начинает преобладать, отложение имеет грубую кристаллическую структуру. Такие металлы, как медь, олово, свинец, серебро, при использовании растворов их простых солей, не содержащих поверхностно активных добавок, выделяются в виде крупных кристаллов, размеры которых превышают 10~3 см. Несколько меньше кристаллы образуются при электро-осаждении цинка и кадмия (10~4 см). Таким, образом, один и тот же металл может быть получен на катоде в различной форме; специальным .подбором условий и режима электролиза можно существенно влиять на структуру электролитических осадков.
В общем случае образованию мелкокристаллических осадков способствуют, кроме рассмотренных выше условий, факторы, увеличивающие электродную поляризацию: а) понижение температуры электролита; б) повышение плотности тока на катоде; в) добавки нейтральных солей, снижающих концентрацию ионов осаждаемого металла у катода; г) разбавление раствора.
А. И. Левин показал, что мелкозернистые осадки меди и серебра, аналогичные покрытиям, образуемым при осаждении из комплексных электролитов, могут быть получены в том случае, если очень сильно разбавить растворы простых солей CuSC>4 и AgNO3 (до 10~6 г-моль/л). Подобные результаты наблюдались и при добавке в испытуемый электролит ионов посторонних солей, участвующих в переносе тока, но не участвующих в разряде и не образующих комплекса с ионами, восстанавливающимися на катоде.
На структуру катодного осадка воздействует также кристаллическая форма основного металла (подкладки). Известно немало примеров, когда образующийся осадок воспроизводит микроструктуру подслоя. Наиболее заметно это проявляется при
1 Как показали исследования К. М. Горбуновой и О. С. Поповой, катодные осадки неявно кристаллического типа составлены из образований со сферической (полусферической) поверхностью, имеющих слоистое строение, сходное со строением осадков, возникающих при коагуляции коллоидов. Форма и размеры этих образований в меньшей степени зависят от природы металла, чем от условий, при которых образуется осадок. К этой группе осадков относится ряд практически важных покрытий, таких 'как хром, марганец, железо, блестящий никель, цинк и др.
Электроосаждение металлов 369
электроосаждении меди, серебра, цинка, которые способны (Повторять и продолжать конфигурацию зерен не только одноименного, но и инородного металла. Ориентирующее действие подкладки сказывается в том случае, если различие в постоянных решетки основного металла и образующегося в процессе электроосаждения не слишком велико (не превышает 15%). Ориентация простирается на относительно большие расстояния в глубь растущего осадка, составляющие десятые доли микрона или даже несколько микронов.
Дефекты поверхности (царапины, язвины, изломы и т. л.) служат обычно участками преимущественного начального кристаллообразования. Наблюдениями М. Фольмера и И. И. Оранского установлено, что кристаллы растут первоначально на недостроенных частях кристаллической решетки. Наиболее быстро растущие грани кристаллов вырождаются в ребра и вершины. При благоприятных условиях роста кристаллы образуются тем большими по величине, чем дольше идет процесс электролиза, пока какая-либо внешняя причина не прекратит их развитие. Если прервать электролиз и возобновить его через некоторое время, то обычно возникают мельчайшие кристаллы, из которых только постепенно со временем разрастаются большие. Следует, однако, заметить, что одновременно с торможением роста отдельных кристаллов концентрация электролита выравнивается, в результате чего концентрационная поляризация снижается, а также возможно обратное растворение мелких кристаллов и рост за их счет крупных. В целом эти процессы протекают сопряженно и приводят обычно к укрупнению структуры осадка.
Такие приемы, как перерыв тока, наложение переменного тока на постоянный, реверсивный ток, дают возможность при постоянных условиях электролиза (/к, t°, составе электролита и электродов) регулировать качество осадка по его характеру и структуре из-за снятия диффузионных ограничений.
Изменяя соответствующим образом характер пульсаций, длительность периодов обращения тока во времени, можно создавать многие варианты режима электролиза и соответствующие этим режимам условия образования осадков по толщине, крупности кристаллов и их структуре.
Подобные режимы в отличие от условий электролиза с постоянной плотностью тока называют нестационарными режимами электролиза; они характеризуются непостоянством величины и направления тока во времени и другими изменениями.
Рассмотрим некоторые примеры нестационарного режима электролиза, позволяющего снять диффузионные затруднения.
Периодическое изменение направления тока. Осадки металлов при этом получаются более светлыми, гладкими и в некоторых случаях менее пористыми. Применение реверсивного тока 24 Заказ 1338
370
Теоретические основы электрохимии
дает возможность значительно повысить плотность тока при электролизе.
Основные преимущества электролитического осаждения металлов с применением реверсивного тока:
а) улучшение качества покрытий—'получение беспористых, плотных, гладких при значительной толщине слоя, блестящих покрытий с повышенной противокоррозионной стойкостью;
б) увеличение скорости осаждения металла;
в) применение электролитов без специальных добавок органических веществ или со сниженной в несколько раз их концентрацией;
г) улучшение рассеивающей способности электролитов;
д) улучшение растворимости пассивирующихся анодов.
К числу преимуществ такого способа ведения процессов относят также то, что получение блестящих покрытий не сопровождается возникновением остаточных напряжений и искажением кристаллической решетки.
Скорость наращивания покрытий, несмотря на то что часть тока затрачивается на анодное растворение металла, выше, чем в процессах при постоянном токе. Это объясняют возможностью ведения электролиза при повышенной плотности тока вследствие обогащения прикатодного слоя электролита ионами металла.
Улучшение свойств покрытий, полученных осаждением на реверсивном токе, обычно приписывают анодной части периода. Причем считается, что выборочное растворение металла покрытия, т. е. предпочтительное растворение микровыступо.в, обусловливает получение гладких и блестящих покрытий.
Процесс осаждения металлов на реверсивном токе не может быть применен для всех электролитов, однако для некоторых кислых и особенно щелочных электролитов он эффективен. К числу процессов, которые получили широкое промышленное применение, следует отнести: меднение, серебрение, кадмирование, цинкование, латунирование и золочение в цианистых электролитах, а также меднение в кислом электролите.
Применение переменного тока. Строение и свойства металлических покрытий удается резко изменить, применяя токи переменного направления. Последовательная смена катодного и анодного процессов на электроде <может осуществляться .при этом изменением постоянного тока, а также одновременным использованием при электролизе постоянного и переменного токов.
За последние годы этот метод стал широко применяться не только при выделении отдельных металлов, но и при получении сплавов. В результате сочетания постоянного и переменного токов разных амплитуд и частот получаются осадки с резко отличными структурами и свойствами.
К. М. Горбунова и А. А. Сутягина опубликовали исследова
Электроосаждение металлов
371
ния с характеристиками процессов, происходящих на катоде, и с данными о строении и свойствах образующихся осадков. Проведенные исследования позволяют сделать вывод, что при кристаллизации никеля, кадмия, частично цинка и меди из электролитов, содержащих и не содержащих добавки поверхностно активных веществ, влияние переменного тока сказывается в укрупнении кристаллов, изменении их ориентации в осадке и часто в уменьшении блеска.
Причины отмеченного влияния переменного тока на процессы кристаллизации перечисленных выше металлов связаны, по-видимому, с изменением условий адсорбции, происходящим при резких изменениях значений потенциала.
Было отмечено, что формы кристаллов, возникающих под влиянием переменного тока в растворах, содержащих поверхностно активные вещества, часто не сходны с отложениями из растворов без добавок. Это является подтверждением того, что наблюдаемое влияние переменного тока нельзя целиком объяснять возникновением десорбции. Можно полагать, что своеобразие форм кристаллов связано с изменением природы поверхностно активных добавок (под воздействием переменного тока).
Закономерности, наблюдаемые в условиях наложения переменного тока в процессе электрокристаллизации металлов, удовлетворительно объясняются при учете концентрационных изменений у катода разряжающихся ионов металла и ионов водорода. Существенное значение имеет происходящее в этих условиях периодическое активирование поверхности в результате либо растворения металла (цинк и кадмий), либо ионизации адсорбированного в осадке атомарного водорода (никель).
Толчки тока. Ранее было установлено, что при резком начальном толчке в медно-цианистом электролите на деталях получаются очень тонкие, беспористые плотные осадки меди, предотвращающие в дальнейшем выделение контактной меди на железе в кислых электролитах. Высокую началыную плотность тока (толчок) применяют также в процессе гальванического хромирования для улучшения кроющей способности хромовых ванн. Значительный эффект, вызываемый изменением направления тока или толчками тока в относительно разведенных растворах простых солей или в комплексных электролитах, оказывается связанным со снижением пассивности анодов. Описанные явления иллюстрируют хорошо известное из гальваностегии правило, согласно которому внезапное повышение плотности тока и уменьшение времени выдержки (толчки тока) благоприятствуют образованию мелкокристаллических осадков.
Применение электролиза с толчками тока позволяет получить осадки с минимальной пористостью в весьма мелкокристаллической форме. Характер кристаллизации и структура осадка при 24*
372
Теоретические основы электрохимии
таком режиме электролиза будут, естественно, отличными для разных металлов. Применение очень высоких плотностей тока :в кратковременные периоды заметно повышает химическую поляризацию и приводит к измельчению зерна осадка. Малая продолжительность импульса и относительно значительная величина паузы влияют на переходные процессы, наблюдаемые при электролизе, в результате чего устраняются концентрационная поляризация и явления, связанные с предельным током.
§ 4. Особенности электрокристаллизации из растворов, содержащих катионы одного металла различной валентности
Некоторые металлы образуют в растворах катионы различной, валентности. Сюда, относятся, например, медь (Cu2+, Си+), олово (Sn4+, Sn2+), железо (Fe3+, Fe2+), хром (Сг6+„ С г34-, Сг2+) и др.
Почти все металлические ионы, существующие в растворе элекртолита в нескольких степенях окисления, а также ряд кислородсодержащих анионов, имеющих в своем составе металлы, способные выделиться на катоде (Сг2О2-, СиР2О2~ и др.), восстанавливаются ступенчато. Например, при восстановлении Со3+ по схеме Со3+->Со2+ и Со2+->-СЬ на поляризационной кривой имеются две (ветви.
В практике встречаются катодные процессы не только с одноэлектронным переходом, но и с участием нескольких электронов в электродной реакции. Наиболее часто, однако, такой акт протекает в две стадии: например, двух1электронный переход делится на два одноэлектронных, трехэлектронный — на одно- и двух-электронный и т. д.
Общее характерное свойство таких электролитов — то, что в присутствии соответствующего металла здесь устанавливается равновесие между ионами окисленной и восстановленной формы:
Cu2+ + Cu5t2Cu+;
2Fe3+ + Fe 2 3Fe2+ и т. д.
В растворах сернокислой меди равновесие между двух- и одновалентными ионами практически полностью сдвинуто в сторону ионов высшей валентности. Так, при 20 °C
л
у Ki = - ~ — = 1429. (XI11,6)
CCu2SO4
В случае железа, наоборот, более устойчивы ионы низшей валентности; здесь при 18 °C ______
Д-о = V CFeS^_. (XIII,7)
V/<CFeI(SO4)3
Электроосаждение металлов
373
При описании окислительно-восстановительных процессов уже отмечалось, что после установления электрохимического равновесия между металлом и раствором его катионов в растворе всегда должны преобладать те катионы, потенциал образования которых менее положителен. Равновесная концентрация катионов другой валентности уже при разности потенциалов (ф1/о—ф2/о) порядка нескольких десятых вольта становится исчезающе малой.
В некоторых случаях это правило нарушается. Так, например, в солянокислых растворах золота равновесные концентрации Аи+ и Аи3+ мало отличаются одна- от другой; так, при 60 °C
САиС1- • ^AuCl- • СС1- • /ci-
К3 =-------1«-----------------= 10°’67 = 4,67. (XIII,8)
CAuCl— ’ /auCI—
Возможно, это связано с тем, что в солянокислом растворе образуются комплексные соединения НАиС14 и НАиС12. При электролизе растворов солей металлов, содержащих ионы неодинаковой валентности, катодные и анодные процессы проходят обычно с такими соотношениями скоростей, при которых сохраняются равновесия типа '(XIII, 6), (XIII, 7) и (XII, 8). Это значит, что в случае меди разряжаются йа. катоде и ионизируются на аноде главным образом ионы высшей валентности (Си2+). Наоборот, железо будет разряжаться на катоде и растворяться на аноде почти исключительно в виде двухвалентного, а золото посылает в раствор ионы Аи3+ и Аи+ почти в равных соотношениях; в таком же близком соотношении ионы обоих видов восстанавливаются на катоде. '
Достижению требуемого равновесия в некоторых случаях препятствует ряд побочных обстоятельств, которые определяют собой тот экспериментальный факт, что катодные и анодные выходы по току металлов рассматриваемой группы электролитов несколько отличаются из-за совместного участия в электродных реакциях ионов различной валентности.
Для примера можно привести систему Cu2+->Cu+->Cu. Двухвалентная медь восстанавливается до одновалентной при потенциале, равном +0,17 в. Так как этот потенциал значительно отрицательнее потенциала восстановления Cu+->Cu (+0,51 й), то ионы одновалентной меди практически не могут существовать возле электрода в заметной концентрации. Однако если ввести в раствор сульфата меди вещество, повышающее устойчивость ионов одновалентной меди вследствие образования с ними комплексного соединения (аммиак или ионы хлора), то потенциал .восстановления такого комплекса станет более отрицательным и на поляризационной кривой получатся две волны,
374
Теоретические основы электрохимии
свидетельствующие о ступенчатом переходе?
Си2+ Си+ -> Си.
Во многих случаях удается создать вполне устойчивые растворы электролитов, содержащие соединения металлов низшей валентности.
Опытные данные показывают, что осадки, образующиеся на катоде в случае использования соединений металлов с низшей валентностью, обладают обычно относительно мелкокристаллической структурой; это в первую очередь относится к растворам одновалентной меди.
§ 5. Совместный разряд ионов и влияние посторонних примесей на электроосаждение металлов
Электролитическое получение металлов достаточно высокой чистоты возможно лишь из растворов, содержащих минимальные количества посторонних примесей.
Технические растворы, полученные, например, :в результате выщелачивания обожженных концентратов, даже после последующей очистки их, содержат некоторое количество примесей. Действие этих примесей может быть многогранным. Они могут влиять на чистоту и качество получаемого металла, на выход по току, прочность сцепления осадка с катодной основой, а также на другие стороны процесса электролиза. Такое существенное влияние сравнительно малых количеств примесей на процесс электроосаждения зависит от закономерностей совместного разряда ионов. Совместный разряд различных по природе ионов возможен по достижении одинаковых величин потенциалов разряда этих ионов, т. е.
= = (XI 11,9)
Здесь ф,к—потенциал, устанавливающийся на катоде при данных условиях электролиза (/к, t и состав электролита) ;
<р, и Т/,— потенциалы разряда ионов соответственно 1-го и 2-го сортов при заданной плотности тока на катоде (zK)-
Потенциал разряда t-того сорта ионов, устанавливающийся при заданной плотности тока на катоде дк, равен:
Ъ = + In CJt - Д?/, (X111,10)
zF
где.фг° —стандартный (нормальный) потенциал.
Таким образом, для оценки возможности совместного разряда того или иного вида ионов необходимо1 знать, помимо величи-
Электроосаждение металлов
‘ 375
ны обратимого потенциала, также значения его катодной поляризации (Д?г) . Однако в действительности процесс совместного разряда протекает сложнее, так как ионы примеси разряжаются на подкладке из чужого металла, что не может не отразиться на кинетике электродного процесса. Действительно, если металл-примесь может образовывать твердые растворы или
химические соединения с основным металлом, то разряд его будет происходить при менее отрицательном потенциале (деполяризация при выделении металлов) и вероятность загрязнения катодного металла возрастет. В общем случае возможность соосаждения нескольких металлов, обладающих различными электрохимическими свой • ствами, можно схематиче ски представить, анализируя ход поляризационных кривых, изображенных на рис. 124. Отрезки 0<pi и Оч2 характеризуют величину потенциалов выделения 1-го и 2-го металлов, один-из которых, например, является ме-таллом-примесью. При
Рис. 124. Кривые потенциал — плотность тока, характеризующие совместный разряд ионов двух металлов
этом подразумевается, что потенциал выделения 1-го металла положительнее; следовательно, на катоде прежде всего будет наблюдаться разряд ионов 1-го металла. По достижении критической плотности тока 7кр потенциал разряда ионов Мех достигнет величины потенциала выделения Л4е2, т. е. станет возможным совместный разряд (соосаждение примеси). Скорости разряда ионов Ме1 и Ме2 будут определяться ходом поляризационных кривых ч>1—Mei и фг—Ме2.
Поскольку скорость электрохимической реакции зависит.от
величины плотности тока, то при потенциале, соответствующем отрезку Or, она будет равна для иона Л4<?1 отрезку Аг и для иона Ме2 — Ьг. Суммарная скорость катодного процесса (разряда основного металла и металла-примеси) в этой точке будет характеризоваться отрезком аг. В зависимости от поляризации картина совместного разряда описывается кривой «PiCCi-2.
376 Теоретические основы электрохимии
Согласно зако1ну Фарадея, количество металлов, выделившихся в осадке, пропорционально количеству электричества, затраченного на разряд ионов Мех и Ме2. При плотности тока /к на катоде выделяется (выход по току для каждого металла принят равным 100%):
mMet = и
Здесь О1—электрохимический эквивалент Л1еь а2— электрохимический эквивалент Ме2; /1 — плотность тока, затраченного на разряд Мег, i2 — то же, для Ме2, т — продолжительность электролиза.
Содержание металла-примеси в основном металле равно:
р = ..... ,Пме*—.. Ю0% (X111,11)
ИЛИ
р = . 100 =----------100%. (XIII,12)
0^1 -р G2I2 и^Аг -р u^br
Таким образом, вероятность попадания примеси в катодный металл зависит от соотношения величин потенциалов выделения основного металла и металлов-примесей, а также от характе-
Д< ра хода поляризационных кривых — величины производных-----—
Афлн. 4/ „ 4/
и -----. В данном случае производная-----выражает зависи-
мость скорости разряда от поляризации и характеризуется наклоном поляризационной кривой. Имея в распоряжении поляризационные кривые разряда основного металла и ионов-примесей из реальных растворов, возможно рассчитать теоретически чистоту получаемого на катоде металла.
Рассмотренные положения теории совместного разряда катионов могут быть весьма полезными для оценки влияния различного рода примесей на основной катодный процесс. Все примеси, сопутствующие разряду основного металла, делятся на две группы: примеси, потенциал выделения которых положительнее соответствующего потенциала для основного металла, и примеси электроотрицательные. Из возможных примесей к первой группе наиболее часто относятся благородные металлы, а также селен, теллур и др. Опыт показывает, что электроположительные примеси осаждаются ib условиях предельного для них тока при потенциале, соответствующем разряду ионов основного металла. Это означает, что практически всегда, при любой концентрации
Электроосаждение металлов 377
в растворе ионов более благородных примесей, они будут со-осаждаться на катоде с основным металлом.
Допустимые содержания примесей этого типа в электролите, при которых еще возможно получение катодного металла достаточной чистоты, определяются соотношением их концентраций и установившейся на катоде плотности тока. Содержание в электролите более электроотрицательных примесей до известного1 предела их концентраций не сопровождается совместным разрядом. Это, однако, имеет место только, если отсутствует деполяризующее действие основного металла, т. е. ib том случае, когда металлы не образуют между собой химических соединений или сплавов.
Совместный разряд ионов-примесей и основного металла — это лишь один из возможных путей попадания их в катодный металл. Катодный продукт может быть загрязнен в результате попадания в него электролита, механического включения взвешенных частиц, шлама, а также вследствие катафоретического перемещения и осаждения коллоидных частиц и т. д.
Наиболее часто катодное выделение металлов происходит при одновременном разряде ионов водорода. Выходы по току металла, (Л) и водорода (1—Л) пропорциональны плотностям тока, потребляемым ионами:
= -^f. (XIII,13>
1 —- Л zH
Как было показано в работах А. И. Левина с сотрудниками, относительное распределение тока между обоими ионами определяется отношением их концентраций в растворе, а также величиной металлического и водородного перенапряжения. Если, например, исходить из того положения, что поляризация при разряде ионов водорода и металла обусловлена недостаточной скоростью перехода электронов с металла на ионы (замедленным разрядом), т. е.
?н = °н +-~г (XIII,14>
анТ
RT
<?Ме = аМе + (X111,15>
aMezt
TO
A _ V in4”»-”»)»7’
’ (XIII,16}
Так как заряд металлического иона, участвующего в совместном разряде, известен, а значение ан близко к 0,5, то зависимость (XIII, 16) может быть использована для экспери
378
Теоретические основы электрохимии
ментального определения коэффициента аме в уравнении
замедленного разряда.
Рис. '125. Поляризационные кривые, полученные в растворе 200 г/л FeSO4 • • 7Н2О при pH = 2,5 (по А. И. Левину ,и С. А. Пушкаревой):
У — общая кривая: 2 — разряд ионов Fe+2; 3 — разряд ионов водорода и зависимость выхода по току от потенциала; 4 — весовой метод; 5 — объемный метод
Далее, определяя выходы по току металла для заданной плотности тока при постоянном значении потенциала (рис. 125, кривые 4 и 5), можно из сум-
марной поляризационной кривой 1 найти в отдельности /— <р-кривые для разряда металлических ионов (кривая 2) и ионов водорода (кривая 3). Соответствующие пересчеты для определения кривых 2 и 3, характеризующих собой разряд металлических ионов и ионов водорода, осуществляются умножением выхода по току металла (Д) и водорода (1—А) на отвечающие им значения общей плотности тока, устанавливающейся при определенном (постоянном) значении потенциала.
§ 6. Особенности электролитического образования сплавов
Существенный технический интерес представляет катодное образование металлических
сплавов. Как отмечалось, одновременное выделение двух и более
ионов при заданной плотности тока возможно при условии равенства потенциалов их разряда. Например, в случае образования латуни необходимо обеспечить:
+J^igOCo-4Tco = ,»„ + A?2S.igazn_A,Pzo.
(XIII, 17)
Заменив величины перенапряжения через
Д<рСи = 0,00017 1g Ki и A<pzn = 0,00017 1g /С2,
легко показать, что для простых солей меди и цинка разница между концентрациями ионов каждого сорта чрезвычайно велика. При 18 °C она составляет величину
Cztl2+ ~ юзе.
ССи2+
Электроосаждение металлов
379
Как видно, совместный разряд с приведенным здесь соотношением концентраций ионов цинка и меди практически немыслим.
Совершенно иное соотношение концентраций ионов обоих металлов может быть получено при использовании комплексных электролитов. В этом случае благодаря комплексообразованию разность между <рСи и <pZn заметно уменьшается (табл. 23). В таком случае электролитически могут быть получены осадки двух и более металлов.
Таблица 23. Электродные потенциалы меди и цинка в цианистых электролитах
Электролит Потенциалы (в) для металла ^Cu-^Zn’ в
Zn Си
Ю,1-н. TMeCN + 0,2-н. KCN —1,03 —0,61 0,42
0,1-н. AJeCN 4- 0,4-н. KCN —1,18 —0,96 0,22
0,1-н. AfeCN + 1-н. KCN —1,23 —1,17 0,06
Сплавы, получаемые электрохимическим путем, по своим физико-химическим свойствам значительно отличаются от сплавов, образуемых термическим и другими способами. При образовании сплава в виде твердого раствора или химического соединения в ряде случаев выделяется значительное количество энергии. Это, как замечают Ю. М. Полукаров и К. М. Горбунова, должно влиять на потенциал разряда ионов металлов, образующих сплав, ибо известно, что
Д<р-—. (XIII,18)
Здесь величина ДФ представляет собой изменение парциальной молярной свободной энергии компонента при вхождении его в сплав.
Рассмотрим зависимость ДФ от состава спл'два. Для простоты возьмем сплав типа АВ (рис. 126), в котором компоненты образуют непрерывный ряд твердых растворов. Пусть компонент А будет электроотрицательнее компонента В. Предположим далее, что катионы обоих металлов имеют одинаковый заряд z. Очевидно, что компонент А начнет разряжаться на катоде ранее компонента В лишь при потенциале, положительнее обратимого потенциала компонента А на величину: дф Ва' zF zF
Сближение потенциала в таком случае оказывается предельно большим. В том случае, когда компонент А уже содержится
(XIII,19)
380
Теоретические основы электрохимии
в сплаве в заметных количествах, потенциал осаждения компонента В также смещен в положительную сторону на величину, пропорциональную изменению парциальной молярной свободной энергии компонента В (равную ДФь). На рис. 126 представлен случай образования сплава, содержащего 20% компонента А и 80% компонента В (точка х).
Величина ДФа компонента А будет в таком случае равна ха, а
Рис. il26. Схематическое изображение кривых зависимости изменения интегральной и 1па1рциальной молярной свободной энергии компонентов сплава от концентрации {(по Ю. М. (Полукарову и К. М.
Горбуновой)
следовательно, заключить,
для В соответственно ДФ& равна xb. Сближение потенциалов будет пропорционально разности (ха — xb). Как нетрудно заметить, эта разница зависит от концентрации компонента А в сплаве. В точке х, т. е. в максимуме интегральной кривой, она достигает значения, равного нулю, после чего меняет свой знак, и, следовательно, при большей концентрации компонента А в сплаве вместо сближения потенциалов можно наблюдать противоположный эффект; тогда для совместного осаждения компонентов А и В понадобится принять дополнительные меры. Можно, что в процессе совместного
осаждения двух металлов потенциал смещается в положительную сторону и для одного и для другого металла на величину, определяемую соотношением обоих компонентов в сплаве; вследствие этого и сближение потенциалов осаждения за счет энергии, выделяющейся при образовании сплава (энергии «смещения»), также (весьма существенно зависит от концентрации сплава. В ;ряде случаев, например при осаждении 'латуни, особенно богатой цинком, сближение потенциалов выделения происходит
не за счет энергии смещения, но вследствие других причин и, в частности, вследствие образования более устойчивых комплексов. в растворе. Так, повышение концентрации цианистого калия в растворе не только сближает равновесные потенциалы ионов меди и цинка (табл. 23), но и обусловливает изменение кинетики совместного разряда этих ионов при заданной плотности тока.
Последнее может быть объяснено, если иметь в виду, что в катодной реакции принимают непосредственное участие ком-
Электроосаждение металлов
381
ллексные анионы типа: Cu(GN)^ и Zn(CN)2—, которые и восстанавливаются до металла по реакциям:
Cu (CN)7 + е -> Си + 2CN*
Zn (CN)4~ + 2е -> Zn + 4CN-
с образованием сплава (латуни).
При катодном образовании твердых растворов выделяются атомы обоих металлов и в результате совместного разряда сложных ионов образуют общую кристаллическую решетку. Поэтому и при растворении твердого раствора или химического соединения двух металлов их ионизация происходит при потенциале, характерном для данного состава твердого раствора или соединения.
§ 7. Условия и особенности образования порошкообразных катодных отложений
Электролитические осадки обычно подразделяют по характеру связи отдельных кристаллитов между собой в основном на два типа: порошки, частицы которых представляют собой дендриты, и плотные — компактные покрытия, состоящие из более или менее четко выраженных кристаллитов. Закономерности роста осадков обоих типов соответствуют основным положениям теории электрокристаллизации.
Экспериментальные данные показывают, что условием начала образования рыхлых осадков на катоде является достижение предельной плотности тока, при которой скорость доставки ионов, участвующих в электродной реакции, к поверхности катода не успевает за разрядом. В таком случае из-за резкого обеднения прикатодного слоя разряжающимися ионами заметно проявляются диффузионные ограничения, при которых нормальное развитие кристаллов затруднено, и тогда начинают развиваться наиболее активные участки катода. Электродная поверхность как бы «прорастает» разрозненными единичными иглами, вытягивающимися в направлении доставки питающей среды.
Характерную картину образования дендритных порошкообразных отложений металлов можно наблюдать мри использовании разведенных растворов электролитов, содержащих простые соли металлов (например, AgNO3), в случае применения особенно высоких плотностей тока.
Уже отмечалось, что процесс развития единичного (нитевидного) кристалла в дендрит связывается обычно с тем, что при высокой плотности тока происходит сосредоточенное потребление ионов у растущей грани. Это способствует обеднению раствора у активной поверхности грани и быстрому нарастанию концентрационной поляризации.
382
Теоретические основы электрохимии
Последние затруднения приводят к сдвигу катодного потенциала до величины, цри которой достигаются условия, обеспечивающие образование зародышей на боковых гранях. Таким образом, у боковых граней кристалла оказываются более благоприятные условия роста, так как концентрация ионов здесь становится выше. Боковые ветви заметно разрастаются, а основной ствол, наоборот, в сечении уменьшается. С течением времени электролиза у боковых ветвей начинается прогрессирующее падение концентрации, что ведет к снижению скорости разряда ионов на активных участках ветви и, следовательно, к уменьшению ее сечения. В то же время концентрация у основного ствола восстанавливается и он вновь начинает расти.
Подобное периодическое изменение концентрации и гидроди-, намических условий, происходящих непосредственно у растущих граней кристаллов, приводит к образованию и росту отдельных хрупких древовидных дендритных кристаллоагрегатов, слабо связанных между собой. Весьма существенно на структуру частиц порошка, помимо резкого обеднения раствора в прикатод-ной зоне, влияют факторы, приводящие к пассивации растущих граней кристаллов.
Чем меньше .материала для роста, т. е. ниже концентрация ионов, находящихся в приэлектродном слое, тем тоньше иглы, из которых состоят ветви дендрита, тем больший сдвиг катодного потенциала в отрицательную сторону и вероятней зарождение новых центров кристаллизации на других направлениях и их развитие в форме игл дендрита. Поэтому дендриты обыкновенно расположены не параллельно, главные оси их перекрещиваются во всевозможных йацравлениях.
Наибольшей величины ветви достигают при отсутствии перемешивания. При перемешивании раствора ветви обламываются. Обломки ветвей не успевают укрупниться из-за потери электрического контакта с катодом, три этом образуются особенно высокодисперсные металлические порошки, падающие на дно электролизера.
На неравномерный рост кристаллов в предельных условиях влияет еще один фактор — повышение вязкости растворов. Образующиеся в прикатодных слоях вследствие защелачивания католита плохо растворимые гидроокиси и основные соли металлов, скапливаясь вблизи граней кристаллов, сильно затрудняют диффузию питательного вещества к отдельным участкам кристаллической решетки и тем самым вызывают прекращение роста участков, находящихся в более «угнетенных» условиях.
Полное прекращение образования новых центров кристаллизации, по-видимому, становится возможным только при очень больших'плотностях тока. Но при этом из-за быстрого нарастания поляризации достигается потенциал, при котором начинается
Электроосаждение металлов
383
разряд других ионов, главным образом водорода. Усиленное барботирование электролита выделяющимися пузырьками газа приводит к активации диффузионных процессов в приэлектродных слоях, и кристаллизация металла на катоде с появлением свежих дисперсных микродендритных образований становится вновь возможной.
В большинстве случаев, однако, в особенности при электроосаждении таких металлов, как серебро, медь, висмут, свинец, олово и др., порошки появляются в области потенциалов, более положительных, чем потенциал выделения водорода.
Поэтому нельзя рассматривать действие водорода, как причину и тем более как обязательное условие образования порошкообразных металлов. Очевидно, что роль водорода, если он выделяется, сводится в основном к интенсификации процесса отложения металлов в дендритной форме из-за образования и концентрирования гидроокисей и основных солей металлов в католите вблизи одних участков поверхности растущих граней и усиленного барботирования раствора в других участках.
Зная заранее наиболее специфические условия, влияющие на величину предельного тока, и учитывая наблюдаемые при электролизе сдвиги в ионных равновесиях в католите ванны (при заданной плотности тока), можно получать порошки с вполне определенной, заранее заданной характеристикой и физической структурой частиц. Этот вывод многократно проверялся на опыте.
Электролитические порошки по важнейшим физико-химическим свойствам отличаются от металлических порошков, полученных другими способами. Катодное отложение порошкообразного металла отличается высокой чистотой и имеет характерную дендритную структуру частиц. Применяя различные составы электролитов и разнообразные условия электролиза, можно получать металлические порошки с предварительно заданными свойствами, что необходимо для решения ряда специфических проблем порошковой металлургии, таких, например, как прочность, коррозионная стойкость, высокая электропроводность, малая истираемость металлокерамических изделий, изготавливаемых из порошков.
§ 8. Особенности получения компактных катодных осадков
Катодное осаждение металлов в компактной форме и с достаточно высоким выходом по току можно производить при плотностях тока, изменяющихся в широких пределах, вплоть до предельной плотности тока. Обычно величина катодной плотности тока для электролитического рафинирования и электроизвлечения металлов в промышленном масштабе определяется экономическими соображениями.
384
Теоретические основы электрохимии
Известно, что электролизеры не располагают какой-то определенной номинальной производительностью, так как она является функцией проходящего через систему тока. Соблюдая некоторые конструктивные и расчетные требования, можно от одного и того же электролизера получить разную производительность, тем большую, чем большая плотность тока установлена на, электродах.
Как замечает В. В. Стендер, в процессах извлечения метал* лов из электролитов с нерастворимым анодом повышение плотности тока увеличивает не только скорость электроизвлечения, но и выход по току. Повышение плотности тока позволяет механизировать выгрузку катодного металла, повысить производительность и улучшить условия труда.
Таким образом, была показана целесообразность использования очень высоких плотностей тока при электролитическом извлечении цинка в электролизерах барабанного, дискового, вертикально-ленточного типа (до 6000—8000 ajM2).
Каждая из названных конструкций ванн имеет свои преимущества и недостатки. Основные недостатки барабанных, щелевых, дисковых и других электролизеров — их чрезмерная громоздкость; кроме того, здесь не устраняется наиболее сложный процесс сдирки катодного цинка. С этой точки зрения интересен предложенный ВНИИцветметом новый тип электролизера с катодом в виде вращающейся непрерывно наращиваемой болванки круглого сечения. Электролизер с торцовым вращающимся катодом может работать полунепрерывно, с периодическим удалением готовой цинковой болванки заданной длины.
Известно, что плохое качество осадка и низкий выход по току металла совпадает с повышенным содержанием в электролите посторонних примесей. Большинство примесей, попадающих в растворы и накапливающихся в них, сильно осложняет электродные процессы.
Катодное осаждение никеля обычно характеризуется весьма значительной химической поляризацией, определяемой замедленной стадией разряда. Большая электродная поляриза-зия при разряде и ионизации этого металла приводит к тому, что анодному растворению подвергается не только основной металл, но и более положительные, чем никель, примеси. На катоде же происходит разряд ионов никеля и большинства примесей, перешедших с анода в раствор.
Практика катодного осаждения никеля одновременно указывает на весьма малый диапазон плотностей тока, при которых металл выделяется с меньшей затратой энергии, чем водород.
Особое внимание уделяется чистоте электролита и сохранению оптимального состава его. В частности, кислотность электролита в процессе электролиза должна оставаться практически
Электроосаждение металлов
385
неизменной в (весьма узких рамках pH. При pH ниже 2 для сульфатного электролита, содержащего 5 г!л NaCl, и при pH примерно 1,5 для электролита, содержащего 50 г!л NaCl, выход по току начинает заметно падать. Более высокий выход по току наблюдается с ростом концентрации ионов С1~ в растворе. А. Л. Ротинян объясняет это явление адсорбцией анионов хлора на катоде, что, по его мнению, облегчает разряд двухвалентных ионов никеля по сравнению с водородом. С повышением pH .выход по току растет, приближаясь к 100%, но на практике установлено определенное значение pH, выше которого в электролите начинают появляться коллоидные частицы. Это приводит к порче катодного осадка и к ухудшению его механических свойств. По достижении точки гидратообразования (pH = 5,2) резко возрастают внутренние напряжения в осадках никеля, увеличивается их твердость, уменьшается плотность и эластичность. Повышение внутренних напряжений и твердости приводит к коротким замыканиям и прорыву диафрагм. Большое внутреннее напряжение ведет к возникновению микротрещин, которые так же, как и поры, заполняются электролитом; в таком случае возрастает вероятность загрязнения катодного осадка. Поэтому в массе электролита и особенно в непосредственной близости к катоду (в католите) должна сохраняться достаточная кислотность.
Однако добавка кислоты, в особенности сильной, может изменить благоприятное для осаждения никеля соотношение концентраций ионов .водорода и никеля. Это поведет к увеличению доли участия ионов Н+ в разряде. Обычно в никелевую ванну вводят слабые, мало диссоциированные кислоты и таким образом сохраняют pH раствора в ограниченных пределах. Процесс катодного Осаждения никеля очень чувствителен к присутствию примесей в растворе. Обычные примеси в черновом (анодном) никеле — медь и железо. Никелю всегда сопутствует кобальт. Реже встречаются цинк, мышьяк, свинец. При анодном растворении никеля эти примеси большей частью переходят в раствор. В дальнейшем они могут отлагаться на катоде, что приведет к загрязнению катодного никеля, ухудшению его структуры. Последнее сопровождается падением выхода по току.
Многочисленными исследованиями было установлено, что, кроме неорганических примесей, в электролит попадают органические вещества, выщелачивающиеся из аппаратуры (дерево, ткани). Органические вещства, присутствуя в растворе даже в ничтожных концентрациях, 'нарушают процесс катодного осаждения никеля — осадок получается очень хрупким и часто чешуйчатым. При этом выход по току резко снижается (на 10— 25%), а расход электроэнергии соответственно возрастает. Поэтому на практике деревянные детали аппаратуры и ткань ди-25 Заказ 1338
386 Теоретические основы электрохимии
афрагмы, соприкасающиеся с раствором, предварительно перед употреблением попеременно обрабатывают слабыми растворами щелочей (соды) и серной кислоты и водой. Загрязненный электролит очищают от органических примесей анодной обработкой током или разрушением органических примесей окислителями (перманганатом, перекисью водорода и т. д.).
Эффективный метод очистки раствора — адсорбция примесей активированным углем. i
Следует иметь в виду, что диафрагма не всегда может полностью предохранить катодное пространство от попадания в него примесей из анодного пространства, и их проникновение через диафрагму вследствие диффузии и миграции должно предотвращаться встречным потоком электролита, циркулирующего через поры диафрагмы *.
Процесс электролитического рафинирования меди за многие годы его практического осуществления не претерпел существенных изменений; система «ящичных» электролизеров с неподвижными стационарными электродами, работающими с относительно небольшой плотностью тока и требующими значительной затраты рабочей силы (для выгрузки катодов и загрузки черновых анодов), сохранилась в большинстве производств. Это свидетельствует о том, что установившиеся технологические показатели электролиза удовлетворяют основным требованиям. По простоте устройства и значительной эффективности (наибольшего использования площади пола цехов) существующее оборудование трудно заменить более рациональным и производительным.
При электролитическом рафинировании меди применяют главным образом сернокислый электролит. Он менее летуч, чем солянокислый, азотнокислый и др. В течение электролиза облегчается освобождение электролита от наиболее вредных примесей (iBi, Sb, As), повышается извлечение благородных металлов (особенно Ag), наблюдается меньшая поляризация на катоде. Сернокислый электролит оказался пригодным в новых производствах, например при электролитическом получений медного порошка и медной фольги. В последнем случае применяют интенсивную циркуляцию электролита и вращающийся барабанный катод. Трудоемкая операция извлечения катодного металла в таком случае облегчается из-за возможности снятия с катода непрерывной ленты.
1 В последние годы в Гипроникеле ведутся исследования с использованием хлорвдного электролита. Большая растворимость хлорида никеля в сочетании с хорошей электропроводностью и большим .коэффициентом активности ионов никеля в таких растворах позволяют надеяться на осуществление электролиза с очень высокой плотностью тока. Отсутствие серы в электролите позволяет получить катодный никель, чистый по содержанию серы.
Электроосаждение металлов
387
В барабанном электролизере была показана полная возможность использования очень высоких плотностей тока на катоде (до нескольких тысяч ампер на квадратный метр).
Многие исследователи показали целесообразность увеличения интенсивности циркуляции электролита. Безусловно приемлем для работы в промышленном масштабе при высоких плотностях тока метод, при котором электролит течет с большой скоростью параллельно электродам. К преимуществам такого способа относится то, что интенсивная циркуляция электролита снимает одновременно диффузионные ограничения и для катодов и для анодов. Во всех случаях, особенно при интенсивной циркуляции раствора, необходимо обеспечить промежуточный отстой и фильтрацию электролита.
Весьма большое влияние на показатели электролиза оказывают поверхностно активные вещества. Качество катодных отложений значительно улучшилось вследствие внедрения в практику электрорафинирования меди добавки тиомочевины. С помощью радиоактивного индикатора меченой серы было установлено, что благоприятное влияние тиомочевины на структуру и свойства катодных осадков меди объясняется специфической адсорбцией ее на углах и ребрах растущих кристаллов.
В последние годы возникло электролитическое производство марганца, который все больше применяется для получения сплавов и заменяет никель, бериллий и другие ценные металлы.
Стоимость электролитного марганца, однако, еще слишком велика. Процесс может быть усовершенствован использованием хлористых растворов вместо сернокислых при условии получения и использования анодного хлора и применения высоких плотностей тока. Электролиз растворов хлористого марганца с получением металлического марганца и хлора потребует большого количества соляной кислоты; при этом оба продукта должны оказаться достаточно дешевыми.
Другим новым производством является гидроэлектрометал-лургия хрома. Из растворов .хромовой кислоты, применяемых в гальваностегии, получают толстые катодные осадки хрома, которые после рафинирования водородом в электрических печах приобретают достаточную чистоту. Но расход электроэнергии велик (около 50 квт-ч!кг). В сульфатных растворах трехвалентного хрома расход энергии составляет 11—14 квт-ч/кг. Здесь достигли выхода до 60%, при pH = 2,5, плотности тока 800 а/м2 и времени наращивания катодного хрома 72—80 ч.
Недостаток процесса — трудность приготовления электролита.
Электролиз растворов хлорного хрома обещает более благоприятные результаты; здесь попутно можно использовать анодный хлор. Электролиз водных растворов применяется для получения рения, индия, таллия и галлия.
25*
388 Теоретические основы электрохимии
Мы рассмотрели несколько примеров получения компактных катодных отложений, но из краткого обзора следует, что производство цветных металлов может быть увеличено не только в результате ввода в действие новых мощностей, но и путем более полного использования электрической энергии, совершенствования аппаратуры и технологии, интенсификации электродных реакций в электролизерах. Более полное изложение поднятых в данном разделе вопросов выходит за рамки учебного пособия по теоретической электрохимии и приводится в специальных курсах технологии электрохимических производств.
ЛИТЕРАТУРА
Баймаков Ю. В. и Журив А. И. Электролиз в гидрометаллургии. Me-таллургиздат, 1963.
Баграмян А. Т. и Петрова Ю. С. Физико-химические свойства электролитических осадков. Изд. АН СССР, 1960.
Горбунова К. М., Ивановская Т. В., Попова О. С. Труды совещания по электрохимии. Изд. АН СССР, 1953.
Горбунова К. М., Попова О. С., Сутягина А. А., Полукаров Ю. М. Рост кристаллов. Изд. АН СССР, 1957.
Есин О. А., Левин А. И., Лошкарев М. А., Левиан Л. Г. Цветные металлы, 1945, 3.
К а д а н е р А. И. Равномерность гальванических покрытий. Изд. Харьковского университета, 1960.
Каишев Р. Спиральный рост и перенапряжение при электрокристаллизации серебра. Труды четвертого -совещания по электрохимии. Изд. АН СССР, ' 1959, 371.
Кунин Л. Л. Поверхностные явления в металлах. Металлургиздат, 1959.
К у Д р а О. К-, Г и т м а н Е. Б. Электролитическое получение металлических порошков. Изд. АН СССР, 1952.
Левин А. И., Власов В. И. Цветные металлы, 1959, 5, 32.
Левин А. И., Н о м б е р г М. И. Цветные металлы, 1962, 9.
Левин А. И., Фаличева В. И. Научные доклады высшей школы, раздел «Химия и химическая технология», 1958, 1.
Л е в и н А. И. Труды УПИ им. С. М. Кирова, сб. 43. Коррозия и металлопокрытия. Машгиз, 1953.
Лошкарев М. А., Озеров А. М., Кудрявцев Н. Т. ЖПХ, 1949, 22, 294.
Натансон Э. М. Коллоидные металлы. Киев, Изд. АН УССР, 4959.
Полукаров Ю. М., Горбунова К. М. Некоторые вопросы механизма электроосаждения сплава. Сб. трудов по защитно-декоративным покрытиям. Машгиз, 1956.
Ротинян А. Л., Хейфец В. Л. Теоретические основы гидрометаллургии, в книге «Основы металлургии», т. I, Металлургиздат, 1960.
Русанов А. И. Термодинамика поверхностных явлений. Изд. ЛГУ, 1960.
Стендер В. В. Прикладная электрохимия. Изд. Харьковского университета, 1961.
Трояновский А. В. Экономия электроэнергии при электролизе цинка и меди. Металлургиздат, 1954.
Трепнел Б. Хемосорбция. ИЛ, 1958.
Федотьев Н. П., Алабышев А. Ф. и др. Прикладная электрохимия. Госхимиздат. 11962.
фрумкии А Н„ Петрий О. А., Николаева-Федорович Н. В. ДАН СССР, 128, 1959, ,1006.
Глава XIV
АНОДНЫЕ ПРОЦЕССЫ
§ 1. Общие сведения
Все многообразие анодных реакций можно разделить на две большие группы—процессы, проходящие на растворимых и нерастворимых электродах. При этом, однако, следует иметь в виду, что и в том и в другом случае на электродах протекают реакции электрохимического окисления. В качестве растворимых анодов обычно применяют металлические электроды, на которых при пропускании тока происходят реакции ионизации:
Me — ze-+ М<?\ (XIV, 1)
Течение (реакции (XIV, 1) обычно сопровождается поляризацией, связанной с замедленной ионизацией атомов металла, а также диффузионными ограничениями. Процессы анодного растворения металла нередко осложняются, кроме того, образованием окислов или труднорастворимых солей на поверхности металла. Особую группу составляют металы, характеризующиеся образованием ионов различной валентности. В случае появления окислов или фазовых пленок определенного состава на поверхности ацода растворение, его частично или полностью прекращается и он может превратиться в «нерастворимый». На нерастворимых анодах идет процесс разряда анионов:
Az — ze -> А.
Если электролизу подвергаются кислородсодержащие кислоты или соли, а также основания, то на аноде идет процесс разряда гидроксильных ионов или молекул воды с выделением кислорода. При электролизе галоидно-водородных кислот и их солей на аноде разряжаются ионы С1~, Вг_ и J~ с выделением соответствующих веществ в молекулярной (газообразной) форме.
Особый случай анодных процессов составляют окислительные реакции, протекающие на инертных электродах. В этом случае металлический анод используется как переносчик электронов — окислитель, обеспечивающий электролитическое окисле-
390 Теоретические основы электрохимии
ние (синтез) органических соединений или протекание различного рода окислительных реакций типа:
МпО^~ — е МпО7,
SO1~ — 2e->S2O|“ и т. п.
Весьма сложную картину можно наблюдать в случае растворения сплавов. Компоненты, содержащиеся в таких анодах, могут образовывать различные фазы, твердые растворы или химические соединения. Поведение многокомпонентного электрода при просхождении тока будет меняться в зависимости от состава сплава и специфических особенностей анодного процесса.
§ 2. Анодное растворение металлов
Ранее, при рассмотрении особенностей катодного процесса, было показано, что наиболее быстро растущие грани кристаллов вырождаются в ребра или вершины. В ходе анодного процесса быстро растворяющаяся грань должна, напротив, непрерывно увеличиваться, пока не достигнет наибольших размеров, характерных для данного растворяющегося кристалла. При этом весь ход явлений оказывается настолько закономерным, что сняв микрокинофильм о зарождении и росте кристаллов и пустив его в обратную сторону, можно с достаточным приближением воспроизвести картину их растворения.
Вместе с тем природа и механизм затруднений, которые встречают ионы при переходе межфазной границы, в деталях своих остаются неясными. Однако независимо от этого из самых общих положений кинетики электродных процессов вытекает, что если при переходе от одной ..грани к другой меняется скорость протекания катодного акта, то в том же направлении должна измениться и скорость протекания обратного анодного акта.
Чтобы убедиться в этом, следует еще раз обратиться к уравнению, связывающему величину плотности тока с перенапряжением и значением равновесного тока обмена:
/ aF>) pFr, X
i = /0 _ e~ J. (XIV,2)
В общем виде это уравнение, как отмечалось, справедливо для любого электродного процесса, который может протекать в прямом и обратном направлении и встречает на межфазной границе тот или иной потенциальный барьер.
Те металлы, которые характеризуются малым перенапряжением при катодном выделении (велик ток обмена), обладают соответственно и малой анодной поляризацией, и, наоборот,
Анодные процессы
391
металлы, отличающиеся при катодном восстановлении малым током обмена, имеют большую анодную поляризацию.
Такая закономерность электролитического растворения правильно ограненных кристаллов, гак же как и их образование, наблюдается при сравнительно малых токах, когда концентрационная поляризация, выражаемая для растворимого анода
уравнением
RT
zF
б .
-----
zFDCn а
(XIV,3)
Д®а
In | Л +
очень мала. Поэтому различия в скоростях роста или растворения отдельных граней должны быть связаны прежде всего с неодинаковостью перенапряжения на разных гранях кристалла.
Очень часто, однако, соотношение (XIV, 2) не соблюдается из-за появления на поверхности окисных или адсорбционных слоев.
Адсорбция поверхностно активных веществ как фактор, влияющий на кинетику электродных процессов, подробно рассматривалась в предыдущих главах. Здесь достаточно указать, что адсорбционный слой, тормозящий разряд металлических ионов, неминуемо должен тормозить и обратную реакцию ионизации, причем каждое поверхностно активное вещество должно по-разному .влиять на электрокристаллизацию и анодное растворение металла. Такой вывод вполне естествен, поскольку сама адсорбция поверхностно активных веществ, а значит, и состояние адсорбционного слоя, как правило, зависят о;т потенциала ионного слоя. Поэтому влияние одного и того же вещества на процессы электрокристаллизации и растворения металлических ионов, особенно при больших поляризациях, может оказаться резко различным. Наиболее вероятно проявление подобных различий в тех случаях, когда равновесный потенциал электрода лежит вблизи его потенциала нулевого заряда.
Окисные пленки на поверхности металла могут образовываться в результате химического взаимодействия с окислителем, а также вследствие анодного окисления. Это в общем виде может быть представлено уравнением
пМе + тН2О~-^еМепОт + 2тН+. (XIV,4)
Вероятность и скорость протекания анодного окисления, так же как и всякой другой анодной реакции, увеличивается при сдвиге потенциала в положительную сторону. Равновесный потенциал этого процесса для большинства металлов лежит положительнее потенциала основной реакции ионизации и разряда металлических ионов:
Ме™Мег+.
392
Теоретические основы электрохимии
Поэтому влияние поверхностных окислов на кинетику указанной реакции наиболее резко проявляется именно в случае растворения, а не осаждения металлов.
По своему характеру и интенсивности это влияние может быть различным в зависимости от конкретных свойств металла и среды. Чаще всего оно сводится к пассивированию, т. е. торможению, либо полному прекращению процесса образования металлических ионов и к соответствующему повышению анодного потенциала. При этом другие анодные процессы: разряд кислорода, перезарядка имеющихся в растворе ионов и т. п., как правило, протекают на том же электроде подобно тому, как они протекают на нерастворимых анодах.
Если металл при растворении способен образовывать ионы различной валентности, то1 стандартные потенциалы различных реакций прямого или ступенчатого образования этих ионов связаны, как отмечалось, правилом Лютера.
После установления электрохимического равновесия между металлом и его ионами в растворе всегда должны преобладать те катионы, потенциал образования которых менее положителен. Равновесная концентрация катионов другой валентности уже при разности потенциалов порядка нескольких десятых вольта становится, как легко подсчитать, исчезающе малой.
Чтобы проиллюстрировать сказанное, рассмотрим два примера.
Медь в растворе сульфата меди. В этом случае в растворе устанавливается равновесие
2Cu+^Cu2+ + Cu. (XIV,5)
Ccu2+
Так как константа этой реакции равна Кр =---------=1,5-106,
ССи+
то равновесие сдвинуто вправо.
На медном аноде в общем случае возможны следующие реакции:
1. Си — е -> Си+ <Рси+/си — 0,51 в
2. Си — 2е-> Си2+ ?си2+/си = 0,34 в
3. Си+ — е -> Си2+ <Рси2+/си+ = 0,17 в.
Легко показать, что из-за весьма малой концентрации ионов в растворе третий процесс не пойдет. Следовательно, наиболее вероятным будет второй процесс, который будет протекать фактически при потенциале, соответствующем реакции ионизации Си — 2е — Си2+. Поскольку между ионом одновалентной и двухвалентной меди существует равновесие, то наряду со вторым процессом частично будет протекать и первый процесс,
Анодные процессы
393
скорость которого легко подсчитать из равенства tPcu+/Cu = ?Си2+/Си
или
0,51 + In aCu+ = 0,34 + In аСц2+.
Таким образом, анод будет растворяться с образованием ионов меди той и другой валентности в соответствии с константой КР, которая равна отношению равновесных концентраций.
Все сказанное справедливо только для обратимого процесса, т. е. для плотностей тока, стремящихся к нулю. При значительных плотностях тока, принятых в практике электролиза, реакции ионизации с образованием ионов Сп+ протекают с меньшими затруднениями — с меньшей поляризацией, чем реакции ионизации Си—2е->Си2+. В результате этого в раствор будут переходить ионы одновалентной меди в количестве, несколько большем, чем это требуется по равновесию (XIV,5). Однако в электролите, где действуют законы термодинамики, а не, электрохимической кинетики, быстро вновь установится соотношение 2Cu+ Си2+ + Си и избыточные против равновесного одновалентные ионы меди будут давать ионы двухвалентной меди и металлическую медь, выпадающую в виде высокодисперсного порошка в шлам.
Железо в растворе собственной простой соли. Здесь в общем случае устанавливаются на аноде потенциалы:
?Fe2+/Fe = ~ °’44 WF/Pe = ~ °>04 б‘-?Fe3+/Fe2+ = + °’78 в'
В противоположность предыдущему случаю <PFe2+/Fe '< <PFe3+/Fe и в равновесии с металлическим железом существуют только ионы низшей валентности Fe24". Второй потенциал <PFe3+/Fe на железном аноде вполне может быть достигнут при достаточно высокой плотности тока. Однако переход в раствор ионов Fe3* при этом не наблюдается. Появление трехвалентного железа становится реально возможным только при приближении к потенциалу <?Fe3+/Fe2+’ чт0 практически маловероятно.
§ 3. Явление пассивности металлов. Строение и толщина пассивных пленок на металлах
Значительные разрушения металлов и сплавов под воздействием окружающей среды привели к необходимости вести борьбу за долговечность металлических изделий и конструкций. Весьма большое значение для увеличения коррозионной стойко
394
Теоретические основы электрохимии
сти имеют пассивные пленки, образующиеся на поверхности металлов.
Поэтому явление пассивности в течение многих лет привлекает внимание исследователей и в данное время является одной из актуальных проблем, требующих дальнейшего изучения.
Впервые явление пассивирования металла было описано М. В. Ломоносовым, который заметил, что в разбавленной азотной кислоте железо растворяется беспрепятственно, а в концентрированной растворение быстро прекращается.
Предположение о том, что остановка растворения в этом и других подобных случаях обусловлена появлением на поверхности химически связанного кислорода, высказал М. Фарадей. Он же применительно к такому инертному состоянию металла ввел в употребление термин «пассивность». К настоящему моменту на механизм пассивирования и природу пассивных пленок установились две основные точки зрения. Согласно одной .из них, торможение процессов на границе фаз металл — раствор наступает в результате образования на поверхности металла фазовой окисной пленки. Согласно другой точке зрения, пассивирование металлов и сплавов обусловлено адсорбцией на поверхности кислорода и некоторых кислородсодержащих соединений.
Гипотеза о существовании на поверхности пассивного металла некоторой пассивирующей пленки развивалась многими исследователями. Тем не менее она очень долго оспаривалась ибо, во-первых, не удавалось показать, что на поверхности пассивного металла имеются какие-либо пленку, а, во-вторых, считалось, что даже безводные окислы, образовавшиеся, например, на железе при высокой температуре, легко растворяются в кислоте; Тем более странным казалось, что защита металла от растворения в кислоте может быть обусловлена окислом, возникшим в водном растворе. Теперь эти возражения опровергнуты рядом экспериментальных работ.
Появление невидимых глазом пленок на чистой поверхности металла, приведенной в соприкосновение с воздухом, было впервые показано Фрейндлихом, а затем Тронстадтом по изменению оптических свойств металла в поляризованном свете.
Вернон нашел, что изменение веса медной пластинки в атмосфере О2 также можно зафиксировать раньше, чем станут заметны какие-либо видимые изменения на поверхности.
Ряду исследователей удалось химическим или электрохимическим подтравливанием металла отделить от поверхности невидимые в обычных условиях окисные пленки в виде самостоятельных прозрачных чешуек. Сначала это было выполнено для алюминия, затем для железа и нержавеющих сталей.
Большое число исследований, посвященных изучению окис
Анодные процессы
395
ных .пленок на металлах, было проведено с использованием рентгене- и электронографических методов. В результате не только был еще раз неоспоримо подтвержден сам факт существования этих пленок, но и получены точные данные об их кристаллическом или аморфном строении и даже о параметрах кристаллической решетки оксида.
Таким образом, наличие окисных или иных, близких по характеру пленок на поверхности металлов, соприкасающихся с окислителями, в настоящее время не вызывает сомнения. Подобные пленки защищают металл от окисления при химическом взаимодействии с окислителем (например, в газовой фазе).
Толщина защитных пленок, возникающих на не слишком нагретом металле при реакции с газом, настолько мала (от нескольких до десятков ангстрем), что они сохраняют прозрачность в проходящем свете. Иногда пленки можно заметить по интерференционным цветам побежалости, но часто, будучи значительно тоньше, чем длины волн видимого света, они не дают и этой картины.
Такая малая толщина пленок говорит об их чрезвычайно высокой защитной способности или, иначе говоря, о способности резко затруднять диффузию окислителя к металлу.
Одно из необходимых условий образования защитной пленки сводится к тому, что объем возникшего окисла должен быть во всяком случае не меньше объема металла, пошедшего на его образование (иначе слой заведомо не будет сплошным).
Другое важное условие — однородность пленки и ее высокая адгезия к металлу. В. А. Кистяковский считал, что это условие выполняется, если окисел образуется в частично гидратированной форме и имеет коллоидную структуру. П. Д. Данков на ряде примеров показал, что причина высокой сплошности и адгезии пленки заключается в способности окисла продолжать или наследовать решетку металла. Такой окисел образуется в результате внедрения атомов окислителя в поверхностные ячейки металлической решетки. При этом вещество пленки не образует самостоятельной кристаллической фазы, а органически срастается с металлом, благодаря чему сама пленка может быть названа нефазовой.
Подобный ход (процесса особенно вероятен, если кристаллическая решетка металла очень прочна, а нормальная решетка оксида по типу и параметрам не слишком отличается от нее или, во всяком случае, при небольшой деформации может быть в нее «вписана». В этом случае работа, которую нужно затратить на деформацию решетки оксида до обычного для нее состояния, унаследованного от металла, оказывается меньше, чем работа разрушения решетки металла и образования самостоятельных зародышей новой фазы.
396
Теоретические основы электрохимии
Напряжения сжатия, возникающие в нефазовой пленке, по П. Д. Данкову, способствуют дополнительному торможению встречной диффузии атомов или ионов окислителя и металла и обеспечивают особенно быструю остановку окислительного процесса.
Изучая химическую стойкость окисных пленок по отношению к агрессивным растворам, Эванс обнаружил, что снятая с железа высокотемпературная окалина (практически не растворяется даже в очень сильных кислотах. Очень медленно растворяются кислотой и прозрачные чешуйки пленок, снятые с железа, окислившегося при комнатной температуре.
Следовательно, сплошная окисная пленка в принципе вполне может защитить металл не только от химического окисления, но и от электрохимического растворения на аноде. В то же время многие окислы металлов, особенно в тонких слоях, обладают достаточной электронной проводимостью для того, чтобы на покрытой ими поверхности могли протекать любые анодные процессы, связанные с передачей электронов от компонентов раствора к металлу, что как раз и характерно для пассивных металлов.
По составу и структуре пленки, возникающие на поверхности металла под действием анодной поляризации или окисления в водных растворах, очень близки к пленкам, образующимся при химическом окислении в газе. Так, на железе, запассивиро-ванном различными окислителями в кислых и нейтральных растворах, неоднократно обнаруживалось соединение с решеткой, характерной для окислов типа Ре^СЦ или y-FegOs, входящих в состав железной окалины.
Однако многие исследователи считают, что пассивирующий окисел, если он существует в действительности, должен обладать какими-то особыми свойствами. Основывается этот взгляд на сопоставлении расчетных потенциалов образования окислов из металла и воды с фактическими потенциалами пассивирования металла.
Наибольшее количество таких сопоставлений проводилось для железа. Уравнение (XIV,4) для случая анодного образования магнетита из железа и воды может быть представлено в следующем виде:
3Fe + 4Н2О — 8е Fe3O4 + 8Н+.
Расчетный потенциал этой реакции
<р = (— 0,095 — 0,058- pH) в. (XIV,6)
Между тем в действительности пассивирование и обратное активирование железа происходят при потенциале
9 = (0,58 — 0,058 pH) в. (XIV,7)
Анодные процессы
397
Значительное расхождение между потенциалами (XIV,6) и (XIV,7) давало повод к предположению о том, что пассивирующий окисел либо является каким-то (высшим кислородным соединением, либо, по крайней мере, пересыщен кислородом с поверхности.
Однако в последнее время в советской и иностранной литературе появились работы, свидетельствующие, что магнетит Fe304—гораздо более сильный электротехнический окислитель, чем считали раньше, и что стационарный потенциал, который он приобретает в водных растворах, очень близок к рассчитанному по формуле (XIV,7).
Таким образом, гипотеза о высших пассивирующих окислах железа оказывается неоправданной, а химическое или электрохимическое образование на его поверхности пленки окисла типа FesO4 представляется бесспорно одним из реально возможных путей пассивирования железа в электролите.
Адсорбционная теория пассивности основывается на том предположении, что на металлической поверхности после взаимодействия с кислородом образуется слой адсорбированного кислорода. При этом часть исследователей считает, что кислород адсорбируется только на активных участках поверхности металла и прочно удерживается силами адсорбции. Таким образом, образуется неактивный поверхностный слой металла, связанный с адсорбированными атомами (кислорода. Другая часть исследователей полагает, что для достижения пассивности необходимо полное заполнение поверхности адсорбированным кислородом. Защитное действие адсорбционного слоя можно представлять двояко. Например, можно полагать, что кислород насыщает активные валентности всех поверхностных атомов металла и тем снижает его активность.
Можно также представить, что адсорбированные поверхностью металла атомы кислорода захватывают его свободные электроны и на поверхности возникает слой Mej О2-. При этом металл заряжается положительно. Такой слой будет в значительной степени повышать работу выхода электрона, что и придает металлу электроположительные свойства.
При определенных условиях закрытие кислородом уже очень малой доли поверхности способно привести к резкому торможению ионизации металлических атомов. Впервые такое явление наблюдалось Б. В. Эршлером, изучившим анодное поведение платины в кислых растворах. Теория Б. В. Эршлера предполагает адсорбцию атомов кислорода на отдельных активных участках поверхности металла, что может тормозить протекание анодного процесса. Атомы кислорода, адсорбируясь на металле, образуют электрические диполи за счет частичной ионизации
398 Теоретические основы электрохимии
кислородного атома электроном из металла. Положительный конец образующегося диполя располагается в металле, а отрицательный— в растворе. Получается сложный адсорбционно-ионный скачок потенциала. В результате общий электродный потенциал металла сдвинется в более положительном направлении. При адсорбции кислорода ионный двойной слой заменяется частично адсорбционным и потенциал электрода при этом как бы облагораживается, работа выхода иона металла в раствор повышается, т. е. стремление металла посылать свои ионы в раствор уменьшается пропорционально числу адсорбированных атомов кислорода. Б. В. Эршлер установил, что скорость растворения платины при заданном потенциале падала в его опытах вчетверо при закрытии кислородом всего 6% поверхности. Вследствие увеличения количества адсорбированного; кислорода скорость растворения также падала (вчетверо на каждые следующие 6%).
Сходные соотношения между скоростью растворения металла и количеством адсорбированного кислорода были отмечены при пассивации железа в щелочных растворах.
Таким образом, явление пассивности состоит в замедлении или полном прекращении процесса ионизации металлов вслед- ' ствие изменения заряда и свойств поверхности, вызванных образованием на ней адсорбционных или фазовых слоев окислов.
§ 4. Рост пленок при анодной поляризации металлов. Нерастворимые аноды
Среди металлов, образующих окисные пленки и обладающих малой растворимостью, следует отметить никель, кобальт, марганец, свинец, серебро, олово и сплавы из этих металлов. Комбинации окислов действуют лучше, чем окислы только одного металла. Окислы образуются обычно на поверхности металлов, при анодной поляризации. Анодно обработанный сплав свинца с серебром образует пленку, состоящую из смеси РЬО2 и Ag2O. Кобальт дает пленку CogOs.
Окислы повышают анодный потенциал, причем металл почти не переходит в раствор, а энергия, расходуемая на аноде, идет почти целиком на выделение кислорода. Медная пластинка в кислом растворе медного купороса может служить нерастворимым анодом, если на ее поверхности имеются прочные пленки из окислов марганца, свинца и олова. Однако без анодной поляризации такая пластинка будет растворяться. Из различных добавок к анодам, имеющих целью уменьшить их растворение и повысить анодный потенциал, наиболее заметное действие оказывает серебро.
В табл. 24 перечислены применяющиеся в промышленности материалы для изготовления нерастворимых анодов.
Анодные процессы
399
Таблица 24. Материалы, применяемые для изготовления нерастворимых анодов
Состав анодов Поверхностная пленка Характеристика и применение
Группа А—для сернокислых электролитов
Магнетит Свинец + 4—12% Sb Сплав Fe—Si (13% Si) Кремнистая медь Сплав РЬ—Ag Fe2O3 • PbO2 -j- Sb2O3 Fe2O3 + SiO2 PbO2, MnO2, SnO2 PbO2 + Ag2O (+Sb2o3) Применение ограничено. Хрупок При электрорафинировании меди При рафинировании меди. Хрупок При получении меди высокой чистоты При производстве цинка, марганца
Группа Б — для различных электролитов
Платина Свинец и сплав РЬ—Sn Никелированное железо Свинец и серебро Сплав РЬ—Sn—Sb—Со® Pto2 PbO2, SnO2 Ni2O3 PbO2, Ag2O, AgCl PbO2, SnO2, Sb2O3, Co2O3 При производстве хлоратов При хромировании При электролизе воды При электролизе хлористых щелочных металлов При производстве марганца
Образование анодных фазовых пленок на поверхности металла может быть результатом кристаллизации на поверхности анода из раствора плохо растворимых солей или гидроокисей данного металла, а также образования фазового окисла в результате электрохимического процесса.
При анодной поляризации свинца в H2SO4, серебра в растворах КС1 и КВг, железа и меди в щелочных растворах окислителей на поверхности металла образуются тонкие и довольно сплошные пленки, представляющие кристаллические осадки, состоящие из плохо растворимых соединений типа PbSO4, AgCl и др. Пленка образуется не сразу после погружения металла в раствор, а через некоторое время, необходимое для пересыщения раствора вблизи анода.
А. Г. Самарцев и Л. Ю. Куртц указывают, что после возникновения на поверхности металла первичной сетки центров кристаллизации вероятность образования новых зародышей сильно уменьшается.
400 Теоретические основы электрохимии
Толщина' пленки в основном •определяется степенью пересыщения раствора, которая зависит от анодной плотности тока. Чем больше кристаллических зародышей образуется на единицу поверхности, тем раньше они сомкнутся в сплошную изолированную пленку, тем меньше будет ее толщина. С ростом га увеличивается пересыщение раствора в прианодном слое, что приводит к возрастанию числа зародышей на единицу поверхности, а следовательно, уменьшает толщину пленки.
Согласно современным представлениям, анодные окисные пленки образуются вследствие электрохимического окисления поверхности металла. Строение и толщина пленки зависят от ряда факторов и в том числе от применяемых электролитов.
По .характеру анодных процессов и получаемой при этом окисной пленки электролиты можно разделить на три группы:
1) электролиты мало или совсем не растворяющие окись металла;
2) электролиты, умеренно действующие на окись,
3) электролиты, сильно растворяющие окись.
В электролитах первой группы образуется тонкая и сплошная пленка, которая быстро достигает максимальной толщины (пленки А12О3 в растворах борной кислоты или нитратов). Толщина таких пленок не превышает 0,5 мк. Они обладают высокими электроизоляциоными свойствами.
В электролитах, сильно действующих на пленку (щелочи и хлориды), она быстро растворяется и поэтому не может достигнуть заметной толщины.
Непременное условие роста пленки — необходимость поддержания окисла в пористом состоянии в течение всего процесса анодного окисления. Это достигается применением электролита, умеренно растворяющего пленку, причем наблюдается продолжительный рост пленки и толщина ее достигает значительной величины.
Анодные пленки больших толщин образуются, следовательно, только в том случае, когда окисел частично растворяется и когда скорость роста пленки значительно преобладает над скоростью ее растворения.
Рост пленок в электролитах, не растворяющих окислы, можно представить следующим образом. В первый момент прохождения тока на поверхности металла в результате электрохимической реакции окисления образуется очень тонкий сплошной слой. Слой образуется достаточно быстро. Но после достижения толщины 0,5—‘1 мк вследствие сильного торможения процесса взаимного проникновения реагирующих частиц дальнейшее образование пленки затрудняется и рост ее прекращается.
Пленка растет не с поверхности, как это наблюдается при гальванических покрытиях, а между металлом и толстой пори
Анодные процессы
401
стой частью пленки. Пограничный слой пленки, периодически разрушаясь и восстанавливаясь, перемещается в глубь металла, так что толщина этого слоя во времени остается приблизительно постоянной.
Верхний слой пленки, напротив, утолщается, однако под действием электролита он оказывается пронизанным густой сетью капиллярных каналов, которые ориентируются преимущественно перпендикулярно к поверхности металла, т. е. по направлению линий тока. Наличие пор в окисной пленке экспериментально установлено рядом работ. Как показал В. Н. Набоков, строение и толщина анодных пленок в общем случае зависят от следующих факторов: 1) природы и концентрации электролита; 2) плотности тока на аноде; 3) температуры электролита; 4) продолжительности анодной обработки; 5) состава металла и среды. Толщина пленки и ее пористость оказывают большое влияние на технологические свойства так называемых нерастворимых анодов. Полностью пассивированные аноды характеризуются высокими значениями потенциала. Это относится в первую очередь к свинцовому аноду. Когда концентрация ионов РЬ2+ у анода превысит произведение растворимости-
{-'pt^+C'SO2- “ 4
которое, кстати, весьма незначительно, начинается образование на аноде твердого PbSO4, так как именно в прианодном слое пересыщение электролита сернокислым свинцом наиболее вероятно. Плотность тока при этом из-за уменьшения активной поверхности анода заметно возрастет. Соответственно увеличится и анодный потенциал, благодаря чему становится возможным процесс дальнейшего окисления ионов РЬ2+ в РЬ4+ по реакции
РЬ2+ —2е-РЬ4+.
Накапливающаяся у анода сернокислая соль четырехвалент-ного свинца легко гидролизуется, образуя малорастворимую двуокись свинца:
Pb (SO4)2 + 2Н2О РЬО2 + 2H2SO4.
Анод, таким образом, пассивируется, делается «нерастворимым». Рассмотрение возможных анодных процессов на электроде, покрытом двуокисью свинца, показывает, что разряд гидроксильных ионов здесь сопровождается особенно высокой поляризацией. При достаточно большой плотности тока на аноде в анолите наблюдается, кроме того, быстрое нарастание кислотности. В таком случае активность ионов ОН- значительно понизится и потенциал разряда их станет очень большим. Именно потому, что анодное окисление гидроксильных ионов в сернокислых растворах сопровождается заметным торможением, 26 Заказ 1338
402
Теоретические основы электрохимии
становится возможным течение других процессов. Можно полагать, что на аноде будет протекать один из следующих процессов:
2Н2О — 4е-О2 + 4Н+;
; 0.00Q2T |glH+Hq.l (xiv,8> Т Т г [HSO]2
2Н2О — 2е - Н2О2 + 2Н+;
?2 = + 0^0002г Ig ; (XIV,9)
Н2О — 2е-> О + 2Н+;
0 , 0,00027 1rT[H+J® [О] /vivinv
?з = <?з + —----1g -дттт2’ (XIV, 10)
z [H2OJ
о которых известно, что <р °, <р 2 и Фз’ вычисленные из значений свободных энергий реакций (XIV,8), (XIV,9) и (XfV,10), равны соответственно 1,229; 1,776 и 2,42 в. Высокое значение равновесного потенциала реакции (XIV,10) показывает, что образование атомарного кислорода в присутствии ионов кислоты возможно лишь в том случае, если такие анионы окисляются с большим перенапряжением. Однако, как это следует из опыта, значение потенциала <ра в электролитной ванне, содержащей серную кислоту, даже при значительной плотности тока не достигает величины <р3. Можно, по-видимому, заключить, что вода на аноде окисляется с образованием молекулярного [реакция (XIV, 8)], а не атомарного [реакция (XIV, 1Q)] кислорода.
По Глесстону, все необратимые процессы анодного окисления протекают через промежуточное образование перекиси водорода, т. е. по схеме (XIV,9). Если следовать этой гипотезе, то можно предполагать, что анодный процесс должен проходить через стадию первичного образования Н2О2. Все же подобный механизм разряда в рассматриваемых условиях маловероятен. Из-за весьма значительной концентрации анионов серной кислоты и уменьшения концентрации Н2О в прианодных слоях скорость образования Н2О2 должна быть весьма незначительной. Вследствие этого потенциал <р2 реакции образования перекиси водорода сдвинут в положительную сторону. Если же учесть далее, что при отсутствии деполяризатора перекись водорода быстро разлагается (на воду и кислород), то можно сделать вывод, что ничтожные количества Н2О2, которые образуются здесь, не успевают оказать заметного окисляющего действия. Таким образом, влиянием перекиси водорода на кинетику анодного процесса можно пренебречь.
Анодные процессы
403
Вместе с тем наблюдаемая при электролизе сернокислых растворов металлов убыль воды в анолите указывает на протекание процесса ее разложения. Это происходит, по-видимому, по реакции (XIV,8).
Потенциалы нерастворимых свинцовых анодов в цинковой, кадмиевой и других электролитных ваннах значительно положительнее потенциала выделения кислорода из воды, который, как известно, равен 1,23 в. Так, например, в цинковой ванне при «а = 500 а/м2 фа = 2,1 в; в кадмиевой при ia = 48 а/м2 <ра = 1,87 в; в медной регенеративной 1,6 в.
Небольшое перенапряжение на анодах ванны обезжиривания обусловлено, по-видимому, более высокой температурой электролита, при которой происходит процесс регенерации (55—60 °C).
§ 5. Перепассивация металлов
Пассивирующие окислы многих металлов не всегда являются соединениями высшей степени окисления. При достаточном повышении анодного потенциала подавляющее большинство этих окислов может быть окислено дальше. Если новое соединение, образовавшееся в результате такого процесса, растворимо, то пассивность металла нарушается и он начинает растворяться, образуя ионы высшей валентности. Нарушение пассивности при весьма сильной анодной поляризации или окислительном воздействии среды получило наименование «пе-репассивации».
На практике повышение анодного потенциала ограничивается начинающимся разрядом гидроксильных или других анионов, скорость которого нарастает с потенциалом. Поэтому случаи окисления пассивирующих пленок и перепассивации наблюдаются сравнительно редко. Однако иногда они оказывают решающее влияние на кинетику растворения металлов и сплавов. Так, например, обстоит дело в случае хрома, анодное поведение которого было исследовано Я. М. Колотыркиным с помощью потенциостатического метода. Кривая анодной поляризации в нейтральных и кислых растворах имеет вид, подобный воспроизведенному на рис. 127. Эта кривая представляет большой интерес, ибо она последовательно охватывает почти все возможные варианты анодного взаимодействия металла с водным раствором.
На кривой можно отметить пять характерных участков: ab, где скорость процесса ионизации растет с увеличением потенциала; Ьс, где зависимость между ф и IgZ меняет знак на обратный, так что с ростом поляризации анодный ток падает; cd, где скорость процесса ничтожна; de, где ток начинает возрастать, хотя потенциал еще не обеспечивает выделения кислорода; е/, 26*
404
Теоретические основы электрохимии
где уже становится возможным разряд кислорода и где анодный ток быстро растет.
Первый участок соответствует нормальному процессу анод-
Рис. 127. Стационарная скорость растворения хрома как функция потенциала в растворах H2SO4 (иго Я- М. Колотьгркину): abcdef - 1,0-н. H2SO4; 1 — 0,1-н. H2SO4; 2 -
0.01-н. H2SO4
ного растворения металла, протекающему с некоторым перенапряжением по закону:
i-Kea\ (XIV,11)
Аномальный ход кривой на участках Ьс и cd Я. М. Колотыркин объясняет, исходя из того положения, что величина коэффициента а в уравнении (XIV, 11) должна зависеть от состояния поверхности.
На основе некоторых теоретических предпосылок он считает вполне вероятным, что в той области, где изменения потенциала приводят к 'изме
нениям в количестве электрохимически адсорбированного кислорода, коэффициент К .является функцией .потенциала:
К = К'е~ь\
(XIV, 12)
Комбинация соотношений (XIV,11) и (XIV,12) дает
i = K'e(a-by‘‘. (XIV,13)
Соотношения а и b при разных потенциалах могут быть различными. В области be по гипотезе Колотыркина, b > а, а в области cdb = а.
Переводя это формальное объяснение на язык .изложенных в предыдущем разделе гипотез, можно сказать, что на участке в результате адсорбции кислорода происходит электрохимическое пассивирование металла.
Участок cd можно с известным упрощением истолковать как отвечающий химическому растворению пассивирующего окисла, скорость которого не зависит от /потенциала.
На участке de начинается перепассивация ^рома, в основе которой лежит, вероятно, процесс:
Сг2О3 + 4Н2О — бе -> Cr2O?~ -| - 8Н+.
Анодные процессы
405
После точки е преобладающая роль в балансе анодных процессов постепенно переходит к часто встречающемуся на практике анодному выделению газообразного кислорода.
Помимо Хрома 'И его сплавов, явление перепассивации наблюдалось у никеля, никелевых сплавов, железа.
С явлением перепассивации в настоящее время чаще приходится сталкиваться в связи с производством и переработкой особо энергичных окислителей (в частности, для жидкостных реактивных двигателей). Поэтому они безусловно нуждаются в дальнейших углубленных исследованиях.
§ 6. Условия устойчивости пассивного состояния
Пассивное состояние—'Часто важнейшее условие для обеспечения длительной работы металлических конструкций или металлов, которые иначе разрушились бы под воздействием внешней среды. К числу таких металлов, как отмечалось, можно отнести свинец, применяемый во многих электрохимических ваннах (хромирование, электроизвлечение цинка, кадмия, меди и т. д.) в качестве нерастворимого анода. В других случаях, наоборот, пассивирование анодов нарушает нормальное течение технологического процесса (электрорафинирование никеля, многие гальванические процессы с частично пассивирующимися ~ анодами).
Поэтому изучение факторов, облегчающих или затрудняющих пассивирование, имеет большое практическое значение.
Пассивная пленка или адсорбционный слой, тормозящие процесс ионизации металла и сообщающие ему свойства инертного электрода, сами .в электрохимическом отношении — нередко довольно неустойчивые образования.
Сдвиг потенциала пассивного электрода в отрицательную сторону может привести к разрушению пленки, к десорбции кислорода и к переходу металла в растворимое активное состояние. С другой стороны, чрезмерное повышение анодного потенциала, хотя и не ликвидирует пленки на поверхности, но зато в некоторых случаях значительно ускоряет ее растворение, •благодаря чему скорость ионизации металла также растет.
Помимо принудительного понижения электродного потенциала в результате наложения внешнего катодного тока или действия восстановителей, причиной растворения окисной пленки и активации пассивного электрода может быть простое механическое повреждение пленки. В результате повреждения немедленно возникает местный гальванический элемент, составленный из обнаженного активного участка металла (анод) и окисла на соседних участках поверхности (катод). Этот элемент во многом подобен заряженному кислотному или щелочному акку
406 Теоретические основы электрохимии
мулятору, который также включает в себя активный металлический и положительный окисный электроды. В нашем случае электроды элемента замкнуты 'накоротко и ток разряда лимитируется только перенапряжением и омическим сопротивлением электролита.
Разряд местного элемента сводится к сочетанию анодного растворения металла с катодным восстановлением окисной пленки. ^Конечным продуктом обоих процессов являются гидратированные ионы металла промежуточной или низшей валентности.
Если этот процесс протекает свободно и если его эффект не перекрывает другие реакции, то он должен привести к расширению активного участка и самоускоряющейся активации запасси-вированного металла.
Электрохимическое разрушение пленки может тормозиться малой электропроводностью электролита или нехваткой водородных ионов, участвующих в реакции восстановления окисла. Причиной торможения может быть также выделение на поверхности труднорастворимой соли металла. Кроме того, результат работы местного элемента может частично или полностью компенсироваться наложенным анодным током или действием имеющихся в растворе окислителей, способных «регенерировать пленку.
Конечный исход процесса — активация или же, наоборот, «залечивание» пленки и репассивация металла — будет в таком случае определяться соотношением скоростей тех электрохимических и химических актов, которые ведут к растворению и обратному образованию пленки.
Очевидно, что анодная поляризация, а также присутствие в растворе окислителей и веществ, дающих с металлами трудно-растворимые соединения, должны способствовать возникновению и сохранению пассивного состояния. Напротив, катодная поляризация, присутствие в растворе восстановителей, электропроводных добавок, ионов водорода, комплексообразователей и другие факторы, облегчающие восстановление и растворение пленки, должны стимулировать процесс активации.
Положение, естественно, изменяется, если металл находится в состоянии перепассивации или близком к нему. В этом случае анодная поляризация и окислители могут только ускорить процесс растворения пленки, а катодная поляризация или восстановители способны его задержать.
Помимо рассмотренных, есть еще одна группа реагентов, которые уже в очень малой концентрации мешают образованию пассивных слоев или пленок и способствуют их разрушению. Это так называемые ионы-активаторы, самыми энергичными из которых являются ионы галогенов: С1~, Вг_, J” и F~.
Сильное ускоряющее действие этих ионов на коррозионный процесс Эванс объясняет малыми размерами ионов и их спо
Анодные процессы
407
собностью проникать в мельчайшие поры окисных пленок и таким образом нарушать их целостность.
По современным воззрениям, активирующие свойства галоидных ионов основаны на очень высокой энергии адсорбции их металлической поверхностью и на вытеснении кислорода, необходимого для пассивации. При этом в концентрированных растворах серной кислоты галоидные ионы, адсорбируясь на поверхности некоторых сталей, сами могут приводить к пассивации. Такой эффект может быть объяснен следующим. При адсорбции галоидов точка нулевого заряда железа смещается в сторону положительных потенциалов; одновременно с этим потенциал саморастворения железа в серной кислоте становится более электроотрицательным. В таком случае из-за изменения ^-потенциала процесс ионизации железа затрудняется.
§ 7. Анодное растворение сплавов
В неоднородном сплаве, состоящем из двух или более кристаллических фаз, все эти фазы электрохимически самостоятельны. Потенциал ионизации каждой из них определяется ее собственными физико-химическими свойствами, и растворяться фаза может только по достижении этого потенциала.
Естественно, что в первую очередь при наложении анодного тока растворяются наиболее электроотрицательные кристаллы, и только после их полного удаления или в случае очень высокой поляризации могут растворяться включения благородных металлов. На практике, если электроотрицательная фаза ионизируется без больших затруднений и ее относительное содержание в сплаве велико, то потенциал часто не достигает величины, необходимой для растворения благородных включений, и они в виде мелких частичек шлама выпадают на дно ванны (например, при растворении медных анодов).
Если электроотрицательный компонент сплава растворяется со значительной поляризацией, то с ростом плотности тока анодный потенциал может сдвинуться ® положительную область столь заметно, что станет возможным анодное растворение и более электроположительного компонента.
Однофазные сплавы, представляющие собой химическое соединение или твердый раствор двух различных металлов, ведут себя при анодной поляризации как единое целое. Активность электроотрицательного металла в таком сплаве, как правило, оказывается ниже, чем в свободном состоянии, а потенциал анодного растворения сплава устанавливается в промежутке между потенциалами основных компонентов (обычно ближе к более отрицательному). В зависимости от особенностей системы и условий электролиза установится и соотношение, в кото-
408 Теоретические основы электрохимии
ррм компоненты твердого раствора будут переходить в электролит. Возможен случай, когда равновесный потенциал одного из компонентов сплава, переходящих в раствор, окажется более положительным, чем потенциал анода, при этом можно наблюдать контактное выделение на аноде более электроположительного металла. Благородный металл будет выделяться в форме губчатого рыхлого осадка. %
§ 8. Реакции анодного окисления
Реакции ионизации металлов — не единственный пример анодных процессов. Большую группу составляют реакции анодного окисления анионов, катионов или молекулярных веществ. Некоторые из этих реакций типа
Fe2+ — e->Fes+ протекают на электроде без больших затруднений и характеризуются в обычных условиях главным образом концентрационной поляризацией.
Другие реакции, среди которых следует в первую очередь назвать выделение кислорода [см., например, (XIV, 9) и (XIV, 10)], безусловно идут с высоким перенапряжением. В практике электролиза такие реакции встречаются не реже, чем реакция электролитического выделения водорода, но изучены они гораздо слабее. Для них также характерна логарифмическая зависимость перенапряжения от плотности тока. Однако воспроизводится эта зависимость значительно хуже и угол наклона соответствующих полулогарифмических прямых не остается постоянным в таких широких пределах изменения плотности тока, как в случае выделения водорода. Связано это, по-видимому, с тем, что при потенциалах протекания реакций с выделением кислорода поверхность металлических электродов в той или иной мере окислена, а перенапряжение сильно зависит от степени окисления.
Поверхностным окислам в процессе выделения кислорода большинство авторов отводит важную роль. Еще Слугиновым была высказана мысль, что этот процесс идет через стадию образования нестабильного высшего окисла. В настоящее время эта теория находит экспериментальное подтверждение, в частности в работах В. И. Веселовского и сотрудников. Сложность подобного механизма анодного окисления делает понятной недостаточную изученность процесса в целом. Лимитирующей стадией подобных реакций является при малых токах распад промежуточного окисла. С повышением плотности тока контроль переходит к стадии разряда молекул воды или ионов ОН'. Весьма интересно наблюдаемое на опыте повышение перенапря
Анодные процессы
409
жения кислорода t)Oj при введении в раствор добавки легко адсорбирующихся анионов (F-, С1~ и т. д.). Такими добавками пользуются, например, для увеличения выхода по току при электролитическом окислении сульфата в персульфат.
Перенапряжение кислорода обнаруживает зависимость и от материала анода. Судя по имеющимся данным, ряд металлов, расположенных в порядке возрастания tjOs, приближенно обратен такому же ряду для реакций электролитического выделения водброда. Так, например, выделение водорода на платинированной платине сопровождается очень небольшим перенапряжением, а кислород, наоборот, выделяется при большом перенапряжении. Этот факт можно сопоставить с тем, что по энергии хемосорбции водорода и кислорода металлы в общем и целом также располагаются во взаимно обратном порядке.
Так как в большинстве случаев процесс выделения кислорода на аноде сопровождается окислением материала анода, при длительном электролизе разряд анионов протекает не на металле, а на окисленной поверхности электрода. Естественно полагать, что на т)Оа существенно влияют особенности и природа поверхностных окислов. С практической точки зрения важна также реакция анодного выделения хлора. В теоретическом отношении эта реакция недостаточно изучена. По данным С. В. Горбачева и Н. П. Жука, перенапряжение выделения хлора в области малых плотностей тока (до 10-3 а]см2) приблизительно линейно связано с логарифмом плотности тока, но затем зависимость усложняется.
ЛИТЕРАТУРА
Бонгхоффер К. Ф. Об активации пассивного железа. Труды четвертого совещания по элёктрохимии. Изд. АН СССР, 1959.
Данков П. Д., Игнатов Д. В., Шишаков Н. А. Электронографические исследования окисных и гидроокисных пленок на металлах. Изд. АН СССР, 1953.
Данков. П. Д. Труды П конференции по коррозии металлов, т. II. Изд. ДН СССР 1044 121
Кабанов Б.’ Н. ДАН СССР, 58, № 8, 1947.
Ко л отырки н Я. М., К н я ж е в а В. М. ЖФХ, 30, 1956, 1990; ДАН СССР, 114, № 6, 1957, 1265.
Колотыркин Я. М., Флорианович Г. М. Пассивация металлов. Химическая наука и промышленность, № 4, 1958, 483.
Левин А. И., Простаков М. Е., Суслошаров Г. Д. ДАН СССР, 129, № 3, 1959, 617.
Левин А. И., Простаков М. Е., Кочергин В. П. ЖФХ 34, № 5, 1960, 1117.
Новые проблемы современной электрохимии, под. ред. Дж. Бокриса, ИЛ, 1962, 284.
Самарцев А. Г. Оксидные покрытия на металлах. Изд. АН СССР, 1944. Эванс Ю. Р. Коррозия и окисление металлов. Машгиз, 1962.
Э р ш л е р Б. В. ДАН СССР, 37, № 6, 1942, 26.
Fink С. G„ Handbook of Nonferrous Metallurgy, New York, 1945.
Глава XV
КОРРОЗИЯ МЕТАЛЛОВ
§ 1. Общие сведения •
Коррозией называется явление самопроизвольного разрушения металлов или сплавов, вызываемого химическим или электрохимическим воздействием на них окружающей среды. Масштабы потерь металлов в результате коррозионного разрушения исключительно велики. Согласно статистическим данным, потери металлов от коррозии составляют одну треть от их производства. Безвозвратные потери металлов приближаются к 10% от ежегодного их выпуска. Использование металлов в современной технике непрерывно расширяется, особенно широко применяются новые конструкционные металлы и сплавы, при этом значительно усложняются условия их использования. Вот почему в настоящее время проблемы защиты металлов и сплавов против коррозии, установления наиболее рационального режима их эксплуатации являются проблемами первостепенного народнохозяйственного значения.
Все многообразие коррозионных явлений в зависимости от механизма их протекания можно разделить на две группы: химическую и электрохимическую коррозию.
Химическая коррозия протекает в газах и неэлектролитах и подчиняется законам химической кинетики. Электрохимическая коррозия обычно возникает при контакте металла с электролитом и подчиняется законам электрохимической кинетики. В реальных условиях эксплуатации металлов химическая и электрохимическая коррозия могут протекать и одновременно.
Электрохимическая коррозия — наиболее распространенный вид коррозионного разрушения металлов и сплавов. Основные законы, по которым протекает электрохимическая коррозия,— законы электрохимической кинетики, поэтому именно этот вид коррозионного разрушения металлов является важнейшим объектом электрохимических исследований.
Коррозия металлов
411
§ 2. Теория коррозии
Согласно классической теории электрохимической коррозии, основоположником которой является швейцарский ученый деля Рив, коррозионное разрушение металла происходит в результате работы многочисленных микроэлементов, возникающих на поверхности металла либо сплава при контакте его с электролитом. Однако следует иметь в виду, что возникновение и работа коррозионных микроэлементов не являются единственной причиной коррозии. Коррозия может протекать при отсутствии микроэлементов на совершенно гомогенной в электрохимическом отношении поверхности. Истинной причиной коррозии является термодинамическая неустойчивость металла в данных условиях. Образование и работа коррозионных микроэлементов — это наименее энергоемкий из возможных путей перехода системы из термодинамически неустойчивого состояния в устойчивое. Большинство используемых в технике металлов находится в термодинамически неустойчивом состоянии; мерой их неустойчивости является изменение свободной энергии.
Однако изменение свободной энергии характеризует лишь потенциальную возможность, меньшую или большую тенденцию перехода металлов в ионное (термодинамически устойчивое) состояние и практически ничего не говорит о действительных скоростях этого перехода. Несмотря на то что повлиять на термодинамическую устойчивость металла не представляется возможным, все же, вскрыв основные закономерности коррозионного процесса, можно показать весьма существенное влияние на скорость коррозии, затормозить ее.
Де ля Рив полагал, что причиной электрохимической гетерогенности поверхности, в результате которой возникают пространственно разделенные катодные и анодные участки коррозионных микроэлементов, является присутствие в металле примесей других более «благородных» металлов. Эти включения совместно с окружающим чистым металлом образуют так называемые местные или микрогальванические локальные элементы, в которых чистый металл служит анодом, а включения — катодами.
Однако, как уже отмечалось, наличие микропар следует рассматривать не в качестве первопричины возникновения коррозионного процесса, а только лишь как один из возможных путей. Г. В. Акимов показал, что поверхность корродирующего металла можно представить себе как более или менее сплошную систему микро- и макрокоррозионных пар. Причиной электрохимической микрогетерогеннбсти может служить любая структурная неоднородность деформации и внутренних напряжений. Коррозия может также возникнуть в результате неод-
412
Теоретические основы электрохимии
породности внешней среды, различия в концентрации электролита и т. п. Поэтому, характеризуя какой-либо металл, нельзя говорить о его коррозионной стойкости, не указывая среду, где он будет служить.
В последнее время была показана термодинамическая вероятность растворения чистых металлов и сплавов без участия микропар, что подтверждается рядом 'Примеров. Так, основываясь на теории коррозионных микропар, особо чистые металлы и амальгамы (натрия, цинка и др.) не должны быть растворимы даже в кислой среде: они однофазны, имеют гомогенно-электрохимическую поверхность. Однако из опытов А. Н. Фрумкина следует, что амальгамы растворяются вполне закономерно. Поэтому катодные и анодные процессы могут быть совмещены на одной однородной фазе и при ее изопотенциальности.
Впервые такая точка зрения на коррозионные процессы была высказана и обоснована А. И. Шултиным. Он предлагает рассматривать растворение .металлов с выделением Н2 как электрохимический обмен, подобный замещению в растворе ионов меди железом, обоснование которого не усложняется представлением о локальных элементах. Шултин предложил следующий механизм растворения: при соприкосновении металла с раствором часть ионов металла, составляющая его кристаллическую решетку, переходит в раствор, оставляя металлическую поверхность заряженной отрицательно; возникающий двойной электрический слой, внешнюю обкладку которого в первый момент составляют перешедшие в раствор ионы металла, через некоторое время может прекратить дальнейшее растворение. Однако в результате кинетического взаимодействия раствора часть ионов металла может быть заменена в двойном слое другими, присутствующими в растворе катионами. Если они имеют менее отрицательную природу, то неизбежно должны будут разрядиться и тем самым вызвать продолжение процесса растворения. Таким образом, роль «постороннего включения» может сводиться не к образованию элементов, а к облегчению катодной реакции вследствие понижения т)н на них.
Дальнейшее развитие этих представлений связано с работами Вагнера и Трауда, которые предложили рассматривать суммарный процесс растворения чистого металла как результат протекания двух или нескольких независимых электрохимических реакций, связанных между собой величиной скачка потенциала и распределенных на поверхности со статистической беспорядочностью.
А. Н. Фрумкин рассмотрел вопрос о роли неоднородности металлической поверхности и условиях нарушения эквипотен-циальности ее и установил возможность применения закона электрохимической кинетики к реакциям, протекающим на по
Коррозия металлов
413
верхности растворимого металла. Это открыло перспективу создания количественной теории коррозии, способной заранее оценить поведение металла в условиях саморастворения на основании его поляризационных характеристик, а следовательно, природы металла и состава электролита. В свете развитых представлений зависимость величины стационарного потенциала <рст и скорости растворения металла от состава раствора и других условий опыта должна быть следствием зависимости катодных и анодных реакций, составляющих сопряженный процесс саморастворения.
Учитывая различную природу этих реакций, естественно ожидать, что они по-разному будут реагировать на изменение состава и концентрации, состояние поверхности металла и других свойств системы. В частности, заранее можно предвидеть, что повышение или понижение концентрации Н+ будет преимущественно сказываться на кинетике реакций, относящихся к ионам водорода, в то время как изменение состояния поверхности металла может приводить к изменению скорости выделения водорода и скорости ионизации металла. Вместе с тем если состав раствора остается постоянным, то кинетика рассматриваемых реакций должна определяться природой металла.
Работа коррозионного элемента схематически 1представлена на рис. 128.
Следовательно, электрохимическое растворение металла состоит из трех основных стадий (процессов):
1) анодного процесса—• перехода ионов корродирующего металла в раствор под влиянием гидратации (сольватации) с освобождением электронов:
Me + пН2О —> Мег+ nH2O + ze;
2) омического процесса—перетекания освободившихся электронов по металлу от анодных участков к катодным и движения ионов в растворе;
3) катодного процесса — ассимиляции освободившихся электронов каким-либо деполяризатором, имеющимся в растворе.
В качестве деполяризатора могут выступать любые ионы и молекулы, находящиеся в растворе и способные восстанавливаться на катодных участках:
D -|- ze [Dze]. (XV, 1)
При детальном изучении коррозии металлов в электролитах есть смысл подразделить деполяризаторы, которые в этих /процессах участвуют, по крайней мере, на две группы. В одну из них входят ионы водорода, металлические ионы высшей валентности (например, Fe3+, Се4+ и др.), а также некоторые анионы, .которые могут отнимать у металла валентные электроны, но,
414
Теоретические основы электрохимии
как правило, не способны вступать с ним в химическое соединение. Основу второй группы составляют кислород и все соединения и ионы, способные его отщеплять, а также галогены.
Вещества первой группы могут быть только деполяризаторами электрохимических коррозионных процессов. Вещества второй группы могут, кроме того, при определенных условиях, реагировать с металлом химически с непосредственным образованием окисных, галоидных и т. п. пленок. Это может существенно повлиять на механизм, а подчас и на кинетику коррозионных процессов.
Металл
Злектролит
Рис. 128. Принципиальная схема работы коррозионного гальванического элемента
Самопроизвольный процесс, сочетающий в себе анодную реакцию растворения металла и катодную реакцию восстановления окислителя-деполяризатора (каждая из которых идет за счет другой), носит название электрохимической коррозии металла.
§ 3. Коррозионные диаграммы
Кинетические характеристики электрохимических коррозионных процессов удобно представлять графически в виде совокупности поляризационных кривых катодного и анодного процессов. Принцип построения коррозионных диаграмм, которые часто называют по имени автора этого метода диаграммами Эванса, легко уяснить из рис. 129. По оси ординат откладывают потенциалы анодных и катодных процессов (<ра и фк), по оси абсцисс — соответствующие анодные и катодные токи.
Основное условие стационарного протекания процесса коррозии заключается в равенстве катодного и анодного токов. Это
Коррозия металлов
415
равенство устанавливается автоматически, ибо если один из процессов протекает быстрее, то баланс зарядов нарушается и потенциал сдвигается так, что этот процесс тормозится, а противоположный — ускоряется.
Нетрудно заметить, что механизм установления стационарного (<рСт), или, как его еще называют, компромиссного потен
циала, во многом сходен с механизмом установления равновес-
ного потенциала. Но это сходство неполное. Если единственным электрохимически активным компонентом раствора являются собственные ионы металла, то заряды через границу раздела могут переноситься только за счет этих ионов и одновременно с балансом зарядов обеспечивается и баланс вещества, т. е. полное электрохимическое равновесие между металлом и электролитом. Если же, помимо ионов металла, в переносе зарядов участвует и другой компонент (окислитель), то при полном балансе зарядов баланс вещества, а значит, и равновесие могут оказаться нарушенными. Вследствие поглощения
Рис. 129. Схема построения коррозионной диаграммы Эванса
электронов металла окисли-
телем будет происходить непрерывный и необратимый переход
ионов металла в раствор.
Если по сравнению с катодной и анодной поляризацией омическое падение напряжения в электролите мало, то стационарный потенциал практически одинаков для всей поверхности и может быть найден простым пересечением анодной и катодной поляризационных кривых (точка С).
Если же омическое сопротивление электролита значительно, а поверхность неоднородна, то потенциалы катодных и анодных участков (точки D и Е) должны отличаться на величину омических потерь, которые можно обозначить символом ~EiR.
В зависимости от того, на что расходуется основная часть свободной энергии или э. д. с. полной коррозионной реакции (?а—Фк)» принято различать процессы по их лимитирующим, или, как чаще говорят, контролирующим стадиям. Контроль может быть анодным т]а > 1]к + tdR, катодным цк > т]а + 'ZiR, омическим SzT? > т]К + ца или же смешанным. В особую группу могут быть выделены процессы с диффузионным контролем, в которых наибольшие затруднения связаны с диффузионной доставкой окислителя или удалением коррозионных продуктов.
416 Теоретические основы электрохимии
Степенью контроля процесса коррозии данной ступени называют долю торможения этой ступени по отношению к суммарному торможению всех ступеней процесса. Для уменьшения скорости коррозии наиболее эффективным является воздействие на контролирующий фактор.
Изучить процесс коррозии — это означает прежде всего вскрыть контролирующий фактор коррозии и установить механизм протекания его. Только при этом условии можно выбрать наиболее действенный путь борьбы с коррозией или оценить коррозионную стойкость какого-либо сплава в той или иной агрессивной среде.
В ряде случаев бывает трудно заранее сказать, какая из ступеней коррозионного процесса является ограничивающей. Поскольку величина поляризации характеризует степень торможения соответствующего электродного процесса, то ограничивающий фактор коррозии можно установить, исследуя характер коррозионных диаграмм поляризации.
Диаграммы Эванса изучаются на модели микроэлемента, представляющей собой короткозамкнутый коррозионный элемент с электродами, имеющими значительно большие размеры, чем у реально 'существующих микропар.
Исследуемый микроэлемент постепенно поляризуется током, который поступает от внешнего источника тока либо возникает в самом коррозионном элементе при замыкании электродов.
С уменьшением омического сопротивления внешней цепи (/?внешн) возрастает величина коррозионного тока и поляризуемость электродов. Однако при этом величина коррозионного тока никогда не достигнет максимума, поскольку даже при /?внешп = 0 еще остается внутреннее омическое сопротивление коррозионного элемента. При исследованиях предпочитают пользоваться внешним источником поляризующего тока, поскольку при этом можно непосредственно из опыта определить максимальный коррозионный ток и получить хорошую воспроизводимость результатов благодаря возможности поддержания постоянной величины тока. Полученные для- исследуемых условий поляризационные кривые наносят на диаграмму поляризации. По мере повышения величины тока в результате явления поляризации потенциалы анода и катода сближаются и, если омичеокое сопротивление коррозионного элемента будет равно нулю (/? = 0), через элемент потечет максимальный коррозионный ток /Макс- При этом потенциалы катода и анода сравняются и поляризационные кривые пересекутся (точка С). В таком случае процесс коррозии идет на изопотенциальной поверхности, т. е. при условии, когда различные участки системы имеют оди-
Коррозия металлов
417
наковый потенциал (<рж). Однако нельзя забывать, что равенство (потенциалов достигнуто при этом благодаря поляризации электродов при работе микроэлемента. Если же изопотёнциаль-ность поверхности есть результат равенства начальных потенциалов, то микроэлементы не возникнут и процесса коррозии не будет.
Графический метод расчета скорости коррозии позволяет в отличие от аналитического рассчитать величину корразии для весьма сложных случаев, соответствующих реальным условиям протекания коррозионного процесса.
Знание потенциала позволяет вычислить величину катодной и анодной поляризации и, следовательно, определить степень контроля.
Рассмотрим с помощью диаграмм Эванса несколько характерных примеров коррозии с водородной деполяризацией, где катодный процесс изображается схемой
2Н+ + 2е -+ Н2, (XV,2)
и с кислородной деполяризацией, которая может быть выражена реакцией
[О] + Н2О + 2е -> 2ОЕГ (XV,3)
или
[О] + 2Н+ + 2е -> Н2О (XV,4)
(здесь символом [О] изображается кислород — элементарный и входящий в состав окислителя).
(При водородной деполяризации на поверхности металла почти всегда имеются пузырьки водорода, в которых давление очень близко к атмосферному. В таких условиях равновесный потенциал водородных катодов выражается уравнением
<Р° = -^1п[Н+]в. (XV,5)
Расчеты показывают, что этим, уравнением без заметной погрешности можно пользоваться тогда, когда пузырьки водорода отсутствуют и он удаляется целиком, растворяясь -в жидкости.
При потенциале (XV, 5) подавляющее большинство металлов не пассивируется. Растворение их в кислых неокислительных растворах идет обычно с относительно небольшой поляризацией. Перенапряжение же водорода редко бывает малым. Поэтому для процессов коррозии с водородной деполяризацией наиболее характерен катодный контроль (рис. 130, катодная кривая /).
27 Заказ 1338
418
Теоретические основы электрохимии
Наиболее общий путь замедления 'процесса коррозии при этом — дальнейшее повышение перенапряжения катодного процесса (катодная кривая 2).
На практике это чаще всего достигается вводом в раствор некоторых органических поверхностно активных веществ, которые, адсорбируясь на катодных участках, повышают энергию активации разряда Н3О+-ионов. Эти вещества, известные под названием ингибиторов коррозии, или правильных присадок, часто позволяют сократить корозионные потери в десятки и сотни раз.
Иногда аналогичный эффект получается в результате контактного осаждения на корродирующей поверхности тончайшего
Рис. 130. Диаграмма Эванса для случая активной коррозии с катодным контролем. Катодная поляризация:
1 — при нормальном состоянии поверхности (ток растворения /^); 2 — прн содержании в растворе веществ, повышающих перенапряжение катодного процесса I ; 3 — при активных катодных включениях на поверхности металла (ток достигает *крит, металл пассивируется н ток падает до ‘с'^
Рнс. '131. Диаграмма Эванса, иллюстрирующая возможность улучшения 1пассивируемости сплава с помощью катодных добавок. Катодные кривые:
1 — для сплава, не содержащего ка-тодных включений (ток растворения i ); 2 — после введения катодной примеси (ток растворения увеличен до
I ); 3 — при большой эффективности катодов (ток достигает *крит, металл пассивируется н ток падает
ДО )
слоя металла, обладающего собственным высоким перенапряжением водорода (ртуть, свинец и т. и.). С этой целью, например, в электролит некоторых гальванических элементов вводят очень малую добавку ртутной соли, обеспечивающую амальгамирование цинковых электродов, а цри катодном травлении
Коррозия металлов
419
стали по методу Булларда — Дана в кислоту вводят примесь ионов свинца.
'Появление в структуре металла включений с малым водородным перенапряжением в общем случае оказывает обратное действие и приводит к усилению коррозии (рис. 130, катодная кривая 3).
Однако из этого правила есть исключения и катодные примеси иногда специально вводят в металл с целью повышения его стойкости в кислоте. Такие, на первый взгляд парадоксальные соотношения наблюдаются в случае нержавеющих сталей, которые растворяются с заметной анодной поляризацией и потенциал пассивации которых (<рПасс) лежит немного отрицательнее равновесного водородного потенциала (рис. 131). Здесь снижение перенапряжения водорода после некоторого повышения скорости растворения металла (катодная кривая 2) может привести к тому, что стационарный потенциал поверхности достигнет точки фпасс (катодная кривая 3) и металл запассиви-руется.
Добавка меди, обладающей сравнительно малым перенапряжением водорода, действительно применяется для тех нержавеющих сплавов, которые должны работать в неокислительных средах. Теоретическое освещение этот вопрос получил в работах Я. М. Колотыркина и Н. Д. Томашова.
Процессы с кислородной деполяризацией отличаются от рассмотренных рядом особенностей. Во-первых, благодаря более высокой окислительной способности эти деполяризаторы могут вызвать коррозию таких металлов, которые не вытесняют из раствора водород. В слабокиолых и особенно нейтральных и щелочных растворах это значительно расширяет круг .металлов, которые могут подвергаться коррозионным воздействиям. Во-вторых, увеличение свободной энергии, а значит, и движущей силы процесса приводит к тому, что скорость растворения металла с 'кислородной деполяризацией бывает во много раз больше, чем с водородной. В-третьих, высокий потенциал катодного процесса делает гораздо более вероятным наступление пассивного состояния, а иногда создает возможность и для перепассивации металлов.
Наконец, для кислородной деполяризации гораздо чаще, чем для водородной, бывают характерны диффузионные ограничения. Это отчетливо видно на примере деполяризации молекулярным кислородом, концентрация которого в водном растворе довольно мала.
На рис. 132 представлен типичный ход катодной кривой для этого случая (катодная кривая /). Здесь, как легко видеть, перенапряжение анодного и катодного процессов практически не влияет на скорость коррозии. Зато она может сильно расти под 27*
420
Теоретические основы электрохимии
влиянием движения жидкости (катодные кривые 2 и 3). Если металл склонен к пассивности, то следствием перемешивания иногда оказывается достижение критического «потенциала фпасс и пассивирование. Такие явления на примере хрома, магния и некоторых других металлов впервые наблюдал В. А. Ки-стяковский, давший им название мотоэлектрохимических.
Подобное влияние на коррозию оказывает и повышение концентрации окислителя. Типичным примером этого служит коррозия железа в азотной кислоте. Сначала с повышением 'концентрации HNO3 коррозия очень сильно растет, а затем резко падает в результате пассивации. Такая же картина наблюдается в растворах хромовой кислоты и некоторых других окислителей.
Рис. 1132. Диаграмма Эванса для случая активной коррозии с диффузионным контролем. «Катодные кривые:
I — при отсутствии принудительного движения; 2 и 3 — при различной интенсивности перемешивания
Рис. 133. Диаграмма Эванса:
1 — малое перенапряжение ка-тодного акта (металл й состоянии перепассивации); 2 — большое перенапряжение катодного акта (металл пассивен)
На рис. 133 представлена диаграмма Эванса для случая, когда металл, корродирующий под действием очень сильного окислителя, находится в состоянии перепассивации (катодная кривая J).
. Видно, что коррозия могла бы быть сильно уменьшена, а сам металл возвращен в нормальное пассивное состояние, если бы в раствор или на поверхность можно было ввести какой-либо ингибитор, повышающий перенапряжение катодного процесса (Катодная кривая 2). Существуют ли подобные ингибиторы, пока неизвестно. Во всяком случае обычные органические поверх
Коррозия металлов
421
ностно активные вещества здесь непригодны, ибо в 'сильных окислительных растворах они разрушаются.
Бороться с явлением перепассивации металла 'можно, понижая окислительные свойства среды или принудительно сдвигая потенциал в область, соответствующую нормальной пассивности.
В любом из рассмотренных примеров вся поверхность металла в равной мере находилась под воздействием агрессивных компонентов раствора. Возможен, однако, случай, когда окислитель распределен в электролите неравномерно, так что к некоторой части корродирующей поверхности он поступает в меньшем количестве, чем к другим, а то и не поступает вовсе. Чаще всего это наблюдается при коррозии с деполяризацией за счет растворенного кислорода, диффузионная доставка которого в углубления, отверстия, щели и т. п. особенно затруднена.
В местах, куда окислитель не поступает, коррозия, на первый взгляд, должна отсутствовать. Однако так как металл и раствор обладают электропроводностью, протекающий с некоторым перенапряжением анодный процесс ионизации металла не локализуется только на тех местах, где идет сопряженный процесс восстановления кислорода, а распространяется/ и на смежные с ними участки. Более того, нередко на местах, легко доступных для кислорода, возникают пассивные пленки; иногда анодный процесс, а значит, и разрушение металла практически целиком сосредоточиваются на участках, не подвергающихся аэрации. Развивается так называемая коррозия с дифференциальной аэрацией, которая возможна при участии не только кислорода, но и других окислителей, способных вызывать пассивность металла.
§ 4. Методы коррозионных испытаний
Лабораторные коррозионные испытания но своей конечной цели могут быть подразделены на следующие группы:
1) исследование механизма корозии;
2) выбор наиболее стойкого и подходящего материала для данной агрессивной среды;
3) определение сред, в которых данный материал может быть применен;
4) оценка свойств новых материалов, сплавов и т. и.;
5) разработка способов контроля при выпуске однотипной продукции.
Для разработки «ускоренного испытания» проводили наблюдения в брызгах раствора соли, в атмосферной камере, при полном погружении в растворы кислот и многие другие. Представ
422
Теоретические основы электрохимии
лялось перспективным найти такое ускоренное испытание, которое давало бы за короткое время характеристику поведения различных материалов, пригодную для оценки их стойкости во время службы.
Значительное ускорение испытаний достигается при переменных условиях коррозии, когда образец некоторое время находится в растворе, а затем извлекается на воздух и коррозия протекает, в контакте с пленкой влаги.
Во влажной камере-образцы непрерывно или периодически обрызгивают 3%-ным раствором NaCl, применяют также камеры тепла и влаги, где воспроизводятся условия субтропиков либо имитируется тропическая атмосфера.
В качестве основных количественных показателей коррозии можно отметить следующие: определение числа коррозионных центров, изменение толщины образца, определение прибыли или убыли в весе.
К количественным методам относятся также количественный анализ пробы раствора, определение изменения механических свойств образца, Определение количества выделяющегося водорода или поглощенного кислорода, определение электрического сопротивления образца и др.
Наиболее простой лабораторный способ определения коррозионной стойкости — испытание образцов в открытом сосуде. В таком случае образцы подвешивают .на стеклянные подвески или нити из инертного материала. Первоначальные, ориентировочные лабораторные испытания выбранных материалов должны быть как можно более простыми, причем желательно по возможности воспроизвести среду и условия, в которых этот материал будет служить. Иногда рекомендуется непосредственно пользоваться существующим оборудованием и агрессивными средами для испытания небольших пробных образцов .в производственных условиях. Часто такие испытания более ценны для предварительного выбора материалов. Следующей ступенью является испытание небольших моделей аппаратуры, изготовленной из материалов, выбранных в результате предварительных испытаний. Все факторы, действующие при коррозии обычной аппаратуры, должны быть тщательно воспроизведены .при испытании модели.
Природа коррозии сложна и условия, с которыми приходится считаться, разнообразны; поэтому особое внимание следует уделять факторам, определяющим воспроизводимость результатов опыта. В основном эти факторы делятся на две группы: связанные с состоянием металлической поверхности и с окружающей средой. Чистота поверхности металла, наличие и природа поверхностных пленок, присутствие различных структурных составляющих в сплаве, подготовка поверхности перед ис-
Коррозия металлов
423
пытанием и поляризуемость металла — это те моменты, на ко-, торые следует обратить внимание. Состав среды, ее однородность, загрязненность и свобода доступа воздуха определяют ее агрессивность. Все, что может повлиять на электрохимический процесс коррозии, будет также влиять и на скорость и характер коррозионного разрушения.
Для получения воспроизводимых результатов следует соблюдать осторожность при приготовлении и испытании образцов и при измерении степени разрушения. Однако даже и при тщательной работе нельзя ожидать .полной воспроизводимости: существует много факторов за пределами контроля экспериментатора. Следовательно, абсолютная ошибка измерения может быть уменьшена только с увеличением числа испытуемых параллельных образцов.
Характер коррозии может сильно влиять на воспроизводимость результатов. При равномерной коррозии воспроизводимость в общем значительно лучше, чем при точечной или местной коррозии.
Для получения большей точности .в последнем случае надо испытывать возможно большее число образцов.
§ 5- Методы борьбы с коррозией
В настоящее время применяются многие методы борьбы с коррозией, главные из них следующие:
1) защитные покрытия (металлические и неметаллические);
2) обработка и изменение состава среды либо состава металла (сплава);
3) электрохимические и электрические методы защиты.
Защитные .покрытия — это слои, искусственно создаваемые на поверхности металла с целью предохранения его от коррозии. Металлические покрытия часто выбирают с таким расчетом, чтобы по отношению к защищаемому металлу их электродный потенциал был более отрицательным (так называемые анодные покрытия). Так, например, цинковое покрытие во влажном воздухе и в воде анодно по отношению к железу. В этом случае поры, трещины, царапины и т. п. оказываются неопасными, так как цинковое покрытие электрохимически защищает железо в местах обнажений.
Разделение покрытий на анодные и катодные зависит от внешних условий. В условиях атмосферной коррозии олово по отношению к железу является катодным покрытием. Однако в присутствии органических кислот (консервы) и без доступа кислорода олово по отношению к железу и его сплавам выполняет роль анодного покрытия.
424
Теоретические основы электрохимии
Наиболее доступный способ защиты от коррозии — неметаллические, в частности лакокрасочные, покрытия. В состав лакокрасочных покрытий часто вводят пигменты, являющиеся замедлителями (ингибиторами) коррозии.
Хорошее защитное действие пленок, получаемых на поверхности металлов в результате их химического воздействия с окружающей средой, привело к применению методов искусственного образования или усиления таких пленок для повышения сопротивления коррозии. Наряду с окисными пленками создают пленки окиснохроматные, фосфатные, сульфидные и др. Оксидирование (воронение) стали и железа осуществляют погружением изделий в ванны с очень концентрированным раствором щелочи, в который добавлены окислители (MnO2, NaNO2). Широкое распространение получило анодное окисление (анодирование), осуществляемое в присутствии окислителя или при последующей дополнительной обработке им. Таким путем достигается, например, усиление окисной пленки на алюминии в изделиях, предназначенных для эксплуатации в более жестких условиях.
Изменение состава среды достигается введением в состав внешней среды таких веществ, которые замедляют коррозию (замедлители коррозии), обработка среды может применяться лишь при ограниченном объеме раствора. Примером этого вида защиты является удаление кислорода из питающей воды для паровых котлов.
Наряду с другими областями применения замедлители коррозии широко .используют при химических методах очистки черных металлов от окалины и ржавчины. Так как замедлители коррозии уменьшают скорость растворения в кислоте самого металла, но не уменьшают скорости растворения ржавчины или накипи, то применение их в этих случаях значительно облегчает процесс.
Действие замедлителей коррозии в этих случаях объясняется тем, что они хорошо адсорбируются на поверхности самого металла, но не его солей или окислов.
Хорошо известно, что различные добавки, вводимые в состав специальных (легированных) сталей, неодинаково влияют на их коррозионную стойкость в различных условиях. Широкое применение получила хромоникелевая сталь и другие марки нержавеющей стали.
Если подвести к корродирующему металлу ток от внешнего источника и подвергнуть его катодной поляризации, то электроны, потребляемые в ходе катодного акта, будут пополняться не только в результате ионизации металла, но и из внешней цепи; в таком случае будет осуществляться электрическая защита от коррозии.
Коррозия металлов
425
Суммарный анодный ток через корродирующую поверхность окажется меньше катодного на величину внешнего катодного тока:
/о — А— /к внешн- (XV,6}
Может показаться, что на-эквивалентную величину должна уменьшиться и скорость растворения металла. Однако в общем случае это неверно. Хотя при самопроизвольной электрохимической коррозии катодный и анодный процессы тесно связаны между собой и идут один за счет другого, они все же в значительной мере самостоятельны. Скорость каждого из них является вполне определенной функцией потенциала и при принудительной поляризации извне обе эти функциональные зависимости должны проявляться. В. М. Новаковский показал, что уменьшение коррозии металла в п раз требует наложения на него катодного тока, в п раз превышающего ток самопроизвольного растворения. Поэтому в условиях активного растворения, когда катодный процесс лимитируется актом разряда (например, при водородной деполяризации), наложенный катодный ток, соизмеримый с ’ током самопроизвольного растворения, практически не может защитить металл от коррозии. Наоборот, катодная защита может быть эффективной, если лимитирующей стадией катодного процесса является диффузия окислителя, например растворенного кислорода. В этом случае наложенный катодный ток практически целиком идет на сокращение анодного тока. На практике катодная защита широко применяется для стальных конструкций в почве, морской воде и других нейтральных растворах солей. Для меди она может с успехом использоваться и в сильнокислых растворах (не обладающих собственными окислительными свойствами). При катодной защите вместо обычных источников тока иногда пользуются так называемыми протекторами, т. е. электродами из электроотрицательного металла (цинка, магния и т. п.). Они электрически соединяются с защищаемой конструкцией и образуют вместе с ней в коррозионной среде гальванический элемент, где защищаемый металл служит катодом. В таком варианте катодную защиту именуют протекторной.
Новой областью применения катодной или протекторной защиты, на которую было обращено внимание в исследованиях В. П. Батракова, является предохранение металла от коррозии в условиях его перепассивации. Возможность такой защиты и притом очень малыми токами подтверждается практическим опытом других исследователей.
В последнее время в работах В. М. Новаковского и А. И. Левина, а также Н. Д. Томаипова было показано, что при определенных условиях защита металла может быть осуществлена
426 Теоретические основы электрохимии
применением не катодной, а анодной поляризации. Подобная анодная защита может обеспечить образование и сохранение на металле пассивной пленки в условиях, когда самопроизвольная пассивация не происходит (железо в концентрированной серной кислоте, нержавеющая сталь в неокислительных кислотах)?
ЛИТЕРАТУРА
Акимов Г. В. Основу учения о коррозии металлов. Металлургиздат, 1946. Бахвалов Г. Т., Турковская А. В. Коррозия и защита металлов. Металлургиздат, 1959.
Наваковский В. М, Левин А. И. ДАН СССР, 94, 6, 1954, 1113.
Новаковский В. М., Соколова Л. А. Труды Уральского научно-исследовательского химического института, № 9, Л. Госхимиздат, 1960.
II о п и л о в Л. Я. Гальванопластика. Машгиз, 1961.
Путилова И. Н., Балезин С. А., Баранник В. П. Ингибиторы коррозии. Госхимиздат, 1958.
Розенфельд И. Л. Замедлители коррозии в нейтральных средах. Изд. АН СССР, 1953.
Фрумкин А. Н. Труды II конференции по коррозии металлов. Изд. АН СССР, 1940, стр. 5.
Федотьев Н. П., Грилехис С. Я. Электрохимическое травление и оксидирование металлов. Машгиз, 1957.
Эванс Ю. Р. Коррозия, защита и пассивность металлов. ИЛ, 1962.
Эванс Ю. Р. Коррозия и окисление металлов. Машгиз, 1962.
ОГЛАВЛЕНИЕ
Стр.
Предисловие ..................... ................................ 3
Введение .......................................................... 5
Глава I Предмет и’ содержание электрохимии.......................... 9
§ 1. Организация электрохимического процесса ................ 9
§ 2. Закон Фарадея ..................................... . . 16
§ 3. Электровесовой метод анализа —кулонометрия............. 24
Глава II Движение ионов под действием электрического поля ... 27
§ 1. Подвижность и электропроводность ионов.................. 27
§ 2. Числа переноса и методика их определения 33
§ 3. Аномальная подвижность Н+ и ОН “-ионов.................. 39
§ 4. Числа перноса и электродные балансы (электрохимическая стехиометрия) .............................................. 41
Глава III Падение напряжения в электролите. Кондуктометрический метод анализа ................................................... 50
§ 1. Электропроводность электролитов......................... 50
§ 2. Методы экспериментального измерения электропроводности 55
§ 3. Кондуктометрический метод анализа....................... 58
Глава IV Свойства растворов электролитов. ...................... 67
§ 1. Слабые и сильные электролиты............................ 67
§ 2. Ступенчатая диссоциация................................ 71
§ 3. Ионное произведение воды и гидролиз .................... 72
§ 4. Теория кислот и оснований............................... 76
§ 5. Амфотерные электролиты (амфолиты) ...................... 80
§ 6. Нейтрализация и буферное действие.................. . 81
§ 7. Индикаторы ............................................ 84
§ 8. Развитие взглядов на свойства растворов электролитов . . 87
§ 9. Причины электролитической диссоциации............<. . . 90
Глава V Основные положения теории растворов электролитов ... 95
§ 1 . Термодинамика растворов электролитов . ............... 95
§ 2. Электростатическая тероия растворов....................102
§ 3. Осмотический коэффициент ..............................113
§ 4. Эквивалентная электропроводность сильных электролитов 116
г § 5. Ионные ассоциации в растворах сильных электролитов ... 121
§ 6. Химическая теория концентрированных растворов сильных электролитов ........................................... 126
428
Оглавление
§ 7. Современное состояние и .развитие теории растворов электролитов ........................................................... 127
Глава VI Электродвижущие силы и электродные потенциалы ... 131
§ 1. Обратимые электрохимические системы......................... 131
§ 2. Изменение свободной энергии обратимых элементов и электродов ............................................................ 13&
§ 3. Уравнение Томсона и Гиббса-Гельмгольца.............138
§ 4. Проблема топливных элементов ........139
§ 5. Скачки потенциалов на границе фаз и механизм их возникновения ............................................................141
§ 6. К®1антовомеханическая теория электродных потенциалов . . 149
§ 7. Измерение электродвижущих сил......................153
§ 8. Полуэлементы (электроды сравнения).................156
§ 9. Окислительно-восстановительные процессы и их потенциалы 160
§ 40. Правило Лютера и окислительно-восстановительные (равновесия ..............................................................166
§ 11. Влияние растворителя на обратимые электродные потен пиалы .......................................................... 168
Глава VII Концентрационные элементы. Потенциометрический метод анализа ...................................................... 173
§ 1. Концентрационные элементы с переносом...................173
§ 2. Диффузионный потенциал .......................................175
§ 3. Концентрационные элементы без переноса ионов.............179
§ 4. Потенциометрический анализ....................................183
§ 5. Индикаторные электроды........................................187
§ 6. Стеклянный электрод; области его применения и теория . . 1901
Глава VIII Строение двойного электрического слоя, электрокапилляр-ные и электрокинетические явления ...............................198
§ 1. Возникновение двойного электрического 'слоя на границе фаз 198
§ 2. Общая теория электрокапиллярных явлений ......................209
§ 3. Поверхностное натяжение и смачивание металлов электролитами ......... .............................................. 216
§4. Потенциалы нулевого заряда и проблема электродвижущей силы гальванического элемента ......................................218
§ 5. Емкость двойного слоя и ее измерение..........................223
§ 6. Электрокинетические явления..................................228
Глава IX Причины возникновения электродной поляризации и методы ее экспериментального изучения.............................. 234
§ 1. Ток обмена. Напряжение разложения водных растворов электролитов. Остаточный ток ......................................234
§ 2. Природа и особенности электродной поляризации.................239
§ 3. Методы экспериментального исследования кинетики электродных процессов ......................................................245
Оглавление
429
.Глава X Диффузионная кинетика и теория концентрационной поляризации ...........................................................269
§ 1. Диффузионные ограничения как причина торможения электродных реакций . . . j . .,. . ..............269
§ 2. Развитие теории концентрационной поляризации . . ... . . • 277
§ 3. Полярографический метод анализа.......................284
§ 4. Амперометрическое титрование .........................293
Глава XI Перенапряжение при разряде ионов водорода...............297
§ 1. Влияние различных факторов на величину перенапряжения водорода . ..................t. . .........................297
§ 2. Некоторые ранние гипотезы водородного перенапряжения . 303
§ 3. Теория замедленной рекомбинации атомарного водорода . . 306
§ 4 Теория замедленного разряда ..........................309
§ 5. Замедленный разряд, по А. Н. Фрумкину . ..............314
Глава XII Перенапряжение при разряде металлических ионов . . 326
§ 1. Общие сведения ...........................,...........326
§ 2. Теория замедленной кристаллизации.....................328
§ 3. Развитие теории перенапряжения металлов...............332
§ 4. Природа ионов, участвующих в электродной реакции . . . 336
§ 5. Явление адсорбции и кинетика электрохимических реакций 342
§ 6. Соотношение кинетики адсорбции и скорости электродных реакций при заданной плотности тока в реальных условиях электролиза ...............................................352
§ 7. Влияние поверхностно активных веществ на смачиваемость поляризуемого катода электролитом .........................358
Глава XIII Электроосаждение металлов ............................361
§ 1. Закономерности образования единичных кристаллов . . .. 361
§ 2. Закономерности образования поликристаллических осадков 363
§ 3. Характеристика катодных отложений и требования к осадкам 367
§ 4. Особенности электрокристаллизации из растворов, содержащих катионы одного металла различной валентности .... 372
§ 5. Совместный разряд ионов и влияние посторонних примесей на электроосаждение металлов................................374
§ 6. Особенности электролитического образования сплавов . . . 378
§ 7. Условия и особенности образования порошкообразных катодных отложений . . ......................................381
§ 8. Особенности получения компактных катодных осадков . . 383
Глава XIV Анодные процессы..................................... 389
§ 1. Общие сведения .......................................389
§ 2. Анодное растворение металлов..........................390
§ 3. Явление пассивности металлов. Строение и толщина пассивных пленок на металлах . ..................................393
§ 4. Рост пленок при анодной поляризации металлов. Нерастворимые аноды . ........................................... 398
430 Оглавление
§ 5. Перепаосивация металлов .............................. 403
§ 6. Условия устойчивого пассивного состояния...............405
§ 7. Анодное растворение сплавов............................407
§ 8. Реакции анодного окисления ...........................408
Глава XV Коррозия металлов........................................410
§ 1. Общие сведения........................................ 410
§.2. Теория коррозии . .................................... 411
§ 3. Коррозионные диаграммы.................................414
§ 4. Методы коррозионных испытании .........................421
§ 5. Методы борьбы с коррозией..............................423
Автор ЛЕВИН Арон Иосифович
Редактор Луковцев П. Д.
Редактор Издательства М. С. Архангельская Технический редактор В. В. Михайлова
Сдано в производство 26/111 11963 г. .
Подписано в печать 17/VII 1963 г.
Бумага 60X90V16-— 13,5 бум. л. = 27 печ. л.
Уч.-изд. л. '25,9 Заказ 4338
Т-08751 Тираж 8600 Цена 1 р. 06 к.
Металлургиздат
Москва Г-34, 2-й Обыденский пер., 14 Типография Металлургиздата, Москва, Цветной б., 30
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр Строка Напечатано Должно быть По чьей вине
37 16 сн. ИОНЫ анионы Кор.
45 7 св. ОН- — F он--д->-|-н2о+
-» — + Н2О + — о2 + т°2 »
48 21 св. (1 - А) н+ -> (1 — А) (1-А)Н+-(1— A)F->
—>(1 — А) Н2 ->(1—А)Нг Ред.
60 13 сн. его находит ее находят Кор.
83 17 св. гн+1-К [СНзСООН1 + [СНзСООН]
1 1 "[CHsCOONaJ—а а [CHsCOONa] а
99 11 и 12 св. (молярность) (моляльность) Ред.
133 12 св. ^r.s 1(?к ’Pajr.s ^Г.я = [(*?к ?а)г.э Кор.
151 6 св. электронного электродного Считч.
187 20 св. рис. 50 рис. 51 Авт.
208 9 св. (см. рис. 67) (см. рис. 68) »
212 7 св. do 8 — —~~ dq do е — — d<p Кор.
218 2 сн. электрода электродного Считч.
269 7 и 8 сн. концентрированных концентрационных В
324 3 сн i = 2FKC х н i = 2FKC х н
- F ^F
„ , 2КТ 27?Г „ 2RT 2RT
хСн.о+ + е е хСн.о+е е Авт.
327 6 и 7 св.
устраняющие ускоряющие
356 7 сн. туг <Р = -^-г1пК + RT <е-=—г1пК +
az^F «ZkF
+ — 1п 1 — RT + -----Ini- Кор.
1 UZkF ^zKF
Заказ 1338
Л.И.ЛЕВИН
ОСНОВЫ
ЭЛЕКТРОХИМИИ