/










































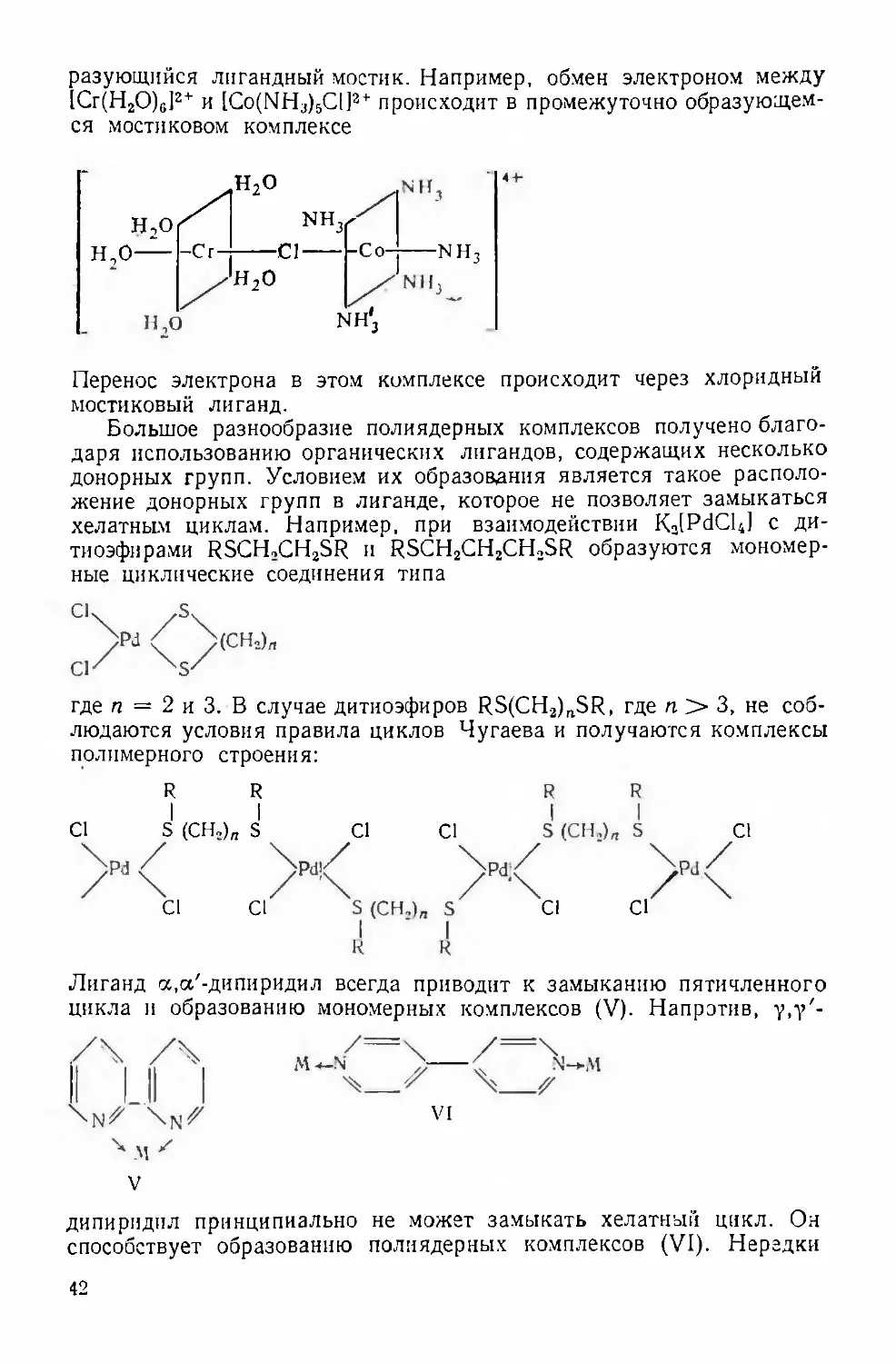
























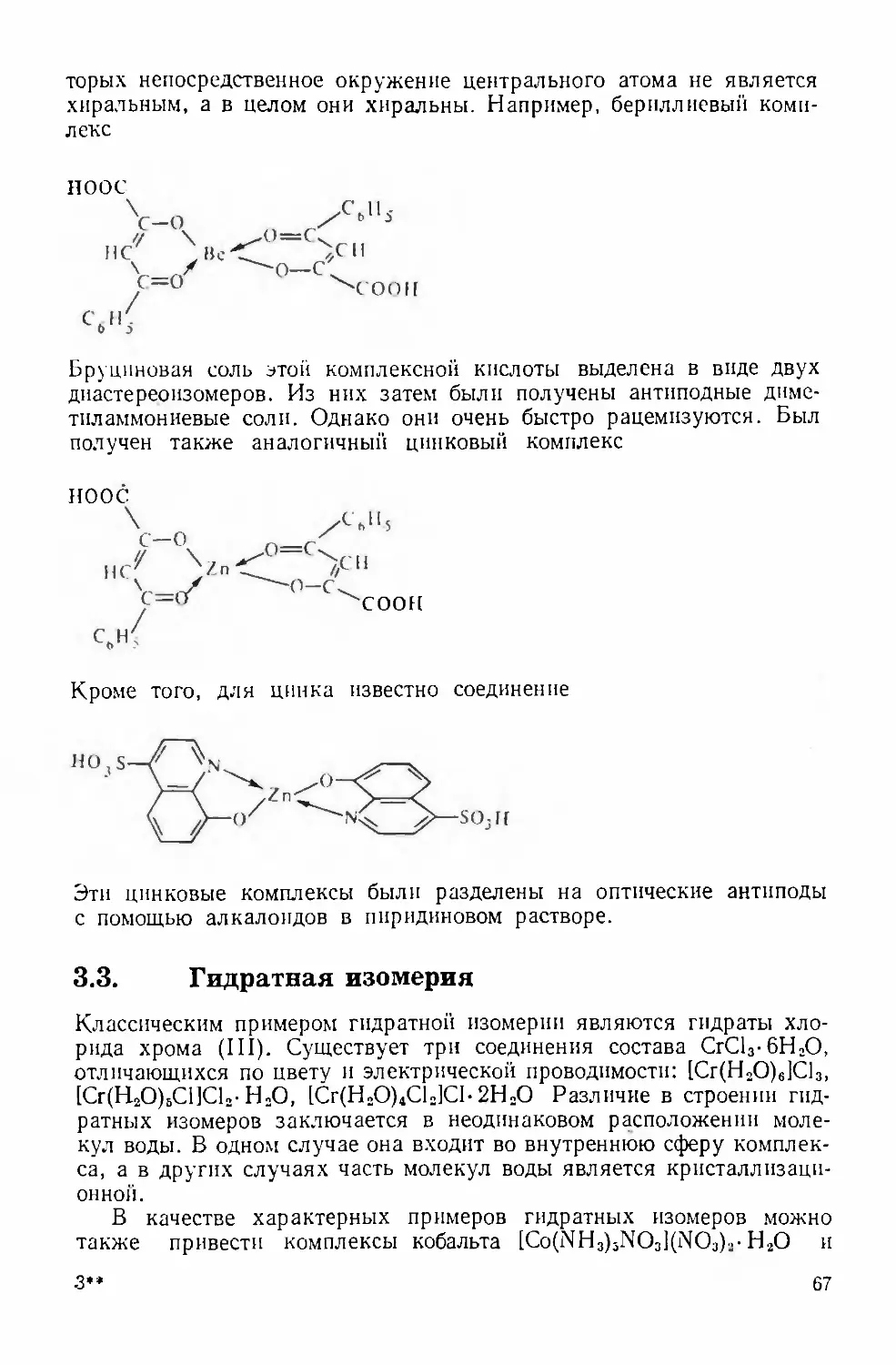





















































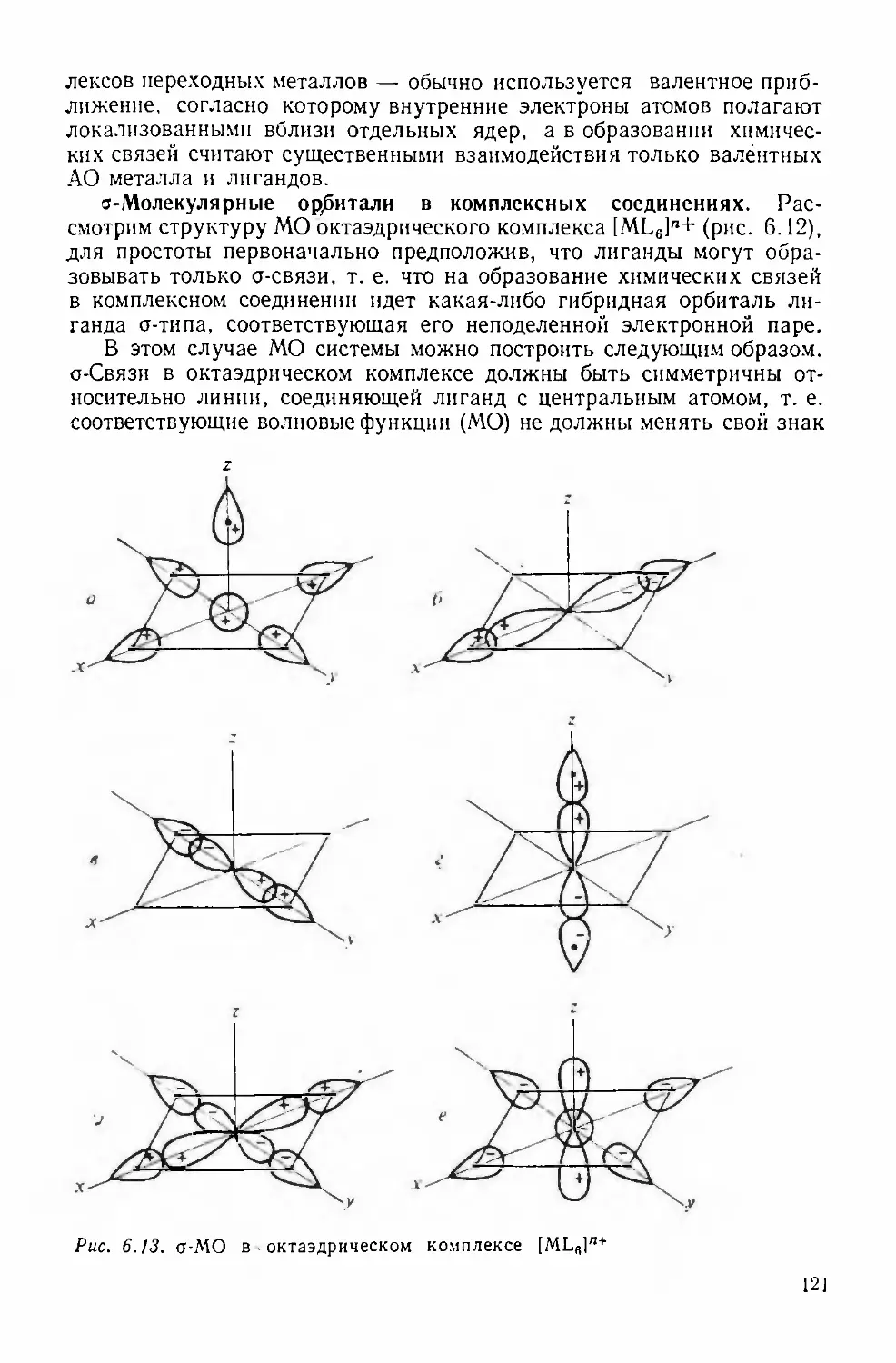
















































































































































































































































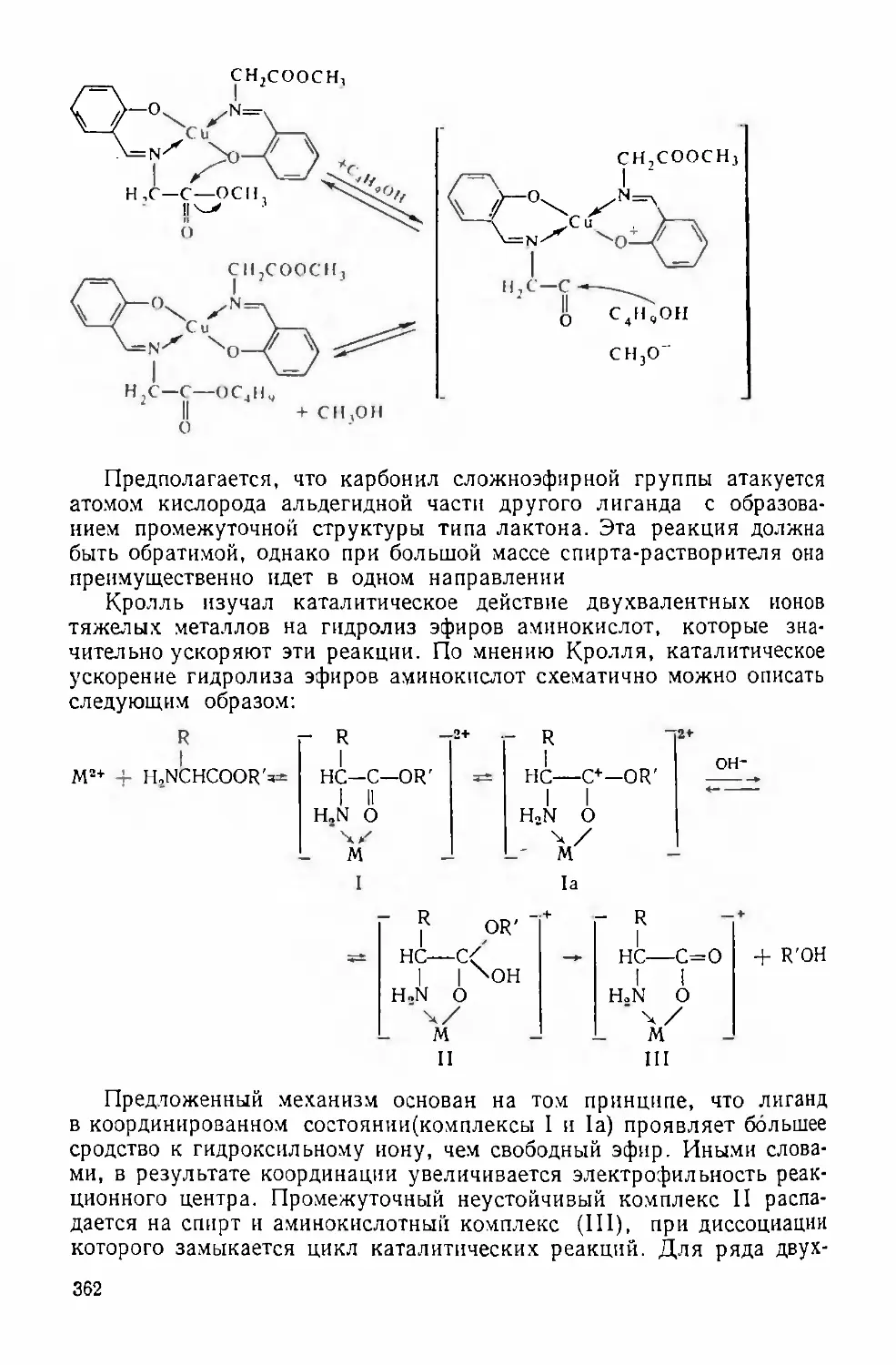
































































































Text
ЮН. Кукушкинхимиякоординационных
СОЕДИНЕНИЙДопущено Министерством
высшего н среднего
специального образования СССР
в качестве учебного пособия
для студентов химических
н химико-технологических
специальностей
высших учебных заведенийМОСКВА «ВЫСШАЯ ШКОЛА»
1985
ББК 24.1
К89
УДК 541.49Рецензенты:кафедра неорганической химии Кишиневского государственного
университета им. В. И. Ленина (зав. кафедрой — проф. Н. М. Са-
мусь), проф. Б. Д. Берёзин (Ивановский химико-технологический
институт)Кукушкин Ю. Н.К89 Химия координационных соединений: Учеб. пособие
для студентов хим. и хим.-технол. спец. вузов. — М.:
Высш. шк., 1985.— 455 с., ил. ’Излагаются свойства координационных соединений — кислотно-основные,
окислительно-восстановительные, особенности их строения и химической свя¬
зи, реакционная способность комплексов и координированных в них лигандов,
условия и механизм комплексообразования, влияние природы растворителя
на эти процессы. Описываются твердофазные превращения комплексов, но¬
менклатура и классификация лигандов и координационных соединении. Рас¬
смотрены равновесия в растворах координационных соединений, взаимное
влияние координированных лигандов.В пер. 1 р. 50 к.1802000000—365К —— 81—85001(01)—85ББК 24.1
540© Издательство «Высшая школа», 1985
«Истинный путь, ведущий длмппым,
но зато верным способом к теорети¬
ческому пониманию сложных явлений,
состоит в опыте и измерении отдель¬
ных частностей сложного явления».Д. И. МенделеевПРЕДИСЛОВИЕНаиболее распространенным учебным пособием по координа¬
ционной химии является книга А. А. Гринберга «Введение в
химию комплексных соединении». Ее последнее прижизненное
издание вышло в 1966 г. Однако за последние десятилетия в хи¬
мии координационных соединении ■получено огромное коли¬
чество новых результатов, разработаны новые концепции и вы¬
явлены новые правила. Несмотря на то что в нашей стране было
издано большое число монографий, посвященных отдельным
разделам координационной химии или отдельным группам ко¬
ординационных соединений, вузам необходимо учебное пособие,
отражающее достижения химии координационных соединений
последних лет.Координационные соединения, закономерности и правила
координационной химии широко используются в различных
отраслях химической науки. Настоящую книгу следует рассмат¬
ривать не только как пособие для вузовского курса «Химия
координационных соединений». Отдельные ее главы могут быть
использованы при изучении различных курсов, например ана¬
литической, металлорганической и бионеорганической химии,
металлокомплексного катализа и т. д.Изложение материала книги основано на конкретных экс¬
периментальных фактах и примерах. Исходя из них развиваются
теоретические положения. Автор пытался излагать материал на
уровне, доступном для' студентов. Однако поскольку книга
может служить пособием для ряда спецкурсов, то некоторые ее
разделы требуют определенной общехимическои подготовки.
В книге не излагаются теоретические основы методов исследо¬
вания координационных соединений. Это привело бы к значи¬
тельному увеличению объема книги или к вынужденному сокра¬
щению основного материала. Однако результаты, полученные3
различными физическими и физико-химическими методами, ши¬
роко используются в ряде глав. Предполагается, что читатель
знаком в общих чертах с этими методами или, при необходи¬
мости, может ознакомиться по специальной литературе.Большую помощь автору в работе над книгой оказали со¬
трудники кафедры неорганической химии Ленинградского тех¬
нологического института им. Ленсовета: Г. Б. Аветикян,
В. Ф. Буданова, Л. И. Данилина, В. К- Крылов, Е. И. Маслов,Н. С. Панина, Е. Ф. Стрижев и В. Б. Украинцев. Автор счи¬
тает приятным долгом выразить нм благодарность. Исключи¬
тельно ценные замечания и пожелания были сделаны рецензен¬
тами: проф. Б. Д. Берёзиным и сотрудниками кафедры неорга¬
нической химии Кишиневского государственного университета
им. В. И. Ленина, руководимой проф. Н. М. Самусь. Автор
также выражает им большую благодарность.В столь большом по объему труде неизбежны недочеты и упу¬
щения. Все замечания и пожелания читателей будут приняты
автором с признательностью.10. Н. Кукушкин
УСЛОВНЫЕ ОБОЗНАЧЕНИЯдмс— диметилсульфиддмсо— диметплсульфоксиддпсо— днпропилсульфоксидДизоПСО— днизопропнлсульфоксидДЭС— диэтилсульфидДЭСО— днэтилсульфоксндТДГ— тиодигликольто— тнокеанАсас— ацетнлацетонатный аннонАпН— аланинАг— арилBirnd— беизимидазолBu— бутилBut— трет-бутлСр— циклопентадиеиильный аннонCod— цнклооктадненDH— депротонированный днметилглиоксимDien— диэтилентрнаминDipy— 2,2'-дипиридилEn— этилеиднаминEt— этилGlyH— глицинHin— ХИНОЛИНMe— метилOxH— окенхннолннPh— фенилPlien— 1,10-фен а нтр олинPic*— ПИКОЛИНPip— пиперидинPn— 1,2-пропнлендиаминPr— пропилi-Pr— изопропилRf— фторированный углеводородный радикThio— тиомочевинаTmen— N.N.N' ,N' -тетраметилэтилен диамин
ВВЕДЕНИЕДолгие годы, вплоть до середины текущего столетия, химия коорди¬
национных соединений считалась частью неорганической химии. Соз¬
датель координационной теории А. Вернер назвал свой основной труд
«Новые воззрения в области неорганической химии» [1]. Примерно
до 50-х годов соединения, которые описывала координационная тео¬
рия, как правило, называли комплексными. Со временем все чаще
их стали называть координационными. Отнесение комплексных, или
координационных, соединений к области неорганической химии вы¬
текало из того, что в роли лигандов чаще всего выступали неоргани¬
ческие ионы или молекулы. Правда, наряду с аммиаком в качестве
лигандов использовались и органические амины, например алкил-
амины или пиридин, а также элементорганические соединения, такие,
как алкил- и арилфосфины, арсины, стибины. Новые органические
соединения, у которых предполагались основные свойства, прежде
всего испытывали платинохлористоводородной кислотой или хлори¬
дом платины на получение солей типа (АН)2[РЬС16]- Так, Вюрц об¬
работкой органических изоцианатов щелочью получил метиламин и
этиламин и выявил их способность образовывать комплексные соеди¬
нения с PtCl2. Андерсон в продукте сухой перегонки костей открыл
пиридин и получил комплекс (PyH)2[PtCI6], Одновременно он уста¬
новил, что при нагревании водного раствора этой соли происходит
перегруппировка с образованием нового вещества:
г(РуН), [PtCI6] -> [PtPy2CI4] + 2НС1Правильную трактовку этой реакции дал Вернер с позиции коорди¬
национной теории. Гофман впервые получил третичные фэсфины, ар¬
сины и стибины. Проводя аналогию между этими соединениями и ами¬
нами, он установил, что они способны взаимодействовать с PtCl2 с об¬
разованием комплексных соединений типа [Pt(3R3):CU].Считают, что первым комплексным соединением, полученным в
лаборатории, было [Co(NH3)6]Cl3. Его случайно открыл Тассер в 1798 г.
Эту дату иногда считают точкой отсчета, от которой ведет свою исто¬
рию координационная химия. До открытия Тассера было известно
типичное координационное соединение' Kj[Fe(CN)6], открытое Дис-
бахом в 1704 г. Однако его рассматривали как двойную соль
4KCN • Fe(CN)2, которая не считалась чем-то необычным. Значение
открытия Тассера состоит в том, что в соединении CoCl3-6NH3,
а позднее в CoCl3-5NH3, CoCl3-4NH3 и других им подобных
химики видели вещества, которые не могли быть объяснены с позиций
существовавших в то время представлений. Правда, так же как жел¬
тую кровяную соль считали двойной солью, так и аммиакаты
СоХ3-гс .\ТН3 рассматривали как аналоги кристаллогидратов.Для объяснения строения аммиакатов солей металлов и им подоб¬
ных соединений потребовался почти столетний напряженный труд6
многих ученых. Пока же не была создана надежная теоретическая ба¬
за группы соединений н отдельные соединения называли по внешним
признакам и прежде всего по цвету: CcCl3-6NH3 лутео (от лат. lute-
us — желтый), две изомерные соли состава СсС13-4NH3 — празео
(от греч. prasinos — зеленый) и виолео (от лат. viola— фиолетовый)
п т. д. Некоторые комплексные соединения, сыгравшие большую
роль в развитии координационной химии, называли по имени авто¬
ров, впервые их получивших, например соль Цейзе K[Pt(C2H4)Cl3],
соль Косса KIPt(NH,)Cl3], хлорид Пейроне *<«c-IPt(NH3)2CI2]. Иногда
название отражало имя автора и цвет соединения, например зеленая
соль Магнуса [Pt(NH3)4] |PtCl4].Выделение комплексных (координационных) соединений в само¬
стоятельную область химической науки в значительной мере связано
с именем Льва Александровича Чугаева. Поскольку Л. А. Чугаев
по образованию был химиком-органиком и имел большой опыт и боль¬
шие достижения в этой области химии, то естественно, что в качестве
лигандов он брал сравнительно сложныеорганическиесоединения, на¬
пример сукцпннмиды, диоксимы и др. С этого времени начинает ин¬
тенсивно развиваться химия координационных соединений, в состав
которых входят сложные органические молекулы. Благодаря рабо¬
там в этом направлении уже в 1906 г. Л. А. Чугаев открыл реакцию
диметилглиоксима на никель. Значение этого открытия состояло не
только в том, что была выявлена эффектная реакция для качествен¬
ного и количественного определения никеля, но и в том, что при этом,
по существу, было сформулировано понятие о конкретной функцио¬
нально-аналитической группировке в органической молекуле. Та¬
ким образом, Л. А. Чугаевым уже в 1906 г. был намечен путь для ре¬
шения одной из наиболее важных проблем современной аналитической
химии ■— использования органических реагентов в качественном и
количественном анализе.Органические реагенты для аналитического определения ионов
металлов начали применяться гораздо раньше. Это были природные
соединения, например морин (для флуоресцентного определения алю¬
миния), ализарин (для колориметрического определения многих эле¬
ментов и прежде всего алюминия). Первое синтетическое соединение в
качестве аналитического реагента на ионы металлов было применено
М. А. Ильинским в 1884 г. Он получил 1-нитрозо-2-нафтол и исполь¬
зовал его в качестве осадителя иона кобальта. Практически в то же
время 3. Скрауп впервые описал соединения ионов металлов с 8-окси-
хинолином («оксином»). О том, что продуктами взаимодействия ионов
металлов с этими веществами являются типичные координационные
соединения, стало известно много позже. Таким образом, координа¬
ционные соединения стали объектами для изучения химпками-ана-
литиками.В настоящее время специалисты самых различных отраслей хи¬
мии считают координационные соединения объектами своих иссле¬
дований. Например, элементоргаников интересуют л-комплексы типа
соли Цейзе и карбонильные соединения. Металлокомплексный ката¬
лиз — актуальное направление современной химии — целиком ос¬7
нован на координационных соединениях. Специалисты по теории
растворов пользуются термином «координационная сольватация»,
рассматривая первую сольватную оболочку катионов как внутрен¬
нюю сферу комплекса. Еще раньше понятие о координационном числе
было введено в кристаллохимию. А. Е. Ферсман по этому поводу пи¬
сал: «Введение в кристаллохимию учения о координационном числе...
представляет очень крупный шаг вперед в понимании законов крис¬
таллохимии. В сущности именно координационное число определяет
структуру соединений и этим путем влияет на радиусы атомов и ио¬
нов»* .В настоящее время координационная химия играет явно интегри¬
рующую роль в химической науке. Подобно связуюш,ему материалу,
она объединяет в единое целое большое здание химии.В середине XIX столетия были получены первые металлорганиче-
ские соединения, характерной особенностью которых является то, что
атом металла в них соединен по крайней мере с одним атомом угле¬
рода. Примерами таких соединений могут служить NaC2H5 и CH3MgI.
Эти соединения, так же как и некоторые координационные, объединя¬
ют в себе органическую и минеральные части. Химическое поведение
таких типичных металлорганпческих соединений определяется в боль¬
шей мере характером металла и в меньшей — характером углеводо¬
родного радикала. Несмотря на это, они отнесены к предмету ис¬
следования органической химии. Одной из причин такого положения
было то, что магнийорганические соединения (реактив Гриньяра)
играют исключительно важную роль в органической химии. Специ¬
фика свойств и реакционной способности металлорганпческих моле¬
кул привела к тому, что в середине XX в. химия этих соединений так¬
же выделилась в самостоятельную отрасль.С самого начала в основу деления соединений, содержащих ме¬
талл и остатки органических молекул, был положен следующий прин¬
цип. Если соединение имеет связь металл — углерод, то это металл-
органическое соединение. С этой точки зрения карбонилы металлов,
например Fe(CO)5 и Ni(CO)4, а также изонитрильные соединения ме¬
таллов, например, Cr(CNPh)0 и Fe(CNBul)5 (в тех и других металл
связан с лигандами через атом углерода), часто считают металлор-
ганическими соединениями.Развитие координационной и металлорганической химии привело
к тому, что они стали все более проникать друг в друга. Появились
принципы, теории, правила, в равной мере справедливые и для коор¬
динационной и для металлорганической химии. Часто трудно одно¬
значно отнести то или иное соединение к координационным или к
металлорганическим. Так, типичное координационное соединение
K2[PtCl0] может быть получено по реакцииРtCI4 — 2КС1 = К2 [PtCI6]Типичные металлорганические соединения (CH3)3PtI и CH3Li взаи¬
модействуют с образованием Li2[Pt(CH3)6]:* Ферсман А. Е. Избранные труды. М., Пзд-во АН СССР, 1955, т. 3, с. 103.8
(СН3)3PtI -f 3CH3Li = Li; [Pt.(CH3)e].+ LilСоединение Li2[Pt(CH3)6], несомненно, следует отнестн к металлорга¬
ническим. Однако принципиальной разницы между K2IPtCl0] и
Li2[Pt(CH3)6] нет. Таким образом, с большим основанием последнее
соединение также можно называть координационным.Хорошо известное координационное соединение золота KLAuCIJ
взаимодействует с металлорганическим соединением CH3Li по урав¬
нению реакцииК [AuCI4] + 4CH3Li =[Li [Au (CH3)4] + KC1 + 3LiClПолучающееся соединение Li[Au(CH3)4] в равной мере можно счи¬
тать как координационным, так и металлорганическим.Положение еще более осложняется, когда рассматриваются соеди¬
нения типа [Pt(CH3)3I] или [Pt(PPh3):I(CH3)J. Какими следует их
считать — металлорганическИмп или координационными? В основу
отнесения можно было бы положить формальный принцип большин¬
ства лигандов. Если' во внутренней координационной сфере комплекса
больше лигандов, связанных через углерод, то это металлорганиче-
ское соединение. Если лигандов, связанных через углерод, меньше,
то это координационное соединение. Однако специалисты по 'металл¬
органической химии высказывают справедливое суждение, что связь
металл — углерод вносит много характерного в свойства соединения
в целом, поэтому, если в1 соединении имеется хотя бы одна связь
металл — углерод, то его следует считать металлорганическим. По¬
скольку любое решение этого вопроса будет условным, то, возможно,
нет необходимости формализовать его до конца.Термин «комплексное соединение» был введен в химическую ли¬
тературу Оствальдом. В литературе на русском языке его впервые
употребил В. А. Кистяковский, который на основе результатов элект¬
ропроводности дал четкое разграничение двойных и комплексных
солей. Всеобщее употребление термина «комплексное соединение»
началось после опубликования Вернером в 1905 г. основополагающего
труда [1]. В этой же книге различаются координационно-насыщенные
и координационно-ненасыщенные соединения. По мере распростра¬
нения координационной теории Вернера на различные типы и классы
соединений термин «координационные соединения» все прочнее вхо¬
дит в химическую литературу.Возражения против использования термина «комплексное соеди¬
нение» основаны на том, что комплексное означает сложное, а сложны¬
ми являются н некоторые соединения, не содержащие металл, напри¬
мер хингидрон С6Н402-СсН4(ОН)2. С другой стороны/ иен HgCl+ не
сложный, хотя принципиально и не отличается от иона HgCl^-.В настоящее время за соединениями, о которых идет речь, закре¬
пилось в основном наименование «координационные». Химия, изу¬
чающая эти соединения, чаще называется координационной. Однако
термин «комплексное соединение» в научной химической литературе9
продолжает широко использоваться. В настоящее время его чаще при¬
меняют к нонам — «комплексный ион». Процесс образования данных
соединений называют процессом комплексообразования.-Многие ученые анализировали и давали определение координа¬
ционному соединению [2, 3|. По мере того как к координационным
относили все новые типы соединений, становилось все труднее давать
нм определение. Безусловно, безупречное определение координацион¬
ному соединению дать очень трудно. Тем не менее автор предлагает
следующее.Координационными называются соединения, существующие как в
кристаллическом состоянии, так и в растворе, особенностью которых
является наличие центрального атома (акцептора электронов), ок¬
руженного лигандами (донорами электронов). Лиганды способны сту¬
пенчато и обратимо отщепляться от центрального атома по гетеро-
литическому типу. В большинстве случаев в молекулярном виде коор¬
динационные соединения могут рассматриваться как состоящие из
простых, способных к самостоятельному существованию молекул.Продукты ступенчатой диссоциации следует рассматривать как
комплексы. Например, наряду с Hgl* комплексами являются Hgl3,
Hgl2 и Hgl+. В растворе эти комплексы координационно насыщены
за счет присоединения к центральному атому молекул растворителя.
Таким образом, в водном растворе правильнее их было бы писать как
lHgI3(H20)]-, [HgI2(H20)2] и [HgI(H20)J+.Продукты ступенчатой диссоциации координационно-насыщенных
по данному лиганду соединений как комплексы были рассмотрены
А. К. Бабко и Силленом. В современной химической литературе име¬
нование комплексами продуктов ступенчатой диссоциации прочно
вошло в лексикон.В предлагаемом автором определении одной из основных характеристик
является наличие координационного центра и координации лигандов. На это
обстоятельство обращали внимание и другие ученые. Ст. Андреев одной из ха¬
рактеристик комплексного соединения считает асимметрическую координацию
лигандов. Он определяет комплексные соединения как соединения с центрами
асимметрической координации и с донорно-акцепторной или двухфазной связью
между центральными атомами и асимметрически координированными по отно¬
шению к ним ионами, атомами или атомными группами [4]. Такое определение
позволяет Андрееву исключить из числа комплексных соединений хлорид нат¬
рия в кристаллическом состоянии, где ионы CI- координированы одинаковым
способом (равнозначно) одновременно к двум ионам натрия. Однако этот кри¬
терий не может считаться безупречным. В настоящее время известно много гало-
генмостнковых комплексов, в которых галогенидные ионы связаны с двумя ко¬
ординационными центрами равноценными связями. Например, одна из крис¬
таллических форм хлорида палладия (II) имеет плоское строение:\ /С\ /С'\ /С'\ /ХсХсХсХВ этой молекуле координация симметричная, но соединение типично координа¬
ционное. Уместно сказать, что хлорид палладия в твердом состоянии обычно
не называют комплексным соединением. Его скорее можно было бы отнести к
простым. Однако изучение «простых» солей многих металлов в твердом и даже
газообразном состоянии показало, что они являются типично координационны¬
ми.!0
В предлагаемом определении подчеркнут акцепторный характер
центрального атома и донорный характер лигандов. Это ни в коей
мере не исключает в отдельных случаях проявления л-донорных
свойств центрального атома (наряду с ст-акцепторнымн) и л-акцептор-
ных свойств лигандов (наряду с ст-донорными).К. Б. Яцимирский считает необходимым признаком комплексного
соединения несовпадение координационного числа и степени окис¬
ления. К комплексным соединениям он относит соединения, образую¬
щие изолированные группы атомов (ионы или молекулы), характе¬
ризующиеся наличием координации, неполной диссоциацией по гетеро-
литическому типу в растворе (или в вакууме) и сложностью состава
(несовпадение координационного числа и степени окисления). У коор¬
динационных соединений металлов, действительно, координационное
число, как правило, выше, чем степень окисления. Однако этот и дру¬
гие выделенные К- Б. Яцимирским признаки характерны, например,
для гидроксиламина: атом азота в гидрокспламине имеет координа¬
ционное число 3, а степень окисления —1. Однако, к комплексным
соединениям его обычно не относят.В химии довольно часто употребляют термин аддукт. Иногда его
применяют к соединениям, которые могут быть отнесены к координа¬
ционным или металлорганическим. Как правило, аддуктами называ¬
ют вещества, состоящие из небольшого числа молекул (двух или трех)
способных к самостоятельному существованию. В формулах моле¬
кулы, объединенные в аддукт, пишут через точку, например
AsC13-SC(NH2)2.Обычно термином «аддукт» пользуются в том случае, если мало что
можно сказать о строении вещества. Поэтому его употребляют чаще
всего применительно к соединениям p-элементов, координационная
химия которых изучена еще незначительно.По мнению автора, термин «аддукт» следует применять лишь к
соединениям без координационного центра, например, к соедине¬
ниям пикриновой кислоты с нафтила'мнном Н0С6Н2(№02)з- C10H7NH».Комплексными иногда называют клатратные* соединения (соеди¬
нения включения). В них молекулы одного компонента («гостя») вклю¬
чены в полости устойчивой структуры другого компонента («хозяина»).
Такие молекулярные соединения образованы не химическими, а меж-
молекулярными связями Ван-дер-Ваальса. В качестве примера можно
назвать соединение гидрохинона с метанолом или с благородными
газами. В данном случае понятие «комплексный» применяется для
обозначения сложности соединения. Соединения включения имеют
очень мало общего с координационными соединениями. Однако иногда
их рассматривают в работах, посвященных координационным соеди¬
нениям. Наряду с координационными иногда рассматривают также
молекулярные органические соединения. Наиболее распространенные
из них следующие: 1) образованные из бензохинона (или его произ¬
водных), а также из нитросоединений, с ароматическими углеводоро¬
дами, аминами, фенолами и ароматическими простыми эфирами;* От лат. clathratus — включенный.11
2) образованные холевыми кислотами с жирными кислотами, сложны¬
ми эфирами и некоторыми парафинами. Эти соединения не имеют ко¬
ординационного центра п они в лучшем случае могут быть отнесены
к аддуктам.Изополп- и гетерополпсоединения здесь не рассматриваются вслед¬
ствие их специфичности. Отнесение их к координационным соедине¬
ниям не вызывает сомнений, как и их большое теоретическое и прак¬
тическое значение.Правила и закономерности координационной химии были выяв¬
лены главным образом при изучении соединений хрома, кобальта и
платиновых металлов. Базой для создания координационной теории
также служили комплексы этих металлов. Это обусловлено относи¬
тельной инертностью комплексов данных металлов, что позволяет
синтезировать соединения и изучать их химическими методами. По
мере появления и внедрения в химическую практику физических и
физико-химических методов в сферу исследования вовлекалось все
большее число элементов: все d-элементы, а также редкоземельные и
актиноидные элементы.С середины XX в. наряду с препаративными методами исследова¬
ния начали широко применяться методы, позволяющие изучать комп¬
лексные соединения без выделения из растворов. С этого времени
химия координационных соединений значительно расширила свои
рамки и стала самостоятельной областью химической науки.Большие возможности для координационной химии появились с
привлечением метода ядерного магнитного резонанса (ЯМР). Метод
ЯМР позволил изучать весьма малоустойчивые комплексы и регист¬
рировать одновременно существование в растворе нескольких соеди¬
нений. С методом ЯМР связан новый этап в координационной химии —
вовлечение в круг исследования координационных соединений р-
элементов. В некоторых случаях поведение и свойства комплексных
соединений p-элементов не укладываются в рамки закономерностей
и правил, сформулированных для d-элементов. Таким образом, в на¬
стоящее время начинает развиваться новый и исключительно важный
раздел координационной химии — координационная химия р-эле-
ментов. Систематические исследования в этом направлении в нашей
стране проводятся научной школой Ю. А. Буслаева.Однако не должно сложиться мнение, что для координационных
соединений p-элементов применимы только методы исследования
в растворах. В настоящее время синтезировано большое число коор¬
динационных соединений, роль центрального атома в которых играют
галогены в положительных степенях окисления. Так, координаци¬
онные соединения 1+ с органическими молекулами в качестве лиган¬
дов были получены еще в 30-х годах Карлсоном. В качестве лигандов
он взял пиридин и его производные. Соединения общей формулы
[ILJX, где X — С!04, N03, СН3СОО~, получались по реакции
[AgL.,]X + Is - [IL2]X + AglИзучению этих и им подобных соединений в настоящее время уделя¬
ется довольно много внимания.
ГЛАВАКООРДИНАЦИОННАЯ ТЕОРИЯ А. ВЕРНЕРА1.1. Предпосылки создания
координационной теорииВ конце XIX в. в органическую химию прочно вошла и получила
широкое распространение теория химического строения. По словам
А. М. Бутлерова, теория химического строения имела своей задачей
выразить «способ химической связи элементарных атомов в частице».
Порядок расположения (взаимодействия) атомов в молекуле
А. М. Бутлеров называл химическим строением. Этот порядок может
быть отражен структурной формулой. Важную роль в разработке
теории химического строения сыграло явление изомерии, которое ши¬
роко распространено среди органических соединений.Следующим этапом в развитии структурных представлений было
стереохимическое учение, развитое Ле Белем и Вант-Гоффом. Оно
позволило судить о пространственном расположении атомов в моле¬
куле. На основе теории химического строения и стереохимического
учения было дано наглядное объяснение явлению изомерии и стало
понятным существование огромного числа органических молекул.Хотя применение теории химического строения принципиально
не сводилось только к органическим соединениям, тем не менее мно¬
гие годы структурные представления применялись главным образом
к соединениям углерода. Одна из причин такого положения заключа¬
лась в том, что для соединений других элементов практически не были
известны случаи изомерии. Таким образом, не было почвы для раз¬
вития теории химического строения на примерах соединений других
элементов, в частности соединений металлов.При определении строения молекул важное значение имеет валент¬
ность , атомов, составляющих эту молекулу. Водородные соединения,
оксиды, кислоты, основания, соли, состав которых соответствовал
классическому учению о валентности, были названы простыми соеди¬
нениями. Необходимость в этом возникла тогда, когда стало ясно, что
имеются соединения, строение которых не может быть объяснено с
позиции существовавших представлений о валентности. К их числу
относятся, например, соединения, валовый состав которых можно
записать следующим образом: CoCl3-6NH3, PtCl3-2NH3, CuS04-4NH3.
В солях СоС13, PtCl3> CuS04 классические валентности насыщены,
однако эти соли могут довольно прочно удерживать строго определен¬
ное число молекул аммиака.В отличие от простых такие соединения, как CoCl3-6NH3 и ему
подобные, были названы комплексными. Их молекулы состоят из не¬
скольких простых, способных к самостоятельному существованию
соединений. Теория, отражающая строение этих соединений, создана13
швейцарским химиком А. Вернером. Она была изложена в статье
«О строении неорганических соединений», опубликованной им в
1893 г.Так же как и органические соединения, первые комплексные со¬
единения, с которыми пришлось столкнуться химикам, были доволь¬
но инертными. Среди них обнаружили и изомерные соединения.
Л. А. Чугаев отметил существование близкой аналогии между соеди¬
нениями углерода и наиболее устойчивыми комплексными соедине¬
ниями. Он считал, что благодаря комплексным соединениям химия
многих элементов периодической системы может стать столь же много¬
образной, как и химия углерода. Первые сведения о комплексных
соединениях появились в конце XVIII в. Естественно, что и до Вер¬
нера многие ученые пытались создать теорию, отражающую строение
этих «необычных» соединений.Вернер при разработке теории строения комплексных соединений опирался
на работы предшественников. Однако он исходил из принципиально новых по¬
зиций. До теории А . Вернера наиболее разработанной была теория строения ком¬
плексных соединений Бломстранда — Иёргенсена. Ее основные положения сле¬
дующие.1. Для некоторых элементов допускалась валентность выше, чем обычно
принятая. Например, считалось, что галогенидные ионы могут быть трех¬
валентными, кислород — четырехвалентным, а азот — пятивалентным.2. Так же как и в органических молекулах, в комплексных соединениях
допускалась возможность цепеобразного сочетания таких атомов, как хлор, кис¬
лород, азот и др. Например, цепи в кристаллогидратах и аммиакатах изобра¬
жались следующим образом:Н Н Н Н\/ \/М—О-О—0 — 0 —/\ /\
н н н нН И К II н н
\/ I \/ Iм — N — N N NI /\ I /\н н н н н нЗдесь кислород четырехвалентен, азот пятивалентен.3. Экспериментально установленное различие между кислотными остат¬
ками объяснялось различным способом их связи в комплексе. Если кислотные
остатки связаны непосредственно с металлом, то при растворении комплекса
они неспособны переходить в раствор в виде ионов. Если же они присоединены
к концу цепи, то при растворении комплекса становятся нонами.Не будем останавливаться на критике этой теории, так как она имеет лишь
историческое значение. Однако обратим внимание на то, чш она содержала
два важных положения: о необычных валентностях и о различном поведении
кислотных остатков в зависимости от их расположения в комплексе.Предположения о наличии центрального атома в комплексных соединениях
также высказывались до Вернера, например Михаэлисом и Гростманом. Двой¬
ную соль РtCli-2KCI они изображали формулой, в которой все атомы хлора
и калия непосредственно связаны с центральным атомом:Cl CI С1\1/K-Pt-K/IVCl Cl Cl
1.2. Основные положения координационной
теорииВернер обратил внимание на одно чрезвычайно важное обстоятельство.
Аналитические данные для большого количества комплексных соеди¬
нений свидетельствовали о том, что число нейтральных молекул, при¬
соединяющихся к молекулам соли металла, чаще всего равно 6 пли 4:СгС13.6ЬШ NiCl,-6NH3СоС1„-6Н20 PtC]4.6NH3CrCI3-6NH3 CuCI,-4NH3CoCl3.6NH3 PdCl2.4NH3CoCI2.6NH3 PtCl,.4NH3Если состав двойных солей записать в виде валовой формулы, то
число кислотных остатков в сумме также равно 6 или 4:Fe (CN)S.4KC\ = K4Fe (CN)eFe (CN)3.3KCN = K3Fe (CN')SPtCl4.2KCl = KoPtClePdCl„.2KCl = K2PdCI4Ni(CN)a.2KCN = K2Ni(CN)4Имеются многочисленные примеры комплексов, сумма нейтральных
молекул и кислотных остатков в которых равна 6 или 4:Со (N02)3.3NH3j PtCl4.2NH3; RhCI3.3NH3; PdCl;.2NH3Из приведенных примеров видно, что числа 6 и 4 характерны для
солей различных металлов и в различных степенях окисления. Вер¬
нер на основании этого пришел к'заключению, что в комплексном со¬
единении имеется центральный атом* (ион металла), вокруг которого
координируются** нейтральные молекулы или кислотные остатки.
Эти группы называют лигандами***. Число координированных
лигандов чаще всего равно 6 или 4. Число лигандов, окружающих
центральный атом, называют координационным числом. Лиганд за¬
нимает около центрального атома координационное место (позицию).
Координационное число и координационное место — важнейшие ха¬
рактеристики комплексного соединения.При координации изменяются свойства как лигандов, так и иона
металла-комплексообразователя. Часто координированные лиганды
и ион металла невозможно обнаружить при помощи химических ре¬* Центральный атом также называют комплексообразователем.** Координация (от лат. со — вместе, ordinatio — упорядочение) до¬
словно означает «совместное упорядочение». В химии слово «координировать»
употребляют в смысле «располагать» около координационного центра.*** Лиганд — связанный. В старой литературе чаще употребляется тер¬
мин адденд — присоединенный. Теперь он почти вышел из употребления.15
акций, характерных для них в свободном (некоординированном) со¬
стоянии.Совокупность иона металла и окружающих его лигандов была на¬
звана Вернером внутренней сферой комплекса. Ее обычно заключают
в квадратные скобки. Все остальное в комплексном соединении сос¬
тавляет внешнюю сферу и пишется за квадратными скобками.Первой задачей на пути исследования строения комплексного со¬
единения является установление состава его внутренней сферы. До¬
пустим, что в результате реакции получено соединение, валовый
состав которого CoCI3-6NH3. Роль центрального атома должен играть
ион кобальта (III). В том, что ион кобальта входит во внутреннюю
сферу комплексного соединения, нетрудно убедиться. Например,
при действии щелочи на раствор данного комплекса не происходит
образования характерного осадка гидроксида кобальта.Нейтральные лиганды, как правило, входят во внутреннюю сферу
комплекса. В данном случае с помощью характерных реакций, на¬
пример с реактивом Несслера или просто с фенолфталеином, можно
убедиться, что свободного аммиака в растворе соединения CoCl3-6NH3
нет.Напротив, хлорид-ионы проявляют все характерные для них свой¬
ства. Например, они осаждаются ионами серебра в виде AgCl так же,
как из раствора хлорида алюминия. По массе осадка можно убедиться,
чтовсе ионы СГ комплекса при действии нитрата серебра переходят
в AgCl.Таким образом, эти простые химические операции позволяют счи¬
тать, что внутренняя сфера данного комплекса, т. е. комплексный
ион, состоит из иона кобальта (III) и шести молекул аммиака
[Co(NH3)0]3+. Поскольку заряженной частицей во внутренней сфере
является только ион металла, то заряд комплексного иона в целом
равен заряду иона металла, т. е. 3—.Количественный опыт по осаждению хлорид-ионов в виде AgCl по¬
казывает, что все три хлорид-иона находятся во внешней сфере. Сле¬
довательно, данное комплексное соединение построено следующим
образом: [Со(МН3)„]С13. В целом соединение нейтрально. Положи¬
тельный заряд комплексного иона компенсирован отрицательными
зарядами трех хлорид-ионов.Если после количественного осаждения ионов СГ нитратом серебра
и отделения осадка выпаривать раствор, выделится комплексное со-,
единение [Co(NH3)c](N03)3. Таким образом, в результате реакции про¬
изошло замещение во внешней сфере комплекса ионов СГ на NOj:
[Со (NH3)6] Cl3 + 3AgN03 = [Со (NH3)J (N03)3 + 3AgClДопустим, что получено комплексное соединение, состав которого
Fe(CN)3-4KCN. В том, что ион переходного металла входит во внут¬
реннюю сферу комплекса, легко убедиться, так как никакими извест¬
ными реакциями на ион Fe2+ не удается обнаружить свободный ион
железа (II). Характерными реакциями на ионы CN~ также не удается
обнаружить их в свободном состоянии. Однако ионы калия в растворе
этого комплекса ведут себя так же, как, например, в растворе K.SOj.16
Они количественно могут быть выделены из раствора. Таким образом,
комплексный ион имеет состав [Fe(CN)cl4_. Его заряд равен алгебраиче¬
ской сумме зарядов составляющих компонентов. Поскольку заряд иона,
железа равен 2+, а шесть ионов CN- в сумме имеют заряд 6—, то за¬
ряд комплексного иона должен быть равен 4—. В том, что состояние
окисления центрального атома не изменяется и остается равным +2,.
легко убедиться. Так же как и свободный ион Fe2+, комплексный ион
легко окисляется перманганатом калия:5Fe"-+ + Mn07+ 8Н+ = 5Fe3+ + Мп=+ + 4Н205 [Fe (CN)e]4~ 4- МпОГ+ 8Н+ = 5 [Fe'(CN)6]3“ + Mn=+ -f 4H20Поскольку ион металла в комплексе окисляется и из состояния
окисления +2 переходит в состояние окисления +3, то заряд комп¬
лексного иона в целом также изменится. Он станет равным 3—•.Допустим, что в результате реакции получилось соединение, состав
которого СоС13-5ЛтН3. Реакциями на кобальт (III) и аммиак убежда¬
емся, что характерные для них в свободном состоянии свойства не
проявляются. Следовательно, они входят в комплексный ион. Нитра¬
том серебра удается осадить в виде AgCl только два иона СГ из трех.
Таким образом, можно сделать заключение, что комплексный ион имеет
строение [Co(NH3)5Cl]2+. Заряд комплексного нона равен 2-f, так
как заряд иона кобальта 3-г, а заряд входящего в него иона СГ равен
1—. Молекулы аммиака на заряд комплексного иона не влияют, так
как они электронейтральны.Во внешней сфере комплексного соединения находятся два хлорид-
иона, следовательно, строение соединения можно выразить формулой
[Co(NH3)5C1]C12. Если после осаждения хлорид-ионов нитратом се¬
ребра получающийся раствор выпарить, то выделится комплексное
соединение [Co(NH3):;Cl](N03)2. Таким образом, комплексный ион
участвует в обменной реакции с нитратом серебра как единое целое.
Реакция может быть выражена уравнением[Со (NH3)6 CI] Cl2 + 2AgN03 = [Со (NH3)6 Cl] (N03)2 + 2AgClВажной особенностью комплексных соединений является способ¬
ность внутренней сферы одновременно заполняться как нейтральными
лигандами, так и кислотными остатками. Поскольку координационное
число данного центрального иона, как правило, величина постоянная,
то для того, чтобы лиганд мог войти во внутреннюю сферу комплекса,
другой лиганд должен покинуть ее. Известны ряды комплексов, ко¬
торые можно рассматривать как продукты постепенного замещения
лигандов одного типа на другие. Так, если действовать аммиаком на
комплексное соединение Na3[Co(N02)6], то можно получить последо¬
вательный ряд соединений в соответствии с реакциями:Na3 [Со <N02)c] + NH3 = Na, [CoNH3 (NO„)5] + NaN02Na3 [CoNH3 (NO„)5] -f NH3 = Na [Co (NH3)2 (NO,)4] + NaN02Na [Co (NH3), (NO:),] 4- NH3 = [Co (NH3)3 (NO,,)3] + NaN02[Co (N'H3)3 (NOJ3J + NH3 = [Co (NH3)4 (NO,).,] N02I/
(Co (NH3)„ (NO,)2] NO; + NH3’= [Co (NH3)5 NOj] (NO.,).
[Co (NH3)6 (NO*)] (N02)2 + NH3'= [Co (NH3)0] (N02)3Если внутренняя сфера комплекса данного иона металла при раз¬
личных лигандах имеет один и тот же состав, то естественно предполо¬
жить, что она имеет и определенное геометрическое строение. До ра¬
боты над координационной теорией Вернер занимался изучением сте¬
реохимии соединений азота и был хорошо знаком с трудами Ле Беля
и Вант-Гоффа — основоположников учения о стереохимии. Он пред¬
положил, что комплексные соединения с координационным числом,
равным 6, имеют конфигурацию октаэдра, в центре которого находит¬
ся ион металла, а лиганды расположены в его вершинах:LКомплексные соединения с координационным числом, равным 4,
имеют тетраэдрическое (I) или плоскоквадратное (II) строение:II IIИзучение изомерных форм комплексных соединений определенного
состава и особенно предсказание и открытие оптической изомерии у
комплексных соединений, не содержащих углерода, позволило пол¬
ностью подтвердить гипотезы о геометрическом строении комплексных
соединений.Одной из трудностей, возникших на пути признания координа¬
ционной теории, было ее разногласие с существовавшей теорией ва¬
лентности. Рамки теории валентности оказались узкими для комплекс¬
ных соединений. Действительно, если соединение PtCl. соответствовало
теории валентности, то строение его комплексного производного
PtCla- 2КС1. т. е. KolPtCU, не могло быть объяснено с тех же пози¬
ций. Ион Pt(II) в этом комплексном соединении связан с четырьмя
кислотными остатками. Существовавшая теория валентности допус¬
кала образование между ним и ионами СГ только двух химических
связей. В связи с этим Вернеру пришлось ввести понятия главной и18
побочной валентностей. В простых соединениях атомы связаны глав¬
ными валентностями. После их насыщения центральный атом спо¬
собен присоединить дополнительные атомы или молекулы за счет
побочных валентностей.Относительно природы главных и побочных валентностей Вернер
предпочитал не высказываться. По сути, это была уступка устояв¬
шимся представлениям. Экспериментальная равноценность всех ли¬
гандов, находящихся во внутренней сфере большинства комплексов*
была доказана значительно позже.1.3. Переходные ряды Вернера — МиолатиВернер одним из первых принял теорию электролитической диссоци¬
ации Аррениуса. Основным методом, который использовал Аррени¬
ус при разработке этой теории, был метод электрической проводимости.
Кистяковский в лаборатории Оствальда выполнил интересное иссле¬
дование (1890), в котором на основании результатов электрической
проводимости водных растворов показал принципиальное различие
между комплексными соединениями и соединениями типа квасцов.Вернер быстро понял, какими огромными возможностями обла¬
дает метод электрической проводимости для исследования комплексных
соединений. В том же 1893 г., когда была опубликована основопола¬
гающая для координационной химии работа, появилась и первая статья
Вернера и Миолати, в которой на основе исследования электрической
проводимости растворов комплексных соединений были даны яркие
доказательства справедливости координационной теории.Известно, что молярная электрическая проводимость слагается
аддитивно из подвижностей отдельных ионов. Ионы одного заряда об¬
ладают примерно одинаковой подвижностью. Подвижности катионов
и анионов, имеющих равные по величине, хотя и противоположные
по знаку заряды, также различаются очень мало. Вследствие этого
стало возможно найти интервалы значений проводимости, характер¬
ных для электролитов определенного типа. Вернер и Миолати опре¬
делили интервалы значений электрической проводимости солен типа
1:1 (например, IVaCl), 1:2 и 2:1 (СаС12 и K2S04), 1:3 (А1С13) и т. д. Из¬
мерение проводимости комплексных соединений в одинаковых усло¬
виях позволило отнести их к тому или иному типу электролитов. Вер-Таблнца 1.1. Значение молярной электрической проводимости
в зависимости от типа электролита (при 25°С)Тнп электролитаИнтервал значений молярной электрической
проводимости. Ом-1 - см2 моль”100—51 : I97—1101:2, 2:1230—2681:3, 3:1352—4271:4, 4:1430—56019
нер составил следующую таблицу значеннй молярной электрической
проводимости для водных растворов электролитов (табл. 1.1).Ряд комплексных соединений, получающийся в результате посте¬
пенного замещения хлорид-ионов в К21РЮ4] на аммиак, выглядит
следующим образом:К, [Р1С14]; К [Pt (NH3) С1Д]; [Pt (NH3)2 Cl2]* [Pt (NH3)3 Cl] Cl; [Pt(NHs)4]Cl2По мер,е того как нейтральные молекулы аммиака замещают во внут¬
ренней сфере комплексного соединения KsIPtCl,] отрицательно заря¬
женные хлорид-ионы,заряд комплексного иона понижается на единицу.
Поэтому на первой ступени комплексное соединение из электролита
типа 2:1 (KJPtClJ) переходит в электролит типа 1:1 (K[PtNH3Cl3l).
Вследствие этого его. электрическая проводимость должна понижать¬
ся. На второй ступени замещения получается соединение [Pt(NH3)2Cl2],
которое не имеет ионов во внешней сфере и потому является неэлектро¬
литом. Дальнейшее замещение внутрисферных хлорид-ионов на ам¬
миак приводит к образованию комплексного иона катионного типа.
Для того чтобы комплексное соединение в целом было нейтральным,
хлорид-ион должен расположиться во внешней сфере: [Pt(NH:,):iCl|Cl.
В результате этого замещения снова получается электролит типа 1:1.
Замещение последнего хлорид-иона на аммиак приводит к образова¬
нию IPt(NH3)4]Cl2, т. е. электролита типа 2:1. Если комплексные
соединения K2[PtCl4] и K[PtNH3Cl3] являются солями комплексных
кислот H2[PtCl4] и H[Pt(NH3)Cl3], то комплексные соединения
|Pt(NH:,)3Cl]Cl и lPt(NH3)JCl2 являются солями комплексных осно¬
ваний [Pt(NH3)3Cl]OH и [Pt(NH3)4](OH)2. Эксперимент показал, что
электрическая проводимость растворов комплексов, входящих в
данный ря'д, изменяется в полном соответствии с координационной
теорией (рис. 1.1).На рис. 1.2 представлена аналогичная диаграмма изменения про¬
водимости в ряду комплексных соединений от K>[PtCl6] к [Pt(NH,)6]Cl4.
Электрическая проводимость соединений, входящих в этот ряд,
также закономерно изменяется, проходя через минимум.Работы Вернера и Миолати по исследованию электрической про¬
водимости комплексных соединений сыграли исключительно важную
роль в утверждении координационной теории. Ввиду большого зна-(PlfNH3)JCIj [PUNHjljClJ KjlPiCI,![PKNHj^ClJCl KlPiNHjClj]Рис. 1.1. Диаграмма молярной электрической проводимости
соединений ряда [Pt(NH3)4]Cl2 — Кг[Р^С14] (по оси абсцисс
отложен заряд комплексного иона)20
iPlfNHjlJCI* [PHN]r,)jCI,]CN • [Pt(NH3),CI4l K2[PICI61
[PKNHjIjCIICIj [Pl(NH3)3Clj]Cl K[Pt(NH3)CI3lРис. 1.2. Диаграмма молярной электрическом проводимости сое¬
динений ряда [Pt(NH3)6]CI4—Ko[PtCle] (по оси абсцисс отложен
заряд комплексного иона)чения диаграмм электрической проводимости ряды соединений, на
основе которых они были построены, и нм подобные называют пере¬
ходными рядами Вернера — Миолати.1.4. Координационная емкость лигандовКоординационной емкостью или дентатностыо* называют число
координационных мест, которое занимает у центрального атома' дан¬
ный лиганд.На основании большого экспериментального материала Вернер
пришел к заключению, что лиганды, состоящие из одного атома, всег¬
да занимают около данного центрального атома только одно коорди¬
национное место, несмотря на то, что валентность и состояние окис¬
ления, проявляемые этими лигандами, могут отличаться довольно
сильно. Например, существует комплексное соединение K3[VF6] и его
производное K2lVOF5]. Формально второе соединение можно рас¬
сматривать как продукт замещения одного из фторид-ионов в K3[VF„]
на лиганд О2". Состояние окисления центральных ионов в этих соеди¬
нениях различно. Его легко можно вычислить, зная состав внутренней
и внешней сфер. В комплексе K3lVFe] заряд комплексного иона равен
3—. Поскольку шесть фторид-ионов имеют суммарный заряд 6—,то
заряд иона ванадия должен быть равным 3+- В соединении K2[VOF5]
заряд комплексного иона равен 2—. Поскольку внутрисферные ли¬
ганды в сумме имеют заряд 7—, то состояние окисления нона ванадия
равно 5-г.Примером комплексного соединения с лигандом (N), занимающим
одно координационное место, но имеющим состояние окисления 3—,
является Ks[OsNCU]. Из алгебраической суммы зарядов следует,
что центральный ион имеет состояние окисления, равное 6+.* От лат. dentatus — имеющий зубы, зубчатый. Монодентатный — одно¬
зубый, бндентатный — двузубый и т. д.
Лиганды, занимающие в комплексе одно координационное место,
называются монодентатными.В октаэдрических комплексах с разнородными лигандами часто
различают два типа координационных мест. Например, в комплексекоординационные места, занимаемые четырьмя лигандами F3, назы¬
вают экваториальными, а координационное место, занимаемое ли¬
гандом Fa, называют аксиальным. Выделение экваториальных и
аксиальных положений в октаэдре весьма условно. Однако для не¬
которых типов комплексов их выделение вполне однозначно и не вы¬
зывает разногласий. Поэтому данная терминология получила в ли¬
тературе довольно широкое распространение. Она применяется и к
комплексам другой конфигурации, в частности к тригонально-би-
пирамидальным.Если лиганд полиатомный, то он может занять более одного коор¬
динационного места во внутренней сфере комплекса. Например, в
комплексном соединении [Co(NH3)4C03]C1 карбонатная группа (СОз )
проявляет координационную емкость, равную двум, т. е. является бн-
дентатной:Аналогичная ситуация наблюдается в оксалатном комплексегде оксалатный ион С2ОГ также бидентатен. Способность того или
иного иона проявлять бндентатность вовсе не исключает того, что
в некоторых случаях он будет в комплексе монодентатным, т. е. свя¬
занным с центральным атомом посредством только одного атома.В органических молекулах часто имеется несколько функцио¬
нальных групп, способных образовывать химические связи с ионами
металлов. Химические связи между центральным атомом и лиган¬
дом, как правило, носят донорно-акцепторный характер. Лиганд
проявляет электронодонорные свойства, а центральный атом электро¬
ноакцепторные. Для образования координационной связи функцио¬
нальные группы должны иметь неподеленные электронные пары.Этилендиамин NH2CH;CH2NH2 содержит две аминогруппы, атомыО?э\ 1FVFFэCIГ\ Г r\ NH3 \т jjС122
азота которых имеют неподеленные электронные пары. Обе они спо¬
собны образовывать координационные связи с ионом металла. Сущест¬
вует этиленднампновый аналог аммиачного комплекса [Co(NH3)0]Cl3,
а именно:NH— СИ— СИ,
- * INH-CH2— СН2.В этом комплексе каждая молекула этилендиамнна эквивалентна двум
молекулам NH3. Молекулы этилендиамнна занимают по два коорди¬
национных места, т. е. являются бидентатными.Функциональные группы органического лиганда могут быть раз¬
личными. Так, простейшая аминокислота глпкокол МН2СН2СООН со¬
держит аминогруппу (iVH2) и карбоксильную (СООН) группу. Обе
эти группы способны вступать в координационное взаимодействие с
ионами многих металлов. Например, известно координационное
соединение меди (II) с гликоколомН:СН,СNH>.'Си'NH,ОоСН2СН,Оно интересно тем, что все координационные места в нем заняты и
заряд центрального нона скомпенсирован кислотными остатками
—СОО". В результате комплексное соединение не имеет внешней сфе¬
ры и является неэлектролитом.Имеется большое число органических лигандов различной ден-
татности. Например, тридентатный лиганд диэтилентриамин
NH2CH2CH™NHCH2CH2NH2 или тетрадентатный лиганд
НООССН2NНСН2СН2NНСН2СООН. Полидентатные органические ли¬
ганды играют важную роль в химии. Одним из широко распростра¬
ненных полидентатных лигандов является этилендиамннтетрауксус-
ная кислотаHOOCCHnvчНООССН.^>nch,ch2n<^,сн,соонСНоСООНПолидентатные лиганды, содержащие электронодонорные функ-
.циональные группы различного типа — кислотные и основные, —23
называют комплсксонами. Наряду с кислотными и основными груп¬
пами некоторые комплексоны содержат нейтральные донорные груп¬
пы, например эфирные атомы кислорода или тиоэфирные атомы серы.Несмотря на то, что понятие координационной емкости было вве¬
дено Вернером в конце прошлого века, до сих пор здесь имеются не¬
определенности. Определим, например, координационную емкость
молекулы этилена в соли Цейзе KlPt(C:H4)Cl3]:Поскольку атомы углерода в этилене расположены на равных рас¬
стояниях от центрального атома, то координационную емкость мож¬
но было бы считать равной 2. Однако координационная емкость ли¬
ганда должна определяться числом монодентатных лигандов, которые
может заместить данный лиганд или быть ими замещенным. Посколь¬
ку соль Цейзе получается замещением хлорид-иона на этилен в
K2[PtCl4], то координационную емкость или дентатность этилена сле¬
дует считать равной единице.1.5. Номенклатура комплексных соединенийНаибольшее распространение имеет номенклатура комплексных (ко¬
ординационных) соединений, рекомендованная Комитетом по номен¬
клатуре неорганических соединений Международного союза по тео¬
ретической и прикладной химии (IUPAC). В соответствии с ней в
соединении вначале называют катион, а затем анион. Если соедине¬
ние неэлектролитного типа, то его называют одним словом. Степень
окисления центрального атома обозначают римской цифрой, заклю¬
ченной в круглые скобки. Нейтральный лиганд называют так же, как
и молекулу, а к лигандам — анионам добавляют на конце суффикс -о.
Для координированной молекулы воды также на конце употреб¬
ляют суффикс -о (акво-). Для обозначения числа одинаковых лиган¬
дов во внутренней сфере комплекса в качестве приставки перед на¬
званием лигандов используют греческие числительные да-, три-,
тетра-, пента-, гекса- и т. д. Приставку моно- не употребляют. На¬
звание комплексного иона пишут в одно слово. Например:[Со (NH3)„] С13 гексамминкобальта(Ш) хлорид[Со (NH3)5 Н,0] С13 аквопентамминкобальта(Ш) хлорид[Со (CH3NH2)5 С1] С12 хлоропентаметиламинкобальта(Ш) хлорид24
[Pt (NH.,), Вго] днбромодиамминплатнна[Со (NH3)3 (N02)a] тринитротриаммннкобальтВ комплексном ионе вначале называют лиганды с отрицательным
зарядом, а затем нейтральные.Слово аммин (с двумя «м») пишется применительно к аммиаку. Для
всех других аминов употребляется только одно «м».Если комплексный ион является анионом, то его название имеет
окончание -ат\<NH4)2 [PdC14] аммония тетрахлоропалладат(П)К [PtNH3Br3] калия пентабромоамминплатинат(1У)Если внутрнсферные лиганды сложные и, особенно, если они уже
включают в свое название приставки сhi-, три-, то для обозначения
их количества употребляют приставки бис-, трис-, тетракис-. На¬
звание сложного лиганда обычно заключают в круглые скобки:[СоЕп2С1=] N03 дихлоро-бис-(этнлендиамин)кобальта(1П) нитрат[Pt (PPh3)3Br]Br бромо-трис-(трифенилфосфин)платнны(П) бромидВ тех случаях, когда лиганд связывает два центральных иона, пе¬
ред его названием употребляется греческая буква Такие лиганды
называют мостиковыми и перечисляют последними:-ОН,К4(С,04)гСг^ \сг(с.04)гЧН0'калия тетраоксалато-^-дигидроксо-
хромат(Ш)NH,(NH3)4Co/ Vo(NH3)4
' 'OH'(N03)4 октаммин-^-амидо-ц-гидроксокобаль-
та(Ш) нитратНоменклатура позволяет отражать пространственное строение
координационных соединений. 05 этом будет сказано в разделе, по¬
священном изомерии комплексных соединений.Координационная теория обобщила большую область химии и дала
объяснение с единых позиций огромному числу соединений, их стро¬
ению и взаимосвязи. При разработке координационной теории Вер¬
нер был вынужден выйти за рамки существовавшей теории валент¬
ности. Введенное им понятие координационного числа сыграло и про¬
должает играть огромную роль в современной науке. В современной
химии оно имеет ничуть не меньшее значение, чем валентность в клас¬
сическом смысле. В настоящее время химики часто отождествляют по:
нятня координационное число и валентность.Огромная ' заслуга координационной теории состоит н в том, что
благодаря ел в неорганическую химию были введены стернческие25
представления. Таким образом, координационная теория способст¬
вовала переводу неорганической химии на качественно новый науч¬
ный уровень.Свою жизненную силу координационная теория неоднократно
проявляла в предсказании фактов и явлений. Как известно, это об¬
стоятельство считается главным критерием научной значимости тео¬
рии.1.6. Электрическая проводимость комплексных
соединений в растворахМетод электрической проводимости продолжает оставаться одним из
важных методов исследования комплексных соединений. Довольно
часто он позволяет определить тип электролита, а следовательно, вы¬
сказать вероятное суждение о внутренней и внешней сфере комплекс¬
ного соединения. В использовании этого метода имеются существен¬
ные ограничения. Он применим к сравнительно прочным комплексным
соединениям, существующим в индивидуальном состоянии. Исследуе¬
мые соединения не должны химически взаимодействовать с раствори¬
телем. Это означает, что склонность растворителя к образованию ком¬
плексного соединения с данным центральным атомом должна быть
значительно ниже, чем склонность к комплексообразованию уже
координированных лигандов.В растворах комплексные соединения диссоциируют на составные
части. Во-первых, по типу сильных электролитов происходит распад
на комплексные ионы и ионы внешней сферы. Например:[Со (NH3),J Br2 = [Со (NH3)6]2+ + 2Вг"К2 [PtCl4] = 2К+ + [PtCUFТакую диссоциацию называют электролитической (раньше называли
первичной), так как она ничем не отличается от диссоциации простых
солей, например СаВг2 или K2S04.Комплексные ионы распадаются на составные части ступенчато
по типу слабых электролитов, например:ptci|— ** ptci;f + cr Ptcio =** ptci+ + cr
PtCljf = Ptcu + Cl- Ptci+ ** Pt-+ + Cl-Как уже было сказано во введении, продукты ступенчатой диссоциа¬
ции координационно насыщены. На освободившиеся во внутренней
сфере координационные места вступают молекулы растворителя. Та¬
ким образом, в растворе (например, в водном) эти равновесия правиль¬
нее писать следующим образом:[PtC14]2- + Н20 ** [Pt (Н20) С13Г + СГ
[Pt (Н20) С13Г + Н.,0 ** [Pt (Н,0)2С12] + С1-
[Pt (Н20)о С12] + Н20 [Pt (H;0)3 C1J+ + С1-
[Pt (Н20)3 C1J+ + Н,0 ^ [Pt (Н20)4]2+ + СГ
26
В связи с этим диссоциацию комплексных ионов называют сольволн-
тической (раньше называли вторичной).Для простоты написания в формулах соединении часто не указы¬
вают молекулы растворителя. Отсюда возникла необходимость наряду
с координационным числом введения понятия л'игандное число — число
координированных лигандов, за исключением координированных мо¬
лекул растворителя.1.6.1. Молярная электрическая проводимость
в органических растворителяхОпределение молярной электрической проводимости комплексных
соединений в водных растворах используется очень широко. Однако
вода как растворитель имеет существенный недостаток. Ее молекулы
обладают большой склонностью к образованию комплексных соеди¬
нений. Вследствие этого молекулы воды вытесняют внутрисферные
лиганды, что осложняет правильную интерпретацию строения изу¬
чаемых соединений. Кроме того, комплексные соединения со сложны¬
ми органическими лигандами часто не растворяются в воде. Однако
такие соединения во многих случаях хорошо растворяются в орга¬
нических растворителях. Поэтому для измерения электрическои про¬
водимости все чаще используют неводные растворители [1].При выборе неводного растворителя с целью его использования
для измерения электрической проводимости комплексных соедине¬
ний исходят из того, что он должен обладать высокой диэлектриче¬
ской постоянной и низкой вязкостью. В табл. 1.2 приведены эти ха¬
рактеристики для наиболее распространенных растворителей.Таблица 1.2. Свойства растворителей, используемых
для измерения электрической проводимости [I]РастворительДиэлектриче¬
ская постоян¬
наяУдельная проводи¬
мость, Ом~1-см2Ацетон20,75,8 -Ю"8Нитрометан35,96,56-10“7Нитробензол34,89,МО"7Метанол32,61,5-10-9Этанол24,31,35-10'°Ацетон и трил36,25,9•10-8Диметнлформамнд36,70,2—0,6- 10-7Диметил сульфок сид46,64,0-10-8Для измерения электрической проводимости используют орга¬
нические растворители соответствующей чистоты, свободные от воды
н растворенных газов. Из нескольких растворителей следует выбрать
тот, который обладает наименьшей склонностью к комплексообразо-
ванию с ионами металлов. Лучшим органическим растворителем счи¬
тается нитрометан. Если комплексное соединение растворяется в
нитрометане, то его нужно предпочесть всем другим растворителям.27
В табл. 1.3. приведены интервалы значений молярной электриче¬
ской проводимости для различных растворителей. В скобках приве¬
дены значения, которые не являются обобщением большого числа
исследований, а отражают лишь отдельные измерения.Таблица 1.3. Интервалы значений молярной электрической
проводимости электролитов различного типа, Ом-,*см2-моль"1РастворительТип электролита1:12 : I (1 : 2)3:1 (1:3)4 : 1 (1 : 4)Нитрометан75—95150—180220—260290—330Нитробензол20—3050—6070—8290—100Ацетон100—1401&0—200(270)(360)Ацетонитрил120—160220—300340—420(500)Диметил форма мнд65—90130—170200—240(300)Метанол80—115160—220(290-350)(450)Этанол35—4570—90(120)(160)Нитробензол удовлетворяет многим требованиям, предъявляемым
к растворителям для целен измерения электрической проводимости.
Однако значения проводимости в нем более низкие, чем в нитрометане.
Диметилсульфоксид в качестве растворителя для измерения электри¬
ческой проводимости имеет два существенных недостатка: высокую
вязкость и склонность к образованию комплексных соединении.Для ацетона часто встречаются резкие отклонения в значениях
электрической проводимости от среднестатистических. Случается, что
электролиты типа 1:1 имеют в ацетоне такие же значения проводи¬
мости, как и электролиты типа 2:1. Если проводимость электролитов
типа 1:1, как правило, все же укладывается в интервал значений
100—140 Ом-• см2-моль"1, то данные для электролитов типа 1:2
и особенно 1:3 значительно менее надежны. Таким образом, ацетон
следует использовать-в случае крайней необходимости, т. е. когда в
других растворителях комплексное соединение не растворяется.Тип электролита комплексного соединения определяют по следую¬
щей схеме. На основе данных элементного анализа рассчитывают
молекулярную массу комплекса для его мономерного состояния, т. е.
его формульную массу. Исходя из формульной массы рассчитывают
концентрацию и измеряют электрическую проводимость комплекса.
Сравнение значений измеренной проводимости с табличными данны¬
ми позволяет отнести электролит к тому или иному типу.1.6.2. Способ идентификации полиядерных комплексовВ случае возможности образования полиядерных комплексов* воз¬
никают осложения с определением типа электролита. Например,* В полиядерных комплексах имеется несколько центральных атомов,
окруженных лигандами. В частности, они получаются, когда лиганды одновре¬
менно координируются около двух ионов металла, образуя мостики.28
принципиально можно представить два типа комплексов, отвечающих
одной и той же формульной массе ML5X2, где L — нейтральные ли¬
ганды, X — кислотные остатки. Один из них может быть мономер¬
ным, построенным по типу тригональноп бнпирамиды (I), а другой —
димерным с октаэдрическим окружением лигандами (II):LLмХ>"НLLL LL—I М 1/М / М /L/ I L ЛL LI IIЭквивалентная масса первого из них равна Ч, формульной массы.
Эквивалентная масса второго равна двум формульным массам, де¬
ленным на 4, т. е. также V2 формульной массы. Следовательно, экви¬
валентные концентрации и эквивалентные электрические проводи¬
мости этих двух комплексов должны быть одинаковы. Казалось бы.
на основании данных электрических проводимостей эти два типа комп¬
лексов различить нельзя. Однако такая возможность все же имеется.Установлено, что изменение, эквивалентной электрической про¬
водимости в зависимости от концентрации определяется зарядом ионов,
электролитов. Фелтхам и Гайтер предложили использовать эго для
установления зарядов ионов различных комплексов [21. Эквивалент¬
ная электрическая проводимость X связана с предельной проводимо¬
стью Ко п концентрацией С простым математическим выражением
X = Хо—С, где а — постоянная величина, зависящая от рас¬
творителя и температуры. Если построить график зависимости —К
от У С, то для различных электролитов получатся прямые линии
с различным наклоном (рис. 1.3) Угол наклона зависит or
заряда ионов электролита. Чем выше
заряд ионов, тем больше угол накло¬
на, несмотря на одинаковую эквива¬
лентную электрическую проводимость.■ Имея серию стандартных электро¬
литов, как, например, на рис. 1.3,
легко оценить заряд комплексного
иона. Это дает возможность отличить
электролит типа [ML5]X2 от электро¬
лита типа [MoL10]X4.Рис. 1.3. Зависимость /.0—?. от У С для
различного типа электролитов:1 — [Co(NH,).(NO:hlCl; 2 — [Co(NH3bNO?lCb; 3 —
[Со(Ж-Ы6]СЬ; 4 - K,[Fc(CN)cl
ГЛАВА2ТИПЫ КООРДИНАЦИОННЫХ СОЕДИНЕНИЙИз переходных рядов Вернера — Миолати следует, что существует
три типа классических вернеровских комплексов: 1) координаци¬
онные ацидосоединения, во внутренней сфере которых содержатся
только кислотные остатки; 2) молекулярные координационные со¬
единения, во внутренней сфере которых содержатся только лиганды
в молекулярной форме; 3) смешанные ацидо-молекулярные коорди¬
национные соединения.Как среди ацидокомплексов, так и среди молекулярных комплек¬
сов существуют соединения, содержащие во внутренней сфере разные
ацидолнганды и разные нейтральные лиганды. Так, для металлов
VIII группы периодической системы нередки случаи, когда во внут¬
ренней сфере находятся кислотные остатки двух или более кислот.
В растворах особенно часто встречаются смешанные молекулярные
координационные соединения, одним из лигандов в которых явля¬
ются молекулы растворителя.Изложение фактического и теоретического материала этой книги
основано главным образом на этих трех типах соединений. Однако
имеются и другие весьма специфические типы координационных со¬
единений, которые и рассматриваются в данной главе.2.1. Внутренние координационные соединенияПолидентатные лиганды, обладающие как нейтральными, так и от¬
рицательно заряженными электронодонорными группами, могут об¬
разовывать особый тип координационных соединений. Если такие
лиганды насыщают внутреннюю координационную сферу и полностью
нейтрализуют заряд иона металла, то получающиеся соединения на¬
зывают внутренними координационными или внутрикомплексными
соединениями. Примером внутреннего координационного соединения
может служить комплекс меди (II) с гликоколем:Бидентатные лиганды, в которых один донорный центр нейтраль¬
ный, а другой заряжен отрицательно, могут образовывать внутрен¬
ние координационные соединения в том случае, если координацион¬
ное число металла равно его удвоенному положительному заряду. Так,сн,—\Н-,0 с=о30
ацетнлацетон в енольной форме СН3—СО—СН=С(ОН)—СН3 с хро¬
мом (III) образует внутреннее координационное соединение, строе¬
ние которого можно отразить классической структурной формулойЕсли заряд иона металла нейтрализован, а координационные места
заполнены не полностью, то они могут быть заняты молекулами раст¬
ворителя, например, в комплексе магния (II) с оксихинолиномПоскольку внутренние координационные соединения являются
неэлектролитами, то они часто плохо растворимы в воде, но хороша
растворимы в органических растворителях, в том числе и в раство¬
рителях с низкой диэлектрической проницаемостью. Последнее об¬
стоятельство позволяет разрабатывать экстракционные процессы с
целью разделения и очистки отдельных элементов.' Малая раствори¬
мость внутренних координационных соединений в воде широко ис¬
пользуется в аналитической химии для выделения ионов металлов
из растворов. Растворимость внутренних координационных соеди¬
нении можно регулировать введением в лиганд гидрофильных или
гидрофобных функциональных групп.Большое число бидентатных лигандов для внутренних координа¬
ционных соединений синтезировано на основе салицилового альдегида
путем реакции конденсации с аминами:Салицилальдимины образуют внутренние координационные со¬
единения с большинством d-элементов, особенно первого переходного
ряда, типаII+ NHoR ** ]-г Н2031
Синтез, таких комплексов часто проводят исходя из салицилальде-
гидных соединении соответствующих металлов с аминами, например+ 2Н,0RRК такому же соединению можно прийти взаимодействием тетра-
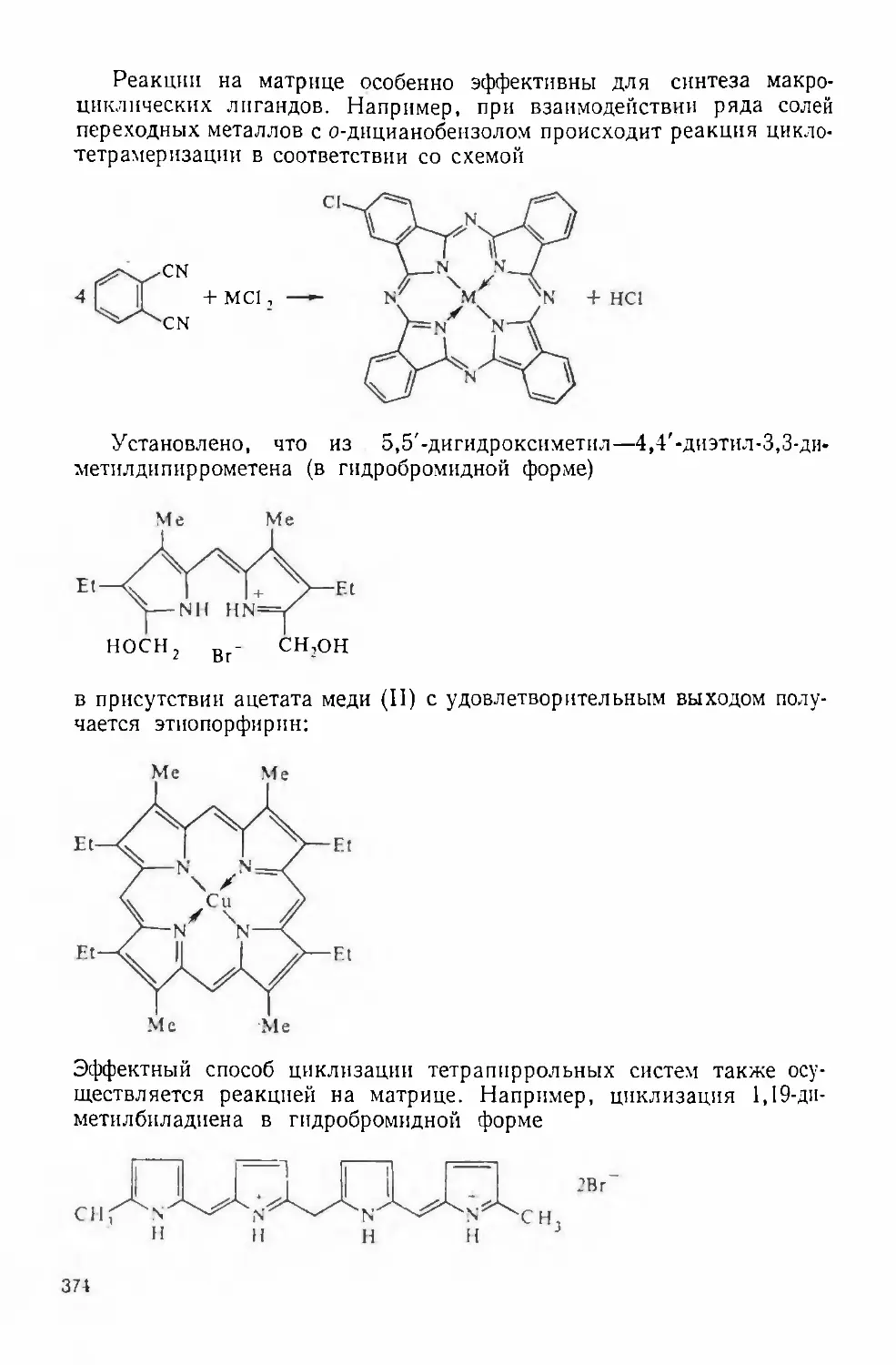
мннного комплекса с салициловым альдегидом по уравнениюТакого рода реакции называют синтезом на матрице или темплатным
синтезом (см. гл. 13).К внутренним координационным соединениям весьма близки ком-
плексонаты металлов.В настоящее время интенсивно исследуются координационные соеди¬
нения с довольно сложными органическими молекулами, содержащими
несколько функциональных групп с электронодонорными свойствами.
К числу таких соединений относятся комплексоны. Классическим
комплексоном является этилендиаминтетрауксусная кислота (ЭДТА)НООС —СН2 ч /СН2—соон•сно+ 2(NH3R)C1 + 2HjC>RR2.2. Комплексонаты металловНООС—сн.2>:N CH., СНо N/\■сн2—сооншироко распространенная в разных странах под торговыми назва-
ниямн трнлон Б, титриплекс III, хелатон III, иргалон, версен. ЭДТА
в растворах существует в виде цвпттер-ионаНООС—СНо + ,сн2-соо~H_^NCH»CH„ N/_h
/ " \ I~оос—сн/ х:н2—соонПри взаимодействии ионов металлов с комплексонами образуются
прочные координационные соединения вследствие замыкания не¬
скольких хелатных циклов. Комплексные соединения металлов с
комплексонами называют комплсксонатами. В отличие от внутренних
координационных соединений комплексонаты металлов могут иметь
заряд, т. е. могут быть катионными или анионными координационными
соединениями.Если хелатный эффект (см. гл. 5) проявляется для бидентатных
лигандов, то для полидентатных лигандов он проявляется в еще
большей степени. Например, в ряду лигандовNH2CH2COO" NH (CH2COO)Т2~ N (CH2COO)|~Г- ИМДА*- НТА’-устопчивость комплексов никеля (II), образующихся по уравнению
Ni2+ + Ln" ^ NiL(2~")+равна: lg Кг — 6,2; lg /Симла = 8,0; Ig /Снта = 11,3.Руководящим принципом в синтезе комплексонов является правило
циклов Чугаева. Оно позволяет планировать чередование в молекуле
электронодонорных групп и создавать потенциальные возможности
для замыкания металл-хелатных циклов (см. гл. 5).В настоящее время промышленностью, выпускается огромный ас¬
сортимент комплексонов с различными функциональными группами.
Широкое распространение получили комплексоны на основе произ¬
водных фосфоновой кислоты, например нитрилотриметилфосфоновая
кислота,СН2—Р03Н2
Н2Р03 — CH., — N<^\сн,-ро3н2Хорошими лигандными качествами обладают фосфоновоуксусные
комплексоны, например(НО)оР (О) СН,. ,СН2Р (О) (ОН),>NCH2CH2N(НООС—СН/ хсн2—соон«Конструирование» молекул различных комплексонов позволяет
в широком диапазоне изменять свойства комплексенатов металлов:
устойчивость к диссоциации, растворимость, термическую стабиль¬
ность. Электронодонорные атомы в комплексоне должны отстоять
друг от друга на такое число «звеньев», чтобы при взаимодействии с2—81733
поном металла имелись условия для замыкания пяти- или шестичлен¬
ных циклов. Так, ЭДТА с ионом металла может образовывать шести¬
координационный комплекс с пятью хелатными циклами:Варьируя функциональные группы, можно создавать комплексоны,
образующие соединения с различными ионами металлов и с различной
прочностью.Комплексоны очень широко применяют в химии, химической тех¬
нологии, технике, медицине и сельском хозяйстве. Применение ком¬
плексонов началось с аналитической химии. Этплендиаминтетраук-
сусную кислоту условно можно обозначить Н4У. В зависимости от
числа замыкаемых циклов при взаимодействии с ионом металла будет
освобождаться различное число ионов водорода, например по ре¬
акцииМя++ Н4У MY"~4 + 4Н+На измерениях pH были основаны первые варианты комплексономет-
рлческих титрований в аналитической химии.Поскольку комплексонаты часто являются очень прочными соеди¬
нениями, в аналитической химии комплексоны используются для мас¬
кировки отдельных ионов. Образование комплексонами прочных со¬
единений с ионами щелочно-земельных металлов уже на первых порах
было использовано в промышленности для умягчения воды. Комп¬
лексоны широко используются в подготовке воды для паросиловых
установок, для очистки промышленных сточных вод, для очистки
трубопроводов от отложений солей, для отмывки теплоэнергетиче¬
ского оборудования, для металлизации диэлектриков. Комплексоны
также применяют для выведения из организма ионов металлов и
т. д.2.3. Комплексы с макроциклическими
лигандамиВ последние годы химики стали проявлять большой интерес к коор¬
динационным соединениям с макроциклическими лигандами. Счи¬
тают, что такие соединения играют важную роль в жизнедеятельности
животных и растительных организмов.Комплексы с макроциклическими лигандами являются объектами
бионеоргапической химии — нового раздела современной химии.
Среди них имеется несколько хорошо исследованных ионофоров —
биологических переносчиков ионов. Иопофоры способны переносить34
небольшие ионы через липидные барьеры и изменять проницаемость
и градиенты биологических мембран. К природным ионофорам отно¬
сят, например, валнномицин и энниатин. В кольцах этих циклических
соединений чередуются амидные и сложноэфнрные группы. Оба они
являются хорошими переносчиками ионов калия.В последние годы внимание ученых привлечено к синтетическим
циклическим полиэфирам, содержащим в остове только эфирные атомы
кислорода. Сравнительно просто удается получать такие эфиры с
различным числом атомов в кольце. Такие циклические эфиры назы¬
вают «коронами» или коронарными эфирами. Например:Гч0 °)3сОО>sJдициклогексил-14-корона-4 бен50-15-корона-5ovодибензо-18-корона-бПервая цифра в названии указывает число атомов или связей в
цикле, а вторая —■ число атомов кислорода. Атомы кислорода по
контуру цикла соединены группами —СН2—СН2— или —СН=СН—.
Коронарные эфиры рассматривают как модели природных ионофоров.
Установлено, что в результате ион-дипольного взаимодействия они
образуют сравнительно устойчивые комплексы со щелочными метал¬
лами. Их устойчивость существенно зависит от размера полости —
внутреннего пространства макроцикла. В табл. 2.1 приведены раз-Таблица 2.1. Диаметр полости и константы устойчивости
комплексов щелочных металловКоронарный эфирДиаметр полости,
нмNa*Is AC
К*Cs*Дицикл огексил-14 -корона-40,12—0,152,21,3 Бензо- 15-корона-50,17—0,223,73,62,8Днбензо-18-корона-60,26—0,324,45,03,6Дибснзо-21-корона-70,34—0,432,44,34,2Дибензо-24-корона-8——3,53,82*•35
меры полостей различных коронарных эфиров и константы устой¬
чивости соответствующих комплексов с ионами щелочных металлов.Используя соответствующие коронарные эфиры, за счет различ¬
ной устойчивости комплексов можно селективно извлекать те пли
иные ионы щелочных металлов.Способность коронарных эфиров связывать ионы щелочных ме¬
таллов используют в органическом сннтезе. Например, они могут
облегчать протекание анионных реакций с участием соответствующих
солей щелочных металлов. Иногда коронарные эфиры способствуют
растворению солей щелочных металлов в органических растворите¬
лях. Так, например, перманганат калия растворяется в бензоле, если
в нем содержится дибензо-18-корона-6. Применение коронарных эфи¬
ров перспективно в качестве катализаторов и ингибиторов реакций,
а также для создания ионообменных материалов нового типа.Среди комплексов с макроциклическими лигандами важное место
занимают соединения с порфиринами и фталоцианином. В общем виде
молекула порфирина имеет вид^7 ^6Сочетание в молекуле различных радикалов R позволяет получать
огромное число различных производных.Ион металла входит в центр порфиринового кольца, замещает
два иона водорода у вторичных аминных групп и образует координа¬
ционные связи с четырьмя атомами азота. По существу, получается
внутрикомплексное соединение.Особенностью металлопорфиринов является большая жесткость
молекулярной структуры, обусловленная плоскостным строением
этого макроциклического лиганда. В нем проявляется уникальное
л-электронное сопряжение по всему макроциклу. Поскольку в жест¬
ком макроцикле размер полости довольно строго фиксирован, то в
зависимости от размера иона металла порфирины образуют комплексы,
резко отличающиеся по прочности. Для порфиринов характерны яр¬
кая окраска, нерастворимость в воде и низкая растворимость в орга¬
нических растворителях.К металлопорфиринам относится хлорофилл, способствующий
фотосинтезу глюкозы из С02 и Н20:36
н2с=снRr'ooc—снсоосн3(здесь R = СН3 или СНО; R' = С20Н39).Из формулы видно, что молекула хлорофилла имеет скелет порфи-
рина. Такой же скелет имеет и тем крови, который входит в состав
белкового комплекса гемоглобина:НООС—сн,—сн2 н2с—сн2—соонВ составе гемоглобина гем крови посредством центрального иона же¬
леза связывает кислород и переносит его из легких к мышцам. Отдав
кислород, гемоглобин использует аминогруппы для связывания моле¬
кул С02 и переноса их в легкие.Порфирины обычно выделяют из природных объектов. Синтетиче¬
ским путем получают лишь простейшие представители этого класса
макроциклических соединений.Близок по структуре к порфиринам фталоцианин:Н,С=СНСН37
Фталоцианины ряда металлов являются прочными и красивыми кра¬
сителями. Их широко применяют для окрашивания искусственных
волокон, бумаги, резины, пластмасс, в больших количествах исполь¬
зуют, в цветной печати. Известен ряд запатентованных методов хлори¬
рования и сульфирования фталоцианина, входящего в комплексные
соединения меди и других металлов. Это позволяет варьировать цве¬
товые оттенки фталоцианиновых соединений и их свойства.Промышленный метод получения самого фталоцианина основан
на получении комплекса меди по реакцииЭтот комплекс без разложения возгоняется в вакууме' при 5703 С.
Ион меди не может быть удален из него действием сероводорода и ни¬
какими другими реагентами.Разнообразные азотсодержащие макроциклы в последнее десяти¬
летие получают конденсацией внутрисфгрных аминов (1,2-, 1,3-, 1,4-
диаминов) с кетонами, в частности с ацетоном. Так, этим способом
были синтезированы комплексы различных З^-элементов с макроци¬
клами типаКомплексы с лигандами типа I и II рассматриваются как модели при¬
родных металлопорфиринов и корриноидов (витамин В12 и его произ¬
водные).Установлено, что макроциклические лиганды образут гораздо
более прочные комплексы, чем аналогичные лиганды нециклического
строения. Эго наглядно вытекает из сравнения констант устойчивости
комплексов Cu(II):38
сн,lUC'' '''СIN"I ! ‘СП—Nil UN СН,\ / 'Си: +СН—Nlf, H^N—СН,IV
Ig А" ”23,9Большая устойчивость комплексов с макроциклическими лиган¬
дами в отличие от аналогичных лигандов с незамкнутым циклом про¬
является довольно широко. Это явление названо макроциклическим
эффектом. По форме оно аналогично хелатному эффекту (гл. 5),
который проявляется в увеличении устойчивости при переходе от
монодентатных лигандов к полидентатным. В данном случае оба
органических лиганда четырехдентатные. Различие заключается лишь
в том, что в свободном состоянии один из них циклический (III), а
другой (IV) нет.Существует мнение, что основные причины этих двух эффектов раз¬
личны. Главной причиной макроциклического эффекта является благо¬
приятная для комплексообразования конфигурация макроцикличе¬
ского лиганда и его меньшая сольватация. Макроцикл представляет
собой уже готовый хелатный узел. Вследствие этого не требуется
затраты энергии на его формирование («сгибание» молекулы). В макро¬
цикле донорные группы располагаются внутри кольца. Это затрудняет
их сольватацию в свободном состоянии лиганда и требует меньшей
затраты энергии десольваташш при комплексообразовании.Для макроциклов типа порфиринов и фталоцианина' важным фак¬
тором, влияющим на устойчивость комплексов, является экраниро-
ва ние хелатного узла л-злектронным облаком сопряженной системы.
Вследствие экранирования затрудняется ‘ атака хелатного узла ре¬
агентами или молекулами растворителя. Экспериментально установ-
ле но, что скорость реакций комплексов с макроциклическими ли¬
га идами типа порфиринов, но без двойных связей в макроцикле, в
103 раз выше скорости реакций комплексов с самими лорфиринами.2.4. Многоядерные комплексыНаиболее распространенной причиной образования многоядерных
комплексов является способность некоторых лигандов связываться
с двумя или тремя ионами металла. Такие лиганды называют мости-
ковыми. Соответственно мостиковыми называют и комплексы. В пре¬
дыдущих разделах уже неоднократно рассматривались мостиковые
комплексы, образованные за счет галогенидных лигандов, например:сн,СН.—С\С ^С(СП-),I I J-СП,—NH UN—СП,- Ч ж" -
Cu:r/ \СН,—{Ш HN—СП,■: I I '(СИ 1C- СН(СН-)' ■ сГг:
ш1ёЛ=:839
ck\ /С1\N Dt / \ |NH/NH3m:iВ данном соединении два типа хлоридных лигандов: мостиковыеи кон¬
цевые (пли терминальные). Эти два типа лигандов легко различаются
по колебательным частотам валентных колебаний связей Pt—'Cl,
проявляющимся в длинноволновых ИК спектрах. Значение vpt—ci
мостикового хлоридного лиганда на 60—90 см'1 меньше, чем конце¬
вого.Следует отметить, что в полиядерных комплексах внутренние
координационные сферы всегда заполнены. Для образования много¬
ядерных комплексов необходимо наличие соответствующего числа
неподеленных электронных пар у лигандов, образующих мостик.
При синтезе мостиковых комплексов не должно быть большого избытка
свободных лигандов. Иначе происходит расщепление мостиковых свя¬
зей в соответствии с уравнениемCk\PtNH,,Ck /NH,
/ \ pt /\ / \
ч:р 'ci4- 2KCI = 2Kг Ck,c\-/L NH;/ \ClЕстественно, что склонность превращения полиядерных комплексов
в моноядерные путем расщепления мостиковых связей зависит от
прочности этих связей и устойчивости комплексов.Принципиально возможны одноатомные мостики, например:F/\
М М•нIо
/\
м мS/\
м мнч ,н
/к\мммIо о
/\ /\
м мм мВ них используются неподеленные электронные пары, принадлежащие
одному и тому же атому.Роль мостиков могут играть полиатомные лиганды, например:М—CN—М М—NCS—МО ОSSS/\
о о/ \Л1мRIС/Чо о
/ \
м мВ таких мостиках используются неподеленные электронные пары,
принадлежащие разным атомам полиатомного лиганда. Мостиковые
лиганды могут связывать различные ионы металлов. Так, А. А. Грин¬
берг осуществил следующую реакцию:NH3SCN 'fNH3 SCNAglPt+ AgN03-+-Pt_NH3SCNNH3 SCNN0,40
Роданидный лиганд в продукте реакции связывает платину (II) и се¬
ребро (I). Ион серебра в этом соединении координационно ненасыщен.
Автором установлено, что в твердой фазе при 135°С происходит поли¬
меризация биметаллического комплекса платина (II) — серебро (I)
за счет координационного насыщения серебра роданогруппой сосед¬
него комплекса. Полимеризации предшествует изомеризация комп¬
лекса. В результате получается соединение строенияNH3 NOT NH3I - I—NCS—Pt—SCN—Ag+—NCS—Pt—SCN—Ag+NOrINH,NH,На основе координационных соединений создано большое число
термокрасок — соединений, изменяющих цвет при определенной
температуре. К числу обратимых термокрасок относится комплекс
состава Ag,HgI4. При температуре около 40°С из светло-желтого
он превращается в оранжевый. При охлаждении первоначальный цвет
быстро восстанавливается.Можно предположить, что Ag2Hgl4 является координационным
полимером, в котором как ион Hg2+, так и ионы Ag+ координационно
насыщены. Все иодидные лиганды в нем, вероятно, мостиковые. По-
видимому, изменение окраски при нагревании связано с ослабле¬
нием или даже разрывом мостиковых связей и образованием Agl и
Hgl2. При охлаждении мостиковые связи восстанавливаются. Напом¬
ним, что HgL имеет яркий оранжево-красный цвет.Существуют мостиковые соединения, в которых лиганд связывает
комплексные соединения одного и того же металла, но в различных
степенях окисления. А. А. Гринберг и Ф. М. Филинов синтезировали
и исследовали соединение состава [Pt(NH3)2Br2] • [Pt(NH3)2Br4]. Они
предположили, что этот комплекс имеет мостиковое строениеN Н,NH,NJhNH,Br-Pt~Br--■■Br-Br-Pt-Br-Br ---PtBr■Br--Pt-BrNH,NH,NH,.JBrNibРентгеноструктурное исследование, выполненное С. Броссе, полностью
подтвердило это предположение. Недавно показано, что молекулярный
комплекс [Pt(NH2C2H6)4]C]2-[Pt(NH2C2H6)4Cl2]Cla имеет аналогичное
строение. Мостиковые полиядерные соединения платина (II) — пла¬
тина (IV) обладают анизотропными полупроводниковыми свойствами,
т. е. их электрическая проводимость зависит от направления в кри¬
сталле, в котором приложено напряжение.Систематические исследования кинетики и механизма окислитель¬
но-восстановительных реакций привели Таубе к заключению, что
перенос электрона между двумя комплексами происходит через об-41
разующийся лигандный мостик. Например, обмен электроном между
[Сг(Н20)612+ и [Co(NH3)5C1]3+ происходит в промежуточно образующем¬
ся мостиковом комплексен,он„о--Сг-Н,0NH-С 1-Н,0-Со-NH,-NH,NH,Н,0NH1,Перенос электрона в этом комплексе происходит через хлоридныи
мостиковый лиганд.Большое разнообразие полиядерных комплексов получено благо¬
даря использованию органических лигандов, содержащих несколько
донорных групп. Условием их образования является такое располо¬
жение донорных групп в лиганде, которое не позволяет замыкаться
хелатным циклам. Например, при взаимодействии K3[PdCI4] с ди-
тиоэфирами RSCH2CH2SR ii RSCH2CH2CH2SR образуются мономер¬
ные циклические соединения типаa s/ '>(СНг)„С Vгде п = 2 и 3. В случае дитиоэфиров RS(CH2)aSR, где п > 3, не соб¬
людаются условия правила циклов Чугаева и получаются комплексы
полимерного строения:R R R RII IICl S (СНо)„ S Cl Cl S (СН,)* S С1/ 'Yphj/ \p,i // \ / \ /Ч / \Cl Cl S (СН„)„ S CI С1I ■ IR RЛиганд а.а'-дипиридил всегда приводит к замыканию пятичленного
цикла и образованию мономерных комплексов (V). Напротив, у.у'-/=\ /=\^ Г \ //\n^ VIдипириднл принципиально не может замыкать хелатный цикл. Он
способствует образованию полиядерных комплексов (VI). НерздкиV42
случаи, когда лиганд имеет возможность замыкать хелатный цикл и
одновременно выступать в роли мостикового. Примером может слу¬
жить 1,6-диоксифеназин, с которым получены устойчивые высоко¬
молекулярные соединения Cu2+, Fe3+, Hg2+ и т. д. типаV. /у,С иОг\л-о/<Qn;v ,n.ЖА. А. Берлин и H. Г. Матвеева [1] наметили следующие пути син¬
теза полимерных полихелатных соединений.1. Полимеризация за счет радикала, содержащегося в мономерном
хелатном соединении, напримерСН=СН,
I ‘/С чпх/ ')У\ч// \—СН—СНо—I ' ■/с\*ч /Y/МЧ .2. Образование полимеров посредством реакции конденсации с
участием координированного лиганда/\2></ "NH2NH:C2H4NHo — 2Н„0 +СНО+)м<■ NHo—SNH„ —-nh2s—NH./ \sCH=NCoH4N = CH'К этому типу реакций можно отнести синтез полимерного соединения
по уравнениюITч /сн,сн,ос,н,43
Такие полимерные вещества обладают рядом полезных свойств, но, к
сожалению, их термостойкость низка.3. «Сшивание» ионами металлов полимерных цепочек—СН—СН ~I 2Rу/ \у
\ /2~СН—СН ~ + М"+—*- Мп+I Y/XXА \7/ \ RX у I~сн—СН2~4. Синтез полимеров, исходя из мономерных ^полидентатных ли¬
гандов и ионов металлов::>\/м\По существу, это также «сшивание», только не полимерных цепей, а
мономерных молекул.Современная координационная химия позволяет прогнозировать
свойства полиядерных хелатных соединений. В зависимости от иона
металла и природы органических лигандов можно получать полихе-
латные соединения, обладающие ценными свойствами: высокой термо¬
стойкостью, заранее заданными электрическими, магнитными и спект¬
ральными характеристиками. Разнообразие свойств может быть дос¬
тигнуто также включением в полимерную цепь ионов нескольких ме¬
таллов,* т. е. синтезом гетероядерных металлополимеров.2.5. Комплексы со связью металл — металлСоединения со связью металл—металл известны давно. К их числу
относятся соли ртути (I). Многие экспериментальные данные свиде¬
тельствовали о существовании двухъядерного катиона Hgjf. Ион
Hg(I) должен иметь неспаренный электрон и, следовательно, должен
быть парамагнитным. На самом деле соединения ртути (I) диамагнитны.
Значит, происходит спаривание электронов. Это может быть лишь
следствием образования двухъядерного катиона Hg2+. Многочис¬
ленные рентгеноструктурные данные указывают на наличие связи
Hg—Hg в соединениях типа Hg2X2, где X—N03 и галогенидные ионы.
Вероятно, связь Hg—Hg образуется вследствие перекрывания sp-гнб-
ри дных орбиталей.Первыми координационными соединениями со связью металл—44
металл, которые стали достоверно известны, были ацетаты меди (II)
и хрома (II). Их димерные структуры со связью Си—Си н Сг—Сг вы¬
явлены в 1953 г. Оказалось, что они имеют строениеСН —С;Н,0Iсн.-с^:м;Ч-О .ус—сн,4-™,''А'Н20Несколько позже И. И. Черняев с сотр. синтезировал ацетат и фор¬
миат родия (II). М. А. Порай-Кошиц и А. С. Анцышкина провели
рентгеноструктурное исследование и установили, что их структура
аналогична димерным ацетатам меди (II) и хрома (II). Впоследствии
такого рода структуры получили название «фонариковых».В настоящее время известно, что большинство металлов способно
образовывать соединения со связью металл—металл. Их образуют как
легчайший металл литий в тетрамерном соединении (LiCH3)4, так и
один из наиболее тяжелых элементов — уран в соединении
(U302)2(0H)4(S04)e. Однако наиболее характерны такие связи для d-
элементов, особенно переходных металлов, расположенных в сере¬
дине рядов.Прочные связи между атомами металла встречаются, например,
в соединениях хрома, вольфрама, рения. Комплексы [Мо2(0,ССН3)4],
[W2C19]3~, IRe,Cls]2_ и их производные являются классическими при¬
мерами соединений с, кратными связями металл—металл.Считают, что образование прочных координационных соединений
со связью М—М наиболее характерно для переходных металлов VI,
VII и VIII групп. У атомов этих металлов число электронов достаточ¬
но для образования связей не только с лигандами, но и между собой.
Следует отметить, что образование таких соединений наиболее харак¬
терно для элементов в низких степенях окисления. Высокий эффектив¬
ный заряд на ионах металлов вследствие электрического расталки¬
вания должен препятствовать образованию связей металл—металл.Часто образованию связи металл — металл способствуют мости¬
ковые лиганды, как в фонариковых соединениях. Однако нередки
случаи образования связи металл — металл и без мостиковых лиган¬
дов. Например, оказалось, что зеленая соль Магнуса построена по
типу бесконечных колонок:NH, Cl NH, С1МГ,^
Pt-N11,ClPt-PlPt-Ml,NH,C!NH,NH,1-45
Мостиковые связи в этом соединении отсутствуют.Известно большое число координационных гетерополиядерных
соединений со связью металл — металл. Например, получены плати¬
новый и палладиевый комплексыРу(СО)6 Мп-М—Мп(СО)6
IРуПо существу, в этих соединениях роль лигандов платины (II) и пал¬
ладия (II) играют группы Mn(CO)s. Хорошо изучен комплекс
[Pt(SnCl3)5]3~. Он имеет строение тригональной бипирамиды:SnCIj\/.CljSn |SnCJjВ большинстве приведенных соединений каждый атом металла связан
с другим только ст-связью.К числу координационных соединений со связью металл — металл
можно отнести PteCI12, структура которого представлена на рис. 2.1.
В этом соединении каждый атом платины связан одинарными ст-свя¬
зями с четырьмя другими идентичными атомами. Шесть таких атомов
составляют октаэдрический остов.Теоретические представления об электронной структуре даже
двухъядерных соединений со связью металл — металл в настоящее
время развиты еще довольно слабо. Описание химических связей в
этих соединениях основано во многом на качественном рассмотрениимолекулярных орбиталей. Соглас¬
но существующим представлениям
одинарная связь металл — металл
состоит из ст-компоненты (перекры¬
вание d3 -орбиталей атомов метал¬
ла). В образование двойной и трон¬
ной связей включаются д-компо-
ненты (перекрывание одной пли
двух napd* -АО металла). Четырех¬
кратная связь, помимо ст- и л-ком-
понент, должна иметь и 6-вклад от
перекрывания do -АО. Считается,
например, что в фонариковых кар-Рис 2.1. Расположение атомов в
(PtCl2)646
боксилатных комплексах родня Rh2(RC00)4-2H20 связь тройная: од¬
на сг-типа и две я-тппа. В двухъядерных комплексах хрома (II), мо¬
либдена (II) и вольфрама (II) связь металл — металл часто проявля¬
ется как четырехкратная связь: одна о-тнпа, две u-типа и одна 3-ти¬
па.Таким образом, связи металл — металл сильно отличаются по
кратности, а следовательно, и по длине.Суждение о наличии в молекуле связи металл — металл обычно
основывается на анализе расстояний М—М. Если они меньше, чем
сумма ковалентных радиусов атомов соответствующих металлов, то
считают, что связь М—М существует. Например, в комплексе W2Clg
три хлоридных лиганда мостиковые, а остальные — концевые (по
три у каждого атома вольфрама). Рентгеноструктурный анализ по¬
казывает, что расстояние W—W в этом комплексе равно 0,241 нм.
Ковалентный радиус атома вольфрама равен 0,140 нм. На этом ос¬
новании было сделано заключение, что наряду с тремя мостиковыми
связями W—Cl—W существует связь W—W.В комплексе Re2Cls мостиковых связей нет. Он имеет следующее
строение!С! С1
ReReCK^^ClСГ ClРасстояние Re—Re в нем равно 0,224 нм, а в решетке металлическо¬
го рения оно равно 0,274 нм. Таким образом, расстояние Re—Re на
0,05 нм меньше, чем сумма атомных радиусов, что свидетельствует о
высокой кратности этой связи. Считается, что в данном комплексе она
максимальна и равна четырем.Наиболее надежным методом исследования, который позволяет
непосредственно судить о наличии связи металл — металл, является
рентгеноструктурный анализ. Однако имеющийся фактический ма¬
териал свидетельствует о том, что длина связей металл — металл не
является простой функцией порядка связей. Она сильно зависит от
числа, размера, окружения и характера лигандов. Поэтому иденти¬
фикация кратности связей требует сравнительного изучения соедине¬
ния подобной стереохимии.Важную информацию о связи металл — металл .дает магнето-
химический метод. Понижение магнитного момента ионов металла в
комплексах по сравнению с их магнитным моментом в изолированном
состоянии является важным указанием на возможность образования
такой связи. Однако результирующее уменьшение магнитного момента
может быть обусловлено и другими причинами. Например, у тяжелых
переходных элементов легко осуществляется внутриионное спарива-47
нне электронных спннов благодаря снятию вырождения энергетиче¬
ских уровней из-за несимметричного окружения ионов металлов.
Для этих металлов большие значения констант спин-орбнтального
взаимодействия также часто приводят к малой магнитной восприимчи¬
вости даже при отсутствии связи металл —• металл. Однако чаще по¬
нижение магнитного момента происходит за счет образования мости¬
ковых связей без непосредственного взаимодействия атомов металла.Связь металл —• металл часто проявляется в виде сильных и ха¬
рактеристических полос в длинноволновой области ИК спектра, а так¬
же в спектрах комбинационного рассеивания (КР)- Одинарная связь
в двухъядерных комплексах характеризуется колебательными часто¬
тами 150—250 см-1 и силовыми константами* 50—150 Н/м, а для
трех- и четырехкратной связи характерны колебательные частоты в
области 300—410 см”1 и силовые константы 300—450 Н/м.Связь металл — металл практически всегда присутствует в клас¬
терных** соединениях (см., например, рис. 2.1). В настоящее
время химики проявляют к ним большой интерес. Одной из причин
этого является использование кластерных соединений в качестве
катализаторов различных процессов.Кластерные соединения рассматривают как особый класс коорди¬
национных соединений. Некоторый парадокс заключается в том, что
координационная теория Вернера, по существу, к ним неприменима.2.6. л-КомплексыСоединения металлов со связью металл — углерод относят к металл¬
органическим. Во введении уже было сказано, что во многих случаях
их с полным основанием можно рассматривать как координационные.
Эти соединения со связью металл — углерод играют исключительно
важную роль в современной химии.Несмотря на то что карбонильные, цианидные и изонитрильные
соединения также содержат связь металл — углерод, в этом разделе
они не рассматриваются. Как типично координационные соединения
они часто описываются во многих разделах.Первыми л-комплексами были соль K[Pt(C2H4)Cl3] и димерное
соединениеОни были получены датским химиком Цейзе в 1827 г.* В колебательной спектроскопии характеристикой прочности связи
является силовая постоянная f. Она соответствует силе, возникающей при из¬
менении межатомного расстояния на единицу длины, и измеряется в ньютонах
на метр. Силовая постоянная связана с частотой валентного колебания связи.
Для ряда связей между атомами с близкими массами иногда сама частота
используется для сравнительной характеристики прочности связи.** От англ. cluster — скопление, пучок, гроздь.48
Представление о характере химической связи ионов металлов с
этиленовыми и ацетиленовыми лигандами дано в гл. 7.Существует формальная классификация углеводородных комплек¬
сов металлов по числу электронов углеводорода, участвующих в об¬
разовании связи с металлом. Углеводородный радикал принимают
электрически нейтральным. Нейтральной, естественно, является и
углеводородная молекула. Эта классификация относится как к ст-,
так и к л-комплексам.Различают комплексы, в которых лнганд передает на связь с ме¬
таллом один, два, три, четыре, пять, шесть и семь электронов. К комп¬
лексам, лиганд в которых передает один электрон, относятся
алкильные и арильные соединения типа R—MLn. Связь металл —
углерод образуется за счет обобщения по одному электрону от атома
углерода и атома (или иона) металла. Для переходных! металлов,
способных наряду с о-связью образовывать л-дативные связи, ариль¬
ные соединения стабильнее алкильных. Это обусловливается пере¬
носом электронной плотности с d-орбиталей металла на разрыхляю¬
щие орбитали ароматических ядер.Широко распространенным способом получения металлорганиче-
ских соединений является взаимодействие галогенопроизводных угле¬
водородов с металлами. В 1849 г. Франкланд получил диалкилцинк
по уравнениям:C:H6I + Zn C„H5ZnI2C2H5ZnI — (С2Н5).. Zn + ZnljИсключительно важное значение имеет осуществленная в 1900 г.
Гриньяром реакция металлического магния с галогеналкилами в эфир¬
ной средеCH3I +Mg CH3MgIАлкильные производные цинка легко воспламеняются, что затрудняет
их использование. Магнийорганические соединения гораздо удобнее
в обращении и поэтому нашли широкое применение для получения
металлорганпческих производных других металлов, в том числе и
переходных.К числу комплексов, лиганд в которых донирует два элект¬
рона, относятся олефиновые и ацетиленовые. Олефиновые комплек¬
сы известны для платины (II), палладия (II), серебра (I), меди (I) и
никеля (0). Другие металлы образуют соединения с низшими олефи-
нами только в том случае, если имеются лиганды л-донорного типа,
например СО или С5Н5. Кроме того, металлы в таких соединениях,
как правило, находятся в низких степенях окислення.Ацетиленовые комплексы менее распространены, чем этиленовые.
В качестве ацетиленовых лигандов л-типа могут выступать лишь
замещенные RC=CR'. Иначе происходит образование ацетиленидов
RCaeC—ML„.К комплексам, лиганды в которых донируют три электрона,
относятся аллильные соединения. Аллильный радикал может быть
связан с атомом металла посредством сг- и л-связи:49
сн2
X \мС_>нн2с'Однако в этой системе возникает делокализованная связь между ат0*
мом металла и трехчленной системой аллильного радикала. Харак¬
терным и хорошо изученным комплексом такого типа является я-ал-
лильный комплекс палладия:ИХСНг\р,/С1\рн/СНгСИ// \с|/ \гнУ;сн,сн,Примером лигандов, которые образуют я-комплексы за счет во¬
влечения в связь четырех электронов, могут служить со¬
пряженные диены, в том числе и циклопентадиенон. Известно большое
число диеновых комплексов железа, например;В продуктах реакции хлорида палладия (II) с дифенилацетиленом
было обнаружено димерное соединениеPh-Ph--Ph-PhCl—Pd—ClCl—Pd—ClPh-Ph--Ph-PhАналогичное соединение известно и для никеля (II).Исключительно интересными являются я-комплексы с п я т и-
электронодонорными лигандами. К их числу от¬
носится аннон С5Н5. В 1958 г. Халлам и Посон получили соедине¬
ние, содержащее циклический сопряженный диолефнн циклопента¬
диен-1,3. я-Комплекс с циклопентадиенилом C5Hs называют ферро¬
ценом:50
Получение ферроцена открыло целую эпоху в элементорганической
и теоретической химии. Формально в ферроцен входит нон С5Н5,
который можно рассматривать как пятиэлектронный донор. Так же
как и в аллильных соединениях, в ферроцене имеет место делокализо-
ванная связь между атомом железа и пятичленной системой цнкло-
пентадиенильного кольца. Молекулу ферроцена можно записать сле¬
дующим образом:Ферроцен легко окисляется до феррициний-иона (C5H5)2Fe+. Соли
этого катиона (феррициния) парамагнитны и имеют голубой цвет.
Они сравнительно хорошо устойчивы. Для системы(С5Н5)2 Fe+ + е~ =д= (С3Н5)2'Ре
голубой коричневыйхарактерна хорошая обратимость. Ее окислительный потенциал
равен 0,649 В. Это позволяет использовать феррициний как мягкий
окислитель в аналитической практике. Введение заместителей в ли*
ганд позволяет изменять окислительно-восстановительный потен¬
циал. В настоящее время известно большое число циклопентадиено-
вых комплексов многих переходных металлов.Бензол и его производные являются лигандами, способными пос¬
тавлять для образования химической связи с атомом металла шесть
электронов. Шесть электронов способен поставлять на обра¬
зование связи циклогептатриен. Хром, молибден и вольфрам обра¬
зуют соединения типа
Производное циклогептатриена С7Н8 — анион С7Н; — является
семи электр онодонорным лигандом.Группировка М(СО)3 наиболее характерна-для я-комплексов пере¬
ходных металлов. Поэтому ее можно взять для иллюстрации зависи¬
мости между числом электронов, поставляемых лигандом, и типом
комплексов, образуемых различными металлами. В основе этой за¬
висимости лежит правило эффективного атомного номера (ЭАН)
(см. гл. 6). Для З^-элементов эффективным атомным номером является
36. Три карбонильные группы вносят 6 электронов. На долю атома
металла и органического лиганда приходится 30 электронов.В табл. 2.2 приведен ряд углеводородных комплексов, которые
существуют или принципиально могут существовать.Таблица 2.2. Углеводородные комплексы, соответствующие
классификации и правилу ЭАНАтомный но¬
мер металла2324252627Число элек¬
тронов от кар¬
бонильных ли¬
гандов66666Число элек¬
тронов от угле¬
водородного ли¬
ганда76543Комплекс(со)39Сг(СО)3М п
(СО)3171L(СО),/ч1Со(С013ОбозначениеVт,5-,3Координационное число всех перечисленных в табл. 2.2 углеводо¬
родных лигандов формально принимают равным трем. Это следует
из того, что комплексы переходных металлов чаще всего имеют коор¬
динационное число 6. Три координационных места приходятся на
карбонильные группы, а остальные — на углеводородные лиганды.
С этой точки зрения железо в ферроцене (C5H6):>Fe также имеет коор¬
динационное число, равное 6.По предложению Коттона металлорганическне соединения и соот¬
ветствующие им лиганды обозначают греческой буквой rj или латин¬52
ской /1*. Цифрой указывают число атомов углерода, с которым свя¬
зан центральный атом. Один и тот же олефиновый углеводород может
быть связан с металлом различными способами. Циклопентадиен-1,3
может осуществлять связь тремя способами:Обозначение такого комплекса: (г)1—С5Н5) (i]3—СЬН5) (v|5—C5H5)MoNO.
Нетрудно убедиться, что его электронный баланс отвечает правилу
ЭАН. Молибден имеет порядковый номер 42 и находится в 5-м периоде
периодической системы. Углеводородные лиганды предоставляют
1 +3+5 электронов, оксид азота — 3 электрона. Таким образом, мо¬
либден оказывается в окружении 54 электронов, что соответствует
правилу ЭАН.Конфигурации соединений, приведенных в табл. 2.2 и анало¬
гичных им, иногда называют «фортепьянной табуреткой». Это назва¬
ние, естественно, не относится к я-аллильным комплексам. Ферро¬
цен и аналогичные ему соединения часто называют «сандвичевымн»
и «двухпалубными».* От греч. haptein — прикрепить.
ГЛАВАИЗОМЕРИЯ КОМПЛЕКСНЫХ СОЕДИНЕНИЙИзомерами (от греч. isos — равный, meros — часть) называются хи¬
мические соединения одного и того же состава, но отличающиеся стро¬
ением и свойствами.Существование изомеров всегда служило для химиков исключи¬
тельно важным аргументом при построении теории строения химиче¬
ских соединений. Так было при разработке А. М. Бутлеровым тео¬
рии химического строения, так было и при разработке координаци¬
онной теории Вернером. Предшественники Вернера, в том числе
Бломстранд и Иергенсен, при создании «цепной теории» также опи¬
рались на факты существования изомерных комплексных соедине¬
ний. Важным шагом в развитии координационной теории была ин¬
терпретация пространственного расположения атомов в комплексных
соединениях.3.1. Геометрическая изомерияГеометрическая изомерия является следствием различного располо¬
жения лигандов в координационном полиэдре. Она преимущественно
изучена на соединениях с координационными числами 6 и 4.3.1.1. Комплексные соединения с координационным
числом 6Будучи убежденным последователем основоположников стереохимии
Вант-Гоффа и Ле Беля, автор координационной теории считал, что,
располагаясь около центрального атома, лиганды образуют опре¬
деленную геометрическую конфигурацию. При симметричном рас¬
положении шести равноценных лигандов вокруг центрального атома
возможны три геометрические фигуры: плоский шестиугольник, три-
гональная призма, октаэдр.Для комплексов типа МА4Х2 каждая из этих трех фигур допуска¬
ет определенное число изомеров. Для плоского шестиугольника их
может быть три:А Л XXXXорто-54мета-пара-
Примером тому могут служить диметилбензолы (ксилолы):/V/сн,\fIсн.оргпо- ксилолл«?та>кснлолСН3I/ч\sIсн,лага ксилолОни различаются по температурам плавления и кипения, по плот¬
ности п показателю преломления.Тригональная призма также допускает существование трех изо¬
меров для комплексов типаXXА/Ч/ I \А X/ 1 \А X/ 1 \;а>-•>*А* \/ \/ \/ \t VА А/А А' ■ ■■ ■ ЧДля октаэдра возможно существование лишь двух изомеров:
А АЭксперимент показал, что для комплексных соединений с коорди¬
национным числом 6 типа МА4Х2 существует не более двух изомеров.
На этом основании Вернер пришел к выводу, что комплексы с коор¬
динационным числом 6 имеют октаэдрическое строение. Изомер, в
котором лиганды X расположены на одном ребре октаэдра, называют
^«с-изомером (от лат. cis — Вхместе). Изомер, в котором, лиганды X
расположены по диагонали, называют транс-изомером (от лат. trans —
напротив). Изомерные соединения, как правило, отличаются по цве¬
ту, форме кристаллов и по отношению к определенным реагентам.
Например, комплекс 4«c-[Co(NH3)4(N02)2]C1 интенсивно-желтого цве¬
та, a m/WHC-[Co(NH3)4(N02)2]CI — оранжевого. Первый из них при
кипячении с концентрированной кислотой образует раствор синего
цвета, а второй — красного.Для комплексов состава МА3Х3 октаэдрическая модель также до¬
пускает существование двух изомерных форм:55
мXмеридиональный
(meridional),
или реберныйлмXлицевой,
(facial),
или граневыЙПримером могут служить изомерные комплексы состава
I Pt(NН3)3С13]С1. Изомер тег-[Р1(ЫН3)зС13Ю1 окрашен в зеленовато¬
желтый цвет. Его растворимость в воде при 20°С составляет 5,64%
(мае.). При восстановлении он образует комплекс [Pt(NH3)3ClJCI.
Изомер /ac-[Pt(NH3)3Cl3]Cl окрашен в бледно-желтый цвет. Его раст¬
воримость 1,49% (мае.). При восстановлении он дает комплекс состава
[PJ(NH3)2C12].Комплексы состава МА5Х не должны давать изомеров при любом
из рассмотренных симметричных полиэдров.Уместно отметить, что конфигурация правильного октаэдра реа¬
лизуется далеко не всегда. Чаще всего октаэдр несколько искажен.
Особенно часто это имеет место при неравноценных лигандах. Иска¬
жения могут быть связаны с различными расстояниями лигандов от
центрального атома. Кроме того, бывают искажения, связанные с
разворотом параллельных граней. Если октаэдр положить одной
гранью на плоскость листа бумаги и смотреть на него сверху вниз, то
станет очевидным, что параллельная грань повернута на угол 6(Г:аТаким образом, бывают искажения, связанные с изменением этого
угла. Если плоскость грани асе развернуть по отношению к грани
bgd на 60°, то октаэдр превратится в тригональную призму. Много¬
численные рентгеноструктурные данные показывают, что искажения,
связанные с разворотом параллельных граней, встречаются довольно
часто.Несмотря на небольшие искажения октаэдрических полиэдров,
химики все же называют такие комплексы октаэдрическими.В 1965 г. было установлено, что рениевый комплекс [Re(S2CoPh3)3l
и молибденовый комплекс [Mo(S2C2H2)3] имеют строение тригональной
призмы:56
Недавно получен комплекс Mo(IV) с 2-меркаптоэтнлсульфидом, так¬
же имеющий строение тригональной призмы [1]. Вероятно, в буду¬
щем число построенных таким образом комплексов увеличится. Умест¬
но сказать, что в органической химии получены соединения, которые
рассматриваются как производные бензола, построенные по типу три-
гональноп призмы.3.1.2. Комплексные соединения с координационным
числом 4Комплексные соединения с координационным числом 4 распростране¬
ны гораздо меньше, чем с координационным числом 6. Однако они
хорошо изучены и сыграли важную роль в развитии координационной
химии. Существуют лишь две симметричные геометрические фигуры,
которые могут отражать строение комплексных соединений с коорди¬
национным числом 4. Это плоский квадрат и тетраэдр. Равноценность
четырех лигандов вытекала бы также из конфигурации комплекса в
виде тетрагональной пирамиды, в вершине которой находится ион
металла. Однако в этом случае лиганды не окружали бы центральный
атом, а были бы по одну сторону от него.Для тетраэдрических комплексов MAmX4_m геометрическая изо¬
мерия не проявляется ни при каком составе комплекса. Комплексы,
имеющие строение плоского квадрата при составе МАХ3 также не
должны иметь изомеров, а при составе МА2Х2 существуют в двух
изомерных формах:XАXмМААтранс-Исходя из существования диаминдигалогенидных комплексов
платины (II) в двух изомерных формах, Вернер пришел к выводу, что
комплексы платины (II) имеют плоскоквадратное строение. Класси¬
ческим примером изомерных комплексов плоскоквадратного типа
являются диамминдихлоридные соединения платины:CINHjClPtPtNH3NH3цмс-транс-NH,CI57
Они отличаются по цвету, растворимости и по отношению к некото¬
рым химическим реагентам.Кроме соединений платины (II) плоскоквадратное строение имеют
комплексы палладия (II), никеля (II), золота (III), меди (III), се¬
ребра (III), а также некоторые комлексы меди (II) и серебра (II).
Однако геометрические изомеры комплексов плоскоквадратного типа,
кроме платины (II), известны лишь для палладия (II).Тетраэдрические комплексы характерны для кобальта (II), нике¬
ля (II), цинка (II), кадмия (II), ртути (II), алюминия (III), галлия(III), индия (III), таллия (III). Иногда встречаются тетраэдрические
комплексы и других металлов.Из перечисленного следует, что комплексы никеля (II) с координа¬
ционным числом 4 существуют как в тетраэдрической, так и в плоско¬
квадратной форме. Устойчивость той или иной конфигурации зависит
от природы лигандов и даже растворителя. Иногда эти формы на¬
ходятся в таутомерном равновесии.Если изомеры находятся в таутомерном равновесии, то полярный
растворитель, как правило, смещает равновесие в сторону более по¬
лярной формы. Поскольку ^«с-изомеры более полярны, чем транс¬
изомеры, то полярные растворители способствуют смещению равно¬
весия в сторону образования комплексов ^«с-конфигурации.Целенаправленный синтез соединений заданного строения и, в
частности, геометрических изомеров является важнейшей проблемой
химической науки. В химии координационных соединений ряда ме¬
таллов, и прежде всего платиновых, руководящим принципом в син¬
тезе соединений определенного строения является закономерность
трансвлияния И. И. Черняева'. Речь о ней пойдет в следующей главе.
Здесь можно сказать, что синтез изомерных соединений удается про¬
вести лишь для сравнительно инертных комплексов. Поскольку изо¬
мерные соединения всегда отличаются по величине свободной энергии,
то для лабильных комплексов будет осуществляться процесс изоме¬
ризации, протекающий в сторону образования одного из изомеров,
а именно имеющего наименьшую свободную энергию. По этой причи¬
не не известны геометрические изомеры плоскоквадратных комплексов
никеля (II), золота (III) и др.Комплексы с координационным числом 4 плоскоквадратного типа,
содержащие симметричный бидентатный лиганд, имеют однозначное
строение:Если бидентатный лиганд несимметричный, то для плоскоквадратных
комплексов уже возможны два варианта:с—м аь58
Путь синтеза отдельных изомеров этого типа зависит от природы цент¬
рального иона, лигандов, находящихся в комплексе, и лигандов,
вступающих в комплекс.Тот факт, что лиганд, замыкающий цикл в комплексе, занимает
два соседних координационных места, широко используется в целе¬
направленном синтезе. Например, комплексный ион [Co(NH3)4C03] +
может иметь лишь одно строение:NH;NHNH,IСоNH,При действии на него соляной кислотой протекает реакция
{Со (NH3)4 С03]+ + 2НС1 = [Со (NTH3)4 CL] + + Н20 + СО,В результате должен получаться комплекс цис-конфигурацииNH,NH,NH3СоNH,который затем может изомеризоваться в соединение транс-конфигу¬
рации. Сразу после проведения этой реакции удается выделить ди-
хлоридный комплекс одного цвета, а после длительного промежутка
времени в твердую фазу выделяется соединение того же состава, но
другого цвета. С большой степенью вероятности можно сказать, что
первый из них имеет цис-, а второй — /пранс-конфигурацию. На
первых порах развития координационной химии, когда было мало
физических методов исследования структуры веществ, о строении
комплексов часто судили на основе такого рода препаративных дан¬
ных.При окислении комплексов плоскоквадратного типа часто образу¬
ются октаэдрические соединения.. Например, при взаимодействии
хлора с комплексами [Pt(NH3)2Cl2] реакции протекают по следующим
схемам:59
CIClClNJJ,NH,' +
Nif,IIn nCl1 ■■' Р1Cl2Pt1ClNH3+ClClNH3ClPtС 1 2Pt1N11,N11,NH,ClClВ окисленном комплексе сохраняется расположение лигандов, ко¬
торое было в исходном квадратном комплексе.Этот путь синтеза комплексов заданного строения очень эффекти¬
вен для комплексов платины (IV) и палладия (IV). Как правило, он
применим и для синтеза октаэдрических соединений родня (III) и
иридия (III) из соответствующих плоскоквадратных соединений ро¬
дия (I) и иридия (I). Чтобы исключить вероятность перестройки кон¬
фигурации комплексов и окисления внутрисферных лигандов, вместо
воды в качестве среды используют инертные растворители, такие, как
бензол или четыреххлористый углерод. Окисление может протекать
гетерогенно.Восстановление октаэдрических комплексов с образованием со¬
единений плоскоквадратного типа часто происходит с удалением од¬
ной из координат октаэдра. Например, восстановление изомерных
комплексов типа [Pt(NH3)4C12]С12 приводит к различным продуктам
реакции. Они могут быть изображены следующими уравнениями:С1NH-NH“Г"PtNH3NH,ClNH,NH,Cl1ClNH,NH,Cl,+ NjH., -2HCI -2Cl,+ N;H4 -2HCI -2NH,NH3NHNH,NH,NH,NIIjCI, + N, + 6HC1Cl + N,+ :NH4C1 + 4HCIЭтот путь также может быть использован для целенаправленного
синтеза плоскоквадратных соединений, если имеется простой способ
получения соответствующих октаэдрических комплексов.
3.2. Оптическая изомерия3.2.1. Понятие о стереохимииЯвление оптической изомерии для соединении углерода было открыто Пасте¬
ром в 1848 г., т. е. еще до создания теории химического строения и стерео-
химического учения. При изучении винной кислоты* Пастер обратил внимание
на то, что винная кислота кристаллизуется в двух энантиоморфных (асиммет¬
рических) формах. Кристаллы этих двух типов можно было без труда разделить
механически с использованием простого оптического прибора. Особенностью
энантиоморфных кристаллов является то, что их формы относятся друг к другу
как предмет и его зеркальное отражение или как левая и правая рука. Никакими
поворотами не удается совместить все элементы таких кристаллов.Растворы энантиоморфных форм вращают плоскость поляризованного све¬
та. Углы вращения одинаковы, однако одна форма вращает плоскость поляри¬
зованного света вправо, а другая влево. В зависимости от направления вра¬
щения энантиоморфные формы называют правовращающей и левовращающей
и обозначают соответственно d и 1.Для сравнения оптически активных веществ пользуются характеристикой,
называемой удельным вращением [а]. Удельное вращение — это угол вращения
плоскости поляризованного света раствором (длина слоя 1 дм), содержащим в
1 мл 1 г оптически активного вещества:100И = а— ,где а — экспериментально определяемый угол вращения плоскости поляриза¬
ции; / — длина кюветы, дм; с — число граммов оптически активного вещества
в 100 мл раствора.Стереохнмическое учение, развитое Ле Белем и Вант-Гоффом, позволило
объяснить сущность различия d- и 1-форм. Асимметрия кристаллов связана в
данном случае с асимметричностью молекулярного строения вещества. Если
атом углерода, находящийся в центре тетраэдра, связан с четырьмя различными
атомами или радикалами, то при одном и том же составе возможны два варианта
пространственного расположения:Никакими поворотами не удается совместить эти фигуры так
всех одинаковых атомов совпадало.Если в вершинах тетраэдра имеются лишь три типа атомов,чтобы положениенапример:* От французского acide racemique — винная кислота — получили на¬
звание рацемические соединения, рацемические смеси и т. д.6L
то такие фигуры могут быть легко совмещены поворотом одной из ннх на угол
180° в плоскости аас. Различие двух вариантов тетраэдров abed и aabc состоит
в том, что в тетраэдре abed нет никаких элементов симметрии. В тетраэдре
aabc можно провести плоскость sbc, относительно которой эта фигура будет
си мметричной:bТетраэдр aabc имеет один элемент симметрии, а именно плоскость симметрии.
Тетраэдрическая молекула, построенная по типу abed, является асимметрич¬
ной. Для нее возможны два изомера, отличающиеся пространственным распо¬
ложением атомов или радикалов.Таким образом, асимметрия кристаллов связана с асимметрией молекуляр¬
ного строения. Молекулы тетраэдрического типа с асимметрическим атомом, на¬
ходящимся в их центре, способны образовывать изомеры, которые по-разному
вращают плоскость поляризованного света. Такие изомеры называют оптиче¬
скими*. Изомерию такого типа называют оптической изомерией. Оптические
изомеры (антиподы) имеют равное по величине и противоположное по знаку
вращение.^Если смешать в растворе равные количества оптических изомеров одного
н того же вещества, то полученный раствор не будет вращать плоскость поляри¬
зации света. При кристаллизации такого раствора могут выделиться кристаллы
в двух энантиоморфных формах. Однако это происходит далеко не всегда. Часто
оптически активные изомеры кристаллизуются совместно, образуя рацемиче¬
ские соединения. Рацемические соединения отличаются по физическим свой¬
ствам (форме кристаллов, плотности, температуре плавления и др.) от состав¬
ных частей. Иногда получаются твердые растворы одного изомера в другом.
Рацемическое соединение обладает физическими константами, отличными от
соответствующих констант аптиподов. В общем эквнмолярную смесь (соединение
или твердый раствор) обозначают d,1 и называют рацематом.В растворе и реже в твердом состоянии оптически активный изомер со вре¬
менем может превращаться в рацемат. На практике это проявляется в падении
оптической активности, а со временем она совсем исчезает. Такой процесс на¬
зывают рацемизацией. Важно подчеркнуть, что при рацемизации сохраняются
состав, строение, химические свойства вещества. Исчезает лишь оптическая ак¬
тивность.3.2.2. Оптическая активность координационных соединенийВсе сказанное по отношению к тетраэдрическим соединениям угле¬
рода, конечно, в полной мере относится и к комплексным соединениям,
построенным по типу тетраэдра.В том случае, если все четыре лиганда тетраэдрического комплекса
разные, то возможно существование оптических изомеров. Такие
комплексные соединения пока неизвестны. Однако известны оптиче¬
ски активные комплексные соединения другого типа, о которых
сказано в разделе 3.2.5.* Иногда оптические изомеры называют также оптическими антпподамн,
зеркальными изомерами.62
Комплексы плоскоквадратного типа не могут иметь оптических
изомеров даже прп четырех различных лигандах, например соединенияаамbbМможно было бы рассматривать как зеркальные отражения. Однако
при переворачивании плоскости одной фигуры на 180° произойдет
полное совмещение ее с другой фигурой. Значит, это одно и то же
соединение. Для плоскоквадратных комплексов все же известны оп¬
тически активные соединения. Однако их оптическая активность свя¬
зана не с асимметрией координационного центра, а с оптической ак¬
тивностью лигандов (см. раздел 3.2.5).Координационная теория предсказывала возможность существо¬
вания оптических изомеров для комплексных соединений октаэдри¬
ческого типа. Так, комплексное соединение [CoEn2NH3Cl]Cl может
существовать в виде двух геометрических изомеров:N11,/ yN II, ./ 7 Е||</ JE°IГClTpjuc-Для комплекса ^«с-конфигурации возможно существование опти¬
ческих изомеров:ЕпГ/EnV^JNH, NHВ 1911 г. Вернер разделил ^Hc-[CoEn.2NH3Cl]2+ на оптические анти¬
поды. Это был триумф координационной теории.В настоящее время получено большое число оптически деятельных
комплексных соединений октаэдрического типа. Как правило, эти
соединения содержат циклические лиганды. Особенно достуцными ока¬
зались соединения с тремя циклическими лигандами типа
Например, [МЕп3]Х3 и М'3[М(С204)3], где М —Co(III), Сг(Ш),
Rh(III), I г( 111) и др.; М' — „он щелочного металла.Для октаэдрических соединений с шестью различными лигандами
теоретически может существовать 15 геометрических изомеров. Каж¬
дый из них может быть расщеплен на оптические антиподы. Таким
образом, для соединений типа [М а b с d е f] должно существовать
30 геометрических и оптических изомеров.Следует отметить, что оптическая активность не всегда связана
с асимметричностью молекул. Геометрическая фигура является асим¬
метричной, если у нее нет ни одного элемента симметрии — ни цент¬
ра, ни оси, ни плоскости. Если взять оптически активную молекулу
винной кислоты, то в ней есть ось симметрии: она проходит через
центр молекулы и перпендикулярна связи С—С:При повороте молекулы вокруг этой оси на 180° ее структура совпа¬
дает с исходной. Оси симметрии имеют и многие оптически активные
комплексные соединения. Например, трис-этилендиаминовые комп¬
лексы октаэдрического типа имеют ось симметрии, проходящую через
центр октаэдра и середины двух ребер:При повороте таких молекул вокруг этой осп на 180° расположение
атомов также совпадает с исходным.Таким образом, оптически активные молекулы не должны иметь
плоскостей и центров симметрии, но могут иметь оси симметрии. По¬
этому термин «асимметричные» молекулы в применении к оптически
активным соединениям оказался неточным. Оптически активные мо¬
лекулы с четырьмя различными заместителями, не имеющие никаких
элементов симметрии, все чаще стали называть хиральными (от греч.
cheir — рука).НО'-'9 соон61
3.2.3. Методы разделения оптических изомеровТрудность разделения оптических изомеров состоит в том, что многие физические
свойства, и в том числе растворимость оптических изомеров одинаковы.Со времени Пастера известны три основных метода разделения оптическихизомеров:1. Метод самопроизвольной кристаллизации с последующим механическим
разделением.2. Метод биохимического разделения.3. Метод химического расщепления.Первый метод не требует большого пояснения. Если в процессе кристалли¬
зации не получаются энантиоморфные формы, размеры которых достаточны для
механического разбора, то этот метод к данной рацемической смеси неприменим.
Методом самопроизвольной кристаллизации был разделен на оптические анти¬
поды Кз[Со(С204)3].Метод биохимического разделения основан на том, что некоторые микро¬
организмы с различной скоростью разрушают компоненты рацемата. Например,
плесневый грибок Penicillium glaucum преимущественно разрушает право¬
вращающую форму винной кислоты. Если этим грибом подействовать на раце¬
мат, то может быть накоплена левовращающая форма. Биохимический способ
успешно используется для получения оптически активных аминокислот. Од¬
нако этот метод почти совершенно непригоден для комплексных соединений.В координационной химии наибольшее распространение получил третий
метод. Он имеет ряд разновидностей. Чаще всего используется способ превра¬
щения оптических изомеров в днастереоизомеры. Диастереоизомерами назы¬
ваются изомеры с несколькими асимметрическими центрами с различной кон¬
фигурацией у некоторых из них и не являющиеся зеркальными изомерами друг
друга.Сущность способа превращения оптических изомеров в днастереоизомеры
заключается в получении соединении с различной растворимостью. Допустим,
необходимо разделить рацемическую смесь комплекса [СоЕп3]Х3. Обозначим
изомерные оптические формы буквами D и L. Добавим к раствору комплекса
d-форму винной кислоты или ее соли. В результате кристаллизации образуются
два днастереоизомера: Dd и Ld. Днастереоизомеры отличаются (хотя и не очень
сильно) по растворимости, поэтому их удается разделить фракционной кристал¬
лизацией. Отделение получаемого оптического изомера от разделяющего реак¬
тива обычно не представляет большой трудности. Эта задача сводится к замене
аниона.Для разделения на оптические изомеры комплексов катионного типа кроме
винной кислоты и ее солей используют а-бромкамфар-я-сульфонат, камфар-я-
сульфонат, а-камфарнитронат и др. Для разделения комплексов анионного типа
используют такие оптически активные вещества, как стрихнин, бруцин, цинхонн-
дин, а-фенилэтиламин, морфин, хинидин и др. 1Другим способом является разделение предпочтительной кристаллиза¬
цией. Его сущность заключается в том, что из пересыщенного раствора при по¬
мощи затравки вызывается кристаллизация одного из геометрических изомеров.
Например, таким путем были разделены на геометрические изомеры комплексы[СгЕп,(С,04)] и [CoEn2(N02)2]+-Для разделения па оптические изомеры комплексов неэлектролитного типа
весьма эффективным оказался способ адсорбции на оптически активированном
мелкораздробленном кварце. Во многих случаях из раствора на кварце преиму¬
щественно адсорбируется один из оптических антиподов. Этим способом обычно
не удается добиться полного разделения, однако он с успехом используется в
тех случаях, когда необходимо убедиться в принципиальной возможности рас¬
щепления комплексных соединений на оптические антиподы.Известен способ разделения на колонке, заполненной крахмалом, и ряд
других.3-817
3.2.4. Зависимость угла вращения от длины волны
облучающего светаЗависимость вращения плоскости поляризации от длины волны света
изучается на спектрополяриметрах. Соответствующий метод иссле¬
дования называется спектрополяриметрией. Для большинства бес¬
цветных оптически активных соединений угол вращения закономерно
возрастает при уменьшении длины волны облучающего света.Это явление называют вращательной (ротационной) дисперсией.
Для окрашенных соединений обычно характерна аномальная враща-
тельнай дисперсия. В этом случае на дисперсионной кривой имеется
пик, кривая проходит через нуль или угол вращения [ос] убывает
с уменьшением длины волны. Для соединений с аномальной вра¬
щательной дисперсией угол вращения зависит от условий измерения:
температуры, природы растворителя и др.У окрашенных соединений дисперсионная кривая обычно прохо¬
дит через пик в той области длин волн, где имеется полоса поглоще¬
ния видимого света. Этот тип аномальной дисперсии был выявлен на
примере щелочных растворов тартрата меди в 1896 г. французским
физиком Коттоном и в литературе получил название эффекта Котто¬
на. Таким образом, поглощение света веществом и его оптическое вра¬
щение взаимосвязаны.Различают положительный и отрицательный эффект Коттона. Если
с уменьшением длины волны кривая вращательной дисперсии изме¬
няется от пика к впадине, это называют положительным эффектом
Коттона. Если с уменьшением длины волны кривая вращательной
дисперсии, наоборот, изменяется от впадины к пику, то это отрицатель¬
ный эффект. Аналогичные, комплексные соединения с одинаковым
эффектом Коттона (положительным или отрицательным) имеют одну
и ту же абсолютную конфигурацию (конформацию). Аналогичные
соединения, но с различным эффектом Коттона имеют противополож¬
ные конфигурации. Таким образом, на основании типа эффекта Кот¬
тона появляется возможность судить о структуре исследуемого со¬
единения.Аномальная дисперсия затрудняет сравнение оптических свойств
различных комплексных соединений. Поэтому если исследование
проводят на поляриметре, то сравнивается удельное вращение при
определенных длинах волн.3.2.5. Оптически активные комплексы с координационным
числом 4Известно довольно много комплексных соединений, которые не могут
давать оптических изомеров, но содержат оптически активный ли¬
ганд, например d- или 1-пропилендиамин. Такие комплексы, естест¬
венно, обладают оптической активностью. Однако независимо от на¬
личия полос поглощения в абсорбционном спектре кривые оптической
дисперсии таких соединений являются нормальными.Известны комплексные соединения тетраэдрического типа, в ко¬66
торых непосредственное окружение центрального атома не является
хиральным, а в целом они хиральны. Например, бериллиевый комп¬
лексНООСY-oНС7 >М<V*iV-V °-с-'X'OOIfБруциновая соль этой комплексной кислоты выделена в виде двух
диастереоизомеров. Из них затем были получены антиподные диме-
тиламмониевые соли. Однако они очень быстро рацемизуются. Был
получен также аналогичный цинковый комплексНООС\ /С.ИС—ос,н{соонКроме того, для цинка известно соединениеЭти цинковые комплексы были разделены на оптические антиподы
с помощью алкалоидов в пиридиновом растворе.3.3. Гидратная изомерияКлассическим примером гидратной изомерии являются гидраты хло¬
рида хрома (III). Существует три соединения состава СгС13-6Н20,
отличающихся по цвету и электрической проводимости: [Сг(Н20)6]С13,
[Сг(Н20)6С1]С12-Н20, [Сг(Н20)4С12]С1-2Н20 Различие в строении гид-
ратных изомеров заключается в неодинаковом расположении моле¬
кул воды. В одном случае она входит во внутреннюю сферу комплек¬
са, а в других случаях часть молекул воды является кристаллизаци¬
онной.В качестве характерных примеров гидратных изомеров можно
также привести комплексы кобальта [Co(iNH3)5N03](N03)3-Н20 и3**67
[Co(NH3)6H20] (N03)3. Установлено, что в одних условиях кристалло¬
гидрат способен переходить в аквокомплекс, а в других, наоборот,
аквокомплекс можно перевести в кристаллогидрат.3.4. Координационная изомерия и полимерияКоординационная изомерия присуща соединениям, состоящим из комп¬
лексного катиона и комплексного аниона, центральные атомы кото¬
рых различны. Координационные изомеры отличаются различным
распределением лигандов между центральными атомами, входящими
в анионный и катионный комплексы. Примером координационных
изомеров могут служить соединения [Pt(NH3)4][PdCl4] и
[Pd(NH3)4] [PtClj. Если соединения образованы ионами одного и
того же металла, но в различных степенях окисления, то и в этом
случае могут существовать координационные изомеры, например
[PtPy4] [PtClel и [PtPy4Cl2] [PtCIJ.Изомерные соединения имеют один и тот же состав, но различное
строение. Их молекулярные массы одинаковы. В координационной
химии весьма распространены случаи, когда одному и тому же ана¬
литическому составу соответствует несколько веществ, отличающихся
молекулярной массой или типом электролитического распада.
Например, комплексы [Pt(NH3)2Cl2], [Pt(NH3)3Cl][PtNH3Cl3],
[Pt(NH3)3Cl]2[PtCl4], [Pt(NH3)4][PtNH3Cl3]2, [Pt(NH3)«] [PtCl4] име¬
ют один и тот же аналитический состав Pt(NH3)2Cl2, но их молекуляр¬
ные массы в 2—3 раза превышают молекулярную массу наиболее лег¬
кого комплекса. Эти соединения называют координационными поли¬
мерами. Хотя, по существу, их нельзя рассматривать как полимер¬
ные формы, однако в литературе до сих пор иногда называют соеди¬
нения |Pt(NH3)3Cl] [PtNH3Cl3] п [Pt(NH3)J [PtClj] димерными, a
[Pt(NH3)3Cl]2[PtCl4] и [Pt(NH3)4][PtNH3Cl3]2 — трнмерными. Пра¬
вильнее было бы первых два соединения называть двухкомплекснымн,
а два последних •— трехкомплексными.Комплекс [Pt(NH3)4] [PtClj в литературе широко известен под
названием «зеленая соль Магнуса». Он был первым платнна-аммиач-
ным комплексом и служил исходным материалом для основополагаю¬
щих в области координационной химии работ Рейзе, Пейроне, Ренне-
ке, Клеве и других ученых, заложивших основы координационной
химии. Комплексы типа соли Магнуса построены в виде колонок, в
которых чередуются плоскоквадратные ионы lPtA4F+ и [PtX4p~. В та¬
ких колонках имеется связь Pt—Pt.3.5. Связевая изомерияСвязевая изомерия комплексов проявляе?ся в тех случаях, когда ли¬
ганд может координироваться к центральному атому несколькими
способами, но является монодентатным. Например, нитритный нон
N02 может координироваться атомом азота или кислорода, а рода-
нидный ион SCN"—атомом азота или серы. Таким образом, прояв¬68
ление связевой изомерии следует ожидать прежде всего для амби-
дентатных* лигандов.Первый случай такой изомерии был обнаружен Иергенсеном.
В процессе синтеза нитрокомплексов [Co(NH3)5I\02]X2 путем взаимо¬
действия солей азотистой кислоты с аквокомплексами [Co(NH3)5H20|X3
Иергенсен выделил светло-коричневые соединения, отличающиеся
от [Co(NH3)5N02]X2 цветом и отношением к разбавленным кислотам.
Этими промежуточными соединениями оказались нитритные комп¬
лексы [Co(NH3),(ONO)]X2.В органической химии хорошо известны изомерные соединения
R—N02 (нитроалканы) и R—ONO (алкилнитриты). Различие изо¬
мерных комплексов [Co(NH3)5N02]X2 и [Co(NH3)5(ONO)|X2 также
состоит в характере связи группы N02 с центральным атомом. По¬
этому в литературе для подобных изомерных комплексных соедине¬
ний было закреплено название связсвых изомеров**.Установлено, что при облучении солнечным светом нитрокомплекс
[Co(NH3)5N02]C]2 превращается в нитритное соединение [Co(NH3)5(ONO)]Cl2.Метод ИК-спектроскопии позволяет довольно просто различать нитрито-
н ннтрокомплексы. Так, нитритные соединения [Co(NH3)5(ONO)]-+,
[Rh(NH3)5(ONO)]2+ и [Ir(NH3)5(ONO)]-+ имеют две характерные полосы погло¬
щения v(NO) в области 1460 и 1065 см-1. Для аналогичного комплекса платины
(IV) [Pt(NH3)5(ONO)]3+ максимумы этих полос имеют значения 1505 и 955 см-1.
В соответствующих нитрокомплексах эти полосы сдвинуты в низкочастотную
область. Они принимают значения для [Rh(NH3)r,NOo]2+ 1422 и 830 см-1, для
[Ir(NH3)6N02p 1410 и 830 см”1 и для [Рt(NН )5N02]3+“ 1330 и 825 см'1.До недавнего времени связевые изомерные пары соединений были
известны только для нитро- и нитритокомплексов. В последние годы
были обнаружены и интенсивно изучались роданидные связевые изо¬
мерные комплексы кобальта. По аналогии с органическими соедине¬
ниями R—SCN и R—NCS комплексные соединения, в которых рода-
нидная группа связана с центральным атомом через серу, называют
тиоцианатными, а связанные через азот — изотиоцианатными. По
классификации Пирсона (см. раздел 6.6) роданидный лиганд можно
одновременно рассматривать как мягкое основание (за счет атома
серы) и как жесткое основание (за счет атома азота). Из этого следует,
что с металлами класса «а», или, что то же самое, с жесткими кисло¬
тами, роданидный ион должен связываться посредством атома азота,
а с металлами класса «&», или, что то же самое, с мягкими кислотами,
посредством атома серы. В полном согласии с этим в комплексах цин¬
ка и кадмия роданидный нон координирован через атом азота, а в
аналогичных соединениях ртути — через атом серы.Для решения вопроса о способе координации роданидного лиган¬
да часто используется метод ИК спектроскопии. Для линейного ро¬
данид-иона имеются две характерные частоты валентных колебаний:
v(CN) « 2050—2060 см'1 и v(CS) » 750 см-1. При координации через* Амбидента".ный лиганд — это лиганд, содержащий несколько (часто два1
э^ектронодонорных центров, но в координации участвует один из них.** Раньше этот тип изомерии координационных соединений называли
солевой изомерией.69
атом азота значение v(CS) повышается примерно до 800 см'1, а при
координации через атом серы, наоборот, v(CS) понижается примерно
до 700 см'1. Значение v(CiV) и в том и в другом случае, как правило,
выше, чем в свободном ионе. В комплексах кобальта роданидный ион
обычно координирован через атом азота.Сравнительно недавно был синтезирован тиоцианатный комплекс
[Co(NHa)5SCNlCl5. Изомерное ему нзотиоцианатное соединение было
известно давно. Строение обоих этих изомеров подтверждено рент-
генострукгурным методом и может считаться вполне надежным.Установлено, что в водном растворе тиоцианатнын комплекс
[Co(NH3)6SCN)2+ нзомернзуется в изотиоцианатный [Co(NH3)5NCS]2+- С по¬
мощью меченного углеродом-14 роданндно*о иона было показано, что при изо¬
меризации в растворе не происходит обмен внутрнсферного роданндного лиган¬
да со свободным роданндным ионом из раствора. Это привело к заключению,
что изомеризация протекает внутримолекулярно. Особенно быстро изомериза¬
ция осуществлялась в щелочной среде:Существует мнение, что депротонирование координированной молекулы аммиа
ка в тракс-положеннн к титщнанатному лиганду лабилизирует связь Со—S и
позволяет перегруппировываться в лабильный интермедиат, в котором роданид-
иая группа связана с кобальтом как атомом азота, так и атомом серы. Считают,
что связь кобальта с расположенной таким образом роданидной группой подоб¬
на связи, которая описана для этиленовых л-комплексов. Возможно, что в об¬
разовании комплекса такой конфигурации одна из ^5/>3-орбиталей кобальта
перекрывается с л-связывающей орбиталью трех атомов нона NCS-. Кроме
того, заполненные dxz- и 4уг-орбнтали центрального атома могут перекрывать¬
ся с вакантными разрыхляющими л-орбиталями роданндного нона.Существует также альтернативное предположение о протекании процес¬
са изомеризации через образование интермедиата димерного тнпа
[(NH3)5CoSCMCo(NH2)(NH3)J1+. Поскольку процесс изомеризации протекает
и в кислой среде, то также было высказано предположение, что интермедиатом
может служить димерОднако концентрационные зависимости скорости изомеризации не подтвержда¬
ют предположений об образовании интермедиатов димерного тнпа.В комплексах платины (II) и палладия (II) роданидные группы
обычно координируются посредством атома серы. Однако было по¬
казано, что роданидный лиганд может координироваться у ионов этих
металлов через атом азота. Примерами таких комплексов являются
соединения типа [M(PEt3)2(NCS)2], где М — Рt(II), Pd(II). Сущест¬
вует мнение, что координация роданидной группы к платине (II) и
палладию (II) через атом азота происходит при наличии в комплексе
лиганцов, обладающих л-акцепторными свойствами. При коордн-он-[(MH3),Co-SCMJJ+ ZT [(MH3)4(NH2)Co-SCN] +медл.N + н+— (NH3)4 (NHj) Со С — [(NH3)5Co-NCS]=*
SSCN70
нации через атом серы достигается стабилизация ее связи с централь¬
ным атомом за счет dT.—dT. -перекрывания. Введение в комплекс
Других лигандов л-акцепторного характера должно ослаблять связь
М—SCN и, следовательно, способствовать образованию связи М—NCS.
Заслуживает внимания тот факт, что в дифосфиновом комплексе
[Pd(Ph2PC2H4PPh2) (SCN) (NCS)] одна из роданидных групп коорди¬
нирована атомом серы, а другая — атомом азота:н,с
-|
н ,сг'.PPh-. уУс/У VеPPhiNАналогично трнэтилфосфину триизопропилфосфит способствует
координации роданндного лиганда через атом азота с образованием
изотпоцианатного комплекса [Pd{P(OC3H7-«3o)3}2(NCS)2]. Интересно,
что в аналогичных условиях триэтилфосфит дает тиоцианатный комп¬
лекс. Возможно, определяющую роль в характере координации рода¬
нидной группы здесь играют стерические факторы. На их роль ука¬
зывали Басоло, Бэдли и Бурмейстер. В диэтилентриаминовом комп¬
лексе палладия роданндная группа связана посредством атома серын,с•|н,е.сн—сн,^
x'Hn; * :>н,'N ИSCNN0,а в устойчивом ^^-тетраэтилднэтнлентриампновом соединении —Н,С'
‘ I
Н,сч.СИ,СИ,
^IIN^ -4N(C^H5)2 ^ncsSCNпосредством атома азота. В растворе и в твердом состоянии происхо¬
дит реакция изомеризации:[Pd {С2Н6)4 Dien ) SCN] X — [Pd [(С2Н6)4 Dien) NCS] Xгде X — SCN", PF6. Это объясняется стерическими препятствиями,
создаваемыми этильными радикалами. Рентгеноструктурные данные
показывают, что связь М—NCS обычно линейная, а угол М —S—CN
значительно меньше 180°. Так, комплёкс 4«c-[Pd(NH3)2(SCN)2] имеет
следующее строение:71
Таким образом, этильные радикалы действительно могут создавать
стерические препятствия для координации роданидной группы через
атом серы в М,1Ч'-тетраэтилдиэтилентриаминовом комплексе
[Pd{(C2H5)4Dien}NCS]SCN.-Подобным же образом ведет себя аналогичный селеноцианатный
комплекс. Полученный при низких температурах изомер
[Pd{(C.H,)1Dien}SeCNJBPhi при повышении температуры переходит
в более устойчивое соединение, в котором селеноцианатный лиганд
связан с палладием посредством атома азота. ^Библиографию по изложенным вопросам связевой изомерии можно
найти в статье [2).3.6. Ионизационная изомерияИонизационная изомерия присуща только комплексам катионного
типа. Примером подобных изомерных соединений могут служить ком¬
плексы [Pt(NH3)iClo]Br3 и [Pt(NH3)4Br2]Cl2. В первом комплексе
ионы С1“ входят во внутреннюю сферу, а ионы Вг~ находятся во внеш¬
ней. Во втором комплексе, наоборот, во внутреннюю сферу входят
ноны Вт', а во внешней располагаются ионы С1".В растворе комплексные соединения подвергаются диссоциации
(ионизации). Первичная диссоциация рассматриваемых комплексов
описывается уравнениями
[Pt (МН3)4 С12] Вг2 = [Pt (NH3)4 СУ* + 2Вг-
[Pt (МН3)4 Вг2] С13 = [Pt (NH3)4 Вг2]2+ + 2С1-В первом случае в растворе качественными реакциями могут быть
легко обнаружены бромид-ионы, а во втором — хлорид-ионы.В растворах часто происходит замещение внугрисферных лиган¬
дов на внешнесферные, например:[Pt(NH3)4CLp Ь Вг~ «= [Pt(NH,)4CIBr]-+-|-C[-
[Pt(NH3)4ClBr]-+ + Br ~ [Pi (NH3)4Br2j-+ +CI-Поэтому длительное существование ионизационных изомеров в раст¬
ворах невозможно. Время установления равновесия в реакции заме¬
щения определяется разницей в устойчивости соответствующих комп¬
лексов и их лабильностью. Поскольку бромидные комплексы платины(IV) устойчивее хлоридных, то в растворе будет наблюдаться преиму¬
щественный переход комплекса lPt(NH3)4Cl2]Br2 в [Pt(NH3)4Br2]Cl2.Таким образом, синтез ионизационных изомеров может быть осу¬
ществлен только для очень инертных комплексов.
3.7. Трансформационная изомерияТрансформационная изомерия связана с химическим превращением
лигандов. Примером трансформационных изомеров может служить
пара комплексов (NH4);lPd(SCN)4] и [Pd{NH2C(S)NH;}2(SCN)o|. Ва¬
ловый состав этих соединений и координационные числа центральных
атомов одинаковы. Однако характер лигандов в них различен. Кроме
того, одно из изомерных соединений является электролитом, а другое
относится к неэлектролитному типу. Роданид аммония и тиомочеви-
на генетически связаны между собой. Так, Сахарова с сотр. пока¬
зали, что в процессе термического разложения тиомочевинных
комплексов редкоземельных элементов получаются роданидные
соединения.Превращение цианата аммония в мочевину(NH4)0CI\t->NH2C(0)NH2 является классическим примером превра¬
щения неорганического вещества в органическое. В связи с этим
принципиально мыслимы изомерные комплексы, например
[Co{NH2C(0)NH2}2(OCN)2] и (NH4)21Co(OCN)4]. Вполне возможно,
что в определенных условиях один из таких изомеров может превра¬
щаться в другой. Процесс изомеризации соединений такого типа пред¬
ставляет большой интерес, хотя и встречается очень редко.К трансформационным изомерам могут быть отнесены следующие
пары соединений: IPtH(C2H4)(PPh3)2lC104 и [PtCoH^PPh^nClOJ;
[PtCl(CCl=CCl2)(PPh3)2] и [Pt(CCU=CCl2)(PPhs)2], а • также
[PtBr(CF=CF2)(AsPh3)2] и [Pt(CFBr=CF2)(AsPh3)2], В процессе пе¬
рехода одного изомера в другой наряду с химическим превращением
лигандов происходит изменение способа их координации (ст—л).3.8. Конформационная изомерияОдинарная a-связь допускает свободное вращение связанных атомов
и групп атомов, поэтому вследствие теплового движения молекулы
меняют свою форму. Неидентичные состояния, через которые прохо¬
дят эти молекулы в процессе свободного вращения, называют конфор¬
мациями. Молекулы, находящиеся в различных конформационных
состояниях, имеют различную энергию. Иногда существует несколько
устойчивых конформационных состояний. Относительно стабильные
соединения, получающиеся в результате вращения вокруг одинарных
связей, называют конформационными изомерами. Например, для
газообразного этана среди бесконечного числа различных конформа¬
ций характерны два крайних положения метильных групп:заторможеннаяформазаслоненнаяформа
Особенно характерно явление конформационной изомерии для
циклических соединений с одинарными сг-связями. Одной из простых
молекул такого типа является циклогексан. Он существует в виде
двух изомеров: «ванны» и «кресла»:конформация «ванны» конформация «кресла»Конформационная изомерия довольно широко проявляется в хе-
латных комплексных соединениях. Например, 1,3-пропилендиами-
новые комплексы существуют в двух формах: «кресла» и «ванны»:Конформационные изомеры мыслимы и для комплексов с пяти¬
членными циклами. Например, этилендиаминовые соединения могут
существовать с конформацией симметричного скошенного пятичлен¬
ного хелатного кольцаМА-хУ'У \ /С' Nу N МуА— NV 'Си с асимметричной конформацией формы «конверта»гI
ILcС-1 чм
/ \N—С , _ ч/I I \/ 1 N L-x N- М :N уN3.9. Формальная изомерияКомплексные соединения [Pt(NH3)(NH2C2H5)Cl2] и [Pt(NH2CH3)3Cl2]
можно рассматривать как изомерные соединения. Они не связаны меж¬
ду собой генетически и не могут переходить одно в другое, но их эле-74
ментный состав одинаков. Поэтому их можно назвать формальными
изомерами.Другой парой формальных изомеров являются комплексыNiNiX'=N/y ^N=C\ Л—N4 /N—(vCH3 4R О OC2H5 Clb" 11 °‘ '0C'HО5По существу, в эти два комплекса входят четыре различных лиганда.
Однако элементный состав комплексов одинаков.Возможно большое число формальных изомерных соединений.
ГЛАВАКОМПЛЕКСНЫЕ СОЕДИНЕНИЯ С НЕОБЫЧНЫМИ
КООРДИНАЦИОННЫМИ ЧИСЛАМИОбобщение Вернера о том, что независимо от природы и валентности
центрального атома его координационное число равно 4 или 6, на пер¬
вых порах утверждения и развития координационной теории сыграло
исключительно важную роль в химии координационных соединений.
Однако постепенно накапливались факты, которые указывали на су¬
ществование комплексных соединений с другими координационны¬
ми числами центрального атома. Во многих случаях соединения, в
которых центральные атомы обладают координационным числом, от¬
личным от 4 и 6, неустойчивы, и их изучение сопряжено с экспери¬
ментальными трудностями. Часто такие соединения не удается вы¬
делить в твердую фазу и их приходится изучать лишь в растворе.
В настоящее время комплексы с необычными координационными чис¬
лами особенно привлекают внимание, так как они играют важную
роль во многих химических превращениях комплексных соединении
как промежуточные соединения — интермедиаты. Библиографию об
этих соединениях можно найти в работе [1].Следует отметить, что обычно химики представляют геометрию
комплекса в виде идеального полиэдра (многогранника). Однако рент¬
геноструктурный анализ показывает, что в большинстве случаев
полиэдры комплексов искажены.4.1. Соединения с координационными
числами 2 и 3Комплексные соединения с координационным числом 2 являются не
столько необычными, сколько малораспространенными. Достоверно
известны такие комплексы для меди (I), серебра (I) и золота (I). При¬
мерами комплексов меди являются аммиачное и тиомочевинное со¬
единения [Cu(NH3):]Cl и [Cu(Thio)2]CI. Они имеют линейную кон¬
фигурацию. Тиомочевина координирована к нону меди посредством
атома серы. Более характерно координационное число 2 для ионов
Ag(I) и Au(I). Для них изучены галогенидные и цианидные комплексы
типа [МХ2]~, а также аминаты типа [M(NH3)2]+. Все они также имеют
линейную конфигурацию.Имеются сведения об интересных комплексных соединениях с
координационным числом 2, где в роли комплексообразователя вы¬
ступают галогенидные ионы, а лигандами являются ионы серебра (!)'•
fAg2Cl]N03, [Ag:Br]N03, [Ag2I]N03. Указания на образование таких
соединений получены при изучении диаграмм плавкости систем76
Рис. 4.1. Октаэдрический полиэдр комплексов соста¬
ва ML3С>онсAgX — AgNO:, на основании изучения раство¬
римости галогенндов серебра в водных раство¬
рах AgiN03 и путем криоскопических исследо¬
ваний.Комплексные соединения с координационным
числом 3 встречаются еще реже. Они известны
для меди (I). Так, существует соединение Y YK[Cu(CN)3], комплексный ион которого имеет
конфигурацию треугольника. Считают, что в
растворе существуют комплексные ионы [СиС13]~ и [AgCU~. Полу¬
чено соединение меди (II) состава CsCuCl3, которое можно было бы
принять за комплекс с координационным числом 3. Однако рент¬
геноструктурное исследование показало, что в кристаллическом со¬
стоянии он имеет структуру, в которой ионы меди находятся в
центре искаженных октаэдров. Таким образом, ионы СГ в этом
соединении являются мостиковыми и связаны с двумя ионами
меди. В результате этого координационное число центрального
атома равно 6. Это весьма распространенный случай, когда стехио-
метрический состав позволяет сделать заключение о комплексе с ко¬
ординационным числом 3, а детальное исследование структуры пока¬
зывает, что вещество полимеризовано за счет образования лигандами
мостиковых связей. В результате этого реальное координационное
число становится гораздо выше. Например, оно может стать равным
6 с октаэдрическим полиэдром (рис. 4.1).В последние годы получен ряд комплексных соединений переход¬
ных металлов третьего периода с интересным лигандом — гексамети¬
лен диснл ил ам идом:_/\Si (СН3)3ИЛИ ,\'RSi (СН,)зКомплексы скандия (III), титана (III), ванадия (III), хрома (III)
и железа (III) имеют состав M(NR2)3. Получены также смешанные
соединения кобальта (II) Co(NR2)2PPh3 и никеля (I) Ni(NR2)(PPh3),5
с трифенилфосфином, а также кобальта (II) и никеля (И) с пиридином
типа M(NR2)2Py: Рентгеноструктурные исследования показали, что
связи металл — лиганд в Co(NR2)oPPh3 и Ni(NR2)(PPh3)2 лежат в
одной плоскости, а углы между ними находятся в пределах 107—130°
Имеются указания на существование комплекса никеля (I)
K2[Ni(CN)3], а также амидных соединений типа K!M(NH2)3], где
М — Be, Са, Sr, Ва, Sn и РЬ. Однако нет уверенности в том, что все
эти соединения являются мономерными.
4.2.Соединения с координационным числом 5В настоящее время бурными темпами развивается химия комплексных
соединений с координационным числом 5. За последние годы появи¬
лись десятки исследований в этом направлении. Особенно большое
число таких комплексов получено и исследовано для железа (II), ко¬
бальта (II) и никеля (II).Геометрической конфигурацией пятикоординационных комплексов
является тригональная бипирамида или тетрагональная пирамида
(рис. 4.2). Во многих случаях эти геометрические фигуры в большей
или меньшей мере искажены.Наиболее распространенными типами пятикоординационных со¬
единений с монодентатными лигандами L являются IML3X2], [ML5]2+
и [ML4X]+, где М — двухвалентный ион металла, X — одновалент¬
ные анионы.В качестве монодентатных лигандов выступают органические арил-
или алкилфосфины, например трифенилфосфин (СеН^зР и триметил-
фосфпн (СН3)ЭР. В этих случаях донорными атомами нейтральных
лигандов являются атомы фосфора.Получено много соединений, в которых нейтральными лигандами
являются оксиды органических фосфинов (СвН5)3РО и арсинов
(CH3)3AsO. В них роль донорных атомов играют атомы кислорода.Если лиганд бидентатный, то наиболее распространенным типом
пятикоординационных комплексов является [PtL2X]+. Донорными
атомами бидентатных лигандов чаще всего служат атомы фосфора,
мышьяка, серы и реже—азота, например:Получены в виде двух модификаций пятикоординационные соеди¬
нения кобальта с (С6Н5)2РСН2СН2Р(С6Н6)2 (Dphp): красного цвета
[Co(Dphp)2Cl] (Sn013) и зеленого цвета [Co(Dphp)2Cl](SnCl3)-СсН5С1.
Рентгеноструктурные исследования показали, что комплексные ионы
в этих соединениях имеют разную геометрическую конфигурацию.
В первом соединении ион имеет строение тетрагональной пирамиды,
а во втором — тригональной бипирамиды.Клойд и Мик получили и исследовали большое число комплексов(CH3)2As As (СНэ),I I✓ \/ЧS/\f7Рис. 4.2. Конфигурация комплексов с
координационным числом 5:а — тригональпая бнпирамнда; 6 — тетраго¬
нальная пирамидаао78
платины (II) п никеля (II) с тетраметилпропилендифосфином типа
[ML2X]+, где X —Cl, Br, I, SCN. Пятикоордпнацнонные соединения
с тридентатными лигандами встречаются практически только одного
типа: [PtLXJ. Тридентатные лиганды, входящие в состав комплексов
пятикоординацнонных, часто содержат донорные атомы азота, фос¬
фора н серы, например:,СН,СНPIi2PHSу' 2. сн.сн.SSH,CH,CH2^ ^CHjCHsn^_ S PPh2^CH2CH2.(СН3).. N NH' N(CH3)oПолучено большое число комплексов кобальта (II) и никеля (II)
с тетрадентатными лигандами и ряд соединений с пентадентатными
лигандами. Примеры таких лигандов:./н —СН—Р(С6Н5-)2сн,/ V
Н,С" NCH,
‘ I ! 2м—сн —СН—Р(С6Н5)2
ХСН,—СН— Р(С6Н5),сн,—S/ 2
сн,С И 2
\>(С6Н5)2S—сн,
Xсн,(С6н5)2р/сн,-Y.>Ov-CH=N^сн -сн,. /СН —СН'ОН но--Хорошо известно, что комплексные соединения платины (II)
и палладия (II) имеют плоскостное строение п координационное
число 4. Это относится и к другим ионам металлов, имеющих
электронную конфигурацию ds.Сейчас накоплено уже много данных, свидетельствующих о том, что
платина (II) и палладий (II) проявляют склонность к образованию
пятикоординацнонных соединений. Непременным условием 'образо¬
вания таких комплексов является л-акцепторный характер лигандов,
окружающих нон металла. Еще в 1960 г. Найхольм с сотр. установил,
что при кондуктометрнческом титровании [Pd(Diars*)2]2+ ионами Г
или СГ наблюдается скачок, соответствующий одному эквиваленту
галогенидного иона. На этом основании был сделан вывод о том, что
в растворе образуется пятикоордпнационный комплекс [Pd(Diars)2X]+./As (CH3)2* Diars — о-фенилен-бис-(диметиларсин)ч/\As (СН3)279
Однако рентгеноструктурный анализ Pd(Diars)2I2 показал, что в кри¬
сталлическом состоянии этот комплекс имеет шестикоординационный
ион металла.Имеются сведения, что комплексные соединения платины (II) и
палладия (II) с трис-(о-дифениларсинофенил)арсином (QAs)QAs[M(QAs)X]X в кристаллическом состоянии имеют тригональноби-
пирамидальное строение:As- ' \I As
XПолучен также комплекс палладия (II) с трис-(о-дифенилфосфино-
фенил)фосфином. Эти пятикоординационные комплексные ионы сохра¬
няются и в растворе. Соединения с тетрадентатными лигандами такого
типа часто называют зонтичными.Третичный фосфин 2-фенилнзофосфинодолин,^\-СН2чll-сн >Р~СбН5
с 2приводит к образованию двух трис-фосфиновых комплексов палладия
(II) типа PdL3Br2. Один из них четырехкоординационный, а другой —
пятикоордннащюнный. Последний построен по типу искаженной
тетрагональной пирамиды, в которой атом брома вышел из плос¬
кости:ВгВестланд показал, что комплексные соединения палладия (II)
и платины (II) типа [ML2]2+, где L — 1,2-бис-(дифенилфосфиноэтан),80
1,2-бис-(дифениларспноэтан) и 1,2-бис-(дифенилстибииоэтан) в не¬
водных растворителях образуют пятикоординационные соеди нения
в соответствии с уравнениемml|+ + х- ^ ml2x+В тех же условиях аналогичные аминные комплексы этой реакции
не подвергаются. Тенденция к образованию пятикоординацнонных
соединений этого тнпа увеличивается в ряду донорных атомов N «
« Р < As < Sb.Известно, что этиленовые и ацетиленовые углеводороды проявля¬
ют л-акцепторные свойства по отношению к ионам переходных ме¬
таллов. Если связывать существование комплексов палладия (II)
и платины (II) с координационными числами больше 4 с наличием
в их составе лигандов л-акцепторного характера, то можно ожидать
существования этиленовых и ацетиленовых комплексов с координа¬
ционными числами 5. В последние годы действительно были получены
такие соединения. НапримерПолучены пятикоординационные комплексы родия (I), иридия (I),.
палладия (II) и платины (II). В качестве лигандов в этих соединениях
выступают эфиры фосфористой кислоты P(OR)3. Комплексные ионы
имеют состав [ML5]n+. В твердую фазу они выделяются комплексными
фтори ными анионами BF4, PF6, AsF6 или SbFe. Необычным в этих
соединениях является то, что все лиганды комплексного катиона оди¬
наковы и монодентатны. Методом ЯМР установлено, что в растворе
эти пятикоординационные комплексы имеют тригонально-бипира-
мидальную структуру.4.3. Соединения платины (II) с шестью лигандамиСуществование комплексов платины (II) с координационным числом
5 в настоящее время не вызывает сомнений. Установлено существо¬
вание комплексов платины (II) с координационным числом 6 [2]. Эти
результаты были получены при изучении обмена тиоэфиров (тиоксана
и диэтилсульфида) в тетрасульфидных комплексах типа [PtLJ2+.Следует отметить, что для ТО удалось выделить из раствора в кри-CI кСМ81
сталлпческом виде гексатиоксаповый комплекс [Pt(T0)e](N03)2 и его
пикратный аналог [Pt(T0)5][C6H20(N02)3]2.Опыты показали, что днэтилсульфид удерживается в комплексе
платины (II) гораздо прочнее, чем тиоксан. Это следует из того, что
при взаимодействии тиоксана с диэтилсульфидным комплексом
[Р1(ДЭС)4]2+ происходит лишь присоединение молекул тиоксана, а
диэтилсульфид на тиоксан не замещается:|Pt (ДЭС)413+ 1-2Ю ^ [Pt (ДЭС)4 (ТО)2]2+Взаимодействие тиомочевины Thio с тиоксановым комплексом
IPt(TO)4]2+ сводится к замещению тиоксана на тиомочевину. Однако
в системе около 50% комплекса содержит пятый лиганд. Таким об¬
разом, реакция схематично может быть изображена в видеlPt(TO)]„P !- 4Thio [Pt(Thio)4(TO)0i5 ] + 3,5ТОТе-же результаты получены, исходя из тетратиомочевинного комп¬
лекса и свободного тиоксана.4.4. Соединения с координационными
числами 7 и 8 [3, 4]Комплексные соединения с координационным числом 7 встречаются
так же редко, как и соединения с координационным числом 3. Пред¬
полагаются три возможные структуры для семикоординационных
комплексов: шапочная тригональная призма, одна из граней которой
является основанием тетрагональной пирамиды; шапочный октаэдр,
одна из граней октаэдра является основанием тригональной пи¬
рамиды; пентагональная бипнрамида (рис. 4.3).Установлено, что комплексный ион гептафторцирконата калия
K3lZrF7] в кристаллическом состоянии построен по типу шапочного
октаэдра (рис. 4.3, б). Октаэдр несколько искажен из-за расталки¬
вания лигандов на грани, над которой расположен седьмой лиганд.
Аналогичная структура приписывается гептафторкобальту (IV) ка¬
лия K3IC0F7], а также {дпбромотрикарбоннл-бнс-(дифениларсино)ме-
тан}вольфраму (II) WBr2(CO)3{(C6H5)2AsCH2As(C6H5)2}. В комплек¬
се молибдена МоС12(СО)2{Р(СН3)2СвН5}3 одна из связей Mo—Р имеет
необычно большую длину, однако все сем,ь лигандов присоединены
к центральному иону.Гептафторидные комп¬
лексы элементов пятой
группы типа [MF;]2' [где
М — Sb(V), Nb(V), Ta(V)]Рис. 4.3. Конфигурация ком¬
плексов с координационным
числом 7:^ а — шапочная тригональная приз*Я 6 в ма; 6 — шапочный октаэдр; в —пентагональная бипнрамида82
Рис. 4.4. Конфигурация комплексов с координационным числом 8:а — архимедова антипризма; б — вертикально двушапочная тригональная призма; в — доде¬
каэдр; г — горизонтально двушапочная тригональная призма; д — гексагональная бипира¬
мидапостроены по типу трнгональной призмы с седьмым лигандом над
одной из граней.Описано получение семикоординационных комплексов
(CH3NH3)3[RuC17], структура которых окончательно не выяснена.
Однозначно установлено, что комплексы уранила КзШ02Р5] и
Cs3[U02(NCS)6] имеют структуру пентагональной бипнрамиды.Комплексы с координационным числом 8 распространены срав¬
нительно широко. Они наиболее характерны для ионов редкоземель¬
ных элементов Э (III) и актинидов Э (IV) и Э (VI). Часто встречаются
восьмикоординационные соединения Y(III), Zr(IV), Hf(IV), Nb(V),
Ta(V), Mo(IV), Mo(V), W(IV), W(V). Наиболее важными факторами,
обусловливающими существование комплексов с координационным
числом 8, являются большой размер центрального иона и сравни¬
тельно высокая степень окисления.Конфигурация восьмикоординационных комплексных соединений
может быть представлена несколькими геометрическими фигурами.
Наиболее простой фигурой является куб. В ранних работах конфигу¬
рация восьмикоординацнонных комплексов обычно предполагалась
кубической. Однако позднейшие рентгеноструктурные исследования
показали, что кубическая конфигурация для комплексов с координа¬
ционным числом 8 почти не встречается. Для них предполагаются
конфигурации, приведенные на рис. 4.4.Эмпирически установлено, что наиболее устойчивыми восьми¬
координационными комплексами являются соединения, лиганды ко¬
торых имеют донорные атомы С, N, О, F или Н. Возможно, одна
из причин заключается в малых размерах этих атомов. Так, в комп¬
лексе тория (IV) [Th(NCS)8]4- из двух возможностей координации
лиганда — через атом серы и атом азота — реализуется последняя.Из восьмикоординационных комплексов наиболее хорошо изуче¬
ны цианиды молибдена и вольфрама типа K4tMo(CN)8] и KJW(CN)8].
Они построены по типу додекаэдра. В этих соединениях угол между
связями металл — углерод — азот равен 180°.СреДи галогенндных восьмикоординационных соединений наи¬
более распространены фторидные комплексы. Известны (NH4)3H[PbF8],
H3[SbF8] и Na3[TaF8j. Ион [TaF8]3- построен по типу тетрагональной
антипризмы, а октафторид осмия OsF8, по-видимому, имеет гране-
центрированную призматическую структуру.83
Наиболее распространенными восьмикоординационными комп¬
лексами лантанидов являются р-дикетонаты общей формулы
M|Ln(RCOCHCOR')d. Установлено, что атомы кислорода в этих
соединениях расположены вокруг центрального иона иногда по типу
ангипризмы, а в некоторых случаях — по типу додекаэдра.Для актинидов в состоянии окисления (VI) чрезвычайно харак¬
терна конфигурация комплекса в виде гексагональной бипирамиды.
Многие соединения, например K3Na[U02(C03)3], Cs[U02(N03)3],
Na[U02(CH3C00)3], где карбонатный, нитратный и ацетатный ионы
бндентатны, построены по этому типу.Большой интерес представляет оксихинолиновый комплекс ура-
нила H[U02(0x)3]. Его можно было бы принять за восьми координа¬
ционный. Однако установлено, что он семикоординационный. Одна
из молекул оксихинолина координирована лишь посредством атома
азота:Атом водорода этой молекулы оксихинолина связан водородной связью
с соседним лигандом.4.5. Соединения с координационным числом 9Для образования комплексных соединений с высокими координаци¬
онными числами весьма важное значение имеют относительные раз¬
меры центрального атома и донорных атомов лигандов. Дей и Хоард
[5] пришли к заключению, что для восьмикоординационных комп¬
лексов отношение межатомных расстояний (М—L)/(L...L) должно
быть около 0,83. Для девяти- и десятикоординационных соединений
это соотношение соответственно 0,87 и 0,92. В зависимости от типа
полиэдра при. одном и том же координационном числе оно может не¬
сколько меняться.Большой экспериментальный материал показывает, что комплек¬
сные соединения переходных металлов имеют максимальное коорди¬
национное число 8. Исключение составляет гидрндное соединение
рения K2[ReH9], координационное число которого 9. Оказалось, что
оно построено по типу трехшапочпой трпгональной призмы (рис. 4.5).Комплексные соединения с координационными числами 9 и выше
известны лишь для /-элементов — лантанидов и актинидов [4].84
Рис. 4.5. Конфигурация комплексов
с координационным числом 9:а — шапочная квадратная антипризма;О — трехшапочная тригональная призмаНаиболее характерной кон¬
фигурацией комплексных соеди¬
нений с координационным чис¬
лом 9 является полиэдр, по ко¬
торому построен нонагидрид ре¬
ния (см. рис. 4.5). Классическим
примером такого типа комплек¬
сов может служить [Nd(H20)9J(Br03)3. Его структура была опреде¬
лена Гельмгольцем еще в 1939 г. Позднее было установлено, что но-
нааквоброматы других лантанидов также имеют аналогичную струк¬
туру.Существуют девяти координационные соединения, построенные по
типу шапочной квадратной антипризмы (см. рис. 4.5, а). К этому
типу относится полимерный комплекс эрбия Ег(С204)(НС;>С)4) -ЗН20.
Оксалатные группы в этом соединении мостиковые, и все их четыре
атома кислорода участвуют в координации. Ион эрбия (III) окружают
восемь атомов кислорода оксалатных лигандов, которые образуют
искаженную квадратную антипризму. Одна молекула воды занимает
«шапочное» положение над одной из плоскостей антипризмы. Среди
комплексов с координационным числом 9 и выше полимерные соеди¬
нения встречаются чаще, чем мономерные.4.6. Соединения с координационным числом 10
и вышеКак уже было сказано, для образования комплексов с координаци¬
онным числом 10 необходимо, чтобы отношение (М—L)/(L...L) имело
значение около 0,92. Из этого следует, что расстояние между сосед¬
ними донорными атомами должно быть намного меньше, чем сумма
радиусов свободных донорных атомов. Поэтому десятикоординаци-
онныз комплексные соединения с десятью монодентатными лигандами
практически неизвестны. Все известные соединения с координацион¬
ным числом- Ю содержат бидентатно связанные ионы N03, СО3 .
Расстояния между атомами кислорода в четырехчленных циклах,
образуемых этими ионами, намного меньше, чем при ван-дер-вааль-
совом взаимодействии двух атомов кислорода.Распространенным полиэдром десятикоэрдинационных соединений
является двухшапочная анти призма. Два других полиэдра основаны
на додекаэдре: двухшапочнын додекаэдр и додекаэдр с раздвоенными
вершинами (рис. 4.6). Примерами комплексов, построенных по типу
последнего полиэдра, являются Ьа(Д ЧСО)4(МЭз)3 и Ш(ДЧС0)4(М03)3.
Все три нитратные группы в них являются бнденгатными. Атомы
кислорода молекул сульфоксидов занимают остальные четыре
вершины. Аналогичное строение имеют ди пиридильные комплексы85
Рис. 4^6. Конфигурация комплексов с координационным числом 10:
а — двушапочная квадратная антипризма; б — двушапочпый додекаэдр; в — до*
декаэдр с раздвоенными вершинамиLa(Dipy)o(N03)3 и Tb(Dipy)2(N03)3. Переход от нитратных к карбо¬
натным комплексам редкоземельных элементов часто связан с обра¬
зованием полимерпых соединений. Карбонатные группы в них играют
роль мостиков.Считают, что в тетраформиатном комплексе [Th(HC00)4(H30)2] ■
■Н20 координационное число тория равно десяти. Структура этого
соединения включает трехмерный каркас, в котором атомы тория свя¬
заны между собой мостиковыми формиатными группами. Атомы кисло¬
рода формиатных групп располагаются по вершинам искаженной анти¬
призмы Архимеда, а атомы кислорода двух молекул воды занимают
координационные места над квадратными гранями этой антнпризмы.В настоящее время достоверно известны только два один¬
надцатикоординационных комплекса: [Th(H20)3(N03)3(0H)]2 и
lTh(H20)6(N03)4]. Комплексных соединений с координационным чис¬
лом 12 известно гораздо больше. К их числу относятся, например,
M[Ce(N03)6] и M[Th(N03)„]-8H20, где М —Mg(II), Mn(II), Co(II),
Ni(II), Zn(II), и полимерное соединение La2(S04)3-9H20. Изучено
соединение U(BH4)4, в котором атом урана связан с 14 атомами водо¬
рода. В соединении U(BH4)4-2(С4Н80) уран связан с 12 атомами водо¬
рода и двумя атомами кислорода, а в соединении U(BH4)40(CH3)2
уран связан с 13 атомами водорода и одним атомом кислорода. Сведе¬
ния о соединениях с координационным числом 13 пока не публико-,
вались.В заключение следует отметить, что установление координацион¬
ного числа часто является сложной задачей. Единственно надежным
методом, при помощи которого удается сделать это однозначно для
любого типа комплексов, является рентгеноструктурный анализ.
Формульный состав комплексного соединения не всегда дает правиль¬
ную информацию о координационном числе. При малом содержании
лигандов во внутренней сфере увеличение координационного числа
может происходить вследствие образования лигандом мостиков между
двумя центральными атомами. В комплексных соединениях с поли-
дентатными лигандами их координационная емкость может быть ис¬
пользована не полностью, и поэтому координационное число оказы¬
вается ниже, чем это следует из формульного состава.
ГЛАВА5ПРАВИЛО ЦИКЛОВ Л. А. ЧУГАЕВА5.1. Историческое развитие правилаВ 1906 г. Л. А. Чугаев опубликовал монографию «Исследованияв области комплексных соединений» [1J. В ней Л. А. Чугаевым были
сделаны открытия, которые вошли в сокровищницу мировой химиче¬
ской пауки. Во-первых, нм было сформулировано правило циклов,
а во-вторых, описана чувствительная и эффектная реакция на никель
с диметилглноксимом.Л. А. Чугаев писал: «Можно высказать в качестве общего прави¬
ла, что те комплексные соединения, в которых наличность циклических
группировок может считаться вероятной, при прочих равных усло¬
виях обыкновенно отличаются большей степенью прочности, чем со¬
единения, не содержащие циклов» [1, с. 111]. На основе фактического
материала он пришел к заключению, что наиболее устойчивыми в
комплексах являются циклы, состоящие из пяти и шести звеньев.Одним из оснований для заключения о правиле циклов послужили
факты взаимодействия никельсукцинимида Ni(Su)где Su — это
СН2-С=0| ^>N~ с диаминами. Л. А. Чугаев установил, что 1,2-
СН2—С=0диамин (этилендиамин) и 1,3-диамин (пропилендиамин) взаимодейст¬
вуют с этой солью никеля с образованием соединений типа
[NiEn3]Su2 и [NiEruSuj]. Реакция с 1,4- и 1,5-диаминами идет со¬
вершенно иначе. Л. А. Чугаев писал: «Разница между триметилен-
диамином и тетраметилендиамином здесь настолько резкая, что про¬
изводит впечатление скачка. Основания эти как будто относятся к двум
совершенно различным классам. Наоборот, отношенне тетраметилен-
диамина и пентаметиленднамина представляется совершенно одина¬
ковым» 11, с. 49]. Л. А. Чугаев связал различное поведение диаминов
с числом звеньев в циклах, образуемых этими лигандами и ионом ни¬
келя. В случае 1,2- и 1,3-диамина соответственно образуются пяти-
и шестнчленные циклы:В случае 1,4- и 1,5-диаминов должны были бы образовываться семи-
и восьмичленные циклы, но они неустойчивы. Вследствие этого тетра¬
метилен и пентаметилендиамин связываются с двумя ионами никеля,
■образуя мостики: NH2CH2CH2CH,CH2NH2 • • •В результате получаются полимерные комплексы.NH,—СН,N< I
NH,—СН.87
Сравнительно недавно установлено [2], что 1,4-диаминобутан на¬
ряду с мостиковым димерным сульфоксидным комплексом
IPtCl2WMCO)NH2CH2CH2CH2CH2NH2Pt(AMCO)Cl2] образует соеди¬
нение [PtWMCO)(NH2CH2CH2CH2CH2NH2)CI]CI, в котором он замы¬
кает семичленный цикл. Фуета и Огино сумели синтезировать комп¬
лекс кобальта (III), в котором циклическими лигандами являются
три молекулы 1,4-диаминобутана. Для этого они заменили воду
(растворитель) на диметилсульфоксид (ДМСО). Более того, в диметил-
сульфоксиде им удалось заместить хлорид-ионы в комплексе цис-
[Со Еп2С12]+ на додеметилендиамин (Don) с образованием комплекса
[Со En2(Don)]3+. Примеры комплексов, в которых бидентатные ли¬
ганды образуют семичленные циклы, пожалуй, этим и ограничива¬
ются.Одной из причин, влияющих на устойчивость циклических орга¬
нических соединений, являются напряжения, возникающие в цикле
при числе звеньев, отличном от пяти или шести. Вероятно, эта же при¬
чина отражается и на устойчивости циклов в комплексных соедине¬
ниях. Образованием устойчивой пятичленной циклической группи¬
ровки Л. А. Чугаев объяснил также склонность к комплексообразо-
ванию а-диоксимов [1, с. 115].В настоящее время считается общепринятым, что даже в диметил-
глиоксиме никеля кроме двух пятичленных циклов за счет водород¬
ных связей образуются еще два дополнительных шестичленных цикла!Большой заслугой Л. А. Чугаева следует считать то, что он впер¬
вые связал характер и устойчивость комплексов с конкретной груп¬
пировкой атомов в органическом лиганде. Эти идеи легли в основу
многочисленных работ по синтезу и аналитическому применению ор¬
ганических реагентов и комплексонов.Правило циклов подтвердилось и при систематических исследо¬
ваниях Чугаевым дитиоэфирных комплексов платины (II). Наиболее
устойчивые и прочные соединения получались в том случае, когда
число метиленовых групп, находящихся между двумя атомами серы
в молекуле тиоэфира, равно двум или трем.ИК спектроскопическое исследование строения продуктов взаимо¬
действия комплексов K2[PtX4], где X — С1 и Вг, с дитиоэфирамнказало, что действительно лиганды с двумя и тремя метиленовыми
группами между атомами серы приводят к образованию мономерныхнч,/ч\о/о'88
циклических соединений типа [PdLCI2] [3J. В случае дитиоэфиров с
одной и четырьмя метиленовыми группами замыкания цикла не про¬
исходит, а образуются полимерные соединения, в которых дитиоэфиры
связывают два атома палладия.При образовании полициклических соединений один из циклов
может быть очень больших размеров. Стабилизация таких комплек¬
сов осуществляется.за счет других пятп- и шестичленных циклов,
образуемых лигандом. Примером соединения, содержащего макро¬
цикл, может служить комплекс=снч /(СН,),На примере лиганда
XH-CH-v /СИ,СН2СН.СН..Кщ/ ‘ nnik ' ' ' N-jh,было показано, что пятичленный цикл стимулирует образование семи¬
членного около того же координационного центра. Тетрадентатный
лиганд сперминXH,CH2CHiv ,СНоСН2СН,СН,х уСН.СНгСН,NH; ‘ 'NIK " ' *\ЧН' "образует комплекс [Си(спермнн)](СЮ4)2, в котором два шестичленных
цикла также стабилизируют семичленный.Правило циклов получило развитие на базе большого фактиче¬
ского материала. В настоящее время можно утверждать, что донорные
группы атомов, несущие на себе отрицательный заряд (чаще всего это
кислотные остатки), способны образовывать устойчивые четырех¬
членные циклы. Это относится как к простым анионам, например
N03, СОГ, SO;", так и к остаткам органических кислот: диалкил
(арил)фосфорных, алкил(арил)фосфоновых, диалкил(арил)дитио-
фосфорных, дитиокарбаминовых, ксантогеновых и т. д.Существуют устойчивые комплексы с трехчленными циклами.
Так, довольно распространены соединения уранила, в которых пер-
оксолиганд Of", имеющий формальный заряд 2—, способен замыкать
трехчленный цикл.Синтезирован комплекс с NC-хелатным лигандом:L пи L\мн.
Продолжительной обработкой цис-\Pt(PMePh2)2Cl2] избытком1 -Li-2-C6H6-1 ,2-В10С2Ню в эфире получено соединение
[(Ph2MeP)Pt(PCH2Ph2)(2-CeH5-l>2-BioC2H1o)]> в котором одна изметальных групп превращена в метиленовую. В результате происхо¬
дит замыкание трехчленного цикла. Структурная формула этого
соединения может быть изображена следующим образом:PhoMeP, у. PPh' >Р< | г (X — 2-СвН6-1,2-В10С2Н” )Xх ХСН2 0Комплексы, содержащие NO-хелатный трехчленный цикл, полу¬
чены реакцией окислительного присоединения иодида диметил(мети-
лен)аммония к соединениям M(CO)3(NCCH3)3, где М •— Мо и W:сн2+ _ 0С\ ! /NMe2[Me2N=CH2] I + M(CO)/NCCH3)3—*- ОС—M + 2CH3CN' oc/|xiNCCH3Можно сделать заключение, что образованию необычных трех¬
и четырехчленных циклов, вероятно, способствует связь металла с
атомом углерода.5.2. Хелатный эффектПравило циклов Л. А. Чугаев сформулировал на основании препа¬
ративных данных. Количественные данные были получены лишь в
40-х годах XX в. Они были частично обобщены Шварценбахом в
1952 г. [4]. На большом экспериментальном материале он показал,
что константа образования циклического комплекса М(АА), где М —
катион металла, а АА — бидентатный лиганд, намного больше, чем
константа образования МА2, где А' — мон одентатный лиганд, близкий
по характеру к лиганду АА.В 1920 г. Морган и Дрю ввели в химическую литературу термин
«хелат» (от англ. chelate — коготь, клешня) для обозначения циклов,
образуемых лигандами при координации около иона металла. Соот¬
ветствующие комплексы, содержащие хелатный цикл, стали называть
хелатными соединениями.Шварценбах предложил называть разность между логарифмом
константы устойчивости хелатного комплекса МАА и нециклического
аналога МА2 «хелатным эффектом». Сравнивая хелатные эффекты
для этидендиаминовых и 1,3-пропилендиаминовых комплексов нике¬
ля (II) и меди (II) (за стандарт были выбраны аммиачные соединения),
Шварценбах пришел к заключению, что пятичленное хелатное кольцо
стабильнее шестичленного.Вычисление хелатного эффекта для комплексов кальция с амнно-
диацетатом (I) и алкилендиаминотетраацетатом (II)90
H—n/СН,—COO- -OOC—CH..ch.—coo-x:h2—coo- -ooc—crIIпоказало, что в зависимости от числа метиленовых групп в лиганде
он равен: 5,2 (п = 2); 1,7 (п = 3); 0,1 (п =; 4);—0,8 (п ^ 5). Таким
образом, хелатный эффект наибольший для пятичленного диаминово-
го кольца, меньший для шестичленного, почти отсутствует для семи¬
членного и отрицательный для восьмичленного.Катталинн с сотр. определили константы равновесия реакций типагде п равно 2 и 3. Они’также пришли к заключению, что наиболее ста¬
билен комплекс с пятичленным хелатным циклом.Считают, что хелатныи эффект связан с изменением энтропии си¬
стемы. При образовании комплексов, например в водном растворе,
происходит замещение молекул воды гидратированного иона металла
на определенный лиганд. Если в комплекс вступают монодентатные
лиганды, то освобождается равное число молекул воды:
lNi(H20)6]2+ + 6NH3=[Ni(NH3)e]2+ + 6НгОЕсли вступает бндентатный лиганд, то число освобождающихся мо¬
лекул воды вдвое больше, чем число лигандов, входящих в комплекс:lNi(HsO),]J* + ЗЕп = [Mi Еп3]2+ + 6Н,0;Это приводит к тому, что возрастает энтропийная составляющая сво¬
бодной энергии TAS в уравненииAG — — RT\nK= АН — T&S,т. е. уменьшается свободная энергия системы. В ряде работ были
сделаны попытки прямой экспериментальной оценки вклада энтропии
и энталйпии в константу устойчивости определенных комплексов.
Весьма наглядные данные были получены в работе [5]. Часть из них
представлена в табл. 5:1.Из табл. 5.1 следует, что для комплексов кадмия.с монодентатными
лигандами (аммиаком и метиламином) и бидентатным (этилендиами-
ном) имеется лишь небольшая разница (или она вообще отсутствует)
в значениях АН0. В то же время разница в значениях AS° достигает
очень большой величины и, что самое главное, для комплексов с би-
дентатным лигандом значение AS° отрицательно.Бьеррум и Нильсен впервые показали, что значение хелатного
эффекта при одних и тех же лигандах, но для ионов различных ме¬
таллов, может отличаться. Следовательно, хелатный эффект может
зависеть не только от энтропийной, но и от энтальпийной составляю¬
щей изобарного потенциала. Чаще всего влияние хелатообразования
на значение АН объясняют отталкиванием лигандов в комплексе,W 441r/91
Таблица 5.1. Константы образования и термодинамические пара¬
метры для комплексов кадмия (II) при 25ХКомплексigP(Моль Л-1)4H”,
кДж-моль 1дс°,к Дж • моль-1AS\Дж-моль^-К-1Cd(NH.,)|+4,9529,7928,245,19Cd(NH.;CH3)22+4,8129,3727,456,46CdEn2+5,8429,4133,30— 13,05Cd(NH3);+7,4453,1442,5135,50Cd(NH2CH3)5+6,5557,3237,4166,94CdEn;+10,6256,4860,67— 13,75например, двух молекул аммиака. Расталкивание двух аминогрупп
в этилендиамине уже существовало. Поэтому при его вхождении в
комплексе дестабилизации, связанной с этим эффектом, не произой¬
дет.Если лиганд с ионом металла способен образовать несколько хе-
латных циклов, то комплексы с полициклическими лигандами ука¬
зываются более устойчивыми, чем с моноциклическими. Например,
из двух комплексов меди (II)сн,—сн,
1 - 1 *2 +"сн,—сн, ‘
1 ‘ 1 -NH, NH,1 1N Н2 NH СН,Ч‘ / ‘\*СиСи СН,/ \/X / -NH, NH,I ' I *NH, NH СН,| * I -1 1_сн2—сн2 __сн2—сн, _второй примерно на четыре порядка устойчивее, чем первый. Причи¬
ной стабилизации в данном случае также является хелатный эффект,
связанный с образованием дополнительного цикла.Хелатный эффект проявляется практически у комплексов всех
металлов. К немногочисленным исключениям относятся соединения
серебра (I) и, вероятно, меди (I). Если сравнить устойчивость этиден-
диаминового Ag Еп+ и аммиачного Ag(NH3)2 комплексов, то оказы¬
вается, что хелатный эффект имеет невероятно большое отрицатель¬
ное значение, равное —2. Это связано с тем, что серебро (I) образует
комплексы линейной структуры, и это должно приводить к очень
большому напряжению цикла. Вероятно, в комплексе Ag Еп+ этилен-
диамин выступает в' роли монодентатного лиганда, т. е. цикл не за¬
мыкается.92
5.3. транс-Хелатные соединенияВ последние годы были предприняты многочисленные попытки полу¬
чения комплексов, в которых хелатный лиганд занимал бы два коор¬
динационных места в транс-положении. Библиографию по данному
вопросу можно найти в статье [6]. Одними из первых о получении
такого соединения заявили Исслейб и Хольфельд. Они сообщили о
получении комплекса [NiLCl,), где L — (CeHn)2P(CH2)5P(CeHn)2,
в котором донорные лиганды располагаются в транс-положении
друг к другу. Дипольный момент этого комплекса равен всего
лишь 7,9 ■ 10-30 К.л-м. Аналогичный комплекс с лигандом
(С6Н11)2Р(СН=)3Р(С!,НП)2, quc-конфигурация которого не вызывает
сомнения, имеет дипольный момент 37,7-10~30 Кл-м. По-видимому,
нужны дополнительные исследования, чтобы доказать реальность
образования транс-хелатных семичленных циклов.Шоу с сотр. получили, вероятно, одно из первых соединений с хе-
латным циклом в транс-положении. Взаимодействием [Pt(NCPh)2Cl2]
с Bib Р(СН2)10Р Ви2 они выделили два комплекса. На основании
данных ЯМР, ИК спектроскопии и результатов измерения молеку¬
лярной массы одному из этих соединений приписана структура 1,
а другому II:Cl'ГрI.PtIрL_(СН2)]0—I
р
I^Pt
СК ICI(СН2),0I_|„С1(СН,)2 40С1 РЧ /•PtIIПозднее были синтезированы и охарактеризованы другие соеди¬
нения платины (II) транс-хелатного типа. Рентгеноструктурное ис¬
следование комплекса транс-[PtLCl2], где L — Ви2 P(CH2)12PBu2,
полностью подтвердило наличие в комплексе транс-хе латного цикла,
состоящего из 14 звеньев.Венанзи с сотр. синтезировали жесткий лиганд/ч/Ч/'ЧСН,
I ■PhoPСН,I '
PPh,9S
который не может образовывать цис-хелатные циклы. С этим лиган¬
дом удалось получить мономерные комплексы типа [MLX2] (где М —
Ni(II), Pd(II) и Pt(II), X — галогенидный ион), которые имеют
транг-конфигурацию. Хелатный цикл этих комплексов состоит из 12
звеньев.Бейлар с сотр. ввели диэтилентриамин в соединение платины (IV),
а затем восстановили его, синтезировав транс-хелатный комплекс
платимы (II):h3nClСЦ I ...,N11
'l^c-iClRI(Cl!,)-,—N-I " IN11,-4CII2)2II,N'H 3N 'Pt■ гCl1,,N11, I
'•Nil—I-NR.p>:III/'N4 з j^Nl/\C13cuКомплекс III неустойчив, и при перекристаллизации из воды аммиак
вытесняется из его внутренней сферы. В процессе этого хелатный ли¬
ганд вместо неустойчивого восьмичленного цикла замыкает два пяти¬
членных с образованием комплексаТаким образом, можно считать несомненным, что существуют комп¬
лексы с транс-хе.латными циклами. Принципиальных ограничений
для максимального числа звеньев не имеется, но должно быть ограни¬
чение для их минимального числа. Вероятно, транс-х&латный цикл
способен образовываться в том случае, если он включает не менее
восьми звеньев.Правило циклов сыграло исключительно важную роль в развитии
координационной химии и постоянно служило руководящим прин¬
ципом в целенаправленном синтезе комплексонов и органических
аналитических реагентов. Важным дополнением к правилу циклов,
сформулированному Л. А. Чугаевым, следует считать установление
способности отрицательно заряженных лигандов образовывать че¬
тырех- и даже трехчленные циклы. Несомненно, что в ближайшие94
годы должен быть сформулирован принцип образования наиболее
устойчивых транс-хелатных циклов.5.4. Изомерия хелатных комплексовОсобый тип изомерии связан с образованием различных хелатпых цик¬
лов. Нередки случаи, когда лиганд в комплексе способен замыкать
как пятичленный, так и шестичленный цикл. Это особенно характерно
для органических оксимов и нитрозосоедпнений. Например, диметил-
иминоглиоксимСН3—С—С—CI13
II II
НО—N NHс ионами Ni(II) может образовать три изомерных соединения:н3сх /СН,с сII IIо' \ / н: Ni :Ik / V>0
Кн3с/ \сн.1I,C. /СН,
X' с11 л(Г \ / н: Ni14 / \N nI! ICH,CH3I IIО N.\ / 11
N1>4 / \II °\AH,C^ "CCH,Этот тип изомерии связан с заменой донорного атома, участвующего
в координации, и с изменением числа звеньев в хелатном цикле. Пер¬
вые два изомерных комплекса стабилизированы за счет водородных
связей.Рассмотренный тип изомерии называют внутримолекулярной хе-
латно-связевой изомерией.
ГЛАВА6ХИМИЧЕСКАЯ СВЯЗЬ В КООРДИНАЦИОННЫХ
СОЕДИНЕНИЯХДля характеристики химической связи в координационных соедине:
ннях в настоящее время наиболее широко используются следующие
теории: валентных связей, кристаллического поля и молекулярных
орбиталей. Все они описывают химическую связь приближенно и
каждая из ннх имеет свои достоинства и недостатки.6.1. Электростатические представленияВажным этапом в развитии теории химической связи в координаци¬
онных соединениях были электростатические представления. Они
во многом связаны с именем В. Косселя. Он считал, что образование
молекул из атомов сводится к образованию ионов с устойчивой вось¬
миэлектронной структурой, характерной для атомов благородных
газов, и с последующим их притяжением в соответствии с законом
Кулона. Так, например, при образовании молекул NaCl из атомов
Na и С1 внешний электрон от атома Na переходит к атому С1. В ре¬
зультате образуется положительно заряженный ион Na+ и отрица¬
тельно заряженный нон СГ, оба имеющие восьмиэлектронные внеш¬
ние оболочки. Ионы притягиваются друг к другу и в результате об¬
разуются молекулы NaCl.Для переходных металлов электронная структура атомов благо¬
родных газов соблюдается далеко не всегда и поэтому в электроста¬
тических представлениях о химической связи в соединениях пере
ходных металлов большую роль отводили зарядам и радиусам ионов.Развивая электростатические представления, Коссель пришел к
заключению, что система, состоящая из /г-зарядного катиона (Мл+)
и п однозарядных анионов (Х“), менее устойчива, чем система [МХп+1]~.
Потенциальная энергия [МХп+1]~ меньше, чем системы МХ„. Следо¬
вательно, образование комплекса [МХп+1]- из МХП и X- выгодно
энергетически. Этот вывод может быть проиллюстрирован на примере
образования комплекса [Agl2]'. Из рис. 6.1 следует, что сила притя¬
жения иона I" к нону Ag+ равна е2/л2, где е — заряд ионов, а силаотталкивания друг от друга двух ионов Г
равна е2/(4г2). Таким образом, сила отталки¬
вания лигандов составляет лишь 0,25 силы ихРис. 6.1. Схема электростатического взаимодейст¬
вия центрального иона и лигандов в комплексе
I AglJ"96
притяжения к центральному нону. Для комплекса [Agl3]2- при рас¬
положении лигандов в вершинах равностороннего треугольника сила
расталкивания лигандов составляет 0,58 силы притяжения к цент¬
ральному нону. Расчеты показывают, что для комплекса [Agl4]3' как
тетраэдрического, так и плоскоквадратного строения сила расталки¬
вания лигандов практически равна силе их притяжения к централь¬
ному иону.С увеличением положительного заряда центральный ион, естест¬
венно, может удерживать около себя большее число противоположно
заряженных лигандов, т. е. с увеличением заряда должно повышаться
его координационное число.Образование комплексов с нейтральными лигандами Коссель рас¬
сматривал с позиции электростатического взаимодействия иона ме¬
талла с отрицательным концом диполя (постоянного или индуциро¬
ванного) молекулы лиганда.6.2. Теории валентных связейВ 1916 г., еще до разработки квантово-химических представлений,
Льюис пришел к заключению, что химическая связь образуется по¬
средством электронной пары. Каждый атом отдает на образование
одной химической связи по одному электрону. Такая двухэлектронная
связь была названа ковалентной. Например,Н Fн: н : n : н в: f• « • •Н FВ 1927 г. Гайтлер и Лондон на основе квантово-механического
расчета молекулы водорода обосновали гипотезу Льюиса и показали,
что ковалентная связь осуществляется парой электронов с антн-
параллельными спинами. Полинг и Слэтер распространили эту кон¬
цепцию на многоатомные молекулы с ковалентными связями. Раз¬
витый в их работах метод получил название метода валентных связей
(ВС), который иногда по имени своих создателей называется методом
ГЛСП (Гайтлер—Лондон—Слэтер—Полинг). Метод ВС в основном
приспособлен для рассмотрения ковалентных молекул. Расчеты по
этому методу довольно сложны, однако качественные оценки, полу¬
чаемые на его основе, широко используют.Известно, что аммиак и трифторид бора реагируют по уравнениюNH3 + BF3 = BF3 • NH3 + 171 кДжОбразование соединения BF3-NH3 происходит за счет свободной
р-орбнтали атома бора и электронной пары атома азота молекулы
аммиака по схемерзв О- :NH34-81797
Такой механизм образования химической связи называют донорно-
акцспторным, а химическую связь — донорно-акцепторной*'. До¬
нором называют атом, который отдает, свою электронную пару для
образования химической связи — в данном случае это атом азота.
Атом, который предоставляет пустую орбиталь, т. е. принимает
электронную пару, называют акцептором. В рассматриваемом случае
это атом бора. Донорно-акцепторная связь является разновидностью
ковалентной связи.Донорно-акцепторный механизм играет важную роль в образова¬
нии координационных соединений. В них акцептором, как правило,
выступают ионы металлов, имеющие вакантные орбитали. Особенно
большое число вакантных орбиталей (за счет d-орбиталей) можно ожи¬
дать у переходных металлов.Разрыв ковалентной связи (процесс, обратный образованию связи)
может протекать гомолитически или гетеролитически. При гемолити¬
ческом разрыве связи каждый атом оставляет за собой один из элект¬
ронов электронной пары, осуществляющей связьМ : L М. + . LПри гетеролитическом разрыве связи электронная пара остается око¬
ло одного из атомов. Возможны два типа гетеролнтического разрыва
связи:м: l -► м+ +: l-м; l м-: -1- L+В комплексах металлов гемолитический разрыв связи и второй тип
гетеролнтического разрыва связи, когда электронная пара остается
на металле, могут рассматриваться как процессы восстановлення
иона металла.Некоторые определения комплексных (координационных) соеди¬
нений прямо отражают важную роль донорно-акцепторного взаимо¬
действия центральный атом — лиганд. Напомним, что Б. В. Некра¬
сов считает, что комплексные соединения — это те, которые обра¬
зуются из составных частей без возникновения новых электронных
пар. Кальвин и Мартел называют координационным соединением
продукт сочетания иона металла с донором электронов.Важное значение в теории валентной связи имеет концепция гиб¬
ридизации атомных орбиталей. Эта концепция развита главным об¬
разом Полингом. Она позволяет простыми способами интерпретиро¬
вать структурные формулы в терминах метода валентных связен.* Часто донорно-акцепторные связи обозначают стрелком, направление-
которой указывает направление смещения электронном пары. Если не акцен¬
тируется внимание на донорно-акцепторном характере связи, ее обозначают,
как и ковалентную связь, черточкой. Иногда этим подчеркивают отсутствие-
принципиального различия между донорно-акцепторной и ковалентной свя¬
зями.98
Полинг постулировал, что число орбиталей центрального атома,
участвующих в образовании координационных связей, равно числу
окружающих его лигандов. Орбитали центрального атома, участвую¬
щие в образовании координационный связей, гибридизованы. Гиб¬
ридные орбитали имеют вполне определенное направление в простран¬
стве. Так, если в гибридизацию вступают одна s- и одна р-орбиталь,
то образующиеся две гибридные s/7-орбитали направлены в простран¬
стве под углом 180°. Они обеспечивают образование молекулы линей¬
ной конфигурации. Если в гибридизацию вовлечены одна d-, одна s-
п две р-орбитали, то они образуют четыре гибридные ds/r-орбитали.
Такие орбитали направлены к вершинам квадрата и, следовательно,
обеспечивают образование молекул или ионов плоскоквадратного типа.
В табл. 6.1 приведены типы гибридных орбиталей и обусловленные
ими конфигурации соединений.Таблица 6.1. Тип гибридизации валентных орбиталей и
пространственная конфигурация молекулТип молекулы
или нонаТип гибридизацииКонфнгурацнямх2sp
sp2ЛинейнаяMX,Треугольнаямх4sp3, sd3
dsp2ТетраэдрическаяПлоскоквадратнаямх5dsp3, sp3d
d'-sp-ТригональнобипирамидальнаяТетрагональнопирамидальнаямх6d2sp3, sp3d-ОктаэдрическаяЛегко представить, какие именно атомные орбитали из нескольких
возможных принимают участие в гибридизации. Если в плоскоквад¬
ратном комплексе провести оси, как это указано на рис. 6.2, то из
трех ^-орбиталей (рх, ру, pj по положению в пространстве орбитали
рх и ру в наибольшей степени соответствуют гибридной ds/r-орбитали.
Из пяти d-орбиталей (dxy, dxz, dyz, dzz, dX2_y,) орбиталь dxt_yi по сим¬
метрии полностью аналогична гибридной. Именно она и включается
в гибридизацию.Таким же образом можно определить атомные орбитали, прини¬
мающие участие в образовании гибридной (^^-орбитали и обуслов¬
ливающие образование октаэдрических
комплексов. Из рис. 6.3 видно, что из
пяти d-орбитален в наибольшей мере
гибридной орбитали соответствуют dZ3И dx 2 у2.Комплексы одной и той же геомет-Рис. 6.2. Положение р-АО металла относи¬
тельно лигандов в плоскоквадратном окруже¬
нии4**99
Рис. 6.3. Положение d-АО металла от¬
носительно лигандов в октаэдрическом
окружении:uuL-pxy — АО dxi_vI и dj.1; внизу — АО d.t.рической конфигурации могут об¬
разовываться гибридными орбита¬
лями с участием внутренних или
внешних d-брбиталей, например
d2sp3 или sp3d2, dsp3 или sp3d (см.
табл. 6.1). Комплексы, образован¬
ные гибридными орбиталями с уча¬
стием внутренних d-орбиталей
(icFsp3, dsp3), называют внутриорби-
тальными, а с участием внешних
с/-орбиталей (sp3d~, sp3d) — внеш¬
неорбитальными.Согласно модели максимального
перекрывания химическая связь
образуется в направлении макси¬
мального перекрывания электрон¬
ных облаков взаимодействующих
атомов. Гибридизация приводит к
образованию орбиталей, ориенти¬
рованных по линии, соединяющей
ядра атомов. Это повышает суммарное перекрывание АО взаимодейст¬
вующих центров и должно приводить к более устойчивой конфигура¬
ции. В координационных соединениях включение в гибридизацию d-AO
центрального атома, близких по энергии к его s- и р-АО, приводит к
существенному возрастанию способности гибридной орбитали к паре-
крыванию. Однако прочность образующихся в комплексе связей
часто сильно зависит от относительных энергий взаимодействующих
уровней металла и лигандов. Если разница в этих энергиях велика,
то даже при возможном большом перекрывании соответствующих
орбиталей образующиеся связи могут быть весьма слабыми. По этой
причине обычно донорно-акцепторньш связи слабее ковалентных.Рассмотрим строение комплексного иона [Ag(NH3)2]+ с точки
зрения метода валентных связей. Электронная конфигурация иона
серебра+Ag4 d5 55 pitItItItwВ пределах валентной оболочки у иона серебра имеются вакантные
орбитали, которые и играют роль акцепторов электронных пар:100
4 d5.т-VАрИиItt1I IH(N NH,5s- и 5р-орбнтали иона Ag+ образуют две гибридные s/7-орбиталн.
Перекрывание орбиталей центрального атома с орбиталями молекул
аммиака можно изобразить следующим образом:Н ,N <3£>Ag<3£> NН ,Рассмотрим строение комплексного иона [Pt(NH3)4]2+. Электрон¬
ная конфигурация свободного иона Pt2+5 d6s6pitItitiiДля образования прочных связей энергетически выгодно освободить
одну из 5^-орбиталей спариванием электронов:5 d6 S6рЭлектронная конфигурация комплексного иона lPt(NH3)4p+5 d6s6pItII♦iH .... \nh.5d-, 6s- и две 6р-орбитали образуют четыре гибридные с^р2-орбитали.
Поскольку они направлены к вершинам квадрата, то конфигурация
комплексного иона будет плоскоквадратной:101
Лиганд, донорный атом которого имеет в валентной оболочке элек¬
тронные пары и вакантные орбнталн, может одновременно быть и до¬
нором п акцептором. Допустим, что атом металла имеет вакантную
с^з-^-орбиталь и заполненную ^-орбиталь. При взаимодействии
атома металла с молекулой аммиака электронная пара аммиака внед¬
ряется в вакантную с^^.-орбиталь металла. Между атомами азота
н металла образуется одинарная связь. В молекуле фосфина РП3
атом фосфора наряду с неподеленной электронной парой имеет ва¬
кантные Зй-орбитали. С металлом кроме обычной донорно-акцепторной
связи фосфоры-металл может образоваться вторая связь металл—«-
-^-фосфор за счет смещения электронной плотности с d-орбитали ме¬
талла на аналогично расположенную в пространстве вакантную
d-орбиталь атома фосфэра. Такая связь, являясь разновидностью до¬
норно-акцепторной, часто называется обратной дативной л-связыо
или просто л-дативной связью:Здесь атом металла является л-донором электронов, а атом фосфо¬
ра л-акцептором. Дативные связи могут быть образованы и за счет
р-орбиталей. Однако d-орбитали вследствие благоприятной про¬
странственной направленности обеспечивают лучшее перекрывание
орбиталей и, следовательно, образование более прочных связей.В четырехкратных связях наряду с ст- и л-компонентами имеется
б-связь. Она образуется за счет ds —атомных орбиталей по схемеS-Связи чрезвычайно характерны дчя координационных соедине¬
ний со связью металл — металл.Рассмотренная теория гибридизации орбиталей центрального атома
полезна при качественном описании локализованного характера хи¬
мических связей во многих соединениях, их направленности в прост¬
ранстве. Однако гибридизация атомных орбиталей — не единственный
фактор, определяющий геометрическую форму молекул. Крэме
электронной составляющей в полную энергию молекулы, минимум
которой соответствует равновесной геометрической конфигурации,
входит энергия межъядерного взаимодействия. Ее вклад может пре¬
обладать.6.3. Концепция отталкивания электронных пар
валентной оболочки102
Найхольм и Гиллеспи предложили концепцию отталкивания элек¬
тронных пар валентной оболочки, которая также не учитывает межъ-
ядерное взаимодействие, но вследствие своей простоты оказалась
весьма полезной для объяснения и предсказания геометрии молекул.
Эта концепция особенно эффективна в применении к молекулам с
лигандами, неравномерно расположенными в пространстве.Идеи Гиллеспи и Найхольма очень близки к описанию образова¬
ния химических связей за счет гибридных орбиталей, но базируются
на несколько иных представлениях. Согласно их концепции связы¬
вающие и неподеленные электронные пары центрального атома рас¬
полагаются в пространстве как можно дальше друг от друга, чтобы
их отталкивание было минимальным.Для предсказания конфигурации молекул с точки зрення Гил¬
леспи и Найхольма необходимо принять, что наибольшее расталки¬
вание имеет место между неподеленными электронными парами. В
меньшей степени расталкиваются неподеленная электронная пара и
связывающая электронная пара. Самсе слабое расталкивание проис¬
ходит между связывающими электронными парами. В пользу этого
Гиллеспи приводит следующие соображения: на неподеленную элек¬
тронную пару влияет только положительно заряженный атомный ос¬
тов, поэтому можно ожидать, что ее облако несколько больше, чем у
связывающей пары, находящейся на той же валентной оболочке, но
в поле двух положительно заряженных атомных остовов.Рассмотрение приложения концепции Найхольма и Гиллеспи нач¬
нем с соединений непереходных элементов. Так, атом бериллия имеет
два валентных электрона. В соединениях бериллия типа ВеХ2 в ва¬
лентном электронном слое центрального атома имеются только две
электронные пары, осуществляющие связь. Эти электронные пары
располагаются в пространстве так, чтобы быть максимально удаленны¬
ми друг от друга, т. е. под углом 180°.Бор имеет три валентных электрона. Молекулы типа ВХ3 имеют
треугольную конфигурацию, так как здесь в валентном электронном
слое находятся три электронные пары, осуществляющие связь. Они
располагаются в пространстве на максимальном расстоянии друг от
друга.Олово (II), например в SnCl2, в валентном слое имеет также три
электронные пары, окружающие центральный атом. Однако химиче¬
ские связи осуществляют только две электронные пары, а третья яв¬
ляется неподеленной. Поэтому вследствие большего расталкивания
неподеленной электронной пары с электронами связей по сравненню
с расталкиванием электронов на связях угол между связями несколь¬
ко меньше 120°:103
Рис. 6.4. Расположение четы¬
рех электронных пар в моле¬
куле:а — СН< — тетраэдр; 6 — NH3 — три-
гопальная пирамида; о — Н20 —
уголiiиРасположение четырех
электронных пар при усло¬вии их максимального удаления приводит к тетраэдру. Тетраэдри¬
ческая направленность связывающих и неподеленнык электронных
пар характерна для центральных атомов в молекулах СН4, NH,,
Н20 (рис. 6.4).В молекуле СН4 четыре связывающие электронные пары. По¬
скольку их расталкивающее действие одинаково, то молекула СН4
имеет строение правильного тетраэдра (углы между связями равныВ молекуле NH3 в валентном электронном слое три связывающие
электронные пары и одна неподеленная. Поскольку неподеленная
электронная пара расталкивает орбитали сильнее, чем связывающая
пара, то угол между связями Н—N—Н в молекуле аммиака несколько
меньше тетраэдрического. Он составляет 106,7°.В молекуле Н20 в валентном электронном слое две связывающие
и две неподеленные электронные пары. Вследствие большого растал¬
кивающего действия неподеленных электронных пар угол между свя¬
зями Н—О—Н становится еще меньше. Он равен 104,5°.Если центральный атом окружен пятью или шестью электронными
парами, как связывающими, так и неподеленными, то их минималь¬
ное взаимодействие возможно в конфигурации тригональной бипира¬
миды или октаэдра соответственно.Конфигурации некоторых молекул, имеющих строение тригональ¬
ной бипирамиды или производного от нее, приведены на рис. 6.5.Вершины тригональной бипирамиды неэквивалентны. На основе
модели жестких шаров Гиллеспи показал, что экваториальные по¬
ложения в ней отличаются большим простором, чем аксиальные.
Поэтому более объемные неподеленные электронные пары будут за¬
нимать экваториальную плоскость.Молекула РС15 имеет строение правильной тригональной бипира¬
миды. Ее остов сохраняется и для молекул с меньшим числом ли-109,5°).а0ьРис. 6.5. Схематичное расположение пяти электронных
пар в молекуле:а — PCU — тригональная бипирамида; б — ТеСЬ — бисфеноид;
в — CIF3 — Т-кокфигурация; г — IC1* — лннеГшая104
Рис. 6.6. Схематичное распо¬
ложение шести электронных
пар в молекулах и ионах:а — SFe и PF, — октаэдр; 6 — IF* —тетрагональная пирамида: в —
1СГ— квадратабвгандов, центральный атом которых имеет соответствующее число не-
поделенных электронных пар. Так, например, в молекуле ТеС14 ли¬
ганды находятся в четырех вершинах тригональной пирамиды, а
к пятой направлена неподелейная электронная пара. Эксперимен¬
тальные данные свидетельствуют о том, что в этой молекуле угол меж¬
ду двумя экваториальными связями С1—Те—С1. близок к 120° и около
90° по отношению к связям, направленным к аксиальным положениям.В молекуле C1F3 в трех вершинах тригональной бипирамиды на¬
ходятся лиганды, а к остальным двум направлены неподеленные
электронные пары. В результате этого молекула имеет Т-конфнгу-
рацию. Вследствие сильного расталкивающего действия неподелен-
ных электронных пар углы между связями F—Cl—F несколько мень¬
ше прямого. Они равны 87,5°.Ион 1С!2 имеет линейную конфигурацию. Лиганды занимают вер¬
шины тригональной бипирамиды, находясь на максимальном расстоя¬
нии друг от друга. Неподеленные электронные пары располагаются
в экваториальном положении.Конфигурации некоторых молекул, характеризующихся окта¬
эдрической направленностью электронных пар, приведены на рис. 6.6.
Молекула SF6 и ион PF6 имеют октаэдрическое строение. Строение
молекулы IF5 а также иона 1С14 является производным от октаэдра.
Первая из них имеет конфигурацию тетрагональной пирамиды, а
IC14—плоского квадрата.При рассмотрении геометрической конфигурации молекул с крат¬
ными связями считается, что л-связи не влияют существенным об¬
разом на расположение сг-связей вокруг центрального атома. В таком
случае геометрическая конфигурация молекулы С02 определяется
расталкиванием двух электронных пар, образующих две ст-связи
углерод— кислород, т. е. она линейная. В то же время атом серы в
молекуле S02 имеет четыре валентных электрона, не образующих
л-связи, т. е. он окружен двумя электронными парами связей сера —
кислород и одной неподеленной электронной парой. Расположение
связей вокруг этого атома определяется расталкиванием трех элек¬
тронных пар, т. е. молекула имеет угловое строение119,5°105
'Молекула РОС13 представляет собой искаженный тетраэдр, так
как атом фосфора образует с кислородом и тремя атомами хлора че¬
тыре сг-связывающие электронные пары.Геометрическая конфигурация молекул с p-элементом в качестве
центрального атома в зависимости от числа и характера валентных
электронных пар систематизирована в табл. 6.2.Таблица 6.2. Конфигурация молекул в зависимости от числа
и характера валентных электронных парЧисло
электрон¬
ных пар
в валент¬
ном слоеЧисло
связываю¬
щих парЧислонеподс-ленныхпарКонфигурация молекулыПримеры220ЛинейнаяBeCI2, С02, COS330ТреугольникВС13. СО*-, NOf,
SO3, СОС1221V-образнаяSnCI2, S02, NOj"440Тетраэдрсн4, so*- ро|-.S02CU, POCI331Тригональная пирамидаNH3,S0^-, SOClo,
CIO72оV-обрачнаяH20. CloO, ClOf50Тригональная бипирамидаPCU. SOF454IИскаженный тетраэдрXe02F232Т-образнаяCIF323Линейная1С1Г60ОктаэдрSFe, PF<r651Тетрагональная пирами¬
даIF542Квадратici—Концепция отталкивания электронных пар валентной оболочки
применима и для рассмотрения геометрической конфигурации коор¬
динационных соединений переходных металлов. Важнейшей чертой106
этого типа соединений является наличие у их центральных атомов
электронов на d-АО, которые могут взаимодействовать с неподеленны-
ми электронными парами и электронами, участвующими в образова¬
нии связей. Если- d-электронная оболочка сферически симметрична
(d°, d5, d10), то, согласно Гиллеспи, она не оказывает влияния на
расположение электронных пар на внешней оболочке. Тогда конфи¬
гурация соединений переходных металлов будет определяться так же,
как и соединений непереходных элементов.Так, комплексный ион [Ag(NH3)2]+ с конфигурацией Ag+ (4d10)
имеет вследствие расталкивания неподеленных пар двух молекул ам¬
миака, осуществляющих донорно-акцепторное взаимодействие ли¬
ганд — металл, линейную структуру, а тетрахлорид цинка [ZnCI4]-~—
тетраэдрическую, так как его центральный ион Zn2+ имеет симметрич¬
ную оболочку d10 и расположение связей вокруг него определяется
взаимодействием четырех неподеленных электронных пар хлорид -
ионов, участвующих в образовании связей Zn—Cl.Геометрия гексафторида железа (III) с электронной конфигурацией
иона железа 3dh определяется взаимным отталкиванием шести элект¬
ронных пар фторид-ионов и соответствует октаэдру. Треугольная
конфигурация цианидного комплекса [Cu(CN)3F* с центральным ио¬
ном, имеющим Зс(10-электронов, обусловлена расталкиванием трех
электронных пар цианид-ионов.Если на d-оболочке электронов мало (d1, d2, d3) либо она мало от¬
личается от симметричной (d4, d6), то взаимодействие ее с электрона¬
ми, образующими связи, считается слабым. Конфигурация молекулы
будет лишь слегка искажена из-за отсутствия у d-оболочки сфериче¬
ской симметрии. Как следствие этого — меньшее разнообразие в
геометрическом строении координационных соединений.Однако если электронная конфигурация центрального атома d7,
d8 и d9, то оболочка из d-электронов достаточно сильно взаимодейст¬
вует со связывающими электронными парами, что отражается на их
расположении. Описание геометрического строения таких координа¬
ционных систем методом Гиллеспи затруднительно. Считается, что
в этом случае d-оболочка имеет приблизительную форму вытянутого
или сплюснутого эллипсоида.В комплексах двухвалентных никеля и палладия с одной и той же
электронной конфигурацией центрального атома (nd8) наблюдается
резкое различие геометрической структуры: комплекс [PdCI4]2- имеет
плоскоквадратное строение, a [NiCl4]2_ — тетраэдрическое. В первом
случае конфигурация центрального атома в диамагнитном комплексе2 о «2 2 Оdry, dyz, dxz, dz2, dxt—yi приводит к тому, что эллипсоидальная
d-оболочка вытянута вдоль оси z и минимальным взаимодействие с
ней связывающих электронов окажется в том случае, если они будут
располагаться в плоскости ху. Таким образом, геометрическая кон¬
фигурация палладиевого комплекса определяется сильным взаимо¬
действием электронных пар на связях с вытянутой эллипсоидальной
d-оболочкой.В парамагнитном никелевом комплексе, возможно, существенную
роль играет неспаренность электронов. Заполненность электронами107
d-оболочки более равномерна. В этом случае геометрическая конфи¬
гурация обусловлена уже не взаимодействием электронов на связях
с электронами d-оболочки, а более сильным расталкиванием четырех
связывающих пар электронов, т. е. является тетраэдрической.Важным фактором, влияющим на величину угла между химиче¬
скими связями, являете^ электроотрицательность центрального ато¬
ма. Так, угол между связями в молекуле НоО равен 104,5°, а в ана¬
логичной молекуле H2S составляет всего лишь 92,5°. Это может быть
объяснено тем, что электроотрицательность атома кислорода (3,5)
значительно выше, чем атома серы (2,5). В соответствии с этим связы¬
вающие электронные пары в Н20 ближе «оттянуты» к центральному
атому, чем в H2S. В результате расталкивание связывающих пар в
молекуле Н20 сильнее, чем в молекуле H2S.6.4. Полимеризация молекул с дефицитом
электроновКак уже было сказано, молекула ВеС12 имеет линейную конфигура¬
цию, а в молекуле А1С13 все атомы расположены в одной плоскости и
угол между химическими связями равен 120°. Такая геометрия моле¬
кул характерна для соединений, находящихся в газообразном состоя¬
нии. При переходе в твердое состояние геометрическое строение этих
молекул резко меняется. Рассмотрим такое изменение на следующем
примере. В пределах валентной оболочки атом бериллия имеет две
вакантные орбитали, которые стремятся заполниться электронами:2s2Р1,11Cl С1В газообразном состоянии, когда взаимодействие между молекулами
практически отсутствует, нет возможностей для заполнения электро¬
нами этих орбиталей. В конденсированном (твердом) состоянии они
могут быть заняты электронными парами хлорнд-нонов соседних
молекул. Действительно, каждый ион хлора в молекуле ВеС12 имеет
по три неподеленные электронные пары, которые могут участвовать
в образовании'дополнительной химической связи по донорно-акцеп-
торному механизму.Вследствие взаимодействия с хлорид-ионами соседних молекул
все вакантные орбитали центрального атома оказываются заполненны¬
ми электронами и каждый атом бериллия имеет четыре 0-связи. Та¬
ким образом, при переходе хлорида бериллия из газообразного состо¬
яния в твердое гибридизация атомных орбиталей изменится от sp-
типа к типу sp3. В результате хлорид бериллия в твердом состоянии
имеет полимерную структуру в виде бесконечных цепей, состоящих
из тетраэдров, в центре которых находится бериллий:108
ClN /\ „
>\ /<
ciClCl4 / \ /-
*\ /"<
CIУ атома алюминия в А1С13 в газообразном мономерном состоянии
также вакантна валентная р-орбиталь:'353/>А1*t*I.Cl С1 С!Уже в газообразном состоянии при понижении температуры и осо¬
бенно при переходе в твердое состояние вакантная р-орбиталь за¬
полняется электронной парой хлорид-иона соседней молекулы. В ре¬
зультате атом алюминия имеет четыре ст-связи и все его валентные
орбитали (s 3р) заняты электронами. Гибридизация sp3-THna обе¬
спечивает образование бис-тетраэдрических димерных молекул А12С1 а
с двумя хлоридными мостиками:Димерные молекулы А12С16 существуют и в органических растворите¬
лях, молекулы которых не являются донорами электронов. В элект-
ронодонорных растворителях вакантная р-орбиталь атома алюминия
будет занята неподеленной электронной парой молекулы растворителя.Электронное строение хлорида палладия PdCla схематично может
быть изображено следующим образом:4 d4 s5 рИиииIt-Cl CIВ пределах валентного слоя у атома палладия имеются три вакантные
р-орбитали. Вовлечение в гибридизацию двух р-орбиталей наряду с
d- и s-орбнталями обеспечивает образование четырех устойчивых
ds/r-орбиталей, расположенных в одной плоскости и направленных
к углам квадрата. Действительно, галогениды палладия в твердом
состоянии имеют строение бесконечных плоских цепей:100
Рентгеноструктурные исследования показали, что хлорид платины
находится в твердом состоянии в виде гексамера PtcCl12. Такого рода
соединения в литературе получили название кластеров. В этом кла¬
стере шесть ионов платины образуют октаэдр, а между собой они свя¬
заны мостиковыми хлорид-ионами.В результате такого построения
каждый ион платины находится в центре квадрата, образуемого че¬
тырьмя мостиковыми ионами хлора (см. рис. 2.1).Три рассмотренные соли ВеС12, А1С13 и PdBr: относятся к простым
солям. Однако в твердом состоянии их строение хорошо объясняется
с позиций координационной теории Вернера. Поэтому с полным
основанием эти соединения в твердом состоянии можно считать коор¬
динационными соединениями. Ионы металлов в них имеют координа¬
ционные числа, равные четырем.Особым случаем молекул с дефицитом электронов являются гид¬
риды бора. Установлено, что простейший представитель гидридов
бора — диборан В2Н6 — имеет димерную структуру, содержащую
водородные мостики:Таким образом, строение В2Нв аналогично строению А12С16. Однако
в отличие от атомов галогенов водород не имеет неподеленных элект¬
ронных пар.Строение диборанов трактуется с точки зрения делокализованных
трехцентровых орбиталей. Концевые связи в диборане образованы
обычными ковалентными связями посредством 5/?3-орбитали бора и
s-орбитали водорода. Дефицит электронов имеет место в мостиковых
связях. Мостики образованы тремя орбиталями (s/73 + s + sp3) и
всего лишь двумя электронами. Таким образом, электронная пара,
предварительно локализованная между атомом водорода и атомом бо¬
ра в ВН32s2 р11Н Н Нв диборане В2Н0 делокализуется. Поэтому такие связи и называют
делокализованными. В результате делокализации электронов все
валентные орбитали атома бора оказываются, хотя и не полностью,
заселенными электронами.Структура более сложных гидридов бора В3Н9, В4Н12 и т. д. ин¬
терпретируется аналогичным образом.6.5. Теория кристаллического поляК началу 30-х годов электростатические представления о характере
связей в координационных соединениях стали явно недостаточны для
описания многих важных свойств комплексов, прежде всего магнит¬
ных и оптических. Одной из приближенных теорий химической свя¬
зи, основанной на квантово-механических законах, явилась теория
кристаллического поля, основные идеи которой были изложены в
работе немецкого физика Бете «Расщепление термов в кристалле»
(1929).Согласно теории кристаллического поля существование неорга¬
нического комплекса обусловлено электростатическим взаимодейст¬
вием центрального атома и окружающих его лигандов, которые в
описываемой теории рассматриваются лишь как точечные заряды или
диполи. Во всех интересующих нас случаях лиганды либо отрицатель¬
но заряжены, либо ориентированы к центральному иону отрицатель¬
ным концом диполя.Одной из важнейших особенностей комплексных соединений пере¬
ходных металлов является наличие у ионов металла d-электронов.
В изолированном газообразном ионе или в сферически симметричном
поле все пять d-атомных орбиталей (АО) имеют одну и ту же энергию,
т. е. уровень d-орбиталей пятикратно вырожден. Однако в комплекс¬
ных соединениях наличие лигандов вокруг центрального иона при¬
водит к понижению симметрии, что, в свою очередь, вызывает рас¬
щепление ранее вырожденных энергетических состояний.Рассмотрим расщепление d-АО металла в октаэдрическом поле
лигандов на примере комплекса MLg+. Простейшей для интерпрета¬
ции является электронная конфигурация d1, например в гексаакво-
комплексе Ti (III) — [Ti(H20)6]3+.При надлежащем выборе системы координат (см. рис. 6.3) орби¬
тали dX!_yi и d2t центрального иона направлены вдоль осей координат
прямо к лигандам, находящимся в центрах граней куба, в который
вписан октаэдр, а орбитали dxz, dyz и dxy расположены между осями
координат симметрично по отношению ко всем шести лигандам. Элек¬
трон, находящийся на АО dx^yl или dzl, должен испытывать большее
электростатическое отталкивание со стороны лигандов, чем если бы
он находился на АО dxz, dyz и dxy. Энергия первых двух АО одина¬
кова и выше последних трех. Таким образом, пять d-состояний, имею¬
щих одинаковую энергию в свободном ионе или в сферически симмет¬
ричном поле, в октаэдрическом поле лигандов расщепляются на вы-
сокоэнергетическсе дважды вырожденное состояние, обозначаемое
eg, и стабилизированное (низкоэнергетическое) трижды вырожден-III
IVd.xz'dyzVРис. 6.7. Расщепление d-АО металла в различных полях ли¬
гандов:/ — тетраэдрическое поле (Td); II — сферическое поле; /// — октаэдриче¬
ское поле (Оь); IV — тетрагональное поле; V—плоскоквадратное поленое t2g (рис. 6.7). Иногда группу орбиталей dxz, dyz, dxy называют
dt -орбиталями, a dx*—yi и dz> — -орбиталями.Разница энергий верхнего и нижнего состояний, обозначаемая 4
или 10 Dq, называется параметром расщепления. Энергии расщеп¬
ленных d-AO таковы, что в сумме они дают среднюю энергию АО в
гипотетическом сферическом поле, принятую за нуль отсчета. Таким
образом, дважды вырожденные ег-орбитали (одноэлектронные состоя¬
ния в отличие от состояния всей системы часто обозначаются малень¬
кими буквами) повышают свою энергию на величину 3/6А или 6 Dq,
а трижды вырожденные i2g понижают на 2/БД (или 4 Dq).При дальнейшем понижении симметрии комплекса вырожденные
уровни расщепляются (рис. 6.7). Удаление аксиальных лигандов,
расположенных вдоль оси z (тетрагональное искажение), вызывает
повышение энергии уровня АО dxи понижение dzt относительно
уровня ее в октаэдрическом поле лигандов. Орбитали tig также пре¬
терпевают расщепление. Уровни АО dx* и dyz понижаются, образуя
дважды вырожденное состояние, а АО аху, соответственно, повышает
свою энергию.Распределение уровней АО в плоском квадрате легко получить,
представив плоскоквадратную конфигурацию лигандов как предель¬
ный случай тетрагонального растяжения октаэдрического комплекса
и удаления двух аксиальных лигандов. Относительные энергии об-112
Рис. 6.8. Положение некоторых-rf-AO ме¬
талла (dz: н dzx) относительно тетраэдри¬
ческого расположения лигандов (а — дли¬
на ребра модельного куба)разовавшнхся уровнен зависят от
конкретного вида волновых функций
металла, однако часто АО dz. понижа¬
ет свою энергию, а АО dxy, располо¬
женная в плоскости лигандов, повы¬
шает ее, т. е. происходит инверсия
указанных АО относительно их рас¬
положения в тетрагональном поле.Дальнейшее понижение симметрии
вызовет расщерление последнего вы¬
рожденного в плоскоквадратном поле
стабилизированного уровня, образо¬
ванного АО dxz,m dyz.В тетраэдрическом окружении
центрального атома большее отталки¬
вание от лигандов должны испыты¬
вать электроны АО dxy, dxz, dyz ме¬
талла, а не и d-., т. к. плоскости первых трех орбиталей пере¬
секают середины ребер куба, а остальные направлены к серединам
его граней (рис. 6.8). В результате образуется стабилизированное,
двукратно вырожденное состояние E(dxt_yt, d2i) и более высокоэнерге-
тнческое трехкратно вырожденное Г2 (dxz, dyz, dx Ситуация очень
напоминает расщепление в октаэдрическом поле лигандов (см. рис. 6.7)
с той разницей, что там вырожденные состояния инвертированы по
отношению к состояниям в тетраэдрическом поле. К тому же поле,
создаваемое шестью октаэдрически расположенными лигандами,
должно быть сильнее поля четырех лигандов тетраэдра, если в обоих
случаях межатомные расстояния достаточно близки. Это приводит
к уменьшению параметра расщепления Д в тетраэдрических комплек¬
сах по сравнению с октаэдрическими: ДТ[] » — Доь.Спектрохимический ряд. Параметр расщепления А является ко¬
личественной характеристикой расщепления и зависит не только от
симметрии комплекса, но и от природы центрального атома и лиган¬
дов. Значение Д для ионов металлов, имеющих различные d-АО, со¬
гласно ряду,' установленному Йоргенсеном, увеличивается в следую¬
щем порядке:Мп^П) < Со (II) « Ni (II) < V (II) < Fe (III) < Сг (III) << Со (II I) < Мл (IV) < Mo (III) < Rh (III) < Ir (III) < Re (IV) < PI (IV)При переходе от комплексов металлов первого переходного ряда
к аналогичным комплексам второго и третьего рядов параметр Д при
одинаковых лигандах возрастает на 30—80% . В некоторой степени это
обусловлено тем, что более тяжелые ионы имеют большие эффектив¬на
ные радиусы и большую поляризуемость. Параметр Д возрастает
и при увеличении степени окисления центрального иона.Изменение лигандов при фиксированном центральном атоме при¬
водит к увеличению параметра расщепления Д в рядуI- < Вг- < С1- ж SCN- < F" < ОН" < НаО < NCS- < CN" < NH3 << HaNC2H4NHo < N07 < CN-(подчеркнут координирующийся с металлом атом).Последний ряд называется спектрохимическим, так как он полу¬
чен из данных спектров поглощения комплексов в видимой и ультра¬
фиолетовой областях. Лиганды, стоящие в конце такого ряда, ока¬
зывают большое воздействие на центральный ион, приводя к значи¬
тельному расщеплению. Их называют лигандами сильного поля.
Лиганды слабого поля расположены в начале спектрохимического ряда.Правило дополнительности. Когда центральный ион имеет более
одного d-электрона, картина расщепления его состояний заметно
усложняется. При этом существенную роль играет межэлектронное
взаимодействие. Однако, согласно правилу дополнительности, элек¬
тронную конфигурацию, содержащую п d-электронов, можно пред¬
ставить как 10—п электронных дырок в замкнутой ^10-оболочке,
что значительно упрощает рассмотрение многоэлектронных состоя¬
ний. Поведение таких электронных дырок аналогично поведению
электронов с той лишь разницей, что дырку следует считать положи¬
тельно заряженной. Конфигурации (1п и d10~n в этом случае имеют вза¬
имно перевернутые схемы расщепления состояний. Так, картина рас¬
щепления орбиталей центрального иона, содержащего девять d-элек¬
тронов (например, Си2+), перевернута относительно рассмотренной
ранее для конфигурации d1. На рис. 6.9 показано соотношение между
октаэдрическими и тетраэдрическими расщеплениями для некоторых
электронных конфигураций. Подобные диаграммы называются диа¬
граммами Оргела. Приведенная на рис. 6.9 простейшая диаграмма
Оргела дает картину расщепления атомных состояний в зависимости
от силы поля лигандов А октаэдрического и тетраэдрического окру¬
жения центрального иона не только с конфигурацией dl, но и d4, d6,
и d9. В других случаях диаграммы Оргела имеют более сложный вид.Энергия стабилизации кристаллическим полем. d-Электроны цен¬
трального атома предпочтительно за¬
нимают положения, при которых их
отталкивание от лигандов минималь¬
но, т. е. более стабилизированные (с
меньшей энергией) уровни. Увеличе¬
ние энергии связи, которое достига¬
ется при этом, называется энергией
стабилизации кристаллическим полем
(ЭСКП). Значение ЭСКП для различ-Рис. 6.9. Диаграмма Оргела
ных конфигураций d-электронов в октаэдрическом поле в единицах
Д приведены в табл. 6.3. ЭСКП, равная понижению энергии основ¬
ного состояния при его расщеплении, тем выше, чем больше электро¬
нов занимает стабилизированный уровень t«g по сравнению ceg.Сильные и слабые поля лигандов. Заполнение d-орбиталей элект¬
ронами происходит в порядке увеличения их энергии и в соответствии
с правилом Хунда. Если число d-электронов начинает превышать
кратность вырождения стабилизированных уровней, например, в
случае электронной конфигурации d1 в октаэдрическом поле лиган¬
дов, то становятся возможными две ситуации: четвертый электрон мо¬
жет расположиться либо на высокоэнергетическом уровне eg, либо,
спариваясь с одним из уже имеющихся электронов, на низкоэнергети¬
ческом t^. Обычно рассматривают два предельных случая.1. Слабое поле лигандов. Разница в энергиях стаби¬
лизированного н дестабилизированного уровня (Д) меньше средней
энергии спаривания электронов (Ег). Состояние с максимальным спи¬
ном в этом случае является основным. Влияние лигандов можно рас¬
сматривать лишь как некоторое возмущение.2. Сильное поле лигандов. Поле литандов с больши¬
ми Д преодолевает электростатическое отталкивание между d-элект¬
ронами: Д > Ер. Образующиеся комплексы являются низкоспино¬
выми .Особый интерес в связи с таким разделением представляют для
октаэдрических комплексов конфигурации d4, d5, d6, d7 и для тетра¬
эдрических d3, d4, d5, d6, так как два предельных случая будут раз¬
личаться спином основного состояния. Распределение d-электронов
центральных ионов в октаэдрических сильных и слабых полях ли¬
гандов в приближении кристаллического поля представлено в
табл. 6.3.Таблица 6.3. Распределение d-электронов в октаэдрических
комплексахКонфи¬гура¬цияПримерСлабое полеСильное полеl2gcgэскп(в ед.
A)f2ge£ЭСКП
(в ед. Д)d°Sc3+, Ca2+00d1Ti3+, V4+40,440,4d2Ti2+, V3+1 10,84 40,8d3v=+, Cr3+I 1 11 ,24 4 |1,2d4Cr2+, ,Mn3+1 4 140,6It 4 41,6d5Mn-+, Fe3+, Os3+1 4 44 40It It 42,0d6Fe-+, Co3+, Ir3+It 4 44 40,4it It it2,4d~Co2+, Ni3+, Rh2+it It 44 40,8It it it41,8d8Ni2+, Pd2+, Pt2+, Au3+It It it4 41 ,2It It It4 41 ,2d*Cu2+, Ag2+it It ItIt 40,6It It ItIt 40,6d10Cu+ , Zn2+ , Cd2+ , Ag+,It It ItIt It0It It ItIt It0Hg;+, Ga3+115
В случае слабого октаэдрического поля лигандов преобладающим
оказывается отталкивание d-электронов между собой. Четвертый элект¬
рон заполнит дестабилизированный уровень es, образуя высокоспи¬
новый парамагнитный комплекс, как, например, в [Сг(Н20)6]г+.При больших значениях Д четвертому электрону выгоднее спа¬
риться с одним из электронов низкоэнергетической трехкратно вы¬
рожденной орбитали t2g, чем преодолевать действие сильного поля ли¬
гандов. Образующийся комплекс будет низкоспиновым и менее пара¬
магнитным (два неспаренных электрона). Увеличение числа d-элект¬
ронов до шести в сильном поле лигандов приводит к диамагнитным
комплексам, примером чему служит соединение [Со(1ЧН3)„]3+Таким образом, в рамках теории кристаллического поля можно во
многих случаях объяснить магнитные свойства комплексных соеди¬
нений, что и было одним из наиболее ранних применений этой теории.
Магнитный момент атома непосредственно связан с числом его неспа¬
ренных электронов. Существует приближенная формула: ц =
= ) ' л (я+ 2) , где и — магнитный момент в магнетонах Бора, п —
число неспаренных электронов. Это соотношение хорошо выполняется
для большинства металлов первого периода, но хуже для более тя¬
желых металлов второго и третьего периодов.Середину спектрохимического ряда составляют лиганды, создаю¬
щие поля промежуточной силы. В этом случае необходимо учитывать
как влияние лигандов, так и межэлектронное взаимодействие. Обыч¬
но в решении таких задач, представляющих наибольшую трудность,
используется одно из предельных допущений, а затем вводится ряд
поправок. Информацию о возможном электронном строении комплек¬
сов в полях промежуточной силы можно получить, используя диа¬
граммы 'Ганабе—Сугано [1].Приложения теории кристаллического поля. ЭСКП составляет
небольшую часть (5—10%) полной электронной энергии комплекса.
Однако при рассмотрении однотипных соединений следует учитывать
эффект кристаллического поля, которое может оказывать влия¬
ние на некоторые свойства комплексов.ЭСКП может стать причиной искажения геометрии координацион¬
ных соединений. Если число электронов на d-АО равно кратности вы¬
рождения (или удвоенной кратности), то облако d-электронной плот¬
ности симметрично по отношению к лигандам и не вызывает искаже¬
ния комплекса. Для октаэдрических соединений такое положение
реализуется в комплексах с конфигурацией центрального нона d°,
d3, d6, dG и d10. В других случаях неравномерное распределение элект¬
ронов по d-АО приводит к тому, что некоторые лиганды испытывают
большее, чем другие, отталкивание. Последствия неравномерного
заселения электронами орбиталей, расположенных между лиган¬
дами tig, гораздо меньше, чем в случае направленных к лигандам tу
орбиталей. В последней ситуации часто наблюдаются значительные
тетрагональные сжатия или растяжения комплексов.' Геометрическая
конфигурация является тогда следствием большей стабилизации си¬
стемы благодаря расщеплению ранее вырожденных состояний.
Этот эффект называется эффектом Яна — Теллера первого порядка.116
Рис. 6.10. Энергия гидратации некото¬
рых двухвалентных ионов
(X — экспериментальные значения)Для двух- и трехзарядных ио¬
нов с конфигурацией dr ЭСКП со-'
ставляет примерно 104,5 и 209
кДж/моль соответственно, т. е. она
соизмерима с энергией обычных хи¬
мических реакций. Поэтому вли¬
яние кристаллического поля может
накладываться на термодинамиче¬
ские проявления свойств комплек¬
сов: энергию гидратации, энергию
образования комплексов, энергию
кристаллических решеток. На рис. 6.10 представлены данные по
энергиям гидратации некоторых двухвалентных ионов, образующих
в растворах октаэдрические аквокомплексы типа [М(Н2О)012+. Из гра¬
фика видно, что теплоты гидратации этих ионов увеличиваются при
переходе от Са2+ к Zn2+. Это вызвано уменьшением радиусов ионов в
рассматриваемом ряду, что приводит к усилению электростатического
притяжения между ионом металла и диполем молекулы воды. Вслед¬
ствие образования более прочных связей выделяется и большее коли¬
чество энергии. Однако вместо ожидаемого монотонного возрастания
энергии гидратации наблюдается кривая с двумя экстремумами. Если
ввести поправки на ЭСКП для каждого из комплексов, то получен¬
ные значения АН лягут на плавную кривую, проходящую через точ¬
ки, где ЭСКП равна нулю, — ионы Са2+, Мп2+, Zn2+. Таким образом,
увеличение энергии гидратации этих ионов вызвано повышенным зна¬
чением ЭСКП.Эффектом кристаллического поля может быть вызвана и допол¬
нительная устойчивость координационных соединений в водных раст¬
ворах. Центральные ионы с большим значением ЭСКП приобретают
в результате комплексообразования повышенную устойчивость. При
этом максимальная дополнительная устойчивость должна наблюдать¬
ся для лигандов, создающих сильные поля. В водных растворах при
замещении молекул воды лигандом с большим значением параметра
расщепления Д »должна наблюдаться дополнительная стабилиза¬
ция.С позиции теории кристаллического поля хорошо объясняются
окислительно-восстановительные свойства некоторых комплексных
соединений.Так, известно, что в водном растворе нон Со3+ проявляет
очень сильные окислительные свойства. Стандартный окислительно¬
восстановительный потенциал системы Со3+ + е~ ^ Со2+ равен
+ 1,82 В. Это свидетельствует о тйм, что в водном растворе гидра¬
тированный ион кобальта (III) [Со(Н20)6|а+ неустойчив: он стремится
присоединить электрон и перейти в аква-ион кобальта (II) [Со(НаО)0]2+.
В присутствии аммиака или цианидных ионов, наоборот, более устой¬
чивыми являются комплексы, в которых кобальт находится в степени117
окисления Со3+. Значения окислительно-восстановительных потен¬
циалов соответствующих систем[Co(NII3)e]3+ + er fCo(NH3)e]J+; £° — + 0,1 В[Co(CN),]»-J-|-:е- « [Co(CN),]«-; £•>.= -0,83 Вуказывают на то, что' комплексные ионы [Co(NH3)e]2+ и [Co(CN)c]4~
обладают довольно сильными восстановительными свойствами, т. е.
они легко отдают электроны и переходят в ионы, где степень окисле¬
ния кобальта -! 3. Такое положение может быть объяснено тем, что
в сильном поле лигандов, которое создают цианид-ионы, седьмой d-
электрон Со2+ должен занимать высокоэнергетический ег-уровень.
Отрыв этого электрона является энергетически выгодным, так как
образующийся ион Со3+ с целиком заполненными /2в-орбиталямн
более стабилизирован (на 0,6Д) полем лигандов. Поскольку параметр
расщепления А цианидного комплекса больше, чем аммиачного, то
стремление перейти в комплекс Со(Ш) в первом случае больше, чем
во втором. Это и отражается в значениях потенциалов. Аммиачный
комплекс [Co(NH3)„]2+ менее сильный восстановитель, чем цианидный
[Co(CN)e]4_.Учитывать эффект кристаллического поля необходимо и при рас¬
смотрении реакционной способности комплексных соединений, так
как различия в энергиях активации могут определяться разницей
в ЭСКП. Например, при замещении лигандов в октаэдрических комп¬
лексах входящий лиганд должен подойти к центральному иону на
расстояние, сравнимое с расстоянием до него шести связанных ли¬
гандов. Если ион металла имеет сферически симметричное распреде¬
ление d-электронов, например db (парамагнитные комплексы Fe3+),
то замещение лиганда не требует больших энергетических затрат и
характеризуется быстрой реакцией. В комплексах же Сг3+ (d3) и низко¬
спиновых соединениях Со3+ (d6) распределение электронной плотностн
на d-АО неравномерно. Это мешает входящему лиганду образовывать
с металлом связи, сравнимые с исходными, и приводит к медленным
процессам замещения с большими затратами энергии на первоначаль¬
ную перестройку электронной конфигурации центрального иона.Естественно, эффект кристаллического поля — не единственный
фактор, влияющий на устойчивость той или иной формы соединений.
Важную роль в смещении равновесий в системах играет природа ли¬
гандов. Это особенно проявляется, когда лиганды образуют л-связи
с центральным ионом.Большое значение теория кристаллического поля имеет при рас¬
смотрении электронных спектров комплекс¬
ных соединений. На этой области применения
описываемой теории следует остановиться
особо.Электронные спектры. Электронные спект¬
ры комплексов в видимой и ультрафиолетовойРис. 6.11. Спектр поглощения раствора комплекса
[Ti(H20)e]3+118
областях обычно обусловлены переходам» d-электронов. Для систем
с несколькими d-электронами спектры усложнены изменениями
межэлектронного отталкивания. Поэтому обратимся снова к про¬
стейшему случаю —■ комплексу с конфигурацией центрального ио¬
на d1 — [Ti(H20)6I3+. Электронный спектр этого соединения имеет
в видимой области один минимум при длине волны 500 нм (рис.
6.11). Этот минимум приписывается d-электронному переходу с
орбитали t2g на орбиталь cg, энергия которого определяется пара¬
метром расщепления: ДЕ — Д. Появление плеча при 575 нм может
быть следствием наложения двух полос поглощения, обусловленных
некоторым расщеплением возбужденного ег-уровня, на который пере¬
ходит только один электрон, вызывая тем самым неравноценность
АО dxt_y2 и dzt (расщепление основного состояния t2g, как отмечалось
ранее, несущественно).Спектральные данные позволяют судить о разнице в энергии щели
между занятыми и свободными d-орбиталями в комплексах, т. е. дают
важную информацию о силе поля лигандов в различных координа¬
ционных соединениях. С другой стороны, интерпретация электронных
спектров комплексов является одной из важных химических задач.
Большинство экспериментальных значений Д соответствует 1—4 эВ.
Таким образом, длина волны поглощенного света при возбуждении
электрона, с одного d-уровня на другой часто лежит в видимой об¬
ласти, что обусловливает окраску многих комплексов (см. табл. 6.4):Таблица 6.4. Окраска акво-ионов [М(НгО)в]'1+, образовавшихся в
водных растворах сульфатов и перхлоратов переходных элементовЧисло
d- электроновЧисло неспарен¬
ных электроновИоны МЛ+Окраска раствора00К+, Са2+, Sc3+Бесцветный11Ti3+Розово-фиолетовый22Зеленый33Cr3+Фиолетовый44Сг2+Синий55Mn2+Бледно-розовый64Fe2+Зеленый73Co2+Розовый82Ni2*Зеленый91Cu2+Синий100Cu+ , Zn2+, Ga3+Бесцветныйкомплексы, содержащие Ni2+, часто зеленые, Си2+ — синие и т.д.
В то же время другие соединения (Sc3+, Zn2+) бесцветны. Чтобы мо¬
лекула могла поглотить излучение,оно должно иметь частоту v, удов¬
летворяющую условию ДЕ = hV. Различия в энергиях занятых и
свободных d-орбиталей в комплексах переходных металлов приводит
к тому, что они поглощают свет в разных частях видимой области.
Окраска соединений определяется пропускаемым светом (т. е. допол¬
нительным к поглощаемому). Причиной отсутствия окраски некото¬119
рых ионов является невозможность d—d-переходов: электроны на
d-АО либо отсутствуют, либо полностью их заселяют.Для комплекса (Ti(H20)e]3+ электронный переход, характеризую¬
щийся длиной волны около 500 нм, соответствует поглощению зеле¬
ной компоненты видимого света. Дополнительным к зеленому является
красный и синий. Сочетание этих цветов определяет пурпурную (ро¬
зово-фиолетовую) окраску комплекса.Теория кристаллического поля дает мощный аппарат для качест¬
венного, а иногда и количественного исследования свойств комплек¬
сных соединений. Однако один из ее существенных недостатков со¬
стоит в том, что она рассматривает лишь электрический характер
влияния лигандов на центральный ион. Поэтому с помощью теории
кристаллического поля можно решать только те вопросы, которые
связаны с изменением электронной конфигурации металла под воздей¬
ствием поля лигандов и нельзя касаться перераспределения электрон¬
ной плотности при комплексообразовании, которое обусловливает
прочность образующихся химических связей. Модифицированной
теорией, приспособленной к рассмотрению ковалентных связей б
координационных соединениях, явилась созданная в 50-х годах Ор-
гелом, Йоргенсеном и Бальхаузеном теория поля лигандов, объединив¬
шая в себе достижения теории кристаллического поля и метода моле¬
кулярных орбиталей.6.6. Метод молекулярных орбиталейВ теории молекулярных орбиталей предполагается, что движение
электронов описывается одноэлектронными волновыми функциями,
охватывающими все ядра системы, — молекулярными орбиталями.
В окрестности ядра атома молекулярная орбиталь (МО) предполагается
близкой к атомной орбитали (АО). Тогда одноэлектронные волновые
функции молекул ^ можно построить в виде линейной комбинации
(ЛК) АО фп входящих в них атомов (приближение МО ЛКАО):)М= 2 с. иsO,"7’”где фп — АО, a cin — искомые коэффициенты разложения, отражаю¬
щие участие данной АО в МО, т. е. определяющие конкретный вид
МО. В МО входят не любые линейные комбинации АО, а только те,
которые разрешены соответствующими правилами симметрии, су¬
щественно упрощающими построение МО, но
изложение которых выходит за пределы дан¬
ной книги.Для изучения электронной структуры и
свойств комплексных соединений метод МО
был впервые использован Ван Флеком. В слу¬
чае больших молекулярных систем — комп-Рис. 6.12. Выбор системы координат для октаэдри*
ческого комплекса [MLe]n+120
лексов переходных металлов — обычно используется валентное приб¬
лижение, согласно которому внутренние электроны атомов полагают
локализованными вблизи отдельных ядер, а в образовании химичес¬
ких связей считают существенными взаимодействия только валентных
АО металла и лигандов.а-Молекулярные орбитали в комплексных соединениях. Рас¬
смотрим структуру МО октаэдрического комплекса [МЬ6]Л+ (рис. 6.12),
для простоты первоначально предположив, что лиганды могут обра¬
зовывать только а-связи, т. е. что на образование химических связей
в комплексном соединении идет какая-либо гибридная орбиталь ли¬
ганда a-типа, соответствующая его неподеленной электронной паре.В этом случае МО системы можно построить следующим образом.
о-Связи в октаэдрическом комплексе должны быть симметричны от¬
носительно линии, соединяющей лиганд с центральным атомом, т. е.
соответствующие волновые функции (МО) не должны менять свой знак712]
при вращении вокруг связи металл—лиганд. Поэтому они могут быть
образованы только s, рх, ру, рг, dxi__yi н d2>-валентными АО централь¬
ного атома. Линейная комбинация а-орбиталей лигандов (групповая
орбиталь ГО) должна иметь те же свойства симметрии, что и рассмат¬
риваемые АО металла.В соответствии с этим s-A(i металла, имеющая одинаковый знак
во всех направлениях, может комбинироваться с групповой орби¬
талью лигандов, указанной на рис. 6.13, а, образуя МОФ1 (°.s) = -f- Сг (а, -1- а2 + о3 -f о4 + а6 + о,) ,обычно обозначаемую как орбиталь alg. При нахождении остальных
МО необходимо учитывать знаки р- и d-АО центрального атома. рх-АО
меняет знак при переходе через начало координат вдоль оси х.Такими же свойствами характеризуется и линейная комбинация
о-орбиталей лигандов (cFj—о3) (рис. 6.13,6), т. е. соответствующая
МО имеет видЬ Ы = C3tPje + С4 (з, — 03).Аналогично'Ь (av) = c3'fn Ч- (a2 — а4);У'h (а*-) = сзтр_ + Q (а5 — а6)(рис. 6.13, в, г).АО с/х»_у, может комбинироваться с ГО (<у1—о2 -г ст3—а*)
(рис. 6.13, д), обладающей теми же свойствами симметрии по отноше¬
нию к вращению вокруг связей лигандов с центральным атомом, т. е.V5 ( ^jt*— V*j) = Съ^Х2—уг "Ь Сб(31 °j) •АО dzг центрального атома, так же как и его s-AO, дает a-связи со
всеми шестью a-орбиталями лигандов. Однако по требованиям сим¬
метрии их линейная комбинация будет иной (рис. 6.13, е):( 0Z«) = cbd * + Ce (s6 -f- а, — ax — о2 — о3 — а4).Из теории кристаллического поля следует, что в октаэдре рх-,
ру-, Pz-AO и dxi-yt, dzг-АО центрального атома имеют одинаковые
энергии, т. е. с точки зрения симметрии принадлежат к вырожденному
уровню. Это означает, что они имеют один тип симметрии. То же са¬
мое относится и к МО — '^4 и (^5, ф6, т. е. указанные МО в окта¬
эдрическом комплексе будут иметь одну и ту же энергию. Обозначают
эти МО tlu и eg соответственно.Три валентные орбитали dxz, dyz, dxy, которые могут образовывать
я-связи с лигандами, в рассматриваемом a-приближении являются
несвязывающими (МО ф7—<^9) и имеют одинаковую энергию — ор¬
битали типа t2g-Диаграмма уровней МО октаэдрического комплекса [ML6]n+ пред¬
ставлена на рис. 6.14. Энергия уровней АО газообразного нона ме¬
талла, как правило, возрастает в ряду End < £(„+1} s < Е(п+,, р. Орбн-122
Рис. 6.14. Диаграмма уровней
МО октаэдрического комплекса F->[A\Lfi]'7+, где L—гт-донорные ли¬
ганды (звездочкой отмечены раз¬
рыхляющие МО)тали свободных лигандов,
обычно более электроотрица¬
тельные, чем АО металла,
располагаются на энергетиче¬
ской шкале ниже.Относительные энергии
трех нижних уровней МО
можно получить на основании
расчетов электронной струк¬
туры соединений или исполь¬
зуя спектроскопические дан¬
ные. Все эти МО будут засе¬
лены электронами, тогда как
далее количество занятых МО
будет зависеть от электрон¬
ной конфигурации централь¬
ного атома. Так, если мы рас¬
сматриваем комплексный ион
|Ti(H20)e]3+ с конфигурацией
Ti3+ [АгШ1, то наличие одного электрона на З^-валентной оболочке
приводит только к частичному заполнению трижды вырожденного уров¬
ня МО i2g, соответствующего несвязывающим dx -АО металла. Энергети¬
ческая разница между верхней занятой МО комплекса и нижней сво¬
бодной МО (например t2g, и еg в случае гексааквокомплекса титана)
соответствует параметру теории кристаллического поля Д, а электрон¬
ные d—d-переходы с указанных МО описывают появляющиеся полосы
в спектрах поглощения комплексных соединений. Если же происхо¬
дит переход электрона с МО, состоящей преимущественно нз АО ме¬
талла, на МО в основном лигандного характера или наоборот, в зави¬
симости от электронной конфигурации комплекса, то в электронном
спектре будут проявляться высокоинтенсивные электронные пере¬
ходы с переносом заряда.я-Молекулярные орбитали в комплексных соединениях. Для об¬
разования зт-связеп в комплексных соединениях, как уже отмечалось,
пригодны dxy-, dxz-, dyj-орбитали центрального атома. В соответст¬
вии с требованиями симметрии, применявшимися ранее для ст-МО,
АО dxy комбинируются с ГО, состоящей из я-орбиталей лигандов
(рис. 6.15, а), образуя МО:?10 {хху) = с:^ху '!' с8 ( Ру1 "Ь PXt Ру, Р х< )•Аналогично (рис. 6.15, б, в)Чи (-.гг) = Ci<lxz -|- £•„( pZi -I- pXi - pZi - pXiy,V. (*,*) = Cjdy. + c8 ( pZ: + pyt - pZi - Pyt).АО металла МО комплекса/123
%\s>Puc. 6.15. Схематичное представление я-ЛЮ в октаэдрическом
комплексе [ML6]n+1Эти МО имеют одинаковую энергию в октаэдрическом комплексе и
называются ^-молекулярными орбиталями.Слабые связи л.-типа могут образовывать и р-кО металла. Соот¬
ветствующие МО (/1и-типа) будут иметь^вид (рис. 6.15, г—е):Ъз = Wx + с10 (РХг + Ри-.+ Рх,-+ Рх,)\+14 = с»Ру + с,« (рл + Pyti+ PUt + Ру,У,+1» = c>Pz + Сю (PZl + PZl’+ p\,;+ P2t).Каждый лйтанд может образовывать л.-связи с металлом посред¬
ством двух рж -АО; таким образом, общее число групповых л-орбита-
лей должно быть равно 12. Оставшиеся шесть из них не имеют по сим¬
метрии аналогов среди АО центрального атома, в результате чего
остаются несвязывающими (МО tig и /2ц).я-Связи лигандов с центральным атомом могут образовываться
также посредством низколежащих я*-орбиталей лигандов С учетом
этих возможных взаимодействий диаграмма уровней МО октаэдриче¬
ского комплекса представлена на рис. 6.16.Для большинства правильных геометрических конфигураций про¬
ведено построение МО в соответствии с требованиями теории симмет¬
рии. Разработаны также и вероятные схемы уровней МО многих ко¬
ординационных соединений. В случае неопределенного порядка рас¬
положения отдельных уровней МО иногда пользуются данными по
молекулярным потенциалам Ионизации систем, значения которых в
соответствии с теоремой Купманса должны быть приближенно равны
энергиям соответствующих МО, взятым с обратным знаком.Коэффициенты cin, указывающие, какой вклад данная АО вносит124
Рис. 6.16. Диаграмма уровней г
МО октаэдрического комплекса '
[MLuln+ с учетом возможных л-
взаимодействий металл — лигандв рассматриваемую МО, на¬
ходят на основе вариацион¬
ного принципа. С помощью
этих коэффициентов можно
построить карты распределе¬
ния электронной плотности в
системах, представляющие
большой интерес при рассмот¬
рении координационных со¬
единений.В настоящее время суще¬
ствует много методов расчета
электронной структуры со¬
единений, основанных на схе¬
ме МО ЛКАО, отличающихся
друг от друга точностью и
адекватностью результатов по
отношению к эксперименталь¬
ным данным. Каждый из этих методов может вполне удовлетворитель¬
но описывать одни характеристики молекулярных систем и быть не¬
пригодным для других. Поэтому при рассмотрении результатов рас¬
четов электронной структуры соединений методом МО ЛКАО необ¬
ходимо учитывать возможности использованного расчетного метода
и границы его применимости.Определение структуры уровней МО в координационных соедине¬
ниях часто бывает полезным и на качественном уровне. Особый интерес
представляет выяснение характера высшей занятой МО (ВЗМО) сис¬
темы и низшей свободной (НСМО), так как именно эти орбитали часто
обусловливают устойчивость определенных геометрических конфигу¬
раций, являются ответственными за протекание тех или иных хими¬
ческих процессов, определяют спектры и другие физические свойства
соединений.Теория МО смогла выработать свой язык, широко используемый и
пригодный для описания химических структур различной степени
сложности. Использование понятия молекулярных орбиталей помога¬
ет интерпретировать результаты физико-химических исследований
соединений. Расчеты по методу МО менее трудоемки, чем по методу
ВС. Простой метод МО применим как для ковалентных, так и для
ионных систем, в то время как простой метод ВС приспособлен для
описания ковалентных молекул. Вместе с тем традиционный метод
МО хуже описывает энергии связей. Однако если улучшить метод МО
с учетом наложения различных конфигураций, а в метод ВС ввести
ионные состояния, то оба метода дадут эквивалентные результаты./, у (о *• П •)✓ . • . Nalv(o )(п + ] )рJ\1'А Ч/«(К'у(\и( **)•V—:-г'к
1\ /InU 1 \ ‘У' / ' , /+ !.)j /'w c,(oVWx-’-,V.:/ \\ / ( /г-71 ; WК<гл \
\\\\ \АО металла МО комплекса ГО лигандов125
6.7.Жесткие и мягкие кислоты и основания6.7.1. Понятие о поляризации и поляризуемости
ионов и молекулВ электрическом поле ион или молекула деформируются, т. е. в них
происходит смещение ядер и электронов относительно друг друга.
Такая деформируемость ионов и молекул называется поляризуемостью.
Поскольку наименее прочно в атоме связаны электроны внешнего слоя,
они испытывают смещение в первую очередь.При одинаковом радиусе и заряде наибольшей поляризуемостью
обладают ионы, имеющие внешнюю 18-электронную оболочку. Мень¬
шей поляризуемостью обладают ионы с незаконченной 18-электронной
оболочкой и еще меньшей — ионы с 8-электроннои структурой, ха¬
рактерной для атомов благородных газов.При одинаковой структуре электронных оболочек поляризуемость
иона уменьшается по мере увеличения положительного заряда, на¬
пример, в рядуF- > Ne > Na+ > Mg:+ > Als+ > Si4+Для ионов электронных аналогов поляризуемость увеличивается с
ростом числа электронных слоев, например:Li+ < Na+ < К+ < Rb+ < Cs+F-<Cl-<Br-<I-Поляризуемость молекул и сложных ионов определяется поляри¬
зуемостью входящих в них атомов, геометрической конфигурацией,
количеством и кратностью связей и т. п. Поэтому априорный вывод
по поляризуемости молекул можно с успехом делать только для ана¬
логично построенных молекул, отличающихся одним из атомов. В этом
случае о различии в поляризуемости молекул можно судить по разли¬
чию в поляризуемости именно этих атомов.Электрическое поле может быть создано как заряженным электро¬
дом, так и ионом. Таким образом, ион сам может оказывать поляри¬
зующее действие на другие ионы или молекулы. Поляризующее дей¬
ствие нона возрастает с увеличением заряда и уменьшением радиуса.
При одинаковом заряде и радиусе поляризующее действие усилива¬
ется при переходе от катионов с электронной оболочкой атомов бла¬
городных газов к ионам с незаконченной 18-электронной оболочкой.
Наибольшим поляризующим действием обладают ионы с- законченной
18-электронной структурой.Поляризующее действие анионов, как правило, значительно мень¬
ше, чем поляризующее действие катионов. Это объясняется большими
размерами анионов по сравнению с катионами и меньшим локальным
зарядом.Молекулы обладают поляризующим действием в том случае, если
они полярны; поляризующее действие тем выше, чем больше диполь¬
ный момент молекулы.126
■«"Х-’Ч -промежуточные элементxvyvj -элементы класса „а* : ^У/У | -элементы класса „б";Рис. 6.17. Ионы — акцепторы электронов6.7.2. Классификация льюисовских кислот по Арланду,
Чатту, ДэвисуВ 1958 г. Арланд, Чатт и Дэвис [2] разделили все ионы — акцепторы
электронов на два класса. Ионами—акцепторами электронов прежде
всего являются ионы металлов. К классу «а» они отнесли ионы, об¬
разующие наиболее стабильные комплексы с лигандами, донорный
атом которых относится ко второму периоду системы Д. И. Менделее¬
ва, например, N, О, F. К классу «б» отнесены ионы, образующие наи¬
более стабильные комплексы с лигандами, донорный атом которых
относится к третьему и следующим периодам, например Р, S, Se, Cl,
Br, I.Склонность ионов металлов к комплексообразованию зависит от
их состояния окисления. Свою классификацию С. Арланд, Дж. Чатт
и Н. Р. Дэвис строили по отношению к ионам с наиболее распростра¬
ненными состояниями окисления. На рис. 6.17 показано положение
в периодической системе элементов, ионы которых относятся к клас¬
сам «а» и «б». Авторы классификации обращают внимание, что ионы
класса «б» занимают треугольник, располагающийся в середине пе¬
риодической системы.Способность донорных атомов образовывать координационные
связи изменяется в следующем порядке:V группа
VII группаVI группаN»P>As>Sb>Bi для ионов класса «а»
N Р > As > Sb > Bi для ионов класса «б»F» Cl > Br > I
F <хС1 < Вг < 1
О» S > Se > Те
0«S sk Se да Тедля ионов класса «а»для ионов класса «б»для ионов класса «а»для нонов класса «б»Для ионов класса «б» способность к образованию координацион¬
ной связи с атомом кислорода намного меньше, чем с атомом серы.
Однако различия в способности к образованию связей с S, Se и Те
почти нет.127
6.7.3. Принцип жестких и мягких кислот и основанийПирсонаВ 1963 г. Пирсон [3] ввел новую терминологию в классификацию ио¬
нов металлов по Арланду—Чатту—Дэвису и сформулировал пра¬
вило, которое дало возможность связать между собой различные яв¬
ления в координационной и органической химии.ИоньУ металлов класса «а» Пирсон назвал жесткими кислотами.
Это ионы, характеризующиеся высоким положительным зарядом, ма¬
лым размером, малой поляризуемостью и не имеющие легко возбуж¬
даемых внешних электронов. Ионы металла класса «б» он назвал мяг¬
кими кислотами. Это ионы с низким положительным зарядом, боль¬
шим размером, высокой поляризуемостью. Такие ионы имеют не¬
сколько легко возбуждаемых внешних электронов (см. табл. 6.5).Таблица 6.5. Классификация кислот по ПирсонуЖесткис кислотыМягкие кислотыН+, Li+ , Na+ , к+Cu+, Ag+ , Au+, ТГ, Hg4-Ве-+, Mg2*, Са2+, Sr2+, Мп2+Pd3+, Со2+ , Pt2\ Hg2+А13+, Sc3+. Ga3+, In3+, La3+, Nd3+.CH3Hg+, Co(CN)r-, Pt4+, Te4+Се3+ , Gd3+ , Lu3+, Cr3+, Co3+, Fe3+,As3+, CH3Sn3+Tl3+ , T1(CH„)3 . BH3 , Ga(CH3)3,Si4+, Ti4+ . Zr4+, Tli4+. U4+ , Pu4+,GaCls, Gal3, InCI3Ce4+, HI4+, W04+, Sn4+R+, RSe+, RTe+uof+, (CH3hSn=\ vo*\ mo3+I+, Br+, HO+,RO+Be(CH3)2. BF3 , B(OR)3 , A1(CH3)3,Io, Br2, ICNAlClg, AIH3Тринитробензол и др.RPO^, ROPO^, ROSO^, S03Хлоранил, хиноны и др.,
(CN)2C=C(CN),I7+, I5+, Cl7+, Cre+О, С1, Вг, I, N, RO-, R03RCO+, CO;, NC+Атомы металлов. Металлы в пассеHX (молекулы, образующие водородные
связи)СН2, карбеныКислоты, занимающие промежуточное положение:Fe2+, Ni2+, Cu2+, In-*, Pb2+, Sn2+, Sb3+, Bi3+, Rh3+, Ir3+, B(CH3)3, SO;,
NO+, Ru2+, Os=+, R3C+, C6H+, GaH3Мягкими основаниями были названы лиганды, донорные атомы
которых обладают высокой поляризуемостью, низкой электроотрица¬
тельностью и легко окисляются. Жесткими основаниями были названы
лиганды, донорные атомы которых обладают низкой поляризуемостью,
высокой электроотрицательностью и трудно окисляются (см. табл. 6.6).Правило Пирсона формулируется так: жесткие кислоты предпо¬
читают связываться с жесткими основаниями, а мягкие кислоты —
с мягкими основаниями.128
Таблица 6.6. Классификация оснований по ПирсонуЖесткие основанияМягкие основанияНоО,ои-F", СН3СОО-, POj-.R.S, RSH, RS", I-, SCN-, Se0§“,SO^-. CI-СО?-, С. Ю^, NOf, ROH ,R3P, R3As, (RO)3P, CN-, RNC, СО,RO-,r2o.NH3, RNHo, N2H4C2H4. CGH0, Н-, R-Основания, занимающиепромежуточное положение:CcH6NH,, C6H5N, N7, Вг-, N07. 5,0 , N,Таблица 6.7. Классификационные признаки жестких
и мягких кислотКислота (акцептор электронов)Характеристикажесткаямягка яПоляризуемостьНизкаяВысокаяЭл ектрополож ител ь н остьВысокаяНизкаяПоложительный заряд, или состояние
окисленияБольшойМалыйРазмер ионов (атомов)МалыйБол ьшойОбычные типы связи с основаниемИонная, элек¬КовалентнаяЭлектроны на внешних орбиталяхтростатическая
Таких электро¬Несколько элек¬электронодонорных атомовнов мало, возбуж¬тронов, возбужда¬даются с трудомются легкоТаблица 6.8. Классификационные признаки жестких
и мягких основанийОснование (донор электронов)ХарактеристикажесткоемягкоеПоляризуемость
Электроотрнцательность
Отрицательный заряд
Размер ионов (атомов)Обычные типы связи с кислотойВакантные орбитали электронодонор¬
ных атомовНизкаяВысокаяБольшойМалыйИонная, элек¬
тростатическая
Высоколежащне,
практически недос¬
тупныеВысокаяНизкаяМалыйБольшойКовалентнаяНизколежащне,
доступные5-817129
Правило Пирсона следует рассматривать как удобный принцип
для качественной оценки относительной прочности комплексных со¬
единений, имеющих один и тот же центральный атом и различные ли¬
ганды, н, наоборот, комплексных соединений, имеющих одинаковые
лиганды, но различные центральные атомы.В связи с этим целесообразно привести сводку классификацион¬
ных признаков жестких и мягких кислот и оснований, впервые данную
Уильямсом в 1975 г. (табл. 6.7 и 6.8). В этой классификации мера
жесткости или мягкости реагента определяется лишь его химическим
поведением, причины которого не разъясняются. Клопманом [4] раз¬
вит теоретический подход к этой проблеме, который, по мнению ав¬
тора, может служить теоретическим базисом принципа Пирсона.
Согласно этому подходу взаимодействие жесткое основание — жест¬
кая кислота обусловлено главным образом благоприятным электро¬
статическим взаимодействием между донором с высокой орбитальной
электроотрнцательностыо и акцептором с низкой орбитальной элект¬
роотрицательностью. С другой стороны, взаимодействие между мяг¬
кими кислотами и мягкими основаниями является результатом ко¬
валентной координации донора с низкой орбитальной электроотри¬
цательностью и акцептора с высокой.В реакциях первого типа донор с высокой электроотрицатель¬
ностью, т. е. с низколежащими занятыми орбиталями, не имеет тен¬
денции отдавать свои электроны. Примерами таких доноров являются
F", ОН", вода, амины. Акцептор же с низкой электроотрицатель¬
ностью имеет малое сродство к электрону. Примерами подобных ак¬
цепторов являются ионы Al3+, Mg2+, протоны. В этом случае сущест¬
вует достаточно большая разница в энергиях граничных МО реаген¬
тов и реакция пойдет между атомами, несущими наибольшие противо¬
положные плотности зарядов.В реакциях второго типа (мягкое основание — мягкая кислота)
доноры с относительно низкой электроотрицательностью имеют высоко-
лежащне занятые орбитали (I", RS", Н~), а акцепторы с относительно
высоким сродством к электрону — низколежащие свободные орби¬
тали (Hg2+, Ag+, Pt2+). Реакция будет протекать между центрами
с наивысшей плотностью заряда на этих граничных орбиталях.Примером реагентов, вступающих в тот или иной тип реакции,
может служить амбидентатный роданидный ион SCN”. Расчет элек¬
тронной структуры по методу Хюккеля показал, что в ионе SCN'
электронная плотность на атоме серы значительно меньше, чем на
атоме азота:—0.79 +0.09 —о.зоN — С — SВысшая занятая МО роданидного иона состоит преимущественно из
АО серы. Именно эти два обстоятельства, согласно рассматриваемому
подходу, являются определяющими в реакционной способности рода¬
нид-иона. В том случае, когда разница в энергии уровней МО реаген¬
тов велика, взаимодействуют атомы, несущие наибольшие противо¬
положные заряды, т. е. происходит координация к металлу этого130
лиганда атомом азота. Если же МО реагентов энергетически близки,
реакция будет протекать между двумя центрами, обладающими наи¬
высшей электронной плотностью на граничных МО, т. е. катион ме¬
талла будет иметь тенденцию координироваться с атомом серы.Орбитальные электроотрицательности рассчитаны из потенциалов
ионизации, сродства к электрону и эффективных ионных радиусов ре¬
агирующих атомов. Полученные величины для серии доноров и акцеп¬
торов с учетом энергии сольватации (табл. 6.9) хорошо согласуются
с классификацией в терминах жесткости и мягкости, эмпирически
найденной Пирсоном на основании их химического поведения.Таблица 6.9. Орбитальная электроотрицательность (эВ)
катионов и анионов в газовой фазе и в водеКатионВ газовой
фазеВ подеАммонВ газовой
фазеВ водеЛ13+26,046,01 tF-6,69-12,га 1La3+17,244,51 [Н„015,80— 10,73 =Ti4 +39,464,35 щон-5,38—10,45 £Ве2+15,983,75 Sа-6,02—9,94 Л,Mg2+13,182,42 5Вг-5,58—9,22 жСа2+10,432.33 £CN-6,05—8,78 £Fe3+26,972,22 •'SH"4,73—8,59 £Sr-+9,692,21I-5,02—8,31 5
—7,37 ТOr3*27,332,06н-3,96Вя2+8,801 ,89Ga3+28,151,45 оCr-+13,080,91 3Fe2+14,110,69 gAln:+13,590,66 £Oo2+14,470,52 XLi +4,250,49 £H+10,380,42 §.Ni2+15,000,29 СNa+3,970Cu'-,+15,44—0,55Zn2+15,82— 1,02T| +5,08—1,88 аCd2+14,93—2,04 £Cu+6,29—2,30 ^Ag+6,23—2,82 <Tl3+27,45—3,37 IAu+7,59—4,35 1Hg2+16,67—4,64 \6.8. Концепция эффективного атомного номераВ некоторых случаях для определения состава химических соединений
оказывается полезной концепция эффективного атомного номера
(ЭАН). При образовании комплексных соединений центральные ионы
принимают от лигандов электронные пары. Количество этих элек¬
тронных пар определяется координационным числом. Сиджвик обратил
внимание на то, что в некоторых случаях в комплексах число электро¬5 *131
нов центрального нона равно числу электронов в атомах ближайших
благородных газов. Например, в комплексном ионе |Co(i\H3)B|3+
нон кобальта (III) содержит 24 электрона и от шести лигандов до¬
полнительно принимает 12 электронов. Таким образом, общее числа
электронов, которое окружает ядро кобальта, равно 36. Тркое число
электронов имеет атом ближайшего к кобальту благородного газа—
криптона. Это число электронов Спджвик назвал эффективным атом¬
ным номером (ЭАН).Концепция ЭАН охватывает комплексы не очень большого числа
металлов. Она сравнительно хорошо применима к карбонильным и
ннтрозильным соединениям d-элементов. Использование этой кон¬
цепции позволяет предсказывать их состав, а иногда н строение.Степени окисления металлов в карбонильных и нитрозильных
комплексах часто равны нулю. Определим составы карбонилов желе¬
за и никеля с позиции представления ЭАН. Каждая молекула СО
предоставляет металлу два электрона. Эффективный атомный номер
как железа, так и никеля в соединениях должен быть равен 36. По¬
рядковый номер никеля равен 28. До достижения ЭАН атому никеля
не хватает 36 — 28 = 8 электронов. Такое число электронов может
быть получено атомом никеля от четырех молекул СО. Таким образом,
устойчивым карбонилом никеля должен быть Ni(CO)4.Порядковый номер железа 26 и, следовательно, атом железа дол¬
жен присоединить пять молекул оксида углерода 26-г 2-5 = 36.
Действительно, устойчивым карбонилом железа является Fe(CO)5.Карбонилы хрома, молибдена и вольфрама имеют состав Сг(СО)5,
Мо(СО),-, и W(CO)c, что также соответствует эффективным атомным
номерам 36, 54 и 86.Считается, что молекула ЛО предоставляет в распоряжение цент¬
рального атома три электрона. С этой точки зрения понятно сущест¬
вование смешанного нптрозилкарбонильного комплекса кобальта
CoNO(CO)3. В этом соединении общее число электронов равно 27 —
-f- 3+ 2-3 = 36, что соответствует ЭАН.В соединениях, содержащих анноны, например Мп(СО)5Вг, число
электронов можно подсчитать двумя способами.1. Марганец находится в степени окисления 1 + , бромидный нон
предоставляет ему два электрона. Тогда число электронов равно 24 т
+ 2-5 -f 2 = 36.2. При подсчете числа электронов можно рассматривать марганец
и бром как бы находящимися в атомарном состоянии. В этом случае
бром для образования химической связи с марганцем предоставляет
один электрон. Суммарное число электронов, конечно, не изменится:
25 4- 2-5 + 1 = 36.Существует чисто карбонильное соединение марганца. Попробуем
представить его состав. Порядковый номер марганца равен 25. До
эффективного атомного номера марганцу не хватает 11 электронов.
Десять электронов могут быть получены от пяти молекул СО. В ре¬
зультате в молекуле Мп(СО)5 до ЭАН, равного 36, марганцу недостает
одного электрона. Для достижения ЭАН две такие группы объеди¬
няются в молекулу с образованием связи Мп—Мп. В результате обоб¬132
ществления электронов от двух атомов марганца будет достиг¬
нут ЭАН. Действительно, карбонил марганца существует в виде
(СО) 5Мп—Л\п (СО) 8.В последние годы многие химики все чаще пишут о «правиле 18
электронов». По существу, это то же самое правило эффективного
атомного номера Сиджвика. По правилу 18 электронов лишь облегча¬
ется подсчет электронов и определение координационного числа. Это
правило гласит, что в комплексах переходных металлов часто общее
число валентных электронов на (п—1 )d-, ns- и пр-орбиталях централь¬
ного атома равно восемнадцати.Действительно, в комплексах lNi(CO)4], [Pd(PPh3)4], [Pt(PPh3)4]
центральные атомы имеют с(1о-электронпые конфигурации. Четыре
лиганда дают восемь электронов. В сумме у каждого комплекса в ва¬
лентном слое имеется по восемнадцать электронов.Комплексы [Co(NH3)6]3+, [RhCl6]3-, [Ir(NH3)6ClP+, [Pt(NH3)0]4+
также отвечают правилу 18 электронов. Электронные конфигурации
центральных атомов в этих комплексах de. Шесть лигандов в каждом
случае дают 12 электронов. В сумме получается 18 электронов.Нетрудно видеть, что такие характерные комплексы, как [PdClJ2-,
[Iг(СО)(РРh3)^С1 ], [Pt(NH3)4]2+, не отвечают правилу 18 электронов.
Естественно, они не отвечают и правилу ЭАН. Электронными конфи¬
гурациями центральных атомов в этих комплексах являются da. Че¬
тыре лиганда дают еще 8 электронов. В сумме в валентном слое полу¬
чается 16 электронов. Для того чтобы комплексы Pd(II), Iг(I), Pt(II)
соответствовали правилу 18 электронов (или правилу ЭАН), необ¬
ходимо окружение центрального атома 5 лигандами.Концепция ЭАН (правило 18 электронов) хотя и носит формаль¬
ный характер, однако в некоторых случаях она имеет предсказа¬
тельную силу. Пока не создана общехимическая теория строения со¬
единений, по-видимому, не следует пренебрегать любыми представ¬
лениями, способными обобщать и предсказывать хотя бы ограничен¬
ный круг фактов. К таким представлениям относится концепция
эффективного атомного номера или ее видоизменение — правило 18
электронов.
ГЛАВАЛИГАНДЫ КООРДИНАЦИОННЫХ СОЕДИНЕНИЙСуществует много разнообразных лигандов, сильно отличающихся
по свойствам и строению. Они относятся к различным классам хими¬
ческих соединений. Иногда весьма трудно найти что-либо общее меж¬
ду отдельными лигандами, но, несмотря на это, имеется принцип их
деления на два класса.Лиганды первого класса имеют электронодонорные
атомы высокой электроотрицательности. На внешнем электронном
уровне этих атомов находится неподеленная электронная пара, но
отсутствуют вакантные орбитали, способные принимать электронную
плотность. Такие лиганды являются только ст-донорами. Некоторые
из них образуют комплексы почти со всеми ионами металлов. Химиче¬
ские связи этих лигандов с ионами металлов имеют значительную
ионную составляющую. Стабильность связи увеличивается с ростом
ионного потенциала центрального иона, т. е. отношения заряда иона
металла к его радиусу. К числу характерных лигандов такого типа
относятся аммиак, вода, спирты, фторид- и хлорид-ноны.Лиганды второго класса имеют донорны? атомы низ¬
кой электроотрицательности. На внешней электронной оболочке на¬
ряду с электронной парой имеются вакантные орбитали. Такие ли¬
ганды одновременно являются a-донорами и л-акцепторами. Они об¬
разуют с переходными металлами преимущественно координационные
связи. Наиболее типичными представителями этого типа лигандов яв¬
ляются оксид углерода (II) и оксид азота (И), изоцианиды, цианид-
ный ион.Некоторые лиганды наряду с неподеленной электронной парой,
обеспечивающей образование a-связи, имеют л-электроны, которые
могут участвовать в образовании л-связей, например ОН", NH3", F'.
Таким образом, данные лиганды являются о- и я-донорами электро¬
нов.В особый класс можно выделить лиганды, не имеющие свободной
электронной пары, но имеющие л-электронную систему, например
этилен, бензол, циклопентадиеннл-ион. Такие лиганды способны от¬
давать электроны центральному атому со связывающих молекуляр¬
ных орбиталей и принимать электроны от центрального атома на
разрыхляющие молекулярные орбитали.Конечно, такое деление весьма условно. Существует много ли¬
гандов, которые невозможно однозначно отнести к тому или иному
классу. Тем не менее эта классификация помогает ориентироваться
в выборе лигандов и предсказании некоторых свойств комплексов.134
7.1.Молекула воды и гидроксильный ионНесмотря на то что в настоящее время специалисты по теории раство¬
ров все больше внимания уделяют неводным растворителям, водные
растворы продолжают играть очень важную роль в химии и химиче¬
ской технологии. Известно, что научные основы современных теории
растворов были заложены Д. И. Менделеевым. Одним из наиболее
важных явлений процесса растворения является сольватация (или,
для водных растворов, гидратация). Представления о сольватации
были введены в науку и развиты Каблуковым и Кистяковскпм в Рос¬
сии, а за рубежом — Фпцпатрпком и Чамичаном.Боркис ввел понятие о первичной и вторичной сольватации. Пер¬
вичная сольватация приводит к образованию сольватной сферы, вклю¬
чающей молекулы растворителя, находящиеся в непосредственном
контакте с ионом. Молекулы первичной сольватной сферы лишены
поступательных степеней свободы и совершают лишь броуновское
движение вместе с ионом как единое целое. В результате вторичной
сольватации образуется вторичная сольватная сфера. Она включает
молекулы растворителя, не принимающие участия в первичной соль¬
ватации, т. е. не вступающие в непосредственный контакт с ионом.
Однако электростатическое взаимодействие молекул растворителя,
входящих во вторичную сольватную сферу, достаточно сильное. Благо¬
даря ему молекулы вторичной сольватной сферы довольно сильно от¬
личаются от молекул растворителя, не участвующих в процессах
сольватации.О. Я. Самойлов также разделяет сольватацию на две составляю¬
щие, но использует для них термины «ближняя» и «дальняя» сольва¬
тация. Он рассматривает сольватацию как влияние ионов на транс¬
ляционные движения ближайших к ним молекул растворителя.Таким образом, в настоящее время имеются два подхода к истол¬
кованию явления сольватации. Один из них основан на преимущест¬
венном учете взаимодействия ион—растворитель. Второй учитывает
преимущественно взаимодействие растворитель—растворитель. Пос¬
ледний связан главным образом с представлениями о кинетической
устойчивости агрегата ион — молекулы растворителя.Для координационной химии наиболее интересным является пер¬
вый подход. Он основан на том, что при сольватации ионы прочно
удерживают определенное число молекул растворителя (первичная
сольватная сфера). Эти числа называют сольватными. Совершенно
очевидно, что в терминологии химии координационных соединений
сольватное число равнозначно координационному числу. Более того,
в настоящее время в теории растворов получил] широкое распростра¬
нение' термин координационная сольватация (гидратация).Таким образом, явления- сольватации) и комплексообразования
ионов металлов с молекулами; растворителя рассматриваются в не¬
разрывной связи. Нужно лишь помнить, что сольватация — более
сложное явление, чем комплексообразование. Комплексообразование
ионов металлов с молекулами растворителя адекватно первичной соль¬
ватации. Следовательно, сольватированные ионы металловтвграство-135
pax являются комплексными соединениями, в которых в качестве
лигандов выступают молекулы растворителя. В водных растворах
это аквокомплексы.Вопрос о числе ближайших молекул растворителя, связанных с
ионом металла, имеет важное значение как для науки о растворах,
так и для химии координационных соединений. Однако их определе¬
ние наталкивается на значительные трудности. В зависимости от вы¬
бранного метода исследования получающиеся сольватные числа силь¬
но отличались. Это и понятно, так как большинство методов не дает
возможности надежно разделить первичную и вторичную сольва¬
тацию.С. И. Дракнн выдвинул предположение, что аквокомплексы в
кристаллогидратах имеют те же структурные группы ион—вода, что
и в водных растворах. Это предположение вытекало из обобщения
большого фактического материала. ■ Так, было установлено, что ок¬
ружение ионов металла молекулами воды в богатых водой кристалло¬
гидратах определяется не столько требованиями плотнейшей упаков¬
ки частиц в кристалле, меняющимся в зависимости от размеров и
конфигурации анионов, сколько особенностями взаимодействия ион-
вода. На основе систематизации данных о структуре кристаллогид¬
ратов, полученных в результате рентгенографического и нейтроно¬
графического исследований, С. И. Дракин предложил координаци¬
онные числа для аквокомплексов ионов различных металлов в вод¬
ном растворе (табл. 7.1).Таблица 7.1. Координационные числа аквокомплексовионов металловИэн■I Ion■ИонnLi+5Cd-+7Pr3+9Na+6Xj8 +fiNd3+9К+7Sn=+f.Sm3+9Rb +8Pb"-+8Eu:)+9Cs+8Mn-+6Gd3+9Ве2+4Fe2+fiTb3+9Mg2+6Co-+6Dv3+9Са'2+JNj2 +fiHo3+9Sr2+8V-+6Er3+9Ва2+8Ga3+6Tm3+9' Al3+6In3+6Yb-3+9уз+9Т|з+6Lu3+9La3+9Cr'l+fiAm3+8Zr1+8Mn3+6U4+8Cua+fiFe3+6Zn- +fiCe3+9Геометрия молекулы воды изучена хорошо. Если исходить из по¬
ложения ядер в пространстве, то молекула Н20 представляет собой
треугольник с углом Н—О—Н, равным 104,5°. В 1951 г. Н. Бьеррум136
высказал идею о тетраэдрическом строении молекулы воды. С этой
точки зрения атом кислорода молекулы воды находится в центре
тетраэдра. К двум его вершинам направлены связи О—Н, а к двум
другим — орбитали с неподеленными электронными парами:Отсюда становится понятным тот факт, что угол между валентными
связями (104,5°) близок к тетраэдрическому.Неэмпирический квантово-химический расчет молекулы воды по¬
казал, что распределение электронной плотности в свободной моле¬
куле в значительной степени определяется влиянием двух неподелен-
ных электронных пар атома кислорода и обладает почти тетраэдри¬
ческой симметрией.Полярность связи О—Н довольно мала. Ее электрический момент
диполя раЕен 0,534-10_3оКл-м. В тоже время электрический момент
диполя молекулы воды составляет 6,14-10~30 Кл-м. Считается, что
столь высокое значение его обусловлено вкладом неподеленных пар,
вытянутых в пространстве.В комплексных соединениях вода бывает двух типов: координа¬
ционно-связанная и кристаллизационная*. В первом случае она
является лигандом, а во втором не имеет непосредственной связи с
ионом металла. 'Изолированные молекулы воды обладают тремя колебательными
частотами в области ИК спектров: симметричные валентные колеба¬
ния связи О—Н (~3650 см-1), антисимметричные валентные коле¬
бания связи О—Н (~3750 см-1) и деформационные колебания угла
Н—О—Н (~1590 см-1). Кристаллизационная вода поглощает в ин¬
тервале частот 3550—3200 см-1 (симметричные и антисимметричные
валентные колебания) и в интервале 1630—1600 см'1 (деформацион¬
ные колебания). Она также имеет полосы поглощения в области 600—
300 см-1, связанные с вращательными движениями молекулы воды
в кристаллической решетке, которые ограничены водородными свя¬
зями. В результате возникают крутильные колебания молекулы в це¬
лом, которые называются либрационными.В результате координации молекулы воды около иона металла
наряду с валентными и®‘деформационными колебаниями становятся* Кристаллизационная воДа чаще всего занимает пустоты в кристалличес¬
кой решетке соли. При ее удалении молекулярный объем, как правило, не из¬
меняется. Примером этого типа может служить комплекс |Pt(NH3)4]CI2-Н«0.
Встречаются кристаллогидраты, в которых воды много, например Na2S04-
■ ЮНоО. Такие кристаллогидраты имеют структуру льда, в полостях которо¬
го находятся молекулы соли или ее ионы. Включенные в структуру льда мо¬
лекулы и ионы резко ее стабилизируют. Кристаллогидрат Na2S04- ЮН20 пла¬
вится лишь при 65° С.Н137
активными веерное, крутильное и маятниковое. Валентные колебания
связей О—Н в аквокомплексах проявляются в области 3200—3550 см-1.
Частота деформационного колебания мало зависит от природы метал¬
ла и обычно проявляется в области 1580—1650 см-1. По этой полосе,
как правило, судят о наличии в веществе координированной или кри¬
сталлизационной воды. Наилучшим критерием прочности связи коор¬
динированной воды с ионом металла считается частота маятникового
колебания (650—800 см-1), которая возрастает с увеличением проч¬
ности этой связи. Отнесение полос облегчается снятием спектров дей-
терированных комплексов. Дейтерирование 'вызывает сдвиг полосы
в область низких волновых чисел. Так, колебательная частота маят¬
никового колебания дейтерированного комплекса в 1,30—1,37 раза
ниже, чем комплекса с легкими атомами водорода.Характер гидроксильной группы как лиганда лучше всего может
быть объяснен с позиции 5/;3-гнбридизации валентных орбиталей
атома кислорода. При удалении протона от молекулы воды к трем
вершинам тетраэдра будут направлены орбитали, занятые неподе-
ленными парами электронов, а к четвертой — связь О—Н:Неэмпирический расчет гидроксильной группы показал, что за¬
ряды на атоме кислорода и водорода соответственно равны —0,8191
и —0,1809.В ИК спектрах щелочей валентные колебания гидроксильной груп¬
пы проявляются в виде узкой полосы в области 3500—3700 см'1. При
координации эта полоса расширяется и смещается в область более
низких волновых чисел вплоть до 3200 см"1. Кроме того, в облает»
800—1100 см"1 проявляется полоса деформационного колебания
О—Н. Наличие полосы колебаний Н—О—Н в области 1580—1650 см"1
рассматривается как свидетельство в пользу гидроксо-, а не аквокомп-
лекса.Колебательные частоты металл — ОН в спектрах моноядерных
гидроксокомплексов находятся в области 300—500 см"1. Важно от¬
метить, что колебания связей металл—кислород в гидратированных
ионах металлов лежат в той же области 330—475 см"1.Водородная связь в ИК спектрах акво- и гидроксокомплексов про¬
является смещением полос валентных колебаний связей О—Н в об¬
ласть более низких частот. Характерным признаком образования
водородной связи могут служить искажения в колебательных спект¬
рах: изменение интенсивности, смещение и уширение полосы. Иногда
интегральная интенсивность полосы валентных колебаний оказыва¬
ется очень чувствительной характеристикой. При образовании водо¬
родной связи она может увеличиваться на порядок. Полуширина
полос валентных колебании О—Н изменяется в очень широких преде¬138
лах, поэтому также может служить критерием образования водород¬
ной связи.Принципиально и молекула воды, и гидроксильный ион могут вы¬
ступать в роли мостиковыл лигандов: молекула воды с образованием
двух координационных связей (имеются две неподеленные электрон¬
ные пары), а гидроксильная — двух или трех связей (имеются три
неподеленные электронные пары). Протоны мостиковой молекулы
воды подвергаются столь сильному электростатическому влиянию
двух положительно заряженных ионов металла, что это, как правило,
приводит к отщеплению одного из них с образованием мостикового
гидроксильного лиганда. В отличие от гидроксосоединений соедине¬
ния, в которых группы ОН" связывают два пли более ионов металла,
называют оловыми соединениями. Процесс образования оловых со¬
единений из аквасоединений называют олЩией.При высоких положительных зарядах ионов металла может от¬
щепляться протон и от мостиковой гидроксогруппы с образованием
оксомостика:нQ—/0\ 5* /ич -f н+П+/ \П + Л+/ +М М М МТакие процессы называют реакциями оксоляции.В комплексах, центральный ион которых обладает высоким поло¬
жительным зарядом и имеет пустые валентные орбитали, может проис¬
ходить отщепление от координированного гидроксильного лиганда
протона с образованием кратной связи, например:F5\v/,+—ОН ** [h5\V=0](n~1)4' f Н+Эта реакция также относится к реакциям оксоляции.Известны мостиковые гидроксокомплексы типа :(ОН)ь 2(ОН)2,
г(ОН)з*, но нет достоверных сведений о существовании соединений
типа 2(ОН)4. Примером мостикового комплекса тнпа 2(OH)! может
служить димерное соединение/ОН\[(NH3)6Cr Cr(NH3)5]C]5Угол между связями Сг—О—Сг в этом комплексе равен 135°. Из¬
вестно тетрамерное соединение [(СН3)..,АиОН]4. Оно имеет строение,
близкое к плоскостному:* В таком обозначении цифры, стоящие перед скобкой, означают число
координационных мест, занимаемых мостиковой группой, а цифра, стоящаяпосле скобки, — число мостиковых групп, связывающих одни и те же ионы
металла. С этой позиции обычные монодентатные группы можно было бы
обозначать i(OH)0.139
Этот тетрамерный комплекс символически можно записать следующим
образом {o(OH)i}.;. Угол между связями Au—О—Au равен 111°.Считается, что в гидроксокомплексах типа 2(ОН)! мостиковая
связь не очень прочная. Для образования прочных полиядерных
гидроксосоединений необходима дополнительная стабилизация за
счет образования л-связей, циклической конфигурации или обра¬
зования дополнительной связи металл—металл.Наиболее распространены гидроксомостиковые комплексы типа
2(ОН)2. Они известны для ионов большинства переходных металлов
в состоянии окисления +2 и выше. Эти мостнковые соединения со¬
держат конфигурацию четырехчленного циклаМ—ОН1 I
1 НО—iM’Образование цикла приводит к значительной стабилизации димер¬
ного комплекса. Об этом свидетельствуют высокие значения констант
димеризации (обычно порядка 102—105), отвечающих равновесиям2МОНл\>= [М(ОН)2М]2В*Интересный случай мостикового комплекса типа 2(ОН)2 пред¬
ставляет соединение [Co1(OH)e(NH.J)12]6+. Считают, что в этом соеди¬
нении один из четырех ионов кобальта отличается от трех других.
Структуру комплекса изображают следующим образом:Co(NH,),/\НО ОН\/
но—Со—он/ /\ V(NH3)4Co—НО ОН—Co(NH3)4Ее можно обозначить символом {2(ОН)2}3.Комплексы типа 2(ОН)3, т. е. соединения с тройными гидроксо-
мостиками, встречаются довольно редко. Их примером может слу¬
жить полученный Вернером комплекс
/он\(NHj)jCo—■—ОН——Co(NH3)j.Структура этого комплекса изучена Андерсоном.Существует несколько надежно наследованных соединении, в ко¬
торых гидроксильная группа является трндентатной. Так, соедине¬
ние [(СНлЬРЮНЬ имеет строение искаженного куба, в вершинах
которого чередуются атомы платины и гидроксильные группы. Угол
между связями О—Pt—О составляет 77,6°, а между связями
Pt—О—Pt 101,2°:Это соединение можно отнести к типу 3(ОН), и символически записать
в виде (slOH)!}). ^Представляет интерес структура тримерного иона [5пл(ОН)4]2+.
В нем одна из гидроксильных групп является трндентатной 3(ОН)!.
а три других — бидентатными 2(ОН)3:Структуру его символически можно записать следующим образом:В заключение следует подчеркнуть, что акво- и гидроксокомп-
лексы образуют практически все элементы периодической системы.
Можно сказать, что они являются наиболее распространенными ко¬
ординационными соединениями.Современная стереохимия соединении азота, по существу, создана
Ганчем и Вернером. Использовав идеи основоположника стереохимии
Вант-Гоффа, они логично и последовательно развили их для соеди¬
нений азота.Электронографически и спектроскопически установлено, что в
соответствии с представлениями Ганча и Вернера молекула аммиакаSn 2+ОН"',(ОН)1{г(ОН)1}3.7.2. Амины141
имеет форму трехгранной пирамиды с атомом азота в вершине и ато¬
мами водорода, расположенными по углам треугольного основания.Орбитали атома азота находятся в состоянии 5/?3-гибридизации.
Они направлены к вершинам тетраэдра. Три s/?3-ni6piwibie орбитали
перекрываются с s-орбиталями атомов водорода, а четвертая содер¬
жит неподелеиную электронную пару:Н'" | '"'Н
НУглы между валентными связями Н—N—Н равны 106,7°. Алкил-
замещенные аммиака также имеют пирамидальное строение. Углы
между валентными связями в МН2СН3, NH(CH3)2 и i\(CH3)3 находятся
в пределах 105—111°, т. е. тоже близки к тетраэдрическим.Наличие неподеленной электронной пары на атоме азота обус¬
ловливает донорные свойства аммиака и органических аминов. Ос¬
новность аминов в определенной мере является характеристикой их
способности к образованию комплексных соединений. Во многих слу¬
чаях донорные свойства аминов по отношению к протону и ионам ме¬
таллов снмбатны, хотя имеются и исключения. Согласно значениям
рАГь основность аминов в воде возрастает в рядах:Увеличение основности аминов с введением в аммиак первых двух
алкильных групп часто объясняется положительным индуктивным
эффектом последних. Понижение основных свойств при введении треть¬
ей алкильной группы связывают со снижением энергии сольватации
ониевого иона водой вследствие уменьшения числа атомов водорода
в сопряженном катионе, способных образовывать устойчивые водо¬
родные связи с водой. Данные по основности аминов в газовой фазе
указывают на монотонное возрастание сродства к протону у аминов
при переходе от аммиака к триметиламину.На основании смещения полосы валентных колебании О—D в ИК
спектрах при растворении алкиламинов в дейтерированном мета¬
ноле установлено, что протоноакцепторная способность увеличива¬
ется в ряду аминов: первичный < вторичный < третичный.При координации молекулы аммиака к иону металла полосы его
валентных колебаний в ИК спектрах становятся шире, а их частоты
лежат ниже, чем в свободной молекуле. Значення частот валентных
колебаний связей в свободной молекуле аммиака равны 3336 и
3414 см-1. При координация также понижаются деформационные
колебания молекулы аммиака. В свободном аммиаке они имеют сле¬
дующие значения: 6(£) = 1628 см'1 и 6(Л() = 950 см -1.Образование координированной молекулой аммиака водородных
связей проявляется следующим образом: максимумы полос погло¬N.NH3 (4,75) < N(CH3)3 (4,2) < NH2CH3 (3,38) < NH(CH3)2 (3,23)
NH3 < NH2C2H6 (3,37) < N(C2H5)3 (3,13) < NH(C,H5)2 (3,07)142
щения N—Н смещаются в область низких частот; полуширина этих
полос увеличивается; интегральная интенсивность полос возрастает.Полосы колебания связи металл—азот лежат ниже 600 см"1.
Обычно они широкие и довольно слабые. Считают, что с увеличением
прочности связи металл—аммиак частоты деформационных колеба¬
ний 6(Л() и маятниковых колебаний (>(£) закономерно возрастают.При отрыве протона от молекулы аммиака образуется амидный
нон, который имеет две неподеленные электронные пары:$-(S7n\"hнИсходя из этого амидный нон должен обладать электронодонорными
свойствами даже в большей мере, чем аммиак. Первый амидокомп-
лекс, а именно [Pt(NH3)4(iVH2)C!lJC12, был получен Л. А. Чугаевым.
Это соединение сыграло важную роль в координационной химии.
В настоящее время известно много амндокомплексов. Большой инте¬
рес представляет гексаамндосоеднненне Na2[Pt(NH»)0].Поскольку амидный ной имеет две неподеленные электронные
пары, то он может выступать в роли мостикового лиганда.
Примером такого мостикового комплекса может служить
l(NH3)5Co—NH2—Co(NH3)5]X5.Иминный ион NH2~,TaK же как п гидроксильный ион, имеет три
неподеленные электронные пары:$-
<S7 \^2>НРис. 7.1. Расположение атомов в
молекуле анилинаПоэтому он также может выступать в качестве мостикового лиганда.
Иминолнганд может быть тридентатным, однако комплексы такого
типа достоверно не известны.Пирамидальную структуру сохраняет атом азота и в ари- / /ламинах. Так, у анилина угол /00 '/Чмежду плоскостями, в кото- / V У w/ \рых расположены бензольное / С]А\ U\f) I/ \ядро и группа NH,, составля- / у/ \ет 45° (рис. 7.1). При таком / А\П О/Л// \ Н /
угле неподеленная электрон- / \/ / н /ная пара атома азота ориенти- / f\ [\ /\ /143
Рис. 7.2. Расположение неподелен- Рис. 7.3. Схема ст-донорного ил-акцеп-
ной электронной пары атома азота торного взаимодействия пиридина с ио-
и л-системы в молекуле пиридина ном металларована почти параллельно л-электронной системе ароматического ядра.
Это обеспечивает ее сопряжение с л-системой. В результате электрон¬
ная пара может вступать в сопряжение с бензольным кольцом и пони¬
жать основность анилина по сравнению с аммиаком и алифатическими
аминами, что подтверждается экспериментальным ходом изменения
рКа- Однако данные,по основности аминов в газовой фазе указывают
на большее сродство к протону анилина, чем аммиака. Такое противо¬
речие снимается предположением, что образующий ион анилиния
C6H5NHj гораздо более, чем нон аммония, стабилизирован за счет
делокализации ониевого заряда. При переходе же этих ионов в воду
выигрыш в энергии сольватации для иона аммония приводит к тому,
что в растворе аммиак оказывается более сильным основанием, чем
анилин.Явление, при котором неподеленная электронная пара лиганда,
ответственная за донорные свойства, сопряжена с л-электронами
ароматической системы, называют рл-сопряжением.Основность третичного гетероциклического амина пиридина
C5H5N по сравнению с его гидрированным производным пиперидином
C5H10NH в воде резко понижена. Причина этого в том же, что и в слу¬
чае анилина. Неподеленная электронная пара атома азота вступает
в сопряжение с л-системой гетероциклического ядра, в результате
чего понижается электронная плотность на атоме азота (рис. 7.2). Во¬
влечение неподеленной электронной пары пиридина в донорно-акцеп-
торное взаимодействие с ионом металла исключает /?л-сопряжение.
Однако в этом случае в сопряжение с л-электронной системой может
вовлекаться заполненная d-орбиталь (подходящей симметрии) цент¬
рального атома, вследствие чего пиридин и его производные прояв¬
ляют л-акцепторные свойства (рис. 7.3).Среди полос ПК спектра, характеризующих колебания пиридино¬
вого кольца, имеется полоса 1578 см-1, которая чувствительна к
взаимодействию этого лиганда с акцептором электронов. При коор¬
динации пиридина около иона металла происходит смещение этой
полосы поглощения в высокочастотную область на 10—25 см-1. Это
обстоятельство широко используется для суждения об участии как144
самого пиридина, так и его производных в донорио-акцепторном вза¬
имодействии.Рассмотренные амины содержат один донорный атом азота, у ко¬
торого имеется одна неподеленная электронная пара. Следовательно,
все они могут выступать только в роли монодентатных лигандов.При мысленном замещении водорода в аммиаке на группу NH2
получается гидразин. Поскольку электроотрицательность этой груп¬
пы выше, чем атома водорода, основность гидразина по сравнению с
аммиаком должна быть понижена. Действительно, значение (p/(b)i =
= 6.07; (рКь)г =- 15,05.Наличие дипольного момента (6,0—6,3-10~30 Кл-м) свидетельст¬
вует о несимметричном строении молекулы гидразина. Считают, что
она имеет цмс-конфпгурациюв которой аминогруппы развернуты относительно друг друга на опре¬
деленный угол.Несмотря на наличие в молекуле двух донорных атомов, гидразин,
как правило, выступает в роли монодентатного лиганда. Известны
случаи, когда он играет роль мостиков, образуя комплексы типаНН нV-N^H/ \
м мОднако в соответствии с низкой склонностью гидразина к протонизации
по второй ступени его бидентатные свойства выражены весьма слабо.В литературе не известно ни одного комплекса, в котором гидразин
образовывал бы трехчленный цикл типаПри замене водорода в аммиаке на группу ОН получается гндро-
ксиламин. Поскольку электроотрицательность группы ОН выше,
чем группы NHo, то основные свойства гндрокснламнна должны быть
выражены слабее, чем гидразина. Действительно, его значение р/Сь
для водных растворов равно 7,77.Рентгеноструктурные данные свидетельствуют о /пранс-конфигу-
рацин молекулы гидроксиламина:145
В гидрокснлампне имеются три неподеленные электронные пары:
две на атоме кислорода и одна на атоме азота. При комплексообразо-
ванни гидроксиламин всегда выступает в роли монодентатного ли¬
ганда за счет донорного атома азота.Амины выступают в роли лигандов по отношению к большому
числу ионов d-элементов. Аминокомплексы сыграли исключительно важ¬
ную роль в разработке Вернером координационной теории и продол¬
жают служить объектами для развития теоретических положении
координационной химии. Большее значение в современной координа¬
ционной химии имеют полидентатные лиганды, в которых наряду с
аминогруппой содержатся другие донорные группы, например амино¬
кислоты, аминоспирты, ампносульфиды.7.3. Органические нитрилыОрганические нитрилы имеют два донорных центра: неподеленную
электронную пару на атоме азота и тройную связь углерод—азот. За
счет первого образуются комплексы типаRC = N: — М(торцевые комплексы), а за счет второго образуются л-комплексы:
RC=NIМКвантово-химические расчеты и экспериментальные данные, полу¬
ченные методами фотоэлектронной п рентгеноэлектронной спектро¬
скопии, свидетельствуют, что верхней заполненной орбиталью ацето¬
нитрила CH3C=N является л-молекулярная орбиталь, а орбиталь,
заполненная неподеленной электронной парой, расположена ниже.
В бензонитриле CcH5C=N самыми высокими по энергии являются
л-орбитали фенильного кольца. Несмотря на это, нитрильные комп¬
лексы с металлами образуются не за счет л-молекулярной орбитали,
а за счет неподеленной электронной пары атома азота.Проявление донорной способности молекулой посредством того
или иного донорного центра зависит от ряда обстоятельств. Одним
из основных является принцип максимального перекрывания орбита¬
лей донора и акцептора. Согласно ему донорно-акцепторная связь
образуется с участием того донорного центра, который обеспечивает
максимум перекрывания. Вероятно, в случае нитрилов этот принцип
в основном и определяет участие донорного центра.Для большинства изученных комплексов угол между связями
М—N—С близок к 180°. Рентгеноструктурные данные показывают,
что при координации нитрилов к нону металла длина связи N—С
уменьшается, т. е. увеличивается ее прочность. Это находит отраже¬
ние в колебательных частотах группы v(CN). При координации про¬
исходит сдвиг максимума соответствующей полосы поглощения в
высокочастотную область. Для свободных алкилнптрплов полосы
колебания связи лежат в интервале 2260—2240 см-1.Известны торцевые нитрильные комплексы, значение v(CN) в ко¬
торых понижено по сравнению с v(CN) свободного нитрила. Считают,
что причиной этого является ослабление связи C=N координиро¬
ванного нитрила в результате проявления нитрильной группой л-
акцепторных свойств. Электронная плотность с d-орбиталей металла
передается на т:*-орбитали нитрильной группы. Таким образом, орга¬
нические нитрилы кроме донорных свойств иногда проявляют л-
акцепторные свойства.Большой интерес представляет комплекс рутения с цианопириди-
ном [Ru(NH3)s(NCC5H4X)]2+- Установлено, что цианопирндин пред¬
почитает координироваться к рутению посредством нитрильной груп¬
пы. Неожиданным оказалось то, что несмотря на положительный
заряд комплексного иона, основные свойства координированного
цнанопиридина выше, чем свободного. Это может означать, что при
протонированнн пиридинового атома азота происходит делокализа¬
ция положительного заряда с вовлечением л-электронов нитрильной
группы. Большее участие л-электронов нитрильной группы в комп¬
лексе по сравнению с нитрильной группой, находящейся в свободном
лиганде, вероятно, обусловлено я-дативной связью металл—лиганд.
Таким образом, этот пример также свидетельствует о способности
нитрильной группы акцептировать электроны с орбиталей атома ме¬
талла.В литературе описано мало комплексов, в которых органические
нитрилы координированы по тройной связи по л-типу. При таком
способе координации значения v(CN) понижаются более чем на
100 см'1. Характерным примером комплексов данного типа может
быть соединение [Pt(PPh3)2(NCCF3)l. Значение v(CN) для него равно
1734 см-1. Это означает, что связь азот — углерод, по су¬
ществу, является двойной и структуру комплекса можно изобразить
формулой,CF3Ph3Px X/><11
Ph-iP/ XN7.4. N-Оксиды аминовОкисление аминов пероксидом водорода или перкислотами приводит
к образованию оксидов аминов. Атом кислорода с шестью валентными
электронами акцептирует неподеленную пару электронов аминного
атома азота с образованием N-оксндов. Оксиды первичных и вторичных147
алифатических аминов неустойчивы и быстро перестраиваются в
соответствующие гидроксил амины:.. :0:
RNH, >' о- '
IR—N+—Н
IНОНIR—N—IIR4 .. Ю:>N—Н ►R/+ у Оr/ \нN?/N—ОНГидроксиламины окисляются дальше до нитросоедннений.Третичные алифатические амины дают устойчивые N-оксиды:R R\ :U: \ ..R—N: >- R—N -*■ :Q:/ / "R RОни разлагаются лишь при высокой температуре по схеме
СН3СН2 + уо-/СН3
СН,СНо/ хсн/СН3СН,Ч .он-\N/ + с2Н4
С Н 3С Н 2 ••Весьма устойчивыми и характерными соединениями являются
N-оксиды ароматических аминов — пиридина и его производных.
Свойства N-оксида пиридина описываются пятью каноническими
структурами:о-о-о-о-о‘JNNNNN1_IIII11ОООООI11IIIJVVПервая структура соответствует донорно-акцепторному образованию
сг-связи азот—кислород. Почти в чистом виде она присутствует в N-
оксидах третичных алифатических аминов.Установлено, что в отличие от пиридина, который подвергается
элекгрофильному замещению лишь с трудом и в иге/па-положении,
оксид пиридина взаимодействует с электрофильными реагентами лег¬
ко, и реакция направляется в орто- и пара-положения. Это говорито вкладе канонических форм II и III в мезомерный* гибрид молекулы.* Электронное сопряжение в ненасыщенной молекуле, находящейся в нор¬
мальном (невозбужденном) состоянии, называют мезомернен. Согласно тео¬
рии мезомерного эффекта распределение связывающих электронных пар в
мезомерной молекуле является промежуточным между распределением, ко¬
торое требуется по теории валентных связей (например, RiN—С=С—С=0),148
Проявление канонических форм II и III свидетельствует о том, что
2рл-электроны атома кислорода взаимодействуют с л-электронной сис¬
темой ароматического кольца. Указание на такое сопряжение, в част¬
ности, дают значения дипольных моментов. Лайнтон установил,
что разница в дипольных моментах (в Кл-м) N-оксида пиридина (14,2х
X 10'3°) и самого пиридина (7,41-10'30) составляет 6,79-10'3°). Она
значительно меньше разницы в дипольных моментах N-оксида три-
метиламииа (16,77- 10~30) и самого трпметиламина (2,17- 10~30), кото¬
рая равна 14,6- 10"30.Экспериментально установлено, что кроме л-донорного влияния
на ароматическое кольцо N-окисная группировка может проявлять
л-электроноакцепторные свойства. Это обусловлено вкладом форм
IV и V.В N-оксидах алифатических аминов валентные орбитали атома
азота находятся в состоянии 5/?3-гнбридизации. Поэтому их структура
близка к тетраэдрической. В N-оксидах ароматических аминов атом
азота находится в состоянии sp'’-гибридизации. Связь N—О распо¬
ложена в тон же плоскости, в которой лежит ароматическое кольцо,
вследствие чего 2р* -электроны атома кислорода имеют возможность
вступать в сопряжение с л-электронной системой кольца.Поскольку в N-оксидах аминов неподеленная электронная пара
атома азота занята, то в качестве донора электронов они могут высту¬
пать только за счет атома кислорода. Избыток электронной плотности
на атоме кислорода обусловливает его способность к взаимодействию
с протонами и другими акцепторами электронных пар, в частности,
с ионами переходных металлов.При координации N-оксидов аминов около ионов металлов полоса
колебания связи N—О смещается в низкочастотную область. Допи¬
рование электронной плотности приводит к понижению участия 2рт. -
орбиталей атома кислорода в сопряжении с л-системой ароматическо¬
го кольца и, следовательно, к понижению частоты колебания связи
N—О.В свободных оксидах ароматических аминов колебания связей
N—О характеризуются частотами 1220—1270 см-1, колебательная
частота связи N—О оксида пиридина в растворе СС14 — полосами
1265 и 1271 см-1, а в таблетках КВг — полосой 1243 см-1. При коор¬
динации N-оксида пиридина эта полоса поглощения сдвигается в низ¬
кочастотную область на 12—60 см'1.Полоса колебания связи металл—кислород проявляется в широ¬
ком интервале частот от 200 до 500 см-1.В ИК спектре N-оксида пиридина, снятом в растворе СС14, имеется
полоса 1610 см-1, а в спектре, снятом в таблетках КВг, наряду с по-и распределением по альтернативной днполярной структуре
R2N=C—С=С—О", т. е.
. 1. iВС-всN0FNeNaMg;AIW//,PS/.W.ClArКСуScTi'vСгM n L Fc/. Y ';Con,;m\'////.
/ Zn/OaЫASScBrKrRb: Sr. v /ZrNbMoTc. | RuRhPU :Ж.'////■Sn;-'////SbЩ'////.XeCs' Ba' LaIlfТа• WRe 1 Os ,\ ...lr, Pt;'A uШ-.: ti./РЬ/BiPoAtRnFrRjAcК u: CePr'.'Nd;PmSmf Eu: Ci d'TbOyHo'■5/Er/'T mYbX/Lu/’ ThPayV'yNpГ>иAmС mBkcrEsFmMdNoLrРис. 7.4. Элементы, для которых получены комплексы с N-оксидами
аминов (заштрихованы)лосой 1610 см-1 (средней интенсивности) имеется еще очень слабая
полоса 1564 см"1.Обращает на себя внимание отсутствие сильной полосы в районе
1578 см"1, весьма характерной для пиридина. Вероятно, это связано
с тем, что координация пиридина около нона металла и образование
N-оксида с точки зрения электронного смещения — процессы ана¬
логичные. Оба они связаны со смещением этой полосы в высокочастот¬
ную область,т. е. к 1600 — 1610 см-1.В литературе описано большое число комплексов различных ме¬
таллов с N-оксидами ароматических аминов. На рис. 7.4 указаны эле¬
менты, для которых получены и исследованы такие соединения. Сле¬
дует отметить, что большинство комплексов с N-оксидами ароматиче¬
ских аминов разлагается водой. Молекулы воды, являясь хорошими
лигандами, оказывают сильную конкуренцию N-оксндам аминов за
координационное место около иона металла. Поэтому для приготов¬
ления упомянутых координационных соединений обычно используют
неводные растворители. Если для синтеза берут кристаллогидраты
солей металлов, то реакцию часто приходится вести в присутствии
дегидратирующих агентов. Выделенные в кристаллическом состоя¬
нии комплексы с N-оксидами аминов, как правило, устойчивы на воз¬
духе.7.5. Фосфины, арсины, стибиныФосфиновые комплексы сыграли исключительно большую роль в раз¬
витии представлений о координационной связи, о геометрическом
строении и структуре комплексов, а также о взаимном влиянии внутри-
сферных лигандов. Важным свойством фосфинов, арсинов и стибинов
является их способность стабилизировать комплексы с необычными
степенями окисления ионов металлов. Можно сказать, что стабилиза¬
ция комплексов, в которых металл находится в состоянии окисления,150
равном нулю, для фосфиновых лигандов характерна почти в такой же
мере, как п для карбонильных. Встречаются случаи, хотя и гораздо
реже, когда фосфиновые лиганды стабилизируют необычно высокие
степени окисления металлов, например i\Ti(PEt3)2Br3.Фосфиновые комплексы широко используются в качестве катали¬
заторов разнообразных реакций. На их основе уже действуют много¬
тоннажные производства органического синтеза. Все это привело к
тому, что фосфиновые координационные соединения привлекают ог¬
ромный интерес исследователей.Первые фосфиновые комплексы были получены еще в 1857 г. Гоф¬
фманом. Это был1Г соединения платины и золота с триэтилфосфином.
В 1870 г. Кахоурс и Гал установили существование а- и fJ-изомеров
состава [Pt(PEt3)2Cl2|. Однако интенсивное развитие химии фосфн-
новых комплексов началось лишь с конца 30-х годов XX в. В большой
степени этому способствовали работы Манна и Чатта (Великобрита¬
ния) и Иенсена (Дания).В отличие от аминов аналогичные фосфины обладают гораздо
меньшей основностью. Это связано с большей диффузностью валент¬
ных орбиталей атома фосфора и его меньшей электроотрпцательпостью
по сравнению с атомом азота.Взаимосвязь между основностью органических фосфннов и приро¬
дой радикалов была изучена Хендерсоном и Строили. Значения р
они получили потенциометрическим титрованием в нитрометане. Было
показано, что первичные, вторичные и третичные фосфины сильно
отличаются по основным свойствам. Основность третичных фосфинов
в водно-этанольной среде была потенциометрически определена Ис-
■слайбом и Брухлосом. Соответствующие данные приведены в табл. 7.2.Таблица 7.2. Значения рКл фосфониевых ионов R'R2PH+ФосфинРКЯ в
ннтроме-
танер/Са в
водно-эта-
нольной
средеФосфинрка в
кнтроме-
танеР Ка вводно-эта -
нольной
среде(СН3)3Р8,65. 7,20(с;н5)2ре6н36,254,00(СНзЬРС-Н,8,62—(С6Н5)3Р2,732,60(СН3)Р (С2Н6)28,62—(С4Н,,),РН4,51—(С2Н5)3Р8,696,68(изо-С4На)„РН4,11—(С3Н,)3Р8,646,24(СН,)„РН3,91—(С4Н9)3Р8,436,00(СвН5).РН0,03—(изо-СэН,)зР7,97—(ызо-С4Н9)РН„—0,02—(СН3),РСвН56,494,18РН3— 14Из табл. 7.2 видно, что фосфины значительно более слабые осно¬
вания, чем амины. Молекула фосфина РН3, так же как и молекула ам¬
миака, имеет пирамидальное строение. Однако угол между связями
Н—Р—Н составляет 93°. Это означает, что в молекуле РН3 связи
образованы не гибридными 5/?3-орбнталями, а почти чистыми р-орби¬
талями фосфора. При переходе к замещенным фосфинам углы увели¬151
чиваются. Например, в молекуле трехфтористого фосфора они сос¬
тавляют 97,8°, в молекуле триметилфосфпна — 98,6, а в молекуле
трифенилфосфина — 103°. Таким образом, в замещенных фосфинах
орбитали атома фосфора гибридизованы в большей мере, чем в самом
фосфине.Этим объясняется очень большая разница в основных свойствах
фосфина и его алкил- и арилзамещенных. При протонировании фос¬
фина угол между валентными связями изменяется от 93 до 109°, для
чего требуется большая энергия возбуждения. У замещенных фос-
финов изменение углов значительно меньше, поэтому их основные
свойства выражены сильнее. Другой причиной может быть разница
в сольватации протонированной и непротонированной форм.Основность фосфннов, так же как и аминов, понижается, но менее
резко, если алкильные радикалы замещены на арильные, что часто
объясняется образованием л-связи между атомом фосфора и фениль-
ным кольцом. В результате такой связи может происходить делока¬
лизация электронной плотности на атоме фосфора и, если не учитывать
сольватацпонные эффекты, понижение основности соответствующего
фосфина.Однако как экспериментальные, так и теоретические работы, на¬
правленные на выявление возможности л-взаимодействня между ато¬
мом фосфора и фенильным кольцом в соединениях трехкоордина¬
ционного фосфора, не содержат единого мнения. В ряде работ выдви¬
гается представление о неспособности неподеленной пары фосфора к
/7—л-сопр яжению из-за ее повышенного s-характера, что согласуется
с данными некоторых экспериментальных исследован и й. В других
работах утверждается, что даже фосфин подобно аммиаку обладает
четко направленной неподеленной парой, что противоречит почти
чистому s-тнпу, предсказываемому на основе валентного угла в этой
молекуле. Считают, что отсутствие р—л-сопряжения в ароматиче¬
ских фосфинах независимо от характера неподеленной пары фосфора
обусловлено ортогональностью этой неподеленной пары и плоскости
фенильного кольца, исключающей возможность р—^-взаимодействия.
Однако при таком геометрическом расположении допускается взаимо¬
действие электронов ст-связей фосфора (например, в случае фенил-
фосфина связи Р—Н) с л-системами радикалов, называемое сверх¬
сопряжением.В отличие от аЗота фосфор имеет вакантные d-орбптали. При об¬
разовании координационной связи они могут принимать электроны с
заполненных rf-орбнталей иона металла. Таким образом, кроме о-связи
(как в соединениях с аминами) фосфнны могут образовывать допол¬
нительную связь по л-типу. Такая связь получила название дативной
я-связи. Представление о л-дативной связи развивал Чатт в начале
50-х годов. Аргумент в пользу существования такой связи Чатт видел
в соединениях PF3 с PtCl2 и с BF0. Платина (II) имеет
заполненные .электронами d-орбитали, которые могут быть л-доно-
рами электронов. У атома бора таких орбиталей нет. Этим обстоя¬
тельством Чатт объяснял прочную связь PF3 с платпной (II) и крайне
неустойчивую связь PF3 с BF3.152
Вследствие высокой электроотрнцателыюстн фтора донорные свой¬
ства молекулы PFn должны быть выражены чрезвычайно слабо. Од¬
нако в комплексах переходных металлов недостаток электронной
плотностн на атоме фосфора этой молекулы компенсирован за счет
л-донорных свойств иона металла'Таким образом, недостаток электронной плотности, вызванный одними
связями, компенсируется электронной плотностью другой связи. Это
очень важный момент в теории Чатта. Соответствующий эффект был
назван в литературе синергическим эффектом.Тенденция к передаче электронной плотностн с d-орбиталей ме¬
талла на пустые d-орбитали лиганда должна быть больше для комп¬
лексов металлов в состоянии окисления (0), чем в положительных
состояниях окисления. В данном случае электронная плотность на
d-орбиталях металла выше, а следовательно, выше и склонность к
образованию л-дативной связи. Считается, что это является одной из
причин стабилизации комплексов в низких состояниях окисления
металла лигандами л-акцепторного типа. Вероятно, не случайным
было то, что одно из первых указаний на наличие л-связи было полу¬
чено на комплексах никеля (0). Чатт и Харт изучили дипольные мо¬
менты карбон ил фосфиновых и карбонпларсиновых комплексов ни¬
келя (0) и установили, что они намного меньше тех, которые можно
ожидать, исходя только из сг-донорного взаимодействия фосфора с
никелем. На этом основании сделан вывод, что наряду с ст-связью
имеет место дативная связь металл —фосфор. Чатт и Харг пришли
к выводу, что кратность связи металл—фосфор должна быть в этих
соединениях в пределах от 1,3 до 1,7. Однако имеются работы, в ко¬
торых ставится под сомнение наличие л-датпвной связи металл-'- фос¬
фор в фосфиновых комплексах переходных металлов. Так, при анализе
значений констант спин-спинового взаимодействия 195Pt —3!Р в изо¬
мерных фосфиновых комплексах платины (II) [Pt(PRa)2CU]iH плати¬
ны (IV) IPt(PRa)sCljl Венанзи пришел к заключению, что наиболее
существенный вкладе связи платина — фосфор вносят s-орбнтали ато¬
ма фосфора, следовательно, роль л-составляющей незначительна.Цумдал и Драго на основании расчетов электронной структуры
комплекса /«pawr-|Pl(PH3)(NH3)CI..] пришли к заключению, что л-
составляющая в связи Pt—Р равна всего лишь 15% от ст-составляю-
щей. Следует заметить, что для расчета они использовали модифика¬
цию расширенного метода Хюккеля, предложенную Хоффманом, в
которой, как к в любом полуэмпирическом методе, получаемые ре¬
зультаты в большой степени зависят от выбора используемых пара¬
метров.Несмотря на имеющиеся в литературе расхождения во мненияхо существенной роли л-составляющей в связи металл — фосфор (фос-
финовый), преждевременно полностью отказываться от этих представ¬153
лений. Поскольку фосфиновые комплексы нзвестны для многих ме¬
таллов и в самых различных степенях окисления, то нельзя отвергать
эти представления для всех комплексов. Безусловно, данный вопрос
требует своего дальнейшего изучения.В ПК спектрах первичных фосфинов имеется полоса 2280+6 см'1,
которая приписывается валентным колебаниям связи Р—Н, а полосы
805+10 и 1077+4 см-1 относят к деформационным колебаниям связей
Р—Н. Валентные колебания связей Р—Н вторичных фосфинов рас¬
положены в той же области, что и первичных. Полосы в интервале
650—800 см-1 в спектрах третичных фосфинов относят к колебаниям
связей Р—С, а полосы при 420 и 520 см-1 связывают с колебаниями
связи фосфора с углеродом фенпльной группы.Существует мнение о том, что и с ионами металлов класса «а», и с
ионами металлов класса «б» склонность к образованию координа¬
ционных соединений убывает в ряду PR3 > AsR3 > SbR3.Сведения о висмутиновых комплексных соединениях крайне скуд¬
ны. Вероятно, это главным образом связано с очень слабыми донорны-
ми свойствами висмута, который уже в значительной мере проявляет
свойства металла. Тем не менее, известны комплексы [M(BiPh3)(CO)5],
гдеМ — Сг, Mo, W, [Ni(BiEt3)(CO)3] и [Ir(BiPh3)(CO)(PPh3)3]CI04.Следует обратить внимание, что все указанные соединения содер¬
жат металлы в низких степенях окисления. Вероятно, это способст¬
вует образованию л-дативной связи, и, следовательно, стабилизации
комплекса.Поскольку основные свойства вторичных и третичных фосфинов
выражены слабо, то для них становятся заметными кислотные свой¬
ства. При отщеплении протона от вторичного фосфинаR;PH « [r,p':]' t№на атоме фосфора появляется дополнительная электронная пара. Та¬
ким образом, ион R2P" может выступать в качестве мостикового ли¬
ганда, связывающего два иона металла.Образование мостиковых комплексов, содержащих остатки вто¬
ричных фосфинов, впервые было доказано Хейтером. Он установил,
что при взаимодействии [Pd(PEt3)2C]2] с диэтилфосфином в присутст¬
вии л-толуидина протекает следующая реакция:2 [Pd(РЕу;СL] + 2EtPH + 2n-CH3CeH4NH„ = [Pd(PEt3) (PEt2)Cl]2 ++ 2n-CH3CeH4NH2 • MCI + 2PEt3Димерный комплекс имеет строениеEl з РEt,ч Р ' ClXXPEt,El-)154
Мостиковые комплексы могут быть получены и прямым взаимодей¬
ствием дигалогенпда палладия с дифенилфосфином:PdBrj + 2PhоРН = [Pd(P|i?PH)2 Вг,]2 [Pd(Ph2PH)2Br2] n-CH3C4HeNH2 = [Pd(Ph,PH) (PhaP) Br]2 +-f n-CH3CeH4NH2 • HBr
Строение этого димерного комплекса аналогично предыдущему:Ph,HPh:P4 уПг>\ /""чВг Р .. PPh ,НPh: “Получен также комплекс с фосфидной мостиково!! группой~ РЕ t3 PEt3 “I IH-Pt-PH.—Pt-H ClI ' I
. PEt3 PF.t., _взаимодействием /H/jaHf-[PtCI(H)(PEt.1)il с Me:,SiPH4 пли с H3SiPH2.Улих и Мюллет получили мостпковын комплекс
[(PPh3)2NiPPh^Ni(PPhy)2]Li- 5,5 THF взаимодействием LiPPh2 с
(PPh3)2i\Ti(C2H4) в тетрагидрофуране. В нем содержится только одна
мостиковая группа PPh2.В заключение следует отметить, что фосфиновые, арсиновые и сти-
биновые комплексы, как правило, хорошо растворимы в органиче¬
ских растворителях.7.6. Триалкилфосфиты и фосфиноксидыКроме фосфинов из соединений фосфора (III) в качестве лигандов
сравнительно хорошо изучены триалкилфосфиты (эфиры фосфористой
кислоты) (RO)3P. Поскольку радикал алкокспл RO— по сравнению
с алкильным и арильным радикалами обладает гораздо большей склон¬
ностью к оттягиванию на себя электронной плотности (отрицательный
индуктивный эффект), то электронная плотность на атоме фосфора в
триалкилфосфите оказывается пониженной и основные свойства не
проявляются. Однако по отношению к ионам металлов донорные свой¬
ства фосфора выражены достаточно ярко. Об этом свидетельствует
наличие многочисленных триалкилфосфитных координационных со¬
единений переходных металлов. Важную роль в образовании коорди¬
национных соединений играет л-дативное взаимодействие металл ->
лиганц, приводящее к упрочению координационных связей.Большое значение в качестве лигандов имеют окиси органических
фосфинов. Оксиды тяжелых фосфинов широко используются в ка¬
честве экстрагентов ионов металлов. По экстракционной способности155
кислородсодержащие фосфорорганическпе соединения располагаются
в следующий ряд:ОМI /Р(R0)3P=0 < RO-P О < R2P< < R3P=0фосфаты J. оксидыК фосфиновфосфонаты фосфинатыИз этого следует, что наибольшей экстракционной способностью
обладают оксиды фосфинов. Их экстракционная способность нахо¬
дится в прямой связи со способностью к комплексообразованпю.Нуклеофильным центром в оксидах органических фосфинов яв¬
ляется атом кислорода. Фосфор1>льная группа Р----0 характеризу¬
ется высокой энергией связи (500—630 кДж/моль), малой длиной
связи (0,144—0,156 нм) и небольшим дипольным моментом
(8,88- Ю':,° Кл-м). Все это находит объяснение, если допустить су¬
ществование л-связп в результате перекрывания орбиталей атома
фосфора и 2/>орбнталей атома кислорода. ,Образование связи фосфор—кислород в фосфорнльной группе
можно рассматривать как донорно-акцепторное взаимодействиеR3Р: — О:. rr-Связь образуется вследствие переноса несвязывающих2р. -электронов кислорода на вакантные 3d- -орбитали атома фосфора.
Считается, что особая прочность связи в фосфорнльной группе обус-
ловлёна участием в л-взапмодействнн двух 3d- -орбиталей атома фос¬
фора с двумя '2рг^ -орбиталями атома кислорода, расположенными
перпендикулярно связи Р—О. Обратный перенос электронной плот¬
ности от кислорода к фосфору и объясняет небольшой дипольнын
момент связи Р= О и ее малую длину.Колебательные частоты связей Р—О в фосфиноксидах находятся
около 1170 см'1. При координации фосфинокспдов около ионов ме-; LiBed.СN0FNeNa .MgAt •.1: Si -'
/PsClAr; кС a'.Sc••TiV •С r: M n'.Fe,■xo;V,;Ni>С и-V/'' Zn ,'GaGe,\sSeBrKr;RbSrYZr, Nb"Mo';TcRuRh.PdAgCd/ -' InSnГе/1XeCsBaLa'IlfТа'wRe'OslrPtAu;HgTI :Pb-/' Bi ;PoAtRnFrRaAcKuCePrNdPm- Sm'EuGdTbDyHoEr'T mYbLu.: r,u \ Pa,.u </Np'PuAmCiTiBkСГEsFmMdNoLrРиг. 7.5. Элементы, для которых получены комплексы с оксидами фос¬
финов (заштрихованы)156
таллов частота валентных колебаний связи Р—О уменьшается пример¬
но на 50 см-1. Это является результатом ослабления связи р-—d- в
фосфор ил ыюп группе.В периодической системе, приведенной на рис. 7.5, выделены эле¬
менты, для которых известны комплексные соединения с оксидами
фосфинов. Из этой схемы видно, что в настоящее время известны ко¬
ординационные соединения оксидов фосфинов с ионами большинства
металлов и с соединениями многих неметаллов.Оксиды аминов, фосфинов и арсинов являются хорошими экстра¬
гентами переходных металлов. Их экстракционные свойства определя¬
ются донорнымн свойствами атома кислорода, т. е. склонностью этих
молекул к комплексообразованню. Рентгеноспектральным методом
установлено, что донорные свойства атома кислорода в R3NO и R3AsO
примерно одинаковы, но заметно больше, чем в R3PO. Этот вывод
согласуется с расчетными данными. Уменьшение электронной плот¬
ности на атоме кислорода в R^PO объясняется взаимодействием элек¬
тронов кислорода с вакантными 3d- -орбиталями фосфора. По-видимо¬
му. участие в связях орбиталей азота и мышьяка меньше, чем фос¬
фора.7.7. Сероводород и его депротонированные формыНесмотря на то что молекула H2S является аналогом Н20, комплексы
с сероводородом в качестве лиганда встречаются крайне редко. При¬
чинами этого можно считать следующие:1) сероводород худший и менее удобный для работы раствори¬
тель, чем вода;2) многие металлы, особенно переходные, образуют малораство¬
римые сульфиды, вследствие этого комплексы часто разрушаются
с образованием бинарных соединений сера—металл; (3) сероводород и его депротонированные формы сравнительно
легко окисляются в комплексах центральными нонами и другими
окислителями, в частности кислородом воздуха, что приводит к малой
устойчивости соответствующих соединений.Структура молекулы H2S довольно существенно отличается от
структуры Н20. Угол между валентными связями Н—S—Н равен
92,3°. Он значительно отличается от тетраэдрического и близок к 90°.
Это означает, что в образовании связей S—Н от атома серы участвуют
почти чистые р-орбитали. Энергия связи S—Н в молекуле H2S равна
380 кДж/моль, а энергия связи О—Н в молекуле воды составляет
500 кДж/моль. Вследствие этого кислотные свойства H2S в воде как
растворителе выражены сильнее на 7 единиц р/Са по сравнению с
Н20. Поскольку при координации около центрального иона кислотные
свойства лиганда усиливаются, то существования комплекса с мо¬
лекулой H2S в качестве лиганда можно ожидать только для металлов
в низких степенях окисления. В качестве немногочисленных примеров
можно указать на существование комплексов:(С2Н6) (CO)jMn(HsS), (CO)5W(HsS), [Ru(NH,)5(ILS)1 (BFj),157
Кислотная диссоциация рутениевого комплекса характеризуется зна¬
чением р/\'п 4,0.Несколько чаще встречаются комплексы с гидросульфидным ли¬
гандом SH". Примерами соединений с терминальной группой SH"
являютсяSHIPh.P—Pt—PPh-,
IН(С5Н5)Ч>Ni—SH
Ви3Р/Так же как и для ОН', для гидросульфидного лиганда характерной
чертой является образование мостиковых комплексов, например(CH,),PtPt(CHj)3В мягких условиях при гидролизе координированной около Pt(II)
тиомочевнны получено соединение с мостиковым гидросульфидным
лигандом'.К,■ Cl CI •
I ICI—Pt—SH—Pt—ClI ICl ClПри его взаимодействии с аммиаком получается аммиачный комп¬
лексНCl s CI\/ \// \ / \NH\ С1NH,в котором наряду с гидросульфидным мостиком замыкается и хлорнд-
ный. Интересно, что если вместо аммиака взять пиридин, то хлорид-
ный мостик не возникает, а образуется соединениеНс [Pt2(uS) Ру3С14] • 4Н20В ПК спектре газообразного Н .S имеются три полосы поглощения:
2615, 2354 и 1183 см"1. В спектре комплекса [\V(CO)0(H.,S)I частота
валентного колебания связи S—Н равна 2560 см-1.Считают, что полосы колебания связи S—Н наиболее интенсивно
проявляются в рамановском спектре. Насыщенный раствор H:S в
0.1 М НС1 имеет две полосы поглощения: 1300 п 2600 с.м-1. Таубе
пришел к выводу, что полоса колебания связи S—Н (2547 см"1) в
комплексе |Ru(\H..)-,(HcS)l(BF,)2 очень слабая. Ее отнесение ослож¬
няется тем. что в этой же области присутствуют полосы, обусловлен¬
ные обертонами N—Н.Существуют многочисленные мостиковые комплексы, в которых
наряду с мостиком из атома серы имеется связь металл—металл, на¬
пример(С5Н5) (СО)г Мп-Мп(СО). (CSH5)]Известны комплексы, в которых такие мостики связывают три и даже
четыре атома металла:В органической химии известно огромное число алкил- и арилпроиз-
водных HoS. Это органические сульфиды или тноэфнры. Так же как
при переходе от РН3 к его алкил- и арилзамещенным, при переходе
от HCS к тиоэфирам атом серы приближается к 5/?3-гибридному состоя¬
нию:Наличие двух неподеленных электронных пар на атоме серы обуслов¬
ливает донорные свойства тиоэфиров. Тиоэфиры с одним атомом серы,
как правило, занимают одно координационное место. Тиоэфиры могут
выступать в роли бидентатных мостиковых лигандов, однако в лите¬
ратуре таких комплексов описано сравнительно мало. Так, было
синтезировано соединение [Pt2{S(C2H6)2}2BrJ и с помощью рентгено¬
структурного анализа установлено, что оно имеет мостнковое строе¬
ние следующего типа:S/ \7.8. Тиоэфиры, меркаптаны и сульфоксидыR159
R’DT ~r2Получен комплекс K=[CI3Pt{S(C;H4)20}PtCIJ, в котором роль мости¬
ка играет тиоксан. Его атом серы связывает два фрагмента PtCl3~.Меркаптидные лиганды предпочитают занимать мостнковое, а не
терминальное положение. Поэтому мостиковые комплексы с лиганда¬
ми RS~ весьма распространены. Например, известен комплекс с ме-
тилмеркаптидным лигандом:Получены экспериментальные данные, которые свидетельствуют
о различной скорости обмена хлорид-ионов, расположенных против
мостикового меркаптидного лиганда. Это явление представляет боль¬
шой интерес, но требует проверки другими методами.В настоящее время большое внимание в качестве лигандов привле¬
кают органические сульфоксиды. Интерес к ним особенно возрос,
когда было установлено, что самый легкий из них — диметилсульф-
оксид (ДМСО) — является исключительным по свойствам апротонным
биполярным растворителем: Кроме того, оказалось, что тяжелые
сульфоксиды обладают ценными экстракционными свойствами для
тяжелых и цветных металлов.Важнейшей особенностью ДМСО как растворителя является сла¬
бая сольватация аннона соли. В протонных растворителях анионы
сольватированы вследствие ион-дипольнбго взаимодействия, на ко¬
торое накладывается образование водородной связи. В апротонных
растворителях вообще и в ДМСО в частности водородная связь от¬
сутствует и, следовательно, сольватация анионов сильно ослабляется.
Как правило, катионы по своим размерам гораздо меньше анионов,
что приводит к более сильному ион-днпольному взаимодействию
ДМСО с катионами по сравнению с анионами..Кроме того, на нон-ди-
пол ьное взаимодействие накладывается комплексообразование, ко¬
торое вносит значительный вклад в энергию сольватаций катиона.
Вследствие высокой диэлектрической постоянной ДМСО притяжение
катиона к аниону, т. е. склонность к ассоциации, весьма мало. Таким
образом, ДМСО, избирательно сольваТируя катион соли, способству¬
ет реакциям, в которых активную роль играют анноны.Сульфоксиды имеют пирамидальное строение с атомом серы в вер¬
шине:СН160
V^ s.R /R'Неподеленная электронная пара на атоме серы направлена к вершине
тетраэдра. Если в сульфоксидах RR'SO радикалы R и R' различны,
то они должны существовать в виде двух оптически деятельных форм.Связь сера — кислород в диметилсульфоксиде обычно описыва¬
ется тремя каноническими структурами I—III с преобладанием струк¬
туры II:+ — — -ЬR:S—О — R2S=0 <- R2S=0’ I ’il IIIДвойная связь в этой молекуле обусловлена а- и л-взаимодействнем
сера — кислород. Последнее реализуется благодаря перекрыванию
заполненных рт. -орбиталей атома кислорода h соответствующих пус¬
тых dh -орбиталей серы. Координация сульфоксида около положитель¬
ного иона металла атомом кислорода должна ослаблять связь /?* —dr,
что согласуется с экспериментально наблюдаемым в этом случае по¬
нижением частоты валентного колебания связи S—О.При рассмотрении координации диметилсульфоксида посредством
атома серы, например в случае плоскоквадратных ^-комплексов с
сг-донорно-акцепторной (обозначена стрелкой) и л-дативной связями
ДМСО—М (d'‘d2z„), наиболее важны следующие структуры:( d®) М S—О: ( £) М S -О (4) М ^ S-6:IV " V ” VI "Вклад валентных структур III, в соответствии с которыми о-связь
с металлом за счет неподеленной пары серы невозможна, должен быть
ничтожно малым. В то же время наличие дативной связи М-э-L тре¬
бует заметного вклада структуры VI. Таким образом, при координа¬
ции через серу должно происходить относительное уменьшение доли
структур с кратными связями сера — кислород.Некоторое понижение кратности этой связи можно объяснить так¬
же уменьшением переноса электронной плотности с кислорода на
Зй-орбитали серы вследствие «конкуренции» за заселение данных ор¬
биталей с nd-орбиталей металла. Таким образом, проведенное ка¬
чественное рассмотрение, а также результаты полуэмпирическпх
квантово-химических расчетов указывают на некоторое ослабление
связи S—О при координации ДМСО через серу. Увеличение же час¬
тоты валентного колебания в этом случае следует, по-видимому, связы¬
вать не с усилением указанной связи, а с усложнением силового поля,
действующего на атом кислорода, в частности за счет электростатиче¬
ского взаимодействия металл — кислород. Однако независимо от6-817161
LiВсВСNоFNcN;iMg\ls.PsClArКCaScTiVCrMnFcCo• Ni>u■Zn/■'/ ■
GaGoAsScBrKrRbSrY7.xNbMoTcRu.V. . rШ>Pd'-AgCdIn. SnSbTc7/:IXcCsBjlaMfТа\WRe;i* o s
' • 'm•: Pi <yA иHgTIPbBiPoAtRnFrRaAcKuCeУРг,NUPmSm,F u -GdЯ’ь.Dy-lloErT mYbLu;Th .Pa• u:NpPuAmС mBkCfEsFmMdNoLrРис. 7.6. Элементы, для которых получены комплексы с органическими
сульфоксндамн (заштрихованы)рассмотрения прочности связи S—О ИК спектроскопический критерий
определения донорного центра сульфоксидов, участвующего в коор¬
динации, считается довольно надежным. Так, при координации их
через кислород всегда происходит понижение частоты валентных коле¬
баний связи S—О на 60—70 см-1, а при координации через атом серы—
повышение частоты валентного колебания связи S—О на 60—100 см'1
по сравнению с частотой валентного колебания для свободного ДМСО,
равной 1055 см-1.На рис. 7.6 указаны химические элементы, для которых выделены
в твердую фазу и охарактеризованы сульфоксидные комплексные со¬
единения. Нз нее видно, что сульфоксидные соединения известны для
большинства элементов. Для неметаллов координационные соедине¬
ния малохарактерны. Другие «белые» пятна объясняются не столько
сложностью получения координационных сульфоксидных соедине¬
ний, сколько малой доступностью некоторых элементов и их солен.
Это относится и к большинству актинидов, прометию, технецию, по¬
лонию, францию. Можно надеяться, что в будущем удастся получить
сульфоксидные соединения вольфрама, мышьяка и бериллия. В раст¬
воре, безусловно, существуют сульфоксидные сольваты рубидия,
цезия, стронция и бария. Однако такие соединения пока еще не вы¬
делены в твердую фазу.Органические сульфоксиды являются амбпдентатнымн лигандами
(см. с. 69). По классификации Пирсона, сульфоксиды являются
одновременно жесткими и мягкими основаниями. Атом серы обеспе¬
чивает сульфоксидам свойства мягкого, а атом кислорода — жесткого
основания. В соответствии с этим при взаимодействии с мягкими кис¬
лотами, которыми являются в первую очередь соли платиновых ме¬
таллов, сульфоксиды должны проявлять свойства мягких оснований,
т. е. реагировать по атому серы. При взаимодействии же с жесткими
кислотами они должны прежде всего проявлять свойства жестких ос¬
нований (реагировать по атому кислорода). Имеющиеся эксперимен¬162
тальные данные хорошо согласуются с этим предположением. Плати¬
новые металлы прежде всего координируют сульфоксиды посредством
атома серы. После того как вследствие стерических препятствий ста¬
новится невозможным дальнейшее присоединение молекул сульфо-
ксидов через атом серы, происходит их координация через
атом кислорода. Большинство ионов металлов являются жесткими
кислотами, и в соответствии с этим они координируют сульфоксиды
через атом кислорода.Из характера координационных соединений, рассматриваемых по
группам периодической системы, видно, что с уменьшением устойчи¬
вости солей МХП облегчается доступность соединений с максималь¬
ным числом координированных молекул сульфоксидов. Как правило,
соединения с максимальным числом сульфоксидных молекул во внут¬
ренней сфере получаются для перхлоратных, нитратных и тетрафтор-
боратных солей. Можно проследить закономерность, заключающуюся
в том, что с уменьшением прочности солей МХП (например, в ряду:
фториды, хлориды, бромиды ииодиды) происходит образование комп¬
лексов с все большим числом внутрисферных сульфоксидов. И наоборот,
с увеличением прочности в ряду ацидолигандов происходит образова¬
ние соединений с меньшим числом внутрисферных сульфоксидов.В заключение приведем расчетные данные по эффективным заря¬
дам на атомах серы и заселенности ее З^-атомных орбиталей, полу¬
ченные полуэмпирнческим методом ППДП со специально отработан¬
ной для серосодержащих соединений системой параметров (табл. 7.3).Таблица 7.3. Эффективные заряды на атомах серы
и заселенности ее d- АОСоединение?sPddH2S+0,060,15(CH3)2s+0,010,38(CH.,),SO+0,610,71CH3S-—0,760,17Результаты рентгеноспектральных исследований свидетельствуюто том, что эффективный заряд на сере в органических сульфидах бли¬
зок к нулю, а в сульфоксидах равен от +0,5 до +0,7. Это хорошо со¬
гласуется с результатами расчетов.Значения эффективных зарядов на атомах серы позволяют сде¬
лать заключение о том, что донорная способность днметилсульфоксида
несколько меньше, чем диметилсульфида. К тому же у диметилсульф-
оксида имеется лишь одна неподеленная электронная пара, а у диме¬
тилсульфида— две. Метилмеркаптидный лиганд в ряду рассматри¬
ваемых должен обладать лучшей ст-донорной способностью, чем ди-
метилсульфоксид и диметилсульфид.6*163
Расчетные данные показали, что по характеру неподеленной элек¬
тронном пары к сульфоксидам довольно' близок ион сульфопия
5(СН3)з. Это позволило сделать вывод, что катион триалкнлсульфо-
ний может проявлять свойства лиганда. Недавно были получены
комплексы марганца и хрома(CjH5)Mn(CO)a {S(CH3)3} BF.,; (C0I Ie) Cr(CO), {S(CH3)3} OSO,Fсодержащие внутрпсферный сульфонпн-катион, а также соединение
рутения [Ru(NHn)5S(CH3)3l(PF0)3.7.9. Оксид углерода (II) и изоцианиды*Карбонильные комплексы играют исключительно важную роль в
промышленном синтезе на основе оксида углерода (II). Структура
молекулы оксида углерода (II) может быть изображена следующим
образом:+ _0==С:Об этом свидетельствует невысокое значение дипольного момента
(0,40- 1СГ30 Кл-м) и особенно его направление. В комплексах молеку¬
ла СО связана с ионом металла ст-связью за счет донорно-акцепторно-
го взаимодействия неподеленной электронном пары атома углерода
и пустой орбитали иона металла:(£^МСЗ + <Ш^С=0: —- (+^М СЕХ=0 =Кроме того, образуется л-связь за счет дативного взаимодействия за¬
полненной d-орбнтали металла с л-разрыхляющей орбиталью оксида
углерода:* ^ С> ^ А ё>М + :С=1) ► М <.=Г)^ (•/' >Как и в других случаях при образовании л-дативной связи металл-
лиганд, перенос электронов от металла на разрыхляющую орбиталь
оксида углерода (II) делает этот лиганд более электроотрицательным,
в результате чего увеличивается ст-донорная способность углерода.* Изоцнаииды также называют нзонптриламн и карбиламнмами.
т. е. увеличивается прочность связи углерод—металл. С другой сто¬
роны, передача электронов от углерода к металлу приводит к умень¬
шению электронной плотности на лиганде и, следовательно, будут
увеличиваться его л-акцепторные свойства.Большую информацию о строении карбонильных комплексных со¬
единений дает ПК спектроскоппя, так как частота валентного коле¬
бания карбонильной группы весьма чувствительна к изменениям за¬
селенности ее л-молекулярных орбиталей. Перенос электронной плот¬
ности с металла на разрыхляющие л-орбитали оксида углерода (II)
вызывает уменьшение кратности связи, силовой постоянной и частоты
колебания связи С—О. Частота валентного колебания связи С—О
свободной молекулы равна 2143 см"1. В большинстве карбониль¬
ных комплексов переходных металлов она находится в области
2000 см-1.В соединении Н3В-СО между оксидом углерода и ВН3 образуется
только донорно-акцепторная 0-связь. Частота валентного колебания
С—О равна 2164 см-1. Таким образом, в результате донорно-акцеп-
торного взаимодействия по ст-типу смещение электронной плотности
от атома углерода к акцептору вызывает смещение л-электронной плот¬
ности от кислорода к углероду. Это приводит к увеличению кратности
связи углерод—кислород и к увеличению колебательной частотыИзменение частоты валентного колебания карбонильной группы
в комплексах переходных металлов зависит от следующих факторов:1) природы центрального атома и состояния его окисления;2) присутствия в комплексе других л-акцепторных или л-донор-
ных лигандов;3) cr-донорных свойств других лигандов внутренней сферы комп¬
лекса;4) геометрического строения и заряда комплексного иона.Кроме комплексов с терминальными карбонильными группами из¬
вестны соединения, в которых СО связывают два и даже три атома
металла. Например, в комплексе [Со2(СО)8] два атома кобальта связаны
двумя мостиковыми карбонильными группами*. Кроме того, в этой
молекуле имеется связь кобальт—кобальт:От мостиковой карбонильной группы в каждую связь вносится по од¬
ному электрону.Примером комплекса, в котором карбонильная группа связывает
три атома металла, является соединение [№з(т)6-С5Н5)з(СО)2]:* Структурные формулы карбонильных соединений здесь и далее схема¬
тичны и кратность связи С—О не указывается.v(CO).У165
01сПри описании электронного строения комплекса с такой мостиковой
группой необходим учет многоцентровой связи в рамках метода моле¬
кулярных орбиталей.Коттон в 1966 г. высказал предположение, которое впоследствии
было неоднократно подтверждено, что в карбонильных комплексах
переходных металлов наблюдается быстрый внутримолекулярный
обмен карбонила из мостикового состояния в терминальное и наоборот.В последние годы выявлен еще один тип карбоннлмостикового
комплекса. Оказалось, что молекула СО с одним атомом металла мо¬
жет образовывать связь за счет электронной пары атома углерода,
а с другим — за счет я-электронной кратной связи. Примером может
служить комплекс [Mn2(CO)5(Dpm)2], где Dpm—■ бис-(дифенилфос-
фино)метан, имеющий строениеХНPhPh —Р°\ I СкС\ I сО—С—Мп.'Ph4'PhPhvVC":p.0' Jk,PhPh'' 'VhT XPhИзоцианиды относятся к немногочисленным лигандам, на атоме
углерода которых имеется неподеленная электронная пара. Строение
алкилизоцианидов может быть представлено в виде наложения двух
структурных формул:R — N=C: и R—N= С:Вследствие противоположного индуктивного эффекта арильных ра¬
дикалов по сравнению с алкильными свойства арилизоцианидов
лучше согласуются с формулойAr=N=C:166
Рис. 7.7. Структура изоцпанидпого
комплекса [Pt3(CNBu)0]Молекулы алкилизоцианидов
п арплизоцианндов линейные.
При координации они сохраня¬
ют строение, близкое к линей¬
ному.Один из методов получения
алкилизоцнанидов заключается
во взаимодействии цианидов пе-реходных металлов с алкилгало-генидами. Вследствие этого координационные соединения переходных
металлов с изоцианидамп, например AgCN-CNR, были получены
практически одновременно с разработкой методов синтеза самих изо¬
цианидов.Электронная структура пзоцианидов аналогична структуре оксида
углерода. На этом основании Гибер и одновременно Клагес пришли к
заключению, что в комплексах металлов эти лиганды должны заме¬
щать друг друга. Эту идею им удалось подтвердить экспериментально.Сходство карбонильного и изоцианидных лигандов заключается
не только в том, что они изоэлектронны и могут взаимно замещаться
в комплексах, но и в том, что они образуют мостики одинакового бп-
дентатного типа.Значительный интерес представляет строение изоцианидного комп¬
лекса [Pt3(BuNC)cl. В этом соединении три молекулы трет-бутилизо-
цианида являются монодентатными, а три другие выступают в роли
бидентатных мостиков. Структура этого комплекса приведена на
рис. 7.7.Балч и Беннер осуществили интересную реакцию замещения мос-
тикового карбонила в комплексе [Mn2(CO)5(Dpm)2] на л-толуолизо-
цианид и получили соединениеО—С—Мп Мп—С—ОЭта реакция еще раз подтвердила очень большое сходство карбониль¬
ного и изоцианидных лигандов.Для получения информации о связи между изоцианидными ли¬
гандами и металлом широко используют положение в ИК спектре
полосы валентного колебания связи С—N. При взаимодействии с ак¬
цепторами электронной пары частота v(CN) может как повышаться, такPhPh167
и понижаться. Считают, что повышение v(CN) свидетельствует об от¬
сутствии или очень малом вкладе я-дативной связи металл-лиганд,
а понижение v(CN), наоборот, говорит о её значительной роли. Та¬
кая же картина наблюдается и в карбонильных комплексах.В комплексах переходных металлов колебательная частота v(CN)
обычно ниже, чем в свободном лиганде. Понижение v(CN) тем больше,
чем ниже состояние окисления металла. Это связано с более благо¬
приятными условиями для образования я-дативной связи. В комплек¬
сах с высокими состояниями окисления центрального атома или при
наличии во внутренней сфере других лигандов я-акцепторного типа
(например, СО или PF3) может наблюдаться повышение v(CN). Это
обстоятельство можно рассматривать как следствие уменьшения
вероятности образования я-дативной связи. В свободных изоциани¬
дах v(CN) находится в области 2000—2200 см-1.7.10. Этилен, ацетилен и их производныеХимия координационных соединений с этиленовыми и ацетиленовыми
лигандами развивается весьма интенсивно. Комплексы металлов с
этиленовыми углеводородами (олефинами) играют роль катализаторов
и получаются в качестве промежуточных соединений в промышленно
важных процессах. Накоплен большой экспериментальный материал
по каталитическим превращениям ацетилена и его производных в при¬
сутствии комплексов переходных металлов.Первое этиленовое соединение было получено Цейзе в 1827 г. За
короткий период ему удалось получить три различных этиленовых
комплекса. В частности, при нагревании в спирте с последую¬
щим добавлением к коричневой жидкости хлорида калия Цейзе
выделил соединение K[Pt(C2H4)CI3]- НгО. Им также был получен
димерный комплекс [Pt(C^.Hi)CI2]2 и этиленаммиачное соединение
[Pt(C2H4)(NH3)Cl2].Работы Цейзе вызвали большую дискуссию, в которую включи¬
лись виднейшие химики того времени, такие, как Либих и Берцелиус.
Однако после бурного периода, связанного с синтезом и исследова¬
нием свойств• новых олефиновых соединений, в конце XIX и начале
XX в. наступило относительное затишье. Интерес к этим комплексам
появился вновь после работ И. И. Черняева и А.Д. Гельман. Пер¬
вые комплексы переходных металлов с ацетиленом были получены
лишь в 1941 г.Одним из наиболее интересных результатов, полученных И. И. Чер¬
няевым и А. Д. Гельман при исследовании этиленовых комплексов,
было установление высокого транс-влияния этилена. Весьма приме¬
чательно, что уже в 1945 г. А. Д. Гельман высказала предположе¬
ние, что ненасыщенные молекулы СгН4, СО и другие в комплексах пла¬
тины выступают одновременно как доноры и акцепторы электронов.
В результате этого между металлом и данными лигандами, в частнос¬
ти между металлом и этиленом, образуется двойная ковалентная связь.С точки зрения теории молекулярных орбиталей две атомные р-168
орбитали в результате линейной комбинации образуют связывающую
и разрыхляющую молекулярные орбитали:Поэтому молекулу этилена можно изображать следующей формулой:На рт, -орбиталях атомов углерода в молекуле этилена имеется по од¬
ному электрону. На молекулярных орбиталях оба они занимают свя¬
зывающую молекулярную орбиталь.В 1951 г. Дьюар развил концепцию, согласно которой связь ме¬
талла с олефином осуществляется одновременно за счет связываю¬
щей и разрыхляющей орбиталей. Например, в комплексе иона се¬
ребра (1) с этиленом заполненная рг. -орбиталь этилена перекрывается
с вакантной бв-орбиталью серебра. Одновременно вакантные раз¬
рыхляющие орбитали этилена перекрываются с заполненными 4d-
орбиталями по схеме (рис. 7.8).Чатт и Данкансон использовали идею Гельман и Дьюара для объ¬
яснения связи платины (И) с этиленом в соли Цейзе. Они считали, что
с-связь металл—этилен образуется при перекрывании заполненной
электронами я-связывающей орбитали этилена с вакантной dsp2-
гибридной орбиталью платины. Обратная я-связь образуется за счет
перекрывания заполненной гибридной 5с/6у0-0рбитали металла с раз¬нес — СН2Рис. 7.8. Перекрывание орбиталей Рис. 7.9. Перекрывание орбиталей
этилена и нона Ag+ ■ этилена и нона Pt:+
рыхляющей л-орбигалью этилена (рис. 7.9). В этом случае располо¬
жение атомов в комплексном ионе [Pt(C2Hi)Cl3]~ будет следующее:с
/ \
II нРентгеноструктурные исследования полностью подтвердили этот вы¬
вод. Следует сказать, что в образовании л-связи со стороны металла
может участвовать и просто ci-орбиталь подходящей симметрии.Сдвиг электронной плотности от иона металла на л-разрыхляющие
орбитали этилена приводит к ослаблению связи С=С и, следова¬
тельно, к уменьшению частоты ее колебания. Значение v(C=C) в сво¬
бодном этилене равно 1623 см-1. Считают, что в соли Цейзе v(C=C)
расположена вблизи 1516 см'1. Имеется и другая точка зрения. В со¬
ответствии с ней связь металл—этилен рассматривается как трех¬
членный цикл PtC2, т. е. предполагается, что двойная связь при
координации превращается в одинарную. С ней связывают полосу
1243 см'1. Многие исследоватетн считают, что для объяснения ИК
спектров олефиновых комплексов переходных металлов необходимо
учитывать участие двойной связи не только в колебании v(С — С), но
и в других колебаниях того же типа симметрии, среди которых наи¬
более важны симметричные плоские деформационные колебания
связен Р(=СН2). Колебания v(C=C) и Р(=СН:) имеют сложную фор¬
му, изменяющуюся при образовании координационной связи, при¬
водящей к сдвигу v(C=C) и Р(=СН2) и к изменению интенсивности
колебания. В результате в качестве критерия прочности координа¬
ционной связи металл—олефин пользуются суммой относительных
«■двигов частот v(C=C) и р(=СН2).В молекуле ацетилена и его производных имеются две л-связи. Они
располагаются в двух взаимно перпендикулярных плоскостях. Та¬
ким образом, ацетиленовые лиганды могут образовывать связь с ионом
переходного металла аналогично олефиновому лиганду. Рентгено¬
структурное исследование комплекса транс-1 Pt(Me:iC—С^С—СМе3)-
■ (СН3СсН4МН2)С12] подтверждает эту точку зрения:Ме,СVIlK—СIMe,СПоскольку ацетиленовая связь содержит две л-связи, то ацетилен
и его производные принципиально могут выступать в качестве мос-170
тикового лиганда между двумя атомами металла. Примером комп¬
лексов такого типа являетсяRС(СО)3Со Со(СО)3Наряду с мостиковой связью в нем имеется связь металл—металл.Колебание-V (СжС) в ацетилене и его симметричных замещенных
запрещено правилами отбора в ИК спектрах и разрешено лишь в
спектре комбинационного рассеяния. В несимметричных ацетилено¬
вых производных колебание -v(C=C) разрешено. Однако полоса,
как правило, слабая, а иногда в ИК спектре не проявляется совсем.
В ИК спектрах ацетиленовых координационных соединений полоса
м(С^С),так же как и v(C=C) олефиновых соединений, смещена в
низкочастотную область. Она обычно находится в интервале 1400—
2000 см-1. По сравнению со свободными лигандами сдвиг происходит
на 200—400 см-1. Причиной этого, вероятно, также является сдвиг
электронной плотности с металла на разрыхляющие я-орбитали аце¬
тиленового лиганда. Естественно, что для бидентатного ацетиленово¬
го лиганда сдвиг v(C*?C) в низкочастотную область происходит
сильнее. Некоторые авторы отмечают, что интенсивность полосы
ч(С^С) в ИК спектре тем выше, чем больше ее сдвиг в низкочас¬
тотную область.В отличие от свободного координированный ацетилен не является
линейным. Это справедливо и для производных ацетилена. Считается,
что так же, как и на смещение v(C-=C), на отклонение от 180° угла
между связями R—С=С влияет л-датнвная связь металл—ацети¬
леновый лиганд.Как для олефПновых, так и для ацетиленовых комплексов кроме
структуры, в которой кратная связь расположена перпендикулярно
к плоскости координации, встречается структура, в которой кратная
связь находится в плоскости координации. В этом случае углеводороды
образуют с атомом металла трехчленные циклы, т. е. они ведут себя
как бидентатные лиганды. Такая структура характерна для переход¬
ных металлов в нулевом состоянии окисления, например для Ni(0),
Pd(0), Pt(0). Так, комплекс [ATi(PPh3);(C2H4)] имеет строениеАналогичный дифенилацетиленовый комплекс [Pt(PPh3)2(PhC=CPh>]
построен так же:171
По-видимому, в комплексах такого типа углеводороды образуют
с центральным атомом две a-связи по обменному механизму. Поэтому
в комплексах, где непредельный углеводород занимает два коорди¬
национных места, понижение частоты валентного колебания кратных
связен происходит в большей степени, чем в комплексах, где кратная
связь перпендикулярна к плоскости координации.Получены и исследованы бис-ацетиленовые комплексы платины
типа [PtLjClj], где L — гликоли:СН3 СН3 СН3 СН3 СН3 СН3III II IСН,-С С—С=С—С=СН2 С2Н5—С С—С 2= с-с=снгII IIон он он онУстановлено, что координация таких лигандов происходит через
тройную связь, а двойная связь в координации не участвует. Вероят¬
но, предпочтительная координация тройной связи по сравнению с
двойной — явление закономерное, так как оно проявлялось и в других
случаях.7.11. Галогенидные лигандыДля галогенидных ионов характерна электронная конфигурация
s2p6, т. е. они имеют четыре неподеленные электронные пары. Вслед¬
ствие этого они могут выступать в роли не только монодентатных, но
и полидентатных лигандов. В последнем случае они будут мостнко-
выми.Галогенидные координационные соединения весьма распростра¬
нены. Они известны для большинства элементов периодической сис¬
темы. В этом разделе не будут рассматриваться комплексы с терми¬
нальными галогенидными лигандами, поскольку их строение целиком
зависит от электронной структуры центрального атома. Здесь целе¬
сообразно остановиться лишь на координационных соединениях, в
которых галогенидные лиганды являются мостиковымн.Хлоридные бидентатные мостики способствуют образованию свое¬
образных комплексов колоночного типа с различной степенью окисле¬
ния центральных атомов, например [PtII(XH3)2Br.:l[PtIV(NH3)nBri].
Структура этих соединений состоит из колонок, в которых чередуются
плоскоквадратные и октаэдрические комплексы172
(где а — 0,263; b ~ 0,302; с --- 0,244 нм). Такие соединения весьма
характерны не только для платины, но и для палладия.В тетрамерном соединении [(CH3)aPtCIJi галогенидные ионы яв¬
ляются тридентатными:(CH,),PtУ\С! гI
I,'С1lCHj>,Pi- Строение этого соединения аналогично гидроксопроизводному
(CH:j)aPt( ОН)]4. Известен аналогичный иодиднын комплекс
T[(CH3)3PtI]4, который построен по такому же типу.7.12. Цианидный лигандЦианидный нон является амбидентатным лигандом, так как и на атоме
углерода, и на атоме азота имеются неподеленные электронные пары.
Эти атомы связаны между собой тройной связью: одной сг- и двумя
я-связямп::Ns=C:~Электронная конфигурация цнанидного иона с позиций метода моле¬
кулярных орбиталей следующая:КК (32s)2 (a*2s)2 (a2р)- (-.2р)4Таким образом, цианидный лиганд является изоэлектронным и изо-
структурным карбонильному лиганду.Цианидный лиганд может координироваться следующим образом:М—CN м—NC М—CN—ММч Мч N>CN >CN—М М - )|1М' М' СДва последних случая пока не имеют надежных экспериментальных
доказательств. Если координация цнанидного лиганда происходит
монодентатно, то связь, как правило, осуществляется через атом угле¬
рода. Это одно из проявлении аналогии с карбонильным лигандом.В органической химии имеются два обширных класса: цианиды
R—CN и изоциан иды R—NC. Однако термодинамически более устой¬
чивыми являются цианиды. Это следует из того, что при температуре
около 250’ С изоцнаниды перегруппировываются в цианиды.Преимущественная координация цнанидного лиганда посредством
атома углерода является следствием распределения электронной
плотности. Квантово-химическим расчетом с использованием одной-С1РКСН,),^РЦС Hj),
Cl173
из модификаций метода Хюккеля показано, что электронная плот¬
ность на атоме углерода в три раза больше, чем на атоме азота.Возможность существования комплексов с N-координированным
цианндным лигандом надежно доказана лишь в растворах. Так, при
взаимодействии |Co(iNH3)5CN|2+ с Cr2+aq в растворе происходит окис¬
ление Сг(П), восстановление Со(Ш) и образуется CrNC2+, который
затем изомеризуется в комплекс CrCN2+ с С-связанным цианидным ли¬
гандом.Аргументы в пользу существования двух изомерных форм цианида
серебра в жидком аммиаке(NH3)n AgCN ^ (NH3)„AgNCбыли получены при помощи рамановской спектроскопии.Комплексы с мостиковым цианидным лигандом довольно рас¬
пространены. Так, при обработке диалкплгалогенида золота R,AuX
цианидом серебра получается соответствующее цианопроизводное,
молекулярная масса которого соответствует тетрамерной структуре:R RI IR—Аич-N —С—Au—RI tС NIII IIIN СI IR—Au—C = N->-Au—RI IR RРентгеноструктурный анализ подтвердил это строение.При взаимодействии Ag(PF6) с эквимолярной смесью
/7zpa«c-[Pt(H)(CN)(PEt3)2] и транс- [Pt(H)CI(PEt3)2] в метаноле вы¬
деляется осадок AgCl, а из раствора удалось выделить кристалличе¬
ский комплекс. Спектр ПМР указывает на неэквивалентность гидрид-
ных атомов в полученном комплексе. На этом основании ему была
приписана следующая структура:■ PEt3 P£t3 ■I IH—Pt—C = N—Pt—HPEt, PEt3 —(PFo)Получены два изомерных цпанид-мостиковых комплекса. Вследствие
твердофазно!! термической дегидратации была осуществлена следую¬
щая реакция:[Co(NH3)5(H20)][Co(CN)6] -*■ [(NH3)5Co-NC-Co(CN)5] + Н?0Изомерное соединение было получено путем[^(N^sCNl^ + tCo^NJbfHjO)]2- -н- [(NH3)5Co-CN-Co(CN)6] + H20В комплексах общей формулы Rh2(RCOO)i-/zKCN-mH20 в зави¬
симости от условий получения цианидный лиганд может быть терми¬174
нальным или мостпковым. Мостнковые комплексы термически более
устойчивы. Вероятно, это связано с широко распространенной за¬
кономерностью, что полимерные соединения характеризуются более
высокой термической устойчивостью, чем аналогичные мономерные
соединения.Примером цианид-мостикового комплекса, включающего в мости-
ковую связь только атом углерода, может служить комплекс
ICu(CN).\HJn.Некоординированный цианпдный ион имеет только одно колеба¬
ние, и его частота примерно равна 2080 см-1. При его координации
в И1\ спектрах проявляются три полосы: одна в области
2100—2250 см-1 и две полосы в области от 400 до 600 см-1. Узкие и
интенсивные полосы в области 2100—2250 см-1 обычно относят к ва¬
лентным колебаниям связей CN, а низкочастотные полосы — к валент¬
ным колебаниям связей металл—углерод и деформационным колеба¬
ниям углов металл — углерод—-азот. Для мостиковых комплексов
со связью металл—азот полосы из области 400—420 см'1 смещены в
более низкочастотную область, а полоса v(Ci\T), наоборот, смещена
в высокочастотную примерно на 30—70 см-1.Для комплексов данного металла значение v(CN) возрастает с уве¬
личением состояния окисления. Например, для комплекса [Ni(CN)4]4-
■v(C.\) — 1985 см-1, а для [Ni(Ci\)4]2~ v(CN) = 2135 см-1. Для циани¬
дов железа (II) v(CN) = 2040 см'1, а для цианидов железа (III)
2120 см'1. С увеличением координационного числа значение v(CN)
понижается. Так, для [Ag(CN):]' v(CN) = 2135 см-1, для [Ag(CN)3]2~
оно имеет значение 2105 см-1, а для [Ag(CN)4]3-— всего лишь
2091 см"1.Так же как и молекула СО, координированный цианидный лиганд
обладает л-акцепторными свойствами. Он может принимать элект¬
ронную плотность с d-орбнталей металла на своп свободные разрых¬
ляющие орбитали. Однако в отличие от карбонила цианидный лиганд
имеет заряд —1. Вследствие этого по сравнению с СО его л-акцептор-
ные свойства понижены.7.13. Роданидный лигандРоданидный (тиоцнанатнып) поп является типичным амбидентатным
лигандом. Достоверно установлены следующие способы координа¬
ции роданидного лиганда:М—SCN М—NCS М—SCN—МТеоретическое рассмотрение вопроса о координации роданидного
иона приведено в гл. 6 (с. 130).Угол между связями М—S—С обычно лежит в пределах 90—120°,
а между связями М—N—С — в пределах 150—180°. Характерным
примером, иллюстрирующим расположение внутрисферных роданид-
ных лигандов в комплексе, является175
P PhНельзя считать исключенным образование координационных фраг¬
ментоводнако пока эти случаи экспериментально не выявлены.Для решения вопроса о способе координации роданидного лиганда
успешно используется метод ИК спектроскопии. Для линейного рода¬
нидного иона имеются две характерные частоты валентных колеба¬
ний: v(CN) « 2050—2060 см-1 и v(CS) « 750 см-1. При координа¬
ции через атом азота значение v(CS) повышается примерно до
800 см-1, а при координации через атом серы, наоборот,, v(CS) пони¬
жается примерно до 700 см-1. Значение v(CN) и в том и в другом слу¬
чае, как правило, возрастает по сравнению с частотой этого колебания
в свободном ионе.В сложных органических лигандах полосы колебания связей С—S
часто перекрываются с полосами лигандов. В этой ситуации способ
координации роданидной группы можно выявить по интегральной
интенсивности полосы поглощения валентного колебания связи С—N,
которая в зависимости от способа координации роданидного лиганда
к иону металла различается весьма существенно. Так, для соедине¬
ний с N-связанной роданидной группой она примерно в три раза
выше, чем для соединений с S-связанной. Для соединений первого типа
эти полосы узкие и их ширина на половине высоты пика состав¬
ляет 6—18 см'1, для соединений второго типа 25—50 см'1.Для изучения способа координации роданидной группы по инте¬
гральной интенсивности разработана методика с применением внут¬
реннего стандарта. В качестве стандартного вещества применяется
салициловая кислота, имеющая интенсивную полосу карбонильного
колебания при 1660 см'1. Для анализа навески комплекса и салици¬
ловой кислоты прессуются в виде таблеток с КВг. О характере связи
роданидной группы с ионом металла судят по значению внутреннего
стандартного отношения (ВСО), которое рассчитывается по формулеПлощадь полосы определяется по произведению ширины полосы,
измеренной на половине высоты пика, на высоту пика. Для комплек¬
сов с S-координированной группой ВСО меньше единицы, а для комп¬
лексов с N-координированной группой ВСО больше 1,5. Например,
для соединении K2LPt(SCN)4] и K2[Pd(SCN)J, в которых роданпдныем/Площадь полосы поглощения валентного колебания связи С—N на ] моль SCN”Площадь полосы поглощения при 1660 см-1 на 1 моль салициловой кислоты176
группы координированы через серу, ВСО равно соответственно 0,3 и0,4. Для комплекса [Pt(PPh3)2(NCS)2] в котором роданидная группа
координирована через азот, ВСО равно 1,7.По аналогии с органическими соединениями R—SCN (тиоцианаты)
и R—NCS (нзотиоцианаты) комплексные соединения, в которых рода¬
нидная группа связана с центральным атомом через серу, называют
тиоцнанатнымн, а комплексы, в которых она связана через азот, —
изотиоцианатными. По классификации Пирсона, роданидный лиганд
можно одновременно рассматривать как мягкое основание (за счет
атома серы) и как жесткое основание (за счет атома азота). Из этого
следует, что с металлами класса «а» или, что то же самое, с жесткими
кислотами роданидный ион должен координироваться посредством
атома азота, а с металлами класса «б», т. е. с мягкими кислотами, —
посредством атома серы. Так, в комплексах цинка и кадмия ион NCS-
координирован через атом азота, а в аналогичных соединениях рту¬
ти — через атом серы.Однако это не единственный фактор, влияющий на способ коорди¬
нации роданндного лиганда. На способ его координации могут влиять
также свойства других внутрисферных лигандов (размеры и их
л-акцепторный характер), состояние окисления центрального атома
и даже свойства растворителя.7.14. Нитратный и карбонатный лигандыСвободный нитратный ион имеет симметричную планарную структуру.
Углы между связями О—N—О равны 120°. Полнатомный лиганд
N03 может проявлять различную дентатность. Достоверно известны
комплексы, в которых он является моно- и бидентатным. Возможны
следующие способы координации:/,0 /О м—о.о—м: мГ N—о /N=0О ^сг М—О'моиолснтатнмй биленшныи чосшковыйВсе сказанное справедливо и для карбонатного лиганда.При координации через кислород длина связи N—О увеличива¬
ется, а длина связей, атомы кислорода которых некоординированы,
уменьшается. Квантово-химические расчеты в рамках метода ППДП
показали, что /7-орбнталн атома азота, перпендикулярные к плоскости
иона ЛтОз, имеют дефицит электронной плотности. За счет этих ор¬
биталей можно было бы ожидать проявления нитратным лигандом
л-акцепторных свойств. Однако пока нет никаких доказательств про¬
явления л-связи в нитратных комплексных соединениях.Атомы кислорода в нитратном ионе имеют неподеленные электрон¬
ные пары с низкой энергией гибридных орбиталей. Они обладают
преимущественно s-характером. Из этого следует, что кислород нит¬
ратного иона является довольно слабым ст-донором.О нитратных комплексах известно сравнительно мало. Одной из
причин этого является то, что многие годы комплексообразование
изучалось в водных растворах. Вода каклпганд оказывает сильную
конкуренцию нитратному иону за место во внутренней сфере комп¬
лекса. Поэтому нитратный лиганд часто даже не входит в спектрохи¬
мические ряды. Нитратные комплексные соединения в водных раст¬
ворах характерны лишь для тория, актиния, урана и трансурановых
элементов. Имеется довольно большая информация о нитратных комп¬
лексах редкоземельных элементов в водных растворах.Для свободного нитратного нона характерны следующие колеба¬
ния: 1050 см-1 (симметричное валентное колебание v,), 830 см”1 (вне-
плоскостное деформационное колебание v2), 1390 см'1 (валентное
колебание v3), 720 см'1 (плоскостное деформационное колебание ■/,).
Первое из перечисленных колебаний проявляется только в раманов-
ском спектре, второе — только в ИК. спектре, а два последних — и в
рамановском и в ИК спектрах.При координации нитратного иона симметрия понижается. Для
координированного по любому из возможных способов нитратного
лиганда должно быть шесть колебательных полос одинаковой актив¬
ности. Колебание vt координированного нитратного иона проявляется
в виде сильной полосы в области 970—1035 см'1, a v3 расщепляется
на две полосы: 1480—1530 см'1 и 1250—1290 см'1. Колебание v, про¬
является в области 815—830 см-1. Плоскостное деформационное ко¬
лебание v4 должно проявляться в виде двух полос, однако обнару¬
жена лишь одна в области 760—800 см-1. Считается, что другая по¬
лоса этого дважды вырожденного колебания лежит ниже 700 см'1.Таким образом, при помощи колебательной спектроскопии можно
отличить внутрисферный нитратный ион от внешнесферного (ионного),
однако нельзя определенно судить о типе его координации (моноден-
татной, бидентатной или мостиковой).Эддисон и Логан считают, что бидентатную координацию нитрат¬
ной группы можно ожидать в комплексах с малым числом координи¬
рованных лигандов. Они рассматривают это заключение как правило.Классическим примером комплекса, в котором нитратный ион би-
дентатен, является безводный нитрат меди (II). Нитратные группы
в этом соединении расположены в двух взаимно перпендикулярных
плоскостях:Для этого соединения в газовой фазе определены все структурные
параметры.Инфракрасные спектры карбонатных комплексов очень похожи
на спектры нитратных комплексов.„ОО178
7.15. Нитритный лигандСвободный нитритный ион имеет угловое строение. Для него установ¬
лены шесть способов координации:М—n;IIо
\ /оIIIМ—NV-мм м
\ /01/\
м ^;n—моIVVIВ результате квантово-химического расчета электронной структуры
полуэмпирическим методом ППДП в ионе N02 получены следующие
значения эффективных зарядов на атомах:-0,583Ч"0.166 ,о""о-0.583VРасчет также показал, что в ионе N02 орбитали атома азота, пер¬
пендикулярные к плоскости молекул, имеют дефицит электронной
плотности. Этим могут быть обусловлены хорошие акцепторные свой¬
ства этого лиганда. Атомы кислорода в ионе N0^, так же как и в ноне
N03, имеют неподеленные электронные пары с низкой энергией гиб¬
ридных орбиталей преимущественно s-характера. Таким образом,
атомы кислорода в ноне N02 могут быть лишь слабыми о-донорами,
но могут проявлять хорошие л-донорные свойства за счет гибридных,
орбиталей, перпендикулярных линиям связи N—О, по тем же при¬
чинам, что и в ионе NO3.При присоединении иона водорода к N02 понижается электрон¬
ная плотность на обоих атомах кислорода и меняется направленность
гибридных орбиталей. Центр тяжести гибридной орбитали, соответ¬
ствующей неподеленной электронной паре атома азота, смещается
к атому азота. Характер остальных гибридных орбиталей в целом ме¬
няется незначительно.Чаще других встречаются первые два типа координации иона N02.
Комплексы, в которых этот лиганд координирован по первому типу,179
называют нитрокомплексами, а по второму — нитритокомплексами.
Метод ПК спектроскопии позволяет довольно просто различать эти
два тнпа комплексов. Так, например, для нитрнтных соединений
[Co(NHn)s(ONO)l3+, [ R h (N Н 3) 5 (ONO) 12+ и [Ir(NH,)s(ONO)]2+ имеются
две характерные полосы поглощения в области 1065—1460 см'1.
Для аналогичного комплекса Pt(IV) [Pt(NH3)5(ONO)]3+ максимумы
этих полос имеют значение 955 н 1505 см'1. В соответствующих нитро¬
комплексах эти полосы сдвинуты в низкочастотную область. Они
принимают значение для [Rh(NH3)3(N02)l2+ 830 и 1420 см'1, для
nr(NH,)s(N02)F 830 и 1410 см'1 и для [Pt(NH3)5(NO,)P+ 825 и
1330 см'1.7.16. Карбоксилатные лигандыКзрбокснлатные н особенно ацетатные соединения известны для
большинства элементов периодической системы. Большой интерес к
этим соединениям вызван широким использованием уксусной кислоты
в качестве как реагента, так и растворителя.Карбоксилатные соединения металлов могут быть следующих ти¬
пов:R—С МIR—С^О—М
IIr—с; - мн
ш-мr—с:^о—*-м
ч'0— мIVR—С ГмR—СС'ОСмммVIКарбоксильные группы насыщенных алифатических кислот ха¬
рактеризуются следующими полосами поглощения в ИК спектре:
валентные колебания группы ОН 3500—3560 см-1, колебания карбо¬
нильной группы 1700—1725 см"1, валентные колебания С—О или де¬
формационные (плоскостные) колебания ОН соответственно 1395—
1440 и 1211 —1320 см"1, деформационные колебания ОН 900—950 см'1.
Отщепление протона от карбоксильной группы приводит к тому, что
атомы кислорода становятся равноценными. При этом исчезают полосы
карбонильного поглощения и появляются две новые полосы в области
1550—1610 и 1300—1400 см"1 (антисимметричные и симметричные
колебания группы СОО").На основании ИК спектров трудно судить о структуре карбоксилата
металла, однако наряду с другими методами ИК спектроскопия спо¬
собствует выбору отдельных структур.v180
7.17. Сульфатный и перхлоратные лигандыСвободный сульфатным мон имеет тетраэдрическое строение. Из че¬
тырех колебательных частот в ИК спектре активны две (613 и
1104 см-1). Сульфатным ион во внешней сфере комплекса подвергается
искажению лишь незначительно и, как правило, также характеризу¬
ется двумя полосами в тех же областях ПК спектра. Если сульфатный
ион координирован к иону металла, то его симметрия понижается и в
ПК спектре начинают проявляться две остальные полосы (в области
970—995 м 438—462 см"1), которые для свободного сульфатного иона
проявляются лишь в спектре комбинационного рассеивания.Сульфатный ион может координироваться следующими способами:М—°х М—Ov^ ^о—мДо недавнего времени существовало мнение, что перхлоратные
комплексы не образуются ни в водных растворах, ни при сплавлении
безводных перхлоратов. Однако в последние годы все чаще появля¬
ются сведения о синтезе комплексов, в которых перхлоратные ионы
входят во внутреннюю сферу. Для этой<цели часто используется без¬
водная хлорная кислота. /Перхлоратный ион, так же как и сульфатный, имеет тетраэдри¬
ческое строение. Из четырех его колебательных частот в ИК спектре
проявляются две (в областях 1050—1170 и 630 см'1). В случае внеш-
несферного расположения перхлоратного иона искажения тетраэдри¬
ческой структуры незначительны, и поэтому данные полосы являются
довольно надежной его характеристикой. При вхождении иона СЮ4_
во внутреннюю сферу комплекса происходит расщепление полосы
1050—1170 см'1 и начинают проявляться в ИК спектре полосы в об¬
ласти 460 и 935 см'1.Д.пя перхлоратного иона возможны те же способы координации,
что и для сульфатного. Существующие экспериментальные данные
показывают, что С104 может быть как монодентатным, так и биден-
татным лигандом.Библиография по вопросам, изложенным в данной главе, приве¬
дена в работе [1].
ГЛАВАВЗАИМНОЕ ВЛИЯНИЕ ЛИГАНДОВ
ВО ВНУТРЕННЕЙ СФЕРЕ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙПроблема взаимного влияния атомов в молекуле, как известно,
была выдвинута А. М. Бутлеровым. Он сделал это одновременно с
формулировкой основных положений теории химического строения.
В 1861 г. в докладе «О химическом строении веществ» Бутлеров по¬
ставил задачу выяснить, «какое взаимное влияние могут оказывать
два атома, находящиеся внутри одной и той же химической частицы,
но химически не действующие непосредственно друг на друга».Продолжателем Бутлерова в деле развития учения о взаимном вли¬
янии атомов был В. В. Марковников. Он сформулировал хорошо из¬
вестные правила («правила Марковникова»), которые сыграли важ¬
ную роль в развитии органической химии.Решения вопросов, связанных со взаимным влиянием атомов в
молекуле, можно ожидать только после создания теории, правильно
отражающей строение молекулы. В органической химии с самого на¬
чала развитие теории химического строения и разработка проблемы
взаимного влияния происходили взаимосвязано.Следует особо подчеркнуть, что в создании теории химического
строения большую роль сыграли экспериментальные факты, свиде¬
тельствовавшие о существовании изомерных форм веществ. Можно
сказать, что изомерия явилась одной из причин создания Бутлеровым
теории химического строения. С другой стороны, развитая в стройное
учение теория химического строения позволила дать строгое научное
определение изомерии, предсказала существование неизвестных изо¬
мерных форм и способствовала экспериментальному обнаружению их.В 1893 г. Вернер сформулировал основные положения координа¬
ционной теории. По этой теории в неорганическую химию были вве¬
дены структурные представления. Если Бутлеров параллельно раз¬
вивал теорию химического строения и представления о взаимном вли¬
янии атомов, то в работах Вернера идея о взаимном влиянии лигандов,
по существу, не получила большого отражения. Резко отрица¬
тельное отношение к координационной теории со стороны видных
специалистов того времени, среди которых выделялись Иергенсен н
Классом, вынудили Вернера приступить к углубленному изучению
изомерии, в частности стереоизомерии комплексных соединений, ко¬
торую предсказывала координационная теория. Вероятно, это и по¬
служило основной причиной того, что Вернер не смог уделить долж¬
ного внимания проблеме взаимного влияния координированных ли¬
гандов.182
Л. А. Чугаев первым среди русских химиков по достоинству оце¬
нил значение координационной теории и стал ее горячим сторонником.
Воспитанный на традициях русской органической химии, он стал
основателем нового направления в координационной химии, харак¬
теризующегося тесным слиянием положений теории химического стро¬
ения, в частности положения о взаимном влиянии атомов в молекуле,
с теорией Вернера.Большой интерес с точки зрения проявления взаимного влияния
лигандов представляет исследование Л. А. Чугаева и С. С. Киль-
тыновнча, в котором было установлено несколько интересных фактов
внутрисферного реагирования лигандов, которые позднее сыграли
важную роль в открытии закономерности трансвлняния. Во-первых,
Чугаев и Кильтыновнч показали, что в нптрогрнаммине платины (II)
молекула аммиака, расположенная напротив нитрогруппы, легко от¬
щепляется при нагревании по схемеNH3- NH3
Pt dta[NO.,NH, 'NOj ->Pt 34-NH3NO.,NH3NO..Во-вторых, было установлено, что сама ннтрогруппа в комплексах
триаминового типа, наоборот, удерживается очень прочно. Ее заме¬
щение хлоридным ионом или аммиаком сопряжено с большими труд¬
ностями. Чугаев считал, что эти два явления связаны между собой об¬
щей причиной. Он особенно подчеркивал тот факт, что вступление
группы N03 во внутреннюю сферу комплекса сопровождается вытес¬
нением молекулы NH3 в транс-положении.8.1. Аминокомплексы платины (II)В 1828 г. Магнус подействовал на горячий солянокислый раствор хлорида пла¬
тины (II) аммиаком и получил ярко-зеленое игольчатое вещество. Теперь это
вещество [Pl(NH3)4] [PtCl4] широко известно как соль Магнуса*. Оно было пер¬
вым аминокомплексом платины и долго служило исходным соединением для
получения других аминокомплексов. Так, при действии аммиака на
[Pt(NH3)4] [PtCI.,] Рейзе получил соль (Pt(NH3),]CI2 и соответствующее ей ос¬
нование [Pt(NH3)4](OH)2.Пейроне в 1844 г. при одном из повторений синтеза соли Магнуса заметил,
что на холоду получается смеешь двух веществ. Для разделения смесь была на¬
грета в солянокислом растворе, нерастворпвшаяся зеленая соль отфильтрована,
а из фильтрата при охлаждении выпали желтые кристаллы. Это был комЬлекс
4«c-[Pt(NH3)2CIo], который впоследствии стали называть хлоридом Пейроне.Наряду с этим соединением в той же самой работе Пейроне получил изо¬
мерный комплекс mprtKC-[Pt(NH3)2CI2]. Для этого он нагревал твердый
IPt(NH3)4]CI2 при 220—240° С,а остаток перекристаллнзовал в кипящей воде.
В том же году и тем же способом комплекс mpa«c-(Pl(NH3)2C!2] был получен
Рейзе. В литературе mpartC-[Pt(NH3)2CI2] стали называть хлоридом второго
основания Рейзе. Первым основанием Рейзе называют [Pt(NH3)4](OH)2.* До разработки Вернером координационной теории и соответствующей
ей номенклатуры вновь полученные комплексы часто называли по характерному
цвету (лутео, пурпурео, внолео и т. д.) или по имени химиков, открывших их.183
8.2. Правила Пейроне и ИергенсенаБолее чем за столетний период было показано, что взаимодействие
аминов с комплексами типа [PtXJ2-, где X — СГ, Вг-, I-, SCN",
CN-, NO2, приводит к образованию диаминатных соединений [PtA2X2]
цис-конфигурации. Реакции схематично можно изобразить следующим
образом:Х PtXX X+ 2АА PtXА X+ 2Х-Экспериментально это правило было найдено в конце прошлого века
Иергенсеном. В память Пейроне, получившего первый диаминатный
комплекс [Pt(NH3)oCl2] (<пс-конфигурации, это правило впоследствии
стало именоваться правилом Пейроне.В большом числе работ, выполненных в разное время, показано,
что взаимодействие солей MX, где X — СГ, Вг-, Г, SCN-, CN-, N03",
с аминокомплексами [Р1А412+ приводит к образованию диаминатных
соединений I Pt А2Х 2] троне-конфигурации:+ 2АЕстественно, в кислой среде эти реакции идут легче, так как связы¬
вание кислотой аминрв способствует их протеканию вправо.Если внутрисферные амины двух видов (А и а), то реакции проте¬
кают по уравнениямАА12+А. XIPt+ 2Х- •XPtАААА а
Pt
А аА а
Pt
а А2Х-+ 2Х-A PtX
X аА + аА PtXX А+ 2АЭто правило экспериментально было установлено Иергенсеном и на¬
звано правилом расщепления Иергенсена.Правильную стереохимическую трактовку оба эти правила полу¬
чили лишь с позиции координационной теории Вернера.8.3. Закономерность трансвлияния И. И. Черняева8.3.1. Экспериментальное обоснование и качественное
изучение явления траневлиянияВ 1911 г. Вернер разделил на оптически деятельные антиподы этилен-
диаминовые комплексы кобальта (III). Тем самым он доказал окта¬
эдрическое строение изученных комплексов и, следовательно, пра¬
вильность основных положений координационной теории. Вопрос о184
плоскоквадратном или тетраэдрическом строении комплексов плати¬
ны (II) мог быть однозначно разрешен получением соединений с че¬
тырьмя различными заместителями и расщеплением их на оптиче¬
ски деятельные антиподы.Такая задача была поставлена Чугаевым, а ее решение было на¬
чато нм совместно с Черняевым. Продолжение же этих работ уже после
кончины Чугаева получило несколько иное направление и привело
Черняева к важнейшему открытию в координационной химии.В плане синтеза комплексов платины (И) с четырьмя различными
внутрисфернымн лигандами Черняев стремился получить соединение
[PtA A' A"(NO,)]N02, где А, А', А" — различные амины. Коорди¬
национная теория не указывала на пути получения таких комплексов.И. И. Черняев установил, что замещение лигандов особенно легко
протекает в транс-положении к нитрогруппе:С1NH3ClNH3ClPtPtNH3NO,РуNO,+ РуРуPtNH,-1- NH,OH =NH3 NO,
[NH2OHClPy
Pt 3
NH3 N02PyPt 3
NH,OH NO,+ NH3 =NH,PtPyNO,ClCiОказалось, что в изомерных комплексах tiuc- и
/п/}анс-1РЦМНз)..:(ГЮ,)2) склонность к замещению нитрогрупп резко
отличается. Например, замещение нитрогруппы на аммиак в комплек¬
се mpaHC-[Pt(NH3),(NO,),] с образованием [Pt(NH3)3N02]N0,NO,NH,NH3 Pt N02JNH,NH,з pt
NH3 NO,NO,происходит значительно быстрее, чем в ^uc-[Pt(NH3)2(NO,)3]NH3NH.PtNO,NO,+ NH3 =NH3 NH3
NH3 NO,NO,Молекулы аммиака или пиридина в соединениях ^«c-[PtA2(NO,)2j
довольно лабильны и способны замещаться другими аминами, напри¬
меррУ pt NO,
Ру NO,+ 2NH3 =NH3 NO,
NH3 NO,+ 2PyNH3NH,PtNOjNO,+ En =CH,NH, NOj
I ' Pt
CH,NH, NO,+ 2NH,Таким образом, лабилизация внутрисферных лигандов в транс¬
положении к нитрогруппе проявляется на широком круге реакций.Представления о большом /лранс-лабилизирующем влиянии нитро¬
группы Черняев распространил и на другие ацидолиганды, в част¬
ности на галогенидные ионы. Он сделал допущение, что транс-па-
билизирующее влияние лигандов анионного типа выше, чем транс-185
лабилизирующее влияние нейтральных лигандов. Наибольшим транс¬
влиянием, по мнению Черняева, должен обладать свободный электрон.Намечающаяся закономерность формулировалась следующим об¬
разом: координированные анионы лабилизирующим образом действуют
на группы и радикалы, находящиеся по отношению к ним в транс¬
положении. Это позволило рассматривать правила Пейроне и Иерген¬
сена с единой точки зрения — трансвлияния внутрисферных лиган¬
дов. Логика рассуждения заключалась в следующем. При внедрении
первой молекулы аммиака в ион [PtCI4]2- замещению подвергается лю¬
бой из внутрисферных лигандов, поскольку они равноценны:C1 Pt C1
Cl Cl+ NH3 —NHClз Clз ptCl+ Cl-NH3СГ-NH3СГPt+ NH3 =J Pt.ClClNH3Clm3^ NH3'12+NH3NH31Pt 3+ 2HC1 =Pt 3. NH3NH3NH3ClЗамещение следующего хлоридного лиганда может происходить на
координате Cl—Pt—Cl или на координате NH3—Pt—С1. Поскольку
транс-лабилизирующее влияние хлоридного лиганда больше, чем
аммиака, то взаимная лабилизация лигандов на координате Cl—Pt—Cl
выше, чем лабилизация хлорид-нона аммиаком на координате
NH3—Pt—Cl. Вследствие этого реакция идет по уравнению+ Cl-Так с позиции представлении о трансвлиянин лигандов было объяс¬
нено правило Пейроне.Первая стадия расщепления тетраминных комплексов соляной
кислотой также протекает путем замещения любой молекулы аммиака
на хлорид-ион:+ NH4CIВторая стадия этого процесса включает замещение молекулы ам¬
миака, находящейся в т^анг-положении к лиганду с наибольшим
/и/?анс-лабилизирующим влиянием, т. е. в транс-положении к СГ:+ ХН4С1Таким образом, с позиции представлений о трансвлиянин лигандов
Черняев объяснил правило Иергенсена.На основании большого фактического материала по синтезу комп¬
лексов платины (11) Черняев расположил лиганды по ослаблению
т^анс-лабилизирующего влияния (трансвлияния) в следующий ряд:NO^, SCN-, I-, Br", Cl", F-, ОН- RNH3, iNH3, Н=0ослабление трансвлиянин лигандовВпервые термин «трансвлиянне» Черняев использовал в докладе,
прочитанном 20 марта 1925 г. Опубликован этот доклад был в 1926 г.[1]. Эта дата и считается годом открытия закономерности трансвлня-
ния.NH3™ NIV+Cl NH,NH3Pt '+ 2HC1 =Pt 3ClNHi Cl186
От первоначального допущения, что трансвлияние ацидолигандов
всегда выше,чем трансвлияние нейтральных лигандов, пришлось
отказаться. Еще до открытия явления трансвлияния Курнаков уста¬
новил, что изомерные комплексы [Pt(NH3)2Cl2| по-разному реагируют
с тиомочевиной. В то время как в транс-изомере замещению на тио-
мочевпну подвергаются только хлоридные лиганды:С1,в г^с-изомере на тиомочевину замещаются все внутрисферные ли¬
ганды:NH3™ Cl+ 2Thio =NH3Thio’PtPt. ciNH3.ThioNH3.NH3CllPt+ 4Thio =NH3CIThio Thio'
Thio ThioCl2 + 2NH3Гринберг показал, что характер протекания тиомочевинных реакций
Курнакова сохраняется при взаимодействии тиомочевины с изомерны¬
ми комплексами [PtA2X2], содержащими различные амины А и ацидо-
лиганды X.Черняев был вынужден принять предположение, что трансвлия¬
ние тиомочевины выше, чем трансвлияние хлоридного лиганда. В этом
случае тиомочевинные реакции Курнакова получали объяснение.
После замещения на тиомочевину хлорид-ионов или NH3 в
(<wc-[Pt(NH3)oCl2] получаются комплексыNH3™ Thiol2+"ThiocnPtилиPtNH3Thio.ThioClв которых лиганды в транс-положении к тиомочевине оказываются
сильно лабилизированными и подвергаются замещению на тиомоче¬
вину. В комплексеNH3ThioPtThioNH3молекулы аммиака взаимно лабилизированы слабо и поэтому не под¬
вергаются замещению.Постепенно накапливались экспериментальные факты, вынуждаю¬
щие принять допущение о высоком трансвлиянии и для других нейт¬
ральных лигандов. Исключительно важное исследование на этом пути
было выполнено А. Д. Гельман. Она установила, что взаимодействие
этилена с солью K[Pt(NH3)Cl3] протекает в соответствии с правилом
Пейроне:+ Cl-Причиной такого направления реакции является большая взаимная
лабилизация хлоридных лигандов на координате Cl—Pt—Cl, чем
лабилизация хлоридного лиганда на координате NH3—Pt—Cl.
Напротив, взаимодействие аммиака с солью K[Pt(C2H4)Cl3] неNH3 СГ
3 Pt-NH3. Cll+ c2H4 =PtCl CiCSH4Cl187
соответствует правилу Пейроне. Реакция протекает в соответствии
с уравнениемс,н4сг-Pt+ NH3 =. С1ClС„Н4CIPtClNH,Cl-Такое направление реакции может означать лишь то, что хлоридный
лпганд на координате С2Н4—Pt—Cl лабилпзирован сильнее, чем на
координате CI—Pt—Cl. Это означает, что трансвлияние этилена выше,
чем трансвлияние хлорид-иона*.В последующих работах Гельман идея о повышенном трансвлия¬
нии ненасыщенных молекул была экспериментально подтверждена
на примере бутадиена, оксида углерода (11), оксида азота (II), ацети¬
лена и его производных.Иенсен установил, что при взаимодействии тноэфиров с KJPtCU
реакция протекает следующим образом:CI СП"- , „
С,Р,С1 + R’S“и CI
Cl CIH:SClRjSClPtClCl+ Cl-PtClR,SОбразование бис-тиозфирного комплекса вызвано тем, что моле¬
кула тиоэфира обладает большим трансвлиянием и хлорид-ион, на¬
ходящийся к ней в т/7аяс-положении, оказывается более лабилнзн-
рованным, чем два других. Вывод о том, что тиоэфиры обладают вы¬
соким трансвлиянием, был принят без особых возражений, так как
уже бесспорным был факт высокого трансвлияния тиомочевины. Оба
этих лиганда содержали донорный атом серы, что и явилось причиной
их высокого трансвлияния.Реакция расщепления тетратиоэфирных комплексов платины так¬
же не должна протекать в соответствии с правилом Иергенсена вслед¬
ствие высокого трансвлияния тиоэфиров. Первую стадию расщепле¬
ния тетратиоэфирных комплексов можно изобразить следующим об¬
разом:R2Sr2sPtRnSR2S+ Cl- =R-SR"-s Pt R3SR2S Cl-B комплексном ионе [Pt(R2S)3Cl]+ наиболее лабильными должны быть
лиганды, расположенные на координате R2S—Pt—R2S. Один из них
и подвергается замещению на второй стадии реакции:R:S RnS-
RoS CICl- =R2Sr2sPtClCl-h R:SРеакция образования бис-тиоэфирных комплексов из тиоэфиров
и тетрагалогеноплатиннтов, а также расщепления тетратиоэфирных* Этот подход был использован в лаборатории автора при выявлении вы¬
сокого трансвлияния диалкилсульфоксидов.188
комплексов галогенидными ионами осложнены взаимными перехода¬
ми цис- и mpaHC-[Pt(R;S)2X2) друг в друга. Поэтому с тпоэфирами
получаются результаты обычно не столь четкие, как с аминами.Поведение триалкилфосфннов в комплексах платины (II) анало¬
гично поведению тиоэфпров. Это позволило сделать вывод об их вы¬
соком трансвлиянии.К началу 50-х годов качественный ряд лигандов по трансвлиянию
для комплексов платины (II) был следующим:CN->C2H4>CO, NO“>SC(NH.,).,>R.,S, PR3, I-> Br-> Cl-> F-,NH3 > OH- > H20Закономерность трансвлияния была выявлена главным образом
при изучении комплексных соединений платины (II). Естественно, что
были предприняты многочисленные исследования для обнаружения
этого явления на комплексах других металлов. Автор закономерности
Черняев убедительно показал, что мранс-лабилизирующее влияние
лигандов проявляется и в комплексах платины (IV). Ряд лигандов
по трансвлиянию в основном сохраняется, однако трансвлияние нитро¬
группы в комплексах платины (IV) исключительно низкое.В настоящее время экспериментально установлено, что кроме ком¬
плексных соединений платины трансвлияние лигандов проявляется
в комплексах палладия, родия, иридия, рутения, осмия, рения, ко¬
бальта, железа, хрома, молибдена, вольфрама,Для каждого элемента в ккждой степени окисления существует'
свой ряд лигандов по трансвлиянию. Однако они сохраняют примерно
тот же порядок, что и для комплексов платины (II), отличаясь друг
от друга лишь положением отдельных лигандов.Закономерность трансвлияния была выявлена на основе препара¬
тивных исследований. После этого она стала руководящим принципом
для предсказания путей последовательного синтеза разнообразных
по составу комплексных соединений. В свое время Вернер на основе
координационной теории предсказал существование комплексных
соединений платины (IV) с шестью различными лигандами, однако не
смог указать обоснованных путей их синтеза. Закономерность транс¬
влияния, явившаяся важнейшим дополнением координационной тео¬
рии, позволила указать путь синтеза таких комплексных соединений.Основываясь на закономерности трансвлияния в начале 1950-х
годов, Гельман и Эссен осуществили синтез соединений платины (IV)
с шестью различными лигандами [PtClBrPyNH3I(N02)J:РуCIPtBr UNO,
NH.PvClNO,PtBrNHК IPyClNO,PtBrNH,NO,Особенно плодотворным оказалось использование закономерности
трансвлияния для синтеза геометрически изомерных соединений.
Так, согласно координационной теории, диацидотетрамминплатина189
(IV) состава [Pt(NHa)iCI2]C!2 должна существовать в виде двух гео¬
метрических изомеров. Из них был известен лишь транс-изомер (соль
Гро), получающийся окислением хлором тетрамминплатохлорида по
уравнениюNH,
NH ,PtNH iNH,Cl,Cl,ClNH jNH,PtNH j
NH,ClCl,В 1939 г. Черняев, используя закономерность трансвлияния, тео¬
ретически разработал путь синтеза и получил соответствующий цис-
изомер из триаммина [Pt(NH3)3Cl]CI:-ClNH3NH,NH,Cl,NH,1NH]NH,NHj1NH3PtPtPtN HiCINH3Cl^ iNHjClCl1|_Cl--Cl-С!,Действительно, в комплексном ионе [Pt(NH3)3Cl3]+ два транс¬
расположенных иона хлора взаимно лабилизированы, а третий ион
хлора, расположенный против молекулы аммиака, должен быть го¬
раздо менее подвижным. Поэтому в соответствии с представлением о
трансвлиянин при действии на это соединение аммиаком наиболее
легко заместится один нз лабилизированных ионов хлора с образо¬
ванием i^wc-изомера.В дальнейшем закономерность трансвлияния как руководящий
принцип при выборе путей синтеза была использована для получения
геометрических изомеров комплексных соединений Pt(IV) самого
различного типа. Так, Черняев и Муравейская для диамминов
[Pt(NH3)2(N02)2Cl2] получили все пять теоретически возможных изо¬
меров; Черняев и Адрианова синтезировали пять из шести предска¬
зываемых теорией изомеров для триамина [PtEn(i\H3)(I402)ClBr]CI.
Бабаева и Ушакова получили изомеры различных ннтрохлорокомп-
лексов K.|Pt(N02)xCI6_A:l (х = 2ч-4).8.3.2. Химические способы идентификации изомеров
платины (II)Эффектное объяснение взаимодействия тиомочевины с цис- и транс-
[Pt(NH3)2Cl2] (реакции Курнакова) с позиции закономерности транс¬
влияния стимулировало разработку других химических способов
отличия такого типа изомерных соединений. Гринберг с сотр. показал,
что нагревание диамннных комплексов типа [PtA2X2] с KI в присутст¬
вии фенолфталеина позволяет легко отличить ^«с-изомер от транс¬
изомера. Цис-изомер дает малиновое окрашивание, а в случае транс¬190
изомера наблюдается лишь пожелтение раствора. В растворе
4«c-[PtA2X2] в результате большей лабильности ацидолигандов проте¬
кают следующие реакции:A XIА 11Pt+ 210 =PtА XА IА IА 1Pt+ KI = КPtА II IА XА ГPt .-f 2К1 -PtX АI А+ 2КХВследствие высокого трансвлияния иодид-ионов молекулы аминов
также замещаются иодид-ионами:4- АПоявление в растворе амина делает среду щелочной, что и фиксируется
окрашиванием фенолфталеина:В случае m/?aHc-[PtA2X:l в не слишком жестких условиях реакция
заканчивается на стадии+ 2КХАмины слабо лабилизируют друг друга и поэтому остаются в комплек¬
се.Этот способ пригоден для идентификации изомерных аминокомп-
лексов платины (IV) типа I Pt А2Х 4 ].К лигандам высокого трансвлияния относятся тригалогениды оло¬
ва (II). например SnCh. При взаимодействии SnCb с KJPtClJ или
KJPtClrJ образуется пятикоординационный комплекс платины (II)
[Pt(SnCl3)5P_. Он характеризуется ярко-красной окраской и высоким
коэффициентом экстинкции. Действие раствора SnCl3 в соляной кис¬
лоте на комплексы типа £^c-IPtA2X2] (при нагревании в кипящей
водяной бане) практически мгновенно приводит к появлению красного
окрашивания, характерного для иона I Pt (SnCI 3) 5]3_. По-видимому, на
первых стадиях реакции ацидолиганды в комплексе замещаются на
SnCb:+ 2Х-'х<А „ SnSI3
Pt 3+ 2SnCr =А X3A SnCl3Вследствне высокого трансвлияния SnC!3 амины замещаются
SnCl3 и комплекс достраивается до пятикоордннационного:на3SnCir = [Pt(SnCl3)5F- + 2АА р( SrrCl3
A SnCI3Изомерные комплексы m/?aHC-IPtA2X2] в тех же условиях подвер¬
гаются лишь реакцииSnCl3’АX'АV Pt-г 2SnCl— =PtXА1 ^ ^ 3SnClgА+ 2Х-Получающийся комплекс имеет гораздо менее интенсивную окрас¬
ку— визуально наблюдается лишь пожелтение раствора. Замещение191
Рис. 8.1. Длинноволновые спектры:а — цшМРИтиоксанЬСУ; б — транс-[ Р1(тиок-
сан)2С12]; в — цис-[PtPy2I2]: г — транс-[PtPy,[,]аминов и образование [Pt(SnCl3)s]3'
в данном случае происходит толь¬
ко при длительном нагревании.Следует отметить, что хлорид
олова (II) с одинаковым успехом
может быть использован для иден¬
тификации как геометрических изо¬
меров платины (II) [PtA2X2], так
и для идентификации изомерных
комплексов платины (IV) типа
[PtA2XJ. В последнем случае вна- ;; чале происходит восстановлениеоловом (II) платины (IV) до плати¬
ны (II).Применение химических способов идентификации наиболее эф¬
фективно при наличии обоих геометрических изомеров. Обычно реак¬
ции проводят параллельно в одних и тех же условиях.Наиболее надежным методом идентификации галогенидных гео¬
метрических изомеров типа [PtL2X3] является длинноволновая ИК
спектроскопия. В области колебания связей платина—галоген в
спектре транс-изомер а имеется одна полоса, а в спектре ^«с-изомера —
две (рис. 8.1). Этот способ вполне надежен при наличии одного из
изомеров. Кроме того, он позволяет судить о наличии примеси одного
из изомеров в другом.На тонкослойных хроматограммах транс-изомеры как неполярные
соединения имеют гораздо большие значения Rf, чем соответствую¬
щие цис-изомеры.8.3.3. Количественные характеристики трансвлиянияРазличают трансвлияние в статическом и динамическом состояниях
комплексов. В зарубежной литературе первый случай называют
трансвлиянием, а второй — трансэффектом. Такие методы исследова¬
ния, как рентгеноструктурный анализ, колебательная спектроскопия,
методы ядерного магнитного резонанса и ядерного квадрупольного
резонанса, позволяют исследовать трансвлияние в статическом сос¬
тоянии комплекса.Кинетический метод. Исследование кинетики реакций замещения
и иных превращений лигандов, а также изомеризации комплексов
в зависимости от природы внутрисферных лигандов позволяет иссле¬
довать трансвлияние в динамическом состоянии комплексов. Синтез
комплексных соединений связан с реакциями замещения, присоеди¬
нения или удаления лигандов. Поэтому при препаративных наблюде¬
ниях проявляется кинетический аспект трансвлияния.Явление, открытое Черняевым, единое — это передача влияния
лиганда в /иранс-положение. В зависимости от выбранного метода192
исследуется та или иная его сторона. Поэтому правильнеё говорить нео трансвлиянии и трансэффекте, а о трансвлиянии статическом и
трансвлиянии динамическом.Начало исследованию количественной характеристики лигандов
кинетическим методом было положено Гельман и Карандашевой
(1952). Они изучили взаимодействие пиридина с комплексами
K[Pt(NH3)Cl2Xl. О скорости реакции судили по появлению осадка
[Pt(NH3)PyClX] в соответствии с уравнением“ NH3 "' NH3 -jCl-Pt-х1-f Py =jPy_Pt—X
tCl1ClПозже Звягинцев и Шубочкина-Карандашева кинетику этих реакций
исследовали потенциометрическим методом (см. табл. 8.1). В анало¬
гичных условиях соль K[Pt(CaH*)Cl3] реагирует с пиридином прак¬
тически мгновенно. Таким образом, количественные кинетическиеТаблица 8. 1. Кинетические характеристики взаимодействия
пиридина с комплексами К [Pt(NH3) Y,X]fc-10*. л-МОЛЬ-1-С"1Комплексный ион •20?C36° С• 3(ГС35rCЕ.кДж/мNH3
1 -Cl—Pt—Cl30,663,010818079,81ClNH3
1 -Cl-Pt—Br10817628645071 ,41ClNH3
1 -Cl-Pt—NO,400555753100346,21ClNH3
1 -Cl—Pt—NO,36851572297548,7NO,NH3
1 -N0,—Pt—NO,21632347268858,01Cl* Более жирным шрифтом выделены лиганды, подвергающиеся замещению.7-817 193
данные полностью согласуются с рядом трансвлияния лигандов, по¬
лученным на основании препаративных данных:С2Н4 > N07 > Вг_ > с1"В связи с этими первыми кинетическими работами встал вопрос о
том, что выбрать количественной характеристикой — константу ско¬
рости или энергию активации. Гринберг и Кукушкин отметили, что
реакции, результаты исследования которых приведены в табл. 8.1,
характеризуются различными температурными коэффициентами. Вы¬
числения показали, что для комплексов- NH3 --- NH3 -Cl—Pt—ClIиjCl—Pt—BrJ1_ Cl __1_ ci _константы скорости должны быть одинаковыми при 146° С, для комп¬
лексов- NH3 --- NH3 -|Cl-pt—Cl
1иICl—Pt—N031 “1 ._ ciClпри 80° С, а для комплексов- NH3 -—~ NH3 -jCl-Pt—N021И|Cl—Pt—Br1Cl1CIони должны стать равными уже при 62°С. Таким образом, на основа¬
нии констант скорости реакции, вычисленных при температурах боль¬
ших, чем 62°С, можно было сделать заключение, что трансвлияние
иона Вг' выше, чем иона NO2.Если в определенном температурном интервале механизм реакции
сохраняется, то энергия активации также не должна изменяться.
Следовательно, за меру трансвлияния правильнее принимать энергню
активации (ее относительное значение по сравнению с энергией для
реакции, выбранной за стандартную), чем константу скорости.Не вдаваясь в экспериментальные подробности, можно сказать,
что количественная характеристика трансвлияния лигандов, полу¬
ченная изучением кинетики реакции замещения, практически всегда
хорошо согласуется с результатами, полученными на основе синтеза
комплексных соединений. Несмотря на то что кинетические исследо¬
вания в каждой серии однотипных реакций давали количественную
характеристику, шкала трансвлиянин не была построена. Это связано
с тем, что относительное значение трансвлияния определенного ли¬
ганда несколько меняется в зависимости от тнпа комплекса, в кото¬
ром изучалась кинетика реакции замещения лиганда. Одной из при¬
чин этого было влияние лигандов, находящихся в цис-положенин к
замещаемому.194
Классическая реакция аммиака и K(Pt(NH3)Cl3] с образованием
z<HC-[Pt(NH3)iCl:] и расщепление соляной кислотой [Pt(NH3)3Cl]+ с
образованием mpaHc-[Pt(NH3).,Cl2] были замечательны тем, что про¬
текали только в одном направлении н долго никому не удавалось об¬
наружить ни примеси транс-изомера в хлориде Пейроне, ни приме¬
си цпс- изомер а в хлориде второго основания Рейзе. Еще в 1958 г.
это обстоятельство послужило автору основанием для вывода, что
определяющая роль в направлении данных реакций принадлежит
кинетическому фактору. Кинетика и термодинамика в одной из этих
реакций не находятся в согласии. Это следует из схемы[Pt(NH3) СЫ--— 4«c-[Pt(NH3)aCI2] kt к lPt(NH3)3Cirmpa«<r-[Pt(NH3)„CI:lЧерняев с сотр. показали, что полная энергия TpaHC-[Pt(NH3)2Cl2]
на 12,6 кДж'моль меньше, чем энергия £{«c-[Pt(NH3)2Cl2]. Рас¬
щепление [Pt(NH3)3Cl]+ соляной кислотой с образованием
mpaHC-[Pt(NH3);Cl2] протекает в соответствии с термодинамикой.
Взаимодействие же аммиака с [Pt(NН3)С13]~ с образованием
q«c-[Pt(NH3)2Cl;], а не mpaHC-fPt(NH3)2Cl2] происходит по термоди¬
намически менее выгодному йаправлению, но в соответствии с законо¬
мерностью трансвлияния. Таким образом, главную роль в направле¬
нии последней реакции, а следовательно, и в проявлении трансвлия¬
ния играет кинетика, а не термодинамика.Заметим, что небольшие примеси изомеров в этих классических
реакциях все же были обнаружены. Это было сделано методом тонко¬
слойной хроматографии [2].Рентгеноструктурный анализ. В последние годы появились много¬
численные исследования, позволяющие судить о статическом транс¬
влиянии.Давно предпринимались попытки обнаружить проявление транс¬
влияния лигандов в изменении длины связи М—X, находящейся в
транс-положении к лиганду L, по сравнению с суммой ковалентных
радиусов. Предполагалось, что увеличение трансвлияния лиганда L,
находящегося на координате L—М—X, должно приводить к удлине¬
нию связи М—X. В связи с созданием прецизионной техники повы¬
силась точность рентгеноструктурных данных и появилась возмож¬
ность построения рядов трансвлияния лигандов.Изучение структуры октаэдрических комплексовPhMe2PClС!-| |PMe,r-hМPMs,PhClмеридианного типа, где М — Re(III), Os(III), Ir(III), показало, что7* *195
связи М—Cl и М—Р, расположенные против фосфинов, длиннее тех
же связей, расположенных против хлоридных лигандов (табл. 8.2).
Из этой таблицы однозначно следует, что статическое трансвлияние
фосфинов выше, чем хлоридного лиганда.Таблица 8.2. Межатомные расстояния в соединениях типа
М (PMe2Ph)3CI3, нмСвязьRe(III)Os (III)Ir(III)М — CI (в транс- к CI)0,23530,23470,2361М — С1 (в транс- к Р )0,24540,24390,2429М — Р (в транс- к С1)0,24010,23500,2277М — Р (в транс- к Р )0,24580,24080,2363В табл. 8.3 приведены длины связей Pt—Cl в различных комп¬
лексах.Т а блица 8.3. Длина связи Pt—Cl в комплексах платины (II)КомплексДонорныЙ
атом транс¬
лигандаДлина связи,
нм[Pt (Acac*)2Cl] -о0,228 + 0,001транс- [Pt (PEt3)2Cl4]С10,230+0,001(C12H17)2Pt2Cl,С=С0,231 -0,001цис- [Pt(PEt3)2CI о]р0,237^:0,001транс- [Pt (PMe2Ph)o Cl (CH2SiMe3)]с0,2415+0,0005транс- [Pt (PPhjEt)2Cl (H)]н0,242+0,001транс-lPt (PPhMe2)2(SiPh2Me) Cl]Si0,245+0,001* Асас — СН3СОСН=С<NCH3Таким образом, на основе приведенных в табл. 8.3 рентгенострук¬
турных данных ряд статического трансвлияния выглядит следующим
образом:SiPh2Me- > CH2SiMe~ ~ Н" > PEt3 > -С=С—, С1" > Асас”Колебательная спектроскопия. Как известно, силовые константы
могут служить мерой прочности химической связи. При сравнении
комплексов одного и того же типа вместо силовых констант часто
используют частоты валентных колебаний химических связей. Обыч¬
но принимают, что чем меньше частота колебания связи металл—ли¬
ганд, тем слабее связь.196
Частота колебания связи, которая служит индикатором, должна
зависеть от массы лиганда, находящегося в транс-положении (лиган¬
ды, расположенные в чис'положенин, обычно одинаковы).Поскольку масса фрагмента LM изменяется, то это должно найти
отражение в частоте колебания связи А1—X, даже если силовая кон¬
станта этой связи останется неизменной. Часто этим осложнением
пренебрегают. Для тяжелых атомов-комплексообразователей по¬
правка на изменение массы лиганда L, как правило, небольшая и ею
действительно можно пренебречь. Другое осложнение в изучении
трансвлияния методом колебательной спектроскопии заключается
в возможности взаимодействия между колебаниями индикаторной
связи М—X с другими молекулярными колебаниями. Однако счи¬
тают, что этим взаимодействием также можно пренебречь, если соот¬
ветствующие фрагменты молекулы разделены тяжелыми атомами,
такими, как платина.Использование твердых препаратов для снятия ИК спектров также
может осложнять картину. В этом случае спектры могут содержать
полосы, которые отсутствуют при исследовании тех же веществ в
растворах, что является следствием колебаний кристаллической ре¬
шетки. Иногда же в ИК спектрах твердых веществ имеет место рас¬
щепление отдельных полос поглощения.Однако, как правило, имеется хорошее совпадение спектров комп¬
лексов, снятых и в растворе, и в твердом состоянии. Следует иметь
в виду, что для комплексов электролитного типа внешнесферные ионы
могут оказывать сильное влияние на частоту колебания связи металл—
лиганд.В 1955 г. Дж. Чатт и сотр. выдвинули гипотезу, по которой транс¬
влияние лиганда связывалось с его способностью образовывать да¬
тивную л-связь с центральным атомом. Эту гипотезу авторы основы¬
вали на информации, полученной при изучении ИК спектров погло¬
щения соединений платины, содержащих в координационной сфере
аммиак. Данное исследование явилось не только важным этапом в
разработке теории трансвлияния, оно было практически первым в
прямом изучении трансвлияния методом ИК спектроскопии. Дж.
Чатт, Л. Данкансон и Л. Венанци предположили, что различные
лиганды в транс-позиции к координированной молекуле аммиака
должны вызывать в ней изменения, в частности в характере связей
N—И. По значениям частот валентных колебаний связей N—И и их
интенсивности можно судить о величине влияния, испытываемого ам¬
миаком со стороны лиганда L, находящегося в транс-положении. Ав¬
торы провели серию определений частот валентных колебаний связи
N—И и установили порядок их уменьшения с изменением транс-ли¬
ганда L. Этот ряд выглядит следующим образом:PR3 > SbR3 > P(OR)3 > AsR3 > TeRn > C2H4 > SeR2 > SR2 > Pipгде R — органический радикал, Pip — пиперидин.Было сделано заключение, что понижение частоты валентных
колебаний связи N—Н отражает уменьшение электронной плотности
на атоме азота, которая уменьшается тем сильнее, чем прочнее связь197
N—«-Pt, т. e. чем выше электроноакцепторная способность атома пла¬
тины. Так как акцепторная способность атома платины по отношению
к электронам молекулы аммиака изменялась антнбатно донорной спо¬
собности лиганда L, то приведенный ряд отражал последовательность
понижения электронодонорных свойств лигандов L. Существенным в
этой последовательности было то, что она не совпадала с известным ря¬
дом трансвлияния, полученным кинетическими методами. Получалось,
что сильно шранс-активные лиганды, например С2Н4> незначительно из¬
меняют частоту валентных колебаний связи N—Н. На основании
наблюдаемых слабых и непоследовательных смещений полос погло¬
щения лигандов различной транс-активности Чатт пришел к заклю¬
чению, что связь Pt—N либо совсем не ослабляется под влиянием
транс-лиганда, либо ослабляется только в том случае, когда транс¬
активность аммиака и транс-лиганда приблизительно одинакова.
Другими словами, прочность связи не является определяющим мо¬
ментом в механизме трансвлияния. Однако этот важный вывод был ос¬
нован на интерпретации значений частот v(NH) и поэтому носил кос¬
венный характер. Требовалось дополнительное подтверждение этого
заключения путем измерения каких-либо свойств связей Pt—N, не¬
посредственно зависящих от их прочности.В 1961 г. А. В. Бабаева с сотр. предложили новый подход к оценке
трансвлняния лигандов. Метод, используемый Чаттом, основанный
на изменении валентных колебаний связи N—Н, из-за высокой сте¬
пени характеристичности частот v(NH) и отсутствия прямой зави¬
симости последних от силовых постоянных связи Pt—N не может
служить «индикатором» прочности связи Pt—NH3. Наиболее чувстви¬
тельными к изменению прочности связи Pt—NH3 являются частоты
не валентных, а маятниковых колебаний аммиака Pnh,- Эти частоты
в г<ис-нзомерах диацидоаминов платины (II) оказались ниже, чем в
транс-изомерах, что указывает на ослабление связи Pt—NH3 под
влиянием ацидогруппы. При переходе от хлорида к иодиду в ряду
^ыс-изомеров [Pt(NH3)2Xsl частоты маятниковых колебаний аммиака
уменьшаются в следующем порядке:I- > Rr" > N0“ ~ SCN- > Cl" > NH3Этот ряд, однако, для отдельных лигандов также не совпадал с кине¬
тическим рядом трансвлияния. Так, роданэ- и нитрогруппы, по дан¬
ным ИК спектроскопии, должны обладать сравнительно небольшим
трансвлиянием. Эго несоответствие объяснялось тем, что в реакциях
замещения в соединениях, содержащих данные группы, вероятно,
имеет место не одно только ослабление связей, но и другие эффекты,
усложняющие механизм трансвлияния.Пауэлл одним из первых изучал трансвлияние лигандов по поло¬
жению полосы валентных колебаний связи Pt—NH3 в спектрах комп¬
лексов типа /гаракс-[Р1Ь(МНз)С12]. Он установил, что v(Pt—NH3)
изменяется в ряду изученных лигандов L следующим образом:NH3 (507 см-1) > (C2H5)i S (493 см*1) > С2Н4 (481 см-1)Накамото с сотр. исследовали v(Pt—NH3) в спектрах изомерных198
соединений [Pt(NH3)3X:l. Они установили, что при изменении X от
СГ к Вг" и Г происходит понижение частоты валентных колебании
связи Pt—N как для цис-, так и транс-изомеров. Однако в случае
изомеров цнс-конфнгурации изменение частот выражено сильнее. Ав¬
торы приписали это различному трансвлиянию галогенид-ионов.Аналогичная картина наблюдается для метиламиновых изомерных
соединении [Pt(NH2CH3)2X2l. Для комплексов транс-конфигурации
при изменении X частота колебания связи Pt—N лежит в узком ин¬
тервале (507—513 см'1)- Для соединении (<«с-конфигурации интервал
частот гораздо более широкий (483—517 см-1). Частота колебаний
связи Pt—N уменьшается в зависимости от лиганда X в следующем
порядке:СГ > N07 > Br- > SCN- > ГЗа исключением нитрогруппы, этот ряд хорошо совпадает с кинетиче¬
ским рядом трансвлияния. Исключительное положение группы N02-
возможно, связано с взаимодействием колебаний связей Pt—N02 и
Pt—NH2CH3.В работе Клегга и Холла было установлено, что в КР спектре
водного раствора |Pt(CH3)3(NH3)3]Cl v(Pt—NH3) равно 390 см-1. Оно
значительно ниже частоты аналогичного колебания связи Pt—N в
спектре комплекса [Pt(NH3)e]4+ (545—569 см-1). Это связано с высо¬
ким трансвлиянием СНз.Исследование трансвлияния лигандов по колебанию индикаторной
связи металл—гидрид было начато Чаттом. Впоследствии к этой за¬
даче обращались многие исследователи. Оказалось, что полосы, соот¬
ветствующие колебаниям >(Pt—Н), наиболее чувствительны к раз¬
личного рода осложнениям. В комплексах катионного типа положе¬
ние полосы (Pt—Н) сильно зависит от природы растворителя. Имеет
место значительное колебательное взаимодействие между v(Pt—И)
и близкими по частотам к v(Pt—Н) колебаниями лиганда, находяще¬
гося в транс-положении к гидридному иону. Это особенно характер¬
но для гидридных комплексов,содержащих такие лиганды, как CN-,
СО, RNC. Гидридные комплексы часто подвергаются внутримолеку¬
лярным превращениям.Эпплетон, Кларк и Манзер на основании значений v(Pt—И), по¬
лученных для комплексных соединений типа mpaHC-[Pt(H)XL2] и
транс-1 Pt(H)L'L2]+, дают следующий ряд трансвлияния лигандов X
или L':С10~ < NO- ~ CF3COO- < /!-СН3С6Н4СОО- < NCO” — NCS- — N“ ^ СГ —~ NH3 ~ Ру ~ NHnCHg < I- SCN- < Thio ~ N0“ < СО — SbPh3 << mpem-C4H9NC < SnCl~< PPh3 < P(OPh3)3 S PE t3< P(OCH3)3 — CN- <C=CPlrОн только в общих чертах напоминает кинетический ряд трансвлия¬
ния.Чатт и Гайтер изучили также ряд дифосфиновых комплексов ру¬
тения (И) и осдоия (И) типа mpaHC-[M(H)X(PPh2R)2l. На ос-199
новации значений частот колебаний связи М—Н их ряд лигандов по
увеличению трансвлияния имеет вид
I- < Br- < Cl" < SCN- < N07 < CN- < Н~Если порядок псевдогалогенидных ионов и нитрогруппы совпадает
с порядком лигандов в кинетическом ряду трансвлияния, то для га-
логенид-ионов ряд оказывается обращенным.Чатт с сотр. одним из первых начал изучение трансвлияния раз¬
личных лигандов по частоте валентного колебания связи Pt—С. Он
установил, что частота колебания связи Pt—С в комплексах типа
m/7aHC-[Pt(CH3)X(PR3)2] зависит от природы лиганда X, находящего¬
ся в транс-положении к метильной группе. Значение v(Pt—СН3)
понижается в ряду лигандов X в следующем порядке:N0~ (566 см-1) > NCS" (556 см-*) > CI- (551 см'1) > Вг- (548 см"1) >> N0“ (544 см-1) > I- (540 см"1) > CN" (516 см'1)Этот ряд довольно хорошо согласуется с кинетическим рядом транс¬
влияния лигандов, если принять, что роданидная группа связана с
центральным атомом посредством атома азота.Частота колебания связи Pt—С в £{ttc-[Pt(CH3)Cl(PEt3)2] равна
527 см-1, что значительно ниже частоты аналогичной связи в
mpaHC-[Pt(CH3)Cl(PEt3)2]. Этот факт хорошо объясняется тем, что
трансвлияние триэтилфосфина значительно выше трансвлияния хло¬
рид-иона, находящегося против метальных групп.Сравнение колебательных спектров q«c-[Pt(CH3)2(PEt3)2] и
4«c-[Pt(CH3)2(EtSC2H4SEt)] показывает, что полосы колебания свя¬
зей Pt—С в первом случае имеют значения 526 и 506 см-1, а во вто¬
ром — 555 и 548 см-1. Эти факты свидетельствуют о более высоком
трансвлиянии дитиоэфира по сравнению с триэтилфосфином.Исследования частот колебаний связей Au—С и частот деформа¬
ционных колебаний метильной группы в ионе [Аи(СН3)2Х2Г пока¬
зывают, что для комплексных соединений золота (III) ряд трансвлия¬
ния тот же, что и для платины (II):(CH3)2S > Вг~ > С1“ > Еп > ОН- :> Н2СЗамещение двух галогенов в плоскоквадратном ионе А11Х4 (X —
СГ или Вг-) на два карбаниона СНз приводит к образованию комп¬
лексов только ^«с-конфигурации и понижает силовую постоянную
связи Au—X более чем на 30% . Эти данные также свидетельствуют огораздо более высоком трансвлиянг;.1 группы СНз по сравнению с
СГ или Вг”.Обширный экспериментальный материал, полученный Дж. Чат-
том с сотр. по влиянию внутрисферных лигандов на связь Pt—Cl,
позволил прийти к выводу, что трансвлияние обусловлено по край¬
ней мере двумя причинами: a-электронным смещением от лиганда к
центральному атому и л-электронным смещением от центрального
атома к лиганду. a-Электронное смещение, по-видимому, является
главной причиной высокого трансвлияния гидридов, алкильных и200
Рис. 8.2. Спектроскопическая шкала трансв¬
лияния лигандов, составленная на основе
комплексов тнпа r(Hc-[PtL2Cl2]арильных карбаннонов, тогда как л-
электронное смещение характерно для
олефинов. о-Электронный механизм пе¬
редач» трансвлияния должен способст¬
вовать ослаблению транс-расположеп-
ной связи в статическом состоянии мо¬
лекулы. л-Электронный механизм пере¬
дачи трансвлияния, напротив, должен
приводить к упрочнению противополож¬
ной связи.На основании частот валентных колебаний связей Pt—Cl и Pt—Вг,
полученных Адамсом, Чатт составил спектроскопическую шкалу
трансвлияния лигандов. Дополненная данными, полученными в ла¬
боратории автора, эта шкала приведена на рис. 8.2.По сравнению с аммиаком и пиридином — лигандами слабого
трансвлияния, фосфины и арсины понижают прочность противопо¬
ложных связей. Диэтилсульфид, диэтилселеннд и особенно цикло¬
октадиен (Cod), проявляющие сильное динамическое трансвлияние
по сравнению с фосфинами, приводят не к ослаблению, а, наоборот,
к закреплению противоположной связи. В этом факте Басоло и Пирсон
видят несоответствие с другими экспериментальными данными. Од¬
нако если иметь ввиду, что эта шкала относится к трансвлиянию в ста¬
тическом состоянии молекулы, то никакого несоответствия не будет.
Благодаря л-акцепторным свойствам сульфидов и особенно олефинов
должен повышаться эффективный заряд на атоме платины, а это
должно приводить к усилению кулоновского взаимодействия централь¬
ный атом—лиганд.Экспериментальные данные и результаты квантово-химических
расчетов свидетельствуют о том, что ди ал кил сульфоксиды обладают
довольно сильными л-акцепторнымн свойствами. В связи с этим было
интересно установить их место в шкале трансвлияния Чатта.
В табл. 8.4 приведены значения частот колебаний связей Pt—Cl в
комплексных соединениях типа цис- и mpaHC-tP^RoSObCU.Табл. 8.5 составлена для цис- и транс-[РЩДМСО)С12], где L —
амины или олефины.Таблица 8.4. Значения частот валентных колебаний (в см-1) связей
Pt—Cl в комплексах цис- и транс-[Pt(R2SO)2C 12]^340320300280—Cod-AsMe,-AsEtj |-TcPr,
TcMci
TeEt i-AsPr, *- -PMCj £-ДПСО-ДЭСО-ДМСОL-Dipy-Py~N!f3-EnдмсоДЭСОдпсоДизо-ПСО(цис-)(цис-)цис-т ране-(транс-)333342350355353309322326——• RfSO — это ДМСО, ДЭСО, ДПСО или Дизо-ПСО.201
Таблица 8.5. Значения частот (в см-1) валентных колебаний связей
Pt—Cl в комплексах цис- н транс- [РЩДМСО)С12]LN11-NH,CH,NH,C,H,С,Н,сан,цис-транс-цис-транс-цис-транс-(цис-)(цис-)3463343.38338334312344313348312319311306Шкала трансвлпяния Чатта (см. рис. 8.2) построена на основании
данных для комплексов ^мс-конфигурацин. Из приведенных данных
вытекает следующее.1. По суммарному влиянию cr-донорных и л-акцепторных свойств
сульфоксиды близки к олефинам, в частности, к циклооктадиену.2. Сравнение положения диэтилсульфида и диэтилсульфоксида
показывает, что сульфоксиды в большей степени способствуют за¬
креплению хлора в транс-положении. Наличие кислорода при атоме
серы должно повышать эффективный заряд атома сульфоксидной серы
по сравнению с сульфидной. Методом рентгеновской эмиссионной
спектроскопии действительно показано, что эффективный положи¬
тельный заряд на атоме серы в сульфоксиде выше, чем в сульфиде.
Такой же вывод был сделан на основании кислотных свойств анало¬
гичных сульфидных и сульфоксидных соединений. Более высокий
положительный эффективный заряд может являться причиной не¬
сколько больших л-акцепторных свойств сульфоксидов по сравнению
с соответствующими сульфидами. Однако эффективный заряд не един¬
ственная причина, которая может сказываться на л-акцепторных
свойствах лигандов.3. В смешанных соединениях диметилсульфоксид—амин и диметил-
сульфоксид—олефин г^с-конфигурацин среднее арифметическое час¬
тот колебания связей Pt—CI примерно то же самое, что и для ди-
сульфоксидных соединений. По-видимому, при наличии в комплек¬
сном соединении двух сильных л-акцепторов доля участия каждого из
них во взаимодействии с d-орбиталями платины уменьшается в такой
степени, что их суммарное влияние на центральный атом остается при¬
мерно таким же.Следует отметить, что для соединений транс-конфигурации, не¬
смотря на большое различие в природе лигандов, частоты валентных
колебаний связей Pt—Cl остаются практически постоянными и на¬
ходятся в интервале 339±3 см'1. Поскольку транс-диолефиновые со¬
единения неизвестны, то они не могут быть представлены в этой шкале.
Было бы полезно дополнить данный ряд соединений комплексами ти¬
па /npaHc-[PtA(C2H4)Cl2].В ряду соединений транс-конфигурации дисульфоксидные со¬
единения занимают аномально высокое положение. Смешанные амин-
сульфоксидные соединения характеризуются несколько меньшими
частотами колебании связей Pt—Cl. Необходимо отметить, что во202
всех изученных соединениях частоты валентных колебаний PI—С1
в соединениях транс-конфигурации выше, чем в соответствующих
соединениях г^с-конфигурации. Возможно, это объясняется более вы¬
сокой симметрией распределения зарядов. На основании колебания
связей Pt—Cl в комплексных соединениях m/7aHC-lPt(PMe2Ph):LCl]
лиганды L по трансвлиянию расположены в следующий ряд:С1-, СН“, (CH3)2SiCH-, Ph2MeSi"Прп переходе от СГ к Ph:MeSi“ частота колебания транс-распо-
ложенной связи изменяется от 370 до 242 см-1. Этот ряд хорошо со¬
гласуется с результатами по трансвлнянию данных лигандов, полу¬
ченными методами ПМР и рентгеноструктурными исследованиями.На основании частот валентных колебаний Pt—Cl в комплексных
соединениях [Pt(AsEt3):LCl]C104 построен следующий ряд транс¬
влияния:СО > RNC > ArNC > P(OPh)3 — Р(ОМе)3 > AsEt3 ~ PEf3Методом ПК спектроскопии удалось обнаружить проявление
трансвлияния в комплексных соединениях иридия (III). Исследова¬
ние спектров реберных соединений 11г(РМе:РН)3ЬС1о] показало, что
в ряду L — СГ, PR3, AsR3, Н" происходит закономерное понижение
частоты 1г—С1, что может говорить об относительном уменьшении
прочности данной связи. Это находится в соответствии с изменением
трансвлияния лигандов: Н~> PR3 ~ AsR3 > СГ. В этой же работе
установлено, что в ряду лигандов L — СГ, Н", Вг",Г, SCN-, CN-
практически не происходит изменения частот колебаний связей Ir—С1
в ^ис-гтоложении.При изучении октаэдрических комплексных соединений родия
(III) и иридия (III) установлено, что частоты валентных колебаний
связей Rh—Cl и Ir—Cl зависят главным образом от природы транс¬
расположенных лигандов. Частоты v(M—Cl) увеличиваются в ряду
лигандов в следующем порядке:С1- < Вг- < I- ~ СО < СН~ ^ PR3 ^ AsR3 < Н"Метод ядерного магнитного резонанса. В 1960-х годах в коорди¬
национной химии начал широко применяться метод ядерного маг¬
нитного резонанса. Исследования были начаты с комплексов плати¬
ны. Этому способствовал тот факт, что в природной смеси изотопов
платина-195, ядро которой имеет квантовое спиновое число / = V2,
составляет окрло 34%. Естественно, что одним из первых вопросов,
который пытались решить с помощью метода ЯМР, было проявление
трансвлияния лигандов в таких характеристиках, как константа спин-
спНнового взаимодействия ядер и химический сдвиг.Ядро фосфора-31 также весьма удобно для исследования соедине¬
ний методом ЯМР. Поэтому комплексы с фосфорсодержащими лиган¬
дами были в числе первых, которые подвергались изучению этим ме¬
тодом. При исследовании изомерных фосфиновых комплексов
[Pt(PR3)2Cl2] было установлено, что взаимодействие между 195Pt и
31Р сильнее в изомере цис-конфигурации, чем в транс-изомере. Это203
могло быть связано с более высоким трансвлиянием фосфинов по
сравнению с хлорид-ионом. С этим выводом согласуются и результаты
работы Аллена и Пидкока. Они установили, что имеется большое раз¬
личие в константе спин-спинового взаимодействия ^Pt—р Для атома
фосфора, расположенного против хлоридного иона (3454 Гц), и три-
бутилфосфина (2270 Гц) в комплексе [Pt(PBu3)3Cl]+.В комплексе ((мс-[Р1(СН3)С1(РЕ1л)21 константа спин-спинового
взаимодействия 195Рt и 31Р, находящегося в транс-положении к ме-
тильнон группе, равна 1719 Гц, а в транс- положен и и к хлорид-иону
4179 Гц. Это свидетельствует о большем .статическом трансвлиянии
метнльной группы по сравнению с хлорид-ионом.Анализируя большой экспериментальный материал, полученный
методом ЯМР, Аллен и Зже расположили лиганды по их статическому
трансвлиянию в следующий ряд:С0Н- > СН- > Р(С2Н6)з, Р(«-С4Н0) > Р(СН3)2СвН5 > Р(СвН5)з >> Р(ОС6Н5)3, CN- > As(C2H5)3 > NO" > NHoC*H5 > МН(СаН4), >>Py, Nr, NCO“, NCS" > Cl", Br-, 1- > ONCT3 2Константа взаимодействия Vpi-p гораздо менее чувствительна
к замене лигандов в ^«с-положении к атому фосфора. Тем не менее,
исходя из результатов, полученных для трех серий комплексов:транс- [Pt(H)X(PEt3)2], тра :с-[Рt(CH3)X(РЕt3)2], mra«c-[Pt{(PhO)a PO}X(PEf3)aможно построить следующий ряд цисвлияния лигандов по увеличе¬
нию '/pt-p: 'CN-, 1->NCS-, Br->NCO-, Cl" > N~ > N0“ > 0N07Результаты, полученные для комплексов ч«с-[Р1ЬР(«-С1Нэ)зС12], поз¬
воляют расположить лиганды L в ряд цисвлияния:(PhO)3P > (СН30)3Р > PPh3 > PCH3Ph2 > Р(я-С4Н9)3, PEt3Многочисленные исследования гидридных и метильных производ¬
ных платины (II), а также метильных комплексов платины (IV) поз¬
волили расположить лиганды по значениям 1Ур[_н и Vpt-cH, •
Эти ряды в общем виде напоминают кинетические ряды трансвлияния,
однако имеются и значительные отклонения. Фосфины, фосфиты, изо¬
нитрилы, нон CN- и оксид углерода (II) находятся в числе лигандов,
обладающих наибольшим статическим трансвлиянием. Среди лигандов
низкого трансвлияния находятся галогенидные, ннтратный, перхло-
ратный и азидный ионы, а также ацетон. Алифатические амины, пири¬
дин и его производные занимают в этих рядах среднее положение.Константы спин-спинового взаимодействия мало чувствительны к
электроотрицательности лиганда L, находящегося в транс-положе¬
нии к индикаторной группе Y на координате L—М—'Y. Считают,
что в этом заключается основная причина довольно сильного отли¬
чия рядов трансвлияния, полученных методом ЯМР, по сравнению
с рядами трансвлияния, полученных другими методами.204
Для характеристики явления трансвлияния довольно широко ис¬
пользуются химические сдвиги гидридного иона. Поскольку
химический сдвиг гидридного иона, например в комплексах
mpaHC-[Pt(H)X(PEt3)2], зависит от ядра атома платины, то химический
сдвиг wept также должен иметь отношение к явлению трансвлпяния.Если одним из факторов, от которого зависит трансвлияние ли¬
ганда X, является ковалентность связи М—X, то должна иметь место
корреляция между значением химического сдвига для ядер 19SPt
и трансвлнянием лиганда X. Ряд статического трансвлпяния, постро¬
енный по. этому принципу,RCOj < NO3 < NOj < Cl' < SCN' < CN" < I~весьма похож на кинетический ряд трансвлияния.Пауэлл и Шоу установили, что химический сдвиг тн в комплек¬
сах mpaHC-[Pt(H)X(PEt3)ol хорошо согласуется с кинетическим транс¬
влиянием лигандов X. Исключение составляет лишь случай, когда Х =
= NCVПаршалл использовал химические сдвиги для 19F в фторфенил-
производных для оценки относительного вклада я- и ст-составляющих
трансвлияния лигандов.Подход к решению этой задачи заключался в том, что в .и-заме-
щенных фторфенильных соединениях химический сдвиг для ядер
19F должен зависеть только от а-индуктивного эффекта заместителяВ n-фторфенильных соединениях, химический сдвиг должен за¬
висеть как от о-индуктивного, так и я-нндуктнвного эффекта за¬
местителя Z. Это связано'с наличием в равновесии форм II и III:Таким образом, разность 6F (пара)—SF (меТа) может быть принята
за л-донорную составляющую ароматического кольца.Паршалл взял в качестве заместителя Z-фрагмент Pt(PEt3)oX:Z:—z\=//FF—ZИ111PEt3F—.205
По разности 6р (пара)—6Р(мета) Паршалл определил л-составляю-
щую лиганда X. л-Донорные свойства ароматического кольца были
определены на примере комплекса, где X — СНз. Считается, что этот
лиганд не обладает ни я-донорными, ни л-акцепторными свойствами.
Значение 6F (мета) использовалось в качестве меры ст-индуктивной
составляющей. В результате был получен следующий ряд лигандов
по ст-пндуктивному вкладу:СН3 > СсН5 > PEt3 > n-FCeH4 > Ph—С = С—>'-m-FC6H4 >> NCO' > CN~ > Cl- > Br' > NCS" > I- Sn Cl3Ряд лигандов по л-акцепторному вкладу выглядит следующим об¬
разом:CN' > SnCl, > — CssCPh > CeH“ , CeH4F- > NCS"Использование этого подхода привело к заключению, что галогенид¬
ные ионы должны обладать л-донорными свойствами.В настоящее время большое внимание уделяется изучению взаим¬
ного влияния лигандов в неплатиновых комплексах. Так, по значе¬
ниям химического сдвига и констант спин-спинового взаимодействия
в соединениях вольфрама и молибдена Ю, А. Буслаев и сотр. опре¬
делили прочность связей различно координированных атомов фтора
в комплексном ионе:При этом было найдено, что аксиальный кислород оказывает замет¬
ное влияние на транс- и цис-расположенные лиганды. Это обнару¬
живалось по изменениям значений констант взаимодействия Av-f
при переходе от октаэдрической конфигурации комплекса [WFJ и
пирамидальной [WO..F4]2_ к бипирамидальной [\V0(02)F4p-. Работы
Буслаева показали, что метод ЯМР может быть хорошим индикатором
возмущений, которые претерпевают транс- и цис-связи металл—ли¬
ганд по отношению к трансактивному лиганду.8.3.4. Теоретические толкования явления трансвлиянияЭволюция теоретических воззрений на т*рансвлияние во многом свя¬
зана с эволюцией общих представлений о природе химической связи.
К сожалению, пока еще не разработана единая и всеохватывающая
теория, описывающая это сложное и многообразное явление. Поэтому
целесообразно отразить наиболее важные вехи на пути теоретического
объяснения передачи влияния лиганда в транс-положение.F206
Рис. 8.3. Схема трансвлияния по Некра¬
совуПоляризационные представления х (+Гринберга—Некрасова. В 30-х годах
в химии широко использовались поля¬
ризационные представления. А. А.Гринберг и практически одновремен¬
но Б. В. Некрасов применили поля¬
ризационные представления для объяснения трансвлияния лигандов
в комплексных соединениях.В 1935 г. Некрасов предложил поляризационную модель транс¬
влияния, исходя из представлений о диполе, индуцированном в цент¬
ральном атоме. Если в плоскоквадратном комплексе все четыре ли¬
ганда одина^рвы, то в каждом из них под действием заряда нона
металла индуцируется одинаковый диполь. Это приводит к дополни¬
тельному, но равному «закреплению» всех лигандов. В таком случае
в центральном атоме результирующий диполь не возникает, и поле
его остается изотропным. При замене одного лиганда на другой,
(рис. 8.3) в центральном атоме возникает результирующий диполь, тем
больший, чем сильнее отличаются X и Y по своим поляризационным
свойствам и чем легче деформируется центральный атом. Например,
если взаимная поляризация Y и центрального атома больше, чем цент¬
рального атома и X, то диполь направлен так, как показано на
рис. 8.3, б; если она меньше — в противоположную сторону. Поэто¬
му поле центрального атома становится анизотропным, что, в свою оче¬
редь, сказывается на прочности связи отдельных лигандов с металлом.
Так, оба лиганда X, расположенные в г^с-положении к Y, практиче¬
ски не испытывают действия этого поля, а транс-связъ X с металлом
окажется либо ослабленной, либо, наоборот, усиленной. Б. В. Не¬
красов подчеркивал, что аналогичные соображения полностью при¬
менимы и к комплексам октаэдрической конфигурации.Концепция поляризуемости позволила сформулировать несколько
критериев, необходимых для проявления трансвлияния:а) трансвлияние проявляется только тогда-, когда центральный
атом (ион) обладает сильными поляризационными свойствами;б) трансвлияние разных лигандов определяется их поляризуе¬
мостью;в) трансвлияние сказывается не только на силе связи групп, но
и на всех свойствах комплекса в зависимости от степени поляризации.Эти критерии существования трансвлияния были в согласии с
экспериментальными данными относительно простых лигандов, где
имелась возможность сопоставления способности лиганда к транс¬
влиянию с величиной его рёфракции. Однако при попытке распро¬
странения представлений Гринберга—Некрасова на более сложные
лиганды отчетливый параллелизм между поляризуемостью (рефрак¬
цией) и трансвлиянием оказался не всегда выдержанным. Поляри¬
зационная модель трансвлияния не могла объяснить повышенное
трансвлияние таких лнгандов, как NO , CN", С2Н4 и другие олефнны.207
Гипотеза А. А. Гринберга о взаимосвязи трансвлияния с восста¬
новительными свойствами лигандов. В 1943 г. Гринберг предпринял
попытку объяснить трансвлпяние лигандов с позиции их восстанови¬
тельных свойств. Эта попытка, в сущности, была синтезом гипотезы
Гринберга—Некрасова и концепции лигандрв — «акцепторов элект¬
ронов», предложенной Гельман (см. гипотезу Чатта—Оргела). Грин¬
берг подчеркивал, что его попытка является некоторым возвраще¬
нием к мысли Черняева о большом трансвлиянии свободного элект¬
рона, развитой в виде общего положения.В поисках общего признака у лигандов, обладающих трансвлия¬
нием, Гринберг пришел к заключению, что таким признаком явля¬
ются их восстановительные свойства. Нарастанию восстановитель¬
ных свойств, определяемых подвижностью электронов, соответство¬
вало и нарастание трансвлияния соответствующего лиганда. Приняв
это за правило, Гринбергу удалось объяснить большое трансвлияние
«классического носителя трансвлияния» — N02, и таких сложных
лигандов, как SCN", S20;T> С:Н4, а также CN", СО, N0.Таким образом, Гринберг установил параллелизм между интен¬
сивностью восстановительных свойств (донорных свойств) и величиной
оказываемого лигандом трансвлияния. Этот параллелизм должен был
быть закономерным при условии правильного выбора критерия ин¬
тенсивности восстановительных свойств. Эта концепция находилась
в удовлетворительном согласии с многими экспериментальными дан¬
ными. Она не потеряла своего значения до настоящего времени, хотя
и носит сугубо качественный характер.Идея Гринберга о зависимости трансвлияния лигандов отлх окис¬
лительно-восстановительных свойств использована Д. В. Сокольским
и Я. А. Дорфманом. Они исходят из широко распространенного пред¬
ставления, что реакции замещения в квадратных комплексах платины
(И) протекают через образование пятикоординационного интермеди¬
ата:L L ,А LЗадачу количественной характеристики трансвлпяния Сокольский
и Дорфман сводят к оценке факторов, облегчающих повышение коор¬
динационного числа и облегчающих разрыв связи платины (II) с ухо¬
дящим лнгандом X. Этими факторами, по их мнению, являются «глу¬
бина» донорно-акцепторного взаимодействия и степень дативного пере¬
носа электронов. Первый фактор они оценивают окислительно-восста¬
новительными потенциалами лигандов и центрального атома, а вели¬
чину дативной связи определяют сравнением констант устойчивости
комплексов, образованных центральным атомом в окисленном и вос¬
становленном состояниях. Ряд трансвлпяния, полученный на осно¬
вании выведенной формулы, совпадает с экспериментально установ¬
ленным.208
Гипотеза «цис-закрепления» Я- К. Сыркина. В 1948 г. Сыркнн
рассмотрел некоторые квантово-механические аспекты направлен¬
ности атомных орбиталей в явлении трансвлияния для плоскоквад¬
ратных комплексов платины (II). В 'ионе [Р1СЫ2" все связи Pt—Cl
равноценны и в значительной степени ковалентны. Равноценность
энергий связи и ковалентного состояния нарушается при замене хлора
другим лигандом. Сыркин считал, что в явлении трансвлияния необ¬
ходимо рассматривать неравноценность связей вокруг центрального
атома. Так как центральный атом платины имеет два электрона (s и d),
способных принять участие в ковалентных_ связях, то, по мнению
Сыркина, имеется возможность sd-гибрндизации их орбиталей. Мыс¬
лимы четыре варианта направленности этих гибридных орбиталей,
т. е. четыре резонансные структуры для PtX3Y*:YIX-PtXIXIX Pt —XXIIYX—PtIXIIIX Pt—XXIVИменно способность этих ковалентных связей образовывать прямой
угол позволяет, как полагал Сыркин, понять механизм трансвлня-
ния. Та$, если в комплексном соединении [Pt(NH3)3Cl]+ связь Pt—Cl
более ковалентна, чем Pt—NH3, то в приведенных резонанснык струк¬
турах состояния I и II представлены больше, чем в III и IV. Более ко¬
валентно связанный хлор благодаря угловой гибридизации типа sd
вовлекает в связь и существенно упрочняет аммиаки, расположенные
в г{ис-положении. Отсюда выходит, что из трех молекул NH3 та, кото¬
рая находится в транс-положении к С1, наиболее лабильна.Гипотеза Сыркина явилась принципиально новым объяснением
закономерности трансвлияния. Она указывала, что транс-ослабление
является кажущимся эффектом на фоне стабилизации (упрочнения)
связей в цис-положении. Отрицание прямого транс-ослабления связи
серьезно подрывало укоренившиеся взгляды на трансвлияние. Оно
требовало не только замены формулировки, но и изменения термина
«трансвлияние», как такового, поэтому встретило возражения ученых.Гипотеза que-закрепления Сыркина не согласовалась с экспери¬
ментальными данными Звягинцева и Карандашевой. В этой работе
было количественно показано, что в ряду комплексовГ NH3 ■—г NH3 "I—г NH3jCl—Pt—ClCl—Pt—Br1jCl—Pt—NOoI1Cl -1L ciL ci Jзамещение лиганда именно в транс-положении значительно облег¬
чается с увеличением транс-активности соответствующего лиганда в* Указаны лишь связи, образованные гибридной sd-орбиталью. Другие
составляющие химической связи, в частности ионная, не приведены.209
ряду Cl-, Вг~, N02. На это обстоятельство впервые обратили внима¬
ние Гринберг и Кукушкин.И. Б. Берсукер и А. В. Аблов отмечали, что метод Сыркина не¬
достаточно обоснован и теоретически, так как неясна физическая при¬
рода сил цнс-закрепления. В сущности, весь результат основан на
выборе гибридизации типа sd, который в основном произволен. Дей¬
ствительно, пе ясно, почему атом платины (II) в плоскоквадратных
комплексах участвует в связи двумя орбиталями, в то время как его
ковалентность равна четырем.Однако позже высказывались мнения, что гипотеза Сыркина —
частный случай квантово-механического объяснения трансвлияния.
Так, в 1968 г. Венанци показал, что она имеет вполне реальную ос¬
нову для объяснения высокого трансвлияния лигандов, которые не
могут образовывать л-связи с ионом металла (например, гидрид-ионн-).Гипотеза Чатта—Оргела. В 1939 г. Гельман предложила отка¬
заться от взглядов на ненасыщенные молекулы, как на лиганды,
обладающие только донорными свойствами. Она считала, что такие
лиганды, как, например, этилен, являются донорами и акцепторами
электронов одновременно.Чатт и практически одновременно Оргел связали способность ли¬
гандов к л-акцепторному взаимодействию с центральным ионом (спо¬
собность к образованию л-дативной связи) с их трансвлиянием. Чатт
обратил внимание на аналогию реакций цис- и транс-замещения в
комплексах платины (II) с реакциями замещения в мета- и пара-по¬
ложениях в ароматических соединениях. Эти факты, по его мнению,
несомненно должны были иметь общую причину. Чатт предполо¬
жил, что в комплексах, содержащих двойные связи металл—лиганд,
как и в ароматических ядрах, должны возникать индуктивный и мезо-
мерный эффекты, но выраженные намного сильнее, чем в последних.
Наличие этих эффектов и главным образом мезомерного эффекта обус¬
ловливает /пранс-направленное замещение, т. е. трансвлияние.Известно, что двойная связь имеет сг- и л-компоненты связи.
<т-Связь образуется при передаче электронов атома-донора к атому
металла. Это происходит путем перекрывания заполненной парой
электронов орбитали донорного атома с одной из вакантных гибрид¬
ных &/?2-орбпталей центрального атома. Дативная л-связь образуется
при передаче электронов металла лиганду путем перекрывания за¬
полненных d- или dp-гибридных орбиталей металла с вакантными р-,
d- или yod-гибриднымн орбиталями атома донора. Типичным приме¬
ром двойной связи может служить перекрывание электронных орби¬
талей в связи ^4pt (см. рис. 8.4).Мезомернып эффект выражает перенос заряда от металла к атому
донора и, согласно Чатту, характерен для л-компоненты связи. Пере¬
нос электронной плотности для различных лигандов различен, так210
Рис. 8.4. Схематичное изображение нап¬
равленности мезомерного и индуктивного
эффектов в связи R3P = Pt:/ — о-связь; 2 — дативная я-связь; 3 — вакантная
5с/бр-орбнтальчто лиганды обладают широким диа¬
пазоном проявления мезомерного эф¬
фекта. С этой точки зрения этилен,
обладая большим мезомерным эффек¬
том оттягивания электронного обла¬
ка от центрального атома, является
лигандом с сильной способностью к
образованию двойной связи.Индуктивный эффект характерен
для a-компоненты связи. Он, по мнению Чатта, также обусловлен
переносом заряда, но направлен от лиганда к атому металла — ак¬
цептору. Чатт считал, что индуктивный и мезомерный эффекты в
комплексных соединениях взаимосвязаны.Чатт не считал индуктивный эффект особо ответственным за транс¬
направляющую силу в комплексных соединениях. Изменение заряда
на центральном ионе в результате индуктивного смещения электронов
должно в равной степени влиять на все лиганды, координированные
у металла.Л\езомерный же эффект, по мнению Чатта, всецело воздей¬
ствует на транс-позицию, поэтому способность лигандов к образова¬
нию я-связей должна определять скорость протекания процессов за¬
мещения.Чатт придерживался точки зрения, что частичное удаление элект¬
ронной плотности из области bb' под влиянием я-акцепторноголиган¬
да А (рис. 8.5) способствует введению нуклеофила С со стороны мо¬
лекулы, наиболее отдаленной от лиганда А, и замещению лиганда В,,
находящегося в- транс-положении к А.Оргел более детально рассмотрел вопрос о переходном состоянии
комплексов в реакциях замещения. Соглашаясь с общими выводами
Чатта, он считал, что в механизме трансвлияния определяющее значе¬
ние имеют состояние активированного комплекса и условия, при ко¬
торых происходит понижение энергии активации. Согласно теории
кристаллического поля для понижения энергии активации комплексанеобходимо, чтобы плотность облака
d-электронов была минимальной у
центрального атома в направлении
лигандов. В исходном комплексе че¬
тыре лиганда образуют плоский квад¬
рат. Входящая группа подходит к
комплексному иону с какой-либо сто-
-Y роны плоскости и в критическую ста-Рис. 8.5. Схема электронного оттягива¬
ния лигандом А (/-орбитали' металла М с
образованием л-дативной связи и замеще¬
ния лиганда В донором С! 1 2 : : з211
Рис. 8.6. Схема образования трнгонально-бнпирамидального интер
меднатадню реакции, двигаясь, например, над замещаемой группой, смеща¬
ет ее вниз до тех пор, пока не возникнет переходное состояние
(рис. 8.6).По мнению Оргела, при наличии в исходном комплексе обычных
связей такой подход группы затруднителен, так как повсеместно вы¬
ступают «гантели» электронных облаков dxz или dyz. Но если один из
лигандов способен образовать дативную л-связь, то это приведет к
оттягиванию соответствующего л-облака dxz- или <2у2-орбитали в
сторону этого лиганда. Именно это оттягивание электронов и вызовет
минимальную концентрацию электронной плотностн по критическим
направлениям dxz- или с(уг-орбиталей со стороны лиганда В, необхо¬
димую для понижения энергии активации.Таким образом, образование пятикоординационного комплекса
должно, согласно Оргелу, стабилизировать переходное состояние и,
следовательно, увеличить лабильность исходного комплекса.Представления Чатта—Оргела об электронных эффектах, вызы¬
ваемых л-акцепторными лигандами, плодотворно использовал Гажо
для интерпретации внутримолекулярных окислительно-восстанови¬
тельных взаимодействий лиганда с центральным ионом.Представления И. Б. Берсукера о взаимозависимости а-донорных
и ^-акцепторных свойств лигандов. Берсукер предположил, что
если какой-либо лиганд обладает свободными гс-орбиталями, способ¬
ными акцептировать электроны, то заполнение их приведет к повыше¬
нию отрицательного заряда на лиганде и при отсутствии компенси¬
рующего процесса будет уменьшать его акцепторные свойства. Ком¬
пенсирующим процессом, по его мнению, могла бы служить отдача
а-электронов от лиганда к центральному атому, которая увеличива¬
ется при повышении отрицательного заряда на лиганде.В случае таких электронных смещений получалось, что чем выше
донорные свойства а-электронов лиганда, тем выше его акцепторныеАх Рис. 8.7. Схема перекрывания
орбиталей по а- и л -типу в плос¬
коквадратном комплексе:а — взаимодействие по а-ткпу; б-
вэаимодействие по а* н Л-типуаб212
свойства для я-электронов, и наоборот, акцептирование электронов
повышает донорные свойства ст-электронов. Наличие такой взаимо¬
зависимости позволило качественно объяснить происхождение транс¬
активности лигандов, не обладающих свободными я-орбиталями.
Слабая транс-активность лигандов, связанная с меньшнмн возмож¬
ностями использования свободной л-орбитали, для акцептирования
электронов, была условно принята Берсукером как отсутствие сво¬
бодных я-орбиталей. Так, если слабая трачс-активная группа L на¬
ходится в транс-положении к еще менее транс-активной группе А, то
электронное облако смещено несколько к L (рис. 8.7, а). При приб¬
лижении замещающей группы со стороны лиганда А электронное об¬
лако, ранее смещенное по направлению к L, еще более сместится под
влиянием этой группы (рис. 8.7, б). Это смещение облегчается тем,
что в транс-положении имеется группа, обладающая большей транс¬
активностью и, следовательно, наибольшей (среди всех лигандов)
способностью принимать л- и отдавать ст-электроны.При приближении атакующей группы со стороны любого другого
лиганда этих преимуществ для смещения я-электронного облака не
имеется. Таким образом, создаются условия для образования проме¬
жуточного состояния с низкой энергией активации и для транс-заме¬
щения.Под влиянием Чатта и Оргела с середины 1950-х годов сложи¬
лось мнение, что высоким трансвлнянием характеризуются лиганды,
обладающие я-акцепторными свойствами. Однако в 1961 г. Басоло,
Чаттом и другими было установлено, что гидридный ион Н~, не способ¬
ный к л-взаимодействию с ионом металла, обладает исключительно
высоким трансвлиянием. Это заключение было сделано на основеТаблица 8.6. Константы скорости замещения хлоридного лиганда
на пиридинКомплексk* , C-1Комплексc~lPEt3PEt3|Cl-Pt—Н4,7*10~2|Cl—Pt—CH3cnОо1*PEt31PEt3ClPEt3|Cl-Pt—PEt34*оitojCl—Pt—CeH61,2- Ю-4|PEt31PEt3PEt3|Cl-Pt—Cl3,5-10-®1PEt3•При 0°С. •• При 25°С.213
кинетических данных по замещению хлоридного лиганда на пиридина
комплексах типа [Pt(PEt3)2LCl]. Значения констант скорости соот¬
ветствующих реакций, измеренных в условиях псевдопервого поряд¬
ка, приведены в табл. 8.6.Из табл. 8.6 видно, что трансвлияние гидридного иона приближа¬
ется к трансвлиянию триалкилфосфинов и на четыре порядка выше,
чем трансвлияние хлорид-иона.Лэнгфорд и Грей предложили на основании а- ил-электронныхэф¬
фектов делить трансвлияние на а- и л-трансвлияние. На основании
значений интегралов перекрывания орбиталей лигандов и комплексо-
образователя они построили два ряда:Ряд лигандов по а-эффектун- >PR3>ДМСО -- SCN- >СНз, СО, CN- >Вг~ >0,51690,37700,360 '0,35740,34940,3357Cl->NH,>он- >с2н4О. 33320,31390,28770,189Рядлигандовпо ^-эффектуДМСО >С, н; :> СО> CN-> NO2 >SCN-0,1360,0960,093О. 0880,066О. 0520,124Лиганды в ряду ст-трансвлияния расположены в порядке умень¬
шения значения интеграла перекрывания между бр-орбиталью пла¬
тины и соответствующей ра -валентной орбиталью лиганда J(6р, /пр,).
Относительное положение лигандов внутри ряда л-трансвлияния опре¬
деляется значением интегралов перекрывания между 5d- -орбиталью
платины и свободной л*-молекулярной орбиталью лиганда.При рассмотрении взаимного влияния лигандов в октаэдрических
и плоскоквадратных комплексах переходных металлов с сг-связямн
методом теории возмущений и на основе квантово-химических полу-
эмпирических расчетов серий комплексов В. И. Барановский по¬
казал, что влияние лигандов по транс-координате передается через
ndx,_yi и (п + 1)5-орбитали центрального атома. Усиление донорных
свойств лиганда приводит к уменьшению прочности связи его транс¬
партнера с металлом. При изучении плоскоквадратных платиновых
и палладиевых комплексов Барановский построил теоретическую шка¬
лу а- и л-донорных и акцепторных способностей лигандов в этих комп¬
лексах и получил следующий теоретический ряд трансвлпяния:CN- >СНз > NOj > И' > СГ >БСНз > ОН~>>NH3, СО, ДМС, С2Н4 > ДМСО > Н208.4. Цисвлияние лигандовВ 1952 г. на совещании по закономерности трансвлпяния Черняевг
была принята следующая формулировка этого явления. У соедине-* Приведены данные, полученные разными авторами.214
ннй с квадратным или октаэдрическим строением внутренней сферы,
в центре которой находится комплексообразующий атом, скорость
реакции замещения всякого атома (или молекулы), связанного с этим
центральным атомом, определяется природой заместителя, занимаю¬
щего противоположный конец диагонали. Таким образом, на проч¬
ность связи металла с любым заместителем очень мало влияет природа
соседних (цис-) с этой связью атомов или молекул, в то время как атомы
или молекулы, находящиеся в наиболее удаленной области комплекс¬
ного соединения (в т/;анс-положении) на диагонали квадрата, сильно
влияют на эту связь. Гринберг высказал предположение, что и в цис-
положении должно существовать какое-то взаимодействие атомов.В начале 1950-х годов появились первые экспериментальные дан¬
ные, позволявшие предполагать наличие влияния цис-расположенных
лигандов. Различная скорость акватации комплексов кобальта (III),
напримертране-[Со (АА)г С12]* + Н20 -*■ [Со (АА)г H.OCIp + С1~(где АА— это NH2C„H4NH2, NH2CH(C6H5)CH(CeH5)NH2), отмеченная
в 1953 г. Пирсоном, Бостоном и Басоло, могла быть объяснена только
различным влиянием цмс-расположенных аминов. В данном случае
это влияние могло сводиться к стерическим причинам.Цисвлияние молекулы аммиака на скорость замещения рядом сто¬
ящих лигандов в комплексах платины было надежно установлено в
1957 г. Гринбергом и Кукушкиным. Они показали, что скорость за¬
мещения хлорид-иона на аммиак в тетрахлороплатинате (II) калия■С1СГк'-NH3СПPt+ NH3 -ч- КPtС1CI. ciCIменьше, чем в соли Косса:NH3СГ~NH3ClкPt+ NH3 -Pt+ KCI. ciCl_NH3ClОсновной вывод — увеличение подвижности цггс-расположенных
лигандов под влиянием аммиака — был в следующем году подтверж¬
ден на реакциях гидролиза платиновых комплексов. Скорость гидро¬
лиза, а следовательно, и скорость гидратации тетрахлороплатината
(II) калия оказались заметно меньше скорости гидролиза соли Косса.
Сравнение констант скорости реакций замещения комплексов плати¬
ны (II), образующих переходный ряд Вернера—Миолати, гидро¬
ксильным ионом (рис. 8.8, а) и аммиаком (рис. 8.8, б), показало
аналогию в поведении комплексов. Диаграммы в полном соответствии
с представлением о цисвлиянии аммиака на хлорид-ионы показали,
что скорость процессов в ряду от [PtCU]2" до [Pt(NH3)Cl3]~ и далее к
mpcwc-[Pt(NH3)2Cl„] возрастает.' В m/?a«c-[Pt(NH3)2CI2] такой ^ис-эффект молекул аммиака выражен
намного сильнее, так как хлорид-ионы лабнлиэированы двумя цис-
расположенными молекулами аммиака. Близость констант скорости
взаимодействия [PtC,I4]2- и [Pt(NH3)3Cl]4-, трудно объяснимая с точки215
а бРис. 8.8. Диаграммы скорости гидролиза (а) и взаимодейст¬
вия с аммиаком (б) комплексов ряда К2[РtClj]—[Pt(NH3)3CI]Cl:А — К=[Р1С1,]; В — K[PtNH3€l3]; С — J-paKC-[Pt(NH3hCl,l; D —[P1(NH3)3C1]C1зрения закономерности трансвлияния, также легко объяснялась цнс-
влиянием. Поскольку в комплексе [Pt(NH3)3CI]+ хлорид-ион испыты¬
вает quc-лабилизирующее действие двух молекул аммиака, то, не¬
смотря на меньшее трансвлияние NH3 по сравненню с трансвлиянием
хлорид-иона, взаимодействие с аммиаком комплексовГ С1 12 —Г NH3jCl—Pt—ci
1ИjCl—Pt—NH3CI' |, NH3происходит с близкими скоростями.Внутрисферная молекула пиридина в комплексах платины (II)
обладает еще большим цис-лабилизирующим действием, чем аммиак.В табл. 8.7 приведены константы скорости взаимодействия комп¬
лексов платины (II) с водой*, аммиаком и пиридином. Из данных
этой таблицы следует, что по цисвлиянию изученные лиганды распо¬
лагаются в следующий ряд: Ру > NH3 > СГ.А. А. Гринберг и М. А. Кузьмина шзучили обмен хлорид-ионов
в комплексе [Pt(C;H4)Cl3]_, и показали, что этилен, обладающий боль¬
шим трансвлиянием, закрепляет в комплексном ионе цис-располо¬
женные атомы хлора. Противоположность рядов транс- и цисвлияния
'лигандов С2Н4, СГ, NH3, Ру наглядно вытекает из данных по скорости
изотопного обмена хлора, сопоставленных в табл. 8.8.Кукушкин и Кузьмина изучили цисвлиянпе диалкилсульфоксн-
дов на скорость обмена хлора и установили, что хлорид-ион, распо¬
ложенный против диалкилсульфоксидов, обменивается практически* Механизм взаимодействия комплексов с гидроксильным ионом сводится
к вступлению во внутреннюю сферу молекулы воды с последующим отщепле¬
нием иона Н+. Таким образом скорость гидролиза, по существу, является ско¬
ростью вступления в комплекс молекулы воды (см. гл. 9).216
Таблица 8.7. Константы скорости реакции замещения при 25 СКомплекс*OH-(HtO) Ю4*
c~l*NH, WfrpylO’Л • МОЛЬ-1 • c—1Cl-кCl—Pt—Cl10,390,421,711_ CI_Г NH3-кCl-Pt—Cl
10,601,143,281L ci_- Pyi-кCl—Pt—Cl—1,755,711L ci_гNH3-С1—Pt—NHCl0,220,37—_NH3_ГNH3 -Cl-Pt—С11.012,9—_NH3 .гNH3-С1—Pt—NH30,38——_iCl- Brк2Br—Pt—Br1,623,1—1L Br- NH3-кBr—Pt—Br
1—7,0(15°C)1L Br-мгновенно, т. е. со скоростью «нулевого обмена». Такое поведение комп¬
лексных ионов типа [Pt(R2SO)Cl3r в отношении обмена транс-рас¬
положенного иона хлора аналогично поведению[Pt(C2H4)Cl3]‘. Это так
же указывает на близость трансвлияния диметилсульфоксида и эти¬
лена.Неожиданно большой оказалась скорость обмена цис-располо¬
женных хлорид-ионов в комплексах [Pt(R2SO)CI3]~. Несмотря на217
Таблица 8.8. Константы скорости обмена хлорид-ионов (в с-1)
в комплексах платины (II) [PtLCI]"Комплексный нойОбмен на координатахС!—Pt—С1L-Pt-ClС!ICl-Pt-CIICl- NH3 -
ICl-Pt-Cl
ICl .■ Py ■ICl—Pt-Cl
I. ClC2H4 -ICl—Pt—ClICl .
-(CH3)2so -
ICl—Pt—Cl
I- Cl .- (C:H6)2SOIci—Pt-Cl
ICI-(CHo)4SO -
■ I
Cl-Pt—Cl
ICl .-(C3H,)2SO
ICl-Pt-CI
ICl(u30-C3H7)..SOICl-Pt-CII- Cl1,04-10-52,9- lO-53,8-КГ54,9 10"®4,9-10-*1,5-10-*3,0-10-*1,1-10-*0,7-10-*1,04-10"4,2-10-52,9-10-®Скорость СЛИШ¬
КОМ велика для
определения дан¬
ным методомТо же218
Рис. S.9. Схема взаимодействия ру-ор¬
биталей центрального атома кислорода
координированного сульфокскдабольшое трансвлияние, диалкил-
сульфоксиды, не закрепляют, а ла-
бнлнзируют хлоридные лиганды в
/<нс-положении. Хлорид-ионы на
координате Cl—Pt—Cl в диэтил-
суЛьфоксидном комплексе весьма
подвижны, но примерно в три раза
менее лабильны, чем в диметилсульфоксидном соединении. Еще мень¬
шей подвижностью обладают хлорид-ноны в дипропилсульфоксидных
комплексах (см. табл. 8.8). По-видимому, это связано с увеличением
размеров алкильных радикалов, что должно приводить к «экрани¬
рованию» ^(.-расположенных лигандов.Из расчета электронной структуры комплекса [РЦДМСО)С13]-,
проведенного методом ССП МО ЛКАО в приближении ППДП, выте¬
кает, что группы R2SO, координированные через атом серы, могут
•образовывать непосредственные связи донорно-акцепторного типа с
г^с-расположенными лигандами.Диффузные 2>d-kO серы пространственно ориентированы таким
образом, что способны образовывать связи как с соседними лигандами
в плоскоквадратных комплексах, так и с вступающей группой в интер¬
медиате. В то же время л*-орбитали этилена пространственно лока¬
лизованы в плоскости Pt/1| и их способность к взаимодействию44 Сс другими лигандами невелика. Таким образом, причина различного
динамического цисвлпяния С2Н1 и ДАЮЭ с этой точки зрения заклю¬
чается в особенности ориентации их л-акцепторных орбиталей. Воз¬
можно, что кроме 3d-AO серы диалкилсульфоксидов в образовании
связи с соседними лигандами участвует р„-орбиталь кислорода. Ра¬
счет показал, что эта орбиталь обладает л-донорной способностью и
также может лабилизировать связи платина—хлор в цис-положении
(рис. 8.9).В табл. 8.9 сведены относительные значения цисвлпяния лиган-Таблмца 8.9. Относительное цисвлиянне лигандов в комплексах
платины (II)РеакцияC,H,ci-NH,Py(C,Hs)2SO(CH,bSO[PtLCl3]-+HoO[PtLCl3]-+NH3[PtLCI3]-+En[PtLCl3]-+Py[PtLCI3]-+»Cl-[PtEnLCl] + +NH30,130,560,240,290,300,270,860,650,530,570,760,841111114,01,55136,2219
дов, вычисленные из отношения скоростей реакций замещения или
изотопного обмена при 25 °С. Цнсвлияние пиридина принято за еди¬
ницу.Таким образом, ряд цисвлияния лигандов для комплексов платины
(II) имеет видС2н4 < СГ < NH-i < Ру ~ (C2H5)2S < (С2Н6)2 SO < (СН3)2 soИз данных для реакций [Pt EnLCI]+ с NH3, как и из табл. 8.8,
следует, что цнсвлияние лигандов сильно зависит от стерических
факторов. При переходе от (CH3)2SO к (CaHa^SO резко понижается
скорость реакции замещения.В заключение следует сказать, что цнсвлияние лигандов.осложнено
различными факторами в гораздо большей степени, чем трансвлияние,
в частности стерическими препятствиями. Геометрические размеры
лиганда мало влияют на скорость замещения транс-расположенной
группы. В то же время они сильно сказываются на скорости замеще¬
ния г^г-р-асположенных лигандов. Конечно, трудно ожидать полной
обратимости рядов трансвлиянин и цисвлияния. Так, имеются сведе¬
ния, что фенильный и метильный карбанионы наряду с высоким транс¬
влиянием обладают и высоким цисвлпянием. Несмотря на отдельные
исключения, тенденция к обратимости рядов цис- и трансвлияния,
безусловно, имеется.Одна из первых теоретических трактовок цисвлияния лигандов
была дана И. Б. Берсукером с позиции взаимозависимости о-донор-
ных и л-акцепторных свойств лигандов. Действительно, смещение
сг-электронов от лиганда L к центральному атому должно зависеть от
о-донорных свойств лигандов, находящихся в цис-положенни к этому
лиганду. Если ст-донорные свойства цис-соседей выражены сильно,то
это должно приводить к меньшему смещению ст-электронов от L к
центральному атому, а это, в свою очередь, приведет к уменьшению
л-акцепторного взаимодействия лиганда с центральным атомом и,
следовательно, к уменьшению трансвлияния L. Этот механизм цис¬
влияния хорошо объясняет наблюдающуюся антибатность рядов цис-
и трансвлияния.В. И. Барановский на основе результатов квантово-химических
расчетов электронной структуры комплексов показал, что усиление
донорных свойств лиганда L может оказывать как упрочняющее, так
и разрыхляющее воздействие на связи металла с i/ис-расположенны-
ми лигандами. При этом образование непосредственных связей между
лигандами может существенным образом изменять ряды цисвлияния.8.5. Правило термической изомеризации
комплексов платины (II) и палладия (И)Термоизомеризация в твердой фазе. Закономерность трансвлияния
И. И. Черняева основана на большом экспериментальном материале,
полученном главным образом на реакциях замещения в растворах.
Естественно, что растворитель вносит существенный вклад в меха-220
ннзм и энергетику процессов превращения координационных соеди¬
нений.Трансвлияние, проявляемое в твердом состоянии, широко иссле¬
довано рентгеноструктурным, ПК и ЯА\Р спектроскопическими ме¬
тодами, которые отражают статическую сторону этого явления.Таким образом, динамическая сторона явления трансвлияния
широко изучена на комплексах, находящихся в растворах. Статиче¬
ская сторона трансвлияния исследована в основном на комплексах,
находящихся в твердом состоянии. В 1921 г. Л. А. Чугаев
совместно с С. С. Кильтыновичем показали, что нитротриаммин
[Pt(NH3)3N02]N02 чрезвычайно легко теряет при 100 °С одну молекулу
аммиака с образованием mpaHc-[Pt(NH3)2(N02)2]. Теперь эта реакция
рассматривается как яркий пример, иллюстрирующий проявление
динамического трансвлияния нитрогруппы в комплексах платины
(II).Правило расщепления тетраминов платины (II) было установлено
Иергенсеном для реакций, протекающих в растворах. В ряде лабора¬
торий было показано, что их расщепление в твердой фазе также про¬
текает в соответствии с правилом Иергенсена.Уже более 70 лет для исследования веществ в твердой фазе успешно
применяется метод дифференциального термического анализа. Еще
сравнительно недавно этот метод использовался главным образом
для изучения фазовых переходов н для характеристики термических
свойств веществ. В связи с широким распространением дериватогра-
фа в настоящее время термография широко используется для изучения
химических превращений, протекающих в твердой фазе и расплаве.
Для комплексов платины метод ДТА был впервые использован
А. В. Николаевым. Одним из наиболее интересных, результатов его
исследования было выявление для i<uc-[Pt(NH3)2Cl2] на кривой
ДТА экзотермических пиков, обусловленных изомеризацией
^wc-[Pt(NH3)2Cb] в mpaHC-[Pt(NH3)2Ch].Термодинамическая вероятность протекания процесса цис- транс¬
изомеризации позднее получила обоснование в работе Черняева с
сотр. Они установили, что полная энергия mpaHC-[Pt(NH3);Cl2] на
12,6±0,8 кДж/моль меньше, чем quc-изомера.Ю. Н. Кукушкин и Е. С. Постникова показали, что иодидные и
бромидные комплексы типа q«c-[PtAsX2] (где А ■— амины, X —
Вг', Г) изомеризуются при нагреваний в твердой фазе (без разложе¬
ния) с выделением энергии и образованием соответствующих соеди¬
нений транс-конфигурации (рис. 8.10). Изомеризация изученных
иодидных комплексов протекала при 135—205 °С, а аналогичных бро-
мидных соединений — при температурах на 30—40 °С выше. Тем¬
пература изомеризации хлоридных комплексов должна быть еще вы¬
ше, поэтому разложение таких комплексов наступает раньше, чем изо¬
меризация (табл. 8.10). Оксимные комплексы платины (II) тнпа цис-
[PtA„X2] изомеризуются при более низких температурах, чем амин-
ные. В результате этого удалось показать, что изомеризация хлорид¬
ных оксимных комплексов протекает аналогично изомеризации бро-
мидных и иодидных комплексов.221
Рис. 8.10. Термограммы mpaHC-lP^NH^CHjWo] (а) и
4uc-[Pt(NH,CH3)2I2] (б):1 — дифференциальная кривая изменения массы; 2— диффе¬
ренциальная кривая изменения температуры; 3— изменение
температуры (°С); 4—изменение массыТаблица 8.10. Характеристики термического превращения комплек¬
сов типа [PtА3Х2]АИзомерХ=1Х=ВгХ=С1t.°сAW1I30M •кДж/мольС^^изом >
кДж/ моль*раэ.иi -Снзомерн-зацино*ос;СП *
Я 3a sQ.о —
2 =
О ЯЛ СО— оо 9S.*NH3цис-20528018,42502655,9295транс-—280—_265—26»CH3NH0цис-15021015,517522013,01Мтранс-—225——220—2'25c2h5nh2цис-13521025,11802258,8160транс*—230——235_215c5h5nцис-17530516,32352659,6240транс-—290——280—250Дериватографический метод позволяет оценить изменение энталь¬
пии превращения комплексов ;{«c-[PtA2Xs] в транс-[PtAsXs]. Уста¬
новлено, что изменение энтальпии растет при переходе от хлоридных
комплексов к бромидным и далее к иодидным. Это находится в соот¬
ветствии с калориметрическими данными для изомерных пар комп¬
лексов [Pt(NH3)2X2], где X — СГ, Вг-, 1~, полученными И. И. Чер¬
няевым и В. А. Палкиным.Палкин с сотр. установили, что разность абсолютных энтропий
изомерных комплексов платины [Pt(NH3)2X2] невелика. При 298,15 К222
разность в значениях TAS составляет 1,7 кДж/'моль для хлоридных
комплексов и 2,1 кДж/моль— для бромидных. Это свидетельствуето том, что направление реакции изомеризации цис-[Р[АгХ2\-+транс-
[PtA„X2] определяется главным образом энтальпией реакций.Комплексные соединения палладия (II), как правило, более ла¬
бильны, чем соответствующие соединения платины (II). Поэтому мож¬
но было ожидать, что изомеризация солен типа [PdA„X2] в твердом
состоянии должна протекать легче, чем комплексов’платины (II).
Действительно, Гринберг и Шульман показали, что в водном раст¬
воре' q«c-[Pd(NH3)2Cl2] легко переходит в транс-изомер, поэтому
синтез соединений 4uc-[Pd(NH3)2X2], где X—Cl-, Br", I-, вызывает
затруднения. Трудность получения их в кристаллическом состоянии
увеличивается при переходе от хлорида к бромиду и нодиду.
Лантон с сотр. установили, что свежеприготовленный комплекс
q«t’-[Pd(NH3)2Cl2] полностью изомеризуется в твердом состоянии при
комнатной температуре в соответствующий транс-изомер за 40 ч.Таким образом, многочисленные экспериментальные данные по¬
казывают, что изомеризация комплексов типа [МА2Х2], где
М — Pt(II), Pd(II); А — амины или оксимы; X — галогенидные или
роданидные ноны, протекает в направлении обра¬
зования соединений, в которых амин распо¬
лагается против лиганда с наименьшим
трансвлиянием. Если не наступает разложения, то в соот¬
ветствии с этим правилом комплексы ц«с-[МА2Х2] нзомеризуются в
соединении /лранс-конфигурации:' АXt°' Ах 1М-*■мАX _. хАИмеются указания,
комплексы платинычто аналогичным образом
(II) с глнкоколом инзомеризуются
аланином типаi^c-[Pt(NH2CH2COOH)2X„], где X — СГ, Вг', Г“HOOCCH2NH2 X ■-HOOCCH2NH2 XPt—PtHOOCCH;.NH2 XX NH2CH2COOH■ C!NH2S03 "Г C1NH2S03 1. C1Pt-*■ K2 1Ptnh2so,LS03nh2ClДо сих пор речь шлд об изомеризации соединений неэлект¬
ролитного типа. Изомеризация сульфаминового комплекса
;{uc-[K2[Pt(NH2S03)2CI2] происходит аналогично:К2Сульфаминатный ион координирован к платине посредством атома
азота, поэтому по трансвлиянию он должен быть близок к аминам.
Вследствие этого процесс изомеризации, так же как и в аминатных
комплексах, протекает в сторону образования соединения транс¬
конфигурации.Таким образом, можно допустить, что заряд комплексного иона223
Рис. 8.11. Термограммы 4uc-[Pt(fl3CO)PyCl2] (д) u гпранс-
1Р1(ДЭСО)РуС12] (б) (обозначения с.м. рис. 8.10)не оказывает влияния на процесс изомеризации. Однако для уве¬
ренности изучение изомеризации заряженных комплексов следует
распространить на другие случаи. К сожалению, удобных для этой
цели объектов очень мало. К их числу могут быть отнесены комплек¬
сы с остатками аминокислот типа K2[Pt(NH2CH2COO)2Cl3].Как уже было сказано, алифатические сульфоксиды, координи¬
рованные платиной (II) через атом серы, обладают трансвлиянием,
близким к трансвлиянию этнлена. Дериватографическое исследова¬
ние многочисленных изомерных соединений типа [Pt(R2SO)ACl;]
показало, что комплексы транс-конфигурации при нагревании изо¬
меризуются в соответствующие соединения цшг-конфигурации:' RnSOСПг[RoSO Cl 1PtPtClА>nЭто справедливо как для хлоридных, так и для бромидных комплек¬
сов. Процесс изомеризации проявляется на кривой ДТА в виде ха¬
рактерных спаренных эндо- и экзотермических пиков (рис. 8.11).
Экспериментально установлено, что эндотермический пик связан
с плавлением вещества. В расплаве происходит реакция изомериза¬
ции. Температуры плавления цис-изомеров типа [Pt(R2SO)ACl2]
обычно выше температуры плавления соответствующих транс-нзо-224
меров. поэтому после изомеризации комплексы q«c-[Pt(R2SO)ACI„],
как правило, кристаллизуются из расплава. Таким образом, экзо¬
термические пики вызваны выделением энергии изомеризации и
энергии кристаллизации.Несмотря на то что в случае сульфоксидаминных соединений про¬
исходит не цис- транс-изомеризация, а, наоборот, транс- -*■ цис-
превращение, направление процесса хорошо согласуется с правилом,
а именно, процесс идет в направлении образования комплексов, в
которых амин располагается против лиганда с наименьшим трапс-
влиянием. Хорошо известно, что трансвлияние дналкилсульфокси-
дов выше, чем трансвлияние хлоридного или бромпдного лиганда.Долгое время не удавалось получить иодидных комплексов тнпа
[Pt(R.,SO)AI.,]. Лишь недавно синтезирована изомерная пара комп¬
лексов [Й(ДМСО)(ЫН2С.,Н4ОН)12] и подвергнута дериватографиче¬
скому изучению. Оказалось, что иодидный аналог ведет себя иначе,
чем хлорцдный и бромидный комплексы. Реакция протекает по сле¬
дующему направлению:гдмсо nh,c,h4ohгГДМСО I 1Pt “ ■PtI II NH,C,H4OHЭто объясняется тем, что трансвлияние иодидного лиганда настолько
высокое, что оно больше, чем трансвлияние диметилсульфокспда.
Амин же и в этом случае стремится занять положение против лиганда
с наименьшим трансвлиянием.Эллис, Вайль и Орчин осуществили транс—*- цис-изомериза-
цию карбонильных комплексов типа [Pt(CO)AC!,], где А — амины.
Напомним, что оксид углерода (II) в комплексах платины (II) обла¬
дает высоким трансвлиянием.В настоящее время синтезировано и подвергнуто термоизомериза¬
ции большое число комплексов типа [PtLACl2], где L — лиганд вы¬
сокого трансвлияния, А — амины или оксимы. В качестве лиганда
L были взяты сульфиды, трифенилфосфин, триэтилфосфит. Установ¬
лено, что во всех случаях, за исключением одного, комплексы
транс-fPtLAClo] (где L — лиганд высокого трансвлияния) изомери¬
зуются при нагревании в соответствующие комплексы цыс-конфигу-
рации. Изомеризация всех этих соединений характеризуется свое¬
образной кривой дифференциального термического анализа — на
ней имеется спаренный эндо-экзотермический пик.Исключением из правила изомеризации является тиоксановый
комплекс [Pt(TO)PyCl2]. При нагревании ^uc-[Pt(TO)PyCl2] с вы¬
делением энергии происходит его превращение в транс-изомер. При¬
чина такого положения пока не выяснена. Возможно, в данном слу¬
чае энергетика процесса определяется разностью энергий кристалли¬
ческих решеток исходного и получающегося изомерных соединений.Правило термической изомеризации распространяется не только
на аминокомплексы, но и на фосфиновые соединения. Так, показано,
что транс-[Pt(PPh3)(R2S)Cl2], где R2S — диэтилсульфид или ди-
изопропилсульфид, при нагревании превращается в цис-изомер. Ней¬8-817225
тральные лиганды — трифенилфосфин и тиоэфир (оба лиганда вы¬
сокого трансвлияния) — «стремятся» занять положение против ли¬
гандов с наименьшим трансвлиянияем, т. е. против хлорид-ионов.Термическая изомеризация quc-[Pt(PA'\e2Ph);MeCl] была иссле¬
дована методом дифференциальной сканирующей калориметрии. Ус¬
тановлено, что реакция протекает следующим образом:Me MeI /■ ICl—Pt—PMe.,Ph PhMcoP—Pt—PMc„PhI ‘ ‘lPMe2Ph ClТермограмма 4“f-[Pt(PMe2Ph)2MeCl] оказалась характерной для тер¬
мической изомеризации многих комплексов.На кривой ДТА имеется эндотермический пик пЛавлення. На него
накладывается экзотермический пик, отвечающий изомеризации комп¬
лекса и его кристаллизации. Направление реакции изомеризации
согласуется с трансвлиянием лигандов. Известно, что метил обладает
очень высоким трансвлиянием. Вследствие этого расположенный
в транс-положении к нему фосфин «стремится» переместиться и за¬
нять место против другой молекулы фосфина.Аналогично при нагревании в твердой фазе ведут себя комплексы
4Uf-[PtL„(SnCl3)Cl], где L — PPh3, PPh2Et и PPhEt2. Так, на кривом
ДТА этих комплексов имеются необратимые экзотермические пики,
которые отражают процесс изомеризации. Примером может служить
реакция изомеризации трпфенилфосфинового комплекса:SnCI3 SnCI3I t° ICl-Pt—PPh3 Ph3P-Pt-PPh3Известно, что лиганд SnCl3 обладает высоким трансвлиянием. При
термической обработке фосфнны перемещаются в т/7анс-положенпе
к лиганду с меньшим трансвлиянием, чем SnCb. Подобным
же образом нзомеризуются палладиевые комплексы типаЛапперт с сотр. обнаружили и изучили термическую перегруппи¬
ровку внутренней сферы карбеновых комплексов платины (II) и пал¬
ладия (II). Они установили, что при нагревании как в кристалличе¬
ском состоянии, так и в растворе протекает изомеризация комплексов,
которая схематично может быть выражена уравнениемКарбеновые лиганды, так же как алкильные и арнльные, обладаютPPh3Cl4uc-[Pd(PR3)2(SnCl3)CI].IClRIR226
высоким трансвлиянием. Поэтому данное превращение также на¬
ходится в согласии с правилом. На большом числе примеров показа¬
но, что палладиевые карбеновые комплексы изомеризуются за более
короткое время и при более низкой температуре, чем аналогичные пла¬
тиновые соединения.Установлено, что при взаимодействии тетрахлор платината (II)
калия с диметил- и диэтилсульфоксидом образуются комплексы
[Pt(R2SO)Xl2] (^с-конфигурации. В случае дипропилсульфоксида
вначале образуется транс-[Р1(ДПСО)2С1„], который при нагревании
раствора превращается в соответствующий quc-изомер. При взаимо¬
действии K2[PtCl4] и диизопропилсульфоксида единственным про¬
дуктом реакции является транс-[Р1(ДизоПСО)„С12]. Эти данные
свидетельствуют о большей термодинамической устойчивости дисульфо-
ксидных комплексов quc-конфигурации. Факт образования диизо-
пропнлсульфоксидного комплекса транс-строения, по-видимому, объ¬
ясняется стерическими затруднениями, которые появляются в случае
расположения двух молекул диизопропилсульфоксида в quc-поло¬
жении друг к другу.Дерпватографическое исследование изомерных комплексов
[РМДПСО)2С12] показало, что соединение транс-конфигурации
превращается в г^ис-изомер. Характер кривой ДТА для
транс-[Р1(ДПСО).,С1„] аналогичен характеру кривых ДТА для комп¬
лексов mpaHC-[Pt(R2SO)ACl2]. Таким образом, изомеризация днсульф-
оксидных комплексов [Pt(R;SO)2Cl2] протекает в направлении обра¬
зования соединений, в которых молекулы сульфоксидов распола¬
гаются против лигандов с наименьшим трансвлиянием.Ранее было сказано, что диметилсульфоксид как лиганд по ряду
свойств подобен этилену. Установлено, что взаимодействие
К[Р1(ДМСО)С13] с этиленом или K[Pt(C2Hj)CI3] с диметилсульфокси-
дом приводит к образованию комплекса [Pt(flMCO)(C2H4)CI2] цис-
конфигурации. Аналогичная картина наблюдается в случае пропи¬
лена и дивинила. л-Акцепторные лиганды, какими являются сульф¬
оксиды и олефины, в комплексах типа [PtLL'CU] стремятся занять
^ыс-положение друг к другу, так как в этом случае осуществляется
лучшее перекрывание dr -орбиталей центрального атома и лигандов.
Образование олефинсульфоксидных соединений типа [PILL'CU] имен¬
но (^с-конфигурации согласуется с правилом, что нейтраль¬
ные молекулы стремятся занять места про¬
тив лигандов с наименьшим т р а н с в л и я н и е м.Первое большое исследование фосфиновых комплексов платины
(II) было проведено Иенсеном. На основании значений дипольных
моментов комплексов типа [Pt(PR3)„X2] Ненсен судил об их геомет¬
рической конфигурации. Он заметил, что при нагревании в твердой
фазе /<«c-[Pt(PEt3)2I2] переходит в транс-изомер. Кроме того, было
установлено, что ^uc-[Pt(AsEt3)s(N02)2] плавится при 169—170° С,
а затем также превращается в транс-изомер. После изомеризации
происходит кристаллизация, и вновь вещество плавится уже при
199—200 °С.Изомеризация бнс-фосфиновых комплексов платины (II) в раст¬227
ворах исследовалась во многих работах. Как правило, в растворе
устанавливалось равновесие между изомерными формами. Его по¬
ложение зависело от многих факторов и, в частности, от природы
растворителя. Термические превращения в твердой фазе изучены
гораздо меньше.Аллен с сотр. при изучении двух превращений сделали вывод,
что изомеризация бис-фосфиновых комплексов платины (II) проте¬
кает с поглощением энергии. Для quc-[Pt(PBu3)2Cl2] получена чет¬
кая кривая ДТА с тремя эндотермическими пиками, которые авторы
уверенно относят к определенным процессам. Один из этих эффектов
они связывают с цис- —транс-изомеризацией. Кривая ДТА для комп¬
лекса q«c-[Pt(PEt3)2Cl2] является аналогичной. Однако эндотерми¬
ческий пик изомеризации и пик, связанный с сублимацией изомери-
зованного комплекса, накладываются. Установлено, что бромидный
комплекс ^uc-[Pt(PBu3)2Br2] изомеризуется в расплаве.Качественные наблюдения Иенсена об изомеризации
i(uc-[Pt(PEt3)2I2] были подтверждены методом дифференциальной
сканирующей калориметрии. Было установлено, что при 129 °С про¬
исходит плавление комплекса, затем при 130 °С протекает экзотерми¬
ческий процесс изомеризации с последующей кристаллизацией
mpGHc-[Pt(PEt3)2I2], а при 135 °С эндотермический пик соответству¬
ет плавлению mpaHC-[Pt(PEt3)2I2], Ли и Штоуфер пришли к заклю¬
чению о том, что в таком же направлении в твердом состоянии про¬
текает изомеризация quc-[Pt(PPh3)2I„],Таким образом, в условиях, не осложненных сольватационными
процессами, ряд изученных бис-фосфиновых комплексов платины (II),
подвергается цис- транс-изомеризации. Эти результаты можно
рассматривать как несогласующиеся со сформулированным правилом.
В продуктах изомеризации фосфины занимают положение против
лигандов с наибольшим трансвлияннем.Установление факта сублимации бис-фосфиновых комплексов
позволило Аллену изучить процесс изомеризации при вакуумной пере¬
гонке. Из его работы следует, что с накоплением фенильных групп
в фосфине затрудняется процесс цис- -*• транс-изомеризации.
К. А. Хохряков и Е. И. Григоров недавно установили, что
mpaHc-[Pt(PCF3Ph2)„CI2] при нагревании изомеризуется в цыс-изомер.
Это пока единственный пример изомеризации бис-фосфинового комп¬
лекса платины (II) в соответствии с правилом.В термическом поведении бис-фосфиновых комплексов палладия
(II) типа [Pd(PR3)2X2] нет единообразия. В трех случаях из четырех
изученных происходит цис--стране-изомеризация, а в одном, на¬
оборот, транс- —► г<ис-превращение.Наибольшие затруднения связаны с трактовкой направления
термоизомерпзацин бис-тиоэфирных комплексов платины (II) и
палладия (II). Соединения палладия (II) типа [Pd(R2S)2X2] известны
только в транс-форме. В твердом состоянии не удалось наблюдать их
изомеризации. Для изученных бис-тиоэфирных комплексов платины(II) типа [PtL2X2] (где L — диметилсульфид, диэтил сульфид, тиофан,
тиоксан и пентаметиленсульфпд, а X — СГ или Вг) в четырех слу¬228
чаях комплексы ^ис-конфнгурации нзомеризуются в соответствующие
соединения троне-конфигурации, а в двух случаях, наоборот, наб¬
людается транс- quc-изомеризация.Роул и Барбей изучили кинетику изомеризации бис-диметилсуль-
фидного комплекса платины (II) в хлороформе и установили, что в
присутствии свободного диметилсульфида происходит цис-->транс-
изомеризация. Процесс протекает через промежуточное образование
трис-диметилсульфидного комплекса [Р1(ДМС)3С1]С1. Они устано¬
вили, что изомеризация является эндотермическим процессом и пред¬
положили, что ее направление в растворе обусловливается увеличе¬
нием энтропии, которое вызвано уменьшением сольватации комплёкса
транс-конфигурации по сравнению с комплексом цис-конфигура-
ции.Не исключается возможность того, что в направлении реакции
изомеризации в некоторых случаях существенную роль играет сим¬
метрия исходных и образующихся комплексов. Возможно, в этом кро¬
ется причина исключительно высокой устойчивости бис-тиоэфирных
комплексов палладия (II) транс-конфигурации. Пока ни в одной из
работ не удалось наблюдать присутствия t^uc-изомеров даже в виде
примеси.Изомеризация плоскоквадратных комплексов без признаков раз¬
ложения позволила Кукушкину, Коновалову и Погаревой предполо¬
жить внутримолекулярный механизм, не включающий отрыва лиган¬
дов. Была сделана попытка его экспериментальной проверки. С этой
целью были изучены температурные изменения ИК спектров комплек¬
са i{UC-[PtPy2h] и q«c-[Pt(NH2CH3)2I2].Установлено, что при нагревании ком¬
плексов их ИК спектры в области час¬
тот колебаний связей Pt—I плавно из¬
меняются (рис. 8.12). Например, для
4uc-[PtPy2I2] в области частот v(Pt—I)
имеется две полосы с максимумами око¬
ло 176—177 и 167 см'1. При повышении
температуры постепенно уменьшается
интенсивность полосы 167 см'1. Полоса
с максимумом 176—177 см'1 сохраняет
интенсивность, но плавно смещается в
коротковолновую область. Эти измене¬
ния происходят в области температур,
при которых на дериватограммах про¬
является процесс изомеризации.Коновалов и Погарева провели анализ длин¬
новолновых ИК спектров с точки зрения воз¬
можности образования тетраэдрического ин¬
термедиата при помощи специально модифи-Рис. 8.12. ИК спектры qHC-IPtPvnlo] при раз¬
личных значениях температуры (СС):1 — цис-fPtPy2l3l2; 2 —конечный продукт после ох¬
лаждения до 25°С — гракс-fPtPyЫ229
Рис. 8.13. Схема термоизомеризацни
комплексовплоскоквадратныхцированного метода расчета нормальных колебаний. Матрицы кинематических
коэффициентов [(цс-квадрата, тетраэдра и щраяс-квадрата рассчитывались для
пятиатомной модели по стандартной схеме. Исследование показало, что изме¬
нение численного значения этой матрицы при изомеризации через тетраэдр
невелики, т. е. нехарактеристичность частот v(Pt—I) должна быть обуслов¬
лена в первую очередь особенностями матрицы динамического взаимодейст¬
вия. Они рассчитали сдвиг экспериментально измеряемой «базовой» частоты
'J;is(Pt—I) комплекса транс-конфигурации при переходе от квадрата к гипо¬
тетическому тетраэдру. В результате получили, что если «базовая» частота
v(Pt — I) транс-квадрата равна 181 см-1, то расчетные частоты для тетраэдра
(с вовлечением в гибридизацию различных орбиталей) находятся в интервале
163—171 см”1.Результаты расчетов, проведенных Коноваловым и Погаревой,
удовлетворительно согласуются с предположением о внутримолеку¬
лярном механизме изомеризации с тетраэдрическим ннтермедиатом
(рис. 8.13). Поскольку значительные смещения больших молекул при
изомеризации вряд ли осуществимы, есть основания включить в рас¬
сматриваемый процесс этап образования промежуточного' геометри¬
ческого изомера, обеспечивающего минимальное перемещение лиган¬
дов из исходных положений в конечные. Эгому условию отвечает про¬
межуточная тетраэдрическая конфигурация (см. рис. 8. 13). При
увеличении температуры искажения могут стать настолько велики,
что плоскостной комплекс превращается в тетраэдрический. Обрат¬
ный переход тетраэдрического интермедиата в плоскоквадратный комп¬
лекс может протекать по трем направлениям. Одно из них приведет
к прежнему расположению плоскости квадрата, а два других вызы¬
вают поворот плоскости по отношению к исходному положению на
90°. Одновременно с поворотом плоскости квадрата меняется распо¬
ложение лигандов, т. е. геометрическая конфигурация комплекса.Известно, что четырехкоординационные комплексы никеля (И)
в растворах часто находятся в виде двух равновесных форм: тетра¬
эдрической и квадратной. Можно предполагать, что легкость превра¬
щения квадратного комплекса в тетраэдрический будет увеличивать¬
ся в ряду комплексов: платина (II) < палладий (II) С никель (II).
С этой точки зрения понятна более легкая изомеризация комплексов
палладия (II) по сравнению с соответствующими комплексами пла¬
тины (II).Таким образом, для предсказания направления твердофазной изо¬
меризации комплексов платины (II) и палладия (II) в качестве инди-'
каторного необходимо выбрать нейтральный лиганд низкого трапс-
влияния. Изомеризация протекает в направле¬
нии образования комплекса, в котором этот
лиганд будет располагаться против друго¬
го лиганда с наименьшим трансвлиянием.Изомеризация в растворе. Растворитель, безусловно, играет
важную роль в механизме процесса изомеризации. Иногда он оказы¬
вает существенное влияние на энергетику и даже на направление
этого процесса. Например, установлено, что тетраметиленсулвфидное
соединение транс-[Pt(SC4H8)„Br,,] при нагревании в кристаллическом
состоянии превращается в комплекс 1<ис-конфигурации. Нагревание
же quc-[Pt(SC4H8)2Br2] в водно-ацетоновом растворе приводит к обрат¬
ной реакции — превращению соединения г^ыс-конфигурацни в тганс-
изомер. Однако в большинстве известных случаев направление про¬
цесса изомеризации комплексов платины (II) и палладия (II) в твер¬
дой фазе и растворе совпадают. Таким образом, сформулированное
правило термической изомеризации может быть вполне применимо и
к процессам, протекающим в растворах. На его основе часто можно
предсказывать, какой из двух возможных изомеров должен образовы¬
ваться с наибольшей вероятностью. Как уже было сказано, фосфи¬
новые комплексы платины (II) и палладия (II) типа I в твердой фазе
с выделением энергии изомеризуются в комплексы типа II:SnCl3 SnCl3I ICl-M—PR3 R3P— M—PR3I IPR.3 cii иЭ. H. Юрченко с сотр. получил комплексы [Pt(PR3)„(SnCl3)Cl]
из растворов и пришел к выводу, что образование преимуще¬
ственно транс-комплексов [Pt(PR3)2(SnCl3)CI] при взаимодействии
i<uc-m/?aHc-[Pt(PR3)„Clo] с SnCU является общим свойством, мало за¬
висящим от природы органического растворителя.Вполне возможно, что правило справедливо для плоскоквадрат¬
ных комплексов не только платины (II) и палладия (II), но и иридия(I), родия (I) и золота (III).Известно, что лиганды, образующие связь с металлом посредством
атома углерода, обладают очень высоким трансвлиянием. Это относит¬
ся к карбонилу, изонитрилам и особенно к карбанионам. Следова¬
тельно, явление геометрической изомеризации для плоскоквадратных
комплексов с этими лигандами должно быть особенно характерно.231
Еще в 1959 г. Чатт и Шоу (установили, что ^«c-[Pt(PEt3)2(Ph)Cl] в
кипящем растворе петролейного эфира переходит в т/7анс-изомер:PEt3 PEt3I IPh—Pt—PEt3 -* Ph— Pt-CII ICl PEt3Свободный триэтилфосфин катализирует этот процесс. Поскольку фе¬
нил обладает более высоким трансвлиянием, чем триэтилфосфин, то
процесс протекает в сторону образования транс-изомера.Позднее было установлено, что в метаноле изомеризация данного
комплекса протекает и без каталитических добавок фосфина, а комп¬
лекс 4uc-[Pt(PEt3)2(Et)Cl] полностью изомеризуется, как только он
растворяется в этом растворителе. Самопроизвольная изомеризация
комплексов i<«c-[Pt(PEt3)2(R)Br] изучена на широком круге объектов
с различными лигандами R. Для всех исследованных соединений в
метаноле изомеризация заканчивалась также практически за время
растворения. В получающемся продукте обнаруживались лишь сле¬
ды исходного комплекса. Во многих работах изучалась кинетика и
механизм изомеризации в растворе комплексов этого типа. Боль¬
шинство авторов пришли к выводу, что стадией, определяющей ско¬
рость реакции, является разрыв связи платина-галогенид и обра¬
зование трехкоординационного интермедиата.Недавно установлено, что палладиевый комплекс
i^uc-([Pd(PEt3)2(C6CI5)Cl] в хлороформе изомеризуется в транс-форму.
Небольшие добавки свободного фосфина, так же как и в случае ана¬
логичного платинового комплекса, катализируют процесс изомери¬
зации.Установлено, что комплекс 4uc-[Pt(PPh3)3(CH2CN)Cl] в ацетоне
и бензоле при комнатной температуре количественно превращается
в транс-изомер по схемеPPh3 PPh3I INCCHi —Pt—PPh3 — NCCH— Pt—Cl' I ICl PPh3Самопроизвольная изомеризация этого комплекса также изучена
в дентерохлороформе. Из ее результатов следует, что цис- транс
изомеризация идет не до конца, а устанавливается равновесие, кон¬
станта которого К = транс-нзожр/цис-изошр =4. На основании
данных ЯМР сделан вывод, что трансвлияние цианоалкильного ли¬
ганда больше, чем трансвлияние трифеннлфосфина. Таким образом,
направление изомеризации комплекса t{iic-[Pt(PPh3)2(CH2CN)CI] на¬
ходится в полном согласии с правилом.Поскольку комплексы палладия (II) гораздо лабильнее аналогич¬
ных комплексов платины (II), то препаративно удается получить
палладиевое соединение [Pd(PPh3)2(CH.’CN)Cl] лишь в транс-конфи¬
гурации.232
Как уже было сказано, Эллис, Вайль и Орчин осуществили твердо¬
фазное превращение карбонильных комплексов по схемеС1 АCoJt—А СО—F't—С1Поскольку группа СО обладает гораздо более высоким трансвлия¬
нием, чем амины, то изомеризация идет в сторону образования комп¬
лексов цыс-конфигурации. Трансвлияние фосфинов гораздо выше
трансвлияния аминов, однако было установлено, что изомеризация
аналогичных фосфиновых и арсиновых комплексов протекает в том же
направлении:Cl LI IСО—Pt—L СО—Pt-ClI ICl Clгде L — PBu3, PEt3, РМегРЬ, РМеРЬг, PPh3, AsMePhs, AsPh3. Кроме
того, было показано, что при комнатной температуре в растворе
бензола и хлороформа реакция полностью протекает за несколько
дней. Свободный оксид углерода (II) катализирует процесс. Реакция
также ускоряется под действием видимого и ультрафиолетового света.Недавно методом ЯМР установлено, что при взаимодействии PhCN
с PtCh получается смесь транс- и г^ис-изомеров [Pt(NCPh)sCb]. При
повышении температуры от 26 до 180°С соотношение изомеров трансI
цис изменяется от 0,46 до 5,3. Таким образом, увеличение
температуры способствует цис- -> транс-изомеризации комплекса
4«c-[Pt(NCPh)2Cls] в растворе бензонитрила. Это находится в со¬
гласии с правилом. Интересно отметить, что значение АН изомери¬
зации t^c-fP^NCPh^Ch] в транс-изомер составляет 18 кДж моль,
что хорошо согласуется со значениями АН изомеризации диаминди-
галогенидных комплексов платины (II).Как уже было сказано, направление твердофазной изомеризации
дигалогенид-бис-фосфиновых комплексов платины (II) не всегда
согласуется с правилом изомеризации. Кларк с сотр. показали, что
в дихлорметане комплексы mpaHC-[PtLL'Cb], где -L — PEt3, L' —
различные фосфины, а также AsPh3 и SbPh3, изомеризуются в соот¬
ветствующие соединения t^c-конфигурации. Таким образом, ре¬
зультаты этой работы свидетельствуют о том, что смешанные бис-
фосфиновые комплексы платины (II) в растворе дихлорметана ведут
себя в полном соответствии с правилом изомеризации.Следует отметить, что комплексы родия (I) и иридия (I) типа
[M(PPh3)2(CO)Cl] известны только в транс-форме. Возможно, при¬
чиной этого также является высокое трансвлияние СО, что приводит
к неустойчивости изомера цшг-конфигурации.Правило изомеризации распространяется на все более широкий
круг объектов. В связи с этим на основании экспериментально уста¬233
новленного направления процесса изомеризации можно судить об
относительном трансвлиянии внутрисферных лигандов, местополо¬
жение которых в ряду трансвлияния еще не вполне определенно.
Так, Кукушкин с сотр. показали, что циклогексилизонитрильный
комплекс m/?aHc-[Pt(CNR)2(SCN)2] при нагревании' плавится, а затем
превращается в г{«с-изомер. Это дало основание считать, что транс-
влпяние циклогексилизонитрила больше, чем трансвлияние рода¬
нидного лиганда.В настоящее время не вызывает сомнения, что динамическое транс¬
влияние — явление кинетическое. С этой точки зрения взаимосвязь
трансвлияния внутрисферных лигандов с процессом направленной
изомеризации не должна вызывать сомнения. Однако изомеризация
безусловно, протекает в сторону образования системы (комплекса)
с меньшей свободной энергией. Если причиной протекания изомери¬
зации действительно является трансвлияние внутрисферных лиган¬
дов, то можно сказать, что в основе явления трансвлпяния лежит и
термодинамический аспект.Библиографию по вопросам изомеризации можно найти в рабо¬
тах [3,4].
ГЛАВА9МЕХАНИЗМЫ РЕАКЦИИ КООРДИНАЦИОННЫХ
СОЕДИНЕНИЙМеханизм химической реакции — это совокупность ее отдельных
стадий, приводящих к превращению исходных реагентов в конечные
продукты. Как правило, химические реакции протекают через несколь¬
ко последовательных (или параллельных) стадий с образованием про¬
межуточных продуктов (интермедиатов).В ходе элементарного акта химического превращения система ре¬
агирующих частиц (атомов, молекул, ионов, радикалов) образует
активированный комплекс* (переходное состояние). Активирован¬
ный комплекс характеризуется максимальной потенциальной энер¬
гией для данного пути реакции (рис. 9.1). Однако она меньше, чем
максимальная потенциальная энергия на любом другом пути.Для того чтобы произошла реакция, реагирующие частицы долж¬
ны обладать определенной энергией (энергией активации), образовы¬
вать активированный комплекс, перераспределять между собой энер¬
гию и разделяться на продукты реакции.На рис. 9.1 графически изображен путь реакцииК+ XY= КХ +Yкоторая протекает в одну стадию. Величина Еа есть энергия акти¬
вации.Если реакция протекает в две стадии, например
К+АХ = КАХ
КАХ = КА + Xто образуется промежуточное соединение — интермедиат (рис. 9.2).Детальный механизм требует полного описания активированных
комплексов (состава, геометрии, энер¬
гии) на всех стадиях химической ре¬
акции. В растворах активированный е
комплекс сольватирован, и это допо-,
лнительно усложняет его описание.Время жизни активированного ком¬
плекса обычно очень мало. Оно прак-* Активированный комплекс не име¬
ет отношения к комплексному или коор¬
динационному соединению.К- X—YРис. 91. Энергетический рельеф реакцииКоордината реакция235
Рис. 9.2. Энергетический рельеф реак¬
ции с образованием интермедиата:1 — активированный комплекс К---Л—X; 2 —
интермедиат К—А—X; 3 — активированный
комплекс К—Л--Xтически равно времени акта хими¬
ческой реакции, т. е. 10~12—10‘13 с.Таким образом, изучение акти¬
вированного комплекса экспери¬
ментально крайне затруднительно.Время существования интермеди¬
атов гораздо больше, иногда они доступны для прямого обнаружения.
Поэтому задача выявления механизма реакции часто решается путем
прямого или косвенного идентифицирования промежуточных соеди¬
нений.Наибольшую информацию о механизме реакции получают при
изучении ее кинетики в зависимости от различных факторов: кон¬
центрации реагентов, температуры, природы растворителя, электро¬
литного фона и др.Описание механизма химической реакции проводят на основе де¬
дуктивного метода, т. е. на основании результатов изучения хими¬
ческого явления делают заключение об отдельных стадиях химическо¬
го взаимодействия.Прямой эксперимент практически никогда не дает возможности
однозначно судить о детальном механизме химической реакции. На
основании кинетических, структурных или термодинамических дан¬
ных умозрительно подбирают механизм реакции. Он должен соот¬
ветствовать всем известным экспериментальным фактам. Чем больше
таких фактов, тем вероятнее механизм. Однако никогда нельзя ска¬
зать, что механизм реакции установлен окончательно. С появлением
новых экспериментальных возможностей могут появиться аргумен¬
ты, противоречащие данному механизму, и тогда его следует считать
неверным. С другой стороны, новые экспериментальные данные могут
привести к дальнейшей детализации уже выявленного механизма ре¬
акции. Таким образом, степень детальности механизма химической
реакции зависит от уровня накопленных знаний и эксперименталь¬
ных возможностей.Несмотря на такое положение, учение о механизмах химических
реакций является одним из фундаментов химической науки. Знание
механизма, пусть иногда несовершенное, дает возможность управ¬
лять реакциями, планировать и осуществлять новые химические
процессы, позволяет синтезировать новые вещества и получать из¬
вестные соединения новыми способами.236
9.1. Некоторые понятия в учении о скоростях
и механизмах химических реакций9.1.1. Молекулярность, порядок реакции, закон скоростиДля выявления механизма весьма существенное значение имеет определение
молекцлярности реакции. Молекулярность — это число реагирующих частиц,
входящих в уравнение химической реакции н участвующих в образовании ак¬
тивированного комплекса. Поскольку реакции, как правило, протекают по-
стадинно, то правильнее говорить о молекулярности определенной стадии ре¬
акции. К сожалению, экспериментальное определение молекулярности реакции
столь же сложно, как и определение состава активированного комплекса.Молекулярность стадии реакции определяется числом частиц (ионов, ра¬
дикалов), участвующих в элементарном акте* химического превращения. К со¬
жалению, молекулярность трудно подвергнуть экспериментальному наблюде¬
нию и охарактеризовать количественно.На практике широко пользуются понятием «кинетический порядок реак¬
ции». Иногда имеется корреляция между молекулярностыо и порядком реак¬
ции.Различают порядок реакции подвеем реагентам** в целом и порядок реакции
в отношении каждого из реагентов. Порядок реакции — это показатель сте¬
пени, в которую возведена концентрация в кинетическом выражении скорости
реакции. Например, при взаимодействии [PtClj*- со щелочью в водном раство¬
ре установлено, что скорость реакции прямо пропорциональна концентрации
комплекса и не зависит от концентрации щелочи. Следовательно, выражение
кинетического уравнения имеет видv=k [PtClj-],где v — скорость реакции; k —- константа скорости реакции; [PtCl4 ] — кон¬
центрация реагента, от которого зависит скорость реакции.Таким образом, взаимодействие [PtCI|]"- со щелочью в водном растворе
в соответствии с уравнением[PtCI4]-‘- + OH- [Pt (ОН) С13Г-- + Cl-является реакцией первого порядка относительно концентрации комплекса.
Общин порядок этой реакции также первый, поскольку концентрация щелочи
не влияет на скорость реакции.По существу, скорость данной реакции должна зависеть от концентрации
растворителя. Порядок реакции по концентрации растворителя обычно не учи¬
тывают. Однако участие растворителя отражают в названии. Так, например,
взаимодействие (PtCI4]J" со щелочью в водном растворе называют не просто ре¬
акцией первого порядка, а реакцией псевдопервого порядка. Слово «псевдо»
означает, что порядок по концентрации растворителя не определяли.Выражение скорости данной реакции правильнее было бы записать в сле¬
дующем виде:o = |fc' [PtCl;-] [Н*0].Следует заметить, что выражения скорости реакции тнпа u=£[PtC!^ ] на¬
ряду с кинетическим уравнением часто называют законом скорости.* Элементарный акт — это химическое превращение реагента на отдельной
стадии реакции. Химические реакции, состоящие из одной стадии, называют
элементарными.** Исходные реагенты иногда называют реактантами.237
Экспериментально установлено, что взаимодействие [PtCIi]2- с аммиаком
в водном растворе с образованием [Pt(NH3)CI3]-[PtC]4]=- -I- NH3 = [Pt (NH3) Ci3]- -h Cl-характеризуется законом скоростиu = [PtCl^—] [NH3].Таким образом, эта реакция является реакцией второго порядка — первого
по концентрации комплекса м первого по концентрации аммиака. Весьма ве¬
роятно, что активированным комплексом является {[РtCI- NH3}*. В этом
случае можно было бы сказать, что молекулярность реакции равна двум. Тогда
молекулярность и порядок реакции совпали бы. Однако, как уже было сказано,
доказательство молекулярностн — задача очень сложная.Считают, что наиболее распространенным случаем являются бимолекуляр¬
ные реакции, реже — мономолекулярные реакции и почти не встречаются три-
молекулярпые и реакции более высоких степеней молекулярности.9.1.2. Механизмы нуклеофильного замещения лигандовSni и Sn2Теория механизмов реакций замещения в начале была разработана для соеди¬
нений с насыщенным атомом углерода. В зависимости от характера разрыва
связи С—X, природы замещаемого атома и природы атакующего реагента ре¬
акции замещения в органических соединениях были разделены натри типа: ра¬
дикальный — символическое обозначение* SR электрофильный — SE и нук¬
леофильный Sj^ В радикальном механизме реакции происходит разрыв хими¬
ческой связи по гемолитическому типу с образованием нового радикала,
например, по схемеCf-;-'X + V С : Y г X*При электрофнльном механизме реакции происходит замещение атома с недо¬
статком электронной плотностн, т. е. атома, эффективный заряд которого по¬
ложительный. Атакующим также является атом или группа атомов с недостат¬
ком электронной плотности**. При нуклеофильном механизме реакции проис¬
ходит замещение атома с избытком электронной плотности. В этом случае
атакующим является атом или группа атомов также с избытком электронной
плотности***.В координационных соединениях нон металла является электрофильный
центром, а лиганды, как правило, представляют собой нуклеофильные части¬
цы. Поэтому реакции замещения лигандов в координационных соединениях
практически всегда являются нуклеофильными. В связи с этим имеет смысл
несколько подробнее рассмотреть механизмы нуклеофильного замещения у на¬
сыщенного атйма углерода.В 1935 г. Хьюз и Ингольд пришли к заключению, что существует два типа
механизмов нуклеофильного замещения, которые различаются кинетически,
и можно теоретически предсказать, по какому механизму должно происходить
то или иное замещение.Схематично эти два типа механизмов изображаются следующим образом:* Буква S — первая буква английского слова substitution — заме¬
щение.** Электрофильная группа ■— способная притягивать электроны.*** Нуклеофильная группа — способная притягивать ядра, т. е. обла¬
дающая отрицательным зарядом или избытком электронной плотностн.238
Рис. 9.3. Изменение механизма ре¬
акции R — X 1-Y в зависимости от
природы радикала RРис. 9.4. Изменение механизма реакции
R'—X + Y в зависимости от природы
атакующего реактанта YV - - R—X -*■ Y—R Х“ (SN 2 — бимолекулярное нуклеофильное замещение)медленноR—XбыстроY--:-R+ »- Y-R(SN 1 — мономолекулярное нуклеофильное замещение)Хьюз н Пнгольд предсказали, что если взять серию галогеналкильных со¬
единенииСН3Х, СН3 — СН,Х,СН,СН.СН,СНХ,\СН,-. ->-СХСН.то механизм их гидролиза с образованием соответствующих спиртов по реак¬
цииR—Xон-ROH + X"будет различным. С увеличением числа метильных групп (вследствие индуктив¬
ного эффекта по гг-связи) прогрессивно увеличивается смещение электронов к
атому углерода, непосредственно связанному с радикалом X. Это смещение пре¬
пятствует доступу реагента, необходимому для механизма SN2, н способствует
ионизации, необходимой для механизма SNI. Изменение механизма должно
обнаруживаться в изменении кинетики реакции. Если для данной серии соеди¬
нении изобразить изменение кинетики графически, то оно должно примерно
иметь вид, приведенный на рис. 9.3.Наличие двух механизмов нуклеофильного замещения должно наблюдаться
н в том случае, если взаимодействует одно н то же соединение R — X с различ¬
ными реагентами, нуклеофильный характер которых изменяется. В простейшем
случае за меру нуклеофпльности принимают основность реагента. При данном
R — X чем более нуклеофильный характер имеет Y, тем более благоприятные
условия создаются для замещения по механизму SN2. Поэтому если взять ряд
реагентов и расположить их в порядке уменьшения основности, например, ОН-,
OCfiH~, COf, СН3СОО-, СГ, то в этом ряду слева направо должно наблюдаться
изменение механизма от S^2 к SN1. Оно проявляется в изменении кинетики
соответствующих реакций. Графическое изменение кинетики должно иметь вид,
приведенный на рис. 9.4.Эти представления о механизмах нуклеофильного замещения были перене¬
сены Пнгольдом и Найхольмом на координационные соединения.239
9.1.3. Активационные параметры реакцииВзаимосвязь между константой скорости реакции к и энергией активации Ел
определяется уравнением Аррениусаk = Ae~E а/(ЛГ\где А — предэкспоненциальный множитель, связанный с вероятностью и чис¬
лом столкновений; е — основание натуральных логарифмов; R — газовая по¬
стоянная; Т — температура, К. Из этого уравнения следует, что чем больше
энергия активации, тем меньше константа скорости реакции, т. е. тем меньше
скорость реакции.Уравнение Арренмуса легко прологарифмировать:In к = In Л — •RTЕсли в качестве переменных выбрать In к и 1/Г и отложить их на координатных
осях (рис. 9.5), то графически это уравнение выразится прямой линией. Тангенс
угла наклона прямой tgtp — —EJR позволяет определить графически энергию
активации химической реакции.Во многих случаях зависимость In k от 1/Г выражается не прямой, а кри¬
вой. Это говорит о сложном характере механизма реакции и часто указывает на
протекание реакции по нескольким направлениям.Иногда график в координатах In к — 1/Г имеет вид ломаной кривой. Это
указывает на то, что в температурном интервале, соответствующем излому на
кривой, происходит изменение механизма реакции.Теория активированного комплекса (теория переходного состояния) дает
возможность вычислить для реакции теплоту активации Д Н* и энтропию ак¬
тивации AS*. Эти параметры являются важными характеристиками системы.
В терминах теории активированного комплекса выражение константы скорости
имеет следующий вид:k Т bS'/R —±H*/{RT)к = е е ,hгде к — постоянная Больцмана; h — постоянная Планка.Теплота активации АН* связана с энергией активации выражениемДЯ* = Еа — RT.При комнатной температуре RT « 2,5 кДж/моль.Значение энтропии активации зависит от выбора стандартного состояния
системы или, что то же самое, от выбора концентрационных единиц. Однако
при любом выборе AS* становится более отрицательной для реакции между дву¬
мя полиатомными молекулами, чем для реакции между двумя атомами. Значение
энтропии активации дает возможность судить о характере активированного
комплекса. В координационной химии кинетика и механизм реакций чаще все¬
го изучаются в растворах. Для реакций между ионами одного знака, как прави¬
ло, энтропия уменьшается при переходе от реагентов к активированному комп¬
лексу. Это связано с сольватационными (де-
сольватационными) процессами. Если активи¬
рованный комплекс образуют ионы противо¬
положного знака, то в переходном состоянии
реагирующие ионы будут частично десольва-
тироваться. Это связано с тем, что при объе¬
динении ионов с различными зарядами общийРис. 9.5. Графический способ вычисления
VT энергии активации240
заряд понижается и в связи с этим понижается и общая их сольватация. В ре*
зультате будет «освобождаться» некоторая часть «связанных» молекул раство¬
рителя и это должно приводить к увеличению энтропии.В случае, если взаимодействуют ионы одного знака, активированный комп¬
лекс будет иметь более высокий заряд. Это обеспечивает большую сольватацию
активированного комплекса,чем двух отдельных ионов. В результате при пере¬
ходе от исходных реагентов к активированному комплексу произойдет допол¬
нительное «связывание» молекул растворителя, т. е. уменьшится энтропия.
Ниже приведены значения энтропии активации для некоторых реакций.Энтропия активации реакций между ионами в водном растворе [1]Реагирующие ионы AS*, э. е. Реагирующие ионы AS*, э. е.Сг(НоО)^,+ + SCN" 0,7 CHjClCOO" + — 17,7Со^НзЬВг* + ОН- 20,1 Co(NH3)5Br2+ + Hg"+ -23,6C10--fC10- — 19,6 S204_+S20^ —41,2СЮ- + С102 • • — —19,8 s20j*--fS0§- —31,4CHjBrCOO- + SjO‘3 . . . .—28,3В последние годы в координационной химии начала применяться экспери¬
ментальная техника, позволяющая изучать кинетику реакций в растворах npir
различных гидростатических давлениях [2, 3]. Она позволяет получить значе¬
ния объемов активации (Л V*), на основе которых можно сделать заключение о
механизме реакции. Объемы активации рассчитывают по формулеAV*= — RT(d\nkp/dP)T ,где R и Т — газовая постоянная н температура; kp — константа, скорости ре¬
акции при заданном давлении; Р — гидростатическое давление.Важной характеристикой является парциальный молярный объем реакции
(ДУ'°). Он равен разности между объемом продуктов реакции и исходных ре¬
агентовД1"° ^ ^прод — 2L ^исхи может быть вычислен из формулыAV°=-RT (din К/дР)т )где К — константа равновесия.Для представления о порядке величин укажем, что молярный объем иона
Ni(NH3)|+ равен 138 см3-моль-1, а нона Co(NH3)|+ 55 см3-моль-1.Таким образом, объемы активации рассчитывают по данным изменения
констант скорости реакций при изменении давления, а молярные объемы реак¬
ции вычисляют изданных констант равновесия при различных давлениях пли по
независимо определенным молярным объемам исходных реагентов и продуктов,
реакции.Значение объема активации положительно, если скорость реакции умень¬
шается с увеличением давления, и отрицательно,если скорость увеличивается
с увеличением давления.Рассмотрим подход к выявлению механизма реакции по значению объема
активации на примере октаэдрических комплексов. Можно принять, что пяти¬
координационный интермедиат ML5 тригонально-бипнрамидального типа, полу¬
чающийся в результате диссоциации комплекса ML6, и семикоординацнонный
интермедиат ML.7, получающийся в результате присоединения лиганда L к
комплексу ML6, имеют примерно тот же молярный объем, что и ML„. Расчетные
данные оказались в хорошем согласии с этими допущениями.Если L — нейтральные лиганды, то при диссоциативном механизме в пере¬
ходном состоянии (ML5 + L) молярный объем возрастает по сравнению с MLS241
и, следовательно, значение объема активации положительно. При ассоциатив¬
ном механизме в переходном состоянии ML.7 молярный объем уменьшается по
сравнению с (ML0 -|- L) и, следопательно, значение A V* отрицательно.9.1.4. Солевой эффектВлияние ионной силы раствора на скорость химической реакции довольно хо¬
рошо обосновано теоретически. Бренстед и Н. Бьеррум независимо друг от
друга на основании теории Дебая — Хюккеля вывели выражение, связывающее
константу скорости реакции с ионной силой раствора. Если в реакции участвуют
ионы А и В, то это выражение имеет видгде к — константа скорости реакции; k0— константа скорости при коэффициен¬
тах активности реагентов н активированного комплекса, равных 1; D —диэлект¬
рическая постоянная растворителя; Т — температура; / — ионная сила раст¬
вора, равная полусумме произведений концентраций ионов на квадрат их за¬
ряда. _Если построить график в координатах lg(k/k0) — V!, то при постоянном
температуре зависимость между этими величинами должна быть линейной. Знак
тангенса наклона прямой линии определяется знаками реагирующих ионов
(рис. 9.6).Из рис. 9.6 следует, что в случае взаимодействия одинаково заряженных
ионов знак тангенса угла положительный, а в случае взаимодействия различно
заряженных ионов отрицательный. Если в образовании активированного комп¬
лекса участвует нейтральная частица и ион, то ионная сила сказывается очень
мало или вообще не влияет на константу скорости реакции.Поскольку теория Дебая — Хюккеля применима только к разбавленным
растворам, то линейную зависимость можно ожидать лишь в области малых
значений ионной силы. При большой ионной силе может наблюдаться солевой
эффект и в реакциях с участием незаряженных частиц.Таким образом, на основании характера влияния ионной силы на константу
скорости реакции можно судить о зарядах частиц, принимающих участие в об¬
разовании активированного комплекса. Следует отметить, что имеются случаи,
когда скорость химической реакции с изменением ионной силы не описывается
столь простой зависимостью Ig (k/k0) от У1.Влияние ионной силы на константу скорости реакции называется первич¬
ным солевым эффектом.В координационной химии изменение констант скорости реакций с изме¬
нением солевого фона (концентрации постороннего электролита) довольно часто
связано с образованием внешнесферных комплексов (ассоциатов) [4]. Так, на¬
пример, при взаимодействии [Co(NН3)5Вгр+ с ОН- по реакцииlg к = lg k0 -1-3,65 • 10Ю~’/г TzA zB у I ,11.200.-10О.ЙО0,200VJ 2- S,01~ + 1- -* SO’- H- S07 + I3 — C12H22011 -|- OH- -*■ инверсия
4- [Co(NH3)6Br[-+ -J- OH- ->■— [Co(NH3bOH]2+ + Br242
lCo(NH3)5Br]“+ + OH- -v [Co(NH3)5OH1*- + Br-на сульфатном фоне образуется ассоцнат {|Co(NH3)6Br]S04°}, что сильно ска¬
зывается на скорости реакции. В этом случае сульфатный фон нельзя считать
инертным.Ионная сила влияет на активность ионов, образующихся в результате дис¬
социации слабых электролитов. Например, активность ацетатного нона, обра¬
зующегося в результате диссоциации уксусной кислоты равна/ *а“сН,СООН
ясн3соо- х \/ f ! *V I Н+ ' СНзСОО-С увеличением ионной силы коэффициенты активности /н+ и /сн,с00- Умень'
шаются, а Ясн3сОО- — возрастает. Увеличение же активности ионов приводит к
увеличению скорости реакций, в которых они участвуют. Изменение скорости
реакции при изменении ионной силы раствора, происходящее в результате изме¬
нения активности участвующих в реакции ионов, называется вторичным соле¬
вым эффектом.9.2. Лабильные и инертные координационные
соединенияМеханизм реакции в значительно большей мере связан с кинетиче¬
скими характеристиками, чем с термодинамическими. Термодинами¬
ческий подход к химическому процессу связан только с начальным
и конечным состоянием системы. Путь, по которому протекает реак¬
ция, для термодинамических расчетов не имеет значения. Термо¬
динамические параметры характеризуют систему, находящуюся в
равновесии.Кинетика дает информацию о скорости, с которой протекает реак¬
ция, или о скорости, с которой система приходит к равновесному
состоянию.При рассмотрении состояния комплекса в растворе или газовой
фазе с термодинамической точки зрения принято использовать тер¬
мины «стабильность» (противоположный ему — «нестабильность») или
«устойчивость» (противоположный ему — «неустойчивость»). Отме¬
тим еще раз, что они характеризуют комплексы в равновесных состо¬
яниях.При рассмотрении комплексов с кинетической точки зрения обыч¬
но используют термин «инертность» или противоположный ему «ла¬
бильность». Они характеризуют скорость, с которой комплексы под¬
вергаются превращениям и приходят к состоянию равновесия.Следует подчеркнуть, что стабильность и инертность не всегда
симбатны. Так, при растворении в воде ^[PtCU] и Ki[PtBr4] комп¬
лексные ионы подвергаются реакции акватации:[PtCl4]2- + Н20 ** [Pt(H20) С13]- + CI-[PtBr4]2- + н20 [Pt(H20) Br3]~ + Br-Константа равновесия хлоридного комплекса равна 1,7-10~2, а бро-
мидного 2,4-Ю"3. Таким образом, комплексный ион [PtBn]2- в вод¬243
ном растворе примерно на порядок устойчивее, чем ион [PtCU]- . Кон¬
станта скорости акватации иона [PtCU]2' равна 0,39-10"4 с , а иона
[PtBrj]2- 1,75-10-4 с'1. Из этого следует, что более стабильный с тер¬
модинамической точки зрения ион [PtBr-i]3-, оказывается менее инерт¬
ным (с кинетической точки зрения). Таким образом, в данном случае
проявляется антибатность между стабильностью и инертностью.Впервые четкая антибатность между стабильностью и инертностью
комплексов была обнаружена Гринбергом и Никольской. Было хо¬
рошо известно, что стабильность увеличивается в ряду комплексов
(внизу приведены значения общей константы неустойчивости для
каждого иона)lPtCI4]2- < [PtBr-jl2- < [PtI4P- < [Pt (CN)4F-
2,5-10“17 4-1СГ21 2,5- I0-30 MO-41Изотопный обмен лигандов в этих комплексах показал, что ско¬
рость установления равновесия по уравнениюlPtX4]=- + 4*Х- =г± [PtX2*X.J2- + 2*Х- + 2Х-самая высокая в цианидном соединении. Она уменьшается в ряду
комплексов(Pt(CN)4]-- > [Ptl4]2- > [PtBr4]2- > [PtCI4]2-Обмен цианидных ионов в [Pt(CN)4]2- происходит за время сме¬
шивания растворов комплекса и K*CN. Период полуобмена* хлорид-
ных ионов в [PtCl4]2~ равен примерно 14 ч.Антибатность термодинамической устойчивости и кинетическом
лабильности — явление, довольно распространенное в координацион¬
ной химии переходных металлов, однако оно проявляется не во всех
случаях. Чаще всего оно наблюдается в рядах однотипных соедине¬
ний с одинаковым центральным атомом. Если взять аналогичные комп¬
лексы платины и палладия, например [PtCli]2" и [PdCU]2-, то оказы¬
вается, что более стабильный платиновый комплекс является и более
инертным.Таким образом, на основании данных термодинамической устой¬
чивости нельзя судить о кинетической лабильности. ,Лабильность комплексных соединений зависит ог многих факто¬
ров. Наиболее важными среди них являются: электронная структура,
радиус и состояние окисления центрального атома, природа лиган¬
дов, координационное число, заряд комплексного иона.Хорошо известно, что растворитель часто оказывает весьма су¬
щественное влияние на скорость химической реакции. Например,
изотопный обмен хлорид-ионов в [PtCl4]2- при 25 °С в воде происходит
со скоростью, период полуобмена которой около 14 ч. При тех же
условиях в ацетоне период полуобмена составляет около 170 ч. При¬
чиной этого могут быть различная сольватирующая способность, вяз¬
кость, диэлектрическая проницаемость и др. Часто молекулы раство¬* Время, за которое обмен протекает на 50%.244
рителя принимают непосредственное участие в механизме реакции,
входя в состав активированных комплексов.Роль отдельных факторов, влияющих на лабильность комплек¬
сных соединений, пока четко не выявлена. Во многих случаях при
переходе от одного комплекса к другому изменяется сразу несколько
факторов. В настоящее время существует лишь одна теория, которая
связывает лабильность комплексов с электронной структурой цент¬
рального атома. Ее сформулировал в 1952 г. Таубе [5]. С тех пор не
было других существенных обобщений в этом вопросе.Теория Таубе относится лишь к комплексам октаэдрического типа.
Однако необходимо отметить, что октаэдрические комплексы пере¬
ходных металлов являются наиболее распространенными. Таубе счи¬
тает, что лабильными комплексами являются те, реакции с участием
которых заканчиваются за 1 мин при комнатной температуре и кон¬
центрации раствора 0,1 М.Большой материал по исследованию лабильности комплексных
соединений был получен методом изотопного обмена лигандов и цент¬
ральных атомов. Реакция изотопного обмена лигандов в общем видеML„ -г *L ML* L + LРеакция изотопного обмена центральными атомами
ML6 -1- *М (Н20)6 ч* *ML6 + М(Н30)„Быстрый обмен центральными атомами свидетельствует о лабильнос¬
ти обоих комплексов. Медленный обмен или его отсутствие может оз¬
начать, что по крайней мере один из комплексов является инертным.Существование комплексов в двух изомерных формах, как прави¬
ло, свидетельствует об их инертности. Изомерные соединения должны
отличаться (хотя бы немного) по свободной энергии. Поэтому в слу¬
чае лабильных комплексов изомер с большей свободной энергией дол¬
жен перейти в изомер с меньшей свободной энергией. В результате
для лабильных комплексов синтезировать изомерные соединения в
индивидуальном состоянии невозможно.Метод ЯМР позволяет следить за быстропротекающими реакция¬
ми и часто дает возможность идентифицировать в растворе изомерные
формы лабильных комплексов.Таубе пришел к выводу, что предсказание лабильности или инерт¬
ности октаэдрических комплексов может быть сделано на основании
их электронного строения с позиции метода валентных связей. Окта¬
эдрические комплексы внутриорбитального типа характеризуются
^^-гибридизацией атомных орбиталей центрального атома, а комп¬
лексы внешнеорбитального типа характеризуются s/AP-гибридиза¬
цией.Для того чтобы комплексы внутриорбитального типа были ла¬
бильными, необходимо иметь вакантную d-орбиталь. Для таких комп¬
лексов характерна d°, d1, d? электронная конфигурация. В табл. 9.1
приведены схемы электронного строения внутриорбитальных инерт¬
ных и лабильных комплексов.245
Таблица 9.1. Ионы металлов, образующие внутриорбитальныеоктаэдрические комплексыЭлектронная конфигурацияТипИоны-ком плексообразователи(л-| ) dns прtltltltltltld2sp-ttltltltltltldP1tttltltltltltld2sp'ittttltltltltltlj1tP3tltttltltltltltld1./73titlttltltltltltld2iРУtltltltltltltltltld -\sр}d°d1d‘d3d4d6deSc(III), Y(I1I), РЗЭ(П1),
Ti(IV), Zr(IV), Hf(IV), Ce(IV),
Th(IV), Nb(V), Ta(V), Mo(VI),
W(VI)Ti(III), V(IV), Mo(V), W(V),
Re(VI)Ti(II), V(III), Nb(III),
Ta(III), Mo(IV), W(IV), Re(Vl,
Ru(VI)V(II), Cr(III), iMo(II I),
W(III), Mn(IV), Re(IV)Cr(II), Mn(III), Re(III).
Ru(IV), Os(V)Fe(III), Ru(III), Os(III),
Ir(IV)Fe(II), Ru(II), Os(II), Co(III).
Rh(IlI), Ir(III), Pd(IV), Pt(IV)Согласно теории кристаллического поля октаэдрические комп¬
лексы ионов металлов, имеющих электронные конфигурации d4, d°,
d6, могут быть высокоспиновыми. В этом случае электронная конфи¬
гурация их октаэдрических комплексов будет внешнеорбнтальной.
Внешнеорбитальными также будут октаэдрические комплексы ионов
металлов с электронной конфигурацией d7, d8, а9 и dl°. В этих слу¬
чаях нельзя получить две пустые d-орбитали, необходимые для гибри¬
дизации. Электронная конфигурация внешнеорбнтальных октаэдри¬
ческих комплексов приведена в табл. 9.2.246
Таблица 9.2. Электронная конфигурация внешнеорбитальныхоктаэдрических комплексовЭлектронная конфигурацияТип( п-] f dnsnpndt 1 ftttltltltltltlsp'd21tittttltltltltltlsp-d2flt j ttttltltltltltlsp !d2tltiittttltltltltltlsp^d1tl111tttltltltltltlsp’d2tltltltlttltltltltltlsp^d1tltltltltltltltltltltlspidldld5d6d7dsdsd10Особенно распространены внешнеорбитальные комплексы ионов
металлов ^-конфигурации. Это комплексы Zn(II), Cd(II), Al(III),
Ga(III), In(III), Tl(IIl). Внешнеорбитальные октаэдрические комп¬
лексы в большинстве являются лабильными. Однако их лабильность
уменьшается с увеличением заряда на центральном атоме, например
в ряду А1РГ > SiFf > PFg > SF6. Следовательно, лабильны не
все внешнеорбитальные комплексы.Безусловно, природа лигандов сильно сказывается на лабильнос¬
ти комплексных соединений. В частности, высокоспиновое и низко-247
спиновое состояния ионов металлов d4, йъ и d6 электронных конфи¬
гураций зависит от силы поля лиганда. Следовательно, от силы поля
лигандов зависит и лабильность комплексов. Однако простой взаимо¬
связи между этими характеристиками нет.Лабильность внутриорбитальных комплексов d°, d1, d1 электрон¬
ных конфигураций центральных ионов и большинства внешнеорби¬
тальных комплексов объясняется тем, что наличие вакантных d-орби¬
талей (внутренних или внешних) способствует образованию коорди¬
национных связей со вступающим в комплекс лигандом. Иными сло¬
вами, вакантные d-орбитали способствуют образованию интермедиата
с координационным числом 7. Эти комплексы стремятся вновь вер¬
нуться в шестикоординационное состояние
ML6 + A 5=t ML„A ** ML6A + LСо статистической точки зрения удалению должен подвергаться не
вступивший лиганд, а один из уже находящихся в комплексе. Следо¬
вательно, наличие вакантных d-орбиталей должно способствовать
механизму замещения лигандов по ассоциативному типу.Считается, что внешнеорбитальные комплексы образуют менее
прочные связи с лигандами, чем внутриорбитальные. Поэтому первые
должны быть более склонны к реакции диссоциации с образованием
пятикоординационпого интермедиата. Таким образом, комплексы
внешнеорбитального типа должны обладать относительно большей
склонностью к реакциям замещения по диссоциативному механизму,
чем комплексы внутриорбитальные.9.3. Стехиометрический механизмВ установлении стехиометрического механизма исключительно
важную роль играет экспериментальное обнаружение промежуточ¬
ных продуктов реакции (интермедиатов). На основании их состава
делают заключение о стадиях, по которым протекает реакция. Без¬
условно, число стадий реакции может быть больше, чем число обна¬
руженных интермедиатов. При выявлении стехиометрического ме¬
ханизма путем исследования интермедиатов можно прийти не к са¬
мому верному заключению. Реакции часто протекают по нескольким
параллельным направлениям. Интермедиат же отражает лишь одно
из них. Реакция может протекать через несколько последовательных
стадий, а интермедиат может дать указание лишь на одну. Таким
образом, другие стадии и направления реакции могут выпасть из поля
зрение исследователя. Тем не менее обнаружение промежуточных
продуктов является прямым доказательством по крайней мере одной
из стадий химического процесса.В установлении стехиометрического механизма реакций исключи¬
тельно важную роль сыграла препаративная химия. Большие успехи
достигнуты при изучении реакций замещения в инертных комплек¬
сных соединениях Co(III), Cr(III), Pt(II), Pt(IV) и др. При выясне¬
нии стехиометрического механизма можно обойтись и без кинетиче¬
ских исследований. Для этого нужно лишь выявить промежуточные248
продукты. Методы их обнаружения могут быть самыми разнообраз¬
ными. Тем не менее наиболее распространенным методом доказа¬
тельства стехиометрического механизма является кинетический.При изучении порядка реакции в отношении отдельных реагентов
удается сделать заключение об их участии в стадии, определяющей
скорость реакции. Так, например, Гринберг с сотр. установили, что
скорость обмена бромид-ионов в [PtBri]'2' зависит от «возраста» раст¬
вора комплексного соединения. Обмен в свежеприготовленном раст¬
воре протекает гораздо медленнее, чем в приготовленном за несколько
часов до начала опыта. При изучении скорости обмена в зависимости
от концентрации K:[PtBr4] и К*Вг было установлено, что она не за¬
висит от концентрации бромид-ионов, т. е. порядок реакции в отно¬
шении концентрации ионов Вг~ нулевой. Все эти экспериментальные
данные дали основание считать, что в водном растворе первой стадией
реакции является акватация комплекса
медленно{PtBr4]2-+ Н20 Ч 1 [Pt(H20) Вг3]“ + Вг~Вслед за ней происходит замещение внутрисферной молекулы воды
бромидным ионом*:быстро[Pt(H,0) Вг3]~ + *Вг- [PtBr’Br]2" -I- Н20Эффект «старения» связан с тем, что в растворе Ka[PtBr4] со вре¬
менем накапливаются аквокомплексы [Pt(H20)Br3]~. Скорость взаимо¬
действия с ними бромид-ионов большая и поэтому реакция обмена
заканчивается скорее.Нулевой порядок реакции в отношении концентрации бромид-ионов
свидетельствует о том, что они участвуют в стадии, скорость которой
не является самой медленной.Предпочтительное взаимодействие свободных ионов Вг- с
[Pt(H20)Br3]“ по сравнению с [ptBu]2- понятно с электростатической
точки зрения. В соответствии с законом Кулона отрицательно за¬
ряженному иону Вг-- легче атаковать аквокомплекс [Pt(H20)Br3]_,
так как его заряд на единицу меньше, чем заряд [PtBr4]2-.Гринберг и Кукушкин изучали кинетику взаимодействия со ще¬
лочью комплексов платины (II). В частности, ими установлено, что
в водном растворе реакция
[Р1С!4р- + ОН- — [Pt(OH)CI3]2- + Cl-является реакцией первого порядка в отношении концентрации комп¬
лекса и нулевого в отношении концентрации гидроксильных ионов.
Это может означать, что гидроксильные ионы не участвуют в медлен¬
ной (определяющей скорость) стадии реакции. Медленной стадией
вновь является акватация комплексамедленно[Ptci4]-- + НоО ^—* [Pt(H„0)CI3]- + С1-* Процесс, обратным акватации, называют анацией. Термин анация приме¬
няется не только к воде, но и к молекулам любого другого растворителя.249
Вслед за этой стадией быстро происходит кислотно-основное взаимо¬
действие между аквокомплексом и гидроксильными ионами:
[Pt(M20)CI3]- + он- « [Pt(OH)Cl3]-- + н2оТаким образом, па основании кинетических данных взаимодейст¬
вия комплексов платины (II) со щелочью (щелочного гидролиза*)
оказалось возможным вычислять константу скорости их акватации.
Для этого нужно лишь экстраполировать экспериментально найден¬
ное значение скорости реакции на условия, при которых концентра¬
ция комплекса равна единице. Константа скорости акватации равна
скорости щелочного гидролиза при концентрации комплекса, равной
единице. В частности, из данных кинетики щелочного гидролиза
следует, что константа скорости акватации комплекса [PtCU]2- равна
0,39-10"4 с-1. Прямые экспериментальные данные замещения хлорид¬
ного лиганда на воду в этом комплексе, полученные Грэнтемом, при¬
водят к константе, равной 0,40 ■ 10-4 с-1.Как уже было неоднократно сказано, реакции могут протекать
по нескольким параллельным стадиям. Например, Грей установил,
что замещение хлоридного лиганда в комплексе [Pt(Dien)Cl]+ на бро-
мидный протекает как через стадию акватации**ki[PtDien С!]+ + Н20 я* [Pt Dien (Н30)]2+ + С1“[Pt Dien (Н20)]2+ + Ег~ ^ [Pt Dien Вг]+ + Н20
так и путем непосредственного замещения:
кг[Pt Dien Cl]+ -J- Вг- [Pt Dien Br)+ + С1"Поскольку стадия акватации медленная, то закон скорости, опи¬
сывающий процесс протекания реакции по первому пути, имеет сле¬
дующий вид:i>i = ki [Pt Dien С1+].Закон скорости, отражающий протекание реакции по второму пути,
записывается следующим Ъбразом:v2 = [Pt Dien Cl+] [Вг-].Выражение суммарной скорости этих двух процессов:
v = + v., = (kv + кг [Вг-]) [Pt Dien Cl+)* Гидролиз дословно означает «разложение водой». Поэтому реакция ак-
ватацми комплексных соединений (гидролитическая диссоциация) может быть
названа гидролизом. Гидролиз также может протекать под действием щелочи с
образованием гидроксокомплексов. В литературе по химии координационных
соединений первый из них (акватацию) называют кислотным гидролизом, э
второй -— щелочным гидролизом.** Процесс, который протекает через промежуточную стадию образования
комплекса с молекулой растворителя с последующим замещением молекулы
растворителя на входящий лиганд, иногда называют криптосольволизом или
скрытым сольволизом (от греч. kryptos — тайный, скрытый). Это название при¬
меняется к процессам, в которых сольватокомплекс обладает высокой реакци¬
онной способностью и трудно поддается экспериментальному наблюдению.250
Рис. 9-7. Зависимость Лнасл от концентрации
вступающего лиганда: L—Вг~(/) н ОН- (2)Поскольку скорость замещения зависит
от концентрации входящего лиганда
толь.ко при протекании процесса по вто¬
рому пути, то имеется простой способ
вычисления констант скорости к1 и кг
обоих направлений. Для этого изучают
скорость процесса в зависимости от кон¬
центрации входящего лиганда и строят
график в координатах: экспериментально наблюдаемая константа
скорости — концентрация, входящего лиганда.При уменьшении концентрации свободных ионов Вг- экспери¬
ментально наблюдаемая константа скорости уменьшается (рис. 9.7,
прямая /). Из уравнения этой прямой линии можно вычислить зна¬
чение кг. Прямая 1 отсекает на оси ординат участок, который равен kl.Экспериментально установлено, что аналогичная картина наблю¬
дается, если вступающим лигандом является иоднд-нон или меченый
хлорид-нон. Однако скорость взаимодействия [PtDienCl]+ со щелочью
имеет иной характер. Она не зависит от концентрации гидроксиль¬
ных ионов (рис. 9.7, прямая 2). Это означает, что данная реакция
протекает только по одному пути — через стадию акватацин комп¬
лекса. Сходную ситуацию мы уже рассматривали для щелочного
гидролиза KifPtCU]. Таким образом, кинетические данные показы¬
вают, что прямое взаимодействие гидроксильных ионов с исходным
комплексом не имеет места.Совершенно естественно, что экспериментальные прямые 1 и 2
для двух реакций отсекают на оси ординат практически один и тот же
отрезок. В изученных условиях скорость обеих реакций определя¬
ется одной и той же медленной стадией — стадией акватацин комп¬
лекса.Параллельное протекание реакции по двум направлениям: через
стадию акватацин (сольватации) и путем непосредственного замеще¬
ния — встречается довольно часто. Закон скорости таких процессов
выражается уравнениемti = + k2 [L]) [Комплекс],где и — экспериментально наблюдаемая скорость реакции; к) и кг —
константы скорости соответствующих стадий реакции; [L] и [Комп¬
лекс] — концентрации лиганда и комплекса. При пересчете скорости
реакции на концентрации реагентов, равные единице, можно вы¬
числить экспериментально наблюдаемую константу скорости.Установлено, что соль Цензе К^ЦСзНОСЫ катализирует реак¬
цию акватацин иона [Pt C1j]-~. Мэханпзм каталитического действия
представляют следующим образом: вследствие сильного трансвлия¬
ния этилена хлорид-ионы в т/?анс-положении к нему в комплексе
[Pt(G2H4)Cl3]- сильно лабнлизированы и с большой скоростью за¬
мещаются на воду:251
[Pt(CjH4)CI3]- + H20 ** [Pt(C2H4) (HjO) Clo] + ci-Считают, что каталитическое влияние соли Цейзе сводится к тому, что
аквокомплекс [PtfGiH-i) (НгО)С1г] реагирует с [Pt CU]2- по реакции[PtCI4]2" -I- [Pt (С2И4) (Н20) С12] «= [Pt(H20) С13Г -I- [Pt(C*H4) Cl3]-Кукушкин и Крылов показали, что каталитическим влиянием на
реакцию акватации нона [Pt СЦ]2~ обладает комплекс [РКДМСО)С13]_.
Поскольку трансвлияние диметилсульфоксида близко к трансвлия-
нню этилена, то механизм каталитического действия этого комплекса,
вероятно, тот же самый.9.4. Классификация механизмов реакций
замещения в координационных соединенияхДеление механизмов реакций замещения в комплексных соединениях
на SnI и Sn2 является довольно грубым и требуется более детальная
классификация. Видимо, не все реакции замещения лигандов можно
безусловно отнести к нуклеофильным. Например, этилен в комплек¬
сах является донором и акцептором электронов, поэтому реакцию[PtCI4]2“ + С2Н4 = [Pt (С2Н4) С13]- + Cl-принципиально можно рассматривать как нуклеофильное и электро-
фильное замещение. Однако, поскольку замещению подвергается внут-
рисферный хлорид-ион, который не является акцептором электронов,
все же больше оснований говорить н о данной реакции как о нуклео¬
фильной.Отнесение реакций к типу SnI или Sn2 обычно проводится на
основании порядка стадии, определяющей скорость.Если скорость
реакции имеет первый порядок как в отношении концентрации комп¬
лекса, так и в отношении концентрации вступающего лиганда, то
считается, что механизм реакции ассоциативный, т. е. Sn2. Если ско¬
рость реакции имеет первый порядок в отношении концентрации комп¬
лекса, но не зависит от концентрации входящего лиганда, то меха¬
низм реакции считается диссоциативным, т. е. SnI.В настоящее время классификацию механизмов реакций замеще¬
ния строят на основе наличия или отсутствия интермедиата. Басоло
и Пирсон предложили обозначать реакцию SnI и™, если существуют
доказательства наличия интермедиата с пониженным координацион¬
ным числом, и S'n2jjm» если существуют доказательства интермедиа¬
та с повышенным координационным числом. При отсутствии доказа¬
тельства интермедиатов реакции по прежнему относили к SnI н Sn2.Лэнгфорд и Грей предложили несколько иную классификацию.
В зависимости от природы интермедиата (с пониженным или повы¬
шенным координационным числом) реакции относят соответственно
к типу D-интермедиатный стехиометрический диссоциативный меха¬
низм или А-интермедиатный стехиометрический ассоциативный меха¬
низм. Однако образование интермедиата не является обязательным
условием протекания реакции. Механизм замещения может быть252
синхронным, в котором уходящий лиганд движется из внутренней
сферы во внешнюю, а входящий лиганд одновременно движется из.
внешней сферы во внутреннюю. Такой тип механизма был назван об¬
менным и обозначен символом I (от англ. interchange).Дальнейшая детализация в классификации механизмов относится
главным образом к обменному типу. В обменном механизме энергия
активации может определяться или актом ассоциации входящего ли¬
ганда, или актом диссоциации уходящего лиганда. В зависимости от
этого первый тип реакции обозначают 1а, а второй Id. В табл. Э.З^
дана схема классификации механизмов реакций.Таблица 9.3. Классификация механизмов реакций замещенияКлассификацияИнтермедиат
с пониженным
координацион¬
ным числомНетинтермедиатаИнтермедиат с повы¬
шенным координаци-
нным числомЛэнгфорда —
Греястехиометри-
ческнн меха¬
низмDIАмолекулярныймеханизмDIdlaАИнгольда-Басоло — ПирсонаllimsN 1Sn2S^2iimТаким образом, в современной классификации механизмов реак¬
ций замещения в координационных соединениях определяющее зна¬
чение имеет суждение о наличии или отсутствии интермедиата. Наи¬
большие трудности связаны с отличием в механизмах А и 1а, с одной,
стороны, и в механизмах D и Id, с другой.Для экспериментального отличия механизма А
XR + Y XRY
XRY st X + RY
от механизма /аXR + Y«X - R-..YeX-г RYиспользуют следующую предпосылку. В механизме А на энергетиче¬
ском рельефе реакционного пути (см. рис. 9.2) интермедиат находится
в потенцнальной яме, за которой следует новое переходное состоя¬
ние. Если интермедиат достаточно стабильный, то скорость реакции
не будет увеличиваться беспредельно с увеличением концентрации
входящего лиганда. При большой концентрации интермедиата ско¬
рость зависит не только от его образования, но и от его диссоциации.
Следовательно, при увеличении концентрации вступающего лиганда
скорость реакции достигает предельного значения, а закон скорости253-
изменяется от второго порядка до первого. Такое экспериментальное
проявление должно быть характерно для механизма А. Для механиз¬
ма 1а закон скорости должен быть только второго порядка и должен
отсутствовать интермедиат.Для экспериментального отличия механизма DXR R + XR + S =* RS■от механизма 1аXR -f S ^ X • • • R • ■ • S ^ X + RSтакже стремятся получить доказательства существования интермеди¬
ата. Это делают следующим образом. Если имеется интермедиат R,
то введение лиганда X должно понижать скорость реакции, так как
в результате обратимости стадии диссоциации понижается концентра¬
ция интермедиата. Второй подход основан на увеличении скорости
реакции в зависимости от инертного электролита. Это происходит
вследствие того, что интермедиат на какое-то время может превра¬
щаться в ионную пару с покидающим комплекс лигандом по схемесюГRZ г± R4- ... Z" R+ ■ ' • CI07 + Z-
|Q-
RQЛонная пара находится в равновесии с комплексом и частично ре¬
комбинирует в него. Добавление «инертного» перхлоратного электро¬
лита приводит к частичному изменению ионной пары, а следовательно,
и к уменьшению склонности ионной пары к рекомбинации в ис¬
ходный комплекс. Это должно приводить к увеличению скорости ре¬
акции. Если отсутствуют экспериментальные доказательства наличия
интермедиатов, но порядок реакции первый, то это может свидетель¬
ствовать в пользу Id механизма.Пути и возможности для определения интермедиатов в каждом
конкретном случае могут быть самые различные. Рассмотрим две ра¬
боты, свидетельствующие о наличии интермедиатов. В одной из них
доказывается наличие интермедиата с пониженным координационным
числом, в другой — с повышенным.Тоуб изучал природу интермедиатов в реакции типа
{СоЕп2АС1]-+ + Н,0 — [СоЕп.А (Н20)]:)+ + С1~в зависимости от природы лиганда А. В основу своих исследовании
он положил представление Ингольда о двойственном механизме за¬
мещения в зависимости от электронного распределения в лиганде А.
Если координированный лиганд А обладает неподеленной электрон¬
ной парой, которая может быть предоставлена нону металла для ком¬
пенсации потери электронов с уходящим лигандом, то это должно
благоприятствовать образованию интермедиата с пониженным коор¬
динационным числом. Наличие такого интермедиата также должно
способствовать изомеризации транс-изомера в соответствующий цис-
изомер. В табл. 9.4 приведены некоторые результаты работы Тоуба.254
Таблица 9.4. Кинетические параметры и стерические направления
реакций /иранс-[СоЕп2АС1]2++Н20-*-[СоЕппА(Н20)]3++С1~Адн*кДж/мольД5*, э. е.Степень стсреоиэомсри-
зацпм, %он-108,8+2075CI-1 10,0+ Н35Вг"104,6+350NCS-126,8+960N394,5020NH,97,4— 110с\-94,5 20no287,8 20Из табл. 9.4 видно, что для лигандов, обладающих дополнитель¬
ной электронной парой, действительно характерно положительное
значение энтропии активации и довольно высокий процент изомери¬
зации. Все это вместе взятое может служить доказательством су¬
ществования интермедиатов с пониженным координационным числом.Катталини с сотр. обосновали существование интермедиата с по¬
вышенным координационным числом в реакции[Rh/i,5-HHMooKTaAnen/ci(SbR3)] -|- А — [Rh/i ,5-циклсоктадиен/с1Л] ++ SbRjгде А > амин.Доказательство образования интермедиата производилось спектро¬
фотометрически. Они экстраполировали разность D,—D00 [в коорди¬
натах (D,—Dae), t] на время, равное нулю, и полученные результаты
сравнили со значениями оптической плотности в отсутствие амина.
Несовпадение значения разности (D(=o—£><*,) с оптической плотностью
раствора комплекса в отсутствие амина дало основание сделать выводо наличии интермедиата с координационным числом 5. Кроме этого,
было показано, что при низких концентрациях амина реакция обра¬
зования интермедиата бимолекулярна, а при увеличении концентра¬
ции амина скорость реакции становится независимой от его концент¬
рации, что находится в полном согласии с ассоциативным интермедн-
атным механизмом А.
ГЛАВАКИСЛОТНО-ОСНОВНЫЕ СВОЙСТВА
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ10.1. Развитие представлений о комплексных
кислотах и основанияхКоординационные соединения сыграли большую роль в разработке
теоршГкислот и оснований вообще и протолитнческой теории Бренсте-
да—Лоури в частности. Важным этапом в развитии взглядов на кис¬
лоты и основания была теория электролитической диссоциации Арре¬
ниуса. С позиций этой теории кислоты — это соединения, которые
отщепляют в растворе ноны водорода. Основания — это соединения,
которые отщепляют гидроксильные группы.О комплексных основаниях еще в 1893 г. писал Н. С. Курнаков.
Он же впервые затронул вопрос о свойствах комплексного основания
и основных свойствах координированных молекул.Принципиальное значение для развития теории кислотно-основ¬
ных свойств комплексных соединений имела работа Пфейффера —
ученика Вернера — по исследованию кислотных свойств комплекса
хрома (III) [Сг Ру2(НгО)4]С13. Было установлено, что при действии
аммиака на водный раствор этого комплекса образуется и выпадает
в осадок малорастворимое соединение [Сг Руг(НаО)2(ОН)2]С1. Эта
реакция сопровождается резким изменением окраски раствора от
красно-фиолетовой до серо-зеленой. Аммиак не входит во внутрен¬
нюю сферу комплекса, а лишь играет роль основания. Таким обра¬
зом, данную реакцию можно описать следующим уравнением:1СгРу,(Н20)4] С13 + .2NH3 = [СгРу2(Н20)о(ОН)2] Cl + 2NH4CIКомплекс [Сг Ру2(НгО)2(ОН)2]С1 всесторонне изучен как в твер¬
дом состоянии, так и в растворе. Его характерным свойством являет¬
ся то, что в водном растворе он создает щелочную реакцию, а сам сту¬
пенчато превращается в исходный тетрааквокомплекс. Эти реакции
удобнее писать в ионном виде:[СгРу„(Н,0)2(0Н)2]+ + Н20 « [СгРу, (Н,,0)30Н]-+ -I- ОН~[СгРу, (Н,0)30Н]2+ + Н,0 =*= [CrPy2 (Н,0)4]3+~ ОН-Естественно, что добавление в раствор кислот приводит к смещению
этих равновесий в сторону образования аквоформы:[СгРуг(НаО)2(ОН)2]=+ + 2Н+ =s= [СгРу2(НгО)4]3+Таким образом, Пфейффер в 1906 г. установил обратимое превра¬
щение аквокомплексов в гидроксосоединения. Вернер быстро оценил
огромное значение этого открытия и на его основе развил свою теорию
кислот и оснований.256
Если исходить из реакции[СгРу.,(Н,0)30НР + Н20 [СгРу.,(Н20)4]3+ + ОН~то можно принять, что гидроксильный ион появляется в растворе не
в результате отщепления от комплексного иона [Сг Ру.,(Н20)30Н]2+,
а вследствие отщепления протона от молекулы растворителя. Вернер
высказал мысль о том, что простые основания, как, например, NaOH,
диссоциируют в растворе по аналогичной схеме: к молекуле (к ан-
гмдрооснованию, т. е. к безводному основанию) присоединяется мо¬
лекула воды и образуется Na0H H20 (аквооснование). В аквоосно-
вании протон от молекулы растворителя переходит к связанной с
катионом гидроксогруппе и образующаяся из воды гидроксогруппа
диссоциирует:NaOH -f Н,0 « NaOH • Н,0 « [NaOH;]OH NaOH, + ОН"Аналогично образукл’ся ионы из ангидрокислот:HF 1- н20 ^ HF ■ Н.О =* Н [F • Н20] 5* Н+ + [F ■ Н20]Не рассматривая теорию ангидрокислот и ангидрооснований более
подробно, так как она имеет лишь историческое значение, отметим
только два обстоятельства, на которые обратил внимание Вернер:1) важная роль растворителя в кислотно-основном равновесии;2) основания можно рассматривать как акцепторы протонов. К
последнему выводу Вернер пришел уже в 1907 г. Позже эти пред¬
ставления легли в основу протолитической теории, одновременно
сформулированной Бренстедом и Лоури в 1923 г. .Вслед за комплексом [Сг Ру2(Н20)4]3+ было выявлено большое
число других аквокомплексов, также обладающих ярко выраженны¬
ми кислотными свойствами. Количественной характеристикой кис¬
лотных свойств аквокомплексов начал заниматься Н. Бьеррум. Позд¬
нее этим исследованиям много внимания уделял Бренстед. В выясне¬
нии причины резкого усиления кислотных свойств координированной
молекулы воды большую роль сыграли представления Косселя. Он
высказал мысль, что усилению кислотной диссоциации молекулы воды,
координированной к иону металла, способствует электростатическое
отталкивание положительно заряженного иона водорода от одноимен¬
но заряженного иона металла:Ч-ймл+—о/н « мл+—о- + Н+
н \н-10.2. Факторы, влияющие на кислотные свойства
координационных соединенийВ работах Н. Бьеррума и Бренстеда было установлено, что кислот¬
ные свойства аквокомплексов зависят от заряда центрального иона и
от заряда комплексного иона. Так, например, кислотные свойства9-817 257
молекулы воды, координированной около Pt(IV), выражены значи¬
тельно сильнее, чем молекулы воды, координированной около Pt(II).
Из двух равновесий кислотной диссоциации[Pt" (NH3)3 (Н20)]2+ ^ н+ + [Pt11 (NH3)3OH]+[PtIV (NH3)3 (H20) Cln]2+ « H+ + [Pt,v (NH3)3 (OH) CI2]+второе смещено вправо намного сильнее. Таким образом, константа
кислотной диссоциации комплекса [Pt(NH3)3(H,,C))]2+ должна быть
меньше, чем комплекса [Pt(NH3)3(H20)Cl2]2+. Большей подвижности
протона координированной молекулы воды способствует более высокий
положительный эффективный заряд на атоме платины в комплексе
[Pt(NH3)3(HX>)CI2]2+ по сравнению с комплексом [Pt(NH3)3(H20)]2+.Кислотные свойства аквокомплексов с одинаковым центральным
ионом зависят от заряда комплексного иона. Например, из соедине¬
ний [PtIV(NH3)3(H20)CIo]2+ и [PtIV(NH3)5(H20)]4+ второе является
более сильной кислотой, так как отщепление протона от него облег¬
чается более сильным электростатическим отталкиванием от комп¬
лексного иона с более высоким зарядом. Таким образом, равновесие[Pt (NH3)5 (Н,0)]4+ [Pt (NH3)jOH]3+ + H+смещено вправо сильнее, чем равновесие[Pt(NH3)3 (Н20) С12р « [Pt(NH3)3(OH) С12]+ + Н+Весьма существенным этапом на пути развития теории кислотно¬
основных свойств координационных соединений было открытие Чу-
гаевым амидореакции. Еще в 1915 г. он установил, что при действии
щелочи на комплекс [Pt (NH3)5C1JC13 получается малорастворимое
соединение [Pt(NH3)4(NHa)Cl]Cb. Эту реакцию можно выразить урав¬
нением[Pt(NH3)5C 1] Cl3 + NaOH [Pt(NH3)4(NH2)CI]CU + NaCI+H,0Таким образом, аммиачный комплекс при взаимодействии со ще¬
лочью ведет себя как кислота. При отщеплении протона от координи¬
рованной молекулы аммиака во внутренней сфере комплекса остается
амидогруппа NHe. С точки зрения кислотно-основных свойств амндо-
комплекс [Pt(NH3)4(NH2)Cl]Cl2 напоминает гидроксокомплекс
[Сг Ру2(Н20)г(0Н)2]С1. При его растворении в воде создается щелоч¬
ная среда. В ионном виде эта реакция выглядит следующим образом:[Pt(NH3)1(NH2) С1Р + Н20 =*= [Рt(NН3)5 Cip+ + ОН-В координированном состоянии неподеленная электронная пара
молекулы аммиака вовлечена в образование химической связи с цент¬
ральным атомом, поэтому функция аммиака как основания уже вы¬
полнена. В некоординированном состоянии кислотная функция моле¬
кулы аммиака выражена очень слабо и практически незаметна на
фоне основной. При координации аммиака кислотная функция уси¬
ливается вследствие электростатического отталкивания протона от
положительно заряженного центрального атома.258
Впоследствии Черняев с сотр. синтезировали большое число ами-
до- и имидокомплексов платины (IV).За несколько лет до Чугаева по существу аналогичная реакция была обна¬
ружена Кнрмрентером. Он синтезировал несколько комплексов платины (II)
с сульфамнновон кислотой. Константа кислотной диссоциации сульфамнновон
кислоты NtH2S03H 1\ = 0,1. В комплексы она входит'в виде сульфамипатного
нона NH2SO3.Кирмрентер установил, что при действии щелочи на водные растворы изо¬
мерных комплексов Iv>[Pt(NH3S03)2CU] образуются соответствующие соеди¬
нения KilPUNHSC^Cy, в которых содержатся депротонированные сульф-
аминатные ионы. Значение этой работы заключается в том, что в комплексе пла¬
тины (II) кислотной диссоциации подвергается лиганд по связи N—Н.Большой вклад в изучение кислотных свойств аквокомплексов
внесли Бьеррум и Бренстед. Открытие Чугаевым амидореакции поз¬
волило Гринбергу распространить взгляды Бьеррума и Бренстеда на
комплексные соединения металлов с другими протонсодержащими
лигандами. В частности, в обобщение были вовлечены комплексные
соединения с аммиаком, метиламином, этилендиамином, диметилглио-
ксимом, гидроксиламином и др. Гринберг сформулировал функцио¬
нальную зависимость кислотных свойств комплексных соединений с
протонсодержащими молекулами RH от ряда факторов:А = / (Н, Е, а, р, л, G),где А — кислотные свойства комплекса, в качестве лиганда в кото¬
рый входит молекула RH; Н — сила поля центрального атома, опре¬
деляемая его зарядом q* , радиусом, структурой электронной оболоч¬
ки и поляризационными свойствами; Е — заряд комплексного иона;
а — степень кислотной диссоциации молекулы RH в свободном со¬
стоянии; р —■ степень деформации лиганда RH под влиянием централь¬
ного иона; п — число внутрисферных лигандов RH; G — геометриче¬
ское строение комплекса.Рассмотрим каждый из этих факторов в отдельности.Сила поля центрального атома. По определению Гринберга, сила
поля центрального атома зависит от заряда, радиуса, электронной
структуры и поляризационных свойств. При прочих равных условиях
ион металла тем в большей степени способствует отщеплению протона
от координационной молекулы RH, чем выше его заряд и меньше ра¬
диус. Отношенне заряда к радиусу q!r называют ионным потенциа¬
лом ф.Электростатические представления, основанные на зарядах и ра¬
диусах ионов, были использованы Косселем в его теории кислотно¬
основных свойств гидроксидов. Теория Косселя позволяет определять
тенденцию в изменении кислотных или основных свойств гидроксидов
и водородных соединений элементов, имеющих аналогичное элек¬
тронное строение.В ряду аналогичных соединений, например Ве(ОН)з, Mg(OH)2 и
Са(ОН)з, Коссель рассматривает группировку ионов Эп+—Oi_—Н+.* Заряд центрального иона здесь понимается как синоним степени окис¬
ления.9*«259
Принципиально гидроксиды могут диссоциировать по связи Э—О и
по связи О—Н. Если диссоциация происходит по связи Э—ОНОЭ—он ^цноэ+ + он-то гидроксид является основанием. Если же диссоциация происходит
по связи О—ННОЭО—H ноэо-м- Н+то гидроксид является кислотой. Если диссоциация происходит как
по связи Э—О, так и по связи О—Н, то такой гидроксид является
амфотерным соединением.Поскольку степени окисления бериллия, магния и кальция в рас¬
сматриваемых соединениях одинаковы, то наиболее очевидное измене¬
ние связано с увеличением радиуса ионов при переходе от Ве2+ к Са2+:В соответствии с законом Кулона с увеличением расстояния меж¬
ду одноименно заряженными ионами (Э2+ и Н+) будет ослабляться их
взаимное отталкивание и, следовательно, в той же последователь¬
ности должны ослабляться кислотные свойства. Притяжение разно¬
именных частиц (Э2+, с одной стороны, и О2- или, правильнее, ОН',
с другой) с увеличением радиуса Э2+ должно ослабляться. Следова¬
тельно, основные свойства в ряду рассматриваемых гидроксидов долж¬
ны усиливаться.Если рассмотреть гидроксиды Fe(OH)2 и Fe(OH)3, то с позиции
схемы Косселя кислотные свойства второго гидроксида должны быть
выражены сильнее, чем первого. Это следует из того, что чем выше
степень окисления железа (даже если принять, что радиусы ионов
Fe2+ и Fe3+ одинаковы), тем выше отталкивание протона и тем сильнее
выражены кислотные свойства соответствующего гидроксида. Если
учесть, что радиус иона Fe3+ несколько меньше, чем иона Fe2+, то боль¬
шее отталкивание протона в соединении Fe(OH)3 по сравнению с
Fe(OH)2 становится еще очевиднее.Эти представления Коссель распространил и на бескислородные
кислоты. Например, в ряду кислот Н20, H2S, H.2Se и Н2Те кислот¬
ные свойства должны усиливаться, так как при одинаковой степенн
окисления —2 кислорода, серы, селена и теллура увеличивается их
радиус, а следовательно, ослабляется связь с протоном и усиливаются
кислотные свойства.Окружение иона металла нейтральными лигандамл приводит к
увеличению радиуса катиона. Следовательно, комплексообразованне
должно приводить к усилению основных свойств гидроксидов. На¬
пример, гидроксид AgOH является слабым основанием (Кв =
= 1,1 ■ 10-4). При взаимодействии с аммиаком получается комплекс
[Ag(NH3)]2OH. В результате комплексообразования радиус катиона260
значительно увеличивается и в результате этого значительно усили¬
ваются основные свойства комплексного основания. Комплекс
[Ag(NH3)2]OH является сильным основанием. По этой же причине
слабое основание Со(ОН)3 при комплексообразорании с аммиаком
превращается в сильное основание [Co(NH3)6](OH)3, и слабое осно¬
вание Pt(OH)2 при комплексообразовании с тиомочевиной также пре¬
вращается в сильное основание [Pt{(NH2),CS}4](0H)2.Положения теории Косселя могут быть перенесены на комплексные
соединения, в которых рассматривается группа атомов Мл+—RH.Влияние заряда центрального иона на кислотные свойства ли¬
ганда иллюстрируют следующие примеры. Константа кислотной дис¬
социации синильной кислоты К = 7,2-10_1°. При координации HCN
около иона Fe3+ происходит резкое усиление ее кислотных свойств.
Комплексная кислота H3[Fe(CN)6] является сильной. Аналогичная
картина наблюдается и с фтористоводородной кислотой. Ее константа
кислотной диссоциации К = 7,4 • 10-4. При координации около крем¬
ния (IV) происходит резкое усиление кислотных свойств HF. Кислота
H2[SiF6] по силе сравнима с серной кислотой.Наиболее надежные предсказания об изменении кислотных свойств
комплексных соединений в зависимости от природы центрального иона
могут быть сделаны для элементов с аналогичным или близким элект¬
ронным строением, так как от электронного строения зависят поля¬
ризуемость и поляризующее действие ионов.Важное значение электронного строения центрального иона вы¬
текает из сопоставлений кислотных свойств комплексов [Pt Еп3]4+ и
[Os Еп3]4+. Для платинового комплекса= 7,Ы0“7, /С2 = 9,2-10~п.
Осмиевый комплекс по первой ступени диссоциирует как силь¬
ная кислота, а его К2 — 1,6- 1СГ6. Следует отметить, что радиусы пла¬
тины (IV) и осмия (IV) близки, а заряды центральных ионов и комп¬
лексных ионов одинаковы.Заряд комплексного иона. Заряд комплексного иона является
одним из существенных факторов, влияющих на кислотные свойства.
С понижением заряда комплексного иона ослабляются его кислотные
свойства.Степень кислотной диссоциации лиганда в свободном состоянии.Если в свободном состоянии лиганд PH является более сильной кис¬
лотой, чем R'H, то во внутренней сфере комплекса при прочих рав¬
ных условиях относительная склонность к отщеплению протона со¬
хранится. Так, комплекс [M(NH3)5(H.,S)]r'+ должен быть более силь¬
ной кислотой, чем [M(NH3)6(H20)]n+, потому что в свободном состоя¬
нии кислота H2S сильнее, чем Н20. По этой же причине комплексы с
фосфином РН3 должны быть более сильными кислотами, чем комп¬Константы кислотной диссоциации ам-
минокомплекссв платины (IV) (при 25Х)[Pt(NH3)5Cl]3+ . . .
4uo[Pt(NH3)4Cl3]2+ .
транс-[Pt(NH3)4CI о ]:+261
лексы с NH3. Напомним, что РН3 обладает более слабыми основными
свойствами, а следовательно, более сильными кислотными свойства¬
ми, чем NH3. На многочисленных примерах установлено, что в акво-
амминокомплексах отщеплению протона прежде всего подвергается
внутрисферная молекула воды. Экспериментально установлено, что
константа кислотной диссоциации аквокомплексов платины (IV) в
1(Р—105 раз больше, чем соответствующих аминных соединений. На¬
пример, это следует из сопоставления констант [Pt(NH3)6]1+ и
[Pt(NH3)5(H20)]l+, которые соответственно равны 1,5-10“7 и ~10"4.
Соотношение констант кислотной диссоциации соответствующих акво-
и аминосоединений зависит от природы центрального иона. Например,
К а для комплекса [Rh(NH3)e]3+ меньше 10"13, а для комплекса
[I^h(NH3)3(H20)]:!+ равна 1,38-Ю'8. Таким образом, соотношение кон¬
стант превышает 10е.В общем случае чем сильнее кислотные свойства соединений, тем
слабее их основные свойства. Это обстоятельство часто позволяет
предсказывать относительную силу комплексных кислот. Однако
встречаются случаи, когда такое предсказание не оправдывается.
Так, алифатические амины являются более сильными основаниями,
чем аммиак. Однако аммиачные комплексы иногда проявляют более
слабые кислотные свойства, чем аналогичные соединения с алифати¬
ческими аминами. Например, константа кислотной диссоциации
комплекса m/?a«c-[Pt(NH3)4Cl2]':+ равна 610~13. Аналогичный этил-
аминовый комплекс характеризуется такой же константой. Однако
метиламиновый аналог m/7aHc-[Pt(NH2CH3)4Cl2]“+ является несколько
более сильной кислотой. Его константа кислотной диссоциации рагаа
1,4- 10~и. Он диссоциирует с образованием имидокомплекса по схеме
[Pt (NH2CH3)4 Cl,]2+ « Н*+ [Pt(NH2CH3)3 (NHCH3) С12]+Из табл. 10.1 видно, что этилендиаминовые и пропилендиамино-
вые соединения платины (IV) проявляют более сильные кислотные
свойства, чем аммиачные. Гринберг связывал это с различной «де¬
формацией» лигандов под действием центрального нона. Он сам от¬
мечал, что название данного фактора условное.Таблица 10.1. Константы кислотной диссоциации аминокомплексов(при 25°С)КомплексK,кгK,[Pt(NH3)6]4+1,5-10-71 ,5lO-io[PtEn(NH3)4r7,1- Ю-79,210"n[PtEn3]4+3,5- 10-e1 ,8l0-i°2,0-Ю'11[PtPn3[4f3,9- Ю-62,510-Ю2, МО'11[OsEn2]4+Сильная кислота1 ,610"6[AuEn2]3+'1,5-10-’[AuPrij]34'2,6-10~7[Co(NH3)e]3+<io-12[СоЕп3]3+-d(T12[RhEn3)3+<10"12[IrEn3]3+< 10-12262
Число внутрисферных протонсодержащих лигандов. Бренстед
пришел к заключению, что кислотные свойства зависят от числа про¬
тонсодержащих лигандов RH во внутренней сфере комплекса.С увеличением в комплексе кобальта (III) числа молекул воды за¬
метно увеличивается значение первой константы кислотной диссо¬
циации:Константы кислотной диссоциации
комплексов кобальта (111) (при 25° С)[Co(NH3)5(H20]3+ 2,04-КГ»[Co(NH3)4(H20),p+ 6,03-10-«[Со(ЫН3)3(Н20)з]3+ I ,88- 10-5[Co(NH3)2(H20),p 4,00-10“4Это можно объяснить «расталкиванием» протонов при скоплении про¬
тонсодержащих молекул. В этом случае существенное значение долж¬
но играть геометрическое строение комплексов. При 1(ыс-расположе-
нии протонсодержащих молекул RH усиление кислотных свойств
должно быть большим, чем при их т/?йнс-расположении. Об этом речь
пойдет при рассмотрении следующего фактора, определяющего кис¬
лотные свойства комплексов.Изменение кислотных свойств в ряду комплексных соединений,
приведенном выше, можно дать и другое объяснение. Молекулы ам¬
миака обладают более сильными донорными свойствами по сравнению
с молекулами воды и потому в большей степени понижают эффектив¬
ный положительный заряд центрального нона. С уменьшением в
комплексе числа молекул аммиака происходит относительное увели¬
чение положительного эффективного заряда на центральном атоме
и, следовательно, усиление отталкивания протонов координирован¬
ных молекул воды. Возможно, этим объясняется резкое различие в
кислотных свойствах [Au Еп2]3+ по сравнению с [Со En3]3+, [Rh Еп3]3+
и [Ir Еп3]3+ (см. табл. 10.1). Увеличение числа донорных аминогрупп
понижает положительный эффективный заряд на центральном атоме.
Это должно приводить к ослаблению кислотных сеойств. Таким об-Таблица 10.2. Константы кислотной диссоциации изомерных
диакводиаминных комплексовКомплексСтроениеКхК,к,/к2[Pt(NH3)2(H20)2pцис-2,8- Ю-64,8-10“858транс-4,8- К)”54,2-К)'81 !47[Pt(NH2CH3)2(HAP+цис-2,7- 10-®7,3-10-837транс-3,4- Ю"58,0- КГ8425[Pt(NH2C2H5)2(H20)2]=+цис-2,9- К)-68,2- Ю'835транс-3.4-10~510,0- 10-8340[PtPy2(H20)2]-+цис-3,2-10"65,8.10-’55транс*I ,5- КГ43,7-10-’406263
разом, в функциональную зависимость кислотных свойств комплек¬
сных согдинемий можно ввезти ецэ один фактор — число лигандов,
определяюцих эффективный положитетьный заряд на центральном
атоме.Геометрическое строение комплекса. Изучение влияния геометри¬
ческой конфигурации комплексных соединений на их кислотные свой¬
ства было начато А. А. Гринбергом и Д. И. Рябчиковым. На примере
изомерных комплексов [Pt(NH3)2(H^O)j]'+ они установили, что /(1
транс-изомера больше, чем /<\ г{ис-изомера. Кроме того, было пока¬
зано, что К\1Кг г{ис-изомера много больше, чем /(//Сг транс-изомера.
В дальнейшем подобные же соотношения были установлены для мно¬
гих других диакводиаминных комплексов (табл. 10.2).Меньшую склонность к кислотной диссоциации по первой ступени
комплекса I по сравнению с комплексом IINH3 Н20
NHg^ НоОNH3 НоО
3 Pt *
Н20 NH3I IIГринберг объясняет большим трансвлиянием NH3 по сравнению с
Н20. Резкое различие Ki и К2 в mpaHC-[Pt(NH3),(H20)2]2+ он связы¬
вал с тем,что диссоциация второй молекулы воды происходила на
координате Н20—Pt—ОН, а трансвлияние гидроксогруппы значи¬
тельно выше, чем Н20 и NH3.Аналогичные соотношения констант кислотной диссоциации были
установлены для изомерных диаквокомтексов [Со Еп.,(Н20)„])+ и
[Сг Еп,(Н20)2]:)+Гидроксиламин NH2OH, ацетоксим (CH3)2CNOH и альдоксим
CH3CHNOH координируются около платины (II) посредством атома
азота. При кислотной диссоциации они отщепляют протон от атома
кислорода. Н^ примере большого числа изомерных пар комплексов
типа [Pt(Ox)2A2]2+ (где Ох — гидроксиламин или оксимы, А — амины)
значение Ку для цис-изомеров больше, чем /<\ для соответствующих
транс-изомеров и /<\//С2 (цис) > К^К*, (транс). Такие соотношения
констант противоположны наблюдаемым для диаминдиаквосоедине-
ний (табл. 10.2). А. И. Стеценко объясняет более ярко выраженные
кислотные свойства гидроксиламиновых и оксимных комплексов цис-
конфигурации по сравнению с транс-изомерами стабилизацией де-
протонированных комплексов, например
A NH2OxPt >НA NH3o/за счет образования водородных связей.Более сильные кислотные свойства октаэдрических комплексов
платины (IV) ^uc-конфигурации по сравнению с соответствующими
транс-изомерами были выявлены Гринбергом с сотр. Так, кислотные
свойства пропилендиаминового комплекса цис-[Pt Рп2С12]2+ харак¬
теризуются двумя константами /С2 == 5,4-10-9 и К3 = 3,б"10'и. Д-тя
транс-изомера удалось определить только значение первой констан¬264
ты кислотной диссоциации. Оно оказалось равным 1,8- 10~п. Ана¬
логичная картина наблюдается и для изомерных комплексов
[Pt(NH3)4Cl2]2+. Для цыс-изомера Л', - 3,5-10“10, К2 = 5,6- 10“и.
Для транс-изомера значение первой константы кислотной диссоциа¬
ции находится на грани возможностей потенциометрического метода,
а именно, Л'2 = 6-10_1“.Недавно Таубе с сотр. показали, что значение Кх для
mpaKC-[Os(NH3)4CI2]'-+ равно 10"4, а для ^ыс-изомера =0,10. Для
изомерных нодидных комплексов [Os(NH3)4I2]2+ разница в кислотных
свойствах не столь велика. Для транс-изомера значение Ka -: 1,3 XXIО-4, а для цшг-изомера К-.ю-Гринберг с сотр. объяснили более сильные кислотные свойства
октаэдрических комплексов платины цыг-конфигурации большим
«расталкиванием» протонов по сравнению с транс-w.зомером. На¬
пример, в комплексеNHвNH,FtClCl2-1-ПрОТОНЫ молекулы аммиака, заключенной в кружок, испытывают
расталкивающее влияние девяти протонов трех соседних молекул
NH3. В комплексеNHС1IPiNH,NH,2 +расталкивающее действие оказывают только шесть протонов сосед¬
них молекул NH3.Если связывать кислотные свойства с трансвлиянием лигандов, то
вывод об относительной силе кислот quc-[Pt(NH3)4CI2]2+ и
mpQHC-[Pt(NH3)4Cl2]2+ не будет соответствовать экспериментальным
данным. Действительно, в комплексе *^c-[Pt(NH3)4Cl2]2+ две моле¬
кулы аммиака расположены против хлоридных лигандов, обладающих
относительно большим трансвлиянием. Это должно было бы приво¬
дить к ослаблению кислотных свойств данных молекул аммиака и
комплекса в целом. Наоборот, в m/7aHC'-[Pt(NH3)4Cl2]2+ все молекулы
аммиака расположены против лигандов относительно слабого транс¬
влияния, и они должны были бы проявлять более сильные кислотные
свойства. Следовательно, фактор трансвлпяния лигандов, по крайней
мере в комплексах платины (IV), имеет меньшее значение, чем фактор
«расталкивания» протонов.265
Таким образом, из приведенных экспериментальных фактов сле¬
дует вывод, что геометрическое строение отражается на кислотных
свойствах комплексных соединений. Это явление может быть обус¬
ловлено различными причинами. Поэтому для предсказания относи¬
тельной силы кислот необходимо в каждом отдельном случае учитывать,
какая причина может иметь определяющее значение.Кислотные свойства однотипных комплексных соединений металлов,
принадлежащих к одной подгруппе периодической системы, обычно
отличаются не сильно. В этом случае тенденция в изменении кислот¬
ных свойств неоднозначна и зависит от типа комплекса. В то же время
имеется довольно четкая картина в изменении кислотных свойств
однотипных комплексов в пределах одного периода. Например, комп¬
лекс [Ru(NH3)5(H20)]3+ (электронная конфигурация центрального
иона йъ) характеризуется 4, а аналогичный комплекс[Rh(NH3)5(H20)]3+ (электронная конфигурация центрального нона
de) имеет рК =6,9..}Для комплекса mpaHC-[Os(NH3)4Cl2]2+ (электрон¬
ная конфигурация центрального иона d1) рKjx 4, а для аналогич¬
ного комплекса m/7fl«c-[Pt(NH3)4CI2]2+ (электронная конфигурация
d!') рД" = 11,2. Для комплекса [Os(NH3)6]1+ рК <С 0, а для комплекса
[Pt(NH3)e]4+ р/< = 6,9.Таким образом, кислотные свойства комплексов с не полностью
занятыми орбиталями t2g выражены сильнее, чем комплексов, у ко¬
торых эти орбитали заполнены. Существует мнение, что в первом слу¬
чае происходит стабилизация лиганда в депротонированной форме за
счет проявления им л-донорных свойств.10.3. Взаимосвязь кислотных свойств
координированных аминов с реакциями
замещения их протоновПредставления Гринберга об усилении кислотных свойств комплексов
вследствие «расталкивания» протонов оказались весьма плодотвор¬
ными. С этой точки зрения в комплексных соединенияхнаибольшей подвижностью протонов должны обладать молекулы
аммиака, заключенные в кружок.
В 1957 г. Ю. Н. Кукушкин установил, что в водных растворах
при взаимодействии с хлором внутрнсферные молекулы аммиака в
комплексах платины (IV) превращаются в дихлорамин по схеме
Pt-NH3 + 2С1о » Pt—NCI2H + 2НС1Дихлорампн во внутренней сфере комплексов платины (IV) обладает
ярко выраженными кислотными свойствами. В твердую фазу всегда
выделяются соединения с депротонированным дихлорамином:Pt—NCLH ** Pt—NCI“ + Н+Экспериментально установлено, что превращению в дихлорамин
в первую очередь подвергаются молекулы аммиака, обладающие наи¬
большей подвижностью протонов. В приведенных выше комплексных
катионах это молекулы аммиака, заключенные в кружок. При на¬
гревании водных растворов дихлорамидных комплексов происходит
внутрилигандный окислительно-восстановительный процесс по схеме2(Pt-NCIf) + Н20 — 2 (Pt—CI-) + N2-f НСЮ + НС1В результате этого вместо дихлорамидной группы в комплексе ока¬
зывается хлоридный лиганд. По конфигурации продукта реакции
можно судить о направлении хлорирования и месте образования
дихлорамина.Ципприху удалось провести рентгеноструктурный анализ
комплекса [Pt(NH3)4(NCls)C]]Clz, полученного хлорированием
[Pt(NH3)bCl]Cl3. Оказалось, что дихлорамндное соединение имеет сле¬
дующее строение:аN—CIN11,
NI i jIINH,NH,Cl24-ЭТО находится в полном согласии с результатами химического иссле¬
дования положения в комплексе дихлорамидной группы.Взаимодействие хлора с гидроксокомплексом [Pt(NH3)sOH]3+ про¬
текает в соответствии с уравнением[Pt(NH3)6OHP+ + 4CI, щ* [Pt(NH3)s(NCI:)2Cl]+ + ЗНС1 + 2Н+ + Н80По данным Ципприха, дихлорамидные группы в этом комплексе рас¬
полагаются следующим образом:С1NHNH.>tICI/С1
N—С1N Н,267
Таким образом, и при хлорировании [Pt(NH3)50H]3+ молекула
аммиака с наибольшей подвижностью протонов действительно оказы¬
вается превращенной в дихлорамидный лиганд.При взаимодействии хлора с этилендиаминовыми комплексами
платины (IV) удается превратить в хлораминные одну или обе амино¬
группы:NHXH . NCICHoPt> ‘I + ci2 ptt | H-HCI + H+sNH2CHj sNH2CH2NCICH. .NClCHjPt£ I ' + CU ^ Pt£ I + HCI
4NH„CH, " 4NC1CH2
HЧерняев с сотр. изучали реакции депротонирования и хло-
рамидирования оптически активных этилендиаминовых ком¬
плексов, например цис-\Pt Еп NH3C1 Ру С1]2+ и изомерного ему
[Pt Еп PyCl NH3C1J2+. По значению оптического вращения и по на¬
личию или отсутствию инверсии знака оптического вращения они
пришли к заключению, что реакции амидирования и хлорамидирова-
ния аналогичны. Следовательно, эти данные также подтверждают
заключение о том, что хлорированию подвергаются аминогруппы с
наибольшей подвижностью протонов.10.4. Влияние л-акцепторных лигандов
на кислотно-основные свойства
комплексных соединенийК концу 60-х годов накопилось много экспериментальных фактов,
которые позволили ввести в функциональную зависимость кислотных
свойств комплексных соединений еще один фактор. Этим фактором
является л-акцепторное свойство внутрисферного лиганда.В 1955 г. Леден и Чатт оценили константу кислотной диссоциации
аквоэтиленового комплекса [Pt(C2H0 (НЮ)СЬ]. Порядок константы
оказался неожиданно высоким — 10~5. По существовавшим тогда
представлениям можно было ожидать значения константы на два
порядка меньше. Напомним, что такие высокие константы кислотной
диссоциации (Ка — Ю_4-М0~°) имеют диаквоаминосоединения типа
[Pt А2(Н.,0).,]2+, заряд комплексного иона которых равен 2+
(табл. 10.2). Аквокомплекс [Pt(C2H4) (Н20)С12] отличался от всех ра¬
нее изученных тем, что он содержал лиганд ярко выраженного л-ак-
цепторного характера. Позднее выяснилось, что наличие л-акцептор¬
ных лигандов всегда вызывает резкое усиление кислотных свойств
комплексов. Так, Гринберг с сотр. показали, что замена аммиака на
пиридин или дипиридил в комплексах платины (II) усиливает кис¬
лотную диссоциацию аквокомплексов (см. табл. 10.3). Установлено
также, что пиридин приводит к заметному усилению кислотных свойств
внутрисферной молекулы аммиака в комплексах платины (IV). Гельф-268
ман и Смоленская подтвердили на примере этиленового комплекса
/npaHL'-[Pt(NH3)o(C2H4)HoO]',;+ выводы Ледена и Чатта о влиянии эти¬
лена на кислотные свойства аквокомплексов платины (II).Кукушкин с сотр. синтезировали диметилсульфоксидный комплекс
К[РЦДМСО)С13] и установили, что по многим свойствам он напоми¬
нает соль Цензе K[Pt(C2H4)CI3]. Вероятно, основная причина сходства
этих комплексов в том, что как этилен, так 'и диметилсульфоксид
обладают л-акцепторными свойствами. Константа кислотной диссо¬
циации аквокомплекса [РЦДМСО) (Н„0)С12] оказалась равной
1,4 10"5. Таким образом, по кислотным свойствам комплексы
[Й(ДМСО) (Н20)С12] и [Pt(C2H4) (Н„0)С12] также весьма близки друг
к другу. “Усиление кислотных свойств комплексов, содержащих во внут¬
ренней сфере л-акцепторные лиганды, можно объяснить увеличе¬
нием положительного эффективного заряда на центральном атоме.
В результате этого уменьшается электронная плотность на других
внутрисферных лигандах и, следовательно, облегчается отщепление
от них протонов.Влияние л-акцепторного характера лигандов на кислотные свой¬
ства диаквокомплексов типа ^«c-[PtL2(H20)2]2'1' иллюстрирует
табл. 10.3. По сравнению с аммиаком и этилендиамином пиридин,
проявляющий л-акцепторные свойства, способствует кислотной дис¬
социации координированных молекул воды. Из табл. 10.3 видно так¬
же, что первые константы кислотной диссоциации тиоэфирных со¬
единений на два или три порядка выше, чем первые константы кис¬
лотной диссоциации аналогичных аминокомплексов. Это можно объ¬
яснить увеличением положительного эффективного заряда на платине
вследствие меньших а-донорных свойств тиоэфиров по сравнению
с аминами и наличием у первых л-акцепторных свойств.Таблица 10.3. Константы кислотной диссоциации диаквокомплексов
(<uc-[PtL2(H20)2]2+LЛЧЛ’г-10"NH,2,8- КГ84,8V2(NH2C2H4NHo)2,3-10-“5,1Ру3,2-10"5580(CoH4) 2S>10"33,2±0,8> I0-33,8zt 1,21/2(C2H5SC3HnSCJH5)>10-31,4+0,41/'>(C3H7SC2H4SC1H-)> I0-33,5rt 1,2V o(ii30-C3H7SC3HcSC3H;)>10-30,6±0,2P(OC2H5)3(3,0+0,4)10~30,4rt0,6По кислотным свойствам триэтилфосфитный комплекс
[Pt L„(H.,0).,]2+ близок к аналогичным тиоэфирным соединениям.
Первая константа кислотной диссоциации фосфитного комплекса не¬
сколько меньше соответствующих констант тиоэфирных соединений,269
однако вторые константы практически одинаковы. На основании со¬
поставления констант кислотной диссоциации можно допустить,
что по суммарному влиянию на эффективный заряд центрального ато¬
ма о-донорных и л-акцепторных свойств триэтилфосфит и тиоэфиры
близки друг другу. Еще сильнее кислотные свойства выражены у бнс-
сульфоксидного диаквокомплекса [Pt(C2H5S0C2H4S0C2H5)(H20)2]2+.
По первой ступени он диссоциирует как сильная кислота, а~ ю^-мо-2.Следует отметить, что вторые константы кислотной диссоциации
диакводиаминных соединений, соответствующих тиоэфирным, а также
триэтилфосфитного комплекса типа цис-[Pt L2(H20)2]-'+ — величины
одного порядка. Если связывать кислотные свойства с эффективным
зарядом на центральном атоме или на лигандах (в частности, на атоме
кислорода в молекуле воды), можно сделать заключение, что в моно-
гидроксильных комплексах аминного [Pt А2(Н20)0Н]+ и тиоэфир-
ного, а также фосфитного типов [Pt L2(H20)0H]+ эффективные заряды
практически одинаковы. Это может быть обусловлено тем, что в тио-
эфирных и фосфитном комплексах неподеленная электронная пара
координированной гидроксильной группы смещена к центральному
атому по л-типу. Поскольку амины (аммиак и алифатические амины)
не обладают л-акцепторными свойствами, то в данном случае гидро¬
ксильная группа не проявляет столь сильно л-донорные свойства.
В результате различного поведения гидроксильного лиганда в тно-
эфирных и фосфитном комплексах, с одной стороны, и в аминных,
с другой, происходит «выравнивание» эффективных зарядов на цент¬
ральном атоме (а затем на атоме кислорода координированной моле¬
кулы воды) и константы диссоциации по второй ступени становятся
практически одинаковыми.i Кислотные свойства аквокомплексов палладия (II) изменяются
в зависимости от природы лигандов, аналогично изменению свойств
аквокомплексов платины (II). При изучении диаквокомплексов типа
[Pd L(H,0)„]2+, где L — NH„C„H4NHCoH4OH, NH„CoH4SCH40H,
NH2C2H4SC,H6, C2H5SC„H4SC2H5'. C3H“7SC,H‘SC3H;,C2H6SC2H4SC2H4OH, HOC2H4SC2H4SC2H4OH, установлено, что при
переходе от лиганда с двумя донорными атомами азота к лигандам
с донорными атомами азот—сера и сера—сера происходит закономер-Таблица 10.4. Константы кислотной диссоциации диаквокомплексов
типа [PdL(H20)2]-+ (при 25°С)LK,-HPnh.,c.,h4nhc.,h4oh2,3=t0,119—4NHX„H4SC„H4OH4,6=t0,315rt3NHoCoH4SC,H56,0=t0 ,62.8—0,3CiHjSCoHjSCoHs13-16,4-0,9C3H7SCoI-I4SC3H,12±I5,8-0,4C2H5SC,H4SCoH4OII20-214-2HOC2H4SCoH4SCnIt4OH30±362-9270
ное усиление кислотных свойств (табл. 10.4). Таким образом, увели¬
чение числа л-акцепторных центров в комплексе приводит к усиле¬
нию кислотной диссоциации внутрисферных молекул воды.Анализ результатов исследования диаквотиоэфирных комплексов
показывает, что кислотные свойства соединений палладия (II) выра¬
жены несколько слабее, чем у соединений платины (II) (см. табл. 10.3).
Сравнение данных для днакводиаминных комплексов свидетельствует
о том, что первая константа кислотной диссоциации больше для комп¬
лексов палладия (II), а вторые константы близки по своим значе¬
ниям.Зависимость кислотных свойств аквокомплексов от природы ли¬
ганда, расположенного в ццс-положении к молекуле воды, изу¬
чена на аланиновых комплексах тнпа [Pt AnL(H20)]+, где Ап —
NHoCH(CH3)COO“. Из данных табл. 10.5 видно, что константы уве¬
личиваются в ряду лигацдов L: NH3 < Ру < (C3H5)„S < R„SO.Таблица 10.5. Константы кислотной диссоциации аланиновых
комплексов типа [PtAnLH20)+ (при 25°С)LА'-10»LК ■ 10*NH30,45±0 ,04(C2H5),S1 ,8+0,2Ру1,2 =t0,1(CH3)2SO4I=t5(C,H5)2SO44rt5(CH„)4SO76rt 1<CH3)2SO8Irt30(C„H4)2S090i2Аммиак не обладает л-акЦепторными свойствами, поэтому констан¬
та для аммиачного комплекса наименьшая. Способность к л-акцептор-
ному взаимодействию у сульфоксидов должна быть выражена силь¬
нее, чем у сульфидов. Это связано со смещением электронной плот¬
ности с атома серы сульфоксида на атом кислорода, вследствие чего
акцепторные свойства сульфоксидной серы должны быть выше, чем
сульфидной. Если допустить, что усиление кислотных свойств в ряду
однотипных соединений связано с повышением положительного эф¬
фективного заряда центрального иона, то можно сделать вывод, что
у сульфоксидов л-акцепторные свойства в большей мере преобладают
над о-донорными, чем у аналогичных сульфидов.Все сказанное для аланиновых комплексов типа [Pt AnL НгО]+
справедливо и для циклических этаноламиновых соединений:LН,00—CPiNH.fLILONH\CPtО Г271
Константы кислотной диссоциации этих соединений приведены ниже:L NH3 Ру (CH3),S (CH3)2SO/С I -107 3,0+0,2 4,4±0,3 5,2i0,6 59+5Л'ц-107 0,62:0,1 1 ,4±0,4 — 48+8Усиление кислотных свойств происходит в ряду лигандов следующим
образом: NH3 < Ру < (CH3)2S < (CH3)2SO. Кислотные свойства эта-
ноламиновых комплексов выражены несколько слабее, чем аланиновых.
Это является следствием смещения электронной плотности к карбо¬
нильному атому кислорода в аланине.Возрастание констант кислотной диссоциации при замене внутри¬
сферных сульфидов на сульфоксиды также установлено на комплек¬
сах типа [Pt EnL(H20)]2+ (табл. 10.6). Кроме того, из значенийТаблица 10.6. Константы кислотной диссоциации комплексов
платины (II) (при 25°С)Комплекс/сю1| Комплекс/С ■ 10s[PtEn (ДЭС) Н20]2+0,73±0,08[Pt (1ЧН3ДЭСО) (NH3H„0)]2+0,16±0,06[PtEn(TO) НгО]2+0,93±0,051 [Pt (NH3h (ДМСО) Н„0]2+14±2[PtEn (ДМСО) Н20]2+6,9±0,8[Pt (NH3)3 НоО]2+1 0,212+0,018]PtEn (ДЭСО) Н20]2+5,4±0,8констант, приведенных в этой таблице, следует, что сульфо-
ксид, расположенный в транс-положении к молекуле воды в
[Pt(NH3)2WMC0)H.,0]2+, приводит к значительно большему увели¬
чению кислотных свойств, чем в цис-положении в комплексе
Р^НзДЭСО) (NH3H20)]2+. Это свидетельствует о направленном
влиянии л-акцепторных лигандов на кислотные свойства
внутрисферных молекул воды. Замена же диметилсульфоксида в
[Pt(NH3)2(AMC0)H20]2+ на аммиак, как и следует ожидать, приво¬
дит к резкому уменьшению константы диссоциации.Установлено, что замена в комплексах типа [Pt L(H„0)2]2+ ди¬
амина на аминосульфид приводит к значительному увеличению пер¬
вой константы диссоциации, в то время как порядок вторых констант
диссоциации тот же самый:L NHX2H4NH2 NHaG,H4SCgH6 NHoC,H4SC.H.1OH/Ci-Ю5 . . 0,23 20 30К»-Ю8 . . 5,1 1 2Изучение кислотных свойств моноаквокомплексов типа
[Pt LA(H20)]2+, где L — C;H5SC,H4NHo (LJ и HOC8H4SCaH1NHi(Ls),
а А — анилин, пириднн, хинолнн и пиперидин, показало, что внутри-
сферная молекула анилина по влиянию на кислотные свойства акво¬
комплексов платины (II) близка к пиридину:L Пиперидин Пнридин Хмнолин АнилннA'- 10s (Lj) . . 0,6+0,1 1,3+0,2 1 ,8±0 ,2 1,5+0,2
К■ 105 (Lo) . . 0,8+0,1 2,0+0,5 2,3+0,5 3,8+0,9272
Вероятно, это можно отнести за счет проявления анилином, с одной
стороны, л-акцепторных свойств, с другой — более слабых ст-донор-
ных свойств. Действительно, в ряду изученных аминов анилнн —
самое слабое основание. Наименьшими кислотными свойствами об¬
ладает комплекс с пиперидином. Причиной этого являются отсутствие
у него л-акцепторных свойств и сравнительно высокая сг-донорная
способность.В табл. 10.7 сведены константы кислотной диссоциации изомерны
комплексов [Pt L(NH2OH)(HoO)CI]+. хТаблица 10.7. Константы (Ki) кислотной диссоциации
аквокомплексов (при 25°С)КомплексLNH,(CH,),S(CH,)2SO' L4 /NH,OHl +a/PVo J" L\Pt/Hn0 1+
Cl-7 N'JHoOHj0,8-10-°
5-IQ"54,2-10"6
6-10"53-10-4
2,3-10-“Из нее следует, что при замене аммиака в транс-положении по отно¬
шению к молекуле воды на л-акцепторный лиганд происходит резкое
изменение кислотных свойств. Отношение констант кислотной диссо¬
циации Kish, '■ Каме • Кдмсо равно 1 : 52 : 376. Кислотные
свойства координированной молекулы воды усиливаются и в случае
замены аммиака на лиганды л-акцепторного типа в цые-положении.
В этом случае отношение тех же констант равно 1 : 1,2: 4,6.Таким образом, замена аммиака на диметилсульфид и далее на
диметилсульфоксид как в цис-, так и в транс-положении по отноше¬
нию к протондонорной молекуле приводит к усилению кислотных
свойств комплексов. Однако протондонорная молекула все же более
чувствительна к замене лиганда в /иранс-положении. Это свидетель¬
ствует о направленном влиянии л-акцепторных лигандов на кислот¬
ные свойства внутрисферных молекул гидроксиламина и его произ¬
водных (табл. 10.8).Влияние аммиака, диметилсульфида и диметилсульфоксида на
кислотную диссоциацию внутрисферных оксимов и гидроксиламина
в комплексах платины (II) сложнее, чем влияние на кислотную дис¬
социацию молекул воды. Из табл. 10.8 следует, что в изученных
комплексах quc-конфигурации замена аммиака на ДМС и ДМСО при¬
водит к усилению кислотных свойств. Исключением является только
аммиачно-альдоксимный комплекс. Аммиачный и диметилсульфидный
аналоги ацетоксимных комплексов имеют практически одинаковые
константы. В случае транс-изомеров диметилсульфидные комплексы
обладают более сильными кислотными свойствами, чем диметнлеуль-273
Таблица 10.8. Константы кислотной диссоциации оксимных
н гидрокснламиновых комплексов (при 25°С)LКомплексNHj(Clb)jS(CH.)iSOLС1—Pt—NH2OH1ClL ОН0,7-10-90,8-lO'82- 10-7I ICl—Pt—N=C (CH3)„
1 ‘
ClL OH1 • lO'70,7-!0-71 ■ 10-'| |Cl—Pt—N=CH (CH3)
1ClCl1■10-°001ОTj*1■10-5L—Pt—NH„OH
IClCl OH2- Ю"104,3 lO'71,6-10-9I 1L-Pt—N=CH (CH3)
1ClCl OH4,8-10-86,8- lO"7j 1L—Pt—»N=C (CH3)„
1 ’
Cl■—■4,7 lO”72,0 lO'7фоксидные. Возможно, причина этого различного влияния л-акцептор-
ных лигандов на кислотные свойства аквокомплексов, с одной сто¬
роны, и комплексами с гидроксиламином и его производными, с дру¬
гой, заключается в том, что в отличие от Н2О оксимы и гидроксил¬
амин, имеющие двойную связь N =С, сами способны проявлять л-
акцепторные свойства.Яркое проявление влияния таких л-акцепторных лигандов, как
N2 и СО, на кислотные свойства аминокомплексов осмия (III) следует
из сравнения значений рК [1]. Для [Os(NH3)6]3+, [Os(NH3)5X.,]3+ н
[Os(NH3)5CO]3+ они соответственно равны 16, 6 и 2-1-3.Влияние лигандов на кислотные свойства протондонорных моле¬
кул в цис- или /пранс-положении можно рассматривать с позиции
регибридизованных ds/г-орбиталей платины. Вывод о том, что пе¬
редача трансвлияния происходит по отдельным s-, р-, d-атомным
(регибридизованным) орбиталям, был сделан на основании эксперн-274
Рис. 10.1- Схема смещения центра тяжести электронной плотности на ор-
битали^у относительно лиганда RH 'пментальных данных Пидкока и Ричардса по константам взаимодейст¬
вия i96Pt—31Р [2], а также на основании квантово-химического ра¬
счета [3].Если протондонорная молекула является только ст-донором, то
влияние лигандов L на ее кислотные свойства может быть сведено
к следующим случаям:1) непосредственная передача влияния одного лиганда другому,
расположенному в ^ыс-положении;2) a-доноры передают свое влияние в транс-положение посред¬
ством р-орбитали;3) a-доноры, расположенные в г^ис-положении, осуществляют
передачу электронной плотности через dx2_^-орбиталь.Таким образом, усиление транс- или цисвлияния, приводящее к
увеличению электронной плотности на связи Pt—RH, должно ослаб¬
лять кислотные свойства координированной молекулы RH.Иная ситуация должна иметь место при наличии во внутренней
сфере лигандов с л-акцепторными свойствами. Если принять, что
молекула RH не обладает ни л-акцепторными, ни л-донорными
свойствами, то влиянне на ее кислотные свойства л-лигандов может
носить только электростатический характер. Эффективный заряд
центрального атома слагается из заряда ядра и зарядов электронов
на орбиталях, принадлежащих центральному атому. Поскольку
по л-типу взаимодействует ограниченное число орбиталей, то об из¬
менении эффективного' заряда целесообразно судить по смещению
электронной плотности на соответствующих орбиталях. Если закре¬
пить лиганд L, обладающий л-акцепторными свойствами, на оси х
и менять относительно него местоположение лиганда RH, то во взаи¬
модействии с первым главным образом будут участвовать dxy- и dxz-
орбитали центрального атома (рис. 10.1). При транс-расположении
рассматриваемых лигандов центры тяжести электронной плотности
орбиталей dxy и dxz будут смещены на расстояние I к л-акцептору и
будут находиться на оси а- (рис. 10.1, а, б).При quc-расположении лигандов центры тяжести орбиталей
dxyiidxz также удалены от RH, но на меньшее расстояние, чем в слу¬
чае транс-расположения (рис. 10.1,6, г). Таким образом, л-акцеп-
торные свойства лиганда L должны усиливать кислотные свойства
молекулы RH как в цис-, так и в транс-положении. Однако влияние
должно быть выражено несколько сильнее при транс-расположении
лигандов.275
Таким образом, экспериментальные данные убедительно показы¬
вают, что введение в комплекс лигандов, обладающих ^-акцепторны-
ми свойствами, приводит к усилению кислотной диссоциации протон¬
содержащих лигандов как в транс-, так и в цис-положении. Вывод
о том, что при замене а-донорного лиганда на л-акцепторный проис¬
ходит усиление кислотных свойств комплексов, может использоваться
для качественного предсказания кислотных свойств еще неизученных
соединений. Сущность этого явления заключается в электронных сме¬
щениях в комплексе в целом. Поскольку лиганды, обладающие л-ак-
центорными свойствами, всегда являются слабыми ст-донорами, то
усиление кислотных свойств под влиянием л-акцепторных лигандов,
несомненно, является суммарным эффектом, складывающимся из
меньшего смещения электронной плотности на центральный атом по
о-связи и смещения электронной плотности от центрального атома на
л-акцепторный лиганд по л-связи.К сожалению, учет ст-донорной и л-акцепторной составляющих
экспериментальным способом пока практически невозможен. В на¬
стоящее время делаются попытки оценить эти составляющие расчет¬
ным путем.Библиографию по влиянию л-акцепторных лигандов на кислот¬
ные свойства комплексных соединений можно найти в работе [4].10.5. Расчет констант кислотной диссоциацииДля определения констант кислотной диссоциации комплексных соедине¬
ний широко используются потенциометрический и спектрофотометрический
методы. Имеются работы по применению для этой цели ИК спектроскопии и
метода ПМР. Однако два последних следует считать корреляционными, так как
их использование основано на взаимосвязи определенного свойства комплексно¬
го соединения с его способностью к кислотной диссоциации. Они не дают воз¬
можности непосредственно измерять активность ионов водорода.На практике наибольшее распространение все же имеет потенциометриче¬
ский метод. Он позволяет определять константы кислотной диссоциации, если
они не менее 10-1". Для определения с достаточной точностью констант относи¬
тельно сильных кислот необходимо, чтобы концентрация кислоты численно была
не меньше первой константы кислотной диссоциации. Растворимость комплекс¬
ных соединений не всегда позволяет создать такие условия.Константа кислотной диссоциации вычисляется на основании результатов
титрования кислоты щелочью или сопряженного с ней основания кислотой. В слу¬
чае частичной нейтрализации одноосновной кислоты щелочью выражение кон¬
станты кислотной диссоциации имеет видЛ' = [Н+] —^—. (10.1)С —агде С — общая концентрация титруемой кислоты; а — концентрация прибав¬
ленной щелочи.Если кислота оттитрована наполовину, т. е. а = С/2, то данное выраже¬
ние превращается в К = [Н+]. Таким образом, для того, чтобы оценить значение
константы кислотной диссоциации одноосновной кислоты, нужно лишь измерить
концентрацию ионов водорода наполовину оттитрованного раствора определяе¬
мой кислоты.Для частично нейтрализованной относительно сильной одноосновной кис¬
лоты выражение константы кислотной диссоциации имеет вид276
л- = 1нч _Л±ЩЧ_. (10 2)1 С —а — [Н+] ' 'Для очень слабых кислот, с учетом гидролиза аннона, выражение кон¬
станты кислотной диссоциации<c^,|inV (,М>Все эти выражения легко выводятся из общего уравнения
.. [Н+] [А-][НА]X. И. Гильденгершель для расчета констант кислотной диссоциации ре¬
комендует вначале определить ориентировочное значение константы по точке
на кривой титрования, соответствующей 0,5 эквивалента добавленной щелочи.
ЕслирК < рС Н- 1,62,то расчет константы следует проводить по формуле (10.2). Если
рА' » рКш — рС — 1,58,то расчет константы проводят по формуле (10.3). ЕслирЛ'ц, — рС — 1,58 » рК > рС -|- 1,62,то расчет проводят по формуле (10.1).Для двухосновных кислот по значению pH при добавлении 0,5 эквивалента
щелочи (рHi) и 1,5 эквивалента (рН2) оценивают значения рК\ и рКг- Если раз¬
ность рНо — pHi > 1.48 при точности 10%, то константы кислотной диссоциа¬
ции рассчитываются по формулам для одноосновных кислот. Фактически си¬
туация аналогична той, когда титруются две одноосновные кислоты одинаковой
концентрации.Если разность рН2 — pHi < 1,48, то применяются формулы для двух¬
основных кислот. При условии pHi < р С + 1,62 расчет ведется по формулам
Бриттона для двухосновных кислот:, BlD, — B,D, .к = - "К, =АуВ, — A2Bl
AiD. — A.D,BlD2 — В, Di
гдеД = [Н+р + а [Н] -С [Н+] JВ = 2С — а — [Н+] ;D — [Н+]: (а + [Н+]).Значения С и а те же.Если pHi > рС + 1,62, то применяются формулы Гнльденгершеля:,h [h;j2 (2 - п.) - п.2 [н;]2 (2—щ) .Ai =[Hj] (1 - я,) (2 - я2) - [н2+] (1 - я„) (2 - я,)
(1 - я8) [h;j пt [h;j2 - (1 - п,) [н|] ,и [н:]2
(2-я,) п2 [Н^]2 — (2 — я2) /*! [н7]2277
Рис. 10.2. Зависимость оптической плот¬
ности от pH раствора для одноосновной
кислотыгде щ и п2 — эквиваленты добавленной ще¬
лочи; [Н^] и [Н^] — соответствующие им
концентрации ионов водорода.Значения Ki и Кг рассчитывают по лю¬
бым двум точкам кривой титрования, ха¬
рактеризующимся достаточной буферной
емкостью. Обязательным условием явля¬
ется щ < «2-Распространенным методом определения констант кислотной диссоциации
является также спектрофотометрический. Так же как и потенциометрический
метод, он позволяет исследовать растворы кислот со значениями рКа в интер¬
вале 2-т-П. Для применения этого метода необходимо, чтобы существовала об¬
ласть спектра, в которой молярные коэффициенты погашения кислотной и ос¬
новной форм заметно различались. Кроме того, необходимо иметь эксперимен¬
тальные значения оптической плотности растворов, содержащих только кислот¬
ную форму £>на и только основную форму Da-. Для этого обычно снимают
спектры определяемой кислоты в 0,01 и 0,1 М растворах щелочи, а также в 0,01
и 0,1 М растворах сильной кислоты, например НС1. Если значения рКа на¬
' 10, то спектры при обеих концентрациях щелочи и при” * и характеризуют DA_ходятся в пределах 4
обеих концентрациях кислоты совпадают между собой
и D'НА'Еще одно необходимое условие — это значение pH
Последнее вытекает из выраженийисследуемых растворов.К ,=СН+ Сд-р К3 = pH + lg■'НАТаким образом, спектрофотометрический метод определения констант кислот¬
ности должен применяться в комбинации с потенциометрическим или иным ме¬
тодом, позволяющим определять значение pH. Это, конечно, усложняет при¬
менение спектрофотометрнческого метода.Для одноосновных кислот функция D — /(pH) является монотонной и име¬
ет одну точку перегиба при pH = р/Са и две асимптоты D ~(рис. 10.2).Вычисление значений рД'а для одноосновных кислот проводят по формулеDa- и D = DhaрД'а = pH + lgDa—DD — DНА'где D — оптическая плотность анализируемого раствора при определенных
значениях pH. На практике снимают спектры растворов исследуемого комп¬
лекса в ряде буферных растворов с различными значениями pH при постоянной
длине кюветы и постоянной суммарной концентрации НА и А-.Подробно методика спектрофотометрического определения значений p^fa
описана в (5].А~10.6. Ступенчатая кислотная диссоциация
комплексных соединений и внутрисферных
лигандовОсновность комплексных кислот может быть равной числу коордн*
нированных протонсодержащих лигандов RH. Возможность их экс-
периментального определения обусловливается лишь чувствитель¬
ностью метода. Известны многочисленные примеры, когда координи¬
рованны» лиганд, содержащий несколько протонов, также отщепляет
их ступенчато.Заключение о ступенчатой диссоциации комплексных соединений
вытекало уже из работы Пфейффера по исследованию [СгРу2(Н20)4]С13.
Потенциометрический метод исследования позволяет сравнительно
легко изучать ступенчатую диссоциацию аквокомплексов, так как
они обладают ярко выраженными кислотными свойствами.Аминокомплексы, в частности аммиакаты, проявляют более сла¬
бые кислотные свойства, чем аквосоединения. Однако и для них Грин¬
берг и Гильденгершель потенциометрическим методом установили
многоосновность и определили константы нескольких ступеней дис¬
социации.| Так, для [Pt(NH3)e]CU удалось вычислить константы двух ступе¬
ней диссоциации Ki = 1,5- 10-7, К2 = 1,5* 10-1°. Для более сильных
кислот [PtEn3]CU и [Pt Рп3]СЦ вычислены даже по три последова¬
тельных константы. Используя низкую растворимость, Кукушкину
и Сибирской удалось выделить в твердую фазу диамидный комплеко
[Pt(NH3)4(NH2)„]h. Рассчитывать на получение комплексов платины
(IV) в водных растворах с большим числом амидогрупп нельзя. Для
этой цели нужно использовать растворитель с большей основностью.
Такую задачу успешно решил Хекк. В качестве среды он взял жидкий
аммиак, а в качестве основания KNH2. Действуя рассчитанным
количеством амида калия, он смог получить остальные члены
ряда амидосоединений: [Pt(NH3)3(NH.i)3]'N03, [Pt(NH3)„(NHo)4],
K[Pt(NH3)(NH2)5] и K2[Pt(NH2)6],Особый интерес представляет гексамидный комплекс K2[Pt(NH2)6],
который является продуктом отщепления протонов всеми шестью мо¬
лекулами аммиака. Принципиально для комплекса [Pt(NH3)6]Cl4
можно ожидать основность выше, чем 6. Это осуществимо в том слу¬
чае, если отщепление протонов будет происходить и от амидогрупп.Хекку удалось синтезировать димерный комплекс с мостиковыми
амидогруппами:,NH„,(NH3)4 Pt/
\nh>Pt (NH3)4CLПри изучении его кислотных свойств в водном растворе оказалось,
что диссоциации протона подвергаются не молекулы аммиака, а мос-
тиковые амидогруппы по схемеPt (NH3)4' /NHo4
(NH3)4Pt< \\nh,/✓NH\(NH3)4Pt/ \pt (NH3)4NMH,'H+H+ +/NH\
(NH3)4Pt< >Pt(NH3)4'NH//NH\(NH3)4Pt/ \pt (NH3)4Это стало возможным вследствие того, что протоны мостиковых амидо¬
групп подвергаются лабилизирующему влиянию двух центральных279
атомов платины (IV). Таким образом, отщеплению протонов от коор¬
динированной амидогруппы должен способствовать высокий заряд
центрального атома.Для некоторых переходных металлов в высоких степенях окис¬
ления хорошо известны нитридные комплексы. В них азот в степени
окисления 3—• связан тройной связью с центральным атомом: Mn+sN.
Первое такое соединение осмия (VIII) было получено еще в 1847 г.
Синтез его осуществляется по реакции0s04 + КОН + NH3 = К [0s03N] + 2Н20В настоящее время изучено большое число нитридных комплексов
различных металлов (рен^я, молибдена и др.). Характерной их осо¬
бенностью является высокая степень окисления центрального атома.Ю. А. Буслаев, М. А. Глушкова и А. М. Большаков путем на¬
гревания в твердой фазе комплекса Re(NH3)Cl4 осуществили превра¬
щение координированного аммиака в нитридную группу:250° СRe(NH3)C!4 >- Re(NH2) Cl3 + НС1400° с;Re(NH2) С13 »- ReNCI + 2НС1«Все соединения здесь записаны в виде мономерных форм.Известны комплексы рения (V) с органоимидными лигандами
NR2-. Впервые они были получены Чаттом с сотр. по реакции[ReOC 13(РЕtoPh)2] + NH2R [ReCl3(NR) (PEt2Ph)2] + H„0Чатт показал, что метилимидолиганд в комплексе
[ReCl3(NCH3) (PPh„R)2], где R — Me, Et, Ph, под действием пириди¬
на отщепляет протон по реакции[ReCI3(NCH3) (PPh2R):] + 2Ру -*■ [ReCI2 (N=CHS) Ру (PPh2R)2] + Ру . HCIВозможно, лиганд N =СН„ образует трехчленный цикл Re jЧСН:что и способствует отщеплению протона.Установлено, что при действии НС1 на [ReCl2(NCH2)Py(PPh2Et)J
происходит обратимое присоединение протона с образованием
[Re Cl„(NCH3)Py(PPh„Et)2]Cl.Образованию амидных и тем более имидных и нитридных соедине¬
ний способствует высокий заряд центрального иона. В водных раст¬
ворах при действии щелочи на комплекс [Pt(NH3)4]Cl2 нет никаких
признаков образования амидокомплексов. Причиной этого являются
очень слабые кислотные свойства данного комплекса. Следует отме¬
тить, что взаимодействие солен ртути (II) с аммиаком приводит к об¬
разованию амидосоединений по реакции
HgCl2 + 2NH3 = NH2HgCl -Ь NH4C1Протеканию данной реакции, вероятно, способствует низкая раство¬
римость амидного соединения ртути.280
Амидные комплексы платины (II) и палладия (И) получены и ис¬
следованы Уоттом. Он, как и Хекк, использовал в качестве среды
жидкий аммиак, а в качестве основания —■ амиды щелочных металлов.
Например:[PtEn.] CL + 2NaNH, =es [Pt (En—H)2] + 2NaCI + 2NH3Гринберг на большом числе примеров показал, что депротониро¬
вание координированных аминов и их производных сопровождается
обратимым изменением окраски комплексов. Из бесцветных они ста¬
новятся окрашеннымн в желтый цвет, а из светло-желтых превра¬
щаются в оранжевые или желтые, но более интенсивно окрашенные.
При добавлении к депротонированным комплексам кислоты их спект¬
ры возвращаются к исходным.Еще более яркое изменение окраски наблюдается при образовании
хлораминных комплексов. Они окрашены в интенсивно красные или
оранжевые цвета. В одном из хлораминных комплексов удалось про¬
вести обратимое присоединение протона по реакции{Pt (rsJClC2H4NH2) PyGI3] + HCI ** [Pt (NHCIC.,H4NH2) PyCI3] Clтемно-красная форма светло-желтая формаСветло-желтая форма образуется при смачивании исходного комп¬
лекса крепкой соляной кислотой. При промывании водой это соеди¬
нение вновь превращается в темно-красный комплекс.Черняев с сотр. осуществили реакцию нитрозаминирования внут¬
рисферных аминов в комплексах платины (IV):IPt(NH,C2HjNH;) РуС!3] С1 + НЧЭ, =д= [Pt (NH,C2H4NH>JO) PyCI3] Cl + Н*0Нитрозамины в поле платины (IV) также обладают ярко выражен¬
ными кислотными свойствами, в результате этого в твердую фазу
выделяются комплексы в депротонированной форме:[Pt (N'HjCjHjNHNO) PyCI3] Cl s* HCI + [Pt (NH2C*H4NNO) PyCl3]Реакции внутрисферного нитрозирования аминов во многом на¬
поминают реакции их хлорирования. Комплексы с внутрисферными
нитрозамидами также обладают более яркими окрасками, чем их
протонированные формы.Интересным лигандом с точки зрения проявления кислотно-ос¬
новных свойств в комплексах является сульфаминовая кислота
NH2SO3H. Координация сульфаминовой кислоты около платины(II) осуществляется посредством атома азота. При этом отщепляется
протон сульфогруппы и во внутренней сфере комплекса образуется
сульфаминатный ион NH2SO3.Привлекает внимание тот факт, что сульфаминатный ион даже в
поле платины (II) проявляет большую склонность к отщеплению про¬
тона от аминогруппы. При действии щелочи на растворы какrapaKc-K:[Pt(NH2S03)2CI2], так н qwc-KalPtfNHsSOaJuClj] образу¬
ются комплексы состава [Pt(NHS03).,Cl.,]. Столь ярко выраженные
кислотные свойства координированного сульфаминатного иона яв¬
ляется следствием влияния атома серы (VI). Протоны аминогруппы281
испытывают электростатическое отталкивание как от платины (II),
так и от серы (VI).Ю. Н. Кукушкин и О. В. Стефанова установили, что ступенчатые констан¬
ты кислотной диссоциации m/7aKc-[Pt(NH2S03)2Cl2]2- рав^ы ATi — 10-7—10"*;
А'2 = Ю-9—10~10. Неожиданным оказалось то, что 4uc-[Pt(NH2S03)2C12]-"
в тех же условиях ведет себя как сильная двухосновная кислота. Столь резкое
отличие в кислотных свойствах изомерных комплексных соединений прояви¬
лось впервые. Детальное исследование (6] ИК спектров цыс-Яг!Pt(NH2S03)2CI2|-
•2Н20 показало, что молекулы воды в этом соединении являются необычными
для кристаллогидратов. Наличие в ИК спектре этого соединения трех узких
полос поглощения в области, где проявляются колебания молекулы воды, поз¬
волило сделать заключение о том, что каждая молекула воды связана водород¬
ными связями с аминогруппой одного сульфаминатного иона и с атомом кисло¬
рода другого, как показано на схемеН Н нI I IO^SO-jN—Н О —Н ■ • ■ О—S02 N—НВ результате молекула воды по состоянию приближается к гидроксониевому
иону. Можно предположить, что такое строение комплекса сохраняется и в
растворе, чем и объясняются его сильные кислотные свойства.Следует отметить, что для транс-изомера такое «сшивание» сульфаминатных
ионов невозможно.Комплекс K2[Pt(NH2S03)4]-2Н20 является сильной четырехосновной кис¬
лотой. Вероятно, в данном случае молекулы воды играют ту же самую роль. Ина¬
че трудно представить, что комплекс диссоциирует по типу сильной кислоты да¬
же на последней стадии диссоциации, где отрицательный заряд комплексного
иона [Pt(NH2S03) (NHS03)3]b_ столь высок.С точки зрения кислотно-основных свойств комплексных соеди¬
нений интересными лигандами яеляются гидроксиламин и оксимы.
С гидроксиламином и простыми оксимами наиболее детально кислот¬
ные свойства изучены для комплексных соединений платины (II).
Поскольку гидроксиламин обладает восстановительными свойствами,
до сих пор не удавалось с достоверностью получить определенные
гидроксиламиновые соединения платины (IV) окислением соответст¬
вующих соединений платины (II). В комплексных соединениях пла¬
тины (II) гидроксиламин координируется посредством атома азота.Гидроксиламиновые комплексы платины (II) являются слабыми
кислотами. Аналогичные оксимные комплексы проявляют несколько
более сильные кислотные свойства (см. также табл. 10.8):Первые константы кислотной диссоциации комплексов
типа [P1L4]2+ (при 25СС)L CH3CHNOH C-H5CHNOH (CH3)2CNOH NHX>HКг 1,5-10“2 5-10"3 3,5-10-4 1,3-10"7А. А. Гринберг и А. И. Стеценко доказали, чуо в координирован¬
ном гидроксиламине протон отщепляется от гидроксильной группы.Глдразиновые комплексы изучены главным образом для металлов, находя¬
щихся в низких степенях окисления. Поскольку гидразин является сильным
восстановителем, то получение комплексов, в которых ион металла находится
в высокой степени окисления, является трудной задачей. В литературе отсутству¬
ют определенные данные о проявлении координированным гидразином кислот¬
ных свойств. Однако имеются сведения [7], что фенилгидразин и его произвол-282
ныс в комплексах циркония (IV) находятся в депротонированной форме. Таким
образом, можно ожидать, что в комплексах, ионы металлов которых имеют со¬
стояние окисления 4+ и выше, координированный гидразин должен проявлять
заметные кислотные свойства.Гидразин в комплексных соединениях выступает в качестве монодентат-
ного лиганда. Неподеленная электронная пара второй аминогруппы остается
свободной. Поэтому в гидразиновых комплексах принципиально можно ожи¬
дать своеобразную перегруппировку. Под влиянием поля центрального атома
протон от координированной аминогруппы может переместиться к свободной
аминогруппе по схеме_ +NH, ^ Мп+-*- NH— NH3Интересная реакция диссоциация протона от координированных
о- и л-нитроанилина в комплексах платины (II) описана в работе [8].
Прн действии щелочи на комплексы [Pt^NHaArJaCh], где NHsAr —
о- и п-нитроаиилин, появляется интенсивное красное окрашивание
раствора. Эго обусловлено отщеплением протона от аминогруппы нитро¬
анилина в соответствии с уравнениями[Pt(MH,Ar),Cl,J ^=6 Н+-Ь [Pt(>!H, Ar) (NHAr)CUJ-[Pt (NH;Ar) (NHAr) Cl2]~ ** H++ [Pt (NHAr)3 Cl2]:_Аналогичный комплекс с лг-нитроанилинэм такой реакции не подвер¬
гается. Авторы объяснили проявление кислотных свойств о- и п-
ннтроанилиновых комплексов платины (II) сопряжением между нитро¬
группой и платиной (II), высту11%ющей в роли л-донора электронов.
В результате сопряжения координированные о- и «-нитроанилин
приобретает хиноидное строение и комплексы начинают поглощать
свет в видимой области спектра. Образующиеся соединения могут быть
изображены, например, следующими формулами:°\ ,/ \ ? /=\. N —У х,— H,N — Pt—NH = < X=-N4о \=/ ‘ I \= О”Cl■°\ /=\ ? /=\ *°=' >=N Н—Р t—NH=/ ) = N.</ \=/ ! \=/ 0“. ClДля л(-нитроанилина такое сопряжение невозможно, поэтому и
не образуется хиноидная структура, обусловливающая появление
окраски.На основе реакции образования аналогичных нитроанилиновых
комплексов палладия (II) разработан способ идентификации о- и п-
нитроанилина [9]. Он позволяет селективно определять о- и я-нитро-
анилин в присутствии лг-нитроанилина и других первичных аромати¬
ческих аминов.Этаноламиновые комплексы платины (II) в отличие от других ами-
натных комплексов проявляют гораздо более ярко выраженную кис¬
лотность. При изучении кинетики взаимодействия этаноламинового283
комплекса if^c-fP^NIbCaHiOH^Cb] со щелочью установлено [10],
что этаноламин замыкает цикл с отщеплением протона от спиртовой
группы в соответствии с реакциейцис-[Pt (NH2C2H4OH)2 CIj] + 20Н- *1= [Pt (NH2C2H40)2] + 2H20 + 201-Этим объясняется повышенная кислотность этаноламиновых соеди¬
нений платины (II).10.7. Соли координационных кислотКислотные свойства аммиакатов платины (IV) широко изучены Грин¬
бергом. Он предсказал, что эти аммиакаты, будучи кислотами, должны
давать соли и в том числе аммонийные соли. Соединение такого ти¬
па синтезировано Черняевым при действии этилендиамина на
[Pt(NH3)2N02CI3]. Ему может быть приписано строение“01 "-- 01Cl NHjNH2 01PtNH3CoH4NH3PtCl NH3NH3 CINO, _no2 _Одно из общих определений свойств протонных кислот как класса
химических соединений заключается в том, что их атомы водорода спо¬
собны замещаться на металл с образованием солей. Для координа¬
ционных кислот можно ожидать протекания следующей реакции:[Pt (NН3)с] СЦ + NaOH ч* [Pt (NH3)5 (NH2Na)] Cl4 + Н20Связь между ионами щелочных металлов и координированной
амидогруппой носит ионный характер, поэтому в растворе они гидро¬
лизуются и выделение таких соединений из водного раствора должно
происходить с большими трудностями.Положение резко меняется, если взять ионы металлов, склонных
к образованию донорно-акцепторной связи [серебро, таллий (I),
ртуть]. Амидогруппа имеет свободную электронную пару, благодаря
которой с ионами таких металлов может образовываться ковалентная
связь. В этом случае в водном растворе будут осуществляться реакции
типа[Pt (NH3)5 NH2]3+ + Ag+ ** [Pt (NH3)5 (NH=Ag)]4+О протекании такой реакции в растворе должно свидетельствовать
увеличение растворимости солей серебра, его гидроксида и оксида.
Опыты показали [11], что в 0,5-10-3 М растворе амидокомплекса
[Pt(NH3)6NH2]3+ растворимость свежеприготовленного оксида сере¬
бра действительно увеличивается на два порядка, а в 0,25- Ю'3 М
растворе [Pt(NH3)4(NH2)Cl]2+ примерно также на два порядка увели¬
чивается растворимость хлорида серебра.Образование солей координационных кислот проявляется также
в изменении pH среды. Весьма характерны кривые потенциометриче¬
ского титрования амидокомплексов платины (IV) растворами нитрата284
Рис. 10.3. Кривые титрования раствором
AgNO.4 при добавлении 1 экв. щелочи раст¬
вора fPt(NH3)sCl](N03)3 (/) и раствора
[Pt(NH3)5CI]CI3 (2)серебра. Так, если к раствору комп¬
лекса [Pt(NH3)oCl](N03)3 добавить
один эквивалент щелочи и титровать
раствором AgN03, то при добавлении
первых капель резко снижается pH
титруемого раствора. В конце титро¬
вания значение pH практически оста¬
ется постоянным (рис. 10.3). Для
комплексов, имеющих внешнесферные хлорид-ионы, вначале сереб¬
ро расходуется на связывание хлорид-ионов в виде AgCl-. Изме¬
нение pH на этом участке кривой титрования небольшое. Рез¬
кий скачок pH начинается после связывания всех внешнесферных
хлорид-ионов. Для комплексов с внешнесферными хлорид-ионами в
начальный момент титрования AgCl из раствора не выделяется, что
согласуется с результатами определения растворимости.Протекающие реакции выражаются следующими уравнениями:[Pt(NH3)5CI]3+ + OH- [Pt (NH3)4 (NH;) Cl]:+ + H20
[Pt (NH3)4 (NH2) CI]2+ -p Ag+ « [Pt (NH3)4 (NH2Ag) Cl]3+Важно отметить, что в процессе титрования не выделяется гидроксид
серебра.Склонность к образованию солей координационных кислот пла¬
тины (IV) и устойчивость их в водных растворах должны зависеть от
силы соответствующих кислот. Устойчивость комплексов с внутри-
сферными молекулами амидов серебра и таллия (I) изучена потенцио¬
метрическим методом [12].Экспериментальные данные сведены' в табл. 10.9.Таблица 10.9. Константы диссоциации комплексных солей и
соответствующих кислот (Ссоли — 5,0 • 10~3 М, ц = 2)Комплексный ионKAe**T1 +Ka[Pt (NH3)3 (NH2M) CloF-1.5.10-46,0-10"12[Pt (NH3)5 (NH2M)]4+4,9-I0"e3,6 to-11,2-10"7[PtPy (NH3)3 (NH2M) Cl]3+5,7-10-73,7-10"41,2-10"7[PtPy2 (NH3)2 (NH2M)Cl]3+2,2-I0-75,5-10-41,6-I0"a[PtPy4 (NH2M) Cl]3+3,2-IQ"5■ 3,2-I0-36,0-IO"5Константы неустойчивости серебряных солей аминатов платины
(IV) близки по значениям к константам кислотной диссоциации соот¬
ветствующих аминатов (табл. 10.9): чем сильнее кислотные свойства
аминатов, тем менее стойки их серебряные соли. Вероятно, факторы,285
определяющие кислотные свойства координационных кислот, сохра¬
няют свое значение и в случае диссоциации их серебряных солей. Од¬
нако количественного соответствия между константами кислотной дис¬
социации и константами неустойчивости солей нет. Так, константы кис¬
лотной диссоциации [Pt(NH3)6]4+ и [Pt(NH3)sCl]3l- различаются более
чем на порядок, тогда как константы неустойчивости их серебряных
солей отличаются только в три раза. То же можно сказать о
[Pt Pyi(NH3)3CIJ3+ и [Pt Py4(NH3)Cl]3+ и их серебряных солях.Таллиевые производные менее прочны, чем соответствующие се¬
ребряные. Это находится в согласии с прочностью аммиачных комп¬
лексных соединений серебра (I) и таллия (I). Константа неустойчи¬
вости иона AgNH3 равна 6,3-Ю'4, а иона TINH3 — 8,3. Таким об¬
разом, как и в случае аммиачных комплексных соединений серебра
и таллия, константы неустойчивости комплексных соединений тал¬
лия (I) с амидами платины (IV) на 3—4 порядка выше, чем соответст¬
вующих соединений с ионом серебра (I).10.8. Кислотно-основные равновесия в растворах
гидратокомплексовИоны металлов в растворе сольватированы. Если молекулы раство¬
рителя в свободном состоянии обладают тенденцией к кислотной дис¬
социации, то при образовании связи с ионом металла кислотные
свойства должны обязательно усиливаться. Так, гидратированные
ионы металлов с высоким положительным зарядом являются доволь¬
но сильными кислотами. Константы кислотной диссоциации акво-
комплексов могут быть легко вычислены из значений последней кон¬
станты неустойчивости соответствующих гидроксокомплексов. На¬
пример, ион алюминия (III) в водном растворе находится в виде
гексааквокомплекса [А1(Н20)6]3+. Требуется вычислить константу
кислотной диссоциации этого комплексного иона в соответствии с
уравнением[А1 (НоО)0]3+ « Н++ [AI (Н,0)50Н]3+= [Н+] [А1(Н,0)50Н-Ч
" [AI (НаО)2+] 'В справочнике находим, что константа неустойчивости гидроксо-
комплекса АЮН2+ равна 10~9. Этой константе соответствует процессА10Н-+ ^ AR + ОН-или в более строгой форме[А! (Н.О)з ОН]2+ + Н20 ^ [А1 (НоО)6]3+ + ОН~Г дггн 01 [А1 (Н^°)б+1 [он-1
неуст.6 А I 3°1 [А1(Н,0)5 0Н2+] ** Порядок нумерации констант неустойчивости см. в разд. 11.1.286
Умножив числитель и знаменатель на [Н+], выражение константы
неустойчивости можно преобразовать в следующее:_ [А1 (н:о)36+] [он-] [н* ] ^ К«,Лнеуст.6 - [А1 (Н:0)5 ОН:+] [Н+] Ка ’ОтсюдаК КГ14 = 10-5.а К 10“9нсуст.6 1иТаким образом, при наличии справочных данных по константам
неустойчивости гидроксокомплексов легко вычислить константы кис¬
лотной диссоциации гидратированных ионов.Аналогично может быть вычислена вторая и следующие константы
кислотной диссоциации гидратированных ионов металлов:.. г.- •• ^111Л аз — ,. I Л а4 — 'д* ' м l' д* '^неуст.5 Ансуст.4 неуст.ЗПри растворении в воде солей металлов в растворе образуются
гидратированные катионы и анионы. Если анионы являются остатка¬
ми сильных кислот, то они не будут влиять на характер среды. Гидра¬
тированные катионы металлов, за исключением щелочных и щелочно¬
земельных, являются кислотами. Их кислотные свойства будут за¬
висеть от заряда, радиуса и электронного строения иона металла.Кислотность среды многоосновных кислот практически определя¬
ется первой ступенью диссоциации. Поэтому для вычисления pH сре¬
ды достаточно знать значение первой константы кислотной диссоциа¬
ции гидратированного иона и концентрацию электролита. Допустим,
необходимо вычислить pH 0,001 М раствора А1С13. Подставляя зна¬
чение Ki = Ю"6 в выражение для одноосновных слабых кислот, лег¬
ко вычислить концентрацию ионов водорода, например, по приб¬
лиженной формуле[ н+] = Vkc.10.9. Полимеризация гидроксокомплексовПоскольку вода до сих пор основной растворитель в неорганической
химии, то в координационной химии наиболее распространенными
являются аквокомплексы. Координированная около иона металла
молекула воды по сравнению со свободной имеет гораздо большую
склонность к кислотной диссоциации, поэтому смешанные аквогидр-
оксокомплексы в водных растворах столь же распространены, как и
сами аквокомплексы. Для центральных ионов высокой степени окис¬
ления характерно превращение гидроксокомплексов в оксосоедине-
ния. Не менее характерна полимеризация гидроксокомплексов.Рассмотрим факторы, от которых должна зависеть полимериза¬
ция гидроксокомплексов. Безусловно, склонность комплексных со¬
единений к полимеризации с образованием мостиковых связей должна287
зависеть от их лабильности. Действительно, полимеризация связана
с необходимостью удаления лигандов из внутренней сферы комплек¬
сов, например в соответствии с уравнением2 [ML„ (OH)]m+ ** [M8Ls(n_,)(OH)i]sm+ + 2LДля того чтобы гидроксильная группа стала бидентатной (мостиковой),
необходимо освободить одно из координационных мест во внутренней
сфере комплекса.Таким образом, возможность образования полимеров определя¬
ется не только наличием лигандов, способных образовывать мости-
ковую связь, но и наличием подвижных лигандов, способных осво¬
бождать координационное место во внутренней сфере комплекса.
Например, комплекс [Pt(NH3)3OH]+ не должен обладать склонностью
к полимеризации, так как он инертен и молекулы аммиака прочно
удерживаются во внутренней сфере. Молекулы воды менее прочно
связаны с платиной (II) и поэтому они легко замещаются на другие
лиганды. В связи с этим в комплексах типа [PtL2(H20)0H]+ проте¬
кание процесса полимеризации вполне возможно по схеме2 [PtLo (Н20) ОН]+ г* [Pt2L4(OH),]2+ 4- 2Н20Лабильность внутренней сферы комплексов зависит не только
от природы центрального атома, но и от природы внутрисферных
лигандов. Например, скорость образования дигидроксодимеров типа
[Pt2L4(OH)2]3+ из комплексов [Pt Ьз(НзО)ОН] в значительной мере
должна зависеть от транс-и цисвлпяния лигандов L. Известно, что
для соединений платиновых металлов проявление взаимного влия¬
ния лигандов особенно характерно.Склонность к полцмеризации аквогидроксокомплексов платино¬
вых металлов известна давно. Однако вследствие их инертности и
трудности достижения равновесных состояний имеется мало коли¬
чественных данных об этих процессах. В весьма редких случаях мож¬
но оценить степень полимеризации аквогидроксокомплексов и сделать
заключение о строении полимерных соединений.По-видимому, первые количественные данные о процессах поли¬
меризации гидроксокомплексов платиновых металлов были получены
практически одновременно для палладия [13] и для платины [14].При изучении кислотных свойств [Pd Еп(Н20)2]2+ было показано,
что один эквивалент протонов удаляется из комплекса обратимо, а
дальнейший участок кривой титрования не отвечает простому равно¬
весию, связанному только с депротонированнем. Эти результаты были
интерпретированы как отражение процесса дпмеризации:Аналогичный платиновый комплекс [Pt En(HiO)2]2+ титруется
щелочью как обычная двухосновная кислота. Это служит хорошим
примером влияния' природы центрального атома на процесс поли-2PdEn (Н2о)2+ ** EnPd PdEn2+ + 2Н30+288
мериза'ции. Однако принципиального различия в поведении ана¬
логичных комплексов палладия (II) н платины (II) нет. Отличие за¬
ключается лишь в кинетике. Так, если к комплексу [Pt Еп(НгО)2]2+
добавить один эквивалент щелочи н выдержать раствор при комнат¬
ной температуре в течение нескольких дней, то дальнейшего расхода
щелочи не наблюдается. Это означает, что в данных условиях прои¬
зошла димеризация и платинового комплекса.Депротонирование как палладиевого, так и платинового комп¬
лекса происходит практически мгновенно. Процесс же дпмеризации
идет во времени. Поскольку палладиевые соединения в 105—10° раз
лабильнее аналогичных платиновых, то в первом случае вслед за уда¬
лением одного протона сразу происходит замыкание цикла с образо¬
ванием димерного соединения. В случае платинового комплекса ди-
меризация протекает намного медленнее, поэтому за короткие проме¬
жутки времени процесс димеризацип не проявляется, а наблюдается
обратимость кислотно-основного равновесия.Полимеризация комплексов платины (II) типа [Pt(PPh3)L(H20)2]2+
была обнаружена для L-диметилсульфоксида и тиодпгликоля [14].
Поскольку трансвлияние Р- и S-лигандов выше, чем аминов, то в
комплексах [РЦРРЬ3)(ДМСО) (НЮЬ]2+ и [Pt(PPh3) (ТДГ) (Н20)2]2+
молекулы воды оказываются лабилизованными и создаются условия
для замыкания гидроксомостиков. Это обстоятельство проявилось
при исследовании кислотных свойств данных комплексов.'Оказалось,
что они титруются щелочью, как одноосновные кислоты. Кривые тит¬
рования имеют один резкий скачок, соответствующий только одному
эквиваленту добавленной щелочи. Оценка констант кислотной дис¬
социации обоих этих комплексов привела к величинам порядка 10~г.В последующих работах было установлено, что глубина проте¬
кания процесса полимеризации зависит от природы радикалов в фос-
финах и природы серосодержащего лиганда. Замещение фенильного
радикала на этильный или бутильный в PPh3 препятствует полимери¬
зации. Возможно, это связано с различием в трансвлиянии таких
фосфинов.Полимеризация гидроксокомплексов палладия (II) и платины (II)
была обнаружена также в бензолсульфинатных комплексах типа
[A^CeHjSOj^HoO);,] [15]. Установлено, что как палладиевый, так и
платиновый комплексы четко титруются как одноосновные кислоты.
Следует отметить, что бензолсульфинатный нон в этих комплексах
связан с ионом металла посредством атома серы. Таким образом, мож¬
но предполагать, что этот лиганд обладает высоким трансвлиянием,
что и обеспечивает повышенную лабильность внутрисферных моле¬
кул воды. .В случае сульфинатных комплексов получающиеся при титрова¬
нии щелочью продукты были выделены в твердую фазу. По данным
элементного анализа и значениям молярной электрической проводи¬
мости выделенные комплексы соответствуют формулам К2[М„Ь4(ОН)о].
Таким образом, препаративные данные полностью согласуются с
заключением о протекании в растворе процесса полимеризации.То, что диаквокомплексы титруются, как одноосновные кислоты,Ю-817289
свидетельствует об удалении из комплекса одной из молекул воды п о
протекании процесса полимеризации. Поскольку диаквокомплексы,
содержащие Р- и S-лиганды, являются довольно сильными кислотами
(Ка > 10~:i), то в их водных растворах мольная доля гидроксокомп¬
лексов довольно большая. Поэтому процесс полимеризации с обра¬
зованием гидроксомостиковых комплексов, безусловно, должен про¬
текать и без добавления щелочи. Однако не исключено, что в образо¬
вании мостиков участвуют координированные молекулы воды и вслед
за этим происходит отщепление от них одного из протонов.Полимеризация диаквокомплексов типа [ML2(H20)0HJ+ транс¬
конфигурации должна протекать с образованием линейных полп-
ме’ров. Если же комплекс имеет цг/с-конфигурацию, то наиболее
вероятно образование циклических димеров типаНа примере комплексов платиновых металлов вполне очевидны
факторы, влияющие на процесс полимеризации гидроксокомплексов.
К их числу относятся следующие:1) природа центрального атома;2) лабильность комплексов в целом и лабильность отдельных
внутрисферных лигандов. Последнее связано с необходимостью ос¬
вобождения координационного места и повышения дентатности ли¬
ганда, становящегося мостнковым;3) природа других внутрисферных лигандов, в частности их цис-
и трансвлияние.Безусловно, эти факторы имеют общее значение и справедливы
дтя полимеризации гидроксокомплексов не только платиновых ме¬
таллов.10.10. Основность комплексов переходных металловВ 1955 г. было установлено, что комплексы переходных металлов
могут проявлять основные свойства. Оказалось, что гидридный комп¬
лекс (C5H5)2ReH взаимодействует с протонами по уравнению [16](С5Н5)2 ReH + Н+ ==* [(СьН5)г ReH,] +По основным свойствам этот комплекс лишь немного уступает амми¬
аку.Обработка сухим НС1 эфирного раствора [Ir(PPh3)„(CO)Cl] также
приводит к протоннрованию центрального иона с образованием гид-
ридного комплекса [ 1г(Н) (PPh3) (СО)С1]С1 [17]. Гидридный комплекс
кобальта (I) при взаимодействии с хлорной кислотой превращается
в дигидридный комплекс кобальта (III) [18]:[Со (Н) (Ph,PC2H4PPh2)2] 'г НСЮ4 « [Со (Н), (PhjPCjH^Ph;;):.] СЮ4Обработка этого дигидридного комплекса спиртовым или водным290
раствором NaOH приводит к образованию исходного комплекса:
[Со (Н)о (Ph3PCoH,PPIi2)oJ СЮ4 + NaOH = [Со (Н) (Ph J’CoI^PPh,),]-f NaC104 + H20Водные или спиртовые растворы многих кислот реагируют с трифенил-
фосфиновым комплексом платины (U) с образованием гидридного
комплекса платины (II):[Pt (РРЬз)з ] + НХ = [Pt (Н) (PPh3)3J Xгде X - СЮ;, bf;, HSO;, CH3OSO3.Если анион кислоты обладает склонностью к вхождению во внут¬
реннюю сферу комплексов платины (II), например НС1 и HCN, то
реакция протекает несколько иначе:[Pt (РРЬзЫ + НХ = [Pt (Н) X (PPhahJ 4 PPh3Интересно, что комплекс [Pt(H) (РРЬ3)гС1] при длительном про¬
пускании избытка НС1 способен присоединять еще один нон водо¬
рода с образованием дигидридного комплекса платины (IV):+HCI[Pt (Н) (PPh3)* Cl] — [Pt (H). (PPh„)s Cl,]-наОбразовавшийся дигидридный комплекс платины (IV) постепенно
теряет молекулу НС1 в твердом состоянии. В растворе реакция идет
быстрее. При действии на растворы комплексов [Pt(H) (PPh3)3]X
щелочи реакция идет в обратном направлении с образованием
[Pt(PPh3)3].С кислотой способен взаимодействовать и нитрозильный комплекс
[Ir(NO) (PPh3)3], Его реакция с хлорной кислотой протекает с обра¬
зованием гидридного комплекса:fir (NO) (PPhjbl + HCIO4 [Ir (H) (NO) (PPh3)3] C104При действии на комплекс [Ir(H)(NO)(PPh3)3]+ щелочи также проис¬
ходит образование исходного соединения [Ir(NO) (PPh3)3J.Таким образом, взаимодействие с кислотой всех рассмотренных
комплексов является обратимым процессом. Поскольку присоеди¬
няющийся протон в них превращается в гидридный лиганд, то данные
реакции можно называть как кислотно-основными, так и реакциями
окислительного присоединения.Как правило, основные свойства проявляют л-комплексы или
комплексы, содержащие лиганды л-акцепторного типа. Основность
комплексов, образованных элементами одной и той же подгруппы, но
имеющих одинаковые лиганды, растет с увеличением порядкового
номера переходного элемента. Напомним, что основные свойства со¬
единений элементов главных подгрупп при увеличении порядкового
номера, наоборот, уменьшаются. Считают, что это различие связано
с характером неподеленной электронной пары, акцептирующей про¬
тон. У элементов главных подгрупп эти электронные пары локали¬
зованы на гибридных sp2- или 5/?3-орбиталях, а у комплексов побоч¬10“291
ных подгрупп протоны акцептируются'верхней занятой молекуляр¬
ной орбиталью, которая является почти Пистон d-орбиталью.Комплексы переходных металлов проявляют оспоЕНые свойства
не только по отношению к протонам, но и по отношению к кислотам
Льюиса. При таком взаимодействии образуется донорно-акцепторная
связь, в которой донором электронов является центральный атом
комплекса.Продукты кислотно-основного взаимодействия комплексов пере¬
ходных металлов с кислотами Льюиса часто подвергаются
дальнейшем превращениям. Например, при взаимодействии
транс-[ 1г(РРЬ3)г(СО)С1] со SnCU образуется комплекс
[Ir(PPh3)2(CO)Cl(SnCl4)]. В ИК спектре этого комплекса как в твер¬
дом состоянии, так и в растворе имеются две полосы поглощения, при¬
надлежащие валентным колебаниям СО [19]. Это обстоятельство объ¬
ясняется наличием двух своеобразных изомеров, переходящих друг в
друга:PPh, N SnCUcj ррь,ч S,nCI?CI\ I / \ i уlr. *£ \lr'\pph- rn' JCOX NPPh3 CO/ (i, 4PPh>3С точки зрения образования комплекса [Ir(PPh3)2(CO) (SnCl3)Cl]
взаимодействие транс-[1г(РРЬ3)2(СО)С1] со SnCU можно рассматри¬
вать как реакцию окислительного присоединения.
ГЛАВА11РАВНОВЕСИЯ В РАСТВОРАХ
КООРДИНАЦИОННЫХ СОЕДИНЕНИЙСоздатель координационной теории А. Вернер виел критерий для
отнесення соединений к комплексным — это их существование не
только в твердой фазе, но и в растворе. В конце XIX и начале XX р.
химики в основном исследовали кинетически инертные комплексные
соединения, поэтому Еопрос о вторичной диссоциации почти не возни¬
кал. Тем не менее не было сомнений, что в процессе комплексообразо-
вания образуются промежуточные формы, т. е. реакция протекает
через ряд последовательных стадий. Основанием для этого, в част¬
ности, были соединения, составляющие переходные ряды типа рядов
Вернера—Миолатн. На практике при синтезе комплексных соедине¬
ний создавались условия, при которых образовывалась лишь одна
из нескольких возможных форм.Одновременное существование в растЕоре нескольких форм комп¬
лексов, по-видимому, Епервые было экспериментально доказано в
1915 г. Н. Бьеррумом. При исследовании равновесий в растворах
инертных роданидных комплексов хрома (III) ему удалось разделить
и идентифицировать отдельные формы, а некоторые из них выделить
препаративно. В результате операций осаждения, экстракции и ре¬
экстракции было установлено существование в растворе ионовCr(SCN)f, Cr(SCN)2-, Cr(SCN)“, Cr(SCK')3_, Cr(SCN); и Cr(SCN)2+.Новая область координационной химии — комплексообразование
в растворах — начала бурно развиваться лишь в конце 40-х и начале
50-х годов XX в. Большую роль в этом направлении исследований
сыграли работы Я. Бьеррума (Дания) и И. Ледена (ШЕеция).11.1. Ступенчатый характер равновесийПри смешивании растЕорсв, например, содержащих ионы Hg2+ и I-,
происходит ступенчатое комплексообразование:Hg:+ + r *= Hgl+; /С>"т =Hgl++I- 5= Hgl2; tf>CT =[Hg:+J [i-j
IHgb)IHgi+jin[ Hgirl[Hgl2] [Г]293
Вся система в целом и отдельные ее ступени находятся в химическом
равновесии. Таким образом, каждая из ступеней и весь процесс комп-
лексообразования могут быть описаны количественно константами рав¬
новесия. Константы /\|'ст, /\'уст, KfT, К\ст называют ступенчатыми
константами устойчивости* или ступенчатыми константами обра¬
зования комплексов. Константу /(уст называют общей (суммарной)
константой устойчивости. Нетрудно видеть, что общая константа
устойчивости связана со' .ступенчатыми константами соотношениемA'y"J:= К, К, К3 Л'4Допустим, что в твердую фазу выделен комплекс KifHgU], При
растворении в воде происходит его электролитическая диссоциация наИОНЬ"Кг [Hgl4] -н. 2К+ f[HgI4j--Она протекает по типу сильных электролитов, т. е. полностью. Вслед
за электролитической диссоциацией происходит сольволитическая
диссоциация. Комплексный ион [Hgh]2- диссоциирует на составные
части по типу слабых электролитов, т. е. ступенчато:Этим равновесиям соответствуют ступенчатые константы неустой¬
чивости (константы диссоциации). Константы неустойчивости и кон¬
станты устойчивости связаны соотношением /Суст= 1 !К" ■ Для того
чтобы ступенчатые константы образования и ступенчатые константы
сольволитической диссоциации соответствовали друг другу, нумера¬HgU =*= Hgi+ + I";Hgl+ «с Hg:+ -f Г;= [Hgl+j [I j2 [Hgl2]h_ [Hg-+| [PI1 [Hgi-42— 24-Hgl4 =Ft Hg — 41^ _ [Hg^j [i-r
Lh8 i»-]* Иногда их называют индивидуальными константами.294
цию ступеней диссоциации комплексных соединений дают в обратном
порядке.В настоящее время химики используют главным образом константы устой¬
чивости. Однако в прошлом наибольшую распространенность имели константы
неустойчивости. Это объясняется тем, что вторичная диссоциация комплексных
соединений по форме весьма напоминает диссоциацию многоосновных кислот
или оснований, например:Н3Р04 ^ Н+ + Н,Р04 ; Л’, =Н..РО, ** Н+ + ПРО! ; Л'„ =- 4 q|Н+ ЦН:Р0Г1[Н3РО.,]I Н+ | f НРО^-[HsPOrJ2- з_ IH- j [РОЗ-ПHPO, ^ H+ + P04 ; /с3= ' J L[ нроаПоэтому на первых порах при расчетах равновесий в растворах более привычно
было использовать константы, аналогичные константам кислотной диссоциации,
т. е. константы неустойчивости.Произведения ступенчатых констант устойчивости (р) также яв¬
ляются константами. Так, для системы Hga+—1“?1=Л,ст= [Н«1Ч .р.= Д’уст /Суст= - ;* 1 2 [Hg2+][1-]2С _ Г^УСТ г,уст ст _ [Hgls ]1'3 = Л, Ло ’3 [Hg21*] [I-]3уст Уст Уст Уст устК — Р4 = Ki К2 к3 Kt± >=[Hgi;-][Hg2+] [I-]При написании уравнений диссоциации или образования комплек¬
сов обычно не пишут координированные молекулы растворителя.
По существу же реакции комплексообразования являются реакциями
замещения координированных молекул растворителя на вступающий
лиганд. Например, взаимодействие №(К03)г с аммиаком в водном
растворе следовало бы записать так (полная форма записи):RHoO)6p + NH3 INi(NH3) (Н20)5]=+ + НаО
[№(NH3) (Н,0)5]2+ + NH3 « [Ni(NH3)2 (Н20)4]2+ + Н20
[Ni(NH3)2 (Н20)4]2+ + NH3 ^ [Ni(NH3)3 (Н20)3]г+ + НгО
[Ni(NH3)s (НоО)3]2+ + NH3 **= [Ni(NH3)4 (Н20)2]г+ + Н*0
(Ni(NH3)4 (Н20)2]=+ + NH3 ** [Ni(NH3)s (Н20)]"-+ + Н20
1^»3)5(Н:0)]:* + NH3 ^ [N'i(NH3),]=+ -i- н,0295
Нитрат никеля (II) в водном растворе диссоциирует практически на¬
цело, и нитратные ионы в данном случае участия в комплексообразо-
ванни не принимают. Обычно координированные молекулы раство¬
рителя подразумевают, но в формулах их не пишут. Поэтому при¬
веденные выше реакции, как правило, записывают в краткой форме:Ni-+ -f- NH3 ^ Ni(N'H3)2*Ni(NH3)w + NH3 ^ Ni(NH3)§+Ni(NH3);+ + NH3 ** Ni(NH3)*+Ni(NH3)2+ + NH3 a Ni(NH3)2+Ni(NH,)=+-j-NH3 =*= Ni(NH3)?+Ni(NH3)2> + NH3 =*= Ni(NH3)J+Координационное число никеля (II) во всех комплексах остается по¬
стоянным и равным 6, а лигандное число изменяется от 0 до 6.Изучение комплексообразования в растворах прежде всего начи¬
нается с выяснения количества и типа образующихся комплексов
и определения их констант устойчивости. Количественному изучению
системы, в которой происходит образование комплексных соединений,
как правило, предшествует качественное. На этой стадии исследо¬
вания выясняются вопросы — имеет ли место процесс комплексо¬
образования, и если имеет, то какие компоненты системы принимают
в них участие. Растворитель практически всегда участвует в процес¬
сах комплексообразования и поэтому такой вопрос не ставится. На
качественном этапе исследования о процессах комплексообразования
часто судят по изменению окраски изучаемой системы. Например,
при добавлении к водному раствору медного купороса раствора ам¬
миака окраска изменяется от голубой до глубокой синей, а при до¬
бавлении к раствору FeCl3 ионов SCN~ появляется кроваво-красная
окраска. В других случаях для суждения об участии в комплексо-
образовании используют аналитические реакции на тот или иной нон
пли молекулу. Если ионы или молекулы принимают участие в обра¬
зовании прочного комплексного соединения, то их не удается обнару¬
жить при помощи качественных аналитических реакций. Например,
при взаимодействии ионов Fe3+ с цианид-ионами образуется прочный
комплекс [Fe(CN)6]3~. Характерные реакции на ион Fe3+ со щелочью
или с роданидом калия будут отрицательными.Иногда при сливании реагентов вначале образуется малораство¬
римое соединение, которое растворяется при добавлении избытка ре¬
агента. Это также служит указанием на процесс комплексообразо¬
вания. Например, при добавлении к раствору Hg(N03)2 раствора KI
выпадает ярко-оранжевый осадок Hgh. При дальнейшем добав¬
лении раствора KI осадок растворяется и раствор становится светло¬
желтым. При избытке иодид-ионов в растворе содержатся комплек¬
сные ионы [Hgl-i]2-. При добавлении раствора аммиака к зеленому
раствору Ni(N03)2 в осадок выпадает аморфный гидроксид никеля.296
На этой стадии реакции аммиак проявляет свойства основания. Даль¬
нейшее добавление аммиака приводит к растворению осадка и обра¬
зованию сине-фиолетового раствора. При избытке аммиака ионы ни¬
келя связаны в комплекс [Ni(NH3)6]2+.Константы устойчивости (неустойчивости) являются количествен¬
ной мерой прочноста комплекса или мерой тенденции нона металла
н данных лигандов образовывать комплекс.В литературе используются как термодинамические, так н кон¬
центрационные константы устойчивости. В выражения для термо¬
динамических констант входят активности. Концентрации ионов и их
активность связаны выражениемHi = liCi,где «г — активность; С; — концентрация; /,■ — коэффициент ак¬
тивности. Концентрационная и термодинамическая константы ус¬
тойчивости комплекса A\Ln связаны следующим образом:*KL„/fm.n /M/Lа „ -Лтррм j ■ ЛД MLn.'м /' LПри бесконечном разбавлении растворов коэффициенты актив¬
ности стремятся к единице и, следовательно, концентрационные кон¬
станты приближаются к термодинамическим. Если концентрационные
константы определить при различных концентрациях, то при экстра¬
поляции значений констант на нулевую концентрацию солей можно
вычислить термодинамическую константу. Коэффициенты актив¬
ности берут из справочников или определяют экспериментально. Их
можно рассчитать, используя теорию Дебая—Хюккеля.При постоянной ионной силе активность иона пропорциональна
его концентрации. Ионная сила в соответствии с теорией Дебая—
Хюккеля рассчитывается по уравнениюгде 1 — ионная сила; г — заряд ;-го иона; С; — его концентрация.Часто исследователи вычисляют концентрационные константы,
но указывают, при какой ионной силе проводились измерения кон¬
центраций ионов. Большую ценность представляют термодина¬
мические константы. Получив их при различных температурах, вы¬
числяют стандартные значения изобарного потенциала ДС°, энталь¬
пии ДН° и энтропии Д5° по уравнениямЫ? = — RT \п К\
din К А Н° .
d? ~ RT2 ’AG°=AH°—TAS°.^ML297
Для создания ионной силы используют перхлорат натрия, пер¬
хлорат лития или другие 1:1-электролиты щелочных металлов. Анион
фонового электролита не должен проявлять склонности к комплексе-
образованию с нонами исследуемых металлов.Силлен [1] сформулировал три необходимых условия, которые
должны соблюдаться при исследовании комплексообразования в
растворе:1. Исследование необходимо проводить при постоянной и высокой
ионной силе, чтобы коэффициенты активности оставались постоянными.2. Метод исследования следует выбирать в зависимости от свойств
системы. Он должен обеспечивать максимально возможную точность
измерений.3. Все измерения должны проводиться в максимально широкой
области концентраций.Для изучения комплексообразования исследования рекомендуют
проводить несколькими методами. Надежными считаются константы,
полученные различными методами и совпадающие по значению в
пределах ошибки опыта. Силлен предложил делить методы исследо¬
вания комплексообразования в растворах на три группы.1. Методы, которые позволяют измерять концентрацию опре¬
деленного компонента, участвующего в комплексообразовании. К ним
относятся методы измерения электродвижущих сил гальванических
цепей, с помощью которых определяется концентрация свободных
ионов металла, ионов водорода, а таКже некоторых лигандов, напри¬
мер Cl", Br, CN". В эту группу входит полярографический метод,
метод измерения упругости пара, методы распределения, ЯМР и ЭПР.2. Методы, позволяющие определять сумму концентраций всех
присутствующих в растворе частиц. К ним относятся методы измере¬
ния понижения точки замерзания, повышения точки кипения и изме¬
нения упругости пара растворителя.3. Методы, позволяющие определять функцию 2/С;С,-, где /С* —
индивидуальная'константа, С;—концентрация г-й частицы. К ним
относятся спектрофотометрический метод, методы электрической про¬
водимости и исследования магнитной восприимчивости.Константы, характеризующие устойчивость координационных со¬
единений, можно найти в справочниках [2—5].11.2. Функция образования и кривая образованияФункция образования широко используется для расчета констант
равновесия и реакций комплексообразования. Экспериментальное
определение констант устойчивости во многих случаях связано с
определением в системе концентрации незакомплексованного иона
металла или свободного лиганда. Общая концентрация как иона ме¬
талла, так и лиганда (свободных и закомплексованный) обычно за¬
дается .Общая концентрация пона металла в растворе, в котором при¬
сутствуют все промежуточные формы комплекса ML„, равна298
n=,VСм=[М)Н [ML] ч 1 [MI- v] = [M] + v [MLn] ,Л— 1где N — максимальное координационное число.Общая концентрация лиганда равнаn=NCL = [L] + [MI ] Г 2 [MLa] + ■ • • -f N [ML,,] = [L] -)- V „ [ML,,] .n— 1Функция образования n имеет смысл среднего числа лигандов, свя¬
занных с ионом металла, и рассчитывается по уравнению, v. п [MLn]- CL ~ Ш n“iп—ХIM] : V [MLn]n- !В ряде случаев чрезвычайно полезным параметром является степень
образования а. Она выражается формулойа-- п/М,где п — функция образования.Степень образования комплекса с лигандным числом п равна[ML„[ Кп [М] [L]” Д'л [L]n°Л р п- N n- Nм [М] + V кп [М] [L]» I + V Д'п [L]»п — 1 п=-1Степень образования отдельного комплекса называют также мольной
долей этого комплекса. Сумма мольных долей всех комплексов, вклю¬
чая мольную долю незакомплексованного нона металла, равна 1.11.3. Статистическое соотношение
ступенчатых констант устойчивостиСоотношения между ступенчатыми константами устойчивости зави¬
сят от многих факторов. Одним из них является взаимное влияние
лигандов. Если исходить только из статистического распределения
лигандов, то можно легко вычислить соотношения между отдельными
ступенчатыми константами для комплексов с различным предельным
координационным числом N. Возьмем промежуточный комплекс ML„
с монодентатными лигандами. Выход лиганда из комплекса должен
быть пропорционален числу занятых координационных мест, т е.
числу ти Тенденция вхождения лиганда L в комплекс MLn должна
быть пропорциональна числу свободных координационных мест, т. е.
А'—п. Таким'образом, для равновесия
*-»ч > MLn_! -f- L299
значение Л'„ = пропорционально (N—п + 1 )/п. Для равно¬весияСMLn+1 = ML„ + LСзначение Д'п+1 = kJk'^_ пропорционально (/V—-/г)х(/г +1). Отношение
констант„ .. (ЛГ-я+ 1)(я+1) ,•п:Лл+1‘ n(N-n) -,п-Подставляя соответствующие значения N и п, получим отвечающие
нм соотношения констант (табл. 11.1).Таблица 11.1. Статистические значения отношения ступенчатых
констант устойчивостиNК, /К,К,/К,К,/К.Кг/К,KJK,23433—- -—42,672,252.67— 52.50222,50 62,401,881,781.882,40Геометрическая конфигурация комплекса не сказывается на ста¬
тистическом соотношении констант устойчивости.Из табл. 11.1 видно, что статистические отношения двух после¬
довательных констант устойчивости для комплексов одного и того же
предельного координационного числа примерно одинаковы.Экспериментально полученные соотношения двух последователь¬
ных констант (табл. 11.2), как правило, сильно отличаются от ста¬
тистического соотношения этих величин.Формально причины, приводящие к отклонению экспериментально
найденных соотношений констант от статистического соотношения кон-Таблица 11.2. Значения Кп/Кп+1 Для ряда систем*СистемаNKdK,K./K,KJ KbKi.'K,Ag+-Cl-24.:27Cu+—Cl-25,57 .__ Ag+—NH320,235 Cu+—NH,о4,45 —Hg2+—Cl~45,75- lO’32,32-10s1,39-I02 —Hg2+—I-4831,48- 10735,6 —Zn2+—NH-j4‘0,830,872,24 —Ni2+—NH363,553,233,472,765,25* Значения констант взяты из работы [2].300
стант, учитываются фактором рассеяния л-, введенным Я. Бьеррумом:Фактор рассеяния колеблется в пределах от 0 до оо. Для статисти¬
ческого случая х ~ Фактор рассеяния может отличаться для со¬
отношений констант в ряду последовательных комплексов.Рассмотрим влияние фактора рассеяния на п в зависимости от
концентрации свободного ли ганда для простейшего случая, где N --- 2.
Соотношение А', : Л'3 — 4.vs. Если выразить А\ и 7\.> через среднюю
константу А, тоК1 = 2хК\ А'г = — А'.Подставив значения А\ и Аг в выражение уравнения функции обра¬
зования (для случая N = 2), получим__ 2л-Д- [L] + К- ГLI2
1+ 2хК [L] -j- Л'2 [L]3 ’11.4. Смешанные комплексные соединенияКомплексные соединения, во внутренней сфере которых находятся
различные лиганды, называют смешанными. Если во внутренней сфе¬
ре находятся одинаковые лиганды, такие комплексные соединения
называют однородными. Смешанные комплексы весьма распростране¬
ны для ионов металлов, образующих инертные соединения, напри¬
мер Сг(Ш), Со(Ш), Pt(II), Pt(IV). Так, известны многочисленные
соединения платины (IV) с шестью различными лигандами. Более то¬
го, синтезированы их изомерные формы. Смешанные комплексы ха¬
рактерны и для лабильных соединений. Однако выделение лабильных
комплексов из растворов в твердую фазу вызывает затруднения.
В растворе же хорошо изучены лабильные одноядерные комплексы ти¬
па MX/Y; Весьма распространены смешанные комплексы, в которых
одним из лигандов являются мрлекулы растворителя. Однако в этом
параграфе изложены некоторые положения теории устойчивости сме¬
шанных комплексных соединений без учета равновесий, включаю¬
щих молекулы растворителя в качестве лигандов.Узловой момент этой теории — выявление соотношения между
константами равновесия однородных и смешанных комплексов. До¬
пустим, что в растворе имеется комплекс [Ni(NH3)2Py„]2+.
Константа устойчивости, отвечающая равновесию Ni2+ + 2NH3 +
-f 2Ру [Ni(NH3).,Py2]2+ выражается уравнением_ [Ni(NH,)2Py2-+] (И(N! (NHj)j Py.pt [Ni2+j [NH3]2 [Ру] “301
Этот смешанный комплекс может быть получен в растворе в резуль¬
тате взаимодействия однородных комплексов в соответствии с урав¬
нением~ (Ni (NH3)4]-+ -j- [NiPy4]2+ * [Ni(NH3)2 Py;]2+Константа этого равновесия (константа сопропорционирования*)
равна[Ni (NH3)2Py2+]Лс0ПР= [Ni(NH3)2+],/»[NiPy^+],/! -2)Комбинация уравнений (11.1) и (11.2) приводит к соотношению
_ /Ceonp[Ni (NH3)4+]',g[NiPyS+]1/*?[Ni (NH,), Py2|2+ — [Nit+| [NHja IPvl4— ^conp I f'tNi(NH,),]J+ "tNiPy4]1<--Таким образом, константа устойчивости (константа образования)
смешанного комплекса [Ni(NH3)2Py2]2+ прямо пропорциональна сред¬
нему геометрическому из констант образования однородных комплек¬
сов [Ni(NH,)4]2+ и [NiPy4]2+. В общем виде для комплекса МХ/У;
константа устойчивости выражается уравнениемя к 1,11т ь‘/т / I I о\1 МУ -V — Лсопр -мх„ ‘ MY */■ 1 j т т
где т — / + <•Константа сопропорционирования учитывает статистическое рас¬
пределение и «совместимость» внутрисферных лигандов. «Совмести¬
мость» лигандов определяется их взаимным влиянием в любых прояв¬
лениях. Если взаимное влияние внутрисферных лигандов отсутствует
и они ведут себя независимо друг от друга, то константа сопропор¬
ционирования определяется лишь их статистическим распределением.
Вследствие хаотического движения частиц перераспределение ли¬
гандов должно протекать в сторону образования смешанных комплек¬
сов. В случае образования смешанного комплекса раствор представ¬
ляет более однородную систему, чем в случае образования нескольких
однородных комплексов. Маркус показал [6], что, исходя из статис¬
тики, для равновесия/МХт + IМYm =* mMXyY;константа сопропорционирования равнаm !^сопр —. /I Я* Величина, обратная константе сопропорционирования, называется кон¬
стантой диспропорционнрования.302
Из этого выражения следует, что константа сопропорционирования,
вычисленная на основании статистических соображений, всегда боль¬
ше единицы. Таким образом, статистическое распределение лигандов
действительно должно приводить к преимущественному образованию
смешанных комплексов. Экспериментальные данные показывают, что
в некоторых случаях устойчивость смешанных комплексов соответ¬
ствует рассчитанным значениям, в редких случаях она более низкая.Смешанные соединения обладают повышенной устойчивостью в
растворах по сравнению с генетически связанными с ними однород¬
ными комплексами [7]. В табл. 11.3 приведены экспериментальные
данные, которые показывают, что константы устойчивости смешанных
комплексов выше, чем усредненное [в соответствии с формулой (11.3)]
значение констант устойчивости соответствующих однородных соеди¬
нений.Таблица 11.3. Константы устойчивости (lg [3) комплексов с амми¬
аком и пиридином при 20±0,05°С и ;л = 1 [7]КомплексCo=+Ni2+Cu=+Zn'- +Cd-+M(NH3)*+3,985,187,654,814,60M(NH3)Py-+3 °24,546,604,073,25МР>|+1 ,983,024,302,012,46,M(NH3)®+5,086,8210,547,486,0M(NH3)aPy-+4,206,659,866,055,60M(NH3)Py=+3,505,408,104,074,04МРу*+2,253,425,682,08—M(NH3)5+5,987,9812,679,557,03M(NH3)3Py=+5,307,1011 ,627,696,69M(NH3),Py; +4,506,3010,836,275,90M(NH3)P\'3+3,855,149,094.144,08J4PyJ+2,353,446,582,122,50Превышение констант устойчивости смешанных комплексов над
расчетными свидетельствует о дополнительной стабилизации за счет
взаимного влияния внутрисферных- лигандов. Кида [8], а затем Мар¬
кус и Элизер [6] сделали попытку количественного учета этой допол¬
нительной стабилизации на основе электростатической модели. Мо¬
дель смешанного комплекса, положенная в основу расчета, пред¬
ставлена на рис. 11.1. Методический подход к расчету образования
смешанного комплекса похож на подход Косселя, давшего в свое вре¬
мя энергетическое обоснование существования комплексных соеди¬
нений. Вследствие различия лигандов и несимметричности комплекса
MXY происходит ион-ионное, ион-дипольное и диполь-дипольное
взаимодействие частиц, составляющих комплекс. Располагая значе¬
ниями зарядов центрального атома и лигандов, расстояниями между303
Рис. It.I. Ионно-поляризационная мо¬
дель смешанного комплекса МХУчастицами, а также зная электри¬
ческие моменты диполей и соответ¬
ствующие поляризуемости, можно
вычислить энергию образования
смешанного комплекса. Однако
Маркус и Элизер в расчетах не
учитывают процессов сольватации,
которые могут играть весьма суще¬
ственную роль. Пещевицкий и Белеванцев отмечают, что если у сме¬
шанного комплекса энтальпия сольватации выше, чем полусумма
теплот сольватации соответствующих однородных комплексов, то за
счет этого также должна проявляться стабилизация смешанного
комплекса.хм Y11.5. Мольные доли. Распределение форм
комплексов в зависимости от концентрации
лигандаДля расчета концентраций отдельных комплексов на основе констант
неустойчивости широко используются мольные доли. Мольной долей
отдельной формы комплекса at называют отношение концентрации
данного комплекса A\L; к общей концентрации иона металла См:а; = fML;]/CM ,Здесь индекс (' означает число лигандов, присоединенных к иону ме¬
талла. Если ион металла свободный, то его мольная доля ссо. Если
к иону металла присоединен 1, 2, 3 и т. д. лиганда, то соответствую¬
щие мольные доли означают a,, at, а3 и т. д. Допустим, что в системе,
состоящей из иона металла и лиганда, образуется всего лишь один
комплекс ML. Равновесию
ML =« М + Lсоответствует константа неустойчивостиIM] fLJ
[ML]Концентрация свободного иона металла равна[mj=^L.1 [L]Общая концентрация иона металла во всех формах
С„ = [М] -f [ML].В эксперименте См обычно задается. Мольные доли М и MX выра¬
жаются уравнениями304
[М] ГМ] A [М1.]/[1] ^ кС,°'' См " [М] i- [/ML] *" A' [MI.]/[L]-| [ML] ЛЧ- [L] '[ML] [ML] _ [I ]См " A’ [ML]/[L] + [.ML] K-'r ILJ ’Если предельной комплексной формой является ML2, то имеет место
равновесие ML., ML + L, для которогоЛ, = ШШУ_: [МИ,= ^Ь1.• [ML,] 1 ' [L]Для второй ступени диссоциации
ML 5* М + LIм) [L] . A, [ML] К,К2 [ML,2]Л i =[ML] ‘ ' [ I- ] [Ll-Общая концентрация иона металла
См = [М] [ML] b[ML:].Мольные доли отдельных форм выражаются следующими уравне¬
ниями:[М] [М] AjA’j [ML2]/[L]2ао ~ ' 'м [М] + [ML] + [ML.] AiA2 [ML;] + А„ [ML„] + [МЬ]* [I]2 ' [L]A'l A; .M*.+ AS[L]-|-[L]S[ML] [ML] A; [ML2]/[L]a‘ “ CM “ [M] + [ML] + [ML;] ~ K,Kt [ML,] A; [ML,][LJ- ! [L][L] K2- L[ML;]A'iAj + AsJL] + [ L ] -Комплекс ML3 диссоциирует но трем ступеням. Для первой ступени
диссоциацииML3 = ML, + Lf [ML;] [L] A3[ML3]A3 = . [ ML- = •[ML3] ‘ 21 [L]Для второй ступени диссоциацииML; ML -j- LЛ'..- [ML] fL| • -*<- ■ Л'2 [ML;] А2Ая [ML:)[[ML;] ■ J [L] f L]2Для третьей ступени диссоциации
ML =F* M L„ [M] [LI Ki [ML] KyK.KilML,]Л, = > imi = = •[ML] ' J [L] [L]3Общая концентрация иона металла
См = [М] + [ML] + [ML,] + [ML3].Мольные доли отдельных форм выражаются уравнениями:= _[М2_ = [М] =°° см ]М] + [ML] + (ML,] + [ML3]_ ' KiK2Kz [ML3]/fL]3 =~~ KiK2K3 [ML3] , AV:.,[ML3] K3[ML3]+ ггт;— + —гг;— + 1М1-з1*1*2*3 + [L] К,Кз + [L]2 К3 + [LJ3[ML] [ML]a, = 1 CM [M] + [ML] + [ML,] - [ML3] ‘ [L] K2K3K,K2K3 + [L] КгКз + [L]2 K3 - [L]3 [MLj>] [ML,] CM “ [M] + [ML]+ [ML2] -J- [ML3] “[LJ2K3KiKiKs + [L] K,K3 + [Lj2K3 + HP[ML3] [ML3]
a, = См [M] + [ML] + [ML,] + [ML3] [L ]3КхКгКя + [L] K2K3 + [L]- K3 [L]3Из формул для мольных долей отдельных форм комплексов, находя¬
щихся в данной системе, следует, что знаменатель один и тот же. Чис¬
литель для ао является произведением ступенчатых констант неустой¬
чивости, а для мольных молей всех последующих форм в произведение
вместо соответствующих констант входит концентрация лиганда в
определенной степени. Отметив эту регулярность, можно сразу на¬
писать значения мольных долей для определенных форм предельного
комплекса ML4:[М] [М]см [/И] + [ML] + [ML,] + [ML3] + [ML4] :
Д'ЛЛ'зЛ',a, =K^KzKiKt + [L] K.K3Ki+ [I]' K3Ki + [L]*/f«+ [L]1
[ML] [L] Л',ЛУС, .-M КхКгКзК^ + [L] K,K3Ki + [L]2 K3Kt + [L]* KK + [L]4
306
[ML.][LJr AVviСмA i А 4+ П-J Л'гА3А4 л- [1-]-ЛГэЛ'«-Ь14■> А,П-1‘[ML*]‘^Мf П-] А2ЛяА4 П J" а3л4 +1 и3А4 !- 1L]1[ML4]11-14СмA iA »1\ з А 4+ (L) А2А3Л4 lLJ:A:|/<4 ;[L]3А4Г П-]4Рис. 11.2. Выход комплексов
HgCr2'n в зависимости от
IglCl"]Таким образом, моль¬
ные доли отдельных форм
определяются значениями
констант неустойчивости
соответствующих ступеней
равновесия и концентраци¬
ей свободного лиганда. Ес¬
ли известны ступенчатые
константы неустойчивости,
то, задавая концентрацию
свободного лиганда, мож¬
но построить кривые распределения отдельных форм комплексов.
На графике зависимости мольной доли данной формы от концен¬
трации лиганда площадь между двумя кривыми характеризует
область существования определенного комплекса. Если при опре¬
деленном значении концентрации свободного лиганда на такой ди¬
аграмме провести вертикальную линию, то отрезки этой линии в
области существования определенных комплексов будут равны моль¬
ным долям соответствующих форм.Весьма наглядной является диаграмма, построенная несколько
иначе. Если принять сумму отдельных форм при данной концентрации
за 100%, то можно построить диаграмму, показывающую выход от¬
дельных форм комплексов при заданной концентрации свободного
лиганда (рис. 11.2).Как уже было сказано, на первых порах теория равновесий в раст¬
ворах координационных соединений многое заимствовала из ученияо равновесиях в растворах многоосновных кислот и многоатомных
оснований. Постепенно положение изменилось и теория и методы ра¬
счета комплексообразования в растворе используются для решения
задач, связанных с равновесием в растворах слабых многоосновных
кислот и многоатомных оснований, гидролиза и сольволиза средних,
кислых и основных солей.
11.6. Комплексообразование и растворимостьЕсли лиганды способны образовывать комплекс с ионом металла, то
они приводят к увеличению растворимости малорастворимых солей
этого металла. Это обусловлено тем, что равновесие растворения
МХ (тв) ** Мп+ + Хп-смещается вправо вследствие связывания ионов металла в комплекс.
Знание констант устойчивости позволяют вычислить растворимость
малорастворимых соединений в присутствии лигандов. Рассмотрим
решение такой задачи на конкретном примереДопустим, что требуется вычислить растворимость AgCl в водном
растворе аммиака определенной концентрации. Растворимость A^CI
в воде характеризуется величиной произведения растворимости б
соответствии с уравнениемAgCl (тв) з=е Ag+ + СI-; nPAgC1 = [Ag+J [CI-].Ионы серебра ступенчато присоединяют молекулы аммиака:[Ag (NH3>2] = КхК2 [Ag+] [NH3]=.Общая концентрация ионов серебра в растворе может быть представ¬
лена выражением= [Ag+J + [AgNH*] + [Ag(NH3):j = [Ag+] (I + K, [NH3J - - KtK2 [NH3]2).Для решения данной задачи важное значение имеет концентрация
не связанного в комплекс иона серебра. Ее можно вычислить исходя
из выражения мольной доли. Для удобства выкладок запишем вы¬
ражениеПодставим концентрацию ионов серебра, выраженную через «о,
в выражение произведения растворимости:Растворимость хлорида серебра равна общей концентрации ионов
серебра во всех формах, т. е. CAg+. Тогдасолей металлов11.6.1. Растворимость малорастворимых солей
в присутствии лигандовAT + NHS « Ag(NH3)+; Kt =[AgN^]; [AgN^] = К, [Ag+] [NH3];[Ag+] [NH3] ’[AgNH^] [NH3]ПР= [Ag+] [C1-] =CAe+»0 [Cl-j.308
■S — СдоН-ПРAfr+ «о [С1-]Если хлорид-ионы присутствуют в системе лишь за счет растворенного
AgCl, то CAg+=[Cl-]. В этом случае выражение растворимости упро¬
щается:5 = J/ ПР — = V ПР (1 + К1 [NH3] + К\Ка [NH3]3) .Если в системе наряду с аммиаком присутствуют хлорид-ионы
(например, требуется определить растворимость в буферной смеси
NH^ + NH4C1), то расчет растворимости нужно вести по полной фор¬
муле:5 = (1 + Ki [NH3] + К,К, [NH3]*).Мы рассмотрели наиболее простой случай — когда предельное
координационное число иона металла равно 2. Не вызывает больших
затруднений распространение данного подхода к решению задачи
при образовании комплекса с большими координационными числами.11.6.2. Растворимость малорастворимых солейпри образовании комплексов с ионом-осадителемДопустим, что необходимо определить растворимость AgCl при из¬
бытке хлорндных ионов. В соответствии со значением произведения
растворимости устанавливается равновесие:AgCl (ТВ) St Ag+ -f СГИоны серебра ступенчато образуют комплексы с хлорид-ионами:Ag+ + ci- « Age I (p-p); Kx = : [AgCl (p-p)J = KJ IP;[ Ag ] [Cl ]AfCI(p.p) + CI- = AgCI7; X, = [дес|1^-}— ; |Agar | - K,K, ..P [CI-).Растворимость определяется суммарной концентрацией ионов серебра
во всех формах:s = V = [Ag+] + [AgCl (p-p)] + [AgCl-] ==' + К, ПР + А'ДоПР [C1-] = ПР + h + [CI-]).Произведение растворимости иодида ртути ПР =[Hg2+][I~]2. При
осаждении HgL в избытке иодид-ионов необходимо учитывать четыре
ступени комплексообразования:,, . fHgI + l ПР+ '■ - «■ - |Нв--| iiT; [Нв‘*1 ~к‘ TIT:309
IHglj] |I-|Растворимость рассчитывается по выражениюS - c11glf |Hg2+j ь [HgP] + [Hgl,] + [Ugl-J b [HglJ-] =I j_j -Г f>2 4 fell ]+?«[! ]2)11.7. Ряд Ирвинга — ВильямсаМногочисленные экспериментальные факты свидетельствуют о том,
что устойчивость аналогичных комплексов изменяется для ионов
металлов в следующем порядке:Mn2+ < Fe2+ < Со:+ < Ni-+ < Cu2+ > Zn2+Эта последовательность ионов металлов, отражающая изменение ус¬
тойчивости аналогичных комплексов, в литературе получила назва¬
ние ряда Ирвинга—Вильямса. Ряд справедлив для большого числа
комплексов с различными лигандами, имеющими N- и О-донорные
атомы. Такой порядок изменения устойчивости комплексов объясня¬
ется с позиции теории кристаллического поля (гл. 6). Все ионы ме¬
таллов, входящие в ряд Ирвинга—Вильямса, как правило, образуют
высокоспиновые комплексы октаэдрической конфигурации. В ряду
ионов Mn2+(d5), Fe2+(ci6), Co2+(d7), Nia+(d8), Cu2+(d9), Zn2+(d10) энергия
стабилизации кристаллическим полем равна соответственно 0;
0,4Д; 0,8Д; 1,2Д; 0,6Д: 0. Комплексы Си2+ обычно имеют структуру
вытянутого по одной из осей октаэдра (эффект Яна—Теллера). Вслед¬
ствие этого происходит дополнительная стабилизация и устойчивость
комплексов Сиг+ становится несколько большей, чем устойчивость
аналогичных комплексов Ni2+. По ряду Ирвинга—Вильямса законо¬
мерно изменяется не только устойчивость комплексных соединений,
но и энергия гидратации ионов металлов.Комплексы переходных металлов часто выступают в роли катали¬
заторов различных реакции. Например, гидратированные ионы пере¬
ходных металлов проявляют каталитическую активность в реакциях
гидролиза сложных эфиров, в декарбоксилированин р-кетокнслот и
др. Каталитическая активность для ионов, входящих в ряд Ирвинга-
Вильямса, обычно изменяется вполне закономерно. Считают, что
причиной этого является образование промежуточных хелатных
комплексов с молекулами, подвергающимися превращению. В ре¬
зультате комплексообразовання облегчается атака координированного
лиганда и превращение протекает с большей скоростью.
ГЛАВАОКИС ЛИТЕ Л ЬНО-ВОССТАНОВИТЕ Л ЬНЫЕ
СВОЙСТВА КООРДИНАЦИОННЫХ
СОЕДИНЕНИЙХорошо известно, что металлическое золото химически мало ак¬
тивно. Например, на воздухе оно не окисляется, а лишь образует па
поверхности тончайший слой оксидной пленки. Золото не взаимодей¬
ствует с кислородом даже при высоких температурах. Однако так же
хорошо известно, что в растворах цнанпдов щелочных металлов ме¬
таллическое золото сравнительно легко окисляется кислородом в
соответствии с уравнением4Au +8KCN + О-. + 2Н30 = 4К [Au(CN)2] + 4КОНЭта реакция положена в основу гидрометаллургического способа вы¬
деления золота из руд. Возможность ее протекания обусловлена об¬
разованием прочного цнанидного комплекса золота (I). Под действи¬
ем цианидных ионов тонкая оксидная пленка растворяется и появля¬
ется возможность для дальнейшего окисления золота кислородом.
Этот пример доказывает важную роль комплексообразования в окис¬
лительно-восстановительных реакциях.Окислительно-восстановительные реакции протекают с передачей
электронов Окислитель принимает электроны и переходит в восста¬
новленную форму:Oxj -+- пс~ з=е RedjВосстановитель, наоборот, отдает электроны и переходит в окислен¬
ную форму:Red2 — mr ^ Ox,Таким образом, для каждой окислительно-восстановительной реак¬
ции с окислителем сопряжен восстановитель, а с восстановителем со¬
пряжен окислитель.Любая окислительно-восстановительная реакция может быть пред¬
ставлена в виде двух полуреакций* :С^+ле- л Red!Ох2 -j- тс~ ^ Red,* На XVII съезде Международного союза по чистой и прикладной химии
(ИЮПАК) в 1953 г. было при пято соглашение, что в окислительно-восстанови¬
тельных полуреакциях в левой части уравнения пишут окисленную форму, а
в правой — восстановленную. Значение окислительно-восстановительного по¬
тенциала соответствует именно такой записи полуреакцнн.311
Вычитание одной полуреакции из другой после умножения на коэф¬
фициенты т и п (для сбалансирования отданных и принятых электро¬
нов) дает суммарную окислительно-восстаиовительную реакциюгпОх1 rRcdo st т Red! -|- лОх2Каждая полуреакция имеет свой окислительно-восстановитель¬
ным потенциал. Зависимость потенциала от активности окислителя
и сопряженного с ним восстановителя выражается уравнением Нерн-
стаWed = С/Red 'I' — ’Redгде E° — стандартны!! потенциал, т. е. значение потенциала при ак¬
тивностях окислителя и восстановителя, равных единице.Активность в уравнении Нернста можно заменить произведением
коэффициента активности на концентрацию. В этом случае уравне¬
ние будет выглядеть следующим образом:р _ Г® ■ RT . , ЛИ , t°-xi _-Ox/Red - Ox/Red ‘ ,,f |п , ' пр ,П [RcdJ "RT [Ox], “I 7Г InOx/Red nF [Red]где E0' — величина, называемая формальным потенциалом. Не¬
которые авторы называют ее реальным потенциалом.Для полуреакции, в которой восстановленной формой является
металлМл+ + пе~ ^ М°,уравнение Нернста имеет более простой вид:RT RT 1Е = £мл+/м° + ^7 |п 1М"Ч = 41"*/м" In [м„+] ■ •Поскольку активность металла в твердой фазе постоянная величина,
то она входит в значение Е0'.12.1. Влияние комплексообразования
на электродные потенциалыСвязывание ионов металла в комплексы, как правило, приводит к
изменению электродных потенциалов. Допустим, что ионы металла
Мп+ образуют с лигандами L серию комплексов ML, ML2, .... ML,.
Для простоты написания заряды не обозначены. В гл. 11 было по¬
казано, что^ = тёт = 1 + р1[1] + Рг11]2+'--+'?[Ь]г/-312
Если известны последовательные константы образования комплексов,
то можно рассчитать величину ссм, а затем и электродный потенциал
по уравнению„ RT 1 RTЕ- £мл+/м° — —Г In — 4 — In .7 .14 nF '-м мМожно пойти и по обратному пути. Измеряя электродные потенциалы
при определенных концентрациях лиганда L, определяют ам- Если
в растворе образуется серия из q комплексов, то измерение потенциала
при различных концентрациях L необходимо произвести столько раз,
чтобы получить q уравнений. Решая q уравнений с q неизвестными,
можно вычислить все общие константы образования р, а затем и по¬
следовательные константы образования К-Расчет потенциалов сильно упрощается, если при выбранных
условиях образуется только одно комплексное соединение ML,. Тогда
полуреакция имеет видМЦ -f пег ■»* М + qLОбщую константу образования рд и значение ам находят из выра¬
жений3 Ё1Ы — =(1 -f|l„ [L]«) [L.]7.[Мл+] [L]¥ ам 9 1Подставляя их в уравнение Нернста, получаемRT „ RT , RT I£=£м"+/м- -JT ^ч-д — \п[Ц - —In (МЦ)Из выражения наглядно следует, что чем устойчивее комплекс
и чем выше концентрация лиганда, тем отрицательнее окислительный
потенциал системы.Если первые три члена в уравнении Нернста объединить в видеI?T RTто реальный потенциал полуреакции будет иметь вид
ro, RT . !р г-0/ 1 . _~ ~ „р П [МЦ]Окислительно-восстановительные потенциалы систем, состоящих
из комплексных соединений, можно довольно сильно изменять, свя¬
зывая одну из форм (окисленную или восстановленную) в малораст¬
воримое соединение. В результате нарушается соотношение между
окисленной и восстановленной формами и это скажется на значении
потенциала. Так в растворе, содержащем ионы [Fe(CN)e]3" и
[Fe(CN)6]4- в соизмеримых концентрациях, 3,3-диметилнафтидин
не окисляется, так как потенциал системы £ре(сК)б_/4_ сравнительно
невысок. Если в эту систему ввести избыток ионоз цинка (II), то об¬
разуется осадок малорастворимого соединения Zn.,[Fe(CN)6] и соот¬31.3
ношение между концентрациями комплексных ионов изменится. Это
скажется на величине потенциала. Он резко возрастет, так как воз¬
растет соотношение [Fe(CN)6]3_/[Fe(CN)6]'1_. Эксперимент показывает,
что в присутствии Zn:+ происходит окисление 3,3-диметилнафтидина
ионамн [Fe(CN)ela_.12.2. Стабилизация лигандами нетипичных
состояний окисления центральных атомов
в комплексах переходных металловСоединения с необычно высокими н необычно низкими состояниями
окисления центральных атомов имеют важное значение в координа¬
ционной химии. Тем не менее, термин «нетипичное» состояние окис¬
ления весьма условный. Нетипичными они являются потому, что в
результате окислительно-восстановительного взаимодействия пере¬
ходят в стабильные состояния окисления. Таким образом, о нетипич-
ностн состояния окисления судят по относительной устойчивости.
Устойчивость определяется термодинамическим или кинетическим
факторами. Термодинамическая устойчивость связана с изменением
свободной энергии, т. е. определяется возможностью перехода в со¬
единения с другими состояниями окисления центрального атома. Ки¬
нетическая устойчивость обусловлена высокой энергией активации
процесса переноса электронов. Для решения вопроса об отнесении
состояния окисления к нетипичному проще исходить из того, что при¬
нимать за типичные состояния окисления. Типичными состояниями
окисления можно считать те, которые наиболее распространены. Со¬
единения металлов в таких состояниях окисления способны су¬
ществовать при условиях, близких к стандартным, в твердом состоя¬
нии и в растворе без изменения в течение длительного времени.Стабильность координационных соединений с нетипичными со¬
стояниями окисления металлов в значительной степени зависит от
природы лигандов. Естественно, что лиганды, обладающие ярко выра¬
женными восстановительными свойствами, будут вступать в окисли¬
тельно-восстановительное взаимодействие с ионами металлов, имею¬
щих высокие состояния окисления. Стабильные соединения с необыч¬
но высокими состояниями окисления центральных атомов могут
существовать лишь при окружении лигандами, устойчивыми к окис¬
лению. Такими лигандами в первую очередь являются F" и О2'. По¬
следний имеет особое значенне в стабилизации ионов металлов в вы¬
соком состоянии окисления. Вследствие большой электронной плот¬
ности он проявляет не только а-донорные, но и л-донорные свойства.
Последнее становится возможным благодаря наличию у этого лиганда
невовлеченных в о-связь неподеленных электронных пар. Этим объ¬
ясняется большая распространенность стабильных «иловых» соеди¬
нений металлов в высоких состояниях окисления.В растворах ситуация часто значительно усложняется. Молекулы
растворителя стремятся войти во внутреннюю сферу комплекса.
Если окислительно-восстановительный потенциал комплекса доста¬314
точно большой, то молекулы растворителя будут подвергаться окис¬
лению. Например, в твердом и газообразном состоянии существует
соединение IrF6. В водном растворе оно гидролизуется и происходит
окисление молекул воды. В результате этого иридий переходит в
1г(ОН)4, в котором ион металла имеет стабильное состояние окисле¬
ния 4+-В твердом или газообразном состоянии могут считаться стабиль¬
ными следующие соединения: IrF6, PtF6, K2Cr04,. KMn04, RuOi,
(Ю4. Однако при замене F~ и 0:~ на большинство других лигандов
устойчивость состояний окисления ионов металлов резко уменьшает¬
ся. Координационные соединения Ir(VI), Pt(Vf), Cr(VI), Mn(VII),
Ru(VIII) и Os(V7111) можно считать нетипичными.Комплексы, в которых ионы металлов имеют необычно низкие со¬
стояния окисления, легко отдают электроны п повышают состояние
окисления центрального атома. Стабилизации ионов металлов в низ¬
ких состояниях окисления способствуют лиганды, обладающие л-ак-
цепторными свойствами. Они, как правило, являются слабыми а-
донорами. Принимая на себя электронную плотность от центрального
атома по л-связи, эти лиганды приводят к состоянию «частичного»
окисления. Это находится в согласии с теорией М. И. Усановича,
в которой взаимодействие кислот и оснований Льюса (донорно-ак-
цепторное) рассматривается и как окислительно-восстановительное.
Таким образом, стабилизации комплексов с ионами металлов в не¬
обычно низких состояних окисления способствуют лиганды л-ак-
цепторного типа: фосфины, арсины, оксид углерода, изонитрилы, этиле¬
новые и ацетиленовые производные.Несмотря на то что цианидный нон изоэлектронен молекуле СО
и может рассматриваться как электронный аналог изонитрилов
CaeNR, вследствие отрицательного заряда он является не столь хо¬
рошим л-акцептором. В результате этого по сравнению с СО и CNR
цианидный ион хуже стабилизирует комплексы в низких состояниях
окисления центрального атома.Координационные соединения металлов в состоянии окисления,
равном нулю, можно рассматривать как нетипичные, хотя в настоящее
время они известны для многих элементов периодической системы.В заключение можно сказать, что с понижением состояния окисле¬
ния центрального атома в комплексе (с увеличением заселенности
d-орбитален) должны возрастать его л-донорные свойства (склонность
к образованию л-дативных связей) и уменьшается л-акцепторные свой¬
ства (склонность к образованию двойных связей лиганд-*-металл).12.3. Влияние природы лигандов на окислительно¬
восстановительные потенциалы
координационных соединенийВлияние комплексообразования на значение окислительно-восста¬
новительного (ОВ) потенциала впервые отметил в 1898 г. Петерс. Он
установил, что в водном растворе ОВ потенциал системы, состоящей315
из ионов Fe(III) и Fe(Il), зависит от концентрации соляной кислоты.
Объяснение этому явлению с позиции образования ионами металла
комплексных соединений было дано Картером и Кливсом лишь в
1924 г. Систематическое изучение влияния природы анионов на по¬
тенциал системы Fe(Ill)—Fe(II) провел Михаэлис с сотр. Он показал,
что заменой аниона можно изменять потенциал в очень широких пре¬
делах: от +700 до —200 мВ.Естественно, что комплексообразование смещает положение равно¬
весия между окисленной и восстановленной формами металла. Если
лиганды являются нейтральными молекулами и координационные
числа комплексов одинаковы, то в общем виде эти равновесия можно
записать следующим образом:Если вследствие комплексообразования в большей мере стабилизиру¬
ется восстановленная форма, то в соответствии с уравнением Нернста
ОВ потенциал системы будет увеличиваться, а если в большей мере
будет стабилизироваться окисленная форма, то потенциал будет
уменьшаться.Стабилизация комплексных соединений зависит от многих факто¬
ров. Ряд из них связан с электронной структурой иона металла, а
другие зависят от природы лиганда. Однако многие факторы зависят
как от иона металла, так и от природы лигандов. Важным фактором
стабилизации комплекса является его координационное число. Хотя
оно главным образом определяется электронной структурой иона ме¬
талла, но иногда зависит н от природы лиганда.Как правило, геометрические изомеры отличаются друг от друга
по энергии. Однако это различие обычно невелико. Для лабильных
комплексных соединений при установлении равновесия один из изо¬
меров переходит в термодинамически более устойчивый. Для инерт¬
ных комплексов пёреход одного изомера в другой может быть затор¬
можен. В этом случае становится возможным изучать окислительно¬
восстановительные равновесия в системах, состоящих из комплексов
определенной геометрической конфигурации. К числу таких комплек¬
сов относятся соединения платины и рутения.Относительная инертность комплексов платины и рутения поз¬
воляет синтезировать соединения заданного состава и строения. Ве¬
роятно, этим объясняется тот факт, что именно на данных металлах
получен наибольший экспериментальный материал по окислительно¬
восстановительным равновесиям.Для платины наиболее распространенные состояния окисления
+4 и +2. Соответствующие нм комплексные соединения имеют окта¬
эдрическую и плоскоквадратную конфигурацию. Окислительно-вос¬
становительные равновесия с участием таких комплексов платины
включают процесс отрыва лигандов, например:М ~*~+ те М(«—т)+(МЦ]" те~=^[МЦ](П т)+[Pt (NH3)4 С!а]2+ + 2е~ ^ IPt(NII3)4p + 2Cn316
Большинство экспериментальных данных для систем, состоящих
из комплексов платины (IV) и платины (II), получены путем измере¬
ния потенциалов при определенных соотношениях концентраций
окисленной и восстановленной форм. Поскольку в равновесии участ¬
вуют лиганды, то положение равновесия, а следовательно, и потенциал
системы будут зависеть от их концентрации. Для вышеприведенного
равновесия выражение потенциала будет следующее:RT [ Pt(NH3)4Cl2+lЕ = £° + — In .£ [Pt(NH3C][Cl-Формальные потенциалы таких систем обычно измеряют на фоне1 М растворов соответствующих электролитов. Для хлоридных комп¬
лексов берут 1 М растворы NaCl или HCI. В этом случае уравнение
Нернста упрощается:RT [ Pt(NH3)«Cl|+|Е = Е° + — In nF [ Pt (NH3)J+]Большое разнообразие комплексов и систематические ряды полу¬
чены для рутения (III). Методом циклической вольтамперометрии
определены формальные потенциалы многочисленных систем, состоя¬
щих из комплексов рутения (III) и рутения (II), например:[Ru (NH3)5 Cl J"+ + e~ 0 [Ru (NH3)6 Cl] +В этих системах координационные числа окисленных и восстановлен¬
ных форм комплексов одни и те же. Поскольку отщепления лигандов
не происходит, то уравнение Нернста в данном случае проще:г_ rn 1 RT [Ru (NH3)r,Cl2+]“ Ь nF 'П [Ru (NH3)5C!+] ‘Таким образом, для систем, состоящих из комплексов рутения (III)
и рутения (II), возможности для выбора электролитного фона гораздо
больше.Арланд и Чатт впервые высказали мысль, что окислительно-вос¬
становительные свойства комплексных соединений зависят от л-ак-
цепторной способности координированных лигандов. Основанием для
этого послужила работа Гельман и Рябчикова, в которой было уста¬
новлено, что соль Цейзе K[Pt(C,H4)Cl;!] не окисляется перманганатом,
в то время как окисление соли K[Pt(NH3)CI:] протекает легко и коли¬
чественно.Взаимосвязь окислительных свойств координационных соедине¬
ний с природой координированных лигандов на примере комплексов
меди (II) изучал Гажо с сотр. Он установил, что в водных растворах
протекает реакция2CuCIz + СН3СОСН3 = 2CuCl + СН3СОСН2С1 + НС!Скорость ее сильно увеличивается в присутствии лигандов, обладаю¬
щих л-акцепторными свойствами, таких, как тиомочевнна, трифенил-317
фосфин, органические нитрилы н др. Гажо показал, что эти лиганды
входят во внутреннюю сферу комплексов меди (II). Следующей сту¬
пенью является гемолитический разрыв связи Cu(II)—CI с образова¬
нием свободного радикала С1. Последний и оказывает хлорирующее
действие на ацетон. Гажо полагает, что гемолитическому разрыву
связи Си(П>—С1( способствует образование л-датнвной связи ме¬
талл—лиганд.Гринберг отметил, что установленные им факты трудного окис¬
ления [Pt(N02)4]-_ и [Pt(CN)4]J~ могут быть объяснены способностью
лигандов N0“ и CN- образовывать л-связи с платиной (II).5 Систематические исследования окислительно-восстановительных
свойств комплексных соединений платины в зависимости от природы
ацидолигандов были выполнены Гринбергом с сотр. Он показал, что
окислительная способность комплексов платины (IV) уменьшается
в ряду ацидолигандов С1~ > Вг > Г > SCN'". Потенциалы аммиа¬
катов платины, составляющих переходный ряд Вернера—Миолати,
уменьшаются по мере накопления в координационной сфере молекул
аммиака.В зависимости от ацидолигандов потенциалы систем, состоящих
из комплексов платины (IV) и платины (II), уменьшается в ряду
N07>C|->Br->SCN->CN-> ГТаким образом, самыми высокими потенциалами характеризуются
нитритные комплексы. Для них весьма существенное значение имеет
электролитный фон. Так, на фоне НС1 значения потенциалов гораздо
выше, чем на фоне NaCI. Возможно, это связано с присоединением
протона к координированному нитритному иону.По влиянию на ОВ потенциал пиридин резко отличается от аммиа¬
ка и алифатических аминов. Если замещение в комплексах хлорид-
ионов на аммиак приводит к понижению ОВ потенциала, то замещение
на пиридин, наоборот, приводит к его увеличению (табл. 12.1). Все
значения потенциалов даны относительно стандартного водородного
электрода. Измерения проведены при 25 °С. Погрешность резуль¬
татов, приведенных в табл. 12.1 —12.5, порядка±3 мВ.Таблица 12.1. Формальные ОВ потенциалыОкислительно-восста новительна я
пара комплексов£»IM NaCIвim на|PtCl6]<--!-2e- = (PtCl,]--+2CI-0,734IPt(NH3)CI3]-- 2е- « |Pt(NH.,)CI.<]- j 2C1~0,7.410,694[PtPyCI5]-+2e- * [PtPvCI-,]- -2CI-0,7630,751|Pt(NHr))4Cl,]-+-i-2e- = [Pt(NH;))4r+ ’ 2C1-0,6190,600(PtPy4C!cl-M-2e- [PtPv4]-+----2C1-0,9320,847Интересно отметить, что при замещении одного лиганда на другой
в однотипных комплексах иногда наблюдается картина, близкая к318
Рис. 12.1. Зависимость окислнтельно-восстано-
вительного потенциала в системе|Pt(NH,).vP>vTCl:l-+—[Pt(NH3).TPy4_.T]-+ от со¬
держания амнион на фоне 1 М NaCl (/) и1 М НС1 (2)аддитивной. Например, при постепенной
замене внутрисферных молекул аммиака в
комплексе m/}aHC-[Pt(NHr,)4Cli]2+ на пири¬
дин ОВ потенциалы соответствующих сис¬
тем увеличиваются аддитивно (рис. 12.1).К сожалению, столь четкая корреляция
наблюдается не всегда. Тем не менее, во
многих случаях при замене одного лигандана другой можно оценить ОВ потенциал еще не изученной системы.Вне зависимости от типа изученных комплексов влияние природы
аминов на потенциалы то же самое. В табл. 12.2 приведены потенциалы
для систем типа[Pt (NH3); э2 CI2]:+ + 2f- ^ I Pt (NH3)2 а2]:+ -\- 2С'ГТаблица 12.2. Формальные ОВ потенциалы систем типа
mpaHC-[Pt(NH3)2a..cy2+ + 2е~ « mpa«c-[Pt(NH3)oa2p++2CI- на
фоне 1 М раствора NaCl1Ру 2Ру ЗРу 4РуАмин «а»E. BАмнн «а»E, ВQHsN’Hn0,569(CH„)2NH0,683(HOC.H4),NH0,593v2( Dipy)0,685]'.2(NH2C2H.jNH2)0,597(C2H5)2NH0,701CH,NH30,6151-Pic0,713NHS0,619]/2(Phen)0,718CoHjNH,0,625^-Pic0,726HOC3H6NH,0,636Hin0,746HOC"H4NH„0,641Py0,776C0H5CHoNHo0,6481;2!CH3).,NC,H4N,(CH3)i}0,786C3H-NH,0,654a-Pic0,801C4H,NHj0,661Такие комплексные соединения наиболее доступны для синтеза, от¬
носительно хорошо растворимы и поэтому на их примере удалось полу¬
чить особенно широкий ряд окислительно-восстановительных систем.
Амины в табл. 12.2 расположены по мере увеличения ОВ потенциа¬
лов соответствующих систем. Из таблицы видно, что в зависимости
от амина потенциалы аналогичных систем могут отличаться на доволь¬
но большую величину.Резкое отличие пиридина от аммиака по влиянию на ОВ потенциал
систем, состоящих из комплексов платины (IV) и" платины (II), свя¬
зано с его л-акцепторными свойствами. Таким образом, аммиак и
пиридин как лиганды могут служить своеобразными эталонами для
сравнения. Первый из них практически не обладает л-акцепторными
свойствами. л-Акцепторная способность пиридина изучена довольно319
хорошо. Она проявляется в различных свойствах комплексных соеди¬
нений. В частности, его относительная л-акцепторная способность
широко изучена на кислотно-основных свойствах соответствующих
аквокомплексов.В комплексах платины (II) имеется большее число d-орбиталей, при¬
годных для образования я-дативных связей, чем в комплексе пла¬
тины (IV). Поэтому за счет образования л-дативных связей платины
с л-акцепторным лигандом должна наблюдаться большая стабилиза¬
ция комплекса платины (II), чем комплекса платины (IV). Таким об¬
разам, л-акцепторные лиганды должны приводить к повышению стан¬
дартных окислительно-восстановительных потенциалов систем, состоя¬
щих из комплексов платины (IV) и платины (II).Вследствие л-акцегггорных свойств диалкилсульфоксиЯов можно
было ожидать, что сульфоксидные комплексы будут характеризовать¬
ся высокими ОВ потенциалами. Это предположение было проверено
на системах типа[PtLCI6]-+ 2е~ ^ [PtLCI3]- + 2CrМолекулы сульфоксидов в комплексах платины (IV) довольно легко
замещаются на хлорид-ионы. Поэтому ОВ потенциалы были измерены
не в стандартных условиях, а’на фоне 0,1 М раствора NaCl и пересчи¬
таны на 1 М NaCl. Полученные результаты приведены в табл. 12.3.Т а б л и ц а 12.3. Формальные ОВ потенциалы систем типа
[PtLC!3]-+2<?- 5=* [PtLCI3]"-; 2CI- на фоне 1 М NaClLE. вL£. BCH3NHa0,728c5h10nh0.755NH,0,730C5H5N0,763C6H6CH2NH„0,720(CH3),SO0,825C.H5NHo0,737(C2H5)2SO0.830Для сравнения в нее включены потенциалы аналогичных систем с
аминокомплексами, измеренные в одинаковых условиях с сульфоксид
ными соединениями. Из таблицы видно, что системы, состоящие из
сульфоксндных комплексов платины, действительно обладают более
высоким потенциалом, чем даже соответствующие пиридиновые
системы.На основании данных табл. 12.1 следует, что при замене одного
из хлорид-ионов на аммиак в комплексе [Pt ЕпСЬ] должен понижать¬
ся ОВ потенциал соответствующей системы. Влияние тиоэфиров в
сравнении с аминами на окислительно-восстановительные свойства
комплексов изучено Кукушкиным с сотр. В комплексах платины за¬
мена аммиака на пиколин, пиридин и тноэфиры приводит к значитель¬
ному повышению потенциала (см. табл. 12.4). Таким образом, л-ак¬
цепторные свойства тиоэфиров также проявляются в увеличении ОВ
потенциалов систем, состоящих из комплексов платины (IV) и плати-320
Таблица 12.4. Формальные ОВ потенциалы систем типа
[PtEnLCI3]++2e- [PtEnLCI]++2CI- на фоне I М НС 1L£. ВL£. ВNH30,6090(C2H4)2S0.6906-Pic0,654(C2H6)2S0,700Ру0,657C,H6SC2H.1OH0,721(CH3),S0,669(HOC4I4),S0,731ны (11). Этот же вывод вытекает из данных, приведенных в табл. 12.5.
Они показывают, что потенциалы систем, содержащих аминосульфиды,
значительно выше потенциалов аналогичных систем с диаминовыми
лигандами.Таблица 12.5. ОВ потенциалы систем типа
[PtLL'CI2]2++2e~5=t: [PtLL']2++2CI- на фоне 1 М НС1LL'E. ВNHXoH4NH,0,566NHoC»H4NHC,H60,595nh2g.h4nh.NH3C2H4SC3H50,688NH2C3H4SCjH4OH0,694NHsCnH4SCeHj0,689nh„c,h4sc,h5NH.G.H4SC3H60,757NH2C:H4SCoH4OHnh„c,h4sc,h4oh0,764Отмеченные закономерности в изменении ОВ потенциалов систем,
состоящих из комплексов платина (IV) — платина (II), оказались
применимы и для систем, состоящих из комплексов рутений (III) —
рутений (II). Так, из табл. 12.6 видно, что замена аммиака в комп¬
лексе [Ru(NH3)6]3+ на метиламин, бензиламин или циклогексиламин
сравнительно мало сказывается на ОВ потенциалах. В то же время за¬
мена аммиака на пиридин приводит к их значительному увеличению.
Потенциал растет с увеличением числа внутрисферных молекул пири¬
дина. Дипиридильные и фенантролиновые комплексы также имеют
гораздо более высокие потенциалы, чем аналогичные аммиачные.
Причиной этого являются л-акцепторные свойства пиридина, дипи-
ридила и фенантролина.Установлено, что ОВ потенциалы систем, состоящих из комплексов
рутений (III) — рутений (II) в ацетолитриле, увеличиваются при за¬
мене во внутренней сфере дипиридила на третичные фосфины. Это
связано с большими л-акцепторными свойствами фосфинов по срав¬
нению с дипиридилом.Закономерное изменение потенциала наблюдается при переходе
от аминокомплексов к иминокомплексам и далее к нитрильным, на-11-817321
пример, при переходе от NJ-hCI-hPh к HN=CHPh и iN^CPh, а также
от 1МНг—СвН„ к NH -=С„Н10 и от ЫНгСНгРу к HN =СНРу
(табл. 12.6) объясняется тем, что с увеличением кратности связейТаблица 12.6. Формальные ОВ потенциалы комплексов рутенияОкислительно*восстановительная пара комплексовС, В[Ru(NH3),]*[Ru(NH3)s(NH*CH,)] '*
[Ru(NH3)5Py]5+/2+транс-[ Ru(NH3)4Py2] ’+/"
цис- [Ru (NH3)4 Руг]1+/1+
[Ru(NH3)4(Dipy)J ^',г+[RuPy6]I+/J+[Ru(Dipy)3]s+/2+[Ru(Phen)3],+[Ru(NH3)s(N CCH3) ] **
[Ru(NH3)5(NH2CH2CeH5)]*+/;
[Ru(NH3)5 (NH=CHC,H6)]
[Ru (NH3)5( N-;CC6H6)J3+/2 +
3 + ,'2+4UC-[RU(NH3)4(N-CC6H5)2]3+/2[Ru(NH3)6(NH2C6Hn)]1+^[Ru(NH3)5(NH=C6H10)]3+/3+Ru(NH3)4jRu(NH3)4 ,//N\=\NH,/NNHRu(NH3)4\0=CN)\:h,\/0,05
0,06
0,10
0,31
0,30
0,42
0,49
g ,5o
0,507
1 ,291.531.54
0,43
0,120
0,320
0.510
0,48
0,87
0,110
0,1900,2950 ,5650,552донорного атома возрастает число л-разрыхляющих орбиталей, спо¬
собных акцептировать электроны с d-орбиталей центрального иона.
Так, потенциал системы [Ru(NH3)4(N = CPh),]3+/2+ почти на 0,4 В322
больше, чем системы [Ru(NH3)6:(N е CPh)) + . Следует отметить,
что ацетильная группа близка по влиянию на потенциал к иминогруп-
пе. Высокие потенциалы обусловливают применение комплексов Ru(I П)
в растворе ацетонитрила в качестве стехиометрических окислителей
в неорганическом и органическом синтезах.Как и для комплексов платины, при замене в комплексах рутения
аммиака на галогенидные ноны происходит понижение ОВ потенциа¬
лов (см. табл. 12.7). Однако большого различия между хлоридными,
бромидными и иодидными системами нет. Возможно, это объясняется
малым числом галогенидных ионов в изученных соединениях.Таблица 12.7. Формальные ОВ потенциалы комплексов рутенияОкислительно-восстановительна я пара комплексов/:. u[Ru(.NH3)»OH]№/*—0,42[Ru(NH3)5C1]'+/+—0,04|Ru(NH,)5Br]!+/+—0,03транс- |Ru(NH3)4C12]+/”—0, 1Gцис-[ Ru(NH3)4C12] +/г’’—0 ,1 Iтранс- [Ru(NH3)4PyCI]0,20цис- [Ru(NH3)4PyCI]I+/+0,17[Ru(Dipy)2Cl2]+/“0,65mpaHC-[Ru(NH3)4PyBr]2+/+0,19i(uc-[Ru(NH3)4PyBr]2+/+0,19mpaHf-[Ru (NH3)4Pyl]и"^+0,23^c[Ru(NH3)4PyI]!+/+0,18цис- [Ru(NH,)4(H20)0H]!+/ +-0,04/npa«c-[ Ru(NH3)4(H20)0H]2+/+-0,12[Ru(NH3)6(H20)]*+/!+0,08[Ru(NH3)4(H20)2],+/‘+0,10[Ru(NH3)4(H20)(SMe2)]*+/,2+0 ,48[Ru(NH3)4(H;0)P(0Et)3]’+/2+0 ;70[Ru (\'H3)4 {P(OEt)3}2]'+/'+0,88«uc-[Ru(NH3)4(H20)N2],+/,+0,94транс- [Ru(NH3)4 (HoO)N„],+/,2 +
4i/c-[Ru(NH3)4(H20)C0]rf" +1 ,301,09транс-[Ru(NH3)4(H20)C0] ‘+/‘+1,14Следует обратить внимание на очень низкие потенциалы систем,
состоящих из гидроксокомплексов рутения [Ru(NH3)6OH]2+/+ и
[Ru(NH3)4(H20)0H]2+/+ (см. табл. 12.7). Это объясняется способ¬
ностью гидроксильной группы к л-донорному взаимодействию с
Центральным ионом. Если лиганды л-акцепторного типа повышают
ОВ потенциалы, то лиганды л-донорного типа должны приводить к323
их понижению. л-Донорвым свойством координированной гидроксиль¬
ной группы уже объяснялись некоторые особенности кислотно-основ¬
ных равновесий аквагидроксокомплексов платины (см. гл. 7).Наибольшие потенциалы среди систем, изученных к настоящему
времени, имеют системы, содержащие во внутренней сфере комплек¬
сов лиганды N„ и СО (табл. 12.7). Эти лиганды являются типичными
л-акцепторами. Высокие ОВ потенциалы комплексов, содержащих
молекулу N2, вероятно, являются одной из причин существования
устойчивых соединений в относительно низких степенях окисления
металла.В заключение следует остановиться на имеющейся информации
по ОВ потенциалам в системах, состоящих из комплексов медь (II) —
медь (I). Установлено, что ОВ потенциал увеличивается при замеще¬
нии в лнгандах донорных атомов азота на серу. Это наглядно выте¬
кает из данных, представленных в табл. 12.8. Из нее видно, что приТаблица 12.8. Формальные ОВ потенциалы для систем типаСи L2+ + е~ a* CuL+в среде 80% метанола — 20% воды при 25 С: ц = 0,1 М НСЮ4LE, ВLE, В/HN/ \\1Н,\NH4 /NH2
^NHj\n4 /NH,H x ‘4NH,
/NH;—0,280±0,010
0 ,356±0,0220,361^0,019^_s/ \NHa
X^-Ss4 /NH,
^S—Et
4\—HN^^/S—Et
s/ ^S— EtXx— yS— Et/—^SH
/X-\_/SH0,312^0,020
0,342+0,020
0,892 -0,010
— 0,842переходе от тетратиоэфирных комплексов к аминотиоэфирным потен¬
циал резко понижается. При отсутствии в лиганде серы потенциал
становится даже отрицательным. Несколько неожиданной является
малая чувствительность потенциала к замене лиганда SN3 на S,Na,
а также резкий скачок в значении потенциала при переходе от амино-324
тиоэфирных лигандов к тетраминовому. Возможно, это объясняется
тем, что для комплексов меди (I) характерно координационное число2. В связи с этим стабилизация комплекса лигандом SN3 практически
та же самая, что и лигандом S2N2, так как в координации участвуют
те же самые донорные центры N—Си—S. При переходе от тетратио¬
эфирных лигандов к тетраминному значения потенциалов систем
Cu(Il) — Cu(I) изменяются от +0,842 до —0,280 В.Имеются данные о том, что нейтральные лиганды, обладающие
системой сопряженных связей и содержащие атом азота, для которого
характерны л-акцепторные свойства, стабилизируют титан в степени
окисления 3 +. Если для лигандов не характерны л-акцепторные
свойства, то в большей степени стабилизируется состояние титана
(IV), чем титана (III) Эти факты, вероятно, тоже можно объяснить
образованием л-дативных связей титан (III) — лиганд. Интересно,
что у титана (III) на d-орбиталях находится всего один электрон.Таким образом, приведенные экспериментальные данные свиде¬
тельствуют о том, что комплексные соединения, содержащие лиганды
я-акцепторного типа, характеризуются относительно высокими зна¬
чениями ОВ потенциалов.Безусловно, существенный вклад в значения ОВ потенциалов мо¬
жет вносить сольватация окисленной и восстановленной форм. По¬
этому при изучении влияния природы лигандов на ОВ потенциалы
правильнее сравнивать лишь изозарядные системы комплексов. В этом
случае разности энергий сольватации окисленной и восстановленной
форм будут иметь близкие значения и роль влияния лиганда будет
более отчетливой.Для систем, в которых заряды комплексных ионов не меняются, на¬
пример в переходном ряду Вернера—Миолати[PtClef-- + 2e- =* [PtCl4]2- + 2С1|Pt(NH3)CIe]-+2e- [Pt,(NH3)CI3]-+2Cr и Т. д.о влиянии природы лигандов на ОВ потенциалы можно судить от¬
носительно надежно, так как разница в энергиях сольватации комп¬
лексных ионов одного заряда не должна быть большой.В системах без изменения координационного^исла при переходе
«т окисленной к восстановленной форме заряды комплексов изменя¬
ются, например:IRu (NH3)e]s+ + ^ [Ru(NH3)6]2+[Ru (NH3)6C1]2+ +е- ** [Ru (NH3)6CI]*Разница в энергиях гидратации между окисленной и восстановленной
формами при переходе, например, от системы [Ru(NH3)e]3+/2+ к си¬
стеме [Ru(NH3)5C1]2+/+ может быть значительной. В таких случаях
при сравнении ОВ потенциалов роль характера лигандов проявля¬
ется не столь очевидно.Библиографию по изложенному вопросу можно найти в статье [I].325
12.4. Расчет констант равновесия окислительно¬
восстановительных реакций
из значений ЭДС гальванических элементов
и значений потенциалов электродных реакцийХорошо известно, что на основе двух химических полуреакций можно создать
гальванический элемент с определенным значением ЭДС. Например, можно
построить гальванический элемент из двух полуреакций:Fe3+ + е~ =: Fc-+i С» = 0,77В;Sn4+ + 2f" Sn2+; £° = 0 ,14В.Стандартная ЭДС гальванического элемента равнаEl =4e-/Fe- - £sn-/sn» = 0 ,77 - 0,14 = 0,63В.Эмпирическое правило гласит, что окислительно-восстановительная ре¬
акция может самопроизвольно протекать в таком направлении, в котором элект¬
рохимическая система с более высоким значением электродного потенциала вы¬
ступает в качестве окислительной. Таким образом, реакция2Fe3+ Sn-+ -t=t 2Fe2+ + Sn4+должна преимущественно протекать слева направо.Самопроизвольная реакция в гальваническом элементе, так же как и любая
другая самопроизвольно протекающая химическая реакция, характеризуется
отрицательным значением изобарного потенциала:— AG = nFEon.Если все реагенты находятся в стандартном состоянии, то уравнение имеет вид- ДС° = nFE°bn .Следует отметить, что значение п в этом уравнении является наименьшим об¬
щим кратным отданных и присоединенных электронов. Для рассматриваемого
случая Fe3'*"/2+ и Sn4_*_/2',‘ п — 2-1=2. Если взять полуреакции5 Sn4+ -f 2с- ч* Sn=+о МпО^+5<Г + 8Н+ ^ Мп-+ + 4Н20то значение п = 2-5 = 10.Поскольку константа любого химического равновесия связана сизобарным
потенциалом выражением —AG = RT \п К., то2,3 RT 0,059В справочниках обычно приводятся стандартные значення электродных
потенциалов по отношению к водородному или каломельному электроду. По¬
этому константу равновесия вычисляют по уравненню тнпа„ п (£реЗ+/2++ £sn4+/2+)
lgK~ •В аналитической химии реагентом дня качественного определения нона Fe3t
давно служит красная кровяная соль K3(Fe(CN)6], а дня определения иона Fe3t—
желтая кровяная соль K_i[Fe(CN)e). В обоих случаях в качестве продуктов ре¬326
акций получаются ярко-синие вещества. Первое из них называют турнбулевон
синью, а второе — берлинской лазурью. Впоследствии оказалось, что два послед¬
них вещества, по существу, являются одним и тем же соединением. В соответ¬
ствии со значениями электродных потенциалов £|Ve(CN)Vl/[Fe(CN)4,'] == 0,36 В и £Fes+/Fc!+ — 0,77 В ионы Fe(III) должны окислять [Fe(CN),,]4-.
Константа химического равновесияFe3+ -f [Fe (CN)6]4~ Fe2+ + [Fe (CN)e]3-равнялась бы примерно 10’. Таким образом, равновесие довольно сильно сдви¬
нуто в правую сторону. Смещению равновесия вправо также способствует малая
растворимость получающегося продукта. В осадок выпадают вещества, близкие
по составу к KFe11 [Fe111 (CN)c]. В зависимости от условии проведения реакции
соотношение между нонами К+ и Fe^ во внешней сфере комплекса может не¬
сколько меняться. С этим связано небольшое отличие в окраске берлинской ла¬
зури и турнбулевой сини.12.5. Типы окислительно-восстановительных
превращений координационных соединенийВ координационной химии наибольшее распространение имеют
реакции, связанные с замещением координированных лигандов. На
них основана практически вся препаративная координационная хи¬
мия и процессы комплексообразования в растворах. Вероятно, второе
место занимают окислительно-восстановительные превращения. Не¬
смотря на то что окислительно-восстановительных реакций с участием
координационных соединений описано гораздо меньше, чем реакций
замещения, разнообразие типов реакций в первом случае значительно
, больше.Окисление или восстановление центрального иона в координаци¬
онных соединениях. Реакции окисления или восстановления цент¬
ральных ионов могут быть разбиты на две группы: реакции, идущие
без изменения координационного числа, например:[Со (NH3)e]2+ — е~ 5* [Со (NH3)e]3+(Fe(CN),r + e- [Fe (CN)e]*~и с изменением координационного числа, например![PtC!j]-~ — 2е~ + 2Н:0 ** [PtCI4 (ОН),]2- + 2Н+[PdCl6]2-+2e- [PdCI4]2-+ 2СГРеакции без'изменения координационного числа протекают быст¬
рее и, как правило, являются обратимыми. Реакции с изменением
координационного числа сопряжены с разрывом или образованием
химических связей. Это часто сказывается на кинетике окислительно¬
восстановительных процессов. Кроме того, нередки случаи
когда такие процессы необратимы. Например, восстановление
^c-[Pt(NH3)4Cl2]Cl2 протекает в соответствии с уравнениемHUC |Pt (NH3)4 С12]г+ + 2е~ -> [Pt (NH3)3C1]+ + NH3 + CPОкисление [Pt(NH3)3Cl]+ в аммиачной среде на хлоридном327
фоне приводит к образованию смеси продуктов: [Pt(NH3)3Cl3]+,
mpa«c-[Pt(NH3)4CI2]2+, ^c-[Pt(NH3)4Cl2]2+, [Pt(NH3)8Cl]3+, а воз¬
можно даже и [Pt(NH3)n]4+. Таким образом, окислительно-восстано¬
вительные реакции с изменением координационного числа комплексов
не всегда обратимы. Отсутствие обратимости особенно часто проявля¬
ется для инертных разнолигандных комплексов.В настоящее время реакции окисления комплексов, сопровождаю¬
щиеся увеличением координационного числа, называют реакциями
окислительного присоединения. Так, окисление тетрахлороплатината(II) хлором, хлорноватистой кислотой, пероксидом водорода—все
это реакции окислительного присоединения:[PtCI4]J- + C!., - [PtCl,]2"[PtCl4]-- + HOCI -v [PtCl6OH]2-[PtCI4]s-+ H,03 — [PtCl4(OH)o]2-Благодаря высоким окислительным потенциалам окислителей та¬
кие реакции во многих случаях протекают практически количественно.
Это дало возможность широко использовать указанные окислители
в синтезе разнообразных комплексов, особенно разнолигандных.Процесс, противоположный окислительному присоединению, на¬
зывают восстановительным элиминированием (отщеплением). Естест¬
венно, что он приводит к понижению координационного числа цент¬
рального нона.Окислительно-восстановительные превращения координированных
лигандов. Изучение реакционной способности координированных
лнгандов является одним из важных направлений современной коор¬
динационной химии. В результате координации свойства иона или
мол’екулы инбгда весьма резко изменяются. Например, координиро¬
ванный нитритный ион во многих комплексах перестает окисляться
перманганатом. Однако имеются примеры восстановления координи¬
рованной группы NO2 в аммиак. Так, Черняев установил, что в комп¬
лексах платины (II) внутрисферная нитрогруппа при взаимодействии
с водородом в момент выделения превращается в аммиак:[PtEn (N02)2] + 12 «Н» + 2НС1 -+■ [PtEn (NH3)2] Cl* + 4H20fПодобным же способом в аммиак был превращен гидроксиламин:[Pt (NH2OH)4] С12 -г «8Н» — [Pt(NH3)4]CI2 + 4H,0Окислительно-восстановительное взаимодействие координиро¬
ванных лигандов с внешними реагентами одним из первых начал изу¬
чать Вернер. Он разрушал азотной кислотой, хлором и пероксидом
водорода роданидную группу в комплексах кобальта (III). В ряде
случаев ему удалось показать, что она без разрыва связи кобальт(III) —азот может быть превращена в аммиак. Например,
транс-\Со En2(NCS)2]+ при взаимодействии с пероксидом водорода
превращается в транс-[Со En2(NH3)2]3+.В результате изучения взаимодействия свободного иона NCS' с
пероксидом водорода установлено, что при pH > 4 продуктами окис¬328
ления являются NH3t SO^, СО? и HOCN. В более кислых рас¬
творах (pH < 2) в результате реакции получаются SO4”, HCN и в не¬
больших количествах S(CN)2.Можно предположить, что взаимодействие родано-группы в комп¬
лексах кобальта (III) с пероксидом водорода происходит подобно вза¬
имодействию свободного иона NCS~ в слабокислой или щелочной сре¬
де. Однако вполне возможно, что взаимодействие свободного и коор¬
динированного нона протекает по различным механизмам.Примерами окисления координированных лигандов могут служить
превращения внутрисферных фосфинов в фосфиноксиды под дей¬
ствием кислорода воздуха. Окисление внутрисферных фосфинов в
фосфиноксидные протекает как в комплексах, находящихся в раст¬
воре, так и в твердой-фазе. Например:[Ni (PR3)212] [Ni (OPR3)2I2]Существует распространенное мнение, что эти процессы протекают
через промежуточное образование оксигенильных соединений. Моле¬
кула кислорода в большинстве комплексов переходных металлов
координируется симметричным образом и по существу является пер-
оксогруппой. Пероксогруппа подвергается стёхиометрическому вос¬
становлению газообразными СО, S02, NO, N02. На примере бис-фос¬
финовых комплексов платины реакции схематично могут быть запи¬
саны следующим образом:Гринберг и Птицын предложили объемный метод определения пла¬
тины (II) титрованием комплексов раствором перманганата калия.
Однако они обратили внимание на то, что этот метод не может быть
распространен на все комплексы платины (II). При наличии во внут¬
ренней сфере пиридина наблюдается завышенный расход КМ1Ю4 по
сравнению с тем, сколько его должно пойти лишь на окисление пла¬
тины. В комплексах платины (IV) пиридин не окисляется перманга¬
натом. В свободном состоянии пиридин также не окисляется. .Анало¬
гично пиридину ведут себя алифатические амины и аминосульфоновая
кислота.о,t СОR3P>V,//\0N0 NO R3PОR3P'ONO R3P 4 Оj 2N02
R3Pv /ONO;/ \R3PX x ONO.О,,0N02329
Считается, что окисление внутрисферных аминов индуцируется
окислением платины (II). Иногда это «индуцированное» окисление про¬
исходит в очень сильной степени. Так, Рябчиков установил, что при
титровании [Pt(NH3)2(NH;>S03)CI] вместо двух эквивалентов перман¬
ганата калия расходуется шесть эквивалентов.Подобное явление было отмечено при окислении щавелевой кисло¬
ты. В свободном виде она не окисляется броматом калия, а в комплек¬
се платины (II) оксалатный ион окисляется очень легко. Причиной
этого, вероятно, также является индуцирование процесса окисления
координированных лигандов окислением центрального иона.Способность внутрисферных лигандов &О5 или гидроксиламина
окисляться хлором или бромом не вызывает удивления, поскольку они
относительно сильные восстановители. Однако к этим реакциям было
проявлено большое внимание. Оказалось, что в процессе окисления
SOj или NH2OH, входящих в комплекс платины (II), одновременно
происходит удаление из внутренней сферы /лранс-расположенных ли¬
гандов. Вслед за окислением этих лигандов и миграцией транс-рас¬
положенных происходит окисление платины (II) до платины (IV).
Эти факты являются своеобразным проявлением трансвлияния ли¬
гандов.Окислительно-восстановительное взаимодействие центрального
иона и лиганда. Еще в конце XIX в. было установлено, что карбо¬
нильные комплексы платины в водных растворах подвергаются внут¬
римолекулярным окислительно-восстановительным превращениям.
Соответствующие реакции можно записать в виде уравнений[Pt (СО) С1,]2 + 2Н20 -ч- 2Pt + 2С02 + 4HCI[Pt (СО)2 С12] + Н20 -ч- Pt + С02 + СО + 2HCIПочти тогда же установили, что в водных растворах оксид угле¬
рода (II) восстанавливает хлорид палладия до металла. В настоящее
время не вызывает сомнения, что этот процесс аналогичен превраще¬
нию в растворе платиновых комплексовФиллипс показал, что при взаимодействии этилена с PdCl2 в вод¬
ном растворе палладий восстанавливается до металла и образуется
уксусный альдегид. Природа этой реакции стала ясна гораздо позже.В 1934 г. Дж. С. Андерсон установил, что при нагревании водного
раствора соли Цейзе выше 90 °С платина (II) окисляет Ьнутрисфер-
ный этилен до уксусного альдегида:К [Pt (С2Н4) С13] + Н20 Pt° + СН3СНО -1- КС] + 2НОЧерез четыре года аналогичная реакция была осуществлена с л-комп-
лексом палладия.Гринберг и Кукушкин обнаружили окислительно-восстановитель¬
ное взаимодействие между платиной (IV) и депротонированным
этилендиамином. Оказалось, что в щелочной среде комплекс
транс-[Pt EmCh]2* превращается в [Pt Епг]2+. При этом образуются
свободные радикалы. На первых стадиях реакции от аминогруппы330
отщепляется протон с последующим переходом электрона с образую¬
щейся импдогруппы на центральный ион:[PtEnXIip + OH- ** [Pt ( NHC2H4NH2) ЕпС12]+ + НгО[PtIV (NHC2H4NHj) ЕпС12]+ ^ [Pt,n(NHC2H4NH2) EnCl2]+Повторение Зтого процесса приводит к получению соединения плати¬
ны (II). Такой комплексный ион должен содержать радикалыNH&H4NH2. Не исключено, что эти радикалы взаимодействуют с
водой по схеме[Pt (NHC2H4NH,)2]2+ 4- 2НгО =»* [PtEn2]:+ + 20НОписано своеобразное превращение сульфоксидных комплексов
платины (II). Оказалось, что в солянокислой среде внутрисферные
сульфоксиды восстанавливаются до тиоэфиров, а окислению подвер¬
гается центральный ион комплекса и хлорид-ионы. Например:[Pt (ДМСО)о С12] + 4HCI [Pt (ДМС)оС14] + С12+2НгОХайн с сотр. интересным способом осуществили синтез 2,2'-ди-
пиридила из пиридина. Превращение происходит во внутренней сфере
комплекса железа (III). Ион Fe3+ восстанавливается по Fe2+, а пири¬
дин превращается в дипиридил по схеме/ч /ч /ч2 I j — 2<Г->- I | || | + 2Н+Обмен электронами между центральными ионами двух комплексов.Известно много случаев, когда прочные комплексы подвергаются
быстрому электронному обмену. Например, в водном растворе элек¬
тронный обмен между комплексами•Fe (CN)*-+ Fe(CN)^ ** *Fe (CN)|-+ Fe(CN)£“происходит с большой скоростью. Это объясняется тем, что структуры
цианидных комплексов железа (II) и железа (III) настолько близки,
что при переносе электрона практически не требуется их перестройки.■ Быстрый обмен электронами между комплексами [Pt Епг]2+ и
[Pt ЕпгС1:]2+ в растворе объясняется образованием мостикового со¬
единения в соответствии с уравнениемErv Епо 0*Р1Еп,С1++ PlEn,C|J+ CI—Pi—Cl—Pt—Cl PtEn,С1++‘PtEn,С1^+■ ’ 0 0 ' “En EnМостиковые комплексы Pt(II) — Pt(IV) известны давно. Важную
роль в переносе электронов между центральными ионами в комплек¬
сах играют лиганды, способные быть мостиковыми.В приведенных примерах электронного обмена реакции практиче¬
ски не протекают, так как комплексы образуют одну окислительно¬
восстановительную систему. Происходит лишь обмен электронами,
а концентрации окисленных и восстановленных форм не изменяются.
Если взять комплексы, не входящие в одну и ту же окислительно¬
восстановительную систему, то при соответствующих условиях мо¬
жет происходить реакция и ее направление будет определяться
потенциалами соответствующих систем. Системы, состоящие из комп¬
лексов платина (IV) — платина (II) на фоне 1 М HCI, имеют следую¬
щие значения окислительно-восстановительных потенциалов:[PtCle]2- + 2e- [PtCI4]2- + 2СГ; Е° = 734 мВ[Pt(NHg)4 Cl2]2+ + 2е- ** [Pt(NH3)4p + 2CP; Е° = 600 мВ[PtPy4CI2]2+ + 2<Г ** [PtPy4]-+ ~г 2СГ; £° = 847 мВПоэтому реакция[Pt (NH3)4]2+ г [PtCI,]2' ** [Pt (NH3)4CI2]2+ + [PtCI4]2-
будет протекать слева направо, а реакция
[PtPy4]2+ + [PtCI6]2- =*= [PtPy4CI2]2+ + [PtCI4]2-наоборот, справа налево.Е. С. Постникова и Г. С. Крылова выявили термическое твердо¬
фазное превращение комплекса [Pt Py4Cl2J [PtCl4] в [Pt Py4][Pt Cl6],
которое протекает в том же направлении, что и в растворе.Окислительно-восстановительное взаимодействие двух лигандов
во внутренней сфере комплекса. В химии весьма важное значение
имеет реакция внедрения олефинов в связь металл—водород. В про¬
цессе этой реакции сначала получается л-комплекс, который затем
испытывает л,ст-перегруппировку по схемеRCH=CH„I I ' I
—М—Н -f- RCH=CH, »-М—Н -н- — m-ch,ch2rI 'IIФормально л,а-перегруппировку можно рассматривать как окисли¬
тельно-восстановительное взаимодействие двух лигандов.Кукушкин с сотр. установили, что при нагревании в атмосфере
аргона комплексов [NiL2(N03)2], где L — PEt2Ph, PEtPhs, PPr:Ph,
PBmPh, происходит окисление фосфинов в фосфиноксид, а одна из
нитратных групп превращается в N0. Эти реакции можно рассматри¬
вать как типичные примеры окислительно-восстановительного взаимо¬
действия лигандов, находящихся во внутренней сфере того же самого
комплекса.Библиография по изложенному материалу содержится в статье332
12.6. Реакции окислительного присоединенияВ общем виде реакция окислительного присоединения может быть
выражена уравнением
[AtL„] X't —*■ [XMYL„]Формально атакующая комплекс молекула «раскалывается», и ее
части присоединяются к центральному атому. Молекулы XY имеют
электр'офильный характер, поэтому электроны (как правило, два)
с центрального атома переходят на лиганды X и Y. В результате
реакции увеличивается степень окисления центрального атома (окис¬
ление) и повышается его координационное число. Следовательно, ре¬
акции окислительного присоединения протекают с участием тех комп¬
лексов, центральные атомы которых способны окисляться и проявлять
при этом склонность к увеличению координационного числа, т. е.
изменять полиэдр. Например, комплекс [Pt(NH3)4]2+ способен окис¬
ляться хлором в соответствии с уравнением[Pt(NH3)4]2++CI2 -ч- [CIPtCKNH3)4r-+Эту реакцию можно рассматривать как типичный случай окислитель¬
ного присоединения. Комплекс рутения (II) [Ru(NH3)6]2+ взаимодей¬
ствует с хлором по уравнению2 [Rn (МН3)в]2+ + С12 [Ru (NH3)e]3+ + 2СГВ этой реакции происходит окисление комплекса, однако координа¬
ционное число не изменяется и поэтому ее нельзя отнести к типу окис¬
лительного присоединения.Поскольку галогены обладают высокими окислительными потен¬
циалами, то реакции с их участием широко используют для синтеза
комплексов. Новый этап в изучении реакций окислительного присо¬
единения наступил, когда было установлено, что в ролй окислителей
могут выступать стабильные соединения, обладающие низкими окис¬
лительными потенциалами. Например, СН31 окисляет комплекс ири¬
дия (I) [Ir(PPh3):(CO)Cl] по уравнениюПротекание подобных реакций связано с разрывом одной связи, но
образованием двух, что и вносит определяющий вклад в энергетику
процессов.Особый интерес реакции окислительного присоединения приобре¬
ли после того, как было установлено, что в роли окислителя выступает
молекулярный водород с образованием гидридных комплексов. На¬
пример, изучена следующая реакция:[I г (СО) (PPh3)о С!] + Н2 — [HIr(H)CO(PPh3)aCIJ333
Образующиеся гидридные комплексы во многих случаях являются
активными интермедиатами в реакциях каталитического гидрирова¬
ния непредельных органических соединений.Как уже было сказано, реакции окислительного присоединения
могут протекать с теми комплексами, которые способны при окислении
превращаться в соединения с повышенным координационным числом,
т. е. способны изменять полиэдр. Для комплексов платиновых ме¬
таллов ориентиром в возможности осуществления реакции окисли¬
тельного присоединения является правило эффективного атомного
номера, или правило 18 электронов. Реакции окислительного при¬
соединения осуществляются с комплексами, которые не соответствуют
этому правилу, но в результате получаетс;я комплекс, который этому
правилу отвечает. В табл. 12.9 приведены данные, отражающие
взаимосвязь электронной конфигурации центрального атома, его сте¬
пени окисления и координационного числа.Таблица 12.9. Электронная конфигурация, степень окисления
и координационное числоЭлектроннаяПлатиновыеметаллы вразличных степеняхКоординационноеконфигурацияокислениячислоd10Pd(0)2, 3, 4*Pt(0)d8Ru(0)Rh(I)Pd(II)4, 5*Os(0)Ir(l)Pt(II)d6Ru(II)Rh (III)Pd(IV)6*Os(II)Ir(III)Pt(IV)• Комплексы с этими координационными числами соответствуют правилу 18 электронов.В реакциях окислительного присоединения могут участвовать и
комплексы, которые соответствуют правилу 18 электронов. В этом
случае часть внутрисферных лигандов покидает комплекс, освобождая
координационные места. В р'езультате таких реакций происходит
увеличение степени окисления центрального атома, а координацион¬
ное число сохраняется прежним. Например, при окислении хлором
комплекса [Pt(PPh3)4] реакция протекает по следующему уравне¬
нию:[Pt (PPh3)4] + CI™ — [ClPtd (PPh3)2] + 2PPh3В данном случае наряду с окислительным присоединением двух ато¬
мов хлора происходит отщепление двух внутрисферных молекул три-334
фенилфосфина. Реакцию такого типа в общем виде можно записать
следующим образом:[ML„] + Х^ -► [ХММ.„.?] +«?LПриведенные,в общем виде два типа реакций окислительного при¬
соединения являются наиболее распространенными.Рассмотрим возможные типы реакций окислительного присое¬
динения.Реакции типа [MLJ + XY-4XMYLJ. Многие химики видели в
свойствах комплексов платины (II) аналогию со свойствами этилено¬
вых углеводородов. Одними из первых на это указывали Чугаев и Хло¬
пни, имея в виду способность тех и других соединений к реакциям при¬
соединения. Они провели окисление ряда комплексов платины (II)
пероксидом водорода и получили соответствующие днгидроксокомп-
лексы. Например, окисление соединения [Pt(NH3)2Cl2] происходит по
уравнению(Pt (NH3),C12] + Н202 [(ОН) Pt (ОН) (NH3)2 CIo]Впоследствии окисление пероксидом водорода широко использова¬
лось для получения шракс-дигидроксокомплексов платины (IV).Гринберг и Михелис изучали взаимодействие комплексов платины(II) с хлорноватистой кислотой и установили, что реакции протекают
по схеме[Pt (NH3)4]2+ + HOCl -- [(НО) PtCl (NH3)4]2+В реакции окисления комплексов платины (II) хлорноватистой кисло¬
той они видели полную аналогию с окислением галогенами или пер¬
оксидом водорода.Таким образом, реакции окислительного присоединения известны
давно. Здесь не рассматриваются реакции окислительного присоеди¬
нения с участием галогенов, поскольку они относительно просты. Пред¬
почтение отдано реакциям с несимметричными молекулами и прежде
всего с молекулами типа НХ и RX.Комплёкс родия (I) m/?aHC-[Rh(CO){PMe2(2-MeOCeH4)}2Cl] при¬
соединяет большое число разнообразных молекул RX (НС1, МеС1,
Mel, CCI4, Cl2, PhOCl) с образованием неустойчивых соединений ро¬
дия (III) общей формулы [RRh(X)CO{PMe2(2-MeOC6H4)}2Cl]. Реак¬
ции с участием m/7aHo[RhCO{PMe2(2-MeOC6H4) }2С1] идут быстрее, чем
с транс [RhCO(PMe2Ph)2CI].Для одной и той же кислоты НХ константы равновесия реакции[Ir (СО) ЬУ] + НХ s* [HIrX (СО) L2Y]в зависимости от Y увеличиваются в ряду 1-> Вг" > С1", а в зави¬
симости от L — в ряду РМе3 > PMe2Ph > PMePh2 > PPh3.Хелатные комплексы [Rh(N—N) (СО)Х] (где N—N —• Dipy,
Phen; X — С1“, Вг", Г) в реакции с иодистым метилом дают соответ¬
ствующее органометаллическое производное [CH3RhI(N—N)(CO)X].
На основании ЯМР спектров в дейтерированном диметилформамиде
предполагается сосуществование трех форм:335
где S — молекула растворителя.Реакция между оптически активным бромидным соединением
RCHBrC02C2H5 (R — СН3, С6Н5) и комплексом [Rh(CNR')4]+ (где
R' — Ви‘, л-СН3С6Н4) приводит к образованию оптически неактив¬
ных продуктов mpaHC-[(RCHC02C2H5)RhBr(CNR')4]+. Однако она тре¬
бует фотоинициирования.При обработке хлороводородом раствора фосфинового комплекса
палладия (0) — [Рс1(РВиз)2] в гексане получен /гс/?а«с-[НРс1С1(РВиз)2].
Он устойчив в твердом состоянии, но в бензольном растворе подвер¬
гается внутримолекулярному циклометаллированию в соответствии с
уравнениемНеобычные октаэдрические хелатные олефиновые комплексы гипа
[HMCI2(bdpps)] получены окислительным присоединением НС1 к
[MCl(bdpps)], где М—lr, Rh; bdpps — 2,2'-бис-(дифенилфосфино)-
транс- стильбен (o-Ph2PC6H4CH =CHCeH4PPh2-o). В противополож¬
ность этому пятикоординационные комплексы [MCI(CO) (bdpps)] про-
тонируются хлороводородом по координированной двойной связи с
образованием хелатных а-алкильных комплексов родия (III) и иридия(III) по схемеОкислительное присоединение органических кислот к комплексам
переходных металлов идет тем легче, чем выше значение рД'а. При
взаимодействии комплекса [Ir(CO)L2X] с бензойной кислотой по урав¬
нению[Iг (СО) L»X] -г PhCOOH -* [HIr (PhCOO) (СО) L3X]Cl
склонность к протеканию реакции увеличивается для X в ряду С1 с< Вг < I, а для L — PPh3 < AsPh3 < P(CH3)Ph2 < PEt2Ph << PMe2Ph < AsMe„Ph <; PMe3.В реакциях окислительного присоединения используются ртуть-
органнческие соединения. Алкильные группы этих соединений можно
перенести на переходный металл с образованием устойчивых а-органи-
ческих комплексов.Нульвалентные трифенилфосфиновые комплексы платины и пал¬
ладия реагируют с ртутьорганическими соединениями RHgX и R2Hg
с образованием ст-производных металлов в качестве конечных про¬
дуктов:М (PPh3)„ + RHgX — RMX (PPh3)„ + HgРеакция проходит стадию образования интермедиатных биметалли¬
ческих соединений,, имеющих связь М—Hg. Их устойчивость зависит
от электронного и стерического строения радикала R. В частности,
все фторированные соединения органортути реагируют с [Pt(PPh3)3]
с образованием вполне устойчивых биметаллических соединений:[Pt (PPhahl + RF HgX -*• [RF HgPtX (PPh3)2] + PPh3Таким образом, реакции ртутьорганических соединений с комплек¬
сами переходных металлов в общем виде описываются уравнениемMLn -Г RHgX -+■ RMXL„ + Hgи могут рассматриваться как особый тип окислительного присоеди¬
нения.Реакции типа [ML„] + XY->-[XMYLn_g] + qL. К настоящему вре¬
мени получена большая информация по окислительному присоеди¬
нению галогенов к координационно-ненасыщенным комплексам ро¬
дия (I). Так, плоскоквадратный с^-комплекс родия, содержащий ди¬
фенил- или дициклогексилдитиофосфатный лиганд, реагирует с иодом
и бромом с образованием мостикового'продукта по следующей схеме:К этому же типу реакций относится окислительное присоединение
бензилгалогенидов к комплексу палладия (0). При этом наблюдается
инверсия на бензильном атоме углерода. Реакция протекает по схемеИнтересный пример окислительного присоединения связан с рас¬
щеплением связи углерод—углерод при реакции дикетона с комплек¬
сом платины (0), протекающей в соответствии с уравнениемX, -со[Rh (СО), (S—S)] -*■ [XRhX (СО)2 (S—S)] —^ [XRhX (СО) (S—S)J -»-+.i/2[XRhX(CO) (S—S)]2CflHjIpd (PPh3)4] + PhCH2 X * [PhCH2PdX (PPh3)2] + 2PPh3'^^-pt(pph3)3V~\o337
Окислительное присоединение дициана к |трифенилфосфиновым
комплексам никеля, палладия и платины также протекает с разрывом
связи углерод—углерод:fM (PPh3)4] |- NC-CN -v [(NC) M (CN) (PPh3),] + 2PPh3(здесь M — Ni, Pd, Pt).Наиболее универсальный метод образования комплексов со связью
металл—углерод основан на расщеплении связей углерод—галоген в
атакующей молекуле. Примером может служить присоединение ацил-
галогенпдов:IRhCI (PPh3)3] -I- RCOCI -» [(RCO) RhCI2 (PPh3)2J + PPh3В результате окислительного присоединения дисульфидов RS—SR
(R — алкил или арил) к комплексам палладия (0) и платины (0) об¬
щей формулы [M(PPh3)4] наблюдается расщепление связи сера—сера
и образование бис-(алкилсульфидо)- или бис-(арилсульфидо)фосфи-
новых комплексов:[M(PPh3)4] I-RS-SR [(RS)M(SR)(PPh3)*J +2PPh3Винилгалогениды, содержащие атомы углерода в состоянии sp-¬
гибридизации, инертны в обычных органических реакциях и непо¬
средственное замещение атома галогена в них происходит с трудом.
Однако винил- и арилгалогениды более реакционноспособны по от¬
ношению к комплексам переходных металлов, чем алкилгалогениды
с атомами углерода в состоянии 5/?3-гибридизации. В этом проявляется
одно из характерных свойств комплексов переходных металлов. Так,
винилхлорид присоединяется к [Pd(PPh3)4] с расщеплением связи уг¬
лерод—хлор:/Н^/Н СН2=СЧ^ y'PPh3СН„=СЧ + Pd (PPh3)4 /Pd\ + 2PPh,v ' \ Г1NC1 Ph3P uГидрндные комплексы металлов образуются в результате окисли¬
тельного присоединения не только молекулярного водорода, но и
других водородсодержащих соединений. Для органического синтеза
представляет интерес реакция отрыва водорода от соединений, содер¬
жащих связь углерод—водород.Примером расщепления связи углерод—водород служит реакция
терминальных ацетиленов с образованием гндридокомплексов:НPh3P4 j /Cs=CR. . “~зRc^c/ | ^PPh3
H2RC » CH [Pt(PPh3)4] -* \ni/ + 2PPhВзаимодействие HX (X — Cl", Br , I ) с изоэлектронными соеди¬338
нениями общей формулы [Ir(CO)n(PR3)5_n]+ (п = 1 или 2) дает комп¬
лексы [HlrX(CO) (PPh3)3]+. Реакции идут с потерей СО или PPh3:нх[Ir(COh(PPh3)3r [ НI г X (СО) (РРЬ3Ы+-|-СОнх[Ir (СО) (PPh3)4]+ - [HIrX (СО) (PPli3)3]+ + PPh3Гидридные комплексы могут образовываться в результате окис¬
лительного присоединения соединений со связями сера—водород. На¬
пример, сероводород и тнолы реагируют с комплексом [RhCl(PPh3)3 ] по
схемеIRhCI (PPh3)3) + RSH — [HRh (SR) Cl (PPh3)2] + PPh3Некоторые соединения со связями азот—водород также способны
вступать в реакции окислительного присоединения. Амины и амиды
присоединяются к трифенилфосфнновому комплексу платины (0)
с образованием связей металл—водород и металл—азот:.0+ [Pt (PPh3)4l „NPt (H) (PPh3)2 + 2PPh36 NH О \0R.NH + [Pt (PPh3)4) -ч- [R.NPt (H) (PPh3)2J — 2PPh3'В некоторых случаях после окислительного присоединения гало-
геноводорода протекает дальнейшая реакция. При реакции внутри-
сферного гидридного лиганда со свободными протонами образуется
газообразный водород. В результате реакция идет по стадиям, на¬
пример:[Pd (PPh3)4] + НС1 — [HPdCI (PPh3)2] + 2PPh3[HPdCI (PPh3)s] + HC1 -» H2 + [PdCI2 (PPh3),JПри взаимодействии комплекса платины (0) [Pt(PPh3)4] с трихлор-
силаном были выделены продукты [Pt(SiCl3)2(PPh3)2] и Н2. В суммар¬
ном виде реакция может быть выражена следующим уравнением:|Р‘ №)4] + 2HSiCI3 — [Pt (SiCI3)2 (PPh3)2] + H2 + 2PPh3Реакция этого типа напоминает взаимодействие IВг с [Pt(PPh3)4]:lpt (pph3)4] + 21 Br -к [Pt (PPh3)2 Br2] -f Iг + 2PPh3Интересная реакция окислительного присоединения происходит
при взаимодействии [Rh(PPh3)3Cl] с СН3СОС1. В суммарном виде она
выражается уравнениемпг». СО
Вначале образуется пятикоордннационный комплекс
PPh34 C,0CI I\IRhCV xPPh3который затем перестраивается в результате миграции метальной
группы и образования конечного продукта. Таким образом, в данной
реакции из атакующей молекулы СН3СОС1 получаются три лиганда
сн;, СО и С1“.Реакции типа [QMLJ + XY-+-[(XQ)MYL(n_,)l + qh. В литературе
пока известно мало реакций этого типа. Так, окислительным при¬
соединением йодистого метила к комплексам [Rh(CO)2(S—S)], где
(S—S) — (PhO)2PS2 или (HUC6)2PS2, получен ацильный комплекс
[(MeCO)Rhl(CO) (S—S)]. Эта реакция является усложненным типом
окислительного присоединения, так как иод присоединяется к родию,
а метил — к координированной молекуле СО.Аналогичная реакция протекает между Mel и комплексом
[Rh(CO)PPh3(mnt)]_, где mnt — малеонитрилдитиолат, с образова¬
нием комплекса [(MeCO)RhI(PPh3)(mnt)]~.Присоединение алкилгалогенидов к некоторым карбонильным со¬
единениям металлов приводит к ацильным, а не к алкильным комп¬
лексам. Примером служит реакция [Pd(CO) (PPh3)3] с различными
органическими галогенидами, например винил-, аллил-, бензил-,
метил- и фенилгалогенидами:[Pd (СО) (PPhshJ + RX — [(RCO) PdX (PPh3)2J f- PPh3К реакциям этого типа можно отнести взаимодействие нитрозиль-
ных комплексов[OsCI (NO) (СО) (PPh3)2], [Os (NO)2 (PPh3)2l и [IrNO (PPh3)3]с HCI. К комплексам присоединяются один, два, три моля НС1 с об¬
разованием соответственно[OsC!2 (HNO) (СО) (PPh3hJ, [OsC!2 (NHOH) (NO) (PPh3)2]и [IrCl3(NHaOH)(PPhj),]К реакциям такого типа относится и алкилированпе координиро¬
ванных молекул азота. В общем виде они выражаются уравнением[М (N,)a (Ph2PC2H4PPh2)o] + RX ->■ [(RN2) MX (Ph2PC,H4PPh3)2J + N2где M — Mo, W; X — Br, I; R — алкил.Реакции типа [MLJ + XY-4XMLJY. В результате этих реакций
образуются комплексы катионного типа. Характерным примером мо¬
жет служить окислительное присоединение иода к комплексу
[Pt(CNBu')2(PPh,)2]:[Pt (CNBu')2 (PPh3)a] + I2 — [IPt (CNBu‘)2 (PPh3)2] IИнтересная реакция осуществляется взаимодействием Aiel или
CF3I с lPt(CNBul)3(PPh3)2]:340
[Pt (CNBu‘)2 (PPhahl + RI [(RC=NBu‘) Pt (CNBu‘) (PPh3)2] IПри окислительном присоединении карбкатионов, таких, как ме-
тилфторсульфонат, или гексафторфосфат триметилоксония, к комп¬
лексам иридия, родия и платины могут быть получены катионные
алкильные комплексы металлов, например:fir (СО) (PPh,)гС I] -HCHaSOjF -» [CH3Ir (CO) (PPh3)2CIJ (S03F)Анион S03F~ обладает слабыми донорными свойствами и поэтому не
входит во внутреннюю сферу комплекса. При комнатной температуре
он легко замещается на другой анион, который может входить во внут¬
реннюю сферу, например:[CH3Ir (СО) (PPh3)2CI] (S03F) + [EtaNJ CI-*[CHsIr (СО) (PPh3)2CI2] -|- [Et4Nj S03FОкислительное присоединение молекулярного кислорода. Комп¬
лексы платиновых металлов с низкими степенями окисления централь¬
ных атомов присоединяют молекулярный кислород с образованием
хелатных дикислородных соединений. Например, при взаимодейст¬
вии родиевого и иридиевого комплексов [M(Ph2PC2H4PPh2)2]X, где
X — Cl~, BPlir, dor, PF6“, с молекулярным кислородом образуют¬
ся комплексы [M(Ph2PC,H4PPh2)2(02)]X. Рентгеноструктурным ана¬
лизом установлено, что они имеют следующее строение:Таким образом, образование дикислородных соединений часто можно
рассматривать как реакцию окислительного присоединения с сохра¬
нением одной из двух связей между атомами кислорода. Состояние
окисления центрального атома повышается на две единицы, а моле-Особый тип реакций окислительного присоединения. Необычная
реакция окислительного присоединения протекает в комплексе
flr(PPh3)3Cl]. В фенильном радикале разрывается связь С—Н и об¬
разуется гидридное циклометаллированное соединение, т. е. реакция
является внутримолекулярной:кула кислорода превращается в пероксидный лиганд 0~2 •PPh3Ph3P-Ir—PPI13,^VPPh2ICl341
Однако для трифенилфосфина это не единственный путь реакции
окислительного присоединения. Он участвует в обратимой реакции
по схемеМ—PPh3 = Ph—.М—PPh3где М — Ni(0) и Pd(0).Аналогичная реакция [Pd(PEt3)3] с CeF5PPh2 выражается урав¬
нением[Pd(PEt3)3] C,Fr,PPh2 [(CeF5) Pd (PPh2) (РЕt3)a] -r PEt3Комплекс [Ir(Me„C6H6_n)COL„] в реакции с P(OR)3 образует про¬
дукты, которые являются циклеметаллированными гидридокомплек-
сами иридия (III):-со[1г (МепС,Н5.„) (СО) L2] + ЗР (ОЮз 1-2LHIr(2-CH2C6H5_nMen_,) (Р (OR3)}3где п = 1, 2, 3, a R — Me, Et, Ph.Замещение СО-группы и двух Р-донорных лигандов L в
[Ir(Me„C6H5_n)(CO)L„] тремя Р-донорными лигандами P(OR)3 вызы¬
вает разрыв связи С—Н о-метальных групп и внутримолекулярное
окислительное присоединение к металлическому центру.За последние годы накоплен большой экспериментальный матери¬
ал по кинетике и стереохимии окислительного присоединения, однако
он пока еще не позволяет однозначно сформулировать мнение о меха¬
низмах этих реакций. Наиболее вероятны два варианта механизма.Первый из них предполагает трехцентровое переходное состояние,
образующееся в результате Sn2 механизма для гомополярных окис¬
лителей (где X и Y одинаковые атомы или группа атомов) или для
окислителей, в которых ни X, ни Y не являются сильноотрицатель¬
ными частицами. Этот вариант механизма окислительного присоеди¬
нения можно назвать «присоединение — перегруппировка». Молеку¬
ла XY присоединяется к реакционному центру, затем следует (иногда
одновременно) перераспределение электронной плотности в моле¬
куле с разрывом связи X—Y и образованием связей М—X и М—Y:/VXL„M + XY L„M(XY) —► LnM' 'К —► LnMПрисоединение по такому механизму является стереоспецифичным
и происходит в tjuc-положение.Наиболее типичными примерами являются реакции присоедине¬
ния водорода и непредельных соединений, например:CIОСч yPPh3 ОСч | /РРЬз>.!< + Н2 ^ >1г<Ph3P/ ХС1 Ph3P/ |II342
В другом возможном механизме окислительного присоединения мо¬
лекулы XY (нуклеофильное замещение) предполагается линейное
переходное состояниеэ-f а—М .. - X ... Yи проводится аналогия с реакцией Меншуткина. Скорость определяю¬
щей ступенью реакции является нуклеофильная атака комплекса ме¬
талла на молекулу XY. Ион металла при этом испытывает двух¬
электронное окисление, например:I IРеакции окислительного присоединения иногда рассматривают
как частный случай кислотно-основного взаимодействия комплексов
переходных металлов с апротонными кислотами. Так, например, в
реакции транс-[\т(СО) (PPh3)2Cl] с SnCU вначале образуется аддукт
[Iг(СО) (РРИзЬС!]-SnCli, а затем происходит его перестройка, свя¬
занная с окислительно-восстановительным процессом. В результате
устанавливается равновесие, в котором участвуют две формы:SnCl4 SnCl3Ph3Px I /СО Ph3P4 [ / СОC|/Ir\pph3 ** Cl/V^PPhjCIБиблиографию по реакциям окислительного присоединения мож¬
но найти в статье [3].12.7. Реакции восстановительного элиминирования
с участием комплексов переходных металловВосстановительное элиминирование является важным процессом пре¬
вращения координационных соединений. Формально оно связано с
■отрывом от центрального атома двух лигандов по гемолитическому
типу. В результате этого из лигандов образуются два радикала, а
степень окисления центрального атома понижается на две единицы.
Образовавшиеся радикалы могут объединяться в молекулу или по¬
рознь реагировать с другими молекулами или ионами. В общем виде
наиболее распространенная реакция восстановительного элиминиро-
вання в виде уравнения записывается следующим образом:XMYL„ -► ML„ + XYРеакция восстановительного элиминирования противоположна ре¬
акции окислительного присоединения, и часто эти две реакции взаимо¬СН,
ОСч ! JPPh,/ К
— рьар/ | Vii343
связаны. Наиболее простой пример — реакция комплекса Васка с
водородом:[IrCO(PPh3)oCI] + II2 ^ IHIrH(CO) (PPhd)2CIJРодиевый аналог комплекса Васка обратимо присоединяет хлор:
[RhCO(PPh3)2Cl] -I С1г ^ [CIRhCl (СО) (PPh3)2Cl]Комплекс транс-[РИ(Ме) (PEt3)s] реагирует с избытком метилио-
дида с образованием комплекса [Pth(Me)2(PEt3)2]. При нагревании
последнего в растворе этанола отщепляется Mel и вновь образуется
транс-[Ptl(Me) (PEt3)a],Реакция восстановительного элиминирования часто следует за
окислительным присоединением. Однако эти две реакции не всегда
обратимы. Восстановительное элиминирование может включать раз¬
рыв иных связей, чем те, которые образовались в результате окисли¬
тельного присоединения. В общем виде это обстоятельство записы¬
вается следующим образом:MZn + XY к XMYZ„ >- XZ + MYZn_!окислительное восстановительноеприсоединение элиминированиеНапример, комплекс i<«c-[PhCH2PdBr(Me)2(PPh3)2], полученный из
£{uc-[Pd(Me)2(PPh3)2] и РИСНгВг, отщепляет при комнатной темпера¬
туре этилбензол и дает комплекс mpaHC-lPdBr(Me) (РРЬ3)г] по схемеMe Me CH,Ph PPh3I \ I /PPh3 IMe—Pd—PPh3 + PhCHjBr -*■ Pd' —■ Me—Pd—Br + PhCHoMeI / I 4 IPPh3 Me Br PPh3’ PPh3Окислительное присоединение метилиодида к t{uc-[Pt(Me)2(PMe2Ph)2]
идет с образованием $ac-[PtI(Me)3(PMe„Ph)2], который при пиролизе
отщепляет этан и превращается в транс-[Ptl(Me) (PMe,Ph)J:4«c-[Pt(Me)2(PMe2Ph)2] + MeI фас-[PtI (Ме)3 (PMe2Ph)2] —транс-1 Ptl(Me) (PMe;>Ph)2] +CSH,Встречаются примеры, когда, наоборот, восстановительное эли¬
минирование инициирует последующее внутримолекулярное окис¬
лительное присоединение. Так, комплекс [IrH3(PPh3)3l, теряя водород
при УФ облучении, отнимает его затем у фенильного атома угле¬
рода координированного фосфина. Образующийся таким цикло-
металлированием комплекс родия (III) далее выделяет водород по
схемеh'<[IrH3(PPh3)3] [IrH(PPh3)3] ->/Ч /Ч
phlp/7pph3 РРЬз phjp/ '4ppha344
Процессы окислительного присоединения и восстановительного
элиминирования в присутствии соответствующих лигандов могут за¬
мыкать цикл превращений и делать реакцию в целом каталитиче¬
ской. В качестве примера можно привести реакцию каталитического
декарбонилирования ацилгалогенида в присутствии комплекса
lRhCO(PPh3)sCl]:[RhCO (PPhs)3 Cl] H- RCOX ** [(RCO) RhX (CO) (PPh3)2 Cl] **-COzriT [(RCO) RhX (PPh3)2Cl] [(R) Rh (СО) X (PPh3)2CI] «со** [RhCO (PPh3)2 Cl] + RX
В этой схеме окислительное присоединение ацилгалогенида к комп¬
лексу [RhCO(PPh3)'2Cl] приводит к образованию ацильного комплекса.
При высокой температуре происходит отщепление координирован¬
ного оксида углерода (II). Ненасыщенный ацильный комплекс пре¬
терпевает затем ацил-алкильную перегруппировку. От получающегося
алкильного комплекса восстановительно элиминирует алкилгало-
генид. На этой стадии осуществляется регенерация комплекса
JRhCO(PPh3)„Cl] и замыкание каталитического цикла.Восстановительное элиминирование формально является моно-
молекулярной реакцией. Для него особо важную роль должен играть
растворитель. Известно, что Кг[Рс1С16] в водном растворе подверга¬
ется восстановительному элиминированию в соответствии с урав¬
нениемК, £PdCI6l -» К2 [PdCI4] -|- С1„В хлорированных углеводородах устойчивость комплексов палладия(IV) гораздо выше, чем в воде, поэтому восстановительное элимини¬
рование затруднено.Выделение водорода по реакции{(Н),Со{Р(ОСН3)3}4]+ ** Н2 + [Со {Р (ОСН3)3}4] +в ацетонитриле протекает значительно быстрее, чем в хлористом ме¬
тилене. Одна из возможных причин — образование интермедиата
I(H)2Co(NCCH3) {Р(ОСН3)з}3]+.Экспериментальные измерения энергии и энтропии активации ре¬
акции восстановительного элиминирования этана из цис-бис-(дифенил-
метилфосфин)диметилпалладия (II) в неполярных (бензол) раствори¬
телях привели к более низким значениям энергии активации и более
высоким отрицательным значениям энтропии активации, чем в поляр¬
ных растворителях (диметилсульфоксид, ацетон).Кроме растворителя важную роль в осуществлении реакции вос¬
становительного элиминирования играют внутрисферные лиганды.
Лиганды, способствующие стабилизации комплексов в низких сте¬
пенях окисления, способствуют и реакции восстановительного эли¬
минирования. К их числу относятся, например, оксид углерода (II),
фосфины и олефины. Так, при взаимодействии бис-(тритил)никеля
[Ph3C—Ni—CPh3] с оксидом углерода (II) при комнатной темпера-345
туре путем восстановительного элиминирования с количественным
выходом получаются гексафенилэтан и [Ni(CO)4]:[Ph3C—Ni—CPh3] + 4CO =№ Ph3C—CPh3 + [Ni (CO)4]Активирующее действие фосфина на реакцию восстановительного
элиминирования можно продемонстрировать следующим примером:АгR3P—Ni—PR3 + NaCN -f- 2PR3 ArCN + [Ni (PR3)4] + NaXIXЗдесь R—CeHs, C6Hn; X — Cl, Br, I.Для обратимых процессов восстановительного элиминирования и
окисл ител ьного пр исоединен и я[(Н)о lr (CO)j L2]+ ** [Ir(CO)2L2] + -[- Н2[HIг (OoCR) (СО).LoCIJ *1= [IrCOLs>Cl] + RCOOHсклонность к восстановительному элиминированию в зависимости от
природы L уменьшается в следующем порядке:PPh3 > PMePh2 > PEtPh, > РЕ t,Ph > -РЕ t3 > P(CgН^^)зКатион 1(ис-[(СН3)гАи(РМеР11з)2]+ устойчив в тех же условиях, в ко¬
торых из комплекса qwc-[(CH3)„Au(PPh3)2]+ выделяется этан.Изучение активации связей металл — алкил под действием оле-
финов, вступающих в координацию с атомом металла, на примере
разложения комплексов [(Et)2Fe(Bipy)2] и [(Et)2Ni(Bipy)2] позво¬
лило сделать следующий вывод: активация связи алкил — металл
тем значительнее, чем более электроотрицателен олефин или чем проч¬
нее его связь, образуемая с атомом металла.Процессу восстановительного элиминирования должен способст¬
вовать, в частности, высокий окислительный потенциал комплекса и
большая энергия связи молекулы, образующейся из отщепившихся
лигандов.Механизмы реакций восстановительного элиминирования весьма
разнообразны. Так, в ряде указанных выше примеров восстановитель¬
ное элиминирование проходило лишь в присутствии в растЕоре из¬
бытка свободных лигандов — фосфинов, оксида углерода (11) или
олефинов. Их координация к металлу приводила к образованию не¬
устойчивых координационно перенасыщенных интермедиатов и ос¬
лаблению связей М—И, М—С или М—галоген.Иногда восстановительное элиминирование осуществлялось без
введения дополнительных лигандов, например при термическом раз¬
ложении следующего комплекса:t°[PtCH3Cl, (PMe.Ph)2J — СН3С1 + [PtC!s (PMe,Ph)2]В ряде случаев присутствие свободного лиганда снижает способ¬
ность комплекса к восстановительному элиминированию. Так, при346
исследовании кинетики разложения $ac-[PtI(Me)3(PMe2Ph)2] в 1,4-
дпоксане в соответствии с уравнениемфас-1 PH (Ме)3 (РМе2Рп).>] -v G,H« + транс-1 PtI (Me) (PMe,Ph)2]найдено, что данная реакция (первого порядка) сильно замедлялась
в присутствии свободного лиганда PMe2Ph. Это указывало на то, что
реакция включает стадию диссоциации фосфинового лиганда, кото¬
рая является скоростьопределяющей в элиминировании этана от
пятикоординацнонного интермедиата:
фаг-[PtI (Ме)3 (PMe,Ph),J PMe2Ph [PtI (Me)3 (PMe2Ph)] —— C2He + mpar.c-[PtI (Me) (PMe2Ph),]Замедление восстановительного элиминирования АгСН3 в присут¬
ствии свободного PEt3 наблюдали в никелевом комплексе:[NiАг (СН3) (PEt3)2] РЕta + [NiАг (СН3) (PEt3)J ч*** АгСН3 + [Ni (PFt3)2]Классификация по типам реакций окислительного присоединения
дана в разделе 12.6. Число известных типов реакций восстановитель¬
ного элиминирования гораздо меньше. Анализ позволил выявить
несколько типов таких реакций. Наиболее распространенный из них
включает расщепление двух связей металл —
углерод:RMR'Ln — RR' + MLnЭтот тип превращений позволил получить большое число молекул со
связью С—С (где R — Ph3C, СВН5, СН3, С6Н6, 4-МеС6Н4; R'— Ph3C,
С6Н5, PhCH2, СН3, 4-МеС6Н4, СОСН3). Важное использование таких
реакций связано с образованием циклических соединений из олефи-
нов и некоторых ненасыщенных соединений. Так, в зависимости от
природы лиганда в комплексах никеля были получены циклодекат-
риен-1,5,9, циклооктадиен-1,5, винилциклогексен, 1,2-дивинилцик-
лобутан и 1-винил-2-метиленциклопентан.Приведем еще несколько примеров реакций этого типа. Комплекс
4uc-[Pt(Me)2(PPh3)J при нагревании до 40 °С в хлороформе выделяет
этан:СН3I гCH3-Pd-PPh3 -Н- с,н6 -!- [Pd(PPh3)2]PPh3Выделение этана наблюдали при растворении в бензоле при ком¬
натной температуре комплексов золота (III);[(СН3)3 AuPPh3] ч- [CH3AuPPh3] +С2Н6Выделение водорода из гидридных комп¬
лексов переходных металлов также является одним из распростра¬
ненных типов реакций восстановительного элиминирования:HMHLn Н, + ML„347
Как правило, это реакции, обратные окислительному присоедине¬
нию водорода. Ключевым фактором в стабилизации связи М—Н яв¬
ляется большая электронная плотность на центральном атоме. Сле¬
довательно, равновесие в такой системе будет смещаться вправо при
наличии лигандов, являющихся слабыми о-донорами и сильными л*
акцепторами.Характерной реакцией восстановительного элиминирования яв¬
ляется отщепление углеводородов от алкил-
гидрндных комплексов. Реакция, которую можно за¬
писать в общем виде как
RMHLn RH + ML„протекает, как правило, необратимо. Например![СН3Со (Н) {Р (OEt),}4]+-*- СН4 + (Со {Р (OEt)3}4] +2 [CH3OS (Н) (СО)4] -v СН4 + rHOs (CO)4Os (СО)4 СН3] —2РЕ t3 » 2СН4 + 2 [Os (СО)4 (PEt3)]Выделение метана из ^«c-[Os(H) (СО)4СН3] с образованием
[Н0з(С0)40,;(С0)4СНз] требует участия двух молекул комплексного
соединения, несмотря на 1<ис-расположение метильного и гидридного
лиганда. Введение в раствор нуклеофильных лигандов, таких, как
Р(ОСН3)3 и PEt3, позволяет образовываться метану из каждой моле¬
кулы комплекса.Формиатный комплекс [Ru(R) (r]2-02CH) (СО) (PPh3)2] в присут¬
ствии PPh3 или Ph2PCH2CH2PPh2 в растворе третичного бутанола
подвергается реакции декарбоксилирования с одновременным восста¬
новительным элиминированием и образованием RH:HCOONa PPhs[Ru (R) (CO) (PPh3>.cU * [Ru(R) (т-02СН) (CO) (PPh3)2] -Cl- -C02. -RH[Ru (CO) (PPh3)3JОбразование связи углерод — галоген так¬
же можно рассматривать как отдельный тип реакций восстановитель¬
ного элиминирования:RMXLn RX + MLnВ основном это реакции, обратные окислительному присоединению
алкил- или бензилгалогенидов.При термическом разложении д и а л к и л ь-
н ы х соединений [(CH3)2Co{P(OEt)3}4]+ и [(CH3)20s(C0)4] про¬
исходит отщепление метана. Реакции в общем виде можно выразить
уравнением(СН3)2MLn ->- СН4 + CH2ML„где L — P(OEt)3, СО.Элиминирование альдегида из металлацильных
гидридных комплексов является ключевой ступенью в оксосинтезе:
(RC=0) М (Н) Ln — RCHO + ML„
ГЛАВА13РЕАКЦИОННАЯ СПОСОБНОСТЬ
КООРДИНИРОВАННЫХ ЛИГАНДОВИзучение реакционной способности координированных лигандов яв¬
ляется одним из важных направлений современной координационной
химии. При комплексообразовании лиганда часто утрачиваются ха¬
рактерные для него свойства, в ряде случаев резко изменяется скорость
и механизм тех или иных реакций, а иногда проявляются новые свой¬
ства, совершенно нехарактерные для лигандов в некоординирован¬
ном состоянии. Например, аммиак, войдя в комплекс, теряет основ¬
ные свойства и проявляет кислотные (см. гл. 10).Одним из первых химиков, которые подняли вопрос об изменении
свойств лигандов при их вхождении в комплекс с ионами металлов,,
был Гринберг (1927).Вопрос изменения реакционной способности лигандов при комп¬
лексообразовании имеет прямое отношение к проблемам гомогенного
катализа под действием комплексных соединений. Вероятно, это
является одной из,причин большого интереса к данному направле¬
нию исследований в химии комплексных соединений. В настоящей
главе рассмотрены некоторые типы реакций координированных ли¬
гандов и факторы, влияющие на изменение реакционной способности
лигандов при координации.13.1. Расщепление координированных лигандовЛ. А. Чугаев предложил интересный способ синтеза триаминовых
комплексов платины '(II)- На ^uc-[Pt(NH3)2Cl2] действуют цианатом
калия с последующим подкислением прогретого раствора соляной кис¬
лотой. Чугаев считал, что цнанат-нон входит во внутреннюю сферу
комплекса, а затем превращается в карбаминовую кислоту. Схема¬
тично эти реакции можно изобразить следующим образом:NH,PtClCIKNCONH3 NCO
NH3 CIh:o |"NH3
NH,PtNHCOOHClrtCI"NH3NHoCOOHlrNH,NH3 ф.Pt 'C! —Pt 310NH3CIVNH3CI .Cl + CO*Протекание реакции через стадию гидролиза некоординированной
циановой кислоты с последующим внедрением в комплекс продукта
гидролиза (аммиака) маловероятно, так как в кислой среде аммиак
должен превратиться в ион аммония.Аблов и Самусь получили большое число диоксимных комплексов
кобальта (III), содержащих цианатную группу [Co(Dioxim)2X(NCO)]~34&
<Х — Cl", Вг, Г, N02,NC0”), и показали, что в кислой среде внутри-
сферные группы NCO- количественно могут быть превращены в аммиак.
Балахура и Иордан изучили кинетику превращения цианатного комп¬
лекса по реакции(Со (NH3)5 NCOP + НзО = [Со (NH3)e]3+ + СО-.Сравнение кинетических параметров этой реакции с кинетическими
параметрами гидролиза свободной циановой кислоты показало, что
константа скорости гидролиза координированной группы на порядок
меньше, а энергии активации практически одинаковы.Превращение цианатной группы в аммиак в комплексах родия
(III) и рутения (III) [M(NH3)5NCO]'“+ было осуществлено Фордом.
Ему удалось выделить промежуточный комплекс состава
[Rh(\’H3)5(NH.:COOH)]3+. Таким образом, Форд подтвердил взгляд
Чугаева на механизм гидролиза координированной цианатной группы.Балахура и Иордан показали, что мочевина и ее производные мо¬
гут быть превращены в комплексе кобальта в цианатный нон по ре¬
акцииNH,V(Со (NH3)i Н20]-1+ + С=0 — [Со (NH3)5 NCO]2+ + RH + Н+ -- Н„0Вернер впервые установил, что координированный роданидный ион
в комплексе кобальта (III) под действием пероксида водорода превра¬
щается в аммиак. Он использовал это для аргументирования коор¬
динации роданидной группы к кобальту посредством атома азота.
Более поздние исследования показали, что превращение роданидной
группы в аммиак под действием окислителей является не единствен¬
ным направлением реакции. По другому пути роданидная группа пре¬
вращается в цианндную. В зависимости от природы окислителя соот¬
ношение между продуктами реакции меняется довольно сильно.13.2. Превращения внутрисферной нитрогруппыИзвестно, что в растворе свободной азотистой кислоты имеют место
следующие равновесия:NO+ + Н20 ^ HN02 + Н+HNO, Н+ + N07Одним из нитрозильных комплексов является нитропруссидный ион
[Fe(CN)5NO]2~. Кольтгофф и Торрен показали, что в растворе уста¬
навливается равновесие:Fe (CN)6 (NO)3- + ОН- *s Fe (CN)5 (N02)4- + H+Они экспериментально определили константы скорости прямой и
обратной реакций и вычислили константу равновесия.350
Машек пришел к заключению, что это равновесие включает также
неустойчивый интермедиат [Fe(CN)6(N02H)]3'. Таким образом, коор¬
динированные нитрозильная и нитритные группы связаны между
собой равновесием, характерным для этих лигандов в свободном со¬
стоянии.Черняев с сотр., а также Левитус и Раскован действием соляной
кислотой на изомерные комплексы [Pt(NH3)2(N02)2] получили и оха¬
рактеризовали нитрозосоединения состава [Pt(NH3)2(N02)(N0)Cl2].
При взаимодействии соляной кислоты с [Pt(NH3)2(N02)Cl] получен
комплекс [Pt(NH3)2(NO)CI3]. Позднее Муравейская с сотр.
произвела аналогичные превращения изомерных комплексов
({«c-[Pt(NH2CH3)2(NC>2)2] и m/f70Hc-[Pt(NH2CH3)2(N02)2], а также
[Pt En(NCh)2]. Подобного рода нитрозильные соединения Стеценко
получала легко и с хорошим выходом Взаимодействием аминатов пла¬
тины (II) с нитритом калия в солянокислой среде.В 1970 г. Мейер, Годвин и Уинтертон установили, что превращение
нитритной группы в нптрозильную в дипиридильных комплексах
рутения (II) осуществляется без разрыва связи рутений—азот. Спект¬
рофотометрическим методом они показали, что переход из одной фор¬
мы комплекса в другую происходит обратимо и количественно в за¬
висимости от pH раствора:[Ru(Dipy)2N02X] + 2Н+ ** [Ru (Dipy)3 (NO) X]2+ + H20Кукушкину и Данилиной действием ионами водорода на комплекс
родия [Rh(PPh3)2C0(N02)] удалось превратить внутрисферную нитро¬
группу в нитрознльную. В процессе этого превращения молекула СО
замещается на анион кислоты, имеющийся в растворе. Наиболее ве¬
роятным представляется протекание реакции по следующим стадиям:2Н+[Rh (СО) NO, (PPh3)2] [Rh (CO) NO (PPh,)2]2+ + HaO[Rh (CO) NO (PPh3)2]2+ + 2X- [Rh (NO) X2 (PPh3)2] + COДействием хлорной кислоты на комплекс [Rh(N0)N02(PPh3)2] полу¬
чено динитрозильное соединение [Rh(N0)2(PPh3)2]C104. Этот комп¬
лекс мало устойчив и в этанольной среде в присутствии избытка три-
фенилфосфина легко переходит в мононитрозильный комплекс не¬
электролитного типа [RhN0(PPh3)3].Своеобразную реакцию Зинина на комплексных соединениях
провел Черняев. Он показал, что внутрисферная нитрогруппа в комп¬
лексах платины (II) при взаимодействии с водородом в момент выде¬
ления без разрыва связи платина — азот может быть превращена в
аммиак. Подобным же образом в аммиак был превращен внутрисфер-
ный гидроксиламин.Черняев н Геннинг установили, что нитрогруппа в комплексах
двух- и четырехвалентной платины может быть восстановлена хлори¬
дом аммония до элементарного азота. Реакция идет настолько легко,
что авторы предложили использовать ее для количественного опре¬
деления азота, входящего в комплекс в виде нитрогруппы. Наиболее
вероятным путем протекания этой реакции, по мнению Черняева, яв¬351
ляется промежуточное образование диазосоединения по схеме—2Н,0Pt-N02 |- NH4CI *- Pt—N=NCI — Pt—Cl + N2Однако лучшим способом устранения из комплексов внутрисфер-
ной ннтрогруппы является реакция с сульфаминовой кислотой. Нит¬
ритный ион количественно взаимодействует с сульфаминовой кисло¬
той по уравнениюN0,7 + NH2S03H = N2 -|- HSO“ + Н20Этот путь был использован для удаления заданного числа нитрогрупп
из комплексов Pt(II), Pd(II), Rh(III), например, по реакции[Pt (N02)4]2- + 2NH2S03H = [Pt (H30)2 (N03)j] -г 2N2 2HS0"13.3. Реакционная способность координированной
нитрозильной группыРентгеноструктурные исследования показали, что лиганд N0, коорди¬
нируясь к переходным металлам, образует в основном два типа комп¬
лексов. Первые содержат линейные группировки М—N0 с валентным
углом, близким к 180° (линейные нитрозилы), вторые имеют фрагмент
М—N0 с углом связи порядка 120—130° (изогнутые нитрозилы).
Электронная плотность в изогнутых нитрозильных группах больше,
чем в линейных. В химических свойствах это проявляется в том, что
нитрозильный лиганд во многих комплексах с линейной группиров¬
кой М—N0 реагирует с большим числом нуклеофильных реагентов,
в то время как в комплексах с угловой связью —N0 координиро¬
ванный ннтрозил вступает во взаимодействие с веществами электро-
фильного характера.13.3.1. Электроноакцепторные свойства нитрозильной
группыКак уже было сказано в предыдущем разделе, при взаимодействии с
гидроксильными ионами координированная нитрозильная группа
может быть превращена в нитритную. Еще в 1895 г. Гофман устано¬
вил, что взаимодействие нитропруссида натрия с гидразином в вод¬
ном растворе происходит с бурным выделением азота. Суммарное урав¬
нение реакции:4Na2 [Fe (CN)5 NO] + 3N2H4 + 4NaOH = 4Na3 [Fe (CN)* H,0] + 5N2 + 4Ha0При взаимодействии с другим нуклеофильным реагентом — гндр-
оксиламином реакция протекает по уравнениюNa: [Fe (CN)6 NO] + NH2OH + NaOH = Na3 [Fe (CN)5 H20] + N20 + H20Реакция между нитрозильной группой нитропруссида и аммиаком
приводит к образованию молекулярного азота, который замещается
следующей молекулой аммиака:
Na, [Fe (CN)5 NO) -r 2NH3 + NaOH = Na3 [Fe (CN)6 NH3] + N2 -) 2H.20Интересна реакция между нитропруссидным и азидным ионами.
В водном растворе она протекает в соответствии с уравнением[Fe (CN)6 NO]-- + N- + M..0 = [Fe (CN)6 H20]3- + N20 -[- N,Приведенные примеры иллюстрируют проявление координиро¬
ванной нитрозильной группой электроноакцепторных свойств. Эти
же свойства нитрозильной группы проявляются и в реакциях ее вос¬
становления.Принципиально нитрозильный лиганд может быть восстановлен
до различных соединений: NO-, N,, NH2OH, N2H4, NH3.Восстановление координированной нитрозильной группы в аммиак
осуществлено в комплексе [Ru(NH3)5NO]3+ ионами Сг(П) по реакции
[Ru (NH3)sNO]3+ + 6Cr2+ + 5Н+ = [Ru (NH3)„]2+ -|- 6Cr3+ + H20Электрохимическим путем удалось восстановить нитрозильную
группу в нитропруссидном ноне в гидроксиламин по схемее~ Н+ е~Fe (CN)b NO2" =** Fe (CN)b NO3- Fe (CN)S (NOH)!-s± Fe (CN)5 (NOH)3- —2c- 2H*- ► Fe (CN)6 (NH2OH)3"Боттомлей с сотр. высказал суждение о наличии взаимосвязи меж¬
ду v(NO) или, более точно, силовой постоянной F^o и электроно¬
акцепторным поведением координированного нитрозила по отноше¬
нию к различным нуклеофилам. Если v(NO) выше 1886 см-1, a Fno
1,4-103 Н-'м, то такие комплексы с большой степенью вероятности
должны реагировать с электронодонорными реагентами. При v(NO)
и Fm меньше этих значений взаимодействие с нуклеофильными
реагентами маловероятно. Такая взаимосвязь дает возможность пред¬
полагать проявление электрофильной способности у нитрозильной
группы, хотя и не является совершенно однозначной.13.3.2. Проявление нитрозильной группы
электронодонорных свойствЭлектронодонорные свойства координированного нитрозильного ли¬
ганда проявляются в двух типах реакций: в протонировании с об¬
разованием координированного нитроксильного (HNO) или гидро-
шгламинового лигандов и в окислении. В роли окислителя часто
выступает кислород или оксид азота N0.Протонирование координированного нитрозильного лиганда с
образованием HNO доказано на примере комплексов [Fe(CN)6(HNO)]“,
[Co(Das).,Br(HNO)]2+ (где Das — о-фенилен-бис-диметнларснн) и др.При действии избытка хлороводорода на родиевый комплекс
[Rh(PPh3)3NO] образуется гидроксиламиновый комплекс родия (III):[Rh (PPh3)3NO] + ЗНС1 = [Rh (PPh3)2CI3 (NH..OH)] + PPh3Аналогично ведет себя иридиевый комплекс [Ir(PPh3)3NOj.]/,13—817353
Окисление кислородом нитрозильных лигандов приводит к об¬
разованию либо нитритного, либо нитратного лигандов. Троглер и
Маззилли пришли к выводу, что критерием для предсказания про¬
дуктов окисления нитрозильного лиганда может быть частота ва¬
лентного колебания связи N—О. Если комплексы имеют v(NO)~16(X)—
1700 см'1, то при окислении они дадут нитросоединения. Комплексы
с v(NO)« 1710—1765 см'1 будут приводить к образованию нитрат¬
ных соединений. Однако этот критерий не может рассматриваться как
совершенно строгий, так как уже сейчас имеются факты, которые не
согласуются с ним.Для более подробного ознакомления с фактическим материалом
и библиографией по вопросам, изложенным в данном параграфе, мож¬
но рекомендовать статью [1].13.4. Окислительное дегидрирование
алифатических аминовПринципиально окисление алифатических аминов (окислительное
дегидрирование) может протекать по уравнениям
RCH2—NH2 RCH=NH + Н2
RCH=NH RCe-N + н2Координированные алифатические амины способны подвергаться
таким реакциям в присутствии окислителей. Кукушкин с сотр. по¬
казал. что молекула этилендиамнна в комплексе [PtPy ЕпС!3]С! при
взаимодействии с хлором превращается в аминоацетальдимин по урав¬
нению[PtPy (NH2CH2CH2NH2) Cl3]+ + С12 — [PtPy (NH2CH2CH=NH) Cl3]+ + 2НС1Вслед за этим Басоло с сотр. установил, что взаимодействие с иодом
комплекса [Ru Еп3]2+ описывается схемой[RuEn3]2+ IX [RuEn2 (NH=CHCH=NH)]2+Аналогичному превращению под действием гипохлорита натрия под¬
вергается этнлендиамин в комплексах [Fe(NH2CH2CH2NH2)(CN)4]- и
[Ru(Phen)2(NH2CH2CH2NH2)]2+.Для превращения аминов в имнны может быть использовано элект¬
рохимическое окисление на аноде.В ацетальдегидном растворе осуществлено превращение в
альдимин координированной молекулы аммиака в комплексе
[Pt(NH3)2(NCl2) (N02)2C1]C1 по схемеPtIV-NH3 + СН3СНО — PtIV-NH=CHCH3При наличии в комплексе хлораминного хлора внутрисферный аль¬
димин далее подвергался окислительному дегидрированию с обра¬
зованием ацетонитрила. Дихлорамидный лиганд при этом превра¬
щался в аммиак. В суммарном виде эта реакция может быть записана
следующим' образом:354
[Pt (NH3)2 (NCI*) (NOj), CI] Cl + CH3CHO == [(CH3CN) Pt (NH3), (N02) CI] Cl + неюИзвестны работы, в которых окислительным дегидрированием
метиламиновый комплекс [Ru(NH2CH3)e]2+ превращен в цианидный.
Реакция проводилась путем барботирования воздуха через раствор
амннокомплекса. Координация метиламина к рутению резко облег¬
чает этот процесс.Окислительное дегидрирование метиламина было проведено также
в комплексе [Ru(NH3)6(NH2CH3)]2+. На основании данных ИК спектро¬
скопии сделан вывод, что в процессе окисления образуется комплекс
[Ru(NH3)6NCH]2+, который затем перестраивается в [Ru(NH3)fiCN]+.
Координированный бензиламин в комплексе [Ru(NH3)6NH„CH2Ph]3+
путем окислительного дегидрирования превращается в бензонитрил.
Процесс превращения бензиламина в бензонитрил протекает через
промежуточную стадию образования имина. Интересно, что окис¬
лительное дегидрирование циклогексиламина в комплексе такого же
типа останавливается на стадии образования иминного соединения.Химическим и электрохимическим путем внутрисферные амины
А (аллиламин, бензиламин и бутиламин) в комплексах [Ru(dipy)2A2]2+
превращены в соответствующие нитрилы. Считают, что процесс на¬
чинается с окисления центрального атома до рутения (III) и лишь за¬
тем происходит превращение аминов в нитрилы.Библиографию по изложенным вопросам можно найти в работе [2].13.5. Превращение внутрисферных нитрилов
в амидиныВ 1907 г. Гофманн и" Бугге сообщили о синтезе комплекса
PtCla-2CH3CN. Чугаев и Лебединский воспроизвели синтез этого комп¬
лекса, получили его изомерную форму и изучили взаимодействие
обоих изомеров с аммиаком. В результате были получены необычные
соединения, имеющие состав [Pt 2CH3CN • 4NH3]C12 . Молекулы ацето¬
нитрила и аммиака в этих изомерных соединениях входят во внутрен¬
нюю сферу комплекса. Об этом свидетельствовали их молекулярная
электропроводность и тот факт, что при взаимодействии с хлороплати-
нитом калия или пикриновой кислотой получались комплексы[P(2CH3CN • 4NH3J [PtCI4] И [Pt2CH3CN • 4NH3] (C0H2N3O7)2Работа Чугаева и Лебединского послужила Ганчу и Розенблату по¬
водом для заключения, что комплексы платины (II) тетраминового
типа [PtA4]X2 имеют октаэдрическое строение и в кристаллическом
состоянии комплекса ионы X" занимают две вершины октаэдра.Продолжение исследований нитрильных комплексов платины (II)
и продуктов их взаимодействия с аминами привело Лебединского и
Головню к заключению, что эти «лишние» молекулы аммиака присо¬
единяются к центральному атому через посредство ацетонитрила, т. е.
координационное число платины равно 4. Формулы соответствующих
комплексов они записывали следующим образом:V.13»355
NH3 pt NCCH3 ■ ■ ■ NH3r-+ 'NH, • • ■ CH3CN NH3NH3 NCCH3 ■ ■ ■ NH3J NH3 CH3CN ■ ■ ■ NH3Несмотря на это еще в 1950 г. Сиджвиг, перечисляя свойства комп¬
лекса [Pt(NCCH3)2(NH3)4]Cl2, утверждал, что в этом соединении пла¬
тина имеет координационное число, равное 6 [3].В 1951 г. Гринберг и Гильденгершель высказали мысль, что амино-
нитрильные комплексы платины (I I) представляют собой соединения
с внутрисферными амидинами.Нитрилы взаимодействуют с аммиаком или органическими амина¬
ми по уравнению.NHRCa-=N + NH3 ** RC<fXNH3Положение равновесия зависит от радикала R — чем сильнее он от¬
тягивает на себя электроны, тем больше равновесие смещается вправо.
Например, взаимодействие аммиака с нитрилами ci3CC=N и F3CC3=N
протекает быстро и почти количественно. Однако степень прев¬
ращения алкилнитрилов в амидины в обычных условиях неболь¬
шая. Синтез амидинов из нитрилов и аминов наиболее эффек¬
тивно протекает в присутствии катализатора Фриделя—Крафтса.
Можно допустить, что механизм каталитического синтеза включает
образование амидинов во внутренней сфере комплекса алюминия.Предположение Гринберга и Гильденгершеля представлялось
весьма вероятным. Однако в 1957 г. Харрис и Стефенсон
на основании рентгеноструктурного исследования комплекса
[Pt(CH3CN)2(NH3)4]Cl2- НаО пришли к выводу, что комплексный ка¬
тион имеет октаэдрическое строение, а лигандами являются две мо¬
лекулы ацетонитрила и четыре молекулы аммиака. Молекулы ацето¬
нитрилов связаны не через атом азота, а посредством тройной связи
Q=N по я-типу.Вопрос о строении «аномальных» аминонитрильных комплексов
платины (II) был решен Харитоновым. При исследовании ИК спект¬
ров комплексов состава [Pt(RCN)„A4l [PtCl,] (где R —это СН3, СН5,
С3Н7, С4Н9, С6Н5; А — NH3, CH3NH.,, NHaC2H4NH2) и их дейтери-
рованных производных было установлено, что они полностью согла¬
суются с гипотезой Гринберга и Гнльденгершеля. В спектрах всех
изученных соединений отсутствовала полоса v(C = N). В области
около 3400 см-1 обнаружены интенсивные полосы, которые были
отнесены к валентным колебаниям связей N—Н некоординированных
амино- или нминогрупп. В ИК спектрах были обнаружены новые
полосы, которые отсутствовали как в аммиачных, так и в нитрильных
соединениях, а именно — интенсивный дублет около 1600 см'1. Вы¬
сокочастотная компонента этого дублета мало изменяет свое положе¬
ние и интенсивность после дейтерировання по связи азот—водород.
На этом основании был сделан вывод о наличии связи C=N.После работ Харитонова Стефенсон повторил исследование струк¬
туры комплекса [Pt(CH3CN)o(NH3)4’|CL,■ Н,0 и пришел к заключению,
что комплексный ион имеет следующее строение:356
NH,СН3 -С NI I3
^ IHN-Pt—NH CH3INH3 Сг!наПосле того как на комплексах платины (II) была выяснена природа
продуктов, получающихся при взаимодействии внутрисферных нит¬
рилов с аминами, аналогичные реакции стали осуществлять на комп¬
лексах других металлов. Так, была осуществлена реакция первичных
ароматических аминов с комплексом [Re(NCR).,Cl4]. В неводных раст¬
ворителях при комнатных температурах был получен ряд амидиновых
комплексных соединений [Re{RC(NH)NHAr},C!.,], где Аг — /1-СсН4Ме,
и/-СвН4(С02Е1), и R—Me, а также Аг — л-С6Н4(ОМе) и R^=Ph.В нейтральном или слабощелочном растворе комплекс
цис-[Со En2(NH2CH.,CN)Cl]’+ подвергается превращению, в котором
аминогруппа этилендиамина взаимодействует с некоординированной
нитрильной группой лиганда NH2CH2CN с образованием амидина.
Реакция протекает с очень большой скоростью. Ее полурериод состав¬
ляет около 4 с. В результате внутри комплекса синтезируется три-
дентатный лиганд по схемеН,С-‘IHjN-СН-I ■NH,\ !Со/INC=NNH—СН,HnC-H:N
\ I x'
Co/ I >■CH2 NH2
I /
N=^=CNHa-CHaХарактер виутрисферного превращения этого комплекса подтвер¬
жден рентгеноструктурным методом.Превращение нитрилов в амидины может протекать под дейст¬
вием гидразина. В этанольном растворе при взаимодействии
mpaHC-fPtfNCBu^oClo] с гидразингидратом образуется комплексНNHnNCl Bu‘\ I /C=NH—Pt—NH=C/ I \Bul Cl NNHoHВзаимодействие аминов типа ArNH2, ArRNH, RR'NH с комплексом
qw>[Pt(o-CH2C0H4CN) (PPh3)2].,(BF4)2 при комнатной температуре в
ДихлорМетане протекает количественно с образованием комплекса12-817357
Этим путем было получено большое число амидиновых комплексов.Следует подчеркнуть, что превращение внутрисферных нитрилов
в амидины протекает в мягких условиях и количественно. Можно
думать, что эта реакция имеет широкораспространенный характер.
Облегчение протекания реакции при координации нитрила, вероятно,
связано с уменьшением электронной плотности на нитрнльном атоме
углерода вследствие ее смещения к центральному атому. Атом угле¬
рода становится более доступным для нуклеофильной атаки аминами.Когда вопрос выяснен, то часто становится очевидным, что реше¬
ние напрашивалось само. Так, вслед за работой, в которой был син¬
тезирован комплекс [Pt(NCCH3)2Cl2], Гофман и Бугге изучили его
поведение в воде в присутствии AgN03 и Ag„S04. Они быделили из
водного- раствора соединение, в котором внутрисферный ацетонитрил
был превращен в ацетамид. По существу реакция гидролиза ацето¬
/нитрила до ацетамида аналогична превращению нитрилов в амидины.13.6. Взаимодействие внутрисферных нитрилов
с водой, щелочами и спиртамиГидролиз нитрилов как в щелочной, так и в кислой среде является
важным процессом синтеза карбоновых кислот. Считают, что реакция
идет в две стадии по схемен,о н,оRCN *- RCONH, —► RCOoH + NH3Обычно стадия гидролиза амида протекает быстрее, чем стадия
гидролиза нитрила. Однако разработаны способы получения амидов
из нитрилов с хорошим выходом. Роль катализаторов в этих процес¬
сах выполняют щелочь или кислота. Предполагаемый механизм ката¬
лизируемой щелочью реакциимедленно +НгОRC=N + OH“ RC<f RCr - ОН'хОН Х)Н.NH ,NHRC<f -* RO
хОН<оКак уже было сказано, гидролиз внутрисферного ацетонитрнла
впервые осуществлен Гофманном и Бугге. Они установили, что полу¬
ченный комплекс [Pt(NCCH3)2Cl2] в водном растворе в присутствии
солей AgN03 или Ag2S04 превращается в синее вещество состава358
pt(\HCOCH3)2-Н30. В литературе оно было названо «платиновой
спныо». Вопрос о степени окислення платины в продукте гидролиза
ацетонитрильного комплекса несмотря на многочисленные исследо¬
вания остается нерешенным. Не вызывает сомнения лишь то, что
продуктом гидролиза ^ординнрованного ацетонитрила является ацет-
амид.Как уже было сказано в разделе 13.5, хлорндный комплекс
цис-[Со En2(NH2CH2CN)CI]2+ подвергается быстрому превращению в
амидиновое соединение. Почти одновременно было установлено, что
хлорндный и бромидный аналоги цис-[Со En2(NH2CH2CN) Х]2+ в
водном растворе и в присутствии ионов Hg2+ превращается в комплекс¬
но En2(NH2CH2CONH2)P+. Вероятно, ионы ртути в этой реакции иг¬
рают роль катализатора.Исследования кинетики гидролиза алкил-, арил- и пиридилнит-
рплов в комплексах одного и того же типа [M(NH3)6NCR]:i+ (где М —
Со3+, Ru3+, Rh3+, Ir3+) свидетельствуют о том, что гидролиз протекает
с образованием амидов соответствующих кислот. Скорость гидролиза
внутрисферных нитрилов на несколько порядков выше,чем свободных.
Вероятно, как и в случае образования амидинов, это происходит по¬
тому, что при координации нитрила имеет место смещение электрон¬
ной плотности к центральному атому, и нитрильный атом углерода
становится более доступным для атаки нуклеофильными реагентами
Нг0 и ОН".Каталитическое влияние ионов переходных металлов на гидролиз
органических нитрилов изучалось многими исследователями. Весьма
вероятно, что механизм этих реакций включает образование нитриль-
ных комплексов с последующим превращением внутрисферных нит¬
рилов в амиды соответствующих кислот.Особое значение имеет исследование Бекка. Он синтезировал комп¬
лекс меди (I) с дицианом и установил, что при его кипячении в 1 М НС1
в течение 6 ч в растворе появляется глицин. Реакция может быть
выражена уравнением3NCCN -f- 6НоО = H2NCH2COOH -f 4HNCO -f NH3Пока трудно сказать, идет ли превращение дициана в глицин внутри-
сферно или по иному механизму. Вне зависимости от этого превраще¬
ние дициана в глицин могло быть важным этапом на пути образования
белковых веществ на Земле.Изучено превращение координированных нитрилов в иминоэфиры.
Установлено, что в спиртах и в присутствии AgBF4 реакция протека¬
ет по уравнениютранс-[MePt (NCC6FS) (PhMe.,P).,]+ -f MeOH + AgBF4 ->-*■ nipcHf-[MePt (NH=C (OMe) C0F5} (PhMe2P)2] BF4 -f Ag+В ИК спектре полученного комплекса отсутствует полоса ч (c=N),
но зато имеется v(C=N) up и 1653 см'1. В спектре ПМР имеется сиг¬
нал протона группы NH.12**359
При обработке в бензоле комплекса mpaHC-[(CcCl6)NiL2(NCR)]C104
(где L — PPhMe3, PPh2Me; R — Me, Ph, CH2Ph) метанолом или эта¬
нолом в присутствии трнэтиламина образуются иминные комплексы
типа mpaHC-[(C6Cl6)NiL2(NH =CR0R')]C104. Суждение о строении
получающегося комплекса сделано на основе данных ПМР. Триэтил-
амин в этой реакции играет роль катализатора. Напомним, что тре¬
тичные амины не взаимодействуют с нитрилами с образованием амн-
динов.Подробную библиографию по этим вопросам можно найти в статье[2].13.7. Превращение координированных нитрилов
в N-ацилзамещенные амидыТ. Н. Сумарокова с сотр. установила, что координация нитрилов к
олову (IV), сурьме (V) и цинку (II) способствует их взаимодействию
с карбоновыми кислотами. При этом образуются N-ацилзамещенные
амиды [4, 5]. Реакция описывается уравнениемSnX4 • RCN + R'COOH ->• SnX4 • RCONHCOR'Например, в присутствии SnX4 в бензоле, хлорбензоле или дихлор¬
этане она протекает всего лишь при 70—90° С. Выход N-ацилзаме-
щенных амидов составляет от 50 до 95%. В отсутствие SnX4 макси¬
мальный выход продукта даже при более высоком нагревании не
превышает 15%, причем он загрязнен побочными соединениями.Т. Н. Сумарокова считает, что карбоновая кислота присоединяет¬
ся к координированному нитрилу по тройной связи:НRCs=N • SnCI4 + R'COOH-»• RC (R'COO) = N • SnCI4Вслед за этим протекает стадия изомеризации координированного
изоамида в N-ацилзамещенный амид по схемеНIR N : SnCl4
СI ..О О:\ /С
IR'Органические роданиды RSCN, подобно нитрилам, также коор¬
динируются у олова (IV) посредством атома азота. Аналогичным ока¬
залось и их взаимодействие с карбоновыми кислотами [5]. Реакции
протекают с образованием N-ацилалкилтиокарбаматов. Выходы про¬
дуктов 'лежат в пределах 45—80%.НI— R—С—N—С—R'!l II
О О
\ ✓SnCI4360
13.8. Изменение реакционной способности
в результате смещения электронов от лигандов
к центральному атомуПфейффер с сотр. осуществили переэтерификацию эфирной части
шпффова основания, координированного у иона меди (II). Изученные
реакции переэтерификацин внутри комплекса происходили с гораздо
большей скоростью, чем в случае свободных эфиров. Например, в
комплексесн,о—сосн3
ос—осн3растворенном в бутиловом спирте, в течение 10 мин происходит за¬
мена метоксигруппы на бутоксигруппу.Значительно позже Вертер и Фрост продолжили исследование
Пфейффера с этим и подобными соединениями. Наряду с большим чис¬
лом реакций переэтерификацин внутри комплексов нм удалось осу¬
ществить амидирование кдординнрованных оснований Шиффа, в
частности, заменить метоксигруппу СН30 на бутиламинную группу
C4H9NH.Существует два взгляда на механизм этих реакций. Мартел и Кель¬
вин объясняют такое большое ускорение переэтерификацин смеще¬
нием электронов от карбонильного углерода по направлению к атому
меди вследствие донорно-акцепторной связи лиганда с центральным
атомом. Это смещение должно облегчать взаимодействие электроно-
донорного атома кислорода молекулы растворителя с углеродом кар¬
бонильной группы:I\и \а у + ROH7 N / / / VNo No Not || I I ;—■; I II—CH—C—OR CH—С—]OR;R—О—H R—О—jHor'Механизм реакции, предложенный Вертером и Фростом, предпо¬
лагает взаимодействие лигандов в комплексе по схеме361
Предполагается, что карбонил сложноэфирной группы атакуется
атомом кислорода альдегидной части другого лиганда с образова¬
нием промежуточной структуры типа лактона. Эта реакция должна
быть обратимой, однако при большой массе спирта-растворителя она
преимущественно идет в одном направленииКролль изучал каталитическое действие двухвалентных ионов
тяжелых металлов на гидролиз эфиров аминокислот, которые зна¬
чительно ускоряют эти реакции. По мнению Кролля, каталитическое
ускорение гидролиза эфиров аминокислот схематично можно описать
следующим образом:М2+ 'г HjNCHCOOR'*- R -2+- R чНС-С—OR'з=£Xп-10
+1ояэ1 111 1H3N Оh2n оW\/_ м _мон-1аf OR' -+- R -|НС—с<НС—с=о1 1 ХОН1 1HnN ОH„N Оч/' \/- М -- М -IIIII+ R'OHПредложенный механизм основан на том принципе, что лиганд
в координированном состоянии(комплексы I и 1а) проявляет большее
сродство к гидроксильному иону, чем свободный эфир. Иными слова¬
ми, в результате координации увеличивается электрофильность реак¬
ционного центра. Промежуточный неустойчивый комплекс II распа¬
дается на спирт и аминокислотный комплекс (III), при диссоциации
которого замыкается цикл каталитических реакций. Для ряда двух¬362
валентных катионов Кролль установил соответствие между тенденци¬
ей к комплексообразованию и каталитическим эффектом этих ио¬
нов.Каталитическое влияние Си(II) на гидролиз и изотопный обмен
кислорода в эфирах аминокислот исследовали также Бендер и Тарн-
квист. Меривезер и Вестхеймер изучали каталитическое влияние
ионов меди (II), никеля (II) и кобальта (II) на реакции гидролиза ами¬
дов:СН„-С6Н6
H2NCH,,CONHj И H„NCHCONHCHoCONH2Катализ гидролиза амида аминоуксусной кислоты они представляют
следующим образом:-с—NH,+ м2+ .н,с—с—nh2
-| IIH,N О‘Ч /
м ■2 +^он2 ■2 +' ОН '+■ -Н,С с—NH,1Н,С с— NH,н,с с=о*1 II~ *■‘1 1 ‘—►„ 1 1H,N О02N оH,N ОЧ /\ /ч /■ м -L м JL м .+ NH,н,с+Эйхгорн изучал влияние комплексообразования на устойчивость
по отношению к гидролизу бис-тиофенальэтилендиамина исалицилаль-
глицина. Он установил, что медь облегчает гидролиз первого и затруд¬
няет гидролиз второго. В результате гидролиза медного комплекса
с бис-тиофенальэтилендиамином получается этилендиаминовый комп¬
лексQLн,с-Т‘ JJ. N=CH 5н,о^ \ш,н,с—сн,
-| I •H,N NH,
\/Си/ чн2о он.+ 2асноИ начальный, и конечный комплексы содержат по одному циклу. Фак¬
тором, определяющим относительную устойчивость этих комплексов
является электронное распределение.В результате гидролиза комплекса с основанием Шиффа и сали¬
цилового альдегида и глицина комплекс из бициклического должен
превратиться в моноциклический:363
211,0НО .ОН,
\ / ‘
Си
/ \H,N О' I , Iсн—с=о.онОС^-^Х'СНОВероятно, большая стабильность бициклического комплекса по
сравнению с моноциклическим является причиной того, что комплек-
сообразование затрудняет гидролиз. По-видимому, этот фактор силь¬
нее влияет на реакционную способность исходного соединения, чем
изменение поляризации атома азота. Таким образом, данный пример
указывает на необходимость учета еще одного фактора — устойчи¬
вости исходных комплексных соединений и продуктов реакции.При изучении влияния координации на гидролиз галоген-
алкиламинов установлено, что по сравнению со свободным
р-хлорэтиламином этот лиганд в комплексном ионе
[CoEn2(Cl—СН2—CH2—NH2)H20]3+ гидролизуется в 50 раз быстрее.
Смещение электронной пары атома азота к кобальту в результате об¬
разования донорно-акцепторной связи действует подобно индуктивно¬
му эффекту:н,0:ЛС1-»-СН-сн.н►N—СоIнКукушкин и Сибирская с сотр. показали, что гидролиз метилизо-
тиомочевины в комплексе платины (II) протекает гораздо легче, чем
в свободном состоянии. В результате координации метилизотиомоче-
внны увеличивается электрофильность атома углерода и вследствие
этого облегчается его атака молекулой воды или гидроксильным
ионом. Схематично реакцию можно изобразить следующим образом:/NHCHq S Су + [Ptcui2- —►'NH, -Cl-СН3I *NH
CI3PU-S4-Cf\NH,h,o-H+[Cl3Pt (SCH3)]2- +H!ho|—c/N
■ NH,[PiCI,]2--Ha0; -2CI-[Pt2 (SCH3) CI5]2- ++ 2H2N-C==N—2CI-[Pt2(SCH3) (H,CN2)2C13IКомплекс Ko[Pt2(SCH3)Cl5] и соединение, содержащее цианамид,
[PL(SCH3) (H2CN2)2C13], были выделены препаративно.Опубликовано большое число' работ, посвященных каталитическо¬
му разложению кетокислот под действием как энзимов, так и ионов364
различных металлов. Катализ декарбоксилироваиия ионами металлов
обнаружен в случае ди- н трикарбоновых кетокислот. Установлено,
что декарбоксилированне монокарбоновых кетокислот или нитро-
уксусной кислоты не ускоряется ионами металлов, а иногда даже за¬
медляется.Механизм декарбоксилирования предложен Штеннбергом и Вест-
хеймером. Они считают, что образование координационных связей
дикарбоновой кислоты с ионом металла вызывает оттягивание элект¬
ронов от некоординированной карбоксильной группы. Это и является
причиной облегчения декарбоксилирования:о С(СН3), 0 о С(СН3), с>V/ * V V/ ^ \-с-сн«сн3>2 + мл+о{ }о °\ /° 0 о\< mVВ случае монокарбоновых кетокислот также имеет место образова¬
ние енольного хелата. Однако его образование способствует смеще¬
нию электронов по направлению к карбоксильной группе, что и при¬
водит к замедлению декарбоксилирования.13.9. Влияние заряда комплексного иона
на реакционную способность
координированного лигандаЯрким примером влияния заряда комплексного иона на реакционную
способность лиганда могут служить реакции алкилирования атома
азота в хинолиновой группировке. Так, алкилирование этой группы
иодистым метилом в нейтральном комплексе происходит сравнительно
быстро, а в комплексе катионного типа практически не происходит:НI4N_/\/4\/\o/Cu/2CH,IH+1-NICH3-HA A, /Ч/vN-II IN—>-Cu/5NCH.IРеакция не идетО365
Хей с сотр. показал, что в комплексе кобальта (III) катионного
типа этиловый эфир глицина гидролизуется со скоростью, константа,
которой k = 3,5• 103 л-моль-1-мин'1:Еп - 24- Еп^Д7С| ПЕ7аv 1 'nH,CH2C02C2H5 I 'NH2CEn * En+ C2H5OHyNH2CH2COO~В тех же самых условиях константа скорости гидролиза свобод¬
ного эфира составляет 38 л-моль-1-мин-1.Таким образом, введение эфира в комплекс кобальта (III), имею¬
щий заряд +2, ускоряет реакцию примерно в 100 раз. Этот эффект
может быть объяснен электростатическим взаимодействием: положите¬
льный заряд комплекса облегчает атаку гидроксильного иона.13.10. Влияние я-дативной связи одного лиганда
на реакционную способность другого лигандао-Фенантролиновые комплексы Mn(II) характеризуются гораздо бо¬
лее высокой каталитической активностью при декарбоксилировании
дикарбоновых кислот, чем гидратированный ион марганца. Ана¬
логично декарбокснлирование ацетондикарбоновой кислоты в 2,2-
дипиридильном комплексе Мп(П) примерно в 10 раз быстрее, чем в
соответствующем аквокомплексе:о=с! С-11 б-но УО—иСН: .0осн2 ..о=с^I ф+О\ /Мп/ Xн,о он2СН7 ,0
<?-Считают, что увеличение скорости реакции связано с образовани¬
ем я-дативной связи марганца с о-фэнантролином или 2,2-дипириди-
лом. Образование л-дативной связи должно увеличивать положитель¬
ный эффективный заряд центрального нона и, следовательно, способ¬
ствовать декарбоксилированию.13.11. Стабилизация одной из таутомерных форм
лиганда в результате комплексообразованияр-Дикетоны служат классическим примером кето-енольной таутоме¬
рии. Их енольная форма обладает большой склонностью к комплексо-
образованию. По-видимому, первой работой по изучению реакци¬
онной способности координированных |3-дикетонов (точнее, их еноль-366
ных форм) является работа Рейлена [6]. В ней установлено, что при
действии брома на ацетилацетонат хрома (в спиртовом растворе) про¬
исходит замещение водорода у центрального углеродного атома на
бром:Г Н,с 1" н,с '3 \3 \с-очВг,с—оч// \НС' 'Сг/3
\ч \ВгС 'с г/3с=ооIIо//Н3С _. нзсКомплексы Металлов с р-дикетонами привлекают внимание химиков
с теоретической точки зрения. Структурные данные показывают, что
расстояния С—С, С—О, и М—О в р -дикарбонильных хелатах экви¬
валентны. На этом основании р -дикарбонильные хелаты рассматри¬
вают как псевдоароматические соединения:С этой точки зрения во многих работах изучаются реакции заме¬
щения водорода в псевдоароматическом кольце: галогенирование,
ацетилированне, нитрование и роданнрование ароматического кольца.Клайбер изучил действие дихлорида серы на соединения общей
формулыЭта реакция была изучена на примере ацетилацетонатов А1(Ш),
Сг(Ш), Со(Ш) и Ве(П). В результате было показано, что водород
при центральном углеродном атоме способен замещаться на группу
—SCI. Интересно, что обычными продуктами реакции дихлорида серы
с енолами, подобными ацетилацетону, являются сульфиды. Сущест¬
вует мнение, что сульфиды образуются через промежуточное соеди¬
нение R—SCI. По мнению Клайбера, сохранение в данном случае груп¬
пы —SCI обусловлено стерической защитой этой группы примыкаю¬
щими к ней метильными группами.367
При действии цианидных ионов на полученный таким образом комп¬
лекс хрома происходит превращение группы —SCI в роданидную груп¬
пу по схемен,с н,сV—0 _ \—оCIS—^Сг/з CN ■- NCS—/” уСг/З+СГ
Н,с н3сТо же самое соединение хрома было получено и при прямом взаимо¬
действии ацетилацетоната хрома с роданом.Коллман и Киттельман получили большое число комплексов
Сг(Ш) с р-кетиминами общей формулыН?С\ /Г
С—NН-/ 'у С г/3чс=<//НзсЦентральный атом углерода вр-кетиминах, входящих в комплек¬
сы хрома, так же как и в комплексах Э-дикетонов, подвергается бро-
мированию N-бромсукцинимидом. Однако реакции нитрования и фор-
милирования внутрисферных р-кетиминов не происходят вследствие
меньшей стабильности кетимннных комплексов по сравнению с дн-
кетонными соединениями.Взаимодействие трикарбонилхром гс-тол у ол альдегидного комп¬
лекса с 2-амино-2-метил-1-пропанолом в эквимолярном соотношении
в тетрагидрофуране приводит к образованию двух изомеров, находя¬
щихся в таутомерном равновесии:Н,С СН,3 \ / 3
Н—C=N—С—СН,ОНСг(СО)3СНМолярное отношение циклической формы к цепной форме равно
4,3 : 1. Соотношение между изомерными формами, находящимися в
таутомерном равновесии, включающем свободные лиганды, равно
1,22 : 1. Таким обазом, этот пример указывает на сильное влияние
трикарбонилхрома на положение равновесия.Сг(СО)368
13.12. Стабилизация комилексообразованием
депротонированной формы лигандаИзвестно, что азосочетание производных фенола и ионов диазония
легче происходит в щелочной среде вследствие большой реакционной
способности соответствующих фенолятов. В кислой среде реакция
азосочетапия часто совершенно не имеетместа. Кузнецов [7] приводит
ряд примеров влияния комплексообразования на способность некото¬
рых производных фенола к реакции азосочетания. Так, пирокатехин
в кислых растворах почти не способен сочетаться с диазобензолсульфо-
кислотой. В присутствии солей галлия, алюминия и особенно цирко¬
ния и германия реакция азосочетания происходит даже в присутствии
минеральных кислот. Кроме того, получение азосоединений, на¬
пример из пирокатехина в щелочной среде, затрудняется его окисле¬
нием под влиянием нона диазония, что значительно снижает выход
продукта. В присутствии ионов перечисленных металлов окисление
пирокатехина солями диазония не идет. Влияние металлов на эту
реакцию связано с образованием внутрикомплекснЫх солей, в кото¬
рых протон фенольной группы замещен ионом металла.Аналогично пирокатехину 8-оксихинолин практически не сочетает¬
ся с диазобензолсульфокислотой в минеральнокислой среде, но это
сочетание происходит, если 8-оксихинолин связан в медный комплекс.Однако комплексообразование не с любым металлом облегчает
реакцию азосочетания. Взаимодействие того же 8-оксихинолина с
диазобензолдисульфокислотой замедляется солями алюминия. В при¬
сутствии солей алюминия эта реакция протекает очень медленно, даже
при наличии ацетата натрия. Кузнецов связывает это с различием
природы связей О—Си и О—А1. Однако, может быть, причина столь
резкого различия кроется в более высоком заряде алюминиевого комп¬
лекса,по сравнению с медным. Более высокий положительный заряд
комплекса должен затруднять атаку координированной молекулы
S-оксихинолина диазониевым катионом.Примером влияния иона металла на реакционную способность пу¬
тем стабилизации депротонированной формы лиганда может служить
реакция хлористого 4-днметиламинофенилдиазония с хромотроповой
кислотой. Краситель получается с 58%-ным выходом, причем заме¬
щение происходит в положении 4 хромотроповой кислоты (1,8-днокси-
нафталин-3,6-дисульфокислоты). При использовании кальциевого
комплекса этой кислоты выход может быть увеличен до 90%.Галогенирование (хлорирование и бромирование) 8-оксихинолина
в комплексах с Сг(Ш), Со(Ш), Fe(III) и Cu(II) дает те же продукты,
что и галогенирование свободного 8-оксихннолина (например, 5,7-
дихлор-8-оксихинолин). Джонс указывает, что хлорированием сво¬
бодного 8-оксихинолина не удается получить дихлорпроизводное с
выходом выше, чем 30%. В то же время при хлорировании внутрисфер-
ного 8-оксихинолина дихлорпроизводное получается с выходом
'80—90%. Джонс также объясняет это наличием в комплексе фено-
лятной формы. Кроме того, металл защищает функциональные груп¬
пы от окисления.369
13.13. Устранение характерных свойств лигандов
в результате координации и использование
этого явления для маскировки отдельных
функциональных группИзвестно, что амины могут терять при координации свойства, обус¬
ловленные наличием свободной электронной пары. Нитрогруппа в
комплексных соединениях Pt(IV) перестает окисляться перманганатом.
Ослабляется или совсем исчезает токсичность цианидного нона. Тот
факт, что комплексообразование может полностью устранять спо¬
собность к реакциям некоторых функциональных групп, не изменяя
существенно реакционную способность некоординированных функ¬
циональных групп, особенно если те удалены от места координации,
позволяет использовать это явление в синтезах.Например, цитруллин H2NC0NH(CH2)3CH—(NH„)COOH входит
в некоторые протеины, в составе которых он принимает участие в
образовании мочевины. Куртц осуществил эту реакцию в обратном
направлении с помощью комплексообразования. Он приготовил мед¬
ный комплекс орнитина (в результате этого была замаскирована а-
аминогруппа) и провел реакцию конденсации этого комплекса с мо¬
чевиной:0=С—СН— (СН2)з—nh2 nh2—с—nh2II + II - N’H3 -ГО NHa О\/Си2+0=С—CH(CH,)3NH—С—NH,+11" IIО NH, О\/ 'Си2+Затем медь была удалена из комплекса действием сероводорода, в
результате чего был получен цитруллин. Куртц провел таким обра¬
зом синтезы различных аминокислот. Эффективность маскирующего
действия ионов металлов в сильной степени зависит от стабильности
комплекса.13.14. Проявление лигандом новых химических
свойств в результате координацииВ качестве примера проявления лигандом новых химических свойств
можно указать на реакцию азосочетания о-фенантролина и диазо-
сульфаниловой кислоты. о-Фенантролин не способен сочетаться с
солями диазония, однако в составе комплекса с железом (II) он энер¬
гично реагирует с солями диазония сульфаниловой кислоты
и л-нитроаннлин-о-сульфокислоты [7].Хорошо известно, что при нитрозировании свободных первичных
аминов образуются неустойчивые диазосоединения, которые разла¬370
гаются на спирт и азот, а вторичные амины образуют устойчивые
нитрозосоединен ия:ONOHR_NHo <- R—N.OH -+• R—ОН + N.' -н.о 'ONOHR-NH *■ R—NNOI -Н.О IR RЧерняев и Андрианова показали, что координированные первич¬
ные амины в реакции нитрозирования могут проявлять свойства вто¬
ричных аминов. Например, алифатические амины в комплексе Pt(IV)
дают устойчивые нитрозосоединения:R—NH.. onoh R—NNO- + H30 + H+I IPt (IV) Pt (IV)Координационная связь первичного амина с платиной (IV) препят¬
ствует образованию молекулы Na, т. е. при координации первичный
амин становится «псевдовторичным».Аммиак в водном растворе взаимодействует с диоксидом серы с
образованием сульфита аммония. Армор и Таубе установили, что в
щелочном буферном растворе комплекс [Ru(NH3)6]3+ при взаимо¬
действии с SO., в присутствии кислорода превращается в соединение,
содержащее во внутренней сфере анион сульфаминовой кислоты. Го¬
раздо легче такое превращение координированного аммиака проис¬
ходит в системе, состоящей из кислорода, [Ru(NH3)6]3+ и тиосульфат-
ного иона. Вместо тиосульфатного иона с таким же успехом можно
взять тиофосфатный нон. Эти реакции протекают по схеме[Ru(\’H3)c]3+-i-Sa02- +»/аО* - [Ru(NH3)5 (NH:S03)]2++S02-+H+При повышении pH раствора скорость реакции и выход продукта
увеличиваются. По мнению Армора и Таубе, механизм данной реак¬
ции включает стадию присоединения атома серы к амидогруппе:S:(МН3)5 Ru - NH’4" + :S: В -> (NH3)5Ru - NHs-[- :B
с последующим окислением серы кислородом.13.15. Изменение реакционной способности
координированных оснований ШиффаКомплексные соединения металлов с основаниями Шиффа весьма рас¬
пространены. Они получаются как из готовых оснований Шиффа,
так и при действии альдегидов и кетонов на аминаты металлов. Иногда
комплексообразование приводит к стабилизации таких альдиминов,
которые в свободном состоянии неустойчивы. Так, Пфейффер полу-371
чнл имид салицилового альдегида в составе комплексных соединений
цинка, меди и ннкеля. Синтезированные соединения в общем виде мо¬
гут быть выражены формулойHN=CH—г****-"4*В свободном состоянии имид салицилового альдегида неустойчив
и не может быть получен.Интересный цикл исследований' по изучению взаимодействия цик¬
лических аминокомплексов никеля и меди с ацетоном и алифатиче¬
скими альдегидами провел Куртис с сотр. Он установил, что образую¬
щиеся вначале основания Шиффа испытывают внутрикомплексную
альдольную конденсацию с образованием макроциклического ли¬
ганда. Эта конденсация катализируется основаниями:СН Н Сн3с чр \^/СН,С Сн,сИзс\ /
Н,С—С■ IН,С—N НСИ,.СН,\N• 2 +
IVv,— NNII||СС:/\ГСН,Н3СN—СН,.2 +Ni
S' "VН,С—N UN—СН,"СИН,С'С .С СН,'Ч / \ 3
СН2 СН3На важную роль комплексообразования в изменении реакционной
способности оснований Шиффа обращал внимание Пфейффер. В част¬
ности, он показал, что если получать хелатный комплекс Cu(II) с оп¬
тически активным основанием Шиффа, образованным из эфира амино¬
кислоты и салицилового альдегида, то происходит быстрая рацеми¬
зация. Это можно объяснить существованием внутрикомплексного
таутомерного равновесия:-СН НГ> I *О N С—СО,С,Н5W ‘СиIRСН,I "0 N=C—СОчСтНс\ ^ 1Cu:+/2 RIIПотеря асимметрии оптически активного атома углерода проис¬
ходит при образовании структуры (II). Обратное превращение комп-372
лекса (11) в комплекс (I) должно приводить к равным количествам
обоих оптически активных антиподов.Внутрикомплексное таутомерное равновесие является основной
стадией в механизме каталитического трансаминирования между пи-
ридоксалем и аминокислотой, предложенном Снеллом. По его мнению,
при трансаминированип образуется основание Шпффа, стабилизи¬
рованное координацией с ионом металла, с последующим перемеще¬
нием двойной связи к углеродному атому аминокислоты:Реакции трансаминирования имеют чрезвычайно важное биологиче¬
ское значение, так как они являются весьма вероятным способом, обес¬
печивающим связь между углеводами и белками.Большое значение в препаративной координационной и органической
химии имеют реакции, при которых сложные органические соедине¬
ния образуются в процессе взаимодействия металла с более простыми
соединениями. По крайней мере одно из-этих органических соедине¬
ний должно обладать свойствами лиганда по отношению к иону ме¬
талла, входящему в состав соли. Ион металла способствует стерео-
химической ориентации реагентов, а следовательно, направлению
реакции по определенному пути.В отсутствие солей металлов соответствующие реакции иногда
не протекают вовсе, но чаще идут с малым выходом. В последнем слу¬
чае целевой продукт является не основным, а побочным соединением.
В таких реакциях ион металла является как бы матрицей, на которой
закрепляются исходные реагенты и формируется получающаяся
молекула. Синтез органической молекулы, основанный на этом прин¬
ципе, часто называют темплатным (от англ. templet — матрица,
лекало).НХ^ N.CH..OR0=С CRIIR'COCOOKCH,NlfО N\ /\4 М,0I13.16. Реакции на матрице, или темплатный синтез373
Реакции на матрице особенно эффективны для синтеза макро-
циклнческих лигандов. Например, при взаимодействии ряда солей
переходных металлов с о-дицианобензолом происходит реакция цикло-
тетрамеризации в соответствии со схемойCN+ MCI,CNУстановлено, что из 5,5'-дигидроксиметил—4,4'-диэтил-3,3-ди>
метилдипиррометена (в гидробромидной форме)Me Me‘2 Вг"в присутствии ацетата меди (II) с удовлетворительным выходом полу¬
чается этиопорфирин:MeMeEtEtMeMeЭффектный способ циклизации тетрапиррольных систем также осу¬
ществляется реакцией на матрице. Например, циклизация 1,19-ди-
метилбиладиена в гидробромидной форме
протекает на матрице ионов никеля (II) и кобальта (II). В результате
получается 1,19-диметилтетрагидрокоррин:Реакция между p-меркаптоэтиламином и а-дикетонами в качестве
главного продукта обычно дает тиазолидины и только в небольших
количествах а-диимины. Баш показал, что если ji-меркаптоэтнламин
находится в комплексе Ni(II), то он реагирует с а-дикетонами собра-
зованием внутрисферного а-диимина с 70%-ным выходом:/ \R'—С=0 Н ,N 'S
+ Ni/ чR—C=0 H2N^ /SR.-cW^S\г+/
vVi + 2H ,0—N \
wЗатем Баш использовал этот комплекс как матрицу для дальней¬
шего синтеза полициклического хелата. Он провел следующую ре¬
акцию:R ^ NV+/S ВгСН,—I А + BrCHpJIК—C=N NS 2 ^\ /Rr— С=>/ —СН\2+//NlR—C=N СНV-/Br,которая оказалась возможной благодаря фиксированному положению
атомов серы.Подавляющее большинство реакций на матрице, изученных к нас¬
тоящему времени, относятся к шиффовой конденсации. Первая стадия
процесса Баша также является конденсацией этого типа.Часто конденсация Шиффа на матрице усложнена. Так, этилен-
диаминовый комплекс никеля (II) в присутствии аммиака взаимодей¬
ствует с формальдегидом с образованием макроциклического комп¬
лекса по схеме375
CNiEn ]!+ + 4CH,0 + 2NII,N NАналогичная конденсация имеет место и в системе\ //К + 2СН,0 + HjNRbf XNHV с/Реакции этого типа позволяют получать разнообразные комплек¬
сы с макроциклическими лигандами. Удаление из комплекса иона
металла приводит к выделению в свободном виде иногда довольно
сложных циклических органических соединений.Библиографию по реакциям на матрице можно найти в работе [8].В заключение следует сказать, что изменение реакционной спо¬
собности лигандов в комплексных соединениях часто является ре¬
зультатом проявления нескольких факторов, влияющих на реак¬
ционную способность. Поэтому отнесение реакций к тому или иному
типу, конечно, не бесспорно.Координация к иону металла, как правило, приводит к резкому
изменению его свойств. Ельцов и Кукушкин исследовали трис-хелаты
Fe(II) с 1-нитрозо-2-нафтолом (I) и с его гетероциклическими ана¬
логами (II и III) (соединения записаны в таутомерной хиноноксимной
форме):NOH NOH NOH(Комплексы Fe(II) на основе этих лигандов обладают весьма высо¬
кой термодинамической стабильностью. Однако совершенно неожи¬
данно оказалось, что самые разнообразные характеристики свободных
и координированных лигандов оказались идентичными. Так, внутри-
лигандные полосы поглощения в электронных спектрах комплексов
удовлетворительно совпадают с этими полосами поглощения в ани¬
онных формах лигандов. ИК спектры твердых комплексов в области
1700—1500 см-1, где должны проявляться колебания связей С=0,СН3ИIII376
C=N, C=C, в хелатном комплексе почти идентичны спектрам натри¬
евых солен соответствующих лигандов. Полярографическое восста¬
новление внутрисферных лигандов происходит при тех же потенци¬
алах, что и восстановление натриевых солей этих лигандов. Интерес¬
но также, что при замене одного из внутрисферных лигандов на лиганд
типа 2,2'-бипиридила также не происходит изменения потенциала
восстановления внутрисферных нитрозонафтолятов. Такая ситуация
является исключительной. В литературе пока нет других примеров
такого сохранения характеристик свободных лигандов при вхождении
их во внутреннюю сферу комплексов. Возможно, это является спе¬
цифическим свойством хелатов железа (II).
ГЛАВА14РОЛЬ РАСТВОРИТЕЛЯ
В КООРДИНАЦИОННОЙ ХИМИИВ неорганической химии вода долго оставалась основным, если не
единственным, растворителем. Лишь на рубеже XX в. в неорганиче¬
скую химию был введен жидкий аммиак, а затем и другие неводные
растворители. В органической химии всегда использовались главным
образом неводные растворители, такие, как спирт, эфир, бензол.Поскольку координационная химия долгое время была частью
неорганической химии, она также была связана с водными растворами.
Действительно, Вернер и его многочисленные ученики проводили
свои исследования главным образом в водных растворах. То же мож¬
но сказать о Чугаеве и его научной школе. Однако Чугаева можно
считать одним из первых химиков, которые указывали на новый путь
развития препаративной координационной химии. Он указывал, что
вода в качестве растворителя имеет серьезные недостатки: в водных
растворах часто протекает гидролиз, который препятствует получе¬
нию ряда комплексных соединений. Кроме того, большинство орга¬
нических соединений нерастворимо вводе. В подтверждение идеи о
целесообразности использования органических растворителей Чугаев
синтезировал трипропиламмониевый комплекс [(C3H7)3NH]2[PtCl4],
растворимый в спирте, хлороформе, ацетоне и на примерах показал
преимущества работы с ним в органических растворителях.Важную роль в использовании органических растворителей в
координационной химии сыграл Чатт. Еще в 1937 г. он начал изучать
триариларсиновые и триарилфосфиновые комплексы платины и пал¬
ладия. Эти комплексные соединения оказались совершенно нераство¬
римыми в воде, и ему пришлось использовать органические раство¬
рители. В настоящее время неводные растворы играют очень большую
роль в координационной химии.Со времени классических исследований Меншуткина стало ясно,
что растворитель нельзя рассматривать как инертную среду. В на¬
стоящее время накоплено достаточно экспериментальных данных,
которые свидетельствуют о влиянии растворителя не только на ско¬
рость реакции, но и на ее направление.14.1. Факторы, влияющие на растворимость
координационных соединенийВопрос о растворимости простых солей весьма сложен. Тем более он
сложен по отношению к координационным соединениям. Исследованием
проблемы их растворимости занимался Яцимирский [1]. Его основ-378
ные выводы, касающиеся минимальной растворимости солей вводе,
сводились к следующему.1. Отношение радиусов ионов, составляющих данную соль, должно
приближаться к определенному значению, характерному для данного
типа соли. Эта величина колеблется в пределах от 0,7 до 1,2. Изме¬
нение растворимости при изменении отношения радиусов ионов тем
резче, чем выше заряды ионов, составляющих данную соль.2. Чем выше заряды комплексных катионов, тем меньше раство¬
римость солей при прочих равных условиях.3. Ионы не должны содержать групп, способных к образованию
водородной связи с молекулами воды (гидроксильная и карбоксиль¬
ная группы, аминогруппа).Яцимирский сформулировал эти положения, исследуя классиче¬
ские вернеровские соединения. Однако они вполне могут быть рас¬
пространены и на комплексы со сложными органическими лигандами.
В качестве растворителей можно рассматривать не только воду, но
и другие органические и неорганические вещества.В настоящее время не вызывает сомнений, что наличие в сложном
органическом лиганде ‘гидрофильных или полярных групп способ¬
ствует растворимости комплексов в воде или полярных растворите¬
лях. Такими группами, например, могут быть —S03, — PQi , —COO",
—ОН, —NH2. Наличие в лиганде гидрофобных групп приводит к
уменьшению растворимости в неполярных растворителях.В сложных органических лигандах, как правило, наряду с гидро¬
фильными группами присутствуют гидрофобные. Гидрофобные группы
большого размера могут экранировать гидрофильные и тем самым по¬
нижать растворимость соединения в полярных растворителях.Файгль ввел в аналитическую химию понятие эффгкта утяжеле¬
ния. Его сущность сводится к тому, что утяжеление органического
реагента, связывающего в комплекс ион металла, происходит за счет
гидрофобной части молекулы. Это приводит к уменьшению раствори¬
мости осадков и увеличению чувствительности качественных реакций.Хорошо известно, что органические кислоты с большой молеку¬
лярной массой при нейтрализации щелочами переходят в соли вслед¬
ствие этого растворяются в воде и полярных растворителях. Это свя¬
зано с лучшей сольватацией образующихся анионов по сравнению с
поляризованной формой кислоты. При образовании комплексов с
ионами металлов посредством отрицательно заряженных функцио¬
нальных групп заряд аниона нейтрализуется положительным ионом
металла, в результате растворимость комплекса в полярных раство¬
рителях понижается по сравнению с растворимостью простой соли
этого лнганда.Наличие в координированном лиганде функциональных групп,
характерных дтя растворителя, как правило, способствует раствори¬
мости комплекса. Особенно высокой растворимостью обладают комп¬
лексные соединения, роль лигандов в которых играют молекулы рас¬
творителя.В растворе ионы металлов сольватпровэны. Поэтому чем больше
солей различных металлов способен раствэрягь тот иди инэй расгво-379
рнтель, тем более распространенными являются соответствующие комп¬
лексные соединения. С уверенностью можно сказать, что наиболее
распространенными являются аквокомплексы. Диметнлсульфоксид
также сочетает в себе хорошие свойства растворителя и лиганда, по¬
этому диметилсульфоксидные комплексы известны практически для
всех металлов.Взаимосвязь между природой функциональных групп координи¬
рованного лиганда и растворимостью соответствующего комплекса
основана на сольватационной составляющей в энергетике процесса
растворения.Несомненно, важную роль в растворимости комплексов играет
энергия кристаллической решетки. При прочих равных условиях с
увеличением энергии кристаллической решетки растворимость долж¬
на уменьшаться. Известно, что комплексы типа зеленой сол« Магнуса
очень мало растворимы. Их кристаллическая решетка имеет колоноч¬
ное строение с чередованием плоскостей катиона и аниона. В соли
Магнуса [Pt(NН3)4] [PtClJ расстояние платина—платина равно0,323 нм. Столь малое расстояние означает, что имеет место взаимо¬
действие между атомами платины с образованием химической связи.
Образование химической связи платина—платина приводит к увели¬
чению энергии кристаллической решетки.Соль i<uc-[Pt EnClo] также имеет колоночный мотив, но расстоя¬
ние платина—платина в ее кристаллической решетке равно 0,339 нм.
Гораздо менее прочная связь должна приводить к меньшей энергии
Кристаллической решетки. Вероятно, это и является одной из причин
того, что неэлектролитные соли i{«c-[Pt ASC12] растворяются в воде
лучше, чем электролитные соли типа [Pt А4] [PtCl4].Весьма важный и интересный вопрос — это растворимость коор¬
динационных соединений в смешанных растворителях. Одним из
первых на него обратил внимание Гринберг. Он установил, что комп¬
лекс [Pt(NH3)J [Pt(SCN)4] практически не растворяется ни в воде,
ни в ацетоне, но хорошо растворяется в их смеси. В настоящее время
имеется много примеров этого явления.При изучении сольватации ионов или полярных молекул в бинар¬
ных растворителях было установлено, что соотношение компонентов
смеси растворителей в сольватной оболочке растворенной частицы
обычно иное, чем в массе раствора, т. е. состав сольватной оболочки
отличается от состава растворителя. Это явление получило название
селективной или избирательной сольватации.Проявление специфических свойств смешанного растворителя
может быть также проиллюстрировано на примере комплекса
[Rh Ру3С1(С„04)]. Он не растворяется ни в воде, ни в пиридине, но
растворяется в смеси вода-—пиридин. Считают, что это обусловлено
одновременной сольватацией молекулами пиридина пиридинового
фрагмента комплекса и молекулами воды остальной его части. Сум¬
марной энергии сольватации оказывается достаточно, чтобы компен¬
сировать энергию кристаллической решетки.Часто исходные соли переходного металла растворяются только
в воде, а лиганды, используемые в синтезе, в воде не растворяются.380
Так, например, соли Cu(I) и Cd(II) растворяются в воде, а некото¬
рые производные тиомочевины не растворяются. Поскольку производ¬
ные тиомочевины растворимы в формамиде и диметилсульфоксиде, то
можно их комплексообразование с Cu(I) и Cd(II) осуществлять в сме¬
сях этих растворителей с водой.Растворимости комплексов уделено так много внимания потому,
что в препаративной координационной химии в подавляющем боль¬
шинстве случаев синтез комплексных соединений осуществляется в
растворе и выделение в твердую фазу является одной из самых важ¬
ных операций.14.2. Синтез в неводных растворителяхЧугаев получил растворимый в спирте тетрахлороплатинат (II) три-
пропиламмония, обработав K2[PtCl4] спиртовым раствором
[(m30-C3H7)3NH]C1 при 60 °С в соответствии с уравнениемКо [Р1СЦ] + 2 [(ызо-С3Н7)3 NH] CI ((изо-С3Н7)3ЫН]2 [PtCI4] + 2KCIОдним из условий осуществления этой реакции является раство¬
римость в спирте образующегося комплекса и нерастворимость ис¬
ходного. К сожалению, ониевые тетрахлороплатннаты (II) редко раст¬
воримы в спирте. Однако в большинстве случаев они хорошо раство¬
ряются в диметилформамиде. Соответствующая калийная соль в нем
не растворяется. В. В. Сибирская и Н. В. Воробьев-Десятовский осу¬
ществили синтез большого числа ониевых тетрахлороплатинатов (II)
по схеме ЧугаеваК: [Ptci4] + 2А . HCI — (АНЬ [PtCI4] + 2КС1!с тон лишь разницей, что в качестве среды был взят днметилформ-
амид. Эта реакция протекает не только с солянокислыми аминами, но
и с солями тиурония.Препаративные возможности при синтезе комплексных соедине¬
ний иногда сильно возрастают, если исходный комплекс хорошо раст¬
воряется в растворителе, молекулы которого не проявляют склонности
к вхождению в его внутреннюю сферу. В координационной химии
палладия (II) таким комплексом является бензонитрильное соеди¬
нение [Pd(NCPh).,Cl2]. Оно хорошо растворяется в бензоле, хлорофор¬
ме, ацетоне и в самом бензонитриле. Кроме того, это соединение легко
получается и устойчиво в кристаллическом состоянии на воздухе.
В перечисленных растворителях внутрисферные молекулы бензонит¬
рила легко замещаются на многие другие лиганды по реакции[Pd (NCPh)2 CU] +2L [PdLoClj] H-2NCPhВ литературе описаны многочисленные примеры использования этого
бензонитрильного комплекса для препаративных целей в качестве
исходного соединения.Такой же подход используется для синтеза комплексов меди (I).
Трудности их получения часто связаны со сравнительно большой
склонностью меди (I) к окислению, с тенденцией к реакции диспро-381
порционирования на Си0 и Cuz+ и во многих случаях с малой раство¬
римостью солей типа Си3Х3. Гарновский показал, что весьма удоб¬
ным растворителем для синтеза комплексов меди (I) является ацето¬
нитрил. Он хорошо растворяет многие соединения Си„Х2 с образова¬
нием комплексов CuX-m(NCCH3), которые относительно стабильны
к окислению. При добавлении в раствор лигандов, обладающих боль¬
шей склонностью к комплексообразованию с ионом Си+, происходит
лигандный обмен но реакцииCuX-m (NCCH3) + nL CuX-nL + mCfbCNЭтот путь синтеза называют методом лигандного обмена.14.3. Синтез координационных соединений
в растворителе, обладающем свойствами
лигандаПри синтезе комплексных соединений часто растворитель является
и лигандом, образующим комплекс. В этом случае получение соеди¬
нений сводится к растворению соответствующей соли и последующему
удалению растворителя. Например, таким образом было получено боль¬
шое число диметилсульфоксидных комплексных соединений.Чатт и Роу нашли, что фосфиновые комплексы рения (V) типа,
[ReOX3L2], где L — арил и алкилфосфины, X — галогенидные ионы,
взаимодействуют со спиртами с образованием моноалкоксипроизвод-
ных [RfeO(OEt)X„L,,]. Котеговым и Кобиловым установлено, что
при нагревании с этанолом взаимодействуют только транс-изомеры,
и эта реакция обратима:OEtL Eton xt 1 Lv - Re
X HX L1 f|—'XОСоответствующие г^ис-изомеры в реакцию этанолиза не вступают.
Этанолиз же бромидных комплексов протекает при более мягких ус¬
ловиях, чем хлоридных. Таким путем этанол можно использовать
для разделения и очистки изомерных комплексов рения (V).Еще в прошлом веке Шутценбергер синтезировал комплексные
соединения платины с трихлоридом фосфора. Для реакции был взят
жидкий трихлорид фосфора и твердый хлорид платины (11). В качест¬
ве продуктов реакции получены мономерный и димерный мостиковый
комплексы:3PtCI2 + 4РС13 — [Pt(PCI3)2 Clo] + [Pt (PCI3) Ch]2Координированная молекула PC13 в водной среде без разрыва связи
платины с фосфором превращается в фосфористую кислоту, а в спир¬
тах — в соответствующие эфиры фосфористой кислоты.382
Известны случаи, когда небольшие добавки растворителей, обла¬
дающих электронодонорными свойствами, резко ускоряют реакцию.
Например, взаимодействие металлического никеля с N204 протекает
с гораздо большей скоростью в присутствии ацетонитрила, чем в его
отсутствие. Причиной ускоряющего действия ацетоннтрила явля¬
ется образование ацетонитрильного комплекса никеля. Реакция вы¬
ражается уравнениемNi + 2NI,Oi + 2CH3CN -* Ni (CH3CN)o (N03)2 + 2NOНитрилопентамминовые комплексы кобальта (III) иногда получают
нитрозированием азндопентамминовых соединений в соответствующем
нитриле. В этих реакциях нитрил играет роль растворителя и внед¬
ряющегося в комплекс лиганда.Амминные комплексы переходных металлов при взаимодействии
с ацетоном способны превращаться в соединения с внутрисферными
молекулами основания Шиффа, например, по_ реакции(СН3)2С!1Н2С N NH.,—СН2[М!Еп.р + 2(СН3)пСО* I ^Ni:+v I + 2Н„0Н2С—H2N 4N—сн2С(СН,)2Вероятно, образование этого комплекса является первой стадией
более сложного процесса. Исследования показали, что при разру¬
шении продуктов реакции ацетона с внутрисферным этилендиамином
образуется оксид мезитила (СН3)зС =СН—СО—СН3. На этом основа¬
нии сделано заключение, что в комплексе существовала циклическая
группировка, содержащая шесть атомов углерода. С данной точки
зрения строение получающегося комплекса правильнее записывать
следующим образом:н>С\г/С\:с/СИз
и Г-™»Н,с N 2+ N СН24Ni✓ чн Г—H,N XNH—СН2Подобных примеров одновременного функционирования вещества
как реагента и как растворителя можно привести много.14.4. Влияние растворителя на направление
реакцииВ соответствии с правилом Чугаева прочные хелатные комплексы об¬
разуют 1,2-диметилендиамин и 1,3-триметилендиамнн, но не 1,4-
тетраметилендиамин. Последний может образовывать лишь неустой-383
чивый семичленный цикл. Еще Чугаев отмечал, что образование комп¬
лексов с семи- и восьмичленными хелатными циклами не происходит,
а образуются мостиковые соединения. Фуета и Огино взяли в ка¬
честве растворителя не воду, а ди'метилсульфоксид и сумели синтези¬
ровать комплекс [Со Тп3]С13, где Тл — 1,4-тетраметилендиамин. Более
того, в диметилсульфоксиде им удалось заместить хлоридные ионы в
комплексе q«c-[CoEn2C!2]+ на додекаметилендиамин (Don) с образо¬
ванней комплекса [CoEn(Don)]3+. Создается впечатление, что ди-
метилсульфоксид благоприятствует образованию циклов, которые
неустойчивы в водных растворах.Растворитель может резко менять окислительный потенциал сис¬
тем, состоящих из одних и тех же ионов. Например, в водных раст¬
ворах комплексы палладия (IV) обычно неустойчивы. Вследствие
высокого окислительного потенциала они отщепляют хлор и пере¬
ходят в соответствующие соединения палладия (II):[PdC]6]~- 5* С!, + [PdCl4]2-В литературе описано получение [Pd(NH3)2CI.,], [PdEnClJ,
[PdPy2Cl4], а также некоторых хлоробромо- и хлороиодосоединений
типа [PdPy2Cl2X2], (где X — бром или иод). Основным методом их
синтеза является действие хлора на суспензию соответствующего со¬
единения палладия (II) в хлороформе или четыреххлористом угле¬
роде.Дрю с сотр. получил [Pd(NH3)2Cl4] в результате окисления соли
[Pd(NH3)4] [PdClJ пероксидом водорода в присутствии соляной кис¬
лоты. При действии смеси пероксида водорода с соляной кислотой или
при действии хлора на [Pd(NH3)3Cl2] окисление протекало только до
образования молекулярного соединения [Pd(NH3)2Cl2]-[Pd(NH3),Cl4].
Лишь в CCI4 при энергичном хлорировании [Pd(NH3)2Cb] Дрю уда¬
лось получить [Pd(NH3)„CI4].Распространенным способом получения комплексных соединений
платины (IV) является окисление соответствующих соединений пла¬
тины (II). Процесс идет по типу окислительного присоединения, на¬
пример:[PtL4]2+ С1; -** [PtL4Cl2]2+Трудности при синтезе возникают в том случае, если координи¬
рованный лиганд L обладает ярко выраженными восстановительными
свойствами. Примером таких лигандов могут быть триалкилфосфиты
или гидроксиламин. В такой ситуации окислению прежде всего под¬
вергается этот внутрисферный лиганд, а не центральный ион.Оказалось, что, заменяя растворитель, эту трудность можно
устранить. Так, Троицкая и Шмакова получили комплексы типа
[Pt{P(QR)3}2Cl4], окисляя хлором соответствующие соединения пла¬
тины (II), но не в воде, а в хлороформе. Аналогичным способом Сте-
ценко и Абзаева синтезировали комплексы платины (IV) с гидроксил-
амином. Замена воды на хлороформ, вероятно, приводит к пониже¬
нию окислительного потенциала системы С12 + 2е~->-2С\~ вследствие'
относительного ослабления сольватации хлорид-ионов в хлороформе.384
Значения стандартных окислительно-восстановительных потен¬
циалов серии рутениевых комплексов в хлористом метилене оказались
менее положительными, чем в ацетонитриле [2]. Это связано с разли¬
чием в энергиях сольватации комплексов данными растворителями.Обнаружено существенное различие в направлении реакции цикло-
пентадпенильного комплекса [RuCp(PPh3)2Cl] с изонитриламн в раз¬
личных растворителях [3]. В малополярных растворителях проис¬
ходит замещение на изонитрил внутрисферного трифенилфосфина,
а в сильнополярных растворителях (в присутствии NH4PFC) за¬
мещению подвергается хлорндный лиганд и образуется комплекс
[RuCp(PPh3),(CNR)]PFc.Примером влияния растворителя на направление реакции явля¬
ется также взаимодействие цнклопентадиенильного комплекса
[CpW(NO)3H] с акцепторами гидридного иона Ph3C+X", где X — BF4,
PF6- В ацетонитриле продукт имеет состав [CpW(NO)s(CH3CN)]Xr
а в хлористом метилене получается димерное соединение
[Cp2W2(NO)4H]X [4].При изучении вольтамперометрического поведения 1-пирроли-
дннкарбодитионатных комплексов марганца (III), железа (III) и меди
(II) в неводных растворителях выявлено закономерное изменение
величин разностй потенциала полуволны окисления и потенциала
полуволны восстановления в зависимости от сольватпрующих пара¬
метров растворителей. Изменение значений (Е?* —Е™) удов¬
летворительно объясняется с позиций энергии сольватации комп¬
лексов.14.5. «Принудительное» введение в комплекс
молекул растворителя с образованием
лабильного интермедиатаМногие реакции замещения в водных растворах протекают через
промежуточное образование аквокомплексов (см. гл. 9). Внутрисфер-
ные молекулы воды, как правило, легко замещаются на другие лиган¬
ды, поэтому аквокомплексы обычно являются весьма лабильными
интермедиатами в реакциях замещения. Принцип образования ла¬
бильных интермедиатов широко используется при синтезе координа¬
ционных соединений. Например, комплекс ^uc-[Pt(NH2CH3)2Cl2] не¬
возможно получить взаимодействием метиламина с KJPtClJ. Основ¬
ным продуктом при этом является [Pt(NH2CH3)4] [PtClj. Иодндный
аналог ^uc-[Pt(NHoCH3)2I2] получается при взаимодействии метил¬
амина с K2[PtI4] очень легко и с хорошим выходом. Превращение
4uc-[Pt(NH,CH3)„I2] в хлоридное соединение при действии на него
раствором КС1 препаративно не может быть достигнуто, так как ио-
Дидный комплекс значительно более прочен, чем хлорндный. Однако-
это превращение становится возможным при использовании ме¬
тода «принудительного» введения растворителя. На комплекс
4“c-[Pt(NH2CH3)„L] действуют точно рассчитанным количеством нит¬
рата серебр"а. Редакция протекает в соответствии с уравнением385
ф/c-fPt (NH,CH3)2 I2] -f 2AgN03 + 2H20 [Pt (NH,CH3)2 (H20)2] (N03h+ 2AgIПосле удаления осадка Agl к раствору добавляют КС1, и в результате
в осадок выделяется хлоридный комплекс:4uc-[Pt (NH.,CH3) , (Н20)2] (N03)2 + 2KCI ** i(uc-[Pt(NH2CH3)2 Cla] f 2KN03+2H,0Принцип «принудительного» введения растворителя используется
и при синтезе комплексов из неводных растворителей. Так, нитрат
серебра хорошо растворим в диметилсульфоксиде, а его хлорид плохо
растворим. Поэтому хлоридные лиганды удаляются из комплекса в
диметилсульфоксидных растворах, так же как и в водных. На освобо¬
дившееся координационное место вступает молекула диметилсульф-
оксида, которая затем замещается на другой подходящий лиганд.
Для этой же цели используют метанольные растворы AgPF6.Реагент, связывающий лиганд и удаляющий его из комплекса,
подбирается в каждом отдельном случае индивидуально. Например,
для удаления из комплекса галогенид-ионов не обязательно выбирать
реагент, образующий малорастворимое галогенидное соединение. Это
соединение может быть хорошо растворимым, но оно обязательно
должно образовывать прочное соединение с галогенидным ионом. К та¬
ким реагентам относятся тригалогениды бора. Тетрагалогенидные ани¬
оны довольно прочны и пригодны для образования в растворах ла¬
бильных интермедиатов. Вместо самих тригалогенидов бора иногда
удобно использовать их эфираты, например BF3-0(C2H5)2. Эфнрат
одновременно может играть роль растворителя и связывающего гало-
генидные ионы реагента.В некоторых комплексных соединениях внутрисферную нитро¬
группу можно заместить на молекулу растворителя введением в раст¬
вор сульфаминовой кислоты. Известно, что эта кислота количественно
взаимодействует с азотистой кислотой по уравнениюNH2S020H + HNOo — No + H0SO4 + Н20Действием рассчитанным количеством сульфаминовой кислоты на
нитрокомплекс [Pd(N02)4]2- можно получить аквонитрокомплексы
IPd(H20) (N03)3]“ и [Pd(H20)2(N02)„]. Дальнейшее извлечение нит¬
рит-ионов из комплекса [P"d(H.,0).,"(N02)2] протекает крайне медленно
даже при избытке кислоты. Такое положение является следствием
низкого трансвлияния внутрисферных молекул воды. В результате
этого нитрогруппы оказываются закрепленными в комплексе.14.6. Индуцированное разложение растворителя
и координация его продуктов к иону металлаОдна из первых реакций, в которых имеет место разложение раство¬
рителя и образование комплексов с продуктами разложения, была
открыта Цейзе. В 1830 г. он установил, что при нагревании этаноль-
ного раствора Na„[PtCl6] происходит дегидратация спирта с образова¬
нием этилена и восстановление Pt (IV) до Pt (II). Этилен координи¬386
руется к платине (II), в результате чего образуется л-комплекс.
Суммарная реакция выражается уравнениемNa: [PtCUJ + CjH6OH -► Na [Pt (C2H4) Cl3] + HaO + NaClАналогичные реакции идут и в более сложных спиртах. Например, вы¬
держивание K2[PtCl4] в пропаноле, бутаноле, З-бутен-1-оле при ком¬
натной температуре в течение четырех дней приводит к образованию
соответствующих олефиновых и бутадиенового комплексов.Каталитическое разложение муравьиной кислоты протекает по
двум направлениям. В одном из них образуются С02 и Н2, а в другом
СО и Н„0. В присутствии комплексов платиновых металлов разложе¬
ние муравьиной кислоты в значительной мере идет по пути дегидрата¬
ции. Образующийся оксид углерода (II) приводит к образованию кар¬
бонильных комплексов.В смешанных ацетоновоальдегидных растворах также происходит
образование карбонильных комплексов, например, по реакции[Pt, (PEt3)4 С1г]2+ Н- 2RCHO — 2 траис-[?\. (PEt3)2 (СО) Cl]+ + 2RHСкорость декарбонилирования изученных альдегидов изменяется в
следующем порядке: СС13СНО > PhCHO > СН3СНО.В 1965 г. Русина и Влчек открыли исключительно важную реак¬
цию получения комплекса [Rh(PPh3)2(CO)Cl]. Они показали, что на¬
гревание RhCl3 в растворе диметилформамида приводит к восстанов¬
лению родия и карбонилированню образующего комплекса.Введение в реакцию трифенилфосфина приводит к выделению комп¬
лекса, который широко используется в качестве катализатора в ряде
химических процессов. Диметилформамид распадается в соответ¬
ствии с уравнением(CH3)2NCHO — (CH3)2NH + СОХидекель установил, что некоторые металлы, в том числе и благород¬
ные, способны окисляться диметилсульфоксидом. Диметилсульфоксид
при этом восстанавливается до диметил сульфида. Реакции протекают
в присутствии хлорированных углеводородов и, вероятно, имеют
сложный механизм. Стадия окисления описывается уравнениемСи + (CH3)2SO ч- СиО -f (CH3)2SСвоеобразному превращению подвергается пиридин в' присутствии
солей железа (III). Х^йн показал, что происходит восстановление
железа (III) до железа (II), а пиридин превращается в дипиридил по
реакции2Fe (111) + Ру -*■ 2Fe (I I) + Dipy + 2Н+Смещению реакции вправо способствует образование исключительно
прочного дипиридильного комплекса железа (II).387
14.7. Влияние основности растворителя на синтез
комплексов с депротонированными лигандамиСовершенно естественно, что растворитель оказывает весьма су¬
щественное влияние на получение комплексов с депротонированными
лигандами. Кукушкин с Сибирской выделили из водного раствора в
твердую фазу диамидокомплекс платины (IV) [Pt(NH3)4(NH2)2]I2.
Вторая константа кислотной диссоциации комплекса [Pt(NH3)6]i+
имеет порядок 10~10. Таким образом, получение из водного раствора
комплексов платины (IV) с еще большим числом амидогрупп практи¬
чески невозможно. В растворителе с большей основностью (жидком
аммиаке) и при использовании вместо щелочи амида калия Хекку уда¬
лось получить K3[Pt(NH,)6] по реакции[Pt(NH3)„] (N03)4 + 6KNH, -ч- К2 [Pt (NH2)„] + 4KN03+ 6 NH3В жидком аммиаке синтезированы комплексы с депротонированным
этилендиамином для осмия (IV), иридия (III), родия (III) и даже для
платины (II) и палладия (II), что принципиально невозможно в вод¬
ных растворах.14.8. Смещение равновесия изомеризации
под влиянием растворителяЧатт и 'Уилкинс установили, что комплексы общей формулы
^i;c-[ML„C12] (где М — Pt, Pd; L — AsR3, SbR3, PR3; R — арил)
при растворении в бензоле претерпевают обратимую изомеризацию,
причем равновесие сдвинуто в сторону транс-форм. Оказалось, что
добавление полярных растворителей (диэтиловый эфир, ацетон,
нитрометан, ацетонитрил, метанол) к циклогексановому раствору
^uc-[(Bu3P)2PtCl2] снижает выход транс-изомера. Таким образом,
полярные растворители способствуют смещению цис-транс-рав¬
новесия в сторону полярной формы комплекса.В хлороформе цис- транс-изомерные формы комплекса [Pt(R2S)2Cl2]
находятся в равновесии. При переходе от хлороформа к более поляр¬
ным растворителям равновесие также смещается в сторону более
полярной цис-формы.Аналогично для равновесной системы, содержащей неэлектролит¬
ную и ионную формы комплекса, с увеличением полярности раство¬
рителя равновесие должно смещаться в сторону ионной формы. Так
было установлено, что равновесие
(где р f р — бис-дифенилфосфинометан) смещается в сторону ион¬ной формы в порядке СН3ОН > С2Н2С14 > СН2С12.Котегов и Кобилов установили, что при кипячении в бензоле жел¬
тая форма комплекса [ReOCl3(PPh3)2] в течение трех часов переходит
в зеленую форму, которая выпадает в осадок. Кипячение зеленого комп¬
лекса в водном растворе соляной кислоты вновь приводит к получению
желтого соединенияClPPh,ClPh,PRePPh,Clc„H.HCI, H,0ClClRePPh,JClОж<1лтан фгр: laозеленая формаЗеленая форма должна быть более полярна, чем желтая, поэтому она
менее растворима в бензоле и вследствие этого выделяется в осадок.
Таким образом, неполярный растворитель может смещать равновесие
в сторону более полярной, но менее растворимой формы.Процесс своеобразной изомеризации олефинового комплекса
[Pt(PPh3).,(C2Cl4)] сильно зависит от природы растворителя. В таких
растворителях, как кипящий бензол, ацетон, хлористый метилен, комп¬
лекс существует в олефнновой форме. Добавление к бензольному раст¬
вору спиртов, четыреххлористого углерода или тетрацианоэтилена
способствует изомеризации л-комплекса в сг-внннльнын:сн3он[Pt (PPh3)2 (C.CI,)! [Pt (PPh3)2 (CCI = СС1г)С1]C.H,По данным спектров ПМР и колебательных cr-аллильная форма
комплекса [Pl(CH2CH =CHMe)CI(PPh3)2], существующая в бензоле,
в хлороформе обратимо переходит в ионную л-аллильную форму:PPhМс снс‘зс„нбпб,МеPh,Px/Pt-Ph,PCI14.9. Влияние растворителя на образование
комплекса в определенном координационном
полиэдреДобавлением рассчитанного количества фосфина к спиртовым раст¬
ворам соответствующих солей №Х„-пНЮ были получены комплексы
[Ni(BuPhaP)2Ха], где X — С1", Вг“ и Г. Установлено, что выделенные
из спиртовой среды твердые соединения [Ni(BuPh-’P)‘iX:] являются389
парамагнитными. При растворении в бензоле их магнитная восприим¬
чивость понижается. Считают, что в бензоле парамагнитная форма
комплекса частично переходит в диамагнитную, в результате чего
устанавливается равновесие:[Ni (BuPh2P)2X2] ** [Ni (BuPh2P)2X;]диамагнитный парамагнитныйРасчеты показывают, что в случае хлоридного комплекса в равно¬
весии находится 10% диамагнитной формы, в случае бромидного 60%
и в случае иодидного 90%. Переход парамагнитной формы в диамаг¬
нитную, вероятно, связан с изменением тетраэдрической структуры
комплекса в плоскоквадратную.Аналогичные результаты получены для алкилдифенилфосфнновых
комплексов [Ni(RPh2P)aX2], где R — метил, этил и пропил. Они могут
существовать как в диамагнитной (плоскоквадратной) форме, так и в
парамагнитной (тетраэдрической). Диамагнитная форма имеет темно¬
красный или коричневый цвет, а парамагнитная — голубой или зеле¬
ный. В хлороформе и нитрометане эти комплексы имеют зеленую ок¬
раску, а в бензоле и сероуглероде — темно-красную с примесью зе¬
леной.Природа растворителя иногда оказывает существенное влияние
на число координированных лигандов у одного и того же иона' ме¬
талла. Так, олово (II) в диметилформамиде с роданидными ионами
образует комплекс [Sn(NCS)3]", а в метаноле и ацетонитриле при тех
же условиях получается [Sn(NCS)4]2-. Кадмий (II) в диметилформ¬
амиде и метаноле присоединяет лишь четыре роданидных лиганда,
а в ацетонитриле — шесть. Установлено также, что прочность рода¬
нидных комплексов олова (II) и кадмия (II) возрастает при переходе
от диметилформамида к метанолу и далее к ацетонитрилу.14.10. Образование связевых изомеров
в зависимости от растворителяИзучение поведения комплексов разных типов, содержащих тиоцианат-
ный лиганд, в различных растворителях показало, что в большинстве
случаев способ его координации определяется природой растворителя.
В растворителях различной полярности можно получить связевые
изомеры с этим амбидентатным лигандом. N-Связанные изомеры обра¬
зуются преимущественно в неполярных растворителях, а S-связанные
изомеры, как правило, в полярных растворителях. Некоторые ис¬
следователи связывают стабилизацию отдельных форм связевых изо¬
меров с диэлектрической константой растворителей.В литературе имеется довольно много примеров переориентации
тиоцианатного, селеноцианатного и цианатного лигандов. Так, иссле¬
дование ИК и электронных спектров растворов комплексов типа
[RhL*(CO)X], [IrL(CO) (NCSfl и МЬХа (где М - Pd(II), Pt(II);
L — фосфиновые лиганды; а X'—> SCN", SeCN", OCN") показало, что
все тиоцианатные и селеноцианатные комплексы (за исключением
родиевых и иридиевых комплексов) имеют S- или Se-связанные ли¬390
ганды в таких растворителях, как диметилформамид, диметилсульф-
оксид, имеющих высокую диэлектрическую постоянную. В растворах
бензола и в хлороформе образуются комплексы с S- (Se-) и N-
или только с N-связанными лигандами.Комплекс [Co(CN)6(SCN)]3_ в воде является S-связанным изомером,
а в хлористом метилене он переходит в N-связанный изомер:СН,С1«[Со (CN)6 (SCN)]3' ; * [Со (CN)6'(NCS)]3-н,оНеполярные растворители (CCU, С0Н6) не оказывают влияния на
способ координации тиоцианатных групп с металлом в комплексах
состава [M{P(OR)3}2(NCS)j], где М — Pt(II) и Pd(II). В этих раст¬
ворителях сохраняется тот же тип координации, что и в кристалличе¬
ских соединениях. Метанол, диметилформамид, диметилсульфоксид
и ацетон способствуют образованию S-связанных изомеров.В качестве другого примера можно привести нитро-нитритоизо-
мерию, когда под влиянием растворителя также меняется донорный
центр амбидентатного лиганда. Комплекс никеля [NiD2(NC>2)2] с
М,М'-диэтилэтилендиамином (D) в твердом состоянии содержит нитро¬
группу. В хлороформе он находится в двух равновесных формах:
[NiD2 (N02)2] *2: [NiD2(ONO)2]Аналогичный комплекс с N.N'-дибензилэтилендиамином (Dben)
в твердом состоянии существует, наоборот, в виде нитритокомплекса.
При растворении в хлороформе также устанавливается нитритб-нитро-
равновесие:[Ni (Dben)2 (ONO)2] [Ni (Dben)2'(N02)2]14.11. Влияние растворителя на скорость реакции
и ее механизмВода, используемая в качестве растворителя, часто играет весьма
активную роль в реакциях замещения и изотопного обмена в комп¬
лексных соединениях. Известно, что многие реакции протекают через
стадию образования аквокомплекса с последующим замещением воды
на вступающий лиганд. Это является одной из причин того, что при
замене воды на органический растворитель скорость реакции часто
резко понижается. Этот принцип понижения скорости реакций замеще¬
ния используют в синтезе. Так, известно, что внутрисферные молекулы
диалкилсульфоксидов в комплексах платины (IV) довольно легко за¬
мещаются в водном растворе на хлорид-ионы. Поэтому при окисле¬
нии комплексов типа [Pt(R2SO)ACh] по реакцииС12[Pt (R2SO) АС12] >- [Pt(RsSO) АСЦ]внутрисферный сульфоксид частично замещается на хлорид-ион. Для
устранения этой побочной реакции окисление ведут в хлороформе.
В этом случае продукт получается с хорошим выходом и аналитиче¬
ски чистым.391
Важную роль играет растворитель в процессах рацемизации комп¬
лексов переходных металлов. Оказалось, что трис-фенантролиновый
и трнс-дипиридильный комплексы никеля (II) в воде рацемизуются
значительно быстрее, чем в спиртах или нитробензоле, а аналогичные
комплексы железа (II) — наоборот. Трис-фенантрблиновый комплекс
никеля (II) рацемизуется в диметилформамиде и диметилсульфоксиде
еще быстрее, чем в воде. Таким образом, пока нельзя сказать ничего
определенного о характере влияния растворителя на скорость рацеми¬
зации.В препаративной практике часто приходится «искать» наиболее
подходящий растворитель для проведения синтеза или для ускорения
реакции и увеличения выхода конечного продукта. Весьма нагляден
следующий пример: для получения транс-[PtPy2Cl2] из [PtPy4]Cl2
в воде требуется длительное кипячение в присутствии соляной кис¬
лоты, а в диметилформамиде реакция идет мгновенно и самопроиз¬
вольно. Расщепление аналогичного тетраимидазольного комплекса
платины (II) с образованием транс-диимидазольного соединения также
происходит гораздо быстрее в диметилформамиде, чем в воде.Реакция между mpaHc-[HPt(PEt3)2Cl] и днметилацетилендикарбо-
ксилатом в метаноле идет в течение нескольких минут при комнатной
температуре:RCi=cR'транс-[НР1 (PEt3),CI] ► цис-(транс-)[(HCR' =CR) Pt(PEt3)2 Cl]В бензоле она заканчивается более чем через неделю. Аналогичная
реакция с СН3СэССООСН3 проходит за 24 ч в метаноле, за 7 дней
в хлороформе и практически не идет в ацетоне.Если механизм реакции включает стадию образования пятикоор¬
динационного интермедиата или реакция сама по -себе должна при¬
водить к увеличению координационного числа центрального атома,
то для реакции следует выбирать растворитель с малой склонностью
к комплексообразованию. Так, при изучении реакции внедрения серо¬
углерода в связь Pt—Н комплекса mpaHc-[H:Pt{P(C6Hn)3}2], приво¬
дящей к mpawc-[HPt(S2CH) {Р(С6НЛ)3}2], установлено, что она про¬
текает в таких растворителях, как гептан, бензол, диэтиловый эфир,
однако в тетрагидрофуране и ацетонитриле не идет совсем. Это может
быть объяснено образованием устойчивых (1:1) аддуктов растворите¬
лей с комплексом, препятствующих координации сероуглерода, а
также тем, что механизм внедрения включает предварительную ста¬
дию образования пятикоординационного интермедиата с/последующим
внедрением координированного сероуглерода в связь Pt—Н.Вопрос о влиянии растворителя на скорость и механизм реакции
замещения в координационных соединениях здесь не излагается. Рас¬
смотрим лишь результаты работы Ежовской—Тшебятовской и Кел¬
лера по исследованию механизма обмена цианидного нона в комп¬
лексе [Fe(CN)6NO]2" в растворах формамидов на основе кинетических
данных. Они считают, что молекула растворителя атакует коорди¬
нированную нитрозильную группу с образованием промежуточного
комплекса:392
((NC)5Fe=N = О J-- + : О = C</НNNRoH ■IС —NRo(NC)sFe — N./
'ЧО:2-Внешнесферный цианидный ион взаимодействует с промежуточным
комплексом по уравнениюнI.О =С—NR.,(NQsFe— N2-1JCN_’ НI14CN—С—NR„IОI(NC)sFe N=0з—На следующей стадии происходит обмен меченой цианидной группы
с цианидным лигандом, находящимся в цис-положении:Н -3-i*CN—С—NR,I ‘CN ОI I(NC)4Fe N=014CNI(NC)4 Fe=N=02- HI+ 0=C-NR,+CN'B альтернативном механизме эти авторы допускают вначале образова¬
ние интермедиата по схемен нI IRjN—C=0 + i*CN- -*■ R„N—С—,4CN* Io-Далее происходит атака интермедиатом координированной нитро¬
зильной группы и обмен цианидными группами по приведенному вы¬
ше уравнению.В любом из этих механизмов роль растворителя исключительно
важна.В. В. Скопенко с сотр. получил четкие экспериментальные данные
о соответствии сольватирующей способности растворителей и констант
устойчивости комплексов серебра в этих растворителях [5]. Они изу¬
чали константы устойчивости соединений серебра с трицианметанид-
ным С(СЫ)з, дицианамидным N(CN)j лигандами, а также с гало-
гениднымн ионами в неводных средах. Было установлено, что наибо¬
лее устойчивые комплексы образуются в пропандиол- 1,2-карбонате,
а наименее прочные — в диметилсульфоксиде.К важному заключению о влиянии протонных и апротонных раст¬
ворителей на устойчивость комплексов пришел С. Арланд [6]. По его
мнению, комплексы, лиганды которых склонны образовывать водо¬14—817393
родную связь, должны быть намного стабильнее в апротонвых раст¬
ворителях по сравнению с протонными. Следует подчеркнуть, что
такое утверждение справедливо при условии, если сольватация иона
металла в апротонном растворителе незначительно превышает соль¬
ватацию в протонном растворителе.Иногда при образовании водородных связей протонсодержащнми
молекулами растворителя с реакционным центром комплекса проис¬
ходит его «блокирование» и атака реактанта направляется на другой
центр.А. К. Бабко, А. Т. Пилипенко с сотр. [7, 8] установили сильное
влияние растворителя на квантовый выход люминесценции комплек¬
сов алюминия и ниобия с некоторыми органическими реагентами
(морин, салицилаль-о-аминофенол, сульфонафтолазорезорцнн). До¬
бавление органических растворителей (метанола и др.) к водным
растворам таких комплексов повышает относительную интенсивность
флуоресценции и квантовый выход люминесценции.С дополнительной библиографией по некоторым из изложенных
вопросов можно ознакомиться по статье [9].
ГЛАВАТИПЫ ТВЕРДОФАЗНЫХ ТЕРМИЧЕСКИХ
ПРЕВРАЩЕНИЙ КООРДИНАЦИОННЫХ
СОЕДИНЕНИЙВ настоящее время одннм из наиболее распространенных методов че¬
редования выделенных в твердую фазу координационных соедине¬
нии является термически» анализ. Он занимает второе место после
колебательной спектроскопии. Отчасти это объясняется широким
использованием дериватографов, позволяющих одновременно ре¬
гистрировать массу, скорость нагревания, изменение массы и тепловых
свойств вещества при повышении температуры. Известны и другие
методы термического анализа, которые позволяют следить за скоростью
газовыделення, изменения магнитных свойств, изменения масс-спектра
выделяющихся газообразных продуктов, изменения электрической про¬
водимости исследуемого вещества и др. Однако подавляющее число
исследований твердофазных термических превращений координа¬
ционных соединений сводится к изучению «термической устойчивости».
О ней, как правило, судят по температуре начала разложения соеди¬
нения или по температурному интервалу, в котором осуществляется
процесс. Часто по температуре отщепления лиганда судят о прочности
его связи с центральным атомом. Необоснованность такой интерпре¬
тации термогравиограмм аргументирована В. А. Логвиненко [1].Первые сведения о термических превращениях координационных
соединений были получены препаративно. Указанием на химические
превращения в твёрдой фазе обычно служило изменение внешних
признаков соединения и изменение массы навески. Поскольку перед
анализом вещества высушивали до постоянной массы при определен¬
ных условиях, то наиболее часто встречающимся типом твердофазных
превращений оказалось удаление кристаллогидратнойводы или кри¬
сталлосольватных молекул растворителя.Качественно новый этап в исследовании термических превращений
координационных соединений начался после разработки теоретиче¬
ских основ метода дифференциального термического анализа. В на¬
шей стране это связано с созданием Н. С. Курнаковым пирометра.
Для систематического изучения твердофазных превращений коорди¬
национных соединений пирометр был применен А. В. Николаевым.
В координационной химии пирометр, в частности, широко использо¬
вался для того, чтобы отличать друг от друга изомерные соединения.Большие возможности для изучения термических твердофазных
превращений появились после создания дериватографа. До этого вре¬
мени гравиметрический анализ и дифференциальный термический
анализ проводили на двух различных приборах. Это создавало труд¬
ности при сопоставлении результатов двух различных видов измерений.395
В дериватографе одновременно получаются три характеристики:
изменение массы в зависимости от температуры (ТГ), дифференциаль¬
ная кривая изменения массы (ДТГ) и дифференциальная кривая из¬
менения температуры (ДТА). Поэтому с появлением дериватографа на¬
ступил новый этап в исследовании твердофазных процессов.15.1. Дегидратация и деакватацияМногие координационные соединения кристаллизуются из водных или
водно-органических растворов с молекулами воды. Вода входит в
кристаллическую решетку соединений и поэтому называется крис¬
таллизационной. Процесс удаления кристаллизационной воды назы¬
вают дегидратацией. В отличие от этого удаление внутрисферной воды
из координационных соединений часто называют деакватацией. Эта
терминология принята и использована в книге.Удаление кристаллизационной воды всегда является эндотермиче¬
ским процессом. При дегидратации энергия расходуется на работу
против сил Ваи-дер-Ваальса и на удаление воды. Теплота испарения
воды в температурном интервале 60—100 °С составляет примерно
40 кДж/моль. На кривой ДТА процесс дегидратации обычно проявля¬
ется в виде сильного эндотермического пика.Число молекул кристаллизационной воды может изме¬
няться в широком диапазоне. Например, соединение
[CoEn3]2[Co(DH)oS03(NCS)]3, где DH — остаток диметилглиоксима,
кристаллизуется с двадцатью двумя молекулами воды, а соединение
[CoEn3]2[Co(DH);,S03(NO.,)]3 — с девятнадцатью. Поэтому процесс де¬
гидратации соединений, содержащих более одной молекулы воды, мо¬
жет происходить ступенчато. Классическим примером является де¬
гидратация медного купороса CuSO-s-SHsO. При 45 °С отщепляются две
молекулы воды и еще две молекулы отщепляются при 82 °С. Послед¬
няя молекула воды удаляется из комплекса при 200 °С.Ступенчатое отщепление кристаллизационной воды часто рассмат¬
ривают как аргумент в пользу неравноценности этих молекул в со¬
единении. Однако такого рода интерпретация термогравиограмм не¬
однозначна. В процессе дегидратации может происходить перестройка
кристаллической структуры, что находит отражение на характере
удаления оставшихся молекул воды.Процесс удаления внутрисферной молекулы воды гораздо более
сложен. Он включает разрыв координационной связи ион металла —
вода и освобождение во внутренней сфере комплекса координаци¬
онного места. Это место заполняется или анионом из внешней сферы,
или внутрисферным лигандом путем повышения его дентатности вслед¬
ствие замыкания цикла. Таким образом, в процессе деакватации про¬
исходит не только разрыв координационной связи, но и образование.
В энергетическом балансе эти процессы в определенной мере ком¬
пенсируют друг друга, поскольку их тепловые эффекты имеют про¬
тивоположные знаки. Процесс испарения воды эндотермический и в
целом деакватация, как правило, также характеризуется эндотерми¬396
ческим эффектом. Это утверждение справедливо до тех пор, пока тер¬
молиз проводится в открытом сосуде. Если деакватацию проводить в
закрытой системе (запаянной ампуле), где испарение воды не имеет
места, то картина резко изменяется [2]. Поскольку эндотермический
процесс испарения воды отсутствует, появляется возможность вы¬
явить тепловой эффект суммарного процесса, а именно: разрыва ко¬
ординационной связи вода — центральный ион и образования новой
координационной связи. В частности, деакватация комплексов типа
[Co(NH3)6H„0]X3 с образованием [Co(NH3)5X]X2 (где X — СГ, Вг“,
Г) при одной и той же температуре в открытой системе в целом эндо¬
термический процесс, а в закрытой — экзотермический (табл. 15.1).Таблица 15.1. Энтальпия (в кДж/моль) реакций деакватациикомплексов типа [Со(\Н3)6Н;0]Х3 в открытой (ДНа) и закрытой системе (\Нв )по данным [2]X•ЛЯАд//вС1-26,4±2,5—19,2±2,1Вг-26,4 + 2.5— 15.5+1 ,71-26,8+2,5—7,1+0,8N0-23,4+2,13,8+0,4 (102°С)—5,0±0,4 (120°С)Если принять, что различие в энергиях кристаллических решеток
комплексов [Co(NH3)6H20]X3 и [Co(NH3)5X]X2 невелико, то суммар¬
ный экзотермический эффект деакватации в закрытой системе можно
отнести за счет разницы в энергиях координационных связей Со—ОНг
и Со-Х.Естественно стремление исследователей привлечь термоаналитиче¬
ский метод для решения вопроса о местонахождении воды в комплексе.
Из общих соображений следует, что дегидратация (отщепление крис¬
таллизационной воды) должна протекать при более низких темпера¬
турах, чем деакватация (отщепление координированной воды). Этот
вывод следует из того, что работа против сил Ван-дер-Ваальса
требует меньшей затраты энергии, чем разрыв координационной
связи. Многочисленные экспериментальные факты вполне согла¬
суются с данным положением. Однако отщепление как кристал-
логидратной воды, так и внутрисферной протекает в довольно
широких температурных интервалах. Эти температурные ин¬
тервалы перекрываются и потому практически невозможно на
основании значения температуры начала удаления воды сделать
однозначное заключение о ее местонахождении. Мало надежны для
решения данного вопроса также площади эндотермических пиков.
Хотя из общих соображений площадь эндотермического пика, свя¬
занного с дегидратацией, должна быть больше, чем площадь пика де¬
акватации.К особому случаю можно отнести реакции деакватации двукомплек¬397
сных соединений, например, типа [Co(NH3)5H.,0] [Co(CN)G]- В этом
соединении пет внешнесферного лиганда, способного занять место
уходящей молекулы воды в комплексном катионе. Однако координи¬
рованный цианидный лиганд комплексного аниона имеет свободную
электронную пару, способную вступать в донорно-акцепторное вза¬
имодействие. Поэтому термическое превращение комплекса протекает
с образованием мостикового соединения по уравнению[Со (NH3h НоО] [Со (CN)0] - Н„0 + [(NH3)6 Co-NC-Co (CN)5]Описано немало реакций деакватации в твердой фазе с образованием
мостиковых комплексов.Мостиковые комплексы часто образуются при удалении внутри-
сферной молекулы воды измоноядерных комплексов неэлектролитного,
а также анионного типов. Например:Реакции этого типа довольно характерны для аквогидроксокомп-
лексов. Например, соединение г4ис-[Со(ЫН3)4(НзО)ОН]Вг2 с эндо¬
термическим эффектом при 120 °С отщепляет воду в соответствии с
уравнениемТакие процессы получили название реакций деакватации—оляции.
В настоящее время нельзя определенно предсказать знак термиче¬
ского эффекта этих реакций. Известны как эндотермические, так и
экзотермические реакции деакватации—оляции.Твердофазные реакции деакватации изучены довольно широко
потому, что известно большое число аквокомплексов. Почти столь же
распространены аминокомплексы металлов. Принципиально же нет
большого различия в свойствах между акво- и аминокомплексамн.
Поэтому мы не останавливаемся подробно на рассмотрении термолиза
аминокомплексов. Укажем лишь, что иногда путем термического
твердофазного превращения аминатных комплексов удается полу¬
чить соединения более простым и надежным способом, чем
синтезом в растворах. Например, при разложении в интервале
температур 160—180 °С комплекса [Pt(NH3)5Br]Br3 получается
mpa/ic-[Pt(NH3)4Br-2]Br2. Синтез этого соединения путем окисления
[Pt(NH3)i]Br2 бромом в водной среде наряду с m/?a/ic-[Pt(NH3)4Br.,]Br.,2[Со(\Н3),(Н20)С13] -*• 2Н20 + [Clj(NH3)2Co,\/•Со (NHghCU]Cl/Cl2К [Со (NH3) (НоО) C14J 2Н20 + К2 [Cl3 (NH3) Со:\'Со (NH3)CI3J\CI/он2 цис-lСо (NH3)4 (Н20) ОН] Вг2 2Н20 + [(NH3)4Co;ОН398
приводит к образованию значительных количеств [Pi(NH3)4Br(OH)]Br2.
Иергенсеновское расщепление [PtPy4]Cl„ соляной кислотой
с целью получения транс-[Pt Ру„С1;] проводят при тем¬
пературе кипения раствора. Образующийся в процессе синтеза
осадок приходится периодически удалять фильтрованием, что растя¬
гивает время синтеза до часа н более. Нагревание в твердой фазе
комплекса [Pt Ру4]С12 при 140 °С приводит к образованию
транс-\Pt Ру.,СЬ] за более короткое время и со 100%-ным выходом.Термическое превращение комплексов с удалением из внутренней
сферы нейтральных лигандов широко распространено. Если лиганды
летучие, то отдельные ступени выражены четко. Для менее летучих
лнгандов процессы термолиза протекают сложнее. Они часто сопровож¬
даются термолизом внутрисферных лигандов или лнгандов, покинув¬
ших внутреннюю сферу, но вследствие низкой летучести оставшихся
в исследуемом образце.15.2. Термическая изомеризацияИзомеризация плоскоквадратных комплексов соединений платины
(II) и палладия (II) рассмотрена в гл. 8. На основании большого фак¬
тического материала сформулировано правило, позволяющее пред¬
сказывать направление процесса изомеризации и относительную тер¬
модинамическую устойчивость изомерных соединений.15.2.1. Изомеризация октаэдрических комплексовТвердофазная изомеризация комплексов октаэдрического типа в ос¬
новном изучена на примерах соединений кобальта (III) и хрома (III).
В настоящее время получено большое число экспериментальных фак¬
тов такой изомеризации. К сожалению, несмотря на большой факти¬
ческий материал, не удается найти принципа, позволяющего пред¬
сказывать направление процесса изомеризации октаэдрических
комплексов.Для описания путей изомеризации предложены два механизма:
внутримолекулярный и межмолекулярный. Внутримолекулярный про¬
цесс стереохимических перегруппировок относится к комплексам с
циклическими лигандами. Он предусматривает отрыв одного конца
бпдентатного лиганда с превращением его в монодентатный. Этот ме¬
ханизм относится к типу размыкания — замыкания цикла. Другой
вариант внутримолекулярного механизма не предполагает отрыва
лиганда. Его называют «твистъ-механизмом (механизмом «скручи¬
вания»), Эти два механизма различаются координационным числом
комплекса в переходном состоянии (5 или 6 соответственно). Геомет¬
рия пятикоординационного интермедиата сводится к тригональной
бипирамиде или тетрагональной пирамиде с разомкнутым циклом в
аксиальном или экваториальном положении.Внутримолекулярный механизм с размыканием — замыканием
Цикла весьма характерен для процессов, протекающих в растворах.399
При его осуществлении происходит сольватация по месту отрыва од¬
ного из концов бидентатного лиганда с последующим его замыка¬
нием в другом месте и удалением сольватной молекулы. В твердой
фазе сольватирующую роль иногда выполняют молекулы кристалли¬
зационной воды или другого растворителя.Дтя твердофазной изомеризации более вероятным является внутри¬
молекулярный механизм без разрыва связи. Для стереохимических
перегруппировок трис- и бис-хелатных комплексов октаэдрического
типа «твист»-механизм впервые был предложен Реем и Даттом, а позд¬
нее его подробно рассматривал Бейлар. Схема «твиста-механизма по
Бейлару может быть представлена следующим образом:ьВ переходном состоянии донорные атомы лигандов занимают вершины
тригональной призмы.Межмолекулярный механизм изомеризации предполагает обяза¬
тельный отрыв лиганда от центрального атома и временный выход
его из внутренней сферы комплекса.15.2.2. Связевая изомеризация комплексовИнформация о твердофазных термических процессах, связанных с
переориентацией амбидентатных лигандов, сравнительно невелика.
Наиболее изученной, так же как и в растворах, оказалась нитро-
нитритная изомеризация. Примером такой изомеризации может слу¬
жить обратимый процесс[Со (NH3)5N03J С1а ** [Со (NH3)6ONO] С1аПрямая реакция является экзотермическим процессом. Равновесную
смесь изомеров можно получить при нагревании как одного, так и
другого изомеров в температурном интервале 162—212°С.Интересное термическое превращение в твердой фазе выявлено
Л. К. Шубочкиным при изучении Ks[Pt(NOj)6]. В интервале темпера¬
тур 220—245 °С протекает эндотермическая реакция, в процессе ко¬
торой комплекс Ks[Pt(N02)6] восстанавливается до соответствующего
соединения платины (II). Получающееся соединение по анализам от¬
вечало составу K*[Pt(NOa)i]. Необычным было то, что оно имело не
белую, а ярко-красную окраску. Оказалось, что в этом соединении на¬
ряду с нитрогруппами содержатся координированные нитритогруп-
пы. Суммарная реакция может быть изображена схемойК: [Pt(NO,)e] К-. [Pt (NO,). (ONO)2] + 2N02400
Примером связевой изомеризации роданндного лиганда служит
процесс[Со (NH3)B SCNJ (SCN)., [Со (NH3)5 NCS] (SCN).,Твердофазная изомеризация комплекса в этом случае сопровождается
обменом внутрисферного роданндного лиганда на внешнесферный.
Опыты с меченым углеродом-14 роданидным ионом показали, что по
диссоциативному механизму изомеризуется по крайней мере 45%
комплекса.Превращение изотиоцнанатной группы в тиоцнанатную происхо¬
дит в комплексе K3[Co(CN)5NCS], который при нагревании в твердой
фазе изомеризуется в Кз[Со(СМ)б5С1М]. Термограмма комплекса
K3[Co(CN)sNCS] имеет два необратимых эндотермических пика. На
основании ИК и абсорбционных спектров пришли к выводу, что вто¬
рой эндотермический пик (при 128°С) соответствует необратимому
процессу изомеризации.При взаимодействии K3[Cr(CN)6] с растворами солей железа (II)
образуется комплекс KFeCr(CN)6 кирпично-красного цвета. Считают,
что он состоит из трехмерных ячеек, которые содержат группы
Fe <- Ne С-— Сг. При нагревании кирпично-красного комплекса
KFeCr(CN)6 до 100 °С его цвет переходит в темно-зеленый. На осно¬
вании ИК спектров это превращение относят к связевой изомериза¬
ции, в процессе которой фрагмент Fe-(-N = C—Сг переходит в
Fe-C==N-»Cr.15.2.3. Другие виды изомеризацииПлоскоквадратные комплексы никеля (II) с бензимидазолом,
[Ni(Bimd)4] (N03)2-2,5 С2Н6ОН и [Ni(Bimd)4]I2 при нагревании в
твердой фазе превращаются в соединения октаэдрического строения
[Ni(Bimd)4(N03)2] и [Ni(Bimd)4I2]. Внешнесферные ацидогруппы при
этом входят во внутреннюю сферу. Превращение нитратного комп¬
лекса происходит при 130 °С с потерей кристаллизационных молекул
спирта. Превращение иодидного комплекса осуществляется при 190 °С
без изменения массы.При нагревании полиядерных комплексов [Mo6X8]Y4 (где X и
Y —СГ, Вг“, I", ОН') происходит обмен внутри- и внешнесферных
ацидолигандов. В частности, в температурном интервале 200—300° С
осуществляется реакция обмена[Мо6С18] (ОН)4 - [Мо,С!6 (ОН)2] С12 (ОН)2При температуре около 450 °С происходит экзотермический процесс:
[Мо6С18] Вг4 -*• [Мо(1С!4Вг4]С!4Эти превращения, по существу, являются реакциями изомеризации с
переходом одних ионизационных изомеров в другие.401
15.3. Правило термического превращения
двухкомплексных соединений типа солей
Магнуса и ВокеленаВ 1920 г. основываясь на сравнительно немногочисленных экспери¬
ментальных фактах (известно было около 10 реакций), Л. А. Чугаев
пришел к заключению, что соли типа соли Магнуса [PtL4] [PtX4]
(где X — Cl', Br”, L — амины, тиоэфиры) при нагревании подверга¬
ются твердофазному превращению в соединения неэлектролитного ти¬
па [PtL2X2],В настоящее время изучено более 100 такого рода реакций [3].
Если не происходит разложения комплексов, то во всех случаях с вы¬
делением энергии соединения [MLJ [МХ4] (где М = Pt(II) и Pd(ll),
X = СГ, Вг“, Г) превращаются в соединения типа [МЬ2Х2]. Чем выше
трансвлияние лигандов L, тем при более низких температурах проис¬
ходит превращение. Комплексы типа соли Вокелена [PdL4] [PdX4]
подвергаются термопревращению при гораздо более низких темпера¬
турах, чем соединения типа солей Магнуса. Термопревращение пос¬
ледних протекает без разложения лишь в случае серо-(тиоэфиры) и
фосфорсодержащих (фосфины, фосфиты) нейтральных лигандов. Если
нейтральным лигандом является амин, то термопревращение в комп¬
лекс неэлектролитного типа сопровождается частичным разложением
исходного соединения до металлической платины.Многочисленные попытки получить соединения типа соли Магнуса
с тномочевиной и ее производными путем взаимодействия комплексов
[PtL4]Cl2 и K2[PtCl4] оказались безуспешными. В качестве продуктов
реакции сразу выпадали комплексы неэлектролитного типа [PtL,CL,].
Использование неводных растворителей позволило синтезировать
два комплекса с М,М'-тетраметилтиомочевнной [Pt(Tmethio)4][PtX4],
где X — СГ и Вг-. Установлено, что бромидное двухкомплексное
соединение в интервале температур 150—165°С превращается
в однокомплексное [Pt(Tmethio)2Br2]. Превращение комплекса
[Pt(Tmethio)4] [PtCl4] в аналогичное однокомплексное
[Pt(Tmethio).,CI2] сопровождается разложением, поэтому в четком
виде процесс термопревращения не выявлен.Несколько иначе ведут себя роданидные комплексные соединения
[PtL4] [Pt(SCN)4], где L — NH3 и Ру. При нагревании в твердом со¬
стоянии с отщеплением молекулы аммиака или пиридина они превра¬
щаются в соединения мостикового типа Pt2L3(SCN)4. Однако если в ка¬
честве нейтрального лиганда взяты бидентатные, где L' — 1,2-бис-
дифгнилфосфиноэтан или ji-аминодиэтилсульфид, то двухкомплексные
соединения [PtL'2| [Pt(SCN)J при нагревании четко превращаются
в однокомплексные неэлектролитного типа [PtL'(SCN)2].Для комплексов типа соли Вокелена с серо- и фосфорсодержащими '
лигандами превращение в соединения неэлектролитного типа проис¬
ходит при комнатной температуре. При сливании растворов [PdL4]X3 =
и KJPdXJ в осадок сразу выделяются соединения [PdL2X2]. Однако
соединения типа соли Вокелена с аминами [PdA4] [PdX4] выделяются402
б твердую фазу легко и с хорошим выходом. Они подвергаются термо-
превращенню количественно без каких-либо признаков разложения.Таким образом, изу.чение платиновых комплексов типа соли Маг¬
нуса и палладиевых соединений типа соли Вокелена с разнообразными
нейтральными лигандами в совокупности позволяет значительно
расширить диапазон закономерности, подмеченной Чугаевым.Можно предположить, что структура платиновых и палладиевых
соединений типа [ML4] [МХ4] и [ML'„] [МХ4], где L — монодентатные,
L' — бидентатные, нейтральные лиганды, аналогична структуре зе¬
леной соли Магнуса [Pt(NH3)4] [PtCl4]. В этом случае механизм тер¬
мического превращения можно представить как синхронный сдвиг
лигандов по одной из сторон «этажерки». Рентгеноструктурные дан¬
ные свидетельствуют о том, что в кристаллической структуре комп¬
лекса [PtEnCl2] сохраняется колонный мотив, характерный для
соединения типа соли Магнуса (I). Таким образом, геометрию крис-
сталлической структуры этого соединения можно действительно рас¬
сматривать как результат сдвига лигандов по одной из сторон «этажер¬
ки» (II):Сcl\i^NN cuС\ f СГ(N><N)1ClI IIВ комплексе ^uc-[Pt(NH3)2Clo] также повторяется колонный мотив,
характерный для [Pt(NH3)4] [PtCl4]. Следовательно, превращение
соли Магнуса в q«c-[Pt(NH3)2Cl2] не требует существенной перест¬
ройки кристаллической структуры. Интересно, что структура
m/WHC-[Pt(NH3)2Cl2] не имеет колонного мотива. Плоскости этих моле¬
кул располагаются по паркетному мотиву. Таким образом, непосред¬
ственное превращение [Pt(NH3)4] [PtClJ в mpaHC-[Pt(NH3)2CI2] по¬
требовало бы весьма существенной перестройки кристаллической
структуры.Превращение соединений типа соли Магнуса и Вокелена в комп¬
лексы неэлектролитного типа происходит с выделением энергии. Ес¬
тественно, возникает вопрос: почему это превращение выгодно энер¬
гетически?Ответ на него вытекает из расчетов констант ступенчатого заме¬
щения хлоридных ионов на аммиак в комплексе [PtCl4]2-. Они вы¬
полнены Пещевицким и Белеванцевым на основе экспериментальных
данных Гринберга и Гельфмана. Расчет приводит к следующим зна¬
чениям констант ступенчатого замещения: lg Кх =7,5; lg /0, =5,4;
lg Кз =3,3; lg /С4 =2,5. Таким образом, замещение перЕого и вто¬
рого хлорид-ионов на аммиак в ионе [PtCl4]2- с образованием
[Pt(NH3)Cl3]" и [Pt(NH3)2Clo] энергетически гораздо выгоднее, чем403
замещение хлорид-ионов в [Pt(NH3)2CI2] и [Pt(NH3)3Cl]+ с образова¬
нием [Pt(NH3)4]2+. Из этих данных следует, что процесс[Pt (NH3)4j [PtCUJ—- 2[Pt (NH3)2 Cl2]|действительно должен протекать с выделением энергии. При образо¬
вании двух молей [Pt(NH3)2Cl2] вместо [Pt(NH3)4]2+ и [PtCl4]2- проис¬
ходит выигрыш энергии за счет разницы в ступенчатых константах
устойчивости.На основании такого представления можно предсказать, в каких
комплексах должна осуществляться экзотермическая реакция пере¬
распределения лигандов (реакция пропорционирования). Если при
взаимодействии лиганда L с комплексом [MXn]m— происходит за¬
мещение X" на L, то, как правило, константа замещения каждой сле¬
дующей ступени уменьшается. При этих условиях рассматриваемые
реакции пропорционирования должны быть выгодными энергетически.
Исходя из этого можно сказать, что принципиально должны проте¬
кать, например, следующие реакции пропорционирования:[PtL3X]a [PtX4] - 3[PtLjX2][PtL3X] [PtLX3] - 2[PtL2X2][PtL4X2] [PtX.J - 2[PtL2X4]Эти прогнозы были высказаны в 1980 г. Часть из них уже полу¬
чила подтверждение. Так, например, установлено, что комплексы
[Pt(R2S)4Cl2] [PtCl6], где R—СН3, С2Н5, при нагревании до 115 °С
с выделением энергии превращаются в [Pt(R2S)2Cl4].Термическое превращение соединений типа солей Магнуса и Во-
келена — широко распространенное явление, можно даже сказать,
что оно имеет характер правила. Впервые к такому выводу пришел
Чугаев.Вполне возможно, что для комплексов некоторых металлов на¬
правление такого рода реакций может быть иным. Например, комп¬
лекс меди (II) [Си С)1ру(С204)]-2Н.,0 при стоянии на воздухе в течение1—2 дней превращается в [Cu(Dipy)2] [Си(С.;04)2]. Аналогично ведет
себя комплекс [CuQ(SCNb], где Q — 10-[2-(диметиламино)пропил]-
фенотиазин. При нагревании до 70—80 °С он превращается в двух¬
комплексное соединение [CuQ,] [Cu(SCN)4],15.4. Твердофазная термическая полимеризацияИзучено своеобразное термическое превращение биметаллического
комплекса ijuc-[Pt(NH3).,(SCNAg) (SCN)1N03. Серебро (I) в этом со¬
единении координационно ненасыщенно, поэтому принципиально
возможно «сшивание» моноядерных комплексов в полимер. На кривой
ДТА этого комплексного соединения при 134—144 °С имеется харак¬
терный экзотермический пик. Процесс протекает без изменения массы404
и изменение его энтальпии равно —38±3 кДж/моль. Контрольное
нагревание показало, что при 134 °С происходит превращение жел¬
того комплекса ^uc-[Pt(NH3)o(SCNAg) (SCN)]N03 в темно-красную
модификацию. Продукт реакции резко отличается от исходного по
растворимости и по отношению к нодиду калия и тиомочевине. На
основании исследования ИК спектров было сделано заключение, что
при нагревании комплекса цис-[Pt(NH3)2(SCNAg) (SCN)]N03 в твер¬
дой фазе происходит изомеризация и полимеризация с образованием
соединения типаno;NH,N07NH3 .....I 3 I J-NCS—Pt-SCN—Ag— NCS— Pt—SCN—Ag—I INH3 NHjВыявлена твердофазная полимеризация плоскоквадратного комп¬
лекса [Ni{P(CH„CH2CN)3}2Br2]. В этом соединении имеются свобод¬
ные нитрильные группы, обладающие донорными свойствами, а цент¬
ральный ион способен увеличивать координационное число на две
единицы. Оказалось, что уже при 80 °С протекает процесс полимери¬
зации с образованием продукта голубого цвета. Схематично он может
быть выражен уравнениемВгN I /JiNCCH;CH,—Р—Ni—Р—CH,CH:CN-ВгВЧ1 /Р“
^СН^р/ТЧгС Hi ' NI ‘ JIIс сIII IN х СН,Вг\1 -—РХ1 Вг// NПроцесс полимеризации протекает и в ацетоновом растворе. Однако
в этом случае к голубому продукту примешан зеленый. Таким об¬
разом, полимеризация в твердом состоянии осуществляется проще
н приводит к более чистому продукту.Эти два примера указывают на предпосылки, необходимые для
протекания процесса полимеризации комплексов, а именно, коорди¬
национная ненасыщенность центрального атома и потенциальная
би- или полидентатность координированного лиганда. Полимеризация
комплексов может также осуществляться, если координированные ли¬
ганды способны к реакциям полимеризацииLM ■CHR• II
CHR=LM—CHR—CHR—CHR—CHR—MLконденсации405
ML—ОН + HO—LM=ML—О—LM -|- Н20
ML—NH, + OCH-LM = ML—N = CH—LM + H20
или им подобнымMLCH2C1 + HS—L'M = MLCHjSL'M + HCI15.5. Реакции дегидрогалогенирования15.5.1. Андерсеновская перегруппировкаПрактически сразу после открытия пиридина в продуктах сухой пе¬
регонки костей Т. Андерсон получил пиридиниевый гексахлоропла-
тинат (IV) и наблюдал его превращение в водном растворе. Вернер
дал трактовку этому превращению с позиции координационной тео¬
рии, которая выражается следующими уравнениями:(PyH)2[PtCle] -► (PyH)[PtPyCl6] + HCI(PyH)[PtPyCl6] [PtPy2Cl4] + HCIТакого рода превращения ониевых комплексов позднее стали назы¬
вать «андерсоновской перегруппировкой».В настоящее время изучено большое число реакций координа¬
ционных соединений в твердой фазе. Литература до 1980 г. по этому
вопросу собрана в книге [4].Наиболее широкие и систематические исследования андерсонов¬
ской перегруппировки проведены на комплексах платины и палладия.
Общие заключения о протекании андерсоновской перегруппировки
координационных соединений в твердой фазе сводятся к следующим.1. Андерсоновская перегруппировка в твердой фазе характерна
для координационных соединений многих металлов.2. Для аналогичных соединений того же самого металла с основа¬
ниями одного и того же класса в общем наблюдается тенденция к
понижению температуры начала андерсоновской перегруппировки
с уменьшением основности лиганда, входящего в ониевый катион.3. При наличии различных лигандов во внутренней сфере комп¬
лекса протон с ониевого катиона переносится на лиганд, обладающий
большей основностью. При этом принимается, что соотношение ос¬
новностей свободных и координированных лигандов сохраняется.В начальной стадии выявления корреляции между температурой
начала андерсоновской перегруппировки и р/Са внешнесферного ли¬
ганда сделан вывод, что все изученные соединения типа (AH)JPtCl4]
по температурам перегруппировки делятся на две группы:1) комплексы с температурой начала превращения 180—200 °С
содержат амины, р/Са которых равны 9,0 и больше; 2) комплексы с
температурой начала перегруппировки 130—150°С содержат амины,
р/Са которых равны 6,0 и менее.Сравнение температур начала андерсоновской перегруппировки
ониевых комплексов платины (II) и палладия (II) с бензимндазолом
и его производными и аналогичных комплексов с пиридином и его406
производными показывает, что температуры начала термопревраще-
нпя бензимндазольных комплексов выше, чем пиридиновых, хотя
р/\л этих гетероциклических аминов близки. В связи с этим было
сделано предположение, что на температуру начала андерсоновской
перегруппировки влияет не только основность, но и наличие допол¬
нительных протонакцепторных групп в амине.Как видно из экспериментальных данных, полученных Г. X. Хам-
нуевым (табл. 15.2), андерсоновская перегруппировка в 2-амино-
пнридиновом комплексе начинается при более высокой температуре
(175 °С), чем в пиридиновом (140 °С). В данном случае, вероятно, на
повышение температуры превращения влияет увеличение основности
и наличие в аминопприднне дополнительного протоноакцепторного
центра, который «перехватывает» протон и препятствует протека¬
нию перегруппировки.Таблица 15.2. Температура начала андерсоновской перегруппировки
в комплексах типа (АН)= [PtCl]4 и значения рКа соответствующих
аминов "Амин (А)ФормулаРКа/ °г
пач* ьПиридинм5,21402-Аминопирнднн/чU-nh=/\ м6,9175Бензимидазол\^\NJ1н5,5170БензтриазолЛ> N.II 1 SnII 1 N/н1,6180Бензтиазол/Ч N1 1 1
\/\5/1,21452-Аминобензтиазол/Ч NU\sJ-nh-4,5220407
Следует отметить, что основности пиридина и бензимидазола от¬
личаются мало, но температура перегруппировки в бензимидазольном
комплексе заметно выше. Повышение температуры термического
превращения согласуется с представлением о существенной роли до¬
полнительных протоноакцепторных групп. Обращает на себя внима¬
ние тот факт, что при переходе от бензимидазола к аминобензтиазолу
основность резко уменьшается, но температура начала андерсоновской
перегруппировки в аминобензтиазольном комплексе выше. Вероятно,
повышение температуры перегруппировки связано с появлением в
лиганде третьей протоноакцепторной группы. Высокая температура
термического превращения беизтриазольного комплекса (AH)„[PtCl4]
(более высокая, чем температура твердофазной реакции аналогичного
комплекса с пиридином, 2-аминопиридином и бензимидазолом) при
очень низкой основности бензтриазола может быть объяснена только
присутствием в бензтриазоле трех протоноакцепторных групп. Ос¬
новность бензтиазола по сравнению с пиридином гораздо ниже, и
можно было бы ожидать резкого понижения температуры начала
перегруппировки, однако в случае пиридинового- и бензтиазольного
комплексов типа (AH)2[PtCl4] она протекает примерно при одной и
той же температуре. Этот факт также свидетельствует о влиянии до¬
полнительной протоноакцепторной группы в лиганде на протеканпе
твердофазной реакции замещения.Изучение перегруппировки в комплексах типа (AH)2[Pt(OH)2Cl4]
(где А — аммиак, метиламин, пиридин; ■у-пиколин, пиперидин и хи-
нолин) показало, что она протекает в соответствии с уравнением(AH)j(Pt (ОН)2С14] — IPtA2Cl4]+ 2Н„0Из двух возможных вариантов перенос протона происходит не на ,\ло-
ридный лиганд, а на гидроксильный. По сравнению с комплексами
(AH)2[PtCl6] перегруппировка в комплексах (AH)2[Pt(OH)oCl4] про¬
исходит при гораздо более низких температурах. Таким образом,
можно сделать заключение, что в процессе андерсоновской перегруп¬
пировки перенос протона происходит на лиганд с большей основ¬
ностью. С этим выводом согласуются результаты, полученные Хам-,
нуевым. Он установил, что при нагревании в твердой фазе бензимида-
зольных комплексов типа i4«c-(AH)2[Pt(N02)„X2] (где X — СГ, Вг')
перенос протона имеет место на нитрогруппу и реакция описывается
уравнением(AH)JPt (NO,)2X2] — [PtA2X2] + 2HN02Изучение термического превращения бензимидазольного комплекса
m/7QHf-(AH)2[Pt(N02)2Br2] показало, что после замещения одной из
нитрогрупп с образованием (АН) [PtA(N02)Br2] вторая нитрогруппа
оказывается закрепленной относительно низкого трансвлияния бенз¬
имидазола. Поэтому, несмотря на большую основность нитро¬
группы, вторым замещается бромидный лиганд с образованием
4«c-[PtA2(N02)Br]. Сравнение температур начала превращения бенз-
имидазольных комплексов (AH)2[PtX4], где X = СГ, Вг" и N02,
показывает, что нитрокомплекс подвергается перегруппировке при408
значительно более низкой температуре (120 °С), чем бромидный
(200 °С) и хлоридный (170 °С). Это также может быть связано
с лучшими протоноакцепторнымп свойствами нитрогруппы по
сравнению с хлорндным и бромидным лигандами.Андерсоновская перегруппировка, по существу, является реак¬
цией замещения внутрисферных лигандов на лиганды, вступающие
в комплекс из внешней сферы. Реакциям замещения внутрисферных
лигандов посвящены многие сотни исследований. В отсутствие раст¬
ворителя такие реакции изучены очень мало. Андерсоновская пере¬
группировка в твердой фазе предоставляет такую возможность. Из
имеющегося в настоящее время фактического материала следует,
что реакции замещения в растворе и твердой фазе резко отличаются.
Известно, что замещение внутрисферных лигандов в комплексах
палладия (II) протекает с гораздо более высокими скоростями, чем
в аналогичных комплексах платины (II). Можно также сказать, что
в растворах реакции замещения в бромидных комплексах платины
и палладия протекают гораздо быстрее, чем в аналогичных хлорид-
ных соединениях. В отсутствие растворителя картина резко отличная
(табл. 15.3). Температура начала андерсоновской перегруппировки
свидетельствует о том, что скорость протекания реакции становится
заметной. Следовательно, на основании температуры начала пере¬
группировки можно делать вывод об относительной скорости анало¬
гичных процессов. Из табл. 15.3 следует, что, во-первых, лабильность
аналогичных комплексов платины (II) и палладия (II) почти одина¬
кова, однако андерсоновская перегруппировка в комплексах пал¬
ладия (II) протекает даже при более высокой температуре, чем комп¬
лексов платины (II). Во-вторых, хлоридные комплексы платнны (II) и
палладия (II) несколько более лабильны, чем бромидные. ТакимТаблица 15.3. Температура начала андерсоновской перегруппиров
ки комплексов платины (II) и палладия (II)Аыин(AH)2[PtCl4](AH),[PdCl1](AH)2[PtBr4](АЫ)2[Рс1ВгЛАммиак210216220225Метиламин180205200225V3 (Этилендиамин)185183220240Пиперидин190202220235Пиридин1401401901807-Пиколин135150190190Анилин150135—160л-Толуидин130150—180■и-Нитроанилнн—152—175Хинолин132160190170-8-Оксихинолин130160——Vo, 1, Ю-Фенантролин
Нмидазол—70—105140170190250Бензимндазол1701702002401,2-Диметилбензимидазол2102152402605,6-Бензимидазол1802051852456-Ннтробензимидазол180160190210405
образом, относительная лабильность комплексов в реакциях заме¬
щения в растворах и твердой фазе резко отлична. Эти эксперименталь¬
ные факты представляют большой интерес.Своеобразная реакция, которая может рассматриваться как «ан-
дерсоновская перегруппировка в противоположном направлении» изу¬
чена Ю. Н. Шевченко с сотр. Они показали, что комплексы с 1,2-
дпкарбаундекарборат-анионом[Сг (NHsCHj),] Br (С2В„Н12)2, [Cr(NH,CH3)5 Br] (С2В9Н:2)2,[Сг (NH2CH3bCI](C2B9HI3)2> [Cr(NH2C2H6)5CI](C2B9H13)aв температурном интервале 90—100°С претерпевают передачу про¬
тона, находящегося в анионе в положении 3, к внутрисферному амину
с образованием ониевого катиона и выходом его во внешнюю сферу.
На освободившееся координационное место в комплекс входит анион
С2В9Нп, который связывается с центральным атомом посредством
л-связи. Схема реакции:[Сг (NH2CH3b Вг] (С2В0Н12)2 — (NH3CH3)2[Cr(NH2CH3)3Br (С,ВэНи)2]Подобные превращения были осуществлены Шевченко и с ана¬
логичными комплексами кобальта:50-60’С 70 —80’С[Со (NH2CH3)e] Вг (С2В„Н:2)2, *- [Co(NH2CH3)5Br](C3B9Hl2)2 ■-»— NH;CHj >- mpaKc-(NH3CH3)2 [Co(NH2CH3)3 Вг (С2ВЭНП)2] ►20’С ► wic-fNH,CH,). [Со (NHoCH3)3 Вг (С2В9НП)2]70—80ССВ заключение следует отметить, что в некоторых случаях путем
твердофазной андерсоновской перегруппировки удается просто и с
количественным выходом получать комплексы, синтез которых в
растворах весьма затруднен.15.5.2. Депротонирование внутрисферного лиганда
с замыканием циклаРеакции депротонирования внутрисферных лигандов с замыканием
ими циклов распространены довольно широко. Однако они изучены
главным образом в растворах. Информации о твердофазных реакциях
такого типа значительно меньше. Замыкание лигандом цикла, естест¬
венно, связано с повышением его дентатности.Известно, что комплекс кобальта (II) Na[Co(HL)Br], где HL —
пентадентатный этилендиаминтетраацетат-ион, при нагревании от¬
щепляет молекулу НВг с образованием соединения Na[CoL], где L —
является гексадентатным лигандом. Нагревание комплекса
[CoEn2(GlyH)Cl]Cl2, где GlyH — глицин, приводит к образованию
[CoEri2(Gly)]Cl2. Процессы дегидрогалогенирования обоих комплексов
являются эндотермическими.410
мОкснхинолпновые комплексы палладия (II) и платины (II)
[М(ОхН)2С12], в которых органический лиганд координирован посред¬
ством атома азота, при нагревании в твердой фазе четко превращаются
в хелатные соединения по реакции[М(ОхН)2С12] [МОх2] -I-2HC1Установлено, что отщепление хлороводорода и в этом случае является
эндотермическим процессом.Аналогичные салицилальэтнлендиаминовые соединения палладия
(II) и платины (II) подвергаются андерсоновской перегруппировке и
замыканию цикла в одну стадию в соответствии с уравнениемСН,—СН- си г—СН,I ' I * I 2 I ~CH=N *- +N=CH\^\ /<^.СП=М. N=CHII " JO-он ,_Н°тс\АгВ результате дериватографического изучения комплекса транс-
[Pt(GlyH)„Br2] установлено, что при 250°С он отщепляет два моля
НВг с образованием хелатного соединения [Pt(Gly)„]. Этот процесс
также связан с поглощением энергии. Для других соединений типа
m/Kwc-[Pt(GlyH)2Xa] (где X = СГ, Г) и qiic-[Pt(GlyH) (Gly)X (где
X = СГ, Вг-) наблюдалось постепенное выделение НХ без четких сту¬
пеней убыли массы. Вероятно, что в данных случаях полному раз¬
ложению комплексов также предшествуют стадии дегидрогалогени-
рования с замыканием циклов.Замыкание циклов происходит не только в результате дегидро-
галогенирования. Так, в комплексе родия (III) Na[Rh(HL)2(OH)„]-Н20
остаток иминодиуксусной кислоты (H2L) координирован бидентатно.
При нагревании комплекса происходит удаление кристаллизационной
воды. Кроме того, имеет место внутрисферная нейтрализация за счет
протона свободной карбоксильной группы и координированного
ОН". В результате этого из комплекса удаляется образующаяся мо¬
лекула воды и замыкается еще один цикл.15.5.3. Термическое депротонирование внутрисферного
аммиакаОдним из первых исследований твердофазного термического депрото¬
нирования следует считать работу В. Г. Тронева. В ней установлено,
что комплекс Re(NH3)4Cl3* при нагревании в твердой фазе превраща¬
ется в диамидосоединение в соответствии с уравнениемt°Re(NH3)4CI3 — Re(NH3)3 (NH3), С! + 2НС1* Поскольку строение обсуждаемых аминных соединений рения известно
не вполне определенно, то их формулы пишем в мономерном виде без указания
внутренней и внешней сферы.411
Этот же диамидный комплекс получается при промывании исходного
соединения Re(NH3)iCl3 жидким аммиаком. Считают, что при даль¬
нейшем нагревании амидокомплекса Re(NH3)2(NH2)2Cl образуется
имидосоедпнение и даже нитрид рения, которому приписывают фор¬
мулу (ResN)„.Аналогичные процессы наблюдал Ю. А. Буслаев с сотр. Путем
нагревания в твердой фазе комплекса Re(NH3)CU они осуществили
превращение координированного аммиака в амидную и далее в нит¬
ридную группу в соответствии с уравнениями250°СRe(NH3)CI4 ► Re (NH2)CI3+HC1400’СRe (NH2)C13 >- ReNCI + 2HC1Образование нитридного соединения рения было установлено при
термическом разложении (NHi)2ReFe. При 300°С в атмосфере аргона
протекает следующий процесс:зоо°с<NH4)2ReF6 ► ReNF + NH4F + 4HFАгОдной из первых стадий этой сложной реакции, безусловно, является
андерсоновская перегруппировка, за которой следует депротониро¬
вание координированного аммиака. Аналогичному превращению под¬
вергается комплекс (NH4)2TcF6.Депротонирование аммиака может происходить с замыканием
амидогруппой мостика между ионами двух металлов. Так, показано,
что двухкомплексное соединение [Со(МН3)зН20] [Co(CN)6] в интер¬
вале температур 130—160°С теряет воду с образованием мостикового
комплекса [(NH3)oCoNCCo(CN)5]. При температуре около 250°С
происходит отщепление HCN с образованием амидогруппы,
которая замыкает второй мостик с образованием комплекса
{(NH3)iCo(NHs) (NC)Co(CN)4]. Дегидратация комплекса[Со(МН3)5НЮ] [Fe(CN)6] происходит одновременно с потерей трех
молей HCN. Считают, что при этом образуется соединение
l(NH3)2Co(NHa)3(NC)Fe(CN)2], содержащее четыре мостика. Данное
обстоятельство вызывает сомнение, однако суть дела не в этом. Глав¬
ное, что в процесс депротонирования вовлечено три молекулы аммиа¬
ка. Термическое превращение комплекса [Co(NH3)sHjO] [Cr(CN)6]
протекает аналогично двум предыдущим. Продукт реакции соответ¬
ствует формуле[(NH3)3 5 Co(NH2)1>5 (NC);Cr (CN)5]Разработан интересный способ получения пленок из нитридов
алюминия и галлия. Эти соединения являются полупроводниковыми
материалами, обладающими значительной тугоплавкостью и большой
шириной запрещенной зоны. Способ получения нитридных соедине¬412
ний заключается в термическом разложении аммиакатов A1C13-NH3,
GaCl3-NH3 или GaI3-NH3. Процессы разложения описываются урав¬
нениемA1C13.NH3 -ч- A1N + 3HC1Для получения нитрида алюминия из хлорндного комплекса разло¬
жение ведут в интервале температур 930—1180°С.15.6. ЦтслометаллированиеРеакции, в результате которых образуется связь металла с органиче¬
ской молекулой через посредство атома углерода, называют реакция¬
ми мета.глирования.Реакции комплексов переходных металлов, в которых внутри¬
молекулярному металлированию подвергается лиганд, уже присоеди¬
ненный к металлу посредством донорного атома (О, N, Р, S, As и др.),
называют реакциями циклометаллирования:(здесь Y — О, N, Р, S, As; X — галоген, алкильная группа).Широко распространенным частным случаем реакции циклометал¬
лирования является ортометаллирование, заключающееся в замеще¬
нии водорода ароматического кольца в орто-положении на атом ме¬
талла:В последние годы выполнено огромное число работ по исследова¬
нию реакций циклометаллирования в растворах. Систематических
исследований этих реакций в твердой фазе до сих пор не проводилось,
поэтому информация о них является довольно скудной.Одна из первых реакций, которую удалось осуществить в твердой
фазе, схематично можэт быть выражена уравнениемН250-300°С-2HCIСН, С1 СИPh, Р Pi—PPhPhi Р’PPh 2Cl413
Происходит ортометаллнрование ароматических колец с образова¬
нием двух пятичленных циклов. Этот процесс исследован в атмосфере
азота дериватографическим и масс-спектрометрическим методами.Осуществлено быстрое и количественное циклометаллирование
комплекса платины (II) с о-дифенилфосфинобензальдегидом путем
отщепления НС1 при нагревании в вакууме:Считают, что этот процесс протекает либо путем окислительного при¬
соединения к платине с разрывом связи С—Н и с последующим вос¬
становительным элиминированием НС1, либо путем прямой электро-
фильной атаки платины (II) на формильную группу с замещением
протона.При нагревании в твердой фазе комплекса jMep-[OsL3Cl3], где L —
диметил(1-нафтнл)фосфин, происходит циклометаллирование одного
фосфинового лиганда в положении 8 (peri) также за счет элиминирова¬
ния НС1 с образованием пятичленного цикла:270° Смер-[OsL3Cl3] _НС1Следует отметить, что для комплексов с Р-донорными лигандами
металлирование чаще осуществляется элиминированием алкана. Та¬
кие реакции осуществлены и в твердой фазе. Например, показано, что
метильные комплексы платины (П) при нагревании в твердой фазе об¬
разуют циклометаллированные соединения путем элиминирования
СН4 (Е —Р, As):414
В первом случае металлируется положение 8, во втором — метильная
группа о-толила, что вызвано образованием устойчивого пятичленного
цикла (металлирование в о-положенин к донорному атому привело бы
к образованию напряженного трехчленного цикла).Поскольку замыкание цикла требует освобождения координаци¬
онного места у атома металла, то координационноненасыщенные со¬
единения, например [1гС1Х(СН3) (PPh3)2] (где X — СГ, Br, NCCT),
ортометаллированию подвергаются легко и количественно. Так, при
нагревании этого комплекса в вакууме в интервале температур 170—
189 °С с отщеплением СН4 образуется димерное соединение
[Ir{C6H4PPh2}ClX PPh3]o.При нагревании до 250—270 °С без растворителя 2-алкоксифосфи-
новых комплексов /7ipaHC-[PtL,Cl2] [где L — PPh2(CeH4OMe);
РРЬь(С|;Н4ОЕ1); PBu1 Ме(С6Н4ОМе); РВиг (С6Н4ОМе)] происходит ре¬
акция внутримолекулярного металлирования по кислороду алкоксиль-
ной группы (О-металлирование). От фосфинового лиганда отщепляется
метильная или этильная группа, а от металла — хлор. В результате
выделяются СН3С1 или СгНзС1 и образуются бисхелатные комплексы
с пятнчленными циклами, например:Циклометаллирование еще более объемистых лигандов РВи^ R
и PPh2R, где R — 2,3- пли 2,6-диметоксифенил, в составе комплексов
[PtLjCL] происходит еще быстрее с образованием соединений
r{«c-[Pt{OC6H3(OMe)PPh2}2] и mpaHC-[Pt{OCGH3(OMe)PBu2}2], так¬
же содержащих по два пятичленных цикла.Описан очень редкий случай образования трехчленного цикла
при термическом разложении диэтиламидного комплекса тантала.
В реакции металлируется вторичный углеродный атом этильной
группы:Та (N’Et.)^ -► Е1М—СН—Та (MEt;)3 + NEt,H]
СН3Подавляющее большинство N-донорных и О-донорных лигандов об¬
разуют в реакциях циклометаллирования пятичленные хелатные
структуры. S- и Р-Донорные лиганды показывают возрастающую тен¬
денцию к образованию трех- и четырехчленных циклов. Такие циклы
образуются в тех случаях, когда нет возможности к образованию
пятичленного цикла.15.7. Термическое декарбоксилирование
и диспропорционированиеПирндинкарбоновые кислоты при нагревании в твердом состоянии
способны отщеплять молекулу С02. В связи с этим исследована воз¬
можность протекания процесса термического декарбоксилирования
координационно связанных пнридннкарбоновых кислот в комплексах
платины (II).Объектами исследования служили комплексы с 3-пиридинкарбо-
новой кислотой: K[Pt(NC5H4COOH)Cl3] и £{wc-[Pt(NC5H4COOH);CI2].
При дернватографическом изучении этих соединений декарбоксили-
рования 3-пиридинкарбоновой кислоты обнаружить не удалось. Однако
было установлено, что термическое превращение первого из них осу¬
ществляется по типу реакции диспропорционирования согласно урав¬
нению2К [Pt (NC6H4COOH)Cl3] q«c-[Pt (NC6H4COOH)2 Cl2] + K2[PtCI4]В литературе описан еще один надежно установленный факт дис-
пропорционировання. Так, показано, что при нагревании комплекса
[Ni(Tmen) (Асас)(Н20)2]Вг (где Trnen — N,N, N',N' — тетраметил-
этилендиамин; Асас — ацетилацетонатный анион) происходит дегидра¬
тация и диспропорционирование с образованием соединений
[Ni(Tmen) (Асас)„] и [Ni(Tmen)Br„]. Считают, что термическое пре¬
вращение протекает через три последовательные стадии:—4Н202 [Ni (Trnen) (Асас) (Н20)а] Вг *- 2 [Ni (Trnen) (Асас)] Вг ►(1) (2) *■ 2 [Ni (Trnen) (Асас) Вг] или [Ni (Tmen) (Асас) Br»Ni (Tmen)(Acac)] *-1‘ (3) ‘ ► [Ni (Trnen) (Acac)aJ + [Ni (Tmen)'Br2]Изучением декарбоксилирования иминоуксусной кислоты (HiL)
в комплексах рутения (III) и родия (III) различного типа установлено,
что при температуре около 100°С происходит дегидратация
комплексов Na[Ru(HL)2Cl„]-H20, Na[Ru(H2L) (HL)C13]-8H20,
Na[Rh(H2L) (HL)CI3]-2H„0. Декарбоксилирование не участвующих
в координации карбоксильных групп с выделением С02 протекает
при температуре около 240°С. Декарбоксилирование координиро¬
ванных (депротонированных) карбоксильных групп наблюдается при
температуре 340 °С и выше. В этом случае наряду с С02 выделяется
СО.416
15.8. Превращение и образование тиоамидных
лигандовВ ряде работ Ю. Г. Сахаровой с сотр. показано, что при нагревании
комплексов редкоземельных элементов состава LnX3-/i Thio-mH>0
(где X — СН3СОСГ; СН3СН.,СОО~; СГ) происходит дегидратация и
при температуре около 150°С изомеризация тиомочевины в роданид
аммония. На примере нескольких комплексов показано, что связь
тиомочевины с ионом редкоземельного элемента осуществляется по¬
средством атома серы. Известно, что тиомочевину получают анало¬
гично мочевине путем изомеризации тиоцианата аммония. При 140 °С
существует равновесная смесь тиоцианата аммония и тиомочевины,
содержащая 28,1% последней, а при 180° С 21,8%. Таким образом,
повышение температуры приводит к смещению равновесия тиомоче-
вина тиоцианат аммония вправо. Разложение тиомочевинных комп¬
лексов редкоземельных элементов происходит по нескольким на¬
правлениям и потому при нагревании получается смесь продуктов.
Изомеризация координированной тиомочевины в роданид аммония была
также обнаружена при нагревании комплекса [Co(CH3COO)2(Thio)2.При термической обработке водных растворов комплексов плати¬
новых металлов получаются соответствующие сульфиды. На этом ос¬
новано применение тиомочевины в качестве коллективного реагента
на все платиновые металлы. Следует отметить, что разложению тио¬
мочевинных комплексов до сульфидов платиновых металлов в вод¬
ных растворах способствует гидролиз. Попытки обнаружить рода¬
нид аммония в продуктах термического твердофазного разложе¬
ния тиомочевинных комплексов платины (II) оказались безуспеш¬
ными.Роданид аммония также не был обнаружен при термическом раз¬
ложении комплексов меди [Cu(Thio)Cl] и [Cu(Thio)2]Cl. Вполне воз¬
можно, что для превращения координированной тиомочевины в рода¬
нид аммония ее связь через серу с ионом металла не должна быть очень
прочной. Поскольку платиновыэ металлы и медь (мягкие кислоты)
обладают большой склонностью к образованию прочных связей с се¬
рой, то разложение их тиомочевинных комплексов идет с образова¬
нием сульфидов металлов. Напротив, редкоземельные металлы и
кобальт гораздо менее склонны к образованию связей с серой, поэтому
становится возможной изомеризация.На основе многочисленных экспериментальных фактов был сде¬
лан вывод о взаимодействии координированных аминов с координи¬
рованными роданидными группами. Установлено, что комплексы
типа цис- или m/;a4C-[PtA.,(SCN)2] (где А — первичный или вторичный
амин) имеют на кривых ДТА характерные спаренные эндо-экзотерми-
ческие пики. Эндотермический пик соответствует плавлению исход¬
ного вещества, а экзотермический пик, по-видимому, объясняется меж-
молекулярным взаимодействием координированного амина и рода¬
нидной группы. Известно, что аммиак и амины присоединяются к эфи¬417
рам изотиоциановой кислоты с образованием N-замещенных произ¬
водных тиомочевины в соответствии с реакциейRNCS + R'NH2 — RNHCSNHRОбычно такие реакции даже при комнатной температуре протекают
количественно. Эфиры тиоциановой кислоты реагируют с аминогруп¬
пами, если имеются условия для образования циклических продуктов:R-CH—SCN R—СН Бч
| *■ | X=NH
СНг—NH2 СНо—nh/Вероятно, твердофазное нагревание роданидных комплексов пла¬
тины (II), содержащих аммиак, первичные и вторичные амины, приво¬
дит к подобным превращениям внутрисферных лигандов. По стери-
ческим соображениям взаимодействовать должны лиганды соседних
комплексов с образованием мостиковых производных изотиомочевнны,
например, по схемеN Nс/ с/NH, ч/ NH, о/Н2С^ \( н2сх \ /. I ;ptf ргН2<4 / \ Н2С\ / \NH, \ N Н S—C = NHс HN = C 1„N NS+ — .pi ч"'N s' \ /СН2V г/ NH,\ /И/ \ /СН2с/ nh2l/Такие превращения комплексов обусловливают появление экзо¬
термических пиков на кривых ДТА и появление новых полос погло¬
щения в ИК спектре в области 1500—1700 см'1.15.9. Твердофазное термическое окислениеПри нагревании на воздухе может происходить окисление коор¬
динированных фосфинов в комплексах железа (III), кобальта (II) и
никеля (II), находящихся в твердом состоянии. Примером такого рода
реакций может быть следующая:[Ni (PPh3)212J —-*• [Ni (OPPh3)2 hi
150"C418
По существу, эти реакции являются внедрением в связь металл —
фосфор. Для их изучения особенно удобным оказался совмещенный
метод дифференциального термического анализа и термогравнмет-
рии. В настоящее время этими методами изучено большое число по¬
добных реакций [4].При твердофазном термическом окислении сульфидов палладия
н платины в атмосфере воздуха в интервале температур 400—500 °С
PdS окисляется до PdS04, a PtS., окисляется до Pt(S04)3 при 300—400°С.
Окисление сульфидов является экзотермическим процессом. Безуслов¬
но, это многостадийный окислительный процесс. Формально его так¬
же можно рассматривать как реакцию внедрения кислорода в связь
металл—сера.
ГЛАВАПРИМЕНЕНИЕ КООРДИНАЦИОННЫХ
СОЕДИНЕНИЙ16.1. Аналитическая и органическая химияИспользование координационных соединении в аналитической
практике происходило параллельно с развитием теоретических пред¬
ставлений об этом классе соединений. В ряде случаев аналитические
реакции были выявлены эмпирически и лишь позднее стало ясно, что
они связаны с явлением комплексообразования. Многие колориметри¬
ческие методы количественного определения ионов металлов основаны
на комплексообразовании. Появление при комплексообразовании
окраски связано с d—d-электронными переходами и с переносом за¬
ряда с металла на лиганд и в обратном направлении. Введение хромо¬
форных групп в органические лиганды позволило получать разно¬
образные реагенты для фотометрического определения отдельных
ионов. Современные теоретические представления позволяют интерпре¬
тировать и предсказывать характер абсорбционных спектров в зави¬
симости от природы центрального атома и природы лигандов.В 1835 г. Кан обнаружил, что взаимодействие растворов солен
платины и хлорида олова (II) приводит к появлению ярко-красной
окраски. Реакция оказалась исключительно чувствительной. Значи¬
тельно позже было установлено, что характерная интенсивная окрас¬
ка появляется при взаимодействии хлорида олова (II) с хлоридом зо¬
лота (III). Этот окрашенный раствор был назван «кассиевым пурпу¬
ром». Считалось, что его образование связано с коллоидным золотом
и гидролизованной формой хлорида олова (IV). По аналогии с «кассие¬
вым пурпуром» окрашенный раствор, получающийся в результате вза¬
имодействия солен платины и олова (II), был назван «платиновым пур¬
пуром». На основании этой реакции были разработаны различные
варианты аналитического определения платины. «Платиновый пурпур»
обладает способностью экстрагироваться рядом органических соеди¬
нений. Введение операции экстракции позволило анализировать пла¬
тину в присутствии цветных металлов.Интенсивные окраски появляются при взаимодействии хлорида
олова (II) со всеми металлами платиновой группы. На этом основаны
методики количественного определения палладия, родия и иридия.
Природа окрашенных соединений олова с платиновыми металлами
начала систематически изучаться лишь в 50-х годах XX в. В настоя¬
щее время установлено, что предельными формами биметаллических
комплексов платиновых металлов с оловом (II) на хлоридном фоне
являются следующие:[Ru (SnCI3)6Cl]4_, [Rh (SnCl3)5CI]3-, [Pd (SnCl3)6]3-, [Os(SnCI3)5Cl]1-,
[Ir(SnCl3).Cl]»-, [Pt (SnCJ3)s]3-420
Как следует из гл. 12, лиганды могут существенно влиять на окис¬
лительно-восстановительные потенциалы ионов металлов. На исполь¬
зовании этой стороны комплексообразования основаны многие электро¬
метрические методы определения, выделения и разделения ионов ме¬
таллов [1—3].Особенно важное значение в аналитической практике приобрели
хелатные соединения [4]. В наши дни по крайней мере треть аналити¬
ческих работ связана с хелатами и хелатообразующими реагентами.
Многие растения содержат хелатные соединения металлов или хелато-
образующие соединения. Химики использовали растительные экстрак¬
ты для аналитических целей еще до того, как стало известно об их хи¬
мической сущности.Теоретической основой синтеза хелатообразующих реагентов яв¬
ляется правило циклов Чугаева. Оно же является и одной из основ¬
ных теоретических предпосылок для синтеза комплексонов.В аналитической химии комплексоны используются в титриметри-
ческнх методах практически всех катионов и многих анионов. Они
также широко применяются для маскировки отдельных ионов. Благо¬
даря работам Ластовского, Дятловой и их сотр., в нашей стране на¬
лажен выпуск большого ассортимента комплексонов, которые широ¬
ко применяются в различных областях науки и техники. Например,
в медицине используют комплексоны для выведения из организмов
тяжелых металлов, а работники сельского хозяйства для лечения рас¬
тений от железного хлороза. С помощью комплексонов достигается
умягчение воды и водоподготовка для специальных технологических
процессов. Комплексоны применяются для очистки теплового и
энергетического оборудования от отложения солей и продуктов кор¬
розии. Столь широкое использование комплексонов основано на об¬
разовании ими прочных и хорошо растворимых комплексных соеди¬
нении.В середине XX в. наступила новая эпоха в неорганической химии
и химии комплексных соединений. Это связано с требованиями со¬
временной техники и созданием новых материалов. В производство
стали вовлекаться редкие и рассеянные элементы, а для этого потре¬
бовалось создание технологий их получения, очистки и разработки
новых методов анализа. Многие из этих вопросов решались с по¬
мощью координационных соединений и процессов комплексообразо¬
вания. Так, гидрометаллургические процессы получения редких и
благородных элементов целиком основаны на явлении комплексообра¬
зования. На этом же явлении основана теория и практика экстракции
соединений металлов. При разработке гидрометаллургических и экст¬
ракционных технологий часто использовался опыт химиков-анали-
тиков. Некоторые аналитические методики разделения лишь с неболь¬
шими изменениями были перенесены в схемы технологических про¬
цессов. Например, было известно, что оксиоксимы являются избира¬
тельными реагентами на ионы меди. Поэтому при разработке ныне
действующих технологических процессов выделения меди были ис¬
пользованы реагенты этого класса.В гл. 13 говорилось о высокой эффективности использования ио¬421
нов металлов в качестве матрицы (темплатный синтез) для получения
довольно сложных органических молекул, в частности макроцикли-
ческих. Этим путем удается осуществить синтез продуктов, которые
в отсутствие ионов металла не получаются вовсе или получаются с
малым выходом, так как реакция в основном идет в другом направ¬
лении .В 70-х годах появились работы по использованию координацион¬
ных соединений для асимметрического синтеза. Для этой цели были
взяты оптически активные комплексные соединения. Оптическая
активность может быть обусловлена как асимметричностью централь¬
ного атома комплекса, так и хиральностью лиганда. В результате
проведения реакции координированного лиганда в таком комплексе
можно получить не рацемическую смесь, а преимущественно один из
оптических изомеров. Комплекс HRh(CO)L, где L — оптически ак¬
тивный фосфин (—) 2,2-диметил-4,5-бис-(дифенилфосфинометил)-1,3-
дноксалан, может быть использован в реакциях гидроформилирова-
ния. Так, избутена-1 получается правовращающий 2-метилбутаналь,
а из стирола правовращающий а-фенилпропаналь. Несомненно, что
работы в этом направлении получат существенное развитие.16.2. Металлокомплексный катализБольшое значение координационные соединения приобрели в качестве
катализаторов промышленно важных процессов. Запатентовано ог¬
ромное число различных координационных соединений, катализи¬
рующих разнообразные реакции.Координационные соединения способны активировать молекулы
водорода, кислорода, азота, оксида углерода (II), олефинов, ацетиле¬
нов и др. Они позволяют осуществлять каталитический процесс в
растворах, т. е. гомогенно. Как правило, гомогенные каталитические
процессы протекают в гораздо более мягких условиях, чем гетероген¬
ные. В ряде случаев координационные соединения позволяют прово¬
дить процесс весьма избирательно и стереоселективно.В 1938 г. Роелен открыл реакцию, позволяющую превращать оле-
фины в кислородсодержащие соединения. В качестве катализаторов
использовались карбонильные соединения переходных металлов. В
первую очередь была изучена реакция с этиленом и установлено, что
она протекает по двум направлениям:СН2=СН2 + СО + Н2 -* СН3СН,СНО
2СНп=СНг + со + Н, -> СН3СН2С(0) СНоСНзВ промышленности она получила название «оксо-реакции». Однако
впоследствии было установлено, что кетоны образуются не из всех
олефинов, и поэтому процесс чаще стали называть «гидроформилиро-
ванием». В общем виде его можно записать следующим образом:
2RCH=CH, + 2СО + 2Н, — RCH2CH,CHO + CH3CHRCHO422
Наиболее распространенным катализатором этого процесса является
НСо(СО)4, но с успехом применяется и [Rh(PPh3)s(CO)Cl].В начале 50-х годов в промышленном масштабе начал осуществ¬
ляться процесс полимеризации этилена (а затем и пропилена) в мягких
условиях на катализаторах, впервые предложенных Циглером. Они
основаны на галогенидах и алкнлгалогенидах металлов, например на
TiCl3 и А1(СгН5)гС1. Ранее в отсутствие этих катализаторов процесс
осуществлялся при давлениях порядка 2000 атм (2• 105 кПа). Ката¬
лизаторы циглеровского типа позволили снизить давление более чем
в 100 раз. Не менее важно, что получающиеся при этих условиях полп-
олефины имеют стереорегулярное строение, т. е. для них характерна
значительно меньшая разветвленность макромолекул и высокая сте¬
пень кристалличности. Стереорегулярность этих полимеров была
установлена Натта. За основополагающие работы по каталитической
полимеризации олефинов при низком давлении Циглер и Натта в 1963 г.
были удостоены Нобелевской премии.Практически одновременно Шмидт и Хафнер в ФРГ, а Сыркин и
Моисеев в СССР разработали каталитический способ промышленного
производства ацетальдегида из этилена. При пропускании этилена
через водный раствор, содержащий хлорид палладия (II), идет реакцияСН2=СН2 + НаО + PdC!: -» СН3СНО + Pd + 2HCIПри наличии в растворе хлорида меди (II) палладий регенерируется
в соответствии с уравнениемPd + 2CuCI2 -> PdCl2 + 2CuCIОкисление меди проводят кислородом воздуха:4СиС1 + 02 + 4HCI -* 4СиС12 + 2Н.,0Синтез ацетальдегида осуществляют в одну операцию, пропуская
смесь этилена и кислорода в реактор, содержащий хлориды палладия(11) и меди (II).К процессу окисления этилена до ацетальдегида по химической
сущности близок синтез винилацетата из этилена и уксусной кислоты.
Его суммарная реакция имеет видСНо=СН2 + CHgCOOH + v2 Оо -*■ СН3С (О) осн=сн2 + Н20Процесс протекает по стадиям:С;Н4 -f СН3СООН 4- PdC2 СН3С (О) ОСН=СН2 + Pd + 2HCIPd-f 2CuC12 PdCU + 2CuCl4CuCl -f 02 -f- 4HC1 -»■ 4CuCI2 + 2H40Винилацетат в больших количествах идет на производство поли-
винилацетата. Гидролизом последнего получают поливиниловый спирт.Миллионы тонн уксусной кислоты в мире получают путем ката¬
литического карбонилирования метанола по реакции423
СН3ОН + СО -V СНзСООНКатализатором процесса служит комплекс [Rh(PPh3)3(CO)Cl], а про¬
мотором СН31. Последний можно получать взаимодействием метанола
с иодистоводородной кислотой. Реакция протекает уже при атмосфер¬
ном давлении и температуре 150°С. Однако на практике процесс осу¬
ществляют при 15 атм (1,5-10:1 кПа) 175 °С. Селективность процесса
достигает 99%.В органическом синтезе в качестве катализатора широко исполь¬
зуется хлорид алюминия. Особенно эффективен он в реакции Фри-
деля — Крафтса, т. е. реакции алкилирования и ацилирования аро¬
матических соединений. Например:л -| + С2Н5С1 Считают, что хлорид алюминия взаимодействует с галогеналкилом
с образованием комплексного аниона [A1C1J- и карбкатиона С2Н5.
Последний взаимодействует с бензолом по реакцииС„Н„ + С,Н^ -н- сбн5с2н5+н+Ион водорода регенерирует молекулу катализатора в соответствии с
уравнениемН+ + [А1СЦ]- А1С13 + HCI16.3. Бионеорганическая химияВ последние два десятилетия уделяется очень большое внимание био¬
логическим аспектам координационной химии. Давно известно, что
такие важнейшие вещества, как гемин крови, хлорофилл и витамин
В13 являются типичными координационными соединениями соот¬
ветственно железа, магния и кобальта. В 50-х годах по инициативе
А. А. Гринберга в Ташкенте под руководством М. А. Азизова нача¬
лись широкие и систематические исследования биологической ак¬
тивности координационных соединений. На первых порах была ис¬
пользована идея целенаправленного синтеза биологически активных
координационных соединений. В качестве лиганда выбирали биоло¬
гически активное вещество. Из него получали комплекс с металлом,
который в организме является микроэлементом. Вначале были ис¬
пользованы амиды никотиновой кислоты, а также некоторые амино¬
кислоты. Работы в этом направлении привели к получению ряда
исключительно важных препаратов, которые в настоящее время ши¬
роко используются в медицине. Например, из амида никотиновой
кислоты (витамин РР) и хлорида кобальта (II) получен препарат
(«Коамид»), строение которого может быть выражено формулой424
ClH,NCi^0OyNHClОн оказался весьма эффективным при лечении некоторых форм ане¬
мии. Комплекс железа (III) с амидом никотиновой кислоты («Ферамид»)
используют для лечения железодефицитной анемии, а смесь «Фер-
амида» и «Коамида» — для лечения гипохромной анемии. Комплекс
кобальта (II) с метионином0=С СН—CH,CHaSCH,II”о nh2
\ >'Со/ \о ,мн2
0=С СН—СН2СН2 SCH3применяют как кроветворный препарат при лучевой болезни [5, 6].В 1969 г. было установлено, что ^uc-[Pt(NH3)2Cb] обладает ярко
выраженными противоопухолевыми свойствами. Интересно отметить,
что изомерное соединение m/7aHc-[Pt(NH3)2Ch] такие свойства не
проявляет. С этого времени лаборатории многих стран были захва¬
чены интенсивными исследованиями противоопухолевой активности
координационных соединений платины. С целью поиска эффективных
соединений типаГА XI
Pt.А X J .выбирались разнообразные амины (А) и ацидолиганды (X). Кроме
того, изучались соединения электролитного типа и соединения платины
(IV). К настоящему времени испытано, вероятно, более 1000 комплек¬
сов платины. В результате отобрано около 30 наиболее перспективных
соединений. Естественно, что поиски также направлены на комплексы
других металлов. Эти результаты отражены в обзорной статье [7].Опухолевые клетки характеризуются более активным синтезом
ДНК по сравнению с нормальными, поэтому действие противоопухоле¬
вых препаратов должно быть направлено на молекулы ДНК. Экспе¬
риментально установлено, что комплекс t<MC-[Pt(NH3)aCI3] действи¬
тельно избирательно подавляет синтез ДНК, но мало влияет на син¬тез РНК [7, 8].15-817425
В настоящее время происходит формирование новой отрасли хи¬
мии — бионеорганической. Эта отрасль науки изучает неорганиче¬
ские вещества, входящие в состав живых организмов. Установлено,
что одиннадцать металлов («металлы жизни»): Na, К, Mg, Са, V, Мп,
Fe, Со, Си, Zn, Мо — играют в организмах уникальную биологиче¬
скую роль.Существует мнение, что в организме человека присутствуют прак¬
тически все элементы периодической системы. Это мнение разделяет
большинство специалистов. Некоторые ученые считают, что все эле¬
менты, присутствующие в организме, выполняют определенную био¬
логическую функцию.В живом организме ионы металлов, как правило, связаны в комп¬
лексные соединения. Поэтому бионеорганическая химия, по сущест¬
ву, является биокоординационной. С литературой по данному вопросу
можно познакомиться по работам [9, 10].16.4. КрасителиВ настоящее время комплексы с органическими лигандами широко
используются в качестве красителей и пигментов [11]. К их числу
относятся, например, хромовые, кобальтовые и медные соединения
с о,о'-диоксиазо- (1), о-карбокси-о-оксиазо- (II) и о-амино-о-оксиазо-(III) лигандами, которые сами являются красителями:Наиболее распространенной органической частью металлсодержа¬
щих красителей являются азосоединения. Использование в качестве
красителей комплексов металлов в ряде случаев привело к вытеснению
операции предварительной протравы. Химическая сущность протрав¬
ливания состоит в закреплении на волокне ионов металлов (в частности,
ионов хрома Сг3+) с последующим взаимодействием органических кра¬
сителей с закрепленными ионами и образованием координационныхII111426
соединений. Ион металла играет роль мостика между молекулой бел¬
кового вещества волокна и молекулой красителя. Он увеличивает
светостойкость красителя, так как способствует отводу от пего на
волокно избыточной энергии, поглощаемой из светового потока. Из¬
быточная энергия дает начало фотоокислительному процессу. Дру¬
гая причина повышения светостойкости красителей заключается в
том, что ион металла замещает подвижные ионы водорода в функцио¬
нальных группах, легко поддающихся фотохимическому окислению.Хромовые комплексы с азосоединеииямп типаначали использоваться в качестве красителей в Швейцарии и Герма¬
нии еще в 1919 г. С 1930 г. для окраски нейлона начали применять
хромовые и кобальтовые координационные соединения типаВ красителях наиболее часто используются ионы хрома (III), кобаль¬
та (III) и меди (II). Первые два входят в состав красителей для шерсти,
а последний для хлопка.Одно из правил цветности гласит, что если ион металла акцепти¬
рует электронные пары атомов, входящих в сопряженную систему
двойных связей, обусловливающую поглощение света, то процесс
комплексообразования сопровождается углублением цвета [12]. Если
комплексообразование вовлекает неподеленную электронную пару, неСИ15**427
11Рис. 16.1. Спектр днтизоната ртути (II) в бен¬
золе:1 — исходная форма; 2 — фотоиндуцированная формаО400 500 Й00 Л. НМвходящую в систему сопряженных связей,
то цвет красителя существенно не изменя¬
ется. Отметим, что в абсорбционном спект¬
ре интенсивность полос поглощения, свя¬
занных с d—d-электронными переходами,
на несколько порядков ниже, чем интен¬
сивность полос, связанных с переходами в
хромофорной системе красителя. Полосы,
связанные с переносом заряда в комплексе
(металл лиганд или лиганд—>-металл)иногда имеют интенсивность, сравнимую с интенсивностью полос,
относящихся к хромофорной системе. В последнем случае может
наблюдаться не только усиление интенсивности полос поглощения в
спектре, но и появление новых.Дитнзонатные комплексы ртути (II), цинка (II) и других обладают
фотохромными свойствами. При облучении их светом происходит рез¬
кое изменение спектра. После прекращения облучения исходный
спектр восстанавливается. На рис. 16.1 приведены спектры исходной
и фотонндуцированной форм дитизоната ртути в бензольном растворе.
Фотохромные превращения дитизонатов металлов связаны с тауто-
мерным изменением и геометрической изомеризацией координиро¬
ванного лиганда [13]. В частности, дитизонат ртути испытывает сле¬
дующее обратимое превращение:Это свойство дитизонатов ртути используется для получения фото-
хромных полимерных пленок.16.5. Неорганические пигментыОдним из наиболее старых неорганических пигментов является ти¬
пичное координационное соединение «железная лазурь». Она была
случайно получена 1704 г. в Берлине Дисбахом. Отсюда ее торговое
название «берлинская лазурь». Это соединение было получено прока¬
ливанием бычьей крови с поташом К2СО3. Позже для этой цели стали\phPh428
применять и другие отходы животного происхождения: рога, кожу,
волосы и др. При прокаливании белковых веществ со щелочами по¬
лучаются цианиды, которые взаимодействуют с солями железа с об¬
разованием комплекса I\4[Fe(CN)6] («желтая кровяная соль»). При
взаимодействии этой соли с железом (III) образуется «берлинская
лазурь»:К, [Fe (CN)6] + Fe3+ KFe [Fe(CN)6] + 3K+Эта реакция используется в аналитической химии как реакция на
ион железа (III).Цвет железной лазури в зависимости от условий получения и дис¬
персности осадка колеблется от черного и темно-синего до синего, а
иногда и голубого. Выпускают 5—8 марок железной лазури. Лазури
используют в производстве нитрокрасок, масляных и полиграфиче¬
ских красок.Для защиты подводной части судов, плавучих доков и портовых
сооружений от обрастания морской флорой и фауной используют спе¬
циальные краски. Композиция одной из таких красок включает оксид
ртути (II) и оксид меди (II). При взаимодействии с хлоридом натрия
морской воды образуется сложная соль состава 6NaCI-3HgCh-CuCl2.
Несомненно, что это меднонатриевая соль, содержащая комплексный
анион [HgCl4]2-.Важное значение в технике имеют термочувствительные пигменты.
При определенной температуре такие пигменты изменяют окраску,
сигнализируя о перенагревании трубопроводов или отдельных частей
различного оборудования. Термочувствительные пигменты делятся на
обратимые и необратимые. Почти все известные обратимые, термо¬
чувствительные пигменты являются типичными координационными
соединениями переходных металлов. Принцип действия одних из
них основан на изменении окраски при удалении кристалли¬
зационной воды. Например, бромидное координационное соедине¬
ние кобальта (II) с гексаметилентетрамином кристаллизуется с
10 молекулами воды. При температуре около 40° С про¬
исходит удаление кристаллизационной воды и окраска изменяется
от розовой до голубой. При охлаждении со временем комплекс вновь
поглощает воду и окраска становится исходной. Аналогичное соеди¬
нение никеля (II) теряет 10 молей кристаллизационной воды при тем¬
пературе около 60° С и изменяет окраску от светло-зеленой до голу¬
бой. Термочувствительный пигмент AgJHgU] изменяет окраску от
желтой до темно-коричневой при 45°, a Cu2[HgU] — от карминово¬
красной до шоколадной при 65° С. Изменение окраски этих коорди¬
национных соединении связано с перестройкой кристаллической
структуры.Подробные сведения о неорганических пигментах можно найти
в [14].429
16.6. Химическая технологияТипичное координационное соединение криолит Na3[AlF6] в больших
количествах используется при электролитическом производстве алю¬
миния. Его применение основано на важнейшем свойстве понижать
температуру плавления глинозема. Природный минерал криолит встре¬
чается довольно редко. Искусственный получают в промышленности
взаимодействием HF в водной среде с Na2C03 и А1(ОН)3.Криолит применяется также в стекольной промышленности и для
получения эмалей. Введенный в шихту, он позволяет получать рас¬
сеивающие свет матовые стекла [15]. В значительных количествах
он используется как наполнитель резины и бумаги.Фгоридные соединения оказывают влияние на вязкость стекол и
их кристаллизационную способность. Поэтому при варке стекол в
шихту вводят, в частности, координационное соединение Na2[SiFe]
[15]. Кислота H2[SiF6] и ее натриевая соль Na,[SiF6] получаются в
промышленных масштабах в качестве побочных продуктов при пере¬
работке природных фосфатов в суперфосфат.Окраска стекол соединениями металлов, главным образом пере¬
ходных и редкоземельных, в настоящее время часто интерпретируется
с позиции образования координационных центров с определенным
полиэдром. Например, известно, что окраска стекол соединениями ко¬
бальта (II) может изменяться от красной до синей. Красная окраска
свойственна для кислых боратных стекол, а синяя — для стекол с
большей основностью. Первую связывают с ионом кобальта (II), име¬
ющим октаэдрическое окружение, а вторую — с ионом кобальта (II),
имеющим тетраэдрическое окружение [16].Координационные соединения широко используются при электро¬
литическом осаждении металлов. Покрытия, полученные из растворов
на основе координационных соединений, часто получаются более
мелкокристаллическими, гладкими, менее пористыми и поэтому проч¬
но скрепленными с основой. Такие покрытия обладают лучшими за¬
щитными и декоративными свойствами, чем полученные из растворов
простых солей. Например, золочение изделий традиционно проводят
из цианидных растворов, в которых золото находится в комплексном
ионе [Аи(СМ)2]". Осаждение серебра также проводят из цианидных
растворов. При этом получаются мелкокристаллические и плотные
осадки. Его осаждение из растворов AgN03 приводит к грубокристал¬
лическим дендритным осадкам. При приготовлении цианидных элект¬
ролитов для серебрения взамен опасного цианида калия широко ис¬
пользуется желтая кровяная соль. С этой целью раствор K4[Fe(CN)6],
поташа К3С03 и свежеосажденного хлорида серебра кипятят в течение
1,5—2 ч. Протекают следующие реакции:2AgCl -f К4 [Fe (CN)6] — 2К2 [Ag(CN)3] -f FeCl2FeCU KoC03 + HoO ->- Fe (OH)2 + 2KC 1 + C024Fe(OH)2 + 02 + 2HoO 4Fe(OH)3430
После удаления гидроксида железа электролит готов для использо¬
вания.Цианидный комплекс меди (I) K2[Cu(CN)3] вместе со станпатом нат¬
рия Na2Sn03 в щелочной среде используют для нанесения на поверх¬
ности металлических изделий защитного слоя бронзы или для прида¬
ния им бронзового оттенка. Для этих соединений меди и олова потен¬
циалы электроосаждения близки, что является непременным услови¬
ем образования сплавов.В 1867 г. Лекланше разработал гальванический элемент, который
схематично может быть изображенZn | NH4CI ] Мп02 (С)При работе элемента протекают реакции!2Zn—4е~ 2Zn:+Zn2+ + 2NH4CI ч- [Zn(NH3)2C:2]+2Н+4Мп02 + 4Н+ + 4е~ — 4МпО (ОН)В суммарном виде реакция может быть записана так:Zn + 2NH4CI + 2MnOo — [Zn(NH3)2Cl2] + 2MnO (OH)Из уравнения видно, что явление комплексообразования играет в
этом элементе существенную роль. Те же самые реакции протекают
на цинковом электроде в воздушноцинковом элементе:Zn | NH4CI102 (С)Электрохимические системы, используемые в серебряно-цинковых
и никелево-кадмиевых щелочных аккумуляторах включают образова¬
ние гидроксокомплексов цинка или кадмия.Раствор аммиачного комплекса [Cu(NH3)4] (ОН)2 используется в
производстве гидратцеллюлозного волокна — первого искусственно¬
го волокна, которое нашло практическое применение [17]. Еще в
XIX в. было установлено, то хлопковая и древесная целлюлоза спо¬
собна растворяться в медно-аммиачном растворе. Для получения во¬
локна получающийся вязкий растЕор (содержащий до 10% целлю¬
лозы) пропускают через никелевые сетки, а затем через формоЕочные
воронки, заполненные водой. В воде происходит выделение целлюлозы
и образование нитей. Состав и строение соединения, получающегося
при взаимодействии аммиаката меди и целлюлозы, точно не установ¬
лены. Кроме раствора [Cu(NH3)4] (ОН)2 целлюлоза способна раство¬
ряться в аммиачных и этилендиаминовых растворах никеля (II),
кадмия (II), цинка (II).Для нанесения тонкого слоя серебра на поверхность стекла ис¬
пользуется раствор, содержащий ион [Ag(NH3)2]+. Металлическое се¬
ребро выделяется из комплекса путем Ессстановления сахаром, сег-
нетовой солью и формальдегидом. Для улучшения адгезионных свойств
и ускорения образования на поверхности стекла серебряной пленки
его предварительно обрабатывают 0,05%-ным раствором SnCl., [18].
Существует способ металлизации поверхности стекла путем вжп-431
гания (или впекания). Например, для получения пленки платины го¬
товят пасту, состоящую из спиртового раствора H2[PtCI6], борной кис¬
лоты, лавандового масла и терпентина. Ее наносят на поверхность
стекла и изделие нагревают в муфельной печи. Получается тонкий
зеркальный слой металлической платины. При нагревании стекла до
определенной температуры слой металла «вжигается» в его поверх¬
ность, оставаясь ровным и блестящим [18].Для металлизации химическим способом поверхности диэлектри¬
ков палладийоловянным сплавом используются растворы биметалли¬
ческих соединений типа [Рс1(5пС13)5],_ [19].Многие рецепты химического покрытия материалов слоями метал¬
лов включают координационные соединения [20].Важное применение в технике имеет так называемое «карбонильное
железо». Оно используется для изготовления сердечников катушек и
магнитов, для производства магнитофонных лент, для заполнения
быстродействующих электромагнитных порошковых муфт и тормозов.
Получают же его путем термического разложения карбонильного
соединения Fe(CO)5.16.7. Другие области примененияКоординационные соединения начали эффективно использоваться в
флотационных процессах. В. А. Конев сформулировал принцип поис¬
ка новых депрессоров (реагентов, увеличивающих смачиваемость
водой частиц минералов) на основе координационных соединений.
Они заключаются в следующем.1. Лиганд в составе комплекса должен иметь неподеленные пары
электронов.2. Малоустойчивые комплексы обладают' лучшими депрессирую-
щими свойствами по сравнению с более устойчивыми аналогами. .3. В ряду Вернера —■ Миолати электронейтральные комплексы
и анионы являются более сильными депрессорами, чем комплексные
катионы.4. Если выбор лиганда удовлетворяет первым трем принципам
1—3, то депрессия или флотация минерала в присутствии комплекса
почти полностью определяются природой центрального иона.5. Если депрессия связана с деактивацией минерала, то при вы¬
боре депрессора следует учитывать, что константа устойчивости акти¬
ватора в присутствии минерала может быть выше, чем в отсутствие
минерала.М. М. Сычев развивает представления об образовании структуры
при твердении цементов или связующих материалов с позиции обра¬
зования аквокомплексов и сольватокомплексов. Главным условием
процесса твердения он считает следующее: системы типа цементов или
связок являются связующими, если жидкость затворения (раствори¬
тель) полярна, а образующая фаза содержит полярные группы —
аквокомтексы, сольватокомплексы [21]. Представления с позиций
координационной химии используются также для интерпретации фак¬432
тов увеличения активности, связующих при добавках в затворитель
полидентатных лигандов, в частности этилендиамина [22]. Такие ли¬
ганды могут связывать зерна, играя роль своеобразных мостиков. Име¬
ются сведения, что на прочность связующих сильное влияние могут
оказывать добавки комплексоновНекоторые координационные соединения могут образовывать кла-
траты. Например, при кристаллизации цианида никеля (II) из амми¬
ачного раствора, содержащего бензол, получается соединение состава
Ni(CN)o-NH3 С6Н6. Рентгенографическим методом установлено, что
основным элементом кристаллической решетки данного соединения
является двумерная плоская сетка. Четыре иона никеля в ней свя¬
заны четырьмя мостиковыми цианидными лигандами. Два из этих
ионов никеля окружены атомами углерода цианидных групп, а два дру¬
гих иона никеля окружены шестью атомами азота, два из которых
принадлежат молекулам аммиака, а четыре — цианпдным лигандам:с N NHI I /NH?з-C—Ni—C=N—Ni—N==I NH> NIII IIIN KTIF СI /NH1 I=N—Ni—N=C—NH~C=/| INH3 N СВ кристаллической решетке, образованной такими сетками, име¬
ются пустоты, которые заселяются инородными молекулами под¬
ходящего размера.Вполне возможно, что клатратные соединения на основе коорди¬
национных соединений найдут применение для разделения близких
по свойствам органических молекул.В технике широко используют нагревание и выдерживание при
определенной температуре металлических изделий или заготовок с
целью изменения их пластических свойств, закалки, отпуска. Для
этого, в частности, используются ванны с расплавленными солями.
Для предотвращения окисления обрабатываемых изделий и восста¬
новления образующихся оксидов металлов в солевые ванны вводят
«раскислителн». Одним из распространенных раскислителей являются
желтая кровяная соль и комплекс Mg[BF4]2 [20, с. 74].Для увеличения твердости и износостойкости металлических из¬
делий и придания антифрикционных свойств их поверхность обога¬
щают углеродом, азотом, серой. Сущность процесса заключается в
диффузии неметаллов в массу металла при нагревании. Для обога¬
щения поверхностного слоя углеродом и азотом (процесс цианиро¬
вания) используют составы солей с непременным включением циани¬
дов: NaCN, KCN или KjFe(CN)6] [20, с. 82].433
Обогащение поверхностей только серой применяется редко. Обыч¬
но проводят одновременно сульфидированне и цианирование. Поэтому
в состав многих сульфидирующих смесей также входит желтая кровя¬
ная соль.Диффузионное обогащение поверхности стали титаном повышает
ее стойкость против коррозии в агрессивных средах. В качестве титан¬
содержащего соединения в соответствующие составы включают комп¬
лекс K[TiF4] [20, с. 86].Флюсы, применяемые при пайке, предохраняют соединяемые по¬
верхности от окисления, растворяют имеющиеся на них оксиды и
улучшают смачивание поверхностей припоем. В состав некоторых
сортов флюсов, применяемых для пайки алюминия и его сплавов, вхо¬
дит NHJBFJ или K[TiF4] [20, с. 122]. Для удаления оксидов с по¬
верхностен металлов иногда используют составы, включающие
Na2[SiFe] [20, с. 175].
ИСПОЛЬЗОВАННАЯ ЛИТЕРАТУРАВведение1. Вернер А. Новые воззрения в области неорганической химии. —Л.:
ОНТП, 1936.2. Кукушкин Ю. Н. — Изв. вузов. Химия и хим. технол., 1979, 22, вып. G,
с. 761.3. Крестов Г. А., Березин Б. Д. Основные понятия современной химии. —
Л.: Химия, 1983.4. Андреев Ст. Годишннк хим. технол. ин-т. София, 1964, т. 11, кн. 3,
с. 117.Глава 11. Gear W. J. Coord. Chem. Rev., 1971, v. 7, p. 812. Feltham R. D., Hayter R. G. J. Chem. Soc., 1964, p. 4587.Глава 21. Берлнн А. А., Матвеева H. Г. — Усп. хим., 1960, 29, № 3, с. 277.Глава 31. Hyde J., Magin L., Zubieta J. Chem. Commun., 1980, p. 204.2. Кукушкин Ю. H. — Коорд. хим., 1978, 4, с. 1170.Глава 41. Кукушкин Ю. Н. — Коорд. хим., 1976, 2, с. 152.2. Кукушкин Ю. Н., Голосов В. В;. — ЖНХ, 1975, 20, с. 206.3. Drew М. G. В. Progr. Inogr. Chem., 1977, v. 23, p. 65.4. Drew M. G. B. Coord. Chem. Rev., 1977, v. 24, p. 179.5. Day V. W., Hoard J. L. J. Amer. Chem. Soc., 1970, v. 92, p. 3626.6. Порай-Кошнц М. А., Асланов Jl. А., Корытный E. Ф. Кристаллохимия,1976, т. 11. — М.: ВИНИТИ.Глава 51. Чугаев Jl. А. Избранные труды, т. 1. —М.: Изд-во АН СССР, 1954.2. Romeo R., Miппiti D., Lanza S. Inogr. Chim. Acta, 1977, v. 22, p. 87.3. Кукушкин Ю. H. и др. — Коорд. хим., 1977, 3, с. 619.4. Schwarzenbach G. Helv. Chim. Acta, 1952, — v. 35, p. 2344.5. Spike C. G., Parry R. W. J. Amer. Chem. Soc., 1953, v. 75, p. 2726, 3770.6. Кукушкин Ю. H. — Коорд. хим., 1977, 3, с. 1117.Глава 61. Драго Р. Физические методы в неорганической химии. — М.: Мир, 1967.2. Ahrland S., Chatt J., Devies N. R. Quart. Rev., 1958, v. 12, p. 265.3. Pearson R. J. J. Amer. Chem. Soc., 1963 v. 85, p. 3533.4. Реакционная способность и пути реакций / Под ред. Клопмана Г. —
М.: Мир, 1977, с. 383.435
Глава 7I. Кукушкин Ю. Н. Лиганды координационных соединений. —Л.: Изд.
ЛТИ им. Ленсовета, 1981.Глава 81. Черняев И. И. — Изв. пн-та по изучению платины АН СССР, 1926,
вып. 4, с. 243.2. Кукушкин Ю. Н., Карпейская Е. И., Трофимов В. А. — ЖНХ, 1971, 16,
с. 765.3. Кукушкин Ю. Н. — Коорд. хим., 1979, 5, с. 1856.4. Кукушкин Ю. Н. — Коорд. хим., 1982, 8, с. 201.Глава 91. Frost A. A., Pearson R. О. Kinetics and Mechanism. New York, 1961.2. Palmer D. A., Kelm H. Inorg. Chim. Acta, 1980, v. 39, p. 275.3. Swaddle T. W. Inorg. Chcm. 1980, v. 19, p. 3203.4. Миронов В. E. — Усп. хим., 1970, 39, с. 703.5. Taube Н. Chem. Rev., 1952, vol. 50, p. 69.Глава 10I.lBuhr J. D., Taube H. Inorg. Chem., 1980, vol. 19, p. 2425.2. Pidcock A., Richards R. E., Venanzi L. M. J. Chem. Soc., 1966, p. 1707.3. Zumdahl S. S., Drago R. S. J. Amer. Chem. Soc. 1968, vol. 90, p. 6669.4. Кукушкин Ю. H. — Коорд. хим., 1977, 3, с. 1876.5. Берштейн И. Я., Каминский Ю. Л. Спектрофотометрический анализ
в органической химии. — Л.: Химия, 1975.6. Кукушкин Ю. Н., Душим Р. Б., Стефанова О. В. — Коорд. хим., 1978,4, с. 780.7. Тимофеева В. Р., Новаковский М. С. — ЖОХ. 1979, 49, с. 1193.8. Блюменталь Т. О., Гельфман М. И., Разумовский В. В. — ЖОХ, 1970,
т. 40, с. 1170.9. Андронов Е. А. и др. — Изв. вузов. Химия и хим. технол. 1978, 21,
с. 973. ,10. Кукушкин Ю. Н., Украинцев В. Б., Мохов А. И. —ЖНХ, 1971, 16,
с. 1663.11'. Гринберг А. А., Кукушкин Ю. Н. —ДАН СССР, 1962, 145, с. 97.12. Кукушкин Ю. Н., Рабинович В. А., Голосеева Р. А. — ЖНХ, 1969,
14, с. 1863 и 2820.13. Lim М. С., Martin R. В. J. Inogr. Nu:l. Chem., 1976, vol. 38, p. 1911.14. Кукушкин Ю. R., Соболева М. С. — Коорд. хим., 1976, 2, с. 1538.15. Кукушкин Ю. Н. и др. — Коорд. хим. 1979, 5, с. 1383. _16. Wilkinson G., Birmingham J. М. J. Amer. Chem. Soc., 1955, vol. 77,
p. 3421. „17. Vaska L., Di Luzio J. W. J. Amer. Chem. Soc., 1932, vol. 84, p. 679.18. Sacco A., Ugo R. J. Chem. Soc., 1964, p. 3279.19. Damman С. B. Inorg. Chem., 1973, vol. 12, p. 2206.Глава 111. Sillen L. O. Inorg. Nucl. Chem,, 1958, v. 8, p. 176.2. Яцимирский К. Б., Васильев В. Н. Константы нестойкости комплексных
соединений. М.: Пзд-во АН СССР, 1959.3. BjerrumJ., Schwarzenbach G., Sillen L. G. Stability Constants of Metal-ion
complexes Part I. Organic Ligands. Chem. Soc., London, 1957.4. BjerrumJ., Schwarzenbach G., Sillen L. G. Stability Constants of Metal-ion
complexes. Part II. Inorganic Ligands. Chem. Soc., London, 1958.436
5. Sillen L. G., Martell A. E. Stability Constants of Metal-ion Completes
Special Publication No. 17, Chem. Soc., London, 1964.6. Marcus Y., Elizer I. Coord. Chem. Rev, 1969, v. 4, p. 273.7. Фридман Я. Д. и др. Устойчивость смешанных комплексных соединений
в растворах. — Фрунзе: Изд-во Илим, 1971.8. Kida S. Bull. Chem. Soc. Japan. 1961, v. 34, p. 962.Глава 121. Кукушкин Ю. H. — Коорд. хим., 1981, 7, с. 335.2. Кукушкин Ю. Н. — Коорд. хим.. 1982, 8, с. 596.3. Кукушкин Ю. Н., Данилина Л. И. — Коорд. хим., 1982, 8, с. 8.Глава 131. Кукушкин Ю. Н., Данилина J1. И., Панина Н. С. — Коорд. хим., 1977,3, с. 1451.2. Кукушкин Ю. Н. — Коорд. хим., 1981, 7, с. 323; 1984, 10, с. 291.3. Sidgwick N. V. The Chemical Elements and Compounds. Oxford Univ.
Press., London, 1950, v. 2. p. 1583.4. Сумарокова Т. H., Славинская P. A. —ЖОХ, 1975, II, c. 2516.5. Литвяк И. Г., Воробьева Э. М., Сумарокова Т. Н. — Изв. АН КазССР,
сер. хим., 1977, вып. 4, с. 26.6. Reilen Н., Jelling R., Wittig R. Вег., 1925, Bd. 58, s. 12.7. Кузнецов В. И. — ЖОХ, 1950, 20, с. 802, 809; 1956, 26, с. 3657.8. Гэрбэлэу Н. В. Реакции на матрицах. — Кишинев: Штпиница, 1980.Глава 141. Яцимирский К. Б. Термохимия комплексных соединений. —М.: Пзд-во
АН СССР. 1951.2. Chakravarty A. R., Chakravarty А. Inorg. Chem, 1981, v. 20, p. 3138.3. Bruce М. I., Wallis R. C. Austral J. Chem., 1981, v. 34, p. 209.4. Hames B. W., Legzdiris P. Orpanometallics, 1982, v. 1, p. 116.5. Скопенко В. В., Самойленко В. М., Гарбуз С. В. — ДАН УССР, 1980,
сер. Б.. ,\ь 12, с. 63.6. Ahrland S., Bjork N. О., Coord. Chem. Rev., 1975, v. 16, p. 115.7. Бабко А. К., Волкова А. И., Гетьман Т. E. — ЖАХ, 1967, 22, c. 1004.8. Пилипенко А. Т., Жебентяев А. И., Волкова А. И. — ЖАХ, 1972, 27,
с. 84.9. Кукушкин Ю. Н. — Изв. вузов СССР. Хим. и хим. технол., 1981, 24,
с. 139.Глава 151. Логвиненко В. А. Термический анализ координационных соединений н
клатратов. — Новосибирск: Наука, 1982.2. Wendland W. W., D’Ascenzo О., Gore Н. R. J. Inorg. Nucl. Chem., 1970,
v. 32, p. 3404.3. Кукушкин Ю. H., Седова Г. H., Буданова В. Ф. — ЖНХ, 1980, т. 25,
с. 200.4. Кукушкин Ю. Н., Буданова В. Ф., Седова Г. Н. Термические превраще¬
ния координационных соединений в твердой фазе. — Л.: Изд-во ЛГУ, 1981.Глава 161. Бабко А. К. Физико-химический анализ комплексных соединений в
растворах. — Пзд-во АН УССР, 1955.2. Шле(Ьер Г. Л. Комплексообразование в растворах. —М.—Л.: Химия,
1964.437
3. Инсцеди Я. Применение комплексов в аналитической химии. — М.:
Л1ир, 1979.4. Умланд Ф., Янсен А., ТиригД., Вюнш Г. Комплексные соединения в
аналитической практике. —М.: Мир, 1975.5. Азизов М. А. О комплексных соединениях некоторых микроэлементов
с биоактивными веществами. — Ташкент: Медицина, 1969.6. Хакимов X. X. — В кн. Реакционная способность координационных
соединений. —М.: Наука, 1976.7. Стеценко А. И., Преснов М. А., Коновалова А. Л. — Усп. хим., 1981,
50, с. 665.8. Williams D. R. Inorg. Chim. Acta Rev., 1972, v. 6, p. 123.9. Неорганическая биохимия / Под ред. Эйхрогна Г. М. — М.: Мир,
1978, в 2-х т.10. Биологические аспекты координационной химии / Под ред. Яцимир-
ского К- Б. — Киев: Наукова думка, 1979.11. Химия синтетических красителей / Под ред. Венкатарамана К. Л. —
М.: Химия, 1974, т. 3, с. 1946.12. Степанов Б. И. Введение в химию и технологию органических краси¬
телей. — М.: Химия, 1971, с. 59.13. Meriwether. L. S., Breitner Е. С., Sloan С. L J. Amer. Chem. Soc., 1965,
v. 87, p. 4441.14. Беленький E. Ф., Рискин И. В. Химия и технология пигментов. — Л.:
Химия, 1974.15. Безбородов М. А. Синтез и строение силикатных стекол. — Минск:
Наука и техника, 1968, с. 171.16. Селвуд П. Магнетохпмия. — М.: ИЛ, 1958, с. 203.17. Роговин 3. А. Основы химии и технологии химических волокон. —
М.: Химия, 1974, т. 1, с. 436.18. Технология стекла / Под ред. Китайгородского И. И. —
М.: Госстройиздат, 1967, с. 188.19. Cohen R. L., West К. W. J. Electrochein. Soc., 1973, v. 120, p. 502.20. Советы заводскому технологу. Справочное пособие. — Лениздат, 1975.21. Сычев М. М. Твердение вяжущих веществ. — Л.: Стройиздат, 1974,
С. 15.22. Сватовская Л. Б., Сычев М. М., Фабианская Е. С. — Ж. прикл. хим.,1977, 50, № 8, с. 1714.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРАВведениеСтаросельский П. И., Соловьев Ю. И. Альфред Вернер и развитие коорди¬
национной химии. — М.: Наука, 1974.Выдающиеся советские химики академики А. А. Гринберг и И. И. Черня¬
ев/Под ред. Шелокова Р. Н. —М.: Наука, 1970.Замяткина В. М., Кукушкин Ю. Н., Макареня А. А. Лев Александрович
Чугаев. —Л.: Наука, 1973.Варшавский Ю. С., Гельфман М. И. Александр Абрамович Гринберг. —
Л.: Наука, 1974.Глава 1Чугаев Л. А. Химия комплексных соединений. — Л.: Наука, 1979.
Черняев И. И. Избранные труды. В 2-х т. —М.: Наука, 1973.Гринберг А. А. Физическая химия комплексных соединении. Избранные
труды. —• Л.: Наука, 1972.Глава 2Березин Б. Д. Координационные соединения порфиринов и фталоцианина,—
М.: Наука, 1978.Фишер Э., Вернер Г. ^-Комплексы металлов. —М.: Мир, 1968.
Херберхольд М. я-Комплексы металлов. —М.: Мир, 1975,Глава 3Эссен Л. Н. Геометрические изомеры платины и трансвлияние. — М.:
Наука, 1969.Хокинс К. Абсолютная конфигурация комплексов металлов. —М.: Мир,1974.Глава 5Школьникова Л. М., Шугам Е. А. Некоторые вопросы стереохимии хелат-
ных соединений. — Кристаллохимия, 1977, 12. — М.: ВИНИТИ.Бусев А. И. Синтез новых органических реагентов для неорганического ана¬
лиза. — М.: Изд-во МГУ, 1972.Бургер К. Органические реагенты в неорганическом анализе. — М.: Мир,1975.Хольцбехер 3. и др. Органические реагенты в неорганическом анализе. —
М.: Мир, 1979.Коренман И. М. Органические реагенты в неорганическом анализе: Спра¬
вочник.— М.: Химия, 1980.Глава 6Гиллеспи Р. Геометрия молекул. —М.: Мир, 1975.Берсукер И. Б. Строение и свойства координационных соединений. —Л.:
Химия, 1971.439
Яцимирский К. Б., Яцимирский В. К. Химическая связь. — Киев: Вищэ
школа, 1975.Дяткина М. Е. Основы теории молекулярных орбиталей. —М.: Наука,1975.Чаркин О. П. Строение молекул и химическая связь. 4. —М.: ВИНИТИ,1976.Клименко Н. М. Строение молекул и химическая связь. 6. — М.: ВИНИТИ,1978.Глава 7Харитонов Ю. Я., Саруханов М. А. Химия комплексов металлов с гидро-
ксиламнном. —М.: Наука, 1977.Химия псевдогалогенндов / Под ред. Голуба А. М., Кёлера X.,
Скопе н ко В. В. — Киев: ' Вища школа, 1981.Успехи химии координационных соединений / Под ред. Я ц и м и р с к о¬
го К- Б. — Киев: Наукова думка, 1975.Глава 8Кукушкин Ю. Н., Бобоходжаев Р. И. Закономерность трансвлняния
И. II. Черняева. —М.: Наука, 1977.Трансвлияние в химии координационных соединений / Под ред. Спицы-
н а В. И. — М.: Наука, 1979.Глава 9Басоло Ф., Пирсон Р. Механизмы неорганических реакций. — М.: Мир,
1971.Тоуб М. Механизмы неорганических реакций. —М.: Мир, 1975.Самусь Н. М., Дамаскина О. Н., Лукьянец Т. С. Реакции замещения в ко¬
ординационных соединениях. — Кишинев: Штница, 1979.Кинетика реакций замещения лигандов в комплексных соединениях пла¬
тиновых металлов и золота: Справочник / Под ред. Николаева А. В. —
Новосибирск: Наука, 1974.Глава 11Белеванцев В. И., Пещевицкмй Б. И. Исследование сложных равновесий в
растворах. — Новосибирск: Наука, 1978.Бек М. Химия равновесий реакций комплексообразования. —М.: Мир,
1973.Фридман Я. Д. и др. Устойчивость смешанных комплексных соединеннй в
растворах. — Фрунзе: Илим, 1971.Глава 12Оксредметрия / Под ред. Никольского Б. П, и П а л ь ч е в с к о¬
го В. В. — Л.: Химия, 1975,Глава 14Мищенко К. П., Полторацкий Г. М. Вопросы термодинамики и строения
водных и неводных растворов электролитов. — Л.: Химия, 1968.Крестов Г. А. Термодинамика ионных процессов в растворах. — Л.: Хи¬
мия, 1973.Гутман В. Химия комплексных соединений в неводных растворах. — М.:
Мир, 1971.
Глава 16Цудзн Д. Органические синтезы с участием комплексов переходных ме¬
таллов.— М.: Химия, 1979.Долгоплоск Б. А. и др. Полимеризация дненов под влиянием it-аллильных
комплексов. —М.: Наука. 1968.Темкин О. Н., Флид Р. М. Каталитические превращения ацетиленовых со¬
единений в растворах комплексов металлов. — М.: Наука, 1968.Корнеев Н. Н., Попов А. Ф., Кренцель Б. А. Комплексные металлоргани-
ческне катализаторы. —J1.: Химия, 1969.Харвуд Дж. Промышленное применение металлорганнческих соединений.—
J].: Химия, 1970.Аспекты гомогенного катализа / Под ред. У го Р. —М.: Мир, 1973.Хенрици-Оливэ Г., Олнвэ С. Координация и катализ. — М.: Мир, 1980.Моисеев И. И. ^-Комплексы в жидкофазном окислении олефинов. — М.:
Наука, 1970.Лисичкин Г. В., Юффа А. Я. Гетерогенные металлокомплексные катализа¬
торы.— М.: Химия, 1981.Гейтс Б., Кетцир Дж., Шунт Г. Химия каталитических процессов. — М.:
Мир, 1981.Накамура А., Цуцун М. Принципы и применение гомогенного катализа. —
М.: Мир, 1983.Сокольский Д. В., Дорфман Я. А., Ракитская Т. Л. Протонно-апротонный
катализ. — Алма-Ата: Наука, 1975.Сокольский Д. В., Дорфман Я. А. Катализ лигандами в водных растворах. —
Алма-Ата^ Наука, 1972.Яцим^рский К. Б. Введение в бионеорганическую химию. — Киев: Науко-
ва думка, 1976.Хьюз Г. Неорганическая химия биологических процессов. — М.: Мир*
1983.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬАддукт 11я-Акцеиторные свойства внутрисферных лигандов 268,269, 273—276 олефинов 201, 268 сульфидов 201, 270, 320 сульфоксидов 269 алкнлфосфнтов 270■ пиридина 321— — дипнрндила 321 фенантролнна 321 фосфинов 321Акватация 250
Аквокомплексы 287, 288, 385
Активность 297, 312
Активность каталитическая 310
Актиниды 84, 162
Ализарин 7
Алкилгалогеннды 338
Алкилизоцпаннды 167
Алкоксил 155Аллиламин 355Алюминии 19, 43, 58, 108—110, 247,
286, 287, 367, 369, 394, 412, 413,
423, 424, 430
Амидины 356—360
Амидные соединения 280, 281, 360
Амидореакцпя 258, 259
Аминокомплексы 146, 225
Амины 141, 142, 144, 191, 223, 281,
354—356, 370, 371
Аммиак 201, 349, 35Г, 354—356, 371,
418Аммиакаты 284Анализ гравиметрический 395— термический 395
Анания 249Андерсоновская перегруппировка
406—410, 412
Арплгалогеннды 338
Арилизоцианиды 166, 167
Арснны 150, 155, 157, 201
Асимметрический синтез 422
Ацетамид 358, 359
Ацетилен 168, 170, 171, 188, 338
Ацетонитрнл 358, 359, 382, 383, 385
Ацидокомплексы 30Бензиламин 355
Бензимидазол 407, 408
Бензоннтрнл 355
Берлинская лазурь 327, 428
Бериллий 67, 106, 108—110, 162, 259,
260, 367Биологически активное вещество 424
Бор 81, 97, 106, 110, 111, 152, 165,386, 433, 434Бром 76
Бутадиен 188Валентность 18, 19
Ванадий 21, 22, 119
Версен 33Винилгалогениды 338
Винная кислота 61, 65
Внешняя сфера комплекса 16—18
Внутренняя сфера комплекса 16—18
Внутрикомплексные соединения 30
Волокно гидратцеллюлозное 431
Вольфрам 45, 47, 82, 83, 90, 132, 139,
154, 157, 158, 162, 206, 340, 385
Вращательная дисперсия 66
Восстановительное элиминирование
(отщепление) 328, 343—347Гемоглобин 37
Гидразин 145, 282, 283
Гидроксиламин 145, 148, 282, 351,384Гндроксокомплексы 287, 288
Гидролиз 250, 358, 359, 363, 364, 378,417Гидроформилирование 422
Г нпотеза
Льюиса 97— «цис-закрепленпя» 209, 210— Чатта—Оргела 210—212
Глицин 359, 363, 410
Гомогенный катализ 349
Группы хромофорные 420Дативная я-связь 98, 152, 210
Дегидратация 396, 412, 417
Декарбоксилирование 365, 416
Дентатность 21
Депротонирование 410—412
Дериватограф 395, 396
Диаграмма Оргела 114— Танабе — Сугано 116
Днастереоизомеры 65
Р-Дикетоны 367, 368
Дифенилацетилен 50
Диметилбензол 55
Диметплсульфид 163
Днметнлсульфоксид 160—163, 252,345, 380, 381, 384, 385, 387, 391
Диметилформамид 387, 391, 392
Дипирндил 321, 331, 387
Диспропорционирование 416
Диссоциация комплексов 279, 304, 305
Дицнан 359
Днэтилселеннд 201
Дпэтилсульфпд 201
Донорно-акцепторпый механизм 98442
Донорно-акцепторная связь 146, 148
Донорный центр 146Железо 6. 8, 15, 16, 37, 43, 50, 51,118, 119, 132, 260, 261, 296, 310,313, 314, 316, 326, 327, 331, 346,350—354, 369, 370, 376, 377, 385,387, 392, 393, 401, 412, 418, 425,
429, 430, 432—434Закон Кулона 260— скорости 237, 238, 251
Заряд комплексного иона 26Г
Золото 9, 58, 76, 139, 140, 174, 200,262, 263, 311, 346, 347, 420, 430Изомеризация в растворе 231— комплексов 223, 224— информационная 73, 74— самопроизвольная 232— связевая 69, 70
Изомерия— внутримолекулярная хелатно-свя-
зевая 95— гндратная 67— ионизационная 72— координационная 68— копформацнонная 73, 74— обратимая 388— связевая 68, 69, 390, 400, 401— солевая 69— твердофазная 399, 401— термическая 226, 399— трансформационная 73, 74— формальная 74, 75— хелатно-связевая 95
Изомеры 54—59, 62, 64, 65
Пзонитрил 164Изоцпанпды 164, 166, 167, 173
ИК-спектроскопия 164, 355, 359
Имндиые соединения 280, 372
Имииоэфнры 359
Имнны 354, 355Индивидуальные константы 294
Индуцированное окисление 330
Инертность 243
Иод 12, 49, 76, 104—106
Иодпд-ионы 191
Ионный потенциал 259
Ионофоры 34, 35Ион-днпольное взаимодействие 160
Иргалон 33Иридий 60, 64, 69, 81, 133, 154, 180,
195, 196, 203, 233, 262, 263, 290—292, 315, 333, 336, 339—344, 346,
353, 359, 390, 415, 420
Интермедиат 235, 236, 252—255, 334,
337, 345—347, 351, 385, 386, 392,
393, 399
Иттрий 83Кадмий 58, 92, 247, 303, 381, 390, 431
Калий 19, 119
Карбнлампн 164Карбоннльное железо 432
Кассиевый пурпур 420
Катализатор 422—424
Катализатор Фрпделя — Крафтса
356, 424— Циглера 423Катализ металлокомплексный 422
Каталитическое влияние ионов 359
б-Кетнмнны 368
Кинетический— метод 192— порядок реакций 237
КлассификацияПнгольда — Басоло — Пирсона 253— Лангфорда — Грея 253— Пирсона 69, 162
Кластерные соединения 48, 110
Клатраты 433
Клатратпые соединения 11Кобальт 6, 7, 12, 13, 15—1S, 22—26,
42, 55, 58, 59, 63—65, 67—70, 77—
79, 82, 116—119, 132, 133, 140, 141,
143, 159, 165, 171, 174, 180, 184,
241—243, 248, 254, 255, 261—264,290, 291, 301, 303, 310, 327, 328,
345, 348, 350, 353, 357, 359, 363—
367, 369, 375, 383, 384, 391, 396—
401, 410, 412, 417, 418, 422, 425—
427, 430Колебательная спектроскопия 196
Колебательные частоты 48
Количественное отделение азота 351
Комплексонаты металлов 32, 33
Комплексопы 24, 32, 34, 421, 433
Комплексы— аквонптро 386— аквогидро 324— активированные 301, 303— алкилгидриднын 348— алкильный 345— амндиновые 357— аммиачные 321— аминные 221, 383— амннотноэфирные 324— ацнльпые 345— бнс-фосфиновые 329— бромидные 221, 222— бутадиеновые 387
Комплексы— Васка 344— гидразнновые 282— гидрндные 333, 338, 339— гндроксо- 323— диамидные 412— днгидрпдные 290, 291— дигидроксо- 335— диокенмные 349—■ днпирнднльные 321— железа 385, 387, 392, 418— золота 311, 347, 420, 430
I— нминные 359— иодидные 221, 222, 401— иридия 333, 336, 341, 342, 353443
Комплексы— карбеповые 226— карбонильные 387— кобальта 328, 329, 350, 365, 366,383, 402, 410, 418, 429— марганца 366, 385— меди 363, 370, 372, 381, 382, 385,404, 417— мостнковые 175, 331, 332, 398, 412,418— никеля 338, 347, 372, 375, 389—
392, 401— нптрпльные 146, 147— нитрознльные 351— нитрито- 179— ннтро- 179— нуклеофильные 348— однородные 301, 303— оксимные 221— октаэдрические 172— олефино^ые 387— онпевые 381, 400— палладия 337, 338, 345, 378, 381,384, 403, 406, 409, 411, 419— переходных металлов 310— платины 183, 188, 189, 191, 221,
226, 290, 291, 329—331, 335, 337—
339, 341, 351, 352, 355—357, 364,
370, 371, 378, 381, 384, 385, 387,388, 392, 400, 402, 403, 406, 409,
411, 414, 416—419— рения 382, 389, 412— родня 335, 336, 337, 341, 344, 350,351, 353, 387, 411, 416— рутения 321—323, 333, 350, 351,
355, 385, 416— смешанные 301—303— сульфатные 289— сульфоксидные 331— тетрааминные 325— тетратпоэфирные 188, 324, 325
—: трнамнновые349— трппропиламмониевые 378— фенантролнновые 321— формнатные 348— фосфнновые 227, 332, 336— цианатные 350— цианидмостнковые 174— цианидные 311, 350— цинка 372— хелатные 335, 336, 341— хлорампнные 281— хлорндные 221, 222— хрома 368— этаноламиновые 283, 284
Константа— диспропорционирования 302— кислотной диссоциации 261—263,
269—274, 276, 278, 282, 285, 286,
289— неустойчивости 286, 287, 294, 295,
297, 304, 307Константа— пропорцпопирования 302— устойчивости 297, 298, 299—303
Концепция— гибридизации Полинга 98, 99— Найхольма — Гиллеспи 10,3— эффективного атомного номера
(ЭАН) 131, 132, 153Координационная емкость 21
Координационная сольватация 8
Координационное место 11
Координационные числа 15, 136, 316
Коэффициент активности 297
Кремний 77, 247, 339, 430, 434
Красители 426—428
Криолит 430
Крнптосольволиз 25
Ксенон 106Лабильность 243—245, 247, 248
Лактоп 362
Лантан 85, 86
Лантаннды 84, 417
Лнгандное число 27
Лиганды 15, 21, 22, 49, 50, 52— аксиальные 22— амбидентатные 69, 162, 173, 175— ацндо- 187, 191— второго класса 134— галогенидные 172, 173, 199, 200,
214— гидрндные 339— гидроксилампновый 353— зонтичные 80— карбеновые 226, 227— карбонатные 177, 178— карбонильные 173— карбоксилатные 180— концевые 40— мостнковые 39, 41, 45, 172— нитратные 177, 178, 354— ннтритные 179, 180, 354— нитрозильные 353, 354— нитроксильные 353— первого класса 134— пероксидный 341— перхлоратный 181— полпатомные 122— полпдентатные 23, 30, 39, 141— роданидиые 175—177— сильного поля 114—116— слабого поля 114—116— сульфатный 181— терминальный 40— фосфиновый 347— хлоридные 187, 196— цианидные 173, 17 1— экваториальный 22
Литий 8, 9, 45Магний 8, 31, 37, 49, 259, 260, 433
Магнитная восприимчивость 298
Макроциклическнй эффект 39444
Марганец 46, 117, 119, 132, 133, 157,
159, 164, 166, 167, 310, 315, 366,385Медь 13, 23, 30, 38, 39, 43, 45, 58,
76, 77, 89, 90, 92, 107, 119, 175,
178, 300, 303, 310, 318, 324, 325,
361—365, 369, 370, 372, 374, 381,382, 385, 387, 396, 404, 417, 423,
426, 427, 429, 431
Мезомернып гибрид 148
Меркаптаны 159
Метод— валентных связен (ВС) 97— ГЛСП 97— дернватографический 222, 413— дифференциального термического
анализа 221— изменения упругости пара 298— ИК-спектроскопин 176, 179, 192,
203, 221— калориметрии 226, 228— лигандного обмена 382— магнетохимпческий 47, 298— молекулярных орбиталей (ДАО)
120—126— объемный определения платины329— повышения точки кипения 298— полярографический 298— понижения точки замерзания 298— потенциометрический 276, 278, 279— протонного магнитного резонанса
(ПМР) 359— распределения 298— спектрофотометрпческий 276, 278,
298— термогравнметрнческий 419— тонкослойной хроматографии 195— Хюккеля 174— электрической проводимости 298— электронного парамагнитного ре¬
зонанса (ЭПР) 298— ядерного магнитного резонанса
(ЯМР) 12, 81, 203—204, 221, 233,
245, 298, 335Методика с применением внутренне¬
го стандарта 176
Механизм— декарбоксилирования 365, 366— каталитического синтеза 356— «присоединение — перегруппиров¬
ка» 342— трансаминнровання 373
Молекулярность реакции 237
Молибден 45, 53, 56, 57, 82, 83, 90,131, 154, 340, 401
Молярная электрическая проводи¬
мость 19Мольная доля комплексов 148, 299,
304—306, 308
Морин 7, 394
Мочевина 350Мышьяк 11, 73, 78, 80—82, 150—155,
162Натрий 8, 19
Нестабильность 243
«Нетипичное» окисленне 314
Неустойчивость 243
Никель 8, 15, 32, 49, 58, 75, 77—79,
87, 88, 90, 91, 93—95, 107, 119, 132,
133, 151, 154, 155, 158, 165, 166,
171, 175, 231, 241, 295—297, 300—
303, 310, 329, 332, 338, 342, 345—
347, 359, 363, 372, 375, 376, 383—
392, 401, 405, 416, 418, 419, 431, 433
Ниобий 82, 83, 394
Ниодим 85Нитрилы 146, 356—360, 383
Нитроаннлнн 283
Нуклеофильная группа 238
Нуклеофильное замещение 239Обменный механизм 253
Обратная дативная л-связь 102
Общая (суммарная) константа ус¬
тойчивости 294
Объем активации 241, 242
Окислительно-восстановительные ре¬
акции (равновесия) 311, 312, 316,
319, 326—328
Окислительно-восстановительные
свойства 317, 318
Окислительно-восстановительные
превращения 327, 328
Окислительно-восстановительный по¬
тенциал (ОВ) 312, 313, 315, 316,
318,319—325,328,332
Окислительное дегидрирование 354,
355Оксигенильные соединения 329
Оксид— азота 188— амина 147—150, 157— углерода 164, 165, 188, 330
Окснмы 282
Оксноксимы 421
Оксо-реакцня 422Олово 46, 78, 103, 106, 141, 191, 192,
226, 231, 292, 343, 360, 390, 420,
431, 432
Оловые соединения 139
Оляцня 139Оптическая изомерия 61, 62, 65
Орбитальные электроотрицательности
131Осмий 21, 83, 195, 196, 199, 261, 262,
265, 266, 274, 280, 315, 340, 348,
414, 420Основание Шиффа 361, 363, 371—373,
383Основность комплексов 290Палладий 10, 15, 25, 42, 46, 49, 50,
58, 60, 68, 70—73, 79, 81, 91, 94,445
Палладий 107, 109, 133. 154, 155, 171,
176, 223, 226, 228, 231, 232, 270,
283, 288. 289, 327, 336—340, 342,
344, 345, 352, 381, 386, 388, 390,
391, 402, 403, 409, 411, 419, 420,
423, 432
Параметр расщепления 112
Парциальный молярный объем реак¬
ции 241Переходные ряды Вернера—Мнолатп21, 318
Пигменты 426, 428, 429
Платина 6—9, 12—15, 18, 20, 21, 24—
27, 40, 41, 45, 46, 48, 49, 56, 58—
60, 65, 68—70, 73, 74, 78—82, 88—
90, 93, 94, 101, ПО, 133, 137, 141,
143, 147, 151 — 153, 155, 158—160,
167—174, 176, 177, 180, 183—196,
198—205, 208—211, 213—228, 232—
234, 237, 238, 243, 244, 248—252,
258, 259, 261—274, 279—286, 288,291, 301, 315—321, 325, 327—335,
337—341, 344, 346, 347, 349, 351,352, 354—359, 364, 370, 371, 378,
380—392, 398—400, 402—409, 411,
413—420, 425, 432
Платиновая синь 358
Платиновые металлы 12, 162, 164,334
Платиновый пурпур 420
Порядок реакции 237
Пиридин 6, 144, 148—150, 193, 201,
268, 280, 303, 318, 319, 321, 329,331. 380, 387, 406, 408
Пирокатехин 369
Пирометр Курнакова 395
Полимеризация 404, 405
Полимеризация гидрокомплексов
287—290
Полимерия 68Поляризационные представления
Гринберга — Некрасова 207
Поляризация 126
Поляризуемость 126
Порфирнн 36, 37, 39
Правило— дополнительности 114— Иергенсена 184, 186, 188, 221— Марковникова 182— Пейроне 184, 186, 188— Пирсона 128, 130— термической изомеризации 220,
225, 231, 233— циклов Чугаева 33, 42, 87—90, 94,383, 421— эффективного атомного номера 52,
334Превращения термические 395, 411
Препарат «Коамид» 424, 425
Принудительное введение раствори¬
теля 385, 386
Прометий 162Раскислптелн 433Рацемат 62Рацемизация 62, 392Рацемические соединения 61, 62Рацемические смеси 61Реактанты 237Реакция— азосочетания 369, 370— Зинина 351— Меншуткииа 343■— металлнрования 413, 415— на никель 87— деакватацпн 397, 398— дегидрогалогенпрование 406— Курнакова 187— ннтрозаминирования 281, 371— окислительного присоединения
328, 333—335, 337, 342—346— оксоляцин 139— оляцни 398— ортометаллирования 413, 415— цнклометаллнрования 413— элементарные 237— этанолнза 382
Реальный потенциал 312Рений 45, 47, 56, 57, 84, 195, 196,280.290, 357, 382, 389, 411, 412
Рептгеноструктурный анализ 195, 352
Роданиды 360Роданидный лиганд 175—177
Родий 47, 60, 64, 69, 81, 133, 174,
180, 203, 233, 255, 262—266. 335—341, 345, 350—353, 359, 380, 387,
390, 411, 416, 420, 422, 423
Ротационная дисперсия 66
Ртуть 9, 10, 42—44, 58, 280, 293—
296, 300, 307, 310, 337, 428, 429
Рутений 83, 147, 157, 164, 199, 266,
315, 322, 323, 325, 333, 348, 350,
351, 353—355, 359, 371, 385, 416,
420Ряд Ирвинга — Вильямса 310, 325
Ряд спектрохимнческий 113, 114
Ряд траневлияння 186, 189, 196—200,
203—206Свинец 83Связевые изомеры 69
Селен 260Сера 79, 105, 106, 108, 159, 164, 247,
260, 261Серебро 12, 16, 17, 40, 41, 49, 58, 77,
92, 96, 97, 100, 101, 107, 167, 169,
174, 175, 260, 261, 284—286, 300,
308, 309, 358, 385, 386, 393, 404,405, 429, 430, 431
Сила поля центрального атома 259
Силовая постоянная 48
Силовые константы 48
Синергический эффект 153
Синтез на матрице см. Темплатный
синтез«Совместимость» лигандов 302446
Соединения мостнковые 384
Солевой эффект 242, 243
Со.чь— Вокелена 402—404— Гро 90— координационных кислот 284— Косса 7— Магнуса 7, 45, 68, 183, 380, 402,
403, 404— Пейроне 7— Цейзе 7, 169, 170, 251, 252, 317,330Сольватация 135, 304, 325, 380, 385,
400— селективная (избирательная) 380
Сольволнз скрытый 250Скандий 119
Спектрополярнметрия 66
Спермин 89
Стабильность 243Степень кислотной диссоциации ли¬
ганда 261
Стерсохпмпческое учение 61
Стсхиометрическнн механизм 248
Стнбины 150, 155
Стронций 162Ступенчатые константы неустойчивос¬
ти 294 устойчивости 294Сульфиды 201, 225, 367
Сульфоксндпминные соединения 225
Сульфоксиды 159—161, 163, 202
Сурьма 81—83, 150—155, 255, 360Таллий 58, 247, 285, 286
Тантал 81, 83, 415«Твнст»-механнзм (скручивание) 399,
400Теллур 104, 105, 260
Теория— Бренстеда — Лоури 256, 257— валентных связей 96, 97— Дебая—Хюккеля 242, 297— Косселя 259— кристаллического поля 96, 111,
116—118, 120, 122, 246, 310— молекулярных орбиталей 96, 168— поля лигандов 120— Таубе 245— Усаповнча 315Темплатный синтез (на матрице) 32,
373—375, 422
Тербий 86Термическая изомеризация 220, 226,
230, 399— перегруппировка 226
Термокраскн 41
Технеций 162
Тпоксан 160
Тиомочевнна 188
Тноцианатные ионы 175
Тиоэфиры 159, 188, 226
Типичное состояние окисления 314Титан 111, 118—120, 123, 325, 423,
431Титриплекс III 33
Тории 83, 86Трансвлняипе Черняева 184—190, 192,
207, 214, 220, 221
Транс-хелатные соединения 93, 94
Трапс-хелатпый цикл 94, 95
Трпалкилфосфнпы 189
Триалкилфосфиты 155
Тригалогепиды олова 191
Трилом Б 33Трнфепилфосфнн 225, 226, 341, 387
Триэтилфосфин 225
Турнбулевая синь 327Удельное вращение 61
Уравнение Аррениуса 240— Нернста 312, 313, 316, 317
Уран 45, 83, 84, 86
Устойчивость 243Фактор рассеяния Бьеррума 301
Фенилгидразпн 282
Ферамнд 425
Ферроцен 50, 51, 53
Ферриципнй-ион 51
Флотационные процессы 432
Флюсы 434Фонариковые структуры 45
Формальный потенциал 312, 317
Формамид 381
Формула Бриттона 277
Формулы Гнльденгершеля 277
«Фортепьянная табуретка» 53
Фосфнны 150—155, 196, 201, 204, 225,332, 346
Фосфиты 204Фосфнноксиды 155—157, 332
Фосфор 73, 77—82, 93, 102, 104—
106, 150—157, 211, 247, 261, 262,342, 343, 385, 386
Фотоннпцннрование 336
Фотохромные свойства 428
Фталоцнаннн 37—39Хлор 76, 104—106
Хлорид основания Рейзе 183
Хлорид основания Пейроне 183
Хлорофилл 36, 37
Хелатные соединения 90, 421
Хелатный эффект 39, 90—92
Хелатон III 33
Хелаты железа 376, 377
Хиральные молекулы 64, 67
Хром 8, 12, 15, 25, 31, 42, 45, 64, 67,
116, 118, 119, 132, 139, 154, 164,
174, 241, 248, 256—258, 264, 279,293, 301, 315, 367—369, 399, 401,
410, 412, 426, 427Цвнттер-ион 33
Цезий 162Центральный атом 15447
Цианиды 173, 174
Цнклогептатрнен 51, 52
Циклооктаднен 201Цинк 49, 58, 67, 107, 117, 119, 247,
300, 303, 310, 360, 372, 428, 431
Цирконии 82, 83Цпсвлнянне лигандов 214—216, 219,
220Цнс-хелатные соединения 94
Цнтруллнн 370Ш кала трансвлияния лигандов (шка¬
ла Чатта) 201, 202Электродные потенциалы 312, 313
Электроноакцепторные свойства 352,
353Электронодонорные свойства 353
Электронные спектры 118
Элсктрофильное замещение 238
Электрофильная группа 238Электрическая проводимость 20, 27—
29Элементарный акт 237
Энергетический рельеф реакции 235,
236
Энергия— активации 235, 240— кристаллической решетки 380— стабилизации 114
Энтропия активации 241
Эрбий 85Этилен 168, 169, 187, 188, 210, 211,
224, 252, 386
Этилендиамнн 433
Эффект— Коттона 66— кристаллического поля 117, 118— старения 249— утяжеления Файгля 379— Янна — Теллера 116, 310
ОГЛАВЛЕНИЕПредисловие
Условные обозначения
ВведениеГлава 1356Координационная теория А. Вернера131.1. Предпосылки создания координационной теории131.2. Основные положения координационной теории151.3. Переходные ряды Вернера — Миолати191.4. Координационная емкость лигандов211.5. Номенклатура комплексных соединений241.6. Электрическая проводимость комплексных со¬единений в растворах26Глава 2Типы координационных соединений302.1. Внутренние координационные соединения302.2. Комплексонаты металлов322.3. Комплексы с макроциклическими лигандами342.4. Многоядерные комплексы392.5. Комплексы со связью металл — металл442.6. и-Комплексы48Глава 3Изомерия комплексных соединений543.1. Геометрическая изомерия543.2. Оптическая изомерия613.3. Гидратная изомерия673.4. Координационная изомерия и полимерия683.5. Связевая изомерия683.6. Ионизационная изомерия723.7. Трансформационная изомерия733.8. Конформационная изомерия733.9. Формальная изомерия74449
Глава 4Комплексные соединения с необычными координационными чис¬
лами4.1. Соединения с координационными числами 2 и 34.2. Соединения с координационным числом 54.3. Соединения платины (II) с шестью лигандами4.4. Соединения с координационными числами 7 и 84.5. Соединения с координационным числом 94.6. Соединения с координационным числом 10 и вышеГлава 5Правило циклов Л. А. Чугаева5.1. Историческое развитие правила5.2. Хелатный эффект5.3. транс-Хелатные соединенияt5.4. Изомерия хелатных комплексовГлава 6 Химическая связь в координационных соединениях6.1. Электростатические представления6.2. Теория валентных связей6.3. Концепция отталкивания электронных пар ва¬
лентной оболочки6.4. Полимеризация молекул с дефицитом электро¬
нов6.5. Теория кристаллического поля6.6. Метод молекулярных орбиталей6.7. Жесткие и мягкие кислоты и основания6.8. Концепция эффективного атомного номераГлава 7Лиганды координационных соединений7.1. Молекула воды и гидроксильный нон7.2. Амины7.3. Органические нитрилы7.4. N-Оксиды аминов7.5. Фосфины, арсины, стибины7.6. Триалкилфосфиты и фосфиноксиды767881828485878790939596969710210811112012613!134135141146147150155
7.7. Сероводород и его депротонированные формы 1577.8. Тмоэфиры, меркаптаны и сульфоксиды 1597.9. Оксид углерода (II) и изоциаииды 1647.10. Этнлен, ацетилен и нх производные 1687.11. Галогенидные лиганды 1727.12. Цианидный лиганд 1737.13. Роданидный лиганд 1757.14. Нитратный и карбонатный лиганды 1777.15. Нитрнтный лнганд 1797.16. Карбокснлатные лиганды 1807.17. Сульфатный и перхлоратные лиганды 181Глава 8Взаимное влияние лигандов во внутренней сфере координационных
соединений1828.1.Амннокомплексы платины (II)1838.2.Правила Пейроне н Мергенсена1848.3.Закономерность трансвлияния И. II. Черняева1848.4.Цисвлняние лигандов2148.5.Правило термической изомеризации комплексов
платины (II) и палладия (II)220Глава 9Механизмыреакций координационных соединений2359.1.Некоторые понятия в учении о скоростях и ме¬
ханизмах химических реакций2379.2.Лабильные и инертные координационные соеди¬
нения2439.3. Стехиометрическнй механизм2489.4.Классификация механизмов реакций замещения
в координационных соединениях252Глава 10Кислотно-основные свойства координационных соединений 25610.1. Развитие представлений о комплексных кис¬
лотах и основаниях 25610.2. Факторы, влияющие на кнслотные свойства
координационных соединений 257Ю.З. Взаимосвязь кислотных свойств координиро¬
ванных аминов с реакциями замещения нх
протонов 266451
10.4. Влияние тг-акцепторных лигандов на кислот¬
но-основные свойства комплексных соедине¬
ний 26810.5. Расчет констант кислотной диссоциации 27610.6. Ступенчатая кислотная диссоциация комплекс¬
ных соединений и внутрисферных лигандов 27810.7. Соли координационных кислот 28410.8. Кислотно-основные равновесия в растворах
гидратокомплексов 28610.9. Полимеризация гидроксокомплексов 28710.10. Основность комплексов переходных металлов 290Глава 11Равновесия в растворах координационных соединений 29311.1. Ступенчатый характер равновесий 29311.2. Функция образования и кривая образования 29811.3. Статистическое соотношение ступенчатых кон¬
стант устойчивости 29911.4. Смешанные комплексные соединения 30111.5. Мольные доли. Распределение форм комплексовв зависимости от концентрации лиганда 30411.6. Комплексообразование и растворимость солей
металлов 30811.7. Ряд Ирвинга — Вильямса 310Глава 12Окислительно-восстановительные свойства координационных со¬
единений 31112.1. Влияние комплексообразования на электродные
потенциалы 31212.2. Стабилизация лигандами нетипичных состоя¬
ний окисления центральных атомов в комплек¬
сах переходных металлов 31412.3. Влияние природы лигандов на окислительно¬
восстановительные потенциалы координационных
соединений 31512.4. Расчет констант равновесия окислительно-вос¬
становительных реакций из значений ЭДС галь¬
ванических элементов и значений потенциалов
электродных реакций 32612.5. Типы окислительно-восстановительных превра¬
щений координационных соединений 327452
12.6. Реакции окислительного присоединения 33312.7. Реакции восстановительного элиминированияс участием комплексов переходных металлов 343Глава 13Реакционная способность координированных лигандов13.1. Расщепление координированных лигандов13.2. Превращения внутрисферной нитрогруппы13.3. Реакционная способность координированнойнитрозильной группы13.4. Окислительное дегидрирование алифатических
аминов13.5. Превращение внутрисферных нитрилов в ами-
дины13.6. Взаимодействие внутрисферных нитрилов с
водой, щелочами и спиртами13.7. Превращение координированных нитрилов вN-ацилзамещенные амиды13.8. Изменение реакционной способности в резуль¬
тате смещения электронов от лигандов к
центральному атому13.9. Влияние заряда комплексного иона на реак¬
ционную способность координированного ли¬
ганда13.10. Влияние и-дативной связи одного лиганда на
реакционную способность другого лиганда13.11. Стабилизация одной из таутомерных форм ли¬ганда в результате комплексообразования13.12. Стабилизация комплексообразованием де¬протонированной формы лиганда13.13. Устранение характерных свойств лигандов в
результате координации и использование это¬
го явления для маскировки отдельных функ¬
циональных групп13.14. Проявление лигандом новых химических
свойств в результате координации13.15. Изменение реакционной способности коорди¬нированных оснований Шиффа13.1G. Реакции на матрице, или темплатный синтез349
34»350352354355
358360361365366
366369370370371373453
Глава 14Глава454Роль растворителя в координационной химии14.1. Факторы, влияющие на растворимость коорди¬национных соединений14.2. Синтез в неводных растворителях14.3. Синтез координационных соединении в раство¬рителе, обладающем свойствами Лиганда14.4. Влияние растворителя на направление реак¬
ции14.5. «Принудительное» введение в комплекс моле¬кул растворителя с образованием лабильного
интермедиата14.6. Индуцированное разложение растворителяи координация его продуктов к иону металла14.7. Влияние основности растворителя на синтезкомплексов с депротонированными лигандами14.8. Смещение равновесия изомеризации под влия¬нием растворителя14.9. Влияние растворителя на образование комп¬
лекса в определенном координационном поли¬
эдре14.10. Образование связевых изомеров в зависимости
or растворителя14.11. Влияние растворителя на скорость реакциии ее механизм15 Типы твердофазных термических превращений координационных
соединений15.1. Дегидратация и деакватация15.2. Термическая изомеризация15.3. Правило термического превращения двух¬
комплексных соединений типа солей Магнуса
и Вокелена15.4. Твердофазная термическая полимеризация15.5. Реакции дегидрогалогеннрования15.6. Циклометаллнрование15.7. Термическое декарбоксилпрованне и диспро-
порцпонированне15.8. Превращение и образование тиоамндных ли¬
гандов15.9. Твердофазное термическое окисление378378381382383385386388
Д88389390391395396
399402404406413416417418
Глава 16Применение координационных соединений 42016.1. Аналитическая и органическая химия 42016.2. Металлокомплексныи катализ 42216.3. Бионеорганнческая химия 42416.4. Красители 42616.5. Неорганические пигменты 42816.6. Химическая технология 43016.7. Другие области применения 432Использованная литература 435Дополнительная литература 439Предметный указатель442
Юрий Николаевич КукушкинХИМИЯ КООРДИНАЦИОННЫХ СОЕДИНЕНИИЗав. редакциеЛ С. Ф. КондрашковаРедактор А. В. БородинаМл. редакторы С. М. Ерохина и Л. С. Мак арки па
Художник Н. В. Беланов
Художественный редактор Т. М. Скворцова
Технический редактор А. К. Нестерова
Корректор С. К- ЗавьяловаИ Б № 4785Изд. Хнм-733. Сдано в набор 23.10.84. Подп. в печать 28.06.85. Формат
C0X90'/i6- Бум, тип. JV? 3. Гарнитура литературная. Печать высокая. Объем
28,5 уел. печ. л. + 0,25 уел. печ. л. форзац. 28.75 уел. кр.-отт. 30,20 уч.-изд. л.-ь
+ 0,44 уч.-изд. л. форзац. Тираж 6000 экз. Зак. № 817. Цена 1 р. 50 к.Издательство «Высшая школа», 101430, Москва. ГСП-4, Неглинная ул.,
д. 29/14.Ярославский полиграфкомбинат Союзполиграфпрома при Государственном
комитете СССР по делам издательств, полиграфии и книжной торговли.
150014, Ярославль, ул. Свободы, 97.
Комплекс ОасмзСО PPhСоль ГиббсаNHjN0.Хлорид ГроN НС(;CIСоль КоссаХлорид ПейронеСоль Вольфрама
CIС2Н 5
NH 2NH2сгн5NH гс2н5CIА. Вернер
Л.А. Чугаев
И.И. Черняев
А.А.Гринберг
Дж. Чатт
Дж. БейпарКомплекс Ренруа
— CIн,он гон 2оsoСоль ВоиеленаСоль РеинекеNH .NH3NCSSCNКомплекс Фишера
NO 2N02Соль ЦейзеСоль ДрекселяЗеленая сопь МагнусаNHjNH.Cl4Соль ЖерараN Н •Соль Чугаеоа
~ CICI.NH3 | NH3NH,Первое основание Рейэе(ОН)Второе основание РейзеСоль ЭрдманаNH зОН