/


























































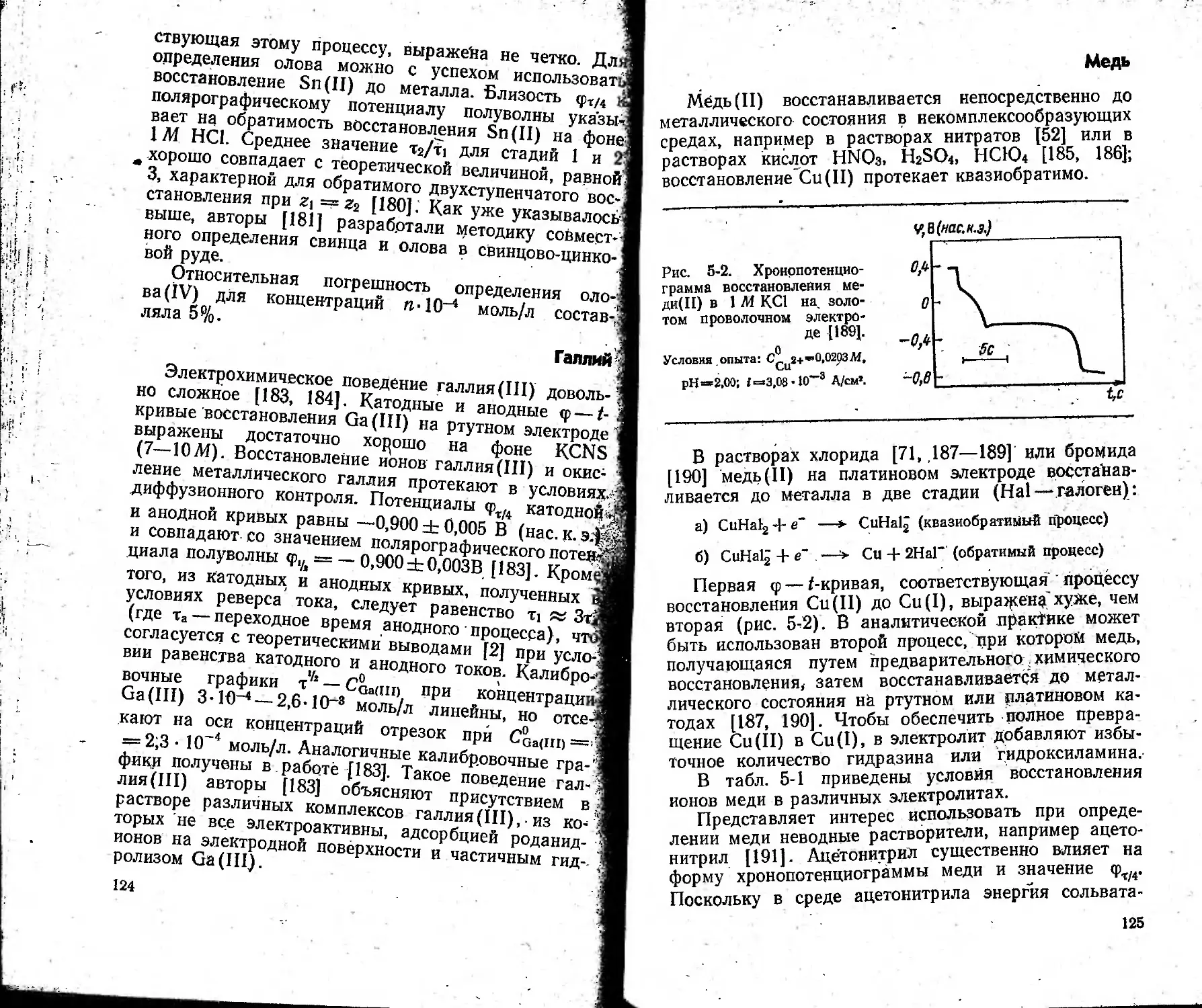




































Author: Захаров М.С. Баканов В.И. Пнев В.В.
Tags: химические методы анализа химия аналитическая химия хронопотенциометрия
Year: 1978




Text
I.
j
3-38
i
Методы
аналитической
химии
М.С.Захаров
В. И. Баканов
В. В. Пнев
Хроно
потенциометрия
М.С.Захаров
В. И. Баканов
В. В. Пнев
Хроно-
• потенциометрия
Моснва;издательстрс« Химия ”,1978
УДК 543.257.5 :541.1: 546
Захаров М. С., Баканов В. И., Пнев В. В.
Хронопотенциометрия (Методы аналитической химии J.
— М.: Химия, 1978. — 200 с., йа
Книга посвящена методу ХронопотенциОметрин, или
гальваносТатическому методу, широко используемому при
электрохимических измерениях. В книге излагаются теоре-
тические основы метода, показаны области его применения,
причем основное внимание уделяется применению хронопо-
тенциометрии в аналитическом контроле, а также для
исследования кинетики электродных процессов, определения
коэффициентов диффузии иоиов и изучения адсорбционных
явлений на электроде.
Книга предназначена для широкого круга научных и
инженерно-технических работников, занимающихся разра-
боткой. и применением электрохимических методов в ана-
литическом контроле. Она может быть также Полезна
преподавателям/ аспирантам и студентам вузов, специали-
зирующимся в области аналитйческой/химии и электро-
химии. *
200 с., 41 рис., 13 табл,, список литературы 435 ссылок.
лад
Редакционн РЭ. ГеЛьман, И. Ф. Дол-
манова, Ю. А. Золотое,(председатель), Ю. А. Кар-
пов, Ю. А. Клячко, Н. М. -Кузьмин, Л. Н. Овсян-
никова (ученый секретарь), Их А. Филиппова
20506-018 -
3 050(61j-78''8-78
© Издательство «Хнмня», 1978 г.
СЕРИЯ МОНОГРАФИИ
«МЕТОДЫ АНАЛИТИЧЕСКОЙ ХИМИИ»
Издательство «Химия», приступило к, выпуску серий
монографий, посвященных' отдельным методам современной
аналитической химии. Цель этой серии — обобщить дости-
жения в теории.и практическом использовании Наиболее
важных и перспективных методов анализа, Особое внима-
ние предполагается уделить физическим и физико-химиче-
ским методам. Серий рассчитана .на ряд лет.
В каждой из монографий серии дается общая характе-
ристика метода, освещаются его теоретические основы, наи-
более важные варианты метода-и их особенности, приемы
работы, аппаратура, области применения, приводятся от-
дельные типичные методики, примеры их использования^
рассматривается возможность использования метода ₽
производственном контроле; Такая структура, книг сделает
их интересными и для-специалистовщсследователей, и для
химиков-аналитиков заводских лабораторий.
Предполагается, что изложение материала должно быть
критическим и обобщающим. Авторы призваны осветить
современный уровень метода и оценить его перспективы, не .
перегружая книги сведениями частного характера. Оформле-
ние монографий однотинное; средний объем 10—15 авторских
листов. Авторы придерживаются современной научной тер-
минологии и системы обозначений (за основу взята терми-
нология, принятая в «Журнале аналитической химии»),
используе’гся международная система единиц СИ.
РЕДКОЛЛЕГИЯ
СОДЕРЖАНИЕ'
Предисловие ; » » :...............-.......................7
Список основных обозначений...............................9
Глава 1. Метод хронопотенциометрии, Основные понятия
о кинетике электронных процессов........- . . II
1.1. Метод хронопотенциометрин.........................11;
1.2. Основы учении о кинетике электродных процессов . . . 15.
Глава 2. Хронопотенциометрия электродных процессов, не-
осложненных химическими реакциями . . . . 26'
2.1. Электродные процессы в условиях линейной диффузии • 26
2.2. Электродные процессы в условиях сферической диффузии 37;
2.3. Электродные процессы в условиях цилиндрической диф-
фузии . .................................................40
2.4. Электродные процессы на вращающемся дисковом элек-
троде ...................................................45
2.5. Хроиопотенциометрия многоступенчатых процессов вос-
становления (окисления) веществ..........................49
2.6. Хронопотенциометрия электродных процессов с участием
нескольких деполяризаторов ................,............54’
2.7. Влияние миграции н конвекции иа переходное время . . 59
2.8. Влияние заряжения двойного электрического слоя на
хронопотенциограммы....................................-601
2.9. Учет влияния заряжения двойного электрического слоя
на переходное время. Определение дифференциальной -
емкости.................................................64
Глава 3. Некоторые разновидности хронопотенциометрнче- й
ского методаг........................................... 6Й
3.1. Хронопотенциометрия С программированным током . . . 6
3.2. Переменнотоковая хронопотеициометрия.............Л . 74.
3.3. Производная хронопотеициометрия ................. 76;
4
Г л а в a 4. Хронопотеициометрия электродных процессов,
Осложненных объемными химическими реакциями 84
4.1. Электродные процессы с быстрыми объемными химиче-
скими реакциями.....................................85
4.2. Электродные процессы с медленными предшествующими
химическими реакциями...............................90
4.3. Электродные процессы с последующими медленными хи-
мическими реакциями............................... 96
4.4. Электродные процессы с медленными каталитическими и
параллельными химическими реакциями.................'04
4.5. Электродные процессы со ступенчатым переносом заряда
и разделяющей электродные стадии медленной химиче-
ской реакцией ........................ ............ 109
Глава 5. Применение хрононотенцнометрин для аналитиче-
ских исследований . . . . . •> ...........IJ6
5.1. Измерение переходного времени.................116
5.2. Аналитический возможности хронопотенциометрии в усло-
виях полубескоиечной диффузии . '............... .1'7
5.3. Исследование поведения элементов и веществ и их анали-
тическое определение...............................120
Таллий........................................ 121
Кадмий ...................: . . . ..............121
Свинец . . . .................................122
Цинк......................................... 123
Олово..........................'................123
Галлий . . . .'...........-. ...................124
Медь............................................125
Золото..........................................126
Никель и кобальт 127
Железо ......................................... 128
Марганец ..............'...........128
Висмут......................................... 129
Серебро....................................... 129
Ртуть ...................................... . 130
Уран.........................................; . 130
Плутоний........................................132
Церий...........................................134
Европий.........................................135
Титан.....................’.....................135
Торий......................................... 137
5
Хром............................................. 13л
Кислород.................................... 13i
Двуокись углерода . ........................ «... 141
Персульфат-ион................................ 14^
Галогениды ........... ........14'
л Щавелевая кислота ............................... 141
Муравьиная кислота и формиат-ионы . ...........14',
Ферроцен, рутеноцен, осмоцеи и их производные . , -.15,
Гидразин и гидроксиламии и нх смесь ....../ 151
Определение элементов на фойе концентрированной фос- .
форной кислоты . ................................ 1Q
( -г
Глава 6. /Применение хронопотеициометрии для физико-хи- J
мнческих исследований .............................15»
• S
6.1. Изучение кинетики электродных процессов . . . ... . 15ft
6.2. Определение коэффициентов диффузии.............. lift
6.3. Изучение адсорбции ............................167
6.4. Применение хронопотеициометрии дЛя физико-химических
исследований в расплавах ..................... 17$
' У
Глава 7. Техника эксперимента . . . . v .............15
7.1. Принципиальные схемы -установок для хронопотенцио-
метрии ........................................ 17s
7.2. Электроды и электролитические ячейки . . '. . ,, . . Г-т
Литература . , . ...................10
ПРЕДИСЛОВИЕ-
Хронопотенциометрический метод был впервые
описан в 1901 г. Сэндом. Тогда же было получено
и основное уравнение хронопотенциометрии — урав-
нение для переходного времени.' Более чем за три
четверти века метод получил значительное развитие.
Он позволяет проводить аналитический контроль, ис-
следовать механизм и кинетику электродных процес-
сов, определять коэффициенты диффузии ионов в рас-
творах и расплавах, исследовать растворимость
веществ в растворах, изучать адсорбцию, исследовать
химические реакции и др. Метод может быть исполь-
зован для исследования как неорганических, так
и органических веществ. Словом, круг задач, решае-
мых с помощью хронопдтенциометрии, таков же, как'
и в полярографии, которой посвящено значительное
число монографий.- Однако в хронопотенциометриче-
ском методе математическая обработка поляриза-
ционных кривых более проста. Последнее значи-
тельно облегчает, по сравнению с полярографией,
исследование сложных электродных процессов и
особенно электродных процессов, осложнённых хими-
ческими реакциями.
До настоящего времени монографии по. хронопо-
тенциометрии не издавалось. Наша работа является
первой попыткой изложить теоретические и экспери-
ментальные результаты хронопотенциометрии в виде
отдельной монографии.
В пределах небольшого объема авторы попы-
тались обобщить современное состояние теории
7
хронопотенциометрического метода для катодных про-
цессов, неосложненных и осложненных химическими
реакциями, рассмотреть некоторые разновидности ме-
тода и применение его для аналитических и физико-
хиьГйческих исследований, описать технику экспери-
мента. Следует отметить, что из-за малого объема
монографий материал излагается кратко, выводы
уравнений не приводятся. Как это принято в поляро-
графии, при логарифмировании исходных уравнений
авторы оставляли под знаком логарифма величины,
имеющие размерности. В тех уравнениях, где отсут-
ствует величина площади электрода, речь идет о плот-
ностях токов,' если это не оговаривается специально.
Авторы не рассматривают один из вариантов хро-
нопотенцнометрии — инверсионную хронопотенциомет-
рию. По инверсионной хронопотенциометрии накоп-
лен большой материал, который может стать предме-
том отдельной книги.
Авторы отдают себе отчет в том, что в книге есть,
по-видимому, недостатки; все критические замечания
будут приняты авторами с благодарностью.
Авторы выражают признательность к. х. н. О. Л. Ка-
бановой за ценные замечания при обсуждении работы.
СПИСОК ОСНОВНЫХ ОБОЗНАЧЕНИЯ'
Cq и Cr — концентрации окисленной и восстановлен-
ной форм деполяризатора в объеме рас-
твора
Со (0, /) и CR (0, /) — концентрации окисленной и восстановлен-
ной форм деполяризатора у поверхности
электрода
Са — дифференциальная емкость двойного элек-
трического слоя
Do и Dr — коэффициенты диффузии окисленной и
восстановленной форм деполяризатора
Е — напряжение ' на электролитической ячейке
F — число Фарадея
f0 и fR — коэффициенты активности окисленной и
восстановленной форм деполяризатора
I — ток (сила тока)
/д — предельный стационарный ток /
i — плотность поляризующего тока
i0 ~ плотность тока обмена .
ip — плотность стандартного тока обмена
iK и г’а — плотность катодного и анодного тока
К — константа равновесия химической реакции
ki, k2 — константы скорости прямой и обратной хи-
мических реакций
kj и —* константы скорости катодного и анодного,
процессов, не зависящие от потенциала
электрода
и kb — константы скорости катодного и анодного
процессов
ks — константа скорости электродного процесса
рри стандартном потенциале
о .
к’’ t""" /? —универсальная газовая постоянная
F,'. '4 tL \ г? r0. — радиус сферы или цилиндра Т — температура, К t— время х — расстояние от поверхности электрода
е г — число электронов о, ₽ — коэффициенты переноса Г — величина адсорбции
4 1 *• ' jj д —толщина диффузионного слоя у поверхн сти электрода т) — поляризация
v < V — кинематическая вязкость раствора
14 L - т — переходное время
<Рк и .фа—потенциал.катода и анода
? Ф° — стандартный потенциал Ф1/2 ~ потенциал полуволны полярограммы
<! ' Фр —равновесный потенциал электрода Ф1 — пси-прим-потенциал (й — скорость вращения электрода
МЕТОД ХРОНОПОТЕНЦИОМЕТРИИ.
ОСНОВНЫЕ ПОНЯТИЯ О КИНЕТИКЕ
ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
1.1. МЕТОД ХРОНОПОТЕНЦИОМЕТРИИ
Сущность хронопотенцйометрии состоит в снятии
кривых зависимости потенциала электрода от вре-
мени при прохождении через электролитическую,
ячейку постоянного тока заданной величины. Бели
говорить точно, то в хронопотенциометрии ток (сила
тока) в течение всего опыта не остается постоянным,
а изменяется скачком от нуля до некоторой постоян-
ной величины, как это показано на рис. 1-1. Поэтому
хронопотенциометрию часто называют методом сту-
пенчатого изменения тока.
Индикаторный электрод, электрод сравнения
и вспомогательный электрод могут быть различными,
и их выбор зависит от характера проводимых иссле- ,
дований. Так, в качестве индикаторного электрода
могут использоваться ртутный капельный, ртутный
стационарный, платиновый, серебряный, графитовый
и другие электроды*, в качестве-электрода сравнения
и вспомогательного электрода — донная ртуть, кало-
мельный, хлорсеребряный, ртутно-сульфатный элек-
троды.
Напряжение Е на электролитической ячейке опре-
деляется соотношением Е — <рк — фа, где фк — потен-
циал индикаторного электрода; фа — потенциал элек-
трода сравнения. Экспериментатора обычно интере-
сует только процесс, протекающий на индикаторном
электроде. Поэтому поверхность электрода сравнения
обычно много больше (например, в 100 раз) поверх-
ности индикаторного электрода, так что электрод
сравнения практически не поляризуется и изменение
напряжения на ячейке в этом случае будет практи-
чески равно изменению потенциала индикаторного
(1-
• Здесь мы рассматриваем катодные процессы; аналогичная
картина будет наблюдаться и при рассмотрении анодных процес-
сов, только в этом случае индикаторный электрод присоединяют
к положительному полюсу источника постоянного тока.
12
Рис. 1-1. Изменение тока
в классической хроно-
потенциометрин.
мый электродный процесс, описывается уравнение
Нернста: ч
электрода (£ = <рк), который и записывается per
рирующим прибором.
Характер изменения потенциала электрода п]
наложении на электролитическую ячейку тока мо
пояснить следующим образом *. Потенциал <р ию
уторного электрода, если на нем протекает обра
Время t
и CR(0, /)—<
деполяриза-
RT Со (0, 0
где <р° — стандартный потенциал электрода; Со (0, t)
концентрации окисленной и восстановленной форм
тора у поверхности электрода в любой момент времени /; Jo
/к — коэффициенты активности окисленной и .восстановленной
форм деполяризатора.
Если в растворе присутствует только окисленная
форма деполяризатора, то, как следует из уравнения
(1-1), перед началом электролиза потенциал элек-
трода будет положительнее потенциала ' восстановле-
ния вещества.
При присоединении индикаторного электрода к от-
рицательному полюсу источника постоянного тока
наблюдается скачок потенциала, соответствующий
падению напряжения в растворе между индикатор-
ным электродом и электродом сравнения (величина
этого скачка не зависит от времени). Далее происхо-
дит заряжение двойного электрического слоя, и на
хронопотенциог'рамме появляется емкостный участок.
При значительном перенапряжении начинается элек-
тродная реакция и наблюдается задержка потен-
циала, при этом заряжение двойного слоя приоста-
Рис. 1-2. Хронопотенциометрическая кривая при восстановлении
одного вещества (а) и нескольких веществ (б).
навливается. В этот момент окисленная форма
деполяризатора восстанавливается, концентрация ее
уменьшается. Через некоторое время концентрация
окисленной формы деполяризатора у поверхности
электрода станет практически равной нулю, и со-
гласно уравнению (1-1) потенциал электрода изме-
нится. В это время вновь заряжается двойной слой,
и электрохимическая реакция протекает в условиях
предельного диффузионного тока.
Потенциал электрода в этих условиях должен
сместиться в область больших отрицательных значе-
ний, но практически он сдвигается до потенциала
начала восстановления Другого деполяризатора. Хро-
нопотенциограммы электровосстановления одного или
нескольких деполяризаторов представлены на
рис. 1-2а и 1-26 соответственно.
Время между двумя емкостными участками хро-
нопотенциограммы называется переходным временем
т. Величина переходного времени зависит от кон-
центрации деполяризатора и тока, проходящего
через электролитическую ячейку [1]: чем больше
концентрация деполяризатора и меньше ток, тем
13
больше переходнре^нремя п₽и прочих равных усло-
виях, и наоборот. Опытным путем/установлено, что
при т< Г мс хронопотенциограммы по форме отли-
чаются от рассчитанных теоретическим путем; при
т > Мин за счет конвекции в растворе происходит
изменение формы хронопотенциограмм [2]. Поэтому
поляризующий ток необходимо подбирать таким,
чтобы 1 мс < т < 1 мин. Экспериментальным путей
установлено, что' при т>2 с хронопотенциограммы
можно регистрировать с помощью самописцев; при
т < 2 с хронопотенциограммы искажаются из-за инер-
ционности самописца, и их регистрируют с помощью
осциллографа; в случае необходимости кривые фото-
графируют с экрана осциллографа [3].
В хронопотенциометрии в условиях полубесконеч-
ной диффузии величина переходного времени не прямо
пропорциональна концентрации вещества {деполяри-
затора) в растворе, поэтому этот метод, открытый
еще в 1901 г. [4], не находил широкого аналитиче-
ского применения. Однако после того как были раз-
работаны варианты хронопотенциометрии с накоп-
лением на ртутном [5—13] и на твердом [14—16]
электродах, .в которых переходное время прямо про-
порционально концентрации деполяризатора, хронопо-
тенциометрию стали широко применять в аналитиче-
ской практике.
Переходное время не зависит от характера элек-
тродного процесса (обратимый или необратимый),
т. е. на него не влияют загрязнения или другие
помехи, вызывающие изменение характера электрохи-
мической реакции. Последнее ^обстоятельство обус-
ловливает более высокую воспроизводимость измере-
ний 'сигнала (переходного времени) в хронопотенцио-
метрии по сравнению с другими вольтамперными
методами. ' «
Широкое Применение хронопотенциометрия нах<м
дит для исследования кинетики электродных процес|
сов [17] и особенно для изучения химических реает
ций, осложняющих электродные процессы. Большй|
возможности хронопотенциометрии для электрохимш
ческих и физико-химических исследований обусл<Яи
лены относительно простой по сравнению с другой
вольтамперными методами математической обработ-
кой хронопотенциограмм.
В работах [18, 19] для исследования кинетики
электродных процессов была предложена хронопо-
тенциометрия с изменением направления тока. Сущ-
ность этого варианта хронопотенциометрии состоит
в том, что в момент достижения переходного времени
на индикаторный электрод накладывают ток проти-
воположного направления. Такой, прием позволяет
получить последовательно катодную и анодную хро-
нопотенциограммы, необходимые для рассмотрения
кинетики электродных процессов. Близка к этому ва-
рианту и циклическая хронопотенциометрия [2, 19,
20], в которой изменения направления тока на инди-
каторном электроде повторяют многократно.
Рядом авторов [21, 22] была предложена хроно-
потенциомётрия с программированным Током.
Применение производной и переменнотоковой
[23] хронопотенциометрии позволило повысить точ-
ность измерения переходного времени.
1.Х ОСНОВЫ УЧЕНИЯ О КИНЕТИКЕ
ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
Электрохимическая поляризация. Рассмотрим сна-
чала процесс, единственной лимитирующей стадией
которого является электрохимическая реакцйя на
электроде. Такие процессы называются необрати-
мыми, и в этом случае говорят об электрохимической
поляризации. Причиной торможения электрохимиче-
ской реакции является достаточно высокая энергия
активации процесса переноса электрона. Последняя
зависит от потенциала электрода. Константы скоро-
сти процессов разряда (kf) и ионизации (kb) в рас-
сматриваемом случае будут достаточно малы, а ста-
дии доставки и отвода реагирующих веществ проте-
кают без заметного торможения.
Рассмотрение будем проводить при следующих
предположениях: 1) в растворе присутствует избыток
индифферентного электролита, достаточный для того,
-Чтобы можно было пренебречь падением потенциала
в диффузной части двойного электрического слоя;
2) отсутствует адсорбция как деполяризатора, так
И.
и нейтральных поверхностно-активных веществ;
3) обе формы деполяризатора растворимы или в рас-
творе, или в материале электрода.
Из теории замедленного разряда — ионизации
[24} плотность тока, проходящего через электрод,
для реакции первого порядка выражается следую-
щим уравнением:
azf (<рк — <р°)
RT
— Сдехр
i= С^ехр
— a) zF (фа — <P°)
RT J J J ’
где ks — константа скорости электродного процесса при стандарт-'
ном потенциале электрода; <р — потенциал электрода; а —коэф-
фициент переноса; Т — абсолютная.температура; R— универсаль-
ная газовая постоянная [8,314 Дж/(моль-К)]; F— число Фара-
дея (96 500 Кл/моль); z— число электронов, участвующих в
электродном процессе; <р°— стандартный потенциал электрода;
Cq и Ср — концентрации окисленной и восстановленной форм
деполяризатора в объеме раствора.
При достаточно большой катодной поляризации
(iK 3* ia) уравнение (1-2) примет вид
i = zFk С° exo Г- (Фк -Ф0)
(1-3)
При достаточно большой анодной поляризации
i’k) можно записать:
ta = zFks(% exp [(1 ~ Q) zFr^ ~ ф0) ] (1-4)
Так как в хронопотенциометрии регистрируются
Ф — /-кривые, то представляет интерес решить урав-
нения (1-3) и (1-4) относительно величины потен-
циала электрода. Прологарифмировав эти уравнения,
получим:
Фк = <Р° + !п zFks + in С°о - -g. In iK (1-5)
q> = <p° - ь In zFk - ~T. b In cl +
T (1 — a) zF s (1 — a)zF R
+ 77-----r—7Г In I
(1 — a) zF a
(1-6)
Концентрационная поляризация. Если константы
скорости электрохимической реакции значительны, то
1?
во время электролиза концентрация деполяризатора
у поверхности электрода может отличаться от объем-
ной концентрации: она может быть меньше или
больше нее. В этом случае лимитирующей стадией яв-
ляется соответственно подвод или отвод деполяриза-
тора посредством диффузии. При протекании элек-
трического тока в рассматриваемом случае равнове-
сие на электроде практически не нарушается, однако
потенциал электрода изменяется с изменением кон-
центрации деполяризатора ,у поверхности электрода.
Наблюдаемое явление называется концентрационной
поляризацией. Замедленный подвод или отвод реаги-
рующего вещества может быть обусловлен замед-
ленностью некоторых стадий: собственно стадии
доставки вещества к электроду; Предшествующих хи-
мических реакций,.в результате которых из электрохи-
мически неактивных форм получаются электрохими-
чески активные формы вещества; последующих реак-'
ций превращения продукта электродного процесса
в другие вещества и др. Токи, лимитируемые ско-
ростью химических реакций, называются кинети-
ческими.
Потенциал электрода в случае концентрационной
поляризации выражается уравнением Нернста, в ко-
торое подставляются активности окисленной и вос-
становленной форм деполяризатора у поверхности
электрода.
Рассмотрим сначала случай, когда лимитирующей
стадией является собственно стадия подвода к элек-
троду (или отвода от электрода) электроактивного
вещества.
Доставка вещества к электроду или отвод его от
электрода могут осуществляться путем свободной
или конвективной диффузии. Для реализации сво-
бодной диффузии при проведении эксперимента
необходимо соблюдать определенные условия: не до-
пускать перемешивания раствора, время электролиза
не должно превышать 1 мин [3]. '
При -рассмотрении вопроса о доставке вещества
к электроду принимают, что раствор в направлении,
перпендикулярном к электроду, бесконечен. В этом
случае диффузию называют полубесконечной. В ус-
ловиях полубесконечной диффузии Со(оо, /) = Со,
Дн«пропетровоки*
НАУЧНАЯ
БИБЛИОТЕКА
17
fc
к
где Со(х, t)— концентрация деполяризатора на расн
стоянии х от поверхности электрода в глубь раствора!
при времени t (время от момента нарушения равно-
весного состояния на электроде); Со — концентрация^
в объеме раствора. При доставке вещества путем)
свободной диффузии концентрация его в заданной)
точке с течением времени будет изменяться, т. е. диф'-з
фузия будет нестационарной. |
Распределение концентрации деполяризатора в!
зависимости от расстоянья до электрода и продол-?
жительности электролиза в условиях свободной не-<
стационарной диффузии для изотропной среды опи-j
. сывается вторым законом диффузии Фика [1]:
дСо (х, f) дгСо (х, О Ц дСо (х, t)
dt “ дх* х дх • 7 1
где П — коэффициент, характеризующий форму электрода (для!
сферического электрода П = 2, для цилиндрического П — 1, для 1
плоского П = 0). я
В дальнейшем мы используем уравнение (.1-7) для!
описания зависимости концентрации деполяризатора!
от времени й от расстоянйя до электрода для, процес-1
сов, протекающих на электродах различных форм при !
наложении на них постоянного тока. 1
... Следует отметить, что уравнение (1-7) является!
точным для плоского и для идеальных сферического !
и цилиндрического электродов. На практике сфериче-1
ский электрод всегда соединен при помощи контакта!
с электрической цепью. Наличие контакта уменьшает 1
диффузионное поле, что приводит к некоторым от- 1
клонениям результатов эксперимента от теоретиче- J
ских расчетов. Верхняя часть цилиндрического элек-1
трода впаяна в стекло, фторопласт или другие мате- I
риалы и поэтому не работает как электрод; нижний I
торец цилиндра покрывают изоляционным материа- J
лом (например, клеем).
Если доставка вещества к электроду (или отвод J
вещества от электрода) осуществляется за счет диф-я
фузии и( конвекции, то общий поток (fs) вещества!
складывается из диффузионного (/а) и конвектив-я
Кого (1ь) потоков:
ЗСО (0, t)
^-^ + ^=-До— +*А(°’° V-8)]
где Do — коэффициент диффузии вещества; vx — скорость кон-
дСо(0, /)
векции; fd — — Do--—-----; fk = vxCo (0, f); Co (0, t) - кон-
центрация вещества у поверхности электрода.
На вращающемся электроде роль конвекции в пе-
реносе вещества весьма существенна. Если прене-
бречь изменением ' концентрации деполяризатора
в растворе во время электролиза, то при достаточно
интенсивном перемешивании на вращающемся элек-
троде вскоре после начала электролиза устанавли-
вается стационарное состояние (т. е. <3Co(0, t)/dt—0),
и уравнение второго закона Фика в этом случае бу-
дет иметь рйд [25, 26]:
дгСо(0,0 dCo(0,t)
D° дх2 ’“°* дх
(1-9)
Если электродный процесс осложнен химическими
реакциями, то необходимо учитывать в расчетах из- ч
менение концентрации деполяризатора. Для примера
рассмотрим более подробно процесс, осложненный
предшествующей мономолекул яр ной химической ре-
акцией, в результате которой неэлектроактивная'
форма деполяризатора превращается в электроактив-
ную. Этот процесс можно представить схемой:
k\
А О + ze~ 4=5= R (Ы0)
«j
где А, О — неэлектроактивная и электроактивная формы деполя-
ризатора соответственно; kt и — константы скоростей прямой
и обратной химических реакций А О.
Следует отметить, что форма А может быть не-
электроактивной лишь в исследуемом интервале по-
тенциалов^ в другом интервале потенциалов она
может быть и электроактивной.
Состояние равновесия между формами А и О
определяется константой равновесия Л:
Cofo
(1-11)
Если константа скорости ki прямой химической
реакции велика (Л13>1), то химическая реакция не
будет оказывать влияния на электродный процесс
19
и электрический сигнал в данном случае будет раве
сигналу для электрохимической реакции, не ослом
ненной химическим процессом.
При k\ С 1 (равновесие химической реакци
сильно смещено влево и концентрация вещества (
в растворе мала) кинетический ток будет мал, и о
будет определяться концентрацией вещества О, пс
лучающегося в результате протекания химическо
реакции. При промежуточных значениях k\ прохоя
дение тока через электрод будет вызывать смещен!
равновесия химической реакции. Слой, в котором Hi
блюдается смещение химического равновесия, наз!
вается реакционным, причем наибольший сдвиг равю
весия будет наблюдаться у поверхности электрода
Для получения уравнений I — «р-кривых для элек
тродных процессов, осложненных химическими реак
циями, применялись два метода: 1) метод, в которо!
используются представления о реакционном ело
[27, 28]; 2) метод, заключающийся в решении урав
нений второго закона Фика с кинетическими членам»
учитывающими протекание химических реакци:
в приэлектродном слое [29J. Более точным являете,
второй метод, получивший в настоящее время шире
кое распространение. Этим методом и были полу
чены уравнения для концентраций деполяризатора,
при проведении процесса в условиях хронопотенч
циометрии.
Для реакции (1-10) уравнения второго закона
Фика, учитывающие протекание наряду с электрод-
ным процессом и химической реакции, в условиях
линейной полубесконечной диффузии имеют следую-
щий вид:
дС. (х, i) д2С. (х, f)
-----fejCA (х, i) + k2Co (x, n (1-12
dCn(x, f) d2Co(x, t)
—%-------~ DO0 - Wo (X, о (1-13
Для нахождения Ca(x, t) и Co(x, t) уравнения (1-12
и (1-13) решаются обычно методом трансформацш
Лапласа с введением определенных начальных и гра
ничных условий.
Система дифференциальных уравнений тип:
(1-12) —(1-13) для электродных процессов, осложнен
них химической реакцией, была впервые сформули-
рована в работах [30, 31]. В случае электродных
процессов, осложненных каталитическими химиче-
скими реакциями, при нахождении выражений для
концентраций веществ необходимо использовать
уравнения (1-12) и (1-13). Решение этих уравнений
будет приведено в гл. 4. Там же будут подробнее
рассмотрены и электродные процессы, осложненные
другими химическими реакциями.
Электродные процессы с двумя лимитирующими
стадиями. При протекании необратимых или квази-
обратимых электродных процессов лимитирующей
стадией, наряду с электрохимической реакцией, мо-
жет быть и процесс доставки деполяризатора к по-
верхности электрода или отвода продукта реакции от
поверхности электрода. В результате приходится учи-
тывать одновременно две Лимитирующие стадии.
В этом случае в уравнения (1-1)—(1-6) вместо кон-
центрации Со и CR нужно подставлять концентрации
деполяризаторов О и R у поверхности электрода,
т. е. Со(0, t) и Cr(0,/). В нестационарных электро-
химических методах, к которым относится и хроно-
потенциометрия, необходимо всегда учитывать это.
Например, уравнения (1-2) — (1-6) в этом случае
примут вид:
I - гИ, { Со (О, I) eq, [-_
-С„(0. /)ехр-*•>]} (1-2а)
/„ _ <0. П «хр [- (I-3-)
/,-ггаА(о. о»р[п(М.)
ф« " ф” + S zFt‘ + S С°° - Sf |П 'к (1-5*’
ф* -ф” - tSSf |п zFk- - oSSrс«<°- '>+
Учет влияния строения двойного электрического
слоя на скорость необратимого процесса. В приве-
21
денных выше уравнениях для скорости необратимыми
электродных процессов не учитывалось влиянии]
строения двойного электрического слоя на концентр Ль
цию деполяризатора у поверхности, электрода.
нако во многих случаях это влияние необходимо уч^М
тывать. Потенциал на границе плотной и диффузнив
частей двойного слоя обозначают ф1 (пси-прим-п-Ж
тенциал). Фрумкин [32] высказал предположений
что в электродном процессе могут участвовать толькИ
ионы, находящиеся в плотной части двойного эле Я
трического слоя, и что на процесс разряда — иониз^Н
ции влияет не общий потенциал (<р) электродгЛ
а только разность потенциалов (<р — ф1). В св Я
с этим при учете влияния строения двойного электриД
ческого слоя на скорость электродных процессов
в уравнений для плотности тока необратимых.и кв ‘Д
зиобратимых электродных процессов потенциал
следует заменить разностью (<р — ф1); тогда получи уж
для квазиобратимых процессов Я
, _ ,Fk, {с0 (0. о «ф [- -уоу-;<-•?’] - ’ - 1
(1^|
для необратимых процессов я
lK = zFksC0 (0, 0 ехр [-. ] (1-ЗбЯ
Влияние адсорбции иа кинетику электродных про-
цессов. Рассмотрим кратко общие вопросы влияния
адсорбции на кинетику электродных процессов.
. При протекании электрохимической реакции могут
адсорбироваться деполяризатор, продукт реакции
или неэлектроактивные вещества, находящиеся в ис-
следуемом растворе. Значительное влияние адсорб-
ции деполяризатора на кинетику электродных про-
цессов обусловливается рядом причин: а) резким
увеличением концентрации деполяризатора на поверх-
ности электрода [33—35], приводящим к увеличению
скорости электродного процесса; б) изменением реак-
ционной способности деполяризатора под влиянием
электрического поля электрода [36], которое также
приводит к увеличению скорости электродного про-
цесса; в) возникновением эффекта больших заполне-
ний [37, 38], что приводит к торможению электро-
химической реакции.
Наиболее правильное объясненйе влияния адсорб-
ции поверхностно-неактивных веществ и продуктов
электродной реакции на кинетику электродного про-
цесса дал Фрумкин [39—41]: адсорбционная пленка
вызывает замедление собственно электрохимической
реакции. Возможны и случаи, когда адсорбционная
пленка увеличивает скорость реакций. Этот эффект
проявляется в присутствии поверхностно-активных
веществ ионного типа, вызывающих изменение фрпо-
тенциала и тем самым увеличивающих константу
скорости электрохимической реакции.'
Рассмотрим количественные соотношения, харак-
теризующие Влияние адсорбции на кинетику элек-
тродных процессов. Если в Процессе обратимой элек-
трохимической реакции адсорбированное вещество
восстанавливается, то уравнение для, плотности тока
будет иметь вид [42]:
i-=zFro (1-14)
где Го — количество адсорбированного деполяризатора на 1 см2
поверхности *. •*
Если адсорбция описывается уравнением Лэнг-
мюра и концентрационная поляризация отсутствует,,
то уравнение (1-14) запишется следующим -Обра-
зом [42]: ;
, рс^
^'15)
1 т₽Со
где р—адсорбционный .индекс, см8/моль; Гом — максимальное
количество адсорбированного вещества при монослойном запол-
нении им 1 см2 поверхности.
При малом заполнении поверхности адсорби-
рованным деполяризатором и в отсутствие кон-
центрационной поляризации плотность тока прямо
* В данном случае Го одновременно является количеством
вещества, участвующего в электродной реакции в единицу вре-
Мещг (моль/с).
пропорциональна объемной концентрации деполяри-
затора [42]:
i — zFrOMCoP0 exp (- а<р2) (1-16}
где ₽о — адсорбционный индекс при потенциале максимальной
адсорбции (фмакс); ф — потенциал электрода ’ относительно фиакс?
а и ро зависят от природы и строения адсорбирующегося веще-
ства; величина а зависит также и от потенциала электрода.
В случае необратимой электрохимической реакции
с участием адсорбированного вещества уравнение
для плотности тока имеет вид [42]:
'• = ^ТОмехр(--^<рк) (1-17)
где k' — константа скорости переноса электрона на адсорбирован-
ное вещество при <р = 0, с-1.
Если адсорбция деполяризатора описывается изо-
термой Лэнгмюра, уравнение (1-17) приводится
к виду [42] (при отсутствии концентрационной по-
ляризации):'
₽с9, / azF \
При малых заполнениях поверхности адсорбирован-
ным деполяризатором плотность4 тока в отсутсТёие
концентрационной поляризации определяется соотно-
шением [42]:
I = zFfe'rOMC((l)p0 exp (- а<р2) exp <р) (1-19)
Влияние адсорбции электрохимически неактивных
веществ на скорость электродного процесса рассмот-
рено в [43, с. 274]. Количественная оценка адсорбции
электрохимически неактивных веществ делается
в предположении, что константа скорости электрохи-
мической реакции в присутствии последних ли-
нейно зависит от степени заполнения поверхно-
сти (0):.
Ч = Ао(1 ~ e) + ^e (1-20)
где fe0, k0 — константы скорости реакции в отсутствие и в присут-
ствии поверхностно-активного вещества.
Если k0 < k0, то электродный процесс замед-
ляется поверхнбстно-активным веществом, в против-
ном случае он ускоряется. Уравнения для тока полу-
чаются подстановкой в кинетические уравнения вме-
сто константы скорости ее выражения из уравне-
ния (1-20).
Следует отметить, что k0 н k0 являются постоян-
ными только в случае незаряженных поверхностно-
активных веществ, в противном случае k0 и ko будут
функциями заполнения поверхности. Влияние адсорб-
ции продуктов реакции на скорость электродных про-
цессов аналогично действию электрохимически неак-
тивных веществ.
Глав;
ХРОНОПОТЕНЦИОМЕТРИЯ ЭЛЕКТРОДН
ПРОЦЕССОВ, НЕОСЛОЖНЕНЫ
ХИМИЧЕСКИМИ РЕАКЦИЯ!
В этой главе мы рассмотрим самый простой пр<
цесс, протекающий под действием постоянного тока г
границе раздела электрод —> раствор и описываемы
уравнением:
О ± ze~ = R * (2-1
Будем полагать: 1) окисленная и восстановлении
формы деполяризатора растворимы в растворе; 2)
растворе имеется избыток индифферентного эле!'
тролита, концентрация которого по меньшей мере н
два порядка превышает концентрацию деполяризЯ
тора; 3) миграционный ток и ^-потенциал отсут
ствуют; 4) доставка вещества к электроду (исключи
ние составляет вращающийся дисковый электрод
осуществляется только свободной диффузией (koi
вективная диффузия отсутствует). Будем рассматр1
вать процесс восстановления, хотя суть математич'
ских выкладок не изменится и для процесса эли
троокисления.
2.1. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ В УСЛОВИ1
ЛИНЕЙНОЙ диффуз>
Уравнения, описывающие зависимость объемнс
концентрации окисленной и восстановленной фор
деполяризатора от времени -электролиза и от ра
стояния от поверхности плоского электрода, имен
следующий вид [44—46]:
n VW'kW ( х2 \
С0(».0-С«о-----2-rap^-_J +
+ A*"IC(W’)
26
-n 2W/ / x2 \
Do ( x \
+ лх---erfc I —п-jr- I (2-3)
PR \2D$th J
где X «= IlzFD.
Если перед началом электролиза восстановленная,
форма деполяризатора отсутствует, то уравнение
(2-3) несколько упрощается:
2W^l/j / х2 \ / х \
Сп (х, 0 =--п— exp I--------14- Xjf erfc I —г-н- I (2-4)
R я7’ \ 4DRt) ' \2D^i'h J
где
erfc (Zi)= 1 — erf (Xi) (2-5)
erf (Xi)—интеграл функции ошибок, определяемый уравнением
Л,
erf (М) = -L f ехр (_ z2) dZ (2-6)
п J .
Z — вспомогательная переменная.
В литературе имеются таблицы функции erf (Xi)
[47]. . . -
Уравнения, описывающие зависимость концентра-
ций окисленной и восстановленной форм деполяри-
затора на поверхности электрода от времени, имеют
вид:
_ . 2№'&'/а
Co(0,t) = C°o------S— (2-7),
л
. 2Wot'!t
. (2-8)
' 2МЯ>!1',г
Cr(0.0 =----(2-9)
л"
В хронопотенциометрии определяют время, через
которое концентрация деполяризатора у поверхнбсти
Электрода становится равной нулю — переходное
^ремя (т). Этот термин впервые был введен Батле-
Зом и Армстронгом [48, 49]. Переходное время, сле-
довательно, определяется из условия Со(0, т)=0.
«7
Приравнивая правую часть уравнения (2-7) нулю, по-
лучим уравнение переходного времени, выведенное
впервые Сэндом [4] и Караоглановым [44]:
* т -------2i~~ (2-10)
Из уравнения (2-10) следует, что т'/> или те1/» прямо
пропорциональны концентрации деполяризатора в
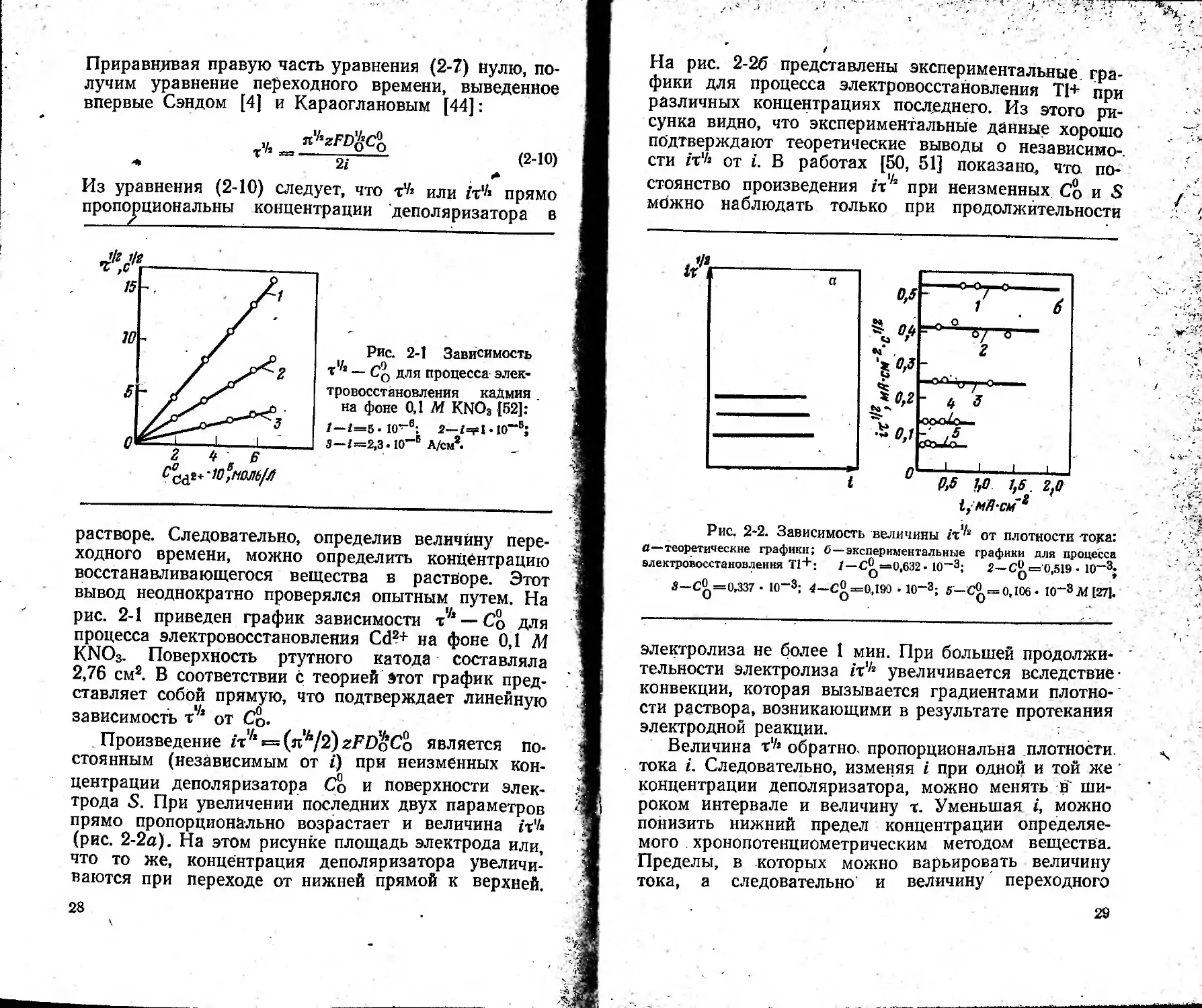
Рис. 2-1 Зависимость
t1/i — Cq для процесса элек-
тровосстановления кадмия
на фоне 0,1 М KNO3 {52]:
z—i=5.io~e-, 2—г=?ыо~®;
3 — I =2,3-10“® А/СМ*.
растворе. Следовательно, определив величину пере-
ходного времени, можно определить концентрацию
восстанавливающегося вещества в растворе. Этот
вывод неоднократно проверялся опытным путем. На
рис. 2-1 приведен график зависимости т^ — Со для
процесса электровосстановления Cd2+ на фоне 0,1 М
KNO3. Поверхность ртутного катода составляла
2,76 см2. В соответствии с теорией Этот график пред-
ставляет собой прямую, что подтверждает линейную
зависимость от Со.
Произведение Zti/*s=(«,/72)zFDoCo является по-
стоянным (независимым от i) при неизменных кон-
центрации деполяризатора Со и поверхности элек-
трода S. При увеличении последних двух параметров
прямо пропорционально возрастает и величина it‘/«
(рис. 2-2а). На этом рисунке площадь электрода или,
что то же, концентрация деполяризатора увеличи-
ваются при переходе от нижней прямой к верхней.
28
На рис. 2-26 представлены экспериментальные гра-
фики для процесса электровосстановления Т1+ при
различных концентрациях последнего. Из этого ри-
сунка видно, что экспериментальные данные хорошо
подтверждают теоретические выводы о независимо-,
сти гт1/! от i. В работах {50, 51] показано, что по-
стоянство произведения /т'4 при неизменных Со и S
можно наблюдать только при продолжительности
Рис, 2-2. Зависимость величины гт^2 от плотности тока:
С—теоретические графики; б—экспериментальные графики для процесса
алектровосстановлення Т1+: 1 —€^==0,632 • 10—3; 2—С^= 0,519 • 10—3;
3—0^=0,337 • IO-3; 4—С3 =0,190 • 10~3; 5—С3 =0,106» 10~3M [27].
электролиза не более 1 мин. При большей продолжи-
тельности электролиза ix'k увеличивается вследствие
конвекции, которая вызывается градиентами плотно-
сти раствора, возникающими в результате протекания
электродной реакции.
Величина т'Л обратно, пропорциональна плотности,
тока I. Следовательно, изменяя i при одной и той же
концентрации деполяризатора, можно менять в ши-
роком интервале и величину т. Уменьшая i, можно
понизить нижний предел концентрации определяе-
мого . хронопотенциометрическим методом вещества.
Пределы, в которых можно варьировать величину
тока, а следовательно и величину переходного
29
Г/.
времени, определяются следующими эксперименталь-
ными условиями: 1) конвекция не должна влиять на '
процесс диффузии; 2) доля тока, расходуемого на за-
ряжение двойного электрического слоя, должна быть ’;
мала по сравнению с общим током, проходящим через
электролитическую ячейку. j
При выводе уравнений зависимости концентрации ]
деполяризатора от времени и расстояния от поверх-
ности электрода не делалось никаких предположений '
о характере электродного процесса, поэтому они оди- 4
' каковы для обратимых, необратимых и квазиобра^- 1
тимых электродных процессов. 1
Для всех/видов электродных процессор соотноше- |
ния для переходного времени будут одинаковыми, 1
поэтому по величине переходного времени нельзя су- Я
дить об обратимости электродного процесса. ' д
Уравнения хронопотенциометрических кривых по- ”1
лучаются подстановкой в уравнение Нернста или д
в кинетическое уравнение теории замедленного раз- 1
ряда — ионизации выражения для концентрации де- "1
поляризатора у поверхности электрода. Подставляя я
соотношения (2-7) — (2-9) в уравнение Нернста (1-1), J
получим уравнение хронопотенциограммы обратимых J
электродных процессов для случая, когда окисленная Ч
и воссстановленная ’
римы в растворе:
ф = ф“ + -—
* zf
формы деполяризатора раство- j
'"4-+^|пЛ?5- (2-п) ]
»«“« !
где
Р
Выразив Со Из уравнения (2-10) и выполнив не-
которые алгебраические преобразования, приведем
уравнение (2-11) к виду.
rt d'J? . rt t4, -t'k
ф фО + _ 1" + __ in , (2-12)
Это уравнение впервые было получено в работе [44].
Сумма первых двух членов в правой части уравне- ;
30
ния (2-12) равна потенциалу полуволны обратимой i
полярографической волны, поэтому: / - -Л
RT х^г_____№
Ф » Ф1Д + — 1П - ?/> (2-13) .<-. у
Из уравнения (2-13) видно, что <pt/4 = <pVj при /=#
= т/4. Следовательно, потенциал хронопотенцио;
граммы при t — т/4 не зависит от концентраций де- ;
поляризатора и плотности тока и может служитьr '
качественной характеристикой состава раствора.
Кроме того, график в координатах ф — 1§[(т’Л-е ’
является прямой линией (рис. 2-3), тангенс
угла наклона которой равен 2,3RT/(zF). По тангенсу
угла наклона прямой можно найти число электронов,
участвующих в электродном процессе. По углу на- 1
клона этой прямой можно судить и об обратимости
электродного процесса. Совпадение расчетного значе-
ния тангенса угла наклона с полученным из опытных
данных будет означать, что рассматриваемый про-
цесс обратим, а расхождение этих значений — что
изучаемый процесс необратим.
Уравнение (2-13) было подвергнуто эксперимен-
тальной проверке. Наиболее надежные результаты
были получены в {20, 52]. Полученные в последней
работе графики зависимости 1g[(т7* — —<р для
процессов электровосстановления TI+ и Cd2+ в кон-
центрации 4-10~3 моль/л на фоне 1 М KNO3 и ферро-
цианида на фоне 1 М КС1 в соответствии с уравне-
нием (2-13) являются прямыми линиями, а значения
31
<рт/4 хорошо согласуются с потенциалами полуволн
<р,А обратимых полярографических волн (рис. .2-4).
Обратные величины коэффициентов наклона прямых
на рис. 2-4 составляют для галлия 0,061; для кад-
мия— 0,032 и для ферроцианида — 0,068 против тео-
ретических значений соответственно 0,060; 0,030 и
ОрО 0)20 -0,1/5 -0,50 -0,55 ^0,60
4>,й(на.с.к.з.)
Рис. 2-4. Графики зависимости Ig [(t’/i — <р для обра-
тимых процессов электровосстаиовления Fe(CN)g~ иа фоне •
1 М КС1 и Т1+ и Cd2+ на фойе 1 М KNO3 [52].
Темные кружки—катодный процесс, светлые—анодный.
0,060 (при 25 9С). Первые два процесса можно счи-
тать обратимыми, третий процесс необратим.
Уравнение переходного’ времени для случая элек-
троосаждения металла на электроде, состоящем из
того же металла, будет таким же, что и для элек-
тродного процесса с участием двух растворимых ве-
ществ [уравнение (2-10)]. Уравнение хронопотенцио-
граммы в этом случае получается подстановкой
в уравнение Нернста значения Со(0, t) из уравнения
(2-7) и значения CR(0,/)= 1:
Ф = Ф° + — In----+ —у In (t‘/s - /Vs) (2-14)
zF zFD$nl‘ zF ’
32
При электроосажденни металла иа электроде гра-
фик в координатах 1g (т% — tty — ср является прямой
линией* тангенс угла наклона которой равен zF!2,3RT.
По тангенсу угла наклона прямой можно определить
величину г.
Уравнение хронопотенциограммы для необрати-
мого электродного процесса восстановления вещества
получают подстановкой значения Со (0,/) из (2-7)
в кинетическое уравнение (1-5а):
RT
+—Г
azaF
In (?£
RT
In I (2-15)
azaF
Выразив Со через переходное время и выполнив ал-
гебраические операции, получим:
Ф « Ф° + in + ~- - tty (2-16)
azaF azaF
На практике для исследования необратимых процес-
сов электровосстановления обычно используют урав-
нение хронопотенциограммы в следующем виде [20]:
RT zFCf^.k'f RT f fty'hl
ф=-----— In----£-£- +---In 1-1— (2-17)
T azaF i azaF L J
где z, — число электронов, участвующих в лимитирующей ста-
дии электродного процесса; kj — константа скорости электродно-
го процесса при потенциале, равном нулю относительного нор-
мального водородного электрода.
Для необратимых процессов потенциал электрода
при <-*0 резко изменяется до величины, определяе-
мой уравнением (рис. 2-5):
RT zFCnok°f
Ф, *= ——,п ~—г—-
azaF i
Зависимость ср — 1п[1 — (Z/tf'] должна быть пря-
мой линией с тангенсом угла наклона, равным
RT/(azBF). По величине тангенса можно определить
ага. Форма хронопотенциограммы для необратимых
процессов зависит от ctza и k°, в результате она ока-
зывается более вытянутой в масштабе потенциалов,
2 Зек. 1078
33
чем в случае обратимых процессов. Потенциал <р,- за-
висит от плотности тока i, и следовательно'хронопо-
тенциограммы могут отчасти сдвигаться по шкале
потенциалов при изменении плотности тока.
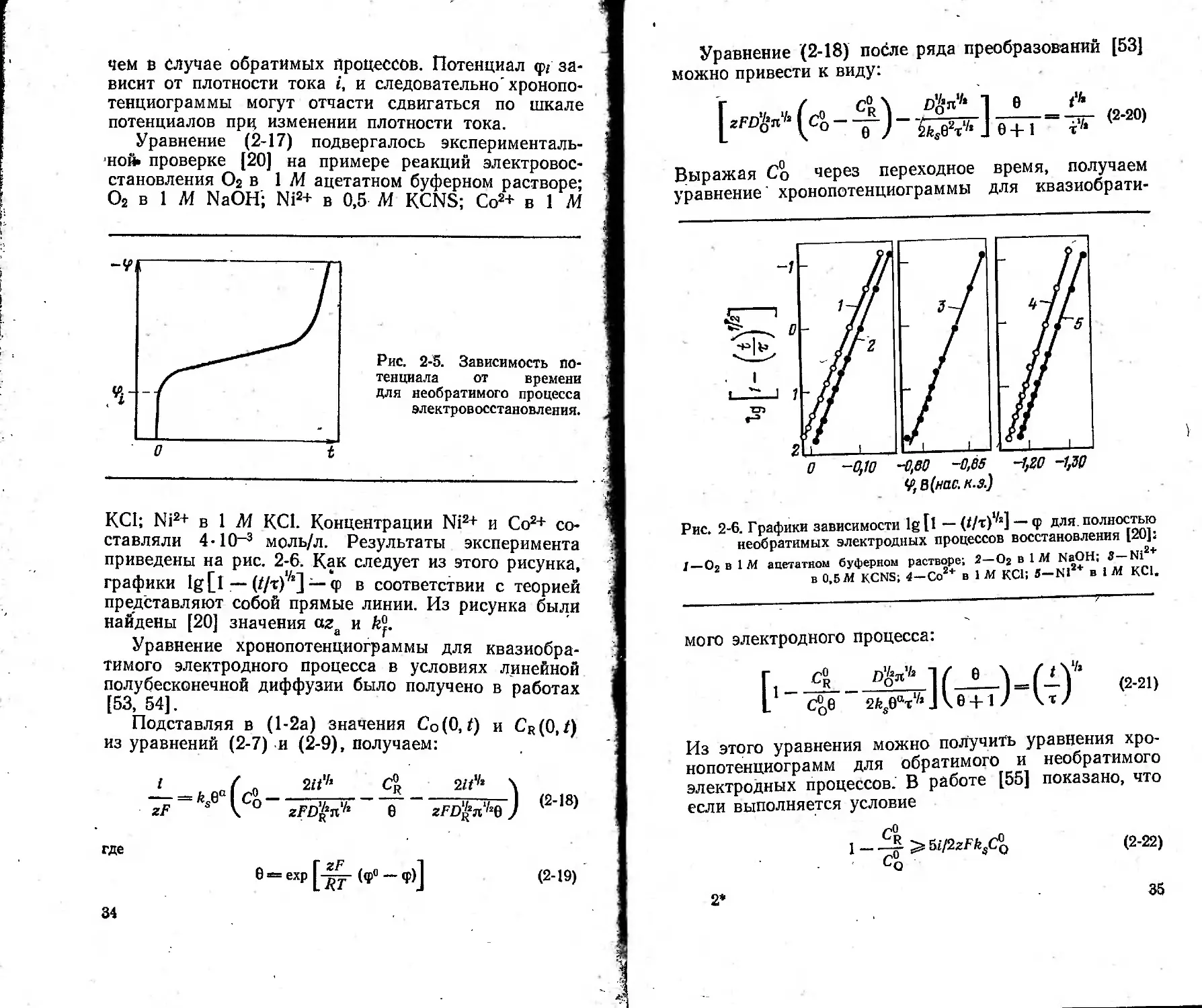
Уравнение (2-17) подвергалось эксперименталь-
ной» проверке [20] на примере реакций электровос-
становления О2 в 1 М ацетатном буферном растворе;
О2 в 1 М NaOH; Ni2+ в 0,5 М KCNS; Со2+ в 1 М
Рис. 2-5. Зависимость по-
тенциала от времени
для необратимого процесса
электровосстановления.
КС1; Ni2+ в 1 М КС1. Концентрации Ni2+ и Со2+ со-
ставляли 4-10_3 моль/л. Результаты эксперимента
приведены на рис. 2-6. Как следует из этого рисунка,
графики 1g [1— (//т)1/г] — <р в соответствии с теорией
представляют собой прямые линии. Из рисунка были
найдены [20] значения aza и k°f.
Уравнение хронопотенциограммы для квазиобра-
тимого электродного процесса в условиях линейной
полубесконечной диффузии было получено в работах
[53, 54].
Подставляя в (1-2а) значения Со(0, t) и CR(0,/)
из уравнений (2-7) и (2-9), получаем:
I ( п 2it'h C°R 2lt4‘ \
= /г Аа СЪ--и-п----- ц-й— (2-18)
zF---------------------------s \ ° zFD^nh 6-zFD^n^Q J
где
б=ехр|д^-(ф0 —<p)J
(2-19)
34
Уравнение (2-18) после ряда преобразований [53]
можно привести к виду:
Выражая Со через переходное время, получаем
уравнение' хронопотенциограммы для квазиобрати-
0 —0,10 -0,80 -0,65 -1,20 -1,30
V, В (нас. к.з.)
Рис. 2-6. Графики зависимости lg [l — (//т)*72] — <р для. полностью
необратимых электродных процессов восстановления [20]:
1—О2 в 1 М ацетатном буферном растворе; 2—Oj в 1 JU NaOH; 3—Ni2+
в 0,5 М. KCNS; 4—Со2+ в 1 М КСГ, «—Ni2+ в 1 М КС1.
мого электродного процесса:
D'^nb 1/ е \ / А*7’
с£0 ” 2kseat'b J Vе-ь 1J — \ т/
(2-21)
Из этого уравнения можно получить уравнения хро-
нопотенциограмм для обратимого и необратимого
электродных процессов. В работе [55] показано, что
если выполняется условие
1----^^5i/2zFksC°o (2-22)
CQ
2*
35
то электродная реакция может рассматриваться как
электрохимически обратимая. Если выполняется ус-
ловие
i
К—г + —дг „ (2-23)
Со zFC°oksQa
то ток будет определяться скоростью переноса элек-
тронов и процесс будет-необратимым.
Уравнения хронопотенциограмм для обратимого
и необратимого электродных процессов анализирова-
лись выше. Остановимся здесь на анализе уравнения
хронопотенциограммы для квазиобратимого элек-
тродного процесса.
На рис. 2-7 представлены хронопотенциограммы,
рассчитанные для различных ks и а при Do =
= Ь10-® см2/с, т = 25 с и Cr/Co=?= Е Из уравнения
(2-21) и рис. 2-7 видно, что каждому значению по-
тенциала соответствует определенное значение (//т)л,
определяемое кинетическими константами ks и а
36
и равновесным-потенциалом <рР. Из рисунка следует
также, что изменение равновесного потенциала при-
водит к сдвигу хронопотенциограммы по шкале по-
тенциалов, не изменяя ее формы. При увеличении а
уменьшается наклон кривой.
2.2. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ В УСЛОВИЯХ
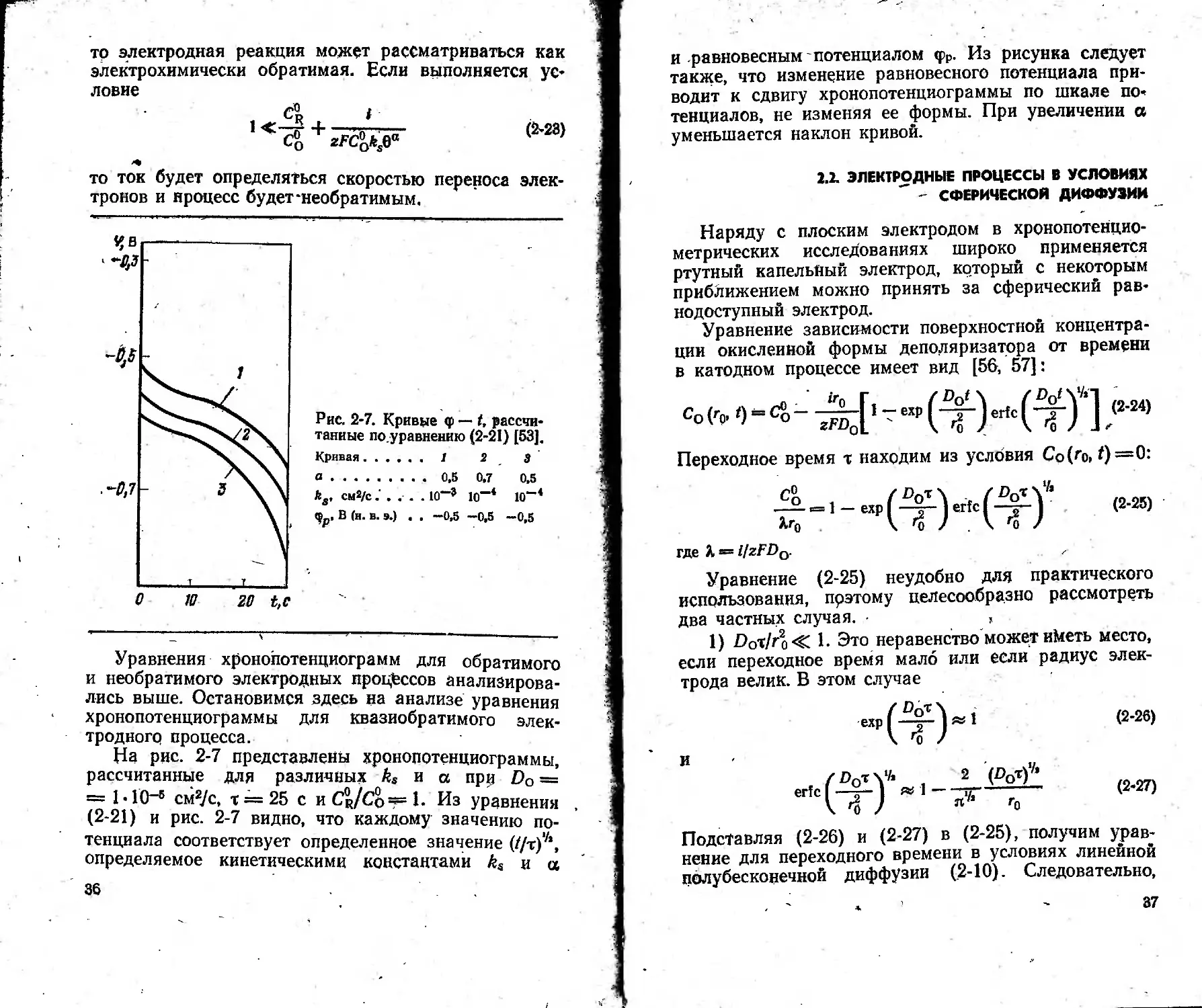
" - СФЕРИЧЕСКОЙ ДИФФУЗИИ
Наряду с плоским электродом в хронопотенцио-
метрических исследованиях широко применяется
ртутный капельный электрод, который с некоторым
приближением можно принять за сферический рав-
нодоступный электрод.
Уравнение зависимости поверхностной концентра-
ции окисленной формы деполяризатора от времени
в катодном процессе имеет вид [56, 57]:
Переходное время т находим из условия Co(r0,t)—Q:
СОТ\
Ъ )
(2-25)
со .
= 1 — ехр
где K^i!zFDa.
Уравнение (2-25) неудобно для практического
использования, прэтому целесообразно рассмотреть
два частных случая. ,
1) £>от/го<С 1- Это неравенство может и^еть место,
если переходное время мало или если радиус элек-
трода велик. В этом случае
и
(2-26)
(2-27)
Подставляя (2-26) и (2-27) в (2-25), получим урав-
нение для переходного времени в условиях линейной
полубесконечной диффузии (2-10). Следовательно,
37
при -малых т и больших го сферичность электрода не
сказывается на характере распределения концентра-
ции деполяризатора и произведение для сфериче-
ской диффузии приближается к значению 1г',г для
линейной диффузии.
2) £>от/го» 1. Этот случай будет наблюдаться
при большом значении переходного времени или ма-
лом радиусе электрода. Разлагая в уравнении (2-25)
функцию ошибок в ряд при большом значении аргу-
мента, можно получить следующее приближенное
уравнение для переходного времени [58]:
,. zFn,2D'^C^ zFnDaC°nT'h
ix =------г2-5 +------<2'28>
Л о
Первый член правой части этого уравнения учиты-
вает вклад линейной полубесконечной диффузии,
а второй член — вклад, обусловленный сферической
формой электрода. Первый член не зависит от вели-
чины поляризующего тока, а второй увеличивается
с уменьшением плотности тока и при i -> 0 стремится .
к бесконечности. Аналогичное влияние на этот член
оказывает и размер электрода (г0).
Для сферической диффузии график зависимости
frl/’ от i не является прямой, параллельной оси абс-
цисс, как это наблюдается для линейной полубеско-
нечной диффузии. Следовательно, в хронопотенциомет-
рии со сферическим электродом при аналитических
исследованиях нужно использовать калибровочный
график, а не метод добавок.
При больших плотностях тока (т мало) второй
член правой части уравнения (2-28) становится ма-
лым и произведение 1тУ2 будет таким же, как для
линейной диффузии. С уменьшением тока и (или)
радиуса электрода (т увеличивается и второй член
возрастает) переходное время будет больше пере-
ходного времени в условиях линейной диффузии. Это
явление можно с успехом использовать для повыше-
ния аналитической чувствительности хронопотенцио-
метрии. При i < nzFDoCoAro переходное время во-
обще не достигается.
Уравнения зависимости поверхностной концентра-
ции восстановленной формы деполяризатора, раство-
38
римой в исследуемом растворе, от времени электро-
лиза получаются решением задачи, аналогичной для
окисленной формы деполяризатора, и имеют следую-
щий вид:
при наличии в первоначальный момент восстанов-
ленной формы деполяризатора в растворе
п ira Г /2М\
при отсутствии в первоначальный момент восста-
новленной формы деполяризатора в растворе
«г0 Г \ /Dr* Va1
Уравнения для концентраций Co(r0,t) и CR(r0, t)
получены без учета характера электродного процесса,
поэтому они справедливы как для обратимых, так
и для необратимых электродных процессов.
Уравнение хронопотенциограммы обратимого
электродного процесса на сферическом электроде для
случая, когда R и О растворимы в анализируемом
растворе, получается подстановкой уравнений (2-24)
и (2-30) в уравнение Нернста (1-1) (принимая
Do — Db = D):
(2-31)
При (ОтДо) < 1 уравнение (2-31) Переходит
в уравнение (2-13).
Из уравнения (2-31) следует, что график ср—1пР
является прямой линией с тангенсом угла наклона,
равным RT/(zF), где
39.
Уравнение хронопотенциограммы для необрати-
мого электродного процесса будет иметь вид:
<р =» ф° + In zFks -j-
* * azar
Заменив Co через переходное время и выполнив
алгебраические операции, получим:
₽=<’)0+ъ5-,п^+^,пР‘
где
График в координатах <р — In Л представляет собой
прямую линию с тангенсом угла наклона, равным
RTfa-z^F. По тангенсу угла наклона можно вычислить
az., а по отрезку, отсекаемому прямой на оси потен-
циалов при lnPft=0, можно вычислить величину ks.
2.3. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ В УСЛОВИЯХ
ЦИЛИНДРИЧЕСКОЙ ДИФФУЗИИ
Широкое применение в электроаналйтической
практике находят электроды, изготовленные из про-
волоки (платиновой, серебряной, углеродной). Они
представляют собой цилиндры, впаянные в стекло
или впрессованные во фторопласт. ,
Зависимость концентрации деполяризатора от вре-
мени электролиза на поверхности цилиндрического
электрода имеет вид [57, 59];
(oa’/i А А^* \
+ 2^“ ”) (2‘34)
Из (2-34) получаем следующее соотношение для
переходного времени:
. У1 ______________zFn'^D'^________________
" gfi м^',г , °от V
X 4г0 4г0 32г^ У
40
Из сравнения уравнения (2-35) с уравнением для
переходного времени (2-10) в условиях линейной
диффузии видно, что знаменатель уравнения (2-35)
учитывает эффект цилиндричности диффузионного
поля у цилиндрического электрода. ЕсЛиО'^О (<>'=
= соотношение (2-35) переходит в уравне-
ние (2-10). Однако расчеты по уравнению (2-35) по-
казывают, что эффект цилиндричности диффузион-
ного поля существен даже в тех случаях, когда зна-
чения т малы. Так, для £>о = 8,6-10~8 см2/с и г0 =
= 0,0255 см даже при т = 1 с эффект цилиндрично-
сти составляет 5% и уменьшается до 1% лишь при
т = 0,04 с.
Из уравнения (2-35) видно, что эффект цилцнд-
ричности диффузионного поля увеличивается с умень-
шением радиуса электрода и увеличением коэффи-
циента диффузии электроактивных веществ. Уравне-
ние (2-35) подвергалось неоднократной проверке
[59, 60]. Результаты показали удовлетворительное
согласие экспериментальных данных с расчетными
значениями (т'^/Со-
Однако оказалось, что уравнение (2-35) не опи-
сывает правильно процесс, когда параметр О прини-
мает большое значение. Авторы работы [61] полу-
чили уравнение для переходного времени, справедли-
вое и при больших значениях О (в нем они взяли
больше членов ряда, учитывающих цилиндричность
диффузии):
(2-36)
где /?1 — члены ряда, учитывающие цилиндричность диффузии.
Авторы [61] рассчцтали на ЭВМ функцию R\ для
переходных времен от 0,09 до 25 с и для Do/fo от
0,004 до 0,4. Результаты приведены в табл. 2-1.
На рис. 2-8 представлены зависимости R\ от т при
различных значениях £>о/го- Из рисунка видно, что
/?1 близко к 1, когда т или Do очень малы. В этих
условиях диффузионный слой очень тонкий, и до-
ставка вещества осуществляется, практически, за
счет линейной диффузии. Величина R\ также близка
41
к единице, когда г0 становится очень большим,
т. е. когда поверхность электрода приближается
к плоской. ‘
Наклон кривых /?1—т зависит от значений РоЛо-
Для малых D'^fro он почти постоянен в интервале
переходных времен от 5 до 25 с, но для Do/r0> 0,2
Рис. 2-8. Зависимость поправочного фактора Ri для хронопотен-
циометрии с цилиндрическим электродом от переходного вре-
мени т при различных значениях (приведены на кри-
вых) [61J.
наклон кривых при т > 5 с становится больше, чем
при т < 5 с. Эта закономерность была подтверждена
в работе [60] на примере электровосстановления
ионов водорода на платиновом цилиндрическом элек-
троде. В этих же исследованиях было установлено,
. что экспериментальные данные в исследованном ин-
тервале т не подчиняются уравнению (2-36) при
D*/r0>0,2.
Уравнение, справедливое при £>о7П)г>0,2, полу-
чено в работе [62]:
/т71 =
n'l>2FD%C°Q
----§—-Ri
(2-37)
43
где
№ “ f 1° 4^" 4
1 + In ф ,
8d3
О«= <р = 4d2/ev
у — постоянная Эйлера; /?2— коэффициент, учитывающий цнлин-
дричность диффузии.
Значения Ri были рассчитаны по уравнению
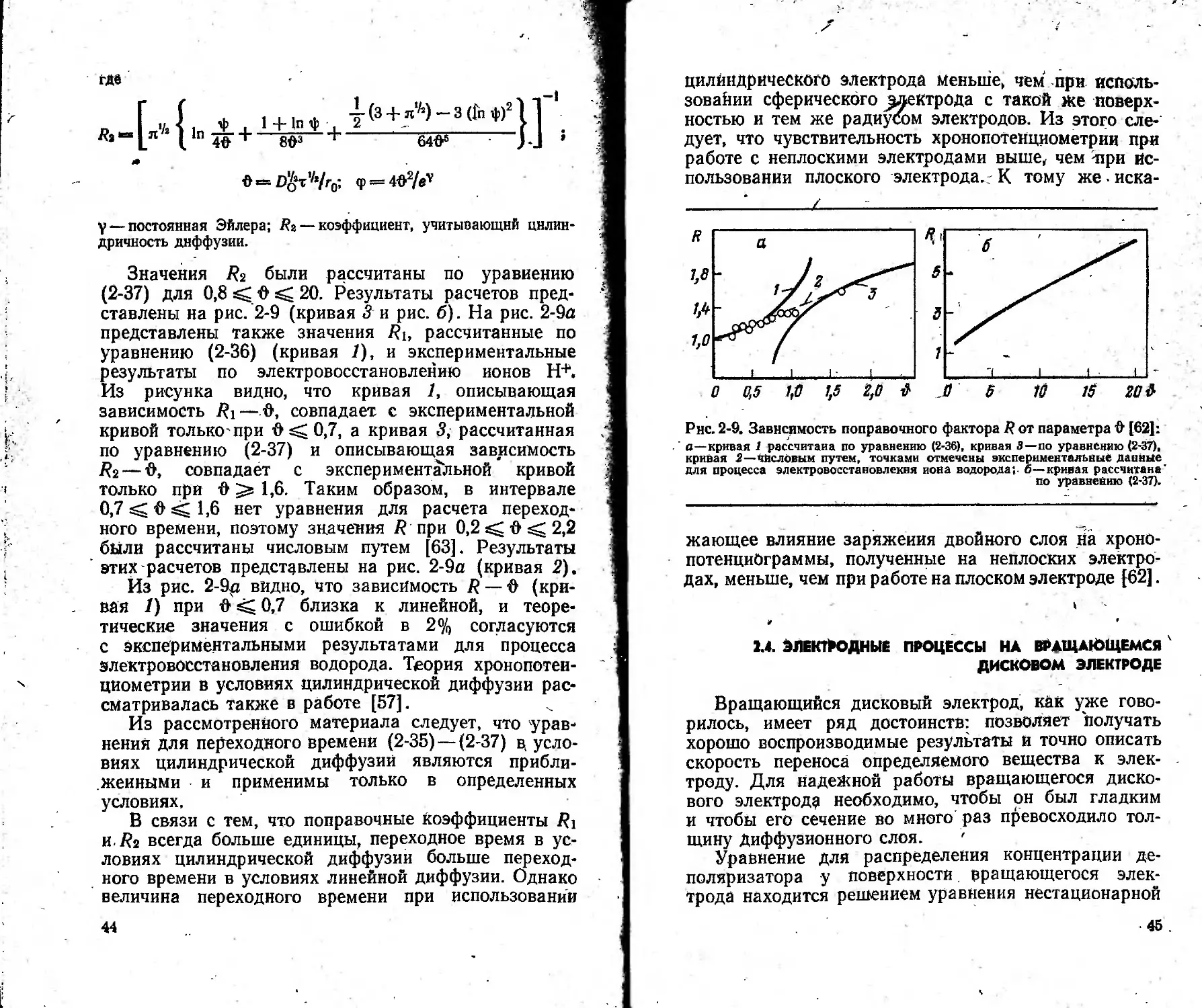
(2-37) для 0,8 С 20. Результаты расчетов пред-
ставлены на рис. 2-9 (кривая 3 и рис. б). На рис. 2-9а
представлены также значения Ri, рассчитанные по
уравнению (2-36) (кривая 1), и экспериментальные
результаты по электровосстановлению ионов Н+.
Из рисунка видно, что кривая 1, описывающая
зависимость Ri— •&, совпадает с экспериментальной
кривой только-при & sC 0,7, а кривая 3, рассчитанная
по уравнению (2-37) и описывающая зависимость
Ri — О, совпадает с экспериментальной кривой
только при О 1,6. Таким образом, в интервале
0,7 О’ 1,6 нет уравнения для расчета переход-
ного времени, поэтому значения R при 0,2 О 2,2
были рассчитаны числовым путем [63]. Результаты
этих расчетов представлены на рис. 2-9а (кривая 2).
Из рис. 2-9ц видно, что зависимость R — О (кри-
вая /) при О 0,7 близка к линейной, и теоре-
тические значения с ошибкой в 2% согласуются
с экспериментальными результатами для процесса
злектровосстановления водорода. Теория хронопотеи-
циометрии в условиях цилиндрической диффузии рас-
сматривалась также в работе [57].
Из рассмотренного материала следует, что урав-
нения для переходного времени (2-35) —(2-37) в усло-
виях цилиндрической диффузий являются прибли-
женными и применимы только в определенных
условиях,
В связи с тем, что поправочные коэффициенты Ri
h.R2 всегда больше единицы, переходное время в ус-
ловиях цилиндрической диффузии больше переход-
ного времени в условиях линейной диффузии. Однако
величина переходного времени при использовании
44
цилиндрического электрода меньше, чем при исполь-
зовании сферического ^лектрода с такой же поверх-
ностью и тем же радиугом электродов. Из этого сле-
дует, что чувствительность хронопотенциометрии при
работе с неплоскими электродами выше, чем -при ис-
пользовании плоского электрода. - К тому же иска-
Рнс. 2-9. Зависимость поправочного фактора 2? от параметра Д [62]:
а—кривая 1 рассчитана по уравнению (2-36), кривая 3—по уравнению (2-37),
кривая 2—числовым путем, точками отмечены экспериментальные данные
для процесса электровосстановлекня иона водорода;, б—кривая рассчитана
по уравнению (2-37).
жающее влияние заряжения двойного слоя на хроно-
потенциОграммы, полученные на неплоских электро-
дах, меньше, чем при работе на плоском электроде [62].
2.4. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ НА ВРАЩАЮЩЕМСЯ '
ДИСКОВОМ ЭЛЕКТРОДЕ
Вращающийся дисковый электрод, как уже гово-
рилось, имеет ряд достоинств: позволяет получать
хорошо воспроизводимые результаты и точно описать
скорость переноса определяемого вещества к элек-
троду. Для Надежной работы вращающегося диско-
вого электрода необходимо, чтобы он был гладким
и чтобы его сечение во много раз превосходило тол-
щину Диффузионного слоя. '
Уравнение для распределения концентрации де-
поляризатора у поверхности. вращающегося элек-
трода находится решением уравнения нестационарной
45.
конвективной диффузии. Для окисленной формы Де-
поляризатора оно имеет вид [64, 65]:
со (0, t) = С°о-2- [1,07 erf J3,\Dot/t>2o -
- 0,73 exp (- 1,65Ро//б^) erf д/1,45Ро</й£ ] (2-38)
где ia — стационарный предельный ток:
id =---т-----
°0
6О== l,61D$v4r,/«
(2-39)
(2-39а)
6о — толщина диффузионного слоя при электровосстановлении
окисленной формы деполяризатора; v — кинематическая вязкость
раствора; ы— угловая скорость вращения электрода.
Уравнения для распределения восстановленной
формы деполяризатора в присутствии и в отсутствие
ее в начальный момент (/ = 0) в растворе имеют со-
ответственно следующий вид [64, 65]:
CR (0, t) = C°R + C°R-/- [1,07 erf Vs.lOfM -
- 0,73 exp (- l,65DRf/dR) erf (l,45DRf/6^)] (2-40)
CR(0, t) = 4 7- [1,07 erf VsJOrWr -
— 0,73 exp (- l,65DRf/6R) erf (1,45DR//S|)] (2-41)
где 6r — толщина диффузионного слоя при элекгроокислении вос-
становленной формы деполяризатора.
В начальный момент времени (/ <С &IDO) влияние
конвекции на изменение концентрации у поверхности
электрода мало, и уравнения (2-38), (2-40), (2-41)
приводятся к уравнениям для линейной полубеско-
- нечной диффузии (2-7) —(2-9), т. е. в начальный мо-
мент времени доставка вещества к поверхности элек-
трода осуществляется только за счет диффузии. '
При больших t устанавливается стационарный ре-
жим доставки вещества, и концентрация на поверх-
ности дискового электрода перестает зависеть от вре-
46
мени. Уравнения (2-38), (2-40), (2-41) в этом случае
соответственно приводятся к виду:
Со (0, П = С°о - 1,07С^ 4- (2-42)
ld
CR (0, 0 = C°R + 1 ,07C°r -4 (2-43)
ld
CR (О, t) =₽ l,07CR 4- (2-44)
id
Соотношение для переходного времени получим из
условия Со(0,т)== 0. Из уравнения (2-38) следуете
= 1.07 erf д/з,1£>от/б^ -
— 0,73 exp (— 1,65Dot/6q) erf aJ1,45£>ot/6q (2-45)
Это уравнение является трансцендентным и в явном
виде относительно т не решается. Подставляя в соот-
ношение (2-45) значение id из уравнения (2-39), по-
лучим:
—4-^- = 1,07 erf д/3,1Рот/д?) -
JVQ * ’ "
- 0,73 exp (- 0,65Пот/6^) erf aJ 1 ,45Dot/6^ (2-46)
Из этого уравнения следует, что, как и в случае ра-
нее рассмотренных электродов, прямой зависимости
между Со и т нет, поэтому в аналитических иссле-
дованиях с вращающимся дисковым электродом
нельзя пользоваться методом. добавок, а следует
пользоваться калибровочным графиком. Расчеты, вы-
полненные по уравнению (2-46) [26, сг 161], показы-
вают, что при малых т (т<;бо/£>о) зависимость т
от i аналогична таковой для линейной полубесконеч-
ной диффузии [см. уравнение (2-10)], когда переме-
шиванием можно пренебречь, а при больших
т (т > бо/Оо) переходное время не зависит от i.
Уравнение (2-45) можно привести к следующему
соотношению [66, 67]:
f, ^о V4 ехр + Ц2 я2дот/4йо]
'л в / ] 1 —7Д (.2k 4 О2 ~
v Л=0
где о(т)—правая часть выражения в фигурных скобках.
= /[!—<г(т)]
(2-47)
47
Из (2-45) и (2-47) следует, что переходное время Я
может быть достигнуто при i 5s В этом случае
независимо от значений I и т произведение 1[1т-(Т(т)] Я
постоянно и равно id, его величина определяется объ- Я
емной концентрацией деполяризатора и скоростью Я
вращения дискового электрода. 1
Верхнее предельное значение переходного вре-
мени, которое можно реализовать на вращающемся ' 4
дисковом электроде, определяется условием i та id, '
что, как это видно из уравнения (2-45), возможно i
при erf д/з»Шот/бо= 1. Отсюда следует: :
^макс<2б2о/Ро (2-48) <
Если принять, что £>о = 1 • Ю-5 см2/с и v = 10-2 см2/с, ’
то, подставляя (2-39а) в (2-48) и произведя вычис-
ления, получим т^500/п, где п — число оборотов ;
диска в 1 с.
Сравнение выражения для стационарного предель-
ного тока диффузии с формулой (2-10) дает:
(2’49) 1
/т'* бр V л
Из уравнения (2-49) следует, что чувствительность . •
аналитических определений на вращающемся диско-
вом электроде может быть на два порядка выше,
чем при определениях в покоящемся растворе (в ус-
ловиях свободной диффузии).
Уравнение хронопотенциограммы для обратимого
электродного процесса, протекающего на вращаю-
щемся дисковом электроде, получают подстановкой
в уравнение Нернста (1-1) выражений для концен-
трации окисленной (2-38) и восстановленной (2-40)
форм деполяризатора. Заменив Со через переходное
время из уравнения (2-46) и выполнив алгебраиче-
ские преобравования, получим (при £>о = — £>):
<Р == т° -L In М
• (2-50)
где
Я в ЬМеНУзДОт/б* -erfV3,lDf/y ) + 0,73
1,07-erf 73,lb</&s - 0.73 *
г / 1,65 Dt\ , /1Л5О7 / 1.65DA , /1,45 6 г
. [ехр ( б—J еНд/ —ехр ( erf д/ -4^-
Х exp (- 1,65 Dt/б1) erf (1,45 Dt/&)
Для необратимого электродного процесса уравне-
ние хронопотенциограммы получают совместным ре-
шением уравнений (1-5а) и (2-38):
RT ksi6o НТ
где
N = 1,07 (erf д/з'1 - erf д/з,1 Dt/62O ) +
4- 0,73 [exp (- 1,65 Df/d^) erf д/1,45ОДб^ -
— exp(— 1,65 Pt/6q) erf -^/1,45 Dt/6q ]
По тангенсу угла наклона прямой ф — InJV можно
определить aza, а по отрезку, отсекаемому на оси
ординат при In N = 0 — величину ks.
Уравнение хронопотенциограммы для квазиобра-
тимого электродного процесса получается подстанов-
кой уравнений (2-38) и (2-40) в (1-2а), но оно имеет
довольно сложный вид и поэтому здесь не при-
водится.
2.5. ХРОНОПОТЕНЦИОМЕТРИЯ МНОГОСТУПЕНЧАТЫХ
"" ПРОЦЕССОВ ВОССТАНОВЛЕНИЯ (ОКИСЛЕНИЯ) ВЕЩЕСТВ
В предыдущих разделах рассматривалась.хроно-
потенциометрия веществ, восстанавливающихся'
(окисляющихся) до конечных продуктов при одном
потенциале. Однако в практике возможны многосту-
пенчатые процессы: исходное вещество О при потен-
циале q>i восстанавливаемся до вещества Ri, которое
затем при более отрицательном потенциале фа восста-
навливается до вещества Rs- Если эти реакции проте-
кают при потенциалах, значительно отличающихся
<9
друг бт друга, то на хронопотенциограмме будут на-
блюдаться две волны.
В литературе рассматриваются три возможные
схемы электровосстановления веществ.
Схема 1.
“ O + zie“ = R! (первая ступень)
Ri-J-zse- — Rs )
O+(z,+z2)e-= Ra J F *
Вещество О при потенциале <pi с участием z\
электронов восстанавливается до Ri (первая волна
на хронопотенциограмме), а затем при потенциале
q>2 вещество Ri с участием Zz электронов восстанав-
ливается до R2. Одновременно со вторым процессом
протекает и восстановление О с участием (?i Д- Z2)
электронов до R2; двум последним процессам на хро-
нопотенциограмме соответствует вторая волна.
Уравнения для концентраций окисленной и вос-
становленной форм деполяризатора у поверхности
электрода и для переходного времени процесса, про-
текающего при «pi, аналогичны уравнениям (2-7),
(2-8), (2-10) соответственно. Для процессов, проте-
кающих при фг. эти уравнения не справедливы. Для
процессов, протекающих при потенциале <р2, плот-
ность тока складывается из двух компонент и опи-
сывается следующей формулой:
Г dCR (0, /') I г дСп (0, t') 1
i = ----J + (2> + Z2) FD° L ' dx J (2-52)
где t' t — t; t — время от начала электролиза.
Авторы работы [2] для процессов, протекающих
при <jp2, получили следующее уравнение, описывающее
распределение концентрации вещества Ri у '-поверх-
ности электрода:
(о’ п ~ т‘! ’(т* + Г),/!1 (2’53)
л L Z1 Jj
Приравнивая нулю концентрацию CR,(0, t'), полу-
чим уравнение для переходного времени (тг) для вто-
рой ступени:
50
(2-54)
Из (2-54) следует, что если. 2i = ?2, то t2 = 3ti,
а при zz!z\ = 2 т2 — 8ть Эти выводы были подтверж-
дены экспериментально для ступенчатого восстанов-
ления кислорода и иона уранила [2].
Уравнения хронопотенциограмм для первой сту-
пени аналогичны уравнениям (2-12) и (2-14).
Для получения уравнения хронопотенциограммы
второй ступени необходимо знать уравнение для кон-
центрации компонента R2 У поверхности электрода.
Вещество Яг является продуктом восстановления О
или Ri. Сумма потоков веществ О, Ri и Ra на поверх-
ности электрода равна нулю:
гас0(х,П1 , _ pcRi(x,ni
D°L—ат—Lo+M—di—_Lp+
Используя это соотношение, авторы [2] получили
уравнение для CR1 (0, t'):
Для обратимого электродного процесса уравнение
хронопотенциограммы получают подстановкой в урав-
нение Нернста (1-1) уравнений (2-53) и (2-56):
, RT - fa + Т»)'А ~ fa + f)'Z*
Ф z2f z (tj + t')'1* — .
(2-57)
где <Pi/2(r,_r,) — потенциал полуволны для окислительно-восстано-
вительной системы Rz/Ri-
Время, при котором потенциал достигает значения
'P'/jCR.-r-h определяют, приравнивая единице дробь под
знаком логарифма в уравнении (2-57):.
z т2 . 2г _
14>ч, 4 2г, 3
Например, если z\ — z2, то т2 — Зть и потенциал по-
луволны ф</2 наблюдается при f = т2/4 4- т2/6 = 5т2/12.
Время, при котором достигается определяется так
же, как и для одноступенчатых процессов.
(2-58)
51
Схема 2.
О + zle~ = Ri (первая ступень) '
О + (zi + za) е~ = Ra (вторая ступень)
этом случае, в отличие от предыдущего, продукт'!
Ri процесса электровосстановления вещества О по]
первой реакции является неэлектроактивным. По-1
скольку на второй ступени вещество Ri не участвует ’
в реакции, переходное время второй волны будет
меньше, чем это было в первом случае. <j
Эта схема рассмотрена в работе [67]. Переходное ,
время для первой ступени определяется уравнением j
(2-10). Выражение для переходного времени, соот-
ветствующее второй ступени, имеет вид:
(т, + т2)’/г - —(2-59)
х ' Z| + Zi
откуда
(2-60)
При zi = z2==l, согласно уравнению (2-60),
т2/ц = 1,78.
В предыдущем случае при Zi = z2 t2/ti = 3.
Схема 3.
О + zte~ = Rt. (первая ступень) '
Ri + z2e— “Ra ] , ’ ' .
„ „ ? (вторая ступень)-
О + (z, + zs) = Rs J
Rs + Zje— = Rj ) 1
„ , .. , . . ) (третья ступень)
O + (z, +zj + zs)e- = Rs J
Теория хронопотенциометрии для такого процесса
рассматривалась в работе [68]. Предполагалось, что
процесс протекает в условиях линейной полубеско-
нечной диффузии и что доставка веществ (О, Ri, R2,
Rs) к электроду подчиняется закону диффузии Фика
(условия, обычные для хронопотенциометрии). Пере-
ходное врейя Для первой и второй ступеней описы-
вается, соответственно, уравнениями (2-10) и (2-54).
Для третьей ступени получается следующее соот-
52
йоШейие, описывающее распределение перечисленных
выше веществ у поверхности электрода:
(г, + г2 + z3) Cq D* - 2lW*F =
== (21 + z2 + z3) Со (0, t) D% + (z2 + z3) CRj (0, t) D# +.
+ z3CR!(0,r)D^ (2-61)
Выражение для переходного времени третьей сту-
пени получают, приравнивая нулю правую Часть
уравнения (2-61):
. т* - (z, + z2 + z3) FC°O (D^/21 (2-62)
Это соотношение является аналогичным случаю од-
ноступенчатой реакции, протекающей с участием та-
кого же числа электронов. Оно не зависит от количе-
ства или характера промежуточных ступеней и может
служить характеристикой концентрации вещества О
в растворе.
Из уравнения (2-62) как частные случаи [68]
можно получить выражения для переходного времени
первой и второй ступеней соответственно:
= z1FCq (пДф^’/Эг (первая ступень) (2-63)
(Т1 + т2)1/’ “ (г1 + %) Fco (nDo)ll‘l2i (вторая ступень) (2-64)
В случае многоступенчатых процессов переходное
время каждой следующей ступени непропорционально
больше переходного времени предшествующей сту-
пени. Например, при m-ступенчатом восстановлении,
если в каждой ступени участвует одно и то же число
электронов, величины переходных времен относятся
как .1:4 :9 ... т2.
Уравнения хронопотенциограммы (<р — /-кривых)
для многоступенчатых процессов очень сложны и мо-
гут -быть получены; например для обратимых про-
цессов, комбинированием уравнения Нернста (1-1)
со следующим соотношением:
c°od% == со (0, о + cRi (0,0 +
+ CR (0, n D& + CR (0, о 2# + .. • + Ср (0, О D'b (2-65)
53
Это уравнение получено в предположении, что сумма'
потоков веществ у поверхности электрода равна
нулю.
Уравнения хронопотенциограмм, выведенные этим
способом, являются общими для обратимых систем
и не зависят от потенциалов отдельных ступеней.
Получение уравнений <р — /-кривых для необрати-
мых электродных процессов является более сложной
задачей. В этом случае плотность тока для второй
ступени может быть определена по уравнению, пред-
ложенному в работе [2]. Такое же соотношение со-
храняется для доли тока, относящейся к третьей сту-
пени. Сочетанием этих соотношений с уравнением
теории замедленного разряда — ионизации и уравне-
ниями (2-61) или (2-65) получают уравнения хроно-
потенциограмм для многоступенчатых необратимых
процессов. Л
2.6. ХРОНОПОТЕНЦИОМЕТРИЯ ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
С УЧАСТИЕМ НЕСКОЛЬКИХ ДЕПОЛЯРИЗАТОРОВ
В исследуемом растворе часто присутствует не-
сколько деполяризаторов, которые могут восстанав-
ливаться по схеме:
01 4- Zie~ = Ri (первая ступень)
0S -|- 22б— = Rs ) , .
— г г (вторая ступень)
О! + Zie~ = R, J
Если окислительно-восстановительные потенциалы
деполяризаторов различны, то на хронопотенцио-
грамме будет несколько волн. Переходное время для
первой ступени рассчитывается по уравнению (2-10).
Потенциал рабочего электрода после достижения пе-
реходного времени Ti сдвигается до потенциала, при
котором восстанавливается 02. Однако вещество Oi
продолжает диффундировать к электроду и восста-
навливаться на нем. В результате постоянный ток,
налагаемый на электрод, расходуется одновременно
на электровосстановление Oi и 02. Переходное время
т2 для второй волны хронопотенциограммы до-
стигается, когда концентрация 02 на поверхности
электрода станет равной нулю, но поскольку проис-
64
ходит одновременное восстановление Oi и О2, то т2
нельзя рассчитывать по уравнению (2-10).
Теория хронопотенциОметрии для процессов элек-
тровосстановления (электроокисления) двухкомпо-
нентных систем в условиях линейной диффузии рас-
сматривалась в работах [2, 70], а многокомпонент-
ных— в работах [21, 52].
Зависимость концентрации компонента О2 на по-
верхности электрода от времени электролиза имеет
вид:
Со, (0, П = С°01 - [fr + - т?] (2-66)
где
/'==/ — Т|.
Уравнение для переходного времени т2 второго
компонента находят из соотношения (2-66) при ус-
ловии Со, (0, т2) = 0:
„ n,3zr.FD'JjC(>
(Ь + *2) '* ~ =----~2i ~~ (2-67)
Подставляя в соотношение (2-67) значение т из урав-
нения (2-10), получим следующее уравнение для пе-
реходного времени второго компонента:
т2 = + '4Do ,С°о,) (2-68)
Отношение t2/ti описывается уравнением:
v-2(t-)+(5-)! (2'а"
Из уравнения (2-67) следует, что график в коорди-
натах [(Т] + т2)'/г — т’/!] — С£а представляет собой
прямую линию. Эту закономерность можно исполь-
зовать для определения концентрации второго ком-
понента (Со,)-
Из уравнения (2-68) видно, что величина пере-
ходного времени т2 второго компонента, восстанав-
ливающегося при более отрицательных ..потенциалах,
зависит от концентрации более электроположитель-
ного компонента. Степень этой зависимости опреде-
ляется концентрацией вещества Oi и его физико-хи-
мическими характеристиками. (zi, До,).
55
Из уравнения (2-69) видно, что переходное врем»
Т2 второго компонента при совместном восстановлен
нии двух веществ больше, чем при восстановлении-
одного этого компонента
[уравнение (2-10)].
Это
позволяет повысить чувствительность определения
элемента, восстанавливающегося при более отрица-
тельном потенциале и в присутствии более электро-
положительного элемента (или окисляющегося при
более положительном потенциале и в присутствии
более электроотрицательного элемента). Регулируя
платность тока после достижения переходного вре-
мени Xi, можно изменять величину т2- В этом случае,
принимая' во вниманйе изменение плотности тока
после достижения ti, уравнение (2-67) запишется
в следующем виде:
n^FD'JiC^.
----°' °' (2-70)
Авторы работы [2] на примере процессов электро-
восстановления Cd2+ и Zn2+ экспериментально под-
твердили правильность уравнения (2-70).
В работе [21] развита обобщенная теория хроно-
потенциометрии обратимого электровосстановления
многокомпонентной системы. Уравнения для концен-
траций у поверхности электрода имеют вид:
где
Cot(0,n^C?>f + zO/(n
/ро YA
CRt <°> — сяг - Гр"11 (О
_ /Л fRtklCR{ - fOiCO{
-------zp у/,
^“(fo/R^^R^O^^Pdi)
' ?г,== (^Г)(<р-<₽1/1Л)
(2-71)
(2-72)
(2-73)
(2-74)
(2-75)
(2-76)
56
Разность переходных времен двух компонентов
описывается соотношением:
nhz,FD'k cl
Т? “ T/L, ------(2-77)
i
гле T,-”XTi
г-1
Из соотношения (2-77) можно получить уравнения
(2-10) и (2-67).
Рис. 2-10. Хронопотенцио-
грамма для процесса электро-
восстановления смеси ионов
свинца, кадмия и цинка
в 0,1 М NaNO3 [52].
Электрод сравнения—ртуть. Кон-
центрация иоиов 1 • 10-’ моль/л.;
кривая 1—1=4,50-Ю-5 А/см*;
кривая 5-1=3,50- 10"'* А/см*.
/
Уравнение хронопотенциограммы для обратимого
электродного процесса имеёт вид:
rt ту* - fV’ ч
(2-78)
Потенциал <p/j наблюдается при времени электролиза
(2-79)
На рис. 2-Ю приведена хронбпотенциограмма
электровосстановления ионов свинца, кадмия и цинка,
находящихся в растворе в одинаковой концентрации
[52]. Из рисунка видно, что при равных концентра-
циях переходное время для элементов, восстанавли-
вающихся при более отрицательных потенциалах,
57
больше, чем для элементов, восстанавливающихся
при более положительных потенциалах, и что соотно-
шение между Xi и т«-1 подчиняется уравнению (2-77).
Рассмотренные соотношения описывают процессы
эдрктровосстановления. Они будут справедливы и для
процессов электроокисления, только в них нужно за-
менить индекс О индексом R.
Электровосстановление смеси двух деполяризато-
ров при постоянном токе в условиях цилиндрической
полубесконечной диффузии изучалось в работах
[57, 71].
Для второй ступени переходное время выражается
уравнением:
I (ti + т2)7’ —
+
L 4г0 4гр 32rg J
______________________________________________________
Г - . DMT-+t2) _ 3”'/<D&(Tl+T2)Vi
L 4r0 4r% 32^ ’ J
(2-80)
В знаменателе уравнения (2-80) все члены, кроме
единицы, учитывают цилиндричность диффузионного
поля, причем эффект цилиндричности будет тем
больше, чем больше коэффициент диффузии, меньше
радиус электрода и больше переходное время.
При наличии в растворе т деполяризаторов бу-
дет справедливо соотношение:
Для цилиндрической диффузии отношение между ве-
личинами переходных времен первой и второй ступе-
58
ней описывается уравнением:
Тг
Т1
PR*1
4г0 4г0
1+т2)1Д *Ш+т2)
о- 4г0
Зл^Л^’т,7’ + ... I2
-I .
3^(г1+т2)7. I2
32г® +]
(2-82)
Из сравнения отношений t2/ti для плоского [урав-
нение (2-69)1 и цилиндрического [уравнение (2-82)]
электродов следует, что t2/ti для цилиндрического
электрода больше, чем для плоского. Для хронопо-
тенциометрии с цилиндрическим электродом отноше-
ние t2/ti растет с увеличением переходного времени,
в то время как для хронопотенциометрии с плоским
электродом оно является постоянным.
2.7. ВЛИЯНИЕ МИГРАЦИИ
И КОНВЕКЦИИ НА ПЕРЕХОДНОЕ ВРЕМЯ
При большом избытке ионов индифферентного
электролита по сравнению с концентрацией деполя-
ризатора (не менее чем в 100 раз) преобладающая
часть тока в растворе переносится ионами индиффе-
рентного электролита. Это наблюдается при низких
плотностях тока, при высоких плотностях часть тока
будет переноситься за счет миграции. Перенос тока
за счет миграции осуществляется и в том случае,
когда концентрация индифферентного электролита
сопоставима с концентрацией деполяризатора: При
наличии миграции переходное время будет больше,
чем при ее отсутствии.
Влияние миграции на переходное время было наи-
более подробно рассмотрено в [72—75].
На величину переходного времени в хронопотен-
циометрии, помимо диффузии и миграции,' может
оказывать влияние и естественная конвекция. Оно про-
является, когда массоперенос за время, равное т, вы-
ходит за пределы диффузионного слоя. Неопределен-
ность толщины диффузионного слоя приводит к тому,
59
что в каждом конкретном случае опытным путем»
нужно подбирать плотность тока, при которой отсут-!
ствует влияние конвективной массопередачи. Иссле-Я
дования по учету влияния конвекции на переходное!
время были выполнены в [26, 64, 76—82]. 1
» В работе [79] получено следующее уравнение для!
переходного времени в условиях линейной диффузии!
с учетом конвекции: !
" zFCnV^o 0,51 / DQn 1
----9Л О- - 1-----л / —2— (о«тз (2-83)!
2i‘Vt 16 V v J
Второй член правой части этого~уравнения учитывает!
влияние конвекции на доставку (отвод) вещества к!
плоскому электроду. Соотношение между переходны-!
ми временами на плоском электроде в отсутствие!
(Тпл) и при наличии (тк) конвекции определяется!
уравнением: 1
/ тТ - / / б \2 |
а/-^ == 1—0,24 А /—Я— = 1 -0,0851 — ] 4
V тк V во kW I
где бх — толщина слоя, в котором наблюдается изменение кон-Я
центрации деполяризатора при прохождении тока. 1
При 6х->6о конвекция приводит к завышенным 4
значениям тк по сравнению с тПл, этого не наблю- ’=
дается при = 0. Практически тпл/тк > 0,99 при
6х/б0 < 0,39. !
2.8. ВЛИЯНИЕ ЗАРЯЖЕНИЯ ДВОЙНОГО
ЭЛЕКТРИЧЕСКОГО СЛОЯ НА ХРОНОПОТЕНЦИОГРАММЫ
При изменении потенциала электрода во времени
некоторая часть тока затрачивается на заряжение
или разряжение двойного электрическот^/слоя. Это
вызывает искажение <р — /-кривых.
Рассмотрим влияние заряжения двойного электри-
ческого слоя на хронопотенциограммы при следую-
щих предположениях:
1) выполняются условия линейной полубесконеч-
ной диффузии,
2) отсутствует специфическая адсорбция деполя-
ризатора,
3) заряжение двойного электрического слоя (для
идеально поляризуемого электрода) и дифференци-
60
(2-85)
(2-86)
альная емкость двойного электрического слоя одина-
ковы при различных потенциалах *.
Плотность поляризующего тока равна сумме плот-
ностей фарадеевского и емкостного токов:
» = if + ie (2-84)
Если емкость двойного электрического слоя в изу-
чаемом интервале потенциалов равна Са, то
, _r rf(-n)
*e—cd dt
где т) — перенапряжение.
Для вывода уравнения <р—-/-кривой необходимо
сначала получить уравнения, описывающие измене-
ние концентрации деполяризатора, используя урав-
нения второго закона Фика со следующим граничным
условием:
5СО1 </(-,))
Do~dT\x-0 l~Cd~dT~
Влияние емкостного тока на хронрпотенциометри-
ческие кривые рассматривалось рядом авторов [83—
86]. Роджерс и Мейтес [85] использовали числовое
решение диффузионных уравнений Фика. Де Вриз
[83], Ольмстед и Никольсон [84] и Драчка [86] по-
лучили решение диффузионных уравнений- с учетом
емкостного тока в виде интегральных уравнений, ко-
торые приводились к числовому виду и решались на
электронно-вычислительной машине.
Для диффузионно-контролируемых процессов урав-
нение <р — /-кривой имеет вид [84]: '
'«)*
1 — ехр (у — Ь)
1 + ехр (у)
(2-87)
о
где . , ___
zF 4i2t С°о ГБ7
«--^(ф-ф^ '
t _ iRTiCd
С° zF(zF(?o^/kD^
(2-88)
(2-89)
* Фактически с изменением потенциала емкость
слоя изменяется, но мы это не рассматриваем.
двойного
61
Искажающее влияние емкости двойного электри-
ческого слоя на <р — ^-кривые характеризуется пара-
метром kc. На рис. 2-11 приведены <р — ^-кривые, рас-
считанные при различных значениях параметра kc.
Изменение доли фарадеевского тока во времени
иллюстрирует рис. 2.12. Согласно расчетам [84],
Рис. 2-11. Теоретические хронопотенциомеТрические кривые для
диффузионно-контролируемого процесса прн различных значе-
ниях kc [84]:
/—0; 2—0,03; 3—0,06.
Пунктиром обозначены реверсные кривые. Значения точек Л и В поясняются
в тексте.
Рнс. 2-12. Изменение доли фарадеевского тока при значе-
ниях kc [84]:
1—0; 2—0,033; 8—0,06. Пунктиром показано изменение поли фарадеевского
тока в условиях реверса тока.
искажение хронопотенциограмм наблюдается, если
параметр kc >= 0,001,
$2
Проверка правильности рассмотренной теоретиче-
ской модели была предпринята в [87]. Результаты
проверки представлены на рис. 2-13 в виде зависимо-
стей х(т/) [ту— рассчитанное (исправленное) значе-
ние переходного времени] от безразмерного пара-
метра kc при различных значениях потенциалов меж-
Рнс. S-13. Зависимость «приведенных» значений переходного вре-
мени в безразмерном виде от параметра kc при различных Л<р- [87];
/—400; 2—360; 3—320; 4—280; 5—240 мВ. Точки соответствуют эксперимен-
тальным значениям.
Рнс. 2-14. Теоретические хронопотенцнограммы для.необратимого
электродного процесса (а =5) при различных значениях пара-
метра
1—0,001; 2—0,01; 3—0,0316; 4—0,1; 5—0,316. Значение Х=1 соответствует
переходному времени при отсутствии влияния заряжения двойного электри-
ческого слоя [86].
ду точками А и В (см. рис. 2-11, кривая 3). Соответ-
ствие между теоретическими и экспериментальными
результатами хорошее, но при высоких плотностях
тока (kc 0,105) наблюдаются отклонения, обуслов-
ленные непостоянством Са при изменении потен-
циала.
63
Для необратимых электродных процессов <р — /-
кривая описывается следующим уравнением [86]: (
*н -1 - [1 - + -§ ka (-^- -7== dgl е^"0*
, z J dx 'fjx — В J
(2-90)
kB = kc/a (2-92)
Хронопотенциометрические кривые, рассчитанные
при различных значениях параметра k„, приведены
на рис. 2-14. Из рисунка видно, что искажающее влия-
ние заряжения двойного электрического слоя мало,
если kB не превышает 0,001. Делая оценку при усло-
виях Со>10~6 моль/см3, —5,35 • 10“3 Ас%/см2,
Cd = 20 мкФ/см2, Do— Ю-5 см2/с, z = 2 и а = 0,5,
получим, что искажающее влияние емкости на хроно-
потенциограмму мало при 1 с.
2.9. УЧЕТ ВЛИЯНИЯ ЗАРЯЖЕНИЯ ДВОЙНОГО
ЭЛЕКТРИЧЕСКОГО СЛОЯ НА ПЕРЕХОДНОЕ ВРЕМЯ.
ОПРЕДЕЛЕНИЕ ДИФФЕРЕНЦИАЛЬНОЙ ЕМКОСТИ
Чувствительность хронопотенциометрического ме-
тода определяется долей тока, расходуемого на заря-
жение двойного электрического слоя.
При приближенном учете влияния заряжения
двойного электрического слоя на величйну переход-
ного времени можно использовать соотношение (2-84).
Совместное решение уравнений (2-10) и (2-85) дает
уравнение [51]:
(2.м)
dt 2х1г
Согласно Гирсту [87]:
о/У (2'94)
64
где Qc — количество электричества, расходуемое на заряжение
двойного электрического слоя.
Подставляя (2-94) в (2-93), получим:
(2-95)
где
kR=^n'llzFD^l2
Из формулы (2-95) следует, что величину Qc
можно найти экстраполяцией прямолинейной зависи-
мости /т — т,/а до tVi —0. Из формул (2-93) и (2-94)
следует еще один вывод: при наличии влияния заря-
жения двойного слоя произведение гс,/а не остается
постоянным, а увеличивается при возрастании i:
На использовании соотношений (2-93) и (2-94)
основан графический метод определения исправлен-
ного значения переходного времени т/, предложенный
Драчкой [86] (см. разд. 5.1).
Согласно сделанной в работе [86] оценке, откло-
нения в определении //т,/а от значений ix'11, рассчитан-
ных при отсутствии влияния емкостного тока, не пре-
вышают 2,5% при kK — 1 и if/i — 0,35. Этот прием
позволяет использовать в исследованиях достаточно
высокие плотности тока — до 38 А/см2.
Устранить влияние емкостного тока на <р—Z-кри-
вые можно, по-видимому, только путем компенсации
его. В работе [88] описана приставка для компенса-
ции емкостного тока. Компенсация емкостного тока
дает возможность снизить минимально определяемую
концентрацию деполяризатора до 10~6 моль/л и ме-
нее. При этом повышается точность анализа — ошиб-
ка в определении хронопотенциометрической констан-
ты, как показано на примере восстановления РЬ2+ и
Т1+, снижается и не превышает 1,5% в интервале
концентраций от 10~5 до 10-3 моль/л [88].
По начальному участку ф— Z-кривой или по кри-
вым переключения при мгновенном изменении плот-
ности тока от 11 до /г можно определять дифферен-
циальную емкость двойного электрического слоя.
Из уравнений (2-84) и (2-85) находим:
3 Зак, 1072
65
В начальный момент времени if = О и весь t
расходуется на заряжение двойного слоя. Опредед
угол наклона кривой — можно найти вед
чину Cd [89—91].
При изменении плотности поляризующего тока
Д/ в области тафелевского участка <р — /-кривой
первый момент времени окажется верным соотнон
ние Cdd(—r])/dt = Дг, из которого также мож
рассчитать емкость двойного электрического слоя.
Г л а в a 3
НЕКОТОРЫЕ РАЗНОВИДНОСТИ
ХРОНОПОТЕНЦИОМЕТРИЧЕСКОГО МЕТОДА
Хронопотенциометрия с постоянным током не на-
ходит столь широкого распространения в аналитиче-
ской практике, как, например, полярография. Это
обусловлено отсутствием прямой зависимости между
величиной переходного времени и концентрацией
определяемого вещества в растворе. Были предло-
жены варианты хронопотенциометрии с другими фор-
мами тока.
Производная хронопотенциометрия и хронопотен-
циометрия с переменным током дали возможность
более точно измерять величину переходного времени.
Хронопотенциометрия с реверсом тока и многоцикли-
ческая хронопотенцйометрия позволили расширить
возможности хронопотеициометрического метода для
изучения кинетики электродных процессов.
В настоящей главе рассматриваются некоторые
разновидности хронбпотенциометрического метода,
представляющие наибольший интерес для аналити-
ческих целей.
3.1. ХРОНОПОТЕНЦИОМЕТРИЯ
С ПРОГРАММИРОВАННЫМ ТОКОМ
Теория хронопотенциометрии с током,- изменяю-
щимся по закону I — pta (р — коэффициент пропор-
циональности; а — любое число больше —1), для
процесса О ± ze~ — R, протекающего в условиях ли-
нейной полубесконечной диффузии, рассмотрена в
[92].
Уравнения, описывающие распределение кон-
центраций у поверхности электрода в любой момент
з*
67
времени, имеют вид [92]: ср<о.о-с°о- ^;иг<°+» zfojT (« + /,) л RV R ZFDV’r(a + 3/2) где Г — гамма-функция [92]. (3-1) (3-2)
Рис. 3-1. Зависимость
Г (а + 3/Я)/Г (а + 1) от
а [83].
(3-3)
[92]:
(3-4)
Уравнение для переходного времени получится при
условии Со (0, t) = 0:
- ^С°0Г(а + 8/а)
рГ(а + 1)
Характер зависимости отношения от вели-
чины а показан на рис. 3-1, из которого следует, что
с возрастанием а это отношение увеличивается.
Уравнения хронопотенциограмм имеют вид
для обратимых процессов
ЯТ та+’/« _ ^+‘/1
+ ^г'"—
для необратимых процессов
RT fefr(a+l) RT td+1/i - ta+'/‘
<B = —--In —ТГ--------1-----In-----—------
azaF Я$Г(а + 72) azaF ta+'^
Наибольший интерес представляет случай,
а = 1/2. Уравнения (3-1)—(3-3) приводятся соответ-
(3-5)
когда
68
ственно к виду [92] г
(3-6)
V>.»-c"R+-^ д/i
2zFD$C°o
рл"
(3-7)
(3-8)
Из уравнения (3-8) следует, что в хронопотенциомет-
рии с током, меняющимся по закону I = суще-
ствует прямолинейная зависимость между переход-
ным временем и концентрацией деполяризатора в
растворе. Этот вариант хронопотенциометрии рас-
сматривался в работах [21, 93—100].
Уравнение хронопотенциограммы для обратимого
процесса с участием растворимых веществ (О и R)
имеет вид:
ч> = <₽•/„ + in * { - (3-9)
Если окисленная форма вещества нерастворима, то
уравнение хронопотенциограммы запишется следую-
щим образом [95] :•
<р;=<1,0 + 4г1просо-(1-т)] (3‘10)
где fo и С°о—коэффициент активности и концентрация веще-
ства О.
Для необратимого электродного процесса с уча-
стием растворимого деполяризатора уравнение хроно-
потенциограммы имеет вид [95]:
(3-11)
Теория хронопотенциометрии с током, изменяю-
щимся по закону I — pta, для процесса О ± zer = R
в условиях сферической и цилиндрической диффузии
рассматривалась в [96]. Распределение концентрации
69
деполяризатора у поверхности сферического и ци-
линдрического электродов описывается уравнениями:
(3-12)
(3-13)
Соотношения, устанавливающие связь переходного
времени с различными параметрами, имеют соответ-
ственно следующий вид:
Из уравнений (3-14) и (3-15) следует, что для хроно-
потенциометрии с током, меняющимся по закону
I == pta, в условиях сферической и цилиндрической
диффузии, так же как и для хронопотенциометрии с
постоянным током, прямолинейная связь между кон-
центрацией и величиной переходного времени отсут-
ствует.
Теория хронопотенциометрии с программирован-
ным током, на плоском электроде при наличии в ис-
следуемом растворе нескольких деполяризаторов, вос-
станавливающихся при различных потенциалах по
схеме
О] ± ze~ = Rj
............ } (3-16)
Om ± ze~ =* Rm '
70
рассматривалась в работе [92]. Уравнение для кон-
центрации деполяризатора у поверхности электрода
имеет вид:
т т 4-V
£ Vfc,(<M) = £ ZmDym т + П (3.17)
т—1. 2 т=1, 2
Знаки минус и плюс относятся соответственно к окис-
ленной и восстановленной формам деполяризатора.
Уравнение для переходного времени при электровос-
становлении любого компонента получится из условия
Ст(0, 0=0:
[т -io+Yj ~ т
Ё Ч - 7/&W Ё (з-,в)
т—1.2 J т=1,2
Из (3-17) можно записать соотношение:
РГ(а-Н)
(3-19)
Из уравнения (3-18) следует, что в хронопотенцио-
метрии с током, изменяющимся по закону I = pta
(кроме случая а = 1/2), так же, как и в обычной хро-
нопотенциометрии, переходное время элемента, вое- _
станавливающегося при более электроотрицательном
потенциале, зависит от концентрации элемента, вос-
станавливающегося при более электроположительном
потенциале.
При а = 1/2 [97] для процесса (3-16) концентра-
ция деполяризатора у поверхности электрода опре-
деляется уравнением: ____
С (0 П = =F р- - А /-Л- (3-20)
(и> Ч 2zmF У Dm
Знаки минус и плюс относятся соответственно к
окисленной и восстановленной формам деполяриза-
тора. Уравнение для переходного времени в этом слу-
чае имеет вид:
2zmFD*C°m
Хт~ .рл*
(3-21)
71
Соотношение между величинами переходных времен
двух компонентов, восстанавливающихся при различ-
ных потенциалах, запишется следующим образом:
• т 2 г№
1т—1 ’-'zn—lzm— 1
т “ C°z D',! 3’22
ЕСЛИ Сщ— l==Cm» Z/n—i=2!m И Dm—l==Dm, TO
t. e. Tm = Tm-i, в то время как в обычной хронопо- <
тенциометрии (i = const) тт = Зтт-ь
Для процесса типа:
01 ± zic = 02 )
02. ± z2e_ = 02 J
(3-23)
для хронопотенциометрии с а — 1/2 распределение
концентрации компонента, восстанавливающегося
(окисляющегося) первым у поверхности электрода,
описывается уравнениями (3-6) и (3-7). Величина
переходного времени определяется уравнение^ (3-8).
Концентрации компонентов О2 и 03 описываются
уравнениями:
с.(0,0-ф 1’,_° (3’24)
(3-25)
Соотношение Нереходных времен процессов (3-23)
определяется равенством:
Ti zi^i^i
т2 z2C“d2
(3-26)
В работе [92] приводится сравнение хронопотен-
циометрических методов с током, меняющимся по за-
кону I — pta, при различных значениях а. Показано,
что хронопотенциометрия с большим значением а
удобна, поскольку можно легко подобрать величину
тока, при которой проводят аналитический контроль
(например хронопотенциометрией с током, изменяю-
щимся по закону I = pfA). Эта рекомендация основана
на том, что с увеличением а переходное время умень-
шается. Выполнив предварительные опыты с примене-
нием хронопотенциометрии с большим а (при этом
величина переходного времени определяется менее
72
точно, чем при малых а, например при а = ’/2).
ориентировочно оценивают величину тока, которую
следует применять в хронопотенциометрии с а = '/2,
для того, чтобы уже при выполнении точного ана-
лиза получались приемлемые величины переходного
времени.
Чем меньше а и чем больше переходное время, тем
меньше ошибка в определении величины переходного
времени и тем меньше искажаются хронопотенцио-
граммы за счет заряжения двойного слоя.
Теория хронопотенциометрии в условия^ линейной
диффуаии с током, изменяющимся по закону I = аеы,
рассмотрена в [98]; в работе [99] рассмотрена тео-
рия хронопотенциометрии с током, изменяющимся по
закону I\= I при 0 < t < ti и I2 = I + Д/ при
t\<t <. t2. Однако эти виды хронбпотенциометрии не
имеют каких-либо преимуществ в аналитической
практике по сравнению с обычной хронопотенциомет-
рией (/ = const во всем интервале времени).
На вращающемся дисковом электроде изучалась
только хронопотенциометрия с программированным
током, изменяющимся по закону I =? р/ [67]. Хроно-
потенциограммы в этом случае являются нестацио-
нарными поляризационными кривыми. Нестационар-
ный предельный ток при данной концентрации опре-
деляется скоростью вращения диска и скоростью
нарастания плотности тока или скоростью съемки по-
ляризационной кривой. Выражение, устанавливающее
связь между нестационарным предельным током /Пр,
переходным временем и скоростью нарастания тока
Р, имеет вид:
При £>от/6о < 1,65 выражение (3-27) приво-
дится к уравнению для переходного времени
73
в неперемешиваемом электролите: -JH
₽т=(-|-2ГС&)’\лР0₽),'« (а!
Если Dot/So > 1,65* то I
1,С7рт = /пр (al
3.2. ПЕРЕМЕННОТОКОВАЯ ХРОНОПОТЕНЦИОМЕ nJ
Теория переменнотоковой хронопотенциометрииЯ
условиях линейной диффузии рассматривалась !
(100—102]. В этом виде хронопотенциометрии на м
стоянный ток накладывается переменный ток. Урл|
нение для концентрации деполяризатора (раствор
мого в электролите) у поверхности электрода име$
вид [100]: $
1 Г 2/ л'!г А Г л Я
Со(0. Г) = С°Ч=---п- —~ + —n-sinl <в/------I (3-Л
° zFSD11 L п1г со Л к 4 7 J "
где А — амплитуда переменного тока; со — угловая частота перс
менного тока; Id — постоянная составляющая тока; минус и плм
относятся соответственно к окисленной и восстановленной фо^
мам деполяризатора в пропессе электровосстановления.
Уравнения хронопотенциограмм для обратимое
[100] и необратимого [101] электродных процессе;
имеют вид:
<Рпб? = Ч>2бр
КТ
zF
(3-31)
2 У® (Ут — У / )
I + A sin со/
необр = „необр > JRT_
*п. т "г azp
(3-32) «
где
' . Ая,а
k=-------п-
2tdco'’
Л кт 2knf (Ут - У/ )
«рнеобр ------|П .. м
azF
]
I
(3-33)
(3-34)
74
Уравнения (3-33) и (3-34) являются уравнениями
хронопотенциограмм в обычной хронопотенциометрии
[см. уравнения (2-13) и (2-17)]. Следовательно,
уравнения для переменнотоковой хронопотенциомет-
рии состоят из двух частей, соответствующих постоян-
ной составляющей тока и переменной составляющей
тока.
Рис. 3-2. Обычные и переменнотоковые хронопотенциограммы [97]:
а-1,2-10-3 М CdSO4 в 0,2 М КС1; б-1,2-10-3Л1 CdSO4 и 0,4-10-3М
ZnSO4 в 0,2 М КНз и NH4C1. Остальные обозначения приведены в тексте.
Величина переходного времени определяется та-
ким же уравнением, как и в обычной хронопотенцио-
метрии [уравнение (2-10)]. Из уравнения (3-32)
видно, что график в координатах
А л/п sin 1га/-—
I_________________L-
<г,необр_ ,п 2-Ую (Ут -7/ )
i + A sin о/
представляет собой прямую, по наклону которой
можно определять величину аг.
Обычные и переменнотоковые хронопотенцио-
граммы представлены на рис. 3-2. На^ нем обычные
75
хронопотенциограммы обозначены цифрой 1, а пере-
меннотоковые— цифрой 2. В исследованиях на элек-
трод накладывался постоянный ток в 150 мкА и пере-
менный ток в 15 мкА. Из рисунка видно, что на пере-
меннотоковых хронопотенциограммах в момент до-
стижения переходного времени получаются острые
пики. Последнее обстоятельство позволяет определять
величину переходного времени в переменнотоковой
хронопотенциометрии значительно точнее, чем это
можно сделать в обычной хронопотенциометрии, что
очень важно в аналитических и физико-химических
исследованиях.
Если восстановленная форма деполяризатора не
растворяется в электролите и ее активность на элек-
троде можно принять за единицу, то уравнение хроно-
потенциограммы для обратимого электродного про-
цесса имеет вид [Ю2]:
(3-3S)
3.3. ПРОИЗВОДНАЯ ХРОНОПОТЕНЦИОМЕТРИЯ
В разд. 2.8 было показано, что искажение хроно-
потенциограмм, обусловленное влиянием емкостного
тока, вызывает определенные трудности при измере-
нии переходного времени. Эти трудности особенно
значительны, когда исследования проводятся при
малых токах и малых концентрациях деполяризатора
и при наличии в растворе нескольких электроактив-
ных веществ, имеющих близкие потенциалы восста-
новления (окисления). В значительной степени эти
трудности удается обойти, применяя производную
хронопотенциометрию, так как при достижении пере-
ходного времени на хронопотенциограмме получаются
острые перегибы (см. рис. 3-3).
Теория производной хронопотенциометрии разра-
ботана для линейной полубесконечной диффузии [23]
в предположении, что окисленная и восстановленная
формы деполяризатора растворимы в электролите.
Дифференцированием уравнения хронопотенцио-
граммы для обратимых электродных процессов (2-13)
76
получаем следующее соотношение:
rf<p = _ RT Г т'&
dt 2zF L
(3-36)
Это выражение представляет теоретическое уравнение
производной хронопотенциограммы для обратимого
электродного процесса. Для реакции окисления знак
первой производной будет положительным.'
Рис. 3-3. Обычная и произ-
водная хронопотенциограм-
мы для электровосстановле-
ния Т1+ [23]:
1—обычная хронопотенциограм-
ма для электровосстановления
6,26 • 10-3 М Т1+ в 0,1 М KNO3
на ртутном электроде (I =
=0,503 мкА); 2—производная
Хронопотеициограмма, соответ-
ствующая кривой Г, 3—произ-
водная хронопотеициограмма,
увеличенная в области экстре-
мума.
Максимум (минимум) на хронопотенциограмме
определяется приравниванием нулю второй производ-
ной по времени и решением ее относительно i:
rf2<p _ RT
dt2 2zF
(3-37)
Уравнение (3-37) будет равно нулю, если числитель
равен нулю, т. е. если — 3/2('1г — 0; отсюда tm ==
= 4/9т. Таким образом, максимум (для процесса
восстановления) или минимум (для процесса окисле-
ния) имеют место, когда время, прошедшее с начала
электролиза, точно равно 4/9 переходного времени.
Если уравнение tm =4/эТ подставить в (3-36), то
можно рассчитать величину dyldt в экстремуме:
/ d<p\ = _ 27/?Г
\ dt /экстр bzFx
(3-38)
77
или, решая относительно т, получим:
27RT 1
8zF ' (d<p/d/)3KCTp
Последнее соотношение дает удобный и точный метод
для- определения переходного времени, так как
((/ф/Л)»кстр можно точно определить по дифферен-
циальной хронопотенциограмме.
Для реакции электроокисления в уравнениях
(3-38) и (3-39) необходимо минус заменить на плюс.
Как уже упоминалось выше, ошибка в определе-
нии переходного времени для обычной хронопотен-
циометрии увеличивается с уменьшением концентра-
ции деполяризатора. В связи с этим на примере элек-
тровосстановления РЬ2+ проведено сравнительное
определение переходного времени обычной и произ-
водной хронопотенциометрией при малых концентра-
циях. Результаты эксперимента приведены в табл. 3-1
[23].
Таблица 3-1. Сравнительные данные по измерению
переходного времени в обычной н производной
хронопотенциометрнн для электровосстановления РЬ2+
при малых концентрациях
Условия опытов: индифферентный электролит 0,1 М KNO3; поверхность
ртутного стационарного электрода 1.38 см2; температура 25 °C
РЬ2+, моль/л /, мкА Обычная хроиопо- тенциометрия Производная хронопотен- циометрия
X, с с» t, с il'l* * С»
1,02 -10“3 188,1 15,69 ± 0,03 530 15,17 ±0,11 520
5,10-Ю-4 90,1 17,96 ± 0,21 542 16,81 ± 0,27 . 525
2,04 • 10~4 38,18 18,3 ± 0,1 581 16,81 ± 0,23 556
1,02-Ю-4 20,27 16,8 ±0,3 591 14,65 ± 0,12 552
4,08 • 10~6 8,45 22,8 717 15,88 ±0,09 598
•Теоретическое значение ^'/г/С0 при D = 0,98-10 6 см2-с—1 и 25 °C
составляет 535 А-с^’-см-а-л-моль“*. (В обычной хронопотенциометрнн
переходное время определялось по методу Куваны 123].)
Из табл. 3-1 видно, что при больших концентра-
циях ионов РЬ2+ экспериментальные значения 1т',21Сй
меньше теоретических. Это расхождение объясняют
78
тем, что истинное значение коэффициента диффузии
меньше того, что было принято в расчетах. Однако
с уменьшением концентрации РЬ2+ отношение [х'ЦСР
увеличивается; его значения, измеренные методом
производной хронопотеициометрии, ближе к теорети-
ческим, чем измеренные в условиях обычной хроно-
потенциометрии. Большая точность измерения т ме-
тодом производной хронопотенциометрии по сравне-
нию с обычной хронопотенциометрией обусловли-
вается тем, что влияние заряжения двойного слоя в
точке экстремума минимально. Несколько завышен-
ные экспериментальные значения ix'11!^, измеренные
методом производной хронопотенциометрии, по срав-
нению с теоретическими, объясняются тем, что вслед-
ствие заряжения двойного слоя скорость изменения
потенциала в экстремуме будет меньше, чем это
должно было бы быть при отсутствии осложнений,
обусловленных заряжением двойного слоя.
Уравнение, производной хронопотенциограммы для
необратимых электродных процессов в условиях ли-
нейной полубесконечной диффузии получится диффе-
ренцированием уравнения (2-17):
d<p RT г 1 1 .„
dt = 2azaF L A (?v* — ?Aj J
Экстремум для производной хронопотенциограммы
находится при t = 'Дт и величина <йр/«//ЭКстр дается
уравнением:
(4г) (з*41)
\ at /экстр агдгт
(для процессов электроокисления знак «минус» нуж-
но заменить на «плюс»). Это уравнение можно ис-
пользовать для определения как т, так и az0. Для.
нахождения одной из этих величин необходимо знать
другую величину. При изучении кинетики элек-
тродных процессов (нахождение aza и переходное
время можно определять методом Драчки, а вели-
чины ага и k°f рассчитывать соответственно по урав-
нениям (3-41) и (2-17). При вычислении по урав-
нению (2-17) <р(0,0) определяют методом экстрапо-
ляции графика ф — 1п[1 — (//т)7’] к t — 0.
- 79
На рис. 3-4 представлены экспериментальные и
теоретические производные хронопотенциограммы
процессов обратимого электровосстановления TI+ и
необратимого электровосстановления иодита, получен-
ные в работе [23]. Из рис. 3-4 видно, что для обра-
тимого процесса ветви производной хронопотенцио-
Рис. 3-4. Хронопотеициограмма восстановления таллия (I) (а) и
йодата таллия (б).
а. Сплошная кривая представляет центральную часть экспериментальной
производной хронопотенциограммы восстановления 6,28 • lfl~s моль/л Т1+
В 0,1 М KNOg при токе 0.480 мкА [23]. Пунктирная кривая соответствует
теоретической производной хронопотенциограмме, рассчитанной по уравне-
‘ пню (3-42) для Т=18.32 с.
6. Сплошная кривая представляет центральную часть экспериментальной
производной хронопотенциограммы для электровосстановления 4,04 • 10—3 М
нодата в 1 М КОН при токе 1,956 мкА. Пунктирная кривая соответствует
теоретической производной хронопотенциограмме, рассчитанной по уравне-
нию (3-47) для т=23,70 с н аг =1,00.
граммы симметричны, а для необратимого процесса —
несимметричны, что может служить критерием обра-
тимости процесса.
Теория производной хронопотенциометрии для
многокомпонентных обратимых электродных систем
разработана в [103]. Дифференцируя уравнение
(2-78) хронопотенциограммы для /-го компонента,
получим:
(3-42)
§9
Для нахождения экстремума необходимо вторую про-
изводную по времени приравнять нулю:
4.5RT/F
а (а2 — а + 1)^’ + 1
Г 1 I................ г
L 2а + (а2 - а + 1)‘/г - 1 а + (а2 - а + 1)'Л - 2
где
( zL Tfn^ i zmCO,mDO.m 2mC0. m
= 77^-4 ----------*7^---------- (3’44>
( X Xm I X 2mC0, tnPo, m S zmCO, m
\m—1 / m—I m—l
Уравнение (3.43) может быть рассчитано на ЭВМ.
Анализ, выполненный авторами работы [93] с
применением этих соотношений, показал, что при
использовании метода производной хронопотенцио-
метрии для многокомпонентных систем происходит
накопление ошибки <при определении переходного
времени для элементов, восстанавливающихся (окис-
ляющихся) последними. Поэтому при аналитических
определениях в этом случае удобнее использовать
метод добавок определяемого элемента (вещества),
а не метод калибровочного графика.
Теория производной хронопотенциометрии с про-
граммированным током, изменяющимся по закону
I = pta, рассматривалась в [104]. Основные соотно-
шения для этого вида хронопотенциометрии приведены
в табл. 3-2, их получают аналогично соотношениям
(3-37) —(3-43).
Приведенные в табл. 3-2 соотношения были рас-
считаны на ЭВМ и протабулированы в {104]. Авторы
этой работы пришли к выводу, что производная хро-
нопотенциометрия с программированным током при
а—>0 удобна для аналитических целей, а при больших
значениях а (1^а^5/г)— Для исследования кине-
тики электродных процессов.
В ряде работ [2, 92, 104, 105—108] разработана
хронопотенциометрия с реверсом тока. Этот вид хро-
попотенциометрии ценен в основном тем, что дает
Таблица 3-2. Соотношения для производной
хронопотенциометрии с программированным током
Ф = [(а4 + 4а3 + аг + а + -Х-)172 - а2 - 2а 4-1];
е-(Ч^)с+3/2<^)а+1^^Ш];
„необр = (JV+3/2Г*Т / Ф + 2а \Г+1/2 Г Г(а+1) 1.
°- KF) 2 I 1 0+З/2 I 1Г (а+3/2)Г
^фа+1/2 _фо+1/2 J
обр = (а + 3/2) Г Г(а + 1) 1. необр 1 [ Г (а 4-1) ]
а F L Г (а + 3/2) ]’ ° OF L Г (а + 3/2) J
МЛ»
п/п
Обратимые электродные
процессы
№№
п/п
Необратимые электродные
процессы
1.
2.
3.
'dm
dt
r RT (а + 1/2) та+1«2
• гр t (тп + 1/2 _ /Я+1/2)
1
t _ТГ—1 1а+1«2
«экстр- т[а + 3/2]
\ dt /экстр
Q 4-3/2
=ч=7тт[а +3/210+1/2
Фэкстр-zp 'п (а-Ы/2)
FT
C° — N°6Pp V
ь0 2а+3/2д1/2 24
Г 1 1a+1/2
L dtp (<«/)экстр J
рЛ ,0+1/2
GO~ zpi/2 1‘sKCTpJ
rf<P - T X
dt + 2azaF A
Г /0+l/2 + 2aTo+I/2
X |_ t (To+l/2 _ za+l/2)
j __________ гд,1 n + 1/2
«экстр — T )Ф)
экстр 2&zFT
Ф + 2a -|
c+3/2 I
ф o+l/2 J
L фа+1/2
RT
фэкстр=^1п
Г O' 1/2 1
+ ln [Фа+1«2 - Фс+'/2]
azaF
уунеобро
'° ыа+112 x
J 1 ]c+1/2
L (с/ф/rf/) экстр J
<“бре г, ,«
z£)1/2 Иэкетр]
c°
co
возможность изучать кинетику электродных процес-
сов, протекающих как в катодном, так и в анодном
циклах.
В последние годы успешно развивается новый ва-
риант хронопотенциометрии — инверсионная хроно-
потенциометрия' с накоплением. В этом варианте
можно использовать все разновидности тока, приме-
няемые в обычной (классической) хронопотенциомет-
рип. К настоящему времени по инверсионной хроно-
потенциометрии имеется большой материал, который
можно обобщить лишь в-отдельной монографии.
Глава 4'
ХРОНОПОТЕНЦИОМЕТРИЯ
ЭЛЕКТРОДНЫХ ПРОЦЕССОВ, ОСЛОЖНЕННЫХ
ОБЪЕМНЫМИ ХИМИЧЕСКИМИ РЕАКЦИЯМИ
Наряду с диффузией деполяризатора к реакцией- I
ной поверхности (или от нее) на изменение его кон- 1
центрации могут влиять химические реакции, проте- 1
кающие у поверхности электрода. Измеряемый в этом 1
случае сигнал несет информацию о кинетике хими- 1
ческих реакций. В случае быстрых химических реак- 1
ций* (контролируемые диффузией процессы) изме- ;|
ряется только диффузионное перенапряжение [76,109], 1
в случае медленных реакций — также и перенапря- Я
жение сопутствующих химических реакций. - j
Метод хронопотенциометрии удобен для изучения J
кинетики химических реакций. Простота математи- 1
ческой постановки задачи позволяет рассмотреть в 1
хронопотенциометрии более сложные реакционные 1
схемы, чем, например, в полярографии. Теоретические <
основы метода были изложены в работе [1]. Приме- •
нение хронопотенциометрии для изучения кинетики .
сопутствующих химических реакций рассматривается ]
в работах [3, ПО, 111]. г
Все медленные химические реакции, сопровож- ’
дающие электродный процесс, можно разделить на 1
группы: 1) предшествующие'электрохимической реак- ч
ции; 2) химические реакции с регенерацией' деполя-
ризатора (каталитические); 3) следующие за элек- • 1
трохимической реакцией; 4) химические реакции, раз- . 1
деляющие электрохимические стадии в случае ступен- I
чатых электрохимических процессов [112—114]. Ч
Большое число сравнительно- простых реакцион- 1
ных схем рассмотрел Рейнмус [ПО, с. 318]; он
предложил следующие критерии, позволяющие раз- I
личать реакционные схемы: 1) существование линей- „
* Имеется в виду, что равновесие между реагентами уста-
навливается быстро.
М
ной зависимости между логарифмом соответствующей
функции времени и потенциалом в сочетании с вели-
чиной наклона линейного участка соответствующей
кривой, 2) изменение потенциала <рт/4 в зависимости
от плотности тока, 3) характер зависимости <рт/4 от
концентрации деполяризатора в растворе, 4) отноше-
ние Т2 (переходное время после реверса тока) к ti
(переходное время до реверса тока).
Кинетику сопутствующих электродному процессу
химических реакций можно изучать, измеряя переход-
ные времена и их зависимости от параметров, ана-
логично тому как в полярографии метод измерения
предельных токов позволил изучить многие сложные
электродные процессы [43].
В этой главе мы обсудим важнейшие достижения
в теории хронопотенциометрии в области электрод-
ных процессов с объемными химическими реакциями.
4.1. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ С БЫСТРЫМИ ОБЪЕМНЫМИ
ХИМИЧЕСКИМИ РЕАКЦИЯМИ
При быстрых химических превращениях скорость
электродного процесса будет зависеть лишь от ско-
рости доставки деполяризатора к реакционной по-
верхности или скорости отвода от нее. Примеры таких
процессов и методы расчета состава комплексных
соединений в полярографии подробно рассмотрены в
[43, 76, 115] для медленных и быстрых электрохими-
ческих реакций. Эти методы расчета без изменений
можно перенести на метод хронопотенциометрии, по-
скольку потенциал в точке хронопотенциограммы, со-
ответствующей значению t — т/4, совпадает с потен-
циалом полуволны [1, с. 219].
Как известно, потенциал полуволны <₽i/2, aq в не-
комплексообразующем электролите равен [115,
с. 117]!
о RT . ПМ
К aq “ %4, aq “ Ъл*+/М + zF pV. (4-1)
где <₽мг+/м ~ стандартный потенциал; Dn — коэффициент диффу-
зии металла в ртути; Do — коэффициент диффузии ионов в рас-
творе.
85
Вследствие небольшой разницы коэффициентов 1
диффузии ионов в растворах при отсутствии комп-
лексообразования и при его наличии их полагают i
равными. Тогда в присутствии избытка лиганда сдвиг
потенциала *рт/4 при переходе от электролита без
компйексообразователя к раствору с комплексообра-
зователем, образующим стабильные комплексные сое-
динения по схеме
м*+ + X MX
мх+х MXs (4.2)
МХП_, + Х мх„
равен: j
n ' •]
Дф1Л = Дфг/4 = фх/4 — фх/4_ aq = — 1п (4-3)
1=0
где ₽, ==^-'мх /(^нг+' С*)—сУммаРная концентрационная кон-
станта устойчивости комплекса МХг; — концентрация лиганда
X, по предположению совпадающая с равновесной; Дф^з — разница
между потенциалами полярографических полуволн в отсутствие '
и при наличии комплексообразования; Дфт/4 — разница между по* 1
тенциалами четвертей хроиопотенциограмм в отсутствие и при Л
наличии комплексообразования. И
С помощью (4-3) можно рассчитать величины р, |
по кривым зависимости Д<рт/4— Сх. Часто эти расчеты J
проводятся по методу Де Форда и Хьюма [43, с. 139; j
115, с. 119]. Можно использовать и другие графиче- |
ские методы расчета [115, 116]. В последние годы .1
развиты методы расчета констант с помощью элек- <
тронно-вычислительных машин [117]. В работе [118]
приводится программа для ЭВМ на языке Фор-
тран-IV, позволяющая рассчитывать константы рав-
новесия для систем, содержащих любое число, реа-
гентов и различные формы комплексов.
Для расчетов констант р, можно использовать
величину сдвига потенциала в точке хронопотенцио-
граммы, соответствующей т/2, поскольку из [1, с. 219]
следует:
Ч>т/2 = Ф^ + 7Г1п(72 -1) (4-4)
86
Поэтому сдвиг потенциала Дфт/2 выражается форму-
лой, аналогичной (4-3):
' ДЧЧ/2 = ЧРт/2 — Фт/2. aq --1п X Р/СХ (4‘Б)
' i-0
В работе [119] рассмотрено влияние различных
видов диффузии на соотношение (4-5).
Для любого комплекса МХ/, диффундирующего к
электроду, справедливо выражение [120]:
(4-6)
где Смхг “ концентрация комплекса в объеме раствора; —
у поверхности электрода; Л = irolzFDi.
Функция f(^i) зависит от вида диффузии:
для линейной полубесконечной
((^,.) = 2Л/57/7^ (4-7)
для сферической полубесконечной диффузии
1 (frj) = 1 — exp erfc д/^7 (4-8)
где О। —D^frQ, r0 — радиус сферического электрода; Di — коэф-
фициент диффузии »-го комплекса.
Если т — переходное время, то, приняв ( = т/2, по-
лучим для <р-г/2 (полагая, что активность атомов ме-
талла не равна 1, например в амальгаме):
. RT С+’=Ч,*/2)
где (йм)г/2 — активность атомов металла в амальгаме у поверх-
ности электрода в момент t = т/2.
В присутствии комплексообразователя:
о . RT V _ Rr
фт/2 = фмг+/м+ ZF " 2j (at) zF
/-о VWt/2
N
in£₽^
i=0
(4-10)
Предположим, что 1) суммарная концентрация
ионов металла в растворах комплексообразователя и
бёз него одинакова, 2) коэффициенты диффузии всех
веществ одинаковы, 3) общий ток равен сумме со-
ez
ставляющих. Вычтя из (4-10) уравнение (4-9), полу-
чим вновь соотношение (4-5).
В [119] рассмотрены и процессы, протекающие в
отсутствие избытка лиганда. В этом случае нужно
учитывать диффузию лиганда, концентрации кото-
рого в объеме Сх и у поверхности электрода Сх
различны. Повторяя все предыдущие выкладки, мож-
но получить:
N
A<₽t/2 = --^ln£Pi(cx)'/2 <4-”>
i-0
Применив уравнение материального баланса [115,
с. 121]:
(С*х - с°х) = ID^ (CSMX[ - C^Xi) (4-12)
и положив i = p, где р— состав преобладающего в
растворе комплекса, получим при Сх С CxZ
(Сх)х/2=-^М^/2) (4-13)
Подставив (4-3) в (4-11), получим:
пУз
Дфг/2 = " I" ₽Р W (*/2)1 (4-14)
Из (4-14) следует, что в этом случае Д<рт/2 зависит от
плотности тока, и по зависимости Афт/2 — In [Xf (0/2)]
можно найти координационное число р и константу 0Р.
Для ступенчатого комплексообразования
N
Дфг/2 = - 7Г ln X ₽г W2)? (4',5)
i~0
Анализ соотношения (4-15) можно проводить ана-
логично (4-5), считая аргументом %f(O/2).
Для процесса изменения заряда вона за счет из-
менения валентного состояния центрального атома в
присутствии избытка лиганда
М% + (г-2')е- Мг'Х/ + (*-/)Х (4-1&)
88
было получено соотношение [119] :•
„ 4. SL
»«! - Тм>+/М>'+ + (г _ У) F С”„Л ± J (V+/2)
- |п (Е f‘c^Dici) <и7>
где z > z7, i = 0, 1, .... N; j = 0, 1, ... > N-
• Если в растворе присутствует преимущественно
одна форма комплекса с координационным числом р,
которое изменяется в процессе реакции на pf, то из
(4-17) следует:
0 — рг /?7*
д^д In Сх = - ~ (4-18)
Это уравнение аналогично выражению, используе-
мому в полярографии [115, с. 126].
Если р — р', т. е. изменение заряда центрального
атома не сопровождается изменением состава внут-
ренней координационной сферы, то потенциал <рт/2 не
зависит от концентрации свободного лиганда.
Электродные процессы с быстрыми объемными
химическими реакциями и замедленной стадией раз-
ряда— ионизации можно анализировать, измеряя за-
висимость тока обмена от концентрации свободного
лиганда. Многочисленные-примеры подобных иссле-
дований приведены в [76, 115, 121, 122].
Для необратимой электрохимической реакции, на-
пример „ ,
MXfe + ze- —> M + ftX (4-19)
плотность тока обмена равна:
= (4-20)
где /0 — измеряемая плотность стандартного тока обмена систе-
мы MXt/M.
Соответствующие порядки реакции по компонен-
там в (4-20) определяются из зависимостей:
(5 In i0/d In С„) = а (4-21)
(д In i0/d In Сх) = аЯ + (1 - а) д In CtAzJd In Сх (4-22)
89
Как следует из (4.22), для нахождения К нужно Изу-
чить зависимость равновесного потенциала электрода
от концентрации свободного лиганда и затем найти
его по уравнению:
Fz
* dlnCMXft/dlnCx = K+-^3<pp/5fnCx (4-23)
Использование приведенных соотношений и их
применение подробно описаны в [76, 115].
4.2. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ С МЕДЛЕННЫМИ
ПРЕДШЕСТВУЮЩИМИ ХИМИЧЕСКИМИ РЕАКЦИЯМИ
Общее рассмотрение электродных процессов с хи-
мическими реакциями в стационарных условиях при-
ведено в [26]. В методе хронопотенциометрии общий
подход разработан в применении к мономолекуляр-
ным реакциям [123].
Простейший случай — предшествующая химиче-
ская реакция первого порядка, протекающая по урав-
нению (1-10), причем предполагается, что вещество
А не восстанавливается при тех потенциалах, при ко-
торых восстанавлйвается вещество О. Вследствие
* fei ~
протекания реакции А О скорость восстановления
вещества О зависит от скорости диффузии Веществ
к поверхности электрода и скорости химической ре-
акции.
Сформулированная задача была решена в [124]
при допущении равенства всех коэффициентов диф-
фузии. Для АС = Со(0,/)—-С0(х, 0) этого процесса
будем иметь [123]:
ДС ------L- f ?.^L------+------*L_erf [(А, + kt} f]%)
I ^л (kt + ka) (k, + kt)'‘ J
(4-2 4
В момент достижения переходного времени тк
концентрация Со-»0 и АС = —С°, тогда из (4-24)
получим:
.‘/а _ TI^zFCPd''2 П!г1 . г. ,7, ..
k 2 2К (kt + kt) crf + Tfe] ^4'25^
где К = k{/k2 ==. CO/CA; C° = C°o + Cfc j = i/zF.
90
Для значения аргумента (Ai + A2)Z>2 функция
ошибок практически равна единице и зависимость
от Z выражается прямой линией согласно урав-
нению:
,л,_ n'l'zF&D'l* п'1ч
к 2 2K(ki + kJ
(4-26)
По тангенсу угла наклона этой прямой можно опре-
делить К (ki 4- k^'*. Из уравнения (4-26) видно, что
для случая, когда отсутствует предшествующая хими-
ческая реакция, в выражении для ix'fr остается лишь
первое слагаемое, т. е. = const и это произведе-
ние не зависит от плотности тока.
Обозначив ___
т'Л = nhzF(?D'/2/2l (4-27)
запишем (4-26) в виде:
=тУ'_ (4‘28)
/Д -j- «2/
Как следует из (4-28), при достаточно большом
значении К переходное время т* « т, т. е.__ кинетиче-
ский эффект не сказывается на величине переходного
времени.
Допустим, что минимальное доступное для изме- .
рения переходное время составляет 10-3 с, а измене-
ние переходного времени составляет 10% этой вели-
чины, тогда из (4-28). следует, что кинетический эф-
фект можно обнаружить, если:
К (ki + Ы7’ < 500с-% С4’29)
При больших плотностях тока (малых значениях
аргумента (Ai 4- kz)Xk) уравнение (4-25) приводится
к виду:
z. 1/Л n^zFC^D^ /Aqm
Из (4-30) видно, что предел произведения не
зависит от тока. Соотношение (4-30) позволяет вы-^
числить константу равновесия К химической реакции.
Если К» 1, то из (4-30) получаем соотношение, ха-
рактерное для процессов, не сопровождающихся ки-
нетическим эффектом [уравнение (4-27)].
91
Если К <£. 1, то из (4-30) получаем:
(,.?) (М1>
X «z/_>oo 2
j. е. переходное время определяется объемной хкон<
центрацией вещества О.
Приведенные соотношения будут справедливы и
для предшествующих реакций псевдомерного порядка,
например:
А + С O + ze- ч=* R (4-32)
A *=? O+C + ze- R (4-33)
fe2
Где C — вещество, не восстанавливающееся на электроде.
В общем случае концентрация вещества С влияет на
скорость образования электроактивной формы О.
Однако если вещество С присутствует в большом из-
бытке, то его концентрацию можно считать постоян-
ной, и тогда справедливы все приведенные выше со-
отношения с соответствующей заменой констант:
k^k'^, k2=*k2; К = К' (4-34)
(4-35)
И
A-, = Kf; k2 = k"Q, К = (4-36)
где
К"^С№>С/С°А (4-37)
Реакция типа (1-10) при неравных друг другу
коэффициентах диффузии рассмотрена в работе [125].
Уравнение для анализа кинетических данных по-
добно уравнению (4-25), только во втором слагаемом
в правой части уравнения (4-25) присутствует мно-
житель (Da/D) (DpJDo), где
D = (DA + WO)/(1 + K) (4-38)
Этот поправочный множитель, например для дис-
социации слабой кислоты в присутствии большого
избытка аниона кислоты, равен ЬН+/£>НА«3 [125].
Умножив и разделив в уравнении (4-26) второй
член правой части на и взяв обратную величину,
получим [126]:
1 2 1 '
ix'fr “ n^zFCPD'1* + zFC°DlllK (^ + k2) (4’39^
Как следует из уравнения (4-39), удобно представить
экспериментальные данные в виде графика.
1//т£ — l/т'/’.По тангенсу угла наклона прямой в этих
координатах можно определить кинетические пара-
метры. Ошибка в этом случае по расчетам, приве-
денным в [126], меньше, чем в случае использования
уравнения (4-26), и не зависит от значения константы
равновесия К. В работе [126] показано также, что
для предшествующей химической реакции можно
представлять данные и в виде графика — 1/г, но
ошибка определения этих параметров того же по-
рядка, как и при определении их по графику ix'fr — i.
В случае ступенчатой диссоциации комплексов,
например, по схеме 27]:
: CdBrJ 4=h CdBr2 CdBr+ Cd2+ (4-40)
электродному процессу восстановления Cd2+ пред-
шествует несколько химических реакций. Качественно
электродный процесс с двумя предшествующими
реакциями рассмотрен в [68], математические соот-
ношения для случая двух предшествующих химиче-
ских реакций первого порядка приведены в [128].
Если константа скорости одной реакции окажется
значительно меньше константы скорости другой реак-
ции, то для электрохимического процесса с двумя
предшествующими химическими реакциями справед-
ливы все математические соотношения, характерные
для электрохимического процесса с одной предшест-
вующей химической реакцией.
Для электродного процесса
As А2 Af+ze- R (4-41)
«23 *
общее уравнение для переходного времени очень гро-
моздко 28], этому примем упрощающее предпо-
93
92
ложение, что аргумент функции ошибок больше двух
и что kz2 "Ь fos foi + ки- Тогда:
,т»_ _______________? (М2)
k 2 2(fe21 + fe12)/jfofo
+f е=
где kt — ^3s/fos> fo = kitlkti.
Как следует из уравнения (4-42), если первая реак-
ция значительно быстрее второй, то в уравнение вхо-
дит лишь ее константа равновесия k\ и кинетические
параметры второй химической реакции. Если, кроме
того, равновесие первой химической реакции сдви-
нуто вправо (fo » 1), то (1 +fo)/fo ® 1 и уравнение
(4-42) совпадает с уравнением (4-26).
Наоборот, если равновесие первой химической
реакции сдвинуто влево (k\ << 1), то из (4-42) по-
лучим:
. у nhzF(^D'h п’ч
it? =--------------------—„ (4-43)
* 2 2(fe2t A-k^ktki
Если предположить, что kw, + fog foi + foa, то по-
лучим для случая, когда аргумент функции ошибок
больше двух [128]: .
- чг nhzFC0D',‘ __________n'hl___________
lXh~ 2 + +fo)
_ л‘/а» (fel + felfez + 1) ,4 44,
" 2(*I24-M‘/’(fo+l)fofo
Как следует из (4-44), на переходное время в этом
случае влияют и первая, и вторая химические реакции.
Рассмотрение влияния двойного электрического
слоя в теории электродных процессов с предшествую-
щей химической реакцией приведено в монографиях
-[121, с. 226, 248], [129, с. 27] и в работах [130—133].
Толщина реакционного слоя ц0 (т. е. слоя, в ко-
тором концентрация деполяризатора отличается от
объемной и деполяризатор не находится в равновесии
с неэлектроактивным веществом) может быть одного
порядка с толщиной диффузионной части двойного
электрического слоя х.
Согласно [132, 133] можно различить два пре-
дельных случая.
94
1) При у = цо/хС 1 реакция протекает в очёИь
тонком слое, в котором потенциал равен постоянной
величине фо = const — потенциалу в плоскости макси-
мального приближения реагирующей частицы к реак-
ционной поверхности.
В предположении, что теория Гун—-Чэпмена
справедлива, истинная поверхностная концентрация
должна быть связана с кажущейся поверхностной кон-
центрацией множителем Больцмана ехр[— (z0F/RT)ty0i,
и тогда для химической реакции первого порядка
[39, 134]:
k = kD exp ( — ф0) (4-45)
где z0 — заряд частицы О; k — кажущаяся константа скорости;
ko — то же, в отсутствие влияния двойного электрического слоя.
2) При у С 1 и при яофо < 0 почти все частицы
образуются за пределами двойного электрического
слоя и k -> k0.
Подробное изложение теории хронопотенциомет-
рии с предшествующей химической реакцией псевдо-
первого порядка с учетом влияния двойного электри-
ческого слоя изложено в [3, с. 319—323].
В работе [86] рассмотрено влияние двойного
электрического слоя на хронопотенциометрические
кривые для процессов с предшествующей химической
реакцией первого порядка в предположении, что про-
текающий через ячейку ток можно представить в виде
суммы фарадеевского и емкостного токов:
l=If + Cddqldt (4-46)
Для упрощения в [86] было принято, что реакция
на электроде протекает медленно, а химическая —
очень быстро, поэтому:
lf = zFkfCs0 exp [- azFy/(RT)] (4-47)
Расчет хронопотенциограммы проводился численно.
Из расчетов следует, что двойной электрический
слой по-разному влияет на диффузионный процесс и
на кинетику химической реакции и, в результате, на
первое и второе слагаемые правой части уравнения
(4-26). Поэтому поправку, учитывающую это влияние,
нужно вводить отдельно в первое и второе слагаемое
95
правой части этого уравнения. Средний диффузион-
ный фарадеевский ток обозначим 7/d, а средний ки-
нетический фарадеевский ток — 7/*.
Поправки на процесс заряжения двойного электри-
ческого слоя вводятся графически [86]. Методика
определения величины т/d показана на рис. 5-1 и
описана в разд. 5.1:
Tfd = ^fd/т (4-48)
Для введения поправки, учитывающей влияние двой-
ного электрического слоя на кинетический ток [86],
проводится касательная 8 к кривой <р—t в точке пе-
ресечения перпендикуляра 5 с кривой. Укажем, что
линия 9 проводится параллельно оси t и проходит
через точку пересечения касательной 8 и перпендику-
лярна 4. Расстояние между точками пересечения
линий 7 и 8 соответствует времени т/*, тогда:
~ lxtk!x <4'49>
Для того чтобы определить константу скорости
предшествующей химической реакции, нужно по-
строить зависимость — Ifk и определить по тан-
генсу угла наклона кинетические параметры анало-
гично тому, как это делалось при отсутствии влияния
двойного электрического слоя [86].
Предложенный метод учета емкости двойного
электрического слоя позволяет расширить возмож-
ности гальваностатического метода. Если верхний
предел измеряемых констант по [1] равен /С(А12-[-'
+ &2i) < 500' c~'h, то, по оценкам работы [86], учет
емкости двойного слоя позволяет повысить .этот пре-
дел до ~6300
4.3. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ С ПОСЛЕДУЮЩИМИ
МЕДЛЕННЫМИ ХИМИЧЕСКИМИ РЕАКЦИЯМИ
Простейший случай — последующая химическая
реакция первого порядка, протекающая по схеме:
fei
Q + ze- R A, (4-50)
к,
ев
рассмотрен в {135]. Для этого процесса величина
ДС равна [123]:
ДС = —Uf- _2fea д/Г fei erf y(fe/+ fej) f 1 ,4 5|.
л/D L Ул (fci + fe2) , (fe( + kifh J
Для диффузионного процесса было получено уравне-
ние ф — /-кривой в виде [135]!
ОТ TVi _ /V» рт
+ ------“Ь£ (4-52)
где
КГ fpD%
. <W)
£ _ 1 . К erf y<fei + M t M 54v
1+K 2 1+K. V(fet + fei) t
К <= fej/fes
Уравнение (4-53) отличается от аналогичного
уравнения для электродного процесса при отсутствии
медленной последующей реакции слагаемым
(RT/zF)\nL в левой части. С увеличением'этого сла-
гаемого кривые смещаются к положительным потен-
циалам (для процесса восстановления). Предложено
[121, с. 243] определять потенциал, в точке, соответ-
ствующей ’Дт, т. е. фт/4, который из уравнения (4-52)'
равен:
= - («S)
где . -
£(,/4)_____1 | У-К ert(y<*,4-W.P) („6)
1 + K 1 + K V(fei + fei)T
Прн высоких плотностях тока (малое т) фУ4 сов-
падает с потенциалом полуволны полярограммы. На-
против, при малых плотностях тока (большое пере-
ходное время) функция ошибок erf(x) w 1 н In Z. (т/4)
будет равен ln(l -f-K)-1. Для К 1
Фг/4 = Ф-/а+^Г1пЛ <4‘57)
Как следует из (4-57), система будет находиться' в
квазиравновесном состоянии, а потенциал полуволны
4 Зак, »072
97
смещен в положительную сторону на величину
(RT/zF)lnK.
Последующие химические реакции удобно изучать
методом, в котором направление тока изменяется в
момент времени ti. Для такого импульса [123]
дс-= Г 2fea . к* erf V(*» + _
L Vn"(^i + k2) (k.i k2) I*
h - 1» Г 2^ (t - tj'‘ , Ь erf V(fei + Ъ) (t - 6) 1 „
----- I —yjj- -------4--*........ — ——— I (4-Oo)
L *\/jt {ki + Afc) k{ + Aj J
Соответствующая этому процессу математическая
модель рассмотрена также в работе [191. Следует
отметить, что уравнение, аналогичное (4-58), можно
получить и методом аддитивных функций отклика
[69, 136].
Для быстрой последующей химической реакции из
уравнения (4-58) для t'(ki 4- k2) > 2,4 (где т' — пере-
ходное время после реверса тока) можно полу-
чить [19]:
(14-*/с')%-£/+1 +
где f = t — ti, U = i2/h.
'у/п U
ЧК (fei 4- k2) %'
(4-59)
В работе [137] приведен пример применения
уравнения (4-59) к изучению химических реакций,
следующих за электродным процессом. В работе [19]
рассмотрена хронопотенциометрия с реверсом тока
для электродного процесса, осложненного необрати-
мой последующей химической реакцией.
Из выражения (4-58) при Л-* О (необратимая хи-
мическая реакция в растворе) можно получить:
erf erf (1 + tji') ' (4-60)
Соотношение (4-60) можно записать в виде:
(ki + А2) Г' = argerf2 (“рр ) (4-61)
где t/^arg erf (2)—функция, обратная z = erf(£7); (kt + k2)ti>-
>6.
Таким образом, для достаточно высоких значений Л
значение т' не зависит от 6. Например, при U == 0,1
98
(ki'-Ук2)т? = 1,43. В работе [138] рассмотрена теория
хронопотенциометрии для процесса
О + ze~ R; mR —> Продукты
(4-62)
т. е. для электродного процесса с последующей хими-
ческой необратимой реакцией m-го порядка с кон-
стантой скорости km превращения деполяризатора R
в электрохимически неактивные продукты. Предло-
жено определять константу km методом хронопотен-
циометрии с реверсом тока в момент времени t\. Для
определения переходного времени / реакции окисле-
ния можно использовать уравнение:
^(I+m)D(,_m)/(1+m) = fm (и) (4 63)
где 1/==/2/й.
Для реакции первого порядка получается ранее
приведенное выражение (4-61), при щ>1 функция
имеет вид:
f /га ( т + 1 y/(m+I) л ,
~) 4([/+1)а +
, (m+l)(m+^(m+1)' л2
2 8(U+1)2
2
(4-64)
в частности, при т — 2 и U 1
f /гп ... Ь028 . 2-425__________1.12
М ' (t/+l)2 + (С7+1)2 (U + 1)ЗЛ*
(4-65)
Например, для V = 3 f2(U) ~ 0,069.
Находя величину Р — dlgz'/^lgii, можно вычис-
лить порядок реакции (4-62) по формуле:
(4’66)
Подробный анализ функции f2([/) и числовые расче-
ты ее для необратимой последующей реакции второго
порядка приведены в работе [139]. -
В работе [140] рассмотрена теория хронопотен-
циометрии для обратимых последующих химических
4*
99
реакций второго порядка, которые можно представить^
в виде следующих уравнений: J
Ъ ;
2R Z; K^kjk» (4-67)-
*1 •
k, ' '
- R Y+Z; K = (4-68)
кг
R *=* 2Z; K = k2lkt (4-69);;
kt
2R V* Y + Z; K = (4-70)
Kl 1
-J
2R 2Z; К = ki/kx (4-71)
«s
где R — продукт электродной реакции О + ze~ R, Y
электрохимически неактивные вещества.
Для хроиопотенциометрии с реверсом тока
чено уравнение в общем виде:
и Z —
{[(1 + <1/T,),/1 - l]/t/ - О V^== G (4-72)'
где G— функция, зависящая от констант скоростей и констант:)
равновесия химических реакций и величин плотностей токов.
В частности, для последующей реакции первого по-?
рядка при /Ст' > 6, как следует из (4-65):
G = 7я/(2К лД7+1Г) (4-73))
Для!реакций второго порядка функция G из урав-
нения (4-72) имеет следующий вид:
1) для реакции (4-67):
G = (у'3- Gy~''3) (Зл/М’/’Л (4-74)
где у = (4Z8/zf Ю (S/Dk^i у > 2,5 • 102; \у1гх' > 24;
k^y-* > 0.59G2 + 180 (172 + U) y-’/a + 1040 (l/2 + U) y~‘h
2) для реакций (4-68) и (4-69):
G = (n/feO'A (y-''‘la + 1 )/2 (4-75)
где^/гК/г/Ч^)*7’; {/<2-10-2/a2; *^'>6; k^y >0,79t/2/e2+' !
+.27 (U» + U) j у/a + 35 (t/2 + U)/y, a = 1 для реакции (4-68)
и a =2 для реакции (4-69).
3) для реакций (4-70) и (4-71):
G « у~41 (Зл/Л1К)‘л (1 + a V-KГ’ {l - 2а [К/3 (1 + а -у/к) X
Х(1+Т^)]Ч}/4а (4-76) ’
100
y = (i2/2zF)(3/DKt)'12; K<0,05/a; ^^"(I+VkVG+a V?(),/s>6;
Kkitiy1' > 0,59U2/a + 3,8(U2 + U) xfx/a + (U2 + 2U), a ='/»
для реакции (4-70) и а = 1 для реакции (4-71).
Для определения типа химической реакции необ-
ходимо изучить зависимость функции G в уравнении
(4-72) от плотности тока. Для реакции первого по-
рядка G не зависит от i2, для реакции (4-67) функция
G пропорциональна для реакций (4-68) и (4-69)
функция G ~ и для реакций (4-70) и (4-71)
G ~ »2-'А.
Последующие реакции могут протекать по не-
скольким направлениям одновременно. Например, в
работе Д141] рассмотрена теория хронопотенциомет-
рии при протекании двух параллельных последующих
реакций для случая, когда одна из них необратимая
реакция первого порядка, а вторая — необратимая
реакция второго порядка. Такой процесс протекает по
схеме:
a. k,
O±ze~ R; R —> Продукты; 2R —*• Продукты (4-77)
Решение этой задачи было получено в виде [141]
= Ь) (4-78)
где т/— переходное время после изменения направления тока в
момент времени G; b^i^zFk^D^' (4-79)
f(U, b)— функция параметров V и Ь.
При &—>0
(4-80)
а при b-> оо ,f(U,b)-+ft(U)/b* (4-81)
и в этом случае решение совпадает с рассмотренными
ранее случаями электродных процессов с одной по-
следующей реакцией.
Для условий:
(6->0) (4-82)
И
6 > бт'/fc (U) (b -> оо) (4-83)
переходное время х' не зависит от величины ti.
101
Последующая химическая реакция, особенно с
участием органических веществ, может протекать в,
несколько стадий. В простейшем случае первое пере-
ходное время после реверса тока может соответство-
вать исчерпанию деполяризатора, второе — окончанию
первой ступени последующей реакции. Если -вторая
стадия необратима и время, при котором происходит
реверс тока, достаточно велико, то переходное
времена обеих ступеней не зависят от и процесс
совпадает с описанной выше необратимой последую-
щей реакцией.
Общее решение для процесса двухступенчатой по-
следующей реакции в хронопотенцнометрии с ревер-
сом тока дано в [142]. Схема электродного процесса
имеет вид:
fel2
O±ze~ R —* At —> Продукты (4-84)
где йц и км — квазимономолекулярные константы скорости.
Предположим, что после реверса тока начинается
окисление вещества R и после достижения переход-
ного времени т' начинает реагировать на электроде
вещество Аь Переходное время т' отсчитывается от
момента реверса t\, а второе переходное время т', со-
ответствующее CSA =0, -- от момента т' + tv
Следовательно, электродная реакция имеет вид:
К tv. o±ze- R; t > tv. R?zJ- —> O' (4-85)
+ Al±ze~->0" ' (4-86)
где вещества О' и О" отличаются потенциалами, при
которых они реагируют иа электроде.
Если + ^)>6 и k23(tl + т[ + > 6, то для
первого переходного времени получаем уравнение
(4-60), которое можно записать в виде:
fei2< = argerf2W(C/+l)]==fi«/)
(4-87)
102
Для второго переходного времени уравнение
имеет сложный вид:
(U + 1) erf ft (U) (1 + т'/<) ^/W'* - 1 + (k23/kl2)’'> X
fj (V)
X J {(С/ + п ft (£/)(!+ </<) - v]*'* +
о
+ [л/i (U) (1 + </<) - л?]-1'* X
X exp [- ft (U) (1 + </<) + v] - 1} X
X exp £—(1 + kia/ku) vj lo (I ~ кгз/к1г) vj dv (4-88)
где v — переменная интегрирования.
В работе f 142] приведены выражения, полученные
из (4-88) в предельных; случаях, а также рассчитан-
ные по уравнению (4-88) отношения т2/Т; при не-
скольких значениях Л2з/^12 и U.
При выполнении неравенств:
it > Br'i/fi (t/); t2 > 6т'/ft (U) (4-89)
из уравнения (4-87) можно определить константу
^12, а затем методом итераций из (4-88) по значению
т2 подсчитать константу Л 23.
Зависимость т2/т[ от U может служить критерием
при выяснении механизмов (4-85) и (4-86), особенно
при больших значениях А23/&12. Эта зависимость
имеет асимптоты:
для (^23/^12)(^23/^12) (^2/^1) 1 (4-90)
для (k23/kl2) -> оо (k23/kl2)y! (t'/tJ) -> g! (U) (4-91)
gi (U) [U/(U + 1)1 exp [f, (L7)l [Л//1 (U)]'b (4-92)
Предельный случай (4-90) соответствует процессу, в
котором вторая стадия реакции (4-84) протекает мед-
леннее, чем первая стадия, поэтому для этого пре-
дельного случая переходное время определяется тем
же уравнением, что и для одностадийной последую-
щей реакции, т. е.
^2-^(1/) (4-93)
103
F
Зависимость от й23/й12 показываем влияние пер-
вой'стадии на переходное время второй для процесса,
в котором первая стадия реакции (4-84) является
медленной.
При малых U значения Л23т2 уменьшаются с ро-
Стом k^/kn, при больших U значение k22x2 увели-
чивается с ростом &2зД12, превышает значение
рассчитанное по равенству (4-93), достигает макси-
мума и затем уменьшается. Уменьшение £23т' отра-
жает тот факт, что концентрация вещества Ai пони-
жается при протекании реакции (4-84) во время
первой остановки потенциала после реверса тока
вследствие разряда только вещества R; уменьшение
концентрации вещества Ai тем значительнее, чем ’
больше Й23/А12, т. е. чем больше скорость второй ста-
дии реакции (4-48).
При больших U к2ях'2 > f ] (17); это объясняется
тем, что во время разряда вещества Ai вещество R
продолжает диффундировать к электроду и разря-
жаться, уменьшая поток вещества Ai и увеличивая
переходное время т'. ~ j
4.4. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ С МЕДЛЕННЫМИ ‘
КАТАЛИТИЧЕСКИМИ И ПАРАЛЛЕЛЬНЫМИ J
ХИМИЧЕСКИМИ РЕАКЦИЯМИ |
Для мономолекулярных каталитических химиче-|
ских реакций схема процесса может быть представ- 1
лена в виде: |
k,
А, А2 (4-94) *
t I ' ]
ze~
Величина АС2 для этого процесса равна [123]:
ДСа --------------- (4-95)
л/» (kt + Ы
При достижении переходного времени т*:
с°“ erf + <4-s&)
'УЬ' («1 -Г «2,
104
При ki s= 0 уравнение (4-96) совпадает с уравне-
нием, приведенным в [135]. Обозначим:
V = WA (4-97)
и введем отношение Тл/т, где -г — переходное время
для диффузионного процесса в отсутствие каталити-
ческой реакции. Это отношение будет равно:
(Tft/T)v* = 2у/д/л erf (у) (4-98) ч
При больших значениях аргумента у функция оши-
бок erf (у)-И, поэтому асимптота зависимости
y — (xkfx)l/1 от у имеет тангенс угла наклона, равный
2у/-\/л. Это значение можно использовать при у > 2.
Поскольку t/(0) = 1, то Xk — x при у->0, т. е. с уве-
личением плотности ток'а каталитический эффект
уменьшается.
Наоборот, с уменьшением плотности тока т* и у
возрастают, что можно использовать в аналитиче-
ских целях. Приведенные соотношения справедливы
для псевдомономолекулярной реакции
А( A2 + Z (4-99)
ze~
(4-100)
при болыпбм избытке вещества Z. В работе [135]
было показано, что уравнением (4-98) можно поль-
зоваться при
фСд; /-2Y-----
' z А‘ Ул erf (у)
где у = (КС^)71.
Теория хронопотенциометрии в условиях линейной
полубесконечной диффузии при сравнимых концен-
трациях веществ Аг и Z рассмотрена в [143]. Для
определения кинетических параметров авторы, полу-
чили уравнение:
1Х,г = — zFC°z ---Yn2ZZ?0\2+
2 Z'V Z zzFkfil (C° )2
. 7 ?FC<>D 7 (^>3Wo
+ -U OO/V^ И4 Z>KDZ
105
которое для не слишком малых токов можно пред-
ставить в виде
irth^N0-Pis (4-102)
ГД*
. No = | zFC° 4- zFC°oDo а/ (4-103)
* 2»
22FkxDl(c^
(4-104)
а — стехиометрический коэффициент, аналогичный введенному
ранее.
Как следует из (4-102), представляя эксперимен-
тальные данные в координатах гг%-—/8, можно по
тангенсу угла наклона прямой определить константу
скорости каталитической реакции ki, а по отрезку на
оси ординат при —Л^— стехиометрический
коэффициент.
Возможен также процесс, в котором образую-
щийся по электрохимической реакции продукт ката-
лизирует превращение исходного деполяризатора в
электрохимически неактивное вещество [144]. Такой
процесс протекает по схеме
А, 4- ге~ А2 4- А[ Z (4-105)
Теория хроиопотенциометрии для такого процесса
при условии ki > рассмотрена в [145] для двух
случаев: 1) вещество А2 адсорбируется на электроде,
и процесс (4-105) протекает на поверхности электро-
да; 2) вещество А2 диффундирует в раствор, и про-
цесс (4-105) протекает в диффузионном слое.
Числовое решение задачи представлено в [145].
Значения (t д/т)0.равны i д/т в отсутствие химической
реакции (£i = /< = 0). Представляя эксперименталь-
ные данные графически в виде зависимости, I
от 1g Cot и сравнивая с приведенным в работе [145]
теоретическим графиком, можно определить кон-
станту скорости k\ = К.
Электродный процесс может протекать по схеме;
Aj-j-ze- А2; А, —> Aj-f-Z (4-106)
106
К такому типу процессов относится, например, реак-
ция [43, с. 368]:
UO|+ + e- UO*
л л
ki п
2UO^+H+ —* UO|* + UOOH+ (4-107)
Теория хроиопотенциометрии для процесса (4-106)
рассмотрена в [146].
Для. определения константы k2 предложено урав-
нение:
I -C^zF
Как следует из (4-108), константу k2 удобно опре«
делить по графику I л/т — /<А- Из (4-108) следует так-
же, что (t Vt)<._>0 = Ca,2F д/лОо-
Обратимое диспропорционирование с последую-
щей электрохимической реакцией согласно схеме
*1
Z *=* А. + С; Ai+ze- А, (4-109)
«1
где Z и С — электрохимически ие активные вещества.
рассмотрено в [147].
Для определения кинетических параметров полу-
чено уравнение:
i Nt- Pt № (4-110)
где
Nt = zFC°z л/лБ/2 (4-111)
Pt -------J—т (4-П2)
K^zFki)1* Ч V D J
K = kt/k2^C°AC0cIC°z
Анализ уравнений (4-110) аналогичен приведенному
выше для (4-102).
В [147] рассмотрена теория хроиопотенциометрии
и для электродного процесса, протекающего по схеме:
Z Ai + Aj; Ai + ze~ Аг 1 (4-113)
«>
Уравнение для анализа экспериментальных данных
имеет вид [147]:
, Vt = Аг - Pil^T4». (4-114)
107
Ni и Pz описываются соответственно уравнениями
(4-111) и (4-112), только в последних нужно заменить
а. на z2.
Для электродного процесса, протекающего по j
схеме
* Z =*=* А, + С; А, + ге~ А2; 2А2 (4-115) J
' Rj Kt
было получено уравнение [147]:
i =2JV1 - ър^'х1*
Для обратимой мономеризации
Z 2Ай 2At + ze~ Аг
Rs
в работе [147] было получено уравнение:
i -yfx = N - Pi'h
где ___
N = zFC° -у/лй [1 + (4КЬ)“,/!]
V \6zFk] ) \Dj
04-116)
(4-117)
(4-118)
(4-119) .i
(4-120)
(4-121)
Все соотношения (4-109) —(4-121) в. [147] были 1
получены с помощью представлений о реакционном^
слое [1, с. 115] по методу, разработанному Коутец- 1
ким в [148, 149].
В работе [150] рассмотрена теория хронбпотен- <
циометрии для процесса:
fe. ' /
^4-ze- As: A2+Q Ч=* R; R — ze :?=> At (4-122)
Rs
причем принималось, что стандартный потенциал си-
стемы A^/R^A^ и вещество R практически пол-
ностью окислено при тех потенциалах, при которых
реагирует деполяризатор Аь Подобный процесс
наблюдается в превращениях комплексов Сг3+ или
Со3+, катализируемых соединениями Сг2+ или Со2+,
которые образуются при электровосстановлении трех-
валентных ионов [151—153].
108
Приняв, что вещество Q присутствует в избытке,
можно положить k\ — k\C° и рассматривать химиче-
скую реакцию как псевдомономолекулярную.
Если значение A(=A2/^i) не слишком мало и
{k\ +. k^t = kt 1, то уравнение для переходного
времени имеет вид [154J:
i л/х - C\zF (лО)‘л {К + 1 )/2А - («т (лА)’л/2А] (4-123)
т. е. зависимость i -у/т от гт является прямой линией
с отрицательным тангенсом угла наклона.
Если А-+0 (необратимая химическая реакция) и
kt 1, то переходное время описывается уравнением:
/т = zFC^ (Do/K)k - (х/2К) (4-124)
В промежуточных случаях (150] :
(/ V* WG W) = [К 4- f +1) (4-125)
где (i Vt)o = ~2 (nDo)'r‘ — значение I 'у/х, соответствующее
разряду Ai без кинетических осложнений;
f (Ат) = 1 + Ат/3 - (Ат)!/30 + (Ат)8/210 • (4-126)
Ряд (4-126) сходится для Кт< 1.
Для случая отсутствия избытка вещества Q по-
правка на бимолекулярное^ химической реакции в
(4-122) была введена решением дифференциального
уравнения методом Коутецкого [148, 149].
4.5. ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ СО СТУПЕНЧАТЫМ
ПЕРЕНОСОМ ЗАРЯДА
И РАЗДЕЛЯЮЩЕЙ электродные стадии
МЕДЛЕННОЙ ХИМИЧЕСКОЙ РЕАКЦИЕЙ
Если электродный процесс протекает в несколько
стадий, то между стадиями переноса заряда воз-
можны химические реакции с участием промежуточ-
ных электроактивных частиц. В наиболее простом
случае это химические реакции первого или псевдо-
первого порядка. В том случае, если промежуточный
продукт первой электродной реакции превращается
в неэлектроактивно? вещество, рассмотрение процесса
аналогично рассмотрению последующей реакции. Са-
109
мостоятельный интерес представляют процессы пре-
вращения продукта первой электродной реакции в
исходный продукт второй стадии, например восста-
новление о-нитрофенола [154].
Теория хронопотенциометрии для процесса:
4* ^23
Ai + z,e- A2 4=^ A3 + z2e~ R (4-127)
для случая &2з > &з2 рассмотрена в [155].
" По значению окислительно-восстановительных
электродных потенциалов двух электрохимических
реакций схемы (4-127) можно различить два случая:
1) электродные реакции протекают при разных по-
тенциалах и на <р — /-кривой появляются две ступени;
2) электродные реакции протекают при близких по-
тенциалах, тогда на ф — /-кривой имеется одна
ступень и этот процесс трудно отличить от процесса
с предшествующей химической реакцией.'
Предположим, что окислительно-восстановитель-
ный потенциал системы Аз/R сдвинут в отрицатель-
ную сторону относительно равновесного потенциала
системы Ai/Аг настолько, что концентрация вещества
Аз вблизи поверхности электрода практически равна
нулю в течение первого процесса с переходным вре-
менем л. Решение этой задачи было получено опера-
ционным методом. Описание первого переходного вре-
мени Ti, соответствующего полному истощению ве-
щества вблизи электрода, не представляет затрудне-
ний. Для переходного времени тг, соответствующего
полному исчерпанию вещества А3 у поверхности
электрода, было получено уравнение:
_ а. (,, + | (£)* ert (Ь + „>,* +
Т1+Т,
+ — ( ?хр (fe2s6/2) _ 0 (4-128)
2 J (t1+t2-0)‘A
Ti
где 0 —переменная интегрирования, /0 — гиперболическая функ-
ция Бесселя нулевого порядка.
По уравнению (4-128) при известных ц и т2 мож-
но рассчитать k23 на ЭВМ. При достаточно большой
скорости химической реакции третьим и четвертым
слагаемыми в уравнении (4-128) можно пренебречь,
в таком случае получим соотношение для ступенча-
того электродного процесса без химической реакции.
При Й2з->0 система ведет себя как однокомпо-
нентная.
При больших значениях аргумента уравнение
(4-128) упрощается. Например, для (Й2зТ1)'Л>8 полу-
чено [155]:
(ti + т»)7’«(ti + Та)« - (za/zi) Mss)“,/a arctg (ti/tj)7* (4-120)
где (Т14-Тг) 00 соответствуют случаю ku -* со.
Для второго случая, когда вещество А3 восста-
навливается при потенциалах, близких к потен-
циалу восстановления вещества Аь при любом зна-
чении константы скорости k23 будет наблюдаться
одно переходное время, соответствующее исчерпанию
у поверхности электрода вещества Ар Для быстрой
химической реакции переходное время будет соответ-
ствовать электродной реакции с (zt+ z2) электро-
нами, а для медленной — реакции с Zi электронами.
Для расчета константы скорости было получено
уравнение [156]:
= 1 + р'(-тг—) Л erf (W)71 +
+ р,"р!?г,,7|(’'^.г1 ip - рчч+
(W)'1 (1 — Р )'
+ ф l(W)7s/(l - p2)1/s]} (4-130)
z
где Ф (z) = exp (t/2) dU; тет соответствует случаю ftS3->oo;
о
р = Zi/(Zi + z2).
Применение уравнения (4-130) к анализу про-
цесса восстановления о-нитрофенола изложено в [154].‘
При низких плотностях тока (высокие значения т)
последним слагаемым в правой части уравнения
(4-128) можно пренебречь; кроме того, при U> 2
erf (И) ял 1, тогда
it* = - pi (л/4^3)7' (4-131)
Как следует из (4-131), константу скорости k23 можно
найти из графической зависимости it1/* от i по тан-
генсу угла наклона прямой в этих координатах. Как
Ш
и в случае процесса с предшествующей химической
реакцией, при i-+0 произведение ix'11 = ix% — const.
Однако при/—>оо величина /Охарактеризует элек-
тродную реакцию с г\ электронами. Если отношение
произведений ix'!» яри i —► 0 и I -> оо есть целое число,
то это может служить доказательством протекания
процесса по механизму (4-127).
Удобнее изучать процесс (4-127) методами хро-
нопотенциометрии с реверсом тока [157, 158] и ци-
клической хроиопотенциометрии ( [159]. При значи-
тельной разности равновесных потенциалов систем
Ai/A2 и Аз/R на <р — /-кривой после реверса появ-
ляется переходное время, соответствующее исчезно-
вению вещества R у поверхности электрода.
Для первого переходного времени х' после реверса
тока от Л (момент реверса принят за начало отсчета)
получено уравнение [158]:
erf [k/fr + ?')]** + р (1 - р2)”1* {L [А (Л + т')/(1 - р2)]1'1 +
+ ехр (- k (й + т')] L [р2К (t, + т')/(1 - р8)]’Л} =
= (U + 1) [erf (Кт')’'* + р (1 - p2)~,/s {L [Кт' (1 - р2)7' +.
+ ёхр (- Кт')] L [р2Кт'/(1 - р2))'/»}] (4-132)
где р = (z2 — crzt)/(zi + Zj); К — kzs + k^; a = kst/kss; L (X) =
X
= e~x' (2/Vn) e0* dv; U = й/й; й> й — плотность тока в прямом
о
и в обратном направлениях.
Из уравнения (4-132) следует, что Кх' зависит от /ь V
и р. Для больших значений Kti величина Ex' не зави-
сит ох, /1 и выражается уравнением:
erf (Кт')'А + р (1 - р2)-'А {Ь1Кт7(1 - р2)]‘/а +
+ ехр (- Кт') L [р2Кт'/(1 - р2)]7»} = 1/([/ + 1) (4-133)
При р = 0 уравнение (4-132) дает соотношение, ана-
логичное полученному для последующей необрати-
мой реакции [19]. Для этого случая х' быстро стре-
мится к предельному значению при /~>оо.
При р #= 0 зависимость х' от t\ проходит через
максимум и медленно приближается к предельному
значению при. й -> оо, так что х' можно считать по-
стоянным задолго до достижения предела,
Ш
Сумма констант скоростей химической реакции мо-
жет быть определена из табл. 4-1 при известном р.
Таблица получена численными расчетами по уравне-
нию (4-132). В этой таблице т'акс — значение пере-
ходного времени в точке максимума Кривой зависи-
мости Кт:' от ti, r'aKC//j — отношение т'акс к /1 при т/ =
= т/макс, — предельное значение т' из уравнения
(4-133). Поскольку 'медленно, то экспери-
ментально трудно установить
Таблица 4-1. Значения некоторых параметров при U — 1
₽ «л' чмакс тмакс/б Тмакс/Тоо
0 0,2275 —
1/4 0,1230 0,1519 0,0701 1,235
1/3 0,09592 0,1310 0,0761 1,365
1/2 0.Q5262 0,09303 0,0862 1,768,
2/3 0,02283 0,05910 0,0931 2,588
3/4 0,01269- 0,04335 0,0961 ‘ 3,415
Для определения Кт'^ можно использовать разложе-
ние в ряд интеграла уравнения (4-132). При больших
значениях аргумента после преобразований получаем:
^>1: лг/ = л:4{1-pln(K<)/dlnl/]x
'X (1 + 1 / Ю Р т)-'11} (4-134)
Как следует из (4-134), Кт'ва можно определить
экстраполяцией к нулевому значению зависимости
т' от Параметр р определяется по зависимо;
сти от отношения токов в прямом и обратном на-
правлениях U либо по зависимости t^aKC/T^ от U,
как следует из табл. 4-1.
В работе [160] рассмотрена теория хронопотен-
циометрии для процесса инактивации промежуточного
продукта.по схеме
Ai Aj 2$е~ ч -X R
(4-135)
Аз
из
Выражение, из которого можно найти переходное
время, имеет вид:
(гВДгГ) (1 + z2/z,) т/« - (2K?V*) (т, + т2)% +
* + (zs/z,) К-7’ erf (Кт, + Кт2)7’ - (1 + Z2/Z1) (лК)-‘А X
Кт,
X е-е/2/о(0/2)[К(т,+ т2)-6Г7зг/6 = 0 (4-136)
о
где К — fesz/fes; К = Aza + ^зг; остальные обозначения те же, что
и в уравнении (4-130).
Для быстрой обратимой химической реакции
(Т(т2^> 1) уравнение (4-136) упрощается:
1 + z2/z, — (1 + т2/т,)7’ + (z3/2z,) (лт2/т,)71 (ККт2)“71 —
— (1 + 22/z,) (t2 + т,)7* (лККт2)-7г arcsin (1 + t,/t2)~7’ = 0
(4-137)
Расчеты по уравнению (4-137) значений t2/ti в зави-
симости от ЛХт2 при разных zz/z\ приведены в [160].
Для ККт2-*оо ,
(т2/т,)м = (z2/z,)2 + 2 (z2/z,) (4-138)
для ККт2 -> 0
(т2/Т1)о == tg2 nz2/(z, + Z2)j (4-139)
В случае необратимой химической реакции К С
1 из (4-136) можно получить:
Kt:
(Zi/Zi) erf (Кт, + Кт2)‘а = (14- z2/z,) л"7’ J е~в,210 (6/2) X
о
X [К (Т, 4- Т2) - 0] ~'l‘de (4-140)
Расчеты по уравнению (4-140) зависимости t2/t, от
Кт2 при нескольких значениях z2/zj также приведена
в [160].
1Н
Из (4-140) можно получить предельные значения
отношения переходных времен:
Для Кт2->0 (t2/t1)0->(z2/zi)2 + 2(z!/zi) (4-141)
Для /Ст2->оо (Та/тОоо->tg2^nza/(z, + z2)j (4-142)
Для Кт2 -> 0 д (т2/т,)/д (Кт2) -> -1 (| + -J-)2 (4-143)
Приведенные в [160] таблицы позволяют по
экспериментальным, данным зависимости (t2/ti) от
т2 находить кинетические параметры процесса (4-139).
Глава 5
ПРИМЕНЕНИЕ ХРОНОПОТЕНЦИОМЕТРИИ
ДЛЯ АНАЛИТИЧЕСКИХ ИССЛЕДОВАНИЙ
5.1. ИЗМЕРЕНИЕ ПЕРЕХОДНОГО ВРЕМЕНИ
Величина переходного времени является характе-
ристикой концентрации деполяризатора в растворе;
она важна и при исследовании кинетики электродного
процесса. Переходное время, определяется по ср—t-
кривой. Было предложено большое число эмпириче-
ских методов определения переходного времени
[20, 50, 51, 83—86, 161—173].
По нашему мнению, наиболее точным методом
определения переходного времени является графиче-
ский метод, предложенный Драчкой [86]. Сущность
метода понятна из построения на рис. 5-1. Прямая 1
является касательной к точке в конце начального
участка ср — /-кривой, прямая 2 — касательной к точ-
ке перегиба, прямая 3— касательной к точке на ко-
нечном участке ф— /-кривой, перпендикуляр 4 опу-
щен на ось времени из точки пересечения линий 1 и 2,
перпендикуляр 5 — из точки пересечения прямых 2
и 3, расстояние между ними равно переходному вре-
мени т. Эта величина переходного времени является
неточной. Для внесения поправки на заряжение двой-
ного слоя проводят линию 6, параллельную оси вре-
мени, которая проходит через точку пересечения пер-
пендикуляра 4 с <р — /-кривой, и линию 7, параллель-
ную касательной 1 и проходящую через точку пересе-
чения перпендикуляра 5 с кривой ф — /. Искомой ве-
личиной переходного времени является величина xjd-
Для определения переходного времени рядом ав-
торов были разработаны электронные установки
[174, 175]. Установка, предложенная в [175], позво,-
ляет определйть переходное время с точностью
± 1 мкс.
В работе [88] для компенсации искажений хроно-
потенциОграмм, вносимых емкостным током, предло-
жена специальная аппаратура.
116
5.2. АНАЛИТИЧЕСКИЕ ВОЗМОЖНОСТИ
ХРОНОПОТЕНЦИОМЕТРИИ
В УСЛОВИЯХ ПОЛУБЕСКОНЕЧНОЙ ДИФФУЗИИ
Хронопотенциометрический метод позволяет про-
водить как качественный, так и количественный ана-
лиз исследуемых растворов.
Рис. 5-1. Графический метод определения переходного времени [86].
Характеристикой качественного состава раствора
является потенциал четверти хронопотенциограммы
который является характерной величиной для
данного иона и раствора электролита в условиях ли-
нейной (плоский электрод) полубесконечной диффу-
зии при обратимом электродном процессе. 1.
Количественной характеристикой раствора (со-
став) является величина переходного времени.
Наиболее простая связь между переходным вре-
менем и концентрацией деполяризатора в растворе
117
наблюдается в случае хронопотенциометрии с по-
стоянным или с программированным током, меняю-
щимся по закону i — <&№, в условиях линейной диф-
фузии. Эти разновидности хронопотенциометрии в ос-
новном и используются для аналитических целей.
- Для хронопотенциометрии с постоянным током
между т и Со существует следующая связь:
„ zFn‘*D'hC^
т’Л =----2Г~“ (М)
Из этого уравнения следует, что
гт’/, = КдСо) .. (5.2)
где йд= n1/22FD1/2/2—хронопотенциометрическая константа.
Для хронопотенциометрии с током, меняющимся
по закону i = р/1/*, эта связь описывается уравнением
По-видимому, впервые экспериментально зависимость
т*7’ —- Со изучалась в работе [52]. Для всех изучен-
ных в этой работе процессов зависимость x’/s — Со
имела линейный характер, как, например, для про-
цесса электровосстановления Cd2+ на фоне 0,1 М.
КЫО3 на ртутном индикаторном электроде (см.
рис. 2-1). Поверхность электрода составляла 2,76 см2.
Ток поляризации для прямых 1, 2 и 3 соответственно
равнялся 2,3- 10~Е; Ы0-5 и 5-10~6 А/см2.
При анализе смеси ионов, восстанавливающихся
при различных потенциалах, на хронопотенциограм-
ме получается ряд ступеней (см. рис. 2-10). Концен-
трация иона, восстанавливающегося первым, опре-
деляется уравнением (5-2). Это уравнение неприме-'
нимо, как указывалось ранее, для ионов, восстанав-
ливающихся при более отрицательных потенциалах,
так как весь ток электролиза будет распределяться
между диффузионными токами первого, второго и т. д.
определяемых ионов. В результате переходное время
элементов, восстанавливающихся при более отрица-
тельных потенциалах, увеличивается. Эффект увели-
чения переходного времени элемента (вещества),
118
восстанавливающегося вторым, с одной стороны,
осложняет количественный анализ, но, с другой сто-
роны, его можно использовать для повышения чув-
ствительности определения второго элемента [52].
При анализе растворов, содержащих несколько
элементов, удобнее всего пользоваться методом до-
бавок. Сначала в раствор вносят добавки самого
электроотрицательного элемента и определяют кон-
центрацию его в исследуемом растворе, затем пооче-
редно вносят добавки менее электроотрицательных
элементов и определяют их концентрацию. При одно-
временном внесении добавок всех определяемых эле-
ментов количественный анализ невозможен, так как
переходное время восстанавливающегося элемента
будет возрастать не только за счет повышения его
концентрации, но и за счет увеличения переходного
времени элемента, восстанавливающегося при более
положительном потенциале.
Из формулы (5-1) видно, что с уменьшением поля-
ризующего тока переходное время будет возрастать.
Однако беспредельно уменьшать ток нельзя, так как
при малых значениях ои будет практически полностью
расходоваться на заряжение двойного электрического
слоя, и аналитическое определение элемента станет
невозможным. Порядок ошибки, обусловленной рас-
ходованием части тока на заряжение двойного слоя,'
определяется из соотношения [1]:
Qr С. Дф
-2£-=«-4-2- (5-4)
<?ф «т
где Qc и Рф = количества электричества, расходуемые на заря-
жение двойного слоя и на электрохимическую реакцию, Кл/см2;
Са — емкость двойного слоя, Ф/см2; Дф — ширина хронопотеицио-
граммы, В.
При ошибке т%, вносимой емкостным током, из
уравнений (5-1) и (5-4) получим:
Г^оу^ 400^.
\гСо) — nF3Dm
(5-5)
При Cd = 25 мкФ/см2, Д<р == 0,1 В, £>=10~5 см2/с и
т » 1 %*
. (zCo)2 = 3,1° 9' (5-6)
Если г «= 2 и i1 • 10-9А/см2, то Ср — 1 • 10“6 моль/л.
U9
Таким образом, методом хронопотенциометрии
можно определять концентрацию элементов порядка "
п-10-6 моль/л.
Как уже отмечалось, методом хронопотенциомет-
рий можно определять одновременно несколько эле-
ыюнтов. Однако, если равновесные потенциалы окис-
лйтельно-восстановительных систем близки друг к
другу, то будет происходить наложение хронопотен-
циограмм, и определение отдельных элементов станет
невозможным. Определение элемента, восстанавли-
вающегося вторым, с ошибкой 1% возможно, если
равновесный потенциал этой системы отличается от
равновесного потенциала системы, восстанавливаю-
щейся при более положительном потенциале, на вели- ;
чину менее Д«р —«родах,— фо.мп (ti — переходное вре- \
мя элемента, восстанавливающегося вторым). ЧемJ
меньше ширина (Д«р) хронопотенциограмм отдельных
элементов, Тем больше элементов можно определять f
по одной суммарной хронопотенциограмме? Величины ?
Д<р для необратимых и обратимых электродных про-(
цессов определяются, из уравнений хронопотенм
циограмм:
Дфнеобр = ^¥ 1g 45 — 0,096В
(при aza=l и Т = 298к);
Дфобр = 1g 440 = В r (Т = 298 К)
Из этих расчетов следует, что если ширина хроно^?
потенциограмм отдельных элементов не превышает-
указанных величин (например при z = 2 — 0,077 4$
4-0,096 В), то на суммарной хронопотенциограмм^
для каждого элемента получаются отдельные ступени?
5.3. ИССЛЕДОВАНИЕ ПОВЕДЕНИЯ ЭЛЕМЕНТОВ'
И ВЕЩЕСТВ И ИХ
АНАЛИТИЧЕСКОЕ ОПРЕДЕЛЕНИЕ4
В настоящем параграфе описываются условий?
хронопотенциометрического определения элементов
веществ, поведение которых изложено в литературе,
При этом имеется в виду, что ионы металлов восстав
навливаются до металлического состояния, кроме спе-
циально оговоренных случаев.
(5-7)
(5-8)
120
Таллий
Ионы таллия (1) легко восстанавливаются на ртут-
ном электроде из кислых, нейтральных и слабоще-
лочных растворов. Чаще всего для хронопотенцио-
метрического определения таллия применяются ней-
тральные растворы 1 М KNO3 [20], 0,1 М KNO3 [23]
и 1 М Na2SO4 [176]. Потенциалы <рт/4 катодных и
анодных хронопотенциограмм мало отличаются меж-
ду собой, что указывает на обратимость процесса
[20, 124]. Катодные и анодные хронопотенциограммы
таллия (I) четко выражены. После внесения поправки
на омическое падение потенциала получены следую-
щие значения <рт/4 (нас. к. э.) при 30 °C [20]: —0,463 В
для катодного и —0,455 В для анодного процесса.
Авторы [23] изучали катодное поведение тал-
лия (I) в растворе 0,1 М KNO3 методом производной
хронопотенциометрии. Значения Jt’VCo = const, най-
денные методами производной и обычной хронопотен-
циометрии, хорошо согласовались между собой и со
значением 388 Ас'/г см3/моль, полученным в работе
[177].
В работе [176] найдено, что для процесса электро-
восстановления Т1(1) на стационарной ртутной капле
экспериментальные и теоретические значения 1х',г/С°о
хорошо согласуются (расхождение не превыщает
±1,1 %). Ошибка в определении т в интервале кон-
центраций (1,89 -г-17,1) -IO-3 моль/л Т1+ не превы-
шала 2%.
Кадмий
Ионы кадмия восстанавливаются на ртутном элек-
троде из кислых и нейтральных растворов. На фоне
1 М KNO.3 [20] или 0,1 М KNO3 [52] и 1 М КС1 +
+10~4 М НС1 [178] кадмий(П) восстанавливается
обратимо. Потенциал <рт/4 катодной хронопотенцио-
граммы на фоне 1 М KNO3 равен —0,580 В, анод-
ной — 0,569 В (нас. к. э.).
При восстановлении Cd (II) на плоском ртутном
электроде [52] соблюдается постоянство отношения
1х1г1Сс в интервале концентраций ионов кадмия
(1-4-6,9) -10~4 моль/л со средним отклонением 4,3%’.
121
Максимальное отклонение при измерении переходного
времени составляло 1,55%. Калибровочные графики
в координатах х'г — представляют собой прямые
линии с началом в нулевой точке.
При восстановлении кадмия (II) на платиновом
электроде хронопотенциограмма наблюдается при бо-
лее отрицательных потенциалах по сравнению с ртут-
ным электродом, при этом верхняя часть ср — /-кривой
сливается с кривой восстановления ионов водорода
{52]. Поэтому кадмий рекомендуется определять на
ртутных электродах.
Свинец
При восстановлении РЬ(П) в нейтральных, кис-
лых и щелочных растворах наблюдаются хорошо вы-
раженные <р — /-кривые. Во многих фоновых электро-
литах восстановление ионов свинца на ртутных элек-
тродах протекает обратимо. Хронопотенциограммы
свинца на фоне 0,1 М КС1 хорошо воспроизводятся
как на ртутном, так и на платиновом электродах [52].
При восстановлении ионов свинца (II) на ртутном
электроде в разбавленном растворе азотной кислоты
(0,2 М НМОз) постоянство 1хь/с'ръз+ соблюдается в
широком интервале концентраций (10*4 4-2-10~2
моль/л) и силы тока (от 10-7 до 10~6А) [179]. Значе-
ние <рт/4 — — 0,40 В (нас. к. э.) совпадает с поляро-
графическим потенциалом полуволны. Авторы [180]
использовали метод хроиопотенциометрии для выяс-
нения некоторых особенностей электрохимического
поведения РЬ(П) и для аналитического определения
его в природных образцах и некоторых полупроводни-
ковых материалах. В качестве фоновых электролитов
использовали растворы КОН различной концентрации
и 1 М НС1. Присутствующий в щелочных растворах
кислород практически не оказывал влияния на пере-
ходное время восстановления свинца (II). Потенциалы
<pt/4 для всех изученных фонов совпадают с поляро-
графическими потенциалами полуволн.
В интервале концентраций (1 10)•10~* моль/л
и при токе 2-10-6 А зависимость — Cpbs+ имела
линейный характер и выражалась уравнением
122
t'/j = 0,9 • 104СрЬ2+. Стандартное отклонение при опре-
делении переходного времени не превышало 2%.
Разработанные методики [181] совместного опре-
деления свинца и олова в карбонатных породах и
свинцово-цинковой руде основаны на предваритель-
ном ионообменном разделении РЬ(П) и Sn(IV), по-
скольку при совместном определении этих элементов
в растворе не удается получить раздельные хронопо-
тенциограммы. Рекомендуемый фоновый электро-
лит — 1М НС1. Относительная погрешность опреде-
ления РЬ(П) в концентрации п-Ю-4 моль/л состав-
ляет 1,8%.
Авторы [180] отмечают высокую точность хроно-
потенциометрического метода.
Цинк
Определение цинка лучше всего проводить в ней-
тральных или подкисленных растворах. На фоне
0,1 М КЫОз при восстановлении ионОв цинка, на ртут-
ном электроде [52] наблюдается хорошо выраженная
<р — /-кривая с <pt/4 — —1,026 В (нас. к. э.). Откло-
нения значений i^/Cznai) в интервале концентраций
ионов цинка (1,15-5-11,6)-IO-4 моль/л не превышает'
4,3%’.
При совместном восстановлении Pb(II), Cd (II) и
Zn(II) на хронопотенциограмме наблюдается три от-
четливых ступени [52]. Первое переходное время п
соответствует восстановлению РЬ(И), второе — вос-
становлению Cd (II), третье — Zn(II).
Олово
Вследствие гидролиза ионов олова (IV) определе-
ние его проводят в кислых растворах. На хронопо-
тенциограмме (фон — 1 М НС1) наблюдаются две сту-
пени [180], соответствующие двум стадиям электро-
химической реакции: 1) Sn(IV) -* Sn(II)—2е“
(фт/4 = ~ °»28в) и 2) Sn (11)4- 2е—> Sn (0) (<Н/4 ==
== 0,46В).
В работе [182] отмечено, что восстановление
Sn(IV) до Sn (II) не может быть использовано в ана-
литических целях, поскольку <р —/-кривая, соответ-
123
ствующая этому процессу, выражейа не четко. ДлйЯ
определения олова можно с успехом использоватЯ
восстановление Sn(II) до металла. Близость <рг/4 ?:Я
: гЛ полярографическому потенциалу полуволны указььЯ
вает на обратимость восстановления Sn(II) на фонеЯ
1М НС1. Среднее значение t2/?i для стадий 1 и 2;Я
>. в хорошо совпадает с теоретической величиной, равной Я
ГЛ: 3, характерной для обратимого двухступенчатого вос-Я
• : становления при Zt — z2 [180], Как уже указывалось Я
‘ выше, авторы [181] разработали методику совмест- Я
(. него определения свинца и олова в свинцово-цинко-Я
,;i р. к вой руде. Я
w й * Относительная погрешность определения оло- Я
' ; > , Ba(IV) для концентраций п-10~4 моль/л состав-‘Я
л ял а 5%. Я
Галлий!
Электрохимическое поведение галлия(III) доволь- Я
/; . но сложное [183, 184]. Катодные и анодные <р — t- Я
: !;i. кривые восстановления Ga(III) на ртутном электроде "!
выражены достаточно хорошо на фоне KCNS 1
- (7—ЮМ). Восстановление ионов галлия(III) и окис- 1
- ление металлического галлия протекают в условиях.;!
диффузионного контроля. Потенциалы <pT/4 катодной>$
и анодной кривых равны —0,900 ± 0,005 В (нас. к.
и совпадают со значением полярографического потен.Й£
Г циала полуволны <р1Д — — 0,900 ± 0,003В [183]. КромйИ
I того, из катодных и анодных кривых, полученных Я
условиях реверса тока, следует равенство ti « ЗтЯ
(где Та — переходное время анодного процесса), что®
согласуется с теоретическими выводами [2] при усло-Я
вии равенства катодного и анодного токов. Калибро-Я
вечные графики т'А — Caatni) при концентрации Я
Ga(III) 3-10~4— 2,6-10—3 моль/л линейны, но отсе-Я
i- кают на оси концентраций отрезок при Сса(Ш)~’Я
= 2;3 • 10~4 моль/л. Аналогичные калибровочные гра-Я
фики получены в работе [183]. Такое поведение гал-Я
, лия(Ш) авторы [183] объясняют присутствием в 1
растворе различных комплексов галлия (III), из ко- Я
торых не все электроактивны, адсорбцией роданид- 1
ионов на электродной поверхности и частичным гид-. 1
релизом Ga(IIl). 1
124
Медь
Медь (II) восстанавливается непосредственно до
металлического состояния в некомплексообразующих
средах, например в растворах нитратов [52] или в
растворах кислот HNOg, H2SO4, НСЮ4 [185, 186];
восстановление Си(II) протекает квазиобратимо.
Рис. 5-2. Хронрпотенцио-
грамма восстановления ме-
ди(П) в 1 М КС1 на золо-
том проволочном электро-
де [1891.
Условия опыта: С^, ^+—0,0203 Л/,
pH =«2.00; i=3,08-10~3 А/см».
В растворах хлорида [71, ,187—189] или бромида
[190] медь (II) на платиновом электроде восстанав-
ливается до металла в две стадии (Hal — галоген):
a) CuHal2 + e“ —> CuHal2 (квазиобратимый процесс)
б) CuHal2 + е~ —> Си + 2НаГ (обратимый процесс)
Первая <р — /-кривая, соответствующая ’ процессу
восстановления Си(II) до Си (I), выраженц хуже, чем
вторая (рис. 5-2). В аналитической практике может
быть использован второй процесс, при котором медь,
получающаяся путем предварительного химического
восстановления, затем восстанавливается до метал-
лического состояния на ртутном или рлатиновом ка-
тодах [187, 190]. Чтобы обеспечить полное превра-
щение Си (II) в Си(1), в электролит добавляют избы-
точное количество гидразина или гидроксиламина.-
В табл. 5-1 приведены условия восстановления
ионов меди в различных электролитах.
Представляет интерес использовать при опреде-
лении меди неводные растворители, например ацето-
нитрил [191]. Ацетонитрил существенно влияет на
форму хронопотенциограммы меди и значение <рг/4.
Поскольку в среде ацетонитрила энергия сольвата-
125
Таблица 5-1. Условия восстановления ионов меди
в различных электролитах
-Фоновый электролит Потенциал восстановле- ния (Фт/4) В (нас. к. э.) Реакции Элект- род Лите- рату- ра
0,Ш KNO3 -0,1 Cu(II) -> Cu(0) HR 52]
1Л1 КС1 (pH = 2) 4-0,070 Cu(II) Cu(l) Pt, Cu 187
-0,240 Cu(I) -» Cu(0) Pt, Cu 187
Ш HNO3,1Л4 HClOj 0 Cu(II) Cu(0) Hg 186
0.05Л1 H2SO4 -0,005 Cu(II) -> Cu(0) Cu 185
0,5Л1 H2SO4 -0,01 Cu(II) -> Cu(0) Hg 186
1МНС1 +0,070 Cu(II) -> Cu(I) Pt 71]
-0,240 Cu(I) Cu(0) Pt(Cu) 71]
1Л1 KBr 4- 0.005Л1 H2SO4 +0,020 Cu(II) -> Cu(D Pt 190]
-0,30 Cu(I) -» Cu(0) Pt(Cu) 190]
ции ионов меньше, чем в воде, то ионы Си (II) вос-
станавливаются при менее отрицательных потенциа-
лах, чем в водных растворах. По данным [191]
<р — ^-кривые восстановления Си (II) до Си(1) на пла-
тиновом дисковом электроде в среде ацетонитрила
хорошо выражены, величина <pt/4 равна +0,72 В отн.
Ag/Ag+ (0,01 моль/л) электрода при 25 °C. Отклоне-
ние при определении /т'^/Ссщп) не превышает 2,6%.
(С°сц(ц) =2,52 • 10 3 моль/л).
Золото
Золото может осаждаться на платиновых электро-
дах как в некомплексообразующих,. так и в комп-
лексообразующих электролитах.
Золото(III) восстанавливается обратимо до метал-
лического состояния, давая четкую <р — /-кривую, на
фоне IM НС1 [192]. Потенциал q>t/4 близок к
+0,42 В (нас. к. э). Величины 7t‘/2/Cau(ih) при концен-
трациях золота (III) в растворе 10-3 и 10-2 моль/л
хорошо согласуются с теоретически рассчитанными
для диффузионно-контролируемых процессов в интер-
вале т от 1 до 20 с.
Ионы золота в растворах соляной кислоты нахо-
дятся в виде комплексов AuCl? и АиСЫ [193]. При
восстановлении смеси AuClJ и AuCli на платиновом
126
электроде в 1 /И НС1 наблюдаются Две. хройопОТей-
циограммы, соответствующие двум процессам:
a) AuClg + e" —> Аи(0) + 2СГ
б) AuCi; + Зе- —> Аи(0) + 4СГ
С течением времени происходит уменьшение ть а
затем ф — /-кривая восстановления AuClJ исчезает,
Рис. 5-3. Зависимость по-
тенциала от времени для
процесса восстановления
Ni(II) в 1 Л/ КС1 (с^+ =
=4» 10“3 моль/л) [20].
поскольку начинается реакция диспропорциониро-
вания [192]:
ЗАиС1г —> AuClJ + 2Аи 4-2СГ
равновесие которой смещено вправо.
Никель и кобальт
Электролитическое восстановление никеля (II) и
кобальта (II) протекает во многих обычных электро-
литах, так что эти процессы вполне применимы для
аналитических целей. Разряд ионов никеля и ко-
бальта происходит при потенциалах более отрицатель-
ных чем —1,0 В и в условиях значительной поляри-
зации [20], тем не менее ф — /-кривые хорошо выра-
жены. На рис. 5-3 показана хронопотенциограмма
восстановления Ni(II) в 1М КС1 на ртутном элек-
троде, а на рис. 2-6 представлены ф — /-кривые вос-
становления Ni(II) и Со (II) на ртутном электроде в
полулогарифмическом виде [20]. Значения фт/4 со-
ставляют: для восстановления Ni(II) в 0,5 М KCNS
127
^0,790 В, Ni (11) в J М КС1 » —1,190 В и Со(Н) й
1 М K.CI —1,170 В (нас. к. э.). Значения ф1/г, рассчи-
танные по <р — /-кривым, согласуются с полярогра-
фическими данными..
Железо
Реакция восстановления Fe(III) до Fe(II) в рас-
творе соляной кислоты [60, 194] является обратимой.
Поэтому для аналитического определения железа ис-
пользуют реакцию Fes++ Fe2+ Обычно восста-
новление Fe(III) проводят на платиновых электро-
дах— дисковых [52, 195] или проволочных [60, 194].
Наиболее подходящим фоном является разбавленный
раствор соляной кислоты. Восстановление Fe(III) на
этом фоне протекает при потенциалах отрицательнее
—0,5 В (нас. к. э.)х Наиболее четкие ф— /-кривые
наблюдаются при концентрации железа (III) в рас--
творе не менее 5-10~4 моль/л. :
Для определения двухвалентного . железа можно
использовать реакцию окисления Fe(II) до Fe(III)
[189], а также реакцию восстановления Fe(II) до
Fe(0) на платиновых или графитовых электродах.
Марганец
Марганец(П) восстанавливается на ртутных элек-
тродах с образованием амальгамы марганца. Хорошо'
изучен этот процесс в 1 М растворе хлористого калия;
[196, 197]. Этот фоновый электролит может быть ре-;
комендован для аналитического определения Мп(II)’.
Для изученных концентраций марганца (II) в интер-
вале от КН до КН моль/л при различных плотно-
стях тока соблюдалось постоянство it'1*, что указывает
на отсутствие в данных условиях предшествующей
химической реакции [196]. Значение фт/4 остается
постоянным и составляет —1,3 В (нас. к. э.). На
основании результатов хронопотенциометрии с ревер-
сом тока [196] делается вывод, что металлический
марганец растворяется в ртути, а не остается на. по-
верхности, так как в противном случае переходное
время для процесса окисления должно быть равно
переходному времени для процесса восстановления,
128
чего йе наблюдается на опыте. Действительно, НО
данным работ [197, 198] марганец растворяется в
ртути и образует с ней интерметаллические соеди-
нения.
Висмут
Ионы висмута(III) восстанавливаются обратимо'
до металла в кислых растворах. Для определения
висмута используют растворы соляной, серной или
азотной кислот [52, 186, 199, 200].
Величины чрг/4 очень близки к значениям поляро-
графических потенциалов полуволн. Значения
для процесса восстановления висмута(III) на различ-
ных фонах приведены в табл. 5-2.
Таблица 5-2. Значения <рт/4 для процесса
электровосстановлення Bi(III) на ртутном электроде
(t = 25°C)
Фоновый электролит ЧЧ/4, В (нас, к. э.) Реакция Литература
0.5А4 H2SO4 Ш НС1 Ш HNO3 —0,01 -0,070 —0,05 Bi(III) -> Bi(0) BiClJ -> Bi(0) + 4СГ Bi(III) -> Bi(0) [186,200] [186,199] [52,186]
Серебро
Электровосстановление серебра (I) из растворов
солей серебра на платиновом электроде является на-
столько хорошо воспроизводимым процессом, что его
часто используют для первичной калибровки (по-
добно процессу восстановления феррицианида). При
восстановлении ионов серебра никаких трудностей не
возникает, если оно протекает в некомплексообразую-
щих средах (например, 1 М KNO3 (pH = 2 4-4) [201];
0,2 М KNO3 + 0,01M HNO3 [87] или 0,1 М HNO3)-
При концентрации ионов серебра 5-10 моль/л
ошибка в определении /т’^/^ак* составляет менее
0,5% [195].
Авторы [202] рекомендуют использовать при р -
делении серебра (I) в качестве растворителя
129
Ь Закм 1072
*
нитрил, a ё качестве фона — 0,1 M LiC10<. В этом
случае серебро (I) дает на ртутном электроде отчетли-
вые <р— /-кривые.
Определение серебра можно проводить также в
комплексных электролитах (хлоридных, роданидных,
аммиачных), но в этих условиях необходимо учиты-
вать образование различных комплексов.
Ртуть
Для определения ртути могут быть использованы
различные фоны. Изучено окисление и восстановле-
ние ртути (I) на фоне 1 М HCIO4 [203, 204] с исполь-
зованием стационарного ртутного электрода. Можно
также применять платиновые электроды. При восста-
новлении Hg(II) на хронопотенциометрической кри-
вой наблюдаются две задержки, соответствующие
ступенчатому восстановлению до Hg(I) и Hg(0).
Авторы [205] для количественного определения
Hg(II) использовали кривую восстановления на пла-
тиновом электроде на фоне Н3РО4.
Уран
Хронопотенциометрию с успехом можно приме-
нять для определения урана, используя процессы
окисления на платиновом'электроде [206] или вос-
становления на ртутном электроде [146, 207]. При
восстановлении U(VI) на платиновом электроде в
кислых средах ф— /-кривая сливается с кривой вос-
становления ионов водорода, поэтому в количествен-
ном анализе используется окисление U(IV) [пред-
варительно восстановленного из U (VI*) ] до U(VI)
[206]. Из исследованных фонов (H2SO4, ,СН3СООН
и НС1О4) наиболее пригодным оказался 1 М. раствор
НСЮ4. Вид ф—/-кривой зависит от состояния поверх-
ности платинового электрода [189]. Хронопотенцио-
грамма окисления U(IV) до U(VI) в 1 М НС1О4 на-
блюдается при потенциалах положительнее --J-0,8B
(нас. к. э.).
Отклонения при определении ixk/Си (iV) в интер-
вале концентраций U(IV) (2,13—12,80)-10-8-моль/л
не превышали 1,4%. Определению урана по указан-
ной методике мешают ацетат никеля, уксусная кисло-
130
та, нитрат никеля и не мешают хром (VI), ванадий (V),
кобальт(П) и молибден(VI).
ypan(VI) восстанавливается на ртутном электро-
де в щелочных средах [207]. На рис. 5-4 показаны
типичные кривые восстановления U(VI) до U(V) в
1 Л1 №гСОз (pH = 11,5) и окисления U(V). Высокое
Рис. 5-4. Кривая ф — t вос-
становления и окисления
урана (реверсионная)' в 1 М
Na2CO3 [207].
Условия опыта: Си^ =
= 10_3 М; /=41,8 мкА/см2;
рН = 11,5; /=25°С.
перенапряжение [207] свидетельствует о том, что эти
процессы являются необратимыми. Это же подтверж-
дается величиной отношения т/та, которое меньше 3
И-
Величина rr'/Cutvi) сохраняет постоянное значе-
ние в интервале плотностей тока от 1,50 • 10~5 до
Ю~3 А/см2, максимальное отклонение от средней вели-
чины не превышает 5%.
Восстановление U(VI) [207] в растворе NaHCQ3
(pH — 8,2 +- 8,4) может осложняться последующей
Таблица 5-3. Условия восстановления или окисления
ионов урана в различных фоновых электролитах
Фоновый электролит Потенциал окисления или восстано- вления, В (нас. к. э.) Реакция Элект- род Лите- ратура
ш нею. +0,90 U(IV) - > U(VI) Pt 206
1М ЫагСОз -1,12 U(VI) - *U(V) Hg 207
1А4 NaHCO3 -1,10 U(VI) - ->U(V) Hg 207
0,5 Af NaHCO3 +0,544 NaClOi -1,09 U(VI) - > U(V) Hg 207
5*
131
реакцией диспропорционирования U(V). В этом слу«
чае величина rr'^/Cucvi) уменьшается при увеличении
плотности тока [55]. Условия определения урана при-
ведены в табл., 5-3.
л Плутоний
Детальное исследование хронопотенциометрическо-
го поведения плутония на платиновом электроде в
различных минеральных кислотах (серной, азотной,
соляной и хлорной) проведено в [208].
В водных растворах для плутония характерны три
устойчивых валентных состояния: Ри(Ш), Pu(lV) и
Pu(VI) [208]. Возможно также и четвертое валентное
состояние — Pu (V). В растворах кислот плутоний на-
ходится в основном в виде Pu(IV). Хронопотенциоме-
трическое поведение плутония чрезвычайно сложно.
Изучено хронопотенциометрическое поведение
Pu(III), Pu(IV) и Pu(VI) на платиновых электродах
в растворах 0,5 М H2SO4, и 1 М HNO3, НС! и НСЮ4
[208].
Плутоний(VI) сначала восстанавливался до
Pu(III) при ф = +0,525 В (нас. к. э.) в 1М НС1,
HNO3 или НСЮ4 и при q> = +0,300 В (нас. к. э.) в
0,5 М H2SO4, затем окислялся до Pu(IV) при ф =
= + 0,895 В (нас. к. э.). Плутоний (IV) восстанавли-
вается на платиновом электроде в растворах перечис-
ленных кислот до плутония (III), давая хорошо выра-
женные ф — /-кривые, если концентрация плуто-
ния (IV) не менее 10~3 моль/л. Характерные ф — /-кри-
вые показаны на рис. 5-5. Близость фг/4 к стандарт-
ному потенциалу указывает на обратимость электрод-
ного процесса. При уменьшении концентрации ионов
плутония значения i-r'VCpu <1 v> увеличиваются, что по-
зволяет использовать хронопотенциометрию для опре-
деления Pu(IV) в концентрациях не ниже 10~3 моль/л.
Плутоний.(Ш) окисляется в растворах кислот до
Pu (IV). Поскольку эта реакция протекает при потен-
циалах окисления платинового электрода, то хроно-
потенциограмма окисления на предварительно восста-
новленной поверхности платины выражена хуже чем
на окисленной. Рекомендуется анодно поляризовать
платиновый электрод непосредственно перед получе-
132
нием <р — /-кривых. Значения фт/4 также близки к
стандартным потенциалам.
На ф— /-кривой восстановления Pu(Vl) наблю-
даются две задержки потенциала: первая соответ-
ствует восстановлению Pu(VI) до Pu(V), вторая —
восстановлению Pu(V) до Pu(III). Значения фт/4 за-
ЦВ(нас.к.з.)
t,c
Рис. 5-5. Хронопотенциограммы восстановления Pu (IV) до Pu (III)
на платиновом проволочном электроде [208]:
О ч
1A1H2SO4; Срц цу^О.526 Л1; 7 = 1,525 • 10”3 А;
2—1A4HNO,; Cpu (IV)=0,0498 Af; /=1,450 - W3 А; 1=25,8 °C;
8-1 Л1 HCJ; СОц ([у)=0,0518 Л1; 7=1,526 - Ю-3 А; 1=26,3 °C;
4-1 М НС1; 7=1,529 • Ю*-3 А; 5=0,370 см2-
висят от плотности тока, что указывает на необрати-
мый характер восстановления Pu(VI)«
Таблица 5-4 Результаты изучения хронопотенциометри-
ческого поведения плутония иа платиновом электроде
в растворах кислот [208]
Электродная реакция Фоновый элект- ролит q>T/4, В (нас.к-s.) % <p°, в (иас.к.э.)
Pu(IV) -> Pu(III) '0,5MH2SO4- +0,51 +0,51
Ш HC1O4 +0,73 +0,740
IM HNO3 +0,68 +0,68
1Л4 HC1 +0,74 +0,728
Pu(III) -* Pu(IV) 0,5M H2SO4 +0,51 +0,51
Pu(VI) -> Pu(V) ш HC1O, +0,56 +0,671
(/= 1,846- 10-3Л/см2)
Pu(V) -> Pu(III) IM HCIO4 +0,835 +0,835
133
Результаты изучения хронопотенциометрического
поведения плутония приведены в табл. 5-4. В этой
таблице для сравнения приведены значения стандарт-
ных потенциалов соответствующих окислительно-вос-
становительных пар [209, 210]. Для аналитического
определения плутония целесообразно использовать
реакцию восстановления Pu(IV) до Pu(III) или же
’ реакцию окисления Ри(1П) до Pu(IV). Возможно, что
применение золотого электрода позволит повысить
чувствительность определения плутония [208].
Церий
Хронопотенциограммы восстановления Се (IV) до
Се(Ш) хорошо выражены [211] на платиновом дис-
ковом электроде в растворах серной, хлорной и азот-
ной кислот умеренной концентрации (1М) [211]. На
серебряном проволочном электроде [212] и платино-
вой фольге [213] <р — /-кривые восстановления Ce(IV)
выражены хуже.
Значение величины ir'^/Co сохраняется постоян-
ным, если концентрация Се(IV) в растворе не ниже
10~4 моль/л. Например, в интервале концентраций це-
рия 2-Ю-3—2-10~2 моль/л отклонение в измерении
хронопотенциометр ической константы не превышает
1,5% [211]. При концентрации ' церия ниже
2-Ю-3 моль/л наблюдается увеличение хронопотен-
циометрической константы. Одной из причин этого мо-
жет быть адсорбция Ce(IV) [214], но основной причи-
ной является, по-видимому, появление платиновой
оксидной пленки вследствие окисления поверхности
платины ионами Се (IV), а также кислородом воз-
духа [2П].
Определению Се(IV) не мешают железо(III), ва-
надий (V), бихромат-ионы и кислород, которые вос-
станавливаются при менее положительных потенциа-
лах [ионы Се(IV) начинают восстанавливаться при
потенциалах —1,1 В (нас. к. э.)], но мешают ионы
MnOi [212]. Адсорбция кислорода не оказывает за-
метного влияния на кинетику реакции восстановления
Се (IV). По данным [215], восстановление Се (IV) до
Се(III) на платиновом электроде протекает необра-
тимо (ks = 4-10”4 см/с, а — 0,65 ± 0,06),
134
Европий
Европий(П1) восстанавливается до европия(II) на
золотом электроде в растворе хлористого аммония
[216]. Авторы [216] подчеркивают преимущества зо-
лотого электрода по сравнению с платиновым. Золото
Рис. 5-6. Катодные хроно-
потенциограммы восстано-
вления иоиов Еи3+ на золо-
том электроде [216]:
/—/=40 мкА; 2—/=48 мкА.
Условия опытов: С^из+“1,61Б X
ХЮ-3 моль/л; фок—0.1Л1 NH1C1.
не сорбирует кислород в заметных количествах [218]
и совсем не сорбирует водород [217]. На золотом
электроде получаются более воспроизводимые резуль-
таты. Для этого рекомендуется предварительно выдер-
живать электроды при анодном (<р —+ 1,4В) и ка-
тодном (<р — 0 В) потенциалах.
На рис. 5-6 показаны типичные ф— /-кривые вос-
становления ионов Еи3+ до Еи2+ на золотом электроде.
Постоянство значения указывает на отсут-
ствие предшествующих химических реакций. Элек-
тродная реакция Еи3++ —> Еи2+ в указанных усло-
виях протекает необратимо [216].
Титан
Титан (IV) на ртутном электроде восстанавливает-
ся до трехвалентного состояния в растворах* кислот
(соляной, щавелевой, винной) при потенциалах отри-
цательнее —0,3 В (нас. к. э.).
В присутствии гидроксиламина увеличивается пе-
реходное время для процесса восстановления Ti(IV)
в растворе щавелевой кислоты [135]. Авторы [135]
объясняют это протеканием на электроде каталитиче-
135
ского восстановления оксалатного комплекса четырех
валентного титана. К такому же выводу пришли ав
торы работ [143, 219, 220] при изучении кинетики это!
реакции обычным хронопотенциометрическим методов
[143] и методом хронопотенциометрии с реверсом ток4
^219, 220]. Согласно выводам, сделанным в [135], сни-
Рис. 5-7. Зависимость (Ткат/т)^2 от плотности тока для катал»
тического восстановления Ti(IV) в присутствии гидроксид!
амина [135]
Кривая Г—в отсутствие гидроксиламина; кривая 2—0,49 М; кривая 3—1,46 Л
0 —3
гидроксиламина. Условия опытов: фон—0,2 М Н2С2О4; Су[ —10 моль/л,
Рис. 5-8. Хронопотенциограмма восстановления и окисления тор*»
в диметнлсульфоксиде [221].
Условия опыта: фон — 0,1 М LiCi; С®, =2,26 • Ю~4 моль/л; 1=3 X
in <1V)
X Ю-4 А/см*.
жая плотность тока, можно достичь очень больших
величин переходного времени, что может быть исполь-
зовано для определения Ti(IV) в малых концентра-
циях. „
На рис. 5-7 приведена зависимость (ткат/т)'л от i
.(где тКат — переходное время в присутствии гидрок-
силамина) при разных концентрациях гидроксилами-
на в растворе. Видно, что с увеличением концентра-
ции гидроксиламина переходное время тКат заметно
возрастает.
136
Торий
В литературе имеется очень мало сведений об оки-
слительно-восстановительных характеристиках тория
в условиях хронопотепциометрии. Можно ожидать,
что поведение тория аналогично поведению урана и
плутония.
Изучено поведение тория(IV) на ртутном элек-
троде на фоне 0,1 М LiCl в диметилсульфоксиде [221].
Кривая восстановления Th(IV) получается при потен-
циалах отрицательнее —1,9 В (нас. к. э.). Типичная ре-
версионная <р — /-кривая электровосстановления то-
рия (IV) и окисления Th (0) приведена на рис. 5-8.
В изученном интервале плотностей тока (0,5-? 1,1) X
ХЮ-3 А/см2 при концентрациях тория(IV) в растворе
(1,13 4-2,26)-Ю-4 моль/л соблюдалось постоянство
значений /т'^/^ть (IV». что указывает на отсутствие ки-
нетических осложнений.
Значения <рт/4 не остаются постоянными с измене-
нием плотности тока и концентрации ионов тория, что
свидетельствует о необратимости электродного про-
цесса. Исходя из хронопотенциометрических [221] и
полярографических [222] данных, можно предполо-
жить, что Th (IV) в диметилсульфоксиде восстанавли-
вается до Th(O).
Хром
Для определения хрома хронопотенциометриче-
ским методом можно использовать различные процес-
сы окисления и восстановления. Возможно непосред-
ственное восстановление комплексных (например хло-
ридных) ионов Сг(Ш) на ртутном электроде до
Сг(П).
Другая методика предполагает определение хрома
в виде хромат-ионов. Хромат-ионы дают хорошо вы-
раженные катодные <р — /-кривые при восстановлении
на ртутном электроде в щелочном растворе [223].
В кислых растворах электродный процесс осложняет-
ся предшествующей химической реакцией [224].
При восстановлений хромат-ионов в карбонатном
растворе (0,1 М Na2CO3) на <р —/-кривой наблюдают-
ся две ступени: первая —при потенциале —0,5 В, вто-
рая—при потенциалах от —0,9 до —1,0 В (нас. к. э.).
137
Значение фт/4 второй ступени зависит от плотности Я
тока, что свидетельствует о необратимом характере Я
этого процесса. Электродный процесс, которому соот- Я
ветствует первая ступень, имеет, очевидно, кинетиче- Я
скую природу — величина 1т'1а уменьшается с увели-Я
Пением плотности тока. Поверхностно-активные ани- Я
оны (например роданид-ионы) «подавляют» первую Я
хронопотенцйометрическую волну. При больших плот- Я
ностях тока (т 1 с) наблюдается только одна сту- Я
пень — вторая, соответствующая реакции: Я
СгО2~ -Ь 4Н2О + Зе~ = Сг(ОН)3 4- БОН" Я
В этих условиях величина Ттг1 /С° остается посто- Я
/ (-гО4 Ж
янной, отклонения не превышают 5% (та* линейно Я
зависит от Сего2')' Независимость этой величины от п
плотности тока и концентрации хромат-ионов указы- I
вает на отсутствие кинетических осложнений (пред- ;1
шествующей химической реакции или адсорбции). Из Я
данных [223] следует, что отклонения при определе- 1
нии концентрации хрома (VI) в интервале концентра- I
ций СтОГ" (1,5-г-8,5)-10~4 моль/л составляет 1%. По
мнению авторов [223] можно снизить определяемую
концентрацию до 5-10-5 моль/л.
Слабая адсорбция хромат-ионов не влияет замет-
но на определение. Возможно определение хрома (III) i
в виде гексаминхром-иона [224] и этилендиаминхром- ,
иона [153]. Хорошо выраженные ф— /-кривые наблю-
даются на ртутном электроде в ацетатном буферном •]
растворе. Величина х'11 пропорциональна 'концентра- ;
ции только при малых концентрациях Сг(Ш): ’
(^1,5-IO-3 тМ).
Кислород !
I
Реакция восстановления кислорода относится к '
числу наиболее хорошо изученных реакций на элек-
тродах различного типа.
Кислород может восстанавливаться либо до пере-
киси водорода (двухэлектронный процесс), либо до
воды (четырехэлектронный процесс). Перекись водо- -
158
рода может восстанавливаться, но крайней мере ча-
стично, до воды и разлагаться каталитически. Воз-
можно параллельное образование Н2О2 и Н2О. Хроно-
потенциометрический метод позволяет устанавливать,
какая из этих возможностей реализуется в том или
другом случае.
Рис. 5-9. Хронопотенциограммы восстановления кислорода на
палладиевом электроде в 1,0 Al NaOH [226]:
/=5,92- 10~4 Л/см2; 1—иа окисленной поверхности, 2—на восстановленной
поверхности, 8—восстановление оксидной пленки при 1=3,70 • 10“* А/см2.
Рис. 5.10. Хронопотенциограммы восстановления Ог на платино-
вом электроде в 1 М NaOH [225].
/=1,1в • 10~4 А/см2; / — на предварительно окисленной поверхности пла-
тины в течение 20 с при /=1,85 • 10~4 А/см2, 2—повторная кривая.
Для ртутных электродов характерно значительное
перенапряжение электровосстановления перекиси во-
дорода. Поэтому на <р — /-кривых, полученных на ртут-
ном электроде, имеются две ступени [2], отвечающие
восстановлению кислорода до перекиси водорода и пе-
рекиси водорода до воды. В каждой из этих стадий
участвуют по два электрона, и следовательно должно
соблюдаться т2 = 3т> [2]. Для восстановления кисло-
рода в 1 М L1C1 получено тг = 2,97ti [2].
При электровосстановлении кислорода на электро-
дах из платиновых металлов (платина, палладий) на-
блюдается одна хронопотенциометрическая волна
[225, 226]. Следует указать, что характер ср —/-кри-
139
вой зависит от предварительной обработки электрод*:
нои поверхности, особенно в кислых средах [225, 227].
На рис. 5-9 представлены хронопотенциограммы
восстановления кислорода на палладиевом электроде
в 1,0 М NaOH, насыщенном кислородом при атмо-
сферном давлении. Кривая 3 характеризует восстано-
вление оксидной пленки в растворе 1 М NaOH, из ко-
торого был удален кислород. Из сравнения кривых 1
и 2 видно, что наличие оксидной пленки приводит к.
некоторому искажению ф— /-кривой и увеличению
переходного времени.
Лингейн [225] подробно исследовал восстановле-;
ние кислорода, а также перекиси водорода, как про-
межуточного продукта на платиновом электроде в
кислых (1 М H2SO4 и 1 М НС1О4) и щелочных раство-
рах (1 М NaOH). В кислых средах, как правило, вос-
производимость определений хуже, чем в 1 М NaOH.
Поэтому для целей анализа более приемлемы щелоч-
ные растворы.
При потенциалах переходного времени наблюдает-
ся ф — /-кривая восстановления окислов платины
[227, 228]. В 1 М NaOH такая волна появляется при
Ф = —0,8 В (нас. к. э.). На рис. 5-10 показаны ф — /-
кривые восстановления кислорода на платиновом
электроде в 1 М растворе NaOH, насыщенном кисло-
родом при атмосферном давлении. Кривая 1 снята не-
посредственно после поляризации электрода при анод-
ных потенциалах в течение 20 с, кривая 2 получалась
после записи кривой /. Задержка потенциала на кри-
вой 1 наблюдается при потенциалах отрицательнее
—0,32 В, а на кривой 2 и последующих — при менее
отрицательных потенциалах (—0,20 В). Уменьшение
перенапряжения обусловлено, очевидно, тем, что вос-
становление кислорода протекает на восстановленной
поверхности электрода. Из сравнения теоретических и
экспериментальных зависимостей /т'^/Со, от т можно
предположить, что восстановление кислорода на пла-
тиновом электроде протекает в одну четырехэлектрон-
ную стадию с образованием Н2О [225].
Восстановление кислорода на золотом электроде
в 1 М NaOH, а также в 1 М H2SO4 протекает до Н2О
в две стадии [229], при этом с увеличением продол-
жительности опыта п увеличивается. Сначала кисло-
140
род восстанавливается до перекиси водорода, которая
в щелочных растворах присутствует в виде НО2> за-
тем ион НО2 диспропорционирует на поверхности
электрода с образованием О2. Вторая ступень соот-
ветствует восстановлению ионов НО2 до ОН~:
а) О2 + Н2О + 2е” == НО2 + ОН' (первая ступень)
fe,
б) 2НО2 О2 + 2ОН"
в) НО2 + Н2О + 2е “ = ЗОН'
(вторая ступень)
Степень увеличения ti зависит от скорости проте-
кания химической реакции б). Если ki = 0, то кисло-
род не будет регенерироваться и первая волна будет
соответствовать двухэлектронному восстановлению
кислорода. С увеличением скорости реакции б) пере-
ходное время ti будет увеличиваться, при больших kt
возможно слияние первой волны со второй; <р— /-кри-
вая в этих условиях будет соответствовать четырех-
электронному восстановлению кислорода до воды.
Константа скорости ki, как показали исследования,
зависит от состояния поверхности электрода, а также
от наличия в растворе поверхностно-активных веществ
[229]. Так, в 0,1 М NaOH наблюдается одна волна
при потенциалах отрицательнее .—0,3 В (нас. к. э.).
Аналитическое определение кислорода лучше всего
проводить на платиновом электроде, хотя возможно
применение и других электродов (ртутного, золотого),
Хронопотенциометрический метод дает возможность
осуществить автоматический контроль содержания
кислорода в различных растворах.
' Двуокись углерода
Реакция восстановления углекислого газа в вод-
ных растворах, находит ограниченное применение в
аналитической практике [230, 231].
Хорошо выраженные <р — /-кривые восстановления
углекислого газа наблюдаются в неводных раствори-
телях (ацетонитриле или диметилсульфоксиде) [232].
Изучено восстановление углекислого газа в диметил-
141
сульфоксиде на амальгамированном платиновом и зо-
лотом электродах [232].
На рис. 5-11 представлена хронопотеициограмма
восстановления СО2 на амальгамированном платино-
вом электроде. Нижний предел обнаружения СО2 со-
ставляет ~ 10-4 М.
Рис. 5-11. Хронопотенцио-
грамма восстановления СО2
на амальгамированном пла-
тиновом электроде в ди-
метилсульфоксиде [232].
Условия опытов; с=4-10~3 н,
СОг
1 = 1,256 • 10“4 А/см* 2, индиффе-
рентный электролит 0,03 н
(Et)4NC104.
Величина /т^/СсогПри восстановлении СО2 на зо-
лотом электроде не зависит от концентрации углекис-
лого газа в растворе, а при восстановлении на амаль-
гамированном платиновом электроде возрастает, если
исходная концентрация СО2 в растворе менее 10“2 М.
Восстановление СО2 протекает через стадию ад-
сорбции промежуточных частиц СО2, которые претер-
певают дальнейшие превращения, образуя формиат-
ионы [232]:
на золотом электроде
СО2 + е —> • COg (аде.)
растворитель
• СО~ (аде.) -------------> • СО~ (сольв.)
растворитель, следы воды
2 • СО2 (сольв.) --------------------------> СО2 + НСО2
на амальгамированном платиновом электроде
СО2 + е~ —> «СО2 (аде.)
растворитель, следы воды
• СО2 (аде.) ч-е -------------------------> НСО2
При высоких концентрациях СО2 электровосстановле-
ние углекислого газа на амальгамированном плати-
142
новом электроде протекает по одноэлектронному ме-
ханизму.
Добавление воды в анализируемый раствор приво-
дит к уменьшению хронопотенциометрической кон-
станты (например при добавке воды в количестве 2%
константа уменьшается на 10%).
t,0 t,c
Рис. 5-12. Хронопотенциограммы восстановления персульфат-
ионов на платиновом проволочном электроде [233].
Условия опытов: С® _ „ =0,005 М; 1=2 мА/см2; рН=7.
Кг=2с>8
Персульфат-ион
На хронопотенциограммах восстановления пер-
сульфат-ионов на платиновом электроде в отсутствие
индифферентного электролита появляются периоди-
ческие осцилляции, которые затухают со временем
(рис. 5-12а). При этом обращает на себя внимание
аномальная форма ф— /-кривых. Авторы {233] по-
дробно исследовали влияние различных условий на
Ф — /-кривые восстановления персульфат-иона: со-
стояния поверхности электрода, pH раствора, концен-
трации фонового электролита, материала электрода.
На полностью окисленной поверхности платинового
электрода ионы S20g’не восстанавливаются (рис. 5-12а,
кривая /). Катодная активация поверхности электро-
да приводит к появлению ф — /-кривых (рис. 5-12а,
кривые 2 и 3) с периодическими осцилляциями. На
начальном участке ф — /-кривой наблюдается острый
минимум, затем потенциал электрода смещается в
сторону положительных значений,
143
При уменьшении pH (от 9,5 до 4) <р — /-кривая не-
сколько изменялась, по осцилляции не исчезали. Ана-
логичные осцилляции на ф — /-кривых наблюдаются
также при восстановлении-ионов SaOg' на золотом и
палладиевом электродах. Осцилляции полностью
устранялись при добавлении фонового электролита
(0,1 М NaNO3), но необычная форма ср — /-кривых
сохранялась (рис. 5-126).
Необычная форма поляризационных кривых вос-
становления персульф ат-анионов в полярографических
условиях хорошо известна и изучалась на протяжении
многих лет [121]. Авторами работы [233] изучалось
электровосстановление персульфат-иона на платино-
’вом электроде.
Для перемешиваемых растворов форма кривой не-
сколько отличается от рассмотренной: потенциал элек-
трода сначала изменяется более резко, затем плавно
смещается к отрицательным значениям (кривая 8).
Аномальная форма <р — /-кривой восстановления
ионов SaOg" на платиновом электроде в [233] объяс-
няется следующим образом: с одной стороны, пер-
сульфат-ионы способны окислять поверхность элек-
трода; с другой стороны, в-процессе катодного восста-
новления происходит частичное восстановление пла-
. типовых окислов. При наложении поляризующего тока
на электрод потенциал сначала резко смещается в сто-
рону отрицательных значений, затем начинает пре-
обладать влияние второго фактора, и потенциал сме-
щается к потенциалу восстановления персульфат-
иона. Более плавное изменение потенциала после мак-
симума в нейтральных растворах авторы [233] объяс-
няют возрастанием концентрации ионов ОН' у поверх-
ности электрода, которые образуются при восстанов-
лении окислов платины на поверхности электрода.
Б кислых растворах и при перемешивании происходит
интенсивный отвод ионов ОН* от поверхности элек-
трода, что приводит к более резкому изменению по-
тенциала электрода.
Появление осцилляций на <р — /-кривых объясняет-
ся двумя причинами: во-первых, как указывалось вы-
ше, ионы SsOe" способны окислять поверхность пла-
тины, и, во-вторых, оксидная пленка ингибирует вос-
144
становление ионов S2Oe" • Очевидно происходит чередо-
вание «активации — пассивации» электрода, что и при-
водит к осцилляциям. Затухание осцилляций можно
объяснить накоплением ионов SO42 (ионов индиффе-
рентного электролита) у поверхности "электрода со-
гласно реакции:
S2O|"4-2e- —> 2SOf"
Возможно появление новой серии осцилляций
(рис. 5-12а, кривая 4). Если осцилляции появляются
на конечном участке <р — /-кривой, то они быстро за-
тухают, поскольку концентрация ионов S2O|- у по-
верхности электрода становится равной нулю (мо-
мент достижения переходного времени).
Галогениды
Прямое определение галогенид-ионов основано на
окислении их на платиновом или графитовом электро-,
дах. До'сих пор не было опубликовано работ по пря-
мому использованию хронопотенциометрии для опре-
деления фторид-иона.
Для определения бромид-ионов лучше использо-
вать платиновый электрод [234], поскольку окисление
на нем протекает обратимо. Электродный процесс на
углеродном пастовом электроде осложняется адсорб-
цией бромид-ионов. Существуют разные точки зрения
на механизм окисления — восстановления галогенидов
на платиновых электродах. •
Постоянство 1х',2/С°цйг, в частности при окислении
бромид-ионов в 1 М H2SO4, в интервале концентраций
(1 Ч- Ю)-Ю8 моль/л указывает на отсутствие пред-
шествующей химической реакции. Отклонение в опре-
делении 1х1г1Сът~ не превышает 0,4%.
По мнению авторов [235, 236] электроактивность
галогенид-ионов на платиновых электродах связана
прежде всего с присутствием в растворе ионов HalJ,
так что электрохимическое окисление должно, оче-
видно, изображаться реакцией:
2Halj 3Halj + 2e“
14^
Таблица 5-5. Хроиопотеициометрические данные
для системы Вг~/Вга
Суммарная реакция (фон Ш H2SO4) Наклон линей- ного участка ф—/-кривой *, электрод azt электрод «тд. В (нас. к. э ). электрод
плати- новый ласто- вый платиновый насто- вый плати- новый ласто- вый
2Вг“ —> Вг2 + 2е~~ 0,056 0,093 z = 1 (обра- тимый про- цесс) 0,36 +0,92 + 1,10
Вг2 + 2е~ —> 2Вг- 0,124 0,320 0,45 0,19 +0,78 +0,43
* В полулогарифмических координатах
Хроиопотеициометрические данные для системы
Вг*/Вг2 приведены в табл. 5-5.
Иодид-ионы окисляются обратимо на пастовом
электроде, на платиновом электроде процесс ослож-
няется адсорбцией иода [237], поэтому определение
иодид-ионов в некоторых случаях лучше всего прово-
дить на углеродном пастовом электроде. Для сниже-
ния определяемой концентрации иодид-ионов авторы
[238] рекомендуют использовать, капельную хронопо-
тенциометрию. Указано на возможность определения
иодид-ионов в концентрации 5-10 * 1 * * * * 6 * В * моль/л в растворе
1 М H2SO4 на платиновом электроде.
Поведение системы Г/12 зависит от типа электрода.
В работе [239] был предложен следующий механизм
окисления иода на пирографитовом электроде:
а) Г —> у12 + е“
б) у 12 + Н2° “* 10' + 2н+ + е~
в) 1СГ + 2Н2О —> IO’ + 4Н+ + е~
В зависимости от тока окисление иодид-ионов может
быть одноступенчатым или двухступенчатым [239].
Так, если Cki=10~3M, то при / = 6-10~4А (s =
= 5,3 см2) на хронопотенциограмме окисления иодид-
ионов в 0,1 М NaC104 на пнрографитовом электроде
|46
появляются две ступени, если / = 16-10~4 А, то одна
ступень. Очевидно, механизм окисления ионов I- вклю-
чает несколько отдельных стадий [240]. Аналитиче-
ское определение иодид-ионов в указанных условиях
необходимо проводить при высоких плотностях тока.
Окисление хлорид-ионов протекает при большом
перенапряжении, при этом молекулярный хлор реаги-
рует с материалом электрода [234]. •
Для определения галогенидов можно использовать
метод, основанный на электролитическом генерирова-
нии ионов серебра (I) [135]. При анодном растворе-
нии серебряного электрода в растворах галогенидов
на его поверхности образуется труднорастворимая
пленка галогенида серебра. В этом случае переход-
ное время соответствует времени, в течение которого
концентрация галогенида у поверхности электрода
достигает нуля (Снаг = 0), Чувствительность метода
ограничивается величиной произведения растворимо-
сти соли. В изученном интервале концентраций хло-
рид-ионов 5-10—4—0,01 моль/л отклонение в измере-
нии хронопотенииометрической константы не превы-
шало 4%. Хорошо выраженные хронопотенциограммы
наблюдаются с использованием электрода, поверх-
ность которого предварительно покрывали тонким
слоем хлористого серебра. Можно определять хлорид-
ионы в различных фоновых электролитах (Na2SO4,
H2SO4, NaNO3). Не рекомендуется применять метод
при больших концентрациях хлорид-ионов (Ссг >
> 0,1 моль/л).
Щавелевая кислота
Окисление различных органических соединений на
платиновых электродах осложняется протеканием хе-
мосорбционных и химических процессов.. Изучено
окисление щавелевой кислоты хронопотенциометриче-
ским методом на платиновом, золотом и палладиевом
проволочных электродах [59, 168, 228, 241—243]. Хро-
нопотенциограмма окисления на платиновом и золо-
том электродах наблюдается при потенциалах поло-
жительнее +0,9 В (нас. к. э.). Как указывалось выше,
характер ср — /-кривой на платиновом и золотом элек-
тродах зависит от предварительной подготовки элек-
147
тродной поверхности. Оксидная пленка, имеющаяся
на электроде, ингибирует окисление щавелевой кис-
лоты. Присутствие веществ, адсорбирующихся на пла-
тиновом электроде, таких, как амиловый спирт, рода-
нид- или хлорид-йоны, также приводит к искажению
<р /-кривых, что свидетельствует об адсорбции щаве-
Рис. 5.13. Хронопотенцио-
граммы окисления 0,01 М
Н2С2О4 в IМ НС1О4 на
палладиевом электроде.
Кривая 1 получена на неокислен-
ном, кривая 2— на окислен-
ном [244] электродах.
левой кислоты на электроде [241]. Аналогичного ин-
гибирования не замечено при окислении щавелевой
кислоты на палладиевом электроде в 1 М НС1О4 или
1 М H2SO4. Характерные <р — /-кривые окисления ща-
велевой кислоты на палладиевом электроде [243] при-
ведены на рис. 5-13. Первая задержка на кривой 1
соответствует образованию оксидной пленки PdO. При
потенциале 4-1,3 В начинается окисление щавелевой
кислоты. Кривая 2 была снята после кривой 1. Как
видно из рис. 5-13, на ней отсутствует первая задерж-
ка. Таким образом, окисление щавелевой кислоты про-
текает на окисленной поверхности палладиевого элек-
трода. Исходя из сравнения теоретических и> экспери-
ментальных результатов, можно предположить, что
окисление щавелевой кислоты протекает по следую-
щему механизму [243]:
a) PdO-J-2OH~ —> PdO2 + Н2О + 2е-
б) PdOa + Н2С2О4 —> 2СО2 + Н2О + PdO
В кислых растворах последующая химическая реак-
ция протекает быстро. В нейтральных или щелочных
растворах высший окисел PdO2 не восстанавливается
или же восстанавливается достаточно медленно, т. е.
появляются кинетические ограничения. В пользу ука-
148
заййого механизма говорит тот факт, что окисление
щавелевой кислоты протекает на окисленной поверх-
ности, а также отсутствие ингибирования процесса по-
верхностно-активными веществами или анионами
(амиловый спирт, ионы С1~, CNS-) [243].
При использовании в качестве растворителя диме-
тилсульфоксида <р — /-кривая окисления щавелевой
кислоты на платиновом электроде наблюдается при
высоких положительных потенциалах <рг/4 =.4-1,25 В
(нас. к.э.) [244]. На фоне гидроокиси тетраэтиламмо-
ния на <р — /-кривой появляются три задержки потен-
циала, что указывает на сложность механизма окис-
ления [244].
Муравьиная кислота и формиат-ионы
Окисление муравьиной кислоты в 1М растворе
H2SO4 на платиновом электроде, так же как и окисле-
ние щавелевой кислоты, сопровождается адсорбцией
самой кислоты и промежуточных веществ. Окисление
ингибируется хемосорбированным кислородом. Хроно-
потенциометрическая кривая окисления наблюдается
[241] при потенциалах положительнее 4-0,9 В (нас.
к. э.).
Формиат-ионы на платиновых электродах в водных
растворах не окисляются [245, 246], поэтому их окис-
ление целесообразно проводить в неводных растворах.
Формиат-ионы дают хорошо выраженную одноэлек-
тронную хронопотенциометрическую волну окисления
на платиновом и золотом электродах на фоне 0,1 н.
раствора перхлората тетраэтиламмония в диметил-
сульфоксиде [247]. Величина <pt/4 равна 4-0,33 В
(нас. к. э.) при окислении на платиновом и —0,07 В
(нас. к.э.)—при окислении на золотом электродах.
Независимость произведения it'/* в изученном интер-
вале значений тока (4—120 мкА) свидетельствует об
отсутствии предшествующих химических реакций.
Исследования с реверсом тока позволяют устано-
вить, что при окислении формиат-ионов образуются
муравьиная кислота и углекислый газ:
НСОО* —> 4- СО2 + ± НСООН + е~
Л
149
Согласно предполагаемому в [247] механизму, окис-
ление формиат-ионов протекает через’ образование
промежуточных активных частиц, распадающихся на
СОа и атомы водорода, которые затем адсорбируются
на электроде. Следует отметить, что тот или иной ме-
ханцзм не полностью объясняет всю сложность про-
Рис. 5-14. Хронопотенцио-
граммы окисления металло-
ценов в 0,2 М LiC104 (в аце-
тонитриле) [251]:
* —1,86 • 10~8 Af осмоцена, 1 —
= 0,338 - 10-8 А/см2; 2-8,18 X
X 10“4 М рутеноцена, i=0,22 X
X Ю“3 А/см2; 3—3,25 • 10-8 М
ферроцена, 1=0,55 • 10—3 А/смХ
текающих процессов при окислении органических ве-
ществ, в частности, формиат-ионов и муравьиной кис-
лоты [248, 249].
Ферроцен, рутеноцен, осмоцен и их производные
Ферроцен, рутеноцен и осмоцен представляют со-
бой соединения циклопентадиена с железом, рутением
и осмием. Известны аналогичные соединения, содер-
жащие атомы других металлов — Со, Mo, Ni, Мп, Сг,
V, Ti, — которые по своим свойствам близки к ферро-
цену. Хронопотенциометрические кривые окисления
[250] ферроцена, рутеноцена и коб.альтоцена на ртут-
ном капельном электроде в растворе этилового спир-
та, содержащего NaClO* и HCIO4, сливаются с кривой
растворения ртути, поэтому измерить характеристики
— /-кривых не удается.
Четкие <р — /-кривые окисления ферроцена, рутено-
цена и осмоцена (рис, 5-14) получены на платиновом
150
(пластинчатом) электроде в ацетонитриле на фоне
0,2 М LiC104 [251]. Окисление ферроцена протекает
по схеме:
(С(Нв)гРе = (CsHs^Fe* + е~
Значение фо,22 — +0,300 В (потенциал ф0,22 соответ-
ствует фт/4 на кривой окисления), найденное из кри-
вой восстановления продукта окисления ферроцена,
приблизительно совпадает с фт/4= 0,307 В, что указы-,
вает на обратимость процесса. Хронопотенциометри-
ческие характеристики электрохимического поведения
ферроцена, рутеноцена, осмоцена и их производных
приведены в табл. 5-6. Результаты изучения показали,
что окисление осмоЦена — двухступенчатый процесс.
Из табл. 5-6 видно, как влияют заместители на реак-
ционную способность производных металлоценов. Най-
дена корреляция между фт/4 и константой заместителя
at [252]. Эта зависимость точнее всего (с коэффициен-
том корреляции 0,979) описывается уравнением фт/4 =
= 0,431 У, + 0,367, где ар — константы Гаммета
р-i
[253]?
Таким образом, на основании хронопотенциометри-
ческих измерений можно получить данные о зависи-
мости между строением и реакционной способностью
металлоценов и их производных.
В работе [254] изучено окисление ферроцена и би-
ферроценила (Fc—Fc) на платиновом электроде в ор-
ганических растворителях — ацетонитриле и хлори-
стом метилене. При добавлении 0,1 М хлорнокислого
тетраэтиламмония к ацетонитрилу ф — Z-кривая окис-
ления ферроцена смещается в сторону положительных
потенциалов (фт/4 = + 0,385В), а окисление биферро-
ценила протекает через две одноэлектронные стадии
согласно следующей схеме [254]:
Fc—Fc —> Fc+—Fc + e- (1-я задержка)
Fc+—Fc —► Fc+—Fc+ + e~ (2-я задержка)
т. e. две ферроценильные, группы окисляются при раз-
личных потенциалах.
Аналогичные результаты получены при окислении
ферроцена и биферроценила в хлористом метилене в
151
Таблица 5-6. Результаты изучения хронопотенциометри-
ческого поведения ферроцена, рутеиоцена, осмоцена
и их производных в ацетонитриле на фоне 0,2М L1C1O< '
Металлоцены и их Фт/4, В (нас. к. э.) <Г0.22.В с« • хокнсл
^производные (нас.,к.э.) А-с‘5-л СМ2 «МОЛЬ ‘’ВОССТ
Ферроцен Производные ферро- цена +0,307±0,002 +0,300 417+5 (2=1) 3,3 4 1
- СН2СНз 4-0,245 +0,235 370+6 3,0
-СНОНСНз +0,298 +0,304 342+6 3,2 1
-Н ' +0,307 +0,300 417+5 3,1 J
-СНОНС6Н5 +0,318 +0,315 335+11 3,8 ’
—CH=CHj +0,325 +0,319 378+10 3,5 J
-соон +0,550 +0,57 +0,35 282 +5 2,8
—COCeHs +0,571 +0,568 342+20 4,7 |
-COCH3 +0,573 +0,570 400+24 * 8 1
1.1'-(СН,СН»), +0,194 +0,186 367+3 3,0 а
Рутеноцен Производные рутено- цен а +0,693+ 0,003 +0,315 807+10 (2=1) V
-СНОНС6Н5 +0,755 4-0,18 577+7
—COCeHs +0,907 4-0,45 666+27 *
l,l'-(COC6H5)j Осмоцен: + 1,089 +0,49 670+59 * —
1-я ступень 2-я ступень’ Производные осмо- цена +0,633± 0,005 + 1,50± 0,01 +0,03 403+10 (2 = 1) 755±8 ** (2=1) 1 j _ 1
- СОСаН6 +0,866 0,0 505+50 * "" i
* Равно значению > (п + Т2>/С°. .
** Приведены значения при низких плотностях тока.
присутствии 0,05 М хлорнокислого тетрабутиламмония
[254].
Гидразин и гидроксиламин и их смесь
Гидразин и гидроксиламин окисляются на плати-
новом электроде на фоне 0,5 М H2SO4; потенциал <рт/4
кривой окисления гидразина равен ф-0,36 В, а гидро-*
ксиламина — -фО,83В (нас. к. э.) [255],
15?
При совместном окислении гидразина (zi = 4) и
гидроксиламина (z2 = 6) [255] переходное время, со-
ответствующее окислению гидроксиламина, в соответ-
ствии с теорией существенно увеличивается в присут-
ствии гидразина. При 50-кратном избытке гидроксил-
амина ф — /-кривая окисления гидразина плохо выра-
жена. При 200-кратном избытке гидроксиламина
Ф— /-кривая гидразина не наблюдается. По мере ро-
ста концентрации гидразина точность определения
гидроксиламина уменьшается — при 50-кратном из-
бытке гидразина ошибка определения -составляет
±16%. Определению мешает присутствие в растворе
хлорид-ионов в концентрации 2-Ю'3 моль/л и выше.
Хронопотенциометрическое поведение гидразина и ги-
дроксиламина изучено недостаточно. На фоне 0,1 н.
серной кислоты гидразин окисляется до азота, а гид-
роксиламин— до нитрата. При окислении гидразина
на платиновом электроде в 1,44 М H2SO4 [256] вели-
чина не остается постоянной, а линейно воз-
растает с увеличением плотности тока, что указывает
на кинетическое осложнение электродного процесса.
При окислении гидроксиламина на платиновом аноде
наблюдается 6-электронная хронопотенциометриче-
ская волна [257]. При увеличении pH на ф — /-кривой
появляется несколько задержек.
Определение элементов на фоне
концентрированной фосфорной
кислоты
В концентрированной фосфорной кислоте при тем-
пературах выше комнатных хорошо растворяются та-
кие вещества, как оксиды металлов, силикаты, метал-
лы, которые практически не растворимы в воде; к то-
му же фосфорная кислота обладает хорошей электро-
проводностью. Благодаря этим свойствам концентри-
рованную фосфорную кислоту можно использовать в
качестве растворителя и фонового электролита при
определении элементов электроаналитическим и мето-
дами.
Серия работ посвящена изучению хронопотенцио-
метрического поведения ванадия(У), селена(IV) и
теллура (IV) [258—260], а также серебра (I), урана (VI)
153
и ртути(И) [261—263] на фоне концентриройан-
ной фосфорной кислоты. Использовалась смесь
кислот следующего состава: ортофосфорная кисло-
та —44%, пирофосфорная кислота —40%, трифосфор-
ная кислота—12% и тетрафосфорная кислота — 4%
(общее содержание Р2О5 — 77%).
Рис. 5-15. Реверсионная
хронопотенциограмма вос-
становления — окисления
Те (IV) — Те (III) в концен-
трированной фосфорной ки-
слоте [260].
Условия опыта: С®, , =1,88 X
le (IV)
ХЮ2 моль/л; /-»5,5Ы0~4 А/см2;
/=100 °C.
Индикаторным электродом служила золотая про-
волока (d — 0,5 мм) с эффективной поверхностью
0,0770 см2. Катодные хронопотенциограммы восстанов-
ления ионов ванадия (V), селена (IV) и теллура (IV)
хорошо выражены. В .качестве примера на рис. 5-15
представлена реверсионная хронопотенциограмма для
Те (IV) [260].
Как правило, ионы V(V), Se(IV) и Те(IV) на фоне
фосфорной кислоты восстанавливаются двухступен-
чато. Для аналитических определений может быть ис-
пользована первая ступень, соответствующая первой
стадии электрохимической реакции. Воспроизводит
мость переходных времен первой ступени составляет
~3%. Для изученных интервалов концентраций
(3,40—19,74) -10—3 моль/л и плотностей тока
(0,935—8,69) -10 4 А/см2 соблюдается линейная зави-
симость между it1* и Со (/ = 100 °C).
Электродная реакция восстановления V(V), соот-
ветствующая первой ступени, обратима, а для Se(IV)
и Те (IV)—необратима. Ионы теллура(IV) восстанав-
ливаются сначала до металлического состояния, а за-
тем до Те(—2), при этом <р — /-кривая, соответствую-
154
щая второй ступени, сливается с кривой выделения
водорода. Возможно одновременное определение
V(V), Se (IV) и Те (IV).
Результаты изучения электровосстановления ионов
ванадия^), селена(IV) и теллура(IV) приведены в
табл. 5-7.
Т а б л и ц а 5-7. Результаты изучения электровосстановления
ионов V(V), Se(lV) и Te(lV) в концентрированной фосфорной
кислоте при 100 °C
Электродная реакция M04, А/см2 Начальная кон- центрация O-IOS.M в (оти. Hg/Hg2SO4 Лите- рату- ра
V(V) -> V(IV) - е- V(IV) -> V(III) - е- 0,935— 3,74 1,41 3,91-19,74 [V(V)] 0,63 0,23 [258]
Se(IV) —> Se(0)-4e~ Se(0) -> Se(—2) - 2e~ 5,24 5,24 3,40 [Se (IV)] 0,19 -0,65 [259]
Te(IV) -> Te(.O) - 4e- Te(0) -> Te(—-2) - 2e- 3,91 3,49 [Te(IV)] -0,09 Выделе- ние водо- рода [260]
Очевидно, в фосфорной кислоте можно также .про*
водить хронопотенциометрическое определение эле-
ментов подгруппы вольфрама, подобно полярографи-
ческим определениям, описанным в [264].
* * *
1
Есть сведения о хронопотенциометрическом по-
ведении о-фталевой [265] и никотиновой [266] кислот
на ртутном капельном электроде, n-метоксифенола на
углеродном пастовом электроде [267], аскорбиновой
кислоты на золотом цилиндрическом электроде [268],
хинина [269] и двуокиси серы [270] на ртутных элек-
тродах.
Глава 6
ПРИМЕНЕНИЕ ХРОНОПОТЕНЦИОМЕТРИИ
ДЛЯ ФИЗИКО-ХИМИЧЕСКИХ ИССЛЕДОВАНИЙ
Хронопотенциометрия, особенно в последнее время,
широко применяется для решения ряда физико-хими-
ческих задач. В этой главе будут показаны возмож-
ности хронопотенциометрии для изучения кинетики
электродных процессов, определения коэффициентов
диффузии и изучения явлений адсорбции.
6.1. ИЗУЧЕНИЕ КИНЕТИКИ ЭЛЕКТРОДНЫХ
ПРОЦЕССОВ
Основной задачей при изучении кинетики элек-
тродных процессов является установление природы
замедленной стадии и определение кинетических па-
раметров io(ks) и a (io — плотность тока обмена).
Плотность тока обмена связана с константой скорости
следующим соотношением:
i0 = zFks(cl)l'a\c^)a (6-1)
Для нахождения'As обычно логарифмируют уравнение
(6-1):
]g Zo = 1g zFks + a 1g C°R + (1 - a) 1g (6-2)
Если теперь построить график lgi0 от IgCo. TO полу-
чим прямую, тангенс угла наклона которой равен
(1— а). Зная плотность тока обмена для каких-либо
концентраций Со и Ср, нетрудно рассчитать констан-
ту скорости ks. t
„ В работах [14, 167] предложен метод анализа
<р — (-кривой для электродных процессов, ограничен-
ных собственно электрохимической реакцией и диф-
фузией [271]. Этот метод позволяет из отдельного
измерения определить плотность тока обмена io и ко-
эффициент переноса а, Влияние заряжения двойного
156
слоя устраняется только путем экстраполяции. Для
таких процессов оказывается справедливым уравне-
ние (2-18), которое удобно представить в координа-
тах т) — t, где т] — поляризация. Запишем его в сле-
дующем виде:
RT , I RT , ,Л
— т1 =—F-ln------=•!/) (6-3)
azF t0 azF ' '
где
то, tr — переходное время соответственно для процессов восста-
новления и окисления.
График зависимости —ц от логарифмической функ-
ции L(t) представляет прямую линию, по наклону ко-
торой можно определить а*, а по отрезку, отсекае-
мому на оси ординат, — плотность тока обмена i0. Для
расчетов, проводимых по уравнению (6-3), необходи-
мо по катодным и анодным <р— /-кривым определить
величины переходного времени т0 и tr, отвечающие
рассматриваемой плотности тока. В упрощенном слу-
чае [205], предполагая Cr = 0 (т. е. в растворе при-
сутствует очень малое количество восстановленной
формы деполяризатора), кинетические параметры мо-
жно определить по отрезку, отсекаемому на оси орди-
нат при t = 0. Из уравнения (6-3) находим:
(6's>
Из зависимости —ц (0) от плотности тока определяют
io и а.
Оценим возможности хронопотенциометричёского
метода, исходя из критерия обратимости электродных
процессов. Согласно [55], условием обратимости яв-
ляется неравенство (при Со — Cr — С0 и а = 0,5):
i/Z0<7s (6-6)
* В общем случае в уравнении (6-3) вместо г более пра-
вильно писать га — число электронов, участвующих в электрохи-
мической стадии. Тогда из уравнения (6-3) определяют не а, а
произведение azo.
157
Отсюда следует, что чем больше плотность поляри-
зующего тока, тем большее значение плотности тока
обмена можно определить из хронопотенциометрйче-
ских измерений. Оценивая значение ks, получим:
ks^5i/zFCD (6-7)
Величину ijzFCG оценим из условия i > tc, когда
можно пренебречь влиянием емкостного тока. Необ-
ходимо, по крайней мере, чтобы i 100tc. Отсюда
плотность поляризующего тока должна быть равна:
1>100СаД<р/то (6-8) '
Выражая т0 согласно уравнению (2-10), получим:
zFC° 400Q ДФ ;
i nzFD0C° '
Если принять Са = 20- 10~6 Ф/см2, Д<р = 0,1 В, z = 2,
Do = 6-10~8 см2/с, С° = 10~6 моль/см8, то из (6-7) и .
(6-9) будем иметь: ks 2,5-10~2 см/с, т. е. в обычных ,
условиях хронопотенциометрическим методом могут
быть изучены электрохимические реакции, константа
скорости которых не превышает 2,5-10~2 см/с.
При высоких перенапряжениях (|т]| Э* RTjzF) ’I
уравнение (6-3) упрощается:
RT , i RT
— т)—
(6-10)
In - ------In I 1---jj-
azcF i0 azaF \ xg
Из уравнения (6-10) видно, что во времени изме-
няется лишь диффузионная составляющая поляриза-
ции, а электрохимическая составляющая должна
мгновенно принимать определенное значение. В дей-
ствительности, как будет рассмотрено ниже, время,
за которое поляризация принимает значение, равное
электрохимической составляющей, определяется вре-
менем заряжения двойного электрического слоя. Тем
. не менее уравнение (6-10) часто используют для рас-
чета величины aza и плотности тока обмена i0. Для
этого строят графики зависимости —т]
in[l-(/AMs)] [1, 20, 135, 272—274]. По тангенсу
угла наклона прямой, равному RT/(azaF), рассчиты-
вают. aza. Экстраполяция этой прямой к значению
t = 0 дает величину RT/(azaF) In (i/i0), по которой рас-
считывают плотность тока обмена i0.
от
168
Проще всего осуществлять экстраполяцию к t = О
для небольших поляризаций (|т]| С RT/zF), не пре-
вышающих, например, 5 мВ, и малого времени элек-
тролиза (/ С tr и t <С т0). При этом поляризацию
можно представить в виде суммы двух составляю-
щих— электрохимической и концентрационной [14].
Из уравнения (6-3) для малых |т)| получим:
_ i [ RT
zF i0 zF
(6-11)
Из уравнения (6-11) видно, что —т] линейно зависит
от А и от плотности тока. Экстраполяция графика
зависимости —т] от t'1* к t — 0 дает величину электро-
химической составляющей поляризации.
Поскольку при выводе уравнения (6-11) емкост-
ный ток не учитывается, то оно не совсем правильно
описывает зависимость т] — t при t—>0. В работе [167]
получено уравнение зависимости —ц от t, которое
учитывает заряжение двойного электрического слоя:
- т] = — 4- + — i М*А “ — №Cdl (6-12)
zF i0 zF [я4 zF J
где N = —
Это уравнение отличается от уравнения (6-11) на ве-
личину емкостной составляющей поляризации. Для
расчета плотности тока обмена по уравнению (6-12)
строят график зависимости г) — fk, который представ”
ляет собой прямую с наклоном
6/
2RTNI
Затем определяют время при котором
RT i
“ zF i0
(6-13)
(6-14)
т. е. когда разность слагаемых в уравнении (6-12)
равна нулю. При этом
= Л2С (6-15)
я* zF
159
откуда
/‘/г== n'^Rt r nCd . ...1
tl -^F-NCd—lF6t (6'16|
Определив с помощью какого-либо метода емкости
двойного электрического слоя Cd, из соотношении
16-16) находят t'[‘. Затем по графику (— т]) — t'h на-Я
ходят — при соответствующем значении t'/1 и из®
соотношения (6-14)' определяют плотность тока об- i
мена. Наконец, зная i0 для различныхСо при условии-1
Cr = const, определяют ks и ас помощью соотно-1
шения (6-1). При Co=10-3Af, Cd =40-10~6 Ф/см2 иЯ
Do = I0~s см2/с указанный прием позволяет оцреде-1
лить значения ks < 1 см/с [275]. ’
Определение кинетических параметров методом^
прямой хронопотенциометрии рассматривалось также)
и в работе [276], а методом хронопотенцио&тетрии c.j
реверсом тока — в работе [53]. В последнем случае ’
для расчета кинетических параметров уравнения
<р-—^-кривых приводились к числовому виду и рас-»
считывались на электронно-вычислительной машине.-
Для нахождения aza авторы [23] использовали .
производную хронопотенциометрию, расчеты прово-
дили по уравнению (3-48).
Хронопотенциометрия широко применяется для
изучения быстрых электродных реакций. Для этого
используются одноимпульсный, двухимпульсный и ци-
клический гальваностатические методы. В одноим-
пульсном гальваностатическом методе электрод поля-
ризуют прямоугольными импульсами тока большой
плотности, при этом зависимость потенциала от вре-
мени изучают в течение очень коротких промежутков
времени (нескольких микросекунд).
Анализ одноимпульсного гальваностатического ме-
тода для электролиза при отсутствии концентрацион-
ной поляризации был дан в [277], а для процессов со
значительной концентрационной поляризацией —в
[167] и.затем в работах [278, 279]. ч
Детальный анализ зависимости —ц от t дан в pa- i
ботах [280, 281] для случая электроосаждения метал-
лов (активность восстановленной формы равна 1). >
Применение одноимпульсного гальваностатического -1
160
метола рассмотрено в [282, 283]. Определение пара-
метров очень быстрых реакций (ks 1 см/с) затруд-
нено тем, что в первый момент после включения тока
происходит заряжение двойного электрического слоя.
Чтобы уменьшить время, затрачиваемое на этот про-
цесс, используют двухимпульсный гальваностатиче-
ский метод, предложенный в [284, 285]. На исследуе-
мый электрод подают последовательно два импульса
тока, причем первый импульс тока большой плотно-
сти заряжает электрод до заданного потенциала за
время порядка 0,1 мкс; затем ток скачкообразно сни-
жают до величины, отвечающей данному перенапря-
жению. Длительность второго импульса составляет
10 мкс. Если соотношение амплитуд выбрано пра-
вильно, то после переключения весь ток расходуется
на фарадеевский процесс, и на кривой т] — t должна
наблюдаться горизонтальная площадка; в этот момент
влиянием емкости двойного слоя можно пренебречь..
Для нахождения приближенных значений iQ и ks,
т. е. при условии отсутствия концентрационной поля-
ризации во время прохождения первого импульса, мо-
жно использовать простое соотношение
^7 г2 /с «7»
(647)
где /а — амплитуда второго импульса.
Для более точных расчетов следует пользоваться
уравнением
+ (6'18)
zF \ i0 Зл» 1 /
полученным в работе [278], в которой показано, что
в момент окончания первого импульса if концентра-
ционной поляризацией пренебрегать нельзя. Из
табл. 6-1 видно, что значения io, найденные для реак-
ции Hg^+ 4- 2е~ 2Hg на фоне 1 М НСЮ4 без учета
и с учетом концентрационной поляризации отличают-
ся в 2 раза. Различаются также и коэффициенты пе-
реноса.
Двухимпульсный гальваностатический метод раз-
вит в [286—291]. Тем не менее он не имеет заметных
преимуществ по сравнению с методикой нахождения
кинетических параметров одноимпульсным методом
1/26 Зак. 1072 161
[282, 290]. В [291] указывается на ограниченность
применения двухимпульсного метода. Из-за несовер-
шенства аппаратуры, а также из-за омического паде-
ния напряжения в растворе начальный участок ф— t-
кривой при t t\ не наблюдается на экране осцилло-
графа. В результате этого трудно подобрать соотно-
шение амплитуд первого и второго импульсов, при
котором выполнялось бы условие =^0. Для
перезаряжения двойного электрического слоя необхо-
димы высокие плотности тока второго импульса, что
приводит к значительному падению напряжения в
растворе.
Теоретическая оценка показывает [291], ,что мак-
симальная константа скорости ks, которая может быть
определена двухимпульсным гальваностатическим ме-
тодом с ошибкой 20%, Составляет 0,5 см/с. Ограниче-
ния двухимпульсного метода, о которых говорилось
выше, приводят к тому, что для быстрых электрод-
ных реакций с плотностями тока обмена выше
(0,150—0,200) А/см2 получаются заниженные значе-
ния i0. Так, для реакции Hg|+ + 2e_ 2Hg на фоне
1 А4 НС1О4 значения io, найденные разными авторами,
не согласуются между-собой (табл. 6-1).
Возможности метода можно расширить, если ис-
пользовать комбинацию гальваностатического импуль-
са тока с последующим кулоностатическим импуль-
сом (порядка 3,5-10-9 Кл). Величина заряда второго
импульса выбирается таким образом, чтобы на кри-
вой ц— t наблюдался горизонтальный участок, т. е.
Таблица 6-1. Значения io для реакции Hg|* q. 2е- 7* 2Hg
в 1М НС1О4
г" GHg2*-108. г-ион/л 2 io, А/см*, по данным
[2781 [284J [291]
2 0,350 0,420 0,700
1 0,200 0,250 0,400
0,6 0,120 0,180 0,200
0,2 0,075 0,080
0,1 0,035
162
так же, как и в двухимпульсном методе. После нало- '
жения такого кулоностатического импульса ток’ не
протекает через ячейку, следовательно, измеряемая
величина поляризации не содержит омической состав-
ляющей. Оценивая возможности импульсного метода
с последующим кулоностатическим импульсом, авто-
ры [290] указывают, что этим методом можно. на*
ходить значения констант скоростей rs « 10 см/с с
ошибкой в 50%.
Вариант гальваностатического метода — так назы-
ваемый циклический гальваностатический метод —
был предложен в работах [292—294]. Принцип этого
метода заключается в том, что на ячейку подаются
прямоугольные импульсы тока определенной частоты
/, и с помощью осциллографа регистрируются значе-
ния —я для четных —т](0") и нечетных —ц (О') пе-
риодов. Для определения кинетических параметров
регистрируют не сами изменения потенциала т) (О') и
т](0") (0 —доля периода), а их разность, так как в
результате дрейфа нуля у генератора прямоугольных
(изменение потенциала для каждого последующего
импульса отлично от изменения потенциала предыду-
щего импульса). Окончательное уравнение можно за-
писать в виде
Г 1 4NL (6) 1
1ч (6') - ч (0")1е - 7=~ (6-19)
zF L «о V z/ J
где L (6) — функция 0, 1Л — амплитуда тока.
Уравнение (6-19) справедливо при малых | т) | и не
учитывает заряжения двойного электрического слоя,
что оправдывается при низких частотах (f 200 Гц).
Условие использования низких частот обусловли-
вает и возможности циклического гальваностатиче-
ского метода. Согласно оценке, сделанной в [295] при
Do = Dr = 1O~S см2/с, f — 200 Гц и Со = Сц — С°, на-
ходим ks ^0,5 см/с, ,т. е. возможности циклического
гальваностатического метода такие же, как у одноим-
пульсного и двухимпульсного гальваностатического
метода.
* Циклическое состояние легко устанавливается,
если на ячейку вместо прямоугольных импульсов тока
наложить синусоидальный ток [296]. В этом случае
V.6*
163
легко, «отфильтровывать» постоянную составляющую
тока, которая вызывает изменение концентрации реа-
гирующих веществ у поверхности электрода. Данный
метод аналогичен циклическому гальваностатическо-
му методу, но определение кинетических параметров
уирощается. Плотность тока юбмена находится из зна-
чения т] при 6 = 0,125;
Че-о-,25=^й; (6'20)
где ai— амплитуда переменного тока.
Зная i0 при различных Концентрациях амальгамы,
можйо определить а по известной методике.
В [297]' предложена разновидность циклических
методов — циклическая хронопотенциометрия, теория
которой рассматривалась на основе принципа «адди-
тивности функций отклика» [69]. В методе цикличе-
ской хроиопотенциометрии направление тока изменя-
ется при достижении переходного времени. Это. значи-
тельно упрощает количественное описание метода.
Полученные уравнения с помощью указанного выше
принципа связывают импульсную плотность тока и
концентрации электроактивных веществ с последова-
тельными, изменяющимися от цикла к циклу, пере-
ходными временами. Эти уравнения являются транс-
цендентными относительно переходных времен и мо-
гут быть решены путем итераций.
Если io = i'r = i, то получаются' следующие зави-
симости:
для нечетных циклов (п = 3, 5..7,
• (at + аг + Яв + ... + an)b — 2 (аз + а3 + ... + +
4- ... + 2a£<=1 (6-21)
для четных циклов (п = 2 4, 6, ...)
Drcr и
д'/а^о " — (д< + + а3 + ... 4- ап)— 2 (аз + ... + ап),г 4-
4- ... 4-2а£ (6-22)
где ап = т„/Т1
Значения ап можно получить путем решения урав-
нений (6-21) и (6-22).
Для одного из циклов кинетические параметры ка-
тодного и анодного процессов можно рассчитать по
164
уравнению (2-17). Константы скорости катодного
и анодного k°b процессов нетрудно определить по вег
личине потенциала при t = 0 в соответствии с урав-
нением (2-17).
Циклическая хронопотенциометрия, теория кото-
рой разработана достаточно полно [159, 298—300],
дает возможность изучать образование промежуточ-
ных продуктов первичных реакций, а также кинетику
химических реакций, протекающих после переноса за-
ряда в первичном электродном процессе. По переход-
ному времени вторичного процесса можно,судить о
скорости химической реакции. Для установления ме-
ханизма электродного процесса применяется главным
образом циклическая хронопотенциометрия.
Хронопотенциометрия широко используется для
изучения электродных процессов с участием комплек-
сов металлов [301]. Постоянство при изменении
плотности трка указывает на отсутствие предшествую-
щей диссоциации комплекса, а в случае кинетически
ограниченного электродного процесса произведение
уменьшается по линейному закону с увеличением
плотности тока.
Выражение для плотности тока обмена на амаль-
гамном электроде в растворе, содержащем' комплекс-
ные ионы металла, имеет вид [301]:
{0 ^K^Mex’^MefHg) (Сх) ₽ (6-23)
где — плотность стандартного тока обмена системы
MeXP|Me(Hg) при стандартном потенциале фр! СМеХркоицентра-
ция комплекса, участвующего в электрохимической реакции
вблизи равновесного потенциала; Сх — концентрация свободного
лиганда.
Подробный анализ выражения (6-23) дан в рабо-
те [301].
Если ионы металла находятся в растворе в виде
комплексов одного типа, то координацйонное число р
можно найти из зависимости 1g t0 — IgCx (при боль-
шом избытке в растворе лиганда):
a ig/о
51g Q
- (I - а) л
(6-24)
165
где « — координационное число комплекса, преимущественно при-
сутствующего в растворе (п находят по зависимости потенциала
от концентрации лиганда).
' Для необратимого восстановления комплексов ме-
таллов при большом избытке лиганда в растворе
уравнение <р — /-кривой имеет вид [301]:
_ 2,3/?Г Г i0KC°rtK„ ( t'>‘ \1
(6-25)
где Кр и Кп — константы устойчивости комплексов МеХр и МеХя
соответственно.
Из уравнения (6-25) получаем:
дф _______ 2,3 (п — р) RT
dlg(?x azaF
(6-26)
Порядок по лиганду реакции необратимого восста-
новления комплекса определяется по формуле:
(и7)
Хронопотенциометрический метод был использован
для изучения многочисленных комплексов [163, 224,
302—308].
6.2. ОПРЕДЕЛЕНИЕ КОЭФФИЦИЕНТОВ ДИФФУЗИИ
Метод хронопотенциометрии может быть использо-
ван для определения коэффициента диффузии [4, 44,
309]. Этот метод является более точным, чем поляро-
графический метод [1], так как математическая трак-
товка процесса диффузии является в этом случае
более строгой, чем для капельного ртутного элек-
трода.
Коэффициенты диффузии находят из выражения
для переходного времени. Если выполняются условия
линейной, диффузии, то из соотношения (2-10) полу-
чим следующую расчетную формулу:
Do = (1,17- 10-5Кд/г)2 (6-28)
Если электродный процесс осложнен предшествую-
щей химической реакцией, то коэффициент диффузии
определяют, из экспериментально полученной зависи-
мости от i по уравнению (4-25).
В условиях цилиндрической диффузии выражение,
для переходного времени не позволяет непосредствен-
но вычислить коэффициенты диффузии [см. уравне-
ние (2-35)]. Для нахождения коэффициента диффу-
зии в этом случае используют метод подбора [59]:
сравнивают теоретически рассчитанные и эксперимен-
тальные зависимости йг/Со от т при различных зна-.
чениях Do. Искомое значение Do соответствует пол-
ному совпадению указанных зависимостей.
Коэффициент диффузии можно также находить по
зависимости т] используя гальваностатический
[282] или циклический методы [294]. В самом деле,
из экспериментальных данных по уравнению (6-12)
нетрудно получить величину N. Величину Do находят
по прямолинейному графику JV — (l/Co) с наклоном
l/(zFDo)- Зная Do и N, можно рассчитать Dr.
Аналогично определяют коэффициенты диффузии,
используя уравнение (6-19). Коэффициенты диффузии
различных ионов гальваностатическим методом опре-
делялись во многих работах [52, 177, 187, 190, 192,
207, 208, 225, 226, 243, 251, 258—260, 282, 294, 307,
310—333].
6.3. ИЗУЧЕНИЕ АДСОРБЦИИ
Метод хронопотенциометрии позволяет получать
информацию о механизме влияния поверхностно-ак-
тивных веществ (ПАВ),.не участвующих в электрод-
ном процессе, на 'электроосаждение и анодное раство-
рение металлов. При наличии в растворе поверхност-
но-активных веществ характер <р — f-кривых сущест-
венно изменяется. Например, в присутствии тетрабу-
тил аммония и бензоидпиперидина на катодных <р — t-
кривых осаждения кадмия из сульфатных и перхло-
ратных электролитов обнаружено скачкообразное
смещение потенциала в отрицательную сторону [315]
(рис. 6-1; т] — перенапряжение, ц ~ <р). Для электро-
лита, содержащего тетрабутиламмоний, • величина
скачка потенциала достигает 0,6 В.
Появление скачка потенциала объясняют образо-
ванием сплошного адсорбционного слоя на поверхно-
167
сти электрода (315], при этом величина скачка соот-
ветствует дополнительному перенапряжению, необхо-
димому для прохождения разряжающихся ионов че-
рез адсорбированный слой. При замене ионов SOI'
ионами CIO4 увеличивается адсорбция поверхностно-
активных веществ вблизи равновесного потенциала
Рис. 6-1. Хронопотенцио-
граммы восстановления Cd2*
из электролита состава 0,1 М
CdSO4 + 2 М Na2SO4 в при-
сутствии различных концен-
траций бензоилпипериди-
на (315]:
1—1,06 • 10-2, 2—0,7 • Ю“2,
3—0,64 - 10~3 М, 4—при отсут-
ствии добавки ПАВ; 1=5 X
' X Ю“2 А/см2.
кадмия и возрастает торможение электродного про-
цесса в этой области потенциалов [334]. Соответст-
венно изменяется и характер т) — /-кривых (рйс. 6-2).
При низких концентрациях бензоилпиперидина
(^4,2-10-3 М) сплошное заполнение поверхности
электрода не достигается, а уменьшение переходного
времени является, по-видимому, следствием экрани-
рования поверхности. Хронопотенциометр и ческий ме-
тод с успехом, может быть использован и для изуче-
ния адсорбции деполяризаторов — исходных веществ
и продуктов реакции. Впервые этот метод под назва-
нием «метода кривых заряжения» был применен для
изучения адсорбции водорода на гладкой Платине в
[335, 336], а затем в работах [165, 337—339]. В даль-
нейшем он неоднократно применялся при изучении
адсорбции и электрохимических реакций в адсорбиро-
ванном слое [269, 340—352].
Гальваностатическое изучение адсорбции осущест-
вляется следующим образом. Электрод выдерживают
некоторое время при начальном потенциале фн, затем
на него накладывают постоянный ток и регистрируют
зависимость потенциала от времени. Измерения по-
вторяют при различных плотностях тока. Длина фа-
168
радеевской задержки на <р — /-кривой пропорциональ-
на количеству вещества, вступившего в реакцию:
' ix — iXi + ix» (6-29)
где ix — длина фарадеевской задержки; fri — количество электри-
чества, израсходованного на снятие с электрода адсорбированно-
Рис. 6-2. Хронопотен-
циограмма восстановления
Cd2+ из растворов пер-
хлоратов [0,1 М Cd(C104)2 +
+ 2 7И NaClOJ в присут-
ствии различных концентра-
ций бензоилпиперидина [315]:
/ — 5,3 • 10“8; 2—4,2 . 10~3;
3—3,2 - 10“3; 4—2,6 • 10“S М;
5—без добавки.
го деполяризатора; 1хг— количество электричества, израсходован-
ного на восстановление (окисление) молекул, продиффуИдировав-
ших к электроду в ходе электролиза.
Величина /п не зависит от плотности тока при
условии, если можно пренебречь десорбцией деполя-
ризатора при регистрации (р — /-кривой. Напротив,
количество, электричества /тг понижается с ростом
плотности тока, так как при этом уменьшается коли-
чество вещества, диффундирующего из объема. По-
этому величину 1Т1, пропорциональную величине ад-
сорбции Г, можно определить путем экстраполяции
длины задержки ix к бесконечной плотности тока. При
этом необходимо исходить из следующих возможных
схем [353, 354]:
1) поверхностная концентрация адсорбата и при-
электродная концентрация диффундирующего из объ-
ема вещества связаны линейной изотермой;
2) сначала восстанавливается адсорбированное ве-
щество, а затем вещество, диффундирующее из объе-
ма (механизм «сначала адсорбат»);
3) сначала восстанавливается диффундирующее
вещество, затем адсорбат (механизм «адсорбат по-
том»);
7 Зак, 1073
169
4) отношение скоростей восстановления адсорби-
рованного заранее и диффундирующего из объема ве-
щества не зависит от времени.
Для схемы 1 величина адсорбции определяется по
формуле, которая при тд/^оСо>» упрощается:
. Fd&~ zF(C^Do
Г = 2С°О 'yj -----—— (6-30)
Схеме «сначала адсорбат» [354—356] отвечает урав-
нение:
z2F2n (C°o)2Do
ix = zFV +-------°- (6-31)
Для схемы «адсорбат потом» точное выражение полу-
чено в [355], которое может быть преобразовано к
виду [353]:
где b ** z2F2nDo (Со)2/4. При zFnVilb 1 для экстра-
поляции следует использовать соотношение:
5Х у i
Для схемы 4 зависимость имеет вид;
ix zF .. zFT
___2______221___ Т72 J
о________________рО
Go Gq
(6-33)
(6-34)
откуда величину Г можно определить из линейной за-
висимости (т от т'Л. В работе [354] при рассмотрении
приведенных схем был объяснен физический смысл
тблько первого случая. В [357] рассмотрены второй
и третий случаи, при этом основная часть работы по-
священа тому, чтобы методами корреляционного ана-
лиза выяснить, отвечает ли зависимость ix от i схеме
«сначала адсорбат» или схеме «адсорбат потом». В ре-
зультате автор [357] приходит к выводу о нецелесо-
образности применения хроиопотенциометрии для
изучения адсорбции,
170
Весьма плодотворным для изучения адсорбции
оказался подход Эршлера с сотр. [358], которые раз-; ”3?
работали теоретические основы хронопотенццометри- .
ческого метода изучения адсорбции органических ве- .
ществ. Авторы рассмотрели различные виды изотерм
адсорбции, в том числе (впервые) — изотерму с двумя
Рис. 6-3. Изотерма адсорбции 5-бром-2-ацетилтиофена на ртут-
ном электроде в ацетатном буфере (pH = 4,7), содержащем 8%
этанола [358].
Фн “-0,500 В (нас. к. 9J.
плато. В качестве примера приводим изотерму 5-бром-
2-ацетилтиофена, построенную на основе хронопотен-
циометрических данных (рис. 6-3).
Преимущества хронопотенЦнометрического метода
измерения адсорбции заключаются в следующем:
1) возможность изучения адсорбции в смешанных
растворителях;
2) измерение непосредственно значений Г;
3) измерение, одновременно с адсорбцией деполя-
ризатора, скорости электродной реакции с его уча-
стием, что делает метод-особенно удобным при изу-
чении электрохимических реакций с участием поверх-
ностно-активных веществ.
Одним из недостатков измерения адсорбции
хронопотенциометрическим методом является то, что
необходимо a priori знать форму изотермы [357].
Практически, однако, достаточно знать форму неболь-
шого участка изотермы, желательно такого, на кото-
ром адсорбция не зависит от концентрации в объеме
[358]. Такой участок всегда можно выбрать, посколь-
ку на изотермах адсорбции обычно имеется хорошо
выраженное «длинное» .плато при высоких объемных
7*
171
концентрациях адсорбата. Поэтому для насыщенных
растворов адсорбата можно применять экстраполя-
ционные графики д/Гг—1/д4 или ix—Xl^T. Если
растворимость деполяризатора великан измерения в
насыщенном растворе невозможны, нужно начинать
измерения адсорбции с разбавленных растворов. При
объемных концентрациях ниже Ifr-4 М и плотностях
тока выше 10 мА/см2 диффузией деполяризатора из
объема за время электролиза можно пренебречь: в
этих условиях длина задержки уже не зависит от
плотности тока и, следовательно, равна zFY.
Читателей, интересующихся изучением адсорбции
органических деполяризаторов хронопотенциометри-
ческим методом, отсылаем к недавно вышедшему об-
зору Эршлера [359].
Применение хронопотенциометрии для изучения
адсорбции ионов металлов на ртутных электродах
[360—364] сопряжено с такими же трудностями, о ко-
торых уже говорилось в этом разделе.
6.4. ПРИМЕНЕНИЕ ХРОНОПОТЕНЦИОМЕТРИИ
' ДЛЯ ФИЗИКО-ХИМИЧЕСКИХ
ИССЛЕДОВАНИИ В РАСПЛАВАХ
В теоретическом отношении’хронопотенциометрия
расплавов не отличается от хронопотенциометрии
растворов — в обоих случаях можно использовать одни
и те же теоретические зависимости. Тем не менее
имеются некоторые особенности, которые необходимо
учитывать в экспериментальных исследованиях рас-
плавов.
Определение коэффициентов диффузии в распла-
вах. Для исследования диффузии в расплавленных
солях хронопотенциометрический метод был впервые
применен в работах [65, 365], а для изучения диффу-
зии в шлаках и жидких сплавах — в работах [366,
367]. Дальнейшее развитие хронопотенциометриче-
ский метод получил в работах [368, 369].
Коэффициенты диффузии рассчитываются по изве-
стному уравнению Сэнда, которое для удобства вы-
числений несколько изменено:
/ Moi V
DO-1,37.10-»^—Jr (6-3S)
172
где то — концентрация ионов, % (масс.); Мо — молекулярная
масса; р — плотность электролита, г/смэ.
На применимость уравнения Сэнда для расплавлен*
ных сред указывалось также в [1].
Некоторые исследователи [370], изучавшие диф-
фузию в расплавленных солях, установили, что так
же как и для растворов коэффициент диффузии мож-
но рассчитывать по уравнению Стокса — Эйнштейна.
В работе [368] проведена проверка этого утвержде-
ния. Значения коэффициентов диффузии некоторых
ионов в различных расплавах приводятся в [65, 365—
379].
Определение толщины диффузионного слоя в рас-
плавах. В условиях принудительного перемешивания
у поверхности электрода существует четко ограничен-
ный диффузионный слой [25]. Как показано в [380],
в солевых расплавленных электролитах всегда возни-
кают интенсивные естественные потоки.
Для определения толщины диффузионного слоя
при плотности тока ниже предельной используется
уравнение хронопотенциограммы в виде [381]:
1 = const “ 1п [ехр (-#*₽)“ ехр (“ ф°°)]
(6-36)
где ср» — величина установившегося потенциала при заданном
токе.
По наклону прямой, выражающей зависимость
(6-36), находят величину 462/(л2£)о). Отношение £>о/6
определяют из значения установившегося потенциала:
6 2^о[! - ех₽ (~ Ф»)]
По величинам 462/(л20о) и Do/S легко найти 6
и Do.
В табл. 6-2 приведены результаты диффузионных
измерений на свинцовом и кадмиевом электродах в
расплавленной эвтектической. смеси КС1—LiCl. Зна-
чения величин Do и 6, полученных при различных
значениях i, отличаются между собой не ''более чем
на 5—6%, что можно считать удовлетворительным
для диффузионных измерений.
171
Таблица 6-2. Результаты диффузионных измерений ионов
свинца(П) и цинка(П) в эвтектической смеси
КС1 — L1C1 (Cq == 1.5 • 10-Ш)
! Плотность трка 1, * А/см» Фоо, мВ £>O’10Scm2/c 6.103, см
РЬ Zn РЬ zn Pb Zn
4 19,6 13,5’ 1,88 2,31 6,33 5,88
6 35,1 23,0 2,11 2,36 6,88 5,95
8 63,7 36,0 2,15 2,44 6,78 6,04
Среднее
2,05 | 2,37 I 6,67 | 5,96
Определение растворимости водорода, хлора и кис-
лорода в расплавах. Электродные процессы в распла-
вах с участием водорода, хлора и кислорода привле-
кают внимание исследователей в связи с развитием
электрохимии топливных элементов. В этом отноше-
нии значительный интерес представляет изучение за-
кономерностей электродных процессов в расплавах
карбонатов, поскольку этот электролит применяется
в высокотемпературных топливных элементах [382].
Растворимость газообразных веществ в расплавах
находят по уравнению Сэнда. Величина произведения
ix'1* однозначно связана с величиной растворимости
водорода, хлора или кислорода в’расплаве, если меж-
ду газами и компонентами расплава отсутствует ца-
кое-либо химическое взаимодействие [383, 384].
Растворимость водорода в расплаве двойной эвтек-
тической смеси карбонатов лития и натрия 'подчиня-
ется закону Генри [384]. Для оценки абсолютного
значения растворимости газов необходимо знать вели-
чину коэффициентов диффузии. Если принять Dnt —
= 5,3 • 10-4 см2/с при Т— 873 К, то растворимость
Н2 будет равна 2,8-10~4 моль/л.
Если в исходном, расплаве содержатся ионы кис-
лорода, то между ‘it1/* и концентрацией ионов О2“ на-
блюдается линейная зависимость [385], на основании
которой можно судить о содержании кислородных
ионов.
174 '
Исследование кинетики электродных процессов и
определение металлов в расплавах. Хронопотенцио-
метриче.ским методом изучена кинетика процессов
электровосстановления ионов SOt" [386, 387], NO3
[388], СОз’ [389] в расплавленной эвтектической смеси
КС1—NaCl, а также анодное поведение нитрит-ионов
[390] в смеси NaNOs—KNO3 (1:1) и ионов кислоро-
да О2- в расплаве КС1—NaCl [385].
Метод хронопотенциометрии использовался также
для исследования адсорбции ионов из расплавов на
твердых электродах [391—396].
Аналитическое определение металлов в расплавах,
как и в растворах, основано.на использовании уравне-
ния Сэнда, применимость которого для расплавлен-
ных систем исследовалась многими авторами. Но, в
отличие от растворов, определение металлов в рас-
плавах возможно только при достаточно высоких их
концентрациях (>10~3 моль/л) ввиду того, что при
высоких температурах заметно проявляется естест-
венная конвекция.'
Глава 7
ТЕХНИКА ЭКСПЕРИМЕНТА
7.1. ПРИНЦИПИАЛЬНЫЕ СХЕМЫ УСТАНОВОК
ДЛЯ ХРОНОПОТЕНЦИОМЕТРИИ
Подробное описание установок для записи кривых
потенциал — время приведено в работах [1, с. 435—
440; 20, 275, 397, 398]. Принципиальная схема одной
из таких установок изображена на‘рис. 7-1.
Трехэлектродная ячейка соединена последователь-
но с регулируемыми сопротивлениями /?i и и при-
соединена к источнику постоянного напряжения Uo.
Ток, проходящий через ячейку, и относительное изме-
нение плотности тока At/i(=A///) равны:
/ = (t/0-£)/(«!+Т?2) (7-1)
AZ/Z = Д£/((70 — £) (7-2)
где £ — напряжение на ячейке.
Из (7-2) следует, что изменение плотности тока,
протекающего через ячейку, тем меньше, чем больше
выходное напряжение • Uo. Ток i регулируется с по-
мощью Ri и измеряется по омическому падению на-
пряжения на сопротивлении R2 с помощью потенцио-
метра Pi. Разность потенциалов между поляризуемым
электродом ei и электродом сравнения е2 подают на
самописец С. Потенциометр Р2 устанавливают на вы-
ходное напряжение, близкое к амплитуде быстрого
начального изменения потенциала. Механизм переме-
щения ленты самописца С запускают до замыкания
ключа S, чтобы не потерять начало отсчета времени.
х Вместо самописца С можно использовать осцилло-
граф [124], при этом электрод е2 присоединяется че-
рез сопротивление («107 Ом’) ко входу усилителя
вертикально отклоняющихся пластин .осциллографа.
Ждущая развертка осциллографа включается за ко-
роткий промежуток времени до включения контура
ячейки для электролиза. В работе [399] на осцилло-
графе с радиальной шкалой времени <р — /-кривая по-
176
лучена в полярных координатах, что увеличивает дли-
ну оси времени по сравнению с длиной ее в ортого-
нальных координатах. Входное сопротивление само-
писца С должно быть достаточно высоким для того,
чтобы ток, идущий через электроды ei и е%, был не-
большим по сравнению с током, идущим чере^ элек-
троды ei и е3, и- им можно было пренебречь. При
очень малых токах .(в аналитической практике) необ-
ходимо применять предусилители с электрометриче-
ской лампой (типа рН-метров).
В качестве самописца можно использовать, напри-
мер, записывающее устройство полярографа ОН-102
(объединение. «Раделкис», Венгрия), Прибор позво-
ляет регистрировать потенциал в пределах от 200 мВ
(чувствительность 0,8 мВ/мм, точность ±2%) до 200 В
(чувствительность 0,8 В/мм, точность ±4%). Входное
сопротивление прибора при этом составляет 107 Ом,
что позволяет использовать токи не менее 5-10~5 А.
Рис. 7-1. Схема для записи кривых потенциал — время [1]:
ц0—источник' постоянного напряжения; 7?1 н Кг—регулируемые сопроти-
вления; Pi и Pj—потенциометры; f i, ез, ез—электроды ячейки; С—само-
писец; S—ключ.
Гальваностатические кривые удобно изучать с по-
мощью потенциостатов П-5827 и П-5848 производства
Гомельского завода измерительных приборов. Потен-
циостатщ позволяют работать в режиме как постоян-
17?
кого, так и линейно меняющегося тока. Особенно удоб-
ны потенциостаты в инверсионной хронопотенциомет-
рии, так как на них можно работать в режиме задан-
ного потенциала в стадии электронакопления. С целью
уменьшения ошибок, обусловленных десорбцией веще-
ства при переключении из потенциостатического ре-
Рис. 7-2. Схема моста для
компенсации омического па-
дения напряжения [124]:
и /?2—сопротивления; 7?з—ре-
гулируемое сопротивление;
Яч—двухалектродная иче&ка;
Ос—осциллограф.
жима в гальваностатцческий, разработаны специаль-
ные устройства, позволяющие исключить колебания
тока и потенциала во время перехода из одного режи-
ма в другой [400]. Время установления заданного то-
ка составляет «5- 10~6 с [400].
Потенциостат П-5827 может обеспечить работу в
режиме «заданный ток», начиная с минимального зна-
чения 10~* 5 А с выходным напряжением на самописце
10, 20, 40, 50 мВ. Диапазон скоростей линейного из-
менения тока составляет 2-Ю-8—0,8 А/мин. Потен-
циостат П-5848 может обеспечивать работу в режиме
«заданный ток», начиная с минимального значения*
5- 10-s А до максимального значения 2 А.
Чтобы избежать омического падения напряжения,
ячейку можно включать в плечо моста, как изображе-
но на рис. 7-2 [124]. Обычно Ri = R2, a R3 подбирают ~
равным сопротивлению раствора в ячейке. Осцилло-
граф Ос регистрирует в этом случае только перена-
пряжение (без падения напряжения на омическом
сопротивлении раствора).
Генератор может подавать в цепь ячейки не толь-
ко одйночные, но и повторяющиеся импульсЬ! раз-
ной полярности (циклическая хронопотенциометрия)
[292—294}. Подробное изложение основных результат
тов этой работы приведено в [398]. Большое значение
для изучения кинетики быстрых электродных процес-
сов имеет двухимпульсный гальваностатический ме-
1од, предложенный в работах [284, 285] и развитый
далее в работах [401, 402]. В этом методе на вход
Рис. 7-3. Схема автомати-
ческой записи переходного
времени [1]:
ci и ег—электроды трехэлектрод-
ной ячейки; Л—реле; Рг—-по-
тенциометр; Т—счетчик времени.
моста подаются последовательно два импульса тока:
первый обеспечивает зарядку двойного электрического
слоя, второй — протекание фарадеевского процесса.
В аналитической практике достаточно записывать
лишь переходное время. Схема установки для авто-
матической записи переходного времени приведена в
[1, с. 438; 403]. В этой установке разность потенциа-
лов между электродами е\ и е2 (рис. 7-3) подается
на потенциометр Р2, на котором установлено напря-
жение, соответствующее потенциалу поляризуемого
электрода, при котором измеряется переходное время.
Замыкая реле, запускают электрический счетчик вре-
мени Т и одновременно подключают к источнику Uo
(см. рис. 7-1) ячейку. При достижении переходного
времени реле 7? размыкает цепь счетчика Т и переход-
ное время отсчитывается счетчиком Т. Чувствитель-
ность реле R должна быть порядка нескольких мВ.
Число публикаций по различным типам хронопо-
тенциометрических утановок значительно. Можно на-
звать прибор для определения свинца в природных
водах [404] и катионов металлов в промышленных
растворах [405, 406]. Простая установка для хронопо-
тенциометрии и кулонометрии, состоящая из стандарт-
ных приборов промышленного производства, описана
в [407]. Установки для определения переходного вре-
мени описаны в [174, 175],
179
М. ЭЛЕКТРОДЫ И ЭЛЕКТРОЛИТИЧЕСКИЕ ЯЧЕЙКИ
При выборе фонового электролита (растворителя)
руководствуются теми же соображениями, что и в по-
лярографии [36, 49, 277, 408—410]. Для уменьшения
омического сопротивления желательно использовать
концентрированные растворы фонового электролита,
для увеличения разрешающей способности исполь-
зуются электролиты, содержащие комплексообразова-
тели. Кислород из раствора удаляют, пропуская через
него водород или инертный газ.
В хроиопотенциометрии используются самые раз-
нообразные электроды. Преимуществом ртутного ка-
пельного электрода является постоянное обновление
поверхности и (связанная с этим) высокая воспроиз-
водимость результатов измерения. Однако этот элек-
трод сравнительно редко применяется в хронопотен-
циометрии [399, 411—413]. Основным недостатком
этого электрода является изменение реакционной по-
верхности во время процесса, поэтому для получения
воспроизводимых кривых необходимо момент снятия
кривой синхронизировать с периодом капания. Удоб-
нее снимать кривые непосредственно перед отрывом
капли, поскольку в этот момент величина ..поверхно-
сти изменяется относительно мало.
В большинстве работ по хроиопотенциометрии ис-
пользовались электроды с постоянной величиной по-
верхности (стационарные и вращающиеся).
Электрод, представляющий собой висящую ртут-
ную каплю, собранную из 1—3 ртутных капель обыч-
ного капельного электрода, был описан в работах
[169, 414]. Чтобы избежать загрязнения ртутр метал-
лом подложки (золото или серебро), можно исполь-
зовать выдавленную из капилляра ртутную каплю.
Подробное описание техники получения такого элек-
трода приведено в [43]. Стационарный ртутный ка-
пельный электрод можно сформировать электролити-
чески на металлическом контакте [415].
Амальгамированные золотые, платиновые и сереб-
ряные электроды описаны в [416—417]. Более под-
робная библиография работ по конструкции и прак-
тическому использованию стационарных электродов
приведена в [418],
180
Особое значение при работе с твердыми рабочими
электродами имеет их предварительная обработка.
Как правило, первоначально электроды подвергают
механической очистке наждачной бумагой. На послед-
ней стадии пользуются полировальной бумагой наи-
меньшей зернистости (например, марки ООО). Затем
электроды подвергают многократной поляризации,
начиная от потенциалов, лежащих в области выделе-
ния водорода, до потенциалов, лежащих в области
выделения кислорода. Следует иметь в виду, что вы-
держивание электрода при электроположительных
потенциалах может приводить к окислению поверхно-
сти электрода выделяющимся кислородом. При замене
раствора для получения устойчивых вольтамперных
кривых иногда достаточно обмыть электрод дистилли-
рованной водой.
С течением времени твердые электроды могут ад-
сорбировать поверхностно-активные вещества, в этом
случае чувствительность определения элементов умень-
шается. Для возобновления «хорошей» работы элек-
трода рекомендуется вновь повторить механическую
обработку и анодно-катодную поляризацию для акти-
вации его поверхности. Таким же образом электрод
приводят в рабочее состояние после его хранения в
нерабочем состоянии на воздухе. Различные способы
хранения электродов в растворах кислот и их после-
дующая обработка описаны в [419].
Специфические требования предъявляются к элек-
тродам при работе в расплавленных электролитах. Все
используемые при исследовании расплавленных элек-
тролитов электроды можно разделить на 2 группы:
электроды, заплавленные в стекло, и незаплавленные
электроды. 3 апл явление в стекло позволяет довольно
точно фиксировать рабочую поверхность катода. Од-
нако уже после двух — трех кратковременных поляри-
заций такого катода при 700—800 °C выделившийся
на нем щелочной металл взаимодействует со стеклом,
образуя на поверхности последнего пленку (вероятно,
силицидов), обладающую электронной проводимостью
[368]. При последующих поляризациях эта пленка ра-
ботает как дополнительный электрод. Поэтому за-
плавленные электроды в условиях электролиза соле-
вых расплавов при высоких температурах (>700°С)
181
можно применять для снятия одной — трех <р — /-кри-
вых.
Ошибки в определении площади незаплавленных
электродов связаны с неточностью измерения глубины
их погружения в расплав. Для уменьшения этих по-
грешностей токоподводы к катодам должны быть воз-
можно малого диаметра. .
Обычно применяют электроды цилиндрической
формы или плоские (в виде пластинки). Поверхность
электрода перед опытом зачищают бумагой, а затем
полируют до зеркального блеска. При такой подго-
товке "поверхности эффективную- площадь катода
можно считать равной площади геометрической по-
верхности, так как размеры шероховатостей полу-
чаются много меньше толщины диффузионного слоя
[420].
При исследовании осаждения металлов нужно учи-
тывать, что при многократном осаждении —растворе-
нии металлов происходит разрыхление , поверхности
электрода, которая не восстанавливается после анод-
ного растворения металла; кривые <р — t становятся,
размытыми, переходы от одного участка к другому
нечеткими. По данным [368] наиболее удобными яв-
ляются катоды из молибдена, вольфрама или плати-
ны. При высоких температурах (>700°C) срок служ-
бы таких катодов ограничен 3—5 циклами.
Для исследований в расплавах широко применяет-
ся платиновый электрод [369, 372, 375, 389, 421—424].
Можно применять электроды и из других металлов
(ниобий, тантал), а также жидкие электроды (напри-
мер, вйсмутовые). Выбор электрода сравнения зави-
сит от изучаемой системы. Часто применяются хлор-
ный, платиновый, а также серебряный электроды [368,
369, 372, 374, 423, 424]. В качестве анода (вспомога-
тельного электрода) используются пластинки из сере-
бра, графитовые стержни и платиновая проволока
или платиновые пластинки (сетки).
Широкое распространение в вольтамперометрии
получили электроды из углеродных материалов:
угольной пасты, стеклоуглерода, углеситалла, графита
[217,419,425—427].
Угольный пастовый электрод изготавливается из
графита (порошка) спектральной чистоты и органи-
182
ческих растворителей, не смешивающихся с водой
(бромнафталин, вазелиновое масло и др.). Приготов-
ленную пасту д тестообразном состоянии запрессовы-
вают в стеклянную трубку либо в отверстие тефлоно-
вого стержня.
Графитовые электроды для. уменьшения пористо-
сти и снижения остаточного тока подвергают специ-
альной пропитке в вакууме [426, 427]. Для приготов-
ления таких электродов используют стержни из гра-
фита спектральной чистоты, боковую поверхность
которых покрывают парафином, эпоксидной смолой,
полиэтиленом и т. д. Пропитка электрода действи-
тельно приводит к уменьшению остаточных токов,
однако вещества, используемые для пропитки графи-
та и покрытия его боковой поверхности, могут влиять
на величину фарадеевских токов.
В связи с этим во многих работах используются
электроды из стеклоуглерода [428]. Этот материал
практически газонепроницаем и химически устойчив.
В работах [429—431] описано применение стеклоуг-
лерода в электроаналитической практике.
Новый тип углеродного материала — углеситалл —
применялся в электроаналитической практике сравни-
тельно мало [432, 433].
Вращающийся электрод представляет собой диск,
укрепленный на вертикальной оси, служащей одно;
временно токоподводом. Детали конструкции электро-
дов и их крепления на вращающемся валу описаны
в [26,434].
Используемые в хронопбтенциометрии плотности
тока могут быть велики, и для уменьшения омическо-
го напряжения в ячейке принимают специальные ме-
ры [1, с. 415; 20, с. 435].
В хронопотенциометрии со стационарными элек-
тродами используются такие же электролитические
ячейки, как и в полярографии, поэтому здесь они не
описываются. Подробное описание ячеек для устано-
вок с вращающимся дисковым электродом и способы
их изоляции от внешней среды (вращающийся шлиф,
гидравлический затвор или торцевое уплотнение) опи-
саны в [26],
ЛИТЕРАТУРА
]. Делахей П. Новые приборы и методы в электрохимии. Пер.
с аигл. Под ред. проф. Б. В. Эршлера. М., Издатинлит, 1957,
509 с.
2 Berzins Т., Delahay Р. J. Am. Chem. Soc., '1953, v. 75, № 8,
р. 4205—4213.
3. Г плюс 3. Теоретические основы электрохимического анализа.
Пер. с польск. Под ред. докт. х. и. Б. Я- Каплана. М„ «Мир»,
1974, 552 с.
4. Sand И. J. S. Phil. Mag., 1901, v. 1, № 1, р. 45—52.
5. Bruckenstein S., Nagai T. Analyt. Chem., 1961, v. 33, № 6,
p. 1201—1203.
6. Neeb R. Z. anal, chem., 1962, v. 198, Ns 1, p. 98—100.
7. Доронин A. H., Кабанова О. Л. ЖАХ, 1965, т. 20, вып. 12,
с. 1321—1323.
8. Захаров М. С., Пнев В. В. Тезисы докладов на Всесоюзной
конференции: Теория и практика амальгамных процессов.
Алма-Ата, изд-во «Наука», КазССР, 1966, с. 163-
9. Захаров М. С., Баканов В. И. Тезисы докладов на Всесоюз-
ной конференции: Теория и практика амальгамных процес-
сов. Алма-Ата, изд-во «Наука» КазССР, 1966, с. 163.
10. Захаров М. С., Пнев В. В. Изв. Томск, политехи, ин-та, 1967,
т. 164, с. 72—98.
11. Kemula W., Strojek J. IP. J. Electroanal. Chem., 1966, v. 12,
№ 1, p. 1—8.
12. Vincent H. A., Wise E. N. J. Elektroanal. Chem., 1966, v. 11,
№ 1, p. 54—61.
13. Доронин A. H., Кабанова О. Л. ЖАХ, 1969, т. 24, вып. 5,
с. 79—81.
14. Агасян П. К., Хамракулов Т. К. ЖАХ, 1968, т. 23, вып. 1,
с. 19—24.
15. Салин А. А., Раннее Г. Г., Коголь И. М. Зав. лаб., 1967,
т. 33, № 12, с. 1361—1364.
16. Захаров М. С., Пнев В. В., Московских Л. А. «Электрохи-
,мия», 1971, т. 7, № 8, с. 1092—1095.
17. Furlani С., Morpurgo G. J. Elektroanal. Chem., 1960, v. 1,
№ 3, p. 351—360.
18. King R. H„ Reilley C. N. J. Elektroanal. Chem., 1960, v. 1,
№ 4, p. 434—445.
19. Dracka O. Coll. Czechoslov. Chem. Comm. 1960, v. 25, № 2,
p. 338—348.
20. Delahay P., Mattax С. C. J. Am. Chem. Soc., 1954, v. 76, №2,
p. 874—878.
21. Kambara Т., Tachl S. J. Phys. Chem., 1957, v. 61, № 5,
p. 1405—1407.
22. Kambara T., Tachi I. J. Phys. Chem., 1961, v. 2, № 1, p. 249—
253
23. Peters D. G., Burden S. L. Anal. Chem., 1966, v. 38, № 4,
p. 530—536.
24. Фрумкин A. H. и др. Кинетика электродных процессов. M.,
изд-во МГУ, 1952, 320 с.
25: Левин В. Г. Физико-химическая гидродинамика. М., изд-во
АН СССР, 1952, ЭЗО с.
26. Плесков Ю. В., Филиновский В. Ю. Вращающийся дисковый
"электрод. М., «Наука», 1972. 344 с.
27. Wiesner К. Z. Elektrochem. 1943, Bd. 49, № 2, S. 164—170.
28. Brdicka R., Wiesner K- Naturwiss., 1943, Bd. 3.1, S. 247—256.
29. Koutecky J., Brdicka R. Coll. Czechosl. Chem. Comm., 1947,
v. 12, № 2, p. 337—346.
30. Kern D. H. H. J. Am. Chem. Soc., 1953, v. 75, № 5, p. 2473—
2480.
31. Kern D. H. H. J. Am. Chem. Soc., 1954, v. 76, № 2, p. 1011—
1018.
32. Frumkin A. N. Z. physik Chem., 1933, Bd. 164A, S. 121—130.
33. Антропов Л. И. ЖФХ, 1950, т. 24, 5, с. 1428—1434.
34. Эршлер А. Б., Тедорадзе Г. А., Майрановский С. Г. ДАН
СССР, 1962, т. 1'45, № 7, с. 1324—1330.
35. Тедорадзе Г. А., Эршлер А. Б., Майрановский С. Г. Изв. АН
СССР, сер. хим. наук, 1963, № 2, с. 235—240.
36. Майрановский С. Г. Каталитические и кинетические волны в
полярографии; М., «Наука», 1966. 288 с.
37. Фрумкин А. Н., Джапаридзе Д. И., Тедорадзе Г. А. ДАН
СССР, 1963, т. 152, № 1, с. 164—170.
38. Тедорадзе Г. А., Эршлер А. Б. Усп. хим., 1965, т. 34,
с. 1866—1891.
39. Фрумкин А. Н. ДАН СССР, 1950, т. 24, № 1, с. 244—250.
40. Фрумкин А. В. ДАН СССР, 1952, т. 85, № 2, с. 373—378.
41. Frumkin A. N. Z. Electrochem., 1955, Bd. 59, S. 807—815.
42, Томилов А. П. и др. Электрохимия органических соединений.
М. — Л., «Химия», 1968. 591 с.
43. Гейровский Я-, Кута Я. Основы полярографии, Пер. с чешек.
Под ред. докт. х. н. С. Г. Майрановского. М., «Мир», 1968.
558 с.
44. Karaoglanoff Z. Z. Elektrochem., 1906, Bd. 12, № 1, S. 5—12.
45. Rosebrygh T. R., Muller W. L. J. Phys. Chem.,. 1910, v. 14,
p. 816—826.
46. Covaci J., Miron C., Bucur R. V. J. Electroanal. Chem., 1965,
v. 2, № 1, p. 86—89.
47. Янке E., Эмде Ф., Лёш Ф. Специальные функции. М., «Нау-
ка», 1964. 344 с.
48. Butler J. А. V., Armstrong G. Trans. Faraday Soc., Proc. Roy.
Soc. (London), 1933, v. A 139, p. 406—416.
49 Butler J. A. (/., Armstrong G Trans. Faraday Soc., 1934, v. 30,
p. 1173—1183.
50 Bard A. 1. Anal. Chem., 1961, v. 33, № 1, p. 11—15.
51. Bard A. J. Anal. СЬещ., 1963, v. 35, № 3, p, 340—343.
Ж
52. Reilley C. N., Everett G. W., Johns К. H. Anal. Chem., 1955,
v. 27, № 4, p. 483—491.
53. Anderson L. B., Macero D. Anal. Chem., 1965, v. 37, № 3,'
p. 322—326.
54. Berzins T., Delahay P. Z. Elektrochem., 1955, Bd. 59, S. 792—
801.
55. Relnmuth W. H. Anal. Chem., 1960, v. 32, № 15, p. 1514—1520.
56. Mamantov G., Delahay P. J. Am. Chem., 1954, v. 76, p. 5323—
5334.
57. Hurwitz H. D. J. Electroanal. Chem., 1964, v. 7, № 4, p. 368—
381.
58. Guminskl C., Gaius Z. Roczn. Chem., 1970, v. 44, № 10,
p. 1767—1775.
59. Peters D. G., Lingane J. J. J. Electroanal. Chem., 1961, v. 2,
№ 1, p. 1—16.
60. Lingane J. J. J. Electroanal. Chem., 1961, v. 2, № 1, p. 46—60.
61. Evans D. H., Price J. E. J. Electroanal. Chem., 1963, v. 5,
№ 1, p. 77—80.
62. Dornfeld D. J., Evans D. H. J. Electroanal. Chem., 1969, v. 20,
№ 3, p. 341—345.
63. Carslaw H. S., Jaeger J. S. In: Conductivity of Heat in So-
lids. 2nd ed. Oxford, London, 1959, p. 338—341.
64. Филиновский В. Ю., Кирьянов В. А. ДАН СССР, 1964, т. 156,
Ns 6, с. 1412—1415.
65. Laitinen Н„ Ferguson N. Anal. Chem., 1957, V, 29, № 1,
р. 4—14.
66. Fetter К. J. Z. Physik. Chem., 1967, Bd. 52, S. 47—56.
67. Черненко В. И., Литовченко К- И. Укр. хим, ж., 1973, т. 39,
№ 4, с. 344—347.
68. Testa А. С., Reinmuth W. Н. Anal. Chem., 1960, v‘. 33, Ns 10,
р. 1320—1324.
69. Murray R. 1^, Reilley C. N. J. Electroanal. Chem., 1962, v. 3,
№ 2, p. 182—202.
70. Bos P. J. Electroanal. Chem., 1971, v. 33, № 2, p. 379—391.
71. Peters D. G., Lingane J. J. J. Electroanal. Chem., 1961, v. 2,
№ 2, p. 249—258.
72. Morris M. D., Lingane J. J. J. Electroanal. Chem., 1963, v. 6,
№ 3, p- 300—313.
73. Morris M. D. J. Electroanal. Chem., 1964, v. 8, № 1, p. 1—4.
74. Okada S. e. a. J. Electrochem. Soc., Japan, 1959, v. 27u p. 140—
154.
75. Mamantov G., Delahay P. J. Am. Chem. Soc., 1954, v. 76,
Ns 11, p. 5342—5343.
76. Феттер К- Электрохимичесокая кинетика. Пер. с нем. Под
ред. Я- М. Колотыркина. М., «Химия», 1967.’ 848 с.
77. Городысский А. В., Делимарский Ю. К. ДАН СССР, 1957,
т. 114, № 5, с. 1261—1265.
78. Рябухин Ю. М. ЖФХ, 1963, т. 37, Ns 3, с. 694—695.
79. Бек Р. Ю., Лифшиц А. С., Бородихина Л. И. Изв. СО АН
СССР, сер. хим. наук, 1970, вып. 7, № 1, с. 37—40.
80. Кирьянов В. А., Филиновский В. Ю. ЖФХ, 1963, т. 37, Ns 10,
с 2122______2126.
81. Meyer R. Е. е.а. З: Electroanal, Chem., 1971, v, 30, Ns 3,
p. 345-358, ‘ ’
82. Posey P. A., Meyer R. Ё. J. Electroanal. Chem., 1971, v. 30,
№ 3, p. 359—373.
83. De Vries W. T. J. Electroanal. Chem., 1968, v. 17, № 1—2, V
p. 31—43.
84. Olmstead M. J., Nicholson R. S. J. Phys. Chem., 1968, v. 72/
• № 5, p. 1650—1656.
85. Rodgers R. S., Meites L. J. Electroanal. Chem., 1968, v. 16,
№ 1, p. 1—11.
86. Dracka 0. Coll. Czechocl. Chem. Comm., 1969, v. 34, № 9,
p. 2627—2644.
87. Olmstead M. L., Nicholson R. S. Analyt. Chem., 1970, v. 42,
№ 3, p. 796—801.
88. Bos P., Van Dalen E. J. Electroanal. Chem., 1973, v. 45, № 2,
p. 165—179.
89. Erdey — Gruz T., Volmer M. Z. phys. Chem., 1930, Bd. 150,
№ 2, S. 203—210.
90. Brandes H. Z. phys. Chem., 1929, Bd. 142, № 1, S. 97—106.
91. Erdey—Gruz T., Kromey G. F. Z. phys. Chem., 1931, Bd;
157 A K’2 S 213____219.
92. Murray R. W., Reilley C, N. J. Electroanal. Chem., 1962, v. 3,
№ 1, p. 64—77.
93. Hoffmann H., Jaenicke W. Z. Elektrochem., Ber. Btinsenges
physik. Chem., 1962, Bd. 66, № 1, S. 7—14.
94. Senda M. Rev. Polarography. Japan, 1956, v. 4, № 1, p. 89—
95 '
95. Hurwitz H., Gierst L. J. Elektroanal. Chem., 1961, v. 2, № 1,
p. 128—141.
96. Hurwitz H. J. Elektroanal. Chem., 1961, v. 2, № 1, p. 142—151.
97. Hurwitz H. J. Elektroanal. Chem., 1961, v. 2, № 2, p. 328—342.
98. Murray R. W. Anal. Chem., 1963, v. 35, № 13, p. 1784—1789.
99. Testa A. C., Reinmuth W. H. Anal. Chem., 1961, v. 33, Ks 10,
p. 1324—1328.
100. Takemori Y. e.a. J. Phys. Chem., 1957, v. 61, № 3, p. 968—
969. . .
101. Gaur H. C., Jindal H. L. J. Elektroanal. Chem., 1967, v. 14,
№ 2, p. 297—301.
102. Bansal N. P., Jindal H. L. J. Indian. Chem. Soc., 1972, v. 49,
№ 5, p. 957—961.
103. Sturrock 'P. E., Anstine W. D., Gidson К. H. Anal. Chem.,
1968, v. 40, № 2, p. 505—508.
104. Shults W. D., Mueller T. R. J. Elektroanal. Chem., 1966, v. 12,
№ 1, p. 154—159.
105. Macero D. J., Anderson L. B. J. Elektroanal. Chem., 1963,
v. 6, № 3, p. 221—226.
. 106. Beyerlein F. H., Nicholson R.S. Anal. Chem., 1968, v. 40,
№ 2, p. 286—288.
107. Hale J. H. J. Elektroanal. Chem., 1963, v. 6, № 2, p. 187—197.
108. Черненко В. И., Литовченко К- И., Кисленко В. Ф. «Электро-
химия», 1971, т. 7, вып. 8, с. 1100—1106. >
109. Guidelli R. In: Elektroanalytical Chemistry. V. 5. Ed.
A. J. Bard. New York, Marcel Dekker, 1971, p. 149—374.
110. Бьюик А., Терек X. В кн.: Современные проблемы электрохи-
мии. Пер. с англ. Под ред. Я. М. Колотыркина. М., «Мир»,
1971, с. 273—344.
187
L
111. Paunovic M. J. Elektroanal. Chem., 1967, v, 14, Ns 3, p. 447—
474. К
112. Losev V. V. In: Modern Aspects of Electrochemistry. V. 7,
Ed. В. E. Conway and Jr O’M. Bocris, London, Butterworths,
1972, p. 314—398. W
113. Ruzic J., Smith D. E. J. Elektroanal. Chem., 1974, v. 52, Ns 2, H
p? 157—192. Я
114. Ruzic J., Smith D. E. J. Elektroanal. Chem, 1975, v. 58, Ns 2, ’Я
p. 145—175. Я
115. Россоти Ф., Россоти X. Определение констант устойчивости Я
и других констант равновесия в растворах. Пер. с англ. Под ;<Я
ред. чл.-корр. АН СССР Д. И. Рябчикова. М., «Мир», 1965. <Я
561 с. .Я
116. Бек М. Химия равновесий реакций комплексообразования. .Я
Пер. с англ. Под ред. И. Н. Марова. М., «Мир», 1973. 359 с. Я
117. Rossotti F. J. С., Rossotti Н. S., Whewell R. J. J. Inorg. Я
Nucl. Chem., 1971, v. 33, Ns 10, p. 2051—2056. Я
118. Sabatini A., Vacca A., Gans P. «Taianta», 1974, v. 21, № 1, \Я
p. 53—77. Я
119. Пнев В. В., Московских Л. А., Захаров М. С. В кн.: Химия Я
и химическая технология. Труды Тюменского индустриально- Я
го института. Тюмень, изд. Тюменск. индустр. ин-та, 1972, Я
с. 90-97. ’1
120. Захаров М. С., Пнев В. В. Изв. Томск, политехи, ии-та,
1967, т. 164, с. 72—78. Я
121. Делахей П. Двойной слой и кинетика электродных процес- Я
сов. Пер. с англ. Под ред. акад. А. Н. Фрумкина. М., «Мир», ЛЯ
. 1967. 351 с. 'Я
122. Tanaka N., Tamamushi R. Elektrochim. Acta, 1964, v. 9, Ns 10, Л
p. 963—989. Ш
123. Ashley J. W., Reiley H. N. J. Elektroanal. Chem., 1964, v. 7, ’’
Ns 4, p. 253—275. -J
124. Delahey P., Berzins T. J. Am. Chem. Soc., 1953, v. 75, Ns 10, j
p. 2486—2496. ?
125. Matsuda H. J. Am. Chem. Soc., 1960, v. 82, Ns 2, p. 329—336. •
126. Reinmuth W. H. J. Elektroanal. Chem., 1972, v. 38, Ns 1,
p. 95—99. i
127. Bocris J. O’M. e. a. J. Elektroanal. Chem., 1963, v. 5, Ns 4,
p. 476—480.
128. Галюс 3. «Электрохимия», 1968, т. 4, Ns 5, с. 553—556. ;
129. Майрановский С. Г. Двойной слой и его эффекты в поляро-
графии. М., «Наука», 1971. 88 с. s
130. Breit er М., Kleinerman М., Delahay Р. J. Am. Chem. Soc.,
1958, v. 80, № 2, p. 511—519.
131. Matsuda H. J. Phys. Chem., 1960, v. 64, Ns 2, p. 336—339.
132. Gierst L, Hurwitz H. Z. Elektrochem., 1960, Bd. 64, Ns 1,
S. 36—48.
133. Hurwitz H. Z. Elektrochem., 1961, Bd. 65, № 1, S. 178—186.
134. Frumkin A. N. In: Advances in Electrochemistry and Electro-
chemical Engineering. Ed. P. Delahay, Interscience, New York,
v. 1, 1961, p. 65—122; v. 3, 1963, p. 287—291.
135. Delahay P., Mattax С. C., Berzins T. J. Am, Chem. Soc., 1954,'
v. 76, № 15, p. 5319—5328,
188
136. Пнев В. В., Захаров М. С В кн.: Химия и химическая тех-
нология. Труды Тюменского индустриального института. Тю-
мень, изд. Тюменск. индустр. ин-та, 1972, с. 64—70.
137. Fischer О., Bezdek J. Coll. Czechosl. Chem. Comm., 1973,
v. 38, Ks 7, p. 1907—1910.
138. Dracka 0. Coll. Czechosl. Chem. Comm., 1961, v. 26, № 7,
p. 2144—2154.
139. DraSka 0, Coll. Czechosl. Chem. Comm., 1970, v. 35, Ks 9,
p. 2480—2484.
140. Dracka O. Coll. Czechosl. Chem. Comm., 1970, v. 36, Ks 7,
p. 1876—1888.
141. Drabka O. Coll. Czechosl. Chem. Comm., 1971, v. 36, № 7,
p. 1889—1897.
142. Drabka O. Coll. Czechosl. Chem. Comm., 1967, v. 32, № 12,
p. 3987—3997.
143. Ficher O., Dratka O., Fischerova E. Coll. Czechosl. Chem,
Comm., 1961, v. 26, Ks 6, p. 1505—1520.
144. Fischerova E., Drabka O., Fischer O. Coll. Czeshosl. Chem.
Comm., 1965, v. 30, Ks 1, p. 10—18.
145. Dracka O., Fischer O. Coll,- Czechosl. Chem. Comm., 1974,
v. 39, № 8, p. 1970—1973.
146. Fischer O., Dralka O. Coll. Czechosl. Chem. Comm., 1959,
v. 24, Ks 11, p. 3046—3056.
147. Fischer O., Dracka O., Fischerova E. Coll. Czeshosl. Chem.
Comm., 1960, v. 25, Ks 1, p. 323—337.
148. Koutecky J. Chem. Listy, 1956, v. 50, Ks 10, p. 1410—1415.
149. Koutecky I. «Nature», 1954, v. 174, p. 233—234.
150. Fischer O., Dratka O., Stenina-Iakovleva 1. V. Coll. Czeshosl.
Chem. Comm., 1968, v. 33, Ks 7, p. 2370—2380.
151. Laitinen H. A., Kivalo P. J. Am. Chem. Soc., 1953, v. 75, Ks 7,
p. 2198—2203.
152. Fischerova E. Coll. Czeshosl. Chem. Comm., 1965, v. 30, Ks 6,
p. 1771—1779.
153. Tanaka N., Yamada A. Bull. Chem. Soc. Japan, 1966, v. 39,
Ks 3, p. 920—928.
154. Testa A. C., Reinmuth W. H. J. Am. Chem. Soc., 1961, v. 83,
Ks 3, p. 784—789. _
155. Testa A. C., Reinmuth W. H. Anal. Chem., 1961, v, 33, Ks 10,
p. 1320—1324.
156. Tanaka N., Ebata K. J. Elektroanal. Chem., 1964, v. 8, Ks 1,
p. 120—128.
157. Herman H. B., Bard A. J. J. Phys. Chem., 1966, v. 70, Ks 2,
p. 396—404.
158. Dracka O. Coll. Czeshosl. Chem. Comm-., 1974, v. 39, Ks 4,
p. 805—811.
159. Herman H. B., Bard A. J. J. Elektrochem. Soc., 1968, v. 115,
Ks 7, p. 1028—1032.
160. Dracka O. Coll. Czeshosl. Chem. Comm., 1963, v, 28, Ks 11,
p. 3194—3205.
161. Reinmuth W. H. Anal. Chem., 1961, v. 33, Ks 4, p. 485—487.
162. Russell C. D., Peterson J. M. J. Elektroanal. Chem., 1963, v. 5,
Ks 6, p. 467—475.
163. Takemori Y. Rev. Polarography (Japan), 1965, v, 12, Ks 5/6,
p. 176—180,
189
164 Laity R. tf'., McIntyre S. D. J. Am. Chem. Soc., 1965, v. 87,,”
№ 7, p. 3806—3812.
165. Pearson I. D-, Butler S. A. V. Trans. Faraday Soc., 1938, v. 34, ;
p. 1163—1170.
166. Делимарский Ю. К.., Макогон В. Ф. «Защита металлов», 1969,
т. 5, с. 128—130. 7
167. Berzins Т., Delahay Р. J. Am. Chem. Soc., 1955, v. 77, № 12,
р. 6448—6453.
168. -Lingane J. J. J. Elektroanal. Chem., 1960, v. 1, № 4, p. 379—
390
169. Delahay P., Berzins T. J. Am. Chem. Soc., 1953, v. 75, № 5,
p. 2486—2493.
170. Эршлер А. Б., Овсянников И. H., Коршунова К. С. «Элек-
трохимия», 1970, ,т. 6, № 6, с. 876—879.
171. Емельяненко Г. А., Симулин Г. Г. ЖФХ, 1965, т. 39, № 7,
с. 1739—1741.
172. Hawley D., Adams N. J. Elektroanal. Chem., 1965, v. 10, Ns 2,
p. 376—382.
173. Рысаков А. А. и др. «Электрохимия», 1974, т. 10, вуп. 12,
с. 1885—1888.
174. Бек Р. Ю., Буренков И. И., Лифшиц А. С. Изв. СО АН
СССР, сер. хим. наук, 1969, № 4, вып. 2, С. 3—8.
175. Буренков И. И., Лифшиц А. С., Бек Р. JO. Изв. СО АН
СССР, сер. хим. наук, 1971, Ns 12, вып. 5, с. 145—147.
176. Mckeon М. G. J. Elektroanal. Chem., 1962, v. 4, Ns 2, p. 93—95.
177. Bruckenstein S., Rouse T. 0. Anal. Chem., 1964, v. 36, Ns 10,
p. 2039—2040.
178. Gierst L., Juliard A. In: Proceeding ol the International Com-
mittee of Electrochemical Thermodynamic and Kinetic 2nd
Meeting. Milan, Tambyrini, 1950, p. 117—133. -
179. Nicholson M. M., Karchmer S. H. Anal. Chem., 1955, v. 27,
Ns 7, p. 1095-1098.
T80. Лунев M. И., Каменев А. И., Агасян П. К- Вести. МГУ.
Химия, 1974, т. 15, Ns 6, с. 686—689.
181. Каменев А. И. и др. В сб.: Тезисы докладов II Всесоюзного
симпозиума по методам определения микроэлементов в при-
родных объектах. Самарканд, изд-во «Фаи», 1973, с. 143.
182. Bard A. S. Anal. Chim. Acta, 1960, v. 22, Ns 5, p. 577—583.
183. Mac Novin W. M., Moorhead E. D. J. Am. Chem. Soc., ‘1959,
v. 81, Ns 12, p. 6382—6386.
184. Moorhead E. D., Furman N. H. Anal. Chem., I960, 32, Ns 11,
- p. 1507—1509.
185. Hurlen T. Acta Chem. Scand., 1961, v. 15, Ns 3, p. 621—629.
186. Isbibashi M. e. a. «Nippon Kagaku Zasshi», 1960, v. 81, Ns 1,
p. 88—92.
187. Peters D. G., Gruser S. A. J. Elektroanal. Chem., 1965, v. 9,
Ns 1, p. 27—40. ,
188. Lingane J. J. In: Elektroanalytical Chemistry. New York, In-
terscience, 1958, p. 623.
189. Lingane J. J. Anal. Chim. Acta, 1958, v. 19, Ns 4, p 394—401.
190 Peters D. G., Franklin K. A. J. Elektroanal. Chem., 1965, v. 9,
Ns 5/6, p. 385-389.
191. Kowalski T. A., Lingane P, S. J, Electroanal. Chem., 1971,
v, 31, Ns 1, p. 1—7.
190.
192. Evans^D. H„ Lingane S^S. J. Electroanal. Chem., 1964, v. 8,
Nj 3, p. 173—180.
193. Lingane J. J. J. Electroanal. Chem., 1962, v. 4, № 3, p. 332—
339.
194. Lingane J. J. «Analyst», 1966, v. 91, № 1, p. 1—9.
195. Lingane J. J. Anal. Chem., 1964, v. 36, № 9, p. 1723—1726.
196. Булина В. А., Зебрева А. И. В кн.: Химия и химическая тех-
нология. Алма-Ата, изд-во «Наука» КазССР, 1973, № 14,
с. 181—183.
197. Wrona Р. К., Krogulec Т., Gaius Z. «Roczniki Chemii», 1974,
v. 48, № 4, р. 693—700.
198. Kemtila IF., Gaius Z. «Roczniki Chemii», 1962, v. 36, № 8,
p. 1223—1230.
199. Nagal T. «Nippon Kagaku Zasshi», 1960, v. 81, № 2, p. 250—
252.
200. Delahay P., Mamontov G. Anal. Chem., 1955, v. 27, № 4,
p. 478-483.
201. Ritagava T. «Japan Analyst», 1966, V. 15, № 12, p. 1311—1315,
202. Hubmann I., Buffle I., Monnier D. Analyt. Chim. Acta, 1972,
V.62, № 4, p. 393—400.
203. Murray R. IF., Gross D. J. J. Electroanal. Chem., 1967, v. 13,
№ 1, p. 132—136.
204. Nagai T. «Nippon Kagaku Zasshi», 1960, v. 81, p. 254—256,
205. Radhakrlshnan T. P., Sundaram A. K. Proc. Indian. Acad. Sci.,
1972, V. A75, № 6, p. 278—293.
206. Davis D. Q. Anal. Chim. Acta, 1962, v. 27, №. 1, p.-2'6—30.
207. Caja I., Pravdil V. J. Electroanal. Chem., 1964, v. 8, № 5,
p. 390-398.
208. Peters D. G., Shults IF. D. J. Electroanal. Chem., 1964, v. 8,
№ 8, p. 200—229. . '
209. Rabidea'u S. IF. J. Am. Chem. Soc., 1956, v. 78, № 5, p. 2705—
2710.
210. Metz C. F. Anal. Chem., 1957, v. 29, № 12, p. 1748—1752.
211, Davis D. G. Anal. Chem., 1961, v. 33, № 13, p. 1839—1842.
212. Davis D. G., Ganchoff I. J. Electroanal. Chem., 1960, v. 1,
№ 3, p. 248—255.
213. Girst I., Mechelynck P. Anal. Chim. Acta, 1955, v. 12, № 1,
, p. 79—91.
214. Anson F. C. J. Am. Chem. Soc., 1961, v. 83, № 5, p. 2387—
2393.
215. Bonewitz R. A., Schmid G. M. J. Electrochem. Soc., 1970,
v. 117, № 10, p. 1367—1372.
216. Dhaneshwar R. G., Kulkarni A. V. Indian Joum. Chem., 1973,
v. 11, № 2, p. 154—157.
217. Adams R. M. In: Electrochemistry at Solid Electrodes. New
York, Marcel Dekker, 1966, p. 23.
218. Hoare J. P. In: The Electrochemistry of Oxygen. New York,
Interscience, 1968, p. 47.
219. Herman H. B., Bard A. J. Anal. Chem., 1964, v, 36, p. 510—
514.
220. Christie I. H., Lauer G. Anal. Chem., 1964, v. 36, № 10,
p. 2037—2038.
221. Choudhury P., Khasgiwale K. A., Sandaresan M. J, Electro-
эда|. Chem-, 1972, v. 4Q, № 2, p. 444—447,
191
I
222 Sancho J., Almagro J., Pujante A. J. Electroanal. Chem., 1968,
v. 16, № 1, p. 77—82.
223. Lingane P. J., Anson F. C., Osteryoung R. A. J. Electroanal.
Chem., 1966, v. 12, № 3. p. 250—253.
224. Tanaka N., Yamada A., Tamamashi R>. Bull. Chem. Soc. Ja-
pan, 1964, v. 37, № 12, p. 1821—1826.
225. Lingane J. J. J. Electroanal. Chem., 1961, v. 2, № 3, p. 296—
ЗГО.
226. Blackburn T. R., Lingane J. J. J. Electroanal. Chem., 1963,
v. 5, № 3, p. 216—235.
227. Peters D. G., Mitchell R. A. J. Electroanal. Chcin.^ 1965, v. 10,
№ 4, p. 306—318.
228. Anson F. C., Lingane J. J. Am. Chem. Soc., 1957, v. 79, № 9,
p. 4901—4910.
229. Evans D. Lingane J. I. J. Electroanal. Chem., 1963, v. 6,
№ 4, p. 283—299.
230. Teeter T. E., Van Rysselberghe P. J. Chem. Phys., 1954, v. 22,
№ 2, p. 759—765.
231. Jordan J., Smith P. T. Proc. chem. Soc., 1960, № 2, p. 246—
252
232. Roberts J. L., Sawyer D. T. J. Electroanal. Chem., 1965, v. 9,
№ 1, p. 1—7.
233. Mark H. B., Anson F. C. J. Electroanal. Chem., 1963, v. 6,
№ 4, p. 251—260.
234. Davis D. G., Everhart M. E. Anal. Chem., 1964, v. 36, № 1,
p. 38—40.
235. Badoz— Lambling J., Dutruc-Rosset C. Anal. Chim. Acta,
1958, v. 19, № 1, p. 43—50.
236. Badoz — Lambling J. J./Electroanal. Chem., 1959/60, v. 1, № 1,
p. 44—52.
237. Osteryoung R. A. Anal. Chem., 1963, v. 35, № 9, p. 1100—1110.
238. Iwamoto R. T., Adams R. N., Lott H. Anal. Chim. Acta, 1959,
v. 20, № 1, p. 84—88.
239. Zittel H. E., Miller F. J. J. Electroanal. Chem., 1967, v. 13,
№ 3, p. 193—207.
240. Evans D. H. J. Electroanal. Chem., 1963, v. 6, № 4, p. 419—
427.
241. Anson F. C., Schultz F. A. Anal. Chem., 1963, v. 35, № 9,
p. 1114—1116.
242. Giner J. Electrochim. Acta, 1961, v. 4, № 1, p. 42—48.
243. Blackburn T. R., Cambell P. C. J. Electroanal. Chem., 1964,
v. 8, № 2, p. 145—150.
244. Jacobsen E., Sawyer D. T. J. Electroanal. Chem., 1968, v. 16
№ 3, p. 361—374.
245. Munson R. A. J. Electrochem. Soc., 1964, v. Ill, № 3, p. 372—
378.
246. Ling C., Franklin T. S. Electrochim. Acta, 1964, v. 9, № 5
p. 517- 525.
247. Jacobsen E., Roberts J. L., Sawyer D. T. J. Electroanal. Chem.,
1968, v. 16, № 3, p. 351—360.
• 248. Багоцкий В. С., Васильев 10. Б. В кн.: Топливные элементы.
Кинетика электродных процессов. М., «Наука», 1968, с. 305.
249. Bagotsky V. S., Vasil’ev Yu. В. Electrochim, Acta, 1966, v, JU,
As 12, p, 1439—145Q,
IS?
250. Page J. A., Wilkinson G. J. Am Chem. Soc., 1952, v, 74, № 10,
p. 6149—6155
251. Kuwana T., Bublitz D. E., Hoh G. J. Am. Chem. Soc., 1960,
v. 82, № 11, p. 5811—5817.
252. Hoh G. L., Mclwen W. E., Kleinberg /. J. Am. Chem. Soc.,
'1961, v. 83, № 8, p. 3949—3953.
253. -Гаммет Л. Основы органической химии. Пер. с англ. 'Под
ред. Л. С. Эфроса. М., «Мир», 1972. 534 с.
254. Matsumoto Т., Sato М., Ichimura A. Bull. Chem. Soc. Japan,
1971, v. 44, № 6, p. 1720—1728.
255. Morris M. D., Lingane I. J. J. Electroanal. Chem., 1964, v. 8,
№ 2, p. 85—92.
256. Bard A. J. Anal. Chem., 1963, v. 35, № 12, p. 1602—1606.
257. Davis D. G. Anal. Chem., 1963, v. 35, № 6, p. 764—770.
258. Goto M., Ishii D. J. Electroanal. Chem., 1970, v, 36/ № 1,
p. 1—10.
259. Goto M., Ishii D. J. Electroanal. Chem., 1972, v. 40, № 2,
p. 303—309.
260. Goto M., Ishii D. J. Electroanal. Chem., 1973, v. 42, № .3,
p. 463—467.
261. Goto M., Ishii D. «Nippon Kagaku Zassahi», 1969, v. 90, № 6,
p. 1250—1254.
262. Goto M., Ishii D. «Nippon Kagaku Zassahi», 1970, v. 91, №3,
p. 343—347.
263. Goto M., Ishii D. «Nippon Kagaku Zasshi», 1971, v. 92, № 2,
p. 234—238.
264. Курбатов Д. И. Докт. дис., Москва, ГЕОХИ, 1974.
265. Buck R. Р. Anal. Chem., 1963, v. 35, № 12, р. 1853—1858.
266. Campanella L., Cignini P., De Angelis G. Rev. Roum, Chim.,
1973, v. 18, № 7, p. 1269—1272.
267. Hawley D., Adams R. N. J. ElectroanaL Chem., 1964, v. 8,
p. 163—166.
268. Holub K., Loucka T. J. Electroanal. Chem., 1973, v. 44, '№ 3,
p. 459—470.
269. Лейбзон В. H. и др. «Электрохимия», 1973, т. 9, № 11,
с. 1689—1692,
270. Jacobsen Е., Sawyer D. Т. J. Electroanal. Chem., 1967, v. 15,
№ 2/3, р. 181—192.
271. Berzins Т., Delahay Р. J. Chem. Phys., 1955, v. 23, № 5,
p. 972—974.
272. Lorenz W. Z. Electrochem.-, 1954, Bd. 58, S. 912—920.
273. Smutek M. Coll. Czechosl. Chem. Comm., 1954, v. 19, p. 31—42.
274. Smutek M. Chem. Listy, 1953, v. 47, p. 963—970.
275. Дамаскин Б. Б. Принципы современных методов изучения
электрохимических реакций. М., изд-во МГУ, 1965. 119 с.
276. Hale J. М., Parsons R. J. Electroanal. Chem., 1964, v. 8, №2,
p. 247—249.
277. Ройтер В. А., Юза В. А., Полуян Е. С. ЖФХ, т. 13, № 4,
с. 605—608, 805—809.
278. Matsuda Н., Oka S., Delahay Р. J. Am. Chem. Soc., 1959, v. 81,
№ 10, p. 5077—5081.
279 Dirske T. P., Hampson N. A. Electrochim. Acta, 1972, v. 17.
№ 2, p. 135—142.
193
280. Birke R. L., Roe D. K. Anal. Chem., 1965, v. 37, № 4, p. 450—
454.
281. Inman D., Bockris J. O’M., Blomberg E. J. Electroanal. Chem.,
1961, v. 2, № 6, p. 506—514.
282. Kooijman D. J., Sluyters J. H. Electrochira. Acta, 1967, v. 12,
№ 6, p. 693—706.
283. Kooijman D. J., Sluyters. J. H. Electrochim. Acta, 1967, v. 12,
№ 12, p. 1579—1592.
284. Gerischer H., Krause M. Z. physik Chem., 1957, Bd. 10, № 2,
S. 264—274..
285. Gerischer H., Krause M. Z. physik. Chem., 1958, Bd. 14, № 1,
S. 184-196.
286. Matsuda H., Delahay p. Phys. Chem., 1960, v. 64, № 2,
p. 322—332.
287. Koijman D. J., Sluyters J. H. Electrochim. Acta, 1967, v. 12,
№ 9, p. 1147—1158.
288. Mizota K. e. a. J. Electroanal. Chem., 1973, v. 44, № 3,
p. 471—480.
289. Slaiman O. J. M„ Lorenz W. J. Electrochim. Acta, 1974, v. 19,
Xs 12, p. 791—798.
290. Van Leeuwen H. P., Sluyters J. H. J. Electroanal. Chem.,
1973, v. 42, № 2, p. 313—317.
291. Koijman D. J., Sluyters J. H. J. Electroanal. Chem., 1967,
v. 13, Xs 1/2, p. 152—156.
292. Wijnen M. D., Smit W. M. Rec. trav. Chim., 1960, v. 79, Xs 1,
p. 22—40.
293. Wijnen M. D., Smit W. M. Rec. trav. Chim., 1960, v. 79, № 2,
p. 203-215.
294. Wijnen M. D., Smit W. M. Rec. trav. Chim., 1960, v. 79, № 3,
p. 289—301.
295. Баканов В. И., Захаров М. С. Усп. хим., 1976, т. 45, № 1,
с. 3-28.
296. Sluyters J. Н. Rec. trav. Chim., 1962, v. 81, Xs 3, p. 297—300.
297. Herman H. B., Bard A. J. Anal. Chem., 1963, v. 35, Xs 9,
p. 1121—1126.
298. Sturrock P. E. J. Electroanal. Chem., 1964, v. 8, Xs 3, p. 425—
432.
299. Черненко В. И., Литовченко К. И., Удовенко Ю. Э. «Элек-
трохимия», 1971, т. 7, Xs 10, с. 1476—1480.
300. Cukman D., Vukovit М., Pravdic V. J. Electroanal. Chem.,
1974, v. 49, Xs 4, p.,421—428.
301. Кравцов В. И. Электродные процессы в растворах комплек-
сов металлов. Л., изд-во ЛГУ, 1969. 190 с.
302. Кравцов В. И., Чамаев В. Н. Вести. ЛГУ, 1966, т. 4, Xs 1,
с. 133—140.
303. Кравцов В. И., Чамаев В. Н. Вести. ЛГУ, 1968, т. 4, Xs 1,
с. 94—101.
304. Кравцов В. И., Зеленский М. И. «Электрохимия», 1966, т 2
Xs 10, с. 1138-1144.
305. Кравцов В. И., Зеленский М. И. Вести. ЛГУ, 1966, т. 4, Ха 1,
с. 127—132.
306. Орехова В. В., Андрющенко Ф. К., Дмитриева Л. Н. Изв. '
ВУЗов. Хим. и хим. технол., 1975, т. 18, Xs 1, с. 90—93.
194
307. Даведова Л. И., Кергши А., Ларионов О. В. «Электррхн-.. ,/S
мия», 1974, т. 10, № 4, с. 653—657. W
308. Кублановский В. С., Литовченко К. И., Кривина Е. И. «Элек- - §
трохимия», 1975, т. 11, № 9, с. 1407—1410. i
309. Stackelberg М., Pilgram М., Toome V. Z. Electrochem., 1963, >£
Bd. 57, № 2, S. 342—350. |
310. Ларионов О. В., Дурдин Я. В. «Электрохимия», 1968, т. 4,
Ks 6, с. 630-635. '
311. Емельяненко Г. А., Симулин Г. Г., Байбарова Е. Я- Укр.'
хим. ж., 1963, т. 29, № 4, с. 404—408.
312. Емельяненко Г. А., Симулин Г. Г. Укр. хим. ж., 1963, т. 29,
№ 5, с. 727—730.
313. Симулин Г. Г., Плесецкая Т. Ф. Укр. хим. ж., 1973, т. 39,
№ 10, с. 978-984.
314. Мацола М. В., Герасименко Л. А., Галинкер В. С. «Электро-
химия», 1974, т. 10, № 3, с. 465—467.
315. Лошкарев Ю. М., Рысаков А. А., Литовка Г. П. «Электро-
химия», 1974, т. 10, Ks 10, с. 1597—1599.
316. Ууаров Л. А. ЖФХ, 1962, т. 36, № 5, с. 981—985.
317. Кравцов В. И., Петрова Г. М. ДАН СССР, 1964, т. 154, №2,
с. 433—436.
318. Орехова В. В., Андрющенко Ф. К- «Электрохимия», 1975,
Т. 11, №7, с. 882—886.
319. Емельяненко Г. А., Симулин Г. Г. ЖФХ, 1965, т. 39, К» 5,
с. 1077—1081.
320. Емельяненко Г. А., Симулин Г. Г. «Электрохимия», 1965, т. 1,
№ 11, с. 1384-1388.
321. Бек Р. К).,-Нечаев Е. А., Кудрявцев Н. Т. «Электрохимия»,
1967, т. 3, № 12, с. 1465—1467.
322. Емельяненко Г. А., Симулин Г. Г. «Электрохимия», 1968,
т. 4, Ks 2, с. 187—191.
323. Lingane S. S., Lingane Р. S. J. Electroanal. Chem., 1963, v. 5,
Ks 1, p. 41—50.
324. Anson F. C„ Lingane S. S. J. Amer. Chem. Soc., 1957, v. 79,
Ks 2, p. 1015—1021.
325. Lingane S. S., Singane P. S. J. Electroanal. Chem., 1963, v. 5,
Ks 6, p. 411—419.
326. Alexander U7. A., Barclay D. S. J. Electroanal. Chem., 1967,
v. 13, Ks 1/2, p. 137—143.
327. Palke W. E., Russell C. D., Anson F. C. Anal. Chem., 1962,
v? 34, Ks 9, p. 1171—1172.
328. Deron S., Laitinen H. A. Anal. Chem., 1966, v. 38, Ks 10,
p. 1290—1297.
329. Nagai T., «Nippon Kagaku Zasshi», 1960, v. 81, p. 252—254.
330. Davis D. G., Everhart M. E. Anal. Chem.,1964, v. 36, Ks 1,
p. 38—40.
331. Ishlbashi M. e. a. «Nippon Kagaku Zasshi», 1960, v. 81,
p. 744—747.
332. Herman H. B., Bard A. S. Anal. Chem., 1964, v. 36, Ks 6,
p. 971—975.
333. Стыркас Д. Д. «Электрохимия», 1969, т. 5, Ks 12, с. 1488—
1491.
334 Лошкарев Ю. М„ Малая Р. В., Снеткова Л. П. ДАН СССР,
1971, т. 199, № 3, с. 650—654.
195
335. Bowden F. P. Proc. Roy. Soc., 1929, v. 125A, p. 446—450.
336. Butler J. A., Armstrong G. Proc. Roy. Soc., 1932, v. 137, № 5,‘
p. 604—612.
337. Фрумкин A. H., Шлыгин А. И. ЖФХ, 1935, t. 3, № 4, c. 791— •
800.
338. Фрумкин A. H., Шлыгин А. И. Изв. АН СССР, сер. хим.
цаук, 1936, № 5, с. 773—780.
339. Фрумкин А. Н., Шлыгин А. И. ЖФХ, 1936, т. 5, № 5, с. 820— ’
829.
340. Тедорадзе Г. А., Хмельницкая Е. 10., Золотовицкий Я- М.
В ки.: Прогресс электрохимии органических соединений. Т. 1.
М., «Наука», 1969, с. 157.
341. Левинсон И. М., Тедорадзе Г. А., Эршлер А. Б. «Электро-
химия», 1968, т. 4, № 4, с. 461—464.
342. Эршлер А. Б. и др. «Электрохимия», 1971, т. 7,'№ 7, с. 953—
960.
343. Herman Н. В., Blount Н. N. J. Electroanal. Chem., 1970, v. 25,
№ 2, р. 165—179.
344. Kiss L., Varsanyi M. Maguar Kern, folyoirat., 1970, v. 76,
№ 3, p. 125—127.
345. Sobkowskl J., Guminski J. Roczn. chemii, 1970, v. 44, № 5,
p. 1135—1139.
346. Подловченко Б. И., Стенин В. Ф., Фрумкин А. Н. «Электро-
химия», 1972, т. 8, № 1, с. 129—132.
347. Ershler А. В. ё. a. J. Electroanal. Chem., 1974, v. 54, № 1,
р. 75—86.
348. Хмельницкая Е. Ю., Тедорадзе Г. А., Золотовицкий Я- М.
«Электрохимия», 1967, т. 3, Ns 2, с. 200—208.
349. Гончарук В. К-, Брыскин И. Е., Тюрин Ю. М. Химия и хим.
технол., 1973, т. 29, № 1, с. 230.
350. Гончарук В. К-, Брыскин И. Е., Тюрин Ю. М. «Электрохи-
мия», 1974, т. 10, № 4, с. 501—509.
351. Эршлер А. Б. и др. «Электрохимия», 1973, т. 9, № 7, с. 952—960.
352. Флёров В. Н., Тюрин Ю. М. «Электрохимия», 1970, т. 6, №9,
с. 1404—1409.
353. Tatwawadi S. V., Bard A. J. Anal. Chem., 1964, v. 36, Ns 1,
p. 2—4.
354. Lorenz W. Z. Electrochem., 1955, Bd. 49, Ns 4, S. 730 —740.
355. Reinmutn W. Anal. Chem., 1961, v. 33, Ns 3, p. 322—325.
356. Anson F. Anal. Chem., 1961, v. 33, Ns 9, p. 1123—1127.
357. Lingane P. J. Anal. Chem., 1967, v. 39, № 4, p. 485—494.
358. Эршлер А. Б. и др. «Электрохимия», 1971, т. 7, Ns 8,
c. 1083-1091.
359. Эршлер А. Б. В кн.: Электросинтез и биоэлектрохимия. М.,
«Наука», 1975, с. 251.
360. Murray R. Р7, Gross D. J. Anal. Chem., 1966, v. 38, Ns 3,
p. 392—404.
361. Gross D. J., Murray R. W. Anal. Chem., 1966, v. 38, № 3,
p. 405—409.
362. Christie J. H., Osteryoung R. A. Anal. Chem., 1966, v. 38,
Ns 11, p. 1620—1622.
363. Osteryoung R. A., Christie J. H. J. Phys. Chem., 1967, v. 71,
Ns 5, p. 1348—1354.
364. Anson F. C. Anal. Chem., 1964, v. 36, № 3, p. 520—523.
196
365. Laitlnen H., Gaur H. C. Anal. Chim. Acta, 1958, v. 18, № I,
p. 1—9.
366. Мусихин В. И., Есин О. А. ДАН СССР, 1961, т. 136, Ns 2,
с. 388—390.
367. Калугин В. Н., Есин О. А., Топорищев Г. А. «Физика не-
металлов и металловедение», 1964, т. 17, № 1, с. 88—92.
368. Барабошкин А. Н., Смирнов М. В., Салтыков Н. А. Труды
ии-та электрохимии Уральск, фил. АН СССР, 1961 № 2,
с. 53—62.
369. Делимарский Ю. К., Городысский А. В., Кузьмович В. В.
Coll. Czechosl. Chem. Cortitn., 1960, v. 25, № 10, p. 3056—
3060.
370. Stein R. B. J. Electrochem. Soc., 1959, v. 106, № 5, p. 528—
536.
371. Van Korman J. D. Anal. Chem., 1961, v. 33, № 7, p. 946—948.
372. Смирнов M. В., Рыжик О. А., Казанцев Г. H. «Электрохи-
мия», 1965, т. 1, № 1, с. 59—62.
373. Van Norman J. D., Egan J. J. J. phys. Chem., 1963, v. 67,
№ 11, p. 2460—2462.
374. Van Norman J. D. J. Electrochem. Soc., 1965, v. 112, № 11,
p. 1126—1129.
375. Делимарский Ю. K-, Макогон В. Ф. Укр. хим. ж., 1968, т. 34,
№ 1, с. 128—130.
376. Буторов В. П. и др. ЖПХ, 1972, т. 45, № 10, с. 2160—2164.
377. Волгин М. А., Львов А. Л., Лоскуткин В. А. «Электрохимия»,
1973, т. 9,. № 3, с. 368—371.
378. Ивановский Л. Е. и др. «Электрохимия», 1975, т. 11, № 9,
с. 1371—1373.
379. Делимарский К- Ю., Гриценко В. Ф., Пархоменко И. И. Укр. '
хим. ж., 1975, т. 41, № 11, с. 11,23-1127.
380. Делимарский Ю. К., Городысский А. В. Электродные про-
цессы и методы исследования в полярографии. Киев-, изд-во
АН УССР, 1960, 253 с.
381. Делимарский Ю. К-, Панов Э. В., Городысский А. В. Теор.
и эксп. химия, 1965, т. 1, № 6, с. 777—784.
382. Филыитих В. Топливные элементы. Пер. с нем. Под ред.
В. С. Багоцкого. М., Издатинлит, 1968, 419 с.
383. Леонова Л. С., Рябухин Ю. М., Укше Е. А. «Электрохимия»,
1969, т. 5, № 1, с. 210—211.
384. Волгин М. А., Львов А. Л. «Электрохимия», 1971, т. 7, № 9,
с. .1388—1390. .
385. Шаповал В. И., Делимарский Ю. К-, Циклаури О. Г. Укр.
хим. Ж., 1974, т. 40, К» 7, с. 734—737.
386. Wrench D. М., Inman D. Electrochim. Acta, 1967, v. 12, № 12,
p. 1601—1608.
387. Шаповал В. И., Василенко В. А. Укр. хим. ж., 1974, т. 40,
№9, с. 986—989.
388. Inman D., Braunstein J. Chem. Czehosl. Comm., 1966, № 6,
p. 148—149.
389.. Делимарский Ю. K-, Шаповал В. И., Василенко В. А. Укр.
хим. ж., 1973, т. 39, № 6, с. 617—619.
390. Marassi R., Bartocci V., Picciarelli F. «Taianta», 1972, v. 19,
№ 2, p. 203—206,
197
391 Petrescu S., Sternberg S„ Petrescu V. Rev. roum. chim., 1973,-
‘ v. 18, № 10, p. 1715—1723.
392. Sternberg S., Petrescu V. Rev. roum. chim., 1974, v. 19, № 1,>
p. 25—36.
393. Sternberg S., Petrescu S., Petrescu V. Rev. roum. chim., 1974,.
v. 19, № 6, p. 955—965. 'J
394. Sternberg S., Petrescu V. Rev. roum. chim., 1975, v. 20, № 2, j
p. 159—168. . i
395. Sternberg S., Galasiu I. Rev. roum. chim., 1974, v, 19, № 4,
p. 523—531. J
396. Sternberg S., Galasiu I. Rev. roum. chim., 1974, v. 19, № 6,;
p. 967—976. '
397. Delahay P. In: Advances in Electrochemistry and Electroche- .i
mical Engineering. V. 1. Ed. P. Delahay. New York, Inter-
science, 1961, p. 233—318.
Васько A. T., Зосимович Д. П. ЖФХ, 1959, т. 33, № 11, •'
398.
с. 2617—2618.
399. Gierst L., Juliard A. J. Phys. Chem., 1953, v. 57, № 2, p. 701—
706.
400. Кноц Л. Л. и др. В кн.: Электрохимические процессы с уча- ,
стием органических веществ. М., «Наука», 1970, с. 47—55. 1
401. Koijman D. I., Sluyte з J. H. J. Electroanal. Chem., 1967,
v. 13, № 1, p. 152—158.
402. Grimes S. Chem. Ihstrum.1973—1974, v. 5, № 2, p. 141—144.';
403. Gierst L., Mechelynck P. Anal. Chim. Acta, 1955, v. 12, № 1,
p. 79-86.
404. Галинкер Э. В. В кн.: Успехи полярографии с накоплением.
Томск, изд-во Томск, гос. ун-та, 1973, с. 182—184.
405. Маковецкий А. Л. В кй.: Успехи полярографии с накопле-
нием. Томск, изд-во Томск, гос. ун-та, 1973, с. 220—222.
406. Салин А. А. и др. В кн.: Автоматическвй контроль при обо-
гащении и гидромета'ллургии цветных металлов. Ташкент,
изд-во «Фан», 1967, с. 54—115. "
407. Вариков В. Г. и др. В кн.: Прикладная и теоретическая <
электрохимия. Вып. 3. Алма-Ата, изд-во «Наука» КазССР,
1971, с. 78-86. • '
408. Крюкова Е. А., Синякова С. И., Арефьева Т. В. Поляро- ':
графический анализ. М., Госхимнздат, 1959: 652 с. i
409. Делимарский Kh К, Скобец Е. М. В кн.: Полярография иа
твердых электродах. Киев, «Технща», 1970, с. 220.
410. Будников Г. К. Принципы и применение вольтамперной по-
лярографии. Казань, изд-во Казанск. гос. ун-та, 1975. 199 с.
411. Takemori У. Rev. Polarography (Japan), 1961. v. 9, p. 246—
256. (
412. Ishibashi M., Fujinaga T. Bull. Chem. Soc. Japan, 1958, v. 31,
№ 3, p. 887—892.
413. Ishibashi M. e. a. J. Chem. Soc. Japan, Pure Chem. Sec., 1959, •
v. 80, № 2, p. 478—486.
414. De-Mars R., Shain I. Anal. Chem. 1957, v. 29, № 10, p. 1825—
1829.
415. Стромберг А. Г., Городовых В. E. Зав. лаб., 1960, т. 26, № 1,
с. 41—45.
416. Cardiner К, Rogers L. Anal. Chem. 1953, v. 25, № 10, p, 1393—
1397,
198
417. Marpl T. L., Rogers L. Anal. Chem., 1953, v. 25, № 9,
p. 1351—1356.
418. Стромберг А. Г., Захарова Э. А. Зав. лаб., 1964, т. 30, № 3,
с. 261—271.
419. Сангина О. А. Амперометрическое титрование. М, «Химия»,
1967. 387 с.
420. Steinberg М., Nachtrieb N. 3. Am. Chem. Soc., 1950, v. 72,
№ 7, p. 3558—3566.
421. Делимарский Ю. К, Грищенко В. Ф. Укр. хим. ж., 1963,
, т. 29, № 5, с. 502—507.
422; Narayan R., Inman D. J. Polarogr. Soc, 1965, v, 11, № 2,
p. 27—30. , \
423. Kisza A., Zabinska G. Bull. L’Academie Polon. scien. Ser. sci.
chim., 1972, v. 20, № 7, p. 719—724.
424. Sternberg S, Herdlicka C. Revue roum. chim, 1974, v. 19,
№ 2, p. 197—206.
425. Волков Г. M., Калугин В. И., Сысков К. И. ДАН СССР,
1968, т. 183, № 2, с. 369—397.
426. Брайнина X. 3., Кива Н. К. В кн.: Методы анализа химиче-
ских реактивов и препаратов. М, изд. ИРЕА, 1963, вьш. 5—6,
с. 134—138.
427. Elving Р. J., Smith D. L. Anal. Chem., 1960, v. 32, № 13,
p. 1849—1854.
428. Чеканова В. Д, Фиалков А. С. Усп. хим, 1971, т. 40, № 5,
с. 777—805.
429. Кабанова О. Л., Бениаминова С. М. ЖАХ, 1971, т. 26, № 1,
с. 111—116.
430. Бениаминова С. М., Кабанова О. Л. ЖАХ, 1975, т. 30, № 1,
с. 68—73.
431. Раннее Г. Г., Волкова В. С., Муртазаев А. М. Зав. лаб,
1970, т. 36, № 12, с. 1446-1448.
432. Анохин Б. А., Игнатов В. И. ЖАХ, 1974, т. 29, ,№ 6,
с. 1221—1222.
433. Гончаров Ю. А. Каид. дис, М, ГЕОХИ, 1975. ч
434. Плесков. Ю. В., Филиновский В. Ю. «Электрохимия»,. 1975,
т. 11, с. 57—108.
435. Ruby W. R., Tremmel С. G. J. Electroanal. Chem, 1966, v. 12,
№ 2, p. 216—219,
Матвей Сафонович Захаров
Вячеслав Иванович Баканов
Владимир Васильевич Пнев
ХРОНОПОТЕНЦИОМЕТРИЯ
Редактор Г. Н. Г ост сева
Художник А. Я. Михайлов
Художественный редактор Н. В. Носов
Технический редактор В. В. Хазикова
Корректоры Л. А. Волкова, Г. М. Гольбиндер
ИБ № 14
Сдано в наб. 5.04.78. Подп. в печ. 10.10.78. Т-19603. Формат
бумаги 84Х108‘/к. Бумага тип. № 2. Гари, литературная. Печать
высокая. Усл. печ. л. 10,50. Уч.-изд. л. 8,65. Тираж 4300 экз.
Заказ № 1072. Цена 1 р. 10 к Изд. № 1064.
Ордена „Знак Почета" издательство «Химия». 107076, Москва,
Стромынка, 13.
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой
«Союзполиграфпрома» при Государственном комитете
СССР по делам издательств, полиграфии и книжной
торговли. 198052, Ленинград, Л-52, Измайловский
проспект, 29.