/













































































































































































































































































































































































































































































































Author: Глушко В.П.
Tags: тепло термодинамика химия физика математическая физика издательство наука химическая термодинамика
Year: 1978
Similar




Text
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ВЫСОКИХ ТЕМПЕРАТУР
ГОСУДАРСТВЕННЫЙ ИНСТИТУТ ПРИКЛАДНОЙ ХИМИИ
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
СПРАВОЧНОЕ ИЗДАНИЕ
В ЧЕТЫРЕХ ТОМАХ
Издание третье, переработанное и расширенное
Согласовано с Государственной службой
стандартных справочных данных
РЕДАКЦИОННАЯ КОЛЛЕГИЯ:
В.П. ГЛУШКО (ответственный редактор),
Л. В. ГУРВИЧ (зам. ответственного редактора),
Г.А.БЕРГМАН, И.В.ВЕЙЦ, В.А.МЕДВЕДЕВ,
Г. А. ХАЧКУРУЗОВ, В. С. ЮНГМАН
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 197S
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
Том I
ЭЛЕМЕНТЫ
О, H(D,T), F, Cl, Br, I, He, Ne, Af,
Кг, Хе, Rn, S, N, Р
И ИХ СОЕДИНЕНИЯ
Книга 1
МЕТОДЫ РАСЧЕТА.
ВЫЧИСЛЕНИЕ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1978
УДК 536+541.1
Термодинамические свойства индивидуальных веществ. Справочное издание: В 4-х т.
/Л. В. Гурвич, И. В. Вейц, В. А. Медведев и др. — 3-е изд., перераб. и расширен. —
Т. I. Кн. 1. — М.: Наука, 1978. — 496 с.
Издание состоит из четырех томов, каждый том — из двух книг: в первой описаны методы
расчета, во второй книге помещены таблицы термодинамических свойств.
В первой книге I тома изложены применявшиеся методы расчета термодинамических функций
веществ в твердом, жидком и газообразном состояниях, а также принципы выбора молекулярных
постоянных и термохимических величин. Основное содержание книги составляют критический
выбор постоянных, необходимых для расчета термодинамических свойств (молекулярных
постоянных, энтальпии образования и фазовых переходов, теплоемкости и т. д.), описание расчета и
оценка достоверности рекомендованных величин для каждого из рассмотренных веществ. Для ряда
газов рассмотрены свойства при повышенных давлениях.
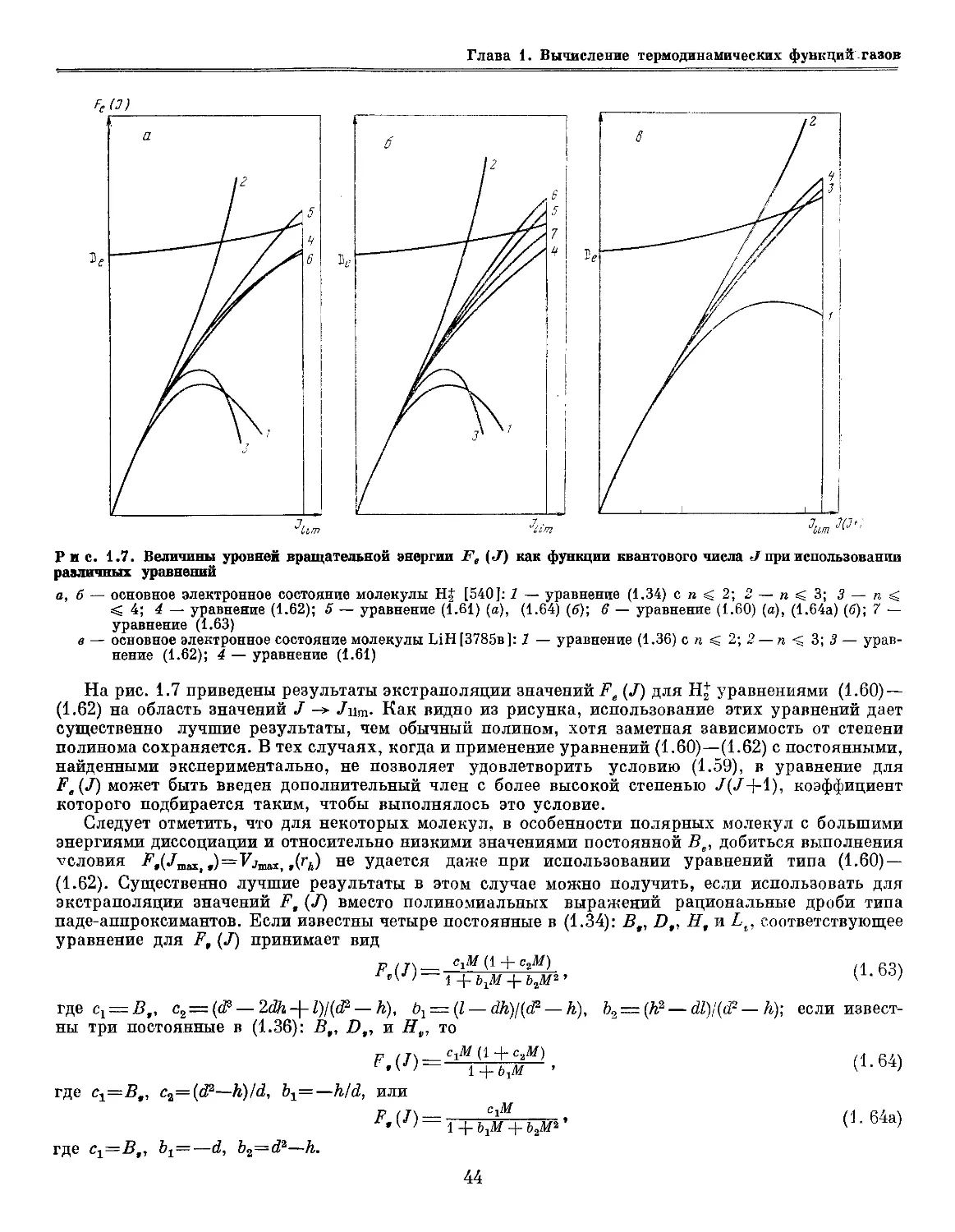
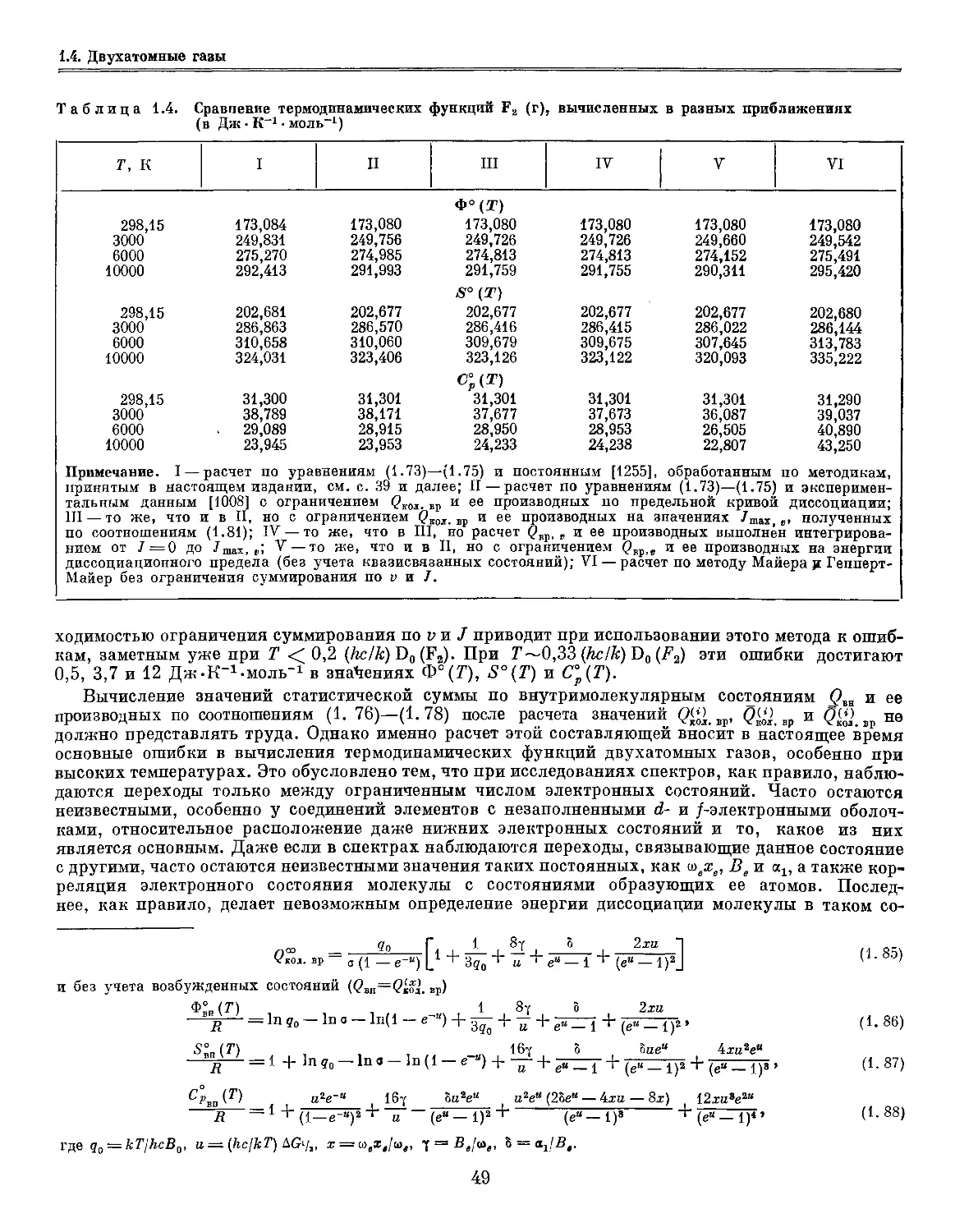
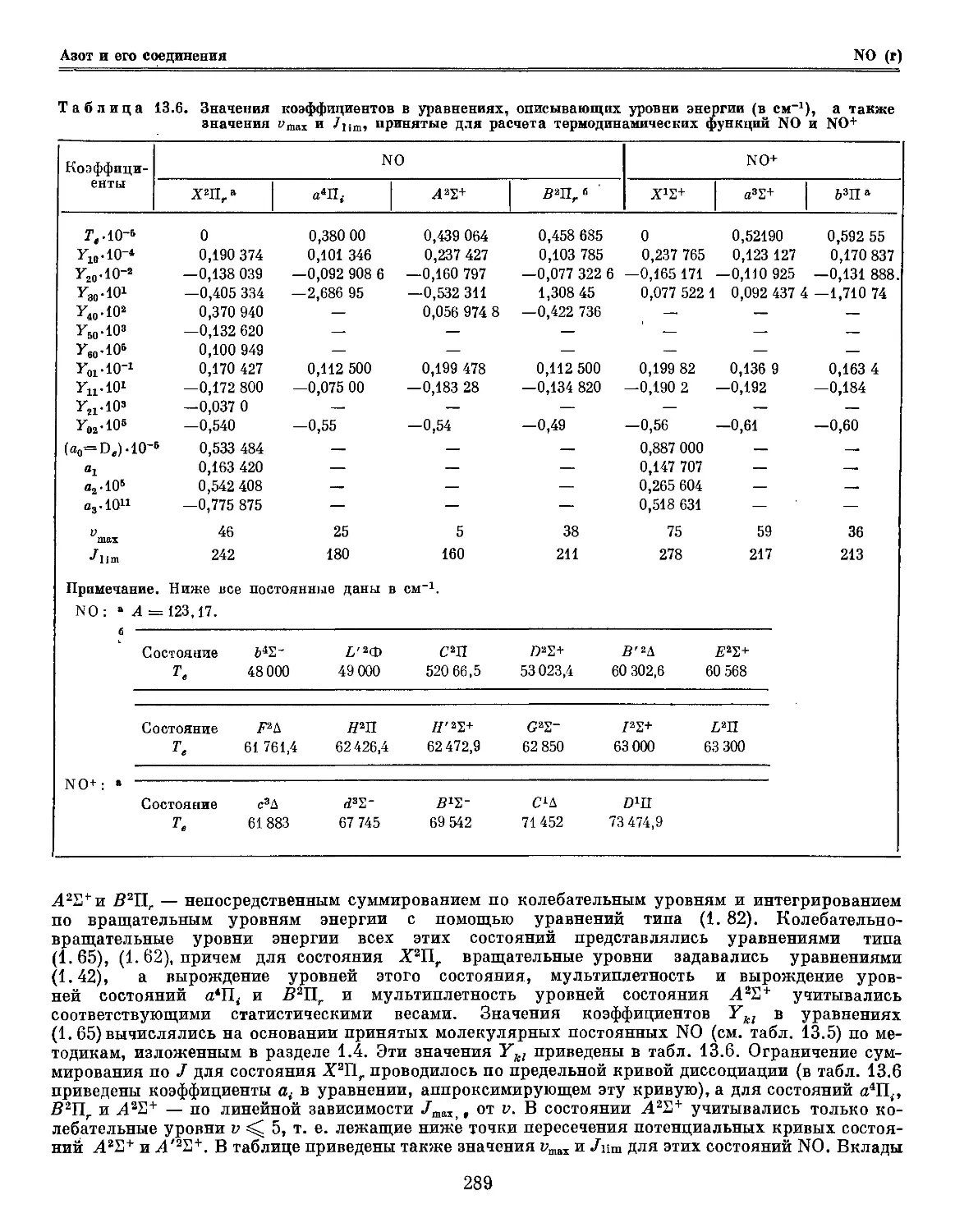
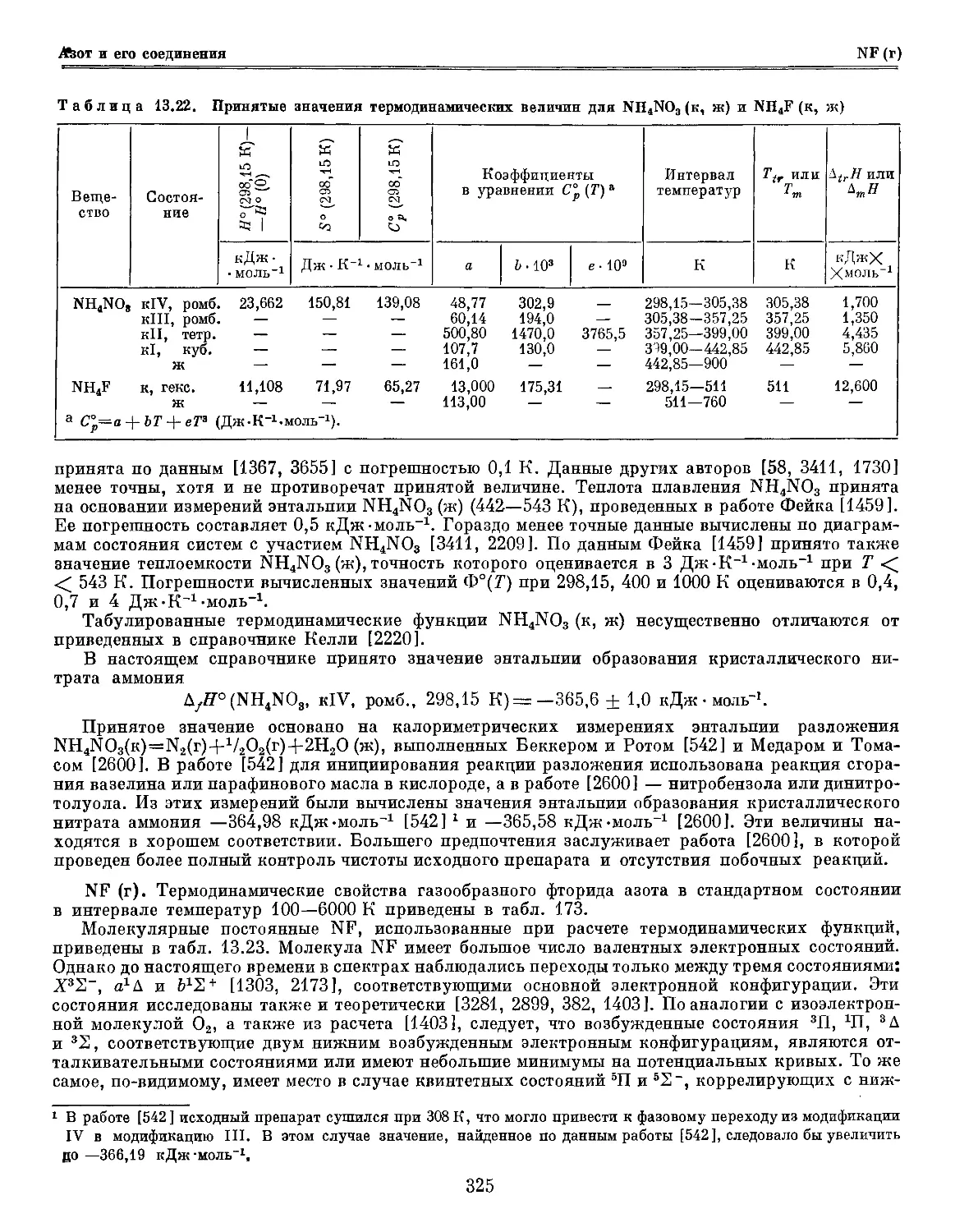
Табл. 144, ил. 13, список лит. 4351 назв.
АВТОРЫ
Л. В. ГУРВИЧ В. С. ИОРИШ В. Я. ЛЕОНИДОВ
И.В.ВЕЙЦ Г.Н.ЮРКОВ Ю.С.ЕЖОВ
В. А. МЕДВЕДЕВ С. И. ГОРБОВ С. Э. ТОМБЕРГ
Г. А. ХАЧКУРУЗОВ Л. Ф. КУРАТОВА И. И. НАЗАРЕНКО
В. С. ЮНГМАН Н. П. РТИЩЕВА А. Л. РОГАЦКИЙ
Г. А. БЕРГМАН И. Н. ПРЖЕВАЛЬСКИЙ О.-В. ДОРОФЕЕВА
В. Ф. БАЙБУЗ В. Ю. ЗИЦЕРМАН М. С. ДЕМИДОВА
Q Издательство «Наука», 1978 г.
Эта книга не может быть полностью или
частично воспроизведена или размножена, введена
в информационно-поисковую систему или
передана по линиям связи в любой форме или
любыми средствами (в том числе электронными
устройствами и на магнитных носителях
информации) без письменного разрешения
издательства «Наука».
ОГЛАВЛЕНИЕ
Предисловие 7
Предисловие к I тому 10
Введение 11
Часть I
МЕТОДЫ РАСЧЕТА ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИНДИВИДУАЛЬНЫХ ВЕЩЕСТВ
Общие соотношения термодинамики 22
Глава 1. Вычисление термодинамических функций газов 28
1.1. Разделение статистической суммы. Вычисление поступательных
составляющих термодинамических функций газов 28
1.2. Особенности вычисления внутримолекулярных составляющих
термодинамических функций газов 28
1.3. Вычисление внутримолекулярных составляющих термодинамических
функций одноатомных газов 31
1.4. Вычисление внутримолекулярных составляющих термодинамических
функций двухатомных газов 35
1.5. Вычисление внутримолекулярных составляющих термодинамических
функций многоатомных газов 52
1.6. Оценка погрешностей вычисленных таблиц термодинамических функций
газов 63
Глава 2. Вычисление термодинамических функций веществ в конденсированном
состоянии 69
2.1. Общие замечания 69
2.2. Вычисление термодинамических функций веществ в кристаллическом
состоянии при Т < 298,15 К 72
2.3. Вычисление термодинамических функций веществ в кристаллическом
состоянии при 298,15 К < Т < Тт 73
?2.4. Учет'фазовых переходов и полиморфных превращений 75
2.5. Термодинамические функции веществ в жидком состоянии 78
2.6. Оценка погрешностей таблиц термодинамических функций веществ в
конденсированном состоянии 79
Глава 3. Выборт рекомендуемых значений термохимических величин 81
3.1. Согласованность термохимических величин 82
3.2. Некоторые критерии ^оценки достоверности экспериментальных
измерений термохимических величин 83
Оглавление
3.3. Вычисление термохимических величин по результатам исследования
равновесий 85
3.4. Приближенная оценка термохимических величин 87
3.5. Оценка погрешности рекомендуемых значений термохимических величин 88
Часть 2
ВЫБОР ИСХОДНЫХ ПОСТОЯННЫХ
И РАСЧЕТ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
Глава 4. Электронный газ и кислород 90
Глава 5. Водород и его соединения 102
Глава 6. Дейтерий, тритий и их соединения 126
Глава 7. Фтор и его соединения 145
Глава 8. Хлор и его соединения 162
Глава 9. Бром и его соединения 181
Глава 10. Иод и его соединения 198
Глава 11. Благородные газы и их соединения . 218
Глава 12. Сера и ее соединения 238
Глава 13. Азот и его соединения 279
Глава 14. Фосфор и его соединения 345
ПРИЛОЖЕНИЯ
Приложение 1. Атомные веса, изотопный состав и спины ядер химических
элементов 390
Приложение 2. Принятые значения фундаментальных постоянных и переводных
множителей для единиц энергии. Температурная шкала 395
Приложение 3. Термодинамические свойства газов при повышенном давлении . . 397
Принятые символы и обозначения 428
Литература 433
ПРЕДИСЛОВИЕ
Первое издание настоящей книги под названием «Термодинамические свойства компонентов
продуктов сгорания» было опубликовано Издательством АН СССР в 1956 г. Работа над справочником,
содержащим таблицы термодинамических функций и констант равновесия компонентов продуктов
сгорания топлив, была начата в связи с тем, что известные в то время в литературе сведения о
термодинамических свойствах продуктов сгорания оказались недостаточными для расчета двигателей
и выбора топлив. Так, таблицы, опубликованные Хаффом и др. [2029], содержали данные только
для 42 компонентов, причем точность большинства таблиц была неудовлетворительна. В книге
Цейзе [4058] были приведены таблицы термодинамических свойств ряда газов, заимствованные
из периодической литературы, однако большинство таблиц охватывало только узкий
температурный интервал и основывалось на неточных или устаревших значениях постоянных.
В первом издании были представлены термодинамические свойства в стандартном состоянии
для температур от 293,15 до 6000 К для 234 компонентов, образованных 21 элементом: Н (D, Т),
Li, Be, В, С, N, О, F, Na, Mg, Al, Si, P, S, Cl, K, Ca, Br, I, Hg, Pb. Таблицы были даны для
основных компонентов продуктов сгорания, соответствующих наиболее вероятным сочетаниям
перечисленных элементов. Принималось, что в продуктах сгорания могут встречаться все
простейшие радикалы, образованные окислителями и горючими, хотя существование некоторых из них
еще не было подтверждено экспериментально. Учитывалась возможная ионизация таких
атомов, как Li, Na, К, Са и образование отрицательных одноатомных ионов F~, С1~, Вг~, 1~, что было
существенно в связи с влиянием ионизации пламени на прохождение радиоволн при определенных
условиях. В то же время не рассматривались многочисленные фтор-, хлор- и фторхлорпроизводные
углеводородов и многие более сложные неорганические соединения, образование которых вероятно
при выходе продуктов сгорания из сопла двигателя и понижении температуры.
Все 207 таблиц термодинамических свойств были рассчитаны на основании наиболее
достоверных значений молекулярных и термохимических постоянных, выбранных в результате
анализа литературы, опубликованной до 1955 г., а при отсутствии таковых — на основании
оцененных постоянных. В справочнике впервые были опубликованы таблицы термодинамических
свойств свыше 100 веществ. Хотя рассмотренный в первом издании перечень компонентов не
охватывал все возможные продукты сгорания, способные образовываться в камере сгорания, он был
намного полнее и точнее известных в то время в литературе. Это позволило существенно повысить
точность разнообразных теплотехнических расчетов.
В последующие годы возникла потребность в термодинамических свойствах более широкого
круга компонентов, а также в данных для ряда газов при температурах выше 6000 К. В связи
с этим в 1962 г. Издательством АН СССР было опубликовано второе издание под названием
«Термодинамические свойства индивидуальных веществ». В нем были представлены данные для 424
компонентов, в том числе для 335 газов (для 22 газов до 20 000 К) и для 45 веществ в
конденсированном состоянии, образованных 31 элементом: Н (D, T), He, Li, Be, В, С, N, О, F, Ne, Na,
Mg, Al, Si, P, S, Cl, Ar, K, Ca, Br, Kr, Rb, Sr, Zr, I, Xe, Cs, Ba, Hg, Pb. Было расширено
число соединений элементов трех первых периодов Периодической системы, включен ряд
соединений более тяжелых элементов, а также ионизированные двухатомные газы. Кроме таблиц
термодинамических свойств веществ в стандартных состояниях, для 34 газов были приведены вириаль-
ные коэффициенты и их производные, что обеспечивало возможность учета отклонения их свойств
при высоких давлениях и умеренных температурах от свойств в стандартном состоянии.
При подготовке второго издания справочник был полностью переработан, заново был
выполнен критический анализ всей литературы, включая опубликованную до 1961 г., выбор
молекулярных и термохимических постоянных, оценки недостающих постоянных и пересчет
данных, полученных в оригинальных работах, для получения системы согласованных
величин. Были существенно уточнены статистические методы расчета термодинамических
Предисловие
функций газов для обеспечения возможности ограничения статистических сумм атомов и молекул,
учета возбужденных электронных состояний молекул и особенностей их вращательных уровней
энергии в мультшшетных и вырожденных электронных состояниях. Во второе издание были
включены 380 таблиц термодинамических свойств; из 207 таблиц первого издания 150 были рассчитаны
заново с использованием новых и более точных значений постоянных и усовершенствованных
методов расчета. Для 45 веществ таблицы термодинамических свойств были опубликованы впервые.
Впервые в литературе были выполнены оценки точности рассчитанных таблиц термодинамических
свойств. Это издание на ряд лет опередило аналогичные зарубежные справочники.
Новое, третье издание должно обеспечить возможность проведения термодинамических
расчетов самых разнообразных процессов и химических реакций, протекающих с участием широкого
круга неорганических и простых органических веществ при температурах от 100 до 20 000 К.
Это издание полностью переработано и существенно расширено (более чем в три раза) по сравнению
с предыдущим. В справочнике представлены термодинамические свойства около 1200
конденсированных и газообразных веществ, образованных 47 элементами: Н (D, T), He, Li, Be, В, С, N,
О, F, Ne, Na, Mg, Al, Si, P, S, Gl, Ar, K, Ca, Ti, V, Cr, Ga, Ge, Br, Kr, Rb, Sr, Zr, Nb,
Mo, In, Sn, I, Xe, Gs, Ba, Hf, Та, W, Tl, Pb, Rn, Th, U, Pu. Среди рассматриваемых
соединений этих элементов — их соединения с кислородом, водородом и галоидами, для ряда
элементов — с серой, азотом и углеродом, включая соли кислородных кислот, большое число
радикалов и ионизированных газов.
Таблицы термодинамических свойств всех веществ приведены в их стандартных состояниях.
Для ряда твердых веществ рассмотрены также свойства их неравновесных модификаций, включая
стеклообразное состояние; для многих газов приводятся данные, позволяющие учесть отклонения
их свойств при высоких давлениях от свойств в состоянии идеального газа. Все таблицы
термодинамических свойств для этого издания были рассчитаны заново на основании постоянных,
критически отобранных в результате анализа литературы, а также теоретических расчетов и оценок,
выполненных при его подготовке.
Для расчетов таблиц термодинамических функций газов были уточнены статистические методы
расчета и создан комплекс программ, обеспечивший вычисление всех таблиц термодинамических
свойств веществ на ЭВМ, их накопление на машинных носителях информации и выдачу на печать
с помощью фотонаборного устройства «ДИГИСЕТ». Разработанные методы обеспечили расчеты
большинства таблиц термодинамических функций веществ в газообразном и конденсированном
состояниях в интервале 100 — 6000 К с точностью, лимитируемой только достоверностью известных
в настоящее время постоянных. Для многих одно- и двухатомных газов, а также некоторых
многоатомных газов достоверность табулированных термодинамических функций в этом интервале
температур ограничивается только точностью газовой постоянной R, и можно предполагать, что они
не потребуют уточнения в течение длительного срока.
В результате пересчета всех экспериментальных данных достигнута полная внутренняя
согласованность всех табулированных термодинамических функций и принятых значений
термохимических величин. Вся система термохимических данных базируется на значениях ключевых
величин, разработанных и рекомендованных КОДАТА—MCHG х при участии авторов этого издания.
По точности табулированных величин, широте температурного интервала и количеству
рассмотренных веществ настоящее издание превосходит другие аналогичные справочники, известные
в мировой литературе. Оно является первым справочником такого рода, полностью основанным
на значениях фундаментальных постоянных и ключевых термохимических величин,
рекомендованных КОДАТА—МСНС.
Предполагается, что материалы этого издания (таблицы термодинамических свойств,
уравнения, аппроксимирующие табулированные величины, и т. п.) послужат основой для создания фонда
данных по термодинамическим свойствам неорганических и простых органических веществ на
машинных носителях информации, который будет доступен потребителям по линиям связи.
Два первых издания этого справочника были подготовлены в 1953—1962 гг. коллективом
сотрудников Института горючих ископаемых (ИГИ) АН СССР и Государственного института
прикладной химии (ГИПХ) под общим научным руководством академика В. П. Глушко. В 1963 г.
эти работы были переведены из ИГИ в Институт высоких температур АН СССР (ИВТАН), где бла-
1 КОДАТА—МСНС — Комитет по численным ^данным для науки и техники Международного совета научных
союзов.
Предисловие
годаря постоянной поддержке директора ИВТАНа академика А. Е. Шейндлина они получили
широкий размах и всестороннее развитие.
Третье издание подготовлено коллективом сотрудников Отдела термодинамики ИВТАНа
(руководитель Л. В. Гурвич) и группы по расчетам термодинамических свойств ГИПХа
(руководитель Г. А. Хачкурузов) под общим научным руководством академика В. П. Глушко.
Научно-методические основы подготовки всех трех изданий справочника, были разработаны
В. П. Глушко, Г. А. Бергманом, И. В. Вейц, Л. В. Гурвичем, В. А. Медведевым, Г. А. Хач-
курузовым и В. С. Юнгманом. В разработке методов расчета таблиц термодинамических
свойств, использованных при подготовке третьего издания, принимали участие В. С. Иоршп,
Г. Н. Юрков и С. И. Горбов.
Основная роль в подготовке этого издания принадлежит сотрудникам Отдела термодинамики
ИВТАНа, которые совместно с сотрудниками группы по расчету термодинамических свойств ГИПХа
выполнили большую творческую работу. Планирование и координация работ по подготовке
справочника в обеих организациях осуществлялась ведущим автором всех изданий Л. В. Гурвичем.
Подготовка нового издания «Термодинамические свойства индивидуальных веществ»
проводилась одновременно с подготовкой в ВИНИТИ и ИВТАНе другого фундаментального
справочника АН СССР «Термические константы веществ», в котором представлены термодинамические
свойства всех изученных неорганических и простых органических веществ при стандартной
температуре и температурах фазовых переходов. Редакционные коллегии и авторские коллективы
обоих справочников работали в тесном контакте, используя общие информационные фонды,
картотеки и уже подготовленные материалы каждого справочника. Это позволило существенно повысить
достоверность рекомендуемых значений и избежать дублирование работы.
В связи с отсутствием в литературе данных, необходимых для расчета термодинамических
свойств многих веществ, в ряде исследовательских организаций и вузов страны начиная с 1953 г.
был поставлен комплекс экспериментальных исследований молекулярных и термохимических
постоянных, данных о теплоемкостях и изменении энтальпии в (твердом и жидком состояниях
и т. п. Среди этих исследований следует отметить электронографические исследования структуры
молекул на Химфаке МГУ и в ИВТАНе, термохимические исследования на Химфаке МГУ,
в ИВТАНе и ИХТИ, исследования спектров простых молекул на Химфаке МГУ, в ИВТАНе
и ГИПХе, масс-спектрометрические исследования процессов сублимации и энергий диссоциации
на Химфаке МГУ и в ИВТАНе, спектрофотометрические исследования энергий диссоциации
в ИГИ, а затем в ИВТАНе, исследования теплоемкостей и измерения энтальпии в широком
интервале температур от 4 до 3000 К, начатые в Институте физических проблем АН СССР и ВНИИ
метрологии (г. Харьков) и продолженные в ИНХе СО АН СССР, ИВТАНе и ИОНХе АН СССР.
Планирование и координация этих работ осуществлялась Отделом термодинамики ИВТАНа.
На протяжении многих лет работы над настоящим справочным изданием авторы имели
возможность творческого общения с членом-корреспондентом АН СССР Я. И. Герасимовым,
профессорами В. А. Киреевым, С. М. Скуратовым, В. А. Соколовым и В. М. Татевским, чьи дружеские
советы и замечания были весьма ценными при подготовке всех трех изданий.
В связи с созданием Рабочей группы КОДАТА—МСНС для разработки международных
рекомендаций по значениям ключевых термохимических величин с 1968 г. были установлены
постоянные научные контакты между Отделом термодинамики ИВТАНа и Центром данных по химической
термодинамике Национального бюро стандартов (НБС) США, а также с другими зарубежными
учеными. Эти контакты, включавшие обмен библиографией и литературными источниками, совместный
критический анализ и выбор ключевых величин, обсуждение противоречивых данных и т. п., были
весьма плодотворными и способствовали подготовке материалов этого издания.
Считаю приятным долгом выразить глубокую признательность дружному коллективу авторов
настоящего издания и многим советским и зарубежным ученым за полезные дискуссии и
предоставление результатов проводимых ими экспериментальных и теоретических исследований.
Редакционная коллегия будет признательна читателям за критические замечания и
предложения, которые помогут в дальнейшей работе по подготовке фундаментальных справочных изданий
АН СССР по термодинамике. Соответствующие замечания и предложения следует направлять по
адресу: 127412, Москва И-412, Коровинское шоссе, Институт высоких температур АН СССР,
Отдел термодинамики.
Академик В. П. ГЛУШКО
ПРЕДИСЛОВИЕ К I ТОМУ
В первом томе рассматривается 247 веществ, включая 15 элементов (О, Н, F, Gl, Br, I, He, Ne,
Аг, Кг, Хе, Rn, S, N, Р), дейтерий, тритий и соединения этих элементов, в том числе
положительные и отрицательные ионы. Для 39 газов таблицы термодинамических свойств публикуются
впервые. Для 50 веществ представлены данные о свойствах при повышенных давлениях и о
критических постоянных.
Подготовка материалов тома была завершена в 1976 г. и при этом была учтена литература,
опубликованная по 1975 г. включительно. Более поздние работы использовались в тех случаях,
когда их результаты могли привести к существенному уточнению соответствующих таблиц
термодинамических свойств.
Первый том подготовлен коллективом сотрудников ИВТАНа (главы 1—8, 11 и 13,
приложения) и ГИПХа (главы 9, 10, 12, 14, материалы глав 5-8 по НО2, НОг, Н2О (г), Н2О2 (г), HF, HG1
и соответствующим D-, Т-замещенным).
Основная авторская работа по первому тому выполнена Л. В. Гурвичем, И. В. Вейц, В. А.
Медведевым, Г. А. Хачкурузовым, В. С. Юнгманом и Г. А. Бергманом. Введение и часть первая
написаны Л. В. Гурвичем, В. С. Юнгманом (раздел 1.5), Г. А. Бергманом (глава 2) и В. А.
Медведевым (глава 3). Кроме того, выбор молекулярных постоянных проводили В. С. Иоршп, Г. Н.
Юрков, Л. Ф. Куратова, И. Н. Пржевальский, Ю. С. Ежов, И. И. Назаренко, при участии Е. А. Ше-
нявской, М. М. Новикова, В. Г. Рябовой, А. Н. Самойловой, Ю. Г. Хаита, Ю. М. Ефремова,
B. А. Кулемзы и В. С. Виноградова; выбор термохимических величин проводили В. Я. Леонидов,
C. Э. Томберг, А. Л. Рогацкий при участии Ю. С. Ходеева, Л. Н. Горохова, А. Г. Муравиной,
М. Е. Ефимова, А. Г. Ефимовой, Л. В. Юркинской и В. Я. Малецкого; расчеты термодинамических
функций газов проводили В. С. Иориш, Г. Н. Юрков, С. И. Горбов, Л. Ф- Куратова, Н. П.
Ртищева, И. Н. Пржевальский, О. В. Дорофеева, М. G. Демидова при участии А. М. Бережковского,
Ю. Г. Хаита, В. Ю. Зицермана и В. А. Кулемзы; расчет термодинамических функций веществ
в конденсированном состоянии проводили С. Э. Томберг, А. Л. Рогацкий при участии В. Н. Вдо-
вина и А. Я. Якобсона. Приложения 1 и 2 подготовлены В. С. Иоришем, Приложение 3 —
В. Ф. Байбузом и В. Ю. Зицерманом при участии А. Ф. Лемберского.
Комплекс программ для обработки экспериментальных данных, расчетов термодинамических
функций и таблиц термодинамических свойств на ЭВМ, а также ряда вспомогательных расчетов
был создан В. С. Иоришем, Н. П. Ртищевой, С. И. Горбовым, В. Ю. Зицерманом при участии
И. Н. Пржевальского, А. М. Бережковского и С. Э. Томберг. Программа для воспроизведения
таблиц на фотонаборном устройстве «ДИГИСЕТ» составлена А. Ф. Ицковичем.
Большую роль в подготовке справочника сыграл созданный в Отделе термодинамики ИВТАН
при содействии ВИНИТИ и Теплофизического центра ИВТАНа фонд библиографических данных
научных публикаций (руководитель Е. G. Маевская).
Основная техническая работа, связанная с проведением расчетов, подготовкой материалов,
оформлением рукописи, составлением списка литературы выполнены Е. С. Маевской, А. И.
Варшавской, О. М. Гайсинской, Е. Л. Осиной, В. С. Шмелевой, Н. Р. Симагиной, В. С. Павловой,
Е. Е. Догадиной, Л. П. Шиловой, И. Г. Байбуз, С. А. Равинской и Е. Ю. Борисовой.
Ряд вопросов, рассмотренных в главах 12 и 14, был обсужден с Д. Д. Уагманом и В. Эвансом
(НБС США), Дж. Берковицем (Аргоннская национальная лаборатория США) и Ж. Дровартом
(Брюссельский университет, Бельгия), чьи ценные замечания и предоставленные материалы
оказались весьма полезными при подготовке этих глав.
Общее редактирование I тома выполнено В. П. Глушко (отв. редактор) и Л. В. Гурвичем (зам.
отв. редактора), разделов по молекулярным постоянным и глав 9, 10, 12 — И. В. Вейц, разделов
по термохимическим постоянным — В. А. Медведевым, глав 11, 13, приложений — В. С.
Юнгманом, разделов по термодинамическим функциям веществ в конденсированном состоянии —
Г. А. Бергманом, предварительное редактирование глав 9, 10, 12, 14 — Г. А. Хачкурузовым.
10
ВВЕДЕНИЕ
Развитие науки и техники за последние 50 лет сопровождается все более широким использованием
методов термодинамики для оценки возможности и перспективности осуществления различных
процессов, в особенности процессов, протекающих при высоких температурах. Успехи,
достигнутые за это время й нефтехимии и металлургии, создании ракетных двигателей и конструкционных
материалов, развитии обычной и атомной энергетики и в ряде других областей, были бы
невозможны без предварительного термодинамического анализа соответствующих процессов и
накопления данных о термодинамических свойствах веществ. Разработка новых методов обеспечения
человечества энергией, выбор оптимальных путей использования сырьевых ресурсов и переработки
отходов промышленности, так же как решение многих других проблем, не смогут быть
осуществлены без предварительных термодинамических исследований. Все это делает необходимым, с
одной стороны, дальнейшее изучение (экспериментальное и теоретическое) термодинамических
свойств чистых веществ, а с другой — накопление уже имеющихся данных, их критический
анализ и оценку достоверности, систематизацию этих данных в виде, доступном ученым и инженерам,
работающим в разных областях науки и техники.
Это предполагает, что совокупность имеющихся данных о термодинамических свойствах
веществ после анализа, обработки и выбора рекомендуемых значений должна издаваться в виде
периодически обновляющихся и легко доступных справочных изданий, а также накапливаться
в специализированных центрах в памяти ЭВМ, откуда эти данные по мере необходимости смогут
быть получены непосредственно по линиям связи.
Современные справочные данные по термодинамическим свойствам веществ, публикуемые
в печати или хранящиеся в памяти ЭВМ, должны удовлетворять следующим требованиям:
1. Рекомендуемые значения рассматриваемых величин должны быть выбраны в результате
критического анализа всех данных, имеющихся в литературе, с использованием строгих и
стандартизованных методов обработки первичной информации и вычисления термодинамических свойств.
2. Рекомендуемые значения термодинамических свойств должны представлять систему взаимно
согласованных величин, включая согласованность термохимических величин и
термодинамических функций.
3. Рекомендуемые значения должны базироваться на фундаментальных постоянных и
ключевых термохимических величинах, рекомендованных соответствующими международными
организациями.
4. Для всех рекомендуемых значений должна быть дана оценка их достоверности.
5. Должны быть доступны краткие тексты, поясняющие выбор и расчет рекомендованных
величин и оценку их достоверности.
6. Должна быть дана исчерпывающая библиография всех работ, использованных при
подготовке справочных данных, из которой читатель мог бы получить представление о полноте анализа,
выполненного для рекомендации каждого конкретного значения, а при необходимости —
ознакомиться с этими работами и проверить анализ.
7. Значения рекомендуемых свойств должны быть представлены для широкого и логически
выбранного круга соединений.
8. Система данных должна быть представлена в таком виде, чтобы она могла регулярно
дополняться включением новых веществ и учетом результатов новых измерений (примерно раз
в 10—15 лет).
Фактически сегодня в мировой литературе нет справочных изданий по термодинамике, которые
удовлетворяли бы полностью всем этим требованиям. Существующие справочники могут быть
разделены на два типа: так называемые критические справочные издания и компилятивные,
вторичные и даже третичные справочники. Критические справочные издания, число которых в мировой
литературе весьма ограниченно, базируются на анализе первичной литературы и самостоятельных
11
Введение
расчетах рекомендуемых значений термодинамических свойств, они в большей или меньшей
степени удовлетворяют приведенным выше первому, второму и шестому требованиям. Подобные
издания, учитывающие также требования пятое и седьмое, получили в советской литературе
название фундаментальных справочных изданий. Первыми подобными справочниками были
«International Critical Tables» [641], а также справочники Быховского и Россини [642] и Келли [2220],
а в отечественной литературе — первое издание настоящего справочника [87].
Компилятивными справочными изданиями, которые сегодня составляют основную часть
литературы по термодинамическим свойствам веществ, здесь и ниже называются справочники,
основанные не на анализе оригинальных работ и последующих самостоятельных расчетах, а целиком
или в значительной степени воспроизводящие материалы других справочных изданий, часто
нескольких, или некритически воспроизводящие разрозненные данные, опубликованные в
литературе. Иногда рекомендации других справочников при этом дополняются данными из более
поздних работ, что, как правило, не улучшает всю совокупность приводимых величин, а иногда даже
ухудшает ее. В таких справочниках не только отсутствует внутренняя согласованность
рекомендуемых значений, но часто приводятся несовместимые величины.
Так, в справочнике под редакцией Брицке и Капустинского [36], который основан на
справочниках Быховского и Россини [642] и Келли [2220], были изменены по сравнению с этими
изданиями рекомендуемые значения энтальпии образования ряда веществ (например, В2О3 (к)) без
изменения энтальпий образования других соединений, основанных на старых величинах. В
результате, если рассчитывать энтальпии реакций по данным справочника [642], можно получить
не только неправильные значения соответствующих величин, но даже знаки этих величин.
Справочник Барина и Кнаке [437а], основанный на материалах таблиц JANAF [2094а] и книги Куба-
шевского и Эванса [2312], содержит десятки ошибок, возникших при переработке и объединении
этих данных. Для расчета таблиц термодинамических свойств авторы справочника [473а]
применили приближенную методику, основанную на аппроксимации значений теплоемкости,
табулированных в справочниках [2094а] и [2040], простыми уравнениями. Некоторые грубые ошибки в
работе [473а] отмечены в рецензиях Скиннера [3437а] и Гальченко [66а], но еще многие остаются
неизвестными широкому кругу читателей 1. В справочнике под редакцией Галкина [187]
объединены таблицы термодинамических функций из книги Галкина и др. [66] и таблиц JANAF [2095].
Однако в этих двух изданиях значения приведенной энергии Гиббса даны с разными началами
отсчета, поэтому их совместное использование должно приводить к грубым ошибкам в расчетах.
Это обстоятельство в справочнике [187] никак не оговорено. В справочнике под редакцией Зефи-
рова [53а], который является, вероятно, наиболее цитируемым отечественным справочником по
термодинамике, также содержатся многочисленные ошибки (см. [32в]). Одной из некорректностей,
допущенных при его подготовке, является то, что при пересчете к единицам системы СИ авторы
не учли различие между термохимической и паровой калориями; в результате приведенные в
справочнике [53а] значения наиболее хорошо изученных термохимических величин отличаются от
измеренных в оригинальных работах существенно больше, чем погрешности самих величин.
Авторы компилятивных справочников обычно не оговаривают эти особенности и часто даже
не указывают, из каких изданий или работ заимствованы конкретно те или иные величины. В то же
время объединение в одном справочнике несогласованных термодинамических величин приводит
к тому, что их совокупность может противоречить основным законам термодинамики и
приводить к большим или меньшим ошибкам в последующих расчетах. Тем, кто не является
специалистом в области численных данных по термодинамике, как правило, трудно обнаружить эту
несогласованность и противоречия.
Не останавливаясь больше на компилятивных справочниках, кратко рассмотрим критические
справочные издания по термодинамическим свойствам неорганических и простых органических
веществ. Такие издания по их содержанию могут быть разделены на два типа. К одному следует
отнести справочники, в которых приводятся данные о свойствах веществ при стандартной
температуре B98,15 К) и температурах фазовых переходов, к другому — содержащие, наряду с этими
1 В дополнении справочника Барина и Кнаке [4736], которое было опубликовано после завершения подготовки
этого тома, был устранен ряд ошибок, а также использованы данные справочников [258, 2039, 2095—2097, 2670,
3375]. Однако в методическом отношении оба издания идентичны и не являются системами согласованных
данных.
12
Введение
данными, таблицы термодинамических свойств в широком интервале температур. Настоящее
издание принадлежит ко второму типу, и поэтому достаточно ограничиться краткой
характеристикой справочников первого типа. В мировой литературе существует ряд хороших критических
изданий этого типа, в которых, однако, как правило, приводятся данные только для узких групп
веществ (см., например, [274, 2632, 3253а]). Известны также фундаментальные справочные
издания этого типа, подготавливаемые в Национальном бюро стандартов США и в Академии наук СССР.
Справочник, подготовленный в НБС США Россини и др. [3256] и опубликованный в 1952 г.,
до настоящего времени остается единственным завершенным фундаментальным изданием первого
типа. Он удовлетворяет всем требованиям, сформулированным выше, кроме третьего, четвертого
и пятого \ С 1962 г. в НБС США готовится новое издание этого справочника и его материалы
регулярно публикуются отдельными частями [3375]. К сожалению, принятая форма публикации
не удовлетворяет не только четвертому и пятому требованиям, но и шестому. Кроме того, в отличие
от издания [3256] в опубликованных выпусках приводятся данные только о свойствах веществ
при стандартной температуре, но нет данных для фазовых переходов. Однако тщательный
анализ всех первичных данных, проводимый авторами этого справочника, обеспечивает высокую
надежность рекомендованных величин, особенно для веществ в конденсированном состоянии и в
водных растворах.
Справочное издание «Термические константы веществ» [258], подготавливаемое под руководством
акад. В. П. Глушко в Отделе термодинамики ИВТАНа при участии ведущих специалистов страны
из разных исследовательских институтов и университетов и издаваемое ВИНИТИ под редакцией
В. П. Глушко, В. А. Медведева и др., станет, когда оно будет завершено, наиболее полным и
строгим изданием первого типа. По охвату веществ (свыше 25 000) и свойств оно существенно
превзойдет справочники НБС США, а при его подготовке были выполнены все требования,
сформулированные выше, кроме третьего и пятого 2. К настоящему времени опубликованы восемь из
десяти частей этого справочника, охватывающие соединения всех элементов Периодической системы,
кроме щелочно-земельных и щелочных. Последние восемь лет авторские коллективы НБС США
и ИВТАНа работают в постоянном контакте, обмениваясь библиографией, литературными данными
и обсуждая наиболее спорные величины, что способствует повышению уровня обоих изданий и
устранению неоправданных расхождений. Справочники [3256, 3375, 258] основаны на
неидентичных наборах ключевых термохимических величин. В связи с этим значения, рекомендованные
в указанных изданиях, строго говоря, несовместимы, хотя в большинстве случаев они близки
между собой.
В настоящем справочнике широко использованы материалы, накопленные при подготовке
соответствующих разделов справочника «Термические константы веществ», хотя принятые
значения практически никогда не совпадают из-за использования разных наборов фундаментальных
постоянных и ключевых величин, а также результатов более поздних исследований, поскольку
издание справочника [258] было начато в 1962 г.
В таблице (см. ниже) представлены основные справочники второго типа, содержащие
термодинамические свойства неорганических и простых органических веществ в газообразном и
конденсированном состояниях для широкого интервала температур, опубликованные после 1955 г.
Рассматриваются только оригинальные справочные издания, в которых все таблицы или, по крайней мере,
их подавляющая часть рассчитаны авторами соответствующего справочника на основании в
большей или меньшей степени критического выбора исходных данных и методов расчета. Помимо
этих справочников известен также ряд публикаций справочного характера, в которых приводятся
таблицы термодинамических функций газов, такие, как [446, 1453а, 1454, 3343, 3946] и некоторые
другие.
Сопоставляя различные справочники этого типа, следует отметить четыре издания, которые
являются результатами многолетних проектов и поэтому выделяются среди других по полноте
и объему информации. Первым из них был справочник Келли [2220], который в течение многих
лет оставался наиболее полным и совершенным изданием по термодинамическим свойствам
веществ в конденсированном состоянии; к сожалению, работа над этим проектом прекратилась
1 К началу подготовки этого издания отсутствовали международные рекомендации значений ключевых
термохимических величин.
а В ИВТАНе и ВИНИТИ в систематизированном виде хранятся тексты, в которых даны обоснования выбора
рекомендованных величин и оценки их достоверности (ом. также сноску 1).
13
Введение
Основные справочные издания, содержащие таблицы термодинамических функций неорганических
и простых органических веществ, опубликованные после 1955 г.
Справочное издание
Вещества, температурный
интервал
Примечания
1. Л. Б. Гурвич, В. С. Юнгман и
др. Термодинамические свой-
ства компонентов продуктов
сгорания. Под ред. В. П. Глушко.
1956 [87]
2. D. R. Stull, G. С. Sinke. Thermo-
dynamic Properties of the
Elements. 1956 [3564]
3. С L. Mader. Ideal Gas
Thermodynamic Properties of Detonation
Products. 1959 [2496]
4. K. K. Kelley, E. G. King.
High-Temperature Heat Content,
Heat Capacity and Entropy
Data for InorganiclCompounds.
1960 [2220]
5. Th. B. Douglas, Ch.W.Bec-
kett. Preliminary Report on the
Thermodynamic Properties of
Selected Light-Element
Compounds. 1960 [1308]
6. L. Haar et al. Ideal Gas
Thermodynamic Functions and
Isotope Exchange Functions for
the Diatomic Hydrides, Deute-
rides and Tritides. 1961 [1783]
7. Л. В. Гурвич, Г. А. Хачкуру-
зов и др. Термодинамические
свойства индивидуальных
веществ. Под ред. В. П. Глушко
и др. 1962 [88]
8. В. J. Me Bride et al.
Thermodynamic Properties to 6000 К for
210 Substances involving the
First 18 Elements. 1963 [2556]
9. R. Hultgren et al. Selected
Values of Thermodynamic
Properties of Metals and Alloys. 1963
[2040]
10. J. Hilsenrath et al. Tables of
Ideal Gas Thermodynamic
Functions for 73 Atoms and their
First and Second Ions to 10 000 K.
1964 [1946]
11. D. R. Stull et al. JANAF Ther-
mochemical Tables. 1965 [2094a]
12. H. L. Schick. Thermodynamics
of Certain Refractory
Compounds. 1966 [3331]
Соединения 21 элемента, в том
числе 29 веществ в
конденсированном состоянии и 178 газов для
температур 293,15—6000 К
92 элемента, вещества в
конденсированном и газообразном
состояниях, включая двух- и
многоатомные газы, до 3000 К
100 газов, включай 15 элементов
и их соединения, от 300 до 6000 К
92 элемента, 490 соединений в
конденсированном состоянии вплоть
до 3000 К и 400 в газообразном
состоянии, в большинстве до 2000 К
18 одноатомных газов и 51
соединение Li, Be, Mg и Al в газообразном
состоянии для температур 50—
6000 К, 28 веществ в
конденсированном состоянии, от 0 до 1500—
4000 К
111 двухатомных газов, соединения
34 элементов, от 50 до 5000 К
Соединения 31 элемента, в том
числе 45 веществ в
конденсированном состоянии и 335 газов, из
них 14—до 4000 К, 299—до 6000 К,
22—до 20 000 К
18 элементов, 192 соединения в
газообразном состоянии от 100 до
6000 К, 28 веществ в
конденсированном состоянии
66 элементов в конденсированном
и газообразном состоянии,
включая атомарные и молекулярные
газы, от 298,15 К до Ть
Приведены таблицы безразмерных
термодинамических функций до
10 000 К нейтральных и
ионизированных одноатомных газов для
всех элементов, кроме
лантаноидов, актиноидов, Fr и At
25 элементов, 254 вещества в
конденсированном состоянии, 605
газов для температур от 100 до 6000 К
Тугоплавкие соединения 35
элементов, 85 веществ в
конденсированном состоянии и 70 газов от
298,15 К до Тъ, газы до 6000 К
Рассчитаны авторами, вся
совокупность данных — система
согласованных величин
В основном рассчитаны авторами
Рассчитаны автором
В основном рассчитаны авторами,
для газов с использованием
приближенных методов, частично
заимствованы
Рассчитаны авторами
В основном рассчитаны авторами
Рассчитаны авторами, все данные
приведены с оцененными
погрешностями и являются системой
согласованных величин
Для газов рассчитаны авторами,
для веществ в конденсированном
состоянии в основном
заимствованы из литературы
Для веществ в конденсированном
состоянии рассчитаны авторами, для
газов в значительной степени
заимствованы
Рассчитаны авторами на основании
уровней энергии, рекомендованных
в [2697]
В основном рассчитаны авторами,
многие расчеты выполнены на
основании грубых оценок
Рассчитаны автором, дана оценка
точности табулированных веществ
14
Введение
Таблица (окончание)
Справочное издание
Вещества, температурный
интервал
Примечания
13. D. R. Stull et al. JANAF Ther-
mochemical Tables. 2nd Ed.
and Supplements. 1971—1975
[2095—2097]
14. H. П. Галкин и др.
Термодинамические свойства
неорганических фторидов. 1972 [66]
15. R. Hultgren, P. D. Desai et al.
Selected Values of the Thermo-
dynamic Properties of the
Elements. 1973 [2039]
16. K. S. Mills. Thermodynamic
Data for Inorganic Sulphides,
Selenides and Tellurides. 1974
[2670]
35 элементов, 328 веществ в
конденсированном состоянии, 699
газов для температур от 100 до
6000 К
88 газообразных соединений 35
элементов, от 298,15 К до 5000—
6000 К
86 элементов в конденсированном
и газообразном состоянии,
включая атомарные молекулярные газы,
от 298,15 К до fj
Соединения 81 элемента, включая
150 веществ в конденсированном
состоянии от 298,15 К до Тт и 205
веществ в газообразном состоянии
от 298,15 К до Т ^ 2000 К
Рассчитаны авторами
Рассчитаны авторами,
значительная часть на основании
приближенных оценок
Для веществ в конденсированном
состоянии рассчитаны авторами, для
газов в значительной степени
заимствованы
Рассчитаны автором, значительная
часть на основании грубых оценок
в 1960 г. Вторым является справочник «Термодинамические свойства индивидуальных веществ»,
первое издание которого было опубликовано под названием «Термодинамические свойства
компонентов продуктов сгорания». Уже в первом издании этого справочника [87] применение
модифицированных вариантов метода Гордона—Барнес и вспомогательных таблиц, рассчитанных на ЭВМ,
позволило существенно повысить точность расчетов термодинамических функций двух- и
трехатомных газов при высоких температурах по сравнению с аналогичными американскими
справочниками, опубликованными в течение следующего десятилетия. Во втором издании [88] был
резко расширен круг веществ, а для ряда газов — увеличен интервал температур. Последнее стало
возможным, благодаря разработке более строгих методов расчета, основанных на
непосредственном суммировании по уровням энергии с ограничением числа учитываемых состояний и на
использовании ЭВМ. Уже в издании 1956 г. впервые в литературе была достигнута полная внутренняя
согласованность всех рекомендуемых значений, включая согласованность термохимических
величин и термодинамических функций, что потребовало пересчета многих экспериментальных данных.
В издании 1962 г. эта обработка и согласование были повторены на существенно более широком
материале. Впервые в литературе была выполнена оценка точности всех рекомендуемых величин,
включая таблицы термодинамических функций. Фактически издание [88] удовлетворяло всем
требованиям, сформулированным выше, хотя в нем были рассмотрены только соединения 31
элемента, необходимые для расчетов высокотемпературных теплотехнических процессов.
Третий справочник «Термохимические таблицы JANAF» вобрал в себя ряд проектов,
осуществлявшихся в США для обеспечения запросов ракетно-космической техники в 1956—1965 гг.,
типа [2496, 1308, 1783, 2556], а также подготовленные в конце 50-х — начале 60-х годов, но не
опубликованные в печати материалы JANAF. В издании 1965 г. — первом издании этих таблиц —
и его дополнениях [2094а] представлены данные для большего числа веществ, чем в справочнике
[88]. Однако рекомендуемые в таблицах [2094а] данные не являются системой согласованных
величин, более того, многие из них противоречат друг другу. Таблицы термодинамических функций
газов рассчитаны приближенными методами, как правило, без учета возбужденных электронных
состояний. Для расчетов приняты не лучшие молекулярные постоянные, а многие оценки выполнены
на основании неверных исходных предположений. В расчетах ряда таблиц допущены ошибки.
Второе издание таблиц JANAF и его дополнения [2095—2097] подготовлены существенно лучше.
По количеству рассмотренных веществ, особенно в конденсированном состоянии, оно существенно
превосходит все другие справочники. Хотя, строго говоря, значения, рекомендуемые в издании
1971—1975 гг., не являются системой согласованных величин, между ними нет существенных
внутренних противоречий. Недостатком этого издания таблиц JANAF является отсутствие оценки
достоверности табулированных величин и использование недостаточно точных методов расчета
термодинамических функций одноатомных и двухатомных газов. В случае одноатомных (а в ряде
15
Введение
случаев и двухатомных) газов весьма случаен выбор числа электронных состояний, учитываемых
в расчетах. Иногда это только валентные состояния, часто к ним добавляются ридберговские
состояния, наблюдавшиеся экспериментально, а иногда и электронные состояния, не наблюдавшиеся
в спектрах и оцененные по методике, аналогичной разработанной в справочнике [88]. Все это
сказывается на неточности табулированных значений функций многих газов при высоких
температурах, особенно на значениях теплоемкости и изменения энтальпии. Для расчетов
термодинамических функций двухатомных газов в таблицах J ANAF, так же как и в других американских
справочниках [1308, 1783, 2496, 2556], использован метод Майера и Гепперт-Майер (см. сноску нас. 48).
В методе не учитывается необходимость ограничения статистических сумм по
колебательно-вращательным состояниям молекул и корректного учета особенностей вращательной структуры термов в
вырожденных и мультиплетных электронных состояниях. Из-за пренебрежения необходимостью
ограничения статистических сумм при высоких температурах термодинамические функции многих
двухатомных газов (имеющих энергии диссоциации меньше 250 кДж -моль) содержат
значительные ошибки, особенно в значениях С°р (Т) и Н° (Т) — Н° @) (см. гл. 1, рис. 1.9 и табл. 1.4).
Термодинамические функции ряда газов, молекулы которых имеют мультиплетные основные
электронные состояния, содержат ошибки в значениях Ф° (Т) и Н° (Т) — Н° @) при низких температурах,
включая 298,15 К.
Четвертый справочник, подготовленный под редакцией Халтгрина, посвящен
термодинамическим свойствам элементов в конденсированном и газообразном состояниях. В издании 1973 г.
[2039] представлены практически все химические элементы (кроме актинидов, начиная с Np). Этот
справочник отличается тщательностью подготовки данных по термодинамическим свойствам веществ
в конденсированном состоянии, включая таблицы термодинамических свойств и сопровождающие
их тексты, и в этом отношении выделяется среди других справочников. Таблицы
термодинамических функций элементов в газообразном состоянии в справочнике [2039] в значительной степени
заимствованы, однако поскольку данные приводятся только для Т ^ 3000 К, они, как правило,
достаточно достоверны.
Таблицы термодинамических функций одноатомных газов, подготовленные Хилзенратом
и др. [1946], являются наиболее полными в литературе по охвату атомов и их ионов
и интервалу температур. Авторы применили приближенный метод расчета (ограничение
статистических сумм на п=5 и пренебрежение уровнями, не наблюдавшимися в спектрах),
который позволил получить достаточно надежные данные для большинства газов в широком
интервале температур. Для атомов с низкими потенциалами ионизации при высоких температурах
точность рассчитанных данных невысока, и авторы попытались оценить соответствующие поправки.
Среди американских справочников [1308, 1783, 2496, 2556] следует отметить справочник Хаара
и др. [1783]. В этом справочнике, благодаря использованию метода непосредственного
суммирования по уровням энергии для расчета термодинамических функций при низких температурах,
приведены более точные данные при Т ^ 500 К, чем в таблицах JANAF [2095 ] для газов, молекулы
которых имеют мультиплетные основные электронные состояния.
Среди остальных справочников, представленных в таблице, следует отметить справочники Шика
[3331], Галкина и др. [66] и Миллза [2670]. Каждый из них посвящен узкому классу веществ,
как правило, только частично представленному в таких изданиях, как [88, 2095], и поэтому они
могут быть весьма полезны читателям, особенно справочник [3331 ]. Этот справочник является
единственным в литературе по термодинамическим свойствам таких тугоплавких веществ, как
окислы, карбиды, нитриды и бориды некоторых металлов в широком интервале температур, хотя
несколько случайный и далеко не полный круг рассмотренных веществ снижает его ценность.
Положительной чертой справочника [3331 ] является тщательный анализ экспериментальных данных,
использованных для расчетов таблиц термодинамических свойств веществ в конденсированном
состоянии. Следует отметить, что это один из немногих зарубежных справочников, где дана оценка
достоверности табулированных величин.
Справочники Галкина и др. [66] и Миллза [2670] также содержат данные для многих веществ,
которые не рассматриваются в других изданиях. К сожалению, подбор веществ в справочнике [66]
весьма случаен, для одних элементов есть все фториды от моно- до гексафторида, для других —
только пентафториды или трифториды, причем выбор рассматриваемых веществ не связан с их
относительной стабильностью или какими-либо другими объективными причинами. В обоих
справочниках подавляющая часть таблиц рассчитана на основании оцененных значений постоянных,
причем во многих случаях оценки выполнены недостаточно корректно и соответствующие таблицы
16
Введение
термодинамических функций содержат значительные ошибки. В справочнике [66], где приведены
таблицы термодинамических функций вплоть до 5000—6000 К, не учтены возбужденные
электронные состояния молекул рассмотренных газов, хотя ненасыщенные фториды переходных металлов
имеют низкие возбужденные состояния. В справочнике [2670] часть данных заимствована из
других справочников и работ и рекомендуемые значения не образуют систему согласованных величин.
Часть рекомендаций основана на устаревших работах и находится в противоречии с такими
справочниками, как [88, 2095]. Более подробную характеристику справочника [2670] см. в работе [76а].
Среди работ, не включенных в таблицу, следует отметить отчеты Фебера и Херрика [1453а,
1454]. В одном из них рассчитаны в интервале 100—6000 К термодинамические функции
газообразных одноатомных лантаноидов и актиноидов, в другом — 14 двухатомных газов — галоиды, га-
лоидоводороды, щелочные металлы и другие. Для двухатомных газов функции были рассчитаны
непосредственным суммированием по уровням энергии с ограничением соответствующих
статистических сумм по предельным кривым диссоциации. По строгости расчетов и точности
табулированных величин данные работы [1454] являются лучшими в зарубежной литературе, хотя
термодинамические функции тех же веществ вычислялись в ряде других справочников, таких как [2095,
2556] и др. В работе Баера и др. [446] для расчета термодинамических функций 30 газов в
интервале температур 10 — 6000 К также были использованы точные методы и тщательный отбор
молекулярных постоянных. Однако если для ряда газов в этой работе были получены достаточно точные
данные, таблицы термодинамических функций других (в том числе таких, как Na, S2, SO и т. д.)
содержат значительные ошибки, причины которых трудно установить из-за отсутствия детального
описания расчетов.
Более подробный анализ всех этих справочных данных, а также многих других компилятивных
изданий может быть найден в обзоре Бергмана и Горбова [766].
Новое издание справочника «Термодинамические свойства индивидуальных веществ»
подготовлено с учетом сформулированных выше требований для обеспечения термодинамических расчетов
самых разнообразных процессов и химических реакций, протекающих с участием соединений
многих элементов.
Построение издания. Материал изложен в четырех томах, в каждом из которых содержатся
данные о термодинамических свойствах группы элементов и их соединений. В справочнике принято
следующее распределение элементов по томам:
I том: О, Н (D, Т), F, С1, Вг, I, Не, Ne, Аг, Кг, Хе, Rn, S, N, Р;
II том: С, Si, Ge, Sn, Pb;
III том: В, Al, Ga, In, Tl, Be, Mg, Ca, Sr, Ba;
IV том: Cr, Mo, W, V, Nb, Та, Ti, Zr, Hf, Th, U, Pu, Li, Na, K, Rb, Gs.
Каждый том состоит из двух книг — текстовой и табличной. В первой книге в сжатой форме
рассмотрены вопросы, связанные с критическим выбором постоянных (молекулярных и
термохимических), принятых для расчета таблицы термодинамических свойств данного вещества в
конденсированном и газообразном состоянии, и оценкой точности этих постоянных, с расчетом
соответствующей таблицы термодинамических функций и оценкой достоверности табулированных
значений, а также дано сравнение рассчитанных термодинамических функций с известными в
литературе. Соответствующий материал сгруппирован в разделы, каждый из которых посвящен одному
веществу в конденсированном или газообразном состоянии. Каждый элемент и его соединения
рассматриваются в отдельной главе в последовательности, соответствующей рис. 1. Исключением
являются дейтерий, тритий и их соединения, выделенные в отдельную главу, а та»же шесть
благородных газов и их соединения, представленные в одной главе.
Значения постоянных, принятые на основании анализа литературы и выполненных оценок
для последующих расчетов таблиц термодинамических свойств, представлены в таблицах отдельно
для веществ, рассматриваемых в каждой главе.
В первом томе в главах 1—3 изложены методы, применявшиеся при подготовке справочника
для расчета таблиц термодинамических функций, оценки точности вычисляемых величин,
принципы выбора постоянных, необходимых для этих расчетов, и оценки постоянных в тех случаях,
когда отсутствуют соответствующие экспериментальные данные. В двух приложениях первого
тома приведены принятые значения атомных масс, фундаментальных постоянных и переводных
множителей для единиц энергии. В приложении 3 каждого тома приводятся данные, позволяющие
учитывать отклонения свойств ряда газов при повышенных давлениях от их свойств в
стандартном состоянии, а также критические параметры ряда веществ.
17
Введение-
0
н
1
в
Ц -]
1
Na
1
К
1
Rb '
1
Cs
1
Frl
Be
1 •
Mg
1
Ca
1
Sr
L j. .J
1
8a
1
- Ra
Sc
La
Ac
IT
Hf
ra
Mo
w
Mn
1c
Re
Fe
— Co[—
Ru
Ni
- Rh
OS
7hJ-[pd"l,
- Ir
Pt
Cuh
1
i
- Аи
Zn -i
1
Cd. -
1
-Hg.
Б -]
1
Al
I
ua
1
In
1
- Tl
с i
!
Si
1
Ge
1
Sn
, 1
N -I
1
p
1
As
1
Sb
1
L вь
5 -]
1
Se j
!
Те
|
L po
HeJ-
1
Ne |
1
Ar
1
Kr
1
Xe|
I
L Rn
, I
T
I
F
1
GL
1
6r
I
г
1
- At
Ce
Pr
-
-
Pm
Sm
-
Ed
ть
Ho
Er
Tm
- Lu
Th -\ Pa - V - Np - Pu - Am - Cm - Bk - Cf - Es - Fm - Md - No - 103
Рис. 1. Последовательность расположения элементов
Во второй книге каждого тома представлены таблицы термодинамических свойств всех веществ г
рассмотренных в данном томе. В таблицах приведены рассчитанные при подготовке справочника
значения С°р (Т) — теплоемкости при постоянном давлении, Ф° (Т) — приведенной энергии Гиб-
бса, S°(T) — энтропии и Н° (Т) — Н°(О) — изменения энтальпии. Для всех газов, кроме
одноатомных незаряженных газов, в таблице приведены также значения lg К°(Г) — константы
равновесия реакции атомизации. Для веществ в конденсированном состоянии в таблице приводятся
значения lg р (Т) — константы равновесия реакции сублимации или испарения с образованием
газа того же молекулярного состава, как и конденсированная фаза х.
Значения термодинамических свойств для веществ в конденсированном состоянии табулированы
для температур 100, 200, 298,15, 300 К и далее через 100 К до температуры, при которой давление
насыщенного пара достигает 100 атм, а также для температур фазовых переходов. Для газов
термодинамические свойства приводятся для температур 100, 200, 298,15, 300 К и далее через 100 К до-
6000 К; для всех одноатомных газов и ряда двух- и трехатомных газов они табулированы до-
10 000 К (выше 6000 К через 200 К), а примерно для 60 газов — до 20 000 К (выше 10 000 К —
через 500 К).
Для заряженных газов и электронного газа таблицы начинаются с 298,15 К.
Помимо табулированных значений термодинамических свойств в таблицы включены: название
вещества и его молекулярная масса, класс точности таблицы, характеризующий погрешность
величин Ф° C000 К) и^ ДГЯ° @), уравнение реакции диссоциации (сублимации), значения Д/Я°@),
AjH° B98,15 К) и ?°д, а также уравнение, аппроксимирующее термодинамические функции
данного вещества в широком интервале температур.
В дополнениях книг, содержащих таблицы термодинамических свойств, приводятся
термодинамические функции некоторых легколетучих веществ в конденсированном состоянии (Н2О, Н2О2,
1 Для некоторых веществ, разлагающихся при сублимации (испарении), приводятся константы равновесия
диссоциации на одноатомные газы.
18
Введение
NH4F и т. п.), неравновесных форм твердых веществ (алмаз, аморфный бор, стеклообразный SiO2,
красный РЬО и т. п.), а также таких газов, как пара- и ортомодификаций водорода и его изотопов
и некоторых других. Соответствующие данные приводятся для более узких интервалов температур.
В эти же дополнения включены таблицы с вириальными коэффициентами и критическими
постоянными для некоторых веществ, рассматриваемых в данном томе.
Номенклатура, термины, символы и единицы. В настоящем издании приняты за основу
номенклатура и символы, рекомендованные Международным союзом теоретической и прикладной химии
(ИЮПАК) (см. [2583а]). В отдельных случаях используются тривиальные названия химических
соединений, что не противоречит рекомендациям ИЮПАК. Все рекомендуемые величины
представлены в Международной системе единиц СИ. Значения фундаментальных физических
постоянных и ключевых термохимических величин приняты по рекомендациям КОДАТА—МСНС
[1000, 1001 ].
Нумерация глав, таблиц, рисунков, литературных ссылок. В настоящем издании принята
сплошная нумерация глав (арабскими цифрами), от главы 1 в первом томе и до последней главы
последнего тома, и аналогичная нумерация приложений. Нумерация уравнений, рисунков и
таблиц в главах и приложениях — двойная, включающая номер главы (приложения) и номер
уравнения (таблицы), рисунка в этой главе; например, уравнение A.38), табл. 7.3 и рис. 10.1 обозначают
уравнение 38 в главе 1, табл. 3 в главе 7 и рис. 1 в главе 10. Нумерация страниц и литературных
ссылок — независимая для каждого тома. Таблицы термодинамических свойств всех
рассматриваемых веществ даны в сплошной нумерации (арабскими цифрами) от первой таблицы I тома до
последней таблицы последнего тома. Таблицы в дополнениях также имеют сплошную нумерацию
по всем томам, но в этом случае номеру таблицы предшествует буква Д.
О числе знаков и точности таблиц термодинамических свойств. В таблицах термодинамических
свойств значения С°р (Г), Ф° (Г) и S° (Т) (в Дж-К^-моль-1) и значения Н° (Т) — Н° @) (в кДжХ
Хмоль'1) приводятся с тремя значащими цифрами после запятой. Число приводимых знаков не
отражает достоверность табулированных величин и, как правило, существенно превышает их точность.
Оно обусловлено необходимостью обеспечения гладкости изменения табулированных и
интерполируемых величин как функции температуры, а также строгостью согласования значений,
вычисляемых по различным термодинамическим циклам. Аналогичным образом значения термохимических
величин, вычисленные по принятым в справочнике значениям, также приводятся с тремя
значащими цифрами после запятой (в кДж-моль), хотя их погрешности могут быть на несколько
порядков больше. Этот подход призван обеспечить полную внутреннюю согласованность всех
термохимических величин и термодинамических функций.
При оценке достоверности всех принимаемых величин, в том числе молекулярных и
термохимических постоянных, а также рассчитанных термодинамических функций, ставилась задача
определения их 95%-ного доверительного интервала с учетом величины возможных систематических
ошибок.
Корректная оценка достоверности, зависящей как от воспроизводимости измерений, так и от
систематических ошибок, как правило, весьма затруднительна и приводимые погрешности в
существенной степени основаны на экспертной оценке последних. Среднеквадратичное отклонение
среднего арифметического вычислялось по формуле о = I/ - ¦ * , где ЕД| — сумма квадратов
отклонений индивидуального ?-го измерения от среднего, п — число измерений. Случайная
погрешность соответствующая 95%-иому доверительному интервалу, находилась как at, где t — фактор
Стьюдента, и складывалась с оцененной систематической ошибкой. Неточность таких оценок
погрешностей может быть значительной; в справочнике погрешности не претендуют на точность
большую, чем 100%. В связи с этим достоверность рассчитанных таблиц термодинамических
свойств охарактеризована классами точности, каждый из которых допускает погрешность
в три раза большую, чем погрешность предыдущего класса. Таблицы свойств характеризуются
по точности термодинамических функций (по погрешности в значении Ф° C000 К) и величины
Аг Н° @), использованной для расчета lg K° (T) или lg p (T).
19
Введение
Ниже приводятся соответствующие характеристики классов точности.
Класс
точности
Ф° (Т)
I
II
III
IV
V
VI
VII
Погрешности
Ф° C000 К)
Дж ¦ К 1 ¦ моль х
<о,оз
<0,1
^0,3
<1
<3
<10
>10
Класс
точности
ДГЯ° @)
А
В
С
D
Е
F
G
Погрешности
ДГЯ° @)
кДж • моль
<0,1
<0,3
<з
<10
^30
>30
Более подробные сведения о точности рассчитанных термодинамических функций (при разных
температурах) и термохимических постоянных (включая значения &М°) приведены для каждого
вещества в соответствующих текстах с обсуждением основных источников их погрешностей, а ме-
воды их оценки —в соответствующих разделах глав 1—3.
Часть 1
МЕТОДЫ РАСЧЕТА
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИНДИВИДУАЛЬНЫХ ВЕЩЕСТВ
ОБЩИЕ СООТНОШЕНИЯ ТЕРМОДИНАМИКИ
Для теплотехнических расчетов и расчетов химических процессов необходимы сведения об энтропии
S (Т) и энтальпии Н (Т) каждого вещества, принимающего участие в этих процессах. Для
расчетов процессов, в которых происходят химические превращения, необходимо также знать константы
равновесия независимых химических реакций, возможных в данной системе. Константа
равновесия К(Т) химической реакции
,vA+vA + v3-43+...^v;^+vX + v^;+..., A)
где v. — число молей вещества А(, связана со значениями энергии Гиббса для веществ,
принимающих участие в реакции A), простым соотношением
RT In К (Т) = -Afi (A (I T) = -{b'fl{A[, T) + ,'2G(A'2, T) + 4G(A'3, Г)+...]-
- KG (Av T) + v2G (A2, T) + v3G {Av T) + ...]}, B)
где ArG(A(, T) — изменение энергии Гиббса в этой реакции1, R — газовая постоянная.
Поскольку
G(T) = H(T) — TS{T), C)
константа равновесия реакции A) может быть записана как
D)
где Дг S (Т) и Дг Н (Т) — изменение энтропии и энтальпии системы при температуре Т в результате
реакции A); ArH (T) часто называют также тепловым эффектом реакции при постоянном давлении
и этой температуре.
Величину Ar H (Т) можно представить как
А,Н (Г) = АГН @) + ДДЯ (Г) - II @)] E)
и
АгН (Т) = АДB98.15К) -f К [# (Т) — Н B98.15К)], F)
поэтому можно ввести функции, получившие название приведенной энергии Гиббса:
_е(т)-н(°) =s(T))- ^>-g(°> . G)
и
Ф(Т)= G(r)-7/B98'15K) =S(T) н (т) - н B^Л5К) _ ф,т> . ДB98,15К)-Я@) ^
и записать
тпК(Т) = АгФ(Т) — АгН{0IТ (8)
и
Я1пК{Т)~АгФ!(Т)~- ДДB98,15К)/7'. (8а)
1 Под изменением какого-либо термодинамического свойства, обозначаемым символом Д, здесь и ниже во всех
случаях понимается разность между суммами соответствующих значений для продуктов реакции и для
исходных веществ.
22
Общие соотношения термодинамики
В уравнениях E)—(8а) АуН @) и Д^й" B98,15 К) — изменение энтальпии в реакции A) при
€ и 298,15 К (тепловой эффект реакции при этих температурах), [Н (Т) — Н @)] и [Н (Т) —
— 2/B98,15 К)] —изменение энтальпии г-го вещества при изменении температуры от 0 или
298,15 К до Г К.
Абсолютные значения ряда термодинамических свойств, таких как энтальпия Н (Т), энергия
Гиббса G (Т) и некоторых других, не могут быть определены ни экспериментально, ни
теоретически. Это не приводит к каким-либо осложнениям, поскольку для решения любых задач достаточно
знать изменение термодинамических свойств при изменении параметров состояния вещества
(давления или температуры) или в химических реакциях. Однако это требует определения некоторого
•состояния веществ как базового или стандартного состояния, от которого могли бы отсчитываться
изменения его свойств.
Комиссия по термодинамике Международного союза теоретической и прикладной химии
{ИЮПАК) рекомендовала в 1975 г. принять следующие определения стандартных состояний
веществ, которые в течение ряда лет уже использовались в большинстве справочных изданий
по химической термодинамике.
Стандартным состоянием для газов является состояние гипотетического идеального газа при
давлении в 1 атм A01 325 Па). Для жидкостей и твердых веществ стандартным состоянием
является состояние чистой жидкости или соответственно чистого кристаллического вещества х при
давлении в 1 атм A01 325 Па). Для веществ в растворах за стандартное состояние принято
гипотетическое состояние, при котором энтальпия одномоляльного раствора A моль вещества в 1 кг
растворителя) равнялась бы энтальпии раствора при бесконечном разбавлении. Свойства веществ
в стандартных состояниях обозначаются значком °.
В любой системе, образованной а элементами и состоящей из Ъ компонентов, возможны только
Ъ—а независимых химических реакций. Поэтому создание системы термохимических величин,
определяющих изменение энтальпии при любых химических реакциях, оказывается невозможным,
если не принять произвольные значения энтальпии для а веществ. В соответствии с практикой,
установившейся в литературе по химической термодинамике, Комиссия по термодинамике ИЮПАК
рекомендовала принять, что энтальпии образования всех элементов в их стандартных состояниях
равны нулю при любой температуре.
Экспериментальное определение термодинамических функций газов при температурах выше
1000 К — задача, практически не выполнимая в настоящее время. Поэтому вся информация о
термодинамических функциях газов в стандартном состоянии основывается на теоретических расчетах.
Как известно (см. [150, 155]), термодинамические функции вещества в идеально-газовом
состоянии, которое является стандартным состоянием газа, могут быть вычислены через
статистическую сумму по состояниям молекул (атомов) газа
»(где s. — энергия ?-го состояния молекулы, е0 — энергия ее нижнего '(основного) состоянияг
р{ — статистический вес i-ro состояния, к — постоянная Больцмана) и ее производные.
Термодинамические функции одного моля газа, состоящего из тождественных молекул и находящегося
в стандартном состоянии, связаны со значениями статистической суммы и ее производных
следующими соотношениями:
S°(T) = RlnQjP+RTdЫ?Т{Т) , A0)
н° (Г)=н° @) -f R т2 д Ы®Т{Т), (И)
A3)
где Н°@) — энтальпия газа при Т=0, N — число Авогадро.
1 Если при данной температуре вещество может иметь несколько кристаллических модификаций, за стандартное
состояние, как правило, принимается его наиболее стабильная модификация.
23
Общие соотношения термодинамики
Из соотношений A0)—A3) можно получить выражения и для других термодинамических
функций, поскольку
A5)
F°[T) = G°{T) — RT, A6)
C°v (Т) = Ср (Т) -R.
Из уравнений A0)—A7) видно, что только для таких функций, как S° (Г), Ф° (Г), С°р (Т) и
С°(Г), через статистическую сумму и ее производные могут быть найдены их абсолютные
значения. Для остальных термодинамических функций теоретические расчеты, так же как и
экспериментальные измерения, позволяют определять только разность между значениями функции при
двух температурах.
Термодинамические функции веществ в конденсированном состоянии, в отличие от газов,.
не могут быть вычислены теоретически с достаточной точностью. Поэтому расчеты таблиц,
термодинамических функций веществ в твердом и жидком состоянии проводятся на основании
III закона термодинамики и результатов экспериментальных измерений теплоемкостей этих
веществ, изменений их энтальпии при изменении температуры, а также при фазовых и полиморфных
превращениях.
Принимая, что при Г=0 теплоемкость и энтропия чистых кристаллических веществ
равны нулю, можно записать:
S°(T) = 2, \ T dT-\- > °"д ''H", A8)
fc=O
Ф° (Т) = 5° (Т) - Н° {Т) ~ Н°@), B0)
где Тк+1 и &trH° (Tk+1) — температура и изменение энтальпии для фазового или полиморфного
превращения.
В тех случаях, когда экспериментальные данные о теплоемкости вещества в некотором
интервале температур отсутствуют или отсутствует часть данных о температурах фазовых переходов,,
полиморфных превращениях или изменении энтальпии в этих переходах, расчеты таблиц
термодинамических функций в конденсированном состоянии могут проводиться на основании оцененных
величин.
Приведенные выше соотношения для термодинамических функций веществ в газообразном
и конденсированном состояниях (соотношения A0)—B0)) справедливы только для чистых
моноизотопных веществ. В действительности большинство элементов, а следовательно, и их
соединений являются смесями различных изотопов. В литературе для всех элементов и их соединений,.
кроме дейтерия и трития, принято приводить и использовать в расчетах термодинамические
свойства природных изотопических смесей. Строго говоря, это предполагает, что в выражения для.
S° (T) и Ф° (Т) должны войти слагаемые, учитывающие смешение различных изотопических
модификаций молекул данного вещества и различие их элементов симметрии (см. ниже, с. 29).
Сумма этих составляющих не зависит от температуры и не изменяется при химических реакциях,.
если они не сопровождают ядерные превращения. В тех случаях, когда спины ядер атомов,
образующих молекулу данного вещества, не равны нулю, энтропия химически чистого
кристаллического вещества, даже моноизотопного, отлична от нуля уже при температурах, близких
к Г=0, так как в нее должна войти составляющая ядерных спинов, практически не зависящая
24
Общие соотношения термодинамики
от температуры:
П
к=1
где п — число атомов, образующих молекулу вещества, г — спин ядра к-то атома (или его
изотопа).
Для одноатомных газов и газов, молекулы которых не имеют элементов симметрии,
составляющая ядерных спинов также не зависит от температуры. У молекул, обладающих плоскостями
или осями симметрии второго или более высокого порядка, статистические веса различных
состояний по-разному зависят от спинов ядер атомов, образующих молекулы. В частности, когда
спины ядер равны нулю, статистические веса части состояний тоже равны нулю. Если разность
энергии соседних состояний молекулы велика по сравнению с кТ, составляющая ядерных спинов,
строго говоря, зависит от температуры. Однако при Т ^ 100 К для всех газов, кроме Н2, D2
и Т2, составляющая ядерных спинов не зависит от температуры.
Поскольку составляющие, обусловленные ядерными спинами и изотопическим составом
элементов, сохраняются постоянными в любых химических реакциях и в большинстве случаев не
зависят от температуры, в химической термодинамике стало общепринятым использовать
значения термодинамических функций без учета ядерной составляющей. Соответствующие значения:
получили название виртуальных или практических термодинамических функций в отличие от
полных термодинамических функций веществ, в которые включается ядерная составляющая. В
дальнейшем в настоящем издании везде, где это специально не оговорено, под термодинамическими
функциями чистых веществ подразумеваются практические значения термодинамических функций'
их природных изотопических смесей. Полные термодинамические функции отличаются от
практических на величину ядерной составляющей. Ядерные составляющие не зависят от температуры,,
тождественны в значениях Ф° (Г) и S° (T) и могут быть вычислены по соотношению
Six = ф1д = R 2 nJ Г2 *,* to B*,* + 1) - 2 xjk In x,k] , B2>
где nj — число атомов элемента / в молекуле данного вещества, Xjk и ijk — мольная доля и ядерный
спин k-то изотопа /-го элемента. Суммирование в B2) ведется по всем элементам, образующим^
молекулы вещества, и их изотопам.
Ядерные составляющие в значениях С° (Т), Н°(Т)—Н°@) и т. п. равны нулю, а в значениях
G° (T) равны TS°n.
Очевидно, что практические значения термодинамических функций не пригодны для расчета
процессов, связанных с изотопическим обменом или процессов, сопровождающих ядерные
реакции.
Как отмечалось, теоретические расчеты и экспериментальные измерения не позволяют
определять абсолютные значения энтальпии, так как остается неизвестной энтальпия веществ при
Т—0 или какой-то другой температуре, которая могла бы быть принята за начало отсчета. В
теплотехнической литературе вместо абсолютной энтальпии Н° (Т), определение которой невозможно,
широко используется функция, получившая название полной энтальпии 1° (Т) г. Величина 1° (Т)
равна энтальпии образования данного вещества при температуре Т из элементов в стандартных
состояниях при температуре То
I°(T) = AfH° (То) + Н° (Т) - Н° (То), B3)
где
Н° (Т) — Н° (То) = [Н° (Т) — Н° @)] — [В0 (Г,,) — Н° @)] B4)'
и kjH° (То) — энтальпия образования вещества при То К.
За начало отсчета полных энтальпий обычно принимается 0, 293,15 или 298,15 К. Введение
полной энтальпии вещества позволяет получить аналогичные выражения для энергии Гиббса
1 Применение термина «полная энтальпия» не может привести к недоразумениям, поскольку ядерная составляющая.
в энтальпии равна нулю.
25
Общие соотношения термодинамики
и других функций, абсолютные значения которых не могут быть определены. Для определения
полной энтальпии 1° (Т) и констант равновесия химических реакций К° (Т), кроме значений
термодинамических функций S° (Т), Ф° (Т) и Н° (Т)—Н° @), необходимы сведения об энтальпиях
образования веществ из элементов в их стандартных состояниях А^Н° (Т) и изменении энтальпии
АуН° @) в реакциях, для которых вычисляются константы равновесия. Значения Afl° @) могут
•быть вычислены, если известны энтальпии образования из элементов в стандартных состояниях
всех веществ, участвующих в данной реакции, по соотношению
ДД°(Г) = Д[ДД°(Л,., Т)] =
= v;A/ff0D;,r) + vJA/fl°D;, T)+...-v1A/H°(A1, T)-^fH°{A2, T)-... B5)
Энтальпия реакции при температуре Тг связана с энтальпией реакции при некоторой
температуре Тх известным соотношением
ДД° (Г2) = АГН° (Гх) + Дг [Н° (Г2) - Н° (Г,)], B6)
где АГ[Н° (Т%)—Н° G\)] — разность изменения энтальпии от температуры Тг до температуры Тг
для продуктов реакции и исходных веществ х.
Для расчета равновесных составов сложных смесей, устанавливающихся в системе в
результате того или иного процесса, необходимы сведения о константах равновесия всех независимых
реакций, возможных в данной системе. Число таких реакций равно числу компонентов, входящих
в систему, минус число элементов, образующих эти вещества (и электрон, если в реакции
участвуют ионизованные газы). При составлении таблиц термодинамических свойств для большого
круга веществ целесообразно вычислять константы равновесия всех соединений для одного из двух
типов реакции: для реакций образования этих веществ из элементов в стандартных состояниях
или для реакции их диссоциации на атомы. В первом варианте существенным преимуществом
является то, что изменение энтальпии в соответствующей реакции тождественно энтальпии
образования вещества из элементов в стандартном состоянии и не зависит от данных для других веществ.
Однако использование констант равновесия такого типа в высокотемпературных расчетах, когда
элементы находятся в состояниях, отличных от их стандартного состояния при 0 и 298,15 К,
требует дополнительных данных о фазовых переходах и давлении насыщенных паров
элементов.
Представление в таблицах термодинамических свойств констант равновесия реакции
диссоциации на одноатомные газы свободно от этих недостатков, однако определение величины Дг#
для реакции диссоциации требует сведений об энтальпиях образования всех одноатомных
газов.
В настоящем издании отдано предпочтение второму варианту 2 и для всех нейтральных двух-
и многоатомных газов в таблицах приводятся константы равновесия их реакции диссоциации
на атомы, а для заряженных газов — константы равновесия реакции диссоциации на атомы
и электрон типа
B7)
1 В отечественной учебной и технической литературе, отдавая дань традиции, обычно используется соотношение
г,
ДГЯ° (Г,) = ДГЯ° G\) + j АгС°р (Т) dT. B6а)
г.
Использование уравнения B6) при наличии в современной справочной литературе табулированных значений
Н°(Т) — #°@) или Н°(Т) — Я°B98 К) безусловно предпочтительнее, так как в расчетах по соотношению
B6а), как правило, всегда приходится использовать интегрирование приближенных выражений для зависимости
С°р от Т или принимать С° (T)=const.
2 Следует отметить, что программы для расчетов на ЭВМ составов и термодинамических свойств сложных
реагирующих смесей, созданные в Советском Союзе, также базируются на константах равновесия реакций атомиза-
ции газов и реакций сублимации (испарения) веществ в конденсированном состоянии в соответствии с
рекомендациями предыдущих изданий настоящего справочника.
26
Общие соотношения термодинамики
Таким образом, константа равновесия реакции диссоциации любого г-го газа, образованного
из j элементов, описывается уравнением
Ко(Т) = р?Лру1Р(, B8)
j
где ctj — число атомов j-го элемента в ?-м соединении, г=0 для нейтральных газов, г=+1 для
положительных ионов и г=—1 для отрицательных ионов, р( — парциальное давление данного
газа, Pj — парциальное давление у-го элемента в виде одноатомного газа, ре — парциальное
давление электронного газа.1
Значения логарифмов констант равновесия вычислялись по соотношению
RhxK°{i, Т) = АГФ°(Т) — ДгЯ°@)/7\ B9)
где ДГФ° (Т) — изменение приведенной энергии Гиббса в данной реакции, &ГН° @) — изменение
энтальпии в этой реакции.
Для веществ в конденсированном состоянии в таблицах термодинамических свойств
приводятся данные для констант равновесия реакции сублимации или испарения с образованием газа
того же молекулярного состава, как конденсированная фаза. В последнем случае
RlnK°(i, T) = Rlnp(i, Т) = Ф°(и г, Т) — Ф°(i, к, Г) —Д,Я°@)/7\ C0)
где Ф° (i, г, Т) и Ф° (г, к, Т) — приведенные энергии Гиббса для ?-го вещества в газообразном
и конденсированном состоянии, А8Й"° @) — энтальпия реакции сублимации при Г=0. Для
некоторых веществ в конденсированном состоянии испаряющихся с разложением, в таблицах
термодинамических свойств приводится константа равновесия реакции диссоциации на одноатомные
газы. Соответствующие значения вычислялись по уравнению B9).
Ниже, в главах 1—3 изложены конкретные методы, применявшиеся в настоящем издании для
расчетов рассмотренных термодинамических свойств веществ в стандартном состоянии.
В уравнении B8) величины р являются безразмерными, так как под ними подразумеваются отношения
парциальных давлений к стандартному давлению р°, равному 1 атм или 101325 Па.
Глава 1
ВЫЧИСЛЕНИЕ ТЕРМОДИНАМИЧЕСКИХ ФУНКЦИЙ ГАЗОВ
1.1. Разделение статистической суммы.
Вычисление поступательных составляющих термодинамических функций газов
Энергия любой частицы газа (атома или молекулы) может быть представлена как сумма двух
независимых частей, одна из которых связана с поступательным движением частицы, а другая —
с ее внутренними (внутримолекулярными) движениями:
Это позволяет записать статистическую сумму по состояниям молекулы (атома) как произведение
суммы по состояниям поступательного движения и суммы по внутренним состояниям
Q(T) = Qmet(T)Qm(T) A.2)
и термодинамические функции газа как суммы двух составляющих:
Н° (Т) - Н° @) = [Н° (Т) - Н° @)]пост + [Н° (Т) - Н° @)]вн, A.5)
с; (Т)=[с; (Г)]поот+[с; (Т)]зв. (i. 6)
Вычисление статистической суммы по состояниям поступательного движения <?Пост не встречает
затруднений; вывод соответствующего уравнения
где М — молекулярный вес газа, р — давление газа, N, h, Аи R — фундаментальные постоянные,
приводится в любом курсе по статистической механике (см., например, [150, с. 147], а также [155,
с. 128]). На основании численных значений фундаментальных постоянных, принятых в настоящем
издании (см. Приложение 2), поступательные составляющие в значениях Ф° (Т), Н° (Т)—Н° @)
и С°р (Т) газа в стандартном состоянии могут быть вычислены по соотношениям1:
Фпост (Т) _ 2<5 in т -f 1,5 In M — 3,664 87, A.9)
[Н° (Т) - н° @)]дост_ \Ср (Л],оеТ _ 2 . ,, 1т
ш — g _ad. A.Щ
1.2. Особенности вычисления внутримолекулярных составляющих
термодинамических функций газов
Уравнения для расчета внутримолекулярных составляющих термодинамических функций газов
через статистическую сумму по состояниям, и ее производные могут быть записаны как
%*P-=biQa, A.11)
1 Согласно уравнениям A0)—A2)
S°(T) _Ф°(Т) Я°(Г)-Д°@)
R ~ R ' RT A-°>
поэтому в настоящем разделе приводятся уравнения для расчета безразмерных значений трех термодинамических
функций: Фа(ТIН, [H°(T)-H°@)]/RT и C°p (T)/R.
28
4.2. Внутримолекулярные составляющие
5 —5T~fc;« AЛЗ)
imax
«=0
• max
(^0(^) AЛ5)
<=0
Здесь
Ei = (&i — su)jhc A.17)
— разность энергии г-го и нижнего (основного) состояний молекул (атомов) газа (в см), р( —
статистический вес ?-го состояния, hclk—c2 — вторая радиационная постоянная (см.
Приложение 2).
В общем случае статистическая сумма по внутримолекулярным состояниям является суммой
по всем связанным электронным, колебательным и вращательным состояниям молекулы (атома),
а также состояниям, обусловленным ориентацией спинов ядер атомов, образующих молекулы газа.
Состояния атомов и молекул, отличающиеся различными ориентациями спинов ядер, обладают
практически одинаковыми энергиями, а спины ядер остаются неизменными во всех случаях, когда
отсутствуют ядерные реакции. Поэтому для атомов и молекул, не обладающих плоскостями или
осями симметрии второго или более высокого порядка, составляющая в статистической сумме,
обусловленная спинами ядер, не зависит от температуры и является дополнительным
статистическим весом для состояний, связанных с другими внутримолекулярными движениями. У молекул,
обладающих плоскостями симметрии или осями симметрии второго или более высокого порядка,
статистические веса вращательных состояний зависят от спинов ядер атомов, образующих
молекулу (см. [155, с. 135] и [149, § 63, 86, 104]). Если разность энергии соседних вращательных уров-
яей симметричной молекулы велика по сравнению с кТ, это оказывает достаточно сложное влияние
на величину статистической суммы по вращательным состояниям. Для всех молекул, кроме
молекулы водорода, ее изотопных модификаций, а также некоторых соединений других элементов
с водородом, например СН4, разность между энергиями соседних вращательных состояний
существенно меньше величины кТ уже при 100 К. Даже для Н3 при температурах выше 500 К
составляющую ядерных спинов можно исключить из значений термодинамических функций, так как
ошибки из-за пренебрежения различием в статистических весах состояний с четными и нечетными
значениями вращательного квантового числа достаточно малы.
В уравнениях A0)—A3), связывающих термодинамические функции газов со статистической
суммой и ее производными, предполагается, что молекулы данного газа тождественны, в частности
состоят из одних и тех же изотопов элементов, образующих эти молекулы. Большинство элементов
имеет несколько стабильных изотопов (см. Приложение 1). Содержание различных стабильных
изотопов одного элемента практически постоянно, и газы, состоящие из молекул, образованных
элементами, имеющими несколько изотопов, имеют практически постоянный состав. Различные
изотопические модификации одной и той же молекулы имеют разный молекулярный вес,
отличающиеся системы уровней энергии и т. п. В настоящем издании вычисляются и табулируются
значения практических термодинамических функций для природной изотопической смеси каждого
рассматриваемого вещества. Для определения практических термодинамических функций газа,
являющегося природной смесью изотопных молекул, необходимо вычислить в отдельности
термодинамические функции каждого газа, состоящего из тождественных молекул, умножить
вычисленные значения на соответствующие молярные доли (пропорциональные процентному содержанию
29
Глава 1. Вычисление термодинамических функций газов
изотопных молекул) и сложить полученные величины г. Описанный путь громоздок и неудобен
при вычислениях таблиц термодинамических свойств. Однако можно прибегнуть к некоторым
упрощениям, которые позволяют свести вычисления к более простым расчетам, аналогичным темг
которые применяются для газов, состоящих из тождественных молекул (см. [88, с. 127]), если
принять, что природная смесь изотопов состоит из молекул, образованных элементами с атомными
весами, соответствующими природной смеси изотопов каждого элемента. В этом
приближении практические термодинамические функции такого газа, как, например, треххлористый бор
в стандартном состоянии, тождественны термодинамическим функциям гипотетического
моноизотопного газа, состоящего из молекул 10'81В 35'453С13, имеющих молекулярную массу 117,169
и уровни энергии гипотетической молекулы с такой массой. Это упрощение не является строгим,
однако в тех случаях, когда атомные массы разных изотопов данного элемента близки между
собой (т. е. для всех элементов, кроме самых легких), а также когда в природной смеси преобладает
один изотоп (т. е. для всех легких элементов), это приближение хорошо выполняется и не приводит
к ошибкам в вычисляемых значениях Ф°(Т) и S°(T), превышающим 0,05 Дж'К~1-моль~1.
Основные сложности расчетов внутримолекулярных составляющих термодинамических
функций газов на протяжении тридцати лет, прошедших с момента создания статистических методов
расчета в начале 30-х и до начала 60-х годов, были связаны с большой трудоемкостью достаточно
корректного расчета статистической суммы и ее производных, в частности для двух- и
многоатомных газов. В связи с этим были развиты различные приближенные методы расчета, позволившие
получать таблицы термодинамических функций многих десятков газов без вычисления значений
(?bhi Qbs и <?вн непосредственным суммированием по уровням энергии по соотношениям A.14) —
A.16). Точность этих методов удовлетворительна при умеренных температурах, когда в
статистической сумме и ее производных должны приниматься во внимание только низкие электронно-
колебательно-вращательные уровни энергии, как правило, только уровни с невысокими
значениями колебательных и вращательных квантовых чисел основного электронного состояния молекул
газа. В литературе по существу отсутствовал критический анализ достоинств и недостатков
разных вариантов используемых приближенных методов расчета вплоть до публикации в 1962 г.
II издания справочника «Термодинамические свойства индивидуальных веществ» [88]. Основные
результаты этого анализа были воспроизведены позднее в книге Н. А. Смирновой [239].
Развитие электронно-вычислительной техники по существу сняло ограничения, обусловленные
трудоемкостью расчетов статистической суммы и ее производных суммированием по уровням
энергии. Это позволило перейти к применению ЭВМ для расчетов термодинамических функций газов
в широком интервале температур с использованием непосредственного суммирования по уровням
энергии уже в конце 50-х—начале 60-х годов, когда подготавливалось предыдущее издание
настоящего справочника (см. [83, 86, 81, 84, 84а]). Однако при этом возникли другие проблемы,
ограничивающие точность вычисления термодинамических функций газов, особенно при высоких
температурах. Они обусловлены тем, что для корректного расчета термодинамических функций
газов в статистической сумме должны учитываться все связанные состояния молекул газа, дающие
заметный вклад при температуре, для которой выполняется расчет. Экспериментальные
исследования строения и спектров молекул, как правило, дают информацию только о небольшом числе
электронно-колебательно-вращательных состояний двух- и многоатомных молекул и электронных
состояний атомов и одноатомных ионов, главным образом, о состояниях с относительно низкими
энергиями возбуждения. При интерпретации соответствующих молекулярных спектров
экспериментаторы обычно определяют коэффициенты в некоторых уравнениях, удовлетворительно
аппроксимирующих низкие колебательно-вращательные уровни энергии молекул. Использование этих
данных (молекулярных постоянных) для описания существенно более высоких уровней может
приводить и, как правило, приводит к грубым ошибкам. Так, если нижние колебательные уровни
молекул могут рассматриваться как уровни гармонического осциллятора, то это приближение
полностью непригодно для высоких уровней, так как число уровней гармонического осциллятора
бесконечно и с ростом колебательного квантового числа его энергия стремится к бесконечности,
а число колебательных уровней молекулы конечно, и их энергия не может существенно превышать
энергию диссоциации или энергию разрыва химической связи молекулы. Таким образом, с раз-
Энтропия смешения изотопов, как и другие слагаемые ядерной составляющей, не входит в практические значения
термодинамических функций.
30
1.3. Одноатомные газы
витием вычислительной техники основным моментом, лимитирующим точность расчетов
термодинамических функций газов, особенно при высоких температурах, стала неполнота современных
данных об энергетических состояниях атомов и молекул, в том числе даже таких, которые
казались хорошо изученными. Методы, применявшиеся в настоящем издании для преодоления этих
трудностей и расчетов внутримолекулярных составляющих термодинамических функций газов,
а также оценки точности этих расчетов, изложены ниже в разделах 1.3—1.5 для одноатомных,,
двухатомных и многоатомных газов соответственно.
1.3. Вычисление внутримолекулярных составляющих
термодинамических функций одноатомных газов
Статистическая сумма по внутримолекулярным состояниям атомов и одноатомных ионов является
суммой по их электронным состояниям:
«шах
() AЛ8)
где Tt — энергия г-го электронного состояния атома (в см), отсчитанная от его основного
состояния. Для ?-го электронного состояния, характеризуемого квантовыми числами суммарного
орбитального момента L, суммарного спинового момента S и полного момента количества движения /,
статистический вес (без учета спина ядра) равен
Pi = BS + i)BL + l). A.19).
Поскольку разность энергии отдельных компонент состояния 2S+1L, характеризуемых разными
значениями квантового числа /, может быть достаточно велика, суммирование в A.18) в ряде
случаев следует проводить по отдельным компонентам таких состояний. Статистический вес каждой
компоненты равен в этом случае
А = 2/ + 1, A.20).
где / принимает значения \L-\-S\, \L-\-S—1[, . . ., \L—S\.
Значения энергии г-го состояния, определяемые из анализа спектров, табулированы для многих
атомов и одноатомных ионов в справочнике Мур [2697], а также в оригинальных работах.
Электронные состояния атома (иона) целесообразно разделить на валентные и ридберговские-
состояния. При этом под валентными состояниями понимаются состояния, соответствующие тем
конфигурациям электронной оболочки, в которых главные квантовые числа п всех электронов не
превышают значения п валентных электронов в основном состоянии атома. Под ридберговскими
состояниями в соответствии с принятой терминологией понимаются состояния, соответствующие
таким конфигурациям электронной оболочки, в которых по крайней мере у одного электрона
главное квантовое число п больше, чем у валентных электронов в основном состоянии атома. Как
известно, энергии ридберговских состояний с удовлетворительной точностью могут быть
представлены соотношением
Tt = RZi\-t * Yi -, л „ I, A. 21)
i(nki+^ki) (ПкО — &к0Г J Ч
где R — постоянная Ридберга, Z — заряд ядра атома, пы — главное квантовое число к-то
электрона в г-м состоянии, Ак{ — квантовый дефект, являющийся функцией квантовых чисел п и I,
в s-м состоянии.
Трудности расчета статистической суммы по электронным состояниям атома обусловлены тем,
что при refc—> оо число состояний атома становится бесконечно большим, а их энергии не
превышают потенциал ионизации данной серии термов, т. е. имеют конечную величину. Это приводит
к расходимости статистической суммы атома и ее производных. Расходимость статистической суммы
при невысоких температурах, когда Т <^ 0,05 10, где /0 — потенциал ионизации атома (иона),
имеет только принципиальное значение, так как при этом вклады отдельных состояний быстро
убывают по мере увеличения энергии этих состояний и суммирование прекращается задолго до того,
как вклады состояний начинают возрастать вновь из-за увеличивающегося статистического веса;
при высоких температурах, когда Т > 0,1 /0, ситуация совершенно отлична и значения Qai, ее
31
Глава 1. Вычисление термодинамических функций газов
S(T), Дж-К'1- г
-'¦>.-!
Рис. 1.1. Зависимость
удельной энтропии смеси
»<Оа, OJ, О, О+, е) и
индивидуальных компонент О
и О2 при 20 000 К от
главного квантового числа п
1 — О2; 2 — 0; 3 — смесь
1>°G\кДж-г~'
160
20
Рис. 1.2. Зависимость
удельной] нолной энтальпии Ю
смеси (О2, Of, О, О+, е) и
индивидуальных
компонент О и О2 при 20 000 К
от главного квантового
•числа п
1 — Ог; 2 — О; 3 — смесь
i i i i i i i i i i
в 10 1Z п
Т ! I I I I I I I ' I I I I
20 z
8 10 11П
производных и термодинамических функций существенно зависят от того, как ограничивается
верхний предел суммы в A.18). На рис. 1.1 и 1.2 приведены изменения удельной энтропии s° (T) и
удельной полной энтальпии i° (T) одноатомного кислорода в зависимости от того, на каком
значении главного квантового числа п валентного электрона ограничена статистическая сумма по
электронным состояниям атома О и ее производная. Из данных, приведенных на рисунках, видно, что
при 20 000 К значения этих функций быстро возрастают с увеличением верхнего предела в
статистической сумме и ее производной от п=2 до ге=12.
Проблема расходимости статистической суммы атомов рассматривалась в десятках работ,
начиная с работ Планка [3037] и Ферми [1468], и были сформулированы различные подходы для
ограничения статистической суммы по электронным состояниям атомов. Предложенные методы
основывались на представлениях об атомах, как о жестких сферах, имеющих ограниченный объем,
который зависит от температуры и давления газа (Ферми [1468], Бете [627], Гурвич и Квли-
видзе [83]), на образовании континуума при энергиях, меньших потенциала ионизации атома
за счет слияния отдельных уровней атома из-за их конечной ширины в результате малого времени
жизни (Бауманн [520]) или штарковского уширения (Инглис, Теллер [2065]), за счет снижения
потенциала ионизации атомов в плазме (Еккер, Вейцель [1377], Фаулер, Гугенхеймер [267], Грим
[1729], Мак-Чесни [2565]), на совместном рассмотрении связанных и несвязанных состояний атома
(Планк [3037], Ларкин [1506]). Обзор этих методов и сравнение их относительных достоинств
может быть найден в работе Фуше, Бароннета и Байарца [1445]. Однако все эти методы, кроме
связанных с ограничением статистической суммы на состояниях, энергия которых на кТ меньше
потенциала ионизации атома [101а, 1376], основаны на учете взаимодействия частиц газа (плазмы)
и, строго говоря, не применимы для вещества в идеально-газовом состоянии. Что касается наиболее
корректных методов, развитых в работах Планка и Ларкина, то полученные в них соотношения
определены только для водородоподобных атомов и должны быть обобщены для многоэлектронных
-атомов, а также молекул.
Следует отметить, что проблема ограничения электронной статистической суммы атомов при
подготовке справочных изданий не может рассматриваться изолированно от аналогичной
проблемы, возникающей при расчете статистической суммы по электронным состояниям молекул. Если
в статистической сумме атома учитываются ридберговские состояния и используется некоторый
метод для ограничения их числа, то аналогичные состояния должны учитываться при расчете
статистической суммы молекул с использованием сходного метода ограничения суммы. В литературе
этот подход до сих пор не развивался, а проблема ограничения статистической суммы по
электронным состояниям молекулы даже не обсуждалась. В то же время, если ридберговские состояния учи-
32
13. Одноатомные газы
тываются в статистических суммах атомов и не учитываются в статистических суммах молекул,
это автоматически вносит ошибки в значения констант равновесия, вычисляемые через
термодинамические функции, поскольку согласно уравнению (8)
\Q (/, Т)
л (I, у;_ехр^ R RT j— Q f exp^ ш j
~~ QnooAu T) QBS(i, т)
где Q (j, T) — статистическая сумма по состояниям /-го атома при Т (в К), Q B, T) —
статистическая сумма по состояниям 2-й молекулы, Cj{ — число атомов /-го элемента в 2-й молекуле.
Учет ридберговских электронных состояний в статистических суммах двухатомных и особенно
многоатомных молекул фактически невозможен, поскольку для большинства молекул данные о
таких состояниях отсутствуют. Для атомов и одноатомных ионов экспериментальные данные о
значительной части ридберговских состояний, особенно возникающих при возбуждении одного из
электронов в состояния с большими значениями орбитального квантового числа I, также
отсутствуют. Тем не менее для большинства атомов число таких состояний может быть установлено на
основании конфигурации электронной оболочки атома, а приближенные значения энергий таких
состояний могут быть оценены с достаточной точностью (см. [83; 88, с. 37]). Однако для молекул,
в том числе простейших двухатомных молекул, даже определение числа связанных ридберговских
состояний затруднительно.
Поэтому в настоящем издании в отличие от предыдущего при вычислении термодинамических
функций одноатомных газов в статистической сумме по электронным состояниям и ее производных
учитываются только валентные состояния атомов (ионов). В соответствующих главах в таблицах
«Уровни энергии атомов. . .» приводятся данные об уровнях энергии каждого атома (иона),
конфигурации электронной оболочки, соответствующей этому состоянию, а также значения р{ и Т{
для этих уровней. Внутримолекулярные составляющие термодинамических функций одноатомных
газов вычислялись на основании этих данных по соотношениям:
[H°(T)-H°@)U_Qal
A.23)
A.25)
где
•'max
A.26)
*шах
г max
Величина погрешности, вносимая пренебрежением ридберговскими состояниями, оценивалась
в каждом расчете в предположении, что более корректное выражение для статистической суммы по
электронным состояниям атома может быть представлено уравнением [58а]
Qtl = ехр (- # /о) 2 2 Р<ъ {eXp["fr(/* ~ Г^] -i—wV*- T**)} > (*• 29)
*=0 ik
33
Глава 1. Вычисление термодинамических функций газов
где /0 — потенциал ионизации атома, к — номер ридберговской серии, Г** — энергия i-то уровня
й-й серии, 1к — ионизационный предел к-й серии. Уравнение A. 29) является обобщением формулы
Планка на случай многоэлектронных атомов.
В табл. 1.1 приведены для примера значения С°р (Т), Ф° (Т) и S0 (Г) для О (г) и Хе (г),
вычисленные для Г=5000, 10 000, 15 000 и 20 000 К с учетом только валентных состояний по уравнениям
A. 26)—A. 28) и с учетом ридберговских состояний по уравнениям типа A.29). Как видно из таблицы,
при низких температурах пренебрежение ридберговскими состояниями практически не влияет на
точность расчета термодинамических функций атомарных газов. Для кислорода, как и для других
атомов с заполненными d- и /-электронными оболочками, учет ридберговских состояний с ростом
температуры приводит к увеличению термодинамических функций. Особенностью атомов с
незаполненными d- и /-электронными оболочками (в том числе атом Хе) является наличие большого
числа валентных электронных состояний, образующихся при переходе электрона с ns-орбитали на
(п—1) d- или (п—2) /-орбиталь. При расчете Qa3, (?эл и EЭЛ по уравнениям A. 26) — A. 28) вклад этих
состояний монотонно возрастает с ростом температуры, а при расчете по уравнению A. 29)
проходит через максимум. Поэтому при высоких температурах расчет (II) приводит к более низким
значениям термодинамических функций, чем расчет (I).
Следует отметить, что зависимость термодинамических функций газов от числа ридберговских
состояний, учитываемых в QBJI, подобная приведенной на рис. 1.1 и 1.2 для кислорода, характерна
только для индивидуальных веществ. В действительности, при температурах, когда этот эффект
начинает сказываться, в результате диссоциации и ионизации исходного индивидуального вещества
возникает сложная многокомпонентная смесь. Как было показано при подготовке настоящего
издания [108, с. 30] (см. также [8076]), свойства реагирующих смесей мало зависят от числа
ридберговских электронных состояний, учитываемых в статистических суммах компонентов смеси, если
для всех компонентов используется одинаковый принцип ограничения статистических сумм.
Таблица 1.1
Г, К
5 000
10 000
15 000
20 000
5 000
10 000
15 000
20 000
Примечание. I
по уравнениям
Результаты расчета значений Ср(Т), Ф
ир У1 1
I
21,799
23,137
22,933
22,500
20,786
21,655
44,826
1 [87,314
II
21,611
23,434
25,474
27,806
20,786
22,185
34,555
41,144
I
О
199,279
214,201
223,178
229,616
Хе
207,397
221,810
230,592
238,804
— учтены только валентные состояния; II -
типа A. 29).
0 (Т) и S° (T) для
Ф°(Т)
II
О
199,279
214,201
223,228
229,866
207,398
221,805
230,586
237,804
- статистическая сумма
(г) и Хе (г) (
S
I
220,468
236,132
245,496
252,034
228,183
242,666
254,345
273,800
в Дж- К • моль >
о{т)
II
220,465
236,158
245,904
253,477
228,181
242,723
253,706
264,998
и ее производные вычислены
Таблица 1.2. Удельные термодинамические свойства кислорода при Я7 = 20000 К (в Дж• К • моль)
Условия
ограничения Qgjs
Поступательная со-
Основное состояние
Ищах —2
3
8
12
s°(O)
14,422
15,259
15,740
15,820
16,226
16,882
*°(О2)
7,481
11,021
11,226
11,280
11,761
12,297
8°(смесь)
23,129
22,983
22,853
22,850
22,848
22,845
*°(О)
41,409
41,409
43,254
44,639
51,798
63,270
ИО2)
12,991
21,648
23,058
23,983
32,154
40,750
г°(смесь)
155,548
152,395
149,465
149,385
149,280
149,084
34
1.4. Двухатомные газы
На рис. 1.1 и 1.2 и в табл. 1.2 наряду с данными об удельной энтропии и удельной полной
энтальпии О и О2, приведены аналогичные свойства равновесной смеси, состоящей из О, О+, О2, 0^ и е,
вычисленные при ограничении суммирования по электронным состояниям атомов и молекул на
состояниях с разным максимальным значением главного квантового числа п. Из приведенных
данных видно, что удельные термодинамические свойства смеси практически не зависят от числа
учитываемых ридберговских состояний даже при 20 000 К, хотя свойства О и О2 очень чувствительны
к этому фактору. В то же время термодинамические свойства смеси, так же как и свойства
индивидуальных компонентов, очень существенно зависят от того, учитываются- или нет в
статистических суммах и их производных валентные состояния атомов и ионов.
1.4. Вычисление внутримолекулярных составляющих
термодинамических функций двухатомных газов
Статистическая сумма по внутримолекулярным состояниям двухатомных молекул является суммой
по их электронным, колебательным и вращательным состояниям. Число колебательных и
вращательных состояний в каждом связанном электронном состоянии обычно так велико, что их энергии
задаются не численными значениями, а аппроксимируются полиномами как функции
колебательного квантового числа v и квантового числа полного момента количества движения молекулы /
(без учета спинов ядер атомов, образующих молекулу). В общем виде статистическая сумма может
быть записана как
»max f max
&» = 2 2 2 HU ^)exp{-^l^' + G"-'H + ^)W-GW@)-^)(/mjn)]}, A.30)
r=X i'=:0 «/mill
где Т\° — энергия г-ro электронного состояния, равная разности энергии минимумов
потенциальных кривых в г-м и основном состоянии молекулы; GU) (v) —уравнение, описывающее
колебательную энергию молекулы в г-м электронном состоянии как функцию квантового числа и, где v=0,
1, 2,. . ., v1^; Flv° (J) — уравнение, описывающее вращательную энергию молекулы в г-м
электронном и у-м колебательном состояниях как функцию квантовых чисел v ж J, где / принимает
ряд целых или полуцелых значений от /^ до /ш'ах,#> ^ Ф) — колебательная энергия молекулы
в ее основном состоянии при v=0; F^~> (JMn) — вращательная энергия в основном электронном
состоянии при v=0, J=Jmi,i; p (г, J) —статистический вес /-го вращательного уровня в i-м элек"
тронном состоянии, равный (без учета спинов ядер) B/+1) с, где с — целое число, зависящее от
типа данного электронного состояния молекулы; X — символ основного состояния молекулы.
Можно ввести обозначения
/max, с
{4H} A.31)
где Qm\ — статистическая сумма по вращательным состояниям г-го электронного и v-то
колебательного состояний и
fmax «/max, p
«а.„=2 2 B/+1)cU)exp{-if[G(<)(y)+^)(;)-G(<)(°)-i;'oi')(^iI,)]}, A.32)
где Q^l вр — статистическая сумма по колебательно-вращательным состояниям г-го электронного
состояния.
Тогда
«max
Q»= 2 #SU
где Et — разность энергии между уровнями i;=0, J—J^ln B J~M и основном электронных
состояниях.
Выражение для Ff (J) существенно зависит от характера взаимодействия механического
момента количества движения молекулы R, орбитального момента ее электронов L и суммарного
35
Глава 1. Вычисление термодинамических функций газов
момента электронных спинов S или взаимодействия момента R с проекциями двух последних
моментов на межъядерную ось (Ли 2). Систематика электронных состояний двухатомных молекул
(см. [71, 1906]) основана на значениях квантовых чисел Л, 2 и Q, где 2=5, S — 1, . . ., —S и
Q= j Л.+ 21. Состояния со значениями Л=0, 1, 2, 3, ... обозначаются как состояния 2, П, Д, Ф
и т. д. При Л>0 вращательные уровни с данным значением квантового числа R расщепляются на
2|компоненты(Л-удвоение); величина этого расщепления мала во всех случаях. Поэтому в расчетах
статистических сумм и термодинамических функций вращательные уровни состояний с Л)>0
могут рассматриваться как дважды вырожденные. Состояния с5>0 являются мультиплетными
и расщепляются на 25+1 компоненту; каждая компонента характеризуется значением квантового
числа Q и обозначается символом 2s+i&Q\ так компонента 2Пз/я состояния 2П соответствует значениям
Л=1, S=1/2, 2=1/2, Q=3/2. При некоторых случаях связи отдельных моментов количества
движения (случай связи Гунда с), квантовые числа Л и 2 теряют смысл и состояния характеризуются
только значениями квантового числа Q, которое для молекул с четным числом электронов
принимает целочисленные значения, а для молекул с нечетным числом электронов — полуцелые
значения 1.
Ниже приведены уравнения для Fv (/), принятые в настоящем издании для электронных
состояний разного типа в соответствии с систематикой, развитой в книгах Герцберга [71, 19061.
Состояния симметрии *2 (*2+, 12~, 12+, . . .):
+ ..., A.34)
где
Вг = Ве- a, (v + V») + Ч (" + lkf -Ч(Р+ %Т + ..; A. 35)
Dt = Dt-^{v + %)-\-^(v + ^-..., A.36)
H, = He-hl{v + ll2) + ..., A.37)
Ве = /г/8гс2с|лг^ — вращательная постоянная молекулы, A.38)
De — постоянная центробежного растяжения; ак, j3fc, hk — постоянные
колебательно-вращательного взаимодействия, ^=77г1та/(те1+та2) — приведенная масса молекулы; г, — равновесное
межъядерное расстояние в данном электронном состоянии молекулы, /=Д.
У гетероядерных молекул квантовое число механического момента количества движения R принимает значения
0, 1, 2, 3,... Статистический вес вращательного состояния определяется величиной квантового числа полного
момента количества движения /, которое в зависимости от характера взаимодействия отдельных моментов принимает
для данного R значения J=R+A или Л + Q. Состояние с квантовым числом / у гетероядерных молекул (без учета
спинов ядер) имеет статистический вес B7+1) и полный статистический вес B/+l)Bi1+l)Bi2-)-l), где it и i2 —
спины ядер атомов, образующих молекулу.
У гомоядерных молекул полный статистический вес уровня не равен B/-)-1)B?+1J, а зависит от симметрии
полной волновой функции молекулы в данном вращательном состоянии и спиновой функции ядра. Для ядер с
целочисленными значениями спина @, 1, 2, . . .) вращательные состояния с симметричной полной волновой функцией
имеют статистический вес B^+l)Bi+l)(i-j-l), а состояния с антисимметричной волновой функцией
статистический вес B7-j-l) i Bi-|-l). Для ядер с полуцелыми спинами A/2, 3/2, . . .) статистические веса
симметричных вращательных состояний равны B/+1) i Bi+l), а антисимметричных — B/+1) Bi+i) (i+l). Это
приводит, в частности, к тому, что если спины ядер равны нулю, у гомоядерных молекул отсутствуют
антисимметричные вращательные состояния (их статистические веса равны нулю). Симметрия полной волновой функции в i-м
состоянии определяется симметрией волновой функции механического момента (симметрична для четных и
антисимметрична для нечетных значений R), симметрией электронной волновой функции по отношению к отражению
в центре симметрии (остается неизменной или меняет знак при этой операции симметрии, что обозначается
индексом (g) или (и) у символа состояния), а также симметрией электронной волновой функции по отношению к
плоскости, проходящей через ядра обоих атомов (для невырожденных электронных состояний, когда A (Q) равно 0,
она симметрична или антисимметрична, что обозначается индексом «+» и «—» у символа состояния; у
вырожденных состояний сЛ(&) > 0 одна компонента Л-удвоения симметрична, а другая антисимметрична). У
гомоядерных молекул с i=0 в состояниях Щ, 2~, OJ, О~ квантовое число принимает только четные значения @, 2, 4, . . .),
а в состояниях 2j, 2+, Oj, OJ — только нечетные значения A, 3, 5, . . .).
36
1.4. Двухатомные газы
Состояния симметрии 2Е BЕ+, 2Е , 2?+, ...):
F"v (I) = BVR (R + 1) - DtR* (R + IJ - • • • - (T/2) R,
где /=7?+V2 для компоненты F' ( E=1/2), J=R—1/2 для компоненты F" ( 2=—V2); у —
постоянная расщепления, обусловленного взаимодействием спина электрона с магнитным моментом,
возникающим при вращении молекулы, уровень с R=0 состоит только из одной компоненты с /=S =
= Vt.
Состояния симметрии 3ЕCЕ+, 3Е-, 3Е+, ...):
F't(R) = B.R(R + 1) -DtR*(R + IJ + . .. + BVBR + 1) -\ +
+ ;. (i? + 1) - ^BД + 3)*-Я,Х+^,
-/?,№(Л + 1)а + ..., A.40)
— DtR2(R-\- IJ — ... —BVBR — 1) — I —
- ^i? + V52 BД - 1) - 2?,
где / принимает значения i?+l, 7? и i?—1 для компонент //', F" и F"' соответственно, X и ix —
постоянные расщепления, обусловленные соответственно взаимодействием спинов двух
электронов и спинов с магнитным моментом, возникающим при вращении молекулы (jn <^ X); при R—0
J=S и уровень с Л=0 одиночный, так же как в состояниях 2Е.
Состояния симметрии Ш, 1Д, 1Ф ... (Ш^, 1Ли, 1Ад, .. .), а также (случай Гунда с) 1, 2, 3, ...:
F.(J) = B,7(J + l)-D.J*(J + i)* + ..., A.41)
где J=Q, Q+l, S+2, . . .
Дублетные состояния симметрии 2П, 2Д, 2Ф, . . . BIIg, гПм . . .) (характер связи промежуточный
между случаями Гунда а и Ь)
F'v (I) = ?.[(/ + V2J - Л2 - 0,5 V4G + V2)a+r(r+4)A«] - i?f/* +...,
где Y=4/J5, Л — постоянная спин-орбитальной связи молекулы, характеризующая
взаимодействие орбитального L и спинового S моментов и величину расщепления компонент F' и F"
данного состояния; для F' /=Л+7?+1/2; для F" /=Л+Л — V2.
Триплетные состояния симметрии 3П, 3Д, . . .:
- 1) -4/9- 27 G + 1)], У = Л/В и .4 -
) 2
постоянная спин-орбитального взаимодействия; для компонент F', 7/" и F"' J принимает
значения A+1+R, A+R и Л+7? — 1 (Д=0, 1, 2, . . .).
Для состояний более высокой мультиплетности (квартетных, квинтетных и т. п.) постоянные
спин-орбитального взаимодействия, а также взаимодействия магнитного и механического момента
в уравнениях типа A. 34), как правило, неизвестны, часто они неизвестны для дублетных и триплет-
ных состояний многих молекул. Во всех этих случаях уровни вращательной энергии молекул в
вырожденных и мультиплетных состояниях могут быть описаны уравнением типа A.34) с учетом того,
что они являются вырожденными из-за Л-удвоения и мультиплетности данного электронного со-
37
Глава 1. Вычисление термодинамических функций газов
стояния. Статистический вес уровня, соответствующего квантовому числу /, для состояний с Л=0
и суммарным спином S равен в этом случае
B7 + 1) B5 + 1), A.44)
для состояний с Л ^> 0 и суммарным спином S
2 B7 + 1) B5 + 1). A.44а)
Уровни колебательной энергии двухатомной молекулы описываются уравнением
G (и) = u), (v + Va) - «>Л (и + V2J + <о.у. (v + VaK - • • •, A • 45)
где ше — частота колебания молекулы, и>ехд, <*>еуе, ... — постоянные ангармоничности, v—Q, 1,
2, • . ., vmax', статистический вес v-то состояния равен 1.
Если энергия колебательных состояний отнесена не к минимуму потенциальной кривой, как
в A.45), а к состоянию с у=0, справедливо уравнение
Go (v) = G(v) — G @) = %v — %xov* + mo?ov* - .... A. 46)
где
(Uozo = 0)A— •••
Разность энергии соседних колебательных состояний, соответствующих значениям v и у+1,
на
равна
Таким образом, разность энергии между состояниями v=l и у=0 (основная частота в
колебательном спектре молекулы) равна
к&12 = и>е — 2шехе-{- ... =ш0—щЛ+ ... A.49)
Постоянные в уравнениях A. 34)—A.49) определяются в результате анализа вращательных,
колебательно-вращательных и электронно-колебательно-вращательных спектров молекул. В
работах, посвященных анализу спектров, как правило, приводятся равновесные значения
молекулярных постоянных (сое, шехе, . . ., Ве, аг и т. д.), а также энергии возбуждения электронных
состояний Те. Значения равновесных постоянных, принятые в справочнике для каждой
рассматриваемой двухатомной молекулы в результате критического анализа всех опубликованных
в литературе экспериментальных и теоретических исследований, приводятся в сводных
таблицах каждой главы, озаглавленных «Молекулярные постоянные. . .». В текстах соответствующих
глав дано обоснование выбора этих значений.
Если известны уравнения для G (v) и Fv (/), описывающие всю совокупность связанных
колебательно-вращательных уровней энергии ?-го электронного состояния, расчет статистической
суммы по колебательно-вращательным состояниям (уравнение 1.30) и ее производных
непосредственным суммированием по уровням энергии не представляет трудностей. Однако практически
в экспериментальных исследованиях удается наблюдать переходы, связанные только с
небольшим (относительно) числом колебательно-вращательных уровней энергии данного электронного
состояния, как правило, переходы между уровнями, соответствующими низким значениям
квантовых чисел v и /, которые существенно меньше величин ymia и Jmst>,. Значения последних, за
редким исключением, вообще не удается определить экспериментально. Исследователи,
занимающиеся изучением спектров молекул, стремятся получить такие значения констант в уравнениях для
G (v) и Fv (/), которые обеспечивают наилучшее описание экспериментальных данных. При этом
степень полиномов по v и / (/+1) обычно мало зависит от особенностей энергетических состояний
данной молекулы, а в основном определяется тем, в какой области изменения квантовых чисел
38
1.4. Двухатомные газы
Рис. 1.3. Уровни колебательной энергии Go (v)
для основного электронного состояния
молекулы F2
1 — расчет по значениям ы>, и ь>ехе, найденным
в работе [1255];
2 — обработка данных [1255] по методике,
принятой в справочнике;
3 — экспериментальные данные [1008]
12000 ~
8000 ~
?000-
16 24 is
v ж J наблюдались спектральные переходы. Экстраполяция уравнений для G (v) и F, (/), с
найденными таким образом постоянными, в область высоких значений квантовых чисел v и / может
приводить к грубым ошибкам, так как эти функции и даже характер их изменения существенно
зависят от степени полинома. Поэтому для расчета значений Qmx^v и ее производных необходимо
уточнить уравнения для G (v) и Fv (J) с тем, чтобы они обеспечивали корректную аппроксимацию
всех колебательно-вращательных уровней молекулы в данном электронном состоянии вплоть до
значений vms_s и /maIi „.
В настоящем издании для экстраполяции уравнения для G (v) на всю область значений v от
О до vmax применялась методика, развитая при подготовке предыдущего издания (см. [88, с. 44])
и изложенная в [84]. Эта методика основывается на том, что для определения постоянных в
уравнении A.46) помимо экспериментальных данных, связывающих значения G (v) (или их функции)
с частотами спектральных переходов, используются два дополнительных условия, определяющих
схождение колебательных уровней данного электронного состояния к его диссоциационному
пределу при
A.50)
где Do — энергия диссоциации молекулы в этом электронном состоянии.
Для решения системы уравнений, включающей частоты спектральных переходов и соотношения
A. 50), и определения значений колебательных постоянных и ушах была составлена программа,
использующая метод наименьших квадратов в сочетании с методом линеаризации [234] г. При этом
оптимизация выбора степени полинома и аппроксимации спектральных данных осуществлялась
с учетом достоверности последних и значения Do. Для иллюстрации на рис. 1.3 приведены
результаты расчета Go (v) для молекул F2: а) по значениям ше и шехе, найденным в работе Ди Ленардо и
Дугласа [1255] на основании изучения переходов между состояниями и=0,1 и 2 (Д&/2 и ^3/2)
(кривая 1); по значениям ше, шехе и ^еуе, вычисленным по экспериментальным данным [1255], величине
Do (F2)=12 896 +100 см с учетом условий A. 50) (кривая 2) и значения GQ (v) для y=0-f-22,
найденные Дугласом с сотрудниками в 1975 г. [1008] (кривая 3). Из рис. 1.3 видно хорошее согласие
(в пределах 1 %) кривых 2 и 3 и их заметное отличие при больших v от кривой 1. Колебательные
составляющие термодинамических функций F2 (г), вычисленные по этим трем вариантам
колебательных постоянных, приведены в табл. 1.3. Из таблицы видно, что уточнение колебательных
постоянных по принятой методике позволяет в 3-4 раза уменьшить ошибки в термодинамических функциях
из-за неточности аппроксимации энергии высоких колебательных уровней. Следует отметить, что
у F2 линейная экстраполяция колебательных уровней по постоянным, найденным в [1255],
приводит к значению Do (F2), относительно близкому к его истинному значению. Такое согласие бывает
редко и уточнение колебательных постоянных по принятой методике может влиять существенно
больше на точность расчета термодинамических функций при высоких температурах.
Была составлена также более общая программа для совместной обработки нескольких систем полос с учетом
схождения колебательных уровней каждого из комбинирующих состояний к соответствующему диссоциационному
пределу.
39
Глава 1. Вычисление термодинамических функций газов
Таблица 1
Г, К
3. Значения колебательных составляющих в п
(в Дж • К • моль)
I
II
III
гермодинамических функциях
I
II
III
F2 (г)
[с; (т)]т
i
II
III
1000 2,734 2,734 2,734 6,934 6,939 6,937 7,604 7,620 7,613
3 000 9,154 9,242 9,207 16,373 16,779 16,633 9,009 10,235 9,829
6 000 14,331 14,875 14,690 21,615 23,094 22,612 5,464 6,823 6,425
10 000 17,724 18,759 18,422 23,590 25,544 24,930 2,495 3,040 2,900
Примечание. I — расчет по постоянным, найденным в работе [1255]; II —расчет по постоянным, полученным
обработкой данных [1255 ] с учетом условия схождения колебательных уровней к диссопиационному пределу; III — расчет
по колебательным уровням, найденным в работе [1008].
Экстраполяция уравнения для Fo (J) в область высоких значений /, а также определение
значений /maXj,, является существенно более сложной задачей. Как известно (см. [1906]), помимо
связанных колебательно-вращательных уровней с энергиями ниже энергии диссоциационного предела
данного электронного состояния, для которых выполняется условие (рис. 1.4)
A.51)
A.52)
молекула АВ имеет квазисвязанные состояния, для которых
[Do (АВ) + G @)] < [G (и) + F, (/)] < Vn (rh).
0@)
"max,umax
max,n
Jmaio=0 JUm J
Рис. 1.4. Соответствие между эффективными потенциальными кривыми, предельной кривой диссоциации
и уровнями колебательно-вращательной энергии G (v)-\-Fe (J)
а — предельная кривая диссоциации A (J) и колебательно-вращательные уровни энергии для р=—У„, 0,
п и vmax;
б — эффективные потенциальные кривые для разных значений /
40
1.4. Двухатомные газы
где Vr (г) — эффективная потенциальная энергия вращающейся молекулы, описываемая выраже ¦
нием
VR (г) = Fo (г) + hR (R + 1)/8*>2с, A. 53)
где Vo (r) — потенциальная энергия (в см) невращающейся молекулы, R — квантовое число
механического момента количества движения молекулы.
При R > 0 функция Vr (г) проходит через максимум в области ге < г < со, который имеет
место при r=rh. С увеличением значения квантового числа R величина rh и глубина потенциальной
ямы уменьшаются, а величина rmln увеличивается, пока при некотором значении i?iim минимум на
эффективной потенциальной кривой не исчезнет вообще. Значение Яцт может быть определено из
условий
= 0.q —тт-Ч =°- С1-54)
Очевидно, что во всех случаях JnmXiV < /iim=-Riim *• Благодаря большой ширине максимумов
на эффективных потенциальных кривых, вероятность диссоциации молекул из квазисвязанных
состояний в результате туннельного прохождения атома сквозь барьер настолько мала, что даже у
соединений водорода эти состояния практически не отличаются от связанных состояний и, как
последние, должны учитываться при расчете Qm и ее производных.
В настоящем издании для определения значений /maXj t использовались представления о
предельной кривой диссоциации, которая является геометрическим местом точек максимумов на
эффективных потенциальных кривых молекулы и ограничивает (см. рис. 1.4, а) область существования
связанных и квазисвязанных состояний молекулы в данном электронном состоянии. Для расчета
предельных кривых диссоциации необходима потенциальная функция невращающейся молекулы
Vo (г). В настоящем издании для расчетов, кроме специально оговоренных случаев, использовался
потенциал Гульберта—Гиршфельдера
Fo (г) = De [1 — exp {—x)f -f- De [ex3 (l-\-bx) exp (—2a;)], A. 55)
где
2 (B.D.I/. г.
Уравнение предельной кривой диссоциации
1J + .--. A-56)
где ao=Do(AB)+(r @)=De(AB), может быть получено, если известна величина ао и рассчитаны
значения Vj (rh) для /цт и нескольких значений / в интервале от 0 до /цт (рис. 1.4, а).
Соответствующие расчеты могут быть выполнены на основании уравнений A.54) и A.55), если кроме
величины D0(AB) известны постоянные (*>в, и>ехе, Ве и аг молекулы АВ в данном электронном
состоянии.
Проблема экстраполяции уравнения F, (/) к предельной кривой диссоциации (см. рис. 1.4, а)
распадается на две части, обусловленные: а) необходимостью экстраполяции уравнения для F,
(J) и, следовательно, уравнений типа A. 35)—A. 37) на область больших значений v,
существенно больших, чем те, для которых имеются экспериментальные данные о постоянных В,,
D, и т. п.; б) необходимостью экстраполяции уравнения для F, (/) на область значений / —> /maXj>.
Установление зависимости постоянных В„ D,, Н„ ... от г; в широкой области изменения
этого квантового числа непосредственно из экспериментальных данных удается только в
исключительных случаях. Для большинства молекул обычно известны значения Bv и иногда Dv для
Для всех типов взаимодействия отдельных моментов количества движения значения квантового числа
механического момента R отличаются от соответствующего ему значения квантового числа полного момента количества
движения / не больше чем на несколько единиц. Поэтому в данной работе принимается, что /1Я=Лцщ. и при
последующем обсуждении экстраполяции F, (/) не проводится различия между квантовыми числами, что
практически не вносит погрешностей в результаты последующих расчетов.
41
Глава 1. Вычисление термодинамических функций газов
Пи-10 ,смш
16
24 V
Рис. 1.5. Зависимость вращательных
постоянных В, от квантового числа v для основного
электронного состояния молекулы F2
1 — расчет по значениям Вв и alt найденным в
работе [1255];
2 — расчет по значениям Ве, ах и а2, найденным
по методике, принятой в справочнике;
3 — экспериментальные данные [1008]
60
Рис. 1.6. Зависимость вращательных
постоянных Dv от квантового числа v для основного
электронного состояния молекул Н2 (а) и С12 (б)
1 — экспериментальные данные [1913, 1301];
2 — расчет по соотношению A.58)
го
42
48
54
SO и
¦ограниченного интервала значений v, как правило для v <^ vma7i. Экстраполяция таких данных
к ушах обычно приводит к неверным значениям. Вычисленные таким образом значения Bt для
ковалентных молекул оказываются равными @,4—0,6) Во, в то время как для тех редких случаев,
когда величина В, была определена для последнего колебательного уровня, она составляла < 0,2 Во.
Так, для F2 (рис. 1.5) экстраполяция значений В, к z;max по уравнению A. 35) на основании значений
Во и аи найденных Ди Ленардо и Дугласом [1255], приводит к -В„тах_ь которое существенно больше,
чем найденное недавно экспериментально в работе Дугласа и др. [1008]. В большинстве других
случаев результаты экстраполяции оказываются даже хуже приведенных на рис. 1.5, в частности
для ионных молекул типа NaCl она может приводить к отрицательным значениям Bv, что
противоречит физическому смыслу этой величины.
В настоящем издании для уточнения зависимости В, от v применялась методика, основанная
на условии, что в области г <^ге потенциальная кривая невращающейся молекулы, построенная
по методу Ридберга—Клайна—Риса, должна монотонно возрастать с уменьшением г (см. [2393,
77]). Это соответствует условиям
dVn <r) ^ JW <г)
-Ч^<0' Т>° A.57)
42
1.4. Двухатомные газы
при г <^ гс, удовлетворение которых может быть обеспечено варьированием некоторой величины
Btt: где г;* больше значений v, для которых имеются экспериментальные данные о Bt.
Полученное значение Bvt совместно с найденными экспериментальными значениями Bv аппроксимируется
уравнением типа A.35) с введением, в случае необходимости, дополнительного коэффициента а,..
Хотя эта методика не позволяет определить точное значение Вьшх, она дает достаточно
удовлетворительные данные для v, весьма близких к г;тах. На рис. 1.5 приведены результаты
экстраполяции величин #„ для F2 по этой методике с использованием значений Во и а,,
найденных Ди Ленардо и Дугласом [1255]. Из рисунка видно, насколько существенно удается
улучшить экстраполяцию экспериментальных данных при использовании этой методики 1.
Зависимость постоянной Dv от v, как правило, неизвестна или установлена ее линейная
зависимость от v при г;=0, 1 и 2. При небольших значениях v постоянная D, монотонно убывает
(в уравнении A.36) 8Х ^> 0). Однако экстраполяция этой зависимости на область v ->уш„ не
корректна, так как из общих соображений можно ожидать, что вблизи диссоциационного
предела Dr должны быть больше значения Do по абсолютной величине. Это получило подтверждение
в результате последних исследований спектров некоторых молекул. На рис. 1.6 приведены
значения D, для Н, и С12 по данным работ [1913,1301], подтверждающие быстрое увеличение этой
постоянной при v ->-ущах. Там же приведены значения D,, вычисленные по соотношению,
предложенному Иоришем [110]:
Д, = 4ЯЗ/(ДС^л)а. A.58)
где значения Bt, G (и) и ДСг»+у2 скорректированы по методикам, описанным выше, для обеспечения
их правильного поведения при v -> ушах. Из рисунка видно удовлетворительное согласие значений
Dt, вычисленных по уравнению A.58), с экспериментальными данными2.
Экстраполяция уравнения Ft (/) на область /-»• Jmaz>, (см. рис. 1.4, а) должна
удовлетворять трем условиям:
а) энергия неколеблющегося ротатора (у=—1/2) при / -*¦ /цт должна приближаться
к энергии в точке перегиба на эффективной потенциальной кривой ротатора с /=/пт, т. е.
должно выполняться условие
" ^(Л,ш);~^лаЫ; A-59)
б) кривые G{v)-\-Ft (/) должны пересекаться с предельной кривой диссоциации A (J)
ПРИ «Лпаж. „"»
в) с ростом и пересечение кривых G(v) -JrFv(J) и А (/) должно происходить при монотонно
уменьшающихся значениях /maXi,.
Экстраполяция полинома по степеням /(/+1) со значениями постоянных Ве, De, He и т. д.,
вычисленными по экспериментальным данным, практически никогда не приводит к выполнению
условия A.59) (рис. 1.7), а экстраполированное значение Fe (J) существенно зависит от степени
полинома. Более удовлетворительный результат получается, если использовать для Fe(J) не
полином по степеням М"=/к(/+1)и, где п ^ 2, 3 или 4, типа A.34), а ввести в соответствующее
уравнение сумму всех последующих членов с и=3, 4, 5, . . . , оо, как это было предложено Вулли [4000].
Соответствующие выражения имеют вид:
F, G) = В,М - D.AP + #,М3 - L.M*+Bj^^uii, A.60)
F, (J) = B.M- D,M* + Я.М8- дJ^+TtM3 > A-61)
F. (J) = B.M - D.BP+B№riin =Bjffr . A. 62)
1 Применение описанной методики часто нецелесообразно из-за низкой точности других постоянных молекулы
в данном электронном состоянии. В этом случае экстраполяция уравнения A.35) либо вообще не
корректировалась, либо выполнялась оценкой дополнительного значения а{ тая условия, что #>шах+1=0.
2 В ряде случаев удовлетворительная точность расчета значений Ft (J) при больших v достигается в
предположении, что D,~~De. Это обусловлено тем, что при больших v значения Jmtx, r достаточно малы, чтобы в уравнении
для Ft (J) член с J'1 (/+1J оставался небольшим по сравнению с членом с / (/+1), несмотря на быстрое
возрастание величины D, (практически во всех случаях В, > 10* D,).
43
Глава 1. Вычисление термодинамических функций газов
Fe(J)
Рис. 1.7. Величины уровней вращательной энергии Fe (J) как функции квантового числа J при использовании
различных уравнений
а, б — основное электронное состояние молекулы Щ [540]: 1 — уравнение A.34) с п < 2; 2 — п < 3; 3 — п <
< 4; 4 — уравнение A.62); 5 — уравнение A.61) (а), A.64) (б); 6 — уравнение A.60) (а), A.64а) {б); 7 —
уравнение A.63)
в — основное электронное состояние молекулы ЫН[3785в]: 1 — уравнение A.36) с п < 2; 2— и < 3; 3 —
уравнение A.62); 4 — уравнение A.61)
На рис. 1.7 приведены результаты экстраполяции значений Fe (J) для Щ уравнениями A.60) —
A.62) на область значений / -> /цт. Как видно из рисунка, использование этих уравнений дает
существенно лучшие результаты, чем обычный полином, хотя заметная зависимость от степени
полинома сохраняется. В тех случаях, когда и применение уравнений A.60)—A.62) с постоянными,
найденными экспериментально, не позволяет удовлетворить условию A.59), в уравнение для
Fe(J) может быть введен дополнительный член с более высокой степенью /(/+1), коэффициент
которого подбирается таким, чтобы выполнялось это условие.
Следует отметить, что для некоторых молекул, в особенности полярных молекул с большими
энергиями диссоциации и относительно низкими значениями постоянной Ве, добиться выполнения
v^obhh F,(Jma%i ,) = FjmaXj ,(rk) не удается даже при использовании уравнений типа A.60) —
A.62). Существенно лучшие результаты в этом случае можно получить, если использовать для
экстраполяции значений F, (J) вместо полиномиальных выражений рациональные дроби типа
паде-аппроксимантов. Если известны четыре постоянные в A.34): В„ D,, Н, и Lt, соответствующее
уравнение для Ft (J) принимает вид
"Г ^2^" ) /Л по\
¦ХТГдГа» (l.bd)
где cx = B,, c2 = (ds — 2dh-\-r)l(d? — h), bx = (I — dh^d2 —,
ны три постоянные в A.36): Bt, D,, и Н„, то
где сх=В„ ca=(d2—h)/d, bx=—hid, или
где сх=В„ b1——d, b2=dz—h.
= (h2 — dl)/(d2 — h); если
известA.04)
A-64a)
44
1.4. Двухатомные газы
В уравнениях A.63), A.64) и A.64а) d——DJB,, h~HJB, и l = —LJB,.
В тех случаях, когда из экспериментальных данных известны только две постоянные E,
и Dt), паде-аппроксимант оказывается тождественным уравнению A.62).
На рис. 1.7, б приведены результаты экстраполяции значений Fe(J) для Н* по уравнениям
A.62)—A.64). Их характерным отличием от экстраполяции по уравнениям типа A.34) или A.60)
является то, что качество экстраполяции слабо зависит от числа известных постоянных, в
отличие от того, что имеет место для полинома A.34).
Изложенные в настоящем разделе методы позволяют получить для аппроксимации системы
колебательно-вращательных термов молекулы в данном электронном состоянии набор постоянных
в уравнении
G (v) + F, (/) = 2 2 Ykl (v + 1/2)* 7^G + 1/, A. 65)
к I
где
yio = aV ^2о=—ША- ^зо = ш.У.
•МЦ~=-"е' •* И == al' ¦*21=:а2' • • ч
YOi = -Dt, У1а = Рх. ...
Использование постоянных Ykl в комбинациях уравнения A.65) с уравнениями типа A.62) —
A.64) и A.39)—A.43) обеспечивает схождение колебательных уровней к диссоциационному
пределу данного состояния и корректный учет всех связанных и квазисвязанных состояний,
ограниченных предельной кривой диссоциаций А G) и кривой Fe{J). ЗначенияYkl, !;„„, и 7нт, а также
коэффициентов а( в уравнении A.56), описывающем предельную кривую диссоциации, полученные
при подготовке настоящего издания х по этим методикам, приводятся в таблицах «Значения
коэффициентов в уравнениях, описывающих уровни энергии (в см), принятые для расчетов
термодинамических функций ...» соответствующих глав. В текстах глав, посвященных расчету
термодинамических функций, отражены специфические особенности выбора и обработки постоянных
для каждого вещества.
Если значения Ykl определены для некоторой изотопической модификации данной молекулы,
их значения для любой другой модификации могут быть получены по хорошо известным
соотношениям:
Yio = ?Y10, Yzo = P2YW, F30 = ?3Y30, • • •»
A. bo)
где p=(fi/^i')'/j, p и tu' — приведенные массы молекул соответствующих изотопических
модификаций.
При подготовке настоящего издания соотношения A.66) использовались для расчета значений
Ykl «эффективных изотопических модификаций» молекул газов, содержащих молекулы различного
изотопического состава. Расчет термодинамических функций природной смеси изотопов по
полученным таким образом постоянным равносилен предположению, что уравнение A.65) с
коэффициентами Ykl для «эффективной изотопической модификации» аппроксимирует колебательно-
вращательные уровни условной молекулы, образованной атомами с массами, равными атомным
массам элементов.
Ограниченность экспериментальных данных часто не позволяет определить значения
постоянных ше, Ве и (или) «!, необходимых для расчета потенциальной функции Vo (r) по соотношению
A.55). В таких случаях эти постоянные оценивались с помощью приближенных теоретических
соотношений, обеспечивающих удовлетворительную точность. Если из спектральных данных
неизвестны постоянные ше и и>ехе, но определена основная частота Д&д и известна энергия
диссоциации молекулы в соответствующем электронном состоянии, постоянные сое и шехе вычислялись
совместным решением уравнений
1 Для проведения соответствующих расчетов был составлен комплекс программ, реализованный на ЭВМ БЭСМ-4.
45
Глава 1. Вычисление термодинамических функций газов
«в2/4(оЛ_Шв/2.
Полученные таким образом постоянные обеспечивают схождение колебательных уровней к диссо-
циационному пределу этого состояния.
Постоянная центробежного растяжения вычислялась по соотношению, предложенному Крат-
цером (см. [1906]),
De = 4Д!/ш2, A.68)
которое приводит к согласию с известными экспериментальными данными в пределах 10%.
Постоянная колебательно-вращательного взаимодействия ах оценивалась по соотношению
Аналогичные приближенные соотношения (см. [1906, с. 108—109]) могут быть использованы
для оценки постоянных J3X и Не.
При подготовке настоящего издания для расчета внутримолекулярных составляющих
статистической суммы, ее производных и соответствующих составляющих термодинамических функций
была составлена программа, в которой вычисление сумм по i, v и / проводится последовательно,
вначале суммированием по / от /mln до /тах , для вращательных уровней v-то колебательного
1-го электронного состояния:
•Лпах, V
A.70)
J41
mm
: 2BB7 + l)c"'[|i^"(/)]exp[-^F<--'(/)], A.71)
•'min
^шах, л
^min
затем суммированием по v для i-го электронного состояния:
A.73)
ff=0
=2
г'"
;.ехр [- тт ]
0=0 «-0
46
1.4. Двухатомные газы
затем суммированием по i электронным состояниям
•тих
____
д?)A • 76>
2<?-- -* (&А/?<)ехр (- ш АЕ>)' A •77)
Х
i~X i=X
A.78)
где Д?,.=Г^''—G<Z)(O)—^'(^ш)' ^«^ — разность энергий между минимумами потенциальных
кривых i-го возбужденного и основного электронных состояний, G(X) @)+FW(/min) — энергия
нижнего колебательно-вращательного уровня молекулы (у=О, /=/min) в основном электронном
состоянии, отсчитанная от минимума потенциальной кривой этого состояния.
У гетероядерных молекул квантовое число / принимает последовательный ряд целых или
полуцелых чисел в зависимости от того, целое или полуцелое значение имеет квантовое число Q в г-м
состоянии (четное или нечетное число электронов имеет молекула). У гомоядерных молекул, если
спины ядер равны нулю, половина уровней отсутствует (уровни с четными или нечетными
значениями квантового числа / в невырожденных электронных состояниях или одна иа
компонент Л-удвоения в вырожденных электронных состояниях). Если спины ядер не
равны нулю, полные статистические веса вращательных уровней таковы, как если бы
отсутствовала половина уровней. Если интервалы между нижними вращательными уровнями
малы по сравнению с температурой, для которой ведется расчет термодинамических
функций, что справедливо для всех молекул, кроме Н2, D2 и Т2 при Т > 100 К, суммы типа A.70)—
A.72) по четным и нечетным значениям / (или по значениям 1/2, 5/2, 9/2, ... и 3/2, 7/2,
11/2,. . . ) равны между собой. В этих случаях для учета различия полных статистических весов
уровней с четными и нечетными значениями / в уравнения для (?вр>, и ее производных вводится
статистический вес 1/а, где а — число симметрии, равное 2 для гомоядерных и 1 для
гетероядерных молекул. Поскольку величина о одинакова во всех электронных и колебательных состояниях
данной молекулы, этот сомножитель может быть вынесен за знак трех сумм в уравнениях типа
A.30) и включен в виде отдельного слагаемого в выражения для термодинамических функций1.
Для молекул в мультиплетных электронных состояниях значения Q<^\ v (или <?W BJ!) должны
вычисляться отдельно для каждой компоненты мультиплета. Однако если величина расщепления
компонент мультиплета (пропорциональная постоянным у, 1 и А в уравнениях A.39)—A.43))
мала по сравнению с температурой, для которой ведется расчет, или с энергией возбуждения
данного электронного состояния Т^ (около 1—2% или меньше), это расщепление может не
учитываться. В таких случаях в выражениях f(l .70)—A.72) для QSVi „, $вр „ и ^, статистический
1 При расчете термодинамических функций газов, состоящих из гомоядерных молекул с невырожденным основным
электронным состоянием, для температур ниже C—5) hcBJk статистическая сумма QeVt f должна вычисляться
с учетом спинов ядер отдельно для пара- и ортосостояний (состояния со статистическими весами B/+1) B/-М) I
и B/+1) B/-J-1) G+1) соответственно). Полная внутримолекулярная статистическая сумма в этом случае есть
сумма ?вн_ орто и ?ВН| лара
9вн == ?вн орто ~г 9вн, пара' (^ • '"/
а практическая статистическая сумма по внутримолекулярным состояниям есть
1J, A.80)
где / — спин ядер атомов, образующих молекулу.
47
Глава 1. Вычисление термодинамических функций газов
вес p(i,J) (см. с. 35) принимается равным (без учета ядерных спинов) B7 -)- l)BS-{- l)c"'\
где c(i) = l для невырожденных состояний с A(Q)=0, cU)=2 для вырожденных состояний cA(Q) ^ 1.
Следует отметить, что точность определения значений уша1 и /тах> „ точность аппроксимации
колебательно-вращательных уровней уравнениями типа A.65) и A.62) в области больших значений
v, а также точность аппроксимации значений Vj (rh) уравнением типа A.56) существенно зависят
от достоверности энергии диссоциации молекулы в г-м электронном состоянии, принятой в
расчетах этих величин. В тех случаях, когда погрешность значения D^'(AB) велика (около 10%) или
?-е состояние имеет достаточно высокую энергию возбуждения ((hc/k)T^ > AT во всем интервале
температур), вычисление QW „ и ее производных, а следовательно, весь расчет
термодинамических функций можно упростить, заменив суммирование по / от /ш1п до /mai_ „ интегрированием
в тех же пределах, если принять линейную зависимость /шах , от v
что соответствует условиям 7Шах,-1Л== ^пш н ^тах, «тах+1 — 0- Тогда, обозначив DJBt = d, вместо
уравнения A. 70) получаем
Аналогичные уравнения могут быть записаны для QSB>, и QBS ,. Поправочные члены в A.82),
существенные только при низких температурах, получены для условия, что F,{J)=B,J(J-\-l) —
—Z)f/2(/+lJ. При подготовке настоящего издания интегралы в уравнениях типа A.82)
вычислялись по квадратурным формулам Гаусса с 25 узлами. Узлы и веса гауссовых квадратур
принимались по таблицам Кронрода [1336]. При низких температурах, когда вклад высоких
вращательных состояний в величину Q^ v мал (!Г<;0,05 Dhc/k), интегрирование в уравнениях типа A.82)
должно быть ограничено при некотором значении /rPi, < Jmax,,' Для нахождения / f
использовалось приближенное соотношение (см. [292а])]
A.83)
В табл. 1.4 сопоставлены результаты расчета термодинамических функций F2(r) с
использованием различной исходной информации, разных методов ограничения суммирования по / при
вычислении QBP , и ее производных и при замене суммирования по / интегрированием в тех же
пределах. Сопоставление колонок III и IV, в которых использован одинаковый метод ограничения
суммирования по v и / (принято, что /шаХ), линейно уменьшаются с ростом v) показывает, что
замена суммирования интегрированием не влияет на результаты расчета, так как при всех
температурах и во всех функциях расхождения не превышают 0,005 Дж-К~1«моль~1. Существенно
большую неточность в расчет термодинамических функций вносит ограничение суммирования
по /, если принять линейную зависимость /ш«,, от v, вместо ограничения по предельной кривой
диссоциации; сравнение данных колонок I и III показывает, что расхождения достигают 0,2 и
0,4 Дж-К-^моль взначениях Ф°(Г) и S°(T) при T~0,3(hc/k) D0(F2). Сравнение колонок II и
V показывает, что при T~0,3(hc/k) Do (F2) вклад квазисвязанных колебательно-вращательных
состояний достигает 0,7 и 2 Дж'К~1-моль~1 в значениях Ф°(Т) и S°(T).
В таблице приведены также результаты расчета термодинамических функций F2(r) по методу
Майера и Гепперт-Майер, применяемому в большинстве зарубежных работ х. Пренебрежение необ-
1 Обсуждение различных приближенных методов см. в справочнике [88, с. 106]. Метод Майера и Гепперт-Майер
основан на предположении, что
G (о) + Р, (/) = <*, (» + 1/2) - а,л {v + 1/2J + [Вв _ ai (у + 1/2)] / (/ + 1) _ ZV2 (-/ + 1)а; A. 84)
экспонентыв уравнениях A.70)—A.76),содержащие и>ехе, UjiB,, можно разложить в ряды с учетом только первых
членов, а верхние пределы в суммах по v и / равны бесконечности. Тогда (для молекул в состоянии Х2)
48
1.4. Двухатомные газы
Таблица 1.4.
Сравнение термодинамических функций F2 (г), вычисленных в разных приближениях
(в Дж • К-1 • моль)
т, к
298,15
3000
6000
10000
298,15
3000
6000
10000
298,15
3000
6000
10000
Примечание. I —
I
173,084
249,831
275,270
292,413
202,681
286,863
310,658
324,031
31,300
38,789
. 29,089
23,945
расчет по
II
173,080
249,756
274,985
291,993
202,677
286,570
310,060
323,406
31,301
38,171
28,915
23,953
III
Ф°(Т)
173,080
249,726
274,813
291,759
S°{T)
202,677
286,416
309,679
323,126
С°р(Т)
31,301
37,677
28,950
24,233
IV
173,080
249,726
274,813
291,755
202,677
286,415
309,675
323,122
31,301
37,673
28,953
24,238
уравнениям A.73)—A.75) и постоянным [1255],
принятым в настоящем издании, см. с. ЗУ и
тальным данным
III — то же, что
по соотношениям
нием от / = 0 до
диссоциационного
V
173,080
249,660
274,152
290,311
202,677
286,022
307,645
320,093
31,301
36,087
26,505
22,807
обработанным
далее; 11 — расчет по уравнениям A.73)—A.75
[МОн] с ограничением (Jmx, вр и ее производных по предельной криво!
а в И, но
A.81); IV-
•* max, p, v
предела (t
с ограничением
— то же, что в
— то же, что и
Укол, вр и ее производных на значениях /шаг.
III, но расчет фвр, „
VI
173,080
249,542
275,491
295,420
202,680
286,144
313,783
335,222
31,290
39,037
40,890
43,250
по методикам,
и эксперимен-
[ диссоциации;
., полученных
и ее производных выполнен интегрирова-
в И, но с ограничением Св„,.
ез учета квазисвязанных состояний
Майер без ограничения суммирования по и а
У.
и ее производных на энергии
; VI — расчет по методу Майера ji Гепперт-
ходимостью ограничения суммирования по v и / приводит при использовании этого метода к
ошибкам, заметным уже при Т < 0,2 (hc/k) Do (F2). При Т~0,33(hc/k) D0(Fz) эти ошибки достигают
0,5, 3,7 и 12 Дж-К-^моль в значениях Ф°(Т), S°(T) и С°р(Т).
Вычисление значений статистической суммы по внутримолекулярным состояниям Qm и ее
производных по соотношениям A. 76)—A.78) после расчета значений Q^ вр, QW и QW в не
должно представлять труда. Однако именно расчет этой составляющей вносит в настоящее время
основные ошибки в вычисления термодинамических функций двухатомных газов, особенно при
высоких температурах. Это обусловлено тем, что при исследованиях спектров, как правило,
наблюдаются переходы только между ограниченным числом электронных состояний. Часто остаются
неизвестными, особенно у соединений элементов с незаполненными й- и /-электронными
оболочками, относительное расположение даже нижних электронных состояний и то, какое из них
является основным. Даже если в спектрах наблюдаются переходы, связывающие данное состояние
с другими, часто остаются неизвестными значения таких постоянных, как ч>ехе, Ве и ах, а также
корреляция электронного состояния молекулы с состояниями образующих ее атомов.
Последнее, как правило, делает невозможным определение энергии диссоциации молекулы в таком со-
QZ*.
вр — о A
дп Г J_ 8f 5 2хи
— е'и) \} + df0 + IT + еи— 1 + (е« —l
без учета возбужденных состояний (<?BH=(?ioJ. вр)
°pBU (T)
hue"
izu2e«
BSe» — ixu — 8x) I2xu*e2a
+
R = 1 + A—e-"J + и ~(ea —
где q0 = kTjhcB0, и = (hc/kT) Д&/„ х = <о.х>„ 7 = В
(eu — IK
A.85)
A.86)
A.87)
A.88)
49
Глава 1. Вычисление термодинамических функций газов
стоянии. Наконец, так же как для атомов, возникает вопрос о том, какие электронные состояния
вообще следует учитывать в расчете Qm и ее производных х.
В настоящем издании в статистической сумме по электронным состояниям двухатомных
молекул и ее производных, так же как у атомов (см. раздел 1.3), учитываются только связанные
валентные электронные состояния, т. е. связанные состояния, соответствующие таким
конфигурациям электронной оболочки молекулы, в которых главные квантовые числа электронов не
превышают их значения в ее основном состоянии. В соответствии с выводами работы [846] в
расчетах до 6000 К учитываются валентные состояния с Те <С 40 000 -^45 000 см, в расчетах до
20 000 К — состояния с Те ^ 75 000-^-80 000 см. Ридберговские электронные состояния
молекул, так же как и атомов, в расчетах не учитываются (исключения обсуждаются в текстах
соответствующих глав). Из рис. 1.1 и 1.2 видно, что пренебрежение ридберговскими состояними О2,
в отличие от ее валентных состояний, не приводят к заметным ошибкам в значениях удельной
энтропии и удельной полной энтальпии равновесной смеси О, О+, О2, О* и е при 20 000 К.
Общее число валентных электронных состояний двухатомной молекулы и тип каждого из этих
состояний могут быть установлены на основании известных правил (см. [71, 149, 1579]) по
конфигурациям электронной оболочки молекулы и на основании корреляции электронных состояний
молекулы и образующих ее атомов. Однако в отличие от атомов только часть валентных состояний
является связанными состояниями, а значительное число (иногда подавляющее) отталкиватель-
ными состояниями2. В данном издании определение числа связанных валентных
электронных состояний и установление симметрии этих состояний для каждой рассматриваемой молекулы
проводились на основании анализа конфигурации ее электронной оболочки в сопоставлении с
результатами экспериментальных исследований спектров и теоретических расчетов как данной
молекулы, так и ее аналогов. Результаты такого анализа кратко излагаются в
соответствующих главах при обсуждении выбора молекулярных постоянных. В тех случаях, когда
в литературе для части электронных состояний отсутствовали как экспериментальные данные,
так и теоретические расчеты, их энергии оценивались по соотношению, предложенному Шифри-
ным [316]. Согласно работе [316] энергии возбуждения идентичных состояний в молекулах
одного класса (АВ и АС) и значения межъядерных расстояний в основных состояниях этих молекул
связаны приближенным соотношением
Те (АВ)/Г. (АС) = ге (АС)/г. (АВ), A.89)
иг
Это соотношение было широко использовано при подготовке предыдущего издания данного
справочника [88], и последующие экспериментальные исследования (в частности, молекул A1F,
А1С1, BF) показали его удовлетворительную точность. Оценки по соотношению A.89)
корректировались на основании сравнения величин Те для других состояний молекул АВ и АС,
полученных по этому соотношению и известных из экспериментальных исследований или квантовомехани-
ческих расчетов.
Вклад s-ro электронного состояния в термодинамические функции газа тем существенней,
чем меньше величина (hc/kT)T(e*\ В тех случаях, когда постоянные в выражении Т(е'} -\-G{i) {v)-\-
-f-.Fi*') (/) известны, a (hcjk) T^ <[ ЗГ, составляющие i-го состояния (значения Q^'l вр и ее
производных) вычислялись по соотношениям A.73)—A.75), а значения Qm, Qxn и GВЯ — по
соотношениям A.76)—A.78). Если для некоторого к-то состояния (hc/k) Tlk) ^>ЪТ или основные постоянные
в выражении для G(k) (v)-\-Fltk) (/) неизвестны (такие, как <о,, Ве и энергия диссоциации),
расчет (^ вр при вычислении таблицы термодинамических функций в справочнике не проводился,
а принималось, что A//?^ (?koAbP = A//V) *??«. вр- В качестве /-го состояния выбиралось такое
электронное состояние, в котором значение ге и энергии диссоциации близки к ожидаемым величинам
для к-то состояния и в то же время известны постоянные Ykl в уравнении A.65). В частном
1 Этот вопрос обходится молчанием в литературе, по-видимому, на основании предположения, что с ростом
температуры молекулы диссоциируют до наступления заметной ионизации. Однако исследования последних лет (см.,
например, [3166а]) показали, что потенциалы ионизации соединений металлов могут быть существенно меньше их
энергий диссоциации и корректный учет ридберговских состояний таких молекул не менее существен, чем учет
высоких связанных и квазисвязанных колебательно-вращательных уровней основного электронного состояния.
я Для О2 в строгом квантовомеханическом расчете Шеффера [3320] было показано, что из валентных состояний
только 12 являются связанными, а 52 отталкивательными.
50
1.4. Двухатомные газы
случав это может быть основное электронное состояние данной молекулы. В этом случае выражения
для (?ви и ее производных вместо соотношений A.76)—A.78) принимают вид
i=X
«=Х
A.91)
t=Jt
где A?j = TW - GW @) - ^W GmJ, pk и Pj — статистический вес к-то и /-го состояний,
учитывающий мультиплетность и вырождение данного состояния и равный B5г("+1)с(<).
Для того чтобы оценить величину ошибок, которые вносятся таким упрощением в расчеты
термодинамических функций, в табл. 1.5 приведены значения Q$Xmtv для семи электронных
Таблица 1.5. Значения —
вр
для семи электронных состояние О2
Состояние
у»)
СМ
10000 К
I
II
20 000 К
I
II
X3S- 0 27 895 — 103 585 —
a4g 7 917,54 30 847 25 306 108 950 96 053
Wb+ 13 195,07 32 796 27 059 103 734 102 986
сгХ~ 33 055,71 67 192 71 847 127 870 279 520
Примечание. I — расчет по уравнению A.73); II — рас
Состояние
СМ
10 000 К
I
II
20000 К
I
II
Л3ДЯ 34 690,91 35 050 65 219 54 343 253 380
B3S+ 35 393,08 33 868 71 613 50 373 278 540
С32- 49 792,11 44 762 89 330 67 430 348 550
!чет по уравнению A.96).
Таблица 1.6 Сравнение термодинамических функций О2 (г), вычисленных с использованием уравнений
A.76)—A.78), A.90)—A.92) и A.96)
Расчет
по формулам
Расчет
по формулам
A.76)—A.78)
A.90)—A.92) *
A.76)—A.78) **
* Для состояний
10 000 К
295,05
295,02
294,88
335,83
335,86
336,18
41,439
42,843
47,379
A.76)-A.78)
A.90)—A.92) *
A.76)—A.78) **
20 000 К
322,47
323,05
324,89
359,58
361,99
369,74
27,681
31,735
47,326
Д 2m m a И ?32« значения QHI. вр и ее производных приняты равными
соответствующим величинам для состояния Xя 2J-
** Для всех возбужденных электронных состояний значения Q[H. Bp. QHL вр и <?ко]. Вр вычислены в
приближении «жесткий ротатор — гармонический осциллятор» (ур-ние A.96)).
51
Глава 1. Вычисление термодинамических функций газов
состояний О2 при 10 000 и 20 000 К, вычисленные по уравнению A.73) и значениям
постоянных из табл. 4.3 (см. гл. 4). Значения QBB, вычисленные с использованием этих данных
по соотношениям A. 76) и A.90) (в последнем случае принято, что для состояний с1!!", А3Аи, В3%*,
С3Е~ выполняется условие A/р{) Q^l.^ — O-IPx) Qifl вр) ПРИ Ю000 К, когда для этих четырех
состояний (hcfkT)T^ ]> 4,5, практически совпадают, несмотря на существенное различие
молекулярных постоянных и энергий диссоциации в этих и основном электронных состояниях. Практически
совпадают также (см. табл. 1.6) значения Ф° A0 000 К) и S°(l0 000 К), вычисленные с
использованием этих методов, однако расхождения в С°(Т) достигают 1,4 Дж • К • моль главным образом
из-за того, что Q^l. вр Для состояний Л3ДМ и Б32+ в 6—8 раз меньше величины Q*ox. вр. При
20000 К, когда для трех состояний (hcjkT) ТУ> ~ 2, расхождения в значениях (?вн, вычисленных
по тем же соотношениям, достигают около 7 %, а соответствующие расхождения в Ф°(Г), S°(T)
и CAT) составляют 0,5, 2 и 4 Дж • К • моль. Следует отметить, что такие заметные
расхождения обусловлены значительной разницей в энергии диссоциации и молекулярных постоянных
(ев, и ге) молекулы О2 в состоянии Х3%~ и состояниях А3Аи, В3Щ и_С3?~ (см. табл. 4. 3), которая
приводит к резкому различию в значениях Qli\ Qu> и особенно в Qli} (в 20—30 раз).
В то же время, если для состояний А3\ и В32* значения (?юм. вр и ее производных принять
по соответствующим величинам состояния С32~ (все три состояния являются изоконфигурацион-
ными и имеют относительно близкие значения постоянных), расхождения в табл. 1.6 уменьшаются
больше чем на порядок. Данные табл. 1.5, 1.6 и других аналогичных расчетов позволяют
оценить (см. раздел 1.6) влияние рассмотренных упрощений на вычисляемые таблицы
термодинамических функций в тех случаях, когда более точные расчеты невозможны из-за
отсутствия сведений о постоянных молекул газа в возбужденных электронных состояниях г.
Значения внутримолекулярных составляющих термодинамических функций двухатомных
газов в настоящем издании вычислялись по соотношениям
^P A.93)
lH°(T)-H°@)]
f
где о — число симметрии, равное 2 для гомоядерных и 1 для гетероядерных молекул; значения
Сип $вн и <?вн находятся по соотношениям A. 76)—A. 78) или A. 90)—A. 92).
1.5. Вычисление внутримолекулярных составляющих
термодинамических функций многоатомных газов
Статистическая сумма по внутримолекулярным состояниям многоатомной молекулы по
аналогии с двухатомными молекулами в общем случае может быть записана следующим образом:
02max vm ma
2 ... 2
2 2 B7 + 1)ехр[-П°(Л #)]• A-97)
1 В литературе для учета возбужденных электронных состояний молекул часто используется расчет QH^. вр и ее
производных в приближении «жесткий ротатор — гармонический осциллятор». В этом случае
«й..р = Л«?4"М1-е-"'). A-96)
где q{," = kTjhcBtf\ u{ = (hc/кТ) о,(".
В табл. 1.5 приведены значения <?кол. вр Для возбужденных состояний О2, вычисленные по уравнению A.96),
а в табл. 1.6 — значения термодинамических функций, полученные в этом приближении. Грубые ошибки,
особенно в значениях 5° (Т) и С°р (Т) (при высоких температурах ошибки в Qf*> и <?с<) превышают их величину в 10—>
20 и 40—100 раз соответственно), делают неоправданным применение этой методики в современных расчетах.
52
1.5. Многоатомные газы
В этом выражении первая сумма берется по всем ?-м связанным валентным электронным
состояниям молекулы, характеризующимся энергией возбуждения Т^ и статистическим весом р(.
Электронные состояния нелинейных многоатомных молекул невырождены, и поэтому
статистический вес таких состояний определяется только мультиплетностыо p4=2S-{-l, указанной
верхним индексом слева от символа типа состояния (гА, SB и т. п.). В случае линейных многоатомных
молекул статистические веса те же, что и для соответствующих состояний двухатомных молекул.
Суммирование по г; в выражении A.97) производится для всех m нормальных колебаний молекулы.
Если молекула, состоящая из N атомов, имеет t вырожденных нормальных колебаний с вырожде-
t
нием dn, то m = 3N— 5 — t для линейных молекул и m = 3N — 6 — 2 (^я—1) Для нелинейных
В=1
молекул. Статистический вес pv состояния (v1, v2, .. -,vm) равен
р. = П P,i(d,-i)V • С1-98)
Энергию колебательных уровней удобно представить в виде (см. Вулли [3998, 3997])
mm m m
Gn(v,, vo, ... v ) = S to v- 4- 51 %ij (и. — 1L- 2 ^_by_yfc 4- 2 V™«f (v., — 1) (у„ — 2) 4-
B=l И=1 B<A »=1
m mm
+ 2 V ^ ^ — 1)^*4- 2 У *м,л;.4-У\ gJnL, A.99)
в^А: в-</с<;у в^&
где ln=vn, vn—2, уя—4, . . . , 0 или 1 для дважды вырожденных колебаний. Для невырожденных
колебаний все 2я=0. Постоянные в уравнении A.99) связаны с частотами колебаний и постоянными
ангармоничности хпп, хпк, ynkJ и т. д., фигурирующими в традиционном выражении для энергии
колебательных уровней (см. Герцберг [69])
m m
+ 2 у- (».+тK+ 2 у-» (у»+тJ ("* + т)+
я=1 я^А
m •»
+ 2 ^
Эта связь выражается следующим образом:
т 2
/« A-103)
Уппп=:Утт' Snkj — Unkj' Уппк~Уппк- A. 104)
В настоящем издании в таблицах молекулярных постоянных многоатомных молекул
приводятся значения основных частот vB, соответствующих переходу между состояниями vn=0 и vn=l.
Очевидно, что в случае невырожденных колебаний vn=<BB, а для вырожденных колебаний уя=б)и+^ик-
Для оценки постоянных ангармоничности трехатомных изотопических молекул одинаковой
53
Глава 1. Вычисление термодинамических функций газов
симметрии в соответствии с работой [280 ] использовалось соотношение
^^фК- AЛ04а)
Суммирование по / в выражении A.97) должно проводиться для каждого значения v всех т
колебаний. Энергия вращательных уровней многоатомной молекулы выражается по-разному
в зависимости от симметрии молекулы.
Для линейных многоатомных молекул и молекул типа сферического волчка, как и для
двухатомных молекул, энергия вращательных уровней описывается выражением
F, G) = B.J (/ + 1) - D.P (/ +1K + ... A.105)
Для молекул типа симметричного волчка
F.(J, K) = B,J(I' + 1) + D,- B,)K2-DjP(J + 1)а- DjzJ G + 1)К- DKK\ A.106)
где К принимает значения от —7 до -f-7 и все уровни, соответствующие К~^>0, дважды
вырождены. В формулах A.105) и A. 106) D,, Dj, Djk и Dk — постоянные центробежного растяжения.
Для молекул типа асимметричного волчка
где Wjz принимает 27 + 1 различных значений для каждого 7 и эта величина соответствует К2
в случае симметричного волчка, но число подуровней в случае асимметричного волчка равно 27 -f- 1
для каждого 7, а не / + 1, как в случае симметричного волчка.
В выражениях A.105)—A.107) величины А„, Вр и Се — вращательные постоянные, связанные
с главными моментами инерции молекулы. В частности,
^ 5 С ' A.108)
где 1л, 1в, 1с соответствуют некоторой средней структуре молекулы в основном
колебательном состоянии.
Следует отметить, что в случае многоатомных молекул индекс v у вращательных постоянных
означает зависимость от BGex колебаний молекулы (поэтому, например, для трехатомной
молекулы запись Аа означает в действительности А000 и т. п.).
Зависимость вращательных постоянных от колебательных квантовых чисел можно выразить
соотношениями [3997]:
т т т
А, = Ао - 2 *ЯЧ - 2 ЧЛ - 2 «>Л. A. Ю9)
«=1 я=1 п<к
)!=1 И=1 «<fc
Если эту зависимость выразить в виде (см. [69])
п=1
И Т. Д., ТО
а»1+22 «-^ t
и=1 и=1 и=1 к<п
и т. д., а постоянные взаимодействия вращения и колебаний связаны следующими соотношениями
54
1.5. Многоатомные газы
Для вычисления постоянных колебательно-вращательного взаимодействия нелинейных
изотопных молекул одинаковой симметрии при отсутствии экспериментальных данных в настоящем
издании использовалось соотношение (см. [88])
<=КК)Х- A.115а)
Число симметрии о в выражении A-97) определяется типом симметрии молекулы (табл. 1.7).
Таблица 1.7. Числа симметрии многоатомных молекул различных типов симметрии
Точечная группа
симметрии
Сц С{, Cs
С3, С3е, Csh
с4, cip, cih
Сб. C5f) C5j,
/1 /~I /"I
1
2
3
4
5
6
Точечная группа
симметрии
D2, D2d, D2h(=Vh)
О„ Dsd, Dlh
Dit Did, Dik
D6, Dbd, Dbh
De, D6i, D%h
4
6
8
10
12
Точечная группа
симметрии
Ccoh
T, Td
oh
3
i
2
12
24
Как и в случае двухатомных молекул, непосредственное суммирование по колебательным
и вращательным уровням энергии является, в принципе, наиболее точным методом вычисления
статистической суммы по внутримолекулярным состояниям многоатомной молекулы A.97) и ее
производных. Однако практическая реализация этого метода возможна только при низких
температурах, когда основной вклад в статистическую сумму вносит небольшое число низколежащих
колебательно-вращательных уровней энергии основного электронного состояния молекулы.
При увеличении температуры применение метода непосредственного суммирования становится
невозможным. Это обусловлено как отсутствием данных о высоких колебательно-вращательных
уровнях энергии даже основного электронного состояния практически всех многоатомных
молекул, так и отсутствием достаточно строгих представлений о зависимости энергии таких уровней
от значений квантовых чисел vn и / и о верхних пределах изменения квантовых чисел vn и / в
связанных и квазисвязанных состояниях многоатомных молекул. Уравнения, приведенные выше,
удовлетворительно аппроксимируют энергию колебательно-вращательных состояний только
в узком интервале изменения (обычно малых значений) квантовых чисел vn и /, и сегодня нет
подходящих идей, как эти уравнения могут быть экстраполированы в область значений vn и /,
существенно выходящих за границы, для которых имеются экспериментальные данные. Весьма
отрывочные сведения имеются о возбужденных электронных состояниях большинства даже простейших
многоатомных молекул, в том числе низколежащих электронных состояний, мультиплетность
которых отличается от мультиплетности основного состояния. В то же время многоатомные
молекулы, в особенности молекулы с не полностью заполненными орбиталями, в основной
конфигурации электронной оболочки должны обладать, по аналогии с атомами и двухатомными молекулами,
рядом связанных валентных электронных состоянияй.
Все эти причины делают невозможным вычисление QBn и ее производных для многоатомных
молекул методом непосредственного суммирования по электронно-колебательно-вращательным
состояниям с помощью уравнений типа A.97). Поэтому современные методы расчета
термодинамических функций многоатомных газов основаны на том, что статистическая сумма по колебательно-
вращательным уровням энергии основного электронного состояния ^] во и ее производные
вычисляются в приближении «жесткий ротатор—гармонический осциллятор», а отклонения
молекул от этой модели, наличие возбужденных электронных состояний и другие эффекты
учитываются, если имеются соответствующие данные, в виде поправок. Это позволяет записать
статистическую сумму по внутримолекулярным состояниям и ее производные в виде
и получить для внутримолекулярных составляющих термодинамических функций соотношения
типа
Ф»°Я(Л_Ф
R
к. р. г. о
(Т)
A. 117)
55
Глава 1. Вычисление термодинамических функций газов
где рх — статистический вес основного электронного состояния и Д — поправка, учитывающая
возбужденные электронные состояния, ангармоничность колебаний, центробежное растяжение
и ряд других эффектов.
Вычисление термодинамических функций в приближении «жесткий ротатор — гармонический
осциллятор» . В этом приближении принимается, что колебания молекулы являются
гармоническими, отсутствуют центробежное растяжение молекулы при вращении и взаимодействие вращения
и колебаний, а верхние пределы по vn и / в статистической сумме и ее производных равны
бесконечности. Тогда
A.119)
и
для линейных молекул и
для нелинейных молекул.
В соответствии с выражением A.118) можно представить термодинамические функции как
суммы соответствующих составляющих. Если обозначить un=(kc/kT)vn, то соответствующие
составляющие термодинамических функций могут быть вычислены по соотношениям:
A.122)
И=1
[Н° (Т) — Н
RT
[Cl^nr.o ^.„„_ (_Ця)[1_ехр(_Ця)]-2 AЛ24)
И
ф».р(г)
1пТ — 1пВ0 — lna-f 0,363 80, A.125)
—-1 A.
для линейных молекул и
ядр '=±ЫТ— \\пА0ВйС0 — 1по +0,026 67, A.127)
з
ЛГ = Л ^T (
для нелинейных молекул.
Если вместо вращательных постоянных использовать главные моменты инерции, то в силу
соотношений A. 108) и значений фундаментальных постоянных, приведенных в Приложении 2,
Ф° (Т)
*'^ = In Т + In (IB ¦ 1039) — In о — 1,393 18 A.129)
56
1.5. Многоатомные газы
для линейных молекул и
R
— In a— 1,517 40
A.130)
для нелинейных молекул (где 1Л, 1В, 10 даны в г • сма).
В тех случаях, когда основные частоты молекул газа неизвестны, для расчета
термодинамических функций в приближении «жесткий ротатор — гармонический осциллятор» может быть
использован полуэмпирический метод, впервые примененный в первом издании настоящего
справочника [87]. В этом случае поступательные и вращательные составляющие термодинамических
функций вычисляются по обычным соотношениям A.9), A.10) и A.125)—A.130), а колебательные
составляющие — по приближенным соотношениям через колебательные составляющие
термодинамических функций других газов, молекулы которых имеют сходную структуру.
Приближенные соотношения, выбираемые для такого расчета, должны удовлетворять двум
требованиям:
а) Колебательные составляющие опорных молекул суммируются с коэффициентами,
определяемыми условиями материального баланса по всем элементам, образующим молекулу
рассматриваемого вещества, например Ф;>0 (PC13F2) г, Г)=0,4 Ф;.о (PFb, г, Г)+0,6 ф;.о (РС18, г, Т).
б) Если расчет может быть выполнен по нескольким наборам опорных молекул, необходимо
проанализировать на рядах однотипных соединений, какой набор может дать наиболее достоверные
величины для рассматриваемого вещества. Этот метод был широко использован во 2-м издании
справочника [88].
Аналогичный метод был предложен в работе [160а], однако без учета условия б).
Поправка на ангармоничность колебаний и взаимодействие колебаний и вращения. При
подготовке настоящего издания на основании работы Вулли [3997, 3998] были получены формулы
для расчета поправок на ангармоничность колебаний и взаимодействие колебаний и вращения
к значениям термодинамических функций, вычисленных в приближении «жесткий ротатор—
гармонический осциллятор». Эти формулы несколько сокращены по сравнению с оригинальной
формулой Вулли для поправки к Ф°{Т)Ш и дополнены отсутствующими в работе Вулли [3997, 3998]
аналитическими выражениями для поправок к [Н"(Т)—H°@)]/RT и C°pIR:
^f1 = 2 КIn \ eg !*!
+ 2
»<*
¦ (Г) - Я" @)I«., __V L Г« .
+ 3^(й„
Д/А+1»пА D, +1)
2 :
n<k<j
. + 2) r
С1-
A + 2dn (dn
. (d. + 1) >2>&V* BSA + «A -
A + 1) ^ («A + 2SA - 1)} +
+ 2
A.132)
57
Глава 1. Вычисление термодинамических функций газов
(
da (dn +1) гуп + 3da (dn +1) (d, + 2) тЗД] gA +
K+l) ^]+ХЙЯЯ Й/А+Ч (de+l) ^+3d. (d.+\) (dn+2)
+ ft- ИЛА + 4 (d.+1) f^]
[-2*rt (ЯА + «A) + *« К A + О ^ + S|
r.) s2+a|
«) #. + Щ B+rk) в\ + 4йАйЛ - 2aA - 4aA]
Л) - 2 IPA + uA + WltSfJSAtyj8*- (*•I33)
В этих формулах
8) йи а Кп = -Jy ^—Хпп + %»,« — "J 8т)»
S ^ ехр(—S,), 5„ = [1 —ехр(—mJ]-1.
Далее,
а =Is.t а =^»Л.1 J».
для линейных молекул и
в\
"яя — 2 [ А2 "Г 2Л0 "Г ?2 "Г 2В0 "г Q ~Г 2С0 J '
[* А А Л R R С1 С О ~\
^7-г^|-+^в7+ ^ ^ c0 ¦+¦ eg J
для нелинейных молекул.
Поправки на центробежное растяжение. Для линейных молекул, энергия вращательных
уровней которых описывается уравнением A. 105), поправки на центробежное растяжение могут быть
вычислены по соотношениям, полученным Мак-Дауэллом [2576]:
Ф(У) j96_ | js.t A. 134)
1—{ > rt Wh-* = 2fT + 20/2Г2 + 296/3Г3, A.135)
где f = kD0lhcB2u.
58
A.136)
1.5. Многоатомные газы
Для молекул типа сферического волчка эти соотношения имеют вид
Ч* V v ' и, | 1*J rrp | ^-J /2Ф2 I luJlJ ^З'ЛЗ | ivjoi ij X&rpi ^ 0
Л 16Г 4 2 '4 '4 4 .Se
A-138)
\c°p (Л1Д. p = 15 jT ^ 135yay2 t зю5/згз_|_ 84 375^^ A.139)
где a = hcDQ/k.
Для молекул типа симметричного волчка, энергия вращательных уровней которых описывается
уравнением A. 106), соответствующие выражения были получены Юнгманом [319] (см. также:
Мак-Дауэлл [2576]):
Ф{Т) ±Bb2-bl)T*; A.140)
—-— ц—= 2&, 71 —[— 3 Bbo ~~ Ь?) 712, A.142)
где
ma) Z)j + Bm + 3/?г2) ?)
Ь = ™' [A28 + 64m + 48m2 + 40m3 + 35m4) Z>3 + 2 A6т + 24т2 + 30т3 + 35т4)
+ 2 (8т2 + 20т3 + 35т4) (р&х + 4 ЯЗх) + 2 (Ют3 + 35т4) ?>лДх + 35т4Д1]
при т = В0/А0.
Наконец, для молекул типа асимметричного волчка на основании работы Вильсона [3959]
(см. также: Хачкурузов и Милевская [285]) можно записать:
»;(*-) [я°(Г)*°@)]ц.Р_г^ AЛ43)
где
[с;(у)] AЛ44)
Здесь х (в см~1) — постоянные центробежного растяжения (см.: Кивельсон и Вильсон [2263]).
Для плоских молекул T{(!jc='Teca(;=O, а также имеют место следующие соотношения:
Постоянные центробежного растяжения обычно находят путем обработки данных по тонкой
структуре спектров многоатомных молекул; в ряде случаев величины х рассчитываются
теоретически из силовых постоянных.
59
Глава 1. Вычисление термодинамических функций газов
Низкотемпературные квантовые поправки. При преобразовании статистической суммы по
вращательным состояниям к виду A.120) или A.121) в результате перехода от суммирования
к интегрированию по формуле Эйлера — Маклорена при низких температурах становятся
заметными поправки, которые обычно называются квантовыми и могут быть записаны как
-5^1 = а1Г-1 + а2Г-2 + °з?т-3, A.147)
(Г) -Н° @)].. „ = _ ^у! + 2^-2 + За8Г-»), A.148)
\СР
где
1 he D 1 / he D \2 . 8 / he „ \2 Do _ 8 / Ac
Q J. "^ U ¦ Si QA V JU "^ 0 / I 41 *S V A" ^ / it
для линейных молекул (в том числе и для двухатомных молекул) и
he Zx / he \2 Zf — 3Z2 / fee Y 4Zi —
A. 149)
8 /fee
~~ 2835 V A
_ Y
A 12' °2~V"F/ 560 ' as~[k) 14175 »
(» = L 2, 3)
для нелинейных молекул (см.: Стрипп и Кирквуд [3560]).
Поправки на кориолисово взаимодействие. В случае молекул типа симметричного волчка,
имеющих вырожденные колебания, в выражении для энергии вращательных уровней A.106)
появляется дополнительный член, учитывающий кориолисово взаимодействие 2AQK 2 ?Jn< где
п
5Я — постоянная кориолисова взаимодействия, соответствующая n-му вырожденному колебанию,
ln=vn, vn—2, vn—4, . . . , —vn. Поправки к термодинамическим функциям на это взаимодействие
(см. Мак-Дауэлл [2577]) имеют следующий вид:
?1 |ад 0-150)
[Но (Т) - Я° @)].. ,_2hcA V-t2rs2/2ars 4-в -1) A 151)
[с; (Г)]... = 2 ы_А^ 4vA {ЫпГА _ 4rA+Un _ 2), A.152)
= 2 _^
и
В формулах A. 150)—A.152) и„=^.п, гй = ехр(-ия) и *я = [1-вхр(-в1,)Г1.
Поправки на резонансное взаимодействие колебательных состояний. При случайном
совпадении энергии различных колебательных состояний имеет место их резонансное взаимодействие,
приводящее к смещению уровней относительно положения, описываемого уравнениями A.99)
или A.100). Изучены взаимодействия состояний (ух, v2, vs) и {vx—1, v2+2, v3) — резонанс Ферми
и состояний (ух ^ 2, v2, v3) и (ух—2, v2, г^з+2) — резонанс Дарлинга—Деннисона. Поправки к
значениям термодинамических функций, обусловленные этими взаимодействиями, могут быть
вычислены по соотношениям, полученным Вулли [3996]:
=
R 2 v2 [1 —ехр(-2«2)][1-ехр(-и2)]2 '
[Да(Г)-Д°@)]р.ф Ф;.ф(Г) о [„ Г, i ехр (-а,) ехр (-2а,) 1
RT = R— • Z \и2L1 + 1 - ехр (-Bl) + 1 - ехр (-2а2) J
60
1.5. Многоатомные газы
Г Л , ехр (-в>) ехр (-2в,) \ ,"»
Л. - R Z Г L 2 V + 1 - ехр (-«,) Т" 1 - ехр (-2b.) J ~ \J
ц2) , 2ехр(-2ц2) -I ,1
(_Ц2)]2+ [l_exp(-2B,)]»J 1/'
где PF0 — постоянная резонанса Ферми (в см), u2 = (hcjkT)^2 (предполагается, что Vj = 2v2);
фр. д (y) _ t2 ехр (-2и) , ,rR
R ~~~ ы2 Ц — ехр (—в)]* ' "" ^
[Я-(D -;О(°Iр.Д =3LU* ехр(-2ц) Г 1+ехР(-ц) _ Л
ЛГ ш2 [1 — ехр (—в)]4 |_ 1 — ехр (—и) J' v '
т2 „а
(-2») 0 f 1 , 1 + ехр (-в) » . 1 + 3 ехр (-и) + ехр (-2ц)
д = ^« ц _ ехр (-в)]* 2 I1 4И 1 - ехр (-и)
где х — постоянная резонанса Дарлинга—Деннисона (в см) и u=(hclkT)<u (u)=(v1+v2)/2).
Поправки на внутреннее вращение. Питцер и Гвинн [3034] разработали метод расчета поправок
к термодинамическим функциям на заторможенное внутреннее вращение для случая, когда это
вращение описывается потенциалом вида
A.159)
где Vo — барьер внутреннего вращения, п — число максимумов (минимумов) на потенциальной
кривой и ф — угол поворота волчка относительно оси вращения.
Метод Питцера и Гвинна основан на решении волнового уравнения с потенциалом A.159)
и последующем вычислении статсуммы для внутреннего вращения. В общем виде выражения для
соответствующих поправок имеют вид [74а]
.,(Т) _ ф°..(Т) (К. A
в.—-—r—{—л
[Я° (Т) - Н° @)].. . _ [Я° (Г) - Я° @)]е. , / [Д° (Г) - Н° @)].. , [Я° (Г) - Я° @)].. \ . „,
RT ~ RT \ RT RT )>"' '
\С°Р{Т)],.Ъ_ [С'р(Т)]втЯ ([С'р(Т)]вшЪ \С°р(Т)]3.в\
^ j (I.
_ _ _ ^ _ _
где величины
R R J' \ RT RT J { д д—
являющиеся разностью соответствующих функций для свободного (F0=0) и заторможенного
внутреннего вращения, находятся по специальным таблицам, составленным Питцером и Гвин-
ном, как функции двух аргументов: V0/RT и l/Qc#Е. Величина <?с „ является статистической
суммой по состояниям свободного внутреннего вращения:
2% BkTI1,vI'' 2,7928697'/» (/пр • 1038I/г
^ °-в nh п ' ^
где /пр — приведенный момент инерции волчка (в г-см2). Соответственно
Ф°^(Г) =Yln Т "Ьу111^ • Ю38) — Inn — 1,0271, A. 164)
\н (Л-Д @I„.,= р ^ c"b=y- A.165)
61
Глава 1. Вычисление термодинамических функций газов
Метод вычисления /пр для общего случая асимметричного волчка разработан Питцером [3034].
Для всей молекулы, имеющей массу М, включая вращающуюся группу в равновесном положении,
находятся главные центральные оси инерции 1, 2, 3 и главные моменты инерции относительна
этих осей Ia, Ib, 1с', затем проводятся координатные оси волчка так, чтобы ось z совпадала с осью
вращения волчка, ось х проходила через центр тяжести волчка и была перпендикулярна оси z
и ось у проходила через точку пересечения осей х, z и была бы перпендикулярна к ним. Атомы
волчка, лежащие на оси вращения z, из дальнейшего рассмотрения исключаются.
Если тк — масса к-то атома волчка, то А = 2 ти (х\ ~Ь 2/1) — момент инерции волчка относи-
к
тельно оси z, В = ^ткхк2к и C = '^imkykzk — произведения инерции, U — ^j,mkxk — фактор не-
к к к
сбалансированности волчка.
Затем находятся направляющие косинусы осей х, у, z относительно осей 1, 2, 3:
а»
а1У
а1*
а2*
а2*
а3*
а33'
а3*
Направление осей выбирается так, чтобы обе системы были либо правыми, либо левыми. При
этом определитель, составленный из направляющих косинусов, равен -)-1, что может служить для
проверки правильности вычисления направляющих косинусов.
Если обозначить вектор из центра тяжести молекулы в начало координат волчка через г,
а его проекцию на главные оси инерции молекулы — через rfl), r<2), гСз), то приведенный
момент инерции одного асимметричного волчка имеет вид
»=1, 2, 3
где рс<) = <Л4 — а*хВ — а.{&С-{-U (a*'1- V+1» _ a<+1. »r{i~n). Индексы i, i — 1 и г+1 могут
принимать значения 1, 2, 3 (в циклическом порядке), поэтому индекс i—1 в случае ?=1 равен 3,
а индекс г+1 в случае ?=3 равен 1.
Следует отметить, что для стабилизированного асимметричного волчка (т. е. волчка, у которого
ось вращения совпадает с одной из главных осей инерции), а также для симметричного волчка
выражение приобретает более простой вид (см. Питцер,Гвинн [3034]):
»=1, 2, 3
Последний член в уравнении A.160) добавляется в том случае, когда число максимумов на
потенциальной кривой не совпадает с числом симметрии волчка ат (относительно оси вращения).
В случае нескольких волчков описанный метод применяется к каждому из них в отдельности,
а составляющие термодинамических функций суммируются по волчкам.
Число колебаний, принимаемых во внимание при расчете составляющей гармонического
осциллятора, уменьшается на число внутренних вращений в молекуле, учитываемых по этому
методу.
Поправки на возбужденные электронные состояния. Если принять, что статистические суммы
по колебательно-вращательным состояниям основного и возбужденных электронных состояний
равны, составляющие возбужденных электронных состояний могут быть вычислены как поправки
к составляющей основного электронного состояния
62
1.6. Оценка погрешностей
RT
A.169)
Л
A.170)
где Г<'> — энергия возбуждения i-ro электронного состояния, р,. — статистический вес t-ro
состояния, рх — статистический вес основного состояния.
В более общем случае, если принять во внимание различие Qmj вр и ее производных для
основного и возбужденных электронных состояний, внутримолекулярные составляющие
термодинамических функций могут быть найдены по соотношениям
\но(т)-но@)]т_-:
RT
2, [
Ас
Ж
+
RT
]й1.чЛ Г
he
кол. вр \ )
л
exp
\С
p \* Лвн
;T ° Л J
[p rp I 6Xp I ~~~ ~~»
A.171)
A.172)
. he кол. вр '
:exp \-wTff>+ д-
(Г) - Я» @)]Й1.
вол. вр
RT
[Clm (T)]m*.
ot. вр
л
л
A. 173)
где-
,(T) [Ho (T) - HO (O)J^i. :
-5
— —колебательно-вращательные составляющие
соответствующих термодинамических функций, вычисленные по соотношениям A.'122) — A.130)
для каждого 1-го электронного состояния отдельно в приближении «жесткий ротатор —
гармонический осциллятор» с учетом всех возможных поправок.
1.6. Оценка погрешностей вычисленных таблиц термодинамических функций газов
Погрешности в значениях термодинамических функций газов, вычисленных статистическими
методами, могут быть обусловлены тремя причинами: неточностью значений фундаментальных
постоянных (-Й, hc/k=c2 и N), принятых в расчете, неточностью использованного метода расчета
внутримолекулярных составляющих термодинамических функций и неточностью (или отсутствием)
данных об уровнях энергии молекул.
63
Глава 1. Вычисление термодинамических функций газов
Несмотря на относительно высокую точность большинства констант в согласованной системе
фундаментальных постоянных, рекомендованной КОДАТА—MGHG [1001 ], значения двух
постоянных, с2 и В., имеют удвоенные стандартные отклонения, равные 0,0062 и 0,0064%. Погрешности
в значениях термодинамических функций из-за неточности фундаментальных постоянных могут
быть оценены в G,0—7,5) -10~3% от их величины, т; е. достигают 0,01—0,03 Дж'К~1-моль~1 в
значениях Ф° (Т) и S° (Т) при величине последних от 140 до 450 Дж-К~1»моль~11. Для ряда газов
при умеренных температурах это является основной причиной, лимитирующей точность расчета
таблиц их термодинамических функций.
Погрешности внутримолекулярных составляющих термодинамических функций одноатомных
газов. Пренебрежение ридберговскими состояниями атомов (ионов) — единственная неточность
использованной методики расчета. Эта неточность начинает сказываться при температурах,
приближающихся к 0,1 (hc/k) /0 и выше, где /0 — потенциал ионизации атомов газа. Оценка
соответствующих погрешностей в значениях Ф° (Т), выполненная на основании A.29), показала (см. табл. 1.1),
что для 0,1 (hc/k) 10 < Г ^ 0,13 (hc/k) 10 они не превышают 0,1—0,3 Дж-К • моль, а для
0,13 (hc/k) Io < Т < 0,18 (hcjk) /0 составляют 0,3—1,0 Дж-К~1-моль~1. Погрешности в значениях
Н° (Т) — Н° @), S° (Т) и С°' (Т) становятся заметными при более низких температурах и имеют
большую величину: в S° (T) — примерно в 5—8 раз больше, чем в Ф° (Г) при той же температуре.
Сведения о валентных электронных состояниях атомов (ионов) с достраивающимися s- и р-
электронными оболочками (элементы главных периодов) достаточно полны и надежны. Поэтому
при Т <^ 0,08 (hc/k) Io погрешности вычисляемых значений Ф° (Т) и S° (T) не превышают 0,03—
0,05 Дж'К~1'МОль~1. У атомов с достраивающейся d-оболочкой (переходные металлы) данные о
валентных электронных состояниях, соответствующих электронным конфигурациям, отличающимся
от конфигурации в основном состоянии, менее полны. Это может приводить к погрешностям в
значениях Ф° (Т) уже при Т ^ 5000 К (Т ^ 0,05 (hc/k) Io), и оценка величины этих погрешностей
требует тщательного анализа в каждом конкретном случае. У атомов (ионов) с достраивающейся
/-оболочкой (лантаноиды и актиноиды) сведения даже о низких валентных электронных состояниях
достаточно отрывочны, что может сказываться на точности расчета термодинамических функций
соответствующих газов уже при температурах в 2000 — 3000 К (примерно 0,04 (hc/k) /0);
корректная оценка погрешностей результатов расчетов для этих газов в настоящее время затруднительна.
Погрешности расчетов внутримолекулярных составляющих термодинамических функций
двухатомных газов. При расчете (?кол. щ> и ее производных непосредственным суммированием по
колебательно-вращательным уровням энергии двухатомных молекул с ограничением числа связанных
и квазисвязанных состояний по предельной кривой диссоциации (уравнения A.73)—A.75))
единственной методической неточностью является пренебрежение конечной шириной квазисвязанных
уровней энергии. Соответствующие погрешности в значениях термодинамических функций растут
по мере уменьшения отношения Бо/Г, где Do — энергия диссоциации молекулы и Г — температура
газа. Как показали расчеты [2296 ] для молекулярного водорода, где этот эффект наиболее
существен из-за того, что вероятность туннельного эффекта максимальна для атома водорода,
соответствующие погрешности не превышают 0,02 Дж-К~1-моль~1 в значениях Ф° (Т) и S0 (Т) при Т ^
^0,13 (hc/k) Do. Можно считать, что для молекул, не содержащих водород, погрешности из-за этого
эффекта не превышают 0,02 Дж-К~1-моль~1 вплоть до Г^ 0,25 (hc/k) Do.
Существенно большими должны быть погрешности при высоких температурах из-за
пренебрежения ридберговскими состояниями молекул при вычислении QBB и ее производных. На основании
оценок для одноатомных газов можно показать, что при Т ^ 0,06 (hc/k) /0, ^0,1 (hc/k) 10 и
^ 0,13 (hc/k) /0 погрешности в значениях Ф° (Т) составляют соответственно 0,02, 0,1 и
0,5 Дж-К^-моль.
Погрешности в значениях термодинамических функций газов из-за неточности молекулярных
постоянных могут быть легко найдены как суммы погрешностей из-за неточности отдельных
постоянных. Однако погрешности молекулярных постоянных, приводимые в экспериментальных
исследованиях для таких величин, как шв, <лехе, . . . Ве, о1г . . . и т. п., как правило, не могут быть
использованы для оценки достоверности вычисляемых термодинамических функций. Эти погреш-
Из соотношений A0)—A3) и (9а) видно, что эта погрешность в основном обусловлена неточностью газовой
постоянной R и, существенно в меньшей степени, постоянной с2.
64
1.6. Оценка погрешностей
ности характеризуют только точность описания экспериментальных данных (совокупности частот
переходов между небольшим числом колебательно-вращательных состояний, обычно состояний
с низкими значениями квантовых чисел и и /), а необходимо знать или оценить точность
аппроксимации энергии всех колебательно-вращательных состояний, ограниченных предельной кривой
диссоциации, уравнением с имеющимся набором постоянных. Последнее является значительно
более сложной, часто неразрешимой сегодня задачей. В связи с этим при подготовке издания в
результате многочисленных расчетов были выработаны следующие принципы.
1. При низких температурах (Т ^ 1000 К) основные погрешности в значениях
термодинамических функций двухатомных газов обусловлены:
а) неоднозначностью данных о типе основного электронного состояния молекулы;
б) отсутствием данных или погрешностями в известных значениях постоянных,
характеризующих мультиплетное расщепление и спин-орбитальное взаимодействие в основном электронном
состоянии;
в) неоднозначностью имеющихся (или отсутствием) данных о существовании о возбужденных
электронных состояний с Д77'*'3 < 4000 см;
г) отсутствием данных об основной частоте А&/2, частоте колебания ше, а также вращательной
постоянной Ве, или неточностью этих постоянных, превышающих 5% от их величины.
Во всех четырех случаях погрешности в значениях термодинамических функций могут быть
определены непосредственным расчетом с учетом неоднозначности имеющейся информации. В
текстах по расчету термодинамических функций соответствующих газов приводятся детали
конкретных оценок.
2. При умеренных и высоких температурах основные погрешности в значениях
термодинамических функций двухатомных газов обусловлены:
а) Отсутствием данных о существовании связанных валентных состояний с энергиями Т^ <^
< 8kT/kc, где Т — температура газа, или неточностью данных об энергии подобных состояний.
Соответствующие погрешности в первом случае могут быть определены на основании оценки вероятного
числа таких состояний с энергиями диссоциации не менее 8000 см. Такая оценка проводилась на
основании рассмотрения конфигурации электронной оболочки молекулы и имеющихся данных для
других молекул с таким же количеством валентных электронов (см. соответствующие тексты) и
их энергий возбуждения. Влияние неточности оцененных энергий возбуждения на значения Ф° (Т)
вычислялось из
8Ф°(Г)_1/у/р< Q&\ вр he 8г7.«,1рхпГ he ()lV A.174)
б) Использованием приближенных методов для оценки значений и1ШХ, /шах и построения
предельной кривой диссоциации из-за отсутствия экспериментальных данных о высоких
колебательно-вращательных уровнях энергии молекул газа в их основном и возбужденных электронных
состояниях. Соответствующие погрешности оценивались на основании модельных расчетов свойств
газов, для молекул которых имеются достаточно полные данные (Н*, Н2, HF и F2) и могли быть
сопоставлены результаты расчетов по этим данным и по небольшой части данных, дополненных
методами, описанными на с. 38—44. Эти расчеты (см., например, табл. 1.4) позволяют утверждать,
что если экспериментальные данные о величинах G (v) и Fc (/) имеются примерно для 10%
колебательных состояний A0% от величины Ушах), погрешности в значениях Ф° (Т) несущественны
« 0,02 Дж-К^-моль-1) при Т < 0,13 {heIk) Do для основного состояния и Т < 0,13 {heIk) GW+
-\-Щр) для возбужденных состояний. При более высоких температурах погрешности становятся
заметными и могут достигать ОДДж • К • моль'1 при Т — 0,25 {he/к) Do (или 0,25 {he/к) (D(<) + Tli)))
и при Г = 0,33(ВДО0 (или 0,33 (hc/k) (DCO + T(i>)) около 0,3 Дж • К • моль.
в) Неточностью принятого значения энергии диссоциации молекулы в ее основном или
возбужденном состоянии. Погрешность в 5—10% в этой величине приводит к погрешностям в
значениях термодинамических функций газа при Т > 0,25 {heIk) Do или Т > 0,25 {heIk) (D^+TW).
Оценка погрешности может быть выполнена расчетом значений итл% и /тах для двух значений со-
1 Это соотношение справедливо только при условии, что ЪТ({> <<; Ти>; в противном случае погрешности
оценивались после расчета значений Ф° (Т), соответствующих двум значениям энергии i-го состояния: Гг'> и 74'1 —bT(i).
65
Глава 1. Вычисление термодинамических функций газов
ответствующей величины и последующим вычислением двух значений Ф° (Т) для нескольких
температур.
г) Введением дальнейших приближений в расчет QBB и ее производных. Замена суммирования
по / интегрированием требует использовать линейную зависимость /maXj,, от v, что приводит к
заметным погрешностям при Т > 0,3 (hcfk) Do (или > 0,3 (hc/k) (ГМ+Щ*'>)). Соответствующие
погрешности в Ф°G7) составляют 0,1 — 0,2 Дж • К • моль при Т = 0,3 (hcjk)D0 (или 0,33 (Ас/к) X
XG""-f D(O)) и 0,2—0,3 Дж-К^-моль при Г = 0,5 (hc/k) Do. Замена <?$. вР и е.& производных
на C'ffi вр и ее производные может приводить к погрешностям при Т^> 0,25 (he/к) 'Г*'. Эти
погрешности достигают 0,1 и 0,2 Дж • К • моль в значениях Ф° (Г) на одно состояние (при р. = рх^
для T = O,Z(hcjk) Г(" и 0,5 (Ас/А;) Г'*'. Особенно значительными эти погрешности оказываются
в тех случаях, когда молекула в возбужденном состоянии имеет более высокую энергию
диссоциации и мультиплетность, чем в основном состоянии.
Погрешности расчетов внутримолекулярных составляющих термодинамических функций
многоатомных газов. В случае многоатомных газов достаточно корректные расчеты
термодинамических функций возможны только при невысоких температурах (Т <[ 0,06 (hc/k)Ec, где Ео —
энергия наиболее слабой связи в молекуле), когда заселено только несколько нижних колебательных
состояний, невысокие вращательные состояния и молекулы газа не имеют низких возбужденных
состояний. В этих условиях погрешности термодинамических функций, вычисленных в
приближении «жесткий ротатор — гармонический осциллятор», определяются главным образом неточностью
молекулярных постоянных (основных частот, вращательных постоянных или произведения
моментов инерции) и могут быть найдены по соотношению:
(Г)
тс
у/
RT
A. 175)
где dk — вырождение А>го колебания, vft — его основная частота, Н°{Т)—Н°@) — энтальпия к-го
гармонического осциллятора.
При более высоких температурах достаточно строгая оценка погрешностей, возникающих
из-за того, что модель «жесткий ротатор — гармонический осциллятор» не пригодна для
описания высоких колебательно-вращательных состояний
многоатомной молекулы, невозможна. Это обусловлено
тем, что отсутствуют как экспериментальные данные, так и
теоретические представления о поведении таких состояний
вблизи диссоциационного предела. Некоторое представление о
величине соответствующих погрешностей может дать
сравнение термодинамических функций двухатомных газов,
вычисленных непосредственным суммированием по колебательно-
вращательным уровням энергии и в приближении «жесткий
ротатор — гармонический осциллятор». Расчеты для ряда
молекул показали, что разность соответствующих величин грубо
пропорциональна величине составляющей гармонического
осциллятора и достигает 5—7% от нее. В настоящем издании
погрешности в значениях Ф°(Г) многоатомных газов,
обусловленные приближенностью модели «жесткий ротатор —
гармонический осциллятор», оцениваются в 5% от
величины Ф°о (Т) и суммируются с погрешностями из-за
неточности молекулярных постоянных, вычисленными по
уравнению A.175). Очевидно, что такая оценка достаточно
условна, так как для низкочастотных деформационных
колебаний, если потенциальная функция последних существенно
отлична от гармонического потенциала, расчет в
приближении «жесткий ротатор—гармонический осциллятор»
может дать ошибку более 20%, как это имеет место для С3 и
С3О3(см. [110]).
Рис. 1.8. Разность значений
приведенной; энергии Гиббса, вычисленной
для двухатомных молекул методом
непосредственного суммирования и
приближенным методом Майера и Гепперт-
Майер
66
1.6. Оценка погрешностей
Расчеты в приближении «ангармонический осциллятор — колеблющийся нежесткий ротатора
могут быть выполнены для немногих газов из-за того, что постоянные ангармоничности,
взаимодействия колебаний и вращения и т. п. известны только для небольшого числа многоатомные
молекул, рассматриваемых в настоящем издании. Учет отклонения от модели «жесткий ротатор —
гармонический осциллятор» в методе, использованном в справочнике, как и во всех других
методах, известных в литературе, дает уменьшение погрешностей расчета только в ограниченном
интервале температур. Это обусловлено тем, что расчетные формулы во всех этих методах получены в
предположении, что в статистических суммах по колебательным и вращательным состояниям верхние
пределы равны бесконечности. Таким образом, эти методы аналогичны методу Майера и Гепперт-
Майер для двухатомных молекул (см., с. 48). Сравнение результатов расчетов ln[QKOX вр методом
непосредственного суммирования и методом Майера и Гепперт-Майер для большого числа
двухатомных молекул в широком интервале температур, выполненное при подготовке справочника,
показало, что разность значений lnQKM вр, вычисленных этими двумя методами (рис. 1.8), является
гладкой функцией величины (hclkf) Do, где Do — энергия диссоциации молекулы иГ — температура
газа. При Т < 0,06 (hc/k) Do результаты расчетов обоими методами совпадают практически во
всех случаях. При 0,06 (hc/k)D0 < Т < 0,2 (hc/k) Do метод Майера и Гепперт-Майер дает заниженные
величины (до 0,3 Дж-К~1-моль~1 в значениях Ф° (Т) для неполярных и до 1,0 Дж-К~1»моль~а
для полярных молекул) из-за того, что в нем не учитывается ряд особенностей изменения энергии
колебательно-вращательных состояний молекулы с ростом квантовых чисел v и /. При Т ]>
>0,25 (hc/k) Do метод Майера и Гепперт-Майер из-за использования условий уш„=оо и /шах „ = оо
дает завышенные результаты, причем ошибка стремительно растет с-ростом температуры*.
Аналогичные ошибки, но ббльшие по величине и при более низких температурах, имеют место в
значениях S° (Т) и С°р (Т) (см. пример для F2 в табл. 1.4).
Оценка погрешностей в значениях Ф° (Т) для многоатомных газов, вычисленных при подготовке
настоящего издания в приближении «ангармонический осциллятор—колеблющийся нежесткий
ротатор», проводилась по кривой типа приведенной
на рисунке. При этом принималось, что
многоатомная молекула состоит из двухатомных
фрагментов, число которых равно числу валентных
химических связей в молекуле. Определив из
термохимических данных энергию каждой связи, из
рис. 1.8 можно найти погрешность в значении
Ф° (Т), приходящуюся на каждое валентное
колебание в молекуле. Сумма этих величия с
удовлетворительной точностью должна описывать
погрешности значений Ф°(Т), обусловленные
неточностью использованного метода расчета Фкол. Bp(^)-
Погрешности из-за неточности молекулярных
постоянных, использованных в расчете в таком.приб-
лижении, оказываются несущественными по
сравнению с погрешностями, обусловленными
применением приближенной методики, и могут не
учитываться в соответствующих оценках.
Трудно поддаются оценке погрешности в
значениях термодинамических функций
многоатомных газов из-за отсутствия данных о
возбужденных электронных состояниях их молекул. В тех
случаях, когда погрешности принятых значений
энергий возбуждения значительны, погрешности
термодинамических функций могут быть оценены
так же, как для двухатомных газов по
соотношениям типа A.174). Однако для подавляющего
большинства многоатомных молекул сведения о
возбужденных электронных состояниях
отсутствуют вообще, что не является свидетельством
отсутствия у них связанных валентных состояний,
40
гооо
чо о о т,и
и С° Т (б) да»
Рис. 1.9. Функции ФС(Т) (а) р
ZrO (г), рассчитанные при значениях Те (а3 А),
1 — 500 см; 2 — 1750 см; 3 — 3500 см3
67
Глава 1. Вычисление термодинамических функции газов
особенно если молекулы в основном электронном состоянии имеют не полностью заполненную
электронную оболочку. Следует отметить, что погрешности в значениях Ф° (Т) таких газов,
обусловленные использованием приближения «жесткий ротатор—гармонический осциллятор»,
а также неточных значений молекулярных постоянных в основном состоянии, обычно настолько
велики, что погрешности из-за пренебрежения неизвестными возбужденными состояниями, как
правило, становятся существенными только при температурах выше 3000 К. Значительно больше
соответствующие ошибки в значениях других термодинамических функций (см. ниже).
Если оценивать в целом точность вычисленных в справочнике таблиц термодинамических
функций, то для всех одноатомных газов (нейтральных и заряженных), кроме актиноидов и
лантаноидов, погрешности в вычисленных значениях Ф° (Т) при Т7^ 5000 К не превышают
0,05 Дж-К • моль, а для подавляющего большинства и 0,02 Дж-К~1-моль~1. Для
двухатомных газов, таких как СО, HF, N2, CN, ОН, Н2, NO, NaCl, ВО, BF и многих других,
у молекул которых можно исключить возможность существования неизвестных связанных
возбужденных электронных состояний с Т1<> < 25 000 см, погрешности в значениях Ф°(Г) при
Т ^ 5000 К также не превышают 0,03—0,05 Дж-К-моль~1. Только у нескольких
многоатомных газов, типа Н2О, СО2, HCN, . . . значения Ф°G7) могут быть вычислены с такой точностью
для температур до 5000 К. Для большинства других многоатомных газов при Т ~ 4000—5000 К
погрешности превышают 0,5 CN — 6) Дж-К«моль, где N — число атомов в молекуле.
В настоящем издании в текстах глав приводятся оценки погрешностей, как правило, только для
приведенной энергии Гиббса Ф° (Т), поскольку именно эти погрешности определяют неточность
вычисленных значений lg K° и равновесных составов реагирующих химических систем, а также
потому, что оценить погрешности этой функции можно точнее, чем погрешности в значениях S°(T),
Н° (Т)—Н° @) и С° (Т). Однако можно связать величину погрешностей в значениях Ф°GТ) и
в других функциях, когда они обусловлены одними и теми же причинами.
Погрешности в значениях S° (Т) и С° (Т) из-за неточности колебательных постоянных шк или
ш4 примерно в 2 раза больше, чем в Ф° (Т) при той же температуре. Погрешности из-за неточности
постоянной Ве или величины IaIbIc одинаковы в значениях Ф° {Т) ж S° (T) и практически равны
нулю в С°р (Т) и Н° (Т) — Н° @). Погрешности из-за пренебрежения необходимостью ограничения
суммирования по колебательно-вращательным уровням энергии (в настоящем справочнике
только у многоатомных газов) в значениях S° (Т) могут быть в 5—10 раз больше, чем в Ф° (Т).
В теплоемкости многоатомного газа, которая должна иметь максимумы на зависимости С°р от Т,
соответствующие погрешности будут еще больше.
Пренебрежение возбужденными электронными состояниями или неопределенность их энергий
возбуждения также приводят к существенно большим погрешностям в значениях S° (Т) и С (Т),
а следовательно, и в Н°{Т) — Н°@), чем в Ф°(Т). Поскольку при высоких температурах
основным источником ошибок в большинстве случаев является отсутствие надежных данных об
энергии связанных возбужденных состояний молекул, на рис. 1.9 приведены зависимости от Т значений
С° (Т) и Ф° (Т) для ZrO (г), вычисленные для нескольких значений энергии возбуждения
состояния а3Д этой молекулы. Из рис. 1.9 видно, насколько существенно влияет неопределенность в
значении Гс*' только одного электронного состояния на точность результатов расчета
термодинамических функций соответствующего газа.
Глава 2
ВЫЧИСЛЕНИЕ ТЕРМОДИНАМИЧЕСКИХ
ФУНКЦИЙ ВЕЩЕСТВ
В КОНДЕНСИРОВАННОМ СОСТОЯНИИ
2.1. Общие замечания
В настоящем издании вычисления таблиц термодинамических функций веществ в твердом и
жидком состояниях проводились на основании результатов экспериментальных измерений
теплоемкости С° (Т), энтальпии Н° (Т2) —Н°(Т^), температур и энтальпий фазовых переходов. Ниже
будут кратко рассмотрены методы обработки экспериментальных данных, использованные в
настоящем справочнике, и некоторые особенности существующих методов измерения этих величин х.
Термодинамические свойства многих веществ изучены недостаточно полно, особенно при
высоких температурах. Для ряда веществ имеющиеся данные либо ненадежны, либо вообще неизвестны.
Тем не менее различные приближенные методы, опирающиеся на эмпирические или
полуэмпирические закономерности, позволяют, во многих случаях с достаточной точностью, оценивать
неизвестные термодинамические величины. Поэтому в настоящем разделе наряду с методами
обработки экспериментальных данных будут также изложены приближенные методы оценки
термодинамических величин, использовавшиеся при подготовке справочника.
В соответствии с общими соотношениями (см. с. 24) термодинамические функции чистых
веществ в стандартном конденсированном состоянии могут быть вычислены по соотношениям:
ТО} ТО)
H°(T) — H°{0)= j C°pm(T)dT-\-btrH°™(Tg)+ j С?» (T) dT + „ .
0 TO)
J c;u+i) (T) dr, B.
J
TO)
TO) TO)
TJ + [
г то)
(T)
B.2)
Ф°(Г)^°(Л-ДО(ГOЯ°@), B.3)
где C°A), C°B), . . ., C°(<+1) — теплоемкости вещества при постоянном давлении в различных
фазовых состояниях, определенные как функции температуры; Т$, Tf), . . ., Щ) — температуры
равновесных фазовых переходов; &trHOii) (ВД, Д<ГЯО(>) (Т$), ..., btrH°U) (TIP) — изменения энтальпии
при фазовых переходах. В выражение B.2) входит величина S @) — энтропия вещества
при Г=0. Согласно третьему закону термодинамики, энтропия чистых веществ в полностью
упорядоченном кристаллическом состоянии равна нулю (без составляющих смешения изотопов).
Однако для ряда веществ упорядочение при Г=0 не достигается, в этом случае при расчетах
термодинамических функций должна учитываться так называемая остаточная энтропия. Величины,
1 Подробное изложение методов измерения можно найти в ряде специальных монографий и обзоров (см.:
М. М. Попов. Термометрия и калориметрия [200]; С. М. Скуратов, В. П. Колесов, А. Ф. Воробьев.
Термохимия, ч. 1 и 2 [236]; Экспериментальная термохимия. Под ред. Ф. Д. Россини [1431]).
69
Глава 2. Вычисление термодинамических функций веществ в конденсированном состоянии
стоящие в правой части уравнений B.1) и B.2), являются исходными для расчета
термодинамических функций веществ в твердом и жидком состояниях.
В общем случае теплоемкость чистого кристаллического вещества при постоянном давлении
¦можно представить в виде суммы ряда составляющих:
+ C. + C,.a + ..., B.4)
где Cq — решеточная составляющая теплоемкости; Ср—С, — составляющая теплоемкости,
обусловленная термическим расширением; Се — электронная составляющая; Ст — магнитная (маг-
нонная) составляющая; Сf — составляющая, связанная с переходами на более высокие
энергетические уровни (эффект Шоттки); Су — составляющая, связанная с процессами упорядочения
(структурное, магнитное, электрическое упорядочение); Съ — составляющая, обусловленная
образованием равновесных вакансий в решетке; Са 0 — ядерная составляющая, связанная со
сверхтонким расщеплением, обусловленным спинами ядер.
Наибольший вклад в теплоемкость твердых тел (см. теорию теплоемкости твердых тел в
монографиях [1689, 97] и обзоре [177]) при не очень низких температурах вносит решеточная
составляющая теплоемкости. Согласно Эйнштейну [1395], колебания атомов в одноатомном твердом теле
могут рассматриваться как колебания квантовых гармонических осцилляторов с частотой ш и
уровнями энергии
B.5)
где е, — энергия осциллятора на v-тл уровне, v — квантовое число, h — постоянная Планка, с —
скорость света. Тогда теплоемкость 1 моля вещества при постоянном объеме равна
СЛТ)~ [ехр(ед/Г)-1]2 =3?(8Д/Г), B.6)
где Е @Е/Т) — функция теплоемкости одномерного гармонического осциллятора, f)E—(hc/k) ш —
характеристическая температура Эйнштейна.
В теории Дебая [1189] одноатомное кристаллическое тело рассматривается как непрерывная
изотропная упругая среда, имеющая бесконечно большое число собственных колебаний с частотами
от 0 до о)шах, причем функция распределения частот в этом интервале имеет вид
/(ш) = Ссо2. B.7)
Теплоемкость 1 моля вещества при постоянном объеме по Дебаю равна
C°q(T)^=3D(^IT), B.8)
где D (9д/Г) представляет собой функцию Дебая для теплоемкости, а 9д = (hc/k) свшах —
характеристическую температуру Дебая. Таблицы значений С° (Т), S° (Г), Ф° (Т) и [0"° (Т) — Н° @)]/Г,
представленных как функции 8л/7\ см. в [1626а].
ji-Яри низких температурах (Г<^0Л) уравнение B.8) принимает простой вид
С;(Г) = 4^G79дK = рГ, B.9)
согласно которому теплоемкость твердого тела пропорциональна третьей степени температуры.
В динамической теории кристаллической решетки Борна и Кармана [706, 707] кристалл
рассматривается как система гармонических осцилляторов, частоты которых соответствуют
собственным частотам кристалла. Теплоемкость сложного соединения, молекулы которого состоят из п
атомов, согласно этой теории может быть представлена в виде комбинации функций Дебая и
Эйнштейна:
С; (Т) = 2 D FВ</Г) + 2 Е (9,./Г), B. 10)
где бд1? бдг и 9д3 — характеристические температуры Дебая, отражающие упругие свойства
анизотропной кристаллической решетки, а 6В< — характеристические температуры Эйнштейна,
связанные с собственными колебаниями решетки.
70
2.1. Общие замечания
Составляющая теплоемкости, обусловленная термическим расширением, численно равна
разности между теплоемкостями при постоянном давлении и постоянном объеме (С°—С°). Из общих
соотношений термодинамики (см., например, [97]) следует:
W)Pl\dP) BЛ1)
и
c;-c;=Ca)*FZ7p, B.12)
где а. — коэффициент линейного расширения, р — коэффициент изотермической сжимаемости.
Ввиду отсутствия экспериментальных данных о величине |3, для выделения составляющей
{С —С°) в значениях теплоемкости при высоких температурах часто применяются приближенные
соотношения
С°р — C°t = kC*T B.13)
или
С°р-С: = к'С1Т, B.14)
а также уравнение Нернста—Линденмана
С" —С° = 0,0214 С» ?-да 0.0214 С\1-. B.15)
v F 1m 1m
Для электронной составляющей теплоемкости твердых металлов справедливо (см., например,
1177]) соотношение
С, = 4-^ = Т7\ B.16)
где f=3A;i?/2em — коэффициент электронной теплоемкости, а ет — максимальная энергия
электронов (ширина заполненной зоны).
При предельно низких температурах (Т < 5 К), когда другие составляющие теплоемкости,
кроме решеточной и электронной, отсутствуют (либо пренебрежимо малы), теплоемкость металла
может быть аппроксимирована двучленным уравнением
С;(Т) = $Т3 + ЪТ. B.17)
Магнитная составляющая теплоемкости имеется только у магнитоупорядоченных веществ
(ферромагнетиков, антиферромагнетиков, ферримагнетиков). Появление этой составляющей
обусловлено возбуждением спиновой системы таких веществ. Магнитная составляющая
теплоемкости для ферромагнетиков (ниже точки Кюри) пропорциональна Т1*-:
СЦТ) = к'Т\ B.18)
а для антиферромагнетиковТ(ниже точки Нееля) — пропорциональна Г3:
С*т(Т) = к"Т\ B.19)
Составляющая теплоемкости, связанная с эффектом Шоттки (электронные переходы между
различными энергетическими уровнями), может дать заметный вклад в теплоемкость некоторых
веществ, в частности лантаноидов, актиноидов и их соединений. Составляющая теплоемкости,
связанная с процессами упорядочения (Су), проявляется в виде так называемой Х-аномалии
теплоемкости. Теория (см. [273, с. 280—285]) дает для Су качественную [зависимость от температуры,
близкую к наблюдаемой на опыте Х-аномалии, и позволяет в ряде случаев рассчитать энтропию
упорядочения. Составляющая теплоемкости, обусловленная образованием равновесных вакансий
в решетке (Св), может быть рассчитана, если известна концентрация вакансий хотя бы при одной
температуре, по уравнению
J(A) B.20)
где Ео — энергия образования вакансий при Т=0. К сожалению, достоверных данных о
концентрациях вакансий, даже для металлов, в настоящее время нет. Данные о концентрациях вакансий,
полученные различными методами, отличаются друг от друга, иногда на несколько порядков.
71
Глава 2. Вычисление термодинамических функций веществ в ¦ конденсированном состоянии
Составляющая теплоемкости, обусловленная сверхтонким расщеплением уровней энергии (так
называемый ядерный эффект Шоттки), проявляется в редких случаях при самых низких
температурах (порядка 1 К и ниже). Вклад этой составляющей в энтропию является по существу ядерной
составляющей полных термодинамических функций и сохраняется одинаковым для элемента во
всех его соединениях, эта составляющая не входит в «практические» значения термодинамических
функций (см. с. 25).
Рассмотрение характера температурной зависимости различных составляющих теплоемкости
твердых тел (а также анализ экспериментальных данных) приводит к выводу, что в общем случае
трудно (а во многих случаях невозможно) подобрать уравнение, которое удовлетворительно
аппроксимировало бы экспериментальные данные и описывало зависимость теплоемкости от температуры
во всем интервале температур от Г=0 до Тт (или Ttr). Ввиду этого целесообразно разбить этот
интервал на два: от Г=0 до стандартной температуры 298,15 К и от последней до Тт. Это выделение
по крайней мере двух интервалов температур оправдано также тем, что в первом интервале
основным экспериментальным методом исследования являются измерения теплоемкости, а во
втором интервале — измерения изменения энтальпии Н° (Т) — Н° B98,15 К). Обычно различаются
также методы обработки соответствующих экспериментальных данных при расчете
термодинамических функций в этих интервалах температур.
2.2. Вычисление термодинамических функций веществ в кристаллическом состоянии
при Т < 298,15 К
Теплоемкость твердых веществ при низких температурах характеризуется резкой зависимостью
от температуры, причем теплоемкости некоторых веществ при одной и той же температуре могут
отличаться на 2—3 порядка. Экспериментальные измерения теплоемкости при низких
температурах проводятся от «азотных» E5 К), «водородных» A2 К) и в некоторых случаях — от
«гелиевых» A—4 К) температур 1. Анализ источников ошибок (см., например, [246]) и сопоставление
данных по низкотемпературной теплоемкости, полученных в различных исследованиях, приводят
к выводу, что погрешность наиболее точных измерений в настоящее время составляет не менее
0,1—0,2% в интервале температур от 20 до 298,15 К, а ниже 20 К повышается до 0,5—1%
и выше. Точность измерений теплоемкости при низких температурах определяется не только
погрешностями метода измерений теплоемкости. Резко увеличивают погрешности определения
низкотемпературной теплоемкости такие факторы, как: а) неблагоприятное соотношение между
теплоемкостями образца и контейнера, которое может ухудшаться при более низких температурах;
б) наличие примесей, особенно тяжелых металлов, имеющих низкую 6л и дающих поэтому
большой вклад в теплоемкость образца при самых низких температурах; в) наличие при низких
температурах фазовых переходов, которые не осуществляются полностью и поэтому могут внести
в измерения теплоемкости и расчеты энтропии существенные погрешности. Дополнительные
погрешности могут быть внесены при температурах ниже 20 К различием между практическими
температурными шкалами, применяемыми в США [1973], СССР [34] и других странах, и
термодинамической температурной шкалой; это различие может достигать нескольких процентов при
температурах ниже 10 К.
Вследствие указанных причин следует осторожно относиться к оценке точности результатов
измерений теплоемкости при низких температурах и последующих расчетов термодинамических
функций при 298,15 К из этих данных. Хотя авторы отдельных работ часто оценивают точность
измерений теплоемкости и вычисленных значений энтропии в 0,1%, в действительности
погрешности этих данных могут оказаться существенно выше вследствие неучтенных источников
ошибок.
При вычислении величин S° B98,15 К) на основании экспериментальных данных
существенная ошибка может вноситься экстраполяцией теплоемкости к абсолютному нулю. Экстраполяция
решеточной теплоемкости может быть сделана достаточно корректно только при самых низких
температурах, когда она подчиняется закону Дебая B. 9). Однако даже в случае простых
кристаллических решеток это имеет место при температурах порядка @,02—0,01) 0^. Сложные и анизо-
1 Использование магнитных свойств некоторых солей дает возможность проводить измерения теплоемкости при
еще более низких температурах, вплоть до 0,03—0,05 К.
72
2.3. Кристаллические вещества при 298,15
тройные решетки (например, слоистые) имеют кривую теплоемкости, которая даже
приблизительно не описывается уравнением Дебая. В этих случаях для экстраполяции теплоемкости к нулю
часто применяется комбинация функций Дебая и Эйнштейна (типа уравнения B.10)). В настоящем
издании для экстраполяции теплоемкости к Т=0 использовалось упрощенное уравнение,
предложенное Келли [2221J:
я—1
где 0в и 9я — усредненные значения характеристических температур Дебая и Эйнштейна г.
Экспериментальные данные по теплоемкости в области низких температур, обычно в интервале
50—100 К, аппроксимировались уравнением B.21) (см. [2221, с. 6]).
Погрешность энтропии, полученной при такой экстраполяции теплоемкости к Т=0, обычно
оценивается в 10%. Однако сопоставление вычисленных таким образом величин с результатами
более поздних экспериментальных измерений, показывает, что даже в случае отсутствия аномалий
теплоемкости погрешность в значениях энтропии, обусловленная экстраполяцией, может
достигать 20—30%. Поэтому существенно, чтобы энтропия данного вещества при температуре нижнего
предела измерений, вычисленная в результате экстраполяции теплоемкости к Т=0, не
превышала величину допустимой погрешности в значении S0 B98,15 К). Для веществ, имеющих при
низких температурах только решеточную составляющую теплоемкости, этот критерий может быть
связан с характеристической температурой Дебая (см. [88, с. 142—143]). В этих случаях только
для немногих веществ, имеющих 6Л порядка 1000 К, термодинамические функции при 298,15 К
могут быть вычислены с достаточной точностью, если измерения теплоемкости проводились выше
55 К (от «азотных» температур). Для веществ с 0^ ^ 200 К необходимая точность может быть
достигнута, если имеются измерения теплоемкости от «водородных» температур A0—12 К), а для
веществ с меньшими характеристическими температурами Дебая — от «гелиевых» температур
A—4 К).
В настоящем справочнике расчет термодинамических функций твердых веществ при
температурах до 298,15 К проводился следующим образом. Анализировались все известные в литературе
данные по низкотемпературной теплоемкости, проводился выбор наиболее надежных из них,
причем при необходимости проводилось усреднение данных, полученных в разных работах с
учетом их различной точности. Экстраполяция теплоемкости к У=0 осуществлялась либо по формуле
Дебая B. 9), либо с использованием комбинации функций Дебая и Эйнштейна B. 21). Выбранные
значения теплоемкости табулировались в интервале 0—100 К с шагом 5 К и в интервале 100—
298,15 К с шагом 10 К. Для веществ с низкими температурами Дебая (бл < 100 К) или в случаях
аномального хода теплоемкости табулирование проводилось с меньшим шагом. Расчет значений
Н° (Г) — Н° @) и S° (Г) проводился численным интегрированием уравнений B.1) и B. 2). В
случае аномалий теплоемкости, связанных с фазовыми переходами второго рода, проводилось
выделение «нормальной» составляющей теплоемкости и ее «аномальной» части.
2.3. Вычисление термодинамических функций веществ
в кристаллическом состоянии при 298,15 К <^ Т <I Tm
Непосредственные измерения теплоемкости веществ с помощью адиабатических калориметров
при высоких температурах наталкиваются на экспериментальные трудности, увеличивающиеся
с повышением температуры. Как правило, такие измерения ограничиваются температурами 1000—
1300 К, причем точность определения теплоемкости при высоких температурах невысока (порядка
2—3%). Практически при температурах выше 1000 К эти методы применяются только для
исследования веществ, имеющих аномалии в изменении теплоемкости или фазовые переходы. В
последнее десятилетие получили развитие модуляционный и импульсный методы измерения
теплоемкости веществ, проводящих ток (методы «нагретой проволоки») (см. [135а]). Эти методы позволяют
определять теплоемкость при существенно более высоких температурах (примерно до 3000 К).
Однако точность результатов измерений невелика и не превышает 5—10% при температурах
1 В случае молекулярных кристаллов значения ЬЕ могут быть вычислены по основным частотам соответствующей
молекулы.
73
Глава 2. Вычисление термодинамических функций веществ в конденсированном состоянии
2000—3000 К. Кроме того, эти методы позволяют исследовать ограниченный круг веществ —
металлы и тугоплавкие соединения, имеющие металлическую проводимость.
Более универсальными и точными являются измерения изменения энтальпии твердых и жидких
веществ с помощью метода смешения. Этот метод позволяет проводить измерения в широком
интервале температур — от комнатных до 2000—3000 К с точностью, которая составляет в
современных работах около 0,5% , а в лучших достигает 0,1—0,3% х. Теплоемкости могут быть найдены
из этих данных путем дифференцирования изменения энтальпии по температуре:
r°iT\— d[H°(T)-H°(T0)] 99
Lp\l> — Щ • (z-zz)
Для аналитического выражения зависимости теплоемкости от температуры при температурах
выше 298,15 К применяются уравнения различного типа. Наиболее часто употребляется
уравнение, предложенное Майером и Келли [2504]
— сТ~2. B.23)
Это уравнение удовлетворительно отражает быстрое изменение производной теплоемкости
по температуре при сравнительно низких температурах и практическое постоянство dC° (T)fdT
при высоких температурах. В интервале температур до 1000—1500 К уравнение B.23) обычно
описывает теплоемкость вещества с точностью 1—2%, а соответствующее выражение для
изменения энтальпии
Н°(Т) — Н° B98,15 К) = а + F/2) Т2 + сТ'1 + d B. 24)
— данные по энтальпии с точностью 0,3—0,5%. Однако в ряде случаев уравнения B. 23) и B.24)
не позволяют аппроксимировать экспериментальные данные по теплоемкости и энтальпии с
удовлетворительной точностью. В связи с этим были предложены более сложные уравнения, например
с экспоненциальным членом типа B.20), который должен отражать быстрое возрастание
теплоемкости твердых веществ при высоких температурах, или паде-аппроксиманты 2.
В настоящем справочнике для аппроксимации теплоемкости твердых веществ (и жидкостей)
применялись полиномы типа B.23), иногда с включением дополнительных членов с более
высокими положительными степенями температуры:
Ср{Т) = а + ЪТ — cT^ + dT2, B.25)
С°р(Т) = а-\- ЪТ — cT~2 + dT2-{-eTs, B.26)
С°р (Т) = а + ЬТ — сТ'2 + dT2 + eTs + fTK B. 27)
При выборе типа уравнения учитывались следующие факторы: 1) ширина рассматриваемого
интервала температур; 2) характер зависимости теплоемкости от температуры в этом интервале;
3) точность исходных экспериментальных данных по энтальпии и теплоемкости; 4) необходимость
экстраполяции теплоемкости в область температур выше того интервала, для которого имеются
экспериментальные данные. Применение четырехчленного полинома B.25), как правило, заметно
улучшает (по сравнению с B.23)) описание экспериментальных данных по энтальпии (и
теплоемкости), полученных в интервале температур до 1500—2000 К. Он может использоваться для
экстраполяции теплоемкости и к более высоким температурам, если интервал экстраполяции
не очень велик. Применение для аппроксимации пяти- и шестичленных полиномов B. 26) и B. 27)
оправдано в сравнительно редких случаях, когда имеет место сложная зависимость теплоемкости
от температуры, получены достаточно точные экспериментальные данные для широкого интервала
1 К числу недостатков метода смешения следует отнести возможность ошибок, связанных с неравновесностью
конечного состояния при быстрой закалке исследуемых образцов во время опыта, а также нечувствительность метода
к'фазовым переходам, энтальпии которых малы.
2 Фокин и Пучкова [272] недавно использовали дробно-рациональную функцию Паде
С, (Т) = ЧТ + 2 а„ТA + ^ W') B- 28)
с добавлением экспоненциального члена B. 20) для аппроксимации теплоемкости нескольких тугоплавких
металлов от Г=0 до температуры плавления.
74
2.4. Фазовые переходы и полиморфные превращения
температур, а уравнения более простого типа не обеспечивают хорошего описания
экспериментальных данных. Использование уравнений типа B. 26) и B. 27) нежелательно для далекой
экстраполяции теплоемкости в область высоких температур.
Очевидно, что уравнения, аппроксимирующие данные по энтальпии и теплоемкости при
высоких температурах, должны быть согласованы с результатами измерений теплоемкости при низких
температурах. Это может быть достигнуто введением граничных условий, например при 298,15 К:
d[H°(T)— H° B98,15 К)]
= С B98,15 К), B.29)
7=298,15 К р\ ' /< \ I
йТ
[/Г(Г)-Я°B98,15К)]г=298,1БК==0 B.30)
(вместо 298,15 К может быть взята любая другая температура).
Для аппроксимации экспериментальных данных по энтальпии полиномами типа B.23),
{2.25)—B.27) использовался метод, предложенный Шомейтом [3413] и видоизмененный
авторами настоящего издания. Этот метод позволяет учесть граничные условия B.29) и B.30) и
состоит в том, что по экспериментальным значениям Н°{Т)—Н°(Тг) и СЛТ-^ вычисляются
значения функции Шомейта
{[Я°(Г)-Я°(Г1I-С;(Г1)[Г-Г1]}Г
F(T) = (Г-Г,)» • (Z-S1>
Затем методом наименьших квадратов с учетом неравноточности значений F (Т) определяются
коэффициенты аппроксимационных уравнений четырех типов:
B.32)
B.33)
B.34)
B.35)
которые позволяют рассчитать коэффициенты уравнений B.23)—B.27). Эти соотношения для
уравнения B. 27) имеют вид *:
g = E< f = D-2gT1, e = C-2fT1-BgTl, c = ATv B.36)
b = B — 2eT1 — ЪЩ — 4gT*,
а = С; G\) - 2ЪТХ + А — 2,еТ\ - kfT\ — bgT{,
При подготовке настоящего издания совместной обработке результатов измерений энтальпии,
полученных в нескольких работах, предшествовала обработка данных каждой работы отдельно.
Такая предварительная обработка позволяет получить сведения о воспроизводимости измерений
в каждой работе, о наличии выпадающих точек (отклоняющихся на величину большую За), о
согласованности этих данных с принятым значением С° B\), а также уточнить опенку достоверности
результатов измерений в каждой отдельной работе. Последующая совместная обработка данных
всех работ проводилась с учетом их различной точности. Детали анализа в каждом конкретном
случае обсуждаются в текстах соответствующих глав.
2.4. Учет фазовых переходов и полиморфных превращений
В ]настоящзм издании приводятся таблицы термодинамических функций равновесных
кристаллических модификаций твердых веществ. По мере повышения температуры кристаллические вещества
могут иметь фазовые переходы. Различаются фазовые переходы первого рода, при которых внут-
Величина g в уравнениях B. 36) — константа интегрирования по Т уравнения B. 27).
75
Глава 2. Вычисление термодинамических функций веществ в конденсированном состоянии
ренняя энергия, энтальпия и плотность вещества изменяются скачком при температуре фазового
перехода, и фазовые переходы второго рода, при которых не происходит скачкообразного
изменения этих величин, однако их частные производные (теплоемкость, сжимаемость и
коэффициент термического расширения) изменяются скачком в точке превращения.
К фазовым переходам первого рода относятся полиморфные превращения и плавление.
Полиморфные превращения твердых веществ сопровождаются изменением их кристаллической
структуры. Почти для всех кристаллических фаз, рассматриваемых в справочнике, известны данные
0 кристаллических структурах.
Полиморфизм весьма распространен среди кристаллических веществ, особенно при высоких
давлениях. Сведения о сингониях и структурных типах рассматриваемых в справочнике веществ
даны в соответствующих текстах, более подробные сведения, в том числе о превращениях при
высоких давлениях, включая параметры тройных точек, приведены в справочнике «Термические
константы веществ» [258].
Фазовые переходы второго рода могут иметь различный характер. В отдельных случаях к
фазовым переходам второго рода относят частично замороженные превращения, протекающие в
некотором интервале температур вследствие кинетических затруднений. В справочнике такие
превращения рассматриваются как фазовые переходы первого рода, а энтальпии превращения
относятся к одной температуре равновесного превращения. К структурным фазовым переходам
второго рода принадлежат явления упорядочения (образование сверхструктур), фазовые переходы,
связанные с началом заторможенного или свободного вращения групп в молекулярных
кристаллах, фазовые переходы, вызванные изменением симметрии решетки вследствие искажений
структуры.
К магнитным фазовым переходам второго рода относятся магнитные упорядочения с
образованием ферромагнетика (точка Кюри) или антиферромагнетика (точка Нееля). При охлаждении
парамагнетик может последовательно оказываться в нескольких антиферромагнитных
состояниях (с разной степенью компенсации магнитных моментов) и затем переходить в ферромагнитное
состояние; соответственно этому у вещества может быть как точка Кюри (при относительно низкой
температуре), так и одна или несколько точек Нееля (при более высоких температурах). Помимо
магнитных, существуют электрические переходы. При охлаждении параэлектриков наблюдаются
случаи их фазовых переходов в антисегнетоэлектрические состояния (точки Нееля) и в сегнето-
электрическое состояние (точка Кюри) 1. В литературе часто употребляется термин «температура
(точка) фазового перехода второго рода». Строго говоря, этот термин противоречит понятию
фазового перехода второго рода, который происходит в конечном, и иногда достаточно широком,
интервале температур (в десятки и даже сотни градусов). Обычно за температуру фазового
перехода второго рода принимают температуру максимума кривой теплоемкости 2. При температуре
фазового перехода второго рода теплоемкость не падает до нормальной (со стороны
высокотемпературной фазы), что объясняется сохранением «ближнего» порядка в некотором интервале
температур.
Точность измерения температур фазовых переходов обычно зависит не столько от точности
измерений температуры, сколько от таких факторов, как наличие примесей, кинетика фазового
перехода, метод фиксации точки фазового перехода и др. Часто данные по температурам фазовых
переходов, измеренные в различных работах, отличаются на несколько десятков градусов, ввиду
чего выбор наиболее надежного значения температуры равновесного фазового превращения
представляет определенные трудности. В литературе встречаются неверные указания на
существование фазовых переходов, полученные, как правило, термографическим методом или при
исследованиях диаграмм состояния соответствующих систем. В ряде случаев эти сведения обусловлены
наличием примесей, образующих низкоплавкие эвтектики с исследуемым вещестгом. Поэтому
данные о наличии фазовых переходов, не подтвержденные структурными исследованиями,
в справочнике не учитывались.
1 В литературе отсутствует унификация терминологии магнитных и электрических фазовых переходов; в частности,
в некоторых работах точки Нееля обозначены, как «антиферромагнитные точки Кюри».
2 Иногда наблюдаются расхождения между температурами магнитных фазовых переходов второго рода,
определенных по максимуму кривой теплоемкости и по результатам магнитных измерений. В справочнике отдается
предпочтение данным, полученным из измерений теплоемкости, поскольку на результаты магнитных измерений
могут оказать влияние небольшие количества магнитных примесей.
76
2.4. Фазовые переходы и полиморфные превращения
Энтальпии полиморфных превращений (так же, как энтальпии плавления) могут быть
определены непосредственным измерением энтальпии превращения в адиабатических калориметрах,
а также по разности энтальпии низкотемпературной и высокотемпературной фаз. Первый метод
дает обычно более точные значения, особенно в случаях, когда энтальпия превращения невелика,
однако он применяется только при температурах не выше 1000 К. Второй метод измерения
энтальпии превращения как разности значений энтальпии двух фаз при температуре превращения
менее точен, однако его достоинством является универсальность и отсутствие ограничений в
отношении температуры фазового перехода, а также то, что в этом случае уравнения для теплоемкости
и изменения энтальпии автоматически согласованы со значением энтальпии превращения, что
весьма важно для последующего расчета термодинамических функций.
В случае фазовых переходов второго рода изотермическая энтальпия перехода отсутствует.
Однако в литературе принято называть изменением энтальпии и энтропии при фазовом переходе
второго рода величины, получаемые интегрированием аномальной части теплоемкости в
некотором температурном интервале (от Тг до Т2 на рис. 2.1) по уравнениям:
= lH°(T,)-H°(T1)]-[H°(T2)-H°(T1)\mVM=
\ о О г ПО / ПП \ о О / гтл \ ] г по / гр \ ГтО / гр \~j 1 Р» 8.Н0М J гр
ЛЬ = [о (У2) — Л (ii)J — [Л G2)—-о (-/1)JH0PM= \ ——j, аТ.
B.37)
B. 38)
Неточность определения «нормальной» части теплоемкости путем экстраполяции
теплоемкости высокотемпературной и низкотемпературной фаз к температуре максимума теплоемкости
вносит значительную погрешность в вычисляемые по уравнениям B. 37) и B.38) значения &Н° и Д5°.
Однако ошибки, связанные с выделением нормальной части теплоемкости, не влияют на точность
расчета термодинамических функций вследствие их компенсации.
Температуры плавления почти всех веществ, рассмотренных в настоящем издании, выбраны
на основании экспериментальных определений. Точность измерения температуры плавления весьма
различна и зависит не только от погрешностей измерения температуры, но и от таких факторов,
как чистота исследованных образцов, стехиометричность состава образцов (в случае соединений
с областью гомогенности), сохранение стехиометрии в процессе измерений, возможное
взаимодействие вещества с материалом контейнера или газовой фазой, точность фиксации момента плавления,
а также от принятой в данной работе температурной шкалы и отклонений этой шкалы от
принятой в настоящее время международной практической температурной шкалы 1968 г.
(МПТШ-68) (см. Приложение 2). При невысоких
температурах температуры плавления измеряются весьма точно
(в лучших работах с точностью 0,01—0,001 К). С
повышением температуры точность измерений падает. Так, точность
точек МПТШ-68 составляет для цинка F92,73 К+0,03 К),
а для серебра A235,08+0,2 К) и золота A337,58+0,2 К).
Реальная точность определения температур плавления
веществ примерно до 1300 К составляет в лучших
современных работах 1—2 К. Для более тугоплавких веществ
(примерно до 2000 К) погрешности возрастают до 5—10 К, а при
темпратурах выше 2000 К могут достигать 20—50 К и выше.
Энтальпии плавления, как и энтальпии полиморфных
превращений (см. выше), определяются по результатам
непосредственных калориметрических измерений и по
разности энтальпий кристаллической и жидкой фаз. Для
тугоплавких соединений с Тm > 1000 К экспериментальные
данные по энтальпиям плавления получены преимущественно рис 2Л. Криваятеплоемкостикристал_
вторым способом. Наибольшие трудности при измерении лического вещества в области фазового
энтальпий плавления вызывает подбор материала контейнера, перехода второго рода (аномалия Х-типа)
77
Глава 2. Вычисление термодинамических функций веществ в конденсированном состоянии
который бы не взаимодействовал с расплавом тугоплавкого соединения. В последние годы для
тугоплавких металлов эти трудности были преодолены применением «левитационного метода» (удержания
расплавленного металла в магнитной ловушке) (см. [32а]). Тем не менее надежные
калориметрические данные по энтальпии плавления имеются только для части рассматриваемых в справочнике
веществ. В некоторых случаях значения энтальпии плавления основаны на измерениях методом
количественной термографии, точность таких данных обычно не выше 10—20%. В тех случаях,,
когда при быстром охлаждении расплава образуются стекла (например, В2О3, SiO2), энтальпия
плавления может быть определена на основании энтальпий образования стекла и равновесной
кристаллической модификации при стандартной температуре и данных по изменению энтальпии
для стекла и кристаллического вещества. Для ряда соединений экспериментальные данные
отсутствуют и значения энтальпии плавления оценивались при подготовке справочника.
Полученные таким образом величины могут иметь погрешности, достигающие 20—30%.
2.5. Термодинамические функции веществ в жидком состоянии
Данные по теплоемкости или энтальпии в жидком состоянии при высоких температурах имеются
только для ограниченного числа неорганических веществ, обычно в сравнительно узком интервале
температур. Точность данных по теплоемкости жидкостей, полученных непосредственно или
дифференцированием результатов измерения энтальпии, не очень высока, обычно не лучше 1—3%.
Рассмотрение имеющегося экспериментального материала показывает, что теплоемкость
расплавленных неорганических веществ в большинстве случаев близка к их теплоемкости в твердом
состоянии около точки плавления. Обычно теплоемкость расплава несколько выше теплоемкости
твердого вещества (в среднем на 5—10%). Однако имеется много исключений из этого правила.
Так, у воды теплоемкость в 2 раза превышает теплоемкость льда вблизи точки плавления, у
тугоплавких металлов разность теплоемкости твердой и жидкой фаз вблизи точки плавления может
иметь разные знаки и достигать 20—30% от величины теплоемкости. С ростом температуры
теплоемкость жидких веществ вдали от критической точки изменяется незначительно. Для многих
жидкостей теплоемкость в пределах точности измерений остается постоянной в интервале
нескольких сот градусов. Однако в ряде случаев теплоемкость жидкостей медленно растет с
повышением температуры (в среднем на 0,5—1% на 100 К). Для некоторых металлов и их соединений
в жидком состоянии (Li, Na, К, Hg, Pb, LiF и др.) теплоемкость сначала уменьшается (примерно
на 1—2% на 100 К в интервале 300—500 К выше точки плавления), а после достижения
минимума начинает медленно возрастать *. Такой характер изменения теплоемкости жидкостей
наблюдается в широкой области температур от точки плавления почти до критической области. Вблизи
критической точки имеет место резкое увеличение теплоемкости жидкой фазы (в несколько раз).
В настоящем справочнике теплоемкость жидкости принималась постоянной:
B.39)
B.40)
если экспериментальные данные свидетельствуют, что в пределах погрешности*"измерений она
постоянна или если она была оценена из-за отсутствия экспериментальных данных. В тех
случаях, когда точность экспериментальных данных достаточна для выявления линейной
зависимости теплоемкости в достаточно широком интервале 'температур (в случае возрастания
теплоемкости), использовались уравнения типа
С°р(Т) = а + ЪТ, B.41)
Н°(Т)-Н'(Тт) = аТ+^Т*-с. B.42)
В тех случаях, когда экспериментальные данные свидетельствуют о нелинейной зависимости
С° (Т), могут быть использованы уравнение типаМайера—Келли B. 23)"или более спожные
уравнения типа B.25)—B.27). При экстраполяции теплоемкости в область высоких температур при-
1 Недавно Хох и др. [1964, 1965] предприняли попытку объяснить такой характер изменения теплоемкости
у'некоторых металлов и их соединений с помощью выделения трех составляющих теплоемкости.
78
2.6. Оценка погрешностей
менение таких сложных уравнений может привести к существенным ошибкам. Поэтому для
определения коэффициентов в соответствующих уравнениях использовался следующий
искусственный прием. Экстраполяция теплоемкости выше Тг (Тг — верхняя температура исследованного
интервала) проводилась при условии, что она постоянная или линейно увеличивающаяся с ростом
температуры. Затем вычислялись несколько значений Н° (Т) — Н° (Т2), обычно через 300—500 К,
включая самую высокую температуру, приводимую в таблице термодинамических функций.
Точность этих значений оценивалась с учетом точности экспериментального значения Н° G\)—
Н° (Т№) и оцененной величины Н (Т)—Н G\).
Совместная обработка экспериментальных и оцененных значений энтальпии методом Шомейта
(см. выше) проводилась с использованием уравнения B. 23), ^которое обычно удовлетворительно
описывает всю совокупность данных.
Эмпирические закономерности. Келли [2220] и несколько позднее Дуглас [1307] обобщили
экспериментальные данные по теплоемкости жидкостей и пришли к выводу, что у расплавленных
элементов теплоемкость приближенно равна 31 Дж-К^-моль. Для неорганических соединений
они рекомендовали значения около 34га Дж-К^-моль, где га— число атомов в грамм-формуле
данного соединения. Дуглас [1307] показал также, что соли некоторых кислородных
кислот (нитраты, сульфаты) в жидком состоянии имеют меньшую теплоемкость (от 21/г
до 29ге Дж-К-моль), гидроокиси и силикаты — около 29га Дж-К^-моль, бораты, титанаты,
хроматы и некоторые другие — от 31га до 38га Дж-К^-моль. Работы последних лет внесли,
однако, существенные коррективы в эти эмпирические правила; это касается в первую очередь
теплоемкости расплавленных металлов. Измерения энтальпии многих металлов, выполненные
«левитационным» методом, показали, что у расплавленных переходных, лантаноидных и
актиноидных элементов теплоемкость существенно больше и достигает 42—50 Дж-К^-моль. Такие
высокие значения теплоемкостей, по-видимому, объясняются наличием у указанных металлов
d- и /-электронов, обусловливающих значительную электронную составляющую теплоемкости.
При подготовке справочника для неорганических веществ, кроме элементов с незаполненными
d- и /-электронными оболочками и их соединений, в отсутствие экспериментальных данных
теплоемкость элементов в жидком состоянии принималась равной 31га Дж-К-моль. Теплоемкость
соединений принималась равной 34га Дж -К -моль, если экспериментальные данные для сходных
соединений не позволяли уточнить эту оценку. Детали оценок приведены в текстах
соответствующих глав.
2.6. Оценка погрешностей термодинамических функций веществ
в конденсированном состоянии
Оценка точности термодинамических величин, выбранных по экспериментальным данным,
проводилась с учетом их случайных и систематических погрешностей. Случайные погрешности
принимались как 95%-ный доверительный интервал, что было возможно далеко не для всех
экспериментальных работ ввиду отсутствия в них необходимых сведений. Существенно труднее оценить
возможные систематические погрешности. Эта оценка проводилась с учетом точности методов,
примененных в каждой работе, чистоты исследованных препаратов, их фазового состояния,
совершенства кристаллической решетки, степени дисперсности, а также с учетом замечаний о
возможных источниках систематических ошибок, сделанных в разделах 2.2—2.5. Важное значение
имеет также сопоставление данных, полученных различными экспериментальными методами,
а также сравнение результатов измерений свойств других веществ, проведенных в этих работах,
с результатами измерений других авторов.
Следует отметить, что авторы экспериментальных исследований часто завышают точность
своих измерений, недооценивая влияние систематических ошибок. Поэтому погрешности
постоянных, принятых для расчетов таблиц термодинамических свойств, как правило, отличаются
от приведенных в оригинальных работах.
При подготовке справочника для расчета таблиц термодинамических свойств многих веществ
пришлось использовать величины, полученные с помощью приближенных оценок. Определение
погрешностей таких величин носит ориентировочный характер. Особенно трудно оценить
погрешность теплоемкости расплавленных веществ в тех случаях, когда она получена далекой экстраполя-
79
Глава 2. Вычисление термодинамических функций веществ в конденсированном состоянии
цией. Если экспериментальные данные для жидкости известны для 2"^ Tv принимается, что в
интервале от Тх до 7\+500 К погрешность оцененного значения составляет 5%, в интервале от
7^+500 К до Г1+1500 К — 10%, а при более высоких температурах — 15%.
Погрешности табулированных значений термодинамических функций веществ в
конденсированном состоянии оценивались с учетом точности величин, принятых в расчете. Следует отметить,
что погрешности измерений теплоемкости при низких температурах существенно больше влияют
на точность определения S° B98,15 К), чем на точность определения Н° B98,15 К)—Н° @),
поскольку функция С (ТIТ проходит через максимум, как правило, при низких температурах
(ниже 298,15 К). Кроме того, погрешность значения S° B98,15 К) существенно зависит от
неточности экстраполяции теплоемкости к Т=0. Поэтому относительная погрешность величины
SD B98,15 К) выше относительной погрешности Н° B98,15 К)—Я0 @). Значения Ф° B98,15 К)
для кристаллических веществ составляют в среднем 2/3 от значения S° B98,15 К), а абсолютные
погрешности этих двух величин близки, особенно в тех случаях, когда погрешность S° B98,15 К)
связана в основном с неточностью экстраполяции теплоемкости к Т=0. В данной работе
погрешность значений Ф° B98,15 К) приравнивалась погрешности величины S° B98,15 К).
В случае расчетов термодинамических функций твердых и жидких веществ на основании
результатов измерений энтальпии оценка точности термодинамических функций представляет более
сложную задачу. Поскольку теплоемкость является первой производной энтальпии по
температуре, ошибки в значениях С (Т), вычисленных из данных по Н°(Т)—Н° B98,15 К), превышают
погрешности последних. Для уменьшения этих погрешностей вывод уравнения для С\ (Т) из
данных по изменению энтальпии при подготовке справочника проводился с учетом измерений
низкотемпературной теплоемкости, см. уравнения B,29) и B.30). Тем не менее погрешность полученных
таким образом значений теплоемкости значительно превышает погрешности в значениях
энтальпии, особенно в области, примыкающей к верхней границе интервала измерений (Т2). При
расчете S°(T) и Ф°(Т) по эмпирическим уравнениям для теплоемкости, выведенным описанным выше
способом, происходит частичная компенсация ошибок.
Погрешности значений Ф° (Т) для твердых и жидких веществ приводятся для температур
298,15, 1000, 1500, 2000 К и выше с интервалом 1000 К. Для их определения рассчитывались
приращения Ф° (Т) между указанными температурами, и относительные погрешности этих
приращений принимались равными относительным погрешностям теплоемкостей (или изменения
энтальпии) в этих же интервалах температур. Полученные погрешности значений Ф° (Т2) — Ф° G\) для
различных интервалов температуры складывались и округлялись.
Глава 3
ВЫБОР РЕКОМЕНДУЕМЫХ ЗНАЧЕНИЙ
ТЕРМОХИМИЧЕСКИХ ВЕЛИЧИН
В таблицах термодинамических свойств веществ, рассматриваемых в настоящем издании,
приводятся константы равновесия диссоциации газообразных веществ на составляющие их атомы и
электрон (в случае ионов), а также давления паров веществ в конденсированном состоянии.
Для их расчетов необходимы значения ряда термохимических величин. В справочнике
рассматриваются следующие термохимические величины:
AfH°@) и ДуЯ0 B98,15 К) — энтальпия образования данного вещества при 0 и 298, 15 К
из элементов в стандартных состояниях;
Д ,G° B98,15 К) — изменение энергии Гиббса при образовании данного вещества из элементов
в стандартных состояниях при 298,15 К (изобарно-изотермический потенциал образования);
АГН° @), ДД° B98,15 К) и ArH° (T) — энтальпия реакции при 0, 298,15 и Г К;
Д8#°@) и Д8#° B98,15 К) — энтальпия сублимации при 0 и 298,15 К;
AVH° B98,15 К) — энтальпия испарения при 298,15 К;
Do — энергия диссоциации газообразного вещества на одноатомные газы при Г=0;
10 — потенциал ионизации;
Ао — сродство к электрону или протону1.
Термохимические величины определяются на основании различных экспериментальных
исследований, таких как калориметрические измерения энтальпии реакции, измерения констант
равновесия и давления паров, изучение оптических спектров, процессов диссоциации и ионизации
молекул, полуэмпирических и эмпирических оценок, а также теоретических расчетов. При
подготовке справочника значения рекомендуемых термохимических величин выбирались на
основании критического анализа всех данных, имеющихся в литературе, а при отсутствии — на
основании оценок. Краткие тексты, обосновывающие выбор принятых значений, приведены при
обсуждении термодинамических свойств каждого вещества.
Приводимые термохимические величины относятся к стандартным состояниям веществ. Для
11 элементов (О2, Н2, D2, T2, F2, Cl2, He, Ne, Ar, Кг, Хе) в качестве стандартного состояния
при всех температурах принято газообразное состояние. Для Вг и Hg в качестве стандартного
состояния при Т=0 принято кристаллическое, а при 298,15 К жидкое состояние. Для всех
остальных элементов, включая 12 и S, в качестве стандартного при 0 и 298,15 К принято кристаллическое
состояние. Обычно за стандартное состояние элемента в кристаллическом состоянии при 298,15 К
принимается его модификация, наиболее стабильная при 298,15 К. Однако в некоторых случаях
приготовление чистых образцов такой модификации затруднительно и проще получить
модификацию, метастабильную при этой температуре. Подобная ситуация имеет место для фосфора и олова,
стабильные модификации которых — черный фосфор (ромбическая модификация) и серое олово
(кубическая модификация) — труднодоступны. В справочнике для этих элементов за стандартные
состояния в соответствии с рекомендациями ИЮПАК и КОДАТА—MCHG [1000] приняты белый
фосфор и белое олово. Стандартным состоянием электрона является идеальный невырожденный
газ при давлении в 1 атм A01 325 Па).
Энтальпии образования и энергии Гиббса для реакции образования элементов в их стандартных
состояниях по определению равны нулю при всех температурах.
1 В литературе сродство к электрону и протону обычно приводится как положительная величина, если
присоединение электрона (протона) сопровождается выделением энергии. Это противоречит правилу знаков изменения
энтальпии, принятому в химической термодинамике. Поэтому в данной работе сродство к электрону (протону),
если соответствующий процесс сопровождается выделением энергии, приводится со знаком минус.
81
Глава 3. Выбор термохимических величие
3.1. Согласованность термохимических величин
Рекомендованные в настоящем издании значения термохимических величин являются системой
взаимно связанных величин. Связи между ними вытекают из основных законов термодинамики.
Взаимная связь между термохимическими величинами обусловливает необходимость их строгого
согласования. Включение, пусть даже более точного, нового значения для какого-то одного вещества
может привести к грубым ошибкам, если оно не согласовано с остальными величинами. Поэтому
все приведенные в справочнике термохимические величины представляют систему взаимно
согласованных значений. Обеспечено также согласование термохимических величин и
табулированных термодинамических функций. Согласованность рекомендованных значений означает, что
между ними строго выполняются все связи, обусловленные уравнениями термодинамики, например;
Д/?°(В2О3, к, 298,15 К) = Д/Я*(В2О3, к, 298,15 К) — 298,15 [S° (B2O8, к, 298,15 К) —
— 25° (В, к, 298,15 К) — 1,5 S° (О2, г, 298,15 К)] C.1)
или
Do (В2О3) = 2ДД» (В, г, 0) + ЗД^° (О, г, 0) - Д^0 (В2О3, г, 0). C. 2}
Для обеспечения взаимной согласованности всех термохимических величин необходимо, чтобы
в расчетах, проводимых в процессе их выбора, использовались одни и те же значения
вспомогательных термохимических величин. Те из этих величин, которые наиболее часто используются
в расчетах, получили название ключевых термохимических величин. В качестве ключевых
величин обычно рассматриваются значения д^0B98,15 К), 5го B98,15 К) и Н° B98,15 К)— #°@)
для таких веществ, как Н2О (ж, г), СО2 (г), HF (г), НС1(г), F" (aq), Cl~ (aq), SOf (aq), Na+ (aq),
S (к, г), О2 (г) и т. п. Выбор этих значений является весьма ответственным, так как они
используются для вычисления рекомендуемых значений для десятков и сотен других веществ. Особое
значение выбор ключевых термохимических величин и проблема создания системы согласованных
величин приобретает при подготовке фундаментальных справочных изданий, таких как [88,
258, 3375, 3256, 2095], охватывающих широкий круг веществ и рассчитанных на использование
в течение длительного времени. Все опубликованные справочные издания, являющиеся
системами согласованных величин, базировались на отличающихся наборах ключевых величин. Это может
приводить к ошибкам при совместном использовании значений, рекомендованных в разных
изданиях. Для устранения этих неоправданных расхождений и создания единой основы для обработки
результатов всех экспериментальных измерений, Комитет по численным данным Международного
совета научных союзов (КОДАТА—МСНС) в 1970 г. начал подготовку международных
рекомендаций значений ключевых величин для термодинамики. Эта работа проводится с участием авторов
настоящего справочника. К 1978 г. закончена подготовка рекомендаций для подавляющей части
элементов, рассматриваемых в справочнике, и эти рекомендации [1000] положены в основу всей
системы термохимических величин настоящего издания. Однако эти международные
рекомендации не исчерпывают всех термохимических величин, потребность в которых возникает при
анализе и пересчетах результатов экспериментальных исследований. Такие недостающие величины
в ряде случаев принимались по справочнику Академии наук СССР «Термические константы
веществ» [258] и справочнику Национального бюро стандартов США «Рекомендованные значения
термодинамических свойств» [3375]; энтальпии растворения и разведения растворов во многих
случаях принимались по рекомендациям Паркер [2959] и Мищенко и Полторацкого [169а].
Для обеспечения согласованности рекомендуемых в настоящем издании термохимических
величин при их выборе использовались только энтальпии реакций, непосредственно измеренные
в работах. Все дальнейшие расчеты (вычисление энтальпий образования, энергий диссоциации
и т. п.) проводились с использованием единых, принятых в настоящем справочнике величин.
Во всех расчетах термохимических величин на основании результатов измерений констант
равновесия (и давления пара) и при пересчетах к стандартной температуре результатов измерений,
выполненных при высоких температурах, использовались значения термодинамических функций,,
приведенные в настоящем справочнике. В тех редких случаях, когда для расчетов были
необходимы термодинамические функции веществ, не рассматриваемых в настоящем издании, они
принимались по справочникам Халтгрена [2039], Келли [2220] и др. В этих случаях в текстах
указывается соответствующий источник.
82
3.2. Критерии достоверности экспериментальных измерений
3.2. Некоторые критерии оценки достоверности экспериментальных измерений
термохимических величин
Рекомендованные термохимические величины были выбраны на основании анализа имеющихся
в литературе экспериментальных исследований или оценены при подготовке справочника. Этот
анализ включал изучение каждой из экспериментальных работ, в которых исследовалось данное
вещество, и затем совместный сравнительный анализ результатов разных работ. Термохимические
величины обычно получают на основании экспериментального определения энтальпии реакции,
в которой участвуют несколько веществ. Эти данные целесообразно использовать для нахождения
энтальпии образования наименее изученного вещества. Во многих случаях сделать такой выбор
достаточно просто. Однако встречаются случаи, когда для правильного решения вопроса
необходимо знание современной термохимической литературы для широкого круга веществ; более того,
появление новых работ иногда заставляет полностью пересмотреть, казалось бы, бесспорный
выбор такого вещества Ч Хорошим примером может служить термохимия соединений бора. В
течение многих лет результаты многочисленных измерений энтальпии сожжения аморфного бора в
кислороде и в хлоре использовались для определения энтальпии образования трехокиси и треххлори-
стого бора. Однако более поздние прецизионные измерения энтальпии реакций с участием
кристаллического бора привели к тому, что результаты измерений с аморфным бором в настоящее время
целесообразно использовать для определения энтальпии перехода кристаллического бора в
аморфный, и именно этот путь использован в справочнике.
При анализе литературных данных по калориметрическому измерению энтальпий реакций
и оценке полученных результатов учитывались конкретные особенности экспериментальных
установок и измерительной аппаратуры. Как показывает опыт, одной из наиболее ответственных
частей такого рода исследований является точное определение характера протекающих реакций
и количества прореагировавшего вещества. В связи с этим первостепенное значение имеют степень
чистоты исследуемых веществ, полнота химического и фазового анализа исходных веществ и
конечных продуктов реакции и тщательный учет возможных побочных реакций, в том числе реакций
с теми частями аппаратуры, которые находятся в непосредственном контакте с реагирующими
веществами. При оценке степени достоверности результатов калориметрических измерений
принимался во внимание опыт, накопленный авторами данной экспериментальной работы, результаты
выполненных ими других исследований, особенно исследований, проведенных с аналогичными
объектами и на той же калориметрической аппаратуре.
Серьезное значение имеет характер представления результатов экспериментальной работы
в соответствующей публикации. Работы существенно обесцениваются, если они публикуются без
достаточных сведений об аппаратуре, методике проведения измерений, без указания точного
уравнения исследуемой реакции с данными о фазовых состояниях всех компонентов, без указания
концентраций и состава растворов, температуры измерений, степени чистоты использованных веществ
и методов ее определения, методов определения полноты протекания реакции, учета примесей
и побочных процессов, а также без указания первичных результатов калориметрических
измерений. При этом не исключается, что такие работы выполнены на хорошем уровне. Детальные
правила публикации данных по термодинамике разработаны КОДАТА—MCHG [227а], и их
выполнение должно стать непременным условием при издании соответствующих работ.
При анализе результатов исследования равновесия химических реакций учитывалась
обоснованность отнесения измерений к конкретной реакции, возможность образования веществ,
не входящих в предполагаемое авторами уравнение реакции (например, димеров или продуктов
диссоциации), возможные изменения конденсированной фазы (образование расплавов, отклонение
от стехиометрии) и т. п. В этом отношении масс-спектрометрический метод выгодно отличается
от таких интегральных методов, как испарение с поверхности, метод Кнудсена, статические
методы, метод протока и т. п., когда отнесение измерений к определенной реакции
основывается на косвенных экспериментальных данных или принимается из общих соображений.
Поэтому в ряде случаев при подготовке настоящего издания результаты таких измерений
исправлялись или уточнялись на основании сведений о молекулярном составе, полученных в других
1 Метод, основанный на совместном решении системы термохимических уравнений, позволяет выбирать
одновременно несколько термохимических величин, входящих в эти уравнения (см. [1758а, 265а], а также текст по
РАо (к)).
83
Глава 3. Выбор термохимических величин
работах или соответствующих термодинамических расчетов. При оценке надежности
использованного в конкретной работе экспериментального метода исследования равновесий или измерения
давления паров принимались во внимание особенности методики, ее преимущества или недостатки
по сравнению с другими, возможность реакции с материалом контейнера, степень отклонения
от равновесных условий, чистота исследованных веществ и т. п. Одной из причин появления
систематических ошибок в таких исследованиях является неточное измерение температуры или
наличие температурных градиентов. Часто наличие таких ошибок трудно установить по приводимым
в работах данным, но о них могут свидетельствовать такие особенности, как температурный ход
энтальпий реакций, вычисленных по методу III закона термодинамики, расхождение
результатов обработки экспериментальных данных по методам II и III законов и резкое отличие значения
д S°, полученного при расчете по методу II закона, от величины, вычисленной по
табулированным значениям термодинамических функций компонентов реакции. Такие расхождения могут
явиться также следствием ошибки при пересчете результатов измерений к абсолютным давлениям
или образования неучтенных побочных продуктов.
Масс-спектрометрический метод исследования равновесий отличается от других методов тем,
что позволяет более надежно индентифицировать газообразные компоненты реакции. Однако
для вычисления парциальных давлений по измеренным интенсивностям ионных токов требуется
соответствующая калибровка аппаратуры. Если в газовой фазе присутствует только одно вещество,
то такая калибровка может быть осуществлена наиболее простым способом — по скорости
потери веса при эффузии. В этом варианте метод по существу не отличается от обычного метода
Кнудсена, приобретая, однако, преимущества надежности идентификации газообразного продукта
реакции. Если в состав газовой фазы входят несколько веществ, такая калибровка невозможна,
а для ее осуществления необходимы сведения о сечениях ионизации компонентов пара. Сечения
ионизации необходимы также и при калибровке прибора с помощью испарения стандартного
вещества (обычно серебра).
Надежность оценок сечений ионизации в настоящее время обычно оценивается множителем
1,5—3 [1326а]. В справочнике принимается, что давления паров и константы равновесия,
вычисленные с использованием данных по оцененным сечениям ионизации, имеют погрешность
порядка множителя 1,5, что для измерений, выполненных при температурах 1000 и 2000 К,
эквивалентно внесению в определяемую энтальпию реакции погрешностей в 3 и 7 кДж -моль
соответственно.
Поэтому в таблицах, суммирующих результаты обработки экспериментальных данных по
константам равновесия реакций, значения энтальпий, основанные на масс-спектрометрических
измерениях, даны с погрешностями, включающими не только 95%-ный доверительный интервал,
но и погрешности, связанные с неточностью сечений ионизации. Эта дополнительная погрешность
приводит к тому, что в случае простого молекулярного состава величины, полученные из масс-
спектрометрических измерений, оказываются во многих случаях менее точными по сравнению
с полученными такими методами, как эффузионный, торзионный или статический.
При анализе масс-спектрометрических измерений следует учитывать, что принятые в
настоящее время методы оценки сечений ионизации существенно отличаются от применявшихся ранее,
Таблица 3.1.
н
Не
Li
Be
В
С
N
О
F
Ne
Na
Mg
Al
Si
0,22
0,21
3,29
3,16
2,60
1,98
1,52
1,27
1,03
0,82
4,02
5,38
6,18
5,35
Сечения ионизации
P
S
Cl
Ar
К
Ca
Sc
Ti
V
Cr
Mn
Fe
Co
Ni
4,45
3,87
3,40
2,83
7,21
10,44
9,51
8,67
7,93
5,10
6,75
6,30
5,96
5,48
Cu
Zn
Ga
Ge
As
Se
Br
Kr
Rb
Sr
Y
Zr
Nb
Mo
атомов (в
3,80
4,65
5,94
5,71
5,02
4,96
4,55
4,05
8,40
12,91
11,91
10,87
7,74
6,91
10-i6
Тс
Ru
Rh
Pd
Ag
Cd
In
Sn
Sb
Те
I
Xe
Cs
Ba
см2), принятые по
9,13
6,73
6,17
6,07
5,05
6,29
7,72
7,70
7,19
7,09
6,75
6,24
10,78
17,26
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
работе
16,07
15,91
16,01
15,63
15,24
14,88
14,60
12,92
12,73
13,90
13,42
13,20
12,99
[2510a]
Yb
Lu
Hf
Та
W
Re
Os
Ir
Pt
Au
Hg
Tl
Pb
12,77
10,92
10,44
9,55
9,24
9,05
8,09
7,71
6,60
5,85
6,43
7,76
7,85
Bi
Po
At
Rn
Fr
Ra
Ac
Th
Pa
U
Np
Pu
Am
8,12
7,91
7,61
7,29
10,21
17,52
16,51
16,74
16,22
15,87
15,38
15,83
14,80
84
3.3. Вычисление по результатам исследования равновесий
и поэтому результаты некоторых более старых работ должны быть пересчитаны. При этом следует
учитывать, во-первых, что в старых работах обычно используются сечения ионизации атомов,
рекомендованные в 1956 г. Отвосом и Стивенсоном [2924а], а в настоящее время общеприняты
сечения ионизации атомов, предложенные в 1970 г. Манном [2510а] (табл. 3.1). Во-вторых,
оценки сечений ионизации молекул в старых работах основывались на постулате аддитивности,
также предложенном Отвосом и Стивенсоном [2924а]. В настоящее время принимается, что
сечение ионизации димера превышает сечение ионизации мономера не в два раза, как это вытекает
из постулата аддитивности, а в 1,5 раза [1326а]. Оценка сечений ионизации многоатомных молекул
более сложна. Можно только предполагать, что для многоатомной молекулы постулат
аддитивности по отношению к сечениям составляющих ее молекулярных фрагментов должен выполняться
существенно лучше, чем по отношению к сечениям атомов. В справочнике принимается, что
сечение ионизации молекулы М связано с сечением ионизации составляющих ее атомов соотношением
а (М)=0,752 а (А). Пересчеты с использованием этого соотношения и приведенных в табл. 3.1
величин проводились лишь в тех случаях, когда они приводили к изменениям в давлениях пара
или константах равновесия, превышающим 20%. Эти пересчеты также не проводились в случаях,
когда выбор величин основывался на результатах измерений другими методами, и результаты
масс-спектрометрических работ имели лишь вспомогательный характер.
Исследования, в которых одновременно измеряются скорость истечения и интенсивность
ионных токов, удачно объединяют преимущества масс-спектрометрического и эффузионного
методов.
3.3. Вычисление термохимических величин
по результатам исследования равновесий
Калориметрические и некоторые другие методы определения термохимических величин основаны
на использовании I закона термодинамики, и измеряемое этими методами значение АГН° связано
со всеми вычисляемыми из него величинами единственным уравнением. В отличие от этого
значение Дг//°, вычисляемое по результатам исследования равновесия реакции, в том числе на
основании измерения ЭДС реакции, может быть найдено двумя независимыми методами, причем
результаты расчетов этими методами в общем случае не совпадают. Оба метода основаны на хорошо
известном соотношении
RlnR°(T) = - ^(Т)=_АГН°^(Т) +А^о(у,^ C.3)
Величины ArH° (T) и ArS° (T) слабо зависят от температуры и в ограниченном интервале
температур могут рассматриваться как константы. Аппроксимация результатов экспериментальных
измерений констант равновесия (или давления пара) уравнением C.3) позволяет найти значения
ArH° (Tcv) и ArS° (Tcv), где Tcv — некоторое промежуточное значение температуры. В справочнике
[п -1
п Х2 ?V • Определение ArH°(Tcv)
•=i * J
как коэффициента при Т L в уравнении C.3) получило в литературе название «расчета по
методу II закона термодинамики»1. Для нахождения значения ArH° (Tcv) по этому методу
достаточно знать только зависимость константы равновесия реакции от температуры, а не
абсолютные значения К° (Т). Это существенно, поскольку некоторые методы, в частности масс-
спектрометрический и спектрофотометрический, в ряде случаев позволяют определять только
величины, пропорциональные константам равновесия реакций, а не значения констант
равновесия. Расчет по методу II закона позволяет определить ArH° (Tcv) из этих данных, однако
при этом вместо значения ArS° (Tcv) получается некоторая произвольная константа. Метод II
закона не может быть использован, если измерения выполнены только при одной
температуре; в тех случаях, когда измерения проведены в узком интервале температур, точность
определения значения АГН° (Гср) невысока. Из серии измерений расчет по методу II закона по-
1 В редких случаях, когда необходимо учесть зависимость Дг Н° от Т, может быть использован уточненный вариант
этого метода, получивший название метода 2-функций (см. [245]).
85
Глава 3. Выбор термохимических величин
зволяет получить только одно значение АГН° (Гор), которое весьма чувствительно к возможным
экспериментальным ошибкам, особенно на границах температурного интервала, и к его величине.
Для вычисления значения АГН° (Гср) этим методом не нужны какие-либо термодинамические
функции, однако для пересчета энтальпии реакции к стандартной температуре То необходимы
значения Н° (Tev)—Н° (То) для всех компонентов реакции.
Значения Arff° из данных о константах равновесия могут быть вычислены также по так
называемому методу III закона термодинамики, основанному на соотношениях
C.4)
или
ArH° @) = Т [ДГФ° (Г) — R In K° (Г)]. C.5)
В этом случае энтальпия реакции может быть получена из каждого значения константы
равновесия независимо, но для расчета нужно располагать значениями термодинамических функций
всех компонентов реакции для температур, при которых проводились измерения. Таким образом,
этот метод расчета требует более полную исходную информацию (абсолютные значения констант
равновесия, термодинамические функции), но и дает более полные данные, которые помогают
оценить достоверность экспериментальных измерений. Так, расчет значений АГН° @) для каждой
из экспериментальных точек позволяет выявить возможный температурный ход этих величин.
Систематические ошибки в измерении температуры или в других величинах, так или иначе свя-
ванных с температурой, встречаются довольно часто (особенно в старых работах) и существенно
искажают результаты вычислений по методу II закона. В меньшей степени такие ошибки влияют
на результаты расчетов по методу III закона, их легче обнаружить, а выпадающие точки могут
быть исключены из дальнейшего рассмотрения. Еще одним доводом в пользу выбора
рекомендуемых значений прежде всего на основании расчетов по методу III закона является то, что он
обеспечивает согласованность между энтальпией данной реакции, термодинамическими функциями
ее компонентов и экспериментально измеренными значениями констант равновесия. За
исключением специально оговоренных редких случаев, когда термодинамические функции одного или
нескольких компонентов реакции оказываются особо ненадежными *, в настоящем справочном
издании отдается предпочтение величинам, вычисленным по методу III закона термодинамики.
Однако результаты экспериментальных исследований во всех случаях, когда это было возможно,
обрабатывались по обоим методам. Сопоставление результатов этих расчетов служило основанием
для оценки надежности измерений.
При подготовке справочника была создана программа, которая позволяет вычислять значения
АГН° по методам II и III законов с нахождением погрешности величин (определяемой как 95%-ный
доверительный интервал), устанавливать экспериментальные точки, статистически несовместимые
с остальными, исключать их из рассмотрения и проводить расчет в следующем приближении.
Программа позволяет также вычислять значения ArS° (Гср) по методу II закона термодинамики,
сопоставлять их со значениями, вычисленными по табулированным термодинамическим функциям,
и тем самым получать дополнительную информацию, характеризующую как степень
достоверности значений ArH° (Tcv) и ArS° (Tcv), вычисленных по методу II закона, так и точность
экспериментальных данных. В текстах и таблицах во всех случаях (если это не оговорено) приводятся
значения термодинамических величин, вычисленные при помощи упомянутой программы, а не
приведенные в оригинальных работах.
Неточность термодинамических функций веществ, участвующих в данной реакции, по-разному влияет на значения
Дг Н° @), вычисляемые по методам II и III законов. Отсутствие достоверных данных о типе основного
электронного состояния молекул одного из веществ или большие погрешности в значениях колебательных или
вращательных постоянных существенно влияют на значения АГН° @), вычисляемые по методу III закона, причем тем
больше, чем выше температура, при которой выполнены измерения К°(Т). В то же время они мало влияют на
значения Дг#° @), вычисленные по методу II закона. Отсутствие данных о низких возбужденных электронных
состояниях (с Ti < kT/2hc) больше влияет на значения этих величин, вычисленные по II закону, и существенно
меньше на результаты вычислений по III закону. (О влиянии этих факторов на значения Ф°(Т) и Н°(Т) —
Я°@) см. раздел 1.6.)
86
3.4. Приближенная оценка термохимических величин
3.4. Приближенная оценка термохимических величин
Для ряда веществ, рассматриваемых в данном справочнике, в литературе отсутствуют
экспериментальные данные, необходимые для выбора термохимических величин. Так, рассматриваются
¦соединения дейтерия и трития с кислородом и галогенами, в то время как экспериментальные
данные известны главным образом для соединений протия или природной смеси изотопов водорода.
Термохимические величины для изотопических модификаций, отличающихся от изученных, могут
•быть определены с высокой точностью, если известны их колебательные постоянные. Поскольку
внутримолекулярное силовое поле практически не зависит от массы ядер атомов, образующих
молекулу, сумма энергии диссоциации и нулевой колебательной энергии одинакова для любой
изотопической модификации молекулы. Для двухатомных молекул это приводит к соотношению
D°o(A<B>) + G@)A,B, = D°o(A''B») + G(O)Ar,B,,, C.6)
где (-г@)а;в,- — колебательная энергия данной изотопической модификации молекулы АВ (см.
уравнение A.45)) при у = 0.
Для многоатомных молекул справедливо соотношение
D0(A'B'C .. .) + G@ .. .)a'b'c... = D0(A"B"C" ...) +G@ .. .)А"В"С"..., C. 7)
где G@ .. .)а<в'ск.. — колебательная энергия данной изотопической модификации молекулы
ABC . . . (см. уравнение A.100)) при 1^=0, v%=0, v3=0 . .. Соотношения C.6) и C.7) позволяют
определять энергию диссоциации любой изотопической модификации данной молекулы с точностью,
которая определяется практически только точностью энергии диссоциации другой модификации
и их колебательных постоянных (см. тексты по соединениям D и Т в главах 6—10).
Существенно менее точными являются оценки термохимических величин, основанные на
различных эмпирических и полуэмпирических методах. Обзор этих методов приведен в книгах
В. А. Киреева [127а] и М. X. Карапетьянца [120а]. При подготовке справочника использовались
главным образом методы, основанные на предположении о равенстве средних энергий связей
в родственных соединениях.
Например,
Do (B-C1)bfci, = Do (B-CI)bci.. Do (B-F)Bfci, = Do (B-F)bf,. C. 8)
Тогда для энтальпии атомизации BFC12 можно записать:
Do (BFCy = Do (B-F)bf3 + 2D0 (B-C1)BC), =1 Do (BF3) +1 DO(BC13). C.9)
Это же уравнение можно записать в иной форме:
ДД° (BFC]2) = IД/Г (BF.) + 4 AfH° (BClg). C.10)
Метод оценки, основанный на предположении о равенстве средних энергий связей, приводит
к хорошим результатам, если сопоставляемые соединения имеют одинаковую структуру и
образующие их атомы близки по своим свойствам. Так, значения средних энергий связей В—Fn В—С1
в молекулах BF3, BFaCl, BFC12 и ВС13 достаточно близки, и их энтальпии образования, измеренные
экспериментально и оцененные по приведенному выше уравнению, различаются примерно на
5 кДж-моль, что не превышает погрешности экспериментальных измерений этих величин.
Расхождения оцененных и экспериментально измеренных энтальпий образования увеличиваются,
если атомы имеют существенно отличающуюся электронную структуру. Так, для ВНС12
расхождение составляет 7 кДж-моль, а для BHF2 — 12 кДж-моль, причем если в случае BF2C1 и BFC12
оценки приводят к величинам, соответствующим большей прочности этих молекул по сравнению
с экспериментом, то в случае ВНС12 и BHF2 расхождения имеют противоположный знак.
Значительно большие погрешности возникают при использовании предположения о равенстве энергий
связей в соединениях, не столь сходных, как рассмотренные выше.
87
Глава 3. Выбор термохимических величии
3.5. Оценка погрешности рекомендуемых значений
термохимических величин
Для всех рекомендованных в настоящем издании термохимических величин приводятся их
погрешности. В тех случаях, когда это было возможно, в качестве погрешности принимался 95%-ный
доверительный интервал, который вычислялся как произведение стандартного отклонения
на коэффициент Стьюдента. Однако достаточно строгое определение 95%-ного доверительного
интервала возможно далеко не для всех экспериментальных данных, поскольку многие авторы
не приводят в публикациях необходимые для этого сведения.
При оценке погрешности каждой из экспериментальных работ принимались во внимание
ее особенности, случайные погрешности эксперимента, характеризующиеся стандартным
отклонением, оценивались возможные систематические погрешности, чистота исследуемых веществ и т. п.
(см. выше, раздел 3.2). При оценке погрешности результатов вычислений тепловых эффектов
реакций по методу III закона или пересчетов к стандартной температуре значений &rH° (Tcv),
полученных при расчете по методу II закона, принималась во внимание достоверность использованных
в расчетах термодинамических функций.
При оценке погрешности термохимической величины, измеренной в нескольких работах и
разными методами, учитывались погрешности каждого из методов измерений, а также
согласованность значений, полученных в разных работах. В тех случаях, когда величины, полученные в
разных работах, совпадали в пределах их погрешностей, вычисление рекомендуемого
средневзвешенного значения проводилось в предположении, что вес каждого измерения обратно пропорционален
квадрату погрешности.
Погрешности величин, являющихся алгебраической суммой нескольких других (например,
погрешность энтальпии образования одного из компонентов реакции, вычисляемой с
использованием значения энтальпии этой реакции и энтальпий образования остальных ее компонентов),
вычислялись как корень квадратный из суммы квадратов погрешностей всех слагаемых:
Погрешности рекомендуемых величин приводятся не более чем с двумя значащими цифрами.
При этом и сами рекомендуемые величины округлялись до знака, соответствующего последней
значащей цифре погрешности. Однако величины, вычисленные на основании принятых значений,
приводятся всегда с большим числом знаков (до трех после запятой), что объясняется
необходимостью точного согласования всех величин, приводимых в справочнике. Как правило, число
значащих цифр в вычисленных таким образом величинах оказывается больше, чем это
оправдывается их погрешностью.
Часть 2
ВЫБОР ПОСТОЯННЫХ
И РАСЧЕТ
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
Глава 4
ЭЛЕКТРОННЫЙ ГАЗ И КИСЛОРОД
е, О, О+, О", О,, О2+, О-, О3
В настоящей главе рассматриваются электронный газ, атомарный и молекулярный кислород, их
ионы и озон в газообразном состоянии. Соединения кислорода с другими элементами обсуждаются
в последующих главах. Термодинамические свойства озона рассчитаны для температур от 100
до 6000 К, шести остальных газов — до 20 000 К. Для всех веществ приведены свойства
для природной смеси изотопов (с атомным весом 15,9994); однако ввиду незначительного
содержания изотопов 17О и 18О в природной смеси, различие молекулярных постоянных отдельных
изотопических модификаций О2, О?, (ХГ и О3 в расчетах термодинамических функций не учитывалось.
В Приложении 3 приведены данные, позволяющие учитывать отклонение термодинамических
свойств О2 (г) и О3 (г) от их свойств в стандартном состоянии в результате межмолекулярного
взаимодействия. Многозарядные ионы типа Оа+ или О^~, полимерные молекулы с числом атомов
кислорода больше трех, а также ион О~ не играют существенной роли в рассматриваемом интервале
температур и не включены в справочник.
е (г). Термодинамические свойства электронного газа в стандартном состоянии в интервале
температур 298,15—20 000 К приведены в табл. 1 (см. наст, том, кн. 2).
Расчет проводился по уравнениям A.3)—A.6), A.9), A.10), в которых внутримолекулярные
составляющие рассчитывались с учетом только статистического веса электрона, равного 2. Строго
говоря, при больших плотностях и низких температурах электронный газ является вырожденным
газом Ферми—Дирака, однако при стандартном давлении и температурах выше 1000—1500 К
его с хорошим приближением можно считать классическим газом Больцмана. Таким образом,
при Т > 1500 К погрешности вычисленных значений термодинамических функций е (г)
определяются только неточностью фундаментальных постоянных, и в значениях Ф°(Т) и S°(T) они будут
составлять 0,01—0,02 Дж-К^-моль. При Т < 1500 К ошибки из-за использования статистики
Больцмана могут быть весьма заметными (см., например, обсуждение в справочнике [88]). Однако
эти ошибки не имеют практического значения, так как при таких температурах в равновесных
условиях содержание электронов всегда будет очень небольшим.
Вычисленные ранее таблицы термодинамических функций е (г) (см., например, [88])
практически совпадают с вычисленными в настоящей работе.
О (г). Термодинамические свойства газообразного одноатомного кислорода в стандартном
состоянии в интервале температур 100—20 000 К приведены в табл. 2 (наст, том., кн. 2).
Таблица 4.1. Уровни энергии атомов О, О+ и О~, принятые для расчета термодинамических функций
Атом
Электронная
конфигурация
Состояние
см
Pi
О Is22s22p* »Pt 0 5
Is22s22p* зрх 158,265 3
Is22s22p4 3Р„ 226,977 1
l*a2s«2p* аДг 15 867,862 5
№2s>2p* ^o 33 792,583 1
О+ №2&2р* *S,lt 0 4
Атом
Электронная
конфигурация
Состояние
Т{
СМ
Pi
О+ ii?2s*2p3 ЮьЫ 26 808,4 6
Is22s22p3 2D3' 26 829,4 4
Is22s22p3 2Р3/' 40 466,9 4
Is22s22p3 *Р^ 40 468,4 2
О" lsa2sa2pe гРз/2 0 4
и*2!?2рь 2Pi*2 180 2
90
Электронный газ и кислород 0+ (г), 0~(г)
Уровни энергии атома О, использованные при расчете термодинамических функций, приведены
Е табл. 4.1. Они приняты по работам Эрикссона [1411, 1413], в которых были получены более
точные данные, чем рекомендованные в справочнике Мур [2697].
Термодинамические функции О (табл. 2) вычислялись непосредственным суммированием
по уровням энергии в соответствии с принятой методикой расчета (см. раздел 1.3). До 10 000 К
погрешности вычисленных значений функций @,02 Дж -К -моль) определяются практически
только неточностью фундаментальных постоянных. При более высоких температурах, особенно
выше 15 000 К, погрешности возрастают из-за того, что в расчетах не учтены состояния ридбергов-
ских серий с конфигурацией . . .2sa2p3 (*?)«?. При 20 000 К погрешности достигают 0,25
и 1,5 Дж-К^-моль в значениях Ф° (Т) и S° (T) соответственно.
Таблицы термодинамических функций одноатомного кислорода представлены во многих
работах. В справочных изданиях и сводках [87, 1673, 1946, 2291, 2029, 2556, 2095] они
вычислялись для температур до 6000 К или ниже. Величины, табулированные в этих работах и табл. 2,
согласуются в пределах различия принятых в расчетах фундаментальных постоянных. В работах
[88, 225, 3848] расчеты были выполнены до 20 000, 12 000 и 10 000 К. Расхождения данных этих
изданий между собой и с табл. 2 обусловлены различием принятых методов ограничения
статистических сумм и их производных. Они достигают 2,5 Дж-К^-моль в значениях Ф°B0 000 К).
Поскольку в отличие от предыдущего издания справочника и некоторых других работ в настоящем
издании в расчетах учитывались только состояния атома О с конфигурацией ls^s^p4 (см. выше,
раздел 1.3), приведенные в табл. 2 значения термодинамических функций ниже, чем в
перечисленных работах.
Энтальпия образования О (г) в соответствии с рекомендацией КО ДАТА—MGHG [1000] принята
равной
AfH°(O, г, 298,15 К) = 249,17 + 0,10 кДж • моль.
Это значение основывается на энергии диссоциации молекулы О2 (см. ниже).
О+ (г). Термодинамические свойства газообразного положительного иона кислорода в
стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 3.
Уровни энергии иона О+, использованные при расчете термодинамических функций, приведены
в табл. 4.1. Они приняты по работе Эрикссона [1413], в которой получены более точные данные,
чем рекомендованные в справочнике Мур [2697].
Термодинамические функции О+ (г) были вычислены непосредственным суммированием по этим
уровням энергии в соответствии с принятой методикой расчета (см. раздел 1.3). Погрешности
табулированных значений термодинамических функций определяются практически только неточностью
фундаментальных постоянных и не превышают 0,02 Дж-К-моль в значениях Ф°(Г) и S°(T).
Ранее термодинамические функции О+ вычислялись в [225, 1723, 1946, 88].- Расхождения
между результатами всех расчетов не превышают 0,01 Дж-К^-моль во всем интервале
температур.
Константа равновесия реакции О+(г)+е (г)=О (г) вычислена на основании значения
АгН° @) — —1313,943 + 0,001 кДж-моль, соответствующего потенциалу ионизации атома
кислорода
/0 (О) = 109 837,02 + 0,06 см = 1313,943 + 0,001 кДж • моль,
принятого по результатам измерений границ ридберговских серий в спектре кислорода в работах
Эрикссона [1413, 1411]. Ему соответствует энтальпия образования О+(г), равная
-1
Д/Я°(О+, г, 0)= 1560,726 + 0,10 кДж • моль
О~ (г). Термодинамические свойства газообразного отрицательного иона кислорода в
стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 4.
Уровни энергии иона О~, использованные при расчете термодинамических функций, приведены
в табл. 4.1. Энергия компоненты 2Pi/2 основного состояния получена на основании экстраполяции
в ряду изоэлектронных атомов и ионов, выполненной при подготовке предыдущего издания
справочника [88]. Она практически совпадает с аналогичной оценкой Хотопа и др. [2015]. Берри
« сотр. [610] при изучении фоторекомбинационного континуума, соответствующего процессу
О+е -> O~+hv, получили более высокую энергию компоненты 2Pi/2 B85+.15 см), однако позднее
91
02(г) Глава 4
в обзоре [609] Берри рекомендовал значение 190 см, практически совпадающее с принятым.
в табл. 4.1. Близкое значение B20 + 50 см) было получено Гофманом при исследовании спектра
испускания дуги в атмосфере кислорода и отнесенного к иону О" [1972].
Термодинамические функции О" (г) вычислялись по уравнениям A.3)—A-6), A.9), A.10),
A. 23)—A. 25) с учетом двух компонент основного состояния этого иона. Погрешности вычисленных
значений термодинамических функций обусловлены практически только неточностью принятой
энергии компоненты 2Рч,, так как ион О~ не имеет связанных возбужденных состояний с энергией
ниже его потенциала ионизации. Если энергия 2Pi/2 компоненты занижена на 100 см, погрешности
в значениях Ф° (Т) при 298,15 и 3000 К составляют 0,6 и 0,15 Дж-К^-моль, а выше 4000 К
не превышают 0,1 Дж-К^-моль.
Ранее термодинамические функции О" (г) вычислялись в предыдущем издании справочника [88 ]
и в справочнике JANAF [2095]. Расхождения результатов этих расчетов с данными табл. 4 не
превышают 0,01 и 0,15 Дж-К-моль соответственно во всем интервале температур.
Константа равновесия реакции О" (г)=О (г)+<? (г) вычислена на основании значения АгН° @) =
=141,2 + 0,6 кДж-моль в соответствии с принятой в справочнике величиной сродства атома
кислорода к электрону
Л0(О) = —11800+ 50 см^—141,2+ 0,6 кДж ¦ моль.
Эта величина получена по практически совпадающим результатам измерений Бранскома и
др. [749], Хотопа и др. [2015] и Демера и Чапки [1204]. В работе [749] значение А0(О) было
определено на основании изучения зависимости сечения фотоотрыва электрона от О~ как
функции энергии фотонов; найденный порог процесса равен А=8460 + 30 А, или 11 817 + 40 см". В
работе [2015] измерялась энергия электронов, образующихся при фотоионизации О" фотонами с Х=
=4880 А (аргон — ионный лазер); найденное таким образом сродство к электрону равно 1,462 +
+ 0,005 эВ, или 11 791 + 40 см. В работе [1204] значение 1,462± 0,003 эВ, или 11 791 + 25 см,
было получено при изучении фотоионизации О2, сопровождающейся образованием ионов О++О~.
Несколько более высокое значение A1921 + 15 см) было получено Берри и др. [610] по границе
непрерывного спектра фоторекомбинационного излучения О+е -> O~-\-hv, возможно, из-за
трудности определения порога процесса в связи с присутствием в спектре посторонних линий.
Значения, найденные в работах [1396, 1236] при изучении фотоионизацииО2и в работе [1972] при
исследовании спектра О", представляются менее надежными. Принятому значению А0(О)
соответствует
ДуЯ°(О-, г, 0)= 105,583 + 0,61 кДж • моль-1.
О2 (г). Термодинамические свойства газообразного двухатомного кислорода в стандартном
состоянии в интервале температур 100—20 000 К приведены в табл. 5.
Молекулярные постоянные О2, использованные при расчете термодинамических функций,,
приведены в табл. 4.2. Молекула О2 согласно теоретическим расчетам Шефера и Гарриса [3320]
имеет 12 связанных валентных электронных состояний. Остальные 52 состояния,
коррелирующие с атомами О в валентных состояниях SP, XD и XS, являются отталкивательными состояниями.
Спектры О2 исследовались в десятках работ (обзор исследований см. в работе [2311]). В спектрах
О2 наблюдались переходы между семью нижними валентными состояниями: Х31,~, агАд &Х2*
с1!!", А3Аи, В22+ и С32~. Наиболее полно изучена система полос Шумана—Рунге С32~—
[2921, 2450, 1136, 1450, 2269, 789, 1892, 2880, 2882, 344, 1448, 1889, 1570, 1449, 1875, 2345, 2668,
2028, 240] (наблюдались переходы с v" <[ 29 и v' < 21; для полос с v" < 27 выполнен анализ
вращательной структуры) и система полос BSI,+—X32~ [1907]. В системе Б32+—Х32~
наблюдались переходы на все колебательные уровни состояния 532*, вплоть до v'=11 и определены
значения В, и D, для всех v'. В системе С32~—Х22~ наблюдались переходы на уровни с i/ <I vmm—l =
=21 и для всех уровней были определены значения этих же постоянных. В четырех остальных
состояниях наблюдались переходы только на отдельные колебательные уровни: на уровни v <I 1
состояния а1Ад [1922, 2156], на уровни с v < 5 состояния Ь1^ [439, 2191, 1894, 1896, 2329, 1892],
на уровни с v=0 и 6—11 состояния сг2~ [1908, 1198, 1199] и на уровни с i>=5 и 6 состояния А3Аи
[1908]. Значения молекулярных постоянных, рекомендованные в обзоре Крупенни [2311] на
основании анализа результатов исследований электронных спектров О2, даны в табл. 4.2. Приве-
92
Электронный газ и кислород
О2(г)
Таб л)
Молекула
яца 4.2.
Состояние
оа хъ-
о!Д„
ЬЩ
с^й
А3\
В3Щ
С32,7
П+ "V2TT
и2 л ii?
л2пм
U
Д2у 4*
с4Б~
С2ФМ
2>2дм
?2Д?
d4Ay
ЯП.
G2^
#22+
е42?
of xmg
Молекулярные постоянные О2,
Т,
в
ше
0 а 1 580,193
7 917,54 1 509,3
13 195,07 1 432,67
33 055,71 794,29
34 690,91 750
35 393,08 799,08
49 792,11 709,309
0 а 1 905,13
32 913 б 1 035,53
40 572 в 898,2
40 600 г 660 г
43 300 г 607 г
46 800 г 522 г
49 500 1 196,91
53 200 900
61 300 г 1 010 г
61 800 930
65 500 г 674 г
66 000 г 973 г
66 668 1 156
69 400 г 1 750 г
72 000 г 1 354 г
0а 11106
О) X
е в
11,981
12,9
13,934
12,736
14
12,16
10,646 8
16,28
10,32
13,57
8,3 Д
—
—
17,135
—
—
—
—
22
—
—
10
Примечание. Ниже все постоянные даны в см.
О2: а Х=1,984,
Д а2=4,294
и для v <
OJ: a .4=195;
;j.=-8,37.10-3; & o>ez
• Ю-6, Pi=—7,7.10-8;
4; к К2=_5,5619-10-4
б а=—40; в Л=8,2;
соотношению A. 67) при ые=660 и D
0^: a Л=—160
ot
и Oj
е е
СМ"
4,747б '
____ /
— 1,43
1,445
Ве
622
1,4263
1,400 479 6
—24,44е 0,915
—
—55,0 0,910
—13,8732 й С
— i
— 1
—
—
—
— 1
—
—
—
—
—
—
—
—
,=1,273-10-3; в „,=6,406-
з o)eze=5,5.
для v < 4.
г принято
,=13100; е
10; ж а2=—7
по результатам
вычислено по A.
1,819
1,689
1,004
1,063
3,667
L.287
1,12
Ю-8,
5
53
02
12
32
0
о
30
о,=—2
,4.10-*;" 3 а2
aj • 10я
1,593 3 в
1,71
1,816 9Д
1,391 ж
—
1,416 3
1,205 5 к
1,958
1,546
2,058
1,0 ж
—
—
2,207
—
—
—
—
—
1,3 в
,85-10-';
De ¦ 10е
5,02 г
4,97
5,356
10,5
—
4,79
5,06 г
5,3
4,88
6,0
2,7 3
—
—
5,8
—
—
—
—
5Г
г приведет
1,207
1,215
1,226
1,517
1,5
1,521
1,604
1,117
1,381
1,408
1,778
1,754
1,743
1,279
1,41
1,403
1,33
1,830
1,561
1,30.
1,553
1,676
1,37
ге
А
536
69
843
4
53
28
123
60
22
г
г
г
65
г
г
г
г
г
значение
=-9,7.Ю-4, ^=0,3.10-»; ?
квантовомеханического расчета;
38);
; « см. текст; в вычислено по соотношению A.69);
ж вычислено по A
г вычислено по A
2=1
Do;
10';
я вычислено по
69); а вычислено по A
68).
68).
денные в этой таблице постоянные 1 и ?л в уравнении A. 40) для состояния X31,ff получены при
анализе системы Ь1^—Х3^ в работе [439] г.
Согласно [3320] (см. также [547]) молекула О2 имеет еще 5 связанных валентных электронных
состояний: -О32~, dxU.g, ехДм, fxAg и g1^*. В спектрах О2, по-видимому, наблюдались переходы в
состояния сР-Ид и ехДм. Как показано в теоретических расчетах [3320] и [547], эти состояния имеют
энергии свыше 50 000 см, неглубокие минимумы на потенциальных кривых и, по-видимому,
небольшое число колебательно-вращательных уровней энергии. Поэтому они не приведены
в табл. 4.2. и не учитывались в расчетах термодинамических функций.
Термодинамические функции О2 (г) вычислялись по уравнениям A. 3)—A. 6), A. 9), A.10), A. 93)—
A.95). Значения Qm и ее производных находились непосредственным суммированием по
колебательно-вращательным уровням энергии семи электронных состояний с использованием
соотношений A.76)—A.78). Энергии. уровней в состоянии Х32~ вычислялись по уравнениям A.40) и
A.65), в остальных состояниях — по уравнениям A.34) и A.65) со значениями коэффициентов
Ykl и других постоянных, приведенными в табл. 4.3. Значения Yk0 для семи электронных состоя-
Исследования микроволновых спектров О2 [3514] (см. [2128, 3831]) дали близкие, но слегка отличающиеся
значения постоянных Во, X и (л.. Соответствующие расхождения не влияют на точность последующих расчетов.
93
Таблица 4.3. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см), а также значения «Шах и <7ilm,
принятые для расчета термодинамических функций О2, О?, Oj
Коэффициенты
Те ¦ 10-'
У,„ • 10-»
У» • Ю'
У» • Ю'
У» • ю»
Ум • Ю'
Ут. • «•
Уео • Ю*
Ум • 10'
Уо1
Уп-Ю'
Уи • 10s
Уз, • Ю'
У„ • ю"
Уа, • Ю'
У., • Ю'
У« • 10»
У.2 • Ю'
У22 • Ю'
Уз„ • Ю»
У« • 10'—
Уоз • Ю'°
X3S-*
0
1,579 55
—11,587 3
-6,547 5
1,291 46
—0,857 221
0,247 155
-0,274 179
—
—
1,446 55
—1,718 35
0,424 316
-0,417 928
0,175 745
-0,279 317
—0,491 3
—
—
(ao=De) • 10-' 4,204 70
о, • 10-*
а2 • 10»
а, • 10'»
v
J?im
0,148 337
0,531 616
—0,155 688
39
230
0,791 754 1,319 507
1,509 39 1,432 30
-12,8 —13,754 5
—9,052 9 —3,252 12
— —0,202 251
_ —
— —
— —
— —
— —
1,426 19 1,400 44
—1,657 4 —1,822 06
—0,277 776 —0,000 218
— —0,105 833
— —
— —
—
—0,486 —0,535 6
- -0,77
— —
— —
—
_ _
3,412 90 2,885 2
0,162 30 0,164 602
0,701 074 0,871116
-0,268 852 —0,492 736
40 34
215 200
о.
с'2-
3,305 571
0,796 11
—13,404 9
-13,935
—0,701 67
0,205 2
—
—
—
—
0,912 792
—1,210 06
—1,414 62
0,852 367
48 225
—
-0,740
—
—
—
0,831 6
0,899 1
0,283 257
0,654 628
—0,368 813
20
155
Примечание. Ниже все постоянные даны в см-1: а X = 1,984
г Состояние
т.
b'sj B'sJ С'Фв D4Ia
43 300 46 800 53 200 61300
АЧи
3,469 091
0,835 75
—12,199
—72,22
—
—
—
—
—
—
0,967 4
-2,62
—
—
—
—
-0,454
—
_
—
—
0,735 6
0,280 526
1,149 42
—1,401 28
14
135
3,539 308
0,805]41
—15,582 2
32,425
—11,730 7
9,151 51
—4,046 86
—
—
—
0,910 62
—1,416
—0,970
—
—
—
—
-0,463
3,0
-1,0
—
—
—
0,665 4
0,284 231
1,213 90
—1,725 52
12
136
, ц = -0,008 37. 6 А =
E4g
61800
d%
65 500
съ-а
4,979 211
0,714 95
—17,509 1
322,681
8,184 58
110,054
-86,965 2
392,928
-0,933 266
0,903 685
0,816 745
-0,763 176
—2,791 03
4,625 59
—4,159 35
15,087 7
—0,186 598
—0,350 3
—0,274 29
-0,802 6
0,012 9
-0,9
—
0,733 4
0,163 983
1,342 65
-1,847 66
23
143
х»п/
0
1,904 663
—16,163 12
0,763 504
_
—
—
—
—
—
1,689 12
—1,953
—
—
—
—
—0,53
_
—
—
—
5,463 55
0,147 098
0,424 012
0,006 140
56
240
195. * А = —200.
F'Ug
66 000
G4-g
66 668
3,291 3
1,033 866
—9,663 97
—6,576 5
—
—
—
—
—
—
1,104 32
1,546
—
—
—
—
—0,488
0,95
_
—
—
—
—
37
192
н'4
69 400
oj
АШи
4,057 2
0,898 498
—13,728 02
2,827 28
—0,161 326
—
—
—
—
—
1,063
2,058
—
—
—
—
—0,60
—
—
—
—
28
156
72 000
4,060
0,660
-8,30
—
—
—
—
—
—
—
0,667
—1,0
—
—
—
—
—0,27
—
—
—
39
191
o?
xmg*
4,950 0 0
1,196 910 1,110 693
—17,135 —10,377 5
- 1,894 37
— —
— —
— —
— —
— —
— —
1,287 2 1,12
2,206 7 —1,3
— —
— —
— —
_ —
-0,581 -0,5
—1,85 —
- 3,355 27
— 0,085 710
— 0,182 827
— 0,093 948
34 65
171 225
Электронный газ и кислород О2(г)
ний были получены совместной обработкой частот начал (или кантов) полос всех переходов,
наблюдавшихся в работах [2921, 2450, 1136, 1450, 2269, 789, 1892, 2880, 2882, 344, 1922, 439, 1908,
1198,1199,1891,1907, 798, 799, 925, 1200, 240] с учетом того, что уровни состояния С32~ сходятся
к пределу О CР)-\-0 (?D), а уровни шести остальных состояний к пределу О CР)-\-ОCР).
Постоянные Ykl для состояния Х3Е~ были рассчитаны по методике, изложенной в разделе 1.4, на
основании значений Bv, найденных в работах [439] (i>=0,l), [2880] (v=2), [2880, 240] (v=3, 4, 5)
[240] (z;=6, 7), [1450] (v=8, 9,10), [2450] (i;=ll-b20), [2450, 1892] (y=21) и [1892] (v=22, 23, 24).
Аналогичная методика была применена для расчета Ykl для состояний а1 А, Ь1!,, сх2, А3А. Для
состояний 2532 + и С32~ постоянные Ykl были вычислены непосредственно по значениям В„,
полученным в работах [1907, 789]. Постоянные Yki были приняты по оригинальным работам, кроме
состояний jB32 и С32, для которых они были рассчитаны по значениям Dt, найденным в работах
[1907, 789]. Мультиплетность и Х-удвоение уровней в состояниях а1 А, А3А, Б32 + и С32~
учитывались статистическими весами 2, 6, 3 и 3 соответственно. В расчете учитывались все уровни энергии
каждого состояния, ограниченные его предельной кривой диссоциации. В табл. 4.3 приведены
значения коэффициентов а{ в уравнениях, аппроксимирующих эти кривые, и значения vmax и Jllm
для каждого состояния, полученные по методике, изложенной в разделе 1.4. Погрешности
вычисленных термодинамических функций О2 при температурах до 6000 К не превышают 0,02 ДжХ
X К-моль и обусловлены неточностью фундаментальных постоянных и в меньшей степени тем, что
в расчете были использованы значения Ykl для молекулы 1вО2. При более высоких температурах
погрешности возрастают из-за отсутствия достаточно полных данных о
колебательно-вращательных уровнях энергии состояний а'Ди &Х2, неточности данных для состояний с^^и А3А, а также
из-за пренебрежения пятью валентными электронными состояниями. При Т > 12s 000 К начинает
также сказываться то, что в принятой методике расчета Qm и ее производных не учитывались
ридберговские состояния О2. Погрешности в значениях Ф° (Т) и S° (T) при 10 000 К могут
достигать 0,05 и 0,1 Дж-К^-моль соответственно, а при 20 000 К — 0,5 и 2 Дж-К^-моль х.
Ранее термодинамические функции О2 вычислялись во многих работах (см. [3960,88]). Из
ранних расчетов можно отметить работы Джонстона и Уокера [2149, 2150, 2151] и Вулли [3994],
выполненные непосредственным суммированием по уровням колебательной энергии состояния
Х3Х~ для Г^ 5000 К. В работах Самуйлова и др. [205, 248, 225] расчеты выполнены для Т=3000—
20 000 К суммированием по колебательным уровням шести электронных состояний при замене
суммирования по / интегрированием с корректным учетом предела интегрирования. В расчетах
не было учтено состояние А3 А, а мультиплетность состояния Х3И~ учитывалась статистическим
весом, что не внесло ошибок, так как расчеты были выполнены для Т > 3000 К. В работах [2412,
1319] методом непосредственного суммирования по уровням семи электронных состояний были
рассчитаны статистические суммы О2 для температур до 15 000 и 35 000 К, однако
термодинамические функции опубликованы не были. Беккетт и Хар [546] рассчитали функции О2 до 25 000 К,
использовав классическое выражение для сумм по состояниям, Мак-Брайд и др. [2556 ] вычисляли
термодинамические функции О2 до 6000 К непосредственным суммированием по колебательным
уровням пяти электронных состояний (Х32~, ахАд, 6*2*, ?32+, С32~). В таблицах JANAF [2095]
функции были вычислены для температур Т <] 6000 К по методу Майера и Гепперт-Майер без
учета возбужденных электронных состояний. Во 2-м издании справочника [88]
термодинамические функции О2 вычисдены по методике, отличающейся от принятой в настоящем издании менее
точным ограничением суммирования по вращательным уровням энергии.
При Т <; 5000 К результаты всех расчетов, кроме приведенных в работе [2095], согласуются
в пределах погрешностей, обусловленных использованием разных молекулярных и
фундаментальных постоянных. Данные JANAF [2095] наименее точны и отличаются от приведенных в табл. 5
при 6000 К на 0,03, 0,22 и 1,1 Дж-К^-моль в значениях Ф°(Т), S°(T) и Ср{Т) соответственно.
При температурах выше 6000 К расхождения с данными разных авторов увеличиваются, особенно
с данными работы [546 ], поскольку в этой работе термодинамические функции О2 вычислялись с
учетом вклада несвязанных состояний (при 20 000 К расхождения достигают 2 и 13 Дж-К^-моль
1 После завершения подготовки этого материала к печати была опубликована работа Крика и Николлса [1113],
посвященная уточнению постоянных О2 в состояниях Х32^, Ь1Е+ и С3Б~. Соответствующие изменения в
термодинамических функциях несущественны.
95
OJ (г) Глава 4
в значениях Ф°(Г) и S°(T)). Расхождения со 2-м изданием справочника обусловлены
прежде всего уточнением числа и энергии высоких колебательно-вращательных уровней
состояний Х3Ъ~, а}Ад и Ь1!,- и составляют 1,2 и 2,7 Дж-К^-моль-1 в значениях Ф° B0 000 К) и
S° B0 000 К).
В настоящем издании принимается, что двухатомный газообразный кислород является
стандартным состоянием этого элемента и в соответствии с этим
AfH°(O2, г)==0.
Константа равновесия реакции О2(г)==2О(г) вычислена по принятой в справочнике энергии
диссоциации
@) = Do(O2) = 493,566 + 0,20 кДж • моль.
Энергия диссоциации О2 была определена в работе Брикса и Герцберга [789] на основании
наблюдаемого схождения уровней состояний С32~ и В32* в предположении, что состояния Х32~
и 2?32+ имеют диссоциационный предел О CРа)+О (*Р2), а состояние С32~ — предел О C-Р2) +
+OAD2). Найденное авторами [789] значение
D0(O2) = 41260 +15 см-1
общепринято и послужило для КОДАТА—МСНС основой для рекомендации величины А..Н°(О, г,
298,15 КI.
OJ (г). Термодинамические свойства газообразного положительного иона двухатомного
кислорода в стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 6.
Молекулярные постоянные OJ, использованные при расчете термодинамических функций,
приведены в табл. 4.2. Ион О* согласно квантовомеханическим расчетам [1271, 1654, 547] имеет 15
связанных электронных состояний с энергиями до 70 000 см, коррелирующих с тремя нижними
диссоциационными пределами О+О+. В оптических спектрах наблюдались переходы, связанные
с пятью такими состояниями (ХШд, а4Па, А2Ии, с42~ и Ga?-). Из десяти остальных состояний
FЕ?, 642J, S2S+, С2Фи, Dmn, Е2Ад, <РД,, Fmg, #2Е+ и е*Е+) два (С2Фи, Е2\) были отождествлены
в фотоэлектронном спектре О2 [1383] (см. [2153, 3299J).
Молекулярные постоянные OJ в состояниях ^2П^, А2П.Ш, а4Пя и с42~ определялись на основании
изучения систем полос А2Ли -> Х2Пд [637, 3532, 3531, 1451, 1405, 727, 726] (переходы с v" < 10
е/< 8), и с42; -^ ати [3920, 2844, 2845, 728, 2842, 1539, 2843] (v" < 6, v' < 4), а также
из фотоэлектронных спектров [1026, 1383]. Постоянные OJ в состоянии Ga2~ определялись из
анализа ридберговских состояний молекулы О2, сходящихся к этому состоянию [2881, 4035, 3622],
и из фотоэлектронного спектра [1383]. Приведенные в табл. 4.2 молекулярные постоянные О?
для состояний X2Rg, а4Пм, А2П.и и с42~ основаны на рекомендации справочника [3591 ], где они были
вычислены с учетом неопубликованных данных Олбриттона и др. (см. [353 ]). Поскольку в спектрах
О? интеркомбинационные переходы не наблюдались, энергии состояний а4Пм и с42^ вычислены
по значениям потенциалов ионизации О2 в состояния иона ХЧ1д (97390 + 32 см~х) и с42~ A46 556 +
+ 2 см) и частоты v00 перехода c4S~-> а4Пи A6 666,74 см). Колебательные постоянные в состоянии
G22~ приняты по работе [1383]. Для состояний С2Фи и Е2Ад молекулярные постоянные,
приведенные в табл. 4.2, приняты в соответствии с результатами исследований фотоэлектронных спектров
О2 [2153, 3299], для остальных состояний — по результатам квантовомеханического расчета
[547].
При подготовке настоящего издания для обеспечения схождения колебательных уровней к
соответствующим диссоциационным пределам постоянные Yk0 были рассчитаны по методике,
изложенной в разделе 1.4. Для состояний Х'П и -42ПИ использовались значения Go(v) по данным [3591]
1 Принятым в справочнике значениям фундаментальных постоянных соответствует D0(Oa)=493,579 +
+ 0,18 кДж -моль, что практически совпадает с величиной, приведенной выше, -котерая была вычислена при
подготовке рекомендаций КОДАТА—MGHG по слегка отличающемуся значению NAhc.
96
Электронный газ и кислород OJ" (г)
(v ^ 10 и 8 соответственно), для состояний а4Пи и с42~ — данные [2756, 728, 1539] х (у ^ 6 и 2
соответственно). Найденные значения Yk0, а также значения Ykl, соответствующие постоянным
табл. 4.2, приведены в табл. 4.3.
Значения QBa и ее производных для О? вычислялись по соотношениям A. 90)—A. 92), при этом
значения <?кол> ,р, Qmi-^ и (?кол;вр для состояния Х2Ид находились непосредственным суммированием
по колебательно-вращательным уровням энергии этого состояния (уравнения A.73)—A.75)),
а для состояний а*Па, Агпи, 62+ и с42~ суммирование по вращательным уровням энергии было
заменено интегрированием с использованием соотношений типа A.82) и линейной зависимости
J'mix.t от °- Уровни вращательной энергии состояния Х2П.д аппроксимировались уравнениями A.42)%
состояний а^П,,, А2Н.и, в2+ и с42~ —уравнением A.34); А-удвоение вращательных уровней в
состояниях Х2П.д, а*П и Л2Па, а также мультиалетнооть уровней в состояниях а4Па, АШи, 6S+ и c4S~
учитывались соответствующими статистическими весами. Значения A//>4) (?^од_ вр и ее производных
для состояний 64S+ В2Ъ+, <21Д?, #2Е+ и e4S+ были приняты равными соответствующим величинам
для состояния 6SJ, а для состояний С2Фи, DzTlu, E2i\g, F2Qg и *232~ —¦ соответствующим величинам
для состояния Л3ПЯ. Этот выбор был обусловлен цримерным равенством значений гt и ш9 для этих
созтояний. В табл. 4. 3 приведены значения коэффициентов Ykl в уравнениях, описывающих
колебательно-вращательные уровни энергии в состояниях Х*П.д, а4Пи, АШи, 6S+ и c4S~, значения
¦''mix и Aim Ддя этих состояний, коэффициентов at в уравнении, аппроксимирующем предельную
кривую диссоциации состояния Х*Н-д, а также значения Те для всех 15 состояний и Ао для
состояния Х*П.д.
Погрешности вычисленных термодинамических функций О?, несмотря на отсутствие
экспериментальных данных об энергии и числе высоких колебательно-вращательных уровней энергии
состояния ХШд, при Т1^ 6000 К не превышают 0,02 Дж-К^-моль благодаря большой
величине D^OJ). При более высоких температурах погрешности возрастают из-за отсутствия
экспериментальных данных о высоких колебательно-вращательных уровнях энергии основного состояния
и об энергии большого числа состояний с Тв > 40 000 см. Общие погрешности в значениях
Ф°B0 000 К) и S°B0 000 К) оцениваются в 0,2 и 0,5 Дж-К^-моль.
Ранее таблицы термодинамических функций О^ вычислялись в работах [205, 225, 546] и
предыдущем издании настоящего справочника [88]. Расхождения данных, приведенных в работах
B05, 225] и в табл. 6, имеют наибольшую величину при 298,15 К из-за приближенного учета
в этих работах мультиплетности состояния Х2Ид (до 0,85 и 1,5 Дж-К^-моль в Ф°(Г) и С°(Т)).
Та же причина приводит к расхождениям с данными, приведенными в работе [546] при низких
температурах (до 1,2 Дж-К^-моль в Ф° A000 К)). При высоких температурах добавляются
расхождения из-за учета в [546] несвязанных состояний О Г, эти расхождения достигают 2,5 и
7 Дж -К-1 -моль-1 в Фэ B0 000 К) и 5° B0 000 К). Данные [88] и табл. 6 при Т < 5000 К
согласуются в пределах 0,1 Дж-К^-моль; при более высоких температурах расхождения растут
из-за применения в справочнике [88] более приближенной методики учета нижних
возбужденных состояний О2Ь и достигают 0,4 и 2,5 Дж -К -моль в значениях Ф° B0 000 К) и S° B0 000 К).
Константа равновесия реакции O2f(r)-)-e(r)=2 О(г) вычислена на основании значения АгН°@) —
= —671,431+0,45 кДж-моль, которое соответствует принятому в настоящем справочнике
потенциалу ионизации молекулы Оа
/0(О2, Х3^) = 12,074 ± 0,004 эВ = 97 393 ±32 см = 1165,0 ± 0,4 кД к • моль.
Определение точного значения /0(Оз) по границе схождения ридберговских серий молекулы
0.2 пока невозможно, так как в спектрах не наблюдались серии, сходящиеся к состоянию Х3П.д(О^);
интерпретация серий, сходящихся к состоянию АгИи, нуждается в уточнении (см. [1383]), а
хорошо изученная граница схождения ридберговской серии к состоянию с42^ A46 556+_2 см)
используется для определения энергий возбуждения квартетных состояний О?.
Приведенное выше значение 10{О2) является средним из полученных в исследованиях
фотоэлектронного спектра О2 Тернером [3743] A2,070±0,005 эВ), Коллином и Натали [1026] A2,075±
1 В работе [2879], опубликованной после завершения расчета функций Of (г), были получены близкие, но
несколько отличающиеся постоянные Of в состоянии c4Sj.
97
О? (г) Глава i
+ 0,005 эВ), Эдквистом и др. [1383] A2,071+ 0,005 эВ) и фотоионизационных измерений Ватанабэ-
и Мармо [3891] A2,078+0,001 эВ) и Самсона и Кэрнса [3297] A2,077 эВ). Следует отметить, что-
в работе [3297] для порога фотоионизации было найдено значение 1027,8+0,1 А, которому
соответствует 97 295 + 10 см, или 12,063 + 0,001 эВ. Однако позже Гийон и Берковиц [1779]
показали, что в данные [3297], как и для многих других фотоионизационных измерений, следует ввести;
поправку, обусловленную совместным влиянием различия вращательных постоянных, О2 в
состоянии Х32~ и Oj в состоянии Х2Лд и «приборной функции» эксперимента. На основании
выполненных расчетов авторы [1779] рекомендовали по экспериментальным данным [3297] значение-
12,077 эВ 1. Близкая величина была получена Макнилом и Куком [2593] при исследовании
фотоионизации О2 в состоянии гАд A2,067 эВ), если ввести в результаты измерений аналогичную
поправку, которая должна иметь величину порядка 0,01 эВ.
В 1975 г. Самсон и Гарднер [3298] повторили исследование фотоэлектронного спектра О2 и
с учетом влияния вращательной структуры на форму полос нашли /0(О2)=12,071 ±0,001 эВ„
Принятому значению /0(О2) соответствуют
Д/Я° (OJ, г, 0) = 1165,0 + 0,4 кДж • моль
и
Do (O+) = 642,509 + 0,44 кДж • моль = 53 710 + 40 см.
07 (г). Термодинамические свойства газообразного отрицательного иона двухатомного
кислорода в стандартном состоянии в интервале температур от 298,15 до 20 000 К приведены в табл. 7.
Молекулярные постоянные О^, использованные при расчете термодинамических функций,,
приведены в табл. 4.2. Молекулярные постоянные иона О^ известны на основании изучения
спектров флуоресценции О^ в кристаллах галогенидов щелочных металлов [3235, 3232, 1994, 1114,,
1422] и изучения сечения рассеяния медленных электронов на молекулах О2 [698, 3491, 1722,
2435, 2961]. Исследования спектров в кристаллах и квантовомеханический расчет, выполненный
Крауссом и др. [2305] (см. также [3919, 2750, 1039, 837]), показали, что ион О2 помимо основного-
состояния Х2Яд (конфигурация . . .Зо|1п* 1 тгр имеет ряд связанных возбужденных состояний;
с неглубокими минимумами на потенциальных кривых. Нижние возбужденные"~состояния
О~ D2~, 22~ и аПв) имеют энергии, превышающие 20 000 см. Поскольку молекулярные
постоянные О^ в состоянии Х*Н.д известны с невысокой точностью, а возбужденные состояния О^ имеют
энергии, превышающие потенциал ионизации иона, в расчете термодинамических функций
возбужденные состояния не учитывались. Спектры О^ исследовались в кристаллах ряда
галогенидов щелочных металлов. Экстраполяция найденных значений Д(?»+>/а привела для свободного иона
к значениям Д&/2=1090 см и шва:в=8,5 см [3235, 1994]. В экспериментах по рассеянию
медленных электронов на молекулах О2 было зарегистрировано от 6 [698, 3491, 1722] до 15 [24351
резонансных пиков. Абсолютная нумерация переходов установлена по совпадению энергий уровня
v=8 иона Ог в состоянии Х2Лд и уровня и=3 молекулы О2 в состоянии Ха2~. Все эти
исследования привели к практически совпадающим значениям постоянных (свв=1089 см, @^=8-^-12 см}
[698, 3491, 2435] за исключением работы [1722], где было получено «^=1169 см и шех,=\2 см.
Приведенные в табл. 4.2 колебательные постоянные О^ приняты на основании значений Д&/г=
•=1090 см, №^=10 см, G0(8)=8150 см (сумма величин A0(Ot) и GoC) для состояния Ха2~
молекулы О2) и D0(O2)c~33 000 см. Погрешность значения а>в оценивается в 20 см, значения шехе —
в 2 см. Межатомное расстояние в ионе О~ оценивалось в ряде работ, результаты этих оценок
лежат в интервале 1,34—1,40 А. В работе [920] значение г„=1<,341 + 0,01 А было получено на
основании сопоставления экспериментально измеренных сечений фотоотрыва электрона от О^ с
относительными вероятностями соответствующих процессов, рассчитанными теоретически для
разных молекулярных постоянных Oj. В работе [2358] при аналогичной обработке данных по
рассеянию электронов было найдено 1,39 А. Значение вращательной постоянной вычислено для
г =1,37 + 0,03 А. Постоянная спин-орбитальной связи О^ в состоянии X41t принята [по работе
1 В справочнике [3591 ] и обзоре Крупени [2311 ] для /0 @2) из данных [3297] принято ошибочное значение 97 265+.
+ 10 см.
98
Электронный газ и кислород О3 (г}
{2351 ], где она была определена при изучении рассеяния монохроматических электронов с
помощью время-пролетного электронного спектрометра. Найденное таким образом значение хорошо
согласуется с рассчитанным теоретически в неопубликованной работе Краусса (см. [2351]) (—150+
+ 30 см-1).
Термодинамические функции О2 (г) вычислены непосредственным суммированием по колебаг
тельно-вращательным уровням энергии состояния Х2Лд. Энергии вращательных уровней нахо-т
дились по уравнениям A.42) с постоянными Yk[, приведенными в табл. 4.3, А-удвоение уровней
учитывалось статистическим весом. В расчете учтены все уровни, ограниченные предельной крит
вой диссоциации состояния Х2Ид. В табл. 4.3 приведены значения коэффициентов а( в уравнении!
аппроксимирующем эту кривую, и значения ршах и /нт, полученные по методикам, изложенным
в разделе 1.4. Погрешности вычисленных значений термодинамических функций достигают 0,7}
1 и 1,5 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000и 6000 К; они обусловлены главным
образом неточностью принятых значений колебательных и вращательных постоянных (от 0,6
до 1,2 Дж -К -моль в значениях Ф° (Т) при 298,15 и 6000 К). При низких температурах к этому
добавляются погрешности из-за неточного значения постоянной спин-орбитальной связи @,2 ДжХ
X К-1 -моль в Ф° B98,15 К) и 0,06 кДж -моль в Н° B98,15 К)—Н° @)).
Ранее термодинамические функции О% вычислялись в таблицах JANAF [2095 ] по методу Майт
ера и Гепперт-Майер и несколько отличающимся значениям постоянных. Расхождения между
данными работы [2095] и приведенными в табл. 7 изменяются от —0,3 до —2,5 Дж -К-моль в знат
чениях S°(T) и от +1,7 до —1,6 Дж-К^-моль в значениях Ф°(Т). Значительные расхождения
в величинах Ф° B98,15 К) и Н° B98,15 К)—#°@), достигающие 1,7 Дж-К^-моль и 0,6 кДжХ
Хмоль, обусловлены тем, что в работе [2095] в расчете не учитывалось расщепление компот
нент состояния X2TLg молекулы OJ.
Константа равновесия реакции Ог(г)=2 O(r)-f-e(r) вычислена по значению АгН°@)=536,066 +,
+ 0,83 кДж-моль, основанному на величине сродства молекулы кислорода к электрону
Л0(О2)=—0,440+0,008 эВ=— 42,5 + 0,8 кДж-моль-1.
Это значение было получено Целотта и др. [920] при изучении фотоотрыва электронов от иона
О~ излучением аргон-ионного лазера с измерением энергии образующихся электронов. Оно
хорошо согласуется с величинами, найденными Паком и Фелпсом [3005, 2935] @,43+0,02 эВ)
при изучении температурной зависимости константы скорости реакции O2+O2=2O2-fe, Берко-
вицем и др. [595] при исследовании реакции 1~-+О2=О~-{-1 0^=0,47 + 0,1 эВ), в работах [2334,
2794] при исследованиях реакций О2 со щелочными металлами в скрещенных молекулярных
пучках @,5 + 0,2 и > 0,46 + 0,05 эВ), а также в работах [2131, 935] (> 0,5+0,1 эВ). Обзоры более
ранних и менее надежных исследований могут быть найдены в работах [920, 609]. Принятому
значению сродства к элекрону молекулы О2 соответствуют
Do (Op = 394,866 + 1,0 кДж • моль'1 & 33 400 ± 85 см
Д/Я° (О~, г, 0) = —42,5 + 0,8 кДж • моль.
О3 (г). Термодинамические свойства газообразного озона в стандартном состоянии в интервале
температур 100^-6000 К приведены в табл. 8.
Молекулярные постоянные О3, использованные в расчете термодинамических функций,
приведены в табл. 4.4. Молекула О3 в основном состоянии Х1А1 имеет структуру, соответствующую
точечной группе симметрии Сг». Молекулярные постоянные О3 в этом состоянии определялись на
основании изучения спектров комбинационного рассеяния [3377,823,1201,422, 2002, 1998, 394],
инфракрасных [3964, 3835, 2647, 2557, 470, 471, 2846, 2192, 986, 3714, 3480, 2270, 468, 765, 3613,
394, 3494], микроволновых [3715, 3987, 2035, 3015, 1688, 2424, 3612, 1164, 1582, 1581, 1216] и
электронных [70, 1863, 1735, 2165, 3913, 3426, 4010, 941, 2031,1865, 323, 2609, 1374, 1168]
спектров, а также электронографических измерений [3400, 2172]. Приведенные в табл. 4.4
колебательные и колебательно-вращательные постоянные приняты по работе Барба и др. [470, 471 ], которые
исследовали ИК-спектры 16О3 и 18О3 и использовали данные [986, 3714, 3480, 3613, 3612]. Эти
значения постоянных v(, xik и Wd описывают частоты начал 18 полос с точностью не хуже 1,1 см.
Вращательные постоянные О3, приведенные в табл. 4.4, получены в работе Лихтенштейна и др.
12424] в результате исследования микроволнового спектра. Они хорошо согласуются^ дав-
99
О, (г)
Глава 4
Таблица 4.4. Значения молекулярных постоянных О, (в си'1), принятые для расчета термодинамических
функций
«11
xM
«12
«18
1103,15
700,95
1042,1
—4,9
-1,0
—10,6
-9,1
—34,8
Примечание.
= 1)
-
Состояние
*Bt
•a\
*«1 —17,0
WD -27,05
Аооо 3,553 664 0
аА -0,002 981
аА —0,053 415
аА 0,053118
Вооо 0,445 276 2
af 0,002 554
аВ 0,001269
af 0,003 992
Сооо 0,394 758
°f 0,002 319
»? 0,002 307
af 0,003 613
т
т«се«
Teaii
Тв4#6
Зриведенн постоянные Og в основном электронном состоянии Ж1А1
. В расчете учитывались также следующие
То
и
10 000
12 500
13 000
13 500
СМ
U1 в1 с*
хЮ117
г3 • смв
600 350 600 48
600 350 600 51
850 500B) — 32
600 350 600 48
3
2
2
6
2
Pi
3
3
1
3
возбужденные состояния:
Состояние
То
СМ
гАг 14 000 600
1В1 16 600 600
яВг 25 000 600
гВ2 28 450 600
350
350
350
333
—8,142 9 • 10-«
—2,386 4 • 10-
—1,269 4-10-
16,947 -10-
—1,666 5-10-
3,271 7 -10-
—9,209 3 • 10"
(Г0 = 0, о = 2, рх =
IAIBIcx
хЮ117
г3 • см*
3
Pi
600 53 2 1
600 47 2 1
600 60 2 3
620 70 1 1
ными, полученными в других исследованиях микроволнового и инфракрасного спектра; им
соответствуют структурные параметры г0(О—О)=1,2717 + 0,0005 А и «^О—О—О=116°47'±47'.
Исследования электронных спектров О3 показали, что эта молекула имеет ряд состояний с
невысокими энергиями возбуждения. Диффузный характер спектров, перекрывание систем полос
и их низкая интенсивность затруднили анализ и полную идентификацию состояний. В работах
13426, 1168, 1863] (см. также [3587]) был выполнен частичный анализ системы полос 1Ват—^1А1
и определены колебательные постоянные верхнего состояния. Анализ остальных переходов до
настоящего времени не выполнен. Более полная информация о возбужденных состояниях О3
получена в квантовомеханических расчетах [1863, 1865, 1228, 4049, 1670, 1864, 1736], в которых были
определены энергии 15 связанных состояний этой молекулы. Для ряда состояний, включая
основное, были определены структурные параметры. Для основного состояния вычисленные значения
г (О—О) завышены на 0,1 А по сравнению с экспериментальными данными. Расчет энергий
возбуждения был сделан для вертикальных и невертикальных переходов (последние в упрощенном
приближении). Расхождения между этими значениями достаточно велики, и авторы работ [1863,
1865] принимают, что они являются верхними и нижними пределами для истинных значений Тв.
В табл. 4.4 приведены энергии восьми нижних возбужденных состояний О3, а также значения v.
в. произведения моментов инерции IaIbIg, принятые по результатам этих расчетов, которые
удовлетворительно согласуются с более поздней работой [1864].
Расчет колебательно-вращательных составляющих основного электронного состояния в
значениях термодинамических функций О3 был выполнен по уравнениям A.122)—A.124) с учетом
ангармоничности колебаний и колебательно-вращательного взаимодействия (уравнения A.131)—
A.133)), центробежного растяжения (уравнения A.143)) и резонанса Дарлинга1—Деннисона
(уравнения A.156)—A.158)). Составляющие восьми возбужденных состояний вычислялись в
приближении «жесткий ротатор—гармонический осциллятор» по уравнениям A.171)—A.173) с учетом
различия постоянных О3 в каждом состоянии. Погрешности вычисленных значений
термодинамических функций обусловлены неточностью учета высоких колебательно-вращательных уровней
основного состояния О3 и пренебрежением необходимостью ограничения суммирования по vk и
../ (энергия связи Оа—О примерно равна 9000 см), а также приближенными данными об энергиях
100
Эяектропный газ ж кжслород О3 (г)
возбуждения электронных состояний О3. Общие погрешности в значениях Ф°(Г) при 298,15f
3000 и 6000 К оцениваются в 0,03, 0,5 и 6 Дж-К^-моль.
Ранее термодинамические функции О3 вычислялись в работе [2771 ] (до 2000 К), предыдущем
издании справочника [88] и таблицах JANAF [2095] (до 6000 К) по близким значениям
постоянных; в [2095] была учтена ангармоничность колебаний О3. Пренебрежение возбужденными
состояниями, центробежным растяжением и другими поправками приводит к тому, что при 2000 К
данные всех этих работ меньше приведенных в табл. 8 примерно на 1 и 3 Дж-К^-моль в
значениях Ф°(Т) и S°(T). При 6000 К расхождения с работами [88] и [2095] достигают примерно 16
и 40 Дж-К^-моль в значениях этих функций.
Константа равновесия реакции О3(г)=ЗО (г) вычислена на основании значения ДгЯ°@)=595,892±
±2,0 кДж-моль, соответствующего энтальпии образования
Д^0 (О3, г, 298,15 К) = 141,8 + 2,0 кДж • моль,
принятой в справочнике по работе Гюнтера и др. [1769]. Авторы этой работы измерили
калориметрически энтальпию разложения озона в смесях с кислородом (О3=3/2 О2). Количество
разложившегося озона определялось с хорошей точностью по изменению давления в калориметре.
В работе Клайна и др. [995] было получено близкое значение A41,0 кДж-моль) на основании
мучения кинетики образования О3 и литературных данных по кинетике разложения О3. Бирли
и Скиннер [654] измерили энтальпии трех реакций окисления О8 в растворах. Однако во всех
случаях из-за протекания побочной реакции О3 -> 8/2 О2 они смогли определить только верхний
предел значения А^Н° (О3, г). Наиболее надежный результат был получен в реакции с AsaO3 (aq)
A42,5 кДж-моль-1). В работах [614, 615, 2736, 2735, 3762, 2184, 2090], выполненных до 1911 г.,
результаты измерений ненадежны из-за недостаточной точности использованной
экспериментальной техники, возможности протекания побочных реакций и недостаточной охарактеризованности
образцов. Следует отметить, что данные работ [2735, 2736 и 2184, 2090], пересчитанные Бихов-
ским и Россини [642], близки к принятому значению A41 и 144,3 кДж-моль).
Глава 5
ВОДОРОД И ЕГО СОЕДИНЕНИЯ
Н, Н% Н-, Н2, о-Н2, и-Н2, Щ, Н+, ОН, ОН+, ОН", НО2, НО", Н2О (к, ж),
Н2О, Н2О+, Н2О2 (к, ж), Н2О2, Н3О+
В настоящей главе рассматриваются термодинамические свойства водорода и его газообразных
соединений с кислородом, а также двух соединений (Н2О и Н2О2) в конденсированном состоянии.
Соединения водорода с другими элементами рассматриваются в последующих главах. Для
большей части веществ в газообразном состоянии (кроме НО2, НО^ и Н2О2) таблицы
термодинамических свойств рассчитаны до 20 000 К. В Дополнении 1 (см. наст, том, кн. 2) приведены также
таблицы термодинамических свойств о-Н2 и тг-Н2 для температур от 10 до 1000 К.
Для всех веществ приведены термодинамические свойства для природной смеси изотопов
водорода с атомным весом 1,0079. Благодаря ничтожно малому содержанию дейтерия и трития в
природной смеси изотопов водорода отличие значений термодинамических функций, вычисленных
с атомным весом 1,007 825, существенно меньше погрешностей, обусловленных неточностью
принятого значения газовой постоянной R. В Приложении 3 приведены данные, позволяющие
учитывать отклонения термодинамических свойств Н2(г), Н2О(г) и Н2О2(г) от их свойств в
стандартном состоянии в результате межмолекулярного взаимодействия.
Н(г). Термодинамические функции газообразного водорода в стандартном состоянии в
интервале температур от 100 до 20 000 К приведены в табл. 9.
Термодинамические функции Н(г) вычислялись по соотношениям A.3)—A.6) с учетом только
основного состояния aS атома Н.
При температурах до 12 000 К погрешности вычисленных значений термодинамических
функций не превышают 0,01 Дж-К^-моль в значениях Ф°(Т) и S°(T) и обусловлены главным
образом неточностью принятых значений фундаментальных постоянных. При более высоких
температурах становятся существенными ошибки из-за пренебрежения возбужденными ридбергов-
скими состояниями атома Н. Они могут достигать 0,07, 0,3 и 1 Дж-К^-моль в значениях Ф°(Т),
S°(T) и С°р(Т) при 20 000 К и 0,012, 0,1 и 0,6 Дж-К^-моль-1 в значениях этих функций
при 15 000 К.
Термодинамические функции атомарного водорода вычислялись в десятках работ. В
справочных изданиях [2095, 2556, 1946, 2029, 2198, 87], так же как в большинстве оригинальных работ,
например [2308, 1673], расчеты были выполнены для температур не выше 6000 К и их результаты
согласуются с данными, приведенными в табл. 9, в пределах 0,008 Дж-К^-моль в значениях
Ф°(Г) и S°(T). В предыдущем издании настоящего справочника [88] расчет был выполнен
до 20 000 К с учетом ридберговских состояний со значениями п ^ 13. Поэтому расхождения между
данными табл. 9 и функциями, приведенными в справочнике [88], достигают 2,5 Дж-К^-моль
в Ф° B0 000 К).
Энтальпия образования одноатомного водорода в соответствии с рекомендацией КОДАТА —
MGHG [1000] принята равной
A/PfH, г, 298,15К) = 217,997 ±0,006 кДж -моль-1.
Она основана на значении энергии диссоциации Н2 (см. ниже).
Н+(г). Термодинамические свойства газообразного положительного иона водорода в
стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 10.
Термодинамические функции иона Н+ вычислены по соотношениям A.3)—A.6), поскольку ион
Н+не имеет электронов и внутримолекулярные составляющие термодинамических функций равны
нулю. Погрешности вычисленных значений определяются только неточностью фундаментальных
постоянных и не превышают 0,01 Дж-К^-моль во всем интервале температур в значениях Ф°(Т)
и S°(T).
102
Водород и его соединения Н~ (г), Н2 (г)
Таблицы термодинамических свойств Н+ вычислялись ранее в справочниках [2095, 19461
((до 6000 К) и [88] (до 20 000 К). Расхождения со всеми этими данными не превышают 0,01 ДжХ
X К-моль в значениях Ф°(Т) и S°(T).
Константа равновесия реакции Н+(г)+е(г)=Н(г) вычислена на основании значения Дг#°@) =
= —1312,049 + 0,001 кДж-моль, равного (с обратным знаком) потенциалу ионизации атома
водорода
/0(Н)= 109 678,764 2 + 0,000 01 см = 1312,049 + 0,001 кДж • моль.
Это значение было вычислено в работе Гарсия и Мака [1566] в рамках релятивистской квантово-
механической модели. В ряде справочных изданий по рекомендации Мур [2697] принималось
значение, полученное в неопубликованной более ранней работе Мака и отличающееся от
приведенного выше примерно на 0,01 см. В работе [1556] погрешность вычисленного значения
оценивается в 10~5 см, однако из-за использования менее точных значений физических постоянных
при переходе к джоулям погрешность возрастает на 3 порядка.
Принятому значению /0(Н) соответствует
Д/ЙГ°(Н+, г, 0) = 1528,083+ 0,006 кДж • моль.
Н" (г). Термодинамические свойства газообразного отрицательного иона водорода в
стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 11.
Основным состоянием иона Н~ является состояние lS. Согласно теоретическим расчетам (см.
13210]) ион Н~ не имеет связанных возбужденных состояний с энергией ниже потенциала
ионизации. Поэтому термодинамические функции Н" вычислялись по уравнениям A.3)—A.6) без учета
внутримолекулярных составляющих. Погрешности вычисленных значений термодинамических
функций определяются только неточностью принятых фундаментальных постоянных и не
превышают 0,01 Дж -К -моль в значениях Фэ (Т) и S° (T) во всем интервале температур.
Ранее термодинамические функция Н~ (г) вычислялись в таблицах JANAF [2095] до 6000 К
и предыдущем издании справочника [88] до 20 000 К, результаты всех расчетов согласуются
в пределах 0,01 Дж-К^-моль.
Константа равновесия реакции Н~ (г)=Н (г)-|-е (г) вычислена на основании значения Arif°@)==
=72,770 + 0,002 кДж'Моль, соответствующего величине сродства атома Я к электрону
40(Н) = — 6083,1 ±0,2 см-1^—72,770 + 0,002 кДж-моль.
Последняя была принята в настоящзм издании на основании кванговомеханичзского расчета,
выполненного Пекерисом [2987]. В приведенной погрешности учтено влияние лэмбовского
сдвига на основании сравнения аналогичного расчета потенциала ионизации атома Не с
экспериментальными данными. Экспериментальные определения величины AQ (Н) по порогу континуума,
связанного с фотоионизацией Н~ [3986, 1461, 3914, 3469], по порогу фотодиссоциации Н2 на Н +
и Н~ [2572] привели к близким, но менее точным значениям, лежащим в иягервале от — 6081 ±
±70 см до —6258 + 160 см.
Принятому значению Ао (Н) соответствует
ДД° (Н-, г, 0) == 143,264 ± 0,006 кДж • моль.
Н2 (г). Термодинамические свойства газообразного двухатомного водорода в стандартном
состоянии в интервале температур 100—20 000 К приведены в табл. 12.
Термодинамические функции Н2(г) были вычислены без учета возбужденных электронных
состояний Н2 для равновесной смеси орто- и параводорода х. Расчет статистической суммы по
колебательно-вращательным уровням энергии состояния X1!,* и ее производных проводился
непосредственным суммированием по соотношениям A.73)—A.75) и уровням энергии, приведенным
в табл. 5.1. Из найденных таким образом значений 1а (?кол< вр для равяозвсной смеси вычиталась
составляющая ядерных ; тинов (Л1п 4). Колебательно-вращательные уровни энергии Н2,
приведенные в табл. 5.1, основана на расчетах, выполненных Уэчем и Бернстейяом [3850] и Ле Роем [2386]
численным дифференцированием уравнения Шредингера с потенциальной функцией состояния
О расчете термодинамических функций о-На и /г-Н2 см. с. 107.
103
Таблица 5.1
/
0
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
р=0
0,0
118,5
354,4
705,6
1168,9
1740,3
2415,1
3187,8
4052,3
5002,4
6031,5
7132,8
8299,4
9524,7
10802,1
12125,0
13487,3
14883,1
16306,5
17752,1
19215,0
20690,1
22172,8
23658,9
25144,2
26624,8
28096,9
29556,8
31001,2
32426,4
33829,0
35205,3
36551,6
37863,5
39136,0
40362,6
41533,7
42630,7
43630,5
. Уровни
колебательно-вращательной энергии молекулы На в
для расчета термодинамических функции
1
4161,1
4273,7
4497,8
4831,4
5271,4
5814,0
6454,5
7187,7
8007,8
8908,8
9884,2
10927,5
12032,2
13191,9
14400,1
15650,6
16937,4
18254,7
19597,0
20958,9
22335,5
23721,8
25113,4
26505,9
27894,9
29276,4
30646,4
32000,8
33335,8
34647,0
35930,0
37179,5
38389,6
39552,0
40653,5
41665,5
2
8087,1
8193,9
8406,5
8722,9
9140,1
9654,5
10261,5
10956,0
11732,6
12585,3
13507,9
14494,3
15538,0
16632,8
17772,6
18951,3
20163,0
21402,1
22663,2
23941,0
25230,5
26526,9
27825,4
29121,5
30410,8
31688,7
32950,8
34192,2
35407,9
36592,0
37737,5
38834,9
39868,5
40802,9
3
11782,3
11883,4
12084,6
12384,1
12778,8
13265,3
13839,2
14495,6
15229,1
16034,1
16904,5
17834,2
18817,3
19847,6
20919,0
22025,7
23161,9
24322,1
25500,8
26692,9
27893,1
29096,5
30298,2
31493,3
32676,6
33843,0
34986,8
36101,6
37179,5
38210,0
39176,2
40038,9
4
15250,4
15345,8
15535,7
15818,3
16190,8
16649,6
17190,5
17808,9
18499,5
19256,7
20074,9
20947,9
21870,0
22835,2
23837,5
24871,1
25930,4
27009,7
28403,6
29206,6
30313,5
31418,9
32517,2
33602,8
34669,4
35710,2
36716,7
37677,8
38575,2
39367,2
5
18491,9
18581,7
18760,4
19026,0
19376,1
19807,1
20315,0
20895,1
21542,6
22251,4
23016,6
23832,2
24692,2
25590,8
26522,2
27480,5
28460,0
29454,8
30459,4
31468,0
32474,6
33473,1
34456,9
35418,6
36349,2
37237,1
38063,3
38784,5
6
21505,6
21589,7
21756,9
22005,5
22332,8
22735,6
23209,8
23750,9
24354,0
25013,8
25724,7
26480,9
27276,8
28106,3
28963,5
29842,6
30737,4
31642,0
32550,0
33454,9
34349,8
35226,7
36076,6
36886,4
37637,6
38285,7
7
24287,8
24366,0
24521,4
24752,4
25056,4
25430,1
25869,6
26370,5
26927,8
27536,3
28190,4
28884,5
29612,6
30368,7
31146,7
31940,3
32742,9
33547,6
34347,2
35133,4
35896,5
36623,6
37294,9
37865,1
основном электронном состоянии 2
8
26831,0
26903,0
27046,2
27258,8
27538,4
27881,5
28284,7
28743,1
29252,0
29806,1
30399,8
31027,2
31682,2
32358,3
33049,1
33747,4
34445,5
35134,9
35805,5
36444,0
37029,4
37516,4
9
29123,9
29189,4
29319,6
29512,7
29766,3
30077,0
30441,0
30853,8
31310,5
31805,5
32333,1
32882,2
33460,6
34046,7
34637,6
35224,4
35796,7
36340,6
36833,4
10
31150,2
31208,6
31324,7
31496,6
31721,8
31997,1
32318,3
32680,9
33079,5
33508,5
33961,5
34431,4
34910,4
35389,6
35858,1
36301,4
36693,0
п
32886,8
32937,5
33037,9
33186,1
33379,7
33615,0
33887,8
34193,1
34525,1
34877,2
35241,8
35609,9
35970,3
36307,0
36588,5
pis; (в
V
i2
34301,8
34343,5
34425,8
34546,8
34703,6
34892,3
35108,2
35345,1
35596,1
35852,2
36101,7
36323,8
см), принятые
13
35351,0
35381,9
35442,5
35530,6
35642,6
35773,7
35917,4
36064,8
36200,4
14
35973,0
35990,1
36022,7
36067,2
36117,2
36153,2
м
и
а
ел
Водород ж его соединения
Н2(г>
, которая была получена Колосом и Вольневичем [2289] в результате неэмпирического
квантовомеханического расчета. В работе [2386] были учтены адиабатическая и нерелятивистская
поправки, полученные Колосом и Вольневичем [2288, 2290].
Значения In Qmi вр и ее производных вычислялгсь по уровням энергии, полученным в квантово-
механических расчетах, в связи с тем, что в экспериментальных доследованиях спектров Н2 не
наблюдались переходы, связанные с колебательно-вращательными уровнями состояния Х1^* со
значениями квантового числа / > 9 (согласно теоретическим расчетам Jma,0=43).
В спектре комбинационного рассеяния Н2 (см. [652, 3161, 3542, 3635]) и квадрупольном спектре
[1904, 1480] изучены только переходы между уровнями су^Зи/^5. В электронном спектре1
+ (см. [629, 2058, 2118, 2121, 3615, 1913])
наблюдав системах полос ЯЧЗ+ — Х'?+ и СШ„ —
лись переходы на уровни состояния
со всеми значениями v
'таж=14 и / <! 9.
Молекулярные постоянные Н2, полученные в этих работах (наиболее полгый набор значений постоянных,
найденный Герцбергом и Хау [1913], приведен в табл. 5.2), плохо описывают
колебательно-вращательные уровни с большими значениями / и не пригодны для их аппроксимации к предельной
кривой диссоциации.
Таблица 5.2. Молекулярные постоянные двухатомных соединений водорода
Молекула
Состояние
в.
e ¦ 10*
СМ
А
Н2
Щ
ОН
ОН+
ОН"
Х22+
(
Л22+
?2Е+
а1 А
а3П
0
0
0а
32 684,5 Д
69 720,7
0а
16 800
28 441,78
29 046,0к
45 000
0
29 000
[4 400,39
2 323,242
3 737,90
3 178,86
659,7и
3 113,37
3 000
2 133,65
3 148Л
1800
3 800
120,815а
67,466а
84,965б
92,917 е
—
78,515
56г
79,55
84л
67
90a
60,864
29,946 35
18,895
17,358
5,519 5
16,794 3
16.7 Д
13,7916
16,686
13.8 Д
19,3
3,076 38 е
1,594 94 е
0,704в
0,786 8»
0,867
0,749 4 е
0,464 е
0,888 9
0,66 е
0,764 е
0,68 е
465,7в
199,06в
1,93г
20,48
9,298
1,917 44°
1,7 ж
2,249 5"
1,92
3,2 ж
20в
0,74158
1,057156
'0,968 7
1,0121
1,794 8
1,028 9
1,032
1,135 4
1,032
1,135 Д
0,96
Примечание. Ниже все величины приведены в см.
Н • 07242 6 6017 Ю2 Е р 2616
р
Н2
ОН
ОН+
ОН":
р
о2 = 6.017 • Ю;
0053 6
; р, 2,616 • 10-3
-0,053; 6 а2 ¦= 3,242 • 10"*, аа =
5,63.10-»; Le = 1,895 • 10"».
ю,!/, = 0,539, <оА= 1,674 • Ю-2, utt.*=—1,637
1,4074-10-', Lo= 1,23-10-"; я-го = О,22; * «
— 0,016; • приведено значение Do;
0 ,210.
1,64 • 10; в В, =8,6255 • 10, В2
10; в а2 = 7
е = -1,7915; e,,
приведено значение ДО,,.
D Н
2,52 • Ю'8, #„ =
10
10-«; Г В, =4,83
— 0,32362, o
ее ;
»Ш(,0« = 0,941, о) Ze
= 1,735-Ю-6, Л, в
а ^ = -139,285; 6
Р2 = 5,3.10-в, #0
= —3,585-Ю-2;
» 1 = 2,1344, 7 = ; 2; B о , ;
по соотношению A.67); * принято по ивоковфигуранвоввому состоянию; е вычислено do соотноше
нию A.69); к вычислено по A.68); в А =—83,83; в приведено вначение Do, Яо==0,104 • 10~в, *
приведено значение То; J вычислено по соотношению A.67).
A67) D (ОН) 38 340
— 0,1478; б а2в=0,01097; в приведено значение DB, Но = 0,123 94 • 10;
A67); * принято по ивоковфигуранвоввому состоянию; е вычислено
вычислено
соотношед о;
вычислено по соотношению A.67) при Do (ОН")
вычислено по A.68).
()
38 340+55 см; 6 вычислено по соотношению A.69);
Сравнение значений энергии колебательно-вращательных уровней, вычисленных в
работах [3850, 2386], с экспериментальными данными для 108 уровней, наблюдавшихся в работе Герц-
берга и Хау [1913], показало, что они согласуются со средним отклонением 3,6 см
(максимальное расхождение 5,5 см). Анализ этих расхождений, выполненный при подготовке настоящего
издания, позволил прийти к выводу, что при /=0 расхождения систематически возрастают с
увеличением v. Введение в рассчитанные авторами работ [3850, 2386] значения G (v)+Ft (J) поправок,
основанных на найденных в работе [1913] экспериментальных значениях G0(v), резко снижает
расхождения экспериментальных и рассчитанных значений Т (v, J) для 108 уровней (среднее
105
Щ (г) Глава 5
отклонение 0,1 см, максимальное 1 см). В табл. 5.1 приведены значения Т (v, J) для всех
связанных и квазисвязанных уровней энергии состояния Х1^, основанные на расчетах Ле Роя
[2386] и таких эмпирических поправках.
Погрешности вычисленных значений термодинамических функций Н2 при температурах
до 5000 К обусловлены только неточностью физических постоянных и не превышают
О,01 Дж-К^-моль в значениях Ф°(Т) и S°(T). При более высоких температурах они
возрастают из-за того, что квазисвязанные колебательно-вращательные состояния Н2 учитывались в
расчете как связанные. При 6000 К погрешности в значениях Ф°(Т) и S°(T) составляют
0,01—0,02 Дж-К-моль, при 10 000 и 20 000 К около 0,02 и 0,05 Дж-К^-моль
соответственно. При температурах выше 10 000 К становятся заметными погрешности,
обусловленные тем, что в расчете не учтены возбужденные электронные состояния Н2. При 20 000 К они
могут достигать 0,1 и 0,4 Дж-К^-моль в значениях Ф° (Г) и S° (Г). Таким образом,
погрешности табулированных значений Ф° F000 К) и Ф° B0 000 К) не превышают 0,02 и
0,1 Дж-К^-моль-1.
Термодинамические функции Н2 в широком интервале температур вычислялись в работах
A612, 1182, 4000, 1783] (до 5000 К), [26, 87, 2296] (до 6000 К), [88] (до 20 000 К). В работах
Джиока [1612] и Джонстона и др. [1182] и основанных на них таблицах [3852] использовались
неточные значения молекулярных постоянных, что обусловило их отличие от данных табл. 12 и
других справочных изданий. В работе Вулли и др. [4000] впервые был сделан тщательный выбор
молекулярных постоянных Н2 и учтена необходимость корректной аппроксимации уровней с
высокими значениями г; и /; результаты этого расчета согласуются с приведенными в табл. 12 в
пределах 0,015 Дж-К^-моль-1 в значениях Ф°(Г) и S°(T). В справочниках [2095, 2029, 2556, 337]
таблицы термодинамических свойств Н2 основаны на расчете [4000] с экстраполяцией их до 6000 К.
Поэтому все функции, кроме С°р (Т), хорошо согласуются с данными табл. 12; расхождения в
€° F000 К) достигают 0,3 Дж-К^-моль для [2095]1. Хар и др. [1783] вычислили
термодинамические функции Н2 методом Майера и Гепперт-Майер. Расхождения с табл. 12 достигают 0,15 и
1,6 Дж-К-моль в значениях Ф° (Т) и С°р (Т). Данные предыдущего и настоящего изданий
согласуются вплоть до 20 000 К в пределах 0,015 Дж^К^-моль, кроме значения S0 B98,15 К)
В работах [26, 2296] термодинамические функции Н2 были вычислены до 6000 К
непосредственным суммированием по уровням энергии, рассчитанным в работе [3850, 2386] соответственно.
Приведенные в работе [26] значения С* (Т) при 6000 К отличаются от данных табл. 12
ла 0,07 Дж-К~х моль. В работе [2296] был использован уточненный метод учета квазисвязанных
колебательно-вращательных уровней На, принимающий во внимание их ширину. Расхождения
данных, приведенных в этой работе и в табл. 12, не превышают 0,02 Дж -К -моль во всем
интервале температур.
Молекулярный водород в газообразном состоянии является стандартным состоянием этого
¦элемента и в соответствии с этим
A/fl^H,. г) = 0.
Константа равновесия реакции Н2 (г)=2Н (г) вычислена при
дгЯ°@)=Бо (Н2)=432,068±0,012 кДж-моль-1.
Первые спектральные измерения Do (H2) были выполнены в работах [3980, 1250] в 20-е годы.
Бойтлер [630] на основании изучения границы континуума в спектре поглощения Н2,
соответствующего переходу В1^ <- ХХ2*, нашел Do (H2)=36 116 ± 6 см, которое принималось во всех
справочных изданиях до 1970 г.2 В 1970 г. Герцберг [1910] вновь исследовал на приборе с
высоким разрешением границу континуума в спектрах Н2, HD и D2, находящихся при температуре
жидкого азота (для предотвращения наложения линий, соответствующих переходам из состоя-
лий с / > 0). Было найдено, что 36 116,3 < Do (H2) < 36 118,3 см.
1 Примечание при корректуре. В 1977 г. авторы работы [2095] предприняли попытку уточнить этот расчет,
в результате чего расхождения в СР(Т) снизились до 0,04 Дж-К-'-моль-1 (при 4500 К).
3 Диссопиационный предел состояния В1?,* соответствует атомам Н (Is 25)-j-H Bs 2S).
106
Шодород н его соединения о-Н,(г), «-II, (г),
Квантовомеханические расчеты Колоса и Вольневича [2290] и Банкера [833] (с учетом неадиа-
;€атической поправки) привели к значениям 36 117,4 и 36 117,9 см соответственно. Поскольку
расчеты могут дать только нижнюю границу значения Do, в справочнике принимается в
согласии с рекомендацией КО ДАТА—МСНС [1000]х
D0(H2)=36 118,3 + 1 см-1=432,068±0,012 кДж-моль-1.
о-Н2 (г). Термодинамические функции газообразного ортоводорода в стандартном состоянии
в интервале температур 10—1000 К приведены в табл. Д.1 (см. наст, изд., кн. 2).
Значения QK0!I вр и ее производных были вычислены непосредственным суммированием по
колебательно-вращательным уровням энергии основного электронного состояния Н2, приведенным
з табл. 5.1, по уравнениям типа
14 Jmax, о
<?¦=«. *р Mb) = 2 2 (J'H + 1} {2is + *>B/ + 1} еХр {- ТГ
«=0 J=l, 3, 5, ...
тде (гн + 1)Bг
Погрешности вычисленных термодинамических функций ортоводорода обусловлены
практически только неточностью фундаментальных постоянных и не превышают 0,01 Дж-К^-моль
в значениях Ф° (Т) и S° (T).
Термодинамические функции ортоводорода ранее вычислялись в работе Вулли и др. [4000];
результаты этих расчетов и табл. Д.1 согласуются в пределах 0,01 Дж-К^-моль; имеющиеся
^расхождения обусловлены различием фундаментальных постоянных.
w-H2(r). Термодинамические функции газообразного параводорода в стандартном состоянии
ж интервале температур 10—1000 К приведены в табл. Д.2.
Значения QK0I, ^ и ее производных были вычислены непосредственным суммированием по
колебательно-вращательным уровням энергии основного электронного состояния молекулы Н2
«(см. табл. 5.1) по уравнениям типа
/шах,«
2 2 ¦ М2*н + 1)B/+ 1)ехр{-^ [<?,(!;) + *•,(')]}. E-2)
t=0 J=0, 2, 4, ...
згде *нB*н+1)=1.
Погрешности вычисленных таким образом значений термодинамических функций обусловлены
только неточностью фундаментальных постоянных и не превышают 0,01 Дж-К^-моль в
значениях Ф°(Г) и S°(T).
Термодинамические функции параводорода ранее вычислялись в работе Вулли и др. [4000]
•(до 1000 К), а также Харом и др. [1783] (до 200 К). Результаты этих расчетов и данные
табл. Д.2 согласуются в пределах 0,01 Дж-К^-моль.
Hj (г). Термодинамические свойства газообразного ионизированного двухатомного водорода
я стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 13.
Термодинамические функции Щ (г) были вычислены без учета возбужденных электронных
состояний HJ для равновесной смеси о-Щ и п-Щ. Расчет статистических сумм по
колебательно-вращательным уровням энергии состояния Х22* и их производных проводился непосредственным
суммированием по соотношениям A.73)—A.75) и уровням энергии, приведенным в табл. 5.3.
Из найденных значений In Qm для равновесной смеси орто- и парамодификаций2 вычиталась
составляющая ядерных спинов (R In 4). Мультиплетность колебательно-вращательных уровней
состояния Х22* учитывалась статистическим весом 2.
1 Принятому в справочнике значению NAhc соответствует 432,071 + 0,009 кДж • моль.
а Значения термодинамических функций о-Н J и п-Щ в справочнике не табулированы, так как при температурах,
когда статистические суммы по четным и нечетным значениям / различны, HJ практически не существует.
107
Таблица 5.3. Уровни колебательно-вращательной энергии молекулы lit в основной электронном состоянии Х2Е+ (в саг1)
принятые для расчета термодинамических функций я '
, К
/
0
1
2
3
к
5
в
7
8
9
10
И
12
13
и
15
1в
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
0,0
58,2
174,2
47,1
575,5
857,6
1191,4
1574,5
2004,2
2477,6
2991,8
3543,6
4129,8
4747,3
5392,8
6063,2
6755,5
7466,7
8193,8
8934,3
9685,4
10444,7
11209,7
11978,3
12748,2
13517,5
14284,3
15046,7
15803,0
16551,5
17290,7
18018,9
18734,6
19436,3
20122,4
20791,2
21441,0
22069,6
22674,7
23253,1
23799,9
24306,1
24740,5
1
2191,3
2246,5
2356,4
2520,2
2736,5
3003,7
3319,7
3642,4
4089,1
4537,0
5023,4
5545,3
6099,5
6683,1
7292,9
7926,0
8579,5
9250,4
9936,1
10633,9
11341,2
12055,6
12774,8
13496,6
14218,9
14939,7
15657,1
16369,2
17074,2
17770,5
18456,2
19129,7
19789,2
20432,9
21058,7
21664,5
22247,5
22804,1
23329,1
23812,1
24213,5
2
4255,5
4307,7
4411,7
4566,6
4771,2
5024,0
5322,9
5665,8
6050,3
6473,5
6933,0
7425,8
7949,0
8499,6
9074,8
9671,6
10287,2
10918,8
11563,9
12219,8
12884,0
13554,3
14228,2
14903,7
15579,7
16251,1
16918,9
17580,4
18233,5
18876,4
19507,3
20124,0
20724,5
21306,3
21866,6
22401,8
22906,5
23370,9
23761,9
3
6196,6
6245,9
6344,2
6490,6
6683,9
6922,6
7204,9
7528,6
891,5
8290,5
8723,9
9188,4
9681,4
10199,9
10741,1
11302,3
11880,8
12473,9
13078,9
13693,5
14315,1
14941,5
15570,4
16199,7
16827,1
17450,7
18068,4
18678,2
19278,0
19865,6
20438,9
20995,3
21531,9
22045,0
22529,4
22976,4
23361,5
4
=8018,2
8064,8
8157,4
8295,5
8477,8
8702,9
8968,9
9273,9
9615,6
-9991,2
10398,9
10835,8
11299,0
11786,0
12293,9
12820,1
13361,9
13916,8
14482,2
15055,7
15634,8
16217,4
16801,0
17383,6
17962,8
18536,7
19102,8
19659,1
20203,1
20732,1
21243,4
21733,1
22196,4
22625,6
23002,3
5
Г9723.4
9767,2
^9854,4
9984,3
10155,8
10367,5
10617,6
10904,2
11225,2
11577,6
11960,2
12369,7
12803,7
13259,5
13734,4
14225,9
14731,3
15248,2
15774,0
16306,3
16842,7
17381,0
17918,7
18453,6
18983,3
19505,6
20017,8
20517,4
21001,4
21466,0
21906,8
22316,7
22681,7
6
11314,3
11355,4
11437,2
11559,1
11720,0
11918,5
12152,9
12421,4
12721,9
13051,4
13409,1
13791,6
14196,6
14621,4
15063,4
15520,2
15989,1
16467,8
16953,7
17444,3
17937,3
18430,2
18920,7
19406,2
19884,2
20351,9
20806,4
21244,0
21660,3
22049,1
22398,9
7
12792,6
12831,0
12907,6
13021,5
13171,9
13357,2
13576,1
13826,6
14106,8
14413,6
14746,4
15102,0
15477,9
15871,6
16280,6
16702,5
17134,6
17574,5
18019,7
18467,6
18915,9
19361,9
19802,9
20236,3
20658,8
21067,1
21456,8
21822,3
22153,8
22424,7
8
14159,4
14195,2
14266,4
14372,4
14512,2
14684,6
14887,9
15120,5
15380,3
15664,2
15972,1
16300,6
16647,2
17009,6
17385,1
17771,4
18165,9
18566,0
18969,2
19372,9
19774,3
20170,7
20558,8
20935,4
21296,2
21618,5
21946,2
22207,7
9-
15414,9
15448,0
15514,0
15612,0
15741,3
15900,5
16088,2
16302,7
16541,9
16802,9
17085,6
17386,5
17703,3
18033,6
18374,9
18724,6
19080,2
19438,9
19798,1
20154,9
20506,2
20848,6
21178,0
21489,5
21775,6
22021,2
10
16558,8
16589,2
16649,8
16739,8
16858,4
17004,4
17176,2
17372,2
17590,6
17828,2
18085,0
18357,8
18644,0
18941,2
19246,8
19558,4
19873,0
20187,8
20499,9
20805,7
21101,4
21382,2
21641,3
21866,1
И
17589,9
17617,6
17672,7
17754,5
17862,2
17994,6
18150,2
18327,3
18524,3
18737,7
18967,9
19211,4
19465,6
19728,1
19996,3
20267,2
20537,8
20804,9
21064,6
21312,4
21541,9
21742,8
12
18506,2
18531,1
18580,5
18653,9
18750,4
18868,7
19007,5
19165,1
19339,7
19528,2
19730,3
19942,9
20163,3
20388,8
20616,4
20843,0
21064,8
21277,4
21474,9
21647,0
13
19304,8
19326,8
19370,3
19434,9
19519,7
19623,3
19744,5
19881,6
20032,6
20194,4
20366,8
20546,3
20730,0
20914,9
21097,3
21273,1
21436,5
21577,8
14
19981,7
20000,6
20037,9
20093,2
20165,5
20253,7
20356,1
20471,2
20596,9
20730,0
20869,9
21012,8
21155,3
21293,2
21421,1
21529,0
15
20531,7
20547,2
20578,0
20623,3
20682,2
20753,6
20835,7
20926,8
21024,6
21125,7
21228,7
21328,8»
21420,4
21492,8
16
20948,3
20960,2
20933,7
21018,1
21062,4
21115,1
21174,6
21238,4
21304,0
21366,9
21422,5
17
21224,2
21232,1
21247,6
21269,7
21297,3
21328,6
21361,1
18
19
21355,5 21378,5
21358,9 21379,0
21365,2
21373,4
и
и
в
водород и его соединения Щ (г)
Приведенные в табл. 5.3 колебательно-вращательные уровни энергии HJ в состоянии
¦основаны на результатах теоретических расчетов, выполненных в работе Винда [3968] (для / ^ 8)
и при подготовке настоящего издания (для / > 8). Винд [3968] рассчитал электронную энергию
Hj для i?=0,05 @,05) 20 а. е. м., а затем с учетом адиабатической поправки по [1005] вычислил
энергии всех связанных состояний Щ для / ^ 8 [3968]. Результаты этого расчета хорошо
согласуются с данными, полученными позднее в работах [2049, 2287, 540]. В более ранних теоретических
работах [3302, 1338, 1005] были получены менее надежные данные. Поскольку ни в одной из работ
яе были определены энергии колебательно-вращательных уровней с / > 8, при подготовке
справочника был выполнен их расчет (см. [110, 111 ]) численным интегрированием уравнения Шредин-
гера по программе [51]. В расчете использовалась потенциальная функция, полученная в
работе [3967], с адиабатической поправкой [2287]. Результаты этого расчета для /=8 согласуются
с данными работ [3968, 540] в пределах 1 см.
Определение колебательно-вращательных уровней HJ или молекулярных постоянных этого
иона с хорошей точностью из спектральных исследований невозможно из-за отсутствия у Н?
связанных возбужденных состояний, способных комбинировать с состоянием Х22+. В работах [3206,
1909, 3598] молекулярные постоянные И.^ были оценены на основании закономерностей в
изменении постоянных Н2 в синглетных и триплетных состояниях ридберговских серий, сходящихся
•к состоянию Х22+ иона Щ. Исследования фотоэлектронного спектра Н2 [1072, 427] и изучение
сечения ионизации Н2 также позволили оценить некоторые постоянные Щ. Результаты этих
измерений согласуются между собой, но существенно менее полны и точны по сравнению с
теоретическими расчетами. Значения молекулярных постоянных, полученные в работе [540],
представляются наиболее надежными и приведены в табл. 5.2.
Погрешности вычисленных значений термодинамических функций при Т ^ 5000 К
обусловлены практически только неточностью принятых фундаментальных постоянных и не превышают
0,01 Дж-К^-моль. При более высоких температурах погрешности несколько возрастают из-за
приближенного учета квазисвязанных колебательно-вращательных уровней и могут достигать
0,03 Дж-К^-моль в значении Ф° B0 000 К). Пренебрежение в расчете возбужденными
электронными состояниями Щ с низкими энергиями диссоциации не влияет на точность рассчитанных
величин.
Таблицы термодинамических функций Н| ранее в литературе не публиковались. В работе
12969] приведен график зависимости In QmXi ^ от Т для Т ^ 25 000 К, рассчитанный по уровням
.•энергии, полученным в ВКБ-приближении с потенциальной функцией из работы [3968]. Данные,
^приведенные в работе [2969], находятся в качественном согласии с табл. 13.
Константа равновесия реакции Щ (г)+е (г)=2Н (г) вычислена с использованием значения
,АГ/Г° @)=1056,292 + 0,013 кДж-моль, которое соответствует принятому в справочнике значению
потенциала ионизации Н2, полученному в работах Герцберга и др. [1909, 1914] при изучении
ридберговских серий в спектре поглощения Н2 на приборе с высокой дисперсией:
/0(Н2)= 124417,2 + 0,4 см = 1488,360 + 0,005 кДж • моль.
Практически совпадающие значения получены при изучении границы схождения ридбергов-
<ских серий в работах Такезава [3599, 3598] A24 418,4+0,6 см) и в результате квантовомехани-
ческих расчетов [2049, 2125, 833] A24 417,3 см). В ряде справочных изданий для /0 (Н2) использо-
.валось менее точное значение, полученное в 1936 г. в работе [631] A24 429 + 13 см).
Принятому значению /0 (Н2) соответствует
Д7/Г (Н?, г, 0) = 1488,360 + 0,005 кДж • моль.
Н3 (г). Термодинамические свойства газообразного ионизированного триводорода в
стандартном состоянии в интервале температур от 298,15 до 20 000 К приведены в табл. 14.
Молекулярные постоянные, использованные для расчета термодинамических функций
Нз (г), приведены в табл. 5.4. Спектры и структура молекулы Щ экспериментально не
исследовались. На основании результатов квантовомеханических расчетов [705, 961, 1038, 1123, 2980,
.3363] и изучения спектров энергетических потерь при рассеянии атомов Ne на молекулах Щ
[3001] в справочнике принимается, что в основном состоянии ХгАх молекула HJ имеет струк-
¦ТУРУ равнестороннего треугольника (симметрия DZh) с г (Н—Н)=0,878+0,005 А и не имеет свя-
109
ОН (г)
Глава 5
Таблица 5.4.
Значения молекулярных постоянных, а также р; и о, принятые для расчета
термодинамических функций HJ, НО2, НО^, Н2О+, Н3О+
Молекула
щ
ног
н2о+
HSO+
Состояние
1^
%*А"
А3А'
&АХ
А~*АХ
Х^А
То
0
0
7029,764
0
0
9400
0
3350
3410
—
3490
3377
3309
3490
^2
с-м
2800 B)
1390
—
1370
1412
644
960
1095 —
— —
1050 —
3447 —
3511 —
3610 B) 1590 B)
г3 ¦ ей6
0,000538 4
0,912 2
—
1,08
0,007 1 8
0,008 7 8
0,028 7
о
6
1
1
1
2
2
3
Р.-
1
2
2
1
2
2
1
занных возбужденных состояний. Значения основных частот Щ, приведенные в таблице, были
рассчитаны в работах [961, 1038, 1123, 3363]; они хорошо согласуются с рекомендованными
авторами работы [3001 ] по результатам исследований рассеяния атомов Ne на Н*. Погрешности
принятых значений vlt v2 и IaIbIc составляют 200 и 150 см и 0,2 -Ю21 г3-см6 соответственно.
Термодинамические функции Щ (г) были вычислены по уравнениям A.3)—A.6), A.9), A.10)ю
A.122) —A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности табулированных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных (до 0,3, 0,5 и 0,7 Дж-К^-моль в значениях Ф° (Т) при 298,15,
3000 и 6000 К) и использованием приближенного метода расчета. Общие погрешности значений
Ф° (Т) могут достигать 0,3, 0,6, 1 и 2 Дж-К^-моль при 298,15, 3000, 6000 и 20 000 К.
Термодинамические функции Н* вычислялись в работе [2968 ] для температур до 7000 К в
приближении «жесткий ротатор — ангармонический осциллятор» на основании постоянных,
оцененных по потенциальной поверхности состояния ХхАг, рассчитанной в работе [1037]. Сравнение ре~
зультатов расчета [2968] с данными табл. 14 невозможно, так как значения термодинамических
функций в работе [2968] не приведены. Однако для частоты v2 в этой работе принято резко
завышенное значение 3601 см, что, по-видимому, должно обесценить применение более точной
методики расчета.
Константа равновесия реакции Hg (г)+е (г)=ЗН (г), приведенная в табл. 14, вычислена с
использованием значения АГН° @) = —455,898 + 10 кДж -моль, соответствующего принятой в
справочнике энтальпии образования Н*
Д/Г^Н*. г, 0)= 1104 + 10 кДж • моль.
Энтальпия образования HJ (г) принята на основании величины сродства молекулярного
водорода к протону:
Л0(Н2)=—4,4 + 0,1 эВ = — 424,5 + 9,6 кДж • моль,
полученной Бертом, Данном и др. [849а] на основании изучения кинетики реакций Н* D,4+
+0,1 эВ), Рошем, Саттоном и др. [3217а] в результате изучения константы равновесия обменных:
реакций XH++Y=YH++X Q>4,20 эВ) и Коттером и др. [1074а] на основании изучения порога
реакций Н2О++Н2-*Щ+ОН и D2O++D2=D++OD D,37±0,05 эВ). Близкие значения были
получены в квантовомеханических расчетах [1123, 2742а, 3291а]. В работах [256а, 272а, 273а,
406а, 964а] на основании изучения кинетики реакций Щ с Н2 и С2Н2, Щ с СН4 и Н2 с Н* Bs
или 2р) были найдены более низкие значения (от 2,65 до 3,7 эВ), по-видимому, из-за того, что в
соответствующих экспериментах ион Н+ образовывался в возбужденных колебательных состояниях
(см. [2395]).
ОН (г). Термодинамические свойства газообразного гидроксила в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 15.
Молекулярные постоянные гидроксила, принятые для расчета его термодинамических
функций, приведены в табл. 5.2. Молекула ОН имеет восемь валентных электронных состояний. Согласно.
110
Водород и его соединения ОН (г>
квантовомеханическим расчетам (см. [1368]), пять из них нестабильны. В спектрах наблюдались
переходы между тремя стабильными валентными состояниями (Х2П{, А2И +, 2?22 +), а также двумя
ридберговскими состояниями (С22 + и .D22~). В колебательно-вращательных [1895, 926, 916,
3865, 510, 2330, 664] и электронных спектрах [1247, 1187, 2147, 3608, 3609, 3614, 1463] ОН изучены
переходы, связанные с колебательными уровнями основного состояния вплоть до v=15 и выполнен
анализ вращательной структуры полос для v ^ 9. В системах полос А22—Х2П и i?22—т!22
[485, 2635, 1462, 1148, 874] была изучена вращательная структура полос, связанных с
колебательными уровнями состояния А22, с v ^ 9, и показано, что в этом состоянии утах=10. В работе-
[874] было также установлено, что в состоянии Б22 молекула ОН имеет только два колебательных
уровня с 1>=0 и 1. Наиболее точные значения колебательных постоянных ОН в состоянии Х2Л
с учетом экспериментальных данных для v ^ 9 получены Чеймберлином и Рослером [926], в
состоянии А2~2 — Барроу [485]. Вращательные постоянные ОН в состоянии Х2П определялись как
при исследованиях электронного и колебательно-вращательного спектров, так и при
исследованиях чисто вращательного спектра [2684, 1334, 1311, 3111, 3079]. Мур и Ричарде [2699] показали,
что ни один из наборов постоянных не позволяет точно описать всю систему
колебательно-вращательных уровней состояния Х2П и что значения постоянных существенно зависят от вида
уравнения. В табл. 5.2 приведены постоянные, полученные в работе Мидзусима [2684. ], так как при
подготовке материалов справочника было установлено, что система термов ОН, полученная в
работе [2699], описывается уравнением типа A.42) с этими постоянными лучше, чем с постоянными,,
найденными в других работах.
При расчете термодинамических функций ОН (г) значения In Qm и ее производных были
вычислены по уравнениям A.76)—A.78) непосредственным суммированием по
колебательно-вращательным уровням энергии состояний Х2П, А2Л, и J522. Энергии уровней рассчитывались по уравнениям
A.42) и A. 65) для состояния Х2П и A.34) и A.65) для состояний Л22 и 522 со значениями
коэффициентов Ykl и А, приведенными в табл. 5.5. Мультиплетность уровней в состояниях Л22, 2?22
и Л-удвоение уровней состояния Х2Н учитывались статистическим весом 2. В расчетах (?кол. вр.
н их производных для состояний Х2Н и .422 учитывались все уровни, ограниченные предельными
кривыми диссоциации этих состояний. В таблице приведены значения коэффициентов а( в
уравнениях, аппроксимирующих эти кривые, а также значения i^ и /цт, полученные по методикам,
изложенным в разделе 1.4. Значения Yk0 для состояния Х2И были вычислены на основании
значений Go (v) при v < 9 по данным [1247, 1895, 3865], &G (v+l/2) при 10 < v < 15 по
[1463] и Do (ОН)=35 420 см. Значения Ykl вычислены на основании значений В„
полученных в работах [1895, 3865, 2684], остальные Ykl — по данным, приведенным в
работе [2684]. Для состояния Л22 аналогичная обработка была проведена на основании постоянных:
Go (v), Bt, D, и Н„ полученных в работах [1247, 874, 2699]. Соответствующие значения Ykl
приведены в табл. 5.5. Поскольку в состоянии 522 существует только два колебательных уровня,,
в таблице для этого состояния приведены значения То, Тх, Во, Вг и Do по работе [874].
Погрешности вычисленных значений термодинамических функций ОН при Т ^ 6000 К не
превышают 0,02 Дж-К^-моль и обусловлены главным образом неточностью фундаментальных
постоянных. При более высоких температурах сказываются неточность определения числа и энергий
колебательно-вращательных уровней с большими значениями / для состояний Х2П и Л22,
приближенный учет квазисвязанных уровней и пренебрежение ридберговскими состояниями ОН. При
20 000 К общая погрешность в значениях Ф° (Т) и S° (T) не превышает 0,2 и 0,5 Дж -К -моль.
Термодинамические функции гидроксила вычислялись в ряде работ и справочных изданий.
Впервые расчет был сделан Джонстоном и Досоном [2146] методом непосредственного
суммирования для Т ^ 5000 К. Результаты этого расчета, а также основанных на нем таблиц [3256, 3255,
3196, 2029], несмотря на использование неточных значений молекулярных постоянных ОН,
согласуются с данными табл. 15 в пределах 0,08 Дж-К^-моль. В работах [1782, 2955],
справочниках [2095,2556,2496, 3946,337], а также [764] расчеты были выполнены приближенными методами^
что обусловливает их значительные расхождения с данными табл. 15 при высоких температурах.
Эти расхождения достигают при 6000 К 0,2, 0,6 и 1 Дж-К^-моль-1 в Ф° (Г), S° (T) и С°р (Г).
В работах [2095, 2496, 337, 764] некорректно учтены вырождение и мультиплетность состояния
Х2П. Соответствующая ошибка в Ф° B98,15 К) достигает 1,2—1,6 Дж-К^-моль, а &
Н° B98, 15 К)—Я°@) примерно 0,4 кДж -моль.
В 1-м издании справочника [87 ] расчет был выполнен для Т <^ 6000 К по
модифицированному методу Гордона и Барнес; расхождения результатов расчета и табл. 15 не превышают-
111
ОН (г),
Гяам 5
Таблица 5.5.
Значения коэффициентов в уравнениях, ончсымкяц ix уровяд эиергян (в см),
а также значения vm« lI ^нш принятые для расчета тс/шид.шам.пз.ммх функции
ОН, ОН+, ОН-
Коэффициенты
те. ю-* о
У,„ • 10-» 3,733 07
Ум • 10-» —0,851 409
Узо 0,591 785
У« • 10* —1,086 19
Уа • Ю3 —6,957 78
У,о • 10* 8,990 33
У,о • 10* —0,577 888
Ую • Ю5 0,117 825
У01 . 10-1 1,891 89
Уи -0,769 694
Уп . 10* 4,536 16
У и • 10s —10,259 7
У„ • 10' 9,521 46
У51 • 10s —3,257 91
У02 • 103 —1,93
У12 • 105 —4,83
Ум . 10s 5,3
У и • 10е 14,074
Уо, . 1013 —12,3
а„ = D, • 1С-* 3,727 0
а, 0,920114
02 • 10* 4,851 985
а3 • 10» 13,800 008
о max 16
Jllm 61
a A = —139,235. 6 X = 2,1344,
он
А^
3,268 45
3,177 99
—0,922 930
-1,918 32
32,789 6
—35,495 7
—
—
—
1,739 2
—0,881 971
4,534 63
—17,112 9
25,535 1
—17,244 7
—2,04
—
—
—
—
2,038 8
0,998 195
18,818 0
—312,297 615
10
47
li. = —0,1478.
6,972 07 0
1,100 0 3,123 06
—2,201 5 —0,862 128
— 1,798 72
— —2,7341
— —
— —
— —
— —
0,551 95 1,679 45
—0,867 —0,750 071
— 1,151 89
— —0,121 984
— —
— —
—0,929 —1,917 44
_ _
— —
— 12,394
_ _
— 4,259 52
— 0,550 812
— 1,502 92
— 0,223 888
1 28
24 75
«'А
1,680 0
3,000
—0,58
—
—
—
—
—
—
1,670
—0,464
—
—
—
—
-1,7
—
—
—
—
4,160 88
0,549 171
1,370 03
10,162 8
26
72
он+
АШ
2,844 17
2,133 65
—0,795 5
—
—
—
—
—
—
1,379 16
—0.8S-J 9
1,730
—
—
—
—2,249 5
—
—
10,4
—
1,411 75
0,6 «885
9,182 07
-51,512 6
12
44
2,904 60
3,148
-J.84
—
—
—
—
—
—
1,663 6
-0,66
—
—
—
—
-1,92
—
—
—
—
2,941 72
0,723 310
4,8>7 79
-3,231 94
18
57
с'П
4,500 0
1,800
-0,67
—
—
—
—
—
—
1,33
—3,74
—3,2
—
—
1,280 63
0,573 209
5.6J8 45
53,593 8
12
44
он-
ХЧ1+
0
3,800
-0,90
_
_
1,930
—3,68
2
_
_
_
_
4,015 61
0,8/9 253
3,234 013
26,418 393
20
62
0,03 Дж-К^-моль в значениях Ф° (Т). В предыдущем издании [88] расчет был выполнен
до 20 000 К ^непосредственным суммированием по уровням энергии с ограничением числа
вращательных уровней. Сходный метод был использован в работе [521 ] для расчета Ф° (Г) при Т ^
^ 10 000 К. До 6000 К результаты этих расчетов и данные табл. 15 согласуются в пределах
О,02 Дж-К^-моль за исключением значений S° (T) выше 5000 К, где расхождения при 6000 К
достигают 0,2 Дж-К^-моль. При более высоких температурах расхождения быстро растут и
при 10 000 и 20 000 К составляют 0,35 и 3 Дж-К^-моль в значениях Ф° (Г). Они обусловлены
учетом меньшего числа квазисвязанных уровней в методиках, использованных в работах [88,
521].
Константа равновесия реакции ОН (г)=О (г) -(-Н (г) вычислена с использованием
ДД° @) = Do (ОН) = 35 420 ± 15 см = 423,72 + 0,18 кДяс • моль,
Это значение было получено Карлоне и Далби [874] короткой экстраполяцией (на 273 см)
величин Д<?,+!/, состояния Л22. Несколько большее значение C5 450 + 100 см~х) было получено
раньше Барроу [485 ] аналогичным методом; разница в данных этих двух работ обусловлена
уточнением в работе [874] значений Д(?,+¦/,, в частности, для последнего наблюдаемого
колебательного уровня. Значение, найденное в работе [485], принималось в ряде справочных изданий в том
числе [88] и др. Ранее значение Do (ОН) определялось на основании изучения равновесия
образования гидроксила [699, 176, 154, 1, 1361, 207, 164], по порогу образования ОН при фотодиссоциации
Н2О и Н2О2 [3387, 1575, 3754, 3207] и далекой экстраполяцией колебательных уровней [514,
.3609, 1578, 2007]. Результаты всех этих измерений имели существенно меньшую точность, хотя
данные [164 и 1361] близки к значению, принятому в настоящем издании.
112
Водород и его соединения ОН+(г)
Принятому значению энергии диссоциации соответствует
, г, 0) = 39,097 + 0,21 кДж • моль.
ОН+ (г). Термодинамические свойства газообразного положительно заряженного иона гидро-
ксила в стандартном состоянии в интервале температур 298,15—20 000 К приведены
в табл. 16.
Молекулярные постоянные ОН+, принятые для расчета термодинамических функций,
приведены в табл. 5.2. Ион ОН+, согласно квантовомеханическому расчету, выполненному Лю и Верха-
геном [2448], имеет пять связанных валентных состояний (Х3Е~, a1 A, b1l1+, А3И и с1!!) и ряд от-
талкивательных квинтетных, триплетных и синглетных состояний. В спектрах ОН+ изучен только
переход А3П—Х32~ [3222, 2461, 3921, 2876, 3122, 3123, 3124, 2616, 2616а]. В работе Мерера
и др. [2616а] впервые удалось получить значения Дб*/з и AG"tl и определить непосредственно
из экспериментальных данных колебательные постоянные, а также уточнить значения
вращательных постоянных ОН+ в обоих комбинирующих состояниях. Значения постоянных ОН+ в этих
состояниях, найденные Мерером и др. [2616а], даны в табл. 5.2. Приведенные в табл. 5.2
постоянные ОН+ в состоянии Ь*2 + получены на основании значений То, АСп/„ Во, с^ и De, найденных
Мерером и др. [2616а] в результате анализа возмущений в состоянии А3И. Значения постоянных
Т и (вв иона ОН+ в состояниях а1 А и сШ получены Лю и Верхагеном [2448] в результате
неэмпирического квантовомеханического расчета; хорошее согласие рассчитанных в этой же работе величин
для состояния ASU с найденными при исследовании спектра ОН+, позволяет предполагать
удовлетворительную точность этого расчета также и для состояний а1 А ж сЧ1. Значения постоянных г,
(и В ) в этих состояниях приняты такими же, как в состояниях b1^,+ и А3П, поскольку
конфигурации электронной оболочки в состояниях а1 А и 6*2+, а также А3П и схЛ одинаковы (. . .За21тса и
. . .30I713 соответственно).
При расчете термодинамических функций ОН+(г) значения Qm, QBa и Qm вычислялись
по уравнениям A.76)—A.78) непосредственным суммированием по колебательно-вращательным
уровням пяти электронных состояний. Энергии вращательных уровней рассчитывались по
уравнениям A.40) для состояния XSH~ и по уравнениям A.34) для состояний a1 A, A3U, 6*2+ и ехП со
значениями постоянных Ykl, X и ц, приведенными в табл. 5.5. Л-удвоение уровней в состояниях ахД,
сШ и А3П и мультиплетность состояния А3И учитывались статистическими весами. В расчетах
#кол. вр и их производных учитывались все колебательно-вращательные состояния, ограниченные
предельной кривой диссоциации. В таблице приведены значения коэффициентов а{ в уравнениях,
аппроксимирующих эти кривые для каждого электронного состояния, а также значения yj^
и /urn- Для состояния Х31,~ коэффициенты Yk0 вычислены на основании значений Ad/,, AG»/,,
Do (ОН+) и величины Go C), полученной по постоянным OD+; коэффициенты Ykl — на основании
величин Во, Вх, Вг и условий A.57). Для остальных состояний они тождественны постоянным,
приведенным в табл. 5.2.
Основные погрешности в табулированных значениях термодинамических функций ОН+
обусловлены отсутствием экспериментальных данных о колебательно-вращательных уровнях энергии
состояния Х32~ с v > 2, а также неточностью порядка 800 см в принятом значении Т, (а1 А).
Общая погрешность в Ф° (Г) при 298,15, 3000, 6000 и 20 000 К составляет 0,02, 0,05, 0,1 и
0,5 Дж-К^-моль.
Термодинамические функции ОН+ (г) вычислялись ранее во 2-м издании справочника [88]
до 20 000 К, таблицах JANAF [2095] до 6000 К и работе Шнейдера [3343] (Г=1000-Н0 000 К).
В [2095] расчет сделан по приближенному методу без учета возбужденных состояний, а
мультиплетность состояния Х32~ учтена статистическим весом. Это предопределило значительные
ошибки в значениях S° (Т) и Ср° (Т) при высоких температурах (при 6000 К до 1,3 и
5,2 Дж -К -моль) и в значении Н° B98,15 К)— Я°@) @,019 кДж -моль). В расчете [88]
учитывалось только одно возбужденное состояние; при 6000 К расхождения составляют 0,2 и
1 Дж-К^-моль в значениях Ф° (Г) и <S° (T), при 20 000 К они достигают 3 и 4 Дж-К^-моль
соответственно. В работе [3343], несмотря на применение метода непосредственного
суммирования по уровням энергии, имеется ряд приближений, что существенно обесценивает результаты
этого расчета; при 10 000 К расхождения с данными табл. 16 достигают 5 и 7 Дж-К^-моль
в значениях Ф° (Т) и 5° Т).
113
ОН- (г) Глава 5
Константа равновесия реакции ОН+(г)+е (г)=О (г)+Н (г) вычислена на основании значения
Дг#°@) =—820,28+5,0 кДж-моль, которое соответствует принятому в справочнике потенциалу
ионизации молекулы ОН
/0(ОН) = 12,90 + 0,05эВ = 104 040 + 400 см~1=1244+5 кДж-моль.
Это значение потенциала ионизации является средним между полученными Дайблером
и др. [1242] по потенциалу появления иона ОН+ при фотоионизации молекулы Н2О A2,94+0,02 эВ)
и Берковицем и др. [591 ] по потенциалу появления ОН+ при фотоионизации HOF A2,88 + 0,05 эВ).
Более старые измерения, выполненные методом электронного удара [1503а, 2510а, 2433а] и
фотоионизации [2621а], привели к существенно менее точным величинам.
Принятому значению /0 (ОН) соответствуют
Do (ОН+)=41 055 + 400 см-1=491,127±5 кДж-моль
и
AfH°(OE.+, г, 0) = 1283,097 + 5 кДж • моль.
ОН" (г). Термодинамические свойства газообразного отрицательного иона гидроксила в
стандартном состоянии при температурах от 298,15 до 20 000 К приведены в табл. 17.
Молекулярные постоянные ОН", принятые для расчета термодинамических функций,
приведены в табл. 5.2. Прямые экспериментальные исследования и строгие теоретические расчеты
систематики электронных состояний и молекулярных постоянных ОН" отсутствуют. Молекула ОН"
изоэлектронна HF и, как последняя, кроме состояния ХХ2 + должна иметь еще три валентных
электронных состояния. Энергия нижнего возбужденного состояния ОН" (а3П) принята по оценке
[2617 ] на основании изучения фосфоресценции ОН" в кристаллах и растворах, а также
неопубликованных теоретических расчетов. Два других состояния А1!! и Й32, по-видимому, являются оттал-
кивательными. В работе [748] показано, что в молекулах ОН и ОН" равновесные межъядерные
расстояния должны быть близки. Значения постоянных ше (или ке) и ге (или Ве) оценивались в ряде
работ [2103, 859, 1549,2443], причем результаты расчетов находятся в удовлетворительном
согласии.
В настоящей работе принимаются значения сое=3800 + 100 см и 5е=19,3±0,4 см,
полученные Кейдом [859] в результате расчета потенциальной функции ОН" в состоянии Х12+ методом
Хартри—Фока. Остальные постоянные ОН", приведенные в табл. 5.2, вычислены с
использованием этих значений и Do (OH")=38 340 см.
При расчете термодинамических функций ОН" (г) значения Qm и ее производных
вычислялись по соотношениям A.90)—A.92) в предположении, что (!//?,•)(?«]. „р = A//>х) <?? вр. и ана"
логичные соотношения имеют место для Q<Jol. вр и Q^o\. вр. Значения Q^l вр и ее производных
были рассчитаны непосредственным суммированием по колебательно-вращательным уровням
энергии состояния Х12+ по уравнениям A.73)—A.75). Энергии уровней состояния Х12+
вычислялись по уравнению A.65) с значениями коэффициентов Ykl, приведенными в табл. 5.5. В расчете
учитывались все колебательно-вращательные уровни состояния XJ2 +, ограниченные его
предельной кривой диссоциации; в табл. 5.5 приведены коэффициенты а{ в уравнении, аппроксимирующем
эту кривую, а также значения vm&x и /цт. Основные погрешности вычисленных значений
термодинамических функций ОН" обусловлены неточностью принятых молекулярных постоянных.
Соответствующие погрешности в значениях Ф° (Т) при 298,15, 3000, 6000 и 20 000 К оцениваются
в 0,2, 0,5, 1,0 и 2 Дж-К^-моль.
Термодинамические функции ОН" вычислялись в предыдущем издании настоящего
справочника [88] и таблицах JANAF [2095]. Данные последней работы и табл. 17 согласуются в пределах
0,8 Дж-К^-моль; имеющиеся расхождения обусловлены некоторым различием принятых в
расчетах постоянных и методом расчета. В предыдущем издании справочника [88] расчет был выполнен
по менее точным значениям постоянных и в приближении «жесткий ротатор — гармонический
осциллятор»; соответствующие значения Ф° (Т) занижены по сравнению с табл. 17 на 1 —
2,5 Дж-К^-моль.
Константа равновесия реакции ОН" (г) = О (г)+Н (г)+е (г) вычислена на основании значения
Д Н° @)=599,85 + 0,26 кДж-моль, соответствующего принятой в данной работе величине
114
Водород и его соединения НО, (г)
сродства гидроксила к электрону
А0(ОЯ) = —14 723 + 15 см = —176,13 + 0,18 кДж • моль.
Эта величина была получена Хотопом и др. [2016] по границе фотоионизации ОН" излучением
лазера с перестраивающейся длиной волны. Практически совпадающее значение A4 754+100 см)
было найдено авторами работы [919] по энергии электронов, образующихся при фотоионизации
ОН" излучением Аг+-лазера D880 А). Близкие, но менее точные значения были получены Бран-
скомом и др. [748] по границе фотоионизации ОН" при использовании монохроматического
излучения A4 760 + 300 см), а также магнетронным методом [2939] A4 360—14 680 см). Принятому
значению Ао (ОН) соответствуют
Д/Г(ОН", г, 0) = —137,033 + 0,27 кДж • моль
и
Do (ОН") = 458,63 + 0,66 кДж • моль == 38 340 + 55 см.
НО2 (г). Термодинамические свойства газообразного диоксида водорода в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 18.
Молекулярные постоянные, использованные для расчета термодинамических функций НО2,
приведены в табл. 5.4. Теоретические расчеты [3872, 724, 2445, 1675] и анализ электронного
спектра [1536, 2050, 1675, 544, 543] показали, что молекула НО2 имеет основное состояние Х2А"
и первое возбужденное состояние Л2А'. В этих электронных состояниях НО2 имеет
несимметричную угловую структуру. В электронном спектре газообразного НО2 [3723, 2947, 1966] известна
также область сильного непрерывного поглощения A860—2600 А) с максимумом при 2050 А
D8 800 см) [1966]. Приведенное в табл. 5.4 произведение главных моментов инерции в состоянии
Х2А" вычислено по значениям вращательных постоянных Ао=20,358+0,003 см, Бо=1,1179 +
+0,002 см, С0=1,0567 + 0,002см~1, найденным Хаугеном и др. [2017] в результате анализа
вращательных переходов в спектре лазерного магнитного резонанса, полученного Рэдфордом и др. [3112].
Эти значения вращательных постоянных хорошо согласуются с результатами анализа
микроволнового спектра НО2 [548,3285] и вращательной структуры полосы 000—000 системы А2А' -> Х2А"
в спектре испускания НО2 [1536]. Им соответствуют межатомные расстояния г (О—Н)=0,977 +
+0,015 А, г (О—О) = 1,335±0,005 А и угол ^ Н—О—0=104,1+2°. Значения основных частот НО2
в состоянии Х2А", приведенные в табл. 5.4, определены Паукертом и Джонстоном [2947]
в результате исследования инфракрасного спектра газообразного НО2 [2152]. Они согласуются
с ИК-спектром НО2 в матрицах инертных газов [2662,2885, 2084, 3464]. Погрешности приведенных
в табл. 5.4 основных частот НО2 не превышают 3 см. Приведенная в табл. 5.4 энергия
состояния 2А' найдена Фридманом и Джонсом [1536].
Термодинамические функции НО2, приведенные в табл. 18, вычислены по уравнениям A. 3)—
A. 6), A. 9), A.10), A.122)—A.124), A.128) и A.130) в приближении «гармонический осциллятор —
жесткий ротатор». Составляющие электронного состояния А2А' вычислялись по соотношениям
A.168)—A.170). Погрешности вычисленных значений термодинамических функций НО2
обусловлены в основном применением приближенного метода расчета и отсутствием данных о высоких
колебательно-вращательных уровнях энергии НО2 в основном электронном состоянии. Они
составляют 0,02, 0,8 и 1,5 Дж-К^-моль-1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций НО2 (г) вычислялись в предыдущем издании
настоящего справочника [88], справочнике JANAF [2095], а также Миллиганом и Джекокс в
работе [2662] (до 3000 К). Все расчеты проводились в приближении «гармонический осциллятор —
жесткий ротатор» без учета возбужденного электронного состояния 2А', по приближенным
значениям молекулярных постоянных НО2 в основном электронном состоянии. В последней работе
не была учтена мультиплетность состояния Х2А", и расхождения с данными табл. 18 достигают
7—8 Дж-К^-моль при 3000 К. В справочнике [88] было принято завышенное значение длины
связи О—О, расхождения между данными табл. 18 и этого расчета проходят через максимум
при 7=2000-^-3000 К и достигают 3 Дж-К^-моль в значениях Ф° (Т) и S° (T).
Термодинамические функции, приведенные в [2095], вычислены с использованием тех же значений постоянных,
что и в работе [2662], расхождения с данными табл. 18 растут с температурой и при 6000 К
достигают 3 и 5 Дж-К^-моль в значениях Ф° (Г) и S0 (Т).
115
HOjfr), Н2О(к, ж) Глава 5
Константа равновесия реакции НО2(г)==2О (г)-)-Н (г) вычислена с использованием значения
АГН° @)=704,6 + 4 кДж-моль, основанного на принятой в справочнике энтальпии образования
НО2
Д^0 (НО2, г, 0) = 5 ± 4 кДж • моль.
Это значение было найдено Кочубеем и Мойным [134, 133] при исследовании кинетики
газофазного окисления HG1, НВг и HI, которое показало, что стадией, определяющей скорости
исследованных процессов в целом, являются реакции типа НХ (г)+О2 (г)=НО2 (г)+Х (г), где
Х=С1, Вг, I. Кочубей и Моин [134] нашли энергии активации этих реакций, которые принимаются
равными их энтальпиям: 215 + 2 кДж-моль (в случае Х=С1 при 7т=1200 К), 152,3 +
+ 1,2 кДж-моль (в случае Х=Вг при Г=800 К), 84,1 + 1,2 кДж-моль (в случае Х = 1 при
7'=400 К). Приведенным значениям энтальпии этих реакций соответствует среднее
значение Do (H—О2)=211+4 кДж-моль и принятая в справочнике энтальпия образования
НОг (г).
Исследования кинетики газофазного окисления бромида водорода в работах [3249, 402 ]
согласуются с данными Кочубея и Моина. Робертсон [3212] и Фонер и Хадсон [1502, 1503, 1506]
определили в масс-спектрометрических исследованиях потенциал появления иона НО2 из Н2Оа,
а также потенциал ионизации НО2. Наиболее точные значения этих величин были определены
Фонером и Хадсоном в работе [1506], они приводят к значению D0(H—О2) ^ 192 + 6 кДж-моль.
Эта величина может быть несколько заниженной, так как при ее вычислении не учитывалась
кинетическая энергия и энергия возбуждения продуктов диссоциативной ионизации Н2О2 под действием
электронного удара.
г (г). Термодинамические свойства газообразного отрицательного иона диоксида водорода
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 19.
Молекулярные постоянные, использованные при вычислении термодинамических функций
НО2 (г), приведены в табл. 5.4. Сведения об экспериментальных исследованиях спектров и
структуры НОГ в литературе отсутствуют. В настоящем издании молекулярные постоянные НО^
в основном электронном состоянии Х1А1 приняты как средние между значениями соответствующих
постоянных молекул HOF и НО2, поскольку молекулярные постоянные ОН~ согласно [859, 748]
имеют промежуточные значения между постоянными молекул HF и ОН. Приведенное в табл. 5.4
произведение главных моментов инерции соответствует значениям г(О—Н)=1,00+0,03 А;
г (О-О) = 1,37 + 0,07А; ^Н-О-О = 102 + 5°.
Термодинамические функции НО2 (г), приведенные в табл. 19, вычислены по уравнениям
A.3)—A.6), A.9), A.10), A.122)—A.124), A.128), A.130) в приближении «гармонический
осциллятор — жесткий ротатор». Погрешности табулированных величин обусловлены неточностями
принятых значений молекулярных постоянных и приближенным методом расчета. Для Ф° (Т) при
298,15,3000 и 6000 К они оцениваются в 1—2 Дж-К^-моль". Ранее термодинамические
функции HOj (г) не вычислялись.
Константа равновесия реакции НО2 (г)=2О(г)+Н (г)+е (г) рассчитана на основании
значения АГН° @)=832,505+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования НО,
г, 298,15 К) = —132 + 40 кДж . моль'1.
Эта величина была рассчитана с использованием значения сродства НО2 к электрону Ао (НО2,
298,15 К) =—134+40 кДж-моль~х, которое было получено Эвансом и Юри [1425, 1424] в
результате оценки суммы сродства к электрону ОН и энтальпии гидратации ОН~ (г) (—617,6 кДж -моль-1)
и аналогичной суммы для НО2 и НО2 (г) (—569 кДж -моль). Эти оценки были сделаны на
основании термохимических циклов, включающих диссоциацию и ионизацию Н2О (г) и Н2О2 (г), а также
корреляции максимумов в спектрах поглощения растворов комплексов Fe+3X~ (где Х~=С1~,
Вг~, ОН~ и НО2) со значениями сумм Ао (Х)+Д801#° (Х~, г).
116
Водород и его соединения
Н2О(к, ж)
Таблица 5.6. Принятые значения термодинамических величин для Н20 и Н202 в кристаллическом
и жидком состояниях
Вещество
Состояние
11° B98,15 К)—
— 11° @)
кДж-
•МОЛЬ
S° B98,15 К)
С° B98,15 К)
Дж • К • моль
Коэффициенты в
уравнении для С°р (Т)*
а
6-Ю3
с • Ю-5
Интервал
температуры
К
т
к
АтН
кДж-
• МОЛЬ
Н2О к1, гекс. — — — — — — — 273,15 6,010
ж 13,293 69,950 75,30 612,73 -4724,535 24,9783» 273,15-600 — —
Н2О2 к1, тетр. — — — _ _ _ _ 272,74 12,500
ж 22,949 109,602 89,33 89,33 — — 272,74—600 — —
Примечание. 'Cl (T)=a+bT—cT-2+dT2+eT3+fT* (Дж-К-^моль); б<М0в=16 675,644; е-109=—27 011,653;
/.1012=16 811,312°
Н2О (к, ж). Термодинамические свойства твердой и жидкой воды в стандартном состоянии
в интервале 5—600 К приведены в табл. Д.З. Значения постоянных, использованных в расчетах
термодинамических функций, приведены в табл. 5.6.
В интервале температур от 0 до тройной точки B73,16 К) термодинамические функции
вычислены для гексагональной модификации льда (структурный тип [3-тридимита), которая является
стабильной при обычном давлении г. Выбор данных по теплоемкости льда при низких
температурах связан с выбором значений стандартной энтропии жидкой воды и остаточной энтропии льда.
Весьма точное значение S° (H2O, ж, 298,15 К)=69,950 + 0,080 Дж-К^-моль вычислено из
энтропии газообразной воды при 298,15 К A88,724 + 0,040 Дж-К^-моль) и энтропии испарения воды
при 298,15 К в идеальный газ при 1 атм A18,784+0,040 Дж-К^-моль). Приведенное значение
5° (Н2О, ж, 298,15 К) рекомендовано КОДАТА—MCHG [1000] в качестве ключевой величины
в термохимии и принято в настоящем издании. Остаточная энтропия льда <S°@)=3,408 +
+0,001 Дж -К-моль принята по расчетам Нагла [2787] 2. Расчет стандартной энтропии воды
по результатам наиболее надежных измерений теплоемкости льда, проведенных Джиоком и
Стаутом [1620] A6—268 К) и Флюбахером и др. [1500] B—27 К), а также с использованием значений
остаточной энтропии льда, теплоты плавления и теплоемкости жидкой воды (при 273,16—298,15 К)
приводит к значению S° (Н2О, ж, 298,15 К) = 70,074 + 0,210 Дж-К^-моль-1, которое на 0,2% выше
рекомендованного КОДАТА [1000]. Поэтому данные, приведенные в работах [1620, 1500],
были скорректированы при подготовке справочника (уменьшены в пределах точности
экспериментальных данных работ [1620, 1500] в среднем на 0,1%), и на основании этих
исправленных данных были вычислены значения термодинамических функций льда, приведенные
в табл. Д.З3.
Принятые в настоящем издании данные по теплоемкости твердого льда весьма близки (в
пределах 0,5%) к полученным в 1974 г. Хайда и Матсуо [1800]. Данные других авторов [2832] (83—
266 К), [3059] B2-87 К), [3422] A0-273 К), [1360] E-10 К), [3570] E6-213 К), [628]
(90—140 К) и [ЗОЮ] (80—230 К) менее надежны и не учитывались при расчете термодинамических
функций.
1 Существует по меньшей мере еще шесть кристаллических модификаций льда, стабильных при высоких давлениях
(см. [258]), а также аморфный лед [3570], который при Т > 130 К необратимо переходит в кубическую
модификацию льда; последняя при Т > 160 К также необратимо переходит в обычный лед (к1, гекс).
2 Согласно Полингу [2975], остаточная энтропия льда равна S° @)=Д1п 1,5=3,372 Дж -К -моль. Позднее эта
величина уточнялась в работах [1258, 3566].
• Отметим, что полученное таким образом значение Н° B98,15 К) —Н° @) = 13,273 кДж-моль на 0,020 кДжХ
Хмоль меньше значения, рекомендованного в качестве «ключевого» [1000],что связано, с некоторой несогласо"
ванностыо выбора в работе [1000] значений S0 B98,15 К) и Н° B98,15 К) — Н° @).
117
Н2О(г) Глава 5
Температура плавления льда при атмосферном давлении, согласно [2787], равна 273,15 К, что
ниже температуры тройной точки воды @,01° С=273,16К) на 0,01° С1. Для энтальпии плавления
льда принято значение Дт/7=6,010 + 0,004 кДж-моль, рекомендованное Бюро стандартов США
[3256, 1620, 2919] на основании рассмотрения 20 работ по экспериментальному определению
теплоты плавления.
Многочисленные и весьма точные данные по теплоемкости и энтальпии жидкой воды были
критически проанализированы Международным комитетом по свойствам воды и водяного пара,
который в «Международных скелетных таблицах» (см. [64]) рекомендовал значения энтальпии жидкой
воды для температур от 0° С до 370° С. Точность этих данных по оценке авторов [64] составляет
около 0,05% до 250° С, 0,1% при 300° С и 0,2% при 350° С. При подготовке справочника эти
данные были аппроксимированы методом Шомейта (см. раздел 2.3) с использованием значения
С° B73,15 К)=75,72 Дж -К -моль [1660] и 36 значений энтальпии Н{Т)—Н B73,15 К) (в интервале
283,15—633,15 К через 10 К). Полученное уравнение (см. табл. 5.6) аппроксимирует эти данные
с точностью 0,1% . Оно использовано для расчета термодинамических функций жидкой воды
в интервале 298,15—600 К.
Погрешность вычисленных значений Ф° (Т) для жидкой воды составляет 0,08 и 0,4 Дж -К моль
при 298,15 и 600 К.
Для энтальпии образования Н2О (ж) в настоящем справочнике в соответствии с рекомендацией
КОДАТА—MGHG [1000] принято значение
AfH°(Hfi, ж, 298,15 К) = —285,830 + 0,042 кДж • моль.
Эта рекомендация основывается на результатах измерений, выполненных в работах Россини
[3250, 3253] и Кинг и Армстронга [2246].
Н2О (г). Термодинамические свойства газообразной воды в стандартном состоянии в интервале
температур 100—20 000 К приведены в табл. 20.
Молекулярные постоянные, использованные при расчете термодинамических функций Н2О,
приведены в табл. 5.7. Значения этих постоянных были выбраны при подготовке второго издания
Таблица 5.7. Значения молекулярных постоянных (в см) в состоянии Я1 А, принятые для расчета
термодинамических функций Н2О (p_j; = l, в = 2)
Vi 3656,65
v2 1594,78
ч3 3755,79
zn —45,18
хгг -17,04
x33 —44,62
Примечание.
*и
х13
г/m
J/222
г/ззз
—15,14
—19,99
—165,48
0,47
—0,60
—0,45
г/иг
г/122
г/223
г/гзз
#113
г/123
Постоянные в уравнениж E
—0,10
—0,10
1,55
—0,81
0,68
—1,72
.4): Pl =
#133
1
¦^000
4
1,17
76,02
27,87
0,89
—2,68
1,26
= 2,24- Ю-5 К,
аА 0,16
оА 0,44
аЛ 0,03
аА —0,29
аА -0,04
р2 = 0,36 • Ю-8
„А
222
•^000
Сооо
к-2.
0,04
14,52
0,20
—0,16
0,11
9,28
аС 0,18
аС 0,14
аС 0,12
настоящего справочника [88] в работах Хачкурузова и др. [277, 283, 286] на основании
результатов исследований вращательной структуры полос молекулы Н2О, выполненных в работах [2598,
522, 1540, 95, 94, 2473, 3048, 3044, 3151, 562, 2857, 2858, 1093, 2690, 566, 2830, 3047, 576, 571, 1156,
568, 577, 575] и вращательного спектра Н2О [4005, 480, 3141, 1592], а также на основании
рассмотрения результатов определений молекулярных постоянных Н2О в работах [2598, 701, 1167,
2858, 1544, 3996, 575]. В последующие годы вращательная структура полос Н2О исследовалась
в работах [3187, 567, 2556, 2648, 1571, 570, 1521, 3100, 1494, 1492, 867, 3954, 3098, 3049, 3156,
1520, 868,1493, 3710, 1493а, 866а], а вращательный спектр Н2О — в работах [325, 324, 1583,3761,
1792, 666, 3157, 3198,2271, 2423, 1807, 1806,1313,1210,1209, 2323, 3975,3526,2036]. Однако
полученные в этих исследованиях уточнения энергии отдельных колебательно-вращательных уровней и
отдельных молекулярных постоянных Н2О не существенны для расчета термодинамических функций.
1 Согласно данным Алиева [15], эта разность составляет 0,0098±0,0002" С.
118
Водород и его соединения Н2О(г}
Поэтому при подготовке настоящего издания были использованы результаты расчетов
термодинамических функций Н2О (г) для второго издания после внесения следующих корректив: 1) были
исключены составляющие возбужденных электронных состояний молекулы Н2О, поскольку все
связанные возбужденные электронные состояния молекулы Н2О являются ридберговскими
состояниями [1671, 3973, 966], 2) результаты расчетов приведены в соответствие с принятыми в настоящем
издании значениями фундаментальных постоянных и атомных масс. Кроме того, по тем же
постоянным были вычислены значения термодинамических функций для Г=10(Н-300 К, а также
значения теплоемкости для всего интервала температур.
Колебательно-вращательная составляющая статической суммы Н2О вычислена
непосредственным суммированием по уровням колебательной энергии
vtm Р2«г Нт
222 Q°*'"'Vs) ехр[-тИ«К vv v,)], E.3)
е.=0 с,=0 г,=0
где Go (vu v2, v3) — уравнение, аппроксимирующее уровни колебательной энергии с учетом
резонанса Дарлинга—Деннисона (для первых ста уровней); vlm, v2m, v3m — предельные значения
соответствующих колебательных квантовых чисел, равные: vlm= 12, y2m = 22, v3m = 13, vx -\-v3^. 12,
в соответствии с условиями G0(vv 0, 0), G0@, 0, v3), G<>(Vi, 0, t>3)^D0(HO—H) и
\dGu(b\, v2, v3)ldv2]t „ =0. Вращательная статистическая сумма Q(^> "*• "«) для каждого
колебательного состояния вычислялась по соотношению
<?&••"'^е*. р а+pi?1+р2п E.4)
где <?ж р — статистическая сумма молекул типа асимметричного волчка в приближении жесткого
ротатора с поправочным множителем Стриппа—Кирквуда и plt p2 — постоянные, учитывающие
центробежное растяжение многоатомной молекулы (см. раздел 1.5).
Погрешности вычисленных термодинамических функций Н2О (г) при Т ^ 3000 К обусловлены
главным образом неточностью фундаментальных постоянных и не превышают 0,02—
—0,03 Дж-К -моль в значениях Ф°(Т) и S°(T). При более высоких температурах становятся
существенными погрешности из-за отсутствия экспериментальных данных о высоких
'колебательно-вращательных уровнях энергии молекулы Н2О и обусловленного этим применения
приближенной методики расчета значений (?вр и ее производных, а также приближенной методики
ограничения суммирования по v(. Выше 10 000 К могут стать заметными погрешности из-за
пренебрежения ридберговскими электронными состояниями Н2О. Погрешности вычисленных значений Ф° (Г)
при 6000 и 20 000 К достигают 0,15 и 2 Дж-К^-моль.
Термодинамические функции Н2О (г) вычислялись в десятках работ и справочников [1693,
1615, 1689, 1690, 1691, 1692, 3852, 2484, 3375, 3717, 3521, 2421, 1544, 1472, 1473, 1666, 1343,
2555, 60, 88, 87, 2029, 2095, 2556, 446,1945 ]. Большинство этих расчетов имеют только исторический
интерес. В работах [1690, 1691 ] Гордон вычислил термодинамические функции Н2О (г) с учетом
ангармоничности колебаний и колебательно-вращательного взаимодействия для Г=298,15-|-
-f-ЗООО К. Позднее эти данные были дополнены поправками, учитывающими центробежное
растяжение [1692], а также изменение фундаментальных постоянных [3852], и экстраполированы до
4000 К [2484], 5000 К [3375] и 6000 К [2094а]. Расхождения данных работы [3375] и табл. 20
увеличиваются с температурой и достигают 1 и 3,5 Дж-К^-моль в значениях Ф°(Г) и S°(T) при
5000 К. Фридман и Хар [1544] рассчитали термодинамические функции Н2О (г) для Т=50—
5000 К по методу, аналогичному изложенному на с. 57—58. Эти данные включены в справочники
[1945, 2095] (в последнем досчитаны до 6000 К). Сходные методики и близкие значения постоянных
были использованы Мак-Брайдом и Гордоном [2555, 2556], Вукаловичем и Артымом [60], Дьюран-
дом и Брандмайром [1343], Бером и др. [446] для расчетов термодинамических функций Н2О (г)
до 6000 К (в [1343] до 7000 К). Данные всех этих авторов и табл. 20 при Т ^ 2000 К практически
совпадают, при более высоких температурах они ниже приведенных в табл. 20; имеющиеся
расхождения увеличиваются с ростом температуры и достигают величин от 0,6 Дж-К^-моль [2556]
до 1 Дж-К^-моль-1 [2095] в значениях Ф°F000 К), а в значениях ?°F000 К) — от 1,5 [2556]
до 2,6 Дж-К^-моль-1 [2095].
Наилучшее согласие с данными табл. 20 имеет место для термодинамических функций Н2О (г),
приведенных в 1-м издании настоящего справочника [87 ], где они были рассчитаны по модифициро-
119
ванному методу Гордона и Варне (расхождения не превышают 0,12 и 0,2 Дж-К^-
чениях Ф°(Т) и S°(T) соответственно). Расхождения с предыдущим изданием [88] объясняются
практически только тем, что в настоящем издании исключены составляющие ридберговских
электронных состояний Н2О; они становятся существенными в значениях Ф°(Т) и S° (T) при
температурах выше 6000 К и достигают 7 и 25 Дж-К^-моль в значениях этих функций при 20 000 К.
В ряде работ термодинамические функции Н2О (г) определялись на основании
калориметрических измерений [1620, 2571 ] и измерений давления взрыва смесей Н2+О2 в сферической бомбе
[3012, 3013, 3014] (см. также [161, 162]). Точность результатов калориметрических определений
ограничена наличием у Н2О (к) остаточной энтропии, в связи с чем значение S° (H2O, ж, 298,15 К)
принимается на основании величины S° (H2O, г, 298,15 К), полученной в статистических
расчетах; точность определения термодинамических функций газов из данных, полученных методом
взрыва, низка из-за ряда ограничений методики (см. [161]).
Константа равновесия реакции Н2О (г) = О (г)+2Н (г) вычислена на основании значения
Дг#°@)=917,764^0,11 кДж-моль, которое соответствует энтальпии образования Н2О (г)
Д/Г0 (НаО, г, 298,15 К) = —241,814 + 0,042 кДж • мать,
принятой в справочнике в соответствии с рекомендацией КОДАТА—МСНС [1000]. Это значение
энтальпии образования Н2О (г) получено из значения &fH° (H2O, ж, 298,15 К) (см. выше) и
энтальпии испарения воды
Д„Я°(Н2О, ж, 298,15К) = 44,016 ±0,010 кДж • моль,
отвечающей результатам измерений давлений пара в работе Кинана и др. [2210].
Н2О+ (г). Термодинамические свойства газообразного положительно заряженного иона оксида
диводорода в стандартном состоянии при температурах от 298,15 до 20 000 К приведены в табл. 21.
Молекулярные постоянные Н2О +в состояниях Х2В1 и А2А1, принятые для расчета
термодинамических функций, приведены в табл. 5.4. Теоретический расчет [1836] и исследование электронного
спектра [2410] показали, что ион Н2О+ имеет основное состояние Х2ВХ и структуру,
соответствующую точечной группе C2t. Анализ электронного спектра позволил определить вращательные
постоянные Н2О+в этом состоянии, которым соответствует значение IaIbIc, приведенное в табл. 5.4,
и параметры ^К—О—Н = 110,5° и г (О—И)=0,999 А. Близкие значения были получены в [1836].
Значение v2 было определено в работе [2410]. Значения частот vx и v3, приведенные в табл. 5.4,
были получены при подготовке настоящего издания для /г=6,5-105 дин-см на основании оценки
по правилу Беджера. Энергия состояния Л2АХ принята по результатам приближенной
экстраполяции значений частот переходов на уровни 0у0 этого состояния, полученных в работе [2410]. Она
удовлетворительно согласуется с величиной, найденной в работе [1836] (около 7950 см).
Структурные параметры Н2О+ в состоянии А2А1 приняты по расчету [1836] с округлением величины
г (О—Н). Сведения о более высоких возбужденных состояниях Н2О+ в литературе отсутствуют.
Термодинамические функции НаО+ (г) были вычислены по уравнениям A.3)—A.6), A.9),
A.10), A.122)—A.124), A.128) и A.130) в приближении-«жесткий ротатор — гармонический
осциллятор». Составляющие состояния А2А1 вычислялись по соотношениям A.71)—A.73); мультиплет-
ность уровней обоих состояний учитывалась статистическим весом 2. Основные погрешности
вычисленных значений термодинамических функций Н2О+ при Т ^ 3000 К обусловлены неточностью
принятых в расчете значений колебательных постоянных Н2О+ в состоянии ХгВг (они составляют
0,1, 0,2, 0,3 и 2 Дж-К^-моль-1 в значениях Ф^при 298,15, 3000, 6000 и 20 000 К). При высоких
температурах начинает сказываться отсутствие достоверных данных об энергии состояния А2Аи
проведение расчета в приближении «жесткий ротатор — гармонический осциллятор» и отсутствие
сведений о других возбужденных электронных состояниях. Общие погрешности в Ф° (Т)
составляют 0,1, 0,5, 1 и 5 Дж-К^-моль-1 при 298,15, 3000, 6000 и 20 000 К.
Таблица термодинамических функций Н2О+ ранее в литературе не публиковалась.
Константа равновесия реакции Н2О+ (г)+е (г) = О (г)+2Н (г) вычислена на основании
значения АгН°@) =—299,449 + 0,51 кДж -моль. Эта величина соответствует энтальпии образования
Н2О +
ДД°(Н2О+, г, 0) = 978,3+ 0,5 кДж • моль,
120
Водород и его соединения Н2О2(к, ж), Н2О2(г)
которая была принята по результатам исследований потенциала ионизации Н2О при изучении
ридберговских серий Н2О в работе Прайса [3086] A2,62 + 0,02 эВ), фотоионизационных измерений
Брема [759] A2,614±0,005 эВ) и изучения фотоэлектронного спектра НаО [824] A2,616 + 0,005эВ).
Ей соответствует значение
/0(Н2О)= 12,615 + 0,005 эВ= 1217,2 + 0,5 кДж • моль.
Несколько более низкие значения были получены при фотоионизационных измерениях Дайб-
лера и др. [1242] A2,594 + 0,012 эВ) и Николсона [2851 ] A2,597 + 0,010 эВ). Исследования,
выполненные методом электронного удара с применением моноэнергетических электронов [1551, 2346,
3434, 2526], дали близкие, но существенно менее точные значения.
Н2О2 (к, ж). Термодинамические свойства кристаллической и жидкой перекиси водорода
в стандартном состоянии при температурах 5—600 К приведены в табл. Д.4. Значения
постоянных, использованных в расчетах термодинамических функций, приведены в табл. 5.6. В интервале
от 0 до тройной точки B72,74 К) термодинамические функции вычислены для тетрагональной
модификации — единственной известной модификации твердой Н2О2 г. При вычислении
термодинамических функций были использованы данные Жигера и др. [1632] по теплоемкости кристаллической
Н2О2 в интервале 12—268 К. В этой работе исследовался образец, содержащий 99,97 мол. % Н2О2;
точность измерений теплоемкости составляла 0,1—0,2% при температурах выше 30 К, заметно
снижаясь при Т < 30 К. Экстраполяция ниже 12 К приводит к S° A2 К)=0,09 Дж-К^-моль.
В интервале 250—272 К авторы [1632] наблюдали аномальное возрастание теплоемкости,
связанное с примесью 0,032 мол. % Н2О; эта аномалия не учитывалась при расчете термодинамических
функций Н2О2 (к). Значения температуры плавления Н2О2 B72,74+0,03 К) и энтальпии
плавления A2,50+0,01 кДж-моль) приняты по данным работы [1632]; результаты других измерений
температуры плавления [1501, 2485, 2487, 912] менее точны.
Теплоемкость жидкой Н2О2 принята постоянной и равной 89,33+0,90 Дж -К -моль по [1632 ]
(со ссылкой на неопубликованную работу Моризе [2711]). С этой величиной хорошо согласуются
полученные позднее [1627] значения средней теплоемкости Н2О2 С° =89,16 B98—318 К) и С° =
=89,45 Дж-К^-моль-1 B98—333КJ. Точность принятых значений 5°B98,15 К) и #°B98,15 К)—
— Я°@) (см. табл. 5.6) составляет 0,40 Дж-К^-моль и 0,08 кДж-моль. Погрешности
вычисленных значений Ф°(Г) составляют при 298,15, 400 и 600 К соответственно 0,4, 0,8 и
2 Дж-К^-моль-1.
В литературе отсутствуют таблицы термодинамических функций Н2О2 (ж), за исключением
справочника Келли [2220], где приводятся значения энтальпии и энтропии до 450 К.
Существенное расхождение этих данных с данными табл. Д. 4 объясняется тем, что Келли для теплоемкости
Н2О2 (ж) использовал данные [3420].
Для энтальпии образования Н2О2 (ж) в справочнике принято значение
Д//°(Н2О2, ж, 298,15 К) = —187,78 + 0,08 кДж • моль'1,
полученное Жигером [1633] в результате калориметрических измерений энтальпии разложения
водных растворов перекиси водорода (содержание Н2О2 от 24,6 до 96,6%). Измерения энтальпии
разложения Н2О2 (ж) в работах [2542, 3259, 1509, 1501а] существенно менее точны и согласуются
с данными [1633] в пределах их погрешностей.]
Н2О2 (г). Термодинамические свойства газообразной перекиси водорода в стандартном
состоянии при температурах 100—6000 К приведены в табл. 22.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 5.8. Молекула Н2О2 в основном электронном состоянии ХгА имеет равновесную гош-
конфигурацию (точечная группа симметрии С2), которая характеризуется, согласно [290], следую-
1 Сведения [1585, 175] о полиморфном превращении кристаллической Н2О2 при —110°С не были подтверждены
позднее [168]. О причинах небольшой аномалии теплоемкости Н2О2 при —110°С см. [1974].
2 Келли [2220] рекомендовал для Г=298+400 Щуравнение С (Н2О2, ж)=53,60+75,31 -Ю Т (Дж-К^-моль),
основанное на расчетных данных [3420]. Это уравнение приводит к неправдоподобно крутому росту теплоемкости
с температурой (от 85,6 до 106,3 Дж -К -моль). По-видимому, это обусловлено использованием в работе [3420 ]
недостаточно надежных данных по давлению пара перекиси водорода [3317, 2486].
121
Н2О2(г) Глава 5
Таблица 5.8. Значения молекулярных постоянных (в см) в состоянии ,
принятые для расчета термодинамических функций Н2О2 (pg=l, a=2)
vx 3598,7
v2 1387,5
875
v6 3610,7
v6 1266
xn —90,9
-11
«13 ~H
«!5
«16
«22
«23
«25
«28
«33
«35
— 167,6
—3
-10
—7
—11,5
—4
—10
—11,1
«36
«55
«56
«68
Ao...
aA
aA
ai
—2
—90,2
О
—3
10,068
—0,234
0,104
0,034
a\
at
в:...
aB
аВ
аВ
аВ
в
—0,234
0,174
0,8740
—0,003
—0,004
-0,007
—0,003
—0,003
Со...
аС
4
DJ
0,8384
0,001
—0,004
—0,013
0,001
-0,007
4,5 -Ю-'
7,5 • Ю-4
DJK
°о
а1
а2
^0
V,
v2
V,
-2 • Ю-6
39,654
0,553
0,0462
802
1014
641
42
щими значениями структурных параметров: г0 (О—Н)=0,965 + 0,005 А; г0 @—0) = 1,464± 0,003 А;
¦4В.—О—0=99,4+1,2°;1<р0=120,3+0,7о. Заторможенное внутреннее вращение в молекуле Н2О2
характеризуется функциями угла поворота <р группы ОН1 : V (<р) — потенциальной энергией
внутреннего вращения и сс(<р) — вращательной постоянной группы ОН. В справочнике эти функции
определяются выражениями, предложенными Хантом и др. [2048]:
V (ср) = Vo + V, cos ? + F2'cos 2? + V3 cos 3?, E. 5)
a (tp) = a0 -j- a2 cosj<p + a2'cos 2cp. \ E. 6)
Приведенные в табл. 5.8 значения постоянных a0, a1? a2 соответствуют указанным выше
структурным параметрам. Эти величины и значения первых пяти уровней энергии внутреннего
вращения Н2Оа, найденные Хантом и др. [2048 J по торзионно-вращательным полосам в далеком ИК-
спектре паров Н2О2, послужили основой для определения приведенных в табл. 5.8 значений
постоянных Vo, Vlt F2, F3. Им соответствуют значения FTpaHC=387 см, F4B0=2499 см, <ре=111,9°,
близкие к полученным в работе [2048]2. Приведенные в табл. 5.8 значения колебательных
постоянных определены Хачкурузовым и Пржевальским [291, 287] на основе критического анализа
результатов исследований колебательных спектров паров Н2О2, полученных в работах [454, 4067,
1623,1626, 952, 3179, 1637]3, при последовательном учете дублетной структуры колебательно-
вращательных полос Н2О2, обусловленной некоторым различием функций V (ср) основного и
возбужденных колебательных состояний 4. Приведенные в табл. 5.8 значения вращательных
постоянных и постоянных центробежного растяжения определены Редингтоном и др. [3179] на основании
анализа вращательной структуры колебательно-вращательных полос в ИК-спектре паров
Н2О2. Эти значения находятся в согласии с результатами анализа микроволнового спектра паров
Н2О2 [2877].
Постоянные колебательно-вращательного взаимодействия, приведенные в табл. 5.8,
вычислены Хачкурузовым и Пржевальским [289] по результатам анализа вращательной структуры
ряда колебательно-вращательных полос в ИК-спектре паров Н2О2, полученным в работах [3179,
4067, 1623], в предположении, что а?=а? (Х=А, В, С).
Термодинамические функции Н2О2 (г) вычислены по уравнениям A.3)—A.10), A.122)—A.124),
A.127), A.128) с учетом ангармоничности колебаний и колебательно-вращательного
взаимодействия по уравнениям A.131)—A.133), центробежного растяжения по уравнениям A.140)—A.142)
и низкотемпературных поправок по уравнениям A.147)—A.149). Составляющие внутреннего вра-
1 Значение ?=0 принято относить к цисконфигурации Н2О2.
2 В работе [292] было показано, что представление E.5) функции V (9) необходимо дополнить членом Vt cos 4 у
Однако это уточнение практически не влияет на значения термодинамических функций Н2О2 (г).
3 Колебательные спектры Н2О2 в конденсированных состояниях исследовались в работах [1455, 3632, 3633, 1628,
2357].
4 При определении значений постоянных ангармоничности использовались также приближенные соотношения,
приведенные в работе [287]. Погрешности принятых в табл. 5.8 значений колебательных постоянных Н2О2
приведены в работе [291].
122
Водород и его соединения Н2О2(г)
щения вычислены на основании использования следующего выражения для статистической
суммы *:
{и* со 1
2 2 ехр[-Й-(^-^о,х)]+ 2 2 2exp[-!f(Z?B,T,-?0I+F0)],E.7)
в=0 т=1, 2, 3, i и=и*+1 т'=1, 2 J
где Е„_т —уровни энергии заторможенного внутреннего вращения; 2?„1Т, — уровни энергии
свободного внутреннего вращения, п* — значение главного квантового числа внутреннего
вращения п, выше которого допустима замена уровней энергии заторможенного вращения уровнями
энергии свободного вращения. Для Н2О2, HDO2 и D,p2 принималось п*=26. Значения Еп т
находились как собственные значения уравнения Матье с использованием выражений E.5) и E.6)
для функций V (<р) и а (<р). Для этого определялись корни соответствующих вековых уравнений 59
и 60 порядков. Погрешности вычисленных значений термодинамических функций Н2О2 (г)
обусловлены неточностями принятых значений молекулярных постоянных и приближенным характером
метода расчета. Суммарные погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются
в 0,05, 1 и 2 Дж-К^-моль-1.
Ранее термодинамические функции Н2О2 (г) вычислялись до 6000 К во втором издании
справочника [88] и до 1500 К в работах [4057, 2638, 1622, 1632, 1631, 208, 3489, 937, 1624]2. Эти расчеты
были выполнены по неточным значениям молекулярных постоянных в приближении «жесткий
ротатор — гармонический осциллятор» с учетом заторможенного внутреннего вращения по
таблицам Питцера и Гуинна [4057, 2638, 1622, 1632, 1631, 88] 3, по приближенной методике [208] и
непосредственным суммированием по уровням энергии внутреннего вращения [3489, 937, 1624].
Расчеты [2638, 1622] являются ошибочными. Вследствие компенсации погрешностей разного
происхождения, различия между соответствующими значениями Ф° (Г), приведенными во втором
издании справочника и в табл. 22, невелики. При 298,15, 3000 и 6000 К они равны 0,56, 0,33 и
1,7 Дж-К^-моль. В то же время различия между соответствующими значениями S° (T) равны
1,5, 1,35 и 6,7 Дж-К^-моль. Наиболее близки к значениям термодинамических функций
Н2О2 (г), приведенным в табл. 22, значения полученные в работах [937, 1624]. В работах [1632,
1623а, 1624] на основании результатов калориметрических измерений [1632, 1633]
предпринимались попытки вычислить S° (H2O2, г, 298,15 К). При этом были получены значения меньшие
вычисленного по молекулярным постоянным на 1,46 и 1,26 Дж-К^-моль. Причиной этого
расхождения, согласно [1624], является использование в расчетах заниженного значения энтальпии
испарения Н2О2 (ж), обусловленного, по-видимому, каталитическим разложением перекиси
водорода при калориметрических измерениях.
Константа равновесия реакции Н2О2 (г)=2 О (г)+2 Н (г) вычислена с использованием значения
ДгЯ°@) = 1055,522 + 0,30 кДж-моль, соответствующего принятой в справочнике энтальпии
образования Н2О2 (г):
ДД°(Н2ОЯ, г, 298,15 К)= — 135,88; + 0,22 кДж • моль.
Эта величина основана на принятом в справочнике значении энтальпии образования Н2О2 (ж)
и на значении Д„#о(Н202, ж, 298,15 К)=51,9±0,2 кДж-моль. Последнее является средним
взвешенным значений, вычисленных по III закону на основании результатов определений давления
пара Н2О2 (ж), полученных в работах [2486, 1393, 3317] (табл. 5.9). Погрешность этого
значения учитывает неточность термодинамических функций, использованных в расчетах. Энтальпия
испарения Н2О2 (ж) определялась также Жигером и др. [1501а, 1633] на основании
калориметрических измерений энтальпии испарения водных растворов Н2О2 различных концентраций при
26,5° С. Экстраполяция полученных данных к чистой перекиси водорода привела к значению
Д„Я° (Н2О2, ж, 298,15 К)=51,63 кДж-моль, которое, как отмечалось выше, является
заниженным.
Множитель 1/2 в выражении E.7) обусловлен тем, что каждому состоянию вращения молекулы Н2О2, как целого,
соответствуют два состояния внутреннего вращения (х=1,^4 при К четном и *=2, 3 при К нечетном).
В справочнике JANAF [2095] приведены вычисленные в работе [1631] значения термодинамических функций
Н2О2 (г), дополненные значениями при 100 и 200 К, вычисленными по принятым в [1631 ] постоянным.
В этих работах функция F(y) аппроксимировалась выражением A.159).
123
Н3О+(г)
Глава 5
Таблица 5.9. Результаты определений энтальпии испарения переписи водорода (в кДж • моль")
Авторы
Метод
Д,Д°(Н2О2, ж, 298,15 К)
II закон
III закон
Мае, Хилберт, 1924 Измерение давления пара над Н202 (ж) ста- 47,95+4,4 51,92+0,15
[2486] тическим методом, 7 точек, 288—315 К
Эджертон, Эмте, Мин- Измерение давления пара над Н2О2 (ж) ста- 47,51 ±3,10 51,75+0,16
ков, 1951 [1393] тическим методом, 11 точек, 278—328 К
Скэтчард и др., 1952 Определение давления пара над Н3О2 (ж) 52,2+10,5 51,925 ±0,073
[3317] в результате термодинамической обработки
данных по давлению пара над
концентрированными водными растворами Н2О2, 3 точки,
333—363 К
Н3О+ (г). Термодинамические свойства газообразного иона оксида триводорода в стандартном
состоянии в интервале температур 298,15—20 000 К приведены в табл. 23.
Молекулярные постоянные, использованные при расчете термодинамических функций Н3О+,
приведены в табл. 5.4. Экспериментальных данных о спектре и структуре иона Н3О+ в свободном
состоянии нет. Приведенное в табл. 5.4 произведение моментов инерции вычислено в
предположении, что в основном электронном состоянии ион Н3О+ имеет пирамидальную симметричную струк-
ТУРУ (точечная группа С3„) с геометрическими параметрами г (О—Н)=0,98±0,05 А и ^гН—О—Н =
= 111°20' + 5°, полученными Лишка и Дизмонсоном [2444] в результате квантовомеханического
расчета методом молекулярных орбиталей с учетом электронной корреляции независимых
электронных пар. С результатами этого расчета удовлетворительно согласуются другие неэмпирические
расчеты структуры свободного иона Н3О+ [662, 984, 2175, 1217, 2284, 1836, 359], в которых для
межатомного расстояния г (О—Н) получены значения от 0,95 до 1,05 А и для угла -^Н—О—Н —
значения от 109 до 114,5°. Практически совпадающие значения структурных параметров Н3О +
в кристаллической решетке были получены при исследованиях кристаллогидратов сильных кислот
HNO3-H2O, НСЮ4-Н2О и H2SO4-H2O методом ЯМР [3199] и методом рентгеноструктурного
анализа [4033], а также при исследовании структуры Н3О+СН3СвН4О7 методом дифракции
нейтронов [2477]. Пирамидальная структура иона Н3О+ в кристаллах и растворах подтверждается также
исследованиями их спектров [1470, 1471, 3634, 266, 3344, 1513, 508], поскольку в инфракрасном
спектре проявляются все частоты. Анализ этих работ показывает, что частоты деформационных
колебаний Н3О+ сохраняются почти постоянными в разных системах (v3 от 1020 до 1200 см и
v4 от 1580 до 1750 см), в то время как в величинах и отнесении частот валентных колебаний vx
и v3 имеется большая неопределенность (vx от 2570 до 3400 см и v3 от 2100 до 3470 см).
Приведенные в табл. 5.4 основные частоты свободного иона Н3О+ вычислены на основании
оцененных значений силовых постоянных /г=7,1 -105 дин-см и /а/г2=0,55-105 дин-см. Они
находятся в разумном согласии с частотами Н3О+ в кристаллах и растворах г. Погрешности
принятых значений частот не превышают 200 см в значениях vx, v3 и v4 и 100 см в значении v.
'2-
Термодинамические функции Н3О+ вычислялись по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128), A.130) в приближении «жесткий ротатор — гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности вычисленных таким образом
термодинамических функций составляют 0,5, 2, 3 и 6 Дж-К^-моль в значениях Ф° (Т) для Г=298,15,
3000, 6000 и 20 000 К и обусловлены при температурах до 3000 К главным образом неточностью
молекулярных постоянных (соответственно 0,5, 1,8, 2 и 2,5 Дж-К^-моль). При более высоких
температурах все большие погрешности вносит приближенный характер расчета.
Ранее таблицы термодинамических функций Н3О+ вычислялись авторами справочника JANAF
[2095] (Т <; 6000 К) и Клифтоном [984] (Т < 5000 К). Оба расчета выполнены в приближении
«жесткий ротатор —гармонический осциллятор» по значениям молекулярных постоянных, оценен-
1 Клифтон [984] оценил /г=8,19-105 дин-см и /а/г2=0,54-105 дин-см и, принимая г (О—Н)=0,95 А, получил
vx=3760 см, v2=1050 см, v3=3870 см и v4=1550 см. Аллавени Леклеч [359] в расчете ab initio получили
для fjr2 значение 0,55-105 дин-см.
124
Водород и его соединения
Н3О+(г)
ным Клифтоном. Расхождения между значениями термодинамических функций, вычисленными
в [2095, 984] и приведенными в табл. 23, составляют от 0,9 до 2 Дж -К-моль в значениях Ф°(Г)
и от 0,9 до 2,6 Дж-К^-моль в значениях <S° (T); они обусловлены некоторым различием в
принятых молекулярных постоянных.
Константа равновесия реакции Н3О+ (r)-f-e (г)=О (г)+ЗН (г) вычислена на основании значения
ДД° @)=296,085+ 15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Aj,H° (Н3О+, г, 298, 15 К) = 598 + 15 кДж • моль.
Эта величина была выбрана на основании анализа результатов исследования сродства
молекулы воды к протону и обменных реакций с участием иона Н3О+, приведенных в табл. 5.10.
Предпочтение было отдано работам Гопкинса и Бона [2003], Хани и Франклина [1821 ] и Бичема и Бат-
рил [539], как наиболее свободным от ошибок, обусловленных образованием продуктов реакции
Таблица 5.10.
Результаты определения сродства к протону молекулы воды и теплоты
образования нона Н3О+ (г) (в кДж-моль)
Авторы
Метод
Л0(Н2О, 298,15 К)
(H3O+, 298,15 К)
Тальрозе, Франке-
вич, 1956 [256]
Ван Роальте, Гари-
сон, 1963 [3777]
Бичем, Батрил, 1968
[539]
Чапка, Расселл, 1968
[964]
Хани, Франклин,
1969 [1821]
Де Паз, Левенталь,
Фридман, 1968—
1969 [1218, 1219]
Лонг, Мансон, 1970
[2458]
Сик, Серлс, 1970
[3418]
Чонг, Майерс,
Франклин, 1972 [954]
Гопкинс, Бон, 1973
[2003|
Исследование ионно-молекулярных реакций
в ионном источнике масс-спектрометра
Измерение потенциалов появления ионов
Н3О+ в масс-спектрах органических
соединений
Исследование ионно-молекулярных реакций
с участием Н3О+ методом ионного
циклотронного резонанса
Исследование зависимости сечения
образования ионов Н30+ по реакции NHJ+H-jO^
=NH2+H3O+ от энергии возбуждения NHJ
Измерение потенциала появления ионов
Н3О+ при ионизации молекул С2НйОН в
сочетании с измерением кинетической энергии
ионов Н3О+
Измерение пороговой энергии процесса
D3O++4He=D2O++D+4He
Масс-спектрометрическое исследование
ионно-молекулярных реакций с участием
Н3О+
Масс-спектрометрическое исследование
ионно-молекулярных реакций с участием
Н3О+
Масс-спектрометрическое исследование
равновесия D3O++D2S=D3S++D2O, 340 К
Масс-спектрометрическое исследование
равновесия реакции H3S++H2O=H3O++H2S,
296—426 К
-Ш>А0 > —720 574< LfHa < 612
-632±13 662±13
—695 ±16»
=5—704
—695
—761 +29«
>-704
598 ±16
==: 590
598
533 ±29
>591
—695+8
о < —678 616< ДуЯ0 < 637
585 ±14
598+8
Авторы работы [539] рекомендуют значение —686 ±16. б Пересчитано на Аа (Н2О).
в возбужденных состояниях, и необходимости использовать другие величины, не измеряемые
в эксперименте. В работах [3777, 3418], по-видимому, были получены завышенные значения Ао
из-за того, что продукты реакции имели избыточную энергию. В работах [1218, 1219] были
получены завышенные пороговые значения ^энергии ионов Н3О+ из-за того, что не были учтены
экспериментальные точки, соответствующие меньшей энергии. Данные работ [256, 964, 2458, 3418,
954] находятся в удовлетворительном согласии с принятой величиной.
Глава 6
ДЕЙТЕРИЙ, ТРИТИЙ И ИХ СОЕДИНЕНИЯ
D, D2, o-D2, n-D2, OD, OD-, DO2, DO~, D2O, D2O2, HD, HDO, HDO2,
T, T2, o-T2, n-T2, ОТ, Т2О, НТ, НТО, DT, DTO
В настоящей главе рассматриваются дейтерий, тритий и их газообразные соединения с
кислородом и протием; термодинамические свойства соединений дейтерия и трития с галогенами
обсуждаются в главах 7—10. Для D, D2, HD, T, Т2, НТ, DT таблицы термодинамических свойств
рассчитаны для интервала температур от 100 до 20 000 К, для остальных 12 газов — от 100
до 6000 К. Кроме того, в Дополнении (см. наст, том, кн. 2) приведены таблицы
термодинамических функций орто- и парадейтерия и трития от 10 до 1000 К. Для семи газов (D2, D2O, D2O2,
HD, T2, НТ и DT) в Приложении 3 обсуждаются данные, позволяющие учитывать отклонения
термодинамических свойств этих газов от их свойств в стандартном состоянии под влиянием
межмолекулярного взаимодействия.
D (г). Термодинамические свойства газообразного одноатомного дейтерия в стандартном
состоянии при температурах 100—20 000 К приведены в табл. 24. Термодинамические функции D(r)
вычислялись по соотношениям A.3)—A.6) с учетом только основного состояния 2S атома D.
Погрешности вычисленных значений термодинамических функций при Т ^ 12 000 К определяются
только неточностью физических постоянных и не превышают 0,01 Дж-К~1-моль~1 в Ф°{Т) и S°(T).
При более высоких температурах погрешности возрастают из-за того, что в расчете не
учитывались ридберговские состояния атома D; при 20 000 К они достигают 0,07 и 0,3 Дж-К~1-моль~1
в значениях Ф°(Г) и S°(T). Термодинамические функции одноатомного дейтерия вычислялись
в работе [1673] и справочниках [88, 337]. Результаты расчетов в этих справочниках согласуются
с данными табл. 24 в пределах 0,01 Дж-К~1-моль~1, данные работы [1673] отличаются на
величины до 0,1 Дж-К~1-моль.
Энтальпия образования одноатомного дейтерия
AfB°(D, г, 0) = 219,807 ± 0,003 кДж • моль'1
принята на основании значения энергии диссоциации D2.
D2 (г). Термодинамические свойства газообразного двухатомного дейтерия в стандартном
состоянии при температурах 100—20 000 К приведены в табл. 25. Термодинамические функции
Da(r) вычислены без учета возбужденных электронных состояний D2 для равновесной смеси орто-
и парадейтерия *. - '
Расчет значений <?код. вр и ее производных для состояния Х1^!* проводился непосредственным
суммированием по соотношениям A.73)—A.75) и колебательно-вращательным уровням энергии
D2. Из найденных таким образом значений R In Qm. вр для равновесной смеси орто- и
парадейтерия вычиталась составляющая ядерного спина (R In 9).
Колебательно-вращательные уровни энергии D2 были получены при подготовке настоящего
издания 2 численным интегрированием уравнения Шредингера с потенциальной функцией,
найденной Колосом и Вольневичем [2290, 2289 ] с учетом адиабатической и релятивистской поправок
[2288] по программе, изложенной в работе [52]. Для упрощения расчета квазисвязанные
колебательно-вращательные уровни D2 вычислялись как связанные (согласно данным [2391 ] это
приводит к неточности в энергии соответствующих уровней, приблизительно равной их полуширине).
Ранее теоретические расчеты уровней D2 проводились в работе [2290 ] и Ле Роем, однако в [2290]
приведены только значения G0(v) и В„ а работа Ле Роя известна по статье [2386], где не
приведены результаты расчета.
1 О расчете термодинамических функций o-D2 и re-D2 см. ниже.
2 Соответствующая таблица будет опубликована отдельно.
126
Дейтерий, тритий и их соединения
D2(r)
Экспериментально молекулярные постоянные D2 определялись при исследовании спектра
комбинационного рассеяния [3635, 3542], систем полос В1^—X1!.* и СШ—ХХ2+ [2120,'3600, 758]
и колебательно-вращательного спектра, индуцированного электрическим полем [747]. Наиболее
полная информация о колебательно-вращательных уровнях и молекулярных постоянных D2
в состоянии Х1!,* была получена в работе Бредохля и Герцберга [758], наблюдавших на приборе
с высокой дисперсией в указанных системах полос переходы на уровни сЦ i>max=21 и / ^ 9.
Молекулярные постоянные D2 в состоянии Х1!!*, найденные Бредохлем и Герцбергом [758],
приведены в табл. 6.1. Однако отсутствие во всех изученных спектрах переходов на уровни с
значениями />9 обусловливает невысокую точность аппроксимации уравнения для G(v)-{-F,(J)'
с постоянными, полученными в этих работах, на область энергии, примыкающую к предельной
Таблица 6.1. Молекулярные постоянные D2, OD, HD, T2, HT, DT
Молекула
Состояние
d2 ;
г.
OX+ 0
OD Xm{ 0a
/
I
hd ;
T2
ht :
DT
Примечание
D2:
OD:
HD:
T2, НТиТ:
L2S+ 32 680,85
?22+ 69 759
f!S+ 0
?!S+ 0
VZ+ 0
f!S+ 0
3 115,78
2 720,24
г 2 316,17
735,9
3 812,924
2 544,74
3 593,65
2 843,24
62,04 a
44,055
50,443
94,5
90,908
40,005
79,08
49,805
Ниже все постоянные даны в см.
а со„г/.= 0,618, со
а Л = —139,089;
г То = 0,1201; я со
ведено значение
а соеуе = 0,504; 6
1 вычислено по
„ze = — 0,0274; «
6 а2 = 0,582 • 10-
,!/, = -0,235; •
?*(>; ^0 = 0,12-
а2 = 3,972-10-2,
соотношению A
а2 = 7,
в.
СМ
30,438 8
10,020 9
л 9,193 6
2,820 8
a 45,662 7
20,327 3
40,563 6
25,380 9
ах • 102
106,9 б
27,57 б
31,81 в
10,2
200,34 б
57,80
161,97
80,51
877 • 10; в приведено значение
3; в приведено значение Da
а2 = —1,187-Ю-3; ж Но =
10-5.
а3 = —
68).
-3,4 • 10~3; в р! = 7,8
, Яо = 1,935
1,645-10"8, i
• ю-*, яо =
Z>, • 10*
113,7 в
5,374 2 в
5,762 6 ж
2,5 3
266,7 в
51,9 а
207 а
80,9 а
J) • и —
•Ю-'8, ?„ =
г,0 = _6,01
2,2-Ю-5.
Ге
А
0,741 65
0,969 8
1,012 5
1,828
0,741 40
0,741 57
0,741 72
0,741 61
3,3-10-'.
—5,19-Ю-13;
• Ю-13; 3 при-
кривой диссоциации. Вычисленные в настоящем издании колебательно-вращательные уровни
D2 для / ^ 9 согласуются с экспериментальными данными [758 ] с точностью лучше 2 см
(расхождение результатов расчета G0(v) в [2290] с теми же данными примерно в 2 раза больше).
Погрешности вычисленных термодинамических функций D2 при Т ^ 5000 К не превышают
0,01 Дж.К~1.моль~1 и обусловлены главным образом неточностью фундаментальных постоянных.
При Т >• 5000 К из-за неточности расчета энергии квазисвязанных уровней D2 и их учета в
статистической сумме и ее производных (см. текст по Н2) погрешности могут достигать ОД Дж-К~1Х
Хмоль в значении Ф° B0 000 К).
Термодинамические функции D2 вычислялись ранее в работах [3755] E0—700 К), [2421]
B98-773 К), [4000] B98-2000 К), [2148] B98-3000 К), [2296] (до 6000 К) и справочниках [88,
337, 1783]. Результаты расчетов [4000, 2296, 337, 88] хорошо согласуются с данными табл. 25,
кроме значений S°{T) для Т > 4000 К в [88], где в расчете была допущена ошибка (до 0,05 ДжХ
XК .моль). Данные справочника [1783] из-за использования метода Майера и Гепперт-Майер
отличаются от приведенных в табл. 25 на 0,15, 0,6 и 1,5 Дж-К~1.моль~1 в значениях Ф°(Г), S°(T)
и С°(Т) при 5000 К (см. сноску на с. 135).
Молекулярный дейтерий в газообразном состоянии является стандартным состоянием этого
элемента и в соответствии с этим
A/ff°(Dlf г) = 0.
Константа равновесия реакции D2(r)=2D(r) вычислялась на основании принятого значения
Дг#° @) = Do (D2) = 36 748,9 ± 0,4 см = 439,614 ± 0,005 кДж • моль.
127
o-D2 (г), n-JJ (г), OD (г) Глава 6
Энергия диссоциации молекулы D2 определена с высокой точностью Герцбергом [1910] по
границе континуума, примыкающего к переходу В1!!*—Х1^ и возникающего при диссоциации D2
на атомы D (ls2S)-\-D Bs2S) (см. текст по Н2). Это значение хорошо согласуется с результатами
квантовомеханических расчетов [2290, 833] C6 748,0 и 36 748,2 см).
o-D2 (г). Термодинамические функции газообразного ортодейтерия в стандартном состоянии
в интервале температур 10—1000 К приведены в табл. Д.5. Значения (?колвр и ее производных
для o-D2 были вычислены непосредственным суммированием по уровням энергии D2 (см. выше),
по уравнениям типа
21 «^шах, ц
<?Кол.вр(о-В2) = 2 2 (^ + i)B^ + 1)BJ + 1)^v{-^[G0(v) + F,(J)]}, F.1)
»=0 J=0, 2, i
где (i,+l) BiB+l)=6.
Погрешности вычисленных термодинамических функций ортодейтерия обусловлены
практически только неточностью фундаментальных постоянных и не превышают 0,01 Дж-К~1-моль~1
в значениях Ф°(Т) и S°(T) во всем интервале температур.
Термодинамические функции o-D2(r) ранее вычислялись Вулли и др. [4000] и в справочнике
[1783]; расхождения данных работ [4000, 1783] и табл. Д.5 не превышают 0,01 Дж-К~1-моль~1.
w-D2 (г). Термодинамические функции газообразного парадейтерия в стандартном состоянии
в интервале температур 10—1000 К приведены в табл. Д.6. Значения (?ш> вр и ее производных
были вычислены непосредственным суммированием по уровням энергии D2 (см. выше), по
уравнениям типа
= 2 ^ is Bгл + 1) B7 + 1) exp { -|f [Go (v) + F, G) - Fo G^)]}, F.2)
«=0 J=l,3,&, ...
Погрешности вычисленных таким образом значений термодинамических функций не превышают
0,01 Дж-К~1-моль~1 в значениях Ф°(Г) и S°(T) во всем интервале температур и обусловлены
практически только неточностью фундаментальных постоянных.
Ранее термодинамические функции ra-D2(r) вычислялись в работе Вулли и др. [4000],
результаты обоих расчетов согласуются в пределах 0,01 Дж-К~1-моль~1.
OD (г). Термодинамические свойства газообразного оксида дейтерия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 26.
В табл. 6.1 приведены молекулярные постоянные OD, принятые на основании анализа
результатов исследований электронных спектров. По аналогии с молекулой ОН в справочнике принято,
что молекула OD имеет 3 связанных валентных электронных состояния. В спектрах OD
наблюдались 2 системы полос Л22+—Х2П{ [2072, 2073, 3310, 3308, 3309, 2926, 2925, 994] иБ22+—А22 +
[3353, 485, 874, 1462, 1148]. Приведенные в табл. 6.1 постоянные OD в состояниях Х2П,. и А22+
вычислены Коксоном [1105] на основании анализа экспериментальных данных, полученных в
работе Клайна, Коксона и Фат [994] и более старых исследованиях [3308, 3309, 2926, 2925]. Эти
постоянные с высокой точностью описывают всю совокупность наблюдавшихся спектральных
переходов для v', !>"^3и/^ 40,5. В состоянии Z?22 + молекула OD (см. текст по ОН) имеет
только три колебательных уровня (v ^ 2), постоянные в этом состоянии приняты по данным [874],
где для перехода 2?22 +—.422 + наблюдались полосы с v' ^ 2, v" ^ 13.
Значения Qm, (?м и EВН вычислялись по уравнениям A.76)—A.78) непосредственным
суммированием по колебательно-вращательным уровням энергии состояний Х2И^ А2Ъ+ и Б22+.
Энергии уровней находились с использованием соотношений A.65) и A.42) для состояния Х2Л( и
соотношений A.34) и A.65), для состояний А21>+ и В2Е+ со значениями коэффициентов Ykl и Ао,
приведенными в табл. 6.2. Мультиплетность состояний А22,+ и 522 + и Л-удвоение уровней в состоянии
ХШ{ учитывались статистическим весом 2. Поскольку для состояния Х2П( экспериментально
изучены переходы только на уровни v ^ 3, приведенные в таблице постоянные Ykl для этого
состояния получены с помощью изотопических соотношений A.66) и аналогичных постоянных ОН
128
Дейтерий, тритий и их соединения
OD(r)
Таблица 6.2.
Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения «шах и Jiim, принятые для расчета термодинамических функций
OD, OD- и ОТ
Коэффициенты
Те ¦ Ю-'
Y,o • Ю-'
Узо
У.о • Ю'
Y50 • 10s
Ym • 10*
Y-o • Ю6
Yeo ¦ 10е
Yo, • Ю-'
Yu
Yn • 102
Уз, • 10s
Yn • 10'
Y5, • 10'
У„, • 10s
Y12 • 105
Y2! ¦ 10»
Уоз • 10»
Yo, • 10»
(<z0 = Be) ¦ 10-'
a,
a2 • 10'
03 • 10»
а А =—139,235
xm
oa
2,721 35
—0,451 246
0,228 337
—0,305 109
—1,422 84
1,338 44
-6,263 33
9,296 86
1,002 7
—0,296 982
1,274 2
—2,098 07
1,417 52
—0,353 103
-0,542
0.9S7 7
—0,789
2,095
-0,97
3,727 0
0,470 534
1,474 189
0,447 126
22
84
6 Те(аЧГ) = 2
OD
AW
3,268 23
2,314 227
-0,488 811
-0,771 016
9,562 07
-7,400 4
—
—
—
0,922 673
—0,390 485
4,126 84
-9,552 31
9,300 72
—3,662 07
0,55
—
—
—
—
2,041 0
0,656 967
4,312 561
-26,242 718
13
65
6,975 76
0,735 9
—0,945
—
—
—
—
—
—
0,282 075
—0,102
—9,9
—
—
—
—0,250
—
—
120
—
0,128 0
0,896 706
11,222 347
—556,041 113
2
36
OD-
0
2,766
—0,48
—
—
—
—
—
—
1,02
-0,26
—
—
—
—
-0,56
—
—
—
—
—
—
—
—
28
73
xm
oa
2,284 878
—0,318 105
0,135 149
—0,151 625
-0,593 677
0,468 89
—1,842 28
2,295 963
0,706 85
—0,175 779
0,633 216
—0,875 413
0,496 591
—0,103 861
-0,269 4
0,412
—0,276 4
0,73
—0,24
3,727 0
0,311 383
0,820 427
—0,704 343
27
100
ОТ
3,268 23
1,942 536
—0,344 845
—0,438 096
4,577 2
—3,028 69
—
_
—
0,649 8
—0,201 42
0,633 003
—1,460 17
1,331 78
-0,549 754
-0,285
—
—
—
—
2,041 0
0,427 387
2,193 231
—9,148 673
16
77
B^
6,975 76
0,672 37
-0,822 528
—
—
_
0,206 22
-0,198
—0,13
.
—
0,128 0
0,420 429
7,285 231 51
—292,341 297
2
39
из табл. 5.5. Уравнение для G (v)-\-Ft(J) с этими значениями Ykl хорошо описывает
экспериментальные данные Клайна и др. [994], обеспечивая схождение колебательных уровней к диссо-
циационному пределу состояния Х2Л( и экстраполяцию колебательно-вращательных уровней
к предельной кривой диссоциации. Значения Уы для состояний A2~Z + и 522+ получены
обработкой данных [874]. В табл. 6.2 приведены также значения а{ в уравнениях, аппроксимирующих
предельные кривые диссоциации OD в состояниях Х2П,., А2!, + и В22 + и значения vmax и /lim в этих
состояниях.
Погрешности вычисленных значений термодинамических функций OD (г) при Т ^ 5000 К
обусловлены главным образом неточностью фундаментальных постоянных и не превышают
0,02 Дж-К^-моль в значениях Ф°(Т) и S°(T). При 6000 К они могут достигать 0,05 Дж-К-1Х
Хмель из-за неточности определения числа и энергии высоких колебательно-вращательных
уровней состояния Х2Л{.
Термодинамические функции OD вычислялись ранее в работах [2574] (Ф°(Т) до 3000 К),
[1782], справочниках [88, 1783, 337]. Данные [2574] при 3000 К отличаются на 1,25 Дж-К^х
Хмоль от приведенных в табл. 26 из-за использования неточных значений молекулярных
постоянных и приближенного метода расчета. Данные, приведенные в работах [1782, 1783],
отличаются от приведенных в табл. 26 при высоких температурах из-за использования метода Майера
и Гепперт-Майер (на 0,1 и 0,4 Дж-К^-моль в Ф°E000 К) и S° E000 К)). Такие же расхождения
имеют место с данными [337], однако в этом справочнике, так же как и для ОН, допущена
неточность в учете мультиплетности и вырождения состояния X2JI.. Соответствующие расхождения в
Ф°B98,15 К) и Н° B98,15 К)—Я°@) достигают 0,7 Дж-К^-моль и более 2 кДж-моль-1.Данные
предыдущего издания справочника [88] и табл. 26 согласуются во всем интервале температур
в пределах 0,06 Дж-К^-моль-1.
129
OD-(r), DO2(r)
Глава в>
Константа равновесия реакции OD(r)=O(r)+D(r) вычислена с использованием значения
АгН° @) = Do (OD) = 35 924 + 15 см = 429,75 + 0,18 кДж • моль-1.
Это значение энергии диссоциации OD было получено Карлоне и Далби [874] на основании
короткой экстраполяции (около 118 см) значений С»+д/, состояния А22,+. Близкое, но менее
точное значение C6 134 см) было получено ранее Барроу [485] при более далекой экстраполяции
(около 1500 см) уровней того же состояния. Расчет с помощью изотопических соотношений C.6)»
на основании значений D0(OH), G @, ОН)=1850 см и G @, OD) = 1347,3 см приводит к
величине 35 923 см.
Принятому значению энергии диссоциации OD соответствует
А//° (OD, г, 0) = 36,84 + 0,21 кДж • моль.
OD~ (г). Термодинамические свойства газообразного отрицательного иона оксида
дейтерия в интервале температур 298,15—6000 К приведены в табл. 27.
Спектры OD" не исследовались. Поэтому термодинамические функции OD~(r) были вычислены
по постоянным, полученным с помощью изотопических соотношений на основании постоянных
ОН". Расчет термодинамических _функций был выполнен с учетом состояний Х1!,+ и а3П (см.
табл. 6.2). Значения Qm, Qw и QBB вычислялись по соотношениям A.90) — A.92) при условии,.
что A//?0
яр
вР
и аналогичные соотношения имеют место для
] вр
и
вр
-
Значения Q[fl вр и ее производных вычислялись непосредственным суммированием по v при замене
суммирования по / интегрированием с использованием соотношений типа A.82). Уровни
колебательно-вращательной энергии состояния Хх.2 + находились по уравнениям A.65) и A.62) со
значениями коэффициентов Ykl, приведенными в табл. 6.2. Там же приведены значения vmax и /н1П»
В расчете учитывались все колебательно-вращательные уровни энергии состояния ХХЪ + со
значениями / <! /„,„,,-
Погрешности вычисленных термодинамических функций OD~(r) обусловлены главным обра-
вом отсутствием экспериментальных данных о молекулярных постоянных этого иона. Они
оцениваются в 1, 1,5 и 2 Дж-К-^моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Термодинамические функции OD" (г) публикуются впервые.
Константа равновесия реакции OD~(r)=O(r)+D(r)+e(r) вычислена на основании значения)
ДгЯ°@)=605,800 + 1,3 кДж-моль. Эта величина соответствует энергии диссоциации OD"
D0(OD~) =38 840 + 100 см=464,6 + 1,2 кДж-моль,
принятой в результате расчета по соотношению C.6) и значению D0(OH~). Соответствующее-
значение энтальпии образования OD~(r) равно
AfH°(OD~, г, 0) =—139,21 + 1,2 кДж • моль.
DO2 (г). Термодинамические свойства газообразного диоксида дейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 28.
Молекулярные постоянные, использованные при расчете термодинамических функций DO3r
приведены в табл. 6.3. Нижними валентными электронными состояниями DO2 являются
состояния %2А" и А2А'. Произведение главных моментов инерции DO2 в состоянии Х2А",
приведенное в табл. 6.3, соответствует вращательным постоянным .40=11,114+0,005 см, 50=1,0541 +
Таблица 6.3. Значения молекулярных постоянных, а также р{, принятые для расчета
термодинамических функций DO2 и DO^ (s = l)
Молекула
DO2
Состояние
Х*А"
AW
Z^A-l
т
•*з
СМ
0
7030
0
2530 1020
2590 1010
1123
1080
г3 • см8
1,945
2,29
Pi
2
2
1
130
Дейтерий, тритий и их соединения DO~(r) D О(г)
+0,002 см~\-С0=0,9628+0,002 см, вычисленным Бирсом и Говардом [549] по значениям
структурных параметров г (D—О) =0,977+0|005 А, г(О—О) = 1,335+0,005 А, ^ D—О—0 = 104,1 ±ТО,
которые были найдены ими при анализе микроволнового спектра DO2. Значения основных частот
DO2 в состоянии ^2Л"приняты по работе Смита и Эндрюса [3464] (см. также [2662,2084]), которые
исследовали инфракрасные спектры НО2 и DO2, изолированных в аргоновой матрице. С учетом
отличия основных частот радикалов НО2 в газовой фазе и изолированных в аргоновой матрице
погрешности значений частот, приведенных в табл. 6.3, составляют 5, 10 и 10 см. Электронный
спектр DO2 исследовали Хунцикер и Вендт [2050], которые нашли для энергии состояния А2А'
величину 7018 + 15 см. Округленное значение принимается в настоящем справочнике.
Термодинамические функции DO2 (г), приведенные в табл. 28, вычислены по уравнениям (\ °,\~
A.6), A.9), A.10), A.122)—A.124), A.128), A.130) в приближении «гармонический осциллятор -
жесткий ротатор» с учетом первого возбужденного электронного состояния и в предположении, что
колебательно-вращательные статистические суммы в состояниях Х2А" и А2А' имеют одинаковые
значения. Погрешности вычисленных значений термодинамических функций DO2 обусловлены
приближенным характером расчета и неточностями принятых значений молекулярных
постоянных. Следует, в частности, отметить наличие взаимного возмущения уровней 001 и 010, которое
по-видимому, искажает значения этих частот. Для Ф°(Т) при 298,15, 3000 и 6000 К погрешности
составляют 0,05, 1 и 2 Дж-К~1-моль~1.
Ранее термодинамические функции DO2(r) не публиковались.
Константа равновесия реакции DO2(r)=2O(r)+D(r) вычислена с использованием принятого
в справочнике значения
ДД° @) = Do (DO2) =712 + 4 кДж • моль,
которое было рассчитано по соотношению C.7), исходя из принятых значений D0(HO2) и величин
G @,0,0; НО2)=2948 см и G @,0,0; DO2)=2367 см". Приведенному значению энергии
диссоциации DO2 соответствует
АуЯ°(БО2, г, 0)= 1,373 ±4 кДж • моль.
DO7 (г). Термодинамические свойства газообразного отрицательного иона диоксида дейтерия
в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 29.
Молекулярные постоянные, использованные для расчета термодинамических свойств DO~(r)
приведены в табл. 6.3. Спектры молекулы DO; не исследовались. Поскольку она является изо-
электронным аналогом молекулы DOF, в основном электронном состоянии 21А1 она должна
относиться к точечной группе симметрии Сх. Значения структурных параметров (г (D—О) = 1 00+
+0,03 А, г (O-O-)=l,37±0,07 A, J$ D-O-O"=102+5°) и основных частот DO" выбраны' как
промежуточные между соответствующими значениями структурных параметров и основных
частот DOF [2981, 404] и DO2, принятых в настоящем справочнике. Возбужденные электронные
состояния DO2 неизвестны.
Термодинамические функции DO;(r) вычислены по уравнениям A.3)—A.6), A.9), A 10)
A.122)—A.124), A.128), A.130) в приближении «гармонический осциллятор — жесткий ротатор'»
Погрешности вычисленных термодинамических функций DO; обусловлены неточностью
принятых значений структурных параметров и основных частот. С ростом температуры погрешности
увеличиваются из-за использования приближенной методики расчета. Погрешности
табулированных значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 1, 2 и 4 Дж-К^-моль, в том числе
из-за неточности принятых значений молекулярных постоянных 0,5, 1 и 2 Дж-К~1-моль~1
Ранее термодинамические функции DO^r) не публиковались.
Константа равновесия реакции DO;(r)=2O(r)+D(r)+-e(r) вычислена с использованием
значения Дг#°@)=840,29+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования DO.,
AfH° (DOj, г, 298,15 К) = -136 + 40 кДж • моль.
Эта величина получена в предположении о равенстве сродства к электрону радикалов НО
и DO2, хотя в действительности различие должно составлять примерно 7 кДж-моль. 2
D2O (г). Термодинамические свойства газообразного оксида дидейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 30.
131
D2O(r)
Глава 6
Таблица 6.4.
Значения молекулярных постоянных (в см), а также р% и а, принятые для расчета
термодинамических функций HDO, D2O, НТО, DTO, Т2О
Постоянная
V2
V3
хп
гзз
Х23
«13
W, Г
Л 000
«и
«2А
«зА
в1«
«2В
?00
«зС
WlQ3
wio3
•wio3
/>х
а
HDO
2725,22
1402,20
3707,46
—41,51
—11,90
—82,34
—16,98
—20,08
—12,91
14,5 (W)
23,410 5
0,253
—1,798
1,087
9,097 5
0,199
—0,147
0,012 5
6,413 7
0,109 8
0,071 0
0,088 1
—56,322
—2,419 2
—0,472 76
5,895 0
—0,757 78
0,830 15
—7,434 8
1
1
D2O
2671,69
1178,33
2788,05
—21,94
—9,46
-24,99
—8,77
—10,17
—85,76
42,5 (г)
15,419 6
0,246
—1,188
0,593
7,270 4
0,095 8
* —0,072 8
0,041 8
4,847 8
0,076 8
0,053 0
0,053 8
—32,153
—2,223 3
—0,255 18
5,667 3
—0,427 97
0,155 17
—1,506 0
1
2
НТО
2297,34
1332,48
3707,93
—29,20
-10,71
—82,29
—13,51
—19,04
—10,82
—
22,607 8
0,149
-1,534
1,086
6,607 2
0,117
—0,125
0,012
5,024 2
0,065
0,061
0,088
—43,365
—1,072 8
—0,318 06
2,535 2
—0,493 76
0,873 83
—5,737 8
1
1
DTO
2291,87
1090,32
2736,02
—28,99
—7,10
—43,70
—10,96
—11,30
-7,86
—
13,681 3
0,148
-0,828
0,420
5,738 8
0,116
—0,068
0,005
3,975 7
0,064
0,033
0,034
—22,683
—1,211 7
—0,175 66
3,268 1
—0,305 26
0,183 15
—1,579 2
1
1
Т2О
2235,18
995,76
2367,55
—15,19
-6,70
—17,81
—6,14
—7,23
—60,25
—
11,301 3
0,142
—0,681
0,357
4,857 7
0,055
—0,031
0,025
3,345 5
0,044
0,028
0,032
—16,844
—0,982 71
—0,123 55
2,728 1
—0,226 93
0,142 48
—0,681 22
1
2
Молекулярные постоянные D2O, принятые для расчета термодинамических функций,
приведены в табл. 6.4. Для колебательных постоянных в таблице приведены значения, полученные
Хачкурузовым [279] на основании анализа результатов исследований структуры колебательно-
вращательных полос D2O в инфракрасном спектре в работах [573, 575, 1244] (см. также [280])
и значений колебательных постоянных Н2О, предложенных в работе [278] и принятых в
настоящем справочнике. Эти значения постоянных находятся в удовлетворительном согласии со
значениями, полученными в работах [3038а, 170, 2957, 1813, 3462а] на основании экспериментальных
данных [575] при использовании потенциальных функций различного типа. Волновые числа
начала полосы v2, найденные в работах [1243, 3954], находятся в соответствии с приведенными
в табл. 6.4 значениями колебательных постоянных. Значения вращательных постоянных и
постоянных центробежного искажения для основного колебательного состояния D2O вычислены при
подготовке настоящего издания из значений 22 постоянных соответствующего гамильтониана,
полученных Стенбикелиером и Белле [3507] на основании частот 36 вращательных переходов,
измеренных в работах [3068, 1415, 2111, 572, 678, 3814, 3506, 557, 3526] в микроволновом и
инфракрасном спектрах D2O. Соответствующие пересчеты были выполнены по методике, описанной
в работах [1041, 1706]. Приведенные в табл. 6.4 значения постоянных аА, а^, а*7 D2O соответству-
132
Дейтерий, тритий и их соединения D2O2 (г)
ют эффективным вращательным постоянным D2O в состояниях @,0,0) и @,1,0), которые были
определены с большой точностью Стенбикелиером и Белле [3507]. Для остальных колебательно-
вращательных постоянных D2O в табл. 6.4 приведены существенно менее точные значения,
полученные Бенедиктом и др. [575]. Погрешности последних могут достигать 0,01—0,02 см.
Термодинамические функции D2O (г) вычислялись по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.127), A.128) с учетом ангармоничности колебаний и
колебательно-вращательного взаимодействия (уравнения A.131)—A.133)), центробежного растяжения (уравнения A.143)—
A.144)), низкотемпературной поправки (уравнения A.147)—A.149)) и резонанса Дарлинга—Ден-
нисона. Погрешности вычисленных значений термодинамических функций D2O(r) обусловлены
неточностью принятых молекулярных постоянных, а выше 3000 К и применением приближенной
методики расчета. Они составляют 0,05, 0,3 и 1,5 Дж-К -моль в значениях Ф°(Г) при 298,15,
3000 и 6000 К.
Ранее термодинамические функции D2O (г) вычисляли Либби [2421, 2422] G1=298,15-^-
773,15 К), Фридман и Хар [1544], авторы первого [87] и второго [88] изданий настоящего
справочника (модифицированный метод Гордона), Абрамович и др. [337] A00—6000 К, на основе
расчета [1544]). Рассчитанные в [1544, 337, 87] значения термодинамических функций D2O
согласуются с приведенными в табл. 30 в пределах 0,1—1,5 Дж»К~1-моль~1; имеющиеся расхождения
обусловлены различиями в значениях молекулярных постоянных, а при высоких температурах —
различиями в методах расчета, принятых в работах [1544, 337, 87] и в настоящем издании.
Расхождения с данными справочника [88] существенно больше из-за некорректного учета
центробежного растяжения D2O.
Константа равновесия реакции D2O (r)=2O(r)-[-D(r) вычислена на основании значения
AJi/°@) =932,647+0,16 кДж-моль, соответствующего принятой в настоящем справочнике
энтальпии образования
A/&°(D2O, г, 298,15 К) = —249,20 + 0,13 кДж • моль.
Это значение основано на измеренных Россини, Ноультоном и Джонстоном [3254]
калориметрическим методом отношениях энтальпий образования и испарения Э20(ж) и Н2О(ж), равных
1,030 69+0,000 29 и 1,031 46+0,000 75, соответственно. Отношение энтальпий образования Б20(ж)
и Н20(ж) было найдено в результате прецизионных опытов по сжиганию водорода и дейтерия
в условиях постоянного давления. Оно хорошо согласуется со значением 1,027+0,003,
полученным ранее Флудом и Тронстадом [1498] на основании измерения энтальпии взрыва смесей
водорода и дейтерия с кислородом, но является более точным.
Приведенным отношениям и принятым значениям ДуЯ°(Н2О, ж) и ABff°(H2O, ж) соответствуют
AfH°(DzO, ж, 298,15 К) = —294,602 ±0,13 кДж • моль
и
Д„#а(О2О, ж, 298,15 К) = 45,401 +0,040 кДж • моль,
сумма которых равна принятому значению энтальпии образования D2O (г). Расчет по
изотопическим соотношениям приводит к практически совпадающей величине (—249,23+0,16 кДж-моль~а).
D2O2 (г). Термодинамические свойства газообразного диоксида дидейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 31.
Молекулярные постоянные D2O2, принятые для расчета термодинамических функций,
приведены в табл. 6.5. Прямых измерений структурных параметров D2O2 не проводилось. В настоящем
справочнике по аналогии с молекулой Н2О2 принято, что в основном электронном состоянии ХхАг
молекула D2O2 имеет неплоскую гошконфигурацию (точечная группа симметрии С2).
Исследования спектра поглощения в далекой инфракрасной области [2047] и микроволнового спектра
[2877] D2O2 в газовой фазе согласуются с этой структурой. Значения вращательных постоянных,
приведенные в таблице, рассчитаны авторами настоящего издания на основании структурных
параметров равновесной конфигурации Н2О2 и колебательно-вращательных постоянных D2O2,
оцененных по изотопическому соотношению A.115а). Приведенные в табл. 6.5 значения
колебательных постоянных D2O2 были вычислены Хачкурузовым и Пржевальским [288, 292] на
основании критического анализа результатов исследований колебательного спектра D2O2 в газовой фазе \
1 Спектр D2O2 в конденсированном состоянии и в матрицах инертных газов исследовался в [3632, 457, 2357].-
133
Глава 6
Таблица 6.5. Значения молекулярных постоянных (в csr1) HDO2 (pg=i, о = 1) и D2O2 (pg=i, g= 2)
в состоянии ХгА, принятые для расчета термодинамических функций
Постоянная
HDO2
D2O2
vj 3620 2667
v2 1332 1028
v3 871 869
v4 a e
v5 2659 2661
ve 984 947
xn —90 —48
*i2 —10 —6
xl3 —10 —8
xn —122 —88
о о
•C^g —z it
Постоянная
HDOa
„ g
х23 —7
Q
25 "—
Д-26 """О
я33 —10
Хз 8
^зб —2
*55 —48
zea —2
Ло... 7,48
a Заторможенное внутреннее вращение
и о2 = 0,0662 (в см-1;
D2O2
-5
—5
—6
—2
— 10
-8
—1
—47
—2
2
4,92
Fo = 805,
постоянные в уравнениях
6 Заторможенное внутреннее вращение
и а2== 0,0869 (в см-1)
Fo = 807
[постоянные в уравнения!
Постоянная
aA
aA
4
aA
О
aA
в'...
aB
аз
aB
aB
6
HDO2
—0,23
0,09
0,03
-0,09
0,08
0,832
—0,003
—0,003
—0,006
—0,001
—0,001
F, = 1022, F2 =
E.5) и
V1 =
. E.5) e
E.6).
1029, F2
t E.6)).
D2O2
-0,09
0,04
0,03
—0,03
0,07
0,788
-0,001
—0,002
—0,006
-0,001
-0,001
= 646, F3
= 651, V
Постоянная
Co-
aC
ЯС
aC
4
a
aC
6
DK
DJK
= 47,
з = 51,
HDO2
D2O2
0,792 0,733
0,001 0,0005
—0,003 -0,002
-0,012 -0,010
0,0005 0,0005
—0,003 —0,003
3•10-« 2•10-»
5 ¦ 10-* 3•10"*
—1,5- 10"B —0,8-10-e
ao = 30,573, в! = 0,551
oq = 21,492, o1 = 0,549
выполненных Жигером и др. [1626,457, 1637]. Приведенные в табл. 6.5 значения постоянных
в уравнениях E. 5) и E. 6), которым соответствуют параметры F^=337 см", Fc=2538 см, сро=
=111,6°, вычислены авторами справочника на основании энергии пяти нижних уровней
внутреннего вращения молекулы D2O2, найденных в работе Ханта и Лекока [2047]. В расчете были
использованы значения структурных параметров, полученные в работе [290] для Н2О2 в основном
колебательном состоянии. При этом рассчитанное значение а0 было увеличено (kx и а2 оставлены
без изменения) для достижения лучшего совпадения вычисленных и экспериментальных
значений энергии пяти нижних уровней внутреннего вращения. Основанием для увеличения а0
послужили работы [2047, 2877], свидетельствующие, что связь О—D в D2O2 может быть короче связи
О—Н в Н2О2, когда обе молекулы находятся в основном колебательном состоянии. Для
постоянной Z)xB табл. 6.5 приведено значение, основанное на определении этой величины в работах [3179,
2048, 2047]; значения D.j и Dje получены в предположении пропорционального изменения
постоянных В к, Dj и Djk при переходе от Н2О2 к D2O2.
Термодинамические функции D2O2 (г) были вычислены по уравнениям A. 3)—A. 6), A. 9), A.10),
A.122)—A.124), A.127), A.128) с учетом поправок на ангармоничность колебаний и колебательно-
вращательное взаимодействие (уравнения A.131)—A.133)), центробежное растяжение (уравнения
A.140)—A.142)) и низкотемпературной поправки (уравнения A.147)—A.149)). Составляющие
термодинамических функций D2O2 (г), обусловленные наличием заторможенного внутреннего
вращения, рассчитаны методом непосредственного суммирования по уровням энергии внутреннего
вращения. Погрешности вычисленных таким образом значений термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных и приближенным
характером расчета колебательно-вращательных составляющих. Общие погрешности в значениях Ф°(Т)
при 298,15, 3000 и 6000 К составляют 0,2, 1 и 2 Дж-К-^моль.
Ранее таблицы термодинамических функций ГJО2 (г) вычислялись Жигером и Лю [1631]
(до 1500 К), во втором издании настоящего справочника [88] (до 6000 К), Чао и др. [937]
(до 1500 К) и Жигером [1624] (до 1500 К). Во всех работах они были вычислены в приближении
«жесткий ротатор—гармонический осциллятор» с учетом заторможенного внутреннего вращения
по методике Питцера и Гуинна [3034, 88] или непосредственным суммированием по уровням
энергии [937, 1624]. Результаты расчета Жигера [1624] и Чао и др. [937] наиболее близки к
приведенным в табл. 31. Имеющиеся расхождения, достигающие при 1500 К величин 0,9 и 1,8 ДжХ
XК -моль в значениях Ф°(Т) и S°(T), обусловлены тем, что в обеих работах не учитывалось от-
134
Дейтерий, тритий и их соединения HD (т)
клонение молекул D2O2 от модели «жесткий ротатор — гармонический осциллятор», в частности
их центробежное растяжение.
Константа равновесия реакции D2O2(r)=2O (r)+2D (г) вычислена на основании значения
Дг^У°@)=1071, 792+0,41 кДж-моль, соответствующего принятой в настоящем справочнике
энтальпии образования
AfH° (D2O2, г, 298,15 К) == —144,3 + 0,4 кДж • моль.
Это значение основано на измерениях Жигера и др. [1633] энтальпии образования (—196,655+
+0,15 кДж -моль) и энтальпии испарения E2,342+0,084 кДж-моль) D2O2 (ж) в
изотермическом калориметре. Энтальпия образования D2O2 (ж) определена по результатам измерений
энтальпии разложения растворов D2O2. Погрешность определения энтальпии образования D2O2 (г)
увеличена до 0,4 кДж-моль, так как на эту величину принятое в справочнике значение энталь-
яии испарения Н2О2 (ж) больше измеренного Жигером [1633] в том же калориметре, в котором
им была измерена энтальпия испарения D2O2 (ж).
Значение A^°(D2O2, г, 298,15 К) =—144,8+0,6 кДж-моль, рассчитанное по
изотопическому соотношению C. 6), в пределах погрешности этой величины согласуется с принятым, однако
менее точно, поскольку известны только приближенные значения нулевых колебательных энергий
молекул D2O2 и Н2О2.
HD (г). Термодинамические свойства газообразного протодейтерия в стандартном состоянии
при температурах 100—20 000 К приведены в табл. 32.
Молекулярные постоянные HD определялись при исследованиях систем полос СШ—Хг"Е и
В1!,— ХХ2+ [2119, 2639, 1553, 3600], спектра комбинационного рассеяния [3635, 3542] и
инфракрасного спектра [1905, 1348, 747J. Наиболее полные данные для нижних колебательных уровней
получены в работах Дьюри и Герцберга [1348] и Стойчева [3542] (соответствующие значения
постоянных приведены в табл. 6.1), для более высоких колебательных уровней (до у=15) — в работе
Танака и др. [3600]. Однако во всех этих исследованиях наблюдались переходы на уровни с
низкими значениями / и поэтому найденные молекулярные постоянные не могут позволить точно
определять число и энергию уровней с высокими значениями /. Следует отметить, что
рассчитанные для настоящего справочника уровни HD лучше согласуются с экспериментальными данными
[1348, 3600], чем значения G0(v), полученные в [2290].
Термодинамические функции HD (г) были вычислены без учета возбужденных электронных
состояний этой молекулы. Статистическая сумма по колебательно-вращательным уровням энергии
для основного состояния X1!,+ и ее производные по температуре вычислялись по уравнениям
*A. 73)—A. 75) непосредственным суммированием по уровням энергии, полученным при подготовке
справочника1 численным интегрированием уравнения Шредингера по программе [52] с
потенциальной функцией Колоса и Вольневича [2290, 2289] при учете релятивистской и
адиабатической поправок [2288]. Аналогичный расчет проводился ранее Колосом и Вольневичем [2289]
и Ле Роем. Однако в работе [2289] приведены только значения G0(v) и В„ а расчет Ле Роя
известен по статье [2386], где отсутствуют численные данные.
Погрешности вычисленных значений термодинамических функций HD (г) при Т ^ 6000 К
не превышают 0,02 Дж-К~1-моль~1 и обусловлены неточностью фундаментальных постоянных,
а выше 5000 К — приближенным учетом квазисвязанных колебательно-вращательных уровней.
Выше 10 000 К становятся существенными погрешности, обусловленные пренебрежением в
расчете ридберговскими состояниями HD. Общая погрешность в значении Ф° B0 000 К) составляет
0,1 Дж-К-^моль-1.
Термодинамические функции HD (г) вычислялись в работах [3755, 2421, 4000, 2148] для
невысоких температур, а также в [2296] и в справочниках [88, 337, 1783]2. Данные [2296, 337, 88]
хорошо согласуются с приведенными в табл. 32, хотя при температурах около 6000 К
расхождения в S°(T) и С(Т) достигают 0,3 Дж-К~1-моль~1 из-за приближенного учета уровней
1 Соответствующая таблица будет опубликована отдельно.
а Примечание при корректуре. В 1977 г. авторы работы [2095] рассчитали функции Б.2(г) и HD(r)
непосредственным суммированием с использованием некоторых упрощений. При удовлетворительном согласии этих
данных с данными настоящего справочника в Ф°(Г) расхождения в S°(T) и С°р(Т) достигают 0,1 и
.
135
HDO (г) Глава 6
с высокими значениями / в [88, 337]. Данные [2296] отличаются от данных табл. 32 примерно
на 0,3 Дж-К~1-моль~1 в значениях Ф°(Т) и S°(T), по-видимому, из-за какой-то ошибки.
Константы равновесия реакции HD (г)=Н (r)+D (г) вычислялись на основании принятого
значения
Arff°@)=:D0(HD) = 36 405,8 + 0,4 см = 435,510 + 0,005 кДж • моль.
Это значение энергии диссоциации было получено Герцбергом [1910] по границе континуума,
соответствующего диссоциации HD на один атом в основном (Is25), а на другой — в
возбужденном Bs2S) состоянии. Найденной границе соответствуют два значения, в зависимости от того,
какой атом, Н или D, образуется в возбужденном состоянии C6 405,8 и 36 406,6 см соответственно).
В справочнике в согласии с рекомендацией [1910] принимается меньшее значение. Квантовомеха-
нические расчеты [2290, 833] привели к близким величинам C6 405,2 и 36 405,5 см). К
практически совпадающему значению C6 406,0+0,6 см) приводит расчет по изотопическим
соотношениям C.6).
Принятой энергии диссоциации HD соответствует
AfH0(BD, г, 0) = 0,331 +0,008 кДж • моль.
HDO (г). Термодинамические свойства газообразного оксида протодейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 33.
Молекулярные постоянные, использованные в расчете термодинамических функций HDO,
приведены в табл. 6.4. Молекула HDO в основном электронном состоянии имеет нелинейную
структуру (точечная группа симметрии С^. Колебательно-вращательный спектр поглощения HDO
исследовался в работах [475, 575, 1921, 574, 3151, 503, 502, 1920, 1558, 1559]. Ранние
исследования колебательно-вращательного спектра НЕЮ [3151, 503, 502, 1920, 1921], выполненные на
приборах с низкой дисперсией, рассмотрены Герцбергом [69]. Колебательные постоянные HDO
определялись в работах [2421, 1544, 575]. В сводках [1571, 570] приведены энергии колебательных
термов, переходы на которые наблюдались в спектрах. В работах [279, 170, 2957, 1813, 169] на
основании различных моделей силового поля и экспериментальных данных [575] для молекул
Н2О, D2O, HDO были вычислены частоты колебаний, постоянные ангармоничности и
колебательно-вращательного взаимодействия молекулы HDO. Колебательные постоянные HDO,
приведенные в таблице, получены Хачкурузовым [279] на основании значений 13 уровней
колебательной энергии НЕЮ, рекомендованных в работе [280] по результатам анализа вращательной
структуры полос HDO в работах [475, 575, 574], и значений колебательных постоянных Н2О,
принятых в настоящем издании. Найденные позднее в работах [1558, 1559] волновые числа
начала полосы v2 хорошо согласуются со значениями постоянных, приведенными в табл. 6.4.
Приведенные в этой таблице вращательные постоянные и постоянные центробежного искажения
вычислены авторами настоящего справочника на основании 22 постоянных соответствующего
гамильтониана Ватсона, определенных Де Лучия и др. [1209] по значениям 53 частот вращательных
переходов в основном колебательном состоянии HDO, найденных при изучении инфракрасного-
и микроволнового спектров HDO в работах [3711, 3551, 3550, 2111, 550, 1415, 1208, 3070, 3912,
3721, 3069, 3646, 679, 3814, 3506, 557]. Соответствующие пересчеты были выполнены по
методике, описанной в работах [1041, 1706]. Значения постоянных колебательно-вращательного
взаимодействия, приведенные в табл. 6.4, получены Бенедиктом и др. [575] по вращательным
постоянным HDO в девяти колебательных состояниях х.
Термодинамические функции HDO (г), приведенные в табл. 33, вычислены по уравнениям
A. 3)—A. 6), A. 9), A.10), A.122)—A.124), A.127), A.128) с учетом ангармоничности колебаник
и взаимодействия вращения и колебаний (уравнения A.131)—A.133)), низкотемпературных
поправок (уравнения A.147)—A.149)), центробежного растяжения (уравнения A.140)—A. 142)) и
резонанса Ферми между колебательными состояниями (vx, v2, v3) и (ух—1, vz+2, v3) (уравнения A. 153)—
A. 155)). Погрешности вычисленных термодинамических функций HDO (г) достигают 0,05, 0,3
и 1,5 Дж -К-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К. Они обусловлены неточностью
принятых молекулярных постоянных, выше 3000 К к ним добавляются погрешности из-за
применения приближенной методики расчета.
1 Вращательные постоянные HDO в состояниях 000 и 010, найденные позднее в работе [1559], хорошо согласуются
с постоянными, приведенными в табл. 6.4.
136
Дейтерий, тритий и их соединения HDO, (г>>
Ранее термодинамические функции HDO(r) вычислялись Либби [2421, 2422] (Ф°(Т) при Т=
=298,15—773,15 К), Фридманом и Харом [1544], Абрамовицем и др. [337] и в первом [87Г
и втором [88] изданиях настоящего справочника.
Авторы работы [337] приводят термодинамические функции HDO(r) по работе [1544],
экстраполируя их до 6000 К. Расхождения между данными [1544, 337] и табл. 33 не превышают 0,4
и 1 Дж-К-моль в значениях Ф°(Т) и S°(T) соответственно. Примерно такую же величину
имеют расхождения между данными, приведенными в справочнике [87] и в табл. 33. Существенно
больше (из-за неточности учета центробежного растяжения) расхождения с данными,
приведенными в справочнике [88]. Они достигают 1,5 и 4,5 Дж-К^-моль в значениях Ф° F000 К) и
S° F000 К).
Константа равновесия реакции HDO (г)=О (г)+Н (r)+D (г) вычислена на основании принятого
в справочнике значения
АгН° @) = Do (HDO) = 924,96 + 0,13 кДж • моль'1,
вычисленного по соотношению C.7) с использованием значений энергий атомизации молекул
Н2О и D2O и величин G @,0,0; Н2О)=4634,0 см, G @,0,0; HDO)=4031,8 см-1 и G @,0,0;
D2O)=3387,5 см. Принятому значению Do (HDO) соответствует
AfH°(RDO, г, 298,15 К) = — 245,27 + 0,16 кДж ¦ моль.
Это значение подтверждается результатами масс-спектрометрических исследований
равновесия реакции H2O(r)-f-D2O(r)=2 HDO(r), выполненных Фридманом, Шанейром [1545]
(—245,4 кДж-моль) и Пайпером, Ньюбери, Бартоном [3105] (—245,4 кДж-моль). Менее
точные значения были получены в результате исследований равновесия реакции Н2О (r)+HD(r) =
=HDO(r)+H2(r), проведенных пикнометрическим методом Фаркаш и Фаркашом [1439]
(—243,0 кДж-моль) и методом измерения микротермопроводности, разработанным Зюэссом
[3567] (-243,4 кДж-моль-1).
В литературе известны результаты определения энтальпии образования HDO (ж). Дёлеман,.
Ланге [1276], Скрипов, Повышев [235] и Дуер, Бертран [1336] калориметрически измерили
энтальпию реакции D2O (ж) с Н2О (ж), откуда было получено значение—290,17 + 0,07 кДж-моль.
Энтальпия испарения НЕЮ (ж) была найдена Уолом и Юри [3857].
Расчет по методу II закона приводит к значению А,Н°(ШH, ж, 298,15 К)=44,6 кДж-моль,
которому соответствует Д^/Г^НБО, г, 298,15 К) = —245,6 кДж-моль. Эта величина больше
вычисленной выше по соотношению C.7) и представляется менее надежной.
HDO2 (г). Термодинамические свойства газообразного диоксида протодейтерия в стандартном1
состоянии при температурах 100—6000 К приведены в табл. 34.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 6.5. Прямые измерения структурных параметров и вращательных постоянных HDO2
отсутствуют. По аналогии с Н2О2 и D2O2 в справочнике принимается, что в основном электронном
состоянии Х1Аг молекула HDO2 имеет неплоскую структуру (точечная группа симметрии Сг).
Значения вращательных постоянных, приведенные в табл. 6.5, рассчитаны в справочнике на основании
равновесных структурных параметров Н2О2, определенных в работе [290], и
колебательно-вращательных постоянных HDO2, приведенных в той же таблице и оцененных по приближенному
соотношению A.115а). Колебательные спектры паров HDO2 не исследовались. Однако при
изучении инфракрасного спектра молекулы D2O2 в газовой фазе [457] и изолированной в аргоновой
и азотной матрицах [2357] была обнаружена полоса с центром при 981 см, отнесенная к частоте
v6 молекулы HDO2. Кроме того, при исследовании спектра комбинационного рассеяния жидкой
смеси Н2О2, D2O2, HDO2 [1455] и инфракрасного спектра поглощения смеси Н2О2, D2O2, HDO2,
растворенной в СС14 [457], обнаружено, что спектр HDO2 аддитивно складывается из спектров
Н2О2 и D2O2. Это позволило Жигеру и Бейну [457] предположить, что значения основных частот
молекул HDO2, H2O2, D2O2 связаны простыми соотношениями. В работах [1631, 937, 3179] эти
соотношения были использованы для определения основных частот HDO2. Значения
колебательных постоянных HDO2, приведенные в табл. 6.5, включая постоянные ангармоничности, получены
на основании значений девяти силовых постоянных, вычисленных в работе [206] по принятым
в справочнике значениям структурных параметров и колебательных постоянных молекул Н2О2;
137
Т(г) Глава 6
и D2O2. Экспериментальные данные об уровнях энергии внутреннего вращения HDO2 неизвестны.
В работе [2541 ] при изучении микроволнового спектра смеси D2O2 и HDO2 обнаружены две серии
линий, одну из которых Хант и др. [2048] отнесли к переходам /, 0, 0, 4 -> /, 1, 0, 2 (/ ^ 1)
молекулы HDO2. В отличие от Н2О2 и D2O2 молекула HDO2 не обладает элементами симметрии
и поэтому для нее невозможно точное отделение общего вращения от внутреннего. Однако Хант
и др. [2048] показали, что уровни внутреннего вращения HDO2 с удовлетворительной точностью
могут быть вычислены без учета их связи с общим вращением, как собственные значения
уравнений E. 5), E. 6), в которых функции V (ср) и a (tp) определяются средними значениями
коэффициентов Vo, Vx, F2, V3 и a0, als a2 из значений соответствующих коэффициентов Н2О2 и D2O2.
Приведенные в табл. 6.5 значения этих постоянных, а также постоянных центробежного растяжения
(Dj, DK, Djk) рассчитаны, как средние арифметические из соответствующих постоянных,
принятых в справочнике для Н2О2 и D2O2.
Термодинамические функции HDO2 (г), приведенные в табл. 34, вычислены по уравнениям
A.3)—A.6), A.9), A.10), A.122)—A.124), A.127), A.128) с учетом поправок на ангармоничность
колебаний и колебательно-вращательное взаимодействие (уравнения A.131)—A.133)),
центробежное растяжение (уравнения A.140)—A.142)) и низкотемпературной поправки (уравнения
A.147)—A.149)). Составляющие термодинамических функций, обусловленные заторможенным
внутренним вращением, рассчитаны непосредственным суммированием по уровням энергии
внутреннего вращения. Погрешности вычисленных таким образом термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных и приближенным характером
метода расчета колебательно-вращательных составляющих. Для Ф°(Т) при 298,15, 3000 и 6000 К
они составляют 0,2, 1 и 3 Дж-К^-моль.
Ранее таблицы термодинамических функций HDO2(r) вычисляли Жигер и Лю [1631 ] (до
1500 К), авторы второго издания справочника [88] (до 6000 К) и Чао и др. [937] (до 1500 К).
Во всех работах они были вычислены в приближении «гармонический осциллятор — жесткий
ротатор» с учетом заторможенного внутреннего вращения по методу Питцера—Гуинна [3034, 88]
или непосредственным суммированием по уровням энергии [937]. Последнее, однако, было
выполнено в работе [937] менее точно, чем в настоящем издании, поскольку учитывались только уровни
внутреннего вращения с энергией до 15 500 см. Использование менее точных значений
постоянных и метода расчета, в частности пренебрежение центробежным растяжением молекул HDO2,
привело к тому, что расхождения между данными табл. 34 и работ [1631, 937] достигают в
значениях Ф°G1) 0,2 Дж -К -моль, а в значениях S°(T) 1,3 Дж-К -моль. Расхождения с
предыдущим изданием в значениях Ф°(Г) составляют от 0,35 Дж-К^-моль при 298,15 К до
1 Дж-К^-моль-1 при 6000 К.
Константа равновесия реакции диссоциации HDO2 (r)=2O (r)+H (r)+D (г) вычислена на
основании принятого в справочнике значения
АгН° @) = Do (HDO2) = 1063,8 + 0,6 кДж • моль.
Это значение было рассчитано по соотношению C. 7) с использованием энергий диссоциации
и колебательных постоянных Н2О2 и D2O2.
Принятому значению Do (HDO2) соответствует
A^HDOj, г, 0) = —134,393+ 0,6 кДж • моль.
Т (г). Термодинамические свойства газообразного одноатомного трития в стандартном
состоянии при температурах 100—20 000 К приведены в табл. 35.
Термодинамические функции Т (г) были вычислены по соотношениям A. 3)—A. 6), A. 9), A.10),
A. 23)—A. 25) с учетом только основного состояния 2S. Погрешности вычисленных значений
термодинамических функций при Т ^ 10 000 К определяются только неточностью физических
постоянных и не превышают 0,01 Дж-К^-моль в значениях Ф°(Г) и S°{T). При более высоких
температурах погрешности возрастают из-за того, что в расчетах QBa и ее производных не учитывались
ридберговские состояния атома Т, при 20 000 К они могут достигать 0,07 и 0,3 Дж-К^-моль
в значениях Ф°(Т) и S°(T). Раньше термодинамические функции одноатомного трития
вычислялись в предыдущем издании [88] (до 6000 К). Результаты обоих расчетов согласуются в пределах
0,01 Дж.-К^-моль-1.
138
.Дейтерий, тритий и их соединения
Энтальпия образования одноатомного трития
AfH°(T, г, 0) = 221,479 +0,006 кДж • моль
принята на основании значения энергии диссоциации молекулы Т2.
Т2 (г). Термодинамические свойства газообразного двухатомного трития в стандартном
состоянии при температурах от 100 до 20 000 К приведены в табл. 36.
Колебательно-вращательные уровни энергии Т2 в состоянии ХХ2 + были получены при
подготовке справочника : численным интегрированием уравнения Шредингера с потенциальной
функцией, полученной в квантовомеханическом расчете [2290, 2289], с учетом адиабатической и
релятивистской поправок [2288]. Раньше Колос и Вольневич [2290] в аналогичном расчете, но без
учета этих поправок, нашли для Т2 значения Д(? (y-f^) и Вг. Экспериментальные данные об
уровнях энергии и молекулярных постоянных Т2 в состоянии Z12+ отсутствовали до 1974 г.
В связи с этим в ряде работ (см. [88]) были выполнены оценки этих постоянных на основании
значений молекулярных постоянных Н2, HD и D2 и известных изотопических соотношений. После
того, как была завершена работа над этим разделом справочника, Эдуарде и др. [1387]
опубликовали результаты экспериментального исследования вращательного и колебательно-вращательного
спектров комбинационного рассеяния Т2. Найденные в работе [1387] значения постоянных
приведены в табл. 6.1. Однако эти постоянные не позволяют рассчитать энергии высоких колебательно-
вращательных уровней Т2 в состоянии Х*2 + и поэтому не были использованы для расчета
термодинамических функций Т2(г).
Термодинамические функции Т2(г) вычислены без учета возбужденных электронных
состояний молекулы Т2 для равновесной смеси орто- и паратрития2. Значения (?кол- вр, QK0X вр и <5КОЛ вр
для основного состояния Т2 были вычислены по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательно-вращательным уровням энергии Т2. Из найденных таким образом
значений In QBa для равновесной смеси орто- и паратрития вычиталась составляющая ядерного
спина (R In 4).
Погрешности вычисленных значений термодинамических функций при Т <^ 5000 К не
превышают 0,01 Дж-К-моль главным образом из-за неточности фундаментальных постоянных.
При более высоких температурах погрешности возрастают из-за приближенного учета
квазисвязанных колебательно-вращательных уровней состояния Х12+, а выше 10 000 К — из-за
пренебрежения ридберговскими состояниями Т2. При 20 000 К погрешности в значении Ф°(Г)
могут достигать 0,1 Дж-К^-моль. Термодинамические функции Т2 вычислялись в работах
[2421] (до 773,15 К), [2148, 2173а] (до 2000 К) и справочниках [88, 1783]. Приведенные в
предыдущем издании [88] значения термодинамических функций Т2, несмотря на использование
оцененных молекулярных постоянных, согласуются с данными табл. 36 в пределах 0,01 Дж-К^-моль,
за исключением значений S°(T) при Т ^> 4500 К. Расхождения в значениях S0 (Т) при Т >
>4500 К растут с температурой и достигают 0,15 Дж-К^-моль при 6000 К. Данные работы
[1783] отличаются от данных табл. 36 из-за использования приближенного метода расчета на
величины до 0,15 и 0,6 Дж-К^-моль в значениях Ф°(Т) и S°(T).
Молекулярный тритий в газообразном состоянии принят за стандартное состояние этого
элемента и в соответствии с этим
Д^ОГ,, г) = 0.
Константа равновесия реакции Т2(г)=2Т(г) вычислена на основании принятого значения
ДД° @) = Do (Т2) = 37 028,4 ± 1,0 см = 442,958 ± 0,012 кДж • моль.
Энергия диссоциации молекулы Т2 экспериментально не определялась. При подготовке
настоящего справочника значение Do (T2) было вычислено при расчете системы
колебательно-вращательных термов этой молекулы численным интегрированием уравнения Шредингера. Аналогичный
расчет энергий диссоциации Н2, HD и D2 привел к величинам, совпадающим с экспериментальными
данными в пределах 0,7 см. В работе [2290] было получено практически совпадающее значение
D0(T2)=37 028,2 см-1.
1 Соответствующая таблица будет опубликована отдельно.
2 О расчете термодинамических функций о-Т2 и п-Т2 см. ниже,
139
о-Т2 (г), те-Т2 (г), ОТ (г), ТаО (г) Глава б
о-Т2 (г). Термодинамические функции газообразного ортотрития в стандартном состоянии при
температурах 10—1000 К приведены в табл. Д. 7. Значения (?км яр и ее производных вычислялись-
по уравнениям типа E.1) непосредственным суммированием по уровням энергии основного
электронного состояния Т2 (см. выше). Погрешности величин, приведенных в табл. Д.7, не превышают
0,01 Дж-К^-моль и обусловлены в основном неточностями фундаментальных постоянных.
Ранее термодинамические функции о-Т2 (г) не публиковались.
*г-Т2 (г). Термодинамические функции газообразного паратрития в стандартном состоянии при
температурах 10—1000 К приведены в табл. Д.8. Значения (?кол вр и ее производных были вычислены
непосредственным суммированием по уровням энергии основного электронного состояния Т2.
(см. выше) по уравнениям типа E. 2). Погрешности вычисленных термодинамических функций
не превышают 0,01 Дж-К^-моль и обусловлены практически только неточностью
фундаментальных постоянных.
Термодинамические функции п-Т2 (г) вычислялись ранее в работе [1783], данные которой
и табл. Д.8 согласуются в пределах 0,01 Дж-К^-моль.
ОТ (г). Термодинамические свойства газообразного оксида трития в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 37.
Экспериментальные данные о молекулярных постоянных ОТ отсутствуют. По аналогии с
молекулой ОН в справочнике принято, что ОТ имеет 3 связанных валентных состояния.
Внутримолекулярная статистическая сумма и ее производные вычислялись по уравнениям A. 76)—A.78)
непосредственным суммированием по колебательно-вращательным уровням энергии состояний
Х2П, А2И и В2И. Энергии уровней рассчитывались с использованием соотношений A. 42) и A. 65)'
(для состояния Х2И) и A. 34) и A. 65) (для состояний Л22 и В2Ъ) со значениями постоянных Ykt
и Ао, приведенными в табл. 6.2. Приведенные в таблице значения постоянных Ykl, Ao и других
величин получены на основании изотопических соотношений и соответствующих постоянных
молекулы ОН. Мультиплетность уровней состояний А22 и -В22 и Л-удвоение в состоянии Х2П
учитывались статистическим весом 2. В расчетах для состояний ХЩ и Л22 учитывались все уровни
энергии, ограниченные предельными кривыми диссоциации этих состояний. В табл. 6.2
приведены значения коэффициентов а{ в уравнениях, аппроксимирующих эти кривые, а также значения
утах и ^ит, полученные по методикам, изложенным в разделе 1.4.
Несмотря на то, что спектр ОТ не исследовался, точность аппроксимации
колебательно-вращательных уровней этой молекулы в состояниях Х2И, Л22 и В2И уравнениями со значениями
коэффициентов Ykn полученными с помощью изотопических соотношений, достаточно велика. Поэтому
погрешности вычисленных термодинамических функций ОТ при температурах до 4500: К
не превышают 0,02 Дж-К^-моль. При 6000 К погрешности в значениях Ф°(Г) и S°(T) могут
достигать 0,05 и 0,1 Дж-К^-моль.
Термодинамические функции ОТ (г) вычислялись в работе [1782] и справочниках [88, 1783].,
Данные, приведенные в работах [1782, 1783], при Т > 3000 К отличаются от приведенных-
в табл. 37 из-за использования метода Майера и Гепперт-Майер. При 5000 К расхождения в Фс'
и S° достигают 0,1 и 0,4 Дж -К -моль. Расхождения между данными предыдущего [88] и
настоящего изданий не превышают 0,05 Дж-К^-моль во всем интервале температур, несмотря на
использование в справочнике [88] приближенного метода расчета.
Константа равновесия реакции ОТ(г) = О(г)+Т(г) вычислена по принятому значению
ДГЯ° @) = Do (ОТ) = 36 139 ± 17 см"^432,32 + 0,20 кДж • моль,
которое было получено на основании следующих величин: Do (OD)=35 924+15 см, G @, OD) =
= 1351,86 см и G @, ОТ) = 1136,22 см. Принятому значению энергии диссоциации
соответствует
AfH° (ОТ, г, 0) = 35,942 + 0,21 кДж • моль.
Т2О (г). Термодинамические свойства газообразного оксида дитрития в стандартном состоянии
при температурах 100—6000 К приведены в табл. 38.
Молекулярные постоянные, использованные в расчетах термодинамических функций Т2О,
приведены в табл. 6.4. Молекула Т2О в основном электронном состоянии имеет нелинейную
структуру (точечная группа симметрии С2с). Значения колебательных постоянных Т2О, приведенные
140
Дейтерий, тритий и их соединения НТ (г)
в табл. 6.4, были определены в работах [279, 280] на основании силовых постоянных молекулы
Н2О и константы ангармоничности D2O. Эти значения колебательных постоянных Т2О хорошо
согласуются с найденными в работах [3499, 367, 884] центрами полос v2, v3, v2+v3 и v1+v3 в
инфракрасном спектре Т2О. В работах [170, 169] были вычислены менее надежные значения частот
колебаний Т2О на основании данных [575] для колебательных постоянных Н2О и D2O и различных
моделей силового поля. Приведенные в табл. 6.4 значения вращательных постоянных и
постоянных центробежного растяжения молекулы Т2О вычислены при подготовке справочника 1 по
значениям 22 постоянных соответствующего гамильтониана, определенных Де Лучия и др. [1212]
на основании весьма точных значений 51 частоты в микроволновом спектре Т2О [1212, 558].
Значения постоянных колебательно-вращательного взаимодействия а(, а?, af, a®, af, a°
вычислены по изотопическому соотношению A.115а) и принятым в справочнике постоянным D2O.
Значения постоянных a?, af, а? вычислены из найденных Карпентером и др. [884] эффективных
значений вращательных постоянных Т2О в состояниях @00) и @10).
Термодинамические функции Т2О (г) были вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.127), A.128) с учетом ангармоничности колебаний и колебательно-вращатель-
лого взаимодействия — уравнения A.131)—A.133), центробежного растяжения — уравнения
A.143), A.144) и низкотемпературных поправок — уравнения A.147)—A.149). Погрешности
вычисленных значений термодинамических функций Т2О (г) обусловлены неточностью принятых
молекулярных постоянных, а выше 3000 К — применением приближенной модели расчета. Они
составляют 0,1, 0,5 и 2 Дж-К^-моль в значениях Ф° (Г) при 298,15, 3000 и 6000 К.
Термодинамические функции Т2О (г) ранее вычисляли Либби [2421, 2422] (Ф° (Т) при Т=
=298,15-^773,15 К), Фридман и Хар [1544] E0—5000 К, по методу, близкому к принятому в
настоящем издании) и авторы предыдущих изданий настоящего справочника [88, 87] по
модифицированному методу Гордона. В работах [2421, 2422] приняты неточные молекулярные постоянные
Т2О, вследствие чего расхождения этих данных и данных, приведенных в табл. 38, достигают
1,2 Дж-К^-моль при 773,15 К. Результаты расчетов Ф°(Т) в [87, 1544] согласуются с данными
табл. 38 в пределах 0,4 Дж-К^-моль. Расхождения с данными справочника [88] достигают
1,4 Дж-К-моль~1 в значениях этой функции и обусловлены использованием в предыдущем
издании обобщенного метода Вильсона для учета центробежного растяжения с постоянной р2.
Константа равновесия реакции Т2О (г) =О (г)+2Т(г) вычислена на основании принятого
значения
ДД° @) = Do (T2O) = 939,11 ±0,18 кДж • моль,
которое было получено по соотношению C.7) и значениям Do (D2O)=932,647+0,16 кДж-моль,
G @, 0, 0, D2O)=3387,5 + 5 см и G @, 0, 0, Т2О)=2847,4+5 см. Приведенному значению Do (Т2О)
соответствует
Д^О^О, г, 0) = —249,369+0,15 кДж • моль'1.
НТ (г). Термодинамические свойства газообразного прототрития в стандартном состоянии
яри температурах от 100 до 20 000 К приведены в табл. 39.
Термодинамические функции НТ (г) были вычислены без учета возбужденных состояний
молекулы НТ. Значения Qmi ^ для состояния ХХ2 + и ее производных по температуре находились
непосредственным суммированием по уравнениям A.73)—A.75) и колебательно-вращательным
уровням энергии, рассчитанным при подготовке настоящего издания 2 численным решением
уравнения Шредингера, так же как для Т2 и других изотопов этой молекулы. Экспериментальные
данные об уровнях энергии и молекулярных постоянных НТ отсутствуют; в некоторых работах
(см. [88]) они были оценены по изотопическим соотношениям и постоянным молекул Н2, HD nD2.
В работе [2290 ] значения AG (y+V2) и В„ для НТ были получены в результате квантовомехани-
ческого расчета.
Значения молекулярных постоянных НТ, приведенные в табл. 6.1, получены обработкой
принятых значений колебательно-вращательных уровней энергии с низкими значениями квантовых
-чисел v и /.
1 Соответствующие пересчеты были выполнены по методике, описанной в работах [1041, 1706].
С оответствующая таблица будет опубликована отдельно.
141
НТО (г) Глава &
Погрешности вычисленных значений термодинамических функций НТ при Т ^ 5000 К не
превышают 0,01 Дж-К^-моль и обусловлены только неточностью фундаментальных постоянных.
При Т > 5000 К погрешности возрастают из-за приближенного учета квазисвязанных
колебательно-вращательных уровней энергии, а выше 10 000 К — из-за того, что в расчете не
учитывались ридберговские состояния НТ. Общая погрешность в значении Ф° B0 000 К) может достигать
0,2 Дж-К^-моль.
Термодинамические функции НТ (г) вычислялись ранее в работе [2421] (до 773 К), [2148,
2173а] (до 2000 К) и справочниках [88, 1783]. Данные предыдущего издания [88] совпадают
с табл. 39 в пределах 0,01 Дж-К^-моль за исключением значений S° (Т) при Т > 5000 К,
где расхождения достигают 0,1 Дж-К^-моль при 6000 К. Данные [1783] отличаются от
приведенных в табл. 39 из-за применения приближенной методики расчета на величины до 0,15 и
0,6 Дж-К^-моль в значениях Ф°(Т) и S°(T).
Константа равновесия реакции НТ (г)=Н (г)+Т (г) вычислена на основании принятого в
справочнике значения
ДДо@) = Б0(НТ) = 36 511,9 + 1,0 см = 436,780 + 0,012 кДж • моль.
Это значение принято на основании расчета системы колебательно-вращательных термов этой
молекулы, выполненного при подготовке настоящего издания. Аналогичный расчет энергий
диссоциации Н2, HD и D2 привел к совпадению с экспериментальными данными в пределах 0,7 см.
Практически совпадающее значение C6 511,7 см) было получено в квантовомеханическом
расчете [2290] и в расчете по изотопическому соотношению C.6) и значению Do (D2). Принятой
энергии диссоциации соответствует
AfH°(HT, г, 0) = 0,733 ± 0,15 кДж • моль.
НТО (г). Термодинамические свойства газообразного оксида прототрития в стандартном
состоянии при температурах 100—6000 К приведены в табл. 40.
Молекулярные постоянные, использованные в расчетах термодинамических функций НТОГ
приведены в табл. 6.4. Молекула НТО в основном электронном состоянии имеет нелинейную
структуру (точечная группа симметрия Cs). Приведенные в табл. 6.4 значения колебательных
постоянных НТО были определены в работах [279, 280] на основании силовых постоянных молекулы Н2О
и констант ангармоничности молекулы HDO. Эти значения колебательных постоянных НТО*
хорошо согласуются с измеренными в работах [3499, 1447] волновыми числами полос vlr v2 и v3
молекулы НТО. Менее точные значения частот нормальных колебаний НТО были получены в
работах [170, 169] на основании колебательных постоянных Н2О и D2O и различных моделей силового
поля. Приведенные в табл. 6.4 значения вращательных постоянных и постоянных
центробежного растяжения молекулы НТО вычислены при подготовке справочника по значениям 22
постоянных соответствующего гамильтониана, найденным Хельмингером и др. [1877] на основании
значений 48 частот вращательных переходов в микроволновом спектре НТО [1877, 558]. Постоянные»
полученные Хельмингером и др. [1877], находятся в хорошем согласии со значениями
соответствующих постоянных, вычисленных Фейтом и Стенбикелиером [1447] из полученных ими данных
о вращательной структуре полос vx и v3 и результатов исследований микроволнового спектра НТО
в работе [558]. Приведенные в табл. 6.4 значения постоянных колебательно-вращательного
взаимодействия НТО вычислены по изотопическому соотношению A.115а) на основании принятых
в справочнике значений аналогичных постоянных HDO. Термодинамические функции НТО (г),
приведенные в табл.40, вычислены по уравнениямA.3)—A.6), A.9), A.10), A.122)—A.124), A.127),
A.128) с учетом ангармоничности колебаний и колебательно-вращательного взаимодействия —
уравнения A.131)—A.133), центробежного растяжения — уравнения A.143)—A.144) и
низкотемпературных поправок — уравнения A.147)—A.149). Погрешности вычисленных значений
термодинамических функций НТО (г) обусловлены неточностями молекулярных постоянных, а выше
3000 К — применением приближенной методики расчета. Они составляют 0,05, 0,3 и1,5Дж*
• К-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции НТО (г) вычисляли Либби [2421] (Ф° (Г) при Г=298,15-|-
-^-773,15 К), Фридман и Хар [1544] E0—5000 К) и авторы предыдущих изданий настоящего
справочника [87, 88]. Расчеты [2421] основаны на неточных значениях молекулярных постоянных
НТО, и расхождения в значениях Ф°(Т), приведенных в работе [2421 ] (с учетом исправлений
142
Дейтерий, тритий и их соединения DT (г), DTO (г>
в работе [2422]) и табл. 40, составляют 0,6 Дж-К~1'Моль~1 при 773,15 К. Результаты расчетов
[87, 1544] близки к данным табл. 40, расхождения в значениях Ф°(Г) становятся заметными выше
3000 К и достигают 0,5 Дж -К -моль при 6000 К. Расхождения с данными, приведенными в
справочнике [88], больше из-за особенностей учета в предыдущем издании центробежного растяжения
молекул Н2О и ее изотопов и, увеличиваясь с температурой, достигают 1,5 Дж-К~1-моль~1 в
значении Ф° F000 К).
Константа равновесия реакции НТО(г)=О(г)+Н(г)+Т(г) вычислена с использованием.
Дгй * @) = Do (НТО) = 928,05 + 0,16 кДж • моль.
Эта величина получена по соотношению C.7) на основании значений Do (D2O) = 932,647 +
+0,16 кДж-моль-1, G @, 0, 0, D2O)=3387,5+3 см и G @, 0, 0, НТО)=3771,4+5 см. Принятой
энергии диссоциации НТО соответствует
Д//°(НТО, г, 0) = —243,754 + 0,16 кДж • моль.
DT (г). Термодинамические свойства газообразного дейтеротрития в стандартном состоянии,
при температурах от 100 до 20 000 К приведены в табл. 41.
Значения термодинамических функций DT (г) были вычислены без учета возбужденных
состояний молекулы DT. Колебательно-вращательная статистическая сумма для состояния Х*2 *"
и ее производные по температуре рассчитаны непосредственным суммированием по уравнениям.
A.73)—A.75) и колебательно-вращательным уровням энергии, полученным при подготовке
настоящего издания1 численным решением уравнения Шредингера, так же как это было сделано для
D2 и других изотопических модификаций этой молекулы. Экспериментальные данные об уровнях
энергии и молекулярных постоянных DT в состоянии Х*2+ отсутствуют; в литературе (см. [88])
были выполнены оценки постоянных DT по изотопическим соотношениям и постоянным молекул
Н2, HD и D2. В работе [2290] значения AG (у-Ь1/^) и В, для DT были получены с помощью квантово-
механического расчета. Значения молекулярных постоянных DT, приведенные в табл. 6.1,
получены обработкой принятых значений колебательно-вращательных уровней энергии с низкими»
значениями квантовых чисел v и /.
Погрешности вычисленных значений термодинамических функций при Т ^ 5000 К не
превышают 0,01 Дж-К~1-моль~1, при Т > 5000 К они возрастают из-за тех же источников ошибок,,
которые определяют величину погрешностей термодинамических функций таких газов, как Ш%
или HD. Общие погрешности значений Ф° A0 000 К) и Ф° B0 000 К) не превышают 0,05 п
0,2 Дж-К~1-моль~1 соответственно.
Термодинамические функции DT(r) вычислялись ранее в работах [2421] (до 773 К), [2148,
2173а] (до 2000 К) и справочниках [88, 1783]. Данные предыдущего издания [88] согласуются
с табл. 41 в пределах 0,01 Дж-К~1-моль~1 за исключением значений S°(T) при Т >• 4500 К,
где расхождения достигают примерно 0,1 Дж-К~1-моль~1 при 6000 К. Так же как для других
изотопов водорода, расхождения данных [1783] и табл. 41 растут с температурой и при 5000 К
составляют 0,15, 0,6 и 1,5 Дж-К^-моль-1 в значениях Ф°(Г), S°(T) и Ср° (Т).
Константа равновесия реакции DT (r)=D (г)+Т (г) вычислена на основании принятого в
справочнике значения
Ar#°@) = D0(DT):= 36 881,2 + 1,0 см = 441,197 + 0,012 кДж • моль.
Это значение было получено при подготовке настоящего издания в результате расчета системы
колебательно-вращательных уровней энергии этой молекулы. Практически совпадающее значение
было получено в квантовомеханическом расчете [2290] C6 880,9 см).
Принятому значению Do (DT) соответствует
Д/Я°(БТ, г, 0) = 0,089 + 0,014 кДж • моль.
DTO (г). Термодинамические свойства газообразного оксида дейтеротрития в стандартном
состоянии при температурах 100—6000 К приведены в табл. 42.
Молекулярные постоянные, использованные в расчете термодинамических функций DTO,
приведены в табл. 6.4. Молекула DTO в основном электронном состоянии имеет нелинейную струк-
1 Соответствующая таблица будет опубликована отдельно.
143
DTO (г) Глава 6
туру (точечная группа симметрии С±). Приведенные в таблице значения колебательных
постоянных DTO были определены Хачкурузовым [279, 280] на основании значений силовых постоянных
молекул Н2О и D2O и констант ангармоничности молекулы HDO. Эти значения колебательных
лостоянных согласуются с центром полосы v3 DTO, найденным в работе [3499]. Менее точные
значения частот нормальных колебаний DTO были вычислены в работах [170, 169] на основании
различных моделей силового поля и частот молекул Н3О и D2O. Приведенные в табл. 6.4
вращательные постоянные и постоянные центробежного растяжения DTO вычислены авторами
справочника, исходя из значений 22 постоянных соответствующего гамильтониана, найденных Хельмин-
гером и др. [1877] по значениям 41 частоты в микроволновом спектре DTO [1877]. Постоянные
колебательно-вращательного взаимодействия DTO, приведенные в таблице, вычислены по
изотопическому соотношению A.115а) и принятым постоянным колебательно-вращательного
взаимодействия молекулы HDO [575].
Термодинамические функции DTO (г), приведенные в табл. 42, вычислены по уравнениям
A.3)—A.6), A.9), A.10), A.122)—A.124), A.127) и A.128) с учетом ангармоничности колебаний
и колебательно-вращательного взаимодействия — уравнения A.131) —A.133), центробежного
растяжения — уравнения A.143), A.144) и низкотемпературных поправок — уравнения A.147)—
A.149). Погрешности табулированных значений термодинамических функций обусловлены
неточностью принятых молекулярных постоянных, а выше 3000 К и применением приближенной
методики расчета. Они составляют 0,1, 0,5 и 2 Дж-К~1-моль~1 в значениях Ф°(Т) при 298,15,
3000 и 6000 К.
Ранее термодинамические функции DTO (г) вычислялись Либби [2421, 2422] (Ф° (Т) при
298,15—773,15 К), Фридманом и Харом [1544] E0—5000 К) и в предыдущих изданиях настоящего
справочника [88, 87]. Расчеты Либби [2421, 2422] выполнены по неточным значениям
молекулярных постоянных DTO; расхождения в значениях Ф°(Г), приведенных в [2421, 2422] и табл. 42,
составляют 1 Дж-К~1-моль~1. Расхождения в значениях Ф°(Г), приведенных в [87, 1544]
и в табл. 42, изменяются от 0,01 до 0,4 Дж-К~1-моль~1 при температурах от 298,15 до 6000 К
и обусловлены различиями в значениях молекулярных постоянных и методах расчета.
Расхождения с данными [88] при 6000 К достигают 1,5 Дж-К~1-моль~1 из-за некорректного учета
центробежного растяжения DTO в справочнике [88].
Константа равновесия реакции DTO (r)=O (r)+D (г)+Т (г) вычислена на основании
принятого в настоящем справочнике значения
АгН° @) = Do (DTO) == 935,77 + 0,20 кДж • моль.
Эта величина была получена по соотношению C.7) и значениям D0(D2O) = 932,647 +
+0,16 кДж -моль, G@, 0, 0, D2O)=3387,5+3 см и G @, 0, 0, DTO)=3126,5 + 10cm-x.
Приведенному значению Do (DTO) соответствует
AfH°(DTO, г, 0) = —247,721+ 0,20 кДж ¦ моль.
Глава 7
ФТОР И ЕГО СОЕДИНЕНИЯ
F, F-, F2, FO, F2O, HF, H2F2, H3FS, H4F4, H6F5,
H6F6, H7F7, HOF, DF, TF
В настоящей главе рассматриваются соединения фтора с кислородом, водородом и его изотопами
в газообразном состоянии, всего 15 веществ. Соединения фтора с другими элементами будут
обсуждены в последующих главах. Для четырех газов (F, F~, F2 и HF) таблицы термодинамических
свойств рассчитаны в справочнике в интервале температур от 100 до 10 000 К; для полиморфных
соединений фтористого водорода, нестабильных при высоких температурах, — для интервала
температур до 3000 К, соответствующие таблицы термодинамических функций приведены в
Дополнении (наст, том, кн. 2); для остальных газов (FO, F2O, HOF, DF и TF) — от 100 до 6000 К.
Для трех газов (F2, F2O и HF) в Приложении 3 приведены данные, позволяющие оценить
влияние межмолекулярного взаимодействия на свойства этих газов при повышенных давлениях. Ионы
F+, F2, F7, FQ~, а также такие соединения, как
FOa и F2O2, не рассматриваются в справочнике,
так как в любых химических системах при
температурах выше 298,15 К их равновесные
концентрации пренебрежимо малы по сравнению с
рассмотренными веществами.
F (г). Термодинамические свойства
газообразного одноатомного фтора в стандартном
состоянии при температурах от 100 до 10 000 К
приведены в табл. 43.
Термодинамические функции F (г) были
вычислены с учетом двух компонент основного
состояния 2Р атома F, приведенных в табл. 7.1. Энергия верхней компоненты 2Pi/2 принята по работе
Лидена [2429], где она была уточнена по сравнению с рекомендацией Мур [2697]. При
температурах до 10 000 К погрешности в значениях термодинамических функций F (г), приведенных
в табл. 43, определяются практически только неточностью фундаментальных постоянных (прежде
всего газовой постоянной R) и не превышают 0,02 Дж-К~1-моль~1.
Термодинамические функции F (г) вычислялись в десятках работ. Среди старых расчетов можно
отметить работы [1013, 1673, 2029, 43], из более новых — справочники [3564, 2496, 2556,
1946], таблицы JANAF [2095] и предыдущие издания настоящего справочника [87, 88].
Результаты всех расчетов, кроме проведенных в работах [43, 1013, 2291], согласуются между собой и
с данными табл. 43 в пределах 0,01 Дж-К~1«моль~1 в значениях Ф°(Т) и S°(T) во всем интервале
температур, а имеющиеся расхождения обусловлены некоторым различием фундаментальных
постоянных. Данные, приведенные в работах [43,1013, 2291 ], отличаются от приведенных в табл. 43
на 0,1—0,05 Дж-К~1-моль~1, по-видимому, из-за ошибок в этих расчетах.
Энтальпия образования одноатомного фтора на основании рекомендации КОДАТА—МСНС
[1000] принята равной
bfH°(F, г, 298,15-К) = 79,38^0,30 кДж • моль.
Таблица 7.1. Уровни энергии
и иона F~
Атом
F
F"
Электрон-
ная
конфигурация
Состояние
Is22s22p5 *Р,и
Is22sa2j95 2Р^
Is22s22pe *S
атома F
CM
0
404,1
0
Pi
4
2
1
Она основана на значении энергии диссоциации молекулы F
2.
F~ (г). Термодинамические свойства газообразного отрицательного иона одноатомного фтора
в стандартном состоянии при температурах от 100 до 10 000 К приведены в табл. 44.
Термодинамические функции F~ (г) были вычислены по соотношениям A.9) A.10), т. е. при
условии, что In <?вн=0, поскольку ион F~ имеет основное состояние Х5 с конфигурацией ls^s^p"
и не имеет связанных возбужденных состояний. Во всем интервале температур погрешности в
значениях термодинамических функций F~ определяются только неточностью фундаментальных по-
145
F2 (г) Глава 7
стоянных и не превышают 0,02 Дж-К~1-моль~1 в значениях Ф°(Т) и S°(T). Термодинамические
функции F" ранее вычислялись в справочниках [2029], JANAF [2095] и [87, 88]. Все расчеты
согласуются между собой и с данными табл. 44 в пределах 0,01 Дж«К~1-моль~1 в значениях Ф°(Т)
и S°(T).
Константа равновесия реакции F~ (r)=F(r)+e(r) вычислена с использованием значения
Дг#°@)=328,01+0,18 кДж-моль в соответствии с принятым в справочнике сродством атома
фтора к электрону
i40(F) = — 27 420 + 15 см = —328,01 +0,18 кДж • моль.
Это значение A0(F) получено в работе Поппа [3064] в результате определения длины волны
границы континуума, возникающего при резонансном захвате электрона атомом фтора.
Совпадающие в пределах точности измерений значения найдены в работе Мильстена и Берри [26731
по границе фотоотрыва электрона от иона F (—27 397+15 см или —327,74+0,18 кДж-моль),
а также авторами работы [3339] на основании изучения сечения отрыва электрона от ионов F~
и Н~ C,39+0,5 эВ или 327+48 кДж-моль). Более ранние и менее точные измерения методом
поверхностной ионизации [27, 456] и магнетронным методом [606] приводили к величинам,
завышенным на 6—10 кДж'Моль. В работе Берри и Рейманд [611] была допущена ошибка (см.
[2673]). Принятому значению AQ(F) соответствует
AfH°(F, г, 0) = —250,735+ 0,27 кДж • моль.
F2 (г). Термодинамические свойства газообразного двухатомного фтора в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 45.
Молекула F2, так же как и молекулы других галогенов, имеет основное электронное состояние
Х2+ (с конфигурацией электронной оболочки . . .За^1тс*1тгр. Сведения о возбужденных
электронных состояниях F2, в том числе нижних с конфигурацией электронной оболочки . . .За^1 t*l ^Заи,
весьма ограничены, так как до настоящего времени в спектрах изучены только переходы,
связанные с высокими валентными и ридберговскими состояниями (с энергией больше 90 000 см, см.
[1008]). Теоретический расчет возбужденных электронных состояний, выполненный Хеем и Карт-
райтом [1862а] с учетом конфигурационного взаимодействия, показал, что все возбужденные
валентные электронные состояния F2, коррелирующие с диссоциационными пределами FBP^2t i/2) +
-\-Ф(*Рз12,Ч2), являются отталкивательными состояниями, а связанные валентные состояния,
коррелирующие с пределами F+ CP, XD, 1S)-\-?~ A5), имеют энергии свыше 90 000 см. К такому же
результату привели более ранние полуэмпирические расчеты [1932, 1933, 1934] г.
В течение длительного времени информация о молекулярных постоянных F2 ограничивалась
результатами анализа вращательной структуры спектра комбинационного рассеяния F2 в работе
[397], где были найдены значения (в см) #„=0,8828+0,0010 и Д&/2=891,85+0,4. В ряде работ
эти данные были использованы для оценки остальных молекулярных постоянных и для
последующих расчетов термодинамических функций F2. В более поздних исследованиях спектра
комбинационного рассеяния газообразного фтора [970], а также инфракрасного спектра и спектра
комбинационного рассеяния жидкого фтора были уточнены значения Во и Д&/2. В 1972 г. Ди Лонардо
и Дуглас [1255] опубликовали предварительные результаты исследования электронного спектра
поглощения F2 на приборе с высоким разрешением. В результате анализа переходов с уровней
v"=0, 1 и 2 на ряд уровней ридберговского состояния были найдены значения (в см) ДСу2=
=893,95+0,10; Д&/з=870,05+0,10; ?„=0,8827+0,0010 и ^=0,0131+0,0005.
Приведенные в табл. 7.2 колебательные и вращательные постоянные F2 вычислены по значениям G @),
G A), G B) и Во, Вх и В2, найденным в работе [1255]. Там же приведено значение Do, найденное
в этой работе. Позднее Колбурн и др. [1008] провели тщательное исследование
спектров испускания и поглощения F2 в вакуумном ультрафиолете. При этом был
выполнен анализ переходов, связанных с колебательными уровнями y=0-f-22 основного состояния Х1^
и определены значения G (v), Bt и D, для всех этих уровней. Соответствующие значения G (v) и
Рис [3182] на основании анализа сплошного спектра F2 в видимой и ультрафиолетовой области высказал
предположение, что состояние 3П . с конфигурацией Зо|1тг^1т:3 3оя может иметь неглубокий минимум при г — 1,8-f-
4-1,9 А. Однако согласно результатам расчетов [1862а] эти спектры связаны с переходами в отталкпвательные-
состояния 3ПИ и 1ПМ.
146
Фтор и его соединения
F2(r)
Таблица 7.2. Молекулярные постоянные F2, FO, HF, DF и TF
Молекула
F2
FO
HF
DF
TF
Состояние
z2na
xw
Те
О О О О О
917,55
1060 б
4138,73
2998,19
2508,5
Примечание. Ниже все постоянные даны в см"
FO: а А =—178; б вычислено при AG1/s=1029,
г вычислено по .соотношению A.68).
HF: а иеуе = 0,932; б а2 = 1,182 • 10~2; bPj = 6
см
11,825
15,5 б
90,05 a
45,76
32,5
D0(FO) =
23-Ю-6; Ь
Вв
-1
0,8902
1,104
20,9555
11,000
7,694
а-102
1,37
0,97 в
79,58 б
29,07
17,6
= 18 000; в вычислено по
ге = 1,68-10-7.
De • 10»
3,3
5Г
2153 в
590
250
Те
А
1,412 7
1,321
0,916 80
0,918 7
0,917 0
соотношению A.69);
Таблица 7.3. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см"),
а также значения vm&% и г/цш, принятые для расчета термодинамических функций
F2, FO, HF, DF и TF
Коэффициенты
(ао=
Те
V
Х7"
Г«
По
у
у
хг
"V
¦ут"
у
а1
а2
а3
• 10~3
•Ю-2
• 101
¦ 103
•ю4
• 104
• 10е
101
102
104
10в
• Ю7
•ю8
• 10е
.10*
• 10
• 105
• 1010
V
F2
Х*Ц
0
0,917 55 а
—0,118 25 а
—
—
—
—
—
.
0,890 2 а
—0,137 а
—
—
—0,033
1,337 1
0,152 477
0,813 142
-_0,704 395
23
178
FO
хт
0
1,060
—0,155
—
—
—
—
—
1,104
—0,097
—
—
—
—
-0,05
1,851 95
0,117 479
0,848 106
—0,304 291
34
179
HF
0
4,138 92
—0,904 808
1,167 54
—0,733 439
7,242 08
-5,689 40
0,221 844
—0,394 921
20,990 262
—8,867 60
7,534 45
—192,868
284,488
—2256,52
0,901 276
—0,144 916
—21,17
4,938 0
0,765 671
43,991 9
—107,542
20
68
a Приводимые постоянные использованы только для построения предельной
Расчет
и
0
1
2
3
4
5
6
7
термодинамических функции проводился ]
G(v)-G @)
0
893,90
1 764,15
2 610,22
3 431,53
4 227,43
4 997,19
5 740,05
В,
0,8833
0,8696
0,8560
0,8423
0,8284
0,8142
0,7996
0,7844
V
G
8
9
10
11
12
13
14
15
то следующим значениям (
(v) - G @)
в.
6 455,17 0,7685
7 141,63 0,7518
7 798,48 0,7343
8 424,67 0,7156
9 019,11 0,6958
9 580,63 0,6747
10108,02 0,6522
10 599,62 0,6282
DF
0
3,001
—0,487
0,815
-0,510
1,948
-0,342
11,003
—3,036
0,384
—1,759
0,916
—3,104
—5,88
4,938
0,457
7,728
21,966
28
93
+
87
953
953
903
20
301
8
2
95
3
87
4
0
666
05
6
TF
0
2,510
—0,341
0,477
89
390
502
—0,250 083
0,797
-0,117
7,698
—1,776
0,188
658
227
64
81
430
—0,720 315
0,313
—0,889
—2,88
4,938
0,337
3,273
11,049
34
111
кривой диссоциации.
?0 (у) и Ь
V
16
17
18
19
20
21
22
23
',:
G
И
11
11
11
12
12
12
12
12
-G@)
053,90
468,96
842,62
172,25
452,98
678,00
830,38
909
998
269
0
497
20
1
в.
0,6025
0,5750
0,5449
0,5094
0,4711
0,4185
0,3365
0,254
F2 (г) Глава 7
В, приведены в табл. 7.3. Точность найденных значений G (v) лучше 0,5 см, погрешности
определения Dv существенно больше, о чем свидетельствует хаотическое изменение значений D, с
ростом V.
Термодинамические функции F2(r), приведенные в табл. 45, были рассчитаны без учета
возбужденных электронных состояний F2. Значения Q^\ вр и ее производных вычислялись
непосредственным суммированием по колебательно-вращательным уровням энергии состояния ХХ2+
по уравнениям A. 73)—A.75). Колебательно-вращательные уровни находились по уравнению
G0(v)+BvJ {J+l)j[i-\-DaJ{J+i)jB,f. Значения G0(v) и В, для у=0-^22 приняты по работе
[1008], значения G0(v) и Bt для утах=23 получены экстраполяцией этих данных (см. табл. 7.3).
В расчете учитывались все колебательно-вращательные уровни, ограниченные предельной кривой
диссоциации состояния Х12+. В табл. 7.3 приведены значения коэффициентов а{ в уравнении,
аппроксимирующем эту кривую, а также значение /llm.
Погрешности вычисленных таким образом значений термодинамических функций F2 при Т =^
^ 2000 К определяются главным образом неточностью фундаментальных постоянных и не
превышают 0,02 Дж-К~1-моль~1 в значениях Ф°(Т) и S°(T). С ростом температуры погрешности
возрастают из-за отсутствия экспериментальных данных об энергии колебательно-вращательных
уровней с большими значениями квантового числа / и использования приближенного метода
расчета предельной кривой диссоциации (для F2 это существенно уже при Т— 4000 К из-за низкой
энергии диссоциации молекулы). Погрешности в табулированных значениях Ф°(Г) при 3000, 6000
и 10 000 К могут быть оценены в 0,05, 0,1 и 0,5 Дж«К~1-моль~1. Погрешности из-за
пренебрежения быстрым изменением постоянной D, при v > 18 существенно меньше этих величин.
Термодинамические функции F2 (г) вычислялись многими авторами [1426, 43, 3072, 3946,
2556, 1308, 2496, 1454, 1013, 125, 2029, 337], в таблицах JANAF [2095] и предыдущих изданиях
справочника [87, 88]. Все расчеты, кроме [1454, 87, 88], проводились по методу Майера и Геп-
перт-Майер или в приближении «жесткий ротатор—гармонический осциллятор». Поскольку
молекула F2 имеет низкую энергию диссоциации, пренебрежение в этих расчетах необходимостью
ограничения суммирования по колебательно-вращательным уровням энергии привело к ошибкам,
превышающим 0,1 Дж-К~1-моль~1 в значениях Ф°(Т) уже при 2000 К и достигающим 1,2, 5 и
13 Дж-К-^моль в Ф°F000 К), ?°F000 К) и С°F000 К) соответственно в таких изданиях, как
[2095, 2556, 337 ]J. В большинстве работ были использованы неточные значения постоянных
Fz, основанные на данных [397] и приближенных оценках. Однако погрешности из-за неточности
постоянных, как правило, примерно на порядок меньше ошибок, обусловленных неточностью
применявшихся методов расчета. В работе Херрика и Фебера [1454] расчет был выполнен по
методике, практически аналогичной принятой в настоящем издании. Соответствующие расхождения,
достигающие 0,4 и 0,7 Дж-К~1-моль~1 в значениях Ф°C000 К) и Ф° F000 К), практически
целиком обусловлены различием молекулярных постоянных F2, использованных в расчетах.
В [87, 88] расчет был выполнен по модифицированному методу Гордона и Барнес, соответствующие
расхождения составляют 0,4 и 2 Дж-К~1.моль~1 в значениях Ф°(Т) при 3000 и 6000 К.
В справочнике принято, что двухатомный газообразный фтор является стандартным состоянием
этого элемента и в соответствии с этим
A/fl'°(F2, г) = 0.
Константа равновесия реакции F2(r)=2F(r) вычислена на основании энергии диссоциации
ДД° @) = Do (F2) = 12 920 + 50 см = 154,55 + 0,60 кДж • моль,
соответствующей энтальпии образования атомарного фтора при 298,15 К, рекомендованной
КОДАТА—МСНС [1000] на основании критического анализа результатов измерений энергий
диссоциации F2, HF и энтальпии образования HF(r).
Энергия диссоциации F2 в течение многих лет была предметом экспериментальных
исследований в десятках работ и противоречивых дискуссий, продолжавшихся вплоть до 1972 г. Наиболее
надежные значения D0(F2) были получены в работах Колбурна и др. [1008] на основании
короткой экстраполяции (на 90 см) колебательных уровней состояния Х12+ A2 920+50 см), Берко-
1 Примечание при корректуре. Результаты расчета JANAF в 1977 г. практически не изменились по
сравнению с расчетом [2095].
148
Фтор и его соединения FO (г)
вица и др. [596] на основании фотоионизационных измерений A53,4 + 1,0 кДж-моль), Дошера
[1277, 1278] в результате измерений давления диссоциации F2 A53,6 + 1,0 кДЖ'МОль) и Уайза
[3976, 3977] на основании эффузионных измерений A56+4 кДж-моль). С ними
удовлетворительно согласуются данные работ [488] (расчет по измеренным энергиям диссоциации фторидов
металлов), [3938] (измерения давления адиабатических взрывов смесей, содержащих F3), [1190]
(измерения методом электронного удара), [2059] (анализ непрерывного спектра поглощения F2).
Менее надежные, в ряде случаев просто неверные значения были получены в более старых работах
[1880, 3500, 3888, 2372, 683, 975, 1221, 3858, 3341, 3342, 914, 915, 3935, 3937, 4003, 1643, 2672].
В работах Дайблера и др. [1240, 1239, 1241 ] при изучении фотоионизации F2 и HF было получено
существенно более низкое значение, однако авторы работ [596, 79] показали, что оно обусловлено
ошибкой в экстраполяции кривых фотоионизации.
Энергия диссоциации фтора непосредственно связана с энергией диссоциации HF и энтальпией
образования HF(r). Результаты измерений этих двух величин (см. ниже) приводят к значению
D0(F2) хорошо согласующемуся с данными работ [1008, 596, 1278, 3977].
FO (г). Термодинамические свойства газообразного фторида кислорода в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 46.
Значения термодинамических функций FO(r) вычислены на основании молекулярных
постоянных, приведенных в табл. 7.2. Электронный спектр молекулы FO до настоящего времени не
изучен, а теоретические расчеты валентных электронных состояний FO не проводились. По аналогии
с изоэлектронными молекулами О^ и СЮ и на основании квантовомеханического расчета [2897]
в справочнике принято, что основным состоянием FO является состояние 2П с конфигурацией
За21тс42тс3, а возбужденные электронные состояния с конфигурациями Зо21п32тс4 и За22-гс42тс24а
имеют энергию возбуждения не ниже 25 000 см и небольшие минимумы на потенциалах кривых
или являются отталкивательными состояниями. Поэтому они могут не учитываться при расчете
термодинамических функций FO.
Спектр FO исследовался только в матрицах инертных газов при фотолизе F2O. Результаты
исследований инфракрасного спектра FO [414, 415, 392] и спектра комбинационного рассеяния [390]
привели к практически совпадающему значению для основной частоты (Д&/2=1028,7+0,2 см).
Квантовомеханический расчет [2897] дал существенно более высокое значение (ше=1211 см,
wexe=5,5i см и AGi/^al20l см) и межъядерное расстояние ге=1,321 А. Приведенные в табл. 7.2
значения молекулярных постоянных FO получены на основании значений Д6?1/2=1029 см,
D0(FO) = 18000 см (см. ниже) и ге=1,321 А. Поскольку основная частота FO найдена по спектру
в матрице, ее погрешность может быть оценена в 10 см, а погрешность в величине ге в 0,03 А.
Постоянная спин-орбитальной связи в состоянии Х2П оценена равной —178+35 см.
Значения QW_ вр и ее производных для FO(r) вычислялись непосредственным суммированием
по колебательно-вращательным уровням энергии состояния Х2Ш по уравнениям A. 73)—A.75).
Колебательно-вращательные уровни состояния Х2П находились по уравнениям A.65) и A. 42)
со значениями коэффициентов Ук1, приведенными в табл. 7.3. В этой таблице приведены также
значения i>max и /цш,'полученные по методикам, изложенным в главе 1, и коэффициентов а{,
аппроксимирующих предельную кривую диссоциации этого состояния. В расчете статистической суммы
FO и ее производных учитывались все уровни энергии, ограниченные предельной кривой
диссоциации. Погрешности значений термодинамических функций FO, приведенных в табл. 46, при
низких температурах определяются главным образом неточностью принятых значений постоянной
спин-орбитальной связи (до 0,5 Дж-К~1-моль~1 в ФсB98,15 К)) и межъядерного расстояния,
при умеренных температурах — неточностью всех молекулярных постоянных FO, которые только
грубо описывают число и энергию вращательно-колебательных уровней состояния Х2И. При
высоких температурах к этому добавляются погрешности из-за возможного вклада возбужденных
состояний FO и приближенного ограничения числа колебательно-вращательных уровней энергии
в состоянии Х2П. Последнее обусловлено, в частности, неточностью принятой энергии
диссоциации FO (до 0,5 и 2 Дж-К-^моль в значениях Ф°(Г) и S°(T) при 6000 К). Общие погрешности
значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 0,7, 0,6 и 0,8 Дж-К^-моль, значения
S° F000 К) — до 4 Дж-К-^моль.
Термодинамические функции FO (г) вычислялись ранее в работе [764] (значения Ф°(Т)
до 3000 К), таблицах JANAF [2095] и предыдущих изданиях справочника [87, 88]. Результаты
149
F20 (г) Глава 7
этих расчетов при низких температурах сильно отличаются от приведенных в табл. 46, особенно
в значениях Ф°(Г), из-за того, что во всех расчетах не учитывалось расщепление компонент
состояния ХШ. Эти расхождения достигают 2 Дж-К~1-моль~1 с данными работ [764, 2095] и
3,2 Дж-К~1-моль~1 с данными [88], где накладываются дополнительные ошибки из-за
неточности оценки постоянных FO. С ростом температуры расхождения уменьшаются (до 0,5 ДжХ
XК-моль с данными справочника [88]), но при 5000—6000 К в данных работы [2095] возникают
ошибки из-за пренебрежения необходимостью ограничения суммирования по
колебательно-вращательным уровням энергии, что особенно существенно для молекул с такой энергией диссоциации,
как у FO. Соответствующие расхождения в С°р(Т) и S°(T) при 6000 К достигают 7 и 1 Дж-К~1х
Хмоль".
Константа равновесия реакции FO(r)=O(r)+F(r) вычислена с использованием значения
Ar#°@)=D0(FO)=215,677+10 кДж-моль, которое соответствует принятой в настоящем
справочнике энтальпии образования
?kfH°(FO, г, 298,15 К) = 109 + 10 кДж • моль.
Это значение принято на основании результатов определений разности потенциалов появления
ионов FO+ из молекул FO и F2O в работе Клайна и Уотсона [997]. Из экспериментальных
данных [997] следует, что Do (F—OF)=38+2 ккал^моль (~159+8 кДж-моль), что вместе с
принятым значением энтальпии образования F2O дает приведенное значение Д JT" (FO,
г, 298,15 К).
Исследования кинетики пиролиза F2O в работах [2433, 3486, 3724, 3728, 2274] и реакции FO
с атомарным водородом в работе [2407] приводят к значениям, совпадающим с принятым в
пределах погрешности измерений, но имеющим меньшую точность.
F2O (г). Термодинамические свойства газообразного дифторида кислорода в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 47.
Значения термодинамических функций F2O(r) вычислены по постоянным, приведенным
в табл. 7.4. Инфракрасный спектр F2O исследовался в работах [1924, 3582, 604, 2157, 2828].
Таблица 7.4. Значения молекулярных постоянных (в см) в состоянии ХгА(р^=1, о = 2),
принятые для расчета термодинамических функций F2O
^2
v3
Ж11
#22
•^33
^13
924,8
461,0
831,0
-4,5
-0,4
—6,3
-6,1
—19,2
*23
¦^000
¦^000
^000
4
4
—11,0
1,960 777
0,363 466
0,305 792
0,001286
—0,023 317
0,019 514
a3
С
«2C
4
^aaaa
0,002 194
0,001413
0,002 320
0,000239
0,001 777
0,003 844
-2.207-10-4
Чьъь
zccce
zaabb
xbboo
^аасс
^abab
w
—3,167.
—1,272-
1,333 ¦
1,928-
4,090-
—5,420-
-6,9
10-6
io-6
lO
10"'
io-6
10"»
В наиболее детальном исследовании Небгена и др. [2828] в области 400—3400 см, помимо
основных частот F2O, было зарегистрировано большое число составных частот. Полученные данные
вместе с постоянной Ферми резонанса W122, найденной в работе Морино и Сейто [2709],
позволили определить колебательные постоянные F2O, описывающие все наблюдавшиеся частоты
с точностью 1 см. Эти колебательные постоянные приняты в настоящем справочнике.
Вращательные постоянные и структурные параметры F2O были определены в результате исследований
микроволнового спектра в работах [1944, 3017, 3016, 2709]. Наиболее тщательное исследование
вращательных переходов в основном колебательном состоянии (для N ^ 39) было проведено Прайсом
и др. [3016], в состояниях 100, 010, 001 и 020 — Морино и Сейто [2709]. На основании полученных
ими данных и данных, представленных в работе [3016], авторы работы [2709] определили
значения вращательных постоянных и постоянных колебательно-вращательного взаимодействия,
приведенные в табл. 7.4. Этим постоянным соответствуют структурные параметры г (F—О) = 1,4053 +
±0,0004 А, -4 F-O-F=103°4'+3'.
Расчет термодинамических функций F2O был выполнен по уравнениям A.3)—A. 6), A. 9), A.10),
A.122)—A.123), A.127) и A.128) с учетом ангармоничности колебаний и колебательно-вращатель-
150
Фтор и его соединения HF (г)
ного взаимодействия — уравнения A.131)—A.133) и резонанса Ферми — уравнения A.153)—
A.155). Основные погрешности табулированных значений термодинамических функций
обусловлены неточностью аппроксимации высоких колебательно-вращательных уровней
уравнениями A.100), A.107) с принятыми значениями постоянных, а при температурах выше 3000 К
дополнительно тем, что в расчете не учитывалась необходимость ограничения числа колебательно-
вращательных состояний. Последнее должно быть существенным для F2O(r) ввиду низкой энергии
связи F—OF. Погрешности в значениях Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 0,05,
0,5 и 8 Дж-К^-моль.
Термодинамические функции F2O(r) вычислялись ранее в работах [3071, 1426, 2766, 3800,
3430, 2417] (Г < 1000-^-2000 К), таблицах JANAF [2095] и предыдущем издании справочника
[88]. В работе [3071] расчет был выполнен по неправильным значениям постоянных, а в работе
[2766] и особенно [3800] допущены ошибки в расчетах. Данные работ [2095] и [88],
вычисленные в приближении «жесткий ротатор — гармонический осциллятор», практически совпадают
между собой, но меньше приведенных в табл. 47. Эти расхождения возрастают с температурой и
«оставляют от 0,1 до 4 Дж-К~1-моль~1 в значениях Ф°(Г).
Константа равновесия реакции F2O(r)=O(r)-|-2F(r) вычислена с использованием значения
Дг//°@)=374,578 + 1,6 кДж-моль, которое соответствует принятой в справочнике энтальпии
•образования
&уН°(?гО, г, 298,15 К) = 24,5 ±1,6 кДж • моль.
Эта величина основана на результатах измерений энтальпии реакций сгорания F2O, F2 и Ог
в водороде в присутствии воды, выполненных Кинг и Армстронгом [2246]. Реакции проводились
в горелке, помещенной в калориметр постоянного давления, с использованием чистых и хорошо
проанализированных образцов F2O и F2. Принятая величина согласуется в пределах погрешности
с результатами измерений энтальпии растворения F2O в растворах КОН, KI и НВг в работе Вар-
тенберга и Клинкотта [3885, 3882], которые с уточнением, сделанным Руффом и Менцелем [3270],
приводят к величине 23 кДж-моль. Данные работ [3269] A9+10 кДж'Моль) и [658, 659]
{—17 кДж-моль), основанные на измерении энтальпии сгорания F2O, содержат ошибки из-за
использования недостаточно чистых образцов, коррозии аппаратуры и некоторых методических
недостатков.
HF (г). Термодинамические свойства газообразного фтористого водорода в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 48.
В табл. 7.2 приведены значения молекулярных постоянных HF в состоянии Х1^*
(конфигурация 1а22а2За21и4). Согласно теоретическим расчетам [2746, 861, 560], которые согласуются с
результатами исследований спектров, молекула HF не имеет связанных возбужденных валентных
состояний с энергиями ниже 80 000 см. Колебательно-вращательный спектр HF исследовался
в работах [2064, 1149, 3319, 2254, 2826, 3603, 569, 2322, 2509, 2510, 1890, 1488, 3905],
вращательный — в работах [3463, 3262, 211, 2539] и электронный — в работах [2127, 1255, 1256]. В
колебательно-вращательных спектрах были изучены переходы, связанные с уровнями,
соответствующими значениям у"=0-^-9, а в электронном спектре — значениям v"=7-f-19. Значения
молекулярных постоянных HF, полученные в работах, выполненных за последние 10—15 лет, хорошо
согласуются между собой. Ди Лонардо и Дуглас [1256] на основании результатов исследования
электронного спектра (у"=7+-19), а также данных Уэбба и Рао [3905] (i/'=0+-2), [1488] (v" =
=3+-5) и Манна с сотр. [2510] (у"=6) рекомендовали наиболее надежные значения G0(v), В„
D,, Н, и Lt для всех колебательных состояний HF с ь>" <[ 19, но не привели равновесных
значений постоянных. Приведенные в табл. 7.2 постоянные HF рекомендованы в работе [3591 ] по
данным работы [2510 ]. При подготовке справочника значения Go (v) и Bt, полученные в работах [2510,
1488, 3905, 1256] были обработаны по методикам, изложенным в разделе 1.4 с использованием
Do (HF)=47 361+64 см. Соответствующие значения Yk0 и YkX приведены в табл. 7.3. Уравнения
с этими коэффициентами описывают экспериментальные значения Go (v) и Bt с точностью
соответственно лучше 0,4 и 0,01 см для всех v <J 19. В табл. 7.3 приведены также значения vm&% и /пт
и коэффициентов а{ в уравнении, аппроксимирующем предельную кривую диссоциации состояния
Х1!,*, рассчитанные по методикам, изложенным в разделе 1.4, а также постоянная Y02, принятая
по [1256].
151
H2F2 (г) Глава 7
Значения (??. вр и ее производные вычислялись по уравнениям A.73)—A.75) непосредственным
суммированием по колебательно-вращательным уровням энергии, которые находились по
соотношению A.65) без учета постоянных Y03 и Yoi. Такое упрощение практически не сказывается
на точности расчета термодинамических функций HF(r). При расчете статистической суммы
HF и ее производных учитывались все уровни энергии, ограниченные предельной кривой
диссоциации.
Погрешности вычисленных значений термодинамических функций при Т ^ 5000 К
обусловлены главным образом неточностью принятых фундаментальных постоянных и не превышают
0,02 Дж«К~1.моль~1 в значениях Ф°(Т) и S°(T). При Т > 5000 К начинает сказываться
неточность аппроксимации колебательно-вращательных уровней энергии со значениями / ^> 40.
При 6000 и 10 000 К погрешности в значении Ф°(Г) могут достигать 0,05 и 0,2 Дж-К~1-моль~1.
Термодинамические функции HF(r) вычислялись во многих работах [2759, 2029, 1013, 3072,
3946, 2556, 1454, 3658], таблицах JANAF [2095] и предыдущих изданиях справочника [87, 88]
для температур не выше 6000 К. Несмотря на различие постоянных и методов расчета,
использованных в разных работах, благодаря большим величинам частоты колебания, вращательной
постоянной и энергии диссоциации HF результаты всех расчетов удовлетворительно согласуются
между собой и с данными табл. 48. Наилучшее согласие имеет место с данными Фебера и Херрика
[1454], которые вычислены методом непосредственного суммирования по уровням энергии и по
постоянным, основанным на данных работы [2127]. Соответствующие расхождения не превышают
0,005 Дж.К~1-моль~1 в значениях Ф°(Т) и S°(T) во всем интервале температур и в С(Т) для
Т ^ 5000 К. Данные [2095, 3658], полученные по методу Майера и Гепперт-Майер, и
предыдущих изданий справочника [87, 88], рассчитанные по методу Гордона и Барнес, согласуются
соответственно в пределах 0,09 и0,04 Дж-К~1-моль~1 в значениях Ф°(Г) и в пределах 0,33 и 0,10 ДжХ
ХК~1-моль~1 в значениях S°(T) с данными табл. 48.
Константа равновесия реакции HF(r)=H(r)+F(r) вычислена на основании значения \Н°@) =
=566,562+0,76 кДж'МОЛь, которое соответствует принятой в справочнике энтальпии
образования HF(r)J
AfH°(EF, г, 298,15 К) = — 273,30 ±0,70 кДж • моль.
Это значение рекомендовано КОДАТА—MCHG [1000] на основании совместного анализа
результатов исследований энергий диссоциации F2 (см. выше), HF и энтальпии образования HF.
Наиболее надежное экспериментальное значение энтальпии образования HF B73,29 кДж-моль)
получено из измерений энтальпии образования жидкой HF в работах Хаббарда с сотр. [2140, 3395]
(—303,55+0,25 кДж-моль) и энтальпии испарения HF в работах Вандерзее и Роденбурга [3771,
3772] C0,26+0,10 кДж-моль"). Существенно менее точные значения A^°(HF, г), в частности
из-за полимеризации паров HF, были получены в более ранних работах [3883, 3268, 3270, 3887].
Энергия диссоциации HF определялась по предиссоциации при вращении в состоянии X1!, +
Ди Лонардо и Дугласом [1255] D7 333+60 см~х=566,2+0,7 кДж-моль) и Джонсом и Барроу
[2127] D7 241+100 см~х=565,1+1,2 кДж-моль"). С учетом точности четырех приведенных
значений и данных по D0(F2) (см. выше) была рекомендована принятая выше величина Д^0 (HF, г).
Ей соответствует
D0(HF)= 566,562 + 0,76 кДж • моль = 47 361 ± 64 см.
Принятое значение энтальпии образования HF(r) согласовано также со значением энтальпии
образования иона F~ в состоянии стандартного водного раствора
AfHo(F-, р-р НД станд. сост., 298,15 К) = —333,71 + 0,54 кДж • моль,
которое рекомендовано КОДАТА—МСНС [1000] в результате расчетов по ряду термохимических
циклов, основанных на измерениях в работах [2140, 3772, 1083, 2246, 1097, 3883, 3431, 3432,
216, 201].
H2F2 (г)- Термодинамические функции газообразного дифторида диводорода в стандартном
состоянии при температурах 100—3000 К приведены в табл. Д.9 (см. наст, том, кн. 2).
Молекулярные постоянные, принятые для расчета термодинамических функций H2F2 (г),
приведены в табл. 7.5. Произведение моментов инерции H2F2 вычислено по структурным параметрам,
152
Фтор и его соединения
H2F2(r)
Таблица 7.5.
Значения молекулярных постоянных и а, принятые для расчета
термодинамических функций H2F2, H3F3, H4F4, H5F5, H,F7 и HOF
Молекула
Состояние
НА &А
H3F3 Ш
ГТ ТГ1 -р-Л А
H6F5 &A
H,F7 &А
HOF ZU
^2
v3
CM
4038
3200 C)
3200 D)
3200 E)
3200 G)
3537
4081
275 C)
275 D)
275 E)
275 G)
1359
171 588
1000 C) 550 C)
1000 D) 550 D)
1000 E) 550 E)
1000 G) 550 G)
886 —
519
—
40
40B)
40D)
—
226
—
20
20B)
20 D)
—
Г3 • CM6
20
2-103
17-103
1,0-Ю5
1,8.10s
1,464
1
3
4
5
7
1
полученным Дайком, Говардом и Клемперером [1362] в результате изучения радиочастотных и
микроволновых спектров этой молекулы и ее дейтерозамещенных. Авторы работы [1362] нашли,
что в основном электронном состоянии H2F2 имеет плоскую изогнутую структуру (точечная группа
симметрии Cs). Предположив, что один атом Н лежит на прямой F. . .F, а межатомное расстояние
Н—F в H2F2 такое же, как в молекуле HF @,917 А), по найденным экспериментально значениям
полусумм вращательных постоянных В ж С молекул H2F2, D2F2 и HFDF Дайк и др. нашли, что
г (F. . .F)=2,79+0,05 Аи 4 Н—F. . .F=108°. Эти значения находятся в хорошем согласии с
результатами неэмпирического квантовомеханического расчета, выполненного Кертиссом и Поп-
лом [1143] (r(F. . .F)=2,81 A, ^ H-F. . .F=115°, r (H-F)=0,903 А), хотя в последнем было-
найдено, что второй атом Н может быть слегка сдвинут от прямой F. . .F («^ F. . .F—Н — 9°).
Результаты других расчетов [2282, 1251, 1207, 2862, 4022, 2383, 2383а] удовлетворительно
согласуются с этими данными. Все теоретические расчеты исключают возможность циклического
строения H2F2 в основном электронном состоянии. Погрешность приведенного значения IaIbIc
оценивается в 1,4-10~117 г3-см6.
В спектре поглощения газообразного фтористого водорода наблюдался ряд полос, которые
принадлежат различным полимерным молекулам, в областях 350—400, 700—800, 900—1200 и
3200—4000 см-1 [3458, 2052, 2373, 3455, 3456, 1978, 1947]. Однако корректное отнесение их к
конкретным молекулам затруднительно. Значения частот H2F2, приведенные в табл. 7.5, приняты
по работе Кертисса и Попла [1143], где они были получены через силовые постоянные, найденные
в результате неэмпирического расчета потенциальной поверхности H2F2 в основном электронном
состоянии. Они удовлетворительно согласуются с частотами, наблюдавшимися в спектрах
фтористого водорода, и другими оценками. Погрешности приведенных частот могут достигать 100 см
в значениях vx, v2, v4, v8 и 50 см в значениях v3 и v6.
Термодинамические функции H2F2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130)
без учета возбужденных электронных состояний. Погрешности вычисленных значений
определяются неточностью принятых молекулярных постоянных и приближенным характером расчета.
Они составляют 3, 5 и 6 Дж -К -моль в значениях Ф°(Г) при 298,15, 1000 и 3000 К. Ранее
термодинамические функции H2F2 в интервале 100—6000 К вычисляли авторы справочника [337].
Расхождения в значениях Ф°(Г), достигающие 29 Дж-К-моль при 3000 К, обусловлены
разницей в принятом числе симметрии молекулы H2F2 (в [337] число симметрии ошибочно принято
равным 2) и различием в оцененных значениях частот колебаний.
Энтальпия образования газообразного димера фтористого водорода принята равной
^"(HgF,, г, 0) = —566,5 + 4 кДж-моль
Эта величина основана на результатах исследования плотности паров фтористого водорода
в интервале 26—56° С и 30—700 мм рт. ст., выполненного Бриглебом и Штромейером [769], и
измерениях теплоемкости ассоциированных паров фтористого водорода в области от —20
до +100° С и 100—700 мм рт. ст., проведенных Франком и Мейером [1523]. Авторы работы [7691,
предположили, что в парах существуют ассоциаты HnFn с п <^ 9, и нашли методом подбора значе-
153
B2F2 (г)
Глава 7
ния констант их последовательной ассоциации, лучше всего соответствующие
экспериментальным данным. При этом результаты расчетов констант равновесия оказались не зависящими от
предполагаемой структуры ассоциатов. Позднее Мак-Лейн и др. [2493] пересчитали заново данные,
полученные в работе [769], и нашли, что они могут быть объяснены в предположении, что в парах
присутствуют только моно-, ди- и гексамерные молекулы. Франк и Мейер [1523] при обработке
экспериментальных данных по теплоемкости паров для получения констант равновесия
ассоциации фтористого водорода принимали, что все ассоциаты, за исключением гексамера, имеют
цепочечную структуру.
При подготовке настоящего издания значения констант равновесия реакции ассоциации HF,
полученные в работах [769, 1523, 2493], а также в работах Лонга и др. [2459], которые
исследовали (Р—V—Г)-свойства фтористого водорода от 0 до 38 °С при давлении 200—639 мм рт. ст.,
и Смита [3456, 3455], который изучал зависимость спектров поглощения фтористого водорода
от температуры B03—346 К) и давления (от нескольких мм рт. ст. до 760 мм рт. ст.), были
обработаны с использованием термодинамических функций, табулированных в справочнике.
Значения энтальпий реакций диссоциации ассоциатов на п молекул HF, рассчитанные по методам II
и III законов термодинамики, приведены в табл. 7.6. Как видно из таблицы, несмотря на то, что
Таблица 7.6.
Результаты расчета энтальпии реакций HnFn (r) = KHF (г) (в кДж'Моль)
по данным разных авторов
Авторы
Лонг и др., 1943,
[2459]
Бриглеб, Штромейер,
1953 [769]
Смит, 1958 [3455]
Степень
ассоциации
6
2
3
4
5
6
7
4
6
ДГЯ
II закон
149
29,7
56,6
88,6
119,5
149,7
177,2
68,1
150,4
°@)
III закон
152,2
17,0
53,7
84,0
115,9
147,7
180,8
—
—
Авторы
Смит, 1959 [3456]
Франк, Мейер, 1959
[1523]
Маклин и др., 1962
[2493]
Степень
ассоциации
2
2
3
4
5
6
7
2
6
II закон
20,6
23,5
46,6
72,7
98,7
151,2
150,9
22,7
152,0
°@)
III закон
—
18,3
52,7
77,5
102,8
150,3
154,5
—
—
при обработке экспериментальных данных в работах [769, 1523, 3456, 2493, 2459, 3455] делались
различные предположения о составе ассоциированных паров фтористого водорода и структуре
молекул ассоциатов, расчет энтальпий реакций по константам равновесия, полученным в этих
работах, привел к согласующимся значениям энтальпий реакции. Особенно хорошее согласие этих
значений имеет место для димера и гексамера. Энергия диссоциации H2F, на 2HF находится также
в хорошем согласии с результатами квантовомеханического расчета Кертисса и Попла [1143]
(примерно 19 кДж-моль).
В табл. 7.7 приведены выбранные в.справочнике в результате анализа перечисленных работ
значения^энтальпий реакций диссоциации ассоциированных молекул фтористого водорода с
оцененными погрешностями этих величин. Следует отметить, что значительная часть приведенных
Таблица 7.7. Значения энергий диссоциации ассоциированных соединений фтористого
водорода (в кДж-моль), выбранные на основании анализа данных табл. 7.6
,
Молекула
НЛ(г) =
= rcHF(r)
Дг//° @)
— HF)
H2F2 20+4 20
H3F3 53+8 33
Молекула
НА (г) =
= «HF(r)
ArH° @)
Do(HK_1F^1 —
— HF)
H4F4 81+12 28
H6F6 109+20 28
Молекула
HA (r) =
= raHF (r)
ArH° @)
Do(H,,_iFn_1 —
— HF)
HeF6 148+8 39
H7F7 168+30 20
154
•Фтор и его соединения H3F3(r), H4F4 (г)
погрешностей, особенно для молекул HnFn e n^4, обусловлена неточностью термодинамических
функций, использованных для расчета значений АГН°(О). Дополнительные погрешности вносит
применение в этих расчетах термодинамических функций газов в стандартном состоянии,
поскольку в условиях отдельных экспериментов отклонения газов от состояния идеального газа
могли быть существенны.
H3F3 (г). Термодинамические функции газообразного трифторида триводорода в стандартном
состоянии в интервале температур от 100 до 3000 К приведены в табл. Д. 10.
Молекулярные постоянные, использованные при расчете термодинамических функций H3F3 (г),
приведены в табл. 7.5. Экспериментальные данные о структуре и частотах колебаний тримера
¦фтористого водорода в литературе отсутствуют. Приведенное в табл. 7.5 произведение моментов
инерции H3F3 вычислено в предположении, что в основном состоянии ХгА молекула Hj,F3 имеет
плоскую циклическую структуру (симметрия C3h) с атомами водорода, лежащими на линиях
F. . .F, с расстояниями г (F .. .F)=2,50+0,10 А и г (H—F)=0,92 + 0,05 А. Выбор циклической
структуры тримера фтористого водорода основывается на результатах работы Дайка, Говарда
и Клемперера [1362], которые исследовали отклонение молекулярного пучка H3F3 в
электрическом поле, и теоретического расчета Дель-Бене и Попла [1207], которые показали, что
циклические полимеры HnFn с п=3, 4,5, 6 более стабильны, чем цепочечные. Значение г (F. . .F) принято
на основании сравнения результатов расчета Дель-Бене и Попла [1207] (г (F. . .F)=2,31 А для
H3F3h2,22A для H6F6) и результатов экспериментального исследования структуры HeFe,
выполненного Янценом и Бартеллом [2105] (г (F. . .F)=2,525+ 0,005 А) г. Значение r(H—F) принято
равным соответствующему значению в молекуле HF. Погрешность величины IjJbIc оценивается
в 5,5-10"15 г3-см6. Значения частот, приведенные в табл. 7.5, оценены по аналогии с частотами
колебаний гексамерных молекул фтористого водорода, вычисленными Сивиным и др. [1147],
а также результатов исследований спектров фтористого водорода в газовой фазе [3458, 2052, 2373,
3455, 3456, 1978, 1947] и в конденсированном состоянии [2259, 3741, 718, 3404]. Погрешности
частот Vj, v2, v3 и v4 оцениваются равными 200, 100, 100 и 150 см соответственно. Принятые
значения частот удовлетворительно согласуются с отнесением частот в спектрах кристаллического
¦фтористого водорода, выполненного Киттельбергером и Хорнигом [2259].
Термодинамические функции H3F3(r) были вычислены в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A.6), A.9), A.10) A.122)—A.124), A.128) и
A.130) без учета возбужденных электронных состояний. Погрешности вычисленных значений
определяются в основном неточностью принятых молекулярных постоянных и в меньшей степени
приближенным характером расчета. Они составляют 7, 13 и 15 Дж-К^-моль в значениях Ф° (Т)
при 27=298,15, 1000 и 3000 К, в том числе из-за неопределенности молекулярных постоянных
6, И и 12 Дж-К^-моль-1.
Ранее термодинамические функции H3F3 (г) вычислялись в справочнике [337] для Т <С 6000 К.
Расхождения между данными [337] и табл. Д.10 не превышают 1 Дж-К^-моль в значениях
Ф°(Г) и S°(T) во всем интервале температур.
Для энтальпии образования газообразного тримера фтористого водорода в справочнике принято
ДД°(Н3Р3, г, 0) = —873 + 8 кДж • моль.
Эта величина основана на результатах исследований плотности паров и теплоемкости
фтористого водорода, выполненных Бриглебом и Штромейером [769] и Франком и Мейером [1523]
(см. H2F2 (г)).
H4F4 (г). Термодинамические функции газообразного тетрафторида тетраводорода в
стандартном состоянии в интервале температур 100—3000 К приведены в табл. Д.11.
Молекулярные постоянные, использованные при расчете термодинамических функций H4F4,
приведены в табл. 7.5. Значение IaIbIc вычислено в предположении, что в основном электронном
состоянии Х1А молекула H4F4 имеет плоскую циклическую структуру симметрии С4Й с атомами
1 Расчет Хойланда и Кира [2021 ] также показывает, что величины г (F.. .F) в H3F3 и HeFe должны отличаться
менее чем на 0,1 А.
155
H5F5(r) Глава 7
водорода, расположенными на линиях F. . .F, с расстояниями г (F. . .F) = 2,50+0,10-A и г (Н—F) =
=0,92 + 0,05 А. Выбор циклической структуры H4F4 основан на теоретических расчетах Дель-
Бене и Попла [1207] и Коллмана и Аллеыа [2281], которые показали, что циклические полимеры
H,,FM с п ^ 3 более стабильны, чем полимеры, имеющие зигзагообразную цепочечную структуру \
Межатомное расстояние г (F. . .F) принято на основании величины г (F. . .F)=2,530 + 0,005 А
в H6F6 [2105], такокак согласно расчетам [1207] и [2021 ] эти расстояния в H4F4 и HeFe отличаются
менее чем на 0,05 А. Величина г (Н—F) принята равной соответствующему значению в HF.
Погрешность вычисленного значения IaIbIc может достигать 5-10~114 г3-см8.
Приведенные в табл. 7.5 значения частот колебаний H4F4 оценены по аналогии с частотами
газообразного циклического гексамера H6Fe и с учетом результатов исследований инфракрасного
спектра и спектра комбинационного рассеяния фтористого водорода в газовой фазе [3458, 2052,
2373, 3455, 4356, 1978] и в кристаллическом состоянии [2259, 3741, 3404] и неупругого рассеяния
холодных нейтронов в жидком и твердом фтористом водороде [718]. Погрешности этих частот
оцениваются в 200, 100, 100, 50, 20 и 10 см соответственно для vl5 v2, v3, v4, v5 и ve.
Термодинамические функции H4F4 (г) были вычислены в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A.122)—A.124), A,128) и
A.130) без учета возбужденных электронных состояний. Погрешности табулированных значений
составляют 12, 20 и 25 Дж -К -моль-1 в значениях Ф° (Т) при Г=298,15, 1000 и 3000 К и
обусловлены неточностью молекулярных постоянных и приближенным характером расчета.
Ранее термодинамические функции H4F4 (г) вычислялись в [337] для Т г^ 6000 К. Расхождения
в значениях функций, вычисленных в [337] и табулированных в табл. Д.11, не превышают
2,5 Дж-К~1-моль~1 в Ф° (Т) и обусловлены различием принятых в расчетах молекулярных
постоянных H4F4.
Энтальпия образования газообразного тетрамера фтористого водорода принята равной
Д,Я"°(Н4Р4, г, 0) = —1174 + 12 кДж • моль.
Это значение основано на результатах исследования плотности паров и теплоемкости фтористого
водорода, выполненных Бриглебом и Штромейером [769] и Франком и Мейером [1523]
(см. ВД(г)).
H6F5 (г). Термодинамические свойства газообразного пентафторида пентаводорода в
стандартном состоянии в интервале температур 100—3000 К приведены в табл. Д.12.
Молекулярные постоянные, использованные при расчете термодинамических функций H5F5>
приведены в табл. 7.5. Экспериментальных данных о спектре и структуре молекулы пентамера
фтористого водорода в литературе нет. На основании теоретических расчетов, выполненных Дель-
Бене и Поплом [1207] и Коллманом и Алленом [2281], а также результатов исследования
отклонения молекулярного пучка в электрическом поле [1362] в справочнике принимается, что в
основном электронном состоянии ХХА молекула H5F5 имеет плоское циклическое строение с атомами
водорода, расположенными на линиях F. . .F (группа симметрии CSh). Приведенное в табл. 7.5
произведение моментов инерции вычислено для г (F. . .F)=2,50+ 0,10 А, основанного на
сравнении результатов расчета Дель-Бене и Попла [1207] для H8F5 и HeFe и экспериментального
исследования структуры H6Fe [2105]. Значение г (Н—F) принято равным соответствующему значению
в молекуле HF. Погрешность в значении Iд1в1с составляет 3 -10~ш г3 -см6. Приведенные в табл. 7.5
значения частот колебаний оценены по аналогии с частотами газообразного циклического
гексамера H6Fe, вычисленными Сивиным и др. [1147], с учетом результатов экспериментальных
исследований спектров фтористого водорода в газовой фазе [3458, 2052, 2373, 3455, 3456, 1978, 1947]
и в конденсированном состоянии [2259, 3741, 718, 3404]. Погрешности оцененных значений частот
составляют 200, 100, 100, 50, 20 и 10 см.
Термодинамические функции H5F5 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A. 9), A.10), A.122)—A.124), A. 128) и A.130)
1 Исследования инфракрасного спектра газообразного фтористого водорода [2052] и спектра комбинационного
рассеяния газообразного и растворенного в жидком SFe фтористого водорода [2373 ] показали, что эти спектры
могут быть интерпретированы, как связанные с валентными колебаниями Н—F в плоских зигзагообразных
молекулах H4F4 и циклических молекулах HeF6. Рентгенографическое исследование, выполненное в работе [434],
свидетельствует о существовании бесконечных плоских зигзагообразных цепей HMFM в твердой фазе.
156
Фтор и его соединения
H6F6 (г)
без учета возбужденных электронных состояний. Погрешности вычисленных термодинамических
функций составляют 20, 30 и 35 Дж-К^-моль-1 в значениях Ф° (Т) при Г=298,15, 1000 и 3000 К
и обусловлены неточностью молекулярных постоянных и приближенным характером расчета.
Ранее термодинамические функции H5F5 (г) вычислялись в справочнике [337]. Расхождения
функций, вычисленных в этом справочнике и табулированных в табл. Д. 12, уменьшаются с ростом
температуры, составляют 12 Дж-К^-моль в значении Ф° B98,15 К) и 6,5 Дж-К^-моль
в Ф° C000 К) и обусловлены различием в оцененных значениях молекулярных постоянных.
Для энтальпии образования газообразного пентамера фтористого водорода в справочнике
принимается значение
AfH° (H5F5, г, 0) = —1475 + 20 кДж ¦ моль.
Эта величина основана на результатах экспериментальных исследований плотности паров
и теплоемкости фтористого водорода, выполненных Бриглебом и Штромейером [769] и Франком и
Мейером [1523] (см. H2F2 (г)).
H6F6 (г). Термодинамические свойства газообразного гексафторида гексаводорода в
стандартном состоянии в интервале температур 100—3000 К приведены в табл. Д.13.
Молекулярные постоянные, использованные при расчете термодинамических функций H6Fe,
приведены в табл. 7.8.
Таблица 7.8. Значения молекулярных постоянных HeFe (в см) в состоянии' Х^А (а = 6, рх = 1),
принятые для расчета термодинамических функций
vt 3065
v, 1029
v3 203
v4 537
ve 26
Примечание. 1AlBlq = 1>
v6 551 B)
v7 322 B)
v8 991 B)
ve 327 B)
v10 33 B)
,95 • Ю-112 г3 • см6.
vu 550
v12 3403
v13 979
v14 356
v15 53
vie
V17
V18
"'ao
3153 B)
1017 B)
252 B)
545 B)
15B)
Б соответствии с результатами электронографического исследования структуры H6F6,
выполненного Янценом и Бартеллом [2105], исследований инфракрасного спектра газообразного
фтористого водорода в работе Хуонг и Кози [2052] и спектра комбинационного рассеяния в работе
Ле Дуфф и Хольцер [2373], а также теоретических расчетов в работах [1207, 2021, 2283, 2281]
в справочнике принимается, что эта молекула имеет плоское циклическое строение (точечная
группа симметрии Ceh). Значение IaIbIc, приведенное в табл. 7.8, вычислено при г (Н—F)=0,973 +
+ 0,009 А и г (F. . .F)=2,530+ 0,005 А по данным работы [2105]. Погрешность этой величины
оценивается в 1 -10~113 г3-см6. Рейхерт и Хартманн [3188] нашли, что полученные ими микроволновые
спектры паров фтористого водорода согласуются с цепочечной структурой HeFe (-4 F. . .F. . .F. . .=
= 104°, г (Н—F)=0,9997 А и г (F. . .F)=2,4995 А). Однако в работе [3188] не обсуждается
возможность согласования или наличие противоречий полученных экспериментальных данных с
циклической моделью H6F6. Поскольку прямые структурные измерения Янцена и Бартелла [2105],
а также теоретические расчеты [1207, 2021, 2281, 2283] противоречат выводу, сделанному в
работе [3188], результаты этой работы не учитывались при выборе молекулярных постоянных H6Fe.
Приведенные в табл. 7.8 значения частот колебаний H6Fe получены Сивиным и др. [1147],
которые, используя силовые постоянные твердого фтористого водорода и его дейтерозамещенных,
рассчитанные Тубино и Зерби [3741], выполнили анализ нормальных координат плоского
циклического гексамера H6Fe с геометрическими параметрами, найденными Янценом и Бартеллом
[2105]. Приведенным в табл. 7.8 частотам H6Fe не противоречат результаты экспериментальных
исследований инфракрасного спектра и спектра комбинационного рассеяния газообразного
фтористого водорода [3458, 2052, 2373, 3455, 3456, 1978, 1947], хотя из-за трудности разделения
поглощения различных полимеров эти исследования неинформативны с точки зрения отнесения
наблюдаемых полос к колебаниям отдельных'полимеров. Погрешности принятых значений частот
оцениваются в Юуо.
157
H7F, (г) Глава 7
Термодинамические функции HeF6 (г) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130)
без учета возбужденных электронных состояний. Погрешности вычисленных таким образом
функций составляют 10, 25 и 30 Дж-К^-моль-1 в значениях Ф°(Г) при Г=298,15, 1000 и 3000 К
и обусловлены неточностью молекулярных постоянных и приближенным характером расчета.
Ранее термодинамические функции H6F6 (г) вычисляли авторы справочника [337] по оцененным
значениям молекулярных постоянных в интервале температур 100—6000 К. Расхождения в
значениях функций, вычисленных в работе [337] и табулированных в табл. Д.13, достигают
24 Дж-К-моль в значениях Ф°(Т) и обусловлены различием принятых значений частот.
Для энтальпии образования газообразного гексамера фтористого водорода принимается
Ayfl°(H6F6, г, 0) = —1788 + 8 кДж • моль~].
Эта величина основана на результатах исследований (Р—V—Г)-свойств ассоциированного
фтористого водорода в интервале 0—38 °С и 200—639 мм рт. ст., выполненных Лонгом и др. [2459],
исследованиях плотности паров и теплоемкости фтористого водорода, выполненных Бриглебом
и Штромейером [769] и Франком и Мейером [1523], и исследованиях зависимости интенсивности
спектров поглощения фтористого водорода от температуры и давления [3456, 3455] (см. H2F2 (г)).
H7F7(r). Термодинамические свойства газообразного гептафторида гептаводорода в
стандартном состоянии в интервале температур 100—3000 К приведены в табл. Д.14.
Молекулярные постоянные, использованные при расчете термодинамических функций H7F7 (г),
приведены в табл. 7.5. Значение произведения моментов инерции вычислено в предположении,
что молекула H7F7 в основном электронном состоянии ХгА, так же как и другие полимерные
молекулы HMFB с п^З, имеет плоское циклическое строение с атомами фтора, расположенными
в вершинах правильного многоугольника, и атомами водорода, расположенными на линиях
F. . .F (точечная группа симметрии C7J. Межатомные расстояния г (Н—F)=0,92 + 0,05 А -и
г (F. . .F)=2,50 + 0,10 А приняты равными соответствующим расстояниям в других полимерных
молекулах с п=3, 4, 51. Погрешность в значении IaIbIc оценивается в 5-10~112 г3-см6. Частоты
колебаний, приведенные в табл. 7.5, оценены на основании принятых значений частот молекулы
H6F6, с учетом имеющихся экспериментальных данных по спектрам фтористого водорода в
конденсированном и газообразном состояниях [3458, 2052, 2373, 3455, 3456, 1978, 1947, 2259, 3741, 718,
3404]. Погрешности в значениях частот v< с i—1, 2, 3, 4, 5, 6 составляют 200, 100, 100, 50, 20 и
10 см соответственно.
Термодинамические функции H7F7 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130)
без учета возбужденных электронных состояний. Погрешности в вычисленных таким образом
термодинамических функциях составляют 35, 50 и 60 Дж-К^-моль в значениях Ф° (Т) при
Т=298,15, 1000 и 3000 К. Они обусловлены неточностью молекулярных постоянных и
приближенным характером расчета. Ранее термодинамические функции H7F7 вычисляли авторы справочника
[337] в интервале температур 100—6000 К по оцененным значениям молекулярных постоянных.
Расхождения между функциями, рассчитанными в настоящем справочнике и в справочнике [337],
составляют 20—30 Дж -К -моль в значениях Ф° (Г) и обусловлены некоторым различием в
принятых значениях частот и структурных параметров.
Энтальпия образования газообразного гептамера фтористого водорода принята равной
A,#°(H7F7, г, 0) = —2080 + 30 кДж • моль.
Рейхерт и Хартманн [3188] исследовали микроволновой спектр паров фтористого водорода и пришли к выводу,
что с наблюдаемыми ими особенностями в спектре лучше всего согласуется предположение о существовании
в паре молекул HeFe и H,F7, имеющих плоское зигзагообразное строение. Авторы работы [3188] нашли следующие
структурные параметры: ^F. . .F. . .F=104°, г (Н—F)=0,9997 A, г (F. . .F)=2,4995 А и ^?. . .F. . .F=103,73°,
т (Ц—F)=0,9640 А, г (F. . .F)=2,5745 А соответственно для HeFe и H,F7. Однако, поскольку эти результаты для
HeFe противоречат результатам структурного исследования Янцена и Бартелла [2105] и теоретического расчета
Дель-Бене и Попла [1207], в настоящем справочнике для H7F7, так же как и дляНвГ6, принимается циклическая
структура.
158
Фтор и его соединения HOF (г), DF (г)»
Эта величина основывается на результатах экспериментального исследования плотности паров
и теплоемкости фтористого водорода, выполненного в работах Бриглеба и Штромейера [769] и
Франка и Мейера [1523] (см. H2F2(r)).
HOF (г). Термодинамические свойства газообразного фторида гидроксила в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 49.
Термодинамические функции HOF(r) вычислены по постоянным приведенным в табл. 7.5.
Колебательный спектр HOF изучен только в матрицах азота и аргона [2868, 1676]. В работе [2868 J
основные частоты HOF были определены по спектру комплекса HF. . .HOF и его дейтеропроиз-
водного. Найденные значения равны v1=3483 см, v2=1393 см и v3=884 см. В работе [1676}
исследовался инфракрасный спектр чистого HOF в азотной матрице при различном разбавлении,
что позволило уточнить значения основных частот этой молекулы. Найденные в [1676] значения
приведены в табл. 7.5. Учитывая возможный сдвиг частот в матрице, их погрешности могут
достигать 10—15 см. Приведенное в табл. 7.5 произведение главных моментов инерции HOF
вычислено по вращательным постоянным Л000=19,68 + 0,20 см, 5000=0,892 550 см, С000=
=0,851 005 см, полученным в результате изучения микроволнового спектра HOF и DOF в
работе [2243]. Этим постоянным соответствуют структурные параметры: г0 (О—Н)=0,964+0,01 А,
Го (О—F) = 1,442+0,001 А и -4Н—О—F=97,2 + 0,6°.
Термодинамические функции HOF (г) были вычислены в приближении «жесткий ротатор—
гармонический осциллятор», значения внутримолекулярных составляющих вычислялись по
уравнениям A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130). Погрешности табулированных
значений термодинамических функций при низких температурах определяются главным образом
неточностью колебательных постоянных. С ростом температуры становятся существенными
погрешности из-за невозможности учета ангармоничности колебаний, центробежного растяжения
и других подобных эффектов, а также необходимости ограничения суммирования по
колебательным и вращательным квантовым числам. Общие погрешности в значениях Ф°(Г) при 298,15,
3000 и 6000 К оцениваются в 0,1, 1 и 3 Дж-К^-моль соответственно.
Термодинамические функции HOF вычислялись ранее в таблицах JANAF [2095] по оцененным
значениям постоянных и в работе [2868] (до 2000 К). Данные работы [2868] отличаются от
приведенных в табл. 49 на 0,6 Дж -К -моль из-за использования в расчете неточных значений
структурных параметров HOF. Расхождения с данными работы [2095 ] увеличиваются с ростом
температуры и составляют от 0,6 до 2,6 Дж-К^-моль в значениях Ф°(Т) и от 0,9 до 3 Дж-К^-моль
в значениях S° (T).
Константа равновесия реакции HOF(r)=O(r)-)-H(r)+F(r) вычислена с использованием
значения АгН°@)=634,092 +5,0 кДж-моль. Эта величина соответствует принятой в
справочнике энтальпии образования
Д^0 (HOF, г, 0) = —94 + 5 кДж ¦ моль,
которая вычислена по измеренным Берковицем, Аппельманом и Чапкой [591 ] фотоионизационным
методом потенциалам появления ионов О"" и ОН+ из молекулы HOF. Эти потенциалы появления
приводят к значениям энтальпии образования —96,1 + 5,0 кДж-моль и —89,8 + 7,0 кДж-моль
соответственно.
DF (г). Термодинамические свойства газообразного ьфторида дейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 50.
В табл. 7.2 приведены значения молекулярных постоянных DF. Так же как молекула HF,
молекула DF не имеет стабильных возбужденных валентных состояний с энергией ниже 70 000 см.
Колебательно-вращательный спектр DF исследовался в работах [3603, 3488] (полосы 1—0 и 2—0),
вращательный — в работах [2990, 1211], электронный — в работе [2127] (переходы на уровни
i?= 15-+-24). Наиболее надежные данные для уровней р=0-+-2 получены при исследовании
инфракрасного спектра DF в работе Спанбауэра и Рао [3488], для высоких колебательных состояний
в работе Джонса и Барроу [2127] при исследовании системы полос F12+—Х1Ъ+. Приведенные
в табл. 7.2 постоянные DF в состоянии Хг2 + рекомендованы в [3591 ] по данным Спанбауэра и
Рао [3488]. При подготовке настоящего справочника для обеспечения схождения колебательных
уровней DF к диссоциационному пределу значения Yk0 были вычислены с использованием
значений Go (v) для v=l и 2 по данным работы [3488], для у=15-+-24 — по данным работы [2127],
159
TF (г) Глава 7
для v=3~li — по значениям величин, рассчитанным по колебательным постоянным,
приведенным в табл. 7.2, и Do (DF)=47 920 + 70 см. Соответствующие значения Yk0 и ушах приведены
в табл. 7.3. Там же приведены значения Ykl и У02, принятые по работе [2127], а также значения
/am и коэффициентов а( в уравнении, аппроксимирующем предельную кривую диссоциации
состояния Х11> + молекулы DF.
Термодинамические функции DF(r) были вычислены без учета возбужденных состояний этой
молекулы. Значения Q^ вр и ее производных вычислялись непосредственным суммированием по
колебательно-вращательным уровням энергии состояния ХХ2+ по уравнениям A.73)—A.75).
Колебательно-вращательные уровни энергии молекулы DF вычислялись по уравнению A.65)
со значениями коэффициентов Ykl, приведенными в табл. 7.3, для всей области энергии,
ограниченной предельной кривой диссоциации. При температурах до 5000 К погрешности вычисленных
значений Ф°(Г) и S°(T) определяются только неточностью фундаментальных постоянных,
принятых в расчете, и не превышают 0,02 Дж -К -моль. При более высоких температурах начинает
сказываться неточность аппроксимации уравнением A.65) с принятыми значениями Ykl энергии
колебательно-вращательных уровней с большими значениями /. Соответствующие погрешности
в значениях Ф°(Г) и S°(T) при 6000 К могут достигать 0,05 и 0,1 Дж-К^-моль.
Термодинамические функции DF (г) вычислялись ранее в работах [3072] (до 5000 К), [3658]
(до 6000 К) и предыдущих изданиях справочника [87, 88] по близким значениям молекулярных
постоянных и методу Майера и Гепперт-Майер в работах [3072, 3658] и модифицированному методу
Гордона и Барнес в справочниках [87, 88]. Расхождения величин, приведенных в работах [3072,
3658], с данными табл. 50 быстро растут при высоких температурах и при 6000 К превышают
0,1 и 0,5 Дж-К^-моль в значениях Ф°G') и S°(T). Расхождения величин, табулированных
в предыдущих и настоящем изданиях, не превышают 0,05 и 0,18 Дж-К^-моль в значениях
Ф°(Г) и S°(T) во всем интервале температур.
Константа равновесия реакции DF (r)=D (r)+F (г) вычислена на основании принятого в
справочнике значения энергии диссоциации
ДД° @) = Do (DF) = 47 920 + 65 см = 573,25 + 0,78 кДж • мать.
Эта величина была получена по соотношению C.6) при Do (HF)=47 361+64 см, G @, HF) =
=2047 см-1 и G @, DF) = 1488 см.
Принятому значению Do (DF) соответствует
AfH°(DF, г, 0) = —276,168 + 0,80 кДж • моль.
TF (г). Термодинамические свойства газообразного фторида трития в стандартном
состоянии при температурах 100—6000 К приведены в табл. 51.
Спектр TF исследовался только в работе [2169], авторы которой получили спектр в
инфракрасной области и изучили вращательную структуру полос 1 — 0и2 — 0. В этой работе были
определены приближенные значения молекулярных постоянных TF, приведенные в табл. 7.2.
По аналогии с молекулами HF и DF в справочнике принято, что TF не имеет стабильных
возбужденных валентных электронных состояний с энергией ниже 70 000 см.
Термодинамические функции TF(r) были вычислены без учета возбужденных электронных
состояний молекулы TF. Значения <?S.Bp и ее производных вычислялись непосредственным
суммированием по колебательно-вращательным уровням энергии состояния Х11,+ по уравнениям
A.73)—A.75). Энергии уровней рассчитывались по уравнению A. 65) с коэффициентами Ykl,
приведенными в табл. 7.3. Ввиду того, что молекулярные постоянные TF, приведенные в табл. 7.2,
получены для переходов между уровнями с значениями квантового числа v <I 2 и низкими
значениями квантового числа /, коэффициенты Ykl для TF были рассчитаны по изотопическим
соотношениям на основании соответствующих коэффициентов для молекулы DF. Уравнение A. 65) с этими
значениями коэффициентов описывает экспериментальные данные [2169] с точностью не хуже
0,5 см, обеспечивает схождение колебательных уровней вблизи диссоциационного предела и
удовлетворительную экстраполяцию значений В, на область больших v. В табл. 7.3 приведены
значения ушм, /цт и коэффициентов а{ в уравнении, аппроксимирующем предельную кривую
диссоциации TF в состоянии X1!,+. В расчете термодинамических функций учитывались все
колебательно-вращательные уровни энергии TF, ограниченные предельной кривой диссоциации
состояния Х12+. При температурах до 4000 К погрешности вычисленных значений Ф°(Т) и S°(T)
160
Фтор и его соединения TF (г)
обусловлены главным образом неточностью фундаментальных постоянных и не превышают
0,02 Дж -К -моль. При более высоких температурах они возрастают из-за неточности
аппроксимации энергии высоких колебательно-вращательных уровней TF, но не превышают 0,1 и
0,2 Дж- К-1-моль-1 в значениях Ф° F000 К) и S0 F000 К).
Термодинамические функции TF (г) вычислялись ранее в предыдущих изданиях справочника
[87, 88] по модифицированному методу Гордона и Барнес и близким значениям постоянных.
Расхождения результатов этих расчетов и данных табл. 51 не превышают 0,05 и 0,15
Дж-К^-моль-1 в значениях Ф°(Т) и S°(T).
Константа равновесия реакции TF (r)=T (r)-j-F (г) вычислена по энергии диссоциации TF
ДД° @) = Do (TF) = 48 162 ± 65 см = 576,15 ± 0,78 кДж • моль,
которая была принята в справочнике на основании значений Do (HF)=47 361 + 64 см, G @, HF) =
=2047 см-1 и G @, TF)=1246 см.
Принятому значению Do (TF) соответствует
Д/ПТР, г, 0) = —277,396 + 0,80 кДж • моль'1.
Глава 8
ХЛОР И ЕГО СОЕДИНЕНИЯ
С1, С1", С12, СЮ, СЮ2, С12О, HCl, HOC1.
DCI, TCI, C1F, C1F3, C1F5
В настоящей главе рассматриваются 13 веществ — хлор и его соединения с кислородом, водородом,,
изотопами водорода и фтором в газообразном состоянии.
Для всех газов приводятся термодинамические свойства для природной изотопической смеси
хлора, поэтому внутримолекулярные составляющие термодинамических функций в тех случаях,,
когда это было возможно, вычислены по «эффективным» значениям постоянных, соответствующих
молекулам, содержащим атомы хлора 35>453С1. Соединения хлора с другими элементами будут
обсуждены в последующих главах. Термодинамические свойства всех соединений хлора, кроме*
С1 и С1~ рассматриваются для температур от 100 до 6000 К, одноатомных газов — до 10 000 К.
Для Cl2, C12O, HC1, C1F3 и C1F6 в Приложении 3 приведены данные, позволяющие оценить
влияние межмолекулярного взаимодействия на
свойства этих газов при повышенных давлениях.
Ионы С1+, С1+, Cl~, СЮ", СЮ;, а также такие
соединения, как НСЮ4, C1F4 и др. не включены
в справочник, так как можно предполагать, что
их равновесные концентрации в любых системах
в газообразном состоянии при 100 ^ Т <^ 6000 К
будут достаточно малы.
Таблица 8.1. Уровни энергии атомов С1 и С1~г
принятые для расчета
термодинамических функций
Атом
С1
С1-
Электрон-
ная
конфигурация
. . . З^Зр5
. . . З^Зр5
. . . З^Зр6
Терм
*РЧг
Х5
СМ
0
882,36
0
Pi
4
2
1
Cl (г). Термодинамические свойства
газообразного одноатомного хлора в стандартном
состоянии при температурах 100—10 000 К приведены
в табл. 52. Значения термодинамических
функций С1 (г) вычислены по уравнениям A. 3)—A.6), A.9), A.10), A.23)—A.25) с учетом двух
компонент основного . . .3s23/>5 2Р состояния атома С1 (табл. 8.1). Энергия компоненты 2Ру2 основного-
состояния атома С1 принята по работе Радзиемски и Кауфмана [3118], которые уточнили
рекомендацию Мур [2697] на основании данных работ [2044, 436]. Погрешности вычисленных
термодинамических функций С1 во всем интервале температур обусловлены только неточностью
принятых значений фундаментальных постоянных и не превышают 0,02 Дж-К^-моль в значениях
Ф°(Т) uS°(T).
Термодинамические функции С1 (г) вычислялись во многих работах, в том числе для широкого-
интервала температур в работах [3196, 1619, 3073] и справочниках [2029, 3564, 2496, 2556, 1946],
таблицах JANAF [2095] и предыдущих изданиях справочника [87, 88]. Данные, приведенные-
в работах [1619, 3564], отличаются от приведенных в табл. 52 на 0,04 и 0,02 Дж-К^-моль.
Расхождения всех остальных расчетов между собой и с настоящим изданием, несмотря на
различия использованных значений постоянных и энергии подсостояния Р^,, не превышают-
0,012 Дж -К-моль в значениях Ф° (Т) и S° (T).
Энтальпия образования одноатомного хлора
Д/Я°(а, г, 298,15 К) = 121,302+ 0,008 кДж • моль
принята по рекомендации КОДАТА—МСНС [1000]. Она получена на основании значения Do (Cl2),.
найденного в работах [1305, 2389, 2388] (см. ниже).
СГ (г). Термодинамические свойства газообразного отрицательного иона одноатомного хлора
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 53.
Термодинамические функции С1~ (г) вычислены при условии, что In Qm и его производные-
равны нулю, поскольку ион С1~ имеет основное состояние XS с конфигурацией . . .3s?3p6 и
неимеет связанных возбужденных состояний. Погрешности вычисленных значений термодинамиче—
162
Хлор и его соединения
ских функций С1~ во всем интервале температур определяются только неточностью
фундаментальных постоянных и не превышают 0,02 Дж-К^-моль в значениях Ф° (Т) и S° (T).
Термодинамические функции С1~ вычислялись в предыдущих изданиях справочника [87, 88] и таблицах JANAF
[2095 ]. При всех температурах значения Ф° (Т) и S° (T), приведенные в этих изданиях и в табл. 53,
согласуются в пределах 0,012 Дж-К^-моль.
Константа равновесия реакции С1~(г)=С1(г)+е (г) вычислена на основании значения
&ГН° @) = 348,70+0,20 кДж-моль в соответствии с принятой величиной сродства атома хлора
к электрону
Ао (С1) = —29 150 + 16 см = —348,70 + 0,20 кДж
моль
Сродство атома С1 к электрону исследовалось с применением различных экспериментальных
методик во многих работах (см. [3091, 609]). Наиболее точные данные были получены Берри
с сотр. [611, 612] на основании определения энергии фотоотрыва электрона от иона С1~ по границе
соответствующего непрерывного спектра поглощения (А=3430 + 1 А, Ао=—29 146 + 6 см) и
Мюкк и Поппом [2731 ] по границе непрерывного спектра испускания, соответствующего
радиационному захвату электрона атомом С1 (А=3428 + 3 А, Ао=—29 163 + 24 см). Принятое значение
Ао (С1) примерно на 15 кДж-моль, или 0,16 эВ, меньше (по абсолютной величине) значений,
полученных ранее при исследованиях методом поверхностной ионизации [456, 27, 29]; оно хорошо
согласуется с менее точными данными [1125, 3339] и рядом теоретических оценок. Принятому
значению соответствует
AfH° (СГ, г, 0) = —229,08 + 0,29 кДж • моль.
С12 (г). Термодинамические свойства газообразного двухатомного хлора в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 54.
В табл. 8.2 приведены значения молекулярных постоянных С12, использованные для расчета
термодинамических функций. Исследования спектров С12 и теоретические расчеты (см. [697])
показали, что молекула С12 имеет только два связанных электронных состояния Х2?+ и А3Ли, кор-
Таблица
Молекула
35С1а
сю
Н35С1
D35C1
Т36С1
SBC1F
8.2. Молекулярные постоянные С12,
ж ^ЧЛ 7flf\ fTW7
иОСЮЯНис
ff
хти
Am
ХЧ+
В3По+
В'0+
Примечание. Ниже
С12: а ие
6
СЮ: » А
НС1: а ш.
DC1: а ш.
Г.
0
17 832,76
0"
31 057 6
0
0
0
0
18 844,87
21 595,10
все величины ]
у. = —3,325 27 • Ю, и,.
—3,9078.
Ю-», а3 = — 7,С
г. = —9,4555 • Ю, ыеие =
3, =0,225
= —318;
.10-', B2 = 0,01J
6 А =—126; в
у, = 0,224 37, в,г, = 0,012
у =0,36-
TCI: a Приведено
C1F: * Вычислено
=
0,0792.
Ю-1; вр1=1д
значение Д&д;
559,750 7
246,7
859
559
2 990,946 3
2 144,77
1 739,10 а
786,147
352,79
107,90
криведены в
s, = 2,273 37.
)811-10-8, а
— 1,5013-10-
)-10-'.
СЮ,
О)
2,694
2,674
6,8
9,3 в
52,818
26,92 а
—
6,161
6,917
14,961
СМ.
10"*,
HCI
271>
7г
56 а
б
г
ы
, DCI, TC1 и C1F
В
см
1 0,244
0,162
0,623
0,480
10,593
5,448
3,745
0,516
0,329
0,209
е
153
54
6
6
416
39
8
48
5
0
«,.10»
0,151 63 б
0,202 2 Д
0,61
0,49 г
30,718 1 б
11,226
6,11
0,435 9
0,38 в
2,21
и, = 2,9204Ы0-в, ш.$.=
4 = — 5,587 5". 10-s;
Шег/е = — 0,05; гс
18; « а2 = 0,177 24.
53.10-в, Яе =
6 В1 = 0,84.
по соотношению A. 68); 6
= 2,31
ю-5.
шеУ» =
= 1
'2 ==
ю-2
10"
,0245 • 10"
4 • 10~4
в h =
-в/д
,«3 = 0,1201. К
9_
0,2161; в я2
= -с
De ¦ 106
0,191 95
0,236 5 с
2,6
1,4
531,936 в
139,0 б
77,0 б
0,89 а
1,15 а
3,1
= 6,029 84.
= 3,1678-Ю-12; г ш.у.
оа = —1,102
)"'; в 31 = 0,
),558 • 10"*;
• 10"*, а3 =
751 • Ю-5, р
Г шеУе — 1
к
в 1,987 2
2,435 5
1,569
1,788
1,274 6
1,274 6
1,273
1,628 31
2,039
2,560
Ю-8; в я2 =
= —0,238 26,
= —0,92.10-*;
= 0,4 - 10"в
,3727, »,,,==
163
Clg (г) Глава 8
релирующих с атомами С1 в 'валентных состояниях 2Р^г, чг (см. гл. 10, рис. 10.1). Все остальные
электронные состояния С12, коррелирующие с этими диссоциационными пределами (см. текст по 12),
являются отталкивательными состояниями. Электронные состояния, соответствующие ионам
C1+CZ), Ф, 1S)-\- CTQS) имеют энергии возбуждения свыше 70 000 см.
В спектрах С12 изучены переходы, связанные с состоянием Х1^ и компонентой 3По+
обращенного состояния А3Ии. Переходы на колебательно-вращательные уровни подсостояний 3П2я, 3111я
и 3П0- в спектрах С12 не наблюдались. Отсутствуют также строгие квантовомеханические
расчеты возбужденных электронных состояний этой молекулы. Расщепление компонент состояния
Л8Па (с конфигурацией ... Ъад2п^п?дЪоы) было оценено Малликеном [2753] и составляет около
250 см между подсостояниями 3П2в и 3По?.
Молекулярные постоянные С12 в состояниях X'Sj и А3П.0+ определялись в результате
исследований системы полос 3П0+ ^ ХХЕ± в спектрах испускания и поглощения [2321, 2791, 1398, 1399,
3200, 1305, 991, 988], где наблюдались переходы на уровни с v" <I 12 и v' ^ 31, а также
изучения спектра резонансной флуоресценции С12, возникающего при переходе из высокого
возбужденного состояния на уровни состояния -Х^2* с z/'=0-j-59 [1301] (см. также [3159]). Приведенные
в табл. 8.2 значения постоянных С12 в состоянии Хг2* приняты по работе Дугласа и Хоя [1301].
Авторы работы [1301 ] измерили в спектре резонансной флуоресценции 35С12 239 вращательных
линий, принадлежащих переходам в колебательные состояния с i;=0-f-59 (энергия уровня G E9)
по данным [1301 ] отличается от энергии диссоциации С12 всего на 16 см). На основании этих
данных были рассчитаны по методу РКР поворотные точки эффективной потенциальной кривой
для /=19, потенциальной кривой невращающейся молекулы и соответствующие им постоянные
в уравнении A.65), приведенные в табл. 8.2. Уравнение с этими постоянными описывает частоты
всех наблюдавшихся переходов для v s^ 40 с точностью не хуже 0,1 см. Получить простые
полиномиальные соотношения для уровней с 41 ^ у ^ 59 авторам работы [1301] не удалось из-за
быстрого изменения значений Go (v), Bt и Dt в этой области изменения квантового числа v, и они
ограничились приведением численных значений этих величин. Постоянные, предложенные
Дугласом и Хоем, хорошо согласуются с экспериментальными данными, полученными при изучении
системы полос ^43П0? — Х1^*, а также спектров комбинационного рассеяния С12 [1992, 1967].
Приведенные в табл. 8.2 молекулярные постоянные С12 в состоянии 3П0+ приняты по работе
Клайна и Коксона [991], которые получили их на основании обработки результатов собственных
измерений и данных [3200, 1305].
Термодинамические функции для природной изотопической смеси С12(г) были вычислены
с учетом двух электронных состояний: Х1^ и А3Пи. Значения Qm и ее производных вычислялись
по соотношениям A.76)—A.78). Статистическая сумма по колебательно-вращательным уровням
энергии состояния -Xй!]? и ее производные были рассчитаны непосредственным суммированием
по уравнениям A.73)—A.75). Уровни энергии со значениями v <^ 40 находились по уравнениям
A.65) и A.62) со значениями коэффициентов Ykl для «эффективного» изотопа 35'453С12,
приведенными в табл. 8.3 и полученными из соответствующих постоянных, найденных в работе [1301 ]
(см. табл. 8.2). Для v J> 41 уровни энергии рассчитывались по тем же уравнениям и значениям
G (v), Вг и D,, приведенным в табл. 8.3. Эти значения были получены на основании
соответствующих величин для молекулы 35С12, найденных авторами [1301] (для y=41-f-59) и экстраполяции
их на значения у — 60 и 61. В расчете @№ вр, Qm_ вр и QW_ Bp учитывались все
колебательно-вращательные уровни энергии, ограниченные предельной кривой диссоциации; в табл. 8.3 приведены
коэффициенты а{ в уравнении, аппроксимирующем эту кривую, а также значения ушах и Jum.
Статистическая сумма по колебательно-вращательным уровням энергии состояния А3П.а и ее
производные вычислялись непосредственным суммированием по колебательным уровням энергии
и интегрированием по значениям / с помощью уравнений типа A.82). В расчете не учитывалось
расщепление компонент состояния 3ПЯ (мультиплетность и вырождение уровней учитывались
статистическим весом 6) и использовались коэффициенты Ykl в уравнениях A.65) и A.34) для
подсостояния 3По?- В статистическую сумму и ее производные включались все уровни энергии
со значениями /<^/шм,,, где для 7maX|t принималась линейная зависимость от v (см.
уравнение A.81)). В табл. 8.3 приведены принятые значения коэффициентов Ykl, ишдх и /1Ш для
«эффективной» молекулы 35-453С12.
164
Таблица 8.3. Значения коэффициентов в уравнениях:, описывающих уровни энергии (в см), а также значения vmsx и
принятые для расчета термодинамических функций С12, СЮ, НС1, DC1, ТС1 и CIF
Коэффициенты
С12
АШа
СЮ
Х2П«
НС1
DC1
ТС1
C1F
»П
0+
Т.- Ю-* 0 1,783 28 0 3,105 69 0 0 0 0 1,884 49 2,159 51
Ую-10-3 0,555 916 0,245 01 0,856 846 0,539 069 2,990 73 2,144 65 1,775 86 0,784 047 0,35194 0,107 64
Уго-Ю -0,265 748 —0,263 82 —0,660 655 — —5,314 58 —2,732 92 —1,873 84 -0,600 296 -0,688 4 —0,148 89
У30-103 —0,325 799 —23,34 —3,013 03 — 36,972 5 13,633 8 . 7,740 58 2,166 19 ' 21,45 136,28
У40-Ю3 -0,221170 9,2 — — —41,7817 —11,048 5 —5,19412 — — —78,44
Уво-106 0,378 793 —14,5 — — 288,416 54,691 21,29 — — —
Ув0.10в —0,057 862 0,982 6 — — —107,607 —14,632 4 —4,716 59 — — —
У01 0,240 8 0,160 32 0,620 8 0,475 9 10,589 5 5,445 43 3,733 68 0,513 997 0,327 9 0,208
Уп.10а -0,148 535 —0,198 1 —0,605 907 — —30,730 7 —11,332 1 -6,433 79 -0,432 8 —0,377 —2,194
У21.104 —0,038 018 —1,072 — — 20,666 9 5,465 04 2,569 22 — —5,53 —
У31-106 0,068 418 0,89 — — —240,522 —45,609 2 -17,754 7 — — —
У41.10« —0,005 362 — — — 21,838 2,969 54 0,957 198 — — —
Y61-W> — — — — —14,756 4 —1,438 92 —0,384 061 — — —
У02-106 -0,186 743 -0,2301 -2,58 — —527,887 —139,589 -65,623 4 -0,888 —1,14 -3,07
K=De)-10-4 2,027 50 — — — 3,724 0 3,724 0 3,724 0 — — —
«VlO1 0,396 980 — — — 6,044 91 3,016 49 1,989 72 — — —
а2.106 0,366 729 — — — 160,243 45,482 3 23,063 3 — — —
а3-1012 -0,510 231 — — — 4310,42 352,978 30,982 4 — — —
ymax 61 32 48 — 20 28 34 50 10 6
/Um 418 227 264 — 81 ИЗ 136 289 148 59
a Приведенные постоянные описывают уровни энергии с и <[ 41. Для уровней с v > 41 использованы следующие данные (в см):
V
G(v)-G @)
в.
Я,-10'
41 17 498,48 0,16149 4,42
42 17 761,51 0,158 28 4,50
43 18 013,47 0,154 96 4,78
44 18 254,12 0,151 45 5,41
45 18 477,49 0,147 54 6,05
46 18 697,70 0,143 44 6,49
47 18 899,46 0,138 94 7,52
б а = -318 см.
V
G(v)-G @)
Б,
D, ¦ 107
48 19 086,08 0,133 82 8,50
49 19 256,56 0,128 20 10,20
50 19 409,27 0,121 68 12,26
51 19 542,48 0,114 24 15,01
52 19 654,81 0,105 76 18,34
53 19 745,79 0,096 63 21,22
54 19 817,21 0,087 30 25,61
V
G (v) - G @)
в.
D, ¦ 107
55 19 871,28 0,077 90 28,90
56 19 911,57 0,069 11 35,09
57 19 940,95 0,060 46 39,93
58 19 962,24 0,053 13 52,68
59 19 977,70 0,045 12 64,13
60 19 991,58 0,037 11 75,6
61 19 997,11 0,0291 87,0
%
СЮ (г) Глава 8
Погрешности табулированных значений термодинамических функций С12 (г) при температурах
до 3000 К определяются главным образом неточностью фундаментальных постоянных и не
превышают 0,02 Дж-К^-моль в значениях Ф°(Г)и5°(Г). При более высоких температурах из-за
пренебрежения расщеплением компонент состояния 3ПЯ погрешности быстро растут с ростом
температуры, особенно в значениях С'(Т) и Н°(Т) — Н°@). При 6000 К погрешности в значениях
Ф° (Т), S0 (Т) и С°р (Т) оцениваются в 0,1, 0,5 и 1 Дж-К^-моль.
Термодинамические функции С12 (г) вычислялись во многих работах, среди которых можно
отметить [1619, 41, 3196, 3408, 1426, 3564, 1308] (до 3000 К) и [3073, 2029, 2496, 2556, 1454],
таблицы JANAF [2095] и предыдущие издания справочника [87, 88]. В расчетах использовались
разные значения молекулярных постоянных и разные методы расчета. При температурах до
3000 К большинство данных согласуются между собой и с табл. 54 в пределах 0,1 и 0,2 Дж-К-1Х
Хмоль^в значениях Ф°(Т) и S°(T), а имеющиеся расхождения обусловлены главным образом
различиями в принятых постоянных 1. При более высоких температурах наилучшее согласие
имеет место с данными Фебера и Херрика [1454], выполнившими расчет непосредственным
суммированием по колебательно-вращательным уровням энергии состояний X1!,*, 3П2, 3И1 и 3По+
(при 6000 К расхождения достигают 0,1, 0,9 и 2,2 Дж-К^-моль в значениях Ф°(Т), S°(T)
и Ср°(Т) прежде всего из-за разного учета вклада состояния 3П и некоторого отличия принятых
молекулярных постоянных), со вторым изданием справочника [88], где расчет был выполнен по
модифицированному методу Гордона и Барнес (расхождения до 0,13 и 0,7 Дж-К^-моль в
значениях Ф°(Т) и S°(T)), и с данными работы [2496] (до 0,3 и 0,8 Дж -К -моль-1 в Ф° (Т) и S° (T)).
Существенно больше расхождения с данными работ [2095, 2556, 1308], в частности, из-за
использования в этих изданиях метода Майера и Гепперт-Майер (до 0,8, 2,5 и 4 Дж-К^-моль в
значениях Ф°(Г), S°(T) и Ср°(Т)).
В настоящем справочнике двухатомный газообразный хлор принят за стандартное состояние
этого элемента и в соответствии с этим
Д/Р(С12, г) = 0.
Константа равновесия реакции С12 (г)=2С1 (г) вычислена с использованием
ДД° @) = Do (С12) =: 19 999 ± 1 см = 239,240 + 0,012 кДж • моль
в соответствии с рекомендованным КОДАТА—MGHG [1000] значением k.fH° (G1, г, 298, 15 К).
Энергия диссоциации С12 определялась в ряде работ на основании изучения схождения
колебательных уровней состояния 3По+ по границе схождения полос системы 3П0+ — Х1^* в спектре
поглощения С12 [1305, 1399, 2321 ] и по границе колебательных уровней состояния Х11,+д из
данных по спектру резонансной флуоресценции [3159, 1301]. Принятое КОДАТА—МСНС значение
основывается на обработке данных Дугласа и др. [1305] по колебательным уровням состояния
3По+ вблизи его диссоциационного предела в работе Ле Роя и Бернстейна [2392], которые
нашли Do C5С12) — 19 997,25 + 0,3 см.
Позднее это значение было несколько уточнено Ле Роем [2386а] A9 997,14+0,14 см), однако
это уточнение несущественно для расчетов термодинамических свойств С13 (г). Практически
совпадающее значение было получено Дугласом и Хоем [1301] по схождению уровней состояния
¦Х^2* из данных по спектру флуоресценции с использованием методики Ле Роя и Бернстейна
[2392] для нахождения дальнодействующей части потенциальной кривой. Существенно более
высокое значение B0 062 + 10 см), предложенное ранее в работе Рао и Венкатесварлу [3159]
на основании экстраполяции уровней состояния ХХ2* в изученном ими спектре флуоресценции,
по-видимому, обусловлено неточностями в анализе и нумерации линий.
Принятое КОДАТА—МСНС значение Do (Cl2) соответствует энтальпии реакции диссоциации
для природной смеси изотопов хлора.
СЮ (г). Термодинамические свойства газообразного оксида хлора в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 55.
1 Существенно больше (до 2 Дж-К -моль) расхождения в Ср (Т) и, следовательно, в Н°{Т) — Я°@).
166
«Хлор и его соединения СЮ (г}
В табл. 8.2 приведены молекулярные постоянные молекулы 35СЮ, использованные в расчете
термодинамических функций. Система валентных электронных состояний молекулы СЮ до
настоящего времени мало изучена. В спектрах СЮ наблюдались переходы между двумя валентными
•состояниями Х2П 5р* А2П [1350, 2950, 3067], единственными принадлежащими двум нижним
электронным конфигурациям , . .а2тт:4тс3 и . . .а2я:3я4и соответствующими пределам С1 BР)+О CР)
и Cl BР)+О A-D). Можно предположить, что остальные валентные состояния СЮ, в том числе
квартетные состояния, являются отталкивательными или слабосвязанными состояниями, так как
¦они соответствуют возбуждению связывающих п- или а-электронов на разрыхляющую о-орбиталь.
Молекулярные постоянные СЮ определялись при исследовании электронных спектров [2950,
5066, 3067, 1350, 506а], инфракрасных спектров СЮ в матрицах [3221, 354, 393], микроволновых
спектров и спектров ЭПР [372, 888, 2133, 374, 3752]. Однако до 1973 г. эти исследования не
позволили получить надежные значения колебательных постоянных СЮ в состоянии Х2П, так как
в спектрах поглощения СЮ [1350, 3066, 3067] наблюдались переходы только с уровня v=0,
в спектре испускания [2950] было трудно дать однозначную нумерацию полос, а все эти данные
противоречили интерпретации результатов исследований спектров в матрицах [3221, 354, 393],
предложенной авторами этих работ.
Приведенные в табл. 8.2 значения колебательных постоянных приняты по работе Баско и Морса
[506а], изучивших в спектре поглощения СЮ шесть ридберговских систем и наблюдавших
переходы на уровни z/=0 и 1. Используя эти данные и данные Портера [3066, 3067] по спектру
испускания, авторы [506а] определили значения ш"в и шех"е с точностью 2 и 1 см соответственно.
Полученное Баско и Морсом [506а] значение ДбЛ/г не противоречит экспериментальным данным [393 ]
по инфракрасному спектру в матрице при изменении интерпретации этих данных. Приведенные
в таблице значения вращательных постоянных СЮ в состоянии Х2И основаны на работах Амано
-с сотр. [372, 374], в которых исследовались микроволновые спектры 35СЮ и 37СЮ, постоянная
спин-орбитального взаимодействия была принята по рекомендации работы [506а]. Последняя
существенно отличается от полученной при исследовании спектра ЭПР [888] (—282 + 9 см).
Колебательные постоянные СЮ в состоянии А2П также основаны на работе Баско и Морса [506а],
которые рассчитали их по измеренным ими кантам полос 2—0 и 3—0, данным для начал полос
прогрессии t/ <- 0, полученным Дьюри и Рамзи [1350] для z/=5-f-25, и канту полосы 4—0 из
работы [3066]. Вращательные постоянные приняты по работе [1350].
При подготовке справочника величины Yk0 и YkX для молекулы СЮ в состоянии Х2И были
получены на основании постоянных, приведенных в табл. 8.2, и значения Do (СЮ)=22 152+
+ 10 см [1350] по методикам, изложенным в разделе 1.4. Соответствующие значения для молекулы
.35,453qo ПрИВедены в табл. 8.3. Там же приведены значения ушах и /iim. Значения QBB и ее
производных вычислялись по уравнениям A.90)—A.92) в предположении, что Ql?}-Vg = (PxlP^)Q%$.ev-
Статистическая сумма по колебательно-вращательным уровням энергии состояния Х2П и ее
производные были вычислены по уравнениям A.73)—A.75) непосредственным суммированием
по у с заменой суммирования интегрированием по значениям / с помощью уравнений типа A.82).
Ограничение суммирования по / осуществлялось в предположении, что с ростом v значения /шах ,
линейно уменьшаются от /цт до 0 при v=vm&x-{-l. Энергии уровней вычислялись по уравнениям
{1.65) и A.42) с коэффициентами Ykl и А из табл. 8.3. Погрешности вычисленных значений
термодинамических функций при низких температурах обусловлены неточностью принятой постоянной
спин-орбитальной связи @,2 Дж-К^-моль в значении Ф° B98,15 К)), а при средних
температурах — отсутствием экспериментальных данных о колебательно-вращательных уровнях основного
состояния с v > 1 и большими значениями / @,1—0,15 Дж-К^-моль в Ф°(Т) и S°(T) при
3000 К); при высоких температурах к ним добавляются погрешности из-за приближенного учета
состояния А2П, возможного вклада других возбужденных состояний, приближенного метода
ограничения числа колебательно-вращательных уровней энергии состояния Х2П; погрешности
в значениях Ф°(Г) и S°(T) при 6000 К могут достигать 0,2 и 0,5 Дж-К^-моль \
1 После завершения расчета термодинамических функций СЮ (г) Коксон и Рамзи [1108] опубликовали новое
исследование спектра поглощения СЮ, в котором в системе АШ—ХШ наблюдались переходы с уровней t>"=0, 1, 2
и был уточнен анализ ряда полос. Найденные значения колебательных постоянных в состоянии Х2Н (*>,=
¦=855,46 см, юеже=5,70 см) удовлетворительно согласуются с приведенными в табл. 8.2, а для-состояния АШ
<7',=31650 см, «,=518,4 см, «^=6,61 см, шеу,=0Л59 см) существенно отличаются от принятым
167
СКМг) Глава в
Термодинамические функции СЮ (г) вычислялись ранее в работах [764] (до 3000 К), [2556,
1308, 2496], таблицах JANAF [2095.] и предыдущих изданиях справочника [87, 88]. [Максимальные
расхождения между данными всех этих работ и табл. 55 имеют место в Ф° B98,15 К) из-за
пренебрежения расщеплением компонент состояния Х2Н (примерно 3,8 Дж-К^-моль). При
высоких температурах расхождения обусловлены некоторым различием постоянных, а также тем,
что расчеты [2095,2556,1308, 2496] проводились без ограничения числа
колебательно-вращательных уровней энергии и без учета состояния А2И. Они достигают 0,5 Дж-К^-моль в Ф° (Т)
и 1 Дж-К^-моль-1 в S°(T) [2095, 2556].
Константа равновесия реакции СЮ (г)=О (г)+С1 (г) вычислена на основании принятого
значения энергии диссоциации СЮ
Дг#° @) = Do (СЮ) = 22 184 ± 3 см = 265,380 + 0,036 кДж • моль.
Это значение получено Коксоном и Рамзи [1108] в результате короткой (на 53 см)
экстраполяции высоких колебательных уровней состояния А2П с использованием теории дальнодей-
ствующих межатомных потенциалов, развитой Ле Роем и Бернстейном. Близкое, но менее точное
значение B2 152 + 10 см) было найдено ранее в работе Дьюри и Рамзи [1350].
Принятому значению Do (CIO) соответствует
AfH°(ClO, г, 0) = 101,023 ±0,11 кДж-моль.
С1О2 (г). Термодинамические свойства газообразного диоксида хлора в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 56.
Термодинамические функции СЮ2 (г) вычислялись по молекулярным постоянным,
приведенным в табл. 8.4. Молекулярные постоянные СЮ2 определялись в ряде работ на основании
результатов исследований электронных [3203, 1044, 1402, 742, 743, 3291], инфракрасных [2856, 3201,
Таблица 8.4. Значения молекулярных постоянных звСЮ2 (в см), принятые для расчета
термодинамических функций
V! 945,3
v2 447,9
v3 1110,8
xn —4,20
2-22 """UjlO
¦^зз —6,75
*ia -4,50
x13 —15,00
Примечание. 5?2ВХ:
va = 289,5, v3 = 783,6
x23
¦00
¦^000
aA
z
„A
T0 = 0, p
CM
—2,40
1,73718
0,331970
0,277 991
—0,013
—0,033 34
0,015
r = 2, a = 2; A2/
.a?
«f
n
аз
aP
«f
»3C
T "H
0,002 6
0,000 29
0,0014
0,001 73
0,000 79
0,0012
—2,843 -10-*
016,34 см, Pi = 2,
хьььь
xceee
Ti4e<!
TaSa4
0 = 2 и
_!
—4
1
—7
1
—2
14
,519.10-»
,628-10-'
,312-10~5
,292-10-'
,920-10-»
,008-10-»
,5
707,9,
3203] и микроволновых спектров [ИЗО, 1129, 1128, 3022, 3706, 2522 и 2315], спектра
флуоресценции [1131] и дифракции электронов [972, 973]. Совокупность колебательных постоянных
СЮ2 в состояниях Х2Вг и Л2А2 была определена Куном и Ортицем [1046], а затем уточнена
Ричардсоном и др. [3203] на основании изучения электронного спектра и результатов исследования
колебательно-вращательного спектра СЮ2 в работе Нильсена и Вольтца [2856]. Авторы работы
[3203] получили значения постоянных (табл. 8.4), описывающие с хорошей точностью
колебательные уровни основного состояния с учетом резонанса Дарлинга—Деннисона. Наиболее
надежные значения вращательных постоянных, постоянных центробежного растяжения и колебательно-
вращательного взаимодействия определены Пиллаи и Керлом [3022] на основании выполненного-
ими исследования микроволнового спектра, исследований электронного, спектра в работе Бранда
в справочнике. Однако достаточно высокая энергия состояния А2Л делает несущественным это уточнение
молекулярных постоянных СЮ для расчета термодинамических функций. Значения Ф° (Т), вычисленные по
постоянным, найденным Коксоном и Рамзи [1108], при 298,15, 3000 и 6000 К отличаются от приведенных в табл. 55
на 0,02, 0,01 и 0,02 Дж-К-^моль.
168
Хлор и его соединения С12О(г)>
и др. [743] и спектра резонансной флуоресценции в работе Керла и др. [1131]. Им соответствуют
структурные параметры молекулы СЮ2 в состоянии Х2Вг: г0 (С1—О) = 1,476 А, ^ О—С1—0 =
=117,5°, и в состоянии А2А2: r0 (C1—О) = 1,619 А, ^ О—С1—0 = 107,0°.
Внутримолекулярные составляющие термодинамических функций С102(г) вычислялись
по уравнениям A.122)—A.124), A.127) и A.128) с учетом ангармоничности колебаний и
колебательно-вращательного взаимодействия — уравнения A.131)—A.133), центробежного
растяжения — уравнения A.143)—A.144) и резонанса Дарлинга—Деннисона — уравнения A.156)—
A.158). Составляющие возбужденного состояния Л2А2 были вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.171)—A.173) с учетом различия
частот СЮ2 в состояниях Х2В1 и А2А2, но без учета различия остальных постоянных. Все расчеты
были сделаны по молекулярным постоянным 35СЮ2. Основные погрешности в вычисленных
значениях термодинамических функций СЮ2 обусловлены при низких температурах использованием
в расчете постоянных молекулы 36СЮ2 вместо эффективных постоянных, соответствующих
35'453СЮ2; при умеренных температурах к ним добавляются погрешности из-за неточности
аппроксимации колебательно-вращательных уровней состояния Х2В1 уравнениями с принятыми
молекулярными постоянными СЮ2; при температурах выше 3000 К начинает сказываться применение
приближенного метода расчета. При 298,15, 3000 и 6000 К погрешности в значениях Ф°(Т)
могут достигать 0,05, 0,3 и 0,6 Дж -К •моль, в том числе из-за использования в расчете
постоянных молекулы 35СЮ2: 0,04, 0,07 и 0,08 Дж-К^-моль соответственно.
Термодинамические функции СЮ2 (г) ранее вычислялись в работах [1426, 1699, 2769, 2778,
3800] ив справочных изданиях [88, 2095, 2556]. В работах [1426, 2769, 2778, 3800] расчеты
были выполнены до 2000 К и без учета мультиплетности вращательных уровней молекулы СЮ2.
Данные этих работ отличаются от приведенных'в табл. 56 на 5,5—6 Дж-К^-моль [главным
образом из-за этой ошибки. Гордон [1699] и авторы работы [2556] выполнили расчет по методу
Пеннингтона и Кобе со значениями колебательных постоянных, близкими к принятым в табл. 8.4,
но без учета колебательно-вращательного взаимодействия, центробежного растяжения и
возбужденного состояния Л2А2. В работе [1699] использованы также неточные значения
вращательных постоянных СЮ2, что частично компенсирует методические ошибки.
Данные последней работы, воспроизведенные в таблицах JANAF [2095], отличаются от табл. 56
на величины до 0,35 Дж-К^-моль в значениях Ф° (Т) (наибольшее расхождение при низких
температурах) и до 1,5 Дж-К^-моль в значениях S° (T). Данные работы [2556] и табл. 56
совпадают в пределах 0,1 Дж-К^-моль при Т ^ 2000 К; при 6000 К расхождения достигают
0,5 и 2 Дж-К^-моль в значениях Ф° (Т) и S° (T). Данные предыдущего издания справочника
[88] были получены с неточными значениями колебательных постоянных СЮ2, соответствующие-
расхождения достигают 1,1 Дж-К^-моль в значении Ф°F000 К).
Константа равновесия реакции СЮ2 (г)=2 О (r)-f C1 (г) вычислена на основании значения
Н° @)=505,714 + 6 кДж-моль, соответствующего энтальпии образования СЮ2(г)
A^fGlOj, г, 298,15 К)= 105 + 6 кДж • моль.
Эта энтальпия образования принята в справочнике по результатам калориметрических
измерений энтальпии реакции разложения СЮ2 в работах Бута и Боуэна [702] и Уоллеса и Гудива
[3864] (соответственно 98,3 и НО кДж-моль). Близкое значение A00 + 10 кДж-моль) было
найдено Флис [268] из сложного термохимического цикла, включающего реакции восстановления
растворов СЮз и СЮ~. Исследования спектров поглощения и предиссоциации СЮ2 приводят
к значениям 102 кДж-моль и 76 кДж-моль [2550, 1484].
С12О (г). Термодинамические свойства газообразного оксида дихлора в стандартном состоянии
при температурах от 100 до 6000 К приведены в табл. 57.
Молекулярные постоянные С12О, использованные для расчета термодинамических функций
С12О (г), приведены в табл. 8.5. Молекула С12О имеет основное состояние ХХА; сведения о
возбужденных электронных состояниях С12О в литературе отсутствуют. Колебательный спектр С12О
исследовался в работах [452, 1925, 1871, 3582, 3220]. Наиболее подробное исследование было
выполнено в работе Рочкинда и Пиментела [3220], в которой были изучены инфракрасные спектры,
шести изотопических модификаций С12О в матрицах из аргона и азота, а также в газовой фазе..
169
€1,0 (г) Глава 8
Таблица 8.5. Значения молекулярных постоянных и ст в состояниях ХгА, принятые для расчета
термодинамических функций С120 и Н0С1 (р_^ = 1)
Молекула
С120
Н0С1
V2
V3
см
640
3609,2
300
1239,9
686
725
111. 10117
lAlBLC 1U
г3 • см8
1168,5
4,357
a
2
1
Ломимо основных частот, авторы наблюдали ряд обертонов и составных частот, что позволило
уточнить отнесение полос и исправить ошибки, допущенные в предыдущих работах. Значения
основных частот 35С12О, найденные в работе [3220] по спектру газообразного С12О, приведены
в табл. 8.5. Поскольку эти значения соответствуют центрам, а не началам соответствующих полос,
их погрешности могут достигать 3—5 см. В табл. 8.5 приведено также произведение моментов
инерции, соответствующее «эффективной» молекуле 35> *5"С12О и вычисленное по вращательным
постоянным молекул 35С12О, 35С137СЮ и 37С12О, полученным в работах Джексона и др. [1886,
2080] в результате изучения микроволнового спектра С12О. Этим постоянным соответствуют
r0 (C1—О) = 1,700 38 + 0,000 69 A, ^i 01—0—0=110, 963 + 0,077°. Близкие значения структурных
параметров С12О были получены в электронографических исследованиях [1339, 526].
Внутримолекулярные составляющие термодинамических функций С12О (г) были вычислены
по уравнениям A.122)—A.124), A.128) и A.130) в приближении «жесткий
ротатор—гармонический осциллятор». В расчете были использованы значения колебательных постоянных
молекулы ЗБС12О, так как учет различия постоянных этой молекулы и молекулы 35>453С12О не имел
смысла из-за невысокой точности принятых колебательных постоянных С12О. Основные
погрешности вычисленных значений термодинамических функций обусловлены неточностью принятых
значений основных частот и применением приближенного метода расчета. Погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,2, 2 и 3 Дж-К^-моль соответственно.
Термодинамические функции С12О вычислялись ранее в ряде работ, в большинстве из них
([2474, 1426, 2417, 2767, 3800]) расчеты были выполнены для температур до 1500—2000 К с
использованием неверных значений основных частот С12О. По неверным значениям основных частот
были выполнены также расчеты в справочнике [2556] и предыдущем издании справочника [88].
Расхождения всех этих данных с величинами, приведенными в табл. 57, достигают 2 Дж-К X
Хмоль-1 в Ф°A000 К) и 3,5—4 Дж-К-моль-1 в значениях Ф° (Т) и S° (T) при 6000 К. В
таблицах JANAF [2095] расчет был выполнен по постоянным, практически совпадающим с принятыми
выше, и результаты расчетов согласуются в пределах 0,03 Дж-К^-моль во всем интервале
температур. В работе [3220] расчет был выполнен для 21=200-j-1400 К по значениям основных
частот С12О в конденсированном состоянии, что обусловило некоторое отличие (до 0,2 Дж-К X
Хмоль) полученных данных от приведенных в табл. 57.
Константа равновесия реакции С12О (г)=О (г)+2С1 (г) вычислена с использованием значения
кгН° @)=405,196 + 10 кДж-моль, которое соответствует принятой в справочнике энтальпии
¦образования С12О г)
AfH°(Cl2O, г, 298,15 К) = 79 + 10 кДж • моль.
Результаты работ, в которых проводилось измерение энтальпии образования С12О (г),
представлены в табл. 8.6. Работы [2551, 1770, 3864, 3676, 2840] выполнены с недостаточно
охарактеризованными образцами и при недостаточном контроле за степенью протекания реакций. На
основании расчетов по сложному термохимическому циклу, основанному на литературных данных,
опубликованных до 1955 г., Эванс и др. [1426] рекомендовали значение 75,7 + 1,5 кДж-моль.
Кустодина, Мищенко, Флис [148, 269] измерили при 0, 25 и 35° С энтальпию восстановления
раствора С12О в СС14 перекисью водорода, откуда для энтальпии образования этого раствора
при 25° С было получено значение 51,6+3,3 кДж-моль. Йост, Фелт [4042] измерили давление
С12О над раствором в СС14 @—25° С) и получили для энтальпии растворения С12О в СС14
значение 27,6 кДж-моль, отнесенное ими к 25° G. Принятое в справочнике значение получено
комбинированием этих двух величин.
170
.Хлор и его соединения
НС1 (г)
Таблица 8.6. Результаты определений энтальпии образования С12О (г) (в кДж-моль)
Авторы
Метод
H° (C12O, г,
298,15 К)
Томсен, 1882 [3676] (см. [1426])
Майер, 1924 [2551 ] (см. [1770, 3864])
Нейман, Мюллер, 1929 [2840]
Уоллес, Гудив, 1931 [3864]
Гюнтер, Векуа, 1932 [1770]
Эванс и др., 1955 [1426]
Флис и др., 1961 [269, 148] Йост,
Фелт, 1934 [4042]
Фишер, 1968 [1489]
Измерение энтальпии гидролиза С12, С12О и других 76,6
реакций
Измерение энтальпии разложения С12О=С12+1/2Оа 82,0+4,2
Измерение энтальпии растворения С12О в воде 74,1
Измерение энтальпии разложения С12О=С12+1/2О2 89,5+2,5
Измерение энтальпии реакции С12 и С12О с подкислен- 75,3
ным раствором KI
Измерение энтальпии разложения С12О=С12+1/2О2 103,3+0,5
Пересчет литературных данных по AG реакций С12 75,7+1,3
и С12О
Измерение энтальпии образования раствора С12О
в СС14 [269, 148] и энтальпии растворения С12О в СС14
[4042|
расчет при 283 К (см. [2095]) 87,9+2,5
расчет при 298,15 К 79,2+4,2
Масс-спектрометрическое измерение потенциалов по- 84+ДЗ
явления СЮ+ из С12О и потенциала ионизации СЮ
Авторы справочника [2095] провели по данным работ [269, 148, 4042] аналогичный расчет
для 283 К и получили, после пересчета к 298, 15 К, значение 87,9 + 2,5 кДж-моль.
Разность объясняется необычно сильной зависимостью измеренной в работах [269, 148]
энтальпии реакции от температуры. Возможно, реакция восстановления С12О протекала недостаточно
¦быстро при низких температурах и это сказалось на результатах измерений. В настоящем
справочнике отдается предпочтение результатам измерений при 298,15 К. Погрешность
рекомендованного значения увеличена с учетом расхождения результатов обработки данных, полученных при
.283 и 298, 15 К.
НС1 (г). Термодинамические свойства газообразного хлорида водорода в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 58.
В табл. 8.2 приведены значения молекулярных постоянных Н35С1, выбранные в результате
-анализа литературных данных.
Исследования электронных спектров НС1 [3686, 3684] показывают, что состояние Хг1,+
является единственным стабильным валентным состоянием этой молекулы с энергией ниже
60 000 см. Состояния а3П, А1П и ?32+, также коррелирующие с диссоциационным пределом
Cl BP)+H BS), являются отталкивательными, а состояние V1!,*, коррелирующее с пределом
С1~ Aj5)+H+, имеет энергию свыше 76 000 см. Спектры НС1 исследовались для определения
молекулярных постоянных в десятках работ, посвященных изучению электронных (см. [3684,
3686]), колебательно-вращательных [2064, 2632, 1919, 982, 2436, 2437, 2826, 2669, 1794, 3050,
.2722, 3146, 3144, 3149, 3148, 3153, 3154, 13, 2406, 2405, 3904, 3905] и вращательных [2569, 1824,
.2161, 2162, 1211] переходов. Наиболее точные значения постоянных молекул Н35С1 и Н37С1 были
получены в исследованиях Ранка с сотр. [3146, 3144, 3149, 3148, 3153] (см. также [3154]),
выполненных на приборах с большой разрешающей способностью. На основании анализа восьми
полос A—0, 2 — 0, 3 — 0, 4 — 0, 5 — 0, 3 — 1, 4 — 2, 5 — 3) включая линии с высокими
значениями / (в полосе 1 — 0 до /=30) были определены постоянные, приведенные в табл. 8.2,
которые с хорошей точностью описывают все наблюдавшиеся переходы. Найденные в этих
работах значения постоянной Во согласуются с результатами исследований микроволнового спектра
Н3бС1 [2161, 2162, 1211] с точностью лучше чем 0,0001 см.
При подготовке настоящего справочника колебательно-вращательные постоянные НС1 были
уточнены по методике, изложенной в разделе 1.4, на основании экспериментальных данных
Рэнка, Рао и Уиггинса [3153] для значений В, и Go (v) при у^5и значения Do (HC1)=35 759 см.
Полученные таким образом постоянные описывают все данные [3153] с точностью лучше, чем
постоянные, найденные в этой работе, а также обеспечивают схождение колебательных уровней
171
Н0С1 (г) Глава &
вблизи диссоциационного предела при уша1=20. Значения Ykl, рассчитанные для молекулы HG1,.
соответствующей природной смеси изотопов хлора, приведены в табл. 8.3. Там же приведены
/lim и коэффициентов а,- в уравнении, аппроксимирующем предельную кривую
диссоциации состояния ХХИ +.
Термодинамические функции НС1 (г) вычислялись без учета возбужденных электронных
состояний этой молекулы. Значения (?)S. „р и ее производных находились непосредственным
суммированием по колебательно-вращательным уровням состояния Х1!,*—уравнения A.73) —
A.75). В расчете учитывались все уровни, ограниченные предельной кривой диссоциации.
Колебательно-вращательные уровни энергии вычислялись по уравнениям A.65) и A.62) х. Погрешности
вычисленных термодинамических функций НС1 при температурах до 4000 К обусловлены
главным образом неточностью принятых фундаментальных постоянных и не превышают 0,02 ДжХ
ХК-моль в значениях Ф°(Т) и S°(T). При более высоких температурах начинает сказываться
отсутствие экспериментальных данных о высоких колебательно-вращательных уровнях HG1
(с v > 5 и / > 25). Соответствующие погрешности могут достигать 0,05 и 0,1 Дж-К^-моль
в значениях Ф° F000 К) и S° F000 К).
Термодинамические функции НС1 вычислялись во многих работах, в том числе при невысоких
температурах в работах [1619, 1694, 1695, 3755, 2440] и до 5000-6000 К в работах [2029, 3073,
1454, 2556, 193, 3946], таблицах JANAF [2095] и предыдущих изданиях справочника [87, 88].
Несмотря на применение разных методов и несколько отличающихся фундаментальных и
молекулярных постоянных, расхождения между результатами расчетов невелики. С данными,
опубликованными после 1955 г., эти расхождения не превышают 0,12 и 0,35 Дж-К^-моль [197, 87,
2095, 2556, 1454] в значениях Ф° (Т) и S° (T). Наилучшее согласие имеет место с расчетами
[88, 1454] (в пределах 0,03 и 0,25 Дж-К^-моль-1 в значениях Ф°(Т) и S°(T)).
Константа равновесия реакции НС1(г)=Н(г)+С1 (г) вычислена на основании значения
Д Н° @)=D0 (Н35> 457С1)=427,78 + 0,13 нДж-моль, соответствующего энтальпии образования
НС1
г, 298,15 К) = -92,31 +0,13 кДж • моль.
Эта величина принята по рекомендации КОДАТА—МСНС [1000], которая основывается
на результатах измерения энтальпии реакции водорода с хлором в работах Россини [3251], Рота
и Рихтера [3260], Вартенберга и Ганиша [3884], Лачера и др. [2333], Файта и др. [1437], Чер-
кетти и др. [923], Кинг и Армстронга [2247].
Принятому значению &jH° (НС1, г) соответствует
Do (НЧЛ) = 427,768 + 0,13 кДж • моль = 35 759 + И см.
КОДАТА—МСНС [1000] на основании приведенного значения AfH° (HC1, г) и измерений
энтальпии растворения НС1 в воде в работах [1766, 1764, 3770, 3258] рекомендовал значение
энтальпии образования С1~ в бесконечно разбавленном водном растворе
AfH°{Cl-, р-р, Н2О, станд. сост., 298,15 К) = —167,080 ± 0,088 кДж • моль,
которое также принимается в настоящем справочнике для последующих расчетов.
НОС1 (г). Термодинамические свойства газообразного гидроксида хлора в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 59.
В таблице 8.5 приведены молекулярные постоянные, использованные для расчета
термодинамических функций НОС1 (г).
Сведения о молекулярных постоянных НОС1 основываются на исследованиях ее
инфракрасного [1872, 3219, 428, 429, 3352а] и микроволнового [2438] спектров. Наиболее детальное
исследование инфракрасного спектра молекул НО35С1 и НО37С1 в газообразном состоянии было
выполнено в работах Ашби [428, 429], проанализировавшего вращательную структуру полос vx и v2
обеих молекул и определившего начала обеих полос и вращательные постоянные. Хотя из-за
1 Применение уравнения типа A.34) с постоянными, найденными в работе [3153] или [13], в области больших /
приводит к неправильным величинам, так как в этом случае функция F, (J) не пересекается с предельной кривой
диссоциации.
172
Хлор и его соединения DC1 (г)
перекрывания полос определить частоту начала полосы v3 не удалось, Ашби [429 ] смог оценить ее
значение с точностью не хуже 3 см. Найденные вращательные постоянные HOG1 (Л000=20,46 +
±0,02 см, 2?000=0,506±0,01 см и СОоо=О,492±0,001 см) хорошо согласуются с результатами
анализа микроволнового спектра HOG1, выполненного в работе [2438], а значения основных
частот— с результатами исследования спектра НОС1 в матрицах из аргона [3352а]. Значения
постоянных НО38С1, найденные в работах [428, 429], приведены в табл. 8.5. Произведение моментов
инерции НОС1, приведенное в таблице, вычислено по вращательным постоянным, найденным
Ашби, с учетом содержания изотопов 35С1 и 37С1 в природной смеси. Этим вращательным
постоянным соответствуют г0 (О—Н)=0,974+0,02 А, г0 (G1-O)=1,689 + 0,006 А, ^ Н—О-С1=104°47' +
±5°.
Термодинамические функции НОС1 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122) — A.124), A.127) и A.128) в приближении «жесткий ротатор — гармонический осциллятор»
без учета различия постоянных молекул НО35С1 и НО37С1. Погрешности табулированных
значений обусловлены неточностью колебательных постоянных, а выше 2000 К отсутствием данных
для использования более точного метода расчета. Погрешности в значениях Ф° (Т) оцениваются
в 0,3, 1 и 2,5 Дж-К^-моль-1 при 298,15, 3000 и 6000 К.
Термодинамические функции HOG1 (г) вычислялись раньше в работах [2474, 2417. 3795] для
¦температур, непревышающих 2000 К, а также в таблицах JANAF [2095] и предыдущем издании
•справочника [88]. Несмотря на некоторое различие в значениях молекулярных постоянных,
использованных в этих расчетах, расхождения между результатами всех расчетов и данными,
приведенными в табл. 59, не превышают 0,4 Дж-К^-моль.
Константа равновесия реакции НОС1 (г) = О (г)+Н (г)+С1 (г) вычислена по значению АгН°@) =
=669,494+15 кДж-моль. Это значение соответствует энтальпии образования HOGl(r)
AfH°(H.OCl, г, 298,15 К) = —90 + 15 кДж • моль,
которая принята в справочнике на основании измерений энтальпии ионизации HOG1 в растворе
и энтальпии восстановления иона СЮ" перекисью водорода в работах Флис, Мищенко и Пахо-
мова [270, 271], получивших для A^^HOCl, р-р, Н2О, станд. сост., 298,15 К) значение
—130,8 кДж-моль. Энтальпия растворения НОС1 в воде была оценена Джельсом [1587]
=(_54 кДж-моль) и авторами таблиц JANAF [2095] (—33 + 12 кДж-моль). При вычислении
AfH° (НОС1, г) использовано значение, более близкое к рекомендации [2095].
DC1 (г). Термодинамические свойства газообразного хлорида дейтерия в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 60.
В табл. 8.2 приведены молекулярные постоянные D35C1, выбранные в результате анализа
литературных данных. Так же как НС1, молекула DG1 не имеет связанных возбужденных валент-
иых электронных состояний с энергией меньше 60 000 см. Колебательно-вращательные
постоянные DG1 в основном электронном состоянии Х1^ + определялись на основании изучения
колебательно-вращательных [1835,3011, 3775, 2722, 3146, 3149, 3148, 3154, 3905] и вращательных [1824,
1090, 1091, 1088, 1211] спектров. В колебательно-вращательных спектрах наблюдались переходы
•с v <^ 4. Наиболее надежные значения постоянных DC1 были получены в работах Моулда и др.
[2722] и Ранка и др. [3149, 3154]. Найденные в этих работах значения вращательной
постоянной Во хорошо согласуются с полученными при исследованиях микроволнового спектра Коуана
л Горди [1090, 1091] и Де Лучия и др. [1211]. В табл. 8.2 приведены значения молекулярных
постоянных D35G1 по работе Моулда и др. [2722].
Поскольку эти постоянные DC1 существенно менее точные, чем постоянные HG1, при
подготовке настоящего справочника значения коэффициентов Ykl для молекулы DG1 были вычислены
по изотопическим соотношениям A.66) и постоянным Yk! молекулы HG1. Значения Ykl для DG1,
соответствующие природной смеси изотопов хлора, приведены в табл. 8.3. Там же приведены
значения i>max, /цт и коэффициентов а{ в уравнении, аппроксимирующем предельную кривую
диссоциации состояния X1!,+ молекулы DG1.
Термодинамические функции DC1 (г) вычислены без учета возбужденных электронных
состояний этой молекулы. Значения (??. вр и ее производных вычислялись по уравнениям A.73) —
A.75) с учетом всех колебательно-вращательных уровней, ограниченных предельной кривой
.диссоциации состояния X1!,+. Колебательно-вращательные уровни энергии вычислялись по урав-
173
TCI (г) Глава &
нениям A.65) и A.62) с коэффициентами Ykl из табл. 8.3. Погрешности вычисленных таким
образом значений термодинамических функций DC1 при температурах до 4000 К обусловлены
главным образом неточностью фундаментальных постоянных и не превышают 0,02 Дж-К^-моль
в значениях Ф° (Т) и S° (T). При более высоких температурах они возрастают из-за отсутствия
экспериментальных данных об уровнях энергии сг;>-4и/|>20и могут достигать 0,05 и 0,1 ДжХ
ХК^-моль-1 в значениях Ф° F000 К) и S° F000 К).
Термодинамические функции DC1 (г) вычислялись ранее в работах [3755] (до 700 К), [3371
и предыдущем издании справочника [88]. Расхождения между данными, приведенными в
справочнике [88] и в табл. 60, не превышают 0,03 Дж-К^-моль в значениях Ф° (Т) и
0,15 Дж-К^-моль в значениях S° (T), однако расхождения с данными, приведенными в
работе [337] достигают 0,4 Дж-К^-моль-1 в Ф° F000 К) и 0,6 Дж-К^-моль в С°р E000 К)
главным образом из-за того, что в работе [337] использован метод Майера и Гепперт-Майер.
Константа равновесия реакции DC1 (r)=D (г)+С1 (г) вычислена на основании принятого в
справочнике значения
Дг#° @) = Do (DC1) = 432,76 + 0,14 кДж • моль = 36 176 ± И см.
Эта величина была вычислена по соотношению C.6) и значениям De (DCl)=De (HC1)=37 241 +
+ 11 см и G @, D 3б'453С1) = 1065 см. Ей соответствуют энтальпия образования DC1
A/S"°(DC1, г, 0) = —93,333+ 0,14 кДж • моль
и энергия диссоциации Do (D35C1)=36 175 + 11 см.
TCI (г). Термодинамические свойства газообразного хлорида трития в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 61.
Молекулярные постоянные ТС1, выбранные в результате анализа литературных данных,
приведены в табл. 8.2. По аналогии с молекулами НС1 и DC1 в настоящем издании принято, что>
ТС1 не имеет стабильных валентных возбужденных электронных состояний с энергией меньше-
60 000 см. Колебательно-вращательный спектр ТС1 исследован в работе Джонса и Робинсона
[2170], которые получили в поглощении и провели анализ вращательной структуры полосы
1—0 молекул Т35С1 и Т37С1. На основании полученных данных и значений шехе и <оеуе,
вычисленных из изотопических соотношений по постоянным НС1, Джонс и Робинсон определили
постоянные Т35С1, которые хорошо согласовались со значениями, рассчитанными по постоянным HCi
и DC1. Микроволновой вращательный спектр ТС1 изучен в работах Горди с сотр. [849, 1211],.
в которых было определено с высокой точностью значение Z?o и с использованием данных работы
[2170] значение Ве. В табл. 8.2 приведены постоянные молекулы Т36С1, основанные на работах
[849, 2170]. При подготовке справочника значения коэффициентов Ykl для ТС1 были вычислены
с помощью изотопических соотношений A.66) и соответствующих коэффициентов НС1. Уравнение
с этими коэффициентами должно лучше описывать энергию колебательно-вращательных уровней
ТС1 со значениями v > 1 и / > 15, чем с постоянными из табл. 8.2, а также обеспечивать
схождение колебательных уровней ТС1 вблизи диссоциационного предела. Значения Ykl,
соответствующие природной смеси изотопов хлора, приведены в табл. 8.3, там же приведены значения уша„
•Aim и а( — коэффициентов в уравнении, аппроксимирующем предельную кривую диссоциации
состояния ХХЕ +.
Термодинамические функции ТС1 (г) вычислены без учета возбужденных электронных
состояний этой молекулы. Значения QW вр и ее производных вычислялись по уравнениям A.73) —
A.75) непосредственным суммированием по колебательно-вращательным уровням энергии,
ограниченным предельной кривой диссоциации. Колебательно-вращательные уровни энергии
вычислялись по уравнениям A.65) и A.62). Погрешности вычисленных значений термодинамических
функций ТС1 при температурах до 3000 К определяются главным образом неточностью принятых
фундаментальных постоянных и не превышают 0,02 Дж-К^-моль в значениях Ф° (Т) и S° (T).
При более высоких температурах начинает сказываться неточность аппроксимации высоких
колебательно-вращательных уровней молекулы ТС1 уравнениями A.65) и A.62) с принятыми
значениями коэффициентов Ykl, и погрешности могут достигать 0,07 и 0,15 Дж-К^-моль в
значениях Ф°F000К) и ?°F000К).
174
Хлор и его соединения ClF(r>
Термодинамические функции ТС1 (г) ранее вычислялись только в предыдущем издании
справочника [88 ] по модифицированному методу Гордона и Барнес и несколько отличающимся
значениям молекулярных постоянных. Расхождения между данными справочника [88] и табл. 61
растут с температурой и изменяются от 0,01 Дж-К^-моль при 298,15 К до 0,1 и 0,4Дж-К~хХ
Хмоль в значениях Ф° (Т) и S° (T) при 6000 К.
Константа равновесия реакции ТС1(г)=Т (г)+С1 (г) вычислена на основании принятого,
в справочнике значения
ДД° @) = Do (TCI) = 36 358 + 14 см = 434,94 ±0,17 кДж • моль.
Это значение получено по соотношению C.6) и значениям Т)е (TC1)=D(( (HC1)=37 241 + 11 см~*
и G @, ТС1)=883 + 2 см. Ввиду неточности колебательных постоянных ТС1 разница между
значениями G @) для Т35С1 и Т35'483С1 не принималась во внимание. Принятому значению Do (TCI)
соответствует энтальпия образования ТС1
ДД^Та, г, 0) = —93,841 +0,17 кДж • моль.
C1F (г). Термодинамические свойства газообразного фторида хлора в стандартном состоянии-
при температурах 100 — 6000 К приведены в табл. 62.
Молекулярные постоянные, использованные для вычисления термодинамических функций
C1F (г), приведены в табл. 8.2. Так же как молекулы других двухатомных интегралогенов (см.
гл. 9, рис. 9.1), молекула C1F должна, помимо основного состояния Х1!,*, иметь четыре связанных,
состояния, соответствующих конфигурации . . .о2л4п3а CП2, 3П1, 3П0- и 3П0+), по крайней мере-
одно слабо связанное состояние 0+, возникающее в результате взаимодействия состояния 3П0+.
с отталкивательным состоянием 0+, а также состояния, образованные ионами Cl+-f-F".
Благодаря большой величине / (С1) последние должны иметь энергии существенно больше 50 000 см'
и не рассматриваются в справочнике. Молекулярные постоянные C1F определялись в результате-
исследовании системы полос 53П0+—ХХ2 + {v" < 2, 3 < v' < 17) [3858, 3340, 3358, 3559, 3558],,
микроволнового [1640, 1639, 2457] и инфракрасного [2960, 2158, 2853, 2852] спектров.
Колебательные постоянные молекулы 35C1F в состоянии ХХ2+, полученные по данным разных авторов,,
существенно отличаются. Это обусловлено тем, что начала полос системы В3П0+—ХХ2 + определены
только для v"=0 и 1 [3858, 3559], а значение Д&/2, вычисленное по этим данным, отличается:
от измеренного в инфракрасном спектре [2852, 2853] на 0,8 см. Приведенные в табл. 8.2
колебательные постоянные 35C1F в состоянии Х12+ вычислены Коксоном [1104] по началам полос.
1 — 0 и 2 — 0, найденным им из данных Нильсена и Джонса [2852]. Принятые значения ше
и шехе отличаются на 1,4 и 0,84 см от вычисленных Шумахером и др. [3358] по кантам полос
системы #3П0+—Z12+ с у^2 ина 2 и 1,7 см — от полученных Штрикером и Крауссом [3559]
по соотношению A.69) на основании найденных ими значений Д&/,, Ве и аг.
Вращательные постоянные 35C1F в состоянии Х12+ приняты по работе Гилберта и др. [1639,,
1640] на основании выполненных ими исследований микроволнового спектра молекул 35C1F
и 37C1F.
Постоянные C1F в состоянии Б3П0+ определялись по данным, полученным при исследовании,
системы 3По+— X1!,*, для которой был выполнен анализ вращательной структуры полос у' =
=3-^-10 и 14-^-17 [3858, 3559]. В этих исследованиях были установлены сильные возмущения
в полосах с у'= 11, 12 и 13 и показано, что уровень г/ = 17 примыкает к диссоциационному пределу
молекулы C1F. Значения постоянных, найденные в разных работах, существенно отличались,
из-за сложного характера изменения G (v) и Bt при увеличении и. Большинство авторов
рекомендовали значения постоянных, описывающих экспериментальные данные для v ^ 10, т. е. до
области возмущений. В справочнике по аналогии с молекулами других интергалогенов (см. рис. 9.1)
принято, что наблюдавшиеся возмущения обусловлены взаимодействием состояния 3П0+ с
отталкивательным состоянием 0+, коррелирующим с атомами F B.Рз/2)+С1 (*P>i.)- В результате
взаимодействия возникает состояние В'0+, которому принадлежат колебательные уровни, относившиеся
в работах [3858, 3559] к уровням v=14-f-17 состояния В3И0+, и которое имеет диссоциационный
предел с энергией 21 512+5 см (граница схождения уровней). Постоянные 35C1F для состояний
В9Л0+ и В'0+, приведенные в табл. 8.2, вычислены при подготовке справочника на основании этого,
предположения и данных Штрикера и Краусса [3559].
175
C1F (г) Глава 8
Сведения о состояниях 3П2, 3П1 и 3П0- молекулы GIF в литературе отсутствуют. Оценка разности
.энергий состояний 3П2 и 3П1 и состояния В3И0+ по соотношениям типа (9.1) показала, что они
составляют около 600 и 300 см соответственно. В связи с небольшой разностью этих энергий и
невысокой точностью постоянных 35G1F в состоянии В3П0+ и колебательных постоянных
в состоянии X1!!+, постоянные этой молекулы в состояниях 3П2, 3П1 и 3П0- в
справочнике не оценивались, и все четыре состояния рассматривались как компоненты состояния 3П.
Термодинамические функции GIF (г) были вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.93)—A.95). Значения QBK, QBB и Qm находились по соотношениям A.76)—A.78), в которых
статистические суммы по колебательно-вращательным уровням энергии состояний Х1Е+, 3П
и В'0+ и их производные были рассчитаны непосредственным суммированием по колебательным
и интегрированием по вращательным уровням энергии с помощью соотношений типа A.82).
Колебательно-вращательные уровни молекулы GIF для «эффективной изотопической модификации
35'453G1F» вычислялись по уравнениям A.65) и A.34) со значениями коэффициентов Ykl,
приведенными в табл. 8.3. Мультиплетногть и вырождение уровней состояния 3П учитывались
статистическим весом 6. В расчетах учитывались все уровни каждого состояния со значениями v ^ ушах
и / г^ /Шах, г> где •Лпах, v находились по соотношениям A.81). Соответствующие значения ушах
и /цт приведены в табл. 8.3. Погрешности вычисленных термодинамических функций C1F (г)
при Т <^ 3000 К обусловлены главным образом неточностью описания
колебательно-вращательных уровней энергии состояния X1!,+ уравнениями с принятыми значениями постоянных.
При более высоких температурах становятся существенными ошибки из-за приближенного учета
возбужденных состояний GIF. Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К могут
достигать 0,05, 0,2 и 0,5 Дж«К~1-моль~1.
Ранее термодинамические функции GIF (г) вычислялись в работах [1426] (Т <^ 1500 К), [1012]
(Т < 2000 К), [3878] (Г < 5000 К) и справочных изданиях [87, 88, 2095, 2556, 1308, 2496, 337].
Данные большинства расчетов при Т ^ 2000 К удовлетворительно согласуются с приведенными
в табл. 62; расхождения не превышают 0,02 и 0,1 Дж-К~1-моль~1 при 298,15 и 2000 К
соответственно. При более высоких температурах расхождения возрастают, наилучшее согласие имеет
место с предыдущим изданием (в пределах 0,2—0,3 Дж-К~1-моль~1 в значениях всех функций,
кроме S0 F000 К)). Расхождения с данными работ [2095, 2556] больше и достигают 0,5 Дж-К^Х
Хмоль в значениях Ф(Т) и свыше 2 Дж-К~1-моль~1 в С° D000 К). В справочнике [337]
в расчете допущена ошибка, расхождения с табл. 62 превышают 0,3 Дж-К~1-моль~1
в Ф° B98,15 К) и достигают 4, 13 и 30 Дж-К^-моль в значениях Ф° (Т), S° (T) и С° (ГI.
Константа равновесия реакции GIF (r)=F (г)+С1 (г) была вычислена на основании принятой
в справочнике энергии диссоциации
АгН° @) = Do (C1F) = 21108 + 5 см = 252,50 ± 0,06 кДж • моль" К
Это значение получено по границе схождения колебательных уровней состояния 5'0+ B1 512 +
+ 5 см) |3858, 3559, 3358] в предположении, что его диссоциационный предел соответствует
¦атомам F BPi/2)+Cl BP*i2). В литературе более 20 лет дискутируются два значения Do (C1F),
одно приведенное выше, и другое, равное 20 630+5 см, или 246,79+0,06 кДж -моль, которое
¦следует из тех же экспериментальных данных, если граница схождения соответствует диссоциаци-
онному пределу F BРз/з)+С1 BPvj)- Исследования спектров C1F не позволяют сделать
однозначный выбор между этими значениями. В ряде работ была сделана попытка решить этот вопрос
на основании измерения энтальпии образования GIF (г). Приведенным выше двум значениям
энергии диссоциации GIF соответствуют значения Ддй"" (GIF, г, 298,15 К), равные —55,7+0,3 и
—50,0+0,3 кДж-моль". В табл. 8.7 приведены энтальпии образования C1F (г), полученные
разными методами.
Несмотря на значительные расхождения между данными разных авторов, результаты
наиболее надежных измерений, выполненных Нордином [2868а] и Армстронгом [416, 2873а]
разными методами, согласуются в пределах их погрешностей с верхним значением Do (GIF).
Следует отметить, что в работах [416, 2873а] был использован метод, позволяющий исключить влияние
1 Примечание при корректуре. Попытка JANAF исправить в 1977 г. расчет [337] привела, по-видимому,
к еще большим ошибкам.
176
Хлор и его соединения
C1F (г)
Таблица 8.7. Результаты определений энтальпии образования C1F (в кДж • моль х)
Авторы
Метод
Д,Я°(С1Р, г,
298,15 К)
Руфф, Лаасс, 1929 [3268]
Викке, 1946 [3936]
Шмутц, Шумахер, 1947 [3342]
Викке, Фриц, 1953 [3938]
Барбери и др., 1969 [472а, 473]
Армстронг, 1971 [416, 2873а]
Нордин [2868а]
Калориметрическое измерение энтальпии реакции —119
C1F (г)+На (r)=HF (г)+НС1 (г)
Калориметрическое измерение энтальпии реакции —48,5
V, Cl.^+Va Fa (r)=ClF (г)
Калориметрическое измерение энтальпии реакций —62,8
NaCl (k)+VA (r)=NaF (k)+V2C12 (г) и NaCl (к)+
+C1F (r)=NaF (к)+С1а (г)
Метод взрыва в сферической бомбе, 1/aFa (г)-Р/аС1а (г, —49,0
избыток)=C1F (г)
Измерение энтальпии реакции C1F (г)+Н2 (г)= —60,8+15
=HF (г)+НС1 (г) в двухкамерной калориметрической
бомбе
Измерение энтальпии реакции C1F (г)+На (г)+ад= —60,2 ±4
=HF (ag)+HCl (ag) в калориметре с горелкой
Исследование равновесия Clj (r)+F (r)=ClF (г)+ _56,7±1
+С1 (г) методом измерения интенсивности хемилюми-
несценции, 306—405 К, 3 точки
полимеризации молекул HF в продуктах реакции, и найденные в аналогичных измерениях
Армстронга данные послужили основой для выбора значений энтальпий образования C1F3 (г) и C1FS (г)
в настоящем издании.
Данные работы Руффа и Даасса [3268], по-видимому, ошибочны из-за использования
недостаточно чистого образца GIF. Определение погрешностей результатов измерений Викке и др.
[3936, 3938] затруднительно, однако точность данных, полученных методом взрыва, для таких
систем не может превышать 6 кДж-моль, а в работе [3936] дополнительная ошибка могла
возникать в результате самопроизвольного начала реакции в однокамерной бомбе. Данные,
приведенные в работах [472а, 473] и [3342], являются недостаточно точными, но также не противоречат
принятой величине.
В литературе в качестве подтверждения нижнего значения Do (C1F) обычно приводится работа
Дайблера и др. [1241 ], в которой был определен порог появления иона С1+ при диссоциативной
фотоионизации C1F. Однако особенности кривой фотоионизации C1F вблизи порога затрудняют
определение его точного значения, аналогично тому, как это имело место в исследованиях
процесса фотоионизации F2 и HF авторами работы [1241 ]. По-видимому, погрешность определения
порога фотоионизации C1F существенно больше указанной в этой работе (+300 см) и эти
измерения не позволяют сделать выбор между двумя возможными значениями Do (C1F). Следует
отметить, что Do (C1F) и Do (F2), полученные Дайблером с сотр., приводят к значению AfH0 (C1F, г) ~
^ _6О кДж-моль (см. [1241]), по-видимому, из-за взаимной компенсации ошибок в
определении порога фотоионизации F2 и C1F.
Следует отметить, что если бы электронное состояние В'0+ молекулы C1F имело предел
F (аР»/2)+С1 BРч), возмущения уровней в области 21 200 см должны были быть более
нерегулярными из-за взаимодействия состояния В3П0+ с двумя состояниями (см. ниже, гл. 9).
Кроме того, в этом случае максимум на потенциальной кривой электронного состояния BSUO+
должен быть примерно на 550 см выше диссоциационного предела F BP»/2)-f-Cl Bi\), в то
время как по оценке [950] он составляет около 100 см в согласии с принятым
значением энергии диссоциации C1F.
Принятому значению Do (C1F) соответствует
A,ff°(ClF, г, 298,15 К) = —55,699 ±0,30 кДж • моль
-1
177
C1F3 (г)
Глава 8
C1F3 (г). Термодинамические свойства газообразного трифторида хлора в состоянии
идеального газа при температурах от 100 до 6000 К приведены в табл. 63.
Термодинамические функции C1F3 (г) были вычислены по постоянным, приведенным в табл. 8.8.
Молекула C1F3 имеет четное число электронов, поэтому принято, что ее основное состояние
является синглетным. Никаких сведений о возбужденных электронных состояниях C1F3 нет. Смит
[3454] в результате исследования микроволнового спектра показал, что в основном состоянии
Таблица 8.8. Значения молекулярных постоянных нов состоянии Х1А, принятые для расчета
термодинамических функций C1F3 и C1F6 (р^ = 1)
Молекула
C1F3
v2
v4
v5
va
v,B)
v8B)
v,B)
CM
752
. 712
529
541
328
486
702
488
442
346
328
375
732
440
302
IAIB1C •
• 10117 r3X
Хсм6
2,7-10'
2,2-104
2
4
молекула C1F3 имеет плоскую «Г-образную» структуру (группа симметрии С2,). Этот вывод
подтверждается исследованиями инфракрасных спектров и спектров комбинационного рассеяния
газообразного C1F3 в работах Классена и др. [971, 3378], а также теоретическими расчетами [56,
1574, 2511 ]. Значения всех основных частот C1F3 были определены в работах [971, 3378] при
изучении спектра C1F3 в матрицах инертных газов [1542]. Результаты этих исследований находятся
в удовлетворительном согласии. Приведенные в табл. 8.8 значения основных частот G1F3 приняты
по работам [971, 3378]. Погрешности в значениях vx и v2 могут достигать 10 см, в остальных
частотах 3 см.
Согласно [3454] молекула C1F3 имеет структурные параметры: -4?г—С1—F,=-^F2—
—Cl-F3=87°29', ^F1-C1-F3=185°2', r(Cl-F1)=r (Cl—F8) = 1,698±0,02 А и r (Cl—F2) = l,598±
±0,02 А. Приведенное в таблице значение ItJidc вычислено по этим параметрам, его погрешность
оценивается в 6-10~116 г3-см6.
Термодинамические функции C1F3 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический осциллятор».
Погрешности вычисленных термодинамических функций обусловлены главным образом
неточностью принятых молекулярных постоянных C1F3, а также проведением расчета в приближении
«жесткий ротатор — гармонический осциллятор». При высоких температурах могут добавиться
ошибки из-за отсутствия сведений о возбужденных состояниях C1F3. Погрешности, обусловленные
пренебрежением различия основных частот разных изотопических модификаций C1F3
несущественны ввиду неточности известных постоянных этой молекулы. Общие погрешности в значениях
Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,3, 4 и 6 Дж-К~1>моль~1 (в том числе из-за
неточности постоянных в 0,1, 0,2 и 0,3 Дж-К~1-моль~1 соответственно).
Термодинамические функции C1F3 (г) вычислялись ранее в справочниках [2556, 2095, 2417]
и работах [3325, 971 ] (для температур до 1500 и 1000 К). Они согласуются с приведенными
в табл. 63 в пределах 0,4—0,6 Дж-К~1-моль~1 в значениях Ф° (Т) и S° (T) во всем интервале
температур. Имеющиеся расхождения обусловлены некоторым различием принятых в расчетах
значений основных частот.
Константа равновесия реакции ClF3(r)=3F(r)+Cl(r) вычислена на основании значения
ЬГН° @)=511,946+5,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования C1F3 (г)
A/F°(ClF3, г, 298,15 К) = —164,6 + 5,0 кДж • моль.
Это значение принято на основании измерений энтальпий реакций сгорания C1F3, Cl2 и F2
в водороде в присутствии воды, выполненных Кинг и Армстронгом [2247, 2246]. Реакции
проводились в горелке, помещенной в калориметрическую бомбу постоянного давления, причем в
экспериментах использовались чистые и хорошо проанализированные образцы. Близкое значение
(—166;6±5,0 кДж-моль), подтверждающее результаты измерений [2246, 2247], получено в ра-
178
Хлор ¦ его соединения C!F5(r)
боте Барбери и др. [473] при измерении энтальпии реакции C1F3 (г)+2Н2 (г)=НС1 (r)+3HF (г)
в двухкамерной калориметрической бомбе. Исследования равновесия реакции C1FS (r)=ClF (r) +
+F2 (г) в работах [3342] E23—620 К) и [3321] E00—800 К) при их обработке по II и III законам
дают более низкое значение (—157,7 кДж-моль). Измерения энтальпии реакции C1F3 (г) +
+3NaCl (K)=3NaF (к)+2С12 (г) в работах [3342] и [3886] привели к резко отличающимся между
собой значениям (—164 и —121 кДж-моль), причем данные работы [3342] практически
совпадают с данными, полученными в работах [473, 2246].
CIF5 (г). Термодинамические свойства газообразного пентафторида хлора в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 64.
Термодинамические функции ClFj (г) были вычислены по молекулярным постоянным,
приведенным в табл. 8.8. Молекула C1F5 имеет четное число электронов, в соответствии с этим в
справочнике принимается, что она имеет синглетное основное состояние. Сведения о возбужденных
электронных состояниях C1F5 в литературе отсутствуют, так же как экспериментальные данные о
структуре молекулы в основном состоянии. По аналогии с молекулами BrF5 и IF6,
подтверждающейся исследованиями колебательного спектра C1F6, можно предполагать, что эта молекула должна
иметь структуру четырехгранной пирамиды (симметрия dv) с атомом хлора в центре основания
или под плоскостью основания. Колебательный спектр G1F3 исследовался в работах Смита с сотр.
[555, 3457] и Готти и др. [1572]. В работе [555] в спектре комбинационного рассеяния и
инфракрасном спектре наблюдались все основные частоты, кроме неактивной частоты v4. В работах
[3129, 811, 958] был выполнен расчет силового поля C1F5 и оценка частоты v4. В табл. 8.8
приведены значения основных частот, основанные на результатах работ [555, 30]. Погрешности
принятых значений всех частот, кроме v4, могут достигать 5—10 см, в значении v4 — до 25 см.
Структурные параметры молекулы C1F5 были оценены на основании сравнения параметров молекул
C1F3, BrF3 и BrFB и приняты равными: -4F—С1—F=90°, r (C1—F1)= ... =r(Cl—F4) = l,70+0.03A,
г (С1—F5)=l,60+0,03 А. Произведение главных моментов инерции C1F5, вычисленное по этим
параметрам, также приведено в таблице. Его погрешность оценивается в 5 -10~114 г3-см6.
Термодинамические функции C1F5 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический осциллятор»
по постоянным, приведенным в табл. 8.8. Погрешности рассчитанных термодинамических
функций C1F5 (г) обусловлены неточностью принятых постоянных и приближенным методом расчета, при
высоких температурах к этому добавляются возможные ошибки из-за отсутствия данных об
электронных состояниях CIF51. Общие погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К
оцениваются в 2, 8 и 11 Дж-К~1-моль~1. Термодинамические функции C1F5 (г) ранее вычислялись
в работе [717] (до 1000 К), и таблицах JANAF [2095]. Результаты расчетов [717, 2095]
согласуются между собой и с данными табл.64 в пределах 0,6 Дж-К "Моль.
Константа равновесия реакции C1F5 (r)=5F (г)+С1 (г) вычислена на основании значения
ЬуН0 @)=735,271+7 кДж-моль, соответствующего принятой в справочнике энтальпии
образования C1F6 (г)
-1
AfH°(ClFb, г, 298,15 К) = —238 ± 7 кДж • моль
Наиболее надежными представляются результаты измерений Армстронга и Кинг [417],
выполненных в калориметрической бомбе постоянного давления с хорошо охарактеризованными
образцами, и исследование равновесия реакции образования C1F6 (г) статическим методом в
работе Бауэра и Шихана [518]. Обработка данных [518] по II и III законам дает хорошо
согласующиеся результаты. С результатами измерений [518, 417] согласуется также величина,
рекомендованная авторами работы [657 ] на основании определения энтальпии двух реакций, хотя обработка
первичных данных [657] приводит к резко отличающимся величинам.
1 Примечание при корректуре. После завершения расчетов термодинамических функций ClF6(r) стали известны
результаты исследования МВ-спектра C1F8[17O9] и электронографического исследования [18а]. Уточнение
структурных параметров C1F5 в этих работах приводит к изменению в значениях Ф°(Т) и S°{T) примерно
на 0,4 Дж-К^'моль.
179
CIF6(r)
Результаты измерений энтальпии образования C1F5 приведены в табл. 8.9.
Таблица 8.9. Результаты измерений энтальпии образования CIF6 (г) (в кДж • моль)
Глава 8
Авторы
Буган и др., 1967 [717]
Бауэр, Шихан, 1Ш7 [518]
Бисби и др., 1968 [657]
Барбери и др., 1969 [473]
Кинг, Армстронг, 1969 [417]
Метод
Изучение равновесия реакции C1F, (r)+F» (г)=
=C1FB (г), 623К;
расчет по III закону
Изучение равновесия реакции C1F3 (r)+F2 (r)=
=C1F5 (г), 486—544 К, 8 точек
расчет по III закону
расчет по II закону
Измерение энтальпии реакций C1F5 (ж)+ЗН« (г)=
= НС1 (r)+6HF (ж)+а (HF)B (г)
C1F6(}k)+8NH3 (r)=NH4Cl (k)+N2 (r)+5NH4F (к)
рекомендовано авторами [657]
Измерение энтальпии реакции ClF6(r)+3H2(r)=
= НС1 W+5HF (г)
Измерение энтальпии сгорания C1F5, F2 и С12 в Н2
в присутствии Н2О
A,H°(C1FS, г,
298,15 К)
—245
—239,3+5
—242 ±12
—269 ±30
—227 ±16
—234+19
—256 ±15
-236,9 ±6,7
Глава 9
БРОМ И ЕГО СОЕДИНЕНИЯ
Вг, Вг~-, Вг2 (к, ж), Вг2, BrO, HBr, DBr, TBr, BrF, BrF3, BrF5, BrCI
Атом
Электронная
конфигурация
Состояние
Tf
C.M-1
Pi
Вг ... isHp* 2P,k 0 4
. . . 4sa4p5 2P1/a2 3685,24 2
Br- ... 4s24p6 1S * 0 1 '
В настоящей главе рассматриваются газообразные соединения брома, а также элементарный
бром в конденсированном состоянии. Для Вг (г) и Вг~ (г) термодинамические свойства рассмотрены
для температур до 10 000 К, для остальных веществ в газообразном состоянии — для температур
от 100 до 6000 К. Критическая температура брома равна 584,16 К, поэтому термодинамические
свойства брома в конденсированном состоянии даны для температур от 5 до 500 К и приведены
в Дополнении (см. наст, том, кн. 2).
Термодинамические свойства всех соединений брома рассчитаны для природной смеси изотопов
этого элемента. Для двух соединений (Вг2 и НВг) в Приложении 3 приведены данные,
позволяющие оценить влияние межмолекулярного взаимодействия на отклонение свойств
соответствующих газов от их термодинамических свойств в
стандартном СОСТОЯНИИ. Таблица 9.1. Уровни энергии атомов Вг
и Вг~, принятые для расчета
т» / \ гп " е термодинамических функций
Вг (г). Термодинамические свойства газообраз- J
ного одноатомного брома в стандартном состоянии
при температурах 100—10 000 К приведены в
табл. 65.
Уровни энергии атома Вг, использованные при
расчете термодинамических функций, приведены в
табл. 9.1. Энергия компоненты 2Pi/2 принята по работе
Теча [3636], в которой было уточнено значение этой
величины, рекомендованное ранее Мур [2697].
Термодинамические функции Вг(г) вычислялись
суммированием по уровням энергии по уравнениям
A.23)—A.25). Погрешности табулированных термодинамических функций Вг(г) обусловлены
только неточностью принятых значений фундаментальных постоянных и не превышают
0,02 Дж-К^-моль-1 в значениях Ф° (Г) и S0 (Г).
Ранее термодинамические функции Вг(г) в широком интервале температур вычислялись в
работах [1426, 2291], в предыдущих изданиях настоящего справочника [87, 88] и в справочниках
[1946, 2095]. Расхождения в значениях термодинамических функций Вг(г), вычисленных в этих
работах и приведенных в табл. 65, не превосходят 0,04 Дж-К~1-моль~1, что связано с различием
принятых в расчетах значений фундаментальных постоянных.
Энтальпия образования одноатомного брома по рекомендации КОДАТА—МСНС [1000]
принята равной
Д/Я°(Вг, г, 298,15 К) = 111,86 + 0,12 кДж • моль.
Эта рекомендация основана на значениях энергии диссоциации молекулы 79Вг 81Вг и энтальпии
образования Вг2 (г) (см. текст по молекуле Вг2(г)). Использование более точного значения
Do G9Br 81Вг), найденного в работе Барроу и др. [489], приведет к увеличению Ay#°(Br, г,
298,15 К) на 0,007 кДж-моль, что несущественно по сравнению с величиной погрешности,
обусловленной неточностью значения Д„/Г°(Вг2, ж).
Вг~ (г). Термодинамические свойства газообразного отрицательного иона одноатомного брома
в стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 66.
Ион Вг~, как и отрицательные одноатомные ионы других галогенов, имеет основное
электронное состояние 15 (см. табл. 9.1). Ион Вг~, по-видимому, не имеет связанных возбужденных
состояний, поскольку энергия первого возбужденного состояния . . . 4s24p55s 3P должна превышать
его потенциал ионизации.
181
Вг2 (к, ж)
Глава 9
Термодинамические функции Вг~ (г) были вычислены по уравнениям A.9) и A.10) без учета
возбужденных состояний. Погрешности рассчитанных термодинамических функций Вг~(г)
обусловлены главным образом неточностью фундаментальных постоянных . и не превышают
0,02 Дж-К~1-моль~1. Ранее термодинамические функции Вг~(г) вычислялись в предыдущих
изданиях справочника [87, 88]. Различия в значениях Ф° (Т) и S° (Г), вычисленных в этих изданиях
и приведенных в табл. 66, не превосходят 0,01 Дж-К~1-моль~1 и обусловлены различием принятых
значений фундаментальных постоянных.
Константа равновесия реакции Вг~(г)=Вг(г)+е(г) вычислена с использованием значения
Д^" @) =324,70+0,20 кДж-моль в соответствии с принятым в справочнике сродством атома
брома к электрону:
Ао (Вг) = —324,70 + 0,20 кДж • моль.
Эта величина основана на измерениях границы непрерывного спектра поглощения,
выполненных Берри и др. [611, 612], и соответствующей фотоотрыву электрона от иона Вг (Х=3685+0,6 А,
или 27 129+5 см~1=324,65+0,06 кДж-моль) и измерениях границы непрерывного спектра
испускания, соответствующего радиационному захвату электрона атомом брома (Х=3682+2 А,
или 27 151+15 см~1=324,80+0,18 кДж-моль), проведенных Франком и др. [1528]. Принятое
значение Ао (Вг) удовлетворительно согласуется с результатами более старых и менее точных
измерений [456, 1124, 1125, 2715, 3091].
Принятому значению сродства к электрону атома брома соответствует
ДД° (Вг~, г, 0) = —206,777 + 0,23 кДж • моль^1.
Вгг (к, ж). Термодинамические свойства твердого и жидкого брома в стандартном состоянии
в интервале 5—500 К приведены в табл. Д.15.
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 9.2. В интервале от 15 К до температуры плавления B65,9 К) приняты данные для
теплоемкости, полученные для особо чистых образцов брома (содержание примесей менее 0,001%)
Таблица 9.2. Принятые значения термодинамических величин для Вг2 в кристаллическом и жидком
состояниях
Состояние
Н° B98,15К)
-Н° @)
кДж • моль"
S°B98,15K) C°B98,15K)
Дж • К • моль
Коэффициенты в
уравнении для С°(Т)а
Ъ ¦ 10s d ¦ 105
Интервал
температур
К
К
кДж-моль
к
ж
24,52
152,21
75,69
— — — — 265,9 10,572
126,5802 —320,2 50,1478 298,15—500 — —
» С {Т) = а + ЪТ + dT2 (Дж • К-1 • моль-1)
Хильденбрандом и др. [1937] с точностью 0,2 %.Ниже 15 К теплоемкость экстраполировалась
по уравнению Cp = D(90/T)Jr2E (llljT), которое воспроизводит экспериментальные значения
С° (Вг2, к) в интервале 15—40 К [1937] с точностью около 0,5% и приводит к значению
S° A5 К)=2,7 Дж-К~1-моль~1. Данные по теплоемкости кристаллического брома, полученные
в работах [2295, 500, 1417, 3571, 2365], менее надежны.
Температура плавления брома также принята по данным работы [1937], ее погрешность не
превышает 0,05 К. Результаты других определений [3185, 3139, 3730, 3740, 4006, 3327, 395, 2602,
3036, 1881, 1882] менее точны, хотя и не противоречат данным [1937]. Значение Д„Д°(Вг2, к),
принятое в справочнике, определено в указанной выше работе как среднее значение из
четырех калориметрических опытов, проведенных с применением методов дискретной и
непрерывной подачи тепла на образец. Точность принятой величины 0,010 кДж-моль. Менее точные
данные Реньо [3185] завышены на 2%. Данные работы [1937] свидетельствуют, что теплоемкость
жидкого брома уменьшается в интервале 266—310 К от Ср B65,9 К)=77,73 до С°р B98,15, К) =
182
Бром ж его соединения Вг2 (г)
=75,684 Дж-К-^моль. Значения ?°B98,15К) и #°B98,15 К) —#°@), приведенные в табл. 9.2,
совпадают с рекомендованными в качестве ключевых величин [1000]. Для теплоемкости жидкого
брома в интервале 298,15—500 К принято уравнение, полученное авторами настоящего издания
с использованием методов теории существенных структур Эйринга [1433]. При этом результаты
расчетов были скорректированы так, чтобы вблизи стандартной температуры они совпадали с
экспериментальными данными [1937].
Погрешности вычисленных значений Ф° (Т) при 298,15, 400 и 500 К оцениваются в 0,4, 0,7
и 1,5 Дж-К~1-моль~1.
Ранее таблицы термодинамических функций твердого и жидкого брома вычислялись авторами
справочника [2095] (Т < 1000 К), Хильденбрандом и др. [1937] (Т < 310 К), Сешарди и др.
[3394] и Чэнгом и др. [928] (до критической точки 584,16 К). Значения термодинамических
функций, приведенные в табл. Д.15, совпадают с результатами расчетов Хильденбранда и др. [1937].
В справочнике [2095 ] теплоемкость жидкого брома при Т > 300 К принята постоянной и равной
75,312 Дж-К~1-моль~1, ввиду чего расхождения с табл. Д.15 в значениях Ф° (Т) при 400 и 500 К
составляют 0,044 и 0,280 Дж-К~1-моль~1.
В справочнике за стандартное состояние брома при 298,15 К принимается Вг2 (ж). В
соответствии с этим
bfH°{Bi%, ж, 298,15 К) = 0.
Вг2 (г). Термодинамические свойства газообразного двухатомного брома в стандартном
состоянии при температурах 100—6000 К приведены в табл. 67.
Молекулярные постоянные Вга приведены в табл. 9.3. Квантовомеханические расчеты [2745,
2747] и результаты исследований электронного спектра Вг2 (см. обзор в работе [3804]) показывают,
что молекула Вг2 имеет семь связанных валентных состояний с энергиями возбуждения до
50000 см: основное состояние Х1^ и возбужденные состояния 3П2м, -43П1я, 8П0-, 53П0+, С32+ и
Z>3S~. Сведения о более высоких электронных состояниях Вг2 можно найти в работах [1827,
1829, 1832, 3802, 3808, 3809, 3815, 3804, 3939]. На рис. 10.1 (см. гл. 10) приведены
потенциальные кривые связанных электронных состояний Вг2, соответствующих электронным
конфигурациям .. .а^тг* и .. .^i^l\.
Изучение систем полос Б3П0+ — ХХ2+ [2321, 2791, 817, 819, 1166, 2009, 1100, 489] и Л3П1а—
—X1!,* [817, 2008, 1166, 1101], резонансной системы полос Вг2 в вакуумной УФ-области [3160]
и резонансного спектра КР паров брома [1992, 450] дало обширную информацию о колебательно-
вращательных уровнях Вг2 в состоянии ХхЪ^д, причем результаты всех исследований находятся
в хорошем согласии друг с другом. Приведенные в табл. 9.3 значения постоянных 79Вг2 в
состоянии X1!,* приняты по работе Барроу и др. [489], в которой была проанализирована вращательная
структура большого числа полос системы -В3П0+ — -Х1^ (г/'^Ю). Информация об уровнях
энергии Вг2 с 36 ^ v" > 10 была получена Рао и Венкатесварлу [3160] при исследовании спектра
резонансной флуоресценции 79Вг81Вг. Однако молекулярные постоянные, полученные в этой
работе, и значения постоянных, рассчитанные по этим данным Ле Роем и Бернстейном [2393],
плохо описывают уровни энергии Вг2 в состоянии X1?,* как при v ^ 10 [2393], так и вблизи дис-
социационного предела [3160].
Переходы, связанные с электронным состоянием 3П2и, в спектрах Вг2 не наблюдались.
Приведенное в табл. 9.3 значение энергии возбуждения состояния 3П2а молекулы Вг2 вычислено по
соотношению
Те f ПОм+) - Те еП2в) = | [То BРЪ) - То BР=/2)] (9.1)
(где То BР*/2), То BP*i,) — энергии атома Вг в состояниях 2i\ и 2i\)> справедливому для
симметричных двухатомных галогенидов [2747]. Неточность приведенного значения Те CП2ц) может
достигать 100 см. Оцененные значения молекулярных постоянных Вг2 в состоянии 3П2а,
приведенные в табл. 9.3, близки по величине соответствующим постоянным в состоянии -43П1м х.
Постоянные 79Вг2 в состоянии -43П1и, приведенные в табл. 9.3, вычислены Коксоном [1102] на основа-
Фебер и Херрик [1454 ] для энергии возбуждения состояния 3П2Я по оценке Малликена [2747 ] приняли
Г0=12 790 см.
183
Глава 9
Таблица 9.3. Молекулярные постоянные 78Вг2,
Молекула
"Вг2
ВгО
Н"Вг
D«Br
TBr
"Br19F
"Br3SCl
Состояние
Те
ВгО, Н81Вг
ыеу, • 102
, D81Br, TBr
ве ¦ ю1
, 79Br19F и 79Вг35С1
а, • 103
D, ¦ 10'
СМ
ХЪ+ 0
3П2м 13 400
А3П1и 13 904
»П0- 15 850
?3п"+ 15 902,47
C3s| 47 000
D3Hj 48 516
xmf оа
Am( 27 871 Д
XX2+ 0
X^+ 0
X!2+ 0
ZJS+ 0
3П2 15 900
43ДХ 16 900
3Щ_ 18210
В3П0+ 18 265,21
B'0+ 23 933
ХХ2+ 0
3П2 15 000
А3Щ 15 400
3П0_ 16 820
Д3П0+ 16 879,91
325,321 3
160
153,8
70
167,606 6
—
162,0
778,7 6
485,9 6
2 648,975 2
1 885,45
1 550,06
670,75
400
400
360
379,3
138,6
444,276
250
250
200
222,68
1,077 42
2,4
2,78
6,2
1,636 08
—
0,29
6,82 б
5,40"
45,217 5
23,08
15,40 а
4,054
8
10
12
5,75
1,95
1,843
4,8
5,5
7,1
2,884
Примечание. Ниже все постоянные даны в см.
79Вг2:
ВгО:
Н81Вг.
ТВг:
79Br19F:
79Вг35С1
^ аа = —1,045-10-
« По ^ —О
6369 ¦ 10-
а А = —980; 6 вычислено по значениям
по соотношению
а а2 = 0,8735-Ю-3
A.68); я л =
, а, = 0,1203
= -100.
—0,229 798
—
—
—
—0,936 87
—
—
—
—7,4 6
—0,29
—1
—
—
—
—
—
—
—
—0,40
—
—
—6,73
s
ыо, «ожо> <°о!/
• 10~3; б приведено Do
а вычислено исходя из определенного
значения ше.
а приведено значение Do.
а а2 = —2,56 • 10"в
числено по соотношению A.(
i ¦ Ю-14; 6
58).
0,821 07
0,57
0,588 0
0,6
0,595 89
—
4,299 е
3,288в
84,648 84
42,875 0
28,993
3,555 84
2,5
2,5
2,5
2,561
1,5
1,524 69
1,07
1,07
1,07
1,077 04
0 из работы
0,318 73а
0,5
0,72
2,8
0,489 095б
—
3,6В
5,1В
233,277а
83,8
45,9
2,61
4
5
6
3,6
2
0,7697а
0,275в
0,275в
0,343 s
—
0,203 2
0,29
0,34
1,76
0,3013
—
—
5Г
6Г
3438,9 6
880
450
4,8а
4
4
5
5
7
0,71836
0,78г
0,78г
1,22 г
—
к
2,28107
2,74
2,695
2,67
2,677 6
—
1,7171
1,963
1,4144
1,414
1,414
1,758 9
2,10
2,10
2,10
2,070 8
2,75
2,136 1
2,55
2,55
2,55
2,5415
[1350]; в для 79Вг0; Г вычислено
в работе [2170] значения Go A) и рассчитанного
[Jj = 3,487¦ 10~10; в вычислено по соотношению A.
там же
69); г вгл-
нии данных работ [989, 1101, 1100] и результатов исследования системы A3U.hl—X1!,*,
выполненного Брауном [817]. Эти постоянные хорошо описывают уровни энергии состояния A3Ulu су<^ 28.
Переходы, связанные с состоянием 3П0-, в спектре Вг2 не наблюдались. Косвенные данные,
свидетельствующие о существовании этого состояния, приводятся в работе [993а]. Принятое в табл. 9. 3
значение Те(*П.А основано на предположении, что разность энергий состояний 3П0+ и 3П0-
составляет 50 + 50 см. Принятому значению Те CП0-) "соответствует De CП0-) = 200 ± 50 см.
Оцененные значения молекулярных постоянных Вг2 в состоянии 3П0- приведены в табл. 9.3. Значения
постоянных молекулы 79Вг2 в состоянии В3Л0+, приведенные в таблице, приняты по работе Барроу
и др. [489]. Авторы [489] выполнили анализ вращательной структуры 88 полос системы #3П0+—Х1^
молекулы 79Вг2 (у'<[55) и 25 полос 81Вг2 (и' ^54) и с использованием данных, полученных ранее
на приборах с высоким разрешением Хорсли и Барроу [2009], Коксоном [1100] и в
неопубликованной работе Калломона, рекомендовали эти значения постоянных. Система полос -63II0+—X*EJ ис-
184
Бром и его соединения Вг, (г)
следовалась также в работах [2321, 2791, 817, 819, 1166], кроме того, в ряде исследований [3808,
3809, 3815, 3939, 770] наблюдались переходы на отдельные уровни состояния ??3П0+ из
высоковозбужденных состояний Вг2. В табл. 9.3 приведены также энергии возбуждения состояний C3S?
и В3Ъ~д, найденные Венкатесварлу и др. [3802, 3809] в результате исследования в спектре
испускания Вг2 группы диффузных полос в области 4200—2000 А и системы полос в области
3150—2970 А.
Термодинамические функции Вг2 (г), приведенные в табл. 67, были вычислены по уравнениям
A. 3)—A. 6), A. 9), A.10), A. 93)—A. 95). Значения QnB, Qm и QSB были получены по соотношениям
A.90)—A.92), в которых статистические суммы по колебательно-вращательным уровням энергии
состояний Х1!^, 8П2а, Л3П1и, 3П0- и B3HQ+ и их производные находились непосредственным
суммированием по уровням энергии Вг2 по уравнениям A. 73)—A.75). Колебательно-вращательные
уровни этих пяти состояний вычислялись по уравнениям A. 65) и A. 62), значения коэффициентов
YM в которых, полученные в соответствии с методиками, изложенными в разделе 1.4, приведены
в табл. 9.4. В расчете учитывались все уровни энергии состояний Х1Т>+д, АШ1и и В3110+,
ограниченные их предельными кривыми диссоциации, и уровни энергии состояний 3П2м и 3П0-,
ограниченные диссоциационными пределами этих состояний. Вырождение вращательных уровней в
состояниях 3П2и и 3П]м учитывалось статистическим весом 2. В таблице приведены также значения
коэффициентов at в уравнениях, аппроксимирующих предельные кривые диссоциации и значения
vmaz и /lim. Составляющие состояний С32„ и Dz~Lg вычислялись в предположении, что Q^ вр= <2?f]. вр.
Погрешности вычисленных значений термодинамических функций при Т ^ 1000 К
определяются неточностью принятых значений фундаментальных постоянных; при более высоких
температурах начинают сказываться, особенно в значениях S° (Т) и С° (Т), неточность оцененных
постоянных состояния 3П2н и неточность аппроксимации уровней колебательно-вращательной
энергии состояний Х1Щ, 3П2а, 3П1и, 3П0- вблизи диссоциационных пределов этих состояний.
Для Ф°(Г) при 298,15, 3000 и 6000 К они могут достигать 0,03, 0,1 и 0,5 Дж • К • моль.
Ранее термодинамические функции Вг2 (г) вычислялись в работах [1697] (Т <! 1600 К), [4055]
(Т < 2000 К), [41, 42], [1426] (Т < 3000 К), [1454, 3944], первом и втором изданиях настоящего
справочника [87, 88] и таблицах JANAF [2095]. В расчетах, приведенных в работах [1967, 4055,
41, 42, 1426, 3944], и в таблицах [2095] учитывалось только основное электронное состояние
Вг2. В расчетах, проведенных в справочниках [87, 88], приближенно учитывался также суммарный
вклад возбужденных состояний 3П2а, АаН1и, 3П0- и В3Н0+ и вклад состояний С3!!» и ZKS?. Расчеты
Фебера и Херрика [1454] были выполнены методом непосредственного суммирования по уровням
колебательно-вращательной энергии состояний Х1Щ, 3П2я, Л3П1к и 53П0+, в остальных работах
использовался метод Гордона и Барнес [1697, 87, 88] и метод Майера и Гепперт-Майер [41, 42,
1426, 3944, 2095]. В работе [4055] термодинамические функции вычислялись в приближении
«гармонический осциллятор — жесткий ротатор». Наилучшее согласие с величинами,
табулированными в справочнике, имеет место для данных работ [1454] и [88] (расхождения в значениях
Ф° (Г) не превышают 0,8 и 0,5 Дж-К^-моль). Расхождения с данными JANAF [2095] достигают
1,7, 4 и 5 Дж-К^-моль-1 в значениях Ф°(Г), ?°E000К) и Ср C000 К). Хилденбранд и др. [1937]
из измерений термодинамических свойств твердого и жидкого брома при низких температурах
нашли S° (Br2, г, 298,15 К)=245,3 + 0,8 Дж-К^-моль, что хорошо согласуется со значением
245,355 Дж-К^-моль, приведенным в табл. 67.
Константа равновесия реакции Вг2 (г)=2Вг (г) вычислена с использованием значения &ГН° @) =
=D0 G9Вг81Вг)=15 894,46 + 0,3 см-1=190,140+0,004 кДж-моль-1. Это значение соответствует
принятым в справочнике энтальпиям образования Вг2(г) и Вг(г). Для энтальпии образования
Вг2 (г) принято значение
AfH°(Br2, г, 298,15 К) = 30,91 ±0,11 кДж • моль,
рекомендованное КОДАТА—MCHG [1000] на основании результатов измерений энтальпии
испарения жидкого брома, выполненных Хилденбрандом и др. [1937].
Энергия диссоциации молекулы Вг2 определялась по схождению колебательных
уровней состояния BSRQ+ в работах [817, 489, 2009, 1100] и вращательной предиссоциации Вга
185
Таблица 9.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (в car1), а также значения vma,* и Jiim,
принятые для расчета термодинамических функций Br2, BrF, Brtl, в различных электронных состояниях
Коэффициент
те ¦ ю-«
У10 • ю-2
У30 • Ю2
У.» • Ю'
У50 • Ю5
У«о • 108
Ум ¦ «•
Т„ • 103
У21 • 10*
Уа1 • 10'
у« • ю»
у« • ю5
Г.! • Юа
^02 ¦ Ю'
У и ¦ Ю»
У22 • 10'
У32 • Юв
Ущ • ю"
Вг2»
ЗТТ
0 1,34
3,23168 1,60
—1,038 59 —2,4
-0,383 212 -
0,509 693 -
-0,121 795 —
0,272 037 —
0,811 102 0,57
-0,316 456 -0,5
-0,894 964 —
0,118 733 —
-0,780 098 —
- -
— —
—0,205 —0,29
_ _
— -
— —
— -
(ao=De)-10-< 1,597 84 0,2660
a, • 101
а2 ¦ 10'
a3 • 10"
«шах
Jlim
0,178 715 —
0,502 954 —
-0,310 480 —
79 33
665 —
а Состояние C3S- D3S-
Г„ см 47 000 48 516
А*ПЫ
1,390 4
1,502 84
-1,815 57
-9,506 26
22,418 7
—
—
0,580 526
-0,677 288
—8,489 96
-9,340 06
—
—
—
-0,346
—
—
—
—
0,213 937
0,189 156
1,894 68
-6,370 23
30
315
1,585
0,70
—6,2
-
-
-
-
0,6
-2,8
-
-
-
-
-
-1,76
-
-
-
-
0,020 7
-
—
-
5
—
в*щ+
1,590 247
1,665 46
-1,61517
-0,753 538
-2,449 89
0,991 688
-6,978 53
0,588 574
-0,487 438
—4,076 72
—2,164 62
2,782 59
-
-
-0,294
-
-
-
-
0,382 811
0,127 359
1,252 60
-2,019 54
60
381
A'XS+
0
6,705 23
-4,243 63
2,212 32
-7,691 57
—
-
3,549 9
-2,610 17
6,148 27
—10,262 6
—
—
—
-4
—
—
-
—
2,084 42
0,484 004
8, 139 73
-15,887 8
50
342
ЗТТ
Х12
1,59
4
-8
-
-
-
-
2,5
-4
—
—
—
—
—
^
—
—
—
—
0,5
—
—
-
.24
—
1,69
4
-10
—
-
—
—
2,5
-5
—
—
—
—
-4
—
—
—
0,4
—
—
-
19
—
BrF
3п0-
1,821
3,6
-12
—
—
—
—
2,5
-6
—
—
—
—
—
-5
—
—
—
—
0,27
—
—
-
14
—
В'П0+
1,826 521
3,793
-5,75
—
—
—
—
2,561
-3,6
—
—
—
-
—
—5
—
—
—
—
0,334 9
—
—
-
10
—
В'0+
2,393 3
1,336
-1,95
-103,7
425
—
—
1,5
-2
—
—
—
-
—
—7
—
—
—
-
0,064 2
—
—
-
7
—
BrCl
хч:+
0
4,413 21
-1,818 10
-0,419 045
0,405 961
—0,204 008
—
1,504 48
-0,754 690
-2,439 49
0,178 910
-2,584 84
—
—
-0,712
-0,34
—
—
-0,153
1,814 39
0,282 520
1,592 55
-1,577 90
65
509
Зп2
1,5
2,5
—4,8
—
—
—
—
1,07
-0,275
—
—
—
—
—
—0,78
—
—
—
—
—
—
—
-
25
—
1,54
2,5
-5,5
—
—
—
—
1,07
-0,275
—
—
—
—
—
-0,78
—
—
—
—
—
—
—
-
22
—
3п0-
1,682
2,0
—
—
—
—
1,07
-0,343
—
—
—
—
—
—1,22
-
—
—
—
—
—
—
-
14
—
ВЗП0+
1,637 991
2,189 18
—3,226 53
-4,813 21
-197,593
283,891
-22 063,5
1,072 09
-1,042 21
-309,980
1 218,45
-26 783,9
0,277 193
-0,119 484
1,183 26
-173,058
0,399 161
—0,323 611
-
—
—
-
-
10
—
Бром и его соединения ВгО (г)
в этом состоянии в работах [2009, 489]. Наиболее точное значение Do G9Br81Br) =
= 15 895,64+0,05 см было найдено Барроу и др. [489] по результатам собственных измерений
и данных Хорсли и Барроу [2009], Коксона [1100] и неопубликованных измерений Коломона.
Практически совпадающее значение A5 895,5+0,5 см) было получено Ле Роем и Бернстейном
[2392] экстраполяцией колебательных уровней Вг2 в состоянии ?3П0+, наблюдавшихся в работах
[817, 2009], с учетом дальнодействующего потенциала.
Энтальпия образования Вг2(г), рекомендованная КОДАТА—МСНС [1000] (см. выше), была
получена на основании значения Do G9Вг 81Вг) = 15 893,5 + 2 см, найденного в работе Хорсли
и Барроу [2009] короткой экстраполяцией колебательных уровней состояния -В3П0+. Разница
между значением энергии диссоциации Do (Br2), вычисленным по энтальпиям образования Вг(г)
и Вг2 (г), рекомендованным КОДАТА—МСНС [1000], и значением, найденным в работе [489],
составляет 0,014 кДж -моль. Таким образом, использование в справочнике энтальпий
образования Вг (г) и Вг2 (г), рекомендованных КОДАТА [1000], практически не влияет на значения lg K°(T)
при Т > 500 К.
ВгО (г). Термодинамические свойства газообразного оксида брома в стандартном состоянии
при температурах 100—6000 К приведены в табл. 68.
Молекулярные постоянные, использованные для расчета термодинамических функций ВгО,
приведены в табл. 9.3. Основным электронным состоянием молекулы ВгО является состояние
X2IL(, первым возбужденным — состояние А2П(. Другие валентные электронные состояния ВгО,
по-видимому, являются нестабильными или имеют низкие энергии диссоциации, так как они
возникают при переходах связывающих тс- или а-электронов на разрыхляющую о-орбиталь.
В спектрах ВгО как в испускании [3758, 1015], так и в поглощении [1350, 4052], наблюдалась
только система полос ^42Пз/2—Х2Л^г. Приведенные в табл. 9.3 значения колебательных
постоянных ВгО в состоянии Х2И{ получены Дьюри и Рамзи [1350] по кантам полос, соответствующим
v" <! И, i/ <; 2 и измеренным Колманом и Гейдоном [1015] в спектре испускания ВгО. Значения
вращательных постоянных в состоянии Х2П,- основаны на изучении микроволнового спектра
ВгО в работах Пауэлла и Джонсона [3076] и Амано и др. [373]. Эти данные были подтверждены
авторами работ [889, 891, 809] в результате анализа спектра ЭПР, и согласуются с менее точными
значениями, полученными Дьюри и Рамзи [1350] из анализа вращательной структуры полосы
4 — 0 системы А2Л*^—Х2П>/2. Принятое значение постоянной А в состоянии X2Ut вычислено
Брауном и др. [809] теоретически. Неточность этого значения, обусловленная в основном
пренебрежением постоянными спин-орбитальной связи d-электронов, оценивается авторами работы
[809] в 30 см1. Значения колебательных постоянных ВгО в состоянии А2П{, приведенные
в табл. 9.3, определены Дьюри и Рамзи [1350] по кантам полос прогрессии v' — 0 (v'=l, 2, ...
.. .,20) системы ^42Пз/, <- Х2Пз/2. На основании анализа полосы 4 — 0 Дьюри и Рамзи нашли, что
В[=6/7 Во. Это соотношение было использовано авторами настоящего издания для вычисления В[
молекулы 79Вг0, что позволило определить значения постоянных Ве и ае ВгО в состоянии A2Ht.
Термодинамические функции ВгО (г) вычислялись непосредственным суммированием по
уровням колебательно-вращательной энергии состояний Х2П,. и А2П8. по уравнениям A. 76) —A.78).
Энергии уровней находились по уравнениям A.65) и A. 42). Значения коэффициентов Ykl в этих
уравнениях, полученные по методике, изложенной в разделе 1.4 без учета различия молекулярных
постоянных 79Вг0 и 81ВгО, приведены в табл. 9.5. Вырождение вращательных уровней состояний
Х2П{ и ^.2nf учитывалось статистическим весом 2. В расчете учитывались все уровни энергии
обоих состояний, ограниченные их предельными кривыми диссоциации. В таблице приведены
значения коэффициентов а( в уравнениях, аппроксимирующих эти кривые, и соответствующие
ушах и /цт. Погрешности вычисленных термодинамических функций обусловлены при низких
температурах пренебрежением различием вращательных постоянных молекул 79Вг0 и 81ВгО,
а также неточностью известной постоянной спин-орбитального взаимодействия в состоянии Х2П{.
При умеренных и высоких температурах к этому добавляются погрешности из-за невысокой
точности аппроксимации колебательно-вращательных уровней энергии с большими значениями
квантового числа / уравнениями с приведенными коэффициентами Ykl. Общие погрешности в
значениях Ф° (Г) при 298,15, 3000 и 6000 К оцениваются в 0,05, 0,1 и 0,3 Дж-К^-моль.
1 Каррингтон и др. [889, 891] определили значение постоянной спин-орбитальной связи ВгО при анализе ЭПР-
спектров. Однако ими было получено, по-видимому, неправильное значение постоянной А.
187
НВг (г)
Глава 9
Таблица 9.5. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения «ша1 и е/цт, принятые для расчета термодинамических функций ВгО,
НВг, DBr и ТВг
Коэффициенты
Т 10-*
iVio-
* 20
Y .10'**
Y • 10^
01
Y .10^
V .10^
~у < л Q4
Y «10®
(a0=De).l0~*
а .101
а2-105
а3-10и
а л=_980 см.
ВгО
А*Щ<
0 2,787 1
7,787 4,859 04
^6,82 —5,401 04
—0,306 115 —0,739 132
0,265 773 —0,002 234 6
— —
0,428 799 0,327 609
—0,324 238 —0,451 548
—0,719 523 —1,140 24
—0,5 —0,6
1,967 94 0,768 97
0,632 681 0,701 827
0,124 303 0,185 090
—0,225 485 —0,991 239
49 27
303 229
6 А=—100 см.
НВг
XW
0
26,474 3
—43,790 8
—4,716 72
65,996 3
—3,337 17
8,468 65
—23,867 5
—35,150 1
—4,753 94
—343,97
3,158 7
5,306 19
19,255 5
—321,421
19
84
DBr
Х^
0
18,844 2
—22,186 4
—1,700 96
16,940 5
—0,609 73
4,290 59
—8,607 21
9,022 65
—0,868 586
—88,3
3,158 86
2,614 87
5,179 96
—58,797 0
26
118
ТВг
Х1Х+
0
15,493 1
—14,997 2
—0,945 32
7,740 55
—0,229 057
2,900 28
—4,783 51
4,122 68
—0,326 302
—40,346
3,157 17
2,110 54
2,405 23
—16,355 5
32
144
Ранее таблицы термодинамических функций ВгО(г) вычислялись во втором издании настоящего
справочника [88] по методу Гордона и Барнес и Брюером и Розенблаттом [764] {Т ^ 3000 К)
в приближении «гармонический осциллятор — жесткий ротатор». Расхождения в значениях
функций, вычисленных в работах [88, 764] и приведенных в табл. 68, обусловлены
преимущественно различиями в учете спин-орбитального расщепления основного электронного состояния
ВгО. Расхождения в значениях Ф° (Т) с данными [88] изменяются от 5 до 0,2 Дж-К^-моль
в интервале 298,15 — 6000 К и с данными [764] — от 0,7 до 3,4 Дж-К^-моль в интервале
298,15 - 3000 К.
Константа равновесия реакции ВгО (г)=Вг (г)+О (г) вычислена на основании принятого
в настоящем справочнике значения
ДД° @) = Do (ВгО) = 19 332 ± 200 см = 231,3 ± 2,4 кДж • моль.
Эта величина была найдена Дьюри и Рамзи [1350] короткой экстраполяцией по 20 уровням
колебательной энергии состояния А2П{ молекулы ВгО. Исследования распределения скоростей
молекул ВгО, образующихся при реакциях в молекулярных пучках [2962, 1263, 3115], приводят
к менее точным значениям, согласующимся в пределах их погрешности со значением,
приведенным выше.
Принятому значению энергии диссоциации соответствует
Д/Я°(ВгО, г, 0)= 133,406 + 2,4 кДж • моль.
НВг (г). Термодинамические свойства газообразного бромидаводорода в стандартном
состоянии при температурах 100 — 6000 К приведены в табл. 69.
Молекулярные постоянные, использованные при расчете термодинамических функций НВг,
приведены в табл. 9.3. Исследования электронного спектра НВг, выполненные в работах Барроу
и Стампера (см. [497а]) и Гинтера и Тилфорда [1661, 1662], показали, что единственным
стабильным валентным состоянием этой молекулы с энергией ниже 67 000 см является состояние X1!,+.
Значения молекулярных постоянных НВг в состоянии X1?,+, приведенные в табл. 9.3, получены
Ранком и др. [3150] на основании анализа вращательной структуры полос 1— 0, 2 — 0, 3 — 1
в ИК-спектре Н81Вг и данных Джонса и Горди [2162] по микроволновому спектру НВг. Значе-
188
Бром и его соединения DBr (г)
ния постоянных, полученные в работах [2064, 3046, 2826, 3671, 3045, 2722, 2094] в результате
анализа полос в ИК-спектре и в исследовании чисто вращательного спектра НВг [1824, 2162],
существенно менее точны. ИК-спектры молекул НВг, изолированных в матрицах инертных газов
при температурах 4—25 К, изучались в работах [721, 720, 477, 478], а электронный спектр
НВг(г) — в работах [1661, 1662, 1681, 515, 1172, 3237, 3236, 2748, 3087, 497а].
Термодинамические функции НВг (г) вычислены без учета возбужденных электронных
состояний непосредственным суммированием по уровням колебательно-вращательной энергии
электронного состояния ХХ2+ по уравнениям A.73)—A.75). Энергии уровней находились по
уравнениям A.65) и A.34) со значениями постоянных Ykl, приведенными в табл. 9.5 и полученными по
методике, изложенной в разделе 1.4 без учета различия постоянных молекул Н79Вг и Н81Вг. В расчете
учитывались уровни энергии, ограниченные предельной кривой диссоциации состояния ХгЛ+.
В таблице приведены значения коэффициентов а{ в уравнении, описывающем эту предельную
кривую, а также значения 1>шах и /нт- Погрешности вычисленных значений термодинамических
функций НВг определяются при низких температурах в основном неточностью принятых
значений фундаментальных постоянных, при Т > 3000 К могут стать заметными погрешности из-за
неточности аппроксимации колебательно-вращательных уровней НВг при больших значениях v
и / уравнениями с принятыми значениями коэффициентов Ykl. Общие погрешности в значениях
Ф° (Т) при Г=298,15, 3000 и 6000 К оцениваются в 0,02, 0,05 и 0,1 Дж -К -моль;
погрешности из-за пренебрежения различием постоянных молекул Н79Вг и Н81Вг не
превышают 0,01 Дж-К^-моль благодаря близости приведенных масс этих изотопов.
Ранее таблицы термодинамических функций НВг (г) вычислялись в работах [1697] (Т ^
^ 1600 К), [3946, 1454], предыдущих изданиях настоящего справочника [87, 88] и таблицах
JANAF [2095] (Т < 6000 К) х. Расчеты [1697, 87, 88] выполнены методом Гордона и Барнес;
в работе [3946] и таблицах JANAF [2095] использован метод Майера и Гепперт-Майер. Фебер
и Херрик [1454] рассчитали таблицы термодинамических функций НВг методом
непосредственного суммирования. Несмотря на различие использованных методов расчета и значений
постоянных, результаты всех расчетов мало отличаются от данных табл. 69. Наилучшее согласи^ данных
табл. 69 имеет место с функциями, вычисленными в работе [1454]. Расхождения с данными [87,
88, 3946] составляют от 0,01 Дж-К^-моль до 0,13 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции НВг (г)=Н (г)+Вг (г) вычислена на основании значения
Дг H° @)=D0 (HBr)=362,491 ±0,21 кДж •моль-1=30 302 + 18 см. Эта величина получена по
принятой в настоящем справочнике в соответствии с рекомендацией КОДАТА—MCHG [1000]
энтальпии образования НВг (г)
г, 298,15 К) = — 36,38 ±0,17 кДж • моль.
Выбор рекомендованного значения энтальпии образования НВг (г) был сделан по результатам
калориметрических измерений энтальпии образования водных растворов НВг [2142а, 21426,
3579а, 663а, 3676], калориметрических измерений энтальпии растворения НВг (г) в воде [3770,
3258, 3676] и измерений энтальпии реакции Н2 (г)+Вг2 (г)=2НВг (г) [2332а].
DBr (г). Термодинамические свойства газообразного бромида дейтерия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 70.
Молекулярные постоянные, использованные при расчете термодинамических функций DBr,
приведены в табл. 9.3. Исследования электронного спектра DBr [1661, 1662] показали, что
состояние Xx1j + является единственным стабильным состоянием этой молекулы с энергией ниже
66 000 см". Колебательные и вращательные постоянные молекул D79Br и D81Br в состоянии
Хг2 + определялись на основании изучения колебательно-вращательных [2722 ] и вращательных
[1706а, 2940, 1091, 1088, 1211] спектров в газе, а также при исследовании инфракрасных спектров
DBr в матрицах инертных газов [721], (см. [2508а]). В спектрах DBr наблюдались полосы ср^З.
Наиболее надежные значения постоянных D81Br, описывающие нижние
колебательно-вращательные уровни энергии состояния X1!! +, были получены Моулдом и др. [2722 ] и приведены в табл. 9.3.
Значения вращательных постоянных, найденные в этой работе, находятся в согласии с
полученными из анализа микроволнового спектра DBr [1706a, 2940, 1091, 1088, 1211].
1 В работе [802 ] методом непосредственного суммирования определены значения коэффициентов в
интерполяционных полиномах, позволяющие вычислять термодинамические функции Н78Вг и Н81Вг при 200—1000 К.
189
ТВг (г) Глава 9
Термодинамические функции DBr (г) были вычислены без учета возбужденных электронных
состояний с использованием непосредственного суммирования по уровням
колебательно-вращательной энергии состояния Х^" по уравнениям A.73)—A.75). Энергия уровней вычислялась
по уравнениям A.65) и A.34) с постоянными Ykl (см. табл. 9.5), полученными по изотопическим
соотношениям A.66) на основании постоянных Ykl молекулы НВг, приведенных в той же таблице.
Так же как и в случае НВг, различие молекулярных постоянных D78Br и D81Br не принималось
во внимание. В расчете учитывались все колебательно-вращательные уровни энергии,
ограниченные предельной кривой диссоциации. Значения коэффициентов а. в уравнении,
аппроксимирующем эту кривую, а также значения ушах и /нш, приведены в табл. 9.5.
Погрешности вычисленных значений термодинамических функций DBr (г) до 2000 К
обусловлены только неточностью фундаментальных постоянных; выше 2000 К добавляются погрешности
из-за неточности расчета числа и энергии высоких колебательно-вращательных уровней состояния
Х12+ по принятым постоянным. Погрешности в значениях Ф° (Т) оцениваются в 0,02, 0,05
и 0,1 Дж-К^-моль-1 при Г=298,15, 3000 и 6000 К, соответственно.
Ранее таблицы термодинамических функций DBr (г) вычислялись в предыдущем издании
справочника [88] по методу Гордона и Барнес х. Значения Ф° (Т) в [88] отличаются от
приведенных в табл. 70 не более чем на 0,10 Дж-К^-моль во всем интервале температур. Имеющиеся
расхождения обусловлены уточнением молекулярных постоянных DBr в настоящем издании,
а также различием использованных методов расчета.
Константа равновесия реакции DBr(r)=D(r)-fBr(r) вычислена на основании принятого
в справочнике значения
ДгЯ° @) = Do (DBr) = 30 678 + 18 см = 367,0 + 0,2 кДж • моль,
которое было найдено по соотношению C.6) и значениям Do (НВг)=30302+ 18 см, G @, НВг) =
=1313±1 см и G@, DBr)=937 + l см.
Принятому значению энергии диссоциации DBr соответствует
ДД0(DBr, г, 0) = —29,27 + 0,24 кДж • моль.
ТВг (г). Термодинамические свойства газообразного бромида згрития в стандартном
состоянии при температурах 100—6000 К приведены в табл. 71.
Молекулярные постоянные ТВг приведены в табл. 9.3. Электронные спектры молекулы ТВг
не исследовались. Так же как у НВг и DBr, основное электронное состояние ТВг должно быть
состоянием ХгЪ +, а все возбужденные электронные состояния иметь энергии свыше 60 000 см.
Колебательные и вращательные постоянные молекул Т79Вг и Т81Вг в состоянии Z12+, найденные
при исследовании колебательно-вращательного [2170] и вращательного спектров [849, 3245,
1211], согласуются между собой. В табл. 9.3 приведены значения молекулярных постоянных ТВг,
определенные Джонсом и Робинсоном [2170], которым не удалось выделить линии,
принадлежащие молекулам Т79Вг и Т81Вг. Эти постоянные существенно менее точны, чем постоянные Н81Вг,
приведенные в той же таблице.
Термодинамические функции ТВг (г) были вычислены без учета возбужденных электронных
состояний ТВг непосредственным суммированием по уровням колебательно-вращательной энергии
состояния ХХ2+ по уравнениям A.73)—A.75). Уровни энергии ТВг вычислялись по уравнению
A.65) со значениями коэффициентов YM, приведенными в табл. 9.5. Значения коэффициентов Ykl
для ТВг были вычислены по изотопическим соотношениям A.66) на основании соответствующих
постоянных молекулы НВг, приведенных в той же таблице. В расчетах учитывались все
колебательно-вращательные уровни энергии, ограниченные предельной кривой диссоциации
состояния Х12+. Коэффициенты ai в уравнении, аппроксимирующем эту кривую, а также значения
ршах и /iim приведены в табл. 9.5. Погрешности табулированных значений термодинамических
функций ТВг(г) при низких температурах обусловлены неточностью принятых значений
фундаментальных постоянных, а выше 2000 К — неточностью молекулярных постоянных ТВг. Они
могут достигать 0,02, 0,08 и 0,2 Дж-К^-моль в значениях Ф° (Г) при Г=298,15, 3000 и 6000 К.
1 В работе [802] непосредственным суммированием по колебательно-вращательным уровням энергии молекул
D79Br и D81Br при 200—800 К определены значения коэффициентов в интерполяционных полиномах,
описывающих термодинамические функции DT-'Br и D81Br.
190
Бром и его соединения
BrF (г)
Vfr)
1 a
А3Л, \
j
/7 +
Рис. 9.1. Схематические изображения кривых потенциальной энергии нижних электронных состояний
двухатомных гетероядерных молекул галогенидов XY (X — атом тяжелого галогенида, Y— атом легкого галогенида)
а — в случае C1F, BrCl, IC1, IBr; б — в случае BrF; в — в случае IF
Ранее таблицы термодинамических функций ТВг (г) вычислялись в справочнике [88] по
методу Гордона и Барнес 1. Расхождения между данными справочника [88] и табл. 71 не
превышают 0,1 Дж-К^-моль во всем интервале температур в значениях Ф° (Г). Имеющиеся
расхождения обусловлены различием молекулярных постоянных, а также использованных методов
расчета.
Константа равновесия реакции ТВг(г)=Т(г)+Вг(г) вычислена по принятому в справочнике
значению
АгН° @) = Do(ТВг) = 30 844 + 18 см = 369,0 + 0,2 кДж • моль,
которое было получено по соотношению C.6) и значениям Do (HBr)=30 302 + 18 см, G @, НВг) =
= 1313 + 1 см-1 и G@, TBr) = 771 + l см.
Принятому значению энергии диссоциации соответствует
г, г, 0) = —29,598 + 0,24 кДж • моль.
BrF (г). Термодинамические свойства газообразного фторида брома в стандартном состоянии
при температурах 100—6000 К приведены в табл. 72.
Молекулярные постоянные, использованные при вычислении термодинамических функций
BrF, приведены в табл. 9.3. Так же как и молекулы других двухатомных интергалогенов,
молекула BrF должна иметь семь связанных электронных состояний, соответствующих атомам Вг и F
в валентных состояниях 2Р%,чг- Четыре состояния (ХХ2, 3П2, А3!!^ 3П0-) имеют общий диссо-
циационный предел Br Bi\)+F BА/,), два состояния 0+ должны коррелировать с пределами
Br Bi\)+F BР'/2) и Br BPVj)+F BA/J, а отталкивательное состояние 0+, коррелирующее
с атомами Br BA/2)+F BA/2), имеет глубокий минимум в результате взаимодействия с двумя
1 В работе [802] непосредственным суммированием по колебательно-вращательным уровням энергии Т79Вг и Тв1Вг
прж 200 и 800 К определены значения коэффициентов в интерполяционных полиномах, позволяющих вычислять
термодинамические функции Т?9Вг и Т81Вг при 200—1000 К.
191
BrF (г) Глава 9
другими состояниями 0+ (рис. 9.1). Энергии возбуждения валентных состояний, образованных
ионами Вг+ CР, W, 1S)+F~ ?S), превышают 50 000 см (см. [794]). В спектрах BrF
наблюдались переходы между основным электронным состоянием Х*2 + и тремя из шести перечисленных
выше возбужденных состояний: АЪП1, BSHO+ и В'0+. Согласно анализу, выполненному в
работах [797, 1104], последнее состояние коррелирует с третьим диссоциационным пределом
молекулы BrF, соответствующим атомам Br (^i/J+F BР»/2).
Молекулярные постоянные BrF в состоянии Х*2 + определялись при изучении системы полос
В3И0+—Х1^" в электронном спектре [793, 794, 795, 796, 797, 993, 1345],
колебательно-вращательного спектра газообразного BrF [3511], а также спектра BrF, замороженного в аргоновой
матрице [3447], и вращательного спектра BrF [3459]. Результаты этих исследований находятся в
удовлетворительном согласии. Приведенные в табл. 9.3 значения колебательных постоянных BrF
в состоянии ХХ2+ приняты по работе Клайна и др. [993], где они были получены в результате
изучения системы полос В3И0+—Х12+ в спектре испускания (v" ^ 14) и данных Бродерсена
и Сикра [797], исследовавших эту систему полос в спектре поглощения (v" ^ 2). Вращательные
постоянные приняты по работе Кэлдера и Руденберга [864а], в которой они рассчитаны на
основании исследования вращательного спектра в работе Смита и др. [3459].
Переходы, связанные с электронными состояниями 3П2 и 3П0-, в спектрах BrF не наблюдались.
Приведенные в табл. 9.3 значения постоянных BrF в этих состояниях оценены на основании
общих качественных представлений о характере потенциальных кривых и значениях молекулярных
постоянных гетероядерных двухатомных галогенов, а также значений постоянных BrF в
состояниях -43nx и 2?3П0+. Погрешности в оцененных значениях Те, ше, швхв и г, могут составлять
соответственно 400, 20, 2 см-1 и 0,05 А.
Сведения о состоянии А3^ молекулы BrF базируются только на весьма ненадежных данных
Бродерсена и Сикра [797], наблюдавших в спектре поглощения слабые полосы, накладывающиеся
на полосы системы BaU0+—Z12+ и на спектр Вг2. Данные работы [797] были критически
рассмотрены Коксоном [1104], показавшим, что только два канта (при 18 836 и 18 159 см) можно с
уверенностью отнести к системе Л3П:—ХХ2+ (полосы 6 — 0 и 6 — 1). Приведенные в табл. 9.3
значения постоянных BrF в состоянии А3^ приняты на основании данных работ [797, 1104],
энергии диссоциации BrF в состоянии А3^ и предположения о близости молекулярных постоянных
BrF в состояниях Л3ПХ и В3Н0+. Погрешности в значениях Те, ше, u>txe и г, могут достигать
соответственно 200, 20, 2 см-1 и 0,05 А.
Молекулярные постоянные BrF в состоянии 53П0* определялись на основании изучения
системы полос B3U0+—X1H+ в спектре испускания [1345, 993] и поглощения [793, 795, 796, 797].
В спектрах во всех перечисленных работах наблюдались полосы cd'^ 10, причем уровни с v ^ 5
возмущены.
Приведенные в табл. 9.3 значения колебательных постоянных BrF в состоянии В3П0ь
вычислены авторами настоящего издания по началам полос z/ ^ 5, полученным в работе
Бродерсена и Кудмана [793]. Значения вращательных постоянных вычислены по найденным в работе
[793 ] значениям постоянных В'± и В'ь. На основании анализа данных, приведенных в работе
Бродерсена и Сикра [797] и с учетом работ [1104, 950] в настоящем справочнике принимается, что
группа слабых полос в области 21 704—22 244 см, наблюдавшаяся в работе [797], относится
к системе В'0+—Х12+. В табл. 9.3 приведены значения постоянных BrF в состоянии 2?'0+ в
предположении, что диссоциационный предел этого состояния B4 282 + 83 см) коррелирует с
атомами Вг BРч2) и F BРз/,). Сведения о состоянии 0+, коррелирующем с атомами Br BP3/2)-(-F BPi/2)
в литературе отсутствуют, но можно предполагать, что в этом состоянии молекула BrF имеет
небольшое число связанных и квазисвязанных колебательно-вращательных уровней.
Термодинамические функции BrF (г) вычислялись по уравнениям A.3)—A.6), A.9), A.10),
A.93)—A.95). Значения (?вн, (?вв и Gва были получены по соотношениям A.76)—A.78), в
которых статистические суммы по колебательно-вращательным уровням энергии состояний ХХ2+,
3П2, ^3П15 3Л0-, -В3П0+ и В'0+ и их производные находились непосредственным суммированием
по уравнениям A.73)—A.75). В расчете учитывались все уровни энергии состояния Х12+,
ограниченные предельной кривой диссоциации, уровни энергии состояний 3П2, А3!!^ 3П0- и 0+,
ограниченные диссоциационными пределами этих состояний, и уровни энергии состояния 53П0+ для
v ^ 10. Колебательно-вращательные уровни энергии определялись по уравнениям A.65) и A.34),
вырождение вращательных уровней состояний 3П2 и 3И1 учитывалось статистическим весом 2.
192
Бром и его соединения BrF3 (г)
Значения постоянных Ykl в этих уравнениях, вычисленные по методикам, изложенным в разделе 1.4,
приведены в табл. 9.4. В той же таблице приведены значения а(, итлх и /нт в уравнении,
аппроксимирующем предельную кривую диссоциации состояния Х1!! + и значения а0 и ушах для остальных
состояний. Погрешности вычисленных термодинамических функций BrF (г) при низких
температурах обусловлены главным образом неточностью фундаментальных постоянных, а выше
3000 К — неточностью принятых значений молекулярных постоянных в возбужденных
электронных состояниях и принятых методик определения числа и энергии связанных и квазисвязанных
колебательно-вращательных уровней энергии вблизи диссоциационных пределов. При Т >
> 5000 К дополнительные погрешности могут быть связаны с тем, что в расчете не учитывалось
состояние 0+, коррелирующее с атомами Br Bi\)+F BЛ/2). Погрешности в значениях Ф° (Т)
при Г=298,15, 3000 и 6000 К оцениваются в 0,02, 0,05 и 0,5 Дж-К^-моль.
Ранее таблицы термодинамических функций BrF(r) вычислялись в работах [1012] (Т7^
^ 2000 К), [1426] (Т ^ 1500 К), [3944], во 2-м издании настоящего справочника [88] и таблицах
JANAF [2095]. Все расчеты, за исключением [88], были выполнены методом Майера и Гепперт-
Майер без учета возбужденных электронных состояний. В справочнике [88] термодинамические
функции BrF вычислялись по методу Гордона и Барнес с учетом суммарного вклада состояний
3П2, ^43ПХ, 3П„- и В3П0+. Значения Ф° (Т), приведенные в табл. 72 и в [88], при 7=298,15, 3000
и 6000 К отличаются на 0,01, 0,2 и 0,6 Дж-К^-моль из-за различия молекулярных
постоянных, принятых в расчетах. Расхождения с данными работ [2095, 3944] достигают 0,4 Дж -К -моль
в Ф° (Т) и 1,5-3 Дж-К^-моль-1 в S0 (Т) и С°р (Т).
Константа равновесия реакции BrF (r)=Br (r)+F (г) вычислена на основании значения
Д(.Яо@)=Б0(ВгР)=246,398 + 1,0кДж-моль-1=20 600 + 80 см, соответствующего принятой в
справочнике энтальпии образования
A/F°(BrF, г, 0) = — 51,2 + 1,0 кДж • моль.
Эта величина принята по результатам исследования равновесия реакции Br2 (r)+BrF3 (г) =
=3 BrF (г) статическим и спектрофотометрическим методами в интервале 328—380 К,
выполненного Штейненбергом и др. [3528]. Обработка по уравнению III закона результатов измерений
[3528], полученных статическим методом, приводит к значению A^°@)=45,46+0,2 кДж-моль,
согласующемуся в пределах 2 кДж-моль с расчетом по II закону. Спектрофотометрические
измерения равновесия этой реакции привели к близкому, но менее точному значению. Учет
возможной димеризации BrF3 не оказывает влияния на результаты расчета.
Исследования равновесия реакций с участием BrF в работах Фишера и др. [1490] и Руффа
и Брайды [3266] приводят к менее точным значениям, согласующимся с принятым выше в
пределах точности их измерений.
Принятое значение согласуется со значением Do (BrF) ^ 20 880 см, найденным по пре-
диссоциации в состоянии B3U0+ (v=8, J ~^> 28) в работе Клайна и др. [994а]. В большинстве
справочных изданий (см. [88, 2095]) для Do (BrF) принималось существенно более низкое значение
A9 230 см или примерно 230 кДж-моль), основанное на оценке энергии диссоциационных
пределов состояний Л3ПХ и В3П0+ в работе [797]. Однако, как показал Коксон [1104], отнесение ряда
полос системы -43ПХ—ХХ2+ в спектре BrF, предложенное Бродерсеном и Сикром [797],
по-видимому, было ошибочным, а экстраполяция уровней состояния В3И0+ к диссоциационному пределу
Br BjPi/2)+F BРз1г), т. е. выше области пересечения потенциальных кривых состояний В3И0+
и В'0+, некорректной. Поэтому значение Do (BrF), основанное на исследованиях спектров BrF
в работах [797, 793, 993], оказалось ошибочным.
BrF3 (г). Термодинамические свойства газообразного трехфтористого брома в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 73.
Молекулярные постоянные, использованные в расчетах термодинамических функций BrF3,
приведены в табл. 9.6. Молекула BrF3 (основное электронное состояние ХХА) относится к
точечной группе симметрии С2, и имеет плоскую Т-образную структуру с неравноценными связями:
одной короткой связью Вг—F', расположенной вдоль оси симметрии, и двумя длинными Br—F",
расположенными под углом, близким к прямому к оси симметрии. Магнусон [2502] на основании
анализа вращательной структуры МВ-спектра газообразного трехфтористого брома определил
вращательные постоянные молекул 79BrF3 и 81BrF3 в основных колебательных состояниях (Ао=
193
BrF5(r)
Глава 9
Таблица 9.6. Значения молекулярных постоянных и а в состоянии X1 Л, принятые для расчета
термодинамических функций BrF3 и BrF6 (p%=i)
Молекула
BrF3
BrF5
v2
v4
v7B)
v.B)
v.B)
CM
675
684
552
584
238
370
614
547
350
277
242
312
644
415
IAIBIC X 10
Г3 • CM6
— 4528
245 2,89 • 10*
2
4
=0,361625 1 и 0,360 482 3 car1, J?0=0,136 013 и 0,136 0011 см, C0=0,098 687 9 и
0,098 60187 см), которым соответствуют значения r0 (Br—F')=1,721 А, г0 (Вг—F")=l,810 A
и -^ F'—Вг—F"=86° 12,6'. Приведенное в табл. 9.6 произведение главных моментов инерции
BrF3 вычислено для природной смеси изотопов брома по вращательным постоянным молекул
79BrF3 и 81BrF3, найденным в работе [2502]. Колебательные спектры BrF3 изучались на приборах
с низкой дисперсией, что не позволило выделить полосы молекул 78BrF3 и 81BrF3. ИК-спектр
паров изучался в работах [971, 3511, 3378], жидкого BrF3 — в работе [1793], а спектр КР
газообразного и жидкого BrF3 — в работах [971, 3378]. Кроме того, в [955, 1542] исследовался ИК-
спектр молекул BrF3, стабилизированных в матрицах инертных газов. Наиболее обстоятельное
исследование колебательных спектров паров BrF3 выполнили Селиг и др. [3378]. Приведенные
в табл. 9.6 значения основных частот основаны на результатах изучения спектров BrF3 в работах
[3378, 1542]. Погрешности в принятых значениях не превышают 3—5 см. Возбужденные
электронные состояния BrF3 неизвестны.
Термодинамические функции газообразного BrF3 вычислены по уравнениям A.3)—A.6),
A.9), A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий
ротатор—гармонический осциллятор» с учетом только основного электронного состояния. Погрешности
табулированных значений обусловлены приближенным характером расчета и в меньшей степени
неточностью молекулярных постоянных. Они составляют 0,5, 4 и 6 Дж-К^-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Ранее термодинамические функции BrF3 (г) в том же приближении вычисляли Классен и др.
[971] (Т < 1000 К), Кристе и др. [955] (Т < 2000 К) и авторы таблиц JANAF [2095] (Т <
<^ 6000 К). Расхождения в значениях Ф° (Т), приведенных в табл. 73 и в работах [2095, 971 ],
достигают соответственно 3 и 3,7 Дж-К^-моль, что связано с использованием в [2095] и [971]
неточных значений основных частот BrF3, предложенных в работе [971 ]. Данные, приведенные
в работе [955] и в табл. 73, совпадают в пределах 0,1 Дж-К^-моль.
Константа равновесия реакции BrF3(r)=3F(r)+Br(r) вычислена с использованием
значения Дг#° @)=594,562+3,1 кДж-моль, соответствующего энтальпии образования
A^° (BrF3, г, 298,15 К) = —255,6 + 3,0 кДж • моль.
Эта величина принята по результатам калориметрического измерения энтальпии реакции
1/2 Вг2 (г)+3/2 F2 (r) = BrFs(r), ДГЯ°B98,15К) = —271,0±3,0 кДж-моль-1, выполненного
Штейном [3512]. Количество BrF5, образующегося совместно с BrF3 в реакции фторирования брома,
было учтено Штейном на основании измерений общего давления продуктов реакции, давления
насыщенных паров BrF3 и BrF5 и результатов изучения кинетики реакции фторирования брома
[3510].
Менее точные значения энтальпии образования BrF3 (г) могут быть получены по данным Ри-
чардса и Волфа [3197] (—272 кДж-моль-1), Руффа и Брайды [3266] (—269 кДж-моль-1) и Берн-
стейна и Каца [605] (—232 кДж-моль).
BrF6 (г). Термодинамические свойства газообразного пятифтористого брома в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 74.
Молекулярные постоянные, использованные в расчете термодинамических функций BrF6,
приведены в табл. 9.6. Молекула BrF5 относится к точечной группе симметрии Cif и имеет
структуру правильной квадратной пирамиды, в вершине которой расположен атом брома, а в углах
194
Бром и его соединения BrF5 (г)
при основании — четыре атома фтора (FpM). Пятый атом фтора (FaKC) расположен на оси симметрии
ниже плоскости основания пирамиды. Связи Br—FpM и Br—Faico неравноценны, а угол между
ними меньше 90°. Эти представления о молекулярной структуре BrF5 основываются на
результатах рентгенографического исследования его кристаллической структуры [835], электронографи-
ческого исследования структуры молекул газа [3213] и измерениях расстояния между
максимумами Р- и .R-ветвей полос \, v2, v3 в ИК-спектре [2579]. Наиболее точные значения
структурных параметров молекулы BrF5 получены Робитте и др. [3213]: г (Br—FaI№)=l,689+0 008А
г (Br—FpM) = l,774±0,003 А; ^ FaKC-Br-FpaI==84,8±0,l°. Они находятся в соответствии со
значениями вращательной постоянной Во молекул 79BrFs и 81BrF6 @,103 380 2 и 0,103 298 8 см),
найденными при анализе вращательной структуры микроволнового спектра [3934, 733, 35831!
Эти значения вращательной постоянной и главный момент инерции 1д, вычисленный по
структурным параметрам, использованы для расчета значения 1А1в1с, приведенного в табл. 9.6. Его
погрешность составляет 1 -Ю15 г3-см6. Молекула BrF5 имеет девять основных частот. Спектр КР
изучался только для жидкого BrF5 [839, 3525, 555], а ИК-спектр для газообразного [839, 2579
555], жидкого BrF8 [3580] и молекул BrF5, изолированных в матрицах из аргона [1542]. Для
частот \, v2, v3, v7, v8 и v9, активных как в спектре КР, так и в ИК-спектре, во всех работах были
получены согласующиеся между собой значения. Наиболее детально на приборе с хорошим
разрешением ИК-спектр газообразного BrF5 исследовали Мак-Дауэлл и Аспри [2579], которые
нашли контуры отдельных ветвей для частот, активных в ИК-спектре, а для полос vx и v3
максимумы ^-ветвей. Приведенные в табл. 9.6 значения частот v1( v2, v3, v7, v8 и v9 приняты но работе
[2579], их погрешности не превышают 1—2 см. Частоты v4, v5 и ve определены значительно
менее точно. Для частоты v4 в таблице приведено значение, вычисленное Биганом и др. [555] по
составным частотам v4+v7 и v2-fv4+v7 , наблюдаемым в ИК-спектре газа. Оно согласуется со
спектром КР [839, 3525, 555] и ИК-спектром жидкого BrF5 [3580]. Полосы, соответствующие
частоте v5 или составным частотам с участием v5, в спектрах BrF5 не наблюдались. В работах
[839, 3525] к частоте v5 была ошибочно отнесена полоса при 310 или 315 см, соответствующая
частоте ve [555]. Расчеты по силовым постоянным привели к значениям v5=281 [555] и 274 см
[7331. В табл. 9.6 для v5 принято среднее значение между этими двумя величинами, для частоты
ve — значение, найденное Биганом и др. [555] *. Погрешности принятых значений v4, v6 и ve
могут достигать 5 см. Возбужденные электронные состояния BrF5 неизвестны.
Термодинамические функции BrF5 (г), приведенные в табл. 74, вычислены по уравнениям
A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор-
гармонический осциллятор» без учета возбужденных электронных состояний. Погрешности
рассчитанных значений обусловлены в основном приближенным характером расчета и в меньшей
степени неточностью молекулярных постоянных. При высоких температурах к этому добавляются
возможные ошибки из-за отсутствия данных о возбужденных электронных состояниях BrFs
Погрешности в значениях Ф°(Т) при 298,15, 3000 и 6000 К могут достигать 1, 9 и 12 Дж -К -моль
соответственно.
Ранее термодинамические функции BrF5 (г) в том же приближении вычисляли Стивенсон
и Джонс [3525], Эванс и др. [1426], Мак-Дауэлл и Аспри [2579], Кханна [2236] (Т <
< 1500 К) и авторы справочников [2095, 66]. В работах [3525, 1426, 2579, 2236] принимались
ошибочные значения частот v5, ve и vg и оцененные значения структурных параметров.
Расхождения в значениях Ф° (Т), вычисленных в этих работах и приведенных в табл. 74, достигают 3 ДжX
X К-моль [1426 и 2579] и более 4 Дж-К^-моль [2236]. Данные работы [2095] и табл. 74
согласуются в пределах 0,5 Дж-К^-моль во всем интервале температур. В работе [66]
приведены значения Ф° (Т) и S° (Т), вычисленные для различных моделей молекулы BrF6 (C3f и Си).
Расчет для квадратной пирамиды был выполнен по оцененным значениям структурных параметро'в,
существенно отличающимся от найденных экспериментально. Авторы работы [66] не решились
рекомендовать один из вариантов как предпочтительный.
Константа равновесия реакции BrF5 (г)=Вг (r)+5F (г) вычислена с использованием значения
Д^0 @)=917,95 + 2,5 кДж-моль, основанного на принятой в справочнике энтальпии
образования
A/F°(BrF5, г, 298,15 К) = — 428,8 ±2,0 кДж • моль.
1 В работах [839, 3525] частоте ve были приписаны значения 480 и 481 см, обусловленные, по-видимому, примесью
195
BrCl (г) Глава 9
Это значение было получено Штейном [3512] в результате калориметрических измерений
реакции фторирования брома, проводимой в условиях избытка фтора при температурах 378—
401 К. В этих условиях, как было показано в кинетическом исследовании реакции фторирования
брома [3510], весь бром превращается в BrF5. Менее точные значения энтальпии образования
BrF5 могут быть получены по данным работ Бернстейна и Каца [605] (—463 кДж-моль) и Руффа
и Брайды [3266] (—406 кДж-моль-1).
BrCI (г). Термодинамические свойства газообразного хлорида брома в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 75.
Молекулярные постоянные, использованные в расчете термодинамических функций BrCl,
приведены в табл. 9.3. Как и молекулы других двухатомных интергалогенов (см. BrF(r) и рис. 9.1),
молекула BrCl должна иметь семь связанных электронных состояний, коррелирующих с атомами
Вг и С1 в валентных состояниях 2Рз/г t/i. Четыре из этих состояний (Х*2 +, 3П2, ^l3!^ и 3П0-) имеют
общий диссоциационный предел Вг (aPs/2)-|-Cl BA/2), два состояния 0+ должны коррелировать
с атомами Вг B.Рз/2)+С1 Bi\) и Вг BР./2)+С1 (ai\), а отталкивательное состояние 0+, также
коррелирующее с атомами Вг Bi\)-f Gl Bi\), в результате взаимодействия с двумя другими
состояниями 0+ имеет относительно глубокий минимум и рассматривается в литературе как
состояние 3П0+. Это взаимодействие приводит к тому, что состояние 0+, имеющее диссоциационный
предел Вг (^«/J-f-Cl BРчг), который расположен всего на 880 см выше предела состояния
В3П0+, является слабосвязанным и имеет небольшое число колебательно-вращательных уровней
энергии. Электронные состояния BrCl, коррелирующие с пределами Br+ CР, XD, ^-f-Cl" (lS),
имеют энергии свыше 50 000 см (см. [1066, 1283]).
Молекулярные постоянные BrCl в состоянии Х1^ + определялись при исследовании
электронного [987, 988, 990, 992, 1103, 1790], микроволнового [3460], инфракрасного спектров [2546,
806] и спектра комбинационного рассеяния [3502, 1992, 3867]. Наиболее полные и точные данные
о состоянии ХХ2+ получены Коксоном [1103] в результате изучения вращательной структуры
полос (v' ^ 8, v" ^ 7) системы В3И0+ <- X1!,+ молекул 79ВгС1 и 81ВгС1. Определенные в работе
[1103] значения постоянных молекулы 79ВгС1 в состоянии Х12+ приведены в табл. 9.3. В
исследованных спектрах BrCl переходы, связанные с состояниями 3П2, 3П0- и 0+, не наблюдались. В
длинноволновой части спектра поглощения BrCl Коксон [1103] обнаружил ряд полос, которые он
предположительно интерпретировал как полосы системы А3Лг—Х1^ +. Приведенные в табл. 9.3
энергии возбуждения состояний 3П2, Л3Пг и 3П0- оценены как средние арифметические из
принятых энергий возбуждения соответствующих состояний молекул Вг2 и BrF, поскольку такое
соответствие имеет место для состояний 53П0+ этих молекул. Погрешности в принятых значениях
энергий возбуждения могут достигать 200 см. Значения тв и Ве молекулы BrCl в состояниях 3П
б 93 53П Д
2,
уд у в е 2,
4П2 и 3П0- в табл. 9.3 оценены по значениям этих постоянных в состоянии 53П0+. Данные о
состоянии В3И0+ основаны на результатах исследования системы полос BZHO+—Х12+ в испускании
[987, 988, 1790] (v' <! 8) и в поглощении [990, 992, 1103] (г/ <! 9). Наблюдаемые аномалии в
энергии колебательных уровней и значениях вращательных постоянных свидетельствуют о
возмущениях, вызванных взаимодействием состояний .В3П0+ и 0+. Наиболее полное исследование состояния
В3Н0+ молекул 79ВгС1 и 81ВгС1 выполнено Коксоном [1103], определившим с высокой точностью
значения Go (v), В„ и D, этих молекул в состоянии В3П0+ для v=2, 3, . . ., 8. Найденные в работе
[1103] значения постоянных молекулы 79Вг3бС1 в состоянии 2?3П0+ приведены в табл. 9.3.
Термодинамические функции BrCl (г), приведенные в табл. 75, были вычислены по уравнениям
A.3) A.6), A.9), A.10), A.93)—A.95). Значения Qm, QBB и QBB рассчитывались по соотношениям
A.76) A.78), в которых статистические суммы по колебательно-вращательным уровням энергии
состояний X1!,+, 3П2, ^43П1; 3П0- и 53П0+ и их производные находились непосредственным
суммированием по уравнениям A.73)—A.75). В расчете учитывались все уровни состояния Х1?,*,
ограниченные его предельной кривой диссоциации, уровни энергии состояний 3П2, Л3П1 и 3П0-,
ограниченные диссоциационными пределами этих состояний, и уровни состояния В3Л0+ со значениями
у <Г 10. Колебательно-вращательные уровни этих состояний вычислялись по уравнениям A.65)
и A.62) со значениями коэффициентов Ykl, приведенными в табл. 9.4 и полученными по методикам,
изложенным в разделе 1.4. Вырождение вращательных уровней состояний 3П2 и у!3Пх учитывалось
статистическим весом 2. В таблице приведены также значения постоянных а{ в уравнении,
аппроксимирующем предельную кривую диссоциации состояния Хг2 +, значения vmax в пяти
электронных состояниях и /um Для состояния Х1^ +. Состояния 0% коррелирующие с диссоциационными
196
Бром и его соединения
ВгС1(г)
пределами Вг B_Рз/2)-[-С1 BPi/J и Вг B^\0+С1 B^Ч)> B расчете не учитывались. Погрешности
термодинамических функций BrCl (г), приведенных в табл. 75, при низких температурах
обусловлены неточностью фундаментальных постоянных, принятых в расчете; при Т ^> 2000 К становятся
существенными погрешности из-за неточности аппроксимации высоких
колебательно-вращательных уровней энергии состояния Х1^ и уровней энергии состояний 3П2, 3П1 и 3П0-, а выше 5000 К —
из-за пренебрежения электронными состояниями 0+. Погрешности в значениях Ф° (Т) составляют
0,03, 0,1 и 0,5 Дж-К^-моль-1 при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций BrCl (г) вычислялись в работах [41, 42] (Т <С
< 1000 К), [1012] (Т < 2000 К), [2546] (Т < 1000 К), [1426] (Т < 1500 К), во 2-м издании
настоящего справочника [88] и таблицах [2095]. Расчеты в работах [41, 42,1012, 1426, 2095] были
выполнены методом Майера и Гепперт-Майер, а в [2546, 88] — методом Гордона и Барнес. Во всех
расчетах учитывалось только основное электронное состояние BrCl и использовались менее
точные, чем в настоящем издании, постоянные. Этими причинами объясняются расхождения между
значениями термодинамических функций BrCl, вычисленными ранее и приведенными в табл. 75.
Расхождения между значениями Ф°(Т) и S°(T), табулированными в работах [88, 2095], с одной
стороны, и в настоящем справочнике — с другой, не превышают 0,03 Дж -К -моль при 298,15 К,
но достигают 1 и 3 Дж-К^-моль при 6000 К.
Константа равновесия реакции ВгС1(г)=С1(г)-1-Вг(г) вычислена по принятому значению
энергии диссоциации
ДГЯ°@) = Do (BrCl) = 215,32 + 0,10 кДж • моль = 17 999 + 8 см.
Это значение получено на основании результатов исследований равновесия реакции 2ВгС1 =
=С12 (г)-}-Вг2 (г), приведенных в табл. 9.7. Поскольку все измерения проводились в узких
интервалах температур, в таблице даны только значения &ГН @), вычисленные по методу III закона.
Погрешности вычисленных величин учитывают дисперсию экспериментальных данных, неточности
термодинамических функций (около 0,04 кДж-моль) и оцененные систематические ошибки.
Таблица 9.7. Результаты определения энтальпии реакции 2ВгС1 (г) = Вг2 (г) -f- Cl2 (г) (в кДж • моль)
Грей, (
Авторы
:тайл, 1930
Йост, 1931 [4038]
Веспер,
Брауэр,
Бисон,
Роллефсон,
[1717]
1934 [3832]
Виктор-Берлин, 1935 [751]
Йост, 1939
[552]
Еллинек, Шютца, 1936 [2110]
Шютца,
Матро
1938 [3361]
и др., 1954
[2546 ]
Метод
Спектрофотометрический, 298 К, 2 точки
Спектрофотометрический, 274—291 К, 16 точек
(измерения в области 361—500 К менее точны и не
принимались во внимание)
Спектрофотометрический, 301 К, 9 точек
Спектрофотометрический, 298 К
Статический, реакция Cl2+Br2+NO, 373—491 К,
21 точка, приближенно учтены поправки на
отклонение от свойств идеального газа
Статический, реакция Вг2+С12 в присутствии КС1
и КВг, 1073 К, 6 точек
Статический, реакция Вг2+С12 в присутствии смеси
хлоридов и бромидов свинца, серебра, калия или
натрия
Масс-спектрометрический, 310 К, 6 точек
ДГД° @)
1,27 ±0,40
1,35 ±0,40
2,03+0,40
1,27+0,20
1,95 ±0,50
6,4
1,1
1,23 ±0,20
Результаты исследований равновесий с участием конденсированных фаз [2110, 3361] менее
точны по сравнению с исследованиями равновесия реакций в газовой фазе. В работе [552] была
сделана попытка учесть отклонения свойств реагирующих веществ от свойств идеального газа,
однако при этом были допущены ошибки, повлиявшие на полученное значение. Основываясь
на наиболее надежных работах [751, 1717 и 2546], при подготовке справочника было выбрано
значение &ГН @)=1,25 + 0,20 кДж-моль, которому и соответствует принятое значение De (BrCl)
и энтальпия образования BrCl (г)
Д/Я°(ВгС], г, 0) = 22,223 ± 0,12 кДж • моль.
Глава 10
ИОД И ЕГО СОЕДИНЕНИЯ
I, Г, 12 (к, ж), 12, 10, HI, DI, TI, IF, IF5, IF7, Id, IBr
В данной главе рассматриваются 12 соединений иода в газообразном состоянии, а также
кристаллический и жидкий иод. Термодинамические свойства I (г) и 1~ (г) рассчитаны для температур до
10 000 К, остальных газов — от 100 до 6000 К. Поскольку давление насыщенных паров иода
достигает 100 атм уже при температурах около 800 К, таблица термодинамических функций
12 (к, ж) в интервале температур 5—800 К приведена в Дополнении (см. наст, том, кн. 2). Для двух
соединений A2 и HI) в Приложении 3 приведены данные, позволяющие учитывать влияние
межмолекулярного взаимодействия на отклонения свойств этих газов от их свойств в стандартном
состоянии.
В настоящем издании не рассматривается ряд многоатомных соединений иода, типа 1С13,
Ю~ и другие, так как в равновесных условиях содержание этих компонентов при Т ^ 1000 К
должно быть пренебрежимо мало.
Таблица 10.1. Уровни энергии атомов I иI",
принятые для расчета
термодинамических функций
Атом
Электронная
конфигурация
Состояние
СМ
Pi
I ... 5s85p6 2P3/2 0 4
. . . 5s25p5 2/>Vj2 7603,15 2
I- ... 5s25p« 15 ' 0 1
I (г). Термодинамические свойства газообразного
иода в стандартном состоянии при температурах
100—10 000 К приведены в табл. 76.
Уровни энергии атома I, использованные при
расчете термодинамических функций, приведены
в табл. 10.1. Энергия компоненты 2Рч, основного
электронного состояния принята по работам Кисса
и Корлисса [2241] и Минхагена [2675], в которых
было уточнено значение этой величины, приведенное
в справочнике Мур [2697].
Термодинамические функции 1(г) вычислялись
по уравнениям A.3)—A.6), A.9), A.10), A.23)—
A.25). Значения QBB и ее производных находились непосредственным суммированием по уровням
энергии, приведенным в табл. 10.1. Погрешности вычисленных значений термодинамических
функций 1(г) во всем интервале температур обусловлены только неточностью принятых значений
фундаментальных постоянных и не превышают 0,02 Дж-К^-моль в значениях Ф° (Г) и S° (T).
Ранее таблицы термодинамических функций I вычислялись в работах [1426, 3055], в
предыдущих изданиях справочника [88, 87] и в справочниках [1946, 2095]. Расхождения в
термодинамических функциях, табулированных в [1426, 3055,88, 87, 1946, 2095] и в табл. 76, не превосходят
0,012 Дж-К^-моль в значениях Ф°(Г) и S°(T) и обусловлены различиями в использованных
значениях энергии терма аРч, и фундаментальных постоянных.
Энтальпия образования 1(г) принята равной
Д/Я°A, г, 298,15 К)= 106,762+ 0,040 кДж • моль
по рекомендации КО ДАТА—MCHG [1000]. Эта рекомендация основана на значениях энергии
диссоциации и энтальпии образования 12 (г).
I" (г). Термодинамические свойства газообразного отрицательного иона одноатомного иода
в стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 77.
Ион 1~ имеет основное состояние XS (см. табл. 10.1), возбужденные состояния 1~ должны иметь
энергии больше потенциала ионизации этого иона. Термодинамические функции 1~ (г) были
вычислены по уравнениям A.9) и A.10), поскольку внутримолекулярные составляющие были
приняты равными нулю. Погрешности вычисленных термодинамических функций во всем интервале
температур обусловлены только неточностью принятых значений фундаментальных постоянных
и не превышают 0,02 Дж-К^-моль в значениях Ф° (Г) и S° (T). Ранее таблицы термодинами-
198
Иод н его соединения
12 (к, ж)
ческих функций 1~ (г) вычислялись только в предыдущих изданиях справочника [88, 87].
Расхождения в значениях Ф°(Г) и S°(T), вычисленных в этих изданиях и приведенных в табл. 77, не
превосходят 0,01 Дж-К^-моль и обусловлены некоторым отличием в принятых значениях
фундаментальных постоянных.
Константа равновесия реакции 1~(г)=1(г)+е (г) вычислена с использованием значения
Дг#° @)=295,59+0,05 кДж-моль в соответствии с принятым в справочнике сродством атома
иода к электрону
АоA) = —295,59 + 0,05 кДж • моль.
Это значение было найдено Берри и др. [612, 611] по границе непрерывного спектра
поглощения, соответствующего фотоотрыву электрона от иона 1~ (Х=4046 + 0,6 А, или 24 709 + 4 см).
Значения Ао (I), найденные в более ранних работах [27, 456, 2715, 3516, 2936], основаны на
существенно менее точных экспериментальных измерениях.
Принятому значению Ао (I) соответствует
AfH°(r, г, 0) = —188,437 ±0,30 кДж • моль'1.
12 (к, ж). Термодинамические свойства твердого и жидкого иода в стандартном состоянии
в интервале 5—800 К приведены в табл. Д. 16.
Значения постоянных, использованные в расчетах термодинамических функций, приведены
в табл. 10.2. При температурах ниже 298,15 К термодинамические функции кристаллического
Таблица 10.2. Принятые значения термодинамических величин для 12 в кристаллическом
и жидком состояниях
Состояние
Н° B98,15К)—
—Я0 @)
кДж • моль
S" B98,15 К)
С B98,15 К)
Дж • К • моль
Коэффициенты в
уравнениях для С° (Г)»
а
6-10»
с•10"»
Интервал
температур
К
Т
К
кДж-моль
к 13,196 116,139 54,438 —133,0993 423,24 —5,44816 298,15—386,75 386,75 15,665
ж — — — 79,555 — — 386,75—800 — —
» С°р(Т) = а + ЬТ—сТ-* (Дж • К • моль).
иода вычислены по результатам измерений теплоемкости иода, проведенных Шерли и Джиоком
[3412]. В этой работе теплоемкость иода высокой чистоты (содержание примесей около 0,01%)
была измерена в интервале 13,6—322 К в адиабатическом калориметре, изготовленном из
материала, пассивного к иоду. Экстраполяция теплоемкости ниже 13,6 К проводилась по
уравнению С°р (T)=D {1ЫТ)+2Е A01/71), которое воспроизводит данные [3412] при Г=13+-36 К
с точностью 0,7% и приводит к значению S" A0 К) = 1,464 Дж-К~1-моль~1. Теплоемкость
кристаллического иода при низких температурах измерялась также в работах [2834, 2831, 2832, 1767,
2354], однако полученные в них данные менее надежны и не учитывались при расчете
термодинамических функций.
В интервале от 298 К до температуры плавления иода принято трехчленное уравнение (см.
табл. 10.2), полученное Линденбергом [2434] на основании результатов измерений энтальпии
кристаллического иода в работе Фредерика и Хильдебранда [1535] C23—386 К) г. Измерения
теплоемкости твердого иода [883] B02—375,7 К) менее точны и, в частности, на 4—5% превышают
значения, полученные в работе [3412]. Более ранние измерения теплоемкости иода при: Т > 298 К
[3184, 3185, 1446] представляют лишь исторический интерес.
Значение Гт=386,75+0,3 К принято по работе Фредерика и Хильдебранда [1535]. Данные,
полученные в других работах, [883, 3139], менее точны, хотя с учетом их погрешности
согласуются с принятой величиной. Значение Дт^Г°=15,665+0,16 кДж-моль выбрано на основании
1 Уравнение Линденберга [2434] скорректировано для учета поправки на систематическую ошибку в работе [1535],
связанную с использованием неточных данных по теплоемкости меди.
199
12 (г) Глава 10
принятых в справочнике уравнений для энтальпии кристаллического и жидкого иода; оно
практически совпадает с значением АтН°=15,65 кДж-моль, полученным на основании прямых
калориметрических измерений [1535]. Более ранние определения энтальпии плавления иода [1446,
2414, 1230, 1939, 2216] основаны на косвенных методах.
Для теплоемкости жидкого иода принято значение С°р (Т)=79,555 ±1,17 Дж-К^-моль,
основанное на измерениях энтальпии жидкого иода в работе [1535] C83—458 К) (с учетом
поправки на теплоемкость меди). Это значение использовано также при расчетах термодинамических
функций жидкого иода при Т > 458 К.
В работе [883] при измерениях теплоемкости жидкого иода в интервале 389—433 К было
найдено, что теплоемкость резко понижается с 83,7 до 76,1 Дж-К^-моль, т. е. примерно на 9%.
Однако точность измерений невысока (около 5%), и в пределах их погрешностей данные работы
[883] не противоречат принятым в справочнике.
Погрешности табулированных значений Ф° (Т) при 298,15, 500 и 800 К оцениваются в 0,4, 1,0
и 3,0 Дж-К^-моль-1.
Ранее термодинамические функции иода вычислялись в работах [37, 1613, 2220, 2095]. В
работах [37, 1613] не были учтены наиболее надежные данные по низкотемпературной теплоемкости
иода [3412]. Результаты расчетов термодинамических функций жидкого иода [2220, 2095]
несколько отличаются от приведенных в справочнике в основном из-за небольшого различия в
принятой теплоемкости жидкого иода. Расхождение в значениях S° (800 К) в работе [2095 ] и табл. Д .16
составляет 0,15 Дж-К^-моль.
Двухатомный кристаллический иод принят в справочнике за стандартное состояние этого
элемента. В соответствии с этим
AfH°(l2, к) = 0.
12 (г). Термодинамические свойства газообразного двухатомного иода в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 78.
Молекулярные постоянные, использованные для расчета термодинамических функций 12,
приведены в табл. 10.3. Молекула 12, как и три других гомоядерных молекулы галогенов Х2,
имеет восемь валентных электронных состояний, коррелирующих с атомами в их основном
электронном состоянии 2Р=/211/2 (.X^SJ, 3ПИ, Ши, 3ILg, Щ , 3S~ 'Д^ и *Щ), а также большое число
валентных электронных состояний, коррелирующих с ионами Х+ CР, W, 1S)~\-X~ (*?). Характер
взаимодействия отдельных моментов количества движения в молекулах галогенов приближается
к случаю Гунда с и поэтому отдельные компоненты их электронных состояний могут
рассматриваться как независимые термы. Теоретические расчеты (см. [2753, 3638]) и исследования спектров
12 показали, что из 23 термов молекулы 12, соответствующих восьми перечисленным выше
состояниям, только четыре терма ХЧ>+ (конфигурация электронной оболочки ... <yt*rc*), 3П2м, 8П1я и
3П0+ (конфигурация электронной оболочки ... o^iz*izsau) являются связанными состояниями. Все
остальные термы, коррелирующие с основными состояниями атомов I, являются отталкивательными
состояниями, включая термы 8П0- и 1ПИ (рис. 10. 1), которые также принадлежат к конфигурации
... о2п%п?аи. У молекул С12 и Вг2 минимум потенциальной кривой состояния 3По+ имеет энергию,
меньшую, чем энергия диссоциационного предела ХBРз/г)-{-ХBРз/2), благодаря чему терм 3П0-
оказывается связанным. Молекула F2 согласно теоретическим расчетам [1862а] в отличие от молекул
других галогенов вообще не имеет связанных возбужденных состояний, коррелирующих с преде-
лами FBP3/2, ?./,) +F^P.,,, 2РЧг).
Помимо четырех связанных термов, приведенных на рис. 10.1, молекула 12, как показали
теоретические расчеты Малликена [2753] (см. также [77а]), имеет 14 связанных валентных ионных
термов с энергией возбуждения меньше 50000 см, которые могут рассматриваться как компоненты
состояний 3Е-, 3Е-, 1Д,, 'Д., 3П^, *?+, *EJ и 41/.
Молекулярные постоянные 12 в состоянии X1!!* определялись в исследованиях систем полос
АШ^^-ХЪ+фЩ, ВШо^Х1^ [816, 2321, 3517, 3518, 487, 498, 3816, 2597, 2460, 814, 3202,
3429, 3908, 3642,1604а], >S+-> JP2+ [3939, 3816,^3889,3940,1828], спектров резонансной флуорес-
1 Сводка данных о ридберговских электронных состояниях 12, полученных разными исследователями, дана в работе
Венкатесварлу [3805].
200
Иод и его соединения
Р н с. 10.1. Схематические изображения кривых потенциальной энергии нижних электронных состояний
двухатомных гомоядерных молекул галогенидов Х2
а — в случае F2; б — в случае С12 и Вг2; в — в случае 12
ценции [3818, 3825, 3145, 3817, 3990, 3143, 2310, 815, 588, 1819, 3290, 2613, 940, 949, 4031, 1810,
3640] и резонансных спектров комбинационного рассеяния паров иода [1992, 2238, 2239, 3952].
Приведенные в табл. 10.3 значения постоянных 12 в состоянии X*Ii* получены Ле Роем [2385]
при совместной обработке результатов исследований спектров резонансной флуоресценции в
работах Ранка и Болдуина [3145] и Верма [3817, 3818]. Ранк и Болдуин [3145] измерили частоты
15 резонансных дублетов в видимой области с точностью 0,001 см г, которые соответствуют
пе' Б3 122 ХХ* [3
р у
реходам с уровня v' состояния Б3П0+ на уровни у"=1-^22 состояния ХХ2*. Верма [3817, 3818]
измерил линии пяти резонансных серий в ультрафиолетовом спектре флуоресценции 12,
обусловленном переходами с колебательно-вращательных уровней высоковозбужденного электронного
1*
состояния2 на колебательно-вращательные уровни состояния
Л [3]
с значениями i/'=0-|-84.
р ур ^ с
Полученные Ле Роем [2385] полиномиальные выражения для Bv и G (v) состояния Хх~2+,
коэффициенты в которых приведены в табл. 10.3, позволяют воспроизводить с высокой точностью
экспериментальные данные вплоть до у=82.
Переходы между первым возбужденным состоянием 3П2я и основным состоянием Х1^
запрещены. Приведенная в табл. 10.3 энергия состояния 3П2м была найдена при подготовке настоящего
справочника в результате обработки данных, полученных при исследовании магнитного кругового
дихроизма в спектрах раствора 12 в циклогексане и в спектре паров иода [785, 784], с учетом
теоретических расчетов Малликена [2747, 2753]. Значение этой энергии согласуется в пределах
погрешности B00 см) с менее точными величинами, полученными Малликеном [2753] и Теллинг-
хьюзеном [3638 ] по спектрам непрерывного поглощения 12. Значения колебательных постоянных
12 в состоянии 3П2а, приведенные в табл. 10.3, получены в результате анализа колебательной
структуры системы полос 3Н2д ->¦ 3П2я [1400, 3940, 3803, 3816, 2753]. Значение Вв для состояния
3П2а оценено по соответствующей постоянной 12 в состоянии ^43П1и. Значения энергии возбуждения
и колебательных постоянных 12 в состоянии -43П1м, приведенные в табл. 10.3, получены Брауном
[81813 на основании измерений кантов 62 полос системы А3Л1а—Х1^ в спектре поглощения (v" =
1 Результаты этих измерений опубликованы Ранком и Рао [3152].
2 Согласно [2753] это состояние D4+.
* Правильность нумерации, принятой Брауном [818], нуждается в дополнительной проверке.
201
Mr)
Глава 10
Таблица 10.3. Молекулярные постоянные 12, Ю, HI, DI и TI
Молекула
Ii
10
HI
DI
TI
^ОиЮННИО
лш1и
оы
«и *
1Д
^2 д > .Li. л™
Е3п0+
»п1д
Х2Щ
АШ.
XW
хч+
XW
т.
оуеув ¦ 10»
вв ¦ ю1
aj • 10s
De ¦ 108
СМ
0 214,5481
10200 110,366
11888 44
15 768,32 126,165
30 000 100я
37 000 100я
39 000 100я
40 000 100й
40 800 100я
41000 100»
41411,8 100
43 600 100й
47 217,8 95,955
0а 681,47
21557,81» 514,57
0 2 309,014
0 1 639,655
0 -
Примечание. Ниже все постоянные даны в
I»: *
Ю: »
ш„ге = 1,263
=4,634 655 4
=—3,64-10-
643 3-Ю-4, о)в9е = 6,1981
• 101, со,v, = 2,933 075 5
0,616 259
1,3603
1
0,867 33
—
—
—
—
0,362 3
4,29
5,52
39,643 5
19,873
—
СМ.
29-10-»,
•108, О).
-11; в а = —1,7 . Ю-», В, = 7 • Ю*;"
е ы.г, = 1,012 324 • Ю-3, шеде = 4 ,~5Е
=2,809 711-
а4 = —1,159
= 1,26-Ю-1
А = —2330;
100, @^ = 1,722 584-10-
-10"8, а5 = 2,195-100,
@ 14 • 10,
а. = -2
', р3 = 1,5-Ю-"; и оценка.
« а2 = —0,97-Ю-5; в А =
= —150.
0,750 7» 0,373 95
—34,5 0,28
80 0,26
107,448 е 0,289 39
— 0,19й
— 0,19я
— 0,19й
— 0,19»
— 0,19я
— 0,19"
— 0,19
— 0,19й
— 0,19й
—130 3,402 6
— 2,763 5
—200 65,108
—459 32,839
— 22,102
ш s = 2 025 597 5 • 10"'
u>' = 7,61-10"le; ба2 =
0,124 35 е
0,Зг
0,1г
0,1204 я
—
—
—
—
—
—
—
—
2,696 е
2-, 73
168,6
60,82
—
0,454в
0,7*
4
0,614»
—
—
—
—
—
—
36
32
2,07-10*
5,31-10s
—
, *),«, = 3,966 2824-10-»
= 4,498-Ю-7,
г вычислено по соотношению A.69);
шезе =1,334 168-10"
= 4,431 776-106; х а,
047-102, о, = 7,579
-в 0) f __ 2
= —5 ГбО8 - 1С
• 10-"; » Вх
а3 = 1,482 •
* вычислено
,499 684-Ю"8
)-», а3 = -2
г.
А
2,665 47
3,1
3,104
3,029 98
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
1,867 6
2,072 3
1,609 1
1,609 1
1,609
, ы и =
ю-», «1=
по A.68);
, шеа,=
,988.10"'
= -1,16-100, 8,=
=2-^-5, г/ ^ 22). Вращательная постоянная соответствует межатомному расстоянию,
найденному Теллингхьюзеном [3638] по непрерывному поглощению в видимой области спектра 12,
связанному с переходом А3Л1и -* Х:2+. Сведения об электронном состоянии Б3П0+ основываются
на изучении системы полос В3Л0+-^Х11>* в спектрах поглощения [498, 3908, 3202,816, 814,2460,
2597, 3429, 3517, 3518, 3642, 4031,1604а] и испускания [3429, 3908] и при резонансной
флуоресценции [588, 815, 940, 949, 1810, 1819, 2310, 2326, 2460, 2596, 2613, 2973, 3143, 3145, 3290, 3990, 4031 ].
Наиболее точные выражения для G (v) и В, в состоянии В3И0+ получены Веем и Теллингхьюзеном
[3908] в результате совместной обработки собственных измерений F полос с 17 ^ v' ^ 26),
нового анализа измерений [3429] A6 полос с 14 <^ v' ^ 38) и данных Барроу и Йи [498] F4
полосы с 4 ^ d' ^ 77). Значения постоянных в уравнениях для G (и) и Bt, найденные в работе
[3908], приведены в табл. 10.3. Приведенные в той же таблице значения коэффициентов в
уравнении для D, (для у' ^ 40), получены в работе [3429]. В спектрах 12 наблюдались также переходы,
связанные с пятью валентными электронными термами, коррелирующими с ионами 1++1~, с
энергиями возбуждения до 50 000 см: D1!,* [822, 1400, 2310, 2326, 2596, 2973, 3639, 3818], F1^*
[3939], 3П2^ [1400, 3803, 3816, 3939, 3940], 3П1д и 3П0+. Выполненные исследования показали, что
в состояниях F1^, sII2j? и 3П0+ молекула 12 имеет близкие значения частот колебания (примерно
100+5 см) и вращательных постоянных (примерно 0,19 см). Энергии диссоциации 12 в этих и
других состояниях, коррелирующих с ионами 1++1~, также должны иметь близкую величину.
202
3
Иод н его соединения 12 (г)
На основании этого в справочнике для всех рассматриваемых ионных состояний приняты
одинаковые значения ше и Ве, близкие к найденным для терма 3П0+ в работах [2753 и 3939]. Энергии
возбуждения 12 в девяти ионных электронных состояниях, приведенные в табл. 10.3, приняты
на основании результатов анализа, выполненного Малликеном [2753], их погрешности могут
достигать 2000 см.
Термодинамические функции 12 (г), приведенные в табл. 78, были вычислены с использованием
уравнений A. 76)—A. 78) для расчета <?вн и ее производных. Значения <?<•]_ вр, ^*>_ вр и Q[H вр для
состояний Х1?+, 3П2а, -43П1а и BSHO+ вычислялись непосредственным суммированием по
колебательно-вращательным уровням энергии этих состояний. Энергии уровней находились по
уравнениям A.65) и A.62). Значения коэффициентов Ykl в этих уравнениях были получены по методикам,
изложенным в разделе 1.4, и приведены в табл. 10.4. Вырождение вращательных уровней состояний
П9а и 3П1я учитывалось статистическим весом 2. В случае состояний Х1^, AsHlu и BSUO+ в расчете
учитывались все уровни, ограниченные предельными кривыми диссоциации этих состояний.
Для состояния 3П2и это ограничение проводилось по диссоциационному пределу. В табл. 10.4
приведены значения коэффициентов а( в уравнениях, описывающих предельные кривые
диссоциации, а также значения ушах и 71Ш. Значения (?(*>. вр и ее производных для восьми электронных
состояний, коррелирующих с ионными пределами I "*"—f-1 , вычислялись в приближении «жесткий
ротатор — гармонический осциллятор», поскольку молекула 12 в этих состояниях должна иметь
энергии диссоциации примерно 35 000 см.
Погрешности вычисленных значений термодинамических функций при температурах меньших
2000 К обусловлены преимущественно неточностью принятых значений фундаментальных
постоянных, при более высоких температурах становятся существенными неточности принятых
значений энергий возбуждения и постоянных 12 в состояниях 3П2я и AsHlu, а выше 4000 К —
неточности, связанные с отсутствием данных, необходимых для корректного учета состояний
с энергиями возбуждения, превосходящими 30 000 см. Общие погрешности вычисленных
значений Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,03, 0,1 и 0,5 Дж-К^-моль.
Таблицы термодинамических функций 12(г) вычислялись ранее Цейзе [4054] (Т ^ 2000 К),
Мерфи [2758] (Т < 1500 К), Эвансом и др. [1426] (Т < 3000 К), в первом и втором изданиях
настоящего справочника [87, 88], справочнике JANAF [2095], а также Фебером и Херриком
A454]. В расчетах, приведенных в работах [1426, 2095, 2758, 4054], учитывалось только основное
электронное состояние 12, а в [87, 88, 1454] — возбужденные состояния, соответствующие
электронной конфигурации . . . °^*т^ам. В работе [2095] расчет был выполнен по методу Майера
и Гепперт-Майер, в [87, 88] — по модифицированному методу Гордона и Барнес с приближенным
учетом вклада возбужденных электронных состояний, в [1454] — непосредственным
суммированием по уровням колебательно-вращательной энергии состояний Х1%+, 3П2я, ^43П1я и B3TiQ+. При
низких температурах (до 1000 К) результаты всех расчетов, несмотря на использование разных
методик и разных значений постоянных, согласуются между собой и с данными настоящего
справочника в пределах 0,03—0,04 Дж-К^-моль. При температурах выше 1000 К расхождения
быстро возрастают, и данные [2095] отличаются от приведенных в табл. 78 на величины до 1, 2
и 7,5 Дж-К^-моль в значениях Ф° (Т), S0 (Т) и С" (Т) главным образом из-за использования
в [2095] метода Майера и Гепперт-Майер и пренебрежения возбужденными электронными
состояниями 12; следует отметить, что соответствующие ошибки частично компенсируют друг друга.
Расхождения с данными [1454] не превышают 0,5, 1,5 и 5 Дж-К^-моль в значениях Ф° (Т),
3° (Т) и С° (Т) и обусловлены тем, что в этой работе не учитывались возбужденные состояния 12
с энергией больше 30 000 см, а также некоторым различием в ограничении числа связанных
и квазисвязанных колебательно-вращательных состояний. Расхождения в значениях Ф° (Т)
и S° (T), табулированных в предыдущем и настоящем изданиях справочника, достигают 0,5 и
4 Дж-К^-моль.
Константа равновесия реакции 12(г)=21(г) вычислена с использованием значения АГН° @) =
= 148,826 + 0,013 кДж-моль, которое соответствует энтальпиям образования 12(г) и 1(г),
рекомендованным КО ДАТА—МСНС [1000]. Значение энтальпии образования
Д/Г^з, г, 298,15 К) = 62,421 ± 0,080 кДж • моль
203
Таблица 10.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см), а также значения vmax и Jlim,
принятые для расчета термодинамических функций 12, 10, HI, DI, TI
Коэффициенты
10
а тF
HI
DI
ТЕ
т.. ю-*
У10-Ю-2
Узо°-Ю2
У40-103
Убо-Ю*
Уво-Ю"
У70-Ю8
У8о-Ю10
Уо-1012
Уюо-Ю16
УогЮ!
Уи.1О2
Ул-10*
УзгЮ"
YiVi0™
У02-10в
У12-Ю'
У2г-Ю8
аг10з
а2-107
а3-1012
Vmxi.
О
2,145 481
—0,616 259
0,007 509
—0,126 364
0,061 981
—0,202 559
0,396 628
—0,463 465
0,293 307
—0,760 905
0,373 95
-0,012 435
0,004 498
-0,014 82
-0,364
-0,004 54
—0,0017
—0,000 7
12,484 8
8,840 2
0,141 711
-0,005 150
103
877
1,02
1,103 66
-1,392 7
0,213
1,188 8
0,44
-0,989 107
0,742 4
0,28
-0,03
0,26
-0,01
—0,007
2,35
43
—0,04
0,656 684
5,218 03
1,774 74
-0,920 59
48
248
Примечание. Все постоянные в см
т ¦ а
1,576 832
1,261 65
-0,867 33
1,074 48
-1,012 324
0,455 014
-1,334 168
2,499 684
-2,809 711
1,722 584
-4,431 776
0,289 39
-0,012 04
-0,056 08
0,298 8
-115,9
2,195
-2,047
7,579
-0,006 14
-0,001 16
-0,000 126
4^369 1
6,211 53
0,290732
-0,022 446
81
590
О
6,818 95
—4,455 60
0,802 055
—0,839 532
2,155 78
5,147 86
-5,616 56
1,116 51
О
23,078 39
—38,679 5
—33,543 6
28,227 6
—22,482 5
О
16,389 85
-19,508 3
-12,014 9
7,180 47
—4,061 57
3,395 06 2,761 71
—0,223 376 —0,255 426
—0,567 386 —0,293 171
—0,36
-0,053
18.800 6
63,495 5
8,075 78
-1,730 55
45
346
—0,32
0,11
13,161 4
40,4917
4,264 57
1,484 14
54
293
—207
25,750 55
487,105 4
1 806,863
-5 418,211
18
87
-52,7
25,736 62
272,269 1
392,110 2
-292,378 1
25
127
О
13,445 52
—13,128 8
—6,633 25
3,252 1
—1,509 06
65,138
—17,591
25,198
—459,696
85
8
8
32,853
-6,301
6,41
-83,045
3
14
9
22,210
-3,478
2,903
—30,855
977
78
14
4
-23,8
25,745 38
170,018 5
200,897
-184,336 5
30
149
Состояние
. 3V-
3,0
Y НО2 1 Q
Ю: а л=-2330; б 4=—150.
3,7
1
1,9]
3,9
1
1,9
4,0
1
1,9
4,08
1
1,9
4,10
1
1,9
4,141 13
1
1,9
4,36
1
1,9
4,721 9
1
1,9
Иод и его соединения 10 (г)
было рекомендовано КО ДАТА—MCHG [1000] на основании измерений давления насыщенных
паров иода в работах Бакстера, Хиккейя, Холмса [524], Бакстера, Гроса [523], Гиллеспи,
Фрейзера [1644] и Штерна [3527] и измерений теплоемкости кристаллического иода в работе Шерли
и Джиока [3412]. Значение энтальпии образования I (г) рекомендовано КО ДАТА—MGHC на
основании этой величины и энергии диссоциации молекулы 12
D0(I2)= 12 440,9 ±1,1 см = 148,826 + 0,013 кДж • моль,
полученной Ле Роем и Бернстейном [2384, 2388, 2390]. Ле Рой и Бернстейн нашли значение
De (I2) с помощью разработанного ими метода экстраполяции колебательных уровней вблизи
диссоциационного предела с учетом дальнодействующего потенциала, используя результаты
измерений кантов полос, выполненных Брауном [816] (в интервале 55 ^ v' ^ 72, изменив
нумерацию v' на единицу). Близкое, но более точное значение диссоциационного предела 20 043,208 +
+ 0,033 см и соответственно Do (I2) = 12 440,06 + 0,05 см было получено впоследствии
аналогичным методом Барроу и Йи [498] по началам полос F4 ^ v' г^ 77).
10 (г). Термодинамические свойства газообразного оксида иода в стандартном состоянии
при температурах 100—6000 К приведены в табл. 79.
Молекулярные постоянные, использованные для расчета термодинамических функций 10,
приведены в табл. 10.3. Основным электронным состоянием молекулы 10 является состояние
Х2И(, а первым возбужденным — состояние A2Ht. Другие валентные электронные состояния 10,
по-видимому, являются нестабильными или имеют низкие энергии диссоциации, так как
соответствуют переходам связывающих п- или а-электронов на разрыхляющую а-орбиталь. В спектре 10
наблюдалась только система полос ^421Ь/г—Х21Ъ/г, как в испускании [668, 1016, 1349],
так и в поглощении [1350]. Наиболее полно эта система исследована Дьюри и др. [1349], которые
наблюдали канты 18 полос, соответствующих v" ^ 10 и v' ^ 4. Приведенные в табл. 10.3 значения
постоянных 10 в состояниях Х2П{ и АШ{ определены Дьюри и др. [1349] в результате анализа
вращательной структуры полос 0—2, 0—3, 0—4, 0—6, 2—0, 3—0, 2—2, 2—9. С приведенными
в табл. 10.3 вращательными постоянными в состоянии Х2П( удовлетворительно согласуются
значения постоянной Во, найденные при изучении спектра ЭПР [809, 886, 889] и микроволнового
спектра [3284].
Принятая постоянная спин-орбитальной связи в состоянии Х2П; вычислена Брауном и др.
[809] теоретически с учетом спин-орбитального возаимодействия /ьэлектронов атомов I и О.
Погрешность этой постоянной из-за пренебрежения взаимодействием d-электронов оценена
в 30 см [809] \
Термодинамические функции 10 (г) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBn и ее производных находились по уравнениям A. 90)—A. 92) с учетом
состояний Х2П и ^12П. Статистические суммы по колебательно-вращательным уровням этих
состояний и их производные рассчитывались по уравнениям A. 73)—A. 75) непосредственным
суммированием по уровням энергии. Энергии уровней находились по уравнениям A. 65) и A. 42).
Значения коэффициентов Ykl в этих уравнениях получены по методике, изложенной в разделе 1.4,
и приведены в табл. 10.4 вместе со значениями Те и А. В расчете учитывались все уровни энергии
каждого состояния, ограниченные их предельными кривыми диссоциации. В табл. 10.4 приведены
значения коэффициентов а{ в уравнениях, аппроксимирующих эти кривые, и значения ушах и /пт
для каждого состояция.
Погрешности значений термодинамических функций при низких температурах обусловлены
практически только неточностью фундаментальных постоянных. Выше 2000 К становятся
заметными погрешности, обусловленные неточностью определения числа и энергии высоких
колебательно-вращательных уровней состояния Х2П{ по уравнениям с принятыми значениями
коэффициентов Ykl, в частности, из-за большой погрешности величины Do A0) (см. ниже). Погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,02, 0,05 и 0,3 Дж-К^-моль-1.
Ранее таблицы термодинамических функций 10 вычислялись во втором издании настоящего
справочника [88] (Г < 6000 К) и Брюэром и Розенблатом [764] (Т < 3000 К). Расхождения
1 Постоянная спин-орбитальной связи А в состоянии Х3К{ определялась экспериментально Каррингтоном и др.
[886, 889] на основании анализа ЭПР-спектра127Ю. Однако при этом было получено неверное значение
постоянной А.
205
HI (г) Глава 10
в значениях функций, вычисленных в [88, 764] и приведенных в табл. 79, обусловлены в основном
различным учетом спин-орбитального расщепления основного состояния и различием методов
расчета функций. Расхождения с данными [88] достигают 5,2 Дж-К^-моль при 298,15 К и
3 Дж-К-моль-1 при 6000 К.
Константа равновесия реакции Ю(г) = 1(г)-[-О(г) вычислена с использованием принятого
в настоящем справочнике значения
= 222 ± 13 кДж • моль = 18 600 ± 1000 см.
Это значение основано на результатах проведенного Рейдлейном, Уайтхедом и Грайсом [3115,
3116] исследования распределения по скоростям молекул 10, образующихся при реакциях IC1
и 12 с атомарным кислородом в пересекающихся молекулярных пучках. В пределах погрешностей
с ним согласуется значение 239+25 кДж -моль, найденное Филлипсом и Сагденом [3007] на
основании спектрофотометрического исследования равновесия реакции диссоциации в пламени, и
верхний предел этой величины B63 кДж -моль), установленный Дьюри и Рамзи [1350] по предис-
социации в состоянии А2П{. Принятому значению Do A0) соответствует
ДДО(ГО, г, 0)= 131,946 ± 13 кДж • моль.
HI (г). Термодинамические свойства газообразного иодоводорода в стандартном состоянии
при температурах 100—6000 К приведены в табл. 80.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 10.3. Исследования электронных спектров HI [1173, 2748, 3087, 3236, 3237,
3685] и анализ систематики электронных состояний HI, выполненный Тилфордом [3685],
показали, что основное электронное состояние ХХ2+ является единственным стабильным валентным
состоянием HI с энергией ниже 50 000 см. Приведенные в табл. 10.3 значения молекулярных
постоянных HI в состоянии ХХ2+ были определены Хойзлером и др. [1795] и Херлоком и др.
[2054] в результате анализа вращательной структуры полос 1—0, 2—0 [1795] и 2—0, 3—0 [2054]
в ИК-спектре HI. Молекулярные постоянные HI, определенные в работах [1795, 2054], описывают
положения нижних уровней колебательной и вращательной энергии молекулы HI в состоянии
X1!!+ с точностью до 0,001 см. Значения постоянных, найденные ранее на основании анализа
колебательно-вращательного [723, 408, 1796, 2826, 2854] и вращательного спектра газообразного
HI [1088, 1089, 1211, 2940], существенно менее точны. ИК-спектры поглощения молекул HI,
изолированных в матрицах инертных газов, исследовались в работах [477, 478, 720, 721 ].
Значение постоянной Во, найденное Бернадтели и др. [599] при изучении структуры полос 5—0 и
6—0 в ИК-спектре поглощения HI совпадает со значением, рекомендованным в работе [1795].
Термодинамические функции Н1(г) были вычислены без учета возбужденных электронных
состояний, значения Q^ вр и ее производных вычислялись непосредственным суммированием по
уровням колебательно-вращательной энергии состояния Z12+ по уравнениям A. 73)—A.75).
Энергии уровней вычислялись по уравнению A. 65) со значениями постоянных Ykl, приведенными
в табл. 10.4 и полученными по методикам, изложенным в разделе 1.4 на основании принятых
молекулярных постоянных. В расчете учитывались все колебательно-вращательные уровни энергии
состояния Х12+, ограниченные предельной кривой диссоциации. Коэффициенты at в уравнении,
аппроксимирующем эту кривую, а также значения tw^/am приведены в табл. 10.4. Погрешности
вычисленных термодинамических функций HI определяются при низких температурах в
основном неточностью принятых значений фундаментальных постоянных, при высоких температурах —
неточностью расчета высоких колебательно-вращательных уровней энергии по уравнению A. 65)
с принятыми значениями Ykl. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К
оцениваются в 0,02, 0,05 и 0,2 Дж-К^-моль-1.
Ранее таблицы термодинамических функций HI вычислялись в работах [802, 2758, 3755,
1454], первом и втором изданиях настоящего справочника [87, 88], а также справочнике
JANAF [2095] х. Во всех этих расчетах использовались менее точные значения молекулярных
постоянных, чем в настоящем издании. Расчеты, проведенные в работах [802,3755,1454], выпол-
1 В работе [802] непосредственным суммированием по уровням колебательной и вращательной энергии HI
определены значения коэффициентов в полиномах, позволяющие вычислять термодинамические функции HI (г) в
интервале температур 200—1000 Kt
206
Иод и его соединения 01 (г)
нены непосредственным суммированием по уровням энергии, в [88] — методом Гордона и Барнес,
а в [2095] — методом Майер и Гепперт-Майер. Расхождения с данными работ [1454, 2095, 88]
не превышают 0,15 Дж-К^-моль в значениях Ф° (Г) во всем интервале температур до 6000 К.
Расхождения в значениях S° (T) и особенно С° (Г) существенно больше и достигают в С" F000 К)
1,7 и 4 Дж-К^-моль для [1454, 2095] соответственно.
Константа равновесия реакции HI (г)=Н (г)+1 (г) вычислена с использованием значения
дд° @)=D0 (Н1)=24 632+21 см-1=294,662+0,26 кДж-моль-1, которое соответствует энтальпии
образования
AfH°(m, г, 298,15 К) = 26,36 + 0,25 кДж • моль,
принятой в настоящем справочнике по рекомендации КО ДАТА—MGHC [1000]. Эта рекомендация
основана на результатах изучения равновесия 2Н1(г)=12(г)+Н2(г) Тэйлором и Кристом [3629],
определения степени разложения Н1(г) Риттенбергом и Юри [3211], фотокалориметрического
изучения равновесия разложения Н1(г) Брайтом и Хейджерти [771 ], теоретических расчетов Марфи
[2758] и калориметрического определения энтальпии реакции 2Н1(г)+С12(г)=2НС1(г)+12(г)
Гюнтером и Векуа [1770].
Принятая энтальпия образования Н1(г) согласована с величиной энтальпии образования
иона 1~ в состоянии стандартного водного раствора
Д/П1". р-рН2О, станд. сост., 298,15 К) = —56,781 + 0,063 кДж • моль,
найденной в результате калориметрического исследования Джонсоном [2137]. Достаточно близкое,
но значительно менее точное значение — 56,90 ±0,84 кДж-моль было рекомендовано КОДАТА—
MCHG [1000] по результатам более ранних экспериментальных исследований.
Вандерзее [3765] уже после опубликования величин, рекомендованных КОДАТА—МСНС
[1000], провел прецизионные измерения энтальпии растворения HI (г) в воде и нашел значение
—83,283+0,084 кДж-моль, что очень близко по величине, но значительно точнее значения
—83,3 + 1,0 кДж-моль", принятого КОДАТА—МСНС [1000]. Комбинация результатов измерений
[2137, 3765] дает AfH° (HI, г, 298,15 К)=26,50±0,10 кДж-моль-1, что близко к величине 26,36 +
+0,80 кДж-моль-1, рекомендованной КОДАТА—МСНС [1000].
С учетом данных, приведенных в работах [2137, 3765], погрешность принятого в справочнике
значения энтальпии образования HI (г) уменьшена до 0,25 кДж -моль.
DI (г). Термодинамические свойства газообразного иододейтерия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 81.
Молекулярные постоянные, использованные при расчете термодинамических функций DI,
приведены в табл. 10.3. Исследование электронного спектра DI, выполненное Тилфордом и др.
[3685], показало, что состояние .Х'12 + является единственным стабильным состоянием молекулы
DI с энергией ниже 50 000 см. Колебательные и вращательные постоянные молекулы DI в
состоянии Х*2+ определялись на основании изучения колебательно-вращательных [2054, 2166,
3099] и вращательных [846, 1088, 1091, 1211, 2264, 2940] спектров. В спектрах наблюдались
переходы с v" ^ 3. В табл. 10.3 приведены значения молекулярных постоянных DI, найденные
в работах Херлока и др. [2054] и Пью и др. [3099]. Полученные в этих работах значения
вращательных постоянных согласуются со значениями, определенными из анализа вращательных спектров.
Термодинамические функции DI(r) были вычислены непосредственным суммированием по
колебательно-вращательным уровням энергии состояния Х12 + по уравнениям A. 73)—A. 75).
Уровни энергии определялись по уравнениям A. 65) и A. 62). Поскольку молекулярные
постоянные DI, приведенные в табл. 10.3, менее надежны, чем постоянные HI, значения коэффициентов
Ykl в уравнении A. 65) для DI были вычислены по изотопическим соотношениям A. 66) и
постоянным Ykl молекулы HI, приведенным в табл. 10.4. Экспериментальные данные по частотам
переходов DI хорошо описываются уравнениями с этими значениями коэффициентов. В расчете
термодинамических функций учитывались все колебательно-вращательные уровни энергии состояния
Хг1>+, ограниченные предельной кривой диссоциации. Значения коэффициентов а4 в уравнении,
аппроксимирующем эту кривую, и значения Ykl, i;max и /цт приведены в таблице. Погрешности
вычисленных таким образом термодинамических функций DI(r) при низких температурах
обусловлены в основном неточностью фундаментальных постоянных, а при высоких температурах —
неточностью определения энергии и числа высоких колебательно-вращательных уровней состоя-
207
Т1(г) Глава 10
ния -Х'12+ по уравнению A.65) с принятыми значениями коэффициентов Уы. Они составляют 0,03,
0,07 и 0,2 Дж-К^-моль-1 в значениях Ф° (Г) при 298,15, 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций DI в состоянии идеального газа рассчитывались
в работах [3755] (Т < 1500 К), [665] (Т < 700 К), и предыдущем издании справочника [88].
Расхождения в значениях термодинамических функций DI, рассчитанных в справочнике [88]
и приведенных в табл. 81, растут с температурой, достигая 0,2 и 1,4 Дж-К^-моль в значениях
Ф° (Т) и S° (T) при 6000 К.
Константа равновесия реакции DI (r)=D (r)+I (г) вычислена с использованием значения
ДГЯ° @)==D0 (DI)=24 955 + 10 см~1=298,53 + 0,11 кДж-моль, соответствующего принятой в
настоящем справочнике энтальпии образования
AfH°(DI, г, 0) = 28,44 + 0,10 кДж • моль.
Принятая энтальпия образования DI (г) получена по энтальпии реакции 2DI (r)=D2 (г)-|-12 (г),
исследованной Тейлором и Кристом [3629] в интервале 667—764 К. Расчет по этим данным и
уравнениям II и III законов термодинамики приводит к значениям 8,83 + 0,60 и 8,612 +
+ 0,030 кДж-моль соответственно.
Близкое значение энергии диссоциации DI, равное 24 960 + 21 см, было получено по
соотношению C. 6) на основании величин Do (HI)=24 632+21 сыт1, G @, HI)=1143 см и G @, DI) =
=815 см-1.
TI (г). Термодинамические свойства газообразного иодотрития в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 82.
Молекулярные постоянные, использованные в расчете термодинамических функций TI,
приведены в табл. 10.3. Ни электронный, ни колебательно-вращательный спектры молекулы TI
не исследовались. По аналогии с DI принимается, что основным электронным состоянием TI
является состояние X1!!+, а связанные возбужденные электронные состояния имеют энергии,
превышающие 50 000 см. Приведенные в табл. 10.3 значения вращательной постоянной и
межатомного расстояния найдены в работах [1211, 3245] в результате исследования вращательного
спектра.
Термодинамические функции TI (г) были вычислены непосредственным суммированием по
уровням колебательно-вращательной энергии состояния Хг1,+ по уравнениям A.73)—A.75).
Колебательно-вращательные уровни энергии вычислялись по уравнениям A.65) и A.62) со
значениями постоянных Ykl, приведенными в табл. 10.4. Эти значения Ykl были получены
по соотношениям A. 66) и постоянным Ykl молекулы HI. В расчете (?W_ вр и ее производных
учитывались все уровни энергии, ограниченные предельной кривой диссоциации состояния Хх?+.
Значения коэффициентов а{ в уравнении, аппроксимирующем эту кривую, а также значения
утах и /нт приведены в табл. 10.4. Погрешности вычисленных термодинамических функций при
низких температурах обусловлены в основном неточностью фундаментальных постоянных; выше
2000 К становятся существенными погрешности, обусловленные неточностью расчета высоких
колебательно-вращательных уровней энергии. Погрешности в значениях Ф°(Т) могут достигать
0,04, 0,1 и 0,3 Дж-К^-моль-1 при Г=298,15, 3000 и 6000 К соответственно.
Ранее таблипы термодинамических функций TI (г) вычислялись в предыдущем издании
справочника [88] г. Значения термодинамических функций TI в справочнике [88] отличаются от
приведенных в табл. 82 не более, чем на 0,3 Дж-К^-моль во всем интервале температур.
Константа равновесия реакции TI (г)=Т (г)+1 (г) вычислена с использованием
АгН° @) = Da (TI) = 25 101 + 12 см = 300,27 + 0,15 кДж ¦ моль.
Это значение было принято на основании расчета по соотношению C. 6) с использованием
величин D0(DI)=24 955±10 см, G@, DI)=815 + 1 см, 6@, Т1)=669+2 см.
Принятому значению Do (TI) соответствует
Д/ПТ!, г, 0) = 28,372 ±0,16 кДж • моль.
В работе [802 ] методом непосредственного суммирования определены значения коэффициентов в
интерполяционных полиномах, позволяющих вычислять термодинамические функции TI в интервале температур 200 — 1000 К.
208
Иод и его соединения
IF (г)
IF (г). Термодинамические свойства газообразного фторида иода в стандартном состоянии
при температурах 100—6000 К приведены в табл. 83.
Молекулярные постоянные, использованные для вычисления термодинамических функций
IF, приведены в табл. 10.5. Исследования спектров IF, а также теоретический анализ позволяют
предполагать, что эта молекула имеет шесть связанных электронных состояний, образованных
атомами I и F в 2Р-состояниях (см. рис. 9.1 и табл. 10.5). Валентные состояния, коррелирующие
с ионами I++F~, должны иметь энергии свыше 40 000 см и в справочнике не рассматриваются.
Приведенные в табл. 10.5 колебательные постоянные IF в состоянии Х1^ + получены Коксоном
[1104] по началам полос системы В3П0+ -> Х1^* (v" ^ 9), найденным в работе Дьюри [1346].
Вращательные постоянные приняты по работеТиманнаидр. [3694], в которой исследован
микроволновой спектр IF. Результаты исследований электронного спектра IF в работах [1345, 993,
656] (Б3П0+-> Х1!,*) и [656] (А3!!!-* ХХ2+) и микроволнового спектра IF [2589] согласуются
с принятыми значениями. Колебательные постоянные IF в состоянии А3Пг получены Бирксом
и др. [656] по кантам полос системы A3R1—X1I,+ (v^10), значение постоянной В, оценено на
основании значений этой постоянной IF в состоянии 53П0+ и постоянных Bt молекул других
интергалогенов в состояниях А3^ и Б3П0+. Исследования системы полос 53П0+—ХХ2 + [656, 993,
1345, 1346] показали, что уровни состояния В3И0+ с v > 7 сильно возмущены взаимодействием
Таблица 10.5.
Молекула
J19F
I35CI
I79Br
Состояние
Молекулярные постоянные I19F, IS5C1 и 179Вг
»п2
лтх
«по_
взп0+
В'0+
х^+
зп2
А3Лг
Зпо_
вщ0+
В'0+
С*2-
Х^+
зд2
Л3ПХ
-ВЗП0+
?'0+
С32-
Примечание. Ниже
J19JT' а
г
IS5CI: a
17эВг: а
я, = —2,7 •
вычислено
лено по A
вычислено
т.
0
14 000
15 706
18 940
19 054,
24 150
0
12 300
13 747,
17 188
17 332
18 156
37 744
0
11100
12 350
16 165
16 8.14
35 427
38 714
39 126
о
610,03
380
380,5
400
53 406,51
300
384,293
255
98 209,111
240
242
37
173,2
268,71
150
138
142,46
65,5
43,0
90,0
77,0
все постоянные даны i
10-в. 6
по A.
со х„
3,096
3,8°
3,8
8.7Д
1,3
3,23»
1,501
4,0
1,886
38,9 г
13
1,25
1,1
0,83
1,5°
1,7
2,57
0,24
0,026
0,15
0,5
1 СМ.
оеу. ¦ Ю1
СМ
—4,95
—
—
—1,98
—
0,2
—0,3558
—
—
—
—
—
—0,2
-1,1
—0,1
—
—
—
вычислено по соотношению A. 67) при '.
68); я вычислено
по A. 67)
.67) при D,E'0+) = 6820+600.
по соотношению A. 68);
* а„ = —2
В. ¦ 101
2
2
2
2
2
1
1
0
0
0
0
0
0
0
0
0
0
0
0
0
0
797 11
3
3
3
2721
5
14159
85
849 974
9
90
532
60 е
,567 88
,45
,447 626
,432
,349 632
,3»
,3»
,3»
ах • 103
1,873 4*
3*
3«
4"
1,398е
2В
0,5354
0,636
0,629 227
2,9*
2,9
3,4
0,4 я
0,199»
0,62 в
0,623 897
0,533 е
0,196 894
—
—
—
(»Д,) = 9500+600; в
при D, CД0_)== 4600+200; е аа =
,3-10-5;Ba2 =
ншо A. 67) при D, (8Пд_) = 370+50; я вычислено по A. 69)
я2 = 7 • 10-'
; б вычислено по A. 67
ношению A. 68);
я а2 = — 5,439 26-
при D, (»
—2,337 27-
П2) = 3700 + 500;
10"e; e вычислено по A
D.
t
2,
3
3
3
2,
2
0
0
- 0
0
0
5
0
0
0
я 0
-0
¦ 0
•107
367
Г
Г
г
8
г
40*
38»
56»
5
5»
а
3»
,08
,16Г
,16Г
,16Г
,40'
—
—
вычислено
-8,21
• 10"";
10; г вычислено по
вычислено по A.
в а2 = —
.69);
5,4- 10-»;
= —8,8302
38); «
1,
2,
2,
2,
2,
2,
2,
2,
2,
2
2
3
3
2
2
2
2
3
3
3
3
по
ж
г.
А
909 79
11
11
11
118 9
6
320 9
7
69
6
6
4
2"
469 9
,8
782
,83
,148
,4
,4
,4
A.69);
вычнс-
соотноше-
оценка.
г вычислено по соот-
10"в;
• оценка
•
209
IF (г) Глава 10
с состояниями 0+, соответствующими диссоциационным пределам I (zP%)+FBPs^) и I B/\)-{-
+F BPi/,). В результате этого взаимодействия потенциальная кривая состояния Л53П0+, пройдя
через максимум, коррелирует с первым пределом, и возникает по крайней мере еще одно связанное
состояние В'0+, коррелирующее с пределом I BA/2)+F BP%). Приведенные в табл. 10.5
постоянные IF в состоянии В3Н0+ получены Дьюри [1346] на основании анализа вращательной структуры
полос с у=11. Дьюри показал, что уровни с р=11, / ^> 11 предиссоциированы.
Поскольку в спектрах IF не наблюдались переходы, связанные с состояниями 3П2, 3П0- и В'0+,
постоянные IF в этих состояниях, приведенные в табл. 10.5, оценены при подготовке справочника.
Энергия возбуждения состояния 3П2 рассчитана по соотношению, аналогичному (9. 1), ее
погрешность оценивается в 500 см. Значение Т, для состояния 3П0- получено в предположении, чта
разность энергий состояний 3П0+ и 3П0- составляет 125 + 50 см. Энергия возбуждения состояния
В'0+ оценена на основании энергии уровня и=11, /=11 B3 341 см) состояния В3Нп+, который
согласно данным работы [1346] предиссоциирован. Погрешность Те достигает 500 см.
Постоянные IF в этих состояниях оценены на основании значений в, и В, в состояниях ^3ПХ и В3Н0+ и
(для состояния В'0+) точки псевдопересечения потенциальных кривых состояний -В3П0+и0+[950].
Термодинамические функции IF(r) были рассчитаны по уравнениям A. 3)—A. 6), A. 9), A.10),.
A. 93)—A. 95). Значения QBB, QBn и Qm находились по соотношениям A. 76)—A. 78), в которых
статистические суммы по колебательно-вращательным уровням энергии шести электронных
состояний и их производные вычислялись непосредственным суммированием по уровням энергии по
уравнениям A. 73)—A.75). Колебательно-вращательные уровни энергии рассчитывались по уравнениям
A.65) и A. 34), коэффициенты Ykl которых приведены в табл. 10.6. Вырождение уровней состояний
3П2 и у!3П1 учитывалось статистическим весом 2, значения Ykl были получены на основании
постоянных из табл. 10.5 по методикам, изложенным в разделе 1.4. В расчетах учитывались все
колебательно-вращательные уровни состояния Х1^+, ограниченные его предельной кривой
диссоциации, состояний 3П2, А3Иг, 3По- и В'0+, ограниченные диссоциационными пределами этих
состояний и состояния В3Л0+ с энергиями меньше энергии уровня у=11, /=11. В табл. 10.6
приведены значения коэффициентов а( в уравнении, аппроксимирующем предельную кривую
состояния X1!,+, а также значения уши и /нт- Погрешности вычисленных термодинамических функций
IF (г) при Т <С 2000 Копределяются главным образом неточностью фундаментальных постоянных
и не превышают 0,03 Дж-К^-моль при 298,15 К При более высоких температурах становятся1
заметными погрешности, обусловленные отсутствием экспериментальных данных о постоянных
IF в состояниях 3П2, 3П0- и других состояниях, коррелирующих с атомами I BР)-\-? (гР).
Погрешности в значениях Ф°(Т) при 3000 и 6000 К могут достигать 0,5 и 1 Дж-К^-моль соответственно^
Ранее таблицы термодинамических функций IF (г) вычислялись в работах [1426] (до 1500 К),
[1012] (до 2000 К), предыдущем издании настоящего справочника [88] и таблицах
JANAF [2095]. Расчеты [1012, 1426, 2095] были выполнены без учета возбужденных
состояний IF, в [88] для приближенного учета четырех компонент состояния 3П состоянию 3П0+
был приписан статистический вес 6. В расчете, выполненном в работе [1012], по-видимому, была<
допущена ошибка. В расчете JANAF [2095] ошибки из-за пренебрежения возбужденными
электронными состояниями и использования приближенного метода расчета при высоких температурах
частично компенсируют друг друга, расхождения между данными табл. 83 и работы [2095] при
3000 К достигают 0,5 Дж-К-моль в значениях Ф° (Т) и S° (T) и 2 Дж-К^-моль в С°Р(Т),
Расхождения между данными табл. 83 и работы [88] не превышают 0,1 Дж-К-моль до 2000 К'
и быстро растут с температурой (до 1 Дж-К^-моль в Ф° F000 К)).
Константа равновесия реакции IF (r)=F (r)-f-I (г) вычислена на основании значения
Дг#° @) = Do (IF) = 22700 + 15 см ^ 271,6 ± 0,2 кДж ¦ моль.
Эта величина была получена Клайном и Мак-Дермидом [9946] по началу предиссоциации
IF в состоянии В3Л0+ при 17=10, /=12. Ранее Коксон [1104] рекомендовал более высокое значенио-
Do (IF)=23 100+120 см на основании экстраполяции уровней состояния Л3П1 и предиссоциации
в состоянии В3И0+, наблюдавшейся Дьюри [1346] на уровне v=ll. Поскольку потенциальная
кривая состояния ВШ0+ проходит через максимум благодаря взаимодействию этого состояния
с состоянием В'0+, в справочнике принимается значение, полученное в работе [9946]. Ему
соответствует
kfH°(IF, г, 0) = —87,162+ 0,36 кДж • моль.
210
Иод и его соединения
IF(r)
Таблица 10.6. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения «шах и *Тцт, принятые для расчета термодинамических функций
IF, IC1, IBr
Коэффициенты
IF
аш, | зПо_ | вт0+ | я'о+
7V10-4 0 1,4 1,570 6 1,894 1,905 453 2,415
Ую-Ю 6,100 63 3,8 3,808 59 4 4,065 1 3
У20 —3,112 001 —3,8 —3,960 491 —8,7 —1,3 —3,23
У30.102 —0,236 845 — 2,385 04 — —19,8 —
У40.103 —0,132 398 — —1,12125 — — —
Уи-101 2,797 44 2,3 2,3 2,3 2,272 1 1,5
уи.Юз —1,869 86 —3 —3 —4 —1,398 —2
Уа1.106 —0,430 228 — — — -8,2 —
У31.Ю' —2,199 15 _____
у .10' —2 4 3 —3 —3 —2 8 2
(a0=D°)-10-s 23J470 05 9,5 7,82 4,6 4^59 6,97
а^Ю2 3,703 86 _____
a2.10' 4,078 69 _____
a3.1013 —4,537 28 — — — — —
iW 66 49 34 22 И 45
Jim 406 _____
Коэффициенты
ICI
зп2
•IV
вт0+
B-V
C3S-
Г,-10"* 0 1,230 0 1,374 798 1,718 8 1,733 2 1,815 6 3,774 4
Ую-10 3,825 59 2,55 2,074 23 2,4 2,444 4 0,362 457 1,732
У20 —1,55154 —4 —1,634 05 —38,9 —13,458 2 —1,158 41 —1,1
У30.102 0,358 954 2 —5,856 08 — — — —
У4О.Ю» —0,123 925 — 0,708 549 — — — —
Уи-101 1,129 30 0,85 0,842 198 0,9 0,9 0,532 0,6
Уц-Ю3 —0,512 587 —0,65 —0,654 505 —2,9 —2,9 —3,4 —0,4
У21.106 —0,591479 —1,5 —1,459 23 — — — —
У31.10' — — —5,056 32 — — — —
У02.10' —0,40 —0,38 —0,58 —0,5 —5 —0,5 —0,3
(а0—De)-10-s 17,499 3 5,258 3,786 69 0,37 0,828 0,283 6,480
а,.1О2 1,962 14 — 2,862 79 — — — —
а2.10' 0,990 358 — 1,818 79 — — — —
а3-1013 —0,766 684 — —4,467 02 — — — —
ига3х 74 53 34 2 4 15 72
/1Im 572 _ 346 - - - -
Коэффициенты
IBr
А*Щ
вт0+
В'0+
ЕЧ.+
f,.10-4 0 1,11 1,235 1,616 5 1,6814 3,5427 3,8714 3,9126
У10-10-2 2,695 22 1,50 1,47102 1,316 89 0,727 334 0,43 0,9 0,77
Ум —1,202 01 —1,5 —2,442 86 —1,896 86 —0,587 986 — — —
У30.102 1,178 94 — —0,813 527 — 0,066 417 4 — — —
у 103 —0,128 018 — 0,371625 — —0,121113 — —
Уот-Ю1 0,563 534 0,45 0,444 222 0,426 563 0,347 815 0,3 0,3 0,3
Уп-103 —0,193 855 —0,62 —0,616 793 —0,443 062 —0,237 858 — — —
У2Г105 0,017 618 8 —0,54 —0,535 684 —0,986 463 —0,419 263 — — —
У81.10' —0,298 672 — — — —1,192 90 — — —
У02.10' —0,08 —0,16 —0,16 —0,18 —0,32 — — —
/^_D.).1O-3 14,762 8 3,700 2,456 47 0,717 1,694 74 — — —
а,.102 0,906 083 — 1,300 12 — 0,734 610 — — —
аа-10' 0,277 326 — 0,992 440 — 1,008 06 — — —
а3-1013 —0,065 752 2 — —2,122 32 — —2,756 59 — — —
i>max 85 49 45 5 38 — — —
/lim 727 — 362 — 332 — — —
211
IF, (г)
Глава 10
IF5 (г). Термодинамические свойства газообразного пентафторида иода в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 84.
Молекулярные постоянные, использованные при расчете термодинамических функций IF6,
приведены в табл. 10.7. Исследования спектров IF5 [732, 2462, 555, 3380, 355, 2918] и электроно-
графические исследования [519, 3213, 2918, 1146] свидетельствуют, что молекула IF5 в основном
Таблица 10.7. Значения молекулярных постоянных и о, принятые для расчета термодинамических
функций IF6 и IF, (p_j = l)
Молекула
Vl
v2
v3
4
v.'
V
v,B)
v.B)
v.B)
vie B)
vii B)
CM
IFS 712
IF, 676
• В случае IF,
615
635
<** =
318
672
it = 2.
60i
257
255
746
275
425
640
363
372
310
189
510
_
352
200
VbVio»'
Г3 • CMe
4,10
12,1
4
10
электронном состоянии ХгА имеет структуру правильной квадратной пирамиды (точечная группа
симметрии С4,), в вершине которой расположен атом иода, а в углах при основании — четыре
атома фтора (FpM). Пятый атом фтора (FaK0) расположен на оси симметрии ниже плоскости
основания пирамиды. Связи I—Fpax и I—Fa]C0 неравноценны и угол между ними меньше 90°. Приведенное
в табл. 10.7 произведение главных моментов инерции молекулы IF5 вычислено на основании
структурных параметров r(I—FpJ=1,867 ±0,003 A, r(I—F.J = 1,827 ±0,007 А, ^ Fpiw—I—FaK0 =
=82,2 + 0,3°, найденных Хевиттом, Робиттом и Шелдриком методом газовой электронографии
(см. [2918, 1146]) и вращательной постоянной 50=(9,098 13 + 0,000 17) -10 см~х, полученной
Брэдли и др. [732] при исследовании микроволнового спектра IF5. Эти структурные параметры
удовлетворительно согласуются с найденными для кристаллического пентафторида иода [2164 Р.
Погрешность в значении IaIbIc составляет 3-Ю"15 г3-см6.
Спектр КР газообразного IF5 изучался в работах [3380, 355], жидкого IF5 — в [2462, 555,
410, 1646, 3380, 355] и кристаллического — в [1646, 355]. ИК-спектр газообразного IF5
исследовался в работах [2462, 555, 410, 2918], жидкого и кристаллического — в работе [1646].
Приведенные в табл. 10.7 значения частот v1? v3, v7, vg найдены Лордом и др. [2462] и Биганом и др. [555]
при исследовании ИК-спектра газообразного IF5, значение частоты v9 принято по работе Осборна
и др. [2918]. Для частот v2, v4 и ve, неактивных в ИК-спектрах, приняты значения, основанные
на результатах исследований спектра КР газообразного IF5, выполненных Селигом и Хольцма-
ном [3380] и Александером и Битти [355]. С принятыми значениями удовлетворительно
согласуются результаты исследований спектров конденсированного пентафторида иода. Для частоты
v5 в табл. 10.7 приведено значение, найденное при подготовке справочника на основании
сравнения энтропии S° (IF6, г, 330 К), вычисленной по молекулярным постоянным и по
экспериментальным данным о теплоемкости IF5 (к, ж) и энтальпиям фазовых переходов [3267, 3231, 2918]. С ним
практически совпадает значение v5=257 см, вычисленное Биганом и др. [555] по силовым
постоянным IF5 2. Погрешности в принятых значениях частот не превышают 3—5 см.
Термодинамические функции IF5(r), приведенные в табл. 84, были вычислены по уравнениям
A.3)—A.6), A.9), A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор —
гармонический осциллятор». Основные погрешности табулированных термодинамических
функций обусловлены использованием приближенного метода расчета и в значениях Ф°(Т) оцениваются
равными 2, 9 и 12 Дж-К^-моль при Г=298,15, 3000 и 6000 К.
Ранее термодинамические функции IF5 (г) вычисляли Эванс и др. [1426], Нагараян [2763],
В работе [836] при рентгенографическом исследовании кристаллического IF5 было найдено г (I—FaEe)=l,75 A,
что не согласуется со значением Во, найденным по МВ-спектру IF6 [732].
В спектре КР газообразного IF5 [3380] вблизи максимума при 272 см, отвечающего частоте ve, имеется плечо
при 250 — 260 см, которое, возможно, соответствует частоте v5.
212
Иод и его соединения
IF7 (г)
Кханна [2236] (Т <[ 1000 К) и авторы справочников [2095, 66]. Все расчеты выполнены в
приближении «жесткий ротатор — гармонический осциллятор». Термодинамические функции,
приведенные в табл. 84 и в справочнике [2095], согласуются в пределах 0,1 Дж-К^-моль. Расчеты,
приведенные в работах [1426, 2763, 2236], основаны на ошибочных данных о структуре IF5 и
неточных значениях частот колебаний1. В справочнике Галкина и др. [66] расчет был выполнен
до 5000 К для моделей молекулы IF5, соответствующих точечным группам симметрии Dilt и С4„
и рекомендованы их среднеарифметические значения. Расхождения в значениях Ф°(Г) достигают
2,5 Дж-К^-моль-1.
Константа равновесия реакции IF5 (r)=5F (r)+I (г) была вычислена с использованием значения
ДгЯ°@) = 1325,922+ 2,3 кДж-моль, которое соответствует энтальпии образования
AjH°(IFb, г, 298,15 К) = — 841,0 + 1,8 кДж • моль.
Это значение было принято на основании анализа результатов измерений энтальпии
образования и испарения IF5 (ж). Энтальпия образования IF5 определялась в работах Волфа [3992]
и Сеттла и др. [3395а]. Волф [3992] измерил энтальпию гидролиза жидкого IF5 в воде и в растворе
едкого кали. Обработка этих измерений приводит к значению AyiP(IF6, ж, 298,15 К) = —878 +
+ 5 кДж-моль. В работе [3395а] методом фторной калориметрии было получено LJI° (IF8, ж,
298,15 К)=— 882,0 + 1,6 кДж-моль. Результаты работ [3992, 3395а] совпадают в пределах их
погрешностей, но измерения методом фторной калориметрии привели к меньшему разбросу
данных, и соответствующая величина принята в справочнике.
Таблица 10.8.
Результаты
Авторы
Py4Si Брайда,
Роджере и др.,
Роджерс и др.,
Осборн и др.,
1934
1954
1956
1971
[3267]
[3231 ]
[3231а]
[2918]
определений энтальпии испарения IFj (ж) (в кДж
Метод
Статический метод с применением кварцевого
манометра, 268—282 К, 4 точки
Динамический метод точек кипения, 283—
343 К, 7 точек
Статический метод с применением ртутного
манометра, 300—330 К, 7 точек
Статический метод с применением манометра
Бурдона, 288 К, 1 точка
Статический метод с применением
нуль-манометра, 285—375 К
• МОЛЬ)
А,Н°
II закон
44,5 ±7,7
40,7 ±4,5
42,4
—
42,2+0,4
B98,15 К)
III закон
41,07 ±0,15
40,8 ±0,2
40,97 ±0,04
44,8
41,0 ±0,05
Результаты расчетов энтальпии испарения IF5 (ж) представлены в табл. 10.8. Наиболее
детальные измерения были проведены Осборном и др. [2918], измерившими также теплоемкость IF5(k, ж),
энтальпию и температуру плавления. Комбинированием полученного в этой работе значения
L,H° (IF5, ж, 298,15 К)=41,0+0,7 кДж-моль (погрешность этой величины увеличена с учетом
неточности термодинамических функций) с приведенным выше значением энтальпии образования
IF8 (ж) и была получена принятая в справочнике энтальпия образования IF5 (г).
IF7 (г). Термодинамические свойства газообразного гептафторида иода в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 85.
Молекулярные постоянные, использованные при расчете термодинамических функций IF7,
приведены в табл. 10.7. На основании результатов изучения колебательных спектров [504, 410,
968, 1434, 2462], электронографических исследований [347, 519, 3668], исследования отклонения
молекулярного пучка IF7 в неоднородном электрическом поле [1438] и изучения ЯМР-спектра
IF7 [1647], а также рентгенографических исследований кристаллической структуры IF7 [834,
1279] и теоретического расчета [2874] в справочнике принимается, что в основном электронном
состоянии %гА молекула IF7 имеет структуру правильной пятиугольной бипирамиды, в центре
которой находится атом иода, а в вершинах — атомы фтора, причем пять из них расположены
В работе [2763] имеются вычислительные ошибки.
213
IC1 (г) Глава 10
в плоскости пятиугольника (Fpas) и два атома — на оси симметрии по обе стороны от этой плоскости
(FSKC) (точечная группа симметрии D5h). Приведенное в табл. 10.7 произведение главных моментов
инерции вычислено на основании структурных параметров, определенных Адамсоном, Томпсоном
и Бартеллом [347] в результате электронографического исследования газообразного IF7: r (I—
-FpJ=1,857±0,004 A, r(I-FaKC)=l,781 + 0,007A, ^ FpaI-I-Fp.5=72o, ^ Fa'KO-I-FpM=90°.
Погрешность значения IaIbIc не превышает 2-10~114г3-смв. Следует отметить, что получен"
ная в работе [347] функция радиального распределения не противоречит моделям, слегка
отличным от правильной бипирамиды и принадлежащим точечным группам симметрии С3 и С,. Этот
эффект может быть объяснен псевдовращением и обусловлен наличием деформационных
колебаний с большой амплитудой [1434]. Молекула IF7 симметрии Dbh имеет одиннадцать основных
частот, из которых семь (v5, ve, v7, v8, v9, v10, vu) дважды вырождены; пять частот (v1} v2, v8, v9, v10)
активны в спектре КР, пять (v3, v4, v5, ve, v7) — в ИК-спектре и одна (vn) неактивна в
колебательных спектрах. Приведенные в табл. 10.7 значения основных частот, активных в ИК-спектре,
приняты по работе Эйзеля и Сеппельта [1434], которые исследовали ИК-спектр газообразного IF7
с большим разрешением и измерили контуры ряда полос. Значения частот v1? v2) v8, v9, v10
приняты по результатам анализа спектра КР, выполненного Классеном, Гаснером и Селигом [968].
Точность определения значений основных частот IF7, измеренных в работах [968, 1434] не ниже
2 см. Приведенное в табл. 10.7 значение частоты vu найдено в работе [1434] по составным
частотам, его погрешность может достигать 10 см1. Поскольку электронный спектр IF7 не
исследовался, сведения о возбужденных электронных состояниях IF7 отсутствуют.
Термодинамические функции IF7 (г) были вычислены по уравнениям A. 3)—A. 6), A, 9), A. 10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности вычисленных термодинамических
функций обусловлены главным образом использованием приближенного метода расчета и в
значениях Ф°(Т) при Г=298,15, 3000 и 6000 К оцениваются в 2, 13 и 16 Дж-К^-моль.
Ранее термодинамические функции IF7 (г) вычислялись в работах Эванса и др. [1426], Венд-
линга и Махмуди [3917], Кханна [2237] (Т < 1000 К), Лавилла и Бауэра [2367] (Т < 5000 К)
и справочнике JANAF [2095]. Рассчитанные в этих работах термодинамические функции
существенно отличаются от приведенных в табл. 85 из-за того, что в них использовались ошибочные
значения основных частот IF7, а в работе [2367], кроме того, число симметрии принималось равным 2.
Расхождения данных табл. 85 и работы [2095] составляют от 3 до 6 Дж-К^-моль и обусловлены
различием в значениях v4, v7, vu и г (I—FaK0).
Константа равновесия реакции IF7(r)=7F(r)+I (г) вычислена с использованием значения
ДгЯ°@) = 1596,226+ 3,0 кДж-моль, соответствующего принятой энтальпии образования
IF7, г, 298,15 К) = — 961,5 ±2,0 кДж • моль.
Эта величина основана на результатах калориметрических измерений энтальпии сгорания
элементарного иода во фторе, выполненных Сеттлом и др. [3395а], и исследований равновесий
с участием IF7 (г), выполненных Бернстейном и Кацем [605]. Измерения [3395а, 605] приводят
к значениям —961,5±2,0 и 961+8 кДж-моль соответственно.
IC1 (г). Термодинамические свойства газообразного хлорида иода в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 86.
Молекулярные постоянные IC1, использованные для расчета термодинамических функций,
приведены в табл. 10.5. На основании общих представлений о систематике электронных состояний
двухатомных молекул интергалогенов и результатов исследований спектров IC1 в справочнике
принимается, что эта молекула имеет семь связанных валентных электронных состояний с
энергиями до 50 000 см (см. рис. 9.1 и табл. 10.5 2). Колебательные постоянные IG1 в состоянии X1!,+
приняты по работе Хультена и др. [2043], в которой исследована система полос А3^—Хг11 +
(v" <; 2) в спектре молекул 135С1 и 137С1; вращательные постоянные получены Хербстом и Штейн-
метцем [1888] в результате изучения микроволнового спектра этих молекул. Принятые постоянные
1 Полоса с максимумом при 174 см в спектре КР жидкого IF7 [2462], отнесенная Вендлингом и Махмуди [3917]
к колебанию vu, в действительности связана с наличием примесей в образце IF7, исследованном в работе [2462]-
2 Известны еще два валентных состояния IC1 с энергиями больше 50 000 см, коррелирующих с пределами 1++
+СГ [1065, 1066, 1830], а также ридберговские состояния IC1 [1283, 1288].
214
Под и его соединения ICI (т)
согласуются с найденными при исследованиях электронного спектра [1165, 1144, 2041, 988],
спектра комбинационного рассеяния [1992], инфракрасного [806] и микроволнового [3911, 3713,
3712] спектров. Молекулярные постоянные 135С1 в состоянии А3^ приняты на основании
результатов исследования системы полос Л3П1—Хг2+ в работах Хультена и др. [2043] (9 <^ v" <^ 18)
и [2041] C «^ у" <[ 35). Они удовлетворительно согласуются с данными, полученными в [1621,
3961, 2972, 1165, 1144, 2042, 1373, 988]. Исследования системы полос ^3ПХ—ZX2 + показали, что
уровни состояния А3!!-! с v ^ 20 сильно возмущены. Хультен и др. [2041 ] пришли к выводу, что
колебательные уровни этого состояния могут быть прослежены только до у=35, и выделили пять
колебательных уровней другого состояния, расположенных выше уровня у=35 состояния А3^
и быстро сходящихся к диссоциационному пределу. Хультен и др. [2041] предположили, что это
состояние, имеющее общий диссоциационный предел с состоянием -43nlt ответственно за
возмущения, наблюдаемые в системе А3^—ХХ2 +. В справочнике принято, что состояние, вызывающее
возмущения, является состоянием 3П2. Приведенное в табл. 10.5 значение ТеC112) вычислено по
соотношению типа (9.1). Колебательные постоянные в этом состоянии были получены на
основании данных [2041 ] по значениям G'(vn) и принятой величины Т, методом подбора. Вращательные
постоянные оценены по постоянным IC1 в состоянии A3TLX. Исследования системы полос В3П0+—
ХХИ + в спектрах поглощения [2041, 821 ] и флуоресценции [695] показали, что в результате
взаимодействия состояния 2?3П0+ и отталкивательного состояния 0+ потенциальная кривая состояния
В3Е0+ проходит через максимум (в области у=4) и возникает связанное состояние В'0+.
Постоянные 135С1 в состоянии В3Л0+ приняты по рекомендации Герцберга [1906], сделанной на основании
данных Брауна и Гибсона [821 ] Ч Постоянные в состоянии В'0+ вычислены по результатам
исследования системы В'0+—Х1^" в работе [821]. Переходы, связанные с состоянием 3П0-, в спектре
IC1 не наблюдались; согласно [2745, 2747] расщепление компонент 3П0- и 3П0+ должно быть
небольшим. Приведенные в табл. 10.5 постоянные IG1 в этом состоянии получены на основании
предположения, что ГвE3П0+)—Г,CП0-)=150+50 см, а значения шв и Ве в этих состояниях
приближенно равны. Постоянные в состоянии С32~ приняты по работе Харанатха и Рао [1830], в которой
исследована колебательная структура системы полос <732 ~—Л3ПХ (у' ^ 26) и выполнена оценка
величины ге этого состояния.
Термодинамические функции 1С1(г) были вычислены по уравнениям A.3)—A. 6), A. 9), A.10) ,
A. 93)—A. 95). Значения Qm, $вн и Qm находились по соотношениям A. 76)—A. 78), в которых
статистические суммы по колебательно-вращательным уровням энергии семи электронных состояний
я их производные рассчитывались непосредственным суммированием по уровням энергии по
уравнениям A.73)—A.75). Колебательно-вращательные уровни энергии вычислялись по уравнениям
^1.65) и A.34) со значениями коэффициентов Ykl, приведенными в табл. 10.6. Вырождение
вращательных уровней состояний 3П2 и А3^ и мультиплетность вращательных уровней
состояния С32~ учитывались статистическими весами. Значения Ykl, приведенные в таблице,
соответствуют природной смеси изотопов хлора и получены на основании молекулярных
постоянных 135С1 из табл. 10.5 с помощью методик, изложенных в разделе 1.4. В расчетах учитывались
все колебательно-вращательные уровни энергии состояний Х1Б + и -48Пг, ограниченные
их предельными кривыми диссоциации, уровни состояний 3П2, 3П0-, В'0+ и С32~,
ограниченные их диссоциационными пределами, и состояния В3П0ь, имеющие энергии ниже энергии точки
запрещенного пересечения потенциальных кривых состояний В3П.0+ и 0+. В табл. 10.6 приведены
значения коэффициентов а{ в уравнениях, аппроксимирующих предельные кривые диссоциации
состояний ХХ3+ и А3ПЪ а также значения ?>„« и /щп (для состояний Х12+ и A3U.j). Погрешности
вычисленных термодинамических функций 1G1 (г) обусловлены отсутствием экспериментальных
данных о колебательно-вращательных уровнях энергии состояния Х12+ со значениями v > 2,
о постоянных в состоянии 3Па и применением приближенной методики ограничения числа
колебательно-вращательных уровней энергии в ряде электронных состояний. Погрешности в
значениях Ф°(Т) могут достигать 0,05, 0,2 и 0,5 Дж -К -моль при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции IG1 (г) вычислялись в работах [1426] (до 1500 К), [1012]
(до 2000 К) и справочниках [88 и 2095]. Во 2-м издании настоящего справочника [88] был учтен
¦суммарный вклад состояний 3П2, 3П1( 3П0- и 3П0+, в остальных расчетах возбужденные состояния
не учитывались. Расхождения в значениях Ф°(Г), табулированных в настоящем и предыдущем
Постоянные IC1 в этом состоянии существенно уточнены в работе [9946], опубликованной в 1977 г.
215
IBr(r) Глава 1&
изданиях, не превышают 0,15 Дж-К^-моль, расхождения в значениях S°(T) больше, но при
Т ^ 5000 К не превышают 1 Дж -К -моль. Расхождения с результатами других расчетов больше,
хотя ошибки из-за пренебрежения возбужденными состояниями и применения метода Майера
и Гепперт-Майер частично компенсируют друг друга. Данные работы [2095] отличаются от данных
табл. 86 на величины до 1, 2 и 7 Дж-К^-моль в значениях Ф°(Т), S°(T) и С°р (Т).
Константа равновесия реакции IG1 (г)=С1 (г)+1 (г) вычислена на основании энергии диссоциации
АгН° @) = Do(IC1) == 17 367,0 + 1,0 cm"x= 207,755 + 0,012 кДж • моль.
Это значение принято по результатам исследования прогрессии v'—0 в системе полос А3Лг *~
«- ХХ2 + молекул 135С1 и Р'С1, выполненного Хультеном и др. [2041 ]. Авторы этой работы
обнаружили в спектре IC1 группу полос, сходящихся к диссоциационному пределу IB.Ps/5)-f C1 BА/2),
которые в настоящем справочнике отнесены к переходу 3П2—X1!, + (см. выше), и нашли Do (I36C1) =
17 366,0 + 0,5 см и DO(I37C1)=17370,1 ±0,5 см". Принятая величина хорошо согласуется со
значениями, полученными в более ранних исследованиях спектров IC1 [821, 1165, 1373, 1621, 3961]
и лежащими в интервале от 17 340 до 17 430 см. Калориметрическое определение энтальпии
реакции синтеза 1С1(ж) из элементов [3675] вместе с данными [258] для энтальпии испарения IC1 (ж)
приводит к значению DO(IC1)=2O8,1 кДж-моль.
Практически совпадающие значения были вычислены также на основании результатов
измерений равновесия реакций 2NO(r)+2ICl(r)=2NOCl(r)+I2(r) и 1С1(г)=0,51г(г)-}-0,5С12(г) [2592].
Равновесия обеих реакций были измерены статическим методом в интервале температур
410—452 К и 641—764 К соответственно. Энергии диссоциации IC1, вычисленные по этим данным
по методу III закона термодинамики, равны 207,7 + 0,2 кДж-моль и 208,1 + 1,5 кДж-моль
соответственно 1. Принятому значению DO(IC1) соответствует
г, 0)= 19,028 + 0,040 кДж • моль.
IBr (г). Термодинамические свойства газообразного бромида иода в стандартном состоянии
при температурах 100—6000 К приведены в табл. 87.
Молекулярные постоянные, использованные для вычисления термодинамических функций
1Вг(г), приведены в табл. 10.5. На основании общих теоретических представлений о систематике
электронных состояний молекул двухатомных интергалогенов (см. [2745, 2747]) и результатов
исследований спектров IBr в справочнике принимается, что эта молекула имеет восемь
связанных валентных состояний с энергиями меньше 50 000 см (см. табл. 10.5). Три состояния
(С32~, ?>1Д и Ег1,+) коррелируют с ионами 1++Вг", остальные — с атомами I (*Р)+ВгBРJ.
Молекулярные постоянные 1'8Вг в состояниях ХХ2 + и А3И1 приняты по работе Селина
13382]3, в которой исследована система полос А3Н1—Х12+ (v" ^ 3, 9 ^ v' ^ 30) в спектрах
1"Вг и 181Вг. Переходы, связанные с состояниями ^Г12+ и Л3Пх, исследовались также
в работах [820, 989, 3381 ] (система полос А3^ — ХХЪ+), [820, 3384] (система полос В3П0+—Х^+),
1820, 1063, 3383] (система полос Б'0+—Хг2+), [3811] (система полос D1A-AsIl1), [2109, 3695}
(микроволновой спектр IBr), [1992] (спектр КР). Экспериментальные данные о
постоянных IBr в состоянии 8Па в литературе отсутствуют, хотя возможно, что возмущение уровней
состояния АаЛг в области v > 31 [1373, 3382] обусловлено взаимодействием с этим состоянием.
Приведенные в табл. 10.5 значения ш, и В, приняты по соответствующим постоянным IBr в
состоянии AaTLu значение ГвCП2) вычислено по соотношению типа (9.1), его погрешность может
достигать 500 см.
Приведенные в табл. 10.5 постоянные IBr в состоянии В3Н0+ приняты по работе Селина и Седе-
берга [3384], в которой исследована вращательная структура полос системы В3И0+ <- ХЧЕ+
B ^ v' <; 4) в спектрах 1'8Вг и 181Вг. Они хорошо согласуются с исследованиями [820, 3383].
Согласно данным этих работ в результате взаимодействия состояния В3Ио+ с отталкивательным состоя-
1 В расчете были испольвованы результаты измерений констант равновесия реакции 2NOC1 (r)=2NO (г)+С1г (г).
в работе [551].
* Поскольку 7',(.В3П0+) существенно больше энергии диссоциационного предела I BP3^)-\-Bv BЛ/г), состояние-
*Пв_ в молекуле IBr должно быть отталкивательным. О ридберговских состояниях IBr см. работы [1065, 1066»
1283, 1289, 1831].
• Постоянные В, и а^ рассчитаны при подготовке справочника по значениям В„ найденным Селиным.
216
Иод и его соединения Шг(г)
нием 0+ потенциальная кривая состояния Б3И0+ проходит через максимум в области между р=5
и i>=4. Одновременно возникает связанное состояние В'0+, коррелирующее с атомами 1B/\)+
4-ВгBАд). Переходы в состояние В'0+ исследовались в спектре поглощения [820,1063, 1373, 3383]
(система полос ?'0+—Z12+, 8<z/< 37) и испускания [3810] (переходы С32~—?'0+ и E1L+ — B'0+,
0 <^ ^"^ 8). Приведенные в табл. 10.5 значения колебательных постоянных в состоянии
J5'0+ приняты по работе Венкатесварлу и Верма [3810], в которой они рассчитаны по кантам
полос. Вращательные постоянные вычислены по значениям В„ найденным в работе Селина [3383] для
12 <; v ^ 27. Колебательные постоянные IBr в состояниях С32~, ?IД и ЕгЪ + приняты по работам
Венкатесварлу и Верма [3810 и 3811] (система полос D1^-*A3H1, v' ^ 31). Вращательные
постоянные IBr в этих состояниях не определялись. Значения, приведенные в табл. 10.5, оценены
на основании сопоставления постоянных Ве в состояниях АъПг, BSUO+ и состояниях,
коррелирующих с ионными пределами, в молекулах других галогенов и интергалогенов.
Термодинамические функции IBr (г) были вычислены по уравнениям A.3)—A.6), A. 9), A.10),
A. 93)—A. 95). Значения (>„„, Qm и QtM находились по соотношениям A. 76)—A. 78), в которых
статистические суммы по колебательно-вращательным уровням энергии и их производные для
состояний Хг2 +, 3П2, АЧ1и В3Л0+ и В'0+ рассчитывались непосредственным суммированием по
уравнениям A. 73)—A. 79), а для состояний С32 ~, DXA и #*2 + вычислялись в приближении «жесткий
ротатор—гармонический осциллятор». Колебательно-вращательные уровни вычислялись по
уравнениям A. 65) и A.34) со значениями коэффициентов Ykl, приведенными в табл. 10.6. Вырождение
вращательных уровней состояний 3П2, А3Пг и ?IД, а также мультиплетность состояния С32~
учитывались статистическими весами. Значения YM, приведенные в табл. 10.6, соответствуют
природной смеси изотопов Вг и получены на основании молекулярных постоянных 179Вг из табл. 10.5г
а в отдельных случаях — первичных экспериментальных данных из соответствующих работ с
помощью методик, изложенных в разделе 1.4. В расчетах учитывались все
колебательно-вращательные уровни состояний ХХ2+, ^43Па и В'0+, ограниченные их предельными кривыми диссоциации,
уровни состояния 3П2, ограниченные его диссоциационным пределом, и уровни состояния Б3Пв+,
имеющие энергии меньше, чем энергия точки запрещенного пересечения потенциальных кривых
состояний BsHq+ и 0+. В табл. 10.6 приведены значения коэффициентов а{ в уравнениях,
аппроксимирующих предельные кривые диссоциации состояний Х1^ +, А9^ и В'0+, а также значения vma
и /ит (для трех состояний). Погрешности вычисленных термодинамических функций IBr (г)
обусловлены отсутствием данных о колебательно-вращательных уровнях состояния Хг2+ со
значениями 1)>3и приближенным учетом возбужденных состояний. Погрешности в значениях Ф°(Г)
при 298,15, 3000 и 6000 К могут достигать 0,05, 0,2 и 0,5 Дж-К^-моль.
Ранее таблицы термодинамических функций IBr (г) вычислялись в работах [1426] (до 1500 К),.
[1012, 4055] (до 2000К), предыдущем издании настоящего справочника [88] и таблицах JANAF
[2095]. Наилучшее согласие с данными табл. 87 имеет место в работе [1426] и предыдущем
издании [88]. С последним расхождения не превышают 1, 2 и 3 Дж-К^-моль в значениях Ф°(Т)
ж S°(T). В работе [2095] допущена ошибка в учете возбужденных электронных состояний IBr
(приняты неправильные статистические веса) и использована приближенная методика расчета.
Это привело к большим расхождениям с данными табл. 87, которые достигают 8и 25 Дж -К -моль
в значениях S°(T) и С°р(Т).
Константа равновесия реакции IBr(r)=Br (r)-fl(r) вычислена на основании принятого:в
справочнике значения
ДJE° @) = Do (IBr) = 14 657 ± 4 см = 175,34 ± 0,05 кДж • моль.
Принятое значение получено короткой экстраполяцией колебательных уровней состояния
АйЛг по данным Брауна [820] и Эберхардта и др. [1373] и состояния В'0* [820] для молекулы
Р'Вг. Оно практически совпадает с вычисленным по уравнению III закона термодинамики на
основании результатов измерений констант равновесия реакции 21Вг (г) = 12 (г)+Вг2 (г), выполненных
Мак-Моррисом и Йостом [2591] статическим методом в интервале температур 388—449 К A75,5 +
§,2 кДж-моль). Близкие значения A76,6± 1,2 и 173,2 кДж-моль) были найдены по
аналогичным данным, полученным в работах [2743 и 688], а также из калориметрических измерений
энтальпии взаимодействия 12 и Вг2, растворенных в СС14 [667] A76,3 кДж-моль).
Принятой энергии диссоциации IBr соответствует
&fHo(lBr, г, 0) = 49,746 ± 0,14 кДж • моль.
Глава 11
БЛАГОРОДНЫЕ ГАЗЫ И ИХ СОЕДИНЕНИЯ
Не, Не+, Ne, Ne+, Аг, Аг+, Кг, Kr+, KrF2, Xe, Хе+, Хе2,
ХеО3, ХеО4, XeF, XeF2 (к, ж), XeF2, XeF4 (к, ж), XeF4,
XeFe (к, ж), XeF6, XeO2F2, XeO3F2, XeOF4, Rn, Rn+
В настоящей главе, наряду с одноатомными благородными газами и их положительными ионами,
для которых термодинамические свойства рассчитаны до Т=20 000 К, рассмотрены также
некоторые наиболее известные в настоящее время устойчивые соединения Кг и Хе в твердом, жидком
и газообразном состояниях. Термодинамические свойства наименее устойчивых двухатомных
Хе2 и XeF даны только до 1000 К, а многоатомных KrFa и окислов, фторидов и оксидфторидов
Хе — до 6000 К. Фториды Хе в конденсированном состоянии представлены до 500—600 К. В
Приложении 3 приведены также данные для пяти газов (Не, Ne, Ar, Кг и Хе) при повышенных
давлениях.
Не (г). Термодинамические свойства газообразного гелия в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 88.
Атом Не имеет основное электронное состояние *S (табл. 11.1). Возбужденные электронные
состояния атома Не являются ридберговскими состояниями [2697] и не учитывались при расчете
термодинамических функций. Погрешности вычисленных величин во всем интервале температур
определяются только неточностью фундаментальных физических постоянных и не превышают
0,02 Дж-К^-моль-1.
Ранее термодинамические функции Не(г) вычислялись неоднократно в широком интервале
температур, в том числе в работе [1946] до 10 000 К, в работе [2291] до 8000 К, в предыдущем
издании настоящего справочника [88] и в справочнике [2556] до 6000 К, а также в работе [2198] до
2000 К. Результаты этих расчетов, за исключением [2198], совпадают с данными, приведенными
в табл. 88 в пределах погрешности последних. В работе [2198] было использовано завышенное
значение Д, что является причиной расхождений с данными настоящего справочника до
0,05 Дж-К^-моль-1 в значениях Ф° (Т).
Газообразный гелий принимается в качестве стандартного состояния этого элемента и в
соответствии с этим
ДД°(Не, г)==0.
Не+(г) Термодинамические свойства газообразного положительного иона гелия в
стандартном состоянии при температурах 298,15—20 000 К приведены в табл. 89.
Ион Не+ имеет основное электронное состояние 2S (см. табл. 11.1). Возбужденные электронные
состояния Не+ являются ридберговскими [2697] и не учитывались при расчете
термодинамических функций. Погрешности вычисленных величин во всем интервале температур определяются
неточностью фундаментальных физических постоянных и не превышают 0,02 Дж-К^-моль.
Ранее таблицы термодинамических функций Не+ (г) вычислялись в работе [1723] до 20 000 К
и работе [1946] до 10 000 К. Результаты этих расчетов совпадают с вычисленными в настоящем
издании в пределах указанных выше погрешностей.
Константа равновесия реакции Не+(г)+е=Не(г) вычислена с использованием значения
АГН° @) = —2372,324 кДж-моль в соответствии с принятым в настоящем справочнике значением
энтальпии образования Не+(г), равным потенциалу ионизации атома Не
ДД°(Не+, г, 0) = /0(Не) = 198 310,76 + 0,02 см = 2372,324 ± 0,001 кДж • моль.
Это значение 10 (Не) рекомендовано Мур [2696] на основании спектральных исследований
Ситона [3369] и Мартина [2535].
Ne (г). Термодинамические свойства газообразного неона в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 90.
Атом Ne имеет основное электронное состояние XS (см. табл. 11.1). Все возбужденные
электронные состояния Ne являются ридберговскими [2697 ] и не учитывались при расчете термодинамиче-
218
Благородные газы и их соединения
Ne(r)
Таблица 11.1. Уровни
Атом
Не
Не+
Ne
Ne+
Аг
Аг+
Кг
энергии атоиов Не, Не+, Ne
, Ne+,
принятые для расчета термодинамических <
Электронная
конфигурация
Is2
Is
...2s22p»
...2s22p5
...2s-2p8
...3s23pe
...3s23p5BPKd
...3s23pBBPKi
...3s23pBBPK<f
...3s23p8BPKd
...3*23p8BPKi
...3s23p8BPKd
...3s23p8BPKd
...3s23p8BPKd
...3s23p8BPKd
...3s23pBBPKd
...3s23p8BPKd
...3s23pBBPKi
...3s23p8
...3s23pB
...3s3pe
... 3s23p* (^jP) 3<2
3s23p* fiP) 3d
...3s23p4CPKd
...3s23p4CPKrf
...3s23p4CPKd
...3s23p4CPKd
.. .4s24pe
...4s24p8BPLd
...4s24p8BPLcZ
...4s24pBBPLd
.. .4s24pB BP) Ы
...4s24pBBPLd
...4s24pBBPLe2
...4s24pBBPLd
...4s24pBBPL<2
...4s24p5BPLd
...4s24p8BPLi
...4s24pBBPLd
Состояние
2S
iS
2Ч
8Л>
зр
з р
3Fm
Зр
SF*
'рг
'pi
*D2
*D3
*Di
iP'l
2D
V.
/2
*F
*P
2F
зр0
3P*
з p
** F
3Fi
'F3
'Pi
SD2
^a
"D3
CM x
0
0
0
0
782
0
111667,87
111818,09
112 138,98
112 750,22
113 020,39
113 426,05
113 716,61
114 147,75
114 641,04
114 805,18
114 821,99
115 366,90
0
1432
108 722,5
132 476,11
142 705,71
144 350,19
147 644,73
149 595,19
150 843,24
0
96 772,31
97 086,00
97 688,58
97 798,15
98 227,08
98 868,22
99 080,19
99 647,02
103 267,12
103 443,46
103 702,239
Pi
1
2
1
4
2
1
1
3
5
9
7
5
7
3
5
5
7
3
4
2
2
20
28
6
12
14
10
1
1
3
5
9
7
5
7
3
5
5
7
Atom
Kr
Kr+
Xe
Ar, Ar+, Kr, Kr+,
функций
Электронная
конфигурация
...4S24pBBPLd
...4s24peBPyJ4/
.. .4s24p8
.. .4s24p8
...4s4p»
...4s24p4CPLd
...4s24p4(«PLi
...4s24p4('PLi
...4s24p4(»PL<i
.. .4s24p4 (»P) U
...4s24p4(»PLi
..As4p*(»PLd
...4s24p4(»PLi
.. • 4s24p* C-P) 4d
... 4s24p* C-P) 4flt
...4s24p4(*PLi
...4s24p*(»PLi
...4s24p*(8PLrf
...4s24p4(»PLci
...4s24p*(»PLd
...4s24p4(»PLi
...5s25pe
...5s25pBBPMi
...5s25p8BPMi
...5s25pBBPMrf
...5s25p8BPMi
.. .5s25pB BP) 5d
...5s25pBBPMi
...5s25p8BPMi
...5s25p8BPMi
...5s25pB(!PL/
...5s25pBBPMi
...5s25pBBPMi
...5s25pBBPMa!
...5s25p8BPM/
...5s25p5BPMd
Xe, Xe+, Rn
Состояние
(*D,*F,*G)
VD^FSG)
2Pa,
ip4
*s '
*4
4 J?
*4
'4
ip4
l4
24
2Z4
2P
*p<>
'Pi
*Ft
3P%
3Fa
8^a
'F3
'Pi
1 ] n 1 IP 1 /~l I
( U, t, lLr )
'D2
3Da
*D
( 3D ^F ^G )
{'D, 1F,1G)
3Di
и Rn+,
T,
CM 1
104 888,114
106 009,90
111380,01
0
5 371
109 002,06
120211,59
120 428,65
121 002,10
121 781,28
126 002,57
127 931,20
129 698,98
129 743,56
130 514,59
130 895,28
131 377,24
131 633,86
132 967,28
134 568.821
134 623,17
137 099,98
0
79 771,798
79 987,16
80 197,16
80 323,28
80 970,93
81 926,04
82 430,72
83 890,47
90 893,23
91153,16
91 447,99
91 747,07
93 394,07
93 618,75
Pi
3
56
28
4
2
2
8
6
4
2
10
8
6
4
2
4
6
8
4
6
6
6
1
1
3
9
5
7
5
7
3
84
5
5
7
84
3
219
Ne+(r)
Глава 11
Таб л
Атом
ица 11.1 (окончание)
Электронная
конфигурация
Состояние
Хе+ ...5s25p* *P,k
...5s25p» 2Л/а
...5s5p* 25
. ..5s25p4(8PMd 4Я./г
...5s25p4(aPM<J *D7^
. ..5s25p*(8PMd *D,U
. ..5s25p4(8PMc2 4ДЛ2
...5s25p*(8PMi 4F7/2
...5s25p4(8PMi *F%
...5s25p4(8PMd *Л/а, 4V,/z
...5s25p4(8PMd 2Ps/j
...5s25p4(8PMd 2Л/а
...5s25p4(8PMd 4Р./г
...5s25p4(8PMd «Pv*
...5s25p4(8PMd «P,^
...5s25p4(8PMd «Z»,^
...5s35p4(8PMi 2ZJ,/it 2F
...5s25p4AJ5Mi *В,'г, *G
...5525р4(х/>M«1 2Ь,/2
...5«25р*(^Mй 2f/
...5,25p4(^Md *Z).J;
CM
0
10 537,3
90 873,83
95 396,68
95 437,60
96 033,44
96 858,14
98 822,66
99 404,90
101 535,65
105 313,33
105 947,54
106 475,17
106 906,11
107 381,75
107 904,50
109 563,15
111326,93
112 703,58
114 751,08
114 913,96
119085,50
Pi
4
2
2
6
8
4
2
8
10
10
4
2
6
2
4
4
20
22
6
6
8
6
Атом
Хе+
Rn
Rn+
Электронная
конфигурация
...5s25p4AOMd
...5si5piADMd
...5s25p4AZ)Md
...5s25p4AZ)M<J
...6s26pe
...6s26pBBPNd
...6s26pBBPNd
.!.6s26pBBPNd
...6s26pBBPNd
...6s26peBPN<2
...6s26pBBPNd
...6s26pBBP) 6d
...6s'26pBBPNd
...6s26pBBPM/
...6s26psBPN/
.. .6s26pB BP) &d
...6s26p*
Состояние
2 P
2 p
1S
*Po
'Pi
'Pi
*F
i/
ip3
3Di »d2, *D3
( *D 3F *G )
\iD iF ig/
Г *D з F 8G |
гр'и
2jPv2
T(
CM
124 302,18
127 010,70
127 527,40
129 248,50
0
67 906,52
68 891,34
69 798,0
70222,82
70 440,42
71790
72 550
73 200
79 608
79 717,18
82 233,3
85170
0
30 895,1
'!
4
2
4
2
1
1
3
9
5
7
5
7
3
15
84
84
5
4
2
ских функций Ne(r). Погрешности вычисленных величин при температурах до 15 000 К
определяются только неточностью фундаментальных постоянных и не превышают 0,02 Дж -К -моль.
При более высоких температурах в значениях S° (Т) и Ср° (Т) возникают ошибки, достигающие
величин порядка 0,1 Дж-К^-моль и связанные с пренебрежением ридберговскими состояниями.
Ранее термодинамические функции Ne (г) вычислялись неоднократно в широком интервале
температур, в том числе в работе [1946] до 10 000 К, в работе [2291] до 8000 К, в предыдущем
издании справочника [88] и в справочнике [2556] до 6000 К, а также в работе [2198] до 2000 К.
Результаты этих расчетов, за исключением [2198], совпадают с вычисленными в настоящем
издании в пределах указанной выше погрешности. В работе [2198] было использовано завышенное
значение R, что является причиной расхождений с данными настоящего справочника до
0,05 Дж-К^-моль-1 в Ф° (Т).
Газообразный неон принимается в. качестве стандартного состояния этого элемента и в
соответствии с этим
A/ff°(Ne, г) = 0.
Ne+ (г). Термодинамические свойства газообразного положительного иона неона в стандартном
состоянии при температурах 298,15—20 000 К приведены в табл. 91.
Уровни энергии иона Ne+, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которую включены только две компоненты основного электронного состояния
2Р [2697, 2996]. Первое возбужденное состояние Ne+BS), относящееся к электронной
конфигурации . . . 2s2pe, имеет энергию свыше 200 000 см, а следующие возбужденные состояния
являются ридберговскими [2697] и не учитываются при расчете термодинамических функций.
220
Благородные газы и их соединения Аг(г), Аг+(г)
Термодинамические функции Ne+(r) вычислены по уравнениям A.3)—A.6), A.9), A.10),.A.23)—
{1.25). Погрешности табулированных значений термодинамических функций во всем интервале
температур определяются только неточностью фундаментальных постоянных и не превышают
0,02 Дж-К-моль.
Ранее таблицы термодинамических функций Ne+(r) вычислялись в работе [1723] до 20 000 К
и работе [1946] до 10 000 К. Результаты этих расчетов согласуются с вычисленными в настоящем
издании в пределах указанных выше погрешностей.
Константа равновесия реакции Ne+(r)+e=Ne (г) вычислена с использованием величины
ДгЯ°@) = —2080,661 ±0,001 кДж-моль в соответствии с принятым в настоящем справочнике
значением энтальпии образования Ne+(r) равным потенциалу ионизации атома неона
+ 0,l см = 2080,661 ± 0,001 кДж.моль.
Это значение Io (Ne) рекомендовано Мур [2696] на основании неопубликованного спектрального
исследования Минхагена.
Аг(г). Термодинамические свойства газообразного аргона в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 92.
Уровни энергии атома Аг, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой даны валентные состояния атома Аг, относящиеся к электронным
конфигурациям . . . З^Зр6 и ... 3s23p53d. Энергии этих состояний приняты по рекомендации
Мур [2697]. Состояния, относящиеся к электронной конфигурации . . . 3s3p63d и имеющие
энергии порядка 200 000 см [3785 а], не принимались во внимание при расчете.
Термодинамические функции Аг (г) вычислены по уравнениям A.3)—A.6), A.9), A.10), A.23)—
A.25). Погрешности вычисленных величин до температур порядка 10 000 К определяются главным
образом неточностью фундаментальных постоянных и не превышают 0,02—0,03 Дж-К^-моль.
При более высоких температурах погрешности возрастают из-за пренебрежения ридберговскими
состояниями и могут достигать 0,1 Дж-К^-моль в значении Ф° B0 000 К).
Ранее таблицы термодинамических функций Аг (г) вычислялись в работах [1946] (до 10 000 К),
[2291] (до 8000 К), [2198] (до 2000 К) и в предыдущем издании справочника [88] (до 6000 К).
Результаты этих расчетов, за исключением [2198], совпадают с вычисленными в настоящем
издании в пределах указанных выше погрешностей для соответствующих интервалов температур.
В работе [2198] было использовано завышенное значение R, что является причиной расхождений
с данными настоящего справочника до 0,05 Дж-К^-моль.
Газообразный аргон принимается в качестве стандартного состояния этого элемента и в
соответствии с этим
Аг+ (г). Термодинамические свойства газообразного положительного ионг аргона в
стандартном состоянии при температурах 298,15—20 000 К приведены в табл. 93.
Уровни энергии иона Аг+, использованные при расчете термодинамических функций, приве-
дены'в табл. 11.1, в которой даны валентные состояния Аг+, относящиеся к электронным
конфигурациям . . . З^Зр5, . . . 3s3pe и ... 3s23p4CPKd Энергии этих состояний приняты по
рекомендации Мур [2697] (см. также [2676]). Состояния, относящиеся к электронным конфигурациям
3s23p4AZ)Kd и ... З^Зр^ЯЩ и имеющие энергии выше 170000 см [2697], а также
состояния, относящиеся к электронной конфигурации . . . 3s3p54<2, энергии которых, по-видимому,
еще выше, не принимались во "внимание при расчете.
Термодинамические функции Аг+ (г) вычислены по уравнениям A.3)—A.6), A.9), A.10), A.23)—
A.25). Погрешности вычисленных значений до температур порядка 15 000 К определяются только
неточностью фундаментальных постоянных и не превышают 0,02 Дж-К^-моль. При более
высоких температурах в значениях S° (Т) и Ср° (Г) возникают ошибки, достигающие величин порядка
0 1 Дж-К^-моль и связанные с пренебрежением ридберговскими состояниями.
Ранее таблицы термодинамических функций Аг+(г) вычислялись в работе [1723] до 20 000 К
и работе [1946] до 10 000 К. Результаты этих расчетов совпадают с вычисленными в настоящем
издании в пределах указанных выше погрешностей.
221
Кг (г), Кг* (г) Глава If
Константа равновесия реакции Аг+(г)+е=Аг(г) вычислена с использованием величины
АГН° @) =—1520,572+0,001 кДж-моль в соответствии с принятым в настоящем справочнике'
значением энтальпии образования Аг+ (г), равным потенциалу ионизации атома Аг
Д/Я°(Аг+, г, 0) = 70(Аг) = 127109,9+0,1 см = 1520,572 ± 0,001 кДж • моль-1.
Это значение /0 (Аг) было рекомендовано Мур [2696] на основании спектрального
исследования Иошино [4034].
Кг(г), Термодинамические свойства газообразного криптона в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 94.
Уровни энергии атома Кг, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой даны валентные электронные состояния, соответствующие электронным
конфигурациям . . . 4s24/?e, . . . 4s4p4d и ... 4s24Jp64/. Энергии этих состояний приняты по
рекомендации Мур [2697]. Состояния, относящиеся к электронным конфигурациям . . . 4s4/>64d'
и ... 4?4р64/, которые не наблюдались в спектрах Кг, имеют, по-видимому, высокие энергии
возбуждения, что позволило не принимать их во внимание при расчете.
Термодинамические функции Кг (г) вычислены по уравнениям A.3)—A.6), A.9), A.10), A.23) —
A.25). Погрешности вычисленных величин до температур порядка 8000 К определяются главным
образом неточностью фундаментальных постоянных и не превышают 0,02 Дж-К^-моль. При
более высоких температурах погрешности возрастают из-за пренебрежения ридберговскими
состояниями и могут достигать 0,3 Дж-К^-моль в значении Ф° B0 000 К).
Ранее таблицы термодинамических функций Кг (г) вычислялись в работах [1946] (до 10 000 К),
[2291] (до 8000 К), [2198] (до 2000 К) и в предыдущем издании настоящего справочника [88].
Результаты этих расчетов, за исключением [2198], совпадают с вычисленными в настоящем
издании в пределах указанных выше погрешностей для соответствующих интервалов температур.
В работе [2198] было использовано завышенное значение R, что является причиной расхождений
с данными настоящего справочника до 0,05 Дж-К^-моль.
Газообразный криптон принимается в качестве стандартного состояния этого элемента и в
соответствии с этим
Д/Г°(Кг, г)==0.
Кг+ (г). Термодинамические свойства газообразного положительного иона криптона в
стандартном состоянии при температурах 298,15—20 000 К приведены в табл. 95.
Уровни энергии иона Кг+, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой даны валентные состояния Кг+, относящиеся к электронным
конфигурациям . . . 4s24p8, . . . 4s4p6 и ... 4s24/LC.PLd. Энергии этих состояний приняты по
рекомендации Мур [2697] (см. также [2677]). Состояния, относящиеся к электронной
конфигурации ... 4s24p*ADLd и имеющие энергии выше 146 000 см [2697], а также состояния,
относящиеся к электронным конфигурациям . . . 4s24/?44/, . . . 4s4p54d и ... 4s4p54/, которые не>
наблюдались в спектрах Кг+, но энергии которых, по-видимому, еще выше, не принимались во
внимание при расчете.
Термодинамические функции Кг+ (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.23)—A.25). Погрешности вычисленных величин до температур порядка 15 000 К
определяются только неточностью фундаментальных постоянных и не превышают
0,02 Дж-К^-моль. При более высоких температурах в значениях S° (Т) и С (Т) возникают
ошибки, достигающие 0,1 Дж-К^-моль и связанные с неучетом ридберговских состоя-
Ранее таблицы термодинамических функций Кг+ (г) вычислялись в работах [1723] (до 20 000 К)
и [1946] (до 10 000 К). Результаты этих расчетов совпадают с вычисленными в настоящем издании
в пределах указанных выше погрешностей.
Константа равновесия реакции Кг+ (г)+е=Кг (г) вычислена с использованием величины
ДДГ3 @) =—1350,757 + 0,001 кДж-моль в соответствии с принятым в настоящем справочнике
значением энтальпии образования Кг+, равным потенциалу ионизации атома Кг
Д/Я°(Кг\ г, 0) = /0(Кг) = 112914,5 +0,1 см = 1350,757 + 0,001 кДж • моль.
Это значение /0 (Кг) рекомендовано Мур [2696] на основании спектрального исследования;
Петерсона [2997].
222
Благородные газы и их соединения
KrF2(r), Хе(г>
Таблица 11.2. Значения молекулярных
постоянных {Pg= 1, о = 2),
принятые для расчета
термодинамических функций
KrF2 и XeF2
Молекула
KrF2
XeF2
Состояние
в
Vl 1
449
514,5
CM
232
213
2)
-l
,6
,2
v,
589,
560,
9
1
I-
г •
11
2A
10s»
см2
,17
,66
KrF2 (г). Термодинамические свойства газообразного дифторида криптона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 96.
Молекулярные постоянные KrF2, использованные для расчета термодинамических функций,
приведены в табл. 11.2. Возбужденные электронные состояния молекулы KrF2 должны иметь,
как и у XeF2 (см. ниже), высокие энергии и могут не учитываться при расчете термодинамических
функций. Исследования колебательных спектров [3743а, 9696, 2019а, 2757а] и электронографиче-
ское исследование [1852а] показали4 что в основном электронном состоянии ^Х2+
молекула KrF2 имеет линейную симметричную структуру (симметрия .Do,а)- Момент инерции KrF2>
приведенный в табл. 11.2, рассчитан по вращательной постоянной (Z?o=0,126 26+0,000 15 см),
найденной из анализа структуры полосы v3 в ИК-спектре высокого разрешения газообразного
KrF2 [2757а]. Этой вращательной постоянной соответствует г (Кг—F)=1,875+0,002 А. Электроно-
графические измерения [1852а] привели к близкому значению г (Кг—F) = 1,889+0,010 А.
Значения основных частот vx и v2, приведенные
в табл. 11.2, приняты по результатам
исследований ИК-спектра и спектра КР газообразного
KrF2 [9696]. Для частоты v3 принято значение
начала полосы, определенное в работе [2757а].
Погрешности приведенных величин составляют
1—2 см для vx и v2 и 0,1 см для v3.
Термодинамические функции KrF2 (г)
вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.129) в приближении
«жесткий ротатор — гармонический осциллятор».
Погрешности рассчитанных значений обусловлены
в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых
молекулярных постоянных. Общие погрешности
в значениях Ф° (Г) при 298,15, 3000 и 6000 К оцениваются в 0,5, 3 и 5 Дж-К^-моль
соответственно; погрешность из-за неточности молекулярных постоянных составляет примерно
0,2 Дж-К^-моль-1 при 6000 К.
Термодинамические функции KrF2 вычислялись ранее в работе Нагараяна [2776а}
(Т <1 2000 К) и в справочнике [187] (Т <1 5000 К). Они согласуются с приведенными в табл. 9&
в пределах 0,2 Дж-К^-моль в значениях Ф° (Т) и S° (T) во всем интервале температур.
Имеющиеся расхождения обусловлены некоторым различием молекулярных постоянных, принятых
в расчетах.
Константа равновесия реакции KrF2 (г)=Кг (r)+2F (г) вычислена на основании значения
Дг#° @)=91,819 + 3,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
LfH°{Kx?^ г, 298,15 К) = 60,2 + 3,3 кДж • моль.
Эта величина была принята по результатам калориметрических измерений энтальпии
разложения KrF2, выполненных Ганном [1763] (см. также [1763в]). Измерения осложнялись частичным
разложением исходного образца еще до начала главного периода калориметрического опыта, что
лимитировало точность найденной величины.
Хе (г). Термодинамические свойства газообразного ксенона в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 97.
Уровни энергии атома Хе, использованные при расчете термодинамических функций, приведены
в табл. 11.1, в которой даны валентные состояния Хе, относящиеся к электронным
конфигурациям . . . 5s25p6, . . . 5s25p55d, . . . 5s25p54/ и ... 5s25p55/. Энергии этих состояний приняты по
рекомендации Мур [2697]. Состояния, относящиеся к электронным конфигурациям . . . 5s5p4f,
. . . 5s5p65d и ... 5s5pe5/, имеют, по-видимому, высокие энергии возбуждения, что позволило
не принимать их во внимание при расчете термодинамических функций.
Термодинамические функции Хе (г) были вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.23)—A.25). Погрешности вычисленных величин до температур порядка 6000 К определяются
главным образом неточностью фундаментальных постоянных и не превышают 0,02 Дж-К-моль
223
Хе+(г), Хе2 (г)
Главя 11
а значениях Ф° (Т). При более высоких температурах погрешности возрастают из-за
пренебрежения ридберговскими состояниями и могут достигать 0,5 Дж -К -моль в значениях Ф° B0 000 К).
Ранее таблицы термодинамических функций Хе (г) вычислялись в работах [1946] (до 10 000 К),
12291] (до 8000 К) и в предыдущем издании настоящего справочника [88] (до 6000 К). Результаты
этих расчетов совпадают с величинами, вычисленными в настоящем издании в пределах указанной
выше погрешности.
Газообразный ксенон принимается в качестве стандартного состояния этого элемента и в
соответствии с этим
Д/Я°(Хе, г) = 0.
Хе+ (г). Термодинамические свойства газообразного положительного иона ксенона в
стандартном состоянии при температурах 298,15—20 000 К приведены в табл. 98.
Уровни энергии иона Хе+, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой даны валентные состояния Хе+, относящиеся к электронным
конфигурациям . . . 5s25p5, . . . 5s5pe и ... 5s25p45d. Энергии этих состояний приняты по
рекомендации Мур [2697]. Состояния, относящиеся к электронным конфигурациям. . . 5s25p44/,
. . . 5s25/>45/, . . . 5s25p45?, . . . 5s5p54/, . . . 5s5p55d, . . . 5s5pb5f и ... 5s5p55g, имеют,
по-видимому, высокие энергии возбуждения, и их можно не принимать во внимание при расчете.
Термодинамические функции Хе+ (г) вычислены по уравнениям A.3)—A.6), A.9), A.10), A.23) —
A.25). Погрешности вычисленных величин до температур порядка 10 000 К определяются главным
образом неточностью фундаментальных постоянных и не превышают 0,02 Дж-К^-моль. При
более высоких температурах погрешности возрастают из-за пренебрежения ридберговскими
состояниями и могут достигать 0,1 Дж-К^-моль в Ф° B0 000 К).
Ранее таблицы термодинамических функций Хе+ (г) вычислялись в работах [1723] (до 20 000 К)
и [1946] (до 10 000 К). Результаты этих расчетов совпадают с вычисленными в настоящем издании
в пределах указанных выше погрешностей.
Константа равновесия реакции Хе+ (г)+е=Хе(г) вычислена с использованием величины
АГН° @) =—1170,355 + 0,001 кДж-моль в соответствии с принятым в справочнике значением
энтальпии образования Хе+ (г), равным потенциалу ионизации атома Хе
Д/йг°(Хе+, г, 0) = /0(Хе) = 97834,0 ±0,1 см= 1170,355 ± 0,3I кДнс • моль.
Это значение /0 (Хе) рекомендовано Мур [2696] на основании спектрального исследования
Петерсона [2997].
Хе2 (г). Термодинамические свойства газообразного двухатомного ксенона в стандартном
состоянии в интервале температур от 100 до 1000 К приведены в табл. Д. 17.
Молекулярные постоянные Хе2 приведены в табл. 11.3. Молекула Хе2 имеет основное
электронное состояние 0^, коррелирующее с атомами ксенона в основном состоянии 1S0. Образование
молекулы Хе2 в этом состоянии обусловлено только ван-д зр-ваальсовым взаимодействием между
Таблица 11.3. Молекулярные постоянные Хе2 и XeF
Молекула
Состояние
Те
Хе2 Z0+» 0
XeF1 Х22+» 0
Примечание. Ниже все величины
21,07
234
(кроме v
= D.(Xe.2)
6 вычислено по соотношению A.38);
найдено из условия Fv — _
VrtTi1. & ti .Q T ¦ 4 9^ n -
-Л.ВГ . ^max — » lim —' liJi "o —
-4,(hm) =
D»(XeF)=
6 вычислено по соотношению A.38);
найдено из условия fv = .
_./, Си*)
0,63
12
Ве
CM
ах ¦ 103
0,01345е 0,3B 0
0,192*
та* и him) ДДНЫ В СМ.
^=195,5, Я1=
в вычислено
= tfmax(/lim)-
= 1144, ог = 5
в вычислено
= С* max (•'lim)
3,3342-10~3,
по A.69);
91579-Ю-2,
по A.69); J
6,5В 5
а2 = 1,12635-Ю-7, а3
г вычислено по A.68);
а2 = 4,481 79-10"», а3
вычислено по A.68);
•10?
,22 г
,2Г
= —6
Я,=
= —9
4
2
,184 16 •
= -2,7-
,95828-
—9,51 •
г.
А
,37
,3
103"
ю-18!
Ю-11;
ю-12,
224
Благородные газы и их соединения
ХеО8(г)
атомами Хе. Возбужденные электронные состояния Хе2 имеют энергии свыше 70 000 см
[1537а] и не учитывались при расчете термодинамических функций Хе2. Приведенные в табл. 11.3
значения колебательных постоянных Хе2 получены в работе Фримана и др. [1537а] в
результате анализа колебательной структуры двух систем полос 0^ -> XQ* и 1а -> Х0+д (y"=0-f-9).
На основании измеренных значений AG>+i/2 и предположения, что отталкивательная часть
потенциальной кривой состояния Х22+ может быть описана уравнением U (r)=De—С6/г6, авторы
работы [1537а] рассчитали энергию диссоциации Хе2 и колебательные уровни этого состояния вплоть
до диссоциационного предела. Соответствующие значения Go (v) приведены в табл. 11.4. Эти
значения хорошо согласуются с полученными при анализе рассеяния молекулярных пучков ксенона
в работах [1272а, 3646а]. Экспериментальные данные о вращательных постоянных Хе2 отсутствуют.
Приведенная в табл. 11.3 вращательная постоянная Ве вычислена по значению ге (Хе2)=4,37 +
+0,02 А, оцененному в работах [475а, 1537а] на основании подгонки различных потенциальных
функций для описания теплофизических свойств ксенона, а также по величине ге (Аг2) и
межатомным расстояниям Аг—Аг и Хе—Хе в соответствующих кристаллах.
Термодинамические функции Хе2 (г) рассчитаны без учета возбужденных электронных
состояний. Значения <?ffi яр и ее производных были вычислены по уравнениям A.73)—A.75)
непосредственным суммированием по колебательно-вращательным уровням энергии. Энергии
уровней вычислялись по соотношению Go (v)-\-BfJ (/+1)—DeJ2 (J-{-lJ-{-HeJ3 (J-\-lK со значениями
Go (v) из табл. 11.4 и вращательными постоянными из табл. 11.3. В расчете учитывались все уровни,
ограниченные предельной кривой диссоциации; в табл. 11.3 приведены соответствующие значения
/нш и коэффициентов я,-.
Таблица 11.4. Уровня колебательной энергии Хе2 (в см)
V
0
1
2
3
4
5
Go И
0
19,90
38,45
55,65
71,82
86,45
V
6
7
8
9
10
Go (*)
100,15
112,78
124,11
134,26
143,21
V
11
12
13
14
15
Go И
151,04
157,83
163,66
168,54
172,70
V
16
17
18
19
20
Go И
176,07
178,77
180,87
182,45
183,59
V
21
22
23
24
25
Go И
184,35
184,81
185,05
185,14
185,15
Погрешности вычисленных термодинамических функций обусловлены главным образом
неточностью энергии диссоциации Хе2 и величины ге. Они составляют 0,7—0,8 Дж-К^-моль
в значениях Ф° (Т) и S° (T).
Ранее таблицы термодинамических функций Хе2 не публиковались.
Значение энергии диссоциации
Do (Хе2) = 185,16 ± 3 см = 2,22 ± 0,04 кДж • моль
принято по работе Фримена и др. [1537а], в которой это значение получено на основании
экстраполяции колебательных уровней Хе2 с приведенным выше потенциалом. Это значение Do (Xe2)
хорошо согласуется с величинами, найденными в различных работах при анализе
теплофизических свойств ксенона (см. [475а]).
Принятому значению Do (Xe2) соответствует
AfH°(Xe2, г, 0) = —2,22 + 0,04 кДж • моль.
ХеО3 (г). Термодинамические свойства газообразного триоксида ксенона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 99.
Молекулярные постоянные ХеО3, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Возбужденные электронные состояния ХеО3 должны иметь высокие
энергии [38766 ] и не рассматриваются в справочнике. Исследования колебательных спектров твердого
и жидкого ХеО3 [3456а, 969г] и рентгеноструктурное исследование [36406] показали, что
молекула ХеО3 имеет структуру тригональной пирамиды (симметрия С3„). Приведенное в табл. 11.5
225
ХеО4(г)
Глава 11
Таблица 11.5. Значения молекулярных постоянных и а, принятые для расчета термодинамических
функций XeOs, ХеО4, XeF4, XeFe, XeO2F2, XeO3F2 и XeOF4 (рх = Ц
Молекула
ХеО3
ХеО4
XeF4
XeF,
XeO2F2
XeOsF2
XeOF4
Состояние
vx
v2
vs
v4
ve
v7
v8
CM
XxAt 780
lUj 775,7
X*Alg 554,3
X M1? 624
X1A1 848
ХгА\ 806,7
ХгАг 926,3
344
267B)
291
630
490
567,4
576,9
833 B)
879,2 C)
218
302
333
631,7
285,9
317B)
305,9 C)
216
156
198
375,4
225
.
—
524
156
324
894 B)
543
_
—
586 B-)
506B)
905
318 B)
219
_
—
161 B)
557 B)
324
190B)
609 B)
_
—
—
48B)
585
361 B)
362 B)
_
— —
— —
252B) 156B
317 —
— —
161 B) —
Г3 • CM*
3,6
9,73
26,8
91
13,5
27,6
41,3
3
12
8
24
2
6
4
произведение моментов инерции вычислено на основании структурных параметров
кристаллического XeOs [36406]: г (Хе—О)=1,76 + 0,03 А и <О—Хе—О=103±2°. Погрешность 1А1В1С
составляет 4-Ю18 г3-см6. Основные частоты приняты по результатам исследования спектра КР
водного раствора ХеО3 [969 г]. Погрешности принятых значений v, оцениваются в 5.—10 см
на основании сопоставления основных частот молекул XeOF4 и XeO2F2 в жидком и газообразном
состояниях.
Термодинамические функции ХеО3 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический осциллятор».
Погрешности рассчитанных значений при низких температурах обусловлены главным образом
неточностью принятых молекулярных постоянных, а при высоких температурах —
приближенным характером расчета. Общие погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
оцениваются в 1, 4 и 6 Дж-К^-моль соответственно; при этом погрешность из-за неточности
молекулярных постоянных не превышает 1 Дж-К^-моль во всем интервале температур.
Термодинамические функции ХеО3 вычислялись ранее в работах [27716] (Т <1 2000 К) и
[2320а] (в статье приводятся данные для 298,15 К). В [27716] допущена ошибка при расчете
моментов инерции и приняты частоты для твердого ХеО3, которые меньше (на 10—30 см)
приведенных в табл. 11.5. Расхождения данных [27716] и табл. 99 не превышают 1,3 Дж -К-моль в
значениях Ф° (Т). В [2320а] использовались те же значения молекулярных постоянных, что и в
настоящем справочнике. Расхождение в 0,5 Дж-К-моль в Ф° B98, 15 К) может быть объяснено
только ошибкой, допущенной в расчетах [2320а].
Константа равновесия реакции ХеО3(г)=Хе(г)+ЗО(г) вычислена на основании значения
ДГЯ° @) = 194,67+45 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/flro(Xe03, г, 298,15 К) = 527 ±45 кДж • моль.
Принятая величина основана на калориметрических измерениях энтальпии взрывного
разложения ХеО3 (к), выполненных Ганном [1763а] (см. [17636]), откуда было получено ^fH° (XeO3,
к, 298,15 К)=401,6 + 8,4 кДж -моль, и на оцененном [17636] значении его энтальпии сублимации
Д,#°(ХеО3, к, 298,15 К) = 125,5±42 кДж-моль.
ХеО4 (г). Термодинамические свойства газообразного тетроксида ксенона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 100.
Молекулярные постоянные ХеО4, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Сведения о возбужденных электронных состояниях молекулы ХеО4
в литературе отсутствуют, однако можно предполагать, что они имеют высокие энергии.
Исследования колебательных спектров [3377а, 2057а, 2579а] и электронографическое исследование
[1762а] показали, что в основном электронном состоянии %.ХАХ молекула ХеО4 имеет тетраэдриче-
скую структуру (симметрия Td). Приведенное в табл. 11.5 произведение моментов инерции ХеО4
вычислено на основании значения г (Хе—О)=1,736+ 0,002 А, найденного методом газовой
электронографии [1762а]. Погрешность IaIbIc оценивается в 8-Ю~11в г8-см6. Основные частоты ХеО4
226
Благородные газы и их соединения XeF (г)
приняты по результатам исследования ИК-спектра и спектра КР газообразного Хе16О4 [2579а}
и находятся в хорошем согласии с более ранними исследованиями спектров ХеО4 [3377а, 2057а].
Частота v2, не наблюдавшаяся в спектрах газообразной ХеО4, оценена из сопоставления
соответствующих частот твердых ХеО4, RuO4, OsO4 и газообразных RuO4 и OsO4. Погрешности
приведенных значений основных частот оцениваются в 0,2 см для vl5 v3 и v4 и 5 см для v2.
Термодинамические функции ХеО4 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 1, 6 и 9 Дж -К -моль соответственно;
погрешность из-за неточности молекулярных постоянных не превышает 1 Дж-К^-моль.
Термодинамические функции ХеО4 вычислялись ранее в работах [2320а] (в статье приведены
только значения при Г=298,15 К) и [2579а] (Т <[ 400 К). Данные, приведенные в работе [2579а]
и в настоящем издании, практически совпадают. Расхождения с данными работы [2320а] (около
1 Дж-К^-моль в Ф° B98,15 К)) обусловлены разницей принятых молекулярных постоянных.
Константа равновесия реакции ХеО4 (г)=Хе (г)+40 (г) вычислена на основании значения
&ГН° @)=337,854+5 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(ХеО4, г, 298,15 К) = 642 ± 5 кДж • моль.
Эта величина была найдена Ганном [17636] в результате калориметрических измерений
энтальпии взрывного разложения ХеО4 (г).
XeF (г). Термодинамические свойства газообразного фторида ксенона в стандартном состоянии
в интервале температур от 100 до 1000 К приведены в табл. Д.18.
Молекулярные постоянные XeF, использованные при расчете термодинамических функций,
приведены в табл. 11.3. Согласно современным теоретическим представлениям, молекула XeF
имеет основное электронное состояние Х22+, коррелирующее с атомами Хе ^Sg) и FBA/2) [2445a,
24456] и образующееся в результате ван-дер-ваальсовского^взаимодействия между атомами
ксенона и фтора. Возбужденные электронные состояния XeF имеют энергии свыше 30 000 см
[3640] и могут не учитываться в расчетах термодинамических функций XeF (г).
В спектре испускания XeF наблюдались две системы полос, обусловленные переходами
А*Щ,-+Х*Щ, и В211-+ХЩ2 соответственно [138а, 2306а, 750а, 3640]. Переход А2Ц,-*Х*Цг
наблюдался также в поглощении в спектре XeF, изолированного в матрице из аргона
[434а]. Приведенные в табл. 11.3 значения колебательных постоянных XeF приняты по работе
Теллингхьюзена и др. [3640], авторы которой выполнили колебательный анализ системы полос
^2 +_Х22 + (у0" ^ 7, г/ <! 6). Экспериментальные данные о вращательных постоянных и величине
re (XeF) в состоянии Х22 + отсутствуют. Приведенная в таблице постоянная Ве вычислена на
основании значения ге (Хе2)=2,3+0,2 А, принятого по результатам приближенного квантовомехани-
ческого расчета [2304а] и оценок, выполненных в работах [4346, 750а].
Термодинамические функции XeF (г) рассчитаны без учета возбужденных электронных
состояний. Значения QW ^ и ее производных были вычислены по уравнениям A.73)—A.75)
непосредственным суммированием по колебательно-вращательным уровням энергии состояния Х22+,
которые находились по соотношению A.65) со значениями постоянных, приведенными в табл. 11.3.
В расчете учитывались все уровни энергии, ограниченные предельной кривой диссоциации
состояния Х22 +; в таблице приведены значения vma% и /цт, а также коэффициентов а,- в уравнении,
аппроксимирующем эту кривую. Мультиплетность уровней состояния Х22 учитывалась
статистическим весом. Погрешности вычисленных термодинамических функций XeF обусловлены
главным образом неточностью принятого значения Ве, а также энергии диссоциации. Общая
погрешность значений Ф° (Т) и S° (T) составляет 1,5—2,0 Дж-К^-моль.
В литературе отсутствуют сведения о расчетах термодинамических функций XeF (г).
Энергия диссоциации молекулы XeF
Do (XeF) = 680 + 30 см'1 = 8,1 + 0,4 кДж • моль
принята на основании ее значения в основном электронном состоянии De (XeF, X22+)=1080+
+ 30 см, вычисленного экстраполяцией колебательных уровней [3640], и энергии состояния
227
XeF2(K, ж) Глава 11
2Рч, атома фтора (см. табл. 7.1). Квантовомеханическая оценка Do (XeF, Z22+), выполненная
Краусом и Лиу [2304а], находится в удовлетворительном согласии с этой величиной.
Принятому значению Do (XeF) соответствует
Afff°(XeF, г, 0) = Ш,т ±0,5 кДж •
XeF2 (к, ж). Термодинамические свойства кристаллического и жидкого дифторида ксенона
в стандартном состоянии при температурах 5—600 К приведены в табл. Д.19.
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 11.6. В интервале от Т=0 до Тт термодинамические функции вычислены для моноклинной
модификации XeF2. При Т < 298,15 К для теплоемкости XeF2 приняты данные Осборна и др.
[2917а] E 348 К), полученные для образца чистоты 99,9% (примесь XeF4 не более 0,1%) с
точностью 5% при 5 К, 1% при 15 К и 0,1% при Т > 25 К. В работе [2917а] учитывалась поправка,
связанная с сублимацией XeF2 в свободный объем калориметра. Экстраполяция теплоемкости ниже
5 К приводит к значению S° E К)=0,0611 Дж-К^-молъ-1. Погрешности принятых значений
5° B98,15 К) и Я9 B98,15 К) — Н° @) (см. табл. 11.6) оценены в 0,12 Дж-К^-моль и
0,015 кДж -моль-1.
Уравнение для теплоемкости XeF2 (к) в интервале 298,15 К—Тт получено на основании
результатов измерений [2917а] в интервале 305—348 К с учетом принятого значения С°р B98,15 К).
Поправка на пересчет С„ к Ср не вводилась ввиду ее незначительности, а также отсутствия в
литературе данных по величине (dV/dT)p.
Температура плавления XeF2 D02,18+0,10 К) принята по данным Шрейнера и др. [3351а].
Результаты более ранних измерений A744а, 1438а, 945а] менее точны и лежат в пределах 400—
413 К. Ввиду отсутствия экспериментальных данных по энтальпии плавления и теплоемкости
жидкого XeF2 в справочнике приняты оцененные величины. В оценках была использована
аналогия в физико-химических свойствах фторидов ксенона (XeF2 и jXeF4) с фторидами хлора, брома и
иода, в частности предполагаемая близость значений энтропии плавления XeF2 (и XeF4) с
соответствующими значениями для G1F3 D6), BrF3 D3), BrF5 C5) и IF5 D0 Дж-К^-моль-1), в
среднем 41,8+8,5 Дж-К^-моль-11. Теплоемкость жидкого XeFa принята постоянной и равной 100±
+ 10 Дж-К^-моль на основании эмпирических закономерностей, обсуждавшихся в разделе 2.5.
Погрешности приведенных в табл. Д.19 значений Ф° (Г) при 298,15 и 500 К оцениваются в 0,1
и 1,0 Дж -К -моль. В литературе отсутствуют таблицы термодинамических функций XeF2 (к, ж),
за исключением таблицы для кристаллического XeF2 в интервале 5—350 К в работе [2917а].
Для XeF2 в'литературе имеются данные по энтальпии образования в кристаллическом и
газообразном состояниях и по энтальпии сублимации. Из этих трех величин энтальпия сублимации
XeF2 (к) определена наиболее точно (см. ниже), что позволяет использовать ее для выбора двух
других величин при совместном анализе данных для обеих фаз. Результаты измерений энтальпии
образования XeF2 представлены в табл. 11.7, где для каждой работы даны энтальпии образования
как в кристаллическом, так и в газообразном состояниях (значение, вычисленное с
использованием энтальпии сублимации, приведено в скобках). Как видно из таблицы, наиболее точные
результаты были получены для XeF2 (к) в калориметрических измерениях Джонсона и др. [2139];
в справочнике принимается ^значение
A/H°(XeF2, к*298,15 К) = —162,76 + 0,90 кДж • моль,
полученное в этой работе.
С этим значением хорошо согласуется энтальпия образования XeF2 (г), вычисленная при
подготовке справочника из данных Вайнстока и др. [3911а] по равновесию реакции образования этого
вещества (в работе [3911а] из тех же данных было получено —ИЗ кДж-моль-1).
Принятое в справочнике значение энтальпии сублимации XeF2 (к)
Л8*5Г° (XoFj;, к, 298,15 К) = 55,74 ±0,20 кДж - лши.,
вычислено по методу III закона на основании измерений давлений насыщенных паров XeF2
статическим методом B73—388 К, 87 точек), выполненных Шрайнером и др. [3351а]. Совпадающее
1 XeF6 имеет аномально низкую энтропию плавления (Дт5°=18 Дж-К'^моль), что вызвано, по-видимому,
наличием полиморфизма и полимеризацией гексафторида ксенона в жидком состоянии.
228
Благородные газы и их соединения
XeF2 (г)
Таблица 11.6. Принятые значения термодинамических величин для XeF2, XeF4 и XeF6
в кристаллическом и жидком состояниях
Вещество
XeF2
XeF4
XeF,
Состояние
к, монокл.
ж
к, монокл.
ж
кШ, монокл
к11, ромб.
к1, монокл
ж
ЬТ-еГ* (Д
Я° B98,15 К)—
—Н" @)
кДж-моль
15,257
—
23,030
—
[. —
—
30,999
—
ж.К-1.моль->
о(о, К)
Дж • K-i
115,090
—
167,000
—
—
210,38
—
С B98Д5К)
Р
• МОЛЬ
75,60
—
118,39
—
—
171,59
—
Коэффициенты в
уравнении для С?а
а
42,767
100,00
66,689
170,00
.—
66,64
4531.241
). 6 сЮ-6=2137,279.
6-103
110,12
—
173,40
352,0 я
—6673.7346
Интервал
температуры
К
298,15-402,18
402,18—600
298,15—390,25
390,25-600
"
298,15—322,63
322,63—500
Ttr или
Тт
К
402,18
—
390,25
—
253,80
291,80
322,63
—
&trH или
АтН
кДжХ
Хмоль
16,8
—
16,3
—
0,972
1,226
5,743
—
Таблица 11.7. Результаты определения энтальпии образования XeF2 (в кДж-моль 1)
Авторы
Метод
AfH° (XeF2, к,
298,15 К)
AfH°(XeF2, г,
298,15 К)
Свек, Флеш, 1963
[3584а]
Вайнсток и др., 1966
[3911а]
Пепекин и др., 1969
[1936]
Берковиц и др., 1971
[596а]
Джонсон и др., 1972
[2139]
Масс-спектрометрическое гзмерение потенци- (—199) —143 ±15
ала появления XeF? из X F4 A4,9 ±0,1 эВ) и
потенциала ионизации Xei''2 A2,6 ±0,1 эВ)
Исследование равновесия реакции Хе(г)+ (—164,0) —108,3
+F2 (r)=XeF2 (г) статическим методом, 774 К,
1 точка
Калориметрическое измерение энтальпии его- —185 (—129)
рания смеси XeF2 (к) и полиэтилена в
кислороде в присутствии воды
Масс-спектрометрический, фотоионивационное (—169,1) —113,4 ±2
измерение порога реакции XeF2=Xe++F~+F,
A1,481 эВ при Г=0)
Калориметрическое измерение энтальпии реак- —162,76 ±0,90 (—107,05)
ции XeF2 (k)+PF3 (r)=Xe (r)+PF6 (г) в
двухкамерной бомбе
значение по этим же данным было получено Осборном и др. [2917а], однако погрешность этой
величины в [2917а] занижена, так как при ее оценке не были учтены погрешности
использованных в расчетах термодинамических функций.
XeF2 (г). Термодинамические свойства газообразного дифторида ксенона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 101.
Молекулярные постоянные XeF2, использованные для расчета термодинамических функций,
приведены в табл. 11.2. Согласно [28616] возбужденные электронные состояния XeF2 имеют
энергии свыше 40 000 см и могут не учитываться в расчетах термодинамических функций XeF2 (г).
Исследования колебательных спектров [348а, 37306, 2019а, 3191а] показали, что в основном
электронном состоянии -X^S* молекула XeF2 имеет линейную симметричную^структуру
(симметрия 2)иI). Момент инерции XeF2, приведенный в табл. 11.2, вычислен на основании вращательной
постоянной .Во=0,113 50 + 0,000 08 см, найденной при анализе вращательной структуры полосы
vs в ИК-спектре высокого разрешения газообразного XeF2 [3191a]. Этому значению вращательной
постоянной соответствует г (Хе—F) = l,9773 + 0,0015 А. Значения основных частот vx и v2, приве-
229
XeF4(K, ж) Глава 11
денные в таблице, приняты на основании исследований ИК-спектров и спектров КР газообразного
XeF2 [37306, 348а]. Для частоты v3 принято значение начала полосы, определенное в работе
[3191а]. Погрешности принятых значений частот оцениваются в 1—2 см для vx и v2 и
0,1 см для v3.
Термодинамические функции XeF2 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.126) иАA.129) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 0,5, 3 и 5 Дж-К^-моль
соответственно; погрешности из-за неточности молекулярных постоянных не превышают 0,5 Дж-К^-моль
при 6000 К.
Термодинамические функции XeF2 вычислялись ранее в работах [3911а, 2777а, 2320а, 2917а]
(Т ^ 2000 К) и в справочнике [187] (Т ^ 5000 К). Расхождения результатов расчетов [3911а,
2777а, 2320а, 187] ив настоящем справочнике меньше 0,7 Дж-К^-моль в значениях Ф° (Т) и
S° (Г) и обусловлены отличием принятых молекулярных постоянных. Термодинамические
функции, вычисленные в [2917а], практически совпадают с приведенными в табл. 101.
Константа равновесия реакции XeF2 (г)=Хе (r)+2F (г) вычислена на основании значения
Д Н° @)=259,133 + 1,1 кДж-моль, соответствующего энтальпии образования газообразного
XeF2*
г, 298,15 К) = —107,05 + 0,92 кДж • моль.
Это значение Д J/° (XeF2, г) вычислено по принятым в справочнике энтальпиям сублимации и
образования XeF2 (к).
XeF4 (к, ж). Термодинамические свойства кристаллического и жидкого тетрафторида ксенона
в стандартном состоянии при температурах 5—600 К приведены в табл. Д.20.
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 11.6. В интервале от Т=0 до температуры плавления термодинамические функции
вычислены для моноклинной модификации XeF4. При Т < 298,15 К для теплоемкости XeF4 приняты
данные Осборна и др. [29176] F—348 К), полученные для образца чистоты примерно 99,8%
(примеси XeF2 и XeFe в количествах менее 0,1%). Точность определения теплоемкости проверена по
результатам измерений теплоемкости бензойной кислоты и составляла около 1 % при Т < 33 К
и 0,15% в интервале 33—298 К. Экстраполяция теплоемкости ниже 5 К приводит к значению
S° E К)=0,101 Дж-К^-моль-1. Погрешности принятых значений S° B98,15 К) и Н° B98,15 К) —
Н° @) (см. табл. 11.6) составляют 0,2 Дж-К^-моль и 0,025 кДж-моль.
Уравнение для теплоемкости|XeF4 (к) в интервале 298,15 — Тт К получено по данным работы
[29176] с учетом принятого значения С B98,15 К) и результатов измерений теплоемкости XeF4
в интервале 304—348 К.
Температура плавления XeF4 (с погрешностью 0,1 К) принята по данным Шрейнера и др.
[3351а]. Значения энтальпии плавления XeF4 и теплоемкости жидкого XeF4 приняты на основании
приближенных оценок (см. гл. 2) с погрешностями 3 и 15 Дж-К^-моль соответственно.
Погрешности приведенных в табл. Д.20 значений Ф° (Т) при 298,15 и 500 К оцениваются в 0,1
и 1,3 Дж-К^-моль. В литературе отсутствуют таблицы термодинамических функций XeF4 (к, ж)
за исключением таблицы для кристаллического XeF4 (см. [29176]).
В .{литературе известны независимые измерения энтальпии образования XeF4 в
кристаллическом и газообразном состояниях и энтальпии сублимации XeF4 (к). Результаты измерений
энтальпии образования представлены в табл. 11.8, где для удобства сопоставления приведены энтальпии
образования XeF4 в кристаллическом и газообразном состояниях, вычисленные по данным,
полученным в каждой из работ. При пересчетах было использовано значение энтальпии сублимации
XeF4 (см. ниже). Наиболее точное значение энтальпии образования, полученное Джонсоном и др.
[2139] в калориметрических измерениях,
A^°(XeF4, к, 298,15 К) = —267,11 +0,90 кДж • моль,
принимается в справочнике. С ним хорошо согласуется значение, вычисленное по
результатам исследования равновесия реакции образования XeF4(r), выполненного Вайнстоком и др.
[3911а].
230
Благородные газы в их соединения
XeF,(r)
Таблица 11.8. Результаты определения энтальпии образования XeF4 (в кДж-моль)
Авторы
Метод
У0 (XeF4, к,
298,15 К)
Н° (XeF4, г,
298,15 К)
Свек, Флеш, 1963
[3584а]
Стейн, Плуриен, 1963
[3512а] (см. [2139])
Ганн, Уильямсон, 1963
[17666, 1766а]
(см. [2139])
Вайнсток и др., 1966
[3911а]
Берковиц и
[596а]
др., 1971
Джонсон
[2139]
и др., 1972
Масс-спектрометрическое измерение
потенциала появления иона Хе+ из XeF4 A2,4 ±0,1 эВ)
Калориметрическое измерение энтальпии
сгорания XeF4(r) в водороде.
Калориметрическое измерение энтальпии
гидролиза XeFi(K). водным раствором KI
Исследование равновесия реакции XeF2(r)+
+F2(r)=XeF4(r) статическим методом, 523—
774 К, 5 точек
Масс-спектрометрический, фотоионизационное
измерение порога реакции XeF4=XeFf+2F
A5,22 эВ) и потенциала ионизации XeF,
A2,35 ± 0,01 эВ)
Калориметрическое измерение энтальпии
реакции XeF4 (k)+2PF3 (r)=Xe (r)+2PF5 (г) в
двухкамерной бомбе
(-265)
! [(-304)
—270
—204 ±10
-243
(—209)
(-269,8 ±3,5) —208,9 ±3,5aj
(-293 ±8)
—232 ±8
—267,11 ±0,90 (-206,19 ±1,0)
Примечание. В скобках приведены значения, вычисленные с использованием величины LSH° (XeF4).
а В работе [3911а] было найдено значение—215 кДж-моль; расхождение объясняется различием
термодинамических функций, использованных в [3911а] и настоящем справочнике. Погрешность приведенной
в таблице величины учитывает погрешности термодинамических функций XeF4 (г).
Для энтальпии сублимации XeF4 (к) в справочнике принято значение
A,ff°(XeF4. к, 298,15 К) = 60,92 ± 0,50 кДж • моль,
вычисленное по данным о давлении насыщенных паров XeF4, измеренным статическим методом
{275—390 К, 58 точек) в работе Шрейнера и др. [3351а].
XeF4 (г). Термодинамические свойства газообразного тетрафторида ксенона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 102.
Молекулярные постоянные XeF4, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Возбужденные электронные состояния молекулы XeF4 имеют энергии
свыше 40 000 см (см. [28616]) и могут не учитываться в расчетах термодинамических функций.
Исследования колебательных спектров [967а, 37306] и дифракционные измерения [694а, 3640а,
841а] показали, что в основном электронном [состоянии ХгА1д молекула XeF4 имеет структуру
плоского квадрата (симметрия Dih) 1. Приведенное в табл. 11.5 произведение моментов инерции
молекулы XeF4 вычислено для г (Хе—F) = l,94 + 0,01 А, найденного методом газовой
электронографии2. Погрешность произведения моментов инерции составляет 0,9-Ю14 г3-см6. Основные
частоты молекулы XeF4 (табл. 11.5) приняты по результатам исследований ИК-спектра и
спектра КР газообразного XeF4 [967а, 37306]. Погрешности приведенных значений vf оцениваются
в 1 см для vx, 5 см для v4 и v7 (определены из предполагаемых обертонов 2v4 и 2v7) и 2 см
для остальных частот. i
Термодинамические функции XeF4 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
{1.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) при 298, 15, 3000 и 6000 К оцениваются в_1, 7 и 10 Дж-К^-моль соответ-
1 Адаме [345а] в спектре КР твердого XeF4 обнаружил значительно больше линий, чем можно ожидать у молекулы
с симметрией Dih, и провел отнесение частот в предположении, что XeF4 кристаллизуется в бимолекулярной
ячейке с симметрией С^.
3 Близкие величины г (Хе—F) получены для XeF4 (к): 1,953+0,002 А [841а] (дифракция нейтронов) и 1,92±0,02 А
[3640а] (дифракция рентгеновских лучей).
231
XeFe(K, ж) Глава II
ственно; погрешности из-за неточности молекулярных постоянных не превышают 1 Дж -К -моль
во всем интервале температур.
Термодинамические функции XeF4 вычислялись ранее в работах [2771а, 3911а, 2320а, 29176 J
(Т < 2000 К) и в справочнике [187] (Т < 5000 К). Расхождения данных работ [2771а, 3911а,
2320а] и настоящего справочника составляют соответственно 2,5, 5 и 1 Дж-К^-моль в
значении Ф° B98,15 К) и обусловлены бблыпим или меньшим различием в принятых молекулярных
постоянных (из-за отсутствия экспериментальных данных в работе [2771а] для большинства
частот использованы основные частоты твердого XeF4 [967а], а в [3911а] эти частоты оценены).
Термодинамические функции, вычисленные в работах [29176, 187], практически совпадают с
приведенными в настоящем издании.
Константа равновесия реакции XeF4(r)=Xe(r)+4F(r) была вычислена на основании
значения АГН° @)=510,492 + 1,6 кДж-моль, соответствующего энтальпии образования
г, 298,15 К) = — 206,19 ± 1,0 кДж • моль.
Это значение &fH° (XeF4, г) основано на принятых в справочнике энтальпиях сублимации
и образования XeF4 (к).
XeF6 (к, ж). Термодинамические свойства кристаллического и жидкого гексафторида ксенона
в стандартном состоянии при температурах 5—500 К приведены в табл. Д.21.
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 11.6. В интервале от Г=0 до температуры плавления термодинамические функции
вычислены для трех энантиотропных модификаций XeF6 *.
Теплоемкости трех указанных кристаллических модификаций XeF6 приняты по работе Шрей-
нера, Осборна и др. [33516] F—320 К), где они были получены для образца чистоты примерно
99,8% XeF6. Точность измерений составляла 5% при 6 К, 1 % при 15 К и около 0,2% при Т >• 25 К
(в области фазовых переходов точность не превышала 0,5%). Экстраполяция теплоемкости ниже
5 К приводит к значению S° E К)=0,422 Дж-К^-моль. Температуры и энтальпии
полиморфных превращений XeF6: b.trH B53,8 К)=0,972+0,010 кДж-моль и &trH B91,8 К)=1,226 +
+0,010 кДж-моль, также приняты по работе [33516]. По этой же работе приняты значения
S° B98,15 К) и Н° B98,15 К) — Н° @); погрешности последних величин составляют
0,3 Дж-К^-моль и 0,05 кДж-моль соответственно.
Для теплоемкости XeF6 (к) в интервале 298,15 — Тт принято уравнение (см. табл. 11.6),
выведенное по данным работы [33516]. По тем же данным приняты значения температуры плавления
XeFe и энтальпии плавления (с погрешностями 0,1 К и 0,005 кДж-моль). Уравнение для
теплоемкости жидкого XeFe (см. табл. 11.6) получено на основании значения С° C22,63 К) =
=324,83 Дж-К^-моль-1 и величин Н° (Т) — Н° C22,63 К), найденных в работе [33516].
Погрешности приведенных в табл. Д.21 значений Ф° (Т) при 298,15 и 500 К оцениваются
в 0,1 и 12 Дж-К^-моль. В литературе отсутствуют таблицы термодинамических функций
XeFe (к, ж), за исключением приведенной в работе [33516] для интервала температур 5—350 К.
В табл. 11.9 представлены результаты измерений энтальпии образования XeFe. Эти измерения
относятся как к кристаллическому, так и к газообразному состояниям XeF6. Для удобства их
сопоставления для каждой из работ приведены энтальпии образования для обоих состояний. При
пересчетах (вычисленные величины даны в скобках) было использовано принятое в справочнике
значение энтальпии сублимации XeFe (к).
Наиболее точные определения энтальпии образования XeF6 выполнены калориметрическим
методом Джонсоном и др. [2139] для XeF6 (к) и Вайнстоком и др. [3911а] в результате
исследования равновесия реакции образования XeF6 (г). Результаты этих работ находятся в хорошем
соответствии, и в справочнике принимается среднее значение
А,Н°(Хе?6, к, 298,15 К) = —338,0 +2,3 кДж ¦ моль.
Принято также значение энтальпии сублимации
As#°(XeFev_K, 298,15 К) = 60,8 + 1,0 кДж • моль.
1 Согласно данным [33516] полиморфные превращения XeF6 протекают в некоторых интервалах температур B45 —
255 К и 290—295 К соответственно), однако при расчетах термодинамических функций XeF6 (к) было принято,
что эти превращения происходят изотермически.
232
Благородные газы и их соединения
XeFg(r)
Таблица 11.9. Результаты определения энтальпии образования XeFe (в кДж-моль)
Авторы
Метод
H° (XeFe, к,
298,15 К)
Я° (XeF6, г,
298,15 К)
Стейн, Плуриен, 1963
[3512а] (см. [2139])
Вайнсток и др., 1966
[3911а]
(-410)
-337,7+2,2)
Селф, 1969
(см. [2139])
Берковиц и
[596а]
[3385]
ДР-, 1971
-588
(-282 ±25)
-349
-276,9 ±2,0 =
(-527)
—343 ±25
Калориметрическое измерение энтальпии
сгорания XeF6 (г) в водороде
Исследование равновесия реакции XeF4(r)+
+F2(r)=XeFe(r), статическим методом, 523—
673 К, 4 точки
Калориметрическое измерение энтальпии
реакции XeFe(K)+3PF3(r)=Xe(r)+3PF5 (г)
Масс-спектрометрический, фотоионизационное
измерение порога реакции XeFe=XeF4+2F
A5,67+0,03 эВ при Т=0) и потенциала
ионизации XeF4 A2,65+0,25 эВ)
Калориметрическое измерение энтальпии
реакции XeF, (k)+3PF3 (r)=Xe (r)+3PF6 (г) в
двухкамерной бомбе
Примечание. В скобках приведены значения, вычисленные с использованием величины Д,Я° (XeF4).
а В работе [3911а] было вычислено значение—295 кДж-моль. Расхождение объясняется различием
использованных в расчетах термодинамических функций. С учетом погрешностей термодинамических функций
указанная в таблице погрешность должна быть увеличена до 9 кДж-моль.
Джонсон
[2139]
и др., 1972
—338,2+2,3 (-277,4 ±2,5)
Принятое значение было вычислено по методу III закона по результатам измерений Шрейне-
ром и др. [33516] давления паров над кристаллическим и жидким XeF6 (статический метод, 254—
350 К, И точек).
XeFe (г). Термодинамические свойства газообразного гексафторида ксенона в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 103.
Молекулярные постоянные XeFe, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Возбужденные электронные состояния молекулы XeFe имеют энергии
свыше 30 000 см [28616, 969а] и могут не учитываться в расчетах термодинамических функций
XeF6 (г). В результате одновременного анализа экспериментальных данных по дифракции
электронов [1574а, 5006, 1873а, 1852а], КР-, ИК- и УФ-спектрам [2242а, 969а, 2861а], отклонению
молекулярного пучка в электрическом поле [1438] и энтропии XeF6 [3351b], а также результатов
теоретического расчета в приближении модели кристаллического поля [3876а], Питцер и Берн-
стейн [3032а] пришли к выводу, что в основном электронном состоянии молекула XeF6 имеет
искаженную октаэдрическую структуру. В результате псевдовращения, обусловленного наличием
деформационных колебаний с большой амплитудой, в молекуле XeF6 происходит переход между
различными конфигурациями (главным образом симметрии C3l и С2,)- Из-за сложности
представления такой динамической модели, Питцер и Бернстейн [3032а] предложили принять для XeFs
статическую модель, соответствующую слегка искаженному октаэдру (симметрия С3с) со
значительными деформационными колебаниями типа flu. Этой структуре XeF6 соответствуют два типа связи
Хе—F A,850 и 1,941 А), три типа коротких расстояний F. . . F B,498, 2,535 и 3,106 А) и один тип
длинных расстояний F. . . F C,757 А). Предложенная в работе [3032а] структура молекулы XeF6
принята в справочнике. Погрешность в произведении моментов инерции, рассчитанном по этим
значениям структурных параметров (см. табл. 11.5), составляет 1,5 Л0~1Ы г3-см6. Основные
частоты молекулы XeF6, приведенные в таблице, приняты (кроме v8) по работе [3032а], где было
предложено новое отнесение спектроскопических данных Классена и др. [969а, 2242а] 1 на осно-
Классен и др. [969а, 2242а ] исследовали ИК-спектры и спектры КР газообразного XeF6 и молекул XeFe,
изолированных в матрице инертного газа. Кристаллические модификации XeFe исследованы методами рентгенострук-
турного анализа и нейтронографии [2164а, 21646, 835а, 8356, 3486]; они являются полимерными структурами,
построенными из тетрамеров и гексамеров с фторными мостиками, в которых отдельные молекулы XeFe теряют
свою индивидуальность. Жидкий XeFe, как следует из данных спектра КР [1570а], является смесью полимерных
и мономерных молекул.
233
XeO2Fa (г) Глава 11
вании принятой структуры искаженного октаэдра. При этом для основных частот vlt v2, v3, v6,
v7 и v9 приняты значения из ИК-спектров и спектров КР молекул XeFe, изолированных в матрице
[969а], а значения неактивной в ИК-спектре и спектре КР частоты v6, а также частот
v4 и v10 оценены из разности между экспериментальным значением энтропии S° C35 К) =
=402,58Дж-К -моль [3351 в] и рассчитанными для Т=335 К вкладами энтропии,
обусловленными поступательным движением и вращением B73,2 Дж-К-моль), колебаниями v±, v2, v3,
V6> V7 и V9 C9,5 Дж-К-моль) и псевдовращением D2,8 Дж-К-моль). Вклад энтропии
псевдовращения был вычислен по потенциалу, предложенному Бартеллом [500а] на основании
анализа данных по рассеянию электронов на XeF6. В настоящем справочнике эффект
псевдовращения учитывался частотой v8=48 см, оцененной из соответствующего вклада энтропии,
вычисленного в работе [3032а] х. Погрешности приведенных в табл. 11.5 значений частот оцениваются в 5—
10 см для экспериментальных величин (что соответствует предполагаемому матричному сдвигу)
и около 20% от соответствующих значений для оцененных частот.
Термодинамические функции XeFe (г) были вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета; они достигают 7, 16 и 20 Дж-К^-моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К (в том числе из-за неточности молекулярных постоянных
6—9 Дж-К^-моль соответственно).
Термодинамические функции XeF6 (г) вычислялись ранее в работах [3911а] (только S° B98,15 К)),
[2320а] (в статье приведены только термодинамические функции при 298,15 К) и в справочнике
[187] (Т ^ 5000 К). В работе [3911а] рассчитаны значения энтропии при 298,15 К для
статических моделей различной симметрии. Наименьшее расхождение со значением S0 B98,15 К),
приведенным в табл. 103, достигает 20 Дж-К^-моль для модели симметрии Сг. В работе [2320а]
для молекулы XeF6 принята симметрия Сх; отнесение частот было проведено на основе имевшихся
спектроскопических данных и оценок самих авторов. Расхождения данных работы [2320а] и
табл. 103 составляют 13 Дж-К^-моль в S° B98,15 К) и 2 Дж-К^-моль в Ф° B98,15 К) и
обусловлены главным образом пренебрежением псевдовращением (т.е. наличием низкой частоты
колебания) в молекуле XeF6. В работе [187] термодинамические функции XeFe рассчитаны по
данным Классена и др. [969а], давших интерпретацию наблюдаемых колебательных спектров
XeFe на основе гипотезы Гудмана [1684в], предполагающей для XeFe существование трех
молекулярных изомеров, отличающихся электронным состоянием и структурой. В настоящее время
показано [2861а, 3876а, 3032а], что эта гипотеза является ошибочной. Расхождения данных работы
{187] и настоящего издания увеличиваются с ростом температуры от 13 до 32 Дж-К^-моль
в значениях Ф° (Т) и от 25 до 34 Дж-К^-моль в значениях S° (T).
Константа равновесия реакции XeFe (r)=Xe (r)+6F (г) вычислена на основании значения
АГН° @)=735,674+3,1 кДж-моль, соответствующего энтальпии образования
A^XeFg, г< 298,15 К) = —277,2 + 2,5 кДж • моль.
Эта величина была рассчитана по принятым в справочнике значениям энтальпий сублимации
и образования XeF6(K).
XeO2F2 (г). Термодинамические свойства газообразного диоксид-дифторида ксенона в
стандартном состоянии при температурах от 100 до 6000 К приведены в табл. 104.
Молекулярные постоянные XeO2F2, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Энергии возбужденных электронных состояний молекулы XeO2F2 должны
быть достаточно велики [38766] и они могут не учитываться в расчетах термодинамических
функций этого газа. Из спектров КР твердого и жидкого XeO2F2 и ИК-спектра молекул XeO2F2,
изолированных в матрице [967в], а также нейтронографического исследования [29966] установлено, что
молекула XeO2F2 в основном электронном состоянии Х1А1 имеет структуру тригональной бипи-
рамиды симметрии С2, (атомы фтора находятся в аксиальных положениях, два из трех
экваториальных положений занимают атомы кислорода, а одно остается свободным). Приведенное в табл. 11.5
1 В табл. 11.5 дано отнесение частот колебаний в предположении симметрии С3„ однако приведено число
симметрии о=24, что связано с псевдовращением молекулы и переходом от одной равновесной конфигурации к
другой через октаэдрическую конфигурацию.
234
Благородные газы и их соединения XeO3F2(r)
произведение моментов инерции вычислено по структурным параметрам, определенным в нейтро-
нографическом исследовании [29966]: г (Хе—О)=1,714+0,004А, г (Хе—F)=1,899+0,003А,
^tO—Хе—0=105,7+0,3°, ^О—Хе—F=91,6+0,l° и -4F—Хе—F=174,7+0,4°; его погрешность
оценивается в 1 -1014 г3- см6. Основные частоты молекулы XeO2F2, кроме v5, приняты по данным
Классена и др. [967в]. При этом значения частот vx, v6—v9 были получены из ИК-спектра
молекул XeO2F2, изолированных в матрице, а не наблюдавшиеся в ИК-спектре частоты v2, v3 и v4 —
из спектра КР жидкого XeO2F2. Результаты расчетов частот колебаний XeO2F2, выполненные
Виллеттом и др. [3950а] в приближении модифицированного силового поля Юри—Брэдли,
ставят под сомнение предложенное в работе [967в] отнесение частоты деформационного колебания v8.
Вычисленное значение v5=324 см" значительно больше найденного в работе [967в] по слабой
полосе 224 см в спектрах КР твердого и жидкого XeO2F2. Поскольку для других частот XeO2F2,
а также для частот молекул XeO3F2 и XeOF4 в [3950а ] было достигнуто хорошее согласие
рассчитанных и наблюдавшихся частот, то для v5 в табл. 11.5 приведено значение, рассчитанное
в [3950а]. Погрешности принятых частот оцениваются в 20 см для v6 и 5—10 см для остальных
частот.
Термодинамические функции XeO2F2 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и в меньшей степени неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) првB98,15, 3000 и 6000 К оцениваются в 1, 6 и 9 Дж -К -моль соответственно,
при этом погрешность из-за неточности молекулярных постоянных не превышает 2 Дж -К -моль.
Термодинамические функции XeO2F2 вычислялись ранее в работе [2320а] (в статье приведены
только значения для 298,15 К). Расхождения данных, приведенных в работе [2320а], с
приведенными в табл. 104 составляют 0,5 Дж -К -моль в значении Ф° B98,15 К) и обусловлены отличием
принятых структурных параметров и частоты v5 в XeO2F2.
Константа равновесия реакции XeO2F2 (г)=Хе (г)+20 (r)+2F (г) вычислена по принятому
в справочнике значению
ДгЯ° @) = 384 + 30 кДж • моль;
эта величина была оценена в предположении о равенстве средних энергий соответствующих
связей в молекулах XeO2F2, XeO3 и XeF6. Принятому значению энергии атомизации соответствует
Д/ИХеО^, г, 298,15 К) = 257,27 + 30 кДж • моль.
XeO3F2 (г). Термодинамические свойства газообразного триоксид-дифторида ксенона в
стандартном состоянии при температурах от 100 до 6000 К приведены в табл. 105.
Молекулярные постоянные XeO3F2, использованные для расчета термодинамических
функций, приведены в табл. 11.5. Сведений о возбужденных электронных состояниях молекулы XeO3F2
в литературе не имеется. Из данных по ИК-спектрам и спектрам КР молекул XeO3F2,
изолированных в матрице [969в], установлено, что в основном электронном состоянии %}А\ молекула XeO3F2
имеет структуру тригональной бипирамиды (симметрия D3h). Экспериментальных данных о
структурных параметрах молекулы XeO3F2 нет. На основании сопоставления с длинами связей Хе—F
и Хе—О в XeOF4 и XeO2F2 в справочнике для молекулы XeO3F2 принято г (Хе—F)=l,90+0,02 A
и г (Хе—О) = 1,71 +0,02 А. Погрешность произведения моментов инерции XeO3F2, вычисленного
по этим параметрам, составляет 2 • 10~114 г3 • см6. Приведенные в табл. 11.5 основные частоты
XeO3F2 приняты по данным Классена и др. [969в], которые исследовали ИК-спектр и спектр КР
XeO3F2 методом матричной изоляции. Погрешности в этих значениях частот оцениваются в 5—
10 см.
Термодинамические функции XeO3F2 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 1, 8 и 12 Дж-К-моль
соответственно; при этом погрешность из-за неточности молекулярных постоянных не превышает
2 Дж-К^-моль.
235
XeOF4 (г) Глава 11
Термодинамические функции XeO3F2 вычислялись ранее в справочнике [187] (Т1^ 5000 К)
по тем же молекулярным постоянным, что и в настоящем издании. Результаты обоих расчетов
совпадают.
Константа равновесия реакции XeO3F2 (г)=Хе (г)+30 (r)+2F (г) вычислена по принятому
в справочнике значению
ДгЯ°@) = 480 + 50 кДж • моль.
Эта величина была оценена на основании сопоставления энергий связей в молекулах XeO2F2,
ХеО3, ХеО4, XeFe, XeF4, XeF2. Принятому значению энергии атомизации соответствует
A^e(Xe03F2, г, 298,15 К) = 405,935 + 50 кДж • моль.
XeOF4 (г). Термодинамические свойства газообразного оксид-тетрафторида ксенона в
стандартном состоянии при температурах от 100 до 6000 К приведены в табл. 106.
Молекулярные постоянные XeOF4, использованные для расчета термодинамических функций,
приведены в табл. 11.5. Энергии возбужденных электронных состояний молекулы XeOF4 велики
[38766] и могут не учитываться в расчетах термодинамических функций этого газа. Исследования
колебательных спектров [9676, 555, 37306] и микроволнового спектра [2536а], а также электро-
нографическое исследование [2080а] показали, что в основном электронном состоянии Х1А1
молекула XeOF4 имеет структуру квадратной пирамиды (симметрия Civ). Произведение моментов
инерции XeOF4 (см. табл. 11.5) вычислено по структурным параметрам, определенным из МВ-
спектра [2536а]: г (Хе-О) = 1,703+0,015 А, г (Xe-F)=1,900 +0,005 А и ^ O-Xe-F=91,8 +
+0,5°. В пределах приведенных погрешностей они совпадают с результатами электронографиче-
ского измерения [2080а]. Погрешность в произведении моментов инерции составляет
1 • 10~114 г3 -см6. Основные частоты XeOF4, наблюдавшиеся в ИК-спектрах и спектрах КР
газообразного и жидкого XeOF4 [9676, 555, 37306], хорошо согласуются друг с другом. Приведенные
в таблице значения приняты по результатам исследования спектра КР газообразного XeOF4
[37306]. Значение ненаблюдавшейся в спектре КР частоты v7 получено в работе [9676] по ИК-
спектру, а частота v6 вычислена по силовым постоянным XeOF4. Погрешности
приведенных значений частот оцениваются в 1 см для \—v3, 10 см для ve и 1—3 см для остальных
частот.
Термодинамические функции XeOF4 (г) вычислены по уравнениям A.3)—A.6), A.9), A.10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных значений обусловлены в основном приближенным характером расчета
и, в меньшей степени, неточностью принятых молекулярных постоянных. Общие погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К оцениваются в 2, 10 и 13 Дж-К^-моль
соответственно; при этом погрешность из-за неточности молекулярных постоянных не превышает
1 Дж-К^-моль при 6000 К.
Термодинамические функции XeOF4 вычислялись ранее в работе [2320а] (в статье приведены
значения только для 298,15 К) и в справочнике [187] (Т ^ 5000 К). Расхождение данных работы
[2320а] и настоящего издания составляет 0,4 Дж -К -моль в Ф° B98,15 К) и вызвано некоторым
отличием принятых молекулярных постоянных. Расхождения данных табл. 106 и работы [187]
C—56 Дж -К" -моль в значениях Ф° (Т)) значительно превышают расхождения из-за различия
принятых молекулярных постоянных и могут быть объяснены лишь ошибкой, допущенной в
расчетах [187].
Константа равновесия реакции XeOF4 (г) = Хе (г) + О (r)+4F (г) вычислена по принятому
в справочнике значению
АгН° @) = 560 + 20 кДж • моль.
Эта величина была оценена на основании предположения о равенстве значений средних
энергий соответствующих связей в молекулах XeOF4, XeO3 и XeF6. Ранее в работе Ганна [17636?
аналогичным методом была получена значительно отличающаяся величина, что было обусловлено
использованием неверных значений средних энергий связей.
Принятому значению энергии атомизации соответствует:
A^°(XeOF4, г, 298,15 К) = —11,458 + 20 кДж ¦ моль.
236
Благородные газы и их соединения Rn (г), Rn+(r)
Rn (г). Термодинамические свойства газообразного радона в стандартном состоянии при
температурах 100—20 000 К приведены в табл. 107.
Уровни энергии атома Rn, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой даны валентные состояния Rn, соответствующие электронным
конфигурациям . . . 6s26pe, . . . 6s26p5BPNd, . . . 6s26p5 BРM/ и ... 6s26/?5BPN/. Энергии этих
состояний приняты по рекомендации Мур [2697]; для ряда состояний, относящихся к
конфигурации . . . 6s26p6BPNd и отсутствующих в работе [2697], были сделаны оценки по аналогии с
другими атомами этой группы. В табл. 11.1 уровни, относящиеся к конфигурациям . . . 6s26pbBPMf
и ... 6s26p5BPN/, были объединены для каждой конфигурации в один уровень с суммарным
статистическим весом.
Термодинамические функции Rn (г) были вычислены по уравнениям A. 3)—A. 6), A. 9), A.10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. При Т <^ 6000 К погрешность
вычисленных значений Ф° (Т) и S° (T) обусловлена прежде всего неопределенностью принятого
атомного веса радона и может составлять 0,1 Дж К -моль. При более высоких температурах
становится существенным пренебрежение в расчете ридберговскими состояниями, а также не-
наблюдавшимися валентными состояниями, относящимися к конфигурациям . . . QsQp6BSNd,
. . . 6s6p6B?M/ и ... 6s6peBiS'N/. Общая погрешность в значении Ф° B0 000 К) может составлять
0,7—1 Дж-К^-моль.
Ранее таблицы термодинамических функций Rn (г) вычислялись Хилзенратом и др. [1946]
до 10 000 К и Поландом и др. [3055] до 8000 К. Результаты этих расчетов удовлетворительно
согласуются (в пределах 0,01 Дж -К -моль для Ф° (Т)) с вычисленными в настоящем
справочнике.
Газообразный радон принимается в качестве стандартного состояния этого элемента и в
соответствии с этим
A/He(Rn, г)==0.
Rn+(r). Термодинамические свойства газообразного положительного иона радона в
стандартном состоянии при температурах 298,15—20000 К приведены в табл. 108.
Уровни энергии иона Rn+, использованные при расчете термодинамических функций,
приведены в табл. 11.1, в которой на основании данных, рекомендованных Мур [2697], дано
дублетное расщепление основного электронного состояния 2Р. Сведения об остальных электронных
состояниях Rn+ в литературе отсутствуют.
Термодинамические функции Rn+(r) вычислялись по уравнениям A.3)—A.6), A.9), A.10),
A.23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т < 5000 К обусловлены прежде всего неопределенностью атомного веса Rn и могут
составлять 0,1 Дж-К-моль в значениях Ф° (Т) и S° (T). При более высоких температурах
становятся заметными погрешности из-за отсутствия данных о возбужденных электронных
состояниях Rn+. Общая погрешность в Ф° (Т) может достигать 0,5 Дж -К-моль.
Ранее таблицы термодинамических функций Rn+(r) вычисляли Грин и др. [1723] до 20 000 К
и Хилзенрат и др. [1946] до 10 000 К. Результаты этих расчетов практически совпадают (в пределах
0,01 Дж-К-моль) с вычисленными в настоящем издании.
Константа равновесия реакции Rn+(r)+e=Rn (г) вычислена с использованием значения
Дг#°@) =—1037,073+0,003 кДж-моль в соответствии с принятым в настоящем справочнике
потенциалом ионизации Rn
Io (Rn) = 86692,5 + 0,2 см = 1037,073 ± 0,002 кДж • моль.
Это значение рекомендовано Мур [2697] на основании спектральных данных [3161а, 1382а].
Глава 12
СЕРА И ЕЕ СОЕДИНЕНИЯ
S (к, ж), S, S", S2, S;, S3, S4, S6, Se, S7, S8, SO, SO", SO2, SO", SO3,
S2O, SH, SH", H2S, H2SO4, SF, SF-, SF2, SFS, SFJ,
SF4, SF5, SF", SF6, SOF2, SO2F2
В настоящей главе рассматриваются 31 газообразное вещество, включая серу и
соединения серы с кислородом, водородом и фтором, а также сера в конденсированном состоянии.
Для S (г) и S" (г) термодинамические свойства рассчитаны для температур до 10 000 К,
для остальных веществ в газообразном состоянии — до 6000 К. Термодинамические свойства всех
соединений серы приведены для природной смеси изотопов этого элемента (атомный вес 32,06),
однако ввиду малой распространенности изотопов серы 33S и 34S термодинамические функции всех
соединений, кроме S2 (г), вычислены по постоянным молекул, содержащих изотоп 32S. В число
рассматриваемых веществ включены практически все соединения серы с кислородом, водородом
и фтором, стабильные в достаточно широком интервале температур.
В Приложении 3 для полимерных соединений серы, SO2, SO3, H2S и SF6 приведены данные,
позволяющие оценить влияние межмолекулярного взаимодействия на отклонения свойств этих
газов от их свойств в стандартном состоянии.
S (к, ж). Термодинамические свойства кристаллической и жидкой серы в стандартном
состоянии при температурах 100—1300 К приведены в табл. 109.
Значения постоянных, использованных в расчете термодинамических функций, приведены
в табл. 12.1.
Известно большое число аллотропических модификаций твердой серы (см. [2623, 1282, 3833,
2623а]). Однако при нормальном давлении термодинамически устойчивыми являются две
кристаллические модификации: ромбическая сера в температурном интервале 0—368,54 К и моноклинная
сера — от 368,54 до 388,36 К (температура плавления). Термодинамические и другие свойства
жидкой серы резко изменяются при 433 К. Согласно современным воззрениям [1456, 3075,
2626], это явление связано с изменениями молекулярного состава жидкой серы. Ромбическая и
моноклинная сера являются молекулярными кристаллами, образованными из циклических молекул
S8. В точке плавления жидкая сера состоит из смеси циклических и цепных молекул S8. С
повышением температуры циклические молекулы S8 в жидкой сере разрываются и превращаются в цепные-
радикалы S8 *. Вблизи 433 К происходит интенсивная полимеризация цепных радикалов S8 с
образованием молекул Sn, где п порядка 10е. При более высоких температурах в жидкой сере
термодинамически устойчивыми оказываются более короткие цепи, причем с ростом температуры
содержание малоатомных молекул серы увеличивается. Спектральными исследованиями [2626}
показано, что при Т > 520 К в жидкой сере содержатся в основном молекулы S3 и S4.
Теплоемкость ромбической серы измерялась Нернстом [2834, 2832] B2,6—202 К), Истманом
и Мак-Гавоком [1370] A5—361,21 К), Уэстом [3924] C03,15—368 К), Мальцевым и Демиденко
[158] E3—305 К) и Пауковым и Березовским [193а] E—306 К) 2. Наиболее надежными являются,
по-видимому, данные Паукова и Березовского [193а], исследовавших хорошо охарактеризованный
образец ромбической серы, содержавший менее 0,001% примесей. Точность данных составляет
1 Исследования термодинамических свойств идеально чистой серы, получаемой методом зонной плавки, и образцов
серы с дозированным содержанием примесей (галогенов) показали, что процесс превращения циклических
молекул S8 в цепные радикалы происходит чрезвычайно медленно в идеально чистой сере, тогда как наличие даж&
незначительных количеств примесей катализирует этот процесс [1456].
а В справочнике Хальтгрена [2039] приводятся результаты измерений теплоемкости серы, выполненных Финке-
в температурном интервале 12 — 432 К. В 1976 г. появилось сообщение Монтгомери [2692а] об измерениях
теплоемкости ромбической и моноклинной модификаций серы. Однако численные данные, полученные в обеих
работах, пока не опубликованы.
238
Сера и ее соединения
S(k, ж)
1% при 5—20 К4ги 0,10—0,15% при Т > 30 К. Приведенные
в табл. 12.1 значения 5° B98,15 К) и Н° B98,15 К) — #°@)
рассчитаны по данным работы [193а], они тождественны
рекомендованным КОДАТА—МСНС [1000], а их погрешности
составляют 0,05 Дж -К -моль и 0,06 кДж -моль
соответственно, причем экстраполяция теплоемкости ниже 5 К дает S°
E К)=0,018 Дж-К'1-моль. Данные Истмана и Мак-Гавока
[1370] лежат ниже данных Паукова и Березовского [193а] на
10% при 15 К, на 1—2% при Т=20^-50 К и на 0,5% при
температурах 250—298 К; в интервале 50—250 К данные
работ [193а] и [1370] согласуются в пределах 0,3%. Данные
Мальцева и Демиденко [158] несколько выше принятых
(в среднем на 0,2%, за исключением интервала Т < 80 К).
Данные Нернста [2834, 2832] существенно менее точны по
сравнению с полученными в других работах.
При температурах выше 298,15 К термодинамические
функции серы вычислены по уравнениям для теплоемкости,
выведенным на основании данных Уэста [3924], полученных
при измерениях теплоемкости образца серы чистотой 99,999 %.
Точность измерений составляла 0,1—0,2%. В этой же работе
были определены принятые в справочнике значения Tir и Тт,
а также энтальпия превращения ромбической серы в
моноклинную и энтальпия плавления моноклинной серы.
Энтальпия превращения ромбической серы в моноклинную
измерялась также в работах [2841, 2691, 2413, 3605, 803, 1456,
2878, 3645, 782]. Наиболее точные результаты были получены
в измерениях Фехера и др. [1456] и авторов'работы [2878],
подтвердивших значение, найденное Уэстом. Энтальпия
плавления моноклинной серы определялась в работах [2841,
2691, 2413, 3605, 1456, 2878, 3943, 2060, 3552, 782]. За
исключением работы [1456], эти измерения проводились
на менее чистых образцах серы, чем у Уэста, и менее
точными методами. Фехер и др. [1456] нашли, что идеально
чистая сера плавится при 392,7 К с энтальпией плавления
1,608+0,008 кДж-моль, тогда как для образца серы,
содержащего 0,04 ат.% хлора, Гт=388,35 К и Д„Д°= 1,739 +
+0,004 кДж -моль. Существенное отличие значений Тт и
ЬтН° идеально чистой серы от значений этих величин для
серы, содержащей хотя бы незначительное количество
примесей, авторы работы [1456] объяснили различием структуры
молекул жидкой серы в этих условиях (циклические и
цепочечные молекулы S8 соответственно).
Наиболее надежные данные по теплоемкости жидкой
серы в интервале 388—673 К получены Уэстом [3924]. Ввиду
сложной зависимости теплоемкости от температуры и
максимума теплоемкости при 433,15 К, данные указанной
работы аппроксимированы при помощи пяти уравнений
(см. табл.12.1). Рассчитанные по этим уравнениям
значения теплоемкости жидкой серы совпадают с измеренными
экспериментально в пределах погрешности последних.
Приведенное в таблице уравнение для теплоемкости
жидкой серы при Т > 550 К основано на
экспериментальных данных Уэста и предположении, что теплоемкость
жидкой серы при Т ^> 433,15 К монотонно убывает с
возрастанием температуры, асимптотически приближаясь
к значению в точке плавления. Теплоемкость жидкой серы
№
ее
Я
S
I
к
в
а
V
се
я
I
о,
ф
н
я
SS
я
о
сё
Я
ю
2
я
се
Я"
а
и
VO
се
Н
со^
со н
m <я
Ф CD
is
S Ф
н
№
ч
н
в
я
«
м
в
СО
Я
1
13
о
00
ел
см
—' I
о
из
й
а
К
ч
о
Ч
§
к
CD
н
§
о
о
О
СМ 00
ss I I I I I
0000 I I I I I
СО 00
со со
-з< со3 'Оюю
iO СО ^^ ^н чН ^^^
сю со оо 2? со со" о
СО 00 CM CO LO Ю СО
Д 'со ii ' '
оо'оооо" сососо
СЛ CD 00 СОЮЮ
см аз
00 щ
I I
297
OCS1
-Тем" |
оо '
СО-^н
'Г I
-^1 CD -"гн 00 СО -чн СМ
СС СО О "^ О CD 00
О^-лЮО СМ* СО*
I I
со см см
¦чч СО О5
СЭ О0 OS СО 1Л О5 Oi
•«rt CM 00 CM !>• О О
О СО CD CD OS o> О
sj* CDO5 OS in 00
CM t> О С- OS GO 00
t— CO 00 CO 00 LO OO
I I
о I || I M
i
5 M II II J
- R
vo a
я 2
О И
РчО
"S
239
S(r)
Глава 12
Таблица 12.2. Уровни энергии атомов S и S",
принятые для расчета
термодинамических функций
измеряли также Брауне и Меллер [755] C93—713 К), Элсен и Элсен [2878] C88—623 К),
Иошиока [4037] C93—703 К) и Фехер и др. [1456] C88—683 К). В работах [755, 2878,
4037] измерения теплоемкости проводились с существенно менее чистыми образцами серы
и менее надежными методами. Результаты измерений Фехера и др. [1456] при температурах
выше 432 К удовлетворительно согласуются с данными Уэста; при более низких температурах
наблюдаются расхождения, вызванные неравновесностью состояния жидкой серы в опытах
[1456].
Погрешности приведенных в табл. 109 значений Ф° (Т) при 298,15, 500 и 1000 К оцениваются
в 0,05, 0,2 и 2 Дж-К^-моль.
Ранее таблицы термодинамических функций кристаллической и жидкой серы были рассчитаны
в справочниках [3564, 2220] до 700 К, [2556] до 2500 К, [2095] до 2000 К и в работе Уэста [3924]
до 717,75 К. Во всех этих таблицах
термодинамические функции ромбической серы при 298,15 К
рассчитывались по данным Истмана и Мак-Гавока
[1370], причем экстраполяция ниже 15 К
проводилась в предположении, что С (Т) — Т3, т. е.
методом, недостаточно точным в данном случае.
В справочнике Келли [2220] теплоемкость жидкой
серы принималась равной 36,53 Дж -К -моль,
в справочнике Сталла и Зинке [3564]
использовались недостаточно надежные данные Брауне и
Мёллера [755], а в справочниках [2556, 2095]
термодинамические функции жидкой серы
рассчитывались по данным Уэста [3924], причем
принималось, что при Т > 700 К теплоемкость жидкой
серы постоянна и равна С°р G00 К). Некоторые
расхождения в значениях термодинамических
функций кристаллической и жидкой серы в этих работах и табл. 109 обусловлены использованием
их авторами данных работы [1370] для Т < 298,15 К и различиями в экстраполяции данных
Уэста [3924] при Т > 700 К.
В настоящем издании расчет термодинамических свойств S (к, ж) доведен до 1300 К, поскольку
Г 1313К
Атом
Электронная
конфигурация
S
s-
Состояние
CM
Pi
. З^Зр4 3Р2 0 5
. З^Зр4 »/>! 396,09 3
. 3s23p4 SPO 573,65 1
. З^Зр4 W2 9238,58 5
. З^Зр4 ^о 22179,99 1
. З^Зр5 2Р3/ 0 4
.3sa3p6 a/>v* 482 2
р
За стандартное состояние элементарной серы в справочнике принимается ромбическая сера.
В соответствии с этим
AfHo(S, ромбич.) = 0.
Парциальное давление одноатомной серы в насыщенных парах серы, S (к, ж)=Э (г), вычислено
с использованием значения kaH° (S, ромбич., 0)=274,785+0,20 кДж-моль~\ основанного на
принятом значении энтальпии образования S (г) (см. ниже).
S (г). Термодинамические свойства газообразной одноатомной серы в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 110.
Уровни энергии атома S, соответствующие его основной электронной конфигурации,
приведены в табл. 12.2. Они приняты по работам Кауфмана и Радзиемски [2201] и Эрикссона [1411],
в которых были получены более точные данные, чем рекомендованные в справочнике Мур [2697].
Термодинамические функции S (г) вычислялись по уравнениям A.3)—A.10), A.23)—A.25)
непосредственным суммированием по уровням энергии, приведенным в табл. 12.2. Погрешности
вычисленных таким образом термодинамических функций определяются практически только
неточностью фундаментальных постоянных и не превышают 0,02 Дж-К-моль.
Ранее таблицы термодинамических функций S (г) вычислялись в работах [2692, 2217, 1673,
1428, 2198, 2291] (до Т < 15004-3000 К) и в справочных изданиях и сводках [87,|88, 2095, 2556].
В расчетах [2692, 2217] учитывалось только основное электронное состояние атома серы ,3Р.
В работах [87, 88, 2556] при расчете термодинамических функций принимались во внимание
уровни энергии, отвечающие основной электронной конфигураций атома S, а авторы справочника
[2095] учли все уровни энергии, приведенные в работе [2697]. Значения термодинамических функ-
240
Сера и ее соединения S~ (г), S2 (г)
ций S (г), приведенные во всех этих работах и табл. 110, согласуются между собой в пределах
0,02 Дж-К-моль-1.
Энтальпия образования одноатомной серы
AfH°(S, г, 298,15 К) = 277,03+ 0,20 еДж-моль
принята в справочнике в соответствии со значениями энтальпии образования S2 (г) и энергии
диссоциации S2 (см. ниже).
S~ (г). Термодинамические свойства газообразного отрицательного иона серы в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 111.
Уровни энергии S", использованные при расчете термодинамических функций, приведены
в табл. 12.2. Энергия компоненты 2А/, основного электронного состояния определена с
погрешностью 2 см Линебергером и Вудуордом [24391 при изучении зависимости сечения реакции
фотоотрыва электрона от S~ как функции энергии фотонов с помощью лазера с перестраивающейся
длиной волны.
Термодинамические функции S~ (г) вычислялись по уравнениям A.3)—A.10), A.23)—A.25)
непосредственным суммированием по уровням энергии, приведенным в табл. 12.2. Погрешности
вычисленных значений термодинамических функций обусловлены в основном неточностью
принятых фундаментальных постоянных и не превосходят 0,02 Дж -К -моль.
Таблица термодинамических функций S~ (г) публикуется впервые.
Константа равновесия реакции S~ (r)=S (г)+е (г) вычислена на основании значения АГН° @) =
=200,43 +0,06 кДж -моль в соответствии с принятой в настоящем справочнике величиной
сродства атома серы к электрону
Ао (S) = —16755 + 5 см = —200,43 ± 0,06 кДж • моль.
Эта величина получена Линебергером и Вудуордом [2439] как порог реакции фотоотрыва
электрона от S". Менее точные значения, согласующиеся в пределах их погрешностей с принятой
величиной, были получены методом фотоотрыва [750] (—199,7+7,0 кДж-моль), магнитронным
методом [401] (—201,7+6,0 кДж-моль), методом электронного удара [2837, 2838, 3242,
2302] (—212+30 кДж-моль) и методом поверхностной ионизации [28, 29] (—205,5 +
+5,0 кДж -моль).
Принятому значению сродства к электрону соответствует
AfH"(S-, г, 0) = 74,355+0,21 кДж • моль.
S2(r). Термодинамические свойства газообразной двухатомной серы в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 112.
Молекулярные постоянные, использованные при расчете термодинамических функций S2 (г),
приведены в табл. 12.3. Согласно классификации электронных состояний S2, предложенной
Барроу и Дю Парком [493], молекула S2 имеет 11 связанных валентных электронных состояний с
энергиями возбуждения, не превышающими 50 000 см1.
О молекулярных постоянных S2 в электронных состояниях, приведенных в табл. 12.3, известно
главным образом на основании изучения систем полос Z?32~—Х3Л~ [3238, 3239, 1519, 962, 1145,
3812, 2910, 2911,912, 2913, 2914, 2594, 1588, 1589, 2821, 2822, 2823, 3962, 493, 2057, 2824, 2825,
2061, 2062, 496, 2229, 2633], /1ДИ—al\ [2229, 3240, 873, 1826, 2230, 2797, 2803, 2804, 2806, 2586,
1284, 494, 3554], ^Д- — а}Ад [494, 3621], НЩ — Ъ1Ц и ?ХЕ (ХД,) — ЪЩ [3621], еШ, — cl2~ [2594],
В'йПд — А'*Аи и B'sJlg — Л3?+ [3239, 2594, 2797, 1225, 2914, 2799, 2798, 2810, 2811]. Данные
об основном состоянии Х32~ молекулы S2 были получены также в результате исследований спектра
комбинационного рассеяния [534, 537, 476, 486, 3030, 1999, 1537], микроволнового спектра [945],
инфракрасного спектра S2 в матрицах инертных газов [2622, 2624, 2625, 762, 761, 729, 509] и
1 Состояние 1ПЯ с энергией примерно 36 000 см, которое вызывает предиссоциацию в состоянии Я32«, как
показали Рикс и Барроу [3208], должно иметь неглубокий минимум при г, — 2,6 А. Известно также 8 ридберговских
состояний с энергиями, превышающими 50 000 см [493, 494, 495, 590, 1284, 1286, 3209, 3620, 3621, 3941, 2497,
2498, 2808].
241
Глава 12
Таблица 12.3. Молекулярные постоянные S2, Sj, SO, SO", SH, SH", SF, SF
Me
>ле-
ла
s2
SO
so-
SH
sh-
SF
SF"
Состояние
хъ-
ахкд
В"*Пи
ВЪИ~
В'аП
еШ
/1Д/
Х2П{
z3s-
ахД
Л'3Д
Л2+
АЧ1Г
В3И~
сзп{
ХЧ1.
Х2Ц.
Л 2Y+
V1Y+
х2п
Aw't
Т.
0»
4 500
8 500
21100
22 200 я
22 600
31500
31 835 е
36 000 *
37 000
41200
0
0»
6 350
725,668
702,35
699,7
533,6в
488,16
484,4
434
533,6в
438,32
600
1 148,19
10 509,97 1067,66
26 900
28 300
28 800
38 455,2
41628,7
42 200
0а
0»
31038,2
0
Оа
25146,8
0
.
.
я 413,3
8 630,4
170
900
2712,9
1979,8
2 600
830
6 488
830
2,844
3,09
3,37
—
2,51
2,25
2,75
—
2,70а
2,8
6,12
7,8
1,6
4,8
5,7
60,7
97,65
52
6,0
3
11,5
Примечание. Ниже все постоянные даны в см:
S2:
so-
so-
SH
sh-
SF
SF-
i
•Х = 11,82, (i =
дено значение
\, = 5,2630 + 0
ния Во и г0;
—0,0066;
D . Д А _
,0319 (v +
• Х = 3,42, A = — 0,0045.
» 4 = -350.
1 Л = —376,96;
1 вычислено по
« а2 = -0
6 вычислено
— 152* е Х-
1/2); (^'=-0
0-10-'; Д А
,022.
соотношению A.68).
Л =—398; 6 4 =—610;
о, = —2,11-10"
в вычислено
в.
см
0,29541
0,29262
—
0,235г
0,2285
—
> 0,2029
0,2244
0,2441г
0,25г
0,22704
0,277
0,72082
0,709 в
0,7026
—
—
—
0,6107
0,502
—
0,63
9,596
8,521
9,45
0,554 715
0,554
0,555
по соотношению A.68); Б
= —4,9, |а =
ах ¦ 103
1,58
1,73
—
—
1,4
—
1,8
—
1,78
1,7
5,74
6,4
19,4е
6,2
5
270
464 6
270
5,09 в
4
4,37»
приведено
= 0,00; ж Л = —130: " вд« =
00557—0,00009 _(»"+V2); e
=162; е я
а = 0,9.10-3; »
по соотношению A. 69).
D, • 10е
0,2148
0,201е
—
0,182 е
0,26
0,231 е
0,233 е
0,22 е
0,243 е
0,24
1,07*
1,15 г
5,33 *
1,28
—
1
480
660
500»
1
2,8
1
значение Дб
= -0,005.
1
1
2
2
<2
2
2
2
2
1
1
1
1
1
1
2
1
1
1
1
1
1
^ = 0,09 -10"; в приведены
вычислено по соотношению
Ге
к
,889
,898 7
,122
,15
,280
,168
,08
,08
,155 5
,95
,48108
,493 в
,500 2
,609 1
,775
,2
,58
,340 7
,423 0
,34
,599
—
,599
приве-
значе-
A.68);
спектра ЭПР [3900]. Приведенные в таблице значения молекулярных постоянных S2 в состоянии
XaIi~ рекомендованы в справочнике [3592] на основании данных, полученных в диссертации
Херста [2057], и частного сообщения Барроу и Рикса. Колебательные и вращательные постоянные
S2 в состояниях о,1 Ад и ЯД,,, приведенные в таблице, определены Карлеером и Колином [873}
на основании исследования вращательной структуры 15 полос системы ^Аи—^Д^,
соответствующих v', v" ^ 11. В спектрах S2 интеркомбинационные переходы между триплетными и синглет-
ными состояниями не наблюдались. Энергия возбуждения состояния а1 А принята по оценке
Барроу и Дю Парка [493], сделанной на основании предположения о пропорциональности энергии
возбуждения состояний а1 А молекул S2 и О2 и состояний W атомов S и О.
242
Сера и ее соединения S2 (г)
Колебательные постоянные S2 в состоянии Ь1!,*, приведенные в табл. 12.3, получены Танака
и Огава [3621] в результате анализа колебательной структуры систем полос h4i+ — Ъ1Ъ+ и
^S+^AJ—bxHi+g, наблюдавшихся в спектре испускания S2 и соответствующих v" ^9 и i/^1; энергия
возбуждения оценена Барроу и Дю Парком [493] так же, как для состояния а}Ад. Молекулярные
постоянные S2 в состояниях сх2~ и е1Пд приняты по работе Микина и Барроу [2594], в которой
проанализирована вращательная структура 0—0 и 1—0 полос системы ёЧ1д—^2", а постоянные
в состояниях AIS&U и В13П.д — по работе Нарасимхама и др. [2811], в которой исследована
система полос В'3Ид — А'3 ки. Значения колебательных постоянных S2 в состоянии Л32* вычислены
авторами настоящего издания на основании результатов исследования системы полос В'3Пд —
—AS2* в работе [2811 ]. Молекулярные постоянные S2 в состоянии 532~, приведенные в табл. 12.3,
рекомендованы в справочнике [3592] ив работе Барроу и Дю Парка [493] на основании
исследований структуры полос системы ?32~— Х32~ в работах [2061, 2062, 2822, 2823, 2914]. Состояние
В3Х~ сильно возмущено и предиссоциировано при z/=8, 9, 10 и при v' ^ 18. Рикс и Барроу
[3208] показали, что предиссоциация при v'=8, 9, 10 вызвана пересечением потенциальных
кривых состояния В3И~ и малоустойчивого состояния 1ПЯ. Предиссоциация при v' ^ 18 обусловлена
взаимодействием с состоянием ВЛи при г < ге (.В32~). Молекулярные постоянные S2 в
состоянии 2?П оценены Риксом и Барроу [3208] в результате анализа возмущений в системе полос
Энергии возбуждения состояния cxS~, а также состояний А'3АЫ и A3~Z* оценены по
соотношению A.89) с использованием принятых в настоящем издании значений энергий возбуждения
и межатомных расстояний в соответствующих состояниях О2. Вычисленные величины приведены
в табл. 12.3, их погрешности не превышают 1000 см. Приведенные в таблице энергии возбуждения
S2 в состояниях е1П, Z1 Дк и В'3П приняты на основании значений частот переходов, связывающих
эти состояния с состояниями ^2", а1Ад и AS2>+ соответственно, и энергий возбуждения последних.
Термодинамические функции S2 (г), приведенные в табл. 112, вычислены по уравнениям A.3)—
A.10), A.93)—A.95). Значения ^и и ее производных вычислялись по соотношениям A.90)—
A. 92), в которых колебательно-вращательная статистическая сумма для состояния Х32,~ и ее
производные находились непосредственным суммированием по уровням энергии этого состояния
(соотношения A. 73)—A. 75)). Составляющие возбужденных электронных состояний вычислялись
в предположении, что <?(']_ вр = (р.//?х) @т_ вр.
Колебательно-вращательные уровни энергии состояния Х311~ находились по уравнениям
A.65) и A.40). Значения коэффициентов Yik, входящих в эти уравнения и приведенных
в табл. 12.4, были получены для эффективной изотопической модификации молекул S2 по
методикам, изложенным в главе 1 (см. раздел 1.4) в результате совместной обработки экспериментальных
данных для 46 полос системы 2?32~—Х32~ (»'^9и v" <[30), полученных в работах Олсона
[2914], Ноде [2822, 2823] и Икеноуэ [2061, 2062]. В расчете учитывались все уровни энергии
состояния X3~L~, ограниченные предельной кривой диссоциации. Коэффициенты ai в уравнении,
аппроксимирующем предельную кривую диссоциации, значения vm!X и /цт для состояния Х32~,
а также значения Те для десяти рассматриваемых возбужденных состояний S2 приведены
в табл. 12.4.
Погрешности вычисленных значений термодинамических функций S2 (г) при Т ^ 500 К
обусловлены главным образом неточностью принятых значений фундаментальных постоянных; при
увеличении температуры становятся существенными погрешности, связанные с неточностью
принятых значений энергий возбуждения состояний a}l\g, b1!,* и других возбужденных электронных
состояний, в особенности в таких функциях, как S° (Т) и С° (Т). Суммарные погрешности в
значениях Ф° (Г) при 298,15, 3000 и 6000 К составляют 0,03, 0,4 и 0,8 Дж-К^-моль-1.
Ранее таблицы термодинамических функций S2 (г) вычислялись в работах [3944, 2692, 75,
75а, 1428] и справочниках [87, 88, 2095, 2556, 2496, 3564, 2670]. Во всех расчетах, кроме
проведенных в справочниках [87, 88], не принимались во внимание возбужденные электронные
состояния молекулы S2. В расчетах, проведенных в работах [2692, 1428, 3564, 87, 3944, 2556,2095, 2670],
мультиплетность состояния Х31,~ учитывалась слагаемым R 1пЗ, что обусловило расхождения
в значениях Ф° B98,15 К), приведенных в этих работах и табл. 112, до 0,65 Дж-К^-моль.
243
Глава 12
Таблица 12.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения vmax и e/lim, принятые для расчета термодинамических функций
S2, Sj, SO, SO", SH, SH~, SF, SF-
Коэффициенты]
s2a
Т, 0
У1в.1О-3 0,723 848
Уао-Ю —0,281396
Узо-Ю2 0,519181
У40.104 —1,708 58
У01 0,295 078
Уи.102 —0,176 641
УМ.1О4 0,159 956
У81.10» —0,350 616
УИ.1О» -0,3
У12.108 —
(ao=D,).lO-* 3,549 27
ах.1О2 3,709 13
аа.1О? 2,798 28
а3-1013 —2,132 26
«max 81
/цп 485
s-
ХЧ1д. i *
0
0,6
—0,28
—
—
0,277
—0,17
—
_-
—0,24
—
—
—i
—
—
107
453
SO»
Х32-«
0
1,149 47
—0,643 271
2,447 40
—5,866 97
0,720 918
—0,588 050
0,303 717
-1,176 63
-1,07
—9
4,372 71
7,014 61
11,872 4
—10,229 5
63
336
Примечание. Ниже все постоянные даны в см.
Состояние а1 Ад 64!+ сх2~
Т. 4500 8500 21100
П= 11,84, fi = —0,007.
SO: *
Состояние ^Д
Т. 6350
612+
10 509,97
6 \ = 5,2789, (л = —0,005 62.
SO": » А = — 350.
SH: » Г, D*2+) = 31 038,2
SF: »Г.И2П<) = 25146,6
А'*Аа А
so-
хт{ •
SH»
ХШ,«
0 0
0,9 2,712 9
—0,57 —6,07
— —
— —
0,63 9,586 30
—0,5 —25,143 4
— —62,233 9
— __
—1,0 —480
— —
— —
— —
— —
— —
74 21
314 740
J В"»Пи ?32-
22 200 22 600 31500 31835
сх2- А'
»Д Л'">2+ АШГ
26 900 28 300 28 880 38 455,2
. « Л = —376,96.
. « Л =—398.
sh-
Z12
0
2,6
-5,2
—
9,45
—27
—
—500
—
—
—
—
—
25
780
В'Шр
36 000
вп-
41628
SF»
хт*6
0
0,83
-0,6
—
0,554 715
—0,509
,
-1,0
—
—i
_
—
68
303
е1Ъд j
37 000 41
cm,
,7 42 200
SF-
ZJ2+
0
0,83
—1,15
—,
—
0,555
—0,437
-2,11
-1,0
—
—
—
—
35
296
200
В расчете [88] была допущена ошибка при учете мультиплетного расщепления состояния Х32~,
достигающая 4,5 Дж-К^-моль в значении Ф° B98,15 К). При более высоких температурах
различия между значениями термодинамических функций S2 (г), приведенными в работах [3564,
87, 2496, 2556, 2095, 2670] и в табл. 112, растут и достигают при 6000 К величин около 2,5, 5 и
5,5 Дж-К^-моль в значениях Ф° (Т), S° (T) и С°р (Г). Эти расхождения обусловлены главным
образом тем, что в этих работах не учитывались возбужденные электронные состояния S2.
Существенно меньше расхождения с данными справочника [88], при Т >• 2000 К они не превышают
0,4 и 0,8 Дж-К^-моль-1 в значениях Ф° (Т) и S° (Г).
Константа равновесия реакции S2(r)=2S (г) вычислена на основании значения Ь.ГН° @) =
=421,277 + 0,030 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования S2 (г) и S (г). Для энтальпии образования Ss(r) принято значение
AfH°(S2, г, 298,15 К) = 128,60+ 0,30 кДж • моль,
244
Сера и ее соединения
S,(r)
Таблица 12.5. Результаты определения энтальпии образования S2 (г) (в кДж • моль)
Автор ы
Метод
ДГЯ°(О)
II закон
III закон
bfH°(Sit г,
298,15 К)
Пройнер, Шапп,
1909 [3085]
Рандалл, Биховский,
1918 [3140]
Брауне и др., 1951
[756]
Уэст, Мензис, 1929
[3926 ]
Дроварт и др., 1967
[1226, 1326]
Pay и др., 1973
[3166]
Исследование равновесия реакции
52 (г)+2Н2 (r)=2H2S (г) в
проточной системе и статическим
методом, 1023—1405 К, 19 точек
Исследование равновесия реакции
Sa (r)+2H2 (r)=2H2S (г) в
проточной системе, 1362—1667 К, 4 точки
Измерение давления и плотности
ненасыщенного пара серы
статическим методом, 623—1273 К, 3 точки
Измерение давления насыщенных
паров серы статическим методом,
373—816 К (см. текст)
Измерение парциального давления
53 в насыщенных парах серы масс-
спектрометрическим методомс
ячейкой Кнудсена, являющейся
одновременно электрохимическим
элементом, 473—673 К, 5 точек
Измерение давления и плотности
насыщенных и ненасыщенных
паров серы статическим методом 823—
1273 К
-162,0 ±2,8 -163,45 ±0,30 128,4 ±1,0
—167 ±15 —163,46 ±0,80 128,4 ±1,3
122 ±14 128,6 ±1,2 128,9 ±1,2
129,1 ±2,7 128,30 ±0,20 128,61 ±0,20
130,5»
Вычислено авторами [3166]. Пересчет с использованием значений энтропии S2 (г) и S8 (г)
образования Sg (г), принятых в настоящем справочнике, приводит к тому же значению.
и энтальпии
основанное на результатах измерений парциального давления S2 в насыщенных парах серы,
определенных в работах Дроварта и др. [1226, 1326]. Оно хорошо согласуется с величинами,
вычисленными по результатам исследований равновесия реакции S2 (г)+2Н2 (r)=2H2S (г) в широком
интервале температур в работах Пройнера и Шаппа [3085] и Рандалла и Биховского [3140], а также
с результатами измерений давления и плотности паров серы в работах Брауне и др. [756] и Уэста
и Мензиса [3926]. В табл. 12.5 приведены результаты обработки экспериментальных данных этих
авторов, а также данных Pay и др. [3166], проведенной при подготовке настоящего издания.
Комбинация эффузионных и электрохимических измерений позволила авторам работ [1226, 1326]
установить значение коэффициента, связывающего интенсивность ионного тока S? с парциальным
давлением. Используя данные работы [3926] по давлению насыщенных паров серы и
электрохимические данные Кьюккола и Вагнера [2260], Дроварт и др. [1226, 1326] вычислили парциальные
давления S2, которым соответствует принятое значение h^H° (S2, г), практически совпадающее с
рекомендованным КОДАТА—MGHG [1000] A28,49 + 0,30 кДж • моль). Следует отметить, что его
погрешность в значительной степени определяется неточностью термодинамических функций S (к, ж).
Погрешности значений AJ7° (S2, г), найденных из данных работы [3140] и особенно [3085] по равновесию
реакции образования H2S (г), в значительной степени обусловлены неточностью принятого в
справочнике значения AfH° (H2S, г). Точность определения b.fH° (S2, г) из данных по давлению и
плотности паров серы, полученных статическими методами, существенно лимитируется
трудностью определения равновесных концентраций молекул S2. Константы равновесия реакций
диссоциации компонентов паров серы, вычисленные по данным работы [756], совместно с данными
по давлению насыщенного пара серы [3926] приводят к значению &jH° (S2, г), хорошо
согласующемуся с найденными другими методами. Однако в работе Pay и др. [3166] таким же методом было
получено более высокое значение, отличающееся на 2 кДж-моль от результатов других
исследований. Это расхожение может быть обусловлено тем, что авторы работы [3166] использовали
для обработки экспериментальных данных существенно отличающиеся значения
термодинамических функций компонентов паров серы и одновременно выбирали значения нескольких величин,
245
Sj-(r) Глава 12
что не могло не сказаться на точности их определения. По-видимому, расхождение в 2 кДж-моль
характеризует погрешность значения, полученного в работе [3166].
Результаты измерений давления и плотности паров серы в работах [3084, 2265, 673, 1774]
представляются менее надежными и не использовались для выбора значения АуН° (S2, г).
Значение энергии диссоциации
D0(S2) = 35 216,4 + 2,5 см = 421,282 + 0,030 кДж • моль,
использованное для выбора энтальпии образования S2 (г), было получено Риксом и Барроу [3208]
на основании анализа предиссоциации при вращении молекул S2S2, 34S2 и 32S 34S в состоянии ?32~.
Рикс и Барроу нашли по предельной кривой диссоциации состояния В3И~ энергию диссоциаци-
онного предела этого состояния C5 999 + 2,5 см) и показали, что состояние 53Е~ коррелирует
с атомами S CP2)+S CРХ). Найденное Риксом и Барроу значение Do (S2) удовлетворительно
согласуется с данными Берковица и Чапки [593], полученными при фотоионизационных измерениях
D22,6+0,8 кДж-моль) и Будининкаса и др. [829], полученными в эффузионно-торзионных
измерениях D26 + 12 кДж-моль). Результаты более ранних измерений были проанализированы
Дровартом и Голдфингером [1325] и Гейдоном [1579], рекомендовавшими значение 423 +
+4 кДж-моль.
S7 (г). Термодинамические свойства газообразного отрицательного иона двухатомной серы
в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 113.
Молекулярные постоянные, использованные при расчете термодинамических функций Sg (г),
приведены в табл. 12.3. Результаты исследований спектров флуоресценции и поглощения
примесных ионов S~ в кристаллах KI, выполненных Ролфом и др. [3234, 3786, 2063], а также аналогия
с изоэлектроннымн молекулами SF и СЮ, позволяют принять, что основным электронным
состоянием молекулы S^ является состояние Х2П(, а первым возбужденным состоянием — состояние
А2ПГ. Приведенные в табл. 12.3 значения колебательных постоянных S% в электронных
состояниях Х2П{ и А2ЛГ, а также величина энергии возбуждения состояния A2tlr оценены на основании
данных, полученных Белла и Ролфом [3786] для примесных ионов Sj в кристаллах KI, с учетом
величины Д6п/2 (S^, Х2П(), найденной при анализе сечения фотоотрыва электронов от ионов S~l
[919]. Погрешность принятого значения ше в состоянии Х2И{ составляет 25 см. Погрешность
значения Те для состояния А2ПГ оценивается в 500 см. Постоянная спин-орбитальной связи А
для Sj в состоянии Х2И{ оценена по величине спин-орбитального расщепления примесных ионов
S^ в кристалле KI [3786] и принятых в справочнике значений постоянной А изоэлектронных
молекул SF и СЮ. Приведенное в табл. 12.3 значение вращательной постоянной Ве в состоянии
Х2П4 вычислено по оцененному значению ге. Оценка была выполнена при подготовке справочника
на основании сравнения колебательных постоянных и энергий диссоциации молекул S2 и Sj,
а также значений г (S—S) в молекулах S2 и S2C12 [1951] и г (S—С1) в молекулах SC12 [2707] и
SF5C1 [2231]. Погрешность оцененного значения не превышает 0,05 А.
Термодинамические функции S~ (г) были вычислены по уравнениям A.3)—A.10), A.93) —
A.95). Значения ^вн и ее производных находились по соотношениям A.90)—A.92), в которых
QkoI вр' Qiox вр и ® вр вычислялись непосредственным суммированием по колебательным уровням
энергии и интегрированием по значениям / от 7mln до /maXi, с помощью соотношений типа A.82).
Колебательно-вращательные уровни энергии S~ в состоянии Х2И( находились по уравнениям
A.65) и A.42), значения коэффициентов Ykl и постоянной А которых приведены в табл. 12.4;
там же приведены значения ушах и /Иш. При расчете значений Qm, Qm и Qm было принято, что
QW vV=={PjPx)Qml.*f Погрешности табулированных значений термодинамических функций при
температурах до 1000 К в основном обусловлены неточностью оцененных значений гв и
постоянных спин-орбитального взаимодействия, в интервале от 1000 до 4000 К неточностью оцененных
значений коэффициентов Ykl, выше 4000 К к этому добавляются погрешности, обусловленные
применением приближенной методики расчета и отсутствием данных о возбужденных состояниях
S^. Общие погрешности в значениях Ф° (Т) оцениваются в 0,8, 1 и 2 Дж-К^-моль при 298,15,
3000 и 6000 К.
Ранее термодинамические функции S^ (г) не публиковались.
Константа равновесия реакции S~ (г) = 2S (г) -\-е (г), приведенная в табл.113, вычислена
на основании значения АГН° @) =581,277 + 4,0 кДж-моль, которое соответствует принятому
246
Сера и ее соединения S, (г)
в справочнике сродству молекул S2 к электрону
Ао (S2) = —1,663 + 0,040 эВ = —160 + 4 кДж • моль.
Эта величина была получена Целотта и др. [919] в результате измерения энергии электронов,
возникающих при фотоионизации Sj излучением аргон-ионного лазера. Ранее приближенное
значение | А0 (S2) | ^ 2 эВ —193 кДж -моль было найдено в работе [2088 ] при исследовании
резонансного захвата электронов молекулами диаллилдисульфида.
Принятому значению Ао (S2) соответствуют
AfH°(S;, г, 0) = —31,707 ±4 кДж-ышнГ1,
Do (S~) = 380,847 + 4 кДж • моль ~ 31 800 + 400 см.
S3 (г). Термодинамические свойства трехатомной серы в стандартном состоянии при
температурах 100—6000 К приведены в табл. 114.
Молекулярные постоянные, использованные при расчете термодинамических функций S3 (г),
приведены в табл. 12.6. Исследования спектров S3 [2002, 2629] показали, что в основном
электронном состоянии ХХА эта молекула имеет симметричную структуру (точечная группа симметрии
Съ). Произведение моментов инерции S3 вычислено по оцененным структурным параметрам
г (S—S) = l,95+0,05 А и Д S—S—S=117+2°. Оценка была выполнена на основании
сопоставления аналогичных величин и энергий связи в рядах молекул О3, SO2, S2O, S3 и О2, О3, S2, S3.
Приведенные в табл. 12.6 значения основных частот vx и v3 молекулы S3 найдены Гопкинсом и др. [2002]
при исследовании ИК-спектров и спектров КР молекул S3, сконденсированных из продуктов
высокочастотного разряда в SO2. Принятое значение vx хорошо согласуется с полученным в работе
[2627] по колебательной структуре электронного спектра поглощения S3 E90 см). В связи с тем,
что другие частоты, наблюдавшиеся в работе [2002], представляются слишком большими для
частоты деформационного колебания S3 при сравнении с частотами О3, SO2 и S2O, значение v2 было
рассчитано по силовым постоянным молекулы S3, соответствующим принятым значениям vlt v3
и Д S—S—S. Погрешности значений vu v2, v3 и 1д1в1с, приведенных в табл. 12.6, могут достигать
5, 40 и 5 см-1 и 5 -Ю-115 г3 -см6.
Исследование электронного спектра [2629] показало, что S3 имеет возбужденное электронное
состояние с Го=23 465 см.
Термодинамические функции S3 (г), приведенные в табл. 114, вычислены по уравнениям A.3) —
A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний S3. Погрешности табулированных
величин при низких температурах обусловлены преимущественно неточностью принятых
молекулярных постоянных S3, а при высоких температурах — применением приближенного метода
расчета и пренебрежением возбужденными электронными состояниями S3. Погрешности значений
Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 1, 3 и 5 Дж-К^-моль-1.
Ранее таблицы термодинамических функций S3 (г) вычислялись в работах [2475] (Т <^ 1000 К)
и [3945] (Т < 3000 К), а также справочнике Милса [2670] (Т < 2000 К) по оцененным значениям
молекулярных постоянных. Расхождения в значениях S° (T), приведенных в табл. 114 и
справочнике [2670], изменяются от 2,8 до 4,6 Дж-К^-моль. В работе [2475] расчет был выполнен для
циклической модели S3 и нелинейной цепной модели с триплетным основным состоянием.
Результаты расчетов для двух моделей отличаются примерно на 24 Дж-К^-моль, причем данные для
циклической модели согласуются с приведенными в таблице в пределах 4,5 Дж-К^-моль.
Дроварт и др. [1326 ] в результате обработки данных по парциальному давлению S3 в парах серы
нашли S° (S3, г, 618 К)=300+6 Дж-К^-моль, в то время как данным табл. 114 соответствует
—311+3 кДж-моль-1.
Константа равновесия реакции S3 (г) = 3S (г) вычислена на основании значения &ГН° @) =
=679,355 + 3,5 кДж-моль в соответствии с принятой в справочнике энтальпией образования
S8(r)
AfH°{S3, г, 0) = 145,0 ± 3,5 кДж • моль.
Эта величина основана на данных о давлении и плотности паров серы, а также равновесии
реакций между отдельными компонентами пара. В табл. 12.7 приведены результаты расчета
энтальпий образования S3 (г), S4 (г), S5 (г), Sc (г) и S7 (г) в четырех сериях работ, позволяющих полу-
247
Глава 12
Таблица 12.6. Значения молекулярных постоянных пав основном электронном состоянии ,
принятые для расчета термодинамических функций S3, S4, S5, Sa, S7, Sg и H2SO4 (p^- =
Молекула
Vl
V2 | V3
V4
V5
V6
V7
V8
V9
v10
VU
V12
V13
V14
V15
CM
Ia^b^c •10Ш
г» • CMS
a
Ss 585 360 651 ____________ 3,6 2
S4 601 270 668B) 320B) — — — — — — _____ 60 6
S5 — — — — — — — — — — _____ 1,10 . 10* 1
S, 483 269 390 312 201B) 451B) 463B) 180B) — — — — — — — 4,80 • 102 6
S, 517 483 402 362 268 237 180 146 510 476 396 288 239 193 153 1,5 . 10s 1
Sg . 483 218 414 237 483B) 187B) 483B) 151B) 76B) 445B) 237B) — — — — 3,9-10» 8
HjSO, 3610 1140 1223 833 550 270 310 360 3610 1160 883 380 1453 310 569 4,06 2
Таблица 12.7. Результаты определения энтальпии образования газообразных S3, S4, S5, S6, S,
(в кДж • моль)
Авторы
Метод
(S,, г, 0)
S7
Брауне и др., Измерения давления
1951 [756] и плотности
ненасыщенных паров серы
статическим методом,
623—1270 К
УэСт, Мензис, Измерение давления
1929 [3926] насыщенных паров
серы статическим
методом, 273—816 К
Берковиц и др., Масс-спектрометриче-
1965 [598, 589, ское исследование
59
146 ±6 (III)
169 ±30 (II)
147 (III)
136 (II)
133 ±15 (III)
212 ±90 (II)
13Г(Ш)
149 (II) а
129 ±4 (III)
131 ±15 (II)
126 (III)
127 (II)
104 ±5 (III)
114±15 (II)
108 (III)
105 (II)
112 ±7 (III)
120 ±10 (II)
113 (III)
116 (II)
144,9 ±3,3 (III) 138,7 ±2,4 (III)
139 ±3A1) 144 ±4 (И)
127,9 ±3 (III)
122 ±5 (II)
104,5 ±3 (III)
110 ±4 (II)
112+4 (III)
121 ±5 (II)
592 ] равновесия реакций
между компонентами
пара над серой C60—
415 К), HgS D30—
580 К), CdS (820—
1030 К), ZnS A110—
1265 К)
Дроварт, Голд- Масс-спектрометриче-
фингер и др., ские измерения пар-
1967 [1226,1326] циальных давлений
компонентов
насыщенных паров серы
в эффузионной
камере с
электрохимическими ячейками,
473-673 К
Pay и др., 1973 Измерения давления
[3166, 3164а] и плотности
насыщенных и
ненасыщенных паров серы
статическим методом,
823—1273 К
а По данным работы [598]. В этой работе было учтено образование иона S4+ в процессах диссоциативной ионизации
и было'получено значение 167 кДж-моль. Позднее Дроварт, Голдфингер и др. [1226,1326 ] рекомендовали по данным
работ [598, 589] значение 130 кДж-моль.
6 По работам Pay и др. [3166].
144 „
A43) б
144 ,
A49) б
121
A13) 6
105
A06)'
115
A18) б
24 8
Сера и ее соединения S3(r)
чить наиболее достоверные значения. Погрешности величин, приведенных в таблице, отражают
как дисперсию экспериментальных данных, так и неточность термодинамических функций Sn (г),
использованных в расчетах.
На основании измерений давления и плотности ненасыщенных паров серы, выполненных
Брауне и др. [756], были рассчитаны константы равновесия реакций диссоциации компонентов пара
Sn (г) = (я/2) S2(r), наилучшим образом удовлетворяющие экспериментальным данным. По этим
константам равновесия и давлению насыщенных паров серы, измеренному в работе Уэста и Мен-
зиса [3926], были найдены парциальные давления компонентов насыщенного пара и
соответствующие им энтальпии образования, приведенные в табл. 12.7.
Берковиц и др. [598, 589] определили отношения интенсивностей ионных токов,
пропорциональные константам равновесия реакций диссоциации S3(r) и S4(r) на S2(r) (измерения
с HgS, CdS и ZnS), а также реакций диссоциации S8(r) на S5(r), S6(r) и S7(r) (измерения с HgS
и серой). Интерпретация экспериментальных данных была затруднена фрагментацией ионов,
которая происходит при ионизации молекул Sn (см. [589, 592]). Значения энтальпий реакций
диссоциации были получены авторами работ [598, 589] по методу II закона термодинамики на
основании зависимости измеренных отношений ионных токов от Т~г. В работе [598] приведены
также относительные интенсивности ионов, которые образуются из компонентов пара над HgS
при 580,9 К. Эти данные позволили авторам настоящего издания рассчитать абсолютные значения
безразмерных констант равновесия для реакций типа S8(r)-j-S2(r)=2S6 (г), S8(r)-j-2S2(r)=3S4(r)
и т. п. и определить по методу III закона термодинамики изменения энтальпии в
соответствующих реакциях. Приведенные в табл. 12.7 значения AfH° (Sn, г) получены на основании найденных
таким образом значений ДГЯ° @) и принятых в справочнике энтальпий образования S2 (г) и S8 (г).
Для учета процесса диссоциативной ионизации компонентов паров серы в масс-спектральных
экспериментах Дроварт, Голдфингер и др. [1226, 1326] применили метод, основанный на
комбинации эффузионных и электрохимических измерений. Пары серы в эффузионной камере
образовывались при пропускании тока через электрохимическую ячейку ~Pt, Ag|AgI |Ag2S|Pt+.
Одновременно определялась ЭДС в идентичной ячейке, также находящейся в эффузионной камере.
Измерения, выполненные при различных силах тока в электрохимической ячейке и различных
температурах эффузионной камеры, позволили авторам работ [1226, 1326] установить условия,
когда ионные токи отдельных компонент пара возникали главным образом в результате прямой
ионизации, а также определить коэффициенты для перехода от интенсивностей ионных токов
к парциальным давлениям. Приведенные в таблице значения AfH° (SK) рассчитаны по
парциальным давлениям соединений Sn, найденным в этих работах.
Pay и др. [3166,3164а] для обработки результатов выполненных ими измерений плотности и
давления насыщенных и ненасыщенных паров серы использовали литературные данные о
термодинамических функциях S2(r), S6(r) и S8(r), энтальпии образования S8 (г), а также оцененные
термодинамические функции S3(r), S4(r), S5(r), S6(r) и S7(r). С учетом поправок на отклонение свойств
газов от их свойств в стандартном состоянии, авторы указанных работ рассчитали значения
энтальпий реакций SB(r) = (n/2) S2(r), наилучшим образом согласующиеся с измеренными ими
величинами, и соответствующие им значения А^Н° (Sn, г). Найденные в работах значения
приведены в табл. 12.7. Следует отметить, что при их вычислении были использованы
термодинамические функции SK(r), которые существенно отличаются от принятых в настоящем издании (для
S4(r) эти расхождения'в энтропии достигают 14 Дж-К-моль, а для S5 (г) — 29 Дж-К^-моль).
Для согласования результатов измерений работ [3166, 3164а] с термодинамическими функциями
настоящего издания значения Дг#° B98,15 К) для соответствующих реакций были вычислены заново
на основании величин ArS°—tb.rH°/T из указанных работ и значений ArS° B98,15 К),
вычисленных по данным настоящего справочника. В табл. 12.7 приведены значения \уН° (Sn,
^^полученные по рассчитанным таким образом значениям Дг#° и принятой в справочнике энтальпии
образования S8(r).
Сопоставление величин, рассчитанных по данным разных авторов, показывает их
удовлетворительное согласие, особенно после пересчета данных работы [598] по уравнению III закона
термодинамики и данных работ [3166,3164а] с использованием термодинамических функций, рассчитанных
в настоящем издании. Наиболее достоверные значения получены по данным Дроварта, Голдфин-
гера и др. [1226, 1326], о чем, в".частности, свидетельствует хорошее согласие величин, найденных
по методам II и III законов термодинамики, и небольшие погрешности значений, полученных
249
S4(г) Глава 12
по каждому из этих методов. Следует отметить, что значительная часть приведенных
погрешностей обусловлена неточностью использованных термодинамических функций Бя(г) (см.
соответствующие разделы).
В настоящем справочнике для энтальпий образования соединений S3(r), S4(r), S5(r), Se(r)
и S7(r) принимаются значения, основывающиеся на данных работ [1226, 1326]. Данные работ
[673, 2265, 3084] по давлению и плотности паров серы, а также результаты более старых масс-
«пектрометрических измерений, рассмотренных в работе [1326], представляются менее надежными
и не учитывались при выборе принятых величин.
S4 (г). Термодинамические свойства газообразной тетрасеры в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 115.
Молекулярные постоянные, использованные при расчете термодинамических функций S4(r),
приведены в табл. 12.6. Результаты теоретических расчетов Мейера и Шпицера [2627] и
исследования колебательных спектров S4 [2002, 2628] показывают, что молекула S4 в основном
электронном состоянии имеет структуру правильного треугольника, соответствующую точечной группе
симметрии D3h г. Такая молекула, согласно теоретико-групповому анализу молекулярных ор-
биталей, должна иметь синглетное основное состояние ХгА. Сравнение энергий разрыва связи
S—S в разных соединениях серы свидетельствует, что 1,89 А=гв (S2)<r (S—S)s4 < г (S—S)HA=
=2,06А, а также, что г (S—S)s, > г (S—S)s3. В настоящем справочнике принимается г (S—S)Sl=
= r(S—S)s2ci2 = 1,97+0,05А [1951]. Этому значению соответствует приведенное в табл. 12.6
произведение главных моментов инерции молекулы S4, его погрешность оценивается в 9 X
ХЮ14 г3-см6. Значения основных частот, приведенные в таблице, основаны на результатах
экспериментальных исследований инфракрасного спектра молекул S4, изолированных в матрицах
инертных газов (Мейер [2628, 2629]), и спектров комбинационного рассеяния продуктов разряда
через SO2, сконденсированных при 20 и 80 К (Гопкинс и др. [2002]), а также на сопоставлении
значений соответствующих частот в молекулах SO2, SO3, S3 и S4. Принятые значения основных
частот S4 определены в работах [2002, 2628, 2629] по спектрам, полученным на приборах с низкой
дисперсией, в матрицах и конденсированной фазе, их погрешности могут достигать 5—10 см~х.
Термодинамические функции S4 (г), приведенные в табл. 115, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128),
A.130). Погрешности табулированных значений термодинамических функций определяются
неточностью молекулярных постоянных и приближенным характером расчета. Они составляют 1,
4 и 6 Дж-К^-моль-1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции S4(r) вычисляли Лафт [2475] (Т ^ 1000 К) и Миле [2670]
(Г ^ 2000 К) по оцененным молекулярным постоянным для структурных моделей молекулы S4,
существенно отличающихся от принятой в справочнике. Вычисленные Лафтом [2475]
термодинамические функции S4(r) для нециклической модели с триплетным основным электронным
состоянием являются ошибочными, так как в них было учтено пять колебательных степеней свободы
вместо шести. Расхождение в значениях Ф° (Т) и S° (T), вычисленных Лафтом для циклической
модели молекул S4 и приведенных в табл. 104, при Т ^ 1000 К не превосходят 3 Дж-К^-моль.
Расхождения в значениях S° (Г) для S4(r), приведенных в^работе [2670] и в табл. 115, достигают
36 Дж-К^-моль. Причины этих расхождений неясны, так как в работе [2670] не указаны
принятые в расчете значения молекулярных постоянных. Дроварт и др. [1326] на основании
измеренного парциального давления S4(r) в парах серы нашли по II закону S° (S4, г, 615 К) = 351 +
+ 10 Дж -К -моль, что в пределах 5 Дж -К -моль согласуется со значением, соответствующим
величинам, приведенным в табл. 115.
Константа равновесия реакции S4(r)=4S(r) вычислена на основании значения t\rH° @) =
=960,14+3,1^кДж-моль, соответствующего принятой в справочнике энтальпии образования
Д^0^, г, 0) = 139 + 3 кДж ¦ моль.
Као [2188а] в результате неэмпирических расчетов для семи возможных конфигураций S4 пришел к выводу, что
наиболее стабильной является незамкнутая бирадикальная структура типа Н2О2 с мультиплетностью, равной трем,
а принятая в справочнике, согласно [2627], конфигурация S4 — наименее стабильной. Однако в работе [2188а]
расчеты проводились'на основе упрощенной неэмпирической методики, не являющейся удовлетворительной для
решения поставленного в ней вопроса.
250
Сера и ее соединения S5 (г), Se (г)
Обсуждение литературных данных и выбор этой величины см. в разделе по энтальпии
образования S3(r).
S5 (г). Термодинамические свойства газообразной пентасеры в стандартном состоянии при
температурах 100—6000 К приведены в табл. 116.
Структура и частоты колебаний молекулы S6 экспериментально не определялись. В настоящем
справочнике принимается, что молекула S5 имеет циклическую структуру, подобно молекулам
S6, S7 и S8. Это предположение опирается на наличие скачка примерно в 100 кДж -моль в
значениях потенциалов ионизации молекул серы [592, 597, 1286, 1326] при переходе от S2, S3 и S4 к S5,
S6, S7, S8. Этот скачок, по-видимому, обусловлен тем, что молекулы Sn при п ^ 4 имеют
нециклическую, а при п ^ 5 — циклическую структуру. Приведенное в табл. 12.6 произведение главных
моментов инерции молекулы S5 вычислено в предположении, что молекула S5 имеет структуру
пятичленного неплоского кольца, состоящего из трапеции SA)SB)SC)SD) и треугольника
SA)SE)SD), расположенных друг по отношению к другу под углом 120+.100. Для межатомных
расстояний приняты значения г (SA)—SB))=r (SD)—SE))=2,10 + 0,10 A; r (SA)—SE))=r (SC) —
—SD))=l,95±0,05 A; r (SB)—Sr3))=2,15±0,05 A; r (SA) . . . SD))=3,43 + 0,05 А. Эта структура
принята по аналогии со структурой молекулы S7 (см. ниже). Погрешность значения IaIbIc
оценивается в 3-10~113 г3-см6.
Термодинамические функции S5(r), приведенные в табл. 116, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A. 124), A. 128)
и A.130). Колебательные составляющие термодинамических функций S5(r) были найдены через
колебательные составляющие S4(r) и S6(r) по приближенным соотношениям вида Ф°м (S5, г) =
=У2Ф°ОЛ (S4, г)+1/2Ф°ол (Se, г). Погрешности вычисленных значений термодинамических
функций обусловлены отсутствием экспериментальных данных о молекулярных постоянных S6 и
применением приближенного метода расчета. Они оцениваются в 3, 8 и 12 Дж-К~1-моль~1 в
значениях Ф° (Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции S5(r) вычислял Миле [2670] (Т ^ 2000 К) по оцененным
молекулярным постоянным, которые в указанной работе не приводятся. Различия в значениях
С°р (Т) при Г=298,15, 1000 и 2000 К составляют 1,05, 0,25 и 0,25 Дж-К^-моль-1, а в значениях
S° (T) они практически постоянны и равны 16,48 Дж-К^-моль. Эти расхождения, по-видимому,
обусловлены отличием структурных параметров S6, принятых в настоящем справочнике и в
работе [2670]. Дроварт и др. [1226, 1326] на основании изучения состава паров серы нашли по
методу II закона S° (S5, г, 592 К)=395 + 17 Дж-К^-моль, что согласуется со значением 395,6 +
+ 7,5 Дж-К^-моль, соответствующим данным табл. 105.
Константа равновесия реакции S6(r)=5S(r) вычислена на основании значения ДГЯ° @) =
= 1245,925±3,2 кДж-моль, соответствующего принятой в справочнике энтальпии образования
AfFI°(Sb, г, 0) = 128 + 3 кДж • моль.
Обсуждение литературных данных и выбор этой величины см. в разделе по энтальпии
образования S3(r).
Se (г). Термодинамические свойства газообразной гексасеры в стандартном состоянии при
температурах 100—6000 К, приведены в табл. 117.
Молекулярные постоянные, использованные при расчете термодинамических функций Se (г),
приведены в табл. 12.6. Рентгенографическое исследование структуры ромбоэдрической серы
(серы Энгеля) [1281 ] в согласии с результатами изучения колебательных спектров паров серы при
низкой температуре [537, 2929], а также спектров ромбоэдрической серы и ее растворов [594,
2864, 2866] показали, что в основном электронном состоянии молекула S6 имеет неплоскую
циклическую структуру (точечная группа симметрии D3d). Произведение моментов инерции,
приведенное в табл. 12.6, вычислено на основании значений г (S—S) ==2,06+0,02 Аи /L S—S—S = 102 + 2°,
найденных Донохью и др. [1281]. Его погрешность не превышает 5-10~114 г3-см6. В соответствии
с принятой структурой молекула S6 имеет восемь основных частот, из которых четыре vx, v2, v6, ve
активны в спектре комбинационного рассеяния, три v4, v7, v8 — в инфракрасном спектре, частота
v3 неактивна в колебательных спектрах газа. Приведенные в таблице значения основных частот
молекулы Se основаны на исследованиях спектров КР и ИК-спектров ромбоэдрической серы и ее
растворов в сероуглероде и в бензоле, выполненных Наймоном и др. [2864, 2866], Берковицем
251
S7(r) Глава it
и др. [594] и Бромельсом [801], спектра комбинационного рассеяния паров серы, полученного
Озиным и др. [537, 2929], и расчетах по силовым постоянным [594, 2865]. С учетом фазового
сдвига частот, погрешности в принятых значениях частот оцениваются в 10 см. Сведения о
возбужденных электронных состояниях S6 отсутствуют.
Термодинамические функции Se(r), приведенные в табл. 117, вычислены в приближении
«жесткий ротатор— гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128)
и A.130). Погрешности вычисленных значений термодинамических функций S6 (г) обусловлены
применением приближенного метода расчета и неточностью использованных в расчете
молекулярных постоянных. Соответствующие погрешности достигают 2, 10 и 15 Дж-К^-моль в значениях
Ф° {Т) при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций S6(r) вычисляли Лафт [2475] (Т <11000 К)„
Берковиц и др. [594] (Т < 3000 К) и Миле [2670] (Т < 2000 К). Лафт рассчитал
термодинамические функции Se(r) для циклической и цепочечной моделей этой молекулы. Расхождения
данных, вычисленных в работе [2475] и приведенных в табл. 117, достигают в значениях Ф° (Т) и
S° (T) около 35 и 60 Дж-К^-моль для этих двух моделей соответственно. Результаты расчетов
работ [594, 2670 ] близки между собой и с табулированными в настоящем справочнике,
расхождения в значениях Ф° (Т) и S° (T) не превышают 0,4 и 0,5 Дж-К^-моль. Дроварт и др. [1226]
при обработке по методу II закона результатов измерений состава насыщенных паров серы нашли
для реакции 0,75 S8(r)=Se (г) значение ArS° E12 К)=31,8 Дж-К^-моль, чему соответствует
S° (S6, г, 512 К)=425 + 14 Дж-К^-моль. Последняя величина находится в согласии со
значением 418 + 4 Дж-К^-моль, вычисленным по данным табл. 117.
Константа равновесия S6(r)=6S(r), приведенная в табл. 117, вычислена на основании
значения &ГН° @) = 1544,21+3,2 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Ay#°(S6, г, 0)= 104,5 + 3,0 кДж-мояь.
Обсуждение литературных данных и выбор этой величины см. в разделе по энтальпии
образования S3(r).
S7(r). Термодинамические свойства газообразной гептасеры в стандартном состоянии при
температурах 100—6000 К приведены в табл. 118.
Молекулярные постоянные, использованные при расчете термодинамических функций S7(r)r
приведены в табл. 12.6. Данные о структуре и частотах колебаний молекулы S7 основываются
на изучении структуры и спектров кристаллической гептасеры, полученной впервые Шмидтом
и др. в 1968 г. [3338]. Масс-спектрометрические [4046], рентгенографические [2203] и
спектроскопические [1567] исследования показали, что основной структурной единицей полученной в
работе [3338] модификации кристаллической серы являются циклические неплоские молекулы S7
с неравноценными связями (точечная группа симметрии Cs). Рентгенографическое исследование
Кавада и Геллнера [2203] позволило определить только проекции межъядерных расстояний
молекулы S7 на плоскость х, у. Расчеты, основанные на результатах работы [2203], при учете данных
о длинах связи S—S в других соединениях серы, позволили оценить структурные параметры
молекулы S7 в основном электронном состоянии Z1./!:
r(S{1) —S(8)) = 2,17±0,06A; г (SF) - SG)) = 2,05 + 0,10 А;
r (SB) — SC)) = 2,13 ± 0,06 A; r (SG, — SA)) =1,96 ± 0,06 A;
r (SC) - SD)) = 2,05 + 0,10 A; Tl = 120 + 4°;
r(SA)-SF)) = ll95±0,06A; ?2=120 + 10°;
r (S«, ~ SF)) = 2,07 ± 0,06 A; r (SG) .. . S(8)) = r (SF) ... SD)) = 3,43 A.
Соответствующее этим параметрам произведение главных моментов инерции молекулы S7
приведено в табл. 12.6, его погрешность оценивается в 2-10~112 г3-см6. Колебательные постоянные
S7 приняты по результатам исследований спектров КР кристаллической S7 и ее раствора в CS2,
а также ИК-спектра поглощения суспензии S7 в парафиновом масле в работе Гарднера и Рогстада
[1567] и спектра КР паров серы в работах Озина и др. [537, 2929]. Они хорошо согласуются
252
Сера и ее соединения S8(r)
с частотами, рассчитанными по силовым постоянным молекулы S8. Погрешности принятых
значений составляют от 3 до 10 см. Сведения о возбужденных электронных состояниях S7 в
литературе отсутствуют.
Термодинамические функции S7 (г), приведенные в табл. 118, вычислены по уравнениям A.3)—
A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности вычисленных значений
обусловлены неточностью молекулярных постоянных S7, применением приближенного метода
расчета и отсутствием данных о возбужденных электронных состояниях этой молекулы.
Погрешности в значениях Ф° (Г) оцениваются в 3, 13 и 20 Дж-К^-моль при 298,15, 3000 и 6000 К.
Ранее таблица термодинамических функций S7 (г) вычислялась Милсом [2670]. В работе [2670]
отсутствуют указания о молекулярных постоянных S7, использованных в расчете. Расхождения
в значениях S° (T), приведенных в указанной работе и табл. 118, достигают 7,4 Дж-К^-моль.
Дроварт и др. [1226] на основании результатов изучения состава насыщенных паров серы нашли
по методу II закона для реакции 0,875 S8 (r)=S7 (г) значение Дг?°E30 К)=30±18 Дж-К^-моль,
чему соответствует S° (S7, г, 530К)=485 + 20 Дж-К^-моль. Эта величина хорошо согласуется
со значением 482+6 Дж-К^-моль, найденным по данным табл. 118.
Константа равновесия реакции S7(r)=7S(r) вычислена на основании значения Дг/7° @) =
= 1811,495 + 3,0 кДж -моль, соответствующего принятой в справочнике энтальпии образования
AfH°(S7, г, 0)= 112 + 3 кДж • моль.
Обсуждение литературных данных и выбор этой величины см. в разделе по энтальпии
образования S3 (г).
S8 (г). Термодинамические свойства газообразной октасеры в стандартном состоянии при
температурах 100—6000 К приведены в табл. 119.
Молекулярные постоянные, использованные при расчете термодинамических функций S8(r),
приведены в табл. 12.6. Рентгенографические исследования структуры ромбической и
моноклинной серы [335, 881, 882, 953, 336, 1047, 1186, 1281, 3078, 3304, 3586, 3876, 3880] *, электронографи-
ческие исследования низкотемпературных наров серы [2020, 2469] и теоретические расчеты [878]
показали, что молекула S8 имеет восьмичленную неплоскую циклическую структуру симметрии
Did с равноценными связями S—S и характеризуется двумя структурными параметрами: г (S—S)
и /?S:—S—S. Приведенное в табл. 12.6 произведение главных моментов инерции молекулы S8
вычислено на основании значений г (S—S)=2,06+0,02 А. иДв—S—S=108+2°, найденных Кароном
и Донохью [882] с учетом данных работ [2469, 878]. Погрешность значения 1д1в1с оценивается
в 5-Ю-112 г3-смв.
Молекула S8 (симметрия Did) должна иметь 11 основных частот; в спектре КР активны частоты
vu V2> v?i V8» V9> vio и Vn> в ИК-спектре — частоты v4, v5 и ve, частота v3 неактивна в колебательных
¦спектрах S8. Основные частоты S8 определялись при изучении спектров ромбической серы [78,
479, 482, 607, 934, 2829, 2928, 2943, 3628, 3788, 3877], растворов ромбической серы в CS2, CeHe
и С10Н8 [537, 607, 934, 1603, 3366] и жидкой серы [1603, 3628, 3788], а также спектров КР
насыщенных паров серы [534, 2929]. Наиболее достоверные значения были предложены Скоттом и др.
[3366] на основании анализа спектров твердой и жидкой серы и ее растворов, результатов
исследования зависимости интенсивности полос в ИК-спектре ромбической серы от температуры [2829 ]
и теоретических расчетов [3365], а также Битти и др. [537], исследовавшими спектр КР паров
серы при 180 °С. Приведенные в табл. 12.6 значения основных частот S8 приняты на основании
сопоставления рекомендаций, предложенных в работе [3366], и данных работы [537], их
погрешности не превышают 2 см для всех частот, кроме v3; для последней погрешность оценивается
в 10 см.
Сведения о возбужденных электронных состояниях S8 в литературе отсутствуют.
Термодинамические функции S8 (г), приведенные в табл. 119, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128) и A.130) без учета возбужденных электронных состояний. Погрешности табулированных
значений обусловлены в основном применением приближенного метода расчета и в меньшей сте-
1 Обзор результатов этих исследований см, в работе Донохью [1280].
253
S8(r)
Глава 12
пени неточностью принятых в расчете молекулярных постоянных. При высоких температурах
могут стать существенными погрешности из-за того, что отсутствуют сведения о возбужденных
электронных состояниях S8. Погрешности в значениях Ф° (Т) оцениваются в 3, 15 и
24 Дж-К^-моль-1 при 298,15, 3000 и 6000 К.
Ранее термодинамические функции S8 (г) вычислялись в таблицах JANAF [2095], работе [1774]
(Т < 1000 К) и справочнике [2670] (Т < 2000 К). Расчеты [2095, 1774] были выполнены по
молекулярным постоянным, предложенным Гатри и др. [1774], допустившими ошибочное отнесение
частот. Расхождения данных, приведенных в работах [2095, 1774], с приведенными в табл. 119
не превышают 5 Дж -К-моль в значениях Ф° (Т) и S° (T). Расхождения с данными справочника
Милса- [2670] существенно больше и достигают 25 Дж-К~1-моль~1 в значениях Ф° (Т). Отсутствие
указаний на принятые молекулярные постоянные S8 затрудняет выяснение ошибки, допущенной
в работе [2670].
Константа равновесия реакции S8 (r)=8S (г), приведенная в табл. 119, была вычислена на
основании значения Д,.1ГО @)=2094,18+2,2 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
kfH°(S8, г, 0)= 104,1 + 1,5 кДж-моль.
Эта величина получена (табл. 12.8) совместной обработкой результатов эффузионных измерений
давления паров серы в работах Неймана и др. [2841], Брэдли и др. [734] и Бриск и др. [782]
в предположении, что в условиях этих экспериментов B98—362 К) насыщенные пары серы состоят,
Таблица 12.8. Результаты определения энтальпии образования S8 (г) (в кДж • моль)
Авторы
Метод
Д/Я°
(S,. г, 0)
Нейман, 1934 [2841 ]
Брэдли и др., 1951 [734]
Бриск и др., 1960 [782]
Брауне и др., 1951
[756]
Уэст, Мензис, 1929
[3926]
Берковиц, 1965 [589]
(см. также [598, 592])
Дроварт и др., 1968
[1326]
Эффузионный, 332—362 К, 8 точек
Эффузионный, 298—306 К, 4 точки
Эффузионный, 303—333 К, 4 точки
Измерение давления и плотности ненасыщенных паров серы |
статическим методом, 623—1273 К I
Измерение давления насыщенных паров серы статическим i
методом, 373—816 К j
Масс-спектрометрическое определение интенсивности
ионного тока SJ при испарении серы, 360—415 К
Масс-спектрометрическое измерение парциального давления
в камере Кнудсена с электрохимическими ячейками, 473—
673 К
104,1+1,5 (III)
105,7 ±1,5 (II)
106+30 (II)
99,5+9,0 (III)
106,3 (II)
101,8+5,0 (III)
110+5 (II)
как было показано Берковицем [589, 598], из молекул S8. В таблице приведены также результаты
расчетов энтальпии образования Sg(r) по данным, полученным другими методами. Как видно из
таблицы, эти величины согласуются с принятым значением в пределах их погрешностей. Следует
отметить значительные погрешности в вычисленных величинах из-за неточности
термодинамических функций S8(r), которые увеличиваются с ростом температуры измерений.
В литературе часто используется значение AfH° (S8, г, 0)=105,6 кДж-моль, вычисленное
Гатри и др. [1774] по данным работ [756, 3926, 734,1518, 3595]. Расчет был выполнен в
предположении, что в условиях этих экспериментов B96—448 К) насыщенные пары серы состоят из молекул
S6 и S8. В расчете использовались константы равновесия реакции Se(r)=0,75S8(r), полученные
в работе [756], и неточные значения термодинамических функций S8(r) (см. выше). Расхождение
между энтальпией образования S8(r), принятой в настоящем издании и вычисленной в работе
Гатри и др. [1774], в значительной степени обусловлено различиями в термодинамических
функциях, использованных в расчетах.
254
Сера и ее соединения SO (г)
SO (г). Термодинамические свойства газообразного оксида серы в стандартном состоянии
в интервале температур 100^—6000 К приведены в табл. 120.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 12.3. Согласно теоретическому анализу, выполненному Колином [1018], молекула
SO имеет 9 валентных электронных состояний (Х32~, ахД, 6*2+, А3ПГ, С3П{, i?32", с1!!", А'3 А
и Л2+) с энергиями, не превосходящими 50 000 см. Известно также (см. [1018])
малоустойчивое валентное состояние 32" с энергией примерно 50 000 см, которое вызывает возмущение
уровней у=15 и 16 состояния 532~, а также 2 ридберговских 2П состояния SO. Молекулярные
постоянные SO определялись в результате исследований переходов А3П—Х32~ [1018, 2800],
?32"-Х32" [2468, 458, 2141, 1883, 2528, 2529, 2527, 2760, 2870, 351, 2585, 2582, 328, 403, 1018,
329, 327], Ь^-'—Х3!,- [1017, 713, 712], микроволнового спектра SO [3077, 3970, 371, 3485, 3692,
3691], спектра ЭПР [2573, 1163, 1162, 1161, 897, 896, 423, 892, 3748, 3901, 1180] и инфракрасного
спектра молекул SO, изолированных в матрицах инертных газов [1997, 2000]. Приведенные
в табл. 12.3 колебательные постоянные SO в состоянии Х32~ вычислены Норршпем и Олдершоу
[2870] по началам полос 0—v F ^ у ^ 11) системы Z?32"—Х32~, измеренным в спектре
испускания SO Мартином [2527]. Значения вращательных постоянных и постоянных мультиплетного
расщепления приняты по работам Амано и др. [371] и Колина [1017] г. Молекулярные
постоянные SO в состояниях ЪХЪ + и A3TLr, приведенные в табл. 12.3, найдены Колином в результате
анализа структуры 4 полос системы ЬХ2 +—Х32~ @ <С z/, v" <^ 2) [1017] и 12 полос системы А3ПГ—
—Х32~ (t/<I8, z/'^2) [1018]. Электронное состояние В3И~ сильно возмущено и предиссоцииро-
вано при v=3, 14, 17 [2527, 328, 1018, 329, 327]. Приведенные в табл. 12.3 колебательные
постоянные в состоянии 532~ получены Норришем и Олдершоу [2870] по кантам полос системы 532~—
—Х32~ C<V<6, i/'<3) [2870] и (i/ < 3, z/'=5,6) [2527]. Вращательные постоянные в
состоянии ?32" приняты по работе Мартина [2527], в которой выполнен анализ вращательной структуры
полос с v'=0,1.
Исследование возмущений состояния i?32~ [2527, 328, 1018, 329, 327] показало, что они
обусловлены взаимодействием с состоянием C3TL{ (предиссоциации при г/=3 и v' = 14) и
с малоустойчивым состоянием 32~. Приведенные в таблице значения постоянных SO в
состоянии С3П{ оценены Колином [1018] в результате анализа возмущений. Переходы, связанные
с состоянием а1 Д, наблюдались в спектре ЭПР Кэррингтоном и др. [894, 895]; в табл. 12.3
приведено значение вращательной постоянной, найденное в этих работах. Энергия возбуждения SO
в состоянии а*Д принята по оценке Колина [1017], в пределах 500 см она согласуется с
величиной, найденной по соотношению A.89). По этому соотношению оценены также энергии
возбуждения состояний ^2", Л'3Д и ^42+, которые по аналогии с молекулами О2 и S2 должны быть
стабильными состояниями, но экспериментально до настоящего времени не наблюдались.
Погрешности в принятых значениях могут достигать 2000—4000 см.
Термодинамические функции SO (г) были вычислены по соотношениям A.3)—A.10), A.93) —
A.95). Значения <2ВН, Q3S и EБН вычислялись по уравнениям A.90)—A.92), в которых
статистическая сумма по колебательно-вращательным уровням энергии состояния Х32~ и ее производные
были получены непосредственным суммированием по уровням энергии этого состояния по
уравнениям A.73)—A.75). Энергии уровней рассчитывались по уравнениям A.65) и A.40) со
значениями коэффициентов Ykl, X и fi, приведенными в табл. 12.4. Соответствующие коэффициенты
были получены на основании постоянных SO, приведенных в табл. 12.3, с помощью методик,
изложенных в разделе 1.4. В расчете учитывались все уровни энергии состояния Х32~, ограниченные
предельной кривой диссоциации этого состояния. В табл. 12.4 приведены значения коэффициентов
af в уравнении, аппроксимирующем эту кривую, а также значения ушах и /нт. Составляющие восьми
возбужденных электронных состояний вычислены в предположении Q^l Бр = (Р</рх) (?^> вр.
Погрешности вычисленных термодинамических функций SO(r) при Т <; 800 К обусловлены
главным образом неточностью принятых значений фундаментальных постоянных, при более
высоких температурах — неточностью принятой энергии возбуждения состояния ахД, при
Т > 3000 К к этому добавляются погрешности из-за отсутствия экспериментальных данных об
энергиях возбуждения состояний ^2"", ^4'3Д, Л2+, а также колебательно-вращательных уров-
1 Они хорошо согласуются со значениями постоянных, полученными при исследовании спектра ЭПР для v
Дейвисом и др. [1180].
255
во- (г) Глава 12
ней состояния а'Ди высоких колебательно-вращательных уровней состояния Х32~. Погрешности
в значениях Ф° (Г) оцениваются в 0,02, 0,3 и 0,5 Дж-К^-моль при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций SO(r) вычисляли авторы справочников [87, 88,
2556, 3944, 337, 2095, 2096, 2670], Монтгомери и Кассель [2692] (Ф° (Т) до 5000 К), Эванс и Уаг-
ман [1428] (Т < 1500 К), Папоушек [2956] (Т < 3000 К), Брюэр и Розенблатт [764]
(Т ^3000 К). Во всех этих расчетах учитывалось основное электронное состояние молекулы
SO, и только во 2-м издании настоящего справочника [88] и справочниках [337, 2096] учитывались
вклады возбужденных электронных состояний. Мультиплетность состояния Х32~ во всех
расчетах учитывалась слагаемым R In 3. В работах [2692, 1428, 87, 3944, 2956] использовались
неточные значения молекулярных постоянных SO в состоянии Х31>~, полученные Мартином [2527].
Расхождения между значениями Ф° (Г), вычисленными в работах [2556, 446, 2095] и
приведенными в табл. 120, изменяются от 0,25 до 1,4 Дж-К^-моль. При низких температурах
расхождения обусловлены пренебрежением мультиплетным расщеплением состояния Х32~, при высоких
дренебрежением возбужденными электронными состояниями молекулы SO. Наиболее близки к
значениям термодинамических функций SO (г), приведенным в табл. 120, данные, полученные в работах
[88, 337, 2096]. Соответствующие расхождения в значениях Ф° (Т) не превышают 0,25 Дж-К X
X моль.
Константа равновесия реакции SO(r)=O(r)+S(r) вычислена с использованием принятого
в справочнике значения
Arff°@) = D0(SO) = 43 219 + 15 см = 517,00 ± 0,18 кДж • моль.
Это значение было предложено Норршпем и Олдершоу [2870] (см. также [1018]) на основании
данных по предиссоциации SO в состоянии В3!,' (v=0, 1, 2 и 3), полученных Мартином [2527].
Авторы работы [2870] уточнили нумерацию полос системы В3Л~—Х32~, исследованной
Мартином, и получили приведенное значение в предположении, что состояние 32~, вызывающее предис-
социацию, коррелирует с пределом S CР2)+О CР2). Принятое значение хорошо согласуется
с оценкой энергии диссоциационного предела состояния 2?32~ E2 500 + 100 см), выполненной
Колином [1018] на основании короткой экстраполяции уровней этого состояния и приводящей
к значению Do (SO) ^ 42 934 см. Оно подтверждается также изучением порога фотодиссоциации
SO2 в работе Окабе [2901 ].
Исследования методом электронного удара [3183] и методом фотоионизации [1234] привели
к существенно менее точным значениям Do (SO), равным соответственно 507 и 535 кДж-моль.
Исследования равновесия реакций с участием SO [3019, 1232] проводились со сложными
системами и при обработке экспериментальных данных были сделаны недостаточно обоснованные
предположения. Поэтому точность вычисленных по данным [3019, 1232] значений E34 и
.522 кДж -моль) невелика, хотя в пределах их погрешностей они согласуются с принятым в
справочнике.
Принятому значению Do (SO) соответствует
AfH°{SO, г, 0) = 4,568+0,30 кДж • моль.
SO" (г). Термодинамические свойства газообразного отрицательного иона оксида серы в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 121.
Молекулярные постоянные, использованные при расчете термодинамических функций SO "(г),
приведены в табл. 12.3. Экспериментально спектр и структура SO" не изучались. В настоящем
справочнике принимается, что основным электронным состоянием SO", как и изоэлектронных
молекул SF и СЮ, является состояние Х2И(, а первым возбужденным состоянием — состояние
А2П.Г Энергия возбуждения состояния А2П{ и постоянная спин-орбитальной связи SO" в
состоянии Х2Л{, приведенные в табл. 12.3, определены как средние арифметические из значений
соответствующих величин для SF и СЮ. Их погрешности могут достигать 3000 и 50 см соответственно.
Значения молекулярных постоянных SO" в состоянии Х2Л{, приведенные в таблице, были оценены
на основании сравнения частот колебаний и равновесных межъядерных расстояний молекул SF,
¦СЮ, Sj, OJT, OF, O2, S2, SO в их основных электронных состояниях. Погрешности оцененных
значений шв и те могут достигать 50 см и 0,05 А.
Термодинамические функции SO" (г) были вычислены по уравнениям A.3)—A.10), A.93) —
A.95). Значения (?„„, Qm и Q№ находились по соотношениям A.90)—A.92), в которых статистиче-
256
Сера и ее соединения SO2 (г)
екая сумма по колебательно-вращательным уровням энергии состояния Х2Л( и ее производные
были рассчитаны непосредственным суммированием по колебательным уровням и
интегрированием по вращательным уровням энергии с помощью соотношений типа A.82). Энергии уровней
рассчитывались по соотношениям A.65) и A.42) со значениями коэффициентов Ykl и А,
приведенными в табл. 12.4. В расчете учитывались все уровни энергии со значениями / ^ Jmn, ,¦, где
/тах , находились по соотношению A.81). В табл. 12.4 приведены соответствующие значения
итал и /llm. Составляющие состояния А2П.{ вычислялись в предположении QiJl.ss = (PA/Px)QifI\ вр.
Погрешности вычисленных термодинамических функций SO~(r) обусловлены отсутствием
экспериментальных данных о молекулярных постоянных этой молекулы. Погрешности в значениях
Ф° (Т) оцениваются в 1, 1,5 и 3 Дж-К^-моль при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций SO~ (г) не публиковались.
Константа равновесия реакции SO" (г)=О (r)+S (г)+е (г), приведенная в табл. 121, вычислена
на основании значения Дг#° @)=622,210+5,0 кДж-моль, соответствующего принятому в
справочнике сродству молекулы SO к электрону
*AQ (SO) = —1,09 + 0,04 эВ = —105,2 + 5,0 кДж • моль.
Эта величина была найдена Фельдманом [1461 ] при масс-спектрометрическом исследовании
зависимости от энергии фотонов сечения фотоотрыва электрона от SO". С ней в пределах
погрешности измерений согласуются менее точные значения, полученные при масс-спектрометрических
определениях потенциалов появления SO" и О" из SO2 [1841] A08+.10 кДж-моль), [2302]
0106+ 10 кДш-моль) и исследованиях ионно-обменных реакций [2302, 2303] «; 105,8 кДж X
X моль), а также данные более старых работ [2839, 3183].
Принятому значению сродства к электрону соответствуют
AfH°(SO-, г, 0) = —100,632 + 5,0 кДж • моль,
D0(SO") = 481,0 ± 5,0 кДж • моль.
SOa (г). Термодинамические свойства газообразного диоксида серы в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 122.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.9. Молекула SOa в основном электронном состоянии ХгА имеет нелинейную
симметричную структуру (точечная группа симметрии Cit). Молекулярные постоянные SO2
определялись на основании изучения спектров комбинационного рассеяния [635, 1245, 1598] и
резонансной флуоресценции [2465, 738, 744, 1725а], инфракрасного [999, 2396, 453, 451, 474, 3405, 2855,
3406, 1791, 1734, 469, 1070, 1071, 1949, 3060, 1635, 472, 1159, 1815а, 1581, 1496, 1859, 360],
микроволнового [3282, 2705, 1152, 3433, 1677а, 1109, 516, 3921а, 517, 3778, 3897] и электронного [3899,
744, 1331, 736, 741, 739, 740, 3871, 1942, 109, 202, 2687, 1815, 3006, 2403, 2404, 1270, 1332, 1330]
спектров, а также электронографических измерений [3350, 1120, 1785, 974]. Приведенные
в табл. 12.9 основные частоты молекулы SOa в состоянии ХгА приняты по работам Хайнкли и др.
[1949] и Корице и др. [1070, 1071], в которых положения начал полос определены с точностью
не хуже 0,10 см. Постоянные ангармоничности вычислены авторами настоящего издания с
использованием наиболее точных экспериментальных данных о колебательных уровнях SO2 [3405,
1070, 1071, 1949, 738] и кубического уравнения для их аппроксимации *. Значения вращательных
постоянных и постоянных центробежного искажения, приведенные в таблице, определены Гебби
и др. [1581], исследовавшими вращательный спектр SO2 в далекой инфракрасной области и
использовавшими результаты микроволновых измерений [2705, 3433, 1109, 516, 3921а, 517, 3778].
Постоянные колебательно-вращательного взаимодействия, приведенные в той же таблице, вычислены
Саито [3282] с учетом результатов работы [556]. Все приведенные в таблице значения
молекулярных постоянных SO2 в состоянии Х1А хорошо согласуются с данными, полученными в других
исследованиях инфракрасного и микроволнового спектров. Сведения о возбужденных
электронных состояниях SO2 можно найти в обзоре Уолша [3871 ], а также в цитированных выше
исследованиях электронного спектра SO2. Поскольку все возбужденные состояния SO2 — ридберговские,
они не приведены в табл. 12.9.
1 Резонанс Дарлинга — Деннисона между уровнями {v% > 2, и2, v3) и (vx — 2, vlt v3-\-2) не учитывался.
257
SO, (г)
Глава
Таблица 12.9. Значения молекулярных постоянных (в см), а также рх в я, принятые для расчета
термодинамических функций SO2 и H2S
Постоянная
V2
^3
*11
*12
*13
Х22
•^23
«33
2/112
2/из
J/122
У123
2/133
У222
2/223
2/233
1/зза
Т
so2
1151,71
517,75
1362,00
—0,80
-3,58
— 12,03
—0,34
—3,54
— 1,78
—0,62
—0,36
-1,13
0,22
4,57
—0,95
0
0,64
—1,29
—0,83
0
HaS
2614,56
1182,68
2627,9
—24,69
— 19,58
—95,10
—5,72
—21,3
—24,28
—
—
—
—
—
. —
—
—
—
—
47,1
Постоянная
-^000
4
«ii
а22
„А
в23
аЗЗ
¦°000
«?
«?
а?
-?1
«5
-?8
«22
<*23
«М
so2
HaS
2,027 356 10,3613
—1,121-10-3 1,057-10-1
—3,761-Ю-2 —2.975.10-1
2,0429-Ю-2 1,703-01-!
—1,53.10-8 —
2,330-10-* —
—2,649-10-* —
0 —
—6,421 • 10-* —
1,004-10-* 9,0162
,0,344 170 1,85-10-1
1,682-Ю-з _2,703-10-1
—7,98-Ю-5 1,392-10-1
1,142-Ю-з _
4,34 • Ю-» —
1,03-10-8 —
1,33-10-6 —
0 —
—8,67 • 10-» —
—8,34 -10-« —
Постоянная
*зС
«S
4
"§
а23
«зСз
вида*
%ьъьь
хсссе
^ааЪЪ
аасе
zabab
Pi
о
so2
0,29353
1,426 • 10-'
5,33 -10-*
1,068-Ю-з
7,01 • Ю-6
2,17-10-8
—3,30-10-8
0
—1,93-10-8
—1,00-Ю-6
—3,3095 • 10-*
—1,331- 10-s
—4,192-10~7
—1,439-10-8
3,418 -10-6
6,767-10-'
—1,775- 10~в
1
2
H2S
4,7314
6,97-Ю-2
6,19.10
5,41 • Ю-2
—
—
—
—
—
—
—5,04 • Ю-3
—8,25-Ю-з
—2,7.10-*
4,12-Ю-3
—5,3-10-*
—5,9.107*
—7,3 -10-*
1
2
Термодинамические функции SO2(r) были вычислены без учета возбужденных электронных
состояний. Составляющие «жесткого ротатора — гармонического осциллятора» находились по>
уравнениям A.3)—A.10), A.122)—A.124), A.128) и A.130), поправки на ангармоничность
колебаний и взаимодействие колебаний и вращения — по уравнениям A.131)—A.133), на
центробежное растяжение — по уравнениям A.143), A.144) и низкотемпературные поправки — по
уравнениям A.147)—A.149). Погрешности вычисленных значений термодинамических функций при
Т < 1000 К обусловлены неточностью принятых молекулярных постоянных, а при более высоких
температурах неточностью аппроксимации энергии высоких колебательно-вращательных уровней
уравнениями с принятыми постоянными и использованием приближенного метода расчета.
Суммарные погрешности в значениях Ф° (Т) при Г=298,15, 3000 и 6000 К оцениваются в 0,05, 0,3
и 1 Дж-К^-моль.
Ранее таблицы термодинамических функций SO2(r) вычисляли авторы справочников [87, 88,
2095, 2556, 446], Гордон [1691а] (Ф° (Г) до 2800 К), Кросс [1119а] (Т < 1800 К), Эванс и Уагман
[1428] (Т < 1500 К), Нагараян [2769] (Т < 1700 К), Гордон [1699], Уилкинс [3944], Венкатес-
варлу и Мариам [3795] {Т < 1000 К) и Миле [2670] (Т < 2000 К). Во всех этих расчетах
учитывалось только основное электронное состояние и не учитывалось центробежное растяжение.
Расчеты, приведенные в работах [1691а, 1119а, 2769, 3795], были выполнены в приближении
«жесткий ротатор — гармонический осциллятор». В работах [1691а, 1119а, 2769, 2670] использовались
неточные значения постоянных, а в [2769] допущены ошибки в расчете. Расхождения в значениях
Ф° (Т) между данными этих работ и табл. 122 достигают 0,7—1,3 Дж-К^-моль. Расчет»
проведенный в работе [3795], выполнен для S18O18O и значения Ф° (Т) отличаются от
приведенных в табл. 122 примерно на 7 Дж-К^-моль из-за различия симметрии молекул
SO2 и S16O18O. В расчетах, проведенных в [87, 88, 2095, 3944, 2556, 446, 1428, 1699],
были учтены постоянные ангармоничности первого порядка {x(j), а постоянные ангармоничности
258
Сера и ее соединения
второго порядка (Уцк)я постоянные колебательно-вращательного взаимодействия (а{, a{j) не
учитывались. Максимальные расхождения в значениях Ф° (Г), приведенных в этих работах и табл. 122,
составляют 0,04 Дж-К^-моль-1 при 7=298,15 К и 0,7 Дж-К^-моль-1 при 6000 К. Джиок и
Стивенсон [1619а] вычислили на основании калориметрических измерений S° B98,15 К) ==
= 247,86 Дж-К^-моль, что удовлетворительно согласуется со значением 248,11 ±0,1 Дж-К~хХ
Хмоль, приведенным в табл. 122.
Константа равновесия реакции SO2(r)=S(r)+2O(r) вычислена с использованием значения
&ГН° @)=1062,615 + 0,35 кДж-моль, соответствующего принятой в справочнике энтальпии
образования SO2 (г)
AfH°(SO2, г, 298,15 К) = 296,81 ±0,20 кДж-моль.
Принятая величина была рекомендована КОДАТА—МСНС [1000] на основании результатов
калориметрических определений энтальпии сгорания ромбической серы, выполненных Экманом
и Россини [1378]. В этих исследованиях сжигание проводилось в условиях избытка серы, что
устраняло возможность образования SO3- В этом отношении работа [1378] выгодно отличается
от более старых работ (библиографические ссылки можно найти в работах [1378, 258, 642]),
в которых состав продуктов сгорания был сложным и не был установлен с достаточной
точностью.
SOj (г). Термодинамические свойства газообразного отрицательного иона диоксида серы
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 123.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 12.10. По аналогии с изоэлектронной молекулой СЮ2 и в соответствии с нечетным числом
Таблица 12.10. Значения молекулярных постоянных, а также а и рх, принятые для расчета
термодинамических функций S2O, SO?, SO3, SF2, SFS, SFr, SF4, SF6, SFr, SFe,
SOF2, SO2F2
Молекула
s2o
so?
so,
SF2
SF3
SFJ
SF,
SFS
SFJ
SF,
SOF,
SQ2F,
Состояние
**в,
JT'A,
X'A,
X2Al
JT'A,
X'A,
*A,
t'At
JT'A,
i'A,
1-A,
1165
965
1068
795
794
750
892
812
800
773,6
1335,8
1270,9
•
388
470
497,55
430
550
530
558
552
540
642,1 B)
808,2
849,0
679
1070
1389,9 B)
830
340
330
356
480
470
947,968 C)
530,4
545,3
4
CM
—
530,18 B)
—
720
700
228
460
450
614,03 C)
377,8
385,0
v8
-1
—
—
450
440
474
270
265
522,9 C)
747,0
389,0
_
_
340
330
730
350
345
346 C)
392,5
1504,2
—
_
—
—
532
610 B)
600 B)
—
552,2
—
867
450B)
435B)
—
886,8
—
350
250B)
245 B)
—
539,5
IAIBIC • 10"'
r* • cm'
6,184 • 10*
1,4 . 101
1,037 • 10»
3,48 . 10'
2,22 • 10s
2,770 • 10»
6,716 • 10s
2,6 • 10*
2,1 • 10*
2,94 • 10'
1,657 • 10»
4,453 • 10»
0
1
2
6
2
2
2
2
4
4
24
1
2
1
2
1
1
2
1
1
2
1
1
1
1
электронов в справочнике принимается, что SO2 имеет основное электронное состояние %2Blt
а возбужденное электронное состояние должно иметь энергию, превышающую потенциал
ионизации SOj.
Приведенное в таблице произведение моментов инерции вычислено в предположении, что
в основном электронном состоянии SOj имеет угловую симметричную структуру (точечная группа
симметрии С2,) и структурные параметры г (S—О) = 1,48+0,02 Аи ДО—Sr— 0=115 + 5°. Эти
параметры, а также основные частоты SOJ, приведенные в табл. 12.10, являются средними между
значениями этих величин в молекуле СЮ2 и значениями, найденными Миллиганом и Джекокс
[2665] при изучении ИК-спектра SOj в матрицах из Аг при осаждении в матрице молекул SO2
с атомами Cs. Авторы работы [2665] отмечают, что найденные ими значения основных частот могут
быть заниженными из-за слабого взаимодействия ионов SOj и Cs+, а также влияния поля матрицы.
259
SO3(r) Глава 12
Погрешности принятых в справочнике основных частот и значения IaIbIg оцениваются в 25 см
и 5-Ю17 г3-см6 соответственно.
Термодинамические функции SO^(r), приведенные в табл. 123, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128) и A.130) без учета возбужденных электронных состояний. Погрешности вычисленных
значений термодинамических функций SO^ (г) обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета. Погрешности значений Ф° (Т), обусловленные
указанными причинами, оцениваются в 1, 3 и 5 Дж-К^-моль при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции SOj (г) не публиковались.
Константа равновесия реакции SO7 (г)=2O(r)+S(r)+e(r) вычислена с использованием
значения &ГН° @) = 1168,415 + 3,5 кДж-моль, соответствующего принятому в справочнике сродству
молекулы SO2 к электрону
Ао (SO2) = —1,097 + 0,036 эВ = —105,8 ± 3,5 кДж • моль.
Принятое значение найдено в работе Целотты и др. [919] из измерений энергии электронов,
возникающих при фотоионизации SO^ излучением лазера с фиксированной длиной волны. Оно
удовлетворительно согласуется с результатами менее точных измерений по сечению реакции
фотоотрыва электрона от SO^ [1461] (^ —96,5 + 5 кДж-моль) и сечению ионно-обменных
реакций с участием SO; [2302, 2303] (—105>Л0>—108), [2034] « —96+10 кДж-моль) и [3261]
(^ —110±15 кДж-моль). Существенно большее (по абсолютной величине) значение было
получено Пиккарди [3009] «[ —193+.10 кДж-моль) при изучении электропроводности пламен
с добавками SO2 и H2S. Однако отнесение результатов этих измерений к конкретным реакциям
не убедительно.
Принятому значению Ао (SO2) соответствует
AfH°{SO-, г, 0) = —400,064 + 3,5 кДж ¦ моль.
SO3(r). Термодинамические свойства газообразного триоксида серы в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 124.
i Молекулярные постоянные SO3, использованные при расчете термодинамических функций,
приведены в табл. 12.10. В согласии с результатами электронографических [2944, 974] и
спектральных исследований в настоящем издании принимается, что в основном электронном состоянии
%.хАг молекула SO3 имеет структуру правильного равностороннего треугольника, в центре
которого расположен атом S (точечная группа симметрии D3h). Приведенное в таблице произведение
главных моментов инерции SO3 вычислено на основании вращательных постоянных Во =
=0,34857 см (найдено Калдором и Маки [2185] при анализе вращательной структуры полосы
2v3 в ИК-спектре газообразного SO3) и Со=0,17402 см (получено Дорнеем и др. [1295] по
значению инерционного дефекта). Этим значениям вращательных постоянных соответствуют
межатомные расстояния г (S—О) = 1,4198 А и 1,4184 А. Практически совпадающее значение 1,4179 А было
найдено Кларком и Бигли [974] в результате электронографического исследования.
Колебательный спектр газообразного SO3 исследовался в работах [1601, 2186, 2466, 586, 2300, 924, 1733,
2671, 3666, 2185, 1600, 1596, 587, 1028], жидкого SO3 —в [634,467,3791, 1601, 1599, 1649, 3774,
1597, 3545, 1733, 3546, 1927, 1600, 1596, 3868] и молекул SO3, изолированных в матрице, в работе
[2466]. Приведенные в табл. 12.10 значения основных частот основаны на результатах работ
[1601, 2186, 3666]. Для частоты vx принято значение, полученное Гердингом и др. [1601] при
исследовании спектра комбинационного рассеяния жидкого SO3. Частоты v2 и v4
найдены Калдором и др. [2186] по началам полос из сравнения рассчитанных контуров полос
с наблюдавшимися Томасом и Томпсоном [36661 в инфракрасном спектре газообразного SO3.
Для частоты v3 принято значение начала полосы, найденное Томасом и Томпсоном [3666 ] с учетом
неточности проведенного в работе [3666 ] анализа ее вращательной структуры. Погрешности
принятых значений оцениваются в 3 см для vx и в 1 см для остальных частот. Сведения о
возбужденных электронных состояниях SO3 в литературе отсутствуют.
Термодинамические функции SO3, приведенные в табл. 124, вычислены по уравнениям A.3)—
A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных значений определяются главным образом приближенным
характером расчета из-за отсутствия данных, необходимых для учета ангармоничности колебаний
260
Сера и ее соединения
SO3 (г)
и других поправок. Суммарные погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К
оцениваются в 0,1, 3 и 5 Дж-К^-моль.
Ранее таблицы термодинамических функций SO3(r) вычислялись в справочниках [88, 2095,
2556] и в работах [3540, 282, 2783, 2670, 2466] (Т < 2000 К), [1428] (Т < 1500 К). Все расчеты,
кроме приведенных в работе [3540], выполнены в приближении «жесткий ротатор —
гармонический осциллятор». Стокмейер и др. [3540] приближенно учли ангармоничность колебаний SO3
по методике, аналогичной принятой в настоящем издании, и оцененным значениям постоянных.
Во всех расчетах принималось несколько завышенное значение произведения главных моментов
инерции; в работах [88, 3540, 282, 2783, 1428, 2670] были приняты неверные значения частот
v2 и v4. Расхождения полученных в этих работах термодинамических функций SO3 (г) с
приведенными в табл. 124 достигают 1,4 Дж-К^-моль в значениях Ф° (Т). В работе [2466] и
справочниках [2095, 2556] использовались значения основных частот, близкие к принятым в настоящем
справочнике. Различия в значениях термодинамических функций, приведенных в работах [2466,
2095, 2556] и в табл. 124, не превышают 0,3 Дж-К^-моль в значениях Ф° (Г) и S° (Т).
Константа равновесия реакции SO3 (r) = 3O (r)+S (г) вь?числена с использованием значения
АГН° @) = 1405,287±0,76 кДж-моль, соответствующего принятой в справочнике энтальпии
образования SO3(r)
Ду#°(?О8, г, 298,15 К) = — 395,9 ± 0,7 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакций с участием
SO3 (г) и калориметрических измерений, приведенных в табл. 12.11 и 12.11а. Погрешности
приведенных значений ДГ?Г° отражают только разброс экспериментальных данных. При вычислении
энтальпии образования SO3 (г) из данных по равновесиям учтены погрешности
термодинамических функций SO3(r), увеличивающие погрешность этой величины. При вычислении энтальпии
образования SO3(r) из данных калориметрических измерений было использовано значение
AtH°(SO3, ж, 298,15 К)=43,1 ±1,2 кДж -моль, рекомендованное в справочнике [258]. Погрешность
этого значения вносит значительный вклад в погрешности значений AfH°(SO3, г), вычисленных
по данным работ [1715, 1716]. Значения энтальпии образования растворов серной кислоты,
рекомендованные в справочниках [258, 3375], заметно отличаются, особенно для разбавленных и
концентрированных растворов. Поэтому в табл. 12.11а приведены результаты расчетов для двух
вариантов этой величины.
Таблица 12.11. Результаты определений энтальпии образования SO3 (г) (в кДж-моль)
SO>() + i/O() SO ()
по исследованиям равновесия реакции SOa>(r) + i/2O2(r) =
Авторы
Метод
SO3 (г)
ДгЯ°@)
II закон
III закон
Д/Я° (SO,, г,
298,15 К)
Нич, 1901, 1903 Метод протока, катализатор—платиновая —84,7 —97,1 ±2,0 —397 ±2
[2272, 2273] сетка, 703—913 К, 6 точек
Бодлендер, Коппен, Статический метод, катализатор — пла- —113,1 —102,0+3,5 —402±4
1903 [689] тиновая сетка, 788—923 К, 8 точек
Лунге, Рейнгард, Метод протока, катализатор — окислы ме- —98,6 —94,4+1,3 —394,4±1,4
1904 [2480] таллов, 898—973 К, 3 точки
Лукас, 1905 [2471] Статический метод, катализатор — пла- —77 —107,4±2,4 —407,4±2,5
тиновая сетка, 838—1143 К, 15 точек
Боденштейн, Поль, Метод протока, исходные продукты S02 —93 ±1 —96,06 ±0,36 —396,07 ±0,65
1905 [686] и О2, платиновый катализатор, 801—
1170 К, 8 серий, 54 точки
Тейлор, Ленер, 1931 Статический метод, катализатор — нагре- —105 ±10 —95,8 ±0,5 —395,8 ±0,75
[3630] ваемая платиновая проволока, 933—
945 К, 5 точек. Температура не
измерялась, а вычислялась по уравнению
из работы [686]
Капустинский, Ша- Статический метод, исследовалась дис- —90 ±5 —95,80 ±0,36 —395,8 ±0,65
мовский 1936 [120, социация паров твердой SO3 в кварце-
119] вом сосуде с раскаленной платиновой
нитью, 850—1000 К, 11 точек
261
SaO(r)
Глава 12
Таблица 12.11а. Результаты определений энтальпии образования
по калориметрическим измерениям
Авторы
Уравнение исследованной реакции
SO3(r)
ДГЯ° B98,15 К)
AfH°(SO3, г, 298,15 К)
[258]
[3375]
Бертло, 1881 [618] SO3 (г)+201Н2О (ж)=Н2ЭО4 (р-р, 200Н2О) —209+4 —393,7 ±4,0 —393,8 ±4,0
Томсен, 1882—6 [3676] SO8 (ж)+1601Н2О (ж)=Н28О4 (р-р, 1600Н2О) —167 ±4 —398,2 ±4,2 —397,7 ±4,2
Жиран, 1913 [1664] SO3 (ж)+40Ш2О (ж)=Н28О4 (р-р, 400Н2О) —169 ±4 —392,2 ±4,2 —392,0 ±4,2
Грау, Рот, 1930 S03 (ж)+500Ш2О (ж)=Н28О4 (р-р, 5000Н2О) —175,60 ±0,25 —393,8 ±1,2 —392,5 ±1,2
[1715, 1716]
Грау, Рот, 1930 [1715, SO3 (ж)+Н20 (ж)=Н2804 (ж) —89,50 ±0,42 —395,7+1,3 —395,5 ±1,3
1716]
Как видно из табл. 12.11, исследования равновесия реакции образования SO3 в наиболее
тщательно выполненных измерениях [686, 120, 119, 3630] находятся в хорошем соответствии и их
погрешность меньше погрешности калориметрических измерений. Принятое значение Д^Я° (SO3, г)
является средним из вычисленных по данным этих работ.
S2O (г). Термодинамические свойства газообразного оксида дисеры в стандартном состоянии
при температурах 100—6000 К приведены в табл. 125.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 12.10. Анализ микроволновых спектров [2621, 1042, 3693] показал, что молекула
S2O в основном электронном состоянии ХгА имеет угловую несимметричную структуру с атомом
серы в вершине угла. Приведенное в таблице произведение главных моментов инерции вычислено
на основании значений вращательных постоянных Аооо—1,398147 6 см, Б000=0,168 751 88 см и
С000=0,15034445 см, найденных Тиманном и др. [3693]. Соответствующие им значения структурных
параметров равны r(S—О)=1,4594+0,0010 A, r(S—S)=1,8845+ 0,0010 А и ^S—S—0=118,08+
+ 0,05°. Значения основных частот S2O определялись при исследованиях инфракрасного спектра
[2155, 677, 3006, 2002, 2001], спектра комбинационного рассеяния [2002, 1999] и электронного
спектра [1064]. Принятые значения частот vx и v3 найдены Джонсом [2155] по (?-ветвям полос
в инфракрасном спектре поглощения продуктов электрического разряда через пары серы и SO2;
принадлежность этих полос молекуле S2O была доказана Меши и Майерсом [2621 ]. Для частоты
деформационного колебания v2 принято значение, найденное Блукисом и Майерсом [677] при
анализе инфракрасного спектра пленок S2O при 77 К, в котором были также зарегистрированы
полосы vx и v3 с центрами при 679 и 1165 см. Погрешности приведенных в табл. 12.10 значений
частот vx и v3 оцениваются в 1 см, частоты v2 — в 10 см. Исследования электронного спектра S2O
[2155, 3329, 1064, 129, 3006, 2344, 2342] показали, что эта молекула имеет два возбужденных
электронных состояния с энергиями 29 262,8 и 44 228,8 см.
Термодинамические функции S2O(r) были вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A. 122)—A. 124), A. 128) и A.130) без
учета возбужденных электронных состояний. Погрешности вычисленных значений определяются
неточностью принятых значений молекулярных постоянных и применением приближенного
метода расчета. Суммарные погрешности в значениях Ф° (Т) составляют 0,2, 2 и 3
Дж-К^-моль при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций S2O(r) вычисляли Блукис и Майерс [677]
(Т < 3000 К), Боттер и др. [711] (Т < 1500 К), Нагараян [2776] и авторы справочников [88,
2095] (Т ^ 6000 К). Все расчеты выполнены в приближении «жесткий ротатор —
гармонический осциллятор». Результаты расчета [2095] наиболее близки к приводимым в табл. 125,
отличаясь от последних не более чем на 0,075 Дж-К^-моль в значениях Ф° (Т)
и S° (T). Расхождения между результатами расчетов [677,711] и данными табл. 125
составляют 1,3 Дж-К^-моль в значении Ф° (Т) при 1000 К и обусловлены неточностью этих расчетов.
Значения Ф° (Т), приведенные в работах [88, 2776] и в табл. 125, отличаются на величины
до 0,4 Дж-К^-моль из-за некоторого уточнения значений молекулярных постоянных.
262
Сера и ее соединения SH (ту
Константа равновесия реакции SaO(r)=2S(r)+O(r) вычислена с использованием значения
АГН° @)=850,353±1,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(S2O, г, 0) = —54,0 + 1,4 кДж • моль.
Эта величина основана на потенциале появления SO+ из S2O A3,745+0,010 эВ после
пересчета к Т=0), измеренном Берковицем и др. [5966] по порогу фотодиссоциации S2O, и на
потенциале ионизации SO A0,29+0,01 эВ, см. [5966]). Заметно отличающиеся значения —43,2 +
+3,0 кДж-моль и —44,5+3,0 кДж-моль были получены при исследовании равновесия
реакции l,5S2(r)+SO2(r)=2S2O(r) в работах Боттера и др. [711] и Дьюинга, Ричардсона [1232]
<(см. пересчет в [677]). В справочнике отдано предпочтение значению, полученному в работе [5966],
так как в исследованиях [1232, 677] равновесие с участием S2O являлось только побочной
реакцией, а метод, использованный в работе [711] (реакция SO2 с парами серы на раскаленной
иридиевой нити и вычисление констант равновесия по интенсивностям ионных токов), не вполне надежен
из-за градиента температуры и связанных с ним неопределенностей в фиксируемом составе
продуктов реакции.
В работах [711, 1797, 1798] приведены результаты измерений потенциалов появления SO+
из SO2 и из смеси SO2+S2O. Эти данные менее надежны по сравнению с измерениями Берковица
[5966], им соответствуют значения энтальпии образования от —29 до —77 кДж-моль. Предис-
социация в спектре S2O, обнаруженная Джонсом [2155], также приводит к неверному значению
энтальпии образования (около —100 кДж -моль), по-видимому, из-за того, что она обусловлена
¦состоянием, коррелирующим с возбужденными продуктами диссоциации.
SH (г). Термодинамические свойства газообразного гидрида серы в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 126.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.3. Молекула SH имеет основное электронное состояние Х2И{ и первое
возбужденное состояние ЛаЕ+, а также ряд электронных состояний с энергией свыше 50 000 см, большая
часть которых ридберговские.
Молекулярные постоянные SH определялись на основании результатов исследований систем
полос Л22 + —Х2П,- и Б22—Х2П,. в электронных спектрах SH [1580, 1366, 3066, 2371, 1365, 345,
2419, 2369, 3131, 2130] и SD [2371]. До настоящего времени в спектрах SH наблюдались только
переходы, связанные с уровнем v—0 состояния Х2ПО а в спектрах SD только переходы с v=0
и 1 этого состояния. Приведенные в табл. 12.3 значения колебательных постоянных SH в
состоянии Х2И{ вычислены при подготовке настоящего издания по соотношениям A. 67), с
использованием величины Д&/, (Х2П„ SD)=1885,5 см [2971], принятого значения Do (SH) и
изотопических соотношений A.66). Найденные значения ше и шехв близки к вычисленным в работах [3066»
2371, 3131, 2971]. Приведенная в таблице постоянная спин-орбитальной связи в состоянии ХЩ{
найдена Рамзи [3131 ] на основании анализа вращательной структуры 0—0 полосы системы А2Ъ —
Х2ПФ. и Морроу [2716] при анализе 0—0 и1—0 полос системы522—Z2nf. Вращательные
постоянные вычислены с использованием данных, полученных Рамзи [3131 ] и Патхаком и Палмером
[2971] при анализе структуры полос в спектрах SH и SD. Молекулярные постоянные SH в
состоянии Л22+ рекомендованы Рамзи и Джонсом [3131, 2130].
Термодинамические функции SH(r) вычислены по уравнениям A.3)—A.10), A. 93)—A.95).
Значения Qm, Qm и Qm находились по соотношениям A. 90)—A. 92), в которых статистическая
сумма по колебательно-вращательным уровням энергии состояния Х2П,- и ее производные были
рассчитаны непосредственным суммированием по колебательным уровням и интегрированием
по вращательным уровням с помощью соотношений типа A. 82). Энергии уровней вычислялись
по уравнениям A. 65) и A. 42) со значениями коэффициентов Ykl и А, приведенными в табл. 12.4.
Л-удвоение вращательных уровней учитывалось статистическим весом 2. В расчете учитывались
все уровни энергии со значениями / ^/тах, »> гДе ^max.t находились по соотношениям A. 81).
В табл. 12.4 приведены соответствующие значения vmax и /цт. Составляющие состояния А2Ъ +
вычислялись в предположении QiiH.sv = (pJPx)Qmx.Bp.' Погрешности вычисленных термодинами,
ческих функций SH (г) при Т ^ 1000 К определяются только неточностью молекулярных
постоянных; они составляют 0,02, 0,1 и 0,3 Дж-К-моль в значениях Ф°(Г)при 298,15, 3000 и
6000 К.
263
SH-(r) Глава 12
, Ранее таблицы термодинамических функций SH(r) вычисляли авторы справочников [87,
88, 2556, 2095] и Хар и Фридман [1782]. Наилучшее согласие с данными табл. 126 имеет место
для 2-го издания настоящего справочника [88], расхождения не превышают 0,15—
0,20 Дж -К -моль в значениях Ф° (Г) и S° (T). Расхождения с данными работ [1782, 2095,
2556] существенно больше: они достигают 0,55 Дж-К-моль в значениях Ф° B98,15 К) из-за
некорректного учета спин-орбитального расщепления уровней состояния Х2П,- и 0,3 и
1 Дж -К -моль в значениях Ф° F000 К) и S" F000 К) из-за применения грубой методики
расчета.
Константа равновесия реакции SH (г)=Н (r)+S (г) была вычислена по принятой в справочнике
энергии диссоциации SH
г-1
ДГЯ° @) = Do (SH) = 351,0 + 3,5 кДж • моль = 29 350 + 300 см'
Это значение принято на основании фотоионизационных измерений порога реакции H2S-\-hv=
=SH+-J-H+e, выполненных Дайблером и Листоном [1234] A376,8+1,9 кДж-моль), значения
Io (SH) = 1003,4+2,9 кДж-моль [2716] и энергии атомизации H2S (см. ниже). Оно хорошо
согласуется с полученным Джонсом и Рамзи [2130] линейной экстраполяцией колебательных
уровней состояния А21,+ B8 480+1000 см). Измерения потенциалов появления ионов SH+ из H2S
[2945] и CaHg из C2H5SH [1534, 2941], а также определение Do (G6H5—SH) толуольным методом
[3374] (см. также [2491]) позволили определить нижний предел Do (SH) ^ 345+20 кДж-моль,
который находится в хорошем согласии с принятой величиной. Близкие, но менее точные значения
Do (SH) были получены в работах [2371, 3066, 3131]. В работе [461] измерения
потенциала появления SH+ из H2S методом электронного удара привели к существенно более низкой
величине нижнего предела Do (SH) ^ 310+20 кДж-моль, по-видимому, из-за ошибки в
интерпретации экспериментальных данных.
Принятому значению Do (SH) соответствует
AfH°(SH, г, 0) = 139.819 + 3,5 кДж • моль.
SH~(r). Термодинамические свойства газообразного отрицательного иона гидрида серы в
стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 127.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.3. Согласно теоретическим расчетам [466, 859], основное электронное состояние
ХХ2+ является единственным стабильным электронным состоянием SH~. Приведенные в таблице
значения колебательных постоянных SH" в состоянии ХХ2+ приняты на основании результатов
исследований колебательных спектров примесных ионов SH" в кристаллах галогенидов К, Na
и Rb, выполненных Чи и Никсоном [947, 946], теоретического расчета Кейда [859] и значения
Do (SH~)=31 350+300 см. Погрешности значений шв и шехд оцениваются в 20 см
и 2 см соответственно. Принятые значения вращательных постоянных SH" получены Кейдом
[859] и Стейнером [3515] в результате теоретических расчетов и измерений сечения фотоотрыва
электрона от SH". Погрешности в значениях Ве и ах не превышают 0,15 и 0,02 см.
Термодинамические функции SH" (г) были вычислены по уравнениям A.3)—A.10), A.93)—
A. 95) без учета возбужденных электронных состояний. Значения Q??, вр и ее производных
находились непосредственным суммированием по колебательным уровням энергии состояния ХХ2 +
по уравнениям типа A.82) и A.73)—A.75). Энергии уровней рассчитывались по уравнениям
A. 65) и A. 34) со значениями коэффициентов Ykl, приведенными в табл. 12.4. В расчете
учитывались все уровни со значениями /<^/шаХ|,, где /maXif находились по соотношениям A.81).
В табл. 12.4 приведены значения vm&% и /цт для SH" в состоянии Z12+. Погрешности
вычисленных термодинамических функций определяются главным образом неточностью принятых
молекулярных постоянных SH". Они составляют 0,2, 0,5 и 1 Дж • К • моль в значениях Ф° (Т) при
298,15, 3000 и 6000 К.
Ранее термодинамические функции SH~(r) не публиковались.
Константа равновесия реакции SH~(r)=H(r)+S(r)+e(r) вычислена на основании значения
Д ,./7° @)=573,179+3,5 кДж -моль, найденного по принятому в справочнике сродству к
электрону молекулы SH
Ао (SH) = —18 563 ± 10 см = —222,06 ± 0,12 кДж • моль.
264
Сера и ее соединения H2S(r>
Это значение Ао (SH) было принято по границе фотоотрыва электрона от SH E387+3А), най-
денной Эйлером и Аткинсоном [1432] при исследовании сечения фотоионизации с помощью
лазера с перестраиваемой длиной волны. Близкие, но менее надежные значения были получены
Стейнером [3515] B24 +1 кДж -моль) при изучении фотоионизации SH~ с помощью
перестраиваемого лазера и Ямдагни и Кебарле [4021] B22,6 кДж -моль) при изучении ионно-обменных
реакций. Более ранние исследования [401, 859, 1900, 2938, 3091, 3241, 3923] приводили к существенно
менее точным данным.
Принятому значению AQ (SH) соответствуют
A/ff°(SH-, г, 0) = —82,241 ±3,5 кДж • моль,
Do (SH-) = 372,63 + 3,5 кДж • моль = 31150 ± 300 см.
H2S (г). Термодинамические свойства газообразного сероводорода в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 128.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.9. Молекула H2S в основном электронном состоянии Х1А1 имеет нелинейную
симметричную структуру (точечная группа симметрии Сг,). Молекулярные постоянные H2S в
основном электронном состоянии определялись на основании исследований инфракрасного [69, 1711,
1118, 2066, 361, 362, 363, 364, 3313, 1388, 1496, 3481, 2569, 1591, 1593, 843, 2917] и микроволнового
спектров газообразного сероводорода [1876, 1878], а также исследований инфракрасного спектра
[2930] и спектра комбинационного рассеяния [3625] молекул H2S, изолированных в матрицах
инертных газов. Приведенные в табл. 12.9 основные частоты vx и va определены Алленом и др.
[361, 364] по началам колебательно-вращательных полос. Для частоты v3 принято значение,
рассчитанное по началам комбинационных полос v2+v3 и \-\-\, найденным Снайдером и Эдуардсом
[34811, и значениям констант ангармоничности, приведенным в таблице. Это значение частоты v3,
находится в хорошем согласии с полученным в спектре комбинационного рассеяния молекул H2S,
замороженных в азотной матрице B631 см) [3625]. Константы ангармоничности молекулы H2S,
приведенные в табл. 12.9, рассчитаны по началам полос v2 [364], vlf 2v2 [361], v1+va, v2+v3 [3481],.
2v2+v3, v1+v3 [3621 и 2^+^, v^Vjj+Vg [1388]. Постоянная резонанса Дарлинга—Деннисона.
f вычислена по началам полос 2v1+v3 и 3v3 [1711], 3vx+v3 [1118] и vx+3v3 [1711J. Для
вращательных постоянных и постоянных центробежного искажения iaaaa, тШ4, imbb и хееев основного
колебательного состояния H2S в таблице приведены значения, найденные Хельмингером и др. [1876]"
из анализа микроволнового спектра. Постоянные ibbc0 и imce вычислены по соотношениям A. 146).
С этими величинами согласуются постоянные центробежного растяжения, вычисленные по
силовым постоянным в работе [285]. Постоянные колебательно-вращательного взаимодействия
вычислены по эффективным вращательным постоянным H2S в колебательных состояниях
@,0,0), A,1,0), @,1,1), B,1,0) и A,1,1), определенным в работах [1876, 1388, 3481].
Принятым значениям вращательных постоянных соответствуют структурные параметры
г (S—Н) = 1,3356 Аи^ Н—S—Н=92,11°. Электронный спектр H2S исследовался в работах
[3090, 3672, 3890, 3086]. Так же как и у молекулы Н2О, все возбужденные электронные состояния
H2S являются ридберговскими.
Термодинамические функции H2S (г) были вычислены без учета возбужденных электронных
состояний. Составляющие «жесткого ротатора — гармонического осциллятора» находились по
уравнениям A.122)—A.124), A.128) и A.130), поправки на ангармоничность колебаний и колебательно-
вращательное взаимодействие — по уравнениям A.131)—A.333), на центробежное растяжение и
низкотемпературные поправки — по уравнениям A.143)—A.144) и A.147)—A.149), резонанс
Дарлинга—Деннисона учитывался по соотношениям A. 156)—A.158). Погрешности вычисленных
термодинамических функций обусловлены главным образом отсутствием данных о колебательно-
вращательных уровнях энергии с большими значениями квантовых чисел vn и / и применением
приближенного метода расчета. Они составляют 0,05, 0,3 и 1 Дж -К -моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций H2S (г) вычисляли авторы справочников [88,.
2095, 446, 2353, 2556, 2670], Кросс [1119] (Г<1800 К), ЭвансиУагман [1428] (Г < 1500 К), На-
гараян [2768] (Т < 2000 К), Хар и др. [1781], Уилкинс [3944]. Наиболее близки к значениям
термодинамических функций, приведенным в табл. 128, данные Мак-Брайда и др. [2556]; соот-
265
Глава 12
ветствующие расхождения не превышают 0,14 Дж -К -моль в значениях Ф° (Г) и S° (T).
Менее точные расчеты выполнены Уилкинсом [3944] и в таблицах [2095]. Во втором издании
справочника [88 ] в расчете была использована неточная методика учета центробежного растяжения
молекул, в связи с чем расхождения с данными табл. 128 достигают 0,2 и 0,8 Дж -К моль в
значениях Ф° C000 К) и Ф° F000 К). Джиок и Блу [1616] рассчитали из калориметрических данных
стандартную энтропию H2S(r). Полученное значение S° (H2S, г, 298,15 К)=205,4+0,4 ДжХ
X К моль в пределах погрешности определения совпадает с приведенным в табл. 128,
равным 205,69+0,05 Дж-К-моль.
Константа равновесия реакции H2S (г)=2Н (r)+S (г), приведенная в табл. 128, вычислена
ю использованием значения АГН° @)=724,53 +0.54 кДж-моль, соответствующего принятой
¦в справочнике энтальпии образования H2S (г)
AfH°(H2S, г, 298,15 К) = — 20,6+0,5 кДж • моль.
Принятая величина основывается на анализе данных, приведенных в табл. 12.12, и является
средней из вычисленных по работам [4059, 118]. Энтальпия образования H2S (г), найденная по
Таблица 12.12. Результаты определений энтальпии образования H2S (г) (в кДж • моль)
Авторы
Метод
ДГЯ° @)
II закон
Ш закон
A,#°(H2S, г,
298,15 К)
Поллитцер, 1909 Исследование равновесия реакции 74,15+0,75 73,58 ±0,03 —19,4 ±1,1
[3058] H2S(r)+2I (к)=2Н1 (r)+S (к) в
проточной системе, 313—354 К; 26 точек
Льюис, Рандалл Исследование равновесия реакции — —159,9 +2,3 —14,7
1918 [2414а] 2H2O(r)+3SM=2H2S(r)+SO2(r)
статическим методом, 717,75 К; 6 точек
Цеймер, Рот, 1934 Калориметрическое измерение энталь- — — —20,50 ±1,0"¦
[4059] пии сгорания H2S(r) в избытке кисло- —20,79±1,06
рода, 20° С, 18 измерений
Капустинский, Кань- Калориметрическое измерение энталь- — — —20,57 ±0,50
ковский 1958 [118] пии сгорания H2S(r) при недостатке
кислорода, 25° С, 10 измерений
Дайблер, Листон Фотодиссоциационное определение энер- — — —21,8+2,1
1968 [1234] гии реакции H2S+Av=S++H2+e,
LrH° @)=129,3 кДж.моль
а Энтальпия разведения H2S04 по [258]. б То же по [3375].
результатам измерений Цеймера и Рота [4059], существенно зависит от данных по энтальпии
разведения H2SO4, принятых для расчетов. В таблице приведены величины, вычисленные с
использованием рекомендации справочников [258, 3375]. В экспериментах Капустинского и Каньков-
ского [118] предполагалось, что свободная сера, образующаяся в продуктах сгорания, находится
в ромбоэдрическом состоянии, что не являлось бесспорным. Однако хорошее согласие величин,
полученных по данным работ [118, 4059] для разных реакций, позволяет исключить возможность
систематической ошибки в принятой величине.
В пределах погрешности с принятым значением совпадают результаты исследования
равновесия реакции [3058] и фотоионизационные измерения [1234]. Измерения [2414а], возможно,
искажены неучтенными побочными реакциями. Данные более ранних исследований b.fH° (H2S, г)
критически рассмотрены в работах [4059, 642, 1428], в настоящее время они потеряли свое
значение.
H2SO4 (г). Термодинамические свойства газообразной серной кислоты в стандартном состоянии
яри температурах 100—6000 К приведены в табл. 129.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.6. Исследования инфракрасного спектра паров H2SO4 [924] и отклонения
молекулярного пучка H2SO4 в неоднородном электрическом поле [827], а также проведенное авторами
2R6
Сера и ее соединения H2SO4(r)
настоящего издания теоретическое изучение структуры H2SO4 [275] показали, что эта молекула
принадлежит к точечной группе симметрии С2,. В соответствии с этим и на основании данных
о структуре молекулы H2SO4 в кристаллической серной кислоте [2963, 2964], а также о структуре
молекул SO2F2, SO3, H2O и Н2О2, в настоящем справочнике приняты следующие геометрические
параметры молекулы H2SO4: r (S—О)=1,41 +0,01 А; г (S—ОН)=1,53+0,01 А; г (О—Н)=0,96 +
±0,01 A; 2L O-S=O=124+2°, 2L HO-S-OH=97+2°, i?S-0-H=95+5o. Произведение
главных моментов инерции H2SO4, приведенное в табл. 12.6, вычислено по этим параметрам, его
погрешность не превышает 7-Ю16 г3 см6. Молекула H2SO4 имеет 15 основных частот. Все частоты
¦активны в спектре комбинационного рассеяния, в инфракрасном спектре активны все частоты,
за исключением v7 и v8. Инфракрасный спектр паров серной кислоты исследован в области 400—
4000 см [924, 3547], спектр кристаллической серной кислоты исследовался в работах [1634,
1710], а результаты исследований инфракрасных спектров жидкой серной кислоты и ее водных
растворов рассмотрены Стопперка [3544]. Спектры комбинационного рассеяния жидкой серной
кислоты и ее растворов изучались в работах [400, 1633а, 3868] (см. также обзор в [400]). Спектр
комбинационного рассеяния кристаллической H2SO4 исследовался в работе [1710]. Приведенные
в таблице значения основных частот vlt v2, v3, v4, v5, v9, v10, vu, v13, v15 молекулы H2SO4 приняты на
основании результатов исследования инфракрасного спектра паров серной кислоты [924, 35471.
Погрешности оцениваются в 2 см для всех частот, кроме v4 и v13 и в 3 см для частот v4 и v13.
Приведенные в табл. 12.6 значения частот крутильных SOH-колебаний v7 и v14 оценены по частоте
крутильного колебания молекулы Н2О2 [2048], значения частот деформационных колебаний
v6, v12 и крутильного колебания v8 группы S (ОНJ оценены на основании сравнения
соответствующих по характеру колебаний основных частот молекул H2SO4 и SO2F2. Погрешности приведенных
в таблице значений частот v7 и v14 оцениваются в 50 см и частот ve, v12 и v8 — в 20 см.
Термодинамические функции H2SO4 (г), приведенные в табл. 129, вычислялись по уравнениям
A.3)—A.10), A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор — гармонический
осциллятор» и без учета возбужденных электронных состояний. Погрешности вычисленных
значений термодинамических функций обусловлены неточностью принятых молекулярных
постоянных и приближенным характером метода расчета. Погрешности в значениях Ф° (Т) при 298,15,
3000 и 6000 К оцениваются в 2, 8 и 12 Дж -К -моль.
Ранее таблицы термодинамических функций H2SO4 (г) вычисляли Жигер и Савуа [1634]
(Т< 1500 К), Гопинатхи Рао [1687] (Т < 1000 К) и авторы справочника [2095]. В работах [2095
и 1687] расчеты были выполнены также как в настоящем справочнике. Жигер и Савуа [1634]
учитывали заторможенное вращение групп ОН по методу Питцера—Гуинна, принимая, что
потенциальная энергия внутреннего вращения H2SO4 имеет три максимума, а высота барьеров для
внутреннего вращения равна 455 см. Для основных частот H2SO4 во всех работах принимались
значения, существенно отличающиеся от приведенных в табл. 12.6 и основанные на результатах
исследований спектров кристаллической и жидкой H2SO4, а также оценках. В связи с этим
вычисленные термодинамические функции существенно отличаются от приведенных в табл. 129.
Расхождения в значениях Ф° (Т) при 298,15, 3000 и 6000 К с данными работы [2095] достигают
6, 11 и 12 Дж-К-моль соответственно, с данными работы [1687] расхождения значительно
больше и достигают 40 Дж -К-моль в S° A000 К), по-видимому, из-за ошибок в расчете.
Константа равновесия реакции H2SO4(r)=2H(r)+S(r)+4O(r) вычислена с использованием
значения Ь.ГН° @)=2412,985+2 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A/Z7°(H2S04, г, 0) = —719 ± 2 кДж ¦ моль
Эта величина основана на результатах исследований равновесия реакции H2SO4 (г) =
= H2O(r)+SO3 (г), представленных в табл. 12.13.
Как было показано в работах [924, 3547, 3548, 65], при 300—500 К в состав продуктов
испарения серной кислоты (до 95%) входят H2SO4, SO3 и Н2О. В парах более концентрированных
растворов и олеумов присутствуют также H2S2O7 и S3O9, учесть образование которых не представляется
возможным из-за отсутствия соответствующих данных. При вычислении по данным табл. 12.13 (III
закон) среднего значения АГН°@) не учитывались 5, 6 и 7-я серии измерений [684, 685], а также 1-я
и 2-я серии измерений [249, 65], в которых количество испарившейся серной кислоты определялось
не по исходной навеске, как в 3-й серии, а менее надежным методом измерения общего давления
267
SF(r)
Глава 12
Таблица 12.13. Результаты исследований равновесия реакции H2SO4 (г) = Н2О (г) + SO3 (г)
(в кДж • моль)
Авторы
Метод
Боденштейн, Ката- Статический метод, измерение общего дав-
яма, 1909 [684, 685] ления ненасыщенного пара
1-я серия, 621—741 К, 94,40% H2SO4,
5 точек
2-я серия, 646—756 К, 94,49% H2SO4,
7 точек
3-я серия, 613—729 К, 85,1%H2SO4, 9 точек
4-я серия, 596-717 К, 85,05% H2SO4,
8 точек
5-я серия, 610—747 К, 100,5% H2SO4
(олеум), 8 точек
6-я серия, 629—750 К, 100,5% H2SO4
(олеум), 8 точек
7-я серия, 611—714 К, 100,4% H2SO4
(олеум), 9 точек
Суворов и др., 1965 Статический метод, измерение общего дав-
[249, 65] ления насыщенного и ненасыщенного пара
1-я серия, 583—801 К, 94% H2SO4, 20 точек
2-я серия, 561—700 К, 65% H2SO4, 9 точек
3-я серия, 613—713 К, 95% H2SO4, 20 точек
ДгЯ°@)
11 закон
77 ±10
83 ±11
91+5
90 ±8
81 ±10
71 ±7
83 ±10
—
135+30
92 ±4
III закон
91,5 ±1,5
91,6 ±0,4
90,0+0,2
89,8 ±0,4
90,9 ±0,8
92,4 ±0,8
94,6 ±0,3
102+4
81+2
92,6+0,2
продуктов диссоциации при высоких температурах. В работах [684, 685, 249, 65] измерения были
выполнены при температурах, достигающих 800 К, т. е. в условиях, когда необходимо учитывать
диссоциацию SO3(r) на SO2(r) и О2(г). -В этих работах диссоциация SO3 учитывалась приближенно.
При подготовке настоящего издания был проведен полный пересчет этих работ с точным учетом
образования SO2 и О2.
Приведенные в табл. 12.13 погрешности отражают только воспроизводимость измерений.
Погрешность среднего взвешенного значения Дг#°@)=90+2 кДж -моль оценена с учетом
погрешностей термодинамических функций, учтенных в расчетах компонентов реакций, и
неточности, обусловленной пренебрежением образования S3O9 и H2S2O7 в парах концентрированной
серной кислоты.
SF (г). Термодинамические свойства газообразного фторида серы в стандартном состоянии
при температурах 100—6000 К приведены в табл. 130.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.3. Так же как и изоэлектронные молекулы СЮ и SO", молекула SF имеет
основное состояние Х2И{ и первое возбужденное состояние А2П(. В литературе имеются данные только
о постоянных SF, полученные при исследованиях микроволнового [370] и электронного спектров
[1257] и спектра ЭПР [887]. Значения вращательных постоянных SF, приведенные в табл. 12.3,
основываются на работе Амано и Хирота [370] и анализе вращательной структуры полос 3—0
и 5—0 системы А2^—Х211{, выполненном Ди Лонардо и Тромбетти [1257]. Частота колебания
SF в состоянии Х2Я{ принята по оценке О'Харе [2892]. Погрешность значения <ое О'Харе
оценил в 20 см. Постоянная спин-орбитальной связи для состояния Х2П,. была найдена в работе
[1257] при анализе колебательной структуры системы полос А2Я{—Х2Я{. Она удовлетворительно
согласуется с менее точными значениями, полученными Каррингтоном и др. [887] при анализе
спектра ЭПР и Брауном и др. [809] в результате теоретического расчета. Значения постоянных SF
в состоянии А2Л{ приняты по данным работы [1257].
Термодинамические функции SF (г), приведенные в табл. 130, были вычислены по уравнениям
A.3)—A.10), A.93)—A.95). Значения QBB, Qm и Qm находились по соотношениям A. 90)—A.92),
в которых статистическая сумма по колебательно-вращательным уровням энергии состояния
Х2П,. и ее производные были рассчитаны непосредственным суммированием по колебательным
уровням энергии и интегрированием по значениям / с помощью уравнений типа A.82). Энергии
268
Сера и ее соединения SF" (г)
уровней вычислялись по уравнениям A. 65) и A. 42) со значениями коэффициентов Ykl и А,
приведенными в табл. 12.4. Вырождение вращательных уровней учитывалось статистическим весом 2.
В расчете учитывались все уровни энергии со значениями / ^/Шах,»> гДе «Лпах,. находились по
соотношениям A.81). В таблице приведены соответствующие значения vmtx и 7ц5. Составляющие
состояния А2И( вычислялись в предположении, что Q^.sp— Qmi. вр- Погрешности
термодинамических функций SF (г) обусловлены отсутствием экспериментальных данных о колебательных
постоянных молекулы SF в основном состоянии и об энергии высоких колебательно-вращательных
уровней этого состояния. Они могут достигать 0,2, 0,5 и 1 Дж -К" -моль в значениях Ф° (Г)
при 298,15, 3000 и 6000 К.
Таблицы термодинамических функций SF (г) вычислялись в справочниках [87, 88, 2556] и
в работе [2670] (Т ^ 2000 К) по оцененным молекулярным постоянным состояния Х2П.(, без учета
¦спин-орбитального расщепления. Последнее объясняет то, что в расчетах, проведенных в
справочниках [87, 88, 2556], были получены завышенные значения Ф° B98,15 К), отличающиеся от
приведенных в табл. 130 на 4,6 Дж -К" -моль. При высоких температурах расхождения в значениях
Ф° (Т) достигают 0,4 Дж-К-моль с данными справочника [2556] и 0,8 Дж-К^-моль с
данными справочника [88].
*' Константа равновесия реакции SF (r)=S (r)-f-F (г) вычислена с использованием значения
Дг#° @) = D0 (SF)=337,26±6,0 кДж-моль ~28 200+500 см, соответствующего принятой в
настоящем справочнике энтальпии образования
AfH°(SF, г, 0)= 14,8 + 6,0 кДж • моль.
Принятое значение основано на результатах исследования реакции GS2 (r)+2SF (r)=CF2 (r) +
+2S2 (г), выполненного Хилденбрандом [1936] масс-спектрометрическим методом A510—1611 К).
Расчет по уравнению III закона привел к величине Д^0 @) = —65,6+8,0 кДж-моль, при оценке
точности которой были учтены погрешности термодинамических функций.
Ди Лонардо и Тромбетти [1257 ] при исследовании электронного спектра SF обнаружили предис-
социацию в состоянии A2U.{ в области 26 500 см и пришли к выводу, что Do (SF) ^ 317 кДж -моль.
В работе [1257] отсутствуют надежные данные о наблюдавшейся предиссоциации, что
затрудняет оценку достоверности значения, полученного ее авторами и установление причин
расхождения данных работ [1257, 1936].
SF~ (г). Термодинамические свойства газообразного отрицательного иона фторида серы в
стандартном состоянии при температурах 298,15—6000 К приведены в табл. 131.
I, Молекулярные постоянные, использованные для расчета термодинамических функций SF" (г),
приведены в табл. 12.3. Спектр SF" экспериментально не исследовался. В согласии с работами
О'Харе [2898, 2892] в справочнике принимается, что SF", как и изоэлектронная молекула C1F,
имеет основное состояние Х1^*, соответствующее электронной конфигурации . .. о2л*л*. Это
состояние не может коррелировать с первым диссоциационным пределом S CP)-\-F~AS) и, по-видимому,
коррелирует с пределом S~ BP)+F BР). Первым возбужденным состоянием SF", как у всех
молекул, имеющих в основном состоянии конфигурацию электронной оболочки ... а2тс4л4,
должно быть состояние 3П с конфигурацией .. .а2тс4v?о, способное коррелировать с пределом SC.P)+
+F~(X<S). Оценка энергии возбуждения этого состояния затруднительна. Если предположить, что
она близка по величине к энергии состояния 3П изоэлектронной молекулы C1F, то это состояние
должно быть расположено вблизи ионизационного предела SF~ A8 000+1000 см и 20 000 +
+4000 см соответственно) и иметь небольшое число колебательно-вращательных уровней
энергии или быть отталкивательным состоянием. Однако нельзя исключить, что SF" обладает
связанными валентными состояниями с энергией —10 000 см. Приведенные в табл. 12.3 молекулярные
постоянные SF" в состоянии Х^Б* оценены. Постоянные шв иВ, приняты равными постоянным
SF, поскольку теоретические расчеты О'Харе и Уола [2898] показали, что потенциальные кривые
SF" и SF имеют минимумы при близких гв. Погрешности принятых значений
оцениваются в 50 см и 0,01 см соответственно. Остальные постоянные в состоянии ХХ2 + оценены на
основания принятых значений ше, Ве и энергии диссоциации SF".
Термодинамические функции SF~(r) вычислены по уравнениям A.3)—A.10), A. 93)—A. 95).
(?мп <2вн ж <?»« вычислялись без учета возбужденных электронных состояний. Статистическая сумма
яо колебательно-вращательным уровням энергии состояния X1S+ и ее производные были рассчн-
269
SFa(r) Глава 12
таны непосредственным суммированием по колебательным уровням и интегрированием по
вращательным уровням энергии с помощью уравнений типа A. 82). Уровни энергии вычислялись no-
уравнениям A. 65) и A.62) со значениями коэффициентов Ykl, приведенными в табл. 12.4. В расчете-
учитывались все уровни со значениями / ^ /тах, ,> где /maXj, находились по уравнениям A. 81).
В таблице приведены соответствующие значения уШ1Ы и -^нт- Погрешности табулированных
значений термодинамических функций SF~(r) определяются отсутствием экспериментальных данных
о молекулярных постоянных и колебательно-вращательных уровнях энергии состояния ХХ2 + ,
неточностью данных об энергии диссоциации SF~ в этом состоянии, а также отсутствием сведений
о возбужденных электронных состояниях. Погрешности в значениях Ф° (Т) при 298,15, 3000 и
6000 К оцениваются в 0,3, 1 и 4 Дж-К~1-моль~1.
Ранее таблица термодинамических функций SF" (г) не публиковалась.
Константа равновесия реакции SF"(r)=S(r)+F(r)+e(r) вычислена на основании значения
Дг#° @)=577,26+50 кДж -моль, соответствующего принятому в справочнике сродству SF к
электрону
Ао (SF) = —2,5 + 0,5 эВ 240 + 50 кДж • моль.
Эта величина была получена О'Харе иУолом [2898] на основании расчетов по методу Хартри—
Фока и приближенной оценки корреляционной поправки. Позднее О'Харе [2892] рекомендовал-
более низкое значение A,8 эВ), полученное в расчете по теореме Купманса. В справочнике отдано
предпочтение более высокой величине, так как она находится в лучшем согласии с принятыми
значениями сродства к электрону молекул SF3 и SF5. Кроме того, оценка корреляционной поправки
для SF в работе [2898] представляется достаточно разумной, поскольку вычисленное с ее учетом
значение потенциала ионизации SF находится в хорошем согласии с экспериментальными
данными [1936].
Принятому значению Ао (SF) соответствуют
A/ffo(SF", г, 0) = —225,2 + 50 кДж • моль-1,
Do (SF-) = 249,25 + 50 кДж • моль ~ 20 800;± 4 000 см.
SF2 (г). Термодинамические свойства газообразного дифторида серы в стандартном состоянии
при температурах 100—6000 К приведены в табл. 132.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. Молекула SF2 в основном электронном состоянии имеет нелинейную
симметричную структуру (точечная группа симметрии С2,). Приведенное в табл. 12.10 произведение
главных моментов инерции SF2 вычислено по значениям вращательных постоянных S2SF2: -4000=
= (838 268,0 +15,7) -10; Бооо=C07 257,9+9,7) -КГ» и С000=B28 354,9+9,7)-Ю см~\
найденным Джонсоном и Пауэллом [2134] в результате анализа микроволнового спектра продуктов
разряда через SFe. Им соответствуют структурные параметры r0 (S—F)=l,589 А. и Д F—S—F= 98°16'.
Для основных частот SF2 в табл. 12.10 приведены значения, вычисленные Блифертом и Ванце-
ком [675] по силовым постоянным SF2, оцененным на основании сравнения структурных
параметров SF2 с соответствующими структурными параметрами и силовыми постоянными молекул SF4r
SOF2, S2F2, SFe, SO2F2, NSF и NSF3. Вычисленное значение частоты v3 практически совпало с
полосой при 830 см, наблюдавшейся Зеелем и др. [3371] в инфракрасном спектре газообразных
продуктов реакции серы с AgF, отнесенной к молекуле SF2, образующейся при диссоциации S2F4.
В инфракрасном спектре продуктов фотолиза молекул SF4, замороженных в аргоновой матрице
при 8 К, наблюдалась полоса при 846,8 см [3446], приписанная частоте v3 молекулы SF2. Данные,
полученные в [3446], не противоречат данным, полученным в работе [675], так как в условиях
экспериментов мог иметь место сдвиг частоты из-за взаимодействия с молекулами SF4 и матрицей.
Погрешности принятых значений частот SF2 оцениваются в 10 см. В справочнике принимается,
что молекула SF2 имеет синглетное основное электронное состояние. Сведения о возбужденных
электронных состояниях SF2 отсутствуют, хотя не исключено, что они имеют низкие энергии.
Термодинамические функции SF2 (г), приведенные в табл. 132, вычислены в приближении,
«гармонический осциллятор — жесткий ротатор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128) и A.130) без учета возбужденных электронных состояний. Погрешности вычисленных
значений термодинамических функций обусловлены неточностью принятых значений основных
частот SF2, отсутствием данных о возбужденных электронных состояниях и применением прибли-
270
Сера и ее соединения SF3 (г>
женного метода расчета. Для Фс (Г) при 298,15, 3000 и 6000 К они оцениваются в 0,3, 2 и
4 Дж К-моль.
Ранее таблицы термодинамических функций SF2 (г) вычисляли авторы справочников [87, 88,
2556, 2670] по оцененным молекулярным постоянным. Наиболее близки к значениям
термодинамических функций SF2(r), приведенным в табл. 132, данные справочника [88], расхождения в
значениях Ф° (Т) и S° (Г) не превышают 0,5 Дж-К~1-моль~1 и обусловлены главным образом
различием принятых структурных параметров SF2. Расхождения с данными [2556] составляют около
2 Дж-К^-моль.
Константа равновесия реакции SF2(r)=S(r)+2F(r) вычислена с использованием значения
ЬГН° @)=720,335 +10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —291 + 10 кДж • моль.
Принятая величина вычислена по результатам масс-спектрометрического исследования
равновесия реакций CS (r)+SF2 (r)=CF2 (r)+S2 (г) и S (r)+SF2 (r)=2SF (г) при 1400—1600 К,
выполненного Хильденбрандом [1936]. С учетом погрешностей термодинамических функций по
уравнению III закона были найдены значения \Н° @), равные соответственно —47,2+8 кДж-моль
и 46,1+8 кДж-моль. Комбинируя значение АГН° для первой реакции со значением Дг.йГ° @) =
=24,7 кДж-моль для реакции CS2 (r)+S (r)=CS (r)+S2 (г), найденным Хилденбрандом [1935],.
было получено AJ7° (SF2, г, 0) = —291+13 кДж-моль. Данные [1936] для второй реакции
приводят к значению—291+14 кДж • моль. Практически совпадающее значение (—290 +
+20 кДж -моль) может быть вычислено по потенциалам появления иона F", найденным Харлан-
дом и Тинне [1843] при изучении ионизации SFe и SF5C1.
SF3 (г). Термодинамические свойства газообразного трифторида серы в стандартном состоянии'
при температурах 100—6000 К приведены в табл. 133.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. В настоящем издании принимается на основании результатов исследования
ЭПР-спектров SF3, выполненного Колусси и др. [1027], и правила Пирсона [2983], что в основном
электронном состоянии молекула SF3 имеет плоскую Г-образную структуру, соответствующую
точечной группе симметрии С2„. Предполагая, что структурные параметры SF3 близки к
соответствующим структурным параметрам молекулы SF4, для расчета произведения главных моментов,
инерции SF3, приведенного в таблице, были приняты структурные параметры: г (S—F')=l,65 +
+ 0,05 А, г (S—F") = l,55 + 0,05 А, Д F'—S—F'=175 + 5°. Погрешность значения 1А1Б1с
оценивается в 5-10~115 г3-см6. Из шести основных частот SF3 две идентифицированы в инфракрасных
спектрах. Ботт [709], исследуя диссоциацию SF4, наблюдал интенсивную эмиссию с максимумом
при 794 см в спектре за фронтом ударной волны с температурой от 2050 до 2830 К и отнес ее
к частоте vx полносимметричного колебания SF3. Смарджевский и Фокс [3446], исследуя спектры
продуктов фотолиза молекул SF4 в аргоновой матрице при 81 К, пришли к выводу, что
интенсивная полоса при 682,2 см принадлежит SF3 и может быть отнесена к его антисимметричному
валентному колебанию v4. Значение частоты vu найденное Боттом [709], близко к частоте
соответствующего колебания молекулы C1FS, несколько превосходя последнюю и занимая промежуточно»
положение между значениями частот полносимметричных колебаний молекул C1F3 и SF4. В связи
с этим для остальных частот SF3 приведенные в табл. 12.10 значения приняты как средние между
частотами молекул C1F3 и SF4. Погрешности этих значений оцениваются в 2 см для vlt 20 см
для v2, v4, v6, v6 и 40 см" для v3. Ввиду наличия у SF3 нечетного числа электронов принято, что
основное электронное состояние SF3 является дублетным. Сведения о возбужденных электронных
состояниях SF3 в литературе отсутствуют.
Термодинамические функции SF3 (г), приведенные в табл. 133, вычислены, в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—1.124),
A.128) и A.130). Расчет проводился без учета возбужденных электронных состояний. Погрешности
вычисленных значений термодинамических функций SF3 (г) обусловлены неточностью принятых
молекулярных постоянных, применением приближенного метода расчета и пренебрежением
возбужденными состояниями. Погрешности вычисленных значений Ф°(Г)при 298,15, 3000 и 6000 К
могут достигать 1, 4 и 7 Дж-К^-моль.
271
SFj (г) Глава 12
Ранее термодинамические функции SF3 (г) вычислял Уилкинс [3945 ] до 3000 К по оцененным
молекулярным постоянным. В заметке [3945] не приведены термодинамические функции SF3 (г),
однако они должны содержать значительные ошибки, так как расчет основывался на неверной
структуре молекулы SF3.
Константа равновесия реакции SF3(r)=S(r)+3F(r) была вычислена с использованием
значения АГН° @)=1006,61+20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfff°(SFa, г, 0) = — 500 + 20 кДж-моль,
основанной на измерениях энергий связей SF2—F и SF3—F. Ботт [709] нашел Do (SF2—F)==
=300 кДж-моль и Do (SF3—F)=331+8 кДж-моль при исследовании кинетики диссоциации
SF4 в ударных волнах A650—1950 К) по спектрам испускания SF3 и SF4. Найденным им величинам
•соответствуют Д J7° (SF3, г, 0) = —514 + 13 и —499 + 22 кДж-моль. Близкие значения были
получены Хилденбрандом [1936] по потенциалу появления иона SF* при ионизации SF4 (более
—503 кДж-моль) и Харландом и Тинне [1844] по порогу реакции SF4-(-e==SF3+F~ (менее
—483 кДж-моль). Расчет энтальпии образования SF3 (ту по потенциалам появления ионов F"
¦и С1" из SFe и SF5C1, найденным Харландом и Тинне в более ранней работе [1843], приводит
к противоречивым результатам.
SF§; (г). Термодинамические свойства газообразного отрицательного иона трифторида серы
в стандартном состоянии при температурах 298, 15—6000 К приведены в табл. 134.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. Поскольку ион SF" изоэлектронен молекуле G1F3, а экспериментальные
данные о структуре и постоянных SFJ отсутствуют, в справочнике принимается, что SF~ имеет синглет-
ное основное электронное состояние и молекулярные постоянные, близкие к постоянным G1F3.
Приведенное в табл. 12.10 произведение главных моментов инерции вычислено при условии, что
в основном электронном состоянии SF3 имеет плоскую Г-образную симметричную структуру
(точечная группа симметрии C2t) и структурные параметры г (S—F')=l,70+0,05 А; г (S—F") =
= 1,60±0,05 А; 4 F'-S-F"=87,5 + 2,5°; Л F'-S-F'=185+5°. Погрешность значения IJBI0
«оставляет 7-1015 г3-см6. Значения основных частот SF" оценены в предположении, что в
пределах 5% они должны совпадать с частотами C1F3. Их погрешности могут достигать 40 см для vx
и v4, 30 см для v2 и 20 см для v3) v5 и ve.
Термодинамические функции SF" (г), приведенные в табл. 134, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A. 10), A.122)—A. 124),
{1.128) и A.130) без учета возбужденных состояний SF". Погрешности вычисленных значений
термодинамических функций обусловлены неточностью принятых значений молекулярных
постоянных, отсутствием данных о возбужденных состояниях SF" и применением приближенного метода
расчета. Для Ф° (Т) при 298,15, 3000 и 6000 К они оцениваются равными 2, 6 и 10 Дж -К -моль.
Ранее термодинамические функции SF" (г) не публиковались.
Константа равновесия реакции SF" (r)=S (r)+3F (г)+е (г) вычислена с использованием
значения АГН° @)=1286,61 + 21 кДж -моль, соответствующего принятому в справочнике сродству
к электрону молекулы SF3:
Ао (SF3) = —2,90 + 0,07 эВ = —280,0 + 7,0 кДж • моль.
Эта величина получена на основании потенциалов появления ионов F" и SFg в реакциях SF4(r)-f-
-j-*(r)=SF3(r)+F-(r) и SF4 (г)+е (r)=SF-(r)+F (г), измеренных Харландом и Тинне [1844]
в масс-спектрометрических экспериментах и равных 19,3+4,8 кДж-моль-1 и 67,5+ 4,8 кДж-моль.
Близкое значение (—260+50 кДж-моль) было получено в работе Пейджа и Гуда [2939]
(см. [3454]) с использованием магнетронной техники.
Измерения потенциалов появления ионов F" и SF* из SF5G1 [1843] не позволяют вычислить
достаточно надежное значение Ао (SF3), так как реакции, приводящие к образованию этих ионов,
неизвестны, а продукты реакции могут обладать избыточной кинетической энергией [1844].
Принятому значению Ао (SF3) соответствует
г, 0) = — 780,0 + 21 кДж-моль.
272
Сера и ее соединения SF4(r), SF5(r)
SF4 (г). Термодинамические свойства газообразного тетрафторида серы в стандартном
состоянии при температурах 100—6000 К приведены в табл. 135.
Молекулярные постоянные, использованные при вычислении термодинамических функций,
приведены в табл. 12.10. Электронографические исследования [2244, 1430], исследования
колебательных [1275, 438, 3178, 2400, 9556, 957, 2401, 1542, 2399, 601] и микроволновых спектров
[1778, 3705, 2066а], а также спектра ЯМР [1075, 2733] и теоретические расчеты [1645, 3117]
показали, что молекула SF4 в основном электронном состоянии ХгА имеет структуру,
соответствующую точечной группе симметрии С2,. Произведение главных моментов инерции, приведенное
в табл. 12.10, рассчитано по вращательным постоянным, найденным Толлесом и Гвином [3705]
из анализа микроволнового спектра SF4 (^40=0,223 078 см, i?0=0,136 317 см, С0=0,107 406 см).
Им соответствуют структурные параметры г (S—F,K0) = 1,646 + 0,003 А, г (S—Fpia)=l,545+0,003 А,
2FaKe—S-FaKO=173°4' + 30', Д FpaJ-S—Fpa=101°53'±30', находящиеся в согласии со
значениями, полученными Кимурой и Бауэром [2244] в результате электронографических
измерений. Приведенные в таблице значения основных частот SF4 рекомендованы Кристе и др. [9556]
на основании изучения спектров КР твердого, жидкого и газообразного SF4 и ИК-спектров
твердого SF4 и молекул SF4, изолированных в твердом азоте, анализа результатов исследований
колебательных спектров SF4 в работах [1275, 3178, 2400, 957, 1542, 2399, 2401, 601, 959], а также
расчетов силовых постоянных и среднеквадратичных амплитуд колебаний молекулы SF4.
Погрешности принятых значений оцениваются в 1—3 см.
Термодинамические функции SF4 (г), приведенные в табл. 135, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128) и A.130) без учета возбужденных электронных состояний молекулы SF4. Погрешности
вычисленных значений термодинамических функций обусловлены преимущественно применением
приближенного метода расчета. При высоких температурах могут стать существенными
погрешности из-за того, что в расчете не учитывались возбужденные состояния SF4. Погрешности в
значениях Ф° (Т) при 298,15, 3000 и 6000 К могут достигать 0,6, 6 и 10 Дж-К^-моль-1. Ранее
термодинамические функции SF4 (г) вычисляли авторы предыдущих изданий настоящего справочника
[87, 88], таблиц JANAF [2045], Мак-Брайд и др. [2556], Радхакришнан [3113] (до 1500 К),
Миллс [2670] и Туманов и др. [264, 265, 66] (до 2000 К). Из-за использования в этих расчетах
неточных значений структурных параметров и основных частот молекулы SF4 их результаты
существенно отличаются от данных табл. 135. Эти расхождения для работ [88, 2095, 2556]
составляют во всем интервале температур 2,5—5 Дж-К^-моль в значениях Ф° (Т) и S° (T).
Константа равновесия реакции SF4 (r)=4F (r)+S (г) вычислена с использованием значения
КГН° @) = 1337,205 + 20 кДж -моль, соответствующего принятой в справочнике энтальпии
образования
bfH°{S>Yv г, 298,15 К) = —760 + 20 кДж • моль.
Эта величина принята на основании результатов калориметрических исследований Николса
[2849а] (см. также [2894]) и Бона и Мюттертиса [3784], пересчитанных с учетом значения
AfH°(F~, aq), рекомендованного КОДАТА—MGHG [1000], и исследования методом электронного
удара, выполненного Хилденбрандом [1936]. В работе [2849а] измерена энтальпия гидролиза
SF4 (г), а в [3784] — энтальпии реакций SF4 (г)+Н2 (г); вычисленные по этим данным значения
Д.#° B98,15 К) равны —760 + 20 и —770 + 20 кДж-моль соответственно. Хилденбранд на
основании измеренного потенциала появления иона SF* из SF6 и потенциала ионизации SF4 нашел
hfH°, равное —753 + 20 кДж-моль. Измерения, выполненные в работах [2204, 1843, 1844
и 2894], приводят к противоречивым результатам и не учитывались при выборе принятого значения.
SF5 (г). Термодинамические свойства газообразного пентафторида серы в стандартном
состоянии при температурах 100—6000 К приведены в табл. 136.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. На основании применения к SF5 правила Пирсона [2983], теоретических
расчетов Грегори [1726 ] и исследования спектра ЭПР [1857а] в настоящем издании принимается,
что в состоянии Х2Аг молекула SF5 имеет структуру квадратной пирамиды (симметрия С4,).
Произведение главных моментов инерции SF5, приведенное в табл. 12.10, вычислено на основании
структурных параметров: г (S-FMC) = 1,68 ±0,03 А, г (S-FpJ = l,72 + 0,03 А, Я Fp4-S-Fpw=
273
Глава 1?
=92 + 1°. Его погрешность оценивается в 3 -1014 г3 -см6. Приведенные в табл. 12.10 значения
основных частот SF5 оценены на основании основных частот SFg и двух частот (vx и v2), найденных Смард-
жевским и Фоксом [3448] в инфракрасном спектре молекул SF5, замороженных в аргоновой
матрице. Погрешности значений Vj и v2 оцениваются в 5 см, а остальных частот — в 10 —30 см.
В литературе отсутствуют сведения о возбужденных электронных состояниях молекулы SF6;:
вероятно, она должна иметь низколежащие возбужденные состояния.
Термодинамические функции SF5 (г), приведенные в табл. 136, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128) и A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных SF5, пренебрежением возбужденными электронными состояниями и
применением приближенного метода расчета. В значениях Ф°(Г) при 298,15, 3000 и 6000 К они могут
достигать 4, 10 и 15 Дж-К^-моль.
Ранее термодинамические функции SF6 (г) вычислялись до 3000 К Уилкинсом [3945 ] по
оцененным молекулярным постоянным, при условии, что молекула SF5 имеет структуру треугольной
бипирамиды (точечная группа симметрии C3t). Однако в заметке [3945] численные значения
термодинамических функций SF5 (г) не приведены.
Константа равновесия реакции SF6(r)=S(r)+5F(r) вычислялась с использованием значения
АГН° @) = 1621,16 + 10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/firo(SF5, г, 0) = —960 + 10 кДж • моль.
Принятая величина основана на результатах измерений энергии разрыва связи SF5—F в
молекуле SFe, представленных в табл. 12.14.
Таблица 12.14. Результаты определений энтальпии образования SF5 (г) и энергии разрыва
связи в SF6 (в кДж • моль)
Авторы
Курран, 1961 [1132]
Кей, Пейдж, 1964 [2204]
Ботт, Джекобе, 1969 [710]
Харланд, Тинне, 1969 [1843
(см. [1936])
Фрост и др., 1968 [1552];
Фезенфельд 1971 [1457]
(см. [1936])
Модика, 1973 [2686]
Лифшиц и др., 1973 [2432]
Метод
Масс-спектрометрическое определение порога
реакции SF6 (г)+е (r)=SF5 (r)+F~ (г) и
кинетической энергии продуктов реакции
Измерение магнетронным методом
Исследование кинетики термической
диссоциации SFe в ударных волнах при 1650—
2050 К
Масс-спектрометрическое определение порога
реакции SFe (г)+е (r) = SF5 (r)+F" (г).
Масс-спектрометрические измерения АР (SF?)
из SF6 и /0(SF5).
Исследование разложения SFe, SF5C1 в
ударных волнах при 1700—2300 К
Масс-спектрометрический, измерение порогов
ряда ионно-обменных реакций
D0(SF5-F)
309 ±15
360+15
318+10а
или
334+10а
< 328
> 318
273
< 347
AfH° (SF6, г, 0)
—974+15
—923+15
—965+10
или
—949+10
==с —955
> —965
—1010
< -936
а В работе [710] были получены два ряда значений Do (SF5—F) и кинетических параметров реакции,
одинаково хорошо соответствующих результатам измерений.
Как видно из таблицы, результаты определений границ возможных значений &^Н° (SF5, г)
в работах [1843, 1936, 1552, 1457] и среднее значение из результатов кинетического
исследования [710] оказались в хорошем соответствии. Принятое в справочнике значение является
средним из полученных в этих работах. Данные работы [2432] также не противоречат принятому
значению.
SF~ (г). Термодинамические свойства газообразного отрицательного иона пентафторида серы
в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 137.
274
Сера и ее соединения SF6 (г)
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. Как было показано Друллингером и Гриффитсом [1329] на основании
анализа инфракрасного спектра и спектра комбинационного рассеяния порошка CsSF6 и его
растворов, ион SF~ в основном электронном состоянии Х1А1 имеет структуру правильной
тетрагональной пирамиды с атомом серы в центре ее основания (точечная группа симметрии Си) подобно
изоэлектронной молекуле C1F5 (а также BrF5 и IF5). Произведение главных моментов инерции SF",
приведенное в табл. 12.10, вычислено по структурным параметрам г (S—FaTC) = 1,60+ 0,05 А,
r (S-FpJ = l,70±0,05 А, Д FaKC-S-Fpa;,=85 + 50, 4FpM-S-Fpia = 120°, оцененным в работах [1329,
956]. Его погрешность составляет 6-1014 г3-см6. Основные частоты SFJ, приведенные в таблице,
приняты на основании результатов изучения инфракрасных спектров и спектров
комбинационного рассеяния твердого CsSF6 и его растворов в работах [1329, 956] с учетом фазовых сдвигов vx,
v2, v3, v4, v7, оцененных по соответствующим данным для BrF5 и IF5. Погрешности принятых
значений частот vx, v2, v4 и v7 оцениваются в 10 см, для остальных частот — в 5 см.
Термодинамические функции SFr (г), приведенные в табл. 137, вычислены по уравнениям
A. 3) —A. 10), A. 122) —A. 124), A.128) и A.130) в приближении «жесткийротатор —
гармонический осциллятор». Погрешности вычисленных значений термодинамических функций обусловлены
неточностью принятых значений молекулярных постоянных и применением приближенного
метода расчета. В значениях Фр (Т) при 298,15, 3000 и 6000 К они могут составлять 3, 8 и
12 Дж-К^-моль-1.
Ранее термодинамические функции SF5 (г) вычисляли Друллингер и Гриффите [1329] до 2000 К
на основании оцененных ими структурных параметров и основных частот, найденных по
спектру CsSF5. Различия в значениях Ф° (Т), приведенных в работе [1329] и в табл. 137 при 298,15
и 2000 К, составляют около 3 и 5 Дж-К~х -моль.
Константа равновесия реакции SF~(r)=5F(r)+S(r)+e(r) вычислена с использованием
значения Дг.йГо@) = 1917,16+ 14 кДж-моль в соответствии с принятым в справочнике сродством
молекулы SF5 к электрону
Ао (SF5) = —296 + 10 кДж • моль-1.
Эта величина основывается на масс-спектрометрическом исследовании сечения обменных
реакций SF-+NO2=SF5+NO; и SF6+X-=SF~+F+X, где Х = О, F, Вг, I и S, выполненном Лиф-
шицем [2432]. В работе [2432] были определены пороговые значения для обеих реакций, из
которых следует, что — 309 + 15 кДж-моль <^0 (SF5) ^ — 283 + 10 кДж-моль. Принятое
значение находится в удовлетворительном согласии с результатами масс-спектрометрических
измерений порога реакции SFe+e=SFj+F в работах Хиккама и Фокса [1928] и Харланда и Тинне [1843]
(см.[1132]) и порога реакции Cs+SFe=SFr+Cs++F в работе Комптона и Купера [1034]. Из
данных этих авторов следует, что —317 +15 и —314 +15 кДж-моль <; ^40(SF6) <; — 277 кДж-моль
соответственно. В работе [2204] магнетронным методом для А0 (SF5) было получено два значения
—280 и —360 кДж -моль; как правило, результаты измерений этим методом неточны, поэтому
данные работы [2204] не учитывались при выборе принимаемого значения.
Принятому значению A0(SF5) соответствует
AfH°(SFi, г, 0) = —1256 + 14 кДж • моль.
SFe (г). Термодинамические свойства газообразного гексафторида серы в стандартном
состоянии при температурах 100—6000 К приведены в табл. 127.
Молекулярные постоянные, использованные в расчете термодинамических функций SF6 (г),
приведены в табл. 12.10. Молекула SFe в основном электронном состоянии имеет структуру
правильного октаэдра с атомом серы в центре симметрии (точечная группа Oh). Произведение
главных моментов инерции SFe, приведенное в табл. 12.10, вычислено при г (S—F) = l,564 + 0,010 А
по работе Ювинга и Саттона [1430] (см. также [754, 791]). Его погрешность оценивается в
l-lO14 г3-см6. Колебательные спектры газообразного SFe исследовались в работах [1418 1380
1205, 2338, 1573, 338, 825, 2018, 1276, 3243, 354а, 25796, 2578а, 2242а, 1762, 969, 708, 1995] и
SF6 в конденсированных состояниях — в работах [326, 1317, 1418, 4041, 1995, 1762].
Приведенные в таблице значения основных частот SFe рекомендованы Мак-Дауэллом и др. [2578а] на
основании результатов исследования спектра КР газообразного SFe в работе Босуэрфа и др. [708]
(для v1? v2, v5, v6) и ИК-спектра высокого разрешения газообразного SF6 в работах [354а, 25796]
275
SOF2(r)
Глава 12
(для v3 и v4). Их погрешности составляют 1 см г (для v6) и ^0,5 см г для остальных частот. В
работе [2578а] были определены также значения констант ангармоничности SF6, которые следует
рассматривать как предварительные и нуждающиеся в проверке и уточнении. Молекула SFe
имеет ряд возбужденных электронных состояний с энергиями превосходящими 90 000 см~х
[2449, 2871, 1002, 3601].
Термодинамические функции SFe(r), приведенные в табл. 138, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A. 124),
A.128), A.130). Погрешности вычисленных значений определяются главным образом
применением приближенного метода расчета; они составляют 1, 8 и 13 Дж-К^-моль в значениях Ф° (Г)
при 298,15, 3000 и 6000 К, причем погрешности, обусловленные неточностью постоянных, не
превышают 0,3 Дж-К^-моль.
Ранее таблицы термодинамических функций SFe (г) вычислялись в справочниках [87, 88, 2095,
2556] и работах [2717, 2896, 66] (до 1000 К), [197] (до 5000 К). Наилучшее согласие с данными
табл. 138 имеет место для справочника J*\NAF [2095] (в пределах 0,25 Дж-К^-моль в
значениях Ф° (Т) и S° (T)). Расхождения со значениями, табулированными в [88, 2556, 197],
достигают 0,5 [2556] и 0,7 Дж-К^-моль-1 [88, 197].
Келли и Кинг [2221 ] рассчитали стандартную энтропию SF6(r) по результатам измерений
теплоемкости SF6, выполненных в работе [1420]. Найденное ими значение 294+3 Дж-К^-моль
удовлетворительно согласуется со значением 291,5 + 1,0 Дж-К^-моль, приведенным в табл. 138.
Константа равновесия реакции SFe(r)=6F(r)+S(r) вычислена с использованием значения
Дг//°@) = 1944,138 + 1,7 кДж-моль~х, соответствующего принятой в справочнике энтальпии
образования
A/F°(SF6, г, 298,15 К) = —1219,4 + 1,5 кДж • моль.
Эта величина основана на результатах измерений энтальпии сгорания серы во фторе,
приведенных в табл. 12.15. Наиболее точные измерения были проведены в работах [2896, 152, 151].
Таблица 12.15. Результаты калориметрических измерений энтальпии реакции
S (к, ромб.) + 3F2 (г) = SF6 (г) (в кДж • моль)
Ав;оэы
Метод
A/H°(SFS, г, 298,15 К)
Йост, Клауссен, 1933 [4041]
Гросс и др., 1959 [1741, 1740]
О'Харе и др., 1966 [2896]
Леонидов и др., 1973 [152, 151]
Шредер, Зибен, 1970 [3352]
Проточный калориметр, чистота F2 и S не оха- —1096 +20
рактеризована
Калориметрический сосуд из пирекса —1207,1 +3,0
Двухкамерная никелевая калориметрическая —1220,8 ±1,За
бомба; 99,999 %S и 99,97 %F2; давление
фтора 6 атм; 6 опытов
Двухкамерная калориметрическая бомба из —1218,0+1,3
монель-металла; 99,97% S и 99,77% F2;
давление фтора 3 атм; 7 опытов
Двухкамерная никелевая калориметрическая —1219,2
бомба; 99,999%S, 99% F2; 3 опыта
а В качестве погрешности приведён 95%-ный доверительный интервал. В оригинале [2896] приведено
значение погрешности, выраженное как 2з.
В работе [4041], по-видимому, был использован фтор, содержащий большое количество примесей.
Работы [1741, 1740 и 3352] изложены в краткой форме, не позволяющей провести их достаточно
полный анализ.
Принятое в справочнике значение является средним из найденных в работах О'Харе и др.
[2896] и Леонидова и др. [152, 151] и практически совпадает с полученным Шредером и Зибеном
13352].
SOF2 (г). Термодинамические свойства газообразного оксид-дифторида серы в стандартном
состоянии при температурах 100—6000 К приведены в табл. 139.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. Электронографические исследования [3593, 1838] и исследования микровол-
276
Сера и ее соединения SO2F2 (г)
нового спектра [1467, 2470] показали, что в основном электронном состоянии молекула SOF2
имеет структуру треугольной пирамиды с атомом серы в вершине (точечная группа симметрии Cs).
Произведение главных моментов инерции, приведенное в таблице, вычислено по вращательным
постоянным молекулы 32S16O19F2 ,40=0,287 359, ?0=0,278 759, С0=0,165 214 см,
полученным в результате исследований микроволновых спектров SOF2 [1467, 2470, 1333, 4019]. Им
соответствуют структурные параметры r0 (S—О) = 1,4127 + 0,0003 A, r0 (S—F) = 1,5854+ 0,0002 А,
ДО—S—F=106,82 + 0,03°, 2?F—S—F=92,83 + 0,02°. Основные частоты молекулы SOF2
определялись в исследованиях спектров комбинационного рассеяния жидкого [4039, 563, 3300, 2933]
и газообразного SOF2 [3300, 2933] и инфракрасного спектра газообразного SOF2 [2909, 3370,
3300, 2933, 3572].
Приведенные в табл. 12.10 значения основных частот молекулы SOF2 основываются на
исследованиях Самуэльсона [3300], Пейса и Самуэльсона [2933] и Сумоди и Пейса [3572]. Для
частоты vx принято значение, среднее между наблюдаемыми в инфракрасном спектре газа
частотами vx и v2+v3 A330,8 и 1340,8 см соответственно), поскольку между ними имеет место
резонанс Ферми. Погрешность значения vx оценивается в 4 см, остальных частот в 1 см.
Термодинамические функции SOF2 (г), приведенные в табл. 139, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128) и A.130) без учета возбужденных состояний молекулы SOF2. Погрешности вычисленных
значений определяются применением приближенного метода расчета, а также отсутствием
данных о возбужденных электронных состояниях SOF2. Погрешности в значениях Ф° (Т) при 298,15,
3000 и 6000 К оцениваются в 0,2, 4 и 6 Дж-К^-моль-1.
Ранее таблицы термодинамических функций SOF2(r) вычисляли авторы справочников [88,
187, 2556, 2095, 2096], О'Лоун и Вильсон [2909] до 1500 К, Нагараян и Миллер [2784] до 2000 К,
Рамасвами и Джаяраман [3127] до 1000 К. Наилучшее согласие с данными табл. 139 имеет место
для таблиц JANAF [2096] (в пределах 0,03 Дж-К^-моль); расхождения с термодинамическими
функциями, приведенными в справочниках [88, 187, 2556], не превышают 0,5 Дж-К^-моль.
В работах [3127, 2784, 2909], по-видимому, были допущены ошибки в расчетах, так как
приведенные в них значения отличаются от данных табл. 139 уже при 1000 К на величины около 1,
1,5 и 30 Дж.К^-моль-1.
Пейс и Тернбулл [2934] на основании измерений теплоемкости SOF2 в интервале 13—230 К
нашли 5°(S0F2, г, 228,84 К)=265,9 Дж-К^-моль, в то время как данным табл. 139
соответствует 264,8 + 0,1 Дж-К^-моль. Поскольку погрешность значения, вычисленного в работе
[2934], может достигать 1,5 Дж-К^-моль, согласие приведенных величин является
удовлетворительным.
Константа равновесия реакции SOF2(r)=O(r)+2F(r)+S(r) вычислена с использованием
значения АГН @) = 1256,118 + 70 кДж-моль1, основанного на принятой в справочнике
энтальпии образования
Дуй"° (SOF2, г, 0) = —580 + 50 кДж • моль.
Принятое значение было оценено сопоставлением энергий связей в молекулах SO2F2, SOC12,
SO2C12, SO2, SO3 и SF4. Близость длины связи S—О в молекулах SO2C12 и SO2F2 [2427, 1837]
является косвенным доводом в пользу допустимости предположения о равенстве энергии,
соответствующей связи в этих молекулах. Поэтому оценке, основанной на этом предположении, был
придан наибольший вес.
SO2F2 (г). Термодинамические свойства газообразного диоксид-дифторида серы в стандартном
состоянии при температурах 100—6000 К приведены в табл. 140.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 12.10. Молекула SO2F2 в основном электронном состоянии состоит из
центрального атома серы, связанного с двумя парами атомов кислорода и фтора, расположенных в двух
взаимно перпендикулярных плоскостях (точечная группа симметрии С2„). Произведение
главных моментов инерции SO2F2, приведенное в таблице, вычислено по вращательным постоянным
молекулы 32SO2F2 Л0=0,171260 см, ?0=0,169218 см~\ С0=0,168684 см, найденным Лайдом
и др. [2428 ] при исследовании микроволнового спектра SO2F2 (см. также [2427]). Этим
постоянным соответствуют структурные параметры r(S—О) = 1,405+ 0,003 A; r(S—F) = 1,530+ 0,003 А;
ДО—S—О=123°58' + 12'; Д F—S—F=96°7' +10'. Молекула SO2F2 имеет девять основных частот,
277
SO2F2 Глава 12
активных и в спектре комбинационного рассеяния и в инфракрасном спектре, за исключением
v5, не активной в ИК-спектре. Инфракрасный спектр SO2F2 исследовался в работах [2991,
2046, 3300, 3572, 3495], в работе [2427]—в матрице из Аг; спектр комбинационного рассеяния
газообразного SO2F2 исследовался в работах [563, 3300 и 3495], жидкого SO2F2 — в работах [563,
644]. Все исследователи не встретили трудностей в отнесении четырех основных частот (v1}
v2, vB и v8). Для остальных основных частот были предложены разные отнесения. Приведенные
в табл. 12.10 значения основаны на исследованиях инфракрасного спектра газообразного SO2F2,
выполненных Самузльсоном [3300] и Сумоди и Пейсом [3572], которые находятся в согласии
с результатами исследования инфракрасного спектра SO2F2 в аргоновой матрице [2427].
Термодинамические функции SO2F2(r), приведенные в табл. 140, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—A. 10), A.122)—A.124),
A.128) и A. 130). Погрешности вычисленных значений обусловлены главным образом применением
приближенного метода расчета, а также неточностью основных частот, использованных в
расчете. Погрешности в значениях Ф° (Г) при 298,15, 3000 и 6000 К оцениваются в 0,3, 5 и
8 Дж-К^-моль-1.
Ранее таблицы термодинамических функций SO2F3 (г) вычислялись в справочниках [88, 2556,
2095, 2096], а также в работах [3793, 3127] (до 1000 К). В справочниках [2556, 2096] расчеты
выполнены по постоянным, которые практически совпадают с принятыми в настоящем издании;
значения термодинамических функций SO2F2 (г), табулированные в этих справочниках,
согласуются с приведенными в табл. 140 в пределах 0,05—0,1 Дж-К^-моль. Расхождения с
данными работ [88, 2095, 3793] составляют от 2 до 6 Дж-К^-моль главным образом из-за
использования в этих работах неверных значений трех основных частот SO2F2. В работе [3127],
по-видимому, допущены ошибки в расчете, так как при тех же молекулярных постоянных
расхождения с данными табл. 140 достигают 4,5 Дж-К^-моль в значениях Ф° (Т) и S° (T).
Маунтфилд и Вейер 12723] рассчитали значение S°(SO2F2, г, 217,78 К)=263,04±
+0,85 Дж -К-моль на основании выполненных ими измерений С° (SO2F2, к) в интервале
температур 2—21 К и данных Бокхоффа и др. [681 ] по теплоемкости кристаллического и жидкого
SO2F2 от 12 до 210 К, а также энтальпиям плавления и испарения SO2F2. Расхождение между этим
значением и значением 264,57 + 0,23 Дж-К~1-моль~1, соответствующим величинам, приведенным
в табл. 140, нуждается в объяснении, которое не является очевидным.
Константа равновесия реакции SO2F2(r)=2O(r)+2F(r)-fS(r) была вычислена с
использованием значения &ГН° @) = 1674,474+10 кДж-моль^1, соответствующего принятой в справочнике
энтальпии образования
A/fl'°(SO2F2, г, 298,15 К) = —760 + 10 кДж ¦ моль.
Эта величина принята на основании значения ДГЯ° B98,15 К) =—101,0+8,4 кДж-моль
для реакции SO2(r)+Cl2(r)+2HF(r)=SO2F2(r)+2HCl(r), вычисленного в таблицах JANAF
[2096] по уравнению III закона термодинамики на основании результатов измерения равновесных
концентраций компонентов реакции в интервале 503—973 К, полученных в неопубликованной
работе Ру и др. A963 г.). Расчет по уравнению II закона привел к значению —87 + 7 кДж-моль.
Поскольку термодинамические функции компонентов этой реакции, принятые в настоящем
издании и в таблицах [2096], отличаются несущественно (соответствующее изменение в ЬГН° не
превышает —0,2 кДж-моль), значение &ГН° было использовано без каких-либо изменений1.
Измерения потенциала ионизации SO2 A200+ 10 кДж-моль) и потенциала появления SO2
из SO2F2 A920 + 30 кДж-моль), выполненные Ризом и др. [3183] методом электронного удара,
приводят к значению AfH° (SO2F2, г, 298,15 К) = — 868^32 кДж-моль, которое, по-видимому,
следует рассматривать, как нижний предел для этой величины; можно предполагать, что продукты
диссоциативной ионизации SO2F2 в экспериментах [3183] обладали избыточной энергией.'
1 Принятое в справочнике значение Д/#° (SO2F2, г, 298,15 К) согласуется со значением — 767,2 кДж-моль х;
полученным Картрайтом и Вулфом [909а] на основании калориметрических измерений энтальпии гидролиза
SO2F2 (г) в водном растворе КОН. Погрешность этого значения в работе [909а] не указана, и не приводятся
дополнительные термохимические данные, использованные авторами [909а] при ее вычислении. В работе [909а]
измерялась также энтальпия гидролиза SO2F2 (г) в спиртовом растворе КОН. Однако полученное на основе
использования этих данных значение энтальпии образования SO2F2 (г) (—795,8 кДж 'моль) является существенно
менее точным.
Глава 13
АЗОТ И ЕГО СОЕДИНЕНИЯ
N, N+, N2, N2+, N3, NO, NO+, NO2, NO2, NO3, N2O, N2O3, N2O4, N2O5, NH, NH+, NH2,
NH3, NHt, N2H2, «jwc-N2H2, mpauc-N2U2, 1,1-N2H2, N2H4, HN3, HNO, HNO2,
транс-ШО2, HNO3, NH2OH, NH2NO2, NH4NO3 (к, ж), NF, NF2,
NP <tnv\ni4t* N F N F innnur-N F pntti-N F FNO FNO
FNO3, F3NO, NHF, NH2F, NH4F (к, ж), NHF2, СШО, C1NO2, NS
NF3, N2F2,
В настоящей главе рассматриваются термодинамические свойства азота и его соединений с
кислородом, водородом, галогенами и серой в газообразном состоянии, а также 2 соединения (NH4NO3 и
NH4F) в конденсированном состоянии. Соединения азота с другими элементами будут рассмотрены
в последующих главах. Для восьми газов (N, N+, N2, N2+, NO, NO+, NH, NH+) в таблицах
термодинамических свойств приводятся данные для температур до 20 000 К, для остальных газов —
до 6000 К. Термодинамические свойства N2H2, HNO2, N2F2 и N2F4 даны как для отдельных
изомеров, так и для их равновесных смесей.
Для всех веществ в настоящем издании
приведены термодинамические свойства в расчете на
природную смесь изотопов азота, однако благодаря
малому содержанию в природной смеси изотопа 15N
внутримолекулярные составляющие
термодинамических функций всех газов вычислялись по
постоянным молекул с изотопом 14N. В Приложении 3
приведены данные, позволяющие учитывать отклонения
термодинамических свойств 14 газов (N2, NO, N2O,
NH NH NF NF Я? NF
Таблица 13.1. Уровни энергии N и N+,
принятые для расчета
термодинамических функций
р
N2O4, NH3, N2H4, NF3,
FN FN N
2, N2F4,
Atom
Электронная
конфигурация
N Is22s22p3
Is22s22p3
Is22s22p3
Is22s22p3
Is22s22ps
N+ Is22s22p2
Is22s22p2
Is22s22p2
ls'22s22p2
Is22s22p2
Is22s2p3
Is22s2p3
Is22s2p3
Is22s2p3
Состояние
T,
CM
4S3/ 0
*Ds'k 19 224,464
2Z>3/[ 19 233,177
tP^ 28 838,920
iP,'i 28 839,306
3P0 0
SP1 48,7
SP2 130,8
1D2 15 316,2
1S0 32 688,8
552 46 784,6
8?K 92 237,2
3?>2 92 250,3
3Z>j 92 251,8
Pi
4
6
4
2
4
1
3
5
5
1
5
7
5
3
FNO, FNO2, F3NO и NHF2) от их свойств в
стандартном состоянии в результате межмолекулярного
взаимодействия.
N (г). Термодинамические свойства газообразного
азота в стандартном состоянии в интервале
температур 100—20 000 К приведены в табл. 141.
Уровни энергии атома N, использованные при
расчете термодинамических функций, приведены
в табл. 13.1, в которой даны валентные состояния
атома N, относящиеся к электронной конфигурации
1s22s22/j3. Энергии этих состояний приняты по
работе Йоханссона [1414], в которой были получены
•более точные данные по сравнению с рекомендацией
Мур [2697]. Состояния, относящиеся к электронным
конфигурациям Is22s2pi и Is22pb, имеют энергии свыше 80 000 см [1412, 2697] и не были
приняты во внимание при расчете термодинамических функций.
Термодинамические функции N(r) вычислены по уравнениям A.3)—A.10), A.23)—A.25).
Погрешности функций до температур порядка 10 000 К определяются главным образом неточностью
-фундаментальных постоянных и не превышают 0,02 Дж-К^-моль. При более высоких
температурах погрешности возрастают из-за пренебрежения более высокими уровнями энергии атома N
и достигают при 20000К величин порядка 0,1 и 0,5 Дж-К-моль соответственно в Ф°(Г) и S°(T).
Ранее таблицы термодинамических функций N (г) вычислялись неоднократно в широком
интервале температур, в том числе во 2-м издании настоящего справочника [88] и в работах [204, 2536]
до 20 000 К, [1946] до 10 000 К, [2291] до 8000 К, в справочниках [87, 2095] и в работах [2556,
2029, 225, 446] до 6000 К, [3946] до 5000 К и в работе [2198] до 2000 К. Результаты этих расчетов
при Т <^ 10 000 К совпадают с вычисленными в настоящем издании в пределах указанных выше
погрешностей, за исключением работ [2291, 2536], для которых соответствующие расхождения
279
N+ (г), N2(r) Глава 13
при этих температурах больше (до 0,2 Дж -К -моль в S° A0 000 К) для [2536 ]), что объясняется
ошибками, допущенными в этих расчетах. При Т >• 10 000 К расхождения с имеющимися в
литературе данными [88, 2536, 204] существенно возрастают из-за того, что в этих расчетах
учитывались более высокие уровни энергии атома N в соответствии с принятыми методиками расчета.
В частности, для расчета, приведенного в справочнике [88], эти расхождения достигают
3,6 Дж-К^-моль-1 в значении Ф° B0 000 К).
Энтальпия образования N(r) в соответствии с рекомендацией КОДАТА—MCHG [1000] и
величиной D0(N2) принята равной
A/fl"°(N, г, 298,15 К) = 472,68 ±0,40 кДж • моль.
N+(r). Термодинамические свойства газообразного положительного иона азота в стандартном
состоянии в интервале температур 298,15—20 000 К приведены в табл. 142.
Уровни энергии иона N+, использованные при расчете термодинамических функций,
приведены в табл. 13.1, в которой даны валентные состояния иона N+, относящиеся к электронным
конфигурациям Is22s22p2 и Is22s2ps и имеющие энергии не выше 100 000 см. Энергии этих
состояний приняты по работе Эрикссона [1410], в которой были получены более точные данные по
сравнению с рекомендацией Мур [2697].
Термодинамические функции N+(r) вычислены по уравнениям A.3)—A.10), A.23)—A.25).
Погрешности этих функций во всем интервале температур, за исключением температур около
20 000 К, определяются главным образом неточностью фундаментальных постоянных и не
превышают 0,02 Дж-К^-моль. При Т=20 000 К погрешности в S° (T) возрастают до
0,03 Дж-К^-моль из-за пренебрежения более высокими уровнями энергии иона N+. В
значениях С°р (Т) погрешности больше и составляют 0,03 и 0,2 Дж-К^-моль при 15 000 и 20 000 К
соответственно.
Ранее таблицы термодинамических функций N+ (г) вычислялись неоднократно в широком
интервале температур, в том числе в работе [1723] до 50 000 К, во 2-м издании настоящего
справочника [88] и в работах [204] до 20 000 К, [1946, 2536] до 10 000 К и [225] до 6000 К. Результаты
расчетов [1723, 88, 1946] при 71 < 10 000 К совпадают с вычисленными в настоящем издании в
пределах 0,01 Дж-К" •моль. Расхождения с остальными расчетами заметно больше и связаны
с ошибками в этих расчетах. При Т > 10 000 К расхождения с имеющимися в литературе данными
[88, 204] возрастают из-за того, что в этих расчетах учитывались более высокие уровни энергии
иона N+ в соответствии с принятыми методами расчета. В частности, для расчета, приведенного
в справочнике [88], эти расхождения достигают 0,2 Дж-К^-моль в значении Ф° B0 000 К).
Константа равновесия реакции N+(r)-fe(r)=N(r) вычислена с использованием значения
АГН° @) = —1402,327 + 0,003 кДж-моль в соответствии с принятым потенциалом ионизации
/0(N) = 117 225,35 ± 0,25 см-1= 1402,327 ± 0,003 кДж ¦ моль,
полученным в работе Мак-Конки и Кернахана [2568] в результате обработки протяженной серии
Ридберга в инфракрасном спектре N с помощью формулы Ритца. Принятому значению /0 (N)
соответствует энтальпия образования, иона N +
AfH°(N+, г, 0) = 1873,145+ 0,40 кДж • моль.
N2 (г). Термодинамические свойства газообразного диазота в стандартном состоянии в
интервале температур 100—20 000 К приведены в табл. 143.
Молекулярные постоянные N2, использованные при расчете термодинамических функций,
приведены в табл. 13.2. В этой таблице на основании результатов экспериментальных
исследований, а также теоретических расчетов [2719, 3763, 2112, 2025, 2091, 1654, 582, 2835, 2916, 2637,
901, 1321, 2430, 3681] х представлены данные для 15 валентных связанных электронных
состояний N2 с энергиями менее 100 000 см. Основное электронное состояние X1?,* изучено до i>=25
главным образом в результате исследований электронного спектра (системы A3Ii* — X1!,*
[4011, 2098, 3948, 2656], аШд — X1!,* [406, 3898, 3492, 3950, 2452, 2657] и В'1^ — Х1^ [3950,
311,1897]), а также спектра комбинационного рассеяния [3541, 564, 1954]. В качестве наиболее
1 В опубликованном недавно обзоре Лофтуса и Крупени [2453а] дана исчерпывающая сводка и критический аналиа
результатов экспериментальных спектральных исследований и молекулярных постоянных N2, а также NJ.
280
Азот и его соединения
Таблица
я
в
к
о
N
Состояние
A3S+
В3П„
W3AU
а'%7
а'ГЬ,
G3Ay
с3п„
С'3ПМ
Я3ФИ
Nj ХЧ+„
А2Па
ь«п
с2п
e'sf
Г>2ФЯ
/*пм
g'*u
E'i-u
Ж*Пд
Н22и
13.2. Принятые значения молекулярных постоянных N2 и
те
0
50 203,51 а
59 619,20 6
59 808
66 272,47 е
68 152,66
69 283,01
72 097,6
77 600 г
85 200
86 800 3
89 136,66 "
93 000 г
97 603,6
104 600 н
0
9 167,5 а
25 461,4
25 500
41 600 6
46 000 б
52 815,7 в
53 000 6
57 000 б
58 500 6
60 000 6
62 000 б
64 000 6
64 100 б
64 595
67 000 6
72 000 6
2 358,027
1 460,518
1 733,391
1 501,4
1 516,883
1 530,254
1 694,208
1 559,236
650 г
1 135
765,9
2 047,178
—
—
924,21
2 207,19
1 903,7
2 419,84
—
1 110 б
1302°
907,71
1342*
851 б
957 б
783 6
937°
1 190 6
909 б
2 099,84
10226
926 б
14,135 1
13,831 3
14,122 1
11,6
12,181 02
12,074 7
13,949 1
11,887 4
—
6,3
11,85
28,445 0
—
—
12,29
16,14
15,02
23,19
—
—
—
11,91
—
—
—
—
—
—
-
14,74
—
—
ве
щ
ах ¦ 102
СМ
-0,017 51 -0,000114 4 1,998 3 1,709
0,005 999 0,001853 1,454 56 1,8017
-0,056 88 -0,003 612в 1,637 62 1,778
— — 1,50 1,7 г
0,041858 0,000 732 3 1,473 59 1,686 1
0,041292 0,000 289 6 1,479 88 1,657 4
0,007 9346 -0,000 2911 1,616 88 1,793 3
0,032 25 — 1,498 1,66
- - 1,00г -
— — 0,90 0,6
— — 0,928 0 1,61
2,088 33 0,535 1,824 73 1,868 3
— — 1,16г —
— — 1,049 6л —
—0,173 — 1,087 3 1,91
-0,04 _ 1,932 2,0
— — 1,748 2,0
-0,537 3 _ 2,085 2,4
— — 2,075 —
- - 1,04* _
- - 1,25 6 -
0,016 _ 1,113 2,0
- - 1,26б -
— — 0,93 б —
- - 1,07 б -
— — 0,71 б —
— — 0,88 6 —
- - 1,21 * -
— — 1,06б —
—0,014 3 -0,009 72 1,510 —
- - 1,286 -
— — 0,88 6 —
Примечание. Ниже все постоянные даны в см.
N
N
2 : а л —
A.68
а4 =
+ :* Л =
-1,330, ¦
); е ь =
Г = —0,0024; 6А =
0,65, т =
= — 0,005
а,-10-»
?>0-10в
—4,06 5,48
-8,6 5,20
-7,29 7,6
— 6,03 я
3,619 я 5,56д
2,408 5,54 Л
—2,929 5,89д
— 5,53д
9,5 «
— 2,3д
— 5,44д
—227,5 в 5,80 д
— —
- 10,9°
— 6,01 д
— 5,9
— 4
— 6,2
_ _
_ —
—
_ 5
_ —
_ —
_ —
_ —
— —
—
— —
_ 4
_ —.
- -
ге
А
1,097 9
1,286 7
1,212 8
1,27
1,278 4
1,275 7
1,220 4
1,268
1,55 д
1,64
1,610 9
1,148 8
1,44 г
1,514 8 м
1,488 3
1,116
1,174
1,074
1,077
1,52 6
1,39 б
1,471
1,38 6
1,61 6
1,50 6
1,84 б
1,65 б
1,41е
1,51 б
1,263
1,37 6
1,65 6
= 42,245; в (oete = — 0,000 110 9; г оценка; Д вычислено по соотношению
« а, = 9,467 -Ю-7; 3 А = — 0,19; аА
—1,50-10"*; ж приведено значение Во; м приведено значение г0; н А =
—74,6; б квантовомеханический расчет; В4 = —16,5.
= 39,2; *а3=-7
= —12,085; °Я, = 1
,33-10-*;
3,3 - 10-10.
надежных могут рассматриваться значения AG'J+y2 для состояния Х1^, полученные в работах
Стойчева [3541] (и=0), Уилкинсона и Хаука [3950] (v—2, 3 и 6—9), Миллера [2656] (у=4, 5)г
Лофтуса [2452] (v=10~U), Чулановского [311] (w=15, 18-f-20) и Германа [1897] (у=16, 17,
21-^-25). Эти данные удовлетворительно описываются колебательными постоянными,
вычисленными Бенешем и др. [582] и приведенными в табл. 13.2. Вращательные постоянные в состоянии
ХХ2+ приведены также из работы [582], в которой были обработаны имеющиеся в литературе
данные по Bv (см. [2453]) с учетом величины Во по данным работ [3764, 3541]. Величина Do взята
по данным работы [854]. Колебательные постоянные в состояниях А3%* и В3Ид, приведенные
в табл. 13.2, вычислены также в работе [582] на основании анализа системы В3Ид — ^432^ в
работах Дике и Хита [1248, 1249] и Тилфорда и др,. [3688]. Вращательные постоянные для этих
281
N, (г) Глава 13
состояний рекомендованы в табл. 13.2 по работе Буллока и Хауса [831], выполнивших по данным
работы [1248] уточненный анализ мультиплетного расщепления в этих состояниях. Близкие
значения вращательных постоянных вычислены в работе [582]. Молекулярные постоянные N2 в
состоянии И/3Дя получены Бенешем и Саумом [581, 3312] из весьма ограниченных данных для
систем IVs Аи — ХХ2* и W3AU—В3ИЯ [3311, 4009]. Наиболее точные значения постоянных N2 в
состояниях 5'32~, аа2~, ах1[д и С3Пи были найдены в работах Тилфорда и др. [3687, 3690, 3764,
3688] из анализа полос, соответствующих переходам из перечисленных состояний в состояние
Х1!,^. Только вращательные постоянные состояния СЧ1и рекомендованы в табл. 13.2 по
работе [1248]. Значения постоянных в состоянии wlku приведены в табл. 13.2 по данным работы
[582] (см. также [2455]). Значения молекулярных постоянных в состояниях G3A.g и Н3Фи
получены Кэрроллом и др. [901 ] из анализа данных по электронному переходу между этими
состояниями (см. также [1576, 1893, 905, 1745, 3830]). Энергии возбуждения этих состояний,
приведенные в табл. 13.2, оценены с точностью 2000 см. Для С3Д„ эта оценка удовлетворительно
согласуется с результатами квантовомеханического расчета [2637]. По данным этого расчета в табл. 13.2
приведены молекулярные постоянные ненаблюдавшегося в спектре состояния Х2^. Постоянные
состояния СПа вычислены Кэрроллом [900] из анализа системы полос С'3Ии — B3tig [1679, 2189,
2190, 3623]. Состояния 62* и 5ПК предсказаны на основании теоретических [2749, 904, 2752] и
косвенных экспериментальных [899, 1808, 525] данных. Рекомендованные в табл. 13.2 величины
для состояния 52* взяты из работы [899], а для состояния 5ПИ — из работ [904, 899, 1745, 1654].
Точность принятых энергий возбуждения составляет 500 см для 52? и 1000 см для 6ПД.
Термодинамические функции N2(r) вычислены по уравнениям A. 3)—A. 10), A. 93)—A. 95),
в которых величины Qm и ее производные по Т вычислялись по уравнениям A.90)—A.92).
Составляющие состояния Х1^* были вычислены непосредственным суммированием по колебательно-
вращательным уровням энергии этого состояния, а состояний A3~L+, B3Ug, В'32~, аа2~, а}11д,
г#хДи и 52* — непосредственным суммированием по колебательным уровням с заменой
суммирования по вращательным уровням интегрированием с помощью уравнения A.82). Колебательно-
вращательные уровни энергии этих состояний представлены уравнениями типа A.62), A. 65).
Коэффициенты YkJ в этих уравнениях (табл. 13.3) были вычислены по методикам, изложенным
в разделе 1.4, с использованием экспериментальных значений AGff+i/2 для состояний ХХЪ^, А3%*
и В3Пд и остальных молекулярных постоянных для этих состояний и состояний В'31Г, аа2~,
аЧ1}, w1^ и 52^, приведенных в табл. 13.2. Мультиплетность и вырождение уровней этих
состояний учитывались в расчете соответствующими статистическими весами. Ограничение
суммирования по колебательно-вращательным уровням для состояния X1!** проводилось по предельной
кривой диссоциации, а для остальных семи состояний — в предположении линейной зависимости
•^шах, f от у. В табл. 13.3 приведены коэффициенты а{ в уравнении, аппроксимирующем
предельную кривую диссоциации для состояния X1!,*, а также значения утах и /нт для рассмотренных
состояний. Вклады остальных возбужденных состояний (И/3ДИ, Х2?, G3kg, С3Ии, 5ПМ, СПа,
Н3Фи) рассчитывались в предположении о равенстве QK0JI вр для состояний С3Пи и Х1^, W3ku
и Ц'аДа, а также всех остальных состояний и 52*. Это предположение основано на близости
колебательных и вращательных постоянных соответствующих электронных состояний и отсутствии
необходимости более точного расчета для этих высоких состояний. Погрешности вычисленных
термодинамических функций N2 (г) при Т < 10 000 К обусловлены главным образом неточностью
фундаментальных постоянных и не превышают 0,02—0,03 Дж К моль. При более высоких
температурах сказывается неопределенность в энергии возбуждения ряда электронных
состояний, пренебрежение в расчете более высокими возбужденными состояниями (см. [23, 24]), а также
неточность аппроксимации высоких колебательно-вращательных уровней основного электронного
состояния. Общая погрешность в значении Ф° B0 000 К) может вследствие этого составить
величину порядка 0,5 Дж-К-моль.
Ранее таблицы термодинамических функций N2 (г) вычислялись неоднократно. Из старых
расчетов следует упомянуть расчеты Джиока и Клейтона [1617] и Джонстона и Дейвиса [2145]
методом непосредственного суммирования, которые позднее были модернизированы Уагманом
282
Таблица 13.3. Значения коэффициентов в уравнениях, описывающих уровни энергии (см !), а также значения г'шах и
принятые для расчета термодинамических функций N2 и NJ
Коэффициенты
Те ¦ Ю-5
У,о • Ю-'
у20 • ю-3
-у 1А[
¦у . 1 Л"
У» • Ю*
Уао • Ю5
У,о • Ю'
У01 • 10-'
У„ • 10'
Y2I • 10'
Ум • Ю5
У„ • 10»
Уо2 • Ю5
(ao=De) ¦ Ю-5
а2 • 105
а3 • 1011
fmax
Jlim
0
0
—0,
—0
0
—о,
0,
-о,
0,
—0,
-о,
0
0,
о,
-о,
Примечание. Ниже
N • a
Состояние
г.
]У+ • а
Состояние
Состояние
Те
235 794
МО 941
307 947
119 025
259 082
—
—
199 85
179 58
467 79
269 53
_
548
798 904
169 67
463 472
472 464
56
273
А
0
0
—0
—0
0
-4
4
0
-0
-0
-0
502 037
145 9S9
134 165
920 349
793 814
017 83
439 744
—
145 456
180 17
860.00
—
_
52
—
—
—
33
200
все постоянные
W3AU
59 808
25 500
ЕЧ»и
64 000
1
85 200
Ь*П,
41
F
64
600
100
втд
0,596 195
0,173 241
—0,136 631
—1,294 03
0,819 695
—2,119 1
—
—
0,163 762
-0,179 78
-0,729
—
—
-0,76
—
—
—
—
35
218
цаны в civ
86 800
46 000
64 595
в
0
0
-0
0
0
—0
0
—0
0
—0
—0
г1.
(
89
с
,662 72Й
,151 643
,119 920
,136 507
,100 619
,376 983
—
—
,147 359
,168 61
,361 9
,094 67
—
,556
—
_
—
—
46
229
136,66
53 000
/
и
67 000
0,681 527
0,153 009
—0,120 106
0,321 986
0,242 251
—0,109 411
—
—
0,147 988
—0,165 74
0,240 8
_
_
—0,554
_
—
—
-
58
244
•п.
93 000
57 000
72 000
0,692 830
0,169 402
—0,133 616
—0,073 092
0,139 573
—0,282 602
—
_
0,161 688
-0,179 33
_
—
—0,539
—
_
—
—
50
233
С Ша
97 603,6
58 500
0,720 976
0,155 938
—0,119 678
0,502 824
-0,130 594
_
—
—
0,149 8
-0,166
—
—
—
-0,553
—
_
—
—
51
236
Н3Фи
104 600
/4пя
60 000
ъч*
0,776 00
0,650
—0,460
—
_
_
—
0,100
-0,530
__
_
—
-0,95
—
_
—
—
6
74
62 000
хщ
0
0,220 808
-0,167 846
1,076 75
-1,172 68
3,482 91
-0,417 173
0,181068
0,193 02
-0,192 4
0,467 179
-0,873 964
0,111 953
—0,59
0,713 770
0,157 893
0,347 551
0,813 041
69
255
А
0,091 676
0
—0
0
0
0
—0
-0
0
0
0
1
,190 411
151 397
,0S5 90S
000 055
_
,174 8
,2
.
—
,40
,622 090
,141 401
,276 475
,021 085
66
251
ч
в^и
0,254 62
0,241 934
—0,231 9
—5,373
.
—
—
0,208 5
-0,12
—
-0,62
0,459 15
0,510 033
0,833 687
—3,030 34
60
224
О
0
528 16
0,090 764
—0
0
0
-0
0,
-0
—0
0
0
0
ИЗ 753
100 356
035 703
036 034
—
—
1113
2
,
—
50
185 609
103 646
432 616
2,462 506
42
172
N+ (г) Глава 1$
и др. [3852] (Т ^ 5000 К) и точность которых до сих пор остается вполне удовлетворительной
(расхождения с данными настоящего издания не превышают 0,03 Дж -К -моль). От этих
данных несущественно отличаются расчеты до 6000 К, выполненные в справочниках [87, 2095, 2556];
в справочнике [2496] точность расчета меньше (расхождения достигают 0,04 Дж-К"-моль
в Ф°F000 К)). Ряд расчетов термодинамических функций N2 (г) был выполнен до более высоких
температур: Беккеттом и Харом [546] и Бристоном и Мак-Чесни [783] до 25 000 К, во 2-м
издании настоящего справочника [88], Дёрингом [1294] и Самуйловым и др. [205] до 20 000 К, Фик-
кеттом и Кауаном [1473] и Мартинеком [2536] до 12 000 К. Расхождения данных настоящего-
издания с результатами расчета работы [546] достигают в Ф° B0 000 К) величины порядка
1 Дж-К-моль, что обусловлено различием методов расчета, скомпенсированным частично
пренебрежением рядом возбужденных электронных состояний в работе [546]. Последняя
причина приводит к тому, что во 2-м издании настоящего справочника [88] значения
термодинамических функций, практически совпадающие с данными настоящего издания до 6000 К, при более
высоких температурах становятся меньше и расхождения достигают 2 и 6,5 Дж-К-моль
в Ф° (Т) и S° (Т) соответственно при Т=20 000 К. Аналогичные расхождения имеют место в
случае расчета [205]. В расчете [783], выполненном тем же методом, что и в работе [546], принято
во внимание максимально возможное число возбужденных связанных электронных состояний N2
с энергией до 104 322 см. Это приводит к увеличению термодинамических функций при высоких
температурах, но расхождения с настоящим изданием составляют всего 0,16 Дж-К-моль
в Ф° B0 000 К). Расчет в работе [1294] выполнен приближенным методом: без ограничения
колебательно-вращательных уровней энергии, но и без учета большинства возбужденных состояний,
что приводит к компенсации ошибок, и расхождения с данными настоящего издания в этом
случае не превышают 1,1 Дж-К-моль в значении Ф° B0 000 К). Расчеты в работах [1473, 2536]
выполнены также приближенными методами.
Двухатомный газообразный азот принят в справочнике за стандартное состояние этого
элемента и поэтому
A/ff°(N2, г) = 0.
Константа равновесия реакции N2(r)=2N(r) вычислена с использованием значения
ArH°@) = Dn(N2) = 941,636 + 0,60 кДж • молЬ-а = 78715 + 50 см,
соответствующего принятой в справочнике по рекомендации КОДАТА—МСНС [1000] величине
энтальпии образования N(r). Эта рекомендация основана на исследовании предиссоциации
в спектре N2, выполненном Бюттенбендером и Герцбергом [8551. Исследованная в работе [855]'
предиссоциация в состоянии С3Пи обусловлена состоянием 5ПИ, коррелирующем с пределом
*S+2D (см. [1579, с. 184]).
Щ (г). Термодинамические свойства газообразного положительного иона диазота в
стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 144.
Молекулярные постоянные N?, использованные при расчете термодинамических функций,
приведены в табл. 13.2. В спектрах N? хорошо изучены переходы между пятью дублетными
состояниями (см. обзоры [2453, 2453а], а также [4004, 3866, 3747]). Приведенные в табл. 13.2 значения:
постоянных NJ в состоянии Х22* основаны на результатах исследований системы В2!,*—Х22^,
(v" ^26) в работах Дугласа [1297], Тайта [3746] и Кэрролла [898]. Они хорошо согласуются;
с результатами более ранних работ [1442, 1902, 1073, 1074, 951, 2958, 1112, 2618], с данными
работы [2268а], а также с результатами исследований других систем полос. Постоянные N? в
состоянии А2Иа определялись при исследовании систем АгХ±и—Х21>* [2603, 2604, 2605, 2606, 1155,
1298, 2544] и С2П?—АЧ1и [1714, 2099, 2100, 3619, 2796, 2101]. Приведенные в таблице
постоянные NJ в этом состоянии, а также в состоянии С2Пу, получены Жаненом и др. [2101] при анализе
полос С^П-д—^42ПЯ (v" ^ 9, v' ^ 9). Принятые для состояния ^42Пи постоянные хорошо согласуются
с найденными Дугласом [1298] при изучении полос А2Ии—Х22+(г/^ 4). Хотя в системе
52Е^—Х2^* изучены и проанализированы переходы на колебательные уровни верхнего состояния
вплоть до v'=29 и определены соответствующие значения Go (v) и В, [1297, 3746, 2102, 898, 1079,.
1074, 951, 2958, 1902, 1442, 2618], наличие сильных возмущений делает невозможным
удовлетворительную аппроксимацию этих величин во всем интервале изменения v. Постоянные N* в состоя-
284
Азот и его соединения N? (г)
нии В2И*, приведенные в табл. 13.2, приняты по рекомендации справочника [3591] и описывают
только уровни, соответствующие малым значениям и. Аналогичная ситуация имеет место для
состояния С?аБ^, постоянные которого определялись в работах [898, 2176, 1297, 3947, 3618, 3597,
3898, 2646, 449, 1996] при исследовании системы полос G22+—X2S+ (г/ <^ 11). Квантовомеханиче-
ские расчеты [1758, 3660, 383, 909, 3681], ряд аномалий в спектрах (см. [1297, 3597]), эксперименты
по рассеянию электронов [922, 1134] и аналогия с изоэлектронной молекулой CN позволяют
утверждать, что в области энергий до 72 000 см наряду с перечисленными пятью дублетными
состояниями, молекула N? имеет еще по крайней мере 8 квартетных и 4 дублетных валентных состояния
•с глубиной потенциальной ямы более 2 эВ г. Анализ систематики электронных состояний NJ"
на основании работ, опубликованных до 1965 г., выполнен Гилмором [1654]. Андерсен и Салструп
C83, 368] и Картрайт и Даннинг [909J выполнили неэмпирические расчеты дублетных и квартетных
состояний NJ с учетом конфигурационного взаимодействия. Найденные ими значения Те
согласуются в пределах 5000 см с экспериментальными данными для четырех дублетных состояний
и результатами единственного пока экспериментального исследования состояния а42 + [1259,
1260] (см. также [322]). Молекулярные постоянные N? в квартетных состояниях, за исключением
«42^, и дублетных состояниях ?>2ФИ, E2hu, F2Hg и Н2Ъ~, приведенные в табл. 13.2, основаны на этих
расчетах. Для состояния а42* в таблице приведены постоянные, найденные Д'Инканом и Топуз-
ханьяном [1259, 1260] при анализе полос 0—0 и 1—1 перехода а*?+ — ^2^?. накладывающегося
на полосу 0—0 перехода S2?+ — X2L+.
Термодинамические функции N^(r) вычислены по уравнениям A. 3)—A. 10), A. 93) — A. 95),
в которых величины QTa и их производных по температуре находились по соотношениям A. 90) —
A. 92). Составляющие состояний Х2И^, АгПи, В2И^ и С2Пу вычислялись непосредственным
суммированием по колебательно-вращательным уровням энергии, задаваемым уравнениями типа A.65)
с добавлением члена A. 62). При этом мультиплетность состояния А2Ии учитывалась по
уравнению A.42). Значения коэффициентов Ykl в уравнениях A. 65), вычисленные на основании
молекулярных постоянных, принятых в настоящем справочнике (см. табл. 13.2), приведены в табл. 13.3.
Мультиплетность уровней в состояниях Х22^, В2!,* и C4Iff, а также вырождение уровней в
состояниях А'*П.и и С'2Ид учитывались статистическими весами. В расчете учитывались все колебательно-
вращательные уровни энергии, ограниченные предельными кривыми диссоциации этих состояний;
в табл. 13.3 приведены значения коэффициентов а{ в уравнениях, описывающих эти предельные
кривые, а также значения угаах и /цт. Составляющие остальных дублетных и квартетных
возбужденных состояний N+ были вычислены в предположении, что QlKM_ вр = (pJpcm ) Q^irsv, поскольку
межатомные расстояния в этих состояниях, за исключением состояния #42^, близки между собой.
Мультиплетность и вырождение этих электронных состояний учитывались соответствующими
статистическими весами. Основные погрешности табулированных термодинамических функций N?
при температурах до 6000 К обусловлены только неточностью фундаментальных постоянных
и не превышают 0,02 Дж-К моль. При более высоких температурах становятся заметными
погрешности из-за неточности принятых значений энергий возбуждения ненаблюдавшихся
экспериментально дублетных и квартетных состояний, а также использования приближенной методики
учета составляющих 13 возбужденных состояний. Они составляют около 0,1 и 1 Дж-К-моль
в значениях Ф° (Т) при Г=10 000 и 20 000 К соответственно. Погрешности из-за неточности
аппроксимации высоких колебательно-вращательных уровней состояния 522* не превышают 0,02 и
0,1 Дж-К^-моль в Ф° (Т) при 6000 и 20 000 К. Общая погрешность значения Ф° B0 000 К)
с учетом неточности использованных данных и пренебрежения ридберговскими состояниями
составляет около 2 Дж -К -моль.
Ранее таблицы термодинамических функций N* (г) вычислялись в работах [546] (до 25 000 К),
[225] (до 6000 К), [2267] (<?вн до 8000 К), [3343] (до 15 000 К) и справочнике [88] (до 20 000 К).
При Т ^ 8000 К результаты всех расчетов согласуются между собой и с данными настоящего
справочника в пределах 0,2 Дж -К -моль в значениях Ф° (Т). При более высоких температурах
расхождения быстро возрастают из-за того, что в работах [546, 3343, 88] было учтено
существенно меньшее число электронных состояний. Эти расхождения достигают в значениях Ф° (Т)
1 Секстетные состояния Щ, коррелирующие с диссоциационным пределом N+ FP)+N D5), являются отталкива-
тельными или слабосвязанными состояниями.
285
N3(r)
Глава 13
5,2 Дж-К-моль при 15 000 К с данными работы [3343]; 5,3 Дж -К -моль" при 20 000 К
с данными [546] и 5,9 Дж-К моль при 20 000 К с предыдущим изданием [88] (во всех
перечисленных расчетах учитывались только пять изученных экспериментально дублетных
состояний N+).
Константа равновесия реакции N2(r)+e(r)=2N(r) вычислена с использованием значения
\Н° @) = —561,674+0,80 кДж-моль, которое соответствует принятому в справочнике
потенциалу ионизации молекулы N2
70 (N2) = 125 667 + 1 см = 1503,31 + 0,01 кДж • моль.
Принятое значение было найдено Огава и Танака [2883 ] при изучении протяженной ридбергов-
ской серии в спектре поглощения N2 (до ?г = 31). Практически совпадающее значение A25 666 +
+1 см) было получено ранее в работе [4001 ] при анализе более короткой ридберговской серии.
Принятому значению Io (N2) соответствуют
A/F°(N+, г, 0)= 1503,31 ±0,01 кДж • моль
и
D0(N+) = 840,65 + 0,80 кДж • моль = 70 270 ± 70 см.
N3 (г). Термодинамические свойства газообразного триазота в стандартном состоянии при
температурах 100—6000 К приведены в табл. 145.
Молекулярные постоянные, использованные при расчете термодинамических функций N3,
приведены в табл. 13.4. Теоретические расчеты [2010, 2927] и результаты исследований электронного-
Та блица 13.4. Значения молекулярных постоянных, а также о и рх, принятые для расчета
термодинамических функций N3, NO^, NOg, NHJ, F3N0, NHF, NH2F и NHF2
Молекула
N,
NOjf
NOj
NHJ
F3NO
NHF
NH2F
NHF,
N3: »
NOj: a
NHF: a
1 f\ /* ГП /~k CT TT Г<Г А
LiUOlU/inllc
VX
^2nS/ a 1400 737 B)
IM,'1 1330 810
IM, 1055 830
X1A1 3250 1700B)
Xх A 1689 740
.?2Л"а 3200 1000
Xх A 3260 3210
Z1^ 3193 1424
Х2П,д, Го = 71,9 см, 0 = 2, px =
o3fi1,Sr0 = 19000 см., o = 2, рй =
^2Л', Го = 20141,26 см-1; о = 1, р^
СМ
2150 —
1245 —
1370B) 720B)
3350C) 1430C)
542 884B)
1432 —
1620 1500
1307 970
2; б приведено значение
3.
4 = 2.
— —
— —
— —
52S B) 398 B)
— —
1280 910
888 500
/•Ю39 г-см2
We-«я»
г3-см6
6,492 6
25
4,8 - 102
0,106
3,25-103
1,221
3,06
1,099-102
2
2
6
12
3
1
1
1
2
1
1
1
1
2
1
1
спектра N3 [3680, 1304] свидетельствуют о линейной симметричной структуре (группа D соА)
молекулы N3 в основном электронном состоянии Х2Пух. Приведенное в табл. 13.4 значение момента
инерции молекулы N3 вычислено на основании вращательной постоянной 50=0,431 17 см, найденной
Дугласом и Джонсом [1304] при анализе 000—000 полосы системы 22^—2Пу в электронном спектре
газообразного N3. Этой вращательной постоянной соответствует r0 (N—N) = l,1815+0,0005 А.
Величина спин-орбитального расщепления состояния ХЧ1д, приведенная в таблице, также
определена в работе [1304]. Значения основных частот N3 приняты по работе Миллигана и др. [2661],
в которой исследован инфракрасный спектр поглощения N3 методом матричной изоляции.
Неактивная в ИК-спектре частота vx была вычислена с использованием найденного в работе [2661 ] зиаче-
1 Расчет [409] приводит к возможности существования более стабильной несимметричной структуры (группа Са,к).
Однако этот вывод нуждается в дополнительной проверке.
286
Азот и его соединения N0 (г)
ния v3 и соотношения /„.=0,12 /г, хорошо соблюдающегося для азидов [1721, 1421]. Погрешности
в принятых значениях частот колебаний могут достигать 100, 70 и 30 см соответственно для vlt
v2x и vg. Энергия возбуждения состояния А2Х + равна 36 774,7 см [1304].
Термодинамические функции N3 (г) вычислены в приближении «жесткий ротатор — гармони--
ческий осциллятор» по уравнениям A. 3)—A. 10), A. 129), A. 126), A. 122)—A, 124) с учетом
поправки на мультиплетное расщепление по уравнениям A.168)—A. 170) в предположении, что
энергия возбуждения подсостояния .Х2П1/2 равна величине А. При этом считалось, что рх=2 как для
Х2Пз/,, так и для Х2П1/„-подсостояний. Погрешности вычисленных термодинамических функций
определяются неточностью принятых молекулярных постоянных, а также приближенным
характером расчета и составляют 0,2, 3 и 5 Дж-К~1-моль~1 в значениях Ф° (Г) при 298,15, 3000 и 6000 К,
в том числе погрешности из-за неточности основных частот 0,2, 2 и 2,5 Дж-К-моль соответственно.
Ранее таблица термодинамических функций N3 (г) вычислялась в предыдущем издании
настоящего справочника [88]. Расчет был выполнен по тем же значениям частот, но с несколько
меньшим значением r0 (N—N) и без учета спин-орбитального расщепления состояния Х2Пд, что
привело к расхождениям,, достигающим 0,9 Дж-К-моль в значениях Ф° (Т).
Константа равновесия реакции N3 (r)=3N (г) вычислена с использованием значения Дг//° @) =
= 973,021+15 кДж-моль, соответствующего принятой в справочнике энтальпии образования
AfH°{Na, г, 298,15 К) = 436 + 15 кДж • моль.
Принятое значение основано на величинах энергии кристаллических решеток азидов щелочных
металлов, вычисленных Диксоном и др. [1264] и Греем и Уоддингтоном [1720], энтальпии
образования азидов щелочных металлов, измеренных Греем и Уодингтоном [1720], и
сродства N3 к электрону, рекомендованного Эвансом и др. [1421]. Результаты выполненного Кларком
и Клайном [978] исследования спектра хемилюминесценции, сопровождающей реакции N3 с N
и О, приводят к нижним границам возможных значений энтальпии образования N3 (г), равным
400 и 423 кДж-моль, что находится в согласии с принятым значением. Рекомендованные ранее,
в литературе отличающиеся значения энтальпии образования N3 (г) были основаны на неточной
величине сродства N3 к электрону.
NO (г). Термодинамические свойства газообразного оксида азота в стандартном состоянии
при температурах 100—20 000 К приведены в табл. 146.
Молекулярные постоянные, использованные при расчете термодинамических функций NO,
приведены в табл. 13.5. Молекулярные постоянные NO в состоянии Х2ПГ определялись на
основании исследований полос систем 52ПГ—Х2П„ (i/ «С 26, v" < 23) [2142, 653, 2114, 2116, 2115, 3334,
3337, 1577, 804, 1197, 1196, 1194, 1195, 2340, 2339, 2382, 3616, 3581, 2644, 1915, 2341, 1408],
А2Я+—Х2П (z/ < 5, v" < 13) [1197, 1196, 2524, 1604, 497, 1408], колебательно-вращательного
[1651, 2850, 3402, 3670, 2006, 2907, 2630, 2208] и вращательного [1.561, 2941, 1804] спектров NO.
Результаты всех этих исследований находятся в хорошем согласии. Приведенные в табл. 13.5.
значения постоянных NO в состоянии Х2ПГ приняты по работе Энглемана и Рауза [1408], в
которой, помимо собственных данных, полученных для v"=4-^-13, использованы результаты
измерений Дженкинса и др. [2114—2116] для г/' = 13-^-16 и предыдущей работы [1409] для
v"=0. Соответствующие колебательные постоянные удовлетворительно описывают все известные
экспериментальные данные вплоть до г;"=23. Постоянные NO в состоянии В2ПГ также приняты
по работе [1408]; они близки к полученным в работах [2114—2116, 3334, 3335—3337]. Из-за
сильных возмущений эти постоянные описывают только колебательно-вращательные уровни с v ^ 6.
Энергия возбуждения состояния Б'2Д принята по данным Джунгена [2181] (система.В'2Д—Х*11г),
состояния G2?~ — по данным Лофтуса и Мишера [2454], состояния '722+ — по данным Дреслера
и Мишера [1323, 2645], состояния L2H — по данным Лагерквиста и Мишера [2341]. Энергия
возбуждения состояния а4П принята на основании исследования системы о4П—Х2Пгв спектре NO'
в матрицах инертных газов [800, 3003, 3028, 3056, 1548], состояния Ь4Е~ — на основании иссле--
дования системы &42~—а4П [804]. Погрешности этих значений могут достигать соответственно,
300 и 1300 см (из-за возможной неточности нумерации полос в работе [804] (см. [1654])).
Колебательные постоянные в состоянии а4П вычислены по значениям Д&/2 и Д&/2 [804]; величина Ве
В работе [101] в результате исследования электронного спектра продуктов фотолиза смесей HN3+C12 было сде-.
лано, по-видимому, ошибочное отнесение наблюдаемой в спектре полосы 205 см к частоте v2 молекулы N8.
287
NO (r)
Глава 13
Таблица 13.Е
Молекула
NO
NO+
Состояние
хтг
a4llf
ВЧ1/
Х^+
a3S+
ЪЧ1
е3Д
d3H~
вч-
С1Д
?41
). Принятые
1\
Оа
38 000
43 906,37
45 868,53 ж
0
52 190
59 255
61 883
67 745
69 542
71 452
73 474,9
значения
<¦>.
1904,40?
1015
2374,307
1037,449
2377,5
1293,4
1710
1313
1284,2
1283
1278
1600,6
молекулярных
«А
постоянных NO и NO+
СМ"
14,187 0
10,5
16,106 0 —0
7,472 4 0,
16,45
15,1
14,2
10,6
10,7
13,5
16
19,94
Примечание. Ниже все постоянные даны в см.
N0:
а А [и) =
[123,26 — 0,1
по соотношению A.
е Состояние
J
Ге
Состояние
35 А (у) =
Те
906 (и +1/2
)— 0,0108 (о + VaJ
69); Д вычислено по A. 68);
48 000
Я2П
62 426,4
?'2ф cm
49 000 52 066,5
62 472,9 62 850
31,32 + 1,152 (и + i/а) + 0,0448-(у + V2J; 3
»еУв
-1
,024 0 б
—
046 45
072 53
—
—
—
—
—
—
—
5302^
1,704 27
1,125
1,994 78
1,125
1,998 2
1,369
1,634
1,377
1,356 8
1,363
1,361
1,586 5
= 0,000 93; в
?'2Л
!,4 60 302,6
Я2+ L41
63 000 63 300
a2 = 0,(
ХЮ 125.
.,.10»
1,728 в
0,75 г
1,8328
1,3482 3
1,902 '
1,92
1,84
1,92
1,92
1,92
1,92
2,401
а2 = —0
60 568
D..10»
5,4
5,5 Д
5,4
4,9
5,6
6,1
6,0
—
—
—
—
6,4
,000037;
F
4 617
А
1,151
1,417
1,063
1,417
1,063
1,284
1,176
1,281
1,290
1,287
1,288
1,192
0
9
0
0
9
' вычислено
2Д
61,4
в этом состоянии принята такой же, как в изоконфигурационном состоянии ВШГ. Энергия
возбуждения состояния Ь'2Ф, которое не наблюдалось экспериментально, принята по квантовоме-
ханическому расчету [2376]; погрешность этой величины может достигать 5000 см.
Помимо рассмотренных девяти электронных состояний, отвечающих нижним электронным
конфигурациям молекулы NO, существуют также еще 7 стабильных электронных состояний,
формально относящихся к ридберговским состояниям, но имеющих низкие энергии возбуждения,
сравнимые с энергиями рассмотренных выше валентных состояний [2340, 497, 2027, 2454, 343,
1452] (см. табл. 13.5). Эти ридберговские состояния из-за взаимодействия с отталкивательными
дублетными состояниями фактически не коррелируют с ридберговскими состояниями атомов
Nh О. Молекулярные постоянные NO в нижнем ридберговском состоянии А21,+ определялись при
анализе полос системы А2?+—Х2П{ [1604, 497, 1196, 1197, 1194, 2340, 2339, 1323 1408].
Приведенные в таблице зна'чения постоянных получены Энглеманом и Раузом [1408] в результате
диализа тонкой структуры полос с у' ^ 5. Переходы на уровни с г/ ^> 5 не наблюдались в спектре
NO, что объясняется пересечением потенциальных кривых состояния Л2?+ и отталкивательного
состояния А'2 2+, которое коррелирует с пределом N (*5)+0 CР). Это же отталкивательное
состояние пересекается также и с другими рассматриваемыми в настоящем справочнике
ридберговскими состояниями С2П, ZJ?, ?'2S+, F2<\, Я2П и Я'2И+. Согласно теоретическим расчетам [1654,
2376] все остальные возбужденные электронные состояния NO в рассматриваемой области
энергий являются отталкивательными или слабосвязанными и не принимаются во внимание при
расчете термодинамических функций.
Термодинамические функции NO (г) вычислены по уравнениям A. 3)—A.10), A. 93)—A.95),
в которых Qm и ее производные по температуре рассчитывались по уравнениям A. 90)—A.92).
Составляющие основного электронного состояния Х2ПГ вычислялись непосредственным
суммированием по колебательным и вращательным уровням энергии, а возбужденных состояний а4П,
288
Азот и его соединения
N0A-)
Таблица 13.6. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см'1), а также
значения итлх и 7iim, принятые для расчета термодинамических функций N0 и N0+
Коэффициенты
т.
Но
Но
Но
Но
Но
Но
Hi
Hi
Hi
Нг
Х2ПГ»
• lO 0
• 10-* 0,190 374
10 —0,138 039
• 101 —0,405 334
10a 0,370 940
103 —0,132 620
105 0,100 949
10 0,170 427
101 —0,172 800
103 —0,037 0
105 —0,540
(ao=De).10-6 0,533 484
ai
«2
as
V
r
J
0,163 420
106 0,542 408
ЮН —0,775 875
46
,» 242
N0
а4П<
0,380 00
0,101 346
—0,092 908 6
—2,686 95
—
—
—
0,112 500
—0,075 00
—
—0,55
—
—
—
—
25
180
Примечание. Ниже все постоянные даны i
NO
N0+
: » A = 123,17.
Состояние Ь42~
Te 48 000
Состояние F2k
Те 61761
. а
Состояние с3Д
Те 61883
Ь'2Ф
49 000
Я2П
4 62 426,4
67 745
Л22+
0,439 064
0,237 427
—0,160 797
—0,532 311
0,056 974 8
—
—
0,199 478
—0,183 28
—
—0,54
—
—
—
—
5
160
i CM.
cm
520 66,5
Я'22+
62 472,9
69 542
Bmr6
0,458 685
0,103 785
—0,077 322
1,308 45
-0,422 736
—
—
0,112 500
—0,134 820
—
—0,49
—
—
—
—
38
211
ZJS+
53 023,4
G2Z-
62 850
СгА
71452
XiS+
0
0,237 765
6 -0,165 171
0,077 522
—
—.
—
0,199 82
—0,190 2
—
—0,56
0,887 000
0,147 707
0,265 604
0,518 631
75
278
Д'2Д
60 302,6
/*Е+
63 000
Dm
73 474,9
N0+
a3S+
0,52190
0,123 127
—0,110 925
1 0,092 437 4
—
—
—
0,136 9
—0,192
—
—0,61
—
—
—
59
217
60 568
Lm
33 300
63П«
0,592
0,170
—0,131
—1,710
—
—
—
0,163
—0,184
—
-0,60
—
—
—
36
213
55
837
888.
74
4
В3ПГ — непосредственным суммированием по колебательным уровням и интегрированием
по вращательным уровням энергии с помощью уравнений типа A. 82). Колебательно-
вращательные уровни энергии всех этих состояний представлялись уравнениями типа
A.65), A.62), причем для состояния Х*ПГ вращательные уровни задавались уравнениями
A.42), а вырождение уровней этого состояния, мультиплетность и вырождение
уровней состояний а4П{ и В2ПГ и мультиплетность уровней состояния A*L+ учитывались
соответствующими статистическими весами. Значения коэффициентов Ykl в уравнениях
A. 65) вычислялись на основании принятых молекулярных постоянных NO (см. табл. 13.5) по
методикам, изложенным в разделе 1.4. Эти значения Ykl приведены в табл. 13.6. Ограничение
суммирования по / для состояния Х2ПГ проводилось по предельной кривой диссоциации (в табл. 13.6
приведены коэффициенты а( в уравнении, аппроксимирующем эту кривую), а для состояний а*Л{,
В2И А2?+ йй /В Л2И+
р фф (
В2ИГ и .А2?+ — по линейной зависимости /П
от v. В состоянии
учитывались только
колебательные уровни v ^ 5, т. е. лежащие ниже точки пересечения потенциальных кривых
состояний Л2Б+ и .4'2?+. В таблице приведены также значения vnmx и /нш для этих состояний NO. Вклады
289
NO+(r) Глава 13
электронных состояний &4?~, Ь'2Ф, .В'2Д, G2?~, /2?+ и L2H вычислялись в предположении, что
для каждого из этих состояний Qm3, щ> и ее производные равны соответствующим величинам для
состояния В2ИГ (без учета мультиплетности и вырождения вращательных уровней).
Составляющие ридберговских состояний С2П, ZJE+, Е12!!", ^2Д, Н2П и Н'2%+ вычислялись в предположении,.
что для них (?Е0Л> вр и ее производные равны этим величинам для состояния Л2 Е+. Мультиплетность
и вырождение этих электронных состояний учитывались соответствующими статистическими
весами. При температурах до 6000 К погрешности вычисленных значений функций практически
обусловлены только неточностью фундаментальных постоянных и не превышают 0,02 Дж-К X
Xмоль в значениях Ф° (Г). При более высоких температурах погрешности возрастают из-за
неточности данных для состояний й*П,. Ь42~ и Ь'2Ф, а также из-за приближенной методики учета
возбужденных состояний и пренебрежения состояниями с энергией, превышающей 64 000 см.
Эти погрешности могут достигать при 10 000 и 20 000 К соответственно 0,1 и 1,0 Дж«К~1«моль~1
в значениях Ф° (Т).
Ранее таблицы термодинамических функций NO (г) вычислялись неоднократно, в том числе
в предыдущем издании справочника [88] до 20 000 К, в справочниках [87,2095, 2556, 2496, 446]
до 6000 К, а также в работах [546] до 25 000 К, [205] до 20 000 К, [1472] до 12 000 К и [1674]
до 5000 К. Расчет Джонстона и Чапмана [1674] является, по существу, одним из первых в
истории создания прецизионных таблиц термодинамических свойств для широкого интервала
температур и лишь использование в нем устаревших данных по молекулярным постоянным является
причиной расхождений (около 0,1 Дж-К-моль) с данными настоящего издания. Кроме того,
в ряде старых изданий [2029, 3196, 3255, 3946] приведены данные по термодинамическим
функциям NO (г) для Т <! 6000 К со ссылкой на неопубликованный расчет Национального бюро
стандартов США; эти данные еще лучше (в пределах 0,04 Дж -К -моль в значениях Ф° (Т))
согласуются с настоящим расчетом. Расхождения расчетов, приведенных в работах [2095, 2556, 2496]
для Ф° (Т), находятся приблизительно в тех же пределах; для S° F000 К) соответствующие
расхождения достигают 0,25 Дж -К -моль, что объясняется как использованием
приближенных методов в этих расчетах, так и пренебрежением в них, частично [2496] или полностью [2095,
2556], возбужденными электронными состояниями. Этими же причинами, по существу,
объясняются и более существенные расхождения с имеющимися в литературе расчетами при более
высоких температурах: 2,6 Дж-Ы1 -моль в S0 A2 000 К) [1472] и 1,6, 7,3 и 8,9 Дж-К-моль
в S° B0 000 К) [546, 88, 205] соответственно.
Константа равновесия реакции NO (г)—N (г)+О (г) вычислена в соответствии с принятым
в справочнике значением
ДД° @) = Do (NO) = 52 400 + 10 см = 626,84 + 0,12 кДж • моль,
полученным Кэллиром, Пилингом [865] и Динглом и др. [1260а] при исследовании прямой и
обратной предиссоциации NO в состоянии С2П.
Наиболее точные калориметрические измерения энтальпии образования NO были проведены
Фриш и Маргрейвом [1546], измерившими энтальпию реакции СО (r)+NO (г)=СО2 (г)+1/2 N2 (г).
Полученное в этой работе значение LjH° (NO, г, 298,15 К)=90,20 +0,40 кДж-моль оказалось
в хорошем соответствии с результатами более старых измерений Бертло [613] и Томсена [3674].
Менее точными являются измерения Кернера и Даниэлса [2275] энтальпии сгорания красного-
фосфора в NO и в смеси N2+O2, которые привели к величине b.fH° (NO, г, 298,15 К)=91,2 +
+1,3 кДж-моль. Результаты измерений констант равновесия реакции 1/2 N2 (г)+1/2О2(г)=
=N0 (г) (см. [2416, 1617]) содержат, по-видимому, систематическую ошибку и приводят к
существенно отличающимся значениям энтальпии образования NO (г). Значению энтальпии
образования NO, найденному в наиболее точных калориметрических измерениях [1546], соответствует
Do (NO)—627,91 +0,57 кДж -моль. Эта величина заметно отличается от полученной в
спектроскопических измерениях [865, 1260а]. Причины этого расхождения, превышающего погрешности
измерений, неясны. В настоящем издании отдается предпочтение прецизионным
спектроскопическим измерениям. Более подробное обсуждение см. в работе Гурвича и Медведева [85].
Принятому значению Do (NO) соответствует
г, 298,15 К) = 91,265 ± 0,43 кДж • моль.
NO+(r). Термодинамические свойства газообразного положительного иона оксида азота
в стандартном состоянии в интервале температур 298,15—20 000 К приведены в табл. 147.
290
Азот и его соединения NO+ (r)
Молекулярные постоянные, использованные при расчете термодинамических функций NO+,
приведены в табл. 13.5. В оптическом спектре NO+ детально изучена только система полос D1!!—
ХХ2+ [448,3617, 446, 2641, 2632]. Сведения о ряде возбужденных электронных состояний NO+
получены в результате исследований фотоэлектронного спектра N0 [1384, 356, 2077, 3296, 1024,
1025, 2026, 864] и квантовомеханических расчетов [2376, 1654]. В 1973 г. Филд [1474] на
основании нового анализа данных, полученных Мишером [2642] для семи полос системы D1!!—Х*2 + ,
уточнил значения постоянных NO+ в этих состояниях. Одновременно, используя данные Эдквиста
и др. [1384] по фотоэлектронному спектру и аналогию с возбужденными состояниями N2 и СО,
Филд рекомендовал значения молекулярных постоянных NO+ для других электронных
состояний NO+. Эти значения для восьми электронных состояний NO+ приведены в табл. 13.5. По оценке
[1474] погрешности в значениях Те для возбужденных состояний, определенных из
фотоэлектронного спектра, составляют от 20 до 75 см, а соответствующих значений ше — от i0 до 30 см
и о)ехе — от 0,8 до 3 см". Другие возбужденные электронные состояния NO+ с энергиями,
превышающими 75 000 см, в расчетах термодинамических функций не учитывались.
Термодинамические функции NO+ (г) вычислены по уравнениям A. 3)—A.10), A. 93)—A. 95),
в которых величины QBJI и их производные по температуре находились по соотношениям A. 90)—
A.92). Составляющие состояния Х1Ъ+ вычислялись непосредственным суммированием по
колебательно-вращательным уровням с ограничением по предельной кривой диссоциации.
Составляющие состояний а32 и Ь3П вычислялись непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням с помощью уравнений типа A. 82) и
ограничением в предположении линейной зависимости /mMi „ от v. Колебательно-вращательные уровни
этих трех состояний представлялись уравнениями типа A. 65) с добавлением члена A. 62).Мульти-
плетность состояний а32 и Ь3П и Л-удвоение уровней состояния bsU учитывались статистическими
весами. В табл. 13.6 приведены значения коэффициентов а{ в уравнении, аппроксимирующем
предельную кривую в состоянии ХХ2+, а также значения ушах и /цт для каждого состояния.
Составляющие состояний с3Д, d32~, 5Х2~ и СХД вычислялись в предположении, что для этих
состояний Qt0It го и ее производные (без учета мультиплетности и вырождения уровней) равны
соответствующим величинам для состояния с32, поскольку они относятся к одной и той же электронной
конфигурации. Аналогичным образом составляющие состояния D1!! были вычислены по QK0Itip
и ее производным для состояния Ь3И. Погрешности табулированных значений термодинамических
функций NO+ (г) до 6000 К практически обусловлены только неточностью принятых значений
фундаментальных постоянных и не превышают 0,02 Дж •К~1-моль~1. При более высоких
температурах начинают сказываться отсутствие экспериментальных данных о
колебательно-вращательных уровнях энергии состояния Z1S+ cp>5, неточность данных об энергиях возбуждения
и молекулярных постоянных возбужденных состояний, а также пренебрежение в расчете более
высокими возбужденными состояниями. Погрешности значений Ф° (Т) при 10 000 и 20 000 К
могут достигать 0,3 и 1 Дж -К -моль соответственно.
Ранее таблицы термодинамических функций NO+ (г) вычислялись в предыдущем издании
справочника [88] до 20 000 К, в справочнике [2095] до 6000 К и в работах [546] до 25000 К, [2051
до 20 000 К и [2267] (Qm до 8000 К). Данные [88], полученные с учетом четырех возбужденных
состояний NO+, согласуются с приведенными в настоящем издании в пределах 0,02 Дж -К -моль
в значениях Ф° (Т) при Т ^ 6000 К; с ростом температуры расхождения достигают 1,2Дж>К~1Х
Хмоль при 20 000 К. В справочнике [2095] не учитывались возбужденные состояния NO+, что
несущественно сказывается при Т ^ 6000 К (расхождения достигают 0,1 и 0,3 Дж -К -моль
в S° (Т) и С (Т) соответственно). Данные работы [205] содержат большие ошибки из-за
неправильной оценки постоянных NO+; расхождения с данными работы [546] при Т ^ 10 000 К
постоянны @,5 Дж -К -моль) из-за использования в этой работе неточного значения Ве; при
более высоких температурах расхождения быстро растут (до 2,5 и 14 Дж -К -моль в значениях
Ф° (Т) и S° (У)) из-за того, что в расчете [546] было учтено только одно возбужденное состояние
(Dm).
Константа равновесия реакции NO+ (r)+e (r)=N (г)+О (г) вычислена с использованием
значения Д^0 @) = —267,01+0,13 кДж-моль, основанного на принятом в справочнике
потенциале ионизации
/0 (NO) = 74 720 + 5 см = 893,85 + 0,06 кДж ¦ моль.
291
NO+(r)
Глава 13
Таблица 13.7. Значения молекулярных постоянных (в см), а также о и р{ 14NleO2, uN2leO, F14NleO
и C114N16O, принятые для расчета термодинамических функций
Постоянные
v3
v3
*11
*12
^13
^22
*23
^33
#22
-^000
^000
Cooo
1
ai
"f
aB
af
aB
3
aC
°f
as
O&Q&
^bbbb
taabb
xabab
^ceee
W
4bee
a
PZ
i4Nieo2: а
14N2leO: *
б
pi4pjl6Q« а
Q]14^16Q. а
14№<Ю2
1319,95
749,60
1616,05
-8,1
—9,7
—29,8
-0,5
—2,7
-15,6
—
8,002 509
0,433 664 6
0,410492 6
—0,075 3
-0,364
0,220 8
0,002 354
0
0,002 718
0,002 82
—0,000 95
0,002 64
—1,136 5-10-2
—1,418-10-6
6,900 ¦ 1СГ5
—8,215 • 10^6
—8,914 • 10"?
3,197 • 10-5
— 1,089.10-*
2
2
Л2Я2, Го = И 956 см-1
o = 2, ра = 4; В*А2,
Б*В%, Го = 40125 см-
Piii=—0,021, j/h2 = -
= 0,390, 2/223 = 0,059,
"N,
№
1276,877
588,771 B)
2223,762
—3,842
0,182
—27,352
—0,271
—14,672
—15,155»
0,366
—
0,419 01136
—
—
—
—
1,917-Ю
-0,577 • Ю-3
3,481 • Ю-3 б
—
—
—
—
—
—
—
—
—
—
1
1
0==2; Р1 = 2;в»в1, то =
Г0 = 27 000 см, о = 2;
1,о = 2, р^ = 2.
-0,152, ^122 = —0,030, 1/2
г/233 = 0,029, 2/ззз = —0,0
= 29,550 (все постоянные даны в см);
au = -l,2.10-s, a12
= —0,3-Ю-5, До = 1
а»Л, Г0 = 15 000 см,
а*А, Г0=10000 см,
= —3,4 • 10-в, a13 = -0,S
795 • 10"' (все постоянные
о = 1, р~ = 3.
о = 1, pg = 3; AU, То =
F14NleO
1843,5
765,8
519,9
—17,5
1,5
2,0
—4,7
—2,0
—1,5
3,175 188
0,395 080
0,350519
0,034 50
—0,019 12
—0,013 54
-0,000 14
0,001 66
0,004 84
0,000 24
0,001 84
0,004 70
—5,20 • Ю-4
—3,32 • 10"»
1,4 • 10
—9,66 • 10"»
—1,89.10"»
4,68 • 10"»
—2,46 • 10"»
1
1
= 14744 см-1, о = 2,
рг = 4; С2А2, То
22 = — 0,007, г/из =
02, ?12а == —0,005,
•10-e, a22=—0,5
даны в см).
= 16 000 см, о = 1
CJ14N«O
1799,7
595,8
331,9
—17,8
0
—0,6
—2,6
—4,3
—1,0
—
2,914 5
0,191 39
0,179 33
0,040 16
—0,038 88
—0,010 61
—0,000 33
0,000 53
—0,001 60
—0,000 13
0,000 69
0,001 53
—5,777 8 • 10-*
—9,750 • 10-'
8,522 2 ¦ 10"»
—3,673 2 • 10-»
—7,032 0 • 10-'
5,294 3 • 10-»
—8,237 4 • 10-?
1
1
pg = 2; a452, Г0 = 26000 см,
= 31000 см, о = 2, рд = 2;
= —0,338, 1/133 = 0,124, i/i23 =
6222 ~~ V,\JX 1, 5322—'^' 122
• 10-», a23 = 3,6-10-5, a33 =
292
Азот и его соединения NOa (г)
Принятое значение основано на найденном Джунгеном и Мишером [2182] пределе схождения
ридберговских состояний N0. Близкие, но менее точные значения /0 (N0) были получены в более
ранних работах Ватанабэ и др. [3891а] G4 675 см) и Хубера [2026] G4 746+40 см). Принятому
значению 10 (N0) соответствуют
ДД°(ГЮ+, г, 0) = 984,611 ±0,43 кДж • моль,
Do (N0+) = 1046,933 + 0,13 кДж • моль'1 = 87 517 + 11 см.
NO2 (г). Термодинамические свойства газообразного диоксида азота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 148.
Молекулярные постоянные NO2, использованные при расчете термодинамических функций,
приведены в табл. 13.7. Молекула NO2 в основном электронном состоянии является нелинейной
и симметричной (симметрия Съ). Основные частоты й постоянные ангармоничности, приведенные
в табл. 13.7, рекомендованы Бланком и Хаус [670] (см. также [2906, 671 ]), исследовавшими спектр
14NO2 и ^NO2 в близкой ИК-области с большим разрешением @,03—0,05 см) и вычислившими по
этим данным ши ш3, хг1, xss и х13, приняв для ш2, х22, х12 и хгз значения, полученные в работе Аракава
и Нильсена [407]. Принятые значения постоянных представляются более точными по сравнению
с полученными на основании меньшего числа экспериментальных данных в работе Уэстона [3927]
(с использованием данных работ [2213, 2214]) и Мура [2700] (с использованием данных работы
[810]), а также с отдельными постоянными, найденными при исследовании спектров NO2 на
приборах с невысокой дисперсией [313, 314, 3289, 1043, 331, 737]. В старых работах, выполненных
до исследований Вильсона и др. [3965, 810] и цитированных в монографии Герцберга [69], были
неправильно определены vx и v2. Определение вращательных постоянных и постоянных
центробежного растяжения NO2 проводилось неоднократно в результате анализа МВ-спектра и тонкой
структуры ИК-полос [810, 2700, 2213, 2214, 645, 646, 648, 1164, 2552, 2553, 647, 1512, 2375, 2583,
2906, 671, 649, 670]. Исследования последних лет [647, 2375, 2906, 649] приводят к
удовлетворительно согласующимся между собой результатам, значительно более точным, чем результаты
более старых работ. Вращательные постоянные и постоянные центробежного растяжения,
приведенные в табл. 13.7, приняты по работе Олмана и Хаус [2906]. Именно эти постоянные были
использованы Бланком и Хаус [670] при определении постоянных колебательно-вращательного
взаимодействия aj'B>G и aj-B-°, которые также приведены в таблице. Для о.\'в<° приняты
величины, найденные Брандом и др. [737]. Ранее в работе [407] были определены постоянные ав, а\
и ав, удовлетворительно согласующиеся с принятыми величинами. Основное "электронное
состояние молекулы NO2 относится к типу гАг (см. монографию Герцберга [70], где дана библиография
работ по исследованию электронного спектра NO2 до 1966 г.). Из большого числа теоретически
предсказываемых возбужденных электронных состояний NO2 [3871, 2580, 1724, 841, 1481, 840,
1482, 1206, 1563, 1862], в настоящее время экспериментально надежно установлено
существование двух низких состояний: 2В2 (работа Бранда и др. [737] по исследованию спектра
флюоресценции при лазерном возбуждении, а также спектра поглощения в области 6400—11500 А (см. также
[3289, 331, 1833,3483,3529,3217]) и 2Вг (работа Хардуика и Бранда [1833] по исследованию спектра
поглощения в области 3700—4600 А (см. также [1302, 1253, 331, 3279, 3529, 433, 850]). Ошибки
в значениях То этих состояний (см. табл. 13.7) могут составлять 50 см-1для2.В2 и 5 см для 2Вг.
Энергии состояний 4Z?2, 442, 2А2 и 252 принимаются по квантовомеханическому расчету Ганги и
Бюрнелля [1563]; их погрешность может достигать 5000 см. Более высокие электронные
состояния NO2 в настоящем справочнике не рассматриваются.
Термодинамические функции NO2 вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A.10), A.127), A.128), A.122)—A.124) с поправками
на ангармоничность колебаний и взаимодействие колебаний и вращения по уравнениям A.131)—
A.133) и на центробежное растяжение по уравнениям A.143)—A.144). Вклад возбужденных
электронных состояний рассчитывался по уравнениям A.168)—A.170), т. е. в предположении, что
Qmi. вр и ее производные для этих состояний и основного электронного состояния одинаковы.
Вычисленные таким образом значения при комнатной температуре имеют погрешности около 0,03 ДжХ
X К-моль, что определяется в основном ошибками исходных молекулярных постоянных и
величины R. При более высоких температурах погрешности быстро возрастают из-за
неопределенности экстраполяции энергий высоких колебательно-вращательных уровней, приближенного
метода расчета, а также из-за неточности в величинах энергий возбужденных электронных состоя-
293
N0 г (г) Глава 13
ний и приближенного метода их учета. Суммарные погрешности могут достигать 0,5 и 1 Дж-К~1Х
Хмель в Ф° (Г) при 3000 и 6000 К.
Ранее таблицы термодинамических функций NO2 (г) рассчитывались в справочниках [88,2095,
2556, 446] и в работе [1699] до 6000 К, а также до более низких температур в работах [3800, 2765,
938] и ряде других (см. [88]). Все эти расчеты, за исключением [88, 2556, 1699, 938], выполнены
в приближении «жесткий ротатор — гармонический осциллятор». Расхождения данных
настоящего издания с результатами расчета работы [2095 ] достигают 3,3 Дж-К~1-моль~1 в значении
Ф°F000К). Расчет, приведенный в справочнике [88], выполнен с учетом поправки на
ангармоничность колебаний, но без учета составляющей, обусловленной взаимодействием колебаний и
вращения (эта составляющая оказывается отрицательной), и с неверно сосчитанной поправкой на
центробежное растяжение (она завышена почти вдвое). Этими причинами объясняется превышение
результатов расчета справочника [88] над данными настоящего издания во всем интервале
температур, достигающее 2,2 и 5,2 Дж-К~1»моль~1 соответственно в Ф° (Т) и S° (Г) при 6000 К. Расчет
Гордона [1699] выполнен по методу, аналогичному использованному в настоящем справочнике
с ограниченным набором постоянных, принятых по работе [407]. Поскольку в работе [1699]
не принимались во внимание центробежное растяжение и возбужденные электронные состояния,
вычисленные значения термодинамических функций меньше, чем приведенные в настоящем
издании, и расхождения достигают 1,1 и 3,7 Дж-К~1-моль~1 соответственно в Ф° (Т) и S° (T) при
6000 К. В справочнике [2556] учитывалась только ангармоничность колебаний, значения
термодинамических функций меньше, чем в настоящем справочнике, и при 6000 К расхождения
достигают 1,5 и 4,9 Дж>К~1-моль~1 соответственно в Ф° (Т) и S° (T).
Константа равновесия реакции NO2(r)=2O(r)+N(r) вычислена на основании значения
ДГД"° @)=927,384+0,67 кДж-моль, которое соответствует принятой в [справочнике энтальпии
образования:
ДД°(гЮ2, г, 0) = 37,0 + 0,5 кДж-моль.
Принятая величина основана на результатах исследования равновесия реакции NO2 (г)=
=N0 (г)-Н/2О2 (г), проведенного Боденштейном и Линдером [682] статическим методом D99—
825 К). Из этих данных по уравнению II закона термодинамики было найдено 37,3 +
+0,5 кДж-моль, а по уравнению III закона 37,04+0,43 К Дж-моль. Менее точными являются
более ранние измерения Боденштейна и Катаяма [684] D92—976 К), из которых были получены
значения 40,1+1,6 кДж-моль и 25,6+0,7 кДж-моль, соответственно по Ни III законам. Дай-
блер идр. [1238] нашли фотоионизационным методом энергию связи D (NO—О), которой
соответствует энтальпия образования 39,4 кДж-моль.
NOj (г). Термодинамические свойства газообразного отрицательного иона диоксида азота
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 149.
Молекулярные постоянные, использованные при расчете термодинамических функций N0^,
приведены в табл. 13.4. Спектр и структура свободного иона NO2 экспериментально не
исследованы. Однако теоретические расчеты [2788, 2982, 3004, 3063, 3142], а также рентгеноструктурные
исследования ионного кристалла NaNO3 [882а, 8826, 3730а] и результаты спектральных
исследований NOj в матрице Аг [2667] и в твердых растворах КС1, КВг, KI [2197,2818], позволяют
принять, что N0^ в основном электронном состоянии ХгА [70] имеет нелинейную симметричную
структуру (точечная группа C2v) с параметрами г (N—О)=1,23+0,05 А и-^О—N—0 = 118+5°.
Вычисленное по этим параметрам произведение моментов инерции NOJ приведено в табл. 13.4
и может содержать погрешность 8 -10~117 г3-см6. Приведенные в таблице значения основных частот
свободного иона N0^ приняты на основании работ [805, 2667], в которых инфракрасный спектр
Ж)г исследован в условиях наименьшего взаимодействия с окружающей средой. В работе [2667]
изучался инфракрасный спектр N07, образующегося непосредственно в матрице [NO2+Cs+Ar]
при ультрафиолетовом облучении или электронной бомбардировке. В работе [805] исследовался
как инфракрасный спектр, так и спектр комбинационного рассеяния водных растворов KNO2.
С принятыми значениями частот NOj" согласуются результаты исследований спектров
кристаллов, растворов и расплавов NaNO2, KNO2 и AgNO2 в различных интервалах температур [3928,
2197, 2818, 3415, 1968, 1151, 826, 2356, 2655, 455, 3627, 2227, 1975, 1976, 700, 2228, 1725, 2703,
3416, 73, 381]. С учетом соответствующего разброса в значениях частот, вызванного влиянием
различного окружения иона NOJ, погрешности принятых значений частот оцениваются равными
294
Азот и его соединения
Таблица 13.8. Результаты определений сродства NO2 к электрону (в кДж-моль)
Авторы
Притчард, 1953 [3091]
Керран, 1962 [1133]
Фезенфельд, Фергусон, 1968 [1458]
Варнек, 1969 [3879]
Фогт, 1969 [3837]
Пейдж, Гуд, 1969 [2939]
Нелли и др., 1970 [2795]
Лифпшц и др., 1970 [2431]
Миллиган и др., 1970 [2667]
Лекман, Хершбах, 1971 [2335]
Берковиц и др., 1971 [595]
Фергусон и др., 1972 [1466]
Данкин и др., 1972 [1340]
Бейд, 1972 [444]
Лефферт и др., 1973 [2377]
Ричардсон и др., 1974 [3205]
Хербст и др., 1974 [1887]
Метод
Расчет по циклу Борна—Габера
Исследование ионно-обменных реакций
Исследование ионно-обменных реакций
Фотоотрыв
Исследование ионно-обменных реакций
Поверхностная ионизация
Исследование ионно-обменных реакций
Исследование ионно-обменных реакций
Фотоотрыв
Исследование зависимости Сечения реакции K+NO2=
=N02"+K+ от кинетической энергии частиц
Исследование ионно-обменных реакций
Исследование реакций в разряде
Исследование кинетики реакций SH"+NO2=NOi"+SH
и Cl2+NOii=NO5+Clsi в разряде
Исследование зависимости сечения реакций K-|-NO2=
=NO2+K+ и Na+NOjs=NO2+Na+ от кинетической
энергии частиц
Исследование зависимости сечения реакции Cs+N02=
=NO2~+Cs+ от кинетической энергии частиц
Фотоотрыв
Фотоотрыв
-Л0(Ш2)
154
> 347
< 347
299
< 234
376
>347
222 ±14
< 376
261 ±19
> 197
< 251
230 ±6
241 ±10
241 ±5
270+35
228+10
20 см г (vx и v2) и 40 см * (v3). Исследования электронных спектров поглощения и люминесценции
кристаллических нитратов [1849, 3624, 3415, 435, 147, 146, 326, 366, 1968, 2518, 2521, 2519],
а также результаты квантовомеханических расчетов [1847, 2581 ] показывают, что в области
энергий ниже 20 000 см ион NOJ имеет только одно возбужденное электронное состояние SBL с
энергией Го=19 000+250 см [366, 1968] К
Термодинамические функции NOj вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.130), A.128), A.122)—A.124) с учетом
возбужденного электронного состояния по уравнениям A.168)—A.170). Погрешности вычисленных
таким образом значений функций составляют 1, 3 и 5 Дж-К~1-моль~1 в значениях Ф° (Т) при
298,15, 3000 и 6000 К и обусловлены неточностью молекулярных постоянных и приближенным
характером расчета.
Ранее таблицы термодинамических функций NOa (г) вычислялись в справочнике [20951
б 149 11
ц р ф a () р
до 6000 К. Расхождения результатов расчета с данными табл. 149 не превышают 1 Дж-К~1>моль~1
в значениях Ф° (Т) и объясняются несколько отличным выбором исходных молекулярных
постоянных.
Константа равновесия реакции N0^ (г) =20 (r)-f-N (г)+е (г) вычислена с использованием
значения Дг#° @)=1155,384+10 кДж'Моль, соответствующего принятому в справочнике
сродству NO2 к электрону
Ао (NO,) = —228 ± 10 кДж • моль.
Принятое в справочнике значение Ао (NO2) основано на приведенных в табл. 13.8 данных.
Из работ, выполненных методом фотоотрыва, наиболее точной и подробной является работа Херб-
ста и др. [1887], на которой основывается принятое в справочнике значение. Лучшая из работ
1 В последние годы появились данные [2982, 1887, 3205] о существовании еще двух изомерных форм иона NOj:
перекисной и кольцевой. Однако поскольку энергии образования этих изомеров на 3,2 и 4,35 эВ превышают
энергию образования обычного изомера [2982], они не учитываются при расчете термодинамических свойств.
295
Ж>з(г) Глава 13
по изучению ионно-обменных реакций [1340] приводит к значению, практически совпадающему
с принятым. Исследования зависимости сечения реакций NO2 со щелочными металлами от
кинетической энергии исходных частиц [2377, 444] также приводят к величинам, совпадающим в
пределах их погрешности с принятой. Однако приведенные в этих работах погрешности, видимо,
занижены, так как экстраполяция при определении порогового значения энергии не может быть
выполнена с достаточной точностью.
Принятому значению Ао (NO2) соответствует
A/flro(NO-, г, 0) = —191 + 10 кДж -моль.
NO3 (г). Термодинамические свойства газообразного отрицательного иона триоксида азота
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 150.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 13.4. Экспериментальных данных о молекулярных постоянных свободного иона NO"
нет. В соответствии с теоретическим расчетом [3316] в настоящем справочнике принято, что ион
NO~ в основном электронном состоянии имеет плоскую симметричную структуру (группа
симметрии D3h). Приведенная в табл. 13.4 величина IaIbIc рассчитана в предположении, что
«30—N—О = 120° и r(N—О) = 1,25+0,05 А. Погрешность в значении IaIbIc составляет 2 -Ю16 г3X
X см6. Выбор таких структурных параметров основан на результатах рентгеноструктурных
исследований ионных кристаллов NaNO3 [1401, 4013, 3307], N2O5 [1738], Pb(NO3J [1816], электроно-
графического исследования паров Cu(NO3J [2368] и квантовомеханического расчета [2788].
Приведенные в таблице значения основных частот va и v2 являются средними из найденных в
работах [805, 2658, 2790, 381, 1725, 3463, 2104, 3839, 3847, 2069, 1923, 1469, 3263, 2092, 830, 2093]
значений для кристаллов нитратов (верхний предел) и расплавов и растворов нитратов (нижний
предел). Возможная погрешность 20 см. Значения дважды вырожденных частот v3 и v4 выбраны
средними по результатам работ [2818, 700, 2197, 2658, 1725, 3453, 3736, 2104, 3847, 2069, 1923,
1469, 3263, 2092, 1181, 2093, 1229], в которых расщепление частот вследствие взаимодействия иона
NOj с окружением минимально; погрешности в значениях v3 и v4 могут достигать; 50 см.
NO" не имеет возбужденных электронных состояний с энергией ниже 20 000 см [3316,
2199, 2634, 4020, 2693, 2694, 2584, 2200, 2530, 3195].
Термодинамические функции NO~ вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A.10), A.130), A.128), A.122)—A.124). Погрешности
вычисленных значений определяются как неточностью принятых молекулярных постоянных,
так и приближенным характером расчета и составляют 1, 5 и 7 Дж-К-моль в значениях Ф° (Т)
при 298,15, 3000 и 6000 К, в том числе 1, 3 и 3 Дж-К~1-моль из-за неточности молекулярных
постоянных.
Таблица термодинамических функций NO3 (г) публикуется впервые.
Константа равновесия реакции NO" (г)=30 (r)+N (r)-j-e (г) вычислена с использованием
значения АгН° @)=1509,167 +5 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
AfH° (NO", г, 0) = —298 ± 5 кДж • моль.
Принятое значение основано в первую очередь на результатах исследования ионно-обменной
реакции NOg (г)+НВг (r)=Br~ (r)+HNO3 (г) в работе Фергусона и др. [1466], в которой было
установлено, что эта реакция сопровождается небольшим тепловым эффектом (—4,3+1,5 кДжХ
Хмоль). Оно подтверждается результатами исследования сечения реакции I" (r)+HNO3 (г) =
=NOg (г)+Ш (г), выполненного Берковицем и др. [595], и исследования реакции NOJ (г)+
-f-NO2 (r)=NOg (r)-(-NO (г), выполненного Фезенфельдом и др. [1458а], из которых следует
—341 <; AfH° (NO", г, 0) ^ —245 кДж-моль. В кратком сообщении Ямдагни, Кебарле [4021]
сообщается значение сродства NO3 к электрону —375 кДж'МОль, полученное при исследовании
ряда равновесий с участием NO".
Близкое значение —374 кДж-моль было ранее рекомендовано Бучельншювой [44]. Если
для энтальпии образования NO3 (г) принять значение, рекомендованное в справочнике [258], это
приведет к величине Ь.^Н° (NOg, г, 0) = —297 кДж-моль,^что практически совпадает с
величиной, принятой в настоящем справочнике.
296
Азот и его соединения N2O (г)
N2O (г). Термодинамические свойства газообразного оксида диазота в стандартном состоянии
в интервале температур 100—6 000 К приведены в табл. 151.
Молекулярные постоянные, использованные в расчете термодинамических функций N2O,
приведены в табл. 13.7. Для основного электронного состояния ZXS+ эти постоянные взяты из
работы Плива [3042], в которой они были определены методом наименьших квадратов с
использованием экспериментальных данных [1911, 3051, 3403, 847, 3683, 3147, 1698, 1744, 3040, 3052,
3041, 2336]. Среди нескольких наборов постоянных, приведенных как в работе [3041], так ив
последующей работе Плива [3043], выбран наиболее оптимальный с точки зрения расчета
термодинамических функций вариант, с помощью которого с хорошей точностью (а=0,011 см)
воспроизводятся экспериментальные данные для 181 колебательного уровня (vx ^ 5, v2 ^ 6, 1г ^ 2, v3 ^ 5)
с энергией до 12 000 см. Из других исследований по определению молекулярных постоянных N2O
[1728, 3039, 3584, 3461, 3462, 3647, 481, 2985, 2314, 3683, 2992, 1342, 378, 379, 380] можно
упомянуть только'работу Тидуэлла и др. [3683], в которой рекомендован столь же полный набор
постоянных, но основанный на меньшем количестве экспериментальных данных A12 уровней с ^ ^ 3,
у2 ^ 5, l2 ^ 3 и vs ^ 2). В остальных работах, а также в многочисленных исследованиях раннего
периода (см. библиографию в монографии Герцберга [69] или Гренье-Бессон [1728])
молекулярные постоянные N2O по точности описания экспериментальных данных существенно уступают
принятым в настоящем издании. Согласно экспериментальным данным [70, 2363, 3106, 1510,
3915] и теоретическим расчетам [3002, 4047, 967, 3971], энергии возбужденных электронных
состояний N2O превышают 45 000 см.
Термодинамические функции N2O вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.125), A.126), A.122)—A.124) с поправками
на ангармоничность колебаний и взаимодействие вращения и колебаний по уравнениям A.131)—
A.133), на резонанс Фермипо уравнениям A.153)—A.155) и на центробежное растяжение по
уравнениям A.134)—A.136). Погрешности вычисленных значений при низких температурах
определяются главным образом неточностью исходных молекулярных и фундаментальных
постоянных, а также пренебрежением различия молекулярных постоянных для изотопной модификации
и природной смеси изотопов. Эти погрешности могут составлять в Ф°.Gт)при ^=298,15 и 1000 К
0,02 и 0,03 Дж-К~1-моль соответственно. При более высоких температурах начинают
сказываться погрешности из-за приближенного метода расчета и связанной с ним неопределенностью
экстраполяции высоких колебательно-вращательных уровней. Суммарные погрешности в Ф° (Т)
составят 0,3 и 1,2 Дж-К^-моль при 3000 и 6000 К.
Ранее таблицы термодинамических функций N2O (г) вычислялись в справочниках [88, 2095,
2556, 446] до 6000 К и в работах Пеннингтона и Коба [2989], Венкатесварлу и др. [3796] до 1000 К
и Нагараяна [2775] до 2000 К. В справочнике [88] расчет был выполнен по табличному методу
Гордона. Результаты этого расчета и данные табл. 151 совпадают при комнатной температуре и,
увеличиваясь с температурой, достигают 0,2 Дж-К~1-моль~1 в Ф° F000 К). Эти расхождения
связаны главным образом с пренебрежением постоянными у^к и а^к в расчете [88]. С данными
справочника [2556], а также [446] расхождения практически те же, но с обратным знаком из-за
различного числа членов в расчетных формулах (расчет в работе [2556 ] выполнен по методу,
аналогичному принятому в настоящем издании). Расчет в работе [2095] выполнен в приближении
«жесткий ротатор — гармонический осциллятор», и поэтому расхождения с настоящим справочником
увеличиваются от 0,05 Дж • К^-моль в Ф° B98,15 К) до 1,4 Дж-К^-моль в Ф° F000 К).
Результаты расчета работы [2989] практически совпадают с полученными в настоящем издании;
существенные расхождения с результатами расчета работы [2775], и, в особенности [3796] (до
15 Дж-К-моль), невозможно объяснить иначе, как ошибками в этих расчетах.
Константа равновесия реакции N2O(r)=O(r)+2N(r) вычислена с использованием значения
АГН° @) = 1103,39+0,94 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/?T°(N2O, г, 298,15 К) = 81,6 + 0,5 кДж • моль.
Эта величина основана на результатах калориметрических измерений, представленных
в табл. 13.9. Как видно из этой таблицы, измерения энтальпии разложения N2O [880, 3982] и
энтальпии сгорания [437, 1464, 880] находятся в хорошем соответствии. Некоторого предпочтения
заслуживают измерения энтальпии разложения в наиболее детальной работе Карлтона-Саттона
297
Таблица 13.9. Результаты определений энтальпии образования N2O (г) (в кДж • моль)
Авторы
Метод
bfH° B98,15 К)
Бертло, 1880 [617] Сожжение N2O в СО в бомбе 86,2
Томсен, 1880 [3674] То же 77,9
Сожжение N2O в На в бомбе 75,4
Карлтон-Саттон, 1932 То же 85,6
[879]
Аубери, Гриффите, 1933 Сожжение N2O и О2 в СО 81,6
[437] при постоянном давлении
Авторы
Метод
HfH° B98,15 К)
Феннинг, Коттон, 1933 Сожжение N2O в СО и в Н2, 82,6 ±0,5
[1464] а также Н2 в О2 в бомбе
Карлтон-Саттон, Амблер, Сожжение N2O и О2 в СО 81,5
Вильяме, 1936 [880] в бомбе
Разложение N2O (r)=N2 (г)+ 81,6 ±0,4
+VA (г)
Виттиг, Шматц, 1959 Разложение N2O (r)=N2 (r)+ 81,5 ±0,4
[3982] +1/2Ог(г) при 460° С в
калориметре постоянного давления
Таблица 13.10. Значения молекулярных постоянных и a(pi=l), принятые для расчета термодинамических функций
N2O3, N2O4, NaO5, N2H4, HNO3, NH2OH, NH2NO2 и FNO3
кула
Моле
N2O3
N2O4
N2O6
N2H4
HNOS
NH2OH
NH2NO2
FNOg
N203:
N2O4:
N2O6:
N2H4:
HN03:
NH2OH:
FNOS:
ф
H
И
к
Q
H
о
о
и
11A
HA
HA
Ж1 А
_м»
НА
НА
• ЛЫ
а Fo =
а Fo =
8 Fo =
а 17 —
^о —
1832
1373
1728
3280
3550
3620
3280
1759,1
. Та = 14
1630
812
1728
3325
1708,2
3297
1613
1300,9
100 см-1,
= 1600 см, /„„ = 0
= 660 см
1305 773
260 »
1338 1247
1587 1275
1330,7 1324,9
1605 1357
1370 1175
927,7 803,7
о = 1, pi=l; 6
322 • Ю-38 г - см2
7 „„ = 0,48 -Ю8 г-см2, с
= 1700 см-1, /пт)==0
= 2730 см"
1 J ___ Q
а Приведены постоянные
а тл
'о —
3510 см-1
0146 • 108 г • см
0135 • 10~38 г • см
V.
414 260
1710 480
860 743
1098 780
878,6 646,6
1115 895
1050 783
633,0 454,5
Fo = 490 см~
, ат = 2, пт-
;т = 2, пт =
2, от=1, пт
2 а 9 П
'¦
СМ'1
160
430
743
3314
579,0
3350
716
302,6
'. 7пр =
= 2.
2.
= 2.
О
н
337
675
645
3350
762,2
765
3400
708,5
v,.
.»
v,,
ХЮ117
r3 • cm'
« ______ 3,562-10»
1758 270 1264 751 — — — 1,05-10*
614 577 353 353 85 a • 2,07-10*
1628 1275 937,2 a - — — 7,0
» ______ 5,9967-102
386,2 ______ 4,878
1540" 800 596 350 — — — 6,37 • 102
a ______ 3,3-103
= 0,1124- Ю-38, г-см2, о,п = 2, гет = 2.
для тракс-]ЧН2ОН; для t;uc-NH2OH: Го =
1709 • 108 г • см2
, ат = 2, пт-
= 2.
= 3800 см-\ o = l, p^=l.
а
1
4
2
2
1
1
1
1
Азот и его соединения N2O3(r)
и др. [880]. В литературе известны исследования энергии связи Nz—О в молекуле N2O [1023,
1135, 3354, 1233]. Наиболее точные измерения, выполненные Дайблером [1233] методом
фотоионизации, приводят к значению энтальпии образования 86,2 кДж-моль.
N2O3(r). Термодинамические свойства газообразного триоксида диазота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 152.
Молекулярные постоянные, использованные в расчете термодинамических функций N2O3r
приведены в табл. 13.10. Молекула N2O3 имеет плоскую несимметричную структуру ONNO2
(точечная группа симметрии Са). Величина IaIbIc, приведенная в табл. 13.10, вычислена по
вращательным постоянным, найденным из МВ-спектра N2O3 Бриттеном и др. [786] (см. также [2316,
3843]). Авторы работы [786] интерпретировали полученные ими данные, исходя из структуры
¦с неэквивалентными связями N—О: г (NA,—ОA)) = 1,217+0,007 A, r (NA)—OB))=l,202+0,006 А,
^(NB)-OC))=1,142+0,004 A, r (NA)-NB)) = l,804+0,007 А, ^ OA)-NA)-OB)=129,81+l,10°,
Д ОA)—NA)—NB) = 117,47+0,42°, ^ NA)— NB)—ОC) = 105,05+0,12°. Основные частоты N2O3,
рекомендованные в таблице, выбраны в результате анализа данных, полученных при исследованиях
ИК-спектров N2O3 в газе [1292, 640], в жидком [1258, 731], в твердом состоянии [1959] и в
матрице [3781], а также спектра КР в растворе СС14 [944]. Величины vlt vs, v4hv5b3hth
непосредственно из измерений соответствующих переходов Байбартом и Юингом [640], причем
погрешность vx и v3 может составлять 2 см, a v4 и v5 5 см. Для v2 принята величина, средняя из
данных, полученных для газовой фазы Д'Ором и Тартом [1292] и Байбартом и Юингом [640], и
близкая к результатам исследования спектра N2O3 в матрице (Веретти и Пиментел [3781 ]) и в твердой
фазе (Брэдли и др. [731]). Погрешность этой величины оценивается в 20 см. Отнесение v6 и v?
дано по работе Брэдли и др. [731] (погрешность ve— 5 см, av, — 10 см). Частота v8
рекомендована с погрешностью 5 см из комбинационных полос [640], хотя к этой частоте отнесена также
менее интенсивная полоса 624 см [731]. Частота крутильного колебания v9 по данным работ
;[640, 731] может быть принята равной 70+7 см; в табл. 13.10 приведена соответствующая этому
значению величина потенциального барьера внутреннего вращения Vo, а также приведенный
момент инерции волчка NO2, рассчитанный по уравнению A.166) на основании принятых в
настоящем справочнике структурных параметров N2O3. Основное электронное состояние N2O3 относится
к типу 1А, а энергия первого возбужденного состояния того же типа принята (с погрешностью
200 см) по данным исследования электронного спектра N2O3 [3845, 3401]. В литературе
имеются сведения [3781, 1444, 1292] о возможности существования симметричного изомера строения
ONONO и даже был предложен набор основных частот этого изомера [3781], однако энергия
взаимного перехода обоих изомеров N2O3 определена настолько неуверенно [3781], что
рассматривать в настоящем справочнике симметричный изомер представляется неоправданным.
Термодинамические функции N2O3 (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.130), A.128), A.122)—A.124) с учетом
вклада внутреннего вращения по уравнениям A.160)—A.162) и вклада возбужденного
электронного состояния — по уравнениям A.168)—A.170). Погрешности вычисленных значений
определяются при низких температурах главным образом неточностью принятого значения
потенциального барьера внутреннего вращения и основных частот, а при высоких температурах —
приближенным характером расчета. Общие погрешности составляют 2, 5 и 8 Дж -К -моль
в Ф° (Г) при 298,15, 3000 и 6000 К, в том числе 1,5—2 Дж -К -моль из-за неточности исходных
постоянных.
Ранее таблицы термодинамических функций N2O3 (г) вычислялись в справочнике [2095]
до 6000 К, а также в работах Чао и др. [938] до 2000 К и Хизатсуне [1957] до 1500 К. Эти расчеты
основаны практически на одних и тех же молекулярных постоянных, отличающихся от принятых
в настоящем справочнике главным образом выбором основной частоты 627 см (см. выше). Кроме
того, в этих расчетах принималось, что внутреннее вращение в молекуле N2O3 является
свободным. Расхождения между результатами этих расчетов и данными настоящего издания составляют
от 2 до 9 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции N2O3 (г)=30 (r)+2N (г) вычислена с использованием
значения Д^° @) = 1590,785+ 1,3 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Яо(Ы203, г, 0) = 91,2 + 1,0 кДж.моль.
299
N204 (г)
Глава 13
Таблица 13.11. Результаты определения энтальпии реакции N2O3 (r) = NO (r)-f-NO2 (г),
на основании исследований ее равновесия (в кДж-моль)
Авторы
Абель, Произл, 1929 [333]
Верхок, Даниэле, 1931 [3813]
Битти, Белл, 1957 [532]
Солк, Пур, 1967 [3484]
Воспер, 1976 [3846]
Метод
Статический, 281—308 К
То же, но при экстраполяции к нулевому
давлению
Статический, 298—318 К, 24 точки
То же, но при экстраполяции к
нулевому давлению
Статический, 278—318 К, 73 точки
То же, но при экстраполяции к
нулевому давлению
Статический, 243—293 К, 5 точек
Статический, 253—323 К
ДГЯ° @)
II закон
39
38
37 ±7
—•
'39,3+1,6
37,5+1,2
41,7+2,8
39,0+0,3
III закон
37,3
37,4
37,07+0,18
36,25+0,71
37,40+0,07
36,75+0,07
37,01+0,37
36,54+0,15
Эта величина основана на результатах исследований равновесия реакции N2O3 (r)=NO (г)-|-
+NO2 (г), представленных в табл. 13.11. В работах [333, 3813, 532] измерялось равновесное
давление, устанавливающееся после смешивания известных количеств N2O4 и NO. Значения
констант равновесия для каждой из температур измерялись при нескольких начальных давлениях
и экстраполяцией определялись значения констант при давлении равном нулю, что устраняло
погрешности, связанные с отклонением свойств газов от идеального состояния. В работе [3484]
для определения концентрации NO2 был использован чувствительный фотокалориметрический
метод, что позволило проводить измерения при низких давлениях и избавиться от необходимости
экстраполяции к нулевому давлению. Воспер [3846] выполнял тщательные измерения констант
равновесия диссоциации N2O3 фотокалориметрическим методом. Он изучил возможные причины
наблюдавшейся в предыдущих работах зависимости констант равновесия от давления NO2 и
подтвердил, что это объясняется адсорбцией на стенках реакционного сосуда. К сожалению,
результаты измерений представлены только графически и в виде значений АгН и &SS, что исключает
возможность полного пересчета. Результаты расчетов по уравнению III закона с использованием
экстраполированных значений констант равновесия по работам [3813, 532] и [3484, 3846]
оказались в хорошем соответствии, что позволяет принять значение АГН° @)=36,6 + 0,5 кДж-моль.
Неточность термодинамических функций N2O3 (г) увеличивает погрешность этой величины до
0,8 кДж -моль.
N2O4 (г). Термодинамические свойства газообразного тетроксида диазота в стандартном
состоянии в интервале температур 100 — 6000 К приведены в табл. 153.
Молекулярные постоянные N2O4, использованные при расчете термодинамических функций,
приведены в табл. 13.10. Электронографические исследования [3465, 2567], а также исследования
инфракрасного спектра [1444, 1959, 3538, 554, 3482, 639, 3644] и спектра комбинационного
рассеяния [554, 1962] согласуются с предположением о плоской симметричной структуре молекулы
N2O4 в основном электронном состоянии ХХА (группа симметрии D2h) с внутренним вращением
волчка NO2 относительно связи N—N. Приведенные в табл. 13.10 значения 1aIbIc и /пр,
вычисленного по уравнению A.167), рассчитаны по структурным параметрам, найденным Мак-Клеллендом
и др. [2567] в результате электронографического исследования N2O4 в газовой фазе: г0 (N—N)=
= 1,782±0,008 A, r0 (N—О) = 1,190 + 0,002 Аи^ О—N—0 = 135,4 + 0,6°. Погрешности в
значениях IaIbIc и /пр могут достигать 4-1015 г3-см6 и 2-10~41 г-см2 соответственно. В таблице
приведены значения основных частот vx, v2, v3, v5, ve, v7, v8, рекомендованные Хизатсуне и др. [1959]
на основании исследований инфракрасного спектра N2O4 в газовой фазе и в матрице, и спектра
комбинационного рассеяния жидкого N2O4, выполненных в работах [1959, 3482, 1962]. С этими
значениями удовлетворительно согласуются частоты (\, v2, v3), найденные Тево и Эндрюсом
[3644] при исследовании спектра комбинационного рассеяния молекул N2O4 с 14N и 18N с исполь-
300
Азот и его соединения N2O5(r)
зованием техники лазерной матричной спектроскопии. Погрешности в этих частотах не превышают
10 см. Значения частот v9, v10, vu, v12, приведенные в табл. 13.10, приняты по работе Байбарта
и Юинга [639], в которой исследован колебательный спектр N2O4 в газовой фазе и проанализиро
звана структура полос. Показано, что полоса 385 см, отнесенная ранее [1959, 3482] к частоте
v10, в действительности связана с составной частотой (v2—v7)+tiv4—rav4. Частота v10 была оценена
авторами работы [639] исходя из экспериментально наблюдаемых частот, частоты крутильного
колебания v4 ~ 79 см, полученной из комбинационных полос, а также с использованием
экспериментального значения энтропии S° (N2O4, г, 294К)=303,30 Дж-К^-моль [1618].
Погрешности в значениях v9, vn и v12 не превышают 1 см. Погрешность в значении v10 может достигать
30 см.
Принятым значениям молекулярных постоянных и приведенному выше
экспериментальному значению энтропии [1618] соответствует барьер внутреннего вращения Fo=1600+300 см
для простейшего потенциала V=VOA—cos 2<p)/2. Значение частоты крутильного колебания,
соответствующее этому барьеру, v4=75 см хорошо согласуется с определенной в [1618]
экспериментальной величиной. Молекула N2O4, по-видимому, не имеет возбужденных электронных
состояний с энергией, меньшей 20 000 см.
Термодинамические функции N2O4 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.130), A.128), A.122)—A.124) с учетом
вклада заторможенного внутреннего вращения по уравнениям A.160)—A.162). Погрешности
вычисленных таким образом значений составляют 2, 6 и 10 Дж-К^-моль в Ф° (Т) при 298,15,
3000 и 6000 К и обусловлены главным образом приближенным характером расчета и неточностью
принятых молекулярных постоянных.
Ранее таблицы термодинамических функций N2O4 (г) вычислялись Хизатсуне [1957] до 1500 К,
Чао и др. [938] до 2000 К, а также в справочниках [2556, 2095] до 6000 К. Во всех расчетах
термодинамические функции вычислялись в приближении «жесткий ротатор — гармонический
осциллятор», причем внутреннее вращение в работе [1957] учитывалось как свободное, в работе [938] —
как заторможенное с барьером F0=550 + 30 см, и в справочниках [2556, 2095] — как
крутильное гармоническое колебание с частотой v4=50 см. Значения структурных параметров и
основных частот N2O4 во всех расчетах близки к приведенным в табл. 13.10, за исключением частоты v10,
для которой обычно принималось значение около 380 см. Расхождения в значениях Ф° (Т),
вычисленных в настоящем справочнике и в работах [1957,2556, 2095], не превышают 1 Дж-К~гХ
Хмоль. Авторы работы [938] ошиблись при вычислении 1Щ, что привело к неверной оценке
величины Fo. Расхождения в значениях функций, приведенных в работе [938] и настоящем
справочнике, достигают 3,6 Дж-К^-моль в значении Ф° B000 К).
Константа равновесия реакции N2O4 (r)=4O (r)+2N (г) вычислена с использованием значения
ЬГН° @) = 1908,368 + 1,3 кДж-моль в соответствии с принятой в справочнике энтальпией
образования1
A/ff°(N2O4, г, 0) = 20,4 + 1,0 кДж • моль.
Эта величина основана на исследованиях равновесия реакции N2O4 (r)=2NO2 (г),
представленных в табл. 13.12. Как видно из таблицы, результаты исследований равновесия диссоциации
N2O4 находятся в хорошем соответствии, и среднее взвешенное из расчетов по уравнению III
закона составляет 53,58+0,05 кДж-моль. Приведенные в табл. 13.12 погрешности отражают
только воспроизводимость данных. Основной вклад в погрешность значения энтальпии
диссоциации @,25 кДж-моль) вносит неточность термодинамических функций N2O4 (г). В принятом
значении Д J/° (N2O4, г) учтены погрешности энтальпии диссоциации и LfH° (NO2, г).
N2O5 (г). Термодинамические свойства газообразного пентоксида диазота в стандартном
состоянии в интервале температур 100—6000 К приведены в |табл. 154.
Молекулярные постоянные N2O5, использованные при расчете термодинамических функций,
приведены в табл. 13.10. Основное электронное состояние N2O5 должно быть синглетным, а
возбужденные электронные состояния с энергией выше 20 000 см неизвестны [70]. Структура
молекулы N2O5 была определена методом газовой электронографии Акишиным и др. [3]. В результате
этого исследования было установлено, что молекула N2O5 имеет неплоскую структуру O2N—О(ц)—
—NO2 с центральным атомом О в вершине угла и внутренним вращением двух волчков NO2
относительно связей N—О(ц). Приведенные в табл. 13.10 значения 1А1в1с и приведенного момента инер-
301
N2O6(')
Глава 1$
Таблица 13.12. Результаты определений энтальпии реакции N2O4(r) = 2NO2 (г) (в кДж-моль)
Авторы
Вурцель, 1919 [4002]
Боденштейн, Бойс, 1962 [682]
Верхок, Даниелс, 1931 [3813]
Сривастава, Баруа, 1961 [3498]
Солк, Пур, 1962 [3484]
Гаррис, Чарни, 1967 [1848]
Воспер, 1970 [3844]
Бленд, 1970 [674]
Метод
Измерение общего давления, 273—360 К,
11 точек
То же, 282—404 К, 67 точек
То же, 298—318 К, 63 точки
То же, 305—363 К, 17 точек
То же, 243—263 К, 3 точкиа
Определение концентрации NOa по
интенсивности поглощения света, 299—309 К,
3 точки
То же, 238—273 К, 20 точек
Определение концентрации NO2 по
измерениям скорости звука 273—323 К,
19 точек j
д
II закон
53,2 ±0,2
53,8+0,3
57,3 ±1,2
54,0±1,9
54+9
52 ±8
52,3+0,7
55,8+1,0
а В работе [3484 ] измерения проводились при очень низких давлениях, когда отклонения от
ного газа были пренебрежимо малы.
ГН° @)
III закон
53,60+0,03
53,61 ±0,03
53,72 ±0,04
53,40+0,10
53,59 ±0,07
53,56 ±0,06
53,56 ±0,04
53.51 ±0,07
Свойств идеаль-
ции волчка /п , вычисленного по уравнению A.166), рассчитаны по структурным параметрам,
найденным в [ЗЬ r(N-O)=l,21+0,01 A, r (N-Ow) = l,46f0,02 А, Д O-N-O = 134 + 5° и Д N-
—О(ц)—N=95 + 3°. Погрешности в значениях I/JbIo и /dpмогут достигать 2-101* г3.см6 и 2- 10~39г-сма
соответственно. Приведенные в табл. 13.10 значения основных частот N2O5 приняты по
рекомендации Хизатсуне и др. [1961 ] на основании исследований инфракрасного спектра
поглощения N2O5 в газообразном [1959, 1961] и твердом [1961] состояниях и спектра комбинационного
рассеяния N2O5 в растворах [944, 1961], а также с учетом результатов исследования
инфракрасного спектра N2O5 в матрице [1444]. Сравнение частот, полученных в газовой фазе и в матрице,
показывает, что их погрешности не превышают 5 см, за исключением частоты деформационного
колебания v13, определенной из комбинационных полос, погрешность в которой может достигать
15 см. Частоты крутильных колебаний (v14 и v15) экспериментально не наблюдались.
Приведенная в таблице величина барьера внутреннего вращения Vo вычислена на основании сравнения
экспериментально измеренной энтропии при298,15 К [3170] C56+21 Дж -К -моль) и энтропии,
рассчитанной по структурным параметрам, найденным Акишиным и др. [3], и частотам,
рекомендованным Хизатсуне [1961]. Погрешность оцененного таким образом значения Vo может
составлять 300 см.
Термодинамические функции N2O5 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.130), A.128), A.122)—A.124) с учетом
вклада внутреннего вращения по уравнениям A.160)—A.162). Общие погрешности
вычисленных термодинамических функций составляют 7, 10 и 15 Дж -К -моль в значениях Ф° (Т) при
298,15, 3000 и 6000 К и обусловлены неточностью молекулярных постоянных, главным образом
неопределенностью величины Vo (соответственно 6, 4 и 4 Дж -К-моль"), а также приближенным
характером расчета.
Ранее таблицы термодинамических функций N2O5 (г) вычислялись в работе Хизатсуне [19591
до 1500 К и в справочнике [2095] до 6000 К. Расхождения результатов расчета в работе [1959]
с данными, приведенными в табл. 154, составляют 10 и 0,5 Дж -К -моль в значениях Ф° (Т)
при 298,15 и 1000 К соответственно и обусловлены в основном тем, что в работе [1959] внутреннее
вращение принимается свободным. Расхождения с функциями, рассчитанными в справочнике
[2095], составляют приблизительно 10,5, 1 и 0,5 Дж-К~1-моль~1 в значениях Ф° (Г) при 298,15,
3000 и 6000 К. Эти расхождения обусловлены главным образом тем, что в этом справочнике для
величины Уо принято значение 1640 см, основанное на неправильно интерпретированных
экспериментальных данных Рея и Огга [3170] (авторы работы [2095] использовали для оценки величины
барьера значение S° (NaO5, г, 298,15 К)=346,4 Дж-К^моль).
302
Азот и его соединения
NH(r>
Константа равновесия реакции N2O5 (г)=5 О (r)+2N (г) вычислена с использованием
значения АГН° @)=2152,672 +1,8 кДж-моль в соответствии с принятой в справочнике энтальпией
образования
A/ff°(N2O6. г, 298,15 К) = 13,3±1,5 кДж
Эта величина основана на результатах калориметрического измерения энтальпии реакции
N2O5 (г)+1,208 NO(r)=0,711NO2 (г)+1,134 N2O4 (г)+0,022 N2O3(r)+0,185 NO (г), АгН°=—68,0±
+0,5 кДж -моль, выполненного Реем и Оггом [3169]. В результате реакции в стеклянной
реакционной камере образовывалась смесь окислов азота, приведенный состав которой вычислен
с использованием принятых в настоящем справочнике данных. При оценке погрешности принятого
значения энтальпии образования N2O5 (г) были учтены погрешности использованных в расчетах
энтальпий образования компонентов реакции.
Совпадающее, но несколько менее точное значение Ь^Н° (N2O6, г, 298,15 К)=13,5+3,1 кДжХ
X моль было вычислено из энтальпий образования и сублимации N2O5 (к). Огг [3167] измерил
энтальпию растворения N2O5 (к) в воде, откуда с использованием значения энтальпии
образования kfH° (NO3, aq) = — 206,35+0,80 кДж-моль (см. [85]), было получено bfH° (N2O5, к,
298,15 К)=—41,4+3,0 кДж-моль. Давление паров N2O5 (к) измерялось Руссом, Покорным
[32751 (статический и динамический методы, 243—284 К, Asi/°B62,4 K)=57,5+0,6 кДж-моль)
и Даниэлсом, Брайтом [1160] (статический метод, 285—288 К, Д8#°B72,8 К)=54,3+1,2 кДжХ
Хмоль). Среднее значение*после пересчета к стандартной температуре составляет Д,//° B98,15 К)
=54,9+1,0 кДж-моль".
NH (г). Термодинамические свойства газообразного гидрида азота в стандартном состоянии
в интервале температур 100—20 000 К приведены в табл. 155.
Молекулярные постоянные, использованные при расчете термодинамических функций NH (г)
приведены в табл. 13.13. Теоретические расчеты [2448, 861, 2051] показали, что из 12 валентных
Таблица 13.13. Принятые значения молекулярных постоянных NH и NH+
Молекула
Состояние
NH Х3?-
ахД
612+
А3И
еШ
NH+ Х2П
а42"
Л22~
?2Д
С22+
Примечание. Ниже все
Те
0а
12 576
21 197
29 806,8 е
44 012
0а
558
22 234,65
23 323,12
35 024,51
3282,1
3314
3347
3231,0
2122,5
3031 б
2621 б
1706,9
2300 б
2102 б
ыех.
Ве
СМ
78,3
63
70,7
98,55
з
54,5 б
50 б
60,7
43 б
49 6
постоянные даны в См.
NH: a Х=0,928, ^=—0,053; б
я pl=2,92-10~5;
- ^=-0,52.10-6,
е Л=-34,
3.=1 3 -К
72; ж
#0=—3-10-'.
)-*, Я0=10,5
^=-2,3-10-6
NH+: а Л=77,8; ° вычислено по соотношению A.63); в
я #0=1,85-10-'.
16,6676
16,77 в
16,7326
16,6901
14,697
15,56
14,90
11,4554
13,71
13,102
Ю-8; в В
вычислено
«г
0,6457
0,66
0,6049
0,7440
1,015
0,41 в
0,42 в
0,6897
0,38 "
0,45 в
о=16,439;
8.10-8;
по A.69);
De • 10»
1,706 б
1,62 г
1,654Д
1,767 ж
1,93 й
1,6 г
1,9 г
2,1 ж
2,0 г
2,0 г
г приведено
8 приведено
г приведено
г.
А
1,0372
1,034
1,0351
1,0365
1,1045
1,073
1,096
1,251
1,143
1,168
значение Z)o;
значение Д<?^;
значение DB;
электронных состояний NH только 5 (Х3И , ахД, Ь1Е+и^43П и схП) являются связанными
состояниями. Остальные 7 состояний отталкивательные или (&!2 и С3П) имеют неглубокие минимумы
(см. [2298, 1850, 2754J). В спектрах NH наблюдались 6 систем полос: A3U—XSI,~ [1556, 1266,
1557, 2757, 2005, 3828, 2507, 2953, 1751, 1752], еЧ1—ахД [2979, 1246, 2792, 1499, 1825, 3409],
с1П-Ь12+ [2481,3932],d12 + -b12+ [3933] d12+-c1n [3931, 2482, 2807], Ь1Е+-Х32~ [3827, 2538,
1642]. Определение молекулярных постоянных NH затруднено тем, что во всех электронных
переходах, как правило, наблюдаются только полосы с i/, у" ^ 1, причем наибольшей интенсивностью
303
NH (г) Глава 13
обладают полосы секвенции Ду=0, а полосы других секвенций удается получить только на
приборах с невысокой дисперсией. Поэтому для определения колебательных постоянных в ряде работ
использовались изотопические соотношения и данные для молекул NH и ND. Приведенные
в табл. 13.13 колебательные постоянные NH в триплетных состояниях приняты по работе Малисе
и др. [2507], которые помимо полос 0—0,] 1—1, 1—0 и 0—1 наблюдали в системе А3Л—Х32"
полосы 1—2 и 2—1 молекулы NH и полосы 1—2, 2—1 и 3—2 молекулы ND. Вращательные
постоянные, а также постоянные мультиплетного расщепления приняты по данным Мураи и Си-
маути [2757 J, хорошо согласующимся с результатами работы [2507]. До 1974 г.
интеркомбинационные переходы в спектрах NH не наблюдались. В связи с этим относительное положение
синглетных и триплетных термов NH оценивалось в ряде работ на основании косвенных
экспериментальных данных (см. [1499, 2903, 3504]), а также с помощью квантовомеханических расчетов
[2051, 2448, 2053, 2915]. Приведенные в таблице энергии синглетных термов NH основаны на
значении v00=21231+10 см перехода Ь1!,*—Х32~, наблюдавшегося в [1642, 3827] при фотолизе
NH3 и ND3. Колебательные постоянные NH в состоянии ахД получены Флорентом и Личем
[1499] при исследовании системы схП—ахД на основании значений AGy, для NH и ND. По оценке
[1499] точность ше составляет 15 см, а шехе — 10 см. Вращательные постоянные приняты
по данным [1499] и Симаути [3409]. Постоянные NH в состоянии Ьг2+ приняты по данным Уит-
такера [3931, 3932] из анализа полос v" <^3 переходов схП—ЬХ2+ и с?-2 +—ЬХ2 + , а в состоянии
сгП — по работам [3931, 2807], в которых исследовалась система полос dx2 +—схП.
Термодинамические функции NH (г) вычислены по уравнениям A.3)—A.10), A.93)—A.95).
Расчет (?вн и ее производных по температуре выполнен по уравнениям A.76)—A.78)
непосредственным суммированием по колебательно-вращательным уровням энергии состояний Х32~,
а1 Д, ЬХ2+ и А3П, а для состояния сЧ1 суммирование по / заменено интегрированием согласно
уравнению A.82). Колебательно-вращательные уровни энергии аппроксимировались
уравнениями типа A.65), A. 61) иA. 62); вращательные уровни для состояния Х32~представлялись
уравнениями A.40); для состояний ахД, А3П и схП мультиплетность и вырождение вращательных
уровней учитывались соответствующими статистическими весами. Коэффициенты Ykl в уравнениях,
аппроксимирующих уровни энергии, приведены в табл. 13.14; они вычислялись на основании
принятых молекулярных постоянных NH по методикам, изложенным в разделе 1.4. Ограничение
суммирования по / для состояний Х32~, ахД, б1!!* и А3И проводилось по предельным кривым
диссоциации этих состояний. В таблице приведены коэффициенты а. в уравнениях,
аппроксимирующих эти кривые. Ограничение суммирования для состояния схП проводилось в соответствии
с условием A.81). Приведены также значения vmax и /нт для всех рассматриваемых электронных
состояний NH. Основные погрешности вычисленных значений термодинамических функций NH
обусловлены неточностью принятых значений молекулярных постоянных и прежде всего энергии
диссоциации NH. Они не превышают 0,02 Дж К -моль в значениях Ф° (Т) при Т <1 3000 К
и могут достигать 0,15 и 1 Дж -К" -моль при 6000 и 20 000 К соответственно.
Ранее термодинамические функции NH вычислялись в справочниках [87, 88, 2095, 2556, 2496,
1783, 337] до 6000 К, а также в работе [3878] до 5000 К. Только в предыдущих изданиях
настоящего справочника [87, 88] и в справочнике [2556] учитывались возбужденные состояния NH.
Расхождения данных настоящего издания и этих двух расчетов не превышают 0,4 Дж -К -моль
в значениях Ф° (Т) (при 298,15 К расхождения составляют 0,03 ДжК моль из-за того, что
во всех расчетах не учтено расщепление компонент состояния Х32~). Расхождения с данными
работ [2095, 2496, 1783, 337, 3878] существенно выше, они достигают 0,55 и 0,80 Дж -К -моль
в Ф° F000 К) для работ [2095] и [2496] соответственно.
Константа равновесия реакции NH (r)=N (г)-(-Н (г) вычислена с использованием принятой
в справочнике энергии диссоциации
ДД° @) = Do (NH) = 348 ± 10 кДж • моль = 29 090 + 800 см.
Принятое значение является средневзвешенным из результатов работ [1532, 3180, 2807, 39311.
В работе Франклина и Дайблера [1532] был измерен потенциал появления иона NJ A6,0+0,1 эВ)
в реакции HN3+e=N2+NH+2e, которому соответствует Do (NH)=346+10 кДж-моль. Рид
и Снедден [3180] измерили потенциалы появления ионов NH+, N+ из NH3 и вычислили по этим
данным Do (NH)=357+25 кДжмоль. Значение Do (NH)=347+12 кДж-моль вычислено при
подготовке настоящего справочника из данных по предиссоциации NH в состоянии схП (у=0 и 1,
¦Лпах=24 и 16), полученных в работе Нарасимхана и Кришнамурти [2807], 'и по предиссоциации
304
Азот и его соединения
NH (г)
Таблица 13.14. Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см-i),
а также значения 2>ша1 и J\im, принятые для расчета термодинамических функций
NH и NH+
Коэффициенты
т,. ю-8
Ую-Ю-*
у20 • ю-2
Узо-Ю1
Уо1 • 10-1
Уи • 101
Уи • Ю'
У02 • Ю8
Уоз-Ю6
(ao=D.) • 10-«
ах
а2 • 108
а3 • 10"
"max
Aim
Коэффициенты
Tt. Ю-8
Ую-Ю-*
У2о • Ю-2
Узо-Ю1
Уох-10
Уц • Ю1
У21 ¦ 10»
Уо2 • Ю6
У оз • 1и
аЧ
0 0,125 771
0,327 877 0,330 915
—0,756 988 —0,598 911
—5,780 44 —5,740 89
1,6650 1,6770
—6,117 —6,600
—12,500 —
—170 —162
0,105 —
0,311200 0,377 663
0,707 842 0,656 360
62,845 0 52,563 5
—2679,63 2703,25
17 20
59 67
а*2"
0 0,005 58
0,3031 0,2621
—0,545 —0,50
— —
1,556 1,490
—4,1 —4,2
— —
-160 —190
_ _
(a0=De)-10-8 0,421578 750 0,340 539
а2 • Ю8
а3 ¦ 10»
^max
Aim
0,548 089 520 0,458467 727
14,403 890 7 20,615 010 7
1028,657 06 357,613 108
27 25
73 67
NH : a ^ = 0,928 и ц = — 0,053 см.
NH+: a Л =7,8
cm~i.
NH
&1S+
0,211 966
0,334 37
—0,681 49
—5,57000
1,673 26
-6,049
—
—164
—
0,351637
0,730 705
46,892 0
330.043
19
63
NH+
АЪ-
0,222 510 5
0,1654
—0,34
—
1,145 54
—6,897
9,08
-210
0,185
—
—
—
23
65
0,298131
0,321006
—0,821 630
—36,4155
1,669 01
—7,44
—
—175,5
0,078
0,205 303
1,093 32
211,174
43022,0
10
49
0,233 2312
0,2300
-0,43
—
1,371
—3,8
—
—200
—
—
—
—
—
26
92
cm
0,439 109
0,212 250
—
— ¦
1,469 70
—10,015
—
—193
—0,3
—
—
—
—
1
37
0,350 2451
0.2J0 2
—0,49
—
1,3102
-4,5
—
—200
—
—
—
—
—
20
52
NH+ (г) Глава 13
ND в этом состоянии (v=0, 1 и 2, /ша1=31, 25 и 18), полученных в работе Уиттакера [3931].
Существенно более низкое значение Do (NH)=310+15 кДжмоль было найдено в работе Сила
и Гейдона [3368] в результате измерений концентрации NH по его спектру поглощения в ударной
волне, распространяющейся в смеси N2+H2+Kr. По-видимому, в работе [3365] была допущена
систематическая ошибка, так как наблюдавшееся в этой работе состояние схП коррелирует
с атомами N BZ?)+H (ZS) и, если принять рекомендованное в этой работе значение Do (NH), то
уровень у=1 состояния с1!! будет иметь энергию, превышающую энергию диссоциационного предела
этого состояния.
Принятому значению энергии диссоциации соответствует
Д/Р^Н, г, 0) = 338,852 + 10 кДж • моль'1.
NH+ (г). Термодинамические свойства газообразного положительного иона гидрида азота
в стандартном состоянии при температурах 298,15 — 20 000 К приведены в табл. 156.
Молекулярные постоянные NH+, использованные при расчете термодинамических функций,
приведены в табл. 13.13. Согласно квантовомеханическому расчету [2447], NH+ имеет 5 связанных
валентных состояний (Х2П, а42"~,Л22~, B2L и С22+) с энергиями диссоциации, превышающими
5000 см. Остальные валентные состояния NH+ являются отталкивательными или имеют
неглубокие минимумы на потенциальных кривых (состояние В!Пс Tt—45 000 см). В спектрах NH+
изучены три системы полос: А^'-Хт, В*Ь-ХШ [1299, 1020] и С22+-ХаП [1453, 1299, 1020].
Во всех системах наблюдались только переходы, связанные с v=0 и 1, причем для состояния Х2П
характерны сильные возмущения, затруднявшие анализ. Приведенные в таблице значения
постоянных NH+ основаны на результатах работы Колина и Дугласа [1020]. Анализ возмущений
в спектрах 14NH+, 15NH+ и 14ND+ позволил авторам работы [1020] определить также значения
постоянных возмущающего состояния а*2~. При подготовке настоящего справочника
колебательные постоянные NH+ в состояниях Х2И, а*2~, 2?2Д и С22+ были вычислены на основании
значений AG/,, найденных в работе [1020], и энергии диссоциации NH+ в каждом из этих
состояний. Значения постоянных мв и швхе для состояния .А22~ приняты по работе [1020], где они были
найдены по значениям Д&д для NH+ и ND+.
Термодинамические функции NH+(r) вычислены по уравнениям A.3)—A.10), A.93)—A.95).
Расчет (?вн и ее производных по температуре выполнен по уравнениям A.76)—A.78)
непосредственным суммированием по колебательно-вращательным уровням энергии состояний Х2П и а*2~,
а для состояний Л22~, 2?2Д и С22+ суммирование по / заменено интегрированием согласно
уравнению A.82). Колебательно-вращательные уровни энергии аппроксимировались уравнениями типа
A.65), A. 61) и A. 62); вращательные уровни для состояния Х2П представлялись уравнениями A.42);
мультиплетность для остальных состояний, а также вырождение уровней всех рассматриваемых
электронных состояний учитывались соответствующими статистическими весами. Коэффициенты
Ykl в уравнениях, аппроксимирующих уровни энергии, вычислялись на основании принятых
значений молекулярных постоянных (см. табл. 13.13) по методикам, изложенным в разделе 1.4.
Значения этих коэффициентов приведены в табл. 13.14. Ограничение суммирования по / для
состояний Х2П и а42 ~ проводилось по предельным кривым диссоциации этих состояний. В табл.
13.14 приведены коэффициенты а{ в уравнениях, аппроксимирующих эти кривые. Ограничение
суммирования для остальных состояний проводилось в соответствии с условиями A.81). В
таблице приведены также значения ушах и Jnm для всех рассматриваемых электронных состояний
NH+. Основные погрешности термодинамических функций NH+ при умеренных температурах
обусловлены неточностью значений ДС^/д в состояниях Х2П и а*2~ и могут достигать 0,03 и
и 0,1 Дж-К-моль в значениях Ф° (Т) при 298,15 и 3000 К. При более высоких температурах
становятся существенными погрешности из-за неопределенности в описании всей совокупности
колебательно-вращательных уровней энергии NH+. Эти погрешности могут достигать
2—4 Дж-К-моль в значении Ф° B0 000 К).
Ранее таблица термодинамических функций NH+ (г) не публиковалась.
Константа равновесия реакции NH+ (г)+е (r)=N (г)+Н (г) вычислена с использованием
значения Д^0 @) = —916+28 кДж -моль в соответствии с принятым в справочнике потенциалом
ионизации
/0(NH)= 13,1 + 0,2 эВ= 1264 + 20 кДж • моль,
найденным Фонером и Хадсоном [1507] методом электронного удара. Это значение совпадает
е вычисленный по потенциалу появления NH+ при диссоциативной ионизации NH3 A3,1 +0,5 эВ
306
Азот и его соединения
NH,(r)
[3180]), но существенно меньше значения, найденного при изучении диссоциативной
ионизации HNCO A3,5+0,4 эВ [2903]). Принятому значению Io (NH) соответствуют
ДД°(ГШ+, г, 0)= 1602,852 + 28 кДж • моль,
Do (NH+) = 396,049 + 28 кДж • моль = 33 110 ± 2300 см.
NH2 (г). Термодинамические свойства газообразного дигидрида азота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 157.
Молекулярные постоянные NH2, использованные при расчете термодинамических функций,
приведены в табл. 13.15. Теоретические расчеты [1206, 979, 561, 3062, 812, 1836] и исследования
Таблица \i
TJ" Ii
!.15 Значения
молекулярных постоянных (в см), а также
j и р,, принятые
термодинамических функций NH2, HN3, HNO, NF2, FNO2, C1NO2
Коэффициенты
V2
VS
Vr
g
С*
•taaii-lO6
T j.108
x .106
¦^iill-lO6
a
PX
NH2: • i^!,
HN3: *¦ Dk =
HNO: а о34",
Djk =
NH2
3173
1497,3
3220
—
—
—
23,692
12,939
8,181
—74 400
-6 000
15 600
—3 500
—9 200
—2 600
—22 000
2
2
Г0 = 10 250; vj
7,67 -lO, Dji
Tn — 6000 см*
= 9,8- 10, Dj =
HN3
3339,9
2139,8
1263,7
1150,5
534,2
607,0
20,380 639
0,401 415 6
0,392 987 8
а
—
—
—
—
—
—
1
1
= 3325, v2 = 633
- = 2,63-10-Б, Dj
= 4,05-10-в см.
HNO
2684,7
1500,4
1565,3
—
—
—
18,476 213
1,411 26
1,306 55
б
—
—
—
—
—
—
1
1
v3 = 3500 см-1
= 1,64.10-' см
; АЫ", То=\
NF2
1>А
1074
573
931
—
—
—
2,351 49
0,396 015
0,338 116
—259
-2,7
9,9
-4,2
-1,3
1,9
-1,8
2
2
FNO2
&А>
1309,6
822,4
567,8
1791,5
559,6
742,0
0,440 348
0,381 805
0,204 107
—1,328
—2,623
0,415
—1,630
—0,224
—0,167
—0,660
2
1
, IAIBIC = 0,002 87 • 10-"? г8
-1.
3 154 см, а =
= 1, Pi=l.
для расчета
СШОа
*А
1286,0
792,6
369,6
1684,6
408,1
652,3
0,443 34
7 0,172 637
5 0,124 069 1
—2,04
—0,59
0,28
—0,86
—0,15
—0,017
—0,284
2
1
•смв, о=2, рх==2.
«Z)x= 4,485-Ю,
электронных спектров [3133, 3134, 3136, 1324, 3138, 1917, 1918, 3132, 3137, 2538, 3216, 1359,
1267, 1178, 2309] показывают, что молекула NH2 в основном электронном состоянии Х2Вг имеет
симм'етричную нелинейную структуру (точечная группа симметрии C2t). Вращательные
постоянные NH2 в этом состоянии, принятые в настоящем справочнике, определены с высокой точностью
[1359] в результате анализа вращательной структуры полос в электронном спектре
газообразного NH2. Этим постоянным соответствуют г (N—Н)=1,025±0,02 А иД Н—N—Н=103,2+0,3°.
Результаты других исследований и в особенности анализ спектра, полученного при лазерном
возбуждении [1178], хорошо согласуются с этими данными. Основные частоты NH2 в состоянии
%2В приняты по работе Дресслера и Рамзи [1324] (v2), в которой был исследован электронный
спектр газообразного NH2, и по работе Миллигана и Джекокс [2664] (vr и v3), в которой был
изучен инфракрасный спектр NH2 в матрице Аг. Значение vx было вычислено в работе [2664] в
приближении простого силового поля на основании частот va и v8. Следует отметить практическое
совпадение величины v2, найденной в матрице [2664] A499 см), с определенной в газе в работе
307
NHs(r) Глава 13
[1324] и работе Кролла [2309] A498 см) в спектре лазерной флуоресценции. В последней этой
работе полоса 3220 см была отнесена не к v3, как в работе [2664], а к vx; тогда в приближении
простого силового поля v1=3270 см. На этом основании рекомендованному в настоящем
справочнике значению vx приписывается погрешность 100 см, в то время как для v2 и v3 она составляет
не более 5 см. Первое возбужденное состояние NH2 относится к типу А*АХ. Диксон [1267] на
основании учета эффекта Реннера показал, что в этом состоянии молекула NH2 имеет в согласии
с теоретическими расчетами [1206, 979, 3062, 812, 1836] нелинейное строение. Приведенное в табл.
13.15 значение IaIbIo Для этого состояния вычислено на основании геометрических параметров
г (N—Н)=1,00 1 и 4 Н—N—Н=144°, рекомендованных Герцбергом [70]. Основные частоты
и энергия возбуждения состояния A2At приняты по данным работы [1324].
Термодинамические функции NH2 (г) вычислены по уравнениям A.3)—A.10), A.171)—A.173),
при этом принималось во внимание различие молекулярных постоянных NH2 в состояниях Х2ВХ
и А2АХ. Составляющие этих состояний вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.122)—A.124), A.127), A.128) с учетом поправки на
центробежное растяжение по уравнениям A.143)—A.145) для состояния Х2Вг. Ошибки из-за неточности
молекулярных постоянных и приближенного метода расчета практически несущественны при
низких температурах: погрешность в Ф° B98,15 К) составляет не более 0,05 Дж-К-моль.
При более высоких температурах погрешности возрастают и составляют 0,7 и 1,5 Дж -К -моль
в Ф° C000 К) и Ф° F000 К) соответственно.
Ранее таблицы термодинамических функций NH2 (г) вычислялись во 2-м издании настоящего
справочника [88] и в справочнике [2095], а также в работах [2307] (до 12 000 К) и [2664]
(до 2000 К). Ни в одном из этих расчетов не была учтена составляющая центробежного
растяжения, что является основной причиной расхождений с данными настоящего издания,
возрастающих с температурой^до 3,1 и 3,8 Дж-К~1*моль~1 в Ф° F000 К) соответственно для справочников
[88] и [2095] (в справочнике [2095] не учитывалось также состояние A*AX). Что-жасается расчета,
проведенного в работе [2307], то использование в нем значения р,=6 для основного электронного
состояния NH2, наряду с другими, менее существенными отличиями в выборе молекулярных
постоянных, приводит к ошибкам в данных, достигающим 7—9 Дж -К -моль.
Константа равновесия реакции NH2 (r)=N (г)+2Н (г) вычислена с использованием значения
Д^0 @)=710,021+10 кДж-моль, в соответствии с принятой в справочнике энтальпией
образования
A/fl"°(NH2, г, 298,15 К) = 190±10 кДж-моль.
Эта величина основана на кинетических исследованиях реакций с участием NH2 [1677, 691,
2327, 2224] и на измерениях потенциалов ионизации NH2 и потенциалов появления иона NH?
[1505]. Результаты работ, в которых были выполнены эти и другие исследования, представлены
в табл. 13.16.
Измерения констант скорости реакции пиролиза гидразина [3589, 2224] и бензиламина [3590,
2224, 1677] оказались в хорошем соответствии и различие вычисленных на их основании энергий
активации реакции пиролиза и соответствующих им энтальпий образования NH2 объясняется
почти полностью тем, что в них были приняты разные методы вычисления энергии активации.
В работе [1677] на основании оцененных молекулярных постоянных и энергии компонентов
реакции и переходного комплекса были вычислены значения предэкспоненциальных множителей,
значительно отличающиеся от более ранних расчетов, что привело к пересмотру рекомендованных
ранее значений энтальпии образования NH2.
Принятое в справочнике значение существенно отличается от общепризнанного ранее A75 +
+8 кДж -моль), что объясняется появлением новых работ и изменением интерпретации
результатов прежних кинетических исследований.
NH3 (г). Термодинамические свойства газообразного аммиака в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 158.
Молекулярные постоянные, использованные в расчете термодинамических функций NH3,
приведены в табл. 13.17. Молекула NH3 в основном электронном состоянии %}АХ имеет
пирамидальную структуру (точечная группа С3в) и для нее характерна инверсия атома N относительно
плоскости Н3. Геометрические параметры определялись в ряде работ из анализа вращательной
308
Таблица 13.16. Результаты определений энтальпии образования NH2 (г) (в кДж • моль 1)
о
п
о
а
[Авторы
Метод
298,15 К)
г,
Шварц, 1949 [3589]
Шварц, 1949 [3590]
Фонер, Хадсон, 1958 [1505]
Керр и др., 1963 [2224]
Гхош, Бэр, 1966 [1607]
Курило, 1969 [2327]
Голден и др., 1972 [1677]
Бёме и др., 1973 [691]
Исследование кинетики пиролиза гидразина толуольным методом, 903— 1055 К 173+8
Исследование кинетики пиролиза бензиламина толуольным методом, 922—1070 К 149
Измерение потенциала ионизации NH2 и потенциала появления NH+ из NHS 185+10
То же, но с использованием потенциала появления NH+ из NaH4 172+20
Исследование кинетики пиролиза гидразина толуольным методом, 887— 1034 К 167
То же, для бензиламина, 829—1061 К 152
То же, но при расчете энергий активации с использованием вычисленных теоретиче- 194
ски значений предэкспоненциальных множителей (см. [1677])
Исследование кинетики реакций N2H4+H=NH3+NH2 методом измерения интенсив- 160+10
ности люминесценции, 293—349 К
Исследование кинетики реакции O-bNH3=NH2+OH методом ЭПР, 361—677 К 195+8
Исследование кинетики пиролиза бензиламина масс-спектрометрическим методом, 194
1053—1258 К. Расчет энергии активации с использованием вычисленных теоретически
значений предэкспоненциальных множителей
То же, но при вычислении энергии активации по зависимости констант скорости реак- 142
ции от температуры
Исследование кинетики и равновесия реакции NH2+H2=H"+NH3 масс-спектроме- 185+10
трическим методом
Таблица 13.17. Значения молекулярных постоянных и о в состоянии %}А, принятых для расчета термодинамических функций
NH8hNFs(Px=1)
Молекула
NH3
NF3
3323,06
1031,91
v2
а
647,16
Примечание. Ниже все
NH3:
0'
оа
1»
Iе
2'
а Уровни
0
0,793
932,4
968,2
1602
в аА==0
•10-*, О
v,B)
v*B)
3443,38 1626,8
908,4 492,62
х1г
—51,3
—2,8
постоянные даны в см.
энергии и вращательные
К
6,196
6,198
6,050
6,130
5,900
078; аВ
87 -10-*,
, = 4,85
9,9443
9,9390
10,07
9,89
10,26
= 0,135; а
2я
3'
3е
4»
4я
4. —; (
в? = —1,438.10-
X
20
—3
постоянные
Еъ
1882,3
2383,5
2895,5
3442
4050
),009; аВ =
475 • 10-'
6
6
в
6
6,
= 0,
73-
и /
12
2
,5
для
130
160
260
21
21
176,
ю-*
«14
СМ""
—171,8 —7,
о о о
колебания у
9,73
9,50
9,20
9,32
9,35
аА = 0,066,
, аВ = 1,288
= 3,27-10-'.
^22
L
1
^23
• 32,0
3 —2,5 —6,5
2-
5'
Ъа
10'
10"
lB =
-ю-'
4675
5326
11985
12788
-0,230, L
6
6
6
6
22-
«24
-ю,
-2,
,21
,21
,21
,21
= 8,
10
*з
3
7 —18,5
4 —3,5
9
9
9
9
00-10
(
35
35
35
35
-4
= 2,
«34
-17
— 1
DTK
629-
«44
,6-18
,5 -0
20»
20я
40s
40я
= -14
10"*, см
А
0
0 6,1966
6 0,1948*
Еъ
29213
30116
66373
67317
,18-10-
1 = 6,5
А
6,
6,
6,
6,
L -10-
0
9,9443 е
0,35628л
•ь
21
21
21
21
^ = 7
о
6
3
Bv,
9
9
9
9
,36
= 1
,35
,35
,35
,35
ю-*.
,493-
2!
NH8(r) Глава 13
структуры ИК- [578, 2721, 3738, 1568, 1312] и MB- [1879, 136, 3080] спектров, а также методом
газовой электронографии [511, 2313]. Результаты всех работ хорошо согласуются друг с другом.
В табл. 13.17 приняты вращательные постоянные, полученные Бенедиктом и др. [578]. Принятым
вращательным постоянным соответствуют структурные параметры г (N—Н)=1,0173 +0,001 А
и -^ Н—N—Н=107,78+0,1°. Колебательный спектр молекулы NH3 изучался многими
исследователями. Обзор работ, выполненных до 1946 г., дан в монографии Герцберга [69]. Позднее в
работах Бенедикта и др. [578, 579, 580] и ряда других авторов [1568, 1126, 3410, 3863, 943] был
выполнен тщательный анализ колебательно-вращательного спектра. Данные, полученные в этих
работах, находятся в хорошем согласии. Авторами работ [578, 579, 580, 1568, 3863] было показано,
что инверсионное расщепление частот vx, v3 и v4 не превышает 1 см, и рекомендуются средние
значения для двух инверсионных уровней, которые и принимаются в настоящем издании. Для
колебательных уровней частоты v2 инверсионное расщепление значительно и быстро растет с
увеличением квантового числа. Для этих уровней принимаются значения, найденные в работах
[578, 579, 1568, 1045, 4044] для Р8^4и рассчитанные по потенциальной функции из работы
[1045 ] для v2 ^> А1. Несколько отличающиеся экспериментальные данные и способ
экстраполяции приводятся в работах [3874, 74]. Для постоянных ангармоничности принимаются значения,
полученные в справочнике [88] совместной обработкой данных работ [572, 1568] и таблицы
значений частот составных полос и обертонов, приведенной в монографии Герцберга [69].
Постоянные колебательно-вращательного взаимодействия для колебаний vl5 v3, v4 принимаются по
работе [578]. Для колебания va вращательные постоянные изменяются нерегулярно с изменением
квантового числа vt, поэтому величины Btl и AVl для ьг ^ 3 принимаются по работам [578, 579,
1568], а для уровней cd2^4 принято, что Вн = (Bs3 + В%)/2 и Аи = (Л|-f- A%)/2. Постоянные
центробежного растяжения Dj, Djk и Dz для всех v принимаются равными соответствующим
средним значениям для двух инверсионных уровней 0е и 0" по данным работы [578]. Энергия
возбуждения первого стабильного электронного состояния NH3 составляет 46 136 см [70].
Термодинамические функции NH3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3) — A.10), A.127), A.128), A.122)—A.124) с учетом
низкотемпературной поправки по уравнениям A.147)—A.149), поправок на ангармоничность
колебаний и колебательно-вращательное взаимодействие по уравнениям A.131)—A.133) и
центробежное растяжение по уравнениям A.140)—A.142). Вклад колебания v2 в статистическую сумму
вычислялся по уравнению, полученному Харом [1780]:
1)=0
где
= 2 "ВТ ФГ <* +^Т + Р'Г2) П A - Л) ехр (-
= G(°« v>
v — номер колебательного уровня v2, /=1, 3, 4 и v2=950 см. Поскольку суммирование
проводится как по четным, так и по нечетным состояниям, число симметрии молекулы NH3 принимается
равным 6, что соответствует квазиплоской молекуле. При температурах до 6000 К,; в расчете
принимались во внимание уровни v2, лежащие ниже 60 000 см. Погрешности вычисленных
термодинамических функций составляют 0,04, 0,4 и 2 Дж -К -моль в значениях Ф° (Т) при 298,15
3000 и 6000 К соответственно и обусловлены главным образом неточностью экстраполяции
уровней v2 и постоянных ангармоничности для других колебаний.
Расчеты термодинамических функций NH8 (г), производившиеся ранее, можно разделить на две
группы. В одну из них входят работы, в которых вклад колебания v2 вычислялся в приближении
гармонического осциллятора (Томпсон [3669], Хлебникова и Морозов [293], Сундарам и др.
[3578] до 1000 К, Стивенсон и Мак-Махон [3520] до 2000 К, Уилкинс и др. [3946] до 50Э0 К,
Шабур и др. [312] и Мак-Брайд и др. [2556] до 6000 К), причем в ряде из них учитывались
низкотемпературная поправка, а также поправка на центробежное растяжение [3520, 2556]. Ряд
расчетов, выполненных в том же приближении до 1938 г., упоминается в обзоре Вильсона [3960].
Расхождения между значениями функций, вычисленными в этой группе работ и приведенными
1 Уровни энергии вычислялись численным шггегрированяем уравяеняя Шредингера по программе,
представленной в работе [52].
310
Азот и его соединения NHJ (г)
в настоящем издании, обусловлены главным образом различием в методах расчета. Во вторую
группу входят работы, в которых вклад колебания v2 вычислялся непосредственным
суммированием по уровням энергии обертонов этого колебания (Хаупт и Теллер [1860] до 150 К, Дин и др.
[3656] до 1000 К, Гаррисон и Коб [1851] до 1500 К, Хар [1780] до 5000 К, справочники [88, 2095,
337] до 6000 К). Различия в значениях Ф° (Т), приводимых в справочнике [2095] и табл. 158,
составляют 0,1, 0,05 и 0,4 Дж К -моль при 298,15, 3000 и 6000 К соответственно и
объясняется тем, что авторы справочника [2095] не учли низкотемпературную поправку и использовали
несколько отличающиеся значения энергий для уровней колебания v2. Значения Ф° (Т),
полученные в работе [1780] и принятые в справочнике [337], совпадают с данными настоящего
издания при комнатной температуре и различаются на 0,3 и 0,4 Дж -К -моль при 3000 и 5000 К
соответственно, что объясняется различием в принятых постоянных ангармоничности и способе
экстраполяции уровней v2. Расхождения со значениями Ф° (Г), приведенными в предыдущем
издании справочника [88], составляют 0,004, 0,3 и 1,6 Дж-К-моль при 298,15, 3000
и 6000 К соответственно и объясняются различием принятых молекулярных постоянных (в том
числе уровней v2), а также более точным способом учета взаимодействия колебания v2 с вращением
и другими колебаниями.
Константа равновесия реакции NH3 (г) = N (г)+ЗН (г) вычислена с использованием значения
LrH° @)=1157,87 +0,53 кДж-моль, соответствующего энтальпии образования
A/ff°(NH3, г, 298,15 К) = — 45,94 + 0,35 кДж • моль.
Эта величина принята в настоящем справочнике по рекомендации КО ДАТА—MGHG [1000 J,
основанной на измерениях ряда авторов. Вандерзее и др. [3766, 3767, 3768, 3769] провели
прецизионные калориметрические измерения энтальпий реакций сожжения и растворения янтарной
кислоты и ее моноаммонийной соли, а также ионизации водного раствора NH3 и получили
значение —45,89+0,24 кДж -моль. Беккер и Рот [542] измерили энтальпии сгорания и
растворения в воде кристаллогидратов щавелевой кислоты и ее аммонийной соли и реакции
нейтрализации щавелевой кислоты аммиаком и получили значение —46,10+0,50 кДж-моль. Хабер
с сотр. [1788, 1787] и Виттиг, Шмуц [3982] измерили энтальпию разложения аммиака и
получили значения —46,3+0,6 кДж -моль, —45,9+0,6 кДж -моль и —45,94+0,42 кДж-моль
соответственно. При выборе величины энтальпии образования NH3 (г) были также учтены
результаты работ [1786, 1789, 2361, 2360, 3355] по исследованию равновесия реакции образования NH3,
критически рассмотренные и пересчитанные в работе [3766], а также при подготовке настоящего
издания, где по результатам этих работ было получено значение —45,56+0,25 кДж-моль.
Связанное с энтальпией образования NH3 (г).значение энтальпии образования иона NH* в
состоянии стандартного раствора
A/#°(NH+, р-р, Н2О, станд. сост., 298,15 К) = —133,26 + 0,25 кДж • моль,
необходимое для проведения ряда расчетов для настоящего справочника, принято по
рекомендации КОДАТА-МСНС [1000]. -
NH| (г) • Термодинамические свойства газообразного иона аммония NH* в стандартном
состоянии в интервале температур 298,15—6000 К приведены в табл. 159.
Молекулярные постоянные NHJ, использованные при расчете термодинамических функций,
приведены в табл. 13.4. Экспериментальные данные о структуре и частотах колебаний NH* в
литературе отсутствуют. В настоящем справочнике по аналогии с изоэлектронной молекулой СН4
принимается, что NH^ в основном электронном состоянии ХгАх имеет тетраэдрическую структуру
(точечная группа симметрии Td) с межатомным расстоянием г (N—Н) = 1,03+0,02 А, которое
оценено на основании результатов исследований ряда солей аммония методом дифракции
нейтронов [2408, 2409, 3855]. Эта структура иона NH* подтверждается теоретическими расчетами [1836,
2685, 1549, 690, 1227, 1369, 2012, 600, 1855, 1727, 661, 352, 2614, 4032, 3061]. Соответствующее
произведение моментов инерции, приведенное в табл. 13.4, имеет погрешность около 1,3-10~119 г3Х
X см6. Приведенные в этой таблице значения основных частот оценены на основании результатов
спектральных исследований кристаллических NH4C1 и NH4Br [1857, 3359] (v2 и v4) и
кристаллического NH4 PFe [3359]. Многочисленные исследования спектров различных солей аммония
в твердой фазе, расплаве и водном растворе [3855, 3849, 2789, 1229, 1085, 3088, 719, 3467, 3673,
3130, 1857, 3573,1086] дают для v2 и v4 значения, практически совпадающие с принятыми, а для
311
N,H2(r)
Глава 13
CN CN ffi
fa
о
& о
s
ев ч*
If
Hi
HO
1
я 5
SO
»o
in
В «
О
тн тН « CD r* 00 СО
«^ ч}1 CD -чн
СО
I I II I MSS
м м м isl
I I I I II l^s
I II II I III
I II II II ||
II II II I 22
CO CO CO
CO
CSJ ч(( CO Ol Ю О CO lQ 00
чгн О fO I> CO ГО CO CO lO
со со со
go i>- со со со ^ о о со
LO ^ О ^О C^J {^ч| СО СО f г*> ?^
^Н -^ -чН "^ -*Н
!
OOCDCDCOCMCNICNlcoCN!
О О О 00 Q
° El.0 5.° §.° *
о о о о о
3
Я
CO
ев
a
a
ч
to
ее
H
и
и
о
н
о
ей
I
1
-г-0"
1 s
S §
о ^
о
«
№
1
Е- «
О S
И
со И
Я 2
Я °
И о
се о
ii
8
,-?
« m CN io
з- II II °-
So II С
в ¦« ~* *^ 8
ы «rt *H t^
<а из
Я. а. о
Сч Сч
vi и vs — меньше примерно на 200 см.
Однако теоретический анализ [2545 ]
взаимодействия иона NH^ с окружением показывает,
что уг > 3250 см и v3 ;> 3350 см, что
прекрасно согласуется со значениями,
принятыми в табл. 13.4. Погрешность принятых
основных частот может достигать 70 см в v2
и v4 и 200 см в vx и v3. По аналогии с СН4
можно ожидать, что возбужденные
электронные состояния NH^" имеют энергии свыше
20 000 см.
Термодинамические функции NH? (г)
вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям
A.3)—A.10), A.122)—A.124), A.128)иA.130).
Погрешности вычисленных величин
составляют 0,5, 3 и 6 Дж -К -моль в значениях
Ф° (Г) при 298,15, 3000 и 6000 К. Они
определяются в основном неточностью принятых
молекулярных постоянных, а также
приближенным характером расчета.
Ранее термодинамические функции NH^(r)
вычислял Олтшуллер [368] в том же
приближении до 1000 К. Поскольку принятые
в указанной работе молекулярные
постоянные NH? мало отличаются от постоянных,
приведенных в табл. 13.4, расхождения
данных работы [368] и настоящего издания не
превышают 0,2 Дж -К -моль в значениях
Ф° (Г).
Константа равновесия реакции NH^ (r)-f-
+ е(г)=4Н (г)+N (г) вычислена с
использованием значения ДГЯ°(О)=684,954+20 кДжХ
Хмоль, основанного на принятой в
справочнике энтальпии образования
~KjH° (NH+, г, 0) = 650 ± 20 кДж • моль.
Принятая величина была выбрана по
результатам исследований равновесия и
направления протекания ионно-молекулярных
реакций F57 кДж-моль [4021а] и
<^ 664 кДж -моль [964]); квантовомехани-
ческих расчетов F57 кДж-моль [690]);
расчетов энергии кристаллических решеток
NH4C1 [16846, 2549а, 2335а, 128а, 54, 326а],
которые приводят к величинам 620—
643 кДж -моль; измерений потенциала
появления NH!" из C2H5NHa F27 кДж -моль
[1821]).
NaH2 (г). Термодинамические свойства
газообразного дигидрида диазота
(равновесная смесь изомеров) в стандартном
состоянии в интервале температур 100—6000 К
приведены в табл. 160.
Молекулярные постоянные N2H2,
использованные при расчете термодинамических функ-
312
Азот и его соединения N2H2(r)
ций, приведены'в табл. 13.18. Теоретические расчеты [2388, 863, 1867, 3318, 3701, 3989, 2984, 4015,
3856, 3214, 3783, 3602, 1590, 3974, 350] показывают, что молекула N2H2 может существовать в трех
изомерных формах: транс-, цис-, и 1,1-N2H2. На основании этих расчетов, а также
экспериментальных исследований спектров N2H2 [3725, 3726, 672, 875, 2848, 3956, 3955, 441, 2849], в настоя-
щем^справочнике принимается, что наиболее стабильной является трансконфигурация этой
молекулы (точечная группа C2h), а энергии цис- и 1,1-N2H2 относительно mpaKC-N2H2 равны
соответственно 3000 ±1000 см [863, 3318, 3701, 3989, 3856, 3214, 350] и 6000+2000 см [3989, 3856
350]. Теоретические расчеты свидетельствуют также о том, что у всех изомеров N2H2 имеются
возбужденные электронные состояния с низкими энергиями. В табл. 13.18 приведены состояния,
энергии возбуждения которых не превышают 20 000 см для каждого изомера. Погрешности в
принятых значениях То составляют 2000 см на основании сравнения результатов различных расчетов
[3989, 3856, 3214, 3501. Приведенное в таблице значение IaJbIc в основном электронном
состоянии ХгАд mpawc-N2H2 вычислено из вращательных постоянных Л0=10,000 21, Б0=1,304 194
и Со=1,149 861 см, найденных Карлотти и др. [875] в результате анализа вращательной
структуры полосы, связанной с симметричным валентным колебанием N—Н связи г. Этим постоянным
соответствуют структурные параметры г (N—N)=1,252+0,002 А, г (N—H)=1,028+0,005 А
и -3 Н—N—N = 106° 51' +28'. Исследования колебательного спектра /npaKC-N2H2 в газовой
фазе [3725, 672, 714], в матрице [714, 3246] и твердой фазе [3726], а также расчет силового поля
[3989, 2848, 2640, 2849] позволяют достаточно надежно определить все основные частоты, за
исключением частоты неплоскостного колебания. Приведенные в табл. 13.18 значения основных частот
найдены Бондибеем и Найблером [714] в результате исследования инфракрасного спектра и
спектра комбинационного рассеяния 7n/><zKc-N2H2, замороженного в аргоновой матрице (v1; v2, v3 и v4)
и Карлотти с соавторами [875] при исследовании инфракрасного спектра тга/?амс-гТ2Н2 в
газообразном состоянии (v5). Частота крутильного неплоскостного колебания ve принята по рекомендации
Карлотти др. [875], в которой они ссылаются на неопубликованное исследование Тромбетти.
Тромбетти наблюдал в области 1250—1350 см полосу, сложная структура которой, возможно,
связана с наличием кориолисова взаимодействия между колебаниями v5 и ve 2. Погрешности
в частотах vx v2, v3 и v4 с учетом возможного матричного сдвига оцениваются равными 10 см,
погрешность в v5 не превышает 1 см, а погрешность в v6 может достигать 150 см.
Термодинамические функции равновесной смеси изомеров N2H2 (г) вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—AЛ0), A.122)—A.124),
A.128), A.130) с использованием молекулярных постоянных состояния Х1АЙ тракс-1Ч2Н2 (см.
табл. 13.18). Состояния а3В„ 7repawc-N2H2, ХЩж as52 ^uc-N2H2 и Х3А2, а1А1 и ЪхАг l,l-NaH2
учитывались в расчете как возбужденные электронные состояния mpaMc-N2H2 по уравнениям
A.168)—A.170). Мультиплетность возбужденных состояний учитывалась при этом
соответствующими статистическими весами. Величины То для состояний, соответствующих цис- и 1,1-N2H2
и отсчитанных от основного состояния »ipaKc-N2H2, приведены в табл. 13.18 в скобках.
Погрешности вычисленных таким образом функций определяются как неточностью молекулярных !по-
стоянных, так и приближенным характером расчета и составляют 0,2, 4 и 7 Дж -К -моль в
значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности, связанные с неточностью молекулярных
постоянных (и прежде всего энергий возбуждения электронных состояний), преобладают при всех
температурах.
Таблица термодинамических функций смеси изомеров N2H2 (г) публикуется впервые.
Константа равновесия реакции N2H2 (r)=2N (г)+2Н (г) для равновесной смеси транс-, цис-
и 1,1-изомеров N2H2 вычислена с использованием значения Дг#°@)=1154,704+10 кДж-моль,
соответствующего принятой в справочнике энтальпии образования
Д^-Я0 (N2H2, г, равновесная смесь, 0) = 219 + 10 кДж • моль.
Эта величина вычислена на основании измерений потенциала появления N2H+ из N2H4 и
потенциала ионизации N2H2, выполненных Фонером и Ходсеном [1508а] (см. также более старые
измерения [1504, 3975]). Уиллис и др. [3975] по измеренному потенциалу появления Щ из N2H2
1 В работе [875] определены также постоянные центробежного растяжения.
2 Теоретический расчет [3318] дает силовые постоянные, которым соответствует слишком высокое значение-v,=
=1500 см, а Минквитц [2674] предположительно отнес к крутильному колебанию mpa»e-N2Ha полосу с
максимумом при 972 см.
313
«ipa«c-N2H2 (r), «|mc-N2H2 (г) Глава 13
вычислили значительно меньшую величину 153 кДж -моль, однако в работе [1508а] эти
измерения не удалось воспроизвести. При У=0 энтальпия образования равновесной смеси совпадает
с энтальпией образования трансизомера.
Tptmc-N2H2 (г). Термодинамические функции газообразного тпракс-дигидрида диазота в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 161.
Молекулярные постоянные mpawc-N2H2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Обоснование выбора молекулярных постоянных в основном
электронном состоянии см. выше в разделе по N2H2. В табл. 13.18 приведено только одно
возбужденное электронное состояние аъВд [3856].
Термодинамические функции rapaHc-N2H2 вычислены в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A. 124), A.128), A. 130). Вклад
состояния а?Вд рассчитывался по уравнениям A. 168)—A.170). Погрешности вычисленных
значений обусловлены неточностью молекулярных постоянных (в основном неопределенностью в
значении ve), а также приближенным характером расчета. Суммарная погрешность в значениях
Ф° (Т) при 298,15, 3000 и 6000 К составляет 0,2, 2 и 4 Дж К'1 моль.
Таблица термодинамических функций mpaKC-N2H2 (г) публикуется впервые.
Ранее Шаад и Кинзер [3318] на основании грубо оцененных молекулярных параметров
вычислили значение стандартной энтропии mpanc-'N^H^, которое на 4,4 Дж-К-моль превышает
рассчитанное в настоящем справочнике значение.
Константа равновесия реакции транс-^2Л2 (r)=2N (г)+2Н (г) вычислена с использованием
значения ArZf° @) = 1154,704+10 кДж-моль, соответствующего принятой энтальпии
образования
AfH° {транс-N^, г, 0) = 219 + 10 кДж • моль-'.
Эта величина совпадает с энтальпией образования равновесной смеси изомеров N2H2 при Т—0
(см. выше).
Д"мс-1М2Н2 (г). Термодинамические функции газообразного tyuc-дигидрида диазота в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 162.
Молекулярные постоянные tywc-N2H2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Существование стабильной цисмодификации N2H2 предсказано на
основании теоретических расчетов [2380, 3318, 3989, 3856, 3214, 3602, 1590]. Спектр и структура
tyuc-N2H2 экспериментально не исследовались 1. Теоретический анализ показал, что основное
состояние tyKc-N2H2 синглетно (ХгА). Приведенное в табл. 13.18 произведение моментов инерции
вычислено на основании оцененных структурных параметров г (N—Н) = 1,028А (как в тпранс-
N2H2) и г (N—N)=l,24+0,02 А и -? Н—N—N = 115,5+3° (приняты по теоретическому расчету
Вонга и др. [3989]). Погрешность в IaIbIc составляет 0,2 -10~117 г3 -см8. Частоты плоскостных
колебаний tyuc-N2H2 вычислены по силовым постоянным, найденным в работе [3989]. Погрешности
в вычисленных таким образом значениях частот не превышают 5 %. Частота крутильного
колебания v6 рассчитана по силовой постоянной, соответствующей потенциальной функции внутреннего
вращения, вычисленной в работе [3318]. Погрешность в значении v6 может достигать 10—15%.
Приведенное в таблице значение энергии возбуждения электронного состояния а?Вг принято
согласно результатам расчета [3856].
Термодинамические функции ^uc-N2H2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A.128), A. 130). Вклад
состояния а3Б2 рассчитывался по уравнениям A.168)—A. 170). Общие погрешности рассчитанных
таким образом термодинамических функций не превышают 0,2, 3 и 6 Дж -К -моль в значениях
Ф° (Т) при 298,15, 3000 и 6000 К. Эти погрешности обусловлены в основном неточностью оцененных
значений молекулярных постоянных, а также приближенным характером расчета.
Ранее таблица термодинамических функций i^c-N2H2 (г) вычислялась в справочнике [2095]
до 6000 К в приближении «жесткий ротатор — гармонический осциллятор». Расчет был выполнен
по оцененным значениям молекулярных постоянных, которые несколько отличаются от приведен-
1 В работе [672] ряд частот в спектре газообразного N2H2 отнесен к tyue-N2H2, однако, как было показано в работе
[3726], эти частоты принадлежат транс-^^Н.2. Кроме того, в спектре N2H2, полученном в матрице [3246], полосы
1279 и 3074 см с большими сомнениями были отнесены к основным частотам
314
Азот и его соединения l,l-NJI2(r), N2H4(r)
ных в табл. 13.18. Соответствующие расхождения не превышают 1,7 Дж-К-моль в значениях
Ф° (Т). Значение стандартной энтропии для ty«c-N2H2, вычисленное Шаадом и Кинзером [3318]
по грубо оцененным молекулярным постоянным, превышает на 4,4 Дж -К -моль значение,
вычисленное в настоящем справочнике.
Константа равновесия реакции ^uc-N2H2 (r)=2N (г)+2Н (г) вычислена с использованием
значения Дг/Г° @) = 1118,704+12 кДж-моль, соответствующего принятой энтальпии
образования
AfH° (i^c-N2H2, г, 0)= 255+ 12 кДж • моль.
Эта величина получена на основании принятых в настоящем справочнике значений энтальпий
перехода mpaHC-N2H2 в yuc-N2H2 C6+12 кДж -моль) и энтальпии образования mpaKc-N2H2.
1,1-N2H2 (г). Термодинамические функции газообразного 1,1-дигидрида диазота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 163.
Молекулярные постоянные 1,1-N2H2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Спектр и структура 1,1-N2H2 экспериментально не исследованы,
однако теоретические расчеты [2380, 3318, 3989, 3856, 3214, 3602, 1590] показывают, что этот
изомер N2H2 является стабильным и имеет триплетное основное электронное состояние. Значение
произведения моментов инерции, приведенное в табл. 13.18, вычислено на основании оцененных
структурных параметров. Величина ?(N—Н)=1,03+0,02 А принималась такой же, как в пгранс-
N2H2, a r (N—N)=l,28+0,02 А и Д Н—N—N=122+3° получены теоретическим расчетом [3989].
Погрешность IaIbIc составляет 0,2-10~117 г3-см6. Частоты плоскостных колебаний 1,1-N2H2,
приведенные в табл. 13.18, вычислены по силовым постоянным, найденным в работе [3989]. Их
погрешности не превышают 5%. Частота неплоскостного колебания ve принята равной частоте
соответствующего колебания в молекуле Н2СО. Погрешность в v6 может достигать 10—15%.
Теоретический расчет показывает, что у молекулы 1,1-N2H2 в области энергий меньше 20 000 см
должно быть два стабильных возбужденных электронных состояния. Найденные в работе [3856]
энергии возбуждения состояний а1А1 и Б1А% приведены в табл. 13.18. Их погрешность оценивается
в 15%.
Термодинамические функции 1,1-N2H2 (г) вычислены в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.128), A.130). Вклады
состояний а1А1 и ВгА2 рассчитывались по уравнениям A.168)—A.170). Общие погрешности
рассчитанных таким образом термодинамических функций не превышают 0,2, 4 и 6 Дж -К -моль
в значениях Ф° (Г) при 298,15, 3000 и 6000 К. Эти погрешности обусловлены в основном
неточностью оцененных значений молекулярных постоянных, а также приближенным характером
расчета.
Таблица термодинамических функций 1,1-N2H2 (г) публикуется впервые.
Константа равновесия реакции 1,1-N2H2 (r)=2N (г)+2Н (г) вычислена с использованием
значения Ь.ГН° @)=1082,704 +24 кДж-моль, соответствующего принятой энтальпий
образования
¦Aftf^l.l-NaHj, г, 0) = 291 +24 кДж • моль.
Эта величина получена на основании принятых в настоящем справочнике значений энтальпии
перехода mpaKc-N2H2 в 1,1-N2H2 G2+24 кДж-моль) и энтальпии образования j mpaKc-N2H2.
NaH4 (г). Термодинамические свойства газообразного гидразина в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 164.
Молекулярные постоянные, использованные в расчете термодинамических функций N2H4,
приведены в табл. 13.10. В результате исследований методами газовой электронографии [1636,
2704], а также MB- [2195, 2196] и ИК- [4017, 4018, 1814] спектроскопии установлено, что
наиболее стабильной является гошконфигурация молекулы N2H4, в которой две группы NH2
развернуты друг относительно друга по оси N—N на угол ср~90° (точечная группа симметрии С2).
К этому же выводу приводят и теоретические расчеты [1483, 1042, 2986, 3785, 365, 2108].
Величина IaIbIc, приведенная в табл. 13.10, была вычислена в предыдущем издании [88] на
основании геометрических параметров г (N—N) = l,4365 A, r (N—Н)=1,016 А, <р=91° 30', рассчитанных
в [88] из найденных Ямагути и др. [4017] вращательных постоянных А—?=3,981 см и
315
N2H4(r) Глава 13
В=0,809 см с учетом результатов измерений, выполненных Морино и др. [2704], и в
предположении, что углы Н—N—НиН—N—N тетраэдрические. Погрешность принятой [величины IaIbIg
составляет 0,1 -1017 г3-см6. Величина /пр для группы NHsj-вычислена по уравнению A.166) на ос-
новании приведенных выше значений структурных параметров. Молекула N2H4 имеет 12
основных частот, и их отнесение, принятое в таблице, выполнено Ямагути [4016] на основании
теоретического расчета силового поля молекулы и результатов измерения ИК-спектра N2H4 в газовой
фазе в работе Жигера и Лю [1629]. Для частоты v12 приведено значение, полученное Хамада и др.
[1814]. Каталано и др. [911] в результате исследований ИК-спектра N2H4 в матрице азота
предложили существенно иное отнесение деформационных частот N2H4, лежащих согласно данным
работ [1629, 4016] в области 1600 см. Однако исследования ИК-спектра и спектра КР в твердой,
жидкой и газовой фазе, выполненные Дюригом и др. [1351, 1354], так же как и более ранние
работы [3854, 4064, 131], противоречат выводам работы [911]. Следует отметить, что после
проведения расчета термодинамических функций N2H4 для настоящего издания, была опубликована
работа Дюрига и др. [1354], в которой в результате анализа спектра КР N2H4 в газовой фазе были
предложены отличающиеся значения ряда основных частот (v1=3398, v2=3329, vs=1642, и v5=
=1076 см). По-видимому, данные этой работы еще будут уточняться в дальнейшем. Основная
частота v7=376,74 см, относящаяся к крутильному колебанию N2H4, была определена в работе
Ямагути и др. [4017, 4018]. Этой частоте соответствует барьер внутреннего вращения 1850 см.
Однако для вычисления термодинамических функций был выбран барьер (см. табл. 13.10),
рассчитанный на основании определенной экспериментально в работе Скотта и др. [3367] величины
S° (N2H4, г, 298,15 К)=238,36+1,30 Дж-К-моль. Погрешность Vo оценивается в 500 см.
Определение величины Vo из анализа МВ-спектра [2195, 2196] и расчетным путем [1483, 1092,
2986, 3785, 365, 2108] дает довольно большой разброс, и оценка по экспериментальной величине
энтропии представляется в настоящее время наиболее надежной. Основное электронное состояние
N2H4 синглетное, а возбужденные электронные состояния с энергиями ниже 20 000 — 30 000 см
неизвестны [39].
Термодинамические функции N2H4 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.128), A. 130) с учетом
вклада внутреннего вращения по уравнениям A.160)—A.162). Погрешности вычисленных
значений определяются при комнатной температуре неточностью экспериментального определения
стандартной энтропии, а при более высоких температурах — главным образом приближенным
характером расчета. Общие погрешности составляют 1, 4 и 6 Дж-К-моль в Ф° (Т) при 298,15,
3000 и 6000 К, в том числе около 1 ДжК^-моль из-за неточности исходных постоянных при
Т > 3000 К.
Ранее таблицы термодинамических функций N2H4 (г) вычислялись в предыдущем издании
справочника [88], справочнике [2095] и работах [4064] (до 1000 К) и [3367] (до 1500 К).
Молекулярные постоянные и метод расчета, принятые в справочнике [88] ив настоящем издании,
практически не отличаются и соответствующие расхождения в Ф° (Т) не превышают 0,05 Дж •К~1Х
Хмоль. В справочнике [2095] принято менее точное отнесение основных частот, а составляющая
внутреннего вращения рассчитывалась как гармонический осциллятор; это приводит к
возрастающим с температурой расхождениям в термодинамических функциях по сравнению с настоящим
справочником, достигающим BfE>° F000 К) величин порядка 8,5 Дж -К -моль. ьВ работе [3367]
использованы основные частоты по данным уже устаревших исследований и несколько
отличающийся способ учета внутреннего вращения, что приводит к расхождениям с данными настоящего
издания до 3—4 Дж-К-моль в Ф° (Т). Расчет [4064] выполнен для структуры NH—NH3
(точечная группа симметрии Cs). Так как основные частоты и произведение моментов инерции
для этой структуры мало отличаются от принятых в настоящем справочнике, отличие в
термодинамических функциях, вычисленных в работе [4064] и приведенных в табл. 161, не превышает
0,3 Дж-К^-моль-1 в Ф°(Г).
Константа равновесия реакции N2H4 (r)=2N (г)+4Н (г) вычислена с использованием значения
Дг/Г°@) = 1696,438+0,94 кДж-моль, соответствующего принятой в справочнике величине
A/ff°(N2H4, г, 298,15 К) = 95,18 + 0,50 кДж • моль.
Эта величина вычислена на основании измерений энтальпий образования и испарения N2H4 (ж).
Энтальпия образования N2H4 (ж) была определена методом сожжения N2HU (ж) в кислороде в работе
Хьюза и др. [2033] (см. исправления к этой работе в статье Коля и Гилберта [1014] E0.38 +
316
Азот и его соединения HN3 (г), HNO (г)
+ 0,ЗЗкДж-моль-1),Астонаидр. [432] E0,12 кДж-моль-1), Хьюза [2032] (см. [2033] E0,08 кДжХ
X моль)) и комбинированием энтальпии сгорания N2H4>H2О [2033,1014] с энтальпиями
растворения N2H4 и N2H4-H2O [851] E0,88 кДж •моль). Для дальнейших расчетов принимается
средневзвешенное значение A,iTo(N2H4, ж, 298,15 К)=50,38±0,30 кДж-моль. Энтальпия
испарения N2H4 при 298,15 К, равная 44,8+0,4 кДж«моль, принята по результатам измерения
давления паров N2H4 статическим методом B73—343 К) в работе Скотта и др. [3367]. Близкое
значение D4,4 кДЖ'МОль) было получено по результатам измерений Хибера и Вёрнера [1930].
HN3 (г). Термодинамические свойства газообразного гидрида триазота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 165.
Молекулярные постоянные HN3, использованные при расчете термодинамических функций,
приведены в табл. 13.15. Исследования электронного [2575], инфракрасного [1435, 2402, 2301, 876,
4174, 1436, 1316, 2695, 1718, 3246, 3026] и микроволнового [3228, 376, 2232, 3969, 565] спектров,
а также результаты электронографического исследования [3349] показывают, что молекула HNg
в основном электронном состоянии %гА' имеет плоскую структуру с линейным фрагментом NA)—
NB)—NC) г (точечная группа симметрии С,) и параметрамиr0 (H—N(l))=0,975+0,015 A,ro(N(l)—
NB))=1,237±0,002 A, ro(NB)-(NC))=1,133±0,002 А и Д H-N(l)-NB)=114°7,8'±30'. Эти
структурные параметры соответствуют приведенным в табл. 13.15 вращательным постоянным и
постоянным центробежного растяжения, найденным в работах Винневиссера и др. [565, 3969]
при анализе спектра H14N3, выполненного в приближении слабоасимметричного волчка.
Приведенные в таблице значения основных частот HN3 получены в работах Карлотти и др. [876] (vx),
Мур и Розенгрена [2695] (v2) и Левина и Дауса [2402] (v3, v4, v8, ve) при исследовании
инфракрасного спектра газообразного HN3. Первое возбужденное состояние HN3 {А1 А') имеет энергию
37 900 см [2575] и не учитывалось в справочнике.
Термодинамические функции HN3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.127), A.128) с поправкой
на центробежное растяжение по уравнениям A.140)—A.142). Основные погрешности вычисленных
величин обусловлены приближенным характером расчета и составляют 1, 3 и 5 Дж-К~1'моль~1
в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции HN3(r) вычисляли Даус и Пиментел [1316] до 1500 К и
Айстер и Джиллет [1436] до 600 К в том же приближении. Значения функций, вычисленные в этих
работах, меньше значений, вычисленных в настоящем издании. Расхождения в значениях Ф°(Т)
достигают 0,7 Дж-К~1«моль~1 для данных работы [1316] при Г=1500 К и обусловлены
использованием в этой работе неточных значений частот v5 и ve. Константа равновесия реакции HN3(r)=
H(r)+3N(r) вычислена с использованием значения Дг#°@)=1328,198±4,2 кДж-моль,
соответствующего принятой в справочнике энтальпии образования
"" г, 298,15"К) = 294|±4 кДж • моль.
Энтальпия образования HN3(r) принятана основании результатов работ [1768, 1719, 1293].
Калориметрические измерения энтальпии разложения газообразного HN3, выполненные Гюнте-
ром и др. [1768], дают значение 294,1 кДж-моль. Результаты работ Грея и Уоддингтона [1719]
(измерялась энтальпия образования N~(aq) и энтальпия ионизации HN3) и Дорацио и Вуда [1293]
(измерялась энтальпия испарения HN3 из водного раствора) приводят к значению
293,7 кДж-моль, совпадающему с найденным при измерениях энтальпии разложения [1768].
HNO (г). Термодинамические свойства газообразного оксид-гидрида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 166.
Молекулярные постоянные HNO, использованные при расчете термодинамических функций,
приведены в табл. 13.15. Теоретические расчеты [3951, 3292, 3293, 1562, 4007], исследования
электронного [985, 1154, 465, 983, 910, 2076], инфракрасного [807, 1856, 2666, 2885, 2886, 2085, 2086]
и микроволнового [3287, 3286] спектров показывают, что молекула HNO в основном ХХА' и
возбужденных а?А" и АгА" состояниях имеет нелинейное строение (точечная группа симметрии
Сг). Приведенные в табл. 13.15 значения вращательных постоянных HNO в состоянии %ХА' опре-
1 Квантовомеханический расчет, выполненный Гаррисоном и др. [1852], указывает на небольшую нелинейность
этого фрагмента (^N A)—N B)-N C)=s=172°).
317
HNO (г) Глава 13
делены в результате исследования МВ-спектра в работе Сайто иТакаги [3286, 3287]. Этим
постоянным соответствуют структурные параметры [2888] ro(N—Н)=1,090 2бА, ro(N—О)=1,2090Аи
Д Н—N—0=108,047°. Практически совпадающие значения вращательных постоянных получены
Далби [1154] при анализе тонкой структуры полос системы АгА"—X1^'; в этой же работе в
приближении симметричного волчка вычислены постоянные центробежного растяжения, которые
были использованы авторами работ [3286, 3287] при анализе МВ-спектра и приведены в таблице.
Основные частоты HNO приняты по работе Клафа и др. [985], в которой исследованы
инфракрасный и электронный спектры испускания газообразного HNO. С найденными в этой работе
значениями vt. практически совпадают частоты, измеренные в инфракрасном спектре поглощения HNO
в матрице Аг [2085, 2086]. В более ранних работах принимались неверные значения 3300—3600 см
[983, 807, 1856, 2666, 2885, 910] и 1100 см [807, 1856, 2666, 2885, 2886]. Погрешности в принятых
значениях частот, по-видимому, не превышают 5 см. Существование стабильного триплетного
состояния а3А" было предсказано в работах [1154, 465, 983] на основании возмущений,
наблюдаемых в электронном спектре HNO. Приведенная в табл. 13.15 энергия возбуждения этого
состояния основывается на теоретических расчетах [3951] E484 см), [3293] E860 см) и [4007|
E730 см), а также исследовании спектра хемилюминесценции HNO [2076] F450 см);
погрешность принятого значения оценивается в 500 см. Энергия возбуждения состояния АгА",
наблюдавшегося экспериментально, принята по работам [1154, 465, 1983, 985]. Следующее возбужденное-
состояние имеет энергию около 50 000 см [866].
Термодинамические функции HNO (г) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.127), A.128) с поправкой
на центробежное растяжение по уравнениям A.140)—A.142) и с учетом двух возбужденных (а3А'г
и АХА") электронных состояний по уравнениям A.168)—A.170).
Погрешности рассчитанных значений обусловлены главным образом неопределенностью в
значении То (а3А") и приближенным характером расчета. В значениях Ф°(Т) общие
погрешности составляют 0,02, 1,0 и 1,5 Дж-К~1>моль~1 при 298,15, 3000 и 6000 К. Погрешности
из-за неточности молекулярных постоянныхв состоянии ХгА' не превышают 0,6 Дж-К~1-моль~1.
Ранее термодинамические функции HNO(r) вычислялись во 2-м издании настоящего
справочника [88] и справочнике JANAF [2095], а также в работах Шурвелла [3414] (до 2000 К) и Дже-
кокс и Миллигана [2085] (до 1500 К). Все эти расчеты выполнены в приближении «жесткий
ротатор—гармонический осциллятор» без учета центробежного растяжения и возбужденных
состояний (кроме расчетов в справочнике [88], где учитывалось состояние А1 А"), а также по отличающимся
значениям основных частот. Это приводит к расхождениям между результатами этих расчетов
и данными табл. 166, достигающим в значениях Ф°(Т) 0,2 Дж-К~1-моль~1 [2085], 0,6 Дж-К*
• моль-1 [88] и 4,9 Дж-К-^моль [2095].
Константа равновесия реакции HNO (г)=Н (r)+N (г)+О (г) вычислена с использованием
значения Дг#°@)=828,630+3,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^^НШ, г, 0)= 105 + 3 кДж • моль.
Эта величина принята на основании результатов исследований, приведенных в табл. 13.19.
Как видно из таблицы, результаты приведенных в ней работ хорошо согласуются между собой.
В справочнике принимается значение, среднее из результатов работ [983, 996, 1983].
Таблица 13.19. Результаты определения энтальпии образования HNO (г) (в кДж-моль)
Автор
Метод
(HNO, г, 0)
Кашион, Полани, 1959 [910] Исследование хемилюминесценции при реакции Н+ < 114,3
+NO=HNO
Клемент, Рамзи, 1961 [983] Предиссоциация в спектре испускания HNO, обра- > 103,5
зующегося при реакции H+NO
Клайн, Траш, 1961 [996]; То же > 103,5
Холмс, 1962 [1983] Кинетическое исследование реакции NO+HI=HNO+ 105,8
-f I и определение ее энергии активации
318
Азот и его соединения HNO2 (г)
HNO2 (г). Термодинамические свойства газообразной азотистой кислоты (равновесная смесь
изомеров) в стандартном состоянии в интервале температур 100—6000 К приведены в
табл. 167.
Молекулярные постоянные HNO2, использованные при расчете термодинамических функций,
приведены в табл. 13.18. Молекула HNO2 существует в двух стабильных конфигурациях (транс
и цис), разность энергий которых невелика A40 см [3782, 3588]), а барьер внутреннего вращения
высок (около 3800 см [2168, 1809, 2588, 3362, 583, 3435]). Величины 1А1в1с для обеих
конфигураций, приведенные в табл. 13.18, рассчитаны на основании вращательных постоянных Ло=3,0783,
?0=0,416 63 и С0=0,366 98 см для транс-HNO^ и Ло=2,7899, Я0=0,437 96 и С0=0,378 56 см
для z/uc-HNO2, определенных Финниганом и др. [1485] из МВ-спектра (см. также [1094, 1095]I.
Этим постоянным соответствуют структурные параметры г (О—Н)=0,969 и 0,989 А, г (N—ОН) =
= 1,442 и 1,399 А, г (N—О)=1,169и1,186А;-3O-N-O=110,6°h113,6°,-$H—О—N=102,1° и 103,9°
(для транс- и цис-ШЯО^ соответственно). Основные частоты приняты по работе Мак-Грау и др.
[2588], в которой исследован ИК-спектр транс- и tyuc-HNO2 в газообразном и твердом состояниях,
используя изотопное замещение, и выполнено отнесение всех шести основных частот для каждой
конфигурации по ^-ветвям (кроме vx для mpaHC-HNO2). Результаты других исследований ИК-
спектра HNO2 [1291, 2168, 1809, 1761] удовлетворительно согласуются с данными работы [2588],
за исключением v8 и v5 для цис- HNO2 в работе [2168], где было сделано неправильное отнесение
этих частот. Основное электронное состояние обоих изомеров HNO2 синглетное, а возбужденные
электронные состояния с энергиями возбуждения ниже 20 000 см не наблюдались [2610, 2245].
Приведенное в табл. 13.18 в скобках значение То ^uc-HNO2 представляет собой разность энергий
между цис- и трансконфигурациями HNO2, определенную в работах [3782, 2588] по температурной
зависимости интенсивности спектральных переходов двух изомеров (см. также [2168]).
Погрешность этой величины согласно [3782] составляет 35 см.
Термодинамические функции равновесной смеси изомеров HNO2 (г) вычислены по уравнениям
A.3)—A.10), A.171)—A.173), причем ^wc-HNO2 рассматривалась как возбужденное электронное
состояние с энергией возбуждения 140 см над наинизшим энергетическим уровнем m/?a«c-HNO2.
Колебательно-вращательные составляющие каждого изомера рассчитывались в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.122)—A.124), A.128), A.130).
Составляющая крутильного колебания для обоих изомеров рассчитывалась в приближении
гармонического осциллятора, поскольку барьер внутреннего вращения достаточно высок.
Погрешности вычисленных значений термодинамических функций составляют 0,6, 2 и 4 Дж-К«моль~1
в Ф°(Г) при 298,15, 3000 и 6000 К соответственно. При низких температурах эти погрешности
обусловлены главным образом неточностью разности энергий транс- и цисизомера, а при более
высоких температурах — приближенным характером расчета.
Ранее таблица термодинамических свойств равновесной смеси двух изо'меров HNO2 (г)
рассчитывалась Олтшуллером [369] до 1000 К и Варма, Кэрлом [3782] до 2000 К в приближении
«жесткий ротатор—гармонический осциллятор». Расчет в работе [369] выполнен по несколько
отличающимся значениям молекулярных постоянных. Расхождения результатов расчета этой работы
и настоящего справочника, доходящие до 3 Дж-К~1-моль~1 в Ф° B98,15 К), обусловлены
пренебрежением в расчете [369] разницей энергий транс- и цисизомеров HNO2. В расчете [3782]
использованы практически те же молекулярные постоянные, что и в настоящем справочнике и
соответствующие различия в значениях термодинамических функций малы.
Константа равновесия реакции HNO2 (г)=2 О (г)+Н (r)+N (г) для равновесной смеси транс-
is. цис-1ШО2 вычислена с использованием значения ДгЯ°@) = 1253,218+0,75 кДж-моль,
соответствующего принятой в справочнике энтальпии образования
A/F°(HNO2, г, 0) = — 72,8 ±0,6 кДж • моль.
Принятое значение основано на результатах исследования равновесия реакции NO(r)+NO2(r) +
+Н O(r)=2HNO (г), представленных в табл. 13.20. Исследования равновесий [3902, 431, 3860,
3846 ] были выполнены статическим методом, причем равновесная концентрация NO2 определялась
В работах [1094, 1095] приведены также постоянные центробежного растяжения
319
mpa«e-HNO2 (г)
Глава 13
Таблица 13.20. Результаты определений энтальпии реакции NO(r) + NO2
и энтальпии образования HNO2 (г) (в кДж • моль)
Авторы
Метод
(г) + Н2О(г)_
ДГЯ° @)
II закон
III закон
2HNO2 (г)
{HfH° HNO2,
г, 0)
Уэйн, Йост, 1951 [3902] Исследование кинетики B98 К), — —34,5 —72,8
1 точка
Ашмор, Тайлер, 1961 [431] Исследование равновесия B93— —31,4±4,2 —33,96±0,13 —72,56±0,60
354 К), 58 точек
Уолдерф, Бабб, 1963 [3860, Исследование равновесия — —33,89 ±0,20 —72,53 ±0,60
3861 ] B94,4—297,7 К), 9 точек
Воспер, 1975 [3846] Исследование равновесия B83— —33,2 ±0,9 —34,59 ±0,08 —72,87 ±0,60
323 К)
спектрофотометричёски. Наиболее тщательно эти измерения выполнены в работе [3846], однако
в этой работе приведены не константы равновесия, а коэффициенты уравнения, описывающего
зависимость констант равновесия от температуры. Погрешности значений Д^^О), приведенных
в таблице, отражают только разброс экспериментальных точек. Погрешности из-за неточности
термодинамических функций HNO2 и энтальпии перехода транс -> ^mc-HNO2 составляют 0,18
и 0,4 кДж-моль соответственно. При вычислении энтальпии образования HNO2 были также
учтены погрешности энтальпии образования NO и NO2. Равновесие реакции N2O3 (г)+Н2О (г) =
=2HNO2(r) было изучено Варма и Керлом [3782], измерившими интенсивности вращательных
переходов всех компонентов равновесной смеси. Авторы вынуждены были проводить измерения
при температуре адсорбционной камеры —49° С, так как при более высоких температурах
поглощение N2OS не наблюдалось, и вместо Н2О использовать DHO и D2O. Полученному из этих данных
значению ДгЯ°@)=4,4+1,3 кДж-моль соответствует Д^ЛГ0 (HNO2, г, 0) = — 71,7+1,0 кДж -моль.
В работе отмечена некоторая ненадежность измерений, которая привела к заметному ходу
измеряемых величин во времени. Полученные крайние значения соответствуют величинам —71,5
и —72,4 кДж-моль. Как видно из табл. 13.20, энтальпия образования HNO2 (г), вычисленная
по результатам измерений в работах [431, 3860, 3846], оказалась в достаточно хорошем
соответствии. Принятое значение является средневзвешенным из этих измерений. Это значение относится
к равновесной смеси двух изомеров. При Т=0 эта равновесная смесь состоит только из
трансизомеров.
Т;Р<ше-1Ш02 (г). Термодинамические функции газообразной трансазотистой кислоты в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 168.
Молекулярные постоянные mpa«c-HNO2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Обоснование выбора молекулярных постоянных дано выше
в тексте по HNO2.
Термодинамические функции mpanc-HNO^ (г) вычислены в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.128), A.130). Общие
погрешности вычисленных величин составляют 0,2, 2 и 4 Дж-К~1-моль~1 в значениях Ф°(Т) при
298,15, 3000 и 6000 К соответственно и обусловлены главным образом приближенным характером
расчета. Погрешности, определяемые неточностью молекулярных постоянных, не превышают
0,2 Дж-К~1-моль~1 в значениях Ф°(Г) во всем интервале температур.
Ранее таблица термодинамических функций mpaHC-HNO2 (г) вычислялась в справочнике [2095]
до 6000 К и в работе [369] до 1000 К. Расхождения между результатами расчетов
термодинамических функций в работах [2095, 369] и в настоящем справочнике не превышают 0,2 Дж-К-1 -моль
в значениях Ф°(Т).
Константа равновесия реакции транс-ТШО^ (г) =20 (г)+Н (r)-}-N (г) вычислена с
использованием значения АгН°@)=1253,218+0,75 кДж-моль, соответствующего принятой энтальпии
образования
Д^0 (mpaHC-HNO2, г, 0) = —72,8 + 0,6 кДж • моль.
320
Азот и его соединения цис-ВЛО2 (г), HNO3 (г)
Эта величина совпадает с принятой энтальпией образования равновесной смеси изомеров
HNO2 при Г=0 (см. выше).
Дмс-НЗЧО2(г). Термодинамические функции газообразного цисизомера азотистой кислоты в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 169.
Молекулярные постоянные tywc-HNO2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Обоснование выбора молекулярных постоянных дано выше в тексте
но HNO2.
Термодинамические функции tyuc-HNO2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.128), A.130). Суммарные
погрешности вычисленных величин составляют 0,2, 2 и 4 Дж-К~1-моль~1 в значениях Ф°(Т) при
298,15, 3000 и 6000 К соответственно и обусловлены главным образом приближенным характером
расчета. Погрешности, определяемые неточностью молекулярных постоянных, не превышают
0,2 Дж'К~1-моль~1 во всем интервале температур.
Ранее таблицы термодинамических функций tywc-HNO2 (г) вычислялись в справочнике [2095 ]
до 6000 К и в работе [369] до 1000 К в приближении «жесткий ротатор—гармонический
осциллятор» по завышенному по сравнению с принятым в настоящем справочнике значению IaIbIg
и основным частотам, отличающимся выбором неверных значений v3 и v5 по работе [2588] (см.
выше текст по HNO2). Расхождения в значениях функций, вычисленных в работах [2095, 369J
и в настоящем справочнике, обусловлены этими причинами и достигают 1,3 Дж«К~1«моль~1.
Константа равновесия реакции tyuc-HNO2 (r)=2O (г)+Н (r)+N (г) вычислена с использованием
значения ДГ?Г°(О)=1251,518+0,85 кДж-моль, соответствующего принятой энтальпии
образования
bfH°(iiuc-ENO2, г, 0) = — 71,1 ±0,7 кДж-моль.
Эта величина получена на основании принятых в настоящем справочнике значений энтальпии
перехода mpawc-HNO2 в цис-КТЯО2 A,7+0,4 кДж-моль) и энтальпии образования тпранс-1ШОг.
Н NO3 (г). Термодинамические свойства газообразной азотной кислоты в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 170.
Молекулярные постоянные HNO3, использованные при расчете термодинамических функций,
приведены в табл. 13.10. Молекула HNO3 имеет плоскую структуру (точечная группа симметрии
Cs). Вращательные постоянные (Л0=0,434 005, Яо=0,403 610 и С0=0,208 831 см) были
определены Милленом и Мортоном [2649] из МВ-спектра. Величина IaIbIc, приведенная в табл. 13.10,
вычислена по этим постоянным. Им соответствуют (см. [1096]) структурные параметры г(О—Н)=
=0,964 A, r(N-OH)=l,406A, r(N-O4He)=1,211 А, ^N-0^^=1,199 А, ^Н-О-Н=102°9',
^ О—N—Овде=И5°53', -3 О—N—Отранс=113°5Г (г„ — структура). Результаты менее точных
электронографических исследований [2, 2549] согласуются с этими данными в пределах ошибок
эксперимента. Рекомендованные в таблице значения основных частот HNO3 определены Мак-
Грау и др. [2587] в результате исследования ИК-спектра и спектра КР четырех изотопных
модификаций HNO3 в твердом и газообразном состояниях. Эти значения соответствуют положению
ф-ветвей в спектре газа. В более ранних исследованиях [1543, 1538, 1007] основные частоты HNO3
определены с меньшей точностью. Частота крутильного колебания v9 и ее обертон 2v9 также были
измерены в работе [2587] D55,8 и 895,4 см соответственно); на основании этих данных авторы
работы [2587] вычислили величину потенциального барьера внутреннего вращения Fo, которая
приведена в табл. 13.10; ее точность составляет 35 см. Другие определения Vo менее надежны из-за
использования неточного значения v9 [1007], а также других основных частот при расчете Vo
из термодинамических данных [2942,1511 ]. Величина 1^ в таблице вычислена по уравнению A.166)
на основании приведенных выше структурных параметров молекулы HNO3. Основное электронное
состояние молекулы HNO3 является синглетным и невырожденным; сведения о наличии низколе-
жащих возбужденных электронных состояний этой молекулы в литературе отсутствуют.
И|Термодинамические функции HNO3 (г) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.122)—A.124), A.128), A.130) с учетом вклада
заторможенного внутреннего вращения по уравнениям A.160)—A.162). Погрешности вычисленных таким
образом значений составляют 0,2, 3 и 6 Дж-К^-моль в Ф°(Г) при 298,15, 3000 и 6000 К
соответственно и обусловлены главным образом приближенным характером расчета (ошибки из-за
неточности молекулярных постоянных не превышают 0,2 Дж-К~1-моль~1).
321
NH2OH (г)
Глава IS
Ранее таблицы термодинамических функций HNO3 (г) вычислялись в справочнике [2095],
а также в работах [2587] до 1500 К и [2942, 1511 ] до 500 К. Из всех имеющихся в литературе
расчетов лишь в работе [2587] использованы те же молекулярные постоянные, что и в настоящем
издании, однако вклад внутреннего вращения в этом расчете принимался равным составляющей
гармонического осциллятора с частотой vg=455,8 см. Это приводит к расхождениям, доходящим;
до 3 Дж-К~1-моль~1 в Ф°(Г) при 1500 К. В остальных расчетах были приняты менее точные
значения молекулярных постоянных, а в расчете, приведенном в справочнике [2095], кроме того,
внутреннее вращение также рассматривалось как гармоническое колебание (v9 = 465 см).
Расхождения между расчетами в справочнике [2095] и данными табл. 170 (до 2 Дж -К -моль в Ф°(Г))
обусловлены различием в основных частотах и произведении главных моментов инерции, а также
способом учета составляющей заторможенного внутреннего вращения.
Константа равновесия реакции HNO3 (г) = Н (r) + N (г)+30 (г) вычислена с использованием
значения Дг//°@) = 1551,401 +0,78 кДж-моль, соответствующего принятой в справочнике энтальпии:
образования
A/ff°(HNO3, г, 0) = —124,2 +0,6 кДж • моль.
Эта величина основана на исследованиях равновесия реакций с участием HNO3 (г) (табл. 13.21)..
При вычислении погрешностей энтальпии образования были учтены вклады из-за неточности
термодинамических функций, которые оказались существенными для кристаллической и жидкой.
Таблица 13.21. Результаты определения энтальпии образования HNO3 (г) из данных
по равновесиям (в кДж • моль)
Авторы
Метод
AfH° @)
11 закон
III закон
Форсайт, Джиок, 1942 [5111] Расчет константы равновесия реакции — —124,3+1,0
3NO2(r)+H2O(r)=NO(r)+2HNO8(r) при 293,1 К
по результатам измерений [926а, 3963 ]
Джонс, 1943 [2160] Измерение констант равновесия реакции —127+7 —124,2+0,6
2NO2(r)+1/2O2(r)+H2O(r)=2HNO3(r) по
интенсивности спектра поглощения HN03 C43—
393 К, 5 точек)
Фейк, 1954 [1459] Измерение констант равновесия реакции —124+7 —124,5+1,5
NH4NO3^)=NH3(r)+HNO3(r) методом
определения точек кипения D63—543 К, 6 точек)
Бранднер и др., 1962 [746] Измерение констант равновесия реакции —124+6 —124,3+1,2
NH4NO3(K)=NH3(r)+HNO3(r) методом протока
C49—438 К, 12 точек)
Тоже, но для NH4NO3 (ж) D43—513 К, 8точек)а —120+7 —124,5+1,5
а В опытах с NH4NO3 (ж) было отмечено необратимое разложение NH4NO3, составляющее от 0,12% при
443 К до 15% при 513 К. Эта побочная реакция, очевидно, исказила результаты измерений, что особенно
заметно по величине, вычисленной по II закону.
HNO3 @,5 и 1,0 кДж-моль х соответственно). Как видно из таблицы, результаты измерений
находятся в хорошем соответствии. Принятое значение получено в наиболее точной из
рассматриваемых работ [2160]. Результаты других работ в пределах их погрешностей совпадают с принятой
величиной.
NH2OH (г). Термодинамические свойства газообразного гидроксиламина в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 171.
Молекулярные постоянные NH2OH, использованные при расчете термодинамических функций,,
приведены в табл. 13.10. Наиболее устойчивой является трансконфигурация молекулы NH2OH;;
энергия цисконфигурации на 3800 + 400 см выше [1702, 2986, 1483, 365]. Рекомендованная
в табл. 13.10 величина IaIbIo Для транс-ТЯН2ОН. вычислена по вращательным постоянным Ао—
=6,370 312, i?0=0,841 238 и С0=0,839 105 см, определенным в работе Цунекава [3739] из МВ-
спектра с использованием изотопного замещения. Этим постоянным соответствуют структурные-
322
Азот и его соединения NH2NO2 (г)
параметры г (О-Н)=0,962 A, r(N-H) = l,016 A, г (N-O)=l,453 A, ^ N-O-H=101°22',
^ Н— N—О = Ю7°06' и <$. Н—N—Н=103°15', удовлетворительно согласующиеся с менее
точными данными, полученными из анализа вращательной структуры ряда полос в ИК-спектре
[1630, 3604]. Значения основных частот транс-NH^OH приняты (за исключением частоты
крутильного колебания v9) на основании работы Жигера и Лю [1630], в которой исследован ИК-спектр
NH2OH в газообразном состоянии. Отнесение частот [1630] подтверждается также исследованием
спектра КР жидкого NH2OH [2599] и ИК-спектра твердого NH2OH [2863], а также результатами
теоретического расчета [276]. Ошибки в принятых значениях частот составляют 30 см (vj),
20 см (v2 и v7), 15 см (v3h v4)h10 cm (v5, v6 и v7). Величина ^принятапо работеТамагакеи др.
[3604], в которой исследована вращательная структура соответствующей полосы в ИК-спектре
газа. Авторы этой работы исследовали также ряд обертонов и «горячих» полос, связанных с
крутильным колебанием, и пытались определить потенциальную функцию внутреннего вращения
в молекуле NH20H. Полученные ими данные неоднозначны и не позволяют сделать окончательный
вывод о величине потенциального барьера внутреннего вращения, во всяком случае он должен
быть достаточно высоким (около 4200 см), что находится в согласии с результатами
теоретических расчетов [1702, 2986, 1483, 365]. В соответствии с этими расчетами, а также данными работ
[1630, 276], в справочнике принимается, что ifuc-NH2OH имеет те же основные частоты и
произведение моментов инерции, что и mpaHc-NH2OH. Основное электронное состояние NH2OH является
синглетным, а возбужденные электронные состояния с энергией ниже 20 000 см, по-видимому,
не существуют.
Термодинамические функции NH2OH (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.128), A.130) с учетом
tyMC-NH2OH как возбужденного электронного состояния по уравнениям A.168)—A.170), т. е.
в предположении, что Qxox вр для обоих изомеров одинаковы. Погрешности вычисленных
термодинамических функций составляют 0,3, 3 и 5 Дж-К^-моль в Ф°(Т) при 298,15, 3000 и 6000 К;
эта погрешность определяется главным образом приближенным характером расчета. Ошибки из-за
неточности молекулярных постоянных составляют 0,2—0,7 Дж-К^-моль.
Ранее таблицы термодинамических свойств NH2OH (г) вычислялись в работах Жигера и Лю
[1630] до 1200 К и Бедриной и др. [32] до 1000 К. В работе [1630] расчет выполнен с учетом вклада
внутреннего вращения по таблицам Питцера (F0=1790 см). Расхождения результатов этого
расчета с данными настоящего справочника лежат в пределах 0,3 Дж -К -моль. Кроме того, в работе
[1630] приведена таблица функций для z/kc-NH2OH, а в [32] — для m/>aKC-NH2OH, причем в обоих
расчетах приняты практически одни и те же значения молекулярных постоянных, совпадающие
с принятыми в настоящем справочнике, за исключением v9, для которой авторы работ [1630, 32]
приняли 430 см. Однако это различие не может объяснить те"значительные расхождения, которые
имеют место между результатами этих двух расчетов с данными настоящего справочника и между
собой. По-видимому, в этих расчетах допущены ошибки.
Константа равновесия реакции NH2OH (r)=N (г)+ЗН (г)+О (г) вычислена с использованием
значения Д 7/°@) = 1405,561 + 10 кДж-моль, соответствующего принятой в справочнике величине
Д//°(Ш20Н, г, 298,15 К) = — 50 ± 10 кДж • моль.
Принятая величина основана на результатах измерений Бертло и Матиньона [624], Бертло
и Андре [622] и Томсена [3676], из которых было вычислено значение энтальпии образования
кристаллического гидроксиламина (—113,4+10 кДж-моль). При вычислении этой величины
было использовано значение энтальпии образования водного раствора гидроксиламина
( 97,5 кДж-моль), вычисленное из термохимического цикла, включающего результаты работ
[367б', 1397, 616, 2476, 2734, 229, 622]. Значение энтальпии сублимации гидроксиламина F3,8+.
+ 0,6 кДж-моль), использованное для вычисления теплоты образования NH2OH (г), получено
^результате обработки данных Бака и Беттса [440] по давлению пара гидроксиламина (торзион-
ный метод, 261—280 К).
NH2NO2 (г). Термодинамические свойства газообразного аминил-нитрита в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 172.
Молекулярные постоянные NH2NO2, использованные при расчете термодинамических
функций, приведены в табл. 13.10. Структура молекулы NH2NO2 в основном электронном состоянии ис-
323
NH4NO3 (к, яг) . Глава 13
следована Тайлером [3745] методом МВ-спектроскопии с использованием изотопного замещения.
Величина IaIbIc, приведенная в табл. 13.10, вычислена по определенным в этой работе
вращательным постоянным (^„=0,422 182, Я0=0,396 691 и Со=0,205 612 см). Согласно [3745],
молекула NH2NO2 имеет неплоское строение (точечная группа симметрии Cs). Угол между плоскостями,
в которых лежат группы NH2 и NO2, равен 51°47', а структурные параметры г (N—О) = 1,206 А,
r(N—N) = 1,427A, r(N—Н) = 1,005А, ^ Н—N—Н=115°11' и ^O-N-O=130°8'. Отнесение
10 (v2—v9 и vu) из 12 основных частот NHaNO2 было выполнено в предположении о плоском
строении молекулы на основании анализа спектра КР в работе Кольрауша и Виттека [2277 ] и ИК-
спектра в твердой фазе и в растворах в работе Дейвиса и Джонатана [1175]. Тем не менее, выбор
этих частот, рекомендованных в таблице, был сделан на основании найденных в работах [2277,
1175] величин, так как их отнесение удовлетворяет характеристичности частот групп NH2 и NO2.
Частоты v10 (NH2, маятниковое колебание) и v12 (NH2, крутильное колебание) были оценены по
аналогичным частотам в молекулах N2H4 и NH2OH. Ошибки в принятых значениях частот
составляют 100 см (v8), 50 см (vx, v10 и v12) и 20 см (остальные частоты). Оценка величины барьера
внутреннего вращения относительно связи N—N не представляется в настоящее время надежной
[1483, 40]. Основное электронное состояние молекулы NH2OH является синглетным, а первое
возбужденное состояние имеет энергию возбуждения выше 30 000 см~х [3501, 2206, 1847] и поэтому
не рассматривается в настоящем справочнике.
Термодинамические функции NH2NO2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A.10), A. 122)—A.124), A.128), A.130). Погрешности
вычисленных значений определяются как ошибками в исходных молекулярных постоянных,
так и приближенным характером расчета и составляют 1, 6 и 10 Дж-К^-моль в значениях
Ф°(Г) при 298,15, 3000 и 6000 К. Ранее таблицы термодинамических функций NH2NO2(r)
не публиковались; в работе [3104] сообщается о расчете, однако численные данные не
приведены.
Константа равновесия реакции NH2NO2 (r)=2N (г)+2Н (г)+2О (г) вычислена с использованием
значения ДгЯ°@) = 1879,614+10 кДж-моль, соответствующего принятой в справочнике величине
A/ff'°(NH2NO2, г, 298,15 К) = — 26 + 10 кДж • моль.
Принятая величина основана на измерениях энтальпии образования кристаллического NH2NO2
[3168] и оцененного значения энтальпии сублимации F0,7 + 10 кДж-моль).
Рей и Огг [3168] измерили энтальпию разложения NH2NO2 в растворе щелочи с образованием
К2Оиводы. Принимая во внимание уточненное значение энтальпии растворения N2O вводе [2714],
из этих данных можно вычислить значение энтальпии образования NH2NO2(k), равное —87,0 +
+ 2,0 кДж -моль.
43 (к, ж). Термодинамические свойства кристаллического и жидкого нитрата аммония
в стандартном состоянии при температурах 5—900 К приведены в табл. Д.22 (наст, том, кн. 2).
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 13.22. В интервале от Г=0 до температуры плавления D42,85 К) термодинамические
функции NH4NO3(k) вычислены с учетом четырех равновесных полиморфных превращений (при 256,2,
305,38, 357,25 и 399,0 К). Для температур до 298,15 К расчет проводился на основании
результатов измерений теплоемкости в работах Нагатани [2785] A5—410 К) и Стивенсона и др. [3519]
A5—315 К). Данные этих авторов приводят к значениям ?"B98,15 К) и #°B98,15 К)— #°@)
(см. табл. 13.22), точность которых составляет 0,4 Дж-К^-моль и 0,06 кДж-моль
соответственно. Результаты измерений теплоемкости NH4NO3 в работах [1115] A83—273 К) и [1394]
A1—280 К) менее точны и не принимались во внимание при вычислении термодинамических
функций. При температурах выше 298,15 К расчет проводился для равновесных кристаллических
модификаций NH4NO3 с использованием данных работы [2785] для теплоемкостей четырех
различных модификаций NH4NO3 и значений температур х и теплот фазовых переходов, определенных
в этой же работе и в работах ряда других авторов (см. [258]). Температура плавления NH4NO3
1 Литературные данные о температурах фазовых превращений NH4NO3 противоречивы, что объясняется, в
частности, каталитическим влиянием следов влаги. Удаление следов влаги из образцов NH4NO3 повышает
температуры полиморфных превращений NH4NO3, как правило, на 10—20°. В справочнике принимаются данные,
относящиеся к неосушенным образцам NH4NO3, содержащим небольшие примеси влаги (около 0,1%).
324
JC3or и его соединения
NF(r)
Таблиц
Вещество
NH4NO3
NH4F
а С°р=а-
а 13.22. Принятые
Состояние
kIV, ромб
кШ, ромб
к11, тетр.
к1, куб.
ж
к, гекс.
ж
_ ЪТ + еТ3
1
К
ю
С*1 О
кДж-
• МОЛЬ
23,662
—
—
—
—
11,108
—
значения
Е2-
о
Дж-К-
150,81
—
—
—
—
71,97
—
Дж-К^.моль).
[ термодинамических величин для N1
3,15
29!
о а.
L • МОЛЬ
139,08
—
—
—
—
65,27
—
Коэффициенты
в уравнении С°р (Т) *
а
,-ЮЗ
е-109
48,77 302,9 —
60,14 194,0 —
500,80 1470,0 3765,5
107,7 130,0 —
161,0 — —
13,000 175,31 —
113,00 — —
[4N03(k, ж) и
Интервал
температур
К
298,15—305,38
305,38—357,25
357,25—399,00
3)9,00-442,85
442,85-900
298,15—511
511—760
NH4F (к,
Ttr или
К
305,38
357,25
399,00
442,85
—
511
—
ж)
^trH или
кДжХ
Хмоль 1
1,700
1,350
4,435
5,860
—
12,600
—
принята по данным [1367, 3655] с погрешностью 0,1 К. Данные других авторов [58, 3411, 1730]
менее точны, хотя и не противоречат принятой величине. Теплота плавления NH4NO3 принята
на основании измерений энтальпии NH4NO3 (ж) D42—543 К), проведенных в работе Фейка [1459].
Ее погрешность составляет 0,5 кДж -моль. Гораздо менее точные данные вычислены по
диаграммам состояния систем с участием NH4NO3 [3411, 2209]. По данным Фейка [1459] принято также
значение теплоемкости NH4NO3 (ж), точность которого оценивается в 3 Дж-К^-моль при Т <
< 543 К. Погрешности вычисленных значений Ф°(Т) при 298,15, 400 и 1000 К оцениваются в 0,4,
0,7 и 4 Дж-К^-моль-1.
Табулированные термодинамические функции NH4NO3 (к, ж) несущественно отличаются от
приведенных в справочнике Келли [2220].
В настоящем справочнике принято значение энтальпии образования кристаллического
нитрата аммония
A/^°(NH4NO3, kIV, ромб., 298,15 К) = —365,6 + 1,0 кДж • моль.
Принятое значение основано на калориметрических измерениях энтальпии разложения
NH4NO3(K)=N2(r)+1/2O2(r)+2H2O (ж), выполненных Беккером и Ротом [542] и Медаром и
Томасом [2600]. В работе [542] для инициирования реакции разложения использована реакция
сгорания вазелина или парафинового масла в кислороде, а в работе [2600] — нитробензола или динитро-
толуола. Из этих измерений были вычислены значения энтальпии образования кристаллического
нитрата аммония —364,98 кДж-моль [542] * и —365,58 кДж-моль [2600]. Эти величины
находятся в хорошем соответствии. Большего предпочтения заслуживает работа [2600], в которой
проведен более полный контроль чистоты исходного препарата и отсутствия побочных реакций.
NF (г). Термодинамические свойства газообразного фторида азота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 173.
Молекулярные постоянные NF, использованные при расчете термодинамических функций,
приведены в табл. 13.23. Молекула NF имеет большое число валентных электронных состояний.
Однако до настоящего времени в спектрах наблюдались переходы только между тремя состояниями:
Х32~, а1 А и ЬХ2+ [1303, 2173], соответствующими основной электронной конфигурации. Эти
состояния исследованы также и теоретически [3281, 2899, 382, 1403]. По аналогии с изоэлектрон-
ной молекулой О2, а также из расчета [1403], следует, что возбужденные состояния 3П, 41, 3Д
и 32, соответствующие двум нижним возбужденным электронным конфигурациям, являются от-
талкивательными состояниями или имеют небольшие минимумы на потенциальных кривых. То же
самое, по-видимому, имеет место в случае квинтетных состояний 5П и 62~, коррелирующих с ниж-
1 В работе [542] исходный препарат сушился при 308 К, что могло привести к фазовому переходу из модификации
IV в модификацию III. В этом случае значение, найденное по данным работы [542], следовало бы увеличить
go —366,19 кДж -моль,
325
NF'(r)
Глава f3
Таблица
Молекула
NF
NS
NF: a X=l,
лено
13.23. Принятые значения молекулярных постоянных NF и
Состояние
те
ы
Х31- 0 а 1141,37
а1*. 11 369 1274в
Ь^+ 18 877,05 1197,49
Х2П 0а 1219,02
а4П 29 000 790
АЧ1 30 230,03г 780,05
64S" 37 000 950
?2Ф 38 000 790
СЧ 39 801,1 Д 950
215 и fi=—0,0048 см; б приведено
по A.67); Д вычислено по A.69).
ШеХе
ве
СМ
8,99
13 г
8,64
7,31
13,03 б
3,66
—
значение D
NS: a 4 = 223,15 см; б вычислено по соотношению A.67);
дено
1,2056
1,23
1,2377
0,7754
0,60
0,5988
0,69
0,60
0,6850 е
NS
аг ¦ Ю2
1,492
1,6Д
1,448
0,64
1,39 в
0,47
.
—
De ¦ 106
к
4,6б 1,3173
4,5б 1,304
5,28б 1,3001
1,2 1,4942
1,3г 1,70
1,3 1,7003
— 1,58
— 1,70
— 1,5897 ж
-,; в вычислено по соотношению A.67); г вычис-
в вычислено
значение То для компоненты 2 Д3/ ; е приведено значение Во; ж
по A.69); г
4=90,4 см; Д приве-
приведено значение т-0.
ним диссоциационным пределом N(*5) и FBP). Приведенные в табл. 13.23 значения постоянных
NF в состояниях Х32~, а1 к. и 6г2 + получены Дугласом и Джонсом [1303, 2173] при исследовании
спектра испускания NF. Принятые значения колебательных постоянных в состоянии Х32~
хорошо согласуются с величиной Д&/2 = 1115 см, найденной в работах [2663, 1030] из спектра
NF в матрицах инертных газов. Точность постоянных, приведенных в таблице, невелика, так
как в состоянии ахД наблюдался только переход, связанный с v=0, а в других состояниях —
с у<2.
Термодинамические функции NF(r) вычислены по уравнениям A.3)—A.10), A.93)—A.95),
в которых величины Qm и ее производные по температуре рассчитывались по уравнениям A.76)—
A.78). Величины Qms вр для состояний Х32~, ахД и 612+ вычислялись непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням по уравнению
A.82). Колебательно-вращательные уровни этих состояний задавались уравнениями A.65), A.62)
Таблица 13.24.
Значения коэффициентов в уравнениях, аппроксимирующих уровни энергии (в см),
а также значения vms,x и e/iim, принятые для расчета термодинамических функций
NF и NS
Коэффициенты
Ге-10~5 0
Ую-10"* 0,114 056
у20.Ю-2 -0,083 742 0
У30 —0,132 347
Ущ-Ю 0,120 56
УХ1-10 —0,149 200
У02-105 — 0,406
^шах 36
/lim 201
Примечание. Ниже все постоянные
NF: а X = 1,215 и ц = — 0,0048.
NS: a A =223,15.
Состояние 42П
Те 30 230,03
NF
0,113 693
0,126 44
—0,127 30
—
0,122 60
-0,070 00
—0,45
49
221
данм в см.
6*2- ?2Ф
37 000 38 000
0,188 772
0,119 637
—0,777 570
-0,185 942
0,123 77
—0,144 800
-0,528
33
204
Сг1
39 801,1
NS
0 0,290 00
0,121 819 0,079 0
—0,069 832 9 —0,133
—0,032 678 4 —
0,077 54 0,060
—0,034 —0,139
—0,12 —0,13
60 29
319 203
326
Азот и его соединения NF2 (г)
ж A.40) (для состояния Х32~), а вырождение уровней состояния а*Д учитывалось статистическим
весом. Значения коэффициентов Y!cl в уравнениях, аппроксимирующих уровни энергии,
вычислены по методикам, изложенным в разделе 1.4, и приведены в табл. 13.24. Ограничение числа
вращательных уровней каждого электронного состояния проводилось в предположении о
линейной зависимости величин /maXi „ от v. Значения fmai и /цт для каждого состояния также приведены
в табл. 13.24. Основные погрешности вычисленных таким образом термодинамических функций
NF обусловлены неточностью принятых молекулярных постоянных в состояниях Х32~ и а*Д.
При температурах выше 3000 К к ним добавляются погрешности из-за использования
приближенной методики экстраполяции и ограничения числа колебательно-вращательных уровней энергии,
а также пренебрежения высокими возбужденными состояниями NF. Общие погрешности
значений Ф°(Г) при 298,15, 3000 и 6000 К не превышают 0,05, 0,3 и 1 Дж-К^-моль-1.
Ранее термодинамические функции NF(r) вычислялись в справочниках [87, 88, 2095, 2556].
Все эти расчеты были выполнены по оцененным значениям постоянных и без учета возбужденных
состояний NF. Расхождения данных настоящего издания и предыдущего [88] не превышают
0,15 Дж -К -моль в значениях Ф°(Т) и S°(T), а расхождения с данными справочников [2095,
2556] составляют 2,3 и 2 Дж-К^-моль соответственно.
Константа равновесия реакции NF(r)=N(r)+F(r) вычислена с использованием значения
Дг#°@)=В0(ОТ)=290,093 + 15кДж-моль-1яь;24250±1300см-1, соответствующего принятойв
справочнике энтальпии, образования
г, 0)=:258 ± 15 кДж • моль'1.
Эта величина принята на основании результатов измерений потенциалов появления ионов
в реакциях N2F4-fe -+ NF++NF2+2e и N2F4+e -+ NF2++F-+NF+e, выполненных Фонером
и Хадсоном [1508] методом электронного удара.
Измерения потенциала появления NF+ при фотоионизации NF3 в работе Дайблера и Уокера
{1237] привели к более низкому значению B30 кДж-моль). Однако точность величин,
полученных в этой работе, невысока из-за отсутствия надежных данных о потенциале ионизации NF.
NF2 (г). Термодинамические свойства газообразного дифторида азота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 174.
Молекулярные постоянные NF2, использованные при расчете термодинамических функций,
приведены в табл. 13.15. Исследования электронного спектра NF2 [1683, 70, 138] и теоретические
расчеты [3063, 1206, 1704, 3677, 2558, 4048] показывают, что в основном электронном состоянии
XtB1 молекула NF2 имеет симметричную нелинейную структуру (точечная группа симметрии
¦С2„), а первое возбужденное состояние имеет энергию выше 36 000 см. Значение вращательных
постоянных NF2 в состоянии Х2Вг определены Брауном и др. [810а] при исследовании
микроволнового спектра. Этим постоянным соответствуют геометрические параметры ro(N—F) = 1,3494 А
и ДР—N—F=103°20'. Найденная Боном и Бауэром [бФЗ] методом газовой электронографии
величина г (N—F) = 1,363+0,008 А относится, очевидно, к r^-структуре. Постоянные
центробежного растяжения также были получены в работе [810а]. Значения основных частот v± и v2 приняты
ло данным исследования спектра комбинационного рассеяния света в газе, выполненного Селигом
и Холлоуэйем [3379] (практически совпадающее значение v2 получено Гармони и др. [1846] при
исследовании инфракрасного спектра газа). Значение vg принято по результатам исследования Мил-
лиганом с сотр. [2086, 2087] и Гармони и Майерсом [1845] инфракрасного спектра NF2 в
матрицах N2. Поскольку частоты v1 и v2 претерпевают относительно небольшой матричный сдвиг, можно
полагать, что погрешность принятой величины v3 составляет 10 см.
Термодинамические функции NF2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.127), A.128) с учетом по-
лравки на центробежное растяжение по уравнениям A.143), A.144). Погрешности вычисленных
значений обусловлены в основном приближенным характером расчета и, в меньшей степени,
неточностью принятых молекулярных постоянных. Общие погрешности в значениях Ф° (Т) при
298,15, 3000 и 6000 К составляют 0,1, 1,5 и 3 Дж-К-моль; при этом погрешности из-за
неточности молекулярных постоянных не превышают 0,5 Дж-К-моль~1.
Ранее термодинамические функции газообразного NF2 (г) вычислялись в предыдущем издании
справочника [88], в справочниках [2095, 2556] до 6000 К, а также в работе Гармони и Майерса
327
NF2(r)
Глава 13
[1845] до 3000 К. Все расчеты выполнены в приближении «жесткий ротатор — гармонический
осциллятор». Расхождения в значениях функций, вычисленных в настоящем издании и в
работах [2095, 2556, 1845] при комнатной температуре, составляют 0,6—0,7 Дж-К~1-моль~1 в
значениях Ф° (Т) и S° (T). Это связано с использованием в настоящем справочнике более точных
вращательных постоянных. При увеличении температуры расхождения уменьшаются из-за учета
составляющей центробежного растяжения в настоящем справочнике и практически исчезают при 6000 К.
Расхождения с результатами предыдущего издания справочника [88] несколько больше из-за
различия принятых значений основных частот.
Константа равновесия реакции NF2(r)=2F(r)-)-N(r) вычислена с использованием значения
Дг#° @)=588,368+5,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/F°(NF2, г, 0) = 37,0 ± 5,0 кДж • моль.
Принятая величина основана на определениях энергии связи NF2—NF2 и NF2—F. Измерения
потенциала появления ионов NF2+ из N2F4 и потенциала ионизации NF2, выполненные Кристи,
Мамоновым [1117] и Фонером, Хадсоном [1508] привели к значениям Do (NF2—NF2)=81+12 и
91+10 кДж-моль соответственно. Харланд и Франклин [1839] исследовали распределение
избыточной энергии по внутренним степеням свободы переходного комплекса, образующегося
при резонансном захвате электрона молекулой NF3, и избыточную кинетическую энергию
образующихся ионов F~. Из этих данных было вычислено значение Aj,H° (NF2, г)=39+20 кДж-моль.
Таблица 13.25. Результаты определения энтальпии реакции N2F4 (r) = 2NF2 (г) (в кДж-моль)
Авторы
Метод
ДГЯ° @)
II закон
III закон
Херрон, Дайблер, 1961 [1899] Масс-спектрометрический C30—451 К) 87,6 ±7,0 —
Джонсон, Колбурн, 1961 Спектрофотометрический B98—353 К) 87,2 86,4
[2136] Зависимость суммарного давления от темпера- 70,0 ±2,5 86,6 ±1,1
туры C73—423 К)
То же, но при экстраполяции значений К к ну- 81,0±0,2 87,0±0,2
левому давлению
Пиетт и др., 1961 [3020] По интенсивности сигнала ЭПР радикала NO2 78,4 ±4,2 —
C42-435 К)
Даренбос, Лой, 1963 [1290] По интенсивности сигнала ЭПР радикала NOa 79,2+5,4 —
B43—298 К)
Элленридер и др., 1967 Зависимость суммарного давления от темпера- 84,0 ±0,4 87,8 ±0,2
[3841 ] туры A23—523 К)
Чуйков-Ру, Мак-Фадден, Исследование кинетики и равновесия в удар- 78,0 87,1а
1973 [3732] ных волнах C51—453 К)
а В работе [3732 ] не указаны численные значения найденных констант равновесия. Однако приведенный в этой
работе график иллюстрирует прекрасное соответствие измеренных значений и уравнения для lg К" из
работы [2136]. Поэтому было принято, что значения hrH° @, III закон), соответствующие результатам
работ [3732, 2136], одинаковы.
Более точные значения могут быть вычислены по результатам исследования равновесия N2F4 (г) =
=2NF2 (г), приведенным в табл. 13.25. Как видно из этой таблицы, результаты расчетов по
уравнению III закона по данным наиболее детальных работ [2136, 3841], а также работы [3732],
оказались в хорошем соответствии. Элленридер и др. [3841 ], так же как и Джонсон, Колбурн [2136],
измеряли общее давление частично продиссоциировавшего N2F4, однако в работе [3841 ]
реакционная камера была изготовлена из алюминия (а не пирекса), который предварительно
пассивировался фтором. Это позволило избежать взаимодействия N2F4 с материалом камеры. Принято©
в справочнике значение энтальпии образования NF2 было вычислено с использованием
значения D0(NF2—NF2)=87,6+0,5 кДж-моль. В погрешность этой величины включен вклад
@,3 кДж-моль), обусловленный погрешностью термодинамических функций N2F4.
Погрешность принятой энтальпии образования NF2 определяется в основном неточностью
использованного при вычислении значения энтальпии образования N2F4 A0 кДж-моль).
328
Азот и его соединения NF3 (г)>
NF3 (г). Термодинамические свойства газообразного трифторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 175.
Молекулярные постоянные NF3, использованные при расчете термодинамических функций,
приведены в табл. 13.17. Молекула NF3 имеет пирамидальное строение с атомом N в вершине и
принадлежит к точечной группе симметрии СЪг. Наиболее точные значения структурных
параметров r(N—F)=l,371 А и 4F—N—F=102°10' получены Отаке и др. [2923] при исследовании
МВ-спектра. Результаты других работ [2878, 3407, 3348, 3069] хорошо согласуются с этими
данными. Основные частоты NF3 были найдены в результате исследования ИК-спектра [3065, 3066,
2932] и спектра КР [2932, 130, 3398]. Наиболее полное исследование ИК-спектра газа с высоким
разрешением @,2 см) было выполнено Поплвеллом и др. [3065], определившими как нулевые
значения основных частот, так и ряд других постоянных, в том числе вращательные постоянные,,
постоянные взаимодействия вращения и колебаний, постоянные ангармоничности колебаний и т. д.
Позже в работах [2923, 2922, 3273, 3543, 198, 3192, 3190, 2540, 358] были детально изучены
отдельные полосы колебательного спектра, а также чисто вращательный спектр NFg. Результаты
этих работ хорошо согласуются между собой и с результатами работы [3065], за исключением
величин af и af, для которых в работах [2922, 3273, 3543] были получены более точные
значения. В табл. 13.17 значения основных частот и постоянных кориолисова взаимодействия
рекомендованы по работе [3065], постоянных ангармоничности и центробежного растяжения — по [198]г
вращательных постоянных и постоянных взаимодействия вращения и колебаний — по [2923].
В работах [2922, 3543] были найдены также постоянные Z-удвоения. Основное электронное
состояние молекулы NF3 — синглетное и невырожденное. Сведения о возбужденных электронных
состояниях NF3 в литературе отсутствуют и можно предполагать, что состояния с энергиями
возбуждения ниже 20 000—30 000 см не существуют.
Термодинамические функции NF3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.127), A.128), A.122)—A.124) с
поправками на ангармоничность колебаний и взаимодействие вращения и колебаний по уравнениям
A.131)—A.133), на центробежное растяжение — по уравнениям A.140)—A.142) и на кориолисово
взаимодействие по уравнениям A.150)—A.152). Поправки на Z-удвоение не принимались во
внимание ввиду их малости во всем рассматриваемом интервале температур. Погрешности
вычисленных значений термодинамических функций при комнатной температуре не превышают
0,05 Дж-К~1-моль~1 и определяются ошибками исходных молекулярных постоянных и величины
R. При более высоких температурах погрешности быстро возрастают из-за приближенного метода
расчета и связанной с ним неопределенности экстраполяции высоких колебательных и
вращательных уровней. Общие погрешности могут составлять 1 и 3 Дж>К~1-моль~1 в Ф° (Т) при 3000 и
6000 К.
Ранее термодинамические функции NF3(r) вычислялись в работах [3966, 3574, 2774, 3315]
до 1500—2500 К и в справочниках [88, 2095, 2556, 337] до 6000 К. Все расчеты были выполнены
в приближении «жесткий ротатор — гармонический осциллятор» по основным частотам и
структурным параметрам, несущественно отличающимся от принятых в настоящем издании. Поэтому
при комнатной температуре расхождения указанных расчетов между собой и с данными
настоящего справочника лежат в пределах 0,05—0,1 Дж«К~1-моль~1, что обусловлено только различием
принятых молекулярных постоянных; при возрастании температуры расхождения результатов
этих расчетов с данными табл. 175 увеличиваются и достигают 6 Дж«К~1-моль в Ф° F000 К),
это обусловлено тем, что в настоящем справочнике расчет выполнен точнее. В расчете Нагараяна
[2774] неверно вычислены главные моменты инерции, что привело к ошибке в значениях Ф° (Т) и
S° (T) примерно 5 Дж-К~1-моль~1.
Константа равновесия реакции NF3(r) = N(r)-f 3F(r) вычислена с использованием значения
Д,.#°@)=828,628+1,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 298,15 К) = —131,7 + 1,0 кДж • моль.
Результаты различных измерений этой величины приведены в табл. 13.26. Измерения
энтальпии сгорания NF3 в водороде в присутствии воды [422, 3431, 103] привели к плохо
согласующимся между собой величинам. Наиболее точное значение этим методом было получено Зинке [3431].
Рекомендованное в справочнике значение основано в первую очередь на результатах измерений
[3862, 3432, 3431], находящихся в прекрасном соответствии между собой.
329
N?F2 (г) Глава 13
Таблица 13.26. Результаты определений энтальпии образования NF3 (г) (в кДж-моль)
Авторы
Руфф и др., 1931 [3271а]
Армстронг и др., 1959 [422]
Людвиг, Купер, 1963 [2472]
Зинке, 1965 [3431]
Зинке, 1967 [3432]
Уокер, 1967 [3862]
Зерчанинов и др., 1969 [103]
Метод
Измерение энтальпии взрыва смеси NF3 с водородом
Измерение энтальпии сгорания NF3 в водороде в
присутствии воды. Определение количества
прореагировавшего вещества титрованием образовавшейся HF
То же, но при определении количества
прореагировавшего вещества по расходу NF3
Измерение энтальпии реакции NF3 с аммиаком с
образованием NH4F(k)
Измерение энтальпии сгорания кристаллического
бора в NF3
Измерение энтальпии сгорания NF3 в водороде в
присутствии воды
Изменение энтальпии разложения избыточного NF3
при его реакции с водородом
Измерение энтальпии реакции NF3 с серой
Измерение энтальпии реакции NF3 с водородом в
присутствии воды
ДуЯ0 B98,15 К)
—124
—134,1+4,2
—160,5±6,3
—133 ±10
—133,9+5,0
—131,6 ±2,2
—131,4+1,3
—131,5+0,8
—146,7+5,0
N2F2 (г). Термодинамические свойства газообразного дифторида диазота (равновесная смесь
изомеров) в стандартном состоянии при температурах 100—6000 К приведены в табл. 176.
Молекулярные постоянные N2F2, использованные при расчете термодинамических функций,
приведены в табл. 13.18. Исследование дифракции быстрых электронов [694, 692] и исследование
микроволнового спектра [2319] показывают, что молекулы N2F2 в газовой фазе существуют в двух
изомерных плоских формах: ^wc-N2F2 (симметрия С2„) и mpawc-N2F2 (симметрия C2h), из которых
более стабильной является цисформа. Этот вывод подтверждается результатами теоретических
расчетов, выполненных в различных приближениях [1703, 3700, 1029, 1655, 1892, 2984, 3742],
а также исследованием спектра ядерного магнитного резонанса [2786]. Поскольку электронный
спектр N2F2 экспериментально не исследовался, в настоящем справочнике принимается на
основании теоретических расчетов, что основные электронные состояния цис- и mpa,Hc-N2F2 синглетны и
относятся к типу ХА. Возбужденные электронные состояния цис- и транс-N2F2 согласно
теоретическим расчетам имеют энергии, превышающие 20 000 см, и при вычислении термодинамических
функций не учитывались. Разность энергий цис- и mpaHC-N2F2 принимается (см. табл. 13.18) с
погрешностью 80 см на основании результатов термодинамических исследований (см. ниже).
Квантовомеханические расчеты дают существенно менее точные значения. Приведенные в таблице
значения произведений моментов инерции цис- и mpaHc-N2F2 вычислены из вращательных
постоянных (в см) А0=0,65558, #„=0,265075, Со=0,18851 для i^c-N2F2 и Л0=2,6, Я0=0,1487,
С0=0,14 для mpauc-N2F2, найденных в результате исследования микроволнового спектра [2319]
для z/we-N2F2 и анализа вращательной структуры полос в инфракрасном спектре поглощения [2249,
2251 ] для транс-№2?2. Этим значениям вращательных постоянных соответствуют следующие
структурные параметры: г (N—N) = l,214+0,05А, г (N—F) = l,384±0,010А и 4N—N—F=
= 114,5+0,5° для 9mc-N2F2 и г (N-N) = l,230±0,010 A, r (N—F) = l,396±0,008 А и 4N-N-F=
= 105,6+0,7° для mpaHc-N2F2- С этими данными прекрасно согласуются результаты электроно-
графических измерений [694]. Молекула N2Fa имеет шесть невырожденных основных частот, пять
из которых связаны с плоскостными колебаниями, и одно —¦ с деформационным крутильным
колебанием, сопровождающимся выходом из плоскости. Спектр комбинационного рассеяния и
инфракрасный спектр поглощения N2F2 изучались в работах [3301, 2249, 2250, 225, 141, 142]. За
исключением работы [3301], в которой полосы в инфракрасном спектре были ошибочно
интерпретированы, как относящиеся к колебаниям 1,1-N2F2, частоты, полученные в перечисленных выше
работах, хорошо согласуются друг с другом. Правильность отнесения подтверждается совпадением
рассчитанных и наблюдаемых основных частот цис- и mj>aKc-N2F2 [142]. Приведенные в табл. 13.18
значения основных частот ^uc-N2F2 получены Москвитиной и Кузяковым [142] при изучении ин-
330
Азот и его соединения i{MC-N2F2 (г)
•фракрасного спектра газообразного N2F2. Частота крутильного колебания v4 экспериментально
не наблюдалась и вычислена авторами работы [142]. Это значение v4 представляется более
разумным, чем значение 550 см'1, предлагаемое в работе [2250], поскольку сравнение с N2H2 и анализ
потенциальной функции внутреннего вращения [3742] показывают, что'частота крутильного
колебания tyuc-N2F2 должна быть близка или ниже соответствующей частоты в 7?ipa«c-N2F2. Частоты
mpawc-N2F2, приведенные в таблице, приняты по данным Кинга и Оверенда [2250]. Практически
те же значения частот рекомендуются в работах [3397, 142]. Погрешности во всех
экспериментально наблюдаемых частотах не превышают 3 см~х. Погрешность в частоте v4 для ij,uc-N2F2 может
достигать 50 см.
Термодинамические функции равновесной смеси изомеров N2F2 (г) вычислены в приближении
•«жесткий ротатор — гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130), причем трансизомер рассматривался как возбужденное состояние и его
составляющие вычислялись по уравнениям A.168)—A.170), т. е. в предположении, что (?км ^ одинаковы
для цис- и wipaHC-N2F2. Погрешности вычисленных величин составляют 1, 4 и 6 Дж -К-моль
в значениях Ф° (Т) при температурах 298,15, 3000 и 6000 К. Они обусловлены в основном
приближенным характером расчета, а также неопределенностью в величине разности энергии транс-
и цисизомеров и неточностью v4 для ty«c-N2F2.
Ранее термодинамические функции равновесной смеси изомеров N2F2 (г) не публиковались.
Константа равновесия реакции N2F2(r)=2N(r)+2F(r) для равновесной смеси изомеров
вычислена с использованием значения АГН° @) = 1029,186 + 10 кДж-моль", соответствующего
принятой в справочнике величине
AfH°(N2F2, г, 0) = 67 + 10 кДж- моль'1.
Эта величина основана на результатах измерений, выполненных Армстронгом и Маранцем
методом сожжения N2F2 в аммиаке [419]. Сожжение образцов с разным содержанием изомеров
позволило найти значения энтальпий образования при 298,15 К для цис- и /rapaHc-N2F2, равные
соответственно 61,7+5 и 74,2+5,0 кДж-моль. Энтальпия образования N2F2 определялась также
Панкратовым и др. [188, 189] на основании измерения теплового эффекта реакции N2F2 с
раствором йодистого калия. Авторы указанных работ нашли, что результаты измерений не зависят
от содержания изомеров в исходных образцах N2F2 и получили для энтальпии образования обоих
изомеров одинаковые значения 96,5+6,5 кДж-моль. Как видно, результаты работ [419], [188,
189] находятся в плохом соответствии друг с другом. Указанные работы выполнены с не очень
чистыми образцами, химический анализ в них проведен не с исчерпывающей полнотой, поэтому
сделать обоснованный выбор между этими работами невозможно. Сопоставление измерений с
другими веществами (например, NF3), выполненных тем же методом сожжения в аммиаке, что и в
работе [419], показывает лучшее соответствие с результатами других работ, чем результаты
измерений авторов работ [188, 189]. Поэтому и в случае N2F2 в справочнике отдается предпочтение
данным работы [419]. Принятое значение энтальпии образования N2F2 относится к равновесной смеси
цис- и трансизомеров, которая при Т=0 состоит только из цисизомеров. По данным работы [419]
энергия перехода из цис- в трансформу составляет 12,5 + 1,5 кДж-моль. Более надежное
значение этой величины может быть получено на основании исследования равновесия реакции
изомеризации. Соответствующие измерения, выполненные Колбурном и др. [1010], Панкратовым,
Соколовым [192, 188] и Веденеевым и др. [50], при обработке по III закону привели к значениям
энтальпий перехода цис- в mpaKc-N2F2 при Т=0, равным 6,53, 5,48 и 4,64 кДж-моль. В справочнике
принимается средняя величина 5,6 + 1,0 кДж-моль.
Jj(mc-N3F2 (г). Термодинамические функции газообразного цисдифторида диазота в стандартном
состоянии при температурах 100—6000 К приведены в табл. 177.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 13.18. Обоснование выбора молекулярных постоянных см. выше в тексте для N2F2.
Термодинамические функции tyuc-N2F2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130).
Погрешности вычисленных величин при низких температурах обусловлены главным образом неточностью
молекулярных постоянных (в основном неопределенностью значения v4). С ростом температуры
растут погрешности, связанные с приближенным методом расчета. Суммарная погрешность в
значениях Ф° (Г) при 298,15, 3000 и 6000 К составляет 1, 4 и 6 Дж-К^-моль.
331
•mpawe-N2F2 (r), N2F4 (г) Глава 13^
Ранее термодинамические функции ^mc-N2F2 (г) вычисляли Нагараян и Дюриг [2780] до 2000 К,
Кузяков и Москвитина [142] до 1000 К, а также авторы справочника [2095] до 6000 К. Во всех
работах термодинамические функции рассчитывались в приближении «жесткий ротатор —
гармонический осциллятор». В работе [2780] и в справочнике [2095] для v4 принято значение 550 вместо-
300 см, рекомендуемого в настоящем справочнике. Кроме того, в справочнике [2095] для частоты
симметричного деформационного колебания также принято завышенное значение. Этими
причинами объясняются расхождения в функциях, вычисленных в работах [2780, 2095] и в настоящем
издании, которые составляют 3—9 Дж-К~1-моль~1 в Ф° (Т). В работе [142] приняты те же
значения постоянных, что и в настоящем справочнике, однако расхождения в значениях Ф° (Т) при
1000 К достигают 5,6 Дж-К~1-моль~1, что, по-видимому, объясняется ошибкой в расчете [142].
Константа равновесия реакции tyMC-N2F2(r)=2N(r)+2F(r) вычислена с использованием
значения Д^Н @) = 1029,186+10 кДж-моль, соответствующего принятой энтальпии образования-
AfHo(quc-N2F2, г, 0) = 67 + 10 кДж ¦ моль.
Эта величина совпадает с принятой энтальпией образования равновесной смеси изомеров
при Т=0 (см. выше).
TpaHC-N2F2 (г). Термодинамические функции газообразного трансдифторида диазота в
стандартном состоянии при температурах 100—6000 К приведены в табл. 178.
Молекулярные постоянные mpaHc-N2F2, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Обоснование выбора молекулярных постоянных см. выше в разделе1
по N2F2.
Термодинамические функции mpaKC-N2F2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130).
Погрешности вычисленных величин при низких температурах обусловлены главным образом неточностью
молекулярных постоянных. При температурах выше 1000 К погрешности в основном связаны
с приближенным характером расчета. В значениях Ф° (Г) при 298,15, 3000 и 6000 К погрешности
составляют 0,5, 3 и 5 Дж-К~1-моль~1.
Ранее термодинамические функции mpaKC-N2F2 (г) вычисляли Ратнер [3276], Мак-Брайд и др..
[2556], авторы справочника [2095J до 6000 К, Нагараян и др. [2782, 3323] и Крейг [1110] до 2000 К
и Кузяков и Москвитина [142] до 1000 К. Во всех работах приняты молекулярные постоянные,,
близкие к рекомендуемым в настоящем справочнике. Расхождения в значениях функций,
вычисленных в работах [2095, 2556] и в настоящем издании, не превышают 0,5 Дж'К~1-моль~г для
Ф° (Г) и обусловлены в основном незначительным различием в принятых значениях 1а, 1в> 1с,
так как геометрические параметры mp<mc-N2F2 в работах [2095, 2556] приняты по результатам
старых электронографических измерений. Расхождения с результатами расчетов в работах [3276] и
[2782, 3323] значительно больше и достигают соответственно 8 и 2,5 Дж-К~1-моль~1, по-видимому,.
из-за ошибок в этих расчетах.
Константа равновесия реакции ?repaHC-N2F2(r)=2N(r)+2F(r) вычислена с использованием
значения A,J70 @) = 1023,586+10 кДж'Моль, соответствующего принятой энтальпии образования
Д/ЙгО {mpa.HC-N2F2, г, 0) = 72,6 + 10 кДж • моль.
Эта величина получена на основании принятых в настоящем справочнике значений энтальпии
перехода ^uc-N2F2 в mpaHC-NtF2 и энтальпии образования z^mc-N2F2.
N2F4 (г). Термодинамические свойства газообразного тетрафторида диазота (равновесная смесь
изомеров) в стандартном состоянии в интервале температур 100—6000 К приведены в табл. 179.
Молекулярные постоянные N2F4, использованные при расчете термодинамических функций,
приведены в табл. 13.18. Молекула N2F4 существует в двух изомерных конфигурациях: 2ozw-N2F4
с углом поворота групп NF2 друг относительно друга ср=65° (точечная группа симметрии С2) и
более устойчивая nzpaKc-N^ (ср=180°, группа симметрии C2ft). Разность энергий этих
конфигураций определялась из электронографического исследования состава пара [871, 1641], по спектру
ЯМР [2135, 1009] и температурной зависимости отношения интенсивностей полос в ИК-спектре
[3379], а также в квантовомеханическом расчете [3853]. В настоящем справочнике разность
энергий принята (см. табл. 13.18) на основании экспериментальных данных, удовлетворительно
согласующихся между собой (возможная погрешность 70 см), но существенно отличающихся от
результатов расчета [3853] (согласно этому расчету возможно также существование tywc-N2F4 со зна-
332
Азот и его соединения wpawc-N2F4 (г), zowt-N2F4 (г)
чительно более высокой энергией). Определение геометрических параметров N2F4 проводилось
методом газовой электронографии [871, 1641, 1353, 190] и по МВ-спектру [2426]. Величина 1л1в1о
в табл. 13.18 для mjoaKC-N2F4 вычислена из данных, полученных Кардилло и Бауэром [871 ] и
Гилбертом и др. [1641]. Согласно [1641] г (N-N) = l,492 A, r (N-F) = l,372 A, 4F-N—F=103,l°,
«=$N—N—F=101,4°. Для soui-N,F4 величина IaIbIc вычислена по вращательным постоянным,
найденным Лайдом и Манном [2426] D0=0,186 00, ?0=0,106 39 и Со=0,093 83 см). Основные
частоты N2F4 определялись в ряде работ [2920, 1353, 131, 172, 1356, 3379, 2297, 1357], однако их
отнесение до последнего времени было неоднозначным и часто противоречивым из-за трудностей
интерпретации спектров при наличии двух изомеров. Эти противоречия были почти полностью
устранены в результате тщательного исследования ИК-спектра (Оскам и др. [2920]) и спектра КР
(Дюриг, Мак-Нами [1357]) газообразного N2F4. Рекомендованные в табл. 13.18 значения
основных частот обоих изомеров приняты по работе [1357], в которой на основании данных по
поляризуемости линий в спектре КР уточнено отнесение ряда частот, предложенное в работе [2920].
Погрешности в принятых значениях не превышают 5 см. Основное электронное состояние N2F4
является синглетным, а возбужденные состояния с энергией ниже 20 000—30 000 см,
по-видимому, не существуют.
Термодинамические функции равновесной смеси изомеров N2F4 (г) вычислены по уравнениям
A.3)—A.10), A.170)—A.173). Изомер 2oz«-N2F4 рассматривался как возбужденное электронное
состояние. Колебательно-вращательные составляющие термодинамических функций для каждого
изомера рассчитывались в приближении «жесткий ротатор — гармонический осциллятор» по
уравнениям A.122)—A.124), A.128), A.130). Общая погрешность рассчитанных таким образом
величин достигает 2, 9 и 12 Дж-К-^моль в Ф° (Т) при 298,15, 3000 и 6000 К. При низких
температурах, наряду с приближенным характером расчета, основной причиной погрешности
является неточность разности энергий двух изомеров; при 3000 К и выше основной причиной
погрешностей остаются методические ошибки, а также неточность молекулярных постоянных.
Ранее таблицы термодинамических свойств для равновесной смеси N2F4 (г) не публиковались.
Константа равновесия реакции N2F4(r)=2N(r)+4F(r) для равновесной смеси изомеров N2F4
вычислена с использованием значения АгН° @) = 1264,228+10 кДж-моль, соответствующего
принятой в справочнике энтальпии образования
A/ff°(N2F4, г, равновесная смесь, 298,15 К) = —22 + 10 кДж • моль.
Эта величина основана на результатах измерения энтальпии реакции N2F4 с избытком NH3,
полученных Армстронгом и др. [421] и известных по работам [418, 419] и справочнику [2095].
Энтальпии образования mpaHC-N2Ft и равновесной смеси N2F4 при Т=0 совпадают.
p2F4 (г). Термодинамические свойства газообразного тракс-тетрафторида диазота
в стандартном состоянии в интервале температур 100—6000 К приведены в табл. 180.
Молекулярные постоянные mpanc-N^^ использованные при расчете термодинамических
функций, приведены в табл. 13.18. Выбор этих постоянных обсуждался в разделе по N2F4.
Термодинамические функции mpaKC-N2F4 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130).
Погрешности вычисленных величин обусловлены главным образом приближенным характером расчета и
в меньшей степени — неточностью молекулярных постоянных. Суммарные погрешности в Ф° (Т)
при 298,15, 3000 и 6000 К составляют 1, 9 и 12 Дж-К^-моль.
Таблицы термодинамических функций mpaHc-N2Fi (г) публикуются впервые.
Константа равновесия реакции 7npawc-N2F4(r)=2N(r)-j-4F(r) вычислена с использованием
значения Дг#° @) = 1264,228 + 10 кДж-моль, соответствующего принятой энтальпии
образования
AfH° (m/?OKC-N2F4, г, 0) = —13,492 + 10 кДж • моль.
Эта величина совпадает с принятой энтальпией образования равновесной смеси изомеров
N2F4 при Г=0 (см. выше).
/Tota-N2F4 (г). Термодинамические свойства газообразного гош-тетрафторида диазота в
стандартном соетоянии в интервале температур 100—6000 К приведены в табл. 181.
Молекулярные постоянные sou{-N2F4, использованные при расчете термодинамических
функций, приведены в табл. 13.18. Выбор этих постоянных обсуждался в разделе по N2F4.
333
FNO (г); Глава 13
Термодинамические функции eoz«-N2F4(r) вычислены в приближении «жесткий ротатор -—
гармонический осциллятор» по уравнениям A.3)—A.10), A.122) —A.124), A.128), A.130). Общие-
погрешности вычисленных величин Ф° (Т) при 298,15, 3000 и 6000 К составляют 1,5, 9 и
12 Дж-К-моль и обусловлена главным образом приближенным характером расчета.
Ранее таблица термодинамических функций eoz«-N2F4 (г) вычислялась в справочнике [20951
до 6000 К в том же приближении, но по значительно отличающимся значениям основных частот,
выбранных в справочнике [2095] по старым работам. Соответствующие расхождения результатов
расчета [2095] с данными настоящего справочника для Ф° (Т) растут с температурой от 6 до
19 Дж-К^-моль".
Константа равновесия реакции eoitt-N2F4(r)=2N(r)+4F(r) вычислена с использованием
значения А.ГН° @) = 1262,736 + 10 кДж-моль, соответствующего принятой энтальпии образования
AjH° COM-N2F4, г, 0) = —12 + 10 кДж • моль.
Эта величина основана на принятых в настоящем справочнике значениях энтальпии перехода
/?i/?aKc-N2F4 в 3oiw-N2F4 и энтальпии образования wipaKc-N2F4.
FNO (г). Термодинамические свойства газообразного оксид-фторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 182.
Молекулярные постоянные, использованные в расчете термодинамических функций FNO,
приведены в табл. 13.7. Вращательные постоянные для изотопной модификации F14N16O приняты
по результатам исследования МВ-спектра, выполненного Куком [1040]. Этим постоянным
соответствуют структурные параметры г (N—O) = l,136 A, r (N—F) = l,512 А и ^F—N—О=110с5"
[828]. В более ранних работах по исследованию MB- [2499, 2500] и ИК- [1355] спектров были
получены близкие результаты. Постоянные центробежного растяжения приняты также по
работе [1040]. Значения основных частот и постоянных ангармоничности взяты из работы Уолтца
и др. [3988], в которой исследован ИК-спектр FNO в газообразном состоянии (см. также [2159Г
2167]). Постоянные взаимодействия вращения и колебаний экспериментально не определялись
и были рассчитаны Шпирко и Спирсом [3493 ] по силовым постоянным, основанным на принятых
в настоящем справочнике колебательных постоянных, с использованием численного метода Монте-
Карло. Эти постоянные также даны в табл. 13.7. Основное электронное состояние FNO относится
к типу гА', а первое наблюдаемое возбужденное состояние 1А" имеет энергию выше 30 000 cm~l
[2144, 1682]. Вместе с тем по аналогии с молекулами HNO и C1NO можно предположить у FNO
наличие состояния й3А с энергией 15 000 + 5000 см. Известно также [3442, 3445] о
существовании таутомера, имеющего строение FON. Однако теоретический расчет [2996а] показал, что
стабильность этого таутомера на 102 кДж-моль меньше, чем FNO. Поэтому в справочнике молекула
FON не рассматривается.
Термодинамические функции FNO (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.127), A.128) с поправками
на ангармоничность колебаний и взаимодействие колебаний и вращения по уравнениям A.131)—
A.133), на центробежное растяжение по уравнениям A.140)—A.142) и на возбужденное
электронное состояние по уравнениям A.168)—A.170). Погрешности вычисленных значений при низких
температурах несущественны и определяются прежде всего неточностью фундаментальных
постоянных и молекулярных постоянных. При более высоких температурах неточность
молекулярных постоянных, и в первую очередь энергии возбуждения состояния а3А и постоянных
взаимодействия вращения и колебаний, становится определяющей, наряду с неопределенностьюf
связанной с приближенным характером расчета и неопределенностью экстраполяции высоких
колебательно-вращательных уровней. Общие погрешности могут достигать 0,04, 0,5и 1,5 Дж • К-1Х
Хмоль-1 в Ф° (Г) при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций FNO (г) вычислялись в справочниках [88, 20951
до 6000 К и в работах Стивенсона и Джонса [3524] до 1500 К и Венкатесварлу и Мариама [3795]
до 1000 К. Все эти расчеты выполнены в приближении «жесткий ротатор — гармонический
осциллятор» по мало отличающимся значениям основных частот и вращательных постоянных.
Расхождения результатов этих расчетов с данными настоящего справочника закономерно возрастают
с температурой (от 0,05 Дж-К^-моль-1 при 298,15 К до 3,5 Дж-К^-моль при 6000 К в Ф° (Т)
для справочников [88, 2095]) и обусловлены учетом в настоящем издании возбужденного электрон-
334
Азот и его соединения FNO3 (г)
ного состояния, а также поправок на ангармоничность колебаний, взаимодействие колебаний и
вращения и центробежное растяжение.
Константа равновесия реакции FNO (r)=F(r)+N(r)+O(r) вычислена с использованием
значения &rH° @)=857,508 + 2,1 кДж-моль, соответствующего принятой в справочнике
величине
г, 298,15 К) = —65,0 + 2,0 кДж • моль.
Эта величина является средним взвешенным из совпадающих в пределах погрешности
результатов калориметрических измерений Джонстона, Бертина [2143] и Чеснокова и др. [309, 310].
Джонстон, Бертин измерили тепловой эффект реакции 2NO (r)+F2 (r)=2FNO (г). Чтобы избежать
побочных реакций, авторы работы [2143] использовали фтор, разбавленный азотом. По данным
этой работы было вычислено значение —65,2 + 1,7 кДж-моль. Чесноков и др. [309, 310]
измерили энтальпию щелочного гидролиза FNO. Измерения были затруднены тем обстоятельством,
что реакция гидролиза протекала с образованием не только нитрит-иона, но и NO (г). В основных
опытах использовалась смесь FNO с кислородом, что позволило окислить NO до нитрат-иона.
По результатам работ [309, 310] было найдено значение —64,0 + 6,0 кДж-моль.
FNO2 (г). Термодинамические свойства газообразного диоксид-фторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 183.
Молекулярные постоянные FNO2, использованные при расчете термодинамических функций,,
приведены в табл. 13.15. Поскольку молекула FNO2 имеет четное число электронов, в настоящем
справочнике принимается, что ее основное электронное состояние синглетно. Анализ
микроволновых спектров [3610, 3611, 980, 2379, 2679], а также результаты квантовомеханических расчетов
[3700, 3346] показывают, что молекула FNO2 имеет плоское строение (симметрия C2v). Наиболее
тщательное исследование МВ-спектра FNO2 в различных колебательных состояниях было
выполнено Танака и Морино [3610, 3611]. Рекомендованные в работе [3611] значения вращательных
постоянных и постоянных центробежного растяжения приведены в табл. 13.15. Принятым
значениям вращательных постоянных соответствуют следующие структурные параметры FNO2:
г (N-F) = l,467 + 0,015 A, r (N—О) = 1,1798 + 0,0035 А, ДО—N-—0 = 136+1,5°. Приведенные
в таблице значения основных частот FNO2 приняты по рекомендации Бернитта и др. [603],
которые выполнили тщательное исследование инфракрасного спектра газообразного FNO2 с
использованием изотопического замещения по азоту. Эти значения основных частот согласуются с данными,,
полученными из спектра комбинационного рассеяния жидкого FNO2 [3173, 960], а также
инфракрасного спектра газообразного [1271] и изолированного в матрице инертного газа [3444] FNO2.
Сведения о возбужденных электронных состояниях FNO2 в литературе отсутствуют.
Термодинамические функции FNO2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.127), A.128) с учетом
поправки на центробежное растяжение по уравнениям A.143):—A.144). Погрешности вычисленных
величин обусловлены главным образом приближенным характером расчета и составляют в
значениях Ф° (Т) 0,1, 2 и 3 Дж-К^-мо.ль-1 при 298,15, 3000 и 6000 К. Погрешности, связанные с
неточностью молекулярных постоянных, не превышают 0,1 Дж -К -моль-1 в значениях Ф° (Т) во всем
интервале температур.
Ранее термодинамические функции FNO2 (г) вычислялись в работах Пураника и Рао [3103],
Чуйкова-Ру [3731], Венкатесварлу и Раджилакшми [3797], Пиллаи [3021] и Бернитта и Миллера
[603] до 1000—1500 К, а также в справочнике [2095] до 6000 К. Все расчеты выполнены в
приближении «жесткий ротатор — гармонический осциллятор» (за исключением работы Пиллаи
[3021 ], в которой учитывалась поправка на центробежное растяжение в приближении
симметричного волчка) по молекулярным постоянным, несколько отличающимся от приведенных в табл. 13.15.
Так, например, в работах [3103, 3731, 3797, 3021, 2095] для v3 ошибочно принято значение 460 см.
Структурные параметры FNO2, использованные во всех расчетах, также отличаются от принятых
в настоящем издании. В силу этих причин, а также из-за неучета центробежного растяжения,
расхождения в значениях Ф° A000 К) с данными настоящей работы составляют 1,2 Дж -К -моль^1
для расчетов [3731, 3103, 3797, 2095] и 0,2 Дж-К^-моль для расчета [603], который выполнен
по тем же значениям основных частот, что и принятые в настоящем справочнике. Что касается
расчета [3021], то в нем, по-видимому, допущена ошибка, в результате которой значения Ф° (Т)~
и S° (T) оказались заниженными почти на 20 Дж-К-моль.
335
FNO3 (г) Глава 14
Константа равновесия реакции FNO2(r)=F(r)+N(r)-f2O(r) вычислена с использованием
значения АГН° @)=1144,590+ 20 кДж-моль, соответствующего принятой в справочнике
величине
A/Fe(FNOa, г, 298,15 К) = —109 + 20 кДж • моль.
Эта величина принята на основании результатов неопубликованных работ Никольса и
Робинсона (—109 + 20 кДж-моль, измерение энтальпии растворения, см. [2095, 1971]) и Макларена и
и др. (—108 кДж-моль, измерение энтальпии реакции NO2 (r)+V2 F2 (r)=FNO2 (г) (см. [ 2095,
3731]).
FNO3 (г). Термодинамические свойства газообразного триоксид-фторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 184.
Молекулярные постоянные FNO3, использованные при расчете термодинамических функций,
приведены в табл. 13.10. Молекула FNO3 имеет четное число электронов, поэтому принимается,
что ее основное электронное состояние является синглетным. Сведения о низколежащих
возбужденных состояниях FNO3 в литературе отсутствуют. Геометрические параметры молекулы были
определены в результате электронографического исследования Паулингом и Брокуэем [2977] для
структуры F-O'—NOa:r(N—O) = l,29+0,05 A, r (N—О') = 1,39+0,05 А, г (F—О')=1,42±
±0,05 А, Д О—N—0=125+5° и Д F—О'—N = 105+5°. При этом авторы работы [2977]
предположили, что плоскости F—О'—N и О'—NO2 взаимно перпендикулярны. Однако Диксон и
Вильсон [1272] на основании результатов исследования микроволнового спектра нитрометана
сделали вывод, что молекула FNO3 является плоской. К этому же выводу пришли Миллер и др.
[2659], показавшие, что контуры полос в ИК-спектре FNO3 согласуются с плоской структурой
симметрии С„ и исключают неплоскую структуру симметрии Си [2977]. Поэтому приведенное
в табл. 13.10 значение IaIbIc вычислено на основании геометрических параметров,
рекомендованных [2977], но для плоской структуры молекулы. Погрешность вычисленного значения не
превышает 1 -10~115 г3 -см6. Колебательный спектр FNO3 исследован в работах [960, 2659, 745, 426, 3836],
в которых были получены согласующиеся результаты, отличающиеся лишь отнесением частот
по формам колебаний. В таблице приведены рекомендуемые значения основных частот, найденные
Миллером и др. [2659], выполнившими наиболее детальное исследование колебательного спектра
FNO3 и его изотопозамещенных. Эти значения соответствуют положению ^-ветвей, и ошибка
в их определении не превышает 1 см". В табл. 13.10 приведены также величины барьера
внутреннего вращения Vo, определенного в работе [2659] на основании измеренного значения частоты
крутильного колебания v9=151,6 см и вычисленного по уравнению A.166) и приведенного
момента инерции волчка /.. Погрешности в значениях VQ и /др равны соответственно 350 см и
0,05-Ю8 г-см2.
Термодинамические функции FNO3 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130) с учетом вклада
заторможенного внутреннего вращения по уравнениям A.160)—A.162). Погрешность принятой
величины IaIbIc и отсутствие точных данных о форме потенциальной кривой внутреннего
вращения являются основным источником погрешностей в функциях при низких температурах (в Ф° (Т)
порядка 2 Дж -К -моль). С ростом температуры увеличивается доля погрешностей,
обусловленных приближенным характером расчета. Общие погрешности в значениях Ф° (Т) равны 2, 6 и
9 Дж-К-моль при 298,15, 3000 и 6000 К.
Ранее термодинамические функции FNO3 (г) вычислялись в справочнике [2095 ] в приближении
«жесткий ротатор — гармонический осциллятор» без учета внутреннего вращения. В указанном
справочнике расчет функций выполнен по оцененным значениям основных частот, а структурные
параметры FNO3 приняты такими же, как и в настоящем справочнике, но в предположении
симметрии Си. Расхождения в значениях термодинамических функций, рассчитанных в справочнике
[2095] и в настоящем издании, обусловлены указанными причинами и в значениях Ф° (Т)
достигают 2 Дж-К^-моль.
Константа равновесия реакции FNO3(r)=F(r)+N(r)+3O(r) вычислена с использованием
значения LrH° @) = 1266, 114+3,0 кДж-моль, соответствующего принятой в справочнике
величине
A,ff°(FNOg, г, 298,15 К) = 15 + 3 кДж-моль.
336
Азот и его соединения F3NO(r>
Эта величина основана на калориметрических измерениях энтальпии реакции гидролиза FNO3
в щелочном растворе, проведенных Талакиным и др. [255], и на измерениях энтальпии реакции
NaNO3 (k)+F2 (r)=FNO3 (r)+NaF (к), выполненных Ревиком и др. [3193]. Соответствующие
значения энтальпии образования FNO3 равны 12,0+3,6 и 17,3+2,4 кДж-моль, если использовать
уточненное значение энтальпии образования NOg (р-р Н2О, станд. сост.), равное —206,5 +
+1,0 кДж-моль. Менее точное значение энтальпии образования FNO3 было получено Скайенсом
и Кейди [3436] в результате исследования кинетики термического разложения FNO3(r).
F3NO (г). Термодинамические свойства газообразного оксид-трифторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 185.
Молекулярные постоянные F3NO, использованные при расчете термодинамических функций,
приведены в табл. 13.4. Поскольку молекула F3NO имеет четное число электронов, ее основное
электронное состояние принимается в настоящем справочнике синглетным (ХгА), что согласуется
с данными теоретического расчета [23321. Исследования МВ-спектра [2253], дифракции
электронов [3038] и вращательной структуры в ИК-спектре [1140] позволили надежно установить
строение молекулы F3NO в этом состоянии (точечная группа симметрии С3„). Плато и др. [3038],
используя при анализе полученных ими электронографических данных величину Бо=0,191 800 +
+0,000002 см, определенную Кирххоффом и Лайдом [2253] из МВ-спектра, вычислили
следующие значения геометрических параметров: г (N—О)=1,159+0,002 А, г (N—F)=1,432+0,002 А,
^ О—N—F=117,4 +0,6° и Д F—N—F=100,5 +1,6°. Величина ЫВ1С, приведенная в табл. 13.4,
вычислена по этим параметрам; ее погрешность составляет 1 -10 115 г3-см6. Колебательный спектр
F3N0 исследован как в газообразном состоянии [1440, 1516, 505, 1955, 377, 1515, 955а], так и в
матрицах инертных газов при низких температурах [339, 3443]. Рекомендованные в таблице значения
vx, v2 и ve приняты по работе Хиршмана и др. [1955] (см. также работу Фокса и др. [15161, в
которой исследован ИК-спектр поглощения газообразного F3NO). Значения v3, v4 и v5 приняты по
совпадающим между собой результатам исследования спектра комбинационного рассеяния
газообразного F3NO Кристе и др. [955а] и Аминадова и др. [377]. На основании данных по поляризации
линий авторы работ [955а, 377] выполнили однозначное отнесение деформационных основных
частот v3 и v5, для которых ранее интерпретация была неверной. Погрешности принятых значений
основных частот не превышают 5 см. Сведения о возбужденных электронных состояниях F8NO
в литературе отсутствуют.
Термодинамические функции F3NO (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130). Основные
погрешности вычисленных значений функций обусловлены приближенным характером расчета.
Ошибки из-за неточности принятых молекулярных постоянных существенно меньше (порядка
0,2—0,4 Дж -К -моль) и суммарные погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К
равны 0,5, 5 и 8 Дж-К^-моль.
Ранее таблицы термодинамических функций F3NO (г) вычислялись в справочнике [2095]
до 6000 К, в работах Мюллера и др. [2740] и Кертиса и др. [1140] до 2000 К и Аминадова и др. [377]
до 1000 К. Все эти расчеты выполнены также в приближении «жесткий ротатор — гармонический
осциллятор», и расхождения с данными настоящего справочника объясняются только различием
принятых молекулярных постоянных. Для расчетов, проведенных в работах [2740, 1140], эти
расхождения не превышают 0,3 Дж-К-моль в Ф° (Т); результаты расчета в работе [377]
практически совпадают с рассчитанными в настоящем справочнике, а значения Ф° (Т) и S° (T) в
справочнике [2095] завышены на 0,7—0,8 Дж -К -моль главным образом из-за неточного значения 1А1в1с
Константа равновесия реакции F3NO(r) = 3F(r)+N(r)+O(r) вычислена с использованием
значения Дг#0 @) = 1128,21+7,0 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
^Я (F3NO, г, 298,15 К) = —187 + 7 кДж • моль.
Эта величина принята на основании измеренных Армстронгом и Кинг [417] энтальпий реакции
сгорания газообразных F3NO, F2 и О2 в водороде. Реакции проводились в калориметре с горелкой,
причем образующийся фтористый водород растворялся в воде, помещаемой в реакционный сосуд.
Исследования равновесий реакции FNO (r)+F2 (r)=F3NO (г), выполненные Буганом и др. [716]
E33—640 К, 100—350 атм), приводят к значительно отличающейся величине —144 кДжмоль,
или, при пересчете констант равновесия к нулевому давлению, —142 кДж -моль, что, по-види-
337
NHF (г), NH2F (г) Глава 13
мому, указывает на возможность протекания в исследуемой системе NO+F2 каких-либо побочных
реакций.
Фотоионизационные измерения Дайблера и Уокера [1237] дают значение —262 кДж-моль,
что ставит под сомнение принятую в работе [1237] интерпретацию измерений.
NHF (г). Термодинамические свойства газообразного гидрид-фторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 186.
Молекулярные постоянные NHF, использованные при расчете термодинамических функций,
приведены в табл. 13.4. Исследования электронного спектра [3991, 1684, 2083] и теоретические
расчеты [2793, 1701, 813, 907, 3596, 3872] показывают, что молекула NHF в основном Х2А' и
возбужденном А2 А' состояниях имеет нелинейную структуру (точечная группа симметрии Cs).
Произведение моментов инерции NHF в состоянии Ё2А', приведенное в табл. 13.4, вычислено
по вращательным постоянным >40=17,688, Б0=1,0389 и С0=0,9777 см, найденным Вудманом
[3991] в результате анализа электронного спектра NHF в газовой фазе. Этим постоянным при
г (N—Н) = 1,06 А (оценка) соответствуют г (N—F)=l,37 Аи ^ Н—N—F=105°. Значения
основных частот приняты по работе Джекокс и Миллигана [2083], в которой исследован спектр
поглощения NHF и его изотопных модификаций в матрице Аг при 14 К. Частоты v2 и v3 наблюдались
в спектре, частота v2 (колебание связи N—Н) оценена авторами работы [2083]. Погрешности
приведенных в таблице значений частот v2 и v3 оцениваются в 40 см, частоты vx — в 100 см. Энергия
состояния А2А' принята по работе [3991] с погрешностью порядка 10 см.
Термодинамические функции NHF (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130) с учетом
возбужденного электронного состояния А2А' по уравнениям A.168)—A.170), т. е. в предположении,
что QmXm „р для состояний А2А' и Х2А' одинаковы. Общие погрешности вычисленных значений
термодинамических функций равны 0,1, 1,5 и 3 Дж-К-моль в значениях Ф° (Т) при 298,15, 3000
и 6000 К. Эти погрешности обусловлены в основном приближенным характером расчета, а также
неточностью принятых молекулярных постоянных.
Ранее термодинамические функции NHF (г) вычисляли Джекокс и Миллиган [2083] в
интервале температур 100—1500 К. Небольшие расхождения в функциях, рассчитанных в указанной
работе и в настоящем издании (около 0,1 Дж -К-моль), обусловлены тем, что авторы работы
[2083] использовали несколько отличающиеся от принятых в справочнике значения структурных
параметров NHF.
Константа равновесия реакции NHF(r)=N(r)+H(r)+F(r) вычислена с использованием
значения &ГН° @)=649,175 +15 кДж-моль, соответствующего принятой в справочнике величине
г, 298,15 К)=112±15 кДж • моль.
Эта величина оценена в предположении, что средние энергии связей N—Н и N—F в молекуле
NHF те же, что и в молекулах NH2 и NF2 соответственно.
NH2F (г). Термодинамические свойства газообразного дигидрид-фторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 187.
Молекулярные постоянные NH2F, использованные при расчете термодинамических функций,
приведены в табл. 13.4. Экспериментальные данные о структуре и частотах колебаний молекулы
NH2F отсутствуют. Поскольку результаты теоретических расчетов [1704, 2381] свидетельствуют
о совпадении структурных параметров молекул NH2F и NHF2, в настоящем справочнике
принимается, что молекула NH2F в основном ХгА состоянии имеет структуру неправильной пирамиды
(группа симметрии Cs), а ее геометрические параметры равны соответствующим параметрам NHF2,
найденным из МВ-спектра Лайдом [2425]: г (N—Н) = 1,02+0,01 А, г (N—F) = 1,40+0,02 А,
-? Н—N—Н=103+2°, -3 H—N—F=100+2° и ср=74+2° (угол между биссектрисой угла H—N—H
и связью N—F). Погрешности в значениях структурных параметров оценены на основании
сравнения результатов теоретических расчетов структуры молекул NHaF и NHF2 [1704, 2381] и
экспериментальных данных для NHF2 [2425]. Принятым значениям структурных параметров NH2F
соответствует произведение моментов инерции, приведенное в табл. 13.4. Его погрешность не
превышает 0,2-1017 г3-см6. Принятые значения основных частот NH2F оценены на основании
данных о силовых полях NH2, NH3, NHF и NHF2 [3577, 3023, 2252, 3576, 2262]. Погрешности в при-
338
Азот и его соединения NH4F (к, ж), NHF,(r)
нятых частотах составляют 30 см" для va, v2, v3 и ve, 40 см для v4 и 50 см для v5 и отражают
разброс значений, полученных для разных наборов силовых постоянных. Сведения о
возбужденных электронных состояниях NH2F отсутствуют.
Термодинамические функции NH2F (г) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130). Погрешности
вычисленных величин при низких температурах определяются в основном неточностью оцененных
молекулярных постоянных. При температурах, превышающих 1000 К, сказывается
приближенный характер расчета. Суммарная погрешность в значениях Ф° (Г) при 298,15, 3000 и 6000 К
составляет 0,5, 3 и 5 Дж -К -моль (в том числе из-за неточности молекулярных постоянных
0,2-1,2 Дж-К-моль-1).
Термодинамические функции NH2F (г) в настоящем справочнике публикуются впервые.
Константа равновесия реакции NH2F(r)=N(r)+2H(r)+F(r) вычислена с использованием
значения &г Н° @) = 1048,051 +15 кДж -моль, соответствующего принятой в справочнике
энтальпии образования
, г, 298,15 К) = — 75 ± 15 кДж • моль.
Это значение энтальпии образования NH2F оценено в предположении, что средние энергии
связей N—Н и N—F в молекуле NH2F те же, что и в молекулах NH3 и NF3 соответственно.
NH4F (к, ж). Термодинамические свойства кристаллического и жидкого фторида аммония
в стандартном состоянии в интервале температур 5—760 К приведены в табл. Д. 23.
Значения постоянных, использованных в расчетах термодинамических функций, приведены
в табл. 13.22. В интервале 5—298,15 К термодинамические функции гексагональной модификации
NH4F x вычислены по результатам измерений теплоемкости, проведенных Бенджаминсом и Уэстру-
мом [584] в интервале 6—309 К (чистота образца примерно 99,9%, точность измерений Ср (Т)
при Т >25 К-равна 0,1 %). Погрешности принятых значений S° B98,15 К) и Н° B98,15 К) — Н° @)
(см. табл. 13.22) составляют 0,20 Дж -К -моль и 0,012 кДж -моль соответственно. Данные
Симона и др. [3423] G1=203-^284 К) менее надежны и не учитывались в расчетах. При высоких
температурах какие-либо экспериментальные данные по термодинамическим свойствам NH4F (к, ж)
в литературе отсутствуют, за исключением данных по температуре плавления NH4F [2325, 3716,
3423], приводящих к значению (см. таблицу), точность которого составляет, по-видимому, 2 К.
Приведенные в табл. 13.22 линейное уравнение для теплоемкости NH4F (к) выше комнатной
температуры, значение теплоты плавления (его точность оценивается в 3 Дж -моль) и значение
теплоемкости NH4F (ж) были получены при помощи приближенных оценок, выполненных с
использованием известных экспериментальных данных для других солей аммония (NH4C1, NH4Br, NH4I
и NH4NO3).
Погрешности вычисленных значений Ф° (Т) при 298,15, 500 и 700 К оцениваются в 0,2, 1,0
и 4 Дж-К-моль.
В литературе таблица термодинамических функций NH4F(k) приводится только в интервале
5-300 К [584].
В справочнике принято значение энтальпии образования NH4F(k)
/1, к, 298,15 К) = — 467,56 + 0,85 кДж-моль,
основанное на измеренной Хиггинсом и Уэструмом [1931] энтальпии реакции взаимодействия
NH, (г) с HF (ж).
Измерения энтальпии растворения NH4F (к) в воде и энтальпии нейтрализации водных
растворов аммиака и фтористого водорода, выполненные еще в прошлом веке (см. [642, 3256]), приводят
к значительно менее точным значениям.
NHF2 (г). Термодинамические свойства газообразного гидрид-дифторида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 188.
1 Гексагональная модификация NH4F (структурный тип вюрцита) устойчива при обычном давлении во всем
интервале температур от'2'=0 до температуры плавления. При высоких давлениях существуют несколько других
модификаций NH4F (см. [2761, 3030, 2325]).
339
UNO (г) Глава 13
Молекулярные постоянные NHF2, использованные при расчете термодинамических функций,
приведены в табл. 13.4. Молекула NHF2 в основном электронном состоянии ХХА имеет структуру
неправильной пирамиды и относится к точечной группе симметрии Cs. Приведенное в табл. 13.4
произведение моментов инерции вычислено по вращательным постоянным Ао=1,768 4603, 2?0=
=0,3634323 и С0=0,3104554 см, найденным Лайдом [2425] в результате исследования
микроволнового спектра NHF2 и NDF2. Этим постоянным соответствуют следующие структурные
параметры: г (N—H)=1,026 ±0,002 А, г (N-F) = 1,400+0,002 А, ^ F-N-F=102,9+0,2°,
-3 Н—N—F=99,8±0,2° и ср=74,1° (угол между биссектрисой угла F—N—F и связью N—Н).
Теоретические расчеты [1704, 2381] подтверждают такую структуру молекулы NHF2. Основные
частоты NHF2, приведенные в таблице, приняты по работам Коумфорда и др. [1031] и Москвитиной
и Кузякова [173], которые исследовали инфракрасный спектр NHF2 и NDF2 в газовой фазе.
Ошибки в значениях частот не превышают 5 см для vx, v2 и v3 (определены положения Р- и R-вет-
вей) и 1 см для v4, v5 и v6 (выполнен анализ вращательной структуры). Частоты v2, v3 и v6,
измеренные в матрице [2083], согласуются с рекомендованными в табл. 13.4 в пределах возможного
матричного сдвига. Сведения о возбужденных электронных состояниях NHF2 в литературе отсутствуют.
Термодинамические функции NHF2 (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128), A.130). Погрешности
вычисленных значений обусловлены в основном приближенным характером расчета и в
значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 0,4, 2 и 4 Дж -К -моль. Погрешности,
связанные с неточностью молекулярных постоянных, не превышают 0,1 Дж-К моль в значениях
Ф° (Г).
Ранее термодинамические функции NHF2 (г) вычислялись в работах [425, 3575] до 1000 К
[2770] до 2000 К и [171] до 5500 К в приближении «жесткий ротатор — гармонический
осциллятор» по тем же молекулярным постоянным, что и в настоящем справочнике (за исключением
работы [3575], где постоянные не указаны). Значения Ф° (Т), рассчитанные в работах [171, 425],
практически совпадают в пределах 0,05 Дж -К -моль со значениями, приведенными в табл. 188.
Отличия функций, вычисленных в работе [3575] A,1 Дж-К-моль) и, в особенности, в [2770]
A9,3 Дж-К-моль), свидетельствуют о наличии ошибок в этих расчетах.
Константа равновесия реакции NHF2 (r)=N (r)+H (r)+2F (г) вычислена с использованием
значения Дг Н° @)=937,816+15 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A/E°(NHF2, г, 298,15 К) = —103 + 15 кДж • моль.
Эта величина оценена в предположении, что средние энергии связей N—Н и N—F в молекуле
NHF2 те же, что и в молекулах NH3 и NF3 соответственно. Экспериментальные измерения
энтальпии реакции NHF2 с кислым раствором иодида калия, выполненные Панкратовым и др. [190,
102, 188], приводят к существенно меньшему значению —73+10 кДж-моль. Следует отметить,
что исследованная в этих работах реакция сложна и протекает по двум уравнениям, что серьезно
затрудняет измерения. При определении таким же методом энтальпии образования N2F2 было
получено значение, отличающееся от принятого в настоящем справочнике также примерно на
30 кДж -моль. Поскольку метод оценки по средним энергиям связей используется в
справочнике для вычисления энтальпии образования ряда соединений, в том числе NHF и NH2F, то в
целях согласованности рекомендуемых термохимических величин для энтальпии образования NHF2
также принимается оцененное значение.
CINO (г). Термодинамические свойства газообразного оксид-хлорида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 189.
Молекулярные постоянные C1NO, использованные при расчете термодинамических функций,
приведены в табл. 13.7. Молекула C1NO имеет нелинейное строение (точечная группа симметрии Cs).
Вращательные постоянные и постоянные центробежного растяжения, приведенные в табл. 13.7,
о-пределены Каззоли и др. [918] в результате исследования МВ-спектра (с использованием данных
Мирри и Маззароли [2681 ]) и находятся в удовлетворительном согласии с величинами,
рекомендованными в ряде работ, посвященных исследованию МВ-спектра C1NO [2680, 1746, 1355]. Несколько
отличающиеся результаты получены в ранних исследованиях МВ-спектра [3227, 2650, 3229, 3903],
а также при электронографическом исследовании [2226]. Основные частоты и постоянные
ангармоничности рекомендованы в таблице согласно работе Джонса и др. [2171], в которой измерены
340
Азот и его соединения СШ02(г1
ИК-спектры изотопных модификаций молекулы C1NO. Ландау и Флетчер [2352] при исследовании
ИК-спектра получили несколько отличающиеся значения постоянных ангармоничности (в см):
х±1——18,3, х22=—8,0, х33=—2,0, ж12=—8,0, х13=—3,9 и х23=—4,9. Основные частоты,
найденные в указанной работе и в более старых работах по Исследованию ИК-спектра C1NO [3978, 842,
3102, 1372], удовлетворительно согласуются с рекомендованными в табл. 13.7. Постоянные
взаимодействия вращения и колебаний C1NO экспериментально не определялись и были рассчитаны
Шпирко и Спирсом [3493] по силовым постоянным, основанным на принятых в настоящем
справочнике колебательных постоянных, с использованием метода Монте-Карло. Эти постоянные также
даны в таблице. Исследования электронного спектра [3089, 1680] и теоретические расчеты [3606,
3121] показывают, что молекула C1NO имеет возбужденное синглетное состояние АХА1 с энергией
16 000+100 см. По аналогии с молекулой HNO можно предполагать, что молекула C1NO имеет
также стабильное электронное состояние а?А с энергией 10 000+3000 см. Основное электронное
состояние C1NO является синглетным ХгА' [3089, 1680, 3606, 3121].
Термодинамические функции C1NO (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124), A.127), A. 128) с поправками
на ангармоничность колебаний и взаимодействие колебаний и вращения по уравнениям A. 131)—
A. 133) и центробежное растяжение — по уравнениям A.143), A.144). Учет возбужденных
электронных состояний а?А в. А1 А проводился по уравнениям A.168)—A. 170). Погрешность
вычисленных таким образом значений может достигать 0,05, 1 и 3 Дж-К-моль для Ф° (Т) при 298,15,
3000 и 6000 К соответственно. При комнатной температуре эта погрешность обусловлена главным
образом неточностью R и исходных молекулярных постоянных (сюда входит также ошибка из-за
того, что в расчете использовались вращательные постоянные для изотопной модификации
35C114N16O). При 3000 К и выше основную погрешность в расчет вносят прежде всего неточность
в величине энергии возбуждения состояния а3А, а также неопределенность, связанная с
неточностью описания высоких колебательных и вращательных уровней молекулы C1NO.
Ранее таблицы термодинамических свойств C1NO (г) вычислялись неоднократно, но эти расчеты
в значительной степени устарели, так как выполнены в приближении «жесткий
ротатор—гармонический осциллятор», а также с использованием неточных значений молекулярных постоянных
[2089, 842, 3102, 3795, 2777]. Учет поправок на ангармоничность колебаний был выполнен только
в расчетах Гордона [1700] (данные этой работы воспроизведены в справочнике [2095]) и сводке
[2097] до 6000 К. При низких температурах (до 1000 К) результаты этих расчетов отличаются
от данных настоящего справочника в пределах 0,2—0,3 Дж'К~1-моль~1, что обусловлено главным
образом использованием в работах [1700, 2097] менее точных значений вращательных постоянных.
При более высоких температурах расхождения возрастают и достигают в Ф° F000 К) величин
0,6 Дж-К~1-моль~1 [1700, 2095] и 2,1 Дж-К~1-моль~1 [2097]. В последнем случае расхождения
обусловлены исключительно неучетом в этом расчете возбужденных электронных состояний,
центробежного растяжения и взаимодействия вращения и колебаний. В расчете [1700]
расхождения из-за пренебрежения перечисленными эффектами в значительной степени скомпенсированы
тем, что приняты менее точные значения колебательных постоянных по работе [2352].
Константа равновесия реакции C1NO (г)=С1 (r)+N (r)-f-O (г) вычислена с использованием
значения АГН° @) = 782,621 +0,65 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д^^СЩО, г, 0) = 54,6 + 0,5 кДж.моль.
Принятое значение основано на результатах исследований равновесия 2G1NO (r)=2NO (г) +
+С12 (г), представленных в табл. 13.27. Работа Бисона, И оста [551 ] выполнена наиболее тщательно,
с использованием современной аппаратуры и методики. В дополнительных измерениях были
определены значения поправок, учитывающих отклонения свойств компонентов реакции от свойств
идеального газа. По результатам работы [551] принимается значение Дг Н° @)=72,37 +
+0,10 кДж'Моль. Близкие значения были получены в работах Диксона [1265] и Траутца, Ва-
хенгейма [3720]. Погрешности использованных в расчетах термодинамических функций
увеличивают погрешность значения Дг Н° @) до 0,16 кДж-моль. Измеренная Бринером, Пилковым
[774а] энтальпия гидролиза СШО (г) в щелочном растворе приводит к значению энтальпии
образования около 32 кДж-моль. Ошибочность этих измерений была показана еще в работе [3720].
Cl NO2 (г). Термодинамические свойства газообразного диоксид-хлорида азота в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 190.
341
CINO2(r)
Глава 13
Таблица 13.27. Результаты определения энтальпии реакции 2C1NO (г) = 2NO (г) -+ С12 (г)
(в кДж ¦ моль)
Авторы
Ваго, 1911 [3757]
Хинк, 1913 [1948]
Траутц, Вахенгейм, 1916
[3720]
Диксон, 1931 [1265]
Бисон, Йост, 1939 [551]
Метод
Статический, 450—572 К, 12 точек
Статический, 434—593 К, 17 точек
Статический, 503—733 К, 9 точек
Статический
избыток С12, 494—760 К, 14 точек
избыток N0, 501—763 К, 18 точек
стехиометрия, 468—757 К, 14 точек
Статический, избыток С12, 373—472 К, 11 точек
То же, избыток NO, 372—462 К, 10 точек
То же, стехиометрия, 372—491 К, 14 точек
ДГЯ°(О)
II закон
92 ±7
65+6
71,0+4,8
70,4+2,4
69,9+1,6
69,9+1,7
71,2+0,8
74,6+1,4
71,8 ±0,3
III закон
77,25 ±0,80
71,48 ±0,52
72,3 ±0,61
73,45+0,47
73,19+0,34
73,48 ±0,36
72,42+0,07
72,39+0,13
72,31 +0,04
Молекулярные постоянные СШ02, использованные при расчете термодинамических функций,
приведены в табл. 13.15. Экспериментальные данные о типе основного электронного состояния
молекулы СШ02 в литературе отсутствуют. В настоящем справочнике принято, что основное
электронное состояние G1NO2 синглетно. В результате исследования микроволнового спектра G1NO2
[2710, 2900, 980, 981, 2651, 1486] установлено, что молекула C1NO2, так же как и молекула FNO2,
имеет плоскую симметричную структуру и относится к точечной группе симметрии С2,.
Приведенные в табл. 13.15 значения вращательных постоянных и постоянных центробежного растяжения
найдены в наиболее тщательно выполненных исследованиях Морино с соавторами [2710, 2900]
(см. также [I486]). Принятым значениям вращательных постоянных соответствуют структурные
параметры г (N-Cl) = l,83+0,01 A, r (N-O) = 1,202+0,005 А и -? O-N-O=130,8±l,5°.
Колебательный спектр газообразного C1NO2 исследовался в работах [3173, 1274, 603]. Приведенные
в таблице значения основных частот СШО2 найдены в работе Бернитта и др. [603], в которой
исследован инфракрасный спектр поглощения G1NO2 с изотопным замещением N и G1. Отнесение частот,
предложенное в работе [603], подтверждается результатами исследования микроволнового спектра
[2710], спектра комбинационного рассеяния жидкого G1NO2 [960], а также расчетом по силовым
постоянным [241]. Сведения о возбужденных электронных состояниях в литературе отсутствуют.
Термодинамические функции C1NO2 (г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.127), A.128) с поправкой
на центробежное растяжение по уравнениям A.143), A.144). Погрешности в вычисленных таким
образом значениях термодинамических функций G1NO2 обусловлены в основном приближенным
характером расчета и, в меньшей степени, неточностью принятых молекулярных постоянных
(значения молекулярных постоянных принимались для изотопа 35C114NO2). Суммарные погрешности
в значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 0,1, 2 и 3 Дж-К^-моль-1. Погрешности,
обусловленные неточностью молекулярных постоянных, не превышают 0,1 Дж«К~1-моль~1 в
значениях Ф° (Т) во всем интервале температур.
Ранее термодинамические функции C1NO2 (г) вычислялись в приближении «жесткий ротатор—
гармонический осциллятор» в работах [1586, 3103, 3797, 603] до 1000 К и [2359] до 1500 К,
а также справочнике [2095] до 6000 К. Все расчеты выполнены по молекулярным постоянным,
мало отличающимся от принятых в настоящем издании. Функции, вычисленные в работах [3103,
603, 2359], согласуются с приведенными в настоящем справочнике в пределах от 0,01
до 0,2 Дж-К~1-моль~1 в значениях Ф° (Т) в интервале 298,15—1000 К. Разница со значениями
Ф° (Т), вычисленными в справочнике [2095], растет с температурой и достигает 1,4 Дж-К~1-моль~1
при 6000 К. Эти расхождения обусловлены в основном пренебрежением в расчетах [3103, 603,
2359, 2095] поправкой на центробежное растяжение молекулы G1NO2. Расхождения с функциями,
вычисленными в работах [1586, 3797], значительно больше и составляют соответственно 1,6 и
9,1 Дж-К~1-моль~1 в значениях Ф° (Т) при 1000 К. Они могут быть объяснены только ошибками,
допущенными в этих расчетах.
342
Азот и его соединения NS (г)
Константа равновесия реакции СШО2 (г)=С1 (r)+N (г)+2О (г) вычислена с использованием
значения Аг Н° @)=1066,118 + 1,1 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я°(СЩО2, г, 298,15 К) = 12,5 + 1,0 кДж • моль.
Принятая величина является средневзвешенным из результатов измерений, выполненных Реем,
Оггом [3171] (табл. 13.28). В термохимических измерениях этих авторов в реакционную камеру,
Таблица 13.28. Результаты определения энтальпии образования C1NO2 (г) (в кДж-моль)
Авторы
Метод
B98,15 К)
Шумахер, 1938 [3357] Исследование кинетики реакции C1NO2=NO2+C1, 373— 25,2
423 К
Кордес, Джонстон, 1954 То же, 453—523 К 27,8
[1069]
Рей, Огг, 1959 [3171] Спектрофотометрическое исследование равновесия реакции 12,2 ±1,2
NO2+C1NO=NO+C1NO2, 298 Ка
Измерение энтальпии реакции 1,000 СШО2+1,086 N0= 12,8+1,0
=0,259 NO2+0,366 N2O4+0,0778 NO+0,008 N2O3+
+1,000 NOC1 (давление СШ02 585 мм рт. ст.)
То же при давлении СШ02 240 мм рт. ст. 12,7+2,0
Измерение энтальпии реакции N2O5+C1NO=C1NO2+2NO2 11,5 ±2,0
а В этой же работе сообщаются результаты исследования равновесия реакции 2NO2+C12=2C1NO2 и обратной
реакции. Однако эти измерения оказались неточными из-за низкой скорости установления равновесия и
наложения полос N2O4 и СШО2 в спектре поглощения.
содержавшую СШ02, вводился NO (избыток). Состав продуктов реакции авторы работы [3171]
определяли термодинамическим расчетом. Проведенный нами расчет этого состава позволил
уточнить уравнение исследованной реакции: 1,000 СШО2(г)+1,086 NO(r) =0,259 NO2(r) +
+0,367 N2O4(r) +0,078 NO(r)+0,008 N2O3(r)+1,000 NOCl(r). Отсутствие необходимых данных
не позволило провести такие расчеты для опытов с пониженным давлением C1NO2 и с N2O5; в этих
¦случаях величины из работы [3171 ] были приближенно скорректированы. Исследования кинетики
реакции C1NO2=NO2+G1 [3357, 1069] привели к существенно более высоким значениям, что,
возможно, свидетельствует о неточности принимаемого механизма реакции.
NS (г). Термодинамические свойства газообразного сульфида азота в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 191.
Молекулярные постоянные NS, использованные при вычислении термодинамических функций,
•приведены в табл. 13.23.
В спектрах NS, анализ которых затруднен из-за сильных возмущений, наблюдались переходы
между дублетными состояниями 22,2П и 2Д [1514, 4053, 891, 893, 885, 890, 3750, 375, 2523, 2814,
2815, 3467, 2951, 2952, 2948, 2949, 1708, 3834, 3008, 491, 492, 2812, 2177, 2817, 2816, 2802, 2813,
2117, 2970, 2809]. По аналогии с изоэлектронной молекулой NO молекула NS должна иметь еще
три стабильных состояния с невысокими энергиями возбуждения: В2Ф, а4П и й*2~. Энергии этих
состояний приняты (см. табл. 13.23) по данным квантовомеханического расчета Биальского и
Грейна [638], скорректированным с учетом принятого в настоящем справочнике значения энергии
диссоциации молекулы NS. Погрешности принятых величин можно оценить в 10 000 см.
Значения колебательных и вращательных постоянных в состояниях 4П и 42 приняты равными
соответствующим постоянным в изоконфигурационных состояниях А2Л и С2Д. Приведенные в таблице
значения постоянных NS в дублетных валентных электронных состояниях Х2Л, Л2П и С2 Д приняты
по работе Женувриер и Паска [2117], авторы которой на приборе с высокой дисперсией
исследовали несколько систем и выполнили анализ вращательной структуры 55 полос, в том числе с v ^ 6
и v ^ 12 для состояний Х2П и А2Л. Полученные в работе [2117] значения вращательных
постоянных NS в состоянии Х2Н хорошо согласуются с результатами исследований микроволнового спектра
в работе [375]. Остальные возбужденные электронные состояния NS имеют энергии возбуждения
343
NS (г) Глава 13
выше 40 000 см и не принимаются во внимание при расчете термодинамических
функций.
Термодинамические функции NS (г) вычислены по уравнениям A.3)—A.10), A.90)—A.92).
Колебательно-вращательные составляющие термодинамических функций для состояний ХаП,а4ПиЛ2П
вычислялись непосредственным суммированием по колебательным уровням и интегрированием
по вращательным уровням. Колебательно-вращательные уровни энергии этих состояний
представлялись уравнением типа A.65) с добавлением члена A.62). При этом мультиплетность состояния
Х2П учитывалась по уравнению A.42). Значения коэффициентов Ykl в уравнениях A.65) приведены
в табл. 13.24. Вырождение уровней состояния Х2П, а также мультиплетность и вырождение
уровней состояний а4П и А2Н учитывались статистическими весами B, 8 и 4 соответственно).
Суммирование по / ограничивалось в предположении линейной зависимости /шгя>, от v. В таблице приведены
также значения ушгя и /нт в этих состояниях. Вклады возбужденных состояний 642~, В2Ф, С2Д
вычислялись в предположении, что (?Кол. вР Ддя каждого из этих состояний и ее производные равны
соответствующим величинам для состояния Л2П (без учета мультиплетности и вырождения
вращательных уровней). Основные погрешности вычисленных термодинамических функций при Т ^
^ 3000 К не превышают 0,02 Дж-К~1-моль~1 и обусловлены неточностью постоянной R. При более
высоких температурах начинает сказываться неточность оцененных значений энергий состояний
а*П, 642~ и Б2Ф (до 0,6 Дж«К~1-моль~1 в значении Ф°F000 К)), пренебрежение ридберговскими
состояниями (до 0,03 Дж-К~1-моль~1 в значении Ф° F000 К) и неточность принятого значения
Do (NS). Общая погрешность в значении Ф° F000 К) оценивается в 1.0 Дж-К~1-моль~1.
Ранее термодинамические функции NS (г) вычислялись в предыдущем издании справочника [88}
и справочниках [2095, 2556]. Несмотря на применение менее точных методов расчета, результаты
этих расчетов и данные настоящего издания хорошо согласуются благодаря большой величине
энергии диссоциации NS в основном состоянии. Расхождения с предыдущим изданием [88?
не превышают 0,04 и 0,3 Дж-К-моль в значениях Ф° (Т) и S° (T), а с данными справочников
12095, 2556] — 0,1, 0,6 и 2,5 Дж-К^-моль-1 в значениях Ф° (Г), S° (Т) и С°р (Т).
Константа равновесия реакции NS (r) = N (r)+S (г) вычислена с использованием принятой
в справочнике величины
LrH° @) = Do (NS) = 479 ± 50 кДж • моль = 40 000 ± 4000 см.
Это значение было рекомендовано Гейдоном [1579], уменьшившим на 20% величину,
полученную экстраполяцией Берджа-Шпонер уровней состояния ХШ. Практически совпадающее значение
получается при оценке по различным эмпирическим методам, а также в результате неэмпирического
квантовомеханического расчета, выполненного О'Харе [2891].
Принятому значению Do (NS) соответствует
ДД°(NS, г, 0) = 266,603 + 50 кДж • моль.
Глава 14
ФОСФОР И ЕГО СОЕДИНЕНИЯ
Р (к, ж), Р, Р2, Р3, Р4, РО, РО-, РО2, РО-, Р2О3, Р2О4, Р2О5, Р3О6, Р4О6, Р4О7
Р4О8, РА, РАо (к, ж), Р4О10, РН, РН2, РН-, НРО, PF, PF2, PF~, PF3, PF6,
POF3, PCI, PCI2, PCI", PC13, PC15, POCI3, PFC1, PFCr, PF2CI, PF4C1,
PFC12, PF3C12, PF2CI3, PFC14, POF2CI, POFC12, PS, PN
В настоящей главе рассматриваются 45 соединений фосфора в газообразном состоянии, в число1
которых входят соединения фосфора с кислородом, водородом, фтором, хлором, серой, азотом,
а также фосфор и Р4О10 в конденсированных состояниях.
Для Р (г) термодинамические свойства рассчитаны до 10 000 К, [для всех остальных газов —
до 6000 К. Термодинамические свойства всех соединений фосфора приведены для природной смеси
изотопов этого элемента (атомный вес 30,97376) на основании молекулярных постоянных,
соответствующих изотопу 31Р.
В Приложении 3 для паров фосфора, PF3, PFaCl, PFC12 и РС13 приведены данные,
позволяющие оценить влияние межмолекулярного взаимодействия на отклонения свойств этих газов от их
свойств в стандартном состоянии.
Р (к, белый; ж). Термодинамические свойства кристаллического и жидкого фосфора в стан-
дартнЪм состоянии при температурах 5—900 К приведены в табл. Д. 24.
Значения постоянных, принятых для расчета термодинамических функций, приведены
в табл. 14.1. Известно несколько аллотропических модификаций твердого фосфора:
кристаллические модификации белого фосфора, кристаллические и аморфные формы красного и черного
Таблица 14.1. Принятые значения термодинамических величин для Р и Р4О10 в твердом и жидком
состояниях
Вещество
Состояние
1
К
ю
CN] о
кДж-моль
осГ
см_
о
со
2*
а:
см
о а.
Хмоль
Р к, бел., ромб. — —
к, бел., куб. 5,360 41,09
зк ~~~
Р4О10 к, гекс. 34,220 231,00
ж — —
а с°р=а + ЪТ + dp (Дж-К^-моль)
23,82
—
215,57
Коэффициенты
в уравнении для С°р а
а
г> - ю3
12,862 5 0,495 89
26,12 —
45,413 671,66
370,00 —
<М0в
—0,432 83
—
—338,6
Интервал
температур
К
298,15—317,3
317,3—900
298,15-699
699-1000
или
Т
К
195,4
317,3
—
699
AirH или
кДж-моль
0,5213
0,659
—
21,0
фосфора и другие. В качестве стандартного состояния твердого фосфора в справочнике принят
кристаллический белый фосфор, образующийся при конденсации паров фосфора.
Кристаллические формы красного фосфора термодинамически более стабильны, чем белый фосфор. Еще
большей термодинамической стабильностью характеризуется кристаллический черный фосфор.
Однако превращение белого фосфора в эти формы протекает чрезвычайно медленно даже при
высоких температурах и давлениях 1. При Т < 298,15 К термодинамические функции белого фосфора
1 Обзор свойств и методов получения различных модификаций твердого фосфора дан в монографии Ван Везера [45 ]•
Результаты калориметрических измерений теплоемкости красного и черного фосфора при Т < 298,15 К, а также-
вычисленные по этим данным таблицы термодинамических функций см. в работах [3522, 1936].
345
V (г) Глава 14
вычислены по результатам калориметрических измерений Стивенсона и др. [3522] и Мапла [2512].
В этих работах была измерена теплоемкость белого фосфора высокой чистоты (примеси 0,0002 моля
As на моль Р4) с точностью 5% при 15—20 К, 1 % при 20—35 К и 0,2% при Т > 35 К. Для
экстраполяции теплоемкости ниже 15 К использовались следующие уравнения, хорошо
аппроксимирующие данные [3522] в интервале 15—35 К: С° {T)=D A29,7/Г)+3?гE4,9/Г)+5,3.10-57'Сг2 и
С°р (T)=DA35,15/T)+D G3,95/Т). Были приняты средние значения С° (Т) из вычисленных
по этим уравнениям, которые приводят к величине S° A5 К) = 1,13 Дж-К~1-моль~1, близкой к
полученной в работе [3522] A,117 Дж-К~1-моль~1). По данным работы [3522] приняты также
значения температуры и энтальпии превращения низкотемпературной модификации белого фосфора
в высокотемпературную (Tir=195,4+0,1 К; AirH°=0,5213+0,0008 кДж-моль). Приведенные
в табл. 14.1 значения S° B98,15К) и#° B98,15 К)—#°@) вычислены по данным [3522], их
погрешности оцениваются в 0,25 Дж-К~1«моль~1 и 0,015 кДж-моль соответственно. Эти значения
совпадают с принятыми КО ДАТА—MGHG в качестве ключевых термодинамических величин [1000].
Результаты более ранних измерений теплоемкости белого фосфора в работах [1231] E0 К), [1429]
A37-282 К) и [4045] B98-317 К), а также определений Ttr в работах [766, 767, 3226, 3842,
3569, 3568] являются существенно менее точными. Для энтальпии превращения белого фосфора
в работах [766, 3226] были получены неверные значения.
В интервале температур 298,15—317,3 К термодинамические функции белого фосфора
вычислены по уравнению для теплоемкости, приведенному в табл. 14.1. Это уравнение основано на
измерениях Стивенсона и др. [3522] при 253—313 К, которые воспроизводятся им с точностью 0,1 —
0,4%. Для температуры и энтальпии плавления белого фосфора в таблице приведены значения,
определенные в работе [3522] соответственно с точностью 0,1 К и 0,003 кДж-моль. Принятое
значение Тт подтверждается результатами менее точных измерений [2037, 2495, 2762]. Значения
Тт, полученные в работах [766, 3029, 2483], являются ненадежными. Значение Дт Н (Р, к, бел.),
полученное в работе [3522], точнее найденных ранее в работах [766, 3000,1222, 3605, 2995].
Термодинамические функции жидкого фосфора рассчитаны на основании приведенного в таблице
значения теплоемкости, измеренного Стивенсоном и др. [3522] при 320,15 К. Данные работ [4045,
1668] для энтальпии и теплоемкости жидкого фосфора являются ненадежными. Погрешности
значений Ф° (Т) при 298,15; 500 и 900 К, приведенных в табл. Д.24, оцениваются в 0,25; 0,6 и
1,4 Дж-К-^моль.
Таблицы термодинамических функций белого фосфора в твердом и жидком состояниях,
приведенные в справочниках [2220, 3564], основаны на неточных данных ранних экспериментальных
исследований. В справочнике JANAF [2095] приведены термодинамические свойства белого
фосфора до 1500 К, основанные на данных [2512], полученных при 15—315 К, а также таблица
термодинамических свойств жидкого фосфора при 298—3000 К, основанная на предположении, что
теплоемкость жидкого фосфора равна 26,326 Дж-К~1-моль~1. Это значение определено авторами
[2095] из данных о давлении паров фосфора. Приведенные в табл. Д.24 термодинамические
свойства фосфора ограничены температурой 900 К, поскольку его критическая температура
равна 968 К.
В справочнике за стандартное состояние элементарного фосфора принят белый фосфор и в
соответствии с этим
Д/Й"°(Р, к, бел.)Е=0.
Давление пара фосфора в реакции Р (к, ж)=Р (г) вычислено с использованием значения
~&ГН° @) =315,563+1,0 кДж-моль в соответствии с принятой в справочнике энтальпией
образования Р (г) (см. ниже).
Р (г). Термодинамические свойства газообразного одноатомного фосфора в стандартном
состоянии при температурах 100—10 000 К приведены в табл. 192.
Уровни энергии атома Р, приведенные в табл. 14.2, приняты по работе Мартина [2534] и
являются более точными, чем приведенные в справочнике Мур [2697].
Термодинамические функции Р (г) вычислялись по уравнениям A.3)—A.6), A.9), A.10), A.24),
A.25). Значения QBB и ее производных были рассчитаны на основании данных, представленных
в табл. 14.2. Погрешности в значениях термодинамических функций определяются неточностью
прилитых фундаментальных постоянных и для Ф° (Т) и S° (T) не превосходят 0,02 Дж.К~1-моль~1.
346
Фосфор и его соединения
Р2(г)
Ранее таблицы термодинамических функций Р(г)
вычислялись в справочниках [87, 88, 2220, 2556,
1946, 2095, 3564] и в работах [3535, 3650, 2198,
2291, 3074]. В ранних расчетах [3535, 3650]
учитывалось только основное электронное
состояние атома фосфора *?з/а (в расчетах, приведенных
в работе [3535], была допущена арифметическая
•ошибка). Остальные расчеты производились с
учетом электронных состояний атома фосфора по
данным Мур [2697]. При этом отличия от значений
<Ф° (Т) и S° (T), приведенных в табл. 192,
обусловленные в основном различиями в принятых
значениях фундаментальных постоянных, не
¦превосходят 0,01 Дж-К~х>моль~1.
Энтальпия образования одноатомного фосфора,
¦принятая в справочнике:
Таблица 14.2. Уровни энергии атома Р,
принятые для расчета
термодинамических функций
Электронная
конфигурация
Is22s22pe3s23p3
Is22s22pe3s23p3
Is22s22p«3s23ps
Is22s22pe3s23pa
Is22s22pe3s23p3
Состояние
*\
2Ч
2Ч
2Ч
2Ч
CM
0,00
11360,80
11376,40
18 722,65
18 747,95
Pi
4
4
6
2
4
AJP(P, г, 298,15 К) = 316,4+ 0,7 кДж • моль,
«соответствует принятым значениям энтальпии образования и энергии диссоциации Р2 (см. ниже).
Р2 (г). Термодинамические свойства газообразного дифосфора в стандартном состоянии при
температурах 100—6000 К приведены в табл. 193.
Таблица 14.3. Молекулярные постоянные Р2, РО, РО"
Молекула
р2
РО
ро-
Состояние
Х1Ц
ьза
и
хтг
а*П,-
?22+
В'2П
В"Щ.
ь4и-
С'2Д
С 2S"
?>'2Д
хч-
агА
Т
(О
е
0 780,89
18 798В 565,17
28 197,11s 645,076
28 507,74 н 604,48
31965 395
34 515,34 618,88
46 945,57 472,806
47158,19л 393,554
0» 1233,34
24 000 в 623
30 730,88 1164,51
33 120,80Д 758,967
35000 е 611
37 410 863
43 742,74 825,74
44 831,75 779,22
46ОО0* 616
49 879,83 850
0 1000
4 480 1 020
8 000 г —
Примечание. Ниже все постоянные даны в см
р • '•
¦
РО:
РО-:
' шеув = —5,
5 приведено
1 А = 18.35;
1 Л =244;
1 вычислено
11-Ю-3; б а2 = -3,2-10; '
г0; « А = 131,11; ' *>ey
" шеув = —9,73 - 10~2.
s шеУе = —5,10; в А = —34
по соотношению A.67) при
нено по соотношению A.68); г оценка
Ш X
е е
СМ"]
2,82»
2,75
3,493»
2,2
4
2,97
2,246к
3,766 м
6,56«
—
13,456г
3,733
7,75
6,93
5,14
—
7,49
5,3»
—
—
-1
Х = -3,29,
9 = 2,94-10-2
• г veyf = —
De = 46740 <
Be
0,303 59
0,2516r
0,280 5
0,258 38
0,20
0,275 2
0,242 08
0,220
0,733 7
0,494
0,746 26
0,539 6
0,487
0,633
0,640
0,590 3
0,479
0,608 2
0,674
—
—
ах • 102
De ¦ 10е
г
А
0,147 7» 0,18 1,893.1
— 0.28Я 2,080 0 е
0,16 0,20 1,969 9
0,14 0,16 2,0525
— — 2,33
0,169 0,22 1,9882
0,171 0,257 2,120 5
0,26 0,25 2,224
0,55 1,3 1,476
— 1,2 1,799
0,885 1,2 1,463
0,49 1,0 1,720
— 1,2 1,810
0,7 1,4 1,583
0,49 1,5 1,580
0,56 1,4 1,645
— 1,2 1,825
0,45 1,2 1,621
0,5 е 1В 1,54
— — —
— — —
j. = —2,3-Ю"8; г приведено Во; я приведено Do;
; н 31 = 3,2
5-10~2; М =
, д = 1 • 10 , в юл* = 4,122 • 10;
-—13,26; «Л =-43; «Л =-139.
зм; 6 оценено по соотношению A.69); в оце-
347
Р2(г) Глава 14
Молекулярные постоянные Р2, использованные для расчета термодинамических функций,
приведены в табл. 14.3. В результате исследований электронного спектра Р2 в настоящее врем»
известно 16 стабильных электронных состояний, из которых восемь валентных состояний (Z12+,.
а32+, Ь3Т1д, fr's?~, 5S+, АШд, C'EJ, с8Пя) имеют энергии возбуждения до 50 000 см.
Экспериментально наблюдались следующие переходы: А1П0-^ Х^^ЗОб, 780, 1756, 3821], С1!,*^ X1!,* [1606,
322, 430, 1912, 1903, 2113, 3155, 2513, 2514, 2515, 1116, 1261, 780, 1756, 2730, 3497], b3llff -> а32;
[777, 777а, 2505а], с3Пи -* Ь*Пд [2730, 777а, 780а, 7806, 904а, 3760] и Ь'32" -^ X1!,* [2730, 776,
779 J1. Электронные переходы, связанные с состояниями, энергии возбуждения которых
превосходят 50 000 см, исследовались в работах [780, 1116, 903]. Приведенные в табл. 14.3 значения
молекулярных постоянных Р2 в состояниях X1!,* и А1!!^ найдены Дугласом и Рао [1306] на
основании анализа структуры полос АгИд—Х1^ с р'^1 и d"^38, Значения молекулярных
постоянных Р2 в триплетных состояниях вычислены Брионом и др.: в состоянии а32+ — в
работах [777а, 2505а, 780а], в состояниях Ъ 3Ид и с3Пм — в работе [780а] и в состоянии 6'32~ — в
работе [779]. Молекулярные постоянные Р2 в состоянии 52+ найдены в работе [2505а] на основании
анализа возмущений в системе полос bsHg—а32*. Для состояния СХ2* приняты постоянные,
также рекомендуемые Брионом и др. [780а]. Энергии возбуждения триплетных состояний и
состояния 52* определены из анализа возмущений, наблюдаемых в состояниях С1^, с3Пя и Ъ3Пд. Их
погрешности не превышают 5—10 см.
Термодинамические функции Р2 (г) были рассчитаны по уравнениям A.3)—A.6), A.9), A.10),
A.93)—A.95). Значения (?вн и ее производных вычислялись по уравнениям A.90)—A.92) с учетом
семи возбужденных электронных состояний, приведенных в табл. 14.3. Статистическая сумма
по колебательно-вращательным уровням энергии состояния Х1^ находилась по уравнениям.
A.73)—A.75) непосредственным суммированием по колебательно-вращательным уровням энергии,
для остальных состояний принималось, что (?*0J[ vt = {pJPx)Q^m, вр- В расчете учитывались все уровни
состояния Хг2+д, ограниченные предельной кривой диссоциации этого состояния. В табл. 14.4
приведены значения z>max, /цт и коэффициенты а{ в уравнении, аппроксимирующем предельную
кривую диссоциации. Колебательно-вращательные уровни энергии состояния Xy~L+g
рассчитывались по уравнениям A.65) и A.62) со значениями коэффициентов Ykl, представленными в табл. 14.4-
Погрешности рассчитанных термодинамических функций Р2 (г) при Т ^ 3000 К обусловлены
в основном неточностью принятых фундаментальных постоянных. При более высоких
температурах начинает сказываться неточность в аппроксимации высоких уровней
колебательно-вращательной энергии состояния Х1^. Суммарные погрешности в значениях Ф° (Т) не превышают 0,02;
0,05 и 0,3 Дж-К -моль при 298,15; 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций Р2 (г) вычислялись в справочных изданиях [87,.
88, 2095 и 2556] и работах [3944, 2890, 3074] для Т < 6000 К. В справочниках [3564, 2220] и
работах [76, 3535] термодинамические функции Р2 (г) рассчитаны для Т <^ 3000 К. Во всех расчетах
использовались близкие значения молекулярных постоянных, поэтому при Т <^ 4000 К
расхождения в значениях Ф°(Т), приведенных в табл. 193 и указанных работах, не превышают-
0,1 Дж-К~1-моль~1. При более высоких температурах расхождения возрастают и достигают 0,5
и 2,5 Дж>К~1-моль соответственно в значениях Ф° F000К) и S0 F000 К) в связи с тем, что во всех
предыдущих расчетах либо вообще не учитывались возбужденные состояния Р2 [2095, 3944, 2890],
либо учитывались лишь состояния ^41ПЙ, и С1!.* [87, 88, 2556].
Константа равновесия реакции Р2(г)=2Р(г) вычислялась с использованием значения
ДГЯ° @) = Do (P2) = 485,606 ± 0,36 кДж • моль,
соответствующего принятым в справочнике значениям энтальпий образования Р(г) и Р2(г).
Значение AfH° (Р2, г) было принято на основании результатов измерения равновесия реакции Р4 (г) =
=2Р2 (г), приведенных в табл. 14.5. При вычислении констант равновесия этой реакции по резуль,-
1 Состояние 62+ известно по возмущениям, наблюдаемым в системе полос^Ь'П^, — a3IJ [2505а].
В работе Бриона и др. [7776], опубликованной после завершения подготовки этой главы, получены
молекулярные постоянные Р2 в состояниях XJ2+ и А1!!^, близкие к принятым в справочнике.
348
Фосфор и его соединения
Р2(г)
Таблица 14.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (см), а также
значения г>шах и */цт, принятые для расчета термодинамических функций Р2, РО, РО",
РН, PF, PCI, PS, PN
Коэффициенты
Р »
2
ХЧ+
Т„ 0
3V10-2 7,808
У20 -2,823
узо.Ю2 —0,443
у40.Ю* —0,491
У01 0,303
Уц-102 —0,158
У21-108 0,560
У31.10в —0,187
У41-107 —
уО2.1О7 —1,84
(ao=De).lO-4 4,089
fll.10l 0,363
95
03
094
171
792
735
074
631
23
12
а2.106 0,272 377
а3-1013 —2,125
Ушах 91
/11П 513
Примечание. Ниже
2 ' Состояние
Те
fu : •
Состояние
6 4=224,0
РО" : »
Состояние
т.
РН : »
Состояние
т.
PF : »
Состояние
т.
РС1 : »
Состояние
т.
PS : *
Состояние
т.
6 4=321,93
PN : a
Состояние
т.
27
все
РО»
мг«
0
12,339
—6,901
2,954
—3,913
0,736
—0,573
0,485
—0,384
—
—10
5,004
0,703
0,940
0,339
58
352
рО-а
X*Z-*
0
9 10
91 —5,3
73 —
01 —
672 0,674
574 —0,5
152 —1,4
785 —
—
—10
327 —
817 —
452 —
РН»
x3s-«
0
23,652
—43,648 2
—52,417 5
—
8,355 2
—24,851 8
—83,263 8
—
—
—4 360
2,625 76
4,684 68
228,154
537 — —55 715,8
93
350
постоянные даны в см
18 798 28197,11 28 507,
а4П<
24 000
4 480
о1Д
7 660
в*
7 090,43
агА
6 500
о*П
20 000
а3Е+
30 000
В22+ В'2
19
77
52 +
74 31 965
П В"*Ф(
30 730,88 33 120,80 35000
б1!
8 000
6*2+ 43П
15 200 29 484
ЬЧЗ+
13 353,91
612+ 43П
12 084 25 914
42П В2Ф
22 894 28 000
63П АШ
35 000 39 805
6
сЧ1
37 500
43П,
29 481,8 ;
31 000 t
6*2-
29 000 :
с3П
,7 46 000
PF»
Х32-'
0
8,467 55
-4,492 458
0,077 391
-0,559 87
0,563 6
-0,391 387
—2,207 38
—
—
-10
3,707 855
0,452 93
0,600 589
6,823 97
81
343
АШд
34 515,34
РЗВС
Х*1
0
5,505
—2,233
4 0,185
—1,342
0,249
—0,138
—0,235
—
—
—2
2,521
0,303
0,292
—2,712
-6
19
92
514
28
99
826
599
51
081
062
484
76
445
С1
t 46 945,57
с2д
37 410 43 742,74
1 = 2,212;
ет '
55 808,54
6 х
g1!!
i5 000
С2Д D
jj. —• —0,
6 \ = 2,{
— 7,0
22" 1
С
PS
хт
7,391
—2,969
0,173
—0,986
0,297
—0,159
—0,307
—
—
—2
3,708
0,339
0,277
-1,894
86
490
сЧ1
47 158
г
19
789
886
45
325
079
281
144
335
536
976
,19
D'4
44 831,75 46 00С
0738
N23;
/2П
51 000 34 000 34 000
—(i = C
РХ
34 68"
PN»
XW
13,370 4
—6,911 37
—0,185 316
—3,036 82
0,786 19
—0,554 035
-1,170 43
1,383 13
-0,424 32
—0,109
5,181 334
0,782 828
0,882 834
10,145 3
67
342
I /?22+
49 879,83
,0018
7,9
349
Р3(г) Глава 14
татам измерений давлений и плотности паров принималось, что основными компонентами пара
являются Р4 и Р2 [3535, 3539, 159, 1529]. Приведенные в таблице погрешности отражают только
воспроизводимость экспериментальных данных. В справочнике для энтальпии образования Р2 (г)
принимается значение
Д/Я°(Р2, г, 298,15 К) = 143,7 + 1,3 кДж • моль,
основанное на величине АГН° @)=224,7+2,5 кДж-моль, вычисленной по результатам
измерений Штока и др. [3539] и подтвержденной результатами измерений Pay [3164] (см. табл. 14.5).
При оценке погрешности принятого значения АгН° @) учтена неточность термодинамических
функций Р4 (г). Значения Аг Н° @), основанные на результатах масс-спектрометрических
исследований, как правило, несколько выше принятого. В пределах погрешностей определения они
согласуются с принятым значением, но менее надежны, о чем свидетельствуют большие различия
результатов расчетов по II и III законам и худшая воспроизводимость.
Использованное для вычисления энтальпии образования Р (г) значение Do (Р2)=40 590 +
+ 30 см~1=485,56+0,36 кДж-моль основывается на данных Герцберга [1903] о предиссоциа-
ции Р2 в состоянии С1!!^ и соответствует предположению, что предиссоциация обусловлена
состоянием, коррелирующим с пределом Р DiS)+PBZ)). Его погрешность связана с неточностью
определения предиссоциации в состоянии С1!]* E1 969+24 см) и неопределенностью корреляции
состояния, вызывающего предиссоциацию с термом 2Zb/2 или 2D^2. Основанное на спектральных
данных значение Do (Р,) подтверждено результатами масс-спектрометрических исследований-
[1658, 1657, 2293, 2294].
Р3 (г). Термодинамические свойства газообразного трифосфора в стандартном состоянии при
температурах 100—6000 К приведены в табл. 194.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. В справочнике принимается, что молекула Р3 в основном электронном состоянии:
Х2Пд имеет линейную симметричную структуру в соответствии с правилами Уолша [3870 ] и по
аналогии с молекулами N3 и NCO. Длина связи Р—Р молекулы Р3 в состоянии X2Uff принята равной
1,95+0,05 А. Ей соответствует значение /, приведенное в табл. 14.6, погрешность которого
оценивается в 2-Ю9 г-см2. Принятое значение гР-Р (Р3) оценено на основании значений ге (Р2)?
в состояниях X1!,*, с3Пи и Аг11д, а также значений rN_N (N3) и re (N2) в состояниях X1!,*, с3Ии и
А1!!^. Приведенные в таблице основные частоты Р3 вычислены в приближении поля валентных
сил по оцененным силовым постоянным х. Их погрешности составляют от 30 до 60 см. Энергии,
возбужденных электронных состояний Р3, по-видимому, превосходят 20 000 см и не
рассматриваются в справочнике.
Термодинамические функции Р3 (г), приведенные в табл. 194, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.126),
A.129). Погрешности табулированных значений обусловлены неточностью принятых
молекулярных постоянных, приближенным методом расчета, а также пренебрежением возбужденными
состояниями Р3. Суммарные погрешности в значениях Ф° (Т) при 298,15; 3000 и 6000 К составляют'
2, 5 и 8 Дж-К^-моль, в том числе из-за неточности молекулярных постоянных 2—4 Дж-К^Х
X моль.
Ранее таблицы термодинамических функций Р3 (г) не публиковались.
Константа равновесия реакции Р3 (г)=ЗР (г) вычислена с использованием значения АГН° @) =
= 732,614+20 кДж-моль, соответствующего принятой в справочнике энтальпии образования
Д/Я°(Р8, г, 298,15 К) = 210 + 20 кДж • моль.
Эта величина основывается на результатах масс-спектрометрического исследования
диссоциативного захвата электронов молекулами Р4 в работе Беннетта и др. [585], в которой она была
получена в предположении, что потенциал появления иона Р" при энергиях электронов в 464 +
+ 10 кДж-моль соответствует процессу P4-f е"=Р"A/))+Р3 и что Aj,H°(P~, г, *D, 0)^^Д/Н°(Р„
г, 0). Эти предположения основывались на том, что соответствующее им значение энтальпии атоми-
зации Р3(г) близко к величине, оцененной Кордесом и Гингеричем [2294]. Принятое в
справочнике значение А* Н° (Р3, г) находится в удовлетворительном согласии с величиной Do (Р3—Р)=
=/г=4,0-106 дин-см; /а/г2=0,2-105 дин-cm.
350
Таблица 14.5. Результаты определений энтальпии реакции Р4 (г) = 2Р2 (г) (в кДж-моль)
Авторы
Метод
@)
Шток, Гибсон, Штамм, 1912 [3539]
Пройнер, Брокмеллер, 1913 [3083]
Маринова и др. 1971 [159]
Pay, 1975, [3164]
Дроварт, Голдфингер, 1958 [1325]
Гутбир, 1961 [1773]
Панич, Артур, 1970 [2946]
Сму, Дроварт, 1973 [3476]
Фарроу, 1974 [1441 ],
Фоксон и др., 1974, [1517]
Алиханян и др., 1975 [16]
Смете и др., 1977 [3449]
Измерение статическим методом давления и плотности пара фосфора, 773—
1473 К, 0,08—1,25 атм, 29 точек
То же, расчет по 20 высокотемпературным точкам, 973—1473 К, 0,1-*
1,25 атм
Измерение статическим методом давления и плотности пара фосфора, 773—¦
1473 К, 0,03—1,17 атм, 84 точки
Измерение статическим методом давления и плотности насыщенного и
ненасыщенного пара фосфора, 373—1273 Ка
Измерение статическим методом давления и плотности паров фосфора,
1000—1375 К, 12 серий 0,17—49 атм, 83 точки
То же, 1000—1356 К, 3 серии, 0,17—0,8 атм, 22 точки
Масс-спектрометрическое исследование состава пара над InP, 870^
950 К, 4 точки
Масс-спектрометрическое исследование Состава пара над InP, 1020—1120 К
Масс-спектрометрическое исследование состава пара над InP, 751—943 К,
10 точек
Масс-спектрометрическое исследование пара над смесью МоР—МоО3,1032 К,
1 точка
Масс-спектрометрическое исследование пара над InP, 818—897 К, 11 точек
Масс-спектрометрическое исследование пара над смесью ВР—ВвР (к)
214 + 17A1)
225+3 (Ш)
223+5A1)
224,7+0,6A11)
125+11 (И)
205+3 (III)
232,8+0,2A1)
210,8±0,8(Ш)
210+11 (II)
223,7±1,0(Ш)
216±14(И)
225,3±0,7(Ш)
119 (И)
229±7(Ш)
261+29 6 (II)
176+27 (И)
221,6±2,8(Ш)
231+7A11)
265+31 (II)
239 + 1A11)
218+19 в (II)
231+8В(Ш)
229,2±4,8
Фотоионизационное исследование пара над белым фосфором. Определение
порога реакции P4+Av=P3+-r-Pa+e
• В работе [159] приведено только уравнение lgKp (атм) = —12 655,7/Г— 1,142 lg T-f-0,142-10~4- 71 + 0,37/Т+ 12,657, которое было
получено на основании определения параметров точек перехода от насыщенного к ненасыщенному пару при 1073—1273 К.
6 Получено Дровар'том после исправления ошибки в оригинальной работе.
в Получено авторами работы [16].
Таб л
.
Молекула
иц а
[4.6. Значения молекулярных постоянных, а также о
Р2О4, Р2О6, Р3О6, Р4О6, Р4О„ Р4О8, Р4О„ Р4О10
v2
V3
и Px,
принятые для
расчета термодинамических функций Р3, Р4
V14
СМ
Til .Kins
Г3 • CM*
p
a
.O»i
Px
Р3 470 260 B) 810 _ _ _______ _ _ _ —а 2 4
Р4 606,5 367,2 B) 464,5 C)---------- - - 0,16 12 1
Р2О3 1500 1300 1100 650 500 300 250 250 200 — — — - — — 0,16 1 1
Р,О4 1500 1300 1100 B) 650 B) 500 400 300 250 250 200 - — — - — 0,44 4 1
P90R 1500 1200 520 350 450 250 500 40 250 40 1500 1150 650 520 250 0,80 2 1
Р3О6 _ _ _____________ 6 36
Р4Ов 620 562 285 B) 691 B) 832 C) 283 C) 959 C) 305 C) 408 C) 642 C) — — — — — 4,6 12 1
Р4О, - - - ____________ 8,2 31
Р4О8 __ _____________ 14 21
ТУ Q , , 20 3 1
P4Ojo 553 717 1440 254 B) 300 B) 770 B) 400 C) 240 C) 950 C) 264 C) 411 C) 573 C) 760 C) 1010 C) 1390 C) 30,3 12 1
» / = 39 ¦ Ю"89 г • см2
«•*(!•) Глава 14
=480 + 90 кДж-моль, вычисленной по данным работы Каретта и Кервина [872], исследовавших
диссоциативную ионизацию молекул Р4 методом электронного удара. В этой работе было найдено
значение Do (Р3—Р)=290 кДж-моль, которое, видимо, является ошибочным, поскольку оно
противоречит приведенным выше данным работ [585, 2294], а также работ Штока и др. [3539]
и Гингерича [1656], из которых следует, что Do (Р3—Р) > 390 кДж-моль.
Р4 (г). Термодинамические свойства газообразного тетрафосфора в стандартном состоянии
при температурах 100—6000 К приведены в табл. 195.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. Молекула Р4 в основном электронном состоянии ХХА имеет структуру правильного
тетраэдра (точечная группа симметрии Тй), характеризующуюся значением г (Р—Р)=2,21 +0,02 А,
найденным Максвеллом и др. [2548] по электронографическим данным. Соответствующее
значение IaIbIc, приведенное в табл. 14.6, имеет погрешность 9-Ю15 г3-см6. Основные частоты Р4
определялись при изучении колебательных спектров белого фосфора и его растворов [633, 3787,
3789, 3788, 1775, 1049, 607, 2578] и колебательных спектров паров фосфора [3787, 2929, 537, 708,
1775]. Приведенные в таблице значения основных частот Р4 определены Босуэрфом и др. [708]
по спектру КР паров фосфора (vj и v2) и Гутовским и Гофманом [1775] по ИК-спектру паров
фосфора (v3). Погрешности приведенных в таблице значений основных частот Р4 не превышают 1 см.
Термодинамические функции Р4 (г), приведенные в табл. 195, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124), A.128),
A.130). Погрешности вычисленных значений обусловлены преимущественно приближенным
методом расчета. Погрешности значений Ф° (Т) при 298,15; 3000 и 6000 К составляют 0,5; 3 и
6 Дж-К^'Моль, при этом погрешности из-за неточности принятых постоянных не превышают
0,2 Дж-К^-моль-1.
Ранее таблицы термодинамических функций Р4 (г) вычислялись в справочниках [87, 88, 3564,
2220, 2556, 2095] и в работах [3535, 3682, 3074, 2890]. В расчете [3682] приняты
неправильные значения основных частот и допущена грубая ошибка. Различие примерно
на 0,2 Дж-К^-моль в значениях Ф° (Т) и S° (T), приведенных в табл. 195 и в работах [87, 88,
3564,2220, 2095,2556,2890], обусловлено главным образом тем, что во всех расчетах для
частоты v2 было принято более низкое значение C63 см по данным работы [3787]), чем в настоящем
издании.
Константа равновесия реакции Р4 (г)=4Р (г) вычислена с использованием значения кгН° @) =
=1196,012±2,8 кДж-моль, соответствующего принятой в справочнике энтальпии образования
Д/Я°(Р4, г, 298,15 К) = 58,9 ±0,3 кДж • моль.
Результаты определения AfH° (P4, г) на основании данных по давлению пара фосфора
приведены в табл. 14.7 с погрешностями, которые отражают только воспроизводимость
экспериментальных данных. Неточность использованных в расчетах термодинамических функций Р (к) и
Р4 (г) увеличивает погрешность значения &fH° на 0,3 кДж -моль. В расчетах предполагалось,
что насыщенные пары фосфора состоят только из молекул Р4. Это предположение основано на
определении среднего молекулярного веса паров фосфора в работах [1963, 3083, 3535, 3539] из
измерений их давления и плотности, а также на масс-спектрометрических исследованиях состава паров
фосфора [2225, 872, 585].
Наиболее точные прямые измерения давления пара белого фосфора при температурах около 293 К
были выполнены методом протока Центнершвером [921 ] и Фишбеком и Эйхом [1487].
Основываясь на результатах этих исследований, было найдено, что давление пара белого фосфора при
293,15 К равно C,42 + 0,13)-10 атм. Дейнтон и Кимберли [1153] провели прецизионные
измерения интенсивности радиоактивного излучения паров белого фосфора, содержащего изотоп 32Р.
Для вычисления давления насыщенного пара из этих данных было использовано приведенное
выше значение давления паров фосфора при 293,15 К, что позволило получить уравнение,
описывающее давление пара белого фосфора в интервале 266—314 К (lg р (атм) = —3529,4/Т7—3,5 lg T-\-
+ 16,2086), которому соответствуют приведенные в табл. 14.7 величины по данным работы [1153].
Принятое значение А.Н° (Р4, г) основывается в первую очередь на результатах работ [1153, 1487,
921, 2495].
352
Фосфор и его соединения
PO(rl
Т аблица 14.7. Результаты определений энтальпии образования Р4 (г) (в кДж-моль)
Авторы
Метод
4, г, 298,15 К)
Жубер, 1874 [2180 J Измерение давления пара над твердым фосфором
статическим методом, 278—313 К, 6 точек
Джолибуа, 1910 [2068] Измерение давления пара над жидким фосфором
статическим методом, 446—585 К, 26 точек
Центнершвер, 1913 [921 ] Измерение давления пара над твердым фосфором методом
протока, 293,15 К, 22 точки
То же при 298—313 К, 4 точки
Смит, Бокхорст,
[3470]
1916
Мак-Рей, Ван Вурис,
1921 [2495]
Фишбек, Эйх, 1937
[1487]
Измерение давления пара статическим методом над
жидким фосфором, полученным плавлением красного фосфора,
777—905 К, 21 точка
Измерение давления пара методом точек кипения над
жидким фосфором, полученным плавлением белого фосфора,
454—682 К, 16 точек
Измерение давления пара над жидким фосфором методом
изотенископа, 373—423 К, 7 точек
Измерение давления пара над твердым фосфором методом
протока, 273—293 К, 3 точки
Дэйнтон, Кимберли, 1950 Измерение давления пара над твердым фосфором, содер-
Менк, 1964 [2612] (по
данным [1068])
Маринова и др., 1971
[159]
жащим радиоактивный изотоп 32Р, 266—314 К, 20 точек
Измерение давления пара над твердым фосфором
статическим методом б
Измерение давления пара над твердым фосфором
статическим методом, 300—316 К 6
То же, над жидким фосфором, 317—677 К б
43 (Щ
55,4 ±0,2 (III)
61.2 ±0,6 (II)
59,17 ±0,06 (III)
58,91 ±0,09 (III)
54 (II)
58,8 ±0,2 (III)
59.8 ±0,5 (II)
57,48 ±0,05 (III)
61.9 ±1,5 (И)
59.3 ±0,2 (III)
58,3 ±0,4 (II)
58,88 ±0,06 (III)
76 (II)
59,9 ±2,7 (III)
58,86 ±0,10 (II)
58,86 ±0,09 а (Ш)
58,7 (II)
58,81 (III)
59.2 (II)
58,42 (III)
58,1 (II)
59.3 (III)
а Погрешность определяется в основном неточностью использованного в расчете значения давления паров
фосфора при 293,15 К (см. текст).
6 Результаты этих работ представлены только в виде уравнений, что исключает возможность вычисления
погрешности их воспроизводимости.
РО (г). Термодинамические свойства газообразного оксида фосфора в стандартном состояния
при температурах 100—6000 К приведены в табл. 196.
Молекулярные постоянные РО, используемые для расчета термодинамических функций,
приведены в табл. 14.3. Молекула РО имеет 10 стабильных валентных электронных состояний с
энергиями возбуждения до 50000 см (ХШГ, a4n<t В22+, 5'2П, 5"аФ,., D'2n, &42", СД, С22~, F22+)
и ряд ридберговских состояний с энергиями возбуждения 40 000—60 000 см (Л22+, ?)аП, G22+,
?2Д, #22 + , /22+). В электронном спектре РО наблюдалось 10 систем полос, связанных с
переходами между дублетными состояниями: 522+—ХШГ [1320, 3873, 1344, 2608, 2607, 3821, 3119, 3427,
3126, 3125,1138,1082, 1262, 2688, 2689, 2998, 2999,1606,1749,1610, 3826], А22?—Х2ПГ [3873,1062,
3158, 1608, 3389, 2998, 2999, 1757, 1750, 1749, 1051, 1747, 1053], Я'2П-Х2ПГ [3305, 1079, 3822,
3819, 1080, 3821, 3826, 1610], Л22 + -?22+ [3824], #'2П-522+ [1078, 3822, 1080, 1052, 16101,
<7'2Д-Х2ПГ [3305, 3306, 1748, 3821, 1262, 1747, 2805, 3093, 1050], С22"-Х2ПГ [1320, 3305, 1050,
3821, 1061, 3093], F22 +—Х2П, [2847], Я2+—522+ [3823, 3824, 1759, 2847], F22 + —422 + [3824,
2847]. Переходы, связанные с квартетными состояниями, в спектре РО не наблюдались.
Приведенные в табл. 14.3 значения молекулярных постоянных РО в состояниях Х2ПГ и 522+ получены
Верма и Сингхалом [3826] в результате анализа вращательной структуры 23 полос (v' ^ 9, v" ^
^ 11) системы В22 +—Х2ПГ. Эти постоянные хорошо согласуются с данными более ранних работ
и с результатами анализа других систем полос. Теоретические расчеты [3218, 3734], возмущения,
наблюдаемые во вращательной структуре полос систем Л22+— 522+ и Я22+— Х2ПГ [3119а, 3824,
3826], а также предиссоциация в состоянии В'Щ [3823, 1610] свидетельствуют о существовании
стабильных квартетных состояний а*П{ и &*2~ и состояний В"гФ{ и D'2H, не обнаруженных в
спектре РО. Приведенные в таблице значения колебательных и вращательных постоянных РО в
состояниях а4По ВФ( и D'2n вычислены Рошем и Лефебр-Брион [3218]. Поскольку энергия воз-
353
РО (г) Глава 14
буждения состояния В'2И, найденная экспериментально [3819], на 3000 см превышает
значение, вычисленное в работе [3218], в табл. 14.3 для состояний а4П,-, 5Ф и D'2U приведены
энергии возбуждения, увеличенные на 3000 см по сравнению с рассчитанными теоретических. Их
погрешность оценивается в 4000 см. Значения молекулярных постоянных в состоянии 642~
найдены Верма и др. [3824, 3826] в результате анализа возмущений во вращательной структуре полос
систем Л22 +—/?22 + и i?22 +—X2U. Приведенные в таблице молекулярные постоянные РО в
состояниях С 2Д и С22~ определены Прюдоммом и др. [3093] из анализа вращательной структуры полос
систем СД—Х2ПГ и С22~—Х2ПГ, а постоянные РО в состоянии .F22 + найдены Нго и др.
[2847] при анализе вращательной структуры полос системы F2~E—Б22+ (i/ ^ 11).
Термодинамические функции РО (г), приведенные в табл. 196, рассчитаны по уравнениям
A.3)—A.10), A.93)—A.95). Значения Qm и ее производных вычислялись по уравнениям A.90)—
A.92) с учетом девяти возбужденных электронных состояний (а*П,-, Б22+, В'2И, ВФ(, Ь42~,
СД, С22~, 1)'2П и F21i+). Статистическая сумма по колебательно-вращательным уровням энергии
основного состояния X2tlr и ее производные находились по уравнениям A.73)—A.75)
непосредственным суммированием по колебательно-вращательным уровням энергии. В расчете
учитывались все уровни, ограниченные предельной кривой диссоциации состояния Х2ИГ. Колебательно-
вращательные уровни состояния Х2ПГ вычислялись по уравнениям A.65), A.42) и A.61) с
коэффициентами Ykl, приведенными в табл. 14.4. Значения коэффициентов Ykl были рассчитаны по
постоянным молекулы РО, приведенным в табл. 14.3 с учетом условий A.50) и A.59). В табл. 14.4
приведены также значения vm!a, /цш и коэффициентов а,- в уравнении, аппроксимирующем предель-.
ную кривую диссоциации состояния Х2ИГ. Составляющие девяти возбужденных состояний РО
вычислялись в предполо, ении, чт( ОЩ.^ — (PilP^QmJ. щ' Погрешности рассчитанных
термодинамических функций при Т ^ 3000 К обусловлены в основном неточностью фундаментальных
постоянных и не превышают 0,02 Дж -К -моль. При более высоких температурах погрешность
возрастает из-за неточности аппроксимации уровней колебательно-вращательной энергии
основного состояния и неопределенности в энергии возбуждения состояния а4П<. В значении Ф° F000 К)
погрешность может достигать 0,1 Дж-К~1-моль~1.
Ранее таблицы термодинамических функций РО(г) вычислялись в справочниках [87, 88,
2095, 942а, 3944, 2556], а также в работах [3074] (Т < 6000 К) и [764] (Т < 3000 К). Во всех этих
работах термодинамические функции РО(г) вычислялись приближенными методами с менее полным
учетом возбужденных электронных состояний, чем в настоящем издании. Расхождения данных
работ [88, 2095, 2556, 3074, 942а] и табл. 196 не превышают 0,1 и 0,6 Дж-К^-моль в значениях
Ф° (Т) и S° (Т) соответственно. Расхождения в значениях С° (Т) при Т > 4000 К более
значительны и для С F000 К) достигают 2,8 Дж-К~1-моль~1 с данными [2095].
Константа равновесия реакции РО(г)=Р(г)+О(г) вычислена с использованием значения
энергии диссоциации
Д,#с@) = Do (РО) = 590 ± 3 кДж • моль,
принятого в справочнике на основании предиссоциации РО в состоянии 5'2П, изученной Верма
и др. [3823], которая определяет верхнюю границу значений D0(PO) ^ 49 536 см-1=
=592,58 кДж-моль, и найденного Дровартом и др. [1327] по масс-спектральным данным
значения D0(PO)=593+8 кДж-моль.
7 Наблюдавшийся в работах [1320, 1344, 1078, 1079, 1080, 3822, 3823, 1610] обрыв вращательной
структуры полос систем В'2И—Х2ПГ и В'2Н—Б22+ при 49 536 см относительно уровня ^=0
состояния Х2ПГ является очень резким, в связи с чем в работе [3823] принималось, что
действительное значение Do (РО) близко к величине 49 536 см. Дроварт и др. [1327] в результате масс-
спектрометрического изучения равновесия реакций в парах фосфидов иттрия и гадолиния B360—
2613 К) и смеси МоР—МоО2—Sn A408—1466 К) рассчитали значения констант равновесия ряда
реакций с участием РО (г) и нашли приведенную выше величину. С учетом того, что предиссоциа-
ция дает верхнюю границу для энергии диссоциации, в справочнике принято округленное значение
Do (РО). Ему соответствует
Д/Я°(РО, г, 0) = —27,654 ± 3 кДж • моль.
1 Рей и др. [3119а] на основании сравнения молекулярных постоянных NO в состояниях а*П и В2Г и РО в состоянии-
В2П оценили следующие значения постоянных РО в состоянии а*П : Ге=27 550 см, @^=800 см ], ?„=0,5918 см
354
Фосфор и его соединения РО~ (г), РО2 (г/у
РО~ (г). Термодинамические свойства газа, состоящего из отрицательных ионов оксида
фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 197.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.3. В соответствии с аналогией с изоэлектронными молекулами PF и SO,
результатами теоретического расчета Бойда и Липскомба [722] и экспериментального исследования Цит-
теля и Линебергера [4065] в справочнике принимается, что РО~ имеет основное электронное
состояние Х32~ и два возбужденных электронных состояния а1 А и ЬХ2. Приведенные в табл. 14.3
значения постоянных Те и ше в состояниях Х32~ и с^Д, а также ге (РО~) для состояния Х32~
определены Циттелем и Линебергером [4065] по спектру электронов, образующихся при
фотоионизации РО~, их погрешности составляют 80 см (Г, и ше) и 0,01 А (ге). Приведенное в таблице
для РО~ значение Те Fг2+) оценено на основании предположения, что отношение значений Тв
состояний а1 А и б1!!" у РО~ близко к среднему от величины этих отношений для PF и SO, его
неточность оценивается в 1000 см.
Термодинамические функции РО~ (г), приведенные в табл. 197, рассчитаны по уравнениям
A.3)—A.10), A.93)—A.95). Значения QBB и ее производных вычислялись с учетом двух
возбужденных состояний по уравнениям A.90)—A.92) в предположении, что <2<*>_ щ = (pjpx) Q^m вр-
Значения QW Jp находились непосредственным суммированием по уровням
колебательно-вращательной энергии состояния Х32~, по уравнениям A. 76)—A. 78). В расчете учитывались все уровни
состояния Х32~ со значениями /<; /max, е, последние находились из условий A.81). Уровни
энергии состояния Х32~ вычислялись по уравнению A.65) с коэффициентами Ykl, приведенными
в табл. 14.4. Мультиплетность и вырождение уровней состояний Х32~ и а1 А учитывались
статистическими весами. Погрешности вычисленных термодинамических функций обусловлены
преимущественно неточностью принятых молекулярных постоянных РО~ в состоянии Х32~, а также
приближенным учетом мультиплетности этого состояния. Погрешности значений Ф° (Т) при 298,15; 3000
и 6000 К оцениваются в 0,5; 1 и 2 Дж-К^-моль.
Ранее таблица термодинамических функций РО~(г) не публиковалась.
Константа равновесия реакции РО~ (г)=Р (г)+О (г)-\-е~ (г) вычислялась с использованием
значения Дг#° @)=695,400+3,3 кДж-моль, соответствующего принятой в справочнике
величине сродства к электрону РО
,40(РО) = —1,092 + 0,010 эВ = —105,4 + 1,0 кДж-моль,
найденной Циттелем и Линебергером [40651 по спектру электронов, возникающих при
фотоионизации РО~. Принятому значению Ао (РО) соответствует
AfH°(PO-, г, 0) = —133,054 + 3,2 кДж-моль'1.
РО2 (г). Термодинамические свойства газообразного диоксида фосфора в стандартном состоянии
при температурах 100—6000 К приведены в табл. 198.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.8. По аналогии с NO2 в справочнике принимается, что молекула РО2 в основном
электронном состоянии Х2Аг относится к точечной группе симметрии С2„ и характеризуется
параметрами г (Р—О) = 1,50+0,03 А и ^0—Р—0 = 130+5°. Последние были оценены на основании
сравнения структурных параметров молекул РО, НРО, NO, NO2 и HNO. Принятым значениям
соответствует приведенное в табл. 14.8 значение IaIbIg, погрешность которого составляет
1 • 10~116 г3-см6. Приведенные в таблице значения основных частот РО2 в состоянии XaAlt а также
энергия первого возбужденного электронного состояния Л2В2 определены Кордесом и Витше-
лом [1068] на основании анализа колебательной структуры полос, наблюдавшихся Румпфом
[3272] в спектре холодного свечения фосфора, и Лундлэмом [2478] в спектре фосфорно-водород-
ного пламени. В работе [1068] эти полосы были отнесены к переходу между основным и
возбужденными электронными состояниями РО2. Это отнесение не является надежно установленным,
так как известно иное объяснение происхождения холодного свечения фосфора (см. [3780]).
Однако определенные в работе [1068] основные частоты РО2 согласуются с вычисленными по
силовым постоянным, а найденная энергия возбужденного А2В2 электронного состояния РО2
находится в согласии с данными об энергии возбужденных состояний молекул NO2, PH2, NH2, НРО
и HNO. На основании сравнения с этими молекулами в справочнике принимается, что РО2 имеет
355
Глава 14
Таблица 14.8. Значения молекулярных постоянных, а также а и р{, принятые для расчета
термодинамических функций РО„ РО2, PH2, РЩ, HPO, PF2, PF2, PC12, PC
PFC1, PFC1-
Молекула
Состояние
РП &2А
А*ВЛ
Б?ВХ
РП - 1?1А
А3ВХ
РН, -?2ЯХ
АЫХ
РН," ХЫХ
НРО ^А
АЫ
PF, -Х^
PF2- 2»Лг
PCL> .^.В
PCI,- i'Ul
PFC1 J1»^
PFC1" J^
To
0
15 447
19 000
0
15 000
0
18 276,6
0
0
19 032,8
0
0
0
0
0
0
vx
H
CM
1 195 530
—, —
— —
1 200 500
—
2 320 1102,0
— —
2 600 1 200
2 330 1 188,08
— —
831 400
800 430
525 300
530 200
840 350
820 300
1400
—
—
1100
2 340
—
2 650
985
—
852
830
452
530
500
530
IAIBIC ¦ 101"
r* • см»
1,1.10»
—
—
1.4-10»
—
7,05.10"»
—
4,9.10"»
5,437
—
3,3-10»
3,4.10я
5,7.10»
5,7.10»
1,4-10»
1,4.10»
a
2
2
2
2
2
2
2
2
1
1
2
2
2
2
1
1
Pi
2
2
2
i
3
2
2
1
1
1
2
1
2
1
2
1
второе возбужденное электронное состояние 2Вг с энергией 19 000 +2000 см. Погрешности
приведенных в табл. 14.8 значений основных частот РО2 в состоянии %2А1 оцениваются в 5—10 см.
Термодинамические функции РОа (г), приведенные в табл. 198, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130) с учетом двух возбужденных состояний по уравнениям A.168)—A.170).
Погрешности вычисленных термодинамических функций обусловлены неточностью принятых
молекулярных постоянных, неполнотой сведений о возбужденных состояниях и приближенным
характером расчета. Погрешности значений Ф° (Т) при 298,15; 3000 и 6000 К оцениваются в 1, 2 и
5 Дж-К^-моль.
Ранее термодинамические функции РО2(г) вычислялись в таблицах JANAF [2095] по
оцененным молекулярным постоянным и без учета возбужденных электронных состояний. Различия
в значениях Ф° (Т), приведенных в табл. 198 и справочнике [20951, составляют от 0,6 до
Г.-1
3 ДжК 1моль
Константа равновесия реакции
ДД° @) = 1087,129+10 нДж-моль,
зования
РО2(г)=Р(г)+2О(г) вычислена с использованием значения
соответствующего принятой в справочнике энтальпии обра-
b.fH° (РО2, г, 0) = —278 + 10 кДж • моль,
которая основана на результатах исследований равновесия реакций с участием РО2(г),
проведенных Дровартом [1327] и Сму и Дровартом [3476] (табл. 14.9) г. Ранее энергия атомизации РО2
приближенно оценивалась в работах [763, 1068, 1179].
РО2 (г). Термодинамические свойства газа, состоящего из отрицательных ионов диоксида
фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 199.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.8. В справочнике принимается, что РО2 в основном электронном состоянии S.1Al
имеет симметричную угловую структуру (симметрия С2,) и возбужденное электронное состояние
* В работе [1759а] на основании масс-спектрометрического исследования пара над твердым СеРО4 при 1700—1850 К
была вычислена энтальпия реакции РО2 (г)=РО (г)+1/2 О2 (г), однако расчет основывался на ненадежных
данных о парциальном давлении РО (г), вследствие диссоциативной ионизации компонентов пара СеРО4.:
356
Фосфор и его соединения
А (г).
Таблица 14.9. Результаты исследований энтальпии образования Р02 (г) (в кДж• моль)
Авторы
Метод
@)
Дроварт и др., 1972 Масс-спектрометрическое исследование равновесия РО2 (г)+
[1327] +2Sn(r)=2SnO(r)+1/2P2(r), 1272—1436 К, 14 точек
То же, а также исследованное в этой работе равновесие
РО (r)+Sn (r)=1/2P2 (r)+SnO (г), на основе которого
рассчитаны константы равновесия реакции РО2 (гL/вР« (г)=
=2РО (г), 1272—1436 К, 13 точек.
Сму и Дроварт, 1973 Масс-спектрометрическое исследование равновесия РО2(г)+
[3476] +1/2Р2(г)=2РО(г), 1200—1380 К
а Погрешность указана по оценке авторов работ.
—247+25 (II)
—278,6 ±10а (III)
—283 ±20 (II)
—279,6 ±8,2 (III)
—287 (II)
-276,1 ±8а (III)
А3Вг с энергией 15 000+3000 см. Основанием для этого является аналогия с ионом NO2, а также
данные о структуре РО^, полученные Быстровым и др. [44а] при изучении характера изменения
колебательного спектра примесного иона РО^ в галогенидах калия при изотопном замещении
кислорода. С учетом этих данных и принятых в справочнике постоянных молекул NOj, SO2 и
РО2 структурные параметры РО^ в состоянии %}АХ были оценены равными г (Р—О)=1,50 +0,05 А|
¦^О—Р—0=120+5°. Им соответствует приведенное в табл. 14.8 значение IaIbIc, погрешность
которого составляет 2 -10~11в г3 -см6. Для основных частот POj в таблице приведены значения,
определенные по спектру примесного иона РО^ в кристаллах КС1, КВг, KI [44а] и по основным
частотам молекулы SO2, изоэлектронной РО,\ Погрешности принятых основных частот
оцениваются в 20—50 см.
Термодинамические функции POj(r), приведенные в табл. 199, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130) с учетом возбужденного электронного состояния ASBX по уравнениям A.68)—A.70).
Погрешности вычисленных значений обусловлены неточностью молекулярных постоянных,
отсутствием данных о возбужденных состояниях РО^ и приближенным характером расчета.
Погрешности в значениях Ф° (Т) при 298,15; 3000 и 6000 К могут достигать 1, 3 и 6 Дж •К -моль.
Ранее таблица термодинамических функций РО2~(г) не публиковалась.
Константа равновесия реакции РО2 (г)=Р (г)+2О(г)+е~ (г) вычислена с использованием
^0 @I3671293 Д1 соответствующего принятой в справочнике
велир рц 2 ()
значения Д^0 @)=1367,129+30 кДж-моль,
чине сродства РО2 к электрону
Ао (РО2) = —3,2 + 0,3 эВ = — 310 ± 30 кДж • моль.
Эта величина была оценена авторами настоящего издания на основании найденного в работе
[1887] методом фотоотрыва значения Ао (NO2) = —2,36+0,10 эВ и сравнения значений сродства
к электрону N и Р, РН и NH, NH2 и РН2, NO и РО [4065а, 919, 4065, 3419].
Р2О3 (г). Термодинамические свойства газообразного триоксида дифосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 200.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. В справочнике принимается, что молекула Р2О3 имеет плоскую асимметричную
структуру и образована соединением связью Р—Р групп РО2 и РО (точечная группа симметрии Са).
Расчеты энергии атомизации Р2О3, основанные на данных работы [3476], показали, что
асимметричная структура молекул Р2О3 существенно более стабильная, чем симметричная. Об этом
свидетельствует и аналогия с молекулой N2O3. Приведенное в табл. 14.6 значение IaIbIc вычислено по
структурным параметрам Р2О3: г(—Р=О) = 1,43+0,05 А; г(Р—Р)=2,05+0,05 А; г(> Р=О)=1,48 +
+0,05 А; ^О=Р=О = 134+5°; -?Р—Р=О=110+5°, оцененным в работе [3476]. Его погрешность
составляет 4-10~114 г3-см6. Для основных частот Р2О3 в табл. 14.6 приведены значения, оцененные
Сму и Дровартом [3476] для асимметричного изомера Р2О3 на основании аналогии с N2O3 и
требования, чтобы расчеты по II и III законам, основанные на найденных в [3476] значениях констант
равновесия реакций с участием Р2О3(г), приводили к совпадающим значениям энтальпий этих
реакций. Погрешности этих постоянных могут достигать 20—100 см.
357
Р2О4 (г), Р2О5(г) Глава 14
Термодинамические функции P2Os(r), приведенные в табл. 200, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных и приближенным методом расчета и составляют 3, 7 и
12 Дж-К-^моль в значениях Ф° (Т) при 298,15; 3000 и 6000 К.
Ранее термодинамические функции Р2О3(г), соответствующие двум возможным изомерным
формам молекулы Р2О3, вычисляли авторы работы [3476] (Т ^ 1400 К). Значения термодинамических
функций Р2О3(г), полученные в этой работе для несимметричного изомера, находятся в
соответствии со значениями, приведенными в табл. 200.
Константа равновесия реакции Р2О3(г)=2Р(г)+ЗО(г) вычислена с использованием значения
&гН°@)=2048,475+15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д,ЙГв(РаО„ г, 0) = —677 + 15 кДж • моль.
Эта величина основана на результатах масс-спектрометрического исследования реакции
Р2О3(г)=РО(г)+РО2(г), выполненного Сму и Дровартом [3476] E серий измерений, 1160—1381 К).
Расчеты по методам III и II законов привели к значениям \Н°@), равным —371,8 + 1,1 и —366 +
+24 кДж-моль соответственно. При оценке погрешности принятой величины, помимо указанной
выше воспроизводимости измерений, были учтены также погрешности термодинамических
функций и энтальпии образования РО (г) и РО2(г). В работе [3476] была также найдена константа
равновесия реакции Р2О3(г)=0,ЗР4О10(г)+0,4Р2(г) при 1032 К, которой соответствует Д/Я°(Р2О3, г, 0)=
=—680 кДж-моль, что находится в согласии с принятой величиной.
Р2О4 (г). Термодинамические свойства газообразного тетраоксида дифосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 201.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. По аналогии с молекулой N2O4 в справочнике принимается, что молекула Р2О4 имеет
симметричную структуру (симметрия D%k). Приведенное в табл. 14.6 значение IaJbIc молекулы
Р2О4 рассчитано по структурным параметрам: r(P—P)=2,05 A; rQ? Р=О)=1,48 А; ^ О=Р=О =
134°, оцененным авторами работы [3476]. Для основных частот Р2О4 в таблице приведены
значения, оцененные Сму и Дровартом [3476 ] для симметричной конфигурации Р2О4 исходя из аналогии
с N2O4 и требования, чтобы расчеты по II и III законам,'основанные на найденных в работе [3476]
значениях констант равновесия реакций с участием Р2О4(г), приводили к совпадающим значениям
для энтальпий этих реакций. Обоснованная оценка погрешностей принятых постоянных
затруднительна.
Термодинамические функции Р2О4(г), приведенные в табл. 201, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета. Погрешности значений Ф°(Т) при
298.15, 3000 и 6000 К оцениваются в 3, 10 и 15 Дж-К^-моль-1.
Ранее термодинамические функции Р2О4(г), соответствующие симметричной (D2h) и
несимметричной (Са) конфигурациям молекулы Р2О4, вычисляли авторы работы [3476] до 1400 К.
Значения термодинамических функций Р2О4(г), приведенные в указанной работе для симметричной
формы молекулы, находятся в соответствии со значениями, табулированными в настоящем
издании.
Константа равновесия реакции Р2О4(г)=2Р (г)+4О(г) вычислена с использованием значения
АгН°@)—2541,258+25 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(Р2О4, г, 0) = — 923 + 25 кДж • моль.
Эта величина принята на основании масс-спектрометрического исследования равновесия
реакции Р2О4(г)=2РО2(г), выполненного Сму и Дровартом [3476] в интервале 1215—1324 К. Расчет
по методу II и III законов привел к совпадающему значению ДгЯ°@)=367 кДж-моль.
РгО5 (г). Термодинамические свойства газообразного пентаоксида дифосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 202.
358
Фосфор и его соединения Р3Ов (г)
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. В справочнике принимается, что молекула Р2О5, подобно молекуле N2O5, состоит
из двух плоских групп РО2, связанных друг с другом через атом кислорода и расположенных под
углом, близким к прямому (точечная группа симметрии С2,). На основании сравнения принятых
значений структурных параметров молекул N2O5, NO2, РО2, Р4О6, Р4О10, а также структурных
параметров молекулы F2POPF2, определенных в работе [2024], принимается, что в молекуле РаОБ
Г(Р_О)=1,50±0,04А; ^О-Р-О=130±5°; г(Р-О')=1,64+0,04А; ^ Р-О'-Р=95±5°, где
через О' обозначен атом кислорода, связывающий две группы РО2. Эти значения использованы
для расчета приведенного в табл. 14.6 значения IaIbIg, погрешность которого оценивается в 2х
ХЮ"Ш г3-см6. Спектры молекулы Р2О6 не изучались. Приведенные в таблице основные частоты
молекулы Р2О5 оценены на основании сравнения принятых в справочнике значений
соответствующих частот молекул N2O5, NO2, N2O4, РО2 и Р4О10, их погрешности оцениваются в 30—50 см.
Термодинамические функции Р2О5(г), приведенные в табл. 202, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных ж приближенным характером расчета. Погрешности
значений Ф°(Г) при 298,15; 3000 и 6000 К могут достигать 10, 15 и 20 Дж-К-^моль.
Ранее термодинамические функции Р2О5(г) вычисляли Сму и Дроварт [3476] до 1400 К по
оцененным молекулярным постоянным, ошибочно приняв, что молекула Р2О5 имеет 14 основных
частот. Приведенные этими авторами значения термодинамических функций Р2О5(г)
существенно ниже значений, табулированных в настоящем издании. Так, различия в значениях Ф°(Т)
лри 298,15 и 1400 К составляют 22 и 32 Дж-К-^моль.
Константа равновесия реакции Р2О3(г)=2Р(г)+5О(г) вычислена с использованием значения
Лг#°@)=2979,041+25 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
ДД°(Р2Об, г, 0) =—1114 + 30 кДж «моль-1.
Принятое значение основано на результатах масс-спектрометрического исследования
равновесия реакции Р2О5(г)=0,5 Р4О10(г), проведенного Сму и Дровартом [3476] в интервале 1272—
1374 К. В этой работе методом III закона для указанной выше реакции было получено ДгЯ°@) =
=—280+18 кДж -моль". В работе не приведены найденные константы равновесия, что не позволяет
провести пересчет с принятыми в настоящем издании термодинамическими функциями. С учетом
разности термодинамических функций компонентов исследованной реакции, принятых в
настоящем справочнике и в работе [3476], было найдено значение АгН°@) =—322+25 кДж-моль,
по которому вычислена принятая энтальпия образования Р2О5(г).
Р3Ов (г). Термодинамические свойства газообразного гексаоксида трифосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 203.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. В справочнике принимается, что молекула Р3Ов имеет циклическую неплоскую
симметричную структуру, в которой атомы фосфора связаны друг с другом через атомы кислорода,
а также двойными связями с другими атомами кислорода (точечная группа симметрии С3с), и сек-
стетное основное электронное состояние. Приведенное в табл. 14.6 значение IaIbIo вычислено
в предположении, что г (Р—О) = 1,62+0,05 А; г (Р=О)=1,42+0,05 А; г (Р.. .Р)=2,90+0,05 А
и что углы между связями равны 109°28'+10. Погрешность этого значения может достигать
5.10-пз г3-см6.
Термодинамические функции Р3О6(г), приведенные в табл. 203, вычислены в приближении
«жесткий ротатор—гармонический осциллятор». При этом поступательные и вращательные
составляющие находились по уравнениям A.3)—A.10), A.128), A.130), а колебательные
составляющие — по приближенным соотношениям вида: ФКОл(РзОб) — ^зФюЛРА^^вФко-ЛРЛо) через
колебательные составляющие Р4Ов и Р4О10, вычисленные по основным частотам, приведенным в табл.
14.6. Погрешности рассчитанных термодинамических функций Р3Ов(г) обусловлены неточностью
принятых структурных параметров и приближенным характером расчета, они могут достигать
10, 20 и 30 Дж-К-^моль в значениях Ф°(Г) при 298,15; 3000 и 6000 К.
Ранее термодинамические функции Р3Ов(г) вычисляли Сму и Дроварт [3476] до 1400 К для
ллоской циклической структуры молекулы Р3Ов с использованием оцененных значений основных
359
Р4О„(г) Глава 14
частот. Расхождения между значениями Ф°(Г), приведенными в табл. 203 и в работе [3476],
составляют 5—19 Дж-К-моль~1.
Константа равновесия реакции Р3О6(г)=ЗР(г)-Ь6О(г) вычислена с использованием значения
ДгЯ°@)=3984,387+35 кДж-моль, соответствующего принятой в справочнике энтальпии обра-
вования
Д/Я° (Р3О6, г, 0) = —1557 ± 35 кДж • моль.
Принятое значение основано на выполненном Сму и Дровартом [3476] масс-спектрометриче-
ском исследовании равновесия Р3Ов(г)=ЗРО2(г) в интервале 1293—1344 К. В указанной работе
не приведены измеренные константы равновесия, что исключает возможность их пересчета с
использованием принятых в настоящем издании термодинамических функций Р3О6 и РО2. Авторы-
работы [3476] приводят значение Дг#°@) = 753±20 кДж-моль для этой реакпии, вычисленное
по уравнению III закона. Учитывая различие термодинамических функций, принятых в настоящем
справочнике и в работе [3476], было найдено значение Дг/7°@)=723+20 кДж-моль, на
основании которого вычислено принятое значение энтальпии образования Р3Ов(г). ;
Р4Ое (г). Термодинамические свойства газообразного гексаоксида тетрафосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 204.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. Молекула Р4Ов в основном электронном состоянии Х1А1 имеет тетраэдрическую
структуру (точечная группа Td), характеризующуюся структурными параметрами г (Р—О)=1,638 +
±0,003 А; -? Р—О—Р=126,4±0,7°; -? О—Р—0=99,8+0,8°, определенными Бигли и др. [5311
в электронографическом исследовании. Близкие, но менее точные значения были получены
ранее в работах [2547, 1817]. Приведенное в табл. 14.6 значение IaIbIc вычислено по этим
параметрам, его погрешность равна 3-10"15 г3-см6. Основные частоты Р4О6 определялись по
колебательным спектрам [1602, 536, 939, 233] и расчетным путем по силовым постоянным [4060, 939].
В таблице для частот v15 v2, v7, v9 и v10 молекулы Р4О„ приняты значения, определенные Битти
[536] по спектру КР газообразного Р4Ое, для частот v3 и v4 — значения, определенные Чапманом
[939] по спектру КР жидкого Р4Ов, и для частот v8 и ve — значения, вычисленные Чапманом [939}
по силовым постоянным. Погрешности принятых значений составляют от 1 до 15 см.
Термодинамические функции Р4Ое(г), приведенные в табл. 204, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений определяются неточностью принятых
молекулярных постоянных и приближенным характером расчета. Погрешности значений Ф°(Т) при
298,15; 3000 и 6000 К составляют 2, 15 и 25 Дж-К~1-моль, в том числе из-за неточности
постоянных 1—3 Дж-К^-моль.
Ранее таблицы термодинамических функций Р4Ов(г) вычислялись в справочниках [87, 88, 2095 }
и в работе [1068] (до 1500 К) на основании неверных значений основных частот и менее точных
структурных параметров. Вследствие этого вычисленные ранее термодинамические функции
Р4О„(г) существенно меньше приведенных в табл. 204. Расхождения между значениями Ф°(Г),
приведенными в табл. 204 и в справочниках [88, 2095], составляют 5—11 и 5—13 Дж.К~1-моль~1
соответственно.
Константа равновесия реакции Р4О6(г)=4 Р(г)+6О(г) вычислена с использованием значения
Дг#°@)=4326,261 +40 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
Д/Я°(Р40в, г, 298,15К) =—1606 + 40 кДж ¦ моль.
Принятая величина является суммой энтальпии образования Р4Ов в жидком состоянии и е&
энтальпии испарения. Энтальпия образования Р4О„ в кристаллическом состоянии (—1668+
+ 40 кДж-моль) была измерена Хартли и Мак-Каубри [1854] методом ее сожжения до Р4О10(к).
Температура плавления Р4О6 составляет 297,1 К [1854]. Энтальпии плавления и испарения равны
соответственно 14,1 кДж-моль (Келли [2216]) и 48,0 кДж-моль (Фарр [1440]). Принятое
значение подтверждается результатами масс-спектрометрических исследований равновесия реакций
с участием Р4О6(г), выполненных Сму и Дровартом [3477] (—1665±60 кДж-моль), и
потенциалов появления, выполненных Муэновым и др. [2732] (—1759+70 кДж-моль с учетом
избыточной кинетической энергии продуктов диссоциативной ионизации). Энтальпия образования Р4Ов(к)
360
Фосфор и его соединения Р4О7 (г), Р4О8 (г)
была также измерена Бертло [617], Отье [2884] и Кернером и Даниэлсом [2275]. Методики
измерения в работах [617] и [2884] не указаны. Кернер и Даниэле измеряли энтальпию сгорания
фосфора в окиси азота, однако сложность состава продуктов сгорания и неточность его определения
не позволили найти достоверное значение энтальпии образования Р40в(к).
Р4О7 (г). Термодинамические свойства газообразного гептаоксида тетрафосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 205.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.6. Бигли и др. [531 ] на основании сравнения данных о структуре молекул Р4О„,
Р4О8, Р4О9 и Р4О10 нашли, что основу структуры Р4О, составляет несколько искаженный тетра-
эдрический каркас Р40в, один из атомов фосфора которого соединен двойной связью с атомом
кислорода. Согласно данным [531 ] молекула Р4О7 характеризуется структурными параметрами
г (Р'-О')=1,637 А; г (Р'-О")=1,67 А; г (Р"-О")=1,59 А; г (Р"-О'")=1,424 А, где Р' и Р"—
трехвалентные и пятивалентные атомы фосфора, О' и О" — атомы кислорода, связывающие
соответственно атомы Р' и Р' и атомы Р' и Р", О'" — атом кислорода, соединенный двойной связью
с атомом Р". Определенные в работе [531] значения длин связей Р'—О' и Р'—О" в Р4О, близки
к длине связи Р—О в Р4О„, равной согласно данным этой работы 1,638+0,003 А, а углы между
связями в Р4О7 близки к углам между соответствующими связями в Р4Ов и Р^Оц, и в пределах
+10° могут быть приняты равными 109°28'. Приведенное в табл. 14.6 значение IjJbIo вычислено
на основании тех же значений г (Р. . ,Р) и г (Р—О), что и для Р40в B,925 и 1,638 А), и значения
г (Р=О)=1,424 А, определенного в работе [531 ] для Р4О7; углы между связями в Р4О7 принимались
равными 109°28' (точечная группа симметрии С8,). Погрешность приведенного значения IJbIc
оценивается в 3-10~113 г3«смв. Сведения об основных частотах Р4О7 отсутствуют.
Термодинамические функции Р4Ог(г), приведенные в табл. 205, вычислены в приближении
«жесткий ротатор—гармонический осциллятор». При этом поступательные и вращательные
составляющие рассчитывались по уравнениям A.3)—A.10), A.128), A.130), а колебательные
составляющие — по приближенным соотношениям вида Фт1(Р4О7)=3^ Qm^iO^^lfivaJPfiw) через
колебательные составляющие Р4Ов и Р4О10, вычисленные по основным частотам, приведенным
в табл. 14.6. Погрешности табулированных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных и приближенным характером расчета. Погрешности
значений Ф°(Т) при 298,15; 3000 и 6000 К оцениваются в 5, 15 и 25 Дж-К^-моль.
Ранее Сму и Дроварт [3476] вычислили значения Ф°(Т) при 298,15—1400 К, а также
#"B98,15)— Й°@) и Н°A300)—Н°@) для Р4О7(г) посредством интерполирования между
соответствующими величинами для Р4О„(г) и Р4О10(г), приведенными в справочнике [2095], с учетом
различия в числах симметрии. Значения Ф°(Т), приведенные в табл. 205 и в работе [3476], при 298,15
и 1400 К отличаются на 4 и 8,5 Дж-К~1-моль~1.
Константа равновесия реакции Р4О7(г)=4Р(г)-1-7О(г) вычислена с использованием значения
Дг#°@)=4948,733 + 15 кДж.моль", соответствующего принятой в справочнике энтальпии
образования
Д/Я°(Р4О7, г, 0) = —1959 + 15 кДж • моль.
Принятая величина основана на масс-спектрометрическом измерении константы равновесия
реакции Р4О7(г)=.0,7 Р4О10(г)-|-0,3 Р4(г) при 1032 К, выполненном Сму и Дровартом [3476]. Эта
величина подтверждается также ыасс-спектроыетрическими измерениями констант равновесия
реакции Р4О„(г)-{-Р4О7(г)=2Р4О8(г), проведенными Муэновым и др. [2732], которые приводят
к значению энтальпии образования —1961 ±40 кДж-моль (расчет по методу II закона).
Р4О8 (г). Термодинамические свойства газообразного октаоксида тетрафосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 206.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. Структура молекулы Р4О8 в газовой фазе не изучалась. Кристаллический Р4О8
согласно рентгенографическому исследованию Йоста [2179] состоит из искаженных тетраэдров
Р4Ов, два атома фосфора которых соединены двойными связями с двумя атомами кислорода.
Значения структурных параметров молекулы Р4О8 были определены Бигли и др. [531 ] по данным
Йоста [2179]. Согласно этим расчетам в Р4О8 г (Р'—O')=l,633 A, r (P'—О")=1,644 и 1,672 А;
г(Р"-О")=1,556 и 1,596 А; г (Р"-О'")=1,596 А; г (Р"-О1Т)=1,414 А, где Р' и Р" - трех-
361
РА (г)
Глава 14
валентные и пятивалентные атомы фосфора, О', О", О'" — атомы кислорода, связывающие
соответственно Р' и Р', Р' и Р", Р" и Р", OIV — атомы кислорода, соединенные двойными связями
с атомами Р". Определенные в работе [531] длины связей Р'—О', Р'—О", Р"—О' и Р"—О'"
близки к длине связи Р—О в молекуле Р4О10, а значение г (Р"—OIY) близко к длине связи Р=О
в этой молекуле (см. ниже). Углы между связями в молекуле Р4О8 согласно данным работ [531,
530] близки (в пределах +10°) к 109°28'. Поэтому приведенное в табл. 14.6 значение IaIbIg
молекулы Р4О8 рассчитано на основании тех же значений междуядерных расстояний, что в
молекуле Р4О10, и в предположении, что углы между всеми связями равны 109°28' (точечная группа
симметрии С2г?). Погрешность принятого значения IaIbIg оценивается в 1 -10~т г3-см6. Спектры
молекулы Р4О8 не изучены и частоты ее колебаний не определялись.
Термодинамические функции Р4О8(г), приведенные в табл. 206, вычислены в приближении
«жесткий ротатор—гармонический осциллятор». Поступательные и вращательные составляющие
были рассчитаны по уравнениям A.3)—A.10), A.128), A.130), а колебательные составляющие —
по приближенным соотношениям вида Фкох("Р^О^==11г ^aa(^'i0e)-{-1/2 Фтл(Р^ю) через
колебательные составляющие Р4Ов и Р4О10. вычисленные по основным частотам, приведенным в табл. 14.6.
Погрешности табулированных термодинамических функций обусловлены приближенным
характером расчета и неточностью принятых постоянных. Погрешности значений Ф°(Т) при 298,15;
-3000 и 6000 К могут достигать 10, 20 и 30 Дж-К^-моль-1.
Ранее таблицы термодинамических функций Р4О8(г) вычислялись в первом и втором изданиях
справочника [87, 88] тем же методом, что и в настоящем издании. Различия соответствующих
значений термодинамических функций Р4О8(г), приведенных в табл. 206 и в предыдущем издании
справочника, составляют 5—10 Дж-К~1-моль~1 в значениях Ф°(Т). Сму и Дроварт [3476]
вычислили для Р4О8(г) значения Ф°(Т) при 298,15—1400 К посредством интерполирования между
значениями термодинамических функций газообразных Р4Ов и Р4О1в из справочника [2095 ] с
учетом различия в числах симметрии. Расхождения в значениях Ф°(Т), приведенных в табл. 206
и в работе [3476], составляют 2,2 и 6 Дж-К'^моль при 298,15 и 1400 К.
Константа равновесия реакции Р4О8(г)=4Р(г)+8О(г) вычислена с использованием значения
)=5510,516+16 кДж-моль, соответствующего принятой в справочнике энтальпии обра-
A^^Og, г, 0) = —2274 ± 16 кДж • моль-1.
зования
Эта величина основана на результатах исследования равновесия реакций с участием Р4О8(г),
представленных в табл. 14.10. Наиболее точное значение энтальпии образования Р4О8(г)
получено по данным Сму и Дроварта [3476], которые провели масс-спектрометрическое исследование
паров над смесями МоР с СоО и FeaO3. Данные работы [1407 ] менее точны, так как ее авторы не
располагали достаточно точным методом определения состава продуктов исследовавшихся реакций
(см. [3661]). Расчет Д/Я°(Р4О8, г) по масс-спектрометрическим измерениям в работе [2732] привел
к близкому, но менее точному значению.
Таблица 14.10. Результаты определения энтальпии образования Р4О8 (г) (в кД;к • моль х)
Авторы
Метод
Д/Я° (Р40„ г, 0)
Эмметт, Шульц, 1939 Исследование равновесия реакции Р4 (гL-8СО2 (г)=8СО (г)+
[1407] +Р4О8(г) в проточной системе, 1273 К, 1 точка
Исследование равновесия реакции Р4О8(г)+2СО2(г)=
=Р4О10 (г)+2СО (г) в проточной системе, 1271—1292 К,
68 точек
Исследование равновесия реакции Р4О10(г)-|-2СО(г)=
=Р4О8(г)+2СО2(г) в проточной системе, 1278—1288 К,
11 точек
Муэнов и др., 1970, Масс-спектрометрическое исследование равновесия реак-
J2732] ции Р4О8(г)+Р4О10(г)=2Р4О9(г), Гор=529 и 678 К
Сму, Дроварт, 1973 Масс-спектрометрическое исследование равновесия реак-
[3476] ции Р4О8(г)=0,8 Р4О10(г)+0,2 Р4(г), 1032 К, 1 точка
—2287 (III)
—2305 ±6 (III)
—2295 ±10 (III)
—2278 ±35 (II)
—2274 ±16 (III)
362
Фосфор и его соединения РА (г)) Р*О10 (к? ж)
Р4О9 (г). Термодинамические свойства газообразного ноноксида тетрафосфора в стандартном
¦состоянии при температурах 100—6000 К приведены в табл. 207.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.6. Структура молекулы Р4О9 в газовой фазе не изучалась. Рентгенографическое
исследование кристаллического Р4О9, выполненное Постом [2178], показало, что это вещество
образовано из молекул Р4О9, имеющих структуру искаженного тетраэдрического каркаса Р4Ов, три
атома фосфора которого соединены двойными связями с тремя атомами кислорода (симметрия С3е
или С3). Структурные параметры молекулы Р4О9 в Р4О8(к) были определены Бигли и др. [530]
по данным Поста [2178] и найдены равными г (Р'—О")=1,657+0,013 А; г (Р"—О')=1,599 +
±0,017 А; г (Р"-О") = 1,588±0,012 А или 1,584+0,012А и г(Р"-О"')=М15±0,014А, где
Р' и Р" — трехвалентные и пятивалентные атомы фосфора, О', О" — атомы кислорода, связывающие
Р" и Р", Р' и Р", О'"—атомы кислорода, соединенные двойными связями с атомами Р". Значения
г (Р'—О"), г(Р"—О') и т- (Р"—О") оказались близки к длине связи Р—О в молекуле Р4О10, а
значение г (Р"—О'") близко к длине связи Р=О в той же молекуле (см. ниже). Углы между
соответствующими связями в молекулах Р4О9 и Р4О10 согласно данным [5301 равны 109°28'+10°. Поэтому
приведенное в табл. 14.6 значение IaIbIo молекулы Р4О9 было рассчитано на основании значений
длин связей Р—О и Р=О в молекуле Р4О10 и в предположении, что углы между всеми связями
равны 109°28'. Погрешность рассчитанного значения IaIbIc оценивается в 1 -10~112 г3 -см6. Спектры
молекулы Р4О9 не изучались и частоты ее колебаний не определялись.
Термодинамические функции Р4О9(г), приведенные в табл. 207, вычислены в приближении
«жесткий ротатор—гармонический осциллятор». При этом поступательные и вращательные
составляющие находились по уравнениям A.3)—A.10), A.128), A.130), а колебательные
составляющие—по приближенным уравнениям вида Фкол(Р4О9)=0,25 ^KM(P4Oe)+0,75-Фкы(Р4О10) через
колебательные составляющие Р4Ов и Р4О10, вычисленные по основным частотам, приведенным
в табл. 14.6. Погрешности табулированных термодинамических функций обусловлены неточностью
принятых молекулярных постоянных и приближенным характером расчета. Погрешности
значений Ф°(Т) при 298,15; 3000 и 6000 К оцениваются в 10, 20 и 30 Дж -К -моль.
Ранее Сму и Дроварт [3477] рассчитали Ф°(Т) при 298,15-1400 К, а также Я°B98,15К)—
—#°@) и#°A300 К)—#°@) для Р4О9(г) интерполированием между соответствующими значениями
термодинамических функций Р4Ов(г) и Р40ю(г), приведенными в таблицах [20951, с учетом
различия в числах симметрии. Значения Ф°(Г), приведенные в табл. 207 и в работе [3477], согласуются
в пределах 2 Дж -К -моль.
Константа равновесия реакции Р4О9(г)=4Р(г)+9О(г) вычислена с использованием значения
АгН°@)=6066,299 +17 кДж -моль, соответствующего принятой энтальпии образования
ДД° (Р4О9, г, 0) = —2583 + 17 кДж ¦ моль.
Эта величина основана на значении энтальпии D+16 кДж-моль) реакции Р4О9(г) =
=0,9 Р4О10(г)+0,1 Р4(г), рассчитанной по уравнению III закона из константы равновесия при
1032 К, найденной Сму и Дровартом [3476] масс-спектрометрическим методом. Менее точное
значение —2444+100 кДж-моль соответствует потенциалу появления Р4О^ из Р4010 B0,0+0,5 эВ)
и потенциалу ионизации Р4О9 A3,0+0,5 эВ), найденным Муэновым и др. [2732].
Р4О10 (к, ж). Термодинамические свойства кристаллического и жидкого декаоксида
тетрафосфора в стандартном состоянии при температурах 100—1000 К приведены в табл. 208.
Значения постоянных, принятых для расчета термодинамических функций, приведены в табл.
14.1. В настоящем издании за стандартное состояние Р4О10(к) принимается гексагональная
модификация Р4О10 \ а при Т > 699 К — жидкость, образующаяся при плавлении Р4О10 (к, гекс.) 2.
1 Гексагональная модификация Р4О10 (к) образуется при непосредственном окислении фосфора и согласно данным
Хилла и др. [1941] практически стабильна до 600—630 К. Термодинамически более стабильными, по крайней
мере при высоких температурах, являются ромбическая и тетрагональная модификации, в которые при
температуре около 650 К превращается гексагональная модификация [1941]. Области стабильности этих модификаций
Р4О10 (к) точно не установлены. Р4О10 известен также в аморфном состоянии.
2 При плавлении Р4О10 (к, гекс.) образуется, по-видимому, мономолекулярная жидкость. При плавлении ромби.
¦ческой и тетрагональной Р4О10 (к) образуются две полимерные формы жидкой Р4О10 (см. [3474,1969, 3472, 3471
3473]).
363
Р4О10(к, ж) Глава 14
Этот выбор обусловлен тем, что Р4О10 (к, гекс.) является наиболее распространенной и изученной
модификацией Р4О10, а для ромбической и тетрагональной модификаций Р4О10 отсутствуют какие-
либо термодинамические данные (включая энтальпии образования, температуры и энтальпии
равновесных превращений и др.). Следует отметить, что Р4О10(ж), для которой в табл. 208 приведены
термодинамические функции, является термодинамически неустойчивой и легко переходит в
полимерные формы, обладающие на 3—4 порядка меньшим давлением пара (по сравнению с давлением
пара мономерной жидкости Р4О10). При температурах Т < 298,15 К термодинамические функции
Р4О10 (к, гекс.) вычислены по значениям теплоемкости, измеренным Сталлом и др. [3563] в
интервале 15—298 К на образце, содержащем 99,2% Р4О10. Экстраполяция теплоемкости к Т=0 no-
уравнению Дебая— Эйнштейна приводит к S°A5 К)=4,9 Дж -К -моль в согласии с данными
работы [3563]. Погрешности принятых по работе [3563] значений 5°B98,15 К) и Н° B98,15 К) —
—Н°@) (см. табл. 14.1) оцениваются в 1,6 Дж -К -моль и 0,1 кДж -моль соответственно.
Теплоемкость Р4О10 (к, гекс.) измерена также Андоном и др. [388] A2—324 К); полученные в этой работе-
значения теплоемкости на 0,5—1% ниже найденных Сталлом и др. [3563], что, по-видимому,
обусловлено неточностью калибровки калориметра в работе [388]. При температурах
318—608 К Франдсеном [1527] были проведены измерения энтальпии образцов Р4О10 (к), фазовое-
состояние которых не было охарактеризовано. Кроме того, результаты калориметрических
измерений [1527] не могут считаться надежными, как об этом свидетельствуют существенные
отличия величины Д„Я° (Р4ОА0, к, гекс), полученной автором этой работы, от вычисленных по
результатам измерений давления пара (см. ниже). Для теплоемкости Р4010 (к, гекс.) при
Т > 298,15 К в справочнике принято уравнение, приведенное в табл. 14.1, которое было
получено на основании следующих условий: 1) согласования с данными Сталла и др. [3563] при Т ^
^ 298,15 К; 2) идентичности значений b.fla (P4O10, к, гекс), вычисленных по II и III законам
по данным для давления пара Р4О10 (к, гекс); 3) соответствия характера температурной
зависимости Су(Р4,О10, к, гекс.) измерениям Франдсена [1527] энтальпии Р4О10(к). Температура
плавления 699 +10 К выбрана на основании значений, найденных экспериментально в работах
[3475] G03 К) и [1941] F95 К), и значения 699 К, определенного по пересчету кривых давления
пара Р4О10 (к, гекс.) [1969, 3473, 3487, 143] и Р4О10(ж) [347]. Энтальпия плавления Р4О10 (к, гекс.)
B1 +4 кДж моль'1) принята как разность энтальпий сублимации и испарения, рассчитанных
по указанным выше данным. Значение теплоемкости жидкого Р4О10 принято по оценке,
основанной на сравнении данных по теплоемкости Р4О10 и V2O5: С°(Р4О10, ж)=С°(Р4О10, к, гекс,
Tm)-\-2kC°p(VzOb, Тт), где ДС°(У2О5, Тт) — разность теплоемкостей кристаллической и жидкой
V2O5 в точке плавления, равная согласно данным работы [238а] 9 Дж К моль.
Погрешности, приведенных в табл. 208, значений Ф°(Т) оцениваются в 0,6; 3 и 10 ДжК моль
при 298,15; 500 и 1000 К соответственно.
Ранее таблицы термодинамических функций Р4О10(к) вычислялись в работах [388, 3563] до
3000 К и до 1500 К в справочнике JANAF [2095]. Таблица термодинамических функций,
приведенная в справочнике [2095], при Т ^ 298,15 К основана на данных работы [388], а при Т ^
^ 298,15 К — на графической экстраполяции значений С°(Т), определенных в этой работе.
Расхождение в значениях Ф°G00 К), приведенных в табл. 208 и справочнике [2095], составляет 2 ДжХ
X К моль.
В справочнике принята энтальпия образования
AfH (Р4010, к, гекс, 298, 15К) = — 3010,1 + 3,2 кДж • моль.
Это значение основано на проведенном при подготовке настоящего справочника согласовании
методом наименьших квадратов [3588, 294] результатов измерений энтальпии сгорания белого
фосфора в кислороде и в хлоре, энтальпии гидролиза РС16(к) и Р4О10(к) и энтальпии сгорания
белого фосфора в присутствии воды. Наиболее точное значение энтальпии сгорания белого фосфора
в кислороде Д^Я°(Р4О10, к, гекс, 298,15 К) = —3010,5+2,0 кДж-моль было получено Иганом
и Луффом [1390]. Менее надежные значения этим методом были найдены в работах Холмса [1988}
(—2984,0+4,2 кДж-моль), Кернера и Даниэлса [2275] (—3001+17 кДж-моль), Рота и др.
[3259а] (—3097 кДж-моль) и Жирана [1663] (—3015+17 кДж-моль, см. [3360]). Наиболее
точные измерения энтальпии сгорания белого фосфора в хлоре были проведены Шаммом и др.
[3360] (см. ниже), а энтальпии гидролиза РС15 — Бирли и Скиннером [655], результаты более
ранних измерений [623, 36761 значительно менее надежны (см. [642, 3360]). Херд и Льюис [18681
364
Фосфор и его соединения
P4OJ0 (к, ж>
измерили энтальпию сгорания белого фосфора в кислороде в присутствии воды. Энтальпия
растворения Р4О10(к) в воде была измерена Ирвингом и Мак Кэроллом [2071] и Холмсом [1988].
Результаты перечисленных выше работ согласуются между собою не вполне удовлетворительно.
Рекомендованное значение основано в первую очередь на результатах измерений энтальпий сгорания
фосфора [1390], энтальпии растворения Р4О10 [2071, 1988], а также энтальпии образования [3360]
и гидролиза [655] пятихлористого фосфора. При оценке степени надежности результатов
экспериментальных измерений, а также при выборе рекомендуемых значений были учтены результаты
аналогичного анализа, проведенного Эвансом [1425а] и Хердом [1867а]. В результате проведенного
согласования, помимо приведенного выше значения Д^/Г" (Р4010, к), было выбрано также значение
Д,Яо(Н,Р04, р-р, 100Н2О, 298,15 К)=-1296,2+1,0 кДж моль,
которое было использовано при вычислении энтальпии образования РС15(г) и РОС13(г).
Давление пара Р4О10(г) вычислено с использованием принятой в справочнике энтальпии
сублимации
ДД°(Р4010, к, гекс, 0) = 107 + 5 кДж • моль.
Эта величина является средневзвешенной из приведенных в табл. 14.11 значений (за
исключением данных работы [3474]), рассчитанных по уравнению III закона. Принималось, что пары
содержат только молекулы Р4О10(г), так как в работах [3473, 3697, 788, 1858, 2732] было показано,
Таблица 14.11. Результаты определения АХЯ° (Р401в, к, гекс, 0) по данным о давлении пара
(в кДж-моль)
Авторы
Смите, Ратгерс,
[3474]
Хафлейк, Шеффер
[1969]
Смите, Дейнум,
[3472]
Саузард, Нелсон,
[3487]
Смите и др. 1938
Кукушкина и др.
[143]
1924
,1926
1930
1937
[3473]
, 1974
Статический
метод,
650 К, 10 точек
Статический
изотеннскопа
метод,
, 533-
То же, 543—635 К,
Статический
таты опытов
18 точек
Статический
метод,
Метод
мембранный
мембранный
-674 К, 40 т
20 точек
мембранный
с двумя основными
метод,
634 К, 30 точек
Статический
9 точек
Статический
9 точек
Статический
23 точки
метод
метод,
метод,
мембранный
кварцевый
стеклянный
нуль-манометр, 569—
нуль-манометр и метод
очек
нуль-манометр (резуль-
образцами), 561—676 К,
нуль-манометр, 489—
манометр, 533—630 К,
манометр, 533—630 К,
кварцевый нуль-манометр, 503—628 К,
Д,Я»@)
108,7 ±9,8
105,4 ±0,6
110,5 ±0,7
106,8 ±0,08
108,4 ±1,2
106,96 ±0,06
110,6 ±3,7
106,9 ±0,2
107,5 ±0,8
106,95 ±0,06
107,9 ±0,5
106,81 ±0,07
110,9 ±0,9
107,1 ±0,2
107,6±1,7
106,76 ±0,1
(И)
(III)
(И)
(III)
(II)
(III)
(И)
(III)
(И)
(III)
(И)
(III)
(II)
(III)
(II)
(III)
•что присутствие других молекул связано только с примесями' в исходных образцах. Влияние
примесей и взаимодействия пара с материалом мембраны обсуждается в работе Кукушкиной и
др. [143]. Как показал Хилл и др. [1941], при температурах выше 630 К с заметной скоростью
происходит превращение гексагональной Р4О10 в ромбическую. Поэтому при обработке измерений,
выполненных в некоторых работах, результаты, полученные при более высоких температурах,
ле принимались во внимание. Приведенные в таблице погрешности отражают только
воспроизводимость измерений. Основной вклад в погрешность принятого значения вносит неточность
термодинамических функций.
Менее точные значения энтальпии сублимации были получены в калориметрическом
измерении энтальпии растворения Р4О10(к) и Р4О10(г) в воде [1527] и масс-спектрометрическим методом
'[1858, 27321. Из этих измерений только измерения Муэнова и др. [2732] при 313—423 К привели
sK величине 105,5 +5,0 кДж -моль, удовлетворительно согласующейся с принятой в справочнике.
365
PjOieW, PH(r) Глава 14
Р4О10 (г). Термодинамические свойства газообразного декаоксида тетрафосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 209.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.6. Молекула Р4О10 в основном электронном состоянии Х1А1 имеет тетраэдриче-
скую структуру (точечная группа симметрии Td), характеризующуюся структурными
параметрами г (Р—О)=1,604±0,003 А, г (Р=О) = 1,429±0,004 А, -? О-Р-О = 101,6±0,8°, ^О-Р=О =
=116,5+0,3°, найденными в электронографическом исследовании Бигли и др. [530].
Близкие, но менее точные значения были получены в работах [1817, 4]. Приведенное в табл. 14.6
значение IaIbIc рассчитано по этим структурным параметрам, его погрешность составляет 3 -1013 г3 -см6.
Колебательные спектры паров Р4О10 исследовались в работах [536, 3288 *¦] и кристаллического
гексагонального Р4О10 — в работах [1594, 939, 233, 1150]. Приведенные в таблице значения
основных частот Р4О10 приняты по следующим данным: частоты v1? v2, v3, v4, v10 и vu определены
Битти и др. [536] по спектру КР пара Р4О10, частоты v6, v6, v7, v9 рассчитаны по силовым
постоянным, полученным в работах [939, 4060, 174] 2, значение частоты v8 оценено при подготовке
справочника на основании частот однотипных колебаний молекул Р4О10 (v4, v8, v10) и Р4О6 (v3, ve, vg),
частоты v12, v13, v14, v15 определены в работах [233, 939] по колебательным спектрам Р4О10 (к, гекс).
Наименее достоверны значения частот vB, v6, v7, v9, погрешности которых составляют 30—50 см^
Погрешности остальных частот оцениваются в 2—10 см.
Термодинамические функции Р4010(г), приведенные в табл. 209, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности табулированных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным методом расчета. Погрешности значений Ф°(Т)
при 298,15, 3000 и 6000 К могут достигать 5, 20 и 35 Дж -К -моль, в том числе из-за неточности;
молекулярных постоянных 2—5 Дж -К -моль.
Ранее таблицы термодинамических функций Р4О10(г) вычислялись в справочниках [87, 88,
2095] и в работах [3707,1068] (Т ^ 2000 К). Различия в значениях термодинамических функций
Р4О10(г), приведенных в этих работах и в табл. 209, обусловлены главным образом различиями
в принятых значениях основных частот. Термодинамические функции Р4О10(г), приведенные
в справочнике JANAF [2095], согласуются в значениях Ф°(Т) в пределах 2,5-Дж-К-моль
с данными табл. 209.
Расхождения с данными справочника [88] составляют 4—6 Дж-К-моль.
Константа равновесия реакции Р4О10(г)=4Р(г)+Ю О(г) вычислена с использованием
значения Дг#°@)=6602,552 ±5,9 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации Р4О10 (к, гекс). Им соответствует также
Д/Я°(Р4О10, г, 0) = —2872,47 ± 5,1 кДж • моль.
РН (г). Термодинамические свойства газообразного гидрида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 210.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.12. Как показывают теоретические расчеты [2174, 862, 860, 2446], спектральные
исследования [1606, 2978, 2074, 2075, 2560, 2378, 460, 2967, 1753, 3257, 1176] и изучение спектра
лазерного магнитного резонанса РН [1176], молекула РН имеет основное электронное состояние Х32
и четыре стабильных валентных электронных состояния с энергиями возбуждения до 50 000 см:
а1^, Ь1!!4", А3Т1, сЧ1. По данным работы [460] известны два ридберговских электронных
состояния CП и !Ф) с энергиями возбуждения, превосходящими 60 000 см. Приведенные
в табл. 14.12 значения молекулярных постоянных РН в состояниях Х32~ и А3Н
определены Ростосом и др. [3257] на основании анализа вращательной структуры полос системы
ASU—Х32~ РН (г/=0; v" <^ 1) и PD (i/, v" <^ 1). Энергия возбуждения состояния а1 Д,
приведенная в таблице, определена Циттелем и Линебергером [4065 ] с погрешностью 80 см при
изучении фотоионизации РН". Приведенные в табл. 14.12 значения вращательных постоянных РН
1 Данные, полученные в работе [3288], остались не известными авторам настоящего издания.
2 Принятое значение частоты v6 согласуется с наблюдаемыми в спектре КР Р4О10 (к, гекс.) полосами при 329 [1594]
и при 330 см [939].
366
Фосфор и его соединения
РН(г>
Т аб л ив
Молекула
РН
PF
РС1
PS
PN
а 14.12. Молекулярные
Состояние
x3s-
охД
Ъ1^
АШ
сШ
хъ-
б1!*
А'Щ е
#41
x3s-
ахД
Ь1^
А3П
gm
Х2П
аЧ1
42П
В2Ф
Ь42~
С2Д
Я2 S-, ЯЩ
/2S+
X1S+.
а32+
Ь3П
с3П
Те
0а
7 660
15 200
29 484 б
37 500
0а
7 090,43
13 353,91
29 481,80 ж
35 808,54 ж
0а
6 500Д
12 084,6
25 914,3
31 000 Д
45 000 Д
0а
20 000 Д
22 894,0
28 000 Д
29 000 Д
31 000Д
34 000 Д
34 687,9
0
30 000 а
35 000 а
39 805,70
46 000 а
Примечание. Ниже все постоянные
РН: а
PF: a
PCI: a
PS: a
PN: a
Хо=2,212, [io=—0,0738; ° ,
Х„=2,9623, [io=O,OO18; б «
дено значение
значение Дби
постоянные РЕ
2 365,2
—
2 030,6
—
846,75
858,79
866,14
436 8
413 3
522,8
—
584,22
299,6
—
739,1
512,2
__
—
534,8
1 337,24
—
1 103,09
—
даны в см.
1=—115,71; в
, PF, PC1,
«Л
PS, PN
СМ
44,5
—
—
98,5
—
4,489 б
4,438 г
4,51
—
—
2,24
2,67
3,17
—
2,96
2,15
_
—
3,31
6,983
—
—
7,222
—
Р,=0,8.10-*.
ег/(,=0,019; в вычислено по
Do; е То D3П0)=29 338,68 и
Ве
8,537
8,5
8,259
—
0,566 5
0,569 9
0,572 5
0,466 3
0,484 8
0,25 б
—
—
0,297 3 б
_
_
—
0,264 4
0,786 21
—
0,730 71
—
<*! • 102
25,1
12
47,3
—
0,456
0,467
0,45
0,38
0,62
0,14 в
—
—
0,16 в
0,126
0,557
0,663
—
соотношению A.68);
Т„ (Л3П2)=29 623 06; »
Х0=7,0 (оценка); *> вычислено через re; B оценено по соотношению
4=321,93; 6
дено значение
Оценка.
вычислено на <
Do', Д оценка
к приведено .
D.-10»
436
418
525 в
—
1,0 в
1,0 Д
1,1 Д
—
2,6 Д
0,2 г
—
—
0,2 г
—
0,25 в
1,09
—
1,29
—
г аеУе—0,0147
значение Т„; а
A.69); г оценено по A.68);
Ге
А
1,422 1
1,43
1,445 8
—
1,589 6
1,585
1,581
1,752
1,721
2,01 Д
—
2,27
—
1,900 7
—
2,013
1,486 9
—
1,542 4
—
Д приве-
приведено
Д оценка.
основании значения В0=0,2Ш5; в вычислено по соотношению A.69); г приве-
в состоянии а^Д определены Бальфуром и Дугласом [460] в результате анализа вращательной
структуры полос 0—0, 1—1 и 2—2 системы гФ—^Д. Для состояний ЬХ2 + и с1!! в таблице
приведены значения энергий возбуждения, полученные в теоретических расчетах [2174, 2446, 3257].
Их погрешности могут достигать 1000 см.
Термодинамические функции РН (г), приведенные в табл. 210, вычислены по уравнениям A.3)—
A.10), A.93)—A.95). Значения (?„„ и ее производных находились по уравнениям A.90)—A.92)
с учетом четырех возбужденных электронных состояний и в предположении, что <?<?> вр =
= (PilPx) <?S. вр- Статистическая сумма по колебательно-вращательным уровням энергии
состояния Х32~ и ее производные рассчитывались непосредственным суммированием по уравнениям
367
РН2 (г) Глава 14
A.73)—A.75). В расчете учитывались все уровни энергии, ограниченные предельной кривой
диссоциации состояния Х32~. Уровни энергии состояния Х32~ вычислялись по уравнениям A.65),
A.40), A.62) со значениями коэффициентов Yik, приведенными в табл. 14.4 и полученными из
постоянных, приведенных в табл. 14.12. Коэффициенты а(, приведенные в табл. 14.4, определяют
предельную кривую диссоциации РН в состоянии Х32~. Погрешности вычисленных
термодинамических функций при низких температурах обусловлены преимущественно неточностью
фундаментальных постоянных и постоянных мультиплетного расщепления, а при высоких —
неточностью постоянных, принятых для аппроксимации уровней колебательно-вращательной энергии
и предельной кривой диссоциации состояния Х32~, а также неточностью энергий возбуждения
состояний ахД и 6ХЕ+. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 0,03,
0,1 и 0,5 Дж-К^-моль.
Ранее таблицы термодинамических функций РН(г) вычислялись в справочниках [87, 88, 2095,
2556] на основании использования менее точных значений молекулярных постоянных.
Расхождения данных табл. 210 и справочников [88, 2095, 2556] достигают 0,4, 0,2 и 1,8 Дж-К^-моль
в значениях Ф°(Т) соответственно.
Константа равновесия реакции РН(г)=Р(г)-}-Н(г) вычислена с использованием принятой в
справочнике энергии диссоциации
Do (РН) = 300 + 13 кДж • моль ~ 25 000 ± 1100 см,
оцененной по верхней C14 кДж-моль) и нижней B88 кДж'Моль) границам ее возможных
значений.
Верхняя граница была найдена по соотношению D0(PH, Х32~) < 0,5 D0(PH2), принятому
в связи с тем, что гв(РН, Х32~)=1,422 А >го(РН2, Ха51)=1,418 А. Нижняя граница определена
равной 0,8 от величины, найденной Ростосом и др. [32571 линейной экстраполяцией постоянных
состояния Х32~. Принятому значению D0(PH) соответствует
Д/Я°(РН, г, 0) = 231,597 ± 13 кДж.моль.
РН2 (г). Термодинамические свойства газообразного дигидрида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 211.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.8. Как показали исследования электронного спектра [3135, 2560, 1754, 2966, 1755,
2965, 1269, 1268, 626], спектра лазерного магнитного резонанса [1177] и теоретические расчеты
[2174, 2242,3579,2027], молекула РН2 в основном электронном состоянии Х2ВХ и в состоянии
А2АХ имеет угловую симметричную структуру. Структурные параметры РН2 в состояниях %2ВХ
(го(Р-Н)=1,418А; ^ Н-Р-Н=9Г42') и А*АХ (го(Р-Н)=1,389 A; J$ Н-Р-Н=123°12')
определены Берту и др. [626] на основании анализа вращательной структуры полос системы
А2АХ—%2ВХ. По найденным в работе [626] вращательным постоянным в состоянии ЁгВх D000=
=9,1332+0,0018; Я000=8,0820 ±0,0019, С000=4,2134 +0,0012 см) вычислено приведенное в табл.
14.8 значение IaIbIc- Приведенное в таблице значение частоты v2 в состоянии Ё2ВХ вычислено
по постоянным @^=1107,26 см ж х?22=—5,24 см, определенным Паска и др. [2965] по началам
полос @, 0, 0)—@; v2=0, 2, 3; 0) системы А2АХ—Х2ВХ. Для частот vx и v3 приняты значения,
основанные на сравнении основных частот РН2, вычисленных по силовым постоянным РН3 [1188],
и известного по спектральным данным [2965] значения v2 Ч Погрешности принятых в табл. 14.8
значений основных частот РН2 оцениваются в 0,5 (v2) и 30 см (vx, v3).
Термодинамические функции РН2(г), приведенные в табл. 211, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130) с учетом возбужденного электронного состояния А2АХ по уравнениям A.168)—
A.170). Погрешности вычисленных термодинамических функций обусловлены неточностью
молекулярных постоянных, неполнотой данных о возбужденных состояниях РН2 и приближенным
характером расчета. Погрешности значений Ф°(Т) при 298,15; 3000 и 6000 К составляют 0,05;
1 и 2 Дж -К -моль; при этом погрешности, обусловленные неточностью постоянных в состоянии
Х2ВХ, не превышают 0,5 Дж -К -моль.
1 Циттель и Линебергер [4065] по спектру фотоионизации РН2 нашли vt (PHa)=2270;|;80 см.
368
Фосфор и его соединения РН2 (г), НРО (г)
Ранее термодинамические функции РН2(г) вычислялись в таблицах JANAF [2095] по
оцененным значениям молекулярных постоянных. Различия в термодинамических функциях РН2(г),
приведенных в [2095] и в табл. 211, составляют от 0,05 до 0,5 Дж -К-моль в значениях Ф°(Г).
Константа равновесия реакции РН2(г)=Р(г)+2Н(г) вычислена с использованием значения
Дг#°@)=624,291+6,2 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д Д° (РН2, г, 0) = 123,4 + 6,2 кДж • моль.
Эта величина основана на значении D0(H—РН2)=326+6 кДж-моль, найденном Дьюером и
Сетсером [1337], и значении Д/Я°(РН3, г, 0)=13,4 + 1,7 кДж-моль, рекомендованном в
справочнике [258] по калориметрическим данным Ганна и Грина [1765]. Дьюер и Сетсер [1337]
в спектре свечения, сопровождающего реакцию атомарного фтора с фосфином, нашли
колебательно-вращательные полосы HF, свидетельствующие о том, что реакция идет по уравнению F+
-f-PH3=HF4-PH2. При этом было установлено, что максимальная энергия молекул HF,
образующихся в этой реакции, составляет 330 кДж-моль и что D0(H—РН2) ^ 326 кДж-моль. Эта
величина подтверждается верхней границей D0(H—РН2), найденной методом электронного удара
C64+25 кДж-моль [3278], 351+5 кДж-моль [2554]) и электронного захвата C35+12 кДжХ
Хмоль-1 [1812, 1375]).
РН~ (г). Термодинамические свойства газа, состоящего из отрицательных ионов дигидрида
фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 212.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.8. Ион РН2 изоэлектронен молекуле H2S, в связи с чем в справочнике
принимается, что в электронном состоянии Х1А1 он имеет симметричную угловую структуру (симметрия
С2с). Приведенное в табл. 14.8 значение IaIbIc вычислено по структурным.параметрам РН2,
оцененным на основании принятых в справочнике параметров молекул H2S и РН2 равными г (Р~—
—Н)=1,34+0,05 А; ^ Н—Р~—Н=92+5°. Его погрешность оценивается в 2-10~119 г3-см6.
Приведенные в таблице значения основных частот РН2 оценены по принятым в справочнике
основным частотам H2S и РН2, их погрешности составляют до 50 см.
Термодинамические функции РН2(г), приведенные в табл. 212, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности табулированных термодинамических функций обусловлены
неточностью принятых молекулярных постоянных и приближенным характером расчета. Погрешности
значений Ф°(Г) при 298,15; 3000 и 6000 К могут достигать 1; 1,5 и 2 Дж-К~1.моль~1, при этом
погрешности, обусловленные неточностью принятых постоянных, не превышают 1 Дж-К~1-моль~1.
Ранее таблица термодинамических функций РЩ"(г) не публиковалась.
Константа равновесия реакции РН7(г)=Р(г)+2Н(г)+е~(г) вычислена с использованием
значения ДгЯ°@)=746,831+6,4 кДж-моль, соответствующего принятому в справочнике сродству
к электрону
40(РН2) = —1,271 +0,010 эВ = —122,6 + 1,0 кДж • моль.
Эта величина определена Циттелем и Линебергером [4065] по спектру электронов,
образующихся при фотоионизации, и согласуется со значением 1,25+0,03 эВ, найденным Смитом и др.
[3478, 3479] по порогу фотоотрыва электрона от иона РЩ. Существенно менее точные значения
А0(РН2) были получены в масс-спектрометрическом исследовании [1375, 1812], в исследованиях
с помощью магнетронной техники [2939] и техники циклотронного резонанса [1991], а также
в приближенных теоретических расчетах [1560].
Принятому в справочнике значению Л0(РН2) соответствует
ДД° (РН~, г,. 0) = 0,8 + 6,4 кДж • моль.
НРО (г). Термодинамические свойства газообразного оксигидрида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 213.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.8. Как показали исследования спектра испускания НРО, выполненные Лам Тханг
Маем и Пейроном [2347, 2348, 2349, 2350], молекула НРО имеет угловую несимметричную струк-
369
PF(r) Глава 14
туру (симметрия Cs) в основном ^А и возбужденном АгА электронных состояниях. Приведенное
в табл. 14.8 значение 1А1в1с в состоянии ХХА вычислено по вращательным постоянным Ао=
=8,8546+0,0010, 50=0,702 36+0,00030, С0=0,6488+0,0003 см~\ определенным в
работах 12349, 2350] на основании анализа вращательной структуры полосы @,0,0)—@,0,0) системы
ЛгА—Х1А. Ему соответствуют структурные параметры г (Р—Н)=1,433А, г (Р—О) = 1,512А,
-3t Н—Р—О=104°44'. Приведенные в таблице значения основных частот НРО в состоянии Ё.гА
определены в работах [2349, 2350] (v2 и v3) в результате исследования полос @,0,0)—@,1,0) и
@,0,0)—@,0,1) системы АгА—ХМ, частота vx оценена на основании соответствующих основных
частот РН и РН3. Погрешности приведенных значений оцениваются в 30 (va), 0,1 (v2) и 2 см (v3).
Термодинамические функции НРО (г), приведенные в табл. 213, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130) с учетом возбужденного электронного состояния А~ХА по уравнениям A.168) —
A.170). Погрешности вычисленных значений обусловлены неточностью принятых молекулярных
постоянных, неполнотой данных о возбужденных состояниях НРО и приближенным характером
расчета. Погрешности в значениях Ф°(Т) составляют 0,05; 1 и 3 Дж-К~1-моль~1 при 298,15; 3000
нбООО К, в том числе из-за неточности молекулярных постоянных в состоянии XхА не превышают
0,5 Дж-К~1-моль~1.
Ранее таблица термодинамических функций НРО (г) не публиковалась.
Константа равновесия реакции НРО(г) = Н(г)+Р(г)-|-О(г) вычислена с использованием
значения Дг//°@)=831,380+40 кДж-моль, соответствующего принятой в справочнике величине
энтальпии образования
Д7#° (НРО, г, 0) = —53 + 40 кДж • моль.
Эта величина вычислена по приближенному значению D0(HP=O) = 531 кДж-моль,
оцененному Струковым и др. [247] по методу Липпинкотта и Шредера [2441, 2442] с использованием
вычисленного ими по данным работ [2347, 2348] значения силовой постоянной растяжения связи
Р=О в молекуле НРО. Оценки, основанные на сопоставлениях энергий связей в РН, РО, НРО
ш NH, NO, HNO, подтверждают принятое значение энтальпии образования НРО(г).
PF (г). Термодинамические свойства газообразного фторида фосфора в стандартном
состоянии яри температурах 100—6000 К приведены в табл. 214.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 14.12. Молекула PF, изоэлектронная молекулам О2, S2 и SO, должна иметь 13
валентных состояний, принадлежащих конфигурациям . . . (wn)i(xaJ(w^J CE~, ХД, 12+),
. . ,(от)« (хо) (wkK (Щ,Щ) и ... (wkK (zaJ (wkK (!2+, Х2-, !Д, 32+, 32", 3Д) [1300]. В спектре PF
наблюдались переходы между шестью состояниями, отвечающими первым трем конфигурациям:
A3R-X32- (i/< I, v"<6), dm-^A (v' < 3, v" < 6), #Ч1-агД (г/=0, v" < 2), <WI-&12 +
(p*<4, y"<2), gm-b1^ (y'=0, w"<l), V2-X*2- (v', v" < 3) [1300, 101911.
Переходы, связанные с состояниями, отвечающими конфигурации . . . (wkK (хоJ (wkK, в спектре
PF до настоящего времени не обнаружены. Приведенные в табл. 14.12 значения молекулярных
постоянных PF во всех экспериментально наблюдаемых состояниях найдены Дугласом и Фра-
ковиаком [1300] в результате исследования отмеченных выше систем. Энергии возбуждения
синглетных состояний получены по началам полос систем ^П—ЬХ2, <WI—а^Д с учетом
результатов анализа системы Ь1!!—Х3И~ в работе Колина и др. [1019]. Электронные состояния, не
наблюдавшиеся экспериментально CД, 32+, Х2~, 32~, ХД, 22+ и 3П), должны иметь энергии
возбуждения около 40 000—50 000 см. Эти состояния в справочнике не учитывались при дальнейшем
рассмотрении.
Термодинамические функции PF(r) рассчитаны по уравнениям A. 3)—A.10), A. 93)—A.95).
Значения QEB и ее производных вычислены по уравнениям A.90)—A.92) с учетом четырех
возбужденных электронных состояний (а'Д, /:Я?+, Б3И, d1!!) в предположении, что QW Бр =
z=={PilPx)Qiol.*t' QiS. вр вычислялась по уравнениям A.73)—A. 75) непосредственным
суммированием. В расчете учитывались все уровни, ограниченные предельной кривой диссоциации этого
* В работе [3438а] в спектре испускания в области 5600 А наблюдались полосы с триплетпой структурой,
предположительно отнесенные авторами к переходу 3Д—3ГГ.
370
Фосфор и его соединения PF2 (г)
состояния. Колебательно-вращательные уровни энергии состояния Х32~ вычислялись по
уравнениям A. 65), A. 40), A. 62); коэффициенты Ykl в этих уравнениях, найденные в соответствии с
методами, изложенными в разделе 1.4 по молекулярным постоянным PF, представлены в табл. 14.4
вместе с коэффициентами а,- в уравнении, аппроксимирующем предельную кривую диссоциации,
и значениями г;ши и /нш.
Погрешности вычисленных значений термодинамических функций PF(r) при Т < 2000 К
обусловлены в основном неточностью принятых значений фундаментальных постоянных, при
более высоких температурах становятся существенными погрешности из-за неточности
аппроксимации высоких колебательно-вращательных уровней энергии состояния Х32~ и неполноты данных
о возбужденных электронных состояниях PF. Погрешности значений Ф°(Г) при 298,15; 3000 и
6000 К составляют 0,03; 0,1 и 0,3 Дж-К.моль.
Ранее таблицы термодинамических функций PF(r) вычислялись в справочниках [87, 88, 2095]
и в работах [3944, 2890] без учета возбужденных электронных состояний. В работах [87, 88,
3944] расчеты были выполнены по оцененным постоянным, в [2890, 2095] постоянные были
приняты по работе [1300]. Расхождения данных справочника [88] и табл. 214 достигают 2 Дж-К-1Х
Хмель в значениях Ф°(Т), расхождения с данными справочника [2095] не превышают 1,2 ДжХ
XК •моль'" в значениях Ф°(Т), но достигают 2,5 и 4,8 Дж-К~1.моль~1 в значениях С°р C000 К)
и С°р F000 К).
Константа равновесия реакции PF(r)=P(r)-|-F(r) вычислена на основании принятой в
справочнике энергии диссоциации PF
ДДо @) = Do (PF) = 440 + 42 кДж • моль = 36 700 + 3500 см.
Это значение было получено Гейдоном [1579] и О'Харе [2890] линейной экстраполяцией
колебательных уровней энергии состояния Х32~ (v" <^ 6) с введением поправок [1938, 541; 1579]
на систематическую погрешность этого метода.
Принятому значению D0(PF) соответствует
Д^0 (PF, г, 0) = —47,162 ± 42 кДж • моль.
PF2 (г). Термодинамические свойства газообразного дифторида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 215.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.8. На основании аналогии с NF2, теоретических расчетов [1033, 2027а, 998]
и исследования ИК-спектра [838] в справочнике принимается, что молекула PF2 в основном
электронном состоянии Ё2Вг имеет нелинейную симметричную структуру (симметрия С2„).
Значение JAIBIc, приведенное в табл. 14.8, рассчитано по структурным параметрам г (Р—F)=l,58±
+0,03 А, 2}. F—Р—F=98±3°, оцененным на основании данных о структуре молекул H2PPF2,
PHF2 и PF3 [2318, 2317, 1953], значения re(PF) в состоянии Х2П и результатов теоретических
расчетов [2027а]. Его погрешность оценивается в 3-10~116 г3-см6. Приведенные в таблице значения
основных частот PF2 определены Бердеттом и др. [838] (ух и v3) по ИК-спектру продуктов
фотолиза PF2H, изолированных в аргоновой матрице, и вычислены (v2) по уравнениям поля
валентных сил при подготовке настоящего издания. Погрешности принятых значений оцениваются в 10
(vlt v3) и 50 см (v2). На основании сравнения данных о возбужденных электронных состояниях
NF2, NH2 и РН2 можно предполагать, что первое возбужденное электронное состояние PF2 имеет
энергию, превышающую 30 000 см.
Термодинамические функции PF2(r), приведенные в табл. 215, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности табулированных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета, они составляют 1, 3 и 5 ДжХ
хК~1-моль~1 в значениях^1)оB') при 298,15; 3000 и 6000 К, при этом погрешности из-за неточности
постоянных в состоянии Х2ВХ изменяются от 1 до 2,5 Дж-К~1.моль~1.
Ранее таблицы термодинамических функций PF2(r) вычислялись в справочниках [87, 88, 2095]
и в работе [2890]. Во всех расчетах принимались значения молекулярных постоянных, отличные
от принятых в настоящем издании; расхождения в значениях Ф°(Т) и S°(T), приведенных в
справочниках [88, 2095] и в табл. 215, достигают 1,2 и 2 Дж-К~1.моль~1 соответственно.
371
PFj-(r), PF3(r) Глава 14
Константа равновесия реакции PF2(r)=P(r)+2F(r) вычислена с использованием значения
Ar.ffo@)—980,113+40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования PF2
AfH° (PF2, г, 0) = —510 ± 40 кДж • моль.
Эта величина основана на значении D0(F—PF2) = 520+40 кДж-моль, вычисленном по
потенциалу появления иона PFJ из PF3 A5,4+0,2 эВ) и потенциалу ионизации PF2 A0,0+0,3 эВ),
найденным Харландом и др. [1842] методом электронного удара. Потенциал появления иона
PFJ из PF3 A3,5 эВ), найденный Дином и др. [1188а], является, по-видимому, неверным, так как
соответствующее ему значение энергии атомизации PF2 находится в противоречии с энергией
диссоциации PF и средней энергией связи в молекуле PF3.
PF~ (г). Термодинамические свойства газа, состоящего из отрицательных ионов дифторида
фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 216.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.8. На основании аналогии с изоэлектронной молекулой SF2 в справочнике
принимается, что PF^ имеет синглетное основное электронное состояние с нелинейной симметричной
структурой (симметрия С2„). Приведенное в табл. 14.8 значение IaIbIo рассчитано на основании
структурных параметров г (Р~—F)=l,59+0,05 A, ^t F—Р—F=98+5°. Эти структурные
параметры и основные частоты PF7 были оценены по соответствующим постоянным молекул SF2 и PF2.
Погрешность значения IaIbIc оценивается в 1.10~118 г3-см6, основных частот в 50 см. В
справочнике принимается, что PFj не имеет стабильных возбужденных электронных состояний.
Термодинамические функции PF^r), приведенные в табл. 216, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности табулированных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета. Погрешности значений Ф°(Т)
при 298,15; 3000 и 6000 К оцениваются в 2, 4 и 6 Дж-К~1-моль~1, причем погрешности из-за
неточности постоянных могут составлять 2—4 Дж-Й~1-моль~1.
Ранее таблица термодинамических функций PF^ (г) не публиковалась.
Константа равновесия реакции PF~(r)=P(r)-|-2F(r)+e~(r) вычислена с использованием
значения Дг#°@) = 1170,113+55 кДж-моль, соответствующего принятому в справочнике сродству
PF., к электрону
AQ (PF2) == —2,0 + 0,4 эВ == —190 ± 40 кДж • моль'1.
Эта величина оценена на основании сравнения сродства к электрону молекул NH2, PH2 и NF2
и находится в согласии со значением ^40(PF2) = —1,71 эВ, полученным в приближенном
теоретическом расчете [1033]. Попытка определения .40(PF2), предпринятая Харландом и др. [1842],
по потенциалу появления иона PF~ при диссоциативном захвате электрона молекулой PF2CN,
привела к заниженному значению (—1,2 эВ), ненадежность которого отмечена авторами этой
работы. Измеренный ранее Мак-Нилом и Тинне [2494] потенциал появления иона PFj при
диссоциативном захвате электрона молекулой PF3 (9,4 эВ) в комбинации с принятым в справочнике
значением D0(F—PF2) приводит к чрезмерно большой величине Ао (PF2) ~ —4 эВ.
Принятому значению ^40(PF2) соответствует
ДД° (PF-, г, 0) = —700 + 55 кДж • моль.
PF3 (г). Термодинамические свойства газообразного трифторида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 217.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.13. Молекула PF3 в основном электронном состоянии имеет структуру
правильной треугольной пирамиды, в вершине которой расположен атом фосфора (симметрия С3„).
Структурные параметры PF3 исследовались в работах [1953, 792, 2976, 2706, 1901, 1652, 3953, 2256].
Значение постоянной Во в работах [1705, 1652, 1953, 2132, 2682] определялось из анализа
микроволнового спектра и в работе [3191] — из анализа полос в ИК-спектре PF3. Приведенное
в табл. 14.13 значение IaIbIg вычислено по структурным параметрам PF3: г (Р—F)=1,5700 +
+0,0012 А, -2} F—Р—F=97,8+0,2°, найденным Морино и др. [2706] по результатам электроно-
графического исследования, его погрешность составляет 2-10~11в г3-см6. Колебательные спектры
372
Фосфор и его соединения
(г)
PF3 исследовались в работах [1776, 3966, 3191,
3189, 4040, 976]. Для основных частот PFS
в таблице приведены значения, определенные
по ИК-спектру газообразного PF3 Рейхманом
[3191] (vx и v2) и Вильсоном и Поло [3966] (v3
и v4). Погрешность принятых значений не
превышает 1 см.
Термодинамические функции PF3(r),
приведенные в табл. 217, вычислены в
приближении «жесткий ротатор—гармонический
осциллятор» по уравнениям A. 3)—A.10), A.122) —
A.124), A.128), A.130). Погрешности
табулированных значений обусловлены
преимущественно приближенным характером расчета,
они составляют 0,3, 3 и 5 Дж-К^-моль
в значениях Ф°(Т) при 298,15; 3000 и 6000 К,
в том числе погрешности из-за неточности
молекулярных постоянных не превышают
0,2 Дж-К^-моль.
Ранее таблицы термодинамических функций
PF3(r) вычислялись в справочниках [87, 88,
2095] и в работе [2890] (до 6000 К), в работах
[3074, 66, 2556] (до 5000 К) и в работах [3966,
2742, 2773] (для Т < 2000 К). В расчетах
[3966, 2742, 3074, 2890, 87, 88, 66] были
приняты заниженные значения г (Р—F). В работе
[2773], кроме того, использовались неверные
значения частот v3 и v4. Расхождения в
значениях Ф°(Т), приведенных в табл. 217 и
предыдущем издании справочника [88],
составляют 0,35 Дж-К~1-моль~1, данные табл. 217 и
справочника [2095] согласуются в пределах
0,05 Дж-К-моль. Пейс и Петрелла [2931]
на основании результатов низкотемпературных
калориметрических измерений нашли ?°(РР3, г,
177,77 К) =244,68+0,63 Дж-К^-моль-1, что
находится в хорошем согласии со
значением 244,43+0,15 Дж-К~1-моль~1,
вычисленным по молекулярным постоянным PF3,
принятым в настоящем справочнике.
Константа равновесия реакции PF3(r) =
=P(r)-|-3F(r) вычислена с использованием
значения Дг#°@) = 1499,127+ 1,7 кДж-моль,
соответствующего принятой в справочнике
энтальпии образования
AfH°(PF3, г, 298,15 К) =
— —957,4 + 1,3 кДж ¦ моль.
Эта величина основана на энтальпии реак-
ции PF3(r)+F2(r) = PF6(r), ДГЯ° B98,15 К) =
=—635,88+0,29 кДж-моль х, измеренной
Джонстоном и др. [2139] методом фтор-
ной калориметрии. Практически совпадающее
значение (—635,93+0,67 кДж -моль") было
получено в более ранних измерениях, вы-
о
fa
Сч
fa*
о.
«о
fa
Рч
а
и
X
л*
а и
35 "
?Q ^
V ^
Я ^
й^>
се ^
L?
но
cd «
w ^^
I*
з©
S*
5 "
|з
Bo
Xfa
в ?
So
Sh
2P-
a
So
g*
К Он
4 ^
4fa*
о д^
gfa
S3 Cu
So
(Ob
CO
14.
cs
sr
a
ч
vo
C3
H
D
О
•
S
**"
I-H
;*
О
OS
;>
QO
?¦
CO
со
s
о
m
1
О
4
1"
О
CO
,20
О
CO
3
о
CD
CO
CM
^r
OO
vf
CM
O3
OO
fa5
Рч
,72
о
I
1
1
о
со
<м
s
ОО
со
ОО
ЧТИ
vf
С<1
о
1С
vf
,С1
fa
ОО
см
со
см
см
CS1
1С
со
о
см
ОО
см
со
ОО
со
ОО
€
fa
Рч
со
S
со
1
1
с?
со"
00
3
1С
о
1С
1С
о"
со
см
см
1С
со
и
Рч
СО
оз
тч
1
3
о
см
3
vf
о?
ю"
см"
со
1С
,4B)
со
о
75,г
1С
со
со"
аз
00
со
СО
тЧ
ОО
fa
Рч
см
356
560
со
vf
vf
^н
о
vf
см
аз
СО
1С
со
vf
•чч
со
vf
о
1С
.^
со
1С
ОО
ч.
fa
Рч
см
см
оз
1
427
500
1С
СМ
а>
00
СО
со
§8
vf
00
со
1С
1С
СО
vf
см
о
vf
00
со
со
СО
со
со
СО
аз
ОО
о.
fa'
Рч
СО
аз
¦^ч
1
3
о
со
со
3
см
см
3
vf
о
vf
B)
см
СО
00
см
СО
S
1С
S
00
со
СО
те
о
fa"
Рч
со
vf
со
1
1
1
1
3
о
3
аз
см
3
аз
со
со
3
о
СО
1С
со
см
00
ОО
СО
1^
CN1
vf
00
с—
и
fa
Рч
CD
1С
1
1
1
3
ОО
CD
CM
с?
8
3
СО
см
3
СО
1С
S
со
о
со
vf
СО
ОО
см
см
аз
СО
о
Рч
СО
со
о"
1
1
1
1
1
1
3
СО
1С*
со
СО
3
см
00
vf
3
аз
аз
vf
см"
t-
vf
со
см"
ОО
21
fa"
о
Рч
•ЧЧ
СО-
1
1
1
vf
vf
о
CD
аз
CM
vf
о
vf
аз
vf
со
CM
CO
о
о
аз
vf
00
со
5
О
Рч
vf_
vf"
1
1
1
t—
vf
Cvl
vf
:o
CD
CM
CD
1С
О
s
CO
382
CO
vf
1С
8
00
1С
CO
¦^4
€
fa
о
Рч
CO
CM-
о"
1
1
1
3
t—"
OO
3
1С
cm"
CO
CO
cm"
CO
1С
1С
ic"
CO
CM
1С
o"
vf
1C_
¦^4
CM
CO
о
о
Рч
373
PF5 (г), POF3 (г) Глава 14
полненных в той же лаборатории Рудзитисом и др. [3265]. Аналогичные измерения
Селфа [3385] привели к ошибочной величине \Н° B98,15 К) = —559+3 кДж-моль" (см.
[2139]). Дюс и Микитюк [1358] измерили энтальпию реакции РС13(г)+1,5 CaF2(K)=PF3(r)+
+1,5 СаС12(к), которой соответствует значение b.fH° (PF3, г, 298,15 К) = — 952+9 кДж-моль,
совпадающее в пределах погрешности с принятым в справочнике. Измерения энтальпии
гидролиза PF3(r), выполненные Бертло [619, 620, 621], не могут быть использованы для вычисления
энтальпии образования PF3(r) вследствие неопределенности состава продуктов этой реакции.
PF5 (г). Термодинамические свойства газообразного пентафторида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 218.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.13. Молекула PF5 в основном электронном состоянии имеет структуру
правильной треугольной бипирамиды с атомом фосфора в центре (симметрия D3h). Структурные параметры
PF6 определялись в электронографических исследованиях [757, 790, 5, 1901, 1823, 226], а также
в исследованиях спектров [4012, 3918]. Наиболее точные значения величин г (Р—FalM.) =
=1,577+0,005 А и г (Р—FPM)=1,534+0,004 А определены Хансеном и Бартеллом [1823] по элек-
тронографическим данным. Принятое в табл. 14.13 для PFB значение IaIbIg вычислено на
основании этих данных и значения постоянной Z?0=0,104 70+0,00004 см, полученного Шацем и Рейх-
маном [3324] при анализе вращательной структуры полос в ИК-спектре PF5. Его погрешность
равна 6-10~116 г3-см6. Колебательные спектры PF8 исследовались в работах [4012, 1776, 2988,
178,1732, 2013, 1731, 2014, 1202, 2451, 2398, 3324, 535, 536, 2654, 2726, 605а]. Приведенные в табл.
14.13 значения основных частот PF5 определены по спектру КР газообразного PF5 Миллером и
Капвеллом [2654] (vx, v2, v8) и Битти и др. [535] (v7) и по ИК-спектру газообразного PF6 Шацем
и Рейхманом [3324] (v3, v4) и Гриффитсом и др. [1732], Хоскинсом и Лордом [2013] (v5, ve).
Погрешности принятых значений основных частот PF5 оцениваются в 0,5—1 см. В работе [605а 1
было показано, что для колебания v7, связанного с выходом атома F из экваториальной плоскости,
характерна большая ангармоничность.
Термодинамические функции PF5(r), приведенные в табл. 218, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены преимущественно
приближенным характером метода расчета. Погрешности в значениях Ф°(Г) при 298,15, 3000 и 6000 К
составляют 2, 7 и 12 Дж-К~1.моль~1, при этом погрешности из-за неточности молекулярных
постоянных не превышают 0,5 Дж-К~1-моль~1.
Ранее таблицы термодинамических функций PF5(r) вычислялись в справочниках [87, 88,
2095], в работах [2890], [69] (до 5000 К) и [1732, 1731, 2893, 2779] (Т < 2000 К). Все эти расчеты,
кроме [2095], основывались на молекулярных постоянных PF5, значительно отличающихся
от принятых в настоящем издании, чем объясняются большие различия приведенных в них
значений термодинамических функций PF6(r) от данных табл. 218, достигающие 15 Дж-К~1.моль~1
в значениях Ф°(Г) и S°(T). Различия между значениями Ф°(Т) при 298,15; 3000 и 6000 К,
приведенными в табл. 218 и в справочнике JANAF [2095], не превышают 0,2 Дж-К~1-моль~1.
Константа равновесия реакции PF6(r)=P(r)+5F(r) вычислялась с использованием значения
Д^^О)=2284,353+2,1 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
A^°(PF5, г, 298,15 К) = —1593,3 + 1,3 кДж • моль.
Эта величина основана на результатах измерений энтальпии сгорания белого фосфора,
проведенных О'Харе и Хаббардом [2893]. Сожжение проводилось в двухкамерной фторной бомбе,
причем в некоторых опытах была обнаружена коррозия никелевого диска, на котором помещался
сжигаемый образец. Однако авторы тщательно учли все погрешности, связанные с этим
эффектом. В полном соответствии с данными этой работы находятся результаты калориметрических
измерений Гросса и др. [1743] (—1595,8+3,4 кДж-моль"), проводившие сожжение фосфора во
фторе в кварцевом сосуде.
POFg (г). Термодинамические свойства газообразного оксид-трифторида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 219.
374
Фосфор и его соединения РСЦг}
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.13. Молекула POF3 в основном электронном состоянии относится к точечной
группе симметрии С3„ и характеризуется структурными параметрами г (Р—F) = 1,436 +0,006 А,
г (Р—О) = 1,524+0,003 А, -3 F—Р—F=101,3+0,2°, определенными Моритани и др. [27121
в результате электронографического исследования. Эти данные находятся в
прекрасном согласии со значением 50=0,153 248 06 см, найденным Беррусом и Горди [8481
по микроволновому спектру. Структурные параметры POF3 определялись также в работах [790,
1861, 3953, 2183а]. Вращательные постоянные и постоянные центробежного растяжения РОР3
определялись также на основании изучения микроволнового спектра в работах [1861, 3953, 3386,
2132, 848, 2183а]. Принятое в табл. 14.13 значение 1А1в1с вычислено на основании приведенных
структурных параметров и постоянной Во, его погрешность оценивается в 4-10~lie г3-см6.
Основные частоты POF3 определялись на основании результатов исследований его колебательных
спектров [1213, 977, 1776, 3376, 317] и расчетов по силовым постоянным [2764, 2248, 4061, 3794J.
Приведенные в таблице значения основных частот POF3 определены по максимумам Q-ветвей
полос в ИК-спектре [3376] (vx, v2, v6) и в спектре КР [977] (v3, v4, v5) газообразного POF3.
Погрешности этих значений не превышают 1 см.
Термодинамические функции POF3(r), приведенные в табл. 219, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A. 130). Погрешности табулированных термодинамических функций определяются
преимущественно приближенным методом расчета. Погрешности значений Ф°(Т) при 298,15; 3000 и
6000 К составляют 0,3; 4 и 8 Дж-К~1-моль~1, при этом погрешности из-за неточности принятых
молекулярных постоянных не превышают 0,2 Дж-К~1.моль~1.
Ранее таблицы термодинамических функций POF3(r) вычислялись в справочниках [88, 20951
и в работах [2764, 4061] при Т < 1500 К. В расчетах [2764, 4061, 88] были приняты неточные
значения молекулярных постоянных, однако расхождения данных [88] и табл. 219 не превышают
0,5 Дж-К~1.моль~1 в значениях Ф°(Г). В расчете, приведенном в справочнике [2095], были
использованы значения молекулярных постоянных POF3, близкие к принятым в настоящем издании,
поэтому расхождения в значениях Ф°(Т), приведенных в табл. 219 и в справочнике [2095], не
превышают 0,1 Дж-К~1-моль~1. Джепсон и Пейс [2122, 2974] на основании результатов
низкотемпературных калориметрических измерений нашли iS^POF,,, г, 236 К)=265,5 +0,6 Дж-К~1.моль~1,
расчет по молекулярным постоянным, принятым для POF3, в табл. 14.13 приводит к значению
270,16+0,3 Дж-К~1-моль~1. Это различие авторы работы [2974] объяснили наличием
остаточной энтропии, обусловленной неупорядоченным расположением молекул в POF3(k) при низких
температурах.
Константа равновесия реакции POF3(r)=P(r)+O(r)+3F(r) вычислена с использованием
значения Дг#°@) =2037,399+8 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
Д^° (POF3, г, 298,15 К) = —1252 + 8 кДж • моль.
Эта величина основана на калориметрических измерениях энтальпии реакции PF3(r)+0,5O2(r)=
= POF3(r) в работе Эбеля и Бречера [1371] (Дг#° B98,15 К) = —295+8 нДж-моль).
Погрешность значения &.ГН° принята равной 8 кДж-моль, поскольку в работе [1371] не проводился
анализ конечных продуктов реакции.
РС1 (г). Термодинамические свойства газообразного хлорида фосфора в стандартном состоянии
при температурах 100—6000 К приведены в табл. 220.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.12. На основании данных работ [1108а, 1300] для PF и PG1 в справочнике
принимается, что молекула РС1 имеет шесть стабильных валентных электронных состояний
с энергиями возбуждения до 50 000 см: Х32~, а1 Д, ЬХ2+, Л3Пг, <М1, g4I. Колебательные
постоянные и энергии возбуждения Р35С1 в состояниях Х3Е~, Ь1!,* и А311Г найдены Коксоном и
Викмаарачи [1108а] в результате анализа колебательной структуры полос А3ЛГ — X3L~ (г/ <^2,
3 < v" < 18) и &ls+ — XSIr lv'< v" < 10)- Значение ге(РС1) для состояния X3Z~ принято по оценке
Спиридонова и Татевского [242], неточность которой они определили в 0,05 А. Постоянная
мультиплетного расщепления состояния Х32~ оценена на основании сравнения значений этой
постоянной для состояний Х32~ молекул S2, SO и PF. Энергии состояний а]Д, ЙЧЕ и g'll вы-
375
РС12 (г) Глава 14
числены по соотношению A.89) на основании энергий возбуждения соответствующих состояний
PF и сравнения результатов аналогичных расчетов для состояний б1!! и А3ЛГ с
экспериментальными значениями [1108а]. Погрешности в оцененных значениях То составляют 500—2000 см.
Термодинамические функции РС1(г), приведенные в табл. 220, вычислены по уравнениям
A.3)—A.10), A. 93)—A.95). Значения Qm и ее производных были рассчитаны по уравнениям
A.90)—A.92) с учетом пяти возбужденных электронных состояний РС1 ив предположении, что
Q^d.Bv=(Pi^Px)Qml. Ер- Статистическая сумма по колебательно-вращательным уровням энергии
состояния Х3Ъ~ и ее производные находились непосредственным суммированием по уравнениям
A.73)—A.75), в расчете учитывались все уровни состояния XSZ~, ограниченные его предельной
кривой диссоциации. Колебательно-вращательные уровни энергии состояния Х3%~ вычислялись
по уравнениям A. 65), A. 40), A. 62). Значения коэффициентов Ykl в этих уравнениях, полученные
по методам, изложенным в разделе 1.4, и постоянным, приведенным в табл. 14.12, представлены
в табл. 14.4 вместе с коэффициентами а{ в уравнении, аппроксимирующем предельную кривую
диссоциации, и значениями vm&% и /цт. Погрешности вычисленных термодинамических функций
РС1(г) обусловлены неточностью принятых молекулярных постоянных в состоянии Х32~ и
энергий возбужденных электронных состояний. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К
не превышают 0,3 Дж-К~1-моль~1.
Ранее таблицы термодинамических функций РС1(г) вычислялись в справочниках [88, 2095].
В этих расчетах учитывалось только основное электронное состояние XSH~ по оцененным
постоянным и без учета мультиплетного расщепления. Расхождения данных справочников [88, 2095]
и табл. 220 достигают 1,8 и 2,3 Дж-К~1-моль~1 соответственно в значениях Ф°(Т). Расхождения
в значениях С°р(Т) с приведенными в справочнике [2095] существенно выше и составляют
примерно 3,5 Дж-К~1-моль~1 при 3000 К.
Константа равновесия реакции РС1(г)=Р(гL-С1(г) вычислена с использованием принятой
в справочнике энергии диссоциации РС1
ДД° @) = Do (PG1) = 330 + 40 кДж • моль.
Это значение оценено на основании сопоставления энергий связей в молекулах PFB и РС1Я
(п = 1-^-3). Принятому в справочнике значению DO(PC1) соответствует
Д/Я°(РС1, г, 0)= 135,183+ 40 кДж • моль.
Ранее попытка определения энтальпии образования РС1(г) предпринималась Зандовалом и
др. [3303] на основании данных, полученных при изучении диссоциативной ионизации РС13
под действием электронного удара. Полученное в работе [3303] значение Д/Я°(РС1, г) =
= 140 кДж-моль в пределах погрешности совпадает с принятым.
РС12 (г). Термодинамические свойства газообразного дихлорида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 221.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.8. В справочнике на основании аналогии с PF2 и РН2 принимается, что РС12
имеет дублетное основное электронное состояние и нелинейную симметричную структуру
(симметрия С2с). Это подтверждается результатами теоретического расчета Хадсона и Виффена [2027а]
и характером ИК-спектра молекул РС12, изолированных в аргоновой матрице [391]. Принятые
структурные параметры г (Р—Cl)=2,01+0,05 A, ^ C1—Р—С1=100+5° оценены на основании
сравнения структурных параметров PF, PF2, PF3 и PCI, PC13 с учетом результатов теоретического
расчета [2027а]. Соответствующее им значение 1А1в1о приведено в табл. 14.8, его погрешность
оценивается в 1.10~114 г3-см6. Приведенные в таблице значения основных частот РС12 основаны
на результатах исследования ИК-спектра молекул РС12, изолированных в Аг матрице в работе
Эндрюса и Фредерика [391], и на расчетах, выполненных при подготовке справочника (v2).
Погрешности принятых основных частот РС12 оцениваются в 10 (v1? v3) и 50 см (v2). Можно
предполагать, что возбужденные электронные состояния РС12 имеют энергию свыше 20 000—30 000 см.
Термодинамические функции РС12(г), приведенные в табл. 221, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых молеку-
376
Фосфор и его соединения PCljf (г), РС13 (г)
лярных постоянных, отсутствием сведений о возбужденных электронных состояниях и
приближенным характером расчета. Погрешности в значениях Ф°(Т) при 298,15; 3000 и 6000 К
оцениваются в 1, 3 и 5 Дж-К~1-моль~1, в том числе из-за неточности постоянных 1—2 Дж-К~1-моль~1.
Ранее таблица термодинамических функций РС12(г) не публиковалась.
Константа равновесия реакции РС12 (г)=Р (г)+2С1 (г) вычислена с использованием значения
Дг Н° @)=606,803+6 кДж-моль, соответствующего принятой в справочнике величине
энтальпии образования
ДД°(РС18, г, 0) = —52 + 6 кДж-моль.
Эта величина основана на энергии связи D (C1—РС12)=353,6+5,0 кДж-моль, найденной
Хальманом и Клейном [1811] в результате измерений потенциала появления С1~ @,05+0,05 эВ)
в реакции РС13 (г)+е = РС12 (г)+С1~ (г). Она находится в удовлетворительном согласии с принятым
в справочнике значением энергии диссоциации PG1. Масс-спектрометрические исследования
продуктов диссоциативной ионизации молекул РС13 и Р2С14 электронным ударом, выполненные Зандо-
валом и др. [3303], не привели к надежным результатам.
1^ (г). Термодинамические свойства газа, состоящего из отрицательных ионов дихлорида
фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 222.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.8. В справочнике принимается, что, подобно изоэлектронной молекуле SC12,
ион РС1^ в основном электронном состоянии ХгА имеет нелинейную структуру, соответствующую
точечной группе симметрии С2г. Приведенное в табл. 14.8 значение IaIbIc рассчитано на основании
структурных параметров г (Р—С1)=2,01 +0,05 А, ^ С1—Р—С1 = 103+5°, которые были оценены
по структурным параметрам молекул РС12 и SC12 (см. [3314]). Его погрешность оценивается
в 2 -10~114 г3 -см6. Для основных частот PClj в таблице приведены значения, оцененные по основным
частотам SG12 [483, 1530]. Погрешности этих значений составляют 50—100 см.
Термодинамические функции PG1J (г), приведенные в табл. 222, вычислены в приближении
«жесткий ротатор—гармонический осциллятор». Погрешности вычисленных значений обусловлены
неточностью принятых молекулярных постоянных и приближенным характером расчета.
Погрешности в значениях Ф° (Т) при 298,15, 3000 и 6000 К составляют 2, 4 и 6 Дж К моль, при этом
погрешности из-за неточности постоянных достигают 1,5—3 Дж-К моль.
Ранее таблица термодинамических функций PClj (г) не публиковалась.
Константа равновесия реакции РС1г=Р (гL-2С1 (г)+е~ (г) вычислена с использованием
значения Дг Н° @)=9002,803 +40 кДж-моль", соответствующего принятой в справочнике величине
сродства к электрону
Ао (РС12) = —296 + 40 кДж • моль.
Эта величина основана на найденном Хальманом и Клейном [1811 ] значении потенциала
появления иона РС1~ @,4+0,4 эВ) в реакции РС13 (r)+e-=PCl2 (г)+С1 (г).
Принятому значению Ао (РС12) соответствует
AfH0(PG\-, г, 0) = —348 + 40 кДж • моль.
РС13 (г). Термодинамические свойства газообразного трихлорида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 223.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула РС13 в основном электронном состоянии ХХА имеет структуру правильной
пирамиды (симметрия С3с). Приведенное в табл. 14.13 значение 1А1в1с рассчитано по структурным
параметрам г (Р—С1)=2,043 +0,003 А, -3 G1—Р—С1=100° 6' +20', найденным Кислюком и Таун-
сом [2257, 2258] из анализа микроволнового спектра. Его погрешность составляет 2 -10~114 г3-см6.
Менее точные значения структурных параметров РС13 были получены в работах [3942, 792, 2976,
1873, 918]. Колебательные спектры газообразного РС13 исследовались в работах [2908, 2464, 1531,
3274, 2861, 976] и РС13 в конденсированном состоянии — в работах [1183, 1531, 633, 3790, 1665,
2860, 753, 2276, 2861, 37291. Приведенные в табл. 14.13 значения основных частот РС13 определены
Руоффом и др. [3274] в результате исследования ИК-спектра газообразного РС13. Погрешности
этих значений оцениваются в 1—3 см.
377
РС16(г)
Глава 14
Термодинамические функции РС13 (г), приведенные в табл. 223, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены преимущественно приближенным
методом расчета. Погрешности в значениях Ф° (Т) при 298,15; 3000 и 6000 К составляют 1, 4
и 7 Дж -К -моль, в том числе из-за неточности молекулярных постоянных не более
0,2 Дж -К -моль.
Ранее таблицы термодинамических функций РС13 (г) вычислялись в справочниках [88, 2095,
25561 и в работах [3535, 2218, 2219, 2463, 2742] (при Т < 2000 К). Во всех расчетах для
молекулярных постоянных РС13 использовались значения, близкие к принятым в настоящем издании,
вследствие чего полученные в них значения Ф° (Т) и S° (T) согласуются с приведенными в табл. 223
в пределах 0,3 Дж -К-моль.
Константа равновесия реакции РС13 (г)=Р (г)+ЗС1 (г) вычислена с использованием значения
Дг Н° @)=960,725 +2,1 кДж-моль", соответствующего принятой в справочнике энтальпии
образования РС13 (г)
Д^0 (РС13, г, 298,15 К) = —289,5 + 2,0 кДж ¦ моль'1.
Результаты определений энтальпии образования РС13 в жидком и газообразном состояниях
представлены в табл. 14.14. В расчетах использовано значение &VH° (PC13, ж, 298,15 К) =
=32,9+0,5 кДж -моль, вычисленное по уравнению II закона из данных по давлению паров РС13,
Таблица 14.14. Результаты определений энтальпии образования РС13 (г) (в кДж ¦ моль)
Авторы
Метод
Д/ЙГО(РС13, 298,15 К)
РС13(ж)
РС13(г)
Бертло, Лугинпн, Калориметрическое измерение энтальпии гидро- —329,1 —296,2
1875 [623] лпза РС13(ж)+ЗН2О(ж)=ЗНС1(водн. р-р)+
+Н3РО3 (водн. р-р)
Томсен, 1882 [3676] То же —322,6 —289,7
Холланд, 1912 Исследование равновесия РС15 (г)=РС13 (г)+С1, (г) — —278,4+6,5A1)
[1977]; Нернст, 1916 статическим методом, 439—634 К, 11 точек " —290,3+3,0 (III)
[2833 ]
Фишер, Юберман, То же, 422—502 К, 12 точек — —283,7+6,5 (II)
1938 [1491] —289,5 ±2,6 (III)
Нил, Вильяме, 1952 Калориметрическое измерение энтальпии гидро- —312,7 —279,8
[2827] лиза РС13(ж)+(га+3)Н2О(ж)= ЗНС1+Н3РО3
(р-р в тгН2О) при 4500 < п < 7500
Чарнли, Скиннер, То же при 4250 < п < 9040 —311,9 —279
1953 [942]
Нил, Вильяме, 1954 Калориметрическое измерение энтальпии окисли- —320,6+2,5 —287,7+2,5
[2827а] тельного гидролиза РС13 (ж)+Вг2 (водн. р-р)+
+ (п+4)Н2О(ж)=Н3РО4+ЗНС1+2НВг(р-р
в п Н2О), при 3500 < п < 6300
Поляченок Л. Д., Исследование равновесия РС16 (г)=РС18 (г)+С12(г) — —292,1+3,0 (II)
Поляченок О. Г., статическим методом, 408—601 К, 24 точки —290,7+2,6 (III)
1972 [196]
полученных в работах Реньо [3186] B70—340 К, 14 точек), Нисельсона, Серякова [184] B92—
358 К, 11 точек) и Холмса [1982] B73—297 К, 6 точек). Пересчет от средней температуры
измерений к 298,15 К проводился с использованием оцененного значения ДС°=—42 Дж-К-моль.
Результаты калориметрических измерений [623, 3676, 2827, 942] недостаточно надежны,
так как образующийся в исследованных реакциях раствор Н3РО3 неустойчив и легко окисляется.
Более надежное значение было получено в работе [2827а], где исследовался окислительный
гидролиз с образованием вполне устойчивого раствора Н3Р04. Исследования равновесия реакции РС15=
=РС13+С12 [196, 1491, 2833, 1977] привели к согласующимся результатам. Принятая величина
является средневзвешенной из указанных выше исследований равновесий и работы [2827а].
РС15 (г). Термодинамические свойства газообразного пентахлорида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 224.
378
Фосфор и его соединения РОС13(г)
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.13. Молекула РС1Б в основном электронном состоянии имеет структуру
правильной треугольной бипирамиды с атомом фосфора в центре (симметрия D3h). Приведенное в табл. 14.13
значение 1А1в1с рассчитано на основании структурных параметров г (Р—С1ако)=2,124 +0,009 А,
г (Р—С1рад)=2,020+0,07 А, найденных Адамсом и Бартеллом [346] в результате электронографи-
ческого исследования. Его погрешность составляет 2 -10~113 г3-см6. Менее точные значения длин
связей в РС15 были получены в электронографических исследованиях [3237а, 32376, 3535, 5, 6, 226].
Колебательные спектры газообразного РС15 исследовались в работах [3958, 536а] иРС15 в
конденсированных состояниях — в работах [3958, 538, 877, 533, 2653, 3776, 3631, 1036]. Приведенные
в табл. 14.13 значения основных частот РС15 приняты по данным, полученным в работах Битти
и Озина [536а] (vl7 v2, v6, v6, v7) по спектру КР газообразного РС1Б, Уильмсхурста и Бернстейна
[3958] (v3) по ИК-спектру газообразного РС16, Битти и др. [538, 2653] (v4) по ИК-спектру растворов
РС16 в CS2 и С6Нв, Ван Хуана и Десета [3776], Тейлора и Вудуорда [3631] и Битти и др. [533]
(v8) по спектрам КР жидкого РС15 и его растворов в СвНв, СС14 и СН2С12. Погрешности принятых
частот оцениваются в 1—5 см.
Термодинамические функции РС15 (г), приведенные в табл. 224, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений определяются преимущественно
приближенным характером расчета, они составляют 2, 10 и 15 Дж -К -моль в значениях Ф° (Т)
при 298,15; 3000 и 6000 К, при этом из-за неточности молекулярных постоянных погрешности
не превышают 1—2 Дж-К-моль.
Ранее таблицы термодинамических функций РС15 (г) вычислялись в справочниках [88, 2095]
и в работах [3535, 3958, 2772] (при Т ^ 2000 К). Расчет [2772] является ошибочным, в работах
[3535, 3958] использованы устаревшие значения молекулярных постоянных. Расхождения в
значениях Ф° (Г), приведенных в табл. 224 и в справочниках [88, 2095], составляют 1,5—
—3,5 Дж -К -моль. В основном они обусловлены неточными значениями частот v2, v4, v6 и
структурных параметров РС16, принятыми в справочниках [88, 2095].
Константа равновесия реакции РС15 (г)=Р (г)+5С1 (г) вычислена с использованием значения
Дг Н° @) = 1284, 658+2,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Й"°(РС1Б, г, 298,15 К) = — 376,0 + 2,0 кДж • моль.
Эта величина вычислена как сумма энтальпий образования и сублимации РС15 (к). Шамм и др.
[3360] измерили энтальпию сгорания белого фосфора в хлоре в проточном калориметре и нашли
Д. Н° (РС16, к, 298,15 К) = —443,85+0,30 кДж •моль. Это значение принимается в настоящем
издании. Оно находится в хорошем соответствии с измерениями энтальпии гидролиза РС15 и
другими термохимическими данными для соединений фосфора (см. Р4О10 (к)). Менее точные измерения
энтальпии хлорирования фосфора были выполнены в работах [340, 396, 623, 3676].
Принятое в справочнике значение Д„ Н° (РС15, к, 298,15 К)=67,9+2,0 кДж-моль было
вычислено по уравнению II закона на основании измерений парциального давления РС15 (г) в парах
над РС15 (к) статическим методом, проведенных Фишером и Юберманом [1491] C74—431 К),
Поляченком и Поляченок [196] C37—418 К). Измерения методом изотенископа (Смит, Калверт
[3451], 371—436 К) уступают по точности работам [1491] и [196].
РОС13 (г). Термодинамические свойства газообразного оксид-трихлорида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 225.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 14.13. Молекула РОС13 в основном электронном состоянии ^А относится к точечной
группе симметрии C3t. Приведенное в табл. 14.13 значение IaIbIc вычислено на основании
структурных параметров г (Р—О) = 1,455+0,005 А, г (Р—С1) = 1,989+0,002 A, J$. С1—Р—С1 = 103,7 +
+0,2°, найденных Ли и др. [2420] из анализа микроволнового спектра РОС13. Его погрешность
равна 1 -1014 г3 -см8. Менее точные значения структурных параметров РОС13 были получены в
работах [790, 443, 55, 2712, 3953]. Колебательные спектры газообразного РОС13 исследовались в
работах [317, 1318, 977] и РОС13 в конденсированных состояниях — в работах [1150, 1884, 2366,
3790, 2355, 858, 3421, 317]. Принятые значения основных частот Р0С13определены Кларком и Рип-
поном [977] по спектру КР газообразного Р0С13, их погрешности не превышают 1 см.
379
Р0С13 (г)
Глава 14
Термодинамические функции РОС13 (г), приведенные в табл. 225, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A. 128), A. 130). Погрешности вычисленных значений обусловлены преимущественно
приближенным методом расчета, они составляют 1, 6 и 10 Дж -К -моль в значениях Ф° (Т) при 298,15;
3000 и 6000 К, при этом погрешности из-за неточности молекулярных постоянных не превышают
0,5 Дж-К^-моль.
Ранее таблицы термодинамических функций РОС13 (г) вычислялись в справочниках [88, 20951
и в работах [3535, 2924, 4061, 2764] (при Т ^ 1000 К). Всо расчеты основывались на неточных
значениях основных частот РОС13, найденных по колебательным спектрам жидкого РОС13, причем
в расчетах [2924, 2095] было принято обратное отнесение частот v3 и v5. Расхождения в значениях
Ф° (Т), приведенных в табл. 225 и в справочниках [88, 2095], составляют 0,4—0,5 и 0,5—
1,5 Дж-К-моль соответственно. Отт и Джиок [2924] на основании низкотемпературных
калориметрических измерений вычислили S° (РОС13, г, 298,15 К)=325,3 Дж-К~х-моль, что хорошо
согласуется с приведенным в табл. 225 значением этой величины C24,347+1,5 Дж-К-моль).
Константа равновесия реакции РОС13 (г) = Р (г)+О (г)+ЗС1 (г) вычислена с использованием
значения Дг Н° @) = 1483,87+2,6 кДж -моль, соответствующего принятой в справочнике
энтальпии образования
Д/Я°(РОС13, г, 298,15 К) = —568,4 ±2,5 кДж • моль.
Эта величина вычислена как сумма энтальпий образования и испарения Р0С13 (ж). В табл. 14.15
приведены результаты определения энтальпии образования РОС13 (ж). Принятое значение
Д^ Н° (РОС13, ж, 298,15 К) = —606,9+2,5 кДж -моль основано на измерениях, проведенных Эга-
ном и Луффом [1391], результаты которых подтверждаются работами Нила и Вильямса [2827]
и Чарнли и Скиннера [942]. В работе [1391] были измерены также входящие в термохимический
цикл энтальпии разбавления растворов Н3РО4 и HG1. Плохое согласование этих измерений с
результатами других работ явилось основанием для увеличения погрешности значения
Д,, #° (РОС13, ж) по сравнению с оригиналом [1391].
Таблица 14.15.
Результаты определения энтальпии образования РОС13 (ж) по калориметрическим
измерениям энтальпии гидролиза РОС13 (ж) + C + п) Н2О (ж) =
=(Н3РО4 + ЗНС1) (р-р иН20) (в кДжмоль-1)
Авторы
Число молей Н2О (ге) в растворе,
температура измерения
Д,Я°(РОС13, ж,
298,15 К)
Бертло, Лугинин, 1875 [623]
Томсен, 1882 |3676]
Нил, Вильяме, 1952 [2827]
Чарнли, Скиннер, 1953 [942]
Эган, Луфф, 1966 [1391]
Не указаны
га=1100,
/г=2600-4300,
га=2650—5230,
ге=4800,
Г=291,85 К
Г=298,15 К
7=298,15 К, 6 опытов
Г=298,15 К, 8 опытов
—630
—640
-607,6+3,0
—605,8+3,0
—606,9+2,5
Таблица 14.16. Результаты определения энтальпии испарения РОС13 (ж) (в кДж-моль)
Авторы
Метод
Д,#°B98,15К)
Арии, 1929 [411]
Нисельсон, Серяков,
1960 [184]
Отт и Джиок,
[2924]
То же
1960
Измерение давления пара статическим методом, 303—
378 К, 9 точек
Измерение статическим методом давления пара Р0С13 (ж)
при изучении рециркуляционным методом фазовых
равновесий в системе SiCl4—POC13, 314—386 К, 10 точек.
Измерение давления пара статическим методом, 280—
298 К, 4 точки
Измерение энтальпии испарения калориметрическим
методом, 298,15 К, 3 опыта
36,92+0,18 (II)
38,00+0,06 (III)
39,44+0,37 (II)
38,28+0,06 (III)
37,1+1,9 (II)
38,26+0,05 (III)
38,58+0,21
Примечание. Погрешности значений А„Н° B98,15 К), вычисленных на основании измерений давления пара,
соответствуют только разбросу этих данных.
380
Фосфор и его соединения PFC1 (г), PFQ- (г)
Для энтальпии испарения РОС13 (ж) принято значение Av H° (РОС13, ж, 298,15 К)=38,5 +
+0,2 кДж -моль, являющееся средневзвешенным из приведенных в табл. 14.16 данных работ
1184, 2924]. При этом учитывалась неточность термодинамических функций, приводящая к
дополнительной погрешности в 0,5 кДж моль" в энтальпии испарения, вычисленной из данных по
давлению пара. Необходимые для расчета термодинамические функции РОС13 (ж) были получены по
данным работы [2924].
PFC1 (г). Термодинамические свойства газообразного фторид-хлорида фосфора в стандартном
состоянии при температурах 100—6000 К приведены в табл. 226.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.8. По аналогии с молекулами PF2 и РС12 в справочнике принимается, что PFC1 имеет Х2Вг
основное электронное состояние со структурными параметрами г (Р—F) = l,58+0,03 А, г (Р—С1) =
=2,01+0,05 А, -^ F—Р—С1=100+10°. Приведенное в табл. 14.8 значение IaIbIc рассчитано
по этим параметрам, его погрешность составляет 2-10~115 г3-см6. Приведенные в табл. 14.8
значения основных частот PFC1 оценены на основании принятых в справочнике основных частот PF2
и РС12, их погрешности оцениваются в 50—100 см. Сведения о возбужденных электронных
состояниях PFC1 отсутствуют.
Термодинамические функции PFC1 (г), приведенные в табл. 226, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A. 122)—A.124)
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных, отсутствием данных о возбужденных электронных состояниях PFC1 и
приближенным характером расчета. Погрешности в значениях Ф° (Т) при 298,15; 3000 и 6000 К
могут достигать 2, 4 и 7 Дж -К" -моль".
Ранее таблица термодинамических функций PFC1 (г) не публиковалась.
Константа равновесия реакции PFC1 (г)=Р (r)+F (г)+С1 (г) вычислена с использованием
принятой в справочнике энергии атомизации
ДгН° @) — Do (PFG1) — 793 + 30 кДж • моль,
оцененной в предположении о равенстве средних энергий связей в молекулах PFC1, PF2 и РС12.
Принятому значению соответствует
AfH°(PFCl, г, 0) = —280,542 + 30 кДж • моль.
PFC1" (г). Термодинамические свойства газа, состоящего из отрицательных ионов фторид-
хлорида фосфора, в стандартном состоянии при температурах 298,15—6000 К приведены
в табл. 227.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.8. По аналогии с ионами PF^" и PCl^ в справочнике принимается, что PFC1~ имеет синг-
летное основное электронное состояние Х1А1 со структурными параметрами г (Р—F) = l,60 +
+0,05 А, г (Р—С1)=2,00+0,05 А, -^ F—Р—С1 = 100+10°. Приведенное в табл. 14.8 значение IJBIo
рассчитано по этим структурным параметрам, его погрешность составляет 3-10~115 г3-см8.
Приведенные в таблице значения основных частот PFC1T оценены на основании принятых основных частот
PFg и PGlj, их погрешности оцениваются в 100 см.
Термодинамические функции PFG1" (г), приведенные в табл. 227, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124),
{1. 128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых значений
молекулярных постоянных и приближенным характером расчета. Погрешности значений Ф° (Т)
при 298,15; 3000 и 6000 К составляют 2, 4 и 7 Дж К -моль, в том числе из-за неточности
молекулярных постоянных 2—4 Дж-К-моль.
Ранее таблица термодинамических функций PFC1~ (г) не публиковалась.
Константа равновесия реакции PFC1~ (r)=P (r)+F (г)+С1 (г)+е~ (г) вычислена с
использованием значения Дг Н° @) = 1033+60 кДжмоль, соответствующего принятому в справочнике
сродству PFC1 к электрону
Ао (PFC1) = —2,5 ± 0,5 эВ = —240 + 50 кДж • моль.
381
PF2C1 (г), PF4C1 (г) Глава 14
Эта величина была оценена по аналогии с принятыми в справочнике значениями сродства
к электрону молекул PF2 и РС12.
Принятому значению Ао (PFG1) соответствует
Д/ЯО(РРСГ, г, 0) = —520,542 ± 50 кДж • моль.
PF2C1 (г). Термодинамические свойства газообразного дифторид-хлорида в стандартном
состоянии при температурах 100—6000 К приведены в табл. 228.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PF2C1 в основном электронном состоянии ХгА имеет структуру
треугольной пирамиды с атомом фосфора в вершине (симметрия Са) с параметрами г (Р—F) = 1,571 +0,003 А,
г (Р—С1) =2,030 +0,006 А, -3 F—P—F=97,3+0,2°, J} F—Р—С1=99,2+0,3°. Эти значения были
получены Бриттеном и др. [787] по вращательным постоянным PF235C1 (^40=0,2535100, Во =
=0,1252306, Со=0,0970091 см) и PF237C1 (Л „=0,2534290, #„=0,1213366, С0=0,0946661 см),
найденным в результате анализа микроволнового спектра. Приведенное в табл. 14.13 значение
IaIbIo вычислено по этим вращательным постоянным с учетом содержания изотопов в природном
хлоре. Колебательные спектры PF2C1 исследовались в работах [2738, 2742, 1215]. Приведенные
в таблице значения основных частот PF2C1 определены Мюллером и др. [2742] по ИК-спектру
газообразного PF2C1, их погрешности оцениваются в 2—3 см.
Термодинамические функции PF2C1 (г), приведенные в табл. 228, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A.124),
A. 128), A. 130). Погрешности вычисленных значений обусловлены преимущественно
приближенным характером расчета, они составляют 0,5; 3 и 6 Дж-К моль в значениях Ф° (Т) при
298,15; 3000 и 6000 К. Погрешности из-за неточности молекулярных постоянных не превышают
0,2 Дж-К^-моль.
Ранее таблицы термодинамических функций PF2C1 (г) вычислялись в работах [21, 2738, 2781,
2742] для {Т <^ 2000 К). В расчетах [21, 2738, 2781] принимались неточные значения
молекулярных постоянных PF2C1, соответствующие расхождения с данными табл. 228 достигают
3,5 Дж -К -моль. Данные работы [2742] и табл. 228 согласуются в пределах 0,2 Дж -К -моль.
Константа равновесия реакции PF2G1 (г) = Р (г)+2F (r)-f-Cl (г) вычислена с использованием
принятого в справочнике значения
\Н° @) = Do (PF2C1) = 1320 + 20 кДж • моль,
оцененного в предположении о равенстве средних энергий соответствующих связей в молекулах
PF3, PC1S и PFgCl.
Принятому значению энергии диссоциации PF2G1 соответствует
A/Bro(PF2Cl, г, 0) = — 730,267 + 20 кДж • моль.
PF4C1 (г). Термодинамические свойства газообразного тетрафторид-хлорида фосфора в
стандартном состоянии при температурах 100—6000 К приведены в табл. 229. '
' Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PF4C1 в основном электронном состоянии ХгА имеет структуру
треугольной бипирамиды с атомом хлора в экваториальной плоскости (симметрия C2v), как это было
показано в работах [1984, 535] на основании изучения колебательных спектров PF4C1. Возможно
также существование других изомеров с атомом хлора на оси третьего порядка и структурой
четырехгранной пирамиды. Приведенное в табл. 14.13 значение IaIbIc вычислено по
структурным параметрам г (Р—F1Ftc) = 1,58+0,05 А, г (Р—Fm) = 1,53+0,05 А, г (Р—С1рад)=2,02+0,05 А,
-^ FaK0—P—FPM=90°, -^ FPM—Р—Fw= ДРрад—Р—С1рад=120°, оцененным по принятым в
справочнике постоянным молекул PF5 и РС15. Его погрешность оценивается в 5 -10~т г3 -см6. Колебательные
спектры PF4C1 исследовались в работах [908, 1984]. Приведенные в таблице значения основных
частот определены Холмсом [1984] на основании изучения колебательных спектров и сравнения
со значениями соответствующих частот аналогичных молекул. Погрешности этих значений
оцениваются в 5—10 см и 20—40 см для частот v5, ve, ve, v12.
Термодинамические функции PF4G1 (г), приведенные в табл. 229, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых молеку-
382
Фосфор и его соединения PFCI2 (г), PF3C12 (г)
лярных постоянных, пренебрежением существования изомеров PF4C1 и приближенным методом
расчета, в значениях Ф° (Т) они составляют 4, 9 и 15 Дж -К-моль при 298,15; 3000 и 6000 К.
Ранее таблица термодинамических функций PF4C1 (г) не публиковалась.
Константа равновесия реакции PP4G1 (r) = P (r)+4F (г)+С1 (г) вычислена с использованием
принятого в справочнике значения энтальпии атомизации PF4C1
± 100 кДж • моль'1.
Эта величина оценена по средним энергиям связи Р—F и Р—G1 в молекулах PF8 и РС15 D57
и 257 кДж-моль соответственно), рассчитанным без учета различия энергии экваториальных
и аксиальных связей в этих молекулах.
Принятому значению энтальпии атомизации PF4C1 соответствует
A^°(PF4C1, г, 0) = —1355,717 ± 100 кДж • моль.
PFCla (г). Термодинамические свойства газообразного фторид-дихлорида фосфора в
стандартном состоянии при температурах 100—6000 К приведены в табл. 230.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PFC12 в основном электронном состоянии ХгА имеет структуру
треугольной пирамиды с атомом фосфора в вершине (симметрия С,). Приведенное в табл. 14.13 значение
IaIbIc соответствует структурным параметрам г (Р—F) = l,56+0,03 А, г (Р—С1)=2,04+0,03А,
-^ F—Р—Cl= -^ C1—Р—С1=100+5°, оцененным на основании структурных параметров
молекул PF3, PF2C1, PC13 (см. выше). Близкие значения были получены в электронографическом
исследовании [790]. Погрешность этого значения IaIbIc составляет 5-10~114 г3-см6. Колебательные
спектры PFC12 исследовались в работах [2741, 2742, 1214]. Приведенные в таблице значения
основных частот PFC12 определены Мюллером и др. [2742] в результате исследования ИК-спектра
газообразного PFC12, их погрешности составляют от 2—3 до 10 см.
Термодинамические функции PFC12 (г), приведенные в табл. 230, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A.3)—A.10), A.122)—A. 124),
A.128), A. 130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным методом расчета. Погрешности в значениях Ф° (Т) при 298,15;
3000 и 6000 К составляют 1, 4 и 7 Дж-К моль.
Ранее таблицы термодинамических функций PFC12 (г) вычислялись в работах [3798, 21, 2781,
2742] при Т ^ 2000 К. Расхождения в значениях термодинамических функций PFC12 (г),
приведенных в этих работах и в табл. 230, обусловлены различиями принятых в расчетах молекулярных
постоянных и не превышают 3,5 Дж -К-моль в значениях Ф° (Т).
Константа равновесия реакции PFC12 (r)=P (r)+F (г)+2С1 (г) вычислена с использованием
принятого в справочнике значения
ДД° @) = Do (PFC12) = 1140 + 20 кДж • моль,
оцененного в предположении о равенстве средних энергий соответствующих связей в молекулах
PF3, PC13 и PFC12.
1 Принятому значению энергии диссоциации PFC12 соответствует
г, 0) = —507,952+ 20 кДж • моль.
PF3C12 (г). Термодинамические свойства газообразного трифторид-дихлорида фосфора в
стандартном состоянии при температурах 100—6000 К приведены в табл. 231.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PF3C12 в основном электронном состоянии ХгА имеет структуру
треугольной бипирамиды, соответствующую точечной группе симметрии C2v, как это было показано
исследованиями ее колебательных спектров [1732, 3294] и спектра ЯМР [1987, 2503]. Возможно
существование других изомеров этой молекулы, в которых один или оба атома хлора находятся
в аксиальном положении. Приведенное в табл. 14.13 значение IaIbIc вычислено на основании зна-
чений г (P-F8KC)= 1,58 ± 0,03 А, г (Р - FpaJ= 1,53 + 0,03А, г(Р-С1рад) = 2,02 + О.ОЗА, ^ F«c -
—Р—FPM = 90°, J$ Fpw—Р—С1рад = J$ С1рад—Р—С1рад = 120 + 5°, оцененных по принятым в
справочнике структурным параметрам молекул PF5 и РС15. Его погрешность оценивается в 1 -10~ш г3 -см6.
Основные частоты PF3C12 определялись в работах [1732, 3294, 1985, 535] на основании изучения
383
PF2CI3 (г) Глава 14
колебательных спектров, полученных в работах [1732, 3294], а также исследований спектров
молекул PF4G1 и CH3PF4 в работах [1984, 1314]. Приведенные в таблице значения основных частот
PF3C12 определены Гриффитсом и др. [1732] и Солтхаузом и Уоддингтоном [3294], а отнесение
vx—v6, v8, v10, vn принято по Холмсу [1985], v7 по Битти и др. [535] и v6, v9, v12на основании
сравнения значений основных частот PF3C12 с принятыми в справочнике соответствующими основными
частотами PF5, РС15 и C1F3. Погрешности принятых значений частот не превосходят 5—10 см.
Термодинамические функции PF3C12 (г), приведенные в табл. 231, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A. 124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных, пренебрежением существованием изомеров PF3G12 и приближенным
характером расчета. Погрешности в значениях Ф° (Т) при 298,15; 3000 и 6000 К могут достигать 4, 8 и
15 Дж-К^-моль-1.
Ранее таблица термодинамических функций PF3G13 (г) не публиковалась.
Константа равновесия реакции PF3C12 (r) = P (r)+3F (г)+2С1 (г) вычислена с использованием
принятого в справочнике значения
Ar#°@) = D0(PF3Cl2) —1900 + 100 кДж • моль,
оцененного по средним энергиям связей Р—F и Р—С1 в молекулах PF5 и РС15 (см. выше PF4G1).
Принятому значению энтальпии атомизации PF3C12 соответствует
A//°(PF3GI2, г, 0) = —1113,372 + 100 кДж • моль.
PF2CI3 (г). Термодинамические свойства газообразного дифторид-трихлорида фосфора в
стандартном 'состоянии при температурах 100—6000 К приведены в табл. 232.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PF2G13 в основном электронном состоянии ХХА имеет структуру
треугольной бипирамиды, соответствующую точечной группе симметрии D3h, как это было показано
исследованиями колебательных спектров [1732] и спектра ЯМР [1987]. По-видимому, существуют
другие изомеры молекулы PF2C13 с одним и двумя атомами хлора в аксиальном положении.
Приведенное в табл. 14.13 значение IaIbIc вычислено по структурным параметрам г(Р—FaBe)= 1,58 +
+ 0,03 А, г(Р-С1рад) = 2,02 + 0,03 А, ^ FaBe-P-ClpM = 90°, ^ С1рад-Р-С1рад = 120°, оцененным
по принятым в справочнике данным для PF5 и РС15. Погрешности величины IaIbIc оцениваются
в 2-10~113 г3-см6. Основные частоты PF2C13 определялись Гриффитсом и др. [1732] по ИК-спектру
газообразного и спектру КР жидкого PF2G13. Приведенные в таблице значения v3, v4, v5, v6
определены в работе [1732] по ИК-спектру, av, — по спектру КР. Для частот v1? v2, v8 в табл. 14.13
приняты значения, основанные на данных [1732] по спектру КР жидкого PF2C13 с учетом поправок
на фазовый сдвиг, оцененных по колебательным спектрам жидких и газообразных PF5 и РС15.
Погрешности принятых значений основных частот оцениваются в 2—10 см.
Термодинамические функции PF2Gl3(r), приведенные в табл. 232, вычислены в приближении
«жесткий ротатор — гармонический осциллятор» по уравнениям A. 3)—-A.10), A.122)—A.124),
A. 128), A. 130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных, пренебрежением существованием изомеров PF2G13 и приближенным
характером расчета. Погрешности в значениях Ф° (Т) при 298,15; 30Э0 и 6ЭЭ0 К составляют 4, 10 и
15 Дж • К • моль.
Ранее таблицы термодинамических функций PF2C13 (г) вычислялись в справочнике [66]
до 5000 К и в работе [1732] до 1000 К. Расхождения данных, приведенных в табл. 232 и работах
[66, 1732], составляют 0,25 и 2,1 Дж-К~1-моль~1 в значениях Ф° (Г) соответственно.
Константа равновесия реакции PF2G13 (r)=P (r)+2F (г)+ЗС1 (г) вычислена с использованием
принятого в справочнике значения
ДгЯ° @) = Do (PF2G13) = 1700 ± 100 кДж • моль,
оцененного по средним энергиям связей Р—F и Р—G1 в молекулах PF5 и РС15 (см. выше PF4C1).
Принятому значению энтальпии атомизации PF2C13 (г) соответствует
A/F°(PF2C13, г, 0) = — 871,027 ± 100 кДж • моль.
384
Фосфор и его соединения PFCI4 (г), POFaCl (г)
PFC14 (г). Термодинамические свойства газообразного фторид-тетрахлорида фосфора в
стандартном состоянии при температурах 100—6000 К приведены в табл. 233.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула PFC14 в основном электронном состоянии ХгА имеет структуру
треугольной бшшрамиды (симметрия С3е), как это было показано исследованиями ее колебательных
спектров [1732]. Возможно существование изомера PFG14 с атомом фтора в экваториальном положении.
Приведенное в табл. 14.13 значение IaIbIg вычислено по структурным параметрам: г (Р—F4K0) =
= 1,58 ± 0,03 А, г (Р-С1акс) = 2,12 ± 0,03 А, г (Р-С1ИД) = 2,02 ±0,03 А, ^ FaKe-P-Glw = ^ С11КС-
—Р—С1рад = 90°, -=$ С1рад—Р—С1рад = 120°, оцененным по аналогичным постоянным молекул PF5 и РС15.
Погрешность этого значения IaIbIg оценивается в 5 -10~113 г3-см6. Приведенные в табл. 14.13
значения основных частот PFG14 определены Гриффитсом и др. [1732] по ИК-спектру
газообразного и спектру КР жидкого PFG14, их погрешности составляют 5—10 см.
Термодинамические функции PFC14 (г), приведенные в табл. 233, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A. 122)—A.124),
A.128), A. 130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных, пренебрежением изомерами молекулы PFG14 и приближенным характером
расчета. Погрешности значений Ф° (Т) при 298,15; 3000 и 6000 К составляют 4, 10 и
15 Дж-К~1'Моль~1.
Ранее таблица термодинамических функций PFG14 (г) не публиковалась.
Константа равновесия реакции PFC14 (r)=P (r)+F (r)-j-4Cl (г) вычислена с использованием
значения
АгН° @) = Do (PFG14) = 1500 + 100 кДж • моль,
оцененного по средним энергиям связей Р—F и Р—С1 в молекулах PF5 и РС15 (см. выше PF4C1).
Принятому значению энтальпии атомизации PFC14 (г) соответствует
14, г, 0) = —628,682 ±80 кДж • моль~х-
POF2C1 (г). Термодинамические свойства газообразного оксид-дифторид-хлорида фосфора
в стандартном состоянии при температурах 100—6000 К приведены в табл. 234.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула POF2C1 в основном электронном состоянии Х1А имеет структуру,
соответствующую точечной группе симметрии С,. Приведенное в табл. 14.13 значение IaIbIo вычислено
по структурным параметрам г (Р—О)=1,44+0,03 А, г (Р—F)=1,52 +0,03 А, г (Р—С1)=2,01 +
+0,03 А, .3 F—Р—F=^ Cl—P—F=102+5°, оцененным на основании структурных параметров
молекул PF3, PF2G1, PFG12, PC13, POF3, POG13 с учетом результатов электронографического
исследования [790] и данных, полученных в работе [3215]. Погрешность значения IaIbIg оценивается
в 5-10~ш г3-см6. В работе [1022] по микроволновому спектру были определены значения
Ao—1U (Во+Со) Для молекул POF235C1 и POF237C1. Колебательные спектры POF2C1 исследовались
в работах [1213, 2737, 2739]. Приведенные в таблице значения основных частот POF2C1 найдены
Мюллером и др. [2737, 2739] в результате исследования ИК-спектра газообразного POF2G1, их
погрешности составляют от 2 до 10 см.
Термодинамические функции POF2C1 (г), приведенные в табл. 234, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета, они могут достигать 2, 6 и
10 Дж'К~1.моль~1 в значениях Ф° (Т) при 298,15, 3000 и 6000 К, в том числе из-за неточности
молекулярных постоянных 1—3 Дж.К~1«моль~1.
Ранее таблица термодинамических функций POF2C1 (г) вычислялась в справочнике JANAF
{2095]. Расчет был выполнен на основании неточных значений частот и структурных параметров,
соответствующие расхождения данных справочника [2095] и табл. 234 составляют от 1 до
6 Дж-К~1-моль~1 в значениях Ф° (Г).
Константа равновесия реакции POF2C1 (г)=Р (г)+О (r)+2F (г)+С1 (г) вычислена с
использованием принятого в справочнике значения
@) = D0(POF2G1) = 1851 ± 25 кДж • моль,
25 Заказ П 295 385
POFCL, (г), PS (г) Глава 14
оцененного в предположении о равенстве средних энергий связей в молекулах POF2G1, POFS
и РОС13.
Принятому значению энтальпии атомизации соответствует
AfH° (POF2C1, г, 0) = —1014,484 + 25 кДж • моль.
POFC12 (г). Термодинамические свойства газообразного оксид-фторид-дихлорида фосфора,
в стандартном состоянии при температурах 100—6000 К приведены в табл. 235.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.13. Молекула POFC12 в основном электронном состоянии ХгА имеет структуру,
соответствующую точечной группе симметрии Cs. Приведенное в табл. 14.13 значение IaIbIc вычислено
по структурным параметрам г (Р—О) = 1,45±0,03 А, г (Р—F) = l,51+0,03 А, г (Р—С1) = 1,99±
+0.03 А, -==? F—Р—С1=-$ С1—Р—С1=103+5°, оцененным на основании принятых в справочнике
структурных параметров молекул PF3, PF2C1, PFC12, PC13, POF3, POC13 с учетом результатов элек-
тронографического исследования [790 ]. Погрешность значения IaIbIc оценивается^ 5 -10~114 г3 -см6.
Колебательные спектры POFC12 исследовались в работах [1213, 2737, 2739]. Приведенные
в табл. 14.13 значения основных частот POFC12 определены Мюллером и др. [2737] в результате
исследования ИК-спектра газообразного POFC12, их погрешности оцениваются в 2—10 см.
Термодинамические функции POFC12 (г), приведенные в табл. 235, вычислены в приближении
«жесткий ротатор—гармонический осциллятор» по уравнениям A. 3)—A.10), A.122)—A.124),
A.128), A.130). Погрешности вычисленных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета, они составляют 2, 6 и 10 Дж -К -моль
в значениях Ф° (Т) при 298,15, 3000 и 6000 К. Погрешности из-за неточности молекулярных
постоянных могут достигать 1—3 Дж-К~1.моль.
Ранее таблицы термодинамических функций POFC12 (г) вычислялись в справочнике JANAF
[2095] и в работе [3027] (до 1000 К). Данные справочника [2095] и табл. 235 согласуются в
пределах 0,3 Дж-К^-моль в значениях Ф° (Т).
Константа равновесия реакции POFG12 (г) = Р (г)+О (r)+F (г)+2С1 (г) вычислена с
использованием принятого в справочнике значения
АгН° @) = D0(POFG12) = 1666 ± 25 кДж • моль,
оцененного в предположении о равенстве средних энергий связей в молекулах POFC12, P0F3
и РОС13.
Принятому значению энтальпии атомизации соответствует
AfH° (POFC1Z, г, 0) = —787,139 + 25 кДж • моль.
PS (г). Термодинамические свойства газообразного сульфида фосфора в стандартном состоянии:
при температурах 100—6000 К приведены в табл. 236.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 14.12. В спектрах PS наблюдались переходы между тремя электронными состояниями
этой молекулы А2Т1 -> Х2П и /22+->Х2П, которые были изучены в работах Дреслера [13201.
и Нарасимхама и др. [2801, 2817]. На основании известных данных о валентных электронных
состояниях молекул NS и РО (см. табл. 13.23 и 14.3) и результатов исследований электронного
спектра PS в справочнике принимается, что молекула PS имеет девять связанных валентных
состояний с энергиями до 50 000 см: Х2П, аШ, А2П, В2Ф, Ь^~, С2 A, D2!,-, Н2П, F2+. Приведенные-
в табл. 14.12 значения молекулярных постоянных PS в состояниях Х2П, А2Н и /22 + определены
в работах [2817, 2801] на основании анализа колебательной структуры систем полос А2П—X4J
{р' ^ 4, v" <^ 8) и /22+—Х2П (г/ <^ 3, v" <^ 7) и анализа вращательной структуры полос 2—0»
и 1—о системы /2Е+—Х2И. Приведенные в таблице для PS энергии состояний а*П, В2Ф, й42~,
С2 A, ZJ2~ и Н2П оценены с помощью уравнения A.89) на основании принятых в справочнике
энергий возбуждения соответствующих состояний NS и РО с учетом расхождений между
оцененными таким же образом энергиями состояний -42Пи /22+и соответствующими экспериментальными
данными. Погрешности оцененных энергий возбуждения не превышают 2000—4000 см.
Термодинамические функции PS (г), приведенные в табл. 236, были рассчитаны по уравнениям
A.3)—A.10), A.93)—A.95). Значения QBB, Qm и QBB вычислялись по уравнениям A.90)—A. 92>
с учетом восьми возбужденных электронных состояний в предположении, что ОД] вр = {pjpx) Q^l
386
Фосфор и его соединения PN (г)
Статистическая сумма по колебательно-вращательным уровням энергии состояния ХЧ1 и
ее производные находились непосредственным суммированием по колебательным уровням и
интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались все
уровни энергии состояния Х2П со значениями J ^ /max, t, последние находились из условий A. 81);
в табл. 14.4 приведены соответствующие значения vmax и /цт. Колебательно-вращательные уровни
энергии этого состояния вычислялись по уравнениям A. 65), A. 42) иA. 62) с коэффициентами Ykl,
приведенными в таблице, эти значения Ykl были получены на основании постоянных молекулы PS,
представленных в табл. 14.12, и условий A. 50) и A.57). Вырождение уровней состояния Х2П
за счет Л-удвоения учитывалось статистическим весом 2. Погрешности вычисленных
термодинамических функций при Т > 2000 К обусловлены неточностью аппроксимации
колебательно-вращательных уровней энергии состояния Х2П и отсутствием экспериментальных данных о большинстве
возбужденных валентных состояний PS. Погрешности в значениях Ф° (Т) оцениваются в 0,03,
0,1 и 1 Дж-К^-моль-1 при 298,15; 3000 и 6000 К соответственно.
Ранее таблицы термодинамических функций PS (г) вычислялись в справочниках [88, 2095,
2556] и в справочнике [3585] (до 5000 К). Расчеты проводились приближенными методами по
постоянным PS, отличающимся от принятых в данном справочнике. Расхождения результатов этих
расчетов с данными табл. 236 составляют 0,2—0,8 и 0,2—1,3 Дж-К~1-моль~1 в значениях Ф° (Т)
и S° (Г). Существенно больше расхождения в значениях С° (Т), которые достигают 3,5 и
5 Дж-К^-моль, с данными [2095, 3585] и [2556].
Константа равновесия реакции PS (r)=P (r)-f-S (г) вычислена с использованием принятой
в справочнике энергии диссоциации
АгН° @) = Do (PS) = 440 + 12 кДж • моль'1 да 36 800 + 1000 см.
Принятое значение Do (PS) основано на результатах масс-спектрометрического исследования
реакции PS (r)+Y (r) = YS (г)+Р (г), выполненного Дровартом и др. [1328] при 2382—2420 К.
Расчет по методам II и III закона привел к значениям ДГЯ° @), равным соответственно —90,3 +
+ 9 и —83,4+0,5 кДж -моль. Принятое значение основано на второй величине. В расчетах
использовались термодинамические функции Y (г) и YS (г) из работ [3331, 1328] и значение Do (YS) =
=522,9 + 10 кДж-моль [1048] (см. также [3508]). Дроварт [1328] предположил, что отсутствие
в системе 222+—ХаП переходов ci;'>4 связано с предиссоциацией более высоких уровней, что
согласуется со значением D0(PS)=440 + 10 кДж-моль~х. Погрешность принятого значения
обусловлена главным образом неточностью величины D0(YS) и термодинамических функций YS(r).
Принятому значению Do (PS) соответствует'
A//°(PS, г, 0)= 150,348 + 12 кДж ¦ моль.
PN (г). Термодинамические свойства газообразного нитрида фосфора в стандартном состоянии
при температурах 100—6000 К приведены в табл. 237.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 14.12. В спектре PN изучена только одна система полос АЧ1—Х11,+ [1137, 1138,
1609, 1757, 2124, 2727]. На основании сопоставления связанных валентных состояний молекул N2
и Р2 (см. табл. 13.2 и 14.3) и результатов исследований спектра PN в справочнике принимается,
что молекула PN имеет пять связанных валентных состояний с энергиями возбуждения меньшими
50 000 см: X1!,+, а32 +, Ь3Н, АЧ1, сЧ1. Приведенные в табл. 14.12 значения молекулярных
постоянных PN в состояниях Х1И + шАЧ1 определены Керри и др. [1137, 1138] на основании исследования
системы полос ^441—Х1И+ (v' <I 9, v" <^ 10), они согласуются с результатами анализа
вращательного спектра PN в работах [4014, 1970]. Приведенные в таблице энергии состояний а32, bsIL, с3П
PN оценены на основании сравнения энергий возбуждения состояния АШ молекул PN, Р2 и N2
и энергий возбуждения состояний а32, Ь3П и с3П молекул Р2 и N2. Погрешности оцененных величин
могут достигать 4000 см.
Термодинамические функции PN (г), приведенные в табл. 237, были рассчитаны по
уравнениям A. 3)—A. 10), A.93)—A.95). Значения QBB,QW и Qm вычислялись по уравнениям A.90)—A.92)
с учетом четырех возбужденных электронных состояний в предположении, что Ql^J,.BV = (PilPx)X
X Qix] B • Статистическая сумма по колебательно-вращательным уровням энергии состояния X1??
и ее производные находились непосредственным суммированием с помощью уравнений A. 72)—
A.75), в расчете учитывались все уровни, ограниченные предельной кривой диссоциации этого
387
I'N (г) Глава 14
состояния. Колебательно-вращательные уровни рассчитывались по уравнению A. 65) с
коэффициентами Ykl, полученными на основании данных [1137, 1138] по началам полос системы АЧ1—
X1S+, вращательных постоянных, представленных в табл. 14.12 и условий A. 50) и A.57).
Соответствующие значения Ykl, коэффициенты а{ в уравнении, описывающем предельную кривую
диссоциации состояния X1!!+, а также величины утах и /цт приведены в табл. 14.4. Погрешности
вычисленных термодинамических функций обусловлены неточностью фундаментальных постоянных,
отсутствием данных о высоких колебательно-вращательных уровнях состояния XХ2 + и о
состояниях а3!, и 63П. Погрешности в значениях Ф° (Т) оцениваются в 0,02, 0,05 и 0,3 Дж-К~1-моль
при 298,15, 3000 и 6000 К.
Ранее таблицы термодинамических функций PN (г) вычислялись в справочниках [87, 88, 2095,
2556] и работах [3074, 3944] (до 6000 К), [2559] (до 1000 К). Несмотря на то, что в этих расчетах
не учитывались возбужденные состояния и был сделан ряд упрощений, их результаты согласуются
с данными табл. 237 в пределах 0,1 и 0,5 Дж-К"-моль~1 в значениях Ф° (Т) и S° (T). Существенно
больше расхождения в значениях С° (Т); они быстро растут с температурой и достигают 1
и 2,5 Дж-К^-моль-1 при 5000 и 6000 К с данными работ [20Э5, 2556] и др.
Константа равновесия реакции PN (г)=Р (r)+N (г) вычислена с использованием принятой
в справочнике энергии диссоциации
АгН @) = Do (PN) = 614 ± 15 кДж • моль = 51 300 ± 1300 см.
Принятое значение Do (PN) основано на величине 200+30 кДж-моль для энтальпии реакции
Р2 (r)+N2 (r)=2PN (г), вычисленной по методу III закона и значениям константы равновесия
при 1918—2194 К, найденным Гингеричем [1658] в результате масс-спектрометрического
исследования (расчет по II закону привел к значению 171+36 кДж-моль). В работе [1658] были
определены также значения константы равновесия реакции 2Р (r)+N3 (r)=2PN (г), которые, однако,
привели к менее точным значениям энтальпии соответствующей реакции, равным —270 +
+34 кДж-моль" (III) и —331+42 кДж-моль" (II). Принятое значение Do (PN) находится в
согласии со значениями, полученными в работах [1137, 1138] F01 кДж-моль), [1609]
F30 кДж-моль), [1757] F02 кДж-моль) линейной экстраполяцией уровней колебательной
энергии PN в состоянии ^4ХП. Измерения давления пара над PN (к), выполненные в работе [2030]
статическим методом при 1173 К, и масс-спектрометрическое исследование [3756] термического
разложения P3N5 (к) при 970—1170 К не позволяют определить надежные значения Do (PN)
вследствие ряда методических недостатков в этих работах (см. [1658]).
Принятому значению Do (PN) соответствует
Л J7°(PN, г, 0)= 172,381 + 15 кДж • моль.
ПРИЛОЖЕНИЯ
Приложение 1
АТОМНЫЕ ВЕСА, ИЗОТОПНЫЙ СОСТАВ
И СПИНЫ ЯДЕР ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
В настоящем издании при вычислении таблиц термодинамических свойств веществ использовались
следующие величины, характерные для каждого химического элемента:
1) атомный вес элемента';
2) содержание (распространенность) стабильных изотопов в природном элементе;
3) атомные массы изотопов;
4) ядерные спины изотопов.
В табл. П1.1 и П1.2 приведены принятые значения указанных величин.
Значения атомных весов элементов в табл. П1.1 основываются на рекомендациях Комиссии
по атомным весам ИЮПАК [2076а, 20766, 2076в] и соответствуют шкале, в которой атомная масса
изотопа 12С равна 12 (точно). С учетом примечаний приведенные величины считаются надежными в
пределах одного последнего десятичного знака или в пределах трех последних десятичных знаков,
если таковой напечатан ниже других. В скобках приведены значения атомных весов радиоактивных
элементов, которые не могут быть точно установлены без данных о происхождении этих элементов.
Приведенные в табл. П1.2 данные о процентном содержании изотопов в природном элементе
и величины ядерных спинов приняты в соответствии со значениями, рекомендованными в 1968 г.
Ледерером и др. [2371а], а также Комиссией по атомным весам в 1975 г. [2076 г]. Атомные массы
изотопов, приведенные в той же таблице, приняты по рекомендации Кравцова [134а]. Они также
основаны на шкале 12С = 12 (точно). Обоснование выбора указанных величин и соответствующие
библиографические данные могут быть найдены в упомянутых работах [2371а, 134а].
Таблица 111.1. Атомные веса химических элементов
Атомный
номер
1
2
3
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
21
22
23
Символ
Н
Не
Li
Be
В
С
N
О
F
Ne
Na
Mg 1
Al
Si
P
S
Cl
Ar
К
Ca
Sc
Ti
V
Наименование
Водород
^елий
Литий
Зериллий
Зор
Углерод
А.зот
кислород
Фтор
1еон
Затрий
Магний
Алюминий
Кремний
Фосфор
Сера
Хлор
Аргон
Калий
Кальций
Скандий
Титан
Ванадий
Атомный вес
1,007 9 6> г
4,002 60 «• в
6,94х в- г' д- х
9,012 18 а
10,81 *• г. Д
12,011 б> г
14,006 7 6- в
15,9994 б> *¦ г
18,998 40*
20,179 *¦ Д
22,989 77 а
24,305 в- ж
26,981 54 а
28,0855 г
30,973 76 а
32,06 г
35,453 в
39,948 б. Б> г> ж
39,0983
40,08 *
44,9559 а
47,90
50,9414б. в
Атомный
номер
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
Символ
Сг
Мп
Fe
Со
Ш
Си
Zn
Ga
Ge
As
Se
Br
Kr
Rb
Sr
Y
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Наименование
Хром
Марганец
Железо
Кобальт
Никель
Медь
Цинк
Галлий
Германий
Мышьяк
Селен
Бром
Криптон
Рубидий
Стронций
Иттрий
Цирконий
Ниобий
Молибден
Технеций
Рутений
Родий
Палладий
Атомный вес
51,996 в
54,938 0 »
55,847
58,933 2 а
58,70
63,546 в- г
65,38
69,72
72,59
74,921 6 а
78,9 ,
79,904 в
83,80 о
85,4678 Е
87,62 ж
88,905 9 а
91,22
92,906 4 а
95,94
(97)
101Л
102,905 5 а
106,4
1 В соответствии с рекомендацией Комиссии по атомным весам ИЮПАК [2076 г] термин «атомный вес»
используется для характеристики элемента, а термин «атомная масса» — для характеристики отдельных изотопов.
390
Атомные веса химических элементов
Таблица
Атомный
номер
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
III
1 (окончание)
Символ
Ag
Cd
In
Sn
Sb
Те
I
Xe
Cs
Ba
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Hf
Та
W
Re
Наименование
Серебро
Кадмий
Индий
Олово
Сурьма
Теллур
Иод
Ксенон
Цезий
Барий
Лантан
Церий
Празеодим
Неодим
Прометий
Самарий
Европий
Гадолиний
Тербий
Диспрозий
Гольмий
Эрбий
Тулий
Иттербий
Лютеций
Гафний
Тантал
Вольфрам
Рений
1 Мононуклидный элемент.
6 Элемент
" Элемент,
г Элемент,
атомного
д Элемент,
изотопов
с одним преобладающи*
для
ДЛЯ
которого атомный i
которого вариации
веса.
для
Атомный вес
107,868 в
112,41
114,82
118,6,
121,75
127,60
126,904 5 »
131,30 Д
132,905 4 а
137,33
138,9055 б
140,12
140,907 7 »
144,2,
A45)
150,4
151,96
157,25
158,925 4 •
162,50
164,930 4 •
167,2в
168,934 2 »
173,04
174,97
178,49
180,947 „ 6
183,8»
186,207 *
Атомный
номер
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
[ изотопом (распространенность
Символ
Os
Ir
Pt
Аи
Hg
Tl
Pb
Bi
Po
At
Rn
Fr
Ra
Ac
Th
Pa
U
Np
Pu
Am
Cm
Bk
Cf
Es
Fm
Md
No
Lr
Наименование
Осмий
Иридий
Платина
Золото
Ртуть
Таллий
Свинец
Висмут
Полоний
Астат
Радон
Франций
Радий
Актиний
Торий
Протактиний
Уран
Нептуний
Плутоний
Америций
Кюрий
Берклий
Калифорний
Эйнштейний
Фермий
Менделевий
Нобелий
Лоуренсий
99-100%).
юс основан на калиброванных измеренияз
изотопного состава
которого большие вариации в атомном
в имеющихся коммерческих образцах.
Атомный вес
190,2 а
192,2,
195,09
196,966 5 а
200,59
204,37
207,2 г- ж
208,980 4 а
B09)
B10)
B22)
B23)
226,025 4 е- *
B27)
232,038 1 е' *
231,035 9 е- ж
238,029б> в' л> *
237,048 2 е
B44)
B43)
B47)
B47)
B51)
B54)
B57)
B58)
B59)
B60)
в геологических образцах ограничивают точность данного
весе обусловлены небрежным или
е Данные относятся большей частью к изотопу с большим периодом полураспада.
ж В некоторых
геологических образцах этот элемент имеет в высшей степени аномальный
ответствующий атомной массе, отличной от данного
атомного веса.
неполным разделением
изотопный состав, со-
Таблица Ш.2.
Атомные массы изотопов химических элементов, распространенность изотопов
и ядерные спины
s
5 ф
°i
< я
1
2
3
4
5
в
леме
го
н
D
Т
Не
Li
Be
В
вое
8|
Й в-
1
2
3
4
6
7
9
10
11
Распространенность
(в %)
99,985
0,015
1,0.10-»
100*
7,5
92,5
100
20
80
* Распространенность изотопа
Атомная
масса
1,007 825 22
2,014 102 22
3,016 049 72
4,002 603 26
6,015 123 4
7,016 004 8
9,012 182 8
10,129 385
11,009 305 33
О-в «
V,
V,
0
1
3/2
и
3
3/2
6
7
8
9
й
ф
ч
СО
С
N
О
F
вое
1§
§8
12
13
14
15
16
17
18
19
Распространенность
(в %)
98,89
1,11
99,64
0,36
99,76
0,04
0,20
100
данного элемента с другой атомной массой меньше 0,01%
Атомная
масса
12 (точно)
13,003 355 08
14,003 074 40
15,000 109 3
15,994 915 02
16,999 133 3
17,999 159 96
18,998 404 6
°а S"
к
SS-
0
V,
1
V,
0
8/2
0
V,
391
Приложение 1
Таблица
ИН]
а&
°§
< и
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
м
а>
S
О)
к
го
Ne
Na
Mg
Al
Si
P
s
Cl
Ar
К
Ca
Sc
Ti
V
Cr
Mn
Fe
Co
Ni
Cu
Zn
П1.2
вое
ассо
1СЛО
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
37
36
38
40
39
40
41
40
42
43
44
46
48
45
46
47
48
49
50
50
51
50
52
53
54
55
54
56
57
58
59
58
60
61
62
64
63
65
64
66
67
68
70
(продолжение)
Распространенность
(в %)
90,51
0,27
9,22
100
78,99
10,00
11,01
100
92,23
4,67
3,1
100
95,02
0,76
4,22
75,77
24,23
0,34
0,07
99,59
93,26
0,01
6,73
96,941
0,647
0,135
2,086
0,004
0,187
100
8,0
7,5
73,7
5,5
5,3
0,25
99,75
4,35
83,79
9,50
2,36
100
5,8
91,8
2,1
0,3
100
68,27
26,10
1,13
3,59
0,91
69,2
30,8
48,6
27,9
4,1
18,8
0,6
Атомная
масса
19,992 440 5
20,993 847 4
21,991 384 8
22,989 770 3
23,985 044 3
24,985 838 5
25,982 594 4
26,981 540 6
27,976 928 6
28,976 496 9
29,973 772 2
30,973 763 3
31,972 072 8
32,971 459 1
33,967 870 1
34,968 853 59
36,965 903 02
35,967 546 5
37,962 733 0
39,962 384 2
38,963 708 9
39,964 000 1
40,961 827 0
39,962 592 1
41,958 618 1
42,958 777 4
43,955 487 5
45,953 689
47,952 526
44,955 917 4
45,952 629 6
46,951 767 0
47,947 949 1
48,947 872 1
49,944 784 3
49,947 164 3
50,943 964 4
49,946 048 8
51,940 510 2
52,940 651 0
53,938 881 3
54,938 046 4
53,939 612 0
55,934 933 9
56,935 390 7
57,933 274 5
58,933 187 9
57,935 335 8
59,930 779 5
60,931 050 2
61,928 339 6
63,927 956
62,929 589 8
64,927 789 0
63,929 140 0
65,926 039 5
66,927 132 2
67,924 848 1
69,925 325 4
=я d
3 •«
и
d< W ее
0
8/2
0
3/2
0
5/2
0
0
V,
0
V,
0
3/2
0
3/
3/2
0
0
0
8/2
4
3/2
0
0
7/.
0
0
0
7/
0
6/2
0
7/2
0
6
71%
0
0
3/2
0
6/2
0
0
а/2
0
7/2
0
0
3/2
0
0
3/2
3/2
0
0
8/2
0
0
3
ТОМЕ
шер
<q И
31
32
33
34
35
36
37
38
39
40
41
42
44
45
46
47
и
з
СО
Ga
Ge
As
Se
Вг
Кг
Rb
Sr
Y
Zr
Nb
Mo
Ru
Rh
Pd
Ag
вое
a ceo
1СЛО
69
71
70
72
73
74
76
75
74
76
77
78
80
82
79
81
78
80
82
83
84
86
85
87
84
86
87
88
89
90
91
92
94
96
93
92
94
95
96
97
98
100
96
98
99
100
101
102
104
103
102
104
105
106
108
110
107
109
Распространенность
(в %)
60
40
20,5
27,4
7,8
36,5
7,8
100
0,9
9,0
7,6
23,5
49,8
9,2
50,69
49,31
0,35
2,25
11,6
11,5
57,0
17,3
72,17
27,83
0,5
9,9
7,0
82,6
100
51,4
11,2
17,1
17,5
2,8
100
14,8
9,3
15,9
16,7
9,6
24,1
9,6
5,5
1,9
12,7
12,6
17,1
31,6
18,6
100
1,0
11,0
22,2
27,3
26,7
11,8
51,83
48,17
Атомная
масса
68,925 579 5
70,924 704 4
69,924 252 0
71,922 082 3
72,923 464 4
73,921 178 6
75,921 404 2
74,921 600 3
73,922 477
75,919 211 7
76,919 913 0
77,917 309 3
79,916 525 3
81,916 708
78,918 332 0
80,916 292
77,920 401
79,916 376
81,918 482
82,914 131
83,911 505 3
85,910 616
84,911 799 0
86,909 187 0
83,913 430 5
85,909 276 3
86,908 893 5
87,905 628 3
88,905 866 7
89,904 710 5
90,905 643 4
91,905 038 6
93,906 320 2
95,908 292
92,906 380 3
91,906 808 3
93,905 089 1
94,905 836 9
95,904 674 8
96,906 022 7
97,905 410 0
99,907 477 5
95,907 598
97,905 289
98,905 936 9
99,904 217 3
100,905 576 6
101,904 348 1
103,905 428
102,905 512
101,905 607
103,904 014
104,905 036
105,903 487
107,903 891
109,905 164
106,905 091
108,904 754 6
3 **-
и
К §.2.
3/2
3/
0
0
9/2
0
0
3/2
о
0
1/
0
0
0
з/г
о2
0
0
8/
0
0
5/
/2
3/2
0
0
9/2
0
V,
0
6/2
0
0
0
»/
0
0
8/2
0
*/
0
0
0
0
в/
0
6/.
0
0
V,
0
0
6/2
0
0
0
V,
V,
392
Атомные веса химических элементов
Таблица
°в
Я ш*
< Я
48
49
50
51
52
53
54
55
56
57
58
59
60
и
я
гсеме
со
Cd
In
Sn
Sb
Те
I
Xe
Cs
Ba
La
Ce
Pr
Nd
Ш.2
вое
О о
О м
||
106
108
110
111
112
113
114
116
ИЗ
115
112
114
115
116
117
118
119
120
122
124
121
123
120
122
123
124
125
126
128
130
127
124
126
128
129
130
131
132
134
136
133
130
132
134
135
136
137
138
138
139
136
138
140
142
141
142
143
144
145
(продолжение)
Распространенность
(в %)
1,2
0,9
12,4
12,8
24,0
12,3
28,8
7,6 •
4,3
95,7
1,0
0,7
0,4
14,7
7,7
24,3
8,6
32,4
4,6
5,6
57,3
42,7
0,1
2,5
0,9
4,6
7,0
18,7
31,7
34,5
100
0,1
0,1
1,9
26,4
4,1
21,2
26,9
10,4
8,9
100
0,1
0,1
2,4
6,6
7,9
11,2
71,7
0,09
99,91
0,2
0,3
88,4
11,1
100
27,2
12,2
23,8
8,3
Атомная
масса
105,906 463
107,904 189 4
109,903 010 1
110,904 185 5
111,902 762 8
112,904 407 4
113,903 366 8
115,904 761 5
112,904 089
114,903 875
111,904 834
113,902 776
114,903 353
115,901 748 3
116,902 960 6
117,901 612 6
118,903 315 9
119,902 207 3
121,903 451 1
123,905 283
120,903 822 3
122,904 220 3
119,904 024
121,903 056 0
122,904 281 7
123,902 830 2
124,904 426 3
125,903 312 4
127,904 467 5
129,906 231 9
126,904 475 5
123,906 12
125,904 279
127,903 532 3
128,904 784
129,903 510 8
130,905 084 7
131,904 156 8
133,905 398
135,907 222
132,905 436
129,906 284
131,905 045
133,904 493
134,905 671
135,904 559
136,905 815
137,905 235
137,907 161
138,906 403
135,907 180
137,906 027
139,905 484
141,909 300
140,907 698
141,907 766
142,909 856
143,910 129
144,912 610
'я 5"
Н .
^ И ее
ЬЧ о w-
0
0
0
V,
0
V,
0
0
8/2
оа
0
V,
0
V,
0
а/2
0
0
0
Б/
'2
о3
0
V,
0
V,
0
0
0
8/2
0
0
0
V,
0
8/
0
0
0
If
0
0
0
з/
0
3/2
0
5
0
0
0
0
5/2
0
V,
0
я
a e.
g Я
62
63
64
65
66
67
68
69
70
71
72
73
74
75
н
н
пеме
го
Sm
Ей
Gd
ТЬ
Dy
Но
Ег
Тт
Yb
Lu
Hf
Та
W
Re
вое
OlfOI
OOOB
2 в-
146
148
150
144
147
148
149
150
152
154
151
153
152
154
155
156
157
158
160
159
156
158
160
161
162
163
164
165
162
164
166
167
168
170
169
168
170
171
172
173
174
176
175
176
174
176
177
178
179
180
181
180
182
183
184
186
185
187
Распространенность
(в ¦%)
17,2
5,7
5,6
3,1
15,1
11,3
13,9
7,4
26,6
22,6
47,8
52,2
0,2
2,2
14,8
20,5
15,7
24,8
21,8
100
0,06
0,1
2,34
18,9
25,5
24,9
28,2
100
0,1
1,6
33,4
22,9
27
15,0
100
0,1
34
14,3
21,9
16,2
31,7
12,7
97,4
2,6
0,2
5,2
18,5
27,1
13,8
35,2
100*
0,1
26,3
14,3
30,7
28,6
37,4
62,6
Атомнзя
масса
145,913 153
147,916 929
149,920 721
143,912 074
146,914 925
147,914 851
148,917 211
149,917 303
151,917 755
153,922 222
150,919 883
152,921 260
151,919 817
153,920 891
154,922 636
155,922 143
156,923 972
157,924 123
159,927 071
158,925 386
155,924 326
157,924 440
159,925 231
160,926 970
161,926 838
162,928 770
163,929 218
164,930 357
161,928 826
163,929 235
165,930 329
166,932 079
167,932 402
169,935 491
168,934 245
167,933 925
169,934 792
170,936 354
171,936 405
172,938 234
173,938 881
175,942 582
174,940 796
175,942 705
173,940 140
175,941 429
176,943 245
177,943 723
178,945 840
179,946 575
180,948 028
179,946 70
181,948 248
182,950 266
183,950 975
185,954 402
184,953 007
186,955 791
* ?i
я •«
н
Ьч О-2-
0
0
0
0
7/2
0
7/
0
0
0
Б/2
0
0
3/2
0
8/
0
0
8/
0
0
0
6/2
0
Б/
0
7/
0
0
0
7/2
0
0
V,
0
0
V,
0
6/«
0
0
7/„
7
0
0
7/2
0
8/
0
"'/»
0
0
V,
0
0
Б/„
6/„
393
Приложение 1
Таб л
>s
2
S ^
|S
< §
76
77
78
79
i 80
и ц а
Е-
И
CD
S
CD
ГО
Os
Ir
Pt
Аи
Hg
Ш.2
iBoe
ассо
1СЛО
* ft-
184
186
187
188
189
190
192
191
193
190
192
194
195
196
198
197
196
(окончание)
Распространенность
(в %)
0,02
1,58
1,6
13,3
16,1
26,4
41,0
37,3
62,7
0,01
0,79
32,9
33,8
25,3
7,2
100
0,2
Атомная
масса
183,952 595
185,953 883
186,955 788
187,955 877
188,958 183
189,958 482
191,961 514
190,960 631
192,962 964
189,959 965
191,961 078
193,962 713
194,964 804
195,964 965
197,967 895
196,966 548
195,965 822
=з ?!.
2 •«
и
Ш ц CD
0
0
1/
0
з ¦
о2
0
3/
1
3/
0
о ¦
0
V,
0
0
8/
0
'я
Р S
< §
81
82
83
90
92
неме
го
Т1
РЬ
Bi
Th
и
вое
ассо
1СЛО
5? в*
198
199
200
201
202
204
203
205
204
206
207
208
209
232
234
235
238
Распространенность
(в %)
10,1
16,9
23,1
13,2
29,7
6,8
29,5
70,5
1,4
24,1
22,1
52,4
100
100
0,005
0,720
99,275
Атолтнэя
X *- X *—' XIL -1J. 1^4. JX
масса
197,966 748
198,968 275
199,968 321
200,970 304
201,970 643
203,973 498
202,972 348
204,974 438
203,973 049
205 974 475
206,975 903
207,976 658
208,980 401
232,038 074
234,040 975
235,043 944
238,050 816
:Е! ем
Я •?
0
1/
0
з/
0
0
V,
V
0
0
1/
0
f/a
0
0
41
0
Приложение 2
ПРИНЯТЫЕ ЗНАЧЕНИЯ
ФУНДАМЕНТАЛЬНЫХ ПОСТОЯННЫХ
И ПЕРЕВОДНЫХ МНОЖИТЕЛЕЙ ДЛЯ ЕДИНИЦ ЭНЕРГИИ.
ТЕМПЕРАТУРНАЯ ШКАЛА
Постоянная
Единица
Значение постоянной
"л
к
R
—г см-К
he
~к
h
Дж-с
моль J
Дж • К
Дж • К ¦ моль
кал • К • моль
•физические постоянные. При применении соотношений для расчета термодинамических функций
по молекулярным постоянным необходимо знать численные значения следующих физических
постоянных: h — постоянной Планка, с — скорости света в вакууме, к — постоянной Больцмана,
NA — числа Авогадро, R — универсальной газовой постоянной. Кроме того, для расчета
термодинамических функций и в особенности для определения численных значений термодинамических
величин по результатам экспериментальных измерений необходимо знать численные значения
переводных множителей для различных единиц энергии, соответствующие принятым значениям
основных физических постоянных.
Все расчеты, проводившиеся при под- Таблица П2.1. Физические постоянные
готовке настоящего издания, основывались
на единой системе значений физических
постоянных, приведенных в табл. П2.1 и
принятых в соответствии с рекомендацией
КОДАТА—MGHG [1001] (см. также [274а]).
Помимо основных постоянных в таблицу
включены множители, часто используемые
в расчетах как постоянные.
Переводные множители для единиц
-энергии, соответствующие принятым
фундаментальным постоянным, приведены
в табл. П2.2. Кроме множителей,
включенных в табл. П2.2, при сравнении
результатов квантовомеханических расчетов и
экспериментальных спектроскопических
данных использовалось соотношение
1 ат. ед. энергии=219 474,65 см.
В настоящем справочнике (как и в дру-
тих справочниках по термодинамическим
•свойствам веществ) за стандартное
давление принято давление в одну стандартную физическую атмосферу A атм=1,013 25-Ю5 Па).
Температурная шкала. В настоящем издании принята термодинамическая (абсолютная) шкала
¦температур (шкала Кельвина). Эта шкала определяется посредством одной лишь фиксированной
¦точки — тройной точки воды, температура которой принята равной 273,16 К (точно).
Поскольку температура плавления льда @° С) при стандартном давлении на 0,01 К ниже
температуры тройной точки, эта температура @° С) соответствует температуре 273,15 К, а
стандартная температура 25° С соответственно составляет 298,15 К.
В экспериментальных исследованиях обычно принимается Международная практическая
температурная шкала (сокращенно МПТШ), которая является достаточно хорошим приближением
ж термодинамической шкале. Положение о МПТШ периодически подвергается пересмотру и
уточнению с учетом новейших метрологических исследований. В настоящем справочнике используется
шкала МПТШ-68 [2067]; представление о соответствии этой шкалы с термодинамической шкалой
можно получить из табл. П2.3, в которой приведены «определяющие постоянные точки МПТШ-68»
•с оценкой погрешности их определения по термодинамической шкале.
В табл. П2.4 приведены поправки к температурам, которые следует учитывать при пересчете
экспериментальных значений температур от МПТШ-48 к МПТШ-68. Очевидно, что
целесообразность внесения этих поправок определяется соотношением величин поправок с погрешностью
исправляемой величины.
г-см
Г • СМ2 • С
Г • СМ • МОЛЬ"
299 792 458 + 1,2
F,626 176+0,000036) -10*
F,022 045+0,000 031) ¦ 102S
A,380 662 +0,000 044). 103
8,314 41+0,00026
1,987 19+0,00006
1,438 786 ±0,000 045
B,799 320+0,000015) • 109
(8,392 150+0,000046) ¦ 10"а»
A6,857 630+0,000028) • 10в
395
Таблица П2.2. Переводные множители для единиц энергии
Единица
эрг•молекула"
Дж-моль"
кал-моль"
эВ-молекула
см
эрг-молекула 1
Дж • моль-1 A,660 565 5±0,000 008 5) • 10-"
кал ¦ моль-' F,947 806+0,000 035) • 10-"
эВ-молекула-1 A,602 189 2+0,000 004 6) • 10-'1
СМ-' A,986 478+0,000 011) • 10~"
F,022 045+0,000 031)
1
4,184
96 484,55+0,27
11,962 657+0,000 016
10" A,439 303 3±0,000 007 31 • 10м
0,239 005 7
1
23 060,362+0,065
2,859 143 7+0,000 003 9
F,241 46О±О,ООО 018) • 10"
A,036 435 3+0,000 002 9) • 10"»
D,336 445+0,000 012) • 10~5
1
A,239 852 0+0,000 003 2) • 10т*
E,034 036+0,000 027) . 10"
(8,359 347+0,000 011) • 10""
C,497 551+0,000 048) . 10
8 065,479+0,021
1
Таблиц а П2.3. Определяющие постоянные точки МПТШ-68
Вещество
Тип фазового
равновесия
Р
атм
Г68
К
°с
Но, равн. Тройная точка 6,939-10-2 13,81+0,01 —259,34
Точка кипешш 0,328 95 17,042+0,01 —256,108
То же 1,000 20,28+0,01 —252,87
Ne Точка кшения 1,000 27,102+0,01 —246,048
О, Тройная точка 1,54-10-3 54,361 + 0,01 —218,789
Точка кипения 1,000 90,188+0,01 —182,962
Вещество
Тип фазового
равновесия
Р
атм
7б8
К
*68
°с
Н2О Тройная точка 6,03 .Ю"» 273,16 0,01
Точка кипения 1,000 373,150 100,000+0,005
Sn Точка плавле- 505,1181 231,9681 + 0,015
ния
Zn To же 692,73 419,58+0,03
Ag » » 1235,08 961,93+0,2
Аи » » 1337,58 1064,43+0,2
Таблица П2.4. Поправка к температурам при переходе от МПТШ-48 к МПТШ-68
'68 ^
— 100
0
0
100
200
300
400
500
600
700
800
900
«68 °С
1000
t 2000
3000
0
10
20
0,022 0,013 0,003
0,000 0,006 0,012
0,000 -0,004 -0,007
0,000 0,004 0,007
0,043 0,047 0,051
0,073 0,074 0,075
0,076 0,075 0,075
0,079 0,082 0,085
0,150 0,165 0,182
0,39 0,42 0,45
0,67 0,70 0,72
0,95 0,98 1,01
0
100
200
1,2 1,5 1,7
3,2 3,5 3,7
5,9 6,2 6,5
Примечание. Величины <в8—1№ даны в
30
—0,006
0,018
—0,009
0,012
0,054
0,076
0,075
0,089
0,200
0,47
0,75
1,04
300
1,8
4,0
6,9
келышнах.
40
—0,013
0,024
—0,010
0,016
0,058
0,077
0,074
0,094
0,23
0,5'J
0,78
1,07
400
2,0
4,2
7,2
50
-0,013
0,029
—0,010
0,020
0,061
0,077
0,074
0,100
0,25
0,53
0,81
1,10
500
2,2
4,5
7,5
60
—0,005
0,032
-0,010
0,025
0,034
0,077
0,074
0,108
0,28
0,53
0,84
1,12
600
2,4
4,8
7,9
70
0,007
0,034
—0,008
0,029
0,067
0,077
0,075
0,116
0,31
0,58
0,87
1,15
700
2,6
5,0
80
0,012
0,033
—0,006
0,034
0,069
0,077
0,076
0,126
0,34
0,61
0,89
1,18
800
2,8
5,3
8,6
90
0,029
—0,003
0,038
0,071
0,076
0,077
0,137
0,36
0,64
0,92
1,21
900
3,0
5,6
9,0
100° С
0,022
0,000
0,043
0,073
0,076
0,079
0,150
0,39
0,67
0,95
1,24
1000° С
3,2
5,9
9,3
Приложение 3
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ГАЗОВ
ПРИ ПОВЫШЕННОМ ДАВЛЕНИИ
В таблицах настоящего справочника приведены термодинамические функции, вычисленные в
приближении идеального газа. Эти данные используются для расчетов в ограниченном диапазоне
параметров, когда плотность вещества достаточно мала для того, чтобы можно было пренебречь
влиянием межмолекулярных взаимодействий. При низких температурах и высоких давлениях
отступления от законов идеального газа становятся заметными и проявляются как в изменении
величин соответствующих функций, так и в общем усложнении термодинамического описания.
При высокой степени сжатия (реальный газ или жидкость) термодинамический расчет требует
привлечения большой совокупности данных: по />УГ-свойствам вещества, термодинамическим
функциям и фазовому переходу жидкость—пар.
р^-свойства задаются в виде уравнения состояния, которое определяет в аналитической форме
связь трех параметров состояния: давления, объема (или плотности) и температуры.
Термодинамические функции (энтальпия, энтропия, теплоемкость и др.) вычисляются через уравнение
состояния посредством дифференциальных соотношений термодинамики. При этом уравнение состояния
позволяет рассчитать избыточные значения термодинамических функций, т. е. изотермические
приращения функций при переходе от стандартного состояния к состоянию реального газа при
той же температуре Т и давлении р.
Для характеристики линии фазового равновесия жидкость—пар задаются: 1) уравнение,
определяющее давление насыщенного пара, 2) уравнение или табличные данные для ортобарических
плотностей, т. е. плотностей насыщенной жидкости р' и пара р"; 3) положение узловых точек
на линии фазового равновесия: критической, тройной, а также нормальной точки кипения; 4)
таблицы термодинамических функций на линии насыщения с обеих сторон пограничной кривой.
Таким образом, общий объем справочных данных для реального газа или жидкости настолько
велик, что с достаточной полнотой он представлен только в специальных справочных изданиях,
посвященных свойствам одного или группы родственных соединений. Подобные издания
существуют для большинства технически важных газов. Однако для подавляющего большинства веществ
имеются крайне ограниченные данные по свойствам реального газа, не позволяющие построить
надежные таблицы термодинамических функций. В этих случаях могут быть использованы методы
приближенного термодинамического расчета (называемые также корреляциями), которые
позволяют с умеренной точностью предсказать все термодинамические свойства на линии насыщения
и в однофазной области. В качестве исходных данных для такого расчета используются
критические постоянные, температура нормальной точки кипения, дипольный момент молекулы и т. д.
Обобщенные корреляции целесообразно применять также и для технически важных газов, если
не требуется слишком высокая точность расчета. Преимущество использования приближенных
методик состоит в их относительной простоте и унификации расчетной схемы. В разделах П3.1 —
П3.5 Приложения рекомендованы некоторые из методов приближенного расчета
термодинамических свойств: обобщенные уравнения состояния, методы расчета второго вириального
коэффициента, термодинамических функций и свойств на линии насыщения. В разделе П3.6 дан краткий
обзор важнейших справочных изданий, содержащих данные по термодинамическим свойствам газов
и жидкостей в широком диапазоне параметров, а также по методам расчетов.
В разделе П3.7 приведены обзоры для отдельных веществ. В обзоры включены рекомендации
по выбору основных справочных изданий, таблиц термодинамических свойств, уравнений
состояния, методик расчета и т. д. Библиография не претендует на полноту — указаны только основные
справочные издания и статьи обзорного характера. Даны также ссылки на публикации, в которых
представлены новые опытные данные, не отраженные в справочных монографиях. В текстах часто
упоминаются уравнения состояния, на основе которых рассчитаны свойства веществ (уравнение
Мартина—Хоу, Бенедикта—Вебба—Рубина, Стобриджа и др.). Ознакомиться с различными ти-
397
Приложение $
пами уравнений состояния, применяемыми в современной литературе, можно по обзорным статьям
[2530, 3735], а также по монографии [222J.
Для некоторых технически важных газов приведена также таблица термодинамических свойств
при повышенных давлениях: для нескольких опорных значений Тир рассчитаны величины
мольного объема V и поправок на неидеальность к энтальпии и энтропии
ЬН = Н{Т, р) — Н°(Т),
bS = S{T,p)-S°{T) + Rln(p/p°).
При небольшой степени сжатия можно использовать упрощенное уравнение состояния,
содержащее только второй вириальный коэффициент В (Т):
Z=1 + B(T)/V,
где Z=pV/RT — фактор сжимаемости, V — мольный объем.
В этом случае все поправки к термодинамическим функциям идеального газа вычисляются
через В (Т) и его производные по температуре. В табл. Д.25 (наст, изд., кн. 2) даны коэффициенты
уравнений, аппроксимирующих температурную зависимость В (Т), что позволяет их использовать
непосредственно для расчета на ЭВМ. В табл. Д.26 приведены критические постоянные веществ.
Материал, изложенный в разделе П3.7, относится к свойствам индивидуальных соединений.
Исключение составляют ассоциированные вещества HF, S, Р, пары которых содержат полимерные
молекулы, а также четырехокись азота — реагирующая система N3O4 <^ 2NO2 ** 2NO+O2.
Применительно к этим веществам оценка неидеальных свойств отдельных компонентов лишена
физического смысла. Для ознакомления с методами инженерного расчета смесей можно рекомендовать
монографию Рида и Шервуда [222] и справочное руководство [167]. Основы статистической
термодинамики смесей и фазовых равновесий изложены в монографии Нраузница [30811.
П3.1. Уравнение состояния
В литературе предложен ряд обобщенных уравнений состояния, константы которых вычисляются
по известным критическим постоянным и некоторым дополнительным параметрам. Для так
называемых нормальных веществ, к которым относят неполярные или слабополярные соединения,
применима трехпараметрическая корреляция Питцера [3032, 3033, 3035], в которой исходными
параметрами для расчета являются критические постоянные TKf, ркр и фактор ацентричности,
определяемый соотношением
тг=о,т
Я \ с
где pr=p/pKV, Tr=T/Tsv, p —давление на линии насыщения. Vt. ~ ~"
На основе корреляции Питцера разработан ряд обобщенных уравнений состояния, применяемых
к широкому классу веществ. Ли и Кеслер [2374] предложили уравнение состояния в форме
"л о v - = V Г'с I К "\ t
Здесь о)л=0,3978, a Z0 и ZR —¦ два идентичных уравнения состояния типа Бенедикта—Вебба—
Рубина с различными константами v . , \f i; т с -
где B = \—b<LlTT—hJT\—bJT\, С=сх—с%1Тг—съ1Г1г, Д=^1+й2/Гг. В уравнении (П3.2) фактор-
сжимаемости Z=prVrlTr, где Vr=VpKV/RTKV (отметим, что в других уравнениях в качестве
параметров приведения используются FKp и ркр, так что Vr=V/VKV, рг=р/ркр — см. уравнения (П3.11,
12, 46, 47). Константы уравнений (ПЗ.З) приведены в табл. П3.1.
С помощью уравнения (П3.2) можно вычислить фактор сжимаемости Z, приведенный объем Vг
и все термодинамические свойства в интервале параметров: 0,3 < Тг < 4; 0 < рг < 10. Процедура
расчета изложена в разделе ПЗ.З. Параметр ш может быть определен из уравнения для давления
пара или по формуле (П3.4) [23741
398
Свойства газов при повышенном давлении
Таблица П3.1. Константы уравнений состояния (ПЗ.З)
Уравнение
Z°
Уравнение
ZR
с\ 0,118 119 3 0,202 657 9
62 0,265 728 0,331 511
Ъ3 0,154 790 0,027 655
64 0,030 323 0,203 488
Уравнение
Уравнение
ZR
сх 0,023 674 4 0,031 338 5
с2 0,018 698 4 0,050 361 8
с3 0 0,016 901
с4 0,042 724 0,041 577
Уравнение
Уравнение
ZR
dr104 0,155 488 0,487 36
<V104 0,623 689- 0,074 033 6,
р 0,653 92 1,226
Г 0,060167 0,037 54
Таблица
Вещество
о2
О3
N2
N2O
П3.2. Фактор ацентричности
со
0,021
0,185
0,04
0,1752
Литература
[167]
[222]
[222]
[167]
Вещество
NO
F3
Cl2
Вг2
со
0,5976
0,0712
0,0825
0,1333
Литература
167]
167]
167]
167]
Вещество
I.
SO,
H2S
SFe
со
0,229
0,510
0,1
0,257
Литература
[222]
[167]
[222]
[167]
—In рур (атм) — 5,927 14 + 6,096 048 б + 1,288 62 In 6 — 0,169 347
15,2518 — 15,6875а-1 — 13,4721 In 6 + 0,435 77 6°
где 6=Г4/77кр (Тъ — температура нормальной точки кипения).
Можно использовать и более простое соотношение [222]
з е
' = -1 + ТГ=в18Л
(П3.4)
(ПЗ. 5)
Данные по температуре нормальной точки кипения приведены в справочнике [258]. В табл. П3.2
приведены значения фактора ацентричности для ряда газов; для инертных газов и так называемых
квантовых газов (Не, Ne, Н2 и т. д.) и>=0.
Для более широкого класса веществ, включающего, наряду с нормальными, также полярные
и ассоциированные газы, может быть рекомендовано уравнение состояния Мартина—Хоу [2531,
2532]
где Fj(T) = Aj + BjTr + С,- exp (-KTr).
В работе Алтунина [17] предложена методика вычисления констант уравнения (П3.6) по
ограниченным опытным данным, которая обеспечивает возможность использования уравнения в
широком диапазоне параметров:
Исходные данные для расчета: критические постоянные Тщ, ркр, VKV, температура Бойля ТБг,-
наклон Mxv=MpKV/TKV критической изохоры и наклон Мп изохоры Vn=VKi/l,8.
Параметр М определяется из уравнения Риделя (П3.44) для давления пара (если известна,
хотя бы одна точка, кроме критической, например нормальная точка кипения) или из формулы
Z-] = 3,72 + 0,238 (М— 7), (П3.7)
рекомендованной в работе [17].
Мартин и Станфорд [2533] предложили для оценки М соотношение:
(П3.8>
'кр
1 Температура, при которой В (Г)=0.
399
Приложение 3
Наклон Мп изохоры Vn определяется соотношением
)^. (П3.9)
p
Температуру Бойля можно оценить с помощью соотношения [222]
ГБ == 30 — 2,42Гкр — 5,67 • \0гЧ\9, (П3.10)
После этого вычисляются все константы уравнения (П3.6) в последовательности, указанной
в табл. ПЗ.З.
Таблица ПЗ.З. Формулы для вычисления констант уравнения состояния Мартина—Хоу (П3.6)
ZKp
ь
Pi (Гкр)
B0,533-
V (i
i
{„,,,
-31,883ZKp).ZKp
Р \
15ZKp /
^2 (Лр)
} PKpFSp +
IUl 3
+ Р ^ Гз-1
(FKp — Ь)
(р(^кр —6K
cp(FKp — ЬK
(FKp —ЬM
18,75+ P — 15ZKn)
15Zjp JJ
В2 R (VJSZKp -
Р р | (^>р
3 а (F
Bi [(Ич-6)-
С5 — [C2(FKp-
-6)
(Vn Ь)\ "В КР
ЪK + С3 (FKp — ЬJ]
Новый усовершенствованный вариант методики построения приближенных уравнений
состояния предложен в работе Мартина и Станфорда [2533].^
Для ориентировочных расчетов можно использовать двухконстантные уравнения состояния
Редлиха—Квонга и Мартина
р _ Тг 0^2748
*'' — ZKp(yr-0,08664/ZKp) rV2FrZ2p(Fr + 0,08664/ZEpJ ^ " ;
и
п ?г -9 D — 7у)
р
где УГ=У/УКР.
В обзорах [2530, 1098] уравнение состояния Мартина (П3.12) рекомендуется как одно из
наиболее точных уравнений этого класса.
П3.2. Второй вириальаый коэффициент
В области малых плотностей можно использовать уравнение состояния в простой форме:
Z=i-\-BjV. (П3.13)
Применение уравнения (П3.13) возможно, если плотность р не превышает примерно
что для большинства веществ в области температур, примыкающих к критической, соответствует
условию р ^ 1,5 МПа. При повышении температуры область применимости расширяется, достигая
при Т > 1000 К величины порядка 10—15 МПа. В справочнике Ривкина [2131 уравнение (П3.13)
рекомендуется использовать при р ^ 5МПа везде, кроме околокритической области.
400
Свойства газов при повышенном давления
' Для нормальных веществ корреляция Питце-
ра—Керла [3033] позволяет вычислить значения В
по заданным значениям Т„, рхд и ш:
Таблица П3.4.
Параметры для квантовых
газов
0,0121Г;3
; - 0,13857^ -
ш @,073 + 0,46 Ту—
0,0073т1-8). (П3.14)
Область применимости этого соотношения
0,75 < Тг <[ 4. Обобщение данной корреляции на
случай квантовых газов (Не, Ne, Нг, D2 и т. д.)
дано в работах [963, 1763]. Фактор ш в этом
случае полагается равным нулю, а вместо
критических параметров используются эффективные
параметры, зависящие от температуры:
Be-
во
Ne
4Не
3Не
н2
JKP
К
45,5
10,47
10,55
43,6
43,6
Примечание.
с цринятыми i
МПа
2,726
0,676
0,601
2,047
2,037
Ве-
во
HD
та
нт
DT
Параметры 7^р,
s табл. J-
ческих постоянных.
•"кр
К
42,9
43,8
42,3
43,5
МПа
1,986
2,077
1,935
2,057
р*р не совпадают
.26 значениями
крити-
т
1 вр
1 -f CJMT'
С2/МТ '
где М — молекулярный вес соединения, Сг=21,8, Са=44,2 и параметры Г*р, р*р для девяти кван"
товых газов приведены в табл. П3.4.
Для широкого класса веществ, включающего наряду с нормальными также полярные,
ассоциированные и квантовые газы, в работе Штейна [3513] предложена корреляция, использующая
в качестве исходных данных Гкр, 7кр и М — параметр Риделя:
= 2 Вч (М — 7)V, (ИЗ. 14а)
Константы уравнения
(П3.17)
т = 1 — Т;1.
Константы этого уравнения приведены в табл. П3.5.
Таблица П3.5. Константы И,- г уравнения
(П3.14а)
Таблица П3.6.
г=0
j=2
0
1
2
3
4
—1,277 618
3,085 930
—2,163 235
1,442 157
-0,360 539
-0,114 085
0,880 066
-1,342 943
0,895 296
-0,223 824
-0,008 399
0,099 631
-0,214 468
0,142 978
-0,035 745
i
0 0,471 587
1 3,434 82
2 —66,328 8
3 476,748
4 —2 224,88
i
bJ
5 6 573,78
6 —11865,6
7 11 917,5
8 —5 096,57
Параметр М определяется из соотношений (П3.7) или (П3.8). Для нормальных веществ можно
также выразить М через фактор ш:
М=5,824+4,83 <¦>.
Уравнение (П3.14а) применимо в интервале Т^ от 0,5 до 20.
Для грубой оценки величины второго вириального коэффициента можно использовать
соотношение, полученное из уравнения состояния Мартина (П3.12):
Tr, (П3.15
где Ь0=0,2256, Ь1== -0,5625.
Соотношение (П3.15) целесообразно использовать для веществ, по которым чрезвычайно
ограничены опытные данные и даже значения критических постоянных получены оценкой.
В последние годы предложено несколько новых корреляций для второго вириального
коэффициента, которые можно пг)именять к соединениям с различными физическими свойствами [1866
2872].
401
Приложение
Важное преимущество уравнения состояния (П3.13) состоит в возможности теоретического
расчета второго вириального коэффициента через межмолекулярный потенциал V (г)
со
В = —2tzNa j (e-frw _ 1) гЧг, (П3.16)
о
где Na — число Авогадро.
Межмолекулярные потенциалы, наиболее часто используемые для расчетов вириальных
коэффициентов: Леннарда—Джонса 12 : 6, Штокмайера (для полярных газов), Кихары,
сферической оболочки т : 6 : 8 и др. Подробный обзор современного состояния проблемы дан в
монографии Мейсона и Сперлинга [166]. Для практических расчетов необходимо располагать
значениями потенциальных параметров и таблицами приведенных величин В* (Т*), тогда
В(Т) = Ь0В*(Т*),
где Т* =kT/s, е и Ъо — параметры потенциала.
Для потенциалов 12 : 6 и Штокмайера соответствующие таблицы приведены в монографии [72$
и приложении V ко 2-му изданию настоящего справочника [88]. В интервале температур Т* =
= 2-^-20 можно использовать аппроксимационное уравнение, полученное в работе [105]:
В* = 2>/Г-'', (П3.17),
j
константы которого приведены в табл. П3.6.
Для потенциала Кихары соответствующие таблицы приведены в работе [1310], для
потенциала сферической оболочки — в [1220], для потенциала m : 6 : 8 — в работе [1822а].
Параметры потенциалов находятся путем специальной обработки по методу наименьших
квадратов опытных данных по сжимаемости и транспортным свойствам. Значения параметров для
многих веществ приводятся в ряде справочников и монографий [72, 166, 167, 222]. В обзорной
статье Полака и Лю [3054] приведены параметры потенциала Штокмайера для всех
полярных газов, которые включены в справочник Даймонда и Смита по вириальным коэффициентам
[1364].
Следует учитывать, однако, что только многопараметрические потенциалы, данные по которым
получены совместной обработкой равновесных и транспортных свойств, эффективно
воспроизводят температурную зависимость второго вириального коэффициента. Этим требованиям
удовлетворяет, в частности, потенциал 12 : 6 с температурно-зависящими константами. Кессельман.
[124] рассчитал на основе этого потенциала вириальные коэффициенты и транспортные
свойства для большой группы веществ (компоненты воздуха, галогены и галоидоводороды, водяной-
пар, аммиак и др.). Для Н2О Зубаревым и Крупиной [106] рекомендован потенциал Кеезома,
который также сводится к потенциалу 12 : 6 с переменными параметрами.
Использование традиционных потенциалов (Леннарда—Джонса и Штокмайера) для
экстраполяции В (Т) в область высоких температур должно проводиться с известной осторожностью,
поскольку в различных температурных интервалах оказываются применимы различные наборы
потенциальных параметров. Более надежная оценка может быть выполнена с помощью обобщенных;
корреляций.
ПЗ.З. Термодинамические свойства
Уравнение состояния позволяет рассчитать избыточные значения для всех термодинамических
функций. Вычисляется изменение в величине термодинамической функции X (Т, р) при переходе-
от стандартного к состоянию реального газа при той же температуре Т и заданном давлении р„
Процедура расчета состоит в итерационном решении уравнения
Р = Р(Р, Т)
402
Свойства газов при повышенном давлении
для нахождения плотности р через р и Т и последующем использовании формул:
о
°'~~ J W2/? р2'
(П3.22)
Здесь И — энтальпия, S — энтропия, / — летучесть, Cv и Ср — изохорная и изобарная теплоемкости.
Летучесть определяет изменение в величине изобарно-изотермического потенциала
/ G(T, р) -& (Т) -RT In pjp"
Если дополнительно к уравнению состояния известна зависимость давления насыщения ре
от температуры, то совместное решение уравнений
определяет плотность насыщенного пара р".
По уравнению Клаузиуса—Клапейрона рассчитывается энтальпия испарения
(П3.23)
если известна плотность жидкой фазы р' (У=1/р); энтальпия и энтропия жидкости на линии
насыщения определяются из соотношений
Н' = Н" — \Н, S' = S" — Atff/T, (П3.24)
где Н", S" — энтальпия и энтропия насыщенного пара.
Для нормальных веществ можно использовать обобщенное уравнение состояния (П3.2);
значения избыточных функций определяются по формулам:
— 1 YX ЩП гщуч + З
% =ln Z - ^ + Ь>% + 2»^-'^p__54fe + 2Е. (П3.26)
__ __ 1 _ т
rV 1(дрг
(^)} (П3.29)
403
Приложение 3
Процедура расчета состоит из следующих этапов:
1) По заданным значениям Тг и рг с использованием уравнения состояния
Z = Z°(Tr, Vr)
находим величины Vr и Z°, а с помощью формул (П3.25)—(П3.29) — значения
термодинамических функций. В формулах используются значения констант уравнения Z° (см. табл. П3.1).
2) Повторяются все операции, выполненные на первом этапе, но с использованием уравнения
состояния
Z = Z*(Tr, Vr).
3) Значение фактора сжимаемости находится по формуле
\{ZR — Z°). (ПЗ.ЗО)
Поел© этого определяется окончательное значение объема
V,
Аналогично вычисляются значения термодинамических функций. Например,
ЬН / ДЯ \° , со Vf ДЯ у ( ДЯ \°1
V
'J С
(П3.31)
Для приближенной оценки приращений к термодинамическим функциям можно использовать
табл. П3.7. В более полном виде эти таблицы приведены в работе [2374]; там же приведены
аналогичные таблицы для фактора сжимаемости Z, величины lg flp и изобарной теплоемкости.
При этом
Н°(ТГ)-Н(ТГ, Рг) _( Я° (ТУ) - Н (Тг, Рг) у | , у Д° (Гг) - Н (Гг, Рг
К ; +*{ -
дтж
аналогичное соотношение имеет место для энтропии.
'кр
Таб л
' т
! 1г
0,5
1
2
3
4
и
ца ПЗ.'
Рг = 0.1
5,459
8,870
0,103
0,093
0,028
—0,007
0,011
—0,014
0,005
—0,016
1. Поправка на
1В°(ТГ)
Рг=1
5,401
8,888
2,584
2,471
0,276
—0,085
0,109
-0,142
0,041
—0,158
-Н{ТГ
рг = 2
5,336
8,909
3,706
3,186
0,541
—0,190
0,205
—0,278
0,072
—0,306
неидеальность к энтальпии
. Рг)]/ДГч
Рг=ГО
5,135
8,978
3,774
3,615
1,167
—0,428
0,415
-0,611
0,116
—0,680
Рг=10
4,791
9,111
3,526
4,100
1,577
-0,255
0,545
—0,899
0,061
—1,097
Р, = 0,1
7,841
11,197
0,069
0,086
0,011
0,004
0,004
0,001
0,002
0,001
и энтропии
[S° (TV,
Pr=l
5,608
11,153
2,178
2,399
0,111
0,035
0,041
0,010
0,021
0,006
Pr)-S(Tr,
Pr = 2
4,989
11,107
2,784
3,067
0,221
0,058
0,080
0,020
0,041
0,012
Pr)VR
Pr = 5
4,282
10,985
2,380
3,387
0,497
0,136
0,181
0,058
0,093
0,038
Pr=10
3,899
10,836
2,105
3,717
0,733
0,434
0 ЗЙЗ
0,15»
0,162
0,100
Верхнее число в каждой клетке табл. П3.7, дает значение, соответствующее индексу «0»,
нижнее число дает значение, соответствующее индексу «i?».
Для более широкого класса веществ, включающего полярные и ассоциированные газы, расчет
свойств можно выполнить с использованием уравнения состояния Мартина—Хоу, константы
которого рассчитываются по правилам, изложенным в разделе П3.1:
„-ЛТг
./=2
U -1)(У- б)/
(ПЗ. 32)
404
Свойства газов при повышенном давлении
//-mv-Wi • (П3-33)
У=2
ПТ^1 • (П3.34)
.7=2
Приближенный расчет В# и §5 можно провести также по таблицам Лидерсена, приведенным
в монографии [222], которые применимы для веществ, имеющих ZKp в интервале 0,23—0,29.
Таблицы охватывают интервал температур от линии насыщения до Гг=15 при pr ^ 30.
При умеренной степени сжатия все поправки на неидеальность можно вычислить через второй
вириальный коэффициент с использованием данных табл. Д.25 или методик, описанных в П3.2:
bHjRT = (B — Bj)IV, (П3.35)
bSlR = B1/V, (П3.36)
In (flp) = B/V, (П3.37)
(П3.38)
p (П3.39)
где
n —тАВ p —r*d2B
1— If' 2— dT%'
В табл. Д.25 приведены константы аппроксимационного уравнения для В вида
(П3.40)
где х = TjTQ, Vo, Тй — параметры приведения. Тогда
Соотношения (П3.41) нельзя применять в тех случаях, когда сами параметры приведения
зависят от температуры (HD, DT, Т2, НТ). В этом случае целесообразно рассчитать через
уравнение (П3.40) таблицу значений В и произвести численное дифференцирование.
П3.4. Линия насыщения жидкость—пар
Для нормальных веществ трехпараметрическая корреляция Питцера [3022, 3035, 2374]
позволяет рассчитать давление насыщения, ортобарические плотности и энтальпию испарения.
В интервале приведенных температур Тг от 0,3 до 1 давление насыщенного пара можно определить
из соотношения
1прг = 5,92714 — 6,09648Г-1 —1,28862 1бГг + 0,169347Г« 4-шA5.2518 — 15-687571г1—
— 13,4721 In Tr + 0,43577Г«). (П3.42)
Совместное решение (П3.42) и уравнения состояния (П3.2) определяет два значения плотности:
р' и р" — плотности насыщенной жидкости и пара.
Для непосредственной оценки ортобарических плотностей можно использовать табл. П3.8 [167]
и уравнение (см. [2126])
Р; = 1 + 0,85 A — Тг) + A,6916 + 0,9846ш) A — ТГ)Ч>. (П3.43)
Энтальпия испарения может быть рассчитана по уравнению Клаузиуса—Клапейрона
(П3.23). Питцером [3032, 3035] разработана специальная корреляция для энтропии
испарения AtS (Tr, ш) в интервале Тг от 0,56 до 1. Соответствующие таблицы приведены в работах [222,
167].
405
Приложение 3
Таблица П3.8. Фактор сжимаемости газа на линии насыщения Z"s =
0,56
0,58
0,60
. 0,62
0,64
0,66
0,68
2°,
0,959
0,953
0,947
0,940
0,932
0,922
0,911
2;
0,070
0,077
0,083
0,090
0,097
0,104
0,113
Tr
0,70
0,72
0,74
0,76
0,78
0,80
0,82
22
0,897
0,881
0,864
0,846
0,826
0,804
0,781
2;
0,122
0,131
0,137
0,142
0,144
0,144
0,140
тг
0,84
0,86
0,88
0,90
0,92
0,94
2°,
0,756
0,73
0,70
0,67
0,63
0,59
2;
0,135
0,125
0,110
0,095
0,075
0,055
Tr
0,95
0,96
0,97
0,98
0,99
1
4
0,565
0,54
0,51
0,47
0,43
0,291
2i
0,045
0,035
0,020
0
—0,030
—0,080
Для более широкого класса веществ, включающего полярные и ассоциированные газы, можно
использовать уравнение Риделя для давления пара [222, 17]
\ърг = М In Tr — 0,0838 (М — 3,75) (ЗбГ;1 — 35 — Т\ + 42 In Tr). (П3.44)
Параметр М можно оценить с помощью соотношений (П3.7, П3.8) или вычислить через
уравнение (П3.44) по известной величине Тъ. Грубая оценка давления пара в интервале от нормальной
точки кипения до критической может быть сделана с помощью уравнения Кирхгофа
lgP, = *;(l-Z?). (П3.45)
где
. Плотность насыщенной жидкости р' с хорошей точностью предсказывается корреляцией Лидер-
сёна с использованием ZKp как третьего параметра. Обширные таблицы для р' приведены в
монографии [222]. Соотношение (П3.46) аппроксимирует данные этих таблиц
Р;= 1,2+ E,563 —11,ОЗЯжр)A — Гг)°.8*р+о,з. (П3.46)
Соотношение Риделя [222] определяет плотность жидкой фазы через параметр М,
оцениваемый по формулам (П3.7) или (П3.8)
[ р;= 1 + 0,85 A — Тг) + @,53 + 0.2М) A — ТГ)Ч>. ь(П3.47)
Для полярных соединений в справочном руководстве [167] рекомендуется четырехпараметри-
ческая корреляция Холма и Стила [1802, 1803]. Применение этой методики требует задания
наряду с TKt, ркр, ш еще одного параметра % — фактора полярности, определяемого соотношением
A13.48)
Определив ^ из уравнения для давления пара, можно с помощью таблиц, приведенных
в работе [167], рассчитать основные свойства на линии насыщения.
Подробная сводка различных методик расчета термодинамических свойств на линии
насыщения дана в [222]. Там же приведены обобщенные корреляции для критических параметров.
П3.5. Расчет ассоциированных соединений
Для ассоциированных веществ типа фтористого водорода, серы, фосфора, пары которого содержат
в заметном количестве полимерные молекулы, методика расчета термодинамических свойств
отличается заметным усложнением. Неидеальность вещества, наблюдаемая здесь уже при
небольших давлениях, связана как с химической ассоциацией, так и с ван-дер-ваальсовым («физическим»)
взаимодействием ассоциатов. Влияние химической ассоциации может быть полностью учтено
по данным о термодинамических функциях компонентов, приведенных в настоящем справочнике.
Для реакций ассоциации «га-мера» пА ** Ап вычисляются константы равновесия:
406
Свойства газов при повышенном давлении
после чего рассчитывается состав пара. Неидеальность определяется в этом случае из соотношения
Z = pvM1/RT = v-1,
Meues.JM1 = '^lixi — фактор ассоциации, Л/сиеои, Мх — молекулярные веса смеси и мономера,
*
v — удельный объем.
При слабой ассоциации можно использовать соотношение Вулли [3995]
или
В — — K\RT,
D = (-20К? + 18К\К°3 — ЗК1) (RTf,
где В, С и D — вириальные коэффициенты в уравнении состояния Z=i-\-BJV-\-C/V2J^D/V3.
Оценка «физической» неидеальности может быть проведена по методике, разработанной Pay
« сотр. [3166, 3162, 3163, 3164]. В этом случае
Z = v-4l + B^aplRT), (П3.49)
где «физическая часть» второго вириального коэффициента В^яз принимается одинаковой как для
вещества в целом, так и для отдельных его фракций. В отсутствие pFr-данных она может быт.
вычислена из обобщенных корреляций. Коэффициент летучести принимается независимым от
состава (приближение Льюиса—Рендалла) и одинаковым для всех компонентов:
(П3.50)
Расчет равновесного состава с учетом летучестей проводится путем замены К°п на Щ = К1^п~1.
Соотношения (П3.49), (П3.50) применимы при малых давлениях. В общем случае рекомендуется
использовать обобщенные таблицы фактора сжимаемости и летучести из монографии [222] или
работы [2374].
Для расчета второго вириального коэффициента ассоциированных газов в литературе широко
используется соотношение
В = В$шз — RTK\.
Его применимость ограничена условием слабой димеризации и отсутствием молекул, более
сложных, чем димеры и мономеры. Обобщенная методика расчета свойств димер-мономерной смеси
при произвольном давлении и степени димеризации дается в работе [2872]. Подробный обзор
методов термодинамического расчета ассоциированных соединений представлен в работе [26а].
П3.6. Справочники и руководства
В ряде справочных изданий можно найти данные по термодинамическим свойствам реальных газов
и жидкостей для широкого круга соединений. В основном эти издания охватывают свойства
углеводородов, а из неорганических соединений, как правило, содержат данные для технически
важных газов. Подробные термодинамические таблицы приведены в справочнике Варгафтика [46 ]
для компонентов воздуха, инертных газов, щелочных металлов, Н2, D2, H2O, F2, Cl2, NH3 и др.
В справочнике Канджара и Маннинга [870] приведены таблицы для 20 важнейших соединений,
в том числе для N2, O2, H2, NH3, SO2.
Трехтомная монография под редакцией Дина [3656, 3657] содержит обзорные статьи, в
которых рассмотрены термодинамические свойства важнейших газов, таких как Ar, N2, NH3, GO, GO2,
воздух и ряд углеводородов. В обзоры включены все данные, опубликованные к моменту издания
монографии (середина 50-х годов), а также таблицы свойств и уравнения состояния. К настоящему
времени этот материал в значительной степени устарел и ценность представляет в основном
подробная библиография старых работ.
407
Приложение 3
Следует отметить также справочник Хильзенрата [1945], изданный в 1955 г., который
содержит таблицы первых двух вириальных коэффициентов и термодинамических свойств для ряда
газов (Аг, СО2, CO, Н2, N2, O2, Н2О) и библиографию основных работ.
Приложение V к I тому предыдущего издания настоящего справочника [88 ] включало обзоры
опубликованных работ по реальным свойствам для нескольких десятков газов, а во II томе были
помещены таблицы первых двух вириальных коэффициентов, рассчитанные по потенциалам Лен-
нарда—Джонса и Штокмайера. В справочнике Ривкина [213] приведены интерполяционные
уравнения для второго вириального коэффициента и поправок к термодинамическим функциям
нескольких технически важных газов.
Опубликован ряд справочных изданий, в которых рассматриваются только отдельные свойства
(критические постоянные, вириальные коэффициенты, давление пара) для большого круга
соединений. Подобные издания отличаются полнотой библиографии и тщательным критическим
отбором данных. Широкой известностью пользуется справочник Даймонда и Смита [1364], в котором
собраны почти все опубликованные данные по вириальным коэффициентам индивидуальных
веществ. Данные представлены в виде таблиц экспериментальных значений для каждой из
цитированных работ и таблицы сглаженных значений. Всего справочник охватывает около 200
соединений, в основном органических; из неорганических соединений наряду с технически важными
газами представлены H2S, SO2, NO, N2O, F2, HC1 и ряд других. В обзор Кокса и Лоуренсона [10981
включены данные, не охваченные справочником [1364].
Обзорная статья Матьюса [2543] содержит справочные данные по критическим постоянным
неорганических соединений вместе с подробной библиографией экспериментальных работ и
критическим анализом последних.
Данные по критическим постоянным приведены также в отдельных выпусках справочника
«Термические константы веществ» [258).
В справочниках Сталла [244] и Эдуардса [1386] собраны данные по давлению пара в интервале
от нормальной точки кипения до критической точки для нескольких десятков неорганических
соединений, причем в справочнике [1386] приведены константы интерполяционного уравнения.
Рядом ведущих специалистов по термодинамике опубликованы обзоры современного состояния
исследований рРТ-свойств и методов обработки данных. Различные виды эмпирических
уравнений состояния, применяемых в инженерной термодинамике, рассмотрены в обзорах Мартина [25301
и Цонопулоса и Праузница [3735]. В известной монографии Мейсона и Сперлинга [166] изложены
методы расчета вириальных коэффициентов через межмолекулярный потенциал и дана
подробная библиография экспериментальных работ по сжимаемости газов. Обзор Левельт-Сенгерс и
др. [2394] посвящен детальному анализу термодинамических свойств в окрестности критической
точки для 15 важнейших веществ (О2, Н2О, Не, Хе и др.). Приведены полученные в результате
обработки значения критических постоянных и специального вида уравнения состояния для
критической области (так называемые неаналитические уравнения состояния).
Экспериментальные работы (по сжимаемости, критическим явлениям) и уравнения состояния, применяемые в
современной литературе, рассмотрены в обзорах Кокса и Лоуренсона [1098] и Дуслина
[1309].
В последние годы опубликовано несколько справочных изданий, специально ориентированных
на применение в инженерной практике. Приводимые в них данные, не отличаясь высокой
точностью, охватывают в то же время широкий комплекс теплофизических свойств: давление параг
теплоемкость газа и жидкости, критические постоянные, вязкость, теплопроводность и др.
В справочнике Хорвата [2011] собраны краткие сводки теплофизических свойств для большой
группы неорганических соединений, в том числе для многих малоизученных веществ: галогенов
и галоидоводородов, окислов азота и серы, N2H4, H2O2, H2S и др. В основном приведены
данные для линии насыщения: давление пара, плотность, критические постоянные и т. д. В серии
статей Явса с сотр. приведен справочный материал по теплофизическим свойствам, оформленный
в виде графиков, удобных для инженерных расчетов. Опубликованные к 1976 г. статьи
охватывают многие соединения: О2, Н2, N2 [4029], Н2О, Н2О2 [4028], Не, Ne, Аг [4026], галогены [3396],
галоидоводороды [4023], NH3, N2H4 [4025], окислы азота [4024] и серы [4027], SF6 [348].
Методы инженерного расчета теплофизических свойств газов, жидкостей и их смесей изложены
в руководстве [167], изданном термодинамическим центром при Всесоюзном объединении «Неф-
техим». Хотя основной материал [167] посвящен углеводородам, расчетные методики приложимы
и к неорганическим соединениям.
408
Свойства газов при повышенном давления
Приведенные таблицы включают критические постоянные, фактор ацентричности, дипольньш
момент молекул и параметры межмолекулярных потенциалов для более 400 соединений, в том числ&
для двух десятков неорганических веществ. Данные этих таблиц представляют исходный
материал для расчета всех теплофизических свойств.
Более полный обзор обобщенных методов термодинамического расчета дан в монографии Рида
и Шервуда [222]. Используя эти методики, можно предсказать критические постоянные и все-
термодинамические свойства при минимальной исходной информации.
П3.7. Обзоры данных для отдельных веществ
О2. Подробный обзор опытных данных и опубликованных таблиц по состоянию на 1965 г.
представлен в монографии Вассермана и др. [47]. Уравнение состояния и таблицы термодинамических
свойств, приведенные в этой работе, охватывают интервал температур от линии насыщения
до 1300 К при давлении до 100 МПа. Таблица термодинамических свойств О2 рассчитана по данным
работы [47] (табл. П3.9). Впоследствии Вебер [3906] исследовал термодинамические свойства
О2 в низкотемпературной области (от тройной точки до 300 К): рУУ-свойства, теплоемкость,
свойства на линии насыщения, критические постоянные. Основываясь на этих данных, Якобсен и
Стюарт [3536, 2082] построили уравнение состояния. Те же данные использованы при расчете
таблиц термодинамических свойств [2563]. Детальный анализ свойств в околокритической области
дан в обзоре Левельт-Сенгерс и др. [2394]. Люстерник и др. [2483а] провели согласование и
обработку теплофизических свойств О2 в широком интервале температур G0—2000 К) с помощью-
межмолекулярного потенциала т—6—8 [1822а]
»-т("»-8)
Найденные значения параметров составляют тп.=11, 7=1, е/&=116,637 К, rm=3,8437A.
В табл. Д.25 приведены константы полинома, аппроксимирующего значения второго вириаль-
ного коэффициента, рассчитанные в работе [2483а]. Критические постоянные (табл. Д.26) приняты
по данным работы [3906].
О3. Бердсалл и др. [651 ] обработали полученные ими рУТ-данные с помощью уравнения
состояния Битти—Бриджмена и рассчитали таблицу термодинамических свойств в интервале
температур 200—300 К при давлениях до 1 МПа. Бердсалл и Дженкинс [650] исследовали свойства
на линии насыщения вплоть до критической точки. Обзор данных по термодинамическим свойствам!
О3 содержится в работе Стренга [3557]. Параметры потенциала Леннарда—Джонса 12 : 6 были,
определены во 2-м издании настоящего справочника [88]. В табл. Д.25 приведены константы
полинома, позволяющие рассчитать второй вириальный коэффициент для этого потенциала. Значения
критических постоянных (табл. Д.26) приняты согласно рекомендациям Матьюса [2543].
Н2. В справочной литературе приводятся данные для нормального Н2 G5% о-Н2), равновесного'
при высоких температурах (w-H2) и для Н2, равновесного при 20,4 К @,21 % о-Н2). Последний
отождествляется с п-Н2 по своим свойствам. Результаты исследований низкотемпературных свойств
п-Н2 суммированы в справочном издании [3225]. В более позднем издании [2564] приведены
данные для широкого температурного интервала (до 3000 К) при давлении до 70 МПа. Для Т > 100 К
в работе [2564] получено уравнение состояния в форме расширенного уравнения Стобриджа.
Поскольку при этих температурах различие в рУГ-свойствах обеих модификаций Н2
пренебрежимо мало, это уравнение состояния может быть использовано для расчета свойств как к-Н2, так
и ге-Н2. В низкотемпературной области уравнения состояния п-Н2 были предложены Гудвином
[1684а], Ткаченко и др. [3702]. Для к-Н2 обработка опытных данных выполнена Стюартом и
Джонсоном [3537]. Рабинович [209] предложил уравнение состояния н-Н2 для температур до 4000° С
при давлении до 500 атм. Уравнение состояния к-Н2 и таблицы для широкой области параметров-
построены в работах Казавчинского и Сердюка [114, 231]. Родер и др. [3224] опубликовали
программы расчетов на ЭВМ термодинамических свойств обеих модификаций Н2. Наиболее полная
компиляция всех теплофизических свойств представлена Маккарти [2562]. В обзорах Есельсона
и др. [96] и Родера с сотр. [3223] даны подробные сводки опытных и расчетных данных для
низкотемпературной области.
409
Приложение 3
Таблица П3.9. Термодинамические
Т
К
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
500
600
700
800
900
1000
1100
1200
1300
1400
1500
500
600
700
800
300
400
500
600
700
800
900
1000
700
800
900
1000
p = i МПа
V
2,480
3,325
4,166
5,002
5,837
6,669
7,504
8,336
2,50858
3,34133
4,17337
5,00513
5,83675
6,66827
7,49974
8,33118
3,975
4,885
5,754
6,607
7,452
8,292
9,130
9,967
10,802
11,637
12,472
3,9758
4,8832
5,7525
6,6057
2,482
3,330
4,170
5,007
5,842
6,676
7,510
8,34
5,763
6,619
7,468
8,311
ЬН
—73,60
—41,60
—22,40
—12,80
—3,20
3,20
6,40
12,80
8,3
12,1
13,9
15,1
15,5
16,1
16,5
16,7
—744,7
—393,4
—249,7
—176,5
—132,5
—103,6
—85,51
—69,94
—58,54
—49,96
—43,35
—796,1
—393,9
—251,4
—159,2
—82,17
—42,10
—21,00
—8,40
0,0321
6,268
11,11
14,98
—253,7
—180,0
—127,0
—89,78
85
—0,209
—0,113
—0,049
—0,038
—0,024
—0,016
—0,011
—0,008
—0,020
—0,0075
—0,0054
—0,0033
—0,0022
—0,0015
—0,0011
—0,0008
-1,137
—0,486
—0,259
—0,157
—0,104
—0,078
—0,063
—0,050
—0,041
-0,034
—0,030
—1,1665
—0,4458
—0,2028
—0,1106
—0,232
—0,115
—0,068
—0,044
) —0,031
—0,023
—0,017
—0,013
—0,281
—0,184
—0,124
—0,086
i
V
0,482
0,666
0,839
1,011
1,180
1,348
1,516
1,684
0,51359
0,68110
0,84799
1,01461
1,18112
1,34754
1,51393
1,68027
_
0,883
1,095
1,284
1,465
1,641
1,813
1,985
2,156
2,326
2,495
0,88290
1,09621
1,28409
0,489
0,671
0,845
1,017
1,187
1,355
1,524
1,691
1,107
1,299
1,483
1,660
свойства
газов при повышенной давлении
о = 5, МПа
ЬН
—374,4
—204,8
—112,0
—54,4
—12,8
16,0
38,4
54,4
44,1
61,7
70,6
76,0
79,2
81,4
83,1
84,3
—
—2247
—1318
—906,6
—668,5
—517,6
—426,3
—348,1
—291,1
—248,3
—215,5
—2271,1
—1307,3
—888,3
—402,2
—200,23
—97,19
—36,25
85
о2
—1,004
—0,524
—0,300
—0,204
—0,140
—0,108
—0,053
—0,038
-0,097
—0,044
—0,024
—0,016
—0,011
—0,007
—0,005
—0,004
Н2О
—2,850
—1,399
—0,842
—0,558
—0,404
—0,317
—0,250
—0,204
—0,172
—0,149
—2,8611
—1,3359
—0,7995
—1,152
—0,560
—0,327
—0,215
4,455 —0,152
34,57
58,00
76,78
—1294
—903,2
—629,4
—441,9
Примечание. V даны в дм3.моль, ЬН — в Дя
а При Т > 100 К значения ЬН и
85, так
—0,112
—0,084
—0,065
HF
—1,444
—0,928
—0,619
—0,426
с-моль,
V
0,238
0,334
0,424
0,512
0,598
0,683
0,768
0,859
0,26445
0,34863
0,43231
0,51576
0,59910
0,68235
0,76558
0,84877
0,361
0,509
0,618
0,716
0,809
0,899
0,988
1,075
1,162
1,248
0,36193
0,51055
0,61771
0,242
" 0,340
0,431
0,519
0,605
0,691
0,776
0,860
0,525
0,635
0,736
0,830
в = ЮМ Па
ЬН
—729,6
—390,4
—211,2
-99,2
—22,4
32,0
76,8
108,8
94,1
126,4
143,1
153,0
159,1
163,3
166,5
168,7
—
—5605
—2834
—1875
—1354
—1039
—849,3
—692,3
—578,2
—493,1
—428,0
-5606,4
—2824,2
—1810,1
—773,9
—373,8
—175,4
—58,92
18,88
76,59
121,7
157,9
—2652
—1810
—1243
—864,7
85
—1,993
—0,969
—0,585
—0,361
—0,265
—0,169
—0,103
—0,073
—0,178
—0,083
—0,044
—0,026
—0,016
—0,012
—0,008
—0,006
—
—7,409
—3,054
—1,759
—1,144
—0,814
—0,636
—0,502
—0,411
—0,346
—0,300
—7,3807
—2,9810
—1,7575
—2,257
—1,074
—0,627
—0,413
—0,293
—0,217
—0,163
—0,125
—2,987
—1,871
—1,230
—0,841
85 —в Дж-К^-моль.
же как и pVT-свойства, одинаковы для обеих
р=20 МПа
V
0,119
0,171
0,219
0,264
0,308
0,351
0,394
0,437
0,14012
0,18248
0,22446
0,26625
0,30795
0,34960
0,39123
0,43283
_
0,208
0,284
0,342
0,394
0,442
0,489
0,535
0,580
0,625
0,20911
0,28442
0,122
0,178
0,226
0,271
0,315
0,359
0,402
0,445
0,235
0,305
0,365
0,418
ЬН
—1325
—688,0
—361,6
—160,0
—16,0
86,4
163,2
224,0
206,8
263,1
292,3
309,6
320,5
328,0
333,2
337,1
—6691
—3993
—2780
—2094
—1686
—1369
—1141
—971,9
—843,9
—6637,2
—3863,3
—1381
—640,4
—281,2
—69,54
72,71
179,1
262,8
330,7
—5536
—3600
—2402
—1646
85
—3,685
—1,797
—1,061
—0,709
—0,485
—0,357
—0,229
—0,197
—0,309
—0,146
—0,079
—0,047
—0,031
—0,021
—0,013
—0,011
—7,456
—3,808
—2,371
—1,649
—1,280
—1,013
—0,831
-0,701
—0,606
—7,3832
—3,5702
—4,191
—1,953
—1,145
—0,761
—0,545
—0,405
—0,307
—0,237
—6,393
—3,778
—2,408
—1,627
модификаций водорода.
410
Свойства газов при повышенном давлении
"Таблица П3.9 (продолжение)
т
к
400
500
600
700
800
900
1000
400
500
600
700
800
900
1000
400
500
600
700
800
900
1000
900
1000
1100
1200
300
400
500
600
700
800
900
1000
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
р = 1 МПа
V
3,156
4,052
4,921
5,776
6,624
7,468
8,310
3,258
4,118
4,966
5,808
6,647
7,484
8,320
2,992
3,928
4,823
5,697
6,560
7,416
8,266
7,275
8,145
9,012
9,874
2,349
3,239
4,103
4,954
5,800
6,642
7,481
8,318
3,164
4,051
4,918
5,775
6,623
7,467
8,308
2,50599
3,33701
4,16803
4,99909
5,83019
6,66133
7,49247
8,32365
ЬН
85
—457,8 —0,727
—317,3 —0,427
—234,5 —0,279
—180,5 —0,195
—141,1 —0,142
—110,5 —0,106
—86,92 —0,082
—222,8 —0,389
—144,5 —0,211
—101,7 —0,131
—73,41 —0,088
—53,56 —0,062
—39,70 —0,045
—29,72 —0,035
—795 —1,181
—570 —0,691
—437 —0,456
—348 —0,323
—283 —0,240
—234 —0,185
—194 —0,146
—564,3 —0,397
—485,1 —0,317
—413,4 —0,254
—354,0 —0,209
—358,8 —0,721
—236,3 -0,375
—168,8 —0,229
—127,0 —0,155
—98,13 —0,112
—75,70 —0,083
—58,18 —0,062
—44,78 —0,048
-416,7 —0,643
—299,0 —0,387
—224,9 —0,258
—175,4 —0,186
—139,9 —0,139
—112,8 —0,107
—90,78 -0,084
13,1 0,00424
12,9 0,00392
12,8 0,00360
12,6 0,00336
12,4 0,00312
12,3 0,00292
12,1 0,00268
11,9 0,00252
р = 5 МПа
V
0,472
0,721
0,930
1,121
1,305
1,484
1,660
0,594
0,792
0,975
1,152
1,327
1,498
1,669
0,566
0,819
1,037
1,238
1,430
1.616
1,276
1,488
1,694
1,894
0,573
0,776
0,964
1,145
1,322
1,495
1,667
0,482
0,720
0,927
1,120
1,303
1,482
1,658
0,51053
0,67640
0,84231
1,00821
1,17416
1,34015
1,50614
1,67217
ЬН
85
С12
—2885 —4,969
—1730 —2,393
—1215 —1,465
—911,8 —0,994
—702,8 —0,713
—545,9 —0,529
—426,6 —0,406
НС1
—1183 —2,096
—739,7 —1,088
—511,0 —0,662
—365,3 —0,437
—264,8 —0,306
—195,4 —0,225
—145,7 —0,173
Вг,
—3873 —5,273
—2497 —2,739
—1855 —1,764
—1460 —1,254
—1182 —0,942
—973 —0,734
Т
h
—3120 —2,284
—2634 —1,781
—2137 —1,334
—1797 —1,073
ВВг
.
—1295 —2,131
—868,7 —1,192
—638,7 —0,782
—488,1 —0,560
—374,3 —0,413
—286,5 —0,310
—219,6 —0,239
HI
—2660 —4,510
—1621 —2,167
—1156 —1,343
—881,2 —0,941
—695,8 —0,697
—557,8 — 0.ГЧ5
_446,6 —0,417
не
63,8 0,02205
63,2 0,02037
62,8 0,01881
62,0 0,01741
61,2 0,01613
60,0 0,01500
59,2 0,01396
58,4 0,01304
V
0,299
0,431
0,541
0,642
0,737
0,830
0,258
0,376
0,477
0,571
0,662
0,751
0,838
0,304
0,449
0,572
0,683
0,786
0,511
0,649
0,777
0,897
0,235
0,360
0,466
0,564
0,658
0,748
0,837
0,299
0,428
0,539
0,640
0,736
0,828
0,26105
0,34378
0,42656
0,50933
0,59214
0,67500
0,75785
0,84071
Р = 10 МПа
ЬН
—3954
—2543
—1841
—1394
—1072
—831,4
—2583
—1525
—1027
—725,1
—521,5
—382,8
—284,2
—6312
—4065
—3030
—2392
—1942
—7338
—5960
—4460
—3652
—2995
—1800
—1283
—967,2
—737,0
—561,2
—428,3
_
—3660
—2389
—1765
—1377
—1097
—873,3
128,3
127,3
126,1
124,5
122,9
121,3
119,7
117,7
85
—5,760
—3,133
—2,030
—1,428
—1,048
—0,799
—4,699
—2,269
—1,340
—0,873
—0,607
—0,445
—0,341
—7,700
—4,034
—2,657
—1,930
—1,477
—5,777
—4,251
—2,848
—2,210
—5,257
—2,519
—1,586
—1,117
—0,819
—0,612
-0,471
—5,188
—2,834
—1,905
—1,390
—1,059
—0,824
0,04327
0,04035
0,03739
0,03466
0,03214
0,02990
0,02782
0,02602
р = 20 МПа
V
0,077
0,180
0,253
0,313
0,367
0,418
0,084
0,168
0,229
0,282
0,331
0,378
0,423
—
0,021
0,147
0,238
0,311
0,374
0,105
0,220
0,316
0,401
0,0563
0,151
0,218
0,276
0,328
0,376
0,423
0,078
0,178
0,251
0,311
0,365
0,416
0,13621
0,17744
0,21866
0,25989
0,30112
0,34238
0,38369
0,42500
ЬН
—11089
—5574
—3706
—2710
—2045
—1567
—6334
—3246
—2064
—1421
—1007
—732,2
—539,0
—42703*
—10027
—6483
—4835
—3823
—21735
—16846
—9653
—7434
—8729
—3843
—2558
—1883
—1419
—1072
—811,4
—9368
—5024
—3484
—2664
—2099
—1657
255,6
254,3
252,2
249,4
246,2
243,0
239,8
236,2
85
—21,992
—7,288
—4,205
—2,831
—2,038
—1,539
—12,977
—4,973
—2,734
—1,731
—1,188
-0,867
-0,662
—210,776
—11,895
—6,046
—4,022
—2,964
—30,54
—14,33
—6,609
—4,667
—23,069
—5,691
—3,232
—2,205
—1,600
—1,188
—0,913
—19,012
—6,350
—3,859
-2,739
—2,063
—1,594
0,08198
0,07838
0,07342
0,06841
0,06365
0,05929
0,05528
0,05164
411
Приложение
Таблица П3.9 (продолжение)
т
к
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
300
400
500
600
700
800
900
1000
р=1 МПа
V
ЬН
55
2,505 5,2 —0,021
3,338 9,5 —0,009
4,170 11,7 —0,005
5,001 12,0 —0,003
5,833 12,2 —0,002
6,664 12,5 —0,0014
7,496 13,1 —0,0010
8,328 13,4 —0,0008
2,480 —91,83 —0,310
3,325 —61,18 —0,218
4,162 —42,51 —0,182
5,001 —31,83 —0,177
5,836 —21,16 —0,145
6,667 —18,47 —0,125
7,502 —11,78 —0,013
8,333 —9,10 —0,010
2,444 —162,6 —0,374
3,303 —95,9 —0,153
4,151 —62,7 —0,118
4,991 —38,0 —0,076
5,829 —21,6 —0,052
6,665 —16,3 —0,037
7,499 —8,1 —0,027
8,333 —2,0 —0,021
2,361 —393,5 —0,846
3,254 —240,0 —0,392
4,116 —165,3 —0,172
4,967 —116,8 —0,154
5,811 —81,5 —0,108
6,653 —59,3 —0,078
7,491 —50,2 —0,059
8,327 —41,2 —0,045
2,197 —886,62 —
3,191 —467,41 —0,874
4,070 —270,22 —0,406
4,961 —176,74 —0,246
5,817 —130,00 —0,187
6,665 —102,25 —0,143
7,511 —87,64 —0,129
8,354 —74,49 —0,114
2,490 -64,63 -0,198
3,335 —30,76 -0,099
4,174 —12,И —0,057
5,010 —0,66 —0,036
5,844 6,91 —0,025
6,678 12,19 —0,018
7,511 16,04 -0,013
8,343 18,94 —0,010
V
0,510
0,678
0,844
1,011
1,178
1,344
1,510
1,677
0,485
0,666
0,839
1,010
1,179
1,347
1,515
1,683
0,449
0,645
0,826
1,001
1,173
1,344
1,513
1,682
0,332
0,593
0,793
0,978
1,157
1,333
1,505
1,677
р = 5 МПа
ЬН
31,4
47,8
56,1
60,4
62,7
64,9
65,7
66,9
—386,3
—215,8
—129,2
—74,58
—31,94
—5,28
13,38
28,06
—866,5
—498,1
—314,1
—205,6
—130,5
—72,2
—30,7
—5,88
—2612
—1238
—808,7
—563,3
—409,8
—308,8
—220,9
—159,4
0,524 —2813,23
0,770 —1559,98
0,976
1,164
1,343
1,518
1,691
0,497
0,676
0,849
1,020
1,189
1,357
1,525
1,692
—968,42
-641,23
-452,80
-344,72
-277,53
—302,85
—140,04
—51,59
2,77
38,86
64,15
82,65
96,62
bS
V
Ne
—0,081 0,261
—0,046 0,345
—0,022 0,429
—0,018 0,512
—0,013 0,596
—0,007 0,679
—0,005 0,762
—0,004 0,845
Аг
—1,070 0,238
—0,579 0,334
—0,383 0,424
—0,258 0,511
—0,186 0,597
—0,166 0,682
—0,137 0,767
—0,130 0,851
Кг
—2,077 0,202
—1,017 0,315
—0,544 0,412
—0,395 0,503
—0,247 0,592
—0,173 0,679
—0,137 0,765
-0,104 0,851
Хе
—6,345 0,075
—2,241 0,262
—1,233 0,381
—0,821 0,482
—0,480 0,577
—0,367 0,669
—0,321 0,758
—0,279 0,846
SFe
р= 10 МПа
ЬН
67,7
96,2
110,6
121,0
125,2
129,5
131,8
132,0
—735,0
—400,7
—226,3
—119,7
—37,14
13,49
48,14
74,79
—1813
—984,2
—615,8
—398,3
—247,8
—147,6
—64,2
—5,88
-8114
—2617
—1596
—1102
—790,5
—584,5
—417,9
—303,8
-5,342 0,213 —6032,53
-2,493 0,367 —3095,14
-1,398 0,485 —1876,95
-0,887 0,588 —1167,07
-0,653 0,682
-0,521 0,771
-0,448 0,857
N2
—0,950 0,251
—0,474 0,345
—0,274 0,434
-0,175 0,522
—0,119 0,607
—0,085 0,692
—0,063 0,777
-0,048 0,861
-733,25
-473,25
-316,96
bS
—0,151
—0,076
—0,032
—0,027
—0,022
—0,014
—0,010'
—0,008
—2,059
—1,007
—0,612
—0,407
—0,336
—0,275
—0,206
—0,199
-4,442
—1,958
—1,149
—0,749
—0,517
—0,360
—0,294
—0,222
—21,34
—4,750
—2,429
—1,492
—1,020
—0,775
—0,598
—0,424
—11,717
—5,012
—2,763
—1,667
—1,083
—0,776
—0,616
р = 20 МПа
V
0,137
0,179
0,221
0,263
0,305
0,346
0,388
0,429
0,119
0,171
0,218
0,263
0,307
0,350
0,393
0,436
0,092
0,115
0,208
0,257
0,303
0,348
0,392
0,436
0,063
0,115
0,182
0,239
0,291
0,340
0,387
0,432
0,130
0,193
0,255
0,312
0,363
0,408
0,454
—557,86 —1,797 0,132
—250,09 —0,897 0,182
—84,10 —0,522 0,228
18,3
L —0,335 0,273
86,67 — 0.22S
134,87 —0,Ш
170,26 —0,122
197,12 —0,09<!
) 0,317
i 0,360
г о,4оз
t 0,445
ЬН
146,4
199,2
227,7
242,0
252,4
258,7
262,9
267,5
—1317
—707,5
—361,3
—150,9
—20,36
62,23
124,8
175,5
—3473
—1814
—1119
—708,4
—440,6
—248,2
—106,1
2,50
-8757
—5085
—3001
—2034
—1447
—1044
—759,3
—527,0
—0,281
—0,125
—0,081
—0,036
—0,031
—0,027
—0,020
-0,015
—3,726
—1,916
—1,081
—0,716
—0,525
—0,424
—0,356
—0,309
—8,567
—3,653
—2,090
—1,355
—0,955
—0,630
—0,481
—0,408
—19,88
—9,359
—4,675
—2,819
—1,953
—1,446
—1,006
—0,832
—8411,94—15,828
—5159,0?
—3244,11
—1976,2'
—1094Д
—489,32
—83,26
> —8,495
5 —4,975
1 —3,018
5 —1,835
1 —1,119
i —0,695
—919,05 —3,144
—393,18 —1,606
—103,15 —0,953
78,97 —0,619
202,35 —0,428
290,4^
355,8^
t —0,310
t —0,232
405,90 —0,180
412
Свойства газов при повышенном давлении
Таблица II3.9 (окончание)
т
к
300
400
500
600
700
600
700
800
900
1000
300
400
500
600
1
V
2,212
3,209
4,094
4,949
5,788
4,897
5,761
6,62Ь
7,470
8,325
2,422
3,304
4,164
5,017
о = 1 МПа
ЬН
-1136,26
—419,46
-233,48
—120,57
—36,27
—335,8
—249,1
—196,2
—161,1
—135,1
—276
—163
—113
—96
bS
—2,914
—0,763
—0,342
—0,134
—0,005
—0,384
—0,219
—0,177
—0,136
—0,095
—0,80
—0,34
—0,17
—0,13
V
в = 5 МПа
ЬН
0,535 —2436,86
0,767 —1227,71 -
0,960 —648,34 -
1,137 —228,89 -
0,780
0,967
1,141
1,310
1,475
0,409
0,629
0,821
1,004
—1941
—1146
—879
-708
—588
—1545
—846
—557
—389
55
NH
-4,578
-1,821
-0,758
-0,110
N2H4
—2,474
—1,527
—1,126
—0,876
—0,713
NF3
—3,91
—1,69
—0,98
—0,73
V
о = 10 МПа
ЬН
0,150 —7498,24
0,350 -2594,22
0,463 —1383,54
0,559
0,487
0,603
0,707
0,804
0,295
0,405
0,504
—559,10
—3091
—2233
—1747
—1423
—1729
—1110
—783
85
_
—15,391
—3,904
—1,678
—0,403
—3,209
—2,067
—1,491
—1,120
—3,43
—2,00
—1,42
р = 20 МПа
V
_
0,140
0,215
0,273
0,157
0,264
0,312
0,389
0,095
0,191
0,251
ЬН
_
—5744,82
—3002,09
—1418,62
—10115
—5307
—3815
—2985
—4677
—2294
—1524
55
_
—8,856
—3,790
—1,339
—11,620
—5,044
—3,279
—2,415
—8,97
—4,07
—2,73
В последние годы опубликованы экспериментальные данные при высоких температурах: pVT-
данные [3907] и измерения энтальпии [1185].
Соотношение для второго вириального коэффициента в табл. Д.25 получено выделением из
уравнения состояния [2564] составляющей, линейной по плотности. При низких температурах
приближенный расчет может быть выполнен с помощью корреляции Питцера с параметрами
из табл. П3.4.
Значения критических постоянных (табл. Д.26) приняты для га-Н2 по данным [2564], для к-Н2 —
по данным Вулли и др. [4000]. Таблица термодинамических свойств Н2 рассчитана по данным
.работы [2562] (см. табл. П3.9).
Н2О. К 1968 г. Международным комитетом по формуляциям были подготовлены две системы
уравнений состояния: точная система 1968 г. [3649] и система уравнений 1967 г. [3648] для
промышленных расчетов. При создании систем уравнений, наряду с данными Международных
скелетных таблиц [165], использовались данные Киса [2234], а также новые данные по удельным
объемам, полученные Ривкиным с сотр. [216, 217, 218, 220, 221]. Подробное описание системы
уравнений 1968 г. для научного и общего применения можно найти в работах [63, 64, 215]. На основе этой
системы были подготовлены таблицы термодинамических свойств [64, 212, 215]. Методы расчета,
основанные на системе 1968 г., и соответствующие программы для ЭВМ приведены в работах [8,
181].
Система уравнений 1967 г. [3648] менее точна для описания термодинамических свойств,
в особенности изобарной теплоемкости. Описание системы содержится в работе [7]. Подробные
таблицы [2631] основаны на системе 1967 г.
В последние годы выполнен ряд важных исследований термодинамических свойств воды, как
экспериментальных, так и расчетно-теоретических. Проведены новые измерения удельных
объемов при температурах 150—360° С и давлении до 80 МПа [80] и в области околокритических
температур [10]. Келл и др. [2211] измерили величины вириальных коэффициентов в интервале
температур 150—450° С. Горниост [1569] исследовал рУГ-свойства в интервале температур
от 100 до 300° С при давлении от 9 до 75 МПа. Танишита с сотр. [3626] получили новые pVT-дш-
ные для широкой области параметров: 323,15—773,15 К, 1,673—195,165 МПа. В монографии
Амирханова с сотр. [19] суммированы данные по изохорной теплоемкости и составлена опорная
таблица значений Се при температурах от 0 до 800° С и давлении до 100 МПа. Новые измерения
413
Приложение 3
С„ проведены Керимовым и Алиевой [122]. Шомекер [3347] получил данные по внутренней
энергии и Ct для околокритической области. Бланк [669] получил новые значения критических
постоянных.
В ряде работ приведены уравнения состояния, более удобные для практического
использования, чем системы уравнений [3648, 3649]. Поллак [3057] построил единое уравнение состояния,
охватывающее всю область гомогенного и гетерогенного состояний (до 1200 К при давлении
до 300 МПа). Уравнение состояния в вириальной форме, построенное в работе Зубарева и Крупи-
ной [106, 107], может быть использовано для расчетов свойств водяного пара в области температур-
от 300 до 2500° С. Вириальные коэффициенты, входящие в это уравнение, вычислены через
межмолекулярный потенциал Кеезома, который включает диполь-дипольное взаимодействие,
усредненное соответствующим образом по углам.
В табл. Д.25 приведены константы уравнения для второго вириального коэффициента,
рассчитанного по работе [106].
Анализ рУГ-свойств в околокритической области выполнен в обзоре Левельт-Сенгерс и др.
[2543], по рекомендации которых приняты значения критических постоянных (табл. Д.26).
Величины, приведенные в табл. П3.9 для Н2О, рассчитаны по данным работы [64], а при Т > 1100 К
по уравнению состояния [106].
Н2О2. Цыкало и Табачниковым [305] было построено уравнение состояния для чистой Н2О2.
и рассчитаны термодинамические функции на линии насыщения и в однофазной области.
Уравнение состояния [305] охватывает диапазон температур 273,15—2000 К и плотностей 0—12,5 мольХ
Хдм~3. Экстраполяция в область высоких температур осуществлена с помощью второго
вириального коэффициента, рассчитанного по потенциалу Штокмайера, параметры которого определены
в работе [303]. В табл. Д.25 приведены константы соотношения для второго вириального
коэффициента из работы [305]. В указанных работах также изложена методика расчета
диссоциированной перекиси водорода и приведена таблица термодинамических свойств при различных степенях
диссоциации. Те же авторы получили уравнение для кривой упругости, справедливое в диапазоне
температур от тройной до критической точки [304], и оценили значения плотности жидкости на
линии насыщения в диапазоне температур 273—425 К [254]. Значения критических постоянных
приведены в табл. Д.26 по работам [303, 305].
D2. В обзоре Притца и др. [3097] рУГ-поверхность аналитически представлена с помощью
уравнения состояния Стобриджа—Козмана в диапазоне температур 98—423 К при давлениях
до 300 МПа. Линейное по плотности слагаемое в этом уравнении с высокой точностью описывает
данные по второму вириальному коэффициенту при Т < 423 К. Константы этого уравнения
приведены в табл. Д.25. При высоких температурах можно использовать выражение для второго
вириального коэффициента Н2 [2564]. В обзоре Родера и др. [3223] содержится сводка данных
по свойствам D2 при Т < Гкр. Для D2, так же как и для Н2, различают две модификации: K-D2f
равновесный при высоких температурах и содержащий 66,67% ортодейтерия, и D2, равновесный:
при 20 К и содержащий 97,8% ортодейтерия. Для обеих модификаций несколько отличаются
pFT-свойства, давление пара и критические постоянные. Данные Притца и др. [3097] относятся
к h-D2. В табл. Д.26 приведены значения критических постоянных, рекомендованные в [3223].
D2O. Подробный обзор теплофизических свойств D2O по состоянию на 1963 г. содержится
в справочном издании [113]. Приведенные в этом издании уравнение состояния и таблицы
охватывают интервал температур 3,8—500° С при давлении до 50 МПа. В последующие годы были
выполнены экспериментальные исследования изобарной теплоемкости [219 ] и удельных объемов
[295, 296, 3733, 1406]. Келл и др. [2212] провели измерения второго вириального коэффициента
D2O в интервале температур 150—500° С. Новые данные по второму вириальному коэффициенту
получены в работе [1772]. Амирханов с сотр. [20] провели измерения С,. В работе Бланка [669}<
были измерены Т^ и рщ.
Уравнение состояния для газовой фазы, охватывающее широкий диапазон параметров,
построено в работе Трахтенгерца [263]. Для жидкой D2O уравнение состояния и таблицы свойств
рассчитаны Александровым и Хасаншиным [12]. Файн и Миллеро [1479] получили уравнение для;
жидкой фазы, основываясь на данных по скорости звука. В обзоре Александрова и Ривкина [11J
414
Свойства газов при повышенном давлении
рассмотрены работы по теплофизическим свойствам D2O, выполненные в СССР в течение 1969—
1974 гг.
В табл. Д.25 приведены константы интерполяционного соотношения для В (Т) из уравнения
состояния [113]. Критические постоянные (табл. Д.26) приняты согласно рекомендациям Ле-
вельт-Сенгерс и др. [2394]. Таблица термодинамических свойств D2O рассчитана по данным
работы [ИЗ] (см. табл. П3.9).
D2O2. В работе Цыкало и Табачникова [304] получено уравнение для давления насыщенного
пара, справедливое в широком интервале температур, и оценены критические температура и
давление, значения которых приведены в табл. Д.26. Второй вириальный коэффициент (табл. Д.25)
рассчитан с помощью корреляции Мартина.
HD, Т2, HT, DT. Экспериментальные данные по термодинамическим свойствам изотопов
двухатомного водорода чрезвычайно ограниченны. В обзоре [3223] представлена сводка
имеющихся данных и оценок, полученных для области сосуществования, включая тройную и
критическую точки.
В литературе был предложен ряд схем вычисления термодинамических свойств изотопов
водорода, в том числе pVT-съотксчъ, критических констант и т. д. по ограниченным опытным данным
[2683, 157, 1763]. Миттельхаузер и Тодос [2683], проанализировав данные для Н2, D2, НЕ) и Т2>.
получили для всех изотопов уравнения, описывающие давление пара в интервале температур
от тройной точки до критической, и оценили критические параметры всех изотопов. Малышенко
и Швальб [157] развили специальный вариант теории соответственных состояний с учетом
квантовых эффектов для двухатомных газов. Обработка ограниченных данных позволила этим
авторам оценить недостающие константы и рассчитать давление пара и второй вириальной
коэффициент для всех изотопов. Вириальные коэффициенты рассчитаны для температур 26—150 К.
При более высоких температурах разница в величинах второго вириального коэффициента для
различных изотопов становится соизмеримой с погрешностью расчета. В работах [1763, 963]
предложен вариант расчетной схемы, основанный на использовании трехпараметрической
корреляции Питцера с заменой истинных критических параметров на эффективные (см. раздел. П3.2).
После такой замены для расчета свойств могут быть использованы все таблицы и уравнения,
основанные на корреляции Питцера, причем для квантовых газов фактор ацентричности <о=0.
Чу и Праузнитц [963], основываясь на этой корреляции, предложили метод вычисления третьего-
вириального коэффициента изотопов водорода. Бриф и Иоффе [768] применили для расчета
свойств квантовых газов обобщенное уравнение состояния Бенедикта—Вебба—Рубина.
В табл. Д.25 константы для второго вириального коэффициента получены согласно корреляции
Питцера. Критические постоянные (табл. Д.26) приняты в соответствии с рекомендациями авторов
работы [3223].
F2. Притц и Страти [3094, 3095, 3096, 3553, 1685] исследовали термодинамические свойства
в интервале температур от тройной точки E3,5 К) до 300 К при давлении до 21 МПа. В
справочном издании [3095] уравнение состояния в аналитической форме было построено только для
малых плотностей (р<С 6 моль-дм~3). Расчет свойств для высоких плотностей проведен
непосредственно по 70 экспериментальным изотермам.
Для высокотемпературной области 300—3000 К Кессельман [126] рассчитал два вириальных
коэффициента, используя потенциал 12 : 6 с параметрами, зависящими от температуры. По этим
данным рассчитаны величины, приведенные в табл. П3.9. Коэффициенты полинома,
аппроксимирующего данные работы [126 ] по В (Т), приведены в табл. Д.25. Значения критических постоянных
(табл. Д.26) приняты по работе [3095].
F2O. Критические постоянные F2O, приведенные в табл. Д.26, определены в работе Андерсена
и др. [384]. Рипс [223] рассчитал давление насыщенного пара в интервале температур от
нормальной точки кипения до критической. Оценка второго вириального коэффициента (табл. Д.25)
выполнена с помощью корреляции Мартина.
415
Приложение 3
Фтористый водород. Под этим названием фактически понимается не индивидуальное веще-
•ство, а реагирующая смесь мономера HF и его полимеров (HF),,.
Характерная особенность фтористого водорода — заметное проявление неидеальности уже
при низких давлениях A0—20 кПа). рУГ-свойства при малых давлениях исследованы Штромайе-
ром и Бриглебом [3561 ]. Эти же авторы проанализировали состав пара и рассчитали константы
равновесия реакций
Изохорная и изобарная теплоемкости измерены в работах Франка и др. [1525, 1526, 1523].
Обзор экспериментальных данных приведен в работе Ван-дер-Зее и Роденбурга [3771 ]. При
наличии данных по константам равновесия />УГ-свойства ассоциированного газа можно представить
уравнением состояния (см. раздел П3.5)
.где 7 = 2гажя — фактор ассоциации, хп — мольные доли полимеров (HF),, (здесь также предпола-
п
гается, что можно пренебречь «физической» неидеальностью).
В дополнении к справочнику [337] и в настоящем справочнике рассчитаны
термодинамические функции полимеров (HF)M в стандартном состоянии. На основе этих данных, а также данных
do энтальпиям ассоциации полимеров можно, в принципе, вычислить состав системы. При
достаточно малых давлениях можно использовать формулу Вулли (см. раздел П3.5) и выразить вириаль-
иые коэффициенты через константы равновесия. В табл. П3.10 приведены значения вириальных
коэффициентов В, С и D в уравнении состояния
^вычисленные по данным настоящего справочника.
Таблица П3.10. Вириальные коэффициенты фтористого водорода, рассчитанные по формулам Вулли
Коэффициенты
В (дм3-моль)
С (дм6-моль)
D (дм9-моль"8)
200
—5,960
4,302
—5,227
-102
-109
• 10й
250
—46,786
3,615
—8,711
-10е
• 10»
т, к
300
—8,508
2,455
—9,853-
10е
109
350
—2,575
2,505-
—3,136-
10*
103
400
—1,101
4,720
—2,559-
103
107
Расчет термодинамических свойств в более широком интервале давлений был проведен в
работе [3771 ] без какой-либо молекулярной модели с использованием только дифференциальных
•соотношений термодинамики. Данные работы [3771] приведены в табл. П3.11. Кессельман [126]
рассчитал в широком интервале температур два вириальных коэффициента фтористого водорода,
также без учета ассоциации. Эти данные можно использовать только при высоких температурах
(Т ]> 500-1-600 К), когда вкладом полимеров можно пренебречь. Уравнение для второго вириаль-
ного коэффициента (табл. Д.25) и термодинамические свойства HF (табл. П3.9) рассчитаны по
данным работы [126]. Значения критических постоянных (табл. Д.26) приняты по данным Франка
и Шпальтгоффа [1524].
С12. Подробный обзор термодинамических свойств С12 содержится в работе Капура и
Мартина [2193]. Обработка данных, проведенная в этой работе, основывалась на использовании
уравнения состояния Мартина—Хоу. Таблицы термодинамических свойств, построенные в [2193],
охватывают интервал температур 180—550 К при давлении до 9,5 МПа. Новые рУГ-данные для
жидкого хлора получены в работе [3851]; в работе [299] исследовано равновесие жидкость—пар
¦вблизи критической точки.
Термодинамические расчеты для широкой области параметров, не изученной экспериментально,
¦были проведены в работах Сешадри и др. [3391 ] до 1500 К при давлении до 220 МПа и Кессельмана
[126], который рассчитал два вириальных коэффициента в интервале температур 300—3000 К
416
Свойства газов при повышенной давления
Таблица
Р
кПа
0
15,463
30,664
42,529
50,662
55,994
69,193
83,058
96,124
98,790
101,325
114,655
122,921
127,187
155,984
189,314
229,310
329,300
0
15,463
30,664
42,529
50,662
55,944
69,193
83,058
96,124
98,790
101,325
114,655
122,921
127,187
155,984
189,314
0
15,463
30,664
42,529
50,662
55,994
69,193
83,058
96,124
98,790
101,325
114,655
122,921
127,187
155,984
189,314
П3.11. Избыточные объем,
292,69
3,7577
4,6661
10,5297
17,962
20,440
21,059
21,059
19,408
17,673
17,343
17,013
_
—
—
—
0
0,7950
3,766
8,745
12,385
14,560
18,577
21,213
22,510
22,719
22,886
—
0
2,516
12,294
28,733
40,598
47,688
60,468
68,516
72,133
72,690
73,119
—
296,15
3,3860
3,8402
6,8133
12,099
15,691
17,343
18,788
18,375
16,971
16,682
16,435
15,113
—
—
—
0
0,6694
2,510
6,025
9,226
11,401
15,640
19,079
21,046
21,338
21,589
22,661
—
0
2,077
8,039
19,539
29,965
37,015
51,115
61,273
67,150
67,984
68,690
71,615
—¦
энтальпия и энтропия
298,15
-(v-
3,0970
3,3447
5,2855
9,3735
12,512
14,370
17,137
17,426
16,517
16,269
16,022
14,824
14,122
—
—
•—
—
0
0,6067
1,966
4,782
7,657
9,623
14,100
17,656
19,958
20,292
20,585
21,903
22,527
—
—
—
Г, В
299,15
V дм'-моль
р )
2,9318
3,1383
4,8313
8,2586
11,232
13,214
16,104
16,517
16,146
15,939
15,733
14,659
13,998
13,668
—
—
—
фтористого
305,15
-1
2,3537
2,5189
3,0144
4,1706
5,6158
6,7720
9,8277
12,058
12,925
13,007
13,049
12,966
12,718
12,553
11,190
—
—
—
SH, кДж-моль
0
0,5816
1,757
4,238
6,904
8,724
13,221
16,882
19,330
19,686
20,000
21,485
22,154
22,460
—
—
— 8-S, Дж-К-^моль
0
1,866
6,231
15,394
24,727
31,098
45,426
56,554
62,953
64,497
65,339
69,071
70,755
—
—
0
1,790
5,525
13,567
22,210
28,071
42,462
53,959
61,414
62,463
63,372
67,652
69,498
70,323
—
0
0,4602
1,067
2,197
3,494
4,602
8,075
11,757
14,832
15,313
15,732
17,761
18,828
19,372
22,058
—
-1
0
1,385
3,236
6,801
10,928
14,452
25,476
37,034
46,591
48,044
49,306
55,394
58,561
60,138
67,844
^~
водорода
311,15
1,8995
1,9821
2,1885
2,6015
3,0144
3,4686
4,9551
6,9785
8,7128
8,9606
9,1670
9,9516
10,117
10,117
10,075
9,2909
—
—
0
0,3682
0,8159
1,318
1,841
2,364
4,268
6,987
9,623
10,125
10,586
12,887
14,184
14,853
17,824
21,652
0
1,089
2,420
3,953
5,554
7,181
13,124
21,582
29,718
31,251
32,663
39,641
43,541
45,504
54,151
65,419
(на 1 ноль
317,15
1,5278
1,5278
1,6104
1,7343
1,9821
2,1472
2,6427
3,5519
4,6248
4,8313
5,0377
6,1526
6,7720
7,1024
8,4650
8,5063
7,7218
—
0
0,2929
0,6276
0,9205
1,192
1,423
2,301
3,724
5,439
5,816
6,213
8,243
9,372
10,146
14,477
18,577
0
0,8443
1,821
2,691
3,496
4,195
6,860
11,214
16,451
17,599
18,813
24,987
28,377
30,725
43,680
55,699
HF)
329,15
1,0282
1,0282
1,0282
1,0365
1,0447
1,0530
1,1521
1,2553
1,4246
1,4453
1,4865
1,7343
1,8994
2,0646
3,5099
5,1616
6,1939
5,4507
0
0,1841
0,3933
0,5858
0,7113
0,8159
1,088
1,464
1,874
1,967
2,050
2,510
2,803
2,971
5,439
10,460
0
0,5085
1,093
1,653
1,996
2,301
3,089
4,182
5,377
5,644
5,885
7,220
8,072
8,555
15,813
30,686
417
Приложение
с использованием потенциала 12 : 6 с параметрами, зависящими от температуры.
Термодинамические свойства С12, приведенные в табл. П3.9, рассчитаны по данным работы [126]. Коэффициенты
полинома, аппроксимирующего значения В (Т) из этой работы, приведены в табл. Д.25. Значения
критических постоянных (табл. Д.26) приняты согласно рекомендации Матьюса [2543].
С12О. В работе Рипса и др. [224] вычислены критические параметры с помощью
корреляции [191 ]. Используя эти данные, Рипс [223] произвел расчет давления насыщенного пара для
интервала температур от нормальной точки кипения до критической. Для расчета второго вириаль-
ного коэффициента (табл. Д.25) использована корреляция Мартина.
Значения критических постоянных (табл. Д.26) приняты по [224].
НС1. Обработка экспериментальных данных в низкотемпературной области выполнена
Волошиным [57].
В основу обработки были положены рVГ-данные из работ [1522, 2508] для интервала
температур 313,15—673,15 К и результаты работы Томаса [3667], исследовавшего свойства на линии
насыщения. При построении уравнения состояния по опытным данным предварительно были
Таблица П3.12
Т
К
293,15
303,15
313,15
333,15
353,15
. Параметры дилера (C1F3J
—ArG° (Г)
кДж • моль
ДМ3 • МОЛЬ
9,372 0,512
10,125 0,446
10,837 0,400
12,259 0,325
13,891 0,254
Т
К
373,15
393,15
413,15
433,15
453,15
—ATG°(T)
кДж • моль
15,397
17,531
19,163
21,087
22,217
—вхш
дм3 • моль
0,212
0,151
0,127
0,102
0,101
определены два вириальных коэффициента, которые затем экстраполировались по методике Кес-
сельмана [126] до температур 3000 К. Поэтому уравнение состояния применимо за пределами
опорной сетки опытных данных.
В работе Сешадри и др. [3390] построены уравнения состояния двух типов: Мартина—Хоу
и Бенедикта—Вебба—Рубина.
Термодинамические свойства НС1, приведенные в табл. П3.9, рассчитаны по данным работы [126].
Константы уравнения, аппроксимирующего значения В (Т) из работы [126], приведены в табл. Д.25.
Значения Гкр и ркр приняты в табл. Д.26 согласно работе [3667], а значение FKp — в соответствии
с оценкой, проведенной в работе [57].
C1F3. Наиболее полная сводка данных по теплофизическим свойствам C1F3 содержится в работе
Моризо и др. [2713] (см. также [2501, 3466]). Авторы работы [2713] получили экспериментальные
данные по второму вириальному коэффициенту в диапазоне температур 20—180° С и выделили
вклад, связанный с димеризацией молекул.
В соотношении В=Вхих-\-В^вз «химическая» часть (см. раздел П3.5) определяется константой
равновесия реакции димеризации А2**2А
Bm = -RT/K0(T),
К° (Т) = f (A)lp (A2) = exp (-drG° (T)/RT).
В табл. П3.12 приведены значения ArG° (Т) для реакции (C1F3J=2C1F3, также Вхвн но [2713].
Значения В^яз аппроксимированы с помощью потенциала Леннарда—Джонса 12 : 6 (е/А:=160 К
с=5,07 А). Константы уравнения для В^в приведены в табл. Д.25.
Критическая температура и давление C1F3 определялись многими авторами [224, 1737, 228]-
Принятые в табл. Д.26 значения получены в работе Гризарда и др. [1737].
418
Свойства газов при повышенной давлении
C1F5. Термодинамические свойства C1F5 на линии насыщения исследованы в работе [3230].
В результате обработки экспериментальных данных получена температурная зависимость
давления пара в интервале 193—423 К. Для температур от 193 до 372 К построено уравнение,
описывающее зависимость ортобарической плотности р' от температуры. В работе [3230] получены также
критические постоянные C1F5, приведенные в табл. Д.26.
Оценка второго внриального коэффициента (табл. Д.25) выполнена с помощью корреляции
Мартина.
Вг2. Кессельман [1261 рассчитал вириальные коэффициенты Вг2, основываясь на теории
термодинамического подобия с использованием потенциала 12 : 6. Сешадри и др. [3394] построили
уравнение состояния Мартина—Хоу и рассчитали таблицу термодинамических свойств в однофазной
области и на линии насыщения. В работе [844] получены данные для ортобарических плотностей
и оценено значение критической плотности. С использованием данных [126] рассчитаны
термодинамические свойства Вг2 (табл.ПЗ.9) и получено аппроксимирующее уравнение для В (Т) (табл. Д.25).
Значения критических постоянных (табл. Д.26) приняты согласно рекомендациям Матыоса [2543].
1а. Бурьель-Луна и др. [844] исследовали свойства 12 на линии насыщения (критические
постоянные и значения р').
Расчет вириальных коэффициентов 12 ввиду отсутствия pFT-данных был выполнен Кессельма-
ном [126] на основе теории термодинамического подобия с использованием потенциала 12; 6.
По данным этой работы рассчитаны термодинамические свойства 12 (табл. П3.9) и получено аппро-
ксимационное уравнение для В(Т) (табл. Д. 25). Значения критических постоянных (табл. Д.26)
приняты согласно рекомендациям Матыоса [2543].
НВг, Ш. Расчеты термодинамических свойств были выполнены Кессельманом [1261 на основе
теории термодинамического подобия
Поскольку анализ экспериментальных данных по вязкости показал, что при умеренных
плотностях N2, HBr и HI термодинамически подобны, то по />УГ-данным для N2 были рассчитаны
параметры потенциала Леннарда—Джонса (зависящие от температуры) и два вириальных
коэффициента в температурном диапазоне 300—3000 К.
Имеющиеся данные по давлению насыщенных паров обработал и привел в своем справочнике
Эдуарде [1386].
Термодинамические свойства НВг и HI (см. табл. П3.9) рассчитаны по данным [126].
В табл. Д.25 приведены коэффициенты полиномов, аппроксимирующих данные по второму ви-
риальному коэффициенту [126]. Значения критических постоянных (табл. Д.26) приняты согласно
рекомендациям Матыоса [2543].
Не*. К настоящему времени подготовлены Международные термодинамические таблицы
ИЮПАК [399], охватывающие область газового и жидкого состояния B,2—1400 К, р < 70 МПа),
за исключением сверхтекучей фазы. Это издание включает детальный обзор экспериментальных
данных, а также уравнения состояния и расчетные соотношения для линий фазового равновесия.
При построении таблиц за основу принята корреляция термодинамических свойств, выполненная
в работе Мак-Карти [2561 ]. Для обработки рУГ-свойств была применена система из трех
независимых уравнений состояния, каждое из которых описывало определенную область параметров.
Во втором издании монографии Цедерберга и др. [298] также представлен подробный обзор тепло-
физических свойств и методов расчета. Термодинамические таблицы, приведенные в указанной
работе, рассчитаны по уравнению состояния с двумя вириальными коэффициентами и охватывают
диапазон температур 0—3000° С при давлении р <J 20 МПа. В работах Казавчинского с сотр. [115,
137, 257] были построены уравнения состояния Не для широкой области параметров: Т=20~
-f-1500 К [137]; Г=2-^2000 К для газовой и жидкой фазы при плотности до 0,2 кг-дм~3 [115, 2571.
Аналитическое выражение для второго вириального коэффициента [115, 257] справедливо вплоть
до 8000 К.
В последние годы в ряде работ [2240, 297] получены новые рУГ-данные. Кирстед [2240] провел
прецизионные измерения на 29 изохорах в узкой окрестности критической точки. Им также были
1 В настоящем справочнике рассматривается природная смесь изотопов гелия. Практически ее pVT-свойства
совпадают со свойствами *Не.
419
Приложение 3
уточнены значения критических постоянных и построено неаналитическое уравнение состояния.
Цедерберг и др. [297] получили />УГ-данные в интервале температур 70—273 К при давлении
до 100 МПа. Гаммон [1564] выполнил прецизионные измерения скорости звука в интервале
температур от —175 до 150° С и давлений от 1 до 15 МПа и построил уравнение состояния. Им
рассчитана также таблица значений второго вириального коэффициента и его производных по
температуре в диапазоне от 20 до 1500 К.
В обзоре Левельт-Сенгерс и др. [2394] представлен детальный анализ рУГ-свойств гелия в
околокритической области.
В табл. Д. 25 приведены константы интерполяционного соотношения для второго вириального
коэффициента из работы [137]. Значения критических постоянных в табл. Д.26 приняты по
таблицам [399]. Термодинамические свойства Не (табл. П3.9) рассчитаны по данным работы [298].
Ne. Подробный обзор термодинамических свойств Ne представлен в монографии Рабиновича
и др. [210]. Для расчета свойств газовой фазы авторы указанной работы построили вириальное
уравнение состояния в широком интервале параметров: от тройной точки B4,55 К) до 1300 К
при р ^ 2 ркр. Отдельное уравнение состояния построено для жидкой фазы в интервале температур
от тройной точки до 50 К при р ^ 1,8 ркр (р ^ 100 МПа).
В области температур от 52 до 423,15 К при р ^ 2рЕр Оносовский и др. [186 ] построили
уравнение состояния Казавчинского с одной температурной функцией. Новые измерения критических
постоянных выполнены Пеком [2985а]. В этой же работе исследовалось поведение изохорной
теплоемкости в узкой окрестности критической точки.
Температурная зависимость второго вириального коэффициента для четырех инертных газов
(Ne, Аг, Кг, Хе) представлена в монографии [210] единым соотношением
где 0=ГБ/Г, Гб— температура Боиля, Ув = {Тс1В1AТ)т~тБ — объем Бойля и /(8) — полином,
константы которого даются в табл. Д.251 и одинаковы для всех четырех газов. Это соотношение
справедливо вплоть до 6=11, что для Ne соответствует Т — 1300 К.
Значения критических постоянных, рекомендованные авторами [210], приведены в табл. Д.26.
Термодинамические свойства Ne (табл. П3.9) рассчитаны также по данным работ [210].
Аг. Наиболее надежные данные по аргону содержатся в монографии Рабиновича и др. [210] и
в Международных термодинамических таблицах [398]. Таблицы термодинамических свойств аргона,
приведенные в монографии [210], представляют собой откорректированные Международные
таблицы [398]. Последние составлены на основании хорошо согласующихся расчетных данных,
полученных по независимым уравнениям состояния в перекрывающейся области параметров [47,
1707].
В монографии [210] содержатся уравнения состояния (отдельно для газовой и жидкой
фаз), при выводе которых были учтены экспериментальные результаты, появившиеся в
литературе после публикации [398]. В обзоре Левельт-Сенгерс и др. [2394] предложено неаналитическое
уравнение состояния для узкой окрестности критической точки.
Уравнение для второго вириального коэффициента Аг (табл. Д.25) идентично по форме
соответствующему уравнению для Ne (см. выше). Значения критических постоянных (табл. Д. 26)
приняты согласно рекомендациям Левельт-Сенгерс и др. [2394]. Термодинамические свойства Аг
(табл. П3.9) рассчитаны по данным работы [210].
Кг. Подробный обзор термодинамических свойств Кг представлен в монографии Рабиновича
и др. [210]. Для расчета свойств газовой фазы авторы указанной работы построили вириальное
уравнение состояния для широкого интервала параметров: от тройной точки A15,76 К) до 1300 К
при р ^ 2 ркр. Отдельное уравнение состояния построено для жидкой фазы в интервале температур
от тройной точки до 240 К при р J> 1,8 ркр (р ^ 100 МПа). В работе Юцы и Зифнера [2183]
на основе данных работ [33, 3651 ] получено уравнение состояния Кг для температурного
интервала 120—423,15 К. Новые pVT-данные в интервале температур 120—300 К при давлении
до 50 МПа получены Новиковым [185]. В той же работе приведены измерения давления
насыщенного пара и построено уравнение состояния. В обзоре Левельт-Сенгерс и др. [2394] предложено
неаналитическое уравнение состояния для узкой окрестности критической точки.
420
Свойства газов при повышенном давлении
Уравнение для второго вириального коэффициента (табл. Д.25) идентично по форме
соответствующему уравнению для Ne (см. выше). Значения критических постоянных (табл. Д. 26) приняты
согласно рекомендациям Левельт-Сенгерс и др. [2394]. Термодинамические свойства Кг (табл. П3.9)
рассчитаны по данным работы [210].
Хе. Подробный обзор термодинамических свойств представлен в монографии Рабиновича
и др. [210]. Для расчета свойств газовой фазы авторы этой работы построили вириальное
уравнение состояния для широкого интервала параметров: от тройной точки A67,36 К) до 1300 К при
р ^ 2ркр. Отдельное уравнение состояния построено для жидкой фазы в интервале температур
от тройной точки до 300 К при р ^ 1,8 ркр (р ^ 100 МПа). рУТ-свойства Хе в
низкотемпературной области изучены Тьюисом и Бирманом [3653, 3654], Стритом и др. [3555] и Новиковым [185]
В последней работе впервые получены данные по плотности Хе в газовой фазе при Т < 273,15 К.
Терри [3641 ] провел измерения плотности жидкого Хе на линии насыщения. Прецизионные
измерения рУГ-свойств в узкой окрестности критической точки выполнены в работе [1416]. Питаев-
ская и др. [195] получили данные по скорости звука в интервале температур 25—200 СС при
давлении до 400 МПа.
Детальный анализ рУГ-свойств в околокритической области выполнен в обзоре
Левельт-Сенгерс и др. [2394]. Значения критических постоянных Хе (табл. Д. 26) приняты по данным [2394].
Уравнение для второго вириального коэффициента (табл. Д. 25) идентично соответствующему
уравнению для Ne (см. выше). Термодинамические свойства Хе (табл. П3.9) рассчитаны по данным
работы [210].
Пары серы. Так же как и фтористый водород, сера относится к ассоциированным веществам
со сложным составом пара (SK при п ^ 8). Наиболее полное исследование термодинамических
свойств серы, включающее определение рУГ-свойств и расчет равновесия, выполнен в работах
Pay и др. [3165, 3166 ]. Обзор старых работ по термодинамическим свойствам серы приведен в статье
Уэста [3925]. В работах [3165, 3166] определены критические постоянные, проведены измерения
давления пара и ортобарических плотностей до температуры 1273 К и приведены pFT-данные.
Методика расчета, предложенная Pay и др. [3166] включает совместную обработку рУ2"-дан-
ных и опубликованных в литературе данных по равновесиям с целью одновременного нахождения
энтальпий и энтропии реакций
STl q
и параметров, определяющих неидеальность пара. Для оценки последних был принят ряд
приближений (см. также раздел П3.5):
1) пары серы рассматривались как идеальная смесь неидеальных компонентов;
2) состав системы рассчитывался в приближении Льюиса—Рендалла;
3) фактор сжимаемости Z и коэффициент летучести у полагались одинаковыми для всех
компонентов и рассчитывались из обобщенных зависимостей Z (Тг, рг), у (Тг, рг).
В настоящем справочнике для Sn приняты несколько иные значения термодинамических
функций, энтальпий образования, а следовательно, и констант равновесия. Однако оценку
неидеальных свойств пара [3166], учитывая ее приближенный характер, можно оставить без
изменений. Второй вириальный коэффициент можно оценить, приняв что пары серы и фосфора (см. ниже)
подчиняются принципу термодинамического подобия. Для фосфора постоянные Ван-дер-Ваальса,
связанные с В (Т) соотношением
В(Т) = Ъ — ajRT,
определены Pay [3164] из pFT-данных.
В табл. Д. 25 для обоих веществ приведено одинаковое уравнение, поскольку одинакова зави
симость Вр^1ЯТщ от Тг. При этом учитывается только «физическая» часть вириального коэффи"
циента, определяющая неидеальность компонентов SB. В табл. Д. 26 приведены значения
критических постоянных по данным работы [3165]. Мольвый объем FKp определяется
соотношением
, = 0,263
421
Приложение 3
и относится к смеси в целом
' кр кр />• -*" смеси"
Критическая плотность 0,563 кг -дм~3 определяется соотношением
РкР== MCMeJVKV = vKVMljVKr
Фактор ассоциации в критической точке vKp=2,78.
502. Основные данные по термодинамическим свойствам в однофазной области и на линии
насыщения получены Кангом и др. [2188а]. Данные по сжимаемости перегретого пара в диапазоне
температур 10—350 °С обработаны [2188] с помощью уравнения состояния Бенедикта — Вебба—
Рубина. В работе [2188а] также приведены уравнения для давления насыщенного пара и орто-
барических плотностей в интервале температур 50—157,5 °С. В работе Канджара и др. [869]
для расчетов свойств на линии насыщения рекомендуются уравнения, приведенные в обзоре Рин-
нинга и Херда [3277]. В справочном издании Канджара и Маннинга [870] рассчитаны таблицы
термодинамических свойств SO2 в британской системе единиц.
В работе [2188а] определены параметры потенциала Штокмайера, с помощью которых
рассчитан второй вириальный коэффициент (табл. Д. 25). В табл. Д. 26 приведены значения j критических
постоянных по работе [2188а].
503. Критические постоянные SO3 определялись в работах Бертоде [625] и Аберкромли
и Тиля [334]. Эдуарде привел в своем справочнике [1386] данные подавлению пара в интервале
температур от нормальной точки кипения до критической и дал собственную оценку ркр, обработав
данные работы [334]. Матьюс [2543] приводит среднее значение TSB из работ [334, 625] и среднее
значение рхр из работ [334, 1386].
Свотношение для второго вириального коэффициента (табл. Д.25) получено с использованием
корреляции Питцера. Значения критических постоянных (табл. Д.26) приняты по
рекомендациям, приведенным в работе [2543].
HaS. Старлинг и Квок [3503] построили корреляцию на базе обобщенного уравнения состояния
Старлинга, обработав совместно данные Римера и др. [3175] и Льюиса и Фредерикса [2418] по
сжимаемости и данные Кея и Рамбосека по давлению насыщенного пара [2205]. Область
применимости уравнения состояния от 60 до 280 °С при давлении до 35 МПа.
Холлеран [1980, 1981] рассчитал фактор сжимаемости H2S, использовав предложенный им
трехпараметрический принцип соответственных состояний. Построенное в работе [1981] вириаль-
ное уравнение состояния пригодно для расчетов свойств газовой фазы в интервале температур
350-1850 К при р < 1,5 Рвр.
В табл. Д. 25 приведены константы уравнения для второго вириального коэффициента из
работы Холлерана [1981]. Значения критических постоянных (табл. Д. 26) приняты согласно
рекомендациям Матьюса [2543].
SF6. В работах Жердева и Улыбина [98, 99] представлен обзор экспериментальных работ по
термодинамическим свойствам и проведена обработка, основанная на многопараметрическом ви-
риальном уравнении состояния; там же получены данные по давлению пара и ортобарическим
плотностям, построены интерполяционные соотношения, рассчитаны таблицы свойств на линии
насыщения и плотность SF6 в однофазной области. С использованием закона единичной
сжимаемости [1979] рассчитана таблица экстраполированных значений плотности при температурах
до 500 °С и давлении до 45 МПа. В ряде исследований [156, 463, 464, 1460, 3101 ] подробно изучены
pFr-свойства SF6 в окрестности критической точки. Неаналитическое уравнение состояния для
критической области построено в работе Левельт-Сенгерс и др. [2394].
Наиболее широкая область параметров с хорошей точностью описана интерполяционными
соотношениями, полученными Алтуниным [18] в результате обработки ряда теплофизических
свойств, включая pFr-данные, а также данные по вязкости, теплопроводности, самодиффузии
и др. Второй вириальный коэффициент рассчитан [18] при помощи трехпараметрического
потенциала сферической оболочки, параметры которого получены совместной обработкой данных
по совокупности свойств. Интерполяционное соотношение для второго вириального коэффициента,
полученное в этой работе, приведено в табл. Д. 25. По данным работы [18] также рассчитаны
422
Свойства газов при повышенной давлении
термодинамические свойства SFe (табл. П3.9). Значения критических постоянных (табл. Д. 26)
приняты согласно рекомендациям, приведенным в работе [2394].
N2. Наиболее полные обзоры данных по термодинамическим свойствам азота приведены
в справочных изданиях [250, 398а]. В монографии Сычева и др. [250] предложен новый метод
расчета термодинамических свойств с использованием математического эксперимента на ЭВМ.
Метод заключается в составлении совокупности уравнений состояния, описывающих р7Г-данные
в пределах погрешности эксперимента, с последующим статистическим усреднением по набору
уравнений состояния. По мнению авторов работы [250], данный метод позволяет наилучшим
образом определить pFr-поверхность и оценить погрешности вычисляемых термодинамических
функций. Таблицы термодинамических свойств и уравнение состояния [250] охватывают интервал
температур 65—1500 К и давлений 0,1—100 МПа. Таблицы Ангуса и др. [398а] охватывают
диапазон температур от 64 до 1100 К. Верхний предел по давлению составляет 1000 МПа при Т <С 650 К
и 90 МПа при Т >• 650 К. Обработка данных и построение таблиц основано на уравнении
состояния Якобсена и Стюарта [2081, 2082, 3536].
В последние годы получены новые экспериментальные данные по pFr-свойствам N2. Тимрот
и др. [260, 261 ] измерили плотность жидкого и газообразного N2 в интервале температур 80—200 К
при давлении до 37 МПа. В широкой окрестности критической точки A20—150 К при плотности
от 0,1 до 0,52 кг-дм~3) pFr-CBOHCTBa исследованы в работе Зозули и Благого [140]. Там же
построено неаналитическое уравнение состояния для критической области. рУТ-цшкые газообразного
N2 при давлении дъ 800 МПа и температурах до 1800 К получены Антановичем и Плотниковым [20а].
Пококк и Уэрмолд [3053 ] измерили изотермический коэффициент Джоуля—Томсона при
температурах от 78 до 298,15 К и давлении до 600 кПа.
Термодинамические свойства N2 (табл. П3.9) рассчитаны по данным работы [250]. По этим же
данным принято также интерполяционное уравнение для второго вириального коэффициента
{табл. Д. 25) и значения критических постоянных (табл. Д.26).
NO. Обработка pFr-данных [773, 774, 1678 J для однофазной области выполнена в работе
Табачникова и Сердюка [253]. Уравнение состояния [253] охватывает широкий диапазон
параметров A90—2000 К при плотности р <! 20 моль-дм~?, что соответствует давлению до 100 МПа).
Соотношение для второго вириального коэффициента [253], приведенное в табл. Д. 25,
справедливо до 10 000 К. Таблицы термодинамических свойств, рассчитанные по уравнению состояния
[253], приведены в работе [232]. Эдуарде [1386] обработал и привел в своем справочнике данные
по давлению насыщенного пара NO.
Значения критических постоянных (табл. Д.26) приняты согласно рекомендации Матьюса
12543].
N2O. Термодинамические свойства N2O в интервале температур от —30 до +150 °С и
давлений до 32 МПа исследованы в работах Кауча и Кобе [913], Хирта и Кобе [1956]. Шампидр. [3322]
•определили второй и третий вириальные коэффициенты в диапазоне температур 0—150 °С.
Сглаженные значения второго вириального коэффициента приведены в справочнике Даймонда и Смита
[1364]. Новые данные по второму вириальному коэффициенту получены в работе [2255]. В работе
{1184] проведены измерения энтальпии N2O при высоких давлениях. Сенгерс и др. [3388]
провели исследование кривой сосуществования вблизи критической точки.
Второй вириальный коэффициент N2O в табл. Д.25 рассчитан через потенциал Леннарда—
Джонса с использованием параметров, найденных в работе [3322]. Принятые значения
критических постоянных [913] приведены в табл. Д.26.
Четырехокись азота. В теплофизической литературе принято четырехокисью азота именовать
не индивидуальное вещество, а химически реагирующую систему
которая обладает свойствами индивидуального вещества: имеет постоянные точки плавления и
кипения, критические параметры и единую линию насыщения. Обзор экспериментальных данных
и методик расчета термодинамических свойств четырехокиси азота представлен в монографии [267а].
Таблицы свойств, рассчитанные в этой монографии, охватывают интервал температур от линии
насыщения до 1300 К и давлений от 0,1 до 24 МПа. Новые данные по энтальпии, не отраженные
423
Приложение 3
в монографии [267а], получены в работе Шейндлина и др. [315а]. Измерения проводились в
интервале температур 305—782 К при давлениях 0,5—30 МПа. На основе этих данных рассчитана таблица
термодинамических свойств (плотность, теплоемкость, энтропия). В работе [31а] проведены
измерения скорости звука в парах четырехокиси азота при температурах 290—400 К при давлении
до 0,1 МПа.
В литературе предложено несколько расчетных методик для вычисления термодинамических
свойств четырехокиси азота [252, 38, 127в, 323а, 3390]. Табачников иМежерицкий [252] построили
уравнение состояния смеси четырех компонент в форме
где xt — мольные доли компонентов, Zif — уравнение состояния г-го компонента, Z(j—
перекрестный член, учитывающий неаддитивность термодинамических свойств. Состав рассчитывался с
учетом летучести (в приближении Льюиса—Рендалла).
Бубнов и Моисеенко [38] предложили уравнение состояния в форме
где alt а2 — степени диссоциации реакций разложения N2O4 и NO2, рассчитанные в идеально-
газовом приближении, v — удельный объем смеси. Функция / (Т, р), полученная обработкой
pFT-данных, учитывает неидеальность смеси. Клепацкий и Шанкин [127в] использовали
уравнение состояния
(
В обеих методиках [38,127в ] уравнение состояния учитывает отклонение от свойств идеально-
газовой реагирующей смеси. Уравнение Клепацкого и Шанкина [127в], охватывающее интервал
температур от линии насыщения до 580 К при р <С 25 МПа, использовалось в монографии [267а I
для расчета термодинамических таблиц. Якуб [323а ] применил к расчету неидеальной
реагирующей смеси аппарат теории возмущений. Сешарди [3390] предложил два варианта уравнения
состояния в форме уравнений для индивидуального вещества (уравнение Мартина—Хоу и Бенедикта—
Вебба—Рубина). Оба уравнения воспроизводят опытные pVT-R&ESbie и, таким образом, эффективно
учитывают влияние диссоциации и неидеальность. Расчеты термодинамических свойств на линии
насыщения проведены в работах [144, 145].
Дл ¦ оценки неидеальных свойств при малых давлениях можно использовать второй вириаль-
ный коэффициент (табл. Д. 25), выделенный из уравнения состояния [127в]. При этом следует
учитывать, что второй вириальный коэффициент определен соотношением
<у_ pv«V,ot __, ¦ В(Т)
где v — удельный объем (в дм3-кг).
Степени диссоциации и константы равновесия связаны соотношениями
в
A - «аJ A + «1 + «Л)
Константы равновесия могут быть вычислены по данным настоящего издания.
Значения критических постоянных (табл. Д. 26) приняты по данным Римера и Сейджа [31741
и Шлингера и Сейджа [3333].
424
Свойства газов при повышенном давлении
NH3. Выполненная недавно Харом и Галлахером [1784а] работа содержит обзор эксперимен"
тальных данных по термодинамическим свойствам аммиака, опубликованных до 1976 г., изложение-
оригинальной методики расчета и термодинамические таблицы. При обработке данных авторы
[1784а] строили аппроксимационное уравнение для энергии Гельмгольца А (р, Т), основываясь
только на измерениях pFr-свойств и свойствах в стандартном состоянии. По мнению
авторов [1784а], примененный ими метод обработки позволил с наибольшей точностью предсказать
остальные термодинамические свойства (энтальпия, теплоемкость и др.) и обеспечить
термодинамическое согласование на линии фазового равновесия. Таблицы термодинамических свойств [1784а]
охватывают интервал температур от тройной точки A95,48 К) до 750 К при давлении до 500 МПа.
В литературе прошлых лет опубликовано немало работ, содержащих обзоры и корреляции
термодинамических свойств NH3. Наибольшей известностью пользуется обзор в монографии
Дина [3656], отражающий данные, опубликованные до 1956 г. По методике Казавчинского [112]
уравнения состояния NH3 были построены в работах Табачникова [251 ] (до 3000 °С при плотности
до 20 моль-дм~3) и Цоймана [301] (до 300 °С при плотности до 28 моль-дм~3).
Шульц [3356] построил уравнение состояния для NH3 в диапазоне температур 200—450 К
при давлении до 2,5 МПа. Перелыптейн и Парушин [194], используя разработанный ими метод
расчета термодинамических свойств фреонов, составили для аммиака уравнение состояния и
интерполяционные соотношения для расчета свойств на линии насыщения. По этим уравнениям были
рассчитаны таблицы свойств от —70 до 350 СС при р ^ 11 МПа.
В последние годы выполнен ряд экспериментальных исследований термодинамических свойств
NH3 [2324, 1170, 1171, 445, 344а], большая часть из которых отражена в обзоре [1784а]. pVT-
свойства газообразного и жидкого аммиака изучались в работах [2324, 1170, 1171]. В работе
Дате [1171 ] исследовалась также окрестность критической точки и были получены новые значения
критических постоянных. Вер и др. [445] измерили в широком интервале температур давление
насыщенного пара NH3. Xap [1784] получил новые данные по изобарной теплоемкости. Адам и
Шрамм [344а] измерили второй вириальный коэффициент аммиака в интервале 25—250 СС и
исследовали изотопный эффект при замене NH3 на ND3.
Термодинамические свойства NH3 (табл. П3.9) рассчитаны по данным работы [1784а]. По этим
данным приняты также значения критических постоянных (табл. Д. 26). Интерполяционный'
полином для второго вириального коэффициента (табл. Д. 25) принят по работе Табачникова [251 ].
Это соотношение рассчитано по потенциалу Штокмайера, охватывает широкий интервал
температур (до 3000 СС) и хорошо согласуется с данными работы [1784а].
N2H4. Обработка экспериментальных данных и расчет термодинамических свойств гидразина
выполнены в работе Даса и Кулура [1169]. Авторы вычислили константы уравнения состояния
Мартина—Хоу, охватывающего диапазон температур 273—1500 К при давлении до 60 МПа,
а также уравнения для давления пара в интервале температур 273—653 К. Цыкало и др. [302]
рассчитали второй вириальный коэффициент N2H4 и определили параметры потенциала
Штокмайера.
В табл. Д. 26 приведены значения критических постоянных, рекомендованные авторами
работы [1169]. В табл. Д. 25 приведено соотношение для второго вириального коэффициента,
выделенное из уравнения состояния Мартина—Хоу по данным той же работы [1169]; эти данные
использованы и для расчета термодинамических свойств N2H4 (табл. П3.9).
NF3. Сешадри и др. [3393] провели обработку имеющихся экспериментальных данных по
свойствам на линии насыщения [2107, 2615, 3018] и получили константы уравнения Мартина—Хоу.
Таблицы термодинамических свойств NF3 рассчитаны ими в интервале температур от линии
насыщения до 600 К и по давлению до 20 МПа. Интерполяционное соотношение для второго
вириального коэффициента (табл. Д. 25) выделено из уравнения состояния Мартина—Хоу по данным этой
работы [3393]. По этим же данным в табл. Д. 26 приведены значения критических постоянных,
а в табл. П3.9 рассчитаны термодинамические свойства NF3.
N2F2. Критические температура и давление цис- и трансизомера N2F2 измерены в работе Кол-
берна и др. [1010]. Однако Панкратов [188] предположил, что экспериментальная величина
[1010] для pKV tyuc-N2F2 нуждается в уточнении, и рассчитал ркр по корреляционным формулам..
В работе [188] рассчитаны также значения укр для обеих форм N2F2.
425
Приложение 3
Оценка второго вириального коэффициента N2F2 (табл. Д. 25) выполнена с помощью
корреляции Мартина. Значение Т^ принято по данным работы [1010], а ркр и укр — по оценке [188]
{табл. Д. 26).
N2F4. Колберн и Кеннеди [1011] исследовали свойства N2F4 на линии насыщения, однако не
указали метода исследования и интервала температур. В работах [139, 140] получены
температурные зависимости давления насыщенных паров вплоть до критической точки и ортобарической
плотности в диапазоне температур от —150 до 20 °С, а также значения критических параметров,
которые приведены в табл. Д. 26. Оценка второго вириального коэффициента (табл. Д. 25)
выполнена с помощью корреляции Мартина.
FNO. Термодинамические свойства FNO изучены в работе Руффа и др. [3271]. Авторы
измерили давление пара для температур от —124,4 °С до —62,1 °С и плотность жидкости в диапазоне
—125 °С < t < 67 °С на линии насыщения.
Экспериментальные данные о критических постоянных FNO отсутствуют. В работе
Кузнецовой и др. [140] эти постоянные рассчитаны по корреляционным формулам; соответствующие
значения приведены в табл. Д. 26. Оценка второго вириального коэффициента (табл. Д. 25) выполнена
с помощью корреляции Мартина.
FNO2. Термодинамические свойства FNO2 в области низких температур (до —61 °С)
исследованы в работах [3271, 2836]. В работе [2836] измерена критическая температура.
Экспериментальные данные о критическом давлении и критическом объеме в литературе отсутствуют. Кузнецова
и др. [140] рассчитали значения ркр и FKp по корреляционным формулам.
Оценка второго вириального коэффициента (табл. Д. 25) выполнена с помощью корреляции
Мартина. Найденные в работах [2836, 140] значения критических постоянных приведены
в табл. Д.26.
F3NO. Свойства F3NO на линии насыщения изучены в работе Фокса и др. [1516]. Давление
ларов определялось экспериментально при t <С 82 °С. В работе измерены критическая температура
и плотность, а по данным о давлении пара оценено также р^.
Оценка второго вириального коэффициента (табл. Д.25) выполнена с помощью корреляции
Мартина. Принятые в работе [1516] значения критических постоянных, приведены в табл. Д.26.
NHF2. Кеннеди и Колберн [2222] измерили давление пара, определили нормальную точку
¦кипения и оценили энтальпию испарения и критические постоянные NHF2. Эти значения
критических постоянных приведены в табл. Д. 26. В табл. Д. 25 дана оценка второго вириального
коэффициента с помощью корреляции Мартина.
Пары фосфора. Детальное исследование термодинамических свойств паров фосфора проведено
Pay [3164]. Были получены pFjT-данные в интервале температур 720—1380 К при давлении до
15 МПа, а также данные по давлению насыщенного пара и ортобарическим плотностям. Пары
.фосфора представляют собой ассоциированное вещество, содержащее в основном молекулы Р2 и Р4.
Поэтому обработка данных [3164] включала одновременное нахождение ван-дер-ваальсовых
констант и энтальпии реакции Р4<^2Р2.
Так же как и для паров серы, считалось, что состав пара можно вычислить в приближении
Льюиса-Рендалла, причем уравнение состояния и коэффициенты летучести полагались
одинаковыми для обоих компонентов (см. также с. 421). При плотностях менее 0,060 кг-дм уравнение
•состояния смеси можно представить в форме (см. раздел П3.5)
где v = 2x2 -f 4;r4, B = b — a/RT.
Для более высокой плотности в работе [3164] рассчитана таблица значений фактора
сжимаемости и коэффициента летучести.
В настоящем издании проведена новая обработка с целью нахождения энтальпии реакции,
поскольку были приняты несколько иные значения термодинамических функций. Однако
поскольку вклад межмолекулярного взаимодействия в термодинамические свойства в изученном
диапазоне параметров относительно невелик, второй вириальный коэффициент (см. табл. Д. 25)
426
Свойства газов при повышенном давлении
принят по данным работы [3164] без изменений. Значения критических параметров (табл. Д.26)
также приняты по оценкам Pay [3164]. В этой работе экспериментально подтверждено значение
критической температуры, найденное ранее [2516], а путем экстраполяции кривой насыщения
определено критическое давление. Мольный критический объем найден в предположении, что
фосфор относится к так называемым нормальным веществам, для которых
Критическая плотность 0,577 кг-дм~3 определяется соотношением
Р = •^сеси/^кр = M^JV^.
PF3, PCI3, PF2C1, PFC12. Экспериментальные данные по термодинамическим свойствам
соединений фосфора чрезвычайно ограничены. Нисельсон и др. [183] измерили ортобарические
плотности РС13 и определили значения Т^д и V^ (см. табл. Д. 26). Сладков и др. [238] также провели
измерения критических параметров для РС13 и по данным о диффузии определили [237] параметры
потенциала Леннарда—Джонса. Для PFS параметры потенциала Леннарда—Джонса определены
в работе [236а]. Для трех соединений PF3, PF2G1 и PFC12 в работе [703] определены значения Гкр
и />«»> приведенные в табл. Д. 26.
Оценка второго вириального коэффициента для PF3, PF2G1 и PFG12 (табл. Д.25) выполнена
с помощью корреляции Мартина. Для РС13 из-за отсутствия опытных данных по /?кр оценка
величины второго вириального коэффициента (табл. Д.25) выполнена с помощью уравнения
состояния Ван-дер-Ваальса.
ПРИНЯТЫЕ СИМВОЛЫ И ОБОЗНАЧЕНИЯ
А — энергия Гельмгольца (изохорно-изотерми-
ческий потенциал)
А — постоянная взаимодействия орбитального
и спинового моментов количества
движения электронов
аь, Oj, ... —коэффициенты в уравнении для
предельной кривой диссоциации двухатомной
молекулы
Ав — сродство к электрону или протону при
Г = 0
Ао, Ае, At — вращательные постоянные многоатомной
молекулы соответственно в основном,
равновесном и vlt v2, . . ., vn-u
колебательном состоянии
A, A', Alt A" — симеолы, характеризующие
симметрию электронного состояния нелинейной
многоатомной молекулы
Б — второй вириальный коэффициент
Bt, Bt, Bt — вращательные постоянные двухатомной
или многоатомной молекулы
соответственно в основном, равновесном и vlt v2, . . .,
. . ., vn -м колебательном состоянии
B, В', Blt В" — симеолы, характеризующие симметрию
электронного состояния нелинейной
многоатомной молекулы
с — скорость света в вакууме
С — третий вириальный коэффициент
Св,Се,С, — вращательные постоянные многоатомной
молекулы соответственно в основном, рав-
Еовесном и vlt v2, . . ., vn -м
колебательном состоянии
С1 С2, • .., С,, C2v • • ¦ — точечные группы симметрии
Ср — теплоемкость при постоянном давлении
С°р(Т) — теплоемкость при постоянном давлении
в стандартном состоянии при Т К
С, — теплоемкость при постоянном объеме
Св — решеточная составляющая теплоемкости
твердого вещества
С, — электронная составляющая теплоемкости
Ст — магнитная (магнонная) составляющая
теплоемкости
С/ — составляющая теплоемкости, связанная
с переходами на более высокие
энергетические уровни (эффект Шоттки)
Св — составляющая теплоемкости,
обусловленная образованием равновесных вакансий
в решетке
Су — составляющая теплоемкости, связанна»
с процессом упорядочения
c2(hcjk)—-вторая радиационная постоянная
d — состояние электрона с квантовым числом
1 = 1
D — состояние атома с квантовым числом L = 2
dn — степень вырождения n-го колебания
многоатомной молекулы
Do, De, Dv — постоянные центробежного растяжения,
двухатомной молекулы соответственно-
в основном, равновесном и v-u
колебательном состоянии
D2, D2h, Dzh .... D2d ... — точечные группы
симметрии
Dj, DjK; DK — постоянные центробежного
растяжения многоатомной молекулы
Do — энергия диссоциации молекулы на атомы
при Г = 0
De — энергия диссоциации двухатомной
молекулы в данном электронном состоянии,
отсчитанная от минимума потенциальной
кривой этого состояния
DFj-)/7T) — функция Дебая для теплоемкости
кристаллического тела
е — заряд электрона
Е', Е", 1Е, гЕ — символы, характеризующие
симметрию дважды вырожденных электронных
состояний нелинейных многоатомных
молекул
Ео — энергия образования вакансий в
кристаллической решетке при Т = О
ЕFЕ/Т) — функция Эйнштейна для теплоемкости
кристаллического тела
?j = r(ei> — G$x)@)—/^(/пйп) — энергия i-ro
электронного состояния двухатомной молекулы,
отсчитанная от энергии нижнего
колебательно-вращательного уровня (у = 0, / =
с= /„„„) основного электронного состояния
/ — летучесть
/ — состояние электрона с квантовым числом
1 = 3
fr — валентная силовая постоянная связи
/а — деформационная силовая постоянная
/гг — силовая постоянная взаимодействия двух
связей
/га — силовая постоянная взаимодействия связи
и прилегающего к ней угла
428
Принятые символы и обозначения
F — состояние атома с квантовым числом L = 3
F*(J) — вращательная энергия в i-u электронном
и v-u колебательном состояниях как
функция квантового числа / двухатомной,
линейной многоатомной молекулы и
молекулы типа сферического волчка
F, Fj, F2 — символы, характеризующие симметрию
трижды вырожденных электронных
состояний нелинейных многоатомных молекул
Ft(J, К) —вращательная энергия молекулы типа
симметричного волчка
Ff(J^) — вращательная энергия молекулы типа
асимметричного волчка
F'V(J),F'V{J) — вращательные термы для дублетных
состояний молекулы
F't(R), F"P(R), F''e(R) — вращательные термы для три-
плетных состояний молекулы
g — состояние электрона с квантовым
числом 1 = 4
gfi — постоянная колебательного момента ко*
личества движения для J-го
вырожденного колебания линейной многоатомной
молекулы
G — состояние атома с квантовым числом L = 4
G — энергия Гиббса (изобарно-изотермический
потенциал)
¦G(vi, v.2, . . .). Go(yi> y2> • • •)—колебательная
энергия многоатомной молекулы, отнесенная
соответственно к минимуму
потенциальной энергии и к основному
колебательному состоянию
¦ОЦи), Gg(y) — колебательная энергия двухатомной
молекулы в 1-м электронном состоянии,
отнесенная соответственно к минимуму
потенциальной кривой и к v = О
ДС^,, —разность колебательной энергии в
состояниях D + 1 И U
h — постоянная Планка
А — состояние электрона с квантовым
числом 1 = 5
Я — состояние атома с квантовым числом L = 5
hx — постоянная колебательно-вращательного
взаимодействия в двухатомных молекулах
(коэффициент разложения Я, по
степеням V -\- Х/2)
Н — энтальпия
Но, Н„ Н„ — вращательная постоянная двухатомной
молекулы соответственно в основном,
равновесном и v-u колебательном состоянии
Н°{Т) — #°@) — изменение энтальпии вещества в
стандартном состоянии от 0 до Г
i — спин ядра
/ — момент инерции
— главные моменты инерции многоатомной
молекулы в нулевом колебательном
состоянии
1В< 1С
/пр — приведенный момент инерции волчка в
случае внутримолекулярного вращения
/„ — потенциал ионизации при Т = О
/ц. — ионизационный предел к-й. серии
электронных состояний атома
1°(Т)—полная энтальпия вещества в
стандартном состоянии при Т К
i(T) — удельная полная энтальпия вещества
/ — квантовое число полного момента
количества движения электрона j
/ — квантовое число полного момента
количества движения молекулы J
/ — квантовое число полного момента
количества движения электронов атома J
•^mai, t — максимальное значение квантового числа
/ в колебательном состоянии v
Jllm — предельное значение квантового числа /,
отвечающее гипотетическому состоянию
с v = -4%
/min — минимальное значение квантового числа /
/т — вращательное квантовое число момента
количества движения молекулы типа
асимметричного волчка
к — постоянная Больцмана
к„ — силовая постоянная
К — вращательное квантовое число в
многоатомной молекуле
К°(Т) — константа равновесия химической реакции
при Г К и условии, что парциальные
давления реагирующих веществ выражены
в безразмерных величинах р/р°
I — квантовое число орбитального момента
количества движения электрона 1
1п — квантовое число колебательного момента
количества движения линейной
многоатомной молекулы
L — квантовое число суммарного орбитального
момента количества движения электронов
атома L
Lo, Lt, L, — четвертая вращательная постоянная
двухатомной молекулы соответственно в
основном, равновесном и v-u колебательном
состоянии
m — число нормальных колебаний
многоатомной молекулы
тп, — масса электрона
М — молекулярный вес
п — главное квантовое число электрона
л — число максимумов на потенциальной
кривой внутреннего вращения многоатомной
молекулы
N — число атомов в молекуле
Na — число Авогадро
Ок — точечная группа симметрии
р — давление
рч — критическое давление
429
Принятые символы и обозначения
Pi — парциальное давление компонента I
?г(= р/ркр) — приведенное давление
р{ — статистический вес i-ro электронного
состояния
рх — статистический вес основного
электронного состояния
Р — состояние атома с квантовым числом L = 1
Р-ветвь — серия линий в полосе, соответствующая
переходу Д/ = —1
дг — постоянная ^-удвоения
(?-ветвь — серия линий в полосе, соответствующая
переходу Д/ = О
Q(T) — статистическая сумма по состояниям
атомов или молекул газа
(?ва — статистическая сумма по
внутримолекулярным состояниям
Qax — статистическая сумма по электронным
состояниям атома или молекулы
QT,0 — статистическая сумма по состояниям
гармонического осциллятора
(?ж.р — статистическая сумма по состояниям
жесткого ротатора
Фж.р.г.о — статистическая сумма по состояниям
жесткого ротатора — гармонического
осциллятора
Qnoci — статистическая сумма, соответствующая
поступательным степеням свободы
@с-в — статистическая сумма по состояниям сво-
бодвого внутреннего вращения
@ко2.вр — статистическая сумма по колебательным
и вращательным состояниям в i-м
электронном состоянии
статистическая сумма по вращательным
состояниям молекулы, находящейся в i-M
электронном состоянии и i'-м
колебательном состоянии
первая производная по 1/2" от Q
вторая производная по 1/Г от Q
расстояние между ядрами атомов А и В.
г„, rt — межатомное расстояние в двухатомной
молекуле при v = 0 и —1/2
соответственно
гА — межатомное расстояние в двухатомной
молекуле, отвечающее точке перегиба на
эффективной потенциальной кривой,
соответствующей /=/1ш
R — универсальная газовая постоянная
R — квантовое число механического момента
количества движения молекулы R
Д—ветвь — серия линий в полосе, соответствующая
переходу М =+1
s — спин электрона
s — состояние электрона с квантовым числом
s — удельная энтропия
S — энтропия
энтропия при температтре
?#!.
Q(T)-
r(A-B) -
Тг{ =
ТТ
Тт —
S°(T) — стандартная
Т К
S — квантовое число суммарного момента
спинов электронов атома или молекулы S
S — состояние атома с квантовым числом L = О
t — температура в °С
Т — термодинамическая температура в К
Тв — температура Бойля
Тd — точечная группа симметрии
Г$ — температура нормальной точки кипения
~ критическая температура
— приведенная температура
температура плавления
температура фазового перехода
Т i — энергия i-ro электронного состояния
атома, отсчитанная от его основного
состояния
,", Т'е() — энергия i-ro электронного состоянии
молекулы, равная разности энергий
колебательных уровней v' = 0 и v" = 0 или
минимумов потенциальных кривых в i-M
и основном электронных состояниях
внутренняя энергия вещества
удельный объем
колебательное квантовое число
максимальное значение v в данном
электронном состоянии
колебательное квантовое число re-го
нормального колебания многоатомной
молекулы
объем
приведенный объем
критический объем
U
v
v
V
Vn\ =
V —
\
=тБ\
— объем Бойля
заторможенного внутреннего-
V,. Vt-
V(r).
VR(r)
У Jim (rh)
хпп, хпк,
потенциал
вращения
высота потенциального барьера
заторможенного внутреннего вращения
высота потенциального барьера
заторможенного внутреннего вращения для
молекул цис- и трансформы соответственно-
— межмолекулярный потенциал
— потенциальная энергия невращающейся,
молекулы
— эффективная потенциальная энергия
вращающейся молекулы
— энергия в точке перегиба эффективной
потенциальной кривой вращающейся
молекулы при / = /iim
постоянная ангармонического резонанса
Ферми многоатомной молекулы
— параметр энергии асимметричного волчка
— мольная доля i-ro компонента
. . — постоянные ангармоничности первого-
порядка многоатомной молекулы
430
Принятые символы и обозначения
X, J? — символ основного электронного состояния
двухатомной и многоатомной молекулы
Yki — постоянные в уравнении типа Данхема
для колебательно-вращательных уровней
двухатомной молекулы
y{jk — постоянные ангармоничности второго
порядка многоатомной молекулы
Z — число валентных электронов в атоме
Z = (pV/RT) — фактор сжимаемости
а — коэффициент линейного расширения
Oj, а2, . . . — постоянные взаимодействия колебания
и вращения (коэффициенты разложения
В„ в ряд по степени v + 1j2)
afi af, af — постоянные взаимодействия вращения
и колебаний многоатомной молекулы
(i = га, пк, пп)
plt (За, . . . . —постоянные взаимодействия колебаний
и вращения двухатомной молекулы (коэф.
фициенты разложения Dp в ряд по
степеням v +1/2)
7 — постоянная спиновой связи в случае
Гунда Ь для состояний 22
-у — коэффициент электронной теплоемкости
•у — постоянная резонанса Дарлинга—Деннисона
86', ЬН, 85 — изменение соответствующей
термодинамической функции при переходе от
стандартного состояния газа к состоянию
реального газа
Д — электронное состояние молекулы с Л = 2
ДяХ — изменение свойства А' (С, Н, S и др.)
в процессе, обозначенном индексом га;
соответствует разности значений
термодинамических свойств конечного и
начального состояний
Да — изменение свойства X в процессе
растворения
Д„ — изменение свойства X в процессе
сгорания
Ду изменение свойства X в процессе
образования данного вещества из элементов
дто изменение свойства X в процессе
плавления
Дг —изменение свойства X в химической
реакции
Д, — изменение свойства X в процессе
сублимации
Д<г — изменение свойства X в процессе
полиморфного превращения твердого вещества
д, — изменение свойства X в процессе
испарения
е — постоянная эффекта Реннера — Теллера
с — параметр межмолекулярного
взаимодействия
Ьв — характеристическая температура Дебая
Ье — характеристическая температура
Эйнштейна
% — коэффициент изотермической сжимаемости
X — постоянная в уравнении для
вращательных уровней двухатомной молекулы в^
состоянии 32 (постоянная мультиплетного-
расщепления, обусловленная
взаимодействием спинов электронов)
Л — квантовое число проекции L гна
межъядерную ось
jj, — постоянная в уравнении для
вращательных уровней двухатомной молекулы в
состоянии 32 (постоянная мультиплетного-
расщепления, обусловленная
взаимодействием спинов электронов с магнитным
моментом, возникающим при вращении,
молекулы)
ji — приведенная масса двухатомной молекулы
v0 — волновое число начала полосы
v00 — волновое число начала системы полос
ч„ — основная частота га-го колебания
многоатомной молекулы
veB) — основная частота дважды вырожденного'
га-го колебания многоатомной молекулы
у„C) — основная частота трижды вырожденного-
re-го колебания многоатомной молекулы
Av — изменение числа молей в реакции
% — постоянная кориолисова взаимодействияг
соответствующая n-му вырожденному
колебанию
П — электронное состояние молекулы сА = 1
р — коэффициент при Т в формуле для
расчета поправки на центробежное
растяжение в термодинамических функциях
многоатомных газов
р — квадратный корень из отношения
приведенных масс изотопных молекул
р', р" — ортобарические плотности
а — параметр межмолекулярного потенциала*
а — число симметрии молекулы
ат число симметрии волчка относительно оси.
вращения
2 — проекция суммарного момента спинов
электронов S на межъядерную ось
2 — электронное состояние молекулы с А = О
т — постоянная центробежного растяжения
многоатомных молекул
у _ угол поворота волчка относительно остова
при внутреннем вращении
Ф — электронное состояние молекулы с А = 3
ф = —=— — приведенная энергия Гиббса
Ф°(Г) — приведенная энергия Гиббса в
стандартном состоянии при Т К
Я°B98, 15 К)—Я°@)
ф'G') = Фо(Г)+ s jr2 —
приведенная энергия Гиббса, отсчитанная от
298,15 К
431
Принятые символы и обозначения
<оо, <оохо, сооуо,... — частота колебания и постоянные
ангармоничности двухатомной молекулы
в уравнении для G0(v)
,ы,, ы,хе, шеу*, ... — частота колебания и постоянные
ангармоничности двухатомной молекулы
в уравнении для G (v)
(йя — частота га-го нормального колебания
многоатомной молекулы в уравнении для
G(vlt vit . . .)
Q — квантовое число проекции полного
момента количества движения электронов
L на межъядерную ось линейной
молекулы
О, 1, 2, ... — обозначение электронных состояний
двухатомной молекулы в случае связи
Гунда с
Нижние индексы
вн — внутримолекулярная составляющая
вр — вращательная составляющая
в.с — составляющая возбужденных электронных
состояний
г.о — составляющая гармонического
осциллятора
ж.р — составляющая жесткого ротатора
з.в — составляющая заторможенного
внутреннего вращения
кол — колебательная составляющая
¦кол.вр — колебательно-вращательная
составляющая
гкр — критический
пост — поступательная составляющая
с.в — составляющая свободного внутреннего
вращения
ц.р — составляющая центробежного растяжения
эл — электронная составляющая
яд — ядерная составляющая
я.с — составляющая ядерного спина
е — индекс электрона
е — индекс равновесного состояния
g — индекс четности
i — индекс возбужденного состояния
i — индекс молекулы, атома, вещества
i — индекс обращенного электронного
состояния
п — индекс гс-го колебания многоатомной
молекулы
О — индекс основного (v = 0) колебательного
состояния
г — индекс нормального электронного
состояния
и — индекс нечетности
v — индекс у-го колебательного состояния
X — индекс основного электронного состояния
Верхние индексы
° — символ стандартного состояния вещества
', " — индексы верхнего и нижнего состояний
соответствующего спектрального перехода
+, - — индексы электронных состояний B, 0,
1, . . .) с симметричными и
антисимметричными волновыми функциями
соответственно
1, 2, 3, . ..—индексы, указывающие мультшшет-
ность данного электронного состояния
атома или молекулы
Фазовые состояния
аморф. — аморфное
г — газообразное
ж — жидкое
к — кристаллическое
р-р — раствор
стекл. — стеклообразное
Структурные модификации (сингонии)
гекс. — гексагональная
куб. — кубическая
монокл. — моноклинная
ромб. — ромбическая
тетр. — тетрагональная
трикл. — триклинная
Спектры
ИК — инфракрасный
КР — комбинационного рассеяния
MB — микроволновой
УФ — ультрафиолетовый
ЭПР—электронного парамагнитного резонанса
(электронного спинового резонанса)
ЛИТЕРАТУРА
1. Авраменко Л., Кондратьев В. — «Журн. экспе- 25.
рим. и теорет. физ.», 1937, 7, 842.
2. Акишин Л. А., Вилков Л. В., Росоловский В. Я. — 26.
«Журн. структурной хим.», 1960, 1, 1.
3. Акишин П. А., Вилков Л. В., Росоловский В. Я. — 26а.
«Журн. структурной хим.», 1960, 1, 5.
4. Акишин Л. А., Рамбиди Л. Г., Засорин Е. 3. —
«Кристаллография», 1959, 4, 360. 27.
5. Акишин П. А., Спиридонов В. П., Романов Г. В.,
Сидоров Л. Л., Семилетов Е. С, Щербаков А. М. 28.
Сводный отчет лабор. электронографии
химического факультета МГУ за 1962. М., 1963. 29.
6. Акишин Л. А., Спиридонов В. Л., Рамбиди Л. Г.,
Сидоров Л. Лч Романов Г. В., Засорин Е. 3. 30.
Сводный отчет лабор. электронографии
химического факультета МГУ за 1963 г. М., 1964.
7. Александров А. А.—«Теплоэнергетика», 1967, 31.
№ 6, 87.
8. Александров А. А., Агапов Р. К.—«Тешюэнер- 31а.
гетика», 1972, № 8, 82.
9. Александров А. А., Иванов А. И., Хасаншин Т. С,
Ершова 3. А. — «Тр. Моск. энерг., ин-та», 1975, 32.
вып. 234, 68.
10. Александров А. А., Ларкин Д. К. — «Тепло- 32а.
энергетика», 1974, № 8, 80.
11. Александров А. А., Ривкин С. М. — «Изв. высш. 326.
учеб. заведений. Энергетика», 1974, № 10, 58.
12. Александров А. А., Хасаншин Т. С. — «Изв. 32в.
АН БССР. Сер. физ.-энерг. наук», 1974, № 3, 96.
13. Александров А. П., Катаев Д. И., Алиев М. Р., 33.
Алексанян В. Т. — «Оптика и спектроскопия»,
1969, 27, 688. 34.
14. Алексеевский Е. В., Голъц Р. К., Мусакин А. П.
Количественный анализ. Л., Госхимиздат, 1957.
15. Алиев 3. Ф. — «Тр. ВНИИ метрологии», 1958,
35, 5.
16. Алиханян А. С, Стебелевский А. В., Гринберг 35.
Я. X., Горкораки В. Л. — «Журн. физ. хим.»,
1975, 49, 3125. 36.
17. Алтунин В. В.— «Теплоэнергетика», 1962, № 3, 73.
18. Алтунин В. В. — «Тр. Моск. энерг. ин-та», 1974,
вып. 179, 26.
18а. Альтман А. В. Мякшин И. Л., Суховерхое 37.
В. Ф., Романов Г. В., Спиридонов В. Л. Тезисы
докл. V Всес. симп. по химии неорг. фторидов. 38.
Днепропетровск, 27—30 июня 1978 г. М., «Наука».
1978, с. 30. 39.
19. Амирханов X. И., Степанов Г. В., Алибеков Б. Г.
Изохорная теплоемкость воды и водяного пара. 39а.
Махачкала, «Наука», 1969.
20. Амирханов X. И., Степанов Г. В., Мурсатов 40.
В. А. — «Теплоэнергетика», 1975, № 4, 68.
20а. Антанович А. А., Плотников М. А. — «Докл. 41.
АН СССР», 1976, 226, № 4, 809.
21. Антонов А. А.— «Журн. физ. хим.», 1964, 38,1713. 42.
22. Артым Р. И.— «Журн. физ. хим.», 1962, 36,1129.
23. Артым Р. И.— «Журн. физ. хим.», 1964, 38, 1464. 43.
24. Артым Р. И.— «Журн. физ. хим.», 1964, 38,1734.
Артым Р. И. — «Журн. физ. хим.», 1970, 44»
2380.
Артым Р. И., Матвеев А. Б. — «Теплофиз.
высоких температур», 1970, 8, 749.
Байбуз В. Ф., Зицерман В. Ю. — Обзоры по тепло-
физическим свойствам веществ, № 1. М., 1977
(Ин-т высоких температур АН СССР).
Бакулина И. Л., Ионов Л. И. — «Докл. АН СССР»,
1955, 105, 680.
Бакулина Л. Л., Ионов Л. И, — «Докл. АН СССР».
1957, 116, 41.
Бакулина И. Л., Ионов Н. И. — «Журн. физ
хим.», 1959, 33, 2063.
Бантов Д. В., Даевицкий Б. Э., Константинов
Ю. С, Суховерхое В. Ф. — «Изв. СО АН СССР.
Сер. хим. наук», 1968, вып. 1, № 2, 81.
Баранова А. И. — «Журн. прикл. хим.», 1958.
31, 167.
Беляева О. В., Ликолаев В. А., Тимофеев Б. Д. —.
«Изв. АН БССР. Сер. физ.-энерг. наук», 1976,
№ 1, 65.
Бедрина М. Е., Саруханов М. А., Парпиев Л. А. ¦
«Узб. хим. журн.», 1974, 6, 29.
Березин Б. Я., Чеховской В. Я., Шейндлин А. Е
«High Temper. Sci.», 1972, 4, 478.
Берцев В. В., Коломийцева Т. Д., Цыганенке
Л. М. — «Оптика и спектроскопия», 1974, 37, 463.
Бергман Г. А., Медведев В. А.— «Журн (Ьиз
хим.», 1966, 40, 1980. "
Благой Ю. П., Сорокин В. А. — «Журн. физ.
химии», 1970, 44, 2745.
Боровик-Романов А. С, Орлова М. П.,
Стрелков П. Г. Установление шкалы низких
температур между 90,19° К и 10° К посредством градуи-
ровочной таблицы группы эталонных
термометров сопротивления. М., 1954.
Бриллюэн Л. Квантовая статистика. Харьков >
Киев, ОНТИ, 1934.
Брицке Э. В., Капустинский А. Ф., Веселовский
Б. К., Шамовский Л. М., Ченцова Л. Г., Анваер
Б. И. Термические константы неорганических
веществ. М.—Л., Изд-во АН СССР, 1949.
Броунштейн Б. И., Юдин Б. Ф., Луковский
О. С. — «Журн. физ. хим.», 1959, 33, 2773.
Бубнов В. П., Моисеенко В. В. — «Изв. АН БССР.
Сер. физ.-энерг. наук», 1970, № 2, 72.
Булычев В. П. — «Теорет. и эксперим. химия».
1968, 4, 543.
Бурдун Г. Д. Справочник по Международной
системе единиц. М., «Стандартиздат», 1971.
Бурштейн К. Я., Фундылер И. Л. — «Изв.
АН СССР. Сер. хим.», 1974, № 4, 892.
Бутков К. В. — «Recueil trav. chim.», 1948, 67,
551.
Бутков К. В. — «Журн. физ. хим.», 1949, 23,
232,
Бутков К. В., Розенбаум Р. Б. — «Журн. физ
хим.», 1950, 24, 706.
433
Литература
44. Бучельникова Я. С. — «Усп. физ. наук», 1958, 65,
351.
44а. Быстрое Д. С, Васильев И. А., Заикина Я. Ю.,
Мельникова И. Н. — «Физ. твердого тела», 1976,
18, 1162.
446. Бэрдж Р. Т. — «Усп. физ. наук», 1944, 26, 74.
45. Ван Besep. Фосфор и его соединения. М., ИЛ,
1962.
46. Варгафтик Я. Б. Справочник по теплофизиче-
ским свойствам газов и жидкостей. М., «Наука»,
1972.
47. Вассерман А. А., Казавчинский Я. 3., Рабинович
В. А. Теплофизические свойства воздуха и его
компонентов. М., «Наука», 1966.
48. Введенский А. А. Термодинамические расчеты
нефтехимических процессов. Л., Гостоптехиздат,
1960.
49. Введенский А. А., Масалитинова Т. Я. — «Журн.
физ. хим.», 1966, 40, 1372.
50 Веденеев В. И., Кибкало А. А.^ Челъяков Г. Г.—
«Докл. АН СССР», 1974, 216, 1084.
51. Веденеев Е. П. Отчет ВЦ МГУ № 47-Зк D00).
М., 1969.
52. Веденеев Е. П. Программы для решения
прикладных математических задач, вып. И. М., 1970
(ВЦ МГУ).
53. Венкатесварлу К., Пиллаи К. — «Оптика и
спектроскопия», 1961, 11, 51.
53а. Верятин У. Д., Маширев В. П., Рябцев Я. Г.,
Тарасов В. И., Рогозкин Б. Д., Коробов И. В.
Термодинамические свойства неорганических
веществ. Под ред. А. П. Зефирова. М., Атомиздат,
1965.
54 Ветчинкин С. И., Пшеничное Е. И., Соколов
Я. Д. — «Журн. физ. хим.», 1959, 33, 1269.
55. Вилков Л. В., Хайкин Л. С, Васильев А. Ф.,
Тулякова Т. Ф. — «Журн. структурной хим.»,
1968, 9, 1071.
55а. Вол А. Е. Строение и свойства двойных
металлических систем. Т. 1., 2. М., Физматгиз, 1959, 1962.
56. Волков В. М., Дяткина М. Е. — «Журн.
структурной хим.», 1963, 4, 728.
57. Волощин А. П. — В кн.: Теплофизические
свойства газов. М., «Наука», 1970, с. 92.
58. Вольфкович С. И., Рубинчик С. М., Кожин В. М.~
«Изв. АН СССР. Отд. хим. наук», 1954, № 2, 209.
58а. Воробьев В. С, Юнгман В. С. — «Теплофиз.
высоких температур» (в печати).
59 Вукалович М. П., Артым Р. И. —
«Теплоэнергетика», 1962, № 6, 63.
60. Вукалович М. Л., Артым Р. И. —
«Теплоэнергетика», 1963, № 4, 75.
61. Вукалович М. П., Зубарев В. Я., Александров
А. А. — «Теплоэнергетика», 1962, № 7, 64.
62. Вукалович М. П., Кириллин В. А., Ремизов С. А.,
Силецкий В. С, Тимофеев В. Н.
Термодинамические свойства газов. М., Машгиз, 1953.
63. Вукалович М. П., Ривкин С. Л., Александров
А. А. — «Изв. АН СССР, Энергетика и транспорт»,
1968, № 6, 100.
64. Вукалович М. Л., Ривкин С. Л., Александров
А. А. Таблицы теплофизических свойств воды
и водяного пара. М., Изд-во стандартов, 1969.
65. Гаджиев С. М. Термодинамическое исследование
некоторых газообразных кислородных
соединений серы и селена. Автореф. дис. на соискание
учен, степени канд. хим. наук. 1966 (ЛГУ).
66. Галкин Я. Л., Туманов Ю. Я., Бутылкин Ю. П.
Термодинамические свойства неорганических
фторидов. Справочник. М., Атомиздат, 1972.
66а. Гальченко Г. Л. — «Новые книги за рубежом.
Сер. А.», 1975, № 3, 93.
67. Герасимов Я. И., Древинг В. Л., Еремин Е. Я.,
Киселев А. В., Лебедев В. П., Панченков Г. М.,.
Шлыгин А. И. Курс физической химии. Т. 1.
М., «Химия», 1969.
68. Герцберг Г. Атомные спектры и строение атомов.
М., ИЛ, 1948.
69. Герцберг Г. Колебательные и вращательные-
спектры многоатомных молекул. М., ИЛ, 1949.
70. Герцберг Г. Электронные спектры и строение
многоатомных молекул. М., «Мир», 1969.
71. Герцберг Г. Спектры и строение простых
свободных радикалов. М., «Мир», 1974.
72. Гиршфельдер Дж., Кертисс Ч., Берд Р.
Молекулярная теория газов и жидкостей. М., ИЛ,
1961.
73. Гнатовская В. Я., Халимонова И. Я. — «Укр.
физ. журн.», 1971, 16, 1625.
74. Годнее И. Я. — «Журн. физ. хим.», 1953, 27,
1702.
74а. Годнее И. Я. Вычисление термодинамических
функций по молекулярным данным. М., Гостех-
издат, 1956.
75. Годнее Я. Я., Свердлин А. С. — «Журн. эксперим*
и теорет. физ.», 1935, 5, 864.
75а. Годнее И. Я., Свердлин А. С. — «Z. Physik»,
1935, 97, 124.
76. Годнее И. Я., Свердлин А. С. — «Журн. физ.
хим.», 1936, 8, 904.
76а. Горбов С. И. — «Новые книги за рубежом. Сер. Б»,
1976, № 8, 44.
766. Горбов С. И., Бергман Г. А. Обзоры по тешгофи-
зическим свойствам веществ. М., 1979 (Ин-т
высоких температур АН СССР).
77. Горбов С. Я., Гурвич Л. В., Иориш В. С,
Юнгман ВС. — «Теплофиз. высоких температур»,
1975, 13, 503.
77а. Гордеев Е. Р., Уманский С. Я., Воронин А. И. —
«Chem. Phys. Lett.», 1973, 23, 524.
78. Горелик В. С, Калдаева Л. Т., Сущинский М. М.
«Физ. твердого тела», 1970, 12, 3276.
79. Горохов Л. Я., Дронин А. В. — «Теплофиз.
высоких температур», 1972, 10, 750.
80. Григорьев Б. А., Мурдаев Р. М., Расторгуев
Ю. Л. — «Теплофиз. высоких температур», 1974,
12, 83.
81. Гурвич Л. В., Воробьев Б. А., Квливидзе В. А.,
Прозоровский Е. А., Ртищева Я. П., Юнгмаш
В. С. — «Тр. ГИПХ», 1962, вып. 49, 38.
82. Гурвич Л. В., Карачевцев Г. В., Кондратьев Ю. А.,
Лебедев В. А., Медведев В. А., Потапов В. Н.г
Ходеев Ю. С. Энергии разрыва химических
связей. Потенциалы ионизации и сродство к
электрону. М., «Наука», 1974.
83. Гурвич Л. В., Квливидзе В. А. — «Журн. физ.
хим.», 1961, 35, 1672.
84. Гурвич Л. В., Квливидзе В. А., Прозоровский
Е. А., Ртищева Я. Л. — «Тр. ГИПХ», 1962,
вып. 49, 61.
84а. Гурвич Л. В., Квливидзе В. А., Ртищева Я. П. —
«Журн. физ. хим.», 1962, 36, 219.
846. Гурвич Л. В., Коробов В. В. — «Журн. физ. хим.»,
1956, вып. 12, 30, 2794.
85. Гурвич Л. В., Медведев В. А. — «Теплофиз.
высоких температур» (в печати).
86. Гурвич Л. В., Юнгман В. С. — «Журн. физ. хим.»,
1961, 35, 1927.
434
Литература
86а. Гурвич Л. В., Юнгман В. С, Прозоровский Е. А.,
Воробьев Б. А. — «Тр. Ин-та горючих
ископаемых», 1960, 12, 198.
87. Гурвич Л. В., Юнгман В. С, Хачкуруаов Г. А.,
Бергман Г. А., Медведев В. А., Ртищева Л. П.,
Броунштейн Б. П., Вейц И. В., Коробов В. В.,
Куратова Л. Ф. Термодинамические свойства
компонентов продуктов сгорания. Т. 1—3. М.,
Изд-во АН СССР, 1956.
88. Гурвич Л. В., Хачкуруаов Г. А., Медведев В. А.,
Вейц И. В., Бергман Г. А., Юнгман В. С,
Ртищева Н. П., Куратова Л. Ф., Юрков Г. М., Кане
A. А., Юдин Б. Ф., Броунштейн Б. И., Байбуз
B. Ф., Квливидзе В. А., Прозоровский Е. А.,
Воробьев Б. А. Термодинамические свойства
индивидуальных веществ. Т. 1, 2. М., Изд-во АН СССР,
1962.
89. Диссоциирующие газы как теплоносители и
рабочие тела энергетических установок. Сборник под
ред. А. К. Красина. Минск, «Наука и техника»,
1970.
90. Долин С. П., Дяткина М. Е. — «Журн.
структурной хим.», 1972, 13, 966.
91. Дукельский В. М., Зандберг Э. Я. — «Докл.
АН СССР», 1952, 86, 263.
92. Елема В. А. Автореф. дис. на соискание учен,
степени канд. техн. наук. Одесса, 1967 (Одес. ин-т
инж. морск. флота).
93. Елема В. А. —В кн.: Теплофизические свойства
жидкостей и газов при высоких температурах и
плазмы. М., Изд-во стандартов, 1969, с. 373.
94. Ельяшевич М. А. — «Бюл. КИСО АН СССР»,
1934, № 10—11.
95. Еропкин Д. П., Кондратьев В. П. — «Бюл.
КИСО АН СССР», 1934, № 7.
96. Еселъсон Б. Н., Благой Ю. П., Григорьев В. Н.,
Манжелий В. Г., Михайленко С. А., Неклюдов
Н. П. Свойства жидкого и твердого водорода.
Справочный обзор № 1. М., Изд-во стандартов,
1969.
97. Жданов Г. С. Физика твердого тела. М., Изд-во
МГУ, 1962.
98. Жердев Е. П. Автореф. дис. на соискание учен,
степени канд. техн. наук. М., 1970
(Энергетический ин-т им. Г. М. Кржижановского).
99. Жердев Е. П., Улыбин С. Л. В кн.:
Теплофизические свойства газов. М., «Наука», 1973, с. 99.
100. Жуховицкий А. А., Шварцман Л. А. Физическая
химия. М., «Металлургия», 1968.
101. Заманский В. М., Степанов П. И., Кузяков
Ю. Я., Москвитина Е. И. — «Вестн. Моск.
ун-та. Сер. хим.», 1973, 14, 412.
101а. Зельдович Я. Б., Райзер Ю. П. Физика ударных
волн и высокотемпературных
гидродинамических явлений. 2-е изд., дополн. М., «Наука»,
1966.
102. Зерченинов А. Н., Чееноков В. И., Панкратов
А. В. — «Изв. СО АН СССР. Сер. хим.», 1968,
№ 2, 74.
103. Зерченинов А. Н., Чееноков В. И., Панкратов
А. В. — «Журн. физ. химии», 1969, 43, 390.
104. Зозуля В. Н., Благой Ю. П. — «Физ. низких
\ температур», 1975, 1, 1171.
105. Зубарев В. П., Козлов А. Д., Спиридонов Г. А. —
«Теплофиз. высоких температур», 1971, 9, 943.
106. Зубарев В. И., Крупина Н. Л. —
«Теплоэнергетика», 1973, № 8, 19.
107. Зубарев В. Н., Крупина И. А. — «Тр. Моск.
энерг. ин-та», 1974, вып. 179, 74.
108. Институт высоких температур Академии наук
СССР. Важнейшие результаты
научно-исследовательских работ за 1971 г. М., «Наука», 1972,
с. 30.
109. Ионов С. П., Порай-Кошиц М. А. — «Жури,
структурной химии», 1966, 7, 252.
110. Иориш В. С. Автореф. дис. на соискание учен.
степ. канд. хим. наук. М., ИВТАН, 1976.
111. Иориш В. С, Юнгман В. С. — «Журн. физ.
химии», 1976, 50, 1932.
112. Казавчинский Я. 3. — «Докл. АН СССР», 1954,
95, 1005.
113. Казавчинский Я. 3., Кесселъман П. М.,
Кириллин В. А., Ривкин. С. Л., Сычев В. В., Тимрот
Д. Л., Шейндлин А. Е., Шпилърайн 9. Э.
Тяжелая вода. Теплофизические свойства. М.—Л.,
«Энергия», 1965.
114. Казавчинский Я. 3., Сердюк Л. С. — В кн.:
Теплофизические свойства веществ и
материалов, вып. 3. М., 1971, с. 3.
115. Кававчинский Я. 3., Таран В. Н. — В кн.:
Теплофизические свойства жидкостей и газов
при высоких температурах и плазмы. М., Изд-во
стандартов, 1966, с. 171.
116. Кане А. А. — «Тр. ГИПХ», 1960, вып. 46, 98.
117. Капустинский А. Ф. — «Изв. АН СССР. Отд.
хим. наук», 1948, 568.
118. Капустинский А. Ф., Канъковский Р. Т. —
«Журн. физ. хим.», 1958, 32, 2810.
119. Капустинский А. Ф., Шамовский Л. М. — «Acta
Physicochim. URSS», 1936, 4, 1.
120. Капустинский А. Ф., Шамовский Л. М. — «Тр.
ВНИИ минерал, сырья», 1936, вып. 109, 55.
120а. Карапетьянц М. X. Методы сравнительного
расчета физико-химических свойств. М., «Наука»,
1965.
121. Карапетьянц М. X. Химическая термодинамика.
М., «Химия», 1975.
121а. Карапетьянц М. X., Карапетьянц М. Л.
Основные термодинамические константы
неорганических и органических веществ. М., «Химия», 1968.
122. Керимов А. М., Алиева М. К. — «Тр. Энерг.
ин-та им. Г. М. Кржижановского», 1974, вып. 16,
44.
123. Кесселъман П. М. — «Инж.-физ. журн.», 1963,
6, № 6, 61.
124. Кесселъман П. М. Автореф. дис. на соискание
учен, степени д-ра техн. наук. Одесса, 1966
(Одесский технол. ин-т холодильной пром-сти).
125. Кесселъман П. М., Афанасьев М. М., Бестужев
Л. С, Бланк Ю. И., Горыкин С. Ф., Котлярев-
ский Л. А., Чернышев С. К., Щекатолина С. А.
Тепло-массоперенос. Докл. 3-го Всесоюзного
совещания. Минск, «Наука и техника», 1968, 7,
142.
126. Кесселъман П. М., Горыкин С. Ф., Волошин
А. П., Воронюк А. К. — В кн.:
Теплофизические свойства газов, 1970. М., «Наука», с. 80.
127. Киреее В. А. Курс физической химии. М.,
«Химия», 1975.
127а. Киреее В. А. Методы практических расчетов в
термодинамике химических реакций. М., «Химия»,
1975.
1276. Климов В. Д., Лобиков Е. А. — «Оптика и
спектроскопия», 1971, 30, 48.
127в. Клепацкий П. М., Шанкин В. Ф. — «Изв. АН
БССР. Сер. физ.-энерг. наук», 1975, № 1, 68.
128. Клюев Л. И., Леонидов В. Я., Гайсинская О. М.,
Первое В. С. — «Журн. физ. хим.», 1974, 48,
212.
435
Литература
128а. Кондратьев В. Н., Соколов Н. Д. — «Журн.
физ. хим.», 1955, 29, 1265.
129. Кондратьева Е., Кондратьев В. Н. — «Журн.
физ. хим.», 1940, 14, 1528.
130. Котов Ю. И., Татевский В. М. — «Оптика и
спектроскопия», 1963, 14, 160.
131. Котов Ю. П., Татевский В. М. — «Оптика и
спектроскопия», 1963, 15, 128.
132 Коттрелл Т. Прочность химических связей. М.,
ИЛ, 1956.
133. Кочубей В. Ф., Гаврилов А. П., Моин Ф. Б.
Тезисы докл. Второй Всесоюзной конференции
по кинетике и механизму газофазных реакций.
Тбилиси, 1971.
134. Кочубей В. Ф., Моин Ф. Б. — «Докл. АН СССР»,
1974, 219, 141.
134а. Кравцов В. А. Массы атомов и энергии связи ядер.
М., Атомиздат, 1974.
135. Краснов К. С, Тимошинин В. С. Оценка
электронных термов ряда атомов и молекулярных
газов. Отчет. Иваново, 1968 (ИХТИ).
135а. Крестов Г. А. Термохимия соединений
редкоземельных и актиноидных элементов. М.,
Атомиздат, 1972.
1356. Крафтмахер Я. А. «Исследования при высоких
температурах». Новосибирск, «Наука», 1966.
135в Кронрод А. С. Узлы и веса квадратурных формул
М., «Наука», 1964.
136. Крупное А. Ф., Герштейн Л. И., Шустрое
В. Т.— «Оптика и спектроскопия», 1971, 30, 790.
137. Кудашев В. И., Таран В. Н. — В кн.: Теплофи-
зические свойства веществ и материалов, вып. 2.
М., 1970, с. 75.
137а. Кудрин Л. П. Статистическая физика плазмы.
М., Атомиздат, 1975.
1376. Кузнецов Н. М. Термодинамические функции
и ударные адиабаты воздуха при высоких
температурах. М., «Машиностроение», 1965.
138. Кузнецова Л. А., Кузяков Ю. Я., Татевский
В. М. — «Оптика и спектроскопия», 1964, 16,
542.
138а. Кузнецова Л. А., Кузяков Ю. Я., Шпанский В. А.,
Хуторецкий В. М.— «Вестн. Моск. ун-та. Сер.
хим.», 1964, № 3, 19.
139. Кузнецова Т. В., Егорова Л. Ф., Панкратов
А. В. — «Журн. физ. хим.», 1964, 37, 1860.
140. Кузнецова Т. В., Егорова Л. Ф., Рипс С. М.,
Зерченинов А. Я., Панкратов А. В. — «Изв.
СО АН СССР. Сер. хим. наук», 1968, № 2, 68.
141. Кузяков Ю. Я., Москвитина Е. Н. — «Вестн.
Моск. ун-та. Сер. хим.», 1965, № 1, 15.
142. Кузяков Ю. Я., Москвитина Е. Я. — «Вестн.
Моск. ун-та. Сер. хим.», 1971, 12, 164.
143. Кукушкина Е. Л., Поляченок О. Г., Дудчик
Г. П., Новиков Г. И. Термодинамическое
исследование парообразования пятиокиси фосфора.
ВИНИТИ. Деп. № 5004-72. М., 1972.
144. Кулешов Г. Г. — «Изв. АН БССР. Сер. физ.-
техн. наук», 1967, 3, 53.
145. Кулешов Г. Г., Моисеенко В. В., Шалыгин С. А. —•
«Изв. АН БССР, Сер. физ.-энерг. наук», 1968,
3, 71.
146. Кулюпин Ю. А., Яценко А. Ф. — «Физ. твердого
тела», 1963, 5, 2756.
147 Кулюпин Ю. А., Яценко А. Ф. — «Изв. АН СССР.
Сер. физ.», 1965, 29, 917.
148. Кустодина В. А., Мищенко К. П., Флис И. Е. —
«Журн. прикл. химии», 1962, 35, 1374.
149. Ландау Л. Д., Лифшиц Е. М. Квантовая
механика. М., Физматгиз, 1963.
150. Ландау Л. Д., Лифшиц Е. М. Статистическая
физика. М., «Наука», 1964.
150а. Латимер В. М. Окислительные состояния
элементов и их потенциалы в водных растворах.
М., ИЛ., 1954.
1506. Ларкин А. И. — «Журн. эксперим. и теорет.
физ.», 1960, 36, 1896.
151. Леонидов В. Я., Первое В. С, Гайсинская О. М.,
Клюев Л. И. — «Докл. АН СССР», 1973, 211,
901.
152. Леонидов В. Я., Первое В. С, Гайсинская О. М.,
Клюев Л. И. —i Шестая Всесоюзная
конференция по калориметрии A7—19 сент. 1973 г.).
Расширенные тезисы докл. Тбилиси, 1973. с. 39.
153. Лучинский Г. П. — «Журн. физ. хим»., 1956,
30, 1207.
154. Льюис Б., Эльбе Г. Горение, пламя и взрывы
в газах. М., ИЛ, 1948.
155. Майер Дж., Гепперт-Майер М. Статистическая
механика. М., ИЛ, 1952.
156. Макаревич Л. А., Соколова О. Н., Розен A.M. —¦
«Журн. эксперим. и теорет. физ.», 1964, 67, 615.
157. Малышенко С. П., Швальб В. Г. — «Теплофиз.
высоких температур», 1970, 8, 762.
158. Мальцев А. К., Демиденко А. Ф. — «Тр. Моск.
хим.-технол. ин-та», 1966, 51, 136.
159. Маринова Л. А., Бутылин Б. А., Ратьковекий
И. А., Яглов В. Н., Новиков Г. И. — «Изв.
АН БССР, Сер. хим. наук», 1971, 2, 92.
160. Мослов П. Г. —> В кн.: Термодинамические и
термохимические константы. М., «Наука», 1970,
с. 235.
160а. Мослов Ю. П., Мослов П. Г. — «Журн. физ.
хим.», 1958, 32, 1715.
161. Медведев В. А. Автореф. дис. на соискание учен,
степени канд. хим. наук. М., 1955 (ИГИ
АН СССР).
162. Медведев В. Л. —«Журн. физ. хим.», 1958, 32,
1851.
163. Медведев В. А. — «Журн. физ. хим.», 1963, 37,
1403.
164. Медведев В. А., Коробов В. В., Байбуз В. Ф. —
«Журн. физ. хим.», 1959, 33, 58.
165. Международные скелетные таблицы
термодинамических свойств воды и водяного пара
1963 года. —i «Теплоэнергетика», 1964, № 4, 84.
166. Мейсон 9., Сперлинг Т. Вириальное уравнение
состояния. М., «Мир», 1972.
167. Методы расчета тепло физических свойств газов
и жидкостей. М., «Химия», 1974.
168. Миронов К. Е. — «Изв. сектора физ.-хим.
анализа ИОНХ АН СССР», 1955, 26, 215.
169. Михайленко В. И. — «Укр. хим. журн.», 1969,
14, 1247.
169а. Мищенко К. П., Полторацкий Г. М.
Термодинамика и строение водных и неводных растворов
электролитов. Л., «Химия», 1968; 2-е изд.,
1976.
170. Морозов В. П., К ваша Н. Т., Цауне А. Я.,
Лисовенко В. А. — «Оптика и спектроскопия»,
1963, 15, 617.
171. Москвитина Е. Я. — «Вестн. Моск. ун-та. Сер.
хим.», 1966, 6, 110.
172. Москвитина Е. П., Кузяков Ю. Я. — «Оптика
и спектроскопия», 1964, 16, 769.
173. Москвитина Е. Н., Кузяков Ю. Я. — В кн.:
Колебательные спектры в неорганической химии.
М., «Наука», 1971, с. 101.
436
Литература
174. Мулдагалиев X. X., Миргородский А. П., Лаза- 197.
рев А. Н. — «Изв. АН СССР. Неорг. матер.»,
1972, 8, 96.
174а. Наумов Г. Б., Рыженко Б. Н., Ходаковский И. Л. 198.
Справочник термодинамических величин (для
геологов). М., Атомиздат, 1971. 199.
175. Нейдинг А. Б., Казарновский И, А. — «Докл.
АН СССР», 1950, 74, 735. 200.
176. Некрасов Л. И., Скороходов И. И. — «Вестн.
Моск. ун-та. Сер. матем., мех., астрон., физ., 201.
хим.», 1956, 213.
177. Немнонов С. А. — «Физ. метал, и металловед.», 202.
1965, 19, 550.
178. Непорент Б. С, Василевский К. П., Байков 203.
В. И., Карелина Н. Ф. Получение и исследование
инфракрасных спектров PF5 и PbF2. Отчет. Л., 204.
1960 (Государственный оптический ин-т).
179. Несмеянов Ан. Н. Давление пара химических
элементов. М., Изд-во АН СССР, 1961.
180. Нестеренко В. Б., Сирота А. М., Илъюхин
Ю. Д. — «Теплоэнергетика», 1973, № 7, 85. 205.
181. Никифоров В. А., Ривкин С. Л. —
«Теплоэнергетика», 1972, № 10, 39.
182. Николаев В. А., Тимофеев Б. Д. — «Изв. АН
БССР. Сер. физ.-энерг. наук.», 1974, № 3, 78.
183. Нисельсон Л. А., МогучеваВ. В., Соколова Т. Д. —
«Журн. неорг. химии», 1965, 10, 592. 206.
184. Нисельсон Л. А., Серяков Г. В. — «Журн. неорг.
хим.», 1960, 5, 1139. 207.
185. Новиков В. 3. Автореф. дис. на соискание учен,
степени канд. техн. наук. М., 1975 (Моск. энерг. 208.
ин-т).
186. Оносовский Е. В., Сердюк Л. С, Яковлева Т. А., 209.
Векслер Л. С. — В кн.: Теплофизические
свойства вещества при низких температурах. М., 210.
1972, с. 40.
187. Основные свойства неорганических фторидов.
Справочник. Под ред. Н. П. Галкина. М.,
Атомиздат, 1976.
188. Панкратов А. В. Химия фторидов азота. М., 211.
«Химия», 1973.
189. Панкратов А. В., Зерченинов А. П., Талакин 212.
О. Г., Соколов О. М., Князева Н. М. — «Журн.
физ. хим.», 1963, 36, 1399.
190. Панкратов А. В., Зерченинов А. П., Чесноков 213.
В. П., Жданова Н. Н. — «Журн. физ. хим.»,
1969, 43, 394. 214.
191. Панкратов А. В., Рипс С. М., Зерченинов А. П.,
Кузнецова Т. В. — «Журн. физ. хим.», 1968, 42,
380. 215.
192. Панкратов А. В., Соколов О. М. — «Журн.
неорг. хим.», 1966, 11, 1761.
193. Папоушек Д., Церман О., Травничкова Г., Ку- 216.
чирек Я. — «Spisy prirodoved fak. univ., Brne»,
1962, 26, 19. 217.
193a. Пауков И. Е., Березовский Г. А. — Седьмая o-ia
всесоюзная конференция по калориметрии. Те-
зисы докл. М., 1977.
1936. Пауков И. Е., Стрелков П. Г., Ногтева В. В., 219
Белый В. И. — «Докл. АН СССР», 1965, 162,
543. 220.
193в. Пепекин В. И., Лебедев Ю. А., Апин А. Я. —
«Журн. физ. хим.», 1969, 43, 1564. 221.
194. Перельштейн И. И., Парушин Е. Б. —
«Холодильная техника», 1976, № 1, 27. 222.
195. Питаевская Л. Л., Билевич А. В., Канищев
Б. Э. — «Журн. физ. хим.», 1975, 49, 1292. 223.
196. Поляченок Л. Д., Поляченок О. Г. ВИНИТИ. 224.
Деп. № 4740-72. М., 1972.
Поляченок О. Г. — В кн.: Термодинамические
и термохимические константы. М., «Наука»,
1970, с. 205.
Пономарев Ю. If., Ховрин Г. В. — «Оптика и
спектроскопия», 1969, 26, 1062.
Пономарев Ю. И., Ховрин Г. В. — «Оптика и
спектроскопия», 1971, 30, 230.
Попов М. М. Термометрия и калориметрия. М.,
Изд-во МГУ, 1954.
Попов М. М., Костылев Ф. А., Карпова Т. Ф. —
«Журн. неорг. хим.», 1957, 2, 9.
Порай-Кошиц М. А., Ионов С. П. — «Журн.
структурной хим.», 1964, 5, 474.
Постников Н. Н. Термическая фосфорная
кислота. Химия и технология. М., «Химия», 1970, с. 40.
Предводителев А. С, Ступоченко Е. В., Ллеша-
нов А. С, Самуилов Е. В., Рождественский И. Б.
Таблицы термодинамических функций воздуха
для температур от 12 000 до 20 000° К и
давлений от 0,001 до 1000 атмосфер. М., 1959.
Предводителев А. С, Ступоченко Е. В.,
Самуилов Е. В., Рождественский И. Б.
Термодинамические функции компонент воздуха в интервале
температур от 12 000 до 20 000° К и интервале
давлений от 0,001 до 1000 атм. Отчет. М., 1956
(ЛФГ ЭНИН АН СССР).
Пржевальский И. Н., Хачкурузов Г. А. —
«Оптика и спектроскопия», 1974, 36, 304.
Пурмаль А. П., Фрост А. В. — «Вестн. Моск.
ун-та. Сер. хим.», 1961, № 1, 25.
Путкова В. К., Годнее И. Н. — «Журн. физ.
хим.», 1967, 41, 2689.
Рабинович В. А. — «Инж.-физ. журн.», 1962, 5,
№ 5, 30.
Рабинович В. А., Вассерман А. А., Недоступ
В. И., Векслер Л. С. — В кн.: Теплофизические
свойства неона, аргона, криптона и ксенона. Под
ред. В. А. Рабиновича. М., Изд-во стандартов,
1976.
Ревич В. 9., Станкевич С. А. — «Докл. АН СССР»,
1966, 170, 1376.
Ривкин С. Л. Теплофизические свойства воды
в критической области. М., Изд-во стандартов,
1970.
Ривкин С. Л. Термодинамические свойства газов.
М., «Энергия», 1973.
Ривкин С. Л. — В кн.: Теплофизические
свойства веществ и материалов, вып. 8. М., Изд-во
стандартов, 1975, с. 190.
Ривкин С. Л., Александров А. А.
Термодинамические свойства воды и водяного пара. М.,
«Энергия», 1975.
Ривкин С. Л., Ахундов Т. С. —
«Теплоэнергетика», 1962, № 1, 57.
Ривкин С. Л., Ахундов Т. С. —
«Теплоэнергетика», 1963, № 9, 66.
Ривкин С. Л., Ахундов Т. С, КременевскаяЕ. А.,
Ассадулаева Н. Н. — «Теплоэнергетика», 1966,
№ 4, 59.
Ривкин С. Л., Егоров Б. Н. — «Теплоэнергетика»,
1963, № 10, 75.
Ривкин С. Л., Трояновская Г. В. —
«Теплоэнергетика» 1964, № 10, 72.
Ривкин С. Л., Трояновская Г. В., Ахундов Т. С. —
«Теплофиз. высоких температур», 1964, 2 219.
Рид Р., Шервуд Т. Свойства газов и жидкостей
(определение и корреляция). Л., «Химия», 1971.
Рипс С. М. — «Журн. физ. хим.», 1971, 45, 2323.
Рипс С. М., Зерченинов А. Н., Панкратов А. В, —
«Журн. физ. хим.», 1969, 43, 386.
437
Литература
225. Рождественский И. Б., Самуилов Е. В. — В кн.: 251.
Физическая газодинамика, теплообмен и
термодинамика газов высоких температур. М., Изд-во 252.
АН СССР, 1962, с. 103.
226. Романов Г. В., Спиридонов В. П. «Журн.
структурной хим.», 1967, 8, 159.
227. Ромашко Б. В. — «Изв. высш. учеб. заведений. 253.
Химия и хим. технол.», 1973, 16, 210.
227а. Руководство для публикаций данных по термо- 254.
динамике, подготовленное ИЮПАК. — «Журн.
физ. хим.», 1973, 47, 2459. 255.
228. Рысс И. Г. Химия фтора и его неорганических
соединений. М., Госхимиздат, 1956.
229. Свентославский В. — «Журн. рус. физ.-хим. о-ва», 256.
1909, 41, 587.
230. Семенов Г. А., Францева К. Е., Щетникова 256а,
И. Л., Трешникова М. Г., Устпьянцев В. М.
«Изв. АН СССР. Неорг. матер.», 1971, 7, 1672. 257.
231. Сердюк Л. С. — В кн.: Теплофизические
свойства веществ и материалов, вып. 6. М., Изд-во
стандартов, 1973, с. 3. 258.
232. Сердюк Л. С, Табачников А. Г. — «Инж.-физ.
журн.», 1967, 13, № 1, 114.
233. Сидоров Т. А., Соболев Д. Н. — «Оптика и спек- 259.
троскопия», 1957, 2, 710.
234. Силин И. Н. Стандартная программа решения
задач методом наименьших квадратов. Препринт
№ 11-3362, Дубна, 1967 (ОИЯИ). 260.
235. Скрипов В. П., Повышев Л. В. — «Журн. физ.
хим.», 1962, 36, 325.
236. Скуратов С. М., Колесов В. П., Воробьев А. Ф. 261.
Термохимия, ч. 1, 2. М., Изд-во МГУ, 1964,
1966.
236а. Сладкое И. Б. — «Журн. физ. хим.», 1975, 49, 262.
3014.
237. Сладкое И. В., Петрова Г. В. Рукопись деп.
ВИНИТИ, 25 ноября 1974, Деп. № 2990-74.
238. Сладкое И. Б., Шмелева В. И. — «Журн. физ. 263.
хим.», 1975, 49, 260.
239. Смирнова Н. А. Методы статистической термоди- 264.
намики в физической химии. М., «Высшая школа»,
1973. 265.
240. Снопко В. Н. — «Оптика и спектроскопия», 1970,
29, 885. 265а.
241. Сомова В. Ф., Свердлов В. М. — «Оптика и
спектроскопия», 1972, 33, 642.
242. Спиридонов В. П., Татевский В. М. — «Журн.
физ. хим.», 1969, 43, 841. 266.
243. Справочник по плавкости солевых систем. Под
ред. Н. К. Воскресенской. М.—Л., Изд-во АН 267.
СССР, 1961.
244. Сталл Д. Р. Таблицы давления паров индиви- 267а.
дуальных веществ. М., ИЛ, 1949.
245. Сталл Д. Р., Веструм Э., Зинке Г. Химическая
термодинамика органических соединений. М.,
«Мир», 1971. 268.
246. Стрелков П. Г., Боровик-Романов А. С, Орлова
М. П. —. «Журн. физ. хим.», 1954, 28, 345. 269.
247. Струков О. Г., Шляпочников В. А., Дубов С. С. —
«Журн. физ. хим.», 1967, 41, 669. 270.
248. Ступоченко Е. В., Стаханов И. П., Самуилов
Е. В., Плешанов А. С, Рождественский И. Б. — 271.
В кн.: Физическая газодинамика. М., Изд-во
АН СССР, 1959, с. 3. 272.
249. Суворов А. В., Добротин Р. В., Гаджиев С. М. —
«Журн. неорг. хим.», 1965, 10, 1307.
250. Сычев В. М., Вассерман А. А., Козлов А. Д., 272а.
Спиридонов Г. А., Цымарный В. Ф.
Теплофизические свойства азота. М., Изд-во стандартов, 273а.
1976.
Табачников А. Г. — «Инж.-физ. журн.», 1963,
6, № 7, 54.
Табачников А. Г., Межерицкий С. М. — В кн.:
Теплофизические свойства жидкостей и газов
при высоких температурах и плазмы. М., 1969,
с. 188.
Табачников А. Г., Сердюк Л. С. — «Инж.-физ.
журн.», 1965, 9, № 3, 332.
Табачников А. Г., Цыкало А. Л. — «Теорет.
и эксперим. химия», 1966, 2, 837.
Талакин О. Г., Аханьщикова Л. А., Сосновский
Е. Н., Панкратов А. В., Зерченинов А. Н. —
«Журн. физ. хим.», 1962, 36, 1065.
Тальрозе В. Л., Франкевич Е. Л. — «Докл.
АН СССР», 1956, 111, 376.
Тальрозе В. Л., Франкевич Е. Л. — «J. Атег.
Chem. Soc», 1958, 80, 2344.
Таран В. Н. — В кн.: Теплофизические свойства
веществ и материалов, вып. 2. М., Изд-во
стандартов, 1970, с. 78.
Термические константы веществ, вып. 1—7.
Справочник. Отв. ред. В. П. Глушкв. М., Изд-во
ВИНИТИ, 1965—1973 гг.
Термодинамические и переносные свойства
химически реагирующих газовых смесей, ч. 2.
Под ред. А. К. Красина и В. Б. Нестеренко.
Минск, «Наука и техника», 1971.
Тимрот Д. Л., Люстерник В. Е., Устюжанин
Е. Е. — «Тр. Моск. энерг. ин-та», 1974, вып.
179, 3.
Тимрот Д. Л., Люстерник В. Е., Устюжанин
Е. Е. Рукопись деп. ВИНИТИ. Деп. № 2385-75.
М., 1975.
Торопов Н. А., Барзаковский В. П., Лапин В. В.,
Курцева Н. Н. Диаграммы состояния
силикатных систем. Справочник. Л., «Наука», 1969,
вып. 1; 1970, вып. 2.
Трахтенгерц М. С. — «Теплоэнергетика», 1970,
№ 5, 70.
Туманов Ю. И. — «Журн. неорг. хим.», 1968,
13, 1488.
Туманов Ю. П., Галкин П. Л. — «Журн. физ.
хим.», 1969, 43, 836.
Уагман Д. Д. Проблемы создания системы
согласованных термодинамических данных. Докл. на
VII Всесоюзной конференции по калориметрии.
М., 31 янв. — 3 фев., 1977.
Уваров А. В., Березкина Ю. Ф. — «Журн. физ.
хим.», 1962, 36, 884.
Фаулер Р., Гуггенгейм Э. Статистическая
термодинамика. М., ИЛ, 1949.
Физико-химические и теплофизические свойства
химически реагирующей системы N2O4 jt 2NO2 ^*
5± 2NO+Oa. Под ред. В. Б. Нестеренко. Минск,
«Наука и техника», 1976.
Флис И. Е. — «Журн. прикл. хим.», 1964, 37,
683.
Флис И. Е., Мищенко К. П., Кустодина В. А. —
«Журн. прикл. хим.», 1961, 34, 306.
Флис И. Е., Мищенко К. П., Пахомова Н. В. —
— «Журн. неорг. хим.», 1958, 3, 1772.
Флис И. Е., Мищенко К. П., Пахомова Н. В. —
«Журн. неорг. хим.», 1958, 3, 1781.
Фокин Л. Р., Пучкова Г. А. Шестая Всесоюзная
конференция по калориметрии. Расширенные
тезисы докл. Тбилиси, «Мецниереба», 1973, с. 359.
Франкевич Е. Л., Тальрозе В. Л. — «Докл. АН
СССР», 1958, 119, 1174.
Франкевич Е. Л., Тальрозе В. Л. — «Журн.
физ. хим.», 1959, 33, 1093.
438
Литература
273. Френкель Я. И. Введение в теорию металлов. 301.
Л., «Наука», 1972.
274. Фрост А. В. — «Тр. завода «Химгаз», 1936, 3, 302.
27.
274а. Фундаментальные физические константы.
ГСССД1-76. М.; Изд-во стандартов, 1976. 303.
275. Хаит Ю. Г. — «Тр. ГИПХ по термодинамике
и кинетике» (в печати). 304.
276. Харитонов Ю. Я., Саруханов М. А. — «Журн.
неорг. химии», 1968, 13, 71.
277. Хачкурузов Г. А. — «Оптика и спектроскопия», 305.
1959, 6, 463.
278. Хачкурузов Г. А. — «Тр. ГИПХ», 1959, вып. 42,
51. 306.
279. Хачкурузов Г. А. — «Тр. ГИПХ», 1959, вып. 42,
96.
280. Хачкурузов Г. А. — «Тр. ГИПХ», 1959, вып. 42, 307.
109.
281. Хачкурузов Г. А. — «Оптика и спектроскопия», 308.
1960, 8, 40.
282. Хачкурузов Г. А. — «Тр. ГИПХ,» 1960, вып. 46,
51. 309.
283. Хачкурузов Г. А. — «Тр. ГИПХ», 1960, вып. 46,
66.
284. Хачкурузов Г. А. — В кн.: Работы по
термодинамике и кинетике химических процессов. Л., 310.
1974, с. 3.
285. Хачкурузов Г. А., Милевская И. С. — «Журн.
физ. хим.», 1960, 34, 2554. 311.
286. Хачкурузов Г. А., Милевская И. С. — «Журн.
физ. хим.», 1961, 35, 142. 312.
287. Хачкурузов Г. А., Пржевальский И. Я. —
«Оптика и спектроскопия», 1972, 33, 237.
288. Хачкурувов Г. А., Пржевальский И. П. — «Оп- 313.
тика и спектроскопия», 1972, 33, 786.
289. Хачкурузов Г. А., Пржевальский И. Н. — «Оп- 314.
тика и спектроскопия», 1973, 35, 374.
290. Хачкурузов Г. А., Пржевальский И. Н. — «Оп- 315.
тика и спектроскопия», 1974, 36, 299.
291. Хачкурузов Г. А., Пржевальский И. Н. — «Оп- 315а.
тика и спектроскопия», 1976, 41, 551.
292. Хачкурузов Г. А., Пржевальский И. Н. —
«Оптика я спектроскопия», 1978, 44, 194. 316.
292а. Хачкурузов Г. А., Юрков Г. Н. — «Журн. физ.
химии», 1973, 47, 381. 317.
2926. Хачкурузов Г. А., Юрков Г. Н. — «Оптика и
спектроскопия», 1969, 27, 905.
292в. Хачкурузов Г. А., Юрков Г. Н. — «Оптика и 318.
спектроскопия», 1969, 27, 911.
292г. Хачкурузов Г. А., Юрков Г. Н. — «Оптика и 319.
; спектроскопия», 1970, 28, 822.
293. Хлебникова В. Н., Морозов В. П. «Укр. хим. 320.
журн.», 1958, 24, 3.
294. Худсон Д. Статистика для физиков. М., «Мир»,
1970. 321.
295. Цедерберг Н. В., Александров А. А., Хасаншин
Т. С. — «Теплоэнергетика», 1972, № 10, 65.
296. Цедерберг Н. В., Александров А. А., Хасаншин 321а.
Т. С, Ларкин Д. К. — «Теплоэнергетика»,
1973, № 8, 13. 3216.
297. Цедерберг Я. В., Попов В. Н., Каленков А. В. —
«Тр. Моск. энерг. ин-та», 1975, вып. 234, 65. 322.
298. Цедерберг Н. В., Попов В. Н., Морозова Н. А. 323.
Термодинамические и теплофизические свойства
гелия. М., Атомиздат, 1969. 323а.
299. Цеханская Ю. В., Полякова 3. А., Козловская
Г. м. — «Журн. физ. хим.», 1967, 41, 501. 324.
300. Циклис Д. С, Поляков Е. В. — В кн.:
Теплофизические свойства веществ и материалов, вып. 2. 325.
М., Изд-во стандартов, 1970, с. 21.
Цойман Г. И. — «Изв. высш. учебн. заведений.
Нефть и газ», 1959, № 12, 95.
Цыкало А. Л., Савенков В. К., Селеванюк В. H.t
Якушев А. П. — «Журн. прикл. хим.», 1974,
47, 441.
Цыкало А. Л., Табачников А. Г. — «Теплофиз.
высоких температур», 1966, 4, 810.
Цыкало А. Л., Табачников А. Г. — В кн.: Тепло-
физические свойства веществ. Киев, «Наукова
Думка», 1966, с. 120.
Цыкало А. Л., Табачников А. Г. — В кн.: Тепло-
физические характеристики веществ, вып. 1. М.,
1968, с. 194.
Цымарный В. А. — В кн.: Теплофизические
свойства газов и жидкостей при высоких
температурах и плазмы. М., 1969, с. 133.
Чернобаев Д. А. — «Хим. пром-сть», 1937, 14,
1235.
Чернозубов Ю. С, Кузнецов Б. П., Клименко
А. А., Подмогилъный 9. В. — ВИНИТИ, Деп.
№ 3171-71. М., 1971.
Чесноков В. И., Зерченинов А. П., Панкратов
А. В., Жданова П. П. Пятая Всесоюзная
конференция по калориметрии (Расширенные тезисы
докл.). М., Изд-во МГУ, 1971, с. 27.
Чесноков В. И., Каграманов Я. Д., Панкратов
A. В., Зерченинов А. Я., Жданова Я. П. Рукопись
деп. ВИНИТИ. Деп. № 3318-71. М., 1971.
Чулановский В. М. — «Изв. АН СССР. Сер.
естеств.-мат. наук», 1935, 7, 1313.
Шабур В. Я., Каташинский А. С, Титова
И. С, Ковалъчук Д. С. — «Теплофиз. высоких
температур», 1969, 7, 369.
Швангирадзе Р. Р., Джамогидзе Ш. 3. —
«Оптика и спектроскопия», 1961, 10, 583.
Швангирадзе Р. Р., Джамогидзе Ш. 3. —
«Оптика и спектроскопия», 1962, 12, 364.
Шейндлин А. Е., Горбунова Я. И. — «Теплофиз.
высоких температур», 1974, 12, 666.
Шейндлин А. Е., Горбунова Я. И., Симонов
B. М. — «Теплофиз. высоких температур», 1977,
15, 767.
Шифрин Ф. Ш. — «Докл. АН СССР», 1956, 106,
233.
Шляпочников В. А., Струков О. Г., Дубов С. С,
Шитов Л. Я. — «Журн. неорг. хим.», 1969, 14,
2913.
Юнгман В. С. — «Оптика и Jспектроскопия»,
1960, 8, 281.
Юнгман В. С. — «Журн. физ. хим.», 1961, 35,
319.
Юнгман В. С, Гурвич Л. В., Квливидзе В. А.,
Прозоровский Е. А., Ртищева Я. Л. — «Журн.
физ. хим.», 1961, 35, 2182.
Юнгман В. С, Гурвич Л. В., Квливидзе В. А.,
Ртищева Я. Л. — «Тр. ГИПХ», 1960, вып. 46,
15.
Юнгман В. С, Гурвич Л. В., Ртищева Я. П. —
«Тр. ГИПХ», 1962, вып. 49, 20.
Юрков Г. Я., Хачкурузов Г. А. — «Оптика и
спектроскопия», 1968, 25, 820.
Яковлева А. В. — «Z. Phys», 1931, 69, 548.
Яковлева А., Кондратьев К. — «Z. Physik»,
1936, 9, 106.
Яку б Е. С. — В кн.: Теплофизические свойства
веществ и материалов, вып. 11. М., 1977, с. 119.
Ярославский Я. Г., Станевич А. Е. — «Оптика
и спектроскопия», 1958, 5, 384.
Ярославский Я. Г., Станевич А. Е. — «Изв.
АН СССР. Сер. физ.», 1958, 22, 1145.
439
Литература
326. Яценко А. Ф., Кулюпин Ю. А., Рабкин Л. М. — 354.
«Физ. твердого тела», 1962, 4, 692.
326а. Яцимирский К. Б. — «Журн. общ. хим.», 1956, 354а.
26, 2376.
327. Abadie D. —«Ann. phys.», 1970, 5, 227.
328. Abadie D., Herman L.—«J. Quant. Spectrosc. 355.
and Radiat. Transfer», 1964, 4, 195.
329. Abadie D., Herman L. — «C. r. Acad. sci.», 1963, 356.
257, 2820.
330. Abe if. — «J. Mol. Spectrosc.», 1973, 48, 395. 357.
331. A be K., Myers F., McCubbin T. K., Polo S. R. —
«J. Mol. Spectrosc.», 1971, 38, 552. 358.
332. Abe K., Myers F., McCubbin T. K., Polo S. R.—
«J. Mol. Spectrosc», 1974, 50, 413. 359.
333. Abel E., Proisl J. — «Z. Elektrochem.», 1929, 35,
712. 360.
334. Abercromby D. C, Tiley P. F. — «J. Chem. Soc»,
1963, 4902. 361.
335. Abrahams S. C. — «Acta crystallogr.», 1955, 8,
661. 362.
336. Abrahams S. C. — «Acta crystallogr.», 1961, 14,
311. 363.
337. Abramowitz S., Armstrong G. Т., Beckett C. W.,
Churney K. I., Dibeler V. H., Douglas Т. В., 364.
Herron J. Т., Krause R. F., jr., McCulloh К. Е.,
Reilly M. L., Rosenstock H. M., Tsang W. New 365.
Ideal-Gas Thermochemical Tables (in the Format 366.
of the JANAF Thermochemical Tables).
AFOSR-TR-72-2004. NBS Rept 10904. Washing- 367.
ton, 1 July 1972. 368.
338. Abramowitz S., Levin I. W. — «J. Chem. Phys.»,
1966, 44, 3353. 369.
339. Abramowitz S., Levin I. W. — «J. Chem. Phys.», 370.
1969, 51, 463.
340. Abria M. — «C. r. Acad. sci.», 1846, 22, 372. 371.
341. Ackermann F. — «Canad. J. Phys.», 1971, 49, 76.
342. Ackermann F., Lefebvre-Brion H., Roche A. L. — 372.
«Canad. J. Phys.», 1972, 50, 692.
343. Ackermann F., Miescher E. — «Chem. Phys. Lett.», 373.
1968, 2, 351.
344. Ackermann F., Biaume F. — «J. Mol. Spectrosc», 374.
1970, 35, 73.
344a. Adam G., Schramm B. — «Ber. Bunsenges. phys. 375.
Chem.», 1977, 81, 442.
345. Acquista N., Schoen L. J. — «J. Chem. Phys.», 376.
1970, 53, 1290.
345a. Adams C. J. — «J. Raman Spectrosc», 1974, 2, 377.
391.
346. Adams W. J., Bartell L. S. — «J. Mol. Struct.», 378.
1971, 8, 23. 379.
347. Adams J., Thompson H. В., Bartell L. S. — 380.
«J. Chem. Phys.», 1970, 53, 4040.
348. Adler L. S., Yaws C. L. — «Solid State Technol.», 381.
1975, 18, N 1, 35.
348a. Agron P. A., Begun G. M., Levy H. A., Mason 382.
A. A., Jones C. G., Smith D. F. — «Science»,
1963, 139, 842. 383.
3486. Agron P. A., Johnson G. K., Levy H. A. — «Inorg.
Nucl. Chem. Lett.», 1965, 1, 145. 384.
349. Ahearn A. J., Thurmond C. D. — «J. Phys.
Chem.», 1962, 66, 575. 385.
350. Ahlrichs Д., Staemmler V. — «Chem. Phys. Lett.»,
1976, 37, 77. 386.
351. Akriche J. — «J. chim. phys. et phys.-chim. biol.»,
1963, 60, 732. 387.
352. Albasiny E. L., Cooper J. R. A. — «Proc. Phys.
Soc. London», 1963, 82, 289.
353. AlbrittonD. L., Harrop W. J., Schmeltekopf A. L., 388.
Zare R. N. — «J. Mol. Spectrosc», 1973, 46,
89.
Alcock W. G., Pimentel G. C. — «J. Chem. Phys.»,
1968, 48, 2373.
Aldridge J. P., Filip H., Ficker H., Holland R. F.T
McDowell R. S., Nereson N. G., Fox K. — «J.
Mol. Spectrosc», 1975, 58, 165.
Alexander L. E., Beattie I. R. — «J. Chem. Soc.» A,
1971, 3091.
Al-Joboury M. I., May D. P., Turner D. W. —
«J. Chem. Soc», 1965, 616.
Al-Joboury M. I., Turner D. W. — «J. Chem.
Soc.» B, 1967, 373.
Allan A., Duncan J. L., Holloway J. H., McKeen
D. С — «J. Mol. Spectrosc», 1969, 31, 368.
Allavena M., Le Clech E. — «J. Mol. Struct.»,
1974, 22, 265.
Allavena M., Rysnik R., White D., С alder V.,
Mann D. E. — «J. Chem. Phys.», 1969, 50, 3399.
Allen H. C, Blaine L. R., Plyler E. K., Cross
P. C. — «J. Chem. Phys.», 1956, 24, 35.
Allen H. C, Plyler E. K. — «J. Chem. Phys.»,
1954, 22, 1104.
Allen H. C, Plyler E. K. — «J. Res. NBS»,
1954, 52, 205.
Allen H. C, Plyler E. K. — «J. Chem. Phys.»,
1956, 25, 1132.
Allen L. C. — «Chem. РП343. Lett.», 1968, 2, 597.
Allen W. C, Dixon R. N. — «Trans. Faraday
Soc», 1969, 65, 1168.
Allison R. G. — «Dissert. Abstrs», 1965, 26, 2834.
Altshuller A. P. — «J. Amer. Chem. Soc», 1955,
77, 3480.
Altshuller A. P. — «J. Phys. Chem.», 1957, 61, 251.
Amano Т., Hirota E. — «J. Mol. Spectrosc»,
1973, 45, 417.
Amano Т., Hirota E., Morino Y. — «J. Phys.
Soc. Japan», 1967, 22, 399.
Amano Т., Hirota E., Morino Y. — «J. Mol.
Spectrosc», 1968, 27, 257.
Amano Т., Yoshinaga A., Hirota E. — «J. Mol.
Spectrosc», 1972, 44, 594.
Amano Т., Saito S., Hirota E., Morino Y. — «J.
Mol. Spectrosc», 1969, 30, 275.
Amano Т., Saito S., Hirota E., Morino Y. —
«J. Mol. Spectrosc», 1969, 32, 97.
Amble E., Dailey B. P. — «J. Chem. Phys.»,
1950, 18, 1422.
Aminadav N., Selig H., Abramowitz S. — «J.
Chem. Phys.», 1974, 60, 325.
Amiot С — «J. Mol. Spectrosc», 1976, 59, 191.
Amiot C. — «J. Mol. Spectrosc.», 1976, 59, 380.
Amiot C, Guelachvili G. — «J. Mol. Spectrosc»,
1976, 59, 171.
Anbar M., Holman H., Pinchos S. — «J. Chem.
Soc», 1960, 1242.
Andersen A., Ohm Y.—«J. Mol. Spectrosc»,
1973, 45, 358.
Andersen A., Thulstrup E. W. — «J. Phys. B:
Atom, and Mol. Phys.», 1973, 6, 1211.
Anderson R., Schnizlein J. G., Toole R. C, O'Brien
T. D. — «J. Phys. Chem.», 1952, 56, 233.
Anderson R., Schnizlein J. G., Toole R. C, O'Brien
T. D. — «J. Phys. Chem.», 1952, 56, 473.
Anderson T. F., Yost D. M. — «J. Chem. Phys.»,
1936, 4, 529.
Andon R. J. L., Counsell J. F., Herington E. F. G.t
Martin J. F. — «Trans. Faraday Soc», 1963, 59,
830.
Andon R. J. L., Counsell J. F., McKerrell H.,
Martin J. F. — «Trans. Faraday Soc», 1963, 59,
2702.
440
Литература
389. Andrews D. H., Lynn G., Jonston J. — «J. Amer. 421.
Chem. Soc», 1926, 48, 1274.
390. Andrews L. — «J. Chem. Phys.», 1972, 57, 51. 422.
391. Andrews L., Frederick D. L. — «J. Phys. Chem.»,
1969, 73, 2774. 423.
392. Andrews L., Raymond J. I. — «J. Chem. Phys.», 424.
1971, 55, 3078.
393. Andrews L., Raymond J. I. — «J. Chem. Phys.», 425.
1971, 55, 3087.
394. Andrews L., Spiker R. C. — «J. Chem. Phys.», 426.
1972, 76, 3208.
395. Andrews L. W., Carlton H. A. — «J. Amer. Chem. 427.
Soc», 1907, 29, 690. 428.
396. Andrews T. — «Philos. Mag.», 1848, 32, 426.
397. Andrychuk D. — «Canad. J. Phys.», 1951, 29, 151. 429.
398. Angus S., Armstrong B. International
Thermodynamic Tables of the Fluid State Argon. London, 430.
1971. 431.
398a. Angus S., Reuch К. М. de, Armstrong В., Jacob-
sen R. Т., Stewart R. B. Final Report on IUPAC 432.
Nitrogen Tables. IUPAC Thermodynamic Tables
Project Centre, 1977. 433.
399. Angus S., Reuch К. М. de, McCarty R. D. Final
Report on IUPAC Helium-4 Tables. IUPAC 434.
Thermodynamic Tables Project Centre, 1975.
400. Angus W. R.-, Leckie A. H. — «Proc. Roy. Soc. 434a.
London», 1935, A149, 327.
401. Ansdell D. A., Page F. M. — «Trans. Faraday 4346.
Soc», 1962, 58, 1084.
402. Antonik S., Lucquin M. — «Bull. Soc. chim. 435.
France», 1970, 2861.
403. Apparao K. W. S. R., Narasimham N. A. — «Proc. 436.
Indian Acad. Sci.», 1968, АЙ8, 173. 437.
404. Appelman E. H., Kim H «J. Chem. Phys.»,
1972, 57, 3272. 438.
405. Apple B. A. —Thesis. Univ. Tennesee, 1968.
406. Appleyard E. T. — «Phys. Rev.», 1932, 41, 254. 439.
406a. Aquilanti V., Volpi G. G. — «J.j Chem. Phys.»,
1966, 44, 2307. 440.
407. Arakawa E. Т., Nielsen A. H. — «J. Mol.
Spectrosc», 1958, 2, 413. 441.
408. Areas P., Haensler C, J of fin C, Meyer C, Nquen
Van Thanh, Barchewitz P. — «Appl. Opt.», 1963, 442.
2, 909. 443.
409. Archibald T. W., Sabin J. R. — «J. Chem. Phys.»,
1971, 55, 1821. 444.
410. Arighi L. S. Dissertation. The Univ. of Wisconsin, 445-
1965 (Dissert. Abstrs, 1965, 25, 5578). ...
411. Arii K. — «Bull. Inst. Phys. Chem. Res., Tokyo», 44b-
1929, 8, 545.
412. Arii K. — «Sci. Repts Tohoku Imp. Univ.», 1933,
22, 182. 447
413. Arii K., Kowabata M. — «Bull. Inst. Phys.
Chem. Res. Tokyo», 1938, 17, 299. ,if>
414. Arkell A. — «J. Phys. Chem.», 1969, 73, 3877. Цд
415. Arkell A., Reinhard R. R., Larson L. P. — «J.
Amer. Chem. Soc», 1965, 87, 1016. 450
416. Armstrong G. T. Private communication.
417. Armstrong G. Т., King R. С Proc. Conf. Inter- 451,
agency Chemical Rocket Propulsion Group,
Thermochemistry Working Group. Cleveland, Ohio, Apr. 452.
9—11, 1969, 7-th Meeting, Bull. 1, p. 19—40.
418. Armstrong G. Т., Krieger L. A. — In: Progress 453.
in International Research on Thermodynamic and
Transport Properties. New York—London, 1962. 454.
419. Armstrong G. Т., Marantz S. — «J. Chem. Phys.»,
1963, 38, 169. 455.
420. Armstrong G. Т., Marantz S., Coyle С. Т. —¦
«J. Amer. Chem. Soc», 1959, 81, 3798. 456.
Armstrong G. Т., Marantz S., Coyle С. Т. —*
«Bull. Chem. Thermodyn.», 1960, 3, 7.
Arnau J. L., Giguere P. A. — «Canad. J. Chem »
1973, 51, 1525.
Arndt J. Dissertation. Freie Univ. Berlin, 1968
Arnold D. E. J., Rankin D. W. H. — «J. Fluor
Chem.», 1973, 2, 405.
Arumugan K., Radhakrishan M. — «Z. phvs
Chem.» (BRD), 1963, 39, 262.
Arvia A.J., Cafferata L. F.R., Schumacher H. J. —
«Chem. Ber.», 1963, 96, 1187.
Asbrink L. — «Chem. Phys. Lett.», 1970, 7, 549.
Ashby R. A. — «J. Mol. Spectrosc», 1967, 23r
Ashby R. A.— «J. Mol. Spectrosc», 1971, 40,
639.
Ashley M. F. — «Phys. Rev.», 1933, 44, 919.
Ashmore P. G., Tyler B. J. — «J. Chem. Soc »
1961, 1017.
Aston J. G., Rock E. J., Isserow S. — «J. Amer
Chem. Soc», 1952, 74, 2484.
Atherton N. M., Dixon R. N., Kirby G. H. —
«Trans. Faraday Soc», 1964, 60, 1688.
Atoj'i M., Lipscomb W. M. — «Acta crystallogr.»..
1954, 7, 173.
Ault B. S., Andrews L. — «J. Chem. Phys»
1976, 64, 3075. T
Ault B. S., Andrews L. — «J. Chem. Phys.», 1976
65, 4192.
Avarmaa R., Rebane L. — «Phys. status solidi»^
1969, 35, 107.
Avellen S. — «Arkiv fys.», 1954, 8, 211.
Awberry J. H., Griffiths E. — «Proc. Roy. Soc.
London», 1933, A147, 17.
Aynsley E. E., Dodd R. E., Little R. — «Spectro-
chim. acta», 1962, 18, 1005.
Babcock H. D., Herzberg L. — «Astrophys. J.»,
1948, 108, 167.
Back R. A., Betts J. — «Canad. J. Chem.», 1965,
43, 2157.
Back R. A., Wills C, Ransay A. D. — «Canad
J. Chem.», 1974, 52, 1006.
Badger R. M. — «J. Chem. Phys.», 1934, 2, 128.
Badgley G. R., Livingston R. L. — «J. Amer.
Chem. Soc», 1954, 76, 261.
Baede A. P. M. — «Physica», 1972, 59, 541.
Baehr H. D., Carnfost H., Pollak R. — «J. Chem.
Thermodyn.», 1976, 8, 113.
Baehr H. D., Hartmann H., PohUH.-Ch., Scho-
macker H. Thermodynamische Functionen idealer
Gase fur Temperaturen bis 6000° K. Berlin—
Heidelberg—New York, Springer-Verlag, 1968.
Baer P., Miescher E. — «Helv. phys. acta», 1951,
24, 331.
Baer P., Miescher E. — «Nature», 1952, 169, 581.
Baer P., Miescher E. — «Helv. phys. acta», 1953,
26, 91.
Baierl P., Kiefer W. — «J. Chem. Phys.», 1975,
62, 306.
Bailey C. R., Cassie A. B. D. — «Proc. Roy. Soc
London», 1933, A140, 605.
Bailey С R., Cassie A. B. D. — «Proc. Roy.
Soc. London», 1933, A142, 129.
Bailey C. R., Cassie A. B. D., Angus W. R. —
«Proc. Roy. Soc. London», 1930, A130, 142.
Bailey С R., Gordon R. R. — «Trans. Faraday
Soc», 1938, 34, 1133.
Bailey C. R., Thompson T. W. — «Nature», 1935,
135, 913.
Bailey T. L. — «J. Chem. Phys.», 1958, 28, 792.
441
Литература
457. Bain О., Giguere P. A. — «Canad. J. Chem.»,
1955, 33, 527.
458. В air W. H. — «Astrophys. J.», 1920, 52, 301
459. Baker E. H. — «Trans. Inst. Mining and Metal-
lurg.», Sect. C, 1971, 80, 93.
460. Balfour W. G., Douglas A. E. — «Canad. J. Phys.»,
1968, 46, 2277.
461. Balkis Т., Gaines A. F., Ozgen G., Ozgen I. T.,
Flowers M. С — «J. Chem. Soc. Faraday Trans.
II», 1976, 72, 524.
462. Ballash N. M., Armstrong D. A. — «Spectrochim.
acta», 1974, A30, 941.
463. Balzarini D. A. — «Canad. J. Phys.», 1972, 50,
2194.
464. Balzarini D. A., Ohree K. — «Phys. Rev. Lett.»,
1972, 29, 840
465. Bancroft J. L., Hollas J. M., Ramsay D. A. —
«Canad. J. Phys.», 1962, 40, 322.
466. Banyard K. E., Hake R. B. — «J. Chem. Phys.»,
1964, 41, 3221.
467. Bar R. — «Helv. phys. acta», 1931, 4, 130.
468. Barbe A., Jouve P. — «С. г. Acad. sci.», 1969,
B268, 1723.
469. Barbe A., Jouve P. — «J. Mol. Spectrosc», 1971,
38, 273.
470. Barbe A., Secroun C, Jouve P. — «C. r. Acad.
sci.», 1972, B274, 615.
471. Barbe A., Secroun C, Jouve P. — «J. Mol.
Spectrosc», 1974, 49, 171.
472. Barbe A., Secroun C, Jouve P., Duterace В.,
Monnanteull N., Bellet J., Steenbeckeliers. G. —
«J. Mol. Spectrosc», 1975, 55, 319.
472a. Barberi P. — «Bull, inform, sci. et techn. CEA»,
1973, N 180, 55.
473. Barberi P., Caton J., Guillin J., Hartmanschenn O.
Rapp. CEA, N 3761, 1969, 38 p.
473a. Barin I., Knacke O. Thermochemical Properties
of Inorganic Substances. Berlin—Heidelberg—>
New York, Springer-Verlag, 1973.
4736. Barin /., Knacke O., Kubaschewski O.
Thermochemical Properties of inorganic Substances.
Supplement. Berlin—Heidelberg—New York,
Springer-Verlag, 1977.
474. Barker E. F. — «Revs Mod. Phys.», 1942, 14,
198.
475. Barker E. F., Sleatop W. W. — «J. Chem. Phys.»,
1935, 3, 660.
475a. Barker J. A., Watts R. O., Lee J. K., Schafer
T. P., Lee Y. T. — «J. Chem. Phys.», 1974, 61,
3081.
476. Barletta R. E., Claassen H. H., McBeth R. L. —
«J. Chem. Phys.», 1971, 55, 5409.
477. Barnes A. J., Hallam H. E., Scrimshaw G. F. —
«Trans. Faraday Soc», 1969, 65, 3159.
478. Barnes A. J., Hallam H. E., Scrimshaw G. F. —-
«Trans. Faraday Soc», 1969, 65, 3172.
479. Barnes R. B. — «Phys. Rev.», 1932, 39, 562.
480. Barnes R. В., Benedict W. S., Lewis С. М. —
«Phys. Rev.», 1935, 47, 918.
481. Barrett J. J., Myers S. A. — «J. Opt. Soc.
America», 1971, 61, 1246.
482. Barrow G. M. —«I. Chem. Phys.», 1953, 21, 219.
483. Barrow G. M. — «I. Phys. Chem.», 1955, 59, 987.
484. Barrow G. M., Pitzer K. S. — «Ind. and Engng
Chem.», 1949, 41, 2737.
485. Barrow R. F. — «Arkiv fys.», 1956, 11, 281.
48a. Barrow R. F., Beattie I.R., Burton W.G., Gil-
son т. — «Traas Faraday Soc», 1971, 67 583.
487. Barrow R. F., Broyd D. F., Pedersen L. В., Yee
К. К. — «Chem. Phys. Lett.». 1973, 18, 357.
488. Barrow R. F., Caunt A. D. — «Proc. Roy. Soc.
London», 1953, A219, 120.
489. Barrow R. F., Clark T.C., CoxonJ.A.,Yee К. К.—
«J. Mol. Spectrosc», 1974, 51, 428.
490. Barrow R. F., Downie A. R. — «Proc. Phys. Soc.
London», 1956, A69, 178.
491. Barrow R. F., Downie А. Д., Laird R. K. — «Proc.
Phys. Soc. London», 1952, A65, 70.
492. Barrow R. F., Drummond G., Zeeman P. B. —
«Proc. Phys. Soc. London», 1954, A67, 365.
493. Barrow R. F., DuParcq R. P. — In: Elemental
Sulphur. Chemistry and Physics. Meyer B. (Ed.).
New York—London—Sydney, Intersci. Publ.,
1965, p. 251.
494. Barrow R. F., Du Parcq R. P. — «J. Phys. B:
Atom and Mol. Phys.», 1968, 1, 283.
495. Barrow R. F., Du ParcqR. P., Ricks J. M. —
«J. Phys. B: Atom, and Mol. Phys.», 1969, 2,
413.
496. Barrow R. F., Ketteringham J. M. — «Canad. J.
Phys.», 1963, 41, 419.
497. Barrow R. F., Miescher E. — «Proc. Phys. Soc.
London», 1957, A70, 219.
497a. Barrow R. F., Stamper J. G. — «Proc. Roy.
Soc. London», 1961, A263, 277.
498. Barrow R. F., Yee К. К. — «J. Chem. Soc.
Faraday Trans. II», 1973, 69, 684.
499. Barrow R. F., Yee К. К. — «Acta phys. Acad.
sci. hung.», 1974, 35, 239.
500. Barshall H. — «Z. Elektrochem.», 1911, 17, 341.
500a. Bartell L. S. — «J. Chem. Educ», 1968, 45, 754.
5006. Bartell L. S., Gavin R. M. — «J. Chem. Phys.»,
1968, 48, 2466.
501. Bartell L. S., Hansen K. W. — «Inorg. Chem.»,
1965, 4, 1777.
502. Bartholome E., Clusins K. — «Naturwissenschaf-
ten», 1934, 22, 420.
503. Bartholome E., Clusins K. — «Z. Elektrochem.»,
1934, 40, 529.
504. Bartlett N., Levchuk L. E. — «Proc. Chem. Soc»,
1963, 342.
505. Bartlett N., Passmore J., Wells E. J. — «J. Chem.
Soc. Chem. Communs», 1966, 213.
506. Basco TV., Morse R. D. — «Proc. Roy. Soc.
London», 1971, A321, 129.
506a. Basco N., Morse R. D. — «J. Mol. Spectrosc»,
1973, 45, 35.
507. Basco N., Yee К. К. — «J. Chem. Soc. Chem.
Communs», 1967, 1146.
508. Basile L. J., Labonville P., Ferraro J. R., Wi-
liams J. H. — «J. Chem. Phys.», 1974, 60, 1981.
509. Basile P., Louis H. — «C. r. Acad. sci.», 1967,
B264, 1196.
510. Bass A. M., Garvin D. — «J. Mol. Spectrosc»,
1962, 9, 114.
511. Bastiansen O., Beagley B, — «Acta chem. scand.»,
1964, 18, 2077.
512. Bates D. R. — «Proc. Roy. Irish Acad.», 1947,
51, 151.
513. Bates D. R., MoiseiwitchВ. L. — «Proc. Phys. Soc,
London», 1955, A68, 540.
514. Bates J. R. — «Z. phys. Chem. Bodenstein-Fest-
band», 1931, 329.
515. Bates J. R., Halford J. O., Anderson L. C. —
«J. Chem. Phys.», 1935, 3, 531.
516. Bauer A., Bellet J. — «C. r. Acad. sci.», 1964,
258, 873.
517. Bauer A., Bellet J., Pouzet P., Remy A. — «С. г.
Acad. sci.», 1963, 257, 3148.
442
Литература
518. Bauer H. F., Sheehan D. F. — «Inorg. Chem.», 551.
1967, 6, 1736.
519. Bauer S. H. — «J. Phys. Chem.», 1952, 56, 343. 552.
520. Baumann G. Dissertation. Stuttgart, 1956.
521. Baumann G. — «Z. phys. Chem.» (BRD), 1958, 553.
14, 173.
522. Baumann W., Mecke R. — «Z. Phys.», 1933, 81, 554.
445.
523. Baxter G. P., Grose M. R. — «J. Amer. Chem. 555.
Soc», 1915, 37, 1061.
524. Baxter G. P., Hickey С H., Holmes W. С — «J. 556.
Amer. Chem. Soc», 1907, 29, 127. 557.
525. В ayes K. D., Kistiakowsky G. B. — «J. Chem.
Phys.», 1960, 32, 992. 558.
526. Beagley В., Clark A. #., Hewitt T. G. — «J.
Chem. Soc. A», 1968, 658. 559.
527. Beagley В., Conrad A. R., Freeman J. M., Monag-
han J. J., Norton B. G., Holywell G. C. — «J. 560.
Mol. Struct.»., 1972, 11, 371.
528. Beagley В., CruickshankD. W. /., Hewitt T. G. — 561.
«Trans. Faraday Soc», 1967, 63, 836.
529. Beagley В., CruickshankD. W. J., Hewitt T. G. — 562.
«Trans. Faraday Soc», 1969, 65, 1219. 563.
530. Beagley В., Cruickshank D. W. <?., Hewitt T. G.,
Haaland A. — «Trans. Faraday Soc», 1967, 63, 564.
836.
531. Beagley В., Cruickshank D. W. J., Hewitt T. G., 565.
Jost K. H. — «Trans. Faraday Soc», 1969, 65,
1219. 566.
532. Beattie I. R., Bell S. W. — «J. Chem. Soc», 567.
1957, 1681.
533. Beattie I. R., Gilson Т., Livingston K., Fawcett
V., Ozin G. A. — «J. Chem. Soc. A», 1967, 712. 568.
534. Beattie I. R., Harder J. R. — «J. Chem. Soc.
A», 1969, 2655. 569.
535. Beattie I. R., Livingston К. М. S., Reynolds
D. J. — «J. Chem. Phys.», 1969, 51, 4269.
536. Beattie I. R., Livingston К. М. S., Ozin G. A., 570.
Reynolds D. J. — «J. Chem. Soc. A», 1970, 449.
536a. Beattie I. R., Ozin G. A. — «J. Chem. Soc. A», 571.
1969, 1691.
537. Beattie I. R., Ozin G. A., Perry R. O. — «J. 572.
Chem. Soc A», 1970, 2071.
538. Beattie I. R., Webster M. — «J. Chem. Soc», 573.
1963, 38.
539. Beauchamp J. L., Butrill S. E., jr. — «J. Chem. 574.
Phys.», 1968, 48, 1527.
540. Beckel С L., Hansen B. D. Ill, Peek J. M. — 575.
«J. Chem. Phys.», 1970, 53, 3681.
541. Beckel C. L., Shafi M., Engelke R. — «J. Mol. 576.
Spectrosc», 1971, 40, 519.
542. Becker G., Roth W. A. — «Z. Elektrochem.», 1934, 577.
40, 836.
543. Becker K. H., Fink E.H., Langen P., Schu-
rath U. -~ «Z. Naturforsch.», 1973, 28, 1872. 578.
544. Becker K. H., Fink E. H., Langen P., Schurath
U. — «J. Chem. Phys.», 1974, 60, 4623. 579.
545. Beckett C. W., Cassidy E. C. NBS Rept, N 8628,
1965. 580.
546. Beckett С W., Haar L. Proc. Conf. on
Thermodynamic and Transport Properties of 581.
Fluids, London, 1958, p. 27.
547. Beebe N. H. F., Thulstrun E. W., Andersen A. — 582.
J. Chem. Phys.», 1976, 64, 2080.
548. Beers Y., Howard C. J. — «J. Chem. Phys.», 1975, 583.
63, 4212.
549. Beers Y., Howard С J. — «J. Chem. Phys.», 1976, 584.
64, 1541.
550. Beers Y., Weisbaum S. — «Phys. Rev.», 1953, 585.
91, 1014.
Beeson С M., Yost D. M. — «J. Chem. Phys.»,
1939, 7, 44.
Beeson C. M., Yost D. M. — «J. Amer. Chem.
Soc», 1939, 61, 1040.
Begum A., Symons M. C. R. — «J. Chem. Soc.
A», 1971, 2065.
Begun G. M., Fletcher W. H. — «J. Mol.
Spectrosc», 1960, 4, 388.
Begun G. M., Fletcher W. H., Smith D. F. — «J.
Chem. Phys.», 1965, 42, 2236.
Bellet J. — «Ann. phys.», 1965, 10, 827.
Bellet J., Steenbeckeliers G. — «C. r. Acad. sci.»,
1970, B271, 1208.
Bellet J., Steenbeckeliers G., Stouffs P. — «С. Г.
Acad. sci.», 1972, B275, 501.
Ben-Aryech Y. — «J. Quant. Spectrosc. and Ra-
diat. Transfer» 1973, 13, 1441.
Bender C. F., Davidson E. R. — «J. Chem. Phys.»,
1968, 49, 4989.
Bender C. F., Schaefer H. F. — «J. Chem. Phys.»,
1971, 55, 4798.
Bender D. — «Phys. Rev.», 1935, 47, 252.
Bender P., Wood J. M. Jr. — «J. Chem. Phys.»,
1955, 23, 1316.
Bendtsen J. — «J. Raman Spectrosc», 1974, 2,
133.
Bendtsen J., Winnewisser M. — «Chem. Phys.
Lett.», 1975, 33, 141.
Benedict W. S. — «Phys. Rev.», 1948, 74, 1246.
Benedict W. S. Theoretical Studies of High
Resolution Spectra of Atmospheric Molecules. Final
Report, AFRL-65-573, 1965.
Benedict W. S., Bass A. M., Plyler E. K. — «J.
Res. NBS», 1954, 52, 161.
Benedict W. S., Bullock B. W., Silverman S.,
Grosse A. V. — «J. Opt. Soc. America», 1953,
43, 1106.
Benedict W. S., CalfeeR. F. Environmental Science
Services Administration Profess. Paper, 1967, N 2.
Benedict W. S., Classen H. H., Shaw J. H. «J.
Res. NBS», 1952, 49, 91..
Benedict W. S., Clough S. A., Frenkel L.,
Sullivan Т. Е. — «J. Chem. Phys.», 1970, 53, 2565.
Benedict W. S., Gailar N., Plyler E. К.-Л.
Chem. Phys.», 1953, 21, 1301.
Benedict W. S., Gailar N., Plyler E. K. — «J.
Chem. Phys.», 1953, 21, 1302.
Benedict W. S., Gailar N., Plyler E. K. — «J.
Chem. Phys.», 1956, 24, 1139.
Benedict W. S., Plyler E. K. — «J. Res. NBS»,
1951, 46, 246.
Benedict W. S., Plyler E. K. — In: Energy
Transfer in Hot Gases. NBS Circ. 523.
Washington, 1954, p. 57.
Benedict W. S., Plyler E. K. — «Canad. J. Phys.»,
1957 35 1235.
Benedict V. S.\ Plyler E. K., Tidwell E. D. —
«J. Chem. Phys.», 1958, 29, 829.
Benedict W. S., Plyler E. K., Tidwell E. D. —
«J. Chem. Phys.», 1960, 32, 32.
Benesch W. M., Saum К. А.—Ч.1. Phys. B:
Atom, and Mol. Phys.», 1971, 4, 732.
Benesch W. M., Vanderslice J. Т., Wilkinson
P. G. — «Astrophys. J.», 1965, 142, 1227.
Benioff P., Das G., Wahl A. C. — «J. Chem.
Phys.», 1976, 64, 710.
Benjamins E., Westrum E. F. — «J. Amer. Chem.
Soc», 1957, 79, 287.
Bennett S. L., Margrave J. L., Franklin J. L. —
«J. Chem. Phys.», 1974, 61, 1647.
443
Литература
586. Bent R., Ladner W. R. — «Spectrochim. acta», 621.
1963, 19, 931. 622.
587. Bent R., Ladner W. R., Pankhurst K. S., Waite
B. D. — «Nature», 1962, N 4810, 62. 623.
588. Berjot M.,GazengelJ.,DupeyratR. —«Ann. Univ.
et Assoc. reg. etude et rech. scient.», 1967, 5, 46. 624.
589. Berkowitz J. — In: Elemental Sulfur. Chemistry
and Physics B. Meyer (Ed.). New York — Lon- 625.
don — Sydney, Intersci. Publ., 1965, p. 125. 626.
590. Berkowitz J. — «J. Chem. Phys.», 1975, 62, 4074.
591. Berkowitz /., Appelman E. H., Chupka W. A.—¦ 627.
«J. Chem. Phys.», 1973, 58, 1950.
592. Berkowitz J., Chupka W. A. — «J. Chem. Phys.», 628.
1964, 40, 287. 629.
593. Berkowitz J., Chupka W. A. — «J. Chem. Phys.», 630.
1969, 50, 4245. 631.
594. Berkowitz J., Chupka W. A., Bromels E., Bel-
ford R. L. — «J. Chem. Phys.», 1967, 47, 4320. 632.
595. Berkowitz J., Chupka W. A., Gutman D. — «J.
Chem. Phys.», 1971, 55, 2733. 633.
596. Berkowitz J., Chupka W. A., Guyon P. M., Hoi- 634.
loway J. H., Spohr R. — «J. Chem. Phys.», 1971, 635.
54, 5165. 636.
596a. Berkowitz J., Chupka W. A., Guyon P. M., Hol-
loway J. #., Spohr R. — «J. Phys. Chem.», 1971, 637.
75, 1461.
5966. Berkowitz J., Eland J. H. D., Appelman E. H. — 638.
«J. Chem. Phys.», 1977, 66, 2183.
597. Berkowitz J., Lifshitz С — «J. Chem. Phys.», 639.
1968, 48, 4346.
598. Berkowitz J., Marquart J. R. — «J. Chem. Phys.», 640.
1963 39 275
599. Bernage P., Niay P., Hondart R. — «C. r. Acad. 641.
sci.», 1974, B278, 235.
600. Bernal M. Т. М. — «Proc. Phys. Soc. London»,
1953, A66, 514. 642.
601. Berney С V. — «J. Mol. Struct.», 1972, 12, 87.
602. Berney С V., Cormier A. D. — «J. Chem. Phys.»,
1973, 58, 4709. 643.
603. Bernitt D. L., Miller R. #., Hisatsune I. C. —
«Spectrochim. acta», 1967, A23, 237. 644.
604. Bernstein H. J., Fowling J. — «J. Chem. Phys.»,
1950, 18, 685. 645.
605. Bernstein R. В., Katz J. J. — «J. Phys. Chem.», 646.
1952, 56, 885. 647.
605a. Bernstein L. S., Kim J. J., Pitzer K. S., Abra-
mowitz S., Lewin I. W. — «J. Chem. Phys.»,
1975, 62, 3671.
606. Bernstein R. В., Metlay M. — «J. Chem. Phys.», 648.
1951, 19, 1612.
607. Bernstein H. J., Powling J. — «J. Chem. Phys.», 649.
1950, 18, 1018.
608. Bernstein H. J., Powling J. — «J. Chem. Phys.», 650.
1951, 19, 139.
609. Berry R. S. — «Chem. Rev.», 1969, 69, 533. 651.
610. Berry R. S., Mackie J. C, Taylor R. L., Lynch
R. — «J. Chem. Phys.», 1965, 43, 3067. 652.
611. Berry R. S., Reimann C. W. — «J. Chem. Phys.»,
1963, 38, 1540. 653.
612. Berry R. S., Reimann С W., Spokes G. N. —
«J. Chem. Phys.», 1962, 37, 2278. 654.
613. Berthelot M. — «Ann. chim. phys.», 1875, 6, 178.
614. Berthelot M. — «C. r. Acad. sci.», 1876, 82, 1281.
615. Berthelot M. — «Ann. chim. phys.», 1877, 10, 162. 655.
616. Berthelot M. — «Ann. chim. phys.», 1877, 10, 433.
617. Berthelot M. — «Ann. chim. phys.», 1880, 20, 255. 656.
618. Berthelot M. — «Ann. chim. phys.», 1881, 22, 429.
619. Berthelot M. — «Ann. chim. phys.», 1885, 6, 358. 657.
620. Berthelot M. — «Bull. Soc. chim. France», 1885,
43, 260.
Berthelot M. — «С. г. Acad. sci.», 1885, 100, 81.
Berthelot M., Andre G. — «Ann. chim. phys.»,
1890, 21, 384.
Berthelot M., Louguinine V. F. — «Ann. chim.
phys.», 1875, 6, 305.
Berthelot M., Matignon M. — «Ann. chim. phys.»r
1892 27 303
Berthond'A. — «Helv. chim. acta», 1922, 5, 513.
Berthou J. M., Pascat В., Guenebaut H., Ramsay
D. A. — «Canad. J. Phys.», 1972, 50, 2265.
Bethe H. A. The Specific Heat of Air up to 25 000°C.
Office Sci. Res. Dev. Rept 3691, 1942.
Beukel A. — «Phys. status solidi», 1968, 28, 565.
Beutler H. — «Z. phys. Chem.», 1934, B27, 289.
Beutler H. — «Z. phys. Chem.», 1935, B29, 315.
Beutler H., Junger H. O. — «Z. Phys.», 1936,
101, 285.
Bevan J. W., Legon A. C, Millen D. J., Rogers
S. C—«J. Chem. Soc. Chem. Communs», 1975, 341.
Bhagavantam S. — «Indian J. Phys.», 1930, 5, 35.
Bhagavantam S. — «Indian J. Phys.», 1930, 5, 63.
Bhagavantam S. — «Nature», 1930, 126, 995.
Bhagavantam S., Venkatarayudu T. — «Proc.
Indian Acad. Sci.», 1939, A8, 101.
Bhale G. L., Ramakoteswara Rao P. — «Proc.
Indian Acad. Sci.», 1968, A67, 30.
Bialski M., Grein F. — «J. Mol. Spectrosc», 1976,
61, 321.
Bibart C. H., Ewing G. E. — «J. Chem. Phys.»,
1974, 61, 1284.
Bibart C. #., Ewing G. E. — «J. Chem. Phys.»,
1974, 61, 1293.
Bichowsky F. R. — In: International Critical
Tables. V. 5. N. Y., McCraw-Hillj Book Co.,
1929, p. 169.
Bichowsky R. F., Rossini F. D. The
Thermochemistry of the Chemical Substances. N. Y., Rein-
bold Publ. Corp., 1936.
Biltz #., Meyer V. — «Z. phys. Chem.», 1889,
4, 249.
Birchall Т., Gillespic R. J. — «Spectrochim. acta»,
1966, 22, 681.
Bird G. R. — «Phys. Rev.», 1955, 98, 1160.
Bird G. R. — «J. Chem. Phys.», 1956, 25, 1040.
Bird G. R., Baird J. C, Jache A. W., Hodgesson
J. A., Curl R. F., Kunkle A. C, Bransford J. W.,
Rastrup-Anderson J., Rosenthol J. — «J. Chem.
Phys.», 1964, 40, 3378.
Bird G. R., Darti A., Lord R. C. — «Spectrochim.
acta», 1958, 12, 247.
BirdG.R., Hunt G. R., Gebbie H. A., Stone
N. W. — «J. Mol. Spectrosc.», 1970, 33, 244.
Birdsall C. M., Jenkins A. C. — «J. Chem. Phys.»,
1952, 20, 1158.
Birdsall С M., Jenkins A. C, Di Paolo F. S. —
«J. Chem. Phys.», 1955, 23, 441.
Birge R. Т., Jeppesen C. R. — «Nature», 1930,
125, 463.
Birge R. Т., Sponer H. — «Phys. Rev.», 1926,
28, 277.
Birley G. I., Skinner H. A. Proc. 1-st Intern.
Conf. Calorim. Thermodynam., Warsaw, 1969,
p. 269.
Btrley G. I., Skinner H. A. — «Trans. Faraday
Soc», 1968, 64, 3232.
Birks J. W., Gabelnick S. D., Johnston H. S. —
«J. Mol. Spectrosc», 1975, 57, 23.
Bisbee W. R., Hamilton J. V., Gerhauser J. M.t
Rushworlh R. — «J. Chem. and Engng Data»,
1968, 13, 382.
444
Литература
¦658. Bisbee W. R., Hamilton J. V., Rushworth R., 694.
Houser I. J., Gerhauser J. M. — «Advanc. Chem.
Series», 1965, 54, 215. 694a.
659. Bisbee W. R., Hamilton J. V., Rushworth R.,
Gerhausen J. M., Houser T. J. — «AIAA Bull.»
(U.S.A.), 1965, 2, N 3, 114. 695.
660. Bischel W. K., Kelley P. J., Rhodes Ch. K. —
«Phys. Rev. A: Gen. Phys.», 1976, 13, 1829. 696.
661. Bishop D. M. — «Theor. chim. acta», 1963, 1, 410.
€62. Bishop D. M. — «J. Chem. Phys.», 1965, 43, 697.
4453.
662a. Bjellerup L., Sunner S., Wadso J. — «Acta chem. 698.
scand.», 1957, 11, 1761.
663. Bjerrum N. — «Z. phys. Chem.», 1912, 87, 36. 699.
664. Blackwell D. E., Ingham H. F., Rundll H. N. —
«Astrophys. J.» 1960, 131, 15. 700.
€65. Blagg J. C. L., Murphy G. M. — «J. Chem.
Phys.», 1936, 4, 631. 701.
666. Blaine L. R., Plyler E. K., Benedict W. S. — 702.
«J. Res. NBS», 1962, A66, 223.
667. Blair С M., Yost D. M. — «J. Amer. Chem. 703.
Soc», 1933, 55, 4489.
668. Blake R. C, Iredale T. — «Nature», 1946, 157, 704.
229.
¦669. Blank G. — «Wahrme- und Stoffiibertrag», 1969, 705.
2 53
€70. Blank R. E., Hause C. D. — «J. Mol. Spectrosc», 706.
1970, 34, 478. 707.
671. Blank R. E., Olman M. D., Hause C. D. — «J. 708.
Mol. Spectrosc», 1970, 33, 109.
672. Blau E. J., Hochheimer B. F. — «J. Chem. Phys.», 709.
1964, 41, 1174. 710.
€73. Bleier O., Kohn L. — «Monatsh. Chem.», 1900,
21, 575. 711.
674. Blend H. — «J. Chem. Phys.», 1970, 53, 4497.
675. Bliefert C, Wanczek K.-P. — «Z. Naturforsch.,
1970, 25a, 1770. 712.
676. Blue R. W., Hicks J. F. G. — «J. Amer. Chem.
Soc», 1937, 59, 1962. 713.
677. Blukis U., Myers R. J. — «J. Phys. Chem.»,
1965, 69, 1154. 714.
678. Bluyssen H. J. Thesis. Nijmegen, 1968.
679. Bluyssen H., Verhoelven J., Dymanus A. — «Phys. 715.
Lett.», 1967, A25, 214.
680. Bockhoff F. J., Pace E. L. — «J. Chem. Phys.», 716.
1962, 36, 3502.
681. Bockhoff F. J., Petrella R. V., Pace E. L. — «Nucl. 717.
Sci. Abstrs», 1960, 14, 15.
682. Bodenstein M. — «Z. phys. Chem.», 1922, 100, 68. 718.
683. Bodenstein M., Jockusch H., Sing-Нои Chong. —
«Z. anorg. und allgem. Chem.», 1937, 231, 24. 719.
684. Bodenstein M., Katayama M. — «Z. Elektrochem.»,
1909, 15, 244. 720.
685. Bodenstein M., Katayama M. — «Z. phys. Chem.»,
1909, 69, 26. 721.
686. Bodenstein M., Pohl W. — «Z. Elektrochem.»,
1905, 11, 373. 722.
€87. Bodenstein M., Pohl W. — «Z. Elektrochem.»,
1905, 11, 457. 723.
688. Bodenstein M., Schmidt A. — «Z. phys. Chem.»,
1926, 123, 28. 724.
689. Bodlander G., Koppen K. — «Z. Elektrochem.», 725.
1903, 9, 787.
€90. Bodor N., Dewar M. J. S., Harget A., Hasel- 726.
bach E. — «J. Amer. Chem. Soc», 1970, 92, 3854.
691. Bohme D. K., Hemsworth R. S., Rundle H. W. — 111.
«J. Chem. Phys.», 1973, 59, 77. 728.
692. Bohn R. K. — «Dissert. Abstrs», 1964, 25, 3282.
693. Bohn R. K., Bauer S. H. — «Inorg. Chem.», 1967, 729.
6, 304.
Bohn R. K., Bauer S. H. — «Inorg. Chem.», 1967,
6, 309.
Bohn R. K., Katada K., Martinez J. V., Bauer
S. H. — In: Noble Gas Compounds. H. H. Hayman
(Ed.). Chicago. Univ. Press, 1963, p. 238.
Bondybey V. E., Brus L. E. — «J. Chem. Phys.»,
1975, 62, 620.
Bondybey V. E., Brus L. E. — «J. Chem. Phys.»,
1975, 63, 794.
Bondybey V. E., Fletcher Ch. — «J. Chem. Phys.»,
1976, 64, 3615.
Boness M. J. W., Schulz G. J. — «Phys. Rev.»,
1970, A2, 2182.
Bonhoeffer K. F., Reichardt H. — «Z. phys. Chem.»j
1928, A139, 75.
Bonn R., Metselaar R., Van der Elsken J. —•
«J. Chem. Phys.», 1967, 46, 1988.
Bonner L. — «Phys. Rev.», 1934, 46, 458.
Booth H., Bowen E. J. — «J. Chem. Soc», 1925.
127, 342.
Booth H. S., Bozarth A. R. — «J. Amer. Chem.
Soc», 1939, 61, 2927.
Borje K., Eriksson S. — «Arkiv fys.», 1967, 33,
357.
Borkman R. F. — «J. Chem. Phys.», 1970, 53,
3153.
Born M., Karman Т.— «Phys. Z.», 1912, 13, 297.
Born M., Karman T. — «Phys. Z.», 1913, 15, 65.
Bosworth Y. M., Clark R. J. H., Rippon D. M. —
«J. Mol. Spectrosc», 1973, 43, 240.
Bott J. F. — «J. Chem. Phys.», 1971, 54, 181.
Bott J. F., Jacobs T. A. — «J. Chem. Phys.»,
1969, 50, 3850.
Better R., Hagemann R., Nief G., Roth E. —
In: Advances in Mass Spectrometry. V. 3. W. L.
Mead (Ed.). London, Inst. Petrol., 1966, p. 951.
Bouchoux A. M., Marchand J. — «Spectrochim.
acta», 1972, A28, 1771.
Bouchoux A. M., Marchand /., Janin J. —
«Spectrochim. acta», 1971, A27, 1909.
Bondybey V. E., Nibler J. W. — «J. Chem. Phys.»,
1973, 58, 2125.
Boughan E. C, Evans M. G., Polanyi M. — «Trans.
Faraday Soc», 1941, 37, 377.
Bougon R., Chatelet J., Desmoulin J. P., Plu-
rien P. — «C. r. Acad. sci.», 1968, C266, 1760.
Bougon R., Chatelet J., Plurien P. — «C. r. Acad.
sci.», 1967, C264, 1747.
Boutin H., Safford G. J., Brajovic V. — «J. Chem.
Phys.», 1963, 39, 3135.
Bovey L. F. H. — «J. Opt. Soc. America», 1951,
41, 836.
Bowers M. Т., Flygare W. H. — «J. Mol.
Spectrosc», 1966, 19, 325.
Bowers M. Т., Flygare W. H. — «J. Chem. Phys.»,
1966, 44, 1389.
Boyd D. В., Lipscomb W. N. — «J. Chem. Phys.»,
1967, 46, 910.
Boyd D. R. J., Thompson H. W.— «Spectrochim.
acta», 1952, 5, 308.
Boyd M. E. — «J. Chem. Phys.», 1962, 37, 1317.
Boyd R. J., Whitehead M. A. — «J. Chem. Soc.
Dalton Trans.», 1972, 1, 73.
Bozoky L. — «Math, naturwiss. Anz. ungar. Akad.
Wiss.», 1936, 54, 557.
Bozoky L. — «Z. Phys.», 1937, 104, 275.
Bozoky L., Schmid R. — «Phys. Rev.», 1935, 48,
465
Brabson G. D., Brewer L. — «Bull. Amer. Phys.
Soc», 1965, 10, 1097.
445
Литература
730. Bradley R. S. — «Trans. Faraday Soc», 1954,
50, 1182.
731. Bradley G. M., Siddall W., Strauss H. S., Varetti 765.
E. L. — «J. Phys. Chem.», 1975, 79, 1949.
732. Bradley R. H., Brier P. N., Whittle M. J. — 766.
«Chem. Phys. Lett.», 1971, 11, 192. 767.
733. Bradley R. #., Brier P. N., Whittle M. J. — «J.
Mol. Spectrosc», 1972, 44, 536. 768.
734. Bradley R. S. — «Proc. Roy. Soc. London», 1951,
A205, 553. 769.
735. Brand J. C. D., Chan W. H., Hardurick J. L. —
«J. Mol. Spectrosc.», 1975, 56, 309. 770.
736. Brand J. С D.,Di Lauro C, Jones V. T. — «J.
Amer. Chem. Soc», 1970, 92, 6095. 771.
737. Brand J. С D., Hardwick J. L., Pirkle R. J.,
Seliskar C. J. — «Canad. J. Phys.», 1973, 51, 2184. 772.
738. Brand J. С D., Humphrey D. R., Douglas A. E., 773.
Zanon J. — «Canad. J. Phys.», 1973, 51, 530.
739. Brand J. С D., Jones V. Т., Di Lauro C — «J. 774.
Mol. Spectrosc.», 1971, 40, 616.
740. Brand J. С D., Jones V. Т., Di Lauro C. «J. 774a
Mol. Spectrosc.», 1973, 45, 404.
741. Brand J. C. D., Nanes R. — «J. Mol. Spectrosc.», 775.
1973, 46, 194.
742. Brand J. C. D., Redding R. W., Richardson 776.
A. W. — «J. Chem. Soc.» D, 1969, 618.
743. Brand J. С D., Redding R. W., Richardson 111.
A. W. — «J. Mol. Spectrosc.», 1970, 34, 399.
744. Brand J. C. D., Srikameswaran K. — «Chem. 777a,
Phys. Lett.,» 1972, 15, 130.
745. Brandle K., Schmeisser M., Luttke W. — «Chem. 7776,
Ber.», 1960, 93, 2300.
746. Brandner G., Junk N., Lawrence J. W., Robins J. — 778.
«J. Chem. and Engng Data», 1962, 7, 227.
747. Brannon P. J., Church С H., Peters C. W. — 779.
«J. Mol. Spectrosc.», 1968, 27, 44.
748. Branscomb L. M. — «Phys. Rev.», 1966, 148, 11. 780.
749. Branscomb L. M., Burch D. S., Smith S. J.,
Geltman S. — «Phys. Rev.», 1958, 111, 504. 780a,
750. Branscomb L. M., Smith S. J. — «J. Chem. Phys.»,
1956, 25, 598. 7806.
750a. Bran С A., Ewing J. J. — «J. Chem. Phys.»,
1975, 63, 4640. 781.
751. Brauer G., Victor-Berlin E. — «Z. Elektrochem.»,
1935, 41, 508. 782.
752. Braun Ch., Leidecker H. — «J. Chem. Phys.»,
1974, 61, 3104. 783.
753. Braune #., Engelbrecht G. — «Z. phys. Chem.,
1932, B19, 303. 784.
754. Braune H., Knoke S. — «Z. phys. Chem.», 1933, ¦
B21, 297. 785.
755. Braune H., Moller O. — «Z. Naturforsch.», 1954,
9a, 210. 786.
756. Braune H., Peter S., Neveling V. — «Z.
Naturforsch.», 1951, 6a, 32. 787.
757. Braune H., Pinnow P. — «Z. phys. Chem.», 1937, nQQ
B35, 239. 7b8>
758. Bredohl #., Herzberg G. — «Canad. J. Phys.»,
1973, 51, 867. 789
759. Brehm B. — «Z. Naturforsch.», 1966, 21a, 196.
760. Brewer L. — «Chem. Rev.», 1953, 52, 1. 790.
761. Brewer L., Brabson G. D. — «J. Chem. Phys.»,
1966, 44, 3274. 79!,
762. Brewer L., Brabson G. D., Meyer B. — «J. Chem.
Phys.», 1965, 42, 1385. 792.
763. Brewer L., Rosenblatt G. M. — «Chem. Rev.»,
1961, 61, 257. 793.
764. Brewer L., Rosenblatt G. M. — In: Advances in
High Temperature Chemistry, v. 2. Eyring L.
(Ed.). New York — London, Acad. Press, 1969,
p. 1.
Brewer L., Wang J. L. — «J. Chem. Phys.», 1972f
56, 759.
Bridgman P. W. — «Phys. Rev.», 1914, 3, 153.
Bridgman P. W. — «J. Amer. Chem. Soc», 1914,
36, 1344.
Brief A., Joffe J. — «J. Chem. and Engng Data»,
1970, 15, 114.
Briegleb G., Strohmeier W. — «Z. Elektrochem.»,
1953, 57, 668.
Briggs A. G., Norrish R. G. W. — «Proc. Roy.
Soc. London, 1963, A276, 51.
Bright N. F. H., Hagerty R. P. — «Trans. Faraday
Soc», 1947, 43, 697.
Brill O. — «Z. phys. Chem.», 1907, 62, 721.
Briner E., Biederman H., Rother A. — «Helv.
chim. acta», 1925, 8, 923.
Briner E., Biederman #., Rother A. — «J. chim.
phys. et phys.-chim. biol.», 1926, 23, 157.
Briner E., Pylkoff Z. — «J. chim. phys. et phys.-
chim. biol.», 1912, 10, 640.
Brian J., Da Paz M., Mongin J., Guenebaut H. —
— «C. r. Acad. sci.», 1971, B272, 993.
Brion J., Malicet J. — «C. r. Acad. sci.», 1974,
C278, 223.
Brion J., Malicet J. — «J. Phys. B: Atom, and
Mol. Phys.», 1975, 8, L164.
Brion J., Malicet J. — «J. Phys. B: Atom and
Mol. Phys.», 1976, 9, 2097.
Brion J., Malicet J., Daumont D. — «C. r. Acad.
sci.», 1977, C284, 647.
Brion J., Malicet J,, Guenebaut H. — «C. r. Acad.
sci.», 1967, B264, 622.
Brion J., Malicet J., Guenebaut H. — «Canad. J.
Phys.», 1974, 52, 2143.
Brion J., Malicet J., Guenebaut H. — «Canad.
J. Phys.», 1976, 54, 362.
Brion J., Malicet J., Merienne-Lafore M. F. —
«Canad. J. Phys.», 1977, 55, 68.
Brion J., Malicet J., Merienne-Lafore M. F. —
«C. r. Acad. sci.», 1977, C283, 171.
Brion J., Malicet J., Mongin J. — «C. r. Acad.
sci.», 1971, B272, 127.
Briske C, Hartshorne N. H., Stranks D. R. —
«J. Chem. Soc», 1960, 1200.
Briston M. P. F., McChesney M. — «Proc. Phys.
Soc. London», 1965, 85, 1237.
Brith M., Rowe M. D., Schnepp O., Stephen»
P. J. — «Chem. Phys.», 1975, 9, 57.
Brith M., Schnepp O., Stephens P. — «Chem.
Phys. Lett.», 1974, 26, 549.
Brittain A. H., Cox A. P., Kuczkowski R. L. -~
—«Trans. Faraday Soc», 1969, 65, 1963.
Brittain А. Д., Smith J. E., Schwendeman R. H. —
«Inorg. Chem.», 1972, 11, 39.
Britzke E. W., Hoffmann E. — «Sitzungsber. Akad.
Wiss. Wien. Math.-naturwiss. К.1.», 1938, 146r
763.
Brix P., Herzberg G. — «Canad. J. Phys.», 1954,
32, 110.
Brockway L. O., Beach J. Y. — «J. Amer. Chem»
Soc», 1938, 60, 1836.
Brockway L. O., Pauling L. — «Proc. Nat. Acad.
Sci. U. S. A.», 1933, 19, 68.
Brockway L. O., Wall F. T. — «J. Amer.' Chem.
Soc», 1934, 56, 2373. »
Brodersen P. H., Cudmani L. Y. C. — Dtsch.
Versuchsanstalt Luft und Raumfahrt. Ber. №250,
Koln, 1963.
446
Литература
794
795
796
797
798
799
800
801
802*
803
804'
805
806
807
808
809
810
810a
811.
812
813.
814
815
816
817
818
819
820
821
822
823
824
825.
826.
827.
828
829
830
Brodersen P. H., May 0. S. — «Z. Phys.», 1955, 831. Bullock L. E., Hause C. D. — «J. Mol. Spectrosc.»,
143 477. 1971> 39> 519-
Brodersen P. H., Schumacher H. J. — «Z. Natur- 832. Bunker P. R. — «J. Mol. Spectrosc.», 1971, 39, 90.
forsch », 1947, 2a, 358. 833. Bunker P. R. — «J. Mol. Spectrosc», 1972, 42, 478.
Brodersen P. H., Schumacher H. J. — «An. Asoc. 834. Burbank R. D. — «Acta crystallogr.», 1962, 15,
quim argent», 1950, 38, 52. 1207.
Brodersen P. H., Sicre J. E. — «Z. Phys.», 1955, 835. Burbank R. D., Bensey F. N. — «J. Chem. Phys.»,
141 515. 1957> 27> 982-
BroidaH P., GaydonA. G. — «J. phys. et radium», 835a. Burbank R. D., Jones G. R. — «Science», 1970,
1954 15, 385. 168, 248.
Broida H. P., Gaydon A. G. — «Proc. Roy. Soc. 8356. Burbank R. D., Jones G. R. — «Science», 1971, 171,
London», 1954, A222, 181. 485.
Broida И P , Peyron M. — «J. Chem. Phys.», 836. Burbank R. D., Jones G. R. — «Inorg. Chem.»,
1960 32, 1068. 1Э74, 13, 1071.
Bromels E. — «Dissert. Abstr.», 1968, B29, 562. 837. Burch D. S., Smith S. J., Branscomb L. M. ~
Bron J , Paul S. O. — «J. Chem. Soc. Faraday «Phys. Rev.», 1958, 112, 171.
Trans II», 1974, 70, 1294. 838. Burdett J. K., Hodgen L., Dunning V., Cur-
Bronsted J N. — «Z. phys. Chem.», 1906, 55, 371. rent J. H. — «J. Phys. Chem.», 1970, 74, 4053.
Brook M, Kaplan /.— «Phys. Rev.», 1954, 96, 839. Burke T. G., Jones E. A. — «J. Chem. Phys.»,
1540. 1951, 19, 1611.
Brooker M H Irish D. E. — «Canad. J. Chem.», 840. Burnelle L., Dressier K. P. — «J. Chem. Phys.»,
1968, 46, '229. 1969, 51, 2758.
Brooks W V F , Cranford В. — «J. Chem. Phys.», 841. Burnelle L., May A. M., Gangi R. A. — «J.
1955 23,' 363. Chem. Phys.», 1968, 49, 561.
Brown H W Pimentel G. C. — «J. Chem. Phys.», 841a. Burns J. H., Agron P. A., Levy H. A. — «Science»,
195829 883. 1963, 139, 1208.
Brown /' D ,'Burns G., he Roy R. J. — «Canad. 842. Burns W. G., Bernstein H. J. — «J. Chem. Phys.»,
J Phys.», 1973, 51, 1664. 1950, 18, 1669.
Brown J M Byfleet С R., Howard B. /., Rus- 843. Burnside P. B. — «Dissert. Abstrs», 1959, 19,
sell D. K. - «Mol. Phys.», 1972, 23, 457. 3329.
Brown M. Wilson M. K. — «J. Chem. Phys.», 844. Burriel L. J. A., Cragg С. В., Rowlinson J. S. —
1954 22 ' 955. <(An- Real. Soc. esp. fis. у quim.», 1968, 64, 1.
Brown R. D., Burden F. R., Godfrey P. D., Gil- 845. Burrus C. A., Gordy W. — «Phys. Rev.», 1953,
lard I. R. — «J. Mol. Spectrosc», 1974, 52, 301. 92, 274.
Brown R. D., Pell J. B. — «Austral. J. Chem.», 846. Burrus С A., Gordy W.— «Phys. Rev.», 1953, 92,
Brown R. D.,' Williams G. R. — «Mol. Phys.», 847. Burrus C. A., Gordy W. — «Phys. Rev.», 1956,
1973 25 673 101i 599.
Brown R. D., Williams G. R. — «Chem. Phys.», 848. Burrus С A., Gordy W. — «J. Chem. Phys.», 1957,
1974 3 19 26, 391.
Brown R L James Т. С — «J. Chem. Phys.», 849. Burrus C. A., Gordy W.,. Benjamin В., Living-
1965 42* 33. ston R- — «Phys. Rev.», 1955, 97, 1661.
Brown r' L.,' Klemperer W. — «J. Chem. Phys.», 849a. Burt J. A., Dunn J. L., McEwan M. G., Sutton
1964 4l' 3072 M. M., Roche A. E., Schiff H. I. — «J. Chem.
Brown W. G. - «Phys. Rev.», 1931, 38, 709. Phys.», 1970, 52, 6062.
Brown W G — «Phys. Rev.», 1931, 38, 1179. 850. Busch G. E., Mahoney R., Wilson K. R. — «IEEE
Brown W G. - «Phys. Rev.», 1931, 38, 1187. J. Quantum Electron», 1970, 6, 171.
Brown W G - «Phys. Rev.», 1932, 39, 777. 851. Bushnell V. C, Hughes A. M., Gilbert E. С -
Brown W G — «Phys. Rev.», 1932, 42, 355. «J. Amer. Chem. Soc», 1937, 59, 2142.
Brown W.'G., 'Gibson G. E. - «Phys. Rev.», 1932, 852. Busing W. R., Levy H. A. - «J. Chem. Phys.»,
40 529 1965, 42, 3054.
Browne R /., Ogryzlo E. A. — «J. Chem. Phys.», 853. Butcher R. J., Jones W. J. — «l. Chem. Soc.
i97o 52 5774 Faraday Trans. II», 1974, 70, 560.
Brumant J L , Barbe A., Jouve P. — «C. r. Acad. 854. Butcher R. J., WillettsD. V., Jones W. J. — «Proc.
sci» 1969* B268, 549. Roy. Soc. London», 1971, A324, 231.
Br'undle С R Turner D. W. — «Proc. Roy Soc. 855. Buttenbender G., Herzberg G. — «Ann. Physik»,
London», '1968, A307, 27. 1935, 21, 577.
Brune^H., Pere, M. - «J. Mol. Spectrosc.», 1969, 856. Cabana^ ^^ fr^%\ ? 1902!
Brunem R., Ollano Z.-«Rend, reale Accad. 857. C.b^.^L^M^ Р^С.,^*, FF. /.-
naz. Lincei», 1931, W, 5<i. 85g> Cabannes / _ <,Ann> h 1933) t 229.
Buchler A., Stauffer J. L., Klemperer W. - «J. 859> Cadg p ?-_ «j. chem. phys.», 1967, 47, 2390.
Chem. Phys.», 1967, 46, bO5. 860_ Cade p ? _ «Canad. J. Phys.», 1968, 46, 1989.
BucktonK. S., LegonA.C, Millen O.J. — «Trans. 861. Cade P. E., Huo W. M. — «J. Chem. Phys.»,
Faraday Soc», 1969, 65, 1975. 1967, 47, 614.
Budininkas P., Edwards R. K., Wahlbeck P. G. — 862. Cade P. E., Huo W. M. — «J. Chem. Phys.»,
«J Chem. Phys.», 1968, 48, 2859. 1967, 47, 649.
Buiis К Schutte С. Т. Н. — «Spectrochim. acta», 863. Cadioli В., Patella P., Pincelll U., David D. J.
1962 18 307. tAUi Soc. natur. mat. Modena», 1969, 100, 47.
447
Литература
¦864. Cairns R. В., Harrison #., Schoen R. J. — «Appl. 892.
Opt.», 1970, 9, 605.
•864a. Colder G. V., Ruedenberg K. — «J. Chem. Phys.», 893.
1968, 49, 5399.
«65. Callear А. В., Pilling M. J. —«Trans. Faraday 894.
Soc», 1970, 66, 1618.
«66. Callear А. В., Wood P. M. — «Trans. Faraday 895.
Soc», 1971, 67, 3399.
«66a. Camy-Peyret C, Flaud J. M. — «Mol. Phys.», 896.
1976 32 523
«67. Camy-Peyret C, Flaud J. M. — «Spectrochim. 897.
acta», 1973, A29, 1711.
«68. Camy-Peyret C, Flaud J. M., Guelachvili G., 898.
Amoit С — «Mol. Phys.», 1973, 26, 825. 899.
869. Canjar L. N., Jonas C. R., Manning F. S. —« 900.
«Hydrocarbon Process», 1966, 45, 161.
870. Canjar L. N., Manning F. S. Thermodynamical 901.
Properties and Reduced Correlations for Gases.
Houston, 1967.
870a. Capitelli M. — «J. Plasma Phys.», 1972, 7, 99. 902.
¦8706. Capitelli M., Ficocelli V. E., Milinari E. — «Z.
Naturforsch.», 1971, 26a, 672. 903.
871. Cardillo M. J., Bauer S. H. — «Inorg. Chem.»,
1969, 8, 2086. 904.
872. Carette J. D., Kerwin L. — «Canad. J. Phys.»,
1961, 39, 1300. 904a.
873. Carleer M., Colin Д.— «J. Phys. B: Atom, and
Mol. Phys.», 1970, 3, 1715. 905.
874. Carlone C, Dalby F. W. — «Canad. J. Phys.»,
1969, 47, 1945. 906.
875. Carlotti M., Johns J. W. C, Frombetti A.— «Canad.
J. Phys., 1974, 52, 340. 907.
876. Carlotti M., Lonardo G., Di Galloni G., Trom-
betti A. — «Trans. Faraday Soc», 1971, 67, 2852. 908.
877. Carlson G. L. — «Spectrochim. acta», 1963, 19,
1291. 909.
878. Carlson G. L., Pederson L. G. — «J. Chem. Phys.»,
1975, 62, 4567. 909a.
879. Carlton-Sutton T. — «Philos. Mag.», 1932, 14,
275. 910.
880. Carlton-Sutton Т., Ambler H. R., Williams
G. W. — «Proc. Phys. Soc, London», 1936, A48, 911.
189.
881. Caron A., Donohuc /.—«Acta crystallogr.», 912.
1961, 14, 548.
882. Caron A., Donohuc J. —«Acta crystallogr.», 913.
1965, 18, 562.
882a. Carpenter G. B. — «Acta crystallogr.», 1952, 5, 914.
132.
8826. Carpenter G. B. — «Acta crystallogr.», 1955, 8, 915.
852.
883. Carpenter L. G., Harle T. F. — «Philos. Mag.», 916.
1937, 23, 193.
884. Carpenter R. A., Gailar N. M., Morgan H. W., 917.
Staats P. A. — «J. Mol. Spectrosc», 1972, 44, 197. 918.
885. Carrington A. Mol. Spectrosc. Proc. 4th Conf.
London, 1968, p. 157. 919.
886. Carrington A., Currie G. N., Dyer P. N.,
LevyD. #., Miller T. A. —«J. Chem. Soc. Chem. 920
Communs», 1967, 641.
887. Carrington A., Currie G. N., Miller T. A., Q9,
Levy D. H. — «J. Chem. Phys.», 1969, 50, 2726. УЛ1ш
888. Carrington A., Dyer P. N., Levy D. H. — «J. 922-
Chem. Phys.», 1967, 47, 1756.
889. Carrington A., Dyer P. N., Levy D. H.—«J. 923.
Chem. Phys.», 1970, 52, 309.
890. Carrington A., Howard B. J., Levy D. H., Robert- 924.
son /. C. — «Mol. Phys.», 1968, 15, 187.
891. Carrington A., Levy D. H.— «J. Chem. Phys.», 1966, 925.
44, 1298.
Carrington A., LevyD. H,— «J. Phys. Chem.», 1967.
71, 2.
Carrington A., Levy D. H., Miller T. A.— «Trans.
Faraday Soc», 1966, 62, 2994.
Carrington A., Levy D. H., Miller T. A.— «Proc.
Roy Soc London», 1966, A293, 108.
Carrington A., Levy D. H., Miller T. A.— «J.
Chem. Phys., 1967, 47, 3801.
Carrington A., Levy D. H., Miller T. A,— «Mol.
Phys.», 1967, 13, 401.
Carrington A., Levy D. H., Miller T. A.— «Proc.
Roy. Soc. London», 1967, A298, 340.
Carroll P. K.— «Canad. J. Phys.», 1959, 37, 880.
Carroll P. K. — «J. Chem. Phys.», 1962, 37, 805.
Carroll P. K. — «Proc. Roy. Soc. London», 1963,
A272, 270.
Carroll P. K., Collins C. C, Murnaghan J. T. —
«J. Phys. B: Atom, and Mol. Phys.», 1972, 5,
1634.
Carroll P. K., Doheny A. P. — «J. Mol. Spectrosc»,
1974, 50, 257.
Carroll P. K., Mitchell P. /.— «Proc Roy. Soc.
London», 1975, A342, 93.
Carroll P. K., Mulliken R. S. — «J. Chem. Phys.»,
1965, 43, 2170.
Carroll P. K., Nulty A. T. — «J. Phys. B: Atom.
and Mol. Phys.», 1976, 9, L427.
Carroll P. K., Sayers N. D. — «Proc. Phys. Soc.
London», 1953, A66, 1138.
Carroll P. K., Subbaram K. V. — «Canad. J.
Phys.», 1975, 53, 2198.
Carsky P., Machacen M. — «Collect. Czech. Chem.
Commun.», 1973, 38, 3067.
Carter R. P., Holmes R. R. — «Inorg. Chem.»,
1965, 4, 778.
Cartwright D. C, Dunning Т., jr. — «J. Phys. B:
Atom, and Mol. Phys.», 1975, 8, L100.
Cartwright M., Woolf A. A. — «J. Fluor. Chem.»,
1977, 9, 495.
Cashion J. K., Polanyi J. C. — «J. Chem. Phys.»,
1959, 30, 317. л
Catalano E., Sanborn R. H., Frazer J. W. — «J.
Chem. Phys.», 1963, 38, 2265.
Cathbertson A. C, Matheson G. L., Maass O. —•
«J. Amer. Chem. Soc», 1928, 50, 1120.
Cauch E. /., Kobe K. A. — «J. Chem. and Engng
Data», 1961, 6, 229.
Caunt A. D., Barrow R. F. — «Nature», 1949,
164, 753.
Caunt A. D., Barrow R. F.—«Trans. Faraday
Soc», 1950, 46, 154.
Cawthon T. M., McKinley J. D. — «J. Chem.
Phys.», 1956, 25, 585.
Cazzoli G. — «J. Mol. Spectrosc», 1974, 53, 37.
Cazzoli <?., Cervellati R., Mirri A. M. — «J. Mol.
Spectrosc.», 1975, 56, 422.
Celotta R. J., Bennett R. A., Hall J. L. — «J.
Chem. Phys.», 1974, 60, 1740.
Celotta R. J., Bennett R. A., Hall J. L., Siegel
M. W., Levine J. — «Phys. Rev.», 1972, A6, 631.
Centnerszwer M. — «Z. phys. Chem.», 1913, 85, 99.
Cermak V., Herman Z. — «Collect. Czech. Chem.
Communs», 1962, 27, 1493.
Cerquetti A., Longhi P., Mussini T.— «J. Chem.
and Engng Data», 1968, 13, 458.
Chackalackal S. M., Stafford F. E. — «J. Amer.
Chem. Soc», 1966, 88, 723.
Chamberlain J. W. — «Astron. J.», 1954, 59,
N 5, 183.
448
Литература
926. Chamberlain J. W., Roesler F. L. — «Astrophys.
J.», 1955, 121, 541.
926a. Chambers F. S., Sherwood Т. К. — «J. Amer.
Chem. Soc», 1937, 59, 316.
927. Chang J. C. — «Chem. Phys. Lett.», 1975, 36,
611.
928. Chang S., Pal K. H., Pai K. W., Mu S. J., Ahn
W. S. — «J. Korean Chem. Soc», 1964, 8, 33.
929. Chang S., Pai K. #., Pai K. W., Mu S. J., Ahn
W. S. — «J. Korean Chem. Soc», 1964, 8, 65.
930. Chang S., Pai K. H., Pai K. W., Mu S. J., Ahn
W. S. — «J. Korean Chem. Soc», 1964, 8, 121.
931. Chang S., Pai K. #., Pai K. W., Mu S. J., Ahn
W. S. — «J. Korean Chem. Soc», 1964, 8, 179.
932. Chang S., Pai K. H., Pai K. W., Mu S. J., Ahn
W. S. — «J. Korean Chem. Soc», 1964, 8, 183.
933. Chantry G. W., Ewing V. С — «Mol. Phys.», 1962,
5, 209.
934. Chantry G. W., Gebbie H. A., Anderson A. —
«Spectrochim. acta», 1964, 20, 1223.
935. Chantry P. J. — «J. Chem. Phys.», 1971, 55, 2746.
936. Chao J., Wilhoit R. C, Zwolinski B. J. — «J.
Chem. Thermodyn.», 1971, 3, 195.
937. Chao J., Wilhoit R. C, Zwolinski B. J. — «J.
Chem. Thermodyn.», 1971, 3, 497.
938. Chao J., Wilhoit R. C, Zwolinski B. J. — «Thermo-
chim. acta», 1974, 10, 359.
939. Chapman A. C. — «Spectrochim. acta», 1968,
A24, 1687..
940. Chapman G. D., Bunker P. R. — «J. Chem. Phys.»,
1972 57 2951.
941. Chappuis J. — «C. r. Acad. sci.», 1880, 91, 985.
942. Charnley Т., Skinner H. Л. — «J. Chem. Soc»,
1953, 450.
942a. Chase M. W., Curnutt J. L., Ни А. Т., Prophet
#., Syverud A. N., Walker L. С JANAF Thermo-
chemical Tables, 1974, Supplement. «J. Phys.
and Chem. Ref. Data», 1974, 3, 311.
943. Chassagne C, Grenier-Besson M. L., Вargues D. —
«J. phys.», 1976, 37, 71.
944. Chedin J. — «С. г. Acad. sci.», 1936, 203, 772.
945. Chenappe K. M., Pendlebury J. M., Smith K. F.
Colloq. intern. CNRS, Paris 22—24 juin 1966,
N 164, p. 73, Puhl. CNRS, 1967.
945a. Chernick С L. — «Chemistry», 1964, 37, 6.
946. Chi C. K., Nicol M. — «Chem. Phys. Lett.», 1973,
21 533
947. Chi С. К., Nixon E. R. — «J. Phys. Chem. Solids»,
1972, 33, 2101.
948. Chihara H., Nakamura M., Masukane K. — «Bull.
Chem. Soc. Japan», 1973, 46, 97.
949. Child M. S. — «J. Mol. Spectrosc», 1973, 45,
293
950. Child M. S., Bernstein R. B. — «J. Chem. Phys.»,
1973, 59, 5916.
951. Childs W. H. T. — «Proc. Roy. Soc. London»,
1932, A137, 641.
952. Chin D., Gignere P. A. — «J. Chem. Phys.», 1961,
34, 690.
953. Chivers Т., Drummond J. — «Chem. Soc. Rev.»,
1973 2 233
954. Chong S.-L., Myers R. A. jr., Franklin J. L. —
«J. Chem. Phys.», 1972, 56, 2427.
955. Christe K. O., Curtis E. C, Pilipovich D. —
«Spectrochim. acta», 1971, A27, 931.
955a. Christe K. O., Curtis E. C, Schack C. J. —
«Spectrochim. acta», 1975, A31, 1035.
9556. Christe K. O., Curtis E. C, Schack C. J., Cyvin
S. J., Brunvoll J., Sawodny W. — «Spectrochim.
acta», 1976, A32, 1141.
956. Christe K. O., Curtis E. C, Schack C. J.,
Pilipovich D. — «Inorg. Chem.», 1972, 11, 1679.
957. Christe K. O., Sawodny W. — «J. Chem. Phys.»,
958. Christe K. O., Sawodny W. — «Z. anorg und
allgem. Chem.», 1968, 357, 125.
959. Christe K. O., Sawodny W., Pulay P. — «J. Mol.
Struct.», 1974, 21, 158.
960. Christe K. O., Schack С J., Wilson R.D. — «Inoro
Chem.», 1974, 13, 2811.
961. Christoffersen R. E. — «J. Chem. Phys.», 1964,
41, 960.
962. Christy A., Nande S. M. — «Phys. Rev.», 1931,
37, 903.
963. Chue P. L., Prausnitz J. M. — «AIChE Journal»,
1967, 13, 896.
964. Chupka W. A., Russell M. Ё. — «J. Chem. Phys »
1968, 48, 1527. * '
964a. Chupka W. A., Russell M. E., Refaey K.—<tl.
Chem. Phys.», 1968, 48, 1518.
965. Chutjian A. — «J. Chem. Phys.», 1969, 51, 5414.
966. Chutjian A., Hall R. I., Trajmar S. — «J. Chem
Phys.», 1975, 63, 892.
967. Chutjian A., Segal G. A. — «J. Chem. Phys.»,
1972, 57, 3069.
967a. Claassen H. H., Chernick С L., Malm J. G. —
«J. Amer. Chem. Soc», 1963, 85, 1927.
9676. Claassen H. H., Chernick С L., Malm J. G. —
In: Noble Gas Compounds. H. H. Hyman (Ed.).
Chicago^Uniy.^Press, ^1963^ P^287.
967b. Claassen H. H., Gasner*E. L., Kim .^
J. L. — «J. Chem. Phys.», 1968, 49, 253.
968. Claassen H. H., Gasner E. L., Selig H. — «J
Chem. Phys.», 1968, 49, 1803.
969. Claassen H. H., Goodman G. L., Holloway J. H.,
Selig H. — «J. Chem. Phys.», 1970, 53, 341.
969a. Claassen H. H., Goodman G. L., Kim H. — «J.
Chem. Phys.», 1972, 56, 5042.
9696. Claassen H. H., Goodman G. L., Malm J. G.,
Schreiner F. — «J. Chem. Phys.», 1965, 42, 1229.
969b. Claassen H. H., Huston J. L. — «J. Chem. Phys!»
1971, 55, 1505.
969r. Claassen H. H., Knapp G. — «J. Amer. Chem
Soc», 1964, 86, 2341.
970. Claassen H. H., Selig H., Shamir J. — «Appl.
Spectrosc», 1969, 23, 8.
971. Claassen H. H., Weinstock В., Malm J. G. — «J.
Chem. Phys.», 1958, 28, 285.
972. Clark A. H., «J. Mol. Struct.», 1971, 7, 485.
973. Clark A. H., Beagley B. — «J. Chem. Soc», A,
1970, 46.
974. Clark A. H., Beagley B. — «Trans. Faraday
Soc», 1971, 67, 2216.
975. Clark С H. D. — «Trans. Faraday Soc», 1940,
36, 370.
976. Clark R. J. H., Rippon D. M. — «J. Mol.
Spectrosc», 1974, 52, 58.
977. Clark R. J. H., Rippon D. M. — «Mol. Phys.»,
1974, 28, 305.
978. Clark T. C, Clyne M. A. — «Trans. Faraday Soc»,
1970, 66, 877.
979. Claxton T. A. — «Trans. Faraday Soc», 1970, 66,
1537.
980. Clayton L., Williams Q., Weatherly T. L. — «J.
Chem. Phys.», 1959, 30, 1328.
981. Clayton L., Williams Q., Weatherly T. L. — «J.
Chem. Phys.», 1959, 31, 554.
982. Cleaves A. P., Edwards С W. — «Phys. Rev.»,
1935, 48, 850.
449
Литература
983. Clement M. J. V., Ramsay D. A.— «Canad. 1012.
J. Phys.», 1961, 39, 205.
984. Clifton D. G. — «J. Ghem. Phys.», 1964, 41, 3656. 1013.
985. Clough P. N., Thrush B. A., Ramsay D. A.,
Stamper J. L. — «Chem. Phys. Lett.», 1973, 23, 155. 1014.
986. Clough S. A., Kneizys F. X. — «J. Chem. Phys.»,
1966, 44, 1855. 1015.
987. Clyne M. A. A., Coxon J. A. — «J. Chem. Soc.
Chem. Communs», 1966, 285. 1016.
988. Clyne M. A. A., Coxon J. A. — «Proc. Roy. Soc.
London», 1967, A298, 424. 1017.
989. Clyne M. A. A., Coxon J. A. — «J. Mol. Spectrosc», 1018.
1967, 23, 257. 1019.
990. Clyne M. A. A,, Coxon J. A. — «Nature», 1968,
217, 448. 1020.
991. Clyne M. A. A., Coxon J. A. — «J. Mol. Spectrosc.»
1970, 33, 381. 1021.
992. Clyne M. A. A., Coxon J. A.—«J. Phys. B:
Atom, and Mol. Phys.», 1970, 3, L9. 1022.
993. Clyne M. A. A., Coxon J. A., Townsend L. M.—
«J. Chem. Soc. Faraday Trans. II», 1972, 2134. 1023.
993a. Clyne M. A. A., Coxon J. A., Woon-Fat A. R. —
«Trans. Faraday Soc», 1971, 67/3155. 1024.
994. Clyne M. A. A., Coxon J. A., Woon-Fat A. R.—
«J. Mol. Spectrosc», 1973, 46, 146. 1025.
994a. Clyne M. A. A., Curran A. H., Coxon J. A. — «J.
Mol. Spectrosc», 1976, 63, 43. 1026.
9946. Clyne M. A. A., McDermid I. S. — «J. Chem.
Soc Faraday Trans. II», 1976, 72, 2252. 1027.
995. Clyne M. A. A., McKenney D.Y., Thrush B. A. —
«Trans. Faraday Soc», 1965, 61, 2701.
996. Clyne M. A. A., Thrush B. A. — «Disc. Faraday 1028.
Soc», 1962, N 33, 139.
997. Clyne M. A. A., Watson R. T. — «Chem. Phys. 1029.
Lett.», 1971, 12, 344.
998. Cobb J. C, Hinchliffe A. — «Chem. Phys. Lett.», 1030.
1974, 24, 75.
999. Coblentz W. W. Investigation of Infra-red Spectra. 1031.
Washington, 1905.
1000. CODATA Recommended Key Values for Thermo- 1032.
dynamics: 1971, CODATA Bull., N 5; 1973,
CODATA Bull., N 10; 1975, CODATA Bull., 1033.
N 17; 1976, CODATA Bull., N 22 (см.: «J. Chem.
Thermodyn.», 1976, 8, 603); 1977, CODATA, 1034.
Bull., N 28.
1001. CODATA Recommended Consistent Values of 1035.
the Fundamental Physical Constants, 1973,
CODATA Bull., N 11; Фундаментальные физи- 1036.
ческие константы. М., Изд-во стандартов, 1976.
1002. Codling К. —«J. Chem. Phys.», 1966, 44, 4401. 1037.
1003. CohenE. A., Poynter R. L. — «I. Mol. Spectrosc», 1038.
1974, 53, 131. 1039.
1004. Cohen E. R., Du Mond J. W. M. — «Revs Mod.
Phys.», 1965, 37, 537. 1040.
1005. Cohen S., Hiskes J. R., Riddell R. J. — «Phys. 1041.
Rev.», 1960, 119, 1025.
1006. Cohen-Adad R., Tranquard A., Marchand A. — 1041a.
«Bull. Soc chim. France», 1968, N 1, 65.
1007. Cohn H., Ingold С. К., Poole H. G. — «J. Chem. 1042.
Soc», 1952, 4272.
1008. Colbourn E. A., Dagenais M., Douglas A. E., 1043.
Raymonda J. W.— «Canad. J. Phys.», 1976, 54,
1343. 1044.
1009. Colburn С. В., Johnson F. A., Haney С — «J.
Chem. Phys.», 1965, 43, 4526. 1045.
1010. Colburn С. В., Johnson F. A., Kennedy A.,
McCullum K., Metzger L., Parker C. — «J. 1046.
Amer. Chem. Soc», 1959, 81, 6397.
1011. Colburn С. В., Kennedy A. — «J. Amer. Chem. 1047.
Soc», 1958, 80, 5004.
Cole L. G., Elverum G. W. — «J. Chem. Phys.»,
1952, 20, 1543.
Cole L. G., Farber M., Elverum G. W. — «J.
Chem. Phys.», 1952, 20, 586.
Cole L. G., Gilbert E. С — «J. Amer. Chem.
Soc», 1951, 73, 5423.
Coleman E. H., Gay don A. G. — «Disc. Faraday
Soc», 1947, 166.
Coleman E. H., Gaydon A. G., Vaidya W. M. —
«Nature», 1948, 162, 108.
Colin R. — «Canad. J. Phys.», 1968, 46, 1539.
Colin R. — «Canad. J. Phys.», 1969, 47, 979.
Colin R., Devillers J., Prevot F. — «J. Mol.
Spectrosc», 1972, 44, 230.
Colin R., Douglas A. S. — «Canad. J. Phys.»,
1968, 46, 61.
Colin R., Goldfinger P., Jeunehomme M. — «Trans
Faraday Soc», 1964, 60, 306.
Coll R. J. Dissertation. Wisconsin, 1959. —
«Dissert. Abstrs», 1959, 19, 1930.
Collin J. E., Lossing F. D. — «J. Chem. Phys.»,
1958, 28, 900.
Collin J. E., Natalis P. J. — «Chem. Phys.
Lett.», 1968, 2, 194.
Collin J. E., Natalis P. J. — «Intern. J. Mass
Spectrom. and Ion Phys.», 1968, 1, 483.
Collin J. E., Natalis P. J. — «Intern. J. Mass.
Spectrom. and Ion Phys.», 1969, 2, 231.
Colussi A. J., Morton J. R., Preston K. F., Fes-
senden R. W. — «J. Chem. Phys.», 1974, 61,
1247.
Colwell J. H. Dissertation. Washington, 1961. —
«Dissert. Abstrs», 1962, 22, 3004.
Combs L. L., Holloman M. •— «Spectrosc Lett.»,
1972, 5, 319.
Comeford J. J., Mann D. E. — «Spectrochim.
acta», 1965, 21, 197.
Comeford J. J., Mann D. E., Schven L. J.,
Lide D. R. jr. — «J. Chem. Phys.», 1963, 38, 461.
Companion A. L. — «Theor. chim. acta», 1972,
25, 268.
Companion A. L., Hsia Y. P. — «J. Mol. Struct.»,
1972, 14, 117.
Compton R. N., Cooper CD. — «J. Chem. Phys.»,
1973, 59, 4140.
Compton R. N., Huebner R. H. — «Adv. Radiat.
Chem.», 1970, 2, 281.
Condrate R. A., Nakamoto K. — «Bull. Chem.
Soc. Japan», 1966, 39, 1108.
Conroy H. — «J. Chem. Phys», 1964, 41, 1341.
Conroy H. — «J. Chem. Phys.», 1969, 51, 3979.
Conway D. С — «J. Chem. Phys.», 1962, 36,
2549.
Cook R. L. — «J. Chem. Phys.», 1965, 42, 2927.
Cook R. L., De Lucia F. C, Helminger P. — «J.
Mol. Spectrosc», 1972, 41, 123.
Cook R. L., De Lucia F. C, Helminger P. —
«J. Mol. Struct., 1975, 28, 237.
Cook R. L., Winnewisser G., Lindsey D. C. —
«J. Mol. Spectrosc.» 1973, 46, 276.
Coon J. В., Cesani F. A., Huberman F. P. —
«J. Chem. Phys.», 1970, 52, 1647.
Coon J. В., Cesani F. A., Loyd С. М. — «Disc.
Faraday Soc», 1963, 35, 118.
Coon J. В., Naugle N. W., McKenzie R. D. —
«J. Mol. Spectrosc», 1966, 20, 107.
Coon J. В., Ortiz E. — «J. Mol. Spectrosc»,
1957, 1, 81.
Cooper A. S., Bond W. L., Abrahams S. C. —
«Acta crystallogr.», 1961, 14, 1008.
450
Литература
1048. Coppens P., Smoes S., Drowart J. — «Trans.
Faraday Soc», 1967, 63, 2140.
1049. Copwell R. J., Rosenblatt G. M. — «J. Mol.
Spectrosc», 1970, 33, 525.
1050. Coquart В., Couet C, Guenebant H., Larzilliere M.,
Ngo T. A. — «Canad. J. Phys.», 1972, 50, 1014.
1051. Coquart В., Couet C, Ngo T. A., Guenebant H. —
«J. chim. phys. et phys.-chim. biol.», 1967, 64,
1197.
1052. Coquart В., Da Paz M., Prudhomme J. С —
«Canad. J. Phys.», 1974, 52, 177.
1053. Coquart В., Da Paz M., Prudhomme J. С —
«Canad. J. Phys.», 1975, 53, 377.
1054. Coquart В., Larzilliere M., Ngo T. A. — «Canad.
J. Phys.», 1972, 50, 2945.
1055. Coquart В., Ngo T. A., Couet C— «C. r. Acad.
sci.», 1970, C271, 708.
1056. Coquart В., Ngo T. A., Couet C, Guenebaut H. —
«C. r. Acad. sci.», 1969, B269, 1242.
1057. Coquart В., Ngo T. A., Couet C, Guenebaut H. —
«C. r. Acad. sci.», 1970, C270, 150.
1058. Coquart В., Ngo T. A., Couet C, Guenebaut И. —
«С. г. Acad. sci.», 1970, C270, 776.
1059. Coquart В., Ngo T. A., Couet C., Guenebaut H. —
«C. r. Acad. sci.», 1970, B270, 1227.
1060. Coquart В., Ngo T. A., Couet C, Guenebaut H. —
«С. r. Acad. sci.», 1970, C270, 1702.
1061. Coquart В., Ngo T.A., Larzielliere M., Couet C. —
«C. r. Acad. sci.», 1971, B273, 384.
1062. Coquart В., Prudhomme J. С — «С. г. Acad.
sci.», 1972, B275, 383.
1063. Cordes И. — «Z. Phys.», 1932, 74, 34.
1064. Cordes H. — «Z. Phys.», 1937, 105, 251.
1065. Cordes #., Sponer H. — «Z. Phys.», 1930, 63, 334.
1066. Cordes H., Sponer H. — «Z. Phys.», 1932, 79, 170.
1067. Cordes H., Warkehr E. — «Z. phys. Chem.» (BRD),
1965, 46, 26.
1068. Cordes H., Witschel W. — «Z. phys. Chem.»
(BRD), 1965, 46, 35.
1069. Cordes H. F., Johnston H. S. — «J. Amer. Chem.
Soc», 1954, 76, 4264.
1070. Corice R. J., Fox K., Tejwani G. D. T. ¦— «J.
Chem. Phys.», 1973, 58, 265.
1071. Corice R. J., Fox K., Tejwani G. D. T. — «J.
Chem. Phys.», 1973, 59, 672.
1072. Cornford А. В., Frost D. C, McDowell С. А.,
Ragle J. L., Stenhouse I. A. <— «Chem. Phys.
Lett.», 1970, 5, 486.
1073. Coster D., Brons H.— «Z. Phys.», 1931, 70, 492.
1074. Coster D., Brons H. — «Z. Phys.», 1932, 73, 747.
1074a. Cotter R. J., Rozett R. W., Roski W. S. — «J.
Chem. Phys.», 1972, 57, 4100.
1075. Cotton F. A., George J. W-, Waugh J. S. ~- «J.
Chem. Phys.», 1958, 28, 994.
1076. Cottrell T. L. The strength of chemical bonds.
London, Butterworths Sci. Publ., 1954.
1077. Couet C, Coquart В., Ngo T. A., Guenebaut H. —
«C. r. Acad. sci.», 1967, B265, 134.
1078. Couet C, Coquart В., Ngo T. A., Guenebaut H. —
«C. r. Acad. sci.» 1967, B265, 479.
1079. Couet C, Coquart В., Ngo T. A., Guenebaut H. —
«C. r. Acad. sci.», 1968, B266, 1219.
1080. Couet C., Coquart В., Ngo T. A., Guenebaut H. —
«J. chim. phys. et phys.-chim. biol.», 1968, 65,
1241.
1081. Couet C, Gunebaut H. — «C. r. Acad. sci.», 1966,
B263, 158.
1082. Couet C, Ngo T. A., Coquart В., Guenebaut H. —
«J. chim. phys et phys.-chim. biol.», 1968, 65, 217.
1083. Coughlin J. P. — «J. Amer. Chem. Soc», 1958,
80, 1802.
1084. Coulson C. A. <— «Cambridge Philos. Soc», 1935,
31, 244.
1085. Couture-Mathieu L., Poulet H., Mathieu J. P. ¦—
«С. г. Acad. sci.» 1952, 234, 1761.
1086. Couture-Mathieu L., Mathieu J. P. — [«J. Chem.
Phys.», 1952, 49, 226.
1087. Couzi M., Le Calve J., Van Huong P., Lascombe
J. — «J. Mol. Struct.», 1970, 5, 363.
1088. Cowan M. J. — «Dissert. Abstrs», 1960, 20, 4139.
1089. Cowan M. J., Gordy W. — «Phys. Rev.», 1956,
104, 551.
1090. Cowan M. J., Gordy W. — «Bull. Amer. Phys.
Soc», 1957, 2, 212.
1091. Cowan M. J., Gordy W. — «Phys. Rev.», 1958,
111, 209.
1092. Cowley A. H., White W. D., Damasco M. С. ~
«J. Amer. Chem. Soc», 1969, 91, 1922.
1093. Cowling T. — «Nature», 1943, 152, 694.
1094. Cox A. P., Brittain A. H., Finnigan D. J. ~
«Trans. Faraday Soc», 1971, 67, 2179.
1095. Cox A. P., Kuczkowski R. L. <— «J. Amer. Chem.
Soc», 1966, 88, 5071.
1096. Cox A. P., Riveros J. M. <— «J. Chem. Phys.»,
1965, 42, 3106.
1097. Cox J. D., Harrop D. — «Trans. Faraday Sock
1965, 61, 1328.
1098. Cox J, D., Lawrenson I. J. — «Chem. Thermodyn.»,
1973, 1, 162.
1099. Cox J. ?>., Pitcher G. Thermochemistry of Organic
and Organometallic Compounds.1 London*—New
York, Acad. Press, 1970.
1100. Coxon J. A. — «J Mol. Spectrosc», 1971, 37, 39.
1101. Coxon J. A. — «J. Mol. Spectrosc», 1972, 41,
548.
1102. Coxon J. A. — «J. Mol. Spectrosc», 1972, 41, 566.
1103. Coxon J. A. — «J. Mol. Spectrosc», 1974, 50,
142.
1104. Coxon J. A. — «Chem. Phys. Lett.», 1975, 33, 136.
1105. Coxon J. A. — «J. Mol. Spectrosc», 1975, 58, 1.
1106. Coxon J. A. — «J. Photochem.», 1976, 5, 337.
1107. Coxon J. A., Jones W. E., Skolnik E. G. — «Canad.
J. Phys.», 1976, 54, 1043.
1108. Coxon J. A., Ramsay D. A. — «Canad. J. Phys.»,
1976, 54, 1034. '
1109. Crable G. F., Smith W. V. — «J. Chem. Phys.»,
1951, 19, 502.
1110. Craig N. С — «J. Phys. Chem.», 1971, 75, 1453.
1111. Crowford B. L., Cross P. S. — «J. Chem. Phys.»,
1937, 5, 621.
1112. Crowford F. H., Tsai P. M. — «Proc Amer.
Acad. Arts and Sci.», 1935, 69, 407.
1113. Creek D. M., Nicholls R. W. — «Proc. Roy. Soc.
London», 1974, A341, 517.
1114. Creighton J. A., Lippincott E. R. <— «J. Chem.
Phys.», 1964, 40, 1779.
1115. Crenshaw J. L., Ritter I. — «Z. phys. Chem.»,
1932, B16, 143.
1116. Creutzberg F. — «Canad. J/ Phys.», 1G66, 44,
1583.
1117. Cristy S. S., Mamantov G. >— «Intern. J. Mass
Spectrom. and Ion Phys.», 1970, 5, 319.
1118. Cross P. C. — «Phys. Rev.», 1935, 47, 7.
1119. Cross P. С — «J. Chem. Phys.», 1935, 3, 168.
1119a. Cross P. С — «J. Chem. Phys.», 1935, 3, 825.
1120. Cross P. C, Brockway L. O. — «J. Chem. Phys.»,
1935, 3, 821.
1121. Cruickskank D. W. J. — «Acta crystallogr.»,
1956, 9, 754.
451
Литература
1122. Cmickshank D. W. J. — «Acta crystallogr.», 1158.
1964, 17, 677.
1123. Csizmadia I. G., Kari R. E., Polanyi J. C, 1159.
Roach A. C, Robb M. A. — «J. Chem. Phys.»,
1970, 52, 6205. 1160.
1124. Cubicciotti ?>. — «J. Chem. Phys.», 1959, 31,
1646. 1161.
1125. Cubicciotti D. — «J. Chem. Phys.», 1961, 34,
2189. 1162.
1126. Camming C, Welsh H. L. — «J. Chem. Phys.»,
1953, 21, 1119. 1163.
1127. Cupp R. E., KempfR. A., Gollacherl. I. — «Phys.
Rev.», 1968, 171, 60. 1164.
1128. Curl R. F. — «J. Chem. Phys.», 1962, 37, 779.
1129. Curl R. P., Heidelberg R. F., Kinsey J. L. — 1165.
«Phys. Rev.», 1962, 125, 1993. 1166.
1130. Curl R. F., Kinsey J. L., Baker J. G., Braid J. C,
Bird G. R., Heidelberg R. F., Sugden Т. М., 1167.
Jenkins D. R., Kenney С N. — «Phys. Rev.»,
1961, 121, 1119. 1168.
1131. Curl R. F., Koichi A., Bissinger J., Bennett C,
Tittel F. K. — «J. Mol. Spectrosc», 1973, 48, 72. 1169.
1132. Curran R. K. — «J. Chem. Phys.», 1961, 34, 1069.
1133. Curran R. K. — «Phys. Rev.», 1962, 125, 910. 1170.
1134. Curran R. K. — «J. Chem. Phys.», 1963, 38, 1171.
|2974.
1135. Curran R. K., Fox R. Я. — «J. Chem. Phys.», 1172.
1961, 34, 1590.
1136. Carry J., Herzberg G. — «Ann. Physik», 1934, 1173.
19, 800.
1137. Curry J., Herzberg L., Herzberg G. — «J. Chem. 1174.
Phys.», 1933, 1, 749.
1138. Curry J., Herzberg L., Herzberg G. — «Z. Phys.», 1175.
1933, 86, 348.
1139. Curtis J. B. — «Dissert. Abstrs», 1975, B35, 1176.
5576.
1140. Curtis E. C, Pilipowich D., Moberly W. H. — 1177.
«J. Chem. Phys.», 1967, 46, 2904.
1141. Curtiss L. A., Pople J. A. — «J. Mol. Spectrosc», 1178.
1973, 48, 413.
1142. Curtiss L. A., Pople J. A. — «J. Mol. Spectrosc.», 1179.
1975, 55, 1.
1143. Curtiss L. A., Pople J. A. — «J. Mol. Spectrosc», 1180.
1976, 61, 1.
1144. Curtis W. E., Patkowsi J. — «Philos. Trans. 1181.
Roy. Soc. London», 1934, A232, 395.
1145. Curtis W. E., iTalansky S. — «Proc. Univ. 1182.
Durham Philos. Soc», 1931, 8, 323.
1146. Cyvin S. J., Brunuoll J., Robiette A. G. — «J. 1183.
Mol. Struct.», 1969, 3, 259.
1147. Cyvin S. J., Devarajn V., Brunvoll J., Ra O. — 1184.
«Z. Naturforsch.», 1973, 28a, 1787.
1148. Czarny J., Felenbok P. — «Ann. astrophys.», 1968, 1185.
31, 141.
1149. Czerny M. — «Z. Phys.», 1927, 45, 476. 1186.
1150. Daasch L. W., Smith D. С — «Analyt. Chem.», 1187.
1951, 23, 853.
1151. Dadien A., Jele J., Kohlrausch K. W. F. — 1188.
«Wien. Ber.», 1931, 140, 293.
1152. Dailey B. P., Golden S., Wilson E. B. — «Phys. 1188a.
« Rev.», 1947, 72, 871.
1153. Dainton F. S., Kimberley H. M. — «Trans. 1189.
Faraday Soc», 1950, 46, 912. 1190.
1154. Dalby F. W. — «Canad. J. Phys.», 1958, 36, 1336.
1155. Dalby F. W., Douglas A. E. — «Phys. Rev.»,
1951, 84, 843. 1191.
1156. Dalby F. W., Nielsen H. H. — «J. Chem. Phys.», 1192.
1956, 25, 934.
1157. Dana V. — «Rev. Phys. Appl.», 1974, 9, 1193.
711.
Dana V., Fontanella J. C. — «Nouv. rev. opt »
1973, 4, 237.
Dana V., Fontanella J. С — «Mol. Phys.», 1975,
30, 1473.
Daniels F., Bright A. C. — «J. Amer. Chem. Soc»,
1920, 42, 1131.
Daniels J. M., Dorain P. B. — «Bull. Amer.
Phys. Soc», 1964, 9, 682.
Daniels J. M., Dorain P. B. — «J. Chem. Phys.»,
1964, 40, 1160.
Daniels J. M., Dorain P. B. — «J. Chem. Phys.»,
1966, 45, 26.
Danti A., Lord R. С — «J. Chem. Phys.», 1959,
30, 1310.
Darbyshire O. — «Phys. Rev.», 1932, 40, 366.
Darby shire O. — «Proc. Roy. Soc. London»,
1937, A159, 93.
Darling В., Dennison D. — «Phys. Rev.», 1940,
57, 128.
Darling В. Т., Lui Chung-Wang — «J. Mol.
Spectrosc», 1966, 21, 146.
Das T. R., Kuloor N. R. — «J. Indian Inst.
Sci.», 1968, 50, 13.
Date K. — «Rev. Phys. Chem. Japan», 1973, 43, 1.
Date K. — «Rev. Phys. Chem. Japan», 1973, 43,
17.
Datta S., Chakravarty В.— «Proc. Nat. Inst. Sci.
India», 1941, 7, 297.
Datta S., Kundu D. N. — «Proc. Nat. Inst.
Sci. India», 1941, 7, 311.
Dauies M. M. — «Trans. Faraday Soc», 1939
35, 1184.
Davies M., Jonathan N. — «Trans. Faraday Soc»,
1958, 54, 469.
Davies P. В., Russell D. K., Thrush B. A. —
«Chem, Phys. Lett.», 1975, 36, 280.
Davies P. В., Russell D. K., Thrush B. A. —
«Chem. Phys. Lett.», 1976, 37, 43.
Davies P. В., Russell D. K., Thrush B. A. —
«Chem. Phys. Lett.», 1976, 42, 35.
Davies P. В., Thrush B. A. — «Proc. Roy. Soc.
London», 1968, A302, 243.
Davies P. В., Wayne F. D., Stone A. J.— «Mol.
Phys.», 1974, 28, 1409.
Davis A. R., Macklin J. W., Plane R. A. — «J.
Chem. Phys.», 1969, 50, 1478.
Davis С. О., Johnston H. L. — «J. Amer. Chem.
Soc», 1934, 56, 1045.
Davis P. W., Oetjen R. A. — «J. Mol. Spectrosc»,
1958, 2, 253.
Dawe R. A., Snowdon P. N. — J. Chem. Soc.
Faraday Trans. I», 1974, 70, 1145.
Dawe R. A., Snowdon P. N. — «Physica», 1974,
74, 435.
Dawson B. — «Acta crystallogr.», 1960, 13, 409.
Dawson D. H., Johnston H. L. — «Phys. Rev.»,
1933, 43, 980.
De Alti G., Costa G., Galasso V. —«Spectrochim.
acta», 1964, 20, 965.
Dean С R. S., Finch A., Gardner P. J. —
«J. Chem. Soc Faraday Trans I», 1974, N 11,1921.
Debye P. P. — «Ann. Physik», 1912, 39, 789.
De Corpo J. J., Steiger R. P., Franklin J. L.,
Margrave J. L. — «J. Chem. Phys.», 1970, 53,
936.
Decker H. С J. de. Dissertation. Utrecht, 1941.
Decker H. C. J. de. — «Recueil trav. chim.»,
1941, 60, 413. '
Decker H. С J. de., Mac Gillavry С. Н. —
«Recueil trav. chim.», 1941, 60, 153.
452
Литература
1194. Deezsi I. — «Acta phys. Acad. scient. hung.»,
1958, 9, 125.
1195. Deezsi I. — «Acta phys. Aead. scient. hung.»,
1960, 11, 155.
1196. Deezsi I., Matrai T. — «Acta phys. Acad.scient.
hung.», 1957, 7, 111.
1197. Deezsi /., Matrai T. — «Magyar fiz. folyoirat.»,
1957, 5, 201.
1198. Degen V. — «Canad. J. Phys.», 1968, 46, 783.
1199. Degen V. — «Canad. J. Phys.», 1968, 46, 2850.
1200. Degen V., Nicholls R. W. — «Proc. Phys. Soc.
London», 1968, Bl, 983.
1201. Deglise X., Giguere P. A. — «Canad. J. Chem.»,
1971, 49, 2242.
1202. Deiters R. M., Holmes R. R. — «J. Chem. Phys.»,
1968, 48, 4796.
1203. De Hemptinne M., Van Riet R. — «Bull. cl. sci.
Acad. roy. Belgique», 1964, 49, 856.
1204. Dehmer P. M., Chupka W. A. — «J. Chem.
Phys.», 1975, 62, 4525.
1205. De Lattre A. — «J. Chem. Phys.», 1952, 20, 520.
1206. Del Bene J. E. — «J. Chem. Phys.», 1971, 54,
3487.
1207. Del Bene J. E., Pqple J. A. —«1. Chem. Phys.»,
1971, 55, 2296.
1208. De Lucia F. C, Cook R. L., Helminger P.,
Gordy W. — «J. Chem. Phys.», 1971, 55, 5334.
1209. De Lucia F. C, Helminger P. — «J. Phys. and
Chem. Ref. Data», 1974, 3, 211.
1210. DeLucia F. C, Helminger P., Cook R. L., Gordy
W. — «Phys. Rev.», 1972, A5, 487.
1211. De Lucia F. C, Helminger P., Gordy W. — «Phys.
Rev.», 1971, A3, 1849.
1212. De Lucia F. C, Helminger P., Gordy W., Morgan
H. W., Staats P. A. — «Phys. Rev.», 1973, A8,
2785.
1213. Delwaulle M. L., Frangois F. —«C. r. Acad. sci.»,
1946, 222, 550.
1214. Delwaulle M. L., Francois F. — «C. r. Acad. sci.»,
1946, 223, 796.
1215. Delwaulle M. L., Francois F. — «J. chim. phys.
et phys.-chim. biol.», 1949, 46, 87.
1216. Depannemaecker J. C, Duterage В., Belief J. —
«С. г. Acad. sci.» 1974, B279, 287.
1217. De Paz M., Ehranson S., Friedman L. — «J.
Chem. Phys.», 1970, 52, 3362.
1218. De Paz M., Leventhal J. J., Friedman L. — «J.
Chem. Phys.», 1968, 49, 5543.
1219. De Paz M., Leventhal J. J., Friedman L. — «J.
Chem. Phys.», 1969, 51, 3748.
1220. De Rocco A. G., Hoover W. G. — «J. Chem.
Phys.», 1962, 36, 916.
1221. Desai M. S. — «Proc. Roy. Soc. London», 1932,
A136, 76.
1222. Desains P. — «Ann. chim. phys.», 1848, 22, 432.
1223. De Santes D., I.urio A., Freund R. S., Miller
T. A. — <a. Chem. Phys.», 1973, 58, 4625.
1224. Desirant M., Duchesne J. — «Bull. cl. sci. Acad.
roy. Belgique», 1935, 21, 1062.
1225. Desirant M., Duchesne J. — «C. r. Acad. sci.»,
1935 201 597
1226. Detry ?>.,' Drowart /., Goldfinger P., Keller H.,
Rickert H. — «Z. phys. Chem.» (BRD), 1967, 55,
314.
1227. Deutsch P. W., Kunz A. B. — «J. Chem. Phys.»,
1973, 59, 1155.
1228. Devaquet A., Ryan J. — «Chem. Phys. Lett.»,
1973 22 269
1229. Devlin У. P., Li P. <?., Pollard G. — «J. Chem.
Phys.», 1970, 52, 2267.
1230. Dewar J. — «Chem. News», 1905, 91, 216.
1231. Dewar J. — «Proc. Roy. Soc. London», 1914, 89,
158.
1232. Dewing E. W., Richardson F. D. — «Trans.
Faraday Soc», 1958, 54, 679.
1233. Dibeler V. H. — «J. Chem. Phys.», 1967, 47, 2191.
1234. Dibeler V. H., Listen S. K. — «J. Chem. Phys.»,.
1967, 49, 482.
1235. Dibeler V. H., Walker J. A. — «J. Chem. Phys.»,
1966, 44, 4405.
1236. Dibeler V. H., Walker J. A. — «J. Opt. Soc.
America», 1967, 57, 1007.
1237. Dibeler V. H., Walker J. A. — «Inorg. Chem.»,
1969, 8, 1728.
1238. Dibeler V. H., Walker J. A., Listen S. K. —
«J. Res. NBS», 1967, A71, 371.
1239. Dibeler V. H., Walker J. A., McCulloh К. Е. —
«J. Chem. Phys.», 1969, 50, 4592.
1240. Dibeler V. H., Walker J. A., McCulloh К. Е. —
«J. Chem. Phys.», 1969, 51, 4230.
1241. Dibeler V. H., Walker J. A., McCulloh K. S.—
«J. Chem. Phys.», 1970, 53, 4414.
1242. Dibeler V. H., Walker J. A., Rosenstock H. M. —
«J. Res. NBS», 1966, A70, 459.
1243. Dickey F. P., Hoffman J. M. — «J. Chem. Phys.»,
1955, 23, 1718.
1244. Dickey F. P., Nielsen H. H. — «Phys. Rev.», 1948,
73, 1164.
1245. Dickinson R. G., West S. S. — «Phys. Rev.», 1930,
35, 1126.
1246. Dieke G. H., Blue R. W. — «Phys. Rev.», 1934,
45, 395.
1247. Dieke G. H., Crosswhite H. M. — «J. Quant.
Spectrosc. and Radiat. Transfer», 1962, 2, 97.
1248. Dieke G. H., Heath D. F. Johns? Hopkins ,Spect-
roscopic Report N 17, December 1959.
1249. Dieke G. H., HeathD. F. U. S. Dept. Commerce.
Office. Techn. Serv., P. B. Rept 146—250, 1959.
1250. Dieke G. H., Hopfild J. J.— «Phys. Rev.», 1927,
30, 400.
1251. Diercksen G. H. F., Kraemer W. P. — «Chem.
Phys. Lett.», 1970, 6, 419.
1252. Diercksen G. H. F., Kraemer W. P. — «Theor.
chim. acta», 1975, 36, 249.
1253. Diesen R. W., Wahr J. C, Adler S. E. — «J.
Chem. Phys.», 1969, 50, 3635.
1254. DillJ.D., Allen L. C, Topp W. C, Pople J. A. —
«J. Amer. Chem. Soc», 1975, 97, 7220.
1255. Di Lonardo G., Douglas A. E. — «J. Chem. Phys.»,
1972, 56, 5185.
1256. Di Lonardo G., Douglas A. E. — «Canad. J.
Phys.», 1973, 51, 434.
1257. Di Lonardo G., Trombetti A. — «Trans. Faraday
Soc», 1970, 66, 2694.
1258. Di Marzio N., Stillinger P. — «J. Chem. Phys.»,
1964, 40, 1577.
1259. D'Incan J., Topouzkhanian A. — «Z. Natur-
forsch.», 1974, 29a, 1377.
1260. D'Incan J., Topouzkhanian A. — «J. Chem.
Phys.», 1975, 63, 2683.
1260a. Dingle T. W., Freedman P. A., Gelernt В., Jones
W. J., Smith I. W. M. — «Chem. Phys.», 1975,
8, 171.
1261. Dixit M. N. — «Proc. Indian Acad. Sci.», 1967,
f. l> i A66, 325.
1262. Dixit M. N., Narasimham N. A. — «Proc. Indian
Acad. Sci.», 1968, A68, 1.
1263. Dixon D. A., Pavrish D. D., Herschbuch D. R. —
«Disc. Faraday Soc», 1973, 55, 385.
453
Литература
1264. Dixon H. P., Jenkins H. D. В., Waddington
Т. С. — «Chem. Phys. Lett.», 1971, 10, 600.
1265. Dixon J. K. — «Z. phys. Chem. Bodenstnin-
Festband», 1931, 679.
1266. Dixon R. N. — «Ganad. J. Phys.», 1959, 37, 1171.
1267. Dixon R. N. — «Mol. Phys.», 1965, 9, 357.
1268. Dixon R. N., Duxbury G. — «Chem. Phys. Lett.»,
1967, 1, 330.
1269. Dixon R. N., Duxbury G., Ramsay D. A. — «Proc.
Roy. Soc. London», 1967, A296, 137.
1270. Dixon R. N., Halle M. — «Chem. Phys. Lett.»,
1973, 22. 450.
1271. Dixon R. N., Hull S. E. — «Chem. Phys.
Lett.», 1969, 3, 367.
1272. Dixon W. В., Wilson E. B. —. «J. Chem. Phys.»,
1961, 35, 191.
1272a. Doaken K. K., Schafer T. P. — «J. Mol. Spectrosc.»,
1973, 46, 454.
1273. Doid R. E., Little R. — «Nature», 1960, 188, 737.
1274. Dodi R. E., Rolfe J. A., Woodward L. A. —
«Trans. Faraday Soc», 1956, 52, 145.
1275. Dodd R. E., Woodward L. A., Roberts H. L. —
«Trans. Faraday Soc.,» 1956, 52, 1052.
1276. Doehlemann E., Lange E. — «Z. phys. Chem.»,
1935, A173, 295.
1277. Doescher R. N. — «J. Chem. Phys.», 1951, 19,
1070.
1278. Doescher R. N. — «J. Chem. Phys.», 1952, 20,
330.
1279. Donahue J. — «Acta crystallogr.», 1965, 18,1018.
1280. Donahue J. — In: Elemental Sulfur. Chemistry
and Physics. Meyer В., Kharasch N. (Eds).
New York—London— Sydney, J. Wilav a. Sons,
Intersei. Publ., 1965, p. 13.
1281. Donohue J., Caron A., Goldisch E. —i «J. Amer.
Chem. Soc», 1961, 83, 3748.
1282. Donohue /., Meyer B. — In: Elemental Sulfur.
Chemistry and Physics. Meyer В., Kharasch N.
(Eds). New York—London—Sydney, J. Wiley
a. Sous, Intersei. Publ., 1965, p. 1.
1283. Donovan R. /., Husain D. —> «Trans. Faraday
Soc», 1968, 64, 2325.
1284. Donovan R. J., Husain D., Jackson P. T. —
«Trans. Faraday Soc», 1968, 64, 1798.
1285. Donovan R. J., Husain D., Jackson] P. T. —>
«Trans. Faraday Soc», 1969, 65, 2930.
1286. Donovan R. J., Husain D., Stevenson. С. О. —
«Trans. Faraday Soc», 1970, 66, 1.
1287. Donovan R. J., Little D. J. — «Spectrosc Lett.»
1971, 4, 213.
1288. Donovan R. J., Robertson P. J. — «Spectrosc.
Lett.», 1972, 5, 281.
1289. Donovan R. J., Robertson P. J. —.«Spectrosc.
Lett.», 1972, 5, 361.
1290. Doorenbos H. E., Loy B. R. — «J. Chem. Phys.»,
1963, 39, 2393.
1291. D'Or L., Tarte P. — «Bull. Soc. roy. sci. Liege»,
1951, 20, 478.
1292. D'Or L., Tarte P. — «Bull. Soc roy. sci. Liege»,
1953, 22, 276.
1293. D'Orazio L. A., Wood R. H. — «J. Phys. Chem.»,
1963, 67, 1435.
1294. Doring W. — «Z. Elektrochem.», 1949, 53, 106.
1295. Dorney A. J., Hoy A. R., Mills I. M. — «J.
Mol. Struct.», 1973, 45, 253.
1296. Doty P. M., Mayer J. E. — «J. Chem. Phys.»,
1944, 12, 323.
1297. Douglas A. E. — «Canad. J. Phys.», 1952, 30, 302.
1298. Douglas A. E. — «Astrophys. J.», 1953, 117, 380.
1299. Douglas A. E. Proc. Intern. Conf. Spectrosc.
Bombey, 1967, 1, SI, Sa, 81.
1300. Douglas A. E., Frackowiak M. — «Canad. J.
Phys.», 1962, 40, 832.
1301. Douglas A. E., Hoy A. R. —«Canad. J. Phys.»,
1975, 53, 1965.
1302. Douglas A. E., Huber K. P. — «Canad. J. Phys.»,
1965, 43, 74.
1303. Douglas A. E., Jones W. —«Canad. J. Phys.»,
1966, 44, 2251.
1304. Douglas A. E., Jones W. — «Canad. J. Phys.»,
1965, 43, 2216.
1305. Douglis A. E., Moller C. K., Stoicheff B. P. —
«Canad. J. Phys.», 1963, 41, 1174.
1306. Douglas A. E., Rao K. S. — «Canad. J. Phys.»,
1958, 36, 565.
1307. Douglas T. B. — «Trans. ASME», 1957, 79, 23.
1308. Douglas Т. В., Beckett С W. Preliminary Report
on the Thermodynamic Properties of Selected
Light-Element Compounds. NBS Rapt 6928,
Washington, 1960.
1309. Douslin D. R. — In: Physical Chemistry Series II,
v. 10. Thermochemistry and Thermodynamics.
Skinner H. A. (Ed.). London—Boston, Butter-
worths, 1975, p. 191.
1310. Douslin D. R., Moore R. Т., Waddington G. —
«J. Phys. Chem.», 1959, 63, 1959.
1311. Dousnvinic G. C, Sanders Т. М., Townes С. Н. —
«Phys. Rev.», 1955, 100, 1735.
1312. Dowling J. M. — «J. Mol. Spectrosc.», 1968, 27,
527.
1313. Dowling J. M. — «J. Mol. Spectrosc», 1971, 37,
272.
1314. Downs A. J., Schmutzler R. — «Spectrochim.
acta», 1965, A21, 1927.
1315. Downs J., Johnson R. E. — «J. Chem. Phys.»,
1954, 22, 143.
1316. Dows D. A., Pimentel G. — «J. Chem. Phys.»,
1955, 23, 1258.
1317. DowsD. A., Wieder G. M. — «Spectrochim. acta»,
1962, 18, 1567.
1318. Doyennette L. — «J. chim. phys. et phys.-chim.
biol.», 1961, 58, 487.
1319. Drellishak K. S., Aeschliman D. P., Cambel
A. B. —«Phys. Fluids», 1965, 8, 1590.
1320. Dressier K. — «Helv. phys. acta», 1955, 28, 563.
1321. Dressier K. — «Canad. J. Phys.», 1969, 47, 547.
1322. Dressier K. — «J. Chem. Phys.», 1976, 64, 3493.
1323. Dressier K., Miescher E. —< «Astrophys. J.»,
1965, 141, 1266.
1324. Dressier K., Ramsay D. A. — «Philos. Trans.
Roy. Soc. London», 1959, A251, 553.
1325. Drowzrt J., Goldfinger P.—.«J. chim. phys. et
phys.-chim. biol.», 1958, 55, 721.
1325a. Drowirt J., Goldfinger P. — «Quart. Rev.», 1966,
20, 545.
1326. Drowirt J., Goldfinger P., Detry D., Rickert H.,
Keller H. — «Advanc Mass Spectrom.», 1968, 4,
499.
1326a. Drowart /., Goldfinger P. — «Angew. Chem.
Intern. Ed.», 1967, 6, 581.
1327. Drowirt J., Myers С. Е., Szmirc Д., Van der
Auwera-Mahieu A., Uy О. М. —«J. Chem. Soc.
Faraday Trans. II», 1972, N 10, 1749.
1328. Drowart /., Myers С. Е., Szirnrc R., Van der
Auwera-Mahieu A., Uy O. M. — «High Temperat.
Sci.», 1973, 5, 482.
1329. Drullinger L. F., Griffiths J. E. — «Spectrochim.
acta», 1971, A27, 1793.
454
Литература
1330. Dubois I. — «Bull. Soc. roy sci. Liege», 1964, 33,
190.
1331. Dubois I. — «J. Mol. Struct.», 1969, 3, 269.
1332. Dubois I., Groetsch A. — «Bull. Soc. roy. sci.
Liege», 1964, 33, 833.
1333. Dubrulle A., Destombes J. L. — «J. Mol. Struct.»,
1972, 14, 461.
1334. Ducas T. W., Geoffrion L. D., Osgood R. M.,
Javan A. — «Appl. Phys. Lett.», 1972, 21, 42.
1335. Duchesne /., Rosen B. — «J. Chem. Phys.»,
1947, 15, 631.
1336. Duer W. C, Bertrand G. L. — «J. Chem. Phys.»,
1970, 53, 3020.
1337. Duewer W. H., Setser D. W. — «J. Chem. Phys.»,
1973, 58, 2310.
1338. Dunham J. L. — «Phys. Rev.», 1932, 41, 713.
1339. Dunitz J. D., Hedberg K. — «J. Amer. Chem.
Soc», 1950, 72, 3108.
1340. Dunkin D. В., Feksenfeld F. C, Ferguson E. E. —
«Chem. Phys. Lett.», 1972, 15, 257.
1341. Dunning T. #., Winter N. W. — «J. Chem.
Phys.», 1975, 63, 1847.
1342. Dupre-Maquaire J., Pinson P. — «J. Mol.
Spectrosc», 1975, 58, 239.
1343. Durand J. L., Brandmaier H. E. — In: Kinetics,
Equilibria and Performance of
High-Temperature Systems. Bahn G. S. (Ed.). New York-
London, Gordon a. Breach Sci. Publ., 1962,
p. 115.
1344. Durga K. K., Rao P. T. — «Indian J. Phys.»,
1958, 32, 223.
1345. Durie R. A. — «Proc. Roy. Soc. London», 1951,
A207, 388.
1346. Durie R. A. — «Canad. J. Phys.», 1966, 44, 337.
1347. Durie R. A., Gaydon A. G. — «J. Phys. Chem.»,
1952, 56, 316.
1348. Durie R. A., Herzberg G. — «Canad. J. Phys.»,
1960, 38, 806.
1349. Durie R. A., Legay F., Ramsay D. A. — «Canad.
J. Phys.», 1960, 38, 444.
1350. Durie R. A., Ramsay D. A. — «Canad. J. Phys.»,
1958, 36, 35.
1351. Durig J. R., Bush S. F., Mercer E. E. — «J.
Chem. Phys.», 1966, 44, 4238.
1352. Durig J. R., Carreira L. A., Odom J. D. — «J.
Amer. Chem. Soc», 1974, 96, 2683.
1353. Durig J. R., Clark J. W. — «J. Chem. Phys.»,
1968, 48, 3216
1354. Durig J. R., Griffin M. G., Macnamee R. W. —
«J. Raman Spectrosc.» 1975, 3, 133.
1355. Durig J. R., Lord R. C. — «Spectrochim. acta»,
1963, 19, 421.
1356. Durig J. R., Lord R. С — «Spectrochim. acta»,
1963, 19, 1877.
1357. Durig J. Д., Macnamee R. W. — «J. Raman
Spectrosc», 1974, 2, 635.
1358. Duus H. C, Mykytiuk D. P. — «J. Chem. and
Engng Data», 1964, 9, 585.
1359. Duxbury G. — «J. Mol. Spectrosc.», 1968, 25, 1.
1360. Duyckaerts G. — «Mem. Soc. roy. sci. Liege»,
1945, 6, 193.
1361. Dwyer R. J., Oldenberg O. — «J. Chem. Phys.»,
1944, 12, 351.
1362. Dyke T. R., Howard B. J., Klemperer W. — «J.
Chem. Phys.», 1972, 56, 2442.
1363. Dyke T. R., Muenter J. S. — «J. Chem. Phys.»,
1974, 60, 2929.
1364. Dymond J. H., Smith E. B. The Virial
Coefficients of Gases. A Critical Compilation». Oxford,
Clarendon Press, 1969.
1365. Dyne P. J. — «Canad. J. Phys.», 1953, 31, 453.
1366. Dyne P. J., Style D. W. G. — «Nature», 1951,
167, 899.
1367. Early R. G., Lowry Т. М. — «J. Chem. Soc»,
1919, 115, 1387.
1368. Easson Ian, Pryce M. H. L. — «Canad. J. Phys.»,
1973, 51, 518.
1369. Easterfield I. R., Lennett J. W. — «J. Chem.
Soc. Faraday Trans. II», 1974, 70, 317.
1370. Eastman E. D., McGavock W. C. — «J. Amer.
Chem. Soc», 1937, 59, 145.
1371. Ebel Fr., Bretscher E. — «Helv. chim. acta»,
1929, 12, 450.
1372. Eberhardt W. H., Burke T. G. — «J. Chem.
Phys.», 1952, 20, 529.
1373. Eberhardt W. #., Chenf W. C, Rentier H. —
«J. Mol. Spectrosc», 1919, 3, 664.
1374. Eberhradt W. H., Shi id W. — «J. Chem. Phys.»,
1946, 14, 525.
1375. Ebinghaus H., Kraus K., Muller-D uysing W.,
Neuert H. — «Z. Naturforsch.», 1964, 19a, 732.
1376. Ecker G., Krohll W. — «Phys. Fluids», 1963,
6, 62.
1377. Ecker G., Weizel W. — «Ann. Physik», 1956, 17,
126.
1378. Eckman J. R., Rossini F. D. — «J. Res. NBS»,
1929, 3, 597.
1379. Edelson D., Griffiths J. E., McAfee К. В. — «J.
Chem. Phys.», 1962, 37, 917.
1380. Edelson D., McAfee К. В. — «J. Chem. Phys.»,
1951, 19, 1311.
1381. Edic J. W., Rohrlich F. — «J. Chem. Phys.»,
1962, 36, 623.
1382. Edlen B. — «J. Chem. Phys.», 1960, 33, 98.
1382a. Edlen B. — «Arkiv mat., astron. fys.», 1943,
A29, 4.
1383. Edqvist O., Lindholm E., Selin L. E., Asbrink
L. — «Phys. scr.», 1970, 1, 25.
1384. Edqvist O., Lindholm E., Selin L. E., Sjogren
#., Asbrink L. — «Arkiv fys.», 1970, 40, 439.
1385. Edse R. — «J. Chem. Phys.», 1950, 18, 244.
1386. Edwards D. G. The Vapour Pressures of 30
Inorganic Liquids between one Atmosphere and
the Critical Point. Livermore. Univ. California
Lawrence Radiat. Lab., June 17, 1964.
1387. Edwards H. G. M., Long D. A., Love R. —
Advances in Raman Spectroscopy. Proc. 3rd Intern.
Conf., Reims, 1972, v. 1. London, 1973, p. 504.
1388. Edwards T. H., Moncur N. K., Snyder
L. E. — «J. Chem. Phys.», 1967, 46, 2139.
1389. Egan E. P., Luff В. В. — «J. Phys. Chem.»,
1961, 65, 523.
1390. Egan E. P., Luff В. В. Heats of Formation of
Phosphorus Oxides. Tennessee Valley Authority
Report. Contr. CMLMC-PA-2B-REP-129, 1963,
June-November.
1391. Egan E. P., Luff В. В. — «J. Chem. and Engng
Data», 1966, 11, 92.
1392. Egan E. P., Wakefield Z. I. — «J. Phys. Chem.»,
1957, 61, 1500.
1393. Egerton A. C, Emite W., Minkoff G. J. — «Disc.
Faraday Soc», 1951, N 10. 272.
1394. Eichenauer W., Liebscher D. — «Z. Naturforsch.»,
1965, 20a, 160.
1395. Einstein A. — «Ann. Physik», 1907, 22, 180.
1396. Elder F. A., Villarejo D., lughram M. G. — «J.
Chem. Phys.», 1965, 43, 758.
1397. Ellingson E. O. — «J. Amer. Chem. Soc», 1915,
37, 699.
455
Литература
1398. Elliott A. — «Proc. Roy. Soc. London», 1929, 1432.
A123, 629.
1399. Elliott A. — «Proc. Roy. Soc. London», 1930, 1433.
A127, 638.
1400. Elliott A. — «Proc. Roy. Soc. London», 1940, 1434.
A174, 273.
1401. Elliott N. — «J. Amer. Chem. Soc», 1937, 59, 1435.
1380. 1436.
1402. Elliott P. M., Coon J. B. — «Bull. Amer. Phys.
Soc», 1964, 9, 145. 1437.
1403. Ellis D. /., Banyard K. E. — «J. Phys. B:
Atom, and Mol. Phys.», 1974, 7, 2021. 1438.
1404. Ellison F. O., Huff N. Т., Patel J. С — «J.
Amer. Chem. Soc», 1963, 85, 3544. 1438a.
1405. Ellworth V. M., Hop field J. J. — «Phys. Rev.»,
1927, 29, 79. 14386.
1406. Emmet R. Т., Millero F. J. — «J. Chem. and
Engng Data», 1975, 20, 351.
1407. Emmett P. H., Shultz J. F. — «Ind. and Engng 1439.
Chem.», 1939, 31, 105.
1408. Engleman Д., Rouse P. E. — «J. Mol. Spectrosc», 1440.
1971, 37, 240.
1409. Engleman R., Rouse P. E., Peek H. M., Bala-
monte V. D. The Beta and Gamma Band Systems
of Nitric Oxide. Los Alamos Scient. Lab. Report, 1441.
LA-4364, 1970.
1410. Eriksson К. В. S. — «Arkiv fys.», 1958, 13, 303. 1442
1411. Eriksson K. B. S. — «Arkiv fys.», 1965, 30, 199. 1443.
1412. Eriksson К. В. S. — «Phys. scr.», 1974, 9, 151. 1444.
1413. Eriksson K. B. S., Isberg H. B. S. — «Arkiv
fys.», 1963, 24, 549. 1445.
1414. Eriksson K. B. S., Johansson T. — «Arkiv fys.»,
1961 19 235
1415. Erlandsson G.,' Cox J. — «J. Chem. Phys.», 1956, 1446.
25, 778.
1416. Ertler W. Т., Носкеп R., Charlton Т., Wilcox 1447.
L. R. — «Phys. Rev. A: Gen. Phys.», 1975, 12,
2118. 1448.
1417. Estreicher Т., Staniewski M. — «Krakauer Anz.D, 1449.
1912, 834.
1418. Eucken A., Aherens H. — «Z. phys. Chem.», 1450.
1934, B26, 297.
1419. Eucken A., Bertram A. — «Z. phys. Chem.», 1451.
1936, B31, 361.
1420. Eucken A., Schroder E. — «Z. phys. Chem.», 1452.
1938, B41, 307. 1453.
1421. Evans B. L., Yoffe A. D., Gray P. — «Chem. 1453a.
Rev.», 1959, 59, 515.
1422. Evans I. C. — «J. Chem. Soc. Chem. Communs»,
1969, N 12, 682. 1454.
1423. Evans M. G., George P., Uri N. — «Trans.
Faraday Soc», 1949, 45, 230.
1424. Evans M. G., Hush N. S., Uri N. — «Quart. Rev.»,
1952, 6, 186. 1455.
1425. Evans M. G., Uri N. — «Trans. Faraday Soc», 1456.
1949, 45, 224.
1425a. Evans W. H. Private communication (Материалы, 1457.
подготовленные для КОДАТА МСНС, 1976).
1426. Evans W. H., Munson Т. R., Wagman D. D. — 1458.
«J. Res. NBS», 1955, 55, 147.
1427. Evans W. H., Prosen E. J., Wagman D. D. — 1458a.
In: Thermo dynamic and Transport Properties
of Gases, Liquids and Solids. N. Y., 1959, p. 226. 1459.
1428. Evans W. H., Wagman D. D. — «J. Res. NBS»,
1952, 49, 141. 1460.
1429. Ewald R. — «Ann. Physik», 1914, 44, 1213.
1430. Ewing V. C, Sutton L. E. — «Trans. Faraday 1461.
Soc», 1963, 59, 1241. 1462.
1431. Experimental Thermochemistry. V. 1. F. D. Ros- 1463.
sini (Ed.). N. Y., Intersci., 1956.
Eyler J. R., Atkinson G. H. — «Chem. Phys.
Lett.», 1974, 28, 217.
Eyring H., John M. Sh. Significant Liquid
Structures. N. Y., Wiley, 1969.
Eysel H. H., Seppelt K. — «J. Chem. Phvs.»,
1972, 56, 5081.
Eyster E. H. — «J. Chem. Phys.», 1940, 8, 135.
Eyster E. H., Gillette R. H. — «J. Chem. Phys.»,
1940, 8, 369.
Faita G., Longhi P., Mussini T. — «J. Electro-
chem. Soc», 1967, 114, 340.
Falconer W. E., Buchler A., Stauffer J. L.,
Klemperer W. — «J. Chem. Phys.», 1968, 48, 312.
Falconer W. E., Sunder W. A. — «J. Inorg.
and Nucl. Chem.», 1967, 29, 1380.
Falconer W. E., Marlon 1. R.~In: Noble-Gas
Compounds. H. H. Hayman (Ed.) Chicago, Univ.
Chicago Press, 1963, p. 238.
Farkas L., Farkas A. — «Trans. Faraday Soc»,
1934, 30, 1071.
Farr T. D. Phosphorus. Properties of the
Element and some of its Compounds. Chemical
Engineering Report, N 8. Alabama, Tennessee
Valley Authority, 1950.
Farrow R. F. C. — «J. Phys. D: Appl. Phys.»,
1974, 7, 2436.
Fassbender M. — «Z. Phys.», 1924, 30, 73.
Fast I. D. — «Philips Res. Repts», 1947, 2, 382.
Fataley W. G., Bent H. A., Crawford B. jr.—
«J. Chem. Phys.», 1959, 31, 204.
Fauchais P., Baronnet I.-M., Bayard S. — «Rev.
intern, hautes temperat. et refract.», 1975, 12,
221.
Favre P. A., Silbermann I. T. — «Ann. chim.
phys.», 1853, 37, 469.
Fayt A., Steenbeckeliers G. — «C. r. Acad. sci.»,
1972, B275, 459.
Feast M. W. — «Nature», 1948, 162, N 4110, 214.
Feast M. W. — «Proc. Phys. Soc. London», 1949,
A62, 114.
Feast M. W. — «Proc Phys. Soc. London»,
1950, A63, 549.
Feast M. W. — «Proc. Phys. Soc. London»,
1950, A63, 557.
Feast M. W. — «Canad. J. Res.», 1950, A28, 488.
Feast M. W. — «Astrophys. J.», 1951, 114, 344.
Feber R. C, Herrik С. С Ideal Gas Thermody-
namic Functions of Lanthanide and Actinide
Elements. Report LA-3184. Los Alamos, 1965.
Feber R. C, Herrik С. С An Improved
Calculation of the Ideal Gas Thermodynamic Functions
of Selected Diatomic Molecules. US NBS Report
LA-3597. Los Alamos, 1966/67.
Feher F. — «Z. Elektrochem.», 1937, 43, 663.
Feher F., Gorier G. P., Lutz H. D. — «Z. anorg.
und allgem. Chem.», 1971, 382, 135.
Fehsenfeld F. C. — «J. Chem. Phys.», 1971, 54,
438.
Fehsenfeld F. C, Ferguson E. E. — «Planet.
Space Sci.», 1968, 16, 701.
Fehsenfeld F. C, Ferguson E. E., Bohme D. C. —
«Planet. Space Sci.», 1969, 17, 1759.
Feick G. — «J. Amer. Chem. Soc», 1954, 76,
5858.
Feke G. Т., Hawkins G. A., Lastovka J. В., Вепе-
dek G. B. — «Phys. Rev. Lett.», 1971, 27, 1780.
Feldman D. — «Z. Naturforsch.», 1970, 25a, 621.
Felenbok P. — «Ann. astrophys.», 1963, 26, 393.
Felenbok P., Czarny J. — «Ann. astrophys.»,
1964, 27, 244.
456
Литература
1464. Fenning R. W., Cotton F. T. — «Proc. Roy. 1498.
Soc. London», 1933, A141, 17.
1465. Fenning R. W., Wiffin A. C. — «Philos. Trans. 1499.
Roy. Soc. London», 1939, A238, 149.
1466. Ferguson E. E., DunkinD. В., Fehsenfeld F. C. — 1500.
«J. Chem. Phys.», 1972, 57, 1459.
1467. Ferguson R. C. — «J. Amer. Chem. Soc», 1954, 1501.
76, 850.
1468. Fermi E. — «Z. Phys.», 1924, 26, 54. 1501a.
1469. Ferraro J. R. — «J. Mol. Spectrosc», 1960, 4, 99.
1470. Ferriso C. C, Horning D. F. — «J. Amer. Chem. 1502.
Soc», 1953, 75, 4113.
1471. Ferriso С. С, Horning D. F. — «J. Chem. Phys.», 1503.
1955, 23, 1464.
1472. Fickett W., Cowan R. D. Values of Thermodyna- 1503a.
mic Functions to 12 000° К for Several
Substances. U. S. Atom. Energy Commiss. LA-1727, 1504.
1954.
1473. Fickett W., Cowan R. D. — «J. Chem. Phys.», 1505.
1955 23 1349
1474. Field R.' W. — «J. Mol. Spectrosc», 1973, 47, 1506.
194.
1475. Finch A., Gardner P. J., Hussain K. S., Sen 1507.
Gupta K. K. — «J. Chem. Soc. Chem. Communs»,
1968, 15, 872. 1508.
1476. Finch A., Gardner P. J., Steadman С J. — «Ca-
nad. J. Chem.», 1968, 46, 3447. 1508a.
1477. Finch A., Gardner P. J., Wood I. H. — «J.
Chem. Soc», 1965, 746. 1509.
1478. Finch I., Fraser R. P. — «J. Chem. Soc», 1926,
117. 1510.
1479. Fine R., Millero F. J. — «J. Chem. Phys.»,
1975, 63, 89. 1511.
1480. Fink U., Wiggins T. A., Rank D. H. — «J. Mol.
Spectrosc», 1965, 18, 384. 1512.
1481. Fink W. H. — «J. Chem. Phys.», 1968, 49, 5054.
1482. Fink W. H. — «J. Chem. Phys.», 1971, 54, 2911. 1513.
1483. Fink W. H., Pan Dorothy C, Allen L. C. — «J.
Chem. Phys.», 1967, 47, 895. 1514.
1484. Finkelnburg W., Schumacher H. J. — «Z. phys.
Chem. Bodenstein-Festhand», 1931, 704. 1515.
1485. Finnigan D. J., Cox A. P., Brittain A. H.,
Smith J. G. — «J. Chem. Soc. Faraday Trans.
II», 1972, 68, 548.
1486. Fiqueira R. R., Forti P., Corbelli G. — «J. Mol. 1516.
Spectrosc», 1975, 57, 97.
1487. Fischbeck K., Eich H. — «Z. anorg. und allgem.
Chem.», 1937, 235, 83. 1517.
1488. Fishburne E. S., Rao K. N. — «J. Mol.
Spectrosc», 1966, 19, 290.
1489. Fischer I. P. — «Trans. Faraday Soc», 1968, 64, 1518.
1852. 1519.
1490. Fischer J., В ingle J., Vogel R. С — «J. Amer.
Chem. Soc», 1956, 78, 902. 1520.
1491. Fischer W., Jiibermann O. — «Z. anorg. und all-
gem. Chem.», 1937, 235, 337. 1521.
1492. Flaud J. M., Camy-Peyret С — «Mol. Phys.»,
1973, 26, 811. 1522.
1493. Flaud J. M., Camy-Peyret С — «J. Mol.
Spectrosc», 1974, 51, 142.
1493a. Flaud J. M., Camy-Peyret C, Maillard J. P. —
«Mol. Phys.», 1976, 32, 499. 1523.
1494. Flaud J. M., Camy-Peyret C, Valentin A. —
«J. phys.», 1972, 33, 741. 1524.
1495. Fleming J. W. — «Chem. Phys. Lett.», 1974,
25, 553. 1525.
1496. Fleming J. W. — «Spectrochim. acta», 1976, A32,
787. 1526.
1497. Fleming J. W., Wayne R. P. — «Chem. Phys.
Lett.», 1975, 32, 135. 1527.
Flood H., Tronstad L. — «Kgl. norske vid. sel-
skabs skr.», 1936, 8, 139.
Florent P. R., Leach S. — «J. phys. et radium»,
1952, 13, 377.
Flubacher P., Leadbetter A. J., Morrison J. —
«J. Chem. Phys.», 1960, 33, 1751.
Foley W. Т., Giguere P. A. — «Canad. J. Chem.»,
1951, 29, 123.
Foley W. Т., Giguere P. A. — «Canad. J. Chem.»,
1951, 29, 895.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1953, 21, 1608.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1955, 23, 1364.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1956, 25, 602.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1958, 28, 719.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1958, 29, 442.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1962, 36, 2681.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1966, 45, 40.
Foner S. N., Hudson R. L. — «J. Chem. Phys.»,
1973, 58, 581.
Foner S, N., Hudson R. L. — «J. Chem. Phys.»,
1978, 68, 3162.
Fontana B. J. Atomic Energy Commiss.
Documents MDDC-542, CC-3482, 1946.
Foo V. J., Brion C. E., Hasted I. B. — «Proc.
Roy. Soc. London», 1971, А322Л 535.
Forsythe W. R., Giauque W. F. — «J. Amer.
Chem. Soc», 1942, 64, 48.
Foster P. D., Hodgesson J. A., Curl R. F. — «J.
Chem. Phys.», 1966, 45, 3760. ^
Fournier M., Mascherpa G., Ronsselet D., Potter
J. — «C. r. Acad. sci.», 1969, C269, 279.
Fowler A., Bakker С J. — «Proc. Roy. Soc.
London», 1932, A136, 28.
Fox W. В., Mac Kenzie J. S., Sukornik В.,
Wamser С A., Holmes J. R., Erbeck R. E.,
Stewart В. В. — «J. Amer. Chem. Soc», 1966,
88, 2604.
Fox W. В., Mac Kenzie J. S., McCarthy E. R.,
Holmes J. Д., Stahl R. F., Juurik R. — «Inorg.
Chem.», 1968, 7, 2064.
Foxon С. Т., Joyce В. A., Farrow R. F. C,
Griffits R. M. — «J. Phys. D: Appl. Phys.»,
1974, 7, 2422.
Fouretier G. — «C. r. Acad. sci.», 1944, 218, 194.
Fowler A., Vaidya W. M. — «Proc. Roy. Soc.
London», 1931, A132, 310.
Fraley P. E. Dissertation. Ohio state. — «Dissert.
Abstrs», 1966, B27, 1236.
Fraley P. E., Rao K. N. — «J. Mol. Spectrosc»,
1969, 29, 348.
Franck E. U., Brose M., Mangold K. — In:
Progress in International Research on Thermo-
dynamic and Transport Properties. N. Y., ASME,
1962, p. 159.
Franck E. U., Meyer F. <— «Z. Elektrochem.»,
1959, 63, 571.
Franck E. U., Spalthoff W. — «Z. phys. Chem.»,
1956, 8, 255.
Franck E. U., Spalthof} W. — «Z. Elektrochem.»,
1957, 61, 343.
Franck E. U., Spalthoff W. — «Z. Elektrochem.»,
1957, 61, 993.
Frandsen M. — «J. Res. NBS», 1933, 10, 35.
457
Литература
1528. Frank H., Neiger М., Рорр Н. Р. — «Z. Natur-
forsch.», 1970, 25а, 1617.
1529. Franke W., Meisel К., Jusa Д., Biltz К. — «Z.
anorg. und allgem. Chem.», 1934, 218, 346.
1530. Frankiss S. G., Harrison D. J. — «Spectrochim.
acta», 1975, A31, 161.
1531. Frankiss S. G., Miller F. A. — «Spectrochim.
acta», 1965, A21, 1235.
1532. Franklin J. L., Dibeler V. H., Reese R. M.,
Krauss M. — «J. Amer. Chem. Soc», 1958, 80,
298.
1533. Franklin J. L., Dillard J. G., Rosenstock H. M.,
Herrou J. Т., Droxl K. Ionization Potentials,
Appearance Potentials and Heats of Formation
of Gaseous Positive Ions. Washington, NSRDS-
NBS-26, 1969.
1534. Franklin J. L., Lumpkin H. E. — «J. Amer.
Ghem. Soc», 1952, 74, 1023.
1535. Frederick K. J., Hildebrand J. H. — «J. Amer.
Chem. Soc», 1938, 60, 1436.
1536. Freedman P. A., Jones W. J. — «J. Chem. Soc.
Faraday Trans. II», 1976, 72, 207.
1537. Freedman P. A., Jones W. J., Rogstad A. — «J.
Chem. Soc. Faraday Trans. II», 1975, 71, 286.
1537a. Freeman D. E., Yoshino K., Tanaka Y. — «J.
Chem. Phys.», 1974, 61, 4880.
1538. Frejacques C. — «C. r. Acad. sci.», 1952, 234,
1769.
1539. Frerichs R. — «Z. Phys.», 1926, 35, 683.
1540. Freudenberg K., Mecke R. — «Z. Phys.», 1933,
81, 465.
1541. Freund S. M., Hougen J. Т., Lafferty W. J. —
«Canad. J. Phys.», 1975, 53, 1929.
1542. Frey R. A., Redington R. L., Aljibury A. L. K. —
«J. Chem. Phys.», 1971, 54, 344.
1543. Freymann M., Freymann R. — «С. г. Acad. sci.»,
1946, 222, 1339.
1544. Friedman A. S., Haar L. — «J. Chem. Phys.»,
1954, 22, 2051.
1545. Friedman L., Shiner V. J. — «J. Chem. Phys.»,
1966, 44, 4639.
1546. Frisch M. A., Margrave J. L. — «J. Phys. Chem.»,
1965, 69, 3863.
1547. Fristrom R. M. — «J. Chem. Phys.», 1952, 20, 1.
1548. Frosch R. P., Robinson G. W. — «J. Chem.
Phys.», 1964, 41, 367.
1549. Frost A. A. — «J. Phys. Chem.», 1968, 72, 1289.
1550. Frost D. C, McDowell С A. — «Proc. Roy. Soc.
London», 1956, A236, 278.
1551. FrostD. C, McDowell С A. — «Canad. J. Chem.»,
1958, 36, 39.
1552. Frost D. C, McDowell C. A., Sandhu J. S.,
Vroom D. A. — «Advanc. Mass Spectrom.», 1968,
4, 781.
1553. Fujioka Y., Wada T. — «Sci. Papers Inst. Phys.
Chem. Res. Tokyo», 1935, 27, 210.
1554. Fukui K., Fujita /., Kuwata K. — «Mass Spec-
trosc», 1975, 23, 105.
1555. Fuller E. J., Ree Т., Eyring H. — «Proc. Nat.
Acad. Sci. USA.», 1959, 45, 1594.
1556. Funke G. W. — «Z. Phys.», 1935, 96, 787.
1557. Funke G. W. — «Z. Phys.», 1936, 101, 104.
1558. Ga.il.ir N. M. — «Dissert. Abstr.», 1958, 19,
839.
1559. GailarN. M., Dickey F. P. — «J. Mol. Spectrosc»,
1960, 4, 1.
1560. Gaines A. F., Page F. M. — «Trans. Faraday
Soc», 1966, 62, 3086.
1561. Gallagher J. J., Jonson С- М. —~. «Phys. Rev.»,
1956, 103, 1727.
1562. Gallup G. A.— «Inorg. Chem.», 1975, 14, 563.
1563. Gangi R. A., Burnelle L. — «J. Chem. Phys.»,
1971, 55, 843.
1564. Gammon В. Е. — «J. Chem. Phys.», 1976, 64,
2556.
1565. Ganguli P. S., McGee H. A. jr. — «Inorg. Chem.»,
1972, 11, 3071.
1566. Garcia J. D., Mack J. E. — «J. Opt. Soc.
America», 1965, 55, 654.
1567. Gardner M., Rogstad A. — «J. Chem. Soc. Dal-
ton Trans.», 1973, 599.
1568. Garing J. S., Nielsen H. #., Rao K. N. — «J.
Mol. Spectrosc», 1959, 3, 496.
1569. Gurnjost H. — «Ber. Bunsenges. Phys. Chem.»,
1974, 78, 1002.
1570. Garton W. R. S., Feast M. W. —«Nature», 1950,
165, 281.
1570a. Gasner E. L., Claassen H. H. — «Inorg. Chem.,
1967, 6, 1937.
1571. Gates D. M., Calfee R. F., HansenD. W.,
Benedict W. S. NBS Monogr. 71. Washington,
1964.
1572. Gatti R., Kriger R. L., Sicre J. E., Schumacher
H. J. — «J. Inorg. andjNucl. Chem.», 1966, 28,
655.
1573. Gaunt J. — «Trans. Faraday Soc», 1953, 49,
1122.
1574. Gavin R. M. — «J. Chem. Educ», 1969, 46, 413.
1574a. Gavin R. M., Bartell L. S. — «J. Chem. Phys.»,
1968, 48, 2460.
1575. Gavioli E., Wood R. W. — «Philos. Mag.», 1928,
6, 1191.
1576. Gay don A. G. — «Proc. Phys. Soc. London»,
1944, A56, 85.
1577. Gaydon A. G. —«Proc. Phys. Soc. London»,
1944, A56, 160.
1578. Gaydon A. G. Dissociation Energies and Spectra
of Diatomic Molecules. London. Chapman and
Hall Ltd, 1953.
1579. Gaydon A. G., Dissociation Energies and Spectra
of Diatomic Molecules. 3 ed. London, Chapman
and Hall Ltd, 1968.
1580. Gaydon A. G., Whittingham G. — «Proc. Roy.
Soc. London», 1947, A189, 313.
1581. Gebbie H. A., Stone N. W. В., Topping G., Gora
E. K., Clough S. A., Kneizys F. X. - «J. Mol.
Spectrosc», 1966, 19, 7.
1582. Gebbie H. A., Stone N. W. В., Walshaw С D. —
«Nature», 1960, 187, 765.
1583. Gebbie H. A., Vanasse G. A. —«Nature», 1956,
178, 432.
1584. Gehri D. C. — «Dissert. Abstrs», 1968, B29, 133.
1585. Geib K. #., Harteck P. — «Ber.». 1932, 65, 1551.
1586. Geiseler G., Ratsch M. — «Z. phys. Chem.» (DDR),
1957, A207, 138.
1587. Gelles E. — «Trans. Faraday Soc», 1951, 47, 1158.
1588. Genard J. — «Bull. cl. sci. Acad. roy. Belgique»,
1931, 17, 387.
1589. Genard J. — «Bull. cl. sci. Acad. roy. Belgique»,
1931, 17, 812.
1590. Genson D. W., Christoffersm R. E. — «J. Amer.
Chem. Soc», 1972, 94, 6904.
1591. Genzel Z. — «Z. Phys.», 1956, 114, 311.
1592. Genzel Z., Eckhardt W. — «Z. Phys.», 1954, 139,
578.
1593. Genzel Z., Eckhardt W. — «Z. Phys.», 1954, 139,
592.
1594. Gerding H., Decker H. C. J. — «Recueil trav.
chim.», 1945, 64, 191.
458
Литература
1595. Gerding Н., Houtgraaf Н.—< «Recueil trav. chim.», 1628.
1955, 74, 5.
1596. Gerding H., Lecomte /.—«Nature», 1938, 142, 1629.
718.
1597. Gerding H., Lecomte J. — «Physica», 1939, 6, 1630.
737.
1598. Gerding H., Nijveld W. J. — «Nature», 1936, 1631.
137, 1070.
1599. Gerding H., Nijveld W. J. — «Reicueil trav. 1632.
chim.», 1940, 59, 1209.
1600. Gerding H., Nijveld W. J., Muller G. J. — «Na- 1633.
ture», 1936, 137, 1033.
1601. Gerding H., Nijueld W. J., Muller G. J. — «Z. 1633a.
phys. Chem.», 1937, B35, 193.
1602. Gerding H., Brederode H. van, Decker H. С J. 1634.
de. —. «Recueil trav. chim.», 1942, 61, 549.
1603. Gerding H., Westri R.—«Recueil trav. chim.», 1635.
1943, 62, 68.
1604. Gero Z., Schmid R. — «Proc. Phys. Soc. Lon- 1636.
don», 1948, 60, 533.
1604a. Gerstenkorn S., Luc P., Perrin A.—«J. Mol. 1637.
Spectrosc», 1977, 64, 56.
1605. Geuter P. —• «Z. wiss. Photogr.», 1907, 5, 33. 1638.
1606. Geuter P. — «Z. wiss. Photogr.», 1907, 7, 1. 1639.
1607. Ghosh P. K., Bair E. J. — «J. Chem. Phys.»,
1966, 45, 4738. 1640.
1608. Ghosh P. N., Ball G. N. — «Z. Phys.», 1931, 71,
362. 1641.
1609. Ghosh P. N., Datta A. C. — «Z. Phys.», 1934,
87, 500. 1642.
1610. Ghosh S., Nagaraj S., Verma R. D. — «Canad.
J. Phys.», 1976, 54, 695. 1643.
1611. Giachardi D. J., Harris G. W., Wayne R. P. —
«Ghem. Phys. Lett.», 1975, 32, 586. 1644.
1612. Giauque W. F. — «J. Amer. Ghem. Soc», 1930,
52, 4816. 1645.
1613. Giauque W. F. — «J. Amer. Ghem. Soc», 1931,
53, 507. 1646.
1614. Giauque W. F., Archibald R. C.—«J. Amer.
Ghem. Soc», 1937, 59, 561. 1647.
1615. Giauque W. F., Ashley M. F. — «Phys. Rev.»,
1933, 43, 81. 1648.
1616. Giauque W. F., Blue R. W. —¦ «J. Amer. Ghem.
Soc», 1936, 58, 831. 1649.
1617. Giauque W. F., Clayton T. O. — «J. Amer. Ghem.
Soc», 1933, 55, 4875. 1650.
1618. Giauque W. F., Kemp J. D. — «J. Chem. Phys.»,
1938, 6, 40. 1651.
1619. Giauque W. F., Ooerstreet R. —< «J. Amer. Chem.
Soc», 1932 , 54, 1731. 1652.
1619a. Giauque W. F., Stephenson С. С. — «J. Amer.
Chem. Soc», 1938, 60, 1389. 1653.
1620. Giauque W. F., Stout J. W. — «J. Amer. Ghem.
Soc», 1936, 58, 1144.
1621. Gibson G. E., Rimsperger H. С — «Phys. Rev.», 1654.
1927 30 598
1622. Giguere P. A. — «Canad. J. Res.», 1950, 28, 1655.
485.
1623. Giguere P. A. —• «J. Chem. Phys.», 1950, 18, 88. 1656.
1624. Giguere P. A. — «J. Chem. Thermodya.», 1974,
6, 1013. 1657.
1625. Giguere P. A. -~ «J. Chem. Educ», 1974, 51,
470. 1658.
1626. Giguere P. A., Bain O. — «J. Phys. Chem.»,
1952, 56, 340. 1659.
1626a. Giguere P. A., Boisvert M. Tables des fonctions
thermodynamiques de Debye. Quebec Presses 1660.
Univ. Laval, 1962.
1627. Giguere P. A., Carmichtel J. L.-^«J. Chem. 1661.
and Engng Data», 1962, 7, 526.
Giguere P. A., Harvey К. В. —• «J. Mol.
Spectrosc», 1959, 3, 36.
Giguere P. A., Liu J. D. — «J. Chem. Phys.»,
1952, 20, 136.
Giguere P. A., Liu J. D. —> «Canad. J. Chem.»,
1952, 30, 948.
Giguere P. A., Liu J. D.—«J. Amer. Chem.
Soc», 1955, 77, 6477.
Giguere P. A., Liu J. D., Dugdale J. S.,
Morrison J. A.—«Canad. J. Ghem.», 1954, 32, 117.
Giguere P. A., Marissette B. G., Olmos A. W.,
Knop O. — «Canad. J. Chem.», 1955, 33, 804.
Giguere P. A., Saooie R. — «Canad. J. Ghem.»,
1960, 38, 2467.
Giguere P. A., Savoie R. —«J. Amer. Chem.
Soc», 1963, 85, 287.
Giguere P. A., Savoie R. —• «Canad. J. Chem.»,
1965, 43, 2357.
Giguere P. A., Schomaker V. — «J. Amer. Chem.
Soc», 1943, 65, 2025.
Giguere P. A., Srinivasan Т. К. К. — «J. Raman
Spectrosc», 1974, 2, 125.
Gilbert С — «Phys. Rev.», 1936, 49, 619.
Gilbert D. A., Roberts A. — «Phys. Rev.», 1950,
77, 742.
GilbertD. A., Roberts A., GriswoldP. A. — «Phys.
Rev.», 1949, 76, 1723.
Gilbert M. M., Gundersen G., Hedberg K. —
«J. Chem. Phys.», 1972, 56, 1691.
Gilles A., Masanet /., Vermeil C. — «Ghem.
Phys. Lett.», 1974, 25, 346.
Gilles P. W., Margrave J. L. — «J. Chem. Phys.»,
1953, 21, 381.
Gillespie L., Fraser L. — «J. Amer. Chem. Soc»,
1936, 58, 2260.
Gillespie R. J. — «J. Chem. Phys.», 1962, 37,
2498.
Gillespie R. /., Clase H. J. — «J. Chem. Phys.»,
1967, 47, 1071.
Gillespie R. J., Quail J. W. — «Ganad. J. Chem.»,
1964, 42, 2671.
Gillespie R. J., Robinson E. A.—«Canad. J.
Chem.», 1961, 39, 2171.
Gillespie R. J., Robinson E. A. —«Canad. J.
Chem.», 1961, 39, 2189.
Gillespie R. J., Robinson E. A.—«Canad. J.
Chem.», 1962, 40, 644.
Gillette R. H., Eyster H. — «Phys. Rev.», 1938,
56, 1113.
Gilliam O. R., Edwards H. D., Gordy W. — «Phys.
Rev.», 1949, 75, 1014.
Gillispie G. D., Khan A. V., Wahl A. C, Hosteny
R. P., Krauss M. — «J. Chem. Phys.», 1975, 63,
3425.
Gilmore F. R.—> «J. Quant. Spectrosc. and
Radiat. Transfer», 1965, 5, 369.
Gimarc В. М. — «J. Amer. Chem. Soc», 1970,
92, 266.
Gingerich K. A. — «J. Phys. Chem.», 1964, 68,
768.
Gingerich K. A.— «J. Chem. Phys.», 1966, 44,
1717.
Gingerich K. A.—«l. Phys. Chem.», 1969, 73,
2734.
Gingerich K. A., Lee P. K. —«J. Chem. Phys.»,
1964, 40, 3520.
Ginnings D. C, Furukawa G. T. — «J. Amer.
Ghem. Soc», 1953, 75, 522.
Ginter M. L., Tilford S. G. — «J. Mol. Spectrosc»,
1970, 34, 206.
459
Литература
1662. Ginter M. L., Tilford S. G. — «J. Mol. Spectrosc», 1695.
1971 37 159
1663. Giran H. — «Ann. chim. phys.», 1903, 30, 203. 1696.
1664. Giran H. — «Bull. Soc. chim. France», 1913,
13, 1049. 1697.
1664a. Gire G. — «Ann. chim.», 1925, 4, 186.
1665. Giulotto L. — «Nuovo cimento», 1941, 18, 367. 1698.
1666. Glatt TV., Adams R. A., Johnston H. L. Techn.
Rept, N316-8, Ohio State Univ. Res. Foundation, 1699.
1953.
1667. Glemser 0., Muller A., Bohler D., Krebs B. — 1700.
«Z. anorg. und allgem. Chem.», 1968, 357, 184.
1668. Glicksman M. E., Schaefer R. J. — «J. Chem. 1701.
Phys.», 1966, 45, 2367.
1669. Glockler G. — «Phys. Rev.», 1934, 46, 111. 1702.
1670. Goddard W. A., Dunning Th. H., Hunt W. /.,
Hay P. J. — «Accounts Chem. Res.», 1973, 6, 368. 1703.
1671. Goddard W. A., Hunt W. J. — «Chem. Phys.
Lett.», 1974, 24, 464. 1704.
1672. Goff J. A., Gratch S. — «Trans. ASME», 1950,
72, 741. 1705.
1673. Goff J. A., Gratch S., Voorhis S. W. — «Trans. 1706.
ASME», 1950, 72, 725.
1674. Gohnston H. L., Chapman A. T. — «J. Amer. 1706a.
Chem. Soc», 1933, 55, 153.
1675. Gole J. L., Hayes E. F. — «J. Chem. Phys.», 1707.
1972 57 360
1676. Goleb J.'A., Claassen H. H., Studier M. H.,
Appelman E. H. — «Spectrochim. acta», 1972, 1708.
A28, 65.
1677. Golden D. M., Solly R. K., Gac N. A., Benson 1709.
S. W. — «J. Amer. Chem. Soc», 1972, 94, 363.
1677a. Golden S., Wilson E. B. — «J. Chem. Phys.», 1710.
1948, 16, 669.
1678. Golding В., Sage В. Н. — «Ind. and Engng Chem.», 1711.
1951, 43, 160.
1679. Goldstein E. — «Phys. Z.», 1905, 6, 14. 1712.
1680. Goodeve С F., Katz S. — «Proc. Roy. Soc. 1713.
London», 1939, A172, 437. 1714.
1681. Goodeve С F., Taylor A. W. C. — «Proc. Roy.
Soc. London», 1935, A152, 221. 1715.
1682. Goodfriend P. L., Woods H. P. — «J. Chem.
Phys.», 1963, 39, 2379. 1716.
1683. Goodfriend P. L., Woods H. P. — «J. Mol.
Spectrosc», 1964, 13, 63. 1717.
1684. Goodfriend P. L., Woods H. P. — «J. Mol.
Spectrosc», 1966, 20, 258. 1718.
1684a. Goodwin R. D. — «J. Res. NBS», 1967, A71, 207.
16846. Goodliffe A. L., Jenkins H. D. В., Martin S. V. — 1719.
«Mol. Phys.», 1971, 21, 761.
1684b. Goodman G. L. — «J. Chem. Phys.», 1972, 56, 1720.
5038.
1685a. Goodman G. L., Brus L. E. — «J. Chem. Phys.», 1721.
1976, 65, 3808.
1685. Goodwin R. D., Prydz R. — «J. Res. NBS», 1722.
1970, A31, 499.
1686. Gopal E. S. R. Specific Heats at Low Tempera- 1723.
tures. London, Heywood Books, 1966.
1687. Gopinath С R., Rao K. S. R. — «Current Sci.», 1724.
1973, 42, 164.
1688. Gora E. K. — «J. Mol. Spectrosc», 1959, 3, 78. 1725.
1689. Gordon A. R. — «J. Chem. Phys.», 1933, 1, 308.
1690. Gordon A. R. — «J. Chem. Phys.», 1934, 2, 65. 1725a.
1691. Gordon A. R. — «J. Chem. Phys.», 1934, 2, 549.
1691a. Gordon A. R. — «J.Chem. Phys.», 1935, 3, 336. 1726.
1692. Gordon A. R. — «Mech. Engng», 1941, 63, 126.
1693. Gordon A. R., Barnes С — «J. Phys. Chem.», 1727.
1932, 36, 1143. 1728.
1694. Gordon A. R., Barnes С — «J. Phys. Chem.»,
1932, 36, 2292. 1729.
Gordon A. R., Barnes С — «Trans. Roy. Soc.
Canada», 1932, 26, 171.
Gordon A. R., Barnes C. — «J. Chem. Phys.»,
1933, 1, 297.
Gordon A. R., Barnes' C. — «J. Chem. Phys.»,
1933, 1, 692.
Gordon H. R., МсСиЪЫп Т. К. jr. — «J. Opt.
Soc. America», 1964, 54, 956.
Gordon J. S. — «J. Chem. and Engng Data»,
1961, 6, 390.
Gordon J. S. — «J. Chem. and Engng Data»,
1962, 7, 82.
Gordon M. S. — «J. Chem. Phys.», 1968, 49,
4643.
Gordon M. S. — «J. Amer. Chem. Soc», 1969,
91, 3122.
Gordon M. S., Fischer H. — «J. Amer. Chem.
Soc», 1968, 90, 2471.
Gordon M. S., Pople J. A. — «J. Chem. Phys.»,
1968, 49, 4643.
Gordy W. — «Revs Mod. Phys.», 1948, 20, 668.
Gordy W., Cook R. L. Microwave Molecular
Spectra. N. Y., Willey Interscience, 1970.
Gordy W., Burrus С. А. — «Phys. Rev.», 1954,
93, 419.
Gasman A. L., McCarty R. D., Hust J. G.
NRSDS-NBS Publ., N 27. Washington, U. S.
Dept Commerce, 1969.
Gondmand P., Dessaux O. — «J. chim. phys. et
phys.-chim. biol.», 1967, 64, 135.
Goulet P., Jurek R., Chanussot J. — «J. phys.»,
1976, 37, 495.
Goypiron A., Villepin J. de., Novak A. —
«Spectrochim. acta», 1975, A31, 805.
Grady H. K., Cross P. C, King G. W. — «Phys.
Rev.», 1949, 75, 1450.
Grahn R. — «Arkiv fys.», 1961, 19, 147.
Grahn R. — «Arkiv fys.», 1962, 21, 1.
Grandmontagne R., d'Incan J., Janin J. — «J.
phys. et radium», 1959, 20, N 10, 59.
Grau R., Roth W. A. — «Z. anorg. und allgem.
Chem.», 1930, 188, 173.
Grau R., Roth W. A. — «Z. anorg. and allgem.
Chem.», 1930, 188, 186.
Gray L. T. M., StyleD. W. G. —«Proc. Roy. Soc.
London», 1930, A126, 603.
Gray P., Waddington Т. С — «Trans. Faraday
Soc», 1955, 53, 901.
Gray P., Waddington Т. С — «Proc Roy. Soc.
London», 1956, A235, 106.
Gray P., Waddington Т. С — «Proc. Roy. Soc
London», 1956, A235, 481.
Gray P., Waddington Т. С — «Trans. Faraday
Soc», 1957, 53, 901.
Gray R. L., Haselton H. H., Krause D., Soltysik
E. A. — «Chem. Phys. Lett.», 1972, 13, 51.
Green J. W., Poland D. E., Margrave J. L. —
«J. Chem. Phys.», 1960, 33, 35.
Green M., Linnett J. W. — «Trans. Faraday Soc»,
1961, 57, 1.
Greenberg Т., Hallgren L. T. — «J. Chem. Phys.»,
1960, 33, 900.
Greenough K. F., Duncan A. B. F. —«J. Amer.
Chem. Soc», 1961, 83, 555.
Gregory A. R. — «Chem. Phys. Lett.», 1974, 28,
552.
Grein F. — «Theor. chim. acta», 1962, 1, 52.
Grenier-Besson M. L. — «Cahiers phys.», 1956,
10, 44.
Griem H. R. — «Phys. Rev.», 1962, 128, 994.
460
Литература
1730. Griffith Е. J. — «J. Chem. and Engng Data», 1762.
1963, 8, 22.
1731. Griffiths J. E. — «J. Chem. Phys.», 1965, 42, 1762a.
2632
1732. Griffiths J. E., Carter R. P., Holmes R."R. — 1763.
«J. Chem. Phys.», 1964, 41, 863.
1733. Grigg E. C. M., Johnston G. R. — «Austral. J. 1763a.
Chem.», 1966, 19, 931.
1734. Grigg E. С M., Johnston G. R. — «Austral. J.
Chem.», 1966, 19, 1147. 17636.
1735. Griggs M. — «J. Chem. Phys.», 1968, 49, 857.
1736. Grimbert D., Devaquet A. — «Mol. Phys.», 1974, 1763b.
27, 831.
1737. Grisard J. W., Bernhardt H. A., Oliver G. D. — 1763г.
«J. Amer. Chem. Soc», 1951, 73, 5725. 1764.
1738. Grison E., Eriks K., Devries J. L. — «Acta cry-
stallogr.», 1950, 3, 290. 1765.
1739. Grosh /., Mu Shik J., Ree Т., Eyring H. — «Proc.
Nat. Acad. Sci. USA.», 1967, 57, 1566. 1766.
1740. Gross P. — «Bull. Chem. Thermodyn.», 1960,
3, 14. 1766a.
1741. Gross P., Hayman C, Levi D. L. 17th Intern.
Congr. Pure and Appl. Chem. — «Abstr.», 1959
1, 90. 17666.
1742. Gross P., Hayman C, Levi D. L., Stuart M. С
Fulmer Res. Inst. Techn. Rept. 146/4/23, 1960; 1767.
«Trans. Faraday Soc», 1966, 62, 2709. 1768.
1743. Gross P., Hayman C, Stuart M. С — «Trans.
Faraday Soc», 1966, 62, 2716. 1769.
1744. Grosso R. P., McCubbin Т.К. — «J. Mol.
Spectrosc», 1964, 13, 240. 1770.
1744a. Groz P., Kiss J., Revesz A., Sipos T. — «J. Inorg.
and Nucl. Chem.», 1966, 28, 909. 1771.
1745. Gran A. E. — «Z. Naturforsch.», 1954, 9a, 1017. 1772.
1746. Guarnieri A., Favero P. —«Nuovo cimento»,
1965, 39, 76. 1773.
1747. Guenebaut H., Couet C, Coquart B. — «C. r. 1774.
Acad. sci.», 1965, 261, 2324.
1748. Guenebaut H., Couet C, Coquart B. — «J. chim. 1775.
phys. et phys. chim. biol.», 1966, 63, 969.
1749. Guenebaut H., Couet C, Houlon D. — «C. r. Acad. 1776.
sci.», 1964, 258, 3457.
1750. Guenebaut H., Couet C, Houlon D. — «C. r. 1777.
Acad. sci.», 1964, 258, 6370.
1751. Guenebaut H., Pannetier G. — «C. r. Acad. sci.», 1778.
1960, 250, 3613.
1752. Guenebaut H., Pannetier G., Goudmand P. — 1779.
«C. r. Acad. sci.», 1960, 251, 1166.
1753. Guenebaut H., Pascat B. — «C. r. Acad. sci.», 1780.
1962, 255, 1741. 1781.
1754. Guenebaut H., Pascat B. — «C. r. Acad. sci.»,
1964, 259, 2412. 1782.
1755. Guenebiut H., Pascat В., Berthou J. M. — «J.
chim. phys. et phys.-chim. biol.», 1965, 62, 1783.
867.
1756. Guenebaut H., Pascat В., Brion J. — «C. r. Acad.
sci.», 1964, B259, 3545.
1757. Guenebaut H., Pascat В., Couet C, Marsigny L. —
«C. r. Acad. sci.», 1963, 257, 135. 1784.
1758. Guerin F. — «Theor. chim. acta», 1970, 17, 98.
1758a. Guest M. F., Pedley J. В., Horn M. — «J. Chem. 1784a.
Thermodyn.», 1969, 1, 345.
1759. Guha S., Jois S. S., Verma R. D. — «Canad. J. 1785.
Phys.», 1972, 50, 1579.
1759a. Guido M., Balducci G., De Maria G., Gigli G. — 1786.
«J. Chem. Soc Faraday Trans. II», 1977,73, 121.
1760. Guillory W. A., Bernstein M. L. — «J. Chem. 1787.
Phys.», 1975, 62, 1058.
1761. Guillory W. A., Hunter С. Е. — «J. Chem. 1788.
Phys.», 1971, 54, 598.
Gullikson С W., Nielsen J. R., Stair A. T. —
«J. Mol. Spectrosc», 1957, 1, 151.
Gundersen G., Hedberg K., Huston J. L. —«J.
Chem. Phys.», 1970, 52, 812.
Gunn R. D., Chue P. L., Prausnitz J. M. —
«AIChE Journal», 1966, 12, 937.
Gunn S. R. — In: Noble Gas Compounds.
H. H. Hyman (Ed.). Chicago, Univ. Press, 1963,
p. 149.
Gunn S. R. — «J. Amer. Chem. Soc», 1965, 87,
2290.
Gunn S. R. — «J. Amer. Chem. Soc», 1966, 88,
5924.
Gunn S. R. — «J. Phys. Chem.», 1967, 71, 2934.
Gunn S. R. — «US Atom. Energy Commiss.
Rept», UCRL, 7992, 1964.
Gunn S. R., Green L. G. — «J. Phys. Chem.»,
1961, 65, 779.
Gunn S. if., Green L. G. — «J. Chem. and Engng
Data», 1963, 8, 180.
Gunn S. R., Williamson S. M. — In: Noble Gas
Compounds. H. H. Hyman. (Ed.). Chicago,
Univ. Press, 1963, p. 133.
Gunn S. R., Williamson S. M. — «Science»,
1963, 140, 177.
Giinther P. — «Ann. Physik», 1916, 51, 828.
Gunther P., Meyer R., MUler-Skjold F. — «Z.
phys. Chem.», 1935, A175, 154.
Gunther P., Wassmuth E., Schryver L. A. — «Z.
phys. Chem.», 1932, A158, 297.
Gunther P., Wekua K. — «Z. phys. Chem.»,
1931, A154, 193.
Guntz A. — «Ann. chim. phys.», 1884, 3, 5.
Gupta R. N., Jain P. S., Nanda V. S. —
«Indian J. Pure and Appl. Phys.», 1973, 11, 308.
Gutbier H. — «Z. Naturforsch.», 1961, 16a, 288.
Guthrie G. В., Scott D. W., Waddington G. —
«J. Amer. Chem. Soc», 1954, 76, 1488.
Gutowsky H. S., Hoffman C. J. — «J. Amer.
Chem. Soc», 1950, 72, 5751.
Gutowsky H. S., Liehr A. D. — «J. Chem. Phys.»,
1952, 20, 1652.
Gutowsky H. S., McCall D. W., Slichter С. Р. —
«J. Chem. Phys.», 1953, 21, 279.
Gwinn W. D., Tolles W. M. — «Bull. Amer.
Phys. Soc», 1961, 6, 152.
Guyon P. M., Berkowitz J. — «J. Chem. Phys.»,
1971, 54, 1814.
Haar L. — «J. Res. NBS», 1968, A72, 207.
Haar L., Bradley I. C, Friedman A. S. — «J.
Res. NBS», 1955, 55, 285.
Haar L., Friedman A. S. — «J. Chem. Phys.»,
1955 23 869
Haar L.,'Friedman A. S., Beckett Ch. W. Ideal
Gas Thermodynamic Functions and Isotope
Exchange Functions for the Diatomic Hydrides,
Deuterides and Tritides. Washington, NBS,
1961.
Haar L., Gallagher I. S. Proc. 6th SymposJTher-
mophys. Properties, 1973, 228.
Haar L., Gallagher J. S. —«J. Phys. and Chem.
Ref. Data», 1978 (in press).
Haase J., Winnewisser M. — «Z. Naturforsch.»,
1968, 23a, 61.
Haber F., Maschke A. — «Z. Elektrochem.»,
1915, 21, 128.
Haber F., Tamaru S. — «Z. Elektrochem.», 1915,
21, 191.
Haber F., Tamaru S., Ocholm L. W. — «Z.
Elektrochem.», 1915, 21, 206.
461
Литература
1789. Haber F., Tamaru S., Ponmz С. — «Z. Elektro- 1823.
chem.», 1915, 21, 89.
1790. Hadley S. G., Bina M. J., Brabson G. D. — «J. 1824.
Phys. Chem.», 1974, 78, 1833.
1791. Hadni A. — «J. phys. et radium», 1954, 15, 417. 1825.*
1792. Hadni A., Janot С — «Rev. opt.», 1960, 39, 451.
1793. Haendler H. M., Bukata S. W., MillardB., Good- 1826.
man E. I., Liftman J. — «J. Chem. Phys.», 1954,
22, 1939. 1827.
1794. Haeusler C, Barchewitz P. — «C. r. Acad. sci.»,
1958, 246, 3040. 1828.
1795. Haeusler C, Meyer C, Barchewitz P. — «J. phys.»,
1964, 25, 961. 1829.
1796. Haeusler C, Nguen Van Thanh, Barchewitz P. —
«J. phys.», 1963, 24, 289. 1830.
1797. Hagemann R. — «С. г. Acad. sci.», 1962, 255,
899. 1831.
1798. Hagemann R. — «C. r. Acad. sci.», 1962, 255,1102.
1799. Hagstrum H. D. — «J. Chem. Phys.», 1955, 23, 1832.
1178.
1800. Haida 0., Matsuo T. — «J. Chem. Thermodyn.», 1833.
1974, 6, 815.
1801. Hainer R. M., King G. W. — «J. Chem. Phys.», 1834.
1947, 15, 89.
1802. Halm R. L., Stiel L. I. — «AIChE Journal», 1835.
1967, 13, 351.
1803. Halm R. L., Stiel L. I. — «AIChE Journal», 1836.
1970, 16, 3.
1804. Hall R. Т., Dowling J. M. — «J. Chem. Phys.», 1837.
1966, 45, 1899.
1805. Hall R. Т., Dowling J. M. — «J. Chem. Phys.», 1838.
1967, 47, 2454.
1806. Hall R. Т., Dowling J. M. — «J. Chem. Phys.», 1839.
1970, 52, 1161.
1807. Hall R. Т., Dowling J. M. — «J. Chem. Phys.», 1840.
1971, 54, 4968.
1808. Hall R. Т., Mazean J., Reinhardt J. — «J. Phys. 1841.
B: Atom, and Mol. Phys.», 1970, 3, 991.
1809. Hall R. Т., Pimentel G. С — «J. Chem. Phys.», 1842.
1963, 38, 1889.
1810. Halldorsson Th., Menke E. — «Z. Naturforsch.»,
1970, 25a, 1356. 1843.
1811. Hallmann M., Klein Y. — «J. Chem. Soc», 1964,
4324. 1844.
1812. Hallmann M., Platzner I. — «J. Phys. Chem.»,
1969, 73, 4376. 1845.
1813. Haloua O. — «Czechosl. J. Phys.», 1965, B15, 267.
1814. Hamada Y., Hirakawa A. Y., Tamagake K., 1846.
Tsiboi M. — «J. Mol. Spectrosc», 1970, 35, 420.
1815. Hamada Y.. Merer A. J. — «Canad. J. Phys.»,
1974, 52, 1443. 1847.
1815a. Hamada Y., Merer A. J. — «Canad. J. Phys.», 1848.
1975, 53, 2555.
1816. Hamilton W. G. — «Acta crystallogr.», 1957, 10, 1849.
103.
1817. Hampson G. C, Stosick A. J. — «J. Amer. Chem. 1850.
Soc», 1938, 60, 1814.
1818. Handbook of Chemistry and Physics. A Ready- 1851.
Reference Book of Chemical and Physical Data.
33 ed. Cleveland, Ohio, 1951. 1852.
1819. Hanes G. R., Dahlstrom С. Е. — «Appl. Phys.
Lett.», 1969, 14, 362. 1852a.
1820. Haney M. A., Franklin J. L.—«J. Chem.
Phys.», 1968, 48, 4093. 1853.
1821. Haney M. A., Franklin J. L. — «J. Chem. Phys.»,
1969, 50, 2028.
1822. Haney M. A., Franklin J. L. — «J. Phys. Chem.», 1854.
1969, 73, 4328.
1822a. Hanley H. J. M., Klein M. — «NBS Techn. Note», 1855.
1973, 628.
Hansen K. W., Bartell L. S. — «Inorg. Chem.»,
1965, 4, 1775.
Hansler R. L., Oetien R. A. — «J. Chem. Phys.»,
1953, 21, 1340.
Hanson H., Kopp I., Kronekvist M., Aslund N. —
«Arkiv fys.», 1965, 30, 1.
Haranath P. B. V. — «Z. Phys.», 1963, 173,
428.
Haranath P. B. V., Rao P. T. — «Indian J.
Phys.», 1955, 29, 205.
Haranath P. B. V., Rao P. T. — «Current Sci.»,
1956, 25, 151.
Haranath P. B. V., Rao P. T. — «Current Sci.»,
1956, 25, 256.
Haranath P. B. V., Rao P. T. — «Indian J.
Phys.», 1957, 31, 156.
Haranath P. B. V., Rao P. T. — «Indian J.
Phys.», 1957, 31, 368.
Haranath P. B. V., Rao P. T. — «J. MoL
Spectrosc», 1958, 2, 428.
Hardwick J. L., Brand J. C. D. — «Chem. Phys.
Lett.», 1973, 21, 458.
Hardwick J. L., Brand J. CD. — «Canad. J.
Phys.», 1976, 54, 80.
Hardy J. D., Barker E. F., Dennison D. M.—
«Phys. Rev.», 1932, 42, 279.
Hariharan P. C, Pople J. A. — «Mol. Phys.»,
1974, 27, 209.
Hargittai I. — «Acta chim. Acad. sci. hung.»,
1969, 60, 231.
Hargittai I., Mijlhoff F. С — «J. Mol. Struct.»,
1973, 16, 69.
Harland P. W., Franklin J. L. — «J. Chem.
Phys.», 1974, 61, 1621.
Harland P. W., Franklin J. L. — «J. Chem.
Phys.», 1974, 61, 1621.
Harland P. W., Franklin J. L., Carter D. E.—
«J. Chem. Phys.», 1973, 58, 1430.
Harland P. W., Rankin D. W. H., Thyn-
ne J. C. J. — «Intern. J. Mass Spectrcm. anct
Ion Phys.», 1974, 13, 395.
Harland P. W., Thynne J. C. J. — «J. Phys.
Chem.», 1969, 73, 4031.
Harland P. W., Thynne J. C. J. — «J. Phys.
Chem.», 1971, 75, 3517.
Harmony M. D., Myers R. J. — «J. Chem. Phys.»,
1962, 37, 636.
Harmony M. D., Myers R. J., Schoen L. /.,
Lide D. R. jr., Mann D. E. — «J. Chem. Phys.»,
1961, 35, 1129.
Harris L. E. — «J. Chem. Phys.», 1973, 58, 5615.
Harris L. E., Churney K. L. — «J. Chem. Phys.»,
1967, 47, 1703.
Harris L. E., Maria J. H., McGlynn S. P.—
«Czechosl. J. Phys.», 1970, B20, 1007.
Harrison J. F. — «Dissert. Abstrs», 1966, B27,
1853.
Harrison R. H., Kobe K. A. — «Chem. Engng
Progr.», 1953, 49, 349.
Harrison S. W., Fischer С R., Kemmey P. J.—
«Chem. Phys. Lett.», 1975, 36, 229.
Harshbarger W., Bohn R. K., Bauer S. H,—
«J. Amer. Chem. Soc», 1967, 89, 6466.
Hartley S. В., Holmes W. S., Jacques J. K.,
Mole M. F., McCoubrey J. C. — «Quart. Rev.», 1963,
17, 204.
Hartley S. В., McCoubrey J. C. — «Nature»,
1963, 198, 476.
Hartmann H., Gliemann G. — «Z. phys. Chem.»,
(BRD), 1958, 15, 109.
462
Литература
1856. Harvey К. В., Brown Н. W. — «J. chim. phys. 1887.
et phys.-chim. biol.», 1959, 56, 745.
1857. Harvey К. В., McQuaker N. R. — «J. Chem. 1888.
Phys.», 1971, 55, 4390.
1857a. Hasegawa A., Williams F. — «Chem. Phys. 1889.
Lett.», 1977, 45, 275.
1858. Hashizume A., Wasada N., Tsuchiya T. — «Bull. 1890.
Chem. Soc. Jap.», 1966, 39, 150.
1859. Hastie J. W., Hauge R., Margrave J. L. — «J.
Inorg. and Nucl. Chem.», 1969, 31, 281. 1891.
1860. Haupt R. F., Teller E. — «J. Chem. Phys.», 1892.
1939, 7, 925.
1861. Hawkins N. J., Cohen V. W., Koski W. S. — 1893.
«J. Chem. Phys.», 1952, 20, 528. 1894.
1862. Hay P. J. — «J. Chem. Phys.», 1973, 58, 4706.
1862a. Hay P. J., Cartwright D. С — «Chem. Phys. 1895.
Lett.», 1976, 41, 80.
1863. Hay P. J., Dunning T. H. jr., Goddard W. A.— 1896.
«Chem. Phys. Lett.», 1973, 23, 457.
1864. Hay P. J., Dunning Т. Н. jr., Goddard W. A. — 1897.
«J. Chem. Phys.», 1975, 62, 3912.
1865. Hay P. /., Goddard W. A.— «Chem. Phys. 1898.
Lett.», 1972, 14, 46.
1866. Hayden J. G., O'Connell J. P. — «Ind. and 1899.
Engng Chem. Process. Design and Developm.»,
1975, 14, 209. 1900.
1867. Hayes L. J., Billingsley F. P. II, Trindle C. —
«J. Organ. Chem.», 1972, 37, 3924. 1901.
1867a. Head A. J. Private communication. (Материалы, 1902.
подготовленные для КОДАТА МСНС, 1976). 1903.
1868. Head A. J., LewisG. В. — «J. Chem. Thermodyn.», 1904.
1970, 2, 701. 1905.
1869. Head A. J., Pedley J. В., Kirk A., Seilman S., 1906.
Heath L. G. Computer Analysis of Thermochemi-
cal Data (CATCH Tables) Phosphorous Compounds,
1972, Sept. 1907.
1870. Hebb M. H. — «Phys. Rev.», 1936, 49, 610. 1908.
1871. Hedberg K. — «J. Chem. Phys.», 1951, 19, 509. 1909.
1872. Hedberg K., Badger R. M. — «J. Chem. Phys.», 1910.
1951, 19, 508. 1911.
1873. Hedberg K., Iwasaki M. — «J. Chem. Phys.»,
1962, 36, 589. 1912.
1873a. Hedberg K., Peterson S. H., Ryan R. R., Wein-
stock B. — «J. Chem. Phys.», 1966, 44, 1726. 1913.
1874. Heinz D. — «Z. anorg. und allgem. Chem.», 1965,
336 137 1914.
1875. Helen G. R., Nicholls R. W. — «J. Atmos. and
Terrestr. Phys.», 1961, 21, 213. 1915.
1876. Helminger P., Cook R. L., De Lucia F. C —
«J. Chem. Phys.», 1972, 56, 4581. 1916.
1877. Helminger P., De Lucia F. C, Gordy W., Staats
P. A., Morgan H. W. — «Phys. Rev.», 1974, 1917.
A10, 1072.
1878. Helminger P., De Lucia F. C, Kirchhoff W. H. — 1918.
«J. Phys. and Chem. Ref. Data», 1973, 2, 215.
1879. Helminger P., Gordy W. — «Phys. Rev.», 1969, 1919.
188, 100.
1880. Henglein F. A. — «Z. anorg. und allgem. Chem.», 1920.
1922, 123, 137.
1881. Henglein F. A., Rosenberg G., Muchlinski A. — 1921.
«Z. Phys.», 1922, 11, 1. 1922.
1882. Henning F. — «Z. Phys.», 1927, 40, 775.
1883. 'Henri V., Wolff F. — «J. phys. et radium», 1929, 1923.
10, 81.
1884. Herail F. M., Tartar N. — «C. r. Acad. sci.», 1923a.
1968, B267, 270.
1885. Herbelin J. M., Cohen N. — «Chem. Phys. 1924.
Lett.», 1973, 20, 605.
1886. Herberich G. E., Jackson R. H., Millen D. Y.~ 1925.
«J. Chem. Soc.» A, 1966, 336.
Herbst E., Patterson T. A., Lineberger W. C —
«J. Chem. Phys.», 1974, 61, 1300.
Herbst E., Steinmetz W. — «J. Chem. Phys.», 1972,
56, 5342.
Herczog A., Wieland K. — «Helv. phys. acta»,
1950, 23, 432.
Herget W. F., Deeds W. E., Gailar N. M., Lovel
R. J., Nielsen A. H. — «J. Opt. Soc. America»,
1962, 52, 1113.
Herman L. — «Ann. phys.», 1938, 11, 548.
Herman L., Herman R., Rokotvarijimy D. —
«J. phys. et radium», 1961, 22, 1.
Herman R. Thesis. Univ. Paris, 1945.
Herman R., Herman L. —«C. r. Acad. sci.»,
1949, 229, 931.
Herman R. C, Hornbeck G. A. — «Astrophys.
J.», 1953, 118, 214.
Herman R., Weniger M. С — «С. г. Acad. sci.»,
1950, 230, 940.
Herman-Montagne R. — «Ann. phys.», 1945, 20,
241.
Herndon W. C, Feuer J., Hall L. H. — «Theor.
chim. acta», 1968, 11, 178.
Herron J. Т., Dibeler V. H. — «J. Res. NBS»,
1961, A65, 405.
Herron J. Т., Rosenstock H. M., Shields W. R. —
«Nature», 1965, 206, 611.
Hersh O. L. — «Dissert. Abstrs», 1963, 24, 2286.
Herzberg G. — «Ann. Physik», 1928, 86, 189.
Herzberg G. — «Ann. Physik», 1932, 15, 677.
Herzberg G. — «Canad. J. Res.», 1950, A28, 144.
Herzberg G. — «Nature», 1950, 166, 563.
Herzberg G. Molecular Spectra and Molecular
Structure. 1. Spectra of Diatomic Molecules.
2 ed. Toronto—New York—London, 1950.
Herzberg G. — «Canad. J. Phys.», 1952, 30, 185.
Herzberg G. — «Canad. J. Phys.», 1953, 31, 657.
Herzberg G. — «Phys. Rev. Lett.», 1969, 23, 1081.
Herzberg G. — «J. Mol. Spectrosc», 1970, 33, 147.
Herzberg G., Herzberg L. — «J. Chem. Phys.»,
1950, 18, 1551.
Herzberg G., Herzberg L., Milne G. G. — «Canad.
J. Res.», 1940, A18, 139.
Herzberg G., How L. L. — «Canad. J. Phys.»,
1959, 37, 636.
Herzberg G., Jungen Ch. — «J. Mol. Spectrosc»,
1972, 41, 425.
Herzberg G., Lagergvist A., Miescher E. — «Canad.
J. Phys.», 1956, 34, 622.
Herzberg G., Monfils A. — «J. Mol. Spectrosc»,
1960, 5, 482.
Herzberg G., Ramsay D. A. —«J. Chem. Phys.»,
1952, 20, 347.
Herzberg G., Ramsay D. A.—«Disc. Faraday
Soc», 1953, 14, 11.
Herzberg G., Spinks J. W. T. — «Z. Phys.»,
1934, 89, 474.
Herzberg G., Verleger H. — «Phys. Z.», 1934, 35,
622.
Herzberg L. — «Z. Phys.», 1937, 107, 549.
Herzberg L., Herzberg G. — «Astrophys. J.»,
1947, 105, 353.
Hester R. E., Scaife С W. J. — «J. Chem.
Phys.», 1967, 47, 5253.
Hetherington G., Robinson P. L. — «J. Chem.
Soc», 1955, 2230.
Hettner G., Pohlman R., Schumacher H. J.—
«Z. Phys.», 1935, 96, 203.
Hettner G., Pohlman R., Schumacher H. J. —
«Naturwissenschaften», 1935, 23, 114.
463
Литература
1926. Heyns A. M., Schalkwyk G. Т. van. —«Spectra- 1959.
chim. acta», 1973, A29, 1163.
1927. Hibben J. H. The Raman Effect and its Chemical 1960.
Applications. N. Y., Reinhold, 1939.
1928. Hicham W. M., Fox R. E. — «J. Chem. Phys.», 1961.
1956, 25, 642.
1929. Hicks W. Т., U. S. Atom. Energy Commiss., 1962.
UCRL-3697, 1957, p. 16.
1930. Hieber W., Woerner A. — «Z. Elektrochem.», 1963.
1934, 40, 252. 1964.
1931. Higgins T. L., Westrum E. F. — «J. Phys. Chem.»,
1961, 65, 830. 1965.
1932. Hijikata K. — «Bull. Amer. Phys. Soc. II»,
1959, 4, 106.
1933. Hijikata K. — «Revs Mod. Phys.», 1960, 32, 445. 1966.
1934. Hijikata K. — «J. Chem. Phys.», 1961, 34, 221.
1935. Hildenbrand D. L. — «Chem. Phys. Lett.», 1972, 1967.
15 379
1936. Hildenbrand D. L. — «J. Phys. Chem.», 1973, 77, 1968.
897.
1937. Hildenbrand D. L., Kramer W. R., McDonald 1969.
R. A., Stull D. R. — «J. Amer. Chem. Soc»,
1958, 80, 4129. 1970.
1938. Hildenbrand D. L., Murad E. — «J. Chem. Phys.,
1969, 51, 807. 1971.
1939. Hildebrand J. H. — «J. Amer. Chem. Soc»,
1937, 59, 2083. 1972.
1940. Hildebrand J. H., Rotarin G. I. — «J. Amer.
Chem. Soc», 1951, 73, 2524.
1941. Hill W. L., Faust G. Т., Hendricks S. B. — «J. 1973.
Amer. Chem. Soc», 1943, 65, 794.
1942. Hillier I. H., Saunders V. R. — «Mol. Phys.», 1974.
1971, 22, 193.
1943. Hills G. W., Philen D. L., Curl R. F. jr., Tittel 1975.
F. K. — «Chem. Phys.», 1976, 12, 107.
1944. Hilton A. R. jr., Jache A. W., Beal J. B. jr., 1976.
Henderson W. D., Robinson R. J. — «J. Chem.
Phys.», 1961, 34, 1137. 1977.
1945. Hilsenrath J., Beckett C. W., Benedict W. S., 1978.
Fano L., Hoge H. L., Masi J. F., NuttullR. L.,
Touloukian Y. В., Woolley H. L. Tables of Ther- 1979.
mal Properties of Gases. NBS-Circ. 564.
Washington, 1955. 1980.
1946. Hilsenrath J., Messina C. G., Eyans^W. H. Tables
of Ideal Gas Thermodynamic Functions for 1981.
73 Atom and their First and Second Ions to
10 000 °K, AD 606 163. Washington, NBS, 1964. 1982.
1947. HimesJ. L., Wiggins T. A.— «J. Mol. Spectrosc», 1983.
1971, 40, 418. 1984.
1948. Hinck C. F. Dissertation. Heidelberg, 1913. 1984a.
1949. Hinkley E. D., Calawa A. R., Kelley P. L.,
Clough S. A. — «J. Appl. Phys.», 1972, 43, 3222. 1985.
1950. Hinze J., Jaffe H. H. — «J. Amer. Chem. Soc», 1986.
1962, 84, 540.
1951. Hirota E. — «Bull. Chem. Soc Japan», 1958, 1987.
31, 130.
1952. Hirota E. — «J. Mol. Spectrosc», 1971, 37, 20. 1988.
1953. Hirota E., Marino Y. — «J. Mol. Spectrosc.»,
1970, 33, 460. 1989.
1954. Hirschfelder J. С — «J. Chem. Phys.», 1938, 6,
795. 1990.
1955. Hirschmann R. P., Harnish D. F., Holmes J. R.,
Mackenzie J. C, Fox W. B. — «Appl. Spectrosc», 1991.
1969, 23, 333.
1956. Hirth L. J., Kobe K. A. — «J. Chem. and Engng 1992.
Data», 1961, 6, 233.
1957. Hisatsune I. C. — «J. Phys. Chem.», 1961, 65, 1993.
2249.
1958. Hisatsune I. C, Devlin J. P. — «Spectrochim. 1994.
acta», 1960, 16, 401.
Hisatsune I. C, Devlin J. P., Wada Y. — «J
Chem. Phys.», 1960, 33, 714.
Hisatsune I. C, Devlin J. P., Wada Y. —
«Spectrochim. acta», 1962, 18, 1641.
Hisatsune I. C, Devlin J. P., Wada Y. —
«Spectrochim. acta», 1962, 18, 1653.
Hisatsune I. C, Fitzsimmons R. V. —
«Spectrochim. acta», 1959, 15, 206.
Hittorf W. — «Ann. Phys. Chem.», 1865, 126, 193.
Hoch M. — «Rev. intern, hautes temperat. et
refract.», 1975, 12, 8. ,
Hoch M., Vernardakis T. Quatrieme Conf.
intern, thermodynamique chimique, Montpellier,
France, Aout 1975. V. 2. Thermophysique, p. 126.
Hochanadel C. J., Ghormley J. A., Ogren P. J. —
«J. Chem. Phys.», 1972, 56, 4426.
Hochenbleicher G., Schrotter H. W. — «Appl.
Spectrosc», 1971, 25, 360.
Hochstrasser R. M., Marshetti A. P. —«J. Chem.
Phys.», 1969, 50, 1727.
Hoeflake J. N. A., Scheffer M. E. С — «Recueil
trav. chim.», 1926, 45, 191.
Hoeft J., Tiemann E., Torring T. — «Z. Natur-
forsch.», 1972, 27a, 703.
Hoffman C. J., Neville R. G. — «Chem. Rev.»,
1962, 62, 1.
Hoffmann H. Proc 12th Intern. Conf.
Phenomena Ioniz. Gases, v. 1. Hoelscher J. G. A.,
Schram D. С (Eds). Amsterdam, 1975, p. 23.
Hoge H., Brickwedde F. — «J. Res. NBS», 1939,
22, 351.
Hogg M. A. P., Spice J. E. — «J. Chem. Soc»,
1957, 3971.
Holah G. D. — «J. Phys. C: Solid State Phys.»,
1971, 4, 2557.
Holah G. D., Happ H. — «J. Phys. C: Solid
State Phys.», 1970, 3, 1807.
Holland С — «Z. Elektrochem.», 1912, 18, 234.
Hollenberg J. L. — «J. Chem. Phys.», 1967, 46,
3271.
Holleran E. M. — «J. Chem. Phys.», 1967, 47,
5318.
Holleran E. M. — «J. Phys. Chem.», 1969, 73,
3700.
Holleran E. M., Hammes J. P. — «Cryogenics»,
1975, 15, 95.
HolmesR. R. — «J. Phys. Chem.», 1960, 64, 1295.
Holmes R. R. — «Proc Chem. Soc», 1962, 75.
Holmes R. R. — «J. Chem. Phys.», 1967, 46, 3718.
Holmes R. R.— «J. Chem. Phys.», 1967, 46,
3724.
Holmes R. R. — «J. Chem. Phys.», 1967, 46, 3730.
Holmes R. R., Carter R. P. — «J. Chem. Phys.»,
1965, 43, 1645.
Holmes R. R., Carter R. P., Peterson G. E. —
«Inorg. Chem.», 1964, 3, 1748.
Holmes W. S. — «Trans. Faraday Soc», 1962,
58, 1916.
Holt R. В., McLane С. К., Oldenberg О. — «J.
Chem. Phys.», 1948, 16, 225.
Holt R. В., McLane C. K., Oldenberg O. — «J.
Chem. Phys.», 1948, 16, 638.
Holtz D., Beauchamp J. L., Eyler J. R. —«J.
Amer. Chem. Soc», 1970, 92, 7045.
Holzer W., Murphy W. F., Bernstein H. J. —
«J. Chem. Phys.», 1970, 52, 399.
Holzer W., Murphy W. F., Bernstein H. J. —
«J. Chem. Phys.», 1970, 52, 469.
Holzer W., Murphy W. F., Bernstein H. J.,
Rolfe J. — «J. Mol. Spectrosc», 1968, 26, 543.
464
Литература
1995. Holzer W., Ouillon R. — «Chem. Phys. Lett.», 2028.
1974, 24, 589.
1996. Hopfield J. J. — «Phys. Rev.», 1930, 36, 789. 2029.
1997. Hopkins A. G. Dissertation. Kingston, Univ.
Rhode Island, 1974.
1998. Hopkins A. G., Brown C. W. — «J. Chem. Phys.»,
1973, 58, 1776.
1999. Hopkins A. G., Brown C. W. — «J. Ghem. Phys.», 2030.
1975, 62, 1598.
2000. Hopkins A. G., Brown С W. — «J. Ghem. Phys.»,
1975, 62, 2511. 2031.
2001. Hopkins A. G., Daley F. P., Brown C. W. — «J.
Phys. Ghem.», 1975, 79, 1849. 2032.
2002. Hopkins A. G., Tang Sheng-yuh, Brown С W. — 2033.
«J. Amer. Ghem. Soc», 1973, 95, 3486.
2003. Hopkins J. M., Bone L. I. — «J. Chem. Phys.», 2034.
1973, 58, 1473.
2004. Hopkinson A. C, Holbrock N. K., Yates K., 2035.
Csizmadia I. G. — «J. Chem. Phys.», 1968, 49, 2036.
3596.
2005. Horani M., Rostas J., Lefebvre-Brion H. — «Canad. 2037.
J. Phys.», 1967, 45, 3319. 2038.
2006. Horn E. F., Dickey F. P. — «J. Chem. Phys.»,
1964, 41, 1614.
2007. Hornbeck G. A. 5th Sympos. Intern, on
Combustion. N. Y., 1955, p. 790. 2039.
2008. Horsley J. A. — «J. Mol. Spectrosc», 1967, 22,
469.
2009. Horsley J. A., Barrow R. F. — «Trans. Faraday
Soc», 1967, 63, 32. 2040.
2010. Horsley J. A., Hall J. A. — «Mol. Phys.», 1973,
25, 483.
2011. Horvath A. L. Physical Properties of Inorganic
Compounds. SI Units. London, Edward Arnold, 2041.
1975.
2012. Horvath J. I. — «J. Chem. Phys.», 1948, 16, 2042.
851.
2013. Hoskins L. С — «J. Chem. Phys.», 1965, 42, 2043.
2631.
2014. Hoskins L. C, Lord R. C. — «J. Ghem. Phys.», 2044.
1967, 46, 2402.
2015. Hoiop H., Bennett R. A., Lineberger W. C. — 2045.
«J. Chem. Phys.», 1973, 58, 2373. 2046.
2016. Hotop H., Patterson T. A., Lineberger W. C. —
«J. Chem. Phys.», 1974, 60, 1806. 2047.
2017. Hougen J. Т., Radford H. E., Evenson К. М.,
Howard C. J. — «J. Mol. Spectrosc», 1975, 56, 210. 2048.
2018. Houston P. L., Steinfeld J. J. — «J. Mol.
Spectrosc.», 1975, 54, 335. • 2049.
2019. Houtefeuille P. — «С. г. Acad. Sci.», 1869, 68,
1555. 2050.
2019a. Howard W. F. jr., AndrewsL. —«J. Amer. Chem.
Soc», 1974, 96, 7864. 2051.
2020. Howe J. D., Lark-Horowitz K. — «Phys. Rev.», 2052.
1937, A51, 380.
2021. Hoyland J. R., Kier L. B. — «Theor. chim. acta», 2053.
1969, 15, 1.
2022. HrubeshL. W., CurlR. F. — «J. Mol. Spectrosc.», 2054.
1976, 61, 144.
2023. Hrubesh L. W., Rinehart E. A., Anderson R. E. —
«J. Mol. Spectrosc», 1970, 36, 354. 2055.
2024. Hsinkang Yu Yaw, Rudolph R. W., Bartell L. S.—
«J. Mol. Struct.» 1975, 28, 205. 2056.
2025. Huber L. M., Thorson W. R. — «J. Ghem. Phys.»,
1964, 41, 1829. 2057.
2026. Huber K. P. — «Canad. J. Phys.», 1968, 46, 1691. 2057a.
2027. Huber K. P., Miescher E. — «Helv. phys. acta»,
1963, 36, 257. 2058.
2027a. Hudson A., Wiffen J. T. — «Chem. Phys. Lett.», 2059.
1974, 29, 113.
Hudson R. D., Carter V. L. — «J. Opt. Soc.
America», 1968, 58, 1621.
Huff V. N., Gordon S., Morrell V. E. General
Method and Thermodynamic Tables for
Computation of Equilibrium Composition and
Temperature of Chemical Reactions, Rept. 1037.
Cleveland, Ohio, NASA, 1951.
Huffman E. O., Tarbutton G., Elmore K. L.,
С ate W. E., Walters H. K., Elmore G. V. ~ «J
Amer. Chem. Soc», 1954, 76, 6239.
Huggins W. — «Proc. Roy. Soc. London», 1890,
48, 216.
Hughes A. M. Thesis. Oregon State College, 1938.
Hughes A. M., Corruccini R. J., Gilbert E. С —
«J. Amer. Chem. Soc», 1939, 61, 2639.
Hughes B. M., Lifshitz C, Tiernan T. O. — «J
Chem. Phys.», 1973, 59, 3162.
Hughes R. H. — «J. Chem. Phys.», 1956, 24, 131.
Huiszoon С — «Rev. Scient. Instrum.», 1971
42, 477.
Hullet G. A. — «Z. phys. Chem.», 1899, 28, 629.
Hultgren R., Desai R. D., Hawkins D. Т., Gleiser
M., Kelley К. К. Selected Values of the
Thermodynamic Properties of Binary Alloys. Ohio
ASM, 1973.
Hultgren R., Desai R. D., Hawkins D. Т.,
Gleiser M., Kelley K. K., Wagman D. D. Selected
Values of the Thermodynamic Properties of the
Elements. Ohio, ASM, 1973.
Hultgren R., Orr R. L., Anderson P. D., Kelley
K. K. Selected Values of Thermodynamic
Properties of Metals and Alloys. New York—London,
John Willey and Sons Inc., 1963.
Hulthen H., Jiirlsdter N., Koffman L. —«Arkiv
fys.», 1961, 18, 479.
Hulthen E., Johansson N., Koffman L. — «Arkiv
fys.», 1960, 16, 499.
Hulthen E., Johansson TV., Pilsater U. — «Arkiv
fys.», 1958, 14, 31.
Humphreys C. J., Paul E., jr. — «J. Opt. Soc
America», 1959, 49, 1180.
Hund F. — «Z. Phys.», 1925, 31, 81.
Hunt G. R., Wilson M. K. — «Spectrochim. acta»,
1960, 16, 570.
Hunt R. H., Leacock R. A. — «J. Chem Phvs »
1966, 45, 3141.
Hunt R. H., Leacock R. A., Peters С W., Hecht
К. Т. — «J. Chem. Phys.», 1965, 42, 1931.
Hunter G., Pritchard H. O. — «J. Chem. Phvs»
1967, 46, 2153. '
Hunziker H. E., Wendt H. R. — «J. Chem
Phys.», 1974, 60, 4622.
Huo W. M. — «J. Chem. Phys.», 1968, 49, 1482.
Huong Ph. V., Couzi M. — «J. chim. phys.
et phys.-chim. biol.», 1969, 66, 1309.
Hurley A. C. — «Proc. Roy. Soc. London», 1959,
A249, 402.
Hurlock S. C, Alexander R. M., Rao K. N.,
Dreska S. N. — «J. Mol. Spectrosc», 1971, 37!
373.
Hurlock S. C, Lafferty W. J., Rao K. N. — «J.
Mol. Spectrosc», 1974, 50, 246.
Hurlock S. C, Rao K. N., Weller L.A.,Yin
P. K. L. — «J. Mol. Spectrosc», 1973, 48, 372
Hurst H. J. Thesis. Oxford, 1965.
Huston J. L., Claassen H. H. — «J. Chem. Phvs »
1970, 52, 5646.
Hyman H. H. — «Phys. Rev.», 1930, 36, 187.
Iczkowski R. P., Margrave J. L. — «J. Chem
Phys.», 1959, 30, 403.
465
Литература
2060. Iitaka I. — «Science Repts Tohoku Univ.», 2091.
1919, 18, 99. 2092.
2061. Ikenoue K. — «J. Phys. Soc. Japan», 1953, 8,
646. 2093.
2062. Ikenoue K. — «Sci. Light», 1960, 9, 79.
2063. Ikezawa M., Rolfe J. — «J. Chem. Phys.», 1973, 2094.
58, 2024.
2064. Imes E. S. — «Astrophys. J.», 1919, 50, 251. 2094a.
2065. Inglis D. R., Teller E. — «Astrophys. J.», 1939,
90, 439.
2066. Innes K. K., Cross P. С, В air E. J. — «J. Chem.
Phys.», 1953, 21, 545.
2066a. Inoue H., Naruse A., Hirota E. — «Bull. Chem. 2095.
Soc. Japan», 1976, 49, 1260.
2067. International Practical Temperature Scale of
1968 (IPTS). U. S. A., 1968, 33 p. . 2096.
2068. lolibois P. — «C. r. Acad. sci.», 1910, 151, 382.
2069. Irish D. E., Walrafen G. E. — «J. Chem. Phys.», 2097.
1967, 46, 378.
2070. Irving R. J., McKerrell H. — «Trans. Faraday 2098.
Soc», 1967, 63, 539. 2099.
2071. Irving R. /., McKerrell H. — «Trans. Faraday
Soc», 1967, 63, 2582. 2100.
2072 Ishaq M. — «Proc. Nat. Inst. Sci. India», 1937,
3, 389. 2101.
2073. Ishaq M. — «Proc. Roy. Soc. London», 1937,
A159, 110. 2102.
2074. Ishaq M., Pearse R. W. B. — «Proc. Roy. Soc
London», 1936, A156, 221. 2103.
2075 Ishaq M., Pearse R. W. B. — «Proc. Roy. Soc.
London», 1939, A173, 265. 2104.
2076. Ishiwata Т., Akimoto H., Tanaka I. — «Chem.
Phys. Lett», 1974, 27, 260. 2105.
2076a IUPAC Commiss. on Atomic Weights. — «Pure
and Appl. Chem.», 1970, 21, 99. 2106.
20766. IUPAC Commiss. on Atomic Weights. — «Pure
and Appl. Chem.», 1972, 30, 639. 2107.
2076b. IUPAC Commiss. on Atomic Weights. — «Pure
and Appl. Chem.», 1974, 37, 589. 2108.
2076r IUPAC Commiss. on Atomic Weights. — «Pure
and Appl. Chem.», 1976, 47, 75. 2109.
2077. Iurner D. W., May D. P. — «J. Chem. Phys.»,
1966, 45, 471. 2110.
2078. Jackels Ch. F., Davidson E. R. — «J. Chem.
Phys.», 1976, 64, 2908. 2111.
2079. Jackson С G. — «J. Chem. Soc», 1911, 99, 1066.
2080. Jackson R. H., Millen D. J. — «Proc. Chem. 2112.
Soc», 1959, 10.
2080a. Jacob E. J., Thompson H. В., Bartell L. S. — 2113.
«J. Mol. Struct.», 1971, 8, 383.
2081. Jacobsen R. Т., Stewart R. B. — «J. Phys. Chem. 2114.
Ref. Data», 1973, 2, 757.
2082. Jacobsen R. Т., Stewart R. В., Myers A. F. — 2115.
«Advanc. Cryogen. Engng», 1973, 18, 248.
2083. JacoxM. E., MilliganD.E. — «J. Chem. Phys.», 2116.
1967, 46, 184.
2084. Jacox M. E., Milligan D. E. — «J. Mol. Spec- 2117.
trosc», 1972, 42, 495.
2085. Jacox M. E., Milligan D. E. — «J. Mol. Spec- 2118.
trosc.», 1973, 48, 536. 2119.
2086. Jacox M. E., Milligan D. E. — «Ber. Bunsen- 2120.
ges. phys. Chem.», 1974, 78, 202. 2121.
2087. Jacox M. E., Milligan D. E., Guillory W. A., 2122.
Smith J. J. — «J. Mol. Spectrosc», 1974, 52, 2123.
322
2088. Jdg'er K., Henglein A. — «Z. Naturforsch., 1966, 2124.
21a, 1251. 2125.
2089. Jahn F. P. — «J. Chem. Phys.», 1938, 6, 335.
2090. Jahn S. — «Z. anorg. und allgem. Chem.», 2126.
1908, 60, 337.
Jain D. С — «Proc. Soc. London», 1964, 83, 17.
James D. W., Leong W. H. — «J. Chem. Phys.»
1968, 49, 5089.
James D W., Leong W. H. — «J. Chem. Phys.»,
1969, 51, 640.
James T. C, Thibault R. J. — «J. Chem. Phys.»,
1965, 42, 1450.
JANAF Thermochemical Tables PB-168370.
Stull D. R. (Ed.). 1965; First Addendum,
PB-168370-1, 1966; Second Addendum
PB-168370-2, 1967; Third Addendum
PB-168370-3, 1968.
JANAF Thermochemical Tables. 2 ed. Stull
D. R., Prophet H. (Eds). NSRDS-NBS, N 37,
Washington, 1971.
JANAF Thermochemical Tables, 1974. Suppl. —
«J. Phys. Chem. Ref. Data», 1974, 3, 311.
JANAF Thermochemical Tables, 1975. Suppl. —
«J. Phys. Chem. Ref. Data», 1975, 4, 1.
Janin J. — «Ann. phys.», 1946, 21, 538.
Janin J., d'Incan J. — «Rev. Univ. mines»,
1959, 15, 258.
Janin J., d'Incan /., Marchand J. — «Ann.
Univ. Lyon» 1959, B12, 29.
Janin J., d'Incan J., Stringat R., Magna-
val J. — «Rev. opt.», 1963, 42, 120.
Janin J., Eyraud I. — «J. phys. et radium»
1954, 15, 885.
Janoschek R., Preuss H., Diereksen G. — «Intern.
J. Quant. Chem.», 1967, 1, 649.
Janz G. J., James D. W. — «J. Chem. Phys.»,
1961, 35, 739.
Janzen J., Bartell L. S. — «J. Chem. Phys.»,
1969, 50, 3611.
Jarry R. L., Davis W. J. — «J. Phys. Chem.»,
1953, 57, 600.
Jarry R. L., Miller H. C. — «J. Phys. Chem.»,
1956, 60, 1412.
Jamie J. O., Rank A. — «Canad. J. Chem.»,
1974, 52, 2785.
Jaseja T. S.—«.J. Mol. Spectrosc», 1960, 5,
445.
Jellinek K., Schiitza H. — «Z. anorg. und allgem.
Chem.», 1936, 227, 52.
Jen C. K., Bianco D. R., Massey J. T. — «J.
Chem. Phys.», 1953, 21, 520.
Jencs F.— «Collect. Czech. Chem. Communs»,
1964, 29, 1745.
Jenkins F. A., Ashley M. F. — «Phys. Rev.»
1932 39 552
Jenkins F. A., Barton H. A., Mulliken R. S. —
«Nature», 1927, 119, 118.
Jenkins F. A., Barton H. A., Mulliken R. S. —
«Phys. Rev.», 1927, 30, 150.
Jenkins F. A., Barton H. A., Mulliken R. S. —
«Phys. Rev.», 1927, 30, 175.
Jenouvrier A., Pascat B. — «Canad. J. Phys.»,
1973, 51, 2143.
Jeppesen C. R. — «Phys. Rev.», 1933, 44, 165.
Jeppesen C. R. — «Phys. Rev.», 1934, 45, 480.
Jeppesen С R. — «Phys. Rev.», 1936, 49, 797.
Jeppesen C. R. — «Phys. Rev.», 1938, 54, 68.
Jepson B. E. — «Dissert. Abstrs», 1969, B29, 4613.
Jevons W. Report on Band. Spectra of Diatomic
Molecules. Cambridge, 1932.
Jevons W. — «Nature», 1934, 133, 619.
Jeziorski В., Kolos W. — «Chem. Phys. Lett.»,
1969, 3, 677.
Joffe J., Zudkevitch D. — «AIChE Sympos.
Series», 1974, 70, 22.
466
Литература
2127. Johns J. W. С, Barrow R. F. — «Proc. Roy.
Soc. London», 1959, A251, 504.
2128. Johns J. W. C, Leprad D. W. — «J. Mol. Spec-
trosc», 1975, 55, 374.
2129. Johns J. W. C, McKellar A. R. W., Trombetti
A. — «J. Mol. Spectrosc», 1975, 55, 131.
2130. Johns J. W. C, Ramsay D. A. — «Canad. J.
Phys.», 1961, 39, 210.
2131. Johnsen R., Brown H. L., Biondi N. A. — «Bull.
Amer. Phys. Soc», 1971, 16, 213.
2132. Johnson C. M., Trambarulo R., Gordy W. — «Phys.
Rev.», 1951, 84, 1178.
2133. Johnson D. R., Powell F. X. — «Bull. Amer.
Phys. Soc», 1968, 13, 594.
2134. Johnson D. R., Powell F. X. — «Science», 1969,
164, 950.
2135. Johnson F. A., Aycock B. F., Haney C, Colburn
С. В. — «J. Mol. Spectrosc», 1969, 31, 66.
2136. Johnson F. A., Colburn C. B. — «J. Amer. Chem.
Soc», 1961, 83, 3043.
2137. Johnson G. K. Proc. IV Intern. Conf. Chemical
Thermodynamics, Montpellier, France, v. 1,
1975, p. 23.
2138. Johnson G. K., Feder H. M., Hubbard W. N. —
«J. Phys. Chem.», 1966, 70, 1.
2139. Johnson G. K., Malm J. G., Hubbard W. N. —
«J. Chem. Thermodyn.», 1972, 4, 879.
2140. Johnson G. K., Smith P. N., Hubbard W. N. —
«J. Chem. Thermodyn.», 1973, 5, 793.
2141. Johnson R. C, Cameron W. H. B. — «Proc.
Roy. Soc. London», 1924, A106, 195.
2142. Johnson R. C, Jenkins H. G. — «Philos. Mag.»,
1926, 2, 621.
2142a. Johnson W. H., Ambrose J. R. — «J. Res. NBS»,
1963, A67, 427.
21426. Johnson W. H., Sunner S. — «Acta chem.
scand.», 1963, 17, 1917.
2143. Johnston H. L., Bertin H. J., jr. — «J. Amer.
Chem. Soc», 1959, 81, 6402.
2144. Johnston H. L., Bertin H. J., jr. — «J. Mol.
Spectrosc», 1959, 3, 683.
2145. Johnston H. L., Davis С V. — «J. Amer. Chem.
Soc», 1934, 56, 271.
2146. Johnston H. L., Dawson D. H. — «J. Amer.
Chem. Soc», 1933, 55, 2744.
2147. Johnston H. L., Dawson H. D., Walker M. K. —
«Phys. Rev.», 1933, 43, 473.
2148. Johnston H. L., Long E. A. — «J. Chem. Phys.»,
1934, 2, 389.
2149. Johnston H. L., Walker M. K. — «J. Amer.
Chem. Soc», 1933, 55, 172.
2150. Johnston H. L., Walker M. K. ~- «J. Amer.
Chem. Soc», 1933, 55, 187.
2151. Johnston H. L., Walker M. K. — «J. Amer.
Chem. Soc», 1935, 57, 682.
2152. Johnston H. S., McGrajv G. E., Paukert Т. Т.,
Richards L. M., Bogaerde J. van der—«Proc.
Nat. Acad. Sci. U. S. A.», 1967, 57, 1146.
2153. Jonathan N., Morris A., Okuda M., Ross K. J.,
SmithD. J. — «J. Chem. Soc. Faraday Trans. II»,
1974, 70, 1810.
2154. Jonathan N., Smith D. J., Ross K. J. — «Chem.
Phys. Lett.», 1971, 9, 217.
2155. Jones A. V. — «J. Chem. Phys.», 1950, 18, 1263.
2156. Jones A. V., Harrison A. W. — «J. Atmos. and
Terrestr. Phys.», 1958, 13, 45.
2157. Jones E. A., Kirby-Smith J. S., Woltz P. J. H.,
Nielsen A. H. — «J. Chem. Phys.», 1951, 19,
337.
2158. Jones E. Л., Parkinson T. E., Burke T G —
«J. Chem. Phys.», 1950, 18, 235.
2159. Jones E. A., Woltz P. J. H. — «J. Chem. Phys »
1950, 18, 1516.
2160. Jones E. I. — «J. Amer. Chem. Soc», 1943, 65,
2274.
2161. Jones G. E., Gordy W. — «Phys. Rev.», 1964,
A135, 295.
2162. Jones G. E., Gordy W. — «Phys. Rev.», 1964
A136, 1229.
2163. Jones G. P., LeeD. A. — «J. Chem. Thermodyn.»,
1970, 2, 760.
2164. Jones G. R., Burbank R. D., Barlett N. — «Inorg
Chem.», 1970, 9, 2264.
2164a. Jones G. R., Burbank R. D., Falconer W. E. —
«J. Chem. Phys.», 1970, 52, 6450.
21646. Jones G. R., Burbank R. D., Falconer W E —
«J. Chem. Phys.», 1970, 53, 1605.
2165. Jones I. T. H., Wayne R. P. — «Proc Roy. Soc.
London», 1970, A319, 273.
2166. Jones L. H. — «J. Mol. Spectrosc», 1957, 1, 179.
2167. Jones L. H., Asprey L. В., Ryan R. R — «J
Chem. Phys.», 1967, 47, 3371.
2168. Jones L. H., Bodger R. M., Moore G. E. — «J
Chem. Phys.», 1951, 19, 1599.
2169. Jones L. H., Goldblatt M. — «J. Mol. Spectrosc.»
1957, 1, 43.
2170. Jones L. H., Robinson E. S. — «J. Chem. Phys.»,
1956, 24, 1246.
2171. Jones L. H., Ryan R. R., Asprey L. B. — «J.
Chem. Phys.», 1968, 49, 581.
2172. Jones M. E., Hedberg K., Schomaker V. — «J.
Amer. Chem. Soc», 1955, 77, 5278.
2173. Jones W. E. — «Canad. J. Phys.», 1967, 45, 21.
2173a. Jones W. M. — «J. Chem. Phys.», 1949 17
1062.
2174. Jordan P. C. — «J. Chem. Phys.», 1964, 41,
1442.
2175. Joshi B. D. — «J. Chem. Phys.», 1967, 47, 2793.
2176. Joshi К. С — «Proc Phys. Soc. London», 1966,
87, 285.
2177. Joshi К. С — «Z. Phys.», 1966, 191, 126.
2178. Jost К. Н. — «Acta crystallogr.», 1964, 17,
1593.
2179. Jost К. Н. ~ «Acta crystallogr.», 1966, 21, 34.
2180. Joubert H. — «C. r. Acad. sci.», 1874, 78, 1853.
2181. Jungen Ch. — «Canad. J. Phys.», 1966, 44, 3197.
2182. Jungen Ch., Miescher E. — «Canad. J. Phys »
1969, 47, 1769.
2183. Juza J., Sifner O. — «Acta* techn. (CSAV)»,
1972, 17, 380.
2183a. Kagann R. H., Ozier I., Gerry M. С L. — «Chem
Phys. Lett.», 1977, 47, 572.
2184. Kailan A., Jahn S.—«Z. anorg. und allgem
Chem.», 1910, 68, 243.
2185. Kaldor A., Maki A. G. — «J. Mol. Struct».
1973, 15, 123.
2186. Kaldor A., Maki A. G., Dorney A. j., Mills
J. M. — «J. Mol. Spectrosc», 1973, 45, 247
2187. Kane J. S., Reynolds J. H. — «J. Chem. Phvs »
1956, 25, 342. '
2187a. Rang T. L., Hirth L. I., Kobe K. A., McKetta
J. J. — «J. Chem. and Engng Data», 1961, 6,
2188. Kang T. L. — «J. Chem. and Engng Data».
1961, 6, 227.
2188a. Kao J. — «Inorg. Chem.», 1977, 16, 2085.
21886. Kao J., Allinger N. L. — «Inorg. Chem » 1977
16, 35. '
2189. Kaplan J. — «Phys. Rev.», 1934, 46, 326.
467
Литература
2190. Kaplan J. — «Phys. Rev.», 1935, 47, 193.
2191. Kiplan J. — «Phys. Rev.», 1947, 71, 274.
2192. Kaplan L. D., Migeotte M. V., Neoen L. — «J.
Chem. Phys.», 1956, 24, 1183. 2220.
2193. Kapoor R. M., Mirtin J. J. Thermodynamic
Properties of Chlorine. Ann Arbor, Michigan,
1957.
2194. Kassell L. S. — «J. Chem. Phys.», 1933, 1,
576. 2221.
2195. Kasuya T. —¦ «Sci. Papers Inst. Phys. Chem.
Res. Tokyo», 1962, 56, 1.
2196. Kasuya Т., Kojimi T. —>«J. Phys. Soc. Japan»,
1963, 18, 364. 2222.
2197. Kato R., Rolfe J. — «J. Chem. Phys.», 1967,
47, 1901. 2223.
2198. Katz T. J., Margrave J. L. — «J. Chem. Phys.»,
1955, 23, 983. 2224.
2199. Katzin L. I. — «J. Chem. Phys.», 1950, 18, 789.
2200. Katzin L. I. — «J. Inorg. and Nucl. Chem.», 2225.
1957, 4, 187. 2226.
2201. Kaufman V., Radziemski L. J. jr. —• «J. Opt.
Soc. America», 1969, 59, 227. 2227.
2202. Kaufman V., Ward J. F. —. «Appl. Opt.», 1967,
6, 43. 2228.
2203. Kawada J., Hellner E. —«Angew. Chem. Intern.
Ed.», 1970, 9, 379. 2229.
2204. Kay J., Page F. M. — «Trans. Faraday Soc», 2230.
1964, 60, 1042.
2205. Kay W. В., Rimbosak G. M. — «Ind. and 2231.
Engng Chem.», 1953, 45, 221.
2206. Кауа К., Kunita К., Nagikuri S. — «Bull. 2232.
Chem. Soc. Japan», 1964, 37, 1055.
2207. Кауатг К., Biird J. С —. «J. Chem. Phys.», 2233.
1967, 46, 2604. 2234.
2208. Keck D. В., Hause С D. —. «J. Mai. Spectrosc»,
1968, 26, 163. 2235.
2209. Keenan A. G. —.«J. Phys. Chem.», 1956, 60,
1356. 2238.
2210. Keenan J. H., Keyes F. G., Hill P. G., Moore
J. G. Steam Tables. N. Y., J. Wiley, 1969. 2237.
2211. Kell G. S., McLaurin G. E., Whzlley E. — «J. 2238.
Chem. Phys.», 1968, 48, 3805.
2212. Kell G. S., McLiurin G. E., Whzlley E. —, «J. 2239.
Chem. Phys.», 1968, 49, 2839.
2213. Keller F. L., Nielsen A. H. — «Phys. Rev.», 2240.
1955, 99, 1624. 2241.
2214. Keller F. L., Nielsen A. H. — «J. Chem. Phys.»,
1956, 24, 636. 2242.
2215. Kelley К. К. Contributions to the Data on
Theoretical Metallurgy. III. Critical Evaluation of 2242a.
Vapour Pressures and Heat of Evaporation of 22426.
Inorganic Substances. «Bull. Bur. Mines (USA.)»,
1935, N 383. 2243.
2216. Kelley К. К. Contributions to the Data on
Theoretical Metallurgy. V. Evaluation of the Heats 2244.
of Fusion of Metals from Freezing Point Lowering
(Equilibrium Diagrams.). «Bull. Bur. Mines 2245.
(USA)», 1936, N 393.
2217. Kelley К. К. Contributions to the Data on Theo- 2246.
retical Metallurgy. VII. Thermochemical Data
and Free Energy Calculation for Sulfur, Sulphides 2247.
and Sulfates. «Bull. Bur. Mines» (USA.)»,
1937, N 406. 2248.
2218. Kelley К. К. Contributions to the Data on
Theoretical Metallurgy. VIII. High Temperature, 2249.
Heat-Content, Heat-Capacity and Entropy Data
for Inorganic Compounds. «Bull. Bur. Mines. 2250.
(USA.)», 1949, N 476.
2219. Kelley К. К. Contributions to the Data on Theo- 2251.
retical Metallurgy. IX. Entropies of Inorganic
Substances Revision A948) of Data and Methods
of Calculation. «Bull. Bur. Mines (USA.)»,
1950, N 477.
Kelley К. К. Contributions to the Data on
Theoretical Metallurgy. XIII. High Temperature,
Heat-Content, Heat-Capacity and Entropy Data
for the Elements and Inorganic Compounds.
«Bull. Bur. Mines (USA.)», 1960, N 584.
Kelley K. K., King E. G. Contributions to the
Data on Theoretical Metallurgy. XIV. Entropies
of the Elements and Inorganic Compounds.
«Bull. Bur. Mines (U. S. A.),» 1961, N 592.
Kennedy A., Colburn С. В. —• «J. Amer. Chem.
Soc», 1959, 81, 2906.
Kerr C. M. L., Webster K., Williams P. — «J.
Phys. Chem.», 1975, 79, 2663.
Kerr J. A., Sekhir R. C, Trotman-Dieckenson
A. F. —.«J. Chem. Soc», 1963, 3217.
Keruin L. —i «Canad. J. Phys.», 1954, 32, 757.
Ketelaar J. A. A., Palmer K. J. —> «J. Amer.
Chem. Soc», 1937, 59, 2629.
Ketelaar J. A. A., Schutte С J. H. — «Recueil
trav. chim.», 1961, 80, 721.
Ketelaar J. A. A., Schutte С J. H., Schram
B. L. — «Spectrochim. acta», 1959, 13, 336.
Ketteringhim J. M. Thesis. Oxford, 1964.
Ketteringhim J. M., В arrow R. F. —i «Proc
Phys. Soc. London», 1964, 83, 330.
Kewley R., Murty K. S. R., Sugden Т. М. —
«Trans. Faraday Soc», 1960, 56, 1732.
Kewley R., Sastry K. V. L. N., Winnewisser
M. -~ «J. Mol. Spectrosc», 1964, 12, 387.
Keyes F. G. — «J. Chem. Phys.», 1949, 17, 923.
Keyes F. G., Smith L. В., Gerry H. T. — «Amer.
Acad. Arts and Sci.», 1936, 70, 319.
КЫппг В. P. —i «Proc Indian Acad. Sci.»,
1959, A49, 293.
Khanna R. K. — «J. Scient. and Industr. Res.»,
1962 B21 352
KhmnaR.K. —«J. Mol.Spectrosc.», 1962, 8, 134.
Kiefer W., Bernstein H. J. — «J. Chem. Phys.»,
1972, 57, 3017.
Kiefer W., Bernstein H. J. — «J. Mol. Spectrosc»,
1972, 43, 366.
Kierstead Я. A. —«Phys. Rev.», 1973, A7, 242.
Kiess С. С, Corliss С. Я. —.. «J. Res. NBS»,
1959, A63, 1.
Kilcast D., Thomson С —¦ «J. Chem. Soc.
Faraday Trans. II», 1972, 435.
Klldal Я. —> «J. Chem. Phys.», 1977, 67, 1287.
Kim Я., Claassen Я. H., Pearson E. —> «Inorg.
Chem.», 1968, 7, 616.
Kim H., Pearson E. F., Appelman E. H. —'«J.
Chem. Phys.», 1972, 56, 1.
Kimura K., Bauer S. H. —¦ «J. Chem. Phys.»,
1963, 39, 3172.
King G. W., Mule D. —. «Canad. J. Chem.»,
1962, 40, 2057.
King R. C, Armstrong G. T. -* «J. Res. NBS»,
1968, A72, 113.
King R. C, Armstrong G. T. — «J. Res. NBS»,
1970, A74, 769.
King S. Т., Nyquist R. A. —> «Spectrochim.
acta», 1970, A26, 1481.
King S. Т., Ooerend J.—'«Spectrochim. acta»,
1966, A22, 689.
King S. Т., Overend J. —> «Spectrochim. acta»,
1967, A23, 61.
King S. Т., Ouerend J. —• «Spectrochim. acta»,
1967, A23, 2875.
468
Литература
2252. King S. Т., Overend J. — «J. Phys. Chem.», 2287.
1967, 73, 406.
2253. Kirchhoff W. H., Lide D. R., jr. — «J. Chem. 2288.
Phys.», 1969, 51, 467.
2254. Ktrkpatrik D. E., Salant E. 0. — «Phys. Rev.», 2289.
1935, 48, 945.
2255. Kironak S., Bose T. — «J. Chem. Phys.», 1973, 2290.
59, 3043.
2256. Kisliuk P. — «J. Chem. Phys.», 1954, 22, 86. 2291.
2257. Kisliuk P., Townes С. Н. — «J. Chem. Phys.»,
1950, 18, 1109. 2292.
2258. Kisliuk P., Townes С. Н. — «Phys. Rev.», 1950,
78 347 2293
2259. Kittelberger J. S., Homig D. F. — «J. Chem.
Phys.», 1967, 46, 3099. 2294.
2260. Kiukkola K., Wagner C. — «J. Electrochem.
Soc», 1957, 104, 379. 2295.
2261. Kivelson D. — «J. Chem. Phys.», 1954, 22, 904. 2296.
2262. Kivelson D., Wilson E. В., jr. — «J. Chem. Phys.»,
1952, 20, 1575. 2297.
2263. Kivelson D., Wilson E.B., jr. — «J. Chem. Phys.»,
1953, 21, 1229. 2298.
2264. Klein J. A., Nethercot A. H. — «Phys. Rev.»,
1953, 91, 1018. 2299.
2264a. Klemm W., Henkel P. — «Z. anorg. und allgem.
Chem.», 1932, 207, 73. 2300.
2265. Klemm W., Kilian H. — «Z. phys. Chem.»,
1941, B49, 279. €301.
2266. Kley D., Welge K. H. — «Z. Naturforsch.»,
1965, 20a, 124. 2302.
2267. Kloss H. G. — «Monatsber. Dtsch. Akad. Wiss. 2303.
Berlin», 1964, 6, 93.
2268. Klynning L. — «Spectrosc. Lett.», 1973, 6, 291. 2304.
2268a. Klynning L., Pages P. — «Phys. scr.», 1972,
6, 195.
2269. Knauss H. P., Bollard H. S. — «Phys. Rev.», 2304a.
1935, 48, 796.
2270. Kneizys F. X., Clough S. A. Ahstrs Sympos. 2305.
on Molecular Structure and Spectroscopy, Ohio
State Univ., 1963, p. 35. 2306.
2271. Kneubuehl F. K., Moser J. F., Steffen H. — «J. 2306a.
Opt. Soc. America», 1966, 56, 760.
2272. Knietsch R. — «Ber.», 1901, 34, 4069. 2307.
2273. Knietsch R. Ber. V. Kongr. angew. Chem., Bd.
1, 1903, S. 613.
2274. Koblitz W., SchumacherH. J. — «Z. phys. Chem.»,
1934, B25, 283. 2308.
2275. Koemer W. E., Daniels F. — «J. Chem. Phys.»,
1952, 20, 113.
2276. Kohlrausch K. W. F. Der Smekal-Raman Ef- 2309.
fekt, Berlin, Springer, 1931, S. 195. 2310.
2277. Kohlrausch K. W. F., Witteck H. — «Acta phys.
austriaca», 1948, 1, 292. 2311.
2278. Kokoszka G. F., Brinckman F. E. — «J. Amer.
Chem. Soc», 1970, 92, 1199. 2312.
2279. Kolitowska J. H. — «Roczn. chem.», 1953, 27, 191.
2280. Kolitowska J. H. — «Roczn. chem.», 1953, 27,
205. 2313.
2281. Kollman P. A., Allen L. С — «J. Amer. Chem.
Soc», 1970, 92, 753. 2314.
2282. Kollman P. A., Allen L. С — «J. Chem. Phys.»,
1970, 52, 5085.
2283. Kollman P. A., Allen L. C. — «Science», 1970, 2315.
167, 1442.
2284. Kollman P. A., Bender C. F. — «Chem. Phys. 2316.
Lett.», 1973, 21, 271.
2285. Kollman P., Johansson A., Rothenberg S. — 2317.
«Chem. Phys. Lett.», 1974, 24, 199.
2286. Kollman P., McKelvey /., Johansson A., Rothern- 2318.
berg S. — «J. Amer. Chem. Soc», 1975, 97, 955.
Kolos W. — «Acta phys. Acad. scient. hung.»,
1969, 27, 241.
Kolos W., Wolniewicz L. — «J. Chem. Phys.»,
1964, 41, 3663.
Kolos W., Wolniewicz L.—«J. Chem. Phys.»,
1965, 43, 2429.
Kolos W., Wolniewicz L. — «J. Chem. Phys.»,
1968, 49, 404.
Kolsky H. G., Gilmer R. M., Gilles P. W. — «J.
Chem. Phys.», 1957, 27, 494.
Kong T. L., HirthL. J., KobeK. A., McKetta J. J.
— «J. Chem. and. Engng Data», 1961, 6, 220.
Kordis J., Gingerich K. A. — «J. Phys. Chem.»,
1972 76 2336
Kordis /.', Gingerich K. A. — «J. Chem. Phys.»,
1973, 58, 5141.
Koref F. — «Ann. Physik», 1911, 36, 49.
Kosloff R., Levine R.D., Bernstein R. B. — «Mol.
Phys.», 1974, 27, 981.
RosterD. F., Miller F. A. — «Spectrochim. acta»,
1968, A24, 1487.
Kouba J., Ohm Y. — «J. Chem. Phys.», 1970,
52, 5387.
Kovacs I. Rotational Structure of the Spectra
of Diatomic Molecules. London, 1969.
Krakow В., Lord R. С — «J. Chem. Phys.»,
1966, 44, 3640.
Krakow В., Lord R. C., Neely G. O. — «J. Mol.
Spectrosc», 1968, 27, 148.
Kraus K. — «Z. Naturforsch.», 1961, 16a, 1378.
Kraus K., Muller-Duysing W., Neuret H. —
«Z. Naturforsch.», 1961, 16a, 1385.
Krauss M., Celotta R. J., Mielczarek S. R.,
Kuyatt С. Е. — «Chem. Phys. Lett.», 1974, 27,
285.
KraussM., Liu B. — «Chem. Phys. Lett.», 1976,
44, 257.
Krauss M., Neumann D., Wahl A. C, Das G.,
Zemke W. — «Phys. Rev.», 1973, A7, 69.
Krishnamurti P. —«Indian J. Phys.», 1930, 5,116.
Krishnamachori S. L. N. G., Narasimham
N. A. — «Current Sci.», 1965, 34, 75.
Kroepelin H., Kippling D. E., Pietruck И.
Progress in Intern. Res. on Thermodyn. and
Transport Properties. Paper 2nd Sympos. on
Thermodyn. Properties. N. Y., 1962, p. 626.
Kroepelin H., Neumann Kl.-K., Winter E. —
«Abhandl. Braunschweig, wiss. Ges.», 1958,
10, 164.
Kroll M. — «J. Chem. Phys.», 1975, 63, 319.
Kroll M., Swanson D. — «Chem. Phys. Lett.»,
1971, 9, 115.
Krupenie P. H. — «J. Phys. and Chem. Ref.
Data», 1972, 1, 427.
Kubaschewski O., Evans E. L., Alcock С. В.
Metallurgical Thermochemistry. 4th ed. Oxford,
Pergamon Press, 1967.
Kuchitsu K., Guillory J. P., Bartell L. S. —
«J. Chem. Phys.», 1968, 49, 2488.
Kichitsu K., lijima Т., Morino Y. Intern. Sympos.
Mol. Struct, and Spectroscopy. Tokyo, 1962,
С 308/1.
Kuchitsu K., Morino Y. — «Bull. Chem. Soc
Japan», 1965, 38, 814.
Kuczkowski R. L. — «J. Amer. Chem. Soc»,
1965, 87, 5259.
Kuczkowski R. L. — «J. Amer. Chem. Soc»,
1968, 90, 1705.
Kuczkowski R. L., Schiller H. W., Rudolph
R. W. — «Inorg. Chem.», 1971, 10, 2505.
469
Литература
2319. Kuczkowski R. L., Wilson E. B. jr. — «J. Ghem.
Phys.», 1963, 39, 1030.
2320. Kudchadker A. P., Kudchadker S. A., Agarwal
P. ./If.—«Indian J. Cham.», 1971, 9, 722.
2320a. Kudchadker S. A., Kudchadker A. P. — «Proc.
Indian Acad. ScL», 1971, A73, 261.
2321. Kuhn H. — «Z. Phys.», 1926, 39, 77.
2322. Kuipers G. A. Smith D. F., Nielsen A. H. —
«J. Chem. Phys.», 1956, 25, 275.
2323. Kukolich S. G. — «J. Ghem. Phys.», 1969, 50,
3751.
2324. Kumagai A., Torinni T. — «J. Ghem. and Engng
Data», 1971, 16, 293.
2325. Kuriakose A. K., Whalley E. — «J. Ghem. Phys.»,
1968, 48, 2025.
2326. Kurzel R. В., Steinfeld J. I. — «J. Chem. Phys.»,
1970, 53, 329.
2327. Kurylo H. J. — «J. Cham. Phys.», 1969, 51,
4497.
2328. Raster F. W. — «Z. anorg. und allgem. Ghem.»,
1904, 42, 453.
2329. Kvifte G. — «Nature», 1951, 168, 741.
2330. Kvifte G. — «Geophys. Publ.», 1959, 20, N 12.
2331. Kwan Y. Y., Cohen E. A. — «J. Mol. Spectrosc.»,
1975, 58, 54.
2332. Labarre J. F. — «J. chim. phys. et phys.-chim
biol.», 1971, 68, 1618.
2332a. Lacher J. R., Casali L., Park J. D. — «J. Phys!
Ghem.», 1956, 60, 608.
2333. Lacher J. R., Kianpour A., Oetting F'., ParkJ.D.—^
«Trans. Faraday Soc», 1956, 52, 1500.
2334. Lacmann K., Herschbach D. R. — «Ghem. Phys.
Lett.», 1970, 6, 106.
2335. Lacmann K., Herschback D. R. — «Bull. Amer.
Phys. Soc», 1971, 16, 213.
2335a. Ladd M. F. C. — «Trans. Faraday Soc», 1970,
66, 1592.
2336. Lafferty W. J., LideD. R. — «J. Mol. Spectrosc»,
1964, 14, 407.
2337. Lafferty W. J., Jams R. L. — «Mol. Phys.»,
1974, 28, 861.
2338. Lagemann R. Т., Jones E. A.— «J. Cham
Phys.», 1951, 19, 534.
2339. Lagerqvist A. — «Canad. J. Phys.», 1962, 40,
352.
2340. Lagerqvist A., Miescher E. — «Helv. phys. acta»,
1958, 31, 221.
2341. Lagerqvist A., Miescher E. —«Ganad. J. Phys.»,
1966, 44, 1525.
2342. Lakshminarayana G. — «J. Mol. Spectrosc», 1975,
55, 141.
2343. Lakshminarayana G., Narasimham N. A. —
«Current Sci.», 1967, 36, 533.
2344. Lakshminarayana G., Narasimham N. A.
—«Current Sci.», 1972, 41, 771.
2345. Lai L. — «Nature», 1948, 161, 477.
2346. Lambdin W. J., Tuffly B. L., Varborough V. A. —
«Appl. Spectrosc», 1959, 13, 71.
2347. Lam Thanh My, Peyron M. — «J. chim. phys.
et phys.-chim. biol.», 1962, 59, 688.
2348. Lam Thanh My, Peyron M. — «J. chim. phys.
et phys.-chim. biol.», 1963, 60, 1289.
2349. Lam Thanh My, Peyron M. — «J. chim. phys.
et phys.-chim. biol.», 1964, 61, 1531.
2350. Lam Thanh My, Peyron M. — «J. chim. phys.
et phys.-chim. biol.», 1966, 63, 266.
2351. Land J. E., Raith W. — «Phys Rev.», 1974,
A9, 1592.
2352. Landau L., Fletcher W. H. — «J. Mol. Spectrosc»,
1960, 4, 276.
2353. Landolt-Bornstein. Zahlenwerte und Funktionen
Physik, Chemie, Astronomie, Geophysik und
Technik, Bd. 2. 6 Aufl. 1961, S. 414, 445.
2354. Lange E. — «Z. phys. Chem.», 1924, AUO, 343.
2355. Langseth A. — «Z. Phys.», 1931, 72, 350.
2356. Langseth A., Walles E.—<tZ. phys. Chem.»,
1934, B27, 209.
2357. LannonJ.A., Verderame F.D., AndersonR. W. —
«J. Chem. Phys.», 1971, 54, 2212.
2358. Larkin I. W., Hasted J. 5. — «J. Phys. B:
Atom, and Mol. Phys.», 1972, 5, 95.
2359. Larmann J. P., Martire D. E., Pollara L. Z. —
«J. Ghem. and Engng Data», 1961, 6, 330.
2360. Larson A. T. — «J. Amer. Chem. Soc», 1924,
46, 367.
2361. Larson А. Т., Dodge R. L. — «J. Amer. Chem.
Soc», 1923, 45, 2919.
2362. Larson H. R., Chipman J. —• «J. Metals», 1953,
5, 1089.
2363. Lassetre E. N., Skerbele A., Dillon M. A., Ross
K. J. — «J. Chem. Phys.», 1968, 48, 5066.
2364. Latimer W. M. — Tables of Free Energy
Functions for Elements and Compounds in the
Temperature Range 2000—5000° K. U. S. Atomic.
Energy Commiss., Rept MMDC-1462, 1947.
2365. Latimer W. M., Hoenshel H. D.—«J. Amer.
Ghem. Soc», 1926, 48, 19.
2366. Laulicht I., Pinchas S., Sadeh D., Samuel D. —
«J. Ghem. Phys.», 1964, 41, 789.
2367. Lavilla R. E., Bauer S. H. — «J. Chem. Phys.»,
1960, 33, 182.
2368. Lavilla R. E., Bauer S. H. — «J. Amer. Chem.
Soc», 1963, 85, 3597.
2369. Leach S. — «G. r. Acad. sci.», 1950, 230, 2181.
2370. Leach S. — «Disc. Faraday Soc», 1950, 9, 81.
2371. Leach S. —• «Boll, scient. Fac chim. industr.
Bologna», 1952, 10, 9.
2371a. Lederer С M., Hollander J. M., Perlman I.
Table of Isotopes. 6th ed. New York—London-
Sydney, 1968.
2372. Lederle E. «Z. phys. Ghem.», 1932, B17, 353.
2373. Le Duff Y., Holzer W. «J. Chem. Phys.», 1974,
60, 2175.
2374. Lee В., Kesler M. G. «AIChE Journal», 1975,
21, 510.
2375. Lees R. M., Curl R. F., Baker J. G. — «J. Ghem.
Phys.», 1966, 45, 2037.
2376. Lefebvre-Brion H., Moser C. — «J. Chem. Phys.»,
1966, 44, 2951.
2377. Leffert Ch. В., Jackson W. M., Rothe E. W. —
«J. Chem. Phys.», 1973, 58, 5801.
2378. Legay F. —> «Canad. J. Phys.», 1960, 38, 797.
2379. Legon A. C, Millen D. J. — «J. Chem. Soc.»
A, 1968, 1736.
2380. Lehn, J. M., Munsch B. — «Theor. chim.acta»,
1968, 12, 91.
2381. Lehn J. M., Munsch B. — «J. Chem. Soc» D,
1970, 1062.
2382. Leitson S. W. — «Astrophys. J.», 1926, 63, 73.
2383. Leroy G., Louterman-Leloup G., Ruelle P. —•
«Bull. Soc. chim. belg.», 1976, 85, 229.
2383a. Leroy G., Louterman-Leloup G., Ruelle P. —<
«Bull. Soc. chim. belg.», 1976, 85, 393.
2384. Le Roy R. J. — «J. Chem. Phys..», 1970, 52,
2678.
2385. Le Roy R. J. — «J. Chem. Phys.», 1970, 52,
2683.
2386. Le Roy R. J. —. «J. Ghem. Phys.», 1971, 54,
5433.
470
Литература
2386а. Le Roy R. J. — In: Molecular spectroscopy.
V. 1. London, 1973, p. 113.
2387. Le Roy R. /., Barwell M. — «Canad. J. Phys.»,
1975 53 1983
2388. Le Roy R. /., Bernstein R. B. — «Chem. Phys.
Lett.», 1970, 5, 42.
2389. Le Roy R. J., Bernstein R. B. — «J. Chem.
Phys.», 1970, 52, 2678.
2390. Le Roy R. /., Bernstein R. B. — «J. Chem.
Phys.», 1970, 52, 3869.
2391. Le Roy R. /., Benstein R. B. — «J. Chem. Phys.»,
1971, 54, 5414.
2392. Le Roy R. J., Bernstein R. B. — «J. Mol. Spec-
trosc», 1971, 37, 109.
2393. Le Roy R. J., Burns G. — «J. Mol. Spectrosc»,
1968, 25, 77.
2393a Le systeme International d'unitees SI. Edition
Bur. intern. Poides e Mesures. Paris, 1970.
2394. Levelt Sengers J. M. H., Greet W. L., Sengers
J. V. — «J. Phys. and Chem. Ref. Data», 1976,
5, 1.
2395. Leventhal J. /., Friedman L. — «J. Chem. Phys.»,
1968, 49, 1974.
2396. Levin A., Bronk-Meyer C. F. — «J. Opt. Soc.
America», 1927, 15, 257.
2397. Levin I. W. — «J. Chem. Phys.», 1969, 50, 1031.
2398. Levin I. W. — «J. Mol. Spectrosc.», 1970, 33,
61.
2399. Levin I. W. — «J. Chem. Phys.», 1971, 55,
5393
2400. Levin I. W., Berney С V. — «J. Chem. Phys.»,
1966, 44, 2557.
2401. Levin I. W., Harris W. С — «J. Chem. Phys.»,
1971, 55, 3048.
2402. Levine D. M. — Dows D. A. — «J. Chem. Phys.»,
1967, 46, 1168.
2403. Levitt B. P., Scheen D. B. — «Trans. Faraday
Soc», 1965, 61, 2404. i•« Щ
2404. Levitt B. P., Scheen D. B. — «Trans. Faraday
Soc», 1967, 63, 540.
2405. Levy A., Rossi /., Haeusler C. — «J. Phys.»,
1966, 27, 526.
2406. Levy A., Rossi I., Joffrin C, Nguyen Van
Thanh, —i «J. chim. phys. et phys.-chim. biol.»,
1965, 62, 600.
2407. Levy D. H. — «J. Chem. Phys.», 1972, 56, 1415.
2407a. Levy H. A., Agron P. A. —«J. Amer. Chem.
Soc», 1963, 85, 241.
2408. Levy H. A., Peterson S. W. — «Phys. Rev.»,
1952, 86, 766.
2409. Levy H. A., Peterson S. W. — «J. Amer. Chem.
Soc», 1953, 75, 1536.
2410. Lew H., Heiber I. — «J. Chem. Phys.», 1973,
58, 1246.
2411. Lewis В., Elbe G. — «J. Chem. Phys.», 1935,
3, 63.
2412. Lewis С H., Burgess E. G. — «AIAA Journal»,
1963, 1, 1928.
2413. Lewis G. N., Randall M. — «J. Amer. Chem.
Soc», 1911, 33, 476.
2414. Lewis G. N., Randall M. — «J. Amer. Chem.
Soc», 1914, 36, 2259.
2414a. Lewis G. N., Randall M. — «J. Amer. Chem.
Soc», 1918, 40, 362.
2415. Lewis G. N., Randall M. Thermodynamik und
freie Energie chemischer Substanzen. Wien,
1927.
2416. Lewis G. N., Randall M. — In: International
Critical Tables. N. Y., 1930.
2417. Lewis G. N., Randall M., Pitzer K. S., Brewer
L. Thermodynamics. New
York—Toronto—London, 1961.
2418. Lewis L. C, Fredericks W. J. — «J. Chem. and
Engng Data», 1968, 13, 482.
2419. Lewis M. N., White J. U. — «Phys. Rev.»,
1939, 55, 894.
2420. Li Y. S., Chen M. M., During J. R. — «J.
Mol. Struct.», 1972, 14, 261.
2421. Libby W. F. — «J. Chem. Phys.», 1943, 11, 101.
2422. Libby W. F. — «J. Chem. Phys.», 1947, 15,
339.
2423. Lichtenstein M., Derr V. E., Gallagher J. J. —
«J. Mol. Spectrosc», 1966, 20, 391.
2424. Lichtenstein M., Gallagher J. J., Clough S. A. —
«J. Mol. Spectrosc», 1971, 40, 10.
2425. Lide D. R. jr. — «J. Chem. Phys.», 1963, 38,
456.
2426. Lide D. R. jr., Mann D. E. — «J. Chem. Phys.»,
1959, 31, 1129.
2427. Lide D. R., Mann D. E., Comeford I. I. — «Spec-
trochim. acta», 1965, 21, 497.
2428. Lide D. R., Mann D. E., Fristrom R. H. —
«J. Chem. Phys.», 1957, 26, 734.
2429. Liden K. — «Arkiv fys.», 1949, 1, 229.
2430. Lie G. C, Clementi E. — «J. Chem. Phys.»,
1974, 60, 1288.
2431. Lifshitz C, Hughes В. M., Tiernan Т. О. — «Chem.
Phys. Lett.», 1970, 7, 469.
2432. Lifshitz C, Tiernan T. O., Hughes В. М. — «J.
Chem. Phys.» 1973, 59, 3182.
2433. Lin M. C, Bauer S. H. — «J. Amer. Chem. Soc»,
1969, 91, 7737.
2433a. Lindeman L. P., Guff у J. C. — «J. Chem. Phys.»,
1958, 29, 247.
2434. Lindenberg A. — «C. r. Acad. sci.», 1971, C273,
1017.
2435. Under F., Schmidt H. — «Z. Naturforsch.»,
1971, 26a, 1617.
2436. Lindholm E. — «Naturwissenschaften», 1939, 27,
470.
2437. Lindholm E. — «Arkiv mat., astron., fys.», 1943,
B29, 1.
2438. Lindsey D. C, Lister D. G., Millen D. J. —
«J. Chem. Soc» D, 1969, 950.
2439. Lineberger W. C, Woodward B. W. — «Phys.
Rev. Lett.», 1970, 25, 424.
2440. Linnett J. W. — «Trans. Faraday Soc», 1940,
36, 527.
2441. Lippincott E. R., Schroeder R. — «J. Chem.
Phys.», 1955, 23, 1131.
2442. Lippincott E. R., Schroeder R. — «J. Amer.
Chem. Soc», 1956, 78, 5171.
2443. Lischka H. — «Theor. chim. acta», 1973, 31, 39.
2444. Lischka H., Dyczmons V. — «Chem. Phys. Lett.»,
1973 23 167
2445. Liskow D. H., Schaefer H. F., Bender C. F. —
«J. Amer. Chem. Soc», 1971, 93, 6734.
2445a. Liskov D. H., Schaefer H. F., Bagus P. S., Liu
B. — «J. Amer. Chem. Soc», 1973, 95, 4056.
2446. Liu H. P. D., Legentil /., Verhaegen G. Selected
Topic in Molecular Physics. Clementi E. (Ed.).
Heidelberg, Verlag Chemie, 1972, S. 19.
2447. Liu H. P. D., Verhaegen G. — «J. Chem. Phys.»,
1970 53 735
2448. Liu H. P. D., Verhaegen G. — «Intern. J.
Quantum Chem. Sympos.», 1971, 5, 103.
2449. Liu T.-K., Мое L., Duncan A. B. F. —
«J. Chem. Phys.», 1951, 19, 71.
471
Литература
2450. Lochte-Holtgreven W., Dieke G. H. — «Ann.
Physik», 1929, 3, 937.
2451. Lockett P., Fowler W., Wilt P. M. — «J. Chem.
Phys.», 1970, 53, 452.
2452. Lofthus A. — «Canad. J. Phys.», 1956, 34, 780.
2453. Lofthus A. — Spectroscopic Report N 2. Dept
Phys. Univ. Oslo, 1960.
2453a. Lofthus A., Krupenie P. H. — «J. Phys. and
Chem. Ref. Data», 1977, 6, 113.
2454. Lofthus A., Miescher E. — «Canad. J. Phys.»,
1964, 42, 848.
2455. Lofthus A., Mulliken R. S. — «J. Chem. Phys.»,
1957, 26, 1010.
2456. Lohr L. L., Lipscomb W. N. — «J. Chem. Phys.»,
1962 36 2225
2457. Lojk'o M. S., Beer Y. — «J. Res. NBS», 1969,
A73, 233.
2458. Long J., Munson B. — «J. Chem. Phys.», 1970,
53, 1356.
2459. Long R. W., Hildenbrand J. H., Morrell W. E. —
«J. Amer. Chem. Soc», 1943, 66, 182.
2460. Loomts F. W. — «Phys. Rev.». 1927, 29,
112.
2461. Loomis F. W., Brandt W. H. — «Phys. Rev.»,
1936, 46, 55.
2462. Lord R. C, Lynch M. A., Schumb W. C, Slo-
winski E. J. — «J. Amer. Chem. Soc», 1950,
72, 522.
2463. Lorenzelli V., Lecomte M. J. — «С. г. Acad.
sci.», 1961, 253, 2052.
2464. Lorenzelli V., Moller K. D. — «С. г. Acad. sci.»,
1959, 248, 1980.
2465. Lotmar W. — «Z. Phys.», 1933, 83, 765.
2466. Lovejoy R. W., Colwell J. H., Eggers D. F.,
Halsey G. D. — «J. Chem. Phys.», 1962, 36,
612.
2467. Low M. J. D., Yang R. T. — «Spectrosc. Lett.»,
1972, 5, 245.
2468. Lowater F. — «Astrophys. /.», 1906, 23, 324.
2469. Lu С S., Donohue I. — «J. Amer. Chem. Soc»,
1944, 66, 818.
2470. Lucas N. J. D., Smith J. G. — «J. Mol.
Spectrosc», 1972, 43, 327.
2471. Lucas R. — «Z. Elektrochem.», 1905, 11, 457.
2472. Ludwig I. R., Cooper W. S. — «J. Chem. and
Engng Data», 1963, 8, 76.
2473. Lueg P., Hedfeld K. — «Z. Phys.», 1932, 75,
512.
2474. Luft N. W. — «J. Phys. Chem.», 1954, 58, 928.
2475. Luft N. W. — «Monatsh. Chem.», 1955, 86, 474.
2476. Lumme P., Lachermo P., Tummavuori J. — «Acta
chem. scand.», 1965, 19, 2175.
2477. Lundgren J. O., Williams J. M. — «J. Chem.
Phys.», 1973, 58, 788.
2478. Lundlan E. B. — «J. Chem. Phys.», 1935, 3,
617.
2479. Lunge G., Pollitt G. P. — «Z. angew. und all-
gem. Chem.», 1902, 15, 1105.
2480. Lunge G., Reinhardt K. «Z. angew. und allgem.
Chem.», 1904, 17, 1041.
2481. Lunt R. W., Pearse R. W. В., Smith E. С W. —
«Proc. Roy. Soc. London», 1935, 151, 602.
2482. Lunt R. W., Pearse R. W. В., Smith E. С W. —
«Proc. Roy. Soc. London», 1936, 155, 173.
2483. Lussana S. — «Nuovo cimento», 1903, 5, 153.
2483a. Lusternik V., FockinL., Lavuschev A. 7thSympos.
on Thermophysical Properties, Abstracts, NBS,
1977, p. 168.
2484. Lutz O. — «Ingr.-Arch.», 1948, 16, 377.
2485. Maass O., Hatcher W. H.—«l. Amer. Chem.
Soc», 1920, 42, 2548.
2486. Maass O., Hilbert P. G. — «J. Amer. Chem. Soc»,
1924, 46, 2693.
2487. Maass O., Waldbauer L. — «J. Amer. Chem.
Soc», 1925, 47, 1.
2488. MacGillary С H., Decker H. C. J. de. — «Chem.
weekbl.», 1942, 39, 227.
2489. MacGillary С H., Decker H. С J. de, Nijland
L. M. — «Nature», 1949, 164, 448.
2490. Mackle H. — «Tetrahedron», 1963, 19, 1159.
2491. Mackle H., McClean R. T. B. — «Trans.
Faraday Soc», 1962, 58, 895.
2492. Mackle H., O'Hare P. A. G. — «Tetrahedron»,
1963, 19, 961.
2493. Maclean J. N., Rossotti F. J. C, Rossotti H. S. —
«J. Inorg. and Nucl. Chem.», 1962, 24, 1549.
2494. MacNeil K. A. G., Thynne J. С J. — «J. Phys.
Chem.», 1970, 74, 2257.
2495. MacRae D., Van Vooris С. С. — «J. Amer. Chem.
p. Soc», 1921, 43, 547.
2496. Mader С L. Ideal Gas Thermodynamic
Properties of Detonation Products. U. S. Atom. Energy
Commiss., AECU-4508, 1959.
2497. Maeder R. — «Helv. phys. acta», 1948, 21, 411.
2498. Maeder R., Miescher E. — «Nature», 1948, 161,
393.
2499. Magnuson D. W. — «J. Chem. Phys.», 1951, 19,
1071.
2500. Magnuson D. W. — «J. Chem. Phys.», 1952, 20,
380.
2501. Magnuson D. W. — «J. Chem. Phys.», 1956, 24,
344.
2502. Magnuson D. W. — «J. Chem. Phys.», 1957, 27,
223.
2503. Mahler W., Muetterties E. L. — «Inorg. Chem.»,
1965, 4, 1520.
2504. Maier C. G., Kelley К. К. — «J. Amer. Chem.
Soc», 1932, 54, 3243.
2505. Malbrunot P., Voder B. — «Physics», 1973, 66,
351.
2505a. Malicet J., Brion J., Daumont D. — «C. r. Acad.
sci.», 1977, C284, 175.
2506. Malicet J., Brion J., Guenebaut H. — «C. r.
Acad. sci.», 1973, C276, 991.
2507. Malicet J., Brion J., Guenebaut H. — «J. chim.
phys. et phys. — chim. hiol.», 1970, 67, 25.
2507a. Malm J. G., Sheft J., Chernick C. J. — «J.
Amer. Chem. Soc», 1963, 85, 110.
2508. Mangold K., Franck E. U. — «Z. Elektrochem.»,
1962, 66, 260. |§i
2508a. Mann D. E., Acquista N., White D. — «J. Chem.
Phys.», 1966, 44, 3453.
2509. Mann D. E., Ball J. J., Moore J. — «Spectro-
chim. acta», 1956, 8, 292.
2510. Mann D. E., Thrush B. A., Lide D. R., Ball
J. J., Acquista JV. — «J. Chem. Phys.», 1961,
34, 420. « m wmwy
2510a. Mann J. B. Proc. Intern. Conf. on Mass Spectro-
scopy Kyoto, Tokyo, Univ. Press, 1970, p. 814.
25106. Mann M. M., Hustrulid A., Tate J. T. — «Phys.
Rev.»,l 1940, 58, 340.
2511. Manni R. — «Theor. chim. acta», 1966, 6, 312.
2512. Maple T. G. Dissertation. Massachusetts Inst.
Technol., 1949.
2513. Marais E. J. Thesis. Univ. Stellenbosch, 1945.
2514. Marais E. J. — «Phys. Rev.», 1946, 70, 499.
2515. Marais E. J., Verleger H. — «Phys. Rev.», 1950,
80, 429.
472
Литература
2516. Marckwald W., Helmholtz R. — «Z. anorg. und
allgem. Chem.», 1922, 124, 81.
2517. Margrave J. L. Physico-Chemical Measurements
at High Temperatures. London, 1959.
2518. Maria H. /., Armstrong А. Т., McGlynn S. P. —
«J. Chem. Phys.», 1968, 48, 4694.
2519. Maria H. J., Armstrong А. Т., McGlynn S. P. —
«J. Chem. Phys.», 1969, 50, 2777.
2520. Maria H. J., McDonald J. R., McGlynn S. P. —
«J. Amer. Chem. Soc», 1973, 95, 1050.
2521. Maria H. J., Wahlborg A., McGlynn S. P.—
«J. Chem. Phys.», 1968, 49, 4925.
2522. Marietta R. P., Curl R. F. — «J. Chem. Phys.»,
1970, 52, 757.
2523. Marliere C, Burie J., Destombes J. L. — «C. r.
Acad. sci.», 1972, B275, 315.
2524. Marmo F. F. — «J. Opt. Soc. America», 1953,
43, 1186.
2525. Marsden M. /., Bird G. R. — «J. Chem. Phys.»,
1973, 59, 2766.
2526. Martignoni P., Morgan R. L., Cason С —«Rev.
Scient. Instrum.», 1965, 36, 1783.
2527. Martin E. V. — «Phys. Rev.», 1932, 41, 167.
2528. Martin E. V., Jenkins F. A. — «Phys. Rev.»,
1931 37 226
2529. Martin E. v'., Jenkins F. A. — «Phys. Rev.»,
1932, 39, 549.
2530. Martin J. J. — «Ind. and Engng Chem.», 1967,
59, 34.
2531. Martin J. J., Hou Y. С — «AIChE Journal»,
1955, 1, 142.
2532. Martin J. J., Hou Y. С — «AIChE Journal»,
1959, 5, 125. ¦
2533. Martin J. J., Stanford T. G. — «AIChE Sympos.
Series», 1974, 70, N140, 1.
2534. Martin W. С — «J. Opt. Soc. America», 1959,
49, 1071.
2535. Martin W. С — «J. Res. NBS», 1960, A64, 19.
2536. Martinek F. — In: Thermodynamic and
Transport Properties of Gases, Liquids and Solids.
New York—Toronto—London, McGraw-Hill Book
Co, 1959, p. 130.
2536a. Martins J. F., Wilson E. В., jr. — «J. Mol.
Spectrosc», 1968, 26, 410.
2537. Maryott A. A., Kryder S. J., Holnes R. R. —
«J. Chem. Phys.», 1965, 43, 2556.
2538. Masanet /., Gilles A., Vermeil С — «J. Photo-
chem.», 1975, 3, 417.
2539. Mason A. A., Nielson A. H. — «J. Opt. Soc.
America», 1967, 57, 1464.
2540. Masri F. N., Blass W. E. — «J. Mol. Spectrosc»,
1971, 39, 98.
2541. Massey J. Т., Beard С I., Jen C. K. — «J.
Mol. Spectrosc», 1960, 5, 405.
2542. Matheson G. L., Maas O. — «J. Amer. Chem.
Soc», 1929, 51, 674.
2543. Mathews J. F. — «Chem. Rev.», 1972, 72, 71.
2544. Mathews W. G., Wallace L. — «J. Atmos. and
Terrest. Phys.», 1961, 20, 1.
2545. Mathieu J. P., Poubt H. — «Spectrochim. acta»,
1960, 16, 696.
2546. Mattrow H. C, Pachucki С F., Hawkins N. J. —
«J. Chem. Phys.», 1954, 22, 1117.
2547. Maxwell L. R., Hendricks S. В., Deming L. S. —
«J. Chem. Phys.», 1937, 5, 626.
2548. Maxwell L. R., Hendricks S. В., Mosley V. M. —
«J. Chem. Phys.», 1935, 3, 699.
2549. Maxwell L. R., Mosley V. M. — «J. Chem.
Phys.», 1940, 8, 738.
2549a. May A. — «Phys. Rev.», 1937, 52, 339.
2550. Mayer H. — «Z. phys. Chem.», 1924, A113, 220
2551. Mayer H. Dissertation. Hannover, 1924.
2552. McAfee К. В. — «Phys. Rev.», 1950, 78, 340.
2553. McAfee К. В. — «Phys. Rev.», 1951, 82, 971.
2554. McAllister Т., Lossing F. P. — «J. Phys. Chem.»,
1969, 73, 2996.
2555. McBride B. J., Gordon S. — «J. Chem. Phys.»,
1961, 35, 2198.
2556. McBride B. J., Heimel S., Ehlers J. G., Gordon
S. Thermodynamic Properties to 6000 К for
210 Substancies Involving the First 18 Elements.
NASA SP-3001. Washington, 1963.
2557. McCaa D. J., Show J. H. — «J. Mol. Spectrosc»,
1968, 25, 374.
2558. McCair D. C, Palke W. E. — «J. Chem. Phys.»,
1972, 56, 4957.
2559. McCallum K. J., Leifer E. — «J. Chem. Phys.»,.
1940, 8, 505.
2560. McCarty M., Robinson G. W. — «J. chim. phys.
et phys.-chim. biol.», 1959, 56, 723.
2561. McCarty R. D. — «J. Phys. and Chem. Ref.
Data», 1973, 2, 923.
2562. McCarty R. D. Hydrogen Technological Survey—
Thermophysical Properties. NASA SP-3089.
Washington, 1975.
2563. McCarty R. D., Weber L. A. Thermophysical
Properties of Oxygen from the Freezing Liquid
Line to 600 К for pressures to 5000 psia. NBS
Techn. Note 384. Washington, 1971.
2564. McCarty R. D., Weber L. A. Thermophysical
Properties of Parahydrogen from Freezing
Liquid line to 5000° К for Pressures to 10 000 psia.
NBS Techn. Note 617. Washington, 1972.
2565. McChesney M. — «AIAA Journal», 1963, 1, 1666.
2566. McClatchey R. A., Benedict W. S., Clough S. A.,
Burch D. E., Calf ее R. F. AFCRL Atmospheric
Absorption Line Parameters Complication. U. S.
Nat. Techn. Inform. Serv., AD Rept, No 762904,
1973.
2567. McClelland B. W., Gundersen G., Hedberg K. —
«J. Chem. Phys.», 1972, 56, 4541.
2568. McConkey J. W., Kernahan J. A. — «Phys.
Lett.», 1968, A27, 82.
2569. McCubbin Т. К. — «J. Chem. Phys.», 1952, 20,.
668.
2570. McCullough J. P., Good W. D. — «J. Phys.
Chem.», 1961, 65, 1430.
2571. McCullough J. P., Pennington R. E., Wadding-
ton G. — «J. Amer. Chem. Soc», 1952, 74, 4439.
2572. McCulloh К. Е., Walker J. A. — «Chem. Phys.
Lett.», 1974, 25, 439.
McDonald С. С. — «J. Chem. Phys.», 1963, 39,
2587.
McDonald G. — «J. Amer. Chem. Soc», 1952,.
74, 5539.
2575. McDonald J. R., Rabalais J. W., McGlynn S. P.
— «J. Chem. Phys.», 1970, 52, 1332-
2576. McDowell R. S. — «J. Chem. Phys.», 1963, 39,
526.
2577. McDowell R. S. — «J. Mol. Spectrosc.», 1964,
12, 34.
2578. McDowell R. S. — «Spectrochim. acta», 1971,
A27, 773.
2578a. McDowell R. S., Aldridge J. P., Holland R. F. —
«J. Phys. Chem.», 1976, 80, 1203.
2579. McDowell R. S., Asprey L. B. — «J. Chem.
Phys.», 1962, 37, 165.
2579a. McDowell R. S., Asprey L. B. — «J. Chem.
Phys.», 1973, 57, 3062.
473
Литература
25796. McDowell R. S., Galbraith H. W., Krohn B. /., 2616.
Cantrell CD., Hinkley E. D. — «Opt. Communs»
(in press). 2616a.
2580. McEwen K. L. — «J. Ghem. Phys.», 1960, 32,
1801.
2581. McEwen K. L. — «J. Ghem. Phys.», 1961, 34, 2617.
547.
2582. McGarvey J. J., McGrath W. D. — «Proc. Roy. 2618.
Soc. London», 1964, A278, 490.
2583. McGibley D. — «Dissert. Abstrs», 1966, 26, 4740. 2619.
2583a. McGlashan M. L., Paul M. A. Manual of Symbols
and Terminology for Physicochemical Quantities 2620.
and Units. IUPAG Additional Publication.
London, Butterworths, 1975. 2621.
2584. McGlynn S. P., Kasha M. — «J. Ghem. Phys.»,
1965, 24, 481. 2621a.
2585. McGrath W. D., McGarvey J. J. — «J. Ghem.
Phys.», 1962, 37, 1574. 2622.
2586. McGrath W.D., McGarvey J. J., DempsterD. N.— 2623.
«Canad. J. Phys.», 1967, 45, 2454. 2623a.
2587. McGraw G. E., Bernitt D. L., Hisatsune I. C. — 2624.
«J. Ghem. Phys.», 1965, 42, 237.
2588. McGraw G. E., Bernitt D. L., Hisatsune I. C. —
«J. Chem. Phys.», 1966, 45, 1392. 2625.
2589. McGurk J. C, Flygare W. H. — «J. Chem.
Phys.», 1973, 59, 5742. 2626.
2590. McLaughlin D. R., Eyring H. — «Proc. Nat.
Acad. Sci. USA», 1966, 55, 1031. 2627.
2591. McMorris J., Yost D. M. — «J. Amer. Chem.
Soc», 1931, 53, 2625. 2628.
2592. McMorris J., Yost D. M.—«J. Amer. Chem. Щ
Soc», 1932, 54, 2247. 2629.
2593. McNeal R. J., Cook G. R. — «J. Chem. Phys.»,
1966, 45, 3469. 2630.
2594. Meakin J. E., Barrow R. F. — «Canad. J. Phys.»,
1962, 40, 377. 2631.
2595. Mears W. H., Rosenthal E., Sinka J. V. — «J.
Phys. Chem.», 1969, 73, 2254.
2596. Mecke R. — «Z. Phys.», 1921, 7, 73.
2597. Mecke R. — «Ann. Physik», 1923, 71, 104.
2598. Mecke R. — «Z. Phys.», 1933, 81, 313. 2632.
2599. Medard L. — «G. r. Acad. sci.», 1934, 199, 421.
2600. Medard L., Thomas M. — «Mem. poudres», 1953, 2633.
35, 155.
2601. Medard M. L. — «G. r. Acad. sci.», 1964, 222, 2634.
1491.
2602. Meerum P. С E. — «Z. anorg. und allgem. 2635.
Chem.», 1905, 47, 206. 2636.
2603. Meinel A. B. — «Astrophys. J.», 1950, 112,
562. 2637.
2604. Meinel A. B. — «C. r. Acad. sci.», 1950, 231,
1049. 2638.
2605. Meinel A. B. — «Astrophys. J.», 1951, 113, 583.
2606. Meinel A. B. — «Astrophys. J.», 1951, 114,
431.
2607. Meinel H., Krauss L. — «Z. Naturforsch.», 1966, 2639.
21a, 1520. 2640.
2608. Meinel H., Krauss L. — «Z. Naturforsch.», 1966,
21a, 1878. 2641.
2609. Melcher D. — «Helv. phys. acta», 1945, 18, 72. 2642.
2610. Melvin E. #., Wuld O. R. — «J. Chem. Phys.», 2643.
1935, 3, 755. 2644.
2611. Melwille H. W., Gray S. C. — «Trans. Faraday
Soc», 1936, 32, 271. 2645.
2612. Menk H. Dissertation. Braunschweig, 1964. 2646.
2613. Menke E. — «Z. Naturforsch.», 1970, 25a, 442.
2614. Menna E., Moccia R., Randaccio L. — «Theor. 2647.
chim. acta», 1966, 4, 408.
2615. Menzel W., Mohry F. —¦ «Z. anorg. und allgem. 2648.
chem.», 1933, 210, 257.
Merer A. J., Achter E. K., Horani M., Rostas
J. — «Ganad. J. Phys.», 1969, 47, 1723.
Merer A. J., Malm D. N., Martin R. W., Horani
M., Rostas J. — «Canad. J. Phys.», 1975, 53,
251.
Merkel P. В., Homill W. H. — «J. Chem. Phys.»,
1971, 55, 2174.
Merton J. R., Pilley J. G. — «Philos. Mag.»,
1925, 50, 195.
MeschiD. J., Myers R. J. — «Spectrochim. acta»,
1956, 8, 311.
Meschi D. J., Myers R. J. — «J. Amer. Chem.
Soc», 1956, 78, 6220.
Meschi D. J., Myers R. J. — «J. Mol. Spectrosc»,
1959, 3, 405.
Metzger P. H., Cook G. R. — «J. Chem. Phys.»,
1964, 41, 642.
Meyer B. — «J. Ghem. Phys.», 1962, 37, 1577.
Meyer B. — «Ghem. Rev.», 1964, 64, 429.
Meyer B. — «Chem. Rev.», 1976, 76, 367.
Meyer В., Brewer L. Sympos. Mol. Struct, and
Spectrosc. Columbus, 1963. Columbus, Ohio.
1963, p. 48.
Meyer В., Brewer L. — In: Rept UGRL, N 10706,
1963, p. 18.
Meyer В., Oommen T. V., Jensen D. — «J. Phys.
Chem.», 1971, 75, 912.
Meyer В., Spitzer K. — «J. Phys. Chem.», 1972,
76, 2274.
Meyer В., Stroyer-Hansen T. — «J. Phys. Chem.»,
1972, 76, 3968.
Meyer В., Stroyer-Hansen Т., Oommen T. V. —
«J. Mol. Spectrosc», 1972, 42, 335.
Meyer C, Haeusler C, Thanh Nguyen Van,
Barchewitz P. — «J. phys.», 1964, 25, 337.
Meyer C. A., McClintock R. В., Silvestri G. J.,
Spencer R. C. Thermodynamic and Transport
Properties of Steam. Comprising Tables and
Charts for Steam and Water. N. Y., ASME,
1968.
Meyer С F., Levin A. A. — «Phys. Rev.», 1929,
34, 44.
Meyer K. A., Crosley D. R. — «Canad. J. Phys.»,
1973, 51, 2119.
Meyerstein D., Treinin A, — «Trans. Faraday
Soc», 1961 , 57, 2104.
Michels A. — «Z. Naturforsch.», 1957, 12a, 887.
Michels A., Wassenaar Т., Louwerse P. — «Phy-
sica», 1960, 26, 539.
Michels H. H. — «J. Ghem. Phys.», 1970, 53,
841.
Mickley H. S. Note on Recommended Values
of Some Thermodynamic Properties of
Hydrogen Peroxide. M. I. T. Rept, N 10 DJC. 6351,
Nord-9107 Fask C, Sept. 1946.
Mie K. — «Z. Phys.», 1934, 91, 475.
Mielke Z., Ratajczak H. — «J. Mol. Struct.»,
1973, 19, 751.
Miescher E. — «Canad. J. Phys.», 1955, 33, 355.
Miescher E. — «Helv. phys. acta», 1956, 29, 135.
Miescher E. — «Helv. phys. acta», 1956, 29, 401.
Miescher E. — «J. Quant. Spectrosc. and Radiat.
Transfer», 1962, 2, 421.
Miescher E. Unpubl. paper (цит. по [559].).
Miescher E., Baer P. — «Nature», 1952,- 169,
581.
Migeotte M., Neven L., Vigroux E. — «Physika»,
1952, 18, 982.
Migeotte M., Neven L., Swensson J. — «Mem.
Soc. roy. sci. Liege», 1957, spec, volume 2.
474
Литература
2649. Millen D. J., Morton J. R. — «J. Ghem. Soc», 2684.
1960, 1523. 2685.
2650. Millen D. J., Pannell J. — «J. Chem. Soc», 2686.
1961, 1322. 2687.
2651. Millen D. J., Sinnot К. М. — «J. Chem. Soc»,
1958, 350. 2688.
2652. Miller С. С. — «J. Ghem. Soc», 1928, 1847.
2653. Miller F. A. —«Pure and Appl. Chem.», 1963, 2689.
7, 125.
2654. Miller F. A., Capwell R. J. — «Spectrochim.
acta», 1971, A27, 125. 2690.
2655. Miller F. A., Wilkins С. Н. — «Analyt. Chem.»,
1952, 24, 1253. 2691.
2656. Miller R. E. — «J. Chem. Phys.», 1965, 43, 1695.
2657. Miller R. E. — «J. Opt. Soc. America», 1970, 2692.
60, 171.
2658. Miller R. E., Getty R. R., Treuil K. L., Leroi 2692a,
G. E. — «J. Chem. Phys.», 1969, 51, 1385.
2659. Miller R. H., Bernitt D. L., Hisatsune I. C. — 2693.
«Spectrochim. acta», 1967, A23, 223.
2660. Miller T. A., Cook T. J., Zegarski B. R., Bre- 2694.
kenridge W. H. — «J. Chem. Phys.», 1973, 58,
1548. 2695.
2661. Milligan D. E., Brown H. W., Pimental G. C. —
«J. Chem. Phys.», 1956, 25, 1080. 2696.
2662. Milligan D. E., Jacox M. E. — «J. Chem. Phys.»,
1963, 38, 2627.
2663. Milligan D. E., Jacox M. E. — «J. Chem. Phys.», 2697.
1964, 40, 2461.
2664. Milligan D. E., Jacox M. E. — «J. Chem. Phys.», 2698
1965, 43, 4487. 2699*
2665. Milligan D. E., Jacox M. E. — «J. Chem. Phys.»,
1971, 55, 1003. 2700.
2666. Milligan D. E., Jacox M. E., Charles S. W.,
Pimental G. С — «J. Chem. Phys.», 1962, 37, 2701
2302 *
2667. Milligan D. E., Jacox M. E., Guillory W. A. — 2702
«J. Chem. Phys.», 1970, 52, 3864.
2668. Millon J., Herman L. — «G. r. Acad. sci.»,
1944, 218, 152.
2669. Mills I. M., Thompson H. W., Williams R. L. —
«Proc. Roy. Soc. London», 1953, A218, 29.
2670. Mills К. С Selected Thermodynamic Data for
Inorganic Sulfides, Selenides and Tellurides. 2705,
London, Butterworth, 1974.
2671. Milne J. В., Ruoff A.—«J. Mol. Spectrosc», 2706.
1967, 23, 408.
2672. Milne T. A., Gilles P. W. — «J. Amer. Chem. 2707.
Soc», 1959, 81, 6115.
2673. Milstein R., Berry R. S. — «J. Chem. Phys.»,
1971, 55, 4146. 2708.
2674. Minkwitz R. — «Z. anorg. und allgem. Chem.»,
1975, 411, 1.
2675. Minnhagen L. — «Arkiv fys.», 1962, 21, 415. 2709.
2676. Minnhagen L. — «Arkiv fys.», 1964, 25, 203.
2677. Minnhagen L,, Strihed H., Petersson B. — «Arkiv 2710.
fys.», 1969, 39, 471.
2678. Mirri A. M., Cazzoli G. — «J. Chem. Phys.», 2711.
1967, 47, 1197.
2679. Mirri A. M., Cazzoli G., Ferretti L. — «J. Ghem. 2712.
Phys.», 1968. 49, 2775.
2680. Mirri A. M., Favero P., Guarnieri A., Semerano 2713.
G. — «Boll, scient. Fac. chim. industr. Bologna»,
1962, 20, 110. 2714.
2681. Mirri A. M., Mazzoaroli E.—«Spectrochim.
acta», 1966, 22, 785. 2715.
2682. Mirri A. M., Scappini F., Favera P. G. —
«Spectrochim. acta», 1965, 21, 965.
2683. Mittelhauser Я. М., Thodos G. — «Cryogenics», 2716.
1964, 4, 368.
Mizushima M. — «Phys. Rev.», 1972, A5, 143.
Moccia R. — «J. Chem. Phys.», 1964, 40, 2176.
ModieaA. P. — «J. Phys. Chem.», 1973, 77, 2713.
Mofitt W. — «Proc. Roy. Soc. London», 1950,
A200, 409.
Mohanty B. S., Rai D. K., Upadhya K. N. —
«Proc. Indian Acad. Sci.», 1968, A68, 165.
Mohanty B. S., Upadhya K. N., Singh R. В.,
Singh N. L.—«J. Mol. Spectrosc», 1967, 24,
19.
Mohler O. C, Benedict W. S. — «Phys. Rev.»,
1948, 74, 702.
Mondain-Monval P. — «Bull, soc chim. France»,
1926, 39, 1349.
Montgomery C. W., Kassel L. S. — «J. Chem,
Phys.», 1934, 2, 417.
Montgomery R. L.—Dissert. Abstrs Intern.,
1976, B36, 5063.
Mookherji A., Tandon S. P. — «Indian J. Phys.»,
1962, 36, 211.
Mookherji A., Tandon S. P.—«Indian J.
Phys.», 1962, 36, 344.
Moore С. В., Rosengren К. — «J. Chem. Phys.»,
1966, 44, 4108.
Moore Ch. E. Ionization Potentials and Ioniza-
tion Limits Derived from the Analyses of
Optical Spectra. NSRDS-NBS 34. Washington, 1970.
Moore Ch. E. Atomic Energy Levels, v. 1, 2, 3.
NSRDS-NBS 35. Washington, 1971.
Moore Ch. E. NSRDS-NBS 3, Sect. 5. 1975.
Moore E. A., Richards W. G.—«Phys. scr.»,
1971, 3, 223. f
Moore G. E. — «J. Opt. Soc. America», 1953,
43, 1045.
Moore G. E., IIarisen H. H. — «J. Chem. Phys.»,
1971, 54, 399.
Morenouse R. L., Christiansen J. J., Gordy W. —¦
«J. Chem. Phys.», 1966, 45, 1747.
Morgan W. A., Silberman E., Morgan H. W. —
«Spectrochim. acta», 1967, A23, 2855.
Morino Y., lijima Т., Murata Y. — «Bull.
Ghem. Soc. Japan», 1960, 33, 46.
Morino Y., Kikuchi Y., Saito S., Hirota E. —
1«J. Mol. Spectrosc», 1964, 13, 95.
Morino Y., Kuchitsu K., Moritani T. — «Inorg.
Chem.», 1969, 8, 867.
Morino Y., Murata Y., Ito Т., Nakamura J. —
«J. Phys. Soc. Japan, Suppl. B-II», 1962, 17,
37.
Morino Y., Oka Т., Kikuchi Y. Proc. Intern.
Sympos. on Molecular Structure and Spectros-
copy. Tokyo, 1962, p. 312.
Morino Y., Saito Sh. —< «J. Mol. Spectrosc»,
1966, 19, 435.
Morino Y., Tanaka T.—«J. Mol. Spectrosc.»,
1965, 16, 179.
Morissette B. G. Thesis. Laval Univ. Quebec,
1952.
Moritani Т., Kuchitsu K., Morino Y. — «Inorg.
Chem.», 1971, 10, 344.
Morizot P., Ostorero J., Plurien P. —> «J. chim.
phys. et phys.-chim. biol.», 1973, 70, 1587.
Morkham A. E., Kobe K. A. -~ «J. Amer. Chem.
Soc», 1941, 63, 449.
Morrison J. D., Hurzeler H., Inghram M. G.,
Stanton H. E. -~ «J. Chem. Phys.», 1960, 33,
821.
Morrow B. 4.^.«Canad. J. Phys.», 1966, 44,
475
Литература
2717. MoTsy Т. Е. — «Бег. Bunsenges. phys. Chem.»,
1964, 68, 277.
2718. Morsy T. E. — «J. Chem. and Engng Data»,
1970, 15, 256.
2719. Morton J. R., Preston K. F. — «Chem. Phys.
Lett.», 1973, 18, 98.
2720. Moskowitz J. W., Harrison M. C. — «J. Chem.
Phys.», 1965, 43, 3550.
2721. Mould H. M., Price W. C, Wilkinson G. P. —
«Spectrochim. acta», 1959, 15, 313.
2722. Mould H. M., Price W. C, Wilkinson G. P. —
«Spectrochim. acta», 1960, 16, 479.
2723. Mountfield K. R., Weir R. D. — «J. Chem.
Phys.», 1976, 64, 1768.
2724. Moureu H., Magat M., Wetroff G. — «C. r.
Acad. sci.», 1937, 205, 276.
2725. Moureu H., Magat M., Wetroff G. — «C. r. Acad.
sci.», 1937, 205, 545.
2726. Moureu H., Magat M., Wetroff G. — «Proc.
Indian Acad. Sci.», 1938, A8, 356.
2727. Moureu H., Rosen В., Wetroff G. — «C. r. Acad.
sci.», 1939, 209, 207.
2728. Mrozowski S., Santaram C. — «J. Opt. Soc.
America», 1965, 56, 1174.
2729. Mrozowski S., Santaram C. — «Bull. Amer.
Phys. Soc», 1966, 11, 311.
2730. Mrozowski S., Santaram C. — «J. Opt. Soc.
America», 1967, 57, 522.
2731. Muck G., Popp H.-P. — «Z. Naturforsch.», 1968.
23a, 1213.
2732. Muenow D. W., Uy O. M., Margrave J. L. —
«J. Inorg. and Nucl. Chem.», 1970, 32, 3459.
2733. Muetterties E. L., Phillips W. D. — «J. Amer.
Chem. Soc», 1959, 81, 1084.
2734. Muhammad S. S., Rao D. H., Hallem M. A. —
«J. Indian Chem. Soc», 1957, 34, 101.
2735. Mulder E., Meulen H. G. L. van der — «Ber.»,
1882, 15, 511.
2736. Mulder E., Meulen H. G. L. van der — «Recueil
trav. chim.», 1882, 1, 73.
2737. Muller A., Glemser O., Niecke E. — «Z. anorg.
und allgem. Chem.», 1966, 347, 275.
2738. Muller A., Glemser O., Niecke E. — «Z.
Naturforsch.», 1966, 216, 732.
2739. Muller A., KrebsB., Niecke E., Ruoff A. — «Ber.
Bunsenges, phys. Chem.», 1967, 71, 571.
2740. Muller A., Nagarajan <?., Krebs B. — «Spectrosc.
Mol.», 1967, 16, 31.
2741. Muller A., Niecke E., Glemser O. — «Z. anorg.
und allgem. Chem.», 1967, 350, 256.
2742. Muller A., Niecke E., Krebs В., Glemser O. —
Z. Naturforsch.», 1968, 23b, 588.
2742a. Muller H. — «Z. Chem.», 1970, 10, 478.
2743. Muller W. — «Z. phys. Chem.», 1926, 123, 1.
2744. Mulliken R. S. — «Phys. Rev.», 1930, 36, 1440.
2745. Mulliken R. S. — «Phys. Rev.», 1934, 46, 549.
2746. Mulliken R. S. — «Phys. Rev.», 1937, 51, 310.
2747. Mulliken R. S. — «Phys. Rev.», 1940, 57, 500.
2748. Mulliken R. S. —«Phys. Rev.», 1942, 61, 277.
2749. Mulliken R. S. — In: The Threshold of Space.
London, 1957, p. 169.
2750. Mulliken R. S. — «Phys. Rev.», 1959, 115, 1225.
2751. Mulliken R. S. — «J. Chem. Phys.», 1960, 33,
247.
2752. Mulliken R. S. — «J. Chem. Phys.», 1962, 37,
809.
2753. Mulliken R. S. — «J. Chem. Phys.», 1971, 55,
288.
2754. Mulliken R. S. — «Chem. Phys. Lett.», 1972,
14, 141.
2755.
2756.
2757.
2757a.
2758.
2759.
2760.
2761.
2762.
2763.
2764.
2765.
2766.
2767.
2768.
2769.
2770.
2771.
2771a.
27716.
2772.
2773.
2774.
2775.
2776.
2776a.
2777.
2777a.
2778.
2779.
2780.
2781.
2782.
2783.
2784.
Mulliken R. S., Liu B. — «J. Amer. Chem,
Soc», 1971, 93, 6738.
Mulliken R. S., Stevens D. S. — «Phys. Rev.»,
1933, 44, 720.
Murai Т., Shimauchi M. — «Sci. Light», 1966,
15, 48.
Murchison S., Reichman S., Anderson D., Overend
J., Schreiner F. — «J. Amer. Chem. Soc», 1968,
90, 5690.
Murphy G. M., «J. Chem. Phys.», 1936, 4, 344.
Murphy G. M., Vance J. E. — «J. Chem. Phys.»,
1939, 7, 806.
Myerson A. L
Chem. Phys.»
Nobar M. A.
Chem. Phys.»
, Taylor F. R., Hanst P. L. — «J.
1957, 26, 1309.
Calvert L. D., Whalley E. — «J.
1969, 51, 1353.
Nachtrieb N. H., Handler G. S. — «J. Chem.
Phys.», 1955, 23, 1187.
Nagarajan G. — «Current Sci.», 19.61, 30, 413.
Nagarajan G. — «J. Scient and Industr. Res.»,
1962, B21, 356.
Nagarajan G. — «Bull. Soc. chim. belg.», 1962,
71, 222.
Nagarajan G. — «Bull. Soc. chim. belg.», 1962,
71, 337.
Nagarajan G. — «Bull. Soc. chim. belg.», 1963,
72, 16.
Nagarajan G. — «Bull. Soc. chim. belg.», 1963,
72, 351.
Nagarajan G. — «Bull. Soc. chim. belg.», 1963,
72, 524.
Nagarajan G. — «Indian J. Pure and Appl.
Phys.», 1963, 1, 403.
Nagarajan G. — «Acta phys. austriaca», 1964,
17, 240.
Nagarajan G. — «Acta phys. austriaca», 1964,
18, 11.
Nagarajan G. — «Bull. Soc. chim. belg.», 1964,
73, 665.
Nagarajan G. — «Indian J. Phys.», 1964, 38r
289.
Nagarajan G. — «Indian J. Pure and Appl.
Phys.», 1964, 2, 237.
Nagarajan G. — «Indian J. Pure and Appl.
Phys.», 1964, 2, 238.
Nagarajan G. — «Bull. Soc. chim. belg.», 1965,
73, 187.
Nagarajan G. — «Acta phys. Acad. scient. hung.»,
1966, 20, 331.
Nagarajan G. — «Acta phys. polon.», 1966, 29,
831.
Nagarajan G. — «Indian J. Pure and Appl.
Phys.», 1966, 4, 244.
Nagarajan G. — «Z. Naturforsch.», 1966, 21a,
244.
Nagarajan G. — «Z. phys. Chem.», 1972, 250,
329.
Nagarajan <?., Durig J. R. — «Bull. Soc. roy.
sci. Liege», 1967, 36, 334.
Nagarajan G., Durig J. R. — «Acta phys. polon.»,
1968, 34, 319.
Nagarajan G., Durig J. R., Muller A. — «Mo-
natsch. Chem..», 1967, 98, 1545.
Nagarajan G., Durig J. R., Perumal A. — «Mo-
natsh. Chem..», 1969, 100, 348.
Nagarajan G., Lippincott E. R., Stutman J. M.—
«J. Phys. Chem.», 1965, 69, 2017.
Nagarajan G., Miller A.^-«Z. Naturforsch.»,
1966, 21b, 612.
476
Литература
2785. Nagatani M., Seiyama Т., Sahiyama M., Suga 2817.
#., Seki S. — «Bull. Chem. Soc. Japan», 1967,
40, N 8, 1833. 2818.
2786. Naggele L. #., Balseschwieler J. D. — «J. Chem.
Phys.», 1962, 37, 182. 2819.
2787. Nagle J. N. — «J. Math. Phys.», 1966, 7, 1484.
2788. Nagy /., Hencsei P., Mester Z. — «Periol. 2820.
Polytechn. Chem. Eng.», 1968, 12, 1.
2789. Nakagawa /., Mizushima S. —> «Bull. Chem. Soe.
Japan», 1955, 28, 589. 2821.
2790. Nakagawa I., Walter J. L. — «J. Cham. Phys.», 2822.
1969, 51, 1389.
2791. Nakamura G. —«Mem. Coll. Sci. Univ. Kyoto», 2823.
1926, A9, 315. 2824.
2792. Nakamura G., Shidei T. — «Jap. J. Phys.»,
1935, 10, 5. 2825.
2793. Nakatsuji H. — «J. Amer. Спзт. Soc», 1973,
95, 354. 2826.
2794. Nalley S. J., Compton R. S. — «Chem. Phys.
Lett.», 1971, 9, 529. 2827.
2795. Nalley S.J., StockdaleJ. A.D., ComptonR.N. —
«Bull. Amer. Phys. Soc», 1970, 15, 418. 2827a.
2796. Namioka Т., Joshino K., Tanaka J. — «J. Chem.
Phys.», 1963, 39, 2629. 2828.
2797. Narasimham N. .4.—«Current. Sci.», 1964, 33,
261. 2829.
2798. Narasimham N. A. Bhabha Atomic Research
Centre. Annual Progress Report to 1972—1973. 2830.
Spectrosc Div. Bombay, 1973.
2799. Narasimham N. A., Apparao K. V. S. R. — 2831.
«Nature», 1966, 210, 1034.
2800. Narasimham N. A., Baghuveer K. Bhabha
Atomic Research Centre. Annual Progress Report 2832.
to 1972—1973. Spectrosc. Div. Bombay, 1973, 2833.
p. 87. 2844
2801. Narasimham N. А., Вalasubramanian Т. К. — *"•"•
«J. Mol. Spectrosc», 1971, 37, 371.
2802. Narasimham N. А., В alasubramanian Т. К. —
«J. Mol. Spectrosc», 1971, 40, 511. 2835-
2803. Narasimham N. A., Bhagvat K. M. N. — «Proc 2836.
Indian Acad. Sci.», 1965, A61, 75.
2804. Narasimham N. A., Brody J. K. — «Proc. In- 2837.
dian Acad. Sci.», 1964, A59, 345.
2805. Narasimham N. A., Dixit M. N., Sethuraman 2838.
V. — «Proc. Indian Acad. Sci.», 1965, A62, 314.
2806. Narasimham N. A., Gopal K. S. — «Current Sci.», 2839.
1965, 34, 454.
2807. Narasimham N. A., Krishnamurty G. — «Proc 2840.
Indian Acad. Sci.», 1966, A64, 97.
2808. Narasimham N. A., Lakshminarayana G. Bhabha 2841.
Atomic Research Centre. Annual Progress
Report to 1972—1973. Spectrosc. Div. Bombay, 2842.
1973, p. 86. 2843.
2809. Narasimham N. A., Raghuveer K., Balasubrama-
nian Т. К. — «J. Mol. Spectrosc», 1975, 54, 160. 2844.
2810. Narasimham N. A., Sethuraman V. Proc. Indian
Sci. Congr., 1963. 2845.
2811. Narasimham N. A., Sethuraman V., Apparao
K. V. S. R. —«J. Mol. Spectrosc», 1976,59,142. 2846.
2812. Narasimham N. A., Srikameswaran K. —«Proc.
Indian Acad. Sci.», 1962, A56, 316. 2847.
2813. Narasimham N. A., Srikameswaran K. —«Proc.
Indian Acad. Sci.», 1962, A56, 325. 2848.
2814. Narasimham N. A., Srikameswaran
K.—«Nature», 1963, 197, 370. 2849.
2815. Narasimham N. A., Srikameswaran K. —«Proc.
Indian Acad. Sci.», 1964, A59, 227. 2849a.
2816. Narasimham N. A., Subramanian Т. К. В.
Proc. Intern. Conf. Spectrosc. Bombay, 1967, 2850.
1, p. 108.
Narasimham N. A., Subramanian Т. К. В. —
«J. Mol. Spectrosc», 1969, 29, 294.
Narayanamurti V., Seward W. D., Pohl R. O. —.
«Phys. Rev.», 1966, 148, 481.
Natta G., Passerini L. — «Nature», 1930, 125,
707.
Natta G., Passerini L. —¦ «Atti Accad. naz. Lin-
cei. Mem. Cl. fis. mat. e natur. Sez. II», =1936,
24, 464.
Naude S. M. — «Nature», 1945, 155, 426.
Naude S. M. — «S. Afrie. J. Sci.», 1945, 41,
128.
Naude S. M. — «Ann. Physik», 1948, 3, 201.
Naude S. M., Christy A. —«Phys. Rev.», 1931,
37, 490.
Naude S. M., Verleger E. — «Z. Phys.», 1950,
128, 173.
Naude S. M., Verleger Я. — «Proc. Phys. Soc.
London», 1950, A63, 470.
Neale E., Williams L. T. D. — «J. Chem. Soc»,
1952, 4535.
Neale E., Williams L. T. D. — «J. Chem. Soc»,
1954, 2156.
Nebgen J. W., Metz F. I., Rose W. B. — «J.
Mol. Spectrosc», 1966, 21, 99.
Neff V. D., Walnut Т. Я. — «J. Chem. Phys.»,
1961, 35, 1723.
Nelson R. C, Benedict W. S. — «Phys. Rev.»,
1948, 74, 703.
Nernst W. — «Sitzungsber. Dtsch. Akad. Wiss.
Berlin, Kl. Math, und allgem. Naturwiss.»,
1910, 262.
Nernst W. — «Ann. Physik», 1911, 36, 395.
Nernst W. — «Z. Elektrochem.», 1916, 22, 37.
Nernst W., Koref F., Lindemann F.
—«Sitzungsber. Dtsch. Akad. Wiss. Berlin, Kl. Math, und
allgem. Naturwiss.», 1910, 247.
Nesbet R. K. — «J. Chem. Phys.», 1965, 43, 4403.
Netherington G., Robinson P. L. —> «J. Chem.
Soc», 1955, 2230.
Neuert H. —i «Ergebn. exakt. Naturwiss.», 1956,
29, 50.
Neuert H., Clasen H. — «Z. Naturforsch.», 1952,
7a, 410.
Neuert H., Rosenbaum O. —¦ «Naturwissenschaf-
ten», 1954, 41, 85.
Neuman В., Mailer G. — «Z. anorg. und allgem.
Chem.», 1929, 182, 232.
Neumann K. — «Z. phys. Chem.», 1934, A171,
416.
Nevin Т. Е. — «Nature», 1937, 140, 1101.
Nevin T. E. — «Philos. Trans. Roy. Soc.
London», 1938, A237, 471.
Nevin Т. Е. — «Proc. Roy. Soc. London», 1940,
A174, 371.
Nevin T. E., Murphy T. — «Proc. Roy. Irish
Acad.», 1941, A46, 169.
Nexen W. E. — «Dissert. Abstrs», 1956, 16,
771.
Ngo T. A., DaPaz M., Coquart В., Couet С —
«Canad J. Phys.», 1974, 52, 154.
Nibler J. W., Barnhart D. M. — «J. Mol.
Spectrosc», 1972, 44, 236.
Nibler J. W., Bondybey V. E. — «J. Chem.
Phys.», 1974, 60, 1307.
Nichols M. J. Thesis. University Durham. U. K.,
1958.
Nichols N. L., Hause С D., Nobble R. H. — «J.
Chem. Phys.», 1955, 23, 57.
477
Литература
2851. Nicholson A. J. С. — «J. Chem. Phys.», 1965, 2884.
43, 1171. 2885.
2852. Nielsen A. H., Jones E. A. — «J. Chem. Phys.»,
1951, 19, 1117. 2886.
2853. Nielsen A. H., Jones E. A. — «Phys. Rev.», 2887.
1951, 83, 485.
2854. Nielsen A. H., Nielsen H. H. — «Phys. Rev.», 2888.
1935, 47, 585. 2889.
2855. Nielsen A. H., Shelton R. D., Fletcher W. H. —
«J. phys. et radium», 1954, 15, 604.
2856. Nielsen A. #., Woltz P. J. H. — «J. Chem. 2890.
Phys.», 1952, 20, 1878.
2857. Nielsen H. — «Phys. Rev.», 1941, 59, 565.
2858. Nielsen H. — «Phys. Rev.», 1942, 62, 422. 2891.
2859. Nielsen H. H., Barker E. F. — «Phys. Rev.»,
1931, 37, 727. 2892.
2860. Nielsen J. R., Liang С Y., Smith D. С — «J.
Chem. Phys.», 1952, 20, 1090. 2893.
2861. Nielsen J. R., Ward N. E. — «J. Chem. Phys.»,
1942, 10, 81. 2894.
2861a. Nielsen V., Haensel R., Schwartz W. H. E. —
«J. Chem. Phys.», 1974, 61, 3581. 2895.
28616. Nielsen V., Schwarz W. H. E. — «Chem. Phys.»,
1976, 13, 195. 2896.
2862. Niessen W. — «Theor. chim. acta», 1973, 31, 297.
2863. Nightingale R. E., Wagner E. L. — «J. Chem. 2897.
Phys.», 1954, 22, 203.
2864. Nimon L. — «Dissert. Abstrs», 1968, B28, 4533. 2898.
2865. Nimon L. A., Neff V. D. — «J. Mol. Spectrosc»,
1968, 26, 175. 2899.
2866. Nimon L. A., Neff V. D., Cantley R. E., But-
tlar R. O. — «J.Mol. Spectrosc.», 1967, 22, 105. 2900.
2867. Noble R. H., Nielsen H. H. — «J. Chem. Phys.»,
1950, 18, 667. 2901.
2868. Nobble P. N., Pimentel G. C. — «Spectrochim.
acta», 1968, A24, 797. 2902.
2868a. Nordine P. С — «J. Chem. Phys,», 1974, 61, 224. 2903.
2869. Norrish R. G. W. — «J. chim. phys. et phys.-
chim. biol.», 1964, 61, 1359. 2904.
2870. Norrish R. G. W., Oldershaw G. A.— «Proc.
Roy. Soc. London», 1959, A249, 498. 2905.
2871. Nostrand E. D., Duncan A. B. F. — «J. Amer.
Chem. Soc», 1954, 76, 3377. 2906.
2872. Nothnagel K. H., AbramsD. S., Pransnitz J. M. —
«Ind. and Engng Chem. Process. Design and De- 2907.
velopm.», 1973, 12, 25.
2873. Novick S. E., Howard B. J., Klemperer W. — 2908.
«J. Chem. Phys.», 1972, 57, 5619.
2873a. Nuttall R. L., Armstrong G. T. NBS Rept 10326; 2909.
1 July 1970. Washington.
2874. Oakland R. L., Duffey G. H. — «J. Chem. Phys.», 2910.
1967, 46, 19. 2911.
2875. O'Brien С J., Perrin J. R., Perrine J. — In: 2912.
Kinetics, Equilibria and Performance of High
Temperature Systems. Bohn G. S., Zukoski E. E. 2913.
(Eds). London, Butterworths, 1960, p. 5.
2876. O'Connor S. — «Proc. Roy. Irish Acad.», 1962, 2914.
A62, 73. 2915.
2877. Oelfke W. C, Gordy W. — «J. Chem. Phys.»,
1969, 51, 5336. 2916.
2878. Oelsen W., Oelsen O. — «Z. Elektrochem.», 1956,
60, 157. 2917.
2879. Ogawa H. S., Ogawa M. — «J. Mol. Spectrosc»,
1975, 55, 56. 2917a.
2880. Ogawa M. — «Sci. Light», 1966, 15, N 2, 97.
2881. Ogawa M. — «Canad. J. Phys.», 1968, 46, 312. 29176.
2882. Ogawa M., Chang H. — «Sci. Light», 1968,
17, N 2, 45.
2883. Ogawa M., Tanaka Y. — «Canad. J. Phys.», 2918.
1962, 40, 1599.
Ogier J. — «С. г. Acad. sci.», 1878, 87, 210.
Ogilvie J. F. — «Spectrochim. acta», 1967, A23,
737.
Ogilvie J. F. — «Nature», 1973, 243, 210.
Ogilvie J. F. — «Canad. J. Spectrosc», 1974,
19, 171.
Ogilvie J. F. — «J. Mol. Struct.», 1976, 31, 407.
O'Hare P. A. G. —The Thermodynamic
Properties of Some Chalcogen Fluorides. U. S. Atom.
Energy Commiss. Rept ANL-7315, 1968.
O'Hare P. A. G. — Thermodynamic Properties
of P2, P4 and Some Phosphorus Fluorides. U. S.
Atom. Energy Commiss. Rept ANL-7459, 1968.
O'Hare P. A. G. — «J. Chem. Phys.», 1970, 52,
2992.
O'Hare P. A. G. — «J. Chem. Phys.», 1973, 59,
3842.
O'Hare P. A. G., Hubbard W. TV.—«Trans.
Faraday Soc», 1966, 62, 2709.
O'Hare P. A. G., Hubbard W. N., Glemser O.,
WegenerJ. — «J. Chem. Thermodyn.», 1970, 2, 71.
O'Hare P. A. G., Hubbard W. N., Glemser O.,
Wegener J. — «J. Chem. Thermodyn.» (in press).
O'Hare P. A. G., Settle J. L., Hubbard W. N. —
«Trans. Faraday Soc», 1966, 62, 558.
O'Hare P.A.G., WahlA. C. — «J. Chem. Phys.»,
1970, 53, 2469.
O'Hare P. A. G., WahlA. С — «J. Chem. Phys.»,
1970, 53, 2834.
O'Hare P. A. G., WahlA. C. — «J. Chem. Phys.»,
1971, 54, 4563.
Oka Т., Morino Y. — «J. Mol. Spectrosc», 1963,
11, 349.
Okabe H. — «J. Amer. Chem. Soc», 1971, 93,
7095.
Okabe H. — «J. Chem. Phys.», 1972, 56, 3378.
Okabe H., Lenzi M. — «J. Chem. Phys.». 1967,
47, 5241.
Oldman R. J., Sander R. K., Wilson K. R. —
«J. Chem. Phys.», 1970, 54, 4127.
Oldman R. J., Sander R. K., Wilson K. R. —
«J. Chem. Phys.», 1975, 63, 4252.
Olman M. D., Hause C. D. — «J. Mol. Spectrosc»,
1968, 26, 241.
Olman M. D., McNelis M. D., Hause C. D. —
«J. Mol. Spectrosc», 1964, 14, 62.
O'Loane J. K. — «J. Chem. Phys.», 1953, 21,
669.
O'Loane J. K., Wilson M. K. — «J. Chem.
Phys.», 1955, 23, 1313.
Olsson E. — «Z. Phys.», 1936, 99, 114.
Olsson E. — «Z. Phys.», 1936, 100, 656.
Olsson E. — «Arkiv mat., astron., fys.», 1937,
B25, N 30, 1.
Olsson E. — «Arkiv mat., astron., fys.», 1938,
B26, N 9, 4.
Olsson E. — Dissertation. Stockholm, 1938.
O'Neil S. V., Schaefer H. F. — «J. Chem.
Phys.», 1971, 55, 394.
Opik U., Thomas Т. Н. — «Mol. Phys.», 1966,
10, 289.
Osaka Т., Takahashi Sh. — «J. Phys. Soc. Japan»,
1968, 25, 1654.
Osborne D. W., Flotow H. E., Malm J. G. —
«J. Chem. Phys.», 1972, 57, 4670.
Osborne D. W., Schreiner F., Flotow H. E.,
Malm J. G. — «J. Chem. Phys.», 1972, 57,
3401.
Osborne D. W., Schreiner F., Selig H. — «J.
Chem. Phys.», 1971, 54, 3790.
478
Литература
2919. Osborne N. S., Stimson H. F., Ginnings D. С —
«J. Res. NBS», 1939, 23, 197.
2920. Oskam A., Elst R., Duinker J. С — «Spectro-
chim. acta», 1970, A26, 2021.
2921. Ossenbruggen W. — «Z. Phys.», 1928, 49, 167.
2922. Otake M., Hirota E., Morino Y. — «J. Mol.
Spectrosc», 1968, 28, 325.
2923. Otake M., Matsumura C, Morino Y. — «J. Mol.
Spectrosc.», 1968, 28, 316.
2924. Ott J. В., Giauque W. F. — «J. Amer. Chem.
Soc», 1960, 82, 1308.
2924a. Otvos J. W., Stevenson D. P. — «J. Amer. Chem.
Soc», 1956, 78, 546.
2925. Oura H. — «Low Temperature Sci.», 1951, A6,
41.
2926. Oura H., Ninomiya M. — «Proc. Phys. Soc.
Japan», 1943, 25, 335.
2927. Owens F. J., Adams G. F. — «J. Chem. Phys.»,
1972, 57, 2212.
2928. Ozin G. A. — «J. Chem. Soc», A, 1969, 116.
2929. Ozin G. A. — «J. Chem. Soc. Chem. Communs»,
1969, 1325.
2930. Pacansky I., Calder V. — «J. Chem. Phys.»,
1970, 53, 4519.
2931. Pace E. L., Petrella R. V. J. — «J. Chem. Phys.»,
1962, 36, 2891.
2932. Pace E. L., Pierce L. — «J. Chem. Phys.», 1955,
23, 1248.
2933. Pace E. L., Samuelson H. V. — «J. Chem. Phys.»,
1966, 44, 3682.
2934. Pace E. L., Turnbull B. F. — «J. Chem. Phys.»,
1965, 43, 1953.
2935. Pack J. L., Phelps А. К — «J. Chem. Phys.»,
1966, 44, 1870.
2936. Page F. M. — «Trans. Faraday Soc», 1960, 56,
1742.
2937. Page F. M. — «Trans. Faraday Soc», 1961, 57,
1254.
2938. Page F. M. — «Advanc. Chem.», 1962, 36, 68.
2939. Page F. M., Goode G. C. Negative Ions and the
Magnetron. London, Wiley Interscience, 1969.
2940. Palik E. D. — «J. Chem. Phys.», 1955, 23, 217.
2941. Palik E. D., Rao K. N. — «J. Chem. Phys.»,
1956, 25, 1174.
2942. Palm A., Kilpatrik M.—«J. Chem. Phys.»,
1955, 23, 1562.
2943. Palmer A. B. Dissertation. Syracuse University,
1963.
2944. Palmer K. J. — «J. Amer. Chem. Soc», 1938,
60, 2360.
2945. Palmer T. F., Lossing F. P. — «J. Amer. Chem.
Soc», 1962, 84, 4661.
2946. Panish M. В., Arthur J. R. — «J. Chem. Thermo-
dyn.», 1970, 2, 299.
2947. Paukert Т. Т., Johnston H. S. — «J. Chem.
Phys.», 1972, 56, 2824.
2948. Pannetier G., Dessaux O., Arditi I., Goudmand
P. — «C. r. Acad. sci.», 1964, 259, 2198.
2949. Pannetier G., Dessaux O., Arditi I., Goudmand
P. — «Bull. Soc chim. France», 1966, N 1, 313.
2950. Pannetier G., Gaydon A. G. — «Nature», 1948,
161, 242.
2951. Pannetier G., Goudmand P., Dessaux O., Taver-
nier N. — «C. r. Acad. sci.», 1962, 255, 91.
2952. Pannetier G., Goudmand P., Dessaux O., Taver-
nier N. — «J. Chem. Phys.», 1964, 60, 395.
2953. Pannetier G., Guenbaut H. — «Bull. Soc. chim.
France», 1959, 190, 962.
2954. Pan Wong D., Fink W. #., Allen L. С — «J.
Chem. Phys.», 1970, 52, 6291.
2955. PapousekD. — «Collect. Czech. Chem. Communs»,
1961, 26, 1909.
2956. PapousekD. — «Collect. Czech. Chem. Communs»,
1962, 27, 1.
2957. Papousek D., Pliva J. — «Collect. Czech. Chem.
Communs», 1964, 29, 1973.
2958. Parker A. E. — «Phys. Rev.», 1933, 44, 914.
2959. Parker V. B. Thermal Properties of Aqueous
Uni-univalent Electrolytes (Category 5 — Ther-
modynamic and Transport Properties). NSRDS-
NBS-2. Washington, 1965.
2960. Parkinson T. F., Jones E. A., Nielsen A. H. —
«Phys. Rev.», 1949, 76, 199.
2961. Parlant G., Fiquet-Fayard F. — «J. Phys." B:
Atom, and Mol. Phys.», 1976, 9, 1617.
2962. Parrish D. D., Herschbach D. R. — «J. Amer.
Chem. Soc», 1973, 95, 6133.
2963. Pascard R. — «C. r. Acad. sci.», 1955, 240, 2162.
2964. Pascard-Billy С — «Acta crystallogr.», 1965r
18, 829.
2965. Pascat В., Berthou J. M., Prudhomme J. C,
Guenebaut ff., Ramsay D. A. — «J. chim. phys.
et phys.-chim. biol.», 1968, 65, 2022.
2966. Pascat В., Berthou J. M., Guenebaut Л., Ramsay
D. A. — «C. r. Acad. sci.», 1966, B263, 1397.
2967. Pascat В., Guenebaut H. — «Ann. Univ. et Assoc.
reg. etude et rech. scient», 1963, 2, 100.
2968. Patch R. W. — «J. Chem. Phys.», 1968, 49, 961.
2969. Patch R. W. — «J. Chem. Phys.», 1968, 49, 963.
2970. Patel M. M. — «Z. Phys.», 1963, 173, 347.
2971. Pathak C. M., Palmer H. B. — «J. Mol.
Spectrosc», 1969, 32, 157.
2972. Patkowski J., Curtis W. E. — «Trans. Faraday
Soc», 1929, 25, 725.
2973. Patterson G. D., Dworetsky S. H., Hozack R. S. —
«J. Mol. Spectrosc», 1975, 55, 175.
2974. Раисе E. L., Jepson В. Е. — «J. Chem. Phys.»,
1970, 52, 911.
2975. Pauling L. — «J. Amer. Chem. Soc», 1935, 57,
2680.
2976. Pauling L., Brockway L. O. — «J. Amer. Chem.
Soc», 1935, 57, 2684.
2977. Pauling L., Brockway L. O. — «J. Amer. Chem.
Soc», 1937, 59, 13.
2978. Pearse R. W. B. — «Proc. Roy. Soc. London»,
1930, A129, 328.
2979. Pearse R. W. B. — «Proc. Roy. Soc London»,
1933, A143, 112.
2980. Pearson A. G., Poshysta R. D., Browne J. С —
«J. Chem. Phys.», 1966, 44, 1815.
2981. Pearson E. F., Kim H. — «J. Chem. Phys.»,
1972, 57, 4230.
2982. Pearson P. K., Schaefer H. F., Richardson T. H.r
Stephenson L. M., Brauman T. I. — «J. Amer.
Chem. Soc», 1974, 96, 6778.
2983. Pearson R. G. — «J. Amer. Chem. Soc», 1969,
91, 4947.
2984. Pearson R. G. — «J. Chem. Phys.», 1970, 52,
2167.
2985. Pearson R. G., Sullivan Т., Frenkel L. — «J.
Mol. Spectrosc», 1970, 34, 440.
2985a. Peck J., Fenichel H. — «Phys. Lett.», 1974,
A49, 97.
2986. Pedersen L., Morokuma K. — «J. Chem. Phys.»,
1967, 46, 3941.
2987. Pekeris С L. — «Phys. Rev.», 1962, 126, 1470.
2988. Pemsler J. P., Planet W. G. — «J. Chem. Phys.»,
1956, 24, 920.
479
Литература
2989. Pennington R. E., Kobe K. A. ~ «J. Chem.
Phys.», 1954, 22, 1442.
2990. Perkins A. J. — «Speotrochim. acta», 1968, A24,
285.
2991. Perkins W. D., Wilson M. K. — «J. Chem.
Phys.», 1952, 20, 1791.
2992. Perrizo J., Pugh L. A., Rao K. N. — «J. Mol.
Spectrosc», 1975, 57, 397.
2993. Person С. С. — «С. г. Acad. sci.», 1846, 23, 163.
2994. Person С. С. — «С. г. Acad. sci.», 1846, 23, 336.
2995. Person С. С. — «Ann. chim. phys.», 1847, 21,
295.
2996. Persson W., Minnhagen L. — «Arkiv fys.», 1968,
37, 273.
2996a. Peslak J., Klett D. S., David C. W. — «J. Amer.
Ghem. Soc», 1971, 93, 5001.
2ЭЭ66. Peterson S. W., Willett R. D., Huston J. L. —
«J. Chem. Phys.», 1973, 59, 453.
2997. Petersson B. — «Arkiv fys.», 1964, 27, 317.
2998. Petrikaln A.—nZ. Phys.», 1924, 22, 119.
2999. Petrikaln A.— «Z. Phys.», 1928, 51, 395.
3000. Pettersson O. — «J. prakt. Chem.», 1881, 24, 296.
3001. Petty F., Moran T. F. — «Chem. Phys. Lett.»,
1970, 5, 64.
3002. Peyerimhoff S. D., Buenker R. J. — «J. Chem.
Phys.», 1968, 49, 2473.
3003. Peyron M. — «Advanc. Mol. Spectrosc.», 1962,
2, 509.
3004. Pfeiffer G. V., Allen L. С — «J. Chem. Phys.»,
1969, 51, 190.
3005. Phelps A. V., Pack J. L. —«Phys. Rev. Lett.»,
1961, 6, 111.
3006. Phillips L. F., Smith J. J., Meyer B. — «J. Mol.
Spectrosc.», 1969, 29, 230.
3007. Phillips L. F., Sugden T. M. — «Trans. Faraday
Soc», 1961, 57, 914.
3008. Peyson M., Lam Thanh My. — «J. chim. phys.
et phys.-chim. biol.», 1967, 64, 129.
3009. Piccardi G. —¦ «Z. Phys.», 1927, 43, 899.
3010. Pick M. A., Wenzl H., Engelhardt H. — «Z.
Naturfor3ch.», 1971, 26a, 810.
3011. Pickworth J., Thompson H. W. — «Proc. Roy.
Soc. London», 1953, A218, 37.
3012. Pier M. — «Z. phys. Chem.», 1908, 62, 385.
3013. Pier M. — «Z. Elektrochem.», 1909, 15, 536.
3014. Pier M. — «Z. Elektrochem.», 1910, 16, 897.
3015. Pierce J. — «J. Chem. Phys.», 1956, 24, 139.
3016. Pierce L., Di Cianni N., Jackson R. H. — «J.
Chem. Phys.», 1963, 38, 730.
3017. Pierce L., Jackson R., Di Cianni N. — «J. Chem.
Phys.», 1961, 35, 2240.
3018. Pierce L., Pace E. L. — «J. Chem. Phys.», 1955,
23, 551.
3019. Pierre G. S., Chipman J. — «J. Amer. Chem.
Soc», 1954, 76, 4787.
3020. Piette L. H., Johnson F. A., Booman K. A.,
Colburn С. В. — «J. Chem. Phys.», 1961, 35,
1481.
3021. Pillai M. G. K. — «Z. phys. Chem.», (DDR),
1961, A218, 334.
3022. Pillai M. G. K., Curl R. F. — «J. Chem. Phys.»,
1962, 37, 2921.
3023. Pillai M. G. K., Gnanadesican S. G. — «Z. phys.
Chem.», (DDR), 1965, A228, 157.
3024. Pillai M. G. K., Ramaswamy K., Pichai R. —
«Austral. J. Chem.», 1965, 18, 1575.
3025. Pillai M. G. K., Ramaswamy K., Pichai R. —
«Canad. J. Chem.», 1965, 43, 463.
3026. Pimentel G. C, Charles С W., Rozengren K. —
«J. Chem. Phys.», 1966, 44, 3029.
3027. Piotrowski E. A., Ziomek J. S. —«Phys. Rev.»,
1955, 100, 1267.
3028. Piper T. S., Drago R. S. — «J. Chem. Phys.»,
1962, 36, 241.
3029. Pisati G., De Frenchis G. — «Gazz. chim. ital.»,
1874, 4, 497.
3030. Pistorius С W. F. T. — «J. Chem. Phys.», 1969,
50, 1436.
3031. Pitzer K. S. — «J. Chem. Phys.», 1946, 14, 239.
3032. Pitzer K. S. — «J. Amer. Chem. Soc», 1955,
77, 3427.
3032a. Pitzer K. S., Bernstein L. S. «J. Chem. Phys.»,
1975, 63, 3849.
3033. Pitzer K. S., Curl R. — «J. Amer. Chem. Soc»,
1957, 79, 2369.
3034. Pitzer K. S., Gwinn W. G. — «J. Chem. Phys.»,
1942, 10, 428.
3035. Pitzer K. S., Lippmann D. Z., Curl R. F., Hug-
gins C. M., Petersen D. E. — «J. Amer. Chem.
Soc», 1955, 77, 3433.
3036. Plaats J. D. — «Recueil trav. chim.», 1886, 5,
45.
3037. Planck M. — «Ann. Physik», 1924, 75, 673.
3038. Plato V., Hartford W. D., Hedberg K. — «J.
Chem. Phys.», 1970, 53, 3488.
3038a. Pliva J. — «Collect. Czech. Chem. Communs»,
1958, 5, 777.
3039. Pliva J. — Intern. Sympos. Mol. Struct, and
Spectrosc, Tokyo, 1962, SI, C222/1.
3040. Pliva J. — «J. Mol. Spectrosc», 1964, 12, 360.
3041. Pliva J. — «J. Mol. Spectrosc», 1968, 25, 62.
3042. Pliva J. — «J. Mol. Spectrosc», 1968, 27, 461.
3043. Pliva J. — «J. Mol. Spectrosc», 1970, 33, 500.
3044. Plyler E. K. — «Phys. Rev.», 1932, 39, 77.
3045. Plyler E. K. — «J. Res. NBS», 1960, A64, 377.
3046. Plyler E. K., Barker E. F. — «Phys. Rev.»,
1933, 44, 984.
3047. Plyler E. K., Gailar N., Wiggins T. — «J. Res.
NBS», 1952, 48, 221.
3048. Plyler E. K., Sleator W. — «Phys. Rev.», 1931,
37, 1493.
3049. Plyler E. K., Tidwell E. D. — «Mem. Soc roy.
sci. Liege», 1957, 18, 426.
3050. Plyler E. K., Tidwell E. D. — «Z. Elektrochem.»,
1960, 64, 717.
3051. Plyler E. K., Tidwell E. D., Allen H. С — «J.
Chem. Phys.», 1956, 24, 95.
3052. Plyler E. K., Tidwell E. D., Maki A. G. — «J.
Res. NBS», 1964, A68, 79.
3053. Pocock G., Wormald C. J. — «J. Chem. Soc.
Faraday Trans.», 1975, pt 1, 71, 705.
3054. Polak J., Liu В. С J. — «Canad. J. Chem.
Engng», 1972, 50, 553.
3055. Poland D. E., Green J. W., Margrave J. L. —
«J. Chem. and Engng Data», 1962, 7, 389.
3056. Pollack G. L., Broida H. P. — «J. Chem. Phys.»,
1963, 38, 2012.
3057. PollakR. — «Brennstoff-Warme-Kraft», 1975, 27,
210.
3058. Pollitzer F. — «Z. anorg. und allgem. Chem.»'
1909, 64, 121.
3059. Pollitzer F. — «Z. Elektrochem.», 1913, 19, 513.
3060. Polo S. R., Wilson M. K. — «J. Chem. Phys.»,
1954, 22, 900.
3061. Pople J. A. — «Mol. Phys.», 1975, 29, 599.
3062. Pople J. A., Beveridge D. L., Dobosh P. A. —
«J. Chem. Phys.», 1967, 47, 2026.
480
Литература
3063. Pople J. A., Segal G. A. — «J. Chem. Phys.»,
1966, 44, 3289.
3064. Popp H. P. — «Z. Naturforsch.», 1967, 22a, 254.
3065. Popplewell R. J. L., Masri F. W., Thompson
H. V. — «Spectrochim. acta», 1967, A23, 2797.
3066. Porter G. — «Disc. Faraday Soc», 1950, 9, 60.
3067. Porter G. — «Proc. Roy. Soc. London», 1950,
A200, 284.
3068. PosenerD. W. MIT Res. Lab. Electr. Techn.,
Rept 225, 1953.
3069. Posener D. W. — «Austral. J. Phys.», 1957,
10, 376.
3070. Posener D. W., Strandberg M. W. P. — «J.
Chem. Phys.», 1953, 21, 1401.
3071. Potter R. L. ~ «J. Chem. Phys.», 1949, 17, 957.
3072. Potter R. L. ~ «J. Chem. Phys.», 1957, 26, 394.
3073. Potter R. L. — «J. Chem. Phys.», 1959, 31, 1100.
3074. Potter R. L., Distefano V. N. — «J. Phys. Chem.»,
1961, 65, 849.
3*075. Poulis J. A., Massen С. Н. — In: Elemental
Sulfur. Chemistry and Physics. Meyer В., Kha-
rasch A. N. (Eds). New York — London —
Sydney, Intersci. Publ., 1965, p. 109.
3076. Powell F. X., Jonson D. R. — «J. Chem. Phys.»,
1969, 50, 4596.
3077. Powell F. X., Lide D. R. — «J. Chem. Phys.»,
1964, 41, 1413.
3078. Powley G. S., Rinaldi R. P. — «Acta crystal-
logr.», 1972, B28, 3605.
3079. Poynter R. L., Beaudet R. A. — «Phys. Rev.
Lett.», 1968, 21, 305.
3080. Poynter R. L., Kakar R. K. — «Astrophys. J.
Suppl. Ser.», 1975, 29, 87.
3081. Prausnitz J. M. Molecular Thermodynamics of
Fluid—Phase Equilibria. Englewood Cliffs (New
(Jersey), Prentice-Hall Inc., 1969.
3082. Preuner G. — «Z. anorg. und allgem. Chem.»,
1907, 55, 279.
3083. Preuner G., Brockmoller J. — «Z. phys. Chem.»,
1912, 81, 129.
3084. Preuner G., Schupp W. — «Z. phys. Chem.»,
1909, 68, 129.
3085. Preuner G., Schupp W. — «Z. phys. Chem.»,
1909, 68, 157.
3086. Price W. С. ~ «J. Chem. Phys.», 1936, 4, 147.
3087. Price W. C. — «Proc. Roy. Soc. London», 1938,
A167, 216.
3088. Price W. C, Sherman W. F., Wilkinson G. R. —
«Proc. Roy. Soc. London», 1960, 4255, 1.
3089. Price W. C, Simpson D. M. — «Trans. Faraday
Soc», 1941, 37, 106.
3090. Price W. C, Teegan J. P., Walsh A. D. — «Proc.
Roy. Soc. London», 1950, A201, 600.
3091. Pritchard H. O. — «Chem. Rev.», 1953, 52, 529.
3092. Prudhomme J. C, Coquart B. — «Canad. J.
Phys.», 1974, 52, 2150.
3093. Prudhomme J. C, Larzilliere M., Couet С —
«Canad. J. Phys.», 1973, 51, 2464.
3094. Prydz R., Straty G. С — «J. Res. NBS», 1970,
A74, 747.
3095. Prydz R., Straty G. C. The Thermodynamik
Properties of Compressed Gaseous and Liquid
Fluorine. U. S. Techn. Note, N 392,
Washington, NBS, 1970.
3096. Prydz R., Straty G. C, Timmerhaus K. D. —
«J. Chem. Phys.», 1970, 53, 2389.
3097. Prydz R., Timmerhaus K. D., Stewart R. B. —
«Advanc. Cryogen. Engng», 1968, 13, 384.
3098. Pugh L. A. Dissertation. Ohio State Univ.
1972.
3099. Pugh L. A., Rao K. N. — «J. Mol. Spectrosc»,
1971, 37, 375.
3100. Pugh L. A., Rao K. N. — «J. Mol. Spectrosc.»,
1973, 47, 403.
3101. Puglielli V. G., Ford N. С — «Phys. Rev. Lett.»,
1970, 25, 143.
3102. Pulford A. G., Walsh A.— «Trans. Faraday
Soc», 1951, 47, 347.
3103. Puranik P. G., Rao E. V. — «Indian J. Phys.»,
1961, 35, 177.
3104. Puranik P. G., Sirdeshmukh L. Proc. Intern.
Conf. Spectrosc, v. 2. Bombay, Jan. 9—18.
1967, p. 358.
3105. Pyper J. W., Newbury R. S., Barton G. W. —
«J. Chem. Phys.», 1967, 46, 2253.
3106. Rabalais J. W., McDonald J. M., Scherer V.,
McGlynn S. P. «Chem. Rev.», 1971, 71, 73.
3107. Rabinovitch B. S., Waage E. V. — «Chem.
Rev.», 1970, 70, 377.
3108. Rabinowitch E. — «Rev. Mod. Phys.», 1942,
14, 112.
3109. Rabinowitch E., Stockmayes W. H. — «J. Amer.
Chem. Soc», 1942, 64, 335.
3110. Rabinowitch E., Wood W. C. — «Trans. Faraday
Soc», 1936, 32, 540.
3111. Radford H. E. — «Phys. Rev. Lett.», 1964, 13,
5o4t.
3112. Radford H. E., Evenson K. M., Howard С J. —
«J. Chem. Phys.», 1974, 60, 3178.
3113. Radhakrishnan M. — «Z. Naturforsch.», 1963,
18a, 103.
3114. Radhakrishnan M. «Indian J. Pure and Appl.
Phys.», 1963, 1, 437.
3115. Radlein D. St. A. G., Whitehead J. C, Grice
R. — «Mol. Phys.», 1975, 29, 1813.
3116. Radlein D. St. A. G., Whitehead J. C, Grice
R. — «Nature», 1975, 253, 37.
3117. Radon L., Schaefer H. F. — «Austral. J. Chem.»,
1975, 28, 2069.
3118. Radziemski J., Kaufman V. — «J. Opt. Soc.
America», 1969, 59, 424.
3119. Rai S. В., Rai D. K., Upadhya K. N. — «J.
Phys. B: Atom, and Mol. Phys.», 1972, 5, 1038.
3119a. Rai S. В., Yadov B. R., Rai D. K. — «J. chim.,
phys. et phys.-chim. biol.», 1976, 73, 905.
3120. Rajan K. J. — «Proc. Roy. Irish Acad.», 1974,
A74, 17.
3121. Rajzmann M., Pouzard G., Bouscasse L. — «C. r.
Acad. sci.», 1971, C273, 595.
3122. Rakotoarljimy D.—«C. r. Acad. sci.», 1968,
B266, 778.
3123. Rakotoarijimy D. — «Physica», 1969, 41, 29.
3124. Rakotoarijimy D. — «Physica», 1970, 49, 360.
3125. Ramanadham R., Rao G. V. S. R. — «Current
Sci.», 1949, 14, 230.
3126. Ramanadham R., Rao G. V. S. R., Ramasaatry
C. — «Indian J. Phys.», 1946, 20, 161.
3127. Ramaswamy K., Jayaraman S. — «Indian J.
Phys.», 1973, 47, 177.
3128. Ramaswamy K., Rao B. — «Z. phys. Chem.»
(DDR), 1969, 242, 18.
3129. Ramaswamy K., Muthusubramanian P. — «J.
Mol. Struct.», 1971, 7, 45.
3130. Ramaswamy K., Ranganathan V. — «IndianJ.
Pure and Appl. Phys.», 1968, 6, 651.
3131. Ramsay D. Д.—«J. Chem. Phys.», 1952, 20,
1920.
481
Литература
3132.
3133.
3134.
3135.
3136.
3137.
3138.
3139.
3140.
3141.
3142.
3143.
3144.
3145.
3146.
3147.
3148.
3149.
3150.
3151.
3152.
3153.
3154.
3155.
3156.
3157.
3158.
3159.
3160.
3161.
3161а.
3162.
3163.
3164.
3164а.
3165.
3167.
3168.
3169.
3170.
3171.
Rank D. H., Eastman D. P., Rao B. S., Wig-
gins T. A. — «J. Opt. Soc. America», 1962, 52, 1.
Rank D. #., Fink U., Wiggins T. A.— «J.
Mol. Spectrosc», 1965, 18, 170.
Rank D. H., Larsen K., Bordner E. — «J. Chem.
Phys.», 1934, 2, 464.
Rank D. #., Rao B. S. — «J. Mol. Spectrosc.»
1964, 13, 34.
Rank D. #., Rao B. S., Wiggins T. A. — «J.
Mol. Spectrosc», 1965, 17, 122.
Rao B. S. — «Dissert. Abstrs», 1965, 25, 4972.
Rao K. N. — «Indian J. Phys.», 1943, 17, 135.
Rao K. N., Brim W. W., Sinnett V. L., Wil-
son R. И. — «J. Opt. Soc. America», 1962, 52,
862.
Rao K. N., Vore R. V. de, Plyler E. K. — «J.
Res. NBS», 1963, A67, 351.
Rao K. S. — «Canad. J. Phys.», 1958, 36, 1526.
Rao Y. V., Venkateswarlu P. — «J. Mol. Spec-
trosc», 1962, 9, 173.
Rao Y. V., Venkateswarlu P. — «J. Mol. Spec-
trosc», 1964, 13, 288.
Rasetti F. — «Phys. Rev.», 1929, 34, 367.
Rasmussen E. — «Z. Phys.», 1933, 80, 726.
Rau H. — «J. Chem. Thermodyn.», 1974, 6,
525.
Rau H. — «J. Chem. Thermodyn.», 1975, 7, 27.
Rau H. — «J. Chem. Thermodyn.», 1975, 7,
903.
Rau H. — «J. Chem. Thermodyn.», 1977, 9, 505.
Rau H., Kutty T. R. N., De Carvalho
J. R. F. G. — «J. Chem. Thermodyn.», 1973,
5, 291.
3176.
3177.
3178.
3180.
Ramsay D. A. — «J. Phys. Chem.», 1953, 57, 3166.
415.
Ramsay D. A. — «J. Chem. Phys.», 1956, 25,
188.
Ramsay D. A. — «Spectrochim. acta», 1956, 8,
286.
Ramsay D. 4. —«Nature», 1956, 178, 374.
Ramsay D. A. — «Ann. N. .Y Acad. Sci.», 1957,
67, 985.
Ramsay D. A. — «Mem. Soc. roy. sci. Liege»,
1957, 18, 471.
Ramsay D. A. Proc. 10th Colloq. Spectrosc.
Intern., College Paris, 1962. Washington, 1963, d172-
coo
Ramsay W., Young S. — «J. Chem. Soc», 1886 31?3-
49, 453.
Randall M., Bichowsky F. R. — «J. Amer.
Chem. Soc», 1918, 40, 368.
Randall H. M., Dennison D. M., Ginsburg JV.,
Weber L. R. — «Phys. Rev.», 1937, 52, 160.
Randhawa H. S., Patil К. С — «Current Sci.»,
1971, 40, 187.
Rank D. H. — «J. Opt. Soc. America», 1946,
36, 239.
Rank D. H. — «J. Opt. Soc. America», 1960,
50, 657. „.„„
RankD. #., Baldwin W. M. — «J. Chem. Phys.», d179-
1951, 19, 1210.
Rank D. H., Birtley W. В., Eastman D. P. —
«J. Opt. Soc. America», 1960, 50, 1275.
Rank D. H., Eastman D. P., Rao B. S.,
Wiggins T. A. — «J. Opt. Soc. America», 1961, 51,
929.
Rank D. H., Eastman D. P., Rao B. S., Wig- 3183
gins Т. А. — «Spectrochim. acta», 1961, 17,
3184.
3185.
3186.
3187.
3188.
3189.
3190.
3191.
3191a.
3192.
3193.
3194.
3195.
3196.
3197.
3198.
3199.
Rau H., Kutty T. R. N., De Carvalho
J. R. F. G. — «J. Chem. Thermodyn.», 1973,
5, 833.
Ray J. D. — «J. Chem. Phys.», 1947, 15, 357.
Ray J. D., Ogg R. A.~«J. Phys. Chem.»,
1956, 60, 1460.
Ray J. ?>., Ogg R. A. — <d. Phys. Chem.»,
1957, 61, 1087.
Ray J. D., Ogg R. A. — «J. Chem. Phys.»,
1957, 26, 984.
Ray J. D., Ogg R. A. — «J. Chem. Phys.»,
1959, 31, 168.
Raymonda J., Klemperer W. — «J. Chem. Phys.»,
1971, 55, 232.
Rayson R., Wilson M. R. — «J. Chem. Phys.»,
1954, 22, 2000.
Reamer H. H., Sage В. Н. — «Ind. and Engng
Chem.», 1952, 44, 185.
Reamer H. H., Sage В. Н., Lacey W. N. —
«Ind. and Engng Chem.», 1950, 42, 140.
Rebentrost F. — «Chem. Phys. Lett.», 1973, 21,
368.
Reddy N. M. — «Aeronaut. Quart.», 1974, 25,
287.
Redington R. L., Berneg C. V. — «J. Chem.
Phys.», 1965, 45, 2020.
Redington R. L., Olson W. В., Cross P. С —
«J. Chem. Phys.», 1962, 36, 1311.
Reed R. J., Snedden W. — «J. Chem. Soc»,
1959, 4132.
Refaey K. M. A.—«Intern. J. Mass Spectr.
and Ion Phys.», 1976, 21, N 1/2, 21.
Rees A. L. G. — «J. Chem. Phys.», 1957, 26,
1567.
Reese R. M., Dibeler V. #., Franklin J. L. —
«J. Chem. Phys.», 1958, 29, 880.
Regnault V. — «Ann. chim. phys.», 1840, 73, 3.
Regnault V. — «Ann. chim. phys.», 1849, 26,.
268.
Regnault V. — «Mem. Acad. Sci. Inst. France»,
1862, 26, 1.
Regula W. — «Ber. Dtsch. Wetterdienstes»,1958,
N 44.
Reichert U. V., HartmannH. — «Z. Naturforsch.»,
1972, 27a, 983.
Reichman S. — «J. Mol. Spectrosc», 1970, 35,.
329
Reichman S., Ginn S. G. W. — «J. Mol.
Spectrosc», 1971, 40, 27.
Reichman S., Overend J. — «Spectrochim. acta»,
1970, A26, 379.
Reichman S., Schreiner F. — «J. Chem. Phys.»,
1969, 51, 2355.
Reichman S., SmithD. E. jr., Overend J. —
«Spectrochim. acta», 1970, A26, 927.
Rewick R. Т., Anderson R., McLaren R. O. —
«J. Chem. and Engng Data», 1972, 7, 4.
Rewick R. Т., Anderson R., McLaren R. O. —
«J. Chem. andjEngng Data», 1972, 7, 409.
Rhodes E. R., Ubbelohde A. R. — «Proc. Roy.
Soc. London», 1959, A251, 156.
Ribaud M. G. Constantes thermodynamiques des
gaz aux temperatures elevees. Publ. sci. et techn.
Ministere de l'air, N 266. Paris, 1952.
Richards G. W., Woolf A. A. — «J. Chem. Soc»
A, 1969, 1072.
Richards P. L. — «J. Opt. Soc. America», 1964,
54, 1474.
Richards R. E., Smith Y. A. S. — «Trans.
Faraday Soc», 1951, 47, 1261.
482
Литература
3200. Richards W. G., Barrow R. F. ¦— «Proc. Chem.
Soc», 1962, 297.
3201. Richardson A. W. — «J. Mol. Spectrosc», 1970,
35, 43.
3202. Richardson A. W., Powell R. A. — «J. Mol.
Spectrosc.», 1967, 24, 379.
3203. Richardson A. W., Redding R. W., Brand
J. С D. — «J. Mol. Spectrosc», 1969, 29, 93.
3204. Richardson F. D. — «3. Iron and Steel Inst.».
1948, 160, 261.
3205. Richardson J. H., Stephenson L. M., Brauman
J. I. — «Chem. Phys. Lett.», 1974, 25, 318.
3206. Richardson O. W. Molecular Hydrogen and its
Spectrum. London, 1935.
3207. Richemeier O., Senftleben H., Pastorff H. — «Ann.
Physik», 1934, 19, 202.
3208. Ricks J. M., Barrow R. F. — «Canad. J. Phys.»,
1969, 47, 2423.
3209. Ricks J. M., Barrow R. F. — «J. Phys. B: Atom,
and Mol. Phys.», 1969, 2, 906.
3210. Risely J. S., Edwards A. K., Geballe R. — «Phys.
Rev.», 1974, A9, 1115.
3211. Rittenberg D., Vrey H. С — «J. Chem. Phys.»,
1934, 2, 106.
3212. Robertson A. J. B. — «Trans. Faraday Soc»,
1952, 48, 228.
3213. Robiette A. G., Bradley R. H., Brier P. N. —
«J. Chem. Soc. Chem. Communs», 1971, 1567.
3214. Robin M. В., Hart R. R., Kuebler N. A. — «J.
Amer. Chem. Soc», 1967, 89, 1564.
3215. Robinson E. A. — «Canad. J. Chem.», 1963, 41,
3021.
3216. Robinson G. W., McCarty M. — «J. Chem.
Phys.», 1958, 28, 350.
3217. Robinson G. W., McCarty M., Keetly M. С —
«J. Chem. Phys.», 1957, 27, 972.
3217a. Roche A. E., Sutton M. M., Bohme D. K.,
Schiff H. I. — «J. Chem. Phys.», 1971, 55, 5480.
3218. Roche A. L., Lefebvre-Brion H. — «J. Chem.
Phys.», 1973, 59, 1914.
3219. Rochkind M. M. — «Dissert. Abstrs», 1966, B27,
785.
3220. Rochkind M. M., Pimentel G. C. — «J. Chem.
Phys.», 1965, 42, 1361.
3221. Rochkind M. M., Pimentel G. С — «J. Chem.
Phys.», 1967, 46, 4481.
3222. Rodebush W. H., Wahl M. H. — «J. Chem.
Phys.», 1933, 1, 696.
3223. Roder H. M., Childs G. E., McCarty R. D.,
Anderhofer P. Survey of the Properties of the
Hydrogen Isotopes below their Critical
Temperatures. NBS Techn. Note 641. Washington,
1973.
3224. Roder H. M., McCarty R. D., Ball W. J.
Computer Programsj for Thermodynamic and
Transport Properties of Hydrogen (Tabcode-II). NBS
Techn. Note 625. Washington, 1972.
3225. Roder H. M., Weber L. A., Goodwin R. D.
Thermodynamic and Related Properties of Parahydro-
gen from the Triple Point to 100° К at Pressures
to 340 atm. NBS Monogr. 94. Washington,
1965.
3226. Rodewald H. J. — «Helv. chim. acta», 1960, 43,
878.
3227. Rogers J. D., Pietenpol W. J., Williams D. —
«Phys. Rev.», 1951, 83, 431.
3228. Rogers J. D., Williams D. — «Phys. Rev.», 1951,
82 131
3229. Rogers j. D., Williams D. — «J. Chem. Phys.»,
1961, 34, 2195.
3230. Rogers H. H., Constantine M. Г., Quaglino J.,
Dubb H. E., Ogimachi N. N. — «J. Chem. and
Engng Data», 1968, 13, 307.
3231. Rogers M. Т., Speirs J. L., Thompson H. В.,
Panish M. B. — «J. Amer. Chem. Soc», 1954,
76, 4843.
3231a. Rogers M. Т., Speirs J. L., Panish M. В.,
Thompson H. B. — «J. Amer. Chem. Soc», 1956, 78,
936.
3232. Rolfe J. — «J. Chem. Phys.», 1964, 40, 1664.
3233. Rolfe J. — «Appl. Phys. Lett.», 1965, 6, 66.
3234. Rolfe J. — «J. Chem. Phys.», 1968, 49, 4193.
3235. Rolfe J., Holzer W., Murphy W. F., Bernstein
H. I. — «J. Chem. Phys.», 1968, 49, 963.
3236. Romand J. — «Ann. Phys.», 1949, 4, 527.
3237. Romand J., Vodar B. — «C. r. Acad. sci.», 1948,
226, 890.
3237a. Renault M. — «C. r. Acad sci.», 1938, 207, 620.
32376. Renault M. — «Ann. phys.», 1940, 14, 78.
3238. Rosen B. — «Z. Phys.», 1927, 43, 69.
3239. Rosen В., Bouffioux F. — «Bull. cl. sci. roy.
Belgique», 1936, 22, 885.
3240. Rosen В., Desirant M. — «Bull. Soc. roy. sci.
Liege», 1935, 6—7, 233.
3241. Rosenbaum O., Neuert H. — «Z. Naturforsch.»,
1954, 9a, 980.
3242. Rosenbaum O., Neuert H. — «Z. Naturforsch.»,
1954, 9a, 990.
3243. Rosenberg A., Birnbaum G. — «J. Chem. Phys.»,
1970, 52, 683.
3244. Rosenberg A., Lightman A., Ben-Reuven A. —
«J. Quant. Spectrosc. and Radiat. Transfer»,
1972, 12, 219.
3245. Rosenblum В., Nethercot A. H. — «Phys. Rev.»,
1955, 97, 84.
3246. RosengrenR., Pimental G. С — «J. Chem. Phys.»,
1965, 43, 507.
3247. Rosenqvist T. — «Metallurg. Trans.», 1949, 185,
451.
3248. Ross A. S., Maass O. — «Canad. J. Res.», 1940,
B18, 55.
3249. Rosser W. A., Wise H. — «J. Phys. Chem.»,
1959, 63, 1753.
3250. Rossini F. D. — «J. Res. NBS», 1931, 6, 36.
3251. Rossini F. D. — «J. Res. NBS», 1932, 9, 679.
3252. Rossini F. D. — «Chem. Rev.», 1936, 18, 233.
3253.. Rossini F. D. — «J. Res. NBS», 1939, 22, 407.
3253a. Rossini F. D., Cowie P. A., Ellison F. O., Brown
С. С Properties of Titanium Compounds and
Related Substances. Washington, Gov. print, off..
1957.
3254. Rossini F. D., Knowlton J. W., Johnston H. L. —
«J. Res. NBS», 1940, 24, 369.
3255. Rossini F. D., Pitzer K. S., Arnett R. L., Braun
R. M., Pimentel G. С Selected Values of
Physical and Thermodynamic Properties of
Hydrocarbons and Related Compounds. Amer. Petrol.
Inst., 1953.
3256. Rossini F. D., Wagman D. D., Evans W. H.,
Levine S., Jaffe J. Selected Values of Chemical
Thermodynamic Properties, NBS, Circ. 500.
Washington, 1952.
3257. Rostos J., Cossart D., Bastien J. R. — «Canad.
J. Phys.», 1974, 52, 1274.
3258. Roth W. A., Bertram A. — «Z. Elektrochem.»,
1937, 43, 376.
3259. Roth W. A., Grau R., Meichsner A. — «Z. anorg.
und allgem. Chem.», 1930, 193, 161.
483
Литература
3259а. Roth W. A., Meichsner A., Richter H.
Eisenhuttenwesen», 1934, 8, 240.
3260. Roth W. A., Richter H. — «Z. phys. Chem.»,
1934, A170, 123.
3261. Rothe E. W., Tang S. Y. — «J. Ghem. Phys.»,
1975, 62, 3.
3262. Rothschild W. G. — «J. Opt. Soc. America»,
1964, 54, 20.
3263. Rousseau D. L., Miller R. E., Leroi G. E. —
«J. Chem. Phys.», 1968, 48, 3409.
3264. Rudzitis E. — «Anal. Chem.», 1967, 39, 1187.
3265. Rudzitis E., Van Doxrenter E. H., Hubbard
W. N. — «J. Chem Thermodyn.», 1970, 2, 221.
3266. Ruff O., Braida A.— «Z. anorg. und allgem.
Chem.», 1933, 214, 81.
3267. Ruff 0., Braida A. — «Z. anorg. und allgem.
Chem.», 1934, 220, 43.
3268. Ruff O., Laass F. — «Z. anorg. und allgem.
Chem.», 1929, 183, 214.
3269. Ruff O., Menzel W. — «Z. anorg. und. allgem.
Chem.», 1930, 190, 257.
3270. Ruff O., Menzel W. — «Z. anorg. und allgem.
Chem.», 1931, 198, 375.
3271. Ruff O., Menzel W., Neumann W. — «Z. anorg.
und allgem. Chem.», 1932, 208, 293.
3271a. Ruff O., Wallauer H. — «Z. anorg. und allgsm.
Chem.», 1931, 196, 421.
3272. Rumpf K. — «Z. phys. Chem», 1938, B38, 469.
3273. Ruoff A. — «Mol. Phys.», 1970, 19, 23.
3274. Ruoff A., Burger H., Biedermann S., Cichon J. —
«Spectrochim. acta», 1972, A28, 953.
3275. Russ F., Рокоту Е. — «Monatsh. Chem.», 1913,
34, 1027.
3276. Rutner E. — «J. Chem. and Engng Data», 1962,
7, 398.
3277. Rynning D. F., Hurd C. O. — «Trans. Amer.
Inst. Chem. Engng», 1945, 41, 265.
3278. Saalfeld F. E., Svec H. J. — «Inorg. Chem.»,
1964, 3, 1442.
3279. Sackett P. В., Yardley J. T. — «Chem. Phys.
Lett.», 1971, 9, 612.
3280. Safron S.A., WeinsteinN. D., HerschbachD. R. —
«Chem. Phys. Lett.», 1972, 12, 564.
3281. Sahni R. С — «Trans. Faraday Soc», 1967, 63,
801.
3282. Saito S. — «J. Mol. Spectrosc», 1969, 30, 1.
3283. Saito S. — «J. Chem. Phys.», 1970, 53, 2544.
3284. Saito S. — «J. Mol. Spectrosc», 1973, 48, 530.
3285. Saito S. — «Astrophys. J.» (in press.).
3286. Saito S., Takagi K. — «J. Mol. Spectrosc.»,
1972, 44, 81.
3287. Saito S., Takagi K. — «J. Mol. Spectrosc»,
1973, 47, 99.
3288. Sakano Т. К. Dissertation. Univ. Wisconsin,
1966. «Dissert. Abstrs», 1966, 26, 6395.
3289. Sakurai K., Broida H. P. — «J. Chem. Phys.»,
1969, 50, 2404.
3290. Sakurai K., Broida H. P. — «J. Chem. Phys.»,
1970, 53, 1615.
3291. Sakurai K., Clark J., Broida H. P. — «J. Chem.
Phys.», 1971, 54, 1217.
3291a. Salmon L., Poshusta R. D. — «J. Chem. Phys.»,
1973, 59, 3947.
3292. Salotto A. W., Burnelle L. — «Chem. Phys.
Lett.», 1969, 3, 80.
3293. Salotto A. W., Burnelle L. — «J. Chem. Phys.»,
1970 52 2936
3294. Salthouse J. A., Waddington Т. С —
«Spectrochim. acta», 1967, A23, 1069.
«Arch. 3295. Sams R. L., Lafferty W. J. — «J. Mol. Spectrosc.»
1975, 56, 399.
3296. Samson J.A.R. — «Phys. Lett.», 1968, A28, 391,
3297. Samson J. A. R., Cairns R. B. —. «J. Opt. Soc.
America», 1966, 56, 769.
3298. Samson J. A. R., Gardner J. L. — «Canad. J.
Phys.», 1975, 53, 1948.
3299. Samson J. A. R., Petrosky V. E. — «J. Electron
Spectrosc. and Relat. Phenomena», 1974, 3, 461.
3300. Samuelson H. V. Dissertation. Western Reserve
Univ., 1965, p. 90. «Dissert. Abstrs», 1966,
B27, 443.
3301. Sanborn R. ff.^«J. Chem. Phys.», 1960, 33,
1855.
3302. Sandeman J. — «Proc Roy. Soc. Edinburgh»,
1935, 55, 72.
3303. Sandoval A.A., Moser H. C, Riser R. W. —
«J. Phys. Chem.» 1963, 67, 124.
3304. Sands D. E. — «J. Amer. Chem. Soc», 1965,
87, 1395.
3305. Santaram С V. V. S. N. K., Rao P. T. — «Z.
Phys.», 1962, 168, 553.
3306. Santaram C. V. V. S. N. K., Rao P. T. —
«Indian J. Phys.», 1963, 37, 14.
3307. Sass R. L., Vidale R., Donahul J. — «Acta
crystallogr.», 1957, 10, 567.
3308. Sastry M. G. — «Indian J. Phys.», 1941, 15, 95.
3309. Sastry M. G. — «Indian J. Phys.», 1941, 15, 455.
3310. Sastry M. G., Rao K. R. —«Indian J. Phys.»,
1941, 15, 27.
3311. Saum K. A., Benesch W. M. —¦¦ «Appl. Opt.»,
1970 9 195
3312. Saum K. A.', Benesch W. M. — «Phys. Rev.»,
1970, A2, 1655.
3313. Savage С. М., Edwards Т. Н. — «J. Chem.
Phys.», 1957, 27, 179.
3314. Savoie R., Trembley J. — «Canad. J. Spectrosc»,
1972, 17, 73.
3315. Sawodny W., Ruoff A., Peacock C. J., Muller
A.—«Mol. Phys.», 1968, 14, 433.
3316. Sayre E. V. — «J. Chem. Phys.», 1959, 31, 73.
3317. Scatchard G., Kavanagh G. M., Ticknor L. B. —
«J. Amer. Chem. Soc», 1952, 74, 3715.
3318. Schaad L. J., Kinser H. B.—«J. Phys. Chem.»,
1969, 73, 1901.
3319. Schaeffer C, Thomas M. — «Z. Phys.», 1923,
12, 330.
3220. SchaeferH. F., Harris F. E. — «J. Ctiem. Phys.»,
1968, 48, 4946.
3321. Schdfer K., Wicke E. — «Z. Elektrochem.», 1948,
52, 205.
3322. Schamp H. W., Mason E. A., Su K. —«Phys.
Fluids», 1962, 5, 769.
3323. S chanmugasundaram G., Nagarajan G. —
«Monatsh. Chem.», 1969, 100, 385.
3324. Schatz J., Reichman S. —.«J. Chem. Phys.»,
1972, 57, 4571.
3325. Scheer M.D. — «J. Chem. Phys.», 1952, 20, 924.
3326. Scheer M. D. — «J. Chem. Phys.», 1952, 20, 1497.
3327. Scheffer F. E. C, Voogd M. — «Recueil trav.
chim.», 1926, 45, 214.
3328. Schenck R., Mihr F., Banthien #. — «Ber.
Dtsch. phys. Ges.», 1906, 39, 1506.
3329. Schenk P. W. — «Z. anorg. und allgem. Chem.»,
1933, 211, 150.
3330. Schenk P. W. — «Z. anorg. und allgem. Chem.»,
1936, 229, 305.
3331. Schick H. L. Thermodynamics of Certain
Refractory Compounds, v. 1, 2. New York—London,
Acad. Press, 1966.
484
Литература
3332. Schlapp R. — «Phys. Rev.», 1937, 51, 342.
3333. Schlinger W. G., Sage В. Н. — «Ind. and Engng
Chem.», 1950, 42, 2158.
3334. Schmid R. — «Z. Phys.», 1929, 59, 42.
3335. Schmid R. — «Z. Phys.», 1929, 59, 850.
3336. Schmid R. — «Z. Phys.», 1930, 64, 279.
3337. Schmid R., Konig Т., Farkas D. von. — «Z.
Phys.», 1930, 64, 84.
3338. Schmidt M., Block В., Block H. D., Kopf H.,
Wilhelm E. — «Angew. Chem.», 1968, 80, 660.
3339. Schmidt-Bocking H., Bethge K.—«J. Chem.
Phys.», 1973, 58, 3244.
3340. Schmitz H., Schumacher H. J. — «Z. Natur-
forsch.», 1947, 2a, 357.
3341. Schmitz H., Schumacher H.J.—«Z. Natur-
forsch.», 1947, 2a, 359.
3342. Schmitz H., Schumacher H. J. — «Z. Natur-
forsch.», 1947, 2a, 362.
3343. Schneider J. — «Z. phys. Chem.», 1974, A255,
986.
3344. Schneider M., Gignere P. A. — «C. r. Acad. sci.»,
1968, B267, 551.
3345. Schneider S. J. Compilation of the Melting
Points of the Metal Oxides. NBS, Monogr.
N 68, 1963.
3346. Schoenborn M., Csizmadia J. G. — «Acta phys.
Acad. scient. hung.», 1969, 27, 377.
3347. Schomacker H. Dissertation Ruhr-Univ. Bohum,
1973.
3348. Schomaker V., Lu C. — «J. Amer. Chem. Soc»,
1950, 72, 1182.
3349. Schomaker V., Spurr K. A. — «J. Amer. Chem.
Soc», 1942, 64, 1184.
3350. Schomaker V., Stevenson D. P. — «J. Amer.
Chem. Soc», 1940, 62, 1276.
3351. Schomaker V., Stevenson D. P. — «J. Amer.
Chem. Soc», 1941, 63, 37.
3351a. Schreiner F., McDonald G. N., Chernick С L. —
«J. Phys. Chem.», 1968, 72, 1162.
33516. Schreiner F., Osborne D. W., McDonald G. N. —
«J. Chem. Phys.», 1969, 51, 4838.
3352. Schroder J., Sieben F. J. — «Chem. Ber.», 1970,
103, 76.
3352a. Schueager J., Arkell A. — «J. Amer. Chem.
Soc», 1967, 89, 6006.
3353. Schuler H., Reinbeck L., Michel A. — «Z. Na-
. turforsch.», 1954, 9a, 279.
3354. Schulz G. J. — «J. Chem. Phys.», 1961, 34,
1778.
3355. Schulz G., Schaefer H. — «Ber. Bunsenges. phys.
Chem.», 1966, 70, 21.
3356. Schulz S. A. — «Bull. Inst. intern, froid.», 1973,
N 4, annexe, 21.
3357. Schumacher H. J. — In: Chemische Gasreaction.
Dresden, 1938.
3358. Schumacher H. J., Schmitz H., Brodersen P. H. —
«An. Asoc quim. argent.», 1950, 38, 98.
3359. Schumaker N. E., Garland С W. — «J. Chem.
Phys.», 1970, 53, 392.
3360. Schumm R. H., Prosen E. J., Wagman D. D. —
«J. Res. NBS», 1974, A78, 375.
3361. Schutza И. — «Z. anorg. und allgem. Chem.»,
1938, 239, 245.
3362. Schwartz M. E., Hayes E. F., Rothemberg S. —
«Theor. chim. acta», 1970, 19, 98.
3363. Schwartz M. E., Schaad L. J.- «J. Chem. Phys.»,
1967, 47, 5325.
3364. Scott D. W. — «Spectrochim. acta», 1960, 16,
1276.
3365.
3366.
3367.
3368.
3369.
3370.
3371.
3372.
3373.
3374.
3375.
3376.
3377.
3377a.
3378.
3379.
3380.
3381.
3382.
3383.
3384.
3385.
3386.
3387.
3388.
3389.
3390.
3391.
3392.
3393.
3394.
3395.
3395a.
3396.
3397.
Scott D. W., McCullough J. P. — «J. Mol.
Spectrosc», 1961, 6, 372.
Scott D. W., McCullough J. P., Kruse F. H. —
«J. Mol. Spectrosc», 1964, 13, 313.
Scott D. W., Oliver G. D., Gross M. E., Hub-
bard W. N., Huffman H. M. — «J. Amer. Chem.
Soc», 1949, 71, 2293.
Seal K. E., Gaydon A. G. — «Proc Phys. Soc.
London», 1966, 89, 459.
Seaton M. J. — «Proc. Phys. Soc. London»,
1966, 88, 815.
Seel F., Budenz R. «Chem. Ber.», 1965, 98, 251.
Seel F., Budenz R., Wanczek K.-P. — «Chem.
Ber.», 1970, 103, 3946.
Seel F., Heinrich E., Gombler W., Budenz R. —
«Chimia», 1969, 23, 73.
Sehon A. H. — «J. Amer. Chem. Soc», 1952,
74, 4722.
Sehon A. H., Darwent В. В. — «J. Amer. Chem.
Soc», 1954, 76, 4806.
Selected Values of Chemical Thermodynamic
Properties. Techn. Note 270-3, 270-4, 270-5,
270-6, 270-7. Washington, U. S. Dept Commerce,
NBS, 1968—1973.
Selig H., Claassen H. H. — «J. Chem. Phys.»,
1966, 44, 1404.
Selig H., Claassen H. H. — «Isr. J. Chem.»,
1968, 6, 499.
Selig H., Claassen H. H., Chernick C. L., Malm
J. G., Huston J. L. — «Science», 1964, 143, 1322.
Selig H., Claassen H. H., Holloway J. H. —
«J. Chem. Phys.», 1970, 52, 3517.
Selig H., Holloway J. H. — «J. Inorg. and Nucl.
Chem.», 1971, 33, 3169.
Selig H., Holzman H. — «Isr. J. Chem.», 1969,
7, 417.
Selin L.-E. — «Naturwissenschaften», 1960, 47,
104.
Selin L.-E. — «Arkiv fys.», 1962, 21, 479,
Selin L.-E. — «Arkiv fys.», 1962, 21, 529,
Selin L.-E., Soderborg B. — «Arkiv fys.», 1962,
21, 515.
Selph С. С Thesis. Georgia Institute Technol.,
1969.
Senatore S. J. — «Phys. Rev.», 1950, 78, 293,
Senftleben H., Rehren J. — «Z. Phys.», 1926,
37, 539.
Sengers J. M. H. L., Straub J., Vicentini-Mis-
soni M. — «J. Chem. Phys.», 1971, 54, 5034.
Sen-Gupta A. K. — «Proc. Phys. Soc. London»,
1935, 47, 247.
Seshadri D. .N. — «Indian J. Technol.», 1975,
13, 19.
Seshadri D. N., ViswanathD. S., Kuloor N.R. —
«J. Indian Sci.», 1966, 48, 36.
Seshadri D. N., ViswanathD. S.
«AIChE Journal», 1970, 16, 420.
SeshadriD. N., ViswanathD. S.
«Indian J. Technol.», 1970, 8
Seshadri D. N., ViswanathD. S.
Kuloor N. R. —
Kuloor N.R. ~-
153.
Kuloor N. R. —
«Indian J. Technol.», 1970, 8, 191.
Settle J. L., Grunberg ?., Hubbard W. N. (ia
press) (см.: [2140]).
Settle J. L., Jeffers H. E., O'Hare P. A. G.,
Hubbard W. N. — «J. Inorg. and Nucl. Chem.
Suppl.», 1976, 135.
Settly H. S. N., Smith J. D., Yaws С L. —
«Chem. Engng», 1974, 81, N 12, 70.
Shamir J., Hyman H. H. — «Spectrochim. acta».
1967, A23, 1191.
485
Литература
3398. Shamir J., Ну man H. H. — «Spectrochim. acta»,
1967, A23, 1899.
3399. Shamir J., Yellin D., Claassen H. H. — «Isr.
J. Chem.», 1974, 12, 1015.
3400. Shand W., Spurr R. A. — «J. Amer. Chem.
Soc», 1943, 65, 179.
3401. Shaw A. W., Vosper A. J. — «J. Chem. Soc.
Dalton Trans.», 1972, 961.
3402. Shaw J. H. — «J. Chem. Phys.», 1956, 24, 399.
3403. Shearer J. N., Wiggins T. A., Guenther A. H.,
Rank D. H. — «J. Chem. Phys.», 1956, 25, 724.
3404. Sheft I., Perkins A. J. — «J. Inorg. and Nucl.
Chem.», 1976, 38, 665.
3405. Shelton R. D., Nielsen A. #., Fletcher W. H. —
J. Chem. Phys.», 1953, 21, 2178.
3406. Shelton R. D., Nielsen A. H., Fletcher W. H. —
«J. Chem. Phys.», 1954, 22, 1791.
3407. Sheridan /., Gordy W. — «Phys. Rev.», 1950,
79, 513.
3408. Sherman R. H., Giauque W. F. — «J. Amer.
Chem. Soc», 1953, 75, 2007.
3409. Shimauchi M. — «Sci. Light», 1964, 13, 53.
3410. Shimizu F. O., Shimizu T. — «J. Mol. Spectrosc»,
1970, 36, 94.
3411. Shirai Т., Ishiabashi T. — «Scient. Papers Coll.
Gen. Educ, Univ. Tokyo», 1955, 5, N 2, 131.
3412. Shirley D. A., Giauque W. F. — «J. Amer. Chem.
Soc», 1959, 81, 4778.
3413. Shomate С. Н. — «J. Amer. Chem. Soc», 1944,
66, 928.
3414. Shurvell H. F. — «Canad. J. Spectrosc», 1971,
16, 71.
3415. Sidman J. W. — «J. Amer. Chem. Soc», 1957,
79, 2669.
3416. Sidman J. W. — «J. Amer. Chem. Soc», 1957,
79, 2675.
3417. Siebert H. — «Z. anorg. und allgem. Chem.»,
1951, 265, 303.
3418. Sieck L. W., Searles S. K. — «J. Chem. Phys.»,
1970, 53, 2601.
3419. Siegel M. W., Celotta R. J., Hall J. L., Levine
J., Bennett R. A. — «Phys. Rev.», 1972, A6,
607.
3419a. Siegel S., Gelbert E. — «J. Amer. Chem. Soc»,
1963, 85, 240.
3420. Simkin D. J., Hurd С. О. — «Jet Propulsion»,
1957 27 419
3420a. Simkin D. J., Hurd С. О. — «Chem. Engng»,
1958, 65, 155.
3421. Simon A., Schulze G.—«Naturwissenshaften»,
1937, 25, 669.
3422. Simon F. Handbuch der Physik, Bd 10. Berlin,
J. Springer, 1926, S. 350.
3423. Simon F., Simson C, Ruhemann M. — «Z.
phys. Chem.», 1927, 129, 339.
3424. Simons J. H., Hildebrand J. H. — «J.
Amer. Chem. Soc», 1924, 46, 2183.
3425. Simons J. H., Hildebrand J. H. — «J. Amer.
Chem. Soc», 1924, 46, 2185.
3426. Simons J. W., Paur R. J., Webster H. A., Bair
E. Y. — «J. Chem. Phys.», 1973, 59, 1203.
3427. Singh N. L. — «Canad. J. Phys.», 1959, 37, 136.
3428. Singh R. В., Rai D. K. — «J. Quant. Spectrosc.
Radiat. Transfer», 1965, 5, 723.
3429. Singh S. M., Tellinghuisen J. — «J. Mol.
Spectrosc.», 1973, 47, 409.
3430. Singh Z., Nagarajan G. — «Acta phys. Acad.
scient. hung.», 1974, 36, 415.
3431. Sinke G. С — «J. Chem. and Engng. Data»,
1965, 10, 295.
3432. Sinke G. C. —«J. Phys. Chem.», 1967, 71, 359.
3433. Sirvetz M. H. — «J. Chem. Phys.», 1951, 19,
938.
3434. Sjogren H. — «Arkiv fys.», 1962, 22, 437.
3435. Skaarup S., Boggs J. E.-—«J. Mol. Struct.»,
1976, 30, 389.
3436. Skiens W. E., Cady G. H. — «J. Amer. Chem.
Soc», 1958, 80, 5640.
3437. Skinner H. A. Modern Aspectsfof jThermoche-
mistry. R. J. С Monogr. N 3. London, 1958.
3437a. Skinner H. A. —.«J. Chem. Thermodyn.», 1974,
6, 711.
3438. Skinner H., Pritchard H. O. —. «Trans. Faraday
Soc», 1953, 49, 1254.
3438a. Skolnik E. G., Goodfriend P. L. — «J. Mol.
spectrosc», 1974, 50, 202.
3439. Skolnik E. G., Salesi Д. J,, Russ С. Д., Good-
friend P. L. — «J. Phys. Chem.», 1973, 77, 1126.
3440. Skorko E. —«Nature», 1933, 131, 366.
3441. Skorko E. — «Acta phys. polon.», 1934, 3, 191.
3442. Smardzewski R. R., Fox W. B. — «J. Chem.
Phys.», 1974, 60,12104.
3443. Smardzewski R. R., Fox W. B. —.«J. Chem.
Phys.», 1974, 60, 2193.
3444. Smardzewski R. R., Fox W. B. — «J. Chem.
Phys.», 1974, 60, 2980.
3445. Smardzewski R. R., Fox W. B. — «J. Chem.
Soc Chem. Communs», 1974, 241.
3446. Smardzewski R. R., Fox W. B. — «J. Fluor.
Chem.», 1976, 7, 353.
3447. Smardzewski R. R., Fox W. B.—a,]. Fluor.
Chem.», 1976, 7, 453.
3448. Smardzewski R. Д., Fox W. B. — «J. Fluor.
Chem.», 1976, 7, 456.
3449. Smets J. Coppens P., Drowart J. —> «Chem.
Phys.», 1977, 20, 243.
3450. Smith A. — «Z{ Elektrochem.», 1916, 22, 33.
3451. Smith A., Calvert R. P.—aJ. Amer. Chem.
Soc», 1914, 36, 1363.
3452. Smith A., Lombard R. H. — «J. Amer. Chem.
Soc», 1915, 37, 2055.
3453. Smith D., James D. W., Devlin J. P. — «J.
Chem. Phys.», 1971, 54, 4437.
3454. Smith D. F. — «J. Chem. Phys.», 1953, 21, 609.
3455. Smith D. F. -~ «J. Chem. Phys.», 1958, 28, 1040.
3456. Smith D. F. —< «J. Mol. Spectrosc», 1959, 3, 473.
3456a. Smith D. F. —«Science», 1963, 140, 899.
3457. Smith D. ^.—«Science», 1963, 141, 1039.
3458. SmithD. F. — «J. Chem. Phys.», 1968, 48, 1429.
3459. Smith D. F., Tidwell M., Williams D. V. P. —
«Phys. Rev.», 1950, 77, 420.
3460. Smith D. F., Tidwell M., Williams D. V. P. —
«Phys. Rev.», 1950, 79, 1007.
3461. Smith D. F., jr, Overend J. — «J. Chem. Phys.»,
1971, 55, 1157.
3462. Smith D. F. jr., Overend J. — «J. Chem. Phys.»,
1972, 57, 523.
3462a. Smith D. F. jr, Overend /.— «Spectrochim.
acta», 1972, A28, 471.
3463. Smith D. J., Nielsen A. H. — «Phys. Rev.», 1955,
99, 1624.
3464. Smith D. W., Andrews L. — «J. Chem. Phys.»,
1974, 60, 81.
3465. Smith D. W., Hedberg K. — «J. Chem. Phys.»,
1956, 25, 1282.
3466. Smith H., Schumacher H. J. — «Z. Naturforsch.»,
1947, 2a, 363.
3467. Smith J. J., Meyer B. — «J. Mol. Spectrosc»,
1964, 14, 160.
486
Литература
3468. Smith К. С, Mclver R. J., Brauman J. I., 3502.
Wallage R. W. — «J. Chem. Phys.», 1971, 54,
2758. 3503.
3469. Smith S. J., Burch D. S. — «Phys. Rev.», 1959,
116, 1125. 3504.
3470. Smits A., Bokhorst S. С.—чЪ. phys. Chem.»,
1916, 91, 249. 3505.
3471. Smits A., Deinum H. W. — «Proc. Koninkl.
nederl. Akad. wet.», 1930, 33, 514. 3506.
3472. Smits A., Deinum H. W. — «Z. phys. Chem.»,
1930, A149, 337. 3507.
3473. Smits A., Ketelaar J. A. A., Meyering J. L. —
«Z. phys. Chem.», 1938, B41, 87. 3508.
3474. Smits A., Rutgers A. J. — «J. Chem. Soc»,
1924, 125, 2573. 3509.
3475. Smits A., Stein-Parve E. P., Meerman P. G.,
Decker H. C. J. de — «Z. phys. Chem.», 1940, 3510.
B46, 43. 3511.
3476. Smoes S., Drowart J. — «Faraday Sympos. Chem. 3512.
Soc», 1973, 139. 3512a.
3477. Smoes S., Drowart J. — «Discuss. Faraday Soc»,
(in press).
3478. Smyth K. C, Brauman J. I. — «J. Chem. Phys.», 3513.
1972, 56, 1132.
3479. Smyth K. C, Mclver R. Т., Brauman J. I., 3514.
Wallace R. W. — «J. Chem. Phys.», 1971, 54,
2758. 3515.
3480. Snider D. E., Shaw J. H. — «J. Mol. Spectrosc», 3516.
1972, 44, 400.
3481. Snyder L. E., Edwards Т. H. — «J. Mol. Spec- 3517.
trosc», 1969, 31, 347.
3482. Snyder R. G., Hisatsune I. C. — «J. Mol. 3518.
Spectrosc.», 1957, 1, 139.
3483. Solarz Д., Levy D. H. — «J. Chem. Phys.», 1973, 3519.
58, 4026.
3484. Sole M., Pour V. — «Collect. Czech. Chem. 3520.
Communs», 1967, 32, 3031.
3485. Solomon J. E., Johnson D. R., Lin С. С. — «J. 3521.
Mol. Spectrosc», 1968, 27, 517.
3486. Solomon W. C, Blaner J. A., Jaye F. С — «J. 3522.
Phys. Chem.», 1968, 72, 2311.
3487. Southard J. C, Nelson R. A. — «J. Amer. Chem.
Soc», 1937, 59, 911. 3523.
3488. Spanbauer R. N., Rao K. N. — «J. Mol.
Spectrosc.», 1965, 16, 100.
3489. Spangenberg H. I., Zulicke L. — «Z. phys. 3524.
Chem.» (BRD), 1967, 52, 336.
3490. Speirs G. K., Splrko V. — «J. Mol. Spectrosc», 3525.
1975, 56, 104.
3491. Spence D., Sehulz G. J. —«Phys. Rev.», 1970, 3526.
A2, 1802.
3492. Spinks J. W. T. — «Canad. J. Res.», 1942, 3527.
A20, 1. 3528.
3493. Spirko V., Speirs G. K. — «J. Mol. Spectrosc.»,
1975, 55, 151. 3529.
3494. Spolity M., Cesargo S. N., Mariti B. — «J.
Chem. Phys.», 1973, 59, 985. 3530.
3495. Sportouch S., Clark R. J. H., Gaufres R. — «J.
Raman Spectrosc.», 1974, 2, 153. 3531.
3496. Spragne A. D., Nielsen H. H.—«3. Chem. 3532.
Phys.», 1937, 5, 85. 3533.
3497. Sreeramamurty K. — «Current Sci.», 1948, 17,
119. 3534.
3498. Srivastava B. N., Barua A. K. — «J. Chem.
Phys.», 1961, 35, 329. 3535.
3499. Staats P. A., Morgan H. W., Goldstein J. H. —
«J. Chem. Phys.», 1956, 24, 916. 3536.
0035 Stackel W. — «Z. Elektrochem.», 1930, 36, 375.
35.01. Stals J., Barraclough C. G., Buchanan A. S. — 3537.
«Trans. Faraday Soc», 1969, 65, 904.
Stammreich H., Forneris R. — «J. Chem. Phys.»
1953, 21, 944.
Starling K. E., Kwok J. C. — «Hydrocarbon
Process», 1972, 51, 86.
Stedman D. П. — «J. Chem. Phys.», 1970, 52,
3966.
Steenbeckeliers G. — «Ann. Soc. scient. Bruxel-
les, Ser. 1, 1968, 82, 331.
Steenbeckeliers G., Bellet J. — «C. r. Acad. sci.»,
1970, B270, 1039.
Steenbeckeliers G., Bellet J. — «J. Mol. Spectrosc»,
1973, 45, 10.
Steiger R. A., Cater E. D. — «High Temperat.
Sci.», 1975, 7, 204.
Steiger R. A., Cater E. D. —«High. Temperat.
Sci.», 1975, 7, 288.
Stein L. — «J. Amer. Chem. Soc», 1959, 81,1269.
Stein L. — «J. Amer. Chem. Soc», 1959, 81, 1273.
Stein L. — «J. Phys. Chem.», 1962, 66, 288.
Stein L., Plurien P. L. — In: Noble Gas
Compounds. H. H. Hyman (Ed.). Chicago Univ.
Press, 1963, p. 144.
Stein W. A. — «Kaltetechnik — Klimat.», 1971,
8, 227.
Steinbach W., Gordy W. — «Phys. Rev. A: Gen.
Phys.», 1975, 11, 729.
Steiner B. — «J. Chem. Phys.», 1968, 49, 5097.
Steiner В., Seman M. L., Branscomb L. M. —
«J. Chem. Phys.», 1962, 37, 1200.
Steinfeld J. I., Campbell J. D., Weiss N. A. —
«J. Mol. Spectrosc», 1969, 29, 204.
Steinfeld J. I., Zare R. N., Jones L., Lesk M.,
Klempere W. — «J. Chem. Phys.», 1965, 42, 25.
Stephenson С. С, BentzD. R., StevensonD. A. —
«J. Amer. Chem. Soc», 1955, 77, 2161.
Stephenson С. С, McMahon H. O. — «J. Amer.
Chem. Soc», 1939, 61, 437.
Stephenson С. С, McMahon H. O. — «J. Chem.
Phys.», 1939, 7, 614.
Stephenson C. C, Potter R. L., Maple T. G.,
Morrow J. С — «J. Chem. Thermodyn.», 1969,
1, 59.
Stephenson С. С, Potter R. L., Maple T. G.,
Morrow J. C. 130th Meeting Amer. Chem. Soc,
Sept. 1956.
Stephenson C. V., Jones E. A. — «J. Chem. Phys.»,
1952, 20, 135.
Stephenson C. V., Jones E. A.—«J. Chem.
Phys.», 1952, 20, 1830.
Stephenson D. A., Strauch R. G. — «J. Mol.
Spectrosc», 1970, 35, 494.
Stern Т. Н. Thesis. Univ. Washington, 1958.
Steunenberg R. K., Vogel R. C, Fischer J.
«J. Amer. Chem. Soc», 1957, 79, 1320.
Stevens С G., Swagel M. W., Wallace R., Zare
R. N. — «Chem. Phys. Lett.», 1973, 18, 465.
Stevens C. G., Zare R. N. — «J. Mol. Spectrosc»,
1975, 56, 167.
Stevens D. S. — «Phys. Rev.», 1931, 37, 1711.
Stevens D. S. — «Phys. Rev.», 1931, 38, 1292.
Stevenson D. P., Hippie I. A. — «J. Amer.
Chem. Soc», 1942, 64, 1528.
Stevenson D. P., Russel H. — «J. Amer. Chem.
Soc», 1939, 61, 3264.
Stevenson D. P., YostD. M. — «J. Chem. Phys.»,
1941, 9, 403
Stewart R. В., Jaeobsen R. T. — «Cryogenics»,
13, 526.
Stewart R. В., Johnson V. J. — «Advanc. Cryogen.
Eng.», 1960, 5, 557.
487
Литература
3538. St. Louis R. V., Crawford B. jr. — «J. Chem.
Phys.», 1965, 42, 857.
3539. Stock A., Gibson G. E., Stamm E. — «Ber.», 1912,
45, 3527.
3540. Stockmayer W. H., Kavanagh G. M., Mtckley
H. S. — «J. Chem. Phys.», 1944, 12, 408.
3541. Stolcheff B. P. — «Canad. J. Phys.», 1954, 32,
630.
3542. Stoicheff B. P. — «Canad. J. Phys.», 1957, 35,
730.
3543. Stone J. M. R., Milesl. M. — «J. Mol. Spectrosc.»
1970, 35, 354.
3544. Stopperka K. — «Z. anorg. und allgem. Chem.»,
1966, 344, 263.
3545. Stopperka K. — «Z. anorg. und allgem. Chem.»,
1966, 345, 277.
3546. Stopperka K. — «Z. Chem.», 1966, 6, 153.
3547. Stopperka K., KHz F. — «Z. anorg. und allgem.
Chem.», 1969, 370, 49.
3548. Stopperka K., KHz F. — «Z. anorg. und allgem.
Chem.», 1969, 370, 59.
3549. Stout J. W., Adams H. E. — «J. Amer. Chem.
Soc», 1942, 64, 1535.
3550. Strandberg M. W. P. — «J. Chem. Phys.», 1949,
17, 901.
3551. Strandberg M. W. P., Wentink Т., Hlller R. E.,
Wannier G. H., Deutsch M. L. — «Phys. Rev.»,
1948, 73, 188.
3552. Stratton K., Partington J. R. — «Philos. Mag.»,
1922, 43, 436.
3553. Straty G. C, Prydz R. — «Phys. Lett.», 1970,
A31, 306.
3554. Strausz O. P., Donovan R. J., Sorgo M. de. —
«Ber. Bunsenges. phys. Chem.», 1968, 72, 253.
3555. Street W. В., Sagan L. S., Staveley L. A. K.—
«J. Chem. Thermodyn.», 1973, 5, 633.
3556. Street W. В., Staveley L. A. K. — «J. Chem.
Phys.», 1971, 55, 2495.
3557. Streng A. G. — «J. Chem. and Engng Data»,
1961, 6, 431.
3558. Strieker W. Deutsche Luft — Raumfahrt. For-
schungsber., DLR-FB-66-04, 1966.
3559. Strieker W., Krauss L. — «Z. Naturforsch.»,
1968, 23a, 1116.
3560. Stripp K. F., Kirkwood J. G. — «J. Chem. Phys.»,
1951, 19, 1131.
3561. Stromeier W., Briegleb G. — «Z. Elektrochem.»,
1953 75 1232
3562. Stull D. !ff. — «Ind. and Engng Chem.», 1947, 39,
517.
3563. Stull D. R., Hildenbrand D. L., Oetting F. L.,
Sinke G. С — «J. Chem. and Engng Data», 1970,
15, 52.
3564. Stull D. R., Sinke G. C. Thermodynamic
Properties of the Elements. Advances in Chemistry.
Series, N 18. Washington, Amer. Chem. Soc,
1956.
3565. Stwalley W. C. — «J. Chem. Phys.», 1972, 56,
680.
3566. Sudsuki J., Watanabe K., Furukawa K. — «Low
Temperat. Sci. A: Phys. Sci.», 1966, N 24, 19.
3567. Suess H. E. — «Z. Naturforsch.», 1949, 4a, 328.
3568. Sugawara Т., Kanda E. — «Sci. Repts Res.
Inst. Tohoku Univ.» Ser. A, 1949, 1, 153.
3569. Sugawara Т., Sakamoto Y., Kanda E. — «Sci.
Repts Res. Inst. Tohoku Univ.» Ser. A, 1949, 1,
29
3570. Sugisaki M., Suga H., Seki S. — «Bull. Chem.
Soc. Japan», 1968, 41, 2591.
3571. SuhrmannR., Lude K. — «Z. Phys.», 1924, 29, 71.
3572. Sumodi A. J., Pace E. L. — «Spectrochim. acta»,
1972, A28, 1129.
3573. Sundaram S. — «J. Chem. Phys.», 1960, 33, 708.
3574. Sundaram S. — «Z. phys. Chem.» (BRD), 1963,
36, 376.
3575. Sundaram S. — «Spectrosc. Mol.», 1967, 16, 50.
3576. Sundaram S. — «Proc. Phys. Soc. London»,
1967, 92, 261.
3577. Sundaram S., Sarma Y. A., Piotrowski E. A. —
«Mol. Phys.», 1966, 10, 465.
3578. Sundaram S., Suszek F., Cleveland F. F. — «J.
Chem. Phys.», 1960, 32, 251.
3579. Sundbom M. — «Acta chem. scand.», 1968, 22,
1317.
3579a. Sunner S., Thoren S. — «Acta chem. scand.»,
1964, 18, 1528.
3580. Surles Т., Quarterman L. A., Hyman H. H. —
«J. Fluor. Chem.», 1974, 3, 293.
3581. Sutclieffe L. H., Walsh A. D. — «Proc. Phys.
Soc. London», 1953, A66, 209.
3582. Sutherland G. B. B. M., Penney W. G. — «Proc.
Roy. Soc. London», 1936, A156, 678.
3583. Suzeau P., Jurek R., Chanussot J. — «C. r. Acad.
sci.», 1973, B276, 777.
3584. Suzuki I. — «J. Mol. Spectrosc», 1969, 32, 54.
3584a. Svec H. J., Flesch G. D. — «Science», 1963, 142,
954.
3585. Svehla R. A. Estimated Viscosities and Thermal
Conductivities of Gases at High Temperatures.
Technical Report R-132. Cleveland, Ohio, Lewis
Research Center, 1961.
3586. Swanson H. E., Cook M. L., Isaacs Т., Evans
E. H. — NBS Circ. 539, 1960.
3587. Swanson N., Celotta R. J. — «Phys. Rev. Lett.»,
1975, 35, 783.
3588. Syverud A. N. Investigation and Compilation
of the Thermodynamic Properties of High
Temperature Chemical Species. Final Techn. Rept.
AFRPL-TR-70-20, 1 Jan. 1967 — 31 dec. 1969.
3589. Szwarc M. — «Proc. Roy. Soc. London», 1949,
A198, 267.
3590. Szwarc M. — «Proc. Roy. Soc. London», 1949,
A198, 285.
3591. Tables de constantes et donnees numeriques
constantes selectionnees. Donnees spectroscopi-
ques concernant les molecules diatomiques.
Rosen B. (Eds). Paris, Hermann et Cie, 1951.
3592. Tables internationales de constantes
selectionnees. 17. Donnees spectroscopiques relatives aux
molecules diatomiques. B. Rosen (Ed.). Oxford—
New York—Toronto—Sydney—Brunswick, Per-
gamon Press, 1970.
3593. Table of Interatomic Distances and Configuration
in Molecules and Ions. L. E. Sutton (Sci. Ed.).
Chem. Soc. Spec. Publ., N 11. London, 1958.
3594. Tables of Interatomic Distances and
Configuration in Molecules and Ions. Suppl. 1956—1959.
L. E. Sutton (Sci. Ed.). Chem. Soc. Spec. Publ.
N 18. London, 1965.
3595. Taillade M. — «C. r. Acad. sci.», 1944, 218, 836.
3596. Takahata Y., Eri Т., J'Haya Y. — «Chem.
Phys. Lett.», 1974, 26, 557.
3597. Takamine Т., Suga Т., Тапака Y. — «Sci.
Papers Inst. Phys. Chem. Res. Tokyo», 1939,
36, 437.
3598. Takezawa S. — «J. Chem. Phys.», 1970, 52, 2575.
3599. Takezawa S. — «J. Chem. Phys.», 1970, 52, 5793.
3600. Takezawa S., Innes F. K., Tanaka Y. — «J.
Chem. Phys.», 1967, 46, 4555.
Литература
3601. Ta-Kong Ыа G., Duncan А. В. — «J. Chem.
Phys.», 1951, 19, 71.
3602. Talaty E. R., Schwartz A. K., Simons C. — «J.
Amer. Chem. Soc», 1975, 97, 972.
3603. Talley R. M., Kaylor H. M., Nielsen A. H.
«Phys. Rev.», 1950, 77, 529.
3604. Tamagake K., Hamada Y., Yamaguchi J., Hira-
kawa A. Y., Tsuboi M. —«J. Mol. Spectrosc.»,-
1974, 49, 232.
3605. Tammann G. — Kristallizieren und Schmelzen.
Leipzig, 1903.
3606. Tanaka M., Tanaka J., Nagakura S. — «Bull.
Chem. Soc. Japan», 1966, 39, 766.
3607. Tanaka Т., Field R. W., Harris D. O. — «J.
Mol. Spectrosc.», 1975, 56, 188.
3608. Tanaka Т., Koana Z. — «Proc. Phys. Math.
Soc, Japan», 1933, 15, 272.
3609. Tanaka Т., Koana Z. — «Proc. Phys. Math.
Soc. Japan», 1934, 16, 365.
3610. Tanaka Т., Morino Y. — «J. Mol. Spectrosc.»,
1969, 32, 430.
3611. Tanaka Т., Morino Y. — «J. Mol. Spectrosc»,
1969, 32, 436.
3612. Tanaka Т., Morino Y. — «J. Mol. Spectrosc»,
1970, 33, 538.
3613. Tanaka Т., Morino Y. — «J. Mol. Spectrosc.»,
1970, 33, 552.
3614. Tanaka Т., Sizaisi M. — «Proc. Phys. Soc.
Japan», 1933, 15, 195.
3615. Tanaka Y. — «Sci. Papers Inst. Phys. Chem.
Res. Tokyo», 1944, 42, 49.
3616. Tanaka Y. — «J. Sci. Res. Inst.», 1949, 43, 160.
3617. Tanaka Y. — «J. Chem. Phys.», 1953, 21, 562.
3618. Tanaka Y. — «J. Chem. Phys.», 1953, 21, 1402.
3619. Tanaka Y., Namioka Т., Yursa A. S. — «Canad.
J. Phys.», 1961, 39, 1138.
3620. Tanaka Y., Ogawa M. — «Spectrochim. acta»,
1959, 14, 751.
3621. Tanaka Y., Ogawa M. — «J. Chem. Phys.»,
1962, 36, 726.
3622. Tanaka Y., Takamine T. — «Sci. Papers Inst.
Phys. Chem. Res., Tokyo», 1942, 39,437.
3623. Tanaka Y., Yursa A. S. — «J. Opt. Soc. America»,
1961, 51, 1239.
3624. Tandon S. P., Tandon K. — «Indian J. Pure and
Appl. Phys.», 1969, 7, 245.
3625. Tang Sheng-Yyh, Brown С W. — «J. Raman
Spectrosc», 1974, 2, 209.
3626. Tanischita I., Watanabe K., Kijima J., Ishii H.,
Oguchi K., Uematsu M. — «J. Chem. Thermo-
dyn.», 1976, 8, 1.
3627. Tarte P. — «Ann. soc. scient. Bruxelles,» Ser. I,
1956, 70, 244.
3628. Taylor A., Rideal E. — «Proc. Roy.SSoc. London»,
1927, A115, 589.
3629. Taylor A. H., Crist R. H. — «J. Amer. Chem.
Soc», 1941, 63, 1377.
3630. Taylor G. В., Lenher S. — «Z. phys. Chem.,
Bodenstein-Festhand», 1931, 30.
3631. Taylor M. J., Woodward L. A.— «J. Chem.
Soc», 1963, 4670.
3632. Taylor R. С — «J. Chem. Phys.», 1950, 18, 898.
3633. Taylor R. C, Cross P. С — «J. Chem. Phys.»,
1956, 24, 41.
3634. Taylor R. C, Vidale G. L. — «J. Amer. Chem.
Soc», 1956, 78, 5999.
3635. Teal G. K., McWood G. E. — «J. Chem. Phys.»,
1935, 3, 760.
3636 Tech J. L. — «J. Res. NBS», 1963, A67, 505.
3637. Tellinghuisen J. — Thesis. Univ. Calif., Berkeley,
1969.
3638. Tellinghuisen J. — «J. Chem. Phys.», 1973, 58,
2821.
3639. Tellinghuisen J. — «Chem. Phys. Lett.», 1973,
18, 544.
3639a. Tellinghuisen J. — «Chem. Phys. Lett.», 1974,
29, 359.
3640. Tellinghuisen J., Tisone G. C, Hoffman J. M.,
Hays A. K.~ «J. Chem. Phys.», 1976, 64, 4796.
3640a. Templeton D. H., Zalkin A., Forrester J. D.,
Williamson S. M. — «J. Amer. Chem. Soc»,
1963, 85, 242.
36406. Templeton D. H., Zalkin A., Forrester J. ?>.,
Williamson S. M. — «J. Amer. Chem. Soc»,
1963, 85, 817. »
3641. Terry M. G., Lynch J. Т., Bunclark M., Mansell
K. R., Staveley L. A. K. — «J. Chem. Thermo-
dyn.», 1969, 1, 413.
3642. Tertan A. — «Bui. stiint. Inst. politechn. Cluj»,
1964, 7, 71.
3643. Terwilliger D. Т., Smith A. L. — «J. Mol.
Spectrosc», 1973, 45, 366.
3644. Tevault D. E., Andrews L. — «Spectrochim.
acta», 1974, A30, 969.
3645. Thackray M. — «Nature», 1964, 203, 1278.
3646. Thaddens P., Krisher L. C, Loubser J. H. N. —
«J. Chem. Phys.», 1964, 40, 257.
3646a. Thakker A. J., Smith J. V. В. — «Mol. Phys.»,
1974, 27, 191.
3647. Thanh N.-V., Haeusler C, Barchewitz P. — «J.
phys. et radium», 1962, 23, 348.
3648. The 1967 IFC Formulation for Industrial Use.
A Formulation of the Thermodynamic
Properties of Ordinary Water Substance Prepared by
the International Formulation Committee (IFC).
February 1967.
3649. The 1968 IFC Formulation for Scientific and
General Use. A Formulation of the
Thermodynamic Properties of Ordinary Water Substance
Prepared by the international Formulation Committee
(IFC), April 1968.
3650. The Chemistry and Metallurgy of Miscelaneous
Materials. Thermodynamics. L. L. Quill (Ed.).
Papers 3—7, New York—Toronto—London, 1950.
3651. Theeuwes F., Bearman R.J. — «J. Chem. Thermo-
dyn.», 1970, 2, 171.
3652. Theeuwes F., Bearman R. J. — «J. Chem. Thermo-
dyn.», 1970, 2, 179.
3653. Theeuwes F., Bearman R. J. — «J. Chem.
Thermodyn.», 1970, 2, 501.
3654. Theeuwes F., Bearman R. J. — «J. Chem.
Thermodyn.», 1970, 2, 507.
3655. Thelin J. H., Van der Meulen P. A. — «J. Amer.
Chem. Soc», 1948, 70, 1796.
3656. Thermodynamic Function of Gases. V. 1, 2.
F. Din (Ed.). London, Butterworths Scient.
PubL, 1956.
3657. Thermodynamic Function of Gases. V. 3. F. Din
(Ed.). London, Butterworths Scient. Publ., 1961.
3658. Thermodynamics of Species Important in
Aerospace Technology (including Selected Topics
in Chemical Kinetics), NBS Rept 10904.
Washington, 1972.
3659. Thorburn R., Craggs J. D. — «Proc. Phys. Soc.
London», 1956, B69, 682.
3660. The Threshold of Space. M. Zelikoff (Ed.). N. Y.f
Pergamon Press, 1957.
3661. Thilo E., HeinzD., Jost К. Н. — «Angew. Chem.»,.
1964, 76, 229.
489
Литература
3662. Thomas С. D., Gingrich N. S. — «J. Chem. Phys.», 3697.
1938, 6, 659.
3663. Thomas R. K. «Proc. Roy. Soc. London», 1971, 3698.
A322, 137.
3664. Thomas R. K. — «Proc. Roy. Soc. London», 3699.
1971 A325 133
3665. Thomas R.' K. — «Proc. Roy. Soc. London», 3700.
1975, A344, 579. 3701.
3666. Thomas R. K., Thompson H. — «Proc. Roy. Soc. 3701a.
London», 1970, A314, 329. 3702.
3667. Thomas W. — In: Progress in International
Research on Thermodynamic and Transport 3703.
Properties. N. Y., ASME, 1962, p. 166.
3668. Thompson H. В., Bartell L. S. — «Trans. Amer.
Grystallogr. Assoc», 1966, 2, 190. 3704.
3669. Thompson H. W. — «Trans. Faraday Soc», 1941,
37, 344. 3705.
3670. Thompson H. W., Green B. A. — «Spectrochim.
acta», 1956, 8, 129. 3706.
3671. Thompson H. W., Williams R. L., Callomon
H. J. — «Spectrochim. acta», 1952, 5, 313. 3707.
3672. Thompson S. D., Carroll D. G., Watson F., 3708.
O'Donnel M., McGlinn S. P. — «J. Chem.
Phys.», 1966, 45, 1367. 3709.
3673. Thompson W. K. — «Trans. Faraday Soc», 1966, 3710.
62, 2667.
3674. Thomsen J. — «J. prakt. Chem.», 1880, 21, 449. 3711.
3675. Thomsen J. — «Ber.», 1882, 15, 3021.
3676. Thomsen J. Thermochemische Untersuchungen, 3712.
Leipzig, Barth 1882—1886.
3677. Thomson C, Brotchie D. A.— «Chem. Phys. 3713.
Lett.», 1972, 16, 573.
3678. Thomson C, Brotchie D. A.— «Mol. Phys.», 3714.
1974, 28, 301.
3679. Thorpe T. E., Tutton A. E. — «J. Chem. Soc», 3715.
1890, 545.
3680. Thrush B. A. — «Proc. Roy. Soc. London», 1956, 3716.
A235, 143.
3681. Thulstrup E. W., Andersen A. — «J. Phys. B: 3717.
Atom, and Mol. Phys.», 1975, 8, 965. 3718.
3682. Thyagarajan G., Cleveland F. F. — «J. Mol.
Spectrosc», 1960, 5, 210. 3719.
3683. Tidwell E. D., Plyler E. K., Benedict W. S. —
«J. Opt. Soc. America», 1960, 50, 1243. 3720.
3684. Tilford S. G., Ginter M. L. — «J. Mol.
Spectrosc», 1971, 40, 568. 3721.
3685. Tilford S. G., Ginter M. L., Bass A. M. — «J.
Mol. Spectrosc», 1970, 34, 327. 3722.
3686. Tilford S. G., Ginter M. L., Vanderslice J. T. —
«J. Mol. Spectrosc», 1970, 33, 505. 3723.
3687. Tilford S. G., Vanderslice J. Т., Wilkinson P. G. —
«Astrophys. J.», 1965, 141, 1226. 3724.
3688. Tilford S. G., Vanderslice J. Т., Wilkinson P. G. —
«Astrophys. J.», 1965, 142, 1203. 3725.
3689. Tilford S. G., Wilkinson P. G. — «J. Mol.
Spectrosc.», 1964, 12, 347. 3726.
3690. Tilford S.G., Wilkinson P. G., Vanderslice J. T. — 3727.
«Astrophys. J.», 1965, 141, 427.
3691. Tiemann E. — «J. Phys. and Chem. Ref. Data», 3728.
1974, 3, 259.
3692. Tiemann E. — «J. Mol. Spectrosc», 1974, 51, 3729.
316. 3730.
3693. TiemannE., Hoeft J., LovasE. J., JohnsonD.R.—
«J. Chem. Phys.», 1974, 60, 5000. 3730a.
3694. Tiemann E., Hoeft J., Torring T. — «Z. Natur- 37306.
forsch.», 1973, 28a, 1405.
3695. Tiemann E., Moller T. — «Z. Naturforsch.», 3731.
1975, 30a, 986.
3696. Tiernan T. O., Hughes B. M., Lifshitz C. — «J. 3732.
Chem. Phys.», 1971, 55, 5692.
Tilden W. A., Barnett R. E. — «J. Chem. Soc»,
1896, 69, 154.
Tinkham M., Strandberg M. W. P. — «Phys.
Rev.», 1955, 97, 937.
Tinkham M., Strandberg M. W. P. — «Phys.
Rev.», 1955, 97, 951.
Tinland B. — «J. Mol. Struct.», 1969, 3, 256.
Tinland B. — «Spectrosc. Lett.», 1970, 3, 51.
Titov E. V. — «J. Mol. Struct.», 1973, 19, 301.
Tkachenko E. A., Holleran E. M., Moses R. A. —
«Cryogenics», 1964, 4, 12.
Tocho J. O., Ranea Sandoval H. F., Tagliaferri
A. A., Garavaglia M., Gallardo M., Massone
С. А. — «Nuov. rev. opt.», 1974, 5, 319.
Toennies J. P., Green E. F. — «J. Chem. Phys.»,
1957, 26, 655.
Tolles W. M., Gwinn W. D. — «J. Chem. Phys.»,
1962, 36, 1119.
Tolles W. M., Kinsey J. L., Curl R. F.,
Heidelberg R. F. — «J. Chem. Phys.», 1962, 37, 927.
Topley B. — «Chem. and Ind.», 1959, 431.
Topp W. C, Allen L. C. — «J. Amer. Chem.
Soc», 1974, 96, 5291.
Torreson Y. G. — «Arkiv fys.», 1960, 18, 417.
TothR.A., MargolisJ. S. — «J. Mol. Spectrosc»,
1975, 55, 229.
Townes С H., Merrit F. R. — «Phys. Rev.»,
1946, 70, 558.
Townes C. H., Merritt F. R., Wright B. D. —
«Phys. Rev.», 1948, 73, 1334.
Townes С H., Wright B. D., Merritt F. R. —
«Phys. Rev.», 1948, 73, 1249.
Trafmar S., McCaa D. J. — «J. Mol. Spectrosc»,
1964, 14, 244.
Trambarulo R., Ghosh S. TV., Burrus C. A.
Gordy W. — «J. Chem. Phys.», 1953, 21, 851.
Tranquard A., Riviere M., Coffy G., Cohen-
Adad R. — «C. r. Acad. sci.», 1970, C270, 2056.
Trautz M., Adler H. — «Z. Phys.», 1934, 89, 12.
Trautz M., Hinck C. F. — «Z. anorg. und allgem.
Chem.», 1915, 93, 177.
Trautz M., Hinck С F. — «Z. anorg. und allgem.
Chem.», 1916, 97, 127.
Trautz M., Wachenheim L. — «Z. anorg. und
allgem. Chem.», 1916, 97, 241.
Treacy E. В., Beers Y. — «J. Chem. Phys.»,
1962, 36, 1473.
Trivedi V. M., Gohel V. B. — «J. Phys. B: Atom,
and Mol. Phys.», 1972, 5, 138.
Troe J. — «Ber. Bunsenges. phys. Chem.», 1969,
73, 946.
Troe J., Wagner H. G., Weden G. — «Z. phys.
Chem.» (BRD), 1967, 56, 238.
Trombetti A. — «Canad. J. Phys.», 1968, 46,
1005. '
Trombetti A. — «J. Chem. Soc», A, 1971, 1086.
Troost Т., Hautefeuille P. — «Ann. chim. phys.»,
1876, 9, 56.
Trotman-Dickenson A. F. — Gas Kinetics. N. Y.
Acad. Press, 1955.
Trumpy B. — «Z. Phys.», 1931, 68, 675.
Trussewitsch A. — «Fortschr. Phys.», 1893, 49,
478.
Truter M. R. — «Acta crystallogr.», 1954, 7, 73.
Tsao P., Cobb С. С, Claassen H. H. — «J. Chem.
Phys.», 1971, 54, 5247.
Tschuikow-Roux E. — «J. Phys. Chem.», 1962,
66, 1636.
Tschuikow-Roux E., Mac Fadden K. O. — «J.
Phys. Chem.», 1973, 77, 734.
490
Литература
3733. Tsederberg N. S., Alexandrov A. A., Hasanshin 3766.
T. S., Larkin D. K. Proc. 8th Intern. Gonf.
on Properties of Water and Steam, France, 1974. 3767.
3734. Tseng T. J., Grain F. — «J. Ghem. Phys.», 1973,
59, 6563. 3768.
3735. Tsonopoulos C, Prausnitz J. M, —«Cryogenics»,
1969, 9, 315. 3769.
3736. Tsuboi M., Hisatsune I. G. — «J. Chem. Phys.»,
1972, 57, 2087. 3770.
3737. Tsuboi M., Overend J. — «J. Mol. Spectrosc»,
1974, 52, 256. 3771.
3738. Tsuboi M., Schimanouchi Т., Misushima S. —
«Spectrochim. acta», 1958, 13, 80. 3772.
3739. Tsunekawa S. — «J. Phys. Soc. Japan», 1972,
33, 167. 3773.
3740. Tsuruta K. — «Phys. Z.», 1900, 1, 417.
3741. Tubino R., Zerbi J. —«J. Ghem. Phys.», 1969, 3774.
51, 4509.
3742. Turner A. G. — «J. Mol. Struct.», 1971, 9, 211. 3775.
3743. Turner D. W. — «Proc. Roy. Soc. London», 1968,
A307, 15. 3776.
3743a. Turner J. J., Pimentel G. C. — «Science», 1963,
140, 974. 3777.
3744. Tursi A. J., Nixon E. R. — «J. Ghem. Phys.»,
1970, 52, 1521. 3778.
3745. Tyler J. K. — «J. Mol. Spectrosc», 1963, 11, 39.
3746. Tyte D. C. —«Proc. Phys. Soc. London», 1963, 3779.
81, 163.
3747. Tyte D. C, Nicholls R. W. Identification Atlas 3780.
of Molecular Spectra. III. The JVJfi2S+—Х*Ц:
First Negative System of Nitrogen (Univ. 3781.
Western Ontario, London, Canada) NASA
Accession N N66-25320, 1965. 3782.
3748. Uchara H. — «Bull. Ghem. Soc. Japan», 1969,
42, 886. 3783.
3749. Uchara H. —«Mol. Phys.», 1971, 21, 407.
3750. Uchara #., Morino Y. — «Mol. Phys.», 1969, 17,
239. 3784.
3751. Uchara H., Morino Y. — «Bull. Chem. Soc.
Japan», 1970, 43, 2273. 3785.
3752. Uchara H., Morino Y. — «J. Mol. Spectrosc»,
1970, 36, 158. 3785a.
3753. Uematsu M., Watanabe K., Tanishita I. Proc.
6th Sympos. on Thermophys. Properties, Aug. 37856.
6—8 1973. P. E. Liley (Ed.). N. Y., ASME,
1973, p. 219. 3786.
3754. Urey H. C, Dawsey L. #., Rice F. O. — «J.
Amer. Chem. Soc», 1929, 51, 1371. 3787.
3755. Urey H. C, Rittenberg D. — «J. Chem. Phys.»,
1933 1 137. 3788.
3756. Uy 6. M., Kohl F. J., Carlson K. D. — «J. Phys.
Chem.», 1968, 72, 1611. 3789.
3757. Vago J. Dissertation. Budapest, 1911.
3758. Vaidya W. M. — «Proc. Indian Acad. Sci.», 3790.
1938, A7, 321.
3759. Vaidyan V. K., Santaram C. — «J. Phys. B: 3791.
Atom, and Mol. Phys.», 1970, 3, 16.
3760. Vaidyan V. K., Santaram С —«Indian J. Pure 3792.
and Appl. Phys.», 1971, 9, 1022.
3761. Vanasse G. A., Strong J., Loewenstein E. — «J. 3793.
Opt. Soc. America», 1959, 49, 309.
3762. Van der Meulen H. G. L. —« «Recueil trav. chim.», 3794.
1883, 2, 69.
3763. Vanderslice J. Т., Mason E. A., Maisch W. G., 3795.
Lippincott E. R. — «J. Ghem. Phys.», 1960, 33,
614. 3796.
3764. Vanderslice J. Т., Tilford S. G., Wilkinson P. G. —¦
«Astrophys. J.», 1965, 141, 395.
3765. Vanderzee С E., Gier L. J. —, «J. Ghem. Thermo- 3797.
dyn.», 1974, 6, 441.
Vanderzee C. E., King D. L. — «J. Ghem.
Thermodyn.», 1972, 4, 675.
Vanderzee C. E., KingW. L., Wadso I. — «J.
Ghem. Thermodyn.», 1972, 4, 685.
Vanderzee C. E., Mansson M., Sunner S. — «J.
Ghem. Thermodyn.», 1972, 4, 533.
Vanderzee C. E., Mansson M., Wadso /., Sunner
S. — «J. Chem. Thermodyn.», 1972, 4, 541.
Vanderzee C. E., Nutter J. D. — «J. Phys.
Ghem.», 1963, 67, 2521.
Vanderzee С E., Rodenburg W. W. — «J. Ghem.
Thermodyn.», 1970, 2, 461.
Vanderzee С E., Rodenburg W. W. — «J. Chem.
Thermodyn.», 1971, 3, 267.
Van Doormaal P. M., Scheffer F. E. C. —
«Recueil trav. chim.», 1931, 50, 1100.
Vandorpe В., Migeon M. — «Rev. chim. miner.»,
1965, 2, 303.
Van Home B. H., Hause С D. — «J. Ghem.
Phys.», 1956, 25, 56.
Van Huong P., Desbat B. — «Bull. Soc. chim.
belg.», 1972, 7, 2631.
Van Roalte D., Harrison A. G. — «Canad. J.
Chem.», 1963, 41, 3118.
Van Riet R. — «Ann. Soc scient. Bruxelles»,
Ser. 1, 1963, 77, 18.
Van Riet R. — «Ann. Soc. scient. Bruxelles»,
Ser. 1, 1964, 78, 237.
Vanzee R. J., Khan A. U. — «Chem. Phys.
Lett.», 1975, 36, 123.
Varetti E. ?., Pimentel I. C. — «J. Chem. Phys.»,
1971, 55, 3813.
Varma R., Curl R. F. — «J. Phys. Chem.», 1976,
80, 402.
Vasudevan K., Peyerimhoff S. D., Buenker R. /.,
Kammer W. E., Hsu Hsiang-lin. ¦— «Chem.
Phys.», 1975, 7, 187.
Vaughn J. D., Muetterties E. L.—4.J. Phys.
Chem.», 1960, 64, 1787.
Veillard Л. — «Theor. chim. acta», 1966, 5,
413.
Veillette P., Marchand P. —«Canad. J. Phys.»,
1974, 52, 930.
Velasco R., Rivero F. — «Opt. рига у apl.»,
1974, 7, 45.
Vella G. J., Rolfe J. — «J. Chem. Phys.», 1974,
61, 41.
Venkateswaran C. S. —¦ «Proc. Indian Acad.
Sci.», 1935, A2, 260.
Venkateswaran С S. — «Proc. Indian Acad.
Sci.», 1936, A4, 345.
Venkateswaran С S. — «Proc. Indian Acad.
Sci.», 1936, A4, 414.
Venkateswaran S. — «Indian J. Phys.», 1931, 6,
275.
Venkateswaran S. — «Philos. Mag.», 1933, 15,
263.
VenkateswarluD. K., Thirugnanasambandam P. —
«Trans. Faraday Soc», 1959, 55, 1993.
Venkateswarlu K., Devi V. M. —> «Indian J. Pure
and Appl. Phys.», 1965, 3, 120.
Venkateswarlu K., Joseph P. A. — «Indian J. Pure
and Appl. Phys.», 1971, 9, 235.
Venkateswarlu K., Mariam S. —«Indian J. Pure
and Appl. Phys.», 1965, 3, 117.
Venkateswarlu K., Mariam S., Rajalakshmi
K. V. — «Bull. cl. sci. Acad. roy. Belgique»,
1965, 51, 359.
Venkateswarlu K., Rajalakshmi K. V. —• «Acta
phys. polon», 1962, 22, 417
491
Литература
3798. Venkateswarlu К., Rajalakshmi К. V. — «J.
Scient. Res. Inst.», 1962, B21, 349.
3799. Vankateswarlu K., Rajalakshmi K. V. — «Indian
J. Pure and Appl. Phys.», 1963, 1, 62.
3800. Venkateswarlu K., Thanalakshmi R. — «Indian
J. Pure and Appl. Phys.», 1963, 1, 377.
3801. Venkateswarlu K., Thanalakshmi R. — «Proc.
Indian Acad. Sci.», 1963, A57, 181.
3802. Venkateswarlu P. — «Proc. Indian Acad. Sci.»,
1947, A25, 138.
3803. Venkateswarlu P. — «Phys. Rev.», 1951, 81, 821,
3804. Venkateswarlu P. — «Canad. J. Phys.», 1969, 47.
2525.
3805. Venkateswarlu P. — «Canad. J. Phys.», 1970, 48,
1055.
3806. Venkateswarlu P. — «Canad. J. Phys.», 1975, 53,
812.
3807. Venkateswarlu P., Khanna B. P. — «Proc. Indian
Acad. Sci.», 1959, A49, 117.
3808. Venkateswarlu P., Verma R. D. — «Proc. Indian
Acad. Sci.», 1957, A46, 251.
3809. Venkateswarlu P., Verma R. D. — «Proc. Indian
Acad. Sci.», 1957, A46, 416.
3810. Venkateswarlu P., Verma R. D. — «Proc. Indian
Acad. Sci.», 1958, A47, 150.
3811. Venkateswarlu P., Verma R. D. — «Proc. Indian
Acad. Sci.», 1958, A47, 161.
3812. Ven Shih, These, Nancy, 1932.
3813. Verhoek F. H., Daniels F. — «J. Amer. Chem.
Soc», 1931, 53, 1250.
3814. Verhoeven J., Bluyssen H. J., Dymanus A. —
«Phys. Lett.», 1968, A26, 424.
3815. Verma R. D. — «Proc. Indian Acad. Sci.», 1958,
A47, 196.
3816. Verma R. D. — «Proc. Indian Acad. Sci.», 1958,
A48, 197.
3817. Verma R. D. — «Spectrochim. acta», 1959, N 9,
751.
3818. Verma R. D. — «J. Chem. Phys.», 1960, 32, 738.
3819. Verma R. D. — «Canad. J. Phys.», 1970, 48,
2391.
3820. Verma R. D. — «Canad. J. Phys.», 1971, 49, 279.
3821. Verma R. D., Broida H. P. — «Canad. J. Phys.»,
1970, 48, 2991.
3822. Verma R. D., Dixit M. N. — «Canad. J. Phys.»,
1968, 46, 2079.
3823. Verma R. D., Dixit M. N., Jois S. S., Nagarai S.,
Singhal S. R. — «Canad. J. Phys.», 1971, 49,
3180.
3824. Verma R. D., Jois S. S. —«Canad. J. Phys.»,
1973 51 322
3825. Verma R. D., Le Roy R. J. — «J. Chem. Phys.»,
1974, 61, 438.
3826. Verma R. D., Singhal S. R. — «Canad. J. Phys.»,
1975, 53, 411.
3827. Vermeil C, Masanet J., Gilles A. —«Intern. J.
Radiat. Phys. Chem.», 1975, 7, 275.
3828. Veseth L. — «J. Phys. B: Atom, and Mol. Phys.»,
1972 5 229
3829. Veseth L. — «J. Phys. B: Atom, and Mol. Phys.»,
1972, 5, 2168.
3830. Veseth L. — «Mol. Phys.», 1973, 26, 101.
3831. Veseth L., Lofthus A. — «Mol. Phys.», 1974, 27,
511.
3832. Vesper H. G., Rollefson G. K. — «J. Amer. Chem.
Soc», 1934, 56, 620.
3833. Vezzoli G. C, Dachille F., Roy R. — «Science»,
1969, 166, 218.
3834. Vidal В., Dessaux O., Marteel J. P., Goudmand
P. — «C. r. Acad. sci.», 1969, C268, 2140.
3835. Vigroux E., Miglotte M., Neven L. — «Physica»,
1957, 19, 140.
3836. Viscido L., Sicre J. E., Schumacher H. J. — «Z.
phys. Chem.» (BRD), 1961, 29, 188.
3837. Vogt D. — «Intern. J. Mass Spectr. Ion Phys.»,
1969, 3, 81.
3838. Vogt K., Konigsberger J. — «Z. Phys.», 1923, 13,
294.
3839. Vollmar P. M. — «J. Chem. Phys.», 1963, 39,
2236.
3840. Volmer M. — «Ann.», 1924, 440, 200.
3841. Von Ellenrieder G., Costellano E., Shumacher
H. J. — «Z. phys. Chem.» (BRD), 1967, 55, 144.
3842. Vorlander D., Selke W.. Ктеф G. — «Ber.»,
1925, 58, 1802.
3843. Vosper A. J. — «J. Chem. Soc», A, 1966, 1759.
3844. Vosper A. J. — «J. Chem. Soc.» A, 1970, 4497.
3845. Vosper A. J. — «J. Chem. Soc.» A, 1971, 1589.
3846. Vosper A. J. — «J. Chem. Soc. Dalton Trans.»,
1976, 135.
3847. Vratny F. — «Appl. Spectrosc», 1959, 13, 59.
3848. Wachman #., Unevsky M.. Lyon R. — «J. Chem.
and Engng Data», 1960, 5, 456.
3849. Waddington Т. С — «J. Chem. Soc», 1958, 4340.
3850. Waech T, G., Bernstein R. B. — «J. Chem. Phys.»,
1967, 46, 4905.
3851. Wagenbreth H. — «PTB-Mitt.», 1968, 78, 91.
3852. Wagman D. D., Kilpatrick J. E., Taylor W. J.,
Pitzer K. S., Rossini F. D. — «J. Res. NBS»,
1945, 34, 143.
3853. Wagner E. L. — «Theor. chim. acta», 1971, 23,
115.
3854. Wagner E. L., Bulgozdy E. L. — «J. Chem.
Phys.», 1951, 19, 1210.
3855. Wagner E. L., Homing D. F. — «J. Chem.
Phys.», 1950, 18, 295, 305.
3856. Wagniere G. — «Theor. chim. acta», 1973, 31, 269.
3857. Wahl M. H., Urey H. C. — «J. Chem. Phys.»,
1935, 3, 411.
3858. Wahrhaftig A. L. — «J. Chem. Phys.», 1942,10,
248.
3859. Waidya W. M. — «Proc Indian Acad. Sci.»,
1937, A6, 122.
3860. Waldorf D. M., BabbA. L. — «J. Chem. Phys.»,
1963, 39, 432.
3861. Waldorf D. M., Babb A. L. — «J. Chem. Phys.»,
1964, 40, 1165.
3862. Walker L. С — «J. Phys. Chem.», 1967, 71, 361.
3863. Walker R. E., Hochheimer B. F. — «J. Mol.
Spectrosc», 1970, 34, 500.
3864. Wallace J. J., Goodeve С F. — «Trans. Faraday
Soc», 1931, 27, 648.
3865. Wallace L. — «Astrophys. J.», 1961, 132, 894.
3866. Wallace L. — «Astrophys. J.» Suppl. Ser., 1962,
6, 445.
3867. Wallart F. — «Canad. J. Spectrosc», 1972, 17,
128.
3868. Walrafen G. — «J. Chem. Phys.», 1964, 40, 2326.
3869. Walrafen G., Dodd D. M. — «Trans. Faraday
Soc», 1961, 57, 1286.
3870. Walsh A. D. — «J. Chem. Soc», 1953, 2260.
3871. Walsh A. D. — <u. Chem. Soc», 1953, 2266.
3872. Walsh A. D. — «J. Chem. Soc», 1953, 2288.
3873. Walsh A. D. — In: Threshold of Space. Proc.
Conf. Chem. Aeron., 1956, p. 165.
3874. Walsh A. D., Warsop P. A. —«Trans. Faraday
Soc», 1961, 57, 345.
3875. Wan J. K. S., Morton J. R.. Bernstein H. J. —
«Canad. J. Chem.», 1966, 44. 1957.
492
Литература
3876. Wanatabe Y. — «Acta crystallogr.», 1974, B30,
1396.
3876a. Wang S. Y., Lohr L. L. jr. — «J. Chem. Phys.»,
1974, 60, 3901.
38766. Wang S. Y., Lohr L. L. jr. — «J. Chem. Phys.»,
1974, 60, 3916.
3877. Ward A. T. — «J. Phys. Chem.», 1968, 72, 744.
3878. Ward J. J., Hussey M. A. 3rd Sympos. on
Combustion and Explosion Phenomena, Baltimore,
1949 p. 599.
3879. War neck P. — «Chem. Phys. Lett.», 1969, 3, 532.
3880. Warren B. E., Burwell J. T. — «J. Chem.
Phys.», 1935, 3, 6.
3881. Warren D. T. — «Phys. Rev.», 1935, 47, 1.
3882. Wartenberg H. — «Z. anorg. und allgem. Chem.»,
1931, 200, 235.
3883. Wartenberg H., Fitzner O. — «Z. anorg. und
allgem. Chem.», 1926, 151, 313.
3884. Wartenberg H., Hanisch K. — «Z. phys. Chem.»,
1932, A161, 413.
3885. Wartenberg H., Klinkott G. — «Z. anorg. und
allgem. Chem.», 1930, 193, 409.
3886. Wartenberg H., Riteris С — «Z. anorg. und
allgem. Chem.», 1949, 258, 356.
3887. Wartenberg H., Schutza H. — «Z. phys. Chem.»,
1933, A164, 386.
3888. Wartenberg H., Sprenger G., Taylor J. — «Z.
phys. Chem., Bodenstein—Festband», 1931, 61.
3889. Waser J., Wieland K. — «Nature», 1947, 160, 643.
3890. Watanabe K., Jursa A. S. — «J. Chem. Phys.»,
1964, 41, 1650.
3891. Watanabe K., Marmo F. F. — «J. Chem. Phys.»,
1956, 25, 965.
3892. Watson J. K. G. — «J. Chem. Phys.», 1966, 45,
1360.
3893. Watson J. K. G. — «J. Chem. Phys.», 1967, 46,
1935.
3894. Watson J. K. G. — «J. Chem. Phys.», 1968, 48,
181.
3895. Watson J. K. G. — «J. Chem. Phys.», 1968, 48,
4517.
3896. Watson J. K. G. — «J. Mol. Spectrosc», 1973,
45, 99.
3897. Watson J. K. G. — «J. Mol. Spectrosc», 1973,
48, 479.
3898. Watson W. W., Koontz P. G. — «Phys. Rev.»,
1934, 46, 32.
3899. Watson W. W., Parker A. E. — «Phys. Rev.»,
1931, 37, 1484.
3900. Wayne F. D., Davis P. В., Thrush B. A. —
«Mol. Phys.», 1974, 28, 989.
3901. Wayne F. D., Stone A. J. — «Ber. Bunsenges.
phys. Chem.», 1974, 78, 201.
3902. Wayne L. G., Yost D. M. — «J. Chem. Phys.»,
1951, 19, 41.
3903. Weatherly T. L., WilliamsD. — «J. Chem. Phys.»,
1956, 25, 717.
3904. Webb D. U., Rao K. N. — «Appl. Opt.», 1966, 5,
1461.
3905. Webb D. U., Rao K. N. — «J. Mol. Spectrosc»,
1968, 28, 121.
3906. Weber L. A. — «J. Res. NBS», 1970, A74, 93.
3907. Weber L. A. Thermodynamic and Related
Properties of Para-Hydrogen from the Triple Point
to 300 К at Pressures to 1000 Bar. NASA SP-3088,
1975.
3908. Wei J., Tellinghuisen J. — «J. Mol. Spectrosc»,
1974, 50, 317.
3909. Wei M. S., Current J. M., Gendell J. — «J.
Chem. Phys.», 1970, 52, 1592.
3910. Wei M. S., Current J. H., Gendell J. — «J.
Chem. Phys.», 1972, 57, 2431.
3911. WeidnerR. T. — «Phys. Rev.», 1947, 72, 1268.
3911a. WeinstockB., Weaver E. E., Knop C. P. — «Inorg.
Chem.», 1966, 5, 2189.
3912. Weisbaum S., Beers Y., Hermann G. — «J. Chem.
Phys.», 1955, 23, 1601.
3913. Weisbeek R., Volkner A. — «Z. angew. Phys.»,
1971, 32, 258.
3914. Weisner J. D., Armstrong В. Н. — «Proc. Phys.
Soc. London», 1964, 83, 31.
3915. Weiss M. J., Mielczarek S. R., Kuyatt С. Е. —
«J. Chem. Phys.», 1971, 54, 1412.
3916. Welch W. #., Mizushima M. — «Phys. Rev.»,
1972, A5, 2692.
3917. Wendling E., Mahmoudi S. — «Bull. Soc. chim.
France», 1972, N 1, 33.
3918. Wendling E., Mahmoudi S., Mac Cordick H. J. —
«J. Chem. Soc.» A, 1971, 1747.
3919. Weisseck R., Volkner A.—«Z. angew. Phys.»,
1971 32 258
3920. Wen'iger 's. — «J. phys. et radium», 1962, 23, 225.
3921. Weniger S., Herman R. — «J. phys. et radium»,
1958, 19, 582.
3921a. Wertheimer R. — «C. r. Acad. sci.», 1959, 248,
1640.
3922. West С — «J. Chem. Soc», 1902, 81, 923.
3923. West С W. — «J. Chem. Phys.», 1935, 39, 493.
3924. West E. D. — «J. Amer. Chem. Soc», 1959, 81, 29.
3925. West J. R. — «Ind. and Engng Chem.», 1950, 42,
713.
3926. West W. A., Menzies A. W. — «J. Phys. Chem.»,
1929, 33, 1880.
3927. Weston R. E. — «J. Chem. Phys.», 1957, 26,
1248.
3928. Weston R. E., jr., Brodasky T. F. — «J. Chem.
Phys.», 1957, 27, 683.
3929. Wetroff G. — «С. г. Acad. sci.», 1939, 208, 903.
3930. White D., Ни J. H., Johnson H. L. — «J. Chem.
Phys.», 1953, 21, 1149.
3931. Whittaker F. L. — «Proc Phys. Soc. London»,
1967, 90, 535.
3932. Whittaker F. L. — «Proc. Phys. Soc. London»,
1968, 1, 977.
3933. Whittaker F. L. — Canad. J. Phys.», 1969, 47,
1291.
3934. Whittle M. J., Bradley R. H., Brier P. N. —
«Trans. Faraday Soc», 1971, 67, 2505.
3935. Wicke E. — «Angew. Chem.», 1948, A60, 65.
3936. Wicke E. — «Nachr. Akad. Wiss. Gottingen,
math.-phys. Kl. Ha», 1946, 89.
3937. Wicke E. — «Z. Elektrochem.», 1949, 53, 212.
3938. Wicke E., Friz H. — «Z. Elektrochem.», 1953,
57, 9.
3939. Wieland K., Tellinghuisen J. В., Nobst A. —
«J. Mol. Spectrosc», 1972, 41, 69.
3940. Wieland K., Waser J. — «Phys. Rev.», 1952, 85,
385.
3941. Wieland K., Wehrli M., Miescher M. — «Helv.
phys. acta», 1934, 7, 843.
3942. Wierl R. —«Ann. Phys.», 1931, 8, 521.
3943. Wigand A. — «Z. phys. Chem.», 1908, 63, 273
3944. Wilkins R. L. Theoretical Evaluation of Chemical
Propellents, New Jersey, Englewood Cliffs,
Prentice-Hall Inc., 1963.
3945. Wilkins R. L. — «J. Chem. Phys.», 1969, 51, 853.;
3946. Wilkins R. L., Lodwig R. M., Greene S. A.
8th Sympos. on Combustion, Pasadena,
California, 1960. Publ. 1962, p. 375.
493
Литература
3947. Wilkinson P. G. — «Canad. J. Phys.», 1956, 34, 3986.
250.
3948. Wilkinson P. G. — «J. Chem. Phys.», 1959, 30, 3987.
773.
3949. Wilkinson P. G. — «Astrophys. J.», 1963, 138, 3988.
778.
3950. Wilkinson P. G., Houk N. B. — «J. Chem. Phys.», 3989.
1956, 24, 528.
3950a. Willett R. D., LaBonville P., Ferraro J. R. — 3990.
«J. Chem. Phys.», 1975, 63, 1474.
3951. Williams G. R. — «Chem. Phys. Lett.», 1975, 30, 3991.
495.
3952. Williams P. F. — «Phys. Rev. Lett.», 1973, 30, 3992.
951. 3993.
3953. Williams <?., Sheridan J., Gordy W. — «J. Chem.
Phys.», 1952, 20, 164. 3994.
3954. Williamson J. G. Dissertation. Ohio State Univ., 3995.
1969. 3996.
3955. Willis C, Back R. A. — «Canad. J. Chem.», 3997.
1973, 51, 3605.
3956. Willis C, Back R. A. —«Nature», 1973, 241, 43. 3998.
3957. Willis C, Lossing F. P., Back R. A. — «Canad. 3999.
J. Chem.», 1976, 54, 1. 4000.
3958. Willmshurst J. K., Bernstein H. J. — «J. Chem.
Phys.», 1957, 27, 661. 4001.
3959. Wilson E. B. — «J. Chem. Phys.», 1936, 4, 526. 4002.
3960. Wilson E. B. — «Chem. Rev.», 1940, 27, 17. 4003.
3961. Wilson E. D. — «Phys. Rev.», 1928, 32, 611.
3962. Wilson F. J. Thesis. Univ. Saskatchewan, 4004.
Canada, 1941.
3963. Wilson G. L., Miles F. D. — «Trans. Faraday 4005.
Soc», 1940, 36, 356.
3964. Wilson M. K., Badger R. M. — «J. Chem. Phys.», 4006.
1948, 16, 741.
3965. Wilson M. K., Badger P. M. — «Phys. Rev.», 4007.
1949, 76, 472. 4008.
3966. Wilson M. K., Polo S. R. — «J. Chem. Phys.»,
1952, 20, 1716. 4009.
3967. Wind H. — «J. Chem. Phys.», 1965, 42, 2371.
3968. Wind H. — «J. Chem. Phys.», 1965, 43, 2956. 4010.
3969. Winnewisser M., Cook R. L. — «J. Chem. Phys.»,
1964, 41, 999. 4011.
3970. Winnewisser M., Sastry K. V. L. N., Cook R. L.,
Gordy W. — «J. Chem. Phys.», 1964, 41, 1687. 4012.
3971. Winnewisser M., Winnewisser G., Gordy% W. —
«Bull. Amer. Phys. Soc», 1965, 10, 490. 4013.
3972. Winter N. W. — «Chem. Phys. Lett.», 1975, 4014.
33, 300.
3973. Winter N. W., Goddard W.A., Bobrowicz F. W. — 4015.
«J. Chem. Phys.», 1975, 62, 4325.
3974. Winter N. W., Pitzer R. M. — «J. Chem. Phys.», 4016.
1975, 62, 1269.
3975. Winton R. S. Dissertation. Duke Univ., 1972. 4017.
3976. Wise H. — «J. Chem. Phys.», 1952, 20, 927.
3977. Wise H. — «J. Phys. Chem.», 1954, 58, 389.
3978. Wise J. H., Elemer J. T. — «J. Chem. Phys.», 4018.
1950, 18, 1411.
3979. Wise S. S., Feder H. M., Hubbard W. N. — «J.
Phys. Chem.», 1961, 65, 1337. 4019.
3980. Witmer E. E. — «Phys. Rev.», 1926, 28, 1223.
3981. Witt J. D., Carreira L. A., Durig J. R. — «J.
Mol. Struct.», 1973, 18, 157. 4020.
3982. Wittig F. E., Schmatz W. — «Z. Elektrochem.»,
1959, 63, 470. 4021.
3983. Wohl K., Elbe G. — «Z. phys. Chem.», 1929, B5,
241. 4021a.
3984. Wohl K., Magat M. — «Z. phys. Chem.», 1932,
B19, 117. 4022.
3985. Wolf L., Yung W. — «Z. anorg. und allgem.
Chem.», 1931, 201, 337.
Wolf M. G., Schwarzschild M., Rose W. K. —
«Astrophys. J.», 1964, 140, 833.
Wolfe P. N., Williams D. — «Phys. Rev.»,
1954, 93, 360.
Woltz P. J. #., Jones F. A., Nielsen A. H. —
«J. Chem. Phys.», 1952, 20, 378.
Wong D. P., Fink W. H., Allen L. С — «J.
Chem. Phys.», 1970, 52, 6291.
Wood R. W., Kimura M. — «Philos. Mag.», 1918,
35, 252.
Woodman С. М. — «J. Mol. Spectrosc», 1970, 33,
311.
Woolf A. A. — «J. Chem. Soc», 1951, 231.
Woolley H. W. — «J. Chem. Phys.», 1941, 9,
470.
Woolley H. W. — «J. Res. NBS», 1948, 40, 163.
Woolley H. W. — «J. Chem. Phys.», 1953, 21, 236.
Woolley H. W. — «J. Res. NBS», 1955, 54, 299.
Woolley H. W. Dissertation. Univ. Michigan,
1955.
Woolley H. W. — «J. Res. NBS», 1956, 56, 105.
Woolley H. W. — «J. Res. NBS», 1958, 61, 469.
Woolley H. W., Scott R. В., Brickwedde F. J. —
«J. Res. NBS», 1948, 41, 379.
Worley R. E. — «Phys. Rev.», 1943, 64, 207.
Wourtzel E. — «C. r. Acad. sci.», 1919, 169, 1397.
Wray K. L., Hornig D. F. — «J. Chem. Phys.»,
1956, 24, 1271.
Wright A. N., Winkler С A. Active Nitrogen.
New York—London, Acad. Press, 1968.
Wright N., Randall H. M. — «Phys. Rev.»,
1933, 44, 391.
Wroczynski A., Guye P. A. — «J. chim. phys.
et phys.-chim. biol.», 1910, 8, 206.
Wu A. A. — «Chem. Phys. Lett.», 1975, 35, 316.
Wu Ching-Hsien, Birky M. M., Hepler L. G. —
«J. Phys. Chem.», 1963, 67, 1202.
Wu H. L., Benesch W. M. — «Phys. Rev.»,
1968, 172, 31.
Wulf O. R. — «Proc Nat. Acad. Sci. U. S. A.»,
1930, 16,» 507.
Wulf O. R., Melvin E. H. — «Phys. Rev.», 1939,
55, 687.
Wyatt R., Roberts J. Т., WentzR. E., Wilt
P. M. — «J. Chem. Phys.», 1969, 50, 2552.
Wyekoff R. W. G. — «Phys. Rev.», 1920, 16, 149.
Wyse F. C, Manson E. L., Gordy W. — «J. Chem.
Phys.», 1972, 57, 1106.
Yamabe H., Kato Д., Yonezawa T. — «Bull.
Chem. Soc. Japan.», 1971, 44, 22.
Yamagouchi A. — <lJ. Chem. Soc. Japan», 1959,
80, 1109.
Yamaguchi A., Jchishima J., Schimanouchi Т.,
Mizushima S. — «J. Chem. Phys.», 1959, 31,
843.
Yamaguchi A., Jchishima J., Schimanouchi Т.,
Mizushima S. — «Spectrochim. acta», 1960, 16,
1471.
Yamaguchi J., Ohashi O., Takahashi H., Sakai-
zumi Т., Onda M. — «Bull. Chem. Soc. Japan»,
1973, 46, 1560.
Yamashita H., Kato R. — «J. Phys. Soc. Japan»,
1970, 29, 1557. \r
Yamdagni R., Kebarle P. — «Ber. Bunsenges.
phys. Chem.», 1974, 78, 181.
Yamdagni R., Kebarle P. — «J. Amer. Chem.
Soc», 1976, 98, 1320.
Yarkony D. R., O'Neil S. V., Schaefer H. F.,
Baskin С P., Bender С F. — «J. Chem. Phys.»,
1974, 60, 855.
494
Литература
4023. Yaws С. L., Adler L. S. — «Chem. Engng», 1974,
81, N 23, 113.
4024. Yaws С L., Hopper I.E. — «Chem. Engng»,
1974, 81, N 17, 99.
4025. Yaws С L., Hopper I. R., Rojas M. G. — «Chem.
Engng», 1974, 81, N 25, 91.
4026. Yaws С L., Hopper T. R., Rojas M. G., Setty
H. S. N. — «Chem. Engng», 1975, 82, N 4, 87.
4027. Yaws С L., Li K. Y., Кио С. Н. — «Chem.
Engng», 1974, 81, N 14, 85.
4028. Yaws C. ?., Setty H. S. N. — «Chem. Engng»,
1974, 81, N 27, 67.
4029. Yaws С L., Setty H. S. N., Hopper I. R., Luna
V. — «Chem. Engng», 1975, 82, N 2, 99.
4030. Yee K. K., Barrow R. F., Rogstad A. — «J.
Chem. Soc. Faraday Trans. II», 1972, 10, 1808.
4031. Yee K. K., Miller G. J. — «J. Chem. Soc. Chem.
Communs», 1972, N 19, 1054.
4032. Yeranos W. A. — «Theor. chim. acta», 1969, 13,
346.
4033. Yoon Y. K., Carpenter G. B. — «Acta crystallogr.»,
1959, 12, 17.
4034. Yoshino K. — «J. Opt. Soc. America, 1969, 59,
1525.
4035. Yoshino K., Tanaka Y. — «J. Chem. Phys.»,
1968, 48, 4859.
4036. Yoshino K., Tanaka Y., Carroll P. K., Mitchell
P. — «J. Mol. Spectrosc», 1975, 54, 87.
4037. Yoshioka T. — «Sci. Repts Tohoku Univ.», 1960,
44, 135.
4038. Yost D. M. — «Z. phys. Chem.», 1931, A153, 143.
4039. Yost D. M. — «Proc. Indian Acad. Sci.», 1938, 8,
333
4040. Yost D. M., Anderson T. F. — «J. Chem. Phys.»,
1934, 2, 624.
4041. Yost D. M., Claussen W. N. — «J. Amer. Chem.
Soc», 1933, 55, 885.
4042. Yost D. M., Felt R. C. — «J. Amer. Chem. Soc»,
1934, 56, 68.
4043. Yost D. M., Russel H. Systematic Inorganic
Chemistry of the 5 and 6 Group Nonmetallic
Elements. N. Y., Prentice-Hall Inc., 1944.
4044. Young В. Т. Thesis. Texas A. & M. Univ., 1964.
4045. Young F. E., Hildebrand J. H. — «J. Amer.
Chem. Soc», 1942, 64, 839.
4046. Za'horsky U. J. — «Angew. Chem.», 1968, 80,
661.
4047. Zahradnik R., Carsky P. — «Collect. Czech.
Chem. Communs», 1971, 38, 1876.
4048. Zahradnik R., Carsky P. — «Theor. chim. acta»,
1972, 27, 121.
4049. Zahradnik R., Carsky P. — «Collect. Czech.
Chem. Communs», 1973, 38, 1876.
4050. Zaidi H. R., Verma R. D. — «Canad. J. Phys.»,
1975, 53, 420.
4051. Zeach H. F., Roberts H. Z. — «J. Chem. Soc».
1960, 4693.
4052. Zeelenberg A. P. — «Nature», 1958, 181, 42.
4053. Zeeman P. B. — «Canad. J. Phys.», 1951, 29, 174.
4054. Zeise H. — «Z. Elektrochem.», 1934, 40, 662.
4055. Zeise H. — «Z. Elektrochem.», 1935, 41, 267.
4056. Zeise H. — «Z. Elektrochem.», 1942, 48, 494.
4057. Zeise H. — «Z. Elektrochem.», 1942, 48, 693.
4058. Zeise H. Thermodynamik. Bd 3/1, Tahellen;
Bd 3/2, Grafische Darstellungen und Literaimr.
Leipzig, S. Hirzel Verlag, 1954—1957.
4059. Zeumer H., Roth W. A. — «Z. Elektrochem.»,
1934, 40, 777
4060. Zijp D. H. — In: Advances Molecular Spectro-
scopy», V. 1. Oxford—London—New York-
Paris, Pergamon Press, 1962, p. 345.
4061. Ziomek J. S., Piotrowski E. A. — «J. Chem.
Phys.», 1961, 34, 1087.
4062. Ziomek J. S., Piotrowski E. A., Walsh E. N. —
«Bull. Amer. Phys. Soc», 1954, 29, Abstr. S 11.
4063. Ziomek J. S., Piotrowski E. A., Walsh E. N. —
«Phys. Rev.», 1955, 98, 243.
4064. Ziomek J. S., Zeidler M.D. — «J. Mol. Spectrosc.»
1963, 11, 163.
4065. Zittel P. F., Lineberger W. С — «J. Chem.
Phys.», 1976, 65, 1236.
4065a. ZollingR. J. — «J. Chem. Phys.», 1969, 50, 4251.
4066. Zulicke L., Spandenberg H. J. — «Theor. chim.
acta», 1966, 5, 139.
4067. Zumwalt L. R., Giguere P. A. — «J. Chem.
Phys.», 1941, 9, 458.
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
Том I
Книга 1
Утверждено к печати
Институтом высоких температур
Академии наук СССР
Редактор издательства В. П. Сироткина
Художник В. Г. Виноградов
Художественный редактор Т. П. Поленова
Технический редактор Э. Л. Кунина
Корректоры А. М. Журавлева, Е. И. Кореневская
ИБ № 7611
<]дано в набор 04.04.78. Подписано к печати 30.11.78.
Т-17562. Формат 84Х108»/1в. Бумага типографская № 1.
Гарнитура обыкновенная. Печать высокая
Усл. печ. л. 52,08. Уч.-изд. л. 59,8. Тираж 8000 экз.
Тип. зак. 295
Цена I т. (кн. 1 и 2) 7 р. 90 к.
Издательство «Наука» 117485, Москва, В-485,
Профсоюзная ул., 94а
Ордена Трудового Красного Знамени
Первая типография издательства «Наука»
199034, Ленинград, 9-я линия, д. 12
ИСПРАВЛЕНИЯ И ОПЕЧАТКИ
Страница
16
38
145
145
199
227
396
Строка
20 св.
5 св.
8 св.
5 сн.
16 сн.
17 сн.
табл. П2.4,
1-я колонка
Напечатано
Np
B2/+1)
полиморфных
от 100
5-800 К
Хеа
0
0
Должно быть
Cm
2B7 + 1)
полимерных
от 298,15
5-760 К
XeF
—0
0