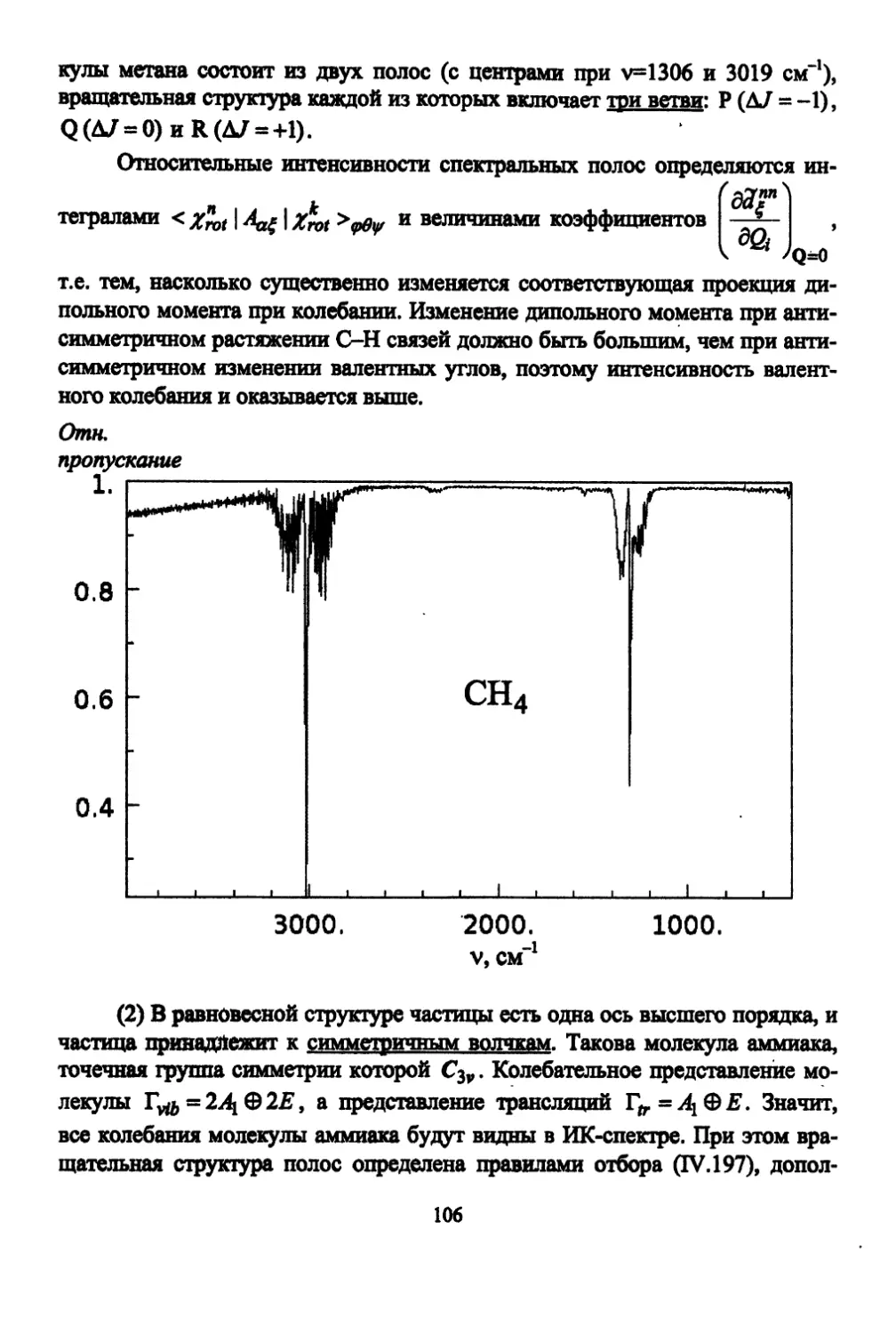
/






























































































































































Text
ББК 22.36 22.311 22.336 22.3я73 24.5
Новаковская Ю. В.
Молекулярные системы. Теория строения и взаимодействия с излучением.
Ч. Ш: Полуклассическая теория взаимодействия молекул с излучением.
М.: Едиториал УРСС, 2005. — 160 с.
ISBN 5-354-01071-3
Оригинал-макет предоставлен автором,
текст опубликован в авторской редакции.
Издательство «Цдиториал УРСС». 117312, г. Москва, пр-т 60-летия Октября, 9.
Лицензия ИД №05175 от 25.06.2001 г. Подписано к печати 17.11.2004 г.
Формат 60x90/16. Тираж 200 экз. Печ. л. 10. Зак. Nil 2-16&/819.
Отпечатано в типографии ООО «РОХОС». 117312, г. Москва, пр-т 60-летия Октября, 9.
ISBN 5-354-00930-8 (Полное произведение)
ISBN 5-354-01071-3 (Часть III)
© Ю. В. Новаковская, 2004
ИЗДАТЕЛЬСТВО УРСС
НАУЧНОЙ И УЧЕБНОЙ ЛИТЕРАТУРЫ
E-mail: URSSQURSS.ru
Каталог изданий
■ /rttemet http://URSS.ru
Тел./факс: 7 (095) 135-42-16
Тел./факс: 7 (095) 135-42-46
3019 ID 25402
Содержание
стр.
Глава IV. Молекулярная спектроскопия 5
§ 1. Электромагнитное поле 6
§2. Электрические и магнитные характеристики молекул
в постоянных полях 12
Классическая модель 12
Молекула в однородном поле 12
Классическая функция Гамильтона молекулы в поле 14
Электрический и магнитный дипольные моменты молекулы 16
Квантовая (полуклассическая) модель 19
Дипольный момент и электрическая поляризуемость молекулы 19
Магнитный момент и магнитная восприимчивость молекулы 21
§3. Молекула в переменном электромагнитном поле 28
Возмущение состояния молекулы полем: одно- и многофотонные
процессы 28
Дипольное приближение 34
Первый порядок теории возмущений: однофотонные процессы 35
Второй порядок теории возмущений: двухфотоиные процессы 36
Поглощение излучения 38
Рассеяние излучения 46
Коэффициенты Эйнштейна 49
§4. Правила отбора 54
Строгие правила отбора 55
Приближенные правила отбора 58
§5. Переходы между электронно-колебательно-вращательными
состояниями молекул 69
Двухатомные молекулы 69
Спектры поглощения 70
Вращательные спектры 71
Колебательно-вращательные спектры 74
Электронные спектры 78
3
Спектры рассеяния 87
Вращательные спектры 89
Колебательно-вращательные спектры 94
Электронные спектры 97
Многоатомные молекулы 99
Спектры поглощения 101
Вращательные спектры 101
Колебательно-вращательные спектры 104
Электронные спектры 113
Спектры рассеяния 116
Вращательные спектры 116
Колебательно-вращательные спектры 118
Электронные спектры 121
§6. Спектроскопия ЯМР и ЭПР 122
Общие теоретические основы 122
Электронный эффект Зеемана 124
Ядерный Эффект Зеемана 126
Спиновые состояния атома в постоянном магнитном поле 127
Спин-спиновые взаимодействия и «полный» гамильтониан
молекулы в магнитном поле 128
Электронный парамагнитный резонанс 133
Модельные системы и реальные спектры молекул 136
Анизотропия g-фактора и g-тензор 140
Ядерный магнитный резонанс 142
Спектры низкого разрешения 142
Спектры высокого разрешения и спин-спиновое взаимодействие
ядер 148
Модельные системы и реальные спектры молекул 151
Литература 160
4
Глава IV. Молекулярная спектроскопия
Все молекулярные спектры - результат взаимодействия молекулы с
полем электромагнитных волн. Внешнее поле может не только инициировать
переходы между электронно-колебательно-вращательными состояниями
молекул, но и изменять структуру самих состояний, в частности расщеплять
вырожденные состояния. Для описания всех этих процессов, а точнее для
описания состояний молекулы, находящейся во внешнем поле, необходимо
дополнить проанализированное нами в предыдущих двух главах уравнение
Шредингера свободной молекулы гамильтонианом поля и членами,
описывающими взаимодействие молекулы и этого поля:
Здесь первое слагаемое - молекулярный гамильтониан, приближенные
методы определения собственных функций и собственных значений которого мы
обсуждали в главах П и Ш; второе слагаемое - гамильтониан поля; третье
слагаемое - взаимодействие поля с молекулой. Есть различные варианты
квантовой теории поля, в частности представление поля электромагнитной
волны совокупностью фотонов разной частоты, причем поглощение или
испускание молекулой определенного кванта отвечает исчезновению или
рождению фотона соответствующей энергии. Но во всех вариантах описание
процесса взаимодействия молекулы с излучением и изменения ее
внутреннего состояния представляет весьма сложную задачу.
Поэтому обычно используют так называемый полуклассический
подход: состояние молекулы описывают в рамках квантовой теории, а поле - с
использованием классических уравнений Максвелла.
5
§1. Электромагнитное поле
Описание электромагнитного поля в терминах наблюдаемых
экспериментально величин - напряженностей магнитного и электрического полей
(обозначаемых как Е и Н) и векторов электрической (D = е£) и магнитной
(В = цН) индукции - было дано Максвеллом в виде системы 4-х уравнений1:
с Ы
(закон электромагнитной индукции Фарадея:
изменение во времени плотности магнитного потока В
приводит к появлению электрического поля с вектором
напряженности Е, ортогональным вектору В)
_ 16D An.
гоШ — = —j
с dt с
(электрический ток с плотностью j,
как и изменение во времени электрического поля Е,
создает магнитное поле Н)
divD = 4яр
(закон Гаусса-Кулона: поток электрического поля
из малой сферы пропорционален заключенному
в ней электрическому заряду)
AVB = 0
(отсутствие магнитного заряда по причине
замкнутости линий магнитного поля,
постулированное Максвеллом)
Л
}
flV.l)
J
Здесь р - объемная плотность заряда; j - вектор плотности электрического
тока, б - диэлектрическая постоянная среды; ц - относительная магнитная
проницаемость (заметим, что в общем случае е и ц суть тензоры второго
ранга, но в изотропной среде это скаляры, а их значения в вакууме приняты за
единицы: е = 1 и ц = 1).
1 Уравнения записаны в системе СГС, часто используемой в теории электромагнитных
взаимодействий. При переходе к системе СИ в этих уравнениях «исчезают»
коэффициенты У и 4;г.
6
Напомним, что, по определению, дивергенция вектора а
diva = —*-
dav
да2
+—-
дх ду dz
есть суммарный поток этого вектора из окрестности данной точки через
поверхность единичной сферы, ее окружающей.
По определению же, ротор (вихрь) вектора а,
rot* =
i
дх
J
д^
ду
к
д^
dz
а
ZJ
- это вектор с компонентами (
д д ч fd д ч (д д ч
Отметим, что в системе уравнений Максвелла первые два уравнения
векторные, т. е. записаны для компонент трехмерных векторов (так что
система (IV. 1) - это 8 уравнений). Общее число неизвестных величин в системе
- шесть: компоненты векторов Е \Ex,Ey9Ez) и В [ВХ9Ву9В2). Однако
величинами, определяющими поле, являются всего четыре: скалярная плотность
зарядов р и 3 компоненты вектора плотности тока j \Jx>Jy>Jz)- Яри этом два
уравнения Максвелла не содержат величин, описывающих конкретную
систему зарядов и токов. Поэтому рациональным был бы переход к некоторым
новым характеристикам поля, в которых первое и последнее уравнения
Максвелла, не включающие величины р и j, были бы тождествами. Такое
преобразование существует, а характеристики носят названия векторный (A(r,f)) и
скалярный (<p(r9t)) потенциалы электромагнитного поля. Наблюдаемые
величины Е и В выражаются через них так:
B = rotA (IV.2)
V = -~^-grad<p (IV.3)
с ot
Получены эти уравнения связи очень просто:
(1) Известно, что для любого вектора а выполнено условие divrota = 0;
следовательно, если согласно последнему уравнению Максвелла divB = 0, то
можно считать вектор В ротором некоторого вектора А: В = rot A;
(2) Подставим этот результат в первое уравнение Максвелла:
гоШ + — = rof (E + ——) = 0. Поскольку для любой скалярной функ-
с dt с dt
ции Ь выполнено равенство: rotgradb = 09 можно считать, что
Е + ——- = -gradq> (знак здесь можно выбрать любой).
с dt
Введя таким образом векторный и скалярный потенциалы А и <р, мы
определили их неоднозначно: точно определены только их производные
(соответственно, rot А и grad<p). В них же самих, как в первообразных, есть
неопределенность: их согласованное изменение вида
А' = А + gradf Л
(где/- некая скалярная функция) не изменяет уравнения Максвелла.
Функцию / называют калибровочной функцией. В зависимости от ее
выбора получается более или менее простая форма потенциалов А и <р.
Например, если потребовать, чтобы для системы в вакууме (т.е. при е = 1 и
ц = 1) было выполнено условие
divA+I^=o,
с dt
(IV.5)
то получится удобная калибровка, называемая лореныевой калибровкой, При
этом функция/определена волновым уравнением
<-v4^,/=o.
Уравнения Максвелла в переменных {А, <р) при использовании
калибровки Лоренца можно переписать следующим образом:
1 а2
divD = Ляр => (-V2 + -=- —z)q> = Ляр
c'dt*
rota-
iao 4ж,
с dt
-}
(-V4
Jlsl
c2dt''
)A = ^j
(IV.6)
При отсутствии свободных зарядов и токов (случай свободного поля)
они упрощаются до опять-таки волновых уравнений, теперь уже
относительно векторного и скалярного потенциалов:
(IV.7)
8
Решения этих хорошо известных в математической физике уравнений
имеют вид:
(IV.8)
где к - волновой вектор, длина которого в вакууме
с
(о?-частота поля).
В дальнейшем нас будет интересовать только поле свободной
электромагнитной волны (без всяких дополнительных источников), что позволяет
использовать удобную в этом случае кулонову калибровку поля
<йуА = 0
р = 0
(IV.9)
В указанной калибровке скалярный потенциал поля отсутствует, а векторный
имеет тот же вид, что и в случае калибровки Лоренца (IV.8).
Подставляя плоскую монохроматическую волну (TV.8) в уравнения
(IV.2) и (IV.3) при условии <р = 0, легко убедиться, что
B = B0e',(kr-e>'>, B0 = -*IA0xk]
E = E0eI'<kr-"'>, E0=/|k|A0,
(TV.10)
т. е. векторы В и Е одинаковы по абсолютной величине и взаимно
ортогональны (напоминаем, что мы работаем в системе единиц СГС, поэтому
|ВНЕ|;всистемеСИ|В|=-|Е|).
с
Как мы видим, поля В и Е, вообще говоря, комплексные. Нас же
интересуют действительные (измеряемые) поля, поэтому далее мы будем
рассматривать волны вида
А = Ао^-"0 + А^Г,(кг-в") = 2А0 cos(kr -a>0. (TV.11)
Заметим, что речь идет пока о волне определенной частоты.
9
Векторы В и Б ортогональны направлению распространения
электромагнитной волны, задаваемому вектором Пойнтинга:
ввакууме
4я An
[ExB]L
(IV.12)
который удобен еще тем, что его длина определяет количество световой
энергии, переносимое волной в единицу времени через единичную
площадку, перпендикулярную его направлению:
(IV.13)
А это не что иное, как плотность светового потока, средняя величина которой
и есть интенсивность излучения (света), регистрируемая измеряющей
аппаратурой (приемником):
if.
I=<S>T=-jS(t)dt.
(IV.14)
(т - характеристическое время приемника). В вакууме в соответствии с
(IV.10)
Н = [пхЕ]
(где п = •— - единичный вектор в направлении распространения волны), и
плотность электромагнитного потока
S = — пЕ2.
Таким образом, измеряемая интенсивность излучения
пропорциональна средней за время измерения величине Е2:
I~<E2>t
(IV.15)
Помимо плотности потока бывает удобно использовать плотность излучения.
т.е. количество световой энергии в единице объема:
аЕ2 + иН2ввакуумеЕ2+Н2 Ё2
р- el— - = — t
8яг 8яг 4я-
(IV. 16)
которая связана с S уравнением
S = nc/7.
10
В силу (TV.3) при кулоновой калибровке (TV.9) приведенные выше
выражения для плотности, плотности потока и интенсивности излучения могут быть
переписаны с использованием векторного потенциала А:
1Г 1 г1
/ = - (S(t) Л =< S >т =< р >т с = [
ri 4яст1
0А|
Л. (IV.17)
Можно получить явную связь всех указанных величин с амплитудой
векторного потенциала А0, представив комплексный вектор А0 в виде
А0=пл|А0|е^,
где пА - единичный вектор в направлении А; | Aq I — длина вектора Aq; а ф-
его фаза - собственно комплексная часть вектора, квадрат модуля которой
равен единице.
Подставляя это выражение в (IV. 11), получаем
А = пА | А01 (е**-*"^ +еч(кг-^+^) = 2ъА | А01 cos(kr-a>t + ф)
и
SA
dt
= 2аякА | А0 | sin(kr - cot + ф).
Соответственно, интенсивность излучения
4жт{\
дА
2* = ^|А0|2
ж
-Jsm2(lo--a>f + ^>fc
Если характеристическое время измерения т - это период колебаний ^%,
то выражение в фигурных скобках равно VL, и формула (TV. 17) приобретает
вид
I=<S>T=<p>Tc = ^-\A0\2\
L7X
(IV.18)
11
§2. Электрические и магнитные характеристики молекул
в постоянных полях
Классическая модель
Молекула в однородном поле
Если внешнее поле однородно и достаточно слабо, то классическую
энергию молекулы е можно разложить в ряд Тейлора по степеням
напряженности приложенного поля. При этом в каждом порядке реакцию молекулы на
внешнее поле определяет та или иная ее собственная характеристика.
В электрическом поле:
*=4
де
Е=0
P=x,y,z
дЕ
fi)
Е=0
* 0>r=x,ytz
дЕадЕу
EpEY+... (IV.19)
Е=0
где Б - вектор напряженности электрического поля;
^Е=о s e° " энеРгия молекулы при отсутствии поля;
де
дЕ
*Ле-о
= dp,
(IV.20)
по определению, - компонента вектора электрического дипольного момента
молекулы;
Г л
^авг
=<^,
(IV.21)
Е=0
также по определению, - компонента тензора электрической поляризуемости
молекулы.
Таким образом, выражение для энергии молекулы в электрическом
поле
fl=x,y,z 0,Г=х>У>*
(IV.22)
где d - вектор электрического дипольного момента; a - тензор
поляризуемости.
12
Аналогично в магнитном поле
*=4
н=о
fi=x,ytz
де
tifi +
д2е
н=о
2Р,Г=х,уЛдН0дНг)
НрНу+...,
н=о
где Н - вектор напряженности магнитного поля;
«I-
н=0 s e - энергия молекулы при отсутствии поля;
1эя^н=о
=/</?>
(IV.23)
(IV.24)
по определению, - компонента вектора магнитного дипольного момента:
( St* \
~Z», 0V.25)
д*е
ая/?аяу;н=о
также по определению, - компонента тензора магнитной восприимч^СТИ
Соответственно энергия молекулы в магнитном поле,
*=£°чм*)4н+хн+...|
(IV.26)
Симметричные тензоры электрической поляризуемости и магнитной
восприимчивости соответствующим поворотом координатных осей могут
быть приведены к диагональному виду:
а =
О
О
а
уу
о
<*zzj
х =
Ххх U
О Хуу
. о о я
о
При этом скалярные величины
(х = У3(ахх^ауу^агг) и * = ,Ц (*»+*»>+*«)
называются соответственно средней поляризуемостью и средней
восприимчивостью молекулярной системы.
Явные выражения дипольных электрического и магнитного моментов
молекулы могут быть получены непосредственно по формулам (IV.20) и
(IV.24) дифференцированием ее энергии в поле, определенной в рамках
классической электродинамики.
13
Классическая функция Гамильтона молекулы в поле
Если частица к с массой тк и зарядом qk движется со скоростью v* в
электромагнитном поле, она подвергается действию силы Лоренца
Щ = 1k№k) + "[V* х Щ* )]), (IV.27)
с
где Е(г^) и Щгк) - напряженности электрического и магнитного поля в
точке с радиус-вектором 1%, задающим положение частицы. Энергия частицы
(ее функция Гамильтона) в этом случае определена выражением (IV.33),
которое можно получить следующим образом.
Если перейти от напряженностей к векторному и скалярному
потенциалам (в соответствии с (IV.2) и (IV.3)), то выражение лоренцевой силы (в
вакууме) приобретет вид:
F* .^gwrf^^-St^) + &[ xr<*A(r*)]). (IV.28)
с ot с
Воспользовавшись хорошо известным выражением двойного векторного
произведения
[[а х Ь] х с] = Ь(ас) - c(ab)
для a = V£,b = VHC = A(r^), можно в (TV.28) векторное произведение
представить следующим образом:
[v* х rot\(rk)] = grad(vk9 A(r*)) - (v*, V)A(r*)
и переписать соответственно лоренцеву силу как:
F* =-qkgradv(rk)-&^& + ^grad{vkMrk))-(4>V)Mrk)}. (IV.29)
С Ot С
Будем рассматривать эту силу как отвечающую некоторому
обобщенному потенциалу. В классической механике обобщенный потенциал (U),
зависящий не только от положения частиц, но и от скоростей их движения,
связан с обобщенными силами (Fz) следующим образом:
где rt - обобщенная координата (радиус-вектор) частицы, a v,- - отвечающая
ей скорость.
14
Сопоставляя выражения (TV.29) и (IV.30), можно «угадать» вид
обобщенного потенциала, отвечающего силе Лоренца, действующей на данную
частицу:
ик=дк<р(тк)-ЩукМгк)).
с
(IV.31)
Тогда функция Лагранжа системы заряженных частиц (ядер и электронов)
-К, (IV.32)
L = T-(U + V) = Z\^-qk<p(rk)-&(vkMrk))
к \ L c
где Г- кинетическая энергия, a (U+V) - потенциальная энергия, включающая
помимо энергии заряженных частиц во внешнем поле их взаимодействие (V)
между собой по закону Кулона. От этой функции Лагранжа можно перейти к
функции Гамильтона (которая есть классический аналог квантовомеханиче-
ского оператора Гамильтона), пользуясь тем, что, по определению,
обобщенный импульс
Ы
Р* =
dvk'
а функция Гамильтона есть
В нашем случае
и соответственно
к
Р*=™*П+ — А(Г*)
с
(рк-&А(гк))2
#=L f +Ел**)+И
к 2mk к I
(IV.33)
есть классическая энергия системы заряженных частил в электромагнитном
поле.
Используя это выражение, получим явные формулы, определяющие
электрический и магнитный моменты системы заряженных частиц.
15
Электрический и магнитный дипольные моменты молекулы
В постоянном электрическом поле (т.е. когда — = 0) частный вид
at
уравнений Максвелла (IV. 1) таков:
гоШ = 0
divD = 4ягр.
Соответственно вместо уравнений (TV2) и (W.3) имеем
Е = —gradq>.
Следовательно, скалярный потенциал в точке с радиус-вектором г* можно
определить как
0К1Ь) = -(ВД, (IV.34)
а функция Гамильтона (TV.33) есть
Я = 2^—Е^(Е^) + Г. (IV.35)
Дифференцируя эту функцию по напряженности электрического поля, в
соответствии с определением (TV.20) получаем хорошо известное выражение
для вектора дипольного момента:
(IV.36)
к
Аналогично частный вид уравнений Максвелла в постоянном
однородном магштшмидодш (когда — = 0) в вакууме:
dt
С
Он задает простую связь напряженности магнитного поля и векторного
потенциала:
H = rotA.
Последнее выражение, что легко проверить, позволяет выразить векторный
потенциал в точке с радиус-вектором г* следующим образом:
A(r*) = -i[r*xH]. (IV.37)
16
(IV.38)
Соответственно функция Гамильтона (IV.33) приобретает вид
* 2т* Л 2ткс2 к ткс
= Е р* ' Уg^xH]21 yftftbfo*H]), v
к2тк ^ %ткс2 к 2щс
Рассчитывая ее первую производную по напряженности магнитного поля (Н)
при условии Н = 0 с учетом того, что
<jfc,[ifc х Щ) = (Н,[Р* х rk]) = -(H,L*)
(где L* - момент импульса частицы), получаем в соответствии с
определением (TV.24) выражение магнитного дипольного момента:
k2m*c
(IV.39)
Оба дипольных момента - и электрический, и магнитный - векторы,
причем первый определяется пространственным распределением зарядов
системы, а второй - угловыми моментами этих зарядов. Таким образом,
электрический момент - статическая характеристика, тогда как магнитный
момент характеризует динамику зарядового распределения молекулы. Обе
величины, будучи свойствами молекулы, должны быть инвариантны
относительно преобразований симметрии. Это значит, что их компоненты
(проекции на оси системы координат) должны преобразовываться по
неприводимым представлениям точечной группы молекулы. При этом, поскольку
изменения декартовых координат ядер (Stk) можно описать в базисе векторов
единичных смещений (трансляций) вдоль координатных осей, компоненты
вектора электрического дипольного момента должны иметь ту же
симметрию, что и соответствующие трансляции. Компоненты же векторов
орбитального количества движения ядер hk определяют вращение вокруг
координатных осей. Поэтому симметрия компонент (проекций^ вектора магнит-
нот дипольного момента та же. что и вращений молекулы.
Приводимые представления и трансляций, и вращений мы обсуждали
при построении полного приводимого представления всех смещений ядер
молекулы в §3 главы Ш. Напомним характеры этих представлений:
г*
г**
Е
3
3
i
-3
3
а
1
-1
сг
2cosy+1
2cosy+ l
S,
2cosy-l
-2cosy+ 1
17
Инвариантность дипольных моментов относительно операций
симметрии означает, что эти векторы не изменяются при выполнении
соответствующих операций. Это существенно в случае статической характеристики
молекулы - ее электрического дипольного момента, определяемого
исключительно пространственным расположением заряженных частиц. В частности,
если в структуре есть плоскость симметрии или поворотная ось, то вектор
дипольного момента должен лежать на этой плоскости или оси. Бели есть
несколько элементов симметрии, то вектор должен лежать на их пересечении.
Например, в молекуле NH3 вектор дипольного момента должен лежать на
пересечении трех плоскостей симметрии и поворотной оси третьего порядка,
т.е. должен быть направлен вдоль этой оси. Бели же элементы симметрии
имеют только одну общую точку или в структуре есть центр инверсии (при
инверсии, напоминаем, координаты каждого вектора изменяют знак), то
вектор момента может быть только нулевым, т.е. молекула неполярная. Таковы
системы С2Н4 (симметрии />2*)> СН4 (Та) и С02 (/>«>*)• Молекулы же NH3
(с3у)инач(Сооу)-шдящше.
Что касается орбитальных моментов частиц, определяющих магнитный
момент молекулы, то поскольку они есть всегда, кажется разумным
предположение о наличии магнитного момента у любой молекулы.
18
Квантовая (полуклассическая) модель
Перейдем от классической функции Гамильтона (IV.33) к квантовому
оператору, придерживаясь выбранной полуклассической схемы, т.е. заменяя
операторами физические величины, характеризующие состояние молекулы, и
оставляя классическими характеристики поля:
й=£—f +5>**1*)+v
(IV.40)
Рассмотрим вновь два простейших случая: постоянное электрическое и
постоянное магнитное поля.
Дипольный момент и электрическая поляризуемость молекулы
В ПОСТОЯННОМ электрическом поле оператор (IV.40), отвечающий
классической функции (TV.35), имеет следующий частный вид:
J?—2^-vl+F-Sftftii). (IV.41)
к 2тк к
Будем считать последний член возмущением гамильтониана свободной
молекулы:
#' = -I>*(E,r*). (IV.42)
к
Тогда поправки к энергии свободной молекулы, которая есть величина е в
(IV.22), можно интерпретировать как соответствуюпще члены в разложении
энергии молекулы в ряд по степеням напряженности приложенного поля.
В первом порядке теории возмущений поправка к энергии л-го
стационарного состояния молекулы, согласно (1.144),
к к
(IV.43)
Эта линейная по полю поправка в соответствии с (IV.22) должна определять
электрический дипольный момент молекулы в данном квантовом состоянии:
к
(IV.44)
19
По смыслу эта величина должна быть средним значением оператора диполь-
ного момента в состоянии %
d.^T.iaiY^.
И хотя процедура восстановления вида оператора по его среднему значению
некорректна, поскольку не может дать однозначный результат, в данном
случае подынтегральное выражение
к
(IV.45)
в точности соответствует замене координат в классическом выражении ди-
польного момента их квантовыми операторами. Следовательно, мы можем
считать оператор (IV.45) квантовым оператором электрического диполъного
момента.
Во втором пррдщк? трррвд врзмуцдащй, согласно (1.148), поправка к
энергии выбранного состояния системы есть
£ &-*р '
что с возмущением (IV.42) дает
Ф-Ь
К^ЕЛда*)!?/^
J*n E^-Ef
_FiT<%iai^x>Fyiai%>l,
(IV.46)
Этот квадратичный по полю член должен в соответствии с (IV.22) определять
статическую поляризуемость молекулярной системы в данном квантовом
состоянии:
_<Yl,|a|Y/xY/|a|Y>l>
]ФП
40)-40)
(IV.47)
Воспользуемся тем, что, определив вид оператора диполъного момента по
выражению для его среднего значения, мы получили корректный результат,
согласующийся с принципом соответствия. Будем считать это достаточным
основанием для применения такого подхода и в данном случае и назовем
20
следующий оператор оператором поляризуемости:
*~ 2£п Йо-Ef)
(IV.48)
где Й0 - гамильтониан свободной молекулы, собственное значение которого
в состоянии Ч?п равно Е®\
Магнитный момент и магнитная восприимчивость молекулы
В постоянном магнитном поле частный вид оператора (IV.40),
отвечающий классической функции (IV.38),
Будем рассматривать последние два члена как возмущение, включающее
линейную и квадратичную по напряженности приложенного поля
составляющие:
Й' = -iftY ** (V*'[r* хЩ) = Гн,У »t"*V* x-uJ>) = -(Н,Уfl&) (iv.50)
it 8w*c
(IV.51)
(trjt"~ оператор орбитального момента количества движения &-й частицы).
Линейную по напряженности поля поправку к энергии свободной
молекулы в первом порядке теории возмущений даст только линейная (первая)
составляющая возмущения (IV.50):
В® =< Тп | Й'|Чп >= (Н,< ¥„ | -£?^ | % », (IV.52)
jt 2m*c
и поправка эта, согласно (IV.26), должна быть пропорциональна среднему
значению магнитного дипольного момента молекулы в состоянии %:
k=<*nilf^i%4
(IV.53)
21
«Восстановим» вид оператора по его среднему значению:
Ш Г2ткс
(IV.54)
и назовем его оператором магнитного дипольного момента, поскольку он
(как и электрический дипольный момент) получается просто заменой в
выражении (TV.39) орбитальных моментов частиц, составляюпщх систему, их
квантовыми операторами. Именно для того чтобы подчеркнуть, что речь идет
только об орбитальной составляющей момента у символа оператора Д стоит
индекс L. Действительно, в классической механике есть только угловой
момент количества движения, а в релятивистской квантовой теории
элементарные частицы (такие как электрон, протон, нейтрон) имеют еще собственный
момент количества движения - спин (в нерелятивистской теории
существование спина, как мы помним, постулировано). Поэтому в релятивистском
гамильтониане, представляемом рядом по степеням у , в первом порядке
помимо слагаемых
к
2ткс
(IV.55)
Eft"
к
(IV.56)
присутствуют еще члены
ft(H,Sfc)t
2ткс
где символ $£ использован для обозначения оператора спина А>й частицы
(обычно спин электрона обозначают S, а спин ядра /-го типа - I*); а
коэффициент gk, называемый g-фактором, определен природой соответствующей
частицы и является феноменологическим параметром модели.
Значения g-факторов ряда частиц приведены ниже:
частица
g-фактор
своб.эл-н
2.0023
'н
5.5854
" 2Н
0.8574
,3С
1.4042
,5N
-0.5660
l9F
5.2546
З.р
2.2610
В этом приближении возмущение Й{9 линейное по напряженности
приложенного поля, должно включать оба выражения (IV.55) и (TV.56).
Дифференцирование второго по Н даст еще один, спиновый вклад в магнитный
пипптплпдй момент системы в состоянии Уп:
к 2ткС
(IV.57)
22
являющийся средним значением оператора спиновой компоненты
магнитного дипольного момента, который можно определить так:
W f 2W*C
(TV.58)
Таким образом, оператор полного магнитного момента в квантовой модели
(IV.59)
A = Ai + As=I^)4t<* + g*M
Магнитный момент электрона, т.е. частицы с зарядом -е и массой те,
записывают с использованием величины
he
Мв =
2тес9
(TV.60)
называемой магнетоном Бора и определяемой как выбранный за единицу
магнитный момент свободного электрона:
H, = -^(L + geS)
(IV.61)
(здесь L и S - орбитальный момент электрона и его спин, a ge - g-фактор
электрона).
Аналогично можно определить ядерный магнетон
как магнитный момент протона, т.е. частицы с зарядом +е и массой тр.
Тогда магнитный момент ядра i-го типа будет записан следующим образом:
m-^(L/+*A)-^Li+rA.
(IV.63)
где L; - суммарный пространственный угловой момент протонов и
нейтронов ядра, 1^ - суммарный ядерный спин, gt - ядерный g-фактор, a yt = —g,-
- так называемое гиромагнитное отношение ядра.
Перейдем к поправкам к энергии, пропорциональным квадрату
напряженности приложенного магнитного поля. В отличие от электрического поля,
где был единственный оператор возмущения, пропорциональный дшюльно-
Щ моменту системы, в магнитном поле есть и линейная по полю составляю-
23
щая возмущения (TV.50), и квадратичная (IV.51). Поэтому интересующие
поправки к энергии даст как первый порядок теории возмущений с оператором
возмущения Й*, так и второй порядок, но с возмущением Й'. Рассмотрим
оба выражения.
Сначала оценим поправку, возникающую благодаря возмущению Й\
которое модифицируем соответственно наличию спиновой составляющей
(IV.56):
Й' = У ft <Н> ** + £ А) = (н у ft Фк + gА)1
2ткс
2ткс
-(№»)•
где ц - полный магнитный момент молекулярной системы.
Согласно (1.148),
40)-40)
=н+
1:
<%|А|'Р/Х^|А|Ч'Я>
£(0)_£(0)
(IV.64)
И если (как обычно) интерпретировать этот результат как компоненту
поправки к энергии, определяемой магнитной восприимчивостью (IV.25), то
соответствующее среднее значение компоненты магнитной восприимчивости
молекулы в состоянии я будет
,<У,|А1^хУу1А1%>
x£=-2L-
j*n
E^-Ef
(IV.65)
а отвечающая ей часть оператора магнитной восприимчивости может быть
определена так:
A|¥,xV,lA
Й0-Е^
(IV.66)
Эта компонента восприимчивости называется парамагнитной (верхний
индекс <<р»).
Парамагнитная восприимчивость молекулы в основном состоянии
положительна, так как суммирование ведется по всем возбужденным
состояниям молекулы (Е$® <Е^) и потому все знаменатели (Е^-Еу)
отрицательны, а в числителях стоят неотрицательные скалярные квадраты
матричных элементов < х¥п | р. | ^ >. Поскольку магнитные моменты частиц
обратно пропорциональны их массам, основной вклад в парамагнитную восприим-
24
чивость вносит электронная подсистема - ее динамика (напоминаем,
магнитный момент частицы пропорционален ее орбитальному моменту количества
движения и спину). Если в выражении магнитного момента пренебречь
ядерными вкладами (что возможно благодаря различвдо на три-четыре порядка
между массами электронов и ядер), то в нем можно выделить суммарный
орбитальный момент (Le) и суммарный спин (S) электронной подсистемы:
Следовательно, можно ожидать, что двухатомные молекулы, находящиеся в
синглетном состоянии (с нулевым спином) и имеющие нулевой орбитальный
момент (основное состояние £ характерно для гомоядерных молекул)
должны иметь нулевой парамагнитный вклад в восприимчивость. Ненулевой
является парамагнитная составляющая восприимчивости у триплетной
молекулы кислорода (ее основное состояние 3Е~) и у гетероядерной молекулы
монооксида азота N0 (2П). Парамагнитная реакция электронов на
приложенное поле фактически усиливает его.
Вторую компоненту можно получить, оценив поправку первого
порядка к энергии, обусловленную возмущением Й":
Вр =<*, \Й*\ЧГЯ >=<%[Z^(H+(^2l7^)HV, >, (IV.67)
к *ткс
где квадраты векторных произведений [гк х Н]2 были преобразованы с
использованием тождества Лагранжа:
[г**Н][г*хН] = Н2^
Соответственно виду поправки (IV.67) вторая компонента средней
магнитной восприимчивости молекулы в состоянии х¥п есть
%i —^„IZ^'^V, >. (IV.68)
и отвечающая ей часть оператора магнитной восприимчивости
(IV.69)
к *Щс2
носит название диамагнитной (верхний индекс V). Как и парамагнитная
составляющая, она определяется в основном электронной подсистемой, только
25
теперь не динамической, а статической ее характеристикой - распределением
электронной плотности, что позволяет упростить выражение (IV.69):
(здесь 1е - тензор инерции электронной подсистемы). Диагональные
элементы этой составляющей тензора восприимчивости (Zfifi) определяются (с
точностью до положительного коэффициента) взятой с обратным знаком суммой
положительных величин, например, при fi-x
(Ч?1-ЧЛ+)*-0£ + *£).
так что диамагнитная восприимчивость отрицательна. И определяемая ею
реакция молекулы на внешнее поле такова, что эффективное поле,
действующее на молекулу, оказывается меньше приложенного.
Таким образом, оператор полной магнитной восприимчивости
молекулы может быть записан как
| Рп #0-4 к 4"*** 1
Диамагнитная составляющая всегда будет ненулевой, поскольку определена
моментом инерции электронной подсистемы, но по абсолютной величине она
уступает парамагнитной составляющей, будучи обратно пропорциональной
квадрату скорости света. Поэтому молекула будет диамагнитной по существу
только при нулевом парамагнитном вкладе, что характерно для двухатомных
частиц в синглетном основном состоянии с полносимметричной орбитальной
электронной функцией. Например, упоминавшиеся выше молекулы
кислорода и окиси азота парамагнитны, а потому «втягиваются» в магнитное поле, а
молекулы водорода и азота диамагнитны и «выталкиваются» из поля.
Итак, квантовомеханические электрический и магнитный дипольные
моменты молекулы в любом ее состоянии определены точно так же, как и
классические, с той лишь разницей, что в магнитном моменте дополнительно
учтена спиновая составляющая, обусловленная наличием релятивистской
характеристики элементарных частиц - их собственного момента количества
движения (спина). Правда, будучи операторами, они позволяют определять
не только соответствующие характеристики конкретных состояний (как их
средние значения), но и характеристики переходов между состояниями (как
их матричные элементы на функциях начального и конечного состояний). С
26
точки зрения симметрии все выводы, сделанные выше относительно
классических величин, остаются верными и в квантовой модели,
В то же время электрическая поляризуемость и магнитная
восприимчивость являются существенно квантовомеханическими характеристиками
молекул, поскольку определяются совокупноегью их стационарных
электронно-колебательно-вращательных состояний. Тем не менее, в классической
теории также можно формально получить выражения для электрической
поляризуемости и магнитной восприимчивости молекул на основании условий
баланса сил, действующих на заряженную частицу в электрическом или
магнитном поле. Эти величины определяются частотой поля, зарядом частицы,
ее массой, скоростью движения (в случае магнитного поля) и собственной
частотой, которая при переходе к квантовомеханической модели может быть
интерпретирована как частота со^ перехода между основным и
возбужденным стационарными состояниями данной частицы.
Симметрия оператора поляризуемости определена произведением
операторов дипольного момента, а магнитной восприимчивости -
произведением операторов магнитного момента или операторов координат. Последнее
наводит на мысль, что симметрия поляризуемости и восприимчивости
должна быть одинаковой. Это действительно так. Если мы посмотрим на
приведенные выше характеры представления трансляций (описывающего
преобразования d) и вращений (описывающего ц). то увидим, что их элементы либо
совпадают, либо различаются знаком. Значит, их квадраты совпадают.
Поэтому в таблицах характеров точечных групп указывают обычно только
симметрию трансляций (Tx,Ty,Tz)9 вращений (RX9Ry9R2) и компонент тензора
поляризуемости (axy,aXZ9ayz.ax2,а 29azi). Определить симметрию
последних легко. Если декартовы координаты J3 и у преобразуются по
неприводимым представлениям Т\ и Г2 соответственно, то элемент ару преобразуется
так же, как произведение /Зу9 т.е. по представлению Г\ ® Г2. Заметим еше, что
одинаковая симметрия компонент тензоров а и % означает, что к
диагональному виду они приводятся в одной и той же системе координат.
Вид операторов электрического и магнитного дипольных моментов и
тензора поляризуемости будет нам необходим уже в следующем разделе,
поскольку именно эти операторы в простейшем приближении определяют
вероятности переходов в молекулах под действием электромагнитных полей
различной частоты. А знание симметрии проекций векторов электрического
и магнитного моментов и компонент тензора поляризуемости помогает
ответить на вопрос, реализуется ли соответствующий переход между парой
уровней известной симметрии.
27
§3. Молекула в переменном электромагнитном поле
Возмущение состояния молекулы полем:
одно- и многофотонные процессы
В предыдущем разделе магнитные и электрические характеристики
молекул были определены в постоянных полях. При этом мы предполагали, что»
наличие поля оказывает лишь небольшое возмущающее действие на
молекулярную систему, состояния которой в первом и втором (по энергии) порядках
теории возмущений суть стационарные состояния свободной молекулы (в
отсутствии полей). Бели мы переходам от постоянных полей к полю
электромагнитной волны, то идеологию теории возмущений (теперь уже временнбй)
по-прежнему можно использовать, если интенсивность поля невелика. Тогда
справедливо допущение, что возмущенное полем состояние молекулы Ч?„
«незначительно» отличается от исходного стационарного состояния *¥JP:
¥n=¥<0) + A. (IV.71a)
После выключения поля состояние молекулы вновь становится
стационарным, причем либо исходным, либо каким-то другим. Последнее обусловлено
тем, что в возмущенном состоянии молекулы в поле как бы «смешаны»
различные стационарные состояния:
д=Ес»*(о^0). (iv.7i6)
кФп
В момент отключения поля молекула «выбирает» одно из них с
вероятностью, определяемой весом этого состояния в возмущенной волновой
функции. Это и есть идеология временнбй теории возмущений, о которой мы уже
говорили в §5 главы I. Согласно проделанным тогда выкладкам, эти веса в
первом порядке теории возмущений определены как
,2
*ЛН$(г)|Ч
;}<^0)|#(0Р^Щ>«
(IV.72)
где fi\t) и есть оператор возмущения.
Переход ко второму порядку требуется тогда, когда в первом порядке
теории возмущений процесс (переход из одного состояния в другое)
запрещен, т.е. его вероятность W$ равна нулю.
28
В этом случае появляющиеся во втором порядке поправки
к
ȣ><*> H'ffwM
f Me) < *f 1^(01^0) > M av.73)
Qm*tt
могут служить оценками вероятностей соответствующих ^переходов.
Фактически матричный элемент
<^0)|i?'(0l^0)>
определяет вероятность прямого перехода молекулярной системы из
состояния 4?jp в состояние Ч^°\ Но помимо такого непосредственного изменения
состояния возможны и какие-то «окольные» пути. В некотором смысле
сумма по стационарным состояниям Ч^°\ отличным и от начального состояния
Ч?®), и от конечного состояния 4?f®
тФп
и есть вариант учета всех таких путей. Фигурирующие в этой сумме
стационарные состояния Ч^ называют виртуальными Гили промежуточными).
Представление о них оказывается удобным при обсуждении
оптических переходов (процессов, сопровождаемых рождением и (или)
уничтожением определенного числа фотонов), которые являются многофотонными.
Процессы вообще классифицируют соответственно числу участвующих
фотонов. И л-фотонный процесс - это процесс, в котором т фотонов рождается
и (п-т) уничтожается, причем эти процессы рождения и уничтожения
фотонов нельзя разделить во времени. Мы не можем рассматривать суммарный
процесс как последовательность разделенных во времени стадий поглощения
и испускания молекулой фотонов. Если бы это было возможно, то процесс
просто был бы совокупностью отдельных однофотонных переходов.
Однрфптотные процессы могут быть всего двух типов - это
поглощение или испускание молекулой кванта определенной .домны волны, причем
испускание может быть как спонтанным (в отсутствии поля)* так й
индуцированным (происходить только при наличии поля);
29
fico
£щ
Ek=En + h(a
поглощение
Eh=En-ha>
испускание
Многофотонные дропессы осуществляются (согласно обычной
интерпретации формул теории возмущений) благодаря наличию виртуальных
состояний, пребывание молекулы в которых нельзя зарегистрировать.
Поскольку при виртуальном переходе молекула не успевает локализоваться на
соответствующем уровне, согласно принципу неопределенности «энергия-
время», дисперсия ее энергии в этом состоянии чрезвычайно велика; и в1
соответствующий энергетический интервал может попасть значительное число
реальных уровней молекулы. Поэтому при рассмотрении перехода молекулы
между реальным и виртуальным уровнями теряет смысл и закон сохранения
энергии.
В частности, если речь идет о двухфотонном процессе, это значит, что
молекула одновременно поглощает фотон одной энергии и испускает фотон
другой энергии, что можно изобразить, например, так:
-4 *—%
ha>i
"THrft
^Nfttf*2
ho
...ат
h&i-hcb^Ek-En
стоксоео
tia)2-ti(Di=En-Ek
антистоксоео
комбинационное рассеяние
рэлеево
рассеяние
Начальный и конечный уровни энергии указаны сплошными линиями и
обозначены соответственно п и *, а виртуальный промежуточный уровень т -
пунктиром. Поскольку энергия виртуального уровня имеет очень большую
неопределенность, его положение на такой энергетической диаграмме
условно. Комбинационное рассеяние (КР), в результате которого система
переходит на более высоко лежащий уровень (стоксово рассеяние) или на более
низкий уровень (антистоксово рассеяние) - типичные двухфотонные процес-
30
сы, описание которых невозможно в первом порядке теории возмущений и
Tj^fiYftT перехода ко второму порядку. Рэлеево рассеяние - тоже двухфотон-
ный процесс, причем значительно большей интенсивности. Это простейший
упгерентный процесс, т.е. процесс, в результате которого состояние системы
не изменяется. Кроме того, он может служить основой для интерпретации
понятия стационарных состояний молекул. Действительно, пребывание
молекулы в стационарном состоянии при наличии внешних полей (которые
всегда есть в реальных условиях) можно рассматривать как непрерывные
виртуальные переходы, в результате которых система каждый раз возвращается в
исходное состояние.
Помимо изображенных выше двухфотонных процессов, существует
также двухфотонное поглощение и двухфотонное испускание:
—г
На>
-*и
~Ет
—я»
2ha> = Ek-En
двухфотонное
поглощение
—Чк
2Па> = Еп-*Ек
двухфотонное
испускание
В качестве примера когерентного процесса можно еще привести трехфотон-
ный переход, используемый в лазерах и называемый генерацией гармоник:
+*inl
En
2fta\ - ha>2
В результате такого процесса монохроматическое излучение с частотой щ
преобразуется в излучение с вдвое большей частотой а^- При этом уничто-
31
жаются два фотона и рождается один. Создается впечатление, что в этом
случае структура уровней квантовой системы уже несущественна, так как в
выражении закона сохранения энергии фигурируют исключительно энергии
фотонов. Однако без квантовой системы такое «слипание» двух фотонов в
один невозможно; да и не каждая квантовая система может обеспечить
нужное преобразование частот. Значит все-таки свойства конкретной
молекулярной (атомной) системы имеют значение.
Перейдем теперь к описанию оптических переходов в молекулах.
Гамильтониан молекулы в поле электромагнитной волны получается из
классической функции Гамильтона (TV.33) согласно принципу соответствия в
полуклассическом варианте - заменой на операторы только динамических
характеристик молекулы:
N+K < Q N+K
A=jL~(Pj -^Mrj)? + * + 1>Л). (IV .74)
Jm\ Zmj C ]=\
Здесь у нумерует и электроны, и ядра (общее количество которых N + К)\
оператор V включает все взаимодействия электронов и ядер между собой;
qj - заряд частицы с номером./ (для электронов qj=-e, для ядер qj = Zje);
А(г.) и <p(Tj) - соответственно векторный и скалярный потенциалы поля в
точке с радиус-вектором Гу. При этом мы (как и в предыдущем разделе)
работаем не в атомной системе единиц, а в обычной. Разница между
гамильтонианом (TV.74) и гамильтонианом свободной молекулы
N+К а?
*о=£о+* 0V-75)
у-1 2mJ
и есть создаваемое полем возмущение данной молекулярной системы:
N+K
а а1 } N+K
tmjc ZntjC J yei
(IV.76)
Учтем, что для любой частицы Mrj)Pj ф РуА(гу), поскольку
[A(rj),Pj]s A(rj)Pj -pjA{Tj) = 4*(A(tj)Vy -VjAfrj)) = ih divjA.
Это различие исчезает, если мы работаем в кулоновой калибровке
(IV.9). Кроме того, такая калибровка поля предполагает отсутствие
скалярного потенциала.
32
Соответственно оператор возмущения (IV.76) упрощается:
^ = ^fLj^A(r)p +AL |A(ry)|2L (IV.77)
% [ mjc 2mjC2 J
Далее мы рассматриваем влияние поля на молекулярную систему как
возмущение. В этом возмущении слагаемыми, пропорциональными
интенсивности поля, отнесенной к квадрату скорости света ^—J-, можно пренеб-
с
речь в сравнении с членами, линейно зависящими от —.
с
Тогда окончательное выражение оператора возмущения будет таким:
(IV.78)
I % [ mJc \
Фигурирующий здесь векторный потенциал характеризует поле
некоторого электромагнитного излучения, которое никогда не бывает строго
монохроматическим, а выше мы выписывали формулы только для волн
определенной частоты. Однако любую волну можно представить в виде наложения
монохроматических волн с различными частотами. Это так называемое
спектральное разложение. И если поле содержит непрерывный ряд спектральных
частот, его можно представить в виде интеграла Фурье:
1 +°°
А(0 = — J А^ ехр(-/й*) da>9
2п
-00
где
А„= |А(ОехрО**)Л.
Если накладываемое поле имеет небольшую спектральную ширину, т.е.
включает частоты из небольшого интервала (ю, a>¥dc*\ то ненулевыми в этом
разложении будут лишь А^, отвечающие этому интервалу. При этом вместо
средней за период колебаний величины р (плотности излучения), надо будет
использовать так называемую спектральную плотность излучения р(&).
33
Дипольное приближение
Для простоты будем пока считать электромагнитную волну строго
монохроматической с частотой со. Кроме того, будем рассматривать простей-
шую модельную систему: единственная частица, на которую воздействует
волна, - это электрон, находящийся в поле бесконечно тяжелого
молекулярного Гатомного^ остова с эффективным потенциалом U. Очевидно, от этой
модели легко перейти к системе, представляющей собой совокупность ядер и
электронов, просто просуммировав результирующее выражение по всем
заряженным частицам системы.
Итак, коэффициенты, определяющие вклад стационарных состояний
у/^ в функцию возбужденного полем состояния электрона, исходно
находившегося в состоянии у/^, в первом порядке теории возмущений таковы:
<$(г) = ~)< rf> 1#'(01^0) > *, (IV.79)
а во втором порядке:
Зависящие от времени функции стационарных состояний щ'9 щ'ъ Ут
могут быть представлены в виде
^0)=е» J \J>, (IV.81)
где | j > - решение стационарного уравнения Шредингера с невозмущенным
гамильтонианом Й0:
UQ\j>=Ej\j>. (IV.82)
Поскольку подынтегральные матричные элементы в (IV.79) и (IV.80) суть
интегралы по пространственным переменным, их можно переписать так:
< <40) |Я'(01^0) >=< * | #'(') I п > е*{Ек~Еп)' =< * | fr(t) | п > еш>«', (IV.83)
где а>ъп " частота перехода между состояниями y/f' и у/^.
34
Оператор возмущения определен выражением (TV.78):
-*-А(г*«-£
пес те\
и его подстановка в (IV.83) дает
# = Л- А(г)р = -*- {АъРе**е~ш + А^Л""},
<t|^(0|ii>^eW=-5-{Ao<ifc|»e*r|ii>^(e|""*)r +
m«c (IV.84)
+ Aj<Jt|pe~*r|w>e1
/(дали+^г
}.
Используя этот вид оператора возмущения, проанализируем выражения
(IV.79)h(IV.80).
Первый порядок теории возмущений:
однофотонные процессы
Интегрируя по времени уравнение (IV.79) с выписанным
подынтегральным вьфажением (IV.84), мы приходим к оценке коэффициентов в
разложении возмущенной волновой функции в первом порядке временнбй
теории возмущений:
^) = ^|а0<*1Р^
/!>-
*>**-*>
+ А0<к\ре'11а\п>- -
(IV.85)
Это сложное на первый взгляд выражение описывает поведение молекулы
под влиянием полей различной частоты, в том числе и в двух предельных с
физической точки зрения случаях:
(1) При cDfabG) первое слагаемое должно значительно превосходить
(IV.86)
Это условие означает, что частота внешнего поля приблизительно
совпадает с частотой перехода между стационарными состояниями щ* и
эеи
ст(тлх е a <k\tu/krin>^lak"~(0)T~1
c"t(r)ttBediAudlpc |л5 Фъ-Ф
35
у/п, причем конечное состояние у/\* имеет более высокую энергию*
Ек*Еп+Па> - это индуцированное электромагнитным полем
поглощение молекулой кванта указанной частоты.
(2) При cofa * -а> второе слагаемое существенно больше первого, и
(*)■
.-/кг |
mech
А0<к\ре'1КГ\п>
,г-1
<*>кп+<»
(IV.87)
Это условие означает, что Ек*Еп-На>, т.е. под воздействием
внешнего поля, имеющего частоту, близкую к расстоянию между уровнями
щ и ^ > происходит индуцированное испускание молекулой
кванта соответствующей частоты и переход ее на более низкий уровень.
Условия и технику проведения реального эксперимента подбирают так,
чтобы можно было изучать процессы определенного типа. Поэтому если
объектом исследования является поглощение, следует анализировать выражение
(IV.86), если испускание - (TV.87), а не общее выражение (IV.85).
Второй порядок теории возмущений:
двухфотонные процессы
Если теперь в формулу (TV.80) подставить выражение (IV.85) с учетом
матричного элемента возмущения (IV.84), можно получить следующее, еще
более громоздкое выражение коэффициента с\^{г):
•JZ
Qm*n
A0<m\p^a\n>- - +
to.
+ А0</п|ре"'"|и>
<0mn-<0
e»mn+<t>
{+Al<k\pe-aa\m>eKa,^+a,yr\
\dt.
(IV.88)
Интегрировать это выражение в таком общем виде мы не будем, поскольку
уже по аналогии с первым порядком теории возмущений можно определить,
какие слагаемые должны отвечать каким двухфотонным процессам.
36
(1) Первое слагаемое в каждой из фигурных скобок отвечает переходу с
поглощением кванта излучения На> (в первой скобке Е$ » EJp + hco,
во второй Е*Р *Е$ + ha>). Следовательно, их произведение должно
описывать двухфотонное поглощение:
ie2
X
c%4t)=№)2
X
f2;fAo<m|p^|»>^^^}{Ao<*l^l*>^^^)r)* =
= -^ Ag У < m |»g^ | ^ х ^ | р^ | m >Г|^("-"")Г "^Ь^ьп-^^
OV*) м« ol d^""e J
(IV.89)
(2) Аналогично, вторые слагаемые в каждой скобке отвечают
переходам с испусканием кванта излучения (в первой скобке:
Effi *Efp-ha>9 во второй: EJp *Е$-hw)9 и их произведение
должно описывать двухфотонное испускание:
. 1 Т ( i(m 4-/а\т -1
ie2
гА?2<^1^"*Г|"Х*1^"ЛГ|^>1£ 1
£п Ol ^«^ J
dv»)2
(IV.90)
(3) Наконец, «перекрестные» произведения первого и второго
слагаемых из разных скобок отвечают комбинации испускания и поглощения
кванта, т.е. процрррам рарсряния:
(IV.91)
Очевидно, в случае и одно-, и двухфотонных процессов достаточно
рассмотреть только один из возможных вариантов. Для оставшихся результат будет
аналогичным. Поэтому далее мы проанализируем однофотонный процесс
поглощения и двухфотонный процесс рассеяния излучения.
37
Поглощение излучения
Согласно (W.86), вероятность перехода молекулы из состояния у/*® в
состояние у/$ в результате взаимодействия с полем электромагнитной
волны, имеющей частоту со, определена так:
2 -I ,/-_ _-w |2
"$ w н 4? w i2=(~)! Ао < * i ** i - >|
2\еК<Оь,-о>)т _у
| «Ли-®
(IV.92)
Вводя обозначение а = —k , можно преобразовать последний
сомножитель в этом выражении:
e^'-l
(X
2а
Прежде чем рассчитывать квадрат скалярного произведения
A0<A:|^/kr|n>, (IV.94)
оценим типичный порядок величины скалярного произведения (к, г) в
надежде попытаться разложить в первом приближении экспоненту в ряд и тем
самым упростить решение задачи.
Радиус-вектор г, проведенный из центра масс системы в начало
координат, имеет длину, не превышающую размеров молекулы, т.е. величину
порядка 2-100 А. Волновой же вектор имеет длину ^пА (Л - длина волны
излучения), так что произведение (к, г) будет значительно меньше единицы,
если длина волны существенно превышает размеры молекулы. Примерные
границы спектральных диапазонов таковы:
рентгеновский 0.1 - 100 А
ультрафиолетовый и видимый 1000 - 8000 А
инфракрасный 8000 А -10 мкм
микроволновый 10 мкм -1 см
радиочастотный 1 см - 100 м
Каковы частоты переходов между электронными, колебательными и
вращательными уровнями молекул и атомов? Расстояния между основным и
первым возбужденным электронными термами обычно не менее 1.5 эВ
38
(12 000 см"1), что отвечает Я * 8 000 А. Это значит, что электронные
переходи лежат в ультрафиолетовой и видимой области спектра.
Внутримолекулярные колебания (а, следовательно, и переходы между соответствующими
уровнями) имеют частоты примерно от 500 до 4 000 см"1, что отвечает
инфракрасному диапазону. Наконец, расстояния между вращательными
уровнями имеют порядок вращательной постоянной с небольшим целым
коэффициентом, т.е. порядка 5-50 см"1, что отвечает микроволновой области
спектра. Во всех этих диапазонах выполнено условие (к,г)«1. Неприменимо
оно только в рентгеновской области спектра, где частоты превышают 10б см"
\ т.е. излучение имеет энергию более 100 эВ. Воздействие с такой энергией
разрушает молекулярную систему.
Итак, во всех спектральных диапазонах, применяемых при неразру-
шающем исследовании молекул, частота поля такова, что в первом
приближении можно разложить экспоненту в ряд
eto*l+ikr+-(ikr)2+... (IV.95)
и ограничиться первым членом этого ряда, т.е. единицей: g'1"' «1.
По физическому смыслу это означает постоянство векторного потенциала А
в предела^ мрлгеулы.
Тогда интеграл в (IV.94) превращается просто в матричный элемент
оператора импульса данной частицы, а соответствующее скалярное
произведение может быть переписано так:
A0<k\p\n>=<k\A0p\n>=<k\A0xpx+A0yPy + A02pz\n>. (IV.96)
Учтем, что оператор проекции импульса частицы равен, с точностью до
постоянного коэффициента, коммутатору оператора Гамильтона частицы и ее
координаты:
[д,#0]Е[х,-^-У2 + Р] =—Рх. (IV.97)
1те те
Используем это представление оператора импульса для расчета матричных
элементов (TV.96), помня, что функции \к> и \п> являются собственными
Для оператора Й0 (см. (TV.82)):
А0х<к\рх\п>=^А0х<к\[х,Й]\п>=Г!£А0х<к\хЙ-Йх\п>=
in in
= ^4)x(E„-Ek)<k\x\n>=-imeJ0xa>rt<k\x\n>.
39
(IV.99)
Подставляя этот результат в (IV.96) и возвращаясь от суммы
произведений декартовых компонент векторов к скалярному произведению,
получаем
A0<k\p\n>=-imea>nkA0<k\r\n>. (IV.98)
Теперь, с учетом выражений (IV.93) и (1V.97), оценим по формуле (IV.92)
вероятность лерехода молекулы из состояния yt^ в состояние у/^:
Присутствующий в этом выражении квадрат скалярного произведения
векторов А и < к | ег | п > можно оценить, основываясь на следуюпщх
соображениях. При случайной взаимной ориентации двух1 векторов & = (ах,ау,а2)и
b=(jbX9by,b2) верно
|(a,b)|2H^|2|UJ2+t«y|2|^|2+l^|2|b2|2.
Бели при этом
1^12Н^12Н^12=^|а|2,
что в случае векторного потенциала А означает изотропность излучения, то
|(a,b)|2=i|a!2|b|2,
т.е. в нашем случае
|A0<it|er|/i^2=i|Ao|2|<*|er|W>|2. (IV.100)
Фигурирующий здесь скалярный квадрат амплитуды векторного потенциала,
как мы помним, определяет плотность излучения (TV. 18):
|A0|2=^</». OV.101)
40
С учетом (IV.100) и (IV.101) выражение вероятности перехода (TV.99)
принимает вид:
пЛП/ ч (®пк\ 2* i li i |2sin2(ar)
(IV. 102)
Как видим, вероятность зависит от длительности действия внешнего
поля на молекулу. Посмотрим, каким будет результат, если частота
электромагнитного поля точно совпадет с частотой перехода между и-м и *-м
состояниями молекулы, т.е. со = со^, или а = 0. Очевидно,
limsin^ = r2> 0V.103)
а->0 а2
т.е. формально получается, что с увеличением длительности воздействия
вероятность перехода неограниченно возрастает. Но этот результат мы
получили, использовав для описания взаимодействия молекулы с полем теорию
возмущений, которая предполагает незначительное изменение состояния
молекулы в поле, т.е. малость кояАФштеитов <$(*") = ^W$(r). Значит,
примененный подход не позволяет рассматривать резонансные (т.е.
происходящие при точном совпадении частоты поля с частотой перехода в молекуле)
процессы поглощения или испускания. Более того, строго
монохроматическое излучение вообще получить невозможно, и работать приходится со
спектральными интервалами вполне определенной ширины (я, оИч/о).
Последнее требует замены в выражении (TV. 102) средней плотности излучения
на спектральную плотность с учетом ширины интервала:
<p>->p(a>)da). (IV.104)
Какова допустимая ширина этого интервала и соответственно
неопределенность в измерении частоты перехода между двумя стационарными
состояниями молекулы? Для ответа на этот вопрос учтем, во-первых, принцип
неопределенности, характеризующий соотношение между длительностью
воздействия поля на молекулу и шириной энергетического интервала, в который
она в результате может попасть и, во-вторых, характер функции
/(в.г)-^Й. (IV.105)
па г
определяющей зависимость вероятности перехода (огУ = —^—)отсоот-
m ..*$&),
41
ношения между временем воздействия поля (г) и отклонением частоты поля
от частоты перехода в молекуле (а).
(1) В соответствии с принципом неопределенности энергия конечного
состояния системы Ек зависит от длительности действия
электромагнитного поля г, и
так что из
(Ek+AEk)-En*ha>
следует
Таким образом, при г- 1 не неопределенность частоты перехода
порядка 109 с"1* 0.03 см"1 - вполне приемлемая точность измерения
(если учесть, что типичные энергии электронных, колебательных и
вращательных переходов не менее 12 000, 500 и 5 см"1 сЬответст-
венно). Заметим, что такая длительность воздействия поля на
молекулу значительно превышает характеристические времена всех
переходов, которые составляют примерно 10"15,10""13 и 10""11 с в случае
изменения соответственно электронного, колебательного и
вращательного состояния.
(2) Функция /(а, т) отлична от нуля фактически только в пределах
интервала (-я/г, л/т):
Таким образом, она играет роль своего рода фильтра: если частота
внешнего поля отличается от частоты перехода не более чем на
42
±2л/г (напоминаем, что а = —— ), вероятность осуществления
перехода наибольшая. Это значит, что соответствующая
неопределенность в измерении частоты перехода в молекуле имеет порядок:
Д(«Ъ-«)-*#.
т.е. открываемое этим фильтром «окно» примерно на порядок шире
того, которое «создает» принцип неопределенности. Поэтому той
же точности определения частоты можно достичь, увеличивая
длительность импульса электромагнитного поля до 10 не.
Заметим, что при увеличении длительности импульса до бесконечности
функция f(a-i т) превращается в дельта-функцию Дирака:
?^——►*(*), (IV.106)
т.е. становится «идеальным фильтром»; однако такой фильтр нам, как мы
выяснили выше, не нужен,, поскольку с ним оказывается неприменимой
теория возмущений. Но его можно формально использовать для того, чтобы
упростить выражение (IV. 102), а затем вернуться к «широкому фильтру»,
допустив существование спектрального интервала (IV. 104).
Итак, в соответствии с (TV. 106) преобразуем выражение (TV. 102):
(IV.107)
где
(а) учтено свойство «^функции
6(Х/а) = а6(Х),
(б) осуществлен переход к вероятности, отнесенной к длительности
импульса электромагнитного поля,
г
(в) принято во внимание наличие спектрального интервала излучения
(а>9 a^-dai).
^.йШ, flvjog)
43
Учитывая, что по определению ^-функции,
Ъ
lf(&6(X-t)dt = f(X) прка<Х<Ь,
интегрируем выражение (TV. 107):
^-^rMl^M2/**.)!
3ft
(IV.109)
Это и есть окончательная формула вероятности перехода в нашей модельной
«одноэяектронной» системе.
Теперь винемся к попнпй wmt, - я взаимодействии ммекупм (как
совокупности ядер и электронов) с П0Л8М ЖКШМаГНИЩЙ ВШПМ, где
оператор возмущения (TV.78) есть сумма одночастичных операторов:
N+K
-1L
ntjC
A(ry)py.
При такой записи оператора возмущения скалярное произведение в (TV.92)
превращается в сумму скалярных произведений:
N+K
ZL
to/
<*|A0£^iv '!">•
j=\ mj
(IV.110)
Поскольку для любой точки в пределах молекулярной системы верна оценка
(k,ry)«l,
можно по-прежнему ограничиться только первым членом (единицей) в
разложении экспоненты в ряд. Учитывая далее, что для любой частицы
молекулярной системы выполнено коммутационное соотношение (TV.97),
N+K h2 ifk
М 2mi mj
(IV.lll)
можно преобразовать выражение (TV.110) к виду, аналогичному (TV.98)
N+K
-й^Ао<*| Лям\п>. (TV.112)
А это есть скалярное произведение амплитуды электромагнитного поля и
44
матричного элемента дипольного момента (IV.45):
N+K N К
7=1 *=1 а=1
Окончательное выражение вероятности перехода молекулярной системы из
состояния Ч^0) в состояние WJp в поле электромагнитной волны таково:
Зя
(IV.113)
Поскольку вероятность зависит от матричного элемента дипольного
момента, получаемая таким образом оценка носит название электрического
дипольного приближение (которое в англоязычной литературе часто
обозначается £1).
Плотность излучения зависит от его частоты, но если регистрируемый
сигнал спектра нормировать с соответствующим частотным фактором, то по
соотношению квадратов матричных элементов < к | й | п > для одного и того
же начального состояния 4?f® и различных конечных состояний Ч^°* можно
судить об относительной интенсивности сигналов, регистрируемых
аппаратурой и характеризующих поглощение молекулой кванта данной длины
волны. При этом условие < к | й | п >= 0 означает запрешенность перехода (Е\-
запрещенный процесс). Но! Запрещен переход только в дипольном
приближении. Если бы мы учли при разложении экспоненты е1*1* в ряд следующие
члены, в частности ikr, то велика вероятность того, что переход,
запрещенный в дипольном приближении, оказался бы разрешенным. Однако,
поскольку /кг «1, пропорциональный этой величине матричный элемент оператора
возмущения должен быть тоже малым. Поэтому термин «переход запрещен»
означает в действительности, что интенсивность данного перехода,
например, на порядок меньше, чем у тех переходов, которые в дипольном
приближении разрешены (являются El -разрешенными). Соответственно, в
зависимости от чувствительности используемой нами регистрирующей аппаратуры,
мы этот переход можем либо заметить, либо вообще потерять на фоне
шумов.
45
Рассеяние излучения
Если речь идет о рассеянии излучения молекулой, изначально
находящейся в основном или одном из низших возбужденных состояний, можно
считать, что в выражении (IV.80) коэффициенты с££(0 формально отвечают
поглощению излучения с переходом на более высоко лежащие уровни, а
матричные элементы < к \ ft* \ т > - испусканию. При этом следует помнить, что
эти процессы неразделимы во времени, и нарисованная картина весьма
условна. Тем не менее, она облегчает восприятие отдельных сомножителей в
выражении (IV.91), определяющем коэффициенты в разложении волновой
функции молекулы в поле электромагнитной волны, а, следовательно, и
вероятность рассеяния этой молекулой излучения.
Рассмотрим, как и в случае поглощения, простейшую модельную
систему - электрон в поле бесконечно тяжелого атомного (или молекулярного)
остова - и перепишем выражение (TV.91) следующим образом:
ftr ikr г 0V.114)
х V <т\р/"\нхк\&- \т>ш(а>шт+а>>т)т_е/(«ь,+«0*)<й.
^ ГЛ.._ — Л) *
m*n w* 0
С учетом того, что
возьмем в этом выражении интеграл по времени:
/(л)=Л * i-^ U. (1УЛ15)
Выписывать выражение | с$(т) |2 и детально анализировать его мы не
будем. При желании это можно сделать аналогично тому, что было
выполнено в предыдущем разделе для величины | с$(г) |2. Рассмотрим лишь
входящие в него величины на «полуколичественном» уровне.
46
Используем, как и ранее, приближение (k,r) « 1. Ограничимся в
разложении экспоненты еР* в ряд только единицей и учтем соотношение
(IV.97), согласно которому рх =—[х,#0]. Тогда интеграл по пространст-
ih
венным переменным в (TV.114) можно преобразовать следующим образом:
<к\ре'Па\тхт\р^кг\п>»<к\р\тхт\р\п>=
(TV.116)
па)тк<к\г\тхт\т\п>9
.(»>.
где
С учетом (IV. 116), вьфажение (IV. 114) для коэффициента,
определяющего поправки второго порядка к волновой функции, принимает вид:
с$<г>""7^2 I AoI2 Z<*l«r|»xm|«r|n>7Qp),
(с») W ^
где
yim)mtm**^m±^ (TV.117)
Руководствуясь теми же соображениями, что и в случае переходов с
поглощением кванта излучения, можно выяснить, что функция ?2(со) также
создает своего рода фильтры электромагнитного излучения на частотах:
(1) сотп « со, что соответствует частоте поглощаемого фотона;
(2) tufa * -о>, что соответствует частоте испускаемого фотона; и
(3) (Ofo * 0, что отвечает рэлееву рассеянию, не зависящему от частоты
приложенного поля и сопровождающемуся возвращением частицы в
начальное состояние (* - л).
Что же касается функции g(r), то при переходе от простейшей модели
(один электрон в поле молекулярного остова) к совокупности ядер и
электронов (общее число которых N+K) она приобретает вид
N+K N+K
g(r)=<k\ ^qjTj\mxm\ ^qfj |я>=<*|д|тхт|4|и>.
47
В результате коэффициент перед комбинацией «фильтрующих
частоту» функций оказывается пропорциональным поляризуемости системы,
только не статической (TV.48), а динамической, характеризующей «реакцию»
молекулы не на постоянное, а на переменное поле. Она отличается от
статической поляризуемости наличием зависимости от частоты поля.
Таким образом, вероятность квантового перехода в молекуле,
сопровождаемого рассеянием излучения,
.taj'fffr)!2
Т
пропорциональна квадрату интенсивности приложенного поля (поскольку
согласно (TV. 18) интенсивность прямо пропорциональна | А012) и квадрату
матричного элемента оператора поляризуемости <к\й\п> и достаточно
сложным образом зависит от частоты поля:
яг
^ Н А0141< А: 1А | п >|2 7(^)1- (IV.118)
Это выражение, определяющее относительные вероятности процессов
рассеяния, тоже можно отнести к дипольному приближению, поскольку к
оператору поляризуемости мы пришли через оператор именно дипольного
момента.
Итак, в дипольном приближении вероятность процессов рассеяния
излучения значительно существеннее зависит от интенсивности световой
волны, чем при поглощении или испускании. Кроме того, поскольку «фильтр»,
отвечающий ф^ » 0 не зависит от частоты излучения (и в этом смысле
универсален), интенсивность рэлеева рассеяния должна значительно
превосходить интенсивность комбинационного рассеяния, как стоксова, так и анти-
стоксова. Именно это и наблюдается в спектрах. Что же касается собственно
комбинационного рассеяния, то здесь относительные вероятности переходов
с одного и того же исходного уровня (л) на различные конечные (*)
определены превде всего интегралами <к\й\п>. Конечно, как и в спектрах
поглощения, нулевые значения этих интегралов означают не отсутствие
соответствующих линий в спектрах, а лишь их существенно меньшую
интенсивность, которая может и не позволить обнаружить сигнал, обусловленный
данным переходом, в реальном спектре на фоне шума, определяемом
чувствительностью аппаратуры.
48
Коэффициенты Эйнштейна
При анализе однофотонных процессов поглощения и испускания
излучения часто используют так называемые коэффициенты Эйнштейн*,
характеризующие ту составляющую вероятности перехода, которая не зависит от
плотности внешнего излучения и от температуры системы (о которой мы
пока ничего не говорили и которая приобретает значение, когда система
{молекула + излучение} находится в состоянии равновесия). Согласно (IV. 114)
вероятность перехода (помимо плотности излучения) определяется дипольным
моментом перехода между данными состояниями. Условия, при которых
<*|д|л>*0,
называются правилами отбора. Их обсуждению посвящены следующие
параграфы. Но прежде чем рассматривать их применительно к конкретным типам
молекулярных спектров, проанализируем коэффициенты Эйнштейна и
соотношения между ними. Как мы помним, поглощение излучения может быть
только индуцированным, тогда как испускание может быть как
индуцированным, так и спонтанным:
к
1
4ь, )
Сплошными линиями обозначены индуцированные полем переходы
между уровнями кип, а. волнистой линией - спонтанное испускание
молекулой кванта излучения с переходом с уровня к на уровень л. Величины Впк,
Вып Afa суть коэффициенты Эйнштейна. При этом
есть вероятность индуцированного поглощения в единицу времени (о-
absorption, /-induced),
&Ы = вкпР(®кп)
- вероятность индуцированного испускания в единицу времени (r-radiation),
- вероятность спонтанного испускания в единицу времени (^-spontaneous).
49
Тогда суммарная вероятность поглощения в единицу времени
та шmaJ = B^PdtOkn), (IV.l 19)
а суммарная вероятность испускания
*т = *т* + *г,з = 4* + *JbiP(«fci). (IV.120)
Если ансамбль молекул данного вещества (в газовой фазе) равновесен,
то число молекул, находящихся в г-м квантовом состоянии, можно оценить
по формуле Больцмана
Ж
Щшщ g*ekTEj, 0V.121)
J
где N0 - полное число молекул, Nt - число молекул в i'-m квантовом
состоянии, Ej - полная энергия молекулы bj-m состоянии (решение стационарного
уравнения, включающее и ядерную, и электронную энергию), gj -
вырожденность данного состояния; в знаменателе стоит сумма по все возможным
электронно-колебательно-вращательным состояниям молекулы.
Зная количество молекул, находящихся в состоянии л, и суммарную
вероятность поглощения молекулой энергии с переходом на уровень ку
можно определить число переходов молекул с уровня п на уровень к в единицу
времени:
Аналогично, зная количество молекул в состоянии к и суммарную
вероятность испускания такими молекулами излучения с переходом на уровень л,
можно определить число переходов молекул из состояния к в состояние п
тоже в единицу времени:
mrNk.
Если система {(ансамбль молекул)+(излучение)} в целом находится в
равновесии, то
maNn=mrNk.
С учетом формулы (IV. 121) это можно переписать как
Еп
*т = 8п* кТ = gn c кТ
*а -fjt gk
gke *т
50
или, в соответствии с (IV. 119) и (TV. 120), - через коэффициенты Эйнштейна:
__]
Ъ. = Ап+Вкпй(<*>кп) ж &в1Г (IV.122)
Найдем соотношение между коэффициентами Эйнштейна и попробуем
связать их с матричным элементом оператора дипольного момента перехода
между данной парой уровней, пользуясь полученной нами формулой
вероятности перехода (EV.114).
Поскольку вероятность спонтанного испускания существенно меньше
вероятности индуцированного,
^«BbPiatn), (IV.123)
будем полагать, что в первом приближении
Лкп+ВкпРОЩп) увкп (TV.124)
ВпкР(&кп) Впк
Рассмотрим предел выражения (IV. 122) при бесконечно большой
температуре:
\im8n_e кт =1« (IV.125)
T-*»gk 8к
Комбинируя оценки (TV. 124) и (IV. 125), приходим к следующему
соотношению:
впк 8к
(IV.126)
называемому первым срртнрщдаизм МАЙДУ крэффщрдантаму Эйнштейн
Используем его для исключения коэффициента Впк из выражения (TV. 122):
вкп
Если фигурирующую здесь ПЛОТНОСТЬ УШАСТОГО ИЗЛУташЯ
аппроксимировать хорошо согласующейся с экспериментом формулой Планка
/>(*>) =
fte?3 1
ж с ехр(-—)-1
51
то можно записать соотношение между коэффициентами спонтанного и
индуцированного испускания
(IV. 127)
называемое часто вторым соотношением мркду ^Ффщшентами
ЭйнштейСоотношения (IV. 126) и (IV. 127) позволяют оценивать относительные
вероятности процессов спонтанного и индуцированного испускания и
поглощения излучения. При этом сами коэффициенты Эйнштейна тоже можно
оценить, зная характеристики молекулярной системы. Вероятность
поглощения (TV.119) можно оценить в дипольном приближении согласно (IV. 114):
Л
шп =
--2
4/Г 2
*,*/>(<»**) = —5-|<*|4|я>| /Каь,),
Ъп
так что
Зя
(IV.128)
С учетом первого соотношения (TV.126) это дает оценку коэффициента Вь,:
(IV. 129)
Bh, = ¥l\<k\*\n>\
2gn
3ftz
8к
а второе соотношение (TV. 127) позволяет оценить и вероятность спонтанного
испускания:
^А<*гаи
ЗПс'
28п
gk
(IV.130)
Теперь проверим, насколько корректным было приближение (IV. 123).
Иначе говоря, в каком случае (в каком спектральном диапазоне) нужно
учитывать спонтанное излучение, а в каком им действительно можно
пренебречь? Если дипольный момент перехода между двумя данными состояниями
<к\&\п> равен, например, 5 Д, а частота излучения лежит в радиодиапазоне
tofa ~ 10 см"1, то Afa ~ 10~5 с~\ Если при том же дипольном моменте
перехода частота излучения соответствует видимой части спектра, например, а>^ ~
IO'cm^to^-IOV1.
52
Величина, обратная коэффициенту А^ - это время жизни молекулы в
состоянии L если единственным процессом, переводящим ее в другое
состояние, является переход на уровень п:
I П
га
Следовательно, без внешнего поля время жизни молекулы в возбужденном
вращательном состоянии 4?f® (когда излучение кванта отвечает переходу на
другой вращательный уровень) на Ц порядков больше времени жизни в
возбужденном электронном состоянии (когда потеря кванта приводит к
переходу в другое электронное состояние) при условии, что дипольный момент
перехода в обоих случаях примерно одинаков. При этом вероятности
индуцированных переходов, определяемые коэффициентами Впк и В^ (согласно
(IV. 128) и (TV. 129)), явно от частоты поля не зависят - неявная зависимость
заключена в дипольных моментах перехода. Если допустить, что для
интересующих нас пар вращательных и электронных состояний они сопоставимы
по величине, оказывается, что относительная вероятность спонтанного
испускания возрастает на несколько порядков при переходе от процессов,
связанных с изменением вращательного состояния молекулы, к процессам
изменения электронного состояния.
Поэтому при анализе микроволновых спектров (вращательных
переходов) спонтанным испусканием действительно можно пренебречь; а в
видимом и ультрафиолетовом диапазонах приходится подчас учитывать
самопроизвольное возвращение молекулы в исходное (невозбужденное) состояние.
Отметим, что применяя теорию возмущений к описанию
взаимодействия молекулы с полем и оценивая таким образом вероятности переходов
молекулы из одного состояния в другое, мы не могли определить вероятность
спрнтг^тптю испускания, происходящего без «индуцирующего» воздействия
поля. Учет такого процесса оказался возможным лишь в рамках
термодинамического анализа в предположении существования равновесия в
совокупной системе {молекула + излучение}.
53
§4. Правила отбора
Строго говоря, правила отбора - это условия, при которых есть
ненулевая вероятность перехода между квантовыми состояниями молекулы в поле
электромагнитной волны заданной частоты. В принципе, запрещенных
переходов в природе, по-видимому, не существует. Есть переходы, происходящие
с очень малыми вероятностями. Поэтому говорить о правилах отбора можно
только в рамках того или иного приближения, использованного для описания
интересующего процесса. Такими приближениями в рассмотренном нами
варианте теории являются
(а) применимость теории возмущений для моделирования отклика
молекулярной системы на электромагнитное поле и
(б) постоянство векторного потенциала А в пределах молекулы (т.е.
превышение длиной волны размеров системы).
Эти допущения привели к формулам электрического дипольного,
приближения, в которых вероятность однофотонных процессов поглощения или
испускания пропорциональна
|<£|д!л>|2, (IV.131)
а вероятность двухфотонного процесса рассеяния пропорциональна
|<*|d|w>|2, (IV. 132)
где | п > и j к > - соответственно начальное и конечное стационарные
состояния молекулярной системы, й - оператор дипольного момента, d -
оператор поляризуемости. Во всем последующем изложении говоря о правилах
отбора в спектрах поглощения или рассеяния излучения, мы будем иметь в
виду условия отличия от нуля интегралов <к\&\п> и <к\&\п>
соответственно. И будем при этом помнить, что равенство нулю этих интегралов
означает не невозможность процесса вообще, а его очень малую интенсивность,
для оценки которой требуется переход к приближению другого уровня,
прежде всего к учету линейного члена разложения е1кт в ряд:
^«1 + rkr.
В результате вероятности переходов будут определяться уже матричными
элементами не оператора электрического дипольнош момента, а магнитного
дипольного момента или электрического квадрупольного момента молекулы.
Итак, не выходя за рамки дипольного приближения, рассмотрим
наиболее общий подход к оценке матричных элементов (IV. 131) и (IV. 132) для
54
процессов перехода между электронными, колебательными и
вращательными состояниями молекул. При этом термин «строгие правила отбора» будет
означать условия отличия от нуля соответствующих матричных элементов,
рассчитанных на точных волновых функциях молекул. А приближенные
правила отбора будут характеризовать ситуацию, когда состояния молекул
описывают функции, полученные в том или ином приближении (адиабатическом
или в пренебрежении взаимодействиями того или иного рода).
Строгие правила отбора
Строгие правила отбора можно определить, основываясь на знании
симметрии молекулярной системы. Согласно (1.47), интеграл в (TV. 131)
отличен от нуля, если
Г^ГЙ®Г^4 (IV.133)
а интеграл в (TV. 132) - при условии
Г^Гй®Г,з4 (IV.134)
где Гк и ГЛ - представления, по которым преобразуются функции
стационарных состояний молекулы | £> и | л >, а Г^ и Га - представления, по
которым преобразуются соответственно проекции дипольного момента и
компоненты тензора поляризуемости. В действительности для того, чтобы одно-
или двухфотонный переход между состояниями \к> и \п> был разрешен.
достаточно отличия от нуля хотя бы одного интеграла
\<k\dff\n> или <к\ару\п>\
соответственно (где 0fy = x9y9z).
Общее для всех молекул основание для классификации их состояний -
полный момент У. Это величина, сохраняющаяся у свободных молекул: как
мы помним, вращательный гамильтониан молекул, являющихся волчками
любого типа, коммутирует как с оператором J29 так и с оператором
проекции момента на ось Oz лабораторной системы координат Jz (см. (Ш.34) и
(Ш.35) в §2 главы Ш). Соответственно, вращательные состояния любой
молекулы определены ее полным моментом У и его проекцией М: \ JM > и
могут быть классифицированы по неприводимым представлениям группы
вращений (о которой шла речь при обсуждении теории кристаллического
поля в §7 главы П). Проекции вектора дипольного момента и компоненты
тензора поляризуемости тоже преобразуются по неприводимым представлениям
DJ у соответственно, трехмерному D1 и пятимерному D2.
55
Таким образом, подынтегральные выражения в (IV. 131) и (TV. 132)
можно формально интерпретировать как результат взаимодействия трех
квантовомеханических систем с различными полными моментами. Этот
результат будет включать полносимметричную компоненту (как того требуют
условия (IV. 133) и (TV. 134)), если такое взаимодействие может привести к
появлению системы с .7=0, описываемой неприводимым представлением
Z>° s^j. Согласно обсуждению в §4 главы I, при взаимодействии систем,
каждая из которых характеризуется своим полным моментом (Jx и J2), полу-'
чается система с целочисленным моментом из интервала
\Jl-J2\ - ^1+^2»
а точнее с определенной вероятностью может получиться система с любым
из этих моментов. Иначе говоря, при перемножении двух функций,
отвечающих полным моментам J\ и J2> получается линейная комбинация
функций с моментами от | J^ - J2 | до J\ + J2:
/' V2 =^1-/2|/!У1"У2|+^,-/2+1|/|У1",/2+1|+-+^.^)/(У1+У2)- С™5)
В терминах неприводимых представлений это записывается так:
DJl ®DJl = nH*~Jde...ez>(4/l4"y2). (IV.136)
В действительности не все функции /l-/i-^lj...>/W+^2) B (iv.135) имеют
ненулевые коэффициенты. Вообще коэффициенты с называются
коэффициентами Клебша-Гордана, и равенство их нулю означает невозможность
появления системы с соответствующим угловым моментом. Чем это
обусловлено? Тем, что, как мы сказали выше, каждое состояние свободной молекулы
характеризуется не только полным моментом, но и его проекцией на ось Oz,
и объединенная система должна иметь согласованные значения момента и
его проекции. Проекции же при сложении векторов складываются
алгебраически. Допустим, были два вектора 15,2 > и 13,2 > (записанные в формате
| JM>). По правилу сложения векторов результирующее значение J может
быть от 2 до 8; а по правилу сложения проекций результирующая проекция
может быть только 4. Но у вектора с длиной 2 или 3 проекция не может быть
равна 4. Следовательно, минимально возможное значение J в данном случае
4. Если бы векторы имели иные проекции, например, 15,2 >и 13, - 2 >, то
результирующая проекция, равная нулю, допускала бы любое из значений
полного момента от 2 до 8. В общем случае минимально возможное значение
момента определяется как максимальное цз двух величин \J\-J2\ и
56
Мх+М2. Кроме того, могут оказаться нулевыми и некоторые другие
коэффициенты Клебша-Гордана, но в детали их расчетов мы вдаваться не будем.
Итак, с учетом (IV. 135) и того, что TdsD}f правила отбора (IV. 133)
для однофотонного перехода | п >->| к > будут такими:
DJk®Dl<8DJ»z>D°,
причем
(DJk ®Dl)®DJ» ггф^'е//* ®DJ*+l)®DJ» =
= ф'к-'п-Ч $ м ф дЛ-Ч+Л ) $
ФфМ*-л1 е...Ф£)(,/*+Ул))е
е (Di/*+1~y«i e... е ^(у*+1+Уя))
(здесь в трех последних строках даны результаты перемножения
последовательно /Э^*"4, DJk и Z)(/*+I) на z/»). По условию (IV. 133) полученная
прямая сумма представлений должна включать D0. Ноль (как минимальное
возможное значение в индексе представления) может отвечать только
минимальному результирующему J в каждой из скобок, т.е.
I'*-4.1-0
|^-Уя+1|=0,
что эквивалентно условию
AJ = Jk-Jn=09±ll
(IV.137a)
В зависимости от типа молекулы (типа волчка) переход с tJ = 0 может
оказаться запрещенным; переходы же с А/ = ±1 разрешены всегда.
В случае двухфотонного перехода | п >-»| к > аналогичное
произведение представлений (TV. 134)
DJ* ®D2®DJ"
(где TasD2) будет включать 5 представлений, отвечающих возможным
минимальным значениям J:
|^-Уй-2|=0, I/4-J.-1I-0
|/*-Л+1|=0,|У*-Уя + 2Н0, J
57
что эквивалентно условию:
А^ = Л-Л=0>±1>±21- (IV.1376)
Как и в предыдущем случае, при более детальном анализе оказывается, что
для ряда молекул переходы с Д/ = 0,±1 не реализуются, а правило Д/ = ±2
работает всегда.
Приближенные правила отбора
В зависимости от того, как аппроксимированы волновые функции на-
-Ent -£**
чального (WJ® Цп>ен * ) и конечного стационарных
состояний молекулы, можно сформулировать более или менее точные
дополнительные правила отбора.
В рамках стандартной схемы решения стационарного уравнения Шре-
дингера дня молекулярных систем (рассмотренной в главах П и Ш) сначала
вводят адиабатическое приближение, т.е. представляют волновую функцию в
виде произведения электронной Ф(г,о* | R) и ядерной #(R) функций:
|t>=0^(r,a|R)^(R).
Затем при решении электронного уравнения (П.7), определяющего
возможные электронные состояния молекулы, в функции Ф(г,<т | R) выделяют
пространственную и спиновую части:
Oik(r,cT|R) = Odt(r|R)Odt(a).
Строго говоря, это приближение хорошо лишь для элементов второго
периода и для валентных электронов более тяжелых элементов.
При решении ядерного уравнения (ЦЛ1) или (ПЛ2) после отделения
свободного движения молекулы в пространстве предполагают отсутствие ко-
риолисова взаимодействия, т.е. отсутствие заметного искажения молекулы,
влияющего на характер ее вращения. Это возможно, если амплитуды
молекулярных колебаний малы. Тогда ядерную функцию можно представить
произведением вращательной функции Xroti9>^>¥) (зависящей от углов
Эйлера) и колебательной функции Х^ь(Я\>Я2>->Яък-б) (зависящей от
внутренних естественных координат молекулы):
58
гДе ХгоА<РьОУу/)ЦЖМ> для сферических и симметричных волчков и
Xrot(9>9>V) И JM > Дл* асимметричных волчков.
Кроме того, малость отклонений ядер от равновесных положений дает
возможность разложить все функции ядерных координат (в том числе и
потенциальную энергию) в ряды Тейлора по степеням этих отклонений и
ограничиться в разложениях первыми ненулевыми членами (квадратичными в
случае энергии). Получающаяся квадратичная форма записи потенциальной
и кинетической энергии позволяет перейти к нормальным координатам {Qj},
в которых молекула представляется совокупностью невзаимодействующих
гармонических осцилляторов (см. §3 главы Ш):
ЗК-6
ZvuMxAi ?з*-б)= Ylz*j(Qj)>
где ZvjiQj) ~ волновая функция одномерного гармонического осциллятора,
описываемого нормальной координатой Qj и находящегося в колебательном
квантовом состоянии vj.
Таким образом, ядерная функция приобретает вид:
ЗК-6
**(R) = zUvM EUto- dV.138a)
7-1
В случае линейных молекул, у которых 2 вращательных и ЗК-5
колебательных степеней свободы, ядерная функция
ък-s
ft(R)-*£,<**> П4<0/)' (IV.1386)
где Zrot(<P>0) Н ^0А/ >э YjAffaG) ~~ сферические гармоники.
В итоге, в рамках этой стандартной схемы полная волновая функция
стационарного состояния (нелинейной) молекулы является произведением
следующих функций:
злг-б !
* >= Ф*(г,сг | R)*>(R) - Фе*(г | И)ФЛ(|Т)Л(ЛЛГ) П4<В/)!
>1
0V.139)
При необходимости (в частности, при обсуждении состояний двухатомных
молекул и переходов между ними) мы будем использовать модифицирован-
59
ный вариант этой формулы, полученный заменой ядерной функции (IV. 138а)
функцией (IV. 1386). Но основное рассмотрение правил отбора будет
выполнено для общего случая (IV. 139).
С волновой функцией молекулы, аппроксимируемой выражением
(TV. 139), правила отбора в дипольном приближении выглядят так:
зк-б зя-б
< * | А | п >=< <ben<bmxnrot Tlzij I * I ф*ф*л& П4 >\ С™-140)
м м
где Л = 3 для однофотонных процессов, и Л = й для двухфотонных.
Поскольку и оператор дипольного момента, и оператор поляризуемости не
зависят от спина, интеграл (IV. 140) можно преобразовать следующим образом:
ЗЯ-6 ЭК-6
<k\k\n>=<O„Z?0t]Jz!j\A\(bekZMnXvj><(bon\<bok>-
В силу ортогональности собственных функций эрмитова оператора с
различными собственными значениями, интеграл А будет отличен от нуля, только
если функции Ф^ и Ф^ совпадают, т.е. описывают состояния с
одинаковым значением спина S (одной и той же мультиплетности (25+1)) и
одинаковой его проекцией Ms.
Итак, первое общее приближенное правило отбора:
Переходы с поглощением или рассеянием излучения происходят
между состояниями одной спиновой мультиплетности:
AS = 0.
(IV.141)
Правила отбора, определяющие отличие от нуля матричного элемента
гк-в злг-6
< ©«лиг ПхЪ IАI ф*хЬ П4 >• (1УЛ42>
различаются для одно-и двухфотонных переходов.
При этом общие закономерности существуют только для молекул,
имеющих центр инверсии. В этом случае все неприводимые представления
соответствующей точечной группы классифицируют по четности
относительно инверсии. Дипольный момент, как и любой вектор, является нечет-
htjmt поскольку все его проекции меняют знак при инверсии. А компоненты
тензора поляризуемости, преобразуемые, как квадратичные формы
декартовых координат, четны. Чтобы произведение трех представлений было четным
(и таким образом, могло включать полносимметричное представление), либо
60
все эти представления должны быть четными, либо два из них - нечетными.
Поэтому в случае дипольного момента одна из функций | к > или | п >
должна быть четной, а другая нечетной, т.е. разрешены однофотонные переходы с
изменением четности состояния;
g-
и и-*£
(IV.143a)
В случае же поляризуемости обе функции \к> и |л> должны быть либо
четными, либо нечетными, так что при двухфотонном рассеянии разрешены
пзретюды тодьуо с адкрарйщем ЧУЩОрти:
' (IV.1436)
g-*g и tf-ttiL
Эта закономерность носит название [правила альтернативного запрета!,
согласно которому переходы, наблюдаемые в спектре поглощения, не видны в
спектре рассеяния и наоборот. При этом может оказаться, что какие-то
переходы не регистрируются ни в том, ни в другом спектре. Обычно это правило
фигурирует при обсуждении колебательных спектров, которые будут
рассмотрены в следующем параграфе.
Прежде чем перейти к рассмотрению остальных правил отбора,
уточним, от каких переменных зависят все подынтегральные функции и
операторы.
Вращательные функции зависят от углов Эйлера (определяющих
переход от лабораторной системы координат к молекулярной - см. §1 главы Ш);
колебательные функции - от нормальных координат (в молекулярной
системе); пространственные электронные функции - от координат электронов, а
также - как от параметров - от нормальных координат ядер, однозначно
задающих ядерную конфигурацию молекулы при известной равновесной
структуре. Операторы же дипольного момента и поляризуемости определены
в лабораторной системе координат. Следовательно, чтобы взять интеграл
(IV. 142), необходимо выразить в молекулярной системе координат и
проекции вектора дипольного момента, и компоненты тензора поляризуемости.
Преобразование координат из неподвижной (лабораторной) системы к
вращающейся (молекулярной) можно задать матрицей поворота (см. (Ш.23) в
главе Ш), называемой также матрицей направляющих косинусов и
представляющей собой произведение трех матриц поворота на эйлеровы углы <р9 в и
уг.
to
to
= А
to
— A^A^AJ
(со$(аОх) cos(aOy) cos(aQz)Yx\
cotfbOx) cos(bOy) cos(bOz) 1 у
[^cosicOx) cos(cQy) cos(cQ2r)J^zJ
61
Здесь %0а - угол между осью 0% вращающейся системы координат (£ = а, Ь,
с) и осью Оа лабораторной системы (а * х, у, z). Обратный переход задает
обратная матрица поворота А^А^А^.1, элементами которой являются
косинусы углов о0£
Будем использовать следующие обозначения элементов матриц
поворота:
А§а = cos(£Oa) для преобразования л.с.к.=>м.с.к. и
А^ = cos(a0£) для преобразования м.с.к.=>л.с.к..
Компоненты (проекции) любого вектора преобразуются точно так же,
как и координаты некоторой точки пространства, поэтому проекция диполь-
ного момента на ось Оа л.с.к. (а = *, у, z) выражается через его щюекции на
оси м.с.к. следующим образом:
rfa=4»rfa +4*6^ + 4*^= Х4*^£- (IV.144)
£=а9Ъ,с
Тензор же поляризуемости (элементы которого ведут себя, как произведения
декартовых координат) преобразуется так:
<*>аЬс = AaxyzA" •
Поэтому его компоненты в л.с.к. выражаются через компоненты в м.с.к.
<*fiy = (A^a^A)^ = J^A^a^A^ (IV.145)
а,Ь,с
(здесь учтено, что матрица поворота - ортогональная матрица: А4 = А+).
Например,
а,Ъ,с
В результате проекции дйпольного момента и компоненты тензора
поляризуемости записаны как линейные комбинации произведений, в которых
элементы матрицы А являются функциями эйлеровых углов (как и
вращательные функции молекулы), а проекции d% и компоненты а^ (&tj = a, 6, с)
зависят от электронных и ядерных (например, нормальных) координат,
определенных во вращающейся системе'. Следовательно, интеграл (IV. 142) можно
записать в виде произведен!» интегралов. Для примера рассмотрим
проекцию dz и компоненту аа. Тогда
62
в случае А = й
ЗАГ-6 3*-6
3*-6 , . 3*-6
- Е<;&14{1 «*>**< Ш&К<Фе«1^1Ф^>г)1Г14^
6=а,Ь,с у=1 ./«1
(IV.146a)
а в случае к^й
3*-6 3*-6
< •«*£* TlZvj I <*« I *dt^L TlZvj >r,^,Q »
з*-б з*-б
= E<*« 14*4|J «*>**< Пху\\<феп\<*&\Фёк>г)\Пх*)><*
(IV.1466)
(г и Q обозначают соответственно совокупности всех пространственных
координат электронов {г,}, i=l...N9 и всех нормальных координат ядер
молекулы {Qj}9 j - 1...3Я-6).
Эти варианты записи означают, что в общем случае электронно-
колебательно-вращательного перехода сначала рассчитывают интегралы по
электронным переменным
fe^^i^i^*.^ ^.....^^^i!^i^^ (1УЛ47)
которые определяют правила отбора для перехода между электронными
состояниями Феп и Ф^. Заметим, что при п = к величины 2g и 8^ суть
средние значения проекции дипольного момента и компоненты тензора
поляризуемости в данном электронном состоянии (л) при заданной ядерной
конфигурации. А при пФк эти величины характеризуют переход между
электронными состояниями и называются соответственно дипольным моментом и
поляризуемостью перехода.
Затем находят матричные элементы
3*-6 ЗК-6 ЗК-6 ЗК-6 !
<n^i*fin4>Q ™ <n#i*Sin*W (IV148)
_>4 _JW l± l± j
определяющие переходы с изменением колебательного состояния молекулы.
63
И наконец (или, наоборот, в самом начале - поскольку это
независимый расчет) находят интегралы
l^l^fJi!^!^_™ fJ^fs^J^^f^ С™-149)
определяющие правила отбора для врашателытих переходов молекулы (в
качестве нижнего индекса здесь вместо z может быть любая из декартовых
координат а=х, у, z лабораторной системы).
Интегралы (IV. 147) и (IV. 149) рассчитывают непосредственно, а
интегралы (IV. 148) обычно упрощают, используя в очередной раз приближение
малых отклонений ядер от положения равновесия при колебаниях. Величины
Zf и Otfy, как функции ядерных координат, можно разложить в ряд
af=af|
ЗК-6
+ У
<н> ft
3*-6|
т Qjtft
{SQ,
'Q=0
«л* ,
шпк\
гк-ь
>1
да:
пк\
•y.*=i
3Jt-6f
бу&+-
Q=0
(IV.150a)
Q«0
(IV.1506)
Если ограничиться в этих разложениях членами, линейными по изменениям
Qj, то интегралы (IV. 148) можно еще упростить. Например,
зя-б зя-б
<П41^1П4>ов
>1 У=1
ЗК-61
. ЗК-6 ЗК-6 „„-л
а#1о=о<П41П4>о+Е
IQ=0 м м м
(ф
ъ
ЗК-6
ЗК^б
<U^(Qj)\Qj\Uxii^j>Q
'J )<&> J* J'1
(IV.151)
Соответственно, интегралы в (IV. 146а) и (IV. 1466) можно переписать и
определить общие правила отбора (IV. 142):
64
гк-6 З-аг-6
£=аДс Q=0 y«l >1
3A:-6f ajj* "\ з*-б зя-6
(a=x,yfz) (IV.152)
- условие того, что разрешен однофотонный
электронно-колебательно-вращательный переход.
И совершенно аналогично
3*-6 3*-6
(a=x,y,z)
ЪК-6 3*-6
<n4iQin4>Q<^i^«47/?i^>^'to
'<Н> M У"*
(IV.153)
- условие того, что разрешен двухфотонный
электронно-колебательно-вращательный переход.
Можно определить некоторые общие для всех молекул особенности
интегралов (матричных элементов), фигурирующих в выражениях (IV. 152) и
(IV.153).
(1) Интегралы
а? =<<*>«,I**|ф*>г и ag-<фш|лЛ|фл>г
отличны от нуля, если
ГИ®Г^®Г^41
Tn9Tk9t^^A^ (IV.154)
соответственно. Здесь Гй и Г^ - представления, по которым преобразуются
электронные функции молекулы; Г^ и Г^ - представления, описывающие
соответственно проекцию дипольного момента на ось 04 (преобразующуюся
так же, как трансляция вдоль этой оси) и компоненту тензора поляризуемо-
65
ста agjj (преобразующуюся как произведение координат grj). Фактически
условия (IV. 154^ - это и есть правила отбора для электронных переходов.
(2) Значения интегралов, включающих колебательные функции
з*-б ък-в зк-6 гк-6
<П^1П4>д и <n^iaiII4>Q' ^Л55)
зависят от того, какого типа переход рассматривается.
(а) Если речь идет о переходе между электпотшими состояниями, то
фигурирующие в этих интегралах фушсции Zvj и Xvj описывают различные
по форме нормальные колебания, т.е. различные осцилляторы. Причина в
том, что вид нормальных координат определяется потенциалом, а
потенциальные поверхности в двух различных электронных состояниях различны. В
результате получаются функции, которые не являются взаимно
ортогональными, т.е. в общем случае оба интеграла (IV. 155) могут быть ненулевыми.
(б) Если дстехдд - колебагельво-вршательшй а пределах одгогр
электронного терма, то в бра- и кет-векторах мы имеем произведение
функций одних и тех же гармонических осцилляторов. Следовательно, первый
интеграл в (IV. 155) может быть представлен произведением интегралов по
каждой из нормальных координат:
Поскольку собственные функции гармонического осциллятора образуют ор-
тонормированный набор, каждый из интегралов в этом произведении
отличен от нуля, только если функции Zvj(Qj) и XvjiQj) совпадают. А
произведение отлично от нуля, только если все сомножители ненулевые, что
соответствует совпадению исходного и конечного колебательных состояний
молекулы. Это значит, что при переходах в пределах одного электронного
состояния первый член в выражениях (ТУ. 152^ и (ТУ. 153^ описывает чисто
вращательный переход, без изменения колебательного состояния.
Второй интеграл в (IV.155) может быть представлен аналогичным
произведением. При этом в одном из сомножителей в подынтегральном
выражении появится дополнительно соответствующая координата:
ЗАГ-6
< ХыШ I Qt I XvtiQi) >Qi П< xnvj(Qj) I XvjiQj) >Qj •
66
Для всех интегралов под знаком произведения верно все сказанное выше.
Интеграл же
<Z$i(Qi)\Qt\zUQt)>Qi>
в силу рекуррентных соотношений для полиномов Эрмита (входящих в
собственные функции гармонических осцилляторов), отличен от нуля, если
колебательные числа осциллятора в состояниях Zvi(Qi) и ZviiQi) отличаются
на единицу:
\Шт±1\ (IV.156)
(см. обсуждение колебательно-вращательных состояний многоатомных
молекул в §4 главы Ш).
Таким образом, второй член в выражениях (ТУЛ52) и (ТУ. 153^ в данном
электронном состоянии описывает переходы, при которых изменяется на
единицу колебательное квантовое число только одного осциллятора. Бели
исходное состояние молекулы было основным (vj = О V/), то результатом
такого перехода может быть только фундаментальное состояние (vj = О V/V i и
vi = 1). Такие переходы называют фундаментальными.
Не каждый одно- или двухфотонный фундаментальный переход может
реализоваться. И зависит это от коэффициентов
(63f
и
Q-0
да(ч
Q-0
соответственно. Если они отличны от нуля (что определяется симметрией
нормальной координаты Qt и симметрией координаты £ или произведения
координат grj), то в используемом нами приближении переход разрешен.
(3) Наконец, интегралы
Y,<x^ot\A^\xkrot>iPeyf и Z<*k 14*4^ !*£*>**
(a,j3=x,y,z) (IV.157)
определяют возможность или невозможность переходов между втуАщаТ?Л1г
тдми состояниями молекулы. Как и в случае колебательных состояний, если
начальное и конечное электронные состояния молекулы различны, то в
общем случае можно полагать эти интегралы ненулевыми.
В пределах же одного электронно-колебательного состояния всё
определяется симметрией начальной и конечной вращательных функций (Xrot и
67
Xrot) и симметрией элементов А^ матрицы направляющих косинусов А
(а=х9 у, z и £= а, Ь, с). Возможная симметрия вращательных состояний
зависит от типа волчка, описывающего данную молекулу. Что же касается
элементов 4г£, то они при преобразованиях симметрии ведут себя так же, как
проекции полного момента на соответствующие оси координат О а, а=х,у,
z. Причина в том, что элемент А^ - это косинус угла между осью О а
лабораторной системы координат и осью 0£ молекулярной системы координат
(£= а, 6, с). Преобразования симметрии затрагивают именно молекулярную
систему. Например, в результате отражения может измениться направление
оси 0%. В этом случае вместо cos(a0£) будет сов(я - о0£) = -cos(a0£), т.е.
элемент А^ изменит знак. Но то же самое произойдет и с направлением
Зращения вокруг оси 0£ А поскольку каждая из проекций момента J% как
раз и описывает вращение вокруг соответствующей оси, симметрия
элементов Ад£ совпадает с симметрией проекции J%.
Тогда в зависимости от того, выполнено ли условие
Гп,,,л®Г™а®*^4| "™ \Trottn®^rat^^®^y^A\ (IV.158)
(где Ттп и Twtk - представления, по которым преобразуются начальная и
конечная вращательные функции молекулы, а^- представление вращения
вокруг оси Оф, вращат^т^Агый переход мзжду срртряниями пик будут ра?-
рущрнилцзапрущ^н.
Применим теперь знание всех этих особенностей к анализу электронно-
колебательно-вращательных переходов в двух- и многоатомных молекулах.
68
§5. Переходы между электронно-колебательно-вращательными
состояниями молекул
Двухатомные молекулы
В приближении «жесткий ротатор-гармонический осциллятор»
колебательно-вращательная функция двухатомной молекулы в некотором
состоянии п (см. (Ш.70)) имеет вид:
где
Х$(Я) = МтНт(Ш)е~ 2 (IV.159)
(R - межъядерное расстояние в молекуле) описывает колебательные
состояния молекулы, являющейся линейным осциллятором (vn - колебательное
квантовое число), и
*£»(**) ■ xif* (.9,0) = bjt. (9>,ff) =
"z/.+gci-JQifim (aaffjp" (IV160)
An(Jn+\Mn\)\ Ъ {C°Sff)e
(0и p- сферические углы, определяющие расположение м.с.к. двухатомной
молекулы относительно л.с.к.) описывает вращательные состояния,
отвечающие наличию двух вращательных степеней свободы (J„ и Мп - полный
момент молекулы и его проекция на ось Oz л.с.к.).
Электронные же состояния двухатомных молекул классифицируют (см.
§6 главы П) по неприводимым представлениям группы D^h (если молекула
гомоядерная) или С^ (если молекула гетероядерная).
Учитывая все это, проанализируем правила отбора для однофотонных и
двухфотонных переходов молекул (IV. 152) и (IV. 153).
Схема анализа всегда (в том числе и для рассматриваемых далее
многоатомных молекул) будет такая: сначала обсуждаются обшие
теоретические ппиштапы и определяемые им# правила отбора, а затем
рассматриваются практические аспекты ¥етода/в том числе спектры
конкретных молекулярных систем и способы оценки по ним различных
характеристик молекул.
69
Спектры поглощения
Заметим, что конфигурация двухатомной молекулы при любых
деформациях остается линейной. Поэтому у нее ненулевой является только
проекция дипольного момента на ось Ос - ось молекулы: da = d^ = 0 и dc=d,
причем d ф 0 у гетероядерных молекул и d = 0 у гомоядерных.
Тогда определенные преобразованием
М
.dz)
= А- А$
*ъ\
\dc)
COS0>
-sinp
О
sin
cosp
О
9 OVl
<p 0\\0 с
1Д° -
о
cos0
sin0
0 i<la)
sin0 I db
™e\dc)
компоненты вектора дипольного момента в лабораторной системе координат
будут
dx =Axcd = dsm<psin0
dy = Aycd
Jcosi>sind
dz=Azcd-dcos0.
Выражение (TV. 152) для однофптпниых переходов в двухатомных
молекулах будет выглядеть так:
I Sti p <Xnv\Xkv>K<Xnrot\A^\x^>^
£=atbtc
dR
1 /д-i^
<zSW\R\&R)>R<Zrt\M\Znt>0*o\
(a=x9y9z). (IV.161)
С учетом этого рассмотрим все типы переходов:
вращательные
колебательно-вращательные
электронно-колебательно-вращательные.
При этом будем помнить, что в выражении (IV.161) члены с df9 $=a, Ь9
важны лишь при процессах, сопровождаемых изменением электронного
состояния молекулы. А в любом заданном электронном состоянии среднее
значение дипольного момента может иметь только одну ненулевую проекцию -
на ось молекулы (Ос). И любое изменение межъядерного расстояния не
может привести к появлению компонент вектораг d в направлениях Оа или Ob.
70
Вращателрные спектры
Тшшшкт тшяшшц
Если изменяется только вращательное состояние молекулы, то в
выражении (TV. 161)
Xv ~" Xv *
и (в соответствии с не раз уже упоминавшимися рекуррентными
соотношениями для полиномов Эрмита, а потому и для собственных функций
гармонического осциллятора)
(IV.162)
<Zi\Zv>R-l
<z№)\R\z№)>r=o.
Последнее означает, что второй член в ГГУ.16П равен нулю, и вероятность
перехода в выбранном нами приближении определяет первый член. Один из
сомножителей в нем - интеграл
а2"-<ф«,1<*с1Ф-.>>.
который есть среднее значение дшюльного момента в данном электронном
состоянии при соответствующей ядерной конфигурации. Если молекула го-
моядерная. то эта величина равна нулю, что означает равенство нулю всего
выражения (IV.161), т.е. отсутствие чисто вращательного спектра у
молекулы.
У гетероядерной молекулы, имеющей ненулевой дипольный момент,
вращательный спектр существует, и вероятность перехода зависит от
матричного элемента
< Xrot I Лю I Xrot >ф$>
который с учетом (TV. 160) с точностью до нормировочного коэффициента
принимает вид
< ]№"\(совв)ё*Мн* 14* | l№k\co*ff)ebMk9 ><%> (IV.163)
где {</„, Мп} и {.7^, Мк) - полный момент молекулы и его проекция в
начальном (я) и конечном (к) состояниях.
71
Наиболее простое выражение получается в случае о = £, поскольку
Azc = cos0 и интеграл в (ГУЛ 63) может быть представлен произведением
интегралов
< ^й1(со80) |cos^| /^(cosfl) >в< е*Мп91 &М" >-.
Интеграл по углу р отличен от нуля только при \Мп-М^[ а интеграл по углу
в определяется ортогональностью полиномов Лежандра при данном М й
разных J и их рекуррентным соотношением:
co8^l(cos^) = ^^^(cos5) + ^/fl(co8e), (IV.164a)
из которого следует, что данный интеграл отличен от нуля при \J^ =УЯ±1|.
Очевидно, Jk=Jn+\ отвечает поглощению излучения, a J^=Jn-l -
испусканию.
При рассмотрении матричных элементов, отвечающих а = х и а = ;>,
удобно перейти к комплексному представлению тригонометрических
функций:
&-*** е^ + е4*
smp- . и cos^> = .
Кроме того, зависимость dx и dy от sin0 требует использования другого
рекуррентного соотношения для полиномов Лежандра:
sm0i^l(cos0) = -J—(/^j+1(cos^) ~ /^+1(cos0)), (IV.1646)
которое показывает, что, при том же условии для полного момента системы,
возможны переходы с изменением его проекции на единицу:
Итак, основной вывод:
\Мь=Мп±\
при отличном от нуля дипольном моменте молекулы
у нее есть чисто вращательный спектр поглощения (испускания),
переходы в котором определены правилом А/ = +1 (А/ = -1)
72
Пвикда/шй йшкт
В отсутствии колебательно-вращательного взаимодействия уровни
энергии жесткого ротатора
а частота переходов между ними при условии AJ = 1:
у«£Д/ + 2Х/ + 1)-ДО' + 9в20#(У + 1). (IV.165)
Это значит, что расстояние между линиями в спектре постоянно и равно 2Ве,
и есть возможность оценить вращательную постоянную молекулы и
определяющее ее равновесное межъядерное расстояние R^,:
Bzz h
(р- приведенная масса молекулы).
Условия реального эксперимента таковы, что молекула находится в
основном или одном из низколежащих колебательных состояний, и
необходимо учитывать колебательно-вращательное взаимодействие, в простейшем
случае выражаемое зависимостью (Ш.74) вращательной постоянной от
колебательного квантового числа v:
Bv=Be-ae(v + fy.
Располагая данными о частотах вращательных переходов в ряде
колебательных состояний, определяемых аналогично (IV. 165) как
у-ЗД/+1),
можно найти вращательные постоянные Bv и определить Вешае.
Кроме того, определив постоянную В0 для нулевого, колебательного
уровня, обычно оценивают и среднее межъядерное расстояние в основном
колебательном состоянии молекулы Rq.
73
Колебателъно-врашателъные спектры
1шюпкки8 тиидииы
Колебательные переходы в пределах данного электронного состояния
определены условиями:
Zv*Xv
На вращательные состояния изначально никакие ограничения не наложены.
Посмотрим, какие переходы возможны. Различие колебательных функций в
начальном и конечном состояниях означает равенство нулю первого члена в
выражении (TV.161) по причине ортонормированности собственных функций
гармонического осциллятора. Тогда всё определяется вторым членом в
(TV. 161). В нем зависимость от колебательных состояний заключена в
сомножителях
SM и <z№)\R\Z$(R)>r.
{ dR )**.
Матричный элемент отличен от нуля при [Ар = ±Ц (в силу рекуррентных
соотношений для функций гармонического осциллятора - см. (IV. 156) и §4
главы Ш). А то, возможны ли переходы с таким изменением колебательного
квантового числа или нет, определяет производная дипольного момента. У
двухатомной молекулы, ориентированной по оси Ос, межъядерное
расстояние и дипольный момент преобразуются по одному и тому же
неприводимому представлению - представлению трансляции по этой оси. Следовательно,
в общем случае производная должна быть отлична от нуля (см. (1.48)). Но
если молекула гомоядерная, никакие изменения ее межъядерного расстояния
не приведут к появлению дипольного момента, т.е. v гомоядерных
двухатомных молекул нет однофотонных колебатап^ип-ир^щательных спектров
(напоминаем, в рамках используемого дипольного приближения).
Бели же молекула гетероядерная, изменение расстояния между ее
ядрами изменяет дипольный момент, а значит у молекул типа НС1, НВг или СО
есть колебательно-вращательные спектры поглощения (испускания). При
этом возможные вращательные переходы определены тем же интегралом, что
и в случае чисто вращательных спектров:
< Xrot I Л» I Xrot ><рв •
74
Этот интеграл, как мы выяснили, отличен от нуля, если [А/ = ±11 Только
теперь поглощение излучения с переходом на возбужденный колебательный
уровень может сопровождаться изменением J как на +1, так и на -1.
Схематически переходы между парой колебательных уровней можно изобразить
так:
v' = l<
v' = 0
t, ТТ
1 А . -JuLL
А А
I I I A MM
Д/ = -1
Д/ = +1
В спектроскопических обозначениях низшее состояние отмечают двумя
штрихами, а более высоко лежащее - одним.
Полоса спектра поглощения молекулы НС1, отвечающая переходу между
основным и первым возбужденным колебательными состояниями выглядит
приблизительно так:
Р-ветвь
Д/ = 0
Д-ветвь
адо део дм
Частота, Гц
Я&*№*
75
Переходы с AJ = -1 образуют так называемую Р-ветвь. а с AJ = +1 - /{-ветвь
полосы. Отдельные линии в полосах обозначаются Rj и PJy где У- полный
момент в исходном вращательном состоянии. Интенсивность этих линий
определяется заселенностью вращательных состояний в условиях
эксперимента. Оценить ее можно на основании распределения Максвелла-Больцмана,
фигурировавшего выше при обсуждении коэффициентов Эйнштейна. Судя
по приведенной структуре спектральной полосы, у молекулы НС1 в основном
состоянии наиболее заселены уровни с J = 2 - 5.
Бели молекула может быть описана моделью «жесткий ротатор
гапмонический оспиллятор». ее колебательно-вращательные состояния
имеют энергаю (Ш.67)
2?w = 2*e/(J + l) + a^i; + }/).
Легко убедиться, что в этом случае расстояния между линиями в Р- и R-
ветвях должны быть одинаковыми:
*/+i -Rj = 2Be=Pj -PJ+1. (IV.166)
Считая вращательную постоянную одинаковой в обоих колебательных
состояниях, ее можно, таким образом, определить по расстояниям между
линиями Р~ и /{-ветвей. Бели вращательную постоянную полагать зависящей от
колебательного квантового числа, то аналогично чисто вращательному
спектру, можно (при некотором усложнении задачи) определить величины Ве и
ае.
Кроме того, расстояние между первой линией в /{-ветви (Rq) и Первой
линией в Р-ветви (/?) такое:
Посередине между этими линиями нет никаких сигналов, поскольку согласно
правилам отбора вращательное квантовое число J не может сохранить свое
значение при переходе:
Д/*0.
Это значит, что чисто колебательные пдр^хпдм в спектре двухатомной
молекулы в дипольном приближении запрещены (если колебания и вращения
молекулы независимы). Сигнал такого отсутствующего перехода должен был
бы находиться точно посередине меящу первыми линиями Р- и /{-ветвей. Его
частота была бы
v(0-»0) = a>e,
76
так что точка посередине между сигналами Rq и Pi дает гармоническую
частоту молекулы.
В приведенной выше спектральной полосе молекулы НС1 линии в?-и
Л-ветвях не эквидистантны. Причина в том, что молекула не является
жестким волчуом. и ее вращательная энергия (согласно (Ш.79)):
В этом случае частоты переходов, например, в /{-ветви:
vt-{^(^-l) + (^(/ + l)}-{J^(i/-CO + iSwir(J)}-
= те + 2Be(J +1) - 4De(J +1)3.
И расстояния между линиями в обеих ветвях уменьшаются с ростом J, т.е. с
увеличением частоты.
Еще одна особенность приведенного выше экспериментального
спектра - «расщепленные» сигналы, или дублеты. Причина в том, что этот спектр
получен для естественной смеси молекул с пачличным имтппним составом
Н 35С1 и Н 37С1. Поверхности потенциальной энергии молекул,
различающихся изотопным составом, как мы помним из обсуждения в главах П и Ш,
совпадают, поскольку в электронное уравнение в приближении Борна-
Оппенгеймера не входят массы ядер. Различие в состояниях таких молекул
появляется на стадии решения ядерной задачи. Частота колебаний молекулы
зависит от приведенной массы ядер и силовой постоянной:
е 1м
Приведенная масса ц = —-—— очевидно зависит от изотопного состава.
Mi +М*±
Силовая же постоянная, которая есть вторая производная потенциальной
энергии по нормальной координате колебания (т.е. межъядерному
расстоянию), одинакова для обеих молекул .по причине совпадения их
потенциальных поверхностей.
Обозначим отношение приведенных масс двух молекул,
различающихся изотопным составом, //д * 0.972 и jui2 « 0.974 как
р2 = Яй- = 0.998.
Ми
77
Тогда гармонические частоты этих молекул соотносятся следующим
образом:
-fr-p. ОУЛвТ)
а.
Это значит, что гармоническая частота второй молекулы (Н 37С1) немного
меньше, т.е. центр соответствующей полосы смещен влево, как и все линии
Р- и Д-ветвей. Бели бы влияние изотопозамещения было этим ограничено, то
спектр был бы просто наложением двух идентичных вращательных структур,
слегка смещенных друг относительно друга. Но расстояние между линиями
зависит от вращательной постоянной, которая обратно пропорциональна
моменту инерции 1е
определяемому равновесным межъядерным расстоянием (одинаковым для
двух молекул) и их приведенной массой. Последнее приводит к следующему
соотношению для вращательных постоянных изотопозамещенных молекул:
\ = Р2- GV.168)
Это значит, что в случае молекулы Н 37С1 расстояния между линиями
вращательной структуры немного меньше, и с увеличением У линии легкой и
тяжелой молекул все больше расходятся.
Электронные спектры
Правила отбора для переходов между электронными состояниями
определены отличием от нуля матричного элемента
Яр~<Фт\Ле\Ч*>т ffV.lW)
(£= а, ft, с). Когда мы рассматриваем состояния ядер, находящихся в
усредненном по пространству потенциале, создаваемом электронами, существует
действительно только одна ненулевая компонента дипольного момента
двухатомных молекул - вдоль оси Ос, т.е.
всегда < Фт \ йа \ Феп >г = 0, а = х, у.
78
Но если речь идет о разных электронных состояниях, то
может быть < Ф^ | йа | Фек >г* 0, а = x,y,z.
Последние интегралы как раз и определяют правила отбора для электронных
переходов, поскольку согласно выписанному выше преобразованию
координат,
da =dxcosp-dysmp
db = dx sinpcostf + dy cos<pco$Q - dz sin0
dc = dxsm<pb\n6 + dyCospsmO + dzQO&0.
(IV.170)
Как мьгпЬмним (см. §6 главы П) электронные волновые функции
двухатомных молекул можно выбрать собственными для оператора проекции
электронного углового момента Lz = -i —-, а потому в них можно выделить
д<р
зависимость от угла поворота вокруг оси Oz следующего вида:
Фе=Ф'ее-*А*.
При таком представлении электронных функций начального и конечного
состояний и с учетом (Гу.ПОУв интегралах (IV.169) можно выделить
слагаемые, в которых оператор не зависит от угла <p(dzco$0 и d2 sin#). Они
определяют правило отбора АЛ = 0. В остальных операторах зависимость от угла
(р заключена в сомножителях sinp или cos^. При использовании
комплексной формы представления эдас функций,
$iato<= z-^i 1 и cose) =
r ' ^2*'.--,■ 2
становятся очевидными два дополнительных правила отбора: АЛ = ±1.
Итак, общее правило отбора для электронных переходов двухатомных
молекул в дипольном приближении:
МтМУ* (IV.171)
Например, из исходного состояния Е молекула может перейти либо в
состояние £ (с тем же' Л = 0), либо П (А = 1);
' При'ЭФом, напоминаем, есть еще общее правило (IV. 142) для всех
молекул, согйаснб которому мультипЛе!1н6стЬ при переходе должна
сохраняться (если мы пренебрегли сШ(-$рШшьвым взаимодействием):
79
и разрешены переходы 11-> 11,to или 3Е-> 3Е, 3П,
но запрещены, например, переходы *Х -> 31 или to -> 3Е .
Кроме того, если молекула имеет центр инверсии, то однофотонные
переходы в ней возможны только с изменением четности состояния (см.
(IV.143a)):
разрешены 3UU -> 3Ig, 3Ug или lZg -* 1Ъи, lUu
и запрещены llg -» tog или to,, -» tott.
Относительно изменения колебательно-вращательных состояний
молекул при электронном переходе можно лишь утверждать, что поскольку
определенные разными потенциалами состояния не будут взаимно
ортогональными, в общем случае существует достаточное количество ненулевых
матричных элементов в (TV.161), отвечающих переходам с изменением
колебательного и вращательного состояний.
При этом предполагается, что переход из одного электронного
состояния в другое происходит настолько быстро, что положения ядер не успевают
измениться. Сначала такую гипотезу на основании полуклассических
соображений высказал Франк, а затем квантовомеханически обосновал Кондон.
Поэтому данное утверждение и носит название принципа Франка-Кондона.
Ограничимся первым членом в (IV.161) и рассмотрим коэффициенты
при интеграле djP:
<Zv\Zv >R< Xrot \Aas\Xkrot><pe- (IV-172)
По причине достаточно большой плотности вращательных состояний можно
считать, что основное значение в выражении (IV. 172) играет величина
<Zv\Zv>R-^ квадрат
\<Zv\Zv>r\\ 0V.173)
называемый фактором Франка-Кондона. и определяет относительные
интенсивности переходов между разными колебательно-вращательными уровнями
двух электронных состояний Фт и Ф^: чем больше перекрывание
начальной и конечной колебательных функций, тем выше вероятность (т.е.
интенсивность) перехода. Это легко понять с помощью следующего рисунка. На
нем схематично изображены потенциальные кривые основного (Ф"е) и
возбужденного (Ф^) электронных состояний некоторой двухатомной молекулы.
80
Точки минимума кривых отвечают энергиям El и Е'е, соответственно. Для
каждой кривой указано положение низших колебательных уровней молекулы
и вид соответствующих колебательных волновых функций (|Xv Г)-
Символами De и Dq обозначены энергии ди^^р^т отсчитываемые
соответственно от точки минимума ттотештальттой immintt (De) И ОТ АУЛШГО Ifflflgfa-
тельного уровня {D0). В основном электронном состоянии показан только
нулевой колебательный уровень, поскольку при обычных условиях тепловое
возбуждение колебаний невозможно и, согласно распределению Максвелла-
Больцмана, заселен только основной колебательный уровень. А в
возбужденном состоянии показаны основной и первые три возбужденных
колебательных уровня молекулы.
При вертикальном переходе (при неизменном межъядерном
расстоянии, что на схемах изображают вертикальной стрелкой) в данном случае
интеграл <Xo\zb >r практически нулевой,
<Zo\zi >r имеет небольшое положительное значение,
<Хо\%2 >r существенно отличен от нуля,
<хЬ\Хъ >R вновь имеет заметное ненулевое значение.
Это значит, что наиболее вероятен переход с колебательного уровня Vя = О
основного электронного состояния на колебательный уровень t/ = 2, и
несколько ниже вероятность переходов в состояния v' = 1 и v' = 3, а переход
системы в основное колебательное состояние v* = О весьма маловероятен.
81
Шиклапнпй я<ж кт
В зависимости от относительного расположения потенциальных
кривых основного и возбужденного состояний возможны различные профили
спектра поглощения. Например, если равновесные расстояния в этих двух
электронных состояниях близки, то наиболее интенсивным будет переход
Vя = 0 -> vf = 0 (или сокращенно 0 -> 0), и с увеличением vf интенсивность
сигнала будет уменьшаться:
1L
(схематическое распределение
интенсивностей полос
поглощения в спектре)
Если в возбужденном электронном состоянии молекулы равновесное
расстояние между ее ядрами несколько больше, чем в основном состоянии
(как в примере на предыдущей странице), то наиболее интенсивными будут
переходы на один или несколько возбужденных колебательных уровней v\
например
.ni
и
(схематическое распределение
интенсивностей полос
поглощения * спектре)
82
И, наконец, если равновесные межъядерные расстояния в основном и
возбужденном электронных состояниях молекулы существенно различны, то
очень «рано» проявляются переходы в область непрерывного спектра
колебательных уровней возбужденного состояния, которая отвечает диссоциации
молекулы:
(переходы в область
(схематическое распределение
интенсивностей полос
поглощения в спектре)
Ряд переходов с одного колебательного уровня Vя на различные
колебательные уровни v1 называется v' -прогрессией. Варьируя условия
эксперимента, можно наблюдать переходы не только с Vя = 0, но и с возбужденных
колебательных уровней основного электронного состояния и, таким образом,
выделить различные г'-прогрессии, отвечающие Vя = 0,1,2,... Можно
аналогично выделить и v' -прогрессии, отвечающие переходам с различных
колебательных уровней основного электронного состояния Vя на один и тот же
колебательный уровень v' возбужденного электронного состояния:
*з
-2
1
А
А
i i
i
i
\
i
i
J
J
t
ь
г-
/ \
и
и
{
7
н
Л
А
Г
^
j
v
P* = l
о' = 0
и'=1
v' -прогрессии Vя -прогрессии
83
Ниже приведен электронный спектр молекулы йода, в котором отмечены
линии нескольких v* -прогрессий (по оси ординат отложено пропускание):
Частоты переходов в электронно-колебательных спектрах (для
простоты о вращательных уровнях мы не упоминаем) определены разницей
электронных энергий состояний в точках минимума потенциальных кривых
Те-Т*е и разницей энергий колебательных состояний в возбужденном и
основном электронном состоянии G\ff) - G"(v"):
(IV.174)
Здесь использованы принятые в спектроскопии обозначения электронных
термов молекулы (Те = -^) и колебательных термов (G(v) = щу ').
Нале he
пример, для х/ -прогрессии (при v" = const) имеем
V = ve-G\v' = const) + o>W + У2)~ atXiv* + ]/$, (IV.175a)
const
а для v" -прогрессии (при vf = const)
V - vg + g'(p' = const) - coKv" + %) + a'Xiv' + )$ • 0V.1756)
const
84
Хорошо видно, что расстояния между линиями в v' -прогрессии равны
расстояниям между колебательными уровнями в возбужденном электронном
состоянии:
ПС
а расстояний в v" -прогрессии, соответственно, - расстояниям между
колебательными уровнями в основном электронном состоянии:
AV = V+i - V = -*>« + Щ?шФ +1) • (TV.1766)
В спектроскопии эти расстояния называют ПЕРВЫМИ КОМбйНШПГСШШМИ
разностями и обозначают AG
Уг
Суммируя эти разности для всех v" (или г/) от нуля до г?^ (г4шх)>
отвечающего диссоциационному пределу, можно получить оценку энергии
диссоциации Dq (отсчитываемую от нулевого колебательного уровня) в
основном (или, соответственно, возбужденном) электронном состоянии:
^0= "Е ЬВ^у и Db = °Y AGv,+ y. (1УЛ77)
Поскольку экспериментально измерить все vv> до v^ сложно (а часто
практически невозможно), существуют различные способы экстраполяции
величин AGp,+ i/ к v^. Один из вариантов - графическая экстраполяция,
или метод Берджа-Шпонер (Birge-Sponer):
AG,
Уг
площадь под кривой
и есть энергия диссоциации
в возбужденном электронном состоянии
85
Этот рисунок передает суть метода. Точки - это экспериментальные
значения, сплошная линия - их корректная (квадратичная) экстраполяция, а
пунктирная линия - линейная экстраполяция. Метод основан на предположении,
что формулы (IV. 176а) и (IV. 1766) корректно отражает изменение
расстояний между линиями в спектре в зависимости от колебательного числа v. В
действительности же при больших v эти расстояния уменьшаются быстрее:
не по линейной, а по квадратичной зависимости, - и для корректного
воспроизведения положений колебательных уровней молекулы следует учесть
члены, кубические по v (т.е. использовать формулу (Ш.74) - см. главу Ш).
Однако, в первом приближении этим пренебрегают, используя линейную
экстраполяцию имеющихся экспериментальных данных.
Рассчитывая помимо первых разностей AGr,+ у еще и вторые разности
д2е'=де,,+^-д<ц^ =-2*>;*;, (iv.ne)
можно определить ангармоническую поправку оэ*ех'е в возбужденном
электронном состоянии. Затем, по зависимости первых разностей от (v' + Y) (по
формулам (TV. 176)) можно найти и гармоническую частоту колебаний
молекулы а)'е в этом состоянии.
Заметим, что, записывая спектры молекул, утттШШЯ ИЗРТОШШМ
составом, можно оценить также гармоническую частоту и ангармоническую
поправку к ней для каждого из них: со*е и со^е. При этом, как мы выяснили
при рассмотрении колебательных спектров (TV. 167),
Зная теперь энергию диссоциации De, которая для осциллятора Морзе имеет
следующее выражение
D п-&-
и (в приближении Борна-Оппенгеймера) одинакова для изотопозамещенных
молекул, можно оценить и соотношение ангармонических поправок к
частотам их колебаний:
mi2J2
<*ехе
86
Спектры рассеяния
Правила отбора для переходов в спектрах рассеяния отличаются от
рассмотренных выше для спектров поглощения тем, что
(1) изменение вращательных состояний определяет теперь не матрич-
ный элемент <Хш \Ах4 \Znt >^,аинтеграл <зЛл\^аАф\3^л ><№'>
(2) вероятность возбуждения колебаний молекулы зависит от измене-
ния не дипольного момента
6R
, а поляризуемости
R=Re
8R
;и
R=R<
(3) наконец, вероятность изменения электронного состояния молекулы
определена матричным элементом не проекции дипольного момента
3^ =< Феп | й^ | Фек >г, а компоненты тензора поляризуемости
*$-<*m\*to\**>f
Учтем, что для двухатомной молекулы тензор поляризуемости диаго-
нализуется в системе главных осей инерции, т.е.
aaftc =
Г<*аа 0 (И
О аьь О
О 0 а„
Тогда выражение, связывающее компоненты тензора поляризуемости в
лабораторной и молекулярной системах координат (TV. 145)
ар?
aj>,c
упрощается до
(IV.180)
Элементы матрицы направляющих косинусов А$р = cos(40/?), а значит и
компоненты тензора поляризуемости в лабораторной системе координат
легко могут быть выражены через эйлеровы углы ви <р. Следующие несколько
громоздкие выкладки можно пропустить и перейти сразу к разделу,
посвященному вращательным спектрам рассеяния. Однако для того, чтобы ясно
представлять себе, почему в этих спектрах правила отбора оказываются
именно такими, желательно ознакомиться с этими преобразованиями.
87
Итак, компоненты тензора поляризуемости в м.ск. и л.с.к. связаны
посредством матрицы поворота:
ааЬс = А" ахугА = Ар А0 axyz^0^q>»
ИЛИ
Подставляя явный вид матриц поворота на углы ви <р, можно придти к
следующему выражению:
ахх аху *дг
<*ух <*уу <*yz
azx azy azy J
aeecos2^+a^sin2^ \(piaa -tfM,)sin2pcos0 \(aaa -aw>)sin2psin0
Ua^-a^mltpcose («aaf*2<P+<*bb*o*2<P)* ^aMsm:l<p+abb cos2 <p-
Ua^-a^smlipsme i(a^V^coiV (a sm2<p+abbcos2<p)x
(IV.181a)
Таким образом, например,
<*xx = aoA cos2 9+«to sin2 P
2
а согласно (IV. 180)
axy - ^(aofl " «6^)sin 2?COS0,
2 9 2
«jcx = aaaAax + <*ЪЪАЬх +<*ссАсх
axy = аааЛахЛау + <*ыЛ*Лу + <*сеЛ*Лгу •
Следовательно,
42r = cos2^
^=sin2p
4=0
A^Agy = sinpcos0>cos0
88
Л^Л^у = - sin 0> cos p cos 0
А^Асу = 0 и так далее.
При этом, в силу все той же аксиальной симметрии двухатомной молекулы
ааа =аЬЬ>
так что
аху ах
ОС ух (Хуу (Zyz
azy azy J
ам о и
0 а^, cos2 0+0^ sin2 0 (a^-a^sintfcosfl
0 (fl^-a^sinflcosfl a^s^^+a^cos2^
(IV.1816)
Зная выражения величин А%а через эйлеровы углы, можно определить
правила отбора (IV.153) для двухфотонных переходов в двухатомных
молекулах, общий вид которых в дипольном приближении для электронно-
колебательно-вращательных спектров такой
£<(* « i7< Xv \Xv>R< ХПтог J ^6ai,P I Xkrat >& +
\£,ri=atb>c
£,tj=a,b,c
dR
<ZvWIRI zt(R)>R< Xm I A^A^p \Хкгы><рв*<>j
Л=Л,
(IV.182)
Впашатепыше спектоы
Как и в спектрах поглощения, при чисто вращательном переходе в
выражении (IV. 182) ненулевым может быть только первый член по причине
ортогональности колебательных волновых функций
<Xv\Xv>R=Snk.
Кроме того, для гармонического осциллятора
<xS\R\x$>R=*nk±i-
(IV.183a)
(IV.1836)
89
Тогда вероятность двухфотонного рассеяния зависит от среднего значения
компонент тензора поляризуемости в данном электронном состоянии при
равновесном межъядерном расстоянии
и при ненулевой поляризуемости возможные изменения вращательного
состояния молекулы определены интегралами
< хт 14*4//? I Хм >*в • 0V.184)
Заметим, что в тензоре поляризуемости (в м.с.к.), характеризующем реакцию
молекулы на внешнее поле (перераспределение ее электронной плотности),
ненулевыми являются все диагональные элементы, а в л.с.к. нулевыми
оказываются только бГду и а^. Это значит, что для двухфотонных переходов в
двухатомных молекулах нет столь жестких ограничений, как для однофотон-
ных переходов. В частности, есть все основания ожидать наличия чисто
вращательных спектров у гомоядерных молекул. Так оно и есть.
Пары вращательных уровней, переходы между которыми разрешены,
определены интегралами (TVЛ 84), причем соответствующие произведения
элементов матрицы А (А^Ацр) нами уже найдены (см. (IV. 1816)). В
окончательном выражении элементов а^ присутствует лишь зависимость от угла
ft Следовательно, в (TV. 184) можно разделить интегрирование по углам ft и
<р.
<Xnrot\^a\p\Xkrot>90^
=< /^(cosft) | а^а^ | i^(cosft) >в< е*Мп9 \ *"" >9.
Интеграл по углу <р сразу определяет правило отбора по проекции полного
момента:
Мп =Мк, т.е. 1АМ = 01. (IV.185)
Принимая во внимание это условие, рассмотрим интегралы
<^l(cosft)|F(ft)|^l(cosft)>(?,
где (согласно (IV.1816)) F(ft) = cosftsinft, cos2 ft или sin2 ft. Их можно
преобразовать, используя рекуррентные соотношения для полиномов Лежандра
(1У.162.а)и(Р/.162.б).
90
При F(0) = cos0 sin0, получим
npHF(0) = cos2ft
:<btf:l+^i\<^il+^£>*
nnpHF(0) = sin20
< <*e#f -*T)i cjdjja*1 -ф|+Ь >,.
Первый интеграл отличен от нуля при \Мп \=\Мк |+1, что не согласуется с
условием (IV. 185). Два других интеграла (в силу ортогональности полиномов
P)M| с одинаковым Ми различными J) отличны от нуля при условий
Jn+l = Jk±l\
Л-1 = Л±1
о Jn=Jk,Jk±2 Ь [А/^0Д21.' (IV.186)
Условие Л/ = 0 при AM = 0 означает не отсутствие процесса перехода
(как в спектрах поглощения), а возвращение системы в исходное^^ состояние,
т.е. рэлеево рассеяние: Условия же А/= ±2ойредейяют комбинационное
В совокупности это дает три ветви в спектре рассеяния: Оветвь
(А/ = -2), Q-ветвь (А/ = 0) и S-ветвь (А/ = +2), причем собственно спектр
КР состоит из двух ветвей, О и S. Обозначения ветвей вращательных
переходов в спектре запомнить очень легко: располагая буквы в алфавитном
порядке, а числа А/ - в порядке увеличения:
0(А/ = -2), Р(Д/ = -1), Q(A/ = 0), R(A/ = +l), S(A/ = +2).
Пример чисто вращательного спектра рассеяния двухатомной
молекулы (14N,5N) приведен ниже.
Если не принимать во внимание центральный сигнал рэлеева
рассеяния, то две ветви спектра комбинационного рассеяния выглядят почта так
же, как вращательная структура колебательного перехода при поглощении
излучения гетероядерной двухатомной молекулой. Только теперь центр
полосы отвечает не гипотетическому чисто вращательному переходу, а частоте
91
возбуждающего излучения. В показанном спектре это v = 20500 см4
(аргоновый лазер), что соответствует типичным энергиям электронного возбуждения
частиц. Эта частота по существу является началом отсчета, т.е. нулем. И,
формально, регистрируемая частота испускаемого молекулой кванта либо
положительна (больше частоты возбуждающего излучения), если ДУ = -2:
(IV.187a)
(IV.1876)
v<P> = Oj = F(J)-F(J-2) = 2B(2J + 3),
либо отрицательна, если bJ = +2:
v^ =Sj = F(J)-F(J + 2) = -2B(2J + 3).
При этом расстояния между линиями в обеих ветвях одинаковы:
Av^Oj-Oj+^Sm-Sj^AB
и, как и следовало ожидать, вдвое больше, чем в колебательно-вращательном
спектре поглощения. Расположены эти ветви симметрично относительно
центрального сигнала рэлеева рассеяния:
S0-O0=\2B.
Интенсивности отдельных линий в О- и S-ветвях опять-таки зависят от засе-
ленностей вращательных уровней, с которых совершаются переходы. В
молекуле 14N15N, как следует из вида спектра, в условиях эксперимента
наиболее заселены уровни с J = 2 -12.
WW
+100
"Г"
-100
Av, см"1
92
Вклад в сигнал рэлеева рассеяния вносят переходы со всех
вращательных уровней, а в сигналы КР - только с уровня с соответствующим J:
N
т
\А (\(\Г\ Г\(
II 1 1 г
т III
_i Ml
III III
It 111
? 1
V '
\f\
V
V
антистоксово
КР
A/ = 0
рэлеево
рассеяние
Д/ = +2
стоксово
КР
Бели к описанию ядерных состояний молекулы применима модель щ&
сткого потатопа то выписанные выше выражения для частот переходов и
расстояний между линиями в О- и S-ветвях позволяют определить
равновесную вращательную постоянную молекулы В-Ве. Если необходим учетко-
лебательн^-^ращят^|у|у<)]^ дщрмодействия. то можно в первом приближении
оценить вращательную постоянную в основном колебательном состоянии
(В0) и постоянную колебательно-вращательного взаимодействия (ае):
В = В0=Ве -Угае. При наличии заметного пемтаобежно™ мжяжвиия.
формулы (IV. 187) следует скорректировать соответственно выражению для
энергии вращательных уровней
F(J) = BJ(J + l)-DJ2(J+l)2.
В этом приближении по расстояниям между линиями в спектре можно
оценить не только Во, но и D0. Заметим, что при существенном центробежном
искажении уже нельзя пренебречь колебательно-вращательным
взаимодействием й считать, что B = Bju D = De.
Величины Ве и В0 позволяют оценить равновесное межъядерное
расстояние ге и среднее межтущерное расстояние в основном колебательном
состоянии молекулы ф. Работа с естественной смесью изотопов или запись
последовательно серии спектров молекул, различающихся изотопным составом,
дает возможность определить В*е и г[ в соответствии с (IV. 168).
93
Колебательно^ращательные спектры
Теоретические ппиипит^
Как и при однофотонных переходах, различие начальных и конечных
состояний молекулы:
Xv * Xv 9
предопределяет равенство нулю первого члена в (IV. 182) и, таким образом,
правило отбора в виде
да\
&L
6R
<x№)\R\x5(R)>R*o.
Последний интеграл отличен от нуля при [Ар = ±lL т.е. и в спектрах рассеяния.
наиболее интенсивными должны быть переходы между основным и
фундаментальным уровнями молекулы (менее интенсивными будут переходы,
разрешенные не в электрическом дипольном, а в последующих приближениях,
например, магнитном дипольном). Такой переход будет наблюдаться в
эксперименте, если
dR
Координата R (межъядерное расстояние) имеет ту же симметрию, что и
трансляция по оси Ос, т.е. полносимметрична (4). В точечных группах C^v
и Дяд, описывающих соответственно гетеро- и гомоядерные двухатомные
молекулы, полносимметричными являются компоненты а^ +(Хуу и аа
тензора поляризуемости. При этом, поскольку изменение межъядерного
расстояния приводит к перераспределению электронной плотности в любой
двухатомной молекуле, и соответственно к изменению ее реакции на
внешнее поле, можно ожидать, что (в отличие от спектра поглощения) спектр
рассеяния существует как v гетеро-. так и v гомоядерных молекул.
Сопровождающее же колебательный переход изменение вращательного состояния
молекулы будет определяться теми же матричными элементами, что и в случае
чисто вращательного спектра, т.е. правила отбора по J будут |А/ = 0,±2[.
Условие AJ = О определяет чисто колебательный переход v = 0-> v = 1,
т.е. наличие Q-ветви в спектре. Изменение J на ± 2 проявляется в
присутствии ветвей О и S. Таким образом, в отличие от спектра поглощения, в спек-
94
тре рассеяния двухатомных молекул вращательная структура колебательной
полосы имеет всегда три ветви.
Прн1шляой аспект
Ниже приведен спектр молекулы монооксида .углерода СО.
О
2200 2100 см
Переходы, образующие ветви спектра, можно схематично изобразить так:
Л
Л
Л
Л
Л
ё
Д/ = -2
О-ветвъ
v
д/=о
Л/ = +2
S-ветвь
Частоты переходов
v = [G{v*) + F( Г)] - [G(v>) + F( J") ]
(IV.188a)
в «идеальном» приближении «жесткий потатор--гапмпничесуий осттштлятоп»
для v" = 0-* if -1 таковы:
95
(IV.1886)
В этом приближении все переходы, образующие Q-ветвь (ДУ = 0),
имеют одну и ту же частоту Vq = a>e, и потому эта ветвь, будучи
суперпозицией сигналов от всех таких переходов, должна выглядеть как один очень
интенсивный пик - напоминающий сигнал рэлеева рассеяния в чисто
вращательном спектре.
Сигналы, образующие О- и S-ветви, в этом приближении будут
симметрично расположены по обе стороны от Уд:
vs=a>e + 2Be(2J + 3)
v0 = a>e-2Be(2J + 3)
при постоянном расстоянии между ними в каждой ветви:
Av5=Av0 = 45e.
В реальных молекулах надо учитывать, во-первых, яттармпнюм колебаний.
который приводит к сдвигу всего спектра на -2<оехе, и, во-вторых, шк6&
ТШНО-ВрашагеЛЬНОе B3aHMQflgfiCTBHS. из-за которого расщепляются сигналы
в Q-ветви:
vq = Qj =К-2а>Л) + (2*; -Л£)/(/+1) (IV.189)
и
А Уд = fi/+l" Qj = ЧЩ, - W +1) - -2ae(J+1),
а сигналы в О-и S-ветвях сближаются с увеличением J:
bvs=Sj+l-SJ=4Be-2ae(J + 4).
В итоге, по одной полосе КР-спектра, отвечающей переходу между основным
(р" = 0) и фундаментальным (v' = \) колебательным уровнем двухатомной
молекулы, можно определить Фе-2техе (или <ое для молекулы с
гармоническим колебанием), Ве к ае. Если есть возможность зарегистрировать еще
и полосу, отвечающую переходу vw = 1-* v' = 2, с частотами линий
то измеряемые величины (а>е -а>ехе) и (а>е -4с»ехе) позволяют оценить уже
гармоническую частоту сое и ангармоническую поправку к ней соехе, а не
только их разность.
Колебательно-вращательные КР-спектры молекул с различным
изотопным составом различаются так же, как и однофотонные спектры поглощения,
96
поскольку различие определяется теми же соотношениями (IV. 167), (IV. 168)
и (IV. 179) для величин afet o^jtj и B\ молекулы.
Эщкурднцце дпуктру
1ашшпищ пуддпдпы
Изменение электронного состояния молекулы в результате двухфотон-
ного перехода определяется матричным элементом
при расчете которого, как и в случае однофотонных переходов, удобно
вернуться к лабораторной системе координат, где в электронной волновой
функции молекулы явно выделяется зависимость от угла поворота (q>) вокруг
оси Oz:
При этом компоненты тензора поляризуемости в м.с.к. могут быть записаны
через компоненты в л.с.к.
fit/=x,ytz
и фигурирующие здесь произведения Л^рА^ либо не зависят от угла <ру либо
включают sin20>, cos2p или sin^cos^ (преобразование, обратное
(TV. 181а)). В комплексном представлении эти тригонометрические функции
выглядят так:
sm^cos^> =
X * к
4/
' . 2 2-е2*-е"2* 2 2+ «*»+«-*»'
sm <р = и сое д> = ,
4 4
Соответственно, правила отбора:
<e~iK*9\const\e~i^4'>*Q, если Л* = Л„,
«ce-'^le^le-^^O, если Лк=Л„+2,
^-^l»-2** !«"***># О, ^ Лл=ли-2,
97
так что
[ДЛ = 0,±2|. (IV.190)
Это значит, что в результате двухфотонного перехода молекула, исходно
находившаяся в состоянии £ (т.е. Л = 0), может перейти либо в состояние Z (с
той же проекцией момента Л = 0), либо Л (т.е. Л = 2).
При этом выполняется общее правило сохранения мультиплетности:
и разрешены переходы
12->11,1А или 32-^32,3Д.
Кроме того, в молекулах с центром симметрии (гомоядерных молекулах)
переход может осуществляться только между уровнями одинаковой четности:
% -► %, lAg или 32и -* 32и, \.
Что же касается относительных интенсивностей переходов между
различными колебательно-вращательными состояниями, то они оцениваются точно
так же, как и при однофотонных процессах - по скалярным квадратам
произведений матричных элементов
l< Xv I Zv >R< Zrot I ^a^fl I *** >9в\2
и, прежде всего, по величине интегралов перекрывания начальной и
конечной колебательных функций молекулы:
Очевидно, при возвращении системы (молекулы) в результате
двухфотонного перехода в исходное состояние, этот интеграл будет равен 1 (т.е.
максимален), а потому и интенсивность рэлеева рассеяния, как и во всех остальных
случаях (вращательных и колебательно-вращательных переходов) будет
заметно превосходить интенсивность комбинационного рассеяния света.
В заключение заметим, что электронные спектры рассеяния
значительно реже используют для оценки энергетических характеристик молекул.
98
Многоатомные молекулы
Анализ спектров многоатомных молекул принципиально не отличается
от выполненного для двухатомных. Разница лишь в том, что
(а) колебания молекулы моделируются совокупностью нескольких в
первом приближении невзаимодействующих осцилляторов соответственно
числу колебательных степеней свободы и
(б) характер вращения определяется типом описывающего молекулу
волчка: сферический, симметричный или асимметричный.
Колебательно-вращательные состояния молекул разного типа были
проанализированы в главе Ш. Наповшим, как выглядят ядерные функции и
соответствующие им энергии молекул в простейшем приближении «жесткий
волчок - совокупность невзаимодействующих гармонических
осцилляторов».
— Линейная молекула:
3J£-5 "
j=i
> (IV.191)
ЗГ-5
- Сферический волчок:
ЗЛГ-6
— Симметричный волчок:
3JT-6
I-
*=1
> (IV.192)
ЪК-6
z^ivAtfZvibiQi.Qi бз*-б)Н-ЖА/> UzjiQj)
j-i
3*-6
Е„ = {&/(У +1) + (С- В)К2}+ ^(ok(vk + y$ (сплюснутый)
ы\
Е„ = }BJ(J +1) + {А - В)К2)+ £ufc(9* + Уд (вытянутый)
KIV.193)
Аг=1
99
— Асимметричный волчок:
ЗАГ-6
м
3JC-6
Е„=ЕЛА,В,С)+ Цщ{рк + У2).
> (IV.194)
Проанализируем правила отбора (IV.152) и (IV.153) для одно- и двухфотон-
ных переходов, учитывая конкретный вид вращательных функций (см. главу
Ш) и связь проекций вектора электрического' дипольного момента и
компонентов тензора электрической поляризуемости в л.с.к. и м.с.к.,
as
\dcJ
— АуАдАр
dx
™<ш **ob ™uc
aba abb abc
aca acb acc)
A
f
\dz)
(IV.195a)
— A^ Ag A^
«» <*xy <*xz
(Xyx CCyy (Zyz
\k¥A$K99 (IV.1956)
где
A =
fcos^ -sinIP 0\
sin у
0
cos^
0
0 cos0 -sin^Isinp cosp 0
0 sin0 cos0 1 о 0 1
-cos^sinp-sin|pcos0cosp 0 ^
-sin|psinp + cos^cos0cosp 0
sin^cosc) cos0
cos|^cos^~sm
sinycosp + cos
sinflsinp
(IV.195b)
Заметим, что симметричный тензор поляризуемости всегда можно привести к
диагональному виду, причем соответствующие оси координат не совпадают с
главными осями инерции только в случае асимметричных волчков.
Общий анализ правил отбора для одно- и двухфотонных переходов в
спектрах поглощения и рассеяния многоатомных молекул был выполнен
выше (на стр. 65-68). Рассмотрим теперь особенности, присущие молекулам,
описываемым разными типами волчков.
100
Спектры поглощения
Вращательные спектры
Теоретические гщищщпы
В дипольном приближении возможные изменения вращательных
состояний молекул при однофотонных переходах определены отличием от нуля
матричных элементов
<ХПгог\Аа$\Хкгог>9Ьг CV.196)
(а = х9 у, z; £ = а, Ъ9 с; элементы матрицы направляющих косинусов А
определены выражением (ГУЛ95в)) и коэффициентами при них:
зк-б з*-б
*Г <П4'П^>д>
1
т.е. средним значением дипольного момента молекулы в данном электронном
состоянии при равновесной конфигурации ядер.
Если волчок сферический, то у него, как у всякой системы,
описываемой одной из высших групп симметрии, дипольный момент отсутствует:
аТ\ =0.
Это означает, что чисто ЩМШИЯЫЮге СТШТО Y ШИК МОЛЕКУЛ НСТ - точнее,
нет переходов, разрешенных в дипольном приближении.
Если молекула - симметричр^ д^ртпк то ее вектор дипольного
момента направлен по главной поворотной оси, соответственно Ос в
сплюснутом волчке и Оа - в вытянутом. Рассмотрим сплюснутый волчок (для
вытянутого все будет аналогично с точностью до циклической замены координат
(а9Ь9с) -» (Ъ9с9а)). Итак, при равновесной конфигурации молекулы вектор ее
дипольного момента в м.с.к. d = (0,0,dc). Следовательно, его компоненты в
л.с.к. зависят только от углов <рив:
dx=dc
dy =dc
dz =dccos0.
101
Вращательная волновая функция молекулы есть функция Вигнера (Ш.37):
IJKM >- DmW,V) = **"*» (V*.
так что правила отбора определены интегралами
(1) <Р*\Р*>¥
(3) <еш*\*ш<р\еш«* ><р, <еш*\со*<р\еш»г >9 ъ<еш*\еш«* >,.
(1) Интеграл по углу у/ отличен от нуля только при условии Кк = KL
то есть при однофотонных переходах проекция полного момента на ось О
молекулярной системы координат должна оставаться неизменной:
АК = 0.
(2) Интегралы по углу <р (при использовании комплексного представлю
ния тригонометрических функций) дают правило отбора по проекции
полного момента на ось Oz лабораторной системы координат:
ДЛ/ = 0,±1.
(3) Наконец, интегралы по углу в определяют, как при поглощение
(испускании) излучения может изменяться полный момент системы /.В сил&
рекуррентных соотношений для функций </ja/(0) эти интегралы отличны от
нуля при следующих условиях:
если К Ф О, М ф О, то AJ = 0, ± 1;
если хотя бы одна из проекций АТ,М = 0,то Д/~±1.
Однако не все комбинации выписанных величин AJ > АК и AM
отвечают спектральным переходам. Причина в том, что уровни энергии
симметричного волчка вырождены по М. Следовательно, изменение состояния
молекулы AJ = О, АК = О, AM = ±1 не сопровождается Поглощением или
испусканием энергии. Поэтому правила отбора, определяющие в дипольном
приближении возможные однофотонные переходы, выглядят так
Д/ = +1, ДАГ = 0, ДА/ = 0,±1
Д/--1, AK = 0, AM = 0,±l
-поглощение _ _
(IV.197)
- испускание.
102
Если молекула - асимметричный волчок, то ее вращательные волновые
функции \JM> представляют обычно линейной комбинацией функций
| JKM > симметричного волчка, искажением которого можно получить
данный асимметричный волчок:
J
\JM>= %СК\ЖМ>.
Следовательно, правила отбора по величинам полного момента J и его
проекции на ось Oz лабораторной системы координат М должны быть теми же,
что и у симметричных волчков:
Д/ = 0,±1, ДЛ/ = 0,±1
При этом отсутствие во вращательном гамильтониане молекулы оператора
Jz вновь приводит к вырождению состояний по проекции М при данном«/, а
значит к тому, что Л/ = 0 не отвечает переходу с изменением энергии
системы. В итоге основные правила отбора в спектрах поглощения и испускания
молекул типа асимметричного волчка такие:
Д/ = +1, Mf=0,±l
Л/ = Ч, ДА/ = 0,±1
-поглощение ^ ш)
- испускание.
Есть еще дополнительные правила отбора, связанные с К, поскольку
состояния асимметричного волчка классифицируют обычно по проекциям полного
момента \К\ и \К*\ состояний вытянутого и сплюснутого симметричных
волчков, с которыми они коррелируют: J\k\\k'\ • В самом общем виде согласно
этим правилам при переходе должна измениться четность хотя бы одной из
проекций, | К | или \К'\.
Если молекула - линейная, ее вращательный гамильтониан подобен
гамильтониану двухатомной молекулы, а собственные функции -
сферические гармоники. Поэтому и правила отбора для однофотонного поглощения
или испускания излучения те же, что и у двухатомных молекул:
А/ = ±1, ДМ = 0,±1. (IV.199)
По-прежнему (из-за вырождения состояний по проекции момента на ось Oz
л.ск.) неизменность полного момента означает отсутствие перехода, а
потому условие 6J = 0, формально отвечающее ненулевому значению интеграла
(ГУ.196)при ДА/=±1, не включено в правило (IV.199).
103
На основании частот вращательных переходов можно, пользуясь
выражениями (TV.191>-(IV.194), определить вращательные постоянные молекул, а
значит, их главные моменты инерции.
Колебательно-вращат^рные спектры
Тедршпсшк припщщц
Правила отбора для колебательных переходов имеют вид
злг-б ък-ь
у z 1
<n^iain4>Q"0-
/Q=0 ■/"» >»
Они означают, что нормальное колебание с номером i может быть
возбуждено излучением с подходящей длиной волны, если
ЗАГ-б ЗЛГ-6
"О И < WXvj\Qi\Y[Xvj>Q*Q,
/Q=0 J=l J°X
faffQ
30
тронном состоянии Ф^,, а
за
где З^" есть среднее значение дипольного момента молекулы в данном элек-
- его изменение при небольшом иска-
жении геометрии молекулы (соответственно форме 1-го нормального
колебания) по сравнению с равновесной конфигурацией. Согласно правилам,
установленным в разделе «Интегрирование и дифференцирование симмётризо-
ванных функций» главы I, такая производная отлична от нуля, если функции
■ У Ду^ дреобразуются по одному и тому же неприводимому представле-
[ этом условии в спектре молекулы, находящейся в основном
колебательном состоянии, будет виден переход pf «0-»'pf =L поскольку (см.
(TV. 156) и предшествующее обсуждение) произведение интегралов
3*-б 3*-б
< П^'бЛП^^ отлично от нуля, если A^=l, a At?y= 0 Vy*i.
Таким образом, в спектре поглощения (ИК-спектре) будут видны
сигналы, отвечающие однократному возбуждению колебаний, форма которых
имеет ту же симметрию, что и одна из проекций дипольного момента моле-
104
кулы. При этом вращательный контур полосы (ее форма) определен типом
волчка, описывающим молекулу. В зависимости от того, какие изменения
полного углового момента молекулы разрешены в дипольном приближении
(см. предыдущий раздел), полоса может иметь дв§ (если Д/ = ±1) или три
(если tJ = 0, ± 1) ветви.
Заметим, что речь идет о невырожденных электронных состояниях
молекул, когда Г(3™) = Г(Фея) ® ЦФ^) <8> Г(ф = Г(^). В случае вырожден-
' А '
ных электронных состояний прямой квадрат представления Г(Фея) является
прямой суммой нескольких неприводимых представлений, одно из которых
всегда полносимметрично. Это значит, что помимо сформулированного
основного правила отбора Г(б,) = Г(^) могут появиться еще какие-то
дополнительные, т.е. окажутся разрешенными переходы с возбуждением каких-то
иных колебаний.
Прикладной аспект
(1) Равновесная конфигурация частицы описывается одной из высших
точечных групп, т.е. это сферический волчок. Рассмотрим молекулу метана с
симметрией Td. Колебательное представление молекулы (построенное в
соответствии со схемой, которая была рассмотрена в главе Ш - см. стр. 147—
152) такое: Гу^ = Ах Ф 2Е Ф 2F2. Представление трансляций любой системы
симметрии Td: Ttr=F2. Это значит, что в дипольном приближении при од-
нофотонном поглощении разрешено возбуждение только трехкратно
вырожденных колебаний молекулы метана. Таких колебаний два - с изменением
межъядерных расстояний С-Н и углов Н-С-Н, соответственно.
Следовательно, спектр должен состоять из двух полос, вращательная структура которых
определена правилами отбора (IV. 196): |А/ = 0,±1|. Отметим сразу два
момента. Во-первых, хотя чисто вращательных спектров у молекул типа
сферических волчков нет (по причине нулевого дипольного момента при
равновесной конфигурации!), колебательно-вращательные переходы возможны,
поскольку для них требуется не наличие дипольного момента, а его изменение
(в данном случае - появление) при соответствующем колебании, Во-вторых,
в отличие от вращательных спектров, когда неизменность величины полного
момента при изменении квантового состояния означала неизменность
энергии системы, т.е. отсутствие спектрального перехода, в колебательно-
вращательных спектрах постоянство J допустимо: помимо вращательного
изменяется еще и колебательное состояние молекулы. Итак, ИК-спектр моле-
105
кулы метана состоит из двух полос (с центрами при v=1306 и 3019 см"1),
вращательная структура каждой из которых включает три ветви: Р (ДУ = -1),
Q(A/ = 0)HROb/ = +l).
Относительные интенсивности спектральных полос определяются ин-
dQt
тегралами < Xwt 14zf I Xrot ><рву и величинами коэффициентов
I \J\Ji L
т.е. тем, насколько существенно изменяется соответствующая проекция ди-
польного момента при колебании. Изменение дипольного момента при
антисимметричном растяжении ОН связей должно быть большим, чем при
антисимметричном изменении валентных углов, поэтому интенсивность
валентного колебания и оказывается выше.
Отн.
пропускание
3000.
2000.
V, СМ""1
1000.
(2) В равновесной структуре частицы есть одна ось высшего порядка, и
частица принадлежит к симметричным волчкам. Такова молекула аммиака,
точечная группа симметрии которой C3v. Колебательное представление
молекулы Г^ = 2^©2£, а представление трансляций Г^=АхФЕ. Значит,
все колебания молекулы аммиака будут видны в ИК-спектре. При этом
вращательная структура полос определена правилами отбора (IV. 197), допол-
106
ненными условием А/ = 0, поскольку, как и в случае с молекулой типа
сферического волчка, неизменность полного момента молекулы теперь не
означает отсутствие поглощения излучения - изменяется колебательное
состояние частицы. В итоге ИК-спектр молекулы аммиака состоит из четырех
полос с центрами при 950,1627, 3337 и 3444 см"1, причем первая и третья
отвечают полносимметричным колебаниям, а вторая и четвертая - двукратно
вырожденным.
Отн.
пропускание
0.75
0.45
3000.
2000.
1000.
V, СМ
-1
В молекулах типа симметричного волчка есть одна ось высшего
порядка. Соответствующее направление является в некотором смысле
выделенным, а в ортогональной ему плоскости любые пары взаимно ортогональных
осей равноправны. Поэтому при анализе колебательно-вращательных
спектров таких молекул используют понятия ЩЩШЩ и ПеРПейДИКУДЯраой
полос. Параллельной (||) называется полоса, отвечающая колебанию, при
котором изменяется проекция дипольного момента на ось Ос лабораторной
системы координат. Перпендикулярные полосы (1) отвечают возбуждению
тех колебаний, при которых изменяются проекции вектора дипольного
момента на оси Оа и Ob.
При этом у симметричных волчков разрешены переходы,
удовлетворяющие следующим условиям:
107
— в параллельной полосе
I Д/ = 0,±1,если АГ,М*0 I
Л/ = ±1, если хотя бы одна из проекций К, М = О
— в перпендикулярной полосе
|А/ = 0,"±1].
В итоге, параллельные полосы в спектре могут иметь как две, так и три
ветви, а перпендикулярные полосы всегда имеют три ветви.
У молекулы аммиака проекции дипольного момента на оси Оа и Ob
изменяются при вырожденных колебаниях, а на ось Ос - при
полносимметричных. Невырожденное (полносимметричное) деформационное колебание с
частотой 950 см"1 дает типичную параллельную полосу, а пара вырожденных
деформационных колебаний с частотой 1627 см"1 -перпендикулярную.
Полосы валентных колебаний значительно менее интенсивны и, будучи близки по
частоте, заметно перекрываются.
Говоря об относительных интенсивностях спектральных полос,
заметим, что дипольный момент молекулы аммиака сильнее всего изменяется при
«зонтичном» колебании. Причина в том, что это колебание с очень большой
амплитудой, в пределе приводящее к инверсии (выворачиванию) молекулы.
При таком движении дипольный момент изменяется от значения,
превышающего 1.47 Д (при валентных углах меньше равновесного значения), до
нуля при плоской конфигурации.
(3) Равновесная ядерная конфигурация молекулы не имеет осей
симметрии высшего порядка, т.е. молекула относится к асимметричным волчкам.
например молекула воды, описываемая точечной группой C2v.
Колебательное представление молекулы Н20 Tvib = 2А^ Ф В\, а
представление трансляций Г^ = Ах Ф В\ Ф В2. Следовательно, в дипольном
приближении разрешено возбуждение как полносимметричных валентного и
деформационного колебаний, так и антисимметричного валентного колебания с
частотами соответственно 3657, 1595 и 3756 см"1. При этом вращательная
структура каждой полосы достаточно сложна. Однако если в первом
приближении считать, что состояния этого асимметричного волчка можно
представить линейной комбинацией функций некоторого симметричного волчка,
то любая полоса в спектре асимметричного волчка должна состоять из трех
вращательных ветвей Р, Q и R.
108
0.4 И
3000.
2000.
1000.
V, CM
(4) Молекула - линейная. Точечная группа симметрии, описывающая
равновесную конфигурацию, либо Д^, либо С^ в зависимости от того,
имеет молекула центр инверсии или нет.
Рассмотрим молекулу диоксида углерода. Ее колебательное
представление Гуф = £+®Ej ФПм, а представление трансляций Г^. = ££ ФПМ.
Следовательно, активными в ИК-спектре будут пара двукратно вырожденных
деформационных колебаний (667 см"1) и антисимметричное относительно
инверсии валентное колебание (2349 см"1).
Четное же валентное колебание, при котором синхронно изменяются
межъядерные расстояния С-О (1333 см-1), не видно, поскольку оно не
приводит к возникновению дипольного момента молекулы.
Спектральная полоса, отвечающая асимметричному растяжению
связей, является параллельной, поскольку при таком колебании появляется
ненулевая проекция дипольного момента на ось Ос. Деформационные же
колебания порождают перпендикулярную полосу, поскольку при таких
колебаниях молекула теряет линейность, и у нее возникает ненулевая составляющая
дипольного момента в плоскости, ортогональной оси Ос,
109
Для параллельной полосы правила отбора 1А/ = ±Ц (см. рекуррентное
соотношение (IV. 164а)), т.е. такая полоса имеет тппккп дне 1уятт^гатп.шлв
ветви (Р и R). Для перпендикулярной полосы условие | А/ = 0, ± 11 обусловли-
вает наличие трех ветвей (Р, Q и R).
Отн.
пропускание
1
3000.
2000.
1000.
V, СМ
-1
Спектры молекул воды и диоксида углерода весьма наглядно
показывают, как можно определить конфигурацию молекулы на основании ее ИК-
спектра. В зависимости от того, какая точечная группа описывает
равновесную конфигурацию молекулы, у нее будет разное количество колебаний
различной симметрии (как вырожденных, так и невырожденных). Например, у
трехатомной линейной молекулы (С02) есть два невырожденных и два
вырожденных колебания, а у изогнутой молекулы (Н20) - три невырожденных
колебания, т.е. формально у обеих молекул есть три различные частоты
нормальных колебаний.
Далее, для каждой точечной группы существуют свои правила отбора,
определяющие число линий в ИК-спектре. В частности, мы выяснили, что у
молекулы воды должны быть разрешены переходы с возбуждением любого
из колебаний, а у молекулы диоксида углерода возбуждение одного из
колено
баний в дипольном приближении запрещено. Это значит, что если
равновесная конфигурация трехатомной молекулы имеет симметрию C2v в ее ИК-
спектре должно быть три полосы, а если симметрию D^h - только две.
Правда, спектр молекулы воды тоже выглядит как совокупность двух полос,
только очень широких (по причине близости частот валентных
симметричного и антисимметричного колебаний). И здесь возникает еще одно условие:
необходимо знать, каковы типичные диапазоны частот колебаний.
Ранее, в главе Ш при решении задачи о колебаниях молекулы, мы
выяснили, что, зная точечную группу симметрии, можно определить формы
нормальных колебаний, и указали типичные диапазоны частот валентных,
деформационных и торсионных колебаний. Можно дать и более точные
оценки. Дело в том, что частоты колебаний одного типа (например,
растяжение связи, образованной данной парой атомов, или, реже, изменение угла в
одной и той же функциональной группе) в разных молекулах различаются,
как правило, не очень сильно. Колебания такого типа называют jfjffMTfflT-
стическими. и вот некоторые из них:
3300-3500 см"1
3200-3500 см"1
3000-3100 см"1
2850-3000 см"1
1760-1670 см"1
1640-1680 см"1
N-H связь в аминах
О-Н связь в спиртах
С-Н связь в алкенах
С-Н связь в алканах
С=0 связь в кетонах и альдегидах
ОС связь в алкенах
Вообще понятие характеристических колебаний возникло в результате
обобщения большого количества экспериментальных данных, прежде всего по
спектрам соединений гомологических рядов, в которых присутствовали одни
и те же мало отличающиеся частоты. Каково их происхождение? Допустим,
есть свободный метальный радикал. Это частица с пирамидальной
равновесной конфигурацией, спектр которой похож на спектр молекулы аммиака: два
симметричных и две пары вырожденных колебаний. При объединении двух
метальных радикалов образуется молекула этана, которая тоже является
симметричным волчком, причем ее ось третьего порядка совпадает с
локальными осями СНз-фрагментов. У молекулы С2Нб, например, в заторможенной
конформации, описываемой точечной группой 2>з</, колебатедьное
представление Туй = 3Aig Ф A2g Ф А^ Ф ЗА2и Ф AEg Ф 4EU. Для решения
колебательной задачи (которое, напоминаем, сводится к диагонализа!щи матрицы
колебательного гамильтониана Й^ь) можно в качестве базиса выбрать
координаты, описывающие колебания изолированных фрагментов СН3 (это 2x6=12
111
координат), и дополнить их координатами, описывающими изменение
взаимного расположения этих фрагментов (еще 12 координат). Если после диа-
гонализаций матрицы Й^ь окажется, что часть новых координат
незначительно отличается от исходных координат, описывавших колебания
отдельных фрагментов СН3, это будет означать, что соответствующие колебания
явйяются локальными - наличие окружения на них почти не влияет. Именно
такие колебаний и являются характеристическими. Справедливости ради
заметим, что в этане колебания двух метальных групп, очевидно, сильнее
«взаимодействуют» друг с другом, чем, например, в ацетоне или более
тяжелых алканах, в которых они пространственно разделены и для которых
поэтому можно ожидать даже большей характеристичности частот.
Итак, зная.атомный состав молекулы, можно построить (на основании
интуитивных соображений) ее вероятные устойчивые конфигурации.
Точечная группа симметрии позволяет определить количество и форму
нормальных колебаний, а также правили отбора для соответствующих переходов в
ИК-спектре. Частоты некоторых переходов можно приближенно оценить по
известным характеристическим частотам родственных соединений.
Сравнивая получающиеся модельные спектры с экспериментальным, можно
выяснить, какова в действительности структура молекулы.
Например, есть молекула состава XY3, которая может иметь как
плоскую, так и пирамидальную равновесную конфигурацию. Плоскую структуру
описывает точечная группа симметрии />3д, в которой колебательное
представление указанной молекулы есть Т^ь = А[ 0 А2 © 22?', а представление
трансляций - Г#=А2®Е'. Следовательно, у такой молекулы ИК-спектр
должен состоять из трех полос; Полносимметричное растяжение связей X-Y
не приводит к появлении* диполытого момента молекулы, а потому это
колебание не дает сигнала в ИК-спсйктрё. Если же устойчивая структура молекулы
пирамидальная, т.е. хфинадДёяЬгг точечной группе C3v, то колебательное
представление молекулы Ц$ - 1А\ 0 2Е, а представление трансляций
Г^ = 4 0 Е, так что в ИК-сйёйре должны быть видны все четыре полосы.
Соответственно, если экспериментальный спектр состоит из трех полос, то
молекула XY3 плоская, если же из четырех полос, то пирамидальная.
Анализ спектров помогает и в определении типа изомера молекулы.
Рассмотрим молекулу 1,2-дихлорэтена, которая существует в виде и цис-, и
транс-изомера. Первый имеет симметрию C2v и колебательное
представление rwb = 6^0 ЪА2 0 6Вг 0 ЪВ2. Представление трансляций в груттё C2v
Г# = Ai 0 В\ 0 2?2, поэтому в ИК-спектре этого изомера должно быть 15
полос. Второй изомер имеет симметрию C2h и колебательное представление
112
Tvib=6Ag®3Bg®3Au®6Bu. Но поскольку представление трансляций в
группе С2н IV = Л ® 22?м (только нечетные неприводимые представления),
то в ИК-спектре этого изомера будет только 9 полос. Как видим, уже по
числу полос в спектре можно определить тип изомера. Если же в системе
предположительно присутствуют оба изомера, то по относительным интенсивно-
стям полос, отвечающих возбуждению их колебаний, можно оценить их
процентное содержание в смеси.
Однако подчас информации только о спектре поглощения оказывается
недостаточно для решения вопроса о строении молекулы. Тоад приходится
привлекать дополнительные данные, в частности спектры рассеяния, которые
мы рассмотрим ниже.
Электронные спектры
Тадвтчгсюгс пмяииш
Поверхности потенциальной энергии разных состояний заметно
различаются. Различны ядерные конфигурации, отвечающие точкам минимума на
этих потенциальных поверхностях, а потому различаются и колебательно-
вращательные волновые функции молекулы в разных электронных
состояниях. Это значит, что в общем случае интегралы
ък-ь з*-б
{а = х9у, z\ £= a, b, с) отличны от нуля; и вероятность однофотонного
перехода между парой электронных состояний зависит от величины матричного
элемента
Ф =<Ф*,1^1*е*>г.
Как уже было отмечено при обсуждении общих правил отбора, этот
матричный элемент отличен от нуля при условии (IV. 154)
Тп®Гк®Г(^Ц (IV.200)
где Гл и Г* - неприводимые представления, по которым преобразуются
электронные функции молекулы Ф^ и Ф^ соответственно, аГ^-
представление, которое описывает проекцию дипольного момента на ось 0£,
(преобразующуюся так же, как трансляция вдоль этой оси).
113
Напоминаем, что мы по-прежнему полагаем возможным представлять
электронные волновые функции в виде произведения пространственной и
спиновой компонент
что порождает дополнительное условие: переходы разрешены только в
состояния той же мультиплетности: AS = 0. В широкой электронно-
колебательной полосе поглощения соотношение интенсивностей отдельных
линий определяется, как и в случае двухатомных молекул, интегралами
перекрывания колебательных функций в начальном и конечном электронном
состояниях.
Приклшпгой ашкт
Основное электронное состояние молекулы с замкнутой электронной
оболочкой чаще всего полносимметрично, т.е. функция Феп преобразуется
по представлению Ах. В этом случае условие (IV.200) можно переписать как
Ц^ЗГ^э^ (IV.201)
что выполнено, если возбужденное электронное состояние имеет ту же
симметрию, что и данная проекция дипольного момента:
г*=г>1
Причина в том, что прямой квадрат любого одномерного представления есть
полносимметричное представление, а прямой квадрат любого многомерного
неприводимого представления всегда содержит полносимметричное.
Например, в точечной группе Tdi описывающей равновесную
конфигурацию метана, проекции дипольного момента (как и трансляции)
преобразуются по трехмерному представлению F2. Это значит, что в дипольном
приближении разрешен электронный переход из основного
полносимметричного состояния этой молекулы только в состояния F2.
В точечной группе C3v, описывающей устойчивую структуру
молекулы аммиака, проекция дипольного момента на ось Oz имеет симметрию AXi a
на оси Охи Оу- симметрию Е. Следовательно, при поглощении излучения
разрешены переходы молекулы аммиака из состояния А[ либо в состояние
такой же симметрии (Ах\ либо в двукратно вырожденное электронное
состояние (Е).
114
В точечной группе C$v, определяющей симметрию равновесной
конфигурации молекулы воды, проекции дипольного момента на оси Ох9 ОуиОг
преобразуются по неприводимым представлениям 4» ^ и 52. Поэтому из
основного состояния молекулы разрешены переходы в полносимметричные
электронные состояния (4) или в состояния, антисимметричные
относительно поворотов вокруг оси второго порядка (Bi и В2).
Если РШШЕ СОГГСЯНИе МОЛеКУЛЫ Hg ПШЖИММЕШШЪ анализ про-
блемы, в какие возбужденные состояния может перейти система в результате
поглощения кванта излучения, в целом аналогична с единственным
отличием. Представление, по которому преобразуется функция конечного состояния
(фе*)> должно совпасть с представлением Г„ ® Г^ (если хотя бы одно из этих
представлений, Гл или Г^, одномерно) или с одним из неприводимых
представлений, прямой суммой которых является прямое произведение Гп ® Г^.
115
Спектры рассеяния
Впашательмые спектры
Хвдшашшшшшшш
В рассматриваемом нами приближении разрешены такие двухфотон-
ные переходы между вращательными состояниями молекул, при которых
отличны от нуля матричные элементы
<Xrot\^aAlp\Xkrot>^ 0V.202)
и стоящие при них коэффициенты
4п\
Q-0
ЗК-6 ЗДГ-6
.Н № ,
v
1
(TV.203)
(где по-прежнему а - х, у, z; £; = а, 6, с; а элементы матрицы направляющих
косинусов А определены выражением (TV.195b)). В силу отличия от нуля
тензора поляризуемости молекулы найдется хотя бы один ненулевой
коэффициент (TV.203). Поэтому в данном случае правила отбора для молекул
любого типа определяются матричными элементами (IV.202).
У молекул типа сферического волчка тензор поляризуемости в системе
главных осей кратен единичному:
*abc =
(аа 0 O'l
О аа О
0 0 а,
a J
и не изменяется при переходе от молекулярной к лабораторной системе
координат:
&xyz ~"
аа 0 0
0 аа 0
10 0 аа)
Это значит, что все элементы Л^аА1)р - константы: либо нуль, либо единица.
Поэтому интегралы (IV.202) либо нулевые, либо суть интегралы
перекрывания вращательных функций
<Xrot \Xrot ><р6уг>
116
которые в силу ортонормированности этих функций отличны от нуля лишь
при условии
л rot -~ Arot •
Таким образом, у сферических волчков ИСТ ВИШтеЛЬНШ СТОПИВ PfitfCffl-
<*аас =
ав О
О а,
О
а
О а.
Бели молекула принадлежит к симметричным вптткам. ее тензор
поляризуемости, диагональный в системе главных осей инерции, выглядит так:
О
(ось Ос совпадает с главной поворотной осью молекулы). Преобразование
его элементов при переходе к лабораторной системе координат определено
лишь двумя углами Эйлера, 0и цг.
Независимость элементов А^аА^р от угла <р обусловливает правило
отбора по проекции полного момента на ось Oz: АЛ/ = 0.
Зависимость элементов А^^р от утла в в виде sin20, cos2в и
sintfeostf (в силу рекуррентных соотношений для функций dj^{9))
накладывает следующее ограничение на изменение полного момента системы:
Д/ = 0,±1,±2.
В результате вращательные двухфотонные переходы в молекулах типа
симметричного волчка разрешены, если
Д/ = 0,±1,±2иДЛ/ = 0
При этом по причине вырождения уровней симметричного волчка по М пе-
? выро:
реходы|А/ = 0 и ДА/= 0| определяют сигнал оэлеева рассеяния, а переходы
А/ = ±1, ± 2] дают в общем случае четыре ветви полосы упм^ттяттиояиот
Бели молекула - асимметричный в^пу ^ тен3ор поляризуемости
может быть приведен к диагональному виду только в системе координат, не
совпадающей с главными осями инерции, поэтому
ааЬс =
йй oh ^*ac
аЬа аЬЬ аЬс
Каса асЬ асс
111
Элементы Л^аАпр зависят от функций sin2^, cos2;' и sin^cos^, где углом
у может быть любой из трех эйлеровых углов: <р, в и у/. Если при этом
состояния молекул можно аппроксимировать линейными комбинациями
функций симметричных волчков с данными значениями J и М и различными.
значениями А', то к правилам отбора, определенным для симметричных волчков,
надо добавить еще АЛ/ = ±2.
Таким образом, вращательные двухфотонные переходы в молекулах
типа асимметричных волчков разрешены, если
|Д/-0,±1,±2 и ДМ = 0,±2|.
Если молекула - ^пн^ау. то условия, при которых разрешены
вращательные переходы между ее состояниями с рассеянием излучения, те же, что
и в случае двухатомных молекул, а именно
lA/ = 0,±2,AM = 0l
так что спектральная полоса имеет интенсивный центральный сигнал рэлеева
рассеяния (Д/ = 0) и две ветви собственно комбинационного рассеяния
(Л/ = ±2).
Капебатепъно-впашателъные спектры
ТедгетичЕские готтм ы
Правила отбора, характерные для двукфотонных процессов рассеяния,
в целом аналогичны рассмотренным выше для однофотонных переходов с
поглощением излучения. Надо только во всех выражениях «заменить»
проекции электрического дипольного момента компонентами тензора
электрической поляризуемости. Колебательный переход разрешен, если
ЪпТ \ зк~6 зк~6
а£Ч <IWiain4>Q*°,
то есть разрешено однократное возбуждение 1-го нормального колебания
(v* = 0 -> v\ = 1) при условии, что функции fljf" и Of преобразуются по од-
зк-б\
118
ному и тому же неприводимому представлению:
*0.
dQt
/Q=0
Здесь #|£ - среднее значение ^компоненты тензора поляризуемости моле-
( я*пп\
кулы в электронном состоянии Ф^, а
да\
а
dQt
- ее изменение при
искало
жении геометрии молекулы соответственно форме /-го нормального
колебания.
Вращательная структура полос различных молекул может быть
определена точно так же, как и в случае ИК-спектра, - на основании выписанных
выше правил отбора по У (полному моменту системы). Поэтому обсуждать ее
в деталях для молекул, описываемых волчками различных типов, мы не
будем. Скажем лишь несколько слов о КР-спектрах тех молекул, ИК-спектры
которых фигурировали выше. ..
Пршиаднвймтект
У молекулы метана СЕЦ есть одно полносимметричное (4)» Два
двукратно вырожденных (Е) и два трехкратно вырожденных (F2) колебания.
Судя по симметрии компонент тензора поляризуемости тетраэдрических
систем (^, Е и F2), в спектре рассеяния будут присутствовать сигналы все^
этих колебаний. Напомним, что в спектре поглощения были видны только
сигналы трехкратно вырожденных колебаний: Таким образом, если цель
работы - определить частоты всех (или максимально возможного числа)
колебаний, то в случае молекул тетраэдрической симметрии рациональнее авали,
зировать спектр рассеяния, который предоставляет более полную
информацию.
У молекулы аммиака NH3 симметрия компонент тензора
поляризуемости та же, что и проекций вектора электрического дипольного момента: А± и
Е. Колебания этой молекулы тоже либо полносимметричны, либо двукратно
вырождены, так что ее спектры ИК и КР «дублируют» информацию о
частотах колебаний молекулы. Что же касается вращательной структуры полос, то
она различна соответственно рассмотренным выше правилам отбора по У, и
потому при оценке вращательных постоянных молекулы, ее геометрических
характеристик и заселенности вращательных уровней эти два спектра суще-
119
ственно дополняют друг друга, позволяя получать более точные величины
упомянутых параметров.
У молекул галидов бора ВНа13, имеющих плоскую равновесную
конфигурацию, количество полос в ИК- и КР-спектрах различно. Причина в том,
что полносимметричное валентное колебание молекулы не дает сигнала в
однофотонном спектре поглощения (поскольку не приводит к
возникновению дипольного момента системы), но дает сигнал в спектре рассеяния.
Таким образом, с одной стороны, КР-спектр дает более полную информацию о
частотах колебаний таких молекул. С другой стороны, по КР-спектру
различить плоскую и пирамидальную конфигурации молекул ХУз невозможно: в
обоих случаях разрешены переходы с возбуждением всех колебаний
молекулы, и оба спектра состоят из четырех полос.
Наиболее интересна ситуация, когда равновесная структура молекулы
имеет центр симметрии. Как мы уже отмечали выше при обсуждении общих
правил отбора, все проекции дипольного момента нечетны
(антисимметричны относительно инверсии), а все компоненты тензора поляризуемости
четны. Поэтому колебания, по форме симметричные относительно инверсии,
могут давать сигнал только в спектре рассеяния, а антисимметричные -
только в спектре поглощения, так что ИК- и КР-спектры таких молекул «не
перекрываются», а дополняют друг друга. Справедливости ради, заметим, что это
взаимное дополнение не всегда позволяет определить частоты всех
колебаний молекулы. Достаточно характерна ситуация, когда какие-то колебания не
дают сигнала ни в том, ни в другом спектре. Это легко понять. Допустим,
равновесная структура молекулы имеет симметрию D4h. У этой группы есть
по 5 неприводимых четных и нечетных представлений (по четыре
одномерных и одному двумерному). При этом представление трансляций
Г*г -Аи®Еи включает лишь два из пяти нечетных представлений, а
различные кошюненты тензора поляризуемости преобразуются по четырем из
пяти четных представлений. Соответственно, если колебания молекулы
описываются какими-то из четырех оставпгахся неприводимых представлений
этой точечной группы, то их возбуждения запрещены правилами отбора, и
мы не увидим их сигналов ни в ИК-, ни в КР-спектре.
120
Электронные спектры
О правилах отбора в электронных спектрах двухфотонного рассеяния
можно сказать почти то &е, что и в случае однофотонных спектров
поглощения, — вновь с точностью до замены матричных элементов проекций ди-
польного момента матричными элементами компонент тензора
поляризуемости.
Матричный элемент
«f -<*«.l*fl**>r
отличен от нуля при условии
[Ги®Га®Г^Ги®Г^®Г7з41 (IV.204)
где Г„ и Г* - неприводимые представления, по которым преобразуются
электронные функции молекулы Ф^ и Ф^ соответственно, а Г^ = Г^ ® Г7
- представление, описывающее симметрию ^компонента тензора
электрической поляризуемости.
Аналогично спектрам поглощения, разрешены переходы из
полносимметричного электронного состояния молекулы в состояния Ф^,
преобразующиеся по представлению [Гд. = I^J; из неполносимметричного состояния
Фет разрешены переходы в состояния, симметрия которых совпадает с
одним из неприводимых представлений, прямой суммой которых является
прямое произведение Г„ $ Г^.
121
§6. Спектроскопия ЯМР и ЭПР
Общие теоретические основы
Магнитно-резонансные методы отличаются от рассмотренных ранее
тем, что в них изучают характеристики спектральных переходов между
уровнями, полученными расщеплением спиновых (и орбитальных) состояний
молекул в магнитных полях. Как было показано в самом начале этой главы, в
первом приближении энергия взаимодействия некоторой частицы с
постоянным полем напряженности Н может быть записана (см. (IV.26)) как
* = -(И,Н), (IV.205)
где ц - магнитный дипольный момент частицы, определяемый ее
пространственным угловым моментом и спином.
Магнитный момент электрона
^ = -^(L + ^S), (IV.206)
где /ив - магнетон Бора (IV.60), L и S - орбитальный момент электрона и его
спин, ge -g-фактор электрона.
Магнитный момент ядра /-го типа
,i/=s^L,+rA, (IV.207)
п
где Ly - суммарный пространственный угловой момент протонов и нейтро-
нов ядра, I, - суммарный ядерный спин, yi = ^g,- - гиромагнитное отноше-
h
ние, ju0 - ядерный магнетон (IV.62), gt - ядерный g-фактор.
Спиновое квантовое число и протона, и нейтрона, и электрона равно
Ц. Если речь идет об атоме или молекуле, то ненулевой вклад в суммарный
спин соответствующей подсистемы (электронов или совокупности протонов
и нейтронов, составляющих ядро) вносят лишь «неспаренные» частицы
(представление о которых для электронов корректно лишь в случае
элементов первых периодов, а применительно к ядрам вообще необоснованно и
используется «по аналогии»). В частности, у молекулы кислорода в основном
состоянии есть два неспаренных электрона, и S = 1. У молекулы N0, как и
множества органических и неорганических радикалов и ион-радикалов, есть
122
один неспаренный электрон, и S = \L. У большинства же стабильных
молекул электронная оболочка замкнута, и S = 0.
Совершенно аналогично можно определить суммарный спин ядра, зная
число протонов и нейтронов в нем. Правда, не всегда удается априори
предсказать, какое из возможных (согласно правилу сложения моментов)
значений спина реализуется в данной системе. Но можно указать общие правила,
которые справедливы всегда.
(1) Бели числа протонов и нейтронов в ядре четны, его суммарный
спин /нулевой, как у ядер кислорода О и углерода С.
(2) Бели числа протонов и нейтронов разной четности, то суммарный
ядерный спин полуцелый. Например, при четном числе протонов и нечетном
числе нейтронов в ядре 13С его спин равен •К. Аналогично, при нечетном
числе протонов и четном (или нулевом) числе нейтронов спин ядер Н, N,
19F и31р равен J^9 аспинядра ив~^/.
(3) При нечетном числе и протонов, и нейтронов, суммарны* спин ядра
- небольшое целое число. Например, 7(14N)=7(2Н) «1, а 7(10В) « 3.
Заметим, что спины ядер бора оказываются неожиданно большими -
втрое больше, чем можно было бы ожидать. Но вернемся к обсуждению
энергии систем элементарных частиц в магнитном поле.
Полагая в первом приближении, что при помещении молекулы в поле
ее волновая функция не изменяется, т.е. рассматривая энергию
взаимодействия (TV.205) как возмущение некоторого стационарного состояния системы
| п > и ограничиваясь первым порядком теории возмущений, оценим
соответствующую поправку к энергии этого состояния:
*(1) =< п | -<А,Н) | п >. (IV.208)
При этом в волновую функцию (в дополнение к спин-орбитальной
электронной функции Ф^Фап и пространственной колебательно-вращательной
функции ядер хп ) включена спиновая ядерная функция %т:
\п>^Феп^апХпХап'
Для простоты возьмем одноэлектронную частицу - атом водорода»
ядро которого имеет спин / = 1^, а состояние единственного электрона со
спином S = \С описывают атомные орбитали, отвечающие определенному
значению орбитального моментах и его проекции ML.
123'
Электронный эффект Зеемана
Будем считать внешнее магнитное поле направленным по оси Oz
лабораторной системы координат H = (0,0,#z) и посмотрим сначала на
электронную составляющую поправки (TV.208):
«?)-<я|-(Й*.Н)|||>=
=<*еп*оп I^(t + *АН|Ф^Ф^ ><^^|^^> = /тл/
Л * v ' (IV.209)
-^яж(<Фж|4|фв1>+*.<фв,1*,|фв1>)
В электронном состоянии Фл = Ф^Ф**» с определенной проекцией
орбитального момента электрона ML среднее значение оператора Lz равно
величине этой проекции, т.е.
<Феп\12\Феп>^Шь.
Аналогично, среднее значение оператора проекции спина на ось Oz в
состоянии Фп равно одному из двух возможных значений Ms = ± ХС:
<Фов|5г|Ф0И>=ЙЛ/5.
В итоге поправка к энергии состояния | п > атома водорода, обусловленная
взаимодействием его электрона с внешним магнитным полем,
e<P=MBHz(ML + geMs). (IV.21U)
Это значит, что при наложении внешнего поля (2Ms+l)(2ML+l)-кратно
вырожденное (в отсутствии поля) электронное состояние атома
расщепляется Зтп и есть aneirrpnmnjft аААвгг Зеемана.
Расщепление уровней соответственно возможным проекциям
орбитального момента определяется напряженностью приложенного поля и
магнетоном Бора (константой), и для некоторого состояния с определенным
угловым моментом L пропорционально ML. Расщепление же, определяемое
наличием ненулевого спина, зависит еще дополнительно от величины g-
фактора. Этот фактор будет различным для свободного электрона, для
электрона в поле одного ядра и для электрона в поле системы ядер и других
электронов. Следовательно, измеряя частоту перехода между расщепленными
уровнями, можно определить g-фактор интересующей системы, и,
сопоставляя результат с эталонами, определить характер состояния электрона. Спра-
124
ведливости ради надо сказать, что в релятивистской теории g-фактор точно
определен только для свободного электрона. Во всех остальных случаях
просто используют ту же функциональную зависимость энергии взаимодействия
электронов с магнитным полем, рассматривая ge как параметр, зависящий от
особенностей конкретной системы.
Для состояния р-типа (с L ж 1) расщепление уровней в магнитном поле
можно схематично изобразить следующим образом (учитывая, что g-фактор
электрона в атоме водорода незначительно отличается от 2):
Ms
ML
-1
МвИг
V
X
V2
"Н^О"
VI
*-
^
ч
ч
ч
ч
f
MBgeBz
*
\
1/2
i 1/2
-1/2
ч
ч . -.
Ч ) , '. 4, S
ч
-1/2.
ч
ч
ч
,. -1/2
Н*0
Переходя к системе электронов, надо в выражении (Г/.208) заменить
магнитный момент одного электрона моментом всей совокупности
электронов, определяемым их суммарным спином и суммарным орбитальным
моментом. При этом существование ненулевой средней проекции суммарного
орбитального момента электронов на ось Oz всегда будет приводить к
расщеплению электронных состояний в магнитном поле. По расстояниям между
расщепленными уровнями 1ЛвН2{(Мь ^geMs)-(M'L^geMts)} можно
попытаться определить не только электронный g-фактор частицы, но и
суммарный орбитальный момент ее электронной подсистемы. Заметим, что
выделение орбитальной и спиновой составляющих в суммарном угловом моменте
электронов вообще корректно лишь при отсутствии заметного спин-
орбитального взаимодействия. Таким образом, задача описания
энергетических состояний электронной подсистемы в поле весьма сложна. И для того,
чтобы в первом приближении понять суть метода, давайте «забудем» про
орбитальный вклад в магнитный момент электронной подсистемы и будем
анализировать только спиновую составляющую.
125
Ядерный эффект Зеемана
Посмотрим теперь на ядерную составляющую поправки (TV.208).
Описывая совокупность нуклонов (протонов и нейтронов), мы, очевидно, не
затрагиваем проблему их пространственного движения в ядре, а потому
единственное, что нас интересует, это их спиновое состояние. Соответствующая
ядерная компонента магнитного момента атома 1-го типа
И поправка первого порядка к энергии атома, обусловленная
взаимодействием его ядра с постоянным магнитным полем, направленным по оси Oz,
*P=<*|-(fo,H)|/i>=
= -^&#х <Хап \1* \Хоп ><ФепфопХп 1 *шРтХп > = -№iHzMi>
I
(TV.211)
пропорциональна значению проекции ядерного спина (Mt) в состоянии \п>.
В качестве модели мы выбрали атом водорода с ядерным спином \i. При
помещении его в постоянное магнитное поле его изначально двукратно
вырожденное по спину ядерное состояние расщепится на два. Это и есть ядер-
Mj
—-1/2
№iH2
" 1/2
н=о н*о
В целом получилась картина, аналогичная поведению электрона атома,
но есть два существенных отличия. Во-первых, из-за различия знаков в
выражениях, связывающих магнитный момент частицы с ее спином в случае
протона и электрона (обусловленного различием знака их зарядов), в
магнитном поле расщепленные уровни этих частиц упорядочены по-разному. У
электрона ниже по энергии лежат состояния типа | JS > (с проекцией спина
- %)> а У ялРа " состояния |а > (с проекцией спина \С). Во-вторых, хотя
зависимость расщепления (расстояния между уровнями) от характеристик
126
частиц и напряженности поля в обоих случаях выглядит одинаково (MbSJ^z
и MogiHz)> величина этого расщепления при одинаковой напряженности
поля различается на три порядка. Дело в том, что при сопоставимых величинах
ядерного и электронного ^-факторов (см. §1 этой главы), ядерный магнетон
меньше магнетона Бора приблизительно в 1836 раз (соответственно
соотношению шсс протона и электрона).
Спиновые состояния атома в постоянном магнитном поле
Обозначая спиновые функции электрона \ае > и | 0е >, а ядра \ап > и
| Рп > соответственно, можно суммировать результаты выполненного нами
анализа электронной и ядерной подсистем атома в постоянном магнитном
поле. Мы допустили, что в первом приближении изменения в состоянии
атома, индуцированные внешним полем, можно учесть по теории возмущений,
ограничившись поправкой первого порядка по напряженности приложенного
поля и рассматривая только спиновые вклады во взаимодействие:
еР = 41) + */(1) = MBgeKzMs - miHzMt
(IV.212)
Общая схема энергетических уровней и их расщепления соответственно
рассмотренным выше взаимодействиям выглядит так:
' > Г \аЛ> ^
|а*ал>.|а«А>,'
\<*еан>,\ае/3„>_
\Р&п>ЛМп>
IWiHz
|«««|»>
МвЬВж
1ДЛ>
А», >* I АА>^
/WA
\№п>
Реальное соотношение расщеплений 103:], изображено как 3:1. Вверху указан
оператор энергии, приводящий к соответствующему расщеплению уровней.
127
Спин-спиновые взаимодействия и
«полный» гамильтониан молекулы в магнитном поле
Полученную выше схему можно было бы принять в качестве базовой
для последующего анализа возможных спектральных переходов между
получающимися невырожденными уровнями энергии, если бы в релятивистском
квантовомеханическом гамильтониане частицы в электромагнитном поле
можно было ограничиться рассмотренными нами членами, обратно
пропорциональными скорости света. Однако заметные вклады в энергию и характер
взаимодействия частиц с полем вносят и следующие члены,
пропорциональные у г • Из всех таких членов будем учитывать только те, которые
определены исключительно спинами составляющих систему частиц (пренебрегая,
как всегда, всеми вариантами спин-орбитального взаимодействия). Это
^cijy^Sj^i)v^~Se^j) " спин-спиновое взаимодействие электронов,
Sfy( *Гft'fX ъ 8jtj) - спин-спиновое взаимодействие ядер,
К/
Й
J]-^lyS - спин-спиновое взаимодействие ядер и электронов,
J h
где S -суммарный электронный спин системы;
коэффициенты Сц и Ьц определены одинаковыми функциональными
зависимостями от радиус-векторов соответствующих частиц:
1 Щ-rjXrj-rj)
|г,-г,.|3 |г,-г,|5 •
а коэффициент aj9 называемый константой сверхтонкого взаимодействия
(СТВ), или константой контактного ферми-взаимодействия, определен
электронной плотностью на ядре и произведением g-факторов и магнетонов
электронов и ядер.
Отметим сразу, что вклад спин-спинового взаимодействия электронов
надо учитывать тогда, когда в исследуемой системе есть более одного неспа-
ренного электрона, т.е. при изучении триплетных систем. В то же время
спин-спиновое взаимодействие ядер надо учитывать, если в системе есть
несколько ядер с ненулевым суммарным спином. При рассмотрении самой
простой модельной системы - атома водорода - оба эти вклада в энергию отсут-
128
ствуют, и единственная поправка, которую следует учесть, - это спин-
спиновое взаимодействие одного ядра (протона) и электрона: -4-1/S. Оче-
А
видно, эта добавка к оператору энергии Й2 (см. последнюю энергетическую
диаграмму) ни к какому дополнительному расщеплению уровней привести не
может (уровни уже все невырождены), она может только изменить их
относительные энергии:
I
#2=^&SH-^ft!,H Й3=Й2+^
-Л.
*~~,—а N
\ав0«>
wA
/ /
// f \ajzn>
Дй,' >,\РеРп> X
W
W
(jtBge-№l)Hj
\\ \\РеРп>
№iHz
\Р&п>
fi^iHz-aill
0*B8e-№i)Hz
Pogfo + Oi/l
Показанные на этой диаграмме изменения энергий электрон-ядерных
спиновых состояний атома водорода в постоянном магнитном поле были
оценены следующим образом. В базисе функций {\а^ап >, \аеР„ >, \Р&п >,
I РеРп > }> собственных для операторов /2 и S2, при расчете среднего
значения оператора
Й й
129
не ноль дают только матричные элементы оператора 1ZSZ:
<aean\-^fzSz\aeaH>=-^<ae\Sz\ae><an\t2\an>=a--^^
</W4'AlAA>=i
ft 4
<aefin\^l2Sz\aej3n>^/3/zn\^lJ2\jSeqn>=-^
ft ft 4
Следовательно, при учете спин-спинового взаимодействия протона и
электрона энергетическая щель между верхней парой уровней уменьшается, а
между нижней - увеличивается.
Бели переходить к более сложным системам, в которых есть несколько
ядер с ненулевым спином и более одного неспаренного электрона,
необходимо учесть еще изменения в относительном положении электрон-ядерных
спиновых уровней, обусловленные спин-спиновыми взаимодействиями
внутри соответственно электронной и ядерной подсистем. При этом мы уже
заметили, что расщепления ядерных уровней на три порядка меньше, чем
электронных. Аналогично, эффекты спин-спинового взаимодействия ядер
примерно на пять порядков меньше спин-спинового взаимодействия электронов,
а энергия взаимодействия ядер и электронов в логарифмической шкале
находится примерно посередине между ними. Следовательно, в зависимости от
того, состояния какой системы, ядер или электронов, мы хотим изучать, т.е. в
какой области шкалы энергетических разностей собираемся работать,
следует оставлять не все, а только часть членов оператора энергии взаимодействия
молекулы с магнитным полем
где Ву=(мв8ф)2с11 и Jij =PQbij8i8j ~ константы ршш-сшшовргр
взаимодействия соответственно электронов и ядер. Подчеркнем, что константы
спин-спинового взаимодействия, как и константа СТВ, не являются
универсальными постоянными. Они зависят от расстояния между
взаимодействующими частицами и их g-факторов, а потому суть просто параметры
феноменологического гамильтониана (TV.213), определяемые строением молекулы
(взаимным расположением ядер) и соответственно состоянием ядерной и
электронной подсистем.
130
Слагаемое 2 iT£/'./**/ отвечает взаимодействию ядер разного типа с
магнитным полем. Индекс у* у символа напряженности поля Н отражает тот
факт, что реальное поле, действующее на ядро, слабее приложенного
внешнего поля благодаря окружающим ядро электронным оболочкам, создающим
собственное магнитное поле. В общем случае используют аппроксимацию
Ну=Н(1-ау),
где <Tj - постоянная экранирования ядра/-те типа.
Будем рассматривать стандартный вариант, когда поле направлено по
оси Oz. Тогда
fgt$n=fgeHz$z
J п J я
Введем еще одну стандартную величину:
Vj^WSjHA-Oj) (IV.214)
- так называемую собственную частоту ядра /-го типа, и перепишем оператор
взаимодействия молекулы с постоянным магнитным полем:
tf=^A+I^M,+l£t,s-I^VLjM,
*</* j Л j h i<jk
(IV.215)
В этом выражении константы Dy, J у, aj и Vj обычно измеряют в Гц (с-1).
Поэтому собственные значения так записанного гамильтониана тоже имеют
размеримте частота-
Итак, помещение молекулы в постоянное магнитное поле приводит к
снятию вырождения ее спиновых электрон-ядерных состояний, причем
расстояния между расщепленными уровнями зависят от индивидуальных
характеристик молекулы: g-фактора электрона, констант экранирования ядер с
ненулевым спином (их собственных частот), констант спин-спинового
взаимодействия элементарных частиц и, наконец, константы сверхтонкого
взаимодействия. Возбуждая переходы между расщепленными уровнями и измеряя
их частоты, можно оценшъ все эти параметры; а поскольку они зависят от
атомного изотопного состава молекулы и взаимного расположения ее
функциональных групп или заместителей, можно определить строение молекулы.
131
Если нас интересуют спектральные переходы между спиновыми
электронными состояниями молекул, имеющих ненулевой суммарный спин,
можно пренебречь взаимодействием системы ядерных спинов с внешним
полем и йрутри себя, но учесть их влияние на электронную подсистему и
анализировать iWEiwroiniaH вида
*anp-^«A*. + Z3FMy-
i<J
h'
■хзм
J П
(IV.216)
Соответствующая экспериментальная техника носит название электронного
парамагнитного резонанса (ЭПР\ или электронного спинового резонанса. В
русскоязычной литературе более распространен первый вариант названия, а в
англоязычной - второй: electron spin resonance, ESR.
При изучении пфеходов между расщепленными спиновыми
состояниями ядерной подсистемы в молекулах с замкнутыми электронными
оболочками, можно «забыть» об эффектах в электронной подсистеме и работать
с гамильтонианом
гЯМР
■-£?У
i<jn
(IV.217)
в котором, напоминаем, зависимость от напряженности постоянного
магнитного поля заключена в величинах Vj (см. (IV.214)). Метод исследования
молекулярных систем, основанный на проявлении таких эффектов, называется
ядерным магнитным резонансом (ЯМР). Здесь расхождений между языками
нет, и в англоязычной литературе метод называется nuclear magnetic
resonance. NMR. Заметим, что этим методом можно исследовать и частицы в
электронных состояниях с ненулевым спином, но это сопряжено с заметными
трудностями, поскольку для интерпретации получаемых спектральных
данных необходимо анализировать собственные состояния гамильтониана,
включающего также взаимодействие неспаренного электрона с ядрами и с
внешним полем. Этот вариант метода мы рассматривать не будем.
В обоих методах изучают переходы между расщепленными в
магнитном поле уровнями, поэтому эксперимент можно организовать двумя
способами: (1) изменять частоту со при Я = const до достижения максимума
поглощения или (2) варьировать напряженность Н при постоянной частоте со.
Первый вариант называют разверткой по частоте, а второй - разверткой по
полю. Обычно в ЭПР-спектрометрах реализован второй вариант, а в ЯМР-
спектрометрах бывает и тот, и другой. В ЭПР диапазон рабочих частот, при
которых происходит поглощение, 10-100 МГц, а в ЯМР 1000-30000 МГц.
132
Электронный парамагнитный резонанс
Обычно методом ЭПР изучают частицы, имеющие один неспаренный
электрон, т.е. всевозможные радикалы и ион-радикалы. С триплетными
системами, в которых есть два неспаренных электрона, работают заметно реже.
Поэтому мы ограничим наше рассмотрение дублетными молекулярными
системами, полагая, что в них надо учитывать взаимодействие только одного
электрона с внешним полем и с ядерной подсистемой. Это позволяет еще
больше упростить оператор энергии (IV.216), исключив из него член спин-
спинового взаимодействия электронов:
Яэпр=fgeS*H,+2$*А с™-218)
Тогда в поле энергия молекулы в состоянии с проекциями спина электронов
Ms и спинов ядеру-го типа Mj имеет вид
в = txBg^dsH2 + ^MsajMj. (IV.219)
J
Чтобы инициировать переходы между такими уровнями, надо подействовать
на систему еще одним полем, теперь уже переменным, т.е. электромагнитной
волной с частотой а>, изменяющейся в пределах ожидаемых расщеплений
уровней:
Hjcosa*. (TV.220)
Энергия взаимодействия с этим дополнительным полем записывается так же,
как и с постоянным:
-pHtCOSO*, (TV 221)
где
(т.е. ядерным вкладом в магнитный дипольный момент мы пренебрегаем).
Вероятности переходов между спиновыми состояниями молекулы в
дипольном приближении определяются, как и в случае электронно-
колебательно-вращательных состояний, скалярными квадратами матричных
элементов пространственной части возмущения (TV.221) на стационарных
функциях исходного и конечного состояний:
< <*>onZon I & I ФокХок >=< ЬопХоп I ЧА^НО | ФаьХок >> (IV.222a)
133
где Фтх<т ~ спиновая-составляющая волновой функции молекулы в
стационарном состоянии ! п >, Поскольку нас интересуют переходы между
состояниями, характеризующимися (прежде всего) определенными проекциями
спина электронной подсистемы, матричные элементы (IV,222a) должны быть
ненулевыми, если проекции спина в начальном и конечном состояниях
различны. Это возможно, если функции Фт не являются собственными для
оператора ($е>Щ). Следовательно, в скалярном произведении (IV.221)
должна доминировать проекция на направление, ортогональное оси Oz.
Будем считать, что поле (IV.220) направлено по оси Ох. Тогда матричные
элементы перехода
-<фопХт\Аех\ФокХок>Н1х =
Un a (IV.2226)
= ь*еН\х <^оп%ап \$х l^akZok >
определяют правила отбора
«bonZonlSxlQokXok^Q-
Поскольку оператор Sx не действует на спиновые переменные ядер, правило
отбора по проекции ядерного спина:
[АМ/^О]. (IV.223)
При этом в силу того, что Sx = — , а лестничные операторы при дейст-
'г. '
вии на функцию с определенной проекцией спина дают функцию с
проекцией, на единицу большей или меньшей соответственно, правило отбора по
проекции спина электронов будет
lA%=±lj (IV.224)
Тогда наблюдаемые экспериментально частоты переходов должны отвечать
разностям энергий, определяемых выражением (IV.219) и соответствующих
неизменному спину ядер и изменяющемуся на единицу спину электронной
подсистемы:
A*=>bS*#z((Ms ±l)-Ms) + ^ajMj((Ms ±V)-MS).
j
В случае поглощения, когда AMS = 1, .
(IV.225)
■ J
134
Поскольку константы сверхтонкого взаимодействия aj малы по сравнению с
величиной pBgeHz, спектр - совокупность линий вблизи базовой частоты
v0 = Мв8еНг (в зависимости от напряженности используемого магнитного
поля лежащей в диапазоне от 1000 до 30 000 МГц). Расстояния между
линиями определены величинами ^ajMj, а число линий называется мульти-
J
тщетностью сигнала, которая, судя по формуле (TV.225), содержит большую
часть информации о наличии в молекуле определенного числа групп
неэквивалентных ядер.
Бели число линий велико, а расстояния между их центрами малы, то по
контуру полосы поглощения (т.е. зависимости суммарной интенсивности
поглощения от частоты поля) не удается надежно определить мультщшетность
сигнала. Поэтому ЭПР-спекгры обычно записывают не в интегральном виде
(зависимость интенсивности от частоты), а в дифференциальном -
зависимость производной интенсивности поглощения по частоте поля от частоты -
который четко выделяет экстремумы поглощения. Заметим, что аналогичный
прием используют и при регистрации спектров поглощения в
микроволновой, инфракрасной и ультрафиолетовой областях.
Рассмотрим в качестве примера квинтет (сигнал, состоящий из пяти
линий). При хорошо разрешенной структуре полосы поглощения
Отн.
поглощение
дифференциальная кривая позволяет лишь уточнить центры линий мульти-
плета:
В, Тс
135
В случае же плохо разрешенной структуры, когда сигнал ЭПР
выглядит, например, так
Отн.
поглощение
ЛГс
дифференциальная кривая дает возможность не только определить мульти-
плетность, но и оценить расстояния между центрами линий, по которым
можно рассчитать соответствующие константы СТВ aj:
Модельные системы и реальные спектры молекул
Допустим, есть одно ядро со спином ^ - атом водорода *Н или
углерода 13С. В этом случае выражение (IV.225) упрощается до
Поскольку спину /= XL отвечают две проекции Af/ =±j^, спектр должен
состоять из двух линий одинаковой интенсивности (в силу равенства
матричных элементов (IV.2226) для этих двух значений проекции ядерного
спина) с частотами
136
Теперь допустим, что неспаренный электрон делокализован по т
эквивалентным ядрам («взаимодействует» с ними), как в метальном радикале
12СН3, где есть три эквивалентных ядра водорода (т = 3). В такой системе
энергии переходов (TV.225) определены выражением
te = MBgeHz + <t£MJy
J
так как для эквивалентных ядер константа СТВ одна и та же. Суммарный
спин системы трех одинаковых ядер со спинами Ij = \С может быть
/ = ^,%. Соответственно четырем его проекциям
М7= ^Mj = ±%,±% спектр состоит из четырех линий с частотами
[П92 =MBgeHz±Y2
4A=VBgeHz±f2-
Расстояния между этими линиями равны а. Интенсивности линий уже
различны, поскольку определены тем, сколькими способами можно получить
данное значение проекции Mj. Бели число ядер в спиновом состоянии | а >
(т.е. с проекцией спина Mj = %) равно та, а число ядер в состоянии \0>
(с проекцией Mj = ~%) - тр, то проекция спина всей совокупности ядер
равна Mj = %(ma - nip). И число способов получения этой проекции
определено биномиальным коэффициентом С = . Для получения
проекту т^!
ции Mj - ± % все ядра должны находиться в одном и том же спиновом
состоянии (|а> или |р> соответственно), т.е. например, ma=m и /и^ = О,
так что С = 1. Для получения же суммарной проекции Mj = ±\i два ядра
31
должны быть в одном состоянии, а третье - в другом, так что С = — = 3.
Это значит, что интенсивности линий с частотами v3,4 должны быть втрое
больше, чем линий v12. Это ясно демонстрирует ЭПР-спектр метального
радикала.
137
Таким образом, в случае m эквивалентных ялеп (со спином Ij)
проекция суммарного ядерного спина молекулярной системы имеет 2mlj+l
значений, что приводит к появлению в спектре мультиплета с числом линий
и центром при частоте v0 = MbS^z • Ищ^цррвнррт^ <угдел>щд(; ли-
2т/у+1
ний мультиплета соотносятся как биномиальные коэффициенты ]С$л (к - О,
... т). При этом линии эквидистантны: поскольку проекция спина изменяется
через единицу, Av = e.
Г
сш
JI
I ... .1t .... i . . . . i . . .1. и . . . . 1 . . i i I i i iliill i i i i I i i i i I ii л i ill i i i i I i
333 3834 Э335 3336 3357 3358 3379 3360 3381 3402 3403 3404
В, Те
Классическим примером молекулы, в которой есть две группы
неэквивалентных ядер, взаимодействующих с неспаренным электроном, является
анион нафталина. В этом анионе есть две совокупности по 4 эквивалентных
протона, которые традиционно обозначают аир:
а а
Константы СТВ для этих двух групп должны различаться (аа Ф ар). Больше
должна быть константа тех ядер, где больше сшшовая плотность, т.е. тех, где
преимущественно локализован избыточный электрон. Априори кажется
более вероятной его преимущественная локализация на ядрах а-типа.
138
Энергии переходов (TV.225) в этом анионе должны быть записаны так:
4 4
Л* = ДО«Я«' + <*a(L МJ ) + */?(£>*) •
./=1 *=1
Каждая из сумм (в отсутствии другой) породила бы сигнал из пяти линий,
расстояния между которыми были бы равны соответственно аа или ар.
Наличие же обеих сумм одновременно в выражении для частоты переходов
приводит к появлению 5x5 = 25 линий, поскольку каждому возможному
значению проекции ядерного спина подсистемы ядер а отвечает пять
возможных проекций спина подсистемы р (или наоборот, что одно и то же). ЭПР-
спектр аниона нафталина действительно является мультиплетом, состоящим
из 25 линий.
В-В09Гс
В общем случае при наличии д групп неэквивалентных ядер (число
ядеру-го типа равно nip j = l...q), с которыми может взаимодействовать
электрон (т.е. вблизи ядер, которых есть ненулевая спиновая плотность),
мультиплетность сигнала ЭПР определена произведением мультиплетностей,
которые наблюдались бы для каждой группы ядер отдельно,
П(2т/,+1)
М
ив
случае ядер с Ij = ХС равна
|ft(^+1)l-
м
139
Как было отмечено, константы СТВ зависят от электронной плотности
на ядре: чем выше электронная плотность, тем больше а. Следовательно, по
экспериментальным расстояниям между линиями в ЭПР-спектре можно
судить не только о числе групп неэквивалентных ядер, с которыми
взаимодействует электрон, т.е. о степени его делокализации в молекуле, но и об
относительной «силе» взаимодействия электрона с ядрами разных групп. Ниже
приведены экспериментальные оценки констант СТВ для взаимодействия
электрона с протонами в некоторых типичных органических радикалах (для
указания типа протонов использована стандартная нумерация ядер
углеродного скелета, с которыми они связаны):
радикал
•СНз
•СН2СН3
•СН2СН2СН3
•СН^СНз)2
•С(СНз)з
[СбН«г
а.МГц 1
аа = 64.6
^=62.7, ад =75.3
аа =61.9, ар =93.0, ау = 1.1
аа =61.9, ар =69.1
ад = 63.7
аа=10.5
Анизотропия g-фактора и g-тензор
На рассмотренные нами общие закономерности формирования сигнала
ЭПР накладываются, как правило, многие усложняющие картину эффекты,
наиболее существенным из которых является анизотропия g-фактора
(различные величины g-фактора, получаемые при различной ориентации
молекулы в магнитном поле). Она возникает из-за заметного спин-орбитального
взаимодействия (т.е. взаимодействия спина и пространственного углового
момента электронов) и проявляется обычно в спектрах радикалов, имеющих
относительно низкую симметрию (не являющихся сферическими волчками) и
находящихся в твердом состоянии (монокристаллов). Этот эффект наиболее
важен в случае комплексов ионов переходных металлов, лантаноидов и
актиноидов. Анизотропия g-фактора требует перехода от скалярной величины g к
g-тензору
£ =
( \
8 хх 8ху 8xz
8 ух 8уу 8yz
8zx 8zy 8a)
140
и соответственно замены члена ^-gJ$EL в операторе ЙЭПР (IV.218) его мат-
ричным аналогом:
х *у
«J
V
8хх 8ху Sxz
8ух 8уу Syz
ySzx Szy 8zz)\"z/
При этом эффективная величина g-фактора при произвольной ориентации
кристалла в магнитном поле определена следующим выражением:
8эфф = (AHx>AHy>AHzi Zyx 8yy 8yz I ARy
где ЛНа = cos(HOa) - косинусы углов между наблюдаемыми осями
монокристалла (a = jc, у у z) и направлением магнитного поля Н (так называемые
направляющие косинусы). Вращая монокристалл в магнитном поле в
плоскостях хОу9 xOz и yOz и определяя напряженность поля Н, отвечающую
максимуму поглощения при каждом угле, можно получить достаточный набор
данных для оценки элементов g2 -тензора и, таким образом, найти
абсолютные величины компонент g-тензора, но не их знаки.
141
Ядерный магнитный резонанс
Идеология метода ЯМР во многом близка к ЭПР. Эта техника обычно
применяется для исследования состояний ядер с полуцелым спином, прежде
всего 13С, 19F, 31P и !Н, причем в последнем случае метод носит название
протонного магнитного резонанса (ПМР).
Спектры низкого разрешения
Рассмотрим сначала ситуацию, когда в гамильтониане (TV.211)
отсутствует член спин-спинового взаимодействия ядер. Физически это отвечает
условиям, когда на фоне взаимодействия ядер с внешним магнитным полем
эффекты от их взаимодействия между собой неразличимы:
#W = £tV (IV.226a)
7 .
Напомним, что величина напряженности поля входит в собственные частоты
ядер (IV.214):
y]=№]HzQ-°j)>
В выражении (TV.226a) изменен (в сравнении с (IV.217)) знак перед суммой,
что соответствует просто изменению направления вектора магнитного поля
на противоположное: H = (0,0,-#z). Именно такое направление выбирают
как стандартное в технике ЯМР.
Будем в первом приближении считать, что в отсутствии поля спиновые
состояния ядер независимы. Это допущение останется вполне корректным и
при наличии взаимодействия (IV.226a). Поэтому спиновую функцию
молекулы Хап можно представить в виде произведения спиновых функций
отдельных ядер ХыАМ\>> характеризующих их состояния с определенными
проекциями спина Мх\
Хап НЛ*1 >\М2 >...|MN >ЦМХМ2..ММ >
(N- число ядер с полуцелым спином).
Энергии этих состояний, определяемые как среднее значение
гамильтониана Й%мр
еп=<Хоп\Яямр\Хоп>>
будут таковы
142
en -<M$i1.J£jt\Y%-ti\M&2.Mi, >=
J n
Vl (IV.2266)
= ,Z<MJ\^\Mj><YlMi\Y[Mi>^^J-
J n t*J i*j j
1
Инициировать переходы между этими состояниями можно, как и в
методе ЭПР, накладывая на систему второе, переменное, магнитное поле -
электромагнитную волну Щсозв*, вектор напряженности которой
ортогонален вектору Н (например, направлен по оси Ох). Вероятность перехода
(как во всех рассмотренных ранее спектральных методах) пропорциональна
квадрату матричного элемента возмущения на функциях начального
| М{..М*х > и конечного | Mi„MN > состояний:
<M1..MN\£'\Mi..M'N>,
где возмущение - это пространственная часть оператора взаимодействия
ядерных спинов с постоянным полем:
#'=-А„Н, =-^Я„5>у^. (IV.227)
я /
Следовательно, правила отбора определены условием
/ j i
которое выполнено, если в начальном и конечном состоянии проекции спина
одного из ядер различаются на единицу, а проекции спинов всех остальных
ядер неизменны:
AMjmMj-M)=±l, АМ{=М1-М1=0 Vi*j\. (TV.228)
Тогда энергии разрешенных переходов в ЯМР-спектре молекулы,
содержащей N ядер (с ненулевым спином), будут определены выражением
A*n=vy(A/y-(A/y±l)), у-l..JV;
и в случае спектра поглощения, когда Мj - Mj = 1,
143
Теперь понятно, почему величины Vj были названы собственными
частотами ядер. Они суть частоты переходов в ЙМР-спектрах. Переход
наблюдается, когда частота v = coc электромагнитной волны Hjcostfrt совпадает с
частотой vJy определяемой (помимо особенностей строения самой молекулы)
напряженностью постоянного поля Н. Частоты, при которых происходит
поглощение энергии в поле 10 000 Гс, имеют порядок 10-100 МГц.
Отметим, что те спектры, на интерпретацию которых мы пока можем
рассчитывать, - это так называемые спектры низкого разрешения, поскольку
мы пренебрегли в операторе возмущения спин-спиновым взаимодействием
ядер. Иначе говоря, мы предположили, что наш прибор недостаточно
чувствителен, чтобы улавливать столь малые эффекты. К каким изменениям
приведет учет этого взаимодействия, мы посмотрим чуть позже. Сначала
выясним, какую информацию можно извлечь из частот Vj.
Величины Vj пропорциональны (1-сгу), где Оу- константа
экранирования ядра, определяемая его электронным окружением. Очевидно,
локальное распределение электронной плотности в областях пространства,
отвечающих различным функциональным группам молекул, различно.
Соответственно, константы экранирования, а следовательно, и собственные частоты
ядер, входящих в разные группы, будут различаться. Например, для протонов
в молекуле этанола СН3СН2ОН должно быть три различные частоты -
характеризующие электронное распределение в группах СН3, СН2 и ОН.
Действительно, ПМР-спектр низкого разрешения этанола выглядит примерно так:
В,Гс
Интенсивности этих трех сигналов (площади пиков) соотносятся как 1:2:3
соответственно числу протонов в функциональных группах. Это понятно. В
ОН-группе поглощение происходит при изменении спинового состояния
единственного протона. В метиленовом фрагменте есть уже два эквивалент-
144
ных протона, т.е. возможны два перехода с одинаковой частотой,
удовлетворяющих условию (TV.228), а в метальной группе таких переходов уже три.
Сигналы переходов в каждом случае накладываются друг на друга, что и
приводит соответственно к вдвое и втрое большей площади двух последних
пиков.
Очевидно, важны обе характеристики пика: его положение (частота) и
площадь. Площадь, как мы выяснили, пропорциональна числу
эквивалентных ядер, а частота определяет тип функциональной группы. Действительно,
есть достаточные основания полагать, что соседние структурные фрагменты
заметно искажают локальное распределение электронной плотности данной
группы лишь в тех областях пространства, где формируется электронная
связь между ними и этой группой. Более того, это искажение должно быть
характеристическим для соседей разного типа и практически не зависеть от
типа функциональных групп, расположенных в углеродном скелете
молекулы через одну от рассматриваемой. Тогда можно ожидать почти совпадения
значений Vj для ядер одной функциональной группы в разных соединениях,
если их окружение одинаково, и близких значений Vj, если окружение
различается. Например, собственные частоты Vj протонов фрагмента СН3 в
молекулах, где ближайшим соседом этой группы являются СН2, СООН,
СН(ОН), должны различаться, но попадать при этом в небольшой
«характеристический» интервал значений.
В результате по тому, в каких интервалах частот обнаружены сигналы
ЯМР в спектре низкого разрешения, можно выяснить, какие функциональные
группы присутствуют в молекуле, но каковы соседи у каждой группы, т.е.
какова структура молекулы, точно определить не удается. Для этого надо
перейти к более точному приближению - включить в гамильтониан член,
отвечающий спин-спиновому взаимодействию ядер. Но прежде выясним, как
экспериментально определить собственные частоты ядер.
Точно измерять абсолютное значение напряженности магнитного поля
сложно. Поэтому шкалу частот калибруют по стандартному веществу и
измеряют не сами частоты ядер молекулы, а разницу между ними и частотой
эталона:
А у as у(эталона) - Vj.
В качестве стандартного вещества в ЯМР-спектроскопии используют тетра-
метилсилан (ТМС), который удобен тем, что химически неактивен и
поглощает энергию в той области спектра, где не поглощает большинство
исследуемых молекул. Но здесь возникает еще одна сложность. Собственные
частоты зависят от напряженности приложенного поля. Для того чтобы сделать
145
экспериментальную технику более гибкой, а результаты независимыми от
конкретных экспериментальных условий (от того, использовали мы поле с
частотой 60 или 100 МГц) необходимо ввести «нормировку» частот, а точнее
их разностей А;-, на напряженность поля. Соответствующая величина
называется химическим сдвигом и определяется так
S,^-1 , (IV.229)
где v-wc - фиксированная частота поля Hj (в МГц). Коэффициент 106 в
числителе обусловлен тем, что собственные частоты ядер - порядка МГц, а
разности между ними Ау- - порядка десятка или сотни Гц. Коэффициент
просто позволяет получать химические сдвиги Sj в удобном интервале значений
-примерно от0до15иот0до 200 так называемых миллионных долей (м.д.,
или в англоязычной литературе, parts per million, ppm) в случае ПМР и 13С-
ЯМР соответственно. О характерных химических сдвигах ядер 13С и *Н в
органических соединениях дает представление следующая таблица.
Тип функциональной группы
"С -СН3
алканы -СН?-
-СН<
спирты или простые эфиры R3C-OR
алкины R-Cs
алкены R2C=
ароматич. углеводороды Аг
амиды R'C-NR2
альдегиды, кетоны >C=Q
?ишшя~'^шшяшшяш^£тшяшш
алканы -СНг1-'
-СН<
спирты -О-Н
спирты или простые эфиры О-С-Н
алкины -С-Н
алкены =С-Н
ароматич. углеводороды Аг-Н
альдегиды -С(0)Н
метил-кетоны -С(0)СН3
аллильный метил -ОС-СН3
ароматич. метил Аг-СН3
5, м.д.
5-30
15-55
20-60
40-80 !
65-85,
10<Ы50
110-160
150т180
np^ip^
0.7-1.3
12-1.4
1.4-1.7
2.5-5.0
3.3-4.0
2.5-2.7
5.0-6.5
6.5-8.0
9.7-10:0
2.1-2.4
1.6-1.9
2.5-2.7
146
Заметим, что в случае С интервалы химических сдвигов существенно
больше для каждой функциональной группы и заметно перекрываются для
разных групп. Протонные же сдвиги более характеристичны. При этом ядра
углерода расположены как бы «в центре» соответствующих групп, а протоны
- «на их периферии», где изменения в распределении электронной плотности
при образовании связей с другими группами более ощутимы. Все это делает
метод ПМР более предпочтительным в сравнении с ЯМР на ядрах 13С.
Скажем несколько слов о принятых в ЯМР-спектроскопии
обозначениях. Эквивалентные ядра (т.е. ядра с одинаковыми собственными частотами)
обозначают одной буквой латинского алфавита. Например, СН4 - это
система А4. При наличии нескольких групп неэквивалентных ядер используют
различные буквы, например, А2В3 или A2X3. И вот тут мы должны
вернуться к обсуждению давно обещанного вопроса о том, к каким изменениям
в спектре приводит учет спин-спинового взаимодействия. Дело в том, что
оно отражено уже в обозначениях спиновых систем. Так, буквы В, С, ...
используют, если разница собственных частот ядер данных групп имеет
порядок константы их спин-спинового взаимодействия:
Бели же различие частот заметно превосходит константу взаимодействия, то
используют буквы X, Y,...:
\va-vx\»Jax-
Сразу надо сказать, что обозначения эти во многом условны, так как в
зависимости от условий эксперимента одна и та же молекула может быть
обозначена и А2Ва, и А2Х3, поскольку собственные частоты, а значит и их
разности, пропорциональны напряженности поля, а константы спин-спинового
взаимодействия от поля не зависят. Как мы увидим дальше, удобнее
работать, когда выполнено второе условие, поскольку в экспериментальном
спектре получается более четкая картина разделенных сигналов от различных
функциональных групп, а в теоретическом рассмотрении оказывается
возможным использовать теорию возмущений. Такие спектры называют
спектрами первого порядка. Их мы и рассмотрим в следующем разделе.
147
Спектры высокого разрешения
и спин-спиновое взаимодействие ядер
Оператор «полной» энергии взаимодействия ядерной подсистемы с
полем (IV.217) есть
Ям* = 7%10 * I&Uj - Aft,+Ag*.
] п i<jn
Как мы выяснили ранее, спин-спиновое взаимодействие ядер заметно меньше
их взаимодействия с внешним полем. Поэтому определить энергии
состояний ядерной подсистемы, описываемые-гамильтонианом (IV.217) можно с
помощью вариационного метода, используя в качестве базиса функции,
собственные для оператора Йдмр (IV.226a), т.е. функции
\mxm2..mn>^m1>\m2>...\mn>.
Мы не будем полностью решать задачу нахождения коэффициентов
Qi,*2,..jw> определяющих разложение искомых функций по выбранному
нами базису
Zon= Z CkU^M\MklMk2..Mw> (IV.230)
к\ук2,...Ш
и рассчитывать энергии этих состояний еп. Нам достаточно выяснил», какие
функции | MkxMk2"^kN > могут быть «смешаны» в соответствующих
линейных комбинациях. Для этого рассмотрим два коммутатора: [ЙЯМРу11\ и
№щр>ЯяЬр] (где ?z = Z^s* "" иРоекц^а суммарного спина ядер на ось Oz).
к
(1) Qirepaipp Йдмр = Й%р + Й%р К9ммугиру?г с одаратрром 1^
В случае первого члена гамильтониана (]£ ?zj ) это очевидно:
J П
■Z^V*i-°-
U п *
Л*А
Для второго члена (Z 2 ^'у ) ^^ легк0 Дока3*1"1»- Рассмотрим оператор 1Ду.
К/й
Он коммутирует с любым оператором Izk 9k * ij. Значит, надо проверить
перестановочность оператора tttj с оператором 1^+1^. А она следует из ра-
148
венства нулю коммутатора [I Jz] для любой системы, в том числе и для
системы двух частиц:
o=[(i<+Iy)2,(4+^)]=[l?,(/2j+^)]+[i3,(4+^)]+2[l/ly,(/2<+^)].
Следовательно,
О О
и значит,
к
il4[M,.I'*]=o
\i<jn к
Точно так же доказывается перестановочность оператора спин-спинового
взаимодействия с любой проекцией суммарного спина системы:
! i<JH i \
(IV.231)
Это свойство нам потребуется при обсуждении спектральных переходов в
спиновой системе ядер, а сейчас вернемся к характеристике собственных
состояний гамильтониана Й
ямр-
Итак,
№пл»Лг1=0
(IV.232)
Следовательно, можно работать с ядерными функциями молекулы,
собственными одновременно и для оператора продацш суммарного спина. Это
значит, что каждое состояние молекулы характеризуется определённой
проекцией суммарного ядерного спина М7, т.е. в разложениях функций (TV23Q)
по базису могут фигурировать только такце функции \MkiMk2..MkN >, для
которых
' ' (IV.233a)
1Ци+Л**2+" + Циг"М/1
Поэтому ядерные спиновые функции молекулы можно обозначить так
ЕН5В (IV.2336)
И поскольку каждое значение Мг (за исключением максимального Мд^ = /
и минимального М^ « -/) можно получить не одним способом, спиновые
состояния ядерной подсистемы являются вырожденными. В случае АГядер со
149
спином у^ кратность вырождения состояния с проекцией Mj равна
CjJf/+Afmax.
(2) Для того чтобы уточнить «состав» функций этих вырожденных
состояний, проанализируем коммутатор [Й^.Й^]. представив его суммой
коммутаторов:
ZJJL
*2
*<J
к
В каждом слагаемом этой суммы оператор 1Д; коммутирует с любым
оператором y^l^k > к * Uj- В случае же двух оставшихся слагаемых воспользуемся
доказанным выше свойством
ИМ**+/*>]-о
и преобразуем коммутатор [1Ду, v^ + Vjlzj] следуюпщм образом:
Как дадим, он равен нулю, если vj = vt, т.е. ядра i mj - одного типа.
Последнее означает, что собственные функции оператора Йщр + Йщр можно
представить линейными комбинациями функций, каждая из которых есть
произведение функций подсистем неэквивалентных ядер. И если спиновая
ядерная подсистема молекулы включает две группы неэквивалентных ядер,
например, типа А и X, то спиновая ядерная функция молекулы,
i * j
где МАк и Мхк - проекции суммарного ядерного спина соответственно
подсистем А и X, причем
(IV.2346)
^Ак+^Хк^Щ
Инициировать переходы между этими состояниями можно, как всегда,
полем электромагнитной водны Hjcosu* , ортогональной постоянному полю
Н. Матричные элементы возмущения будут выглядеть аналогично (TV.227):
<А//|-1«/л<|М}>— <MI\^lxl\M'I>
i n i n
(IV.235)
150
Модельные системы и реальные спектры молекул
Сначала посмотрим на «простейшую систему», состоящую из ядер
только одного типа, т.е. А„, причем эти ядра эквидистантны. Последнее
условие существенно, ведь константы спин-спинового взаимодействия зависят
от расстояний между ядрами, убывая с расстоянием по закону У 3 *
Поэтому системой А„ является молекула метана (п = 4), но не этана, в котором на
первый взгляд все протоны одинаковы. В действительности это (A3>2, т.е.
это две группы эквивалентных протонов.
Вернемся к системе Ал. Бе гамильтониан имеет вид:
^=I^VI^M,-^4+^EM,, (iv.236)
j П i<j П П П i<j
где vA - собственная частота эквивалентных ядер А, а /^ - их константа
спин-спинового взаимодействия.
Обозначим собственные значения оператора Йямру т.е. энергии
ядерных состояний еМл, где нижний индекс МА указывает проекцию
суммарного ядерного спина в данном состоянии (напоминаем, что в состояниях
| МА >, собственных для Йямр9 выполнено условие (IV.233)). Эти энергии -
средние значения оператора Й ямр на функциях | МА >:
= ул¥а + ^Ъ<мл\ Щ \МА>.
Заметим, что второй член в выражении энергии пропорционален константе
спин-спинового взаимодействия ядер (лежащей обычно в интервале от 0 до
500 Гп\ .тогда как первое слагаемое пропорционально собственной частоте
ядер, превосходящей это взаимодействие на три-четыре порядка. Это значит,
что этот второй член можно рассматривать как небольшую поправку к
уровням энергии, которые получаются при пренебрежении спин-спиновым
взаимодействием ядер (TV.2266).
(IV.237)
151
Правило отбора (IV.235) для переходов между этими состояниями в
поле электромагнитной волны подходящей частоты будет таким:
-^<М^|/х|М;,>*0, (IV.238)
п
где уА - гиромагнитное отношение ядер А, а 1Х - проекция суммарного
ядерного спина на ось Ох. Интересно, что это соотношение помогает найти
частоты переходов между состояниями (IV.237), энергии которых мы пока
точно не определили. Сделать это можно следующим образом. Как было
отмечено выше, оператор X~f My ^^яЬр коммутирует с любой проекцией
/<уЛ
суммарного ядерного спина (IV.231), в частности с 1Х:
Матричные элементы этого нулевого оператора на любых функциях равны
нулю. Посмотрим, что будет, если этими функциями являются начальное
| МА > и конечное | МА > состояния системы, отвечающие спектральному
переходу, разрешенному правилом отбора (TV.238):
о=<мА\[й%„1х]\м-А>= ^2Щ
=< МА | Й%р1х\ U'А > - < МА11ХЙ% | М'А >.
Заметим, что
и аналогично
Я%р\М'а >=(еми -vAMA)\M'A >,
где еМ'А и еМл - энергии исходного и конечного состояний ядерной системы
с полными проекциями спина соответственно М'А и МА. Тогда выражение
(IV.239) можно преобразовать так:
0 = ((^ -vAMA)-(eMU -vAM'A))<MA\lx\MA>.
Интеграл в правой части этого равенства отличен от нуля по правилам отбора
(TV.238), следовательно, должно быть выполнено условие:
*мА ~ем'л =*аМа-уаМ'а.
152
Но это не что иное, как частота спектрального перехода из начального
состояния системы в конечное, которая при условии (IV.235), т.е. при
Мл -М'л = 1 (что отвечает поглощению излучения) оказывается равна
просто 0. Значит, спектр системы Ап (например, CEU, которому соответствует
П « 4) QQgTQMT ИЗ рДВДЦ ЩИт.
Таким образом, в случае спиновой системы Ап учет спин-спинового
взаимодействия не изменяет оцениваемую частоту перехода: она совпадает с
оценкой, полученной для спектров низкого разрешения. Принимая во
внимание сделанный выше вывод, выражаемый формулой (IV.234a), этот
интересный результат можно обобщить и сформулировать общее правило:
спектр системы спинов не зависит
от взаимодействия внутри подсистем эквивалентных ядер.
Это утверждение заметно упрощает задачу анализа спектров систем,
включающих несколько групп неэквивалентных ядер, к которой мы и переходим.
Гамильтониан такой системы, состоящей из двух групп
неэквивалентных ядер, можно записать следующим образом:
(TV.240)
где
*ЙрМ>-^. * Й%>{Х)*у-£-1ъ
- операторы взаимодействия подсистем А и X с внешним полем
(УАИУХ~ собственные частоты ядер подсистем А и X,
Iaz и ?Xz ~ операторы проекций полных спинов этих подсистем на ось Oz)\
Й%р(АА) = ^ £М, и Й^ХХ)^ IM,
п (i<j)eA n (k<l)eX
- операторы спин-спинового взаимодействия внутри подсистем ядер А и X
(^ал и Jхх - константы взаимодействия ядер одного типа,
условия (z <j)eA и (к<1)еХ означают суммирование по ядрам в пределах
соответствующих подсистем);
153
n ieAkeX n
- оператор спин-спинового взаимодействия ядер А и X между собой
(1^ и t;r - полные спины соответствующих ядерных подсистем).
В силу сформулированного выше общего утверждения о
независимости спектра от взаимодействий эквивалентных ядер между собой, в
операторе (IV.240) можно опустить вклады Й^р(АА) и Й^р{ХХ) и
анализировать оператор вида
(IV.241)
Мы выбрали модельную систему АпХт. Такое обозначение ядер
соответствует условию (см. выше)
Следовательно, оператор Й^р(АХ) мал, и его можно рассматривать как
возмущение системы, описываемой гамильтонианом
в$Ь> = Яйв»^) + *%>Ю, (IV.242a)
соответствующим сйеюру низкого разрешения.
Оператор (TV 242) является суммой операторов, каждый из которых
действует на переменные только указанной подсистемы ядер. Поэтому его
собственные функции суть произведения собственных функций операторов
&ямр(Л) и Й^р(Х)у а собственные значения - суммы собственных
значений этих операторов. И те, и другие были определены нами ранее (см.
выражение (TV.2266) и предшествующее обсуждение), так что
Ха = \Mv.Mn>\Mn+i:Mn+m>^MA >\МХ>ЦМАМХ > (IV.2426)
система Ап система Хт
и
ем\мх в *иА + *мх = ул¥а + vxMx > (IV.242B)
гДе 8мл и емх -энергии спиновых ядерных подсистем Али Хт.
Заметим, что мы ранее выяснили (см. (IV.234a)), что при учете спин-
спинового взаимодействия ядерная спиновая функция оказывается линейной
154
комбинацией функций (IV.2426), каждая из которых отвечает определенному
значению проекции суммарного спина соответствующей подсистемы,
причем проекции спина подсистем связаны условием (IV.2346).
Оценим в первом порядке теории возмущений поправки к энергиям
(IV.242b), порождаемые оператором Й^р(АХ):
*млмх =<МАМХ | ^Млг \MJtx >=
^^<MA\tAz\MA><Mx\lx,\Mx>^JAXMAMx.
Здесь мы в очередной раз учли, что в среднее значение оператора
ненулевой вклад вносит только последнее слагаемое.
Итак, с учетом поправки на взаимодействие ядер двух подсистем,
энергии состояний спиновой системы АпХт
емлмх = улмa +vxMx + JaxmaMx
(IV.243)
Переходы между этими состояниями разрешены, если отличен от нуля
матричный элемент (IV.235), который в случае двух подсистем неэквивалентных
ядер может быть преобразован так:
< MjM'x) Y-±1m + ^/Л | M'jM'x >=
= ^<МА\1Ах\М'АхМх\М'х>+^-<МА\МАхМх\1Хх\М'х>
Первое слагаемое отлично от нуля при условии
\МА=М'А±1, МХ=М'Х[
а второе - при условии
МА=М'А, МХ*М'Х±1\.
Как следует из (IV.243), частоты спектральных переходов определены
выражением
*емлмх -Ул(МА-Ми)^х(Мх-М'х)^^(МАМх-МгАМ'х).
155
Следовательно, переходы первого типа имеют частоту
A4fAMx = у л + Jax^x , (IV.244a)
а второго - частоту
Ь*млмх =vx+ JaxMa . (IV.2446)
В используемом нами приближении \vA - Vx\» J ax это значит, что спектр
состонгг из двух групп линий с центрами при vA и vx. Расстояния внутри
каждой группы линий равны У1Y и заметно меньше расстояния между
группами, равного \vA -vx\- Число линий в первой группе (около частоты vA)
определено числом возможных проекций полного момента подсистемы X,
т.е. 2т1Х\ +1, где 1Х\ - спин частицы типа X. Число же линий во второй
группе (около частоты vx) равно числу возможных проекций спина
подсистемы А: 2/1/^+1.
Обычно анализируют спектры ядер с полуцелым спином (в основном
протонов),
поэтому струщура едектра спиноврй риртрмц АяХт будет такая:
мультиплет с центром у а и числом линий т + 1
I мультиплет с центром vx и числом линий л + 1. |
Соотношение интенсивностей линий мультидлета. как и в случае ЭПР-
спектров, определяется биномя? тплтими коэффициентами, а общие плошади
мультиплетных сигналов пропорциональны числу атомов данного типа.
Реальные системы
Простым примером системы типа АпХт является 1,3-дахлопропан, в
котором протоны хлорметипьных групп образуют одну подсистему
эквивалентных ядер (Х4), а два протона метиленового фрагмента- вторую (А2):
156
У системы А2Х4 должно быть 2 пика: квинтет с частотой vA и
соотношением интенсивностей линий 1:5:10:5:1 и триплет с частотой Vx и
соотношением интенсивностей 1:2:1. В полном соответствии с этими предсказаниями
протонный ЯМР-спектр 1,3-дихлорпропана такой:
СзНбСЬ
-С(С1)НГ
-€Нг-
ш
5, м.д.
Формально диэтилкетон тоже может быть представлен формулой
А6Х4, где 6 ядер одной группы - это протоны обоих метилов, а 4 ядра
второй группы - это протоны обоих метиленовых фрагментов.
Н-
•Н
Однако по тем же причинам, по которым этан - это система (А3)2»а не
А6, протонную систему диэтилкетона правильнее отображает запись
(А3Х2)2. Пренебрегая спин-спиновым взаимодействием ядер,
принадлежащих не соседним функциональным группам, мы должны рассмотреть лишь
взаимнбе влияние метальной и метиленовой групп в пределах одного
стильного фрагмента. Поэтому ПМР-спектр кетона состоит из двух сигналов с
мультиплетностью 3 и 4. А его отличием, например, от спектра молекулы
CH3CH2ONa (т.е. системы А3Х2) будут лишь вдвое большие площади пиков
(соответственно наличию двух этильных групп в молекуле).
157
EtC(0)Et
-CHr
Ш
-CH3
1111
Tfo 8, м.д.
Аналогично можно описать ЯМР-спектры спиновых систем,
включающих более двух групп неэквивалентных ядер. Например, рассмотренная
ранее молекула этанола может быть представлена формулой A3X2Y, где А3
- три метальных протона, Х2 - два протона фрагмента СН2, a Y - протон
гидроксильной группы:
Ближайшим соседом метила является метилен, благодаря которому
сигнал при частоте v(CH3) будет триплетным. Сигнал самого метилена -
октет: благодаря взаимодействию с тремя протонами метила формируется
квартет, каждая линия которого дополнительно расщепляется на две из-за
взаимодействия с протоном гидроксильной группы. Сигнал же протона ОН-
группы - триплетный благодаря наличию соседней метиледрвой группы.
Однако в спектре для ОН-группы наблюдается синглет, а для СНг-группы -
квартет. Причина в том, что «/що)-(С)Н существенно меньше, чем /н(С)-(С)н>
и расщепления, обусловленные взаимодействием протонов гидроксильной и
метиленовой групп, не видны на фоне расщепления, вызванного
взаимодействием протонов СНз и СН2-групп.
158
С2Н5ОН
-СНз
-он
-СНг
-Л_
л
А
7 6 5 4 3 2 10. 0,М.Д.
На этом мы заканчиваем обсуждение магнитно-резонансных методов.
Их существенное отличие от рассмотренных ранее спектральных методов
заключается в том, что сами уровни индуцированы взаимодействием с
внешним постоянным магнитным полем, а частоты переходов на несколько
порядков ниже (поскольку речь идет о релятивистских эффектах, имеющих
малую величину).
159
Рекомендуемая литература
Основная
1. А. С. Давыдов. «Квантовая механика», Москва: Гос. Изд. Физ.-мат.
Лит., 1963.
2. Н. Ф. Степанов. «Квантовая механика и квантовая химия», Москва:
Мир, Изд. Моск. Унив., 2001.
3. А. Б. Болотин, Н. Ф. Степанов. «Теория групп и ее применения в
квантовой механике молекул», Вильнюс: Элком, 1999.
4. И. В. Абаренков, В. Ф. Братцев, А. В. Тулуб. «Начала квантовой
химии», Москва: Высшая школа, 1989.
5. В. И. Минкин, Б. Я. Симкин, Р. М. Миняев. «Теория строения
молекул», Ростов-на-Дону: Феникс, 1997.
6. В. М. Татевский. «Строение молекул», Москва: Химия, 1977.
7. А. А. Мальцев. «Молекулярная спектроскопия», Москва: Изд. Моск.
Унив., 1980.
8. У. Флайгер. «Строение и динамика молекул», в двух томах, Москва:
Мир, 1982.
Дополнительная
1. П. В. Елютин, В. Д. Кривченков. «Квантовая механика с задачами»,
Москва: Физматлит, 2001.
2. Л.Д. Ландау, Е.М. Лифшиц, «Квантовая механика» (том 3) и «Теория
поля» (том 2). М: Наука, 1988,1989.
3. Р. Фларри. «Квантовая химия», Москва: Мир, 1985.
4. С. Фудзинага. «Метод молекулярных орбиталей», Москва: Мир, 1983.
5. Ф. Банкер, П. Йенсен. «Симметрия молекул и спектроскопия»,
Москва: Мир, Научный мир, 2004.
6. Р. Драго. «Физические методы в химии», М: Мир, 1981.
7. Л. В. Вилков, Ю. А. Пентин. «Физические методы исследования в
химии», в двух томах, Москва: Высшая школа, 1989.
160