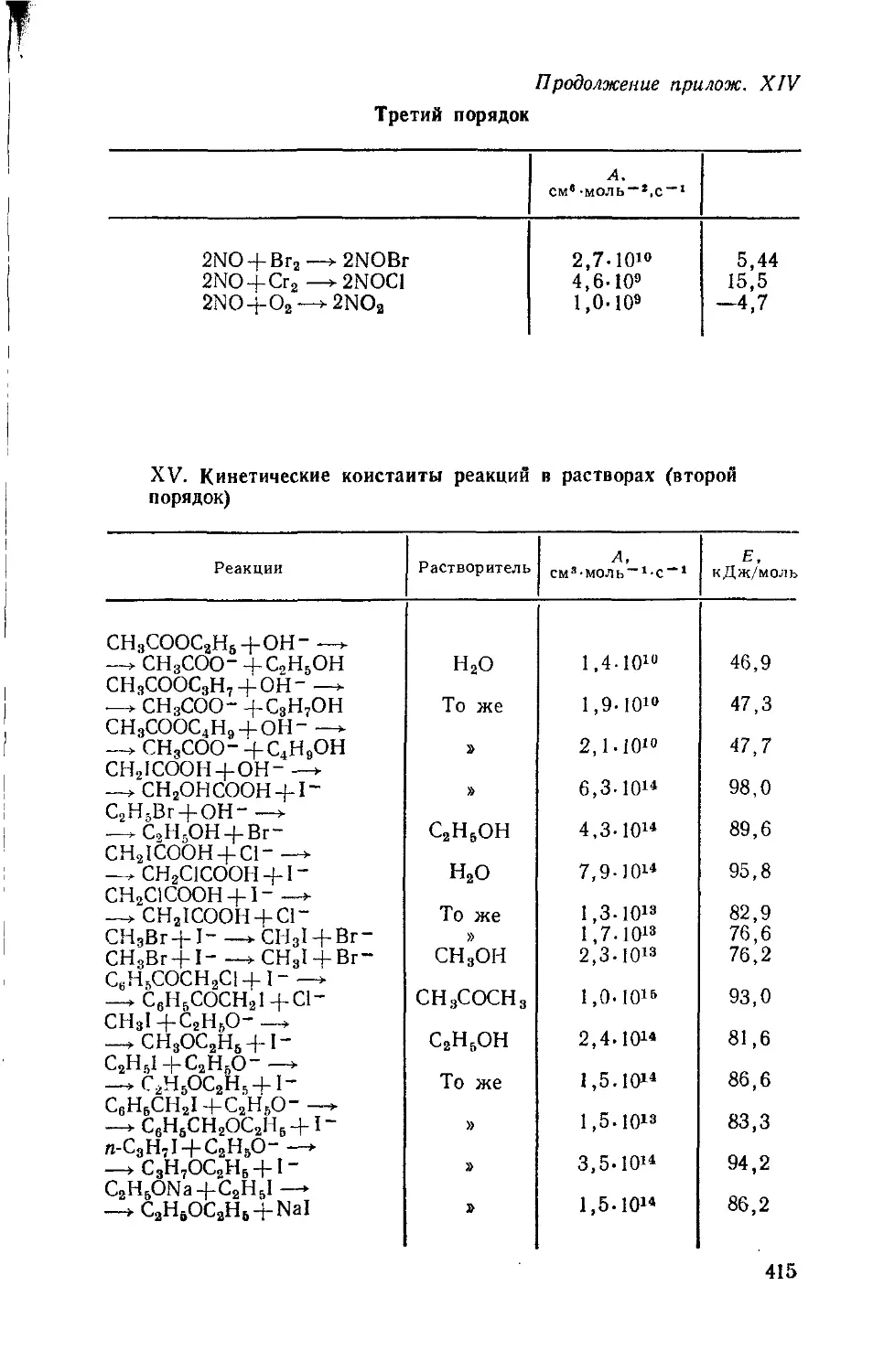
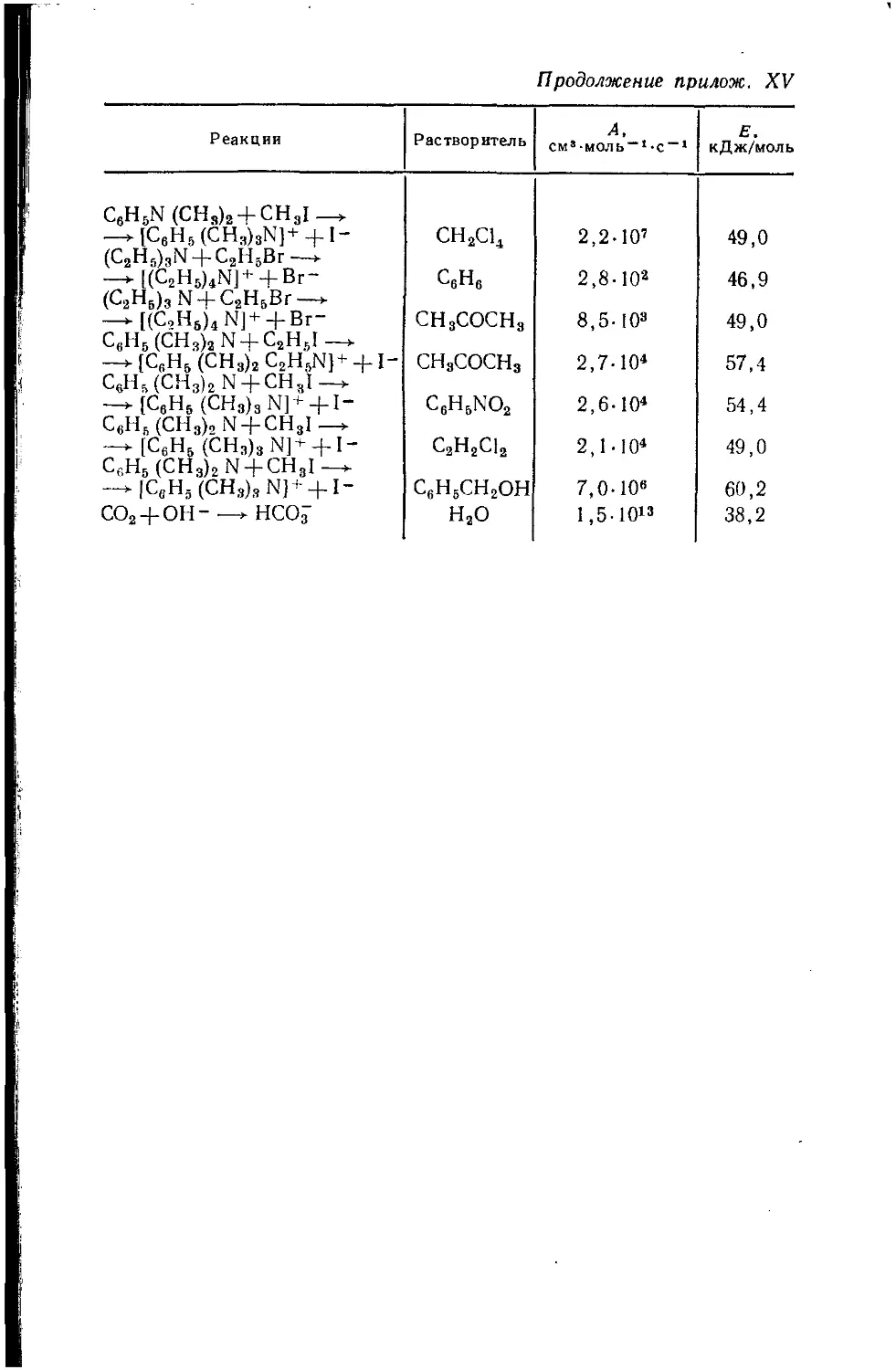
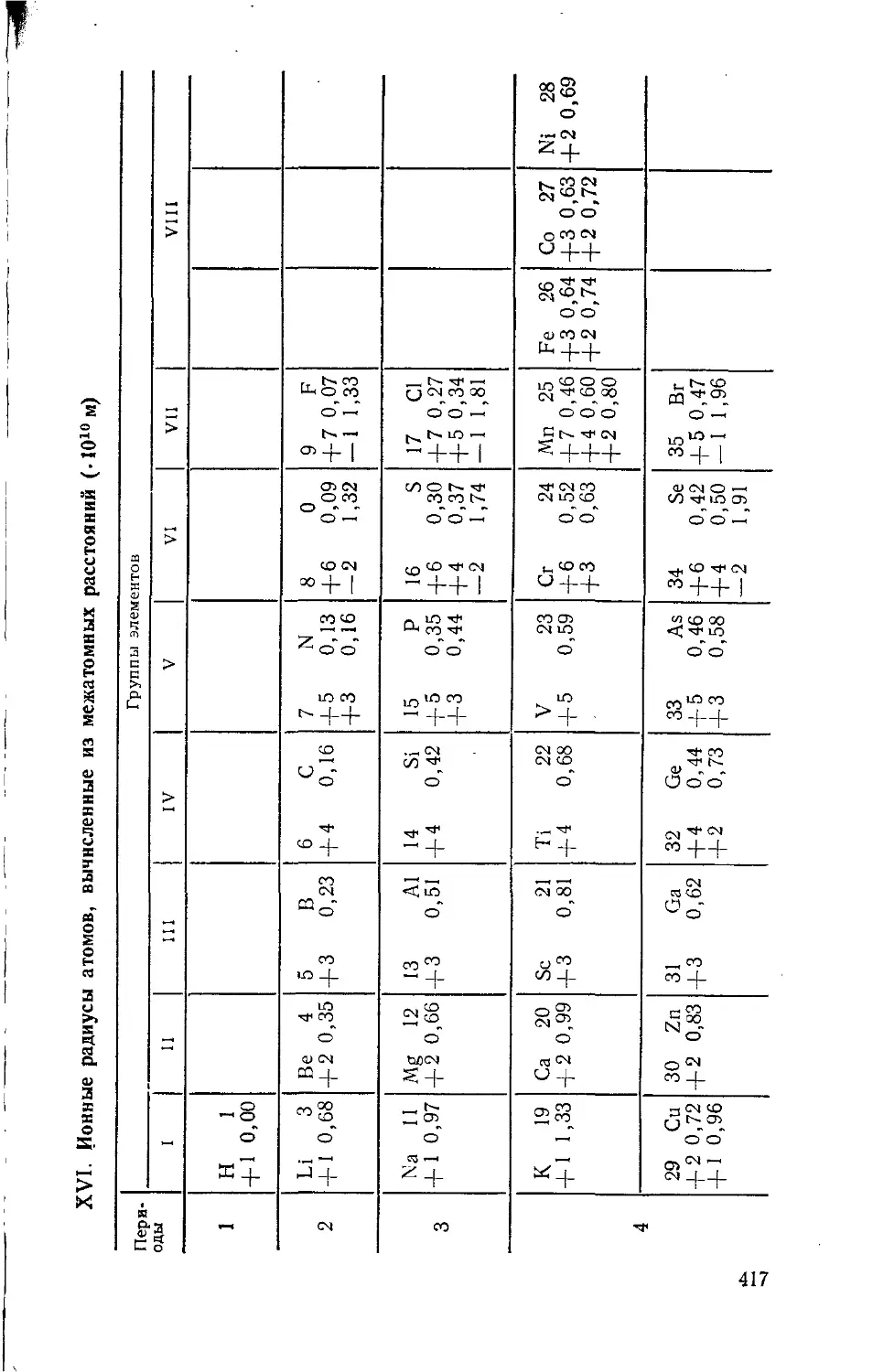
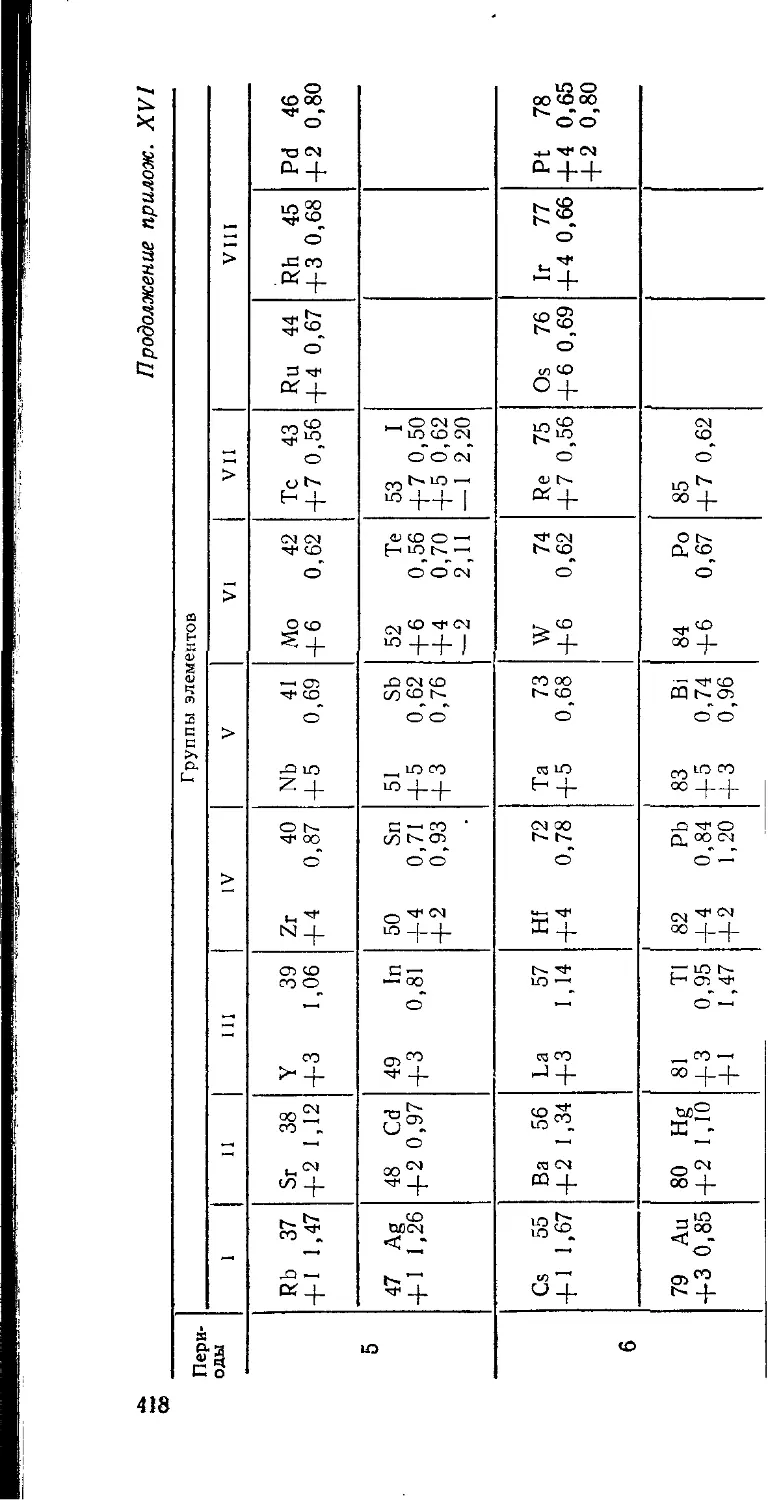
/























































































































































































































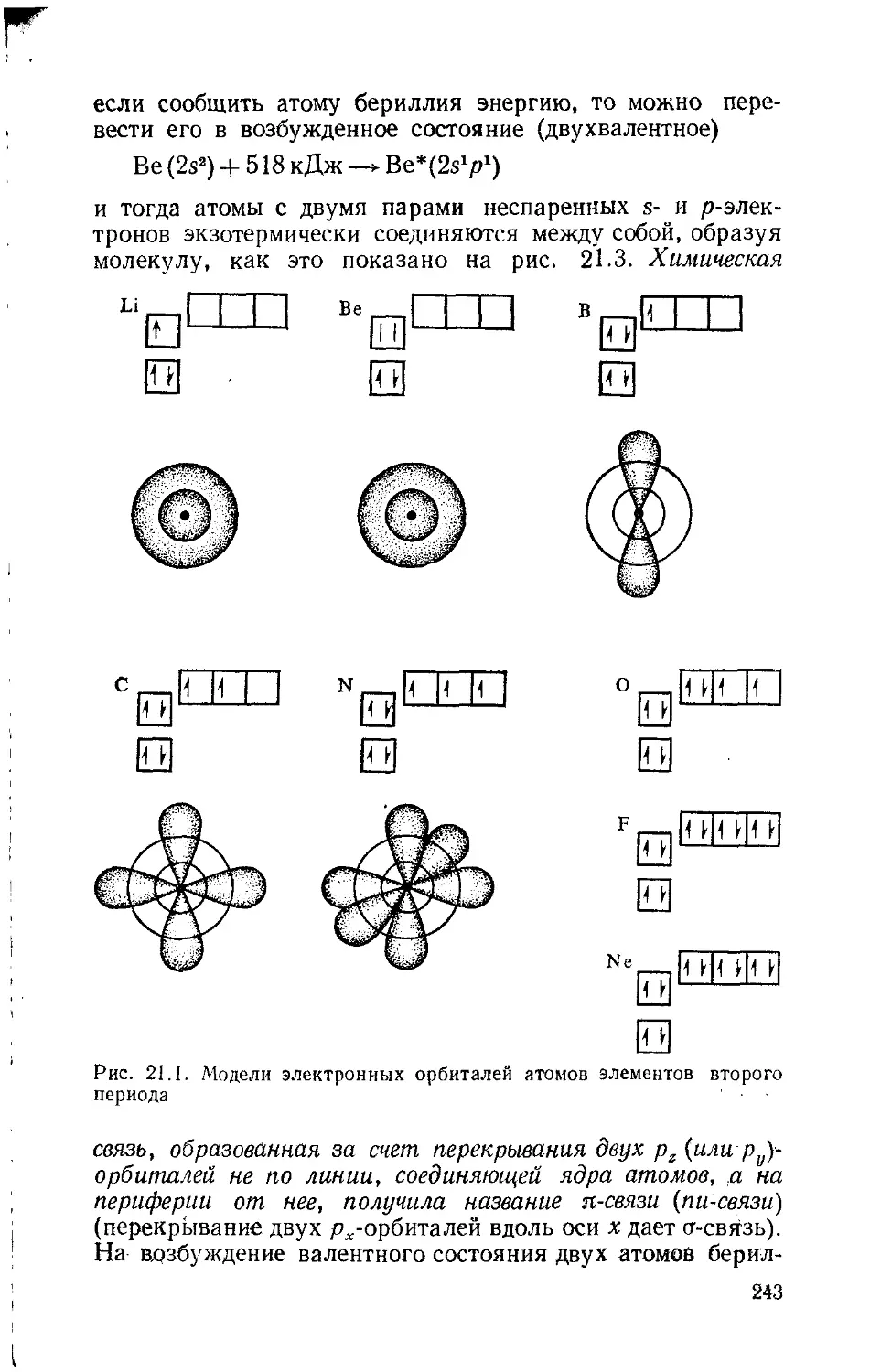
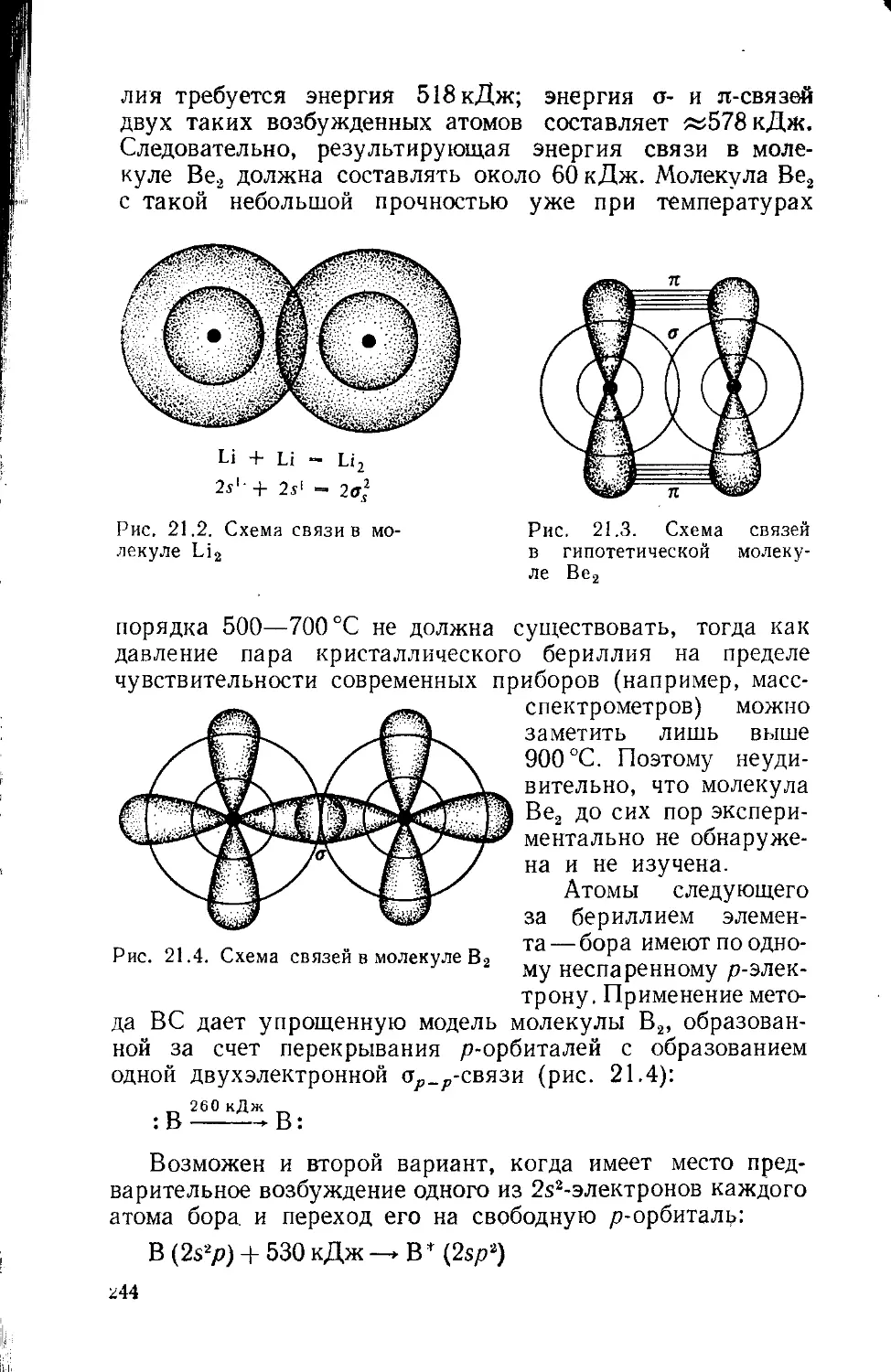





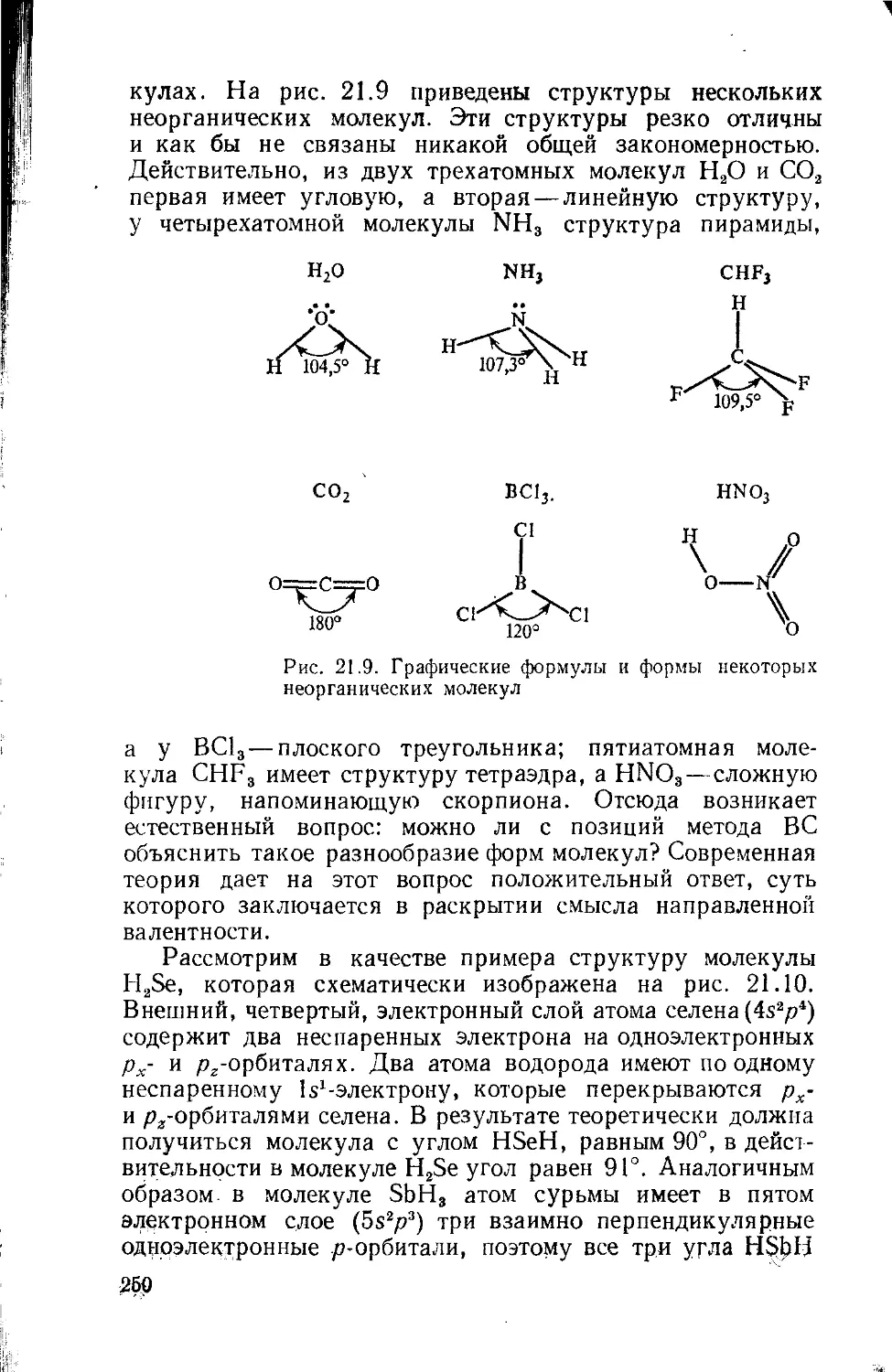


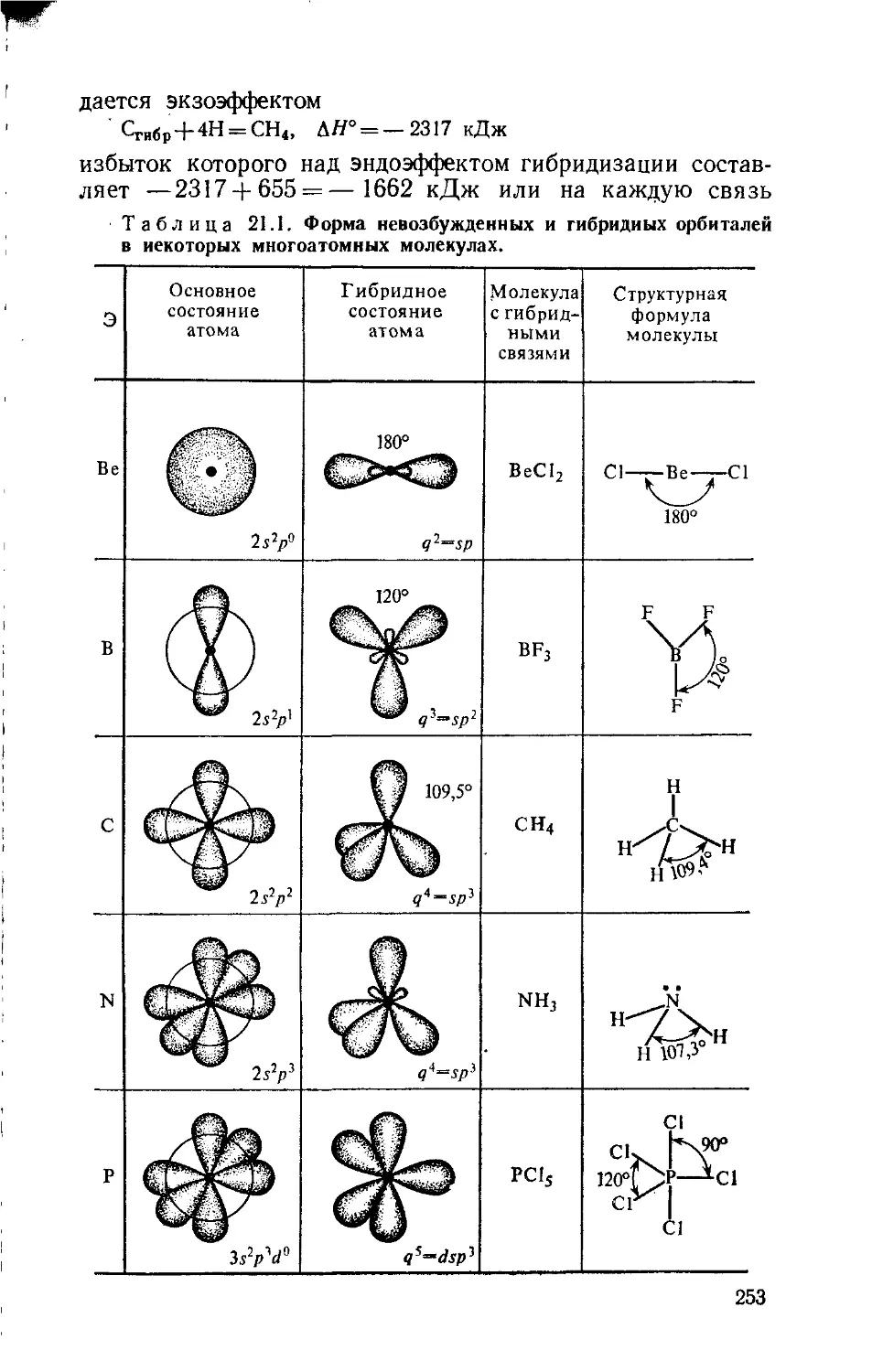



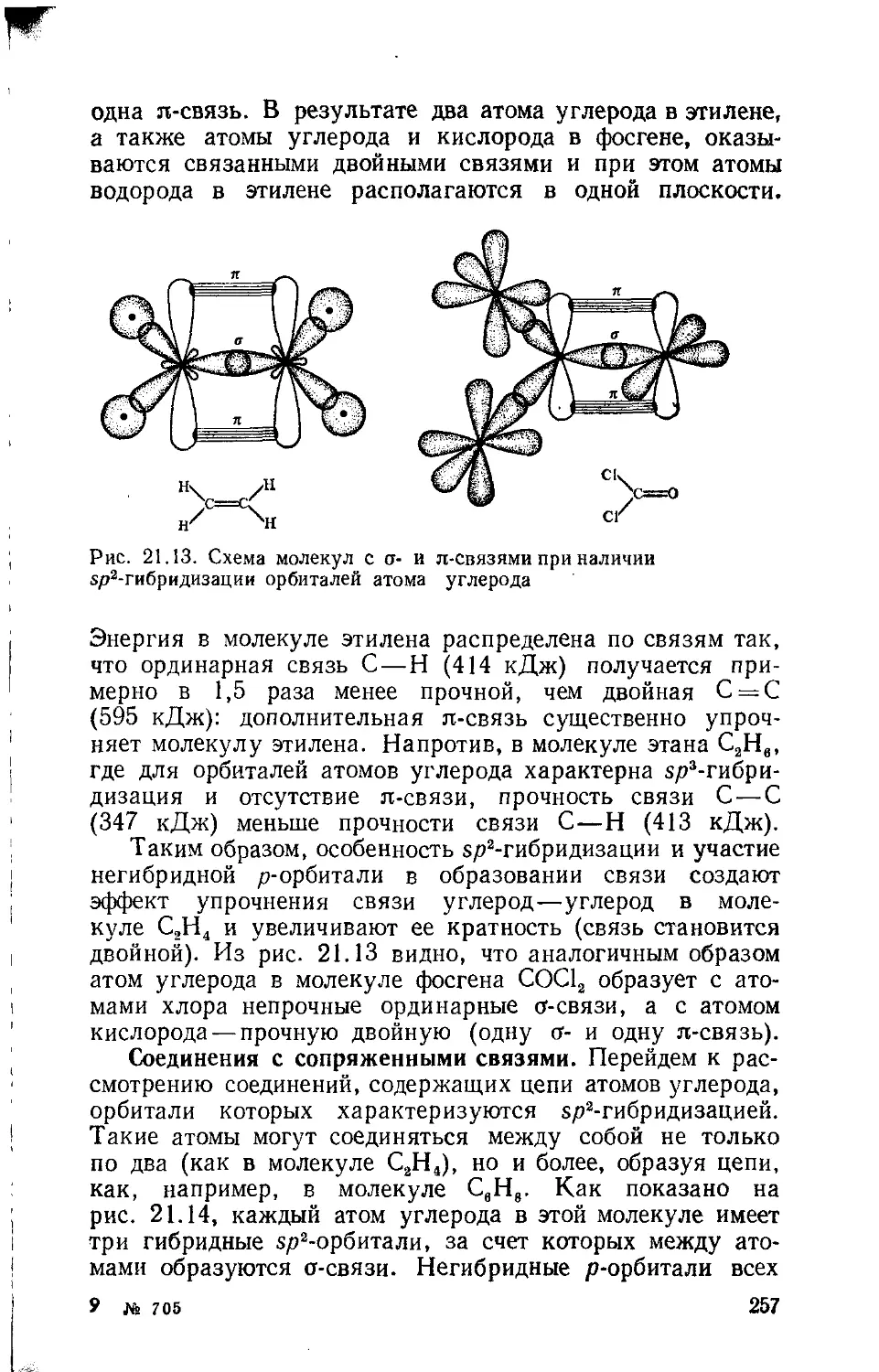
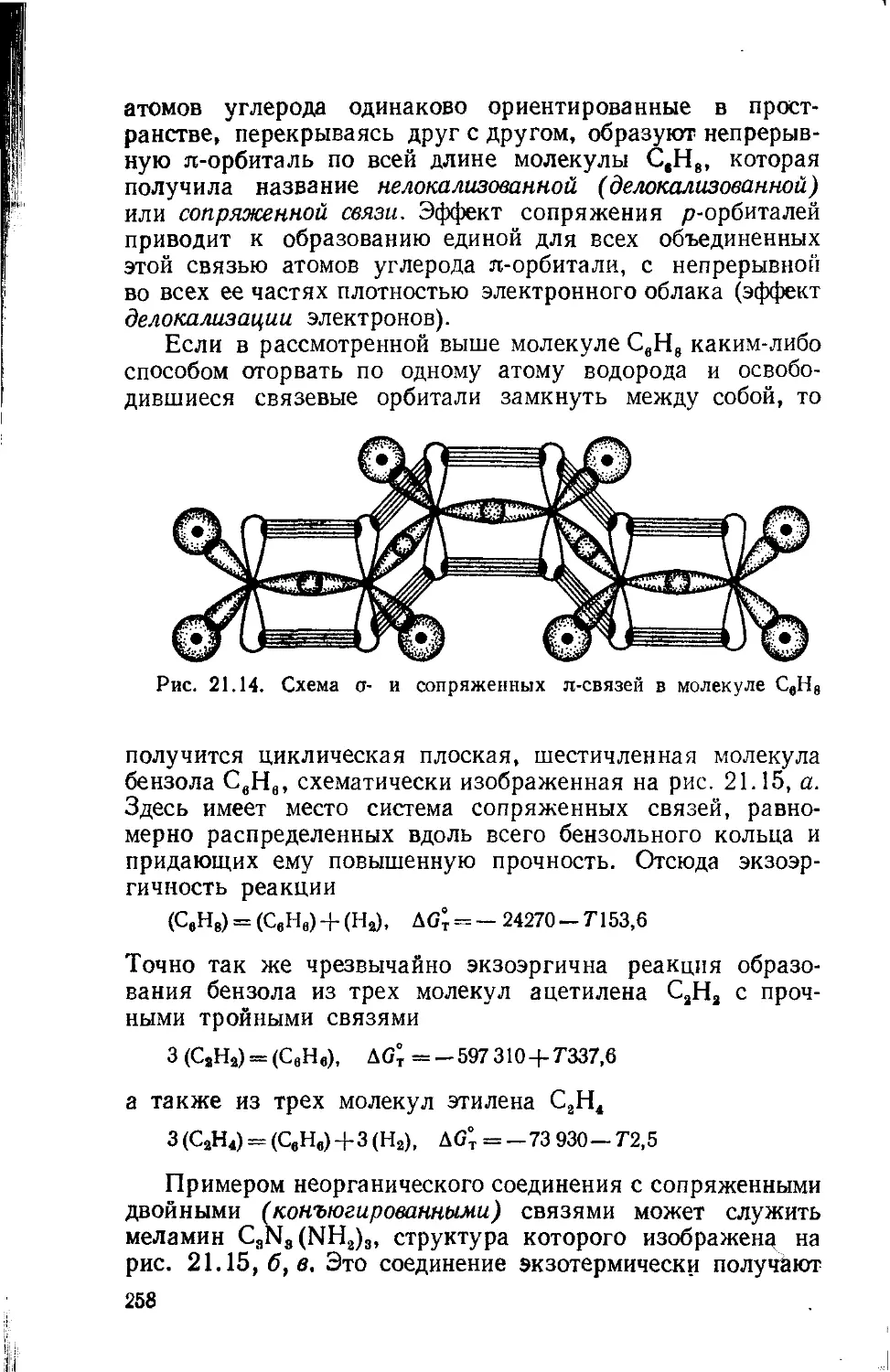
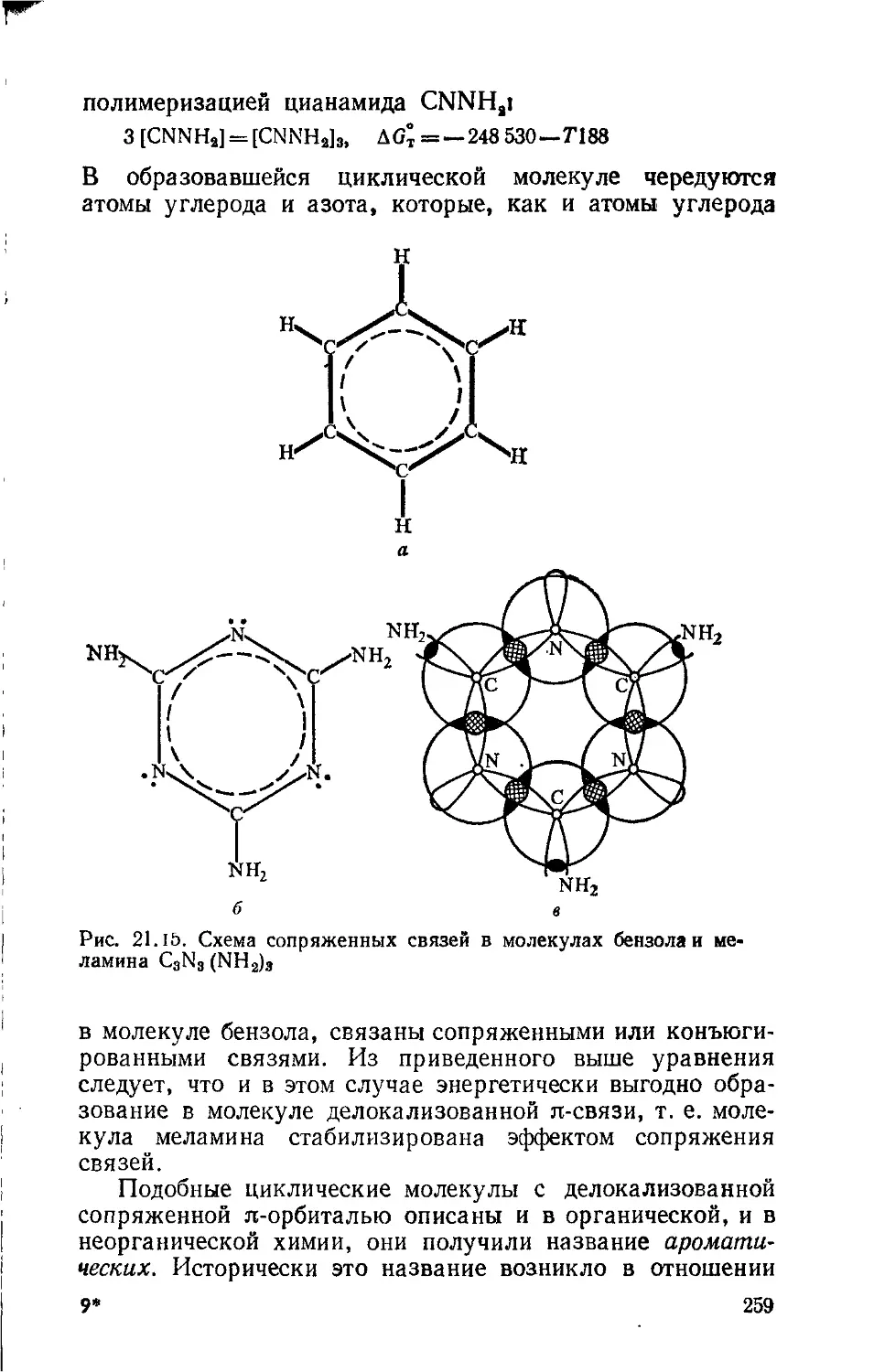
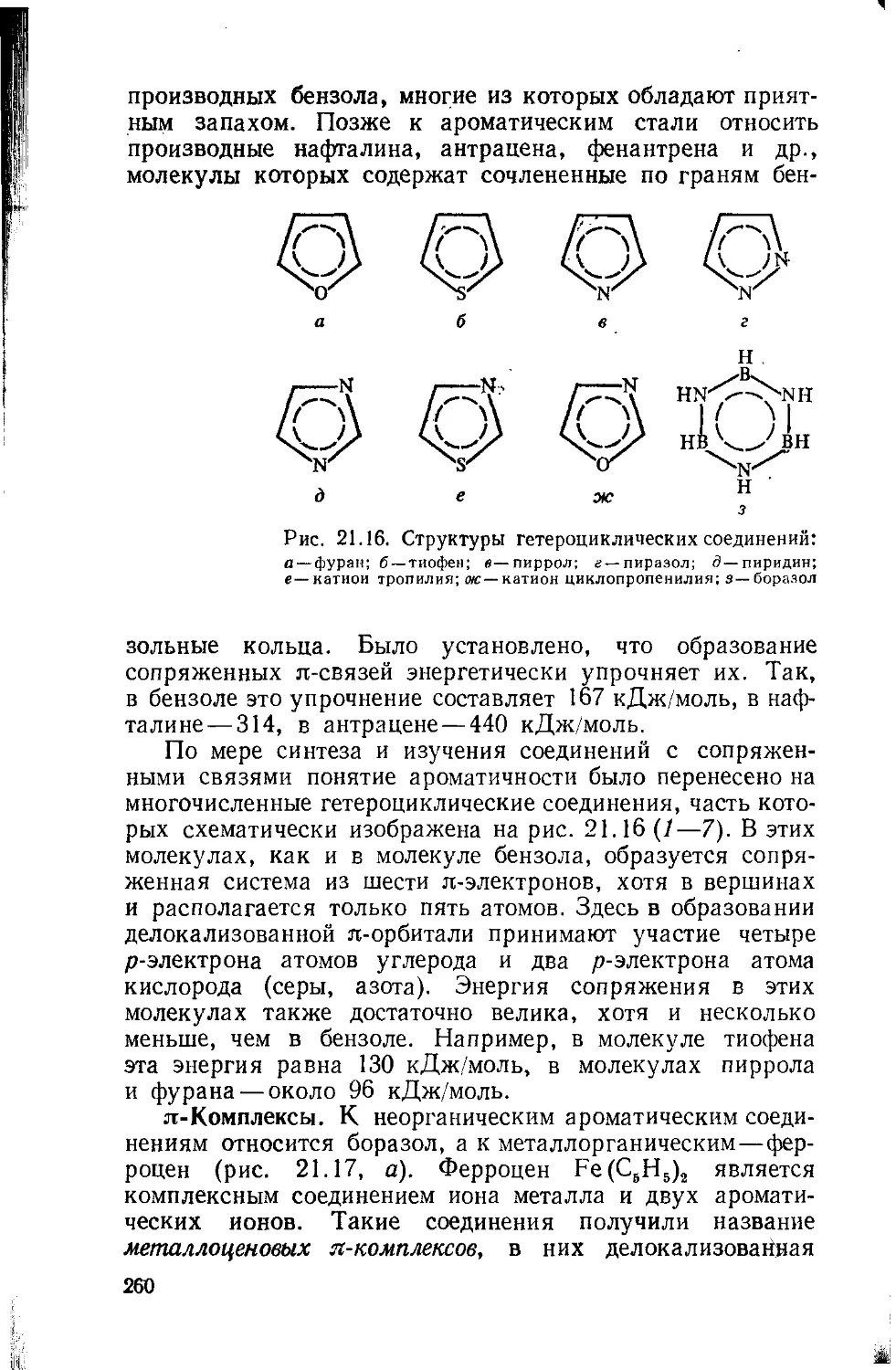








































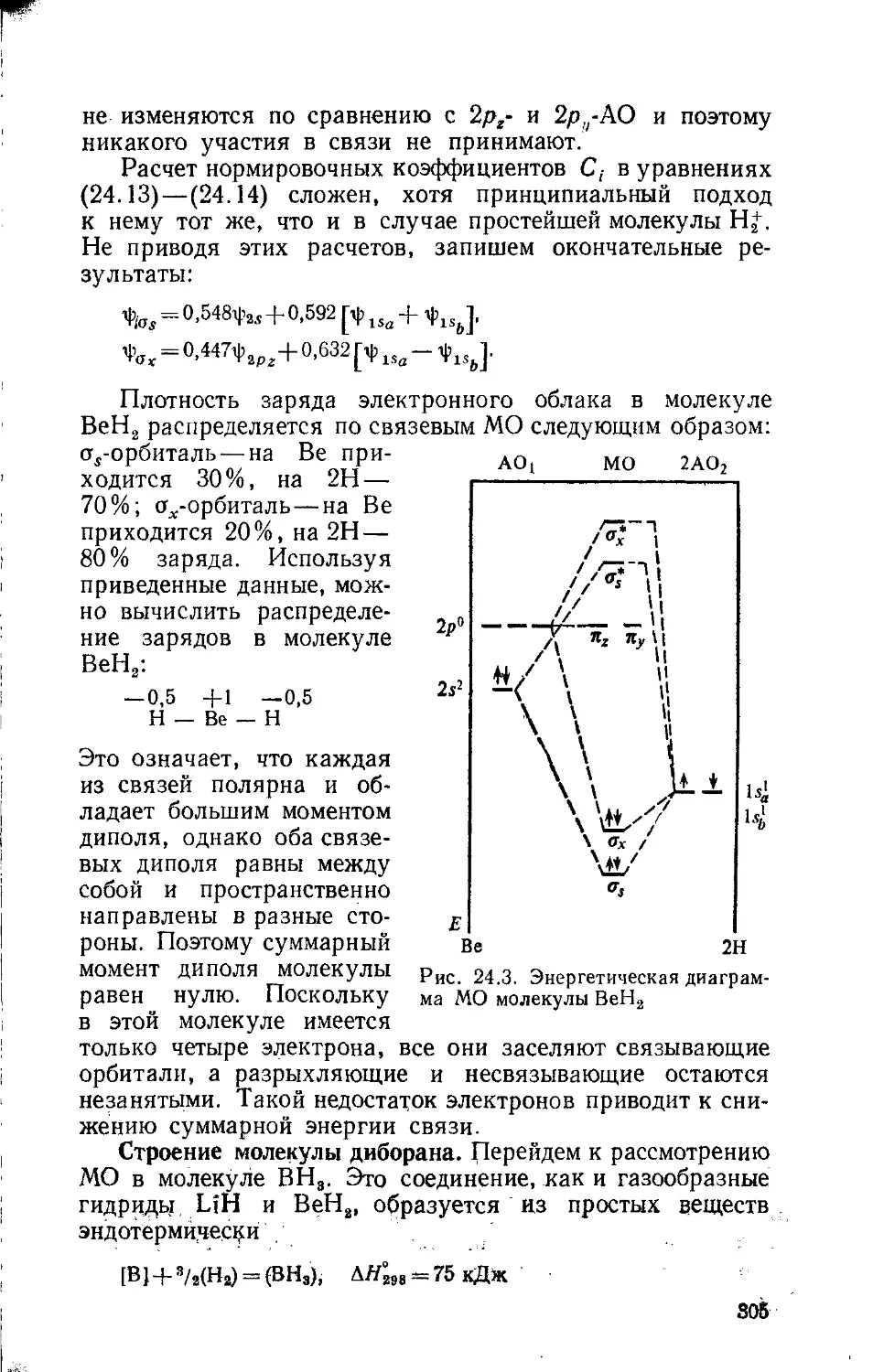


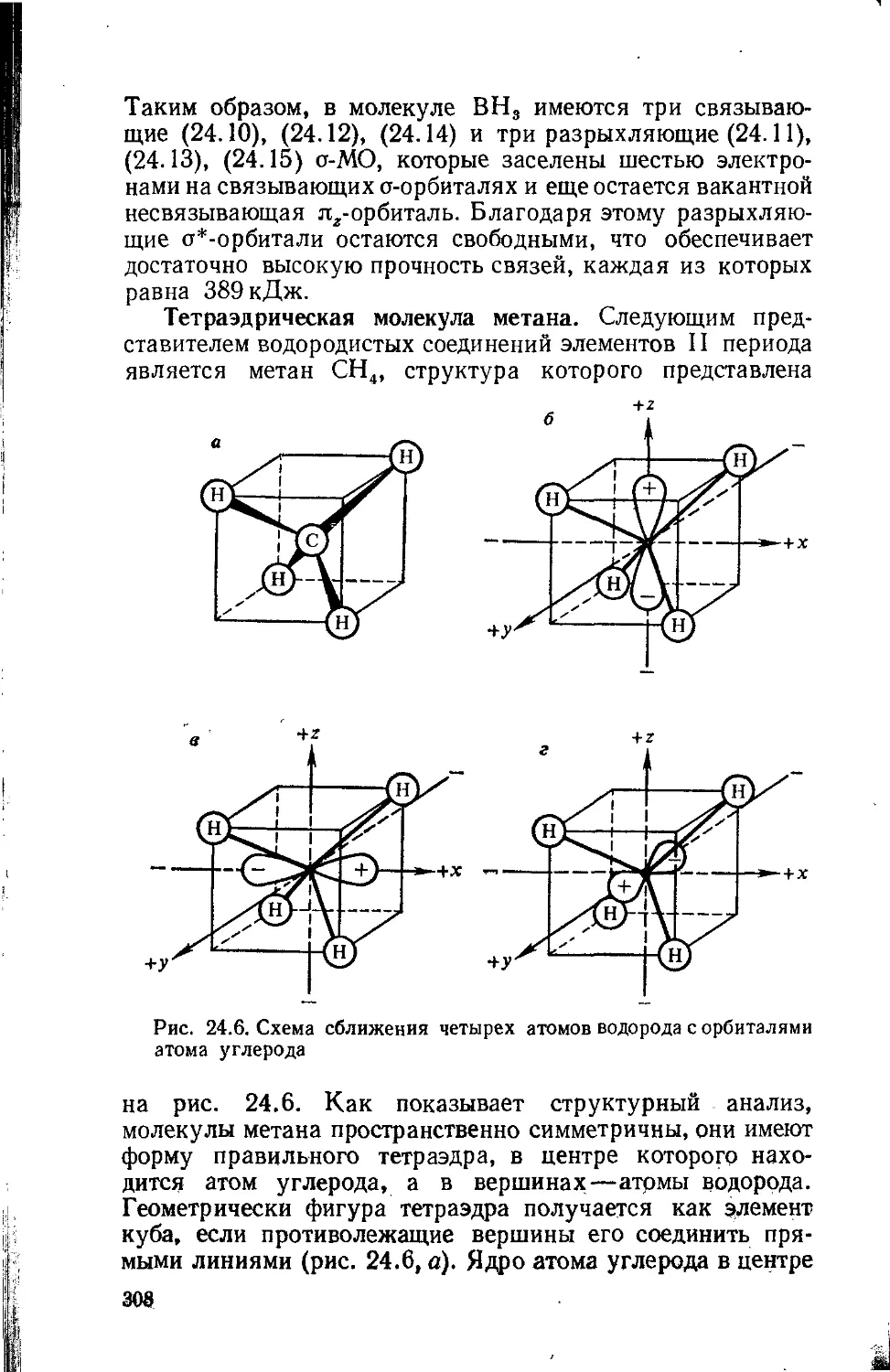


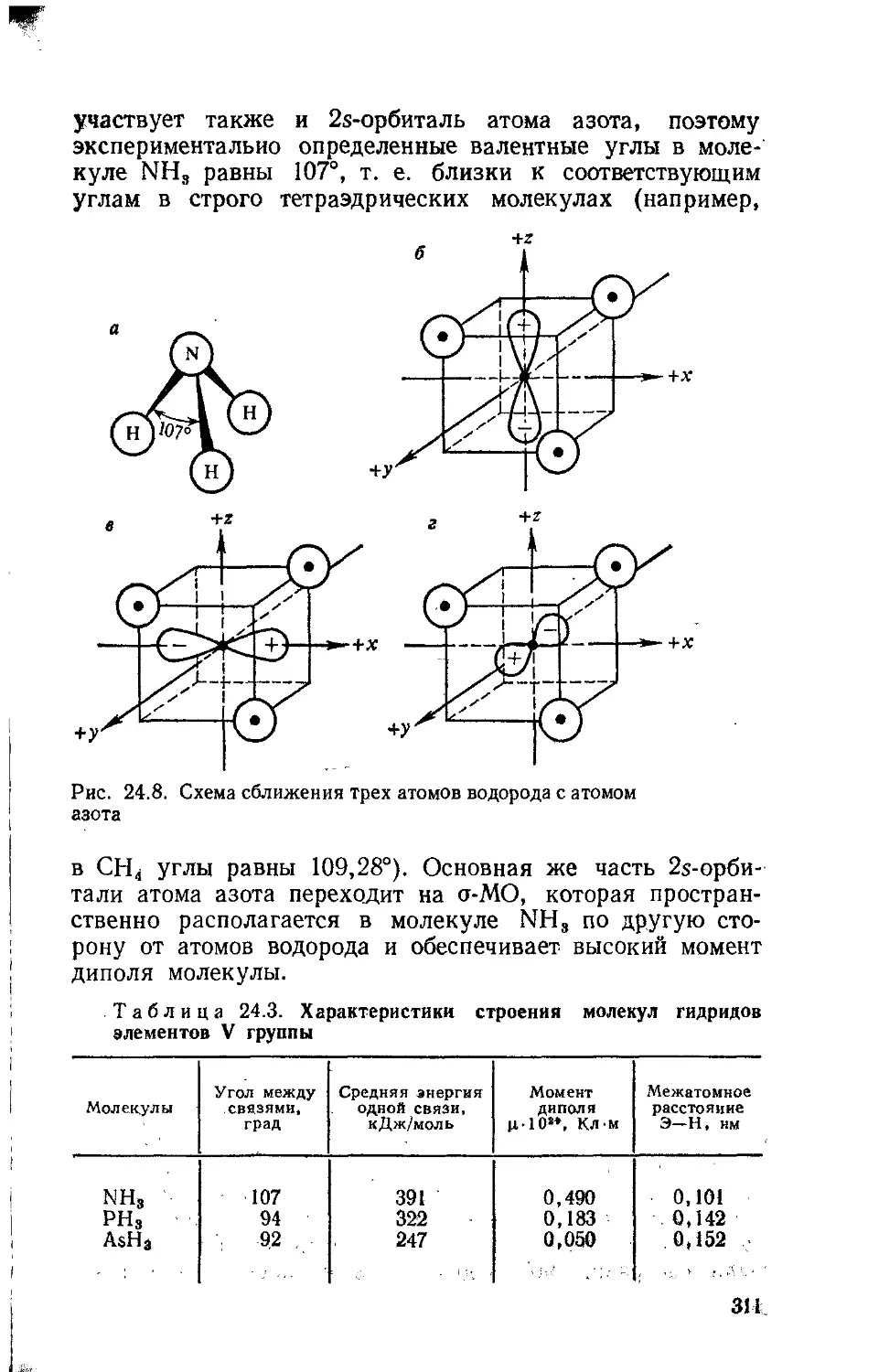


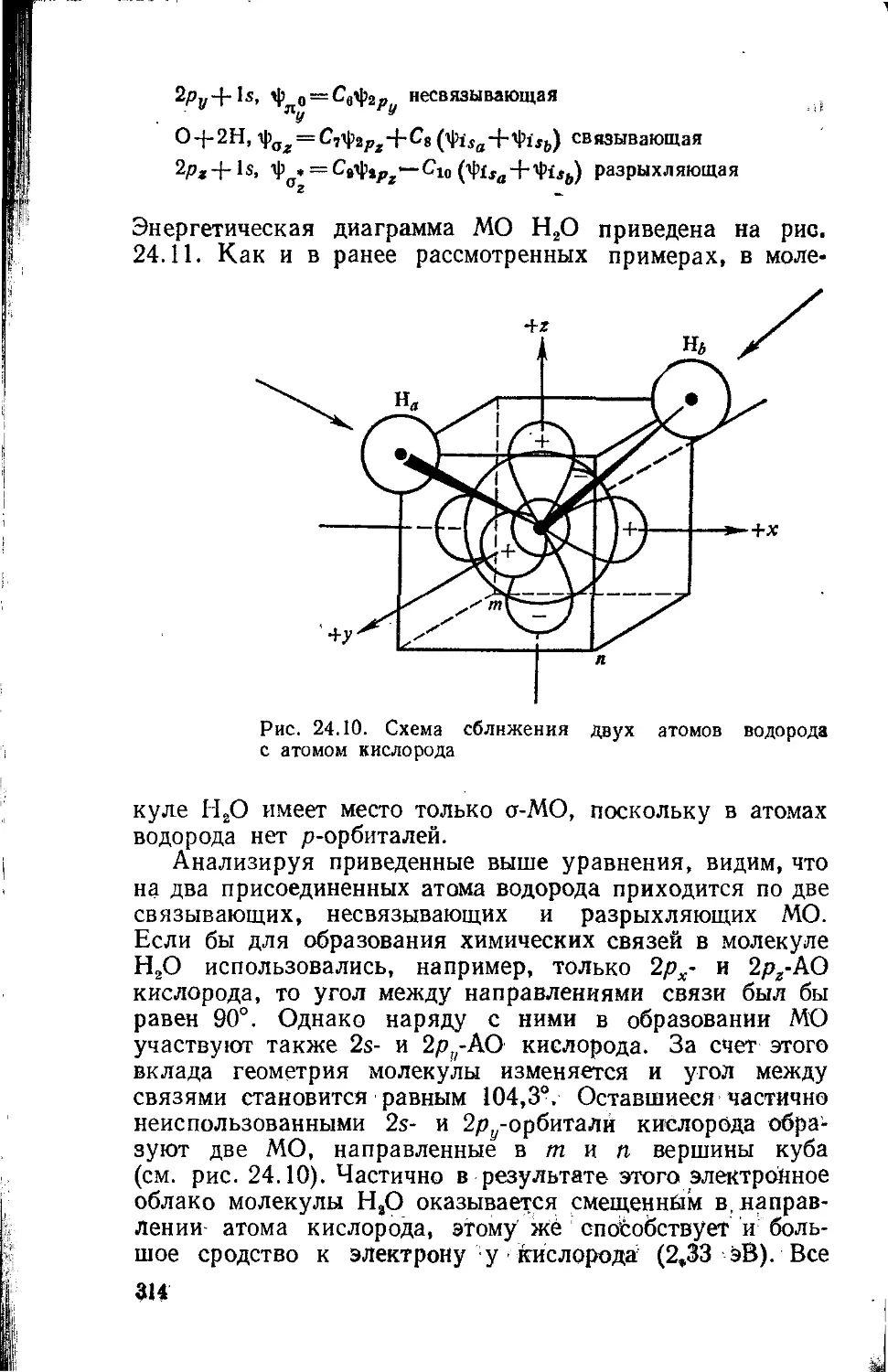

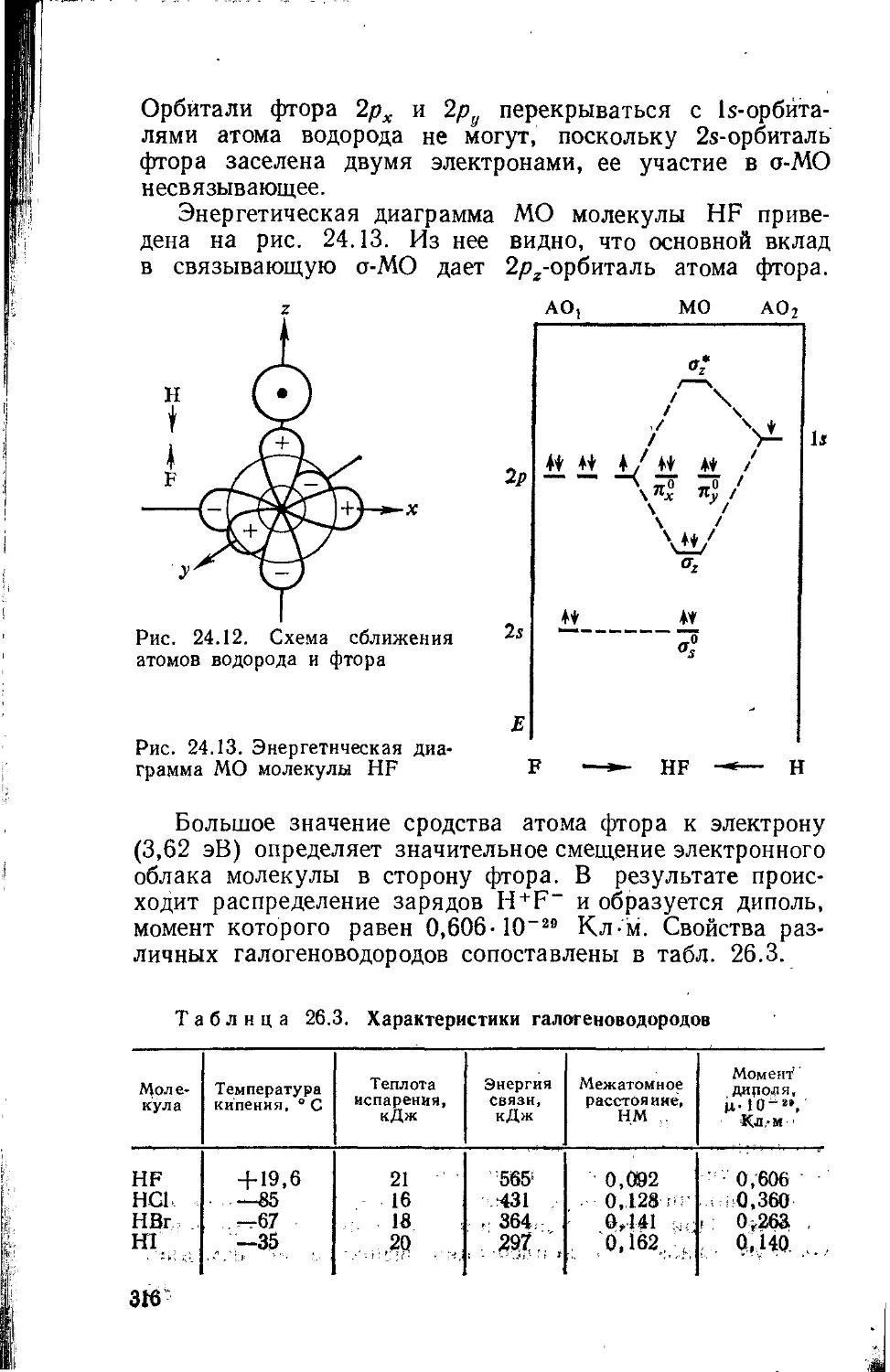







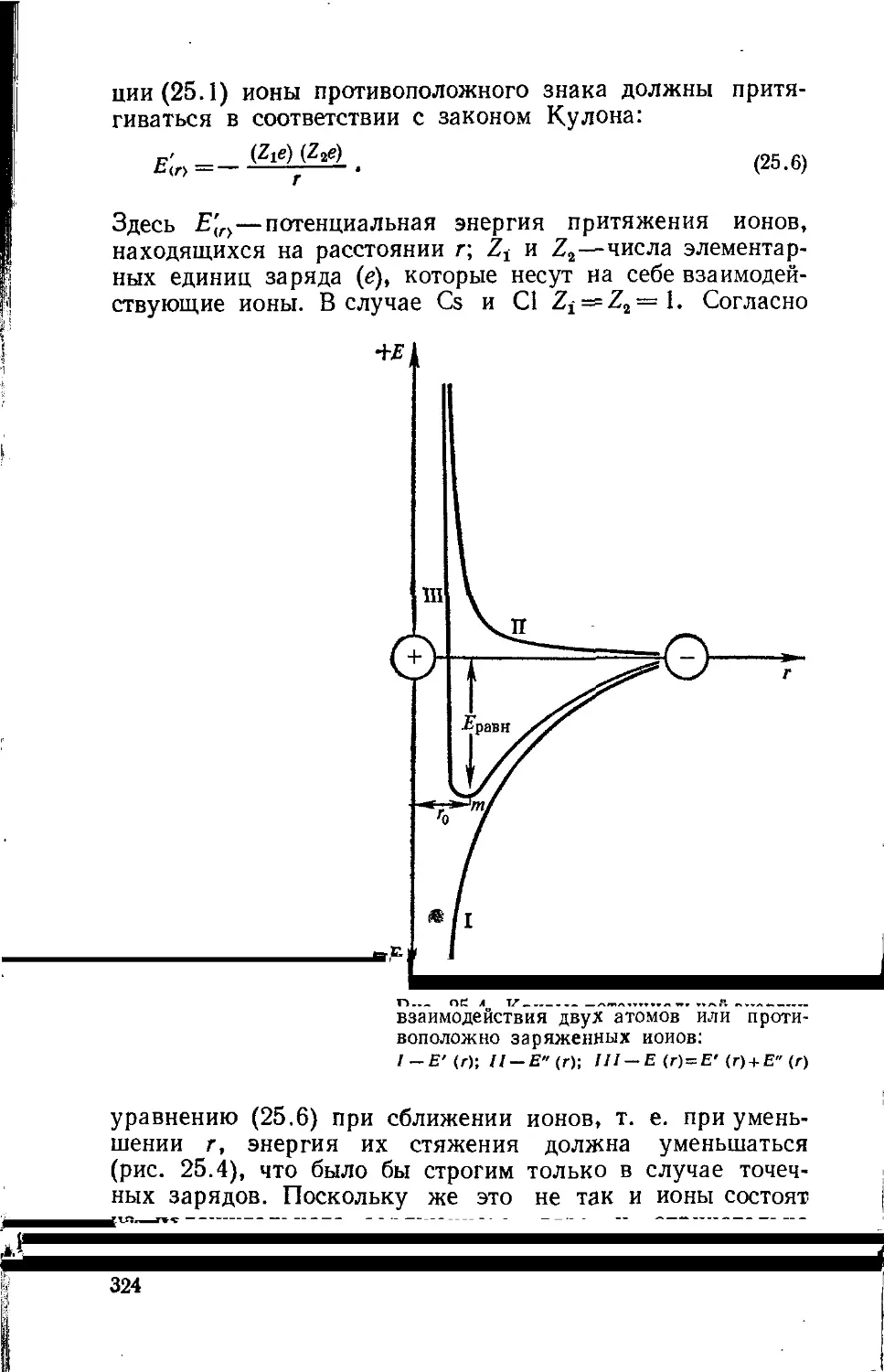



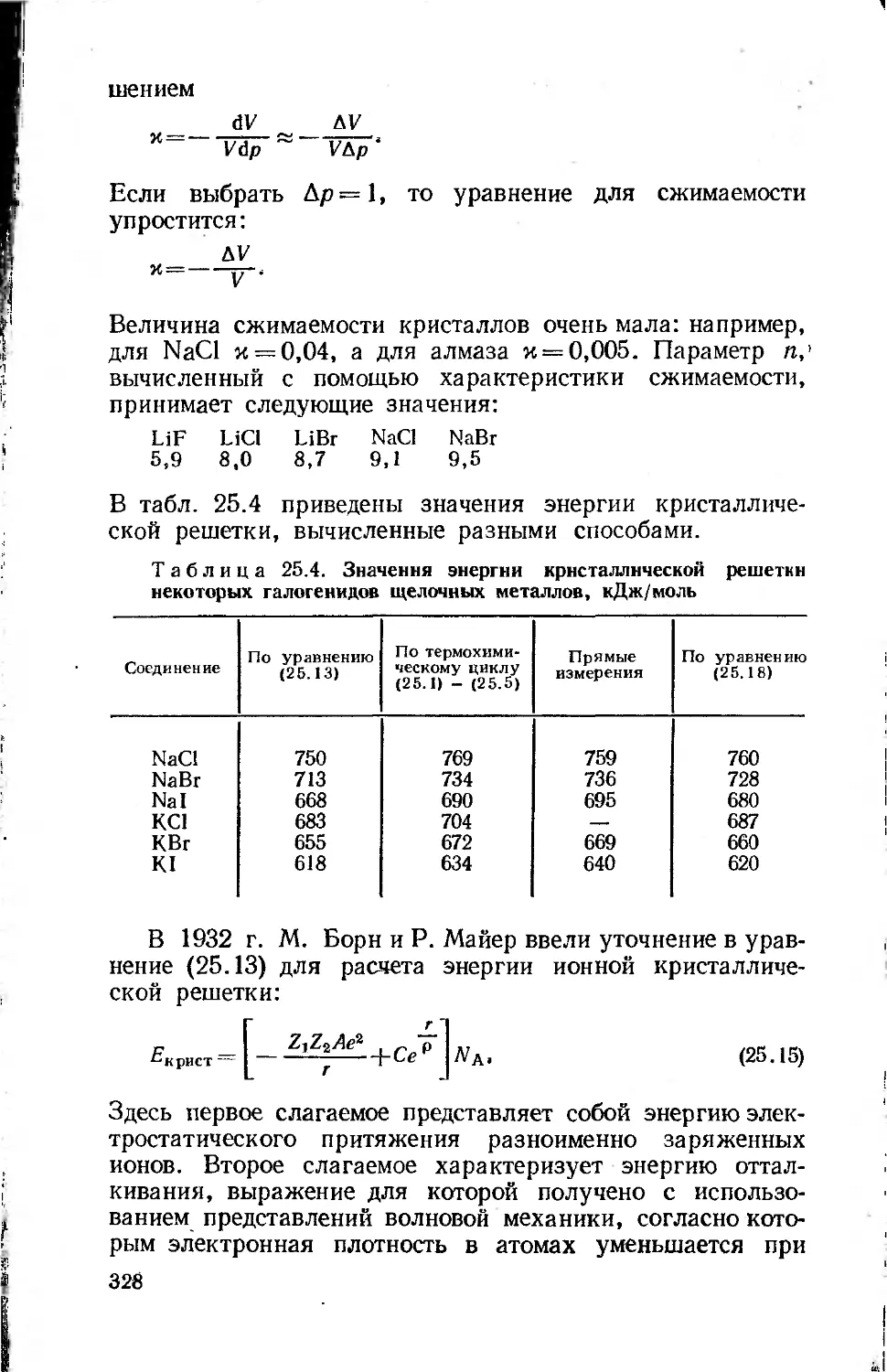




































































Text
Г.И.Новиков
ОСНОВЫ ОБЩЕЙ ХИМИИ
Учеб. пособие для химико-технолог. спец. вузов.—М.: Высш. шк., 1988.— 431 с.
Изложены современные представления о стехиометрии, термохимии,
эргохимии, основах химической кинетики и начала учения о строении атомов
молекул, жидкостей, кристаллов и соединений с невалентными связями в свете
фундаментальных законов естествознания: сохранения массы-энергии,
сохранения заряда и периодического закона элементов Д. И. Менделеева.
Большое внимание уделено вопросам термодинамики и стехиометрии.
Теоретический курс основ общей химии подкреплен решением практических
задач.
Оглавление
Предисловие 3
I. Основные законы химии 7
Глава 1. Фундаментальные законы и законы стехиометрии 8
II. Классы химических соединений и типы реакций 17
Глава 2. Основные классы простых и сложных соединений 18
Глава 3. Типы химических реакций 28
III. Основы химической термодинамики и кинетики 38
Глава 4. Термохимия 39
Глава 5. Химическое равновесие 49
Глава 6. Смещение химического равновесия 60
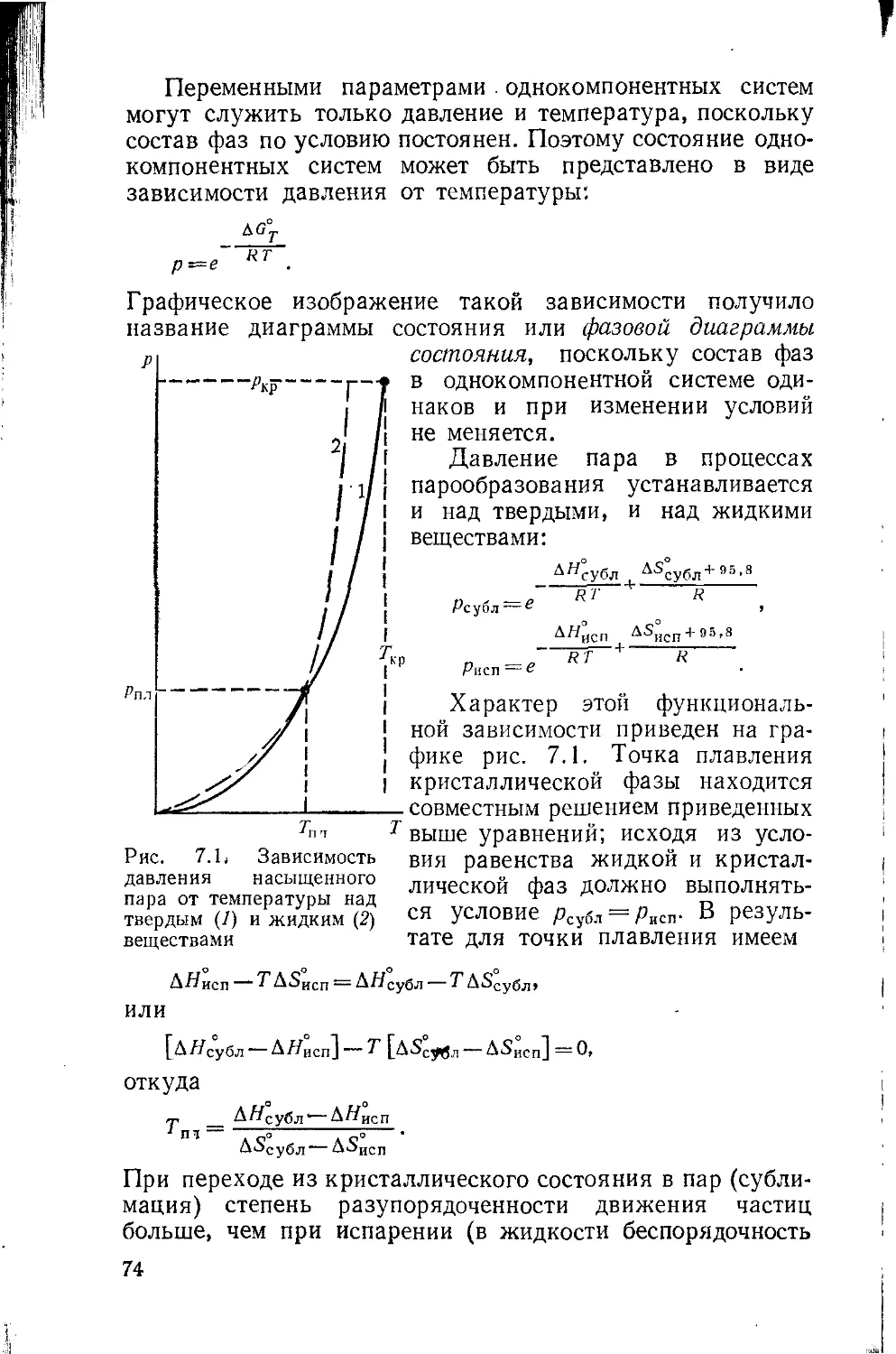
Глава 7. Правило фаз Гиббса 71
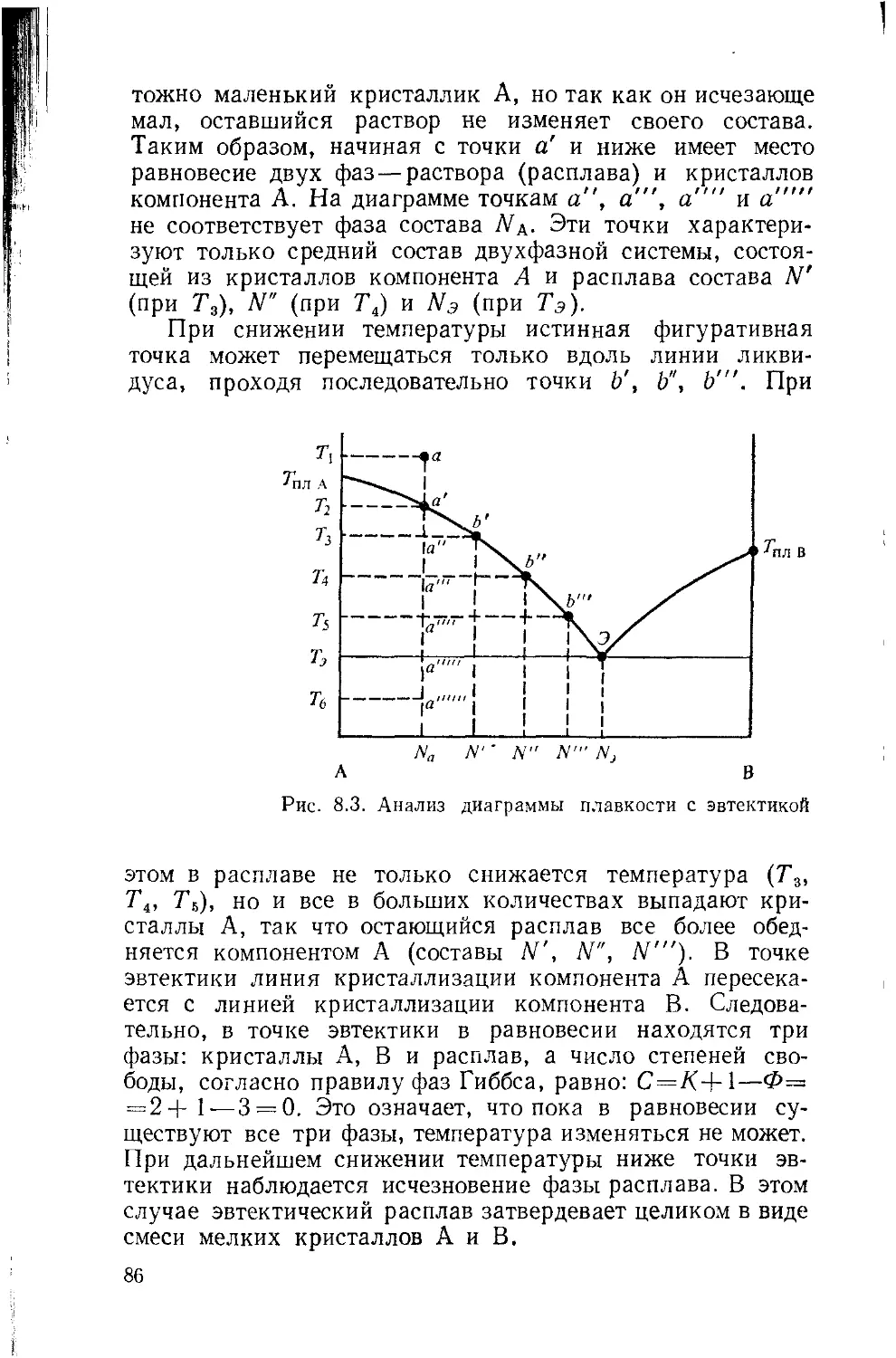
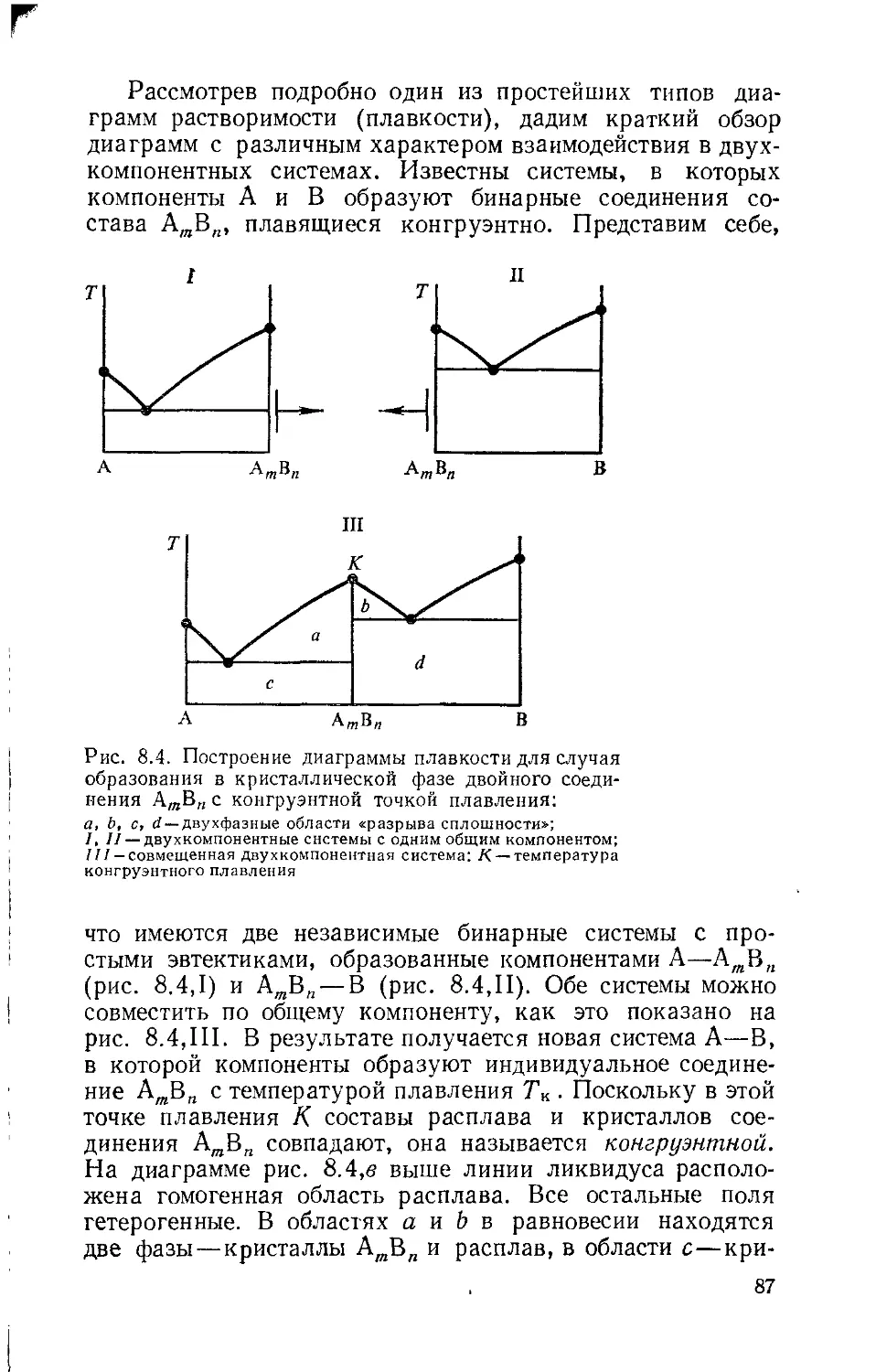
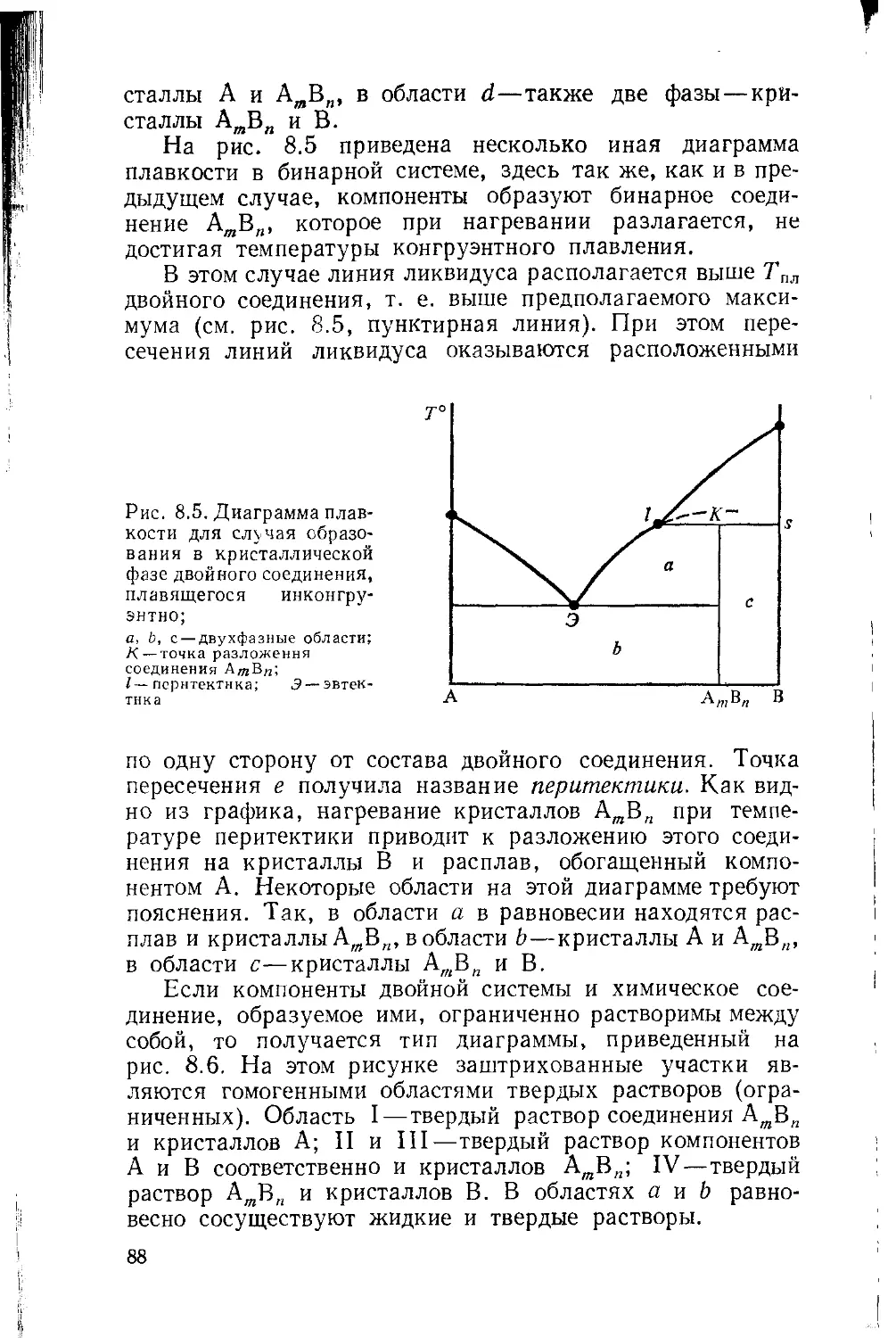
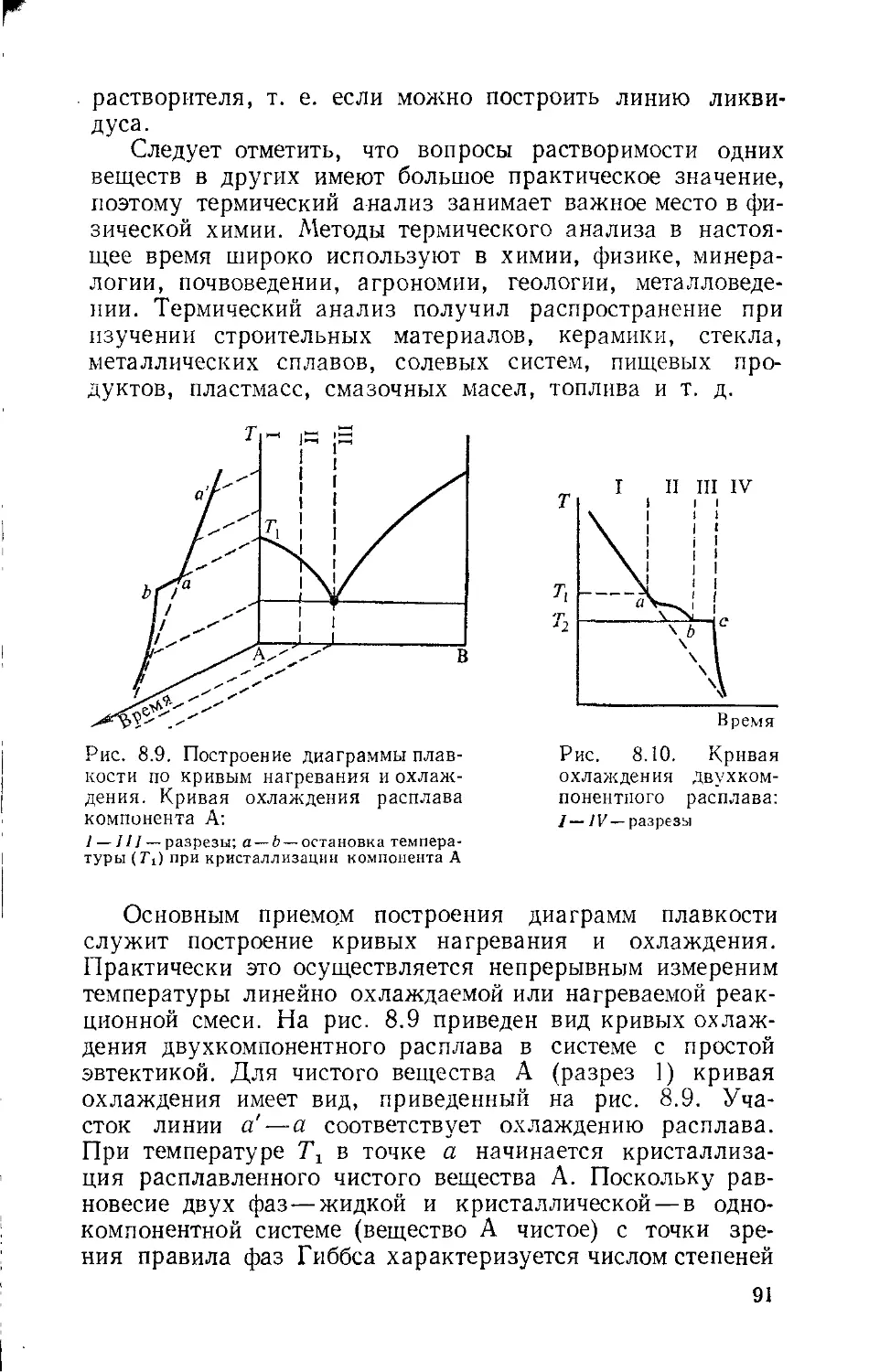
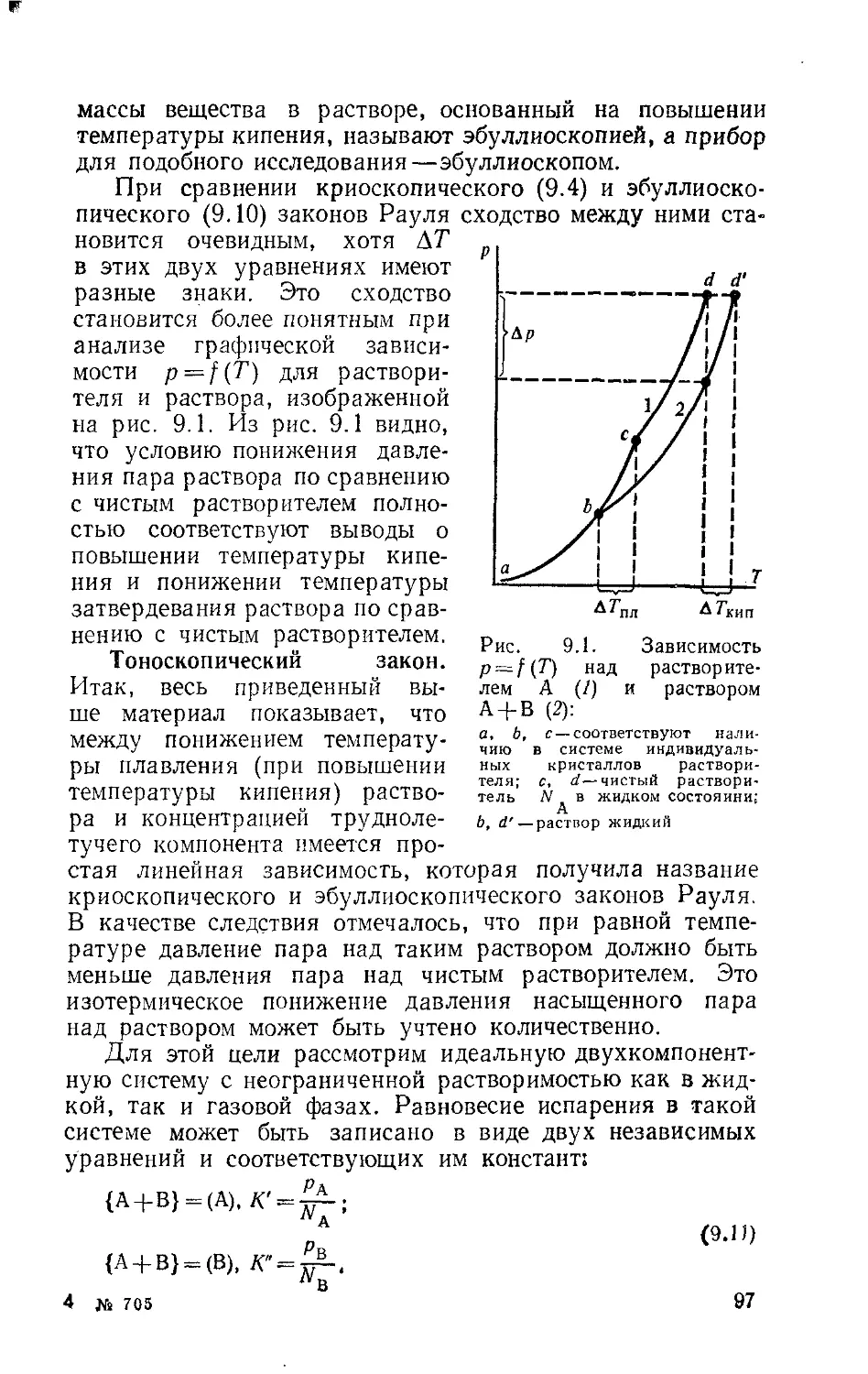
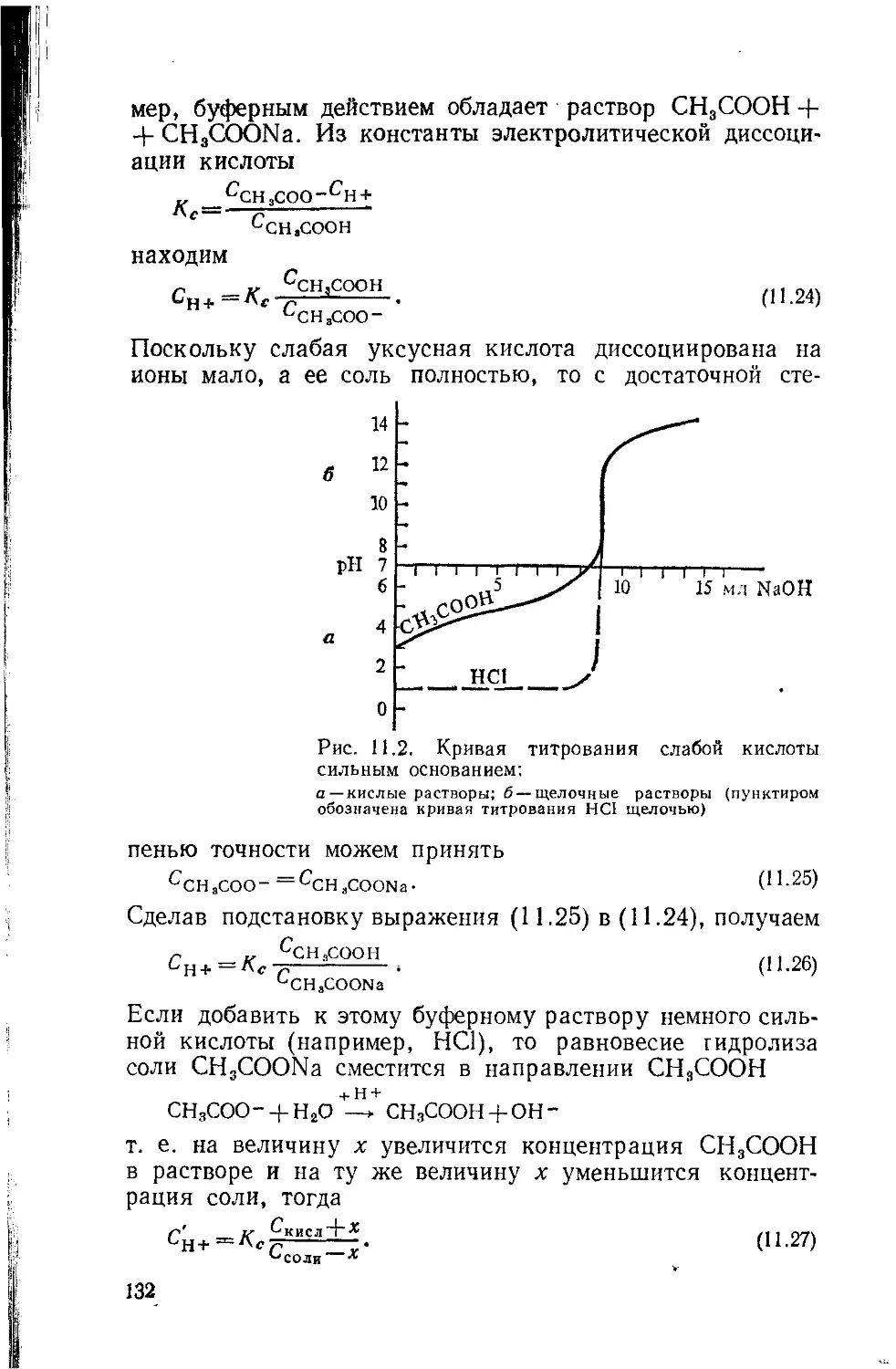
Глава 8. Диаграммы растворимости 79
Глава 9. Законы Рауля 93
Глава 10. Растворы электролитов 107
Глава 11 Гидролиз солей и нейтрализация 120
Глава 12. Растворимость электролитов 133
Глава 13. Электрохимические процессы 140
Глава 14. Прикладная электрохимия 151
Глава 15. Кинетика химических реакций 163
Глава 16. Катализ 178
IV. Строение вещества 187
Глава 17. Исторические предпосылки теории строения 188
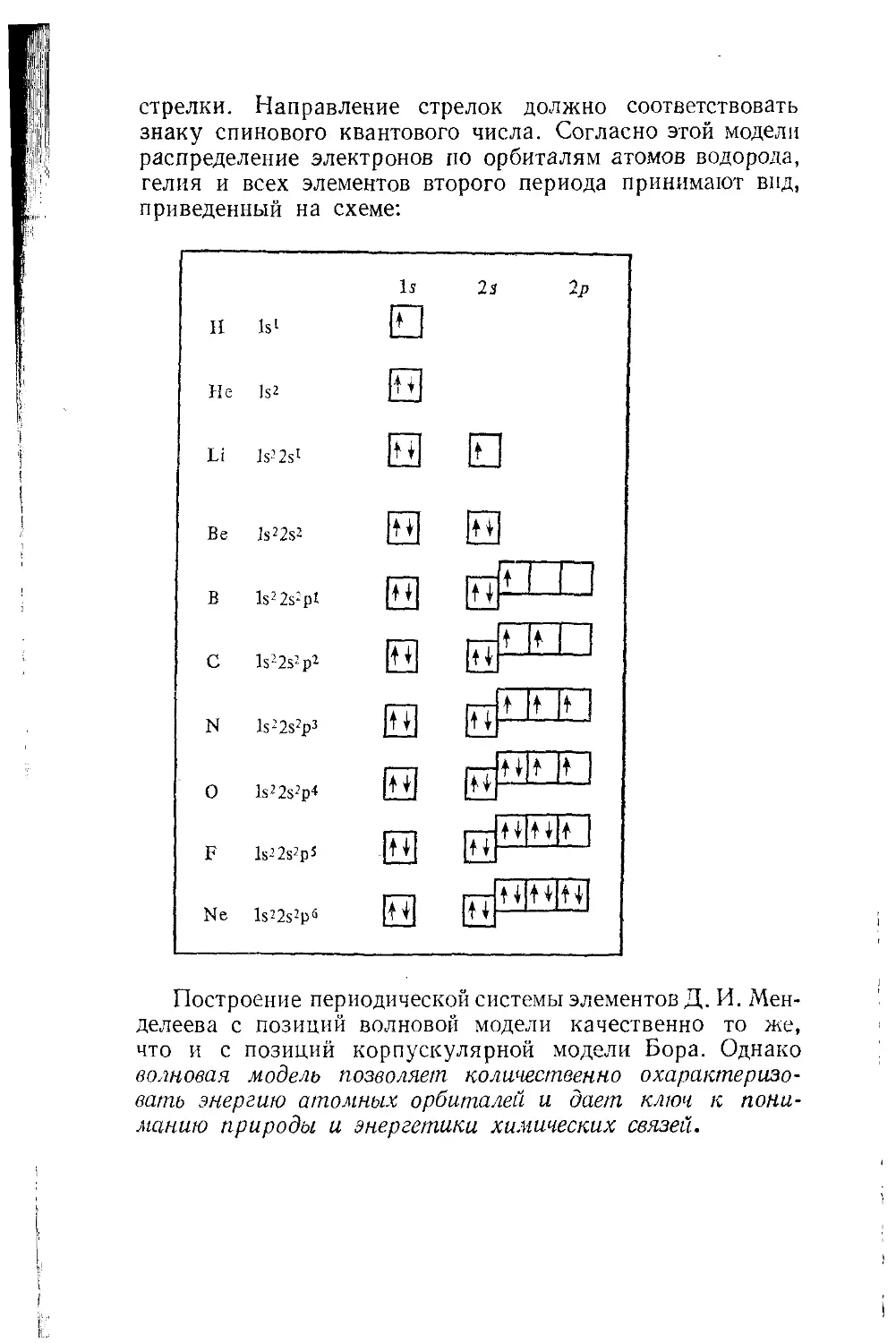
Глава 18. Волновая модель атома 199
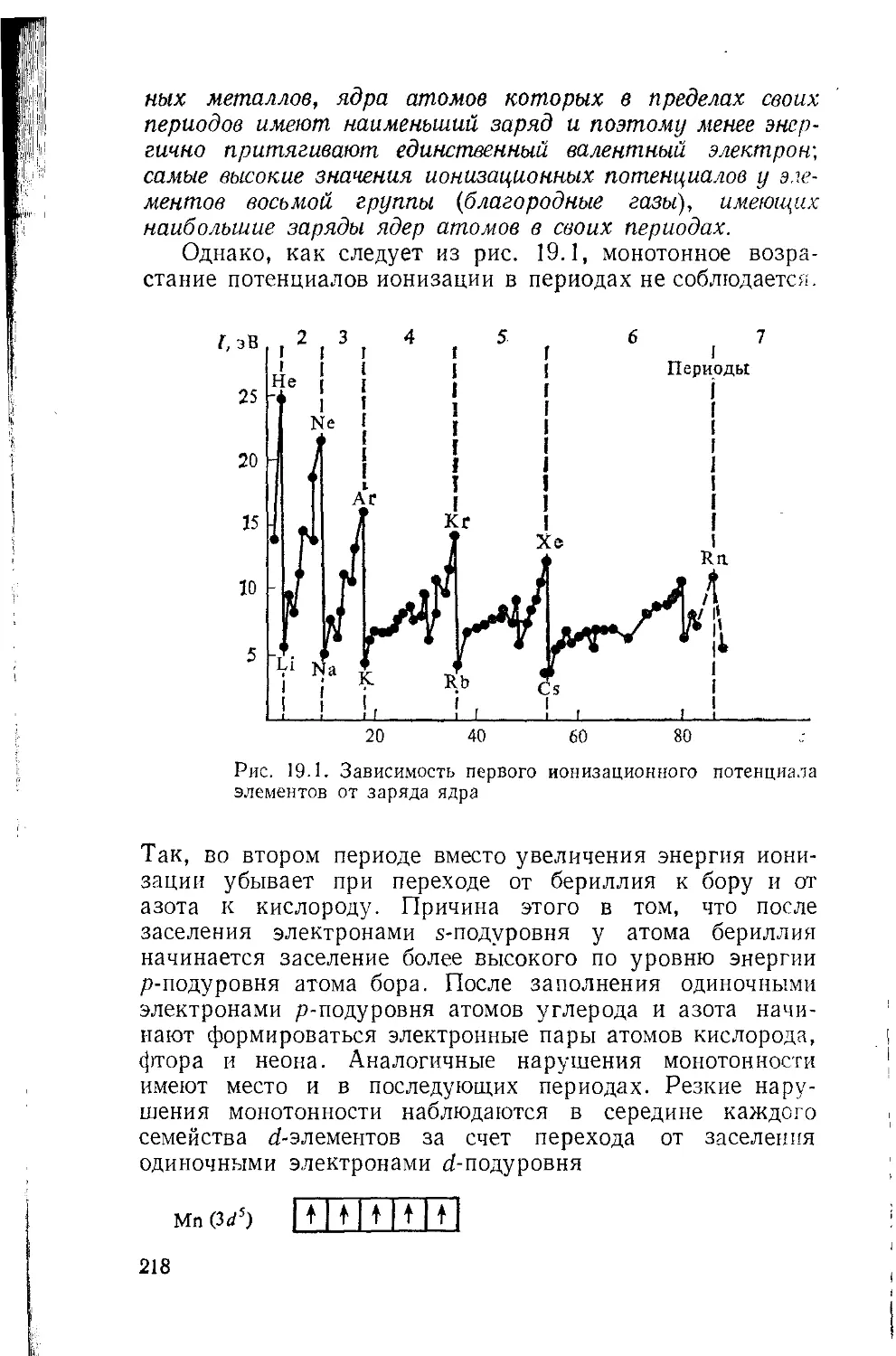
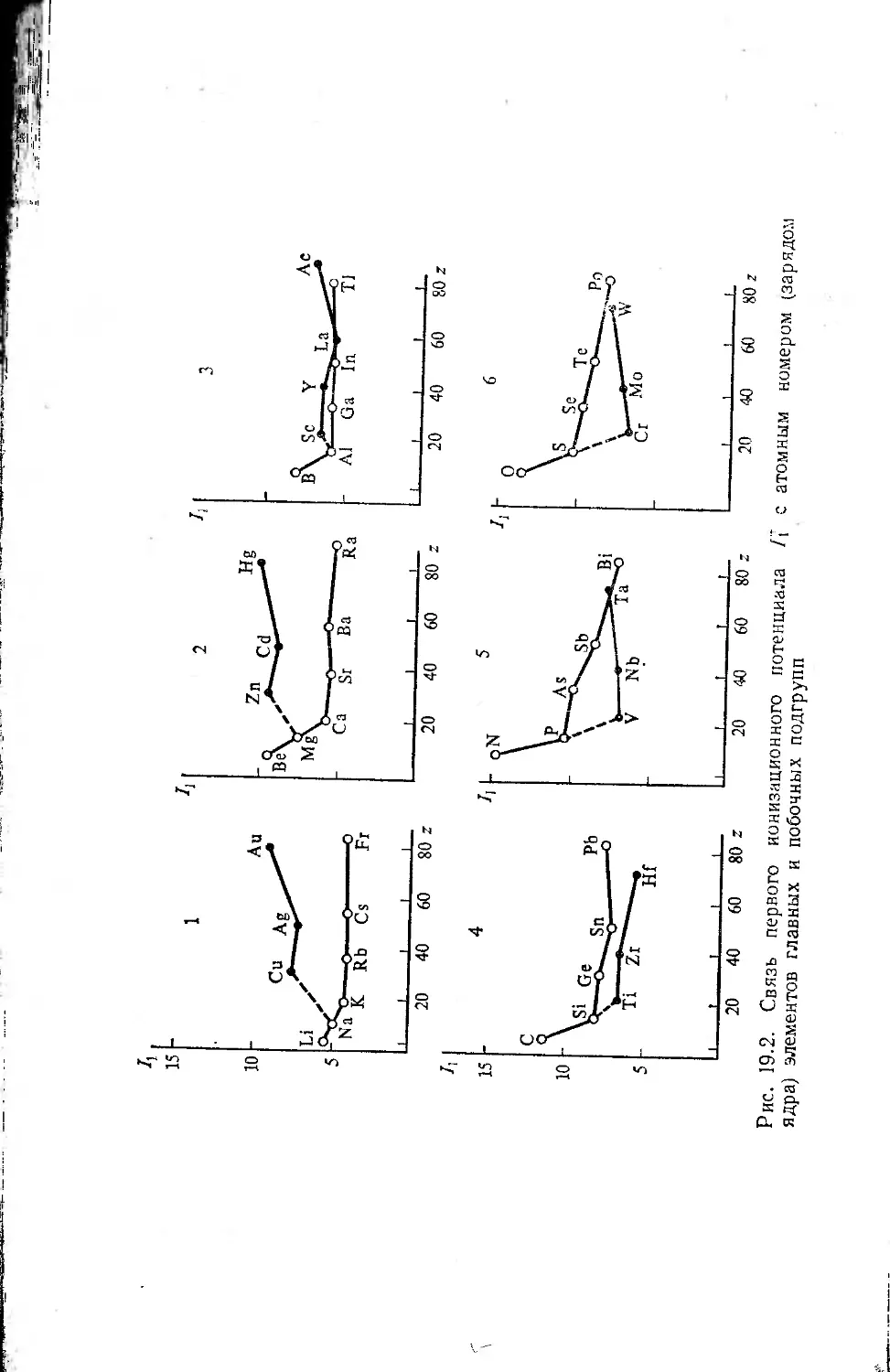
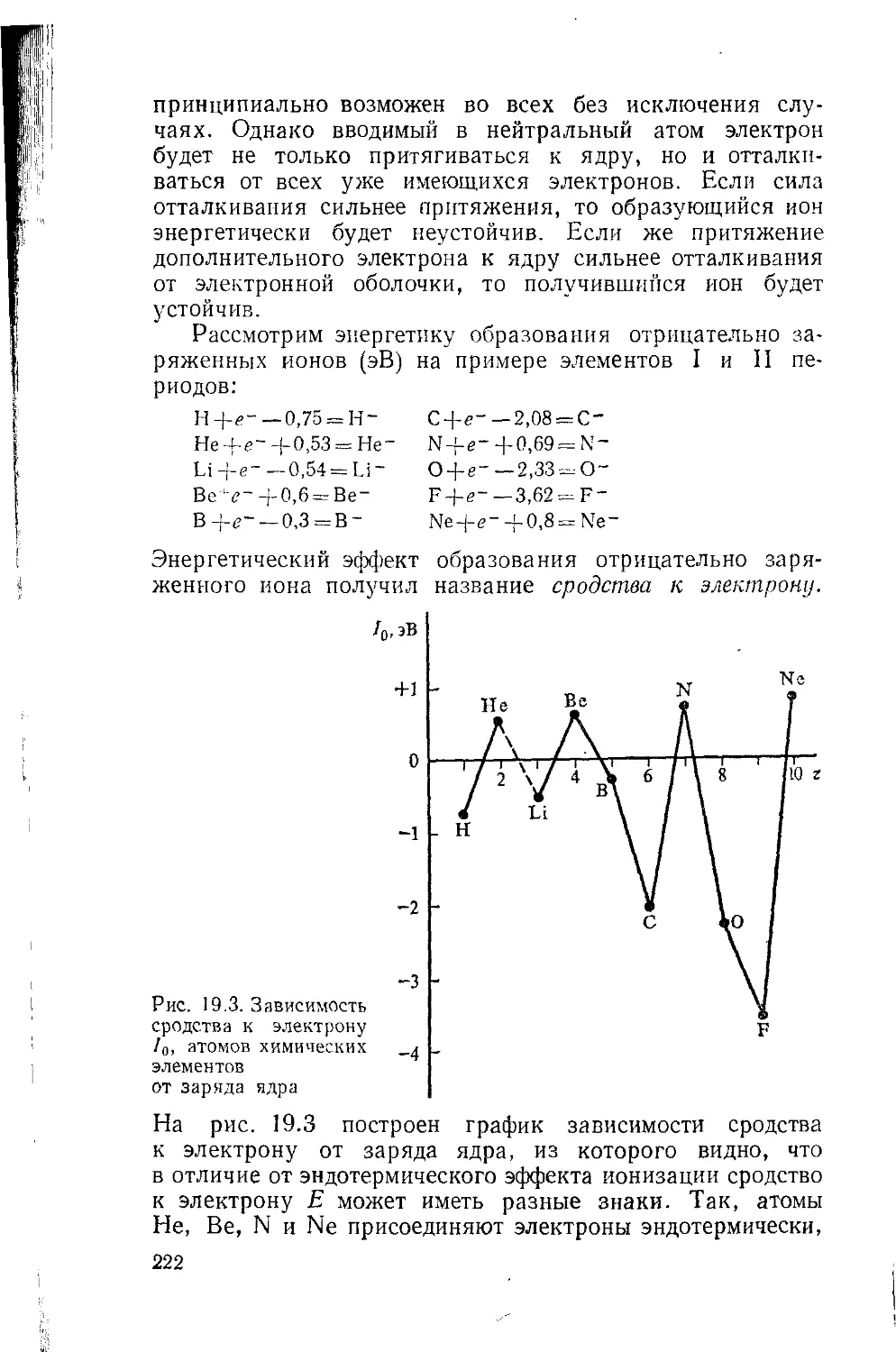
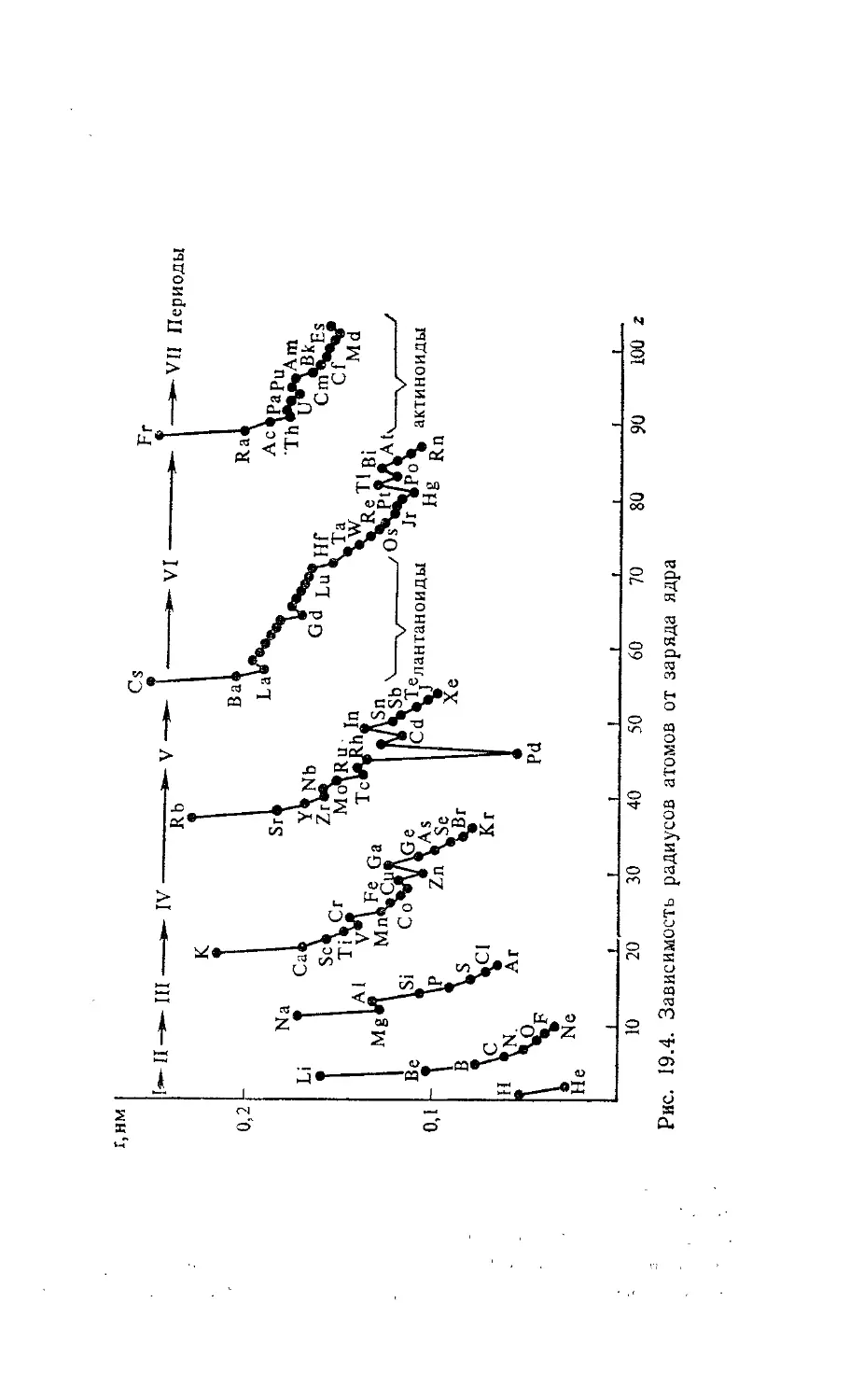
Глава 19. Потенциалы ионизации и эффективные параметры атомов 215
Глава 20. Теория валентных связей (ВС) 233
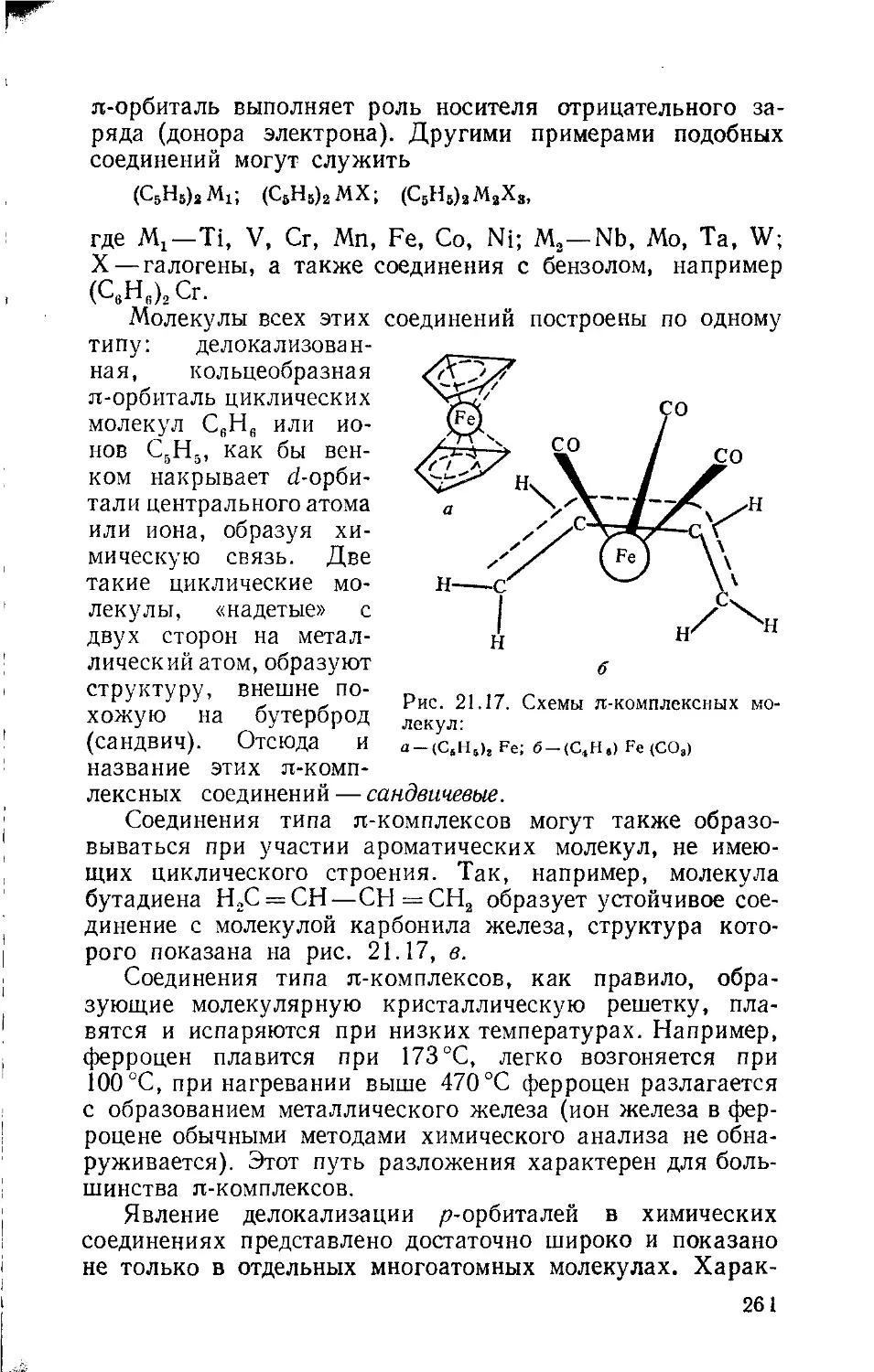
Глава 21. Молекулы с кратными связями и гибридными орбиталями 242
Глава 22. Комплексные соединения и координационный тип химической 264
связи
Глава 23. Теория молекулярных орбиталей (МО) 284
Глава 24. Молекулярные орбитали в молекулах гетеросоединений 298
Глава 25. Строение ионных кристаллов 317
Глава 26. Кристаллические решетки с дефектами 331
Глава 27. Соединения с невалентными связями 346
Приложения 360
Рекомендуемая литература 426
Предметный указатель 427
Предисловие
Основные направления экономического и социального
развития СССР на 1986— 1990 годы и период до 2000 года
предусматривают поднять на качественно новую ступень
производительные силы и производственные отношения
в нашем обществе. Для достижения этой цели необхо-
необходимы улучшение всей системы подготовки инженерных
кадров и активизация преподавания основ естественных
дисциплин в вузах в плане усиления проблемности обу-
обучения, компьютеризации учебного процесса и максималь-
максимального сближения его с производством. Одной из состав-
составных частей этого процесса является подготовка учебников
и учебных пособий, отвечающих указанным требованиям.
Изложение общей и неорганической химии студентам
химико-технологических специальностей требует некото-
некоторых особенностей, поскольку знакомство с этой дисцип-
дисциплиной происходит в первый год обучения до прохожде-
прохождения курсов математики и физики. В связи с этим
обязательной составной частью курса общей и неоргани-
неорганической химии должен быть теоретический раздел, который
состоит из основ учения о строении вещества и теории
химических процессов. Очевидно, что соотношение между
теоретической и описательной частями, полнота и поря-
порядок изложения материала требуют дальнейшей методи-
методической работы.
Построение данного учебного пособия основано на
последовательности ступеней познания теоретических на-
начал химии: стехиометрия, термохимия, эргохимия (хими-
(химическое равновесие и основы химической термодинамики),
хронохимия (основы химической кинетики), начала уче-
ния о строении вещества (строение атома, молекул, жид-
жидкостей, кристаллов и соединений с невалентными связями).
Изложение ведется в свете фундаментальных законов хи-
химии: сохранения массы—энергии, сохранения заряда,
периодического закона Д. И. Менделеева. Такая после-
последовательность не утверждает исключительную правиль-
правильность изложения материала, хотя многолетний опыт
преподавания подсказывает автору некоторые преимущества
предлагаемого построения. С этих позиций основы теории
строения воспринимаются как естественный переход от
теории химических процессов к химии элементов и их
соединений. В том и другом методическом подходе есть
свои слабые и сильные стороны, но можно с уверен-
уверенностью сказать, что при достаточном педагогическом
такте эти трудности легко преодолимы;
Автор приносит глубокую благодарность рецензен-
рецензентам—доктору химических наук, профессору Н. С. Ахме-
тову и лауреату Государственной премии СССР, доктору
химических наук, профессору Я. А. Угаю, чьи обстоя-
обстоятельные замечания облегчили дальнейшую работу над
рукописью и несомненно способствовали ее улучшению.
Автор
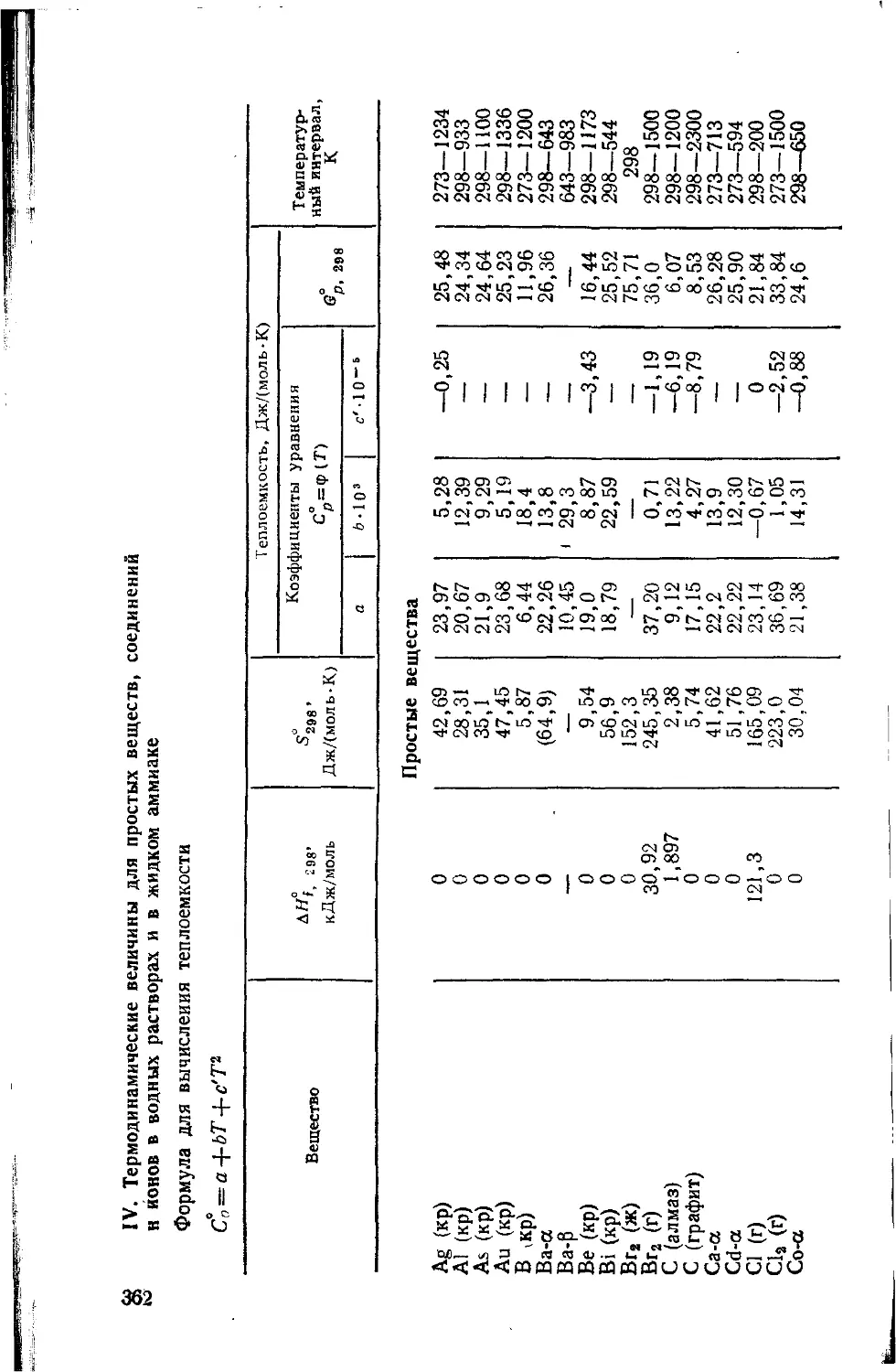
Обозначения
Е ¦—энергия
Е* —энергия активации
е —элементарный заряд, основание натуральных логарифмов
т — масса
те — масса электрона
с —скорость света в пустоте
v —скорость
Q •—тепловой эффект реакции, количество теплоты
А —работа, константа Маделунга, атомная масса
М —молекулярная масса, момент количества движения
р —давление
V —объем, напряжение
ф — потенциал
q —количество электричества
U —полная внутренняя энергия
Н —энтальпия, оператор Гамильтона
ДЯ —изменение (разность) энтальпии
N\ — постоянная Авогадро
R —универсальная газовая постоянная, расстояние между ядрами
атомов
Т —температура, К
С —концентрация, число степеней свободы системы, теплоемкость
S —энтропия, интеграл перекрывания
Дб—энергия Гиббса
К —константа равновесия, число независимых компонентов
k —константа скорости реакции, постоянная Больцмаиа
Д —разность
2 —сумма
Ni —молярная доля
a —степень диссоциации
Ф — число фаз системы
lg —логарифм десятичный
In —логарифм натуральный
п. —главное квантовое число, число молей, число электронов
в элементарном электрохимическом акте
h —постоянная Планка
v — частота колебаний
Я —длина волны
F ¦—постоянная Фарадся, сила
L —произведение растворимости
/ —сила тока, ионизационный потенциал
т — время
Э —химический эквивалент
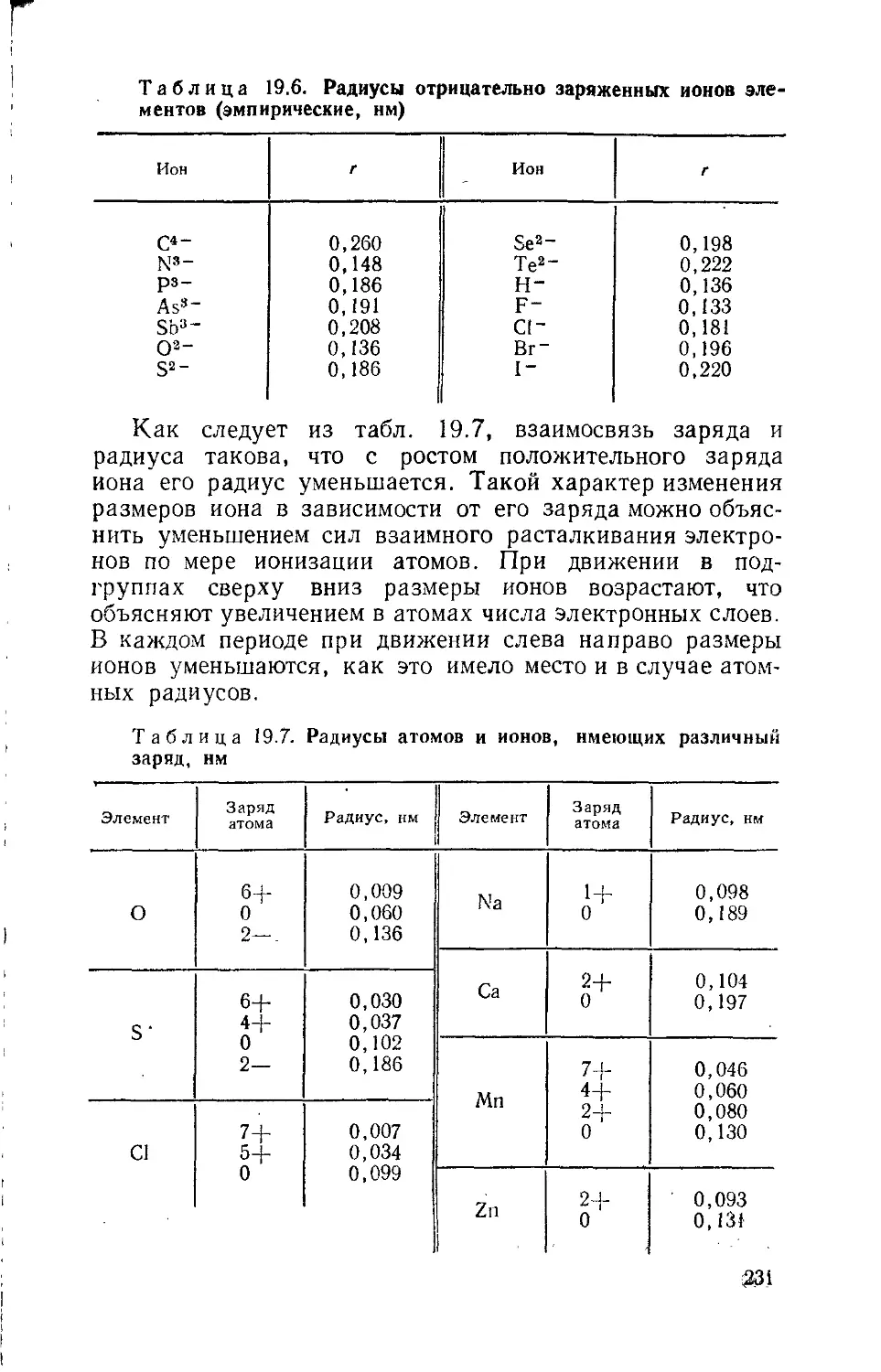
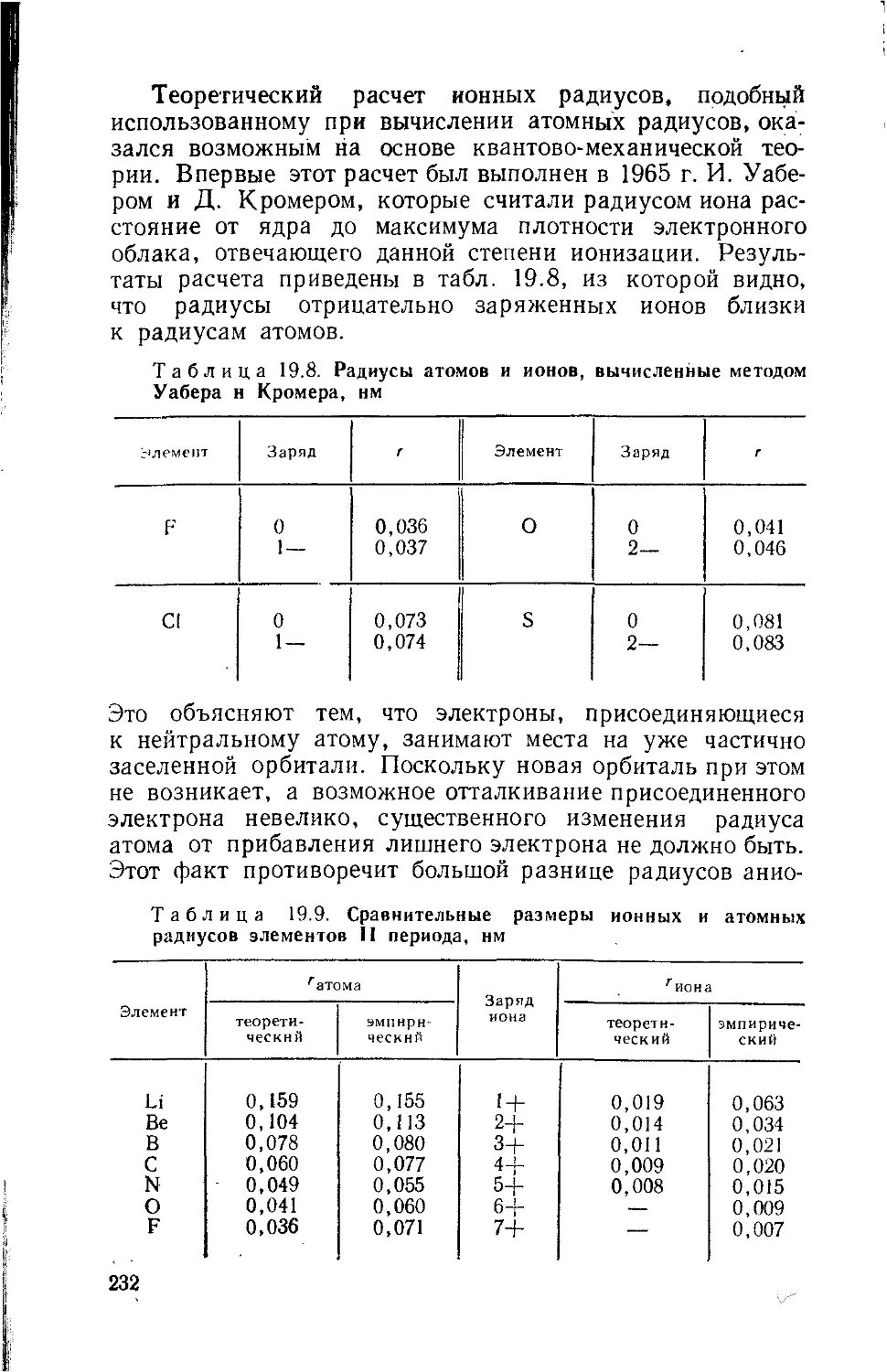
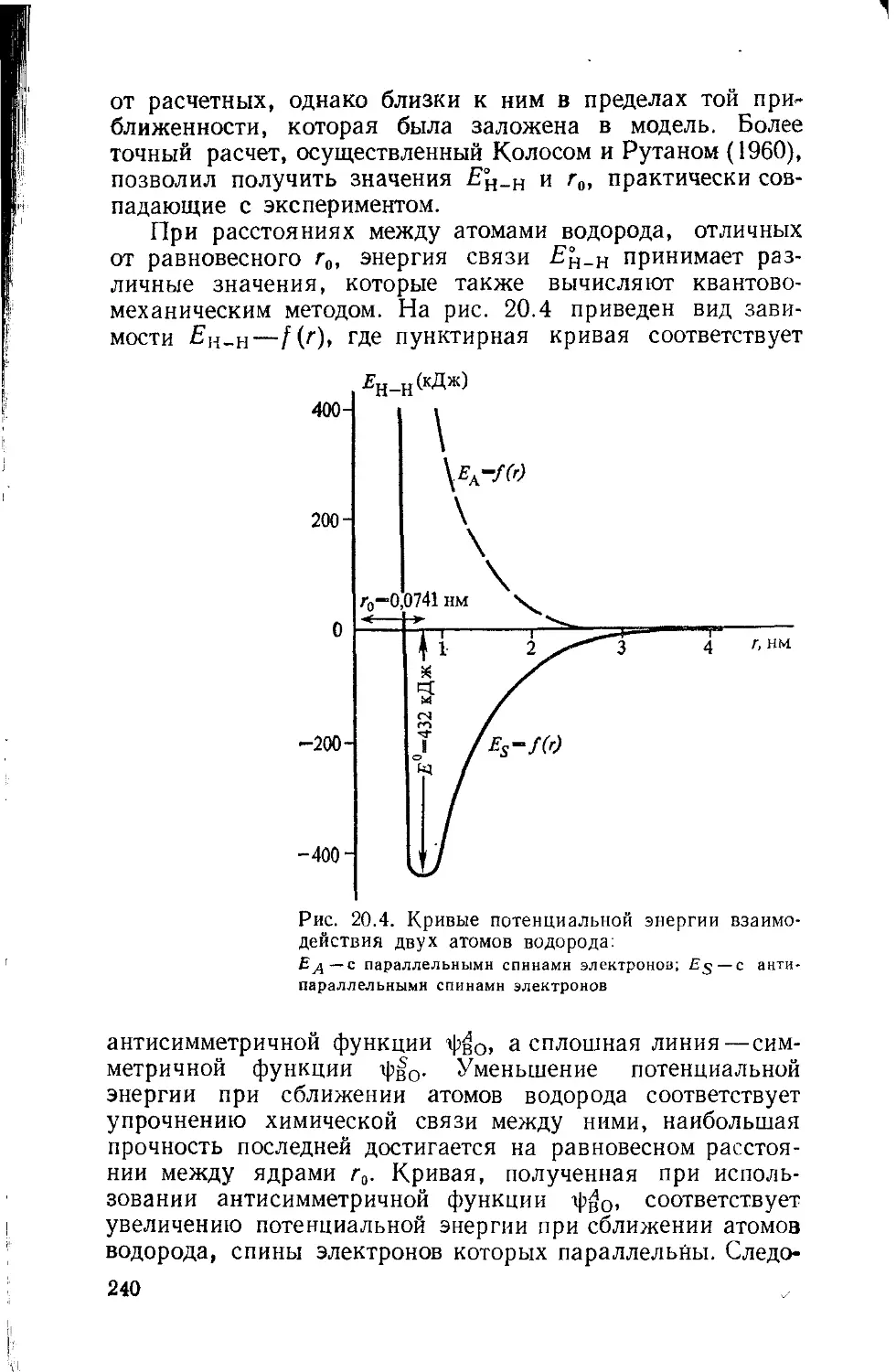
г — радиус атома или иона
Rh —константа Ридберга
mi —магнитное квантовое число
/ —побочное квантовое число
s —спин электрона, энергетический подуровень при 1 = 0
у ¦—гидромагнитное отношение
[X —магнитный момент, момент диполя
\|) •—волновая функция
V —оператор Лапласа
р ¦—энергетический подуровень при 1=\
d —энергетический подуровень при / = 2
f —энергетический подуровень при 1 = 3
g ¦—энергетический подуровень при / — 4
г —заряд
КЧ-—координационное число
Основные
законы
химии
Фундаментальные законы естествознания и химии, как части
естественных наук, являются научно-теоретической базой вскрытия
и количественного описания причинно-следственных связей химической
формы движения. Фундаментальными следует считать законы
сохранения массы-энергии, сохранения заряда, периодичности развития.
Эти законы выполняются в любых условиях и масштабах. Множество
других законов ограничено действием, они носят частный характер,
но в пределах действия не должны противоречить законам
фундаментальным.
Глава 1
Фундаментальные законы
и законы стехиометрии
Законы сохранения массы — энергии, сохранения
заряда, периодичности развития; законы эквива-
эквивалентов, постоянства состава, кратных отношений,
объемных отношений, удельных теплоемкостей.
Химия—одна из древних наук, которая в современ-
современном обществе, все больше смыкаясь с физикой и опираясь
на математику и вычислительную технику, ускоренным
темпом проникает в различные отрасли производства.
Одной из важнейших вех на пути развития химии
явилось открытие Д. И. Менделеевым периодического за-
закона элементов, ставшего основой химической система-
систематики, развития учения о строении вещества и создания
теории химических процессов. Большое значение для
углубления представлений о природе химических превра-
превращений имело их энергетическое, точнее, термодинамиче-
термодинамическое описание. Именно термодинамика внесла в химию
представление о количественной мере направления и глу-
глубине химических процессов, т. е. о химическом сродстве,
или энергии Гиббса. Термодинамический метод позволил
количественно связать химические превращения с влия-
влиянием температуры и давления, с изменением концентра-
концентраций веществ. На этой основе периодический закон обрел
прочную базу для количественного описания свойств еще
неизученных, а иногда и несинтезированных веществ.
Успешно справляясь с задачей количественного опи-
описания химических превращений, термодинамика не вскры-
вскрывает их причинно-следственные связи. В результате на
принципиально новой основе возникла необходимость
создания теоретического фундамента химии—квантовой
механики. Так возникло учение о строении вещества, хи-
химической связи и валентности, которое в существующем
виде играет важную роль в понимании химической формы
движения. Химия как один из важнейших разделов есте-
естествознания в своем многообразии опирается на совокуп-
совокупность законов природы, обладающих огромной познава-
познавательной и преобразующей силой. Однако не все законы
в равной степени всесторонне охватывают причинно-след-
причинно-следственные связи описываемых ими явлений и фактов.
Одни из них, более частные, имеют ограниченную область
действия, другие являются общими для всего естество-
естествознания и в связи с этим получили название фундамен-
фундаментальных—это законы сохранения заряда, сохранения
массы-энергии, периодичности развития.
Имея в своей основе фундаментальные законы, химия
на всех этапах развития остается наукой о веществах и
их превращениях. Под веществом понимают атомы хими-
химических элементов и их соединения во всех состояниях:
твердом, жидком, газообразном, плазменном (при сверх-
сверхвысоких температурах или в электрических разрядах) и
сверхметаллическом (при гигантских давлениях). Однако
структура вещества чрезвычайно многообразна, поскольку
сами атомы состоят из элементарных частиц сложной
природы (см. приложение III). Эти частицы различны
по массе, времени жизни, заряду и таким менее привыч-
привычным характеристикам, как спин, странность, очарование
и др. В 1964 г. М. Гелл-Манн и Дж. Цвейг ввели пред-
представления о кварках — первичных микрочастицах, из ко-
которых строятся все остальные.
Для химии наибольший интерес представляют ста-
стабильные элементарные частицы, такие, как электроны,
протоны и нейтроны, которые энергично взаимодействуют
между собой, образуя первичную форму вещества — ато-
атомы химических элементов. Атомы состоят из положительно
заряженного чрезвычайно плотного протонно-нейтронного
ядра и диффузной отрицательно заряженной электронной
оболочки. Подчиняясь законам квантовой механики, ста-
статистики и электродинамики, атомы взаимодействуют между
собой, образуя бесчисленное множество химических со-
соединений. Атомы одного вида образуют гомосоединения,
атомы разных видов — гетеросоединения, различные по
составу и агрегатному состоянию. Все это и есть химиче-
химическая форма материи — вещество.
Важнейшим свойством вещества является наличие
массы, согласно второму закону Ньютона, определяемой
как мера инерции. Однако это свойство вещества прояв-
проявляется только под действием сил внешнего воздействия
и поэтому является пассивным. К активным свойствам
вещества следует отнести гравитацию и заряд, действие
которых связано с явлениями тяготения масс, притяже-
притяжения (отталкивания) зарядов. Эти свойства указывают на
наличие связи между двумя формами материи—вещест-
материи—веществом и полем. Для характеристики химической природы
веществ особенно важен заряд. Нейтрон имеет заряд.
равный нулю; электрон и протон обладают одинаковыми,
но противоположными по знаку зарядами, величина ко-
которых равна 1,6-К)9 Кл. Заряд протона и электрона
далее неделим и поэтому получил название элементар-
элементарного. При образовании из нейтронов и протонов атомных
ядер и при формировании около них электронных обо-
оболочек заряды элементарных частиц алгебраически сум-
суммируют. Так, если ядро атома гелия, состоящее из двух
протонов и двух нейтронов, имеет заряд, равный +2е, а
его электронная оболочка из двух электронов с зарядом
—2е, то в целом атом гелия (как и атомы других эле-
элементов) электронейтрален — общая сумма положительных
и отрицательных зарядов равна нулю. В общем случае
алгебраическая сумма зарядов любой изолированной си-
системы постоянна—это утверждение, абсолютное без иск-
исключения, является фундаментальным законом, поскольку
не может быть выведено из других законов.
Два точечных заряда Qf и Q2 взаимодействуют между
собой согласно закону Кулона
Е- I QlQ*
где Е—энергия взаимодействия; Qt, Q3—точечные заряды;
/¦—расстояние между ними. Если заряды имеют один и
тот же знак, то при взаимодействии они отталкиваются;
если же заряды разноименны, то притягиваются.
Взаимодействие между атомными ядрами и электрон-
электронными оболочками в атомах и химических соединениях
осуществляется главным образом благодаря наличию у
них зарядов и служит основой при возникновении хими-
химической связи и образовании химических соединений.
В ядрах атомов между протонами и нейтронами дейст-
действуют особые ядерные силы, которые во много раз больше
сил взаимодействия зарядов. Именно поэтому в химиче-
химических реакциях даже при самых высоких температурах
A04 —10? К) атомные ядра остаются устойчивыми, тогда
как электронные оболочки атомов испытывают глубокие
изменения.
Взаимосвязь массы и энергии, вскрытую А. Эйнштей-
Эйнштейном и отражающую фундаментальный закон сохранения
массы-энергии, выражают соотношением
Е = тс\ A.1)
где Е и т—эквивалентные значения энергии и массы;
с—скорость света в пустоте, равная 2,997925 • 10^ м/с.
ю
Это означает, что никакой химический процесс не осуще-
осуществляется без изменения массы вещества.
Например, при сгорании водорода в кислороде с об-
образованием водяного пара по схеме
выделяется и рассеивается в окружающей среде 241 835 Дж
энергии. С этой энергией рассеивается масса вещества:
Е 241 835 Дж о
0
Изменение массы в 102 кг обнаружить даже современ-
современными методами взвешивания невозможно, поскольку луч-
лучшие аналитические весы позволяют заметить разницу
масс около 10~9 кг. Это обстоятельство и послужило ос-
основой формулировки закона сохранения веса веществ,
экспериментально доказанного М. В. Ломоносовым на заре
современной химии в середине XVIII в.
Понятие вещества теснейшим образом связано с. по-
понятием движения. Связь массы вещества с движением
проявляется не только в пассивном сопротивлении изме-
изменению скорости, но и в прямой зависимости ее от скорости:
^ПОКОЯ /| 2\
"'ДВИЖ уг =~ ' ('•*>
здесь /идвиж—масса тела, движущегося со скоростью v;
тпокоя — масса покоя того же тела; с—скорость света в
пустоте. Приведенные в тексте уравнения A.1) и A.2)
означают, что энергия, сообщаемая массе вещества, рас-
расходуется не только на увеличение скорости, но и на при-
приращение массы. С приближением скорости тела к скорости
света почти вся сообщаемая телу энергия идет на увели-
увеличение неограниченно возрастающей массы. Этот эффект,
названный релятивистским, необходим для понимания
строения вещества и играет важную роль в мире элемен-
элементарных частиц и атомов, где электроны могут перемещаться
со скоростями, приближающимися к скорости света. След-
Следствием этого факта является наличие у атомов и молекул
строго определенной массы (с точностью до ничтожных
поправок на изменение массы вещества за счет тепловых
эффектов реакций). Таким образом, масса атомов и моле-
молекул служит специфической характеристикой и заметно
* Здесь и в дальнейшем условимся обозначать жидкое и расплав-
расплавленное состояние веществ фигурными скобками, газообразное—круг-
газообразное—круглыми и твердое — квадратными скобками.
И
не изменяется от формы движения (механическое, хими-
химическое и др.)- В обычном так называемом макромире
скорости движения существенно меньше скорости света,
и поэтому релятивистский эффект практически равен нулю.
Напротив, в отличие от неощутимых при химических ре-
реакциях изменений массы вещества эквивалентные им энер-
энергетические эффекты реакций изменяются в широких пре-
пределах. Так, 1 моль алюминия при окислении кислородом
порождает 837 кДж теплоты, 1 моль жидкого бензола
СвНй при сгорании в кислороде—3275 кДж и т. д. По-
Подобные энергетические эффекты являются специфической
чертой всех химических реакций и количественной мерой
химической формы движения.
В настоящее время доказано, что любые движущиеся
частицы одновременно проявляют корпускулярные и вол-
волновые свойства. В макромире при больших массах и ма-
малых скоростях движения волновая природа вещества
остается незаметной. В случае же движения микроскопи-
микроскопических частиц (электронов, атомов, молекул), движущихся
с большими скоростями, волновая природа становится
определяющей. Все сказанное свидетельствует о важности
закона сохранения массы энергии, его универсальности,
всеобщности и, следовательно, фундаментальности.
Третий фундаментальный закон естествознания — закон
периодичности развития. Примерами периодичности раз-
развития могут служить системы элементарных частиц, ядер
атомов, электронных оболочек атомов, химических соеди-
соединений и т. д. Строгий закон периодичности свойств эле-
элементарных частиц до конца еще не открыт.
Хорошо изученным этапом усложнения структуры
корпускулярной формы материи являются продукты ассо-
ассоциации нуклонов—семейства атомных ядер. Расположен-
Расположенные в последовательности возрастания заряда, они пред-
представляют собой ряд периодических чередований устойчи-
устойчивости ядерных конфигураций. Это означает, что строение
атомных ядер, все более усложняющееся по мере пере-
перехода от легких элементов к тяжелым, периодически
повторяет некоторую структурную комбинацию протонов
и нейтронов. Наиболее устойчивые ядра можно характе-
характеризовать так называемыми «магическими числами»—-2, 8,
20, 28, 50, 82, 126, которые представляют собой числа
протонов (р) и нейтронов (п) в атомных ядрах, например:
He*B/?2n), O"(8/?8n), Ca40B0/?20n), Ni<"> B8/>32п),
Sr88 C8/?50rt), ZrMD0/?50n), Sn119 EQp69n), Ba138 E6p82n),
РЬ208(82р126/г). Ядра этих атомов не только обладают
12
повышенной устойчивостью, но отличаются также боль-
большей распространенностью в природе и повышенным чис-
числом стабильных изотопов. При этом наилучший эффект
достигается в тех случаях, когда совпадают нейтронное
и протонное «магические числа», например, у Не4 Bр2п)
и Оы{8р8п).
Однако все, что связано со строением ядер, изучают
в курсе ядерной физики. Химические же свойства опре-
определены строением и периодическим изменением свойств
электронных оболочек атомов. Эта периодичность была
обобщена Д. И. Менделеевым в 1869 г. в открытом и
сформулированном им периодическом законе, а периоди-
периодическая система элементов явилась количественным вопло-
воплощением этого закона. Периодичность изменения свойств
атомов химических элементов, как известно, обязана
своим происхождением послойному строению электронных
оболочек и строго ограниченной емкости каждого элект-
электронного слоя. Так, первый электронный слой в атоме не
может содержать более 2 электронов, второй—более 8,
третий—более 18, четвертый—более 32 и т. д. Последо-
Последовательному заполнению каждого из этих слоев соответст-
соответствуют семейства элементов, свойства которых изменяются
монотонно. Каждый раз при переходе к новому элект-
электронному слою структура во многом повторяет строение
предыдущего, представляя качественно прежнюю, но ко-
количественно иную монотонность изменения свойств эле-
элементов. Что касается периодичности изменения свойств
различных классов химических соединений, то здесь вся
химия, во всем ее многообразии являет собой торжество
и всеобщность периодического закона.
Помимо фундаментальных законов химии, являющихся
основой количественного описания и вскрытия причинно-
следственных связей химических превращений, химическая
форма движения материи подчиняется большому числу
менее общих (частных) законов. Так, для химии чрезвы-
чрезвычайно важны законы стехиометрии, устанавливающие ко-
количественные соотношения элементов в химических со-
соединениях, и уравнения химических реакций. Открытый
немецким физиком И. Рихтером A792—1794) закон экви-
эквивалентов описывает количественные соотношения хими-
химически взаимодействующих веществ: его формулируют так:
массы (т) реагирующих веществ пропорциональны их эк-
эквивалентам (Э), т. е.
13
Эквивалентом называют реальную или условную частицу вещества,
которая в данной кислотно-основной реакции эквивалентна одному
иону водорода или в дайной окислительно-восстановительной реак-
реакции— одному электрону. Фактором эквивалентности служит число,
обозначающее долю реальной частицы вещества, эквивалентной одному
иону водорода в данной кислотно-основной реакции, или одному
электрону в данной окислительно-восстановительной реакции. Фактор
эквивалентности может быть равен или меньше единицы. Молярная
масса эквивалента (ММЭ) вещества — масса одного моля эквивалента
этого вещества, равная произведению фактора эквивалентности на его
молярную массу. В соответствии с законом эквивалентов массы взаи-
взаимодействующих без остатка веществ пропорциональны их эквивалентам.
Прокаливая, например, чистую металлическую медь (масса mcu) на
воздухе при 400 — 500° С, получаем оксид меди (масса таю). Раз-
Разность mcuO — "iCu дает массу связанного в оксиде кислорода mo-
Используя закон эквивалентов, составляем пропорцию, где ММЭо = 8,
8 то '
откуда находим численное значение ММЭси- Расчет ММЭ основан на
определении соотношений масс, в которых вещества без остатка всту-
вступают в реакцию друг с другом. ММЭ простых веществ вычисляют
при помощи различных методов—весовых, объемных (для газов),
электрохимических (для окислительно-восстановительных реакций).
Применяя закон эквивалентов, можно количественно определить со-
содержание данного элемента в соединении с другим элементом, ММЭ
которого известно. Электрохимическим методом находят массу ме-
металла, выделившегося на катоде в процессе электролиза определенного
количества соединения, содержащего этот металл, и рассчитывают
его ММЭ.
ММЭ сложных химических соединений (гидроксидов, солей и др.)
вычисляют по данным реакций, идущих без изменения степени окис-
окисления элементов, входящих в состав реагирующих соединений. На-
Например, ММЭ кислоты в реакции нейтрализации
НС1 + NaOH = NaCl + Н2О
можно найти по ММЭ щелочи: ММЭ1( = —— 40, где ММЭщ = 40.
Шщ
В общем случае ММЭ кислот, оснований и солей в реакциях обмен-
обменного разложения находят из соотношения
ММЭ = М/п,
где М — молярная масса вещества; п — сумма зарядов функциональ-
функциональных групп, которые замещаются в процессе реакции.
ММЭ сложных веществ в окислительно-восстановительных реак-
реакциях находят из соотношения
ММЭ = М/г,
где z — число элементарных зарядов (электронов), участвующих в
данном окислительно-восстановительном процессе. Например, ММЭ
перманганата калия в реакции
КМпО4 + 8НС1 = MnCl2 + 6/2С12 + КС1 + 4Н2О
где МпО4"-|-8Н+-г-5е = Мп2+ _|_4Н2О, равна 1/5 молярной массы
(здесь 1/5—фактор эквивалентности, который рассчитывают иа осно-
основании стехиометрии данного химического процесса).
14
Закон постоянства состава химических соединений,
открытый в результате многолетнего A801 —1808) науч-
научного спора французских химиков Ж. Пруста и К. Бер-
толле, формулируют следующим образом: каждое хими-
химическое соединение, каким бы способом оно не было полу-
получено, имеет один и тот же постоянный состав.
Развитие химии в последующие годы привело к вы-
выгоду о том, что наряду с химическими соединениями
постоянного состава существуют также соединения пере-
переменного состава, в силу чего закон имеет ограниченное
приложение. По предложению акад. Н. С. Курнакова
соединения переменного состава названы бертоллидами,
а постоянного—дальтонидами. Примерами соединений
переменного состава могут служить карбиды титана (TiC1_!/),
циркония (ZrC,_v).
Закон кратных отношений установлен Дж. Дальто-
Дальтоном A803) и заключается в следующем: в двух соедине-
соединениях, образованных из одних и тех же элементов, на
одно и то же массовое количество одного элемента при-
приходятся такие массовые количества другого элемента, ко-
которые относятся, как небольшие целые числа. Так, на-
например, в оксидах углерода СО и СО.2 на одну массовую
часть углерода приходятся разные количества кислорода
и отношение их между собой равно 1:2.
Если в химических реакциях принимают участие газы,
то, как установил Ж. Гей-Люссак A808), вступает в
силу закон объемных отношений: объемы вступающих в
реакцию газов относятся между собой, а также к объемам
образующихся газообразных продуктов, как небольшие
целые числа.
Исходя из простоты объемных отношений в реакциях
между газами, итальянский физик А. Авогадро сформу-
сформулировал закон: в равных объемах различных газов при
одинаковых внешних условиях содержится одинаковое число
молекул. Из закона Авогадро следует:
1) одинаковое число молекул различных газов при
равных условиях занимает одинаковый объем;
2) один моль различных газов при температуре 273,16 К
и давлении 1,013-105 Па занимает объем, равный 0,0224 м3,
в котором содержится 6,02-1023 структурных единиц
(атомов, молекул), — это постоянная Авогадро (/VA).
Один моль газа связан следующим соотношением с
объемом, давлением и температурой:
PV^RT, A.3)
15
где р—давление (Па); V—объем газа (м3), Т — темпера-
температура (К); R — универсальная газовая постоянная. Выра-
Выражение A.3) названо уравнением состояния идеального газа.
Для п молей идеального газа уравнение состояния при-
принимает вид
ZLRT,
которое называют уравнением Клапейрона—Менделеева.
Закон удельных теплоемкостей предложен француз-
французскими учеными Дюлонгом и Пти A819). По этому закону:
произведение удельной теплоемкости (с) простого вещества
в твердом состоянии на атомную массу (А) этого эле-
элемента является величиной приближенно постоянной:
Л-ся;25,9-10:) Дж/К,
удельную теплоемкость выражают количеством теплоты,
необходимым для нагревания 1 моль простого вещества
на Г С. Использование этого закона позволило опреде-
определить атомные массы ряда элементов, значения которых
в дальнейшем уточнялись. Открытие законов кратных
отношений и удельной теплоемкости сыграло важную роль
в развитии атомной теории. Применение законов стехио-
стехиометрии нашло свое отражение в составлении вещественного
баланса химических реакций, эти вопросы достаточно
подробно будут рассмотрены в последующих главах.
Классы
химических
соединений
и типы реакций
Для понимания и простейшего количественного описания химических
и фазовых превращений необходимо знание законов стехиометрии,
т. е. законов, устанавливающих количественные соотношения масс
элементов, входящих в состав химических соединений, а также
соотношений масс простых и сложных веществ, участвующих
в химических реакциях. В прикладной (инженерной) химии законы
стехиометрии составляют основу вещественного (материального)
баланса производственных процессов.
Часть
II
8ОН
2NO
+ ^
4Н7О
2NO3
8Н,О +
Институт Й70й88й Знергвв
вм. й, В. Начатом
БИБЛИОТЕКА
Глава 2
Основные классы простых
и сложных соединений
Гомо- и гетеросоединения. Простые вещества. Би-
Бинарные гетеросоединения — оксиды, хлориды.
Сложные гетеросоединения —- гидрокснды, соли,
оксохлориды.
Основной особенностью атомов химических элементов
является их способность образовывать химические соеди-
соединения. Причины, объясняющие эту особенность и способы
ее количественного и качественного описания, будут рас-
раскрыты далее, поскольку это возможно лишь на основе
данных о строении атома и современных представлений
о химической связи. Следует лишь отметить, что такая
тенденция для химических элементов чрезвычайно ха-
характерна. Исключение составляют лишь некоторые пред-
представители группы «благородных» газов — гелий, неон и
аргон, которые при всех условиях атомарны и не прояв-
проявляют химической активности. Все остальные элементы
периодической системы в атомарном состоянии химически
активны и образуют многочисленные гомо- и гетеросоеди-
гетеросоединения.
Гомосоединения, простые вещества. Гомосоедимениями
называют продукты взаимодействия атомов одного и того
же элемента. Примерами гомосоединений могут служить
молекулы газов—О8, 02, Р4, Cl2, SB; жидкостей — ртуть,
бром; твердых веществ—железо, кремний.
Среди гомосоединений элементов периодической си-
системы важную роль отводят простым веществам—терми-
веществам—термически наиболее устойчивым гомосоединениям данного эле-
элемента в характерном для него агрегатном состоянии
при давлении 1 атм* и температуре 298 К. Условимся,
что термическую устойчивость определяют численным
значением и знаком теплового эффекта химической реак-
реакции разложения или образования соединения. Тепловой
эффект реакции обычно выражают числом джоулей (Дж),
знак минус (—) перед тепловым эффектом соответствует
* В Международной системе единиц (СИ) давление рекомендовано
измерять не в физических атмосферах, а в Паскалях A атм=1,01325х
ХЮ5 Па, или 1,013 гкПа.
18 '"
L .
экзотермическому процессу, знак плюс (+)—эндотерми-
(+)—эндотермическому.
Если атомы элемента химически пассивны и не обра-
образуют гомосоединений, то простым веществом будет одно-
одноатомный газ (благородные газы). Из двух молекулярных
форм кислорода—О2, О3 — двухатомная молекула экзо-
экзотермически образуется из трехатомной
г/з(Оз)-Н2кДж=(О2)
что свидетельствует об устойчивом существовании моле-
молекул О2. Следовательно, в силу данного определения к
простым веществам можно отнести только молекулы О2.
Если в твердом состоянии гомосоединение существует в
двух и более кристаллических модификациях, простым
веществом также нужно принять только одно—наиболее
термически устойчивое. Например, углерод образует три
кристаллические [ формы — алмаз, карбин и графит. По-
Поскольку алмаз экзотермически превращается в графит
простым веществом считают только графит (карбин не
изучен). Исключением является фосфор, простым веществом
для которого принято считать белый фосфор, хотя красный
представляет более устойчивую модификацию.
Бинарные гетеросоединения. Гетеросоединениями на-
называют продукты взаимодействия атомов различных эле-
элементов. Гетеросоединения, беспредельно разнообразные
по составу и строению, делят на соединения стехиомет-
рического состава (истинные или дальтониды, например
Н2О) и нестехиометрического состава (бертоллиды, напри-
например ZrN0i6i)). Простейшие гетеросоединения, состоящие из
атомов двух видов, называют бинарными. Реальное число
всех бинарных соединений велико, среди них можно вы-
выделить соединения, в которых наиболее отчетливо прояв-
проявляются индивидуальные особенности образующих их
элементов. К таким бинарным соединениям в первую
очередь следует отнести соединения различных элементов
с водородом, кислородом и хлором.
Для водорода характерно простейшее строение атома,
состоящего из ядра и электронной оболочки с одним
электроном. Свободные атомы водорода обладают высокой
реакционной способностью и экзотермически связываются
в молекулы Н2:
2(Н)-217,9
19
Такой молекулярный водород при температуре 298 К и
давлении 1,013 гПа считают простым веществом.
Молекулярный водород энергично взаимодействует с
наиболее активными металлами и неметаллами (тепловой
эффект в кДж):
1/2(Н2)-60 =
[Са} + (Н2)-!74 = [СаН2] l/« (Cl2) + 1/2 (Н2)-92 = (НС1)
[Ва] + (Н2)-178=[ВаН2] V. (F2) + V2 (H2)-269 = (HF)
Степень окисления * водорода в соединениях с металлами
— 1, что соответствует способности атома водорода при-
присоединять экзотермически один электрон
~ —71 кДж = (Н~)
в связи с чем гидриды активных металлов (щелочных,
щелочно-земельных) при обычных условиях представляют
собой ионные кристаллы, состоящие из положительно за-
заряженных ионов металла и отрицательно заряженных
ионов водорода. В соединениях с неметаллами водород
имеет степень окисления +1. что соответствует слабовы-
раженной способности атомов водорода отдавать электрон:
(Н)— е-+ 134 кДж = (Н+)
Поскольку этот процесс эндотермичен, для водорода ион-
ионное состояние с зарядом 1+ в химических соединениях
нехарактерно. Большинство соединений неметаллов с водо-
водородом— полярные.
Среди бинарных водородистых соединений следует
отметить семейство углеводородов с безграничным разно-
разнообразием форм. Многочисленные представители углеводо-
углеводородов объединены в гомологические ряды с общими фор-
формулами С„Н2п+2, С„Н2„, С„Н3„_, и др.
Кислород образует множество соединений со всеми
другими элементами периодической системы (кроме гелия,
неона и аргона). В соединениях он обычно имеет степень
окисления, равную —2. С водородом кислород образует
несколько соединений — Н2О, Н2О,, Н2О3, Н2О4. Тепловые
эффекты образования этих соединений из простых веществ
* В учебной литературе употребляют родственные термины:
формальный заряд, окислительное число, значность, пользуясь кото-
которыми важно помнить различия между ними и понятием валентности.
20
(в кДж)
(Н2) + 2(О2)+25,1 = (Н2О4)
(Н2)+3/2(О2)-56,5=(Н2Оз)
(На) + (Оа) — 136,0 = (Н2О2)
(О2)- 242,7 =(Н2О)
указывают, что наиболее устойчивой формой среди
них является вода, в молекулах которой отсутствует
неустойчивая цепочка атомов кислорода. Для жидкой
воды характерна собственная электролитическая диссо-
диссоциация:
2{Н2О} + 55,9 кДж = {Н3О + } + {ОН-}
Однако процесс этот эндотермичен и количество продис-
социировавшей воды при 25° С составляет 10~е%.
Таким образом, в широком интервале температур наи-
наиболее энергетически устойчивое соединение водорода и
кислорода —вода. Она образует на Земле океаны, моря,
льды, пары и туман, в большом количестве содержится
в атмосфере, в толщах пород вода представлена капил-
капиллярной и кристаллогидратной формами. Такая распро-
распространенность и необычность свойств (аномалия плотности
воды и льда, полярность молекул, способность к электро-
электролитической диссоциации, к образованию гидратов, раст-
растворов и др.) делают воду активным химическим агентом,
по отношению к которому обычно рассматривают свойства
большого числа других соединений.
Сложные гетеросоединения. По своим кислотно-основ-
кислотно-основным свойствам вода занимает промежуточное положение
среди всех остальных соединений, а характер ее взаимо-
взаимодействия с ними чрезвычайно разнообразен. Так, при
экзотермическом взаимодействии воды с оксидами щелочных
металлов экзотермически образуются основания (щелочи)
[Na2O]+{H2O}+151,5
которые представляют собой гидроксиды металлов, хорошо
растворимые в воде и полностью диссоциирующие на ионы:
Символ «aq» обозначает весьма большое количество воды,
в которой растворяется 1 моль NaOH с образованием
ионов, гидратированных молекулами воды. Наличие из-
избытка ионов гидроксида в растворе как раз и определяет
его щелочные свойства.
21
При взаимодействии воды с оксидами неметаллов
экзотермически образуются гидраты, именуемые кислотами:
(SO8)+{H2O} — 132,2 кДж = {Н25О4}
При растворении в избытке воды образовавшийся гидрок-
сид, гидратируясь дальше, диссоциирует на ионы водорода
и кислотного остатка:
Наличие большой концентрации ионов водорода в растворе
и определяет его кислотные свойства.
Кислотой называют соединение, которое, подвергаясь
электролитической диссоциации при растворении в воде,
образует раствор, содержащий избыток ионов водорода.
Основанием называют гидроксид, который, подвергаясь
электролитической диссоциации при растворении в воде,
образует раствор, содержащий избыток ионов гидроксила.
Силу кислот и оснований определяют степенью их
электролитической диссоциации в водных растворах на
ионы. Практически полностью диссоциирующие хорошо
растворимые кислоты и основания называют сильными, в
малой степени диссоциирующие малорастворимые кислоты
и основания—слабыми. В соответствии с этим оксиды,
образующие с водой кислоты, будут кислотными, а оксиды,
образующие щелочи,—основными.
Рассмотрим в качестве примера отношение к воде
высших оксидов элементов третьего периода системы
Д. И. Менделеева. Ниже приведены тепловые эффекты
(в кДж) реакций взаимодействия оксидов с 1 моль воды:
[Na2O]-|-{H2O}— 15I,5 = 2NaOH очень сильное основание
[MgO]+{H20}—39,7 = [Mg(OHJ] слабое основание
1/з[А12О3]+{Н2О}— 15,9=2/3[А1(ОН)з] очень слабое основание
[SiO2]+{H2O}+25,5 = [H2SiO3] {
> очень слабые кислоты
V2[SiO2]+{H2O} + 25,5 = V2[H4SiO4] /
1/з[Р2О5]+{Н2О} — 48,6 = 2/з[Н3РО4] средней силы кислота
[SO3] + {H2O} — 71,5 = [H2SO4] сильная кислота
{С12О7}+{Н2О}—113 = 2 [НС14О] очень сильная кислота
Из этого примера видно, что большие экзотермические
тепловые эффекты наблюдаются при образовании хорошо
растворимых гидроксидов, которые в водных растворах
проявляют свойства сильных электролитов—щелочей
(NaOH) и кислот (H2SO4, HC1O4). Напротив, небольшие
экзотермические и эндотермические тепловые эффекты
имеют место при образовании малорастворимых гидрокси-
22
дов, проявляющих свойства слабых оснований (А! (ОНK,
Mg(OH),, Fe(OHJ) и слабых кислот (H2SiO3, Н2СО3,
H.SiO,)/
Отношение гидроксидов различной кислотно-основной
природы к процессам электролитической диссоциации ил-
иллюстрируют следующие примеры (кДж):
-71,l={Cs+}aQ + {OH-} >
.„ „ ... Л4 I /f)u_i f сильные основания
{NH3}+{H2O} — 7,0={NHi}aq + {OH-}aq слабое основание
слабая кислота
сильные кислоты
Так, сильные основания и кислоты (CsOH, H2SO4 и др.)
растворяются и электролитически диссоциируют на ионы
в водных растворах с большим экзотермическим эффектом;
слабые—NH4OH, Н2СО3 диссоциируют с небольшим экзо-
экзотермическим и даже эндотермическим эффектами.
Однако приведенные примеры не дают ответа на воп-
вопрос, почему в одних случаях при растворении гидроксида
гидратация приводит к образованию кислоты
aq B.1)
а в других случаях—к образованию основания
aq B.2)
Зто скязано с тем, что гидратация по схеме B.1) пре-
преимущественна в том случае, если связь Н—О в гидрок-
сиде будет более полярной, чем связь R—О. Гидратация
же по схеме B.2) преимущественна в том случае, когда
связь R—О гидроксида полярнее связи Н—О. Когда
же полярности обеих связей в соединении ROH сравнимы
по величине, гидроксид приобретает свойства и слабого
основания и слабой кислоты. Это свойство электролитов
называют амфотерностью. Амфотерный гидроксид в кис-
кислой среде ведет себя как основание
а в щелочной — как кислота
ROH+{OH-}aq = {RO-}aq+{H2O}
Например, слабоосновный гидроксид алюминия раство-
растворяется как в растворах кислот, так и щелочей, причем в
23
растворах кислот активней (тепловой эффект в кДж):
Al(OHK + 3{H + }aq-106 = {Al3+}aq + 3{H2O}
Напротив, слабокислый гидроксид кремния со щелочью
взаимодействует экзотермически, с кислотой процесс эндо-
термичен и практически не идет:
Следовательно, гидроксид кремния неамфотерен и может
быть только кислотой, соответственно оксид SiO2—кис-
SiO2—кислотный.
Изменение кислотных и основных свойств можно наблю-
наблюдать у оксидов одного и того же элемента. Так, у ме-
металлов низшие кислородные формы являются основными,
высшие—кислотными, а промежуточные—амфотерными,
например:
МпО Мп2О3, МпО2 МлО3, Мп2О,
основный амфотерные кислотные
В этом семействе слева направо убывают основные и на-
нарастают кислотные свойства, в результате чего основный
оксид МпО экзотермически взаимодействует с кислотой
[MnO]+2{H + }aq-119,6 кДж = {Н2О} + {Мп2+}а()
а кислотный оксид Мп2О7 экзотермически взаимодействует
со щелочами
0,5[Мп2О7] + {OH-}aq-67,2 кДж = {MnO4-}aq + 0,5{Н2О}
Если подобное семейство оксидов образует неметалл
(например, азот), то общая тенденция сохраняется, но
свойства оксидов смещены в направлении увеличения
кислотности в последовательности N2O3, N2O4, N2O6:
0,5(N2O6)+0,5{H2O}-70,0 кДж = {Н + }
Помимо оксидов основного, кислотного и амфотерного
типа, можно выделить так называемые несолеобразующие
оксиды, которые не образуют с водой гидратных форм
и не могут быть ни основными, ни кислотными. Примером
таких соединений служит N0. Обычно к несолеобразую-
щим оксидам относят СО, хотя при высоких давлениях
оксид углерода может взаимодействовать со щелочами,
24
образуя соли муравьиной кислоты, например:
СО + КОН—*НСООК
Перейдем к характеристике солей, которые представ-
представляют собой продукты полного или частичного замещения
атомов водорода в кислотах на атомы металлов или про-
продукты замещения гидроксильных групп оснований на кис-
кислотные остатки. Соли образуются
при взаимодействии кислотных и основных оксидов
кислот и оснований
2[NaOH] + {H2SO4} = [Na2SO4]
металлов с кислотами и щелочами
[Znj + 2(НС1) = [ZnCy + (Н2)
Если кислота имеет более одного атома водорода, спо-
способного замещаться на металл, ее называют многооснов-
многоосновной. Так, HNO3 — одноосновная, H2SO4 — двухосновная,
Н3РО4—трехосновная, а Н4Р2О7 — четырехосновная кис-
кислоты. Атомы водорода в растворах многоосновных кислот
замещаются на атомы металла ступенчато, в результате
образуются кислые соли (гидросоли). Например, кристал-
кристаллический хлорид натрия взаимодействует с концентриро-
концентрированной серной кислотой по схеме
[KaCl] + {H2SO4} = [NaHSO4] + (НС1)
гидросульфат
натрия
Гидросульфат натрия может существовать в виде инди-
индивидуального кристаллического соединения, но при раст-
растворении в воде подвергается электролитической диссо-
диссоциации
и при достаточном большом разбавлении диссоциация
идет до конца
Если основание содержит более одной гидроксильной
группы, способной замещаться на кислотный остаток, то
его называют многокислотным. Так, NaOH—однокислот-
ное, Са(ОНJ—двухкислотное, А1(ОНK — трехкислотное
основания. Гидроксильные группы в многокислотных
25
основаниях также замещаются на кислотный остаток
ступенчато. В результате образуются основные соли (гид-
роксосоли), например:
Са(ОНJ + HNO3 = Ca(OH)NO3 + Н2О гидроксонитрат
кальция
2А1(ОНK + H2SO4 = [A1(OHJ]2SO4 + 2Н2О дигидроксосульфат
алюминия
[A1(OHJ]2SO4 + H2SO4 = 2[A1(OH)SO4] + 2Н2О гидроксосульфат
алюминия
Гидроксосоли при растворении в воде электролитически
диссоциируют, образуя гидроксоионы:
Полное замещение атомов водорода в кислотах на металл
или гидроксильных групп в основаниях на кислотный
остаток приводит к образованию средних солей.
Соединения, образованные хлором с другими элемен-
элементами периодической системы, также многочисленны. В от-
отличие от кислорода хлор может иметь степень окисления
от —I до +7. Однако диагональным аналогом кислорода
в хлористых соединениях является лишь однозарядный
хлор — С1". С водородом хлор образует двухатомные мо-
молекулы хлорида водорода. Их прочность такова, что рас-
распад на атомы не происходит даже при самых высоких
температурах, об этом свидетельствует большой эндотер-
эндотермический эффект реакции распада на атомы — атомизация:
(НС1)+431,4 кДж = (Н)-f (C1)
Однако при взаимодействии хлорида водорода с поляр-
полярными молекулами воды в растворе образуются гидрати-
рованные ионы {H + }aq и {Cl~}aq. Этот процесс экзотер-
мичен и идет самопроизвольно:
(HCl)-f aq-95,0
В результате электролитической диссоциации водный раст-
раствор хлорида водорода обогащается ионами водорода и
приобретает свойства кислоты—это бескислородная, силь-
сильная, хлористоводородная или соляная кислота.
При взаимодействии с другими элементами периоди-
периодической таблицы хлор образует многочисленные соедине-
соединения—хлориды, которые в зависимости от свойств партнера
(металла или неметалла) могут быть либо солями хло-
хлористоводородной кислоты, либо несолеобразными хлори-
хлоридами. Примером монотонного изменения физических и
химических свойств от основных к кислотным и от солей
к несолеобразующим формам может служить семейство
хлоридов, образуемых элементами третьего периода пе-
периодической системы:
NaCl, MgCl2 AlClg SiCl4, РСЦ, SCI4
соли несолеобразующие вещества
Остановимся на растворимости и взаимодействии этих
соединений с водой. Хлорид натрия представляет типич-
типичную соль, которая при растворении в воде образует
нейтральный раствор гидратированных ионов натрия и
хлора:
Водные же растворы MgCl2 и тем более А1С13 приобретают
все более кислую реакцию (подробнее см. гл. 14). Сум-
Суммарный процесс растворения MgCl2 с учетом частичного
гидролиза соли протекает по схеме (кДж)
Растворение SiCl4 и тем более РС15 сопровождается пол-
полным гидролизом и образованием двух кислот, например:
Следовательно, из приведенных реакций видно, что
в рассматриваемом ряду хлоридов при переходе от натрия
к сере взаимодействие с водой все более приобретает
характер гидролиза и становится наиболее энергетически
выгодным. Последнее обстоятельство, а также аналогия
между кислородными и хлористыми соединениями позво-
позволяют считать, что NaCl обладает основными свойствами,
а РС15—кислотными. Аналогично оксидам взаимодейст-
взаимодействуют между собой основные и кислотные хлориды, на-
например:
[NaCl]-f-[AlCl8] — 25 кДж= [NaAlCl4]
Таким образом, при переходе от оксидов к хлоридам
кислотно-основное взаимодействие сохраняется, оно лишь
меняет свои привычные формы.
Процессы замещения кислорода на хлор в бинарных
соединениях служат характеристикой их химической
27
природы:
Na2O+Cl2-391,2
основный
оксид
MgO + Cl2-40,6 кДж =
слабо основ-
основный оксид
/з2 +
амфотерный
оксид
VBP2O5 + С12 + 130 кДж = 2
кислотный
оксид
Из этого примера видно, что замещение кислорода в окси-
оксидах при переходе от основных к кислотным становится
энергетически менее выгодным. Отмеченные выше особен-
особенности кислорода и хлора и их соединений как раз и от-
отражают их диагональное сходство и связанную с этим
конкуренцию в химических превращениях. Как диаго-
диагональные аналоги кислород и хлор могут не полностью,
а частично замещать друг друга в оксидах и хлоридах,
в результате чего образуется особый класс смешанных
соединений, именуемых оксохлоридами. Например, воз-
возможна экзотермическая реакция с образованием оксотри-
хлорида фосфора:
В то же время тенденция к образованию оксохлоридов
уменьшается по мере перехода от кислотных к основным
формам оксида и хлорида. Так, Na2O и NaCl не взаимо-
взаимодействуют между собой.
Глава 3
Типы химических реакций
Фазовые и химические превращения. Реакции об-
обменного разложения. Реакции окислительно-восста-
окислительно-восстановительные.
Фазовые и химические превращения. Превращения
индивидуального вещества, в результате которых изме-
изменяются структура или агрегатное состояние, но неиз-
неизменным остается его химический состав, называют фазо-
фазовыми. К таковым относят процессы сублимации, испа-
испарения, плавления веществ, а также их полиморфные
28
превращения. Фаза—это однородная часть системы инди-
индивидуальных веществ, она отличается от других фаз си-
системы либо агрегатным состоянием, либо строением.
Систему, состоящую из одной фазы, называют гомогенной,
из двух или большего числа фаз—гетерогенной.
Фазовые превращения осуществляются только в гете-
гетерогенных системах, где химический состав всех фаз оди-
одинаков. Например, в гетерогенной системе «вода—лед—пар»
при 0°С сосуществуют три фазы. Они различаются между
собой состоянием (твердое, жидкое и газообразное) и струк-
структурными характеристиками молекулы Н2О. Взаимосвязь
фаз в этой системе описывают совокупностью уравнений
фазовых превращений:
[Н2О] = (Н2О) (сублимация)
{Н2О} = (Н2О) (испарение)
[Н2О] = {Н2О} (плавление при 0°С)
При температурах выше О °С в системе могут сосущест-
сосуществовать только две фазы вода — пар и сохраняется только
одно фазовое превращение
{Н2О}+44 кДж = (Н2О) (испарение)
При температуре ниже 0°С вода замерзает, сосуществуют
фазы лед—пар и сохраняется также одно фазовое пре-
превращение:
[Н2О] + 50 кДж = (Н2О) (сублимация)
Примером фазового превращения одной кристалличе-
кристаллической модификации в другую (полиморфный переход) может
служить превращение алмаза в графит при температуре
выше 1000 °С:
[С]—1,9кДж=[С]
алмаз графит
Эти различные формы гомосоединений одного элемента
получили название аллотропных модификаций. К подоб-
подобным фазовым превращениям относятся и самопроизволь-
самопроизвольный полиморфный переход белого фосфора в красный,
очень медленный при обычной температуре и ускоряю-
ускоряющийся при нагревании или под действием света:
[Р]белый—18.4 кДж = [Р]красный
Превращения, при которых изменяется химический
состав и структура соединений, называют химическими.
Изменение агрегатного состояния может только сопро-
сопровождать химический процесс, но не быть определяющим.
29
Примерами химических превращений могут служить тер-
термическое разложение гидрокарбоната натрия
2[NaHCO3] + 134 кДж = [Na2CO3] + (Н2О) + (СО2)
и взаимодействие перманганата щелочного металла с хло-
хлористоводородной кислотой в водном растворе
Различие в фазовых превращениях для этих двух
реакций несущественно. Принципиальное различие за-
заключается в том, что первая реакция идет без измене-
изменения степени окисления элементов в реагирующих соеди-
соединениях, а вторая—с изменением степени окисления
(Мп7+—*Мп2+, С1~ —>- С1°). Это различие и положено
в основу классификации химических превращений, кото-
которые разделяют на реакции обменного разложения и окис-
окислительно-восстановительные. Представление о степени
окисления атомов в соединениях складывается из сле-
следующих положений.
1. Если в соединении связаны между собой атомы
одного и того же элемента, то их степень окисления
принимают равной нулю, примером служат простые ве-
вещества (О,), (Н2), [Fe], [S].
2. Атомы элементов IA- и ПА-подгрупп (щелочные и
щелочно-земельные элементы) в гетеросоединениях имеют
степень окисления +1 и +2, соответственно (Na2O, СаС12).
3. Атомы кислорода в гетеросоединениях относительно
атомов других элементов (кроме фтора) имеют степень
окисления, равную —2 (НгО, Р2О3, H2Si03). Однако сте-
степень окисления кислорода в пероксидах (Н2О2) равна —1,
в надпероксидах —0,5.
4. Атомы водорода в гетеросоединениях с атомами
неметаллов (углерод, кремний, элементы подгруппы азота,
кислорода, галогенов) имеют степень окисления +1 (СН4
NH,, H2SO4) КОН).
5. Атомы водорода в соединениях с элементами IA- и
ПА-подгрупп имеют степень окисления, равную —1 (NaH,
СаН2, LiAlHJ.
Понятие степени окисления чрезвычайно важно для
составления уравнений химических реакций, поскольку
основой для таких уравнений служит фундаментальный
закон сохранения заряда. Это означает, что какими бы
путями ни шла реакция, какие бы промежуточные и ко-
конечные продукты при этом ни образовывались, суммарный
заряд системы веществ до и после реакции остается неиз-
30
менным. При этом для выполнения закона безразлично,
идет ли речь об истинном заряде ионов или о формаль-
формальном заряде атомов элементов, входящих в состав соеди-
соединений. Рассмотрим каждый из указанных видов химических
реакций на конкретных примерах.
Реакции обменного разложения идут без изменения
степени окисления элементов в соединениях. Примерами
таких реакций могут служить процессы кислотно-основ-
кислотно-основного взаимодействия оксидов различных элементов:
+6-2 + 1-2 +1+6-2
+2-2 +5-2 +2 +5-2
ЗСаО + Р2О5 = Са3(Р О4J
+1-2 +1-2 +1-2+1
Приняв степени окисления атомов кислорода и водорода
равными —2 и +1 соответственно, для остальных атомов
получаем заряды неизменными как до, так и после реак-
реакции. Тот же тип реакции имеет место и в случае про-
процессов
нейтрализации кислот основаниями:
2H2SO4 + Са(ОНK = Ca(HSO4J + 2H2O
гидролиза солей:
2CuSO4 + Н2О = (CuOH) 2SO4 + H2SO4
реакций осаждения:
Na2SO4 -f- ВаС12 = jBaSO4 + 2NaCl
К реакциям обменного разложения относят процессы обра-
образования и разрушения газообразных многоатомных гомо-
соединений. Известно, что подавляющее большинство
элементов периодической системы в газообразном состоя-
состоянии существует не только в атомарном, но и в молекуляр-
молекулярном состояниях. Так, известны формы кислорода'—О2, О3;
серы — от S2 до S8; фосфора—Р2> Р4 и т. д. Между этими
формами в зависимости от условий среды происходят
взаимопревращения, например:
0 0 0 0
2(О,) = 3(О»), 3(S8) = 4(Se).
Разновидностью реакций обменного разложения являются хими-
химические превращения, идущие с участием комплексных соединений, ко-
которые образуются за счет внедрения одной или нескольких молекул
(ионов) некоторых соединений (Н20, NH3, 0H~, F~, CN~) в струк-
структуру другого соединения без изменения степени окисления состав-
31
ляющих их атомов. При этом образующееся комплексное соединение
по свойствам является новым, т. е. отличается от суммы свойств
исходных веществ.
Химические свойства комплексных соединений зависят от его
молекулярной структуры, которая может быть различной у соединений
одного и того же состава. Например, соединение СгС13-6Н2О в кри-
кристаллическом состоянии существует в трех различных комплексных
формах: Сг(Н2О)„С13, Сг[(Н2ОMС1] {С12Н2О} и Сг[(Н2ОLС12]{СЬ2Н2О}
(внутренний, ближний к атомам хрома слой представлен группами
в прямоугольных скобках, а внешний слой — группами в фигурных
скобках). Все структурные разновидности Cr(H2O)(iCl3, растворяясь
в воде, образуют ионы различного состава и строения:
[Cr(H2O)sClClgHsO]+aq = {
Растворы этих трех изомеров различны по электрической проводи-
проводимости, по физическим и химическим свойствам.
Химические превращения комплексных соединений наиболее ин-
интересно и разнообразно осуществляются в растворах. Если слить
растворы с ионами Cd2+ и перхлората натрия NaC104, то никаких
видимых изменений не произойдет, так как образующийся при этом
перхлорат кадмия Cd(C104J — хорошо растворимое вещество. Напро-
Напротив, если в раствор, содержащий ионы {Cd(NH3J}2 + , добавить пер-
перхлорат натрия, то выпадет малорастворимый осадок
{Cd(NH3Lf+ +2{C1O4-} = [Cd(NH3L(C104J]
Другим примером может служить почти бесцветный раствор соли
железа со степенью окисления -f-2. Если к этому раствору добавить
цианистый калий, то первоначально выпадет коричневый осадок циа-
цианида железа
{Fe}2 + +2{CN-}= fFe(CN'J]
но при дальнейшем прибавлении KCN осадок растворится:
-} = {Fe(CN)J-}
Из такого раствора железо (его степень окисления не меняется) не
осаждается ни аммиаком, ни щелочью. Окисление железа в комплекс-
комплексном ионе {Fe(CN)s~} происходит с трудом, тогда как ион {Fe24}
при обычных условиях легко окисляется кислородом воздуха.
Реакции окислительно-восстановительные идут с изме-
изменением степени окисления одного или нескольких элемен-
элементов, входящих в состав реагирующих соединений. В этих
реакциях окислителями называют элементы, которые, вос-
восстанавливаясь, понижают свою степень окисления; вос-
восстановителями—элементы, которые, окисляясь, повышают
свою степень окисления. Эти изменения степени окисле-
окисления атомов можно условно связать с процессами присоеди-
присоединения или отдачи электронов.
К реакциям окисления — восстановления следует прежде
всего отнести процессы, идущие с участием простых
32
веществ, например:
восстановитель окисляется
-II 4-
0-20 /ч П
t
окислитель восстанавливается
Здесь окислителем является молекулярный кислород со
степенью окисления, равной нулю, а восстановителем —
атом иода в HI, имеющий степень окисления —1. Про-
Процесс окисления-восстановления можно изобразить схема-
схематически:
-1 0 -1 О
21 — 2е~—*- 21 восстановитель I окисляется до I
0-2 0 -2
0-f-2e~ —>- О окислитель О восстанавливается до О
Суммируя эти две полуреакции так, чтобы результирую-
результирующий заряд полной реакции был равен нулю, получаем
полную окислительно-восстановительную реакцию C.1).
Такой метод расчета стехиометрических коэффициентов
окислительно-восстановительных реакций получил назва-
название метода молекулярно-электронного баланса.
Рассмотрим окислительно-восстановительные реакции
с участием пероксида водорода Н2О2 и пероксидных соеди-
соединений состава Rj—О—О — R2, содержащих цепочку из
двух атомов кислорода —О—О —, которая является как
бы фрагментом гомосоединения из двух атомов кислорода,
встроенных в гетеросоединение. Степень окисления таких
атомов кислорода относительно друг друга формально
принимается равной нулю, а по отношению к группиров-
группировкам /?, и R, — равной —1. Благодаря этому пероксидные
соединения могут принимать участие в реакциях в каче-
качестве как окислителя, так и восстановителя.
1. Пероксид водорода в качестве окислителя
окисление
K2CrO4-f-K2SO4 C.2)
t t
окисление Сг — Зв~ =
-1 -2
воссга новление О -f- I е ~ = О
восстановление
+ 3 +6
З
1
В качестве коэффициентов в левую часть реакции C.2) вводим
полученные из баланса зарядов окислительно-восстановительные мно-
множители 1 и 3, получая
+ ЗН2О2 + КОН —> КаСгО4 + K2SO4 C.3)
705 83
веществ, например:
восстановитель окисляется
-lj I
I 0 -2 0
окислитель восстанавливается
Здесь окислителем является молекулярный кислород со
степенью окисления, равной нулю, а восстановителем —
атом иода в HI, имеющий степень окисления —1. Про-
Процесс окисления-восстановления можно изобразить схема-
схематически:
-10 -1 О
21 — 2е~—i- 21 восстановитель I окисляется до I
0-2 0 -2
0-|-2е~ —>-0 окислитель О восстанавливается до О
Суммируя эти две полуреакции так, чтобы результирую-
результирующий заряд полной реакции был равен нулю, получаем
полную окислительно-восстановительную реакцию C.1).
Такой метод расчета стехиометрических коэффициентов
окислительно-восстановительных реакций получил назва-
название метода молекулярно-электронного баланса.
Рассмотрим окислительно-восстановительные реакции
с участием пероксида водорода Н2О2 и пероксидных соеди-
соединений состава R}—О—О — R2, содержащих цепочку из
двух атомов кислорода —О—О—, которая является как
бы фрагментом гомосоединения из двух атомов кислорода,
встроенных в гетеросоединение. Степень окисления таких
атомов кислорода относительно друг друга формально
принимается равной нулю, а по отношению к группиров-
группировкам /?! и R? — равной —1. Благодаря этому пероксидные
соединения могут принимать участие в реакциях в каче-
качестве как окислителя, так и восстановителя.
1. Пероксид водорода в качестве окислителя
окисление
2О2 + КОН —* K2CrO4 + K2SO4 C.2)
I t t
восстановление
+ 3 +6
окисление О"—3«~=Сг 1
[ 2
восстановление О-\- \е~ — О
3
В качестве коэффициентов в левую часть реакции C.2) вводим
полученные из баланса зарядов окислительно-восстановительные мно-
множители 1 и 3, получая
Cr2(SO4)s+ЗН2Оа + КОН —*¦ КаСгО4 + KgSOa C.3)
2 ks 705 83
высокотемпературной оксидной н галогенндной металлотермии
3[Fe3O4] + 8{А1} = 4[А12О3] + 9{Fe}
(TiCl) + 2{M} [Ti] + 2{MCl}
Иначе рассчитывают коэффициенты ионных окисли-
окислительно-восстановительных реакций в водных растворах
с помощью метода ионно-электронного баланса.
Рассмотрим в качестве примера окисление сульфида
мышьяка
As2S3 концентрированной азотной кислотой!
восстановление
+ 3 -2 +5 +6 +5 + 2
1 t
окисление
Условно разделим весь процесс на две ионные полуреакции окисле-
окисления и восстановления. Полуреакция окисления As2S3 с участием моле-
молекул воды в качестве дополнительного восстановителя может быть
записана следующим образом:
окисление восстановителя
II 4- 4-
As2S3+H20 —-у 2AsOl" Н-ЗБОГ -(- Н +
I t
окисление восстановителя
Поскольку в правой части схемы имеется 20 атомов кислорода,
в левой части перед Н2О необходимо поставить коэффициент 20,
соответственно в правой части перед ионами водорода следует поста-
поставить коэффициент 40:
As2S3 + 20Н2О —* 2AsO|~ + 3SO|" + 4OH +
Введением этих коэффициентов получают баланс по числу атомов, но
не по зарядам ионов, так как суммарный заряд левой части равен
нулю, а правой —(—28. Следовательно, полный баланс этой полуреак-
полуреакции будет иметь место, если из левой части вычесть 28 единичных
зарядов:
As2S3+20H2O— 28e-=:
Полуреакция восстановления азотной кислоты НЫОз может быть
записана аналогичным образом:
восстановление окислителя
i I
N0JT+4H+-4-Зг- = NO-f-2HaO
I t
восстановление окислителя
Сложение этих двух полуреакций следует производить так, чтобы
суммарный заряд системы веществ стал равен нулю:
As2S8+20HaO —28e-=2AsO4~-4-3SO4"+4OH+ 3
NO8"-f4H+-4-3e-=NO-T-2HaO 28
3As2S3+ 28NO8~-t-4H2 О = 8Н + +9SOr+6AsOj"+28NO
2* 85
Полученное уравнение полностью отвечает требованиям баланса по
числу атомов и по зарядам.
В качестве второго примера расчета стехиометрических коэффи-
коэффициентов ионных окислительно-восстановительных реакций методом
ионно-электронного баланса рассмотрим следующий процесс:
окисление
Эз + KMnO4 + КОН —> K2SO4 + K2Mn04 + H2O
J t
восстановление
Записываем в ионной форме две полуреакции окисления SOl~ и ОН~
и восстановления МпОГ-
- = SO4 Н-2МпОГ+Н2О
Полученное ионное уравнение является сбалансированным и по числу
атомов, и по зарядам.
Для реакций окисления — восстановления с участием
органических соединений характерно то, что в одних
и тех же соединениях атому углерода по отношению к раз-
различным элементам следует приписывать различные сте-
степени окисления. Степень окисления углерода в соедине-
соединениях с азотом, серой, кислородом, галогенами принимается
равной +4, в оксиде углерода СО равна +2, а в соеди-
соединении с водородом от —4 до —1, в соединениях атомов
углерода друг с другом—0.
В ряду хлорпроизводных метана степень окисления
углерода может изменяться от —4 до +4 (считая степень
окисления хлора —1, а водорода +1):
-4 -2 0 + 2 + 4
СН4, СН8С1, СН2С12) СНС13) СС14
В гомологическом ряду замещенных метана формальный
заряд атома углерода в этане С2Н„ равен —3, этаноле
С2Н„О —2, уксусном альдегиде С2Н4О —1, уксусной
кислоте С2Н4О2 —0. Степень окисления углерода зависит
от кратности связи углерод—углерод в соединении. Напри-
Например, в этане СгН„ (ординарная связь С—С), в этилене
-2 • -1
С2Н4 (двойная связь С = С), в ацетилене С2Н4 (тройная
связь С = С).
Следует отметить, что степень окисления углерода
в органических соединениях определяет только соотноше-
соотношение атомов элементов в формуле (углерода, водорода,
кислорода и др.) и не связана с их взаимным расположе-
36
нием. Поэтому степень окисления углерода в изомерах
не изменяется при переходе от одной структуры к дру-
другой. Примером могут служить одинаковые по составу, но
разные по строению и химической природе молекулы эти-
этилового спирта С2Н5ОН и диметилового эфира (СН3JО,
в которых суммарная степень окисления углерода равна —2.
Рассмотрим в качестве примера процесс окисления этилового
спирта до уксусной кислоты перманганатом 'калия, используя для
расчета коэффициентов метод молекулярно-электронного баланса:
с2н
-2
2С-
+ 7
Мп
окисление
+ 7
5ОН + КМпО4-
0
-4е~=2С
+ 4
+-3е~ = Мп
3
4
J
->СН
J
3СО О К + МпО2 + КОН + Н2О
t
восстановление
Расставив окислительно-восстановительные коэффициенты, получим
уравнение реакции
ЗС2Н5ОН + 4КМпО4 = ЗСНзСООК + 4МпО2 + КОН + 4Н2О
В качестве другого примера рассмотрим реакцию окисления глю-
0 +1-2
козы QH12O6 кислородом:
окисление
О О
С„Н12Ов + О2
I
восстановление
О +4
6С-24е~
О -2
—* 20
1
6
Расставим окислительно-восстановительные коэффициенты
^б^12ОеН~ 6О2 —>¦ 6СО2
и закончим решение расчетом коэффициентов процессов обменного
разложения (с учетом образования воды):
С„Н18Ов = 6О2 + 6СО2 + 6Н2О
Основы
химической
термодинамики
и кинетики
Важнейшими разделами химической науки, химической технологии
и техники являются эргохимия (учение о работе химических реакций
и химическом равновесии) и хронохимия (учение о течении химических
реакций во времени, о скорости процессов, т. е. химическая кинетика
и катализ). Знание этих разделов позволяет количественно определить
направление и предельную глубину процессов, расход реагентов во
времени, энергозатраты, коэффициент полезного действия, влияние
катализаторов и ингибиторов на выход продукта. Умение решать
также задачи обязательно для будущего инженера, научного работника
и преподавателя.
Глава 4
Термохимия
Теплота и работа. Тепловой эффект реакции и
энтальпия. Закон Гесса. Термохимические урав-
уравнения.
Теплота и работа. Основным неотъемлемым свойством
материи является движение, без которого немыслимо само
понятие материи. Мерой движения служит энергия, кото-
которая в зависимости от вида движения принимает различные
формы. Взаимодействие тел проявляется в обмене движе-
движением, т. е. энергией. Наиболее важные для химии формы
обмена энергией между телами—теплота и работа.
Теплота представляет собой количественную меру бес-
беспорядочного, хаотического движения частиц, образую-
образующих данные тела или систему (молекул, атомов, электро-
электронов и т. п.). При этом в процессе самопроизвольного
обмена энергией в форме теплоты часть энергии тела,
обладающего более высокой температурой, передается
телам с более низкой температурой вплоть до их выравни-
выравнивания.
О природе теплоты около трехсот лет тому назад были высказаны
две гипотезы, одна из которых была сформулирована Г. Галилеем
A613). Он считал, что теплота —некоторое «вещество», способное бес-
беспрепятственно проникать во все обычные вещества и предметы и выхо-
выходить из них. Позже это «вещество» получило название «теплород».
Изменение количества «теплорода» меняет температуру тел, при пол-
полном его отсутствии достигается «абсолютный нуль». Эта гипотеза была
полностью опровергнута, поскольку точные экспериментальные про-
проверки не позволили обнаружить изменение массы тел при нагревании
и охлаждении. Правда, в XX в. в связи с созданием теории относи-
относительности было доказано, что масса горячего тела больше, чем холод-
холодного, однако эти изменения лежат далеко за пределами возможностей
техники взвешивания.
Согласно другой гипотезе, выдвинутой Ф. Бэконом, теплота про-
проявляется в результате движения частиц, образующих тела. Большая
роль в развитии и дальнейшей разработке этой теории принадлежала
М. В. Ломоносову. Однако и эта гипотеза, хотя и более близкая
к истине, оказалась неверна, так как в соответствии с ней теплота
была свойством системы, в то время как в XIX в. иаука пришла
к весьма важному выводу — теплота ни. в каком виде не содержится
в системе и не является ее свойством.
Однако прн измерении количества теплоты методами калоримет-
калориметрии ее условно считают свойством системы. Такое допущение нару-
нарушает теоретическую строгость, но не влияет на точность и смысл
результатов калориметрических исследований. Прибор, позволяющий
измерять количество теплоты, выделившейся или поглощенной веще-
39
ством при его химическом превращении, называют калориметром.
Современные калориметры позволяют проводить исследования в широ-
широком интервале температур, измерять тепловые эффекты от малых
долей до тысяч джоулей. Существуют типы калориметров, предна-
предназначенные для определения теплоемкости веществ в различных агре-
агрегатных состояниях, теплот сгорания в различных окислительных сре-
средах, теплот растворения веществ и разбавления жидкостей, скрытых
теплот фазовых превращений.
Работа, напротив, является количественной мерой упо-
упорядоченного движения или перемещения частиц в некото-
некотором направленном силовом поле. Например, работу А
расширения системы от первоначального объема V1 до
конечного V2 под действием постоянного давления (/?=const)
выражают соотношением
а работу гальванического элемента — произведением коли-
количества электричества q на разность электродных потен-
потенциалов ф2 и ц>х:
Превращение одной формы движения в другую всегда
осуществляется в строго эквивалентных соотношениях.
Эквивалентность взаимопревращений различных видов
энергии * доказана всем многовековым опытом человече-
человечества и поэтому является естественным законом, известным
как закон сохранения энергии. Это означает, что если
к системе или совокупности веществ подвести некоторое
количество теплоты Q, то в общем случае она может рас-
расходоваться на: 1) изменение внутренней энергии систе-
системы AU (изменение интенсивности поступательного, вра-
вращательного и колебательного движений внутри молекул
и кристаллов; 2) совершение работы А против сил, дей-
действующих извне на данную систему (внешнее давление,
поверхностное натяжение и т. д.).
Тепловой эффект реакции и энтальпия. Математически
закон сохранения энергии можно записать следующим
образом:
D.1)
* В системе единиц СИ единицей энергии (теплота, работа) яв-
является джоуль (Дж), равный работе силы в 1 ньютон (Н) на пути
в 1 м, 1 Дж = 1 Н-1 м. Другая важная единица измерения энергии —
электронвольт/моль. Один эВ/моль равен энергии, приобретаемой N&e
элементарными электрическими зарядами (Л/д — постоянная Авогадро,
е—заряд электрона) при п-охождении через поле с разностью потен-
потенциалов 1В; 1 эВ = 96 487 Дж.
40
Если на систему тел не действуют никакие силы, кроме
постоянного давления, то работа может быть выражена
произведением
Al = p(V2-V1) = PbV. D.2)
Подставив выражение D.2) в уравнение D.1), получаем
Qp = bU + pbV. D.3)
Если АН представить в виде двух энергетических уров-
уровней (U2 — t/j), то соотношение D.3) примет вид
Qp={U2-U1)+p(V2-V1) = {U2 + pV2)~(U1 + pV1). D.4)
Выражение
H D.5)
получило название энтальпии или теплосодержания (Я).
Подставив выражения D.5) в D.4), получаем
Следовательно, изменение энтальпии АН—это тепловой
эффект реакции при условии постоянства давления. При
более конкретном условии, т.е. при р= 1,013-105 Па,
для энтальпии вводится обозначение АН°.
Изменение энтальпии реакции лишь в небольшой сте-
степени зависит от температуры. Зависимость АН от темпе-
температуры (и давления) определяют изменением теплоем-
теплоемкости АСр в процессе химической реакции или фазового
превращения
поскольку значение АСр в процессе реакции обычно мало,
величина АН° в первом приближении практически не
зависит от температуры, AH°Ti — АН°Т2. В расчетах тепло-
тепловых эффектов при любых температурах обычно используют
стандартные значения АН°Ш, полученные при 25°С.
В качестве стандартных термохимических характе-
характеристик индивидуальных веществ, необходимых для расчета
тепловых эффектов химических реакций Л#°98, приняты
следующие.
1. Стандартные теплоты (энтальпии) образования
I моль соединений в заданном агрегатном состоянии из
* Нижний индекс у символов работы, теплоты и теплоемкости
указывает на постоянство давления; в химии обычно приходится
иметь дело с этим условием.
41
простых веществ AH°fi 298 (или AfH2it), например:
0,5(N2) + l,5(H2) = (NH3), &H°U 298 = — 46,0 кДж
[Na] + 0,5(Cl8) = [NaCl], AH°fi 298 = —410,9 кДж
[Cu]H-[S]H-4,5(O2) + 5(H2) = [CuSO4.5H2O], ДЯ°,, 298 = -2278 кДж
2. Теплоты сгорания органических веществ AH°ni
в кислороде с образованием газообразного СО2, жид-
жидкой Н2О и других продуктов реакции при Р= 1,013- 105Па
и температуре 298 К, например:
метан (СН4)Н-2(О2) = (СО2)Н-{Н2О}, ДЯ298 = — 890,4 кДж
диоксан {С4Н8О2}Н-5(О2) = 4(СО2) + 4{Н2О}, ДЯ298 = —2317,7 кДж
ксилит [С5Н12О5]Н-5,5(О2) = 5(СО2) + 6{Н2О},
ДЯ°2е8 = —2563,8 кДж
Наиболее удобны и применимы для расчетов и анализа
химических реакций теплоты образования соединений из
простых веществ. Эти характеристики в дальнейшем
изложении будут служить основой описания и анализа
энергетики химических реакций. Экспериментальные зна-
значения AH°fi 298 исследованных химических соединений
сведены в таблицы и широко используются в качестве
справочных данных (см. приложение IV).
Закон Гесса. Раздел химии, в котором рассматривают
тепловые эффекты химических и фазовых превращений,
а также некоторые другие тепловые свойства веществ,
назван термохимией, а уравнения реакций с указанием
теплового эффекта получили название термохимических.
В подобных уравнениях тепловой эффект реакции либо
непосредственно вводят в уравнение материального ба-
баланса, либо пишут рядом с этим уравнением, например:
[Zn]-f2{HCl}aq-153 кДж
или
[Zn]+2{HCl}aq = {ZnCl2}aq + (H2), ДЯ°2И = —153 кДж
Основа термохимии—закон Г. И. Гесса, являющийся
прямым следствием закона сохранения массы-энергии, его
формулируют следующим образом: тепловой эффект реак-
реакции зависит от природы и состояния исходных и конеч-
конечных веществ и не зависит от числа и характера проме-
промежуточных стадий при р = const или V = const.
Поясним этот закон примерами.
Пример 1. Предположим, нужно получить 1 моль NaOH, если
имеется 1 моль металлического натрия, 0,5 моль водорода и 0,5 моль
кислорода. Существует несколько способов решения этой задачи.
42
Первый способ:
сжигаем натрий в кислороде
[Na] + 1/2@2) = 1/2[Na302], Д//°98 = — 252,3 кДж
сжигаем водород в кислороде
1/2(Н2) + 1/4(О2) = 1/г{Н2О}) ДЯ298 = —142,7 кДж
соединяем воду с пероксидом натрия
1/2{H20} + 1/2[Na202] = [Na0H] + 1/4@2), ДЯ°2е8 = -31,8 кДж
Суммарный процесс образования 1 моль NaOH из простых веществ
[Na]+1/2(H2) + i/2(O2)=[NaOH], ДЯ°;, 298 =-426,8 кДж
идет с выделением энергии, равной сумме ДЯ298 трех стадий.
Второй способ:
сжигаем водород в кислороде
(Н2) + 1/2(О2) = {Н2О}, AH°f: 298 = — 285,8 кДж
даем возможность металлическому натрию взаимодействовать с этой
водой:
= [NaOH]H-1/2(H2), Д#° = —141,0 кДж
Суммарный процесс
1/2(O2)=[NaOH], AtfJ, 2S8 = —426,8 кДж
как и в предыдущем случае, характеризуется тем же изменением
энергии.
Пример 2. Кристаллический хлоралюминат натрия NaAlCl4
может быть получен непосредственным сжиганием натрия и алюми-
алюминия в хлоре:
[Na] + [Al]H-2(CI2) = [NaAlCI4], ДЯ;_ 2S8 =—1142,2 кДж
Тот же процесс можно провести в три независимые стадии;
AH°fi 2S8 = — 412,1 кДж
ДЯ}; 288 = —696,6 кДж
Cl] + [AlCl3]=[NaAlCl4], AH°f, 2e8 = —39,5 кДж
98 = — П48,2 кДж
При этом суммарный тепловой эффект трехстадииного процесса не
отличается от одностадийного.
Оба примера убеждают в том, что независимо от пути реакции
при одних и тех же исходных веществах и конечных продуктах, при
условии постоянства давления или объема, тепловой эффект реакции
остается неизменным.
Рассмотрим способ расчета тепловых эффектов на при-
примере термической диссоциации перманганата калия. Для
этого необходимо написать уравнение реакции и указать
под каждым реагентом значения стандартных теплот
образования из простых веществ:
2[КМлО4] = [K2Mn04j + [МлО2] + (О2)
АН)>т, кДж —808 —1185 —519 0
43
Затем из суммы теплот образования продуктов реакции
надо вычесть сумму теплот образования исходных веществ
Д/Г298 = — 519— 1185—(—808-2) = — 88 кДж,
получившийся результат будет тепловым эффектом дан-
данной реакции.
Приведенное уравнение теплового баланса является
математической формой первого следствия из закона
Гесса: тепловой эффект реакции равен сумме теплот
образования продуктов реакции за вычетом суммы теплот
образования исходных веществ с учетом стехиометрических
коэффициентов.
Тепловой эффект реакции между органическими веще-
веществами, например
{СН3СООН} + {С2Н5ОН} = {СН3СООС2Н5}+{Н2О}, Д#1
может быть вычислен алгебраически суммированием теп-
теплот сгорания участников реакции:
{СН3СООН}+2(О2) = 2(СО2) +2{Н2О}, Д#° = _ 871,1 кДж
{C2H5OH}-f 3(O2) = 2(CO2)-f3{H2O}, АН°з = —1367 кДж
{СН3СООС2Н5} + 5(О2)=4(СО2)+4{Н2О}, АН1 = — 2284 кДж
Суммируя теплоты сгорания, находим тепловой эффект
реакции: АН\ = АЯ°2 + ДЯ3—Д#; = —871,1 — 1367,0 +
+ 2284,0 = 46 кДж. Из приведенного примера вытекает
второе следствие закона Гесса: тепловой эффект реакции
между органическими веществами равен сумме теплот
сгорания исходных органических веществ за вычетом суммы
теплот сгорания продуктов реакции.
Тепловые эффекты пропорциональны массе веществ,
участвующих в химической реакции, что следует из за-
закона сохранения массы-энергии. При удвоении стехио-
стехиометрических коэффициентов реакции тепловой эффект
удваивается (условие экстенсивности).
Фазовые и химические процессы, их тепловые эффекты.
Остановимся на сравнительной характеристике тепловых
эффектов фазовых и химических процессов.
Как уже указывалось, к фазовым превращениям отно-
относят сублимацию, испарение и полиморфные превращения,
например:
сублимация
[12] = A2), Д#°г98 = 62,42 кДж
испарение
{HSO}=(H2O), Д#»6 = 41,02 кДж
44
плавление
[А1Вг3] = {А1Вг3}, ДЯа«8= 11,34
переходя из аморфного состояния в кристаллическое
[В2О3] = [В2О3], ДЯ29в = -18,4 кДж
аморфная кристалли-
форма ческая форма
полиморфный переход
[As2O4] = [As2O4], Д#°298 = 5,9 кДж
октаэдриче- моноклинная
екая форма форма
Все фазовые превращения в последовательности пере-
перехода из твердого состояния через жидкость в газ являются
процессами эндотермическими. Причина этого в том, что
для каждого последующего состояния по сравнению
с предыдущими характерно увеличение хаотичности вза-
взаимного расположения и движения частиц, образующих
вещество. Такая возрастающая подвижность частиц (ато-
(атомов, ионов, агрегатов) связана с разрывом части хими-
химических связей в структуре веществ, что требует затраты
энергии. Фазовые превращения при зафиксированном
давлении осуществляются, как правило, при строго опре-
определенных температурах. Так, при барометрическом давле-
давлении в 1013 гПа вода затвердевает (или плавится лед)
при 0°С, а при 100°С—кипит.
Химические превращения, как известно, в от-
отличие от фазовых обязательно сопровождаются измене-
изменением состава химических соединений.
Приведем несколько примеров химических превра-
превращений.
1. Процессы электролитической диссоциации. Разновидностью
химических реакций служат процессы электролитической диссоциации
и химические превращения в водных растворах электролитов. Про-
Процессы растворения ионных соединений подобны обычным химическим
реакциям. Примеры таких термохимических характеристик гидрати-
рованных ионов в водных растворах приведены в приложении V, где
теплота образования иона водорода {Н + }ач по схеме
принята равной нулю при рн = 1013 гПа и концентрации
Сн+ = 1 кмоль/м3. Теплоты образования остальных ионов вычислены
относительно этой величины.
Рассчитаем по закону Гесса значение Д#298 растворения NaCl,
NaOH и Na2S2O,n ПРИ образовании ими одномолярных растворов,
используя для ионов и кристаллических соединений величины, взятые
45
из таблиц:
q
, 298, кДж/моль —410,9 —239,7 —167,5
= 3,8 КДЖ
[] + q { }aq + {}aq
298, кДж/моль —426,8 —239,7 —228,4
г98 = —41,3 кДж
q q
ДЯ/, 298, кДж/моль —117,1 —479,4 —609,6
ДЯ°29з = 29,1 кДж
Вычисленное значение ДЯ2оз характеризует процесс растворе-
растворения NaCl как слабо эндотермический. Растворение NaOH — экзотер-
экзотермический процесс, растворение Na2S2O3— эндотермический. Опыт
показывает, что растворение в воде твердой NaOH действительно
сопровождается разогреванием раствора, а растворение Na2S2O3 —
охлаждением.
2. Процесс растворения известняка в разбавленных кислотах
можно представить из нескольких этапов. С одной стороны, проис-
происходит термическое разложение карбоната кальция — известняка
[СаСО3] = [СаО] + (СО2)
ДЯ^, 298, кДж 1186,2 —634,7 —393,3
которое, согласно закону Гесса, сопровождается тепловым эффектом
ДЯ°!=:— 634,7 — 393,3— A186,2)= 158,2 кДж,
такой эндотермический эффект определяет высокую термическую
прочность СаСО3 (начинает заметно разлагаться лишь выше 600°С).
С другой стороны, один из продуктов этой реакции—оксид кальция
(негашеная известь) СаО, как и любой другой основный оксид, экзо-
экзотермически растворяется в кислотах:
[СаО] +2{H + }aq = {Ca« + }aq +{H2O}
ДЯ;, 298, кДж —634,7 0,00 —543,1 —285,8
с тепловым эффектом ДЯг = —543,1—285,8 + 634,7 = —193,3 кДж.
Суммарный процесс растворения известняка сопровождается экзотер-
экзотермическим тепловым эффектом ДЯ3 = ДЯ1 + ДЯ2 = 158,2— 193,3 =
= —35,1 кДж. Такой процесс самопроизвольно идет при обычной
температуре.
3. Реакция восстановительного обжига
[Cas(PO4J] + 5[С]+3[SiO2] = 3[CaSiO3] + 5(СО) + (Р2)
Эндотермический тепловой эффект этой реакции, равный ДЯ2эз =
= 1697,9 кДж, может быть преодолен лишь за счет высоких темпе-
температур, достигающих 1400—1600°С. В этой реакции SiO2 экзотерми-
экзотермически связывается с СаО, образуя силикат CaSiO3, который сравни-
сравнительно легкоплавок, в виде расплавленной массы его удаляют из
реакционной части печи. Экзотермический эффект такого связывания,
ДЯ289 = —43,1 кДж, несколько снижает эндотермичность про-
46
цесса, идущего без участия SiO2, сравните:
[Са3(РО4J] + 14[С] = 3[СаО] +5(СО) + (Р„)
ДЯ/>ги —4119,1 0,00 —634,7 —110,5 +144,3 (кДж)
ДЯ°29в= 1807,5 кДж
4. Реакция окислительного обжига
Cu2S + (O2) = 2{Cu} + (SO2)
ДЯ2»в, кДж —60,7 0,00 +13,0-2 —296,2
Как показывает расчет, эта реакция экзотермична (ДЯ2эк =—-209,6 кДж)
и идет самопроизвольно с сильным разогреванием. Следует добавить,
что рассмотренный процесс положен в основу одного из промыш-
промышленных способов получения медн из сульфидных руд.
5. Процессы смешения жидкостей. Тепловой эффект растворения
характеризует химическое взаимодействие молекул растворителя
с растворенным веществом, он зависит от количества и природы
растворителя, в котором растворяют 1 моль данного вещества. Так,
растворение 1 моль H2SO4 в различных количествах воды сопровож-
сопровождается следующими тепловыми эффектами:
{H2SO4}+l{H2O} = {H2SO4}i, ДЯ298 = -28,07 кДж
{H2SO4}+100{H2O}=={H2SO4}100, ДЯ29в = — 73,39 кДж
{H2SO4}+10 000{H2O} = {H2SO4}10000, ДЯ20в = —86,32 кДж
6. Процессы атомизации и разрыва связей. Энергия атомиза-
цни ДЯат — это энергия, необходимая для разрушения молекул газооб-
газообразных веществ на свободные атомы, она представляет собой энергию
разрыва химических связей. Простейшими примерами процессов ато-
атомизации служат реакции диссоциации двухатомных молекул гомо-
соединепий, например,
(N2) = 2(N), ДЯат = 946 кДж
(О2) = 2(О), ДЯаТ = 495 кДж
(F2) = 2(F), ДЯ°ат = 157,3 кДж
Для трех- и более атомных молекул гомосоединений суммарная
энергия разрыва связей должна быть отнесена к числу пар связанных
атомов. Получившийся при этом результат характеризует энергию
разрыва связи каждой пары атомов. Так, суммарная энергия разрыва
связей в молекуле озона (О3) = 3(О), равна ДЯат = 562,7 кДж.
В молекуле озона присутствуют две пары связанных атомов (три
атома кислорода соединены последовательно, образуя угол в 117°,
и концевые атомы не связаны между собой), тогда энергия разрыва
химической связи каждой пары атомов будет соответствовать
562,7:2 = 281,3 кДж. Сравнивая прочность связи в молекулах О2
и О3, видим, что значительно меньшая прочность связей в моле-
молекуле О3 определяет энергетическую выгодность образования двух-
двухатомной молекулы кислорода из озона:
2@3) = 3{02), ДЯ209 = —360 кДж
Аналогично решен вопрос об энергии разрыва химических связей
в молекулах бинарных многоатомных ге/геросоединений, например
47
в Н2ОТ NH3 и СН4. Приводим значения энергии их атомизации:
(Н2О) = 2(Н) + (О), ДЯ°ат = 925,5 кДж
(NH3) = 3(Н) + (N), ДЯат = 1172,8 кДж
(СН4) = 4(Н) + (С), ДЯат= 1665,2 кДж
Согласно структурам этих молекул в молекуле Н2О —две химические
связи, в NH3 — три и в СН4 — четыре. Следовательно, энергия раз-
разрыва одной О—Н, N —Н и С — Н связей в рассматриваемых моле-
молекулах будет равна:
ДЯо_н = ^=^р=426,8 кДж,
= 416,3 кДж.
Расчет энергии разрыва отдельных связей в молекулах, состоящих
более чем из трех различных элементов, также возможен, но менее
надежей и связан с большими трудностями. Рассмотрим в качестве
примера соединение СН2С12, которое можно представить как продукт
взаимообмена молекул СН4 и СС14:
i/2(CH4) + V2(CC14) = (СН2С12), ДЯ°298 = 2,9 кДж
Суммарная энергия разрыва химических связей в молекулах исходных
соединений:
(СН4) = (С) + 4(Н), ДЯ°ат= 1665,2 кДж
(СС14) = (С) + 4(С1), ДЯоат = 13Ю,4 кДж
(СН2С12) = (С) + 2(Н) + 2(С1), ДЯ^Т = 1484,9 кДж
Энергия разрыва связи С — Н в молекуле СН4 уже приведена ранее
D16,3 кДж). Энергия разрыва связи С—>С1 в молекуле СС14 равна
АН'с-гл = 1310,4/4 = 327,6 кДж. Если предположить, что при переходе
от СН4 и СС14 к новому химическому соединению СН2С12 значения
энергии разрыва связи С—Н и С — С1 не изменяются, то для моле-
молекулы СН2С12 получаем ДЯ;т = 2.416,3 + 2-327,6= 1487,8 кДж. Вели-
Величины суммарной энергии разрыва связей и молекуле СН2С12, вычислен-
вычисленные двумя способами, близки, поскольку в рассмотренном примере
энергия разрыва связи атомов данной пары элементов не зависит
от энергии связи других пар. Так, в приведенном выше примере
энергия разрыва связи С —Н остается практически неизменной
и в СН4, и в СН2С12. Однако такое совпадение не характерно и стро-
строгий расчет энергии разрыва связей в молекулах гетеросоединений,
состоящих из трех и большего числа элементов, представляет слож-
сложную теоретическую задачу.
7. Энергия кристаллической решетки. Энергия кристаллической
решетки — это энергия образования соединения кристаллической формы
из свободных атомов (энергия атомной решетки) или из газообразных
ионов (энергия ионной решетки). Так, в ионном кристалле NaCI
каждый ион взаимодействует с шестью ионами противоположного
знака. Сумма этих взаимодействий равна энергии ионизации кристалла,
которая выражается уравнением
(Na+) + (Cl-J = [NaCl], ДЯ298 = —777,8 кДж.
48
В этом простейшем случае расчет энергии одной связи в ионных ре-
решетках производится делением суммарной энергии на число ионов,
окружающих ион противоположного знака, т. е. энергия связи ионов
777 Я
Na+ и С1~ равна ^—=—129,6 кДж.
Помимо веществ с ионными кристаллическими решетками (гало-
гениды наиболее активных металлов, щелочи, некоторые соли и др.)
встречаются вещества, кристаллические решетки которых построены
из атомов (атомные решетки) (оксиды, карбиды, силициды, нитриды
тяжелых металлов). Энергию атомной решетки относят к процессу
образования из нейтральных атомов, например:
(Si) + 2(O) = [SiO2j, ДЯ2вв = 1874кДж
Вычисленная таким образом энергия атомной решетки представляет
собой суммарную энергию связей в соединении. Приближенный
расчет энергии связей в атомной решетке возможен только для про-
простейших веществ при условии, если известна структура кристаллов.
В отношении кристаллов SiO2 известно, что каждый атом кремния
связан с 4 атомами кислорода. Если других взаимодействий между
атомами в решетке SiO2 нет, то A#si-O = 1874/4 = 469 кДж.
Глава 5
Химическое равновесие
Обратимые и необратимые реакции. Константа
химического равновесия, ее зависимость от темпе-
температуры и природы реагирующих веществ. Энтропия
и изменение энтропии. Энергия Гиббса.
Обратимые и необратимые реакции. Термохимические
уравнения реакций являются количественным выражением
закона сохранения массы-энергии и позволяют глубже
проанализировать характер химических превращений,
чем это доступно при использовании уравнений баланса
массы веществ, вступающих в реакцию и образующихся
после нее. По величине и знаку теплового эффекта реак-
реакции, вычисленного по таблицам, можно, не проводя опыта,
установить, будет ли идти при обычной температуре тот
или иной процесс.
Мысль о том, что в системе из всех возможных реак-
реакций преимущественной будет та, которая сопровождается
наибольшим экзотермическим эффектом, высказывалась
еще Ю. Томсеном A854) и М. Бертло A867). Этот кри-
критерий осуществимости процессов был назван принципом
наибольшей работы или принципом Бертло, отождествляю-
49
щим тем самым теплоту и работу. В ряде случаев прин-
принцип Бертло действительно подтверждается экспериментом.
Например, при образовании галогенидов углерода
[С] + 2 (F2) = (CF4), ДЯ°298 = - 920,5 кдЖ
[С] + 2 (С12) = (СС14), ДЯ298 = — Ю7,1 кДж
[С]-Ь2{Вг2} = (СВг4), ДЯ°298 = + 75,ЗкДж
[C]+2[J2] = (CI4), ДЯ°298 = +251,0кДж
экзотермичность взаимодействия переходит в эндотерми-
чность, что согласуется с активностью этих процессов
и с термической прочностью галогенидов. Так, С14 можно
разложить на 12 и С214 при 170°С, а полностью — при
1700 °С; четырехфтористый углерод практически не раз-
разлагается до 3000°С.
Наиболее отчетливо принцип Бертло получил подтвер-
подтверждение на примерах необратимых реакций, идущих до
конца и не способных изменить направление при изменении
температуры и давления. Примером таких реакций может
служить экзотермическое разложение нитроглицерина
С.,Н«,(ОКЮ2K по схеме
{C3H5(ONO2K} — 3(СО2) + 2,5(Н2О) + 0,5(NO) + 1,25(N2)
Этот процесс имеет характер взрыва, сопровождается
выделением теплоты (ДЯ°9в = —1427кДж)и образованием
большого объема газов. Обратный ему эндотермический
процесс невозможно осуществить никакими изменениями
давления и температуры, что является основным призна-
признаком химической необратимости.
Однако наряду с необратимыми превращениями су-
существует бесчисленное множество химически обратимых
реакций. Эти реакции, не объясняемые с позиций прин-
принципа Бертло, при одних условиях температуры и давле-
давления идут в одном направлении, а при других — в обрат-
обратном. Эти процессы ни в одном из направлений не идут
до конца, а лишь до строго определенных соотношений
исходных и получившихся продуктов. Такое состояние
получило название химического равновесия.
Примерами химически обратимых процессов, идущих
при постоянной температуре до состояния равновесия,
могут служить термическое разложение гидроксида каль-
кальция (химический процесс)
[Са(ОНJ] = [СаО] + (Н2О), AHl9B = 109,1 кДж
и испарение воды (фазовое превращение)
50
В этих процессах каждому значению температуры соот-
соответствуют строго определенные значения давления водяного
пара. При диссоциации Са(ОНJ давление водяного пара
при температуре 450 °С равно 133 гПа, при 524 °С —
666 гПа; испарению воды при температуре 100 °С соот-
соответствует давление водяного пара 1013 гПа. Обратимость
процессов испарения и конденсации воды при изменении
температуры повседневно можно наблюдать в виде при-
природных явлений (выпадание росы ночью и испарение ее
днем), в быту (выкипает чайник и сразу же запотевает
окно, в лаборатории (дистилляция воды) и т. д.
Аналогичным примером обратимого процесса может
служить растворение в воде азотнокислого аммония:
[NH4NO3]±aq=={NHl}aq + {NO3-}aq, Д#°298 = 25,8 кДж
Этот процесс эндотермический и, согласно принципу
Бертло, не должен идти. Опыт же показывает, что рав-
равновесная растворимость NH4NO3 в воде достаточно велика
B14 г в 100 мл воды при 25°), при этом растворение со-
сопровождается понижением температуры вплоть до —50 °С.
Эти и подобные им примеры иллюстрируют истинное
химическое равновесие, которое характеризуется следую-
следующими признаками:
1) при отсутствии внешних воздействий состояние
равновесия системы сохраняется во времени неизменным;
2) при изменении внешних воздействий (давления, тем-
температуры, концентрации веществ и т. д.) состояние системы
изменяется, но при восстановлении исходных условий
восстанавливается и исходное состояние.
Кроме истинного часто встречается так называемое
ложное или замороженное равновесие. Примерами могут
служить экзотермические процессы, лежащие в основе
синтезов аммиака и серной кислоты:
(N2) +3(Н2) = 2(NH3), ДЯ298 = — 92 кДж
(SOа) + V2(O2) = (SOa),
Эти процессы при обычных условиях должны были бы
идти, но не идут, создавая впечатление равновесных
газовых смесей. Однако с введением соответствующих
катализаторов эти реакции интенсивно развиваются вплоть
до установления истинного химического равновесия.
Константа химического равновесия. Все приведенные
выше примеры убеждают в том, что тепловой эффект
реакции не может служить количественным критерием
осуществимости химических реакций, мерой работы хими-
61
ческих процессов. В поисках такого критерия было
создано и развито стройное учение о химическом равно-
равновесии, основные положения которого будут изложены
в этой и последующих главах. Одним из примеров рав-
равновесных химических превращений в смеси газообразных
веществ может служить взаимодействие
(СО2) + (Н2) = (СО) + (Н2О), ДЯг,8 = 41,2 кДж E.1)
Очевидно, для акта химического взаимодействия необхо-
необходимы столкновения молекул СО2 и Н2, число таких столк-
столкновений можно подсчитать.
Положим, имеется псо, молекул углекислого газа
и пнг молекул водорода. Максимальное число однократ-
однократных столкновений Wt молекул СО2 с каждой из молекул
водорода равно ясо,- Отсюда общее число столкновений
молекул СО2 с одной молекулой водорода и между собой
будет пропорционально псо,:
wi — асо„пссу i
Точно так же общее число столкновений W2 для молекул
водорода будет равно
Здесь асо, и аНг— коэффициенты пропорциональности.
Общее число столкновений Wlt 2 будет равно произведению
Вместо числа частиц «со, и nHi удобнее пользоваться
величинами концентраций
"го. _ "н.
где jVa — постоянная Авогадро, V—объем газа. В этом
случае выражение E.2) примет вид
где Рсо^^ ^/
Однако столкновения частиц еще не определяют воз-
возможности их химического взаимодействия. Большинство
таких столкновений носит упругий характер, похожий на
столкновения биллиардных шаров. И только соударения
наиболее энергичных молекул, которые за счет энергии
столкновения возбуждаются, приводят к химическому
взаимодействию и химической перестройке молекул.
52
Каждый сорт молекул требует для такого возбуждения
определенную энергию Е*. Схематически возбуждение
молекул при соударении можно изобразить следующим
образом:
Если кинетическая энергия соударений молекул угле-
углекислого газа и водорода будет меньше суммы Есог + ?н2, то
столкнувшиеся молекулы не перейдут в достаточно актив-
активное состояние и произойдет простое упругое столкновение
частиц. Лишь те столкновения, энергия которых равна
или больше суммы ?со, + ЕНг, приводят к образованию
продуктов реакции, т. е. молекул Н2О и СО.
Из физики известен закон Максвелла — Больцмана,
который в количественной форме характеризует распре-
распределение частиц по величинам их энергии. Согласно этому
закону число частиц п*, обладающих энергией Е*, выра-
выражается соотношением
еь"и, E.4)
"оби
где п*-—число частиц, обладающих энергией Е*, п —
общее число частиц, R — газовая постоянная, Т — темпе-
температура, е—основание натуральных логарифмов.
Объединив уравнения E.2), E.3) и E.4), получим
выражение для числа активных столкновений:
К * = Рсо, Lcco/ RT J Рн.
RT J Рн Lch/ RT J =
RT
приводящих к образованию продуктов реакции E.1).
Поскольку эта реакция обратима, с появлением конечных
продуктов Н2О и СО становится возможным их обратное
взаимодействие (Н2О) + (СО)—>-. Число активных соуда-
соударений, приводящих к химическому взаимодействию, в этом
обратном процессе составит
Ен,о+Ясо
RT
где ?нго и ?со—энергии активации молекул Н.,0 и СО.
Следует 'заметить, что реальные химические процессы
63
в основном определяются совокупностью двойных соуда-
соударений реагирующих молекул, тройные соударения весьма
редки.
Если общее количество вещества в рассматриваемой
системе постоянно, то число химически активных соуда-
соударений молекул, характеризующих прямую реакцию, со
временем должно уменьшаться из-за снижения концен-
концентраций СО, и Н2; число же активных столкновений
молекул Н26 и СО с увеличением концентрации будет
возрастать. При этом обязательно наступит такой момент,
когда Wli3 станет равной W*34. Начиная с этого момента
времени и далее устанавливается химическое равновесие,
т. е. в системе устанавливается такое состояние, при
котором соотношение равновесных концентраций ужб не
зависит от времени. Такое равновесие называют динами-
динамическим.
Таким образом, равновесное состояние характери-
характеризуется условием
Wl, 2=Гз, 4,
?н,о+?со
следовательно,
Рсо2Рн2исо2ин2е —Рн2оРсоин2оьсое
Перенесем символы равновесных концентраций в одну
сторону от знака равенства, а все остальные — в другую:
Рн2оР
со
В таком случае левая часть уравнения E.5) для данной
системы веществ при заданной температуре представляет
собой постоянную величину, получившую название кон-
константы химического равновесия (К). Константа равнове-
равновесия зависит только от природы реаёирующих вешаете
и температуры и не зависит от давления и концентра-
концентрации реагирующих веществ. Следовательно, правая часть
уравнения E.5) может быть приравнена к величине кон-
константы химического равновесия, т. е.
К= "г0„с0 , E.6)
°CO2LH,
Уравнение E.6) отражает объективный закон природы,
получивший название закона действующих масс, который
54
эмпирически был установлен К. Гульдбергом и П. Вааге
A864 —1867). Этот закон является фундаментом учения
о химическом равновесии и важнейшей составной частью
дисциплины, называемой термодинамикой.
Одно из следствий закона действующих масс—правило
смещения равновесий Ле Шателье A884), которое форму-
формулируется следующим образом: если на равновесную систему
произвести внешнее воздействие, то равновесие будет
смещаться в направлении этого воздействия до тех пор,
пока нарастающее в системе противодействие не станет
равным внешнему действию.
Энтропия и изменение энтропии. Возвратимся к ана-
анализу выражения E.5), его логарифмирование приводит
к виду
-=Д1пр,—
RT
E.7)
Умножив правую и левую части уравнения E.7) на —RT,
получим
Рассмотрим подробнее слагаемые правой части уравне-
уравнения E.8). По условию АЕ* характеризует сумму значе-
значений энергии активации прямого и обратного процессов
равновесия E.1), каждый из которых приводит к образо-
образованию энергетически возбужденного активного комплекса:
—прямой процесс
О---С •••о/| *
ЧН
обратный процесс (Н2О)+(СО)+[?^2О+?^О].
Активным комплексом называют некоторую комбинацию
атомов взаимодействующих веществ, обладающих макси-
максимумом анергии, которая с энергетическим выигрышем
самопроизвольно распадается на продукты реакции.
Сочетая прямую и обратную реакции, получаем урав-
уравнение
(СО,) + (Н2) + [Е'СОг + ?Нг- ?нго~ ?со] = (Н.О) + (СО) E.9)
Сравнение равновесия E.9) с термохимическим уравнением
(СО2) -\- (Н2) + Л# ив = №0) + (СО) E.10)
показывает, что [ЕСОа + Ен2 — ?н2о—ЕсО] — АЕ* есть не
55
что иное, как тепловой эффект реакции E.1), т.е.
Д?*= АЯ°2в8.
Вторым слагаемым в правой части уравнения E.8)
является произведение TRA In Р,-. Смысл символа Р в вы-
выражении E.3) становится понятным, если задать усло-
условие Ссо2 = Сн2 = Сн2о = Ссо = 1 кмоль/м3, откуда
^1, 2 = РсО.Рн,- Wa, 4 = Рн2оРсО-
Следовательно, произведение р,- представляет собой общее
число соударений частиц газовой смеси при условии равен-
равенства всех концентраций 1 кмоль/м3. Поскольку число
соударений частиц характеризует беспорядок в системе,
выражение типа R In р принято считать количественной
мерой беспорядочности движения молекул в газах, жид-
жидкости, атомов в молекулах, атомов и ионов в решетках
кристаллов и т. д. Такая мера неупорядоченности полу-
получила название энтропии (S), т. е.
R In p = S.
Энтропия имеет размерность — Дж'(моль-К) (для крат-
краткости з. е.).
Неупорядоченность движения частиц, образующих
данное вещество, принято характеризовать с помощью
вероятности состояния (W), под которой понимают число
осуществляющихся в каждый данный момент времени
мимолетных состояний. Между вероятностью данного со-
состояния вещества и его энтропией установлена количест-
количественная взаимосвязь (формула А. Больцмана):
где/г—постоянная Больцмана: W — вероятность состояния.
Само по себе число соударений характеризует лишь
поступательное движение частиц газа и отвечающую ему
энтропию поступательного движения. Внутри молекул
образующие их атомы всегда находятся в относительном
колебательном движении (даже при абсолютном нуле
температуры сохраняются «нулевые» колебания). Этому
внутримолекулярному колебательному движению соответ-
соответствует колебательная составляющая энтропии, которая
существенно зависит от числа атомов в молекуле, харак-
характера связей- атомов в молекуле, и геометрии их взаимного
расположения. Кроме этого, молекулы в целом или их
части могут вращаться, этому вращательному движению
соответствует вращательная составляющая энтропия.
56
Полная энтропия складывается из поступательной, коле-
колебательной и вращательной составляющих (в валентно-нена-
валентно-ненасыщенных соединениях есть еще так называемая электрон-
электронная энтропия)
*-*подн ~4^пост~г4^колеб г^вращ»
т. е. каждое вещество в соответствующем агрегатном состоя-
состоянии наделено определенным, присущим только ему значе-
значением энтропии. Наиболее упорядоченное состояние веще-
вещества—кристаллическое, для него характерно наименьшее
значение энтропии. Для жидкого и парообразного состоя-
состояний соответствуют более высокие значения энтропии, так
как в этом случае беспорядочность движения частиц воз-
возрастает. Например, энтропия 1 моль льда равна 47,95 э. е.
при 0°С; жидкой воды—69,96 э. е., а водяного пара —
188,74 э. е. при 25 °С
Энтропия как мера хаоса связана не только с движе-
движением, но и с числом частиц вещества, их массой и харак-
характером взаимного расположения. Так, 1 моль газообраз-
газообразного двухатомного фосфора (Рг) имеет энтропию 218,4 э. е.,
а четырехатомного (Р4) — 279,9 э. е.
С увеличением массы одинаковых по форме газообраз-
газообразных молекул в ряду СН4, CF4, СВг4, возрастают и зна-
значения энтропии этих соединений—186,2; 261,5 и 358,2 э. е.
соответственно. Энтропия не может быть отрица-
отрицательной. При полной упорядоченности структуры кри-
кристаллической решетки (при температуре ОК) энтропия может
быть равна нулю. Значения стандартных энтропии инди-
индивидуальных веществ представлены в приложении I.
Таким образом, в уравнении E.8) выражение ^Д\
представляет собой алгебраическую сумму энтропии
участников равновесия E.1), которую называют измене-
изменением энтропии и обозначают символом AS0, т. е.
подставив стандартные значения энтропии, получаем
AS°= 188,7 + 197,4 — B13,6+ 130,5) = 42,0 э. е.
Если стехиометрические коэффициенты в уравнении
реакции отличаются от единицы, то значение энтропии,
относящееся к 1 моль, следует умножить на стехиометри-
ческий коэффициент.
Например, для процесса
(Н2О)+ (С12) =
188,7 223,0 2-186,6 0,5-205,0 э. е. ('
57
изменение энтропии ASj88 равно
AS°m=[2-186,6 + 0,5.205,0]-[188,7 + 223,0] = 64,0 э. е.
Энергия Гиббса. Учитывая все изложенное выше,
равенство E.8) можно записать так:
—RT In К = bH° — TAS°. E.12)
Левая часть уравнения E.12) — RTlnK — имеет раз-
размерность энергии (Дж/моль) и характеризует уровень
энергии системы, отвечающий равновесным концентра-
концентрациям взаимодействующих веществ, т. е. состоянию равно-
равновесия. Чтобы получить выражение для работы химической
реакции E.11), необходимо иметь два уровня энергии
или состояния системы, соответствующих различной глу-
глубине химической реакции при заданной температуре. По-
Поскольку одно из этих состояний равновесное с уровнем
энергии
/-2 /-0,5
RT In нс' °2 = —RT In Kc, E.13)
СН2ОСС12
второе состояние должно быть неравновесным и ему дол-
должен соответствовать другой, исходный уровень энергии
E.14)
здесь Ц;—неравновесные значения концентраций. Пере-
Переход с произвольного энергетического уровня E.14) на
равновесный E.13) должен характеризоваться изменением
энергии, за счет которой осуществляется работа хими-
химической реакции:
14O RT In Kc- E.15)
Символ AG представляет собой изменение энергии системы
или работу реакции при переходе от произвольных кон-
концентраций к равновесным, С;, при р = const.
Следовательно, величина работы зависит не только от
равновесных концентраций, строго определенных приро-
природой реагирующих веществ и температурой, но и от зна-
значений исходных концентраций, которые могут быть выб-
выбраны произвольно. Для исключения такой неопределен-
неопределенности вводят условие стандартности исходных концент-
концентраций реагирующих веществ q,= l кмоль/м3. С учетом
этого условия стандартная величина ДС связана с кон-
58
стантой равновесия соотношением
AG°T = — RTlnKc
сравнивая которое с уравнением E.12) и вводя условие
температурной зависимости А#° и AS°, получаем
или приближенно (так как зависимость от температуры
невелика)
AG°r = Д//298 — Т AS°298 = —RTlnKc- E.16)
Выражение E.16) называют основным уравнением
термодинамики, оно количественно связывает между собой
закон действующих масс (К), тепловой эффект реакции
(Л#298), меру хаотичности движения и расположения
частиц (ASaeg) и температуру (Г) с величиной работы AGor.
Величина G характеризует уровень энергии при совер-
совершении работы химической реакции в условиях постоян-
постоянства давления, название этой величины—энергия Гиббса,
в память об известном американском физике Джозайя
Вилларде Гиббсе A839—1903).
Если равновесие устанавливается в газовой фазе или
с участием газов, то при небольших давлениях, исполь-
используя уравнение Менделеева — Клапейрона, можно перейти
от парциальных молярных концентраций С,- к парциаль-
парциальным давлениям компонентов:
= 0,00831 CjT,
где С; — концентрация, кмоль/м3, р;—давление, Па. При
атмосферном давлении 1,013:105 Па и температуре 273 К
молярная концентрация газа будет равна
C>=~RT=lSw=0'0447 кмоль/м3-
При условии С,= 1 кмоль/м3 давление газа будет равно
/?,.=8,31 Т-1000=8,31 -273.1000=2269 кПа = 2,27-106 Па.
Если в качестве стандартной величины исходного пар-
парциального давления каждого газообразного компонента
равновесной, газовой смеси принять /?, = 1,013-105 Па, то
уравнение изотермы E.15) примет вид
ДО°Г= 19.138Г lg A,013- 105)Дл!- 19.138Г lg Kp =
958rA— 19Д38Г Igtf^, E.17)
где Av—алгебраическая сумма коэффициентов, стоящих
69
в уравнении реакции перед газообразными компонентами
[для реакции E.11) Av = B + 0,5 —1 —¦ 1) = 0,5].
Следовательно, для газовых реакций (равновесий)
основное уравнение термодинамики E.12) принимает вид
95,8ГМ> — 19.138Г \gKp = kH°—TAS°, E.18)
или
Глава 6
Смещение химического равновесия
Термодинамический анализ химических превраще-
превращений. Смещение равновесия в системах при изме-
изменении температуры, давления и концентраций
реагирующих веществ.
Учение о химическом равновесии, некоторые положе-
положения которого были изложены в предыдущей главе, яв-
являются составной частью термодинамики. В настоящее
время наряду с термодинамикой равновесных систем
успешно развивается термодинамика необратимых процес-
процессов, приобретающая большое значение в самых различ-
различных областях науки и техники, в частности при изуче-
изучении биологических процессов. Химическая термодина-
термодинамика—один из разделов термодинамики равновесных
систем — вскрывает основные законы энергетики хими-
химического равновесия.
Термодинамический анализ химических и фазовых пре-
превращений позволяет, не прибегая к эксперименту, из
всей совокупности возможных реакций сделать выбор
реально осуществимых процессов и количественно опре-
определить их направление и глубину. Связь между тер-
термодинамическими характеристиками и равновесными кон-
концентрациями реагирующих веществ осуществляется с по-
помощью константы химического равновесия (см. гл. 5).
Для понимания смысла константы химического или
фазового равновесия и правильного использования этой
характеристики необходимо ввести некоторые дополни-
дополнительные понятия. Совокупность всех веществ, принимаю-
принимающих участие в химическом равновесии, принято называть
системой. Систему называют равновесной, если в ней при
одних и тех же температуре и давлении в течение дли-
60
тельного времени не происходят видимые химические или
фазовые превращения, и если это состояние равновесия
достигается в результате как прямой, так и обратной
реакции.
Если вся совокупность свойств строго одинакова во
всех частях системы, ее считают гомогенной. Например,
равновесие
С2 С0'6
2(О2), К= "С' °г
ь1
устанавливается в гомогенной газовой системе. Точно так
же для уксусной кислоты {СН3СООН}, растворенной
в воде, характерно равновесие электролитической диссо-
диссоциации:
сн+ссн3соо-
Л = —г • F.1)
LCH3COOH
Этот раствор представляет единую равновесную, гомо-
гомогенную систему молекулярных и ионных форм.
Систему, состоящую из двух и более фаз, называют
гетерогенной. Например, термическое разложение кристал-
кристаллического карбоната кальция
[СаСОз1=[СаО1 + (СО2), Кр = рСО! F.2)
приводит к образованию новых веществ, которые в си-
системе строго обособлены и образуют самостоятельные
фазы с характерными для них наборами свойств, это
одна газообразная фаза СО2 и две твердые—СаСО3 и СаО.
Фазы, входящие в состав любой системы, могут быть
постоянной и переменной концентрации. Фазы постоянной
концентрации — это индивидуальные химические соедине-
соединения в твердом или жидком состояниях, для которых
характерно постоянство относительных концентраций
составных частей данной фазы (при постоянной темпера-
температуре). С этих позиций индивидуальное вещество в газо-
газообразном состоянии СО2 в равновесии F.2) не является
фазой постоянной концентрации, так как не имеет собст-
собственного объема, и поэтому его общая концентрация
в объеме газа может изменяться в широких пределах.
В соединениях СаО и СаСО3 относительные массы свя-
связанных кислорода, углерода и кальция остаются прак-
практически неизменными— это фазы постоянной концентрации.
Фаза переменной концентрации—это твердый, жидкий
или газообразный раствор, концентрации составных частей
61
которого переменны в результате реакций, протекающих
в растворе, или за счет внесения их извне, при условии
неизменной гомогенности. Так, в гомогенной системе F.1)
в зависимости от температуры будут изменяться соотно-
соотношения между концентрациями ионов Н + , СН3СОО~ и
молекул СН3СООН. При постоянной температуре можно
произвольно изменить концентрацию ионов {H + }aq и
ионов {CH3COO~}aq, добавляя в раствор сильную кис-
кислоту или хорошо растворимую соль [CH3COONa]. Однако
такая произвольность ограничена, она регламентируется
законом действующих масс. Как уже отмечалось, одно из
положений учения о химическом равновесии утверждает,
что константа равновесия не зависит от концентраций.
Поэтому в выражении для константы F.1) произвольному
изменению концентраций одной или двух равновесных
форм соответствует обязательное изменение концентраций
остальных форм за счет дополнительного прохождения
реакции.
Другим примером гетерогенного равновесия может слу-
служить восстановление твердого хлористого железа водоро-
водородом при температурах 300—600 °С:
[FeCl2] + (H2) = [Fe] + 2(HCl). F.3)
Металлическое железо и дихлорид железа ие образуют
твердых растворов, поэтому они индивидуальны. Являясь
постоянными величинами, концентрации этих соединений
не учитываются в величине константы равновесия. Сле-
Следовательно, для процесса F.3)
г2
к— НС1
Численное значение константы равновесия зависит от спо-
способа выражения концентрации веществ:
1. Молярную концентрацию определяют числом молей
растворенного вещества в единице объема раствора, в этом
случае константу равновесия выражают через молярные
концентрации и обозначают Кс.
2. Молярную долю N/ определяют числом молей п{
растворенного вещества, отнесенным к общему числу молей
фазы переменного состава
62
а для идеального газового раствора
N=4*- = -EL- F.5)
2? Робщ
Константу равновесия выражают через молярные доли,
обозначая Kn-
3. Состав идеального газового раствора чаще
характеризуют парциальными давлениями р;, сумма кото-
которых в газовой фазе равна общему давлению (закон
Дальтона):
Робщ
= 2 Л'-
Константу равновесия, выраженную парциальными давле-
давлениями реагирующих веществ, обозначают Кр. Так, равно-
равновесие F.3) правильнее характеризовать константой
Рна
K
а равновесие F.2)—Кр = рсо,-
Между константами Кс, Кр и KN можно установить
количественное соотношение, для этого рассмотрим равно-
равновесие в гомогенной газовой системе:
где х, у, т, п—стехиометрические коэффициенты; А, В,
D, Е — химические соединения. Согласно закону дейст-
действующих масс
САСВ
Если система представляет собой идеальный газовый раст-
раствор и на нее распространяется закон Менделеева — Кла-
Клапейрона, то парциальные давления и концентрации свя-
связаны соотношением
p = CRT. F.7)
Подставив в выражение F.6) значения С из уравне-
уравнения F.7), получим
_ /_pd_y (Je_\" i/_^а_У ( %.Y
~\RT J \ RT J I\RT/ \ RT J '
63
а при вынесении общего множителя —
РАРВ
ИЛИ
Kc = Kp(RT)x+y-m-n. F.8)
К системам, представленным жидкими или твердыми
растворами, уравнение газового состояния F.7) неприме-
неприменимо, показатель степени в уравнении F.8) теряет смысл,
становясь равным нулю. Таким образом, для равновесий
в конденсированных системах КР = КС-
Соотношение между Кс и Кы определяют совместным
решением уравнений F.6) и F.4), которое приводит к вы-
выражению
отсюда
ttl + n-X-y
ш
Решим несколько конкретных задач.
Задача 1, Пользуясь табличными значениями для энтропии
иДЙ/, •!,» образования соединений из простых веществ, определим
температуру, при которой давление термического разложения каль-
кальцита равно атмосферному A013 гПа).
Запишем уравнение реакции и выражение для константы равно-
равновесия:
[CaCO3] = [CaOj + (CO2), Kp = pca>, Av=I.
Теплоты образования и энтропии всех индивидуальных соединений
этой системы, найденные из таблиц, имеют следующие значения:
АН},
¦Sase»
298
э.
, кДж...
е
СаСО,
—1206,7
92,9
СаО
—635,5
39,7
СО2
—393
219
,3
,8
Исходя из этих значений, вычислим АН2д$ и Д52э8 разложения
кальцита:
AH'ms=— 393,3 — 635,5—(—1206,7) =177,9 кДж,
ASlss =219,8 + 39,7 — 92,9= 166,6 э.е.
Подставив численные значения ДЯ2е8, ASaes и давление, равное
1,013-105 Па, в уравнение
AG\ =9b,8T—RT InpCOa = b.H\tb — TAS^g = 0,
64
получаем температуру разложения кальцита:
АЯ°2!)8 177 900 42 500
(ИЛИ °35 С),
Экспериментальное определение температуры дает величину 989 °С,
т. е. на 63° выше. Причина этой ошибки кроется в том, что в реше-
решении задачи не учтена температурная зависимость ДЯ298 и AS298. ко-
которая хотя и невелика, но все же искажает результаты расчета.
Задача 2. Определить значения ДЯ298 и AS298 процесса испаре-
испарения воды при температуре 20 °С и давлении насыщенного пара воды
2,Ы03Па
{Н2О} = (Н2О), Кр = рНа0-
Известно, что в нормальной точке кипения, т. е. при 373 К,
давление насыщенного пара воды равно 1,013-105 Па, это означает,
что, как и в задаче 1,
8 = 0. ' F.9)
Работа, необходимая для осуществления процесса испарения при
заданном условии, будет равна
AG293 = 95,8-293 =—19,14-293 lg 2,Ы03 = 9,45 кДж. F.10)
Решая совместно систему уравнений F.9) и F.10) для процесса ис-
испарения воды при температуре 20 °С, находим ДЯ^В8 = 43,9 кДж,
AS298=H8 э. е.
Задача 3. Определить, при каком общем давлении выход аммиака
при его синтезе из азота и водорода составит молярных 90%, считая газ
идеальным при любых давлениях. Температура реакционной смеси
400 °С. Соотношение парциальных давлений водорода и азота в ис-
исходной смеси равно 3.
Напишем уравнение реакции синтеза аммиака и выражение его
константы равновесия:
2
(N,)+3(H2) = 2(NH»), Кр = PnH' , Av = -2.
Pn2 Ph.
Используя термодинамические характеристики всех участников про-
процесса :
(NH.) (Н,) (N.)
ДЯО2В8, кДж... —46,0 0 0
Д5298, э. е. ... 192,2 134,8 191,5
получаем ДЯ°298 = 2 (—46,0) = —92,0 кДж,
Д5гв8 = 2-192,2 — C-134,8 + 191,5)= —211,5 э. е.
Подставив значения ДЯ298, Д52д8 и температур F73 К по условию)
в уравнение Гиббса, получаем
д0о673 = -95,8-673-2-19,14-673 lg/Cp = —92 000 + 673.211,5 =
= 50,1 кДж.
№ 705 65
Значение Л'р находим из решения уравнения
-178840 _2
р ' — - > • • \ • )
Выход аммиака по условию составляет 90%, следовательно,
По условию задачи в исходной смеси отношение парциальных давле-
давлений водорода рн и азота pN равно
С образованием аммиака равновесные значения давления водорода
и азота будут меньше исходных, т. е. рн =Pjj —3/2Pnh > Pn =
C 3)B 3) = 3- F.13)
Решая систему уравнений F.11), F.12) и F.13), находим парциаль-
парциальные давления pN =329,2 гПа, рн =987,7 гПа, Pjy-н =13 169гПа
и искомое общее давление газа робщ= 14 486 гПа.
Смещение равновесия в системах. Ранее было сформу-
сформулировано правило смещения равновесия Ле Шателье, оно
справедливо в отношении всех равновесных систем. Из
основного уравнения термодинамики —RT In Кс = ^Н°—¦
— TASlss при использовании математических методов вы-
выводится выражение для константы равновесия как функ-
функции температуры:
/Сс = 101в-14Г К)"'14, F.14)
где второй сомножитель—бестемпературный коэффициент.
Знак (+ или —) величины ДЯ^,, в первом множителе
определяет характер изменения константы равновесия.
Если знак положителен (эндотермический процесс), то
с ростом температуры константа равновесия увеличивается
и равновесие смещается в сторону продуктов реакции.
Напротив, если знак отрицателен (экзотермический про-
процесс), то константа равновесия уменьшается, т. е. равно-
равновесие смещается в направлении исходных веществ. Мно-
AS,
298
житель 10 т в уравнении F.14) выполняет роль коэф-
66
фициента, определяющего величину, но не направление
смещения.
Степень и направление температурного смещения рав-
равновесия, как это видно из уравнения F.14), зависит
только от знака и величины AH°29i. Действительно, если
Д#298 = 0. то константа равновесия не зависит от темпе-
температуры. Чем больше отличается AH°2Sa от нуля, тем
в большей степени величина константы зависит от темпе-
температуры и тем значительнее равновесие смещается при
одном и том же изменении температуры.
В качестве примера рассмотрим процесс разложения карбоната
кадмия. Этот процесс эндотермичен и сопровождается ростом энтро-
энтропии (реакция идет с увеличением числа газовых молей):
298 = 95,0 кДж
[CdCO3] = [CdO] + (CO2) Д5°298 = 168,2 э. е. F.15)
Используя основное уравнение термодинамики E.12), находим /Ср:
_ 49 70
95,87" — ДПп/(р = 95 000 — Т.168,2, /Ср= 101».'8 • 10 т ,F.16)
При подстановке численных значений температуры в уравнение F.16)
получаем значения константы равновесия (в Па): Т = 515/(— Кр =
= 0,135-Ю6; Т" = 615 К-—Кр = 4,90-105. Сростом температуры равно-
равновесие F.15) смещается вправо.
Для сравнения рассмотрим процесс, характеризующийся близким
к процессу F.15) значением теплового эффекта, но резко отличной
величиной Д52В8:
Л#298 = 91,6кДж
H2) + 1/2(CIa) AS°2B8 = -10,0 э. е. F.17)
Av = 0
Используя имеющиеся данные, напишем основное уравнение термо-
термодинамики и определим величину константы равновесия:
-4790
#р = 0,3-10 т , F.18)
В процессе F.17) значение &S°iss составляет небольшую величину
(поскольку реакция идет без изменения числа газовых молекул), по-
поэтому и постоянный коэффициент в уравнении F.18) очеиь мал. В ре-
результате при подстановке численных значений температуры в выраже-
выражение F.18) получают значения констант равновесия F.17): при 515 К—
—/Ср= 1,5- Ю-10; при 615 К — /Ср = 48- Ю0, на пятнадцать поряд-
порядков меньше, чем в предыдущем примере.
Рассмотрим, как смещаются равновесия под воздейст-
воздействием изменения давления и концентраций. Выше отмеча-
отмечалось, что в пределах не очень больших давлений и концен-
концентраций константа равновесия не зависит от их изменения.
Но входящие в выражение константы равновесия значения
з* 67
равновесных давлений газообразных соединений могут
существенно изменяться, т. е. равновесие может смещаться
в ту или иную сторону.
Рассмотрим в качестве примера реакцию получения водяного
газа:
ДЯ2,8= 131,4 кДж
[С] + (Н2О) = (СО) + (Н2) д5; = 116,5 э. е. F.19)
Av=l
Запишем для этого процесса уравнение 95,8 Г — 7?Т In /Ср= 131 400 —
— Г-116,5 и константу равновесия
Общее давление в системе идеальных газов согласно закону Даль-
Дальтона выражается суммой парциальных давлений:
Робщ=Рсо+Рн, + Рн,О- F.21)
Из стехиометрии процесса F.19) следует, что
Рсо = Рн,- F.22)
Решая систему уравнений F.20), F.21) и F.22), определим парциаль-
парциальные давления всех компонентов системы и вычислим степени взаимо-
взаимодействия:
Рсо+Рна
а = ».
Робщ
После подстановки вычисленных значений парциальных давлений
получается выражение
/+^ F.23)
Робщ L ' Кр J
Как видно из уравнения F.23) степень прохождения реакции а
должна зависеть от роащ. Так, для равновесия F.19) при 1127 К и
Кр = 1 имеет место следующая взаимосвязь:
Робщ. па 1,013-10' 1,013- 10е 1,013-106 1,013-103
а 1,3- Ю-3 4,0-Ю-3 1,3-Ю-2 1,3-Ю-1
Из примера следует, что с ростом давления уменьшается степень
(или глубина) прохождения реакции и равновесие смещается в на-
направлении исходных веществ.
Особенностью реакции F.19) является то, что она идет
слева направо с увеличением числа газообразных молекул.
Именно поэтому возрастание общего давления в системе
приводит к смещению равновесия в направлении умень-
уменьшения числа газовых частиц.
Если реакция идет без изменения числа газовых мо-
молекул, то степень ее прохождения не зависит от общего
68
давления, например:
(HC1) = V2(H,) + V2(C12), Kv=Pk"& • F.24)
Здесь, как и в реакции F.19),
Рн ~ЬРа
Роб1я = Рнс1+Рн, + рс1, и а = —J— ». F.25)
гобщ
Решая совместно три уравнения F.24) и F.25), находим
2ЛГ + 1 '
т. е. в выражении для степени прохождения реакции F.24)
отсутствует характеристика общего давления.
Если реакция идет не в газовой фазе, а в растворе,
то вместо характеристики исходного давления в системе
следует использовать исходную концентрацию вещества Со.
Например, равновесие электролитической диссоциации
уксусной кислоты в водном растворе
{СН3СООН} + aq = {СН.СОО-},, + {H+}aq
Ссн,соон = A—°с)С0> Ссн3соо-=аС0, Сн+ = осС0F.26)
характеризуется следующим выражением для константы
равновесия:
с ссн3соон СоA—а) 1 —а'
Поскольку уксусная кислота является слабым электро-
электролитом и процесс F.26) идет неглубоко, можно прибли-
приближенно считать, что A—а) —>- 1. В этом случае Кс = Соа2,
откуда а = УКС1'Со, т. е. процесс, идущий с возрастанием
числа частиц в растворе, с увеличением общей концент-
концентрации растворенного вещества будет смещаться в сторону
образования исходного вещества.
Необратимые процессы. Разновидностью химических
реакций являются необратимые процессы, которые будучи
проведены в прямом направлении не могут быть смещены
в обратном только за счет изменения температуры и дав-
давления. В качестве примера рассмотрим широко известную
необратимую реакцию горения угля в смеси с бертоле-
бертолетовой солью:
V, [КС1О,] + [С] — 2/3 [КС1] + (СО2)
Для этого процесса, согласно данным таблиц,
ДЯ° = —422,6 кДж, AS0 =167,8э. е. Уравнение энергии
69
Гиббса для этой реакции AGJ. = —422 600 — Т-167,8, в ко-
котором одинаковые знаки слагаемых показывают, что при
всех температурах этот процесс остается экзоэргичным
п константа его равновесия Кр = рсо2 даже при 0°С равна
огромной, нереальной величине /Ср=1090. Из приведен-
приведенного примера следует, что необратимые процессы описы-
описывают характеристиками AH°2SS и AS°2Sa разного знака, т. е.
необратимыми будут все экзотермические реакции, идущие
с увеличением энтропии. Примерами необратимых реакций
могут служить горение порохов, взрыв бризантных ве-
веществ и детонаторов, разложение перекисных соединений
и т. д.
С необратимыми реакциями не следует смешивать про-
процессы обратимые, но при данных условиях практически
до конца смещенные в одну из сторон. Например, при
комнатной температуре невозможно заметить даже следов
атомарного кислорода, образующегося за счет реакции
(О2) = 2(О), AG°r = 493 700 —Г-116,7
Однако при высоких температурах порядка 3000 К и выше
это равновесие смещается в направлении образования
атомарного кислорода и процесс этот обратим (/С, = 1
при 4230 К).
Одной из важнейших особенностей химического равно-
равновесия в сложных системах, где одновременно устанавли-
устанавливается не одно, а несколько равновесий, является вза-
взаимная зависимость этих равновесий, их аддитивность
(простое сложение). Например, мало растворимая соль
Ag4Fe(CN)e в незначительной степени растворяется в воде,
диссоциируя на ионы:
}aq F.27)
Константа этого равновесия при 25 °С равна
а энергия Гиббса, вычисленная с использованием этого
значения, составит AG[ = — 19,14-298 lg 1,5-10"" =
= 233 кДж.
Но комплексный анион Fe(CN)g~ при 25 °С подвер-
подвергается дальнейшей электролитической диссоциации:
{Fe(CN)i-}aq = {Fe» + }.q + 6{CN-}.q F.29)
* Процессы могут быть экзо- или эндоэргичными, если они
характеризуются отрицательными или положительными знаками ACj-
соответственно.
70
Константа равновесия F.29) равна:
... с-+с--_ = 5.10_37; фщ
а энергия Гиббса — AG°2 = — 19,14• 298 lg 5-10~3' =
= 207,1 кДж.
Равновесия F.27) и F.29) связаны между собой общей
для них концентрацией Fe(CN)s~ в растворе и в то же
время независимы с точки зрения величин констант рав-
равновесия и значений всех термодинамических характерис-
характеристик. Как независимые равновесия единой системы их
можно суммировать:
.aq F.31)
Константа равновесия F.31) равна произведению констант
равновесий F.28) и F.30):
а энергия Гиббса AG3 равна сумме энергий Гиббса: AG\ —
= AG\ -I- AGl = 440,2 кДж.
Глава 7
Правило фаз Гиббса
Фазы, независимые компоненты и степени свободы
системы.
Формулировка правила фаз. Одиокомпонентные
системы.
Диаграмма состояния воды. Двухкомпонентиые
системы.
Общая характеристика растворов.
Независимые компоненты и степени свободы системы.
Представления о гомогенных и гетерогенных системах и
фазах, изложенных в предыдущей главе, следует допол-
дополнить понятиями о компонентах и степенях свободы. Ком-
Компонентами называют индивидуальные вещества системы,
концентрации которых определяют состав всех ее фаз,
т. е. веществ в системе может быть больше, чем требуется
для описания состава всех ее фаз. Например, при уста-
установлении равновесия
[СаСО3] = [СаО] + (СО2)
71
система состоит из трех веществ, но для описания состава
всех трех фаз достаточно знать любые два, их называют
независимыми компонентами; состав третьей фазы легко
определить, используя уравнение вещественного баланса.
Из приведенного примера следует: число независимых ком-
компонентов равновесной системы равно общему числу инди-
индивидуальных веществ минус число связывающих их уравнений.
При наличии химических реакций число независимых
компонентов системы меньше числа индивидуальных
веществ. Если же между веществами нет химического
взаимодействия, то числа независимых компонентов и
индивидуальных веществ совпадают (например, воздух
состоит из такого же числа независимых компонентов,
сколько сортов молекул входит в его состав).
Степенями свободы (С) равновесной системы называют
те параметры, которые можно произвольно изменять,
не меняя числа и природы фаз системы. Степенями сво-
свободы могут являться концентрации реагирующих веществ
в фазах переменного состава (растворах), давление, тем-
температура.
Число степеней свободы определяют разностью между
общим числом независимых переменных системы и числом
уравнений, их связывающих. В приведенном выше примере
общее число переменных равно двум (температура и дав-
давление СО2 или его концентрация), уравнение одно, сле-
следовательно, число степеней свободы С данной системы
равно: 2— 1 = 1.
Количественное соотношение между числами степени
свободы (С), фаз (Ф) и независимых компонентов (К),
получившее название правила фаз, выведено В. Гиббсом
A876), оно имеет вид
G.1)
Здесь 2—число переменных параметров системы (давление
и температура). Использование этого правила помогает
при изучении химических реакций и равновесных систем.
Рассмотрим некоторые конкретные примеры.
Пример 1. Система представлена равновесием
[СаСО3] = [
Число независимых компонентов К равно разности числа индиви-
индивидуальных веществ и числа уравнений, их связывающих, следовательно,
К = 3—1=2. В системе три фазы (две твердые и одна газообразная).
Используя правило фаз G.1), вычисляем число степеней свободы:
C = /f-f-2 —Ф = 2 + 2 —3= 1. Следовательно, в этой системе без изме-
изменения числа и природы фаз произвольно можно изменять только один
72
параметр! Если произвольно задать температуру, то этим уже будет
использована единственная степень свободы и все другие параметры
примут строго определенное, независимое от нашего желания значение.
Связь Кр с температурой и природой реагирующих веществ выра-
выражается уравнением 95,87" — RT 1прсо =Д#298 — T^AS^e. Единствен-
Единственными переменными величинами в этом уравнении будут рсо и Т.
Задав произвольно одну из них в качестве аргумента, получим строго
определенное значение другой. Это и означает, что данная система
имеет только одну степень свободы.
Пример 2. Пусть система представлена равновесием
(N2)+3(H2) = 2(NH3), КУ=
Число независимых компонентов в ней равно /С=3— I =2, т. в,
система двухкомпонентна. Все вещества системы газообразны, следо-
следовательно, фаза одна. Используя правило фаз G.1), вычисляем число
степеней свободы С = /С + 2—l=2-j-2—1=3. Это означает, что три
любых параметра этой системы могут быть заданы произвольно
(всего таких параметров четыре: температура и три концентрации нли
парциальных давления) в следующих комбинациях: а) Т, робщ, р^ ;
б) Т, Робщ, Рнг\ в) Т, Ро6щ, pNHs; г) pNi, рНг, pNHj; д) робщ, pNj>
Рн2> е) Робщ. PNt, Pnh3; ж) Р°6ш-> рН2> Рмн3- Эт0 легк0 понять, если
записать выражение температурной зависимости константы равновесия:
°,8-2
Следовательно, чтобы определить значение любой из Переменных,
нужно задать значения остальных трех.
Пример 3. Пусть система представлена ненасыщенным водным
раствором хлористого натрия NaCl. В растворе, помимо молекул
воды {Н2О}, имеются ионы {Cl~}aq и {Na + }aq. Единственное связы-
связывающее их уравнение —CNa+=СС]-.
Эта однофазная система представлена тремя индивидуальными
формами соединений и одним уравнением, отсюда число независимых
компонентов в этой однофазной системе равно /( = 3—1»е2.
Поскольку газовая фаза (пар над раствором) не входит в рассмат-
рассматриваемую систему (по условию), то в выражении правила фаз Гиббса
число параметров должно стать на единицу меньше, т. е. С-\-Ф =
= K-j-l; тогда С = 2 + 1 — 1=2. Следовательно, данная система имеет
две степени свободы —температуру и концентрацию раствора.
Однокомпонентные системы. Наиболее просты системы,
состоящие из одного компонента. Каждая однокомпонент-
ная система представлена единственным веществом, нахо-
находящимся в различных агрегатных состояниях. Так, напри-
например, вода может существовать в парообразном, жидком
и кристаллическом состояниях, каждое из которых устой-
устойчиво в определенных интервалах температуры и давления.
73
Переменными параметрами . однокомпонентных систем
могут служить только давление и температура, поскольку
состав фаз по условию постоянен. Поэтому состояние одно-
компонентных систем может быть представлено в виде
зависимости давления от температуры:
дс°
р-е
Графическое изображение такой зависимости получило
название диаграммы состояния или фазовой диаграммы
состояния, поскольку состав фаз
в однокомпонентной системе оди-
одинаков и при изменении условий
не меняется.
Давление пара в процессах
парообразования устанавливается
и над твердыми, и над жидкими
веществами:
Рп:
+ 95,8
Рсубл
Рис. 7.1, Зависимость
давления насыщенного
пара от температуры над
твердым (/) и жидким B)
веществами
или
откуда
Т —
убл — АЯисп] — Т [Д5с*бл
Характер этой функциональ-
функциональной зависимости приведен на гра-
графике рис. 7.1. Точка плавления
кристаллической фазы находится
совместным решением приведенных
выше уравнений; исходя из усло-
условия равенства жидкой и кристал-
кристаллической фаз должно выполнять-
выполняться условие Рсубл — Рисп- В резуль-
результате для точки плавления имеем
= О,
При переходе из кристаллического состояния в пар (субли-
(сублимация) степень разупорядоченности движения частиц
больше, чем при испарении (в жидкости беспорядочность
74
движения частиц больше, чем в кристаллах), поэтому
величина ASns положительна:
Поскольку температура может принимать только значения
Т > 0, величина АН°ПЛ всегда положительна:
] >0.
Точно так же обстоит дело с полиморфными превраще-
превращениями, если они есть.
Зависимость температуры плавления чистых веществ
от давления p = fT можно определить уравнением Клау-
зиуса — Клайперона (приближенная форма):
Рг—Pi Д#пл
Та-Тг T(Vm-VTB) ¦
В этом уравнении Уж и Утв являются объемами 1 моль
соединения в расплавленном и кристаллическом состоя-
состояниях соответственно. Если Уж > Утв, то правая часть урав-
уравнения положительна, если Уж < УГв —то отрицательна.
р2
г.
Рис. 7.2. Изменение наклона линии p = f (T)
при различных соотношениях удельных
объемов твердого вещества и его расплава:
а —если V^. > I' „, то -rz. > 0; б— если
Ж IB (] /
v < v < °
Ha рис. 7.2, а, б показано, что каждому из условий
соответствует определенный наклон линии p = f{T). Кру-
Крутой ход кривых в равновесии между двумя конденсиро-
конденсированными фазами объясняют незначительной разностью
У —У
Рассмотрим в качестве примера однокомпонентную
систему вода—лед — пар. С точки зрения правила фаз
75
Гиббса для такой системы С + Ф = /( + 2 = 3. Следова-
Следовательно, эта система максимально может быть трехфазной
(Ф = 3) при числе степеней свободы, равном нулю (С = 0).
Этому случаю соответствует равновесие вода—лед—пар
при строго определенных температуре B73 К) и давлении
E,33- Ю2 Па).
Равновесие двух фаз (испарение, сублимация) характе-
характеризуется одной степенью свободы (произвольны давление
или температура). Двухфазные превращения описываются
следующими уравнениями:
[Н2О] = (Н2О) — #Г1прНвО = 49 790 —Г-236,8
{Н2О} = (Н2О) — RT In pHjo = 43 930 — Т- 214,6
[Н„О] = {Н2О} — 0 = 6008 — Т ¦ 22,0
(Кж—Ктв) = —1,62 мл/моль (получено экспериментально).
При использовании этих данных на рис. 7.3 построен
график зависимости р =
=/(Т), получивший на-
название диаграммы": состоя-
состояния воды. Линии (а), ф) и
(с) отвечают равновесиям
двух фаз: твердое веще-
вещество— пар; твердое веще-
вещество— жидкость; жид-
жидкость — пар соответствен-
соответственно. Точка пересечения
трех кривых носит назва-
название тройной точки, она
отвечает равновесию всех
трех фаз, а участки диа-
диаграммы I, II и III отве-
отвечают однофазному равно-
равновесию с тремя степенями
свободы. Линия (Ь) имеет
отрицательный наклон,
соответствующий отри-
отрицательному значению
(Vm—Утв). Следовательно, при замерзании воды ее объем
увеличивается. Этот известный факт объясняется тем, что
кристаллическая структура льда содержит большое число
пустот. При плавлении льда такая структура в значи-
значительной степени разрушается, пустоты заполняются моле-
молекулами воды и ее объем уменьшается. Веществ с такой
аномалией известно немного; кроме воды можно назвать
76-
273,1576
Рис. 7.3. Диаграмма состояния
воды:
/—лед; // — вода; /// — пар
элементарные висмут и галлий; из соединений —оксид
бора. Все остальные вещества имеют положительный
наклон линии Ь на диаграмме состояния.
Двухкомпонентные системы. Перейдем к характерис-
характеристике более сложных, двухкомпонентных систем. С точки
зрения правила фаз число степеней свободы в системах
с К = 2 может максимально быть равным 3, если Ф= 1.
Это означает, что кроме таких переменных параметров
системы, как давление и температура, появляется тре-
третий— концентрация раствора. Примером однофазных си-
систем переменного состава, образованных двумя и более
независимыми компонентами, служат растворы. Растворы
образуются во всех агрегатных состояниях. Они могут
быхь газообразными, жидкими и твердыми.
Все газы в любых соотношениях между собой неогра-
неограниченно растворимы. Только при низких температурах
и очень высоких давлениях в некоторых газовых смесях
наблюдается расслоение. В жидком состоянии ограничение
растворимости наблюдают довольно часто. Однако с повы-
повышением температуры растворимость обычно возрастает,
так что многие жидкие смеси, расслоенные на две фазы,
с повышением температуры становятся однофазными.
Наиболее важные группы жидких растворов следующие:
1) водные, неводные и органические растворы;
2) солевые расплавы галогенидных, кислородных и
сульфидных соединений (при высоких температурах);
3) расплавы металлов (при высоких температурах).
В твердом состоянии ограничение в растворимости
также имеет место. Известны три типа твердых растворов:
замещения, внедрения и вычитания. Твердые растворы
замещения обычно образуются двумя или несколькими
различными элементами, так что в кристаллической струк-
структуре места атомов одного элемента неупорядоченно заме-
замещаются атомами второго элемента. В результате распре-
распределение каждого из элементов оказывается хаотическим,
а соотношение между количествами атомов того и другого
сорта — произвольным. Примерами твердых растворов заме-
замещения служат сплавы меди и никеля или смешанные
кристаллы хлористого и бромистого натрия. Твердые
растворы внедрения получают чаще всего при растворении
небольших по размеру атомов неметаллов в металлической
решетке, например раствор углерода в железе. В этом
случае атомы неметаллов (Н, В, С, О и др.) хаотически
и в произвольных соотношениях располагаются в проме-
промежутках между атомами металла.
77
Твердые растворы вычитания, или дефектные струк-
структуры, характеризуются наличием пустот, которые разме-
размещаются хаотически и создают беспорядок в расположении
мест, занятых атомами элементов. Примером таких струк-
структур служит сульфидный минерал пирротин, монотонно
изменяющий свой состав в пределах FeS^og.j,!,,.
Общая характеристика растворов. Процесс растворе-
растворения— сложный физико-химический акт, а не простое
распределение частиц одного вещества между частицами
другого, которое в какой-то степени применимо для описа-
описания разреженных газовых смесей. В жидких и твердых
растворах частицы растворителя и растворенного вещества
непосредственно взаимодействуют между собой и находятся
на таких коротких расстояниях, как и в химических соеди-
соединениях. Взаимодействие молекул растворителя с раство-
растворяемым веществом зависит от сил разнообразной природы,
за счет которых в растворе образуются устойчивые комп-
комплексные и полимерные соединения, способные сущест-
существовать вне раствора,—сольваты, а в случае водных
растворов — гидраты.
Одна из важнейших количественных характеристик растворов,
связанная через закон действующих масс с энергетикой процессов
растворения,— концентрация раствора. Поэтому целесообразно вспом-
вспомнить некоторые наиболее используемые способы выражения концент-
концентраций, кроме уже упомянутых полярности и молярной доли следует
знать массовую и объемную доли, моляльную концентрацию (моляль-
ность), титр.
Поскольку процессы растворения имеют сложную хими-
химическую природу, законы, определяющие растворение и
отражающие природу растворов, сложны. Для упрощения
введено понятие идеального раствора, простейшими при-
примерами которого могут служить разреженные газы.
В реальных газах соотношение между объемом, давлением
и температурой описывается уравнением Ван-дер-Ваальса
В этом уравнении слагаемые a/V2 и Ъ учитывают силовое
взаимодействие между частицами газа. В разреженных
газах при достаточно высокой температуре эти слагаемые
столь незначительны по величине, что ими можно пре-
пренебречь с большой степенью точности. В результате полу-
получаем уравнение Клапейрона pV = RT, относящееся к раз-
разреженным газам, у которых тепловой эффект взаимодейст-
взаимодействия между частицами равен нулю.
78
Составляя смеси таких газов, можно убедиться в том,
что для их взаимодействия справедливо условие Л#°98 = 0
и полная аддитивность парциальных объемов и давле-
давлений, т. е.
Газовые растворы, которые образуются из составляющих
их частей с нулевым тепловым эффектом, а объем их смеси
в точности равен сумме объемов частей, получили назва-
название идеальных.
Для жидких и твердых растворов также существует
понятие идеальности, в общем виде оно может быть сфор-
сформулировано следующим образом: идеальными называют
растворы, которые образуются из составных частей при
выполнении условий
Д#°293 = 0 и ДК = 0. G.2)
Примерами идеальных растворов могут служить газо-
газовые смеси изотопов, а также все разбавленные растворы.
И действительно, добавление к растворителю небольшого
количества растворяемого вещества мало изменяет свойства
растворителя и раствора, и условие идеальности G.2)
справедливо. Но свойства растворяемого вещества в таком
растворе далеки от идеальных.
Концентрированные растворы по свойствам не близки
к идеальным, однако для получения грубой качественной
картины их можно описывать с помощью модели идеаль-
идеальных растворов. Для строгого решения производственных
и научных задач требуется обязательное введение попра-
поправок на реальность.
Глава 8
Диаграммы растворимости
Ограниченная и неограниченная растворимость
веществ в жидком растворителе. Диаграммы раст-
растворимости (плавкости) двухкомпонентных систем.
Уравнение Шредера — Ле Шателье.
Раствор по сравнению с исходными веществами всегда
обладает большей энтропией, так как беспорядочность
расположения частиц в растворе всегда больше, чем
79
в чистых исходных веществах. Энтропию вещества в иде-
идеальном растворе можно вычислить из соотношения
Si = S°i — R In N;, (8.1)
где S( — энтропия 1 моль вещества i в растворе; S°{—эн-
S°{—энтропия 1 моль этого же вещества в чистом виде; N{ —
молярная доля вещества i в растворе. При определении
энтропии идеального газового раствора величины энтро-
энтропии отдельных компонентов суммируются (аддитивность
энтропии).
Например, при стандартных условиях смешали газообразные
1 моль азота и 3 моль водорода так, что давление исходных компо-
компонентов и полученного газового раствора равно 1,013-10? Па. Тре-
Требуется вычислить энтропию этого раствора.
Зная молярную долю исходных газов в смеси и используя зна-
значения S29? из таблиц, находим сначала энтропию обоих компонентов
в газовом растворе
SHj= 130-5—19,14 lg0,25=130,5+11,5 = 142 э. е.,
SN'=247,7—19,14 lg0,75 = 247,7 + 2,4 = 250,l э. е.,
а затем энтропию раствора заданного состава, которая равна
35Hz + SNa = 3-142,0 + 250,1 =676,1 э. е.
Следовательно, энтропия данного газового раствора на 36,9 э. е.
больше суммы энтропии исходных чистых веществ F39,2 э. е.).
Анализируя уравнение (8.1), находим, что выражение
R\r\N. характеризует возрастание энтропии 1 моль ве-
вещества в идеальном растворе по сравнению с его энтро-
энтропией в" чистом состоянии, т. е.
Умножив правую и левую части этого уравнения на
температуру газа, получаем
RTlnNi. (8.2)
Поскольку при образовании идеального раствора должно
выполняться условие А#° = 0, выражение (8.2) представ-
представляет собой энергию Гиббса этого процесса:
AG} = —TAS} = RT In Ni. (8.3)
По определению N{ ^ 1, значит, правая часть уравнения
(8.3) всегда является отрицательной величиной и, следо-
следовательно, процесс растворения экзоэргичен, причем экзо-
эргичность растет с повышением температуры. Так, в при-
приведенном выше примере работа растворения 1 моль Н2
и 3 моль N2 равна при 298° К AG:m=- — 298-18,7 =
80
= —5573 Дж, при 500 К AG;ot> = — 9350 Дж. Такой
экзоэргический эффект характеризует неограниченную
взаимную растворимость водорода и азота.
Экзотермическое и одновременно экзоэргическое взаи-
взаимодействие компонентов при растворении еще больше
способствует этому процессу и может приводить к обра-
образованию устойчивых форм химических соединений. В ка-
качестве примера можно привести систему
{SO3}+{H2O}={H2SO4}, ЛЯда=—87,36 кДж; ДСг98=-84,68кДж.
Ограниченная растворимость веществ. Напротив, эндо-
эндотермическое взаимодействие компонентов часто, хотя и
не всегда, приводит к ограничению растворимости. Так,
процесс растворения NH4NO3 эндотермичен (AH°2S8 ==
= 25,8 кДж) и в небольшой степени экзоэргичен (AG°m=
= —6,7 кДж), но характеризуется высокой растворимо-
растворимостью. Однако для ряда солей возрастающая эндотермич-
ность растворения сопровождается уменьшением раство-
растворимости: NaCl, NaHCO3, Pbl2.
Растворение хлористого натрия в воде
[NaCl] + aq = {NaCl}aq
слабо эндотермично (Д#298 = 3,76 кДж). Этому соответствует ограни-
ограниченная растворимость NaCl в воде (в 100 г Н2О при 25°С до насы-
насыщения растворяется 36 г, при 100°С — 39,4 г NaCl). Зависимость
растворимости от температуры может быть получена из уравнения
t±G°r = —RT lnCNaCI=3760 —Г.43,1.
Растворение гидрокарбоната натрия в воде
[NaHCO3] + aq = {NaHCO3}aq
более эндотермично (ДН298= 17,2 кДж), этому соответствовует еще
большее ограничение растворимости (при 25°С в 100 г Н2О раство-
растворяется 10,4 г, при 100°С — 24,3 г NaHCO3). Зависимость раствори-
растворимости от температуры
И, наконец, процесс растворения малорастворимого йодистого свинца
в воде
= {PbI2}aq
еще более эндотермичен (Д#298 = 64,9 кДж) чему соответствует его
малая растворимость (при 25°С в 100 г Н2О растворяется 0,076 г,
при 100°С —0,436 г РЫ2), ее температурная зависимость AG" =
ЯПС 64 900 — Г.63,2.
Пределом ограниченной растворимости является рав-
равновесие между кристаллическим веществом (одна фаза) и
его насыщением раствором (вторая фаза) при условии
81
постоянства давления и температуры. Рассмотрим равно-
равновесие растворимости газа в жидкости на примере системы
хлористый метил — вода. Это равновесие и его константа
могут быть записаны следующим образом:
(CH3Cl) + aq = {CH3Cl}aq
Kc = -
(в в°Де)
СН3С1
газе)
(8.4)
(8.5)
В табл. 8.1 приведены значения растворимости СН3С1
в воде, из которой следует, что при увеличении давле-
давления адекватно изменяются концентрации хлористого ме-
метила в воде, в связи с чем константа равновесия при
заданной температуре остается практически постоянной.
Таблица 8.1. Растворимость (СН3С1) в воде при 25°С в кмоль'м3
В газе
0,111
0,309
0,407
0,504
В воде
0,0287
0,0801
0,1059
0,1308
0,258
0,259
0,260
0,259
Константа равновесия (8.5) не зависит от давления,
строго сохраняется при каждой заданной температуре и
является математическим выражением закона Генри. От-
Отклонение от него наблюдается лишь при достаточно высо-
высоких значениях р и С, когда в системе существенно нару-
нарушаются условия идеальности.
Используя значение константы равновесия (8.4), можно
вычислить величину AGj98 равновесия при 25°С:
ДО°293 = —#298 1п-?2- = — 19,14-298 lg 0,259 = 7,7 кДж.
Полученная величина указывает на эндоэргический про-
процесс растворения хлористого метила в воде, что и опре-
определяет ограниченный характер такой растворимости.
Ограниченная взаимная растворимость жидкостей также
наблюдается в тех случаях, когда их смещение сопро-
сопровождается эндотермическим тепловым эффектом. При этом
вся смесь разделяется на два слоя, две взаимно насы-
насыщенные фазы. С повышением температуры взаимная раст-
растворимость обеих фаз возрастает, так что их составы посте-
82
пенно сближаются и при некоторой температуре делаются
одинаковыми, т. е. двухфазная система превращается
в однофазную. Согласно графику, представленному на
рис. 8.1, при температуре 7\
в равновесии находятся две
жидкие фазы состава N' и N".
В точке К, при критической
температуре растворения Ткр,
система из двухфазной превра- т\
щается в однофазную.
Растворимость твердых ве-
веществ в жидкостях в общем
случае описывается схемой
[A]+slv = {A}slv,
3.6)
А Л" Л'"
Рис. 8.1. Диаграмма ограни-
ограниченной взаимной раствори-
растворимости двух жидкостей:
А и В —компоненты, К — крити-
критическая точка растворимости
где slv — растворитель (сокра-
(сокращенное от solvent). Константа
равновесия этого процесса
имеет вид Kiv = NA, где NA —
молярная доля вещества А в растворе.
Температурная зависимость растворимости твердых тел
следует из основного уравнения термодинамики, записан-
записанного применительно к равновесию растворимости (8.6):
— RT In Л'а = АН\ — TAS°a, (8.7)
где ДЯд и AS°A—тепловой эффект и изменение энтропии
растворимости.
Диаграммы плавкости двухкомпонентных систем. Мо-
Молярные доли каждого из веществ А и В в растворе свя-
связаны соотношением jVA + yVB=l. Этот способ выражения
концентраций раствора удобен для графического изобра-
изображения взаимной ограниченной растворимости, представ-
представленной на рис. 8.2. Если некоторый отрезок прямой, при-
принятый за 1, считать характеристикой полного состава
раствора, то отдельные части отрезка будут графическим
выражением молярных долей компонентов в растворе.
Если в качестве функции использовать температуру
равновесной растворимости, а в качестве аргумента —
средний состав системы, то, исходя из уравнения (8.7),
получим соотношение
ГА= . АЯ°* . (8.8)
ASA-R\nNA
В этом уравнении ДЯА представляет собой сумму тепло-
теплоты плавления кристаллов А и теплоты растворения полу-
83
чившегося расплава в компоненте В, т. е.
аналогично этому
Если считать образующийся раствор (расплав) идеаль-
идеальным, то АЯ5]У{а)=0. Значение ASs]v {A) в случае идеаль-
идеального раствора не равно нулю, но при малых содержа-
0,6
-0,8-
Рис. 8.2. Построение диаграммы растворимости
(плавкости) в системе двух компонентов А и В,
образующих одну эвтектику (Э)
ниях компонента В в растворе стремится к нулю. При
выполнении этих условий можно принять, что АЯА —»¦
—* АЯпЛ [А] и ASA —>¦ ASnjl LA]. Исходя из этого, в урав-
уравнении (8.8) сделаем подстановку
ЛС° . А"пл (А)
пл [А]
и получим
т=д#°л
Зададим N,
мер, при N
84
i -I
пл[А]
(8.9)
значения, отличные от единицы. Так, напри-
напри= 0,8 уравнение (8.9) примет вид
пл[А]
, т.е.
при yVA = 0,6
, т. е.
При NA = 0 уравнение (8.9) обращается в нуль. Таким
образом, в интервале от NA=l до AfA = O температура
равновесной растворимости уменьшается от Тпл [А] до нуля
(см. рис. 8.2).
В той же системе, состоящей из компонентов А и В,
в области, обогащенной компонентом В, все приведенные
выше рассуждения должны повториться в отношении рав-
равновесной растворимости кристаллов компонента В:
Т-\Н° Г ПЛ [В]
' — АЛпл [В] И
пл[В]
Точно так же при NB=l
при Л/в = 0,8 Г3= ДЯ°пл в . "л [R] +0,44
U пл[В]
1
J
Тпл1
Г л//°
U пл
иприЛ/в = 0,6 Г4 = ДЯп ^
J L Упл[В]
т. е. Гпл [В] > Г, > Т4.
В результате на графике рис. 8.2 получают две на-
наклонные линии, пересекающиеся в точке Э, которая полу-
получила название точки эвтектики, наклонные линии назы-
называют линиями равновесной кристаллизации или ликвидуса
(от лат. «жидкий»). Отрезки АЭ и ВЭ на этом графике
не реализуются, так как не имеют физического смысла
и не отвечают условиям равновесия. В результате полу-
получается диаграмма растворимости или диаграмма плавкости
эвтектического типа, которая отдельно представлена на
рис. 8.3.
Любая точка, нанесенная на эту диаграмму, имеет
конкретный смысл с позиций функциональной зависимо-
зависимости состав—свойство. Такая точка на диаграмме полу-
получила название фигуративной, ей отвечает строго опре-
определенный состав однофазной системы или средний состав
двух- или трехфазной системы. Рассмотрим движение
фигуративной точки а на графике рис. 8.3. Точка а харак-
характеризует расплав состава AfA при температуре 7Y При
снижении температуры до Т2 фигуративная точка а пере-
перемещается на линию равновесной растворимости в поло-
положение точки а'. При этом из раствора выделяется нич-
85
тожно маленький кристаллик А, но так как он исчезающе
мал, оставшийся раствор не изменяет своего состава.
Таким образом, начиная с точки а' и ниже имеет место
равновесие двух фаз — раствора (расплава) и кристаллов
компонента А. На диаграмме точкам а", а'", а"" и а'""
не соответствует фаза состава NA. Эти точки характери-
характеризуют только средний состав двухфазной системы, состоя-
состоящей из кристаллов компонента А и расплава состава N'
(при Г,), N" (при Г4) и N9 (при Тэ).
При снижении температуры истинная фигуративная
точка может перемещаться только вдоль линии ликви-
ликвидуса, проходя последовательно точки b', b", b'". При
7*1
A
T2
T4
Ть
T,
-^
la" ^\
1 ! N
h la'" Г
v j
1 1
A"
1
J
1
1
1
I
I4
1
1
1
1
!
у
T
i
i
i
Na N'' N" N'" Nj
А В
Рис. 8.3. Анализ диаграммы плавкости с эвтектикой
этом в расплаве не только снижается температура G,
Г4, Ть), но и все в больших количествах выпадают кри-
кристаллы А, так что остающийся расплав все более обед-
обедняется компонентом А (составы N', N", N'"). В точке
эвтектики линия кристаллизации компонента А пересека-
пересекается с линией кристаллизации компонента В. Следова-
Следовательно, в точке эвтектики в равновесии находятся три
фазы: кристаллы А, В и расплав, а число степеней сво-
свободы, согласно правилу фаз Гиббса, равно: С=/С+1—Ф—
= 2+1-—3 = 0. Это означает, что пока в равновесии су-
существуют все три фазы, температура изменяться не может.
При дальнейшем снижении температуры ниже точки эв-
эвтектики наблюдается исчезновение фазы расплава. В этом
случае эвтектический расплав затвердевает целиком в виде
смеси мелких кристаллов А и В.
86
Рассмотрев подробно один из простейших типов диа-
диаграмм растворимости (плавкости), дадим краткий обзор
диаграмм с различным характером взаимодействия в двух-
компонентных системах. Известны системы, в которых
компоненты А и В образуют бинарные соединения со-
состава АтВ„, плавящиеся конгруэнтно. Представим себе,
Рис. 8.4. Построение диаграммы плавкости для случая
образования в кристаллической фазе двойного соеди-
соединения АтВ„ с конгруэнтной точкой плавления:
а, Ъ, с, d — двухфазные области «разрыва сплошности»;
/, //— двухкомпонентные системы с одним общим компонентом;
77/-совмещенная двухкомпонеитная система: К —температура
конгруэнтного плавления
что имеются две независимые бинарные системы с про-
простыми эвтектиками, образованные компонентами А—АтВ„
(рис. 8.4,1) и АОТВ„ — В (рис. 8.4,11). Обе системы можно
совместить по общему компоненту, как это показано на
рис. 8.4,111. В результате получается новая система А—В,
в которой компоненты образуют индивидуальное соедине-
соединение АтВ„ с температурой плавления Тк . Поскольку в этой
точке плавления К составы расплава и кристаллов сое-
соединения АтВ„ совпадают, она называется конгруэнтной.
На диаграмме рис. 8.4,в выше линии ликвидуса располо-
расположена гомогенная область расплава. Все остальные поля
гетерогенные. В областях а и Ъ в равновесии находятся
две фазы — кристаллы АтВ„ и расплав, в области с—кри-
87
сталлы А и АЯВ„, в области d—также две фазы — кри-
кристаллы АтВ„ и В.
На рис. 8.5 приведена несколько иная диаграмма
плавкости в бинарной системе, здесь так же, как и в пре-
предыдущем случае, компоненты образуют бинарное соеди-
соединение АгаВ„, которое при нагревании разлагается, не
достигая температуры конгруэнтного плавления.
В этом случае линия ликвидуса располагается выше Тпл
двойного соединения, т. е. выше предполагаемого макси-
максимума (см. рис. 8.5, пунктирная линия). При этом пере-
пересечения линий ликвидуса оказываются расположенными
Рис. 8.5. Диаграмма плав-
плавкости для сл\чая образо-
образования в кристаллической
фазе двойного соединения,
плавящегося инконгру-
энтно;
а, Ь, с — двухфазные области;
К — точка разложения
соединения АОТВП;
/ — перитектика; Э — эвтек-
эвтектика
А„,В„ В
по одну сторону от состава двойного соединения. Точка
пересечения е получила название перитектики. Как вид-
видно из графика, нагревание кристаллов АтВ„ при темпе-
температуре перитектики приводит к разложению этого соеди-
соединения на кристаллы В и расплав, обогащенный компо-
компонентом А. Некоторые области на этой диаграмме требуют
пояснения. Так, в области а в равновесии находятся рас-
расплав и кристаллы А^В„, в области Ь—кристаллы А и АгаВ„,
в области с—кристаллы АтВ„ и В.
Если компоненты двойной системы и химическое сое-
соединение, образуемое ими, ограниченно растворимы между
собой, то получается тип диаграммы, приведенный на
рис. 8.6. На этом рисунке заштрихованные участки яв-
являются гомогенными областями твердых растворов (огра-
(ограниченных). Область I—твердый раствор соединения АотВ„
и кристаллов А; II и III—твердый раствор компонентов
Аи В соответственно и кристаллов АтВ„; IV—твердый
раствор АОТВ„ и кристаллов В. В областях а и Ь равно-
равновесно сосуществуют жидкие и твердые растворы.
В случае, если неограниченная растворимость наблю-
наблюдается не только в жидком, но и в твердом состояниях,
вид диаграмм плавкости (растворимости) будет таким,
как показано на рис. 8.7. Если на этом графике прове-
провести горизонталь (условие постоянства температуры 7\),
Рис. 8.6. Диаграмма
плавкости
для случая ограниченной
растворимости
компонентов
в твердых фазах:
/ — /V —области твердых
растворов;
а, Ъ — двухфазные области
разрыва сплошности
то в области II интервал составов от N'B до N'% будет
соответствовать двум растворам: жидкому, состава Л^ и
твердому, состава N'B. В областях I и III система харак-
характеризуется наличием жидкого и твердого растворов.
В качестве конкретного примера диаграммы плавкости
двухкомпонентной системы можно привести диаграмму
системы Са—А], изображенную на рис. 8.8. Из диа-
диаграммы плавкости видно, что компоненты системы обра-
образуют два химических соединения определенного состава
А13Са и А]2Са, а также твердый раствор а на основе
алюминия. Соединение А12Са конгруэнтно плавится при
температуре A079°), соединение А13Са плавится инкон-
груэнтно (плавление с разложением) при температуре
около 700°С. В системе имеется две эвтектики и узкая
область твердого раствора а с небольшой растворимостью
А13Са в металлическом алюминии. Выводы, сделанные
на основе вида диаграммы плавкости, важны как в на-
научном, так и в практическом отношении.
Уравнение Шредера — Ле Шателье. Исходя изданных
по растворимости веществ при различных температурах,
можно вычислить теплоту плавления растворенного ве-
вещества. Для этого воспользуемся уравнением (8.7), кото-
которое решим относительно двух составов равновесного раст-
раствора. Для плавления чистого вещества А, т. е. для слу-
случая yvA=l, это уравнение принимает вид
89
Для идеального раствора при условии NА < 1 спра-
справедливо соотношение
1 1
Т
igN
А//ш1 [А]
19,Н
пл [А]
(8.10).
где АЯпл [А]—теплота плавления кристаллов А; Тпл [А] —
температура плавления кристаллов А; Т—температура
Рис. 8.7. Диаграмма
плавкости
с неограниченной
растворимостью
в жидкой (/)
и твердой (///) фазах,
// — область
сосуществования
двух фаз
N'
равновесия раствора состава NA с кристаллами А. Соот-
Соотношение (8.10) получило название уравнения Шредера —Ле
Шателье. Оно описывает равновесие растворимости
Рис. 8.8. Диаграмма
плавкости системы
алюминий — кальций:
/ —жидкость;
// — двухфазные области^
с расплавом; d—твердый
раствор;
остальные поля —
кристаллические,
двухфазные области
t,°c
1000
800
6U0
А
/ж+А12Са
^1ч/ж+А1зСа
L и
Г а+А13Са
Ч,
<
А
\ I
II \
А12Са + ж\ >/ П
\/ж+Са
AljCa+Ca
AI
Са
в двойных системах при условии, что в твердом состоя-
состоянии взаимодействие отсутствует, а в жидком наблюдается
взаимная растворимость веществ при различных темпера-
температурах, если известны АЯ„Л и температура плавления
90
растворителя, т. е. если можно построить линию ликви-
ликвидуса.
Следует отметить, что вопросы растворимости одних
веществ в других имеют большое практическое значение,
поэтому термический анализ занимает важное место в фи-
физической химии. Методы термического анализа в настоя-
настоящее время широко используют в химии, физике, минера-
минералогии, почвоведении, агрономии, геологии, металловеде-
металловедении. Термический анализ получил распространение при
изучении строительных материалов, керамики, стекла,
металлических сплавов, солевых систем, пищевых про-
продуктов, пластмасс, смазочных масел, топлива и т. д.
I п ш IV
Рис. 8.9. Построение диаграммы плав-
плавкости по кривым нагревания и охлаж-
охлаждения. Кривая охлаждения расплава
компонента А:
I — III — разрезы; а — Ъ — остановка темпера-
температуры (Гх) при кристаллизации компонента А
Время
Рис. 8.10. Кривая
охлаждения двухком-
понентпого расплава:
/— IV — разрезы
Основным приемом построения диаграмм плавкости
служит построение кривых нагревания и охлаждения.
Практически это осуществляется непрерывным измерении
температуры линейно охлаждаемой или нагреваемой реак-
реакционной смеси. На рис. 8.9 приведен вид кривых охлаж-
охлаждения двухкомпонентного расплава в системе с простой
эвтектикой. Для чистого вещества А (разрез 1) кривая
охлаждения имеет вид, приведенный на рис. 8.9. Уча-
Участок линии а'-—а соответствует охлаждению расплава.
При температуре 7\ в точке а начинается кристаллиза-
кристаллизация расплавленного чистого вещества А. Поскольку рав-
равновесие двух фаз—жидкой и кристаллической — в одно-
компонентной системе (вещество А чистое) с точки зре-
зрения правила фаз Гиббса характеризуется числом степеней
91
свободы, равным нулю, затвердевание всей массы рас-
расплава А должно происходить при постоянной темпера-
температуре 7\; горизонтальный отрезок аЪ соответствует времени
затвердевания расплава А при температуре 7\.
С точки Ъ и ниже затвердевшая масса начинает моно-
монотонно остывать. Этому соответствует участок Ь—Ъ' кривой.
Разрезу диаграммы по линии II соответствует кри-
кривая охлаждения, приведенная на рис. 8.10. Здесь
участок кривой II отвечает кристаллизации компо-
компонента А из расплава, точка а при 7\ соответствует началу
кристаллизации, весь участок аЪ характеризует движение
фигуративной точки вдоль линии ликвидуса и возрастаю-
возрастающее количество выпавших кристаллов А. Точка Ъ при Т2
соответствует эвтектике (см. рис. 8.2), где начинается
кристаллизация эвтектического расплава по всему объему
(участок III). С точки зрения правила фаз число степе-
степеней свободы системы на участке III, т. е. в эвтекти-
эвтектической точке, равно С = /С+1—Ф = 2+1—3 = 0, что
соответствует равновесию трех фаз (расплав, кристаллы
А и В). Отсюда постоянство температуры на участке be.
После затвердевания эвтектики (участок IV) смесь кри-
кристаллов начинает монотонно охлаждаться (С = 2+1 —
-2=1).
Разрезу диаграммы по составу эвтектики III (рис. 8.9)
соответствует кривая охлаждения чистого вещества, так
как эвтектический расплав затвердевает при строго опре-
определенной температуре.
В современной экспериментальной химии метод изуче-
изучения и построения диаграмм плавкости выступает как
частный случай физико-химического анализа, который
позволяет определить совокупность свойств, являющихся
функцией состава системы. К числу таких свойств, кроме
температур кристаллизации (растворимости), следует от-
отнести кристаллическую структуру фаз (отсюда рентгено-
структурный анализ), электрическую проводимость (кон-
дуктометрический анализ), потерю массы за счет разло-
разложения с образованием легколетучих компонентов (грави-
(гравиметрический анализ), давление насыщенного пара (тензи-
метрический анализ) и т. д.
Идея физико-химического анализа была высказана еще М. В. Ло-
Ломоносовым в 1752 г. в его работе «Введение в истинную физическую
химию». Однако эта работа была опубликована только в 1904 г. и
физико-химический анализ был предложен независимо от нее во вто-
второй половине XIX в. в результате работ Д. К. Чернова, Д. В. Гиббса,
Д. И. Менделеева, Д. П. Коновалова, определивших интенсивное раз-
развитие этого направления в связи с потребностями бурно растущей
92
техники. Дальнейший подъем и огромная его популярность связаны
с именами А. Ле Шателье, Г\ Розебома, Г. Таммана и особенно
с именем Н. С. Курнакова, создавшего теоретическую и эксперимен-
экспериментальную базу физико-химического анализа и целую научную школу,
представители которой успешно продолжают и развивают это важное
направление.
Глава 9
Законы Рауля
Криоскопический закон. Дифференциально-терми-
Дифференциально-термический анализ. Эбуллиоскопический н тоноскопи-
ческий законы. Ректификация.
Криоскопический закон. Из уравнения Шредера —Ле
Шателье (8.10) и вида диаграммы плавкости с одной эвтек-
эвтектикой (см. рис. 8.3) следует, что между концентрацией
раствора AfA и понижением температуры его затвердева-
затвердевания (Тпл [д] — Т) существует количественная взаимосвязь
где А/А— молярная доля растворителя в растворе; AT—
разность температуры замерзания растворителя и раствора;
ДЯпл [А]—теплота плавления растворителя; ГПЛ[А]—тем-
ГПЛ[А]—температура плавления растворителя. Формула (9.1) строга
лишь в области очень разбавленного раствора компонен-
компонента В, так как только такой раствор ведет себя как иде-
идеальный. Для него справедливо условие XfB = (l—NA)<^1
или А/А = A—А/в)—>-1. В этом случае величина AT очень
мала и значения Т и Тпл [А] близки между собой. Это
означает, что в знаменателе дроби уравнения (9.1) про-
произведение температуры может быть заменено квадратом
температуры плавления компонента А ГГПЛ[А] « АГпЛ[А]»
и уравнение (9.1) можно записать следующим образом:
(в) fr>
к ' пл [А]
В уравнении (9.2) по условию величина AfB<^l. в таком
случае левая часть этого уравнения может быть разло-
разложена в ряд Маклорена, известного из курса математики,
и с достаточной степенью точности определяется первым
членом этого ряда. Так, функцию 1пA—х) можно пред-
93
ставить в виде ряда 1пA — х) = х—у + -о—-Т-+*--'
область применимости которого определяется условием
— 1 < х^ 1. Следовательно,
ln(l—-tfB) « —#B> (9.3)
Отсюда, подставив выражение (9.3) в (9.2), получаем
N _А//пл[А] AT
1 пл [А] .
ИЛИ
(9.4)
1-Л#пл[А]-1
Поскольку выражение в скобках есть величина постоян-
постоянная для каждого растворителя, ее называют криоскопи-
й й Е
рр
ческой постоянной Е'кр, т. е.
(9.5)
Уравнение (9.5) является математическим выражением
криоскопического закона Рауля, который формулируется
следующим образом: для разбавленных растворов пониже-
понижение температуры замерзания по сравнению с чистым раст-
растворителем прямо пропорционально молярной доле раство-
растворенного вещества. На этом законе основан криоскопиче-
ский метод определения молекулярной массы веществ
в растворах.
Произведем некоторые преобразования в выражении
(9.4). По определению
где п — число молей соответствующего компонента в рас-
растворе. Поскольку, по условию, раствор разбавленный,
справедливо соотношение пв<$с.Па, тогда
(9.6).
где Шъ—масса растворенного вещества; Мв — его молеку-
молекулярная масса. Используя выражения (9.5 и 9.6), получаем
94
т. е. молекулярная масса вещества в разбавленном рас-
растворе обратно пропорциональна величине понижения тем-
температуры замерзания раствора AT. Таким образом, зная
для каждого растворителя величину ?кр, задав тв, экспе-
экспериментально определив величину ЛТ в приборе (криоско-
пе), находят М растворенного вещества.
Как уже отмечалось (см. гл. 8), помимо термического
анализа, в котором превращения в системах различной
сложности связывают только с температурой, существуют
другие варианты физико-химического анализа, в частно-
частности тензиметрический анализ. В нем в качестве основных
параметров, зависящих от состава системы, выступают
либо давление насыщенного пара (при Т = const), либо
температура кипения раствора (при р = const). Напомним,
что температурой кипения называют такую температуру,
при которой давление насыщенного пара равно внешнему
давлению. Если внешнее давление равно 1013 гПа, то
температуру кипения (ТК1Ш) называют нормальной точкой
кипения (Тнтк).
Эбуллиоскопический закон. Пусть имеется разбавлен-
разбавленный (идеальный) ненасыщенный раствор труднолетучего
вещества В в жидком легколетучем растворителе А. Усло-
Условие разбавленности позволяет рассматривать такую систе-
систему, как идеальную. Насыщенный пар над этим раствором
состоит по условию только из частиц сорта А, поэтому
равновесие испарения может быть записано схемой
{А + В} = (А) или {А}в = (А). Константа этого равновесия
и основное уравнение термодинамики примут вид
^ T2AS°A. (9.7)
При бесконечном разведении NA—+l, т. е. для равнове-
равновесия испарения чистого растворителя А имеем
{А} = (А), К' = р\ (9.8)
— RT1\np°A = AH0 — T1AS°.
Условие рА~р°А (рА—давление над раствором; р°А—дав-
р°А—давление над чистым растворителем) достигается, если равно-
равновесия (9.7) и (9.8) устанавливаются при двух разных тем-
температурах, причем Т2 > 7\. В этом случае уравнения (9.7)
95
и (9.8) можно переписать в следующем виде:
. , ., АН°А
1ПР1пУУ
1ПРа_1пУУа = ___ + __, 1прА = ___ + __
Вычитая из второго уравнения первое, получаем
рА ' ЛА ~ ? ^ Т2 Tl
а при условии р°А=рА
Это и есть уравнение, связывающее температуры кипения
раствора (Т2) и растворителя G\) с составом этого рас-
раствора. Анализируя уравнение (9.9), следует помнить, что
по условию задачи был взят идеальный разбавленный
раствор, т. е. молярная доля растворенного вещества
Л^в-^1. Отсюда, используя соотношения (9.3) и (9.9), на-
находим
_
или, поменяв знак на обратный,
N*=AIRA ПТг
Поскольку раствор разбавленный, температура его кипе-
кипения должна быть близка к температуре кипения чистого
растворителя, т. е. ТгТ2^Т1, откуда
'в = Еэ6~, (9.10)
где Ея6—эбуллиоскопическая постоянная. Это означает,
что повышение температуры кипения AT раствора по
сравнению с чистым растворителем прямо пропорционально
молярной доле растворенного вещества NB — так форму-
формулируется эбуллиоскопический закон Рауля и таково его
математическое выражение.
Уравнение (9.10), подобно уравнению (9.4), исполь-
используется для определения молекулярной массы растворенных
веществ. Определив экспериментальное значение AT для
исследуемого раствора и найдя по таблицам или рассчи-
рассчитав величину Ея6, можно вычислить молекулярную массу
растворенного вещества. Метод определения молекулярной
96
d d'
массы вещества в растворе, основанный на повышении
температуры кипения, называют эбуллиоскопией, а прибор
для подобного исследования—эбуллиоскопом.
При сравнении криоскопического (9.4) и эбуллиоско-
эбуллиоскопического (9.10) законов Рауля сходство между ними ста-
становится очевидным, хотя ДГ
в этих двух уравнениях имеют
разные знаки. Это сходство
становится более понятным при
анализе графической зависи-
зависимости p = f(T) для раствори-
растворителя и раствора, изображенной
на рис. 9.1. Из рис. 9.1 видно,
что условию понижения давле-
давления пара раствора по сравнению
с чистым растворителем полно-
полностью соответствуют выводы о
повышении температуры кипе-
кипения и понижении температуры
затвердевания раствора по срав-
сравнению с чистым растворителем.
Тоноскопический закон.
Итак, весь приведенный вы-
выше материал показывает, что
между понижением температу-
температуры плавления (при повышении
температуры кипения) раство-
раствора и концентрацией трудноле-
труднолетучего компонента имеется про-
простая линейная зависимость, которая получила название
криоскопического и эбуллиоскопического законов Рауля.
В качестве следствия отмечалось, что при равной темпе-
температуре давление пара над таким раствором должно быть
меньше давления пара над чистым растворителем. Это
изотермическое понижение давления насыщенного пара
над раствором может быть учтено количественно.
Для этой цели рассмотрим идеальную двухкомпонент-
ную систему с неограниченной растворимостью как в жид-
жидкой, так и газовой фазах. Равновесие испарения в такой
системе может быть записано в виде двух независимых
уравнений и соответствующих им констант:
(9.1))
97
Рис. 9.1.
р = /(Г) над
лем А (/) и
А + В B):
Зависимость
растворите-
раствором
ных кристаллов раствори-
теля: с, а — чистый раствори-
тель N в жидком состоянии;
ь, <с_раствор жидкий
705
Рассмотрим равновесие при условии Т = const и пере-
переменном составе раствора. Это означает, что в каких бы
соотношениях ни смешивались компоненты А и В, вели-
величины констант равновесия К' и К" должны оставаться
неизменными. Составим два независимых уравнения для
растворов двух разных составов, но при одной и той же
температуре. Первый раствор пусть характеризуется со-
составом Л/д=1 (т. е. представляет собой чистый раство-
растворитель А) и константой равновесия
К! = р'а, (9.12)
где р°А—давление насыщенного пара чистого раствори-
растворителя. Второй раствор характеризуется составом NA < 1
и константой равновесия
Поскольку выражения (9.12) и (9.13) относятся к одной
температуре, то К' = К[, следовательно, р\ = -?— и
Точно так же для парциального давления компонента В
в этой системе можно вывести соотношение
РВ = Р'ЪМВ. (9.15)
Уравнения (9.14) и (9.15) являются математическим
выражением тоноскопического закона Рауля, который
можно сформулировать так: давление насыщенного пара
каждого из компонентов идеального раствора прямо про-
пропорционально молярной доле этого компонента в растворе.
Вычислим величину понижения парциального давления
пара каждого компонента в зависимости от содержания
в растворе второго компонента. Для этого произведем
следующие преобразования:
Используя условие NA + NB= 1, находим, что для ком-
компонента А в растворе ApA = p\Na, а для компонента
В—Дрв=/7вЛ^А- Следовательно, понижение парциального
давления насыщенного пара каждого из компонентов иде-
идеального раствора пропорционально молярной доле второго
98
компонента—такова другая форма записи тоноскоииче-
ского закона Рауля.
Поскольку тоноскопический закон Рауля формули-
формулируется для условия постоянства температуры, для- его гра-
графического изображения следует р
выбрать диаграмму зависимости
р =¦/(#), как показано на рис. 9.2. р°
Здесь прямые наклонные линии,
соединяющие точки р\ и р°ъ с
началами координат, графически
отражают линейные зависимости
(9.14) и (9.15).
Для идеальных газов и их
смесей справедлив закон Даль-
Дальтона: общее давление идеальной
газовой смеси равно сумме пар-
парциальных давлений компонентов,
образующих смесь. Математически
этот закон выражается следую-
следующим образом: ро6щ = р1 + р2 +
4-р3+ . .. =2р,-> гДе 2!~сумми-
2!~суммирование. /
Таким образом, в рассматри- /,
ваемой двухкомпонентной системе
А — В общее давление насыщенного
раствором выражается суммой
А В
Рис. 9.2. Зависимость
парциальных и общего
давлений от состава раст-
раствора при постоянной тем-
температуре (тоноскопиче-
(тоноскопический закон Рауля)
> II-P°BNB:
пара над жидким
Робщ =
(9.16)
которая также является линейной функцией состава рас-
раствора. Следовательно, на рис. 9.2 общее давление также
изображается прямой линией, соединяющей точки р°А и р°3.
Однако конденсированные (жидкие, твердые) системы
в отличие от газовых растворов редко ведут себя как
идеальные. Чаще всего, из-за взаимодействия близко рас-
расположенных частиц в них условия идеальности не реали-
реализуются, т. е, для них справедливо условие образования
реальных растворов AH°2BS^0 и ДУФ0.
В случае, когда компоненты раствора А и В при сме-
смешении не образуют химических соединений, способных
переходить в пар, наблюдается отрицательное отклонение
от закона Рауля, т. е. фактическое давление пара в си-
системе ниже требуемого по условию уравнения (9.16).
Положим, что в растворе компоненты А и В экзотер-
экзотермически взаимодействуют, образуя химическое соединение
4* 99
АИВ„ (тип выбираются так, чтобы т + л=»1)г
{В} = {АтВп}, ДЯ°<0.
В этом случае формальная или аналитическая молярная
доля компонента А (иногда ее называют брутто-молярной
долей NA) будет связана с истинными значениями N; каж-
каждого из индивидуальных веществ в растворе следующим
образом:
n. (9.17)
Для компонента В аналогичный расчет приводит к ре-
результату
Если сделать подстановку уравнений (9.17) и (9.18)
в (9.16), то получим выражение
Робщ = p\Nа + p°BNB = p\NA + p°bNb + [mp\ + пр°в] МАтВп.
которое будет оставаться линейной функцией состава
только при условии, если давление пара соединения АИВ„
окажется равным коэффициенту перед NAmBn, т. е.
при условии, что максимальное значение МАтв„ = —г—.
На самом же деле давление может быть каким угодно и,
в частности, близким к нулю. Тогда справедливым будет
неравенство
и выражение для фактического общего давления в системе
станет меньше требуемого по условию идеальности. В этом
случае наблюдается отрицательное отклонение от закона
Рауля, изображенное графически на рис. 9.3, а, б, в.
На участке т графика (а) реальная кривая сливается
с идеальной. Здесь концентрация компонента В очень
мала, поэтому раствор ведет себя как идеальный. Напро-
Напротив, в области, богатой компонентом В, отклонение от
условия идеальности наибольшее.
Это отклонение от условий идеальности может быть
столь существенным, что на кривой общего давления обра-
образует минимум (рис. 9.3, в). Точка минимума К называется
100
азеотропической и замечательна тем, что в ней жидкость
и пар имеют один и тот же состав. Такой раствор кипит
как индивидуальное химическое соединение.
о 6
р
А В А В
Рис. 9.3. Виды диаграмм p = f(N) с отрицательным
(а, б, в) и положительным (г, д, е) отклонениями от
закона Рауля
Отклонение от закона Рауля может быть положитель-
положительным (рис. 9.3, г, д, ё). Условиями такого отклонения слу-
служат образование летучего двойного соединения в двух-
101
компонентном растворе, т. е.
ип
i
1—
V
I ^
^^
-const
^-^-7
^¦"^ II /
1 III 1
Л""
Л"'
Л''
В
Рис. 9.4. Диаграмма Т = / (N) равновесия
жидкость — пар (р = const):
/ — область существования пара; // — область
равновесия двух фаз —жидкости и пара; /// —
область существования жидкости
Раств„ > '
эндотермический эффект образования раствора и ассоциа-
ассоциация компонентов раствора.
Если давления пара компонентов не сильно разли-
различаются между собой, то при положительном отклонении
от закона Рауля сум-
г'ки„ марное давление па-
пара может проходить
через максимум (на
рис. 9.3, е точка /).
т\ Эта точка также азе-
отропическая, такой
раствор кипит, не
изменяя своего со-
состава, как индивиду-
индивидуальное химическое со-
соединение. В некото-
некоторых случаях макси-
максимум давления пара
действительно связан
с образованием в га-
газовой фазе двойного
соединения.
Однако совершенно не обязательно, чтобы компоненты
А и В соединялись в молекулы АтВ„ или образовывали
полимеры типа Аст и Ви. Наиболее общим для растворов
является лишь стремление к таким видам взаимодействия
(некоторое избыточное притяжение или отталкивание между
частицами), которые вызывают эффекты отклонения систем
жидкость — пар от закона Рауля.
Ректификация. Кроме случаев образования азеотропов,
испарение из растворов протекает таким образом, что рав-
равновесный пар преимущественно обогащается наиболее лег-
легколетучим компонентом раствора. Следовательно, жидкая
фаза в процессе отгонки легколетучей фракции все более
обогащается труднолетучим компонентом. Процесс такого
перераспределения компонентов между жидкой и газовой
фазами и, как следствие этого, разделение исходного
раствора на части с избытком первого и второго компо-
компонентов называется ректификацией. Многократно повторен-
повторенный этот процесс позволяет практически полностью раз-
разделить компоненты, даже если они имеют близкие темпе-
температуры кипения.
102
Для понимания сути процесса ректификации рассмот-
рассмотрим рис. 9.4, на котором представлена диаграмма зави-
зависимости: TKlin = f(N). Как было показано выше (9.10),
повышение температуры кипения растворителя при добав-
добавлении к нему более труднолетучего вещества пропорцио-
пропорционально молярной доле этого вещества в растворе. Та-
Такое направление изменения температуры кипения раст-
раствора в начальной стадии соответствует нижней кривой
Т'кип — я — Т'кип- Эта линия соответствует зависимости тем-
температуры кипения раствора Гкип от его состава N{. Так,
pmnn
Рис. 9.5. Ректификационная колонна и диаграмма
ректификации
точка а отвечает раствору состава N', кипящему при
температуре 7\. При этом пар обогащен более легколе-
легколетучим компонентом А (точка с, состав N'"). Таким обра-
образом, линия Т'кш—С—Т"К11П (верхняя кривая) характери-
характеризует состав пара, находящегося в равновесии с жидкой
фазой. Выше этой линии участок I — гомогенная область
существования пара, участок II между линиями—гетеро-
линиями—гетерогенная область, здесь в равновесии находятся две фазы —
жидкая и газообразная. Так, в фигуративной точке Ъ при
среднем составе N" и температуре 7\ система будет пред-
представлять собой две фазы—пар состава N'" и раствор
состава N'.
На рис. 9.5 схематически изображена ректификацион-
ректификационная колонна, в которой по мере движения пара вверх
103
температура конденсата снижается. Рядом с колонной
приведена диаграмма TKm = f(N), расположенная так,
чтобы температурная ось на ней совпадала по направлению
с соответствующим изменением температуры в колонне.
Положим, что в нижнем резервуаре колонны находится
смесь состава Nt (точка а). При температуре 7\ из смеси
отгоняется пар состава jV2(to4-
т< ка а'), который, поднимаясь
ипвверх по колонне, охлаждается
и конденсируется при Т2 (точ-
(точка Ь). При этом равновесный с
жидкостью пар будет иметь,
согласно диаграмме, состав /V,
(точка Ь'). Этот пар, поднима-
поднимаясь далее по колонне, конден-
конденсируется при температуре Т3
(точка с). Равновесный с этой
жидкостью пар имеет состав
jV4 (точка с'). Поднимаясь далее
по колонне, пар конденсиру-
конденсируется при температуре Тх (точ-
(точка /). Равновесный с этой жид-
жидкостью пар состава N ъ (точка
/') в процессе дальнейшего
движения попадает в холо-
холодильник, конденсируется и выво-
выводится из системы. Таким образом,
Рис. 9.6. Диаграмма Т—
= f (Ni) (при p = const) для
равновесия твердое — жид-
жидкое— пар в двухкомпонент-
ной системе:
/ — пар; // — расплав: ///-смесь
п^Ив-а^"Д+расп^Г+ПкрРи+с?аал"- в процессе ректификации жидко-
лы а или в сти состава N1 получается легко-
летучая фракция состава /V6,
очень близкая по составу к чистому компоненту В. Уве-
Увеличивая число таких ступеней ректификации, можно до-
добиться высокой степени разделения веществ, мало отли-
отличающихся по температурам кипения.
Внутреннее распределительное устройство ректифика-
ректификационной колонны получило название тарелок, а конденсат,
собирающийся в этих тарелках,— флегмах.
Диаграммы зависимости TKen = f(N), характеризующие
равновесие жидкость — пар, являются естественным про-
продолжением ранее рассмотренных диаграмм плавкости
^пл= / (Щ- Они могут быть совмещены в единую диаграмму
T = f(N), как это показано на рис. 9.6. Здесь каждый
из компонентов А и В при повышении температуры про-
проходит последовательно через плавление и кипение (при
р = const). Смеси компонентов сначала взаимно раство-
104
т
•'кип
Гпл'
I
па|
1
1
тв+жу
ТВ
f
' ж+тв
А+В
'ки
В А
В
Рис. 9.7. Влияние давления на вид диа-
диаграммы T = f (Ni), отражающей равновесие
твердое — жидкость — пар в двухкомпонент-
ной системе:
заштрихована
JJ-pa<p1;
область жидкого состояния; l —
Il-pa <pt; IV-pt <pa
ряются, переходя в расплав, а при более высоких тем-
температурах кипят.
Если изменять общее давление в системе, т. е. изобра-
изобразить графически несколько разрезов диаграммы T = f(N)
при ряде зафиксированных давлений, то получится картина,
изображенная на рис. 9.7. Четыре отдельные диаграммы
представляют собой разрезы /—IV одной и той же системы
при разных давлениях (ри рг, р3, р±). Разрез / характе-
характеризует полное разделение областей жидкого, твердого и
парообразного состояний. При снижении давления гра-
граница равновесия жидкость — пар резко снижается, тогда
как граница равновесия твердое состояние—жидкость от
давления почти не зависит. В результате получают раз-
разрез //, на котором диаграмма жидкость—пар начинает
перекрываться с диаграммой плавкости. В результате
область жидкого состояния уменьшается и разрывается
на два участка. При этом появляется область равновесия
твердое тело—пар, т. е. в данном интервале температур
кристаллы компонента В находятся в равновесии с газо-
газовым раствором (A-f В), (сублимация). При дальнейшем
снижении давления (разрез ///) область жидкого состоя-
состояния становится совсем небольшой и нигде не касается
координатных осей. Это означает, что оба компонента
системы при данном давлении с повышением температуры
сублимируют. При еще большем снижении давления вид
диаграммы упрощается (разрез /V). Область жидкого со-
состояния исчезает совершенно, остается лишь один вид
равновесия твердое вещество—пар.
Из сказанного следует, что особенности разделения
смесей компонентов существенно изменяются в зависимо-
зависимости от выбора условий. В частности, разделение жидких
растворов при обычном или слегка сниженном давлении
принципиально отличается от разделения компонентов
в условиях вакуума и более низких температур. Разно-
Разнообразие свойств различных смесей, с которыми прихо-
приходится иметь дело на практике, требует использования
различных технических средств и условий теоретической
разработки процессов дистилляции и ректификации. Успе-
Успехи, достигнутые в этой области, позволяют в настоящее
время производить тончайшую очистку различных про-
продуктов и разделять смеси очень сложного состава.
Глава 10
Растворы электролитов
Структура воды и водных растворов электролитов.
Условия применимости законов Рауля к растворам
электролитов. Ионное произведение воды. рН раст-
растворов. Сила электролитов. Закон разведения сла-
слабых электролитов. Кислоты и основания.
Структура воды и водных растворов электролитов. Как
уже отмечалось выше, растворение—сложный процесс,
при котором частицы растворяющегося вещества активно
взаимодействуют с молекулами растворителя. Энергия
этого взаимодействия обычно столь велика, что оказы-
оказывается достаточной для разрушения прочных кристалли-
кристаллических решеток различных твердых тел (например, раст-
растворение поваренной соли в воде). Одним из самых уни-
универсальных растворителей является вода. Она растворяет
многие твердые, жидкие, газообразные, органические и
неорганические вещества. В отличие от других растворов
водные растворы солей, кислот и оснований обладают
способностью хорошо проводить электрический ток. Ве-
Вещества, проводящие в растворе электрический ток, назы-
называют электролитами, причем электрическая проводимость
электролитов определяется не свободными электронами,
как в металлах, а гидратированными ионами — катионами
и анионами, которые образуются в результате электро-
электролитической диссоциации соединений в воде. Учение, объ-
объясняющее и количественно описывающее такие процессы,
получило название теории электролитической диссоциации.
Важнейшая особенность воды как растворителя заклю-
заключается в том, что ее молекулы полярны и имеют боль-
большой электрический момент диполя * ц—произведение ве-
величины зарядов на расстояние между ними. Три атома
(два водорода и один кислорода) в молекуле воды обра-
образуют равнобедренный треугольник (рис. 10.1, а). Атом
кислорода в этой молекуле имеет восемь электронов
(шесть своих и два от двух присоединившихся атомов
водорода) (рис. 10.1,6). При этом две электронные пары
образуют прочные химические связи с двумя атомами
водорода, которые за счет смещения этих электронных
* Момент диполя для воды равен 0,61 • 10~gs Кл-м; для СН4,
СО2> СС14 —нулю.
107
пар к атому кислорода приобретают результирующий
положительный заряд. Две другие электронные пары оп-
определяют избыточные отрицательные заряды на атоме
кислорода и при конденсации молекул водяного пара
используются для об-
образования слабых, мо-
молекулярных связей с
положительно заряжен-
заряженными атомами водорода
других молекул воды
(так называемые водо-
водородные связи). Способ-
Способность образовывать до-
достаточно прочные водо-
Рис. 10.1. Структура молекул воды: родные связи определяет
о —формальная схема; б —пространственное рЭСТВОрИМОСТЬ В ВОДв
распределение зарядов боЛЬШОГО ЧИСЛЭ ОрГЭНИ-
ческихсоединений(спир-
ческихсоединений(спиртов, кислот и т. д.). Этому способствует также амфотер-
ность воды, которая позволяет ей с кислотами взаимо-
взаимодействовать по механизму основания, а с основаниями —
по кислотному механизму:
= {Н3О+}-НС1-}
кислота
осно-
основание
При взаимодействии полярных молекул воды с поляр-
полярными молекулами или ионными кристаллами растворяемых
в ней веществ (солей, кислот, оснований) процесс раст-
растворения идет с частичной или полной диссоциацией этих
веществ на противоположно заряженные ионы. В качестве
примера можно рассмотреть растворение в воде газооб-
газообразного хлористого водорода. Молекула газообразного
НС1 полярна, чтобы разорвать её на ионы, нужно затра-
затратить огромную энергию:
| A0.1)
которая значительно больше энергии, требуемой для дис-
диссоциации на атомы (диссоциация становится заметной лишь
при температуре 1500°С):
(НС1) = (Н)+(С1), ДЯ;88 = 431кДж
Следовательно, при обычной температуре молекулы газо-
газообразного HQ недиссоциированы. При взаимодействии же
108
с водой энергично идет процесс электролитической дис-
диссоциации:
A0.2)
Работа этой реакции AG°r = —75157 + Г-131,46 при
температуре 25°С равна 36,0кДж, т.е. процесс настолько
экзоэргичен, что равновесие A0.2) оказывается смещен-
смещенным вправо и давление НС1, равное 1013 гПа, достигается
лишь при 41% концентрации раствора НС1. Вот почему
для соляной кислоты при атмосферном давлении и тем-
температуре 25 °С эта концентрация предельна. Сравнивая
процессы A0.1) и A0.2), видим принципиальное различие
между ними с точки зрения их осуществимости. Выгод-
Выгодность процесса A0.2) определяется экзоэргичностью обра-
образования гидратированных ионов водорода и хлора.
Следует отметить, что кроме воды известно огромное
число самых различных растворителей. И так же, как
при образовании водных растворов, центральную роль
играют процессы сольватации—взаимодействие молекул
растворителя с растворяемым объектом. Значение процес-
процессов гидратации при электролитической диссоциации в вод-
водных растворах отмечалось впервые в работах И. А. Каб-
лукова A891) и В. А. Кистяковского A888—1890), поло-
положивших начало развитию теории электролитов, один из
важнейших вопросов которой является изучение структуры
растворов и характера распределения в них ионов. Уста-
Установлено, что не только молекулы воды влияют на струк-
структуру раствора (поляризация, ионизация), но и раство-
растворяемое вещество в свою очередь влияет на структуру
воды (растворителя). Как заряженные частицы, ионы об-
обладают электрическим полем, напряжен юсть которого
достигает величин порядка 106В/см. Это поле определяет
сильное электростатическое взаимодействие между ионом
и полярными молекулами воды. Молекулы воды, нахо-
находящиеся в непосредственной близости к иону, могут свя-
связываться с ним силами химической связи, образуя хими-
химическое соединение. Непосредственно присоединенные к иону
молекулы воды строго ориентированы, их расположение
напоминает структуру кристалла. Следовательно, при
растворении электролита структура воды становится не-
неоднородной. Часть молекул воды, которая далека от иона,
остается в прежнем состоянии, это собственная структура
воды {Н2О}да, другая часть—псевдокристаллическая струк-
структура, характерная для «ионной зоны» {H2O}V. В переход-
переходном слое между этими зонами вода имеет промежуточную
109
структуру, гак называемая деструктурированная вода
{Н2О}О. Между этими структурами имеет место равновесие
которое при заданной температуре смещается в ту или
иную сторону при изменении природы и концентраций
растворенных веществ.
Описанное
слойное
^
Рис. 10.2. Изменение структуры воды
в электростатическом поле ионов
выше по-
изменение
структуры воды около
ионов в растворе графи-
графически представлено на
рис. 10.2.
Поскольку ионы,
лу\\''\^\^/'^^/^1 образующиеся в раст-
^—'^ / . . .1^1 V\_ BOpaX) обязательно по-
положительно и отрица-
отрицательно заряжены, рас-
распределение их в раст-
растворе небеспорядочно.
Чем выше концентра-
концентрация раствора и меньше
среднее расстояние меж-
между ионами, тем сильнее эффект их взаимного влияния и
больше упорядоченность их взаимного расположения.
В концентрированных растворах ионы противоположного
знака могут слипаться своими гидратными оболочками,
образуя ионные пары, тройники, квадруполи, уменьшаю-
уменьшающие число частиц в растворе и его истинную концентра-
концентрацию. При дальнейшем увеличении концентрации наступает
такое состояние, когда сначала исчезают зоны собствен-
собственной структуры воды {Н26}да и деструктурированной воды
{Н2О}Л, а затем начинают разрушаться псевдокристалли-
псевдокристаллические ионные зоны {Н2О}у. На этом этапе становятся
возможными выпадение в осадок кристаллов (NaCl из
насыщенного водного раствора); образование кристалло-
кристаллогидратов (Na2SO4-10H2O, MgCl2-6H2O, CuSO4-5HaO); сво-
свободных молекул, которые могут улетучиваться из раствора
(выделение газообразного НС1 из соляной кислоты); комп-
комплексных соединений и ионов (ионы Cd2+ в присутствии
избытка галоген-ионов образуют комплексные формы CdXj
и CdXf-) и т. д.
Условия применимости законов Рауля к растворам
электролитов. Характер распределения ионов в водном
растворе существенно отличается от распределения ией-
110
тральных молекул. Нейтральные молекулы могут обра-
образовывать раствор, не нарушая заметно тетраэдрического
каркаса льда. Такие частицы внедряются в «пустоты»
кристаллической структуры льда, а молекулы воды вокруг
«захваченной» частицы становятся более упорядоченными,
«замороженными». Образуется нечто вроде «айсбергов»,
которые в некоторых случаях при замораживании могут
быть даже выделены в свободном состоянии в качестве
индивидуальных кристаллических соединений — клатра-
тов. Так были выделены клатратные гидраты ксенона
4Хе-23Н2О, хлора С12-8Н2О и др. Напротив, ноны обра-
образуются за счет энергичного, разрушающего взаимодействия
веществ со структурой растворителя (см. рис. 10.2). Электро-
Электролитическая диссоциация изменяет и фактическую концен-
концентрацию вещества. Так, при растворении в воде хлористый
натрий полностью диссоциирует на ионы Na+ и С1~,
число частиц растворенного вещества при этом удваивается
и концентрация возрастает вдвое. Согласно законам Рауля
АТ'кяп. А7ПЛ и понижение давления пара растворителя
пропорциональны молярной доле растворенного вещества,
поэтому эффект электролитической диссоциации должен
вдвое увеличить эти характеристики. Если же электро-
электролитическая диссоциация веществ идет не до конца, а до
состояния равновесия, то фактическая концентрация будет
больше исходной. Следовательно, исходное количество
вещества Па.ъ , растворенного вещества АЛВ5 и фактиче-
фактическое п* для растворов электролитов можно выразить
соотношением
где i—коэффициент Вант-Гоффа, или изотонический коэф-
коэффициент.
Рассмотрим физический смысл этого коэффициента
на примере равновесного процесса электролитической
диссоциации:
Очевидно, что равновесное количество вещества п* и всех
трех форм в растворе будет следующим:
.в
к s
111
где а — равновесная молярная доля каждого из ионов
в растворе или степень электролитической диссоциации.
Полное число молей всех форм в растворе равно их сумме
(Ю.5)
или, сделав подстановку выражения A0.4) в A0.5), п*=»
= Яа в [1 + (k -j-s— 1)а]. Сумма k + s равна числу ионов Z,
на которые распадаются в растворе молекулы растворен-
растворенного вещества, следовательно,
(Ю.б)
Сравнивая выражения A0.6) и A0.3), находим г —
= 1 + (Z —l)a.
Таким образом, изотонический коэффициент i прямо
связан с числом ионов Z и степенью электролитической
диссоциации а. Если электролит сильный, диссоциация
идет до конца, то а= 1 и i = Z. (В сильных электролитах
аэфф ^ 1 за счет образования ионных пар. Это кажущаяся
или эффективная степень диссоциации, которую опреде-
определяют экспериментально.)
Примерами растворов, для которых Z = / = 2, служат
растворы NaCl, HNO3, NaOH. При растворении таких
веществ, как Na2SO4 и К3РО4, величина i может прибли-
приближаться к значениям 3 и 4.
Если правую и левую части уравнения A0.3) разде-
разделить на сумму количеств веществ, составляющих раствор,
то получится уравнение в молярных долях N* — Na.b i-
Отсюда математические выражения сформулированных
выше законов Рауля применительно к растворам электро-
электролитов будут иметь следующий вид (В—растворяемое трудно-
труднолетучее вещество, А—растворитель) для:
тоноскопического —ApA = pAyVB [I -\-(Z— 1) а],
криоскопического —Д^пл — Екр^в [1+B—1) ее],
эбуллиоскопического —Д7'кип = ?'эбЛ'в [1 -(-B—1) а],
Из приведенных уравнений следует, что такие важные
характеристики растворов электролитов, как Ар, АТПЛ,
ЛТК1Ш, зависят:
1. От величины степени электролитической диссоциа-
диссоциации а, численное значение которой хорошо известно только
в случае разбавленных растворов сильных электролитов
(диссоциация полная), для слабых электролитов значение
а<^1 устанавливают опытным или расчетным путем.
112
2. От величины Z — числа ионов, которые образует
данный сорт электролита. Эта величина также нуждается
в уточнении, так как многие электролиты имеют не одну,
а несколько ступеней диссоциации с разными значениями а.
Например, серная кислота H2SO4 по первой ступени
H2SO4 + Н2О = Н3О + + HSOI
диссоциирует практически полностью, а по второй
шог 4- н2о=н3о+ + sor
лишь частично, как слабый электролит (/Сп = 1,2-10~2).
3. От степени отклонения растворов от идеальности.
Как отмечалось, в растворах электролитов ионы проти-
противоположных знаков чередуются, соблюдая некоторую за-
закономерность расположения, при этом они могут слипаться,
захватывать в свои ионные сферы молекулы растворителя,
образовывать гидратные и комплексные формы соединений,
которые трудно учесть, и т. д.
Многие процессы электролитической диссоциации в
растворах обратимы и подчиняются законам химического
равновесия. Однако наличие у гидратированных ионов
заряда и связанное с этим существенное изменение струк-
структуры воды, а также упорядоченность распределения заря-
заряженных частиц не позволяют рассматривать растворы
сильных электролитов (кроме очень разбавленных) как
идеальные. Поскольку сильные электролиты диссоциируют
в водных растворах до конца, т. е. необратимо, эти про-
процессы не могут быть рассмотрены с позиций химического
равновесия. Напротив, электролитическая диссоциация
слабых электролитов является обратимым процессом, по-
поскольку концентрации ионов малы, подобные системы могут
рассматриваться как идеальные растворы с установив-
установившимся химическим равновесием.
Ионное произведение воды. Анализ экспериментальных
данных показал, что чистая вода сама по себе уже элект-
электролит, хотя и очень слабый. Отдельные ее молекулы за
счет взаимодействия с себе подобными подвергаются элект-
электролитической диссоциации, в результате чего образуются
ионы водорода и гидроксила. Ион водорода Н+ пред-
представляет собой элементарную частицу протон, размеры
которого в 105 раз меньше атома водорода (линейные
размеры протона 10~1? см). При таких ничтожных разме-
размерах и огромном относительном заряде (+1) энергия вза-
взаимодействия изолированного протона с полярными моле-
113
кулами воды велика:
) = (Н3О+), ДЯ288 = — 707 кДж
Положительно заряженный ион Н3О+ взаимодействует
с водой с большим экзотермическим эффектом и при этом
образуется, как правило, гидратированный ион гидро-
ксония *:
(Н3О+) + aq = {Н3О+}aq, Д tf°298 = -460 кДж
(Возможны и более сложные взаимодействия молекул воды
с ионом водорода, например Н6О^, а ион гидроксила более
предпочтителен в форме Н3О^.) Иногда в литературе для
простоты используют символ Н + , называя ионом водо-
водорода. Здесь нет ни ошибки, ни различия в точках зрения;
во всех случаях имеется в виду ион гидроксония.
В растворах (в отличие от газов) невозможно выде-
выделить и отдельно изучить ионы того или иного заряда,
поэтому определить энтропию и теплоту образования иона
гидроксония в растворе из простых веществ невозможно.
В связи с этим были приняты следующие условные харак-
характеристики процессов:
e-={H + }aq, )Д/&8 = 0
> A0.7)
) °
Точно так же из-за невозможности выделить и отдельно
изучить ион гидроксила {OH~}aq ничего не известно о
теплоте его образования в растворе из простых веществ
по схеме
^ A0.8)
Однако электрохимическое исследование равновесия
aq A0.9)
позволило установить для него следующий тепловой эф-
эффект:
ДЯдН_ + ДЯ^0+—2ДЯ„г0 = 55,90 кДж A0.10)
Подставив значения Д#н2о из таблиц, а также величину
А#н3о = 0 (Ю.7) в уравнение A0.10), для реакции A0.8)
получаем Д#он-=—229,94кДж. Это означает, что обра-
образование 1 моль гидроксила {OH~}aq на 229,94 кДж вы-
выгоднее образования 1 моль гидроксония {H3O+}aq (или
иона {H+}aq). Точно так же по экспериментальным дан-
данным для процесса A0.9) AS298 = —80,483 э. е., откуда
114
энтропия иона {OH~}aq равна —10,54 э. е. Поскольку эн-
энтропия ионов не абсолютна и ее отсчитывают от услов-
условного нуля A0.7), то искомые значения могут быть и от-
отрицательными. Значения энтропии нейтральных форм могут
быть только положительными.
В дальнейшем ионное равновесие электролитической
диссоциации воды будем записывать упрощенно:
{H2O} = {H+}aq+{OH-}aq A0.11)
Для характеристики равновесия A0.11) используют вы-
выражение константы равновесия Kw A0.12), которое назы-
называют ионным произведением воды:
Вычислим величину Kw при температуре 25 °С;
55 900 80,48
" . A0.12)
Очень малая величина константы свидетельствует о том,
что концентрация ионов Н+ и ОН" в воде ничтожна.
Следовательно, для водных растворов любых веществ
(будь то растворы кислот, оснований, солей) соотношение
A0.12) справедливо. Так, например, в 0,1 М растворе
сильной соляной кислоты концентрация ионов водорода
будет равна 0,1 кмоль/м3. При температуре 25 °С кон-
концентрация ионов гидроксила в этом растворе может быть
вычислена из соотношения
откуда Сон- = 10~14/0,l =¦ 10~13 кмоль/м3.
Из соотношения A0.12) следует, что с повышением
температуры величина Kw должна экспоненциально воз-
возрастать, например, Kw при 0°С равна 0,13-10~14; при
25°С—Ю-14; при 100°С—74,00-10~14.
При электролитической диссоциации чистой воды по
схеме A0.11) концентрации ионов Н+ и ОН" равны между
собой — Сн+ ==Сон- = V Kw< чт0 соответствует нейтральной
среде. Очевидно, что эти концентрации с увеличением
температуры также возрастают: при 0°С равны
0,36- 1(Н кмоль/м3; при 25 "С—1,00-10"'; при 100°С —
8,60- Ю-31 кмоль/м3.
При избытке ионов водорода, т. е. при условии Сн+ >
> Сои-, раствор характеризуется как кислый; при из-
избытке ионов гидроксила Сн+< Сон как щелочной.
115
Таким образом, при температуре 25°С раствор с соотно-
соотношением (в кмоль/м3)
является нейтральным, с соотношением Сн+ = Ю—кис-
Ю—кислым и Сон-=Ю~11—щелочным.
Для удобства используют соотношение A0.12), лога-
логарифмирование которого приводит к виду
или для температуры 25 °С
Для чистой воды при этих условиях будем иметь
—14 = (—7) + (—7), для децимолярного раствора сильной
одноосновной кислоты 14 = (— 1)-Ь(—13) и т. д. Введя
в формулу A0.13) обозначения — lg/(№=p/(; —lgCH + = pH;
— lgCoH-=pOH, имеем
или для температуры 25 °С
14 = рН+рОН.
С учетом этих обозначений раствор с рН < 7—кислый,
с рН 7—нейтральный, с рН > 7—щелочной.
Закон разведения слабых электролитов. При растворении
в воде слабых электролитов (С„Н6ОН, СН„СООН, NH4OH)
устанавливается равновесие между недиссоциированной
формой и получившимися ионами, например:
{С6Н6ОН}+aq = {C6H6O- }aq+{Н+}aq.
Равновесная концентрация каждой формы в растворе
будет: Cc,h8oh = QjH|iOHA—a), Cc.H6o- = QeH]>OHa, CH+ =
= Сс„нвона- Выражение для константы равновесия примет
вид
к сс.н.о-сн+ г" «2
Дс = = ьснон
с,нвон 177 •
с,н„он 1—a
что является математической формой выражения закона
разведения Оствальда A888). Если электролит достаточно
слаб, то а <^ 1, и тогда
Кс = С°с,ншон<* 00.14)
116
или
Г
V L
Из этих соотношений следует, что степень электролити-
электролитической диссоциации а зависит от разведения 1/С°, а кон-
константа диссоциации пропорциональна а2 и при данной
концентрации может служить мерой силы электролита.
Для слабых электролитов (СН3СООН, NH4OH) величина Кс
лежит в пределах 10~6—10""9; для электролитов средней
силы [Са(ОНJ, первая ступень Н3РО4, вторая ступень
H2SO4) в пределах 10~2—10~4; при Кс= Ю~10 электролит
считается очень слабым (Н2О, С6Н5ОН, HCN, CH3CONH2).
Свое название закона разведения соотношение A0.14)
получило в силу пропорциональности между степенью
электролитической диссоциации а и величиной разведения
1/С°; чем больше разведение (т. е. чем меньше С°), тем
больше степень электролитической диссоциации а. Зна-
Значение закона разведения ограничивается практическим
приложением его к решению задач, связанных с поведе-
поведением растворов слабых электролитов, правильнее было
бы соотношение A0.21) считать одной из частных задач
химии растворов электролитов.
Кислоты и основания. До сих пор под кислотами и основаниями
подразумевали растворы электролитов, которые при растворении
в воде образуют избыток ионов водорода или гидроксила. Такое оп-
определение кислот и оснований дал в конце XIX в. С. Аррениус и
для водных растворов электролитов его в большинстве случаев ис-
используют вплоть до настоящего времени. Однако в процессе развития
химии возникли трудности в определении этих классов соединений.
Стало известно, что основные свойства проявляют многие вещества,
не содержащие гидроксильных групп, например аммиак, а также
многие органические вещества. Точно так же-кислотными свойствами
обладают многие вещества, не содержащие водорода.
В связи с этим Дж. Бренстедом A923, Дания) и Т. Лоури A923,
Англия) была предложена более общая водородная, или протолити-
ческая, концепция кислотности: кислота — вещество, стремящееся
отдать протоны, а основание — вещество, стремящееся присоединить
протоны, т.е. кислота = Н+ -J-основание. Как уже отмечалось, сво-
свободный протон как частица не может существовать в растворах, он
обязательно присоединяется к каким-либо частицам, сообщая им по-
положительный заряд. Так, при растворении HCI в воде устанавли-
устанавливается так называемое протолитическое равновесие:
1**
НС1 + НОН= С1- +Н3О+
основа- осно-
ннс ванне
Это означает, что НС1 может проявить свойства кислоты только
в том случае, если растворитель (в данном случае вода) обладает
117
относительно него свойством основания. Напротив, при растворении
в воде аммиака молекулы воды выступают в качестве донора про-
протона, т. е. кислоты:
I
I
НОН = NH4+ + ОН-
кислота кислота основание
Молекулы воды настолько прочно связывают протон в ион гндрок-
сония НзО+, что по отношению к воде многие неорганические кис-
кислоты выступают в качестве сильных, т. е. нацело диссоциируют на
ионы.
Если же в качестве растворителя взять, например, 100%-иую
уксусную кислоту СН3СООН, которая взаимодействует с протоном
менее энергично, то по отношению к ней неорганические кислоты
проявят себя в качестве слабых электролитов. Тогда процессы типа
I III
HNO3 + СН3СООН + СН3СООН? -f NO3"
кислота основание кислота основание
идут неглубоко, до состояния равновесия, и розволяют составить ряд
относительной силы кислот:
НС1О4 > НВг > H2SO4 > HC1 > HNO3
Однако протолитическая теория ограничена, она не распространяется
на большое число соединений, явно проявляющих свойства кислот и
оснований. Примером могут служить щелочи, для которых схема
обмена протоном теряет свой физический смысл:
I I I I
он- + нго = н2о + он-
основание кислота кислота основание
Эта ограниченность стала очевидной, когда в качестве растворителя
стали использовать не только воду, но и жидкие NH3> COC12, SO2,
SOC12 и др. Одновременно опыт показал сходство кислотно-основных
реакций в водных растворах и неводных (например, в жидком
аммиаке), что определено сходством процессов собственной электро-
электролитической диссоциации молекул растворителей;
2Н2О = ОН
2NH3 = NH4+ +
аммоний- амид-
нон ион
В этом свете реакция нейтрализации сильной кислоты сильным осно-
основанием описывается одинаковыми схемами как в водном растворе,
так и в жидком аммиаке:
OH3Cl+NaOH = NaCl + 2H2O
NH4C1 + NaNHjj = NaCl + 2NHS
т. е. кислота-)-основание = соль-f-растворитель.
Такое представление о кислотно-осиовном взаимодействии стало
основой обобщенной теории сольво-систем, наиболее стройную фор-
формулировку которой дали Г. Кэди и Г. Элсей: растворимое вещество»
118
способное прямой диссоциацией или посредством реакции с раство-
растворителем образовывать катион, характерный для растворителя, явля-
является кислотой, а анион —основанием. Так, если растворителем явля-
является жидкий SO2, то имеет место электролитическая диссоциация:
В этом случае реакция
SOCla+ NaaSO3 =2NaCl + 2SO2
кислота основание растворитель
представляет собой взаимодействие кислоты и основания с образова-
образованием соли и растворителя.
Более универсальной является электронная теория кислот и
оснований Г. Льюиса. Согласно этой теории кислота — вещество,
способное использовать для построения химической связи свободную
пару электронов посторонней молекулы, т. е. акцептор электронной
пары. Основание — это вещество, способное быть донором свободной
пары электронов. Например, в реакции
Н+ + :О:Н-=Н:О:Н
кислота основание
ион водорода присоединяется к паре электронов водорода гидрок-
сильного иона и поэтому является кислотой, а ОН- имеет свобод-
свободную электронную пару, необходимую для образования ковалентной
связи с ионом водорода и потому является основанием. Процесс
электролитической диссоциации серной кислоты по теории Льюиса
изображается так:
н
г н у
[H:O:HJ -
но в общем случае кислотно-основные свойства веществ не связаны
со способностью электролитически диссоциировать в растворе. Так,
продукт взаимодействия BF3 и NH3
Н :F: H:F:
H:N:+ B:F: —¦+ H:N:B:F{
H :F:" H:F:
не распадается на ионы, однако образуется в результате кислотно-
основного взаимодействия.
Теория Льюиса дает более глубокое понимание механизма кис-
кислотно-основных процессов, однако она оказалась совершенно несос-
несостоятельной в решении вопроса о силе кислот. Поэтому обычно для
объяснения кислотно-основных свойств протонных кислот исполь-
используют теорию Бренстеда — Лоури, а в процессах с комплексообразо-
ваиием — теорию Льюиса.
Следует также остановиться на апротонной теории кислот и
оснований М. И. Усановича. По этой теории кислота —вещество,
способное отдавать катионы, которые соединяются с анионами и
электронами или нейтрализуют основания с образованием солн; осно-
основание—вещество, способное отдавать анионы или электроны, кото-
которые соединяются с катионами или нейтрализуют кислоты с образова-
образованием солей. Теория Усановича самая общая из всех приведенных
U9
выше и содержит все предшествовавшие определения кислот и осно-
оснований. Однако, следуя ей, пришлось бы все возможные химические
реакции отнести к разряду кислотно-основных, что существенно сни-
снижает избирательную ценность этой теории.
Глава 11
Гидролиз солей и нейтрализация
Взаимодействие кислот и оснований —
нейтрализация. Гидролиз солей. Кривые
титрования. Буферные растворы.
Взаимодействие кислот и оснований приводит, как
известно, к образованию соли. Процесс такого взаимо-
взаимодействия в общем виде может быть выражен уравнением
MOH + HR = MR+H2O (ll.l)
основаиие+кислота = соль + вода
Поскольку кислоты, основания и соли являются электро-
электролитами, то между всеми участниками этой реакции должно
устанавливаться ионное равновесие. Как отмечалось
выше, кислоты и основания различаются степенью элект-
электролитической диссоциации. Соли почти во всех случаях
в водных растворах полностью диссоциированы на ионы
(малорастворимые соли в растворенной части также дис-
диссоциированы нацело).
В качестве модели примем, что сильные и средние по
силе электролиты диссоциируют полностью, а слабые
электролиты, например вода, не диссоциируют совсем.
В этом случае если основание МОН—слабый электролит,
равновесие A1.1) примет вид
{MOH}aq+{H+}aq = {M+}aq+{H2O} (Ц.2)
если слабым электролитом является кислота, то
{OH-}aq+{HR}aq = {R-}aq+{H2O} (П.З)
Если же электролиты сильные, то
{OH-}aq+{H+}aq = {H2O}, Кс = К? О1-4)
Если и основание и кислота—слабые электролиты, то
{MOH}aq+{HR}aq = {M+}aq+{R-}aq+{H3O} (Ц.5)
Поскольку это процессы обратимы, к равновесному со-
состоянию можно подходить как от избытка исходных реа-
120
гентов, так и от избытка продуктов реакции. Взаимо-
Взаимодействие кислот и оснований называют нейтрализацией.
Совершенно очевидно, что равные значения концент-
концентраций ионов {OH~}aq и {H + }aq могут быть только в слу-
случае равновесия A1.4), поскольку единственной формой,
связывающей эти ионы в нейтральную молекулу, является
вода. Примером такого равновесия может служить взаи-
взаимодействие сильной кислоты НС1 и сильного основания
NaOH, в ионной форме это будет уравнение A1.4). Вос-
пользуясь стандартными значениями термодинамических
характеристик для процесса A1.4), получаем —RT In Кс =
= —55 900—Г-80,5, отсюда для температуры B98 К.)
'55 900 80. 5
Кс==- 1 ==1019.U.298 + T9TT4 = I(I4_
Исходя из этих данных и условия Сон-=СН+ получаем
уже известное соотношение
СОН_=СН+ = 10-' кмоль/м3 (рН7), A1.6)
что справедливо только для нейтральных водных раст-
растворов или для чистой воды. Отсюда следует, что раст-
растворы эквимолярных количеств сильных кислот и основа-
оснований имеют рН 7, т. е. нейтральны (отсюда реакции нейт-
нейтрализации).
Гидролиз солей. Поскольку равновесие A1.4) обра-
обратимо, то возможна и обратная задача определения реак-
реакции (кислая, нейтральная или щелочная) водного раст-
раствора хлорида натрия. Результат будет тот же, что и в
A1.6), т. е. водный раствор NaCl имеет нейтральную
реакцию, как и чистая вода. Иными словами, гидролиз
соли, образованной сильной кислотой и сильным основа-
основанием, не идет.
Рассмотрим случай A1.2), когда соль образуется при
взаимодействии слабого основания NH4OH и сильной
кислоты:
{NH4OH}aq+{H+}aq={NHj}aq + {H8O} A1.7)
Используя значения термодинамических характеристик
из таблиц, для данного равновесия находим
— КГ 1п/Сс = —51354—7-4,67,
откуда вычисляем константу равновесия при 298 К:
С 51 354 4,67
121
Если кислота и основание взяты в эквимолярных коли-
количествах, т. е. Смн,он = Снс1 или Cnh4oh = Сн+> а концентт
рация образовавшейся соли CNH+ равна 1 кмоль/м3, то
CH+=Kl0~9 = 10'6 кмоль/м3 (рН4,5). Это означает,
что водный раствор NH4C1 имеет кислую реакцию
(рН < 7). Следовательно, водный раствор при гидролизе
соли, образованной слабым основанием и сильной кисло-
кислотой, имеет кислую реакцию.
Рассмотрим реакцию между сильным основанием NaOH
и слабой кислотой HCN, которая проходит по типу рав-
равновесия A1.3):
Исходя из стандартных термодинамических данных, имеем
— КГ 1пК«. = —10 290 —Г-69,6,
q 10 290 69,6
отсюда при 298 К Ке= п С1Г—= 1019-14-298+19-14 =
LOH~CHCN
= 106'4. Если кислота и основание взяты в эквимоляр-
эквимолярных количествах, т. е. Coh-=CHcn. и содержание обра-
образующейся соли 1 кмоль/м3, то Сон- =KlO~5'4 =
= 10~2'7 кмоль/м3, откуда концентрация ионов водорода
Сн+ яа Ю-("-V) я* Ю-11.3 кмоль/м3 (рН 11,3).
Полученный результат (рН > 7) свидетельствует о ще-
щелочной реакции среды, возникающей при растворении
NaCN. Следовательно, при гидролизе соли, образованной
сильным основанием и слабой кислотой, водный раствор
приобретает щелочную реакцию.
Примером взаимодействия кислоты и основания,
являющихся слабыми электролитами A1.5), может слу-
служить реакция
{NH4OH}aq +{H2CO3}aq - {NHt}aq + {HCOs-}aq + {НгО}
Поскольку NH4OH и Н2СО3 существуют только в раз-
разбавленных растворах, а при концентрировании распа-
распадаются с образованием NH3, CO2 и НгО, строже этот
процесс можно записать так:
a) + (CO2)+{H2O}={NH+}aq+{HCO3-}aq X^
для которого, — RT InKc = — 98370 4-7-268,2 и при 298К
С С
Кс = — = 103)*. Это означает, что при гидро-
Рин, Рсо,
122
лизе соли NH4HCO3, концентрация которой в растворе
равна 1 кмоль/м3, образуются газообразные NH3 и СО2,
парциальные давления которых равны 25 гПа при 25° С
и 600 гПа при 80° С. Полученные результаты свидетель-
свидетельствуют о том, что гидролиз одномолярного раствора
NH4HCO3 уже при комнатной температуре идет глубоко,
а при удалении газообразных NH3 и СО2—до конца.
Отсюда следует, что гидролиз солей, образованных сла-
слабыми кислотой и основанием, идет глубоко, обычно до
конца.
Что касается реакции среды растворов солей, обра-
образованных слабыми кислотой и основанием, то этот воп-
вопрос решается через сравнение степеней электролитической
диссоциации образующихся при гидролизе кислоты и
основания. Так, в рассматриваемом примере значения
констант электролитической диссоциации
Л:1=1,8Ы0-8
позволяют заключить, что концентрация ионов гидрок-
сила несколько больше концентрации ионов водорода
(Ki > K2)- Следовательно, раствор за счет гидролиза
в нем соли NH4HCO3 должен иметь слабо-щелочную
реакцию.
Таким образом, глубина процесса гидролиза солей
определяется прежде всего природой соли. Количествен-
Количественной мерой гидролиза, как и любого химического взаи-
взаимодействия, является работа реакции или изменение
энергии Гиббса:
ДОрцдр = А "гидр — Т А5ГИдр •
Основным рабочим инструментом этой меры является кон-
константа гидролиза
зная которую можно определить степень гидролиза соли:
й
ИЛИ Р2 =
здесь Pj относится к равновесию A1.3), а Р2—к равно-
равновесию A1.2). Связь между р, константой гидролиза/Сгидр
и исходной концентрацией соли С°сош для двух типов
123
равновесия выражают соотношением
и, если р<^ 1,
^. (П.7)
Из этого соотношения A1.7) следует, что чем более раз-
разбавлен раствор соли, тем больше степень ее гидролиза.
Для практических целей важно знать величину гид-
гидролиза, а также соотношение /(гидр, ^hr и /Croh (кон-
(константы диссоциации слабых основания или кислоты). Рас-
Рассмотрим последовательно примеры равновесного гидролиза.
Так, при гидролизе уксуснокислого натрия, образованного
слабой кислотой и сильным основанием,
{CH3COONa}aq +{НгО} = {CH3COOH}aq + {NaOH}aq
слабая сильное
кислота основание
или в ионной форме
{CH3COO-}aq+{H2O} = {CH3COOH}aq+{OH-}aq
имеет место избыток ионов ОН" по сравнению с-ионами
Н+ и раствор приобретает щелочную реакцию. Запишем
и преобразуем /СГидр этого равновесия:
Лгидр= p p =-p p ^H + ^OH"
CCH3COO~ I -..--- I -
Kw
Лкисл
в общем виде
Л"гидр = Т7
(здесь ROH — гидрат, который является слабой кисло-
кислотой).
Второй пример касается гидролиза соли, образован-
образованной сильной кислотой и слабым основанием:
{NH4Cl}aq + {Н2О} = {NH4OH}aq + {HCl}aq
слабое сильная
основание кислота
в ионной форме
124
Запишем и преобразуем КТЯ№ этой соли:
„ cnh4ohch + сон __ Kw к _ Kw
"гидр — -?¦ 7—~к Агидр 17 ' ("¦")
L °ОН- Лосн AROH
В общем виде соотношение A1.9) подобно A1.8), однако
в случае A1.9) ROH — слабое основание.
Третий пример касается гидролиза соли, образованной
слабыми кислотой и основанием:
{CH,COONH«}aq + {Н2О} = {CH3COOH}aq + {NH4OH}aq
слабая слабое
кислота основание
поскольку соль полностью диссоциирована, можно запи-
записать
{CH3COO-}aq + {NH4+}aq +{H2O} = {CH3COOH}aq +{NH4OH}aq
A1.10)
Запишем и преобразуем ХГИДр этой соли:
„ _CCH3COOHCNH4OH Kw
"гидр— r f —у к •
CCHaCOO-LNH<- ACH3COOHANH4OH
или
v Kw
А гидр — Т> Р '
Акисл^оси
Степень гидролиза р для равновесия типа A1.10) может
быть выражена соотношением
Ьсоли "соля
Концентрации всех участников реакции гидролиза можно
выразить через С°СОЛ1 и р, тогда
ССН,СОО Сш+-Ссоли A-Р);
отсюда
Агидр^ ^ | j
ИЛИ
т. е. не зависит от концентрации соли.
В качестве примеров к рассмотренному материалу
приведем решения нескольких задач.
125
Задача 1. Определить константу гидролиза и степень гидролиза
CHsCOONa в сантимоляриом @,01 М) растворе, если константа дис-
диссоциации уксусной кислоты /(сн соон Равна 1|85-10~5.
Решение.
'О4
^_ -./5,4.10-1°
сот ~ V 0,01 ~
= 2,3- Ю-4 @,023%).
Задача 2. Определить константу гидролиза и степень гидролиза
уксуснокислого анилина CH3COOC6H5NH3, если известны следующие
значения констант диссоциации:
q aq, /С„„сл= 1,85-10-6
{QH6NH3OH}aq = {CeH5NH+}aq + {OH-}aq, Коси = 4,5- 10-ю.
Решение
А* 10~14
Агидр—к к — | ос, ¦ п — 6.4 К 10-10— ' '
Акисл^осн 1,00-1U •¦I.D-IU
:-—-===- = 0,52 E2%).
Рассмотрим вопрос о количественном определении рН
гидролиза солевых растворов на примере гидролиза
NH4C1:
{NH4+}aq+{H2O} = {NH4OH}aq + {H + }aq (H.ll)
Если известны константы гидролиза KTnw и концентра-
концентрация соли Ссоли, то для определения концентрации ионов
водорода Сн+ и рН гидролиза составляем систему урав-
уравнений, неизвестными в которых являются концентрации
реагирующих веществ процесса A1.11).
Итак, записываем выражение для константы гидро-
гидролиза
„ ?nh4ohch +
NH +
A1.12)
и для концентрации соли в растворе:
Четвертое уравнение получаем из условия электронейт-
электронейтральности раствора, т. е. из условия равенства сумм
концентраций ионов одного и другого знаков:
Сын4++Сн+=СОН-+Сс1- (Н-15)
ын
126
пятое уравнение—ионное произведение воды:
KW = CH+COH-. A1.16)
В результате имеем пять уравнений с пятью неизве-
неизвестными. Можно несколько упростить задачу. Если вспом-
вспомнить, что в конкретном примере раствор NH4C1 имеет
кислую реакцию, следовательно, в нем Сн+ >СОн-- Это
означает, что в уравнении A1.15) можно пренебречь чле-
членом СОн- и полностью исключить уравнение A1.16). Ре-
Решая совместно уравнения A1.12), A1.13), A1.14) и A1.15),
получаем
Анализируя уравнение A1.17), можно заключить, что
если константа гидролиза очень мала (/Сгидр <«5 1), а кон-
концентрация соли в растворе достаточно велика, то урав-
уравнение A1.17) примет вид
Cft+ — V Кгиа-дСсолк ¦ A1.18)
Очевидно, что для солей, дающих в растворе щелочную
реакцию, будем иметь
Сон- = V ^гидрСсоли • A1.19)
В рассмотренном примере A1.11) константа гидролиза NH4C1
/СгнДр = 4,3-10—9. Пусть концентрация соли будет Ссоли=0,01 кмоль/м3,
тогда, используя уравнение A1.17), получаем 2Сн+= — 4,3-10~а -j-
-j- y^l,84-10-17 + l,72-10-10= 13-10-6 кмоль/м3. Совершенно оче-
очевидно, что в данном случае можно воспользоваться приближенным
уравнением A1.18)
Сн+ = Y~4,3-10~"• 10~2 =^6,5• 10~6 кмоль/м3, рН 6,8.
Напротив, в приведенной выше задаче 2 концентрация ионов во-
водорода может быть вычислена с использованием точной формулы
A1.17). Так, если концентрация соли равна 0,01 кмоль/м3 и ^Сгидр =
= 0,52, получаем, Сн+ =0,5 (—0,52+|/M22 + 4-0M2-0,1) ==
= Ю-2 кмоль/м3, т. е. этот раствор имеет очень кислую реакцию
(рН2).
В заключение рассмотрим так называемый совместный
гидролиз солей, которые порознь дают кислую и щелоч-
щелочную реакции в водном растворе. Положим, что приготов-
приготовлены одномолярные растворы двух солей, одна из которых
дает кислую реакцию (NH4C1)
ГС С
—RT 1п\ Н* HN<un| =51350 + Г-4,7 (рН4,5), A1.20)
n*oh] =5i
+ J
127
а другая—щелочную (NaCN)
69,6 (рН11,7). A1-21)
Если объединить реакции гидролиза в единую систему,
то химический процесс в ней будет суммой реакций
A1.20), A1.21) и реакции нейтрализации ионов ОН" ио-
ионами Н + :
A1.22)
= 7590 Дж.
В результате получился процесс гидролиза соли, обра-
образованной слабой кислотой и слабым основанием, идущий
практически до конца, где /Сгицр = 21,4, а степень гидро-
гидролиза E = |УТЛ]:[1 + КМ] = 0,82(82%).
Таким образом, совместный гидролиз двух солей, из
которых одна сообщает раствору кислую реакцию, а дру-
другая— щелочную, сводится в конечном итоге либо к реак-
реакции нейтрализации кислоты и щелочи, либо к реакции
гидролиза соли, образованной слабыми кислотой и осно-
основанием. В примере A1.22) образующееся слабое основа-
основание NH4OH устойчиво в растворе лишь в очень небольшой
концентрации. При увеличении концентрации газообраз-
газообразный аммиак будет выделяться и процесс гидролиза пой-
пойдет необратимо.
Кривые титрования. Как уже отмечалось в начале
главы, гидролиз и нейтрализация являются противопо-
противоположно направленными процессами. Прямое взаимодействие
кислот и оснований с образованием соли и воды, назы-
называемое титрованием, широко используется в лабораторной
практике для взаимного определения концентрации и ко-
количества реагентов. Зависимость рН титруемого раствора
от количества добавляемого реагента называют кривой
титрования. Величину рН раствора определяют с помо-
помощью индикаторов или инструментальными методами (по
электрической проводимости, по ЭДС гальванического
элемента, спектрофотометрически и т. д.). Равному числу
эквивалентов кислоты и основания в растворе соответст-
соответствует эквивалентная точка.
Разберем случай титрования сильной кислоты силь-
сильным основанием (или наоборот). Допусти^ соляную кис-
128
лоту НС1 титруют щелочью NaOH. В табл. 11.1 приведены
результаты отдельных этапов такого титрования при ис-
исходном количестве кислоты 10 мл (увеличением объема
раствора для простоты пренебрегаем). Данные табл. Ц.1
можно изобразить в виде графика зависимости рН раст-
раствора от количества добавляемого реактива (см. рис. 11.1).
Ю
15 мл КОН
а 4
2
0
Рис. 11.1. Кривая титрования сильной кислоты
сильным основанием:
а —кислые растворы; б —щелочные растворы
Вблизи эквивалентной точки (рН7) прибавление ничтож-
ничтожных количеств реагентов резко изменяет рН раствора.
Поэтому переход через границу эквивалентности, опреде-
определение эквивалентной точки легко осуществить с помощью
различных средств и, в первую очередь химически, с по-
помощью индикаторов.
Индикаторы—вещества, которые меняют свою окраску
в зависимости от концентрации ионов водорода в растворе.
Каждый индикатор характеризуется определенной узкой
областью значений рН, в которой его цвет изменяется.
Таблица 11.1. Данные по титрованию кислоты щелочью
Добавляемая
щелочь, мл
0
9
0,9
0,09
0,009
0,0009
0,00009
рН раствора
1
2
3
4
5
6
7
Добавляемая
щелочь, мл
0,00009
0,0009
0,009
0,09
0,9
9
*
рН раствора
8
9
10
11
12
13
5 № 705
129
Основоположниками теории индикаторов [Оствальд
A891), Гантч A907), Бьеррум A918)] установлено, что
органические вещества существуют в зависимости от среды
в виде основной или кислотной форм. Например, фенол-
фенолфталеин имеет следующее строение:
ОН ОН О О
кислотная форма
бесцветная
основная форма
малиновая
Кислотная форма фенолфталеина (HL) представляет собой
слабую органическую кислоту, а основная (NaL)—соль
Таблица 11.2. Переходы окраски индикаторов
Индикатор
Метиловый оранжевый
Бромкрезоловый зеленый
Бромкрезоловый пурпур-
пурпурный
Бромтимоловый синий
Крезоловый красный
Фенолфталеин
Ализариновый желтый
Интервал рН
перехода
окраски
3,1—4,4
3,8—5,4
5,2—6,8
6,0—7,6
7,2—8,8
8,3—10,0
10,0—12,0
Окраска
в кислоте
Красная
Желтая
Желтая
»
Бесцветная
Желтая
в щелочи
Желтая
Голубая
Пурпурная
Голубая
Красная
»
Лиловая
этой кислоты, которая полностью диссоциирует в раст-
растворе. Поскольку соль NaL образована сильным основа-
основанием (NaOH) и слабой органической кислотой (HL), она
подвергается гидролизу:
{Li-}aq+{H2O} = {HL}aq+{OH-}aq A1.23)
в щелочной среде
в кислой среде
Равновесие гидролиза A1.23) имеет собственное значение
рН, которое и определяет границу перехода окраски.
Оно смещается вправо или влево в зависимости от из-
избытка или недостатка ионов водорода в растворе.
130
Если вновь вернуться к рис. 11.1, то обращает на
себя внимание факт резкого изменения рН раствора
вблизи эквивалентной точки. Этот участок охватывает
область значений рН от 3 до 11. Следовательно, для
титрования сильной кислоты основанием (и наоборот)
годны почти все индикаторы, приведенные в табл. 11.2
(подробнее см. табл. VI приложения).
Иная картина возникает в случае титрования слабого
основания сильной кислотой или слабой кислоты силь-
сильным основанием, например:
{CH3COOH}aq+{NaOH}aq = {CH3COONa}aq+{H2O}
или в ионной форме
{CH3COOH}aq+{ОН - }aq = {СН3СОО - Ц+{Н2О}
Слабая уксусная кислота лишь незначительно диссоци-
диссоциирована в водном растворе /Cchscooh = 1,85-10"?. Поэтому
если 0,1 М раствора НС1 имеет рН = 1, то для 0,1 М ра-
створа СН3СООН рНЗ, так как Сн+ = К/Ссн3соон С° =
= К1,8510-5-0,1 = 10-2'9. Если к 10 мл 0,1 М раствора
уксусной кислоты добавить 0,1 мл 0,1 М NaOH, то это
будет соответствовать гидролизу 0,1 М раствора соли
CH3COONa. Концентрация ионов гидроксила в этом ра-
растворе получается равной
сон-=УгКг„дрСсол^ = У5,4.10-ю.0,1=7,4-10-е,
откуда рН9, т. е. раствор будет иметь щелочную реак-
реакцию. При этом вспомним, что при смешении равных объе-
объемов 0,1 М растворов сильных электролитов НС1 и NaOH
раствор нейтрален (рН7).
Графически зависимость рН раствора СН3СООН при
постепенном добавлении к нему раствора NaOH представ-
представлена на рис. 11.2. Как видно из этого рисунка, в случае
титрования уксусной кислоты интервал значений рН в
области эквивалентности очень узок—от 7 до 10. Из пе-
перечисленных в табл. 11.2 индикаторов для определения
эквивалентной точки пригодны лишь крезоловый красный
и фенолфталеин. Чем слабее кислота, тем менее отчетлива
эквивалентная точка, тем более узок интервал значений
рН и менее надежен способ титрования.
Буферные растворы составляют из смеси слабой кис-
кислоты или слабого основания и соответствующей им соли,
рН этих растворов почти не изменяется при добавлении
небольших количеств сильных кислот и оснований. Напри-
S* 131
мер, буферным действием обладает раствор СН3СООН -f
+ CH3COONa. Из константы электролитической диссоци-
диссоциации кислоты
кс-
^сн.соон
находим
¦-Н+
-К с
A1.24)
Поскольку слабая уксусная кислота диссоциирована на
ионы мало, а ее соль полностью, то с достаточной сте-
pH 7
6
а 4
2
О
i i i i i i i i
НС1
10
ii
15 мл NaOH
Рис. 11.2. Кривая титрования слабой кислоты
сильным основанием;
а—кислые растворы; б — щелочные растворы (пунктиром
обозначена кривая титрования НС! щелочью)
пенью точности можем принять
С - =С A1 25)
Сделав подстановку выражения A1.25) в A1.24), получаем
A1.26)
Если добавить к этому буферному раствору немного силь-
сильной кислоты (например, НС1), то равновесие гидролиза
соли CH3COONa сместится в направлении СН3СООН
+ н+
сн3соо- + н2о —+ сн3соон+он-
т. е. на величину х увеличится концентрация СН3СООН
в растворе и на ту же величину х уменьшится концент-
концентрация соли, тогда
"Г
^соли —
A1.27)
132
Если Скисл и Ссоли сравнимы и достаточно велики, а зна-
значение х мало, то разница между A1.27) и A1.26) неболь-
небольшая и рН буферного раствора изменится мало.
Пример. Имеем 10 мл раствора, содержащего Ссн соон =
¦=1 кмоль/м3 и CCHsCOoNa=' кмоль/м3 (Кс= 1,85-10~6). Тогда
Сн+= 1,85-10-6 кмоль/м3 (рН4,7).
Добавим 0,1 мл ЮМ раствора соляной кислоты (это равносиль-
равносильно добавлению 1 мл 1 М кислоты, что практически не изменяет
объема раствора). При этом 0,1 часть соли превратится в кислоту,
т. е. концентрация кислоты СН3СООН возрастает на 10%, а концент-
концентрация соли СНзШОЫа уменьшится на 10%, т. е.
Сн+= 1,85-10-» |^°'|=2,26-10-» кмоль/м3 (рН4,6).
Сравнивая значения рН до и после прибавления соляной кисло-
кислоты, видим, что оно уменьшилось от 4,7 до 4,6, хотя количество до-
добавленной кислоты составило 10% от исходного.
В то же время, если к 10 мл чистой воды (рН7) прибавить 0,1 мл
10 М соляной кислоты, то значение рН будет равным нулю.
Буферные растворы используют в лабораторной прак-
практике в качестве эталонов величины рН, при определении
концентрации водородных ионов путем сопоставления
цвета эталона и испытуемого растворов после добавления
индикатора.
Глава 12
Растворимость электролитов
Растворимость гидроксидов и солей. Произведение
растворимости малорастворимых электролитов.
Влияние на растворимость одно- и разноименных
ионов. Солевой эффект.
Растворимость гидроксидов и солей. Растворимость ве-
вещества определяют его равновесным содержанием в еди-
единице объема или массы растворителя или насыщенного
раствора. Чаще растворимость выражают в массовых до-
долях. Форма записи процесса растворения может отражать
либо сам факт перехода в раствор
либо более детальную картину электролитической диссо-
диссоциации
133
Растворимость солей и гидроксидов различна. Так,
соли и гидроксиды щелочных металлов являются силь-
сильными электролитами и в подавляющем большинстве хо-
хорошо растворимы в воде. Хорошо растворимые основания
образуют элементы II группы—барий и стронций; гид-
роксид кальция обладает средней растворимостью, магний
и бериллий образуют малорастворимые формы гидрокси-
гидроксидов. Основания всех остальных металлов представляют
собой малорастворимые соединения.
Гидроксиды кислотного типа типично неметаллических
элементов обычно хорошо растворимы. К ним относятся
высшие кислоты хлора (НС1О4), серы (H2SO4), азота (HNO3),
фосфора (Н3РО4). По мере снижения степени окисления
центральных атомов анионов кислотные свойства гидрок-
гидроксидов уменьшаются и растворимость снижается. Элементы
Б-подгрупп, являющиеся в свободном состоянии метал-
металлами, как правило, образуют малорастворимые гидроксиды
даже в случае высоких степеней окисления, примером
может служить вольфрамовая кислота (H2WO4). Лишь
высшие гидроксиды металлов В-подгрупп VI и VII групп —
хромовые (Н2Сг04 и Н2Сг207) и марганцовая (НМпО4)
кислоты — характеризуются большой растворимостью.
Соли классифицируются по растворимости весьма
сложно. Ниже приведена краткая характеристика хлори-
хлоридов, нитратов, карбонатов и сульфатов щелочных и ще-
лочно-земельных металлов.
Таблица 12.1. Растворимость хлоридов при 20 °С, г/100 г Н2О
Хлорид
LiCl-H2O
NaCl
КС1
RbCl
CsCl
Растворимость
83,2
35,9
34,4
91,1
186,5
Хлорид
ВеС12-4Н2О
MgC!2-6H2O
СаС12-бН2О
SrCl2-6H2O
ВаС12.6Н2О
Растворимость
72,8
54,8
74,5
53,1
36,2
Из табл. 12.1 видно, что хлориды элементов I и II А-подгрупп
хорошо растворимы в воде. Численное значение их растворимости
находится в сложной зависимости от их состояния и положения вто-
второго элемента соли в периодической системе. Так, растворимость
хлоридов щелочных металлов проходит через минимум, достигая наи-
наименьшего значения в случае КС1, а наибольшего — в случае CsCl.
С энергетической точки зрения растворимость хлоридов некоторых
134
щелочных металлов связана с температурой следующим образом;
+ }aq+{Cl-}aq, AG; = 17200-Т 75,1
aq, AG°T= 17890 — Т 90,8
Из сравнения этих процессов следует, что различие в растворимости
солей определяется превышением энтропии растворения CsCl над
энтропией растворения КС1 на 15,7 э. е., что объясняется большим
размером иона цезия по сравнению с ионом калия.
Растворимость дихлоридов элементов II А-подгруппы иосит ион-
ионный характер. Хлориды могут быть не только безводными солями,
но и кристаллогидратами с различным содержанием кристаллизаци-
кристаллизационной воды. Температурная зависимость растворимости трихлоридов
(например, А1С18, FeCl3) дополнительно осложняется гидролизом этих
солей, а также комплексообразованием в растворе.
Как следует из табл. 12.2 сульфаты щелочных металлов хорошо
растворимы. Наличие кристаллогидратов у сульфатов лития и натрия
способствует их растворимости. Однако у сульфатов щелочно-земель-
ных металлов, расположенных в той же последовательности, раство-
растворимость резко уменьшается.
Нитраты —соли азотной кислоты, обладают высокой растворимо-
растворимостью (табл. 12.3), которая следует иной закономерности, чем ранее
рассмотренные хлориды и сульфаты.
Из табл. 12.4 следует, что растворимость карбонатов высока
только в случае щелочных металлов, кроме лития.
Таблица 12.2. Растворимость сульфатов при 20 °С, г/100 г Н2О
Сульфат
LiSO4-H2O
Na2SO4-10H2O
K2SO4
Rb2SO4
CsaSO4
Растворимость
34,7
19,2
11,1
48,2
178,7
Сульфат
BeSO4-4H2O
MgSO4-7H2O
CaSO4-2H2O
SrSO4
BaSO4
Растворимость
40,0
35,1
0,79
1,14-10-"
2.3-10-*
Напротив, карбонаты щелочно-земельных металлов и практически
всех остальных металлов периодической системы —соединения мало-
малорастворимые.
Таблица
Нитрат
LiNO3-3HaO
NaNO3
KNO3
RbNO3
CsNOs
12.3.
Растворимость нитратов при 20
Растворимость
70,0
87,6
31,6
53,5
23,0
Нитрат
Be(NO3J.4H2O
Mg(NO8)a-6H2O
Ca(NO8)a-4H2O
Sr(NO3)a-4HaO
Ba (NO8)a
°C, г/100 г НаО
Растворимость
107,0
73,3
128,8
70,4
9,05
135
Таблица 12.4. Растворимость карбонатов при 20 СС, г/100 мл Н2О
Карбонат
Li2CO3
Na2CO3-10H2O
К2СО3-1,5Н2О
Rb2CO3-H2O
Cs2CO3-3,5HaO
Ра створн мость
1,32
21,8
111,0
69,0
210,0
Карбонат
MgCO3
СаСО3
SrCO3
ВаСО3
Растворимость
9.4-10-3
1,3-10-з
1,0-Ю-з
1,7-Ю-з
Следует отметить, что соли кислые и основные обычно сущест-
существенно отличаются по растворимости от средних солей. Так, кислые
соли щелочно-земельных металлов имеют значительно большую раст-
растворимость, чем средние соли, а основные — наоборот.
Произведение растворимости малорастворимых электро-
электролитов. Рассмотрим малорастворимые соли и гидраты, на-
насыщенные растворы которых весьма разбавлены и потому
их можно квалифицировать как идеальные. Такие веще-
вещества в своей растворенной части нацело диссоциированы
на ионы, т. е. молекулярные формы соединений в раст-
растворе отсутствуют. Используем в качестве примера равно-
равновесие процесса растворения малорастворимого карбоната
серебра
[Ag2CO3] + aq = 2 {Ag}aq + {COl"}aq A2.1)
константа этого гетерогенного равновесия имеет вид
Исходя из стандартных значений термодинамических ха-
характеристик А#298 и A529S процесса растворения Ag2CO3,
находим AG° =41670 + Т 72,6. Памятуя, что AG'T =
= — \9,l4T\gKc для температуры 25 °С, Кс = 7,75-10"".'
Используя это значение, можно вычислить растворимость
соли, которая численно должна быть равна равновесной
концентрации иона СО|". Из стехиометрии уравнения A2.1)
следует, что концентрация иона Ag+ в два раза больше
концентрации СО|-, т. е.
2Ccol-==CAg+> A23>
Сделав подстановку равенства A2.3) в выражение для
константы равновесия A2.2), получаем
со§-
136
откуда находим растворимость соли, численно равную
3 /~1Г~
CAg2CO, = ]/ j = ?/0,25.7,75-10-«= 1,25.10"* кмоль/м**,
или 3,45 • 10~3 г/100 г раствора.
Константа равновесия растворимости малораствори-
малорастворимых солей и гидратов получила название произведения
растворимости L. В общем виде равновесие
}.q A2.4)
характеризуют произведением растворимости
CZy_. A2.5)
Из уравнения A2.4) следует, что
пС х+ =тС у_, A2.6)
т. е.
Подставив концентрации A2.6) и A2.7) в уравнение
A2.5), получаем выражение для растворимости соли
т + п
Ссоли —
Для солей, диссоциирующих на два иона, справедливо
соотношение
^соли= V L •
Значения растворимости и произведения растворимости
некоторых труднорастворимых солей и гидратов при 25 °С
приведены в приложении XIII.
Добавление к растворам малорастворимых солей и
гидратов хорошо растворимых соединений, дающих при
растворении уже имеющиеся в растворе ионы, способст-
способствует уменьшению растворимости исходных солей и гид-
гидратов. Рассмотрим это явление на примере соли AgCl,
для которой L = CAg+Cci- = 1,56• 10~10, откуда, Сдй+ =
= Са- = 1,25-10~5, а растворимость AgCl равна s =
= 1,8-10~4 г/100 г Н2О. Если к 1 л раствора AgCl до-
добавить 1 г хорошо растворимой соли AgNO3 (M = 170),
которая полностью диссоциирует на ионы, то концентра-
концентрация ионов Ag+ возрастает на величину CAg+^-fm^
137
¦=5,9-10 3 кмоль/м3. Тогда произведение растворимости
можно будет записать следующим образом:
отсюда
Растворимость AgCl в растворе AgNO8
Cr,-Mr, 2,6.10-3.35,45
ю Го
=10"т г/юо г
что примерно в 1000 раз меньше, чем растворимость AgCl
в чистой воде.
Солевой эффект. Растворимость малорастворимых солей
и гидратов зависит не только от избыточного содержания
в растворе одного из ионов растворяющегося образца
(одноименных ионов), но также и от содержания «посто-
«посторонних» ионов. Так, например, в присутствии 1 кмоль/м3
KNO3 растворимость PbSO4 увеличивается в 14 раз,
а в присутствии 1 кмоль/м3 A1(NO3K — примерно в 77 раз.
Причина этого явления в том, что «посторонние» ионы,
взаимодействуя своими зарядами с ионами «хозяевами»,
удерживают их в растворе в количестве, превышающем
растворимость малорастворимого образца в чистом раство-
растворителе, т. е. имеет место так называемый солевой эффект.
При введении в раствор «посторонних» ионов концентра-
концентрации С ах и Съу- возрастут и примут значения Сд*+ и CBy-t
так что выражение для L усложнится:
где у — коэффициент активности ионов, для нахождения
которого можно использовать уравнение
lg у = - 0,509**/
у ~
здесь mlt m2, ...,mn—моляльные концентрации каждого
из «посторонних» ионов в растворе; zu za, ..., zn—заряды
этих ионов.
Используя эти соотношения, можно решать практиче-
практические задачи на растворимость с учетом различных «по-
«посторонних» ионов, содержащихся в растворе.
Задача 1. В 1 л раствора содержится 500 мг Ag+ и 5 мг РЬ2+.
Если к этому раствору прилить раствор KssCrO4, то будет выпадать
138
осадок. Малорастворимая соль какого из указанных катионов выпа-
выпадает в осадок первой?
Прежде всего находим концентрации содержащихся в растворе
катионов Ag+ (М = 108) и РЬ2+ (М = 207):
СДг+=щ=4,62.10-' кмоль/м8,
CpbS+==^5=2,42-Ю-» кмоль/м».
При добавлении к этому раствору легкорастворимой соли К2Сг04
становится возможным образование двух малорастворимых солей!
Ag2Cr04 (L1 = 4,05-10-i2) и PbCrO4 (L2= 1,8-10-").
Концентрация ионов CrOf", необходимая для выпадения в осадок
соли Ag2Cr04, будет равна:
„ '.Ц _ 4,05.10- ]910_
C\ + ~ D,62-10-3J -'.—
а для выпадения в осадок соли PbCrO4
С 2_= 2 = '' =7,5-10-" кмоль/м3.
Поскольку минимальной величиной концентрации, необходимой для
начала осаждения, характеризуется РЬСгО4, именно эта соль и будет
первой выпадать в осадок.
Задача 2. Избыток твердой малорастворимой соли РЬСгО4 обра-
обрабатывают 1,5 моль Na2CO3, содержащимися в 1 л раствора. Вычислить
значение концентрации ионов СгО4~ и СО§~ в этом растворе*
В этой системе возможна реакция обмена
[PbCrO4] +{Na,CO,},q = {Na2Cr04}aq + [PbCQs]
или
В результате кроме РЬСгО4 образуется еще одна малорастворимая
1 соль —РЬСО3, на осаждение которой идет немного меньше 1,5 моль
СОаГ. Произведение растворимости солей РЬСгО4 и РЬСО3 находим
из приложения ХШ, тогда для
Коистаита равновесия A2.8) может быть записана следующим образом:
"c<^F~''5-cck.j--3-10"" ' '
Отсюда С 2_ =0,53 кмоль/м3, С 2_= 1,5—0,53 = 0,97 кмоль/м3.
СгО С^з
.139
Глава 13
Электрохимические процессы
Окислительно-восстановительные реакции и галь-
гальванические элементы. Стандартные электродные
и окислительно-восстановительные потенциалы.
Нормальный водородный электрод. Уравнение Нерн-
ста. Электрохимический анализ окислительно-вос-
окислительно-восстановительных реакций.
Окислительно-восстановительные процессы. Рассмотрим
окислительно-восстановительную реакцию вытеснения меди
металлическим цинком из раствора сульфата меди в ион-
ионной форме:
[Zn]+{Cu2+}aq = [Cui+{Zn*+}aq A3.1)
Д^. 298 0,0 64,4 0 -152,4
5°29в 41,6 —98,7 33,3 —106,5
Значения термодинамических характеристик процесса A3.1)
получаются равными Д#298 = —216,8 кДж, ASlts =
= — 16,1 э. е. Отсюда AGT = —216 800 + 16,1 Т и константа
равновесия при температуре 25° С /Г = rZn'+ = 1037. Такое
LCu2 +
нереально большое значение константы равновесия сви-
свидетельствует о практической необратимости процесса A3.1),
идущего слева направо. Эту сложную реакцию можно
представить в виде суммы двух независимых полуреакций:
растворения цинка
+ 2e-, kG°m = —152 420 + Г 148,1 +А'е A3.2)
и растворения меди
[Cu] + aq = {Cu2 + }aq + 2e-, ДО^ = 64 390 + Г 132+ А"е. A3.3)
Здесь А'е и А"е—работы образования в растворе двух сво-
свободных электронов (ее значение неизвестно).
Процесс A3.2), взятый в отдельности (цинковая пла-
пластинка в водном растворе ZnSO4), вполне равновесен;
константа этого равновесия при температуре 25° С Kt = 1019.
Поскольку водные растворы не обладают электронной
проводимостью, освобождающиеся электроны Bе~) не могут
находиться в растворе и накапливаются на металлическом
цинке, создавая строго определенную, равновесную кон-
концентрацию. Эта концентрация избыточна по сравнению
со сбалансированным количеством положительно и отри-
140
II
Zn
Zn+2+SO72
II
Cu
цательно заряженных частиц, характерных для металла,
и поэтому проявляется в качестве отрицательного электри-
электрического заряда металла, или его потенциала ц>т.
Из уравнений A3.2) и A3.3) следует, что чем меньше
концентрация ионов металла в растворе (Zn2+ или Си2+),
тем больше должна быть равновесная концентрация элек-
электронов в металле электрода. Процесс A3.3) (медная пла-
пластинка в водном растворе CuSO4) также равновесен, но
константа этого равновесия /С2
при температуре 25° С очень
мала A0~18). Отвечающая этой
величине избыточная концент-
концентрация электронов в медной
пластине намного меньше, чем
в цинковой. Поэтому потен-
потенциал медной пластины ф„ дол-
должен быть относительно более
положительным по сравнению
с потенциалом цинковой плас-
пластины ц>т. Из-за электростати-
электростатического притяжения электронов
в металле и ионов в растворе
на границе этих двух сред воз-
возникает так называемый двойной
электрический слой.
Если системы A3.2) и
A3.3) объединить в одну, соеди-
соединив цинковую и медную пластины металлическим провод-
проводником с электронной проводимостью, а растворы ZnSO4
и CuSO4—электролитическим проводником с ионной про-
проводимостью, то получится замкнутая неравновесная си-
система-— гальванический элемент, схема которого приведена
на рис. 13.1. Поскольку потенциалы электродов различны,
по соединяющему их металлическому проводнику (II)
перемещается поток электронов—электрический ток. Для
восстановления равновесного потенциала цинкового элек-
электрода цинк должен переходить в раствор. Увеличение же
отрицательности потенциала медного электрода за счет
переместившихся электронов повлечет разрядку части ионов
Си2+ и выделение из раствора металлической меди на
медном электроде. В результате около цинкового электрода
электролит приобретает избыточное число положительно
заряженных ионов Zn2+ по сравнению с исходным, а около
медного электрода образуется недостаток ионов SO2~. Ре-
Результатом различия заряда ионных растворов будет ионный
141
Cu+2+SO~2
Рис. 13.1. Схема
ческого элемента
гальвани.
ток, т. е. перемещение отрицательно заряженных ионов
SO|~ от медного электрода к цинковому по электролити-
электролитическому проводнику I. Таким образом, этот односторонне
направленный процесс A3.1) характеризуется непрерыв-
непрерывным увеличением концентрации ионов цинка и уменьше-
уменьшением концентрации ионов меди в растворе.
Итак, согласно схеме избыточные заряды, создающие
на цинковом электроде отрицательный потенциал ц>т, пере-
перемещаются по металлическому проводнику к медному элек-
электроду, имеющему относительно более положительный по-
потенциал ф„. Если сопротивление внешней цепи гальвани-
гальванического элемента очень велико, то перемещения электронов
практически не происходит и разность потенциалов между
электродами
фл — фш = Лф A3.4)
называют электродвижущей силой (ЭДС), а потенциалы
Фи и Фя—электродными потенциалами. Химические источ-
источники электрического тока, подобные рассматриваемому,
называют гальваническими элементами, а части гальвани-
гальванического элемента I и III — полуэлементами.
Уравнение Нернста. Работа химической реакции A3.1),
лежащей в основе гальванического элемента, может быть
определена двумя способами. Один из них основан на
знании разности потенциалов и количества электричества,
прошедшего во внешней цепи за время действия элемента
где е~ — заряд электрона; JVA—постоянная Авогадро; п —
число электронов, участвующих в окислительно-восстано-
окислительно-восстановительном процессе; F—постоянная Фарадея. Работа элек-
электрического тока равна
A = nF (Асркон — Дфисх), A3.5)
Дфисх и Дфкон—соответственно исходная и конечная раз-
разности потенциалов гальванического элемента в стандартных
условиях при концентрациях электролита в полуэлементах
в 1 кмоль/м3.
С другой стороны, работа химического процесса в галь-
гальваническом элементе от некоторых конечных значений
концентраций ионов Czn*+ и Сси»+ ДО некоторых исходных
Czn'+ и Сси'+ равна убыли энергии Гиббса:
^. A3.6)
142
Если в качестве стандартизации для исходного состояния
ввести условие Czn'+ — Ccu*+ = 1 кмоль/м3, а для конеч-
конечного—равновесное состояние, то уравнение A3.6) примет
уже известный вид:
дОт =—RT In ———. A3.7)
Очевидно, что работа химической реакции тождественна
работе, произведенной электрическим током, полученным
за счет этой реакции. Приняв, что максимальная работа
химической реакции равна убыли энергии Гиббса, т. е.
— А = AG°, получаем
AGt=— RT In f+=nF Аф1 — nF Лф2. A3.8)
Если конечное состояние является равновесным (А(р{ = 0),
а исходное относится к условию С = 1 кмоль/м3, то для
стандартных условий находим
л^Дф2 или AGt = — nF Дф2) A3.9)
откуда, пользуясь данными таблиц для расчета AG^g,
можно вычислить разность стандартных (нормальных)
электродных потенциалов любой окислительно-восстано-
окислительно-восстановительной реакции.
Уравнение A3.8) можно записать и иначе, памятуя, что
В этом случае получаем
RT
In С'Сиг+, A3.10)
отсюда для каждого из электродов величина электродного
потенциала может быть выражена уравнением
RT
4>Zns 4> +
о RT
4)Cu>+/Cu= ФС +
здесь и далее верхний индекс у С опускаем.
Если в этих уравнениях сделать подстановку числен-
численных значений констант и Г = 298 К, то уравнение A3.10)
N3
примет вид
RT
0,059
l
A3.11)
к второму
электроду
Уравнения типа A3.11) для любого электрода получили
название уравнений Нернста. Они характеризуют потен-
потенциалы соответствующих
электродов, абсолютные
значения которых из-за
неопределенности значе-
ния Ае в уравнениях
A3.2) и A3.3) определить
невозможно, тогда как
определение разности по-
потенциалов возможно и име-
имеет наиболее упрощенный
вид, если электрод сравне-
сравнения является нормальным
(т. е. концентрация элект-
электролита в нем равна
1 кмоль/м3, Г = 298 К и
/>=1013 гПа).
Стандартный водород-
водородный электрод. В электро-
электрохимии принято определять потенциалы различных полу-
полуэлементов относительно нормального водородного элект-
электрода, химическая схема которого
A3.12)
Рис. 13.2. Схема нормального водо-
водородного электрода
а потенциал в стандартных условиях условно принят рав-
равным нулю, т. е. Фн+/н2=0. Конструкция нормального
водородного полуэлемента схематически изображена на
рис. 13.2. Основой нормального водородного полуэлемента
(или нормального водородного электрода) служит пластина
черненой платины, которая погружена в 1 М раствор
кислоты (H2SO4 или НС1). Через раствор непрерывно про-
пропускают газообразный водород под давлением 1013 гПа.
Мелкодисперсная платиновая поверхность электрода обес-
обеспечивает хорошую адсорбцию на ней водорода, в резуль-
результате чего между электродом и раствором устанавливается
равновесие A3.12). Такой нормальный водородный электрод
является международным электродом сравнения, относи-
относительно которого определяют потенциалы всех остальных
электродов (полуэлементов).
144
Если раствор испытуемого полуэлемента имеет кон-
концентрацию 1 кмоль/м3 и измерение проводят при стандарт-
стандартных условиях, потенциал такого электрода по отношению
к нормальному водородному называют нормальным или
стандартным электродным потенциалом (ф°). В прило-
приложении VI приведены значения некоторых стандартных
электродных потенциалов ср° в вольтах (В).
Следует более подробно остановиться на вопросе о зна-
значениях и знаках электродных потенциалов. Рассмотрим
реакцию взаимодействия металлического лития с 1 М кис-
кислотой:
[Li]+{H+}aq = {Li+}aq + V2(Hs) A3.13)
ДО°298, кДж 0 0 —293,8 О
Для процесса A3.13) AG;98 = —293 800 Дж; исходя из
соотношения A3.9), находим Дф° = 293 800/( 1 • 96 487) =
= 3,04 В.
С другой стороны, окислительно-восстановительная
реакция A3.13) может быть представлена алгебраической
суммой двух полуреакций:
{Li + },q+e-=[Li] + aq, <p°u+/u =-3,04 В
= °.° В
Отсюда Дф° = фн+/н, —<Pu+/Li = —<Pu+/u=3,04 В. Этим
способом можно вычислять значения нормальных электрод-
электродных потенциалов.
Используя уравнение Нернста A3.11), можно рассчи-
рассчитывать значения Дф° любой окислительно-восстановитель-
окислительно-восстановительной реакции или гальванического элемента, вычитая из
электродного потенциала окислителя электродный потен-
потенциал восстановителя. Согласно уравнению Нернста один
и тот же электрод при различных концентрациях ионов
имеет различное значение потенциала, поэтому возможны
чисто концентрационные гальванические элементы. Напри-
Например, гальванический элемент, составленный из двух мед-
медных полуэлементов
<Pcu*+/Cu = °.34 B
но с различными концентрациями электролита в каждом,
будет иметь потенциалы:
. 0,059
Ф (Р
145
. 0,059
ФСи«+/Си=Фси!1+/СиТ 2~
Разность потенциалов Л<р или ЭДС этого концентрацион-
концентрационного гальванического элемента равна
, „ _0,059 С"си,+
Ф&1«+/Си ~ lS~. •
т. е. зависит только от концентраций полуэлементов, но
не от природы электродов.
К изложенному материалу приводим решение несколь-
нескольких типовых задач.
Задача 1. Гальванический элемент построен из никелевого н нор-
нормального водородного электродов
ЭДС гальванического элемента равна 0,309 В. Определить концентра-
концентрацию ионов Ni2+.
Вспомним, что ЭДС элемента по отношению к нормальному водо-
водородному электроду равна потенциалу второго электрода. Следовательно,
потенциал никелевого электрода в этом элементе
{Ni2+}aq + 2e- = [Ni] + aq, tpN1,+/N, =-0,309 В
Стандартный электродный потенциал никелевого электрода ф
= — 0,25 В. Из уравнения Нернста находим
+ 0'25_. „
lgCNl2+ оТ0295 М295
следовательно, CNi2+ = 10-2 кмоль/м3.
Задача 2. Вычислить константу равновесия окислительно-восста-
иовительной реакции вытеснения водорода цинком:
[Zn] + 2 {HCl}aq = {ZnCl2}aq + (НО
Эта реакция может быть осуществлена в гальваническом элементе,
построенном из двух нормальных электродов — водородного и цин-
цинкового:
= 0,0 В
-0-76 в
{}q + ( ф = 0,76 В
Используя соотношение
Р С
H%Zn'"t"
= — Hi In л, где л =
находим
„ _nF Дф° _ 2 • 96 487 • 0,76
'ёЛ-^^зоз— J9.14-298 '
откуда /С = 102в, т. е. реакция вытеснения водорода цинком практи-
практически необратима.
Задача 3. Определить произведение растворимости i-Agb если
известно, что потенциал полуэлемента, состоящего из серебряного
146
электрода, погруженного в раствор KI, равен —0,151 В. Концентра-
Концентрация 1~-нонов равна 1 кмоль/м3, и в этом растворе взмучен осадок Agl.
Для решения этой задачи необходимо определить концентрацию
ионов серебра в растворе, получившемся за счет реакции растворения
малорастворимой соли:
[AgI] + aq = {Ag+}aq + {I-}aq, LAgI =CAg+C,_ .
Используя условие задачи и взяв из таблиц значение нормального
потенциала серебряного электрода <PAg+/Ag = 0>80 В, по уравнению
Нернста находим
~
или CAg+ = 10-16 кмоль/м3.
Следовательно, произведение растворимости
Выше были рассмотрены гальванические элементы
с металлическими электродами, которые в работе хими-
химически взаимодействуют с электролитом. Однако окисли-
окислительно-восстановительные реакции в растворах можно
проводить так, чтобы перераспределение зарядов происхо-
происходило без химического взаимодействия раствора с материа-
материалом электрода. Так, окислительно-восстановительная
реакция
может быть разделена на две полуреакции:
}+2e-={Sn2+}aq + 6{Cl-}aq, Ф1=0,154В A3.14)
}aq+2e-=2{Fe2+}aq, ф°2 = 0,771 В A3.15)
Дфо = ф;_ф°=0,771—0,154 = 0,617 В.
Представим себе, что каждая из двух полуреакций
осуществляется в отдельном сосуде, как это показано на
рис. 13.3. При прохождении реакции A3.14) в сосуде I
освобождающиеся электроны должны скапливаться на
химически инертном Pt-электроде и увеличивать его отри-
отрицательный потенциал до определенной величины cpl.
Напротив, прохождение реакции A3.15) в сосуде II
способствует потере электронов с Pt-электрода и восста-
восстановлению ионов Fe3+. При этом электрод приобретает
положительный потенциал ц>1.
Если растворы в сосудах I и II соединить электро-
электролитическим III, а платиновые электроды—металличе-
электроды—металлическим IV проводниками, то в результате движения электро-
электронов из области с более высоким отрицательным потенциалом
147
в область с более низким возникает электрический ток,
работа которого в нагрузке V будет представлять собой
работу окислительно-восстановительной реакции. При этом
через электролитический проводник III из сосуда II
в сосуд I передвигаются ионы хлора С1~, необходимые
для образования комплексных ани-
анионов SnClj^.
Потенциалы <pi и ц>\ полуре-
полуреакций A3.14) и A3.15) опреде-
определяют, как и в рассмотренных ра-
ранее случаях, относительно потен-
потенциала нормального водородного
электрода. Потенциал каждого
электрода в этом гальваническом
элементе вычисляют с помощью
уравнения Нернста (концентрации
неравновесные):
! = —0,154
0,059
С
SnCli
Рис. 13.3. Схема гальвани-
гальванического элемента без уча-
участия материала электро-
электродов в окислительно-вос-
окислительно-восстановительной реакции
2 = +0,771 -\
0,059
Работа такого гальванического
элемента может быть вычислена
из соотношения A3.7) (концентра-
(концентрации равновесные). В стандартных условиях
1 К -
& * С ~
nF0,6V
#298
2-96 487-0,617
8,31-298
т. е. /Се = 1021, что характеризует реакцию взаимодейст-
взаимодействия SnCl2 и FeCl3 как идущую практически до конца.
Таким образом, любую электролитическую окисли-
окислительно-восстановительную реакцию можно использовать
в гальваническом элементе. Каждая из них может быть
разделена на полуреакции, которым при стандартных
условиях соответствуют строго определенные нормальные
окислительно-восстановительные потенциалы. В приложе-
приложении VI приведены значения таких потенциалов для наи-
наиболее известных окислительно-восстановительных полу-
полуреакций. Используя эти данные, можно решать различные
задачи, связанные с реакциями окисления-восстановления,
примеры которых приведены ниже.
Электрохимический анализ окислительно-восстановительных
реакций.
148
Задача 1. Можно ли вытеснить медь из раствора медного купо-
купороса CuSO4-5H2O металлическим железом?
Реакция вытеснения меди железом из медного купороса
[Fe] + {Cu*+}aq = [Си] + [Fe*+]aq
может быть представлена двумя полу реакциями:
-°.44 в
Pcu* +/cu = 0,34 В
Отсюда находим
Энергия Гиббса Д029в реакции при температуре 298 К
ДО298 = — 2 • 96 487 • 0,78 = — 150 520 Дж/моль.
Следовательно, данная реакция экзоэргическая и должна самопроиз-
самопроизвольно идти слева направо. Поскольку ДОт =—RT In К.с> можно
определить максимальную глубину протекания реакции, вычислив
величину константы равновесия
150 5 20
4'298 = 102в.
Задача 2. Можно ли в кислом водном растворе с помощью МпОГ
окислить ион NOJ до NO^"?
Записываем две полуреакции:
}, ф2 = 0,84В
Сумма этих полуреакций дает окислительно-восстановительный процесс
q q
+ 3 {Н2О}
с разностью потенциалов Дф = фх—ф2=1,51—0,84 = 0,67В. Работа
этой реакции
ДG298 = — я^Дф = 2• 96 487-0,67 = — 129,3 кДж/моль.
Экзоэргический эффект определяет самопроизвольность реакции. Из
соотношения Д029в = — RT 1п Кс находим значение константы равно-
равновесия— Кс= 1023, которое свидетельствует о том, что реакция идет
практически до конца.
Важным условием протекания окислительно-восстановительных
процессов в растворах служит рН раствора. В качестве примера
можно привести реакцию взаимодействия перманганата с соляной
кислотой
2{MnO4-}aq+16{H + }aq+10{Cl-}aq=5(Cl2)-f2{Mn2+}aq-f8{H2O},
A3.16)
которая идет только в достаточно концентрированных кислых раство-
растворах, т. е. при рН < 7:
}, Ф1=1,51В
=5{Cl-}aq, фа=1,36В
149
в область с более низким возникает электрический ток,
работа которого в нагрузке V будет представлять собой
работу окислительно-восстановительной реакции. При этом
через электролитический проводник III из сосуда II
в сосуд I передвигаются ионы хлора С1~, необходимые
для образования комплексных ани-
анионов SnCl|~.
Потенциалы <р! и q>°2 полуре-
полуреакций A3.14) и A3.15) опреде-
определяют, как и в рассмотренных ра-
ранее случаях, относительно потен-
потенциала нормального водородного
электрода. Потенциал каждого
электрода в этом гальваническом
элементе вычисляют с помощью
уравнения Нернста (концентрации
неравновесные):
0,059
lg
Рнс. J3.3. Схема гальвани-
гальванического элемента без уча-
участия материала электро-
электродов в окислительно-вос-
окислительно-восстановительной реакции
<р2 = +0,771
Работа такого гальванического
элемента может быть вычислена
из соотношения A3.7) (концентра-
(концентрации равновесные). В стандартных условиях
1 к —
nFo,6i:
#298
2- 96 487-0,617
8,31-298
_
' *
т. е. Кс*=10г1, что характеризует реакцию взаимодейст-
взаимодействия SnCl2 и FeCl3 как идущую практически до конца.
Таким образом, любую электролитическую окисли-
окислительно-восстановительную реакцию можно использовать
в гальваническом элементе. Каждая из них может быть
разделена на полуреакции, которым при стандартных
условиях соответствуют строго определенные нормальные
окислительно-восстановительные потенциалы. В приложе-
приложении VI приведены значения таких потенциалов для наи-
наиболее известных окислительно-восстановительных полу-
полуреакций. Используя эти данные, можно решать различные
задачи, связанные с реакциями окисления-восстановления,
примеры которых приведены ниже.
Электрохимический анализ окислительно-восстановительных
реакций.
148
среды, Лф° = 0,44В:
2МпОГ+20Н - + SOl~ = МпОГ + SOf" + Н2О
Поскольку ионы гидроксила в этой реакции являются
одним из исходных веществ (слева от знака равенства),
усиление щелочности среды будет способствовать протека-
протеканию процесса.
Глава 14
Прикладная электрохимия
Электролиз. Потенциалы поляризации, перенапряжения и разложения.
Законы Фарадея. Химические источники тока. Электрохимическая
коррозия и защита от нее.
Ранее рассмотрены окислительно-восстановительные
процессы, протекающие в гальваническом элементе, когда
на электродах элемента возникает электродвижущая сила.
Возможен и обратный процесс. Если подобную систему
(два электрода, помещенные в раствор электролита) вклю-
включить во внешнюю цепь постоянного тока, то на электро-
электродах будет протекать обратный окислительно-восстанови-
окислительно-восстановительный процесс, в котором:
а) положительно заряженный электрод (анод) прини-
принимает электроны от восстановителя, который сам при этом
окисляется;
б) отрицательно заряженный электрод (катод) отдает
свои электроны окислителю, восстанавливая его.
Схематически эти процессы показаны на рис. 14.1,
где анод электрохимической цепи является окислителем,
а катод—восстановителем. Окислительно-восстановитель-
Окислительно-восстановительный процесс, осуществляемый за счет энергии электриче-
электрического тока, называют электролизом.
Потенциалы поляризации, перенапряжения и разложе-
разложения. Рассмотрим в качестве примера систему из двух
платиновых электродов, помещенных в водный раствор 1 М
хлористого никеля (рис. 14.2). Если к электродам на этой
схеме приложить извне небольшую разность потенциалов,
то в первый момент в цепи появится ток, который можно
зарегистрировать измерительным прибором. Однако через
короткий промежуток времени он прекращается в резуль-
результате поляризации электродов. Суть этой поляризации
состоит в следующем. Под действием внешнего электри-
151
ческого поля положительно заряженные ионы никеля дви-
движутся к катоду и разряжаются на нем по схеме
выделяя следовые количества металлического никеля.
Одновременно с этим процессом отрицательно заряженные
Рис. 14.1. Общая схема
электролиза (объяснение
в тексте)
Рис. 14.2. Электролиз
водного раствора хло-
хлористого никеля
движутся к аноду и разряжаются на нем
(CU)
ионы хлора
по схеме
2{Cl-}aq-2e-
покрывая анод тончайшей пленкой адсорбированного газо-
газообразного хлора. В результате платиновые электроды
поляризуются, т. е. превращаются в никелевый и хлорный
электроды, стандартные потенциалы которых фм«+/м =
= — 0,25 В и фс!,/с!- = +1.36 В.
Разность потенциалов суммарного процесса поляризации
выражают величиной
ЛФ°С12/К!=<РС1г/С1--ф№^/№=1.36В-(-°.25) В =1,63 В.
Это означает, что по прошествии короткого времени,
когда на платиновых электродах появляются заметные
следы металлического никеля и адсорбированного моле-
молекулярного хлора, возникшая встречная ЭДС поляризации
никель-хлорного элемента препятствует дальнейшему про-
прохождению электрического тока от внешнего источника.
Как показывает опыт, электролиз раствора хлористого
152
никеля становится возможным лишь в случае, если напря-
напряжение Лфрзд внешнего источника тока будет существенно
больше ЭДС поляризации Афплр.
Экспериментально определяемая величина Афр3л, мини-
минимально необходимая для осуществления процесса электро-
электролиза, получила название разности потенциалов разложе-
разложения. Разница между значениями потенциалов разложения
и поляризации названа перенапряжением'.
Дфпнп = Дфрзл — Дфплр-
Явление перенапряжения сложное по своей природе и зави-
зависит от многих факторов: от материала электродов и харак-
характера их поверхности; агрегатного состояния веществ, выде-
выделяющихся на электродах; плотности тока и температуры.
В первом приближении явление перенапряжения можно
объяснить следующим образом. Каждое вещество в твер-
твердом и жидком состоянии обладает большим поверхностным
натяжением. В процессе образования при электролизе
газообразных веществ обязательна стадия появления газо-
газообразного пузырька, т. е. рождения новой поверхности
раздела газ—жидкость, что требует совершения работы
против силы поверхностного натяжения на этой границе.
В случае выделения твердых веществ, например металлов,
также появляется новая граница раздела, но уже между
двумя конденсированными фазами: металл—металл или
раствор — металл. Разность поверхностного натяжения на
такой границе значительно меньше, следовательно, и
работа, требуемая для ее создания, существенно меньше.
При выделении на электродах газообразных продуктов
величина Афпнп велика, и ее обязательно следует учиты-
учитывать в расчетах; при выделении металлов Афпнп мала,
и в первом приближении ею можно пренебречь.
Значения перенапряжения при выделении водорода
и кислорода в процессе электролиза (для малых плотно-
плотностей тока) приведены в приложении VII, они велики
и имеют тот же порядок, что и величины электродных
потенциалов, для расчета процессов электролиза их учет
обязателен. Однако значения Афпвп не могут быть строго
постоянными и зависят от температуры, интенсивности
перемешивания, плотности тока и других условий электро-
электролиза.
При электролизе водных растворов электролитов всегда
следует учитывать и собственную электролитическую дис-
диссоциацию воды
153
за счет которой при электролизе на электродах возможно
выделение газообразных водорода и кислорода:
Если в растворе присутствуют два и более электро-
электролитов, то в первую очередь на электродах будет проте-
протекать та окислительно-восстановительная реакция, для осу-
осуществления которой необходим меньший потенциал раз-
разложения. На примере электролиза смеси растворов мед-
медного купороса и серной кислоты можно выяснить вопрос
о возможности выделения меди на катоде без водорода.
Согласно принятой схеме разложения на платиновом катоде
выделяется медь:
Перенапряжение меди будем считать равным нулю. На
платиновом аноде в кислом растворе выделяется кислород:
{Н2О}-2е- = V. (О*) + 2 {H+}aq, ФОг/НгО = 1,23 в
Отсюда суммарный процесс поляризации
Дфплр = —0,34 +1,23 = 0,89 В.
Это означает, что для подавления поляризации с внешнего
источника на электроды нужно подать минимальную
Дфплр = 0,89 В. Согласно данным приложения VII пере-
перенапряжение кислорода на платине равно 0,5 В. Следова-
Следовательно, потенциал разложения
Дфрзл = Дфплр + Дфпнп = 0,89+0,5 =1,39 В.
Вычислим потенциал разложения второго компонента
этого электролита—серной кислоты, при электролизе
которой на катоде должен выделяться водород
2{H+}aq + 2e- = (Hs), ДФ°Н+/Н1=ОВ
а на аноде—кислород
ФО/но = !-23 В
Суммарный процесс поляризации Аф„лр
характеризуется разностью потенциалов, которая равна
—1,23 В. Потенциалы перенапряжения водорода и кисло-
кислорода на платине равны соответственно 0,3 и 0,5 В, сле-
154
довательно, потенциал разложения серной кислоты
= Дфплр + [Дфна+ДфЬ1]пнп= 1.23 + [0,3+0,5] = 2,03 В.
Это означает, что при напряжении внешнего источника
постоянного тока в пределах от 1,39 до 2,03 В на катоде
будет выделяться только медь, а при более высоких зна-
значениях напряжения—медь и водород. Точно также, под-
подбирая соответствующие напряжения внешнего источника
тока, производят раздельное выделение веществ, например
из раствора серебра и меди, кадмия и меди.
Если же потенциалы разложения смешанных электро-
электролитов близки между собой, то раздельное восстановление
их провести не удается. В подобных случаях подбирают
электроды с более выгодным Афп„п; либо изменяют рН
раствора (например, в аммиачной среде цинк образуется
без выделения водорода, а в кислой или нейтральной—¦
с выделением), либо используют комплексообразование,
которое приводит к изменению окислительно-восстанови-
окислительно-восстановительных потенциалов и т. д. Техника и теория электролиза
разработаны в настоящее время настолько, что иногда
есть возможность создать условия, необходимые для раз-
раздельного выделения компонентов сложных растворов.
Законы Фарадея. В заключение следует кратко оста-
остановиться на двух основных законах электролиза, сформу-
сформулированных в первой половине XIX в. английским физи-
физиком И. Фарадеем.
Первый закон Фарадея: массы веществ, выделяющихся
на электродах, прямо пропорциональны количеству про-
протекающего через раствор электричества. Математическая
форма выражения этого закона следующая:
Kq? A4.1)
где т—масса выделенного на электроде вещества; / — сила
тока; т—время прохождения тока; q—количество электри-
электричества; К—электрохимический эквивалент. При токе 1 А
за 1 с на электроде выделяется масса вещества:
Второй закон Фарадея: при пропускании через раст-
растворы различных электролитов равного количества электри-
электричества массы веществ, выделенных на электродах, пропор-
пропорциональны их химическим эквивалентам (Э). Отсюда сле-
следует, что отношение химического эквивалента к электро-
электрохимическому есть величина постоянная для любых веществ,
.155
ее называют постоянной Фарадея (F):
^ A4.2)
Таким образом, для восстановления или окисления на
электродах 1 моль любого вещества через раствор электро-
электролита следует пропустить ~ 96 500 Кл электричества.
Решив совместно уравнения A4.1) и A4.2), получаем
т = ^~ или 5=^. A4.3)
Поскольку известно, что валентность X элемента в хими-
химическом соединении связана с атомной массой А соотно-
соотношением
х А
получим еще одно важное для практических целей соот-
соотношение
. mFX
Л=—¦
Обычно, работая с растворами электролитов, эксперимен-
экспериментатор знает не массу вещества в растворе, а его концент-
концентрацию С. По определению, моль-эквивалентную концентра-
концентрацию выражают соотношением
где V—объем раствора; т — масса вещества.
Решая совместно уравнения A4.3) и A4.4), находим
время полного разложения вещества при электролизе:
__CVF
С помощью приведенных соотношений решают электро-
электрохимические задачи, имеющие большое практическое зна-
значение, в таких разделах, как химические источники
электрической энергии, защита металлов от электрохими-
электрохимической коррозии, гальванические покрытия, электрохими-
электрохимическая очистка воды, электрохимический синтез, электро-
электрохимическая обработка металлов.
Химические источники тока. Рассмотрим примеры прак-
практического применения электрохимии. Известно, что источ-
источником электрической энергии может служить любая окисли-
окислительно-восстановительная реакция. Однако практически
156
используют лишь некоторые более экономичные реакции,
дающие значительную разность потенциалов при наимень-
наименьшей поляризации и постоянство ЭДС во времени, позволяю-
позволяющие отбирать большие токи. Среди распространенных
в настоящее время гальванических элементов можно на-
назвать марганцово-цинковые, воздушно-цинковые, медно-
оксидные и оксидно-ртутные.
В воздушно-цинковых элементах отрицательным элект-
электродом служит металлический цинковый электрод, а в каче-
качестве активного вещества положительного электрода исполь-
используют кислород воздуха, который диффундирует через
пористый угольный электрод к поверхности раздела
электрод—электролит. В качестве электролитов исполь-
используются растворы NH4C1 или NaOH. В случае щелочного
электролита в гальваническом элементе протекает следую-
следующая окислительно-восстановительная реакция:
[Zn] + Vi (O2) +2{NaOH}aq = {Na2Zn02}aq + {H2O}
В медно-оксидных элементах (МОЭ) отрицательным
электродом служит цинк, а положительным—оксид меди,
электролит—щелочь. При работе такого элемента метал-
металлический цинк окисляется, а оксид меди восстанавливается:
[Zn] + [CuO] + 2 {NaOH}aq = {Na2Zn02}aq + [Си] + {H2O}
В оксидно-ртутных элементах отрицательным электро-
электродом служит амальгамированный порошок цинка, а положи-
положительным—оксид ртути в смеси с графитом, электролит —
раствор КОН. Основным окислительно-восстановительным
процессом в этом элементе является окисление цинка
и восстановление оксида ртути:
= [ZnO]+{Hg}
В качестве гальванического элемента чаще всего исполь-
используют дешевый, стабильный и емкий марганцово-цинковый
элемент (МЦ). В нем один электрод представляет собой
металлический цинк, а другой—оксид марганца (IV),
электролит—раствор хлорида аммония. При разомкнутой
цепи на электродах устанавливаются электрохимические
равновесия:
q - =4 [МпО (ОН)]
При замыкании внешней цепи процессы, идущие на отри-
отрицательно заряженном цинковом аноде и положительно
заряженном, оксидно-марганцовом катоде, начинают идти
157
необратимо. Энергию суммарного процесса
+4 [МпО (ОН)]
направляют на совершение работы во внешней цепи (на-
(например, вращение якоря электромотора).
Все перечисленные выше гальванические элементы разо-
разового использования, что снижает их практическую цен-
ценность. Их общая особенность — необратимость процессов,
т. е. если частично или полностью использованные элементы
подключить к внешнему источнику постоянного тока и
пропустить через них ток в обратном направлении, то ни
одна из систем не возвратится в исходное состояние.
Существуют и обратимые гальванические элементы,
получившие название аккумуляторов. Пропуская через
разряженный аккумулятор ток от внешнего источника,
можно заставить реакцию идти в обратном, эндоэргическом
направлении, что и отличает аккумуляторы от обычных
гальванических элементов разового действия.
Среди наиболее распространенных в настоящее время
аккумуляторов можно назвать свинцовые (кислотные),
никель-железные, никель-кадмиевые (щелочные) и сереб-
серебряно-цинковые.
В готовом к работе (заряженном) свинцовом аккуму-
аккумуляторе анодом служит металлический губчатый свинец
(обратимый анод), катодом—оксид свинца (IV). Электрод-
Электродные процессы можно выразить уравнениями
[Pb] + {soj"}., = [PbSO4]+2e-
суммарный процесс—
При подключении разряженного аккумулятора к внешнему
источнику тока (зарядка) электрохимические процессы
полностью обращаются.
Кадмиево-никелевые(КНА) и железо-никелевые (ЖНА)
щелочные аккумуляторы имеют между собой много общего.
В этих аккумуляторах положительным электродом служит
NiO(OH), а отрицательным в ЖНА — железо, в КНА —
кадмий; электролит — раствор щелочи КОН. При разомк-
разомкнутой цепи на электродах устанавливается электрохими-
электрохимическое равновесие:
[Cd] + 2{ОН - }aq = [Cd(OH)a] + 2e~
2[
158
При замыкании внешней цепи идет суммарная окисли-
окислительно-восстановительная реакция
iO(OH)] + 2{H2O} = [Cd(OHJ] + 2[Ni(OHJ]
за счет энергии которой возникает в цепи и совершает
работу электрический ток. При зарядке разряженного
аккумулятора процессы на его электродах обращаются,
в результате вновь образуются металлический кадмий и
NiO(OH). Аналогичные процессы протекают на электро-
электродах и в ЖНА.
В менее распространенном серебряно-никелевом акку-
аккумуляторе катодом является Ag2O, анодом — губчатый цинк,
электролитом — раствор КОН. При разомкнутой внеш-
внешней цепи на электродах устанавливается равновесие:
[Ag2O] + {Н2О} + 2е~ = 2[ Ag] + 2{ОН - }aq
[Zn] + 2{OH - }aq = [Zn(OHJ] + 2e~
При работе аккумулятора, когда внешняя цепь замкнута
на нагрузку, в нем протекает суммарная окислительно-
восстановительная реакция:
[Ag2O] + [Zn] + {H2O} = 2[Ag] + [Zn(OHJ]
Наряду с преимуществами аккумуляторы обладают
рядом существенных недостатков—они дорогостоящие,
с небольшой удельной емкостью, источником пополнения
энергии в них может быть только электроток. Во многих
странах мира проводят исследования по созданию эле-
элементов, в которых бы рабочие вещества, участвующие
в реакции токообразования, непрерывно подавались извне
к электродам, а продукты реакции непрерывно отводились.
Такие источники электроэнергии получили название топ-
топливных элементов.
Примером топливных элементов может быть водородно-
кислородный элемент, в основе действия которого лежит
реакция окисления водорода:
(H2) + V2(O2) = {H2O}, AG;es = — 237,2 кДж A4.5)
При обычной температуре эта реакция не идет. Для ее
осуществления необходимы катализаторы, в качестве ко-
которых используют порошки платины, палладия, серебра
и оксидов некоторых металлов. Элемент состоит из порис-
пористых угольных электродов (содержащих катализаторы),
соединенных с резервуарами водорода и кислорода. Элек-
Электроды разделены раствором щелочи. Водород диффундирует
через пористый электрод с катализаторами (Pt и Pd), при
159
этом малоактивный молекулярный водород активируется:
(Нг) A4.6)
Кислород диффундирует через второй электрод, молекулы
О2 также активируются катализаторами (Pt, Ag или оксиды
металлов):
(О,)—.(О!) A4.7)
При разомкнутой внешней цепи на границе электродов
с раствором электролита устанавливаются электрохими-
электрохимические равновесия
A4.8)
,q. A4.9)
При замыкании внешней цепи в ней начинает идти элек-
электрический ток за счет протекания экзоэргической ре-
реакции A4.5), получающейся суммированием процессов
A4.6) — A4.9). Как видно из уравнения A4.8), электроны
генерируются на водородном электроде (аноде), откуда
они по внешней цепи перетекают на кислородный электрод
(катод), где и используются для осуществления реак-
реакции A4.9).
В настоящее время интенсивно разрабатывают и дру-
другие топливные элементы на основе более дешевых и кало-
калорийных видов топлива, таких, как оксид углерода, спирты
и различные нефтепродукты. В качестве окислителя кроме
кислорода используют фтор и хлор. Применение топлив-
топливных элементов в народном хозяйстве открывает большие
возможности на пути прямого превращения химической
энергии топлива в электрическую с высоким коэффициен-
коэффициентом полезного действия.
Электрохимическая коррозия. Образование гальвани-
гальванических пар в металлах и сплавах может носить само-
самопроизвольный характер и быть источником коррозии.
Под электрохимической коррозией подразумевают про-
процессы взаимодействия металлов и сплавов с электролитами
(водными растворами, расплавами солей и др.). Это взаимо-
взаимодействие может быть разделено на следующие самостоя-
самостоятельные процессы:
а) анодный процесс — растворение металла
160
б) катодный процесс—-связывание избыточных элек-
электронов, образующихся в анодном процессе каким-либо
веществом, именуемым деполяризатором. В обычных усло-
условиях деполяризаторами могут служить молекулы воды,
кислорода и др. В качестве примера процесса деполяри-
деполяризации можно привести процессы
Отношение металлов в электрохимической коррозии
определяется значениями их нормальных электродных
потенциалов, согласно которым все металлы делят на
4 группы:
А — металлы повышенной активности, от щелочных до
кадмия (ф°^—0,4 В), корродируют даже в нейтральных
водных средах без кислорода и окислителей;
Б — металлы средней активности, от кадмия до водо-
водорода (ф° = 0,0 В), устойчивы в нейтральных растворах
при отсутствии кислорода и не устойчивы в кислых средах;
В—металлы малой активности — висмут, медь, сурьма,
мышьяк, серебро, ртуть, родий (ср° от 0 до +0,8 В),
в отсутствие кислорода и других окислителей устойчивы
не только в нейтральных, но и в кислых средах;
Г—благородные металлы—золото, платина, иридий,
палладий—устойчивые во всех средах, кроме кислых,
в присутствии сильных окислителей.
Коррозия резко уменьшает сроки жизни металличе-
металлических изделий, что приносит огромный вред народному
хозяйству. С коррозией ведут непрерывную борьбу, в связи
с чем разработаны всевозможные методы защиты. Наибо-
Наиболее применимы защитные металлические (цинковые, хро-
хромовые, никелевые, свинцовые, алюминиевые и др.) и не-
неметаллические (азотированные, фосфатированные, силици-
рованные, лакокрасочные, пластмассовые и гумированные)
покрытия, а также протекторная защита металлов от кор-
коррозии и обработка коррозионной среды ингибиторами.
Протекторная защита заключается в том, что из двух
соединенных между собой и погруженных в электролит
металлов растворяться (т. е. подвергаться электрохимиче-
электрохимической коррозии) будет металл, имеющий более отрицатель-
отрицательное значение ф°. Так, если железо и цинк соединены
6 № 703 161
между собой и опущены в раствор электролита
ф'2п.+/211 = -0,76 В
(pFe,+/Fe = -0,44 В
[Zn] + {Fe2 + }aq = [Fe] + {Zn2+}aq, Д<р° = —0,44- (—0,76) = 0,32 В
то цинк будет вытеснять железо из раствора. Это озна-
означает, что пока есть металлический цинк, находящееся
с ним в контакте железо растворяться, т. е. корродиро-
корродировать, не будет. Вид защитного металлического покрытия
следует выбирать с точным учетом условий, в которых
будет находиться предохраняемый покрытием металл.
Из приведенного примера видно, что цинковое покрытие
железа — хорошая защита против электрохимической кор-
коррозии. Другое распространенное покрытие железа—оло-
железа—оловянное (лужение) — в тех же условиях будет способство-
способствовать усилению коррозии железа, действительно:
<p°Fe,+/Fe = -0,44 В
<Psn»+/Sn = -°>14 B
откуда
+}aq> Дф° = — 0,14 — (—0,44) = 0,30 В
Полученное значение Дф°=+0,30В для процесса вытес-
вытеснения олова и разрушения железа свидетельствует о том,
что этот процесс пойдет с большим выигрышем энергии:
AG° = —nFAq>° = — 2-96 487-0,30= — 57,9 кДж.
Теория и практика электролиза позволяют в настоя-
настоящее время широко использовать гальванические покры-
покрытия, электролиз расплавленных солей, электросинтез,
гидроэлектрометаллургию для создания плотных покрытий
одних металлов другими. Электрохимическим путем осу-
осуществляют также покрытия цинком, кадмием, медью, ни-
никелем, хромом, оловом, серебром, золотом и сплавами —
латунью, бронзой. Окислительные процессы, идущие при
электролизе на аноде, используют для электрополировки
металлов, анодно-механической обработки металлов и на-
нанесения прочных защитных оксидных пленок (оксидиро-
(оксидирование). Методом электролиза расплавленных солей в за-
заводских условиях получают металлический алюминий
(электролизом расплава Al2O3+Na3AlFfl); магний (из рас-
расплава MgCl2); натрий (из расплава NaOH или NaCl);
литий, калий, кальций, бериллий (из расплавов хлоридов)
162
и др. Важные для современной химической промышлен-
промышленности и галогенидной металлургии газообразные хлор и
фтор также получают электролизом расплавленных солей—
фторидов и хлоридов.
Электролизом водных растворов (гидроэлектрометал-
лургический путь) рафинируют медь, серебро, золото,
никель, кобальт, свинец, электроэкстрагируют цинк, кад-
кадмий, марганец, хром. Электролизом водных растворов
получают промышленные количества водорода, кислорода,
пероксида водорода и надсернокислых соединений, щелочи,
гилохлорита натрия, хлорной кислоты, перманганата ка-
калия, свинцовых белил, гидросульфата натрия. Большое
значение имеют электрохимические способы синтеза раз-
различных органических соединений.
Глава 15
Кинетика химических реакций
Константа скорости реакций. Молекулярность и по-
порядок реакций. Цепные реакции. Реакции в закры-
закрытых системах. Односторонние и двусторонние реак-
реакции. Параллельные и последовательные реакции.
Кинетика реакций в открытых системах. Реакторы
идеального смешения и идеального вытеснения.
Константа скорости реакций. Как было показано
в предыдущих главах, равновесное состояние химических
реакций остается неизменным во времени, как бы долго
ни шло наблюдение за системой. Напротив, подход хими-
химической реакции к состоянию равновесия осуществляется
во времени. Закономерности течения химических реакций
во времени составляют предмет химической кинетики. Одна
из задач химической кинетики состоит в количественном
описании скорости химической реакции, ее связи с при-
природой реагирующих веществ, температурой, влиянием
катализаторов (ускорителей) и ингибиторов (замедлителей).
Скорость химической реакции v определяют уменьше-
уменьшением концентраций реагирующих веществ АС за отрезок
времени Ах, т. е.
АС_
Дт
6» Ш
V = — ¦
Дт '
Выше, при описании химического равновесия на примере
взаимодействия
в качестве характеристики осуществимости прямой реак-
реакции СО + Н2О —>¦ было выведено соотношение, характери-
характеризующее число активных столкновений
К * = fWW RT ccoCH!o. 05.1)
а в качестве характеристики обратной реакции ¦<— СО2+
+ Н2 соотношение
2
Есо2+Ен,
К «= Рсо.Рн/ RT СсоСИг. A5.2)
Величины р, стоящие перед экспоненциальными членами,
получили название предэкспоненциальных коэффициентов.
Смысл их будет вскрыт ниже. Величины Е* представляют
собой значения энергии активации реагирующих веществ
(энергия активации прямой реакции равна сумме ?нго+
+ ?со, энергия обратной реакции—?со, + ?н,)- Энергия
активации—это энергия молекул при столкновении, уро-
уровень которой достаточен для осуществления химической
реакции, т. е. это энергия возбуждения сталкивающихся
молекул, необходимая для превращения их в промежу-
промежуточный, неустойчивый активированный комплекс, который,
экзотермически распадаясь, превращается в конечные про-
продукты реакции.
Число активных соударений молекул реагирующих
веществ в единицу времени по своей сути есть скорость
химической реакции, поэтому символ W*—число соударе-
соударений можно заменить на символ v—скорость химической
реакции.
Если концентрации реагирующих веществ Сн2о, Ссо и
Снг в момент времени т принять равными 1 кмоль/м3,
то выражения A5.1) и A5.2) примут вид
ьобр
Эти выражения получили название удельных скоростей
или констант скорости химической реакции:
Е*
k = Ae RT . A5.3)
164
Как видно из соотношения A5.3), константа скорости
химической реакции зависит от температуры процесса и
природы реагирующих веществ (Е*). В понятие природы
реагирующих веществ входят не только состав химиче-
химических соединений (формулы) и их относительные количества
(стехиометрические коэффициенты в уравнении реакции),
но также катализаторы и ингибиторы, т. е. вещества,
которые количественно в реакции не расходуются, но
влияют на ее механизм и тем самым на значение энергии
активации Е*. От Е* зависят значение константы скорости
реакции k и, следовательно, скорость реакции. Допустим,
что некоторая реакция идет при температуре 1000 К и
Е{ = 76 кДж, т. е.
Положим, что за счет введения в процесс катализатора
энергия активации уменьшилась вдвое, т. е. Е*2 = 38 кДж,
тогда
38 000
Соотношение kJk2=l0~*, это означает, что скорость реак-
реакции в присутствии катализатора будет в 100 раз больше,
чем без него.
П редэкспоненциальный множитель А в выражении
константы скорости реакции A5.3), согласно теоретиче-
теоретическому расчету, для простейших мономолекулярных реак-
реакций равен
А «Т Т
NAh
где Nд — постоянная Авогадро; R — газовая постоянная;
h — постоянная Планка; AS*— изменение энтропии реаги-
реагирующих веществ при их переходе в промежуточную, воз-
возбужденную форму, которая образуется в прямом процессе
и очень быстро разрушается, превращаясь в продукты
реакции.
Таким образом, предэкспоненциальный множитель А
имеет размерность порядка 1012—1014 и по своей природе
является температурно-энтропийным. Для некоторых прак-
практических расчетов можно использовать приближенное со-
соотношение
k
165
Следует отметить, что расчет скоростей химических
реакций зависит от степени удаленности системы реаги-
реагирующих веществ от равновесного состояния. Результи-
Результирующая скорость реакции уре8 представляет собой раз-
разность скоростей прямой vt и обратной и2 реакций:
ирез = vl — V2-
Если система реагирующих веществ далека от состояния
равновесия,' то vt^>v2 и ирез да vlt что и составляет основу
приведенных выше расчетов, которые соответствуют усло-
условию необратимости реакции.
Молекулярность и порядок реакции. Химические реак-
реакции можно классифицировать по числу молекул, участ-
участвующих в каждом элементарном химическом акте.
Мономолекулярными называют реакции, в которых
элементарный химический акт представляет собой хими-
химическое превращение одной молекулы за счет избытка
внутримолекулярной энергии. Примером может служить
термическая диссоциация оксида азота (V)
u = ftCN 0 (формально v -s- С1)
Такой способ описания характеризует реакции первого
порядка, так как показатель степени концентрации реаги-
реагирующего вещества в выражении для скорости реакции
равен единице.
Бимолекулярными называют реакции, элементарный
акт которых осуществляется при обязательном столкнове-
столкновении двух молекул, например, реакция омыления эфира
щелочью:
{CH3COOCH(CH3J}+{NaOH}aq->{CH3COONa}aq+{(CH3JCHOH}
Сг С2
v = АС,С2 (формально v -s- С2)
Такой способ описания характеризует реакции второго
порядка, так как сумма показателей степеней концентра-
концентраций в выражении для скорости процесса равна двум.
Тримолекулярными называют реакции, в которых эле-
элементарный акт осуществляется при одновременном столк-
столкновении трех молекул; примером подобных реакций слу-
служит взаимодействие
2(NO) + (O2)—*2(NO2)
v = kC^oCOa (формально v ¦+¦ С3)
166
Такой способ описания характеризует реакции третьего
порядка. Однако вероятность тройных соударений, т. е.
одновременное столкновение трех частиц, чрезвычайно
мала, поэтому в большинстве случаев имеют место реак-
реакции первого и второго порядков. Сложные же процессы
обычно осуществляются как совокупности последователь-
последовательных и параллельных моно- и бимолекулярных реакций.
Одна из наиболее распространенных теорий, объяс-
объясняющих механизм химических реакций,— это теория
активированного комплекса, или переходного состояния.
Активированным комплексом называют некоторую возбуж-
возбужденную конфигурацию связанных между собой атомов,
которая возникает в процессе данной реакции и отвечает
некоторому максимальному значению потенциальной энер-
энергии системы реагирующих веществ.
Допустим, что некоторым исходным веществам АХ и В
соответствует некоторый уровень потенциальной энергии.
Перестройка взаимного расположения частиц АХ и В
в прямом процессе сначала приводит к повышению уровня
их потенциальной энергии вплоть до некоторого предела Е*-.
где Е*АХ+В—энергия активации, отвечает тому взаимному
расположению частиц А, X и В, которое получило на-
название активированного комплекса А — X — В. Дальней-
Дальнейшие изменения в расположении частиц активированного
комплекса снижают потенциальную энергию системы вплоть
до стабильного уровня Екон:
Иными словами реакцию
АХ + В + ЛЯЭ = А + ВХ A5.4)
можно условно расчленить на две составные части:
АХ + В + Е* —•- \А'1 ! , где Е*=Еакт -ЕИСх.сост
Л
А'' 1
- Ej *- А + ВХ , где Ej-l
суммируя их, получаем
AX-f В+[?l—i'l]—* A-fBX A5.5)
167
Сравнение уравнений A5.4) и A5.5) приводит к выводу,
что
т. е. алгебраическая сумма энергий активации участни-
участников прямой и обратной реакций численно равна тепловому
эффекту А#° процесса A5.4). В качестве примера, иллюст-
иллюстрирующего эту схему, можно привести реакцию радика-
радикалов ОН:
О-О .
ОН + ОН—> ; .; ?1 = 121кДж
Н---Н
О-О
¦ •—*Н2+О2 Е2'=188кДж
Н-Н
?'—?¦;= 121 — 188 = —67 кДж = Л//°
Различные реакции су1цественно отличаются по энер-
энергии активации. Так, свободные атомы и радикалы (будем
отмечать их звездочкой), обладающие высокой химической
активностью, имеют минимальную энергию активации,
например:
35 кДж = Н2, Е* = 4 кДж
б — 343 кДж = С4Н,0> ?* = 4 кДж
NOC1* + C1* — 86 кДж=Ш+С12 ?* = 4 кДж
*—63 кДж=Н*+О2> ?*=17 кДж
—27 кДж = С2Н4 + Н2, ?*=29 кДж
При взаимодействии же молекул с валентно-насыщенными
связями энергия активации принимает большие значения:
Н2 + 12—10 кДж = 2Щ, ?* = 163 кДж
Цепные реакции. Особое место в химической кинетике
занимают цепные реакции, которые протекают чрезвычай-
чрезвычайно быстро и часто заканчиваются взрывом. Вначале за
счет энергии извне образуется небольшое число очень
активных частиц типа свободных атомов или радикалов,
а затем в процессе их взаимодействия с невозбужденными
молекулами вновь получаются свободные атомы или ра-
радикалы и т. д. Цепные реакции—это лавинообразные про-
процессы, в которых одни активные частицы порождают воз-
возрастающее число других активных частиц.
Химические превращения с цепным механизмом реак-
реакции чрезвычайно распространены в химии газообразных
веществ. Типичным примером таких реакций может слу-
168
жить взаимодействие водорода с хлором на свету. Смесь
этих двух газов в темноте может очень долго существо-
существовать, не реагируя. Если же эту смесь импульсно осветить
ярким светом (вспышка магния), то произойдет взрыв и
образуется хлористый водород. Первая стадия этого про-
процесса заключается в фотохимическом разложении на атомы
молекулы хлора под действием энергии излучения E — hv:
Эта реакция зарождения цепи служит началом последую-
последующего процесса развития цепи, в каждом звене которой
вновь возникают свободные атомы:
и так далее.
К цепным реакциям относят взрыв гремучего газа,
горение различных газообразных органических веществ
в кислороде, фторе, хлоре. Горение метана в кислороде
(СН4) + 2(О2) —* (СО3) + 2(Н3О), АН" = —802 кДж
на первой стадии описывают следующим механизмом:
локальный нагрев ^
зарождение цепи СН4 *-СН3+Н*
дальнейшее развитие цепи:
СН3ОО*+СН4 —* СН3ООН + СНз
СН3ООН —> СНзО* + ОН*
СН3О* + СН4 —* СН3ОН +СНз
ОН* + СН4—*СНд + Н2О н т. д,
Особенность цепных процессов заключается в зависи-
зависимости скорости реакции от размеров и формы сосуда,
в котором протекает процесс, поскольку для развития
цепи необходима некоторая минимальная протяженность
пространства. Если на пути развивающейся цепи оказы-
оказывается стенка прибора, то цепь обрывается и процесс
прекращается. Кроме того, скорость развития цепного
процесса зависит от давления газа и наличия в нем ча-
частиц постороннего вещества. Совокупность подобных осо-
особенностей служит основой существования верхнего и
нижнего пределов взрываемости газовых смесей. Наличие
таких пределов долгое время оставалось непонятным и
было объяснено лишь после разработки теории цепных
169
реакций. Большой вклад в разработку этой теории внесли
русские ученые и в первую очередь академик Н. Н. Семенов.
Поскольку цепные реакции многостадийны, для каждой
стадии характерны различная скорость и, следовательно,
различное значение энергии активации. В качестве при-
примера можно привести разветвленную цепную реакцию
взрыва гремучего газа:
2(Н2) + (О2) -^ 2(Н2О), ЛЯ° =—485 кДж
Первая стадия этого процесса — зарождение цепи
Н2 + О2 —у 2ОН*, El = 188 кДж
вторая стадия — продолжение цепи
ОН*+Н2—«.Н2О + Н*, ?*2 = 59 кДж
третья стадия — разветвление цепи
Н* + о2 —у ОН* + О* El = 80 кДж
О* + Н2 — ОН* + Н*. El = 38 кДж
и, наконец, обрыв цепи на молекулах инертного газа и
на стенке М аппарата
НО*+М, ?б = 0
?* = 67 кДж
Реакции в закрытых системах. Все варианты процес-
процессов, идущих в газовых или жидких, т. е. в гомогенных
средах, описывают методами кинетики гомогенных реак-
реакций. Процессы в гетерогенных системах, состоящих из
двух и более фаз, значительно сложнее. Примерами гете-
гетерогенных реакций могут служить процессы: восстановле-
восстановления оксидов
[FeO]-HCO]->[Fe] + (CO2)
(СО2) + [С]-+2(СО)
окислительного обжига
3[FeS2J + 8(О2) -+ [Fe3O4] + 6(SO2)
растворения в растворах электролитов
[СаСО31
и многие другие.
Особенность течения гетерогенных реакций состоит
в том, что в них большую роль приобретают: , ¦
170
а) процессы переноса исходных веществ в жидкой или
газообразной фазе из объема к границе твердой фазы и
в обратном направлении;
в) величина поверхности соприкосновения фаз, на ко-
которой происходят химические превращения;
в) химическая активность этой поверхности соприкос-
соприкосновения.
Скорость химической реакции между раствором (жид-
(жидким или газовым) и поверхностью твердого тела без учета
процесса переноса определяют в общем виде соотношением
где v—скорость реакции; k — константа скорости; S—ве-
S—величина активной поверхности твердого тела; С,-—концен-
С,-—концентрация реагентов в жидкой или газовой фазе.
Если скорость подвода реагента из объемов раствора
к поверхности твердого тела больше скорости самой реак-
реакции на этой поверхности, то считают, что реакция про-
протекает в кинетическом режиме. Если же, напротив, ско-
скорость подвода реагента меньше скорости реакции на
поверхности, то реакция протекает в диффузионном режиме.
Односторонние и двусторонние реакции. По кинетиче-
кинетической обратимости химические реакции классифицируют
как односторонние и двусторонние. Односторонними на-
называют реакции термодинамически необратимые (горение
пороха) или обратимые, но при рассматриваемых условиях
далекие от состояния химического равновесия (окисление
водорода кислородом принципиально обратимо, но равно-
равновесие практически полностью смещено в направлении
образования воды вплоть до температур порядка 1500—
2000 К). Двусторонними называют реакции, заканчиваю-
заканчивающиеся достижением химического равновесия, т. е. проте-
протекающие в условиях, когда скорости прямой и обратной
реакций становятся сравнимыми.
В общем случае результирующую скорость реакции
vpea определяют разностью скоростей прямой г/ и обрат-
обратной у" реакций, т. е.
При условии v'^>v" Урез = у', что характерно для одно-
односторонних кинетически необратимых реакций, Реакции,
для которых выполнимо условие v' ~ v", т. е. имеет место
соизмеримость прямой и обратной скоростей, относят к
двусторонним или кинетически обратимым» ¦
171
Рассмотрим в качестве примера одностороннюю реак-
реакцию первого порядка, протекающую в закрытой системе:
А —»• продукты реакции
В этих условиях отсутствует поступление в систему ве-
вещества А, а его расход определен скоростью реакции
v^-~ = k(C0-x) = kC, A5.6)
где k — константа скорости; Со — концентрация Априт = 0;
х—количество А, прореагировавшее в единице объема
к моменту времени т.
Преобразуя выражение A5.6) математически, при усло-
условии, что k не зависит от т и С, находим
A5.7)
o()
Из уравнения A5.7) получаем
In (Ср/С) _2,31g (С0/О
или
Эти уравнения позволяют определить значения k по экспе-
экспериментальным данным и использовать их для решения
основной задачи химической кинетики — расчета скорости
реакции и зависимости скорости от времени.
Часто для характеристики скорости реакции исполь-
используют время половинного превращения исходного вещества,
или период полураспада, значение которого находят, пре-
преобразуя выражение A5.8):
2,3 [lg Co —Ig (Ср/2)] _2,31g2_0,693
То.б Ъ ? ?-. A5.9)
Очевидно, что в реакциях первого порядка период полу-
полураспада не зависит от начальной концентрации Со и опре-
определяется только константой скорости реакции k.
Аналогичным путем математического анализа выраже-
выражения A5.6) можно найти среднее время жизни молекул:
*ср = т. A5.10)
Подставив выражение A5.9) в A5.10), получим
172
откуда следует, что среднее время жизни уменьшается
с ростом температуры.
Уравнение первого порядка характеризует не только
скорости бимолекулярных реакций, но и более сложные
процессы, которые протекают по схемам первого порядка.
Так, например, гидролиз этилацетата
СН3СООС2Н5+ Н2О —> СН3СООН + С2Н5ОН
как бимолекулярная реакция описывается уравнением
первого порядка из-за большого избытка воды в системе,
количество которой в процессе реакции практически не
изменяется.
Рассмотрим одностороннюю реакцию второго порядка
А + В —>¦ продукты реакции.
В этом случае выражение для скорости реакции запишется так:
0 = --^=*САСв, A5.11)
где СА и Св — концентрации компонентов А и В. Если исходные
концентрации (при т —0) этих компонентов Со и COr; x—-количе-
x—-количество молей А и В веществ, прореагировавших к моменту т, то изме-
изменение их концентраций за время т> 0 будет С^=С0 —х и Св =
= сов-х-
В этом случае уравнение A5.11) примет вид
V = k(C°A-X)(C°B~X>
откуда
2 3 (^оа~х) ^"А
c -с
Размерность к для реакций второго порядка—м3/(кмоль-с).
Если исходные концентрации компонентов А и В равны между
собой, то выражения A5.11) и A5.12) примут вид
откуда
или 7-=
1Лг Т' 7т^+
1 -f- кС0 Т С Ьо
Из уравнения A5.13) находим выражение
Я~ С0Ст '
которое удобно для экспериментального определения константы ско-
скорости реакции второго порядка. Период полурейкции второго порядка
для этих условий равен To.5==rj-. т. е. в отличие от реакций пер-
первого порядка т0.5 обратно пропорционален не только константе ско-
скорости, но и исходной концентрации веществ.
173
Следует отметить, что полученные соотношения справедливы
только для простейшей реакции: А + В —>- продукты. Реакции же типа
а А -\- рВ —> продукты
описывают уравнениями более сложными, чем A5.12), хотя в отдель-
отдельных случаях для них остаются применимыми и уравнения второго
порядка.
Рассмотрим односторонние реакции n-го порядка:
A -f- В + С + • • • —* продукты реакции
Скорость таких реакций при условии равенства начальных концен-
концентраций всех компонентов выражают уравнением
« = —^-=АО = А(С0-*)В( A5.14)
где п — число компонентов и характеристика порядка реакции. Ана-
Анализ уравнения A5.14) приводит к выражениям:
1 cj?~'—с»-1
~(«-1)т Сп0-1С"-1
2"-'—1
т
Эти уравнении справедливы для односторонних реакций n-го порядка
при любом значении п, как целом, так и дробном.
Перейдем к рассмотрению двусторонних реакций пер-
первого порядка, в которых имеет место равновесие
где одновременно протекают реакции — прямая
у' = k'C
и обратная
о" = k"C".
Выражение для результирующей реакции скорости при-
принимает вид
ирез — " и — j —к ^ «ь. ^ij.ioj
При условии, что начальные концентрации компонентов
А и В равны Со и Со соответственно, а количество про-
прореагировавшего в единице объема к моменту т вещества
равно х, получаем
,174
подставив эти значения в выражение A5.15), находим
Dpe3 = & Со — k"Cl — (k' + k") X.
Математический анализ этого уравнения приводит к вы-
выражению
In —, *'С°„~*"С° =(*' + k") x
k'C0-k"Cu-(k' + k")x
ИЛИ
In =A-= (*' + *") т.
X— X
где х—равновесное значение х.
Не останавливаясь на двусторонних реакциях второго
и n-го порядков, математическое описание которых еще
более сложно, остановимся на количественном описании
их типовых разновидностей, важных для понимания ха-
характера возможных процессов. Это параллельные одно-
односторонние реакции
А —
V.
примером которых может служить
—*¦ углеводороды
(СО + Н2) —
> спирты
к,
—> альдегиды
Точно так же при нитровании и сульфировании арома-
ароматических соединений могут получаться орто-, мета- и па-
пара-производные; при крекинге нефти — различные газо-
газообразные углеводороды и т. д. Примером последователь-
последовательных односторонних реакций
А — В — С
может служить процесс омыления эфиров двухосновных
кислот щелочью
COOR
*• I к,
Ri(COOR2J + 2NaOH-* Rt -fRuOH + NaOH —*¦
COONa
—* Rx (COONaJ -1-2R2OH
175
а также более сложная реакция гидрирования фенола до
циклогексанола
+ н.
он
где возможны различные комбинации констант скоростей:
&! и k2 соизмеримы по значениям, равны и несоизме-
несоизмеримы.
Во всех случаях кривая зависимости концентрации
промежуточного продукта во времени будет проходить
через максимум, но характер взаимного расположения
этих зависимостей существенно различается.
Кинетика реакций в открытых системах. Перейдем к рассмотре-
рассмотрению химических систем реакций в открытых системах, в которых
происходит обмен веществом с окружающей средой. Примером описа-
описания систем такого типа может служить кинетика реакций в реакто-
реакторах идеального смешения при постоянном объеме.
Пусть в реактор объема V подается непрерывно раствор с кон-
концентрацией реагентов Со, и объемной скоростью подачи раствора U.
С той же скоростью из реактора выводится раствор с концентрацией
реагентов С/, причем раствор так интенсивно перемешивают, что
концентрации С,- всех компонентов одинаковы в любом месте объема
реактора. Если в растворе протекает односторонняя реакция первого
порядка
то выражения для скоростей реакции по компонентам А и В при-
примут вид
"а=Тг(соа~Са)-*с'а- Ч = — (сов-Св)-*св-
Отсюда при СОв = Св = 0, Со = СА и в пределах от т = 0 до т > О
получаем для начальной, нестационарной стадии процесса
с
"-A
С течением времени в проточной системе устанавливается стационар-
стационарный режим, при котором СА и Св стремятся к некоторым постоян-
176
ным значениям
ucOk kvc
0A
так как выражения в скобках с ростом т быстро изменяются, стре-
стремясь к единице. Скорость реакции в установившемся состоянии про-
процесса будет равна:
U+kV '
Помимо реактора идеального смешения, в кинетике реакций от-
открытых систем рассматривают также модель реактора идеального
вытеснения, в которой реагирующая жидкость или газовый раствор
движется по трубкам, между которыми протекает теплоноситель для
охлаждения или нагревания реактора. Пусть объемная скорость раст-
раствора в трубке U; концентрации реагентов С{\ концентрации на входе
и выходе трубки Со и Ck соответственно; температура постоянна;
концентрация раствора изменяется при движении раствора по трубке,
длина которой / и площадь поперечного сечения S. Пусть в растворе
идет односторонняя реакция первого порядка
Если реакция проходит в газовом растворе без изменения числа
газовых молекул, то в реакторе идеального вытеснения скорость реа-
реакции описывают теми же уравнениями, что и в закрытых системах,
т. е.
Здесь Vq — приведенный объем газа, проходящего через трубку дли-
длиной / и сечением S, при заданных значениях р и Т.
Все изложенное свидетельствует о том, что даже в случае про-
простых химических превращений кинетическое описание их представляет
собой достаточно сложную задачу. Реальные же кинетические превра-
превращения в большинстве случаев представляют собой совокупности
параллельных и последовательных реакций, для каждой из которых
необходимо прежде всего рассчитать значения констант скоростей.
В этих расчетах физико-химическая природа констант
скоростей не вскрывается, их значения определяют из
данных эксперимента. Главная же задача кинетики —
вскрытие закономерностей связи константы скорости с тем-
температурой системы, строением реагирующих молекул, влия-
влиянием катализатора или ингибитора. Накопление факти-
фактического материала в химической кинетике, как и в других
разделах химической науки, является важнейшим инстру-
инструментом познания.
177
Глава 16
Катализ
Катализ и катализаторы. Катализ в одно- и мно-
многостадийных процессах. Гомогенно- и гетерогеиио-
каталитические реакции.
Катализ — процесс увеличения скорости химической
реакции при участии катализаторов. Катализаторами
могут быть вещества в состоянии атомов, молекул, ионов
или поверхности раздела фаз, которые взаимодействуют
с исходными химическими соединениями, резко изменяют
скорость реакции и выделяются на последующих стадиях
в химически неизменном виде. Вещества, которые не
ускоряют, а замедляют реакцию (увеличивают Е*), назы-
называют ингибиторами.
Катализ может быть гомогенным, если реагирующие
вещества и катализатор находятся в одной фазе. Приме-
Примером гомогенного катализа может служить реакция
которая идет при высоких температурах и резко уско-
ускоряется в присутствии небольшой примеси паров воды.
Катализ называют гетерогенным, если реагирующие
вещества и катализатор находятся в разных фазах и
имеют границу раздела, например процессы окисления
аммиака на платиновом катализаторе
и разложение пероксида водорода в присутствии твердого
оксида марганца (IV)
[МпО.1
{Н2О2} -*{H,O} + Vi(O,)
Для некоторых веществ термодинамически допустимо
не одно, а несколько направлений превращения. С по-
помощью специально подобранных катализаторов можно
ускорять одни и замедлять другие превращения. Так,
одно и то же вещество—этиловый спирт—в присутствии
оксидов алюминия или тория разлагается с образованием
этилена и воды
(С8Н6ОН) [А'г0"][ТЮ4] (СаН4) + (НаО), AG? = 45 600- Т 126,4
178
а в присутствии серебра и меди разлагается с образова-
образованием уксусного альдегида и водорода
(С2НЬОН)[Ag] [CuI (СНзСОН) + (Н„), ДО? = 68620-Г 11,2
Каталитическая активность различных катализаторов
может резко изменяться в присутствии некоторых веществ
иной химической природы, которые сами не являются
катализаторами, но резко увеличивают его каталитическую
активность—такие вещества называют промоторами или
активаторами. Так, каталитическая активность твердого
оксида V2O5 в отношении реакции окисления SO2 в сотни
раз увеличивается в присутствии сульфатов щелочных
металлов. Вещества, которые, сами не являясь катализа-
катализаторами, снижают их каталитическую активность, назы-
называют каталитическими ядами. Так, для процесса окисле-
окисления SO2 в сернокислотном производстве каталитическим
ядом является ничтожная примесь соединений мышьяка.
Впервые явление катализа было открыто в 1806 г.
Н. Клеманом и Ш. Дезормом в камерном процессе полу-
получения серной кислоты. Они установили каталитическое
действие оксидов азота на скорость окисления SO.,. В кон-
конце XIX в. промышленным методом получения серной
кислоты стал контактный способ, основанный на окисле-
окислении SO2 кислородом в присутствии платинового катализа-
катализатора. В настоящее время вместо дорогостоящих платиновых
катализаторов успешно работают оксидные смеси (напри-
(например, V2O5 с K2SO4). Каталитическим способом проводят
промышленный синтез аммиака (N,) + 3(H,) —> 2(NH3),
где в качестве катализатора используют железо, промо-
тированное оксидами алюминия и калия. Синтез азотной
кислоты осуществляют с помощью каталитического окис-
окисления аммиака в присутствии платинового катализатора.
Первым промышленным производством, в котором был
использован гетерогенный катализ, явился процесс Дикона
(получение хлора)
+2(Н2О)+2(С!2)
хорошо идущий в присутствии солей меди.
В промышленном синтезе газообразного водорода ис-
используют процессы
р
пользуют процессы
(СО) + (НаО) -¦ (СО2) + (На)
Ч» (О,) + (СН4) + (Н,О) —> (СО,) + 3 (НJ
179
в первом из которых катализаторами служат оксиды же-
железа, во втором—различные никелевые катализаторы.
Особенно большие успехи в деле промышленного ис-
использования катализа были достигнуты в процессах
органического синтеза. Каталитическая гидрогенизация
соединений с двойными связами; синтетическое моторное
топливо; крекинг нефти; десульфуризация нефтепродуктов;
синтез каучука, этанола и метанола, окиси этилена, изо-
пропилового спирта, ацетона, акролеина, дивинила,
изопрена, бензола, толуола; получение синтетических
волокон и других высокополимерных веществ; каталити-
каталитическая очистка технологических газов —вот далеко не
полный перечень продуктов, которые получают в про-
промышленном масштабе с использованием широкого ассорти-
ассортимента катализаторов.
Каталитические реакции бывают одностадийными
(слитными) и многостадийными, проходящими через по-
последовательные стадии, из которых одна является лими-
лимитирующей, т. е. стадией с наименьшей скоростью. Рас-
Рассмотрим слитный механизм каталитического процесса на
примере бимолекулярной реакции
А+ X ^± (А- X)* —+ В + X
где X — катализатор; (А — X)* — активированный комплекс.
Скорость этой реакции v — kCkCx, где Сх—концентрация
катализатора. Этот же процесс можно записать, опустив
переходное состояние (А — X)*,
А + Х—+В + Х.
Константа скорости такой каталитической реакции может
быть существенно больше константы той же реакции в
отсутствие катализатора из-за уменьшения энергии акти-
активации.
В случае двустадийной реакции
*1
A-f-X^itAX (первая стадия)
AX-J-B—>-AB-f-X (вторая стадия)
Если k[ > k2, то осуществляется первая стадия в режиме,
похожем на равновесный (двусторонняя реакция). Поэтому
ее можно охарактеризовать константой равновесия
180
Скорость второй односторонней стадии равна
v2 = k2CAXCB. A6.2)
Объединив выражения A6.1) и A6.2), получаем
Вещество АХ, которое иногда называют промежуточным
веществом Аррениуса, реально существует, его можно
экспериментально определить методами спектрального
анализа, ЭПР и др. Обычно концентрация этого вещества
мала (в этом случае мало значение константы k), что и
делает вторую стадию замедленной, лимитирующей. По-
Подобная форма записи многостадийной реакции является
достаточно общей, она обязательно включает произведение
такого числа констант, которое равно числу стадий. Рас-
Рассмотрим наиболее характерные особенности гомогенно- и
гетерогенно-каталитических реакций.
Гомогенно-каталитические процессы в газовой фазе
встречаются редко, так как газообразные катализаторы
почти неизвестны. Примером может служить процесс пи-
пиролиза ацетальдегида, катализируемый парообразным
иодом,
(СН3СОН) —> (СН4) + (СО)
В этом процессе катализатор снижает энергию активации
с 198 до 134 кДж/моль.
Гомогенный катализ наиболее распространен в ра-
растворах. В связи с большим числом конкретных примеров
гомогенно-каталитические реакции этого типа принято де-
делить на кислотно-основные и окислительно-восстанови-
окислительно-восстановительные с участием комплексных соединений. К кислотно-
основному катализу относят процессы изомеризации,
гидратации и дегидратации, гидролиза, этерификации,
алкилирования, деполяризации. В зависимости от типа
основания или кислоты эти реакции условно делят на
четыре группы:
1) специфический кислотный катализ ионами Н3О+;
2) специфический основной катализ ионами ОН"";
3) общий кислотный катализ (любыми кислотами);
4) общий основной катализ (любыми основаниями).
Если процесс, катализируемый кислотой или основа-
основанием, идет в растворе, то общая скорость реакции будет
равна сумме скоростей реакций, катализируемых соответ-
соответствующими катализаторами, а именно:
2^ = CiCii2fe<c< = ftC1Ciil A6.3)
181
где
(здесь k0—константа скорости некаталитической реакции;
остальные k—константы скоростей реакций, катализируе-
катализируемых соответственно ионами водорода, гидроксила и дру-
другими кислотами и основаниями). Для специфического
кислотного катализа уравнение A6.3) записывают так:
k = ko-\- kH + CH + , для основного—& = &о + ?он-Сон-- При-
Примером специфического кислотного катализа может слу-
служить реакция гидролиза сложного эфира:
первая стадия
О г о—Н 1+ ,
,, *' I I (кето-енольное
d Д г> d i н+ —i- Id r n d равновесие)
г\j —— \^¦—-\j—— *\2~т" ч L^i"~~ — *— ^2А
(Ct) к\ (С2)
вторая стадия (промежуточная)
О —Н "|+ Г О—<Н
+ Н2О-
третья стадия
Г о —Н + <*)
R1_C—О —]
6-Н
н
L
О
II
Ri—С —
С — 6-R2
6-Й
Н
(активи-
(активированный
комплекс)
—OH + H +
L
Первая стадия — быстрая, почти мгновенная, идет до
равновесия
=тФ-' A6.4)
вторая стадия — медленная, лимитирующая, для нее
A6.5)
подставив A6.4) в A6.5), получим
и2 = kzKCiC н+С Hz 0;
третья стадия, процесс распада активированного комплекса,
приводит к образованию продуктов реакции.
182
Перейдем к рассмотрению гомогенного катализа комп-
комплексными соединениями переходных металлов. При таком
катализе в присутствии комплексных катализаторов (чаще
всего катионов переходных металлов) осуществляют реак-
реакции восстановления и окисления, гидрирования и гидра-
гидратации, полимеризации и изомеризации. Примером может
служить метод промышленного окисления этилена до
ацетальдегида в водной среде в присутствии палладиевого
катализатора
С2Н4 + PdCli" + Н2О —* СНзСОН + 4С! - + 2Н + + Pd
Окисление образующегося металлического палладия осу-
осуществляется ионом Си2+
Pd + 4С1 - + 2Си2 + —> PdCll" + 2Cu +
и, наконец, Си+ окисляется кислородом воздуха до Си2+
2Cu+ +2H+ +»/2 О2 —>• 2Си2+ +Н2О
Этот процесс в промышленных условиях идет сначала
без доступа воздуха, образовавшийся ацетальдегид отго-
отгоняют, после чего ведут продувку воздуха. Если в этом
процессе заменить хлор на бром, скорость реакции воз-
возрастет в 17 раз; если вести процесс в уксуснокислой
среде, из эгилена образуется винилацетат. Приведенный
пример показывает, что, воздействуя на катализатор,
можно изменить не только скорость, но и химическую
схему каталитической реакции.
Остановимся на характеристике гомогенно-каталити-
гомогенно-каталитического ферментативного катализа, который осуществляется
при использовании биологических катализаторов—фермен-
катализаторов—ферментов, представляющих собой природные белки, входящие
в состав тканей. Ферментативный катализ является осно-
основой управления сложных жизненных процессов в расте-
растениях и животных организмах. Так, фотосинтез, брожение,
дыхание, пищеварение, синтез белков, сокращение мышц
являются каталитическими процессами, использующими
в качестве катализаторов различные ферменты.
Среди других видов каталитических реакций фермен-
ферментативный катализ является самым высокоорганизованным,
поскольку ферменты отличаются высокой избирательно-
избирательностью, специфичностью и каталитической активностью.
Ферменты—это высокомолекулярные белки, состоящие из
различных аминокислот, связанных пептидными связями.
Нативная конформация молекулы фермента образует ак-
активный каталитический центр, содержащий полярные
183
COOH, NH2, NH, OH, SH, а также гидрофобные группы,
способные ориентировать молекулы реагирующих веществ
в определенном положении по отношению к активному
центру. В состав активного центра многих ферментов
входят ионы металлов, причем при удалении иона металла
из металлофермента последний теряет каталитические
свойства. Каталитическая активность ферментов имеет
максимум на шкале рН, в сильнокислых и сильнощелоч-
сильнощелочных средах она, как правило, не проявляется. Катали-
Каталитическая активность ферментов наиболее оптимальна при
температуре от 20 до 40° С, при 60 — 70° С происходит их
денатурация. Активные центры имеют строго определенную
структуру, что позволяет ферменту присоединять только
молекулы определенного строения. Так, например, фер-
фермент уреаза гидролизует карбамид CO(NH2) в 1014 раз
быстрее, чем ион водорода, и не оказывает влияния на
реакции гидролиза других родственных карбамиду соеди-
соединений. В настоящее время известно около тысячи фер-
ферментов, одни из которых катализируют только окислитель-
окислительно-восстановительные процессы, другие—реакции с пере-
переносом групп, третьи — реакции гидролиза и т. д.
Гетерогенно-каталитические процессы занимают особое
место в кинетике, они протекают на границе раздела фаз
твердое тело — газ, твердое тело—жидкость. Эти процессы
широко используют в промышленной практике. В табл. 16.1
приведены примеры таких каталитических процессов и
катализаторов.
Преимущество гетерогенно-каталитических процессов
перед гомогенным катализом объясняется большим удоб-
Таблица 16.1. Примеры промышленных каталитических
процессов
Название
процесса
Окисление
Синтез
Гидрирование
и дегидрирова-
дегидрирование
Каталитическая реакция
NH3—>NC
so2 —>. so3
CO + H2O-
CH3OH —>
N2+3H2 —
СО + 2Н2-
\ /_
/ "~ \ "*"
-*СО2 + Н2
СН2О
+ 2NH3
-* СН3ОН
-* чсн не/
— / ^\
Катализатор
pt
V2O5) V2O4
Fe, Fe3O4
Ag, Ag2O
Fe, Mo, Os
Cu, ZnO, Zn~Cr2Os
Оксиды и сульфиды
переходных металлов
184
ством гетерогенных катализаторов, легкостью их отделе-
отделения от реагирующих веществ. Важнейшая характеристика
гетерогенного катализатора—величина его активной по-
поверхности. Часто катализаторы получают нанесением
активной формы на пористый носитель (трегер) с высоко-
высокоразвитой поверхностью. В качестве таких носителей при-
применяют активированный уголь, силикагель, оксид хрома (III)
и др. Многие катализаторы получают осаждением из ра-
растворов в виде гидроксидов (Fe2O3, Cr2O3, A12O3, ZnO и
др.) с последующей термической обработкой. Повышение
температуры обычно приводит к увеличению скорости ге-
терогенно-каталитического процесса. Гетерогенный катализ
более сложен в плане теоретического описания, чем го-
гомогенный. Если в гомогенном катализе катализатор нахо-
находится в молекулярном состоянии, которое можно строго
описать термодинамически, то в гетерогенном катализе,
как правило, неясно, что принимать за молекулярную
единицу катализатора и как охарактеризовать состояние
молекул, находящихся на границе с твердой фазой ка-
катализатора. Поэтому для физико-химического описания
различных стадий гетерогенно-каталитического процесса
часто прибегают к условным понятиям и приближенным
моделям.
Большая роль в гетерогенном катализе принадлежит
процессам адсорбции—физической адсорбции и хемосорбции.
Физическая адсорбция является результатом межмолеку-
межмолекулярного взаимодействия между частицами (атомами, ио-
ионами, молекулами) поверхностного слоя твердой фазы и
молекулами газовой фазы или раствором. Хемосорбция
(химическая сорбция) завершается химическим взаимодей-
взаимодействием адсорбированного вещества с поверхностью твердой
фазы. Адсорбирующее твердое вещество называют адсор-
адсорбентом; вещество, которое адсорбируется,—адсорбтивом.
Адсорбция—экзоэргический процесс, сопровождающийся
ростом концентрации упорядоченности адсорбтива на по-
поверхности адсорбента. В табл. 16.2 приведены значения
тепловых эффектов хемосорбции. Величину адсорбции (Г),
т. е. концентрацию веществ на адсорбирующей поверхно-
поверхности, измеряют в молях на м2.
Гетерогенно-каталитические процессы идут через не-
несколько стадий: например, процесс
A6.6)
протекающий на железо-оксидных катализаторах, можно
разделить на следующие стадии: подход молекул СО и
185
Таблица 16.2. Энергия хемосорбции газов на металлах,
кДж/моль
Газы-адсорбтивы
н2
о2
^2
со
NH3
Fe
—134
—314
-167
— 134
-368
Металлы-адсорбенты
Ni
—130
—545
—147
— 155
w
—188
—650
—398
—276
Н.2О к поверхности катализатора; проникновение их в
норы катализатора; физическая адсорбция и хемосорбция
молекул СО и Н2О; химическое взаимодействие молекул
СО и Н/), адсорбированных на поверхности катализатора;
десорбция молекул Н2 и СО2 и их отход от поверхности
катализатора. Для описания скорости реакции A6.6) ис-
используют уравнение
О, A6.7)
где k — константа скорости гетерогенно-каталитической
Е*
реакции, которая пропорциональна kaACe RT (здесь kaac —
константа адсорбции, Е*—энергия активации процесса).
Первичной стадией гетерогенно-каталитического про-
процесса является процесс адсорбции (т. е. увеличение кон-
концентрации реагирующих веществ), однако главная сущность
каталитического влияния заключена в химическом взаи-
взаимодействии реагирующих молекул с поверхностью катали-
катализатора по схеме
НаО-
с-о—н
I I
0-H
активирован-
активированный комп-
комплекс на по-
поверхности ка-
катализатора
Гетерогенный катализ—сложное явление, требующее
глубокого теоретического анализа. Наиболее распростра-
распространенные варианты теории были развиты в работах акаде-
академика А. А. Баландина (мультиплетная теория катализа)
и Н. И. Кобозева (теория активных ансамблей).
Строение
вещества
Учение о строении вещества (строение электронных оболочек атомов,
строение молекул, жидкостей, растворов, твердых веществ различной
природы) один из важнейших разделов теоретической
и экспериментальной химии, цель которого—вскрытие первичных
причин химических свойств и превращений. Составными частями
этого учения являются теория химической связи и теория
валентности, а практическими инструментами — приближенные
методы решения волнового уравнения Э. Шредингера—теория
валентных связей (ВС) и молекулярных орбиталей (МО).
Глава 17
Исторические предпосылки
теории строения
Планетарная модель Э. Резерфорда. Постулаты
Н. Бора и модель атома водорода. Строение мно-
гоэлектрониых атомов. Квантовые числа. Принцип
В. Паули. Правила В. М. Клечковского. Построе-
Построение периодической системы элементов Д. И. Мен-
Менделеева.
Первые представления о строении вещества заложены
в атомистике древних, а также «элементах» или «началах»
средневековых алхимиков. Современный этап начинается
с М. В. Ломоносова (XVIII в.), который дал определение
атомам как мельчайшим частицам химических элементов.
Началом новой эры в естествознании стало открытие
Д. И. Менделеевым в 1869 г. периодического закона,
устанавливающего зависимость изменения свойств хими-
химических элементов от их атомных масс. Вслед за этим, в
1886 г., А. М. Бутлеров высказал мысль о том, что атомы
неделимы лишь в известных нам химических превраще-
превращениях, но могут быть разделены в каких-то еще не изу-
изученных процессах. Прошло 10 лет и в 1896 г. А. Бек-
керель открыл радиоактивность, чем заложил первый
кирпич в фундамент современных представлений о слож-
сложной природе атома.
В 1900 г. немецкий физик М. Планк объяснил осо-
особенности распределения энергии в оптических спектрах.
Как известно, атомные спектры состоят из отдельных
спектральных линий (линейчатые спектры), каждая из
которых соответствует определенному уровню энергии и
характеризуется определенными значениями частоты элект-
электромагнитных колебаний v и длины волны %, связанных
соотношением % = —, где с—скорость света в пустоте.
Наиболее простые спектры зарегистрированы у атомов
водорода и ему подобных ионов (Не+, Li2+, Be2+ и т.д.)
Частоты электромагнитных колебаний спектральных ли-
линий описывают общей формулой
v 1 „ ,/ 1 1 \
188
где RH — постоянная величина (Ридберга); Z—заряд ядра
атома; и,-—некоторые целые числа, при подстановке по-
последовательных значений которых и получают серия соот-
соответствующих спектров.
Анализируя спектры, М. Планк предположил, что пе-
переход от одной спектральной линии к другой соответст-
соответствует изменению энергии, пропорциональной частоте
электромагнитных колебаний
?,- — ?ж = Д? = /п>, A7.1)
где h — коэффициент пропорциональности, получивший
название постоянной Планка, численно равной 6,6256х
X 10~34 Дж-с; Е; и Ек—значения энергии двух спектраль-
спектральных линий. Выражение A7.1) явилось количественной
основой гипотезы, согласно которой атомы воспринимают
и излучают энергию квантами, мельчайшими, далее неде-
неделимыми порциями hv, и которая стала базовой в разви-
развитии учения о строении вещества.
В результате фундаментальных исследований в области
развития учения о строении атомов химических элементов
были открыты и количественно охарактеризованы элемен-
элементарные частицы, обладающие массой покоя,— электроны,
протоны и нейтроны. В 1891 г. английским физиком
Дж. Стонеем был введен термин электрон, обозначавший
единичный электрический заряд, а в 1897 г. Дж. Томсон,
изучая катодное излучение в трубке Крукса, доказал,
что оно представляет собой поток отрицательно заряжен-
заряженных частиц. В 1909 г. Р. Малликен установил заряд
электрона, равный 1,60210-10~19 Кл (масса электрона
9,1091-Ю1 кг, размер 10~15 м). Каналовое излучение
в аналогичных опытах представляло, как было установлено
немецким физиком Е. Гольдштейном A886), потоки поло-
положительно заряженных частиц, заряды которых были
кратны заряду электрона или равны ему, но противопо-
противоположны по знаку, а масса совпадала с массой атома водо-
водорода A,67252- 107 кг). Эти частицы были названы про-
протонами (Дж. Томсон, В. Вин). В 1932 г. Дж. Чедвик
при изучении ядерных реакций открыл нейтральную час-
частицу с массой 1,67474-10"*' кг, которая была названа
нейтроном.
Планетарная модель Резерфорда—Бора. Первая модель строения
атома была предложена в 1904 г. У. Томсоиом. Согласно этой модели
положительный заряд равномерно распределяется по всему объему
атома и нейтрализуется электронами, вкрапленными в этот объем.
Но уже в 1911 г. Э. Резерфорд экспериментально установил у атомов
наличие ядер и предложил планетарную модель, согласно которой
189 .
электроны вращаются вокруг положительно заряженного ядра, как
планеты около Солнца, Однако и модель Резерфорда осталась внут-
внутренне противоречивой. По законам электродинамики электроны, дви-
двигающиеся по криволинейным путям, должны непрерывно излучать
энергию и в результате упасть на ядро.
Этот принципиальный недостаток модели в 1913 г. был устранен
датским физиком Н. Бором, который распространил квантовый закон
М. Планка на планетарную модель атома, предположив, что электрон
в атоме не должен излучать энергию, если он движется по некоторым
строго определенным стационарным орбитам. Каждой такой орбите
соовтетствуют строго определенные значения энергии Е±, Е%, Е3 н т. д.,
Рис. 17.1. Спектр неко-
некоторых электронных пере-
переходов в атоме водорода
2000
4000 6000
8000
причем разность энергий двух соседних стационарных орбит должна
быть равной кванту A7.1), переход электрона с более высокой стацио-
стационарной орбиты на более низкую должен сопровождаться излучением
кванта электромагнитных колебаний строго определенной частоты.
Такая модель строения электронной оболочки атома качественно соот-
соответствовала виду атомных спектров, а для атома водорода давала
точное совпадение. Эта взаимосвязь между видом спектра атома водо-
водорода и характером переходов электрона с одного стационарного уров-
уровня на другой схематически изображена на рис. 17.1. Следует отметить,
что Н. Бор разработал модель лишь в отношении атома водорода —
простейшего из элементов, электронная оболочка которого содержит
одни электрон, а орбита электрона по этой модели имеет форму
окружности.
Согласно классической механике Ньютона движение частицы по
круговой орбите характеризуется моментом количества движения ,, ,
M — mevr,
где /Ц — момент количества движения; те — масса электрона; о—ско-
рость движения электрона; г—радиус орбиты. По предположению
Н. Бора, по каким-то причинам существуют устойчивые стационарные
круговые орбиты атома водорода, для которых должно выполняться
условие
_ JL М—п— " 07 2)
mevr— 2я — 2я' .. '
190
Двигаясь по этим орбитам, электрон не излучает энергию. В этом
уравнении коэффициент п, получивший название квантового числа,
принимает только целочисленные значения натурального ряда чисел
A, 2, 3, ...). По своему физическому смыслу квантовое число п опре-
определяет номера стационарных круговых орбит электрона в атоме водо-
водорода, движение по которым не связано ни с потерей, ни с приобре-
приобретением энергии. Анализируя уравнение A7.2), можно заключить, что
каждой стационарной орбите атома водорода характерны дискретные
значения произведения vr—радиуса на скорость. Самая близкая к
ядру первая орбита электрона (п=\) в атоме водорода получила на-
название основной, все остальные — возбужденных. На основной орбите
электрон при отсутствии внешнего воздействия пребывает неограни-
чено долго; на возбужденных — лишь около 10~8 с, после чего само-
самопроизвольно -перескакивает на незанятую, более близкую к ядру ор-
орбиту, излучая квант энергии в виде «пакета» электромагнитных ко-
колебаний строго определенной частоты.
Выражение A7.2) позволяет вычислить значения радиусов стацио-
стационарных орбит в атоме водорода, скорость движения и энергию элек-
электрона. Для такого расчета необходимо дополнительное уравнение, по-
получаемое из условия равновесия силы электростатического притяже-
притяжения электрона к ядру Ft и центробежной силы F2:
*-=?. (.7.4)
где е — элементарный заряд; е0 —абсолютная диэлектрическая прони-
проницаемость вакуума. Приняв условие равенства
/?1 = /72, A7.5)
после подстановки уравнений A7.3) и A7.4) в A7.5) получаем
A76)
Решая систему уравнений A7.2) A7.6), находим скорость электрона
на стационарных круговых орбитах в атоме водорода:
Подставив численные значения констант, входящих в это выражение,
получаем, что
A,602- Ю-19J
2п6,626-10-34-8,854-10-12
_
Следовательно, на первой, самой близкой к ядру орбите скорость
электрона равна 2185 км/с — величине, составляющей 0,73% от ско-
скорости света.
Решая совместно уравнения A7.2) и A7.7), получаем выражение
для радиуса электронной орбиты в атоме водорода;
A7,8)
лтее2
После подстановки значений констант получаем г = 0,529- 10~*°л2 м.
С учетом этого первая стационарная орбита в атоме водорода (п= 1)
181
равна 52,9 пм, вторая (л = 2) — 52,9• 2а = 211,6, третья — 52, 9-За =
= 476 пм и т.д., т.е. по мере удаления от ядра расстояния между
стационарными орбитами делаются все больше. Предложенная модель
позволяет вычислить значения энергии электрона на различных ор-
орбитах в атоме водорода. Как известно, полная энергия системы Е
представляет собой сумму кинетической Яптн и потенциальной Екяа
энергий;
Сделав подстановку уравнений A7.4) и A7.8) в A7.9), получаем
meei
Используя уравнение A7.10), вычисляем значение энергетическо-
энергетического кванта электромагнитного излучения:
AE = hv. A7.11)
При поглощении кванта энергии происходит перескок электрона
с основной орбиты на возбужденную, или же квант излучается при
переходе электрона с возбужденной орбиты на более глубокую. С по-
помощью уравнения A7.10) получаем выражение для кванта возбужде-
возбуждения электрона:
A7.12)
nt /
Энергия (квант энергии), необходимая для ионизации или полного
отрыва электрона от невозбужденного атома водорода, определена
условием п = оо и п=1, тогда уравнение A7,12) принимает вид
/пее4
Д?,,ониз=
4/i%0
Отсюда для процесса ионизации атома водорода
(*VгД^-иоииз ***" {Н )~Г^~
находим энергию или потенциал ионизации
_ 9,109-Ю-" A,602-Ю-19L
Дж,
что соответствует 13,60 эВ. Экспериментально найденное значение
этой величины 13,595 эВ указывает на соответствие теории с экспе-
экспериментом.
Другая проверка теории Бора заключалась в расчете спектраль-
спектральных линий водородных атомов и сравнении вычисленных спектров
с экспериментально полученными. Переход к спектрам, т. е. к часто-
частотам электромагнитных колебаний, осуществляем сравнением уравне-
уравнений A7.11) и A7.12):
1
1 tii
192
\ / 1 1 \
) = ^h(-2 2 1
где Ян — константа Ридберга, которая при подстановке численных
значений постоянных в уравнение A7.13) равна 3,2898-1015 с .
Обычно Ян выражают не в единицах частоты v, а в волновых чис-
числах v, связанных между собой соотношением
v = г v,
где с—скорость света в пустоте. Исходя из этого получаем другое
значение для константы Ридберга: Ян = 10967757,6 м. Экспери-
Экспериментально из спектров было найдено очень близкое к вычисленному
значение (Ян = 10967758,1 м), что свидетельствует о строгости тео-
теории Бора в отношении строения электронной оболочки атома водорода.
Строение многоэлектронных атомов. Описание многоэлектронных
атомов химических элементов с позиций теории Бора было проведено
в 1916—1925 гг. А. Зоммерфельдом и другими исследователями.
Сложность многоэлектронных атомов потребовала учитывать взаимо-
взаимодействие электронных орбит и заставила предположить, что помимо
круговых орбит в атоме могут быть также и эллиптические орбиты.
Эллиптическим орбитам, эквивалентным круговым, соответствуют
более высокие уровни энергии. Движение электронов по эллипсам
характеризуется так называемым угловым моментом количества дви-
движения Мг, который может принимать только строго определенные
значения, пропорциональные h/2n:
a, A7.14)
где / — орбитальное, или побочное, квантовое число, принимающее все
значения натурального ряда чисел от 0 до (п—1), т.е. всего п зна-
значений. В этом случае общий момент количества движения М можно
представить в виде суммы моментов:
М = М„ + М[. A7.15)
Здесь Мп — момент количества движения, относящийся к круговой
орбите A7.9), Mi — угловой момент количества движения. Подставив
значения этих величин в уравнение A7.15), получим
где п —главное квантовое число. Каждому значению главного кванто-
квантового числа п соответствует п значений побочного квантового числа /:
=2
1 = 0, М = А-3;
2я
1=2, м=Аз+|.^=А
7 № 705 193
Следует отметить, что в соответствии с существующей термино-
терминологией электроны, охарактеризованные значением / = 0, принято на-
называть s-электронами; 1=1—р-электронами; / = 2—d-электроиами;
1 = 3— /-электронами. Эти обозначения представляют собой началь-
начальные буквы английских названий серий спектральных линий атомных
спектров: sharp^—резкая; prinsipal — главная; diffuse — диффузия, раз-
размытая; fundamental — основная. Таким образом, каждому значению
главного квантового числа п в многоэлектронном атоме соответствует
несколько электронных орбит разной энергии (или разных подуров-
подуровней энергии).
Далее, в процессе развития спектральных исследований было
установлено, что при действии магнитного или электрического полей
на исследуемые вещества спектральные линии расщепляются. Эти
факты заставили учитывать взаимодействие магнитных полей элек-
электронных орбит в атоме между собой и с внешним магнитным полем.
Суть этого взаимодействия состоит в том, что движущийся по замк-
замкнутым орбитам электрон создает магнитное поле подобно тому, как
это имеет место в соленоиде. Это магнитное поле вращающегося элек-
электрона характеризуется магнитным моментом (я, взаимодействие кото-
которого с внешним полем определяет пространственное расположение
электронной орбиты в атоме, что связано с изменением энергии элек-
электрона. При делении величины магнитного момента электронной орбиты
па так называемое гиромагнитное отношение v = e/Bmec) получают
новую характеристику |x/v = ,1Jm, являющуюся проекцией углового
момента количества движения Mi на направление внешнего магнит-
магнитного поля. Поскольку энергия атома может изменяться только кван-
квантами, величина Мт принимает лишь строго определенные значения,
пропорциональные кванту действия:
Здесь nif—магнитное квантовое число, которое может быть равным
нулю и принимать положительные и отрицательные значения в интер-
интервале от 0 до ±1, т.е. всего 2/+1 значений. Из этого следует, что
каждому значению побочного квантового числа / соответствует 2/-f-1
электронных орбит, которые различаются пространственным располо-
расположением в атоме, например:
1 = 0 mi = 0 соответствует одной орбите,
/=1 /л^ = + 1, 0, — 1—трем орбитам,
1 = 2 тг = +2, -\-1, 0, —1, —2 — пяти орбитам и т.д.
Все рассмотренные выше движения электрона характеризуют его
поступательное перемещение в околоядерном пространстве атома. По-
Поэтому логично предположить, что частицы обладают некоторым собст-
собственным движением н, следовательно, собственным моментом количест-
количества движения. Грубой моделью такого движения может служить
вращение частицы вокруг собственной оси. Такая модель породила
и специфическое наименование внутреннего движения, происходящее
от английского слова spin — волчок, вращение. В соответствии с этим
внутренний момент количества движения микрочастиц называют спи-
спином (s). Понятие спина было введено в 1925 г. Дж. Уленбеком н
Г. Гаудсмитом, которые предположили у электрона наличие спино-
194
вого момента количества движения
и связанного с ним магнитного момента -я--я ¦ В уравнении A7.16)
коэффициент 1/2 представляет собой спиновое квантовое число
которое в зависимости от направления собственного вращения элек-
электрона может принять два значения: /л5 = -}-1/2 и tns=—1/2-
Как уже отмечалось, модель вращающегося электрона очень гру-
груба. Если следовать этой модели и произвести расчет, то скорость та-
такого вращения будет превышать скорость света б пустоте. Спин раз-
различных элементарных частиц не одинаков. Так, ms=±1/2 имеют,
кроме электрона, также нейтрино, мюоны и барионы. Спин фотона
равен нулю и единице. Спин я- и К-мезонов равен нулю. Все это
говорит о сложной природе спина. По значению спина все элементар-
элементарные частицы делят на два семейства: с целочисленным и полуцелым
спином. Понятие спина положено в основу фундаментального кванто-
во-мехаиического принципа естествознання, сформулированного В. Пау-
Паули, согласно которому взаимодействующие частицы микромира с по-
полуцелым спином не могут одновременно находиться в одном и том
же состоянии. Электроны, образующие оболочку атомов и являю-
являющиеся частицами с полуцелым спином, подчиняются принципу В. Пау-
Паули в следующей частной формулировке: в атоме не может быть двух
и волге электронов, движение которых можно характеризовать одина-
одинаковыми значениями всех четырех квантовых чисел.
Принцип Паули определяет емкость каждого электронного слоя
в атоме, т. е. набор электронов с одним и тем же значением главного
квантового числа. И действительно, каждому значению побочного
квантового числа / соответствует 21-\-\ электронных орбит. Каждая
орбита имеет максимальную емкость — два электрона (с противопо-
противоположными спинами). Следовательно, общее число электронов со значе-
значением / равно 2 B/—|— I). Как уже отмечалось, побочное квантовое
число / принимает значения от 0 до п—1; следовательно, для нахо-
нахождения общего числа электронов в слое необходимо величину 2 B/-J-1)
просуммировать по всем значениям /:
2 2«2. A7-17)
1 = 0
Это означает, что максимальная электронная емкость слоев или элек-
электронных уровней составляет:
первый слой — 2-12 = 2 электрона,
второй слой —2-22 = 8 электронов,
третий слой — 2-32 = 18 электронов,
четвертый слой —2-42 = 32 электрона.
Именно такое послойное строение электронных оболочек атомов
химических элементов и объясняет периодическое изменение их физи-
физических и химических свойств. Порядок заполнения электронами ста-
стационарных орбит определен условием стремления системы к миниму-
7* 195
му энергии. В соответствии с этим требованием осуществляются все
самопроизвольные, т. е. энергетически выгодные, процессы, причем
состояние, отвечающее минимуму энергии, является равновесным.
Анализируя уравнение A7.10), видим, что наименьшей энергии систе-
системы ?min соответствует наименьшее значение главного квантового чис-
числа п. Это означает, что заполнение электронных орбит в атомах
химических элементов должно происходить последовательно от орбит
с меньшим значением главного квантового числа к орбитам с большим
значением главного квантового числа.
13-
12-
11-
10-
9-
8-
7 -
6-
5-
4-
3-
2-
1-
—
s
1
¦г P
2
tz
s p d
V
? -
-CD —
\
s p d f
4
? -
¦ ? •-
s p d f g
'5'
CZ
a
—*-o
^ J I ^
s p d f g
1,,,,
С
о
о
о
a
0
.? р (i f g
, , , , ,
-*- Периоды
Рис
ней
17.2. Порядок заполнения электронами энергетических уров-
атомов различных элементов
Однако выше было показано, что энергия электрона в атоме за-
зависит не только от главного квантового числа л, но и от побочного
квантового числа /. Поэтому фактическая последовательность заселе-
заселения электронных оболочек подчинена более сложному правилу, сформу-
сформулированному советским ученым В. М. Клечковским в 195! г. Эта
закономерность получила название (п-{-1)-правила, его формули-
формулируют следующим образом:
1. Заполнение электронных орбит атомов с увеличением заряда
их ядер (атомного номера) происходит последовательно от орбит с
меньшим значением (« + /) к орбитам с большим значением этой сум-
суммы (стремление системы к минимуму энергии).
196
2. При равных значениях (п-\-!) для ди\ х орбит заполнение их
происходит в последовательности значений главного квантового чис-
числа п, от меньшего к большему (и здесь стремление к минимуму энергии
при условии большего вклада п по сравнению с /).
Построение периодической системы элементов. Используя прави-
правило Клечковского, перейдем к объяснению расположения элементов
в периодической системе Д. И. ЛЪенделеева (рис. 17.2). Элементы I
периода периодической системы характеризуются значениями щ = \,
/1 = 0 (s-орбита) и, следовательно, П1~1Л = 1. Согласно уравнению
A7.17) максимальное число элементов в 1 периоде будет равно двум.
Этими двумя элементами являются водород и гелий.
Элементы 11 периода характеризуются значениями п.2 — 2; /t = 0
(s-орбита); /2=1 (/7-орбита), отсюда
Это означает, что заполнение электронных орбит осуществляется в
последовательности от я2+^ = 2 к п2 + /2 = 3. Согласно уравнению
A7.17) максимальная емкость электронных орбит элементов II периода
равна восьми Bп2 = 2-22 = 8). Так появляются элементы:
li Be В С N О F Ne
Элементы III периода характеризуются значениями л3 = 3; /j = 0
(s-орбита); /а = 1 (р-орбиты); /3 = 2 (d-орбиты) и, следовательно,
Сравнивая элементы III и II периодов, видим, что и тем, и другим
свойственна сумма п + / = 3. Однако, согласно пункту 2 правилч
Клечковского, значение главного квантового числа п элементов II
периода меньше и поэтому заполнение электронами орбит этих эле-
элементов энергетически более выгодно.
Следует также отметить, что III период содержит 8 элементом,
тогда как максимальная емкость электронных орбит для п = 3 состав-
составляет 2я2 = 2-32= 18 электронов. Это означает, что из 18 электронных
вакансий 10 не используются. Чтобы установить причину этого, рас-
рассмотрим элементы IV периода, для которых n4 = 4; /t = 0 (s-орбита);
/2=1 (р-орбита); /3 = 2 (d-орбита); /4 = 3 (/-орбита) и, следовательно,
2 = 4+ 1=5,
В первой строчке этой последовательности n4 + /i = 4, тогда как
в предыдущей'—п3 + /3 = 5. Это означает, что заполнение ь-орбиты
в четвертом электронном слое «4 + /i = 4 энергетически более выгодно,
чем заполнение d-орбиты в третьем электронном слое п3 + /3 = 5.
Именно поэтому в III периоде число элементов (от Na до Аг) равно 8,
а не 18. Однако в IV периоде после заполнения s-орбиты я4 + ;г = 4
197
начинается заполнение пропущенных d-орбит яя-|-/д = 5 в третьем
слое. Так появляются в IV периоде элементы первой вставной декады
от Sc до Zn.
Таким же образом в V периоде периодической системы появля-
появляются элементы второй вставной декады от Y по Cd; в IV периоде
сначала появляются 14 /-элементов лантаноидов (за счет заполнения
/-вакансий в 4-м электронном слое) и только после этого элементы
третьей вставной декады от Ш до Hg A-й d-элемент, La, появляется
до элементов /-семейства, т. е. до лантаноидов). Точно так же в VII
периоде между s- и не существующими р-элементами (т. е. в проме-
промежутке между Ra и Ки) появляются /-элементы за счет заполнения /-
вакансий в пятом слое. Далее начинаются элементы четвертой встав-
пой декады, вторым представителем которой является курчатовий Ки,
синтезированный в Советском Союзе в лаборатории акад. Г. Н. Фле-
Флерова.
Более наглядно картина последовательного заполнения п-\-1
энергетических уровней электронами и, следовательно, построения
периодической системы элементов Д. И. Менделеева, представлена на
рис. 17.2. Здесь по оси ординат отложены значения « + /, а по оси
абсцисс — последовательные значения главного квантового числа (но-
(номера периодов) и соответствующие значения побочною квантового
числа / (s, p, d, /, . ..). Горизонтальные площадки соответствуют
иеличинам п-\-1 в каждом периоде, а соединяющие их стрелки ука-
указывают на последовательность заполнения соответствующих орбит
(подуровней), также отчетливо видна закономерность построения
периодической системы элементов в привычном для нас и принятом
но всем мире виде.
В заключение дадим краткую характеристику пределам периоди-
периодической системы элементов. Вопрос о Еерхнем пределе или начале
'•истемы по существу является вопросом о «нулевом», докодородном
элементе, заряд ядра которого равен нулю. Еще в 1920 г. Резерфорд
развил подобную идею, предположив существование частицы с массой,
близкой к массе атома водорода, с нулевым зарядом ядра, не имею-
имеющей никакой оболочки. По размерам эта частица должна быть близка
к ядру атома водорода A0~1-> м), обладать огромной проникающей
способностью и охотно взаимодействовать с ядрами атомов. Этой
гипотезой Резерфорд предвосхитил открытие нейтрона.
Чрезвычайно ннтересным является также вопрос о месте водорода
is периодической системе элементов. Электронная структура атома
водорода такова A электрон при максимальной емкости орбиты в 2
электрона), что он может относиться и к элементам I группы, про-
проявляя в соединениях некоторые свойства, сближающие его со щелоч-
щелочными металлами, и к элементам VII группы (галогенам). Однако
сходство его с элементами атих групп не полное, в силу чего ему
приписывают особое место в системе.
Точно так же положение элемента гелия в равной степени может
быть отнесено н к VIII группе (благородные газы) и ко II группе
(наличие в электронной оболочке атома двух электронов).
Вопрос о нижней (конечной) границе периодической системы
остается неясным и поныне. Однако следует отметить возрастающую
неустойчивость все более массивных ядер, а также неустойчивость
электронной оболочки атомов. Источником этой неустойчивости явля-
является «релятивистский эффект» возрастания массы электрона, скорость
движения которого в очень тяжелых атомах становится сравнимой
со скоростью света.
Глава 18
Волновая модель атома
Недостатки теории Бора—Зоммерфельда. Волны
Луи де Бройля. Принцип неопределенностей
В. Гейзенберга. Уравнение Э. Шредингера. Волно-
Волновые функции орбиталей aTosia водорода. Формы
орбиталей. Волновые функции многоэлектронных
атомов. Правило Ф. Хунда.
Планетарная модель атома Э. Резерфорда, Н. Бора,
А. Зоммерфельда позволяет создать качественную карти-
картину строения электронных оболочек атомов элементов пе-
периодической системы Д. И. Менделеева, объяснить атом-
атомные спектры, количественно рассчитать энергию электрона
в атоме водорода и объяснить эффект расщепления
спектральных линий атомов в магнитном и электрическом
поле. Однако, несмотря на отмеченные достоинства, в про-
процессе разработки и практического использования этой
теории обнаружились принципиальные недостатки, а
именно:
1) условие квантования момента количества движения
электрона в атоме не подчинено законам классической
механики и электродинамики и не доказывает наличие
стационарных электронных орбит;
2) невозможно точно рассчитать интенсивность спект-
спектральных линий, а также энергию электрона во всех ато-
атомах, кроме атома водорода;
3) математический аппарат не позволяет перейти к
расчету энергии химической связи в молекулах, даже
самых простых.
Основной причиной несостоятельности теории Бора —
Зоммерфельда явилось противоречие между микроскопич-
микроскопичностью объектов (электрон, атом) и способом их описания
(классическая механика, все законы которой отнесены к
большим телам или макрообъектам). Это противоречие
возникло потому, что в период создания теории Бора —¦
Зоммерфельда, только рождались ныне очевидные разли-
различия между механикой движения больших тел (макрообъ-
(макрообъектов) и малых, типа атомов, электронов, протонов (мик-
(микрообъектов), а также между механикой движения с малыми
и очень большими скоростями, сравнимыми со скоростью
света.
199
Известно, что механику, как единую науку о движе-
движении тел и взаимодействиях между ними, можно разделить
на три самостоятельных раздела.
1. Классическая механика, основанная на законах
И. Ньютона, т. е. механика макрообъектов, движущихся
со скоростями много меньше скорости света.
2. Движение частиц со скоростями, близкими к ско-
скорости света, рассматривают с позиции релятивистской
механики, на основе теории относительности и законов
А. Эйнштейна.
3. Движение микрочастиц (электронов, ядер, атомов)
рассматривает квантовая механика, основоположником
которой является М. Планк A900), авторы математиче-
математического расчета—Э. Шредингер, В. Гейзенберг, Л. де Бройль,
П. Дирак, М. Бори.
Принцип неопределенностей Гейзенберга. Одним из
важнейших положений квантовой механики следует на-
назвать соотношение неопределенностей В. Гейзенберга,
согласно которому невозможно с одинаковой точностью
определить и координаты движущегося в атоме электрона,
и его импульс (p = mv). Математическая запись этого
соотношения имеет вид
^А или ДддОяЭгЛ
A8.1)
где Ах—неопределенность положения движущегося элект-
электрона вдоль оси х (неточность определения его координат);
i\px — неопределенность проекции импульса электрона;
i\vx — неопределенность скорости электрона, движущегося
вдоль оси х; h — постоянная Планка; те — масса элект-
электрона.
Из соотношения неопределенностей следует, что чем
точнее вычислен импульс (&рх —* 0) или скорость (Avx —+ 0)
электрона в атоме, тем неопределеннее становятся его
координаты (Ах-+ оо). Таким образом, характеризуя орби-
орбитальный электрон в атоме точным значением энергии или
импульса, мы вносим очень большую неопределенность в
характеристику электронной орбиты, т. е. в данный мо-
момент времени можно говорить лишь о вероятности нахож-
нахождения электрона в различных точках околоядерного про-
пространства. Следует отметить, что из-за малого значения
постоянной Планка h эффект, вытекающий из соотноше-
соотношения неопределенностей, выявлен только у микрообъектов
(например, движущийся электрон). Что же касается мак-
макрообъектов, т. е. обычных, окружающих нас тел, то их
200
движение строго описывается законами классической ме-
механики.
Как отмечалось в гл. 17, электроны в атомах дви-
движутся со скоростями, составляющими заметную долю от
скорости света. Следовательно, для описания атомных
систем необходимым оказалось одновременное привлечение
и квантовой механики, и теории относительности. Слия-
Слияние двух важнейших разделов механики привело к рож-
рождению квантовой теории электромагнитного поля — кван-
квантовой электродинамики. Один из важнейших выводов
квантовой электродинамики — представление о двойствен-
двойственной природе быстродвижущихся микрообъектов, кото-
которые проявляют себя и как частицы (корпускулы), и как
волны. Такая двойственная природа впервые была уста-
установлена для света. Разрабатывая теорию света, ученые
первой половины XIX в. доказали, что он представляет
собой электромагнитные колебания и проявлениями его
волновой природы являются преломление, интерференция,
дифракция и др. Однако с позиций волновой природы не
удавалось объяснить открытый в 1889 г. А. Г. Столето-
Столетовым фотоэффект (испускание металлом или полупровод-
полупроводником электронов под действием света). Считалось, что
энергия электромагнитных колебаний накапливается по-
постепенно, по мере поступления, между началом освещения
и моментом вылета электрона должно проходить длитель-
длительное время. Опыт же показывал, что фотоэффект можно
наблюдать в момент освещения металла.
Для объяснения этого явления А. Эйнштейн A905)
предложил рассматривать свет не только как электромаг-
электромагнитные волны с энергией E = h\, но одновременно и как
поток частиц, фотонов, движущихся со скоростью света
(с) и обладающих массой т;. Энергия фотонов может
быть вычислена из соотношения
Е = т,с2. A8.2)
Особенность фотонов как частиц заключается в том, что
они не имеют массы покоя и существуют лишь при дви-
движении со скоростью света.
Таким образом, движущийся по атомной орбите элект-
электрон, испытав столкновение с фотоном, приобретает энер-
энергию кванта
и если этой энергии достаточно для полного отрыва его
от атома — возникает фотоэффект.
201
Решая совместно уравнения A7.11) и A8.2), получаем
выражение для массы фотона:
ni/ = hv/c2 A8.3)
или
A8.4)
Например, для желтого участка видимого спектра (л =
= 6• 10~7 м или v = 5-1014 с) масса фотона
6,626-!0-34-5-1014 „, .. ,.
т/= ЭТГО^ =3,7.10- кг,
т. е. масса движущегося со скоростью света фотона при-
примерно в 10? раз меньше массы электрона.
Волны де Бройля. В 1924 г. Луи де Бройль сделал
важное позже подтвердившееся предположение о двойст-
двойственной, т. е. одновременно волновой и корпускулярной,
природе любых движущихся вещественных частиц. Это
означает, что если в соотношениях A8.3) и A8.4) символом
т обозначить массу покоя любой частицы, а вместо ско-
скорости света поставить соответствующую ей скорость у,
далекую от скорости света, то получится соотношение
де Бройля
X = h/(mv) A8.5)
или
i=A, A8.6,
v tnv
где v и к—частота и длина волны де Бройля; т и v —
масса и скорость частицы.
Как было показано в гл. 17, скорость движения элект-
электрона в атоме водорода на самой близкой к ядру орбите
равна 2,185-10s м/с, масса покоящегося электрона
9,109-10~31 кг. Сделав подстановку этих величин в урав-
уравнение A8.5), получаем Я, = 0,332 нм. Радиус первой бо-
ровской орбиты гг в атоме водорода равен 0,0529 нм, а
длина этой орбиты 2яг! = 2-3,14-0,0529 = 0,332 нм. Сле-
Следовательно, в невозбужденном атоме водорода электрон-
электронная волна Я. в точности равна длине электронной орбиты.
Рассмотрим атом водорода в возбужденном состоянии.
Для этого найдем длины возбужденных электронных орбит
2лг1п2 = 0,332л2 нм A8.7)
202
и скорости электрона на них
vi 2,185-Ю6
Ц" = Т = п М/С
(здесь п—главное квантовое число). Используя последние
соотношения и уравнение A8.5), находим длины элект-
электронных волн возбужденных орбит:
?иП = А,„ = 0,332л нм. A8.8)
Для того чтобы определить, сколько раз длина волны
электрона уложится в длине электронной орбиты 2nr1n%i
разделим уравнение A8.7) на A8.8):
_ 0,332
_
Кхп ~~~kTn~~0M2n~n'
следовательно, длина электронной волны кп в общем слу-
случае п (целое число) раз укладывается в длине электрон-
электронной орбиты 2яг„:
2пгп = пк:1. A8.9)
Из курса физики (раздел колебания и волны) известно,
что уравнение A8.9) выражает условие образования стоя-
стоячей волны, которая при отсутствии сил трения является
устойчивой формой колебательного движения, происходя-
происходящего без рассеяния энергии. Следовательно, такие элект-
электронные орбиты должны быть неизменными во времени.
Если уравнения A8.5) и A8.9) решить совместно, иск-
исключив ?., то получим соотношение
h
являющееся математическим выражением основного посту-
постулата Н. Бора, утверждающего квантованность момента
количества движения электрона в атоме водорода
[(см. A7.4)]. Таким образом, признание волн де Бройля
A8.5), т. е. двойственной природы (волновой и корпус-
корпускулярной) электрона в атоме, позволило отчасти обосно-
обосновать планетарную модель атома Н. Бора, но главное
перейти к новой, более строгой квантово-механической,
или волновой, модели.
Следует отметить, что гипотеза де Бройля о двойст-
двойственной природе движущегося электрона в 1927 г. была
экспериментально подтверждена К. Девиссоном, Л. Джер-
мером, а также Дж. Томсоном и П. С. Тартаковским.
Они обнаружили дифракцию быстролетящих электронов
203
на кристаллической решетке металлов, которая похожа
на дифракцию рентгеновского излучения. В настоящее
время дифракцию на кристаллах и молекулах широко
используют для научных целей, а промышленность
выпускает серийный прибор — электронограф. Помимо
электронов явление дифракции обнаружено на быстро-
летящих нейтронах, атомах гелия, молекулах водорода,
так что справедливость идеи де Бройля не вызывает сом-
сомнений, хотя следует все же отметить, что волны де Бройля
не столько реальность, сколько хороший математический
прием, описывающий движение микрочастиц. Следующим
шагом в развитии волновой модели строения атома были
исследования В. Гейзенберга и Э. Шредингера.
Уравнение Шредингера. Согласно квантово-механиче-
ским представлениям Шредингера, одной из основных ха-
характеристик движущихся микрочастиц является волновая
функция г|э, по своей сущности отдаленно напоминающая
амплитуду пространственного волнового движения, кото-
которое можно графически или аналитически разложить на
три взаимно перпендикулярных направления х, у, z, т. е.
y = f(x, у, г).
В физике пространственное волновое движение описывают
дифференциальным уравнением
!$+l$+lw= 5*-*(*•*- *)• С8-10)
где г|)(дг, у, г) — амплитуда пространственной волны;
-K-j- и т. д. — вторые производные (т. е. производные произ-
производных) функции \Jj по переменным х, у и г; v — частота
колебаний; и—скорость распространения колебаний. Урав-
Уравнение A8.10) для краткости обычно записывают в виде
оператора Лапласа (V):
4jx2v2
V4= -г- Ч> (х, У, г). A8.11)
Если в уравнение A8.11) сделать подстановку выражения
волны де Бройля A8.6), то получим новое соотношение
^(*. У, г). A8.12)
Кинетическая энергия электрона в атоме равна разности
полной Е и потенциальной Епти энергии:
204
или
2me = (E — En-ta) = my. A8.13)
(Подставив уравнение A8.13) в A8.12), получим
^ = ^?-(E-EnTa)yf(x, у, г). A8.14)
Это и есть волновое уравнение Шредингера — основное урав-
уравнение квантовой механики, учитывающее двойственную
природу движущегося электрона — корпускулярную и вол-
волновую.
Потенциальную энергию в атоме определяют электро-
электростатическим взаимодействием орбитальных электронов с
ядром и между собой; для атома водорода, согласно за-
закону Кулона, ее выражают простым соотношением
где е—элементарный заряд; г — радиус орбиты по Бору.
С учетом выражения A8.15), уравнение Шредингера
A8.14) принимает вид
Для того чтобы вычислить энергию электрона в атоме
водорода, необходимо найти вид функции \\> = f(x, у, г),
характеризующей амплитуду электронной волны, которая
для первой круговой орбиты ls-электрона в атоме водо-
водорода имеет следующий вид:
A8.16)
где е—основание натуральных логарифмов; а—коэффи-
а—коэффициент, равный j^—j. Обычно уравнение Шредингера
A8.14) преобразуют
/г2
и записывают в краткой форме
Нг|) = ?.г|з. A8.17)
Поясним каждый из трех символов этого уравнения:
Н—так называемый оператор Гамильтона, указывающий
205
на определенную последовательность операций с Ч-"ФУНК"
цией; г|;— волновая функция электрона, имеющая в пер-
первом приближении смысл амплитуды стоячей электронной
волны (она может быть положительной, отрицательной,
равной нулю и даже мнимой величиной); Е-—энергия
взаимодействия ядра и электронов в атоме, значения ко-
которой находят решением уравнения ?=-^. Однако эти
решения возможны лишь в отношении ^-функций, удов-
удовлетворяющих определенным условиям, а именно, ар-функ-
ар-функция должна быть конечной, однозначной и непрерывной
во всем рассматриваемом пространстве. Реальные состоя-
состояния электрона могут описывать только \|;-функции, удов-
удовлетворяющие этим требованиям.
Относительную плотность заряда электрона в каждой
точке атомного объекта, точнее, интенсивность электрон-
электронной волны в бесконечно малых объемах do = dxdydz око-
околоядерного пространства выражают квадратом волновой
функции, умноженной на величину dv:
t|Jdy. A8.18)
Более строгое название величины A8.18) — плотность ве-
вероятности нахождения электрона в микрообъеме dv
стоячей электронной волны. Эффективный заряд электро-
электрона в каждом микрообъеме dv можно определить из произ-
произведения величины A8.18) на заряд электрона е:
Формы орбиталей. Решение дифференциального урав-
уравнения Шредингера A8.17) осуществляют суммированием
этих плотностей по бесконечному множеству бесконечно
малых объемов do, точнее, его интегрированием. Строгое
решение A8.17) удалось осуществить только для атома
водорода, перейдя от прямоугольной системы координат
(х, у, г) к сферической @, ф, г—широта, долгота и ра-
радиус-вектор). Для расчета энергии более сложных атомов
остальных элементов периодической системы используют
различные методы приближений. В случае атома водоро-
водорода интегрирование приводит к волновой функции вида
4>«,г, Я/ = Л7П, *(»¦)/;, Я/(в, ф, г), A8.19)
где п, I, т1 — главное, побочное и магнитное квантовые
числа; 6, ф, г—координаты в сферической системе; N —
нормировочный коэффициент, значение которого опреде-
определяют из уравнения joj;2do=l, откуда также следует, что
206
ty2dv = e. Оба эти соотношения отражают тот очевид-
очевидный факт, что вероятность нахождения электрона во всем
околоядерном пространстве атома равна единице, т. е. в
результате проявления волновых свойств электрон не исче-
исчезает, он только «размазывается» в виде волны по этому
пространству. Причем в силу особенностей стоячих волн
их «плотность», т. е. ijJ dv, на узловых участках равна
пулю, а в областях пучностей становится максимальной.
Таким образом, вместо строго определенной электронной
орбиты наблюдается электронное облако, состоящее из
сгущений и разряжений, определяющих понятие орбита-
ли. Форма, размеры и пространственное расположение
облака или орбитали определяют сомножителями /и,г(г)
и f'i,m(Q, ф, г) в уравнении A8.19), которые задают чис-
численные значения гр для каждого микрообъема околоядер-
околоядерного пространства в атоме.
Решение уравнения Шредингера всегда содержит не-
некоторые безразмерные параметры, принимающие значения
натурального ряда чисел и представляющие собой кван-
квантовые числа п, I, m (главное, побочное, магнитное), зна-
значение которых определяют числом степеней свободы ча-
частицы-волны. Трехмерное пространство атома характери-
характеризуют тремя степенями свободы частицы-волны, которым
соответствуют три только что упомянутых квантовых чис-
числа. Движение частицы вокруг собственной оси определяет
четвертую степень свободы и требует введения четвертого,
спинового квантового числа ms. В уравнении A8.19)
нижние индексы как раз и есть те параметры, т. е. кван-
квантовые числа.
Множитель fn>i{r) в уравнении A8.19) является ра-
радиальной частью волновой функции и определяет размер
и сферическую форму электронного облака, а [/„, г(г)]а
определяет вероятность нахождения электрона (или плот-
плотность электронного облака) на расстоянии г от ядра. Ра-
Радиальная часть волновой функции включает в себя два
квантовых числа п и /. Значение квантового числа п
определяют числом узловых поверхностей стоячей элект-
электронной волны, вдоль которых ij)du = O и ijJdu = 0. Зна-
Значение квантового числа / определяют числом узловых
поверхностей стоячей электронной волны, проходящих
через ядро атома. Если, например, л=1, т. е. узловая
поверхность волны одна, то она располагается бесконечно
далеко от ядра атома. Тогда через ядро не проходит
узловая поверхность, т. е. 1 = 0. Из этого следует, что
207
максимальное значение / может быть равным я—1, а
минимальное—нулю. Так, при п — Ъ квантовое число I
может принимать значение 2, 1,0.
Множитель /г, га (9, ф, г) представляет собой так назы-
называемую угловую часть волновой функции, с которой свя-
связаны побочное / и магнитное т1 квантовые числа. Маг-
Магнитное квантовое число т„ как и в теории Бора — Зом-
мерфельда, определяет возможные значения проекции
момента количества движения электрона на ось г, харак-
характеризующую направление внешнего магнитного поля; оно
может принимать значения от 0 до ± / и число таких
значений равно 21+ 1.
Паспорт электронной орбитали. Каждая стоячая элект-
электронная волна, разрешаемая квантовой механикой и опи-
описываемая функцией -ф;,, и т' характеризует понятие элект-
электронной орбитали.
Орбиталь полностью определена, если для нее извест-
известны значения главного квантового лA, 2, 3, ... и т. д.),
побочного квантового l(s, p, d, f) и магнитного кванто-
квантового mt чисел. Поэтому паспортом электронной орбитали
служит последовательная запись первых двух квантовых
чисел —
а нижний индекс указывает на характер пространствен-
пространственного расположения орбитали. Например, в атоме азота
паспорт электронов внешнего, валентного слоя записывают
Здесь верхний индекс у символа s указывает на то, что
орбиталь 2s заселена двумя электронами. Если не отра-
отражать пространственного расположения орбиталей в атоме
азота, то паспорт внешнего электронного слоя можно
изобразить так:
2s22p3.
Плотность электронного облака в атоме определяется
величиной г|з2 dv, которая изменяется от t|Jdo = 0 (на уз-
узловых участках) до некоторых наибольших, максималь-
максимальных значений ijJdu = max. Если провести поверхность
через точки максимальной плотности электронного обла-
облака, то получится пространственная фигура, называемая
формой электронной орбитали. Рассмотрим в качестве
примера форму электронных орбиталей основной (л=1)
и возбужденных (и > 1) в самом простом по структуре
208
атоме водорода. Для ls-электрона в этом атоме плотность
электронного облака можно вычислить из уравнения
A8.16):
A8.20)
Если разделить все околоядерное пространство в атоме
водорода на бесчисленное множество сфер, расстояния
между которыми бесконечно малы (равны dr), то вероят-
вероятность нахождения электрона в каждом сферическом слог,
т. е. плотность электронного облака
D в них, будет равна произведению
ijJ на объем сферического слоя Am* dr:
D =. АлгЩ^г,
а если использовать
A8.20), то
D = —„/•%
а3
dr.
уравнение
A8.21)
л /1П „,, Рис. 18.1. Форма Is-
Анализируя уравнение A8.21), ви- орбитали атомг| В0ДС).
дим, что при некотором значении рода
г = г0 значение электронной плот-
плотности максимально, т. е. D = Dmax. Если в пространстве
прямоугольных координат х, у и z через точки, удален-
удаленные от центра на г0, провести поверхность, то получится
сфера (рис. 18.1). Таким образом, ls-орбиталь в атоме
водорода имеет сферическую форму.
Волновые функции орбиталей атома водорода. Соглас-
Согласно известным квантово-механическим расчетам волновые
функции возбужденных орбиталей электрона в атоме во-
водорода для значения главного квантового числа п= 2
выражают уравнениями:
2s(m,=0); t|)w =
j_ 2
по J g a.
A8.22)
{у или г)
х(у, г).
A8.23)
209
Волновой функции ty2s A8.22) соответствует форма орби-
тали сфера в сфере. Каждой из волновых функций
фгр , >f>2P , tyip соответствует похожая на гантель форма,
приведенная на рис. 18.2, а, б, в. Три р-орбитали каждого
электронного слоя атома расположены в трех взаимно
перпендикулярных направлениях, как это показано на
*-.v
Рис. 18.2. Форма р-орбиталей возбужденного атома водорода
(а, б, в, г, д); схема взаимосвязи прямоугольных (х, у, г) и сфери-
сферических (ф, 0, г) координат (д)
рис. 18.2, г, что соответствует наименьшему взаимному
электростатическому расталкиванию одноименно заряжен-
заряженных электронных облаков.
Для случая, когда главное квантовое число равно
трем (п = 3), электронное облако атома должно иметь
одну Зэ-орбиталь, три Зр-орбитали и пять Зй-орбиталей,
волновые функции которых:
Зй-орбиталь (/ = 0, /л = 0)
81
210
г
Зр* (у „ли г,-орбиталь A=1, т1 = ± 1, 0)
-я (г/ или г);
г
<» или
81
3<2г2-орбиталь
^ L 2 6
Зс?лг-орбита ль
3d ^-орбиталь
З^^-орбиталь
Здесь, в третьем электронном слое атома водорода, для
3s- и 3/7-орбиталей характерны сферическая (три сферы
одна в другой) и гантелеобразная (рис. 18.2) формы. Что
касается Зс?-орбиталей, то из приведенных выражений
для волновых г|)-функций следует, что области максималь-
максимальной плотности электронного Зс?-облака располагаются
не вдоль осей х, у и г, т. е. являются функциями не одной
переменной х, у или г, а функциями двух переменных
(л;2—у2, xz, г/г, ху). Исключение составляет Зс?г»-орбиталь,
волновую функцию которой определяют аргументом г2.
Формы d-орбиталей возбужденного атома водорода
приведены на рис. 18.3, где фрагменты а, б, в, г и д
представляют собой графическое изображение отдельных
d-орбиталей, а фрагмент е—совокупность пяти Зо!-орби-
талей возбужденного атома водорода. В четвертом элект-
211
т
ронном слое такого атома (л = 4; / = 0, 1, 2, 3; т1 —
= ±3, +2, ±1, 0) побочному квантовому числу 1 = 3
соответствует 2/+1=7 /-орбиталей. Выражения tyif для
них еще более сложны, чем для Зо!-орбиталей:
¦ф, = Ахуг,
где Л — выражение, подобное стоящему перед скобками
в уравнениях A8.24). Каждая такая 4/-орбиталь про-
У
Ф2
Рис. 18.3. Формы d-орбиталей возбужденного атома водорода
(а, 6, в, г, д) и заполнение ими околоядерного пространства (е)
странственная и имеет сложную конфигурацию. Соответ-
Соответственно этому совокупность семи /-орбиталей в атоме
представляет еще более сложную картину. Как известно,
в многоэлектроиных атомах химических элементов запол-
2J
If
нение электронами далее /-орбиталей не идет, поэтому
рассматривать вид ^-функций и формы g-орбиталей не
имеет смысла.
Волновые функции многоэлектронных атомов. При пе-
переходе от атома водорода к атомам других элементов
условимся, что формы s-, p-, d- и /-орбиталей, изобра-
изображенные схематически на рис. 18.1 —18.3, качественно
остаются теми же. Поэтому при описании формы орби-
орбиталей атомов различных элементов считают их примерно
такими же, как и в возбужденном атоме водорода. Следует
также отметить, что точное решение уравнения Шредин-
гера для многоэлектронных атомов
[%+p(
еще не осуществлено (в этой формуле rk — расстояние от
ядра до ^-электрона, Vk—энергия взаимного расталкива-
расталкивания электронов). Даже для атома гелия, электронная
оболочка которого содержит всего два электрона, анало-
аналогичное уравнение столь сложно, что абсолютно строгого
решения найти пока не удалось.
Однако существует метод последовательных прибли-
приближений (метод самосогласованного поля Д. Хартри и
В. Фока), который позволяет получить волновую функцию
/г-электронного атома в виде произведения fe-волновых
функций отдельных электронов, похожих на волновые
функции атома водорода. Именно это и дает возможность
использовать орбитали атома водорода (см. рис. 18.1 —18.3)
для характеристики орбиталей многоэлектронных атомов.
Как отмечалось выше, спиновое квантовое число может
принимать только два значения (±1/2) и характеризует
четвертую степень свободы электрона. С учетом этой
дополнительной, спиновой степени свободы все орбитали
атома имеют максимальную емкость два электрона, каж-
каждый из которых по отношению ко второму обладает про-
противоположным спином.
Последовательность заселения орбиталей электронами
определяют правилом Ф. Хунда: при данном значении
побочного квантового числа I суммарный спин электро-
электронов 2]s, заселяющих орбитали, должен быть максимален.
Для иллюстрации этого правила условимся, что геомет-
геометрическим символом каждой орбитали атома будет квадрат,
а символом электронов, заселяющих эти орбитали,—
213
стрелки. Направление стрелок должно соответствовать
знаку спинового квантового числа. Согласно этой модели
распределение электронов по орбиталям атомов водорода,
гелия и всех элементов второго периода принимают вид,
приведенный на схеме:
Н Is1
Не Ь2
Li W2sl
Be Js22s2
В Is22s2pt
С Is22s;p*
N ls:2s2p3
О Is22s2p4
F Is22s2p5
Ne
Is
rtTI
77
[п]
FT]
¦ 1»П
ill liil
ul fnl
Построение периодической системы элементов Д. И. Мен-
Менделеева с позиций волновой модели качественно то же,
что и с позиций корпускулярной модели Бора. Однако
волновая модель позволяет количественно охарактеризо-
охарактеризовать энергию атомных орбиталей и дает ключ к пони-
пониманию природы и энергетики химических связей.
Глава 19
Потенциалы ионизации
и эффективные параметры атомов
Потенциалы ионизации и сродство к электрону.
Эффективные квантовые числа и эффективные
заряды ядер. Радиусы атомов и ионов.
Потенциалы ионизации. Сложность уравнения Шредин-
гера не дает возможности строго рассчитать энергию
орбиталей многоэлектронных атомов. В связи с этим раз-
разработан ряд приближенных методов, которые требуют
применения быстродействующих электронных счетных
машин, однако погрешности получаемых результатов
большие. Поэтому по точности теоретический расчет
энергий атомных орбиталей не может пока конкурировать
с экспериментальными способами. Наиболее разработан-
разработанными и универсальными методами экспериментального
определения энергий атомных орбиталей являются спектро-
спектроскопические. Спектры атомов, как известно, представляют
собой совокупности серий спектральных линий, причем
каждая из таких серий отвечает переходам электронов
с различных удаленных орбиталей (в том числе и из бе-
бесконечности) на одну из близлежащих к ядру. При этом
самой коротковолновой границе спектральной серии,
которая характеризует переход электрона из бесконеч-
бесконечности на ближайщую к ядру незанятую орбиту (переход
в основное состояние), будет соответствовать выделение
энергии, численно равной энергии отрыва электрона, т. е.
энергии ионизации, или потенциалу ионизации
где /—¦ энергия ионизации; Е„—энергия электрона, нахо-
находящегося за пределами атома; Е0СИ—энергия электрона,
находящегося на ближайшей к ядру свободной орбитали
(основное состояние).
Если коротковолновой границе такой спектральной
серии соответствует частота электромагнитных колеба-
колебаний v,., то энергия ионизации 1{ может быть вычислена
из уравнения Планка
В случае многоэлектронного атома этим путем можно
получить энергию отрыва каждого из электронов, возбуж-
215
денного (выбиваемого с основной орбиты) в процессе
облучения. Существуют и другие экспериментальные
методы определения энергии ионизации атомов, например
электронный удар, фотоионизация и др.
Величина энергии ионизации является важной харак-
характеристикой химических элементов, поскольку прочность
удержания электронов на атомных орбиталях в значи-
значительной степени определяет химические свойства этих
элементов; знание ее позволяет глубже понять особенности
распределения плотности электронных облаков в моле-
молекулах химических соединений и кристаллических решет-
решетках. Значения энергии последовательного отрыва электро-
электронов от многоэлектронного атома существенно различаются
между собой, хотя энергия притяжения к ядру каждого
электрона данного подуровня одинакова (электростати-
(электростатическое взаимодействие ненасыщаемо). Причина этого
заключается в том, что электроны не только притягиваются
к ядру, но и расталкиваются между собой в силу одно-
одноименности зарядов. Поэтому отрыв первого электрона
требует наименьшей энергии, а каждого последующего —
все более возрастающей. Особенно резкого возрастания
энергии ионизации следует ожидать при переходе от
электронов более удаленного от ядра слоя к менее уда-
удаленному.
В табл. 19.1 приведены примеры значений ионизацион-
ионизационных потенциалов для элементов I и II периодов периодичес-
периодической системы Д. И. Менделеева. Из таблицы видно, что отрыв
даже первого, наиболее слабо удерживаемого электрона
представляет трудную задачу, осуществимую в рамках
химического эксперимента только для атомов щелочных
металлов. Так, ионизация атомов водорода может стать
заметной лишь при температурах порядка 103 К. Более
же глубокая ионизация многоэлектронных атомов может
быть осуществлена лишь в условиях горячей плазмы
A05—106 К). Поэтому для расчета энергии химической
связи существенны лишь первые ионизационные потен-
потенциалы, а все последующие имеют смысл меры стремления
ядер удерживать около себя электронное облако.
Это заключение не противоречит тому известному
факту, что атомы многих металлов могут в растворах,
расплавах и кристаллических решетках существовать
в виде ионов одно-, двух-, трех- и даже четырехзарядных
(например, Na + , Ca2+, Fe3+, Th4+). Однако эти ионы не
индивидуальны, они либо гидратированы (сольватированы)
полярными молекулами растворителя, либо в кристаллах
216
Таблица 19.1. Ионизационные потенциалы элементов
I и II периодов, эВ
Элемент
н
Не
Li
Be
В
С
N
0
F
Ne
.Элемент
н
Не
Li
Be
В
С
N
0
F
Ne
Л
13,595
24,581
5,390
9,320
8,296
11,256
14,540
13,614
18,418
21,559
489,84
551,93
138,08
157,12
157,9
54,
79,
18,
25,
24,
29,
35,
34,
41,
666
739
185
207
и
403
619
206
149
376
60
15
98
07
,83
,11
,14
,2
122,
153,
37,
47,
47,
54,
62,
63,
U
871,
935,
239,
419
850
920
871
426
93
65
5
12
8
1
1,
217,
259,
64,
77,
77,
87,
97,
I,
1101
1195
657
298
48
45
39
23
16
,8
,4
340,127
392,00
97,86
113,87
114,21
126,4
j
1360,2
и расплавах окружены ионами противоположного знака.
При этом экзоэргический эффект электростатического
взаимодействия разноименно заряженных частиц с избыт-
избытком перекрывает эндоэргический эффект ионизации атомов
металлов. Это означает, что гидратированные ионы пред-
представляют собой сложные химические соединения.
Ионизационные потенциалы служат одной из коли-
количественных характеристик химических элементов, опре-
определяя место элемента в периодической системе, характер
и свойства образуемых им соединений. На рис. 19.1
приводится взаимосвязь значений первого ионизационного
потенциала с зарядом ядра, которая показывает, что
ионизационные потенциалы являются периодической функ-
функцией заряда ядра. Так, в каждом периоде ионизационные
потенциалы возрастают, подчиняясь некоторому сложному
закону: самые низкие значения /х характерны для щелоч-
217
ных металлов, ядра атомов которых в пределах своих
периодов имеют наименьший заряд и поэтому менее энер-
энергично притягивают единственный валентный электрон;
самые высокие значения ионизационных потенциалов у эле-
элементов восьмой группы {благородные газы), имеющих
наибольшие заряды ядер атомов в своих периодах.
Однако, как следует из рис. 19.1, монотонное возра-
возрастание потенциалов ионизации в периодах не соблюдается.
Г, эВ
25
20
15
10
5
,2
Не
I Ne
3
!
И
1 *
* г
1
Ы
Г
Г'
1
4
Аг
t
i
J
Г 1
¦ К
i
5
[
1
1
1
f
I
I
Кг
f
/И
'г1
Rb
i
i t
1
f
1
1
1
1
I
1
Xe
t
if
гииг
Cs
I f
6
7
Периоды
i
i
i
Ra
Jf ft
1
I 1
20
40
60
80
Рис. 19.1. Зависимость первого ионизационного потенциала
элементов от заряда ядра
Так, во втором периоде вместо увеличения энергия иони-
ионизации убывает при переходе от бериллия к бору и от
азота к кислороду. Причина этого в том, что после
заселения электронами s-подуровня у атома бериллия
начинается заселение более высокого по уровню энергии
р-подуровня атома бора. После заполнения одиночными
электронами /^-подуровня атомов углерода и азота начи-
начинают формироваться электронные пары атомов кислорода,
фтора и неона. Аналогичные нарушения монотонности
имеют место и в последующих периодах. Резкие нару-
нарушения монотонности наблюдаются в середине каждого
семейства d-элементов за счет перехода от заселения
одиночными электронами d-подуровня
218
к электронным парам (образуется первая пара d-элект-
ронов)
Fe Cd6) |+t|t|t | tit
Такие же изменения монотонности отмечены в конце
каждой декады d-элементов на границах кадмий — индий,
ртуть—таллий, цинк — галлий, например:
GaCd"V) IUHUHI
t
В шестом и седьмом периодах нарушения монотонности
хода потенциалов ионизации наблюдаются в семействах
лантанидов D/-элементы) и актинидов E/-элементы).
Интересным является различие хода потенциалов иони-
ионизации в главных и побочных подгруппах. Из графика,
представленного на рис. 19.2, видно, что в каждой группе
элементы по свойствам делятся на три ветви. Первая
ветвь—это элементы П и III периодов, атомы которых
не содержат d-электронов, в связи с этим они занимают
в каждой из групп несколько обособленное положение.
Две другие ветви характеризуют свойства элементов
главной и побочной подгрупп. Существенно то, что имеет
место закономерная тенденция в характере расположения
линий при переходе от элементов одной группы к эле-
элементам другой. Так, в I и II группах значение энергии
отрыва первого электрона 1г для s-элементов главной под-
подгруппы существенно ниже аналогичных значений для
d-элементов побочной подгруппы, что объясняют значи-
значительно большим зарядом ядер элементов побочной под-
подгруппы (очевидно, сравнивать можно только элементы
одного и того же периода).
Поясним сказанное. Согласно теории Бора потенциалы
ионизации атомов без учета сил взаимного расталкивания
электронов выражают соотношением
Г 2я%1?* I 22
A9.1)
где Z—заряд ядра атома; п — главное квантовое число
(номер периода); т и е—масса и заряд электрона;
h — постоянная Планка. В этой формуле выражение
в скобках представляет постоянную величину, второй
сомножитель — переменную, разную для каждого из эле-
219
15 -
10 -
5 -
-
-
Li
1
Си
К Rb
i i
1
A
Cs
I
Au
***
Fr
i
20 40 60 80 z
Ca Sr Ba
Ai Ga In
20 40 60 80 z
20 40 60 80 z
20 40 60 80 z
Рис. 19.2. Связь первого ионизационного потенциала II с атомным номером (зарядом
ядра) элементов главных и побочных подгрупп
г
ментов. Используя это соотношение, можно сравнить
потенциалы ионизации двух элементов IV периода — калия
и меди:
'к 2к |у
Фактическое соотношение этих потенциалов несколько
меньше из-за различия во взаимном расталкивании элект-
электронов каждого атома:
/Си ..7'724 - 1 о
/к ~4,339
По той же причине при переходе к элементам II, III и
других групп подобный расчет менее верный, поскольку
в данной последовательности энергия взаимного отталки-
отталкивания электронов в атомах элементов главной и побочной
подгрупп становится все более разной. Такая тенденция
определяет увеличение различия в строении оболочек
ятомов элементов главной и побочной подгрупп. Действи-
Действительно, в I и II группах все электронные слои в атомах,
кроме внешнего валентного, полностью заселены, и строе-
строение внешнего электронного слоя одинаково, например
К B, 8, 8, 1) и Си B, 8, 18, 1). В III группе и в после-
последующих группах как глубинные, так и внешние электрон-
электронные слои имеют разную степень заселенности [например,
Ge B, 8, 18, 4) и Ti B, 8, 10, 2)J. В связи с этим резуль-
результаты расчета отношений потенциалов ионизации по
уравнению A9.1) должны все больше расходиться с экс-
экспериментальными данными. Эта тенденция приводит
к тому, что в III группе (см. рис. 19.2) потенциалы
ионизации элементов главной и побочной подгрупп ока-
оказываются близкими по значениям, а в IV и последующих
главных подгруппах больше, чем в дополнительных.
В результате имеет место серьезное различие в соотно-
соотношении свойств элементов главной и дополнительной под-
подгрупп по левую и правую сторону от III группы, что
нашло свое отражение в построении периодической
системы.
Процессы ионизации атомов химических элементов
не могут быть сведены только к отрыву электронов и
образованию заряженных ионов. Возможен и другой
вариант—присоединение одного или нескольких допол-
дополнительных электронов к нейтральным атомам с образо-
образованием отрицательно заряженных ионов. Поскольку
электростатические силы ненасыщаемы, то подобный акт
221
принципиально возможен во всех без исключения слу-
случаях. Однако вводимый в нейтральный атом электрон
будет не только притягиваться к ядру, но и отталки-
отталкиваться от всех уже имеющихся электронов. Если сила
отталкивания сильнее притяжения, то образующийся ион
энергетически будет неустойчив. Если же притяжение
дополнительного электрона к ядру сильнее отталкивания
от электронной оболочки, то получившийся ион будет
устойчив.
Рассмотрим энергетику образования отрицательно за-
заряженных ионов (эВ) на примере элементов I и II пе-
периодов:
Н+е-—0,75 = Н- С + е-—2,08 = С-
Не+е~ + 0,53 = Не~ N+e~ +0,69 = N~
Li+e~— 0,54 = Li- O + e~ —2,33=^0-
BeJ-<?-+0,6 = Be- F + e-—3,62 = F-
B+<?- — 0,3 = B- Ne+e-+0,8 = Ne-
Энергетический эффект образования отрицательно заря-
заряженного иона получил название сродства к электрону.
/0,эВ
+J
0
-1
-г
-з
-4
Не
Л
V
. н
-
-
-
Be
Li ^
6
\/
V
с
N
III I
/ \ 8
Л°
\
Ne
,/
/10 z
J
Ъ
F
Рис. 19.3. Зависимость
сродства к электрону
/о, атомов химических
элементов
от заряда ядра
На рис. 19.3 построен график зависимости сродства
к электрону от заряда ядра, из которого видно, что
в отличие от эндотермического эффекта ионизации сродство
к электрону Е может иметь разные знаки. Так, атомы
Не, Be, N и Ne присоединяют электроны эндотермически,
222
что объясняют высокой симметрией и заселенностью их
электронных орбита лей. В атомах гелия и неона пол-
полностью заселены Is2- и 2$2р6-вакансии и лишний электрон
может найти себе место только в следующем слое, уда-
удаленном и заэкранированном от ядра плотным элек'/ронным
облаком. В атоме бериллия лишний электрон может
занять место только на 2р-орбитали, также более уда-
удаленной от ядра, чем заселенная 25-орбиталь; в атоме
азота электроны образуют очень симметричное электрон-
электронное облако из одной s- и трех р-орбиталей и присоеди-
присоединение нового электрона должно существенно нарушить
эту симметрию, что и вызывает дополнительную затрату
энергии.
Напротив, атомы кислорода присоединяют электроны
экзотермически, причем выгодность этого процесса воз-
возрастает в последовательности увеличения ионизационного
потенциала. Атом неона имеет завершенную электронную
оболочку и потому сродство его к электрону положи-
положительно. Присоединение более чем одного электрона, т. е.
образование многозарядных ионов, является эндотермич-
ным процессом, например (эВ):
е-—2,33 = 0- N + e-+0,69 = N-
+ e- —l,04 = S-
Из этого примера следует, что образование двукратно
отрицательно заряженных ионов кислорода и серы,
трехзарядного иона азота и четырехзарядного иона угле-
углерода энергетически невыгодно, и они в химических реак-
реакциях либо совсем не образуются, либо образуются
в ничтожных концентрациях.
Эффективные квантовые числа и заряды ядер. Выше
отмечалось, что теория Бора дает возможность вычислить
приближенные значения потенциалов ионизации много-
многоэлектронных этомов по уравнению A9.1) без учета энер-
энергии взаимного расталкивания электронов. Однако энергия
такого расталкивания очень велика и пренебрежение ею
приводит к большим ошибкам, которые делают расчет
бесцельным. Для устранения этой трудности были введены
представления об эффективных значениях главного кван-
квантового числа п* и заряда ядра Z*.
Эффективный заряд Z*—часть фактического заряда
ядра, которую воспринимает один внешний электрон
(или посторонний атом) через слой экранирующей его
223
электронной оболочки атома. Рассмотрим в качестве
примера атом лития, электронная оболочка которого
состоит из трех электронов. Истинный заряд ядра атома
лития равен 3+. Однако внешний валентный электрон,
помимо притяжения к ядру, испытывает отталкивание
от двух и более глубоко лежащих, но «размазанных»
по околоядерному пространству электронов, заряд которых
равен 2—. Результирующий, или эффективный, заряд
ядра, действующий на внешний электрон, складывают
сложным путем из зарядов противоположного знака и
получают равным 1,26 вместо 3. С увеличением числа
электронов в атоме эффект экранирования заряда ядра
возрастает, что приводит к относительному уменьшению
эффективного заряда ядра. Например, Z* атома ртути
получают равным 3,68, в то время как истинный заряд
ядра равен 80. В табл. 19.2 приведены истинные и эффек-
эффективные заряды ядер некоторых элементов периодической
системы.
Таблица
19.2. Истинные i
атомов некоторых элементов
Эле-
Элемент ы
н
Li
Be
В
С
N
О
F
Na
%
А1
1
3
4
5
6
7
8
9
11
12
13
2Эфф
1
1,26
1,66
1,56
1,82
2,07
2,00
2,26
1,84
2,25
1,99
Эле-
Элементы
Si
р
S
С1
к
Са
Sc
Ti
V
Cr
Mn
i эффективные
''ист
14
15
16
17
19
20
21
22
23
24
25
2эфф
2,32
2,64
2,62
2,93
2,09
2,48
2,57
2,62
2,61
2,61
2,74
заряды
Эле-
Элементы
Fe
Со
№
Си
Zi
Ga
Ge
As
Se
Br
Rb
ядер
¦^HCT
26
27
28
29
30
31
32
33
34
35
37
2Эфф
2,82
2,81
2,77
2,79
3,08
2,46
2,82
3,14
3,13
3,45
2,22
Необходимость введения п* связана с тем, что реаль-
реальные многоэлектронные атомы и ионы требуют для расчета
энергии использования всех четырех квантовых чисел и
всего аппарата квантовой механики, поэтому главное
квантовое число п в уравнении A9.1) может быть заме-
заменено на эффективное его значение п*, разность
получила название квантового дефекта. Дж. Слетер пред-
предложил следующие значения п*, отвечающие соответствую-
224
хдим значениям главного квантового числа:
п ...
п*...
q ...
1
1
0
,0
2
2,
0
0
3
3,
0
0
4
3
0
,7
,3
5
4
1
,0
,0
6
4,
1,
2
8
С учетом введенных эффективных характеристик Z* и
п* уравнение A9.1) для расчета ионизационных потенциа-
потенциалов атомов принимает вид
A9.2)
Используя это соотношение, можно вычислять фактиче-
фактические значения ионизационных потенциалов атомов, учи-
учитывая не только притяжение данного электрона к ядру,
но и отталкивание его от всех остальных электронов.
Выражение в первых квадратных скобках уравнения A9.2)
представляет собой ионизационный потенциал атома водо-
водорода
"*""'" ¦=ун = 13,595 эВ,
отсюда
Следует отметить, что знание эффективных характе-
характеристик Z* и п* важно при расчете волновых функций \р
электронных орбиталей атомов химических элементов по
формуле, предложенной Слетером A930):
z*
^ = ^•-•«"^,,„F, ф, /•), A9.3)
где fu тф, ф, г)—угловая часть волновой функции.
Кроме того, эффективный заряд ядер и эффективное
квантовое число широко используют для расчета поляри-
поляризуемости атомов и ионов, их радиусов, а также электро-
электроотрицательности элементов. Рассмотрим в качестве при-
примера вопросы, связанные с представлениями об атомных
и ионных радиусах, а также способы их расчета
Радиусы атомов и ионов. Само понятие размера атома
или иона с точки зрения учения о строении неопреде-
неопределенно, поскольку электронная оболочка атома представ-
представляет собой диффузное «электронное облако», не имеющее
четких границ.
Согласно одному из определений радиус атома—это
расстояние от ядра до наиболее удаленного максимума
8 № 7 05 225
г
электронной плотности. Если продифференцировать вол-
волновую функцию Слетера A9.3) и найти области макси-
максимальной плотности электронного облака, то получим со-
соотношение
/•max = const
(«*)а
где гтах и есть искомое расстояние от ядра до максимума
электронной плотности. В табл. 19.3 приведены вычис-
вычисленные значения rmax для атомов некоторых элементов
периодической системы.
Графическая зависимость радиус атома — заряд ядра
представлена на рис. 19.4, где показана периодичность
изменения атомных радиусов. Относительно самые боль-
большие размеры атомов характерны для щелочных металлов,
которые в своих периодах имеют наименьший заряд ядра.
Наименьшие размеры свойственны атомам элементов
Таблица 19.3. Радиусы нейтральных атомов, нм
Атом
н
Не
Li
Be
В
С
N
О
F
Ne
Орби-
таль
Is
Is
2s
2s
2p
2p
2p
2p
2p
2p
Vax
0,053
0,029
0,159
0,104
0,078
0,060
0,049
0,041
0,036
0,032
Atom
Na
Mg
Al
Si
P
S
Cl
Ar
К
Ca
Орби-
таль
3s
3s
3p
3p
3p
3p
3p
3p
4s
4s
rmax
0,171
0,128
0,131
0,107
0,092
0,081
0,073
0,066
0,216
0,169
Atom
Sr
Ti
V
Cr
Mn
Fe
Co
Ni
Cu
Zn
ОрСи-
таль
4s
Ad
U
4d
4d
4d
4d
4d
4d
4d
max
0,157
0,148
0,140
0,145
0,129
0,123
0,118
0,114
0,119
0,107
благородных газов, имеющих в своих периодах наиболь-
наибольшие заряды ядер. Из графика A9.4) отчетливо видно,
что в каждом периоде более резко уменьшаются радиусы
s-элементов, менее—/^-элементов и постепенно—радиусы
d-элементов (дополнительных подгрупп). Наиболее поло-
пологим является ход значений радиусов /'-элементов V пери-
периода, что можно объяснить следующим образом. Электрон-
Электронные оболочки /'-элементов характеризуются заполнением
третьего электронного слоя, т. е. слоя глубинного, близко
расположенного к ядру. В результате они испытывают
такое большое электростатическое притяжение к ядру,
что изменение заряда ядра уже не в состоянии сущест-
существенно изменить их размеры; для d-элементов характерно
226
г,нм
Телантаноиды u*""k актиноиды
Hg Rn
VIJ Периоды
10 20 30 40 50 60 70 80
Рис. 19.4. Зависимость радиусов атомов от заряда ядра
100 z
заполнение второго сверху электронного слоя, более уда-
удаленного от ядра, менее плотного и потому способного
к более заметным деформациям при изменении заряда
ядра; для s- и /^-элементов характерно заполнение внеш-
внешних «рыхлых» электронных орбиталей, еще более удален-
удаленных от ядра, способных сильно деформироваться при из-
изменении заряда ядра. Различие «хода» радиусов s- и р-
элементов объясняют различием формы орбиталей (s—
сферическая, а р—гантелеобразная).
Существует и другое определение атомного радиуса,
за который принимается расстояние от ядра до некото-
некоторого малого значения плотности электронного облака
внешней оболочки атома. Такому определению соответ-
соответствует расчет радиусов атомов по опытным данным. Сле-
тер предложил измерять расстояние от ядра до такой
области периферии атома, где волновая функция гр будет
составлять примерно 10% от значения ее максимума, т. е.
Y 10 '
Для этого предлагается умножить rmax на 3,38 при п*=1;
на 2,49 при п* = 2; на 2,25 при п* = 3; на 2,01 при
п* = 3,7; на 1,96 при п* = 4,0.
Совершенно независимый способ расчета атомных
радиусов основан на использовании данных по между-
междуядерным расстояниям в гомосоединениях различных эле-
элементов. В случае атомов металлов для этой цели исполь-
используют данные по плотности соответствующих металлов.
Если разделить атомную массу меди А на ее плотность D,
то получится значение объема одного моля меди:
„ ЛСи _ 63,46
си Оси 8,93.10е '
В одном моле содержится 6,023-1023 атомов меди. Если
предположить, что атомы в кристалле упакованы плотно,
без пустот, то объем отдельного атома можно найти из
соотношения
TV7~ 6,023- юи»-и</вш М-
Здесь предполагается, что каждый атом имеет форму куба,
поэтому расстояние от центра такого куба до его грани
гСи получают равным
cu У Си 1/а У U78.10-»» = 1,14-10"» м.
228
Если же предположить, что атомы имеют форму шара и
между ними в решетке металла есть пустоты, то объем
отдельного атома может быть вычислен из соотношения
откуда радиус атома
Атомные радиусы неметаллов вычисляют аналогичным
образом, как половину межатомного расстояния в моле-
молекулах или кристаллах простых веществ.
Из сравнения табл. 19.3 и 19.4 видно, что приведен-
приведенные в них численные значения атомных радиусов доста-
достаточно близки, но не совпадают, что свидетельствует как
о недостатках методов расчета, так и о неопределенности
понятия радиуса атома. Несмотря на это, величины атом-
атомных радиусов позволяют успешно решать многие сравни-
сравнительные задачи по определению самых различных свойст г»
химических элементов и их соединений, т. е. служат кор-
коррелятивным параметром.
Таблица 19.4. Радиусы атомов, вычисленные из межатомных
расстояний в простых веществах, нм
Элемент
н
Li
Be
в
с
N
О
F
'аг
0,037
0,155
0,113
0,080
0,077
0,055
0,060
0,071
Элемент
Na
Mg
Al
Si
P
S
Cl
к
Лаг
0,189
0,160
0,143
0,118
0,095
0,102
0,099
0,236
Элемент
Ca
Sc
Ti
V
Cr
Mn
Fe
Co
'at
0,197
0,164
0,146
0,134
0,127
0,130
0,126
0,125
Кроме радиусов нейтральных атомов большое значе-
значение в химии приобрели ионные радиусы, т. е. радиусы
положительно заряженных катионов г^ и отрицательно
заряженных анионов гА, хотя они, как и атомные, яв-
являются условностью, коррелятивным параметром. Ионные
радиусы в первом приближении могут быть вычислены,
если известны расстояния R между ядрами атомов в хи-
химических соединениях ионной структуры. И действительно,
если принять, что межъядерные расстояния равны сумме
радиусов аниона и катиона
то, определив независимым методом радиусы ионов одного
или двух наиболее характерных для ионных соединений
элементов, можно вычислить значения всех остальных.
Так, используя данные оптических методов, были опреде-
определены радиусы анионов F" (г=0,136 нм) и Оа~ (г=0,132 нм).
С помощью этих величин, а также межъядерных расстоя-
расстояний в кристаллических решетках различных соединений
были вычислены ионные радиусы всех остальных элемен-
элементов. Например, в кристаллах NaF расстояние между
ядрами Na и F было найдено равным 0,231 нм. Зная
радиус иона F" и используя уравнение A9.4), находим
радиус иона Na+ :/"Na+ =/?NaF — /"f- = @,231—0,133) =
= 0,098 нм. Этот прием позволяет вычислить значения
формальных ионных радиусов и для элементов, которые
не образуют ионных соединений, например для гипотети-
гипотетических ионов О6+ (/- = 0,009 нм) и С1'+ (г = 0,026 нм).
Формальные ионные радиусы, как и отвечающие им заряды,
хотя и не имеют строгого физического смысла, однако
полезны как коррелятивные параметры. В табл. 19.5 и
Таблица 19.5. Радиусы положительно заряженных ионов эле-
элементов, найденные эмпирически, нм
Ион
U +
Na +
К +
Rb +
Cs+
Fr+
Ве2+
Mg2 +
Са2 +
Sr2 +
Ва2 +
Ra2 +
0,068
0,095
0,133
0,149
0,165
0,175
0,034
0,074
0,104
0,120
0,138
0,144
Ион
в3+
А[3 +
Ga3+
In +
1п3+
Т1 +
Т13+
Si4 +
Ge4+
Ge2+
Sn4+
0,021
0,057
0,062
0,130
0,092
0,136
0,105
0,020
0,039
0,044
0,065
0,067
Ион
Sn2+
Pb4 +
Pb2 +
№ +
рб +
As6 +
As3 +
Sb5 +
Sb3 +
Bi5 +
Bi3 +
Oe +
r
0,102
0,076
0,126
0,015
0,035
0,047
0,069
0,062
0,090
0,074
0,120
0,009
Ион
Se +
Se6 +
Se3+
Ti4 +
V6 +
Cre +
Mn7 +
Fe3+
Co3+
Ni3+
Cu2+
Zn2+
r
0,030
0,069
0,083
0,064
0,059
0,052
0,046
0,073
0,072
0,072
0,080
0,083
19.6 приведены значения ионных радиусов многих эле-
элементов периодической системы (кроме лантанидов и акти-
актинидов), вычисленные из экспериментальных данных по
межатомным расстояниям в гетеросоединениях.
230
Таблица 19.6. Радиусы отрицательно заряженных ионов эле-
элементов (эмпирические, нм)
Ион
с*-
N3-
рз-
Ass~
Sb3-
ог-
s2-
г
0,260
0,148
0,186
0,191
0,208
0,136
0,186
Ион
Se2-
Те2~
н-
F-
CI-
Вг-
I-
г
0,198
0,222
0,136
0,133
0,181
0,196
0,220
Как следует из табл. 19.7, взаимосвязь заряда и
радиуса такова, что с ростом положительного заряда
иона его радиус уменьшается. Такой характер изменения
размеров иона в зависимости от его заряда можно объяс-
объяснить уменьшением сил взаимного расталкивания электро-
электронов по мере ионизации атомов. При движении в под-
подгруппах сверху вниз размеры ионов возрастают, что
объясняют увеличением в атомах числа электронных слоев.
В каждом периоде при движении слева направо размеры
ионов уменьшаются, как это имело место и в случае атом-
атомных радиусов.
Таблица 19.7. Радиусы атомов и ионов, имеющих различный
заряд, нм
Элемент
о
S'
С1
Заряд
атома
6+
0
2—.
6+
4+
0
2—
И
0
Радиус, нм Элемент
0,009
0,060
0,136
0,030
0,037
0,102
0,186
0,007
0,034
0,099
Na
Са
Мп
Zn
Заряд
атома
1 +
0
7+
4+
2+
0
2+
0
Радиус, нм
0,098
0,189
0,104
0,197
0,046
0,060
0,080
0,130
0,093
0,131
231
Теоретический расчет ионных радиусов, подобный
использованному при вычислении атомных радиусов, ока-
оказался возможным на основе квантово-механической тео-
теории. Впервые этот расчет был выполнен в 1965 г. И. Уабе-
ром и Д. Кромером, которые считали радиусом иона рас-
расстояние от ядра до максимума плотности электронного
облака, отвечающего данной степени ионизации. Резуль-
Результаты расчета приведены в табл. 19.8, из которой видно,
что радиусы отрицательно заряженных ионов близки
к радиусам атомов.
Таблица 19.8. Радиусы атомов и ионов, вычисленные методом
Уабера н Кромера, нм
с'лемент
F
CI
Заряд
0
]
0
1—
г
0,036
0,037
0,073
0,074
Элемент
о
S
Заряд
0
2
0
2
г
0,041
0,046
0,081
0,083
Это объясняют тем, что электроны, присоединяющиеся
к нейтральному атому, занимают места на уже частично
заселенной орбитали. Поскольку новая орбиталь при этом
не возникает, а возможное отталкивание присоединенного
электрона невелико, существенного изменения радиуса
атома от прибавления лишнего электрона не должно быть.
Этот факт противоречит большой разнице радиусов анио-
Таблица 19.9. Сравнительные размеры ионных и атомных
радиусов элементов II периода, нм
Элемент
Li
Be
В
С
N
О
F
гатома
теорети-
теоретический
0,159
0,104
0,078
0,060
0,049
0,041
0,036
эмпири-
эмпирический
0,155
0,113
0,080
0,077
0,055
0,060
0,071
Заряд
иона
1 +
2+
3+
4+
5+
6+
7+
ГИОН?
теорети-
теоретический
0,019
0,014
0,011
0,009
0,008
—
эмпириче-
эмпирический
0,063
0,034
0,021
0,020
0,015
0,009
0,007
232
Шв и нейтральных атомов, вычисленных из межатомных
расстояний. Различие между размерами атомов и положи-
положительно заряженными ионами сохраняется, но приобре-
приобретает качественно иной характер, как это видно из
табл. 19.9 на примере элементов второго периода. В при-
приведенном примере у лития теоретический радиус больше
эмпирического, но уже у бериллия и бора картина ста-
становится обратной, а к концу периода эмпирические
радиусы атомов оказываются больше теоретических. Ионные
эмпирические радиусы во всех случаях много больше
теоретических, причем это различие наибольшее в начале
периода.
Таким образом, теоретически вычисленные и эмпири-
эмпирически полученные радиусы атомов и ионов различаются
не только количественно, но и качественно, что подтвер-
подтверждает тезис о неопределенности понятия радиуса. Однако
каждый из способов дает качественно однотипную кар-
картину изменения свойств химических элементов в последо-
последовательности возрастания заряда их ядер, позволяет судить
о строении различных соединений и в отдельных случаях
решать практические задачи.
Глава 20
Теория валентных связей (ВС)
Представления о молекулах. Квантово-механическая
модель молекулы водорода. Сигма-связь. Валент-
Валентность и предел насыщения.
Представления о молекулах. Молекула—наименьшая
частица вещества, обладающая его основными химическими
свойствами и способная к самостоятельному существованию.
Атомы, образующие молекулы, связаны между собой теми
или иными видами химической связи, более прочными,
чем межмолекулярные. По числу атомов молекулы раз-
различаются в чрезвычайно широких пределах: от двух до
многих тысяч (макромолекулы). Наиболее четко молеку-
молекулярная структура проявлена в газах, где связь между
молекулами практически отсутствует. Примерами могут
служить газообразные Н2, NH3, CO2, СН4, суммарная
энергия связей в которых равна 431, 1046, 1406,
1431 кДж/моль соответственно. В молекулярных жидко-
жидкостях и кристаллах прочность межмолекулярных связей
233
Рис. 20.1. Пространственное
чередование атомов кремния и
кислорода в кристаллах кварца
хотя и не равна нулю, но остается небольшой и обяза-
обязательно существенно меньшей, чем прочность связей внутри
молекул. Например, прочность каждой из четырех связей
в молекулах СС14 составляет
234 кДж/моль, а прочность
межмолекулярных связей в
жидком четыреххлористом
углероде—32,6; прочность
связей в молекулах иода
12—151, тогда как энергия
молекулярного взаимодейст-
взаимодействия в кристаллах иода—•
61,9 кДж/моль.
Такой диапазон значе-
значений энергии связей отличает
молекулярные Среды ОТ ИОН-
ных и атомных кристаллов
ИЛИ расплавов, В которых
нельзя выделить какие-либо
чередующиеся внутримолекулярные и межмолекулярные
зоны. Например, в кристаллах кварца SiO2 каждый атом
кремния симметрично окружен четырьмя атомами кисло-
кислорода и это пространственное чередование атомов кремния
и кислорода (рис. 20.1) строго периодично. Все связи
Si-—О одинаковы по прочности и равны 431 кДж, так
что выделить какие-то группы атомов и назвать их моле-
молекулами невозможно. Точно так же в ионных кристаллах
NaCl каждый атом натрия окружен шестью атомами
хлора, а каждый атом хлора — шестью атомами натрия.
И здесь невозможно выделить какие-то обособленные
группы связанных между собой атомов, отвечающих по-
понятию молекулы.
Близкое к современному представление о молекулах сложилось
еще в начале XIX в. и впервые было сформулировано А. Авогадро.
Важным этапом развития учения о химической связи в молекулах
была теория строения А. М. Бутлерова, согласно которой в соедине-
соединениях имеет место строгая закономерность расположения агомов,
а химическая связь ответственна за различия в свойствах соединения
и сумму образующих его атомов. Далее следует отметить развитие
А. Вернером в конце XIX в. представлений о координационных сое-
соединениях и их структурах.
Фундаментом учения о строении вещества явилось открытие
Д. И. Менделеевым периодического закона, пользуясь которым можно
описать свойства соединений, в том числе и молекулярных, основы-
основываясь на свойствах образующих их атомов. Первые варианты теории
химической связи, основанные на представлениях об электронной
природе атомов, принадлежат В. Косселю и Дж. Льюису. В теории
234 ' Ц
Косселя химическая связь объяснена ионным взаимодействием. В эту
модель Льюнс внес уточнение, полагая, что образующие связь элек-
электронные пары могут не только полностью переходить на орбиталь
одного из атомов в соединении (ионная или электрозалентная связь),
но и оставаться в промежуточном положении, т. е. принадлежать
сразу обоим взаимодействующим атсмам (ковалентная или гомеопо-
лярная связь).
В 1925 г. был заложен фундамент квантовой механики и с этого
времени начался период интенсивного развития учения о химической
связи. А в 1927 г. В. Гейтлер и Ф. Лондон, используя квантово-
механическую теорию, вычислили энергию связи в молекуле водорода
и равновесное расстояние атомов в ней. Это событие можно считать
датой рождения нового раздела знания—квантовой химии. В про-
процессе развития этого учения была создана квантово-механическая
теория строения, объяснены направленность, причины возникновения
кратных (двойных, тройных) связей и другие их особенности. Однако
появились и разочарования. Оказалось невозможным дать точное
решение задачи об энергии связей в молекуле более сложной, чем Н2,
а приближенные методы приводили к результатам, существенно
отличающимся от экспериментальных. И тем не менее стало ясно,
что теоретическая химия находится на верном пути и количественное
решение проблемы химической связи является реальной целью буду-
будущего.
Согласно современным воззрениям химические связи
между атомами возникают в результате взаимодействия
электронов и ядер, образующих эти атомы. На рис. 20.2
Рис. 20.2. Схема вза-
взаимодействия двух ато-
атомов водорода (-<—>¦ от-
отталкивание, -*¦ ¦<- при-
притяжение)
изображен как бы моментальный снимок двух сблизившихся
атомов и стрелками указано электростатическое взаимо-
взаимодействие ядер и орбитальных электронов. При этом взаимо-
взаимодействие одноименно заряженных частиц носит характер
отталкивания, а взаимодействие разноименно заряженных
частиц—характер притяжения. Согласно закону Кулона
энергию электростатического взаимодействия заряженных
частиц выражают соотношением
где е—величина элементарного заряда; R — расстояние
между частицами; знак ( + ) или (—) определяет оттал-
235
кивание или притяжение соответственно. Тогда суммарную
энергию взаимодействия атомов, изображенных на рис. 20.1,
можно выразить следующим образом:
АЬ ^ Аа ^ ВЬ =
[ill 4-[ill 4-[il1 — [ill _
I D I I I D I j 1 D 1 I D I
I К JAB \ К lab L " JAb I л J аЯ
ВЬ
i'i-отталкивание Ег-притяжение
B0.1)
Из уравнения B0.1) видно, что при условии Еу < Е2 атомы
будут притягиваться, в результате чего между ними воз-
возникнет химическая связь. Анализ множества химических
соединений показывает, что соотношение B0.1) отражает
факт всеобщей тенденции атомов к химическому взаимо-
взаимодействию, расчет по этому уравнению произвести нельзя,
поскольку значения R во времени быстро изменяются,
так как электроны атомов постоянно находятся в движе-
движении, природа которого волновая и описывается уравне-
уравнением Шредингера. Это означает, что движение электронов
в атомах и их энергия могут быть описаны только на
основе квантовой механики, с учетом их двойственной
(корпускулярной и волновой) природы.
Квантово-механическая модель молекулы водорода. Точ-
Точное значение энергии молекулы, состоящей из N атомов
и п электронов в них, может быть определено лишь путем
решения уравнения Шредингера A8.17). Однако, как уже
отмечалось, возможность такого решения резко убывает
с увеличением числа частиц (электронов и ядер), образую-
образующих соединение. Применив метод квантовой механики,
Гейтлер и Лондон нашли приближенное решение уравне-
уравнения Шредингера для молекулы Я2, причем прибли-
приближенную волновую функцию электронов в молекуле грво
получили из ls-функций изолированных tya- и г|5ь-атомов
водорода
iCbo = C1i()^, B0.2)
где а и Ъ — индексы ядер атомов, штрихи (') и (")—ин-
(")—индексы электронов в них; ВО—валентная орбиталь; С —
постоянный коэффициент. Поскольку электроны и ядра
в атомах водорода совершенно одинаковы, возможен экви-
эквивалентный обмен атомов своими электронами;
¦Уво^С^'ь. B0.3)
236
Истинная волновая функция молекулы водорода должна
учитывать оба варианта движения электронов в молекуле,
т. е. она должна быть линейной комбинацией уравнений
B0.3) и B0.2)
B0.4)
B0.5)
Функция г()|0 получила название симметоичной. Знак
(+ ) в правой части уравнения B0.4) указывает на то,
что при перемене мест ядер атомов или электронов вол-
волновая функция ВО останется неизменной. Функция ty?0
со знаком (—) в правой части уравнения B0.5) получила
название антисимметричной, так как при перемене мест
ядер атомов или электронов знак волновой функции ВО
изменится на обратный. Симметричная волновая функ-
функция г^0 характеризует такую систему орбиталей атомов
водорода (молекул Н2), в которой электроны имеют про-
противоположные, т. е. антипараллельные, спины. Антисим-
Антисимметричная волновая функция ij^0 характеризует систему
атомов водорода с электронами, спины которых парал-
параллельны, т. е. одинаково направлены.
Ранее приведена сокращенная форма уравнения Шре-
дингера:
B0.6)
Умножив правую и левую части уравнения B0.6) на ве-
величину i|)du (здесь dv—бесконечно малый элемент объема
внутримолекулярного пространства), получим выражение
проинтегрировав которое по всему объему молекулы, по-
получим уравнение для расчета энергии молекулы
dv. B0.7)
Множитель \ t|Jdu в правой части уравнения B0.7) харак-
характеризует вероятность нахождения электрона в объеме моле-
молекулы.Очевидно, что такая вероятность равна 1, т. е. при
B0.8)
электрон обязательно должен находиться, где-то в пределах
границ данной молекулы водорода. Это условие получило
название условия нормировки, оно определяет выбор в ка-
качестве единицы той суммарной плотности электронного
237
облака в молекуле, различие в концентрации которого
на различных внутримолекулярных участках и определяет
наличие химической связи.
После подстановки уравнения B0.8) в B0.7) получаем
выражение для энергии молекулы
B0.9)
где г|)-функция характеризует валентную орбиталь, поэтому
возможна подстановка уравнений B0.4) и B0.5) в B0.9),
в результате получаем
= \ Wa^b — ^'a) Н {№'ь — Ш"а
Произведем в правых частях этих уравнений почленное
умножение:
'bVa dw + J ФЖ н УЖ do, B0.10)
й^а dy — $ г^гЙ Я \|)^й da.
Первые два интеграла в каждом из уравнений B0.10)
тождественны, они описывают энергию зарядового взаимо-
взаимодействия электронов со своими ядрами, это так называе-
называемые кулоновские интегралы (/) каждого из атомов водо-
водорода. Вторые два интеграла в каждом из уравнений
характеризуют энергию взаимодействия каждого электрона
с обоими ядрами, т. е. движение каждого электрона около
обоих ядер (обмен электронами), их называют обменными
интегралами (К). Таким образом, уравнения B0.10)
в краткой форме можно записать
ES = I + K, EA = I—K. B0.11)
Кулоновский интеграл / в первом приближении характе-
характеризует энергию изолированных атомов водорода, а обмен-
обменный интеграл /(—энергию химической связи между ними.
238
Из сравнения уравнений B0.11) видно, что в случае
симметричной волновой функции г|)§0 (т. е. когда электроны
в молекуле обладают антипараллельными противополож-
противоположными спинами), знак обменного интеграла соответствует
стяжению атомов и образованию прочной молекулы.
В случае же антисимметричной волновой функции i|^o
(т. е. при параллельных спинах электронов обоих атомов)
знак обменного интеграла соответствует отсутствию хими-
химической связи между атомами водорода. На рис. 20.3 в виде
схемы представлено взаимодействие двух атомов водорода.
Рис. 20.3. Схема перекрывания электронных орбиталей
двух атомов водорода с антипараллельными (а) и парал-
параллельными (б) спинами
Следует отметить, что выражения B0.11) в приведен-
приведенной форме записи не точны, более строгий расчет при-
приводит к выражениям
где S—так называемый интеграл перекрывания, который
показывает степень перекрывания электронных облаков
атомов водорода при образовании молекулы. Если сделать
допущение, что два атома водорода полностью совмести-
совместились, это соответствовало бы условию S=l. Для двух
полностью изолированных, не взаимодействующих атомов
S = 0. Для молекулы Я3 вычисленное значение S = 0,75.
Валентность и предел насыщения. Используя рассмот-
рассмотренный выше квантово-механический способ расчета,
Гейтлер и Лондон вычислили значения энергии химиче-
химической связи и равновесного расстояния между ядрами
атомов в молекуле водорода: Е°н_н = 302,5 кДж; г„ =
= 0,090 нм. Экспериментально определенные значения
?°н_н = 457,3 кД-ж и г0 = 0,074116 нм хотя и отличаются
239
от расчетных, однако близки к ним в пределах той р
ближенности, которая была заложена в модель. Более
точный расчет, осуществленный Колосом и Рутаном A960),
позволил получить значения Е°н_н и г0, практически сов-
совпадающие с экспериментом.
При расстояниях между атомами водорода, отличных
от равновесного г0, энергия связи ?н-н принимает раз-
различные значения, которые также вычисляют квантово-
механическим методом. На рис. 20.4 приведен вид зави-
мости ?н-н—/@> гДе пунктирная кривая соответствует
400-
200-
-200-
-400-
Рис. 20.4. Кривые потенциальной энергии взаимо-
взаимодействия двух атомов водорода:
Ед—с параллельными спннамн электронов; Eg— с анти-
параллельнымн спинами электронов
антисимметричной функции i|)^o, а сплошная линия—сим-
линия—симметричной функции i|)jf0. Уменьшение потенциальной
энергии при сближении атомов водорода соответствует
упрочнению химической связи между ними, наибольшая
прочность последней достигается на равновесном расстоя-
расстоянии между ядрами г0. Кривая, полученная при исполь-
использовании антисимметричной функции г|)$0, соответствует
увеличению потенциальной энергии при сближении атомов
водорода, спины электронов которых параллельны. Следо-
240
вательно, между такими атомами химическая связь воз-
возникнуть не может.
Квантово-механическая модель молекулы получила
количественное подтверждение в экспериментальной химии,
что позволило использовать метод валентных связей (ВС),
или электронных пар, для описания строения и энерге-
энергетики более сложных молекул, образованных из атомов
различных элементов периодической системы. Проведя
расчет энергии химической связи в молекуле Н2, Гейтлер
и Лондон сделали попытку вычислить энергию присоедине-
присоединения к ней третьего атома водорода: (Н2) + (Н) -+(Н3). Расчет
показал, что этот процесс невозможен. Отсюда был сделан
вывод о том, что химическая связь, возникающая в мо-
молекулах за счет появления общей пары электронов, имеет
предел насыщения. Двухэлектронная химическая связь
получила название ковалентной.
Одиночные (или неспаренные) электроны в электрон-
электронных оболочках атомов, за счет спаривания которых воз-
возникает химическая связь в молекулах, называют валент-
валентными. Число общих электронных пар, образующихся при
взаимодействии атомов химических элементов, определяет
их валентность. Так, два атома водорода, имеющие по
одному неспаренному, т. е. валентному, электрону, при
соединении в молекулу Н2 образуют одну общую пару
электронов, т. е. проявляют валентность, равную единице.
Схематически образование молекулы Н2 из двух атомов
водорода можно представить следующим образом:
н + н —*- н2
t
—»-
АОХ АО2 ВО
(АО—внешняя электронная орбиталь изолированного
атома Is1).
Такая связь, которая определена образованием области
повышенной электронной плотности в промежутке между
ядрами атомов, т. е. располагается вдоль линии, соеди-
соединяющей эти ядра, получила название сигма-связи (а-связи).
Атомы следующего за водородом элемента — гелия
имеют строение Is2. Отсутствие неспаренных электронов
в его атомах свидетельствует о том, что валентность гелия
равна нулю (соединений не существует).
241
Глава 21
Молекулы с кратными связями и гибридными
орбиталями
Строение молекул гомо- и гетеросоединеиий с ор-
ординарными а- и кратными л-свяэями. Промотиро-
вание валентного состояния. Гибридизация орби-
орбиталей. Соединения с сопряженными связями, я-
комплексы. Строение кристаллов графита.
Строение молекул гомосоединений. Рассмотрим процессы
образования из свободных атомов двухатомных молекул
гомосоединений элементов второго периода системы
Д. И. Менделеева (значения энергии в кДж):
(Li) + (Li)-107 = (Li,) (N) + (N)-941=(N,)
(Be) + (Be) — F0) * = (Be2) (O) + (O) — 494 = (O2)
(B) + (B)-B59) = (B2) (F) + (F)- 155 = (F2)
(C) + (C) - 598 = (C2) (Ne) + (Ne) - @) = (Ne2)
(энергетические эффекты образования Веа и Ке2 условны,
поскольку эти молекулы не синтезированы). Используя
квантово-механическую модель, можно представить формы
электронных орбиталей атомов этих элементов. На рис. 21.1
показано, что сферические орбитали атомов лития и бе-
бериллия одинаковы, их различает только заселенность
электронами 25-подуровня (Li 2s1, Be 2s2). В атомах бора,
углерода и азота электроны (по одному) последовательно
заполняют гантелеобразные р-орбитали. Формы орбиталей
атомов кислорода, фтора, неона аналогичны атомам азота,
но отличны заселенностью (одноэлектронные р-орбитали
переходят в двухэлектронные).
Рассмотрим механизм образования химических связей
в двухатомных молекулах этих элементов. Два атома
лития соединяются в молекулу по схеме, представленной
на рис. 21.2. Поскольку атомы лития имеют по одному
неспаренному электрону, при их взаимодействии возни-
возникает общая пара электронов. Электронные орбитали атомов
лития перекрываются так, что в промежутке между их
ядрами возрастает плотность электронного «облака» и
образуется единичная (ординарная) химическая а-связь:
Атомы бериллия, не имеющие неспаренных электронов,
химически связываться в молекулу Ве2 не должны. Однако
242
если сообщить атому бериллия энергию, то можно пере-
перевести его в возбужденное состояние (двухвалентное)
Be Bs2) + 518 кДж —* Be*Bs1p1)
и тогда атомы с двумя парами неспаренных s- и р-элек-
тронов экзотермически соединяются между собой, образуя
молекулу, как это показано на рис. 21.3. Химическая
Е
Be
13
11
!
в
С|ГТ|ИШ Nni
(ПЕПЛ
liiJ
Рис. 21.1. Модели электронных орбиталеи атомов элементов второго
периода
связь, образовйнная за счет перекрывания двух pz (или ру)-
орбиталей не по линии, соединяющей ядра атомов, а на
периферии от нее, получила название п-связи (пи-связи)
(перекрывание двух р^-орбиталей вдоль оси х дает ст-связь).
На возбуждение валентного состояния двух атомов берил-
243
лия требуется энергия 518 кДж; энергия о- и я-связей
двух таких возбужденных атомов составляет «578 кДж.
Следовательно, результирующая энергия связи в моле-
молекуле Ве2 должна составлять около 60 кДж. Молекула Ве2
с такой небольшой прочностью уже при температурах
Li + Li - Li2
2s1'+ 2s' - 2<TS2
Рис. 21.2. Схема связи в мо-
молекуле Li2
Рис. 21.3. Схема связей
в гипотетической молеку-
молекуле Ве2
порядка 500—700 °С не должна существовать, тогда как
давление пара кристаллического бериллия на пределе
чувствительности современных приборов (например, масс-
спектрометров) можно
заметить лишь выше
900°С. Поэтому неуди-
вительно, что молекула
Ве2 до сих пор экспери-
ментально не обнаруже-
обнаружена и не изучена.
Атомы следующего
за бериллием элемен-
элемента — бора имеют по одно-
одному неспаренному р-элек-
трону. Применение мето-
метода ВС дает упрощенную модель молекулы В2, образован-
образованной за счет перекрывания р-орбиталей с образованием
одной двухэлектронной <т/!_/,-связи (рис. 21.4):
Рис. 21.4. Схема связей в молекуле В2
Возможен и второй вариант, когда имеет место пред-
предварительное возбуждение одного из 252-электронов каждого
атома бора, и переход его на свободную /?-орбиталь:
В Bs2/?) + 530 кДж — В * Bs/?2)
z44
Для возбуждения, или промотирования, трехвалентного
состояния атомов бора потребуется 1060 кДж энергии
(на молекулу В2), в результате чего в каждом атоме по-
появляется по три неспаренных электрона, которые прини-
принимают участие в образовании связей в молекуле В2. Пере-
Перекрывание р-орбиталей вне лини'и, соединяющей ядра
атомов, соответствует образованию я-связи. Таким обра-
образом, в результате взаимодействия возбужденных атомов
бора образуются одна ст-связь и две я-связи. Суммарная
энергия их составляет 1320 кДж. Отсюда результирующую
прочность связи в молекуле В2 находят из разности
?в ш в = 1320— 1060 = 260 кДж.
Модель с тройной связью в этой молекуле отчасти объ-
объясняет более высокую энергию связи между атомами
бора B60 кДж), чем в молекуле Li2 (энергия связи 107 кДж).
Однако обе модели молекулы В2 не объясняют нали-
наличия у нее магнитных свойств, называемого парамагнетиз-
парамагнетизмом, установленного
экспериментально, и по-
поэтому не могут считать-
считаться строгими.
Атомы углерода име-
имеют в основном состоянии
электронную структуру
CBs2p2), т. е. два не-
неспаренных электрона,
За счет этих электро-
электронов атомы углерода
образуют молекулу
С,, строение которой схематически изображено на
рис. 21.5. В результате перекрывания наполовину засе-
заселенных одноэлектронных р-орбиталей двух атомов угле-
углерода образуются две двухэлектронные связи с энергией
600 кДж (средняя энергия единичной связи 300 кДж).
Исходя из этого, штриховая модель молекулы С2 может
быть изображена следующим образом:
Рис. 21.5. Схема связей в молекуле С,
Для азота характерна электронная конфигурация
атомов — 2s*p3, т. е. наличие в основном состоянии трех
неспаренных электронов (трех одноэлектронных /7-орбита-
лей). За счет перекрывания этих орбиталей образуется
24а
молекула N2, строение которой показано на рис. 21.6у
при этом образуются одна а-связь и две л-связи:
Сумма всех трех связей в этой самой прочной из всех
двухатомных молекул элементов II периода равна 941 кДж,
средняя энергия единич-
единичной связи в молекуле
Ы2341кДж. Это упроч-
упрочнение единичной связи
происходит за счет
уменьшения межъядер-
межъядерного расстояния в мо-
молекуле азота по срав-
сравнению с двухатомной
молекулой углерода в
Рис. 21.6. Схема связей в молекуле N2 1.2 раза и возросшим
перекрыванием элект-
электронных орбиталей.
За азотом в семействе элементов II периода следует
кислород. Строение его внешнего электронного слоя
описывают конфигурацией 2s2p4. Это означает, что из
шести электронов два
являются неспаренны-
ми, т. е. определяющими
валентность атомов кис-
кислорода. Два таких ато-
атома образуют молекулу
О2, строение которой
схематически показано
на рис. 21.7. В соот-
соответствии с представле-
Рис. 21.7. Схема связей в молекуле О2 ниями метода ВС че-
четыре неспаренных элект-
электрона (по два от каждого атома) должны давать одну
сг-связь и одну я-связь:
Суммарная энергия связи составляет 493 кДж, а единич-
единичная—247 кДж, т. е. существенно меньше, чем в молекуле
азота. Межъядерное расстояние в молекуле кислорода
246
больше, чем в молекуле азота, в 1,1 раза, что и опре-
определяет меньшую степень перекрывания его электронных
р-орбиталей и снижение энергии единичной связи. Этому
же способствуют и орбитали спаренных электронов, ко-
которые взаимно расталкиваются, ослабляя связь. Следует
отметить, что изображенная на рис. 21.7 схема строения
молекулы О2 не строга.
Опытным путем было
установлено, что моле-
молекулы кислорода, как и
молекулы В2, обладают
ярко выраженными па-
парамагнитными свойст-
свойствами, т. е. в этих мо-
молекулах должны быть
неспаренные электроны,
которые теорией ВС не Рис' 21 -8- Схема связей в молекуле F2
описываются.
Атомы фтора характеризуются конфигурацией второго
электронного слоя 2s22p5. Это означает, что из семи элек-
электронов внешнего слоя только один остается неспаренным
и определяет валентные возможности фтора. Два атома
фтора за счет образования общей электронной пары со-
соединяются в молекулу, схема которой изображена на
рис. 21.8. Прочность единственной ст-связи в этой моле-
молекуле 155 кДж. Поскольку все остальные орбитали атомов
полностью заселены парами электронов, никаких допол-
дополнительных возможностей для упрочнения связи в моле-
молекуле нет.
Следует отметить, что аналог фтора—хлор образует
двухатомные молекулы С12 с прочностью связи 239 кДж,
такое увеличение прочности (по сравнению с F2) объяс-
объясняют кратностью связи в этой молекуле. В отличие от
фтора атомы хлора имеют свободные d-орбитали:
tilt lit
1 1
d
u
Pi Py Px
В связи с этим химическая связь в молекуле С12 возни-
возникает не только за счет образования ст/,_/)-связи из двух
неспаренных 3^-орбиталей, но и в отличие от молекулы F2
за счет двухэлектроннои 3^-орбитали одного атома хлора
247
и свободной З^-орбйтали другого атома хлора, образуй
две дополнительные я-связи что и объясняет факт упроч-
упрочнения.
Строение молекул гетеросоединений. Дадим краткую
характеристику валентных связей в молекулах гетеросое-
гетеросоединений. В молекуле СО суммарная прочность связи
составляет 1072 кДж, молекула имеет строение
c mi
2s
0 Г77
Pr,
Щ
t
Pv
¦1
Pr
За счет двух неспаренных 2рх- и 2рц-электронов каждого
из атомов в молекуле СО образуются по одной ор_р- и
яр_р-связи. Кроме этого, двухэлектронная ^-орбиталь
атома кислорода и свободная рг-орбиталь атома углерода
за счет донорно-акцепторного взаимодействия образуют
третью, так называемую ковалентную я^^-связь. В связи
с этим структуру молекулы СО можно изобразить следую-
следующим образом:
В результате общее число электронов, участвующих
в образовании тройной связи в молекуле СО, такое же,
как и в молекуле N2. Подобные молекулы называют изо-
электронными.
Рассмотрим строение двухатомной молекулы оксида
азота N0, имеющей прочность связи 627 кДж. Элекрон-
ные орбитали атомов азота и кислорода имеют строение
0
|и
2s
|Й|
¦ 1
Рг
¦¦1
Ру
1
Рх
¦ 1
248
IJ результате перекрывания атомных одноэлектронных рх-
и р^-орбиталей образуются валентные ор_р- и пя_я-с
При этом на орбита ли pz атома азота остается один неспа-
ренный электрон, наличие которого объясняет химическую
активность оксида азота. Так, например, кислородом воз-
воздуха N0 окисляется до N02 при обычной температуре
без участия катализаторов.
Основываясь на кратком анализе энергетики и элект-
электронной структуры некоторых двухатомных молекул, на-
напомним основные положения метода ВС или электронных
пар Гейтлера и Лондона.
1. Химическая связь между атомами возникает за счет
спин-валентного взаимодействия одноэлектронных атомных
орбиталей, число которых определяет валентность элемента
в соединении. Если два таких электрона с противополож-
противоположными спинами (по одному от каждого атома) объединяются
в пару, т. е. образуют валентную орбшпаль, общую для
этих атомов, то получится единичная (или ординарная)
химическая связь. Таких электронных пар (связевых орби-
та"лей) между двумя атомами может быть и более одной.
В этом случае образуются так называемые кратные (двой-
(двойные, тройные) связи.
2. Помимо спин-валентного взаимодействия неспарен-
ных электронов, приводящего к образованию связевых
электронных пар, возможно донорно-акцепторное взаимо-
взаимодействие электронной пары одного атома с вакантной
орбиталью другого. При этом образуется координационная
химическая связь.
3. Образование химической связи сопровождается
интерференцией электронных волн (перекрыванием орби-
орбиталей), т. е. образованием между атомами зон с повы-
повышенной плотностью электронного «облака». Такая связь
имеет пространственную направленность, определяемую
формой и пространственным положением орбиталей вза-
взаимодействующих атомов.
4. Прочность химической связи зависит от степени
перекрывания атомных орбиталей, плотности электронного
облака (плотности вероятности i|j2 dv), а также эффективных
значений заряда и радиуса атома.
Одно из важных достоинств метода ВС—обоснование
явления направленности химических связей в моле-
249
кулах. На рис. 21.9 приведены структуры нескольких
неорганических молекул. Эти структуры резко отличны
и как бы не связаны никакой общей закономерностью.
Действительно, из двух трехатомных молекул Н2О и СО2
первая имеет угловую, а вторая — линейную структуру,
у четырехатомной молекулы NH3 структура пирамиды,
Н2О NH} CHFj
н
109,5°
CO2 BC13. HNO3
11
\
Рис. 21.9. Графические формулы и формы некоторых
неорганических молекул
а у ВС13 — плоского треугольника; пятиатомная моле-
молекула CHF3 имеет структуру тетраэдра, a HNO3—сложную
фигуру, напоминающую скорпиона. Отсюда возникает
естественный вопрос: можно ли с позиций метода ВС
объяснить такое разнообразие форм молекул? Современная
теория дает на этот вопрос положительный ответ, суть
которого заключается в раскрытии смысла направленной
валентности.
Рассмотрим в качестве примера структуру молекулы
H2Se, которая схематически изображена на рис. 21.10.
Внешний, четвертый, электронный слой атома селена Ds2pi)
содержит два нес паренных электрона на одноэлектронных
рх- и рг-орбиталях. Два атома водорода имеют по одному
неспаренному ^-электрону, которые перекрываются рх-
и рг-орбиталями селена. В результате теоретически должна
получиться молекула с углом HSeH, равным 90°, в дейст-
действительности в молекуле H2Se угол равен 91°. Аналогичным
образом в молекуле SbH3 атом сурьмы имеет в пятом
электронном слое Es2p3) три взаимно перпендикулярные
однрэлектронные /?-орбитали, поэтому все три угла Н$1?И
260
должны быть равными 90° и молекула должна иметь вид
тригональной пирамиды. Опыт подтверждает такую струк-
структуру, внося лишь небольшую поправку на угол 91,3°.
Таким образом, если за счет р-орбиталей образуются две
или три ст-связи, то эти связи должны располагаться
между собой под углом, близким к 90°.
Однако из рис. 21.9 видно, что во многих случаях это
условие по каким-то причинам не выполняется. В моле-
молекуле аммиака NH3 вместо ожидаемых углов HNH, равных
90°, опыт дает угол 107,3°. Точно так же и в молекуле
воды Н2О валентный угол получают равным 104,5°. Эти
Se
Н
Рис. 21.10. Схема связей в молекуле H2Se
примеры свидетельствуют о том, что в рассматриваемых
молекулах имеет место иная симметрия, чем это опреде-
определяется строением электронных оболочек центральных
атомов.
Рассмотрим более подробно молекулу ВС13, суммарная
энергия связей которой 1343 кДж. Многочисленные иссле-
исследования показывают, что все атомы хлора в этой молекуле
равноправны, располагаются в одной плоскости и одина-
одинаково удалены от атома бора. Следовательно, энергия
каждой из трех связей равна 448 кДж. Строение электрон-
электронной оболочки нейтрального возбужденного атома бора
с тремя одиночными электронами, т. е. трехвалентного,
таково, что из трех неспаренных электронов Bsp2) один
образует s-орбиталь, а два остальных—р-орбитали, пере-
пересекающиеся под прямым углом. Если три атома хлора
присоединяются к такому атому бора, то два из них,
/7-орбитали которых перекроются с /7-орбиталями атома
251
255 кДж
бора, образуют связевой угол С1 — В — С1, равный 90°.
Третий же атом хлора, ^-орбиталь которого перекроется
со сферической s-орбиталью атома бора, займет на ней
неопределенное положение. Энергии и длины таких связей
должны быть разными, что противоречит эксперименту,
т. е. плоской, симметричной молекуле с тремя совершенно
одинаковыми связями. Аналогично этому опыт приводит
к линейной, а не к уголковой молекуле хлористого берил-
бериллия и строго тетраэдрической молекуле метана.
Гибридизация орбиталей. Для устранения противоре-
противоречия между теорией и экспериментом американские ученые
Дж. Слетер и Л. Полинг предложили модель особым
образом возбужденного цент-
центрального атома в моле-
молекуле. Согласно этой модели
электронные орбитали такого
атома различны по энергии,
а следовательно, по форме
и пространственному распо-
расположению до возбуждения, но
могут быть равными по уров-
уровням энергии после возбуж-
возбуждения. Такие орбитали по-
получили название гибридных,
а сам эффект — гибридизации
орбиталей. Однако следует
отметить, что гибридизация
не физическое явление, это
всего лишь внутренне непро-
непротиворечивая модель, матема-
математический прием, который по-
позволяет количественно опи-
описать реальные структуры
молекул, их энергетику.
На рис. 21.11 приведена
схема возбуждения четырех-
четырехвалентного состояния у атома углерода D00 кДж) и пере-
перехода возбужденных орбиталей в гибридное состояние
B55 кДж), обозначаемое символом q. Эти две энергети-
энергетические ступени возбуждения, требующие для осуществле-
осуществления 655 кДж, могут быть реализованы только при условии,
если последующий экзотермический эффект образования
химических связей с избытком перекроет их. И действи-
действительно, взаимодействие атома углерода, имеющего гибрид-
гибридные орбитали, с четырьмя атомами водорода сопровож-
252
4-
400 кДж
E '
Рис- 21.11. Схема возбуждения
гибридного состояния орбиталей
в атоме углерода
дается экзоэффектом
' Сги6р+4Н = СН4> ДЯ° = — 2317 кДж
избыток которого над эндоэффектом гибридизации состав-
составляет — 23174-655 = —1662 кДж или на каждую связь
Таблица 21.1. Форма невозбужденных и гибридных орбиталей
в некоторых многоатомных молекулах.
Основное
состояние
атома
Гибридное
состояние
атома
Молекула
с гибрид-
гибридными
связями
Структурная
формула
молекулы
Be
180°
ВеС1,
С1-—Be—т-С1
180°
120°
BF,
109,5е
сн4
N
NH,
PCI,
Cl
CI
20°
С I
90°
I
253
С—Н —416 кДж. Этот энергетический выигрыш и стаби-
стабилизирует гибридное состояние орбиталей, в результате
чего образуется прочная симметричная, тетраэдрическая
молекула СН4.
Важным условием гибридизации орбиталей является
близость между энергетическими уровнями исходных атом-
атомных орбиталей. Если различие в уровнях энергии (негиб-
(негибридных орбиталей) велико, то энергия гибридизации (воз-
(возбуждения) будет столь большой, что окажется невозмож-
невозможным перекрыть ее энергией образовавшейся химической
связи.
В табл. 21.1 приведены схемы невозбужденных и гиб-
гибридных орбиталей атомов некоторых элементов в обра-
образуемых ими соединениях.
Из таблицы следует, что гибридные орбитали централь-
центрального атома должны отвечать требованию наибольшего
перекрывания орбиталей соединяющихся атомов. Поэтому
гибридные орбитали атомов в молекулах обеспечивают
наиболее прочные связи, как это видно из табл. 21.2.
Таблица 21.2. Сравнение прочности
молекул и радикалов
Соединение
сн
сна
сн3
сн4
NH
NH2
NH,
ОН
ОНа
Связь
С-Н
С-Н
с—н
с—н
N —Н
N —Н
N —Н
О-Н
О —Н
Характер
связи
Простая
Гибридная
»
»
Простая
Гибридная
»
Простая
Гибридная
гибридных и
Энергия
связи, кДж
347
43!
410
4L
356
368
389
423
462
негибридных
Упрочнение,
кДж
84
63
67
12
33
39
У атомов, заряд ядра которых мал и, следовательно,
валентные электроны занимают близкие к ядру орбитали,
для гибридизации в основном используются лишь s и
^-орбитали (различее в энергии s- и d-орбиталей слиш-
слишком велико). У атомов с большим зарядом ядра валентные
электроны занимают далекие от него орбитали, поэтому
разница в энергии s-, p- и d-орбиталей уменьшается и
они могут принимать участие в гибридизации. Так, в мо-
молекуле ВеС13 имеет место sp-гибридизация орбиталей
атома бериллия. Результирующие волновые функции этих
254
гибридных ^-орбиталей:
Эти две функции являются зеркальным отражением друг
друга. Отвечающие им орбитали пространственно распо-
располагаются так, как показано в табл.
21.1.
В молекуле СН4 у атома углеро-
углерода имеет место яр'-гибридизация. Ре-
Результирующие волновые функции
при этом имеют вид:
% (
4-4 = -g- (*, - Ьх -
Рис. 21.12. Схема ги-
гибридных хр2-орбиталей
в атоме углерода в мо-
молекулах с, сопряжен-
сопряженными связями
Эти гибридные sp'-орбитали направ-
направлены пространственно в вершины
тетраэдра.
Включение d-орбиталей в процесс образования хими-
химических связей делает возможным осуществление плоской
квадратной конфигурации расположения гибридных орби-
талей, в ионе AuCl, четыре гибридные волновые функции
имеют вид:
(здесь нижний индекс х2—уг указывает на то, чтой-орби-
таль располагается в плоскости ху вдоль осей координат).
С участием d-орбиталей возможна и более сложная ком-
комбинация гибридных орбиталей, как это имеет места, на-
255
пример, в ионах PtClf- (сB5/?8-гибридизация) и [Mo(CN)g]*-~
(^^-гибридизация). В первом случае гибридизация орби-
орбиталей приводит к образованию октаэдрической структуры,
во втором—к образованию додекаэдрической структуры
иона. В табл. 21.3 приведены типы симметрии и форм
молекул (ионов), отвечающих различным видам гибри-
гибридизации.
Таблица 21.3. Типы симметрии молекул (ионов)
Вид
гибридизации
sp-
sp*-
sps-
dsp*-
dsp*-
dhps-
dhp3-
Симметрия молекулы
(иоиа)
Линейная
Плоская треугольная
Тетраэдрическая
Плоская
Тригонально-бипира-
мидальная
Октаэдрическая
Додекаэдрическая
ВеС12
BF3,
СС14,
АиСЦ
PCI5,
ptcii
Примеры
, HgCl2, CO2
NOa"
NH4+, POj", BF4"
", Pt(NH3JCl2
NbCl6, MoCl5
-, Co(NH3)?+, Cr(H2O)^+
Mo (CN)I"
Модель гибридизации электронных орбиталей не обя-
обязательно распространяют на все орбитали атома. Известны
случаи, когда модель требует считать часть орбиталей
гибридными, т. е. усредненными по энергиям и дополни-
дополнительно симметризованными по расположению в прост-
пространстве, а часть орбиталей — негибридными. Рассмотрим
подобный случай на примере некоторых производных
углерода. Известно, что строение таких соединений, как
этилен С2Н4 и фосген С0С12, объясняют одинаковым харак-
характером гибридизации орбиталей атомов углерода в этих
молекулах. Из четырех одноэлектронных орбиталей атома
углерода (одной s и трех р) три орбитали считают гиб-
гибридными, они образуют своим расположением фигуру
правильной трехлучевой звезды E/?2-гибридизация), а одна
/7-орбиталь остается негибридной. Она располагается пер-
перпендикулярно к плоскости звезды, как показано на
рис. 21.12. За счет гибридных 5/?2-орбиталей атом угле-
углерода образует три а-связи (две—с атомами других эле-
элементов и одну—с соседним углеродным атомом, имеющим
аналогично расположенную р-орбиталь).
Схемы структур молекул этилена и фосгена приведены
на рис. 21.13, из которых видно, что кроме а-связи между
атомами углерода в молекуле этилена образуется также
256
одна it-связь. В результате два атома углерода в этилене,
а также атомы углерода и кислорода в фосгене, оказы-
оказываются связанными двойными связями и при этом атомы
водорода в этилене располагаются в одной плоскости.
Рис. 21.13. Схема молекул с а- и л-связями при наличии
5р2-гибридизации орбиталей атома углерода
Энергия в молекуле этилена распределена по связям так,
что ординарная связь С—Н D14 кДж) получается при-
примерно в 1,5 раза менее прочной, чем двойная С = С
E95 кДж): дополнительная я-связь существенно упроч-
упрочняет молекулу этилена. Напротив, в молекуле этана С2Нв,
где для орбиталей атомов углерода характерна sp3-ru6pu-
дизация и отсутствие я-связи, прочность связи С-—С
C47 кДж) меньше прочности связи С—Н D13 кДж).
Таким образом, особенность з/?2-гибридизации и участие
негибридной р-орбитали в образовании связи создают
эффект упрочнения связи углерод—углерод в моле-
молекуле С,Н4 и увеличивают ее кратность (связь становится
двойной). Из рис. 21.13 видно, что аналогичным образом
атом углерода в молекуле фосгена СОС12 образует с ато-
атомами хлора непрочные ординарные cr-связи, а с атомом
кислорода-—прочную двойную (одну а- и одну я-связь).
Соединения с сопряженными связями. Перейдем к рас-
рассмотрению соединений, содержащих цепи атомов углерода,
орбитали которых характеризуются з/?2-гибридизацией.
Такие атомы могут соединяться между собой не только
по два (как в молекуле С2Н4), но и более, образуя цепи,
как, например, в молекуле СвН8. Как показано на
рис. 21.14, каждый атом углерода в этой молекуле имеет
три гибридные 5/?2-орбитали, за счет которых между ато-
атомами образуются ог-связи. Негибридные р-орбитали всех
9 № 705 257
атомов углерода одинаково ориентированные в прост-
пространстве, перекрываясь друг с другом, образуют непрерыв-
непрерывную я-орбиталь по всей длине молекулы С,Н8, которая
получила название нелокализованной (делокализованной)
или сопряженной связи. Эффект сопряжения р-орбиталей
приводит к образованию единой для всех объединенных
этой связью атомов углерода л-орбитали, с непрерывной
во всех ее частях плотностью электронного облака (эффект
делокализации электронов).
Если в рассмотренной выше молекуле СвН8 каким-либо
способом оторвать по одному атому водорода и освобо-
освободившиеся связевые орбитали замкнуть между собой, то
Рис. 21.14. Схема а- и сопряженных л-связей в молекуле СвН8
получится циклическая плоская, шестичленная молекула
бензола СвН„, схематически изображенная на рис. 21.15, а.
Здесь имеет место система сопряженных связей, равно-
равномерно распределенных вдоль всего бензольного кольца и
придающих ему повышенную прочность. Отсюда экзоэр-
гичность реакции
(СвН8) = (С„Нв) + (Н2), AG°T = - 24270- Т 153,6
Точно так же чрезвычайно экзоэргична реакция образо-
образования бензола из трех молекул ацетилена СаН„ с проч-
прочными тройными связями
3 (C,H2) = (CeHe), AG; = _597310 + Г337,6
а также из трех молекул этилена С2Н4
3 (С2Н4) = (С„Нв)+3 (Н2), AG°T = -73 930— Г2,5
Примером неорганического соединения с сопряженными
двойными (конъюгированными) связями может служить
меламин C3NS(NH2K, структура которого изображена на
рис. 21.15, б, в. Это соединение экзотермически получают
258
полимеризацией цианамида CNNH2t
AG°T =—248 530—П88
В образовавшейся циклической молекуле чередуются
атомы углерода и азота, которые, как и атомы углерода
NH-
-NH,
б в
Рис. 21.15. Схема сопряженных связей в молекулах бензола и ме-
ламина C3N3 (NH2K
в молекуле бензола, связаны сопряженными или конъюги-
рованными связями. Из приведенного выше уравнения
следует, что и в этом случае энергетически выгодно обра-
образование в молекуле делокализованной я-связи, т. е. моле-
молекула меламина стабилизирована эффектом сопряжения
связей.
Подобные циклические молекулы с делокализованной
сопряженной л-орбиталью описаны и в органической, и в
неорганической химии, они получили название аромати-
ароматических. Исторически это название возникло в отношении
9* 259
производных бензола, многие из которых обладают прият-
приятным запахом. Позже к ароматическим стали относить
производные нафталина, антрацена, фенантрена и др.,
молекулы которых содержат сочлененные по граням бен-
Н
н .
/ >*
вн
d e ж Н
Рис. 21.16. Структуры гетероциклических соединений:
а — фуран; б —тиофен; в—пиррол; г —пиразол; д — пиридин;
е—катион тропилия; о/с—катион циклопропенилия; з—боразол
зольные кольца. Было установлено, что образование
сопряженных я-связей энергетически упрочняет их. Так,
в бензоле это упрочнение составляет 167 кДж/моль, в наф-
нафталине— 314, в антрацене—440 кДж/моль.
По мере синтеза и изучения соединений с сопряжен-
сопряженными связями понятие ароматичности было перенесено на
многочисленные гетероциклические соединения, часть кото-
которых схематически изображена на рис. 21.16 (/—7). В этих
молекулах, как и в молекуле бензола, образуется сопря-
сопряженная система из шести л-электронов, хотя в вершинах
и располагается только пять атомов. Здесь в образовании
делокализованной л-орбитали принимают участие четыре
/7-электрона атомов углерода и два /^-электрона атома
кислорода (серы, азота). Энергия сопряжения в этих
молекулах также достаточно велика, хотя и несколько
меньше, чем в бензоле. Например, в молекуле тиофена
эта энергия равна 130 кДж/моль, в молекулах пиррола
и фурана — около 96 кДж/моль.
я-Комплексы. К неорганическим ароматическим соеди-
соединениям относится боразол, а к металлорганическим—фер-
металлорганическим—ферроцен (рис. 21.17, а). Ферроцен Fe(C6H5J является
комплексным соединением иона металла и двух аромати-
ароматических ионов. Такие соединения получили название
металлоценовых я-комплексов, в них делокализовайяая
260
л-орбиталь выполняет роль носителя отрицательного за-
заряда (донора электрона). Другими примерами подобных
соединений могут служить
(С5Н5JМь (С6Н5JМХ; (С5Н5)аМаХ3,
где Мх—Ti, V, Сг, Mn, Fe, Co, Ni; М2—Nb, Mo, Та, W;
X — галогены, а также соединения с бензолом, например
(С6Н6JСг.
Молекулы всех этих соединений построены по одному
типу: делокализован-
ная, кольцеобразная
л-орбиталь циклических
молекул СвН,, или ио-
ионов С5Н5, как бы вен-
венком накрывает d-орби-
тали центрального атома
или иона, образуя хи-
химическую связь. Две
такие циклические мо-
молекулы, «надетые» с
двух сторон на метал-
металлический атом, образуют
структуру, внешне по-
похожую на бутерброд лекул.
(сандвич). Отсюда и o-(Csf
название этих л-комп-
лексных соединений — сандвичевые.
Соединения типа л-комплексов могут также образо-
образовываться при участии ароматических молекул, не имею-
имеющих циклического строения. Так, например, молекула
бутадиена Н2С = СН—СН=СН2 образует устойчивое сое-
соединение с молекулой карбонила железа, структура кото-
которого показана на рис. 21.17, в.
Соединения типа л-комплексов, как правило, обра-
образующие молекулярную кристаллическую решетку, пла-
плавятся и испаряются при низких температурах. Например,
ферроцен плавится при 173 °С, легко возгоняется при
100 °С, при нагревании выше 470 °С ферроцен разлагается
с образованием металлического железа (ион железа в фер-
ферроцене обычными методами химического анализа не обна-
обнаруживается). Этот путь разложения характерен для боль-
большинства л-комплексов.
Явление делокализации ^-орбиталей в химических
соединениях представлено достаточно широко и показано
не только в отдельных многоатомных молекулах. Харак-
261
Н
Рис. 21.17. Схемы я-комплексных мо-
|, Fe; б-(С4И.) Fe (СО3)
терный эффект сопряжения /?-орбиталей и связанная с этим
делокализация электронов имеет место в графите. Кри-
Кристалл графита построен из атомов углерода, образующих
совокупность плоских сеток (рис. 21.18). Каждый атом
углерода в такой сетке связан с тремя другими углерод-
углеродными атомами 5/?2-гибридными связями. Четвертый же
электрон каждого атома углерода образует /?-орбиталь,
Рис. 21.18. Схема
делокализованных
р-орбиталей (л-свя-
зей) в решетке гра-
графита
перпендикулярную к плоскости рисунка (окружности на
рисунке—проекции этих /?-орбиталей). В результате все
Б/Я-гибридные связи в каждой структурной ячейке графита
упрочнены за счет дополнительных я-связей (на рисунке
области перекрывания /?-орбиталей). Это, с одной стороны,
приводит к упрочнению и укорочению связей (в алмазе
межъядерное расстояние равно 0,154 нм, в графите, в пло-
плоскости сетки—0,142 нм), а с другой—к появлению
эффекта делокализации /^-электронов, которая ответст-
ответственна за электронную проводимость графита. Расстояние
между такими сетками велико—0,335 нм, что определяет
«чешуйчатую» структуру графита и легкость расщепления
его кристаллов по плоскостям спайности на тончайшие
пластинки. Энергия разрыва связей графита между ато-
атомами углерода в плоскости каждой сетки составляет
715 кДж, а между сетками — всего 17 кДж. Сопряжение
/ьорбиталей приводит к образованию эффекта делокализа-
делокализации электронов. В результате такой делокализации (т. е.
равнораспределения) электронов в молекуле теряет смысл
чередования ординарных и двойных связей, которые ста-
становятся равнонапряженными во всех частях и потому
более прочными. ^
262
Весь рассмотренный выше материал по теории химических про-
цессов и теории строения является базой для анализа представлений
о валентности атомов в химических соединениях. В естественно-фило-
естественно-философском смысле валентность является, как известно, количественной
мерой способности атомов к образованию химической связи. Это фун-
фундаментальное, но не однозначное понятие прошло долгий путь эволю-
эволюции, пополняясь различным содержанием по мере накопления факти-
фактического материала. И каждый раз, когда экспериментаторы открывали
новый класс соединений с необычными структурами и свойствами,
понятие валентности существенно изменялось и дополнялось. Поэтому,
чтобы ответить на поставленный вопрос, необходимо вспомнить
историю.
Первый этап развития представлений о валентности обычно назы-
называют формальным. В начале XIX в. Дальтон сформулировал закон
кратных отношений, который позже был распространен также на
бтдельные атомы и молекулы. Именно способность атомов связывать
или замещать определенное число других частиц в соединениях и
получила название валентности, которую измеряли целыми числами.
При этом первоначально в качестве стандарта (шкалы валентностей)
был выбран водород с валентностью, равной единице, а позже — кис-
кислород с валентностью, равной двум.
Во второй половине XIX в. был сформулирован принцип четырех-
четырехвалентное™ углерода, ставший фундаментом органической химии.
В таком виде понятие валентности вошло в теорию строения А. М. Бут-
Бутлерова A861), согласно которой каждую химическую связь считали
направленной и строго локализованной между двумя атомами. Моле-
Молекулы при этом изображали в виде структурных формул, в которых
штрих отождествлял единицу валентности. Периодический закон
Д. И. Менделеева расширил понятие валентности, связав его с поло-
положением элементов в группах. Однако с рождением химии комплекс-
комплексных соединений представления о строго определенной валентности
атомов оказались недостаточными.
Начало XX в. ознаменовалось большими успехами атомной и
и молекулярной физики. Этот период характеризуется переходом от
формальных представлений о валентности ¦ к электронной теории
Льюиса и Косселя A916—1917), согласно которой каждому валент-
валентному штриху в структурной формуле соответствует связевая электрон-
электронная пара и понятие валентности было отождествлено с числом неспа-
ренных электронов внешней оболочки атомов. Эта теория объяснила
насыщаемость ковалентных химических связей и ненасыщаемость
ионных, а также привела к пониманию зависимости валентности не
только от природы атома, но и от его окружения. Однако и элект-
электронная теория оказалась недостаточно строгой, поскольку она не
могла объяснить валентность без дополнительных данных о геомет-
геометрической структуре соединений.
Выход из создавшегося тупика был открыт квантово-мехаиическои
моделью валентности A927), когда В. Гейтлер и Ф. Лондон впервые
описали строение молекулы водорода с позиций квантовой механики.
Основным выводом из результатов этого приближенного расчета
появилась уверенность в том, что химическая связь может быть одно-
однозначно определена на основе законов квантовой механики. Дальней-
Дальнейшее развитие этих идей и распространение их на многоатомные
молекулы привело к созданию теории валентных, или локализованных,
связей. Согласно этой теории все связи в молекуле независимы друг
от друга и строго локализованы в межатомных промежутках. Каждая
такая связь образована двумя электронами с антипараллельными
263
спинами, а валентность атомов определяется не только числом неспа-
ренных электронов, но и состоянием их на атомных орбиталях.
Теория валентных связей сыграла большую роль в развитии
представлений о химической связи, однако ей не хватило внутренней
согласованности, ее математический аппарат оказался слишком гро-
громоздким и не позволил провести расчеты достаточно сложных моле-
молекул. Кроме того, стали известны соединения, строение которых прин-
принципиально не согласуется с теорией валентных связей. Например,
в молекуле диборана ВаНв число межатомных промежутков В — Н (8)
больше числа электронных пар F); в циклопентадиениле железа
Fe(C5H5J атом железа связан с 10 атомами углерода, хотя у них
и нет 10 электронных пар, необходимых для образования таких свя-
связей. Точно так же с точки зрения метода локализованных пар не
могли быть описаны соединения, содержащие связи металл — металл
(кластеры типа Re3CI9), соединения нульвалентных металлов (карбо-
нилы типа Сг (СО6) и т. д.
В поисках объяснения подобных фактов была разработана теория
молекулярных орбитсыей (МО), согласно которой любая молекула
характеризуется набором МО, делокализированных между парами
атомов и охватывающих всю молекулу в целом. Поскольку в рамках
этой теории отпадает необходимость соответствия числа электронных
пар числу межатомных промежутков, с его помощью стало возможным
объяснить и количественно описать строение любых, самых необычных
молекул.
Развитие метода МО и использование его в теории строения при-
привело к существенному сдвигу в представлениях о валентности, кото-
которая утратила предметную четкость и самостоятельность. В результате
в настоящее время существуют несколько отличных друг от друга
понятий валентности: мера реакционной способности свободных ато-
атомов, структурная характеристика атомов в соединении, индивидуаль-
индивидуальная единица сродства связанного атома.
Эволюция представлений о валентности еще не завершена. В на-
настоящее время трудно предсказать, каков будет дальнейший путь
развития этого важного, но сложного естественно-философского понятия.
Глава 22
Комплексные соединения и координационный
тип химической связи
Донорно-акцепторный механизм взаимодействия.
Комплексные соединения в растворах электролитов.
Координационный тип химической связи. Теория
кристаллического поля. Влияние природы лигандов
на расщепление энергетических уровней d-орбита-
лей центрального атома-комплексообразователя.
Донорно-акцепторный механизм взаимодействия. Би-
Бинарные соединения типа Н2О, NH3, SO3, SiCl4 и т. д.
называют соединениями первого порядка. Более сложные
вещества, полученные взаимодействием соединений первого
порядка, но не путем простого замещения в них одних
компонентов на другие, называют соединениями высшего
порядка. К соединениям высшего порядка относятся гид-
роксиды, соли, аммиакаты и кристаллогидраты солей, про-
продукты присоединения органических молекул к неоргани-
неорганическим, двойные соли.
Графическое изображение простейших структур гидро-
ксидов и солей кислородных кислот должно подчиняться
правилу, основанному на представлениях о степени окис-
окисления атомов химических элементов. Так, графические
формулы сульфата меди и гидроксида алюминия, изобра-
изображенные в соответствии со степенями окисления (формаль-
(формальными зарядами) элементов
/°ч /° /О—Н
Си< >&? ,0— А1<
хо7 х) н/ хо—н
отражают структуры этих соединений в свете представле-
представлений о формальной валентности. Структуру сульфата меди
как соли можно изобразить в ионной форме
Cu2+[oXo
Однако опыт показывает, что эти валентно-насыщенные
соединения способны дополнительно присоединять моле-
молекулы воды, образуя новые, более сложные химические
соединения определенного состава — CuSO4-5H2O,
А1(ОН,)-ЗН2О:
Н20 Н20 О Н20 Н20 ОН
Ч/ II Ч/ ОН | 7Н20
— O-Cu-0-S-O Cu-0 >A1<
/Ч II /Ч ОН/ | ХН2О
Н20 Н20 О --- Н20 - Н20 Н20 Н20
Эти соединения высшего порядка называют комплексными,
они имеют в своем составе устойчивые, не укладывающиеся
в рамки формальной валентности комплексные группы
(комплексный катион [Си(Н2ОL]2+ и комплексный анион
[А1@Н)в]3~), которые обладают специфическими химиче-
химическими свойствами и во многих химических реакциях пере-
переходят из соединения в соединение, не изменяясь.
Напротив, группировка (ион) S0|~, хотя по форме
и похожа на комплексную группу, отличается от послед-
последней тем, что структуру ее можно определить из представ-
265
лений о формальных степенях окисления составляющих
ее атомов (формальной валентности). Эта группировка
названа радикалом. Примерами других радикалов могут
служить РО1" и N0^. Как правило, радикалы в виде
ионов в кристаллах и расплавах или в виде гидратиро-
ваииых ионов в водных и неводных растворах обладают
высокой устойчивостью. Например, процессы образования
из простых веществ ионов {SO|~}aq и {NO.f}aq характери-
характеризуются большими экзотермическими эффектами (кДж):
—907,6; —206,6 соответственно. Структура таких ионов
в разбавленных растворах пространственно симметричная.
Так, ионы SO|~ и MnOj" представляют собой тетраэдры,
в центрах которых находятся атомы серы и марганца,
а в вершинах—атомы кислорода; ионы NOj представляют
собой плоский равносторонний треугольник, в центре кото-
которого находится атом азота, а в вершинах — атомы кисло-
кислорода; ион N0^ имеет уголковую форму.
Комплексные соединения образуются донорно-акцеп-
торным способом за счет присоединения комплексообра-
зователем определенного числа других ионов, молекул или
радикалов, именуемых лигандами. Наиболее распростра-
распространенными следует считать следующие лиганды: анионы —
С1-, CN-, SOJ-, ОН", О2", NH,-, NH2-, SCN-; нейтраль-
нейтральные— Н2О, NH3, CO, NO. В качестве атомов-комплексо-
образователей могут быть нейтральные и заряженные
частицы. Нейтральными комплексообразователями служат
атомы многих элементов побочных подгрупп V, VI, VII,
VIII групп периодической системы. Так построены, напри-
например, молекулы карбонилов металлов Сг(СО)„, Мп2(СОI0,
Fe3(CO)l2, Ni(COL. В этих соединениях нейтральные атомы
металлов связаны с атомами углерода карбонильных групп
СО за счет неподеленнои пары электронов атомов углерода
и вакантных орбиталей никеля, образуя структуру типа
ОСч .СО
ОСУ* \СО
Примерами соединений с заряженными атомами-ком-
плексообразователями—могут служить [Cu(NH3L]Cl2,
K3[Fe(CN)e], строение которых графически изображено
следующим образом (стрелка—валентная пара электронов,
возникшая донорно-акцепторным путем):
K+rCN-
K+ CN-FeCN
Cl- K+LCN-/' \CN
-
-J
266
Здесь в качестве лигандов используются нейтральная мо-
молекула NH3 и ион CN~, которые входят в координацион-
координационную сферу комплексообразователей за счет свободных
электронных пар атомов азота и углерода:
Н
N = C-—* H-N—*
Таким образом, образование комплексов из соединений
первого порядка не связано с возникновением новых элек-
электронных пар.
Различные виды комплексных соединений получили
специфические названия. Так, продукты, содержащие
в составе комплексных ионов молекулы воды, называются
кристаллогидратами или аквакомплексами (CuSO4-5H2O);
продукты присоединения аммиака — аммиакатами
(CuSO4• 3NH3); продукты взаимодействия двух солей, кото-
которые в воде диссоциируют на простые ионы,—двойными
солями (например, карналлит KClMgCl26H2O); вещества,
которые при взаимодействии с водой образуют комплекс-
комплексные ионы, а в качестве второго продукта диссоциации ионы
водорода, гидроксила или металла, получили название
ацидокомплексных соединений (например, HBF4 в водном
растворе распадается на ионы Н+ и BFV).
Число, показывающее, сколько лигандов может коор-
координировать около себя данный центральный атом или
комплексообразователь, называется координационным чис-
числом (К.Ч). Наибольшее число лигандов, способное присо-
присоединяться к центральному атому, определяет максималь-
максимальное координационное число. Соединения, в которых исполь-
использовано максимальное координационное число, называют
координационно-насыщенными. Так, СаС1а с аммиаком об-
образуют четыре комплексных соединения, из которых
CaCl2-NH3 содержит ион Ca(NH3J+; CaCl2-2NH3—линей-
CaCl2-2NH3—линейный ион Ca(NH3)^+; CaCl2-4NH3—ион, в котором моле-
молекулы NH3 располагаются в вершинах тетраэдра, а Са2+ —
в центре; CaQ2-8NH3—ион, в котором молекулы NH3
располагаются в вершинах куба, а Са2+ — в центре. Из
приведенного примера видно, что один и тот же комплексо-
комплексообразователь может образовывать несколько координаци-
координационных соединений. При этом координационно-насыщенным
является только октааммиакат хлорида кальция.
Комплексные соединения, центральный атом которых
имеет К.Ч, равное 2, встречаются достаточно редко; эти
267
комплексы чаще имеют линейную структуру; КЧ — 3 можно
наблюдать только у легких элементов. КЧ = 4 соответ-
соответствуют три конфигурации: тетраэдрическая, плоская квад-
квадратная и седловидная (рис. 22.1, /, 2, 3). Из них наиболее
распространена тетраэдрическая; плоская квадратная
и седловидная встречаются реже, их можно наблюдать
в соединениях элементов VIII группы, а также у меди
и золота. Пространственными фигурами при КЧ==5 могут
быть тригональная бипирамида и квадратная пирамида
*\
Рис. 22.1. Наиболее характерные формы расположения ли-
гандов около атома (иоиа)-комплексообразователя:
а—тетраэдрическая; б—плоскоквадратиая; в—седловидная; г—три-
гональио-бнпирамидальиая; д—тетрагонально-пирамидальная; е-сж-
таэдрическая
(рис. 22.1, 4, 5). Комплексные соединения, центральный
атом которых имеет КЧ = 6, многочисленны, пространст-
пространственная конфигурация расположения лигандов в этих со-
соединениях отвечает фигуре октаэдра (рис. 22.1, 6), при-
примерами могут служить Na2SiFe, K2PtCle, [Pt(NH3LCl2]Cl2.
КЧ > 6 характерно для элементов V, VI и VII периодов
(переходных, лантаноидов, актиноидов), т. е. элементов
с большими размерами ионов и сложным профилем элек-
электронных оболочек. Примерами соединений с КЧ > 6 слу-
служат ионы UF?-, ZrFiJ-, TaFI", Mo(CN)i-, ReHS" и др.
268
Комплексные соединения в растворе электролитов. Изо-
Изомерия. Комплексные соединения подразделяют на электро-
электролиты и неэлектролиты. Электролиты при растворении
в воде (или ином растворителе) взаимодействуют с ней,
диссоциируя на ионы. Неэлектролиты электролитической
диссоциации не подвергаются.
Комплексные соединения — электролиты, по специфике
диссоциации делятся на кислоты, основания и соли. На-
Например, растворение в воде кристаллического комплекс-
комплексного гидрата H2PtCle-6H2O описывают схемой
Эта реакция идет с образованием ионов водорода, следо-
следовательно, раствор этого гидрата представляет собой ком-
комплексную кислоту. Растворение в воде кристаллического
(NH4JSiFe описывают схемой
согласно которой (NH4JSiFe — комплексная соль. Ком-
Комплексное соединение Ag(NH3JOH взаимодействуют с водой
по схеме
[Ag(NH3JOH] + aq =
т. е. раствор обогащен ионами гидроксила и приобретает
основные свойства, следовательно, Ag(NH3JOH— осно-
основание.
Поскольку комплексные соединения, как правило,
сложны по составу и структуре, важным и распростра-
распространенным свойством их становится изомерия, т. е. сущест-
существование соединений с одинаковым химическим составом, но
с различным строением и свойствами. Известны два основ-
основных типа изомерии: структурная и пространственная.
Для структурной изомерии характерно различие рас-
расположения атомов или их группировок в комплексном
соединении. Среди комплексных соединений отмечают два
варианта структурной изомерии: а) ионную изомерию
с различным распределением ионов между частями ком-
комплексного соединения (например, соединение состава
СгС13-6Н2О существует в различных структурных компо-
композициях): [Сг(Н2ОN]С1„ [Сг(Н2ОNС1]С12-Н2О и
[Сг(Н2ОLС12]С1-2Н2О); б) координационную изомерию с раз-
различным распределением лигандов между комплексообра-
зователями, например:
[Cu(NH8L]a+.[PtCl4]«- или [Pt(NHsL]2+.[CuCl4]2-
[Co(NH3)e]3+[Cr(CNe)p- или [Cr(NH3)e]s+-[Co(CN)e]?-
269
Для пространственной изомерии характерно различное
чередование атомов или атомных групп в пространствен-
пространственной модели соединения при одинаковой геометрической
Структуре, что может быть проиллюстрировано комплекс-
комплексным соединением Pt(NH3JCl2, которое образует две раз- j
личные плоские изомерные формы
NHS C1
H3N-Pt—C1 и H3N-Pt-NH3
Один из вариантов пространственной изомерии—зеркаль-
изомерии—зеркальная изомерия, в этом случае изомеры соотносятся между
Собой, как предмет со своим зеркальным изображением.
Учение о координационных и комплексных соединениях возникло
в конце XIX в. В 1893 г. А. Вернер сформулировал основной посту-
Лат координационной теории: «Даже тогда, когда согласно учению
О валентности связующая способность определенных атомов уже ис-
исчерпана, атомы эти все же обладают еще способностью участвовать
в дальнейшем построении комплексных молекул с образованием совер-
совершенно определенных атомных сочетаний. Причина этой способности
атомов к дальнейшему присоединению заключается в том, что в них,
кроме главных валентных сил, имеются еще силы — силы побочной
валентности».
Некоторую определенность в представлении о природе главных
и побочных валентных сил внесла теория электронных пар Льюиса,
которая качественно объясняла образование комплексных соединений
с нейтральными молекулами (аммиак, вода).
Теория, объясняющая образование комплексных соединений с по-
позиций электростатического взаимодействия ионов, была разработана
Косселем A916—1922). Согласно модели, лежащей в основе этой тео-
теории, ионы лигандов и комплексообразователя представляют собой
абсолютно твердые сферы с зарядом, сосредоточенным в центре. Ис-
Используя эти представления, а также закон Кулоиа, можно вычислить
энергию связи в любом комплексном ионе. Однако ионная модель,
качественно правильно описывая строение комплексных соединений,
не дает количественного совпадения экспериментальных и теоретиче-
теоретически вычисленных значений энергии связи. Наибольшее расхождение
имеет место в тех случаях, когда лиганды являются не ионами,
а нейтральными молекулами. Расчеты Косселя показали также, что
при увеличении числа лигандов силы взаимного расталкивания их
возрастают, в связи с чем уменьшается прочность связей между ли-
гандами и центральным атомом.
Дальнейшая разработка теории координационной связи была осу-
осуществлена Н. Сиджвиком A927), развившем представления о донорно-
акцепторном взаимодействии центрального атома с лигандами в ком-
комплексных соединениях. Квантово-механическая трактовка химической
связи в комплексных соединениях в своей основе также содержит
донорно-акцепториый механизм образования комплексов и метод ва-
валентных связей для описания их структур и свойств. Применение
метода валентных связей оказалось весьма плодотворным для качест-
качественного описания свойств многих комплексных соединений, ио^ ие
Позволило провести количественных расчетов.
270
Рассмотрим применение метода ВС для описания струк-
структуры и некоторых свойств координационных (комплексных)
соединений, простейшим примером которых является ион
аммония Nhtf, который образуется, если к молекуле NH3
экзотермически присоединяется катион водорода Н+:
(NH8) + (H+) = (NHi-), ДЯ°298 = -862 кДж
Катион водорода не имеет электронов, в процессе присо-
присоединения он использует гибридную парноэлектродную
орбиталь NH3. Выигрыш энергии при образовании NH^
достигается за счет того, что в молекуле NH3 имеется
некоторый избыток электронов (в связях с тремя атомами
водорода используются восемь электронов), что разупроч-
няет молекулу. Напротив, в ионе аммония NH<f те же
восемь электронов приходятся на четыре атома водорода,
что более соответствует условию оптимальной плотности
связевого электронного облака для элементов второго пе-
периода. Структура иона аммония NH^" строго тетраэдриче-
ская. Точно так же при взаимодействии молекулы Вг3
(атом бора —акцептор) с ионом F~ (донор электронной
пары) энергетически выгодно образуется координационный
ион BF*:
(BF3) + {F-}aq = {BF4~}aq> AG°2,8 = — 65 кДж
Перейдем к комплексным соединениям d-элементов.
Среди них в качестве примера активным комплексообра-
зователем можно назвать однозарядный ион меди
Си1
имеющий свободные орбитали для донорно-акцепторного
взаимодействия с неподеленными электронными парами
молекул-доноров, например молекул аммиака. Присоеди-
Присоединение первой молекулы происходит сравнительно с неболь-
небольшим энергетическим эффектом
aq> ДО°98 = -45,2 кДж
поскольку при одном лиганде у центрального иона нет
гибридизации орбиталей и симметрия иона низка. Однако
присоединение двух лигандов
aq> ДО;83:=-72 кДж
делает процесс более выгодным, поскольку в этом случае
образуются sp-гибридные орбитали и ион Ct^NHg)? при-
приобретает высокую симметрию.
Еще существенней эффект комплексообразования про-
проявляется в случае, когда при образовании связей исполь-
используется большое число d-орбиталей. Рассмотрим в качестве
примера процесс образования комплексного иона с шестью
лигандами:
{Nia + }aq + 6(NH3) = {Ni(NH3)^}aq, Д0°2вв = —59,8 кДж
Электронная оболочка никеля в комплексном ионе
Ni(NHs)jj+ имеет строение
Неспаренные
электроны
•5/>3</2-гибридные
орбитали
/\
нпи
ж
3s
.ад
Согласно теории ВС здесь имеет место 5/?3сB-гибридизация
за счет орбиталей четвертого электронного слоя. В резуль-
результате образуется ион с октаэдрическим расположением ли-
гандов, причем на Зс!-подуровне остаются два неспаренных
электрона, что сообщает этому валентно-ненасыщенному
соединению высокую химическую активность.
Рассмотрим вопрос о солеобразных комплексных соеди-
соединениях в водных растворах на примере фторалюмината
натрия. Первая ступень растворения имеет характер обыч-
обычной электролитической диссоциации сильного электролита:
Используя численное значение AG^s процесса, вычислим
в первом приближении растворимость соли (концентрацию
ионов AlFjj-):
4 / 27 600
Дальнейший процесс диссоциации комплексного аниона
осуществляется ступенчато:
= A1F2++F-
B2.1)
272
Суммарному процессу соответствует большой эндоэргиче-
ский эффект AGags = 103,8 кДж, что указывает на неболь-
небольшую глубину диссоциации. Константы равновесия каждой
из ступеней процесса B2.1) получили названия констант
нестойкости Кнест. Значения констант нестойкости ступен-
ступенчатой диссоциации комплексного иона в последовательности
отрыва лигандов, как правило, монотонно уменьшаются,
например:
Cu(NH3)I+=Cu(NHs)r + (NH3), /Cj = 3,0
Cu(NH3)I+ = Cu(NH3)i+ + (NH8), Кл = 0,7-10-*
Cu(NH3)l+ =Cu(NH3)^+ +(NH,), K3= 1,2-10-»
Cu(NH3)i+ = Cu(NH3J++(NH3), ^ = 3,2-10-*
Cu(NH3J+=Cu2+ + (NH3), /E = 7,1-10-*
Теория кристаллического поля. Высокая симметрия
комплексных соединений, их повышенная термодинами-
термодинамическая устойчивость и особенности химического поведения
потребовали более строгого теоретического объяснения.
В результате была разработана теория кристаллического
поля, основные положения которой были сформулированы
X. Бете A929). Эту теорию используют и в настоящее
время. В случае электростатического взаимодействия цент-
центрального иона или атома комплекса с лигандами она по-
позволяет достаточно полно охарактеризовать свойства со-
соединений. Однако, когда это взаимодействие носит кова-
лентный характер, ионная модель соединения становится
недостаточной, в этом случае используют теорию поля
лигандов, являющуюся более высокой ступенью развития
теории кристаллического поля.
Теория кристаллического поля описывает воздействие
лигандов на d-орбитали иона-комплексообразователя.
Основные положения этой теории можно сформулировать
следующим образом.
1. Комплексные соединения устойчиво существуют за
счет электростатического взаимодействия центрального
иона с лигандами. (Это положение перенесено из теории
Косселя и сохранило свою значимость.)
2. Центральный ион рассматривают с учетом его элект-
электронного строения и тех изменений, которые вносят лиганды
своим электростатическим воздействием. При этом лиганды
рассматриваются только как носители определенного за-
заряда, их собственную электронную структуру не учиты-
учитывают.
3. Взаимодействие между центральным атомом и лиган-
273
дами количественно описывают законами и математическим
аппаратом квантовой механики.
Таким образом, теория кристаллического поля рас-
рассматривает комплексное соединение в качестве устойчивой
системы с электростатическим стяжением центральным
ионом симметрично расположенных вокруг него лигандов.
Эти лиганды, как точечные отрицательные заряды, взаимо-
взаимодействуют с центральным ионом, притягиваясь к его ядру
и отталкиваясь от его электронных орбиталей. Такой
эффект отталкивания возбуждающе действует на орбитали
центрального иона, изменяя их энергию. При этом в соот-
соответствии с законом Кулона ближайшие к лиганду орби-
орбитали будут испытывать большее отталкивание (с этим
связано наибольшее их возбуждение), удаленные — мень-
меньшее, и этому будет соответствовать меньшее изменение
энергии.
В нейтральном атоме любого элемента внешние валент-
валентные электроны относят, как правило, к s- и р-подуровням.
Если же этих валентных электронов нет, т. е. атом пре-
превращен в ион, то внешними станут более глубокие орби-
орбитали, доступные для взаимодействия с лигандами. Если
данный атом принадлежит к семейству d-элементов, то при
его ионизации такими внешними будут d-орбитали. Именно
эти d-орбитали и перекрываются с орбиталями лигандов
при образовании комплексных соединений.
Форма d-орбиталей изображена на рис. 18.3, причем
фрагменты а, б, в, г, д дают представление об отдельных
d-орбиталях, а фрагмент е—о совокупности их в атоме.
Из теории строения следует, что в невозбужденном атоме
или ионе орбитали каждого подуровня, в том числе и
d-орбитали, наделены одинаковыми значениями энергии.
В сложной совокупности d-орбиталей имеет место эффект
их сопряжения (орбитали пространственно делокализо-
ваны). За счет перекрывания d-орбиталей с орбиталями
лигандов образуется комплексный ион. При этом, помимо
притяжения лигандов к ядру, между электронами цент-
центрального иона и лигандов должно иметь место отталки-
отталкивание. Пусть энергия этого отталкивания равна Ео.
Если бы суммарный заряд лигандов был равномерно
«размазан» вокруг центрального иона (сферическое поле),
то энергия каждой d-орбитали центрального иона возросла
бы на величину Ео. Однако лиганды занимают строго
определенные места в пространстве вокруг центрального
иона, поэтому наиболее сильно должны возбуждаться
близлежащие к ним d-орбитали и слабее — удаленные.
274
в
Под действием силового поля лигандов ранее единый,
вырожденный, энергетический уровень всех пяти d-орби-
талей центрального иона (его называют терм) расщеп-
расщепляется на два различных уровня энергии, как это пока-
показано на рис. 22.2. Наи-
Наиболее сильному возбуж-
возбуждению должны подвер-
подвергнуться орбитали dx'-y'
и d22, а наименьше-
наименьшему — орбитали dxy, dxz
и dyz.
При этом характер
расщепления исходных
вырожденных термов
определен геометрией
расположения лигандов
около центрального ато-
атома: d-орбитали, распо-
располагающиеся далеко от
лигандов, мало меняют
свой энергетический
уровень, а d-орбитали,
оказывающиеся в тесном
контакте с лигандами,—
существенно ис' Схема расщепления уровня
У ъ ' энергии d-орбиталей центрального
Рассмотрим в качест- атома под действием электростатиче-
ве примера расщепление ского поля лигандов (октаэдрического);
УРОВНеЙ ЭНерГИИ (тер- А~ ПРИ отсутствии лигаидов; Б — при иали-
\ j * « чии внешнего поля, равномерно распреде-
MOBj Ct-ОрОИТаЛеИ При ленного по сфере; Б — в поле лигандов
октаэдрическом
расположении лигандов. Схема октаэдрического располо-
расположения лигандов вокруг центрального иона приведена на
рис. 22.3.
Без эффекта расщепления А энергетических уровней
(термов) d-орбиталей энергия связи центрального иона
с лигандами определялась бы выражением
где Е1 — потенциальная энергия стяжения центрального
атома и лигандов; Ео—энергия расталкивания лигандов
в гипотетическом сферическом слое; ?сф—результирующая
энергия взаимодействия центрального атома со сфериче-
сферическим полем лигандов (см. рис. 22.2). При наличии эффекта
расщепления, а также при условии октаэдрического окру-
275
жения центрального иона лигандами энергия связи равна
где п—числовой коэффициент, характеризующий долю
энергии расщепления каждой d-орбитали. Величина Е*
должна включать в себя дополнительные эффекты: экзо-
экзотермические (по сравнению с уровнем сферического поля) —
за счет использования орбиталей dxy, dxz и dyz и эндотер-
z мические—за счет ис-
использования орбиталей
dZ2 и ^2_у2. Вклад каж-
каждой из пяти d-орбита-
лей в эффект расщепле-
расщепления А находят из со-
соотношения
2х Зу _ „
где х — составляющая
вклада орбиталей dz* и
dX2-yi в эндотермический
эффект расщепления;
Рис. 22.3. Октаэдрическое расположе- ^ТТ? Ж^' Н° °Рбита"
ние лигандов около центрального иона леи dxy, dxz, dyz В ЭК-
зотермический эффект
расщепления. Это означает, что вклад d-орбиталей ху,
xz, yz в величину А составляет 2/5А, а d-орбиталей z2 и
х2—у2—3/5 А.
Энергию связи, в образовании которой принимают
участие различные d-орбитали центрального иона при
октаэдрическом окружении, рассчитывают из соотношений:
d*y l ~ЕсЛ> — УЛ
Из схемы видно, что по мере заселения электронами
d-орбиталей центрального иона энергия связи его с лиган-
лигандами будет сначала возрастать, а затем уменьшаться. Как
уже неоднократно отмечалось в предыдущих главах, каж-
каждая орбиталь может заселяться двумя электронами (с про-
противоположными спинами), и, согласно правилу Хунда,
заселение осуществляется сначала по одному электрону
на каждую орбиталь. Такой характер заселения реаяи-
276
зуется либо при отсутствии внешнего силового поля, либо
в слабом поле, когда энергия межэлектронного отталки-
отталкивания больше энергии расщепления Д (высокоспиновый
порядок заполнения орбиталей). При этом в каждой
декаде d-элементов прохождение через минимум энергии
связи Е* будет повторяться дважды—в первой пятерке
элементов и во второй. Рассмотрим в качестве примера
характер изменения энергии связи двухвалентных ионов
d-элементов первой декады с учетом октаэдрического окру-
окружения их лигандами:
З^-орбитали
(слабое поле)
Ион М2
Са
Sc
Ti
Cr
Mn
F»
Co
Ni
Cu
Zn
При сферическом распределении заряда лигандов вок-
вокруг центрального иона Eei) должна монотонно, почти
линейно, изменяться в этом ряду элементов пропорцио-
пропорционально увеличению заряда ядер атомов. Относительно
уровня энергии Ei + E0 комплексные катионы с КЧ = 6
будут наименее прочными в случае Са2+, Мп2+ и Zn2+,
пА которых равна 0. Наиболее же прочными должны
быть комплексные катионы у V2+, которые имеют наиболь-
наибольшее абсолютное значение пА. Этот вывод полностью под-
подтверждает эксперимент. В качестве иллюстрации рассмот-
рассмотрим характер ионов {M(H2O),+}aq тех же элементов первой
декады периодической системы. Для этой цели на рис. 22.4
приведены значения энергии процесса для элементов пер-
первой вставной декады от Sc до Zn;
xy
t
t
t
¦
t
ti
ti
t +
ti
ti
t
t
t
t
t
ti
ti
ti
t
t
t
t
t
ti
H
t
t
t
¦¦¦
t
ti
ti
t
t
t
t
t
ti
? -
E -
? -
E -
E -
Я -
E -
? -
— Ei
tEi
-Ei
-Ei
-Ei
-Ei
-Ei
-Ei
- E,
+
+
+
+
+
+
+
+
+
Eo -4/5Д
Eo - 6/5 Д
Eo - 3/5 Д
Eo -28 A.
Eo -4/5Д
?o - 6/5 Д
?o -3/5Д
?o
6 {HgO}
aq »{М(Н8О)|+}а11
277
которые находятся в полном соответствии с теорией.
В минимумах кривых располагаются относительно наибо-
наиболее устойчивые ионы V(H2O)|+ и Ni(H2O)jj+, а точки,
отвечающие относительно наименее устойчивым ионам
Са(Н2О)|+, Мп(Н2ОI+ и Zn(H2O)l+, располагаются почти
на прямой линии, лежащей выше всех остальных точек.
Са Ti Cr Fe Ni Zn
-2300
-2500
-2700
Рис. 22.4. Энергия ги-
гидратации двухвалент- _2900
ных ионов М(НгО)в+
d-элементов (высоко-
(высокоспиновое комплексе- —3100
образование)
Sc
Ma Co
Cii
ЛГгидр.хДж/моль
Наряду с высокоспиновым комплексообразованием воз-
возможно и низкоспиновое, которое становится возможным,
если энергия расщепления d-термов А превышает энергию
межэлектронного отталкивания. В этом случае, т. е.
в сильном поле лигандов, заполнение d-орбиталей иона-
комплексообразователя электронами будет проходить в сле-
следующем порядке (октаэдрическое поле лигандов):
Ион М
2+
Зй-орбитали
(сильное поле)
Са
Sc
Ti
V
Cr
Mn
Fe
Co
Ni
Cu
Zn
f
1
I
U
H
u
u
H
n
I
I
¦
u
u
u
tl
¦
¦
u
tl
t
t*
¦
t
E -
j? - Есф - 25 Д
J? - ?сф - *5Д
• E - Есф - 8Z5 Д
E - Есф - 10/5 Д
? _ Ясф _ 15/5 Д
Е - Есф - 9/5 Д
? — Есф — &5 Д
? — ?сф — 3/5 Д
? - Есф
Из приведенной схемы видно, что в случае сильного поля
и низкоспинового варианта заполнения d-орбиталей элект-
278
ронами характер изменения энергии принимает вид, при-
приведенный на рис. 22.5, из которого следует, что в случае
низкоспинового комплексообразователя в рассматриваемом
семействе d-элементов наблюдают только один экстремум
стабилизации (у железа) и величина стабилизации комп-
комплексного соединения за счет расщепления d-термов суще-
существенно возрастает. Так, например, для высокоспинового
комплекса CoFjj" значение Л равно 202,7 кДж, а для
низкоспинового комплекса Co(NHs)jj+ 270,2 кДж.
Са Ti Cr Fe Ni Zn
Рис. 22.5. Энергия комп-
лексообразования ионов d-
элементов в сильном поле
(низкоспиновое комплексо-
образование)
Рис. 22.6. Тетраэдрическое расположе-
расположение лигандов около центрального
иона
Кроме рассмотренного выше октаэдрического окруже-
окружения лигандами центрального иона в координационных
соединениях имеют место также случаи тетраэдрического,
плоскоквадратного, кубического и других видов окруже-
окружения. Остановимся на комплексных соединениях с тет-
раэдрическим окружением лигандами центрального
иона, схема которого приведена на рис. 22.6. Сравнивая
рис. 18.3 и 22.6, можно заметить, что при таком распо-
расположении лигандов наиболее сильному возбуждению под-
подвергнутся dxy-, dxz- и <а^г-орбитали центрального иона
и наименьшему—dd>- и с^г-^-орбитали. Кроме того, ни
один из лигандов не попадает прямо на d-орбиталь,
а лишь в промежутки между ними. Поэтому возбуждение
d-орбиталей и связанное с этим расщепление d-термов цент-
центрального иоиа будет меньше, чем в случае октаэдриче-
октаэдрического окружения. Характер расщепления энергетических
279
уровней d-орбиталей в слабом тетраэдрическом поле лиган-
дов схематически изображен на рис. 22.7, а выражения
для энергии связи между центральным ионом и лигандами
при тетраэдрическом расположении имеют следующий вид:
H
E* = t -к
¦+<**.-
Из этой схемы видно, что расщепление d-термов централь-
центрального иона в тетраэдрическом поле лигандов качественно
сходно с октаэдрическим. Однако видно и различие: отно-
относительно наиболее прочные комплексы при тетраэдриче-
тетраэдрическом окружении должны образовывать второй и седьмой
л к и элементы (Ti и Со), но
не третий и восьмой (V
и Ni), как при октаэд-
рическом окружении.
Сравнивая значения
энергии расщепления Л
в высокоспиновых окта-
эдрических и тетраэдри-
ческих комплексах, сле-
следует иметь в виду, что
при одинаковых катио-
катионах и анионах, а также
при равенстве межатом-
межатомных расстояний анион—
катион имеет место сле-
следующее соотношение
между ними: АТетР =
=4/вДОкт- Поэтому карти-
картина, подобная рис. 22.4 и
22.5, для тетраэдриче-
ских комплексов будет
не столь отчетливой.
Четыре лиганда в
координационной сфере
центрального иона мо-
могут своим расположе-
расположением описать не толь-
только фигуру тетраэдра, ной плоского квадрата. Такая
плоскоквадратная конфигурация наблюдается, например,
280
Рис. 22.7. Схема расщепления уров-
уровней энергии d-орбиталей централь-
центрального иона в тетраэдрическом поле
лигандов:
А — отсутствие лигандов; В —наличие внеш-
внешнего поля, равномерно распределенного по
сфере; В—при тетраэдрическом расположе-
расположении лигандов
в некоторых соединениях двухзарядной меди. В ионе Си2+
имеется 9й-электронов, так что в октаэдрическом окруже-
окружении на орбитали находится только один электрон. В ре-
результате отрицательный
заряд этой орбитали
будет меньше, чем у
других орбиталей. По-
Поэтому четыре отрица-
отрицательно заряженных ио-
иона лигандов, распола-
располагаясь в углах плоского
квадрата (рис. 22.8),
будут сильно притяги-
притягиваться к ядру иона ме-
меди, т. е. в плоскости
этого квадрата хими-
химическая связь будет проч-
прочнее. Два других лиган-
да, которые располага-
располагаются по линии орбита-
орбитали dz*, будут, напротив,
взаимодействовать с ядром иона слабее (эта орбиталь
заселена двумя электронами) и могут легко отщепляться
в процессе химических реакций, что и определяет КЧ = 4
Рис. 22.8. Плоскоквадратное располо-
расположение лигандов около центрального
иона
Рис. 22.9. Переход от октаэдрического (а) к пло-
плоскоквадратному (в) расположению лигандов вокруг
иона с полностью заселенными (а, б) и частично
заселенными (в) d-орбиталями
и плоское строение комплексов иона Си2+. Этот пример
показывает, что симметрия плоского квадрата и симметрия
октаэдра в комплексах связаны между собой. Если все
шесть связей в комплексе одинаковы, получается октаэд-
рическое окружение иона лигандами (КЧ = 6); если четыре
281
связи в основании бипирамиды существенно прочнее двух
других, сохраняется симметрия квадрата (КЧ = 4); если
же эти связи не одинаковы, но прочности их различаются
не очень сильно, то комплекс приобретает форму вытя-
вытянутого, деформированного октаэдра, как это показано на
рис. 22.9.
Перейдем к характеристике расщепления d-термов цент-
центрального иона в кубическом поле восьми лигандов
Рис. 22.10. Сравнитель-
Сравнительная характеристика
расщепления d-термов
центрального иона
в поле лигандов:
А — вырожден ное
стояние
без внешнего поля;
Б — сферическое поле; г,
В — тетраэдрнческое поле;'1
Г —кубическое поле
dX2 dyl
»тетр
уАте/гр
|уДте,т
7Дкуб
(т. е. в комплексах с КЧ = 8). Такое расщепление схема-
схематически изображено на рис. 22.10. Для сравнения на этом
рисунке приведена также схема расщепления d-термов
в тетраэдрическом поле лигандов (В). Из схемы видно,
что характер расщепления в кубическом поле полностью
аналогичен расщеплению в тетраэдрическом поле, только
численное значение расщепления в первом больше, чем
во втором:
> Дтетр-
Причина этого заключается в том, что кубическая сим-
симметрия размещения лигандов является предельным слу-
случаем тетраэдрической (см. рис. 22.8), поэтому увеличение
числа лигандов при переходе от тетраэдра к кубу (от
282
КЧ = 4 к КЧ = 8) лишь усиливает уже имеющийся эффект
расщепления.
Подводя итог, необходимо отметить следующее.
1. При сближении лигандов (ионов, полярных молекул)
с центральным ионом d-орбитали последнего возбуждаются.
Таким образом, ранее пятикратно вырожденный rf-терм
изолированного иона расщепляется (вырождение снимается)
под действием поля лигандов на величину энергии А.
2. Под действием лигандов энергетические уровни
d-орбиталей центрального иона расщепляются так, что
часть их стабилизируется.
3. Энергия стабилизации в поле лигандов (т. е. раз-
разница в энергии комплексного иона, вычисленная с учетом
и без учета расщепления d-термов) для двух- и трехза-
рядных центральных ионов составляет примерно 100 —
200 кДж. Эта величина сравнима с тепловым эффектом
тех химических реакций, в которых участвуют соединения
d-элементов. Следовательно, энергия стабилизации в поле
лигандов оказывает существенное влияние на химические
и физические свойства комплексных соединений.
4. Энергия расщепления d-термов А зависит от при-
природы центрального иона и лигандов (их электронной
структуры, зарядов и т. д.), от координационного числа
центрального иона и от пространственного расположения
лигандов вокруг него.
Способность различных лигандов вызывать расщепле-
расщепление убывает в следующем порядке:
CN- > NOiT > NH3 > Н2О > ОН- > F- > С1~ > Вг~ > 1~
Это означает, что CN~ вызывает наибольшее расщепление
d-термов, 1~ — наименьшее, а НаО—промежуточное. Про-
Промежуточное положение воды в этом ряду имеет большое
значение для химии процессов комплексообразования в вод-
водных растворах, поскольку ионы металлов в водных раст-
растворах обязательно гидратированы, т. е. представляют
собой аквакомплексы. При образовании комплекса в таком
растворе молекулы воды гидратированного иона заме-
замещаются другими лигандами в соответствии с положением
их в ряду. Лиганды, расположенные в этом ряду правее
воды, будут иметь меньшую возможность вытеснить ее из
аквакомплекса, а лиганды, расположенные левее,— боль-
большую. Однако это не означает, что комплексы с лиган-
лигандами, расположенными в ряду слева, будут обязательно
прочнее комплексов, образованных лигандами правой сто-
стороны этого ряда. Речь идет лишь о величине дополни-
283
тельного упрочнения, связанного с расщеплением d-термов
центрального иона.
В заключение остановимся на вопросе о границах при-
применимости теории кристаллического поля. Как отмечалось
в начале этой главы, в основе теории заложено представ-
представление о строго ионном взаимодействии центрального иона
и лигандов, причем электронную структуру лигандов не
принимают во внимание—лиганды рассматривают как
точечные заряды. Отсюда принципиальная невозможность
учитывать природу связи «центральный ион —лиганды»,
а также особенности распределения электронных облаков
в координационных соединениях; невозможность строгого
количественного расчета энергетических и других харак-
характеристик, особенно в тех случаях, когда необходимо учи-
учитывать образование я-связи ион — лиганд.
Усовершенствованным вариантом теории кристалличе-
кристаллического поля стала теория поля лигандов, в которой учтено
электронное строение лигандов и особенности перекрыва-
перекрывания орбиталей лигандов и центрального атома (т. е. кова-
лентность связей). Однако по своему содержанию моди-
модифицированная таким образом теория кристаллического
поля становится неотличимой от широко используемой и
хорошо разработанной в квантовой химии теории МО,
которая достаточно строга и применима к любым много-
многоатомным системам.
Глава 23
Теория молекулярных орбиталей (МО)
Недостатки метода ВС. Приближение МО ЛКАО,
Строение молекулярного иоиа Hf и молекулы Н2.
Строение миогоэлектрониых двухатомных молекул
гомосоединений элементов второго периода.
Современный квантово-механический этап развития
теории строения исторически создавался на базе двух раз-
разных подходов, двух разных теоретических методов при-
приближенного решения уравнения Шредингера. Это теория
ВС, разрабатываемая в 30-е годы XX в. Л. Полингом,
Дж. Слетером и др., и практически одновременно воз-
возникшая теория молекулярных орбиталей (МО), основные
положения которой сформулированы в трудах Р. Малли-
кена, Ф. Хунда, Г. Герцберга, В. Хюккеля и др. Обе
284
теории в своих высших приближениях дают достаточно
верные результаты. В первом приближении каждая из
этих теорий имеет свои преимущества при описании одних
сторон строения и недостатки в описании других.
Теория ВС чрезвычайно удобна для объяснения насы-
насыщаемости химической связи, направленной валентности,
энергии активации химических реакций и др. Однако
оказалось невозможным использовать эту теорию для
количественного описания строения соединений с сопря-
сопряженными связями, с нечетным числом электронов (напри-
(например, молекулярные радикалы), а также для расчета коли-
количественных значений молекулярных характеристик и стро-
строгого описания спектров молекул.
Простейшим примером молекул, строение которых не
может быть описано методом ВС, является газообразный
молекулярный ион Н^, имеющий два ядра и один элект-
электрон. Ион HJ реально существует (он был открыт Дж. Том-
соном в конце XIX в.) и легко получается при бомбар-
бомбардировке молекул водорода электронами. Из сравнения
характеристик нейтральной молекулы Н2 и иона Н?
(Н2) = 2(Н), ДЯг98 = 436,0 кДж
, ЛЯ°298 = 262,8 кДж
видно, что ион HJ имеет достаточно прочную связь, хотя
и меньшую, чем в нейтральной молекуле Н2.
Это не единственный пример соединения с неспарен-
ным электроном, участвующим в образовании химической
связи. Так, на основании изучения магнитных свойств
молекулярного кислорода было установлено, что моле-
молекулы О2 парамагнитны, т. е. заметно притягиваются маг-
магнитом. Это свойство присуще только веществам, в состав
которых входят атомы с неспаренными электронами.
Следовательно, возможность образования двойной связи
в структуре молекулы О2 за счет спаривания двух пар
одиночных электронов, согласно методу ВС, следует счи-
считать ошибочной. Аналогичное противоречие имеет место
и при описании связей в молекулах СЮ2, NO2, NO и
в большом семействе так называемых свободных радика-
радикалов— частиц содержащих неспаренные электроны и обла-
обладающих высокой реакционной способностью: СН3, NH2,
ОН, СН, CN и др.
Решение подобных задач выходит за пределы возмож-
возможностей метода ВС, в связи с чем была разработана тео-
теория МО.
285
Приближение МО ЛКАО. С позиций теории МО моле-
молекулу рассматривают в качестве сложного, многоядерного
«атома», к которому приложимы все квантово-механиче-
ские законы. Каждый энергетический молекулярный уро-
уровень имеет определенный набор квантовых ячеек, как и
в обычном одноядерном атоме. Это означает, что валент-
валентный слой молекулы может быть представлен либо одной
связывающей квантовой ячейкой с максимальной емкостью
2 электрона (Н2), либо четырьмя ячейками с максималь-
максимальной емкостью 8 электронов (CF4), либо девятью ячейками
с максимальной емкостью 18 электронов [Сг(СО)„]. Если
сумма электронов, образующих молекулу атомов, больше
указанной емкости молекулярных орбиталей, все «лишние»
электроны переходят на разрыхляющие орбитали, т. е. на
более высокие уровни или подуровни энергии. Поэтому
электроны, число которых не превышает указанной пре-
предельной емкости, участвуют в образовании химической
связи, их называют связывающими; электроны, число кото-
которых превышает указанную предельную емкость,— раз-
разрыхляющими, они переходят на более высокие уровни
энергии и разупрочняют связь в молекуле (так же на-
названы и соответствующие орбитали).
Точный расчет волновых функций МО в настоящее
время невозможен, поэтому приближенно МО рассмат-
рассматривают в качестве линейной комбинации атомных орби-
орбиталей (ИКАО). Понимать это следует так. Когда элект-
электрон в молекуле находится вблизи одного из атомов, его
молекулярная волновая функция г|)мо близка к атомной
волновой функции ^ао именно этого атома. Таким обра-
образом, в первом приближении молекулярная орбиталь *|эмо
может считаться линейной функцией атомных орбита-
лей я^А0:
*МО = 2 ^АО,
где С — нормирующий множитель. Например, в молекуле
водорода Н2 волновая функция каждой электронной орби-
орбитали имеет вид
= с [Фао,
где знаки (+) и (—) соответствуют iJjmo и г)?мо—связы-
г)?мо—связывающей и разрыхляющей орбиталям соответственно.
Строение молекулярного иона Щ и молекулы Н2. Рас-
Рассмотрим характер МО в наиболее простом соединении,
содержащем один электрон—в молекулярном ионе Щ.
Положим, что это соединение образуется из атома водо-
286
рода и протона по схеме
Н + Н+—262,8 ?
Волновую функцию молекулярной орбитали можно записать
в двух вариантах:
+мо=СA'ао,+1>ао,). B3Л)
B3.2)
В данном случае единственный электрон может быть в поле
каждого из ядер и в равной степени принадлежать им.
Поэтому г|?Ао, и урлог являются волновыми функциями
электрона, находящегося в поле первого или второго про-
протона. Решим в общем виде задачу об энергии молекуляр-
молекулярного иона Н^", используя уравнение Шредингера
Е = С ЦНц dV. B3.3)
Подставив уравнения B3.1) и B3.2) в B3.3), находим
Е = С% [ 5 *A0,W *AOt d" + $ *АО,^АО. dv ±
*А0, dy
В уравнении B3.4) первые два слагаемых характеризуют
энергию электрона в поле «своего» ядра и потому не опре-
определяют энергию химической связи. Их называют куло-
новскими интегралами или валентными потенциалами
ионизации—</. Вторые два слагаемых представляют энер-
энергию взаимодействия электрона с двумя ядрами, т. е.
энергию химической связи. Их называют обменными инте-
интегралами и обозначают р. Тогда уравнение B3.4) прини-
принимает вид
? = 2C»fo±P]. B3.5)
Помня условия нормировки волновой функции
=l, B3.6)
подставим в это выражение уравнения B3.1) и B3.2):
» =1.
\ [ЧЪа.+Ч'ао,]1 dy = ca J Ч'ао,
287
Согласно условию B3.6) интегралы типа J ч|>аоЙу в урав-
уравнениях B3.7) являются нормированными в отношении
атомных орбиталей и равными единице; введя это упро-
упрощение, имеем
B3.8)
В уравнениях B3.8) выражение J i|)ao,^ao, dy характери-
характеризует плотность электронного облака в области взаимного
перекрывания атомных орбиталей, его называют инте-
интегралом перекрывания (S). С учетом этого уравнения B3.8)
примут вид
= 1, 2 (С*J [1—S] = l;
откуда
2A+5)'
с*=
2 A -S)'
B3.9)
Подставив уравнение B3.9) в B3.5), находим для
энергии
B3.10)
B3.11)
+ S'
Таблиц
a 23.1. Интегралы
перекрывания (S) гомо-
н гетеросвязей атомов некоторых элементов
Гомосвязь
в молекулах
Н...Н +
н—н
Li —Li
Be-Be
В —В
с-с
N —N
О —О
F —F
Na —Na
Si-Si
P—P
S-S
Cl —Cl
s
a(s)
0,590
0,750
0,583
0,583
0,459
0,343
0,237
0,155
0,106
0,607
0,375
0,313
0,250
0,207
s
0,225
0,225
0,317
0,329
0,287
0,220
0,166
0,227
0,385
0,363
0,336
0,282
s
я(р)
0,411
0,411
0,300
0,195
0,119
0,070
0,044
0,452
0,222
0,160
0,117
0,097
Гетеросвязь
а молекулах
Li-F
Na —F
K-F
Rb —F
Cs —F
Li —H
Na —H
K-H
Rb —H
Cs —H
H-F
H-Cl
H —Br
H-I
sa
0,061
0,026
0
0
' 0
0,474
0,395
0,262
0,213
0,180
0,297
0,452
0,466
0,441
288
" В настоящее время известны способы расчета инте-
интегралов перекрывания для большого числа молекул. В табл.
23.1 для ряда гомосоединений приведены значения инте-
интегралов перекрывания s-орбиталей (Sa(s)); /?-орбиталей,
образующих cr-связь (Sa(p)); /?-орбиталей, образующих
я-связь (Sn(P))- Для гетеросоединений приводятся инте-
интегралы перекрывания s- и р-орбиталей, образующих
<г-связь E0).
Рис. 23.1. Образование моле-
молекулярного иона водорода Н^;
а —молекула Hj со
связывающей молекулярной
орбиталью;
б —молекула Н+с разрыхляющей
а б молекулярной орбнталью
Из табл. 23.1 следует, что по мере усложнения элект-
электронных оболочек атомов и ростом полярности связей их
интеграл перекрывания в молекулах уменьшается. Так,
в ионных (полярных) молекулах K.F, RbF и CsF инте-
интеграл перекрывания стремится к нулю. Взяв значение S
для Щ из табл. 23.1 и подставив его в уравнения B3.10)
и B3.11), получим
^=¦^1=0,69(9+?),
Анализ выражений B3.13) позволяет схематически изо-
изобразить строение молекулярного иона HJ. Согласно схеме,
представленной на рис. 23.1,6, в молекулярном ионе HJ
общее электронное облако обладает симметрией кокона и
притягивается сразу к двум ядрам (протонам), что дает
значительный выигрыш в энергии по сравнению с изоли-
изолированным атомом и протоном. В промежутке между ядрами
(рис. 23.1, а) это облако достаточно плотно, хорошо экра-
экранирует положительные заряды ядер друг от друга и
уменьшает их взаимное отталкивание. Такого рода МО
называют связывающей а-орбиталью. Возможен и другой
вариант образования МО при взаимодействии протона
с атомом водорода, изображенный на рис. 23.1, б. В этом
случае электронное облако в молекуле сгущается вокруг
каждого ядра, образуя узел в промежутке между ними.
Такие электронные облака не экранируют ядер, их вза-
взаимное отталкивание делает молекулу неустойчивой. По-
10 № 70S 289
добную МО называют разрыхляющей а*-орбиталью. Гра-
Графически образование МО HJ из атома водорода и протона
представлено на рис. 23.2 и 23.3, а.
Поскольку в молекулярном ионе Hi всего один элект-
электрон, из двух возможных энергетических уровней Есв и
?Разр он выбирает наиниз-
АО мо ший, т. е. ?СЕ (стремление
системы к минимуму энер-
энергии). Образовавшаяся в
Н^ (т5-связь достаточно
прочна B62,8 кДж), хотя
и образована одним элект-
электроном.
Аналогичным образом,
но несколько сложнее ма-
математически может быть
решена задача об энергии
МО в нейтральной (двух-
электронной) молекуле во-
водорода Н2. Анализ приво-
св дит к схеме, изображенной
на рис. 23.3,6. Как видно
из этой схемы, при взаимо-
Рис. 23.2. Энергетические уровни действии двух нейтральных
МО иона н2 атомов водорода образует-
образуется двухэлектронная связы-
связывающая МО, так как емкость молекулярной а-орбитали
в Н2 равна двум электронам. Энергия двухэлектронной
связи в молекуле Н2 равна 435 кДж, т. е. связь эта более
чем в полтора раза прочнее одноэлектронной связи в
ионе Н?. Однако размещение двух электронов на одной
МО в соответствии с принципом Паули возможно лишь
в случае, если спины электронов антипараллельны
m
——
р р
—7г)- Если же спины электронов двух
взаимодействующих атомов водорода параллельны
(ms==4-V», flts —+V»)i то один из них займет место на
связывающей орбитали Есв, а другой—на разрыхляющей
?разР. как это показано на рис. 23.3, в. В этом случае
энергия связи двух атомов водорода с параллельными
спинами электронов равна нулю.
Из приведенного примера можно сделать вывод: в мо-
молекулах на МО один разрыхляющий электрон полностью
компенсирует действие одного связывающего электрона.
По аналогии с методом ВС можно считать, что ординар-
ординарной связи в молекуле соответствует связывающая орёи-
290
таль. С этой точки зрения в молекуле Нв имеет место
одна полная cr-связь (два связевых электрона), а в моле-
молекулярном ионе HJ—78а-связи (один связевой электрон).
Величину, подобную числу связей в методе ВС, в методе
МО называют порядком связи (Пс) и определяют полу-
разностью числа связывающих усв и разрыхляющих
электронов:
Пс = -
в — Тразр
Отсюда Пс для молекулы HJ равен 0,5, для Н2—1.
Выше уже отмечалось, что в методе МО молекулы
рассматривают как многоядерные атомы, на которые рас-
распространяются известные по строению атома квантово-
механические законы (квантовые числа, принцип Паули,
АО
а
МО
АО
АО
б
МО
АО
АО
в
МО
АО
н+н-н2
W
Jb
н+н-н+н
¦Ераэр
Рис. 23.3. Распределение электронов по МО:
о —в иоие Н2+; 6 и в—в молекуле Н, о аитипараллельиыми (б) н параллель-
параллельными (в) спинами электронов
правило Хунда и т. д.). Однако сложность молекул по
сравнению с атомами вносит в применение этих законов
и правил определенное своеобразие. Так, энергия элект-
электрона в изолированном атоме зависит от главного п и
побочного I квантовых чисел и не зависит от магнитного
квантового числа mlt которое определяет величину про-
проекции момента количества движения электрона на направ-
направление силового поля, характеризующее положение элект-
электронной орбитали в пространстве. В атоме никаких избран-
избранных направлений нет, в отсутствие внешнего поля они
ю* 291
все энергетически равноценны. В молекуле же имеется
такое преимущественное направление—это линия хими-
химической связи атомов, т. е. энергия электронов зависит от
пространственного расположения МО. Поэтому для харак-
характеристики МО принята несколько иная символика. Ана-
Аналогом ГП[ в молекулах является квантовое число Я, харак-
характеризующее величину проекции момента количества дви-
движения электронов на направлении химических связей.
Так же, как и т1У квантовое число к принимает значения
от 0 до =F U и каждому значению % соответствует МО
(квантовая ячейка). В атоме все 21 + 1 ячеек (орбиталей)
наделены одинаковой энергией (вырожденное состояние).
В молекуле же вырождение энергии снимается и часть
из 2/ + 1 молекулярных ячеек могут стать разными по
уровню энергии (связывающие и разрыхляющие).
Как уже отмечалось (см. гл. 19), для орбиталей атома
принята символика:
/ ... О 1 2 3
АО ... s p d f
Для молекулярных орбиталей принят аналогичный спо-
способ обозначения:
/ ... О I 2 3
МО ... а п Ь ф
Однако из-за того, что энергия связывающих МО частично
компенсирована энергией разрыхляющих МО, в химиче-
химической связи фактически участвуют не все электроны. Так,
на каждой а-орбитали может находиться до двух связы-
связывающих электронов с антипараллельными спинами, на
я-орбиталях—до четырех электронов и т. д. При этом
следует также считаться с различным участием в хими-
химической связи электронов, принадлежащих в атомах раз-
разным слоям (уровням). Очевидно, что наибольшее участие
в образовании связи должны принимать электроны внеш-
внешнего валентного слоя. Участие же электронов более глу-
глубоких слоев должно быть малым, и в первом приближении
им можно пренебречь. Поэтому при записи МО учитывают
только внешние орбитали атомов, считая более глубин-
глубинные неизменным атомным остовом.
Образование молекулярных орбиталей из атомных
подчиняется в общем случае следующему правилу: пере-
перекрывание атомных орбиталей (интерференция электрон-
электронных волн) тем более вероятно, чем ближе они по энерге-
энергетическим уровням и чем более сходны по симметрии.
292
Строение многоэлектронных молекул гомосоединений.
Рассмотрим в качестве примера двухатомные молекулы эле-
элементов II периода, по сложности идущие за молекулой
водорода Н2. Для атомов этих элементов валентными будут
2s-, 2px-, 2ру- и 2рг-орбитали. Поскольку двухатомные
молекулы элементов должны быть предельно симметричны,
выберем для их графического изображения систему коор-
координат, приведенную на рис. 23.4, а. Согласно этой системе
координат а-орбиталь молекул располагается вдоль оси х.
Рис. 23.4. Схемы образования молекулярных ст-орби-
талей в двухатомных молекулах гомосоединений
На этом же рисунке изображены 2s- и 2рл.-орбитали,
которые, перекрываясь, дают молекулярные а-орбитали,
связывающие (б, е) и разрыхляющие (г, д). Если прибли-
приближенно считать интеграл перекрывания этих орбиталей
равным нулю, то нормированные волновые функции их
будут иметь следующий вид:
1
при перекрывании s-орбиталей
при перекрывании рх-орбиталей
293
Орбитали 2pz и 2ры, не обладающие осевой симмет-
симметрией относительно оси х, при взаимном перекрывании
образуют связывающую и разрыхляющую МО, изобра-
изображенные на рис. 23.5. Как видно из рис. 23.5, а, я-орби-
таль характеризуется наличием повышенной плотности
i
г
И
X
1 яК^
Рис. 23.5. Схемы
образования моле-
молекулярных я-орби-
талей в двухатом-
двухатомных молекулах го-
мосоединений
электронного облака в боковых межъядерных промежут-
промежутках. Напротив, я*-орбитали в промежутке между ядрами
имеют узел (рис. 23.5,6), т. е. минимум плотности. Если
принять, что S = 0, то нормированные волновые функции
примут вид
при перекрывании /7г-орбиталей
в плоскости xz
при перекрывании ру-орбиталей
в плоскости ух
Я,*, l/ ^2pl
Исходя из сказанного, рассмотрим распределение элект-
электронов по МО в двухатомных молекулах элементов II пе-
периода. Молекула Li2 образуется из атомов экзотермически:
ДЯо = —106,7 кДж
Эта энергетическая стабильность молекулы Lia связана
с электронным строением атома лития Li(ls22s1) или
Li(K2s1) (здесь К — атомный остов, не принимающий уча-
участия в образовании МО). Образование молекулы Li2 можно
в первом приближении схематически изобразить так:
Li
(о,J]
294
Здесь одиночные 25-электроны двух атомов Li образуют
os = МО, что и обеспечивает прочность связи в молекуле Li2,
а глубоколежащие ^-электроны практически не участвуют
в образовании химической связи, как это видно из
табл. 23.2.
Эта таблица представляет собой достаточно грубую модель, она
не учитывает, что небольшая часть электронного облака (~ 0,02 за-
заряда электрона) занимает также связывающую aip = ftl0. Образо-
Образовавшаяся as- и частично я^-орбитали частично перекрываются друг
другом, несколько упрочняя связь. В этом перекрывании участвует
часть электронного облака, эквивалентная ~ 0,2 заряда электрона*
Как видно из приведенного примера, даже в наиболее простой
молекуле Li2 строение МО является достаточно сложным. Свидетель-
Свидетельством этому служит следующий факт. Экспериментально определен-
определенное межатомное расстояние R в молекуле Li2 равно 0,267 нм. Если
же рассчитать это расстояние, предполагая только ст^-связь (т. е. без
участия 2/7-орбиталей и других взаимодействий), то R = 0,278 нм,
а при чистой Яу-связи R = 0,200 нм, т.е. даже небольшое участие
2р-орбитали в формировании химической связи способствует упрочне-
упрочнению молекулы.
Таблица 23.2. Энергетические диаграммы МО двухатомвых
молекул элементов второго периода
МО
Разрых-
Разрыхляющие
»
Связы-
Связывающие
Разрых-
Разрыхляющие
Пс
R< Ю10 м
Есв, кДж
Li,
*
Ох
Пу Я2
ох
пулг
*
Os
¦¦ и-
Os
1
2,67
107
Be,
*
Ox
Пу Я2
ох
ПуПг
Н
*
ох
н
Os
0
<60
В,
Ох
Пу Я2
ах
t t
Jty Яг
ti
*
CTS
H
OS
1
1,59
261
с,
*
Ox
Пу я2
ох
п и
и
"to
и
OS
2
1,31
600
N2
O*x
пу я2
и
Ox
n н
Пу Яг
*
и
Os
3
1,10
941
o,
*
Ox
t t
Пу Лг
n n
ny nz
H
ox
и
Os
H
Os
2
1,21
494
F,
*
Ox
tin
Я^ Яг
и и
Лу Лг
Л
ох
os
и
Os
1
1,42
155
Ne,
и
*
Ох
и и
Пу Лг
Н tl
пу я,
ОХ
и
Os
_t?
OS
0
—
,295
Рассмотрим строение неизвестной, но, возможно, при
определенных условиях существующей молекулы Ве2. Если
предположить распределение электронов по МО без ис-
использования вакантных р-орбиталей (см. табл. 23.2), то
процесс формирования электронной оболочки молекулы
можно записать следующим образом:
Be [K2s2] + Be [K2S2] — Ве2 [К2 (asJ (ajJ]
а распределение электронов по энергетическим уровням
приведено в табл. 23.2. Это означает, что такая молекула,
имеющая две двухэлектронные—связывающую и разрых-
разрыхляющую— орбитали, существовать не должна. Однако
наличие свободных р-орбиталей делает возможным частич-
частичное их использование за счет возбуждения 2s2/?0—>-2s1p1.
Расчет показывает, что при таком возбуждении возможен
переход приблизительно 0,8 части электронного облака
(заряда) на л-подуровень, что могло бы в небольшой сте-
степени стабилизировать молекулу Ве2 подобно тому, как
это имеет место в молекуле Li2.
Атомы бора при взаимодействии образуют устойчивые
двухатомные молекулы
= (В2), Д#° = — 260 кДж
процесс формирования электронных оболочек которых
можно изобразить следующим образом:
В [K2s12p1] + B [K2s2V] = В2 [К2 (asJ (asJ яу пг]
Из энергетической диаграммы молекулы В2 (см. табл. 23.2)
следует, что неспаренные электроны двух атомов бора
должны занять по одному (правило Хунда) две связы-
связывающие пу- и лг-орбитали. Остальные 4 электрона засе-
заселяют <Ту-связывающие и «^-разрыхляющие орбитали и не
дают вклада в энергию химической связи молекулы. На-
Наличие двух неспаренных электронов в молекуле В2 тре-
требует появления нескомпенсированного магнитного момента
и связанного с этим парамагнетизма. Следует отметить,
что опыт подтверждает парамагнитность молекул В2, а
следовательно, и правильность приведенной выше элект-
электронной структуры.
Атомы следующего за бором углерода образуют при
высоких температурах большое семейство молекул гомо-
соединений, простейшее из которых—двухатомная С,:
щ.
Процесс образования электронной оболочки молекулы из
атомных записывается следующим образом:
С [K2s22p2]+C [K2s22p2] _^ С2 [к2 Ю2 (^J (я„)« (я,)»]
Из энергетической диаграммы молекулы С2 (табл. 23.2)
следует, что связь между атомами обеспечивают две двух-
электронные пу- и лг-орбитали.
Наиболее прочной молекулой является N2:
(N) + (N) = (Na), ДЯ° = —941 кДж
Процесс формирования эзектронных оболочек молекул
азота:
N [K&VI + N [K2s=2p3] _> Щ [к» (a,)i (alY (я„)»(я,)« (а*J]
В этой молекуле os- и с*-орбитали компенсируют друг
друга, а л;у-, пг- и ох-орбитали являются связывающими.
Порядок связи равен трем. Именно это и обеспечивает
молекуле высокую прочность.
Кислород образует уже менее прочную молекулу:
(О) + (О) = (Оа), ДЯ° = —494 кДж
Причина этого отчетливо видна в конфигурации молекулы
(см. табл. 23.2):
] — О2 [К2 Ю* W
здесь as- и а*-орбитали взаимно компенсируют друг друга.
Связывающая орбиталь ах дает наибольший выигрыш
в энергии A84 кДж). Разрыхляющие одноэлектронные
орбитали п,у и л* частично компенсируют две двухэлект-
ронные связывающие орбитали лу и п2, так что энергия
этих связей составляет около 310 кДж.
Еще меньшая прочность химической связи имеет место
в молекуле F2:
(F) + (F) = (Fj), ДЯ° = -155кДж
что также связано с особенностью электронной конфигу-
конфигурации этой молекулы:
- " — Fi [К2 Ю2 (а:)а (а^п^ Ш
Здесь две двухэлектронные орбитали afn<fa, а также jty-,
пг-, Л^-'й йг-орбйтали Взаимно компенсируются,'в ре-
результате чего связь осуществляется только за счет одной
связывающей 0.,-орбитали (Пс = 1).
Молекула Ne2 не существует, поскольку в ней числа
связывающих и разрыхляющих электронов одинаковы.
В заключение следует дать дополнительное пояснение
к таблице 23.2. Можно заметить, что на приведенных
энергетических диаграммах на границе между азотом и
кислородом изменяется взаимное расположение уровней
ах- и л(/1 г-орбиталей. В ряду Li — N в двухатомных мо-
молекулах этих соединений более прочными (более глубоко-
лежащими на диаграммах рис. 23.2) являются связи, об-
образуемые л - и лг-орбиталями. При заполнении электро-
электронами подуровней разрыхляющих п*и- и Лг-орбиталей (О2, Fa)
последние возбуждающе действуют на связывающие лу-
и лг-орбитали, делая их менее энергетически выгодными
по сравнению с уровнем сг^-орбитали.
Глава 24
Молекулярные орбитали
в молекулах гетеросоединений
Моменты диполей связей. МО в двухатомных и
многоатомных молекулах с полярными связями.
Строение молекул гидридов. Строение молекулы
диборана. Тетраэдрнческая пятиатомная молекула
метана. Пирамидальная четырехатомная молекула
аммиака. Уголковая трехатомная молекула воды.
Двухатомная молекула фтористого водорода.
Моменты диполей связей. В молекулах гомосоединений
(например, в Н2, Li3, Cl2) заряды атомных ядер одина-
одинаковы и по отношению к ним молекулярные орбитали
строго симметричны. Напротив, в молекулах гетеросоеди-
гетеросоединений заряды ядер различны и они по-разному притя-
притягивают орбитальные электроны. В результате молекуляр-
молекулярные орбитали смещаются в направлении ядра, обладаю-
обладающего большим положительным зарядом, и молекула
приобретает свойство полярности (превращается в диполь).'
Такое разделение зарядов по длине молекулы определено
значением электрического момента диполя ц, который
равен произведению заряда полюса диполя q на расстоя-
расстояние между центрами тяжести положительного и отри-,
дательного зарядов г;
ц = 9г. .'.,. B4,1).
При этом г обычно выражают в метрах, q—в кулонах
(Кл), а ц—в Клм. Поскольку единичный заряд равен
1,6-10~19Кл, расстояние между полюсами диполя
г~ 10~10м, то момент диполя имеет порядок 10~29Кл-м.
Момент диполя — вектор, направление которого для
каждых двух химически связанных атомов в соединении
совпадает с линией, проходящей через их ядра. Наличие
у молекулы момента диполя часто соответствует упрочне-
упрочнению связи, поскольку появляется дополнительная энергия
электростатического притяжения разноименно заряженных
частей молекулы. В табл. 24.1 приведены данные, из
которых видна симбатность изменения значений ц и энер-
энергии связи в полярных молекулах галогеноводородов.
Однако эта закономерность необязательна, поскольку
эффект перекрывания атомных орбиталей находится
в сложной зависимости от величины ц. Например, в мало-
малополярной молекуле СО (р, = 0,03-10~29Клм) энергия
связи равна 1071 кДж, а в более полярной молекуле CS
(ft = 0,67-10~29 Клм) энергия связи значительно меньше
— 736 кДж.
Таблица 24.1. Моменты диполя и энергии связей
в молекулах галогеноводородов
Молекула
HF
HCI
НВг
HI
ц-10», Клм
0,640
0,347
0,263
0,127
Энергия связи, кДж
565
427
364
293
В более сложных молекулах момент диполя получают
суммированием моментов диполей отдельных связей (век-
(векторов) с учетом их пространственного расположения.
Согласно методу МО в приближении ЛКАО в двухатомной моле-
молекуле ZY ядра атомов Z и Y ивлиются центрами связывающей моле-
молекулярной орбиталии:
, <24.2)
где X—коэффициент свизан с полярностью связи в молекуле и харак-
характеризует различие вклада каждой АО в МО; С—нормирующий мно-
множитель. Если использовать способ определения значения нормирующего
множителя С, примененный при описании МО в молекулярном ионе
Hj (см. гл. 23), то для плотности электронного облака p = *pzY do
бй ZY
Hj (см. гл. 23), то для потос элерого облака p *pzY do
в любой точке околоядерного пространства молекулы ZY получим
2*"
выражение
1+X2+2XS '
B4.3)
где S—интеграл перекрывания, равный \ 1|з21|зу do.
Если ц, = 0, т. е. связь строго ковалентна (например, в молеку-
молекулах На, О2), то Х=1 и уравнение B4.2) обращается в B3.3). В мо-
молекулах же гетеросоединеиий связи поляриы
((д, > 0), и этому соответствует существенное
отклонение значения X от единицы. Если
предположить, что Х^>1, a S = 0, то электрон-
электронная МО сосредоточится в основном у атома Y,
а это соответствует почти полному переходу
связевой МО к атому Y и в предельном случае
отвечает образованию двух иоиов Z+ и Y-,
связанных между собой ионной связью, т. е. толь-
только силой электростатического взаимодействия.
Из сказанного следует, что величина X—
важнаи характеристика МО гетеросоединений.
Именно поэтому необходима количественная
взаимосвизь X с моментом диполи ц,, величина
которого может быть измерена эксперимен-
экспериментальным путем. Представим себе молекулу YZ
в виде схемы, изображенной иа рис. 24.1.
Пусть линия химической связи совпадает с
осью *. В качестве начала координат выберем
точку О, равноудаленную от ядер Z и Y
(OZ = OY); расстояние между Z и Y примем
за R. Если точка О ивляетси центром тяжести
положительных зарядов идер, а точка С—центром тяжести отрица-
отрицательных заридов, то отрезок ОС представляет собой длину диполя г,
который можно найти нз соотношения
Рис. 24.1. Схема
молекулы гетеро-
соединения ZY
' = \ дер do,
B4.4)
где р—плотность электронного заряда МО в каждой точке объема V
молекулы; х—координата на линии связи, вдоль которой н должен
располагаться центр тяжести заряда МО.
После подстановки уравнения B4.3) в B4.4), получим
'=1
л
\+X*+2XS
X2 f *f2Y dv 2S С
г+
dv
B4.5)
В Этом уравнении члены \ *i|>tdt> й \ xtyу do представляют собой
координаты центров электронной плотности Z и Y-атрмиых ррбитаувдй.
Если отсутствует усложняющий картину эффект гибридизаций 6рби-
аей то центры »тих АО должны совпадать с координатами ядер
атомов Z и Y, т. е.
'=-4' B4-б>
v = 4- B4.7)
Третье слагаемое уравнения B4.5) содержит член \ xi^z^Y dV, мало
отличающийся от нуля, поскольку произведение ipzij'Y должно отли-
отличаться от нуля лишь в области перекрывания атомных орбиталей
Z и Y, т. е. там, где х—>¦ 0. Учитывая это, а также после подста-
подстановки уравнений B4.6) и B4.7) в B4.5), получаем
г= (Х2-')/?о. B4 8)
И наконец, подставив B4.8) в B4.1), находим выражение для момента
диполя молекулы
в котором количественно связываются экспериментально определяемая
величина и, с характеристикой X, важной для расчетов по методу
МО ЛКАО.
Представления о моментах диполей широко исполь-
используются в химии. Они необходимы при выяснении типа
химической связи, геометрического строения молекул,
для обнаружения донорно-акцепторного взаимодействия.
Данные по моментам диполя позволили сделать оконча-
окончательный вывод об уголковом строении молекул Н2О, HaS;
SO2; о пирамидальном строении молекул NH3, SbCl3; о
характере и степени ассоциации молекул в растворах
и т. д.
Моменты диполей подразделяются на молекулярные
и связевые. В гетеросоединениях моменты диполей отдель-
отдельных связей всегда больше нуля, однако если молекулы
из трех и более атомов пространственно симметричны, их
ц могут быть равны нулю. Например, молекула ВС13
имеет молекулярный ц = 0, так как все три связи в ней
равны по длине, расположены в одной плоскости и имеют
одинаковые углы между направлениями связей. Напротив,
молекула NH3 имеет ц = 0,486• 10~29, поскольку атом
азота в ней расположен в вершине пирамиды; линейная
молекула СО2 имеет ц = 0.
Строение молекул гидридов. Рассмотрим с позиций
МО ЛКАО особенности строения молекул гетеросоедине-
ний газообразных гидридов элементов второго периода.
Газообразный гидрид лития LiH образуется из двухатом-
301
ных молекул гомосоединений эндотермически:
(Li2) + (Н2) = 2(LiH), ДЯ298 = 69,5 кДж
Это свидетельствует о том, что сумма энергий связи
в исходных молекулах Li2 и Н2 больше, чем в конечном
продукте—молекулах LiH. Используя известные данные
для атомизации Li2 и Н2, находим, что энергия связи
в молекуле LiH равна 234 кДж, а полусумма связей Li2
и Н2 равна 265 кДж. Атом лития имеет орбитали 2s, 2рх,
2р„, 2pz, из которых только 2в-орбиталь заселена одним
электроном. Атом водорода имеет одноэлектронную ls-
орбиталь. Отсюда возможно
образование следующих свя-
связывающих и разрыхляющих
орбиталей:
а) перекрывание ls-орби-
тали атома водорода и 2s-op-
битали атома лития
2s>
АО
МО
АО,
/Г
/ / "*
\ /Hi 71
Л
\ \
\ \
\ \
V \ tJx
Л
^ \
\\
;\\ .
\\
V
4
Li
Н
Рис. 24.2, Энергетическая ди-
диаграмма молекулы LiH
S r i B4.10)
б) перекрывание ls-орби-
тали атома водорода и 2рх-
орбитали атома лития
B4.11)
в) 2pz и 2/?у-орбитали ато-
атома лития не перекрываются
ls-орбиталью атома водорода. Энергетическая диаграмма
молекулы LiH представлена на рис. 24.2.
Поскольку в варианте B4.10) атомы ;Н и Li имеют
на своих s-орбиталях по одному электрону, а образую-
образующаяся сг,-МО может иметь два электрона, то оба электрона
в соответствии с условием стремления системы к минимуму
энергии должны занять наинизщую, связевую МО, кото-
которая, как показано на рис. 24.2, и обеспечивает- значи-
значительную часть энергии связи в молекуле LiH.
В случае B4.11) 25-орбиталь атома Li и Is атома Н
(см. рис. 24.2) могут образовывать связывающую os-
и разрыхляющую о?-МО. В результате близости уровней
6Х и otj небольшая часть электронного облака' @>,26 е~)
переходит на огл-подуровень, В связи с малым потейцйа-
лом ионизации атом Li в молекуле LiH несет на себе
избыточный положительный заряд, а аюм Н — избыточный
отрицательный заряд. Если спин 2/?х-орбитали лития
и ls-орбитали водорода параллельны, то при их взаимо-
взаимодействии один электрон переходит на разрыхляющую
or*-МО и молекула LiH не образуется. Как видно из
рис. 24.2, 2pz- и 2/уорбитали атома Li не перекрываются
с ls-орбиталью атома водорода.
Таким образом, в молекуле LiH за счет перехода
электронов на as и о^-орбитали образуется прочная
химическая связь, требующая для разрыва затраты
234 кДж. Из-за смещения МО в сторону атома водорода
возникает диполь (ц— 1,96- 10~29Кл-м). Согласно расче-
расчетам в этой молекуле имеет место следующее распределе-
распределение зарядов —Li°'8+H°'8~. Это означает, что связь
в молекуле LiH на 80% имеет полярный, близкий к ион-
ионному характер. Заряженный отрицательно ион водорода
Н~ имеет две орбитали, у одной из которых радиус
0,053 нм, а у второй—0,187 нм, а межъядерное расстоя-
расстояние в молекуле LiH равно 0,160 нм.
Аналогичным образом построены двухатомные моле-
молекулы с единичной связью у различных элементов перио-
периодической системы. К этому типу следует отнести моле-
молекулы гидридов щелочных металлов, галогеноводородов
и некоторых двухатомных радикалов. Для некоторых
двухатомных молекул такого типа в табл. 24.2 приведены
значения межъядерных расстояний и энергий химической
связи (кДж/моль).
Таблица 24.2.
гетеросоединеиий
Молекула
LiH
NaH
КН
RbH
CsH
ОН
сн
NH
FH
Характеристики двухатомных
Энергия
связи, кДж
234
197
180
163
176
425
, 335
356
561
молекул
г, им
0,160
0,189
0,224
0,237
0,249
0,097
0,112,
0,104
0,092
Следующий элемент второго периоду—бериллий обра-
образует с водородом гидрид ВеНг—эндотермическое, непррч-
303
ное кристаллическое вещество, необратимо разлагающееся:
при 100°С:
= [ВеНа], ДЯ°г98= 126 кДж
Энергия сублимации ВеН.2 неизвестна, но по аналогии
с галогенидами бериллия может быть оценена примерно
в 170 кДж, отсюда
= (ВеН2), ДЯ298+293кДж
Энергия атомизации бериллия и водорода известна из
таблиц:
[Be] = (Be), ДЯ°298 = 326 кДж
(Н2) = 2(Н), ДЯ288 = 4Э'4 --"-
Используя закон Гесса, находим
Ве + 2(Н) = (ВеН2), ДЯ°гв8 w — 460 кДж
тогда энергия каждой связи Be — Н в молекуле ВеНг
близка к 230 кДж. По своей геометрической структуре
молекула гидрида бериллия является линейной, все три
атома располагаются вдоль прямой линии в последова-
последовательности НВеН.
Электронная формула атома бериллия Is22s2p0, т. е.
полностью заселенную 2з-орбиталь и три вакантные
2рх-, 2ри- и 2/?г-орбитали. С позиций метода МО возможны
две связывающие орбитали:
и две разрыхляющие:
%z = c**px-H*»'-M} B414)
В этих выражениях индексы аи Ъ относятся к двум
разным атомам водорода в молекуле ВеН2.
Приведенные выражения для г)змо в молекуле ВеН2
несколько отличаются от фмо в молекуле LIH и нуж-
нуждаются в пояснении. Из (рис. 24.3) видно, что две
связывающие о,- и огж-орбитали заселяются двумя парами
электронов, а разрыхляющие орбитали o*s и с? остаются
вакантными, что и обеспечивает молекуле ВеНг доста-
достаточно высокие значения энергии связи. Уровни л,- и пу-МО
304
не изменяются по сравнению с 2рг~ и 2pf/-AO и поэтому
никакого участия в связи не принимают.
Расчет нормировочных коэффициентов С,- в уравнениях
B4.13)—B4.14) сложен, хотя принципиальный подход
к нему тот же, что и в случае простейшей молекулы H,J~.
Не приводя этих расчетов, запишем окончательные ре-
результаты:
2рг+ 0,632 р, иа - г|>15 J.
Плотность заряда электронного облака в молекуле
ВеН2 распределяется по связевым МО следующим образом:
оуорбиталь — на Be при-
приходится 30%, на 2Н —
70%; о^-орбиталь—на Be
приходится 20%, на 2Н —
80% заряда. Используя
приведенные данные, мож-
можно вычислить распределе-
распределение зарядов в молекуле
ВеН2:
_0,5 +1 —0,5
Н — Be — Н
2p»
2s2
E
AOt MO
//*
Y~~z П
/\
Л\
\ Ox
*
2AO2
"\
1
\\
;W
л'
4
Be
2Н
Это означает, что каждая
из связей полярна и об-
обладает большим моментом
диполя, однако оба связе-
вых диполя равны между
собой и пространственно
направлены в разные сто-
стороны. Поэтому суммарный
момент диполя молекулы
равен нулю. Поскольку
в этой молекуле имеется
только четыре электрона, все они заселяют связывающие
орбитали, а разрыхляющие и несвязывающие остаются
незанятыми. Такой недостаток электронов приводит к сни-
снижению суммарной энергии связи.
Строение молекулы диборана. Перейдем к рассмотрению
МО в молекуле ВН3. Это соединение, как и газообразные
Рис. 24.3. Энергетическая диаграм-
диаграмма МО молекулы ВеН2
гидриды LiH и
эндотермически
ВеН2, образуется из простых веществ
[Щ + »/,(НJ = (ВН,), ДЯ°288 = 75 кДж
805
но две молекулы ВН„ экзотермически взаимодействуют
между собой, образуя димерные молекулы диборана:
2(ВН3) = (В2Нв), ДЯ°=—117 кДж
Процесс димеризации стабилизирует молекулы В2Н„,
делает их термически более устойчивыми:
в), ДЯ°=16,5 кДж
н
У
н
V
+0,16
н
Суммарная энергия связей в молекуле ВН„ может
быть найдена из уравнения реакции атомизации
(ВН„) = (В) + 3(Н), ДЯ°ат=1142кДж
откуда средняя энергия каждой связи В — Н равна
1142:3 = 381 кДж.
Если в молекуле ВН„ заменить атомы водорода на
атомы фтора, на внешних орбиталях которых распола-
располагаются по семь электро-
-0,05 нов Bs2/?6). то образуется
значительно более прочная
молекула BF3. В ней, по-
помимо а-МО участвует так-
также связывающая л-МО за
счет использования /?-элек-
тронов трех атомов фтора.
Общее число электронов
внешнего слоя в молекуле
BF8paBHo24. Это оптималь-
оптимальное значение, ему соответ-
соответствует значительно боль-
большая прочность связи, рав-
равная 644 кДж. Отсюда
большая термическая
прочность BF3, устойчи-
устойчивого по отношению к разло-
разложению даже при температу-
температурах порядка 2000°. Более
устойчивая по сравнению
Рис. 24.4. Геометрические пара- с ВН3 молекула В2Н„ СО-
метры В2Н„ держит меньше электронов,
чем это требуется для
образования прочных связей. Крайние атомы водорода
в ней связаны с атомами бора обычными двухэлектрон-
ными связями, а два мостиковых атома водорода связаны
с двумя атомами бора только двумя парами электронов
вместо четырех, т. е. каждая из двух пар электронов
306
Принадлежит одновременно трем атомам (ВНВ), образуя
единую трехцентровую орбиталь. Фрагменты ВН4 в такой
молекуле имеют не плоскую, а несколько искаженную
тетраэдр ическую структуру, как это показано на рис. 24.4.
В этой димерной молеку-
молекуле электронами заполнена
только связывающая МО,
на несвязывающей элект-
электронов нет. Такая трехцент-
ровая связь В—Н — В,
образующаяся в резуль-
результате перекрывания двух
5/?3-орбиталей атомов бора
и одной s-орбитали атома
водорода, энергетически
выгоднее двухцентровых
В—Н связей.
В атоме бора имеются
два 2s- и один 2/?-элект-
роны. В атомах водорода
по одному ls-электрону.
За счет 2s, 2рх, 1рц и
2/^-орбиталей атома бора
и 1 s-орбитали трех атомов
водорода при их сближе-
сближении и образуются МО в мо-
молекуле ВН3. На рис. 24.5
показано, что в процессе
2s2
?
АО[ МО
;
/ 1
11
//
к_ ' / _
/\
/а ! 1
Ч \
\ \
\№
\
N
В
(I*
"%
в»
ЗАО2
1
\ 1
\ 1
\ 1
\ 1
\]
//
ъ/
1
1
1
зн
такого сближения 2s-ор-
2s-орбиталь атома бора пере-
перекрывается с ls-орбиталями
Нь и Нс), образуя одну as- и одну a*s-
Рис. 24.5( Энергетическая диаграм-
диаграмма МО молекулы ВН3
трех атомов водорода (На,
, [ab v], . B4.10)
%*s = С,Ъ* ~ Ct [%Sa + %$b + *HJ. 24.11)
В процессе сближения Is-орбитали трех атомов водорода
перекрываются 2рх н 2^-орбиталями атома бора. Харак-
Характеристика этих МО с позиций волновых ¦ф-функций может
быть записана следующим образом:
' B4;12)
¦¦->- B4.13)
B4,16)
807
Таким образом, в молекуле ВН3 имеются три связываю-
связывающие B4.10), B4.12), B4.14) и три разрыхляющие B4.11),
B4.13), B4.15) сг-МО, которые заселены шестью электро-
электронами на связывающих а-орбиталях и еще остается вакантной
несвязывающая лг-орбиталь. Благодаря этому разрыхляю-
разрыхляющие а*-орбитали остаются свободными, что обеспечивает
достаточно высокую прочность связей, каждая из которых
равна 389 кДж.
Тетраэдрическая молекула метана. Следующим пред-
представителем водородистых соединений элементов 11 периода
является метан СН4, структура которого представлена
+ х
+ х
Рис. 24.6. Схема сближения четырех атомов водорода с орбиталями
атома углерода
на рис. 24.6. Как показывает структурный анализ,
молекулы метана пространственно симметричны, они имеют
форму правильного тетраэдра, в центре которого нахо-
находится атом углерода, а в вершинах—атомы водорода.
Геометрически фигура тетраэдра получается как элемент
куба, если противолежащие вершины его соединить пря-
прямыми линиями (рис. 24.6, а). Ядро атома углерода в центре
308
куба (тетраэдра) примем за начало прямоугольных коор-
координат, координатные оси при этом проведем перпендику-
перпендикулярно к граням куба. Такая система координат позволяет
рассмотреть особенности перекрывания атомных и обра-
образования молекулярных орбиталей. Атом углерода имеет
во внешнем электронном слое 2s- и 2/7-орбитали, на кото-
которых расположены четыре электрона. Все эти орбитали
должны быть использованы для построения МО СН4.
Атомные ls-орбитали четырех атомов водорода будут
перекрываться с 2s- и 2р-АО углерода. Перекрывание
с 25-орбиталью приводит к возникновению а-МО в резуль-
результате чего получаются связывающая и разрыхляющая
орбитали:
С + 4Н, ta^i
2s+ls, %* =C,[we 1>„„,]
Перекрывание 2рх-, 2р - и 2/?г-орбиталей атома угле-
углерода с ls-орбиталями четырех атомов водорода, как это
иллюстрирует рис. 24.6, б, в, г, приводит к образованию
связывающих и разрыхляющих очМО:
С+4Н, %г=С6г|>2/г [
2/?, +Is, 4>a, =С,г|>2/,г—С8
C + 4H, %x = Cty2
C + 4H, %l/ =
Таким образом, в молекуле СН4 могут быть четыре свя-
связывающие и четыре разрыхляющие МО. Общее число
электронов, принимающих участие в образовании связей
в молекуле, равно 8 D у атома углерода и 4 от атомов
водорода). Согласно условию стремления системы к ми-
минимуму энергии электроны при образовании МО должны
заселить (рис. 24.7) сначала самые глубокие, т. е. свя-
связывающие, МО, так что разрыхляющие остаются свобод-
свободными. Наличие четырех связывающих МО обеспечивает
высокую энергию связей С—Н, равную 415,5 кДж, что
определяет достаточно высокую термическую устойчи-
устойчивость молекулы:
АО,
МО
4А0,
Из-за высокой пространственной симметрии молекулы
СН4 суммарный момент ее диполя равен нулю, что,
в свою очередь, определяет слабое взаимодействие между
молекулами СН4 и высокую летучесть (/кип = —161,6°С)
этого соединения.
Пирамидальная четырехатомная молекула аммиака. Пе-
Перейдем к описанию молекул, имеющих форму тригональ-
ной пирамиды, примером которых может служить моле-
молекула NH3. Атом азота
имеет во внешнем элект-
электронном слое 5 электронов
Bs22/>3), присоединив 3
атома водорода, он допол-
дополняет свою электронную
оболочку до 8 электронов.
Это оптимальное число,
характеризующее предель-
предельное заселение электронами
всех связевых орбиталей.
Опыт показывает, что
NH3—устойчивое соеди-
соединение, разлагающееся
эндотермически:
(ЛШ3) =
1
1
1
il/
\
\
<
/—
1
1
1
\
\
\х
—i
\
i
\L±±±
5
D
Я
/|
кДж
сн.
4Н
Рис. 24.7. Энергетическая диаграм-
диаграмма МО молекулы СН4
Энергия связи N—Н в
молекуле аммиака равна
391 кДж. Для описа-
описания МО в молекуле NH3
воспользуемся ранее рас-
рассмотренной моделью тетраэдра, в которой одна из четырех
вершин остается незанятой. На рис. 24.8, а—г графически
изображена структура молекулы NHg) стрелками пока-
показаны направления перемещения атомов водорода при их
сближении с атомом азота. При этом ls-орбитали атомов
водорода перекрываются с 2s- и 2/0-орбиталями атома
азота, образуя молекулярные а-орбитали. Всего в моле-
молекуле NH3 в валентном электронном слое находится
8 электронов D пары), три из этих электронных пар за-
заселяют три связывающие cr-МО (рис. 24.9) так, что обра-
образуются три, равноценные связи с тремя атомами водо-
водорода. Если бы в образовании а-МО участвовали только
2/?-орбитали атома азота, то валентные углы Н — N—Н
были бы равны 90°. Однако в этом процессе частично
310
участвует также и 2в-орбиталь атома азота, поэтому
экспериментально определенные валентные углы в моле-
молекуле NHg равны 107°, т. е. близки к соответствующим
углам в строго тетраэдрических молекулах (например,
+х
+х
Рис. 24.8. Схема сближения трех атомов водорода с атомом
азота
в СН4 углы равны 109,28°). Основная же часть 2s-op6n-
тали атома азота переходит на а-МО, которая простран-
пространственно располагается в молекуле NH3 по другую сто-
сторону от атомов водорода и обеспечивает высокий момент
диполя молекулы.
Таблица 24.3. Характеристики строения молекул гидридов
элементов V группы
Молекулы
NH3
РН3
AsHa
Угол между
связями,
град
107
94
, 92 ,
Средняя энергия
одной связи,
кДж/моль
391
322
247
Момент
диполя
ц-10", Кл-м
0,490
0,183
0,050
'•:¦¦• ."¦¦ -¦
Межатомное
расстояиие
Э-Н, нм
0,101
0,142
0,152
АО.
МО
if1
k k ¦' ' ¦¦
"V
if ^
/
/
/
/
/
0
ъ —
N ¦
NH,
¦зн
Кроме пирамидальной структуры для молекуды
аммиака принципиально возможна также плоскотреуголь-
плоскотреугольная структура (подобно BF3). Однако пирамидальная
структура энергетически выгоднее плоскотреугольной на
25 кДж. Такой небольшой энергетический барьер между
пирамидальной и плоской структурой является ответст-
ответственным за особенность моле-
молекул аммиака—структурную
инверсию. Смысл ее заклю-
заключается в способности атома
азота в процессе колебатель-
колебательных перемещений проскаки-
lj вать плоскость основания
пирамиды, т. е. в способности
молекулы аммиака вывора-
выворачиваться наизнанку. В про-
процессе такой инверсии излу-
излучается энергия в виде коле-
колебаний строго определенной
частоты (v = 2,387-1010 с),
используемая для очень точ-
точного измерения времени. Мо-
Молекул, подобных аммиаку,
со строением тригональной
пирамиды, известно довольно
много. В качестве примера
можно привести родственные аммиаку фосфин РН3 и
арсин AsH3, характеристики которых приведены в табл.
24.3.
Из таблицы видно, что при движении в V-A подгруппе
сверху вниз валентный угол Н—Э—Н становится все
меньше, приближаясь к 90°. В той же последовательно-
последовательности на порядок уменьшается и момент диполя, что можно
объяснить наличием в молекуле NH3 аг-орбитали, кото-
которая располагается по другую сторону от атомов водорода
и тем самым определяет повышенное значение момента
диполя.
В молекуле РН3 атом фосфора в отличие от азота
имеет разрешенную Зй-орбиталь, на которую при воз-
возбуждении переходит избыточная часть электронного облака
атома фосфора. Энергия такого возбуждения достаточно
велика, и в совокупности с другими факторами умень-
уменьшает энергию связи в молекуле РН,, C22 цДж). При
этом в три фаза, уменьшается и момент,диполя РН3 из-за
отсутствия гибридной орбитали, аналогичной той, кото-
Рис. 24.9. Энергетическая диа-
диаграмма МО молекулы NH3
рая имеет место в молекуле NHS. Валентный угол
Н — Р—Н также становится меньше, поскольку при
образовании МО в РН3 роль Зя-орбитали атома фосфора
уменьшается. Аналогичные рассуждения можно исполь-
использовать и в отношении молекулы AsH3, но здесь наличие
полностью заселенной Sd-орбитали атома As уменьшает
прочность связи.
Уголковая трехатомная молекула воды. Следующим
за азотом в периоде является кислород Bs2p4), который
экзотермически образует с водородом распространенное
в природе соединение—воду:
(Н„) + 1/2 (О2) = {Н2О}, ДЯ°298 = —286 кДж.
Приняв энергию атомизации Н2 и О2 равной 436 и
498 кДж соответственно, а теплоту испарения воды рав-
равной 44,0 кДж, находим суммарную энергию связей в га-
газообразных молекулах Н2О:
(Н2О) = 2(Н) + (О), ДЯ°298 = 927 кДж
Отсюда средняя энергия каждой связи составляет
463,5 кДж. Экспериментальным путем было доказано,
что молекулы воды имеют уголковое строение. Угол
между направлениями связей 104,3°, близок к тетра-
эдрическому A09,28°) и далек от прямого. Более того,
в кристаллах льда угол между связями практически не
отличается от тетраэдрического. Следовательно, для на-
нахождения МО молекулы воды без большой ошибки можно
воспользоваться схемой, представленной на рис. 24.10.
Здесь координатные оси проведены через центр куба
перпендикулярно к его граням. Размеры куба выбраны
так, что расстояние от центра до любой из его вершин
равно длине связи в молекуле Н2О. Расположим в центре
куба (в начале координат) атом кислорода, а в двух
противолежащих вершинах куба атомы водорода На и Hft.
Отметим положительные и отрицательные направления
координатных осей знаками ( + ) и (—). Как и в ранее
рассмотренных примерах, будем считать, что ls-орбитали.
водорода могут перекрываться с 2s-, 2рх-, 2ру- и 2pz-
орбиталями атома кислорода, образуя а-МО и а*-МО.
В соответствии с этим запишем волновые функции МО:
if 0=?CiHpis несвязывающая
О+2Н,-.'%x=i€^xpx +C»(fWe — %,d) связывающая
2^-Ms, ^o,^C^2^-^Cs(f1,4i--%^);разрыхляющая'
313
2py-\-\s, ty u =
0+2H, %z = C
несвязывающая
связывающая
ft) разрыхляющая
Энергетическая диаграмма МО Н2О приведена на рис.
24.11. Как и в ранее рассмотренных примерах, в моле-
Рис. 24.10. Схема сближения двух атомов водорода
с атомом кислорода
куле Н2О имеет место только сг-МО, поскольку в атомах
водорода нет р-орбиталей.
Анализируя приведенные выше уравнения, видим,что
на два присоединенных атома водорода приходится по две
связывающих, несвязывающих и разрыхляющих МО.
Если бы для образования химических связей в молекуле
Н2О использовались, например, только 2рх- и 2рг-кО
кислорода, то угол между направлениями связи был бы
равен 90°. Однако наряду с ними в образовании МО
участвуют также 2s- и 2ру-к0 кислорода. За счет этого
вклада геометрия молекулы изменяется и угол между
связями становится равным 104,3°. Оставшиеся частично
неиспользованными 2s- и 2/^-орбитали кислорода обра-
образуют две МО, направленные в т и п вершины куба
(см. рис. 24.10). Частично в результате этого электронное
облако молекулы НаО оказывается смещеннйм в, направ-
направлении- атома кислорода, этому же способствует и боль-
большое сродство к электрону у кислорода B,33 эВ). Все
314
перечисленные факторы определяют наличие у молекулы
воды большой полярности (ц — 0,613 • 10~?9 Кл-м).
В связи с наличием большого дипольного момента и
за счет двух гибридных МО молекулы воды могут сое-
соединяться между собой (ис-
(используя водородные связи), ао( мо 2ао2
а также присоединяться к
ионам и молекулам других
соединений, образуя гидра-
тированные ионы и кристал-
кристаллогидраты. Эта особенность
молекул воды и является . ™.±.Л?. — >?•— и
определяющей при объясне-
объяснении эффекта электролитиче-
электролитической диссоциации.
Двухатомная молекула
фтористого водорода. Взаи-
Взаимодействие газообразных фто-
фтора и водорода экзотермично,
протекает со взрывом:
1/2(H2) + 1/2(F2) = (HF),
AW2S8==~268 КДЖ Рис. 24.11. Энергетическая диа-
Температура фтористоводо- грамма МО молекулы °
родного пламени может при
определенных условиях достигать значения свыше 5000° С.
С учетом теплот атомизации молекулярных Н2
D36 кДж) и F2 A59 кДж) тепловой эффект атомизации
HF получается равным
¦1.1,1
Ж-
О -
/ н
?
-*- н2о
У
-*— 2Н
(HF) =
8 = 965 кДж
Это значение и является энергией химической связи
1 моль HF.
При сближении атомов водорода (Is) и фтора Bs2p5)
перекрывание их орбиталей схематически показано на
рис. 24.12. Из этой схемы видно, что ls-орбиталь водо-
водорода может перекрываться только с 2s и 2рг-орбиталями
атома фтора. Исходя из этого, составим возможные вол-
волновые функции МО:
несвязывающая
связывающая
разрыхляющая
... ?s ,
F-I-H, ,%л =
315
Орбйтали фтора 2рх и 2ру перекрываться с ls-орбита-
лями атома водорода не могут, поскольку 28-орбиталь
фтора заселена двумя электронами, ее участие в а-МО
несвязывающее.
Энергетическая диаграмма МО молекулы HF приве-
приведена на рис. 24.13. Из нее видно, что основной вклад
в связывающую а-МО дает 2/^-орбиталь атома фтора.
Рис. 24.12. Схема сближения
атомов водорода и фтора
Рис. 24.13. Энергетическая диа-
диаграмма МО молекулы HF
25
E
АО,
¦¦
/
¦/ ¦¦
\
MO
^\
«J/
-
AO2
HF
Н
Большое значение сродства атома фтора к электрону
C,62 эВ) определяет значительное смещение электронного
облака молекулы в сторону фтора. В результате проис-
происходит распределение зарядов H+F~ и образуется диполь,
момент которого равен 0,606-10~29 Кл-м. Свойства раз-
различных галогеноводородов сопоставлены в табл. 26.3.
Таблица 26.3. Характеристики галогеноводородов
Моле-
Молекула
HF
НС1.
НВг ..
HI
Температура
кипения, • С
+ 19,6
—85
-г67
-35
Теплота
испарения,
кДж
21
16
1в
Энергия
связи,
кДж
'565>
431
, 364;
Межатомное
расстоя ине,
НМ
0,092
0,128 г Г
в, 141 ;,;
0,162, ,
MOMenf
дироля,
Кл-м
¦ 0,606
iQ,360
( о,2ба ,
316"
Значения температуры кипения и теплоты испарения
жидких галогеноводородов, приведенные в табл. 26.3,
свидетельствуют о том, что наименьшая тенденция к ас-
ассоциации имеет место у хлористого водорода. Энергия
связи в ряду HF—HI уменьшается, что обусловлено
резким возрастанием числа электронов в атомах галоге-
галогенов в ряду F — I, а также уменьшением различия в энер-
энергии уровней и подуровней по мере увеличения числа
электронных слоев. В результате этого уменьшается сте-
степень перекрывания орбиталей водорода и галогена и воз-
возрастает межатомное расстояние. Моменты диполей гало-
галогеноводородов в связи с уменьшением тенденции к раз-
разделению зарядов и увеличением межатомных расстояний
в той же последовательности существенно уменьшаются.
Итак, на примерах молекул главным образом водо-
водородистых соединений элементов второго периода кратко
изложено содержание теории МО в приближении ЛКАО,
предложенного Р. Малликеном. В настоящее время эту
теорию считают лучшим способом трактовки химической
связи, хотя по наглядности она уступает теории ВС,
основы которой заложены в работе Гейтлера и Лондона.
Глава 25
Строение ионных кристаллов
Кристаллохимические структуры и их типы. Ион-
иый тип кристаллической решетки. Энергия иои-
ных кристаллов. Поляризация ионов. Правила
Фаянса.
Кристаллохимические структуры и их типы. Тенден-
Тенденция атомов одного или различных элементов к взаимно-
взаимному притяжению приводит к возникновению химической
связи и образованию молекул химических соединений.
Однако этим силы взаимного стяжения частиц не исчер-
исчерпываются. Молекулы соединений обладают способностью
взаимодействовать между собой и образовывать конден-
конденсированные продукты, жидкие или твердые. Нет ни одного
газа, который при достаточно низкой температуре и вы-
высоком давлении не переходил бы в конденсированное со-
состояние. При этом если силы стяжения газообразных
молекул слабы, то конденсация вещестэа не изменяет
317
Life
заметно энергии связей внутри молекул. Если силы стя-
стяжения молекул велики, то в конденсированном продукте
энергии связей в молекулах существенно изменяются.
Увеличение энергии стяжения молекул и уменьшение
энергии внутримолекулярных связей могут привести
к полному их выравниванию. В этом случае в конденси-
конденсированном продукте полностью исчезнут межмолекулярные
границы и получится жидкое или твердое вещество
с большой прочностью всех связей, мало похожее по
совокупности свойства на исходное газообразное. Так,
например, энергия внутримолекулярных связей в жидком
и газообразном кислороде равна 494 кДж/моль, а энер-
энергия межмолекулярных связей в твердом кислороде —
7,3 кДж/моль, т. е. в 68 раз меньше. Этому факту соот-
соответствует малая разница в химических свойствах твер-
твердого, жидкого и газообразного кислорода. В качестве
другого примера рассмотрим хлористый натрий NaCl.
Энергия связей в газообразных молекулах NaCl равна
410 кДж/моль, а суммарная энергия связей в кристал-
кристаллах NaCl составляет 644 кДж/моль. В кристаллах NaCl
нет не только молекул, но также и атомов, поскольку
поляризация связей Na—С1 доведена до 82%-ной степени
ионности, тогда как в молекулах NaCl степень ионности
связи равна 57%. Это означает, что свойства кристаллов
и молекул NaCl существенно различаются между собой.
Если представить, что между газообразными молеку-
молекулами воды нет никаких сил взаимодействия, то для до-
достижения этим газом плотности льда при 0°С потребова-
потребовалось бы давление около 1,27-1013 Па. Если такое же
допущение сделать в отношении молекул NaCl, то для
их сжатия до плотности кристаллов хлорида натрия
(d20 = 2,l7 кг/м3), потребовалось бы давление около
2,23-1013 Па. При таком же допущении и температуре
210° С для конденсации молекулярного кислорода нужно
было бы всего 2,08-1012 Па. Приведенные примеры сви-
свидетельствуют о том, что переход от газообразного состоя-
состояния к конденсированному связан с затратой энергии, а
следовательно, и с изменением строения этого вещества.
Общие положения учения о химической связи остаются
справедливыми и по отношению к жидким и твердым ве-
веществам, но требуют учета ряда дополнительных фак-
факторов, характерных только для конденсированного состоя-
состояния. Поэтому для полноты представлений о строении
вещества необходимо рассмотреть особенности строения
конденсированных сред и в первую, очередь кристаллов.
- Кристаллами называют тела с равновесным, законо-
закономерным, периодически повторяющимся расположением
образующих их частиц — ионов, атомов или молекул. Если
центры тяжести этих частиц мысленно соединить отрез-
отрезками прямых, то образуется пространственная, периоди-
периодическая решетка, получившая название кристаллической.
о
н—о—s—о—н
Рис. 25.1. Структура газообразной молекулы сер-
серной кислоты:
а — формальная (штриховая схема); б —истинная структура
Центры тяжести частиц находятся в пересечениях, или
узлах, кристаллической решетки.
Структуру кристаллов изучают в разделах естество-
естествознания, называемых кристаллофизикой и кристаллохи-
кристаллохимией. Содержанием кристаллохимии является установле-
установление зависимости условий образования и физико-химиче-
физико-химических свойств кристаллов от их структуры и состава,
изучение энергетики и выяснение природы химической
связи в кристаллах. Основным методом исследований
в кристаллохимии является рентгеноструктурный анализ,
использующий явление дифракции рентгеновского излу-
излучения на кристаллах, открытое М. Лауэ и др. A912).
В последние десятилетия получили широкое распростра-
распространение методы электронографии (дифракция быстролетящих
электронов на кристаллической решетке) и нейтроногра-
нейтронографии (дифракция медленных, тепловых нейтронов на крис-
i таллах). Каждый из этих методов обладает спецификой
применения, ввиду чего совокупность их позволяет про-
проводить структурные исследования самых различных обра-
образцов, существенно различающихся по своей природе.
Кристаллические структуры химических соединений
I значительно сложнее структур молекул. Если графиче-
, екая формула газообразной молекулы серной кислоты
достаточно полно передает ее истинную структуру
i (рнс. 25.1)-, то такой же формальный прием в отношении
кристаллических соединений в значительной степени те-
i 319
[Ж
ряет свой смысл. Это легко показать на примере мине-
минерала перовскита СаТЮ3, графическая формула и фраг-
фрагмент истинной структуры которого приведены на рис.
25.2. Поэтому введены понятия кристаллохимических
структурных формул, учитывающих координационные
числа атомов в решетке. Так, например, структурная
формула кристаллического хлористого натрия—[NaCle/e]3°°,
Рис. 25.2. Структура фрагмента кристаллической
решетки перовскита СаТЮ3:
а —формальная графическая схема; б —фрагмент кристал-
кристаллической решетки (атомы кальция — ф, кислорода —О>
титана —.)
здесь нижний индекс F/6) указывает на КЧ натрия и
хлора, верхний индекс (Зоо)—на трехмерную повторяе-
повторяемость этого пространственного рисунка.
В табл. 25.1 приведены примеры других кристаллохими-
кристаллохимических структурных формул.
Таблица 25.1. Кристаллохимические структурные формулы
некоторых соединений
Соединение
Кристалл охимическая
структурная формула
Пояснения к структуре
CsCl
CaF2
PBr5
РС15
СаТЮз
8]3~
[CsC18/8]
[CaFe/4]300
[PBr+] Br-
[PC1-] [PCI
Са[ТЮв/а]з
]
K4Ca =
K4Ca=I2, K4Ti =
кристаллические решетки rto характеру химических
связей образующих их частиц подразделяются на атом-
атомные, ионные, ,.металлические и молекулярные. Принци-
320 ; '
пиальная схема таких решеток показана на рис. 25.3.
Для кристаллических решеток с атомной структурой
характерно наличие в узлах атомов, прочно связанных
между собой валентными связями. Кристаллы такого
типа обладают высокой температурой плавления, боль-
большой твердостью и хрупкостью. Примером кристаллов
с атомной структурой может служить алмаз.
Решетки с ионной структурой содержат в узлах
электростатически взаимодействующие ионы. Кристаллы
000©
©000 0"О©'
Рис. 25.3. Типы кристаллических решеток:
а — атомная; б — ионная; в — металлнческая; г— молекулярная
с подобной структурой обладают несколько меньшей тем-
температурой плавления и твердостью, но большей хруп-
хрупкостью. Примером кристаллов с ионной структурой явля-
является поваренная соль.
Для решеток с металлической структурой характерно
наличие в узлах кроме атомов также и ионов, которые
образуются за счет отрыва электронов. Атомы и ионы
находятся в состоянии непрерывного обмена электронами,
причем процесс этот происходит без затраты или осво-
освобождения энергии (в единицу времени число атомов, по-
потерявших электроны, и присоединивших их ионов равно).
В процессе такого непрерывного обмена электронами
часть их стационарно остается в свободном состоянии,
образуя так называемый «электронный газ». Наличие
свободно перемещающихся электронов и динамически
обменивающихся ими ионов и атомов сообщает металли-
металлическим кристаллам специфические свойства: пластичность,
электронную проводимость, высокую теплопроводность,
металлический блеск, непрозрачность. Специфика струк-
структуры металлических кристаллов создает условия для
большого разнообразия их свойств. Так, например, тем-
температура затвердевания ртути —38,9° С, в то время как
вольфрам плавится лишь при 3380° С; натрий мягок, как
воск, а рений с трудом можно обработать инструментом,
изготовленным из специальных сортов стали.
11
705
321
Решетки с молекулярной структурой в узлах содер-
содержат молекулы, связанные между собой слабыми межмо-
межмолекулярными связями, которые легко разрушаются;
поэтому молекулярные кристаллы, как правило, легко-
легкоплавки и обладают малой твердостью. Примерами таких
кристаллических веществ могут служить кристаллический
иод (^пл = 113,4° С), в узлах решетки которого находятся
молекулы /2; лед, плавящийся при 0°С; кристаллы СО2,
плавящиеся под давлением при —57° С, и др.
Кристаллические вещества, подобно жидкостям и газам, могут
образовывать между собой растворы. Известно несколько видов твер-
твердых растворов: внедрения, замещения и вычитания.
В твердых растворах внедрения атомы растворенного элемента
располагаются между атомами кристаллической решетки раствори-
растворителя. Обычно твердые растворы внедрения получаются при растворе-
растворении в металлах переходных элементов атомов неметаллов с небольшими
атомными радиусами (Н, В, С, N, О). Иногда твердые растворы внед-
внедрения образуются и на основе химических соединений (например,
при растворении Ni в кристаллическом соединении NiSb).
Твердые растворы замещения образуются в результате частичной
замены некоторых атомов кристаллической решетки растворителя
на атомы растворенного вещества. Примером твердых растворов за-
замещения может служить система NaCl-f-KCl, в ней атомы хлора и
атомы металла закономерно чередуются в объеме кристалла. Но узлы
решетки, предназначенные для металлических атомов, хаотически
распределены между атомами натрия и калия.
Твердые растворы вычитания (или дефектные структуры) образу-
образуются только на основе химических соединений, в кристаллической
решетке которых имеются вакансии, т. е. незанятые узлы, которые
хаотически распределены по объему кристалла и создают эффект беспо-
беспорядка, связанного с образованием твердого раствора вычитания. Приме-
Примером может служить оксид титана TiO, который имеет вакансии как по
кислороду, так и по титану. В результате гомогенный твердый раствор
вычитания охватывает широкую область составов от ТЮ0,( до TiOji26.
При этом в растворах с недостатком кислорода (TiO0,e—TiO) наряду
с двухвалентными атомами титана имеется некоторое число одно-
одновалентных. А в твердых растворах с избытком кислорода (TiO—TiOli86)
наряду с атомами двухвалентного титана содержатся трехвалентные.
Подобные кристаллические решетки образуют ТаС, TiC, NbC, ZrC
и VC, где вакантными являются узлы, по закону симметрии предна-
предназначенные для атомов углерода, но не занятые ими. Аналогичная
картина наблюдается и в соединениях NiSb, CoS, FeSe.
Ионный тип кристаллической решетки. Перейдем
к энергетической характеристике твердого состояния кри-
кристаллов, обладающих ионной структурой. Ионные кри-
кристаллы образуются атомами элементов, одни из которых
имеют небольшие значения ионизационного потенциала,
а другие—большим сродством к электрону. Характерными
представителями таких кристаллов могут служить соеди-
соединения атомов щелочных металлов и галогенов, как это
видно из табл. 25.2.
322
Таблица 25.2. Ионизационные потенциалы атомов щелочных
металлов и сродство к электрону атомов галогенов
Li
Na
К
Rb
Cs
Ионизационный
потенциал, эВ
5,39
5,14
4,34
4,18
3,89
Э+е~=Э-
F
С1
Вг
1
At
Сродство к элек-
электрону, эВ
— 3,62
— 3,82
— 3,54
— 3,23
—
Рассмотрим в качестве примера расчет энергии ионной
кристаллической решетки CsCI. Согласно табл. 25.2 энер-
энергия, необходимая для получения 1 моль Cs+ и 1 моль С1~
из свободных атомов этих элементов, равна
х= 3,89 эВ
2 = — 3,82 эВ
B5.1)
эВ
Полученный результат означает, что образование ионов
Cs+ и С1~ из их нейтральных атомов является почти
атермическим процессом, т. е. идущим с тепловым эффек-
эффектом, близким к нулю. Из термохимических данных изве-
известна стандартная теплота образования кристаллического
CsCI из простых веществ
y(Cl2)+[Cs]=CsCl,
= — 432,6 кДж
B5.2)
а также теплота образования из атомов твердого Cs и
газообразного С12
(С1) = 1/2(С12), Д#298 = — 121>2 кДж B5.3)
(Cs) = [Cs], Д Hiss = — 78,2 кДж B5.4)
Суммируя уравнения B5.1)—B5.4), получаем
(Cs+L-(Cl-) = [CsCI], Д#298 = — 638,3 кДж B5.5)
это значение и является энергией образования кристал-
кристаллической решетки CsCI из ионов. Такой термохимический
способ расчета энергии ионной решетки получил назва-
название цикла Борна—Габера. Он является косвенным и тре-
требует обязательного знания теплоты образования данного
кристаллического вещества из простых веществ4
Теоретический метод расчета основан на использова-
использовании электростатической модели. Образовавшиеся по реак-
11» 323
цииB5.1) ионы противоположного знака должны притя-
притягиваться в соответствии с законом Кулона:
?<r>=-
B5.6)
Здесь Е\п—потенциальная энергия притяжения ионов,
находящихся на расстоянии г; Z4 и Z2—числа элементар-
элементарных единиц заряда (е), которые несут на себе взаимодей-
взаимодействующие ионы. В случае Cs и Cl Zj == Z2 = 1. Согласно
П.— ОС Л
й
П.— ОС Л 17 л~~.,....~ ... .,~.Ч .,
взаимодействия двух атомов или проти-
противоположно заряженных иоиов:
1-Е'(г); II-Е" (г); III-E (г)=Е' (г) + Е" (г)
уравнению B5.6) при сближении ионов, т. е. приумень-
приуменьшении г, энергия их стяжения должна уменьшаться
(рис. 25.4), что было бы строгим только в случае точеч-
точечных зарядов. Поскольку же это не так и ионы состоят
324
жении ионов начинают проявляться силы взаимного от-
отталкивания (между ядрами и оболочками). Потенциальная
энергия такого отталкивания E(r), no M. Борну, может
быть выражена соотношением
?ю=-^-, B5.7)
где В—коэффициент отталкивания Борна; п—показатель
степени, обычно близкий к 9.
Потенциальная энергия взаимодействия ионов Е(г)
равна сумме сил притяжения B5.6) и отталкивания B5.7):
-^r. B5.8)
При уменьшении расстояния г между ядрами ионов энер-
энергия Е'1п убывает, а Е"(п возрастает. В результате сумма
их Eir) представляет экстремальную функцию расстоя-
расстояния г, как это показано на рис. 25.4. Минимуму кривой
в точке т соответствует равновесное межъядерное рас-
расстояние г0 и равновесное значение энергии связи Ерюк.
Борновский коэффициент отталкивания В в уравнении
B5.7) можно определить, зная, что в минимуме кривой
(рис. 25.4) тангенс угла наклона касательной равен нулю
или, что то же самое, производная функции в точке ми-
минимума равна нулю
- = 0.
йг
Взяв производную уравнения B5.7), получим
Решая уравнение B5.9) относительно борновского коэф-
коэффициента В, находим
5__Z122eVg'1 • B5.10)
п
И наконец, после подстановки выражения B5.10) в B5.7)
находим энергию связи на равновесном расстоянии между
двумя ионами:
325
Чтобы перейти от энергии связи между двумя ионами
к энергии 1 моль газообразных двухатомных молекул,
необходимо выражение B5.11) умножить на общее число
ионов, т. е. на число Авогадро NA:
B5.12)
©-ООЧЭ©
О-0ОЧЭО
При переходе к энергии кристаллической решетки необ-
необходимо учесть все те дополнительные эффекты электро-
электростатического притяжения и отталкивания, которые возни-
возникают при сближении мо-
молекул и образовании кри-
кристалла. Для понимания
смысла этих эффектов на
рис. 25.5 схематически
изображена гипотетичес-
гипотетическая плоская кристалли-
кристаллическая решетка, образо-
образованная в результате сбли-
сближения 15 двухатомных
ионных молекул, причем
так, что все 34 кратчай-
кратчайшие расстояния между
разноименно заряженны-
заряженными ионами одинаковы и
равны г0, а между од-
одноименно заряженными
ионами по диагонали—•
го]^2. Допустим, что со-
соединение ионных молекул в кристаллическую решетку
происходит без изменения межатомных связей и расстоя-
расстояний, тогда при вычислении энергии плоской кристалли-
кристаллической решетки помимо 15?равн, имевших место в изоли-
изолированных молекулах, необходимо учитывать 34?равн за счет
энергии решетки:
?крист== 15?равн + 34?равн = 49лПрИТЯЖ.
Однако при таком сближении молекул кроме дополни-
дополнительных сил стяжения рождаются и силы расталкивания,
действующие по диагоналям ячеек между одноименно
заряженными ионами. Как показано на рис. 25.5, таких
наиболее коротких промежутков имеется 40. Диагонали
квадратов больше сторон квадратов г0 в У'2 раз, поэтому
энергия расталкивания одноименно заряженных ионов
326
(ЕХЭОЧЭО
Рис. 25.5. Схема конденсации ион-
ионных молекул и образования гипо-
гипотетической плоской ионной кристал-
кристаллической решетки
равна 40?равн:К2 = 28,3?расталк. Отсюда полная энергия
плоской кристаллической решетки равна
— 28,
= 20,
Вспомним, что по условию задачи энергия 15 изоли-
изолированных молекул была равна 15?равн. Отсюда отношение
энергии кристаллической решетки к простой сумме энер-
энергии 15 исходных молекул получается равным 20,7:15 =
= 1,38. Это означает, что кристаллическая решетка в 1,38
раза прочнее разрозненных молекул. Следует отметить,
что в этой модели учитывалось взаимодействие только
близлежащих ионов. Взаимодействие дальних ионов зна-
значительно слабее, оно вносит лишь небольшую поправку
в искомую величину.
Таким образом, зная геометрию расположения ионов
в кристаллической решетке, можно найти коэффициент,
позволяющий перейти от энергии изолированных ионных
молекул к энергии кристаллической решетки. Этот коэф-
коэффициент получил название константы Маделунга (Л),
с его учетом уравнение B5.12) принимает вид
ZlZ2ANAe2
-"кристал '
4)-
B5.13)
В табл. 25.3 приведены значения константы Маделунга
для кристаллических форм некоторых соединений.
Таблица 25.3 Значения константы Маделунга
Соединение
NaCl
CsCl
ZnS
CaF2
ТЮ2
Кристаллохимическая
структура
[NaCl6,6]3-
[CsCls,8]3~
[ZnS4/4p°°
[CaF8/4f~
[TiO6/3]3~
Константа Маделунга А,
(безразмерная)
1,7475
1,763
1,638
2,520
2,408
Величина п в уравнении 25.13 может быть вычислена
из уравнения для сжимаемости кристалла
18г$
Аег (п — i
B5.14)
Сжимаемость кристаллов и определяют изменением их
объема V под действием давления р и выражают соотно-
327
шением
dV
Vdp
V&p
Если выбрать Ар=1, то уравнение для сжимаемости
упростится:
_ AV
%- -у.
Величина сжимаемости кристаллов очень мала: например,
для NaCl и = 0,04, а для алмаза и = 0,005. Параметр и,1
вычисленный с помощью характеристики сжимаемости,
принимает следующие значения:
LiF LiCl LiBr NaCl NaBr
5,9 8,0 8,7 9,1 9,5
В табл. 25.4 приведены значения энергии кристалличе-
кристаллической решетки, вычисленные разными способами.
Таблица 25.4. Значения энергии кристаллической решетки
некоторых галогенидов щелочных металлов, кДж/моль
Соединение
NaCl
NaBr
Nal
КС1
КВг
KI
По уравнению
B5.13)
750
713
668
683
655
618
По термохими-
термохимическому циклу
B5.1) - B5.5)
769
734
690
704
672
634
Прямые
измерения
759
736
695
_
669
640
По уравнению
B5.18)
760
728
680
687
660
620
В 1932 г. М. Борн и Р. Майер ввели уточнение в урав-
уравнение B5.13) для расчета энергии ионной кристалличе-
кристаллической решетки:
B5.15)
Здесь первое слагаемое представляет собой энергию элек-
электростатического притяжения разноименно заряженных
ионов. Второе слагаемое характеризует энергию оттал-
отталкивания, выражение для которой получено с использо-
использованием представлений волновой механики, согласно кото-
которым электронная плотность в атомах уменьшается при
328
удалении от ядра по экспоненциальному закону. В этом
выражении С—постоянная величина, подобная коэффи-
коэффициенту Борна В в уравнении B5.7); р—также постоян-
постоянная величина, отражающая свойства взаимодействующих
частиц. Как и при выводе уравнения B5.13), здесь ис-
используют условие -т— = 0. Следовательно,
отсюда
AW' B5Л6)
р
Подставив уравнение B5.16) в B5.15), при условии г =
= г0 получаем уравнение Борна—Майера:
B5.17)
Из данных по сжимаемости кристаллов B5.14) было
найдено, что величина р для большинства кристалли-
кристаллических веществ примерно одинакова и близка к 0,345 х
х 10~10; произведение NAe2 при подстановке численных
значений числа Авогадро и заряда электрона получается
равным 332,0; величина г„ принимается равной сумме
ионных радиусов, т. е. го = гк + гА. Значение константы
Маделунга А в двухкомпонентных кристаллах (например,
кристаллы NaCl) относится именно к двум компонентам.
В расчете же на один компонент следует брать 1/2Л;
если же кристаллическая решетка такой же кубической
структуры содержит 2/и,- компонентов, то величина кон-
константы Маделунга будет равна для нее Л2;/2.
Путем подобных рассуждений, приняв структуру всех
кристаллических веществ кубической, А. Ф. Капустинский
A943) вывел приближенную формулу для расчета энер-
энергии ионной кристаллической решетки (величина А —
— 1,7476 характеризует кубическую решетку NaCl):
290.1*А Г, 0,345 1 B5 18)
Расчет энергии ионных кристаллов для галогенидов ще-
щелочных металлов с использованием уравнения B5.18),
как это показано в табл. 25.4, приводит к результатам,
близким с экспериментом.
329
Поляризация ионов. Правила Фаянса. Однако ионная
модель строения кристаллов полностью оправдывает себя
только в отношении галогенидов щелочных металлов,
поскольку полная ионизация атомов не достигается даже
в этих соединениях. В результате часть электронного
облака становится общей для них, что создает эффект
частичной валентной связи. Как правило, в соединениях
чем выше энергия ионизации атомов металла, тем менее
ионным становится взаимодействие их с атомами неме-
^-~-«^ талла. В итоге менее
^j >v строгим получается ре-
(+к __ | зультат расчета энер-
^~\ ) гии решетки по урав-
\.У нениям B5.13), B5.16)
&Г и B5.18). В табл. 25.5
г'о<г +г' для сравнения приведе-
'" а ? ны значения решетки
Рис. 25.6. Поляризация ионов: как вычисленные по
а-ионная связь г„ = лк + гА; б-полярная, уравнению B5.16), ТЭК
валентная связь го < гк+гА ' И ПОЛуЧеННЫе ЭКСперИ-
ментально.
Из табл. 25.5 видно, что влияние неионных сил сцеп-
сцепления на энергию решетки достаточно велико. Установле-
Установлено, что степень неионности решетки нарастает в рядах:
F- < С1- < Вг- < I- и О2- < S2" < Se2~
На рис. 25.6, а изображен гипотетический случай обра-
образования соединений из ионов без перекрывания орбитали.
Как правило, в соединениях происходит частичное пере-
перекрывание атомных орбиталей и образование примеси
валентной связи обычно приводит к уменьшению межъ-
межъядерного расстояния между атомами (рис. 25.6,6). Это
формально соответствует искажению сферической формы
ионов, т. е. смещению центра тяжести электронной обо-
оболочки иона относительно заряда ядра. Следовательно,
деформация ионов при их взаимодействии должна сопут-
сопутствовать их поляризации. Очевидно, ионы, обладающие
большим зарядом ядра и «тонкой» электронной оболочкой,
«прижатой» электростатическими силами к ядру, должны
сами деформироваться слабо, но обладать повышенной
деформирующей, поляризующей способностью. Напротив,
отрицательно заряженные ионы, имеющие относительно
меньший положительный заряд ядра и объемистую рыхлую
330
Таблица 25.5. Значения энергии кристаллической решетки
некоторых соединений, кДж/моль
Соединение
AgF
AgC!
AgBr
Agl
MnF2
FeF2
NiF2
SnS
SnSe
Эксперимент
— 967
— 916
— 908
— 895
-2770
— 2912
— 3046
-3615
— 3611
Расчет по урав-
уравнению B5.16)
— 870
— 782
- 757
- 736
— 2745
— 2753
— 2916
— 3427
— 3305
Разность
96
134
151
159
25
159
130
188
305
электронную оболочку, должны легко деформироваться
под действием внешнего поля, т. е. поляризоваться.
Именно этот смысл заложен в четырех правилах Фаянса:
1) поляризующая способность катиона увеличивается с уменьше-
уменьшением его размеров (т. е. числа заселенных электронных орбиталей):
2) поляризуемость аниона растет с увеличением его размеров
(т. е. числа заселенных электронных орбиталей);
3) чем выше заряды катиона и аниона, тем больше поляризую-
поляризующая способность катиона и поляризуемость аниона;
4) наличие у иона электронной конфигурации инертного газа
усиливает экранировку заряда ядра и тем самым уменьшает поляри-
поляризующую способность катиона и поляризуемость аниона.
Из анализа этих правил следует, что в периодической системе
при движении элементов снизу вверх и слева направо увеличива-
увеличивается поляризующая способность катионов и уменьшается поляризуе-
поляризуемость анионов. Так, поляризующая способность катионов элементов
второго периода от Li до F возрастает столь интенсивно, что только
Li и Be выступают в соединениях в качестве ионов, все же осталь-
остальные элементы этого периода катионов не образуют. Что же касается
анионов, то, согласно правилу Фаянса, можно ожидать усиления их
поляризуемости в рядах F~, С1-, Вг~, I" и О2~, S2~, Se2~, Te2~.
Глава 26
Кристаллические решетки с дефектами
Полупроводники п- и />-тнпов. Металлические ре-
решетки. Зонная теория. Энергия металлической ре-
решетки. Интерметаллические соединения.,
Кристаллические решетки с дефектами. Кристалличе-
Кристаллические решетки, в которых все ионы без исключения зани-
занимают места, отвечающие соответствующим структурным
331
типам, считают идеальными. В действительности, в резуль-
результате теплового движения частиц и ряда других причин
любой стехиометрический кристалл при температурах
выше абсолютного нуля обязательно несовершенен. Это
несовершенство проявляется в наличии так называемых
дефектов, которые могут возникать двумя путями.
« >—О-тН I О 1 I О
о—•—о—•—о—»
•-1.О-2
Рис. 26.1. Виды дефектов в ионных кристаллах:
1 — катнон; 2 — анион
1. Некоторые ионы, сместившись по отношению к сво-
своему равновесному положению, попадают в междоузлия
решетки, образуя ион внедрения. В равновесном же поло-
положении остается вакансия, как это показано на рис. 26.1, а.
Такие отклонения от идеальности получили название
дефектов по Френкелю.
2. Некоторые положительно и отрицательно заряжен-
заряженные ионы смещаются вплоть до выхода на поверхность
кристалла, как это показано на рис. 26.1,6. Такие от-
отклонения кристаллов от идеальности получили название
дефектов по Шоттки.
Дефекты обоих типов связаны с переходом ионов из
идеального состояния в разупорядоченное, т. е. процессу
образования дефектов соответствует энергия
С другой стороны, наиболее упорядоченное состояние
идеального кристалла наделено меньшей энтропией 5ЯД, а
разупорядоченное—большей 5разуп, т. е.
А5 = [5разуп-5ИД]> 0.
Используя основное уравнение термодинамики, получаем,
что на образование 1 моль кристалла затрачивается ра-
332
бота
AG° = ДЯ°— rAS° = - RT In К,
откуда
AG°
Рассмотрим дефектную решетку по Френкелю на при-
примере твердого раствора дефектов (т. е, разупорядоченных
катионов и их вакансий) в ионной решетке (рис. 26.1, а).
Обозначим концентрацию разупорядоченных катионов и
равную им концентрацию дефектов через п. Концентра-
Концентрацию упорядоченных ионов в объеме решетки обозна-
обозначим Nlt а концентрацию возможных междоузлий—N2.
Известно, что число дефектов в решетках, близких к иде-
идеальным, очень мало по сравнению с числом упорядочен-
упорядоченных ионов, а также междоузлий в объеме кристалла,
т. е. n<^:N1 и n<^N2. В этом случае процесс образования
дефектов по Френкелю можно схематически записать в виде
равновесия следующим образом:
Ионы в рав-1 [Междоузлия! Г Возбужденные
новесной I-}- равновесной I, катионы
решетке J LPe™eTKH J 1^ежД0УЗ.
денные] ГВакансии-|
в +1 решетке
лиях J L J
Используя эти данные, запишем выражение для константы
равновесия
K=wk=e RT • B61)
где AGj—энергия Гиббса (работа) образования дефектов
по Френкелю; R—газовая постоянная; Т—температура.
Таким образом, из уравнения B6.1) находим число де-
дефектов в объеме 1 моль кристалла:
2RT
Рассмотрим в рамках аналогичной модели процесс
образования дефектов по Шоттки. В этом случае, как
один из вариантов, катион и анион могут создавать в кри-
кристалле парные вакансии, число которых в объеме кри-
кристалла равно п0, а общее число ионных пар в равновес-
равновесной идеальной решетке N. Запишем схему процесса обра-
333
зования дефектов по Шоттки:
Г Ионы в П Г Вакансии иаТ Г Вакансии ~| Г Ионные пары на!
I равновесной -}- поверхности | ч ионных пар \~\~\ поверхности
|_решетке | [кристалла J [_в решетке J |_кристалла J
(N — no)«jV n0
Используя эту модель, получаем приближенное выраже-
выражение для константы равновесия
до:
К = -$-=в RT , B6.2)
где /XG?2—энергия Гиббса образования дефектов по Шоттки.
Из уравнения B6.2) находим число дефектов (концентра-
(концентрацию дефектов в объеме 1 моль кристалла):
Сравнивая два вида дефектов, изображенных на
рис. 26.1, естественно предположить, что катионы, нахо-
находящиеся в междоузлиях решетки (а), из-за большой плот-
плотности окружения испытывают значительно больший эффект
«выталкивания», чем ионы на поверхности решетки (б).
Этот эффект должен зависеть от зарядов ионов и их раз-
размеров. Если катионы и анионы близки по размерам, то
эффект выталкивания из междоузлий решетки должен
быть больше, чем с поверхности, т. е.
тогда справедливо соотношение п^>«„. Это означает, что
в таких решетках дефекты по Шоттки должны быть пред-
предпочтительнее дефектов по Френкелю. Примером может
служить фтористый калий, радиусы ионов которого близки
по величине.
В других случаях, когда радиусы ионов существенно
различаются между собой (обычно катионы меньше анио-
анионов), соотношение сил меняется. Если катионы малы по
сравнению с анионами, то переход первых в междоузлия
уже не требует столь большой затраты энергии. Кроме
того, небольшие по размерам катионы, согласно правилам
Фаянса, обладают высокой поляризующей способностью,
что в случае перемещения катиона в междоузлие приво-
приводит к выигрышу энергии за счет поляризации окружаю-
окружающих анионов. В результате имеют место соотношения
AGJ < AGI и п<п0, что означает предпочтительность де-
дефектов по Френкелю. Примером таких решеток может
334
служить AgBr, в кристаллах которого преимущественным
является хаотическое распределение ионов серебра. Под
действием электрического тока дефекты кристаллической
решетки (как ионы, так и вакансии) могут перемещаться
по кристаллу (вакансия при этом эквивалентна иону про-
противоположного знака). Наиболее важным следствием суще-
существования дефектов кристаллической решетки является
объяснение природы нестехиометрических кристаллов.
Нестехиометрическими называют такие соединения, состав
которых не может быть выражен целочисленными стехио-
метрическими коэффициентами.
В качестве соединений такого рода можно привести
систему титан — кислород. По мере присоединения неболь-
небольшого количества кислорода к металлическому титану вна-
вначале образуется однородная кристаллическая фаза, тип
кристаллической решетки которой остается неизменным
в интервале составов ТЮ0_0,5 (область гомогенности). При
дальнейшем увеличении общего содержания кислорода
появляется новая фаза — состава Ti(\88, масса которой
будет возрастать, а масса фазы TiO0i5 убывать. Как только
в процессе увеличения общего содержания кислорода
исчезает фаза TiO0i5, начинает изменяться состав второй
фазы ТЮ0,88 количество кислорода в которой может мо-
монотонно возрастать вплоть до TiOli20. Во всем интервале
составов TiO0i88_li20 также наблюдается область гомоген-
гомогенности с неизменным типом кристаллической решетки, т. е.
способ расположения атомов титана и кислорода не
изменяется. Причина заключается в том, что оксид титана
не имеет строго определенного стехиометрического состава,
его кристаллы образуют кубическую структуру каменной
соли. Однако в решетке этого оксида, содержащего по
1 моль титана и кислорода, остаются вакантными 15%
позиций, предназначенных для атомов титана, и 15%
мест, предназначенных для атомов кислорода. Преиму-
Преимущественное заполнение первых или вторых вакансий
и приводит к изменению состава от TiO0i88 до TiOli2 при
сохранении неизменности структурного типа решетки.
Другим примером соединений нестехиометрического
состава служит оксид железа FeO, который в строгом
стехиометрическом соотношении компонентов неустойчив.
Было установлено, что оксид представляет собой соедине-
соединение, существующее в интервале гомогенности Fe0,sll_OiMO
(при 1000 °С), причем кристаллы его обладают дефектной
структурой с пространственным расположением атомов
каменной соли. Если бы все вакансии в нем были заняты
335
равным числом атомов железа и кислорода, то получилось
бы соединение FeO. Однако часть вакансий, предназна-
предназначенных атомам железа, остаются незанятыми (например,
при составе Fe0i89O не заняты 11 % позиций атомов же-
железа). В то же время часть имеющихся в структуре атомов
железа имеют степень окисления 2 +, а часть 3 +, при-
причем атомы железа (III) и вакансии образуют комплексы
типа {Fe?" [вакансия]} и даже более сложные {Fe^I! [ва-
[вакансия]},,, которые представляют собой «островки» со свя-
связями металл—металл.
Полупроводники п- и /?-типов. Все нестехиометрические
соединения можно условно разделить на четыре группы
< i о (
О (т\ (
0 1
1 С
>—ц
1 О
)—(т—о—i
1—i
е
о—о—1
1 С
) 1
р—*—?
е
>—*н
О • 6 1
•-1, О-2, ©-3
Рис. 26.2. Типы решеток нестехиометрических соедине-
соединений:
1 — катион; 2 — аннон; 3 — катион 2 +
нестехиометрии (рис. 26.2, а—г) со следующими харак-
характерными особенностями структуры:
1) наличие, катионных и отсутствие анионных вакан-
вакансий (FeO, СоО, №0, Си2О);
336
2) наличие анионных и отсутствие катионных вакан-
вакансий (NaCl, KC1, КВг);
3) присутствие избыточных катионов, которые распо-
располагаются в междоузлиях (ZnO, CdO);
4) наличие избыточных анионов, располагающихся
в междоузлиях (UO2).
Поскольку кристалл электронейтрален, то наличие
катионных вакансий или избыточных анионов в междо-
междоузлиях (рис. 26.2, а, г) должно компенсироваться тем, что
некоторые катионы в узлах решетки имеют больший заряд,
чем остальные.
Точно так же, избыток катионов металла (рис. 26.2, в, г)
требует присутствия свободных электронов. За счет погло-
поглощения энергии извне такие электроны приобретают спо-
способность перемещаться по междоузлиям решетки, создавая
эффект электронной проводимости. С повышением темпе-
температуры число дефектов, а следовательно, и число свобод-
свободных электронов B6.2, в, г) увеличивается, так как
оно пропорционально величине е RT . Кристаллы, обла-
обладающие такой проводимостью, называют полупроводниками.
Полупроводники с подобным способом проводимости обо-
обозначают символом п (тип п).
В нестехиометрических кристаллах (рис. 26.2, а, г) под
действием энергии извне становятся возможными пере-
перескоки электронов от катионов с меньшим положительным
зарядом на катионы с большим зарядом. Эти перескоки
электронов эквивалентны перемещению в обратном на-
направлении положительных зарядов («положительных ды-
дырок»). Полупроводники с такой проводимостью относят
к типу р.
Первые две группы решеток являются преобладающими
и характеризуются дефектами по Шоттки. Для структур
третьей группы характерны дефекты по Френкелю, кото-
которые возникают лишь при больших различиях в радиусах
ионов, что делает решетку подобного типа менее распро-
распространенной. Реже встречаются решетки четвертого типа,
так как вероятность нахождения анионов в междоузлиях
очень мала.
Металлические решетки. Зонная теория. Перейдем
к рассмотрению свойств и строения металлических кри-
кристаллов. Металлическое состояние вещества существенно
отличает металлы от всех остальных веществ в твердом
и жидком состояниях, что послужило основанием для вве-
введения специального понятия металлической связи. И дей-
337
E
АО
a
f/
!'¦
Vs
w
\_
MO
б
ствительно, наличие таких качеств, как металлический
блеск, ковкость (по определению М. В. Ломоносова ме-
металл—светлое тело, которое ковать можно), электронная
проводимость, высокая теплопроводность и т. д., свой-
свойственно только металлам. Природа химической связи
в металлических кристаллах также заметно отличается
от ранее рассмотренных видов связи. Для объяснения
электрической проводимости металлов П. Друде A900)
и Г. Лоренц A916) предложи-
предложили теорию электронного газа,
которая хорошо объясняла за-
закон Ома, соотношение между
теплопроводностью и электри-
электрической проводимости метал-
металлов, отчасти термическую
эмиссию электронов. Однако
представление об электронном
газе содержало в себе ряд
противоречий и не объясняло
удовлетворительно магнит-
Рис. 26.3. Схема расщепления ные свойства, зависимость
вырожденного энергетического электрической проводимости
уровня атома в металлической от температуры И др.
В 1928 г. А. Зоммерфельд
углубил теорию электрон-
электронного газа, связав ее с кван-
квантовой физикой, что позволило
объяснить термоэлектрические потенциалы и устранить
имевшиеся противоречия в представлениях о теплоемко-
теплоемкости, но при этом оставались еще не выясненными зави-
зависимости электрической проводимости и теплопроводности
металлов от температуры, а также причины различия
в свойствах проводников и изоляторов. Эти затруднения
были преодолены в результате работ М. Бриллюэна, ко-
который, положив в основу представление о волновой при-
природе электрона, развил теорию движения электронного
газа в периодическом поле ионов и атомов кристалличе-
кристаллической решетки металлов. Периодическое поле свойственно
атомам многих элементов периодической системы, его су-
существование объясняют наличием в каждом атоме разре-
разрешенных электронных орбиталей и дискретных уровней
энергии. Однако у атомов металлов внешние электроны
сравнительно слабо удерживаются полем ядра. При сбли-
сближении атомов и образовании металлического агрегата
энергетические уровни их валентных электронов расщеп-
338
о —вырожденный энергетический
уровень изолированного атома; б —
расщепление уровня при образовании
металлического агрегата
ляются на ряд подуровней, для которых характерна раз-
различная энергия. Схема такого расщепления изображена
на рис. 26.3. В результате вместо одного уровня появ-
появляется серия подуровней, образующих в совокупности
широкую зону.
Из рис. 26.3 видно, что при расщеплении энергетиче-
энергетического уровня атома образующиеся подуровни распола-
располагаются по зоне выше и ниже исходного уровня. Это озна-
означает, что запас энергии электрона, находящегося на самом
верхнем расщепленном подуровне, должен быть больше,
ГАЯ;
$
™ШШ?®Ш
Рис. 26.4. Схема энергетических зон в атомах:
а — металлов; 6 — полупроводников; б—неметаллов
чем на нерасщепленном, т. е. для отрыва такого электрона
необходимо затратить меньшую энергию, чем если бы он
находился на нерасщепленном атомном уровне. Например,
энергия ионизации (первый ионизационный потенциал)
изолированного атома серебра равна 7,574 эВ, в то время
как энергия отрыва электрона из металлического серебра
составляет только 4,7 эВ.
За счет расщепления двух уровней атома образуются
две зоны. У атомов неметаллов эти зоны располагаются
далеко друг от друга (рис. 26.4, в), а у металлов близко
и даже перекрываются, как это показано на рис. 26.4, а.
Заштрихованные зоны заполнены валентными электронами,
незаштрихованные—«свободными» электронами (зона про-
проводимости). Электроны металла размещаются по подуров-
подуровням зоны так, чтобы сначала заполнялись наиболее глу-
глубоко лежащие подуровни, а по мере их заселения—менее
глубокие (выполнение условия стремления системы к ми-
минимуму энергии). У металлов, имеющих мало валентных
электронов, заполненными оказываются только наиболее
339
глубоко лежащие подуровни (на каждом не более двух
электронов). Однако соседние подуровни очень близки по
энергии, поэтому переход электронов с одного подуровня
на другой осуществляется легко, без затраты большой
энергии.
Неметаллические структуры (рис. 26.4, в) отличаются
от металлических, поскольку у образующих их атомов
заполнены все подуровни валентной зоны, а увеличение
запаса энергии электрона, достаточное для перехода его
в следующую зону (зону проводимости), требует большой
затраты энергии АЕ3. Например, для алмаза эта энергия
составляет 6 эВ. Преодоление такого энергетического барь-
барьера в обычных условиях оказывается невозможным, в связи
с чем неметаллы не обладают электронной проводимостью.
Согласно схеме, представленной на рис. 26.4, б, полу-
полупроводники занимают промежуточное положение между
металлами и неметаллами. У них, как и у неметаллов,
валентная зона полностью заполнена1 электронами, но
энергия АЕг, необходимая для перехода электрона в зону
проводимости, невелика (например, для германия A?2=
= 0,7 эВ, для кремния 1,10 эВ). Поэтому кристаллы таких
веществ при нагревании становятся электропроводными.
При снятии внешнего поля электроны возвращаются в ниж-
нижнюю, валентную зону и кристалл снова становится непро-
непроводником. Рост электрической проводимости с увеличением
температуры—одно из отличительных свойств полупровод-
полупроводников (у металлов наоборот).
Зонная теория наиболее полно раскрывает различные
свойства металлических и полупроводниковых кристаллов
и хорошо описывает электрическую проводимость метал-
металлов. Учет волновой природы электрона приводит к выводу
о том, что всякое нарушение симметрии расположения
атомов в металлическом кристалле должно привести к пони-
понижению электрической проводимости. Такое нарушение
симметрии, искажение периодичности кристаллической
решетки может достигаться увеличением температуры (из-
за усиления колебательного движения атомов), добавками
легирующих элементов, механической обработкой металла
(ковка, протяжка и т. д.).
Зонная теория получила широкое распространение для
описания физических свойств металлов.
Энергия металлической решетки. Рассмотрим вопрос
об энергии кристаллической решетки металла. Сделаем
допущение, что в узлах такой решетки находятся поло-
положительно заряженные ионы (катионы), несущие заряд Ze
340
и имеющие массу т, а в объеме кристалла (в междоузлиях)
равномерно распределен электронный газ (Z — валентность
металла, е—заряд электрона). В такой модели центр тя-
тяжести заряда электронного газа должен находиться между
каждыми двумя катионами, т. е. на расстоянии г/2 от
каждого узла решетки. Исходя из этих условий, энергия
стяжения катионов электронным газом будет равна
AZ2e2N, a
^ 263
где А — константа Маделунга; NA — число Авогадро; а~
= 2AZ*eVNA.
Энергии притяжения в решетке металла противостоит
энергия отталкивания, которую определяют хаотическим
движением электронного газа (средняя скорость электро-
электронов в этом «газе» составляет около 100 км/с, что примерно
в 200 раз превышает среднюю скорость теплового движе-
движения молекул в воздухе). Таким образом, можно принять,
что энергия отталкивания ?отт будет равна кинетической
энергии электронного газа, которая, по данным кинети-
кинетической теории, может быть связана с межатомным расстоя-
расстоянием г следующим образом:
\, B6.4,
где у— постоянная, зависящая от типа кристаллической
решетки металла; h — постоянная Планка; т—масса элек-
электрона; Ь = ——s • Суммируя уравнения B6.3) и B6.4),
получаем в рассматриваемом приближении полную энер-
энергию кристаллической решетки металла:
?=_?+* ' B6.5).
м Т г Г2 \ i
Величина этой энергии в зависимости от изменения рас-
расстояния г будет проходить через минимум. Минимуму
энергии соответствует равновесное расстояние г0 между
ядрами атомов. В точке минимума кривой тангенс угла
наклона касательной равен нулю, или, что то же самое,
— = 0. Используя это условие и взяв производную по
межатомному расстоянию для уравнения B6.5), получим
4^ 4 (),
Го Го Го
341
откуда
=" r°-
Подставив выражение B6.6) в B6.5), получим
?
2г0 ¦
B6.6)
B6.7)
Из уравнения B6.6) после подстановки значений а и b из
выражений B6.3) и B6.4), соответственно, можно вычис-
вычислить радиусы ионов в решетке металлов:
/о ^ ь _ У '
2 2"
= 1,31
B6.8)
Подставив значение г0 из уравнения B6.8) в B6.7), по-
получим выражение для энергии кристаллической решетки
щелочных металлов
AZe*NA
B6.9)
В табл. 26.1 приведены результаты такого расчета.
Там же для сравнения даны экспериментальные значения
Таблица 26.1. Сравнение теоретических ?м
и экспериментальных ?м значений энергии кристаллической
решетки щелочных металлов, нДж
Металл
Li
Na
К
Rb
Cs
г0, им
0,308
0,380
0,470
0,496
0,534
749
607
490
464
431
Первый
нонизацн-
ониый по-
потенциал /,
519
494
418
402
372
ДЯсубл
металла
163
109
84
79
79
= Д«СУ6Л + '.
682
602
502
481
452
\ м к)
67
4
—13
—17
—21
той же энергии решетки, полученные совместным реше-
решением уравнений, характеризующих атомизацию (субли-
(сублимацию) металла АЯ°субл и ионизацию атомов 1г:
[М[ = (М) ДЯ°субл
(M) (M+)+l /
Отсюда энергия образования решетки из газообразных
катионов и электронов
342
Как видно из табл. 26.2, для всех металлов, кроме
лития, значения расчетных величин энергии кристалли-
кристаллической решетки близки к экспериментальным. Это свиде-
свидетельствует о достаточной строгости выбранной модели
ионной металлической решетки с электронным газом, ко-
который равномерно распределен по объему кристалла.
Однако для металлов других групп периодической системы,
кроме щелочных, расчет по формуле B6.9) приводит
к величинам ?м, значительно превышающим эксперимен-
экспериментальные (ЛЯсубл + ^У)- ^3 табл. 26.2 следует, что зна-
значения энергии кристаллической решетки для щелочно-зе-
мельных металлов, вычисленные по ионной модели намного
превосходят полученные экспериментально. Это означает,
что у щелочно-земельных металлов в отличие от щелочных
в ионном взаимодействии принимают участие не все ва-
валентные электроны. Чем выше положительный заряд ядра
и чем меньше электронных слоев имеет атом, тем больше
разница между фактической энергией кристаллической
решетки и вычисленной по ионной модели. Очень большие
расхождения E'm и ?м свидетельствуют о непригодности
ионной модели для этих элементов.
Интерметаллические соединения. Как правило, состав
интерметаллидов не подчиняется правилам формальной
валентности, но эти соединения образуют упорядоченные
структуры с отчетливо выраженной индивидуальностью
свойств. В некоторых случаях два металла могут образо-
образовывать не одно, а несколько соединений, например:
[Mg]-f [Zn]=[MgZn], ДЯ? = + 10,5 кДж/моль
[2Mg]+2[Zn] = (MgZn2], A#2 = +15,l кДж/моль
Zn] = [Mg2Znu], ДЯ^-j-lO кДж/моль
Это примеры эндотермических процессов образования интер-
интерметаллических соединений, которые приобретают устойчи-
устойчивость лишь за счет увеличения энтропии при переходе от
индивидуальных металлов к гетеросоединениям. Однако
чаще процессы образования интерметаллидов экзотермичны:
[%] + [Y] = [MgY], ДЯ2 = -1О,9 кДж
5[Mg] + 2[ Y] = [М&,„Y], AHl = -12,6 кДж
17[Mg]-f 3[Y] = [Mgi,Y8], A//!! =-10,5 кДж
Устойчивость интерметаллических соединений зависит
от степени различия природы составляющих их элементов.
В приведенных выше примерах интерметаллические со-
соединения магния и цинка эндотермичны потому, что эти
343
Таблица 26.2. Сравнение теоретических ?м '
и экспериментальных Ем значений энергии кристаллической
решетки щелочно-земельиых металлов, кДж
Металл
Be
Mg
Са
Sr
Ва
г°, нм
0,224
0,320
0,394
0,430
0,444
982
687
558
511
495
4109
2184
1732
1611
1523
ЛЯсубл
326
151
176
163
192
яМ=лясубл +
+ h + i,
2975
2335
1908
1774
1715
Д? =
= (^-?м)
1134
540
427
364
356
элементы находятся в одной группе периодической системы.
Напротив, соединения магния и иттрия экзотермичны, что
объясняют принадлежностью их к разным группам. С этих
позиций понятно, почему такие комбинации элементов, как
[Mg] + [Tl] = [MgTl], АЯ°298 = -50,2 кДж
[Mg] + [Sn] = [MgSn], ДЯ2°98 = -76,6 кДж
3[Mg]+2[Sb] = [Mg3Sb2], A//°298 =-232,2 кДж
протекают с большой экзотермичностью взаимодействия.
Интерметаллические соединения весьма многочисленны
и разнообразны по свойствам. В основе их классифика-
классификации лежат факторы объемные (наиболее выгодное соотно-
соотношение объемов атомов, образующих соединение), электрон-
электронные (комбинации элементов, обеспечивающие строго опре-
определенную концентрацию электронного газа в металле),
валентные (комбинации металлических атомов, которые
связываются валентными связями) и др.
Среди интерметаллидов обычно выделяют соединения
Лавеса, при образовании которых важное значение имеет
соотношение объемов или радиусов атомов. Наиболее
устойчивые комбинации получают при соотношении атом-
атомных объемов в пределах VA/FB=l/2. Такие комбинации
элементов приводят к образованию наиболее плотно упа-
упакованных структур интерметаллических соединений состава
АВ2. Примерами соединений (фаз) Лавеса могут служить:
MgCu2, MgNi2, MgZn2) TiTaCo,Cu4MnSn,CuMgAl и др.
Другой разновидностью интерметаллидов следует счи-
считать электронные соединения, смысл которых следует по-
пояснить. Считают, что в металлах, а также их сплавах
концентрация свободных электронов определяется числом
валентных электронов, приходящихся на каждый атом
344
металла. Определить эту концентрацию можно, разделив
полное число валентных электронов в кристалле на число
образующих его атомов. Предполагают, что в металличе-
металлических системах, для которых характерна одинаковая кон-
концентрация электронов, одинаковые силы связей должны
обеспечить образование одинаковых структур. Эти пред-
предположения часто оправдываются. Так, например, все ще-
щелочные металлы образуют однотипные кристаллы объемно
центрированных кубов и, как видно из табл. 26.1, энер-
энергия этих решеток мало изменяется в ряду Li — Cs. Такое
же положение имеет место и в металлических сплавах.
Так, например, Р-латунь содержит медь и цинк в отно-
отношении 1:1, и с каждой парой таких атомов в решетке
связаны два электрона цинка и один электрон меди, т. е.
электронная концентрация Р-латуни CuZn равна 3/2- Из-
Известно немало сплавов и интерметаллических соединений
с подобной электронной плотностью, большинство из ко-
которых обладает близкими свойствами и структурой (объем-
(объемно центрированный куб).
Достаточно распространены интерметаллические соеди-
соединения с электронными концентрациями 7/4 (GuZn3),
21/]3(Cu5Zn8), которые получили название электронных или
соединений Юм—Розери. Их образование объясняют зон-
зонной теорией, согласно которой каждой устойчивой ком-
комбинации элементов в данной системе соответствует неко-
некоторая предельная электронная емкость энергетических
уровней. Дальнейшее заселение электронами этих уров-
уровней становится невозможным при достижении определен-
определенной концентрации электронов. После достижения этого
предела возникает другой тип кристаллической решетки.
Третьей разновидностью интерметаллических соедине-
соединений следует считать валентные соединения, которые обра-
образуются при достаточном различии химической и физиче-
физической природы взаимодействующих атомов. Состав таких
соединений обычно определяют правилом валентности; их
свойства сходны со свойствами солеобразных веществ. Для
них характерны большие тепловые эффекты образования,
пониженная электрическую проводимость (часто они бывают
полупроводниками) и наличие узких областей гомогенно-
гомогенности. Примерами валентных интерметаллидов служат LaSb,
LiMgSb, Mg2Pb и т. д. Рассмотренные выше разновидно-
разновидности интерметаллических соединений не исчерпывают всего
многообразия свойств представителей этого класса кри-
кристаллических соединений, более детальное их изучение
требует специальных знаний.
345
Глава 27
Соединения с невалентными связями
Уравнение Ван-дер-Ваальса. Ориентационные,
индукционные и дисперсионные виды межмоле-
межмолекулярного взаимодействия. Уравнение Леннарда—
Джонса. Соединения включения. Цеолиты. Соеди-
Соединения с водородными связями.
Уравнение Ван-дер-Ваальса. В предыдущих главах
рассмотрены соединения атомов одного или разных эле-
элементов, связанные прочными ковалентными, ионными или
металлическими силами химической связи. Пространствен-
Пространственное распределение этих сил определяет расположение
атомов или ионов в соединении, создавая стройную кар-
картину структуры. Однако наряду с этими силами, опреде-
определяющими основу энергетики и структуры вещества, суще-
существуют иные, значительно более слабые межмолекулярные
или остаточные силы невалентного происхождения. Нали-
Наличие таких сил проявляется, в частности, в неподчинении
газов, находящихся под большим давлением, закону Мен-
Менделеева— Клапейрона, в способности всех газов конден-
конденсироваться при соответствующих температурах и давле-
давлениях, в так называемом эффекте Джоуля—Томсона
(изменение температуры газа, продавливаемого через по-
пористую перегородку).
Учитывая эти силы, голландский физик Я.Ван-дер-
Ваальс A873) предложил формулу
где г^—сила взаимодействия молекул газа (сила Ван-
дер-Ваальса), или внутреннее давление газа; Ь—собствен-
Ь—собственный объем молекул газа. В качестве примера в табл. 27.1
приведены значения коэффициентов а и b для некоторых
веществ при давлении 1013 кПа и температуре 273 К.
Вычисленные значения внутреннего давления газов, при-
приведенные в табл. 27.1 очень малы; они свидетельствуют о
незначительном отклонении этих газов от условий иде-
идеальности. Однако именно эти незначительные силы ответ-
ответственны за изменение температуры большинства газов при
продавливании через пористую перегородку, их широко
используют в работе холодильных машин.
346
Таблица 27.1. Константы Ван-дер-Ваальса
Газообразные
соединения
н2
Не
N2O4
NH3
НС1
со2
Cl2
а-1010, м'-Па
2,81
4,93
0,07
10,67
8,42
7,41
7,25
13,11
ft-101», м3
17,47
11,88
10,58
19,75
16,55
18,22
19,05
25,10
i^-10' (при
Робщ=1013гПаЬПа
55,7
98,3
142,0
2,1
1,7
1,5
1,4
2,6
Известно, что любые газы при достаточно низких тем-
температурах и высоких давлениях конденсируются в жид-
жидкости и кристаллы. В табл. 27.2 приведены температуры
кипения и плавления, а также теплоты испарения жид-
жидких и плавления твердых форм элементов восьмой группы
периодической системы. Из этой таблицы видно, что даже
атомы элементов, имеющих полностью заселенные s- и р-
орбитали внешнего уровня (благородные газы), взаимо-
взаимодействуют между собой, образуя конденсированные среды,
и что прочность конденсированных форм от Не к Rn
возрастает, поскольку в той же последовательности воз-
возрастают температуры плавления и кипения, а также теп-
теплоты испарения и плавления.
Таблица 27.2. Значения температур н теплот фазовых
переходов элементов восьмой группы (при 1013 гПа)
Элементы
Не
Ne
Аг
Кг
Хе
Rn
т°,с
кипения
—268,93
—245,9
—185,9
—153,2
—108,1
-63
плавления
—272,6
—248,6
—189,3
— 157,1
—111,8
—71
[АН°
испарения
0,08
1,80
6,53
9,04
12,6
16,4
кДж
плавления
0,021
0,33
1,17
1,63
2,30
2,89
Атомы всех остальных элементов, имеющих недостро-
недостроенные электронные орбитали, образуют прочные гомо- и
гетеросоединения с валентными или ионными связями.
347
Например, атомы хлора, соединяясь, образуют прочные
молекулы:
2(С1) = (С12), Д#2°93 = —243,9 кДж
В этих молекулах в результате возникновения валентных
связей атомы хлора имеют по 8 электронов на каждый
атом, т. е. полностью заселенные валентные электронные
слои. Тем не менее молекулы хлора при снижении тем-
температуры и далее экзотермически взаимодействуют между
собой, образуя жидкий, а затем твердый хлор:
(С12) = {С]2}, ДЯ° = — 20,5 кДж
{С12} = [С12], АН° = — 6,5 кДж
Сравнивая тепловые эффекты процессов, видим, что пер-
первый из них, характеризующий валентное взаимодействие,
более чем на порядок превышает остальные, определяю-
определяющие невалентное, ван-дер-ваальсово взаимодействие.
Виды межмолекулярного взаимодействия. По своей
природе силы Ван-дер-Ваальса можно разделить на три
типа:
а) взаимодействие полярных молекул (ориентационный
эффект);
б) возбуждение (индукция) полярными молекулами
неполярных молекул с последующим взаимодействием их
диполей {индукционный эффект);
в) взаимодействие неполярных молекул, возникающее
в результате дисперсионного эффекта.
Рассмотрим подробнее эти взаимодействия. В 1921 г.
П. Кизом создал электростатическую модель невалентного
взаимодействия молекул. Согласно этой модели энергия
электростатического стяжения молекул, находящихся на
расстоянии г и имеющих моменты диполей [% и р,2, равна
Ьк~ 3kTr<>' (L>
где Т—температура; k—постоянная Больцмана, равная
1,38-10~23 Дж/К. Соотношение B7.1) и есть количествен-
количественная характеристика ориентационного эффекта диполей, из
которого следует, что энергия ориентационного взаимо-
взаимодействия уменьшается с повышением температуры и уве-
увеличением расстояния между молекулами. Вспомним, что
энергия ионного взаимодействия обратно пропорциональна
первой степени межъядерного расстояния, энергия валент-
валентного взаимодействия—третьей степени, ван-дер-ваальсовы
348
же силы—шестой степени межмолекулярного расстоя-
расстояния и проявляются лишь на очень коротких расстояниях.
Энергия индукционного взаимодействия между поляр-
полярной и неполярной молекулами может быть вычислена
исходя из представлений П. Дебая A920), создавшего
модель индуцирования (наведения) диполя в неполярной
молекуле диполем полярной. Согласно этой модели энергию
индукционного взаимодействия ED определяют из соотно-
соотношения
Ч B7.2)
где а — поляризуемость неполярной молекулы; ц.—момент
диполя полярной молекулы. Из соотношения B7.2) видно,
что индукционная энергия также обратно пропорциональна
шестой степени межмолекулярного расстояния, но в от-
отличие от ориентационной не зависит от температуры. Сле-
Следует отметить, что индукционный эффект намного сла-
слабее ориентационного, так как предварительная поляризация
неполярной молекулы требует затраты энергии.
Исходя из представлений Кизома и Дебая оказалось
невозможным объяснить все эффекты ван-дер-ваальсова
взаимодействия. Так, например, полярные молекулы хло-
хлористого водорода НС1 (^, = 0,309-10~29 Кл-м) конденсиру-
конденсируются с незначительным выигрышем энергии:
(НС1) = [НС1], ДЯ°98= —18,16 кДж
в то время как неполярные молекулы хлора С12 (ц- =
конденсируются большим энергетическим эффектом:
(С12) = [С12], АЯ^98=— 26,8 кДж
Из приведенного примера следует, что существует тре-
третий способ межмолекулярного стяжения. Даже при абсо-
абсолютном нуле электроны в атомах находятся в состоянии
интенсивного движения относительно ядер и в различные
моменты времени происходят случайные смещения центров
тяжести положительного и отрицательного зарядов, т. е.
возникает короткоживущий момент диполя. Ориентация
в пространстве каждого мгновенного диполя случайна,
поэтому суммарный эффект такой самопроизвольной, часто
повторяющейся в течение длительного времени поляриза-
поляризации равен нулю. Однако электрическое поле в момент
существования диполя может либо ориентировать, либо
индуцировать диполь одного из соседних атомов. Такие
часто появляющиеся и исчезающие (осциллирующие) диполи
349
атомов или молекул взаимодействуют между собой, вызы-
вызывая стяжение частиц, как и в случае действия ориента-
ционных или индукционных сил. Энергия взаимодействия
осциллирующих диполей, названная дисперсионной (EL),
может быть вычислена из соотношения, предложенного
Лондоном:
3/iv0a2
Аге
B7.3)
где h — постоянная Планка; v0—частота осцилляции при
абсолютном нуле (с).
Следует отметить, что дисперсионная энергия может
составлять значительную долю от вклада других видов
ван-дер-ваальсова взаимодействия. В табл. 27.3 приведены
численные значения отдельных сил, составляющих меж-
межмолекулярное взаимодействие для различных веществ.
Таблица
27.3.
Энергия ван-дер-ваальсова взаимодействия в
молекулярных кристаллах веществ
Вещество
Не
Аг
Хе
СО
НС1
NH,
Н2О
ц-102»,
Кл-м
0
0
0
0,033
0,347
0,50
0,61
ftvo-10»,
кДж
3,92
2,47
1,84
2,29
2,19
2,56
2,89
Энергия, кДж/моль
Ек
0
0
0
ю-4
2,6
17,1
38,4
во
0
0
0
ю-3
0,77
2,1
2,0
El
0,
7,
15,
6,
14,
18,
9,
1
7
0
1
8
9
5
Так, для неполярных и малополярных атомов и молекул
(например, атомы благородных газов, молекулы СО) ос-
основной вклад в энергию ван-дер-ваальсова взаимодейст-
взаимодействия дает дисперсионный эффект. По мере увеличения
полярности молекул (НС1, NH3, H2O) становится все
большим вклад ориентационного эффекта. Удельный вклад
индукционного взаимодействия невелик даже у полярных
молекул. Способность атомов и молекул к поляризации
сказывается на величине энергии дисперсионного взаимо-
взаимодействия.
Из приведенных материалов следует, что энергия меж-
межмолекулярного притяжения обратно пропорциональна
шестой степени расстояния. Суммируя уравнения 27.1 —
27.3, получаем выражение для полной энергии ван-дер-
350
ваальсова стяжения частиц:
B7.4)
Однако при очень сильном сближении частиц силы стя-
стяжения уступают место отталкиванию, энергия которого
Еу в первом приближении обратно пропорциональна две-
двенадцатой степени межмолекулярного расстояния:
Е-у^. B7.5)
Полная энергия ван-дер-ваальсова взаимодействия Ev
равна сумме энергий стяжения B7.4) и отталкивания B7.5):
Ev = Ev + Ev=-^ + ^i. B7.6)
Если построить график зависимости Ev = f (r), то полу-
получится известная кривая потенциальной энергии с мини-
минимумом (см. рис. 25.4). Минимуму энергии Е° на этой
кривой отвечает равновесное расстояние /¦„. Касательная,
проведенная в точке минимума кривой, должна быть па-
параллельна оси абсцисс, т. е. тангенс угла наклона каса-
касательной равен нулю, или, что то же самое, -р. Отсюда,
взяв производную для выражения B7.6), получим
&п 12т _
' 13 ~
'о
ИЛИ
B7.7)
Величину ^/т/п в этом выражении, имеющую размерность
протяженности, интерпретируют как эффектный диаметр
взаимодействующих молекул а, в пределах которого при
столкновении энергия взаимодействия минимальна, тогДа
выражение {27Л) можно записать так:
Продолжая анализ уравнения B7.6), приведем его к виду
B7.8)
Выражение B7.8) получило название уравнения потенциаль-
потенциальной энергии межмолекулярного взаимодействия Леннар-
351
да—Джонса; оно наиболее применимо при расчете энер-
энергии взаимодействия неполярных или малополярных мо-
молекул. .
Силы межмолекулярного взаимодействия ненаправлен-
ненаправленные, поэтому при конденсации пара в молекулярные кри-
кристаллы образуются структуры, основанные на стремлении
молекул более экономно заполнить объем, создать условия
для более тесного сближения (вспомним, что ван-дер-
ваальсовы силы действуют лишь на очень коротких рас-
расстояниях). Поскольку ван-дер-ваальсово взаимодействие
очень слабо, твердые тела, образованные за счет этих
сил, обладают низкой механической прочностью, легко
плавятся, для них характерно высокое давление пара.
Например, кристаллический иод плавится при 113,7 °С,
давление пара над кристаллами—13 гПа уже при 73°С.
Из гетеросоединений аналогичный пример представляет
OsOj, молекулярные кристаллы которого плавятся при
39,5 °С (белая форма) и при 41 °С (желтая форма), а рас-
расплавленный OsO4 кипит при 129,5 °С.
Соединения включения. Ван-дер-ваальсово взаимо-
взаимодействие ответственно не только за процессы конденсации
однотипных атомов и молекул, но также и за химические
превращения. Типичным примером ван-дер-ваальсовых
гетеросоединений могут служить гидраты благородных
газов. Так, Р. Вийяр получил кристаллогидрат Аг-6Н2О
сжатием Аг до 150 атм над переохлажденной водой. Точно
также были получены Кг-6Н2О и Хе-6Н2О.
Позже Б. А. Никитиным были разработаны методы
соосаждения родственных кристаллосольватов:
Ne-6H2O, Rn-6H2O, Ar-3C6H5OH, Rn 3ClCeH4OH и др.
Согласно представлениям Ф. Крамера, образование
таких соединений происходит в результате включения
атомов благородных газов в крупноячеистые пустоты,
образующиеся при кристаллизации воды и ряда органи-
органических соединений, т. е. кристаллогидраты благородных
газов представляют собой типичные соединения включения
(или клатраты). В кристаллической элементарной ячейке
таких гидратов содержится 46 молекул воды и 8 атомов
благородного газа. Молекулы воды располагаются в вер-
вершинах пентагондодекаэдров, а атомы благородных газов —
внутри этих пространственных фигур. Таким образом,
теоретическая формула таких кристаллогидратов должна
быть R-*e/3 H2O (или R-5,75H2O). В случае криптона
такое соединение действительно существует. В некоторых
ячейках кристаллогидратов других благородных газов
352
остаются незаполненные места, что и приводит к соеди-
соединениям состава R-6H2O (R—атом благородного газа).
Помимо соединений, упомянутых выше, благородные газы
могут образовывать молекулярные соединения типа
Ar-/zBF3 (п— 1, 2, 3, 6, 8, 16) и др.
Клатраты являются наиболее типичными ван-дер-
ваальсовыми невалентными соединениями и достаточно
широко представлены в химии. По своей природе они
являются промежуточными между твердыми растворами
и истинными соединениями. Кристаллическая решетка
клатратов построена в виде полостей. Молекулы (или
атомы), попадающие в эти полости, оказываются запер-
запертыми в них и геометрически, и силами их ван-дер-вааль-
сового взаимодействия со стенками полости. Энергия та-
такого взаимодействия невелика, она обычно составляет
около 20—40 кДж. Однако этой энергии во многих слу-
случаях достаточно для стабилизации связей внутри основ-
основного каркаса, образующего полости. Соединения включе-
включения могут быть как определенного состава (с постоян-
постоянными соотношениями каркаса и включений), так и неоп-
неопределенного (типа твердых растворов). Очевидно, что
полости каркаса должны иметь размеры, сравнимые с
включенными молекулами. Характер полостей определяет
специфику соединений включения, которые можно клас-
классифицировать следующим образом:
а) решетчатые соединения, в которых полости возни-
возникают только при образовании кристаллической решетки;
б) молекулярные соединения включения образуются либо
за счет полостей отдельных молекул, либо за счет по-
полостей между цепями высокомолекулярных соединений.
В качестве примера можно привести ряд клатратных
соединений гидрохинона С6Н4 (ОНJ. При кристаллизации
этого соединения из водных и спиртовых растворов под
давлением различных газов (О2, NO, CH4, SO2, HC1, НВг,
Ar, Kr, Xe, H2S) или в присутствии некоторых веществ
(HCN, СН3ОН) образуются соединения включения, кото-
которые даже внешне отличаются от кристаллов чистого гид-
рохинс?-а. Эти соединения появляются в силу того, что
одна из модификаций С6Н4 (ОНJ, а именно |J-гидрохинон,
является кристаллической; в ней каждые три молекулы,
связываясь, образуют сферическую полость диаметром
около 4-Ю? м. Отверстия, соединяющие соседние по-
полости, имеют размеры значительно меньшие, так что мо-
молекулы, попадающие в полости в процессе кристаллизации
Р-гидрохинона, сравнимы по величине с диаметром этих
12 № 705 353
отверстий. Поскольку в образовании каждой полости
участвуют 3 молекулы гидрохинона, то соотношение меж-
между компонентами соединения включения должно быть
равным 3:1. Обычно часть полостей остается пустой, в
результате чего соотношение 3:1 выполнимо. Например,
для клатратных соединений с благородными газами извест-
известны соотношения:
С6Н4(ОНJ:Кг = 3:0,74,
С6Н4(ОНJ:Хе =3:0,88.
Молекулы большей протяженности, чем 4-100 м, не
могут образовать клатратных соединений с гидрохиноном,
именно поэтому молеку-
молекулы С2Н5ОИ не образу-
образуют клатрат с гидрохи-
ноном в отличие от
СН3ОН. Слишком малые
по размерам атомы и
молекулы, свободно про-
проходящие через отвер-
отверстия полостей, соедине-
соединений включения не обра-
образуют (например, атомы
гелия).
Лед, как известно,
в связи с особенно-
особенностью структуры моле-
молекул воды (вспомним те-
тетраэдр ическую симмет-
симметрию электронных орби-
талей атомов кислорода,
входящих в состав льда)
пронизан полостями в
виде шестигранных ка-
каналов (рис. 27.1). Это
определяет возможность
проникновения в ка-
каналы небольших по размерам атомов и молекул и
образования соединений включения. Когда вода затвер-
затвердевает в присутствии некоторых газов или легколетучих
веществ (например, СНС1„), образуется структура льда,
полости которого включают молекулы этих веществ.
Крупные атомы и молекулы искажают структуру льда,
делают ее рыхлой. Переход от структуры чистого льда к
рыхлой структуре клатрата требует затраты энергии. Если
354
• -1.О-2
Рис. 27.1. Схема кристаллической ре
шетки льда:
/ — водород; 2—кислород
этот эндотермический процесс перекрывается экзотерми-
экзотермическим эффектом образования ван-дер-ваальсовых связей,
то получившееся клатратное соединение будет устойчивым.
Среди клатратных соединений выделяют класс гидра-
гидратов газов, частными примерами которых являются рас-
рассмотренные ранее гидраты элементов нулевой группы.
Известны две наиболее распространенные структуры гид-
гидратов газов. В одной из них в элементарной ячейке клат-
ратного соединения содержится 46 молекул воды, которые
образуют 6 больших и 2 малые полости. Эта структура
устойчива, если полости заполнены такими молекулами,
как С12, СН3С1, SO2 и др., при атмосферном давлении
газов. При заполнении полостей могут образовываться
соединения, содержащие 5,76 Н2О, однако обычно наблю-
наблюдается лишь частичное использование полостей (например,
гидрат хлора С12-7,ЗН2О).
Элементарная ячейка второй структуры гидратов газов
состоит из 136 молекул воды, которые образуют 8 боль-
больших и 16 малых полостей. Примером гидратов такого
типа может служить соединение хлороформа СНС13 с во-
водой. Предполагают, что образование жидких гидратных
кристаллов хлороформа в тканях мозга определяет анесте-
анестезирующее действие этого соединения.
Цеолиты. Среди неорганических соединений, имеющих
ячеистую структуру и способных захватывать в свои по-
полости посторонние молекулы, большой интерес представ-
представляют кристаллические цеолиты, которые по своей природе
являются алюмосиликатами общего состава:
где п—заряд катиона или щелочно-земельного металла
Ми+.
Важнейшая особенность цеолитов—наличие в их струк-
структуре ячеистого каркаса, основа которого представляет
собой пространственное повторение группировки А1О4,
SiO4. В каждую такую ячейку, или полость, через имею-
имеющиеся з их стенках просветы могут проникать атомы или
молекулы различных веществ и удерживаться там силами
Ван-дер-Ваальса. Частицы, размеры которых больше ве-
величины просветов, не могут проникать внутрь полости,
что определяет - избирательность захвата, или сорбции,
цеолитами молекул того или иного состава. Благодаря
этому они широко используются в качестве избиратель-
избирательных, или селективных, абсорбентов при очистке от вред-
12* 355
ных примесей газов и жидкостей, при разделении смесей
веществ, близких по свойствам, и т. д.
В качестве примера приведем формулу одного из наи-
наиболее распространенных цеолитов—кристаллического алю-
алюмосиликата натрия 12(Na/llSiO4)-27H2O, который при
нагревании до 350°С в вакууме превращается в безвод-
безводную форму. В этом веществе тетраэдры АЮ4 и SiO4,
соединяясь между собой, образуют кольца, состоящие из
8 атомов кислорода на каждой стороне кубической ячей-
ячейки, а у каждого угла—аналогичные кольца из 6 атомов
кислорода. Образуемая сочленениями тетраэдров каждая
большая полость имеет протяженность 1,14 нм и связана
с шестью аналогичными ячейками и восемью ячейками,
имеющими размеры 0,66 нм. Отверстия в больших по-
полостях имеют размеры 0,42 нм, а в малых—0,20 нм.
Кристаллогидратная вода располагается в этих полостях,
после ее удаления остаются пустоты, которые могут слу-
служить для включения молекул других веществ. Данный
цеолит избирательно адсорбирует молекулы углеводоро-
углеводородов с неразветвленными цепями, очень слабо удерживает
малые молекулы, но не в состоянии адсорбировать моле-
молекулы бензола.
Соединения с водородными связями. Среди сил нева-
невалентного взаимодействия большое значение имеет водород-
водородная связь, образованная взаимодействием положительно
поляризованного атома водорода и отрицательно поляри-
поляризованными атомами (чаще кислородом), которые могут
входить в состав разных или одинаковых молекул. В мо-
молекулярных соединениях с водородной связью атомы
водорода имеют координационное число, равное двум.
Примерами соединений с водородными связями служат
вода и лед. Количественной мерой прочности водородной
связи в кристаллах льда можно считать энергию субли-
сублимации:
[Н2О] = (Н2О), ДНда = 50,0 кДж
Как уже отмечалось, атом кислорода в молекуле воды
имеет четыре гибридные орбитали (я/т'-гибридизация), из
них две связывающие, несущие избыточный положитель-
положительный заряд, и две несвязывающие, несущие избыточный
отрицательный заряд. Как результат этого молекулы воды
имеют большой момент диполя @,613- 10~ав Кл-м).
При конденсации молекул в кристалле льда у каждой
молекулы возникают четыре водородные связи О—Н ...
к
356
... О—Н, как это видно из кристаллической решетки
Н
льда, изображенной на рис. 27.2. Из схемы следует, что
все углы между направлениями связей в кристалле льда
тетраэдрические, длина связей и их прочности — разные.
Так, длина основной ковалентной связи О—Н равна
0,100 нм, а дополни-
дополнительной (межмолеку-
(межмолекулярной, водородной)
почти в два раза
больше — 0,176 нм.
Энергия основной свя-
связи равна 464 кДж,
1,76-1(Г10м
1,<ХН<Г10м
»-1, О-2
Рис. 27.2. Схема водородных связей моле-
молекул воды в кристаллической решетке
льда:
/ — водород; 2—кислород
а водородной 21 кДж.
Таким образом, рас-
расстояние между ато-
атомами кислорода в
кристаллах льда рав-
равно 0,276 нм. При
плавлении это рас-
расстояние увеличивает-
увеличивается до 0,290 нм. В
кристаллах льда ко-
координационное чи-
число молекул воды,
равно четырем, в
жидкой воде при
1,5°С увеличивается
до 4,4 при 83 °С—до
4,9. Причина этого заключается в том, что при плавле-
плавлении структура льда (рис. 27.2) частично разрушается,
появляются «осколочные» молекулы воды, которые запол-
заполняют «пустоты» в структуре. С повышением температуры
возрастают число «осколков» и степень заполнения ими
«пустот» (рост КЧ). Однако одновременно растет и объем
«пустот», поэтому плотность воды уменьшается.
Гипотезы о природе и строении воды составляют обширный
раздел истории химии. Еще в 1882 г. В. Рентген высказал мысль о том,
что вода представляет собой взаимопроникающее образование из льдо-
образной молекулярной структуры и свободных молекул, образующих
«истинную жидкость». В 1933 г. английские исследователи Д. Бер-
нал и Р. Фаулер предложили структуры для двух форм воды.
Русский ученый О. Я. Самойлов A946) предположил, что жид-
жидкая вода—это молекулы свободной воды, связанные молекулярными
силами в «пустотах* льдообразиого каркаса (сходство в клатратной
357
моделью Л. Полннга). Именно поэтому плотность льда меньше плот-
плотности жидкой воды и лед не тонет в воде (это свойство отличает
воду от подавляющего большинства веществ); прн температуре 0°С
лед имеет плотность 0,9168 г/см3, а вода —0,99987 г/см3. Из-за «ажур-
«ажурности» структуры льда возможны и другие, более плотные упаковки
молекул воды в его кристаллической решетке, которые осуществля-
осуществляются прн высоких давлениях и имеют плотность 1,12; 1,03; 1,09;
1,13; 1,5 кг/м3. Седьмая разновидность льда получается при давле-
давлении 3,2-10е Па и имеет температуру плавления -{-192°С.
Возникновение водородной связи имеет место также
и в случае аммиака. Известно, что теплота испарения
жидкого аммиака, равная 23,3 кДж/моль, по существу и
есть энергия разрыва водородных связей, поскольку
каждая молекула аммиака имеет одну неиспользованную
МО. Другим характерным примером соединений с водо-
водородными связями могут служить молекулы фтористого
водорода, которые при конденсации и полимеризации свя-
связываются прочными водородными связями
) = (H2F2), ДН°2в8 = -26 кДж
6(HF) = (H6Fe), ДН°298 =—167 кДж
а в водном растворе образуют устойчивые ионы HFj
aq, ДН°2в8 = -9,6 кДж
Средняя энергия водородной связи HF в газовой фазе
около 33,5 кДж/моль, максимальная — 56,5 кж/Дмоль.
Фтористый водород в отличие от других галогеноводоро-
дов легко конденсируется в жидкость (температура ки-
кипения 19,6°С), что также связано с наличием водород-
водородной связи. В жидком HF энергия водородной связи равна
29 кДж/моль.
В органической химии примером соединений с водо-
водородными связями могут служить карбоновые кислоты
R—СООН. Как в газовой, так и в жидкой фазах они
существуют, как правило, в виде димеров, образованных
за счет водородных связей:
,О ... Н-СХ
ХО—Н... о"
Если энергия ковалентной связи в карболовых кислотах
лежит в пределах 15—30 кДж, то энергия водородной
связи в 15—20 раз слабее.
358
Заканчивая изложение материала о соединениях с не-
невалентными связями, следует отметить, что слабые взаи-
взаимодействия атомов и молекул в отличие от валентных
имеют место в любых химических соединениях между
любыми частицами, расположенными достаточно близко
друг к другу. Это обстоятельство заставляет учитывать
силы Ван-дер-Ваальса во всех случаях, когда необходим
достаточно строгий расчет энергии химической связи в
молекулах, жидкостях и кристаллических структурах.
Приложения
I. Греческий алфавит
Написание
букв
Аа
вр
г?
Дб
Ее
Ч
Нт,
в$9
Ii
Кч
А\
Мц
|
Название
букв
альфа
бета
гамма
дельта
эпсилон
дзета
эта
тэта
йота
кйппа
ламбда
ми (мю)
Написание
букв
Nv
sg
Оо
Пд
Рр
2ag
Тт
Го
Фф
Хх
ЧГЦ
Qa
Название
букв
ни (ню)
кси
омикрон
пи
ро
сигма
тау
ипсилон
фи
хи
пси
оме га
II. Основные физические постоянные
Постоянная
Скорость света в ваку-
вакууме
Элементарный заряд
Постоянная Авогадро
(по углеродной шкале)
Символ
С
е
Na
Значение
2,997925
1,60210
6,02252
Единицы СИ
ХЮ" м-с-*
10-» Кл
102S моль-*
360
г
Продолжение
Постоянная
Масса покоя:
электрона
протона
нейтрона
Постоянная Фарадея
Постоянная Плаика
Универсальная газовая
постоянная
Постоянная Больцмана
Символ
тв
nip
тп
F
h
R
k
Значение
9,1091
1,67252
1,67474
9,64870
6,6256
8,3143
1,38054
Единицы СИ
10"sl кг
Ю-27 кг
10~27 кг
104 Кл-моль
Ю-34 Дж-с
10-83 Дж-К~*
III. Элементарные частицы
:ЙСТВО
г
о
Лептоны
Нукло-
Нуклоны
Мезоны
Гиперо-
Гипероны
§
?
о
е~
е+
V
V
Р
п
ц±
я»
К+
К°
л°
S+
s-
S
ё
к
«
S
со
X
Электрой
Позитрон
Фотон
Нейтрино
Протон
Нейтрон
Мю-мезон
Пи-мезон
Пи-мезон
К-мезон
К-мезон
Ламбда-гнпе-
рон
Сигма-гиперон
Сигма-гиперои
Кси-гиперон
о
о>
§
1
1
0
0
1836
1839
206,8
273,2
264,4
966,6
974,4
2181
2328
2343
2584
д-1,6-10-
к
— 1
+1
0
0
+1
0
±1
±1
0
+1
0
0
+1
—1
я
к
с
о
Vi
ll%
1
V.
Vi
Vi
Vi
о2
0
0
0
*/•
Чг
ч1
I ОСТЬ
л
5
|s"
О я
as
С *
Стабильный
»
»
1,1-Ю3
2,2-10-в
2,5-10-s
~105
~ю-8
^Ю-10 (-7)
2,8-10-1°
~5-10"и
~10~10
361
IV. Термодинамические величины для простых веществ, соединений
н ионов в водных растворах и в жидком аммиаке
Формула для вычисления теплоемкости
Вещество
Ag (кр)
А] (кр)
As (кр)
Аи (кр)
В ,кр)
Ва-а
Ва-р
Be (кр)
Bi (кр)
Вг2 (ж)
Вг2 (г)
С (алмаз)
С (графит)
Са-а
Cd-a
Cl (г)
CU (г)
Со-а
¦, 298*
кДж/моль
0
0
0
0
0
0
—
0
0
0
30,92
1,897
0
0
0
121,3
0
0
298'
ДжАмоль.К.)
Теплоемкость, Дж/(моль-К
Коэффициенты уравнения
0
Простые вещества
42,69
28,31
35,1
47,45
5,87
F4,9)
9,54
56,9
152,3
245,35
2,38
5,74
41,62
51,76
165,09
223,0
30,04
23,97
20,67
21,9
23,68
6,44
22,26
10,45
19,0
18,79
37,20
9,12
17,15
22,2
22,22
23,14
36,69
21,38
р— V
6 • 103
5,28
12,39
9,29
5,19
18,4
13,8
i 29,3
8,87
22,59
0,71
13,22
4,27
13,9
12,30
—0,67
1,05
14,31
¦¦\
/
<?'-10~s
—0,25
—
—3,43
—1,19
—6,19
—8,79
0
—2,52
—0,88
Яр, 288
25,48
24,34
24,64
25,23
11,96
26,36
16,44
25,52
75,71
36,0
6,07
8,53
26,28
25,90
21,84
33,84
24,6
Температур-
Температурный интервал
К
273—1234
298—933
298—1100
298—1336
273—1200
298—643
643—983
298—1173
298—544
298
298—1500
298—1200
298—2300
273—713
273—594
298—200
273—1500
298—650
PJf
00 О CO О О О СМ
-Г^-Г . <>* СМ >> ,-л т-
00 O«00W
№ О NO5
00 00О1
О) СП О
см см ю
о
O
СО
1
0
88^888
. -_ , -- -.-^,--00—«C000CNCO
I I II I I М \Щ М I I I I
ОООСООООООООООООО^ООООСОООСОСО
cnmt^cnt^i^
см см см см см см
*
см со см см
—3,68
OOJC
СМ СО С
СЛ — U
см со с
ою
rf СО
а
оо^.
^ СО 00
1 1
* *
>ет> со о см сг> ¦>
о ее en t^o ¦«
LO CO
1 °"<N" 1 1
J-СП t^C
f CO CDC
If 1
D-* CMLOOCMLOC
CMOi
¦* LO
1 iff 1111
3CDO CM CO
" О) СЛ CO О
-1 CM CO CM CM
^•*
t^-o
f f 11
CM
Cft
CO
CD
1
CM
CO
CO
1
[^- CO Отр tOO CJJCT) CO^f^^t^-COCO CJJCO (NlOO
CO СЧ COoOO CO CM LO 1^. LO CO CJ> CO — •* CM ¦* ^ COO CO— h-
O) I CD I ¦* — — I «O I « ¦*" I loTo— iLOO^LO^CMCjrlcOcolcD — CO
CM — ^f CM CO — — CM CM —
CO Tf OCJILO COCO CMOCO CD lOCOI^-CMCJJ COCO COCOCO
¦4-,CO,"*cOCN .CO , ИСЛ . . — ¦* CM .t^cOCOOCOOiOi ,-*O .ООСОСЛ
¦* ' см ' t^-"-*oT ' со ' t^Tto" ' ' о t~-"o" ' см" см* со" см t^ о cd ' — t^-" ' cjTlo'co
CM CM CMCO —' CM CMCM rt^COCM —iCMCMCMCMCM —¦ CO-* —iCOCM
CDlQOtP lOQ?00 COCO ^-^LO COLOCOCO CMCOLOCO LO , ч
s«niNoiai-.5racDtDr—icnt^-LO — иоюмлю^иоюоога So — en
сО'Фсосо^см^ —^CMr^ococo^cDOco^oocM —'со —¦ — слоюад^сч'со^
CMCOCOCM^OCM-*-* — n»NN-i COLOCDCMCOCOCMOiLOCMCOOCO^CM — CD
— — CM — — — _ — CM — — — CM CM -i-CM
я
^\O Sd -
363
Продолжение прил. IV
Вещество
Pt (кр)
Rb (кр)
S (монокл.)
S (ромб.)
S2 (г)
Sb (кр)
Se (кр)
Si (кр)
Sn (бел.)
Sr (кр)
Те (кр)
Th (кр)
Ti-a
Tl-a
U (кр)
W (кр)
Zn (кр)
Zr-a
4///, 298>
кДж/моль
0
0
0,30
0
A29,1)
0
0
0
0
0
0
0
0
0
0
0
0
0
S298'
Дж/(моль-К)
41,8
G6,2)
32,55
31,88
227,7
D5,69)
42,44
18,72
51,4
54,4
49,71
53,39
30,66
64,22
50,33
32,76
41,59
38,9
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
С°=ф(Г)
а
24,02
14,90
14,98
36,11
23,1
18,95
24,02
18,49
23,43
23,8
21,67
22,09
22,01
14,18
24,02
22,38
28,58
ft- 10s
5,61
29,08
26,11
1,09
7,28
23,01
2,58
26,36
5,73
6,28
19,0
10,04
14,48
33,56
3,18
10,04
4,69
с'-10-'
-3,52
—4,23
2,93
—3,81
°Р, 298
26,57
30,42
23,64
22,60
32,47
25,43
25,36
19,8
26,36
25,1
25,6
27,33
25,0
26,40
27,5
24,8
25,48
25,15
Температур-
Температурный интервал,
К
298—1900
273—312
368—392
273—368,6
273—2000
298—903
273—493
273—1174
273—505
298—508
273—620
298—1500
298—1155
273—505
298—935
273—2000
273—693
298—1135
AgBr (кр)
AgCl (кр)
Agl-a
Неорганические соединения
—99
— 126
(-64
,16
,8
.2)
107
96
114
,1
,07
,2
33,
62,
24,
18
26
35
64
4
100
,43
,18
,8
-11,30
52,38
50,78
54,43
298—691
273—725
273—423
w
COOOONOWNOO
ооо^сл^юс^оо^
О О О 00 О О Ю
_00 OQ WtC^ONOON
^Ю^Ю^Э^^--<^С^иОпГ1--'<—«Ь--*^00-*СО—<^r«-_
¦ :: i i i i i II i 111 м ii i м i 111
ООСОООООСОООООООООСО^ООООООСОСОООООООСООО^ОО
~" — — ¦ — — — — . COCOCOt^t^COCOCOt^l-"- СО
О) <М О) О) (М СЧ СЧ (М О) СЧ СЧ
тс
I
СО
t
i. .
Ю L
)оЬо1>о!>^оо
8сч
1_О СО —^ О LQ
СО LO LO —* (М СЛ LO СЛ СЛ LO LO ("
to coco 01
1 II 1
l^cS^I 1
'7
СО 00 LO СОСЛ О
— -rf Ю О —' ОО 00 СО О СО
- «^ -^ см
LO0000 — "^ СО СО СМ
СОг^СОГ-^^ftNLOCO —О
<SlO^SSloK«Sco^
^Л Г4^" СО СЛ ^Т) 00 ^^
0СОЮСЛ СО
¦^ <м о <м ю
« СО LO СО 00
?J 00 ^J* СЛ
IS
СО
1
СО
LO ¦* СТ>
00О
t^. СО
CD 00 1
7 '
h» СО Г^- LO
СЛОСООСЛГ^СОСО
¦^ LO СО О
¦ LO О LO СЛ
о см со со
ю
-^—«« — — COlOCOCMOOOOlOlO —
^—' см со — — cm oj —
со
со
ю
125
00 •*
t^ to
OJ СЧ
S2
1 1
1 CO
0 со
h» со г- "Ф со
ю со о
h» со 00
^°°
38
oo2c
«-с
1 1
103
2 <M
151
LO CO h» CO Lf
gcoS^tS
CD CO COO
*ф LO О СЧ
000 a
1 1
100
1
rt* C7J Oi 00
— -* to см
00 -* t^. 00
(M ^t* CD ^t4
¦*CO C4LO С
COCO -4-
LO CO CT)
1^ 00 LO
Oi 00 СЧ
CO OO СП
to — —
— CM
О COLO
LO COO
3 00 cj 01
1 CO t^- O5
LO oT—« C
1777 11
77
0СЛЮСЛ^Ю ЮчСОСМ—¦
1 1 1 1717 Mill
* <?
я
JO
" О- -Г-
365
Продолжение прил. IV
Вещество
\Н
I, 298'
кДж/моль
Дж/(моль>К)
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
Ь-103
с'- Ю-5
Температур-
Температурный интервал,
К
СаС12 (кр)
CaF2-a
Ca(NO3J (кр)
СаО (кр)
Са(ОНJ (кр)
СаНРО4 (кр)
СаНРО4-2Н2О (кр)
Са(Н2РО4J (кр)
Са(Н2РО4J-Н2О (кр)
Са3(РО4J-а
CaS (кр)
СаБОз (ангидрит)
CdCl2 (кр)
CdO (кр)
CdS (кр)
CdSO4 (кр)
С12О (г)
С1О2 (г)
СоС12 (кр)
CoSO4 (кр)
СгС13 (кр)
СгО3 (кр)
Сг2О3 (кр)
(—785,8)
—1214
—936,9
—635,1
-986,2
—1820
—2409
—3114,5
—3418
—4125
—478,3
—1424
—389,0
—256,1
— 144,3
—925,9
75,7
104,6
-325,4
—867,9
—554,8
—594,5
—1141
113,8
68,87
193,2
39,7
(83,4)
88
167
189,5
B59,8)
240,9
56,5
106,7
115,3
54,8
71,0
A23,1)
266
251,3
106,6
113,3
122,9
72
81,1
71,88
59,83
122,9
49,63
105,2
201,8
42,68
70,21
61,25
40,38
54,0
77,32
53,18
48,28
60,29
125,9
81,34
119,4
12,72
30,46
154
4,52
12,0
166
15,90
98,74
40,17
8,70
3,8
77,40
3,35
7,53
61,09
41,51
29,41
9,20
—2,5
1,96
—17,28
—6,95
— 19,0
—20,92
—7,78
-7,74
— 15,65
72,61
67,03
149,4
42,80
87,5
97,1*
259,2
231,6
47,40
99,66
73,22
43,43
55,2
9Э,60
45,6
41,8
78,6
138
91,8
104,6
298—1055
298—1000
298—800
298—1800
298—600
293
298
298—1373
273—1000
299—1400
273—841
273—1200
273—1273
298—1273
298—2000
298—1500
298—1000
298—700
286—319
350—1800
ш
00 Tf "
—< оэ с
СЛ 00 Ч
0000 с
СЛО> Г
см ^ч с
о
Э СО 1О
П ?-СМ
1 М
0 СО 00
Ч СМСМ
СО
т
см
з
2
о
О СО
см г^
— СО
oioo
O5CJ5
СМ СМ
00 *^ СО °Ч 00 00 00 00 (
CNO1(>)CSCM CN (MCN
ii'T'TgKi
3 00 00 00 ^ ?J 00
СО t~- 00 CM
CD 00 , — Ю - —
Tf t~-CN COCNO О Tf ¦* СМЮС? CO
ИИЧ'К*ЮЮС»ОО
I I I I I I II I II
5
I
SS8
I —"cp-T
ct> — t o"o o"—
CJ) CO СЧ Ol lO
— COO
-см2сл§
1 1 CM CD~CM00O*
CD О Tf CO —
. (DOONON
Tf Ю O5CD CM
52
1 ю
1 °°
О 00 CO CD
t О Ю — "* |
CO —
, 00 — ТОЮ
Tj- CM CO CM
JO Tf—.
Ol ОЭ t^»
1 NinO
О CNO
' s<oo
CM CM CO
o*t-
OlO
^^ CO
o"co
1 ю
4 nfOiDeg
00 «3 tDlO COCX>I>)-*COOO
—.СОЮ CO t^-OT
—'C0CNtOC0COCTH0CMCS100CT)—i
СЛ —t Tf CD —¦ СУ» —'CONCfJlOCOlOt
OODNOcOtOOO
—f(N —1—-.CM — СЧ-¦ —¦—¦
CD —О — 00 OOCN —. О I>-O op ¦* 00 Tf О О — ¦* —. ¦* 1Л (N
COOCNCOCOCOCOt~--"*lO-"*CO t-~ 0>_lO^CO OO5CDO5000000O
CDOOTfCyTfCD
—t I CN CM b- CNI
I ' I I I I
O
TfCO^ CN I N I CN CM b
I I I I I I ' I I ' I I I
DcN ONSS
NI 00 —' I Ot» '—¦ О
I ' I 17
ODlfoOaJOO
— —>CM CM CM CM —¦
ill Mil
367
Продолокение прил. IV
Вещество
ЛЯ/, 29 8-
кД ж/моль
298'
Дж/(моль-К)
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
с' ¦ 10 ~ •
Ср, 298
Температур-
Температурный интервал,
К
H.S (Г)
HaSO4 (Ж)
Н,РО4 (ж)
Н8РО4 (кр)
HgBra (кр)
Hg2Bra (кр)
HgCla (кр)
Hg2Cla (кр)
HgI2-a
HgO (краен.)
HgS (краен.)
Hg*SO4 (кр)
1пг03 (кр)
In2(SO4)a (кр)
KA1(SO4J (кр)
КВг (кр)
КС1 (кр)
КСЮз (кр)
KI (кр)
КМпО4 (кр)
KNO8-a
КОН (кр)
КгСгО4 (кр)
КС0 (кр)
—20,15
—811,30
(—1271,94)
— 1283,65
—169,45
—206,77
—230,12
—264,85
— 105,44
—90,37
—58,16
—742,0
—926,76
—2907,88
—2465
—392,04
—435,85
—391,20
—327,61
—813,37
—492,71
—425,93
— 1383
—2033
205,64
156,90
200,83
176,15
162,76
212,97
144,35
195,81
176,36
73,22
81,59
200,83
112,97
280,75
204,50
96,65
82,68
142,97
104,35
171,71
132,93
59,41
200,0
291,21
29,37
64,02
92,47
72,84
45,61
234,10
48,37
41,38
50,63
60,88
179,08
15,40
43,10
30,96
16,74
15,27
82,34
13,89
21,76
8,16
118,83
171,54
-58,41
3,22
33,93
137,57
106,10
76,60
101,67
77,82
45,73*
50,21
131,80*
93,72*
280,33*
193,00
53,62
51,49
100,25*
55,06
119,25*
96,27
146,0
219,70
298—1800
298
298
273—553
273—798
273—403
278—371
278—853
273—307
273—373
298—373
298—1000
298—543
298—1000
289—371
273—955
287—318
273—401
298
298—671
69S soz w ?1
Z Z Z Z Z Z Z Z-3 2 Z Z Z 2 2 2 2 2 2 2 2 2 2 2 S Г Г Г Г Г Г *
> пРР °» » Ж«Г» » сГ ел» « о О П П SC?=i Onnc^ZOf2nnon
^je, 4- /^-2w>D ОО» —О Ои sOOOs q?^ О
¦а
2- w w
¦а-а
13
I JLJa I | , I i| I I II col I I LL
CO — CO CO I I N>C0<L0CnC04*.00O<0OCTiO4>->
' ~) Oo Oi —¦ Г --- ..-.
СОСЛ— O0C0COCTi^-J4^*— 0L^000 01-'OoCTi^OOOtOO^COCoOoOoO—'NOJ
О Сл Оо CO СП 00 СО *~- СО СО 00 *~" О Сл Оо О} ^О CTi СО CTi to Оо Ьо Ю СО 00 **J 00 CTi >?*
СО— S Сл^О*^ О О CO"^4COtOOo—'СлСО — Gi Gi 4^ СО *— 00 COO Оо *^J CO 4^
Со to ю to — to ю *— *— —¦ —
--3 О I ОЮ^^-СЛЮ1-^ | СО --J 4^ — СП Gi ^-*
OCOJ ^00000054^ | tOOoQoOCOO""^
СО
> Сл О Ю GO Oi n
j to сос~
2 >?». Gi С! СО Ю Сл
сл to о о о о
. оо —со со -ч
- —' О -J Сл СО
Ю
СО 00
со Оо оо со I >—
I.-1 «I
- СО >— |— СО
СПОООСООО
— Оо
en х>
СП-Ч СП с
4^ СП to О N3 >—" Ю ^О
оосо-чеп — toto~
СПСО — tO
О*>* I I to
•* " II4"
to оооо оо
4^ СП
.И I I
4^ 00 СП О
tD Сл -*4 Сл
СО 4* to
ill I L
иашоно
I 11
СП
со to |
СО СО
<ХЭ-*4'— ООС004^>—-Ч0>*ХЭ-Ч-ЧО0000Сп~СС>4^ОСп •—СЛО4* *ХЭ
Ю —~ ¦— OJOSOOOiOldOOWWOiO Ю •— Coto tooo о О
111 11
i
Продолжение прил. IV
Вещество
AHf, 298'
кДж/моль
-298'
Дж/(моль-К)
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
Ь-10'
"р, 298
Температур-
Температурный интервал.
К
NaBr (кр)
NaC2H3Oa (кр)
NaCl (кр)
NaF (кр)
NaHCO3 (кр)
Nal (кр)
NaNO3-a
NaOH-a
NaOH (ж)
Na,B4O7 (кр)
Na2CO3-a
NajCOg-lOHjO
Na2CO3 (ж)
Na2HPO4.12H2O
NaaO (кр)
Na2O2 (кр)
NaaS (кр)
Na2SOa (кр)
NaaSO4-a
Na2SO4-P
Na2SO4 (ж)
Na2S2O3-a
NaaSiFe (кр)
359,8
—710,4
—410,9
—570,3
—947,4
—287,9
—466,5
—426,6
i = 6,36
—3290
— 1129
—4077
—52297~
—430,6
—510,9
—389,1
—1090
—1384
ш7 = 24,06
1117
2849,7
83,7
123,1
72,36
51
102
91
116
64,18
189,5
136,0
2172
71,7
93,3
94,1
146,0
149,4
214,64
49,66
45,94
43,51
52,30
25,69
7,34
89,58
70,63
65,69
69,87
82,88
65,0
121,6
8,79
16,32
16,23
6,78
225,94
125,0
—5,86
135,6
22,59
65,26
68,61
220,9
80,92
—1,38
13,38
52,3
80,33
50,79
46,82
87,72
54,31
93,05
59,66
186,8
110,0
536
188,3*
557,0
72,43
89,33
103,22
120,1
130,8
197,4*
146,0
298—550
298
298—1073
298—1265
298
298—936
298—550
298—566
595—1000
298
298—723
298
> U27
298
298—1100
298—865
298—1000
298
298—518
518—1157
1157—1850
298
00 OOO 0000
01 (Dm *O1
csi со <N — сч
-Mil
о о о
— С0ООЮОО OO
СОтСООоООО О —<
<NO Ю со ;.
—t —> —¦ CD CD 00 b- to —• —i.
М
УП T I 1ST I I I
iopopopopooopcoopopgpop^oo.
см с
оо ех> оо оо со со
ет> ™< ст> СТ> t*« f*~
CN CM CN CN CM CM
00 —• CD QO О 00
3 Ю Tf 00NOO3 <
5С000100ЮЮС . OO COO <
io о
_» _ —, cj <M 00
U0 СО f~ — О 00
I I
t— * CO Ю
00 00 Tf CD t^ (N
a> —" tC o" to" —Г
ntosmoo
I I I
2 I ITS I I I
5?
со" I
eg
Ma f
CO — CO <N — <J1 O> CO O> CO CM I I Ol
lOf Ю —tOO-^CNCO OJ
LO О CO LO
Ю Ю 00 00 Ю
(N I <J1 CO CO —•
— b- og — t^
CM
00 f^ C71 ^- LO
oooloo"ct>oi-~ —
COl^- COO
Ю Ю ^T f-
2 I S
OO
CO
CM
— tNO—OOCDN
^-OoOOCOCOh^CO
ГО CO CN ~ — — ~
CD —• — NOO5C400SOO0
t^- —'d^COO^^~ —iLOGOCsl
CM —• — CM — CM CM COCM —i —
LO
LO
coco ~ i
00
LO .
COc-4-
I a:
<3
COcOtOiM^O
— — tM S О tX3
O CSlHC4CO
QOLO
COCO
^11411711111114117111111
Продолжение прил. IV
Вещество
SbaO6 (кр)
Sb3Ss (черн.)
SiCl4 (ж)
SiF4 (г)
SiOa (кварц-а)
SiO2 (кварц-Р)
SiO2 (тридимит-а)
SiO2 (тридимит-р)
SiOa (кристобалит-а)
SiO2 (кристобалит-р1)
SnCl3 (кр)
SnCl4 (ж)
SnO (кр)
SnO2 (кр)
SnS (кр)
SrSO* (кр)
ТеС14 (кр)
ТеОг (кр)
Th(OHL (кр)
ТЮЯ (кр)
TiCl4 (ж)
TiCl4 (г)
TiOs (рутил)
TiO2 (анатаз)
&"f, 298'
кДж/моль
—880
—160
—671,4
—1548
—859,3
—856,9
—857,7
—
—349,6
—544,9
—286
—580,8
—101,8
—1444
—323,0
—325,5
—1763,6
—1231
—800
—759,8
—943,9
•*298'
ДжДмоль'К)
125,1
166,6
239,7
281,6
42,09
43,93
43,26
—
136,0
258,5
56,74
52,34
77,0
121,7
217,5
73,7
133,9
65,24
249
352
50,23
49,90
Геплоемкость, ДжДмоль'К
Коэффициенты уравнения
а
_
101,3
91,46
46,94
60,29
13,68
57,07
17,91
60,25
67,78
—
39,96
73,89
35,69
91,2
—
57,95
—
66,27
106,5
71,71
72,01
р-ф
«МО»
55,20
13,26
34,31
8,12
103,8
11,05
88,12
8,24
38,74
14,64
10,04
31,30
55,65
28,74
12,05
—
1,0
4,1
4,52
г'.10-»
—19,66
—11,3
-21,59
3,77
—6,69
—9,87
—14,64
—15,02
)
в'р, 298
117,7
117,75
145,3*
73,37
44,48
44,68
44,18
—
79,4
165,2*
44,31
52,59
149,25
107,8
138,5*
66,48
62,34
156,9*
95,69
56,44
56,44
Температур-
Температурный интервал,
К
298
273—821
298—331
298—1000
298—848
848—2000
298—390
390—2000
298—500
500 -2000
298—520
298—388
298—1273
273—1500
298—875
288—1500
298—497
298—1000
298—1800
285—410
298—2000
298—1800
298—1300
T1C1 (кр)
T1CI (г)
TI2O (кр)
UF4 (кр)
UF6 (кр)
UF6 (г)
U02 (кр)
U8O8 (кр)
UO2F2 (кр)
UO2(NO3J (кр)
ZnCO3 (кр)
ZnO (кр)
ZnS (кр)
ZnSO4 (кр)
ZrCl4 (кр)
ZrfV
Органические соединения
Формула для вычисления теплоемкости
—204,
—68,
— 178
— 1854
—2163
—2113
— 1084,
-3583,
— 1637,
-1377
—810,
—349,
—201
—978,
—982,
— 1094
97
41
5
6
6
,7
0
2
0
111,2
255,6
99,5
151,2
227,8
379,7
77,95
281,8
135,6
276,1
82,4
43,5
57,7
124,6
186,1
50,32
50,21
37,4
149,0
80,33
38,9
48,99
50,88
71,42
133,6
69,62
8,37
8,45
6,78
138,0
5,Ю
5,19
87,03
7,53
—1,05
-19,37
-16,56
—9,12
-5,69
-12,18
-14,06
52,72
36,23
117,6
166,75
129,7
63,76
237,9*
103,2*
80,18
40,25
46,02
97,35
119,9
56,04
298—700
298—2000
298
298
298—1000
298—1500
373—593
273—425
293—573
273—1573
298—1200
298—1000
298—550
298—1478
Вещество
AHf, 298-
кДж/моль
•^298'
Дж/(моль-К)
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
с-108
<М0»
^р, 298'
Дж/(МОЛь-К)
Температур-
Температурный интервал,
Углеводороды
СН4 (г) метан
со С2Н2 (г) ацетилен
со
—74,85
226,75
186,19
200,8
17,45
23,46
60,46
85,77
1,117
58,34
—7,20
15,87
35,79
43,93
298—1500
298—1500
Продолжение прил. IV
Вещество
С2Н4 (г) этилен
С2Н6 (г) этан
С3Н4 (г) пропадиен
С3Н6 (г) пропилен
С3Н8 (г) пропан
С4Н6 (г) 1,3-бута-
диен
С4Н8 (г) 1-бутилен
С4Н8 (г) ццс-2-бути-
лен
С4На (г) транс-
/-оутилен
С4Н8 (г) 2-метил-
пропилен
н-С4Ню (г) я-бутан
из0-С4Ню (г) изо-
бутан
C6Hio (г) циклопен-
тан
CSH1O (ж) цикло-
пентан
h-C6Hi2 (г) я-пентан
CjH12 (г) 2-метил-
бутан
0
U 298"
кДж/моль
52,28
—84,67
192,1
20,41
— 103,9
111,9
1,17
—5,70
—10,06
—13,99
-124,7
— 131,6
-77,24
—105,9
— 146,4
— 154,5
s°
298"
Дж/(моль-К)
219,4
229,5
234,9
226,9
269,9
278,7
307,4
300,8
296,5
293,6
310,0
294,64
292,9
204,1
348,4
343,0
Теплоемкость
Дж/(моль-К)
Коэффициенты уравнения
а
4,196
4,494
13,07
3,305
—4,80
—2,96
2,54
—2,72
8,38
7,08
0,469
—6,84
—54,39
—
1,44
—9,29
с° —
р~1
*-108
154,59
182,26
175,3
235,86
307,3
340,08
344,9
307,1
307,54
321,63
385,38
409,64
545,8
—
476,5
517,7
(Т\
c-10«
—81,09
—74,86
—71,17
— 117,6
— 160,16
-223,7
— 191,28
—111,3
—148,26
—166,07
—198,88
-220,55
—307,7
—
—250,4
—292,9
<мо»
16,82
10,8
—.
22,68
32,75
56,53
41,66
—
27,28
33,50
39,97
45,74
66,59
—
51,24
64,78
с"
Р, 298'
Дж/(моль-К)
43,63
52,70
58,99
63,89
73,51
79,54
89,33
78,91
87,82
89,12
97,78
96,82
82,93
126,73
122,6
120,6
Температур-
Температурный интервал,
К
298—1500
298—1500
298—1000
298—1500
298—1500
298—1500
298—1500
298—1000
298—1500
298—1500
298—1500
298—1500
298—1500
298
298—1500
298—1500
CsHi2 (ж) 2-метил-
бутан
С5Н12 (г) 2,2-диме-
тилпропан (нео-
пентан)
С6Н6 (г) бензол
С6Н6 (ж) бензол
С6Н12 (г) циклогек-
сан
н-СеНц (г) я-гек-
сан
H-CeHi4 (ж) я-гек-
сан
С6Н8 (ж) толуол
С7Н8 (г) толуол
h-C7Hi6 (г) я-геп-
тан
«-C7Hi6 (ж) я-геп-
тан
о-С8Ню (г) о-ксилол
о-С8Н10 (ж) о-кси-
о-ксилол
ж-С8Ню (г) ж-кси-
лол
jm-C8Hio (ж) м-кси-
ЛОЛ
л-С8Ню (г) п-ксилол
л-С8Н10 (ж) п-кси-
п-ксилол
h-C8Hi8 (г) я-октан
С8Н10 (кр) нафта-
нафталин
—179,3
—166,0
82,93
49,04
—123,1
—167,19
—198,8
50,00
8,08
—187,82
—224,4
19,0
—24,4
17,24
—25,42
17,95
—24,34
—208,4
75,44
260,4
306,4
269,2
173,2
298,2
386,8
296,0
319,7
219
425,3
328,0
352,8
246,0
357,2
252,2
352,4
247,4
463,7
167,4
— 15,10
-33,90
59,50
-51,72
3,08
—33,88
5,02
— 14,81
—27,38
—25,92
6,91
548,6
471,87
255,02
598,8
565,8
557,0
653,76
591,1
620,9
609,7
741,9
—322,9
—298,34
—230,0
—300,4
—342,4
—348,7
—339,6
—363,9
—350,6
—397,3
73,54
70,84
62,06
79,87
72,32
74,70
81,38
76,88
82,64
164,9
121,63
81,67
136,1
106,3
146,7
195,0
103,8
166
170,8
224,7
133,3
188,8
127,6
183,2
126,9
183,8
194,9
165,7
298
298—1500
298—1500
281—353
298—1000
298—1500
298
298—1500
298
298—1500
298
298—1500
298
298—1500
298
298—1500
298
298—1500
298
Вещество
гНю (кр) дифенил
CuHio (кр) антра-
антрацен
ОиНю (кр) фенан-
трен
АН1. 298'
кДж/моль
96,65
128,0
113
Дж/(моль-К)
206
207,5
211,7
Теплоемкость, ДжДмолЬ'К)
Коэффициенты уравнения
C°=f (Г)
« 1
.11 1
Ь-10>
—
с.ю.
—
d-10'
—
Продолжение прил. IV
е°р, гее-
Дж/(моль-К)
195
209
231
Температур-
Температурный интервал,
К
298
298
298
СН2О (г) муравьи-
муравьиный альдегид
СН2О2 (ж) муравьи-
муравьиная кислота
СН2О2 (г) муравьи-
муравьиная кислота
СН4О (ж) метило-
метиловый спирт
СН4О (г) метиловый
спирт
СгН2О4 (кр) щаве-
щавелевая кислота
С2Н4О (г) уксусный
альдегид
С2Н4О (г) оксид эти-
этилена
-115,9
-422,8
-376,7
—238,7
-201,2
—826,8
—166,0
—51,0
К и с л
218,8
129,0
251,6
126,7
239,7
120,1
264,2
243,7
ородс одерж ащие соединения
18,82 | 58,38 | -15,61
19,4
15,28
13,00
—9,60
112,8
105,2
153,5
232,1
-47,5
-31,04
—53,7
—140,5
32,90
35,34
99,0
48,7
81,6
43,9
109
54,64
48,5
298—1500
298
298—1000
298
298—1000
298
298—1000
298—1000
С2Н4О2 (ж) уксус-
уксусная кислота
С2Н4О2 (г) уксус-
уксусная кислота
С2Н6О (ж) этило-
этиловый спирт
С2Н6О (г) этиловый
спирт
С2Н6О (г) димети-
ловый эфир
С2Н6О2 (ж) этилен-
гликоль
С2Н6О2 (г) этилен-
гликоль
С3Н6О (ж) ацетон
С3Н6О (г) ацетон
и-С3Н8О (ж) я-про-
пиловый спирт
к-С3Н8О (г) я-про-
пиловый спирт
изо-С3Н80 (ж) изо-
пропиловый
спирт
изо-С3Н8 (г) изопро-
пиловый спирт
С3Н8Оз (ж) глице-
глицерин
С4Н4О4 (кр) малеи-
новая кислота
С4Н4О4 (кр) фума-
ровая кислота
QHeO2 (ж) масля-
масляная кислота
—484,9
—437,4
—277,6
—235,3
—185,4
—454,9
—397,9
—247,7
—216,4
—306,6
—260,4
—318,7
—275,4
—659,4
—787,8
—811,0
—524,3
159,8
282,5
160,7
282,0
266,6
179,5
323,5
200
294,9
192,9
317,6
180
306,3
207,9
159
166
255
5,56
19,07
22,47
243,5
212,7
201,8
-151,9
-108,6
—63,5
36,8
21,9
153,4
223,0
137
142
178
298
298
298
298
123,4
66,5
111,4
73,6
65,94
151
125
74,9
148,6
298
298—1500
298
298—1500
298
298
298
298—1500
298
298
Продолжение прил. IV
Вещество
QHsOis (ж) уксус-
но-этиловый эфир
С4Н8О2 (ж) 1,4-ди-
оксан
С6Н10О (ж) цикло-
пентанол
к-С5Н12О (ж) н-ами-
ловый спирт
«-С5Н12О (г) я-ами-
ловый спирт
С6Н6О (кр) фенол
СвНвО2 (кр) гидро-
гидрохинон
СвН4О2 (кр) хинон
С7НвО2 (кр) бензой-
бензойная кислота
С7Н8О (ж) бензило-
вый спирт
С8Н4О8 (кр) ангид-
ангидрид фта левой кис-
кислоты
CsHeOi (кр) фтале-
вая кислота
f, 298'
кДж/моль
—469,5
—400,8
—300,2
—360,1
—307,2
—162,8
—363,0
— 186,8
—385,2
—161,0
—460,4
—781,9
°298>
Дж/(моль-К)
259
196,6
206
254,8
381,6
142
—
—
167,6
216,7
179,5
207,9
Теплоемкость. Дж/(моль-К)
Коэффициенты уравнения
С° —! (Т\
р ' ^ >
а
—
—
—
—
—
—
—
—
—
—
—
6-10»
—
—
—
—
—
—
—
—
—
—
—
с-10" 1 d-10»
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
—
Р, 298'
Дж/(моль-К)
170
152,9
184
209,2
—
134,7
142
132
146,8
217,8
161,8
188,2
Температур-
Температурный интервал,
К
298
298
298
298
—
298
298
298
298
298
298
298
СцНцА (кр) хин-
т4 IX Tf т\ п и
1 И.Д[Л_Ш
CiaHjaOn (кр) са-
сахароза
CHsF (г) фтористый
метил
CHSC1 (г) хлори-
хлористый метил
СН8Вг (г) броми-
бромистый метил
CHsI (ж) йодистый
метил
СН31 (г) йодистый
метил
CH2Fj (Г) дифтор-
метан
СН2С12 (ж) дихлор-
метан
СН2С12 (г) дихлор-
метан
CHFs (г) трифтор-
метан
СНС13 (ж) трихлор-
метан (хлоро-
(хлороформ)
СНС13 (г) трихлор-
метан (хлоро-
(хлороформ)
379
(—581,6)
—2221
—247
—82,0
-35,6
-8,4
20,5
—441,6
—117,1
—87,9
—680,3
— 131,8
— 100,4
360
Галогенсодержа
222,8 9,75
233,5
245,8
162,7
253,0
246,0
178,6
270,2
259,5
202,0
295,6
15,57
18,53
—
19,67
11,39
—
16.Ю
18,80
—
81,38
_
щие соединения
97,3
92,74
89,40
—
92,67
118,2
—
144,4
127,9
—
16,0
—29,10
—28,31
—27,28
—
—32,28
—46,0
—
—98,60
—55,78
—
—18,7*
—
—
—
—
—
—
25,2
—
—
425
37,40
40,71
42,4
127,2
44,1
43,0
100,0
51,1
51,09
116,3
65,7
298
298—150Э
298—1500
298—1500
298
298—1000
298—1000
298
298—1000
298—1000
298
298—1000
Продолокение прил. IV
Вещество
АН
f, 298>
кДж/моль
Дж/(моль-К)
Теплоемкость, Дж/(моль-К)
Коэффициенты уравнения
b-10*
с-10«
up, 298'
Дж/(моль-К)
Температур-
Температурный интервал,
CF4 (г) тетрафтор-
метан (четырех-
фтористый угле-
род)
СС14 (ж) тетрахлор-
метаи (четырех-
хлористый угле-
углерод)
CClj (г) тетрахлор-
метан (четырех-
хлористый угле-
углерод)
C2H6F (г) фтори-
фтористый этил
С2Н6С1 (г) хлори-
хлористый этил
CeH6F (ж) фторбен-
зол
CeH6F (г) фторбен-
зол
СвН5С1 (ж) хлор-
хлорбензол
СвН6С1 (г) хлорбен-
хлорбензол
—908
—139,3
— 106,7
—297
—105,0
— 145,4
— 109,7
10,65
52,13
262,0
214,4
309,7
364,8
274,8
205,9
323,5
194,1
313,2
85,67
97,65
8,39
13,07
-34,2
-33,9
15,9
9,62
190,2
188,5
532,0
558,0
-26,3*
—15,06*
—67,83
—71,94
—375,8
—445,2
98,0
139,4
61,2
131,7
83,4
58,6
62,3
146,4
94,4
150,1
97,1
298—1000
298
298—1000
298—600
298—700
298
298—1500
298
298—1000
3 (ж) фенил-
трифторметан
C7H6F3 (г) фенил-
трифторметан
-618,6
-581,0
271,5
372,6
—33,4
681,3
—490,5
129,0
Азотсодержащие соединения
188,4
130,4
298
298—1500
~H5N (г) метиламин
CH2N2 (г) диазоме-
тан
CH9N2 (ж) метил-
гидразин
CH6N2 (г) метил-
гидразин
C2H7N (г) диметил-
амин
C8H9N (г) триметил-
амин
C5H6N (ж) пиридин
C6H6N (г) пиридин
C6H,N (ж) анилин
C9H,N (г) анилин
CH4ON2 (кр) моче-
мочевина
C2H6O2N (кр) ами-
ноуксусная кис-
кислота (гликоколь)
CeH6O2N (ж) нитро-
нитробензол
—28,03
192
—
—
—27,61
—46,02
99,95
140,2
29,7
82,4
—333,1
—524,9
11,2
241,6
238,7
165,9
278,7
273,1
288,8
177,9
282,8
192
301
104,6
109,2
224,3
16,34
54,02
—
25,30
—3,10
—11,95
—
—38,60
—
—
—
—
130,6
31,5
—
179,0
283,1
414,2
—
479,5
—
—
—
—
—38,45
— 13,16*
—
—56,4
—152,2
—245,8
—
—326,6
—
—
—
—
—
—
—
—
—
32,0
56,8
—
83,1
—
—
—
—
—
51,7
48,85
131,9
71,13
69,04
91,76
132,7
78,12
191
—
93,14
100,3
187,3
298—1500
298—1000
298
298—1500
298—1500
298—1500
298
298—1500
298
_
298
298
298
* Теплоемкость не зависит от температуры.
V. Ионы в водных растворах
Ион
Ag-ь
А13+
AsOS"
Ва2 +
Вг-
ВгОГ
сн3соо-
CN-
соГ
с2оГ
Са2+
Cd2+
Ci-
cio-
С1О.Г
СЮз
С1ОГ
Со2 +
CrOl~
Cs+
Cu-ь
Cu2+
F-
Fe2 +
рез+
H+
HCOO-
HCO7
Hg2 +
Hgl+
нроГ
HaPO7
hs-
hsoj-
Hsor
I-
кДж/моль
105,90
—524,7
—870,3
—538,36
—120,92
—40,2
—488,87
151,0
—676,26
—824,2
—542,96
—72,38
—167,46
— 107,65
-69,0
—98,32
— 131,42
—67,4
—863,2
—247,7
71,5
64,39
—329,11
—87,9
-47,7
0
—410,0
—691,11
174,01
168,3
—1298,7
—1302,5
—17,66
—627,98
—885,75
—55,94
AC298-
кД ж/моль
77,11
—481,2
—636,0
—560,7
—102,82
45,6
—375,39
165,7
—528,10
—674,9
—553,04
—77,74
—131,17
—38,53
14,6
—2,59
— 10,75
—51,5
—706,3
—282,04
50,2
64,98
—276,48
—84,94
—10,54
0
—334,7
—587,06
164,77
154,18
—1094,1
—1135,1
12,59
—527,31
—752,87
—51,67
Дж/(моль-К)
73,93
—313,4
—144,8
13
80,71
161,1
—
92,0
—53,1
51,0
—55,2
—61,1
55,10
47,53
100,4
163,2
180,7
—111,7
38,5
133,1
39,3
—98,7
—9,6
—113,4
—293,3
0*
91,6
95,0
—22,6
74,1
—36,0
89,1
61,1
132,38
126,86
109,37
382
Продолжение прилож. V
Ион
Юз
к+
Li +
Mg3+
Mn3+
МпОГ
NH4+
ПОг
NO3"
Na +
Ni2+
он-
Pb3+
Rb +
S2-
sor
Sr2 +
u3+
u4+
uoi
Zn2 +
&H}, 298-
кДж/моль
—230,1
—251,21
—278,46
—461,96
—218,8
—518,4
— 132,80
— 106,3
—206,57
—239,66
—64,0
—229,94
— 1284,1
1,63
—246,4
41,8
—907,51
—545,51
—514,6
-613,8
— 1035,1
— 152,42
AG29«'
кДж/моль
— 135,6
—282,28
—293,80
—456,01
—223,4
—425,1
—79,50
—35,35
— 110,50
—261,87
—64,4
— 157,30
— 1025,5
—24,31
—282,21
83,7
—742,99
—557,3
—520,5
—579,1
—994,1
— 147,21
S298'
Дж/(моль-К)
115,9
102,5
14,2
— 118,0
—79,9
190,0
112,84
125,1
146,4
60,2
— 123,0
—10,54
—218
21,3
124,3
—26,8
17,2
—26,4
— 126
—326
50
— 106,48
Br-
ci-
С1ОГ
Cs+
H +
Hg2+
I
K+
Li +
Ионы в жидком аммиаке
108,8
—246,9
—418,4
—274,9
-199,6
—163,2
0,0
189,1
-189,5
-169,4
-205,0
42,3
—67,4
73,6
— 167,4
—418,0
— 184,5
—74,1
— 193,7
0,0
129,3
— 121,3
—196,6
—225,9
141,8
—11,3
96,2
— 126,8
—87,9
— 126,8
62,8
121,3
0,0
146,4
— 104,6
89,5
33,5
—41,8
103,3
383
Продолжение прилож. V
Ион
NOJ
Na+
Rb2+
Rb+
SCN-
кДж/моль
—324,7
— 159,4
87,9
— 163,2
—49,4
ДС2»8-
кДж/моль
—178,6
— 182,4
54,4
— 196,2
—
S298-
ДжДмоль • К)
—20,9
63,2
46,0
121.3
_ .
* Абсолютная энтропия Н+ S°98=-14,2 Дж/(моль-К).
VI. Нормальные (стандартные) окислительно-восстановительные
потенциалы в водных растворах при 25 °С по отношению
к нормальному электроду
Название и
символ элемента
Азот N
Электродный процесс
3N2 + 2Н + + 2е - = 2HN,
N2+4H2O+2e-=2NH2ON + 2ON-
N2 + 2H2O + 4H++2e- = BNH2OH)H +
N2 + 4H3O + 4e-=N2H4 + 4OH-
N2 + 5H++4e- = (NaH4)H +
N2 + 6H++6e- =2NH3
N3 + 8H + +6e-=2NHj
N2O+10H+ +8e~ =2NHj + HaO
2NO + 2H+ +2e- = H2N2Oa
HNO2 + 6H+ +6e~ = NH3 + 2H2O
HNO3+H + +e-=NO2 + H2O
NOJ +2H+ +e~ = NO2+H2O
2HN03+2H++2e- = NaO4+2H20
2N03~ + 4H++2e- = N2O4+2H2O
NOr + 2H++2e- = NOa" + H2O
NO+6H++5e-=NH^ + H2O
HNO2+7H+ +6e- = NHj + 2H2O
NOr + 10H++8e- = NHi"+3H2O
HNO3+2H++2e- = HN02 + H20
Стандарт-
Стандартный элект-
электродный по-
в L П 1^ 11 ell -l 1
Ф°. В
—3,1
—3,04
— 1,87
— 1,16
—0,23
+0,057
+0,275
+0,647
+0,71
+0,755
+0,755
+0,755
+0,803
+0,803
+0,835
+0,836
+0,864
+0,87
+0,934
384
w
Продолжение прилож. VI
Название и
символ элемента
Азот N
Актиний Ас
Алюминий А1
Америций Am
Электродный процесс
ШО3+ЗН+ +3е~ = NO+2H2O
N0i"+4H+ +3e- = N0+ 2Н2О
HNO2 + H++e-=NO + H2O
NO2 + 2H++2e- = NO + H2O
NO2 + 2H+ -4-е- = HN02
2HNO3 + 8H++8e-=N2O+5H2O
2NO3" +1 OH + + 8e - = N20 + 5H2O
2HNOs+10H + + 10e = N2 + 6H2O
2HNO2 + 4H++4e~ =N2O + 3H2O
2NO2 + 8H++8e- — N2 + 4H2O
2HNOs + 6H+ +6e~ =N2 + 4HaO
2NO + 4H+ +4e- =Na + 2H2O
N2O+2H++2e-=N2 + H2O
H2NaO, + 2H4-+2e-=Na + 2H2O
Ac3++3e- =Ac
[AlF6]3-+3e~=Al + 6F~
Al (OHK+3H+ +3e- = Al +3H2O
AlOJT +4H- +3e- = Al +2H2O
Am3++3e- = Am
Am(OHK + 3H+ +3e~ = Am + 3H2O
Am2O3 + 6H + + 6e ~ = 2Am + 3H2O
2Am(OHL + 2H++2e- = Am2O3 + 5H2O
2AmO2 + 2H++2e~ = Am2O3 + H2O
Ат(ОНL + Н++е-=Ат(ОНK + Н2О
AmO2 + H2O+H+ -4-e~ = Am @HK
AmO2b+4H + +e- =Am*++2H2O
Am2O5 + 2H++2e~= 2 AmO2 + H20
Am2O5 + 3H2O+2H+ +2e- =2Am @HL
Am02++e- = Am0f
Am2O5+ 10H+ +4e- =2Am3+ +5HaO
AmO2+ + 4H++3e-=Am3+ + 2H2O
Стандарт-
Стандартный элект-
электродный по-
трип Un п
ЕСПЦпЗЛ
ф°, В
+0,957
+0,957
+ 1,004
+ 1,049
+ 1,093
+ 1,116
+ 1,116
+ 1,246
+ 1,297
+ 1,363
+ 1,454
+ 1,678
+ 1,766
4-2,65
+2,6
-2,07
— 1,663
— 1,471
— 1,262
—2,320
— 1,872
— 1,676
—0,185
—0,072
+0,420
+0,533
+ 1,261
+1,418
+ 1,530
+1,639
+ 1,639
+ 1,694
385
Продолжение прилож. VI
Название и
символ элемента
Америций Am
Барий Ва
Бериллий Be
Берклий Вк
Бор В
Бром Вг
Электродный процесс
kmOt +4Н + +2е~ = Ат3+ +2Н2О
Am (ОНL + 4Н+ +е- = Ат3+ +4Н2О
2АтО2+ + Н 20+2е - = Ат2О5+2Н +
АтО2+4Н + +е- = Ат3+ +2Н2О
2АтО3 + 2Н++2е- = Ат2О5+Н2О
Ат4+ -\-е~ — Ат3+
в..+ +*-=в.
Ве« + +2е- = Ве
Be (ОНJ + 2Н+ +2е- =Ве + 2Н2О
Ве2О|~ + 6Н+ +4е~ =2Ве+ ЗН2О
ВеОГ + 4Н++2в-=--Ве + 2Н2О
Вк4++,-.Вкз+
[BF4]"+3e- - В + 4F-
Н3ВО3 + ЗН++Зе- =В + ЗНаО
ВОз~ + 6Н + +3е~ = В + ЗН2О
2ВгО-+2Н2О + 2,- = Вг2 + 4ОН-
ВгО3" + ЗН2О+6е- = Вг-+6ОН-
ВгО~ + Н2О+2е- = Вг + 2ОН~
Вг|Г + 2е- =ЗВг~
{Вг2}+2е-=2Вг-
ВгС1+2е-=Вг-+С1-
ВгОз" + 6Н+ +6е~ = Вг- +ЗН2О
BrOJ + 5H++4e-=HBrO + 2H2O
2BrOs~ + 12Н + + l(k- = Bra + 6H2O
2НВЮ + 2Н+ +2e~ = Bra + 2H2O
Стандарт-
Стандартный элект-
электродный по-
потенциал
ф°. В
+ 1,721
+ 1,746
+ 1,804
+ 1,856
+ 1,930
+2,181
—2,905
— 1,847
— 1,820
— 1,387
-0,909
1,6
— 1,04
—0,869
-0,165
+0,45
+0,54
+0,61
+0,76
+ 1,05
+ 1,065
+ 1,2
+1,44
+ 1,49
+ 1,52
+ 1,59
386
г
Продолжение прилож. VI
Название и
символ элемента
Ванадий V
Висмут Bi
Водород Н
Вольфрам W
Электродный процесс
V2++2*- = V
VaOa+4Н + + 4е - = 2V + 2Н2О
V3+-j_e-=v? +
VO2++e~=VO+
VO2++2H++e-=V3+ + H2O
HV2O5"+9H+ +4e~ = 2V? + +5H2O
[V(CN)8]3-+e- = [V(CN)8]4-
H3V2O7~ Ч-ЗН+ +2e = V2O4+3H20
V2O5 + 6H+ +2e- =2VO?+ +3H2O
\Юа++2Н+ +e~ = VO?+ +H2O
H3V267" + 7H+ +2e~ = 2VO?+ +5H2O
VOi~ + 6H++2e- = VO++3H2O
H2VO4" + 4H++e-=VO2+ + 3H2O
BiOCl+2H++3e-= Bi + H2O + Cl-
[BiCl4]-+3e- = Bi + 4Cl-
Bi3++3e-=Bi
BiOH?+ + H++3e- = Bi + H2O
BiO + 2H++3e-=Bi + H2O
Bi2O3+6H++6e-=2Bi + 3H2O
Bi4O7 + 2H++2e-=2Bi2O3 + H2O
2Bi2O4 + 2H++2e- = Bi4O7 + H2O
Bi2O5+2H + +2e-=Bi2O4 + H20
Bi2O5 + 10H++4e-=2Bi3++5H2O
H2+2e-=2H~
H + +2e~ = H-
2H++2e- = H2
Hl + H++,?- = H2
2H2O+2e-=H2+2OH-
WO2 + 4H++4e-=W + 2H2O
W2O5+2H++2e-=2WO2 + H2O
2WO3 + 2H++2e-=W2O6 + H2O
WOr+8H++6e-=W+4H2O
Стандарт-
Стандартный элект
родный по-
f?tll (ГЦ n л
ф°, В
—1,175
—0,820
—0,255
—0,044
+0,337
+0,338
+0,51
+0,806
+0,958
+1,004
+ 1,096
+ 1,256
+ 1,314
+0,16
+0,16
+0,215
+0,254
+0,320
+0,371
+1,338
+1,541
+1,607
+1,759
—2,251
—1,125
0,000
+2,106
—0,828
—0,119
—0,031
—0,029
+0,049
387
9Si'O—
Z6'0~
966'6—
996' Г—
696 'S—
s=
000'0
oer'o—
S8J'O—
98S'O—
696' 0—
sog'i—
989'I —
ooz'i—
6ie'o+
eit-'o—
629'0—
108'0+
И 'о*
1/ВИПНЭ1
-ou ед1чн1Л>A
огне+ла = -эе++не+(!(но)'(;а
огне+он = -^6+ + не+8(но) он
ОЕН6~Ь ^0 ^^ — **\ ~^г + Н^ Ч^?О^ОЕН
OsHS~Ь^0 ^^ —Ъ\Л~ +Н?~ЬЕО^О
OEH + 8D = -^S+ +HS+030
ОЕН6+ +гэ9= +9S+ +Hfr + eO3D5H
OEHS + »H = 3t'+ +Ш+ЕОШ
OEH{' + JH;= -9t' + +H{' + OsHS':;OJH
OFH. + PO-4-U™^»
-*[8(ю) л\] = -э+ -еР(ыэ) л\]
эээпоЛи B^Htfodxnaice
ng HHUodag
Xq HHeoduDHj/
OH HHW41fOJ
]H иинфе^
BQ guifb-BJ
pr> HHHUIfOirBJ
//I •Morndti anuaMvoQOdц
Название и
символ элемента
Железо Fe
Золото Аи
Индий In
Продолжение
Электродный процесс
Fe2S3 + 2е - = 2FeS + S? -
Fe?++2e- = Fe
Fe3O4+8Н + + 8е~ = 3Fe + 4Н2О
Fe2O3 + H2O + 2H++2e-=2Fe(OHJ
FeaO3+6H++6e-=2Fe + 3H2O
Fe (OHJ+2H+ +2e~ =Fe + 2H2O
Fe(OHK + 3H++3e-=Fe+3H2O
[Fe(CN),]»-+e-=[Fe(CN)e]*-
FeOH^ + H^ +e- = Fe2++H2O
Fe3Oj + 8H + +2e+=3Fe2++4H2O
FeOf" +5H+ +4e- = HFeO^ + 2H2O
FeOr + 8H-+3(?- = Fe3+ + 4H2O
[Au (CNJj- +e~ = Au + 2CN
AuI + e~ = Au + I~
[Au (SCN)a]~ +e~ = Au+2SCN-
[AuBr4]-+2e- = AuBri"+2Br-
AuO2 + H2O+e- = HAuOl~ + H +
[AuBr4]~ +2e~ = Au + 4Br-
[AuBr2j- +e~ = Au + 2Br-
[AuCl4]-+3e- = Au + 4Cl-
AuCl + e~ ^Au+Cl~
Au3++2e=Au +
Au2O3 + 6H++6e-=2Au+3HaO
Au3++3e~ = Au
H3AuO3+3H++2e- = Au++3H2O
AuO2 + 4H+ +e- = Au3+ +2H2O
In3++3e- = In
InCl+e- = In + Cl-
In (OH)8+3H+ + 3e- = In+3HaO
In++e- = In
InOa4-4H+ +3e = In +2H2O
прилож. VI
Стандарт-
Стандартный элект-
электродный по-
потенциал
<Р°. В
—0,7
—0,440
—0,085
—0,057
—0,051
—0,047
+0,059
+0,271
+0,36
+0,771
+0,914
+0,980
+ 1,001
+ 1,700
—0,60
+50
+0,69
+0,82
+0,822
+0,87
+0,96
+ 1,00
+ 1,17
+ 1,401
+1,457
+1,498
+1,502
+2,507
—0,342
—0,34
—0,172
—0,139
+0,146
389
Продолжение
Название и
символ элемента
Иод I
Иридий Ir
Иттербий Yb
Иттрий Y
Кадмий Cd
Электродный процесс
ЮГ + ЗН„О + 6е- = 1- = 6ОН-
Ю- + НаО + 2е- = 1-+2ОИ-
12 + 2е- = 21-
13+3*- = 31-
IQf+ 2НаО+4е- = Ю- + 4ON-
2ICN + 2H++2e- = I2 + 2HCN
НзЮ|~+2е- = Юз+ЗОН-
2IBr2 + 4e- = I2 + 4Br-
НЮ + Н + +2е- = 1- + НаО
2IBr+2e- = I2+2Br~
21С12+2е- = 12+2С1- :-\
1Оз" + 6Н+ +6е- +1- +ЗН2О
21С1+2е- = 12+2С1-
2IOif + 12H+ + 10e- = I2 + 6HaO
21С12 + 6е- = 12 + 6С1-
2HIO + 2H+ +2в- = 12 + 2НгО
Н5Юв + Н + +2е-=Юз""+ЗН2О
[1г16]2-+е- = 1г1Г
[IrCl6]3-+3e- = Ir + 6Q-
IrO2 + 4H+ +4e- = Ir + 2H2O
[IrBrep-+e-=[IrBr6p-
[IrBre]3-+e- = [IrBre]4-
1гз++3е- = 1г
Yb+e-=Yb2+
уз+-(-Зе- = У
Y2O3 + 6H+ +6e~ = 2Y + 3H2O
Cd + H++e-=CdH
CdS+2«-=Cd+S2"
[Cd(CNL]2-+2e-=Cd + 4CN-
[Cd(NH3L]2++2e-=Cd + 4NH3
Cd2++2e~=Cd
390
г
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
+0,26
+0,49
+0,536
+0,536
+0,56
+0,63
+0,7
+0,87
+0,99
+1,02
+1,06
+1,085
+1,19
+1,19
+1,28
+1,45
+ 1,69
+0,48
+0,77
+0,93
+0,947
+0,99
+1,15
—1,205
-2,372
-1,676
-2,417
-1,24
-1,09
-0,61
-0,403
Продолжение прилож. VI
Название и
символ элемента
if " f1 j
Кадмии Cd
Калий К
Кальций Са
Кислород О
Кобальт Со
Кремний Si
Электродный процесс
Cd (OHJ + 2H+ +2e- = Cd + 2H3O
CdO+2H++2*-=Cd + H2O
Са (ОНJ + 2е ~ = Са+2ON -
О2 + 2Н++2е- =О + Н2О
О2 + Н++2е- = НО;Г
Оа+2Н + + 2в- = НаОа
Оа + 4Н+ + 4*-=2Н2О
О8 + 6Н++6е-=ЗНаО
HO2" + 2H + +2e-=OH-+H2O
Н2О2 + 2Н+ +2е- =2Н2О
О3+2Н++2е-=О2+Н2О
О+2Н+ + 2е- = НаО
p-CoS+2e-=Co + S2-
a-CoS + 2e- =Co + S2~
[Co(CNN]3-+e+=Co(CN)e4-
СоСОз+2e - = Co + COe~
[Co (NH8)e]2+ +2e- =Co+ 6NHS
Co2++2e-=Co
CoO + 2H++2e-=Co + H2O
НСоО2~ + ЗН++2г-=Со+2Н2О
CoO2 + 4H + +e-=Co2++2H2O
Co3++e-=Co2 +
SiO2 + 4H++4e-=Si + 2H2O
Стандарт-
Стандартный злект-
эодиый по-
Ф°, В
+0,005
+0,063
—2,924
—3,03
—2,866
+0,037
+0,338
+0,401
+0,682
+1,228
+ 1,511
+1,706
+1,776
+2,076
+2,421
— 1,07
—0,90
—0,83
—0,64
—0,42
—0,277
+0,095
+0,166
+0,659
+1,416
+ 1,612
+ 1,808
—1,2
—0,857
391
Продолжение прилож* VI
Название и
символ элемента
Кремний Si
Лантан La
Литий Li
Лютеций Lu
Магний Mg
Марганец Мп
Медь Си
Электродный процесс
H2SiO3 + 4H++4e-=Si + 3H2O
H2SiO8 + 4H++4e- = Si + 3H2O
SiOf~+6H++4e- =Si + 3H2O
Si + 4H+ + 4e-=SiH4
La3++3«- = La
La2O3 + 6H++6e-=2La + 3H2O
Li++«- = Li
Lu3++3e- = Lu
LuaO3 + 6H+ + 6e-=2Lu + 3H2O
Mg2++2e-=Mg
Mg (OHJ + 2H+ +2e- =Mg + 2H2O
MnCOg+2e-=Mn+CO|~
Mn2++2e-=Mn
Mn (OHJ+2H++2e-=Mn + 2H2O
[Mn (CN)ef- +e- = [Mn (CN)e]4-
MnO7+e-=Mn|~
МпОГ + 2H2O + 3e ~ = MnO2 + 4OH
MnO3 + 4H + +2e-=Mn2++2H2O
Mn2O3+6H+ +2e- =2Mn2+ +3H2O
MnO7+8H++5e-=Mn2++4H20
Mn3++e-=Mn2+
МпОГ + 4Н++Зе-=МпО2+2Н2О
МпОГ + 4Н++2е-=МпО2 + 2Н2О
Cu+H++e-=CuH
Cu2S+2e-=2Cu + S?-
CuS + 2e-=Cu+S2-
[Cu(CNJ]-+e-=Cu + 2CN-
Стаидарт-
иый элект-
электродный по-
потенциал
Ф°, В
—0,807
—0,789
—0,455
+0,102
—2,522
—1,856
—3,045
—2,255
—1,892
—2,363
— 1,862
— 1,48
— 1,179
—0,727
—0,22
+0,564
+Р.6
+ 1,228
+ 1,443
+ 1,507
+ 1,509
+ 1,692
+2,257
—2,775
—0,93
—0,79
—0,43
392
Продолжение прилож. VI
Название и
символ элемента
Медь Си
Молибден Мо
Мышьяк As
Электродный процесс
CuSCN+г- =Cu + SCN-
CuI+e-=Cu + I~
[Cu(NH8J]++e-=Cu + 2NH3
[Си (NH3L]2+ + 2е~ = Си +4NH3
CuI7+e~=Cu+2I-
CuBr+e- =Cu + Br~
СиС1+е-=Си+С1-
Cu2++e-=Cu+
2Cu2+ + H2O+2e-=Cu2O + 2H +
Cu2++2e- = Cu
Cu2O+2H + +2e-=2Cu + H2O
Cu++e~ =Cu
Cu2++Cl-+e-=CuCl
CuO+2H-+2e-=Cu + H2O
Си (ОНJ + 2Н+ +2e- =Cu + 2H2O
Cu2++Br-+e~ =CuBr
2CuO+2H + +2e-=Cu2O+H2O
Си2++1-+г-=Си1
Cu2+ +CN~ +e- =CuCN
HCuO2"+3H++2e-=Cu+2H2O
Cu022~ + 4H++2e-=Cu + 2H2O
H2MoO4+2H++2e-=MoO2 + 2H2O
Mo3++3e-=Mo
MoO2 + 4H++4e-=Mo + 2H2O
MoOl" + 8H-+6e-=Mo + 4H2O
MoO3+2H++2e-=MoO2 + H2O
MoOi~ + 4H++2e-=MoO2 + 2H2O
[Mo (CN)8]3- +e- = [Mo (CN)8]*-
AsS^+3e- = As + 2S2.-
As+3H++3e- = AsH3
As2O3+6H + +fe- = 2As + 3H2O
AsO++2H++3e-=As + H2O
H,,AsO4+3H+ +2e" = AsO+ +3H2O
H8AsO4+2H+ +2e~ = HAsO2 + 2H2O
AsO*-+8H++5e- = As + 4H2O
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
—0,27
—0,185
—0,12
—0,05
+0,00
+0,03
+0,137
+0,153
+0,203
+0,337
+0,471
+0,520
+0,538
+0,570
+0,609
+0,640
+0,669
+0,86
+ 1,12
+ 1,127
+ 1,515
—1,091
—0,200
—0,072
+0,154
+0,320
+0,606
+0,73
—0,8
—0,608
+0,234
+0,254
+0,550
+0,560
+0,648
393
Продолжение прилож. VI
Название и
символ элемента
Натрий Na
Неодим Nd
Нептуний Np
Никель N1
Электродный процесс
Na + +е = Na
Nd2O3 + 6Н + + 6е~ = 2Nd + ЗН2О
Np3++3e-=Np
Np2O3 + 6Н + + бе- = 2Np + ЗН2О
2NpO2 + 2H++2e-=Np2O8 + H2O
2Np(OHL+2H++2e~ = Np2O3+5H2O
Np4++«- = Np3+
NpO2 + 4H + +e~ = Np3+ +2H2O
Np(OHL + 4H++e-=Np3+ + 4H2O
NpO2" + 4H++2e- = Np3( +2H2O
NpOj+2H2O+e-=Np(OHL
NpOj+e-^NpOa
NpO2b + 4H++e--Np4-+2HaO
NpOl++e- = NpO2
Np2O5+3H2O+2H+ +2г~ = 2Np (OHL
Np2O5 + 2H++2e- = 2NpO2 + H2O
2NpO3 + 2H++2e-=Np2O6 + H2O
NpO, + 2H++e-=NpO2+ + H1O
^-NiS+2e~ = Ni + S2~
a_NiS+2e~ = Ni +S2~
[Ni(CNL]2-+e-=[Ni(CNL]8-
[Ni(NH3)G]2++2e- = Ni + 6NH8
№СО3+2е-=№+СОГ
Ni2++2e-=Ni
№ (OHJ + 2H + +2e- = Ni +2H2O
NiO+2H + +2e-=Ni + H2O
HNiO^" + 3H++2e-=Ni + 2H2O
Ni8O4+2H++2e-=3NiO + H2O
3Ni2O4 + 2H + + 2e~ =2Ni8O4 + H2O
2NiO2+2H++2e-==Ni2O3
NiO2 + 4H+ +2e- = Ni2+ +2H2O
Стандарт-
Стандартный элект-
электродный по
ф°. В
—2,714
—2,431
— 1,811
—1,856
—1,420
—0,962
+0,982
+0,152
+0,337
+0,391
+0,451
+0,530
+0,560
+0,749
+ 1,149
+ 1,219
+ 1,253
+ 1,310
+1,998
—0,99
—0,83
—0,82
—0,49
—0,45
—0,250
+0,110
+0,116
+0,648
+0,897
+ 1,305
+1,434
+ 1,593
394
Продолжение прилож. VI
Название и
символ элемента
Ниобий Nb
Олово Sn
Осмий Os
Палладий Pd
Платина Pt
Электродный процесс
Nb8+ + 3e-=Nb
NbO+2H++2e-=Nb + H2O
Nb02+2H++2e~=NbO+H2O
Nb2O5 + 2H++2e-=2Nb02 + H2O
Sn + 4H + + 4e- = SnH4
SnS+2e"=Sn+S2-
SnFJj~+4e-=Sn + 6F-
Sn2++2e-=Sn
SnO2 + 2H++2e-=SnO+H2O
SnO2+4H++4e-=Sn + 2H2O
SnO+2H+ +2e~ =Sn + H2O
Sn(OH)+2H++2e-=Sn+2H2O
Sn(OHL + 4H+ + 4*?- = Sn+4H2O
Sn1++2e~ = Sn?+
HSnO2" + 3H++2e-=Sn+2H2O
SnOr+3H++2e- = HSnO2~ + H2O
OsO2 + 2H2O + 4e- = Os + 4OH-
H0sOr + 4H2O+&- =Os+9OH-
OsOr + 2H2O+2e- =OsO2 + 4OH-
HOsO5~ + 2e ~ = OsOl~ + OH -
OsO4+8H + + 8g - = Os + 4H2O
OsO4+4H++4e-=Os+2H2O
OsO4+8H+ +C1- +4г- =OsCl2- +4H2O
[PdIep-+2e- = [PdI4]2-+2I-
[PdCl4]2-+2e-=Pd + 4Cl-
PdO + 2H+ +2e- = Pd + H2O
Pd2++2e~=Pd
[PdBr6]2- +2e- = [PdBr4]2- +2Br
PdO2+2H++2«~=PdO + H2O
[PdCleJ2- +2e- = [PdCl4]2- +2C1-
PdO3 + 2H++2e- = Pd03 + H2O
PtS+2e-=Pt+S2-
PtS2+2e-=PtS+S2-
Стаидарт-
иый элект-
электродный по-
потенциал
Ф°, В
— 1,1
—0,733
—0,625
—0,289
—1,074
—0,94
—0,25
—0,136
—0,108
-1,106
-0,104
—0,091
—0,008
+0,151
+0,333
+0,374
—0,15
+0,02
+0,1
+0,3
+0,85
+0,96
+1,00
+0,482
+0,623
+0,896
+0,987
+0,993
+1,283
+1,288
+2,030
—0,95
—0,64
395
Продолжение г
Название и
символ элемента
Платина Pt
Плутоний Ри
Полоний Ро
Празеодим Рг
Электродный процесс
Pt(OHJ + 2e- = Pt + 2OH-
[PtIe]2-+2e- = [PtI4]2--f-2I-
[Pt(SCN)e]2- +2e~ = [Pt (SCNLp~ -J-2SCN-
[PtBr4]2~ +2e- = Pt+4Br-
[PtCl4]2--f-2e-=Pt + 4Cl-
Pt(OHJ + 2H + +2e- = Pt + 2H2O
PtO2+2H + +2e-=Pt(OHJ
Pta++2<?- = Pt
PtO3 + 2H++2e-=PtO2 + H2O
Pu3++3e~=Pu
Pu2O3 + 6H++6e-=2Pu + 3H2O
2PuO2 + 2H++2e-=Pu2O3 + H20
2Pu (OHL + 2H+ +2e~ = PuaO3 + 5H2O
PuO2 + 4H + +e~ = Pu3+ +2H2O
PuO|++e- = PuOt
PuO^+ + 2H2O+2e- = Pu(OHL
Pu4++e~=Pu3 +
PuO2+ + 4H++3e- =Pu3i"+2H2O
PuOf+ + 4H++2e- =Pu1++2H2O
2PuO8 + 2H++2e- = Pu2O6 + HaO
РиОГ + 2е- = РиОа
PuO2+ + 4H+ +e~ = Pu4+ +2H2O
Pu(OHL + 4H++e- = Pu3+ + 4H2O
PuO3 + 2H + +2e- = PuO2 + H2O
Pu2O5 + 3H2O + 2H+ +2e- =2Pu (OHL
Pu2O5-f-2H++2e-=2PuO2 + H2O
РоОГ+ЗНаО+4е-=Ро + 6ОН-
Ро2++2г- = Ро
PoO2 + 4H+-|-2e- = Po2++2H2O
Pr3++3e- = Pr
Pr2O3+6H++6e- =2Pr+3H2O
396
Продолжение прилож. VI
Стандарт-
Стандартный
элект-
родный по-
потенциал
+о
+0
¦ +о
+о
+о
+0
+1
+1
+2
2
j
—0
—0
+о
+о
+0
+0
+1
+1
+1
+1
+1
+1
+1
+1,
+1.
+1,
—о,
+о,
+о,
—2,
— 1,
, Ь
,15
,393
,468
,58
,73
,980
,045
,188
,000
,031
592'
455
135
862
928
935
967
017
042
062
095
157
182
325
485
588
908
5
65
8
462
829
Название и
символ элемента
Празеодим Рг
Прометий Рт
Протактиний
Ра
Радий Ra
Рений Re
Родий Rh
Ртуть Hg
Электродный процесс
2РгО2 + 2Н++2е- = Рг2О3+Н2О
РгО2 + Н2О + Н++е- = Рг(ОНK
Pm3++3e- = Pm
Pm(OHK + 3H + +3e- = Pm+3H2O
РаО2++4Н + +5е- = Ра+2Н2О
Ra2++2e-=Ra
ReO8 + 4H++e-=Re3++2H2O
Re3++3e- = Re
ReO4~ + 4H++&?- = ReO2 + 2H2O
ReO4~ + 8H++3e- = Re3++4H2O
[RhCl,]3- + 3e~ = Rh +6CJ-
Rh2Os + 6H++6e- = 2Rh + 3H2O
RhO2++2H + +e- = Rh3++H2O
RhOr+6H+ +2e- = RhO2+ +3H2O
Hg+H++e-=HgH
HgS + 2e- = Hg + S2-
[Hg (CNL]2- +2e = Hg + 4CN-
Hg2I2 + 2e-=2Hg + 2I-
HgI4 + 4e- = Hg + 4I-
Hg2Br2 + 2e~ =2Hg+2Br~
[HgBr4]2-+2e- = Hg+4Br-
Hg2Cl2 + 2e- =2Hg+2Cl-
[HgCl4]2-+2e- = Hg + 4Cl-
Hg2SO4+2e" = 2Hg + SOr
Hg22++2e-=2Hg
Hg2++2e- = Hg
2Hg2++2e-=Hg^+
Стаидарт-
ный элект-
электродный по-
потенциал
Ф°, В
+0,863
+ 1,431
—2,423
—2,008
—1,0
—2,92
+0,157
+0,300
+0,510
+0,795
+0,44
+0,87
+ 1.4
+ 1,46
—2,281
—0,75
—0,37
—0,041
—0,04
+0,140
+0,21
+0,267
+0,48
+0,615
+0,789
+0,850
+0,920
397
Продолжение п
Название н
символ элемента
Ртуть Hg
Рубидий Rb
Рутений Ru
Самарий Sm
Свинец Pb
Селен Se
Электродный процесс
HgO+2H++2e-=Hg+H2O
Hg (OHJ+2H+ +2е~ = Hg+2H2O
Rb + +e-^Rb
RuOr+!HfRuOr=RU + 4OH"
Sm3++3e-=Sm
Sms++e-=Sm?+
Pb+2H++2e- = PbH2
PbIa+2e-=Pb + 2I-
PbSO4+2e- =Pb+S04~
PbBr2+2e-=Pb+2Br-
Pb?++2e- = Pb
PbO+2H+-f-2e- = Pb + H2O
Pb (OHJ + 2H+ -j-2e~ = Pb+2H2O
HPbOa" +3H+ +2e~ = Pb+2H2O
Pb3O4+2H++2e-=3PbO + H2O
PbO2+4H+ +2e~ = Pb2+ +2H2O
PbOa +SO|~ +4H+ +2e- = PbSO4+2HaO
РЬОз~+4Н+ +2е- = PbO+2H2O
ЗРЬОз" + ЮН ++4e-= Pb3O4+5H2O
Se+2H++2e-=HsSe
398
VI
Продолжение прилож. VI
Стандарт-
Стандартный
элект-
родный по-
потенциал
Ф
+с
+ 1
—2
—0
+0
+о
+о
—3
—1
1
—0
—0
—0
—0
—0
—0
—0
+о
+0
+0
+0
+1
+1.
+1,
+1,
+1,
+2,
+2,
—0,
-о,
, В
,926
,034
,925
,04
,6
,68
,79
,121
,000
,507
,98
,506
,365
356
280
268
126
248
277
702
972
127
449
547
685
694
001
515
92
40
Название и
символ элемента
Селен Se
Серебро Ag
Скандий Sc
Стронций Sr
Сурьма Sb
Таллий Т1
Электродный процесс
SeO3~ +3H2O+2e~ =Se + 6OH~
SeO4~ + Н2О + 2е~ =SeO3~ + 2OH~
H2Se03 + 4H++4e- =Se + 3H2O
Se2Cl2 + 2e-=2Se + 2Cl-
SeOr+4H++2e-=H2SeO3+2H2O
AgBrO3+e- = Ag + BrO3
AgNO2+e- = NOi"
AgCH3COO+e- = Ag + CH8COO-
Ag+_|_e- = Ag
Ag2O + 2H+ +2e- = 2Ag + H2O
2AgO+2H + +2e- = Ag2O+H2O
Ag2O3 + 2H++2e-=2AgO + H2O
Ag2O3 + 6H+ +4e- =2Ag+ +3H2O
AgO++2H++2e- = Ag+ + H2O
AgO- +2H+ +e~ = Ag+H2O
Sc3++3e- = Sc
Sc(OHK + 3H++3e-=Sc + 3H2O
Sc2O3 + 6H + + 6e - = 2Sc + 3H 2O
Sr2++2e-=Sr
SbSt + 2e - = SbS2~ + 2S2 -
Sb+3H+ + 3e-=SbH3
Sb2O3+6H++6e-=2Sb+3H2O
SbO+ + 2H+ +3e~ = Sb + H2O
SbO2" + 4H + +3e-=Sb + 2H2O
Sb2O5 + 4H++4e-=Sb2O3 + 2H2O
5ЬО?+2Н++2е-=5ЬО++Н2О
Tl2S+2e- = 2Tl + S?~
Стандарт-
ный элект-
электродный ПО-
Тру || *? rt J\
Ф°, В
—0,366
+0,05
+0,741
+ 1,1
+1,15
+0,55
+0,562
+0,643
+0,799
+ 1,173
+1,398
+1,569
+ 1,670
+ 1,998
+2,220
—2,077
—1,784
—1,591
—2,888
—0,6
—0,510
+0,152
+0,212
+0,353
+0,446
+0,671
+0,720
—0,93
—0,753
399
Продолжение прилож. VI
Название и
символ элемента
Таллий Т1
Тантал Та
Теллур Те
Сера S
Электродный процесс
Т1Вг+е- = Т1 + Вг-
Т1С1+е~=Т1 + С1-
Т1++е~ = Т1
Т1 (ОНK + 2е- = Т1ОН + 2ОН-
Т1ОН+Н + +е- = Т1 + Н2О
Т1»++2в- = Т1 +
Та2С,+ .0Н+ + 10е-=2Та + 5Н2О
Те + 2е- = Те2-
2Те + 2е- = ТеГ
Те + 2Н + +2г~ = Н2Те
ТеОГ + ЗН2О + 4е- = Те + 6ОН-
ТеО2 + 4Н++4е-=Те + 2Н2О
ТеООН+ + ЗН++4е-= Те + 2Н2О
Те4++4е- =Те
TeOl~ + 2H+ +2е~ = ТеОз~ + Н2О
НлТеО +2Н++ 2е~ = ТеО2 + 4Н2О
SO24- + H2O + 2e-=SOr + 2OH-
Sr + 2e- = 2S2-
2S3^ + 2e-=3S2~
S + 2e~ =S2-
3Sr+2e-=4Si~
2S + 2e- =S2~
S + H++2e-=HS-
S2Or + 6H++8e-=2S2"+3H2O
S2- + 5H++8e- = 5HS-
S2O|" + 2e-=2SOr
S2" + 4H++6e-=4HS-
Sr + 3H++4e = 3HS-
SO4~+8H + +8e- =S?-+4H2O
S + 2H++2e- = H2S
Стандарт-
Стандартный элект-
электродный ПО*
тенцнал
Ф°, В
—0,658
—0,557
—0,336
—0,05
+0,778
+ 1,252
—0,750
— 1,14
—0,84
—0,72
—0,57
+0,529
+0,52
+0,559
+0,568
+0,892
+ 1,02
—0,93
—0,524
—0,506
—0,48
—0,478
—0,476
—0,065
—0,006
+0,003
+0,026
+0,033
+0,097
+0,149
+0,171
400
Продолжение прилож. VI
Название н
символ элемента
Сера S
Серебро Ag
Тербий ТЬ
Электродный процесс
SO2" + 4Н + +2е- = H2SO3 + H2O
SOr + 6H++6e-=S2-+3H2O
HSO4"" + 9H++8e-=H2S+4H2O
S2- + 2H++2e- =2HS~
SO^ + 10H+ +8e- = H2S + 4H2O
SOj- + 10H + + 8e- = H2S + 4H2O
SOl"+8H++6e- = S + 4H2O
_2SO8 + 4H+ +2e~ =S2O4~ + 2H2O
H2SO3 + 4H++4e-=S+3H2O
SO2 + 4H+ +4e- =S+2H2O
2SOl" + 6H+ +4e- =S2Ol"+3H2O
SO + 2H++2e-=S+H2O
S2O82"+2e-=2SO2-
Ag2S+2e.=2Ag+s2_
[Ag(CNJ]-+e-=Ag+2CN-
Agl_|_e- = Ag+-1-
AgCN+e- =Ag + CN-
[Ag (S2O3J]3- +e- = Ag+ 2S2O2"
AgBr+e-=Ag+Br-
AgSCN + e- = Ag + SCN~
Ag4 [Fe (CN)e] +4e- =4Ag + [Fe (CN)e]4~
AgCl+e~ = Ag + Cl~
AgNs+e- = Ag + Ni"
AgIO8+e- = Ag+IOr
[Ag (NH8J]+ +e~ = Ag + 2NH,s
AgCNS + e- = Ag+CNS~
[Ag(SO3J]3-+e-=Ag + 2SOr
Ag2Cr04 + 2e - = 2Ag+СгОГ
Ag2C2O4+2e- =2Ag+C2O|-
Tb8+ + 3e_ = Tb
Tb (OHK+3H+ +3e- =Tb+3H2O
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
+0,17
+0,231
+0,289
+0,298
+0,303
+0,311
+0,357
+0,416
+0,449
+0,451
+0,705
+ 1,507
+2,010
—0,69
—0,31
—0,152
—0,017
+0,01
+0,03
+0,09
+0,194
+0,222
+0,292
+0,35
+0,378
+0,41
+0,43
+0,446
+0,472
—2,391
—1,999
14 № 705
401
Название и
символ элемента
Технеций Тс
Титан Ti
Торий Th
Тулнй Tu
Углерод С
Продолжение
Электродный процесс
ТсО2 + 4Н++2<?- = Тс2++2Н2О
ТсО2 + 4Н++4<?- = Тс+2Н2О
Тс2++2е~ = Тс
ТсО4~ + 8Н+ +5в~ = Тс2+ + 4Н2О
ТсО7 + 2Н++е-=ТсО3 + Н2О
ТсО4" + 4Н++Зе- = ТсО2 + 2Н2О
ТсОя + 2Н++2е-=ТсО2 + Н2О
Ti2++2<?- = Ti
TiO + 2H++2e- = Ti + H2O
TiFr+4e-=Ti + 6F-
TiO2 + 4H+ +e- = Ti3+ +2H2O
TiO2 + 4H+ +2e~ = Ti2+ +2H2O
Ti2O8+6H+ +2e~ = 2Ti2+ +3H2O
Ti3++e- = Tia+
TiO2++2H + 2e- = Ti2++H2O
TiO2+ +2H+ +e~ = Ti3+ +H2O
T\O\+ +2H+ +2e- = TiO2+ +H2O
TiO^+ + 2e- = TiO2
Th»++4e- = Th
ThO2 + 4H+ +4e- = Th + 2H2O
Th(OHL + 4H+ + 4e- = Th + 4H2O
Tu3++3e- = Tu
Tu(OH).,+3H++3e- = Tu + 3H2O
2H2CO3 + 2H++2e-=H2C2O4+2H2O
H2CO8+2H + +2e-=HCO2H + H2O
C+4H++4e-=CH4
H2CO3+4H++4e- = HCOH + 2H2O
С2ОГ +2Н+ +2e- =2HCOi"
HaCO3+6H+ +fe- =CH3OH + 2H2O
HCO2H + 4H+ + ie~ = CH3OH + H2O
прилож. VI
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
+0,144
+0,272
+0,400
+0,500
+0,700
+0,738
+0,757
—1,630
—1,306
— 1,19
-0,666
—0,502
—0,478
—0,368
—0,135
+0,100
—1,800
+2,182
—1,809
—1,789
—1,650
—2,278
—1,913
—0,386
—0,156
—0,132
—0,050
+0,013
+0,044
+0,145
402
г
Продолжение прилож. VI
Название и
символ элемента
Углерод С
Уран U
Электродный процесс
НСО2~+ЗН++2е-=НСОН + Н2О
COl~+6H+ + 4e-=HCON+2H2O
НСОа" + 5Н + + 4е~ = СН3ОН + НаО
СО2 + 4Н++4е-=С+2Н2О
СО^-+8Н++&?- = СН3ОН+2Н2О
СО!~+ЗН + +2е- = НСС>2 + Н2О
Н2СО3 + 4Н++4е-=С + ЗН2О
НСООН + 2Н++2е-=СН3ОН
2COl" + 4H + +2e-=C2O4~+2H2O
Со!~ + 6Н++4е-=С + ЗН2О
СО + 6Н++6е-=СН4+Н2О
СО+2Н++2е-=С + Н2О
U8++3e-=U
2UO2+2H++2e-=U2O3+H2O
U2O3 + 2H++2e-=2UO+H2O
UO2 + 4H++4e~ = U + 2H2O
UO + 2H++2e-=U + H2O
U (OHL + 4H+ +4e- = U + 4H2O
U2O3 + 6H++6e-=2U+3H2O
Ua++3H++6e- = UH3
UO2 + 7H+ +7e- = UH3+2H2O
U (OHL + 7H+ +7e~ = UH3 + 4H2O
и!о8~+Т2Н^+12е- = 2иН3+ЗН2О
UOH3-+H + +e- = U8++H2O
иО+5Н++5е-=иНз + Н2О
Зи05++2Н20+2й- =U3O8 + 4H +
UO2+4H+ +e- = U3+ +2H2O
U (OHL + 4H + +e~ =U3+ +2HaO
3UO3-H2O+2H+ +2e~ = U3O8+2H2O
UOl+ + 2HaO+2e-=U(OHL
UO2++e- = UO2"
UO3-H2O+2H++2e-=UO2+2H2O
UO3 + H2O + 2H++2e- =U(OHL
Стандарт-
Стандартный элект-«
родный по-
потенциал
Ф°, В
+0,167
+0,197
+0,199
+0,207
+0,209
+0,227
+0,228
+0,232
+0,441
+0,475
+0,497
+0,518
—1,798
—1,738
—1,163
—1,444
—1,438
—1,375
—1,353
—1,346
—0,772
—0,716
—0,664
—0,607
—0,545
—0,538
—0,422
—0,403
—0,382
—0,019
+0,038
+0,040
+0,052
+0,368
+0,475
14*
403
Продолжение прилож. VI
Название в
символ элемента
Уран U
Фосфор Р
Фтор F
Хлор С1
Электродный процесс
U30e+4H++4e-=3UO2+2H20
"UO2H+3H++e- = UOHs++H2O
UO2++4H++e- = U4++2H2O
UO3+2H++2e- = UO2 + H2O
3UO3+2H++2e-=U3Oe + H2O
Н2РОг+е- = Р + 2ОН-
НРО|~ + 2НаО+2е- = Н2РО2~ + ЗОН-
Н2РО7 + ЗН2О + 4е = РН3+5ОН-
РО43~ + 2Н2О+2е = НРО|- + ЗОН-
2Н3РО4 + 2Н++2е- = Н4Р2О„ + 2НаО
Н3РО2 + Н++е-=2Н2О
Н3РОз + ЗН+ +3е~ = Р+ЗН2О
Н3РО4+5Н + +5е- = Р + 4Н2О
Н3РО4 + 5Н + +5е~ = Р + 4Н2О
Н3РО4+2Н+ +2е~ = Н3РО3 + Н2О
Р + ЗН + + Зе- = РН3
Н4Р2Ов+2Н + + 2е- = 2Н3РО3
F2O+2H+ +4e- - 2Fe~ +Н2О
F2+2e-=2F~
F2 + 2H++2e-=2HF
СЮ3" + ЗН2О+6е-=С1-+6ОН-
С12+2Н++2е-=2НС1
СЮГ+2Н++2е-=СЮ3~ + Н2О
С12О + 4Н + +4е- = 2НС1 + Н2О
С12+2е-=2С1-
2CIO4~ + 16H+ + 14e-=Cl2+8H2O
С1ОГ+8Н++8е- = С1- + 4На0
ClOa+5H + +6e- = HCl+2H20
2С1ОГ+12Н+ + 10е- =С12+6Н2О
НСЮ+Н + +2в-=С1 + Н2О
С1О2+4Н++5е-=С1-+2На0
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
+0,533
+0,546
+0,612
+0,657
+0,904
—2,05
—1,57
— 1,18
— 1,12
—0,94
—0,51
—0,502
—0,411
—0,383
—0,276
+0,06
+0,38
+2,1
+2,87
+3,06
+0,63
+0,987
+1,189
+1,351
+1,359
+1,385
+1,389
+1,436
+1,470
+1,494
+1,511
404
Продолокение прилож. VI
Название н
символ элемента
Хлор С1
Хром Сг
Цезнй Cs
Церий Се
| Электродный процесс
2СЮ2+8Н++8е- = С12 + 4Н2О
2СЮ2+8Н++8е-=С12 + 4Н2О
НС1О2 + ЗН++4е- = С1-+2Н2О
2НС1О + 2Н++2е-=С12+2Н2О
2НС1О2 + 6Н+ +6е~ =С12 + 4Н2О
2НСЮ + 2Н+ + 2е" = С12 + 2Н2О
2НС1О2 +6Н+ +6е~ = С12 + 4Н2О
С12О + 2Н + +2е- =С12 + Н2О
С12О + 2Н+ + 2е- = С12 + Н2О
С12О + 2Н++4е-=2С1-+Н2О
Сг2++2е-=Сг
Сг3++Зе-=Сг
Сг(ОНK+ЗН++Зе-=Сг+ЗН2О
СЮ + 2Н+ +2е~ =Сг + Н2О
Сг3++е-=Сг2 +
СЮ2~ + 4Н++Зв-=Сг + 2Н2О
Сг2ОГ + 14Н+ + 12е- =2Сг3+ +7Н2О
Н2Сг04+6Н+ +6е- =Сг + 4Н2О
СгОГ + 2Н++Зе-=СгО1~ + Н2О
CrOl" +8H + +6е- = Сг + 4Н2О
СгОз~+6Н + +Зе-=Сг + ЗН2О
СгОГ + 4Н++Зе-=СгО2+2Н2О
СгО.Г + 4Н++е~=Сг2++2Н2О
Сг2О,~ + 14Н++6е- =2Сг3++7Н2О
Н2Сг04+6Н + + Зе ~ = Сг3 + + 4Н2О
СгОГ+4Н++2е-=СЮ2 + 2Н2О
СЮГ+8Н++Зе-=Сг3++4Н2О
СЮ2 + 4Н++е-=Сг3++2Н2О
Cs++e-==Cs
Се8++Зе-=Се
Се (ОНJ2+ +2Н+ +е- =Се3+ +2Н2О
Стандарт-
Стандартный элект-
электродный по-
потенциал
<Г°. В
+ 1,540
+ 1,549
+1,570
+1,594
+ 1,628
+1,630
+1,640
+1,879
+ 1,714
+2,152
—0,913
—0,744
—0,654
—0,588
—0,407
+0,213
+0,294
+0,295
+0,359
+0,366
+0,374
+0,945
+ 1,188
+1,333
+1,335
+1,437
+1,477
+ 1,556
—2,925
—2,483
—1,731
405
Продолжение прилож. VI
Название и
символ элемента
Цннк Zn
Цирконий Zr
Эрбий Ег
Электродный процесс
ZnS+2<?-=Zn+S2-
[Zn (CNL]2~ +2e~ = Zn + 4CN-
ZnCO3 + 2e-=Zn + CO|~
[Zn (NH8L]2+ +2e- =Zn + 4NH3
Zn2++2e-=Zn
Zn (OHJ + 2H + + 2e-=Zn+2H2O
Zn (OHJ+2e~ =Zn+2H2O
HZnO7+3H++2e-=Zn + 2H2O
ZnO2~ + 4H+ +2e~ =Zn + 2H2O
ZrO22+ +2H+ +4e- =Zr + H2O
ZrO2 + 4H++4e~=Zr + 2H2O
Zr4++4e-=Zr
HZrOi"+5H++4e-=Zr + 3H2O
Er3++3e- = Er
Стандарт-
Стандартный элект-
электродный по-
потенциал
Ф°, В
—1,44
— 1,26
— 1,06
— 1,04
—0,763
—0,439
—0,400
+0,054
+0,441
—1,570
—1,553
—1,539
—1,276
—2,296
VII. Перенапряжение водорода и кислорода, В
Электрод
Pt (черненая)
Аи
Fe
Pt (гладкая)
Ag
Ni
Си
Cd
Zn
Hg
ДфПНП водорода
0,03
0,06
0,1
0,3
0,3
0,3
0,5
0,6
0,7
0,9
Афпнп кислорода
0,3
0,5
0,3
0,5
0,04
¦ 0,05
0,4
_.
406
VIII. Типы диаграмм одно-, двух- и трехкомпонентных систем
t,°C
120
110
100
90
HNO3—Н2О
/
7
1 j
у
г
У
с
Ч
S
\
\
N
у
\
V
|
Пар
0 20 40 60 80 100
Мол. доли, °/oHN03
t,°C
t,°C
75
70
65
60
ecu—с2н5он
a
\
\
4
\
s,
4
\
r
/
)
//
/
i
7
800
700
600
500
4П0
a
ч
ч^
Ч
AgCI—NaCI
Ч, '
\
ч
ч,
ч
V
S
ч
s
¦«»
b
t,cc
320
280
240
200
160
0 20 40 60 80 100 0 20 40 60 80 100
Мол. доли, %СС14 Мол. доли, %AgCl
C6H5NH2—H2O
t,°C
160
KNO,—>IaNO3 140
120
Ю0
80
60
4Q
О 20 40 60 80 100 0 20 40 60 80 100
Мол. доли, %KNO3 Mac, доли, %C6H5NH2
a
\S
\
V
4
\
\
^1
1
1
s
s
/
/
/
/
—»
\
ft
/
/
/•
\
/
/
v
\
\
t,°C
1000
900
CaCl2—CsCI
CuCl—KCl
800
a
700
600
\
0
/
1
/
/
с
\
\
\
\
I
J
20 40 60 80
Мол, доли
f, %CsCl
t,°c
700
600
500
400
300
b
200
100 ,™
<
s
\
e
V
\
I
V
\(
с
I
/
f
b
/
20 40 60 80 100
Мол, доли, %CuCl
T12SO4—T12(NO3J—T12C1,
T12SO4
TI2(NOjJ
TI2C12
IX. Криоскопические и эбуллиоскопические коистаиты
Вещество
Анилин
Бензойная кис-
кислота
Бензол
Дифенил
Дифениламин
Диэтиловый эфир
Камфора
Нафталин
Нитробензол
Пиридин
Метиловый спирт
Фенол
Хлороформ
Этиловый спирт
Четыреххло-
ристый углерод
Вода
Формула
СвН6Н2
С6Н5СООН
СвНв
(С6Н5J
(CeH5JNH
(С2Н5J О
CioHi60
СюН8
C6HfiNO2
C5H5N
СН3ОН
с6н5он
СНС13
С2Н5ОН
СС1,
н2о
<пл. °С
—5,96
122,05
5,478
71
50,2
— 117
178,3
80,1
5,82
—40
—97,8
40
—63,2
—117
—23
0,0
?кр
5,87
8,79
5,08
8,0
8,6
1,79
39,8
6,89
8,1
4,97
—
7,27
4,90
29,8
1,86
'кип- °С
184,3
250,0
80,2
254,9
302,0
34,60
204
218
210,9
115,8
67,0
182,1
61,1
77,4
76,7
100,0
3,69
.
2,57
7,06
—
2,12
6,09
5,80
5,27
2,69
0,84
3,60
3,63
1,04
5,3
0,51
X. Цветные индикаторы
Индикатор
Тимоловый голубой
Р-Динитрофенол
Метиловый оранже-
оранжевый
Бромфеноловый го-
голубой
а-Динитрофенол
Бромкрезоловый зе-
зеленый
Метиловый красный
у-Динитрофенол
Бромкрезоловый
пурпурный
Бромтимоловый
голубой
п-Нитрофенол
Феноловый красный
Крезоловый красный
л-Нитрофенол
Тимоловый голубой
Фенолфталеин
1,51
3,69
3,7
3,98
4,06
4,67
5,1
5,2
6,3
7,0
7,1
7,9
8,3
8,35
8,9
9,4
Область
перехода
рН
1,2—2,8
2,2—4,0
3,1—4,4
3,0—4,6
2,8—4,5
3,8—5,4
4,2—6,3
4,0—5,5
5,2—6,8
6,0—7,6
5,6-7,6
6,8—8,4
7,2—8,8
6,7—8,4
8,0—9,6
8,3—10,0
Окраска
Красная
Бесцветная
Красная
Желтая
Бесцветная
Желтая
Красная
Бесцветная
Желтая
»
Бесцветная
Желтая
»
Бесцветная
Желтая
Бесцветная
Желтая
»
»
Голубая
Желтая
Голубая
Желтая
»
Пурпурная
Голубая
Желтая
Красная
»
Желтая
Голубая
Красная
409
XI. Константы электролитической диссоциации слабых
кислот и оснований в водных растворах при 25 °С
Названне
Кислоты:
азотистая
азотистоводородиая
метаалюминиевая
метаборная
ортоборная
бромиоватая
бромноватистая
бромистоводородная
германиевая
йодноватая
селенистая
селенистоводородная
серная
сернистая
сероводородная
сурьмяная
метасурьмянистая
теллуристая
теллуристоводородная
теллуровая
угольная
ортофосфористая
уксусная
муравьиная
фтористоводородная
хлорноватистая
кремневая
мышьяковая
ортомышьяковистая
мета мы шьяковистая
Формула
HNO2
HNO3
НАЮ2
HBO*
Н8ВО3
НВгО3
НВгО
НВг
H2Ge03
ню3
HsSeO3
H2Se
H2SO4
H2SO3
H2S
HjSbO*
HSbO2
H2TeOs
H2Te
HsTeO4
H2CO3
HSPOS
CH3COOH
HCOOH
HF
нею
H2Si03
HsAsO4
H3AsOs
HAsO2
Ступень
диссоциа-
диссоциации
I
II
HI
I
II
11
I
II
I
и
11
I
II
I
II
I
II
I
I
II
I
I
II
I
11
I
II
I
II
I
II
III
I
II
К
4-10-4
2,6-10-5
6-10-"
7,5-10-1°
5,8-10-1°
5.8-10-13
1,6-10-1*
2-10-1
2,1-10-»
1,0-10+»
1,7-10-»
1,7-lO-i
3,5-lO-3
50-10-8
1,7-10-4
1 .in-11
1 ' IV
1-103
1,2-10-2
1,6-10-2
6,3-10-8
6.10-8
1. Ю-"
4.IO-5
1-10-13
3-10-3
2-10-8
l-10-з
2,3.10-8
6,5-10-12
4,4-10-'
4,7-10-11
1,6-10-3
6,3-10-'
1,75-10-*
1,77-10-4
6,6-10-4
3,2.10-8
2,2.10-1°
1,6.10-"
5,98- Ю-3
6-10-'
3,89-10-"
6-10-1°
1.7-10-14
6-10-1°
410
I
Название
оловянистая
оловянная
роданистоводородная
свинцовистая
фосфорноватистая
Гидроксиды:
аммония
алюминия
галлия
железа (II)
железа (III)
кадмия
кобальта
лития
магния
марганца
меди
никеля
свинца
серебра
хрома
цинка
Продолжение
Формула
H2Sn02
H2Sn03
HSCN
H2Pb02
H3PO2
NH4OH
Al (OH),
Ga (OHK
Fe (OHJ
Fe (OH),
Cd (OHJ
Co (OHJ
Li (OH)
Mg (OH),
Mn (OHJ
Cu (OHJ
Ni (OHJ
Pb (OHJ
AgOH
Cr (OH),
Zn (OHJ
Ступень
диссоциа-
диссоциации
1
I
I
III
II
III
11
II
III
II
11
II
II
II
11
I
III
II
прилож. XI
К
6- Ю-18
4-I0-"
1,4-10-*
2-10-16
7,9-10-2
1,79-10-5
1,38-10-e
l,6-10-ii
4-10-12
1,3-10-4
1,8-10-и
1,3-10-12
5-Ю-3
4-10-5.
6,7-10-1
2,5-10-з
5-10-4
3,4-Ю-7
2,5-10-5
9,6-10-4
1,1-10-4
1-10-ю
4-10-5
XII. Значения общих коистаит нестойкости
комплексных иоиов в водных растворах
Комплексный
ион
Ag(NH3)a+
Cd (NH3)!+
Cd (NH8J6+
Cu(NH3)l+
AgBra-
AgBrJ"
CdBi-Г
CuBr-T
Темпе-
Температура,
"С
30
30
30
30
25
25
25
20
к
Комплексный
ион
Комплексы с аммиаком
9,3-Ю-8
ю-'
7,3-10-»
2,14.10-13
Бромидные
7,8-Ю-8
1,3-10-»
2-10-4
1,3.10-»
Hg (NH3)f+
Mg (NH3)S+
Ni (NH3)|+
Zn (NH3)I+
комплексы
HgBrl"
PbBr!~
TiBr4
ZnBri"
Темпе-
Температура,
°C
22
22
30
30
25
25
25
25
к
5,3-10-20
1930,0
1,86-10-»
2,5-10-ю
Ы0-21
ЬЮ-3
1,3.10-2*
50
411
Продолжение прилож. XII
Комплексный
ион
Темпе-
Температура,
"С
Комплексный
ион
Темпе-
Температура,
°С
Agl!"
Bill"
CdlJ"
Ag (SCNJ-
Ag (SCNL~
Au (SCN)i~
Ag (S2O3)i~
Cd (S2O3)*~
Cu (S2O3I-
AgCla~
AgCl43"
AuCli"
BiCIe"
CdCl!"
Ag(CN)^
Au (CN)j"
Fe (CN)«-
Fe (CN)?"
25
20
25
Иодидные
1.8.10-»!
3,1.10-12
8.10-' 1
Роданидные
25
25
25
2,7.Ю-8
8,3.10-и
1,0-10-12
комплексы
Cdl!"
Hgir
рыГ
комплексы
Cd (SCN)I-
Hg (SCN)I"
Ni (SCNK-
Тиосульфатные комплексы
25
25
25
2,5.10-м
3,6.10-'
6,0-10-w
Хлоридные
25
25
18
18
18
1,76.10-5
1,2.10-*
5,0.10-22
3,8.10-'
9,3-10-3
Цианидные
18
25
25
25
1,0.10-21
5,0.Ю-39
1,0.10-2*
1,0.10-31
Hg (S2O3J2"
Hg (SSOS)|-
Hg (S2Os)l~
комплексы
СиС1Г
HgCl32-
HgClI"
рьаГ
PtCl!"
комплексы
Hg (CN)i"
Ni (CN>r
Zn (CN)J-
Cd (CNI~
25
25
25
30
25
20
25
25
25
18,0
25
25
25
25
25
25
20
20
1,0.10-"
1.5.10-3»
1,4.10-*
1,7-10-2
5,9-10-22
1,5-10-2
3,6-10-3»
1,3-10-32
5,8-10-34
5,0.10-e
8,5-10-15
8,5-Ю-"
7.Ы0-3
l.O.lO-i"
4,0-10-12
1,8-10-"
l,3-10-i'
1,4-Ю-"
XIII. Растворимость (S) и произведение
растворимости (L) трудиорастворимых веществ
при 25° С
Кристалли-
Кристаллическая фаза
AgBr
AgCl
Agl
AgCN
Ag2Cr04
•S,
г/lOOr H2O
1,89-10-5
1,8-10-1
2,3-10-'
1,1.10-'
3,3-10-?
1,1
1,56
9,7
7,5
4,10
h
• Ю-"
• lO-io
• 10-1'
¦ Ю-"
•10-12
Кристалли-
Кристаллическая фаза
Mg(OHJ
MnS
Mn (OHJ
Ni (OH),
NiCO3
s,
г/ЮОг H2O
1
2
4
,15.10-8
,96-Ю-з
,3-10-s
3,07-
3,0
5-
1,3
1,3
L,
10-n
Ю-15
0-u
10-ie
• 10-'
412
Продолжение прилож. XIII
Кристалли-
Кристаллическая фаза
Ag2SO4
Ag3PO3
BaCrO4
BaSO4
ВаСО3
СаСО3
CaSO4
CdS
Cd (OH)a
Со (ОНJ
Сг (ОНK
CuS
Fe (OHJ
FeS
Hg2Cl2
Hg2SO4
MgCO3
s,
r/100 г HjO
1,
3
2
1
9
2
6
I
6
4
3
0,84
14-10-*
,9-10-*
,4-10-*
,8-10-3
,8-10-*
0,1
,6-10-4
,8-Ю-6
,3-10-7
,2-10-6
__
,5-10-s
,9-10-2
0,12
7,7
1,46-
2,4
1,08
8,1
9,3
6,1
1,2-
2,3-
1,6-
6,7-
4,0-
1,65-
К
3,5-
:6,2
'2-
10-5
10-21
lO-i»
lO-i»
• m-9
• 10-5
20~28
lO-i*
Ю-"
10-"
J0-S8
Ю-15
)-19
Ю-18
¦ 10-'
ю-*
I
Кристалли-
Кристаллическая фаза
РЬВг2
РЬС12
РЫ2
Pb (OHJ
PbSO4
PbCrO4
Sn (OHJ
SrCO3
SrSO4
TlBr
TIC1
Til
Th (OHL
Zn (OHJ
a = ZnS
ZnCO3
P = ZnS
•S,
г/100 г Н,О
0,49
0,48
6,2-10-2
1,5-10-*-
3,8-10-8
4,3- 10-e
3,3-10-8
6,0-10-*
5,6-10-2
0,33
8,4-I0-3
—
1,4-IO-S
ю-*
—
L
9,2-10-"
2,12-Ю-6
9,8-Ю-9
1,0.10-1»
1,6-Ю-8
1,8-Ю-1*
5,0-10-2«
1,6-10-°
3,5-10-7
3,9- 10-е
1,9-10-*
6,3-Ю"8
Ю-60
1,3-10-1'
7,4-10-2'
6-10-и
4,5-Ю-24
XIV. Кинетические константы гомогенных реакций в газах
k — константа скорости реакции, Е — энергия активации, А
предэкспоненциальный множитель в уравнении k=Ae~El
Реакция
Е,
кДж/моль
Первый порядок
Реакции между молекулами
А. Разложение
24
С2Н4 +
—* СН2 = СНС1 + НС1
СС СН НС1
25г
С2Н6С1—
СН3СНС12
3з2 2 +
СН8СООС2Нб —* СН3СООН -f С2Н4
цикло (CHSCHO)S—ЗСНСНО
N2O5-+N2O4+i/2O2
N2O, -*¦ 2NO2
Б. Изомеризация
/п/кгнс-Дихлорэтилен —>- цис
Циклопропан —* пропилен
Винилалиловый эфир —»• аллилацеталь-
дегид
7,2-1012
4-10*
1,3-1012
3,2-Ю1»
3,2-1012
1,3.10".
4,6-1013
101»
218,0
247,5
207,8
201,0
200,5
185,5
103,5
54,4
4,9
1,5
б.
• 101а
• 104
10"
175,
272,
128,
8
8
3
413
Продолжение прилож. XIV
Реакция
A. c-«
Е,
кДж/моль
Реакции с участием атомов и радикалов
2-Ю13
„ . Зг I
С2Н61—*С2Н5 + 1
СС14 —3 +
СН8С1-+СН3
2СН
Реакции меокду молекулами
СН8СН = СНСН3 + НВг-
—t- CHsCH2CHBrCH3
—*-С2Нв
Hl+CHSI
2HI -
2NO2—*2NO+O2
1,6-101°
4-1013
1,6.10х*
2-10i*
9,2.10i3
9,4-Ю1?
Второй порядок
Реакции с участием атомов и радикалов
356,2
356,2
354,0
225,2
216,0
94,2
180,5
165,5
140,0
186,4
112,6
Вг + СН4-
Вг+С2Н6-
Вг + Н2—*
С1+СН4-
С1 + С2Н6-
С1 + Н,—
н+сн4 —
н+с2н2-
Н + С2Н4-
н+с2н„-
Н+СС14 —
Н + СгН,С1
Н + НВг—,
Na+CH3CI
NO+C12 —
ОН + Н2 —
он+со —
ОН+СН4-
ОН + С2Н6
¦* НВг + СНз
— HBr+CjHj
НВг + Н
*НС14-СН3
¦^HCl+CjHj
НС1 + Н
> Н24-СН3
-*с2н3
¦* с2н6
н.Н. + С.Н,
> НС1 + СС13
—*НС!-4-С2Н5
> На+Вг
—»NaCl + CH3
s-NOCl + Ci
с Н2О+Н
*СО2+Н
¦-H2O-bCHs
-*Н»О + С2Н6
А,
СМ3>МОЛЬ "-С
5-Ю13
—
6,9- Ю13
2,5-1013
1,3-Ю»
9,5- Ю"
3,2-Ю10
2-Ю"
3,2-10"
3,2- Ю12
—
1,3-1013
4-1012
1,4-10"
1,3-10"
—
*—
76,6
58,2
74,2
16,3
4,2
23,0
27,6
75,4
17,2
28,5
14,6
33,5
4,6
42,7
85,0
41,8
29,3
36,2
23,0
414
2NO + Br2—*
2NO+Cr2 —>-
2NO+O2—+5
Третий порядок
2NOBr
2NOC1
и poo
CM«
2
4
1
олжение
A.
моль — *, с
,7-101°
,6-10°
,0-Ю3
прилож.
5
15
4
XIV
,44
,5
,7
XV. Кинетические коистаиты реакций в растворах (второй
порядок)
Реакции
сн3соосан6+он-—*
—-> сн3соо- +с2н5он
сн,соос3н7+он- —*
-*сн3соо-+с3н7он
СН3СООС4Н, + ОН- —¦*
—*CH3COO--fC4H9OH
ch2icooh+oh-—¦*
-^СН2ОНСООН + 1-
^2П5ОГ-риП >¦
—> Cji>ricOii —I— гэг~~
—^2ch2cicooh+i-
СН2С1СООН + 1-—*¦
). СН21СООН-)-С1~
СН5Вг+1- —* СН31 + Вг-
CHsBr-)-I- —* СН31 + Вг-
С6Н6СОСН2С1 + 1-—*
—*СвН5СОСН21+С1-
СН31+С2Н5О-—*
> CH3OC2H5 ~\~ I ~"
с2н51+с2н„о-—*
Лсвн62сн2ос2н6ч-^-
п-С3Н71 + С2Н5О —->
C2H5ONa+C2H5I —
Растворитель
Н2О
То же
»
С2Н6ОН
н2о
То же
»
СН3ОН
CH3COCH3
С2Н5ОН
То же
»
•
А,
см3-моль — '-с~'
1,4.101»
1,9-101°
2,1-iO1»
6,3-10"
4,3-10"
7,9-J0"
1,3-1013
1.7.1018
2,3-Ю!3
1,0.I015
2,4.10"
1,5.10"
1,5.1013
3,5-10"
1,5-10"
Е,
кДж/моль
46,9
47,3
47,7
98,0
89,6
95,8
82,9
76,6
76,2
93,0
81,6
86,6
83,3
94,2
86,2
415
Реакции
A[CeH6(CH3KN]++P
Ь^Йнй^ + вТ^
-Л- [С6Н53(СНз)а C2H6,N]+ +1-
-^fceH63(CH3KN]+ + b
-XfC(lHll(CH8),NJ+8-fiH
СО2 + ОН--*НСОз-
Растворитель
СН2С!4
свнв
СН3СОСН3
СН3СОСН3
CeH5NO2
С2Н2С12
С6Н6СН2ОН
нао
Продолжение
А,
см»-моль~'-с
2,2-10'
2,8-10г
8,5-I03
2,7-10*
2,6-10*
2,1-10*
7,0-10"
1,5-10"
прилож. XV
-1
Е.
кДж/моль
49,0
46,9
49,0
57,4
54,4
49,0
60,2
38,2
XVI. Ионные радиусы атомов, вычисленные из межатомных расстояний (•1010м)
Пери-
Периоды
1
2
Н
+ 1
Li
+ 1
Na
+ 1
К
+ 1
29
+ 2
+ 1
I
1
0,00
3
0,68
11
0,97
19
1,33
Си
0,72
0,96
Be
+ 2
Ms
+ 2
Са
+ 2
30
+ 2
И
4
0,35
12
0,66
20
0,99
Zn
0,83
5
+ 3
13
+з
Sc
+ 3
31
+з
III
E
0
0
0
!
23
Al
51
21
81
Ga
0
62
6
+ 4
14
+ 4
Ti
+ 4
32
+ 4
+ 2
IV
С
0,16
Si
0,42
22
0,68
Ge
0,44
0,73
Группы элементов
7
+ 5
+з
15
+ 5
+ 3
V
+ 5
33
+ 5
+ 3
V
N
0,13
0,16
p
0,35
0,44
23
0,59
As
0,46
0,58
8
+ 6
— 2
16
+ 6
+ 4
-2
Cr
+ 6
+3
34
+ 6
+ 4
2
VI
0
0,09
1,32
S
0,30
0,37
1,74
24
0,52
0,63
Se
0,42
0,50
1,91
VII
9
+ 7
17
+ 7
+ 5
Mn
+ 7
+ 4
+ 2
35
+ 5
F
0,07
1,33
Cl
0,27
0,34
1,81
25
0,46
0,60
0,80
Br
0,47
1,96
VIII
Fe 26
+3 0,64
+ 2 0,74
Co 27
+3 0,63
+ 2 0,72
Ni 28
+ 2 0,69
Пери-
Периоды
5
6
Rb
+ 1
47
+ 1
Cs
+ 1
79
+3
I
1
i
1
1
0
37
,47
\s
,26
55
,67
Au
,85
Sr
+ 2
48
-f-2
Ba
+ 2
80
+ 2
II
1
0
1
1
38
,12
Cd
,97
56
,34
H?
,10
Y
+3
49
_i_3
La
+ 3
81
+ 3
+ 1
ill
39
1,06
In
0,81
57
1,14
Tl
0,95
1,47
Zr
+ 4
50
_j_4
+ 2
Hf
+ 4
82
+ 4
+ 2
IV
0
0
0
0
0
1
40
,87
Sn
,71
,93
72
,78
Pb
,84
,20
Группы элементов
Nb
+ 5
51
+ 3
Та
+5
83
+ 5
+ 3
V
41
0,69
Sb
0,62
0,76
73
0,68
Bi
0,74
0,96
Mo
+ 6
52
_i_g
+ 4
— 2
W
+ 6
84
+ 6
VI
42
0,62
Те
0,56
0,70
2,11
74
0,62
Po
0,67
VII
Tc
+ 7
53
+ 7
+ 5
-1
Re
+ 7
85
+ 7
0
0
0
2
0
0
43
,56
I
,50
,62
,20
75
,56
,62
Продолжение прилож.
Ru
+ 4
Os
+ 6
44
0,67
76
0,69
VIII
Rh
+ 3
Ir
+ 4
45
0,68
77
0,66
Pd
+ 2
Pt
+ 4
+ 2
XVI
46
0,80
78
0,65
0,80
Fr 87
+ 1 1,80
Ra 88
+ 2 1,43
Ac
+ 3
1,18
(Th)
90
1,02
(Pa) 91
+ 4 0,65
(U)
+ 6
92
0,80
XVII. Теоретически вычисленные И. Вабером и Д. Кромером ионные и атомные радиусы (•1010м)
Пери-
Периоды
1
2
3
Л
н
0 0
+1
I
1
,529
0,00
0 Li 3
0 1
+ 1
Na
0
+1
К
+0
1
29
0
+ 1
+2
,586
0,189
11
1,713
0,278
19
2,162
0,592
Си
1,191
0,312
0,308
Be
0 1
+2
Щ
0
+2
Са
0
+2
30
0
+2
п
4
,040
0,139
12
1,279
0,246
20
1,690
0,538
Zn
1,065
0,293
5
0
+3
13
0
+3
Sc
0
+з
31
0
+3
111
0
0
1
0
1
0
1
0
в
,776
,11
Al
,312
,221
21
,570
,493
Ga
,254
,276
6
0
+4
14
0
+4
Ti
0
+4
32
0
+4
IV
С
0,596
0,09
Si
1,068
0,20
22
1,477
0,456
Ge
1,090
0,26
Группы элементов
7
0
+5
15
0
+5
V
0
+3
+5
33
0
+3
+5
V
N
0,488
0,08
Р
0,919
0,18
23
1,401
0,430
0,424
As
0,982
0,826
0,247
8
0
—2
16
0
—2
Ст
0
+2
+3
34
0
2
VI
О
0,414
0,46
S
0,810
0,83
24
1,453
0,414
0,401
Se
0,918
0,92
VI
9
0 0
—1 0
17
0 0,
— 1 0
Мп
0 1
+2 0
+4 0
35
0 0
— 1 0
¦ 1
,360
,369
725
,742
25
,278
,388
,365
Вг
,851
,869
Fe
0 1
+2 0
+3 0
26
,227
,364
,355
VIII
Со
0 1
+2 0,
+3 0
27
181
343
336
Ni
0
+2
+3
28
1,139
0,325
0,319
Продолжение прилож. XVII
Пери-
Периоды
5
6
Rb
0
+1
47
0
+1
+2
Cs
0
+ 1
79
0
+ 1
+3
Fr
0
I
37
2,287
0,734
Ар
1,286
0,536
0,529
55
2,518
0,921
Аи
1,187
0,633
0,600
87
2,447
Sr
0
+2
48
0
+2
Ва
0
+2
80
0
+ 1
+2
Ra
0
II
38
1,836
0,683
Cd
1,184
0,507
56
2,060
0,866
н?
1,126
1,099
0,605
88
2,042
Y
0
+3
49
0
+3
La
0
+3
81
0
+ 1
+3
Ac
0
ill
39
1,693
0,640
In
1,382
0,481
57
1,915
0,819
Tl
1,319
1,049
0,580
89
1,895
Zr
0
+4
50
0
+2
+4
Hf
0
+4
82
0
+2
+4
Th
0
+4
IV
40
1,593
0,603
Sn
1,240
0,997
0,458
72
1,476
0,610
Pb
1,215
0,986
0,558
90
1,788
0,880
Группы элементов
Nb
0
+3
+5
51
0
+3
+5
Та
0
+5
83
0
+3
Pa
0
V
41
1,589
0,703
0,550
Sb
1,140
0,931
0,438
73
1,413
0,589
Bi
1,130
0,933
91
1,804
Mo
0
+3
+6
52
0
—2
W
0
+6
84
0
U
0
+4
+6
VI
42
1,520
0,661
0,542
Те
1,11
1,12
74
1,360
0,570
Po
1,212
92
1,775
0,843
0,811
VII
Тс
0
53
0
—1
Re
0
+2
85
0
43
1,391
I
1,044
1,065
75
1,310
0,73
At
1,146
Ru
0
+3
+4
Os
0
+4
44
1,410
0,598
0,582
76
1,266
0,655
VIII
Ph
0
+3
+4
Ir
0
+4
45
1,364
0,570
0,560
77
1,227
0,649
Pd
0
+2
+4
Pt
0
+4
46
0,567
0,553
0,536
78
1,221
0,628
XVIII. Энергия (потенциал) ионизации для разных ступеней
ионизации
6
S о.
Поря
вый
номе
1
2
3
4
5
6
7
8
9
10
11
12
13
17
18
19
20
21
35
36
37
38
39
53
54
55
56
57
86
Эле-
Элемент
Н
Не
Li
Be
в
с
N
О
F
Me
Na
Mg
Al
С!
Ar
К
Ca
Sc
Br
Kr
Rb
Sr
Y
I
Xe
Cs
Ba
La
Rn
I
/•ю-»,
Дж/моль
13,12
23,72
5,21
8,99
8,01
10,87
14,02
13,13
16,81
20,8!
4,96
7,37
5,77
12,55
15,21
4,19
5,90
6,31
11,43
13,5
4,03
5,49
6,16
10,08
11,71
3,75
5,03
5,41
10,37
/, эВ
13,59
24,58
5,39
9,32
8,30
11,26
14,53
13,61
17,42
21,56
6,14
7,64
5,98
13,01
15,76
4,31
6,11
6,54
11,84
14,0
4,18
5,69
6,38
10,45
12,13
3,89
5,21
5,61
10,75
II
/•Ю-',
Дж/моль
52,2
73,0
17,6
24,2
23,9
28,6
34,0
33,7
39,5
45,6
14,5
18,1
22,9
26,5
30,6
11,5
12,4
18,5
23,6
26,3
10,6
11,9
18,3
20,5
22,6
9,6
11,0
A9)
/, эВ
54,1
75,7
18,2
25,1
24,8
29,6
35,2
34,9
40,9
47,3
15,0
18,8
23,7
27,5
31,7
11,9
12,8
19,2
24,5
27,3
11,0
12,3
19,0
21,2
23,4
10,0
П,4
B0)
II
/•ю-8,
Дж/моль
117,5
148,5
36,6
46,2
45,7
53,0
60,5
61,7
69,2
77,4
27,5
38,5
39,3
43,9
49,2
23,9
34,4
35,5
38,3
D1)
19,7
C0)
31,0
C3)
C6)
18,4
B9)
/.эВ
121,8
153,9
37,9
47,9
47,4
54,9
62,7
63,9
71,7
80,2
28,5
39,9
40,7
45,5
51,0
24,8
35,6
36,8
39,7
D3)
20,4
C1)
32,1
C4)
C7)
19,1
C0)
1\
/¦ю-\
Дж/моль
209,0
250,2
62,2
74,7
74,7
84,2
93,0
95,4
105,5
115,8
51,6
E9)
58,5
65
71,0
48,4
E0)
E1)
55,0
F0)
D0)
D3)
D4)
D7)
E0)
D2)
1
1, эВ
216,6
259,3
64,5
77,4
77,4
87,3
96,4
198,9
109,3
120,0
53,5
F1)
60,6
67
73,6
50,2
E2)
E3)
57,0
F2)
D2)
D5)
D6)
D9)
E2)
D4)
XIX. Сродство атомов к электрону /0
Поряд-
Порядковый
номер
1
2
3
4
5
6
7
8
Эле-
Элемент
Н
Не
Li
Be
В
С
N
О
/о-К)-»,
Дж/моль
—0,721
—0,18
—0,79
—0,18
—0,32
— 1,08
—0,048
— 1,42
/0, эВ
-0,747
—0,19
—0,82
0,19
—0,33
—1,12
—0,05
— 1,47
Поряд-
Порядковый
номер
9
10
11
12
13
17
19
35
53
Эле-
Элемент
F
Ne
Na
Mg
Al
Cl
К
Br
I
/0-10-',
Дж/моль
—3,45
0,55
—0,45
0,31
—0,50
—3,63
—0,79
—3,42
-3,17
'о, эВ
—3,53
0,57
—0,47
0,32
—0,52
—3,76
—0,82
—3,54
—3,29
421
XX. Строение многоатомных молекул в газообразном состоянии
Молекула
со2
csa
NaO
NO2
нао
HaS
so2
NH3
PCl3
CC14
CH4
Форма
Линейная симметричная
O = C = O
Линейная симметричная
S = C—S
Линейная несимметричная
N = N = O
Изогнутая
с/ \>-
^ ONO 140°
Изогнутая
н/Ч
^ НОН 105°3«
Изогнутая
^ HSH 92°20«
Изогнутая
0// Ч)
2 OSO 124°
Симметричная пирамида
Симметричная пирамида
Правильный тетраэдр
Cl\r/Cl
с\/ Xci
^ при С 109°28'
Правильный тетраэдр
Hv /Н
н/ хн
^ при С 109°28«
Межъядерное
расстояние
г-10"м
1,13
1,54
N = 0, 1,22
N = N, 1,10
1,18
О —Н 0,97
Н —Н 1,53
S —Н 1,35
S = O 1,45
H-N 1,01
Н —Н 1,61
Высота пира-
пирамиды А = 0,3
CI —С1 3,1
Р—С1 2,0
С —С1 1,76
С—С1 2,99
С-Н 1,09
422
Продолжение прилож. XX
Молекула
с2н4
С2Н2
СН3С1
СНС13
Форма
Плоская
/НСН~П4°55'
Линейная симметричная
Н—С=С—Н
Тетраэдр
Н\
Н-)С—С1
н/
Тетраэдр
Ск
С|-^С-Н
Cl/
/С1СС1 112°
Межъядерное
расстоянне
л-10>» м
С —Н 1,071
С—С 1,353
С—Н 1,06
С=С 1,20
С-Н 1,1
С-С 1,77
С-Н 1,10
С —С1 1,80
С1-С1 2,93
XXI. Энергия связи (кДж/моль)
Средняя энергия связи ?0 (термохимическая энергия связи) —
величина, приписываемая каждой связи, так что сумма энергий всех
связей равна теплоте атомизации газообразной молекулы при 0 К
в основном состоянии. Длина связи г0 — среднее межъядерное рас-
расстояние при 0 К в основном состоянии. В термохимических расчетах
можно полагать Еа и ?298.
Связь
Соединения или группы
r0-1010, м
(по Полин-
гу —
Сыркину)
(по
Кот-
треллу)
(по Мель-
вин —
Хьюзу)
С-С
с=с
С-С
Многоатомные молекулы.
Парафины (алканы)
Олефины (алкены)
Ацетиленовые (алкины)
Бензольное кольцо
Альдегиды и кетоиы
1,54
1,34
1,20
1,398
1,516
262
423
759
—
,30
,25
,40
277
472
628
,0
,4
.8
331,8
587,8
823,1
487,1
350,5
423
Продолжение прил
Связь
с-н
С-Н
с-н
с-н
С —Вг
C-CI
C-F
C-I
C-N
с=о
С-0
с = о
с = о
с=о
C-S
c=s
As—H
Н—О
Н-0
Н-Р
Н—S
н-с
N —N
N —Н
М .... Г~\
N iii^ О
О —О
О-О
О— С1
O-F
s==o
s=o
Si-Cl
Si-H
Si = O
Теплота
Соединения или группы
Парафины
Олефины
Ацетиленовые
Бензольное кольцо
Галогеналкилы или
галогена рнлы
То же
»
»
Амины, нитропарафины
СО2
Спирты, простые эфиры
Кетоны
Альдегиды
Кислоты, сложные эфиры
= C-S —
CSa
AsH3
Н2О
Спирты
РН3
H2S
HCN
/* * \
NH3, амины
NOa
Нитропарафины
Н2О2
Перекиси
С|2О
FaO
so2
SO,
SiCl4
SiH4
SiO2
возгонки углерода
r0- l О10, м
1,0954
1,07 !
1,064J
1.084J
1,94
1,767
1,381
2,14
1,472
1,160
1,43
1,23
1,23
-1,2
1,554
1,523
0,958
0,96
1,42
1,334
1,066
1,47
1,008
— 1 4
U22
1,48
—
1,70
1,42
1,432
1,43
2,02
1,456
— 1,5
—
Е„.
(по Полин-
гу —
Сыркииу)
357,98
238
293
~435
180
223,8
711—753
314
653
628
1340—
1505
226
492,9
460,2
—
264
343
—
113
348,5
452
146
—
206
,
385,8
-523
Е„
(по
Кот-
треллу)
378,6
—
232,2
732
322,6
678
665
247
506
245,2
462,7
—
322,2
364
403,3
88
352,7
452
527
146
188
205
189,5
535
473
364
318
489
Б77
Двухатомные молекулы
CsO
К—Вг
К—Cl
Na—Вг
Na—Cl
424
CO
KBr
KC1
NaBr
NaCl
1,131
2,94
2,79
2,64
2,51
884,9
366,9
407,9
937
379,0
423,4
366,1
410,3
I
I
I I
1МММП!
I
о
) CO О
¦ on
I I
&
X
X
x
&
s
к
I
Я5
X
X
00
СЧООСООО<МОООО
сосо<м<мсосососососо
00
1 О I
OO ^1" CD О N C
COQOCOO СЧЮЮО
—< со т ¦*$• i спюсооо
OOOOO llOCOCOO
— •— <м<мсчсч
00 N CO CD CO (O 00
CO Tp CO CO OOOO
-и tN ^m i to go — —
¦ф-ооз Ico^cooj
<m сч <m •— сч сч <m сч
О
00О>00ОЮ
C50LOCOQO
„«-чО^^СО
N CJC
ОО
O —
О
CM CO CM 00 CM CO
о •—* —* t^ то •—<
—i ОЭ 00 t^ t^ 00
о о
Ifc I!? I
CM CM
«i
m о со ¦* s« —
cm—Г -^оосо^юслто ю
о см со — ооам-^слто о
•*-* СМ ТО ¦* —' СМ 1^ ТО h-
Рекомендуемая литература
Ахметов Н. С. Общая и неорганическая химия. М., 1981.
Бариард А. Теоретические основы неорганической химии. М., 1968.
Введение в общую химию/Под ред. Г. П. Лучинского. М., 1980.
Дей. М, Селбин Д. Теоретическая неорганическая химия. М.,
1977 г.
Зайцев О. С. Химическая термодинамика к курсу общей химии.
М., 1973.
Кдрапетьяиц М. X. Введение в теорию химических процессов.
М., 1970.
Карапетьянц М. X., Дракин С. И. Строение вещества. М., 1970.
Карапетьянц М. X., Дракин С. И. Общая и неорганическая химия
М., 1981.
Коттон Ф., Уилкинсон, Дж. Современная неорганическая химия.
М., 1970.
Краснов К. С. Молекулы и химическая связь. М., 1977.
Курс общей химии/Под ред. Н. В. Коровина. М., 1981.
Некрасов Б. В. Основы общей химии. М., 1965—1970. Т. 1—3.
Новиков Г. И. Физические методы неорганической химии. Минск,
1976.
Полинг Л. Общая химия. М., 1968.
Реми Г. Курс неорганической химии. М., 1963—1966. Т, 1, 2.
Угай Я. А. Общая химия. М., 1977,
Щука рев С. А. Неорганическая химия. М., 1970— J974. Т. 1, 2.
Яцимирский К. Б., Яцимирский В. К. Химическая связь. Киев,
1975.
Предметный указатель
Адсорбция 185
Азеотропы 101
Аккумуляторы 158, 167
Активный комплекс 55
Аммиакаты 267
Амфотерность 23
— воды 108
Ароматические соединения 259
Атомизация 47
Атомный остов 292
Валентность 20
Взаимная растворимость жид-
жидкостей 83
Водородный электрод 144
Волновая функция 204
Волны де Бройля 202
— стоячие 203
Гальванические элементы 140,
156
Гетеросоединения 19
Гетерогенно-каталитические
процессы 184
Гибридизация 252
Гидролиз солей 121
Гиромагнитное отношение 194
Гомосоединения 18
Гомогенно-каталитические про-
процессы 181
Границы периодической систе-
системы 198
Двусторонние реакции 171
— состояния 74
— состояния воды 76
Делокализация электрона 258
Деполяризация 161
Дефективные структуры 78
Джоуля — Томсона эффект 346
Диаграмма плавкости 171
Дисперсионный эффект 348
Диполь 298
Донорно-акцепторное взаимо-
взаимодействие 248
Закон Генри 82
— Гесса 42
— Дальтона 99
— действующих масс 54
— кратных отношений 15
— Кулона 10
— Максвелла—Больцмана 53
— объемных отношений газов
15
— постоянства состава 15
— разведения Оствальда 116
— Рауля криоскопический 93
— Рауля тоноскопический 97
— Рауля эбуллиоскопический
95
— сохранения заряда 10, 30
— сохранения массы, энергии
10
— стехиометрии 8
— удельных теплоемкостей 16
— Фарадея 155
— эквивалентов 13
Зонная теория 337
427
Изомерия 269
Изотонический коэффициент
Вант-Гоффа 111
Изоэлектронные молекулы 248
Ингибиторы 178
Индикаторы 129
Индукционный эффект 348
Инкогруэнтное плавление 89
Ионная изомерия 269
Ионное произведение воды 113
Интегралы кулоновские 238
— обменные 238
— перекрывания 239
Интерметаллические соединения
343
Испарение 29, 74
Истинные заряды атомов 224
Катализаторы 178
Квантовая механика 200
Квантовые числа 193
Квантовый дефект 224
— химия 235
Кварки 9
Кинетика 163
Кислоты 22
Классическая механика 200
Клатраты (гидраты газов) 111
Комплексные соединения 32,
365
Конгруэнтное плавление 89
Кондуктометрическнй анализ 92
Константа гидролиза 123
— Маделунга 327
— нестойкости 273
— скорости 164
— химического равновесия 54
Координационная изомерия 269
¦— теория Вернера 270
Координационное число 267
Координационные соединения
234
Коррозия металлов 160
Коэффициент активности 138
Кривая потенциальной энергии
240
Кривые титрования 128
Кристаллогидраты 267
Кристаллохимические формулы
320
Кристаллы 319
Лиганды 266
Ликвидуса линия 85
428
Линейная комбинация атомных
орбиталей 286
Ложное равновесие 51
Магические числа 12
Магнитное квантовое число 194
Международный электрод срав-
сравнения 144
Металлическая связь 337
Металлические валентные соеди-
соединения 345
Метод Хартри — Фока 213
Методы валентных связей 240
— молекулярных орбиталей 284
Механика квантования 200
Молекула 233
Молекулы аммиака строение 310
— воды строение 313
— метана строение 308
— фтористого водорода строе-
строение 315
— с кратными связями 245
Молекулярные моменты дипо-
диполей 301
— соединения включения 353
Молярная масса эквивалента 14
Момент диполя 107, 208
Мономолекулярные реакции 166
Направленность ковалентной
связи 249
Насыщенные растворы 81
Независимые компоненты сис-
системы 72
Нейтрализация 120
Нейтрон 189
Несолеобразующие соединения
27
Нестехиометрические кристаллы
335
Низкоспиновое комплексообра-
зование 378
Обменный интеграл 238
Ограниченная растворимость 81
Односторонние реакции 171
Окислители 33
Оксохлориды 28
Основания 22
Операторы 205
Орбиталь 207
Орбитальный момент электрона
190
Орнентационный эффект 348
Очарование 9
Парамагнетизм 245, 285
Паспорт электронной орбитали
208
Период полураспада 172
Периодическая решетка крис-
кристалла 319
Перитектика 88
Пи-комплексы 260
Пи-связь 243
Плавление 29, 75
Планетарная модель атома 190
Побочное квантовое число 193
Полупроводники 336
Порядок реакции 166
— связи 291
Потенциал ионизации 215
— поляризации 151
Правила Фаянса 330
— фаз Гиббса 71
Правило Клечковского 196
— Хунда 213
Превращения необратимые 69
— фазовые 28
— химические 29
Предэкспоненциальный коэф-
коэффициент 165
Произведение растворимости 136
Промотирование валентного со-
состояния 245
Промоторы 179
Простое вещество 18
Протон 189
Процессы восстановительного
обжига 46
— окислительного обжига 47
— смещения жидкостей 47
Работа 40
Равновесие химическое 50
Радиальная часть волновой
функции 207
Радикал 226
Радиус лтома 225
— иона 229
Разведение 116
Разрыхляющие орбитали 286,
290
Растворимость гидроксидов 133
— карбонатов 136
— нейтратов 135
— ограниченная 81
— хлоридов 134
Растворы буферные 131
— идеальные 79
Реакторы идеального смещения
176
— идеального вытеснения 177
Реакции в закрытых системах
17
— обменного разложения 31
— окислительно - восстановите-
восстановительные 32
Релятивистская механика 200
— эффект 11
Ректификация 102
Рентгено-структурный анализ
92
Решетки с дефектами 332
— соединения 353
Сандвичевые соединения 261
Связь водородная 108, 356
Связевые моменты диполей 301
— орбитали 259
Смещение равновесия 66
Сигма-связь 241
Сила электролитов 22
Совместный гидролиз
Соединения включения 352
— внешнего порядка 265
— Лавеса 344
— первого порядка 265
— Юм-Розери 345
Солевой эффект 138
Соли 25
Сольваты 78
Соотношение неопределенностей
200
Сопряжение связи 257
Спин 9
Спин-валентное взаимодействие
249
Спиновое квантовое число 195
Среднее время жизни 172
Сродство к электрону 223
Стандартные электродные потен-
потенциалы 145
Степени свободы системы 72
Степень диссоциации 68
— окисления 20
— окисления углерода 37
Странность 9
Строение графика 262
— молекулярного иона водоро-
водорода 286
Структура воды 107
429
Структура комплексных соеди-
соединений 267
Структурная инверсия 312
Сублимация 29, 74
Твердые растворы 322
Тензиметрический анализ 92, 95
Теории кислот и оснований 117
¦— апротонная Усановича 119
— протолитическая Бенстеда и
Лоури 117
— сольво-систем 118
— электронная Льюиса 119
Теория индикаторов 130
— кристаллического поля 273
— строения Бутлерова 234
— электролитической диссоциа-
диссоциации 107
— электронного газа 338
Тепловой эффект реакции 40
Теплосодержание 41
Теплород 39
Теплота 39
— образования соединения 42
Терм 275
Термическое разложение реак-
реакции 34
Термохимия 39
Тримолекулярные реакции 166
Тройная точка 76
Типы гибридизации 253
Топливные элементы 159
Угловая часть волновой функ-
функции 208
Угловой момент электрона 193
Уравнение Больцмана 56
— Борна 327
— Борна—Майера 329
— Ван-дер-Ваальса 78, 346
— Капустинского 329
— Клаузиса—Клапейрона 75
— Леннарда—Джонса 352
— Менделеева—Клапейрона 16
— Нернста 142
— сжимаемости кристалла 327
— Слетера 225
— Шредера — Ле Шателье 89
— Шредингера 204
Учение о химическом равнове-
равновесии 52
Фазовая диаграмма состояния
74
Фазовые превращения 28
Фазы системы 61
Фактор эквивалентности 14
Ферменты 183
Ферроцен 261
Фигуративная точка 85
Флегма 104
Форма электронной орбитали 208
Фотон 201
Фотоэффект 201
Функции симметричные 237
— антисимметричные 234
Химические источники тока 156
— превращения 29, 45
Химические связи 233
— водородная 356
— ковалентная 241
— координационная 249
— металлическая 337
Химическое равновесие 50
Цеолиты 355
Цепные реакции 168
Цикл Борна—Гебера 323
Эвтектика 85
Экзотермические процессы 19
Экстенсивность 44
Электродные потенциалы 142,
145
Электролиз 151
Электролитическая диссоциация
45
Электрон 189
Электронография 204
Эндотермические процессы 19
Энергетическая диаграмма 290,
295
Энергия активации 164
— Гиббса 58
— металлической решетки 340
— кристаллической решетки 48
Энтропия 55
Эффект Джоуля—Томсона 346
Эффективная степень диссоциа-
диссоциации 112
Эффективный заряд 223
Число независимых компонен-
компонентов 72
Щелочи 22
Оглавление
Стр.
Предисловие . 3
I. Основные законы химии 7
Глава 1. Фундаментальные законы и законы стехиометрии 8
II. Классы химических соединений и типы реакций 17
Глава 2. Основные классы простых и сложных соединений 18
Глава 3. Типы химических реакций 28
III. Основы химической термодинамики и кинетики 38
Глава 4. Термохимия 39
Глава 5. Химическое равновесие 49
Глава 6. Смещение химического равновесия 60
Глава 7. Правило фаз Гиббса 71
Глава 8. Диаграммы растворимости 79
Глава 9. Законы Рауля 93
Глава 10. Растворы электролитов 107
Глава 11. Гидролиз солей и нейтрализация 120
Глава 12. Растворимость электролитов 133
Глава 13. Электрохимические процессы 140
Глава 14. Прикладная электрохимия 151
Глава 15. Кинетика химических реакций 163
Глава 16. Катализ 178
IV. Строение вещества 187
Глава 17. Исторические предпосылки теории строения . . 188
Глава 18. Волновая модель атома 199
Глава 19. Потенциалы ионизации и эффективные параметры
атомов 215
Глава 20. Теория валентных связей (ВС) 233
Глава 21. Молекулы с кратными связями и гибридными
орбиталями 242
Глава 22. Комплексные соединения и координационный
тип химической связи 264
Глава 23. Теория молекулярных орбиталей (МО) .... 284
Глава 24. Молекулярные орбитали в молекулах гетеросое-
динений 298
Глава 25. Строение ионных кристаллов 317
Глава 26. Кристаллические решетки с дефектами .... 331
Глава 27. Соединения с невалентными связями 346
Приложения 360
Рекомендуемая литература 426
Предметный указатель 427
43J
Учебное издание
Георгий Иванович Новиков
Основы общей химии
Зав. редакцией С. Ф. Кондрашкова
Редакторе. С. Трапезникова
Мл. редакторы С. М. Ерохина и Л. С. Макаркина
Художник Н. Ф. Маникало
Художественный редактор С. Г. Абелин
Технический редактор А. К- Нестерова
Корректор С. К. Завьялова
ИБ № 7 201
Изд. № Хим -711. Сдано в набор 26.03.87. Подп. в печать 30.09.87. Формат
84х108'/з2- Бум. тип. № 2. Гарнитура литературная. Печать высокая. Объем
22,68 усл. печ. л. + форзац 0,21 усл. печ. л. 23,10 усл. кр.-отт. 22,14 уч.-изд.
л. + форзац 0,35 уч.-изд. л. Тираж 40 000 экз. Зак. № 705. Цена 1 р. 10 к.
Издательство «Высшая школа», 101430, Москва, ГСП-4, Неглинная ул.,
д. 29/14.
Ордена Октябрьской Революции и ордена Трудового Красного Знамени МПО
«Первая Образцовая типография» имени А. А. Жданова Союзполиграфпрома
при Государственном комитете СССР по делам издательств, полиграфии и книж-
книжной торговли. 113051, Москва, Валовая, 28
пико- нано-
нанометры метры
микро-
микрометры
метры
102 103 101 102 103 Ю1 Ю2 103 104 105 1 10 100
Ii I1||||
Рентгеновское УФ-
излучение излучение
Спентр
ИК-
УКВ KB
Радиоволны
видимого света
СВ
Г
400 500
Fe
Са Н Са Н Н Мд
Са
600
700 ни
ни V
Na О Н Q. О
Солнечный спектр с линиями поглощения
Фиолетовый Синий Зеленый Оранжевый
Желтый Красный
Спектры испускания атомов
I \
400
500
600
700 нм
Спентры испускания и поглощения
10"+
Самая
далекая 10
галантина
10" +
Ю" +
102'-f Ближайшая ю"
галантина
io2
о I Размер
10" +
10'°+
10" +
10'° +
галактики
До
ближайшей
звезды
10' +
10» +
10° +
-О4 + Эверест <0~Ц ^рТ*
До
Плутона
До
Солнца
101 +Дом 10 " +
8 I Размер 2
|и т Юпитера
Размер
Земли
10"' +
Небоскреб
м+ Человек 10 lof Атом
Размер ,. _
Юа + Солнца Ю ' + Мышь 10 " +
Ягода
10"" +
!0~3 4- Блоха 10 |3-|-
10""+ Песчинна 10 '" +
10 5 + Нлетна
крови
Мелкий
вирус
Большая
молекула
Молекула
сахара
Атомное
ядро
Протон
Сравнительные размеры {в метрах) вещественных объектов Вселенной