/



























































































































































































Text
6П7.5
Г67
УДК 678. 048
Горбунов Б. Н., Гурвич Я. А., Маслова И. П.
Химия и технология стабилизаторов полимерных
материалов.— М.: Химия, 1981.— 368 с., ил.
В книге впервые обобщен и систематизирован материал
по методам синтеза и технологии промышленных стабилиза-
торов полимерных материалов. Рассмотрены сырье и перспек-
тивные методы получения стабилизаторов; приведены принци-
пиальные технологические схемы.
Кратко изложены современные представления о механиз-
ме действия, структуре и эффективности стабилизаторов;
впервые предложены их классификация и номенклатура; даиы
основные представления о тенденциях развития производства
стабилизаторов. В сжатой форме описаны основные и возмож-
ные области применения стабилизаторов и их свойства.
Книга предназначена для химиков и технологов, рабо-
тающих в области синтеза стабилизаторов и их применения
в промышленности синтетических каучуков, пластических масс,
синтетических волокон, резиновых технических изделий и дру-
гих полимерных материалов. Она будет полезна преподавате-
лям, аспирантам и студентам старших курсов химико-техно-
логических вузов.
368 с., 7 табл., 55 рис., список литературы 1050 ссылок.
Рецензент — начальник Технического управления
МНХП СССР А. И. ЛУКАШОВ
„ 31407-175
Г 050(01 i-si"79-80-2803010000
© Издательство «Химия», 1981 г.
СОДЕРЖАНИЕ
Предисловие..........................................................7
Глава 1. Общие понятия о стабилизаторах . . 9
1.1. Основные представления о механизме действия, структуре и эффектив-
ности стабилизаторов......................................... ... 10
1.2. Классификация и номенклатура стабилизаторов....................19
1.3. Тенденции развития производства стабилизаторов.................26
Литература..........................................................28
Глава 2. Производные аминов..................................зо
2.1. Сырье для получения аминных стабилизаторов ... 31
2.2. Реакции араминирования. Ароматические амины ... 35
N-ФеНил-Р-нафтиламин ...............................................36
N-Фенил-а-нафтиламин ...............................................40
пТидроксифенил-ф-нафтиламин.........................................43
Дифениламин.........................................................46
п-Гидроксидифениламин...............................................51
М,М'-Дифенил-п-фенилендиамин 55
N.N'-Ди-Р-нафтил-п-фенилендиамин....................................62
Литература..........................................................67
2.3. Реакции нитрозирования. Нитрозопроизводные дифениламина . 69
N-Нитрозодифениламин . . 72
п-Нитрозодифениламин................................................75
Литература..........................................................80
2.4. Каталитическое гидрирование ароматических нитрозосоединений, нит-
росоединений и аминов. Другие способы. восстановления .... 81
4-Аминодифениламин................................................ 86
2-(2'-Гидрокси-5'-метилфеннл)бензгриазол............. . . 94
4,4'-Диаминодициклогексилметан . . ............. ... 99'
Литература........................................ . . . . 103.
3
. 2.5. Каталитическое восстановительное алкилирование ароматических нит-
розосоединений, нитросоединений и аминов. М.М'-Диалкил- и N,N'-aji-
киларил-п-фенилендиамины.............................................
N.N'-Ди-втор-бутил-п-фенилендиамин......................... . . .
у N-Фенил-М'-циклогексил-п-фенилендиамин...............................
N-Изопропил-М'-фенил-п-фениленднамин................................
'• И-(1,3-Диметилбутил)-М'-фенил-п-фенилендиамнн.......................
Литература..........................................................
2.6. Алкилирование-ароматических аминов............................
/ 4,4'-Диалкилдиф£нилаМин............................................
4,4 ’-Ди (1,1,3,3-тетраметилбутил) дифениламин.....................
4,4'-Ди (а-метилбензил) дифениламин...........................
4,4'-Бис (а,а-диметилбензил) дифениламин.....................
Литература.........................................................
105
109
111
116
122
125
127
129
131
134
136
137
2.7. Конденсация ароматических аминов с карбонильными соединениями 138
4,4'-Диаминодифенилметан ...............................................144
Альдоль-а-нафтиламин....................................................152
Полимер 2,2,4-триметил-1,2-дигидрохинолина..............................155
2,2,4-Триметил-6-этокси-1,2-дигидрохинолин..............................157
Продукты конденсации дифениламина с ацетоном . 162
Литература..............................................................166
2.8. Тиоацилирование аминов. Дитиокарбаматы, тиокарбамиды и меркапто-
бензимидазолы ................................................... 169
Диалкилдитиокарбаматы металлов.....................................171
Замещенные тиокарбамиды............................................175
2-Меркаптобензимидазол............................................ 181
Литература.............................................• . . . 184
I
Глава 3. Производные фенола................186
3.1. Сырье для производства фенольных стабилизаторов.....................186
3.2. Алкилирование фенола................................................191
2,6-Ди-трет-бутилфеиол...................................................201
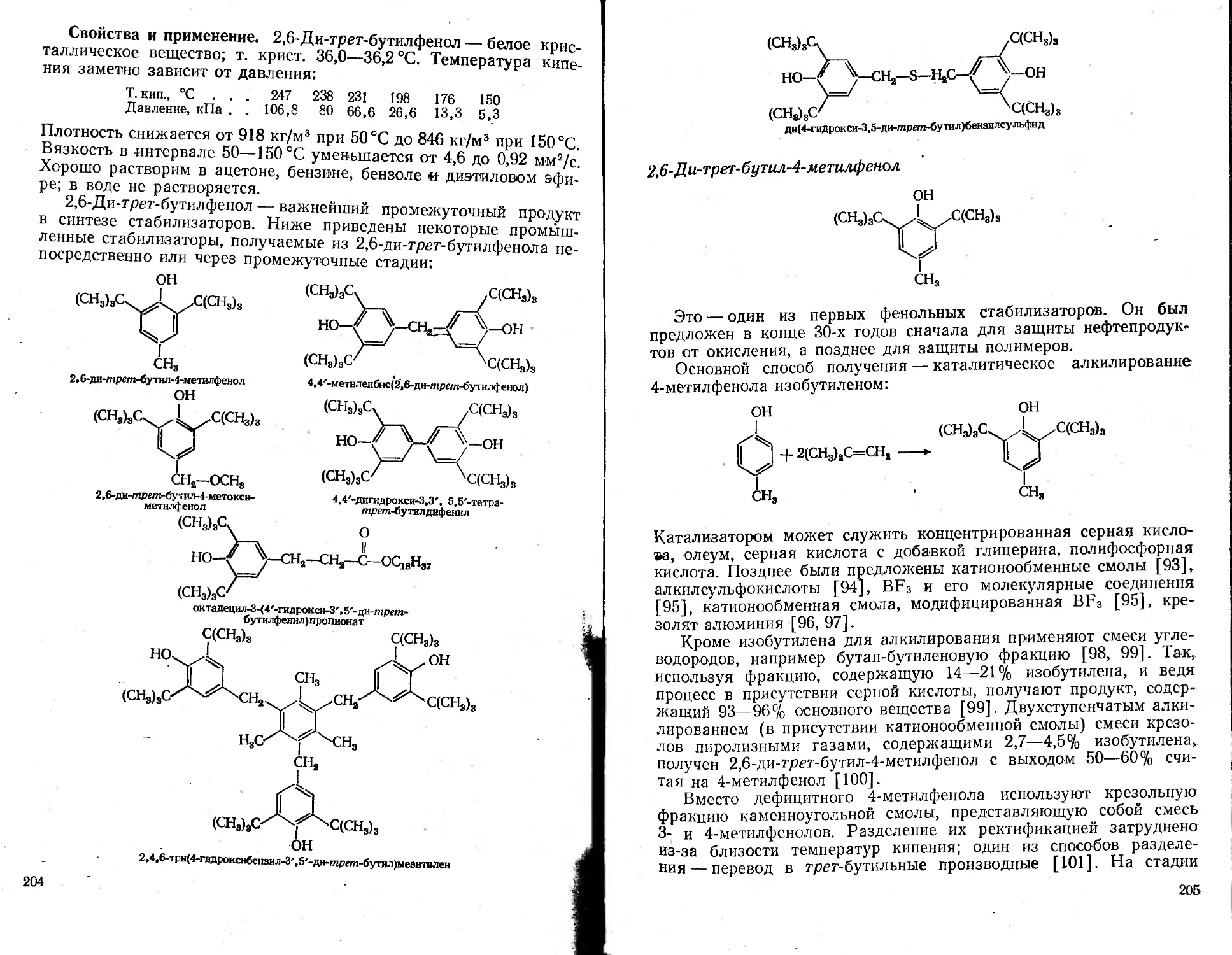
2,6-Ди-грет-бутил-4-метилфенол .... 205
2-трет-Бутил-4-метилфенол................................................212
6-трег-Бутил-З-метилфенол................................................215
а-Метилбензилфеиолы......................................................216
2,6-Ди-трет-бутил-4-метоксиметилфенол....................................220
4-Гидрокси-3,5-ди-трет-бутилбензиловый спирт.............................222
б-трет-Бутил-2-метилфенол................................................223
Эфиры 0-(4-гидрокси-3,5-ди-трет-бутилфенил) пропионовой кислоты . . 224
2,5-Ди-трет-бутилгадрохинон..............................................227
2,5-Ди-трет-амилгидррхинон . ............................................228
Литература...............................................................230
4
3.3. Конденсация алкилфенолов с карбонильными соединениями. Синтез ме-
тиленбисфенолов........................................................... 233 -
2,2'-Метиленбис(6-трет-бутил-4-метилфенол).................................244
2,2'-Метиленбис(6-трет-бутил-4-этилфенол)..................................250
2;2'-Метнленбнс(4,6-диметилфеиол)..........................................251
2;2'-Изобутилиденбис(4,6-диметилфенол).....................................252
2,2'-Метиленбис(4-метил-6-а-метилциклогексилфенол).........................253
4,4'-Метиленбис(2,6-ди-трет-бутилфенол)....................................255
4,4'-Бутилиденбис(6-7ре7-бутил-3-метилфенол)...............................258
Литература............................................................... 259
3.4. Конденсация алкилфенолов с тиохлоридами. Синтез тиобисфенолов 260
2,2'-Тиобис(6-трет-бутил-4-метилфенол).....................................267
2,2'-Тиобис(4-метил-6-а-метилбензилфенол)..................................27о
2,2'-Тиобис(4,6-ди втор-амилфенол).........................................271
4,4'-Тиобис(6-трет-бутил-.2-метилфенол)....................................273
4,4'-Тиобис(6-трет-бутил-3-метилфенол) . . ...........................274
Бис (4-гидрокси-3,5-ди-трет-бутилбензил) сульфид...........................277
Литература.................................................................279
3.5. Синтез стабилизаторов группы трис- и тетрафенолов -..................280
1,1,3-Трис(5'-трет-бутил-4,-гидрокси-2/-метилфенил)бутан...................291
2,4,6-Трис(4/-гидрокси-3',5'-ди-трет-бутилбеизил)мезитилеи.................293
Эфир 4-гидрокси-3,5-ди-трет-бутилпропиоиовой кислоты и пентаэритрита 297
Литература.................................................................298
3.6. Ацилирование фенолов. Синтез о-гидроксифенилкетонов 298
2,4-Дигидроксибензофенон...................................................305
4-Алкокси-2-гидроксибензофенои.............................................309
2-Гидрокси-4-метоксибензофенон.............................................311
Литература.................................................................313
Глава 4. Эфиры фосфористой кислоты . . . з15
Три-н-октилфосфит..................................................318
Три (2-этилгексил) фосфит..........................................319
Трифеиилфосфит.................................................... 320
Три (n-нонилфеиил) фосфит..........................................322
Три (а-метилбензилфенил) фосфит....................................323
Литература.........................................................325
Глава 5. Эфиры тио (дипропионовой) кислоты згт
Тио(дилаурилпропиоиат) . ........................................332
Тио(дистеарилпропионат)...............................\ ’ 333
Тио(димиристилпропионат) ........................................333
Литература.......................................................334
5
Глава 6. Металлсодержащие стабилизаторы 335
6.1. Соли свинца . . . 336 j
Силикат свинца...................................................337
Трехосновный сульфат свинца..................................... 340
Основной карбонат свинца........................................ 342
Двухосновный фосфит свинца ......................................344
Средний стеарат свинца ......................................... 345
Двухосновный стеарат свинца..................................... 347
Двухосновный фталат свинца...................................... 349
6.2. Соли металлов I—III групп ..................................350
Стеарат кальция..................................................350
Стеарат цинка.................................................. 352
Стеарат кадмия................................................... 354 ;
6.3. Оловоорганические соедииеиия................................355
Диалкилкарбоксилаты олова...................................... 358
Оловоорганические стабилизаторы, содержащие серу.................362
Литература.......................................................364
Предметный указатель............................................. 366 /
к
ПРЕДИСЛОВИЕ
Химия и технология стабилизаторов определилась как само-
стоятельное направление химической науки 25—30 лет назад. К на-
чалу этого периода' потребности полимерной промышленности в
стабилизаторах удовлетворялись в основном N-фенил-р-нафтилами-
ном. В настоящее время описано более 2000 соединений, являю-
щихся стабилизаторами полимерных материалов, причем наряду с
представителями «традиционных» классов — ароматическими ами-
нами и фенолами — практическое значение приобрели бензтриазо-
лы, тиокарбамиды, фосфиты, тиодипропионаты и другие соедине-
ния. Работы по синтезу стабилизаторов интенсивно ведутся во всех
технически развитых странах, быстрыми темпами идет организа-
ция промышленного производства, из года в год увеличивается
объем выпуска стабилизаторов. Сегодня только в США выпуска-
ется около 700 марок стабилизаторов.
Возможность значительного улучшения свойств полимеров и
повышения эксплуатационных характеристик изделий из полимер-
ных материалов за счет введения в рецептуру более эффективных
стабилизаторов обусловила и активизировала изучение механизма
их действия, установление взаимосвязи между строением и эффек-
тивностью, создание теоретических основ подбора стабилизаторов
для конкретного-полимера.
Быстрое развитие работ в области химии и технологии стабили-
заторов обусловило в свою очередь резкое увеличение объема науч-
но-технической информации, рассеянной в многочисленных журна-
лах, сборниках и патентных описаниях. Если исследования, связан-
ные с теоретическими вопросами стабилизации и ролью стабилиза-
торов в процессе старения полимеров, нашли отражение в ряде
обзорных статей и монографий, то работы по синтезу, разработке
технологии, изучению физико-химических и санитарно-гигиенцчес-
ких свойств стабилизаторов не обобщены до настоящего времени.
Необходимость такого обобщения для развивающейся отрасли
науки, которой являются химия и технология стабилизаторов, оче-
видна— оно способствует быстрому ознакомлению с новой, тема-
тически подобранной информацией широкого круга специалистов,
повышает эффективность использования средств, выделяемых на
научные исследования*.
Эмануэль Н. М,— Вестник АН СССР, 1978, № 5, с. 72—77.
7
В настоящей монографии впервые систематизирован имеющий-
ся в научной и патентной литературе материал по методам синте-
за и технологии стабилизаторов, получению промежуточных про-
дуктов. Включены данные справочного характера, касающиеся фи-
зико-химических и санитарно-гигиенических свойств. Впервые сде-
лана попытка классифицировать стабилизаторы по свойствам и
применению и упорядочить их номенклатуру.
При написании книги авторы испытывали известную трудность
относительно оптимального варианта расположения материала и
отдают себе отчет, что выбранный вариант — по химическим реак-
циям синтеза — не лишен недостатков, так как способы получения
стабилизаторов, особенно производных ароматических аминов, не
укладываются в рамки одной реакции. За основу классификации
методов получения стабилизаторов были приняты промышленные
способы их получения. Отдельные аспекты технологии носят дис-
куссионный характер. Там, где это представлялось возможным,
были сопоставлены точки зрения различных авторов. Краткость
первой главы, несмотря на важность рассматриваемых в ней во-
просов, обусловлена тем, что они в значительной мере отражены
в отдельных работах, обзорах и монографиях Н. М. Эмануэля,
Е. Т. Денисова, В. В. Ершова, Б. М. Коварской, А. С. Кузьминско-
го, С. Д. Разумовского, Э. Г. Розанцева, В. Я. Шляпинтоха и их
сотрудников, в ряде сборников и переводных изданий. Монография
является первой работой такого плана и, конечно, не свободна от
недостатков. Авторы будут благодарны всем, кто выскажет кри-
тические замечания в ее адрес. %
Считаем приятным долгом выразить признательность профес-,
сору В. В. Ершову, кандидату химических наук М. С. Хараш и
кандидату технических наук С. Т. Кумок за ценные замечания,
высказанные при просмотре рукописи, а также Л. А. Пугачевой
и О. Ф. Стариковой за помощь в оформлении рукописи.
ГЛАВА 1
Общие понятия о стабилизаторах
Объем и темпы роста производства полимерных материалов до-
стигли очень высокого уровня. В настоящее время в мире про-
изводится примерно 50 млн. т пластических масс и химических во-
локон, 9,5 млн. т синтетического каучука и перерабатывается более
4 млн. т натурального каучука. Для защиты этих полимеров тре-
буется «500 тыс. т стабилизаторов. Они увеличивают срок служ-
бы изделий из полимерных материалов, повышают их стабильность,
надежность в работе. Без применения стабилизаторов невозможны
синтез и переработка таких важных для народного хозяйства по-
лимеров, как диеновые каучуки, полипропилен, полиформальдегид.
Стабилизаторы в значительной степени определяют качество и экс-
плуатационные показатели полимеров, существенно влияя на стои-
мость изделий из полимерных материалов. Применение стабилиза-
торов экономически выгодно, и в технически развитых странах их
выработка опережает в своем развитии другие химические произ-
водства.
Промышленное производство стабилизаторов, как и других хи-
мических добавок к полимерам, возникло и традиционно развива-
лось в рамках анилинокрасочной промышленности с использова-
нием методов, аппаратуры, технологии и сырьевой базы этой от-
расли. Однако специфика потребительских свойств, особенности
областей применения, а также необходимость быстрого обновления
и расширения ассортимента потребовали изменения сырьевой ба-
зы, разработки новых методов синтеза и новых технологических
процессов. В настоящее время производство добавок для полимер-
ных материалов постепенно приобретает черты, присущие промыш-
ленности, производящей сами полимеры. Подотрасль химических
добавок идет по пути крупнотоннажных непрерывных автомати-
зированных производств и комплексов на базе единого сырья* и
полупродуктов, по пути унифицированных технологических схем,
позволяющих быстро менять ассортимент вырабатываемых мало-
тоннажных веществ.
Развитие производства стабилизаторов неразрывно связано с
развитием производства полимеров и имеет огромное значение для
научно-технического прогресса, экономического потенциала стра-
ны и для благосостояния населения. У нас в стране этой актуаль-
ной проблеме химии и технологии уделяется большое внимание.
9
В «Основных направлениях экономического и социального разви-
тия СССР на 1981—1985 годы и на период до 1990 года» (проект
ЦК КПСС к XXVI съезду партии) намечено развивать производст-
во высококачественных полимеров и «более полно удовлетворять
потребности народного хозяйства в химических добавках для по-
лимерных материалов...» [1]. Развитию производства стабилиза-
торов уделяется большое внимание и в странах социалистического
лагеря. Заметных успехов достигли в этой области ЧССР и ГДР.
С 1971 г. страны социализма осуществляют специализацию и коор-
динацию работ в области химических добавок для полимерных
материалов в рамках ИНТЕРХИМ.
1.1. ОСНОВНЫЕ ПРЕДСТАВЛЕНИЯ О МЕХАНИЗМЕ
действия, структуре и эффективности
СТАБИЛИЗАТОРОВ
Вещества, способные задерживать «порчу» органических ма-
териалов, в том числе и природных полимеров, применялись еще
в глубокой древности. Туземцы Южной Америки для сохранения
пищевых качеств медвежьего жира добавляли в него экстракт ко-
ры красного ильма. Фармацевты применяли экстракты почек не-
которых деревьев для стабилизации различных мазей на жировой
основе. Позднее было установлено, что в коре и почках деревьев
содержатся эфиры галловой кислоты — эффективные прнродны^
антиокислители.
В самом общем смысле стабилизаторами называют органичес-
кие и неорганические соединения, способные замедлять процессы,
ухудшающие эксплуатационные показатели полимеров под дейст-
вием внешних условий (тепло, свет, действие озона, радиация, ме-
ханические нагрузки). Под влиянием этих факторов снижается
эластичность, ухудшаются электроизоляционные и другие свойства.
Эти явления, называемые в совокупности старением, приводят к
необратимым изменениям свойств полимерных материалов и со-
кращают срок службы изделий из них.
Механизм действия стабилизаторов. В основе современных
представлений о стабилизации полимеров и механизме действия
стабилизаторов лежит теория цепных разветвленных и вырожден-
норазветвленных реакций Н. Н. Семенова, нашедшая дальнейшее
развитие в трудах Н. М. Эмануэля и Е. Т. Денисова [2—4].
При эксплуатации большинство полимерных материалов нахо-
дится в контакте с кислородом воздуха, т. е. в окислительной сре-
де. Реакции, протекающие при старении в естественных условиях,
в большинстве случаев носят характер окислительной деструкции и
представляют собой радикально-цепной окислительный процесс.
Этот процесс активируется различными внешними воздействиями—
тепловым, радиационным, химическим, механическим.
10
Характерная особенность радикально-цепных окислительных
процессов — возможность их резкого замедления путем введения
небольших количеств ингибиторов (стабилизаторов). Добавление
стабилизаторов — наиболее эффективное средство защиты поли-
меров от старения. Стабилизаторы с высокой скоростью реагируют
с пероксидными радикалами, обрывая цепной процесс окисления.
Такне соединения, как тио(диалкилпропионаты) взаимодействуют
с гидропероксидами, разрушая их без образования свободных ра-
дикалов.
Основу макромолекулы большинства полимеров общего назна-
чения составляет углеродная цепь типа:
Н Н Н Н Н Н Н Н
I I I I 1111
—С—С—С—С— или —С—С—С—С—
R R R R R R
R—Н, Aik или Аг
Механизм
мой [3—5]:
окисления углеводородов можно представить схе-
(0) RH --► R- Зарождение цепей
(1) R- +О2 ---► ROO- ]
}Продолжение цепей
(2) ROO.+RH -------> ROOH-j-R. /
(3) ROOH -*- RO. +НО-
(3. 1) ROOH + RH -> ROO-4-Н2ОR*
(3. 2) ROOH + R'CH=CH2-»- RO- + R'CH2—СН2ОН
(3. 3) 2ROOH -► ROO.+H2O+RO-
Разветвление
цепей
(4) R. +R. ----> R—R
(5) R. + ROO- ---> ROOR
(6) ROO- + ROO- ---> ROOR -| O2
Обрыв цепей
Окисление карбоцепного полимера в твердой фазе по сравнению с
жидкофазным окислением углеводородов имеет ряд важных осо-
бенностей— эстафетный механизм передачи валентности за счет
сегментальной диффузии, зависимость скорости окисления от жест-
кости полимера, повышенная стабильность алкильных макроради-
калов и др. Тем не менее эти процессы протекают по единому
механизму цепной автоиницинрованной реакции с участием алкиль-
ных и пероксидных радикалов в реакции продолжения цепи и об-
рывом цепи по бимолекулярной реакции (4).
В результате действия кислорода на углеводород или полимер
образуются активные пероксидные радикалы ROO-, которые ата-
куют полимерную цепь по реакции (2). От концентрации этих ра-
дикалов и скорости реакции (2) зависит процесс окисления в це-
лом. По мере накопления гидропероксида ROOH происходит его
11
распад с образованием свободных радикалов, способных генери-
ровать новые цепи окисления,— реакции (3) — (3.3).
Наиболее эффективные ингибиторы, в частности производные
аминов и фенолов, с высокой скоростью реагируют с пероксидны-
ми радикалами, обрывая цепной процесс окисления. Другие соеди-
нения, например тио(диалкилпропионаты), взаимодействуют с гид-
ропероксидами, разрушая их без образования свободных ради-
калов.
Реакции, протекающие в присутствии ингибитора InH (ингиби-
рованное окисление), можно представить схемой [2]:
(7) ROO.+InH —> ROOH + In-
(7.1) R- -)-InH --> RH + In-
(8) ROO- -|- In- -> Молекулярные продукты
(8.1) ROO- -}- In- -► InH + Молекулярные продукты
(8.2) R- + In- -* Rin
(9) In- + In- -—► In—In
(10) In-+RH ----»- InH + R-
(11) InH -|- ROOH --> Молекулярные продукты
(11.1) InH -|- ROOH --* Свободные радикалы
Взаимодействуя c ROO- по реакции (7), стабилизатор снижает
концентрацию пероксидных радикалов и замедляет окисление. Об-
разующийся при этом радикал In- может вступать в реакцию с
другими свободными радикалами, давая молекулярные продукты?
Вместе с тем этот радикал должен быть мало активным и не дол-
жен вступать в реакцию (10), чтобы не могли возникнуть новые
цепи окисления. Для эффективного стабилизатора отношение кон-
стант скоростей kiolkz реакций (10) и (2) должно быть очень ма-
лым, а отношение £г7//г2 большим. По реакциям (11) гидропероксид
разрушается. Эти реакции уменьшают скорость вырожденного раз-
ветвления цепей окисления и тем самым тормозят процесс.
Практическое значение для защиты от окисления имеют ингиби-
торы, обрывающие цепи при реакции с пероксидными радикалами
(фенолы, ароматические амины), и ингибиторы, разрушающие гид-
ропероксиды (азот-, серо- и фосфорсодержащие органические со-
единения). Молекула, содержащая несколько функциональных
групп, например гидроксильную группу и атом серы, может ока-
заться ингибитором смешанного типа, реагирующим и с пероксид-
ными радикалами, и- с. гидропероксидами. Чаще всего в качестве
разрушителей гидропероксидов используют монофункциональные
соединения, например органические сульфиды, не содержащие дру-
гих функциональных групп, в частности тио(дилаурилпропионат)
S (СН2СН2СООС12Н25)2, широко используемый в сочетании с фе-
нольными антиоксидантами для защиты полиолефинов (эффект
синергизма).
12
На свойства полимеров сильно влияет солнечный свет. Старение
полимеров под его действием получило название фотодеструкции.
Фотодеструкция полимеров в общем случае представляет собой
сложный многостадийный процесс. Важнейший способ повышения
стабильности полимерных материалов — введение в полимер све-
тостабйлизаторов, которые поглощают часть химически активного
света и превращают поглощенную энергию в тепло. В качестве све^
тостабилизаторов находят применение производные о-гидроксибен-
зофенона, бензтриазола, триацетонамина.
Защитное действие о-гидроксибензофенонов и о-гидроксибенз-
триазолов связано с передачей энергии при обратимом кетоеноль-
ном превращении:
При этом непосредственно. поглощенная световая энергия (или
энергия электронного возбуждения макромолекул полимера и при-
месей) превращается в форму, безопасную для полимера [6].
Свойства светостабилизатора^ проявляют о-гидроксифенил-
сульфоксиды, способные трансформировать световую энергию за
счет обратимого тион-тиольного превращения [7]:
В некоторых случаях фотостабилизация может осуществляться
за счет фотохимической реакции в полимерной матрице. Например,
эфиры салициловой или бензойной кислоты, введенные в полимер,
под действием света превращаются в. гидроксифенилкетоны:
В действительности механизм действия эфиров, по-видимому, бо-
лее сложен, так как исходный эфир в некоторых случаях был бо-
лее эффективен, чем гидроксибензофенон [8].
Изучение механизма действия нового класса светостабилизато-
ров — производных триацетонамина — показало, .что сам амин не
13
является светостабилизатором. Его стабилизирующее действие свя-
зано с продуктами окисления — нитроксильными радикалами [9,
10]. Механизм светозащитного действия нитроксильных радика-
лов пока еще мало изучен. Нитроксильные радикалы реагируют с
алкильными радикалами, а с пероксидными не взаимодействуют.
Преимущество нитроксильных радикалов состоит в том, что они
вступают с активными радикалами в реакцию присоединения
R- +ON\-----* RON^
а фенолы и амины, как отмечалось выше,— в реакцию замещения
ROO- + InH —> ROOH + In-
приводящую к образованию гидропероксида — потенциального ис-
точника радикалов. В реакции присоединения гидропероксид не
образуется. В условиях фотоокисления нитроксильные радикалы
также обрывают цепь по реакции с алкильными радикалами. В то
же время электронновозбужденные нитроксильные радикалы спо-
собны инициировать окисление полимеров:
/NO + RH ---> /NOH + R-
При фотоокислении полимеров нитроксильные радикалы могут га-
сить возбужденные состояния и поглощать энергию светового из-
лучения. Вопросам фотодеструкции и защиты полимеров от нее
посвящена монография В. Я. Шляпинтоха [11].
Одна из основных причин сокращения сроков службы резино-
вых изделий — процессы озонного старения. Во всем мире ведутся*
интенсивные поиски способов защиты резин от действия озона. Ос-
новной способ защиты — введение небольших количеств веществ,
способных замедлять растрескивание резин и получивших назва-
ние химических антиозонантов (в отличие от физических антиозо-
нантов — восков, создающих на поверхности резины барьер, пре-
пятствующий доступу озона). Механизм действия химических анти-
озонантов весьма сложен. Для объяснения процесса озонного
старения был предложен ряд гипотез, согласно которым антиозо-
нанты катализируют разрушение озона или вместе с продуктами
озонолиза образуют на поверхности резины пленку, защищающую
двойные связи полимера от атаки озоном, соединяют разорванные
концы макромолекул, препятствуя возникновению и разрастанию
трещин, или реагируют с озоном, выступая конкурентом в реакции
озона с полимером.
Важнейшую группу антиозонантов составляют производные
n-фенилендиамина. Взаимодействие антиозонантов с озоном про-
текает очень быстро, а между скоростью реакции и эффективно-
стью антиозонанта существует определенная зависимость [12]'.
В табл. 1 сравниваются эффективность защитного действия не-
которых соединений (характеризуемого временем до появления
трещин при экспозиции резины в озонной камере) и константа ско-
14
Таблица 1. Эффективность защитного действия антиозонантов
Соединение
Формула
Время до
появления
трещин,
мин
Л-1Ов,
л/(моль-с).
N.N'-Ди-н-октил-
л-фениленди-
амии
М,М'-Диизопентил-
п-фениленди-
амин
N-Изопропил-М'-
фенил-п-фени-
лендиамин
М,ЬГ-Ди-а-метил-
бензил-л-феии-
лендиамин
Ы-а-Метилбензил-
анизидии
Метилолеат
С5Нц—HN
840
870
500
250
7
8
7
5
80 1
(без ан-
тиозонан-
та)
роста реакции соединения с озоном. Видно, что константы скоро-
сти взаимодействия озона с большинством представленных соеди-
нений значительно выше, чем с двойной связью углеродной цепи.
В системах полимер — антиозонант вначале с озоном реагирует ан-
тиозонант, а макромолекулы полимера остаются практически не-
измененными, и только после израсходования большей части анти-
озонанта начинается деструкция полимера [12].
Реальный процесс озонного растрескивания резин осложняется
действием солнечной радиации и кислорода, протекает очень
сложно, и механизм его нельзя считать точно установленным.
При деформации резин под действием механических нагрузок
(«утомление») могут происходить разрывы полимерных цепей с
образованием макрорадикалов быстро окисляющихся до
ROO-. Обычные ингибиторы окислительных процессов .проявляют
при взаимодействии с ROO* такую же активность, как при тер-
мическом окислении полимеров. Обычно не выделяют особый класс
противоутомителей, так как их действие не обусловлено какой-ли-
бо спецификой [13].
Структура и эффективность стабилизаторов. В конце 1940-х—
начале 1950-х годов появилось довольно много работ, посвящен-
ных поиску корреляций между химической структурой стабилиза-
15
тора и эффективностью ингибирования окислительных процессов.
Эти работы обобщены в ряде обзоров и монографий [14—20].
Признано, что структура молекулы антиоксиданта должна прежде
всего обеспечивать его высокую реакционную способность при вза-
имодействии с пероксидными радикалами ROO* и высокую ста-
бильность образующихся радикалов In* (CeHgO* в случае фено-
лов, СеН5М* в случае аминов). Такими свойствами обладают про-
изводные фенолов и ароматических аминов с «подвижным» атомом
водорода НО- или HN-группы.
Большой экспериментальный материал позволил сформулиро-
вать [14] некоторые общие закономерности, согласно которым эф-
фективность фенольных антиоксидантов повышается при введении
в молекулу фенола таких заместителей, которые в силу своих до-
норно-акцепторных свойств или создаваемых ими стерических пре-
пятствий уменьшают полярность связи О—Н, снижают способ-
ность к образованию водородной связи и потенциал окисления (до
0,6—0,8 В) или повышают стабильность феноксильного радикала,
образующегося при окислении фенола. Ингибирующая активность
аминов зависит от эффекта а—л-сопряжения в молекуле. Так, в
ряду дифениламин — фенил-0-нафтиламин — ди-р-нафтил-п-фени-
лендиамин она монотонно увеличивается, что проявляется в удли-
нении индукционного периода. Относительная подвижность водо-
рода аминогруппы в указанном ряду увеличивается, и соответст-
венно уменьшается активность образующего радикала In*. Как и
в ряду фенолов, на ингибирующую активность ароматических ами-
нов оказывают влияние заместители, вводимые в /шрп-положен^е
к аминогруппе [13].
Приведенными данными далеко не исчерпываются факторы,
влияющие в реальных условиях на эффективность стабилизаторов,
которая зависит от летучести стабилизатора, совместимости его
с полимером, способа введения, стабильности самого стабилизато-
ра и, наконец, от свойств полимера и компонентов полимерной си-
стемы [20]. Пока не существует точных экспериментальных дан-
ных о зависимости эффективности стабилизатора от комплекса пе-
речисленных выше показателей для каждого реального случая или
общих закономерностей в целом. Поэтому эмпирический подбор
стабилизаторов до настоящего времени играет решающую роль.
Так, из каждых 1000 синтезированных соединений только 5 по эф-
фективности оказываются конкурентоспособными с серийными. По
технико-экономическим соображениям из этих пяти обычно исклю-
чаются еще 3—4, и только 1—2 находят практическое применение.
Поэтому весьма актуальным остается создание теоретических ос-
нов выбора структуры эффективного стабилизатора и исключение
эмпирического подхода к этой проблеме.
В 1969 г. было экспериментально доказано [21], что ингибиру-
ющая способность различных соединений по отношению к ради-
кальной полимеризации винилацетата может служить критерием
оценки их антиокислительной активности. Оказалось, что порядок
16
изменения ингибирующей способности по отношению к радикаль-
ной полимеризации совпадает с порядком изменения этой величины
по отношению к окислению высокомолекулярных углеводородов.
При исследовании [21] взаимосвязи между антиокислительной ак-
тивностью антиоксиданта и его реакционной способностью, рассчи-
танной квантовохимическим методом МО ЛКАО с помощью ЭВМ,
оказалось, что соединения, обладающие высоким индексом реак-
ционной способности в радикальных реакциях, характеризуются
высокой ингибирующей активностью в процессах окисления или
полимеризации.
Для «утверждения» предложенного метода автор рассчитал
для 22 новых антиоксидантов индексы реакционной способности в
радикальных реакциях и энергетические уровни высших занятых
орбиталей. Из этих 22 синтезировано 7 соединений, характеризую-
щихся относительно высокими индексами и энергетическими уров-
нями, близкими к соответствующим значениям для винилацетата.
Оказалось, что синтезированные соединения обладали антиокисли-
тельной активностью того же порядка или выше по сравнению с
известными антиоксидантами. Показано, что по реакционной спо-
собности между растущими радикалами мономеров и ингибитора-
ми полимеризации имеются значительные различия. Так, если для
винилацетата эффективность ингибиторов возрастает в ряду: мо-
нофенолы < бисфенолы < моноамины < n-фенилендиамины, то
в случае стирола эффективность тех же ингибиторов возрастает в
другом порядке: п-фенилендиамины < моноамины < монофено-
лы < бисфенолы, т. е. обнаруживается тенденция, обратная на-
блюдаемой в случае винилацетата. В то же время для метилмет-
акрилата, например, не обнаружено ни одной из приведенных за-
висимостей.
Для объяснения этих фактов было предположено, что в про-
цессе дезактивирования растущего радикала в результате его реак-
ции с антиоксидантом обе реагирующие частицы сближаются, и
возникает переходное состояние
ROO + он
-->ROOM +
2—1057
17
Рис. 1- Относительное расположение высших занятых
энергетических уровней различных соединений:
1 — мономеры; 2 и 3 — антиоксиданты (2 — амины, 3 — фе-
нолы) [21].
энергию стабилизации которого (А£) мож-
но рассчитать квантовохимическим мето-
дом. При сопоставлении величин А£ и эф-
фективности ингибирования полимеризации
была получена прямолинейная зависи-
мость, причем значения А£ увеличиваются
в ряду: п-фенилендиамины < моноами-
ны < фенолы.
Таким_ образом было установлено, что
антиокислительная активность исследован-
ных соединений возрастает с увеличением
стабильности переходного состояния, образованного этим соедине-
нием и соответствующим растущим радикалом. Для достижения
значительной энергии резонансной стабилизации необходимо, что-
бы мономер и антиоксидант имели близкие значения энергии выс-
шего занятого уровня.
На рис. 1 условно изображены значения высших занятых энер-
гетических уровней аминов и фенолов для винилацетата (ВАЦ),
стирола (СТ) и метилметакрилата (ММА). Как следует из приве-
денных данных, по отношению к полимеризации винилацетата эф-
фективны многие соединения аминного типа, тогда как по отно-
шению к полимеризации стирола более эффективны фенолы, a не
амины; для метилметакрилата мало эффективны и амины .и фено-
лы, что хорошо согласуется с экспериментальными данными.
На основании этого был сделан вывод, что изменение типа оп-
тимального антиоксиданта в зависимости от вида полимера зако-
номерно, а существование антиоксиданта, одинаково эффективного
для всех полимеров, теоретически невозможно (по крайней мере,
для антиоксидантов — акцепторов свободных радикалов).
Такйм образом, на основании расчета высших занятых энерге-
тических уровней полимеров, казалось бы, можно выбирать опти-
мальные антиоксиданты не только для существующих полимеров,
но и для тех, которые будут получены в будущем. Однако на прак-
тике все обстоит гораздо сложнее. Дело в том, что теоретический
метод расчета, предложенный в работе [21], очень громоздок, труд-
но проверяется экспериментально, плохо сопоставим и должен учи-
тывать предпочтительные конформации молекул антиоксиданта.
Ошибка в таких расчетах очень велика, а в некоторых случаях она
не позволяет сделать однозначного вывода о характере и поведе-
нии антиоксиданта.
Вопросы тестирования и прогнозирования эффективности ста-
билизаторов исследованы в работах советских ученых [22, 23].
Некоторые из разработанных ими методов уже применяются для
предварительной оценки эффективности новых стабилизаторов.
18
] 2. КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
СТАБИЛИЗАТОРОВ
Классификация стабилизаторов. Стабилизаторы находят при-
менение в самых разных областях народного хозяйства. Сведения
об их ассортименте, свойствах, возможных областях использова-
ния необходимы специалистам практически всех отраслей промыш-
ленности, занимающихся получением и переработкой полимерных
материалов. Поэтому сегодня, когда на мировом рынке представ-
лены сотни разных марок стабилизаторов, остро ощущается необ-
ходимость в классификации, которая позволила бы лучше ориен-
тироваться при выборе областей их применения. Известный в на-
стоящее время ассортимент стабилизаторов наиболее удобно
классифицировать по химическим признакам (химическая класси-
фикация) и по техническим свойствам и областям применения
(техническая классификация).
В основу химической классификации положен тип функциональ-
ных групп, играющих основную роль при ингибировании процессов
деструкции полимеров. Химическая классификация удобна для спе-
циалистов, работающих в области синтеза, изучения свойств, меха-
низма действия и эффективности стабилизаторов, а также при
изучении химии и технологии стабилизаторов.
По химическому строению стабилизаторы можно разделить на
7 основных классов.
1. Производные ароматических аминов характе-
ризуются наличием в молекуле атома азота, связанного с арома-
тическим ядром. Это — N-фенил-р-нафтиламин, N-фенил-а-нафтил-
амин, 4-гидроксифенил-р-нафтиламин, М,|М'-дифенил-п-фениленди-
амин, М,ЬГ-ди-р-нафтил-п-фенилендиамин, 4,4'-ди (1-метилбензил) -
дифениламин, 4,4/-ди-трет-октилдифениламин, 4,4'-ДИ (1,1 -диметил-
бензил) дифениламин, М-изопропил-П'-фенил-, М-циклогексил-N'-
фенил-, Й-(1,3-диметилбутил)-М'-фенил)-, N.N'-диалкил-п-фени-
лендиамины.
2. Гетероциклические азотсодержащие соеди-
нения характеризуются наличием гидрохинолинового, бензтри-
азольного или бензимидазольного циклов, связанных с ароматичес-
ким ядром. Это — 2,2,4-триметил-1,2-дигидрохинолин, 2,2,4-триме-
тил-б-этокси-1,2-дигид рохинолин, 2-меркаптобензимидазол, 2- (2'-
гидрокси-5'-метилфенил) бензтриазол.
3. Производные тиокарбамида и дитиокарбами-
новой кислоты характеризуются наличием группировки
—C(=S)N^ Это — дифенил-, трибутилтиокарбамид, дибутилди-
тиокарбаматы цинка и никеля, диметилдитиокарбамат висмута.
4. Производные фенолов характеризуются наличием в
молекуле одной или нескольких пространственно затрудненных
гидроксильных групп. Это — одноядерные алкилированные фенолы
(2,6-ди-трет-бутил-4-метилфенол), замещенные бисфенолы [2,2'-ме-
2‘ 19
тиленбис(6-трет-бутил-4-метилфенол), 2,2'-метиленбис(4-метил-6-
а-метилциклогексилфенол), 4,4'-метиленбис(6-т’рет’-бутил-2-метил-
фенол), 2,2'-тиобис(6-трет-бутил-4-метилфенол), 4,4'-тиобис(6-трет-
бутил-3-метилфенол) ], многоядерные фенолы [1,1,3-три(5'-трет-бу-
тил-4/-гидрокси-2'-метилфенил) бутан, 2,4,6-три (4-гидрокси-3,5-ди-
трет-бутилбензил) мезитилен], о-карбонилзамещенные фенолы (2-
гидрокси-4-метоксибензофенон, 2-гидрокси-4-октоксибензофенон).
5. Фосфорсодержащие соединения. Важнейшие из
них — фосфиты, которые характеризуются наличием атома фосфо-
ра, связанного с группировкой —О—Аг (или Aik): трифенил-, три-
октил- и тринонилфенилфосфит, смесь а-мегилбензилфенилфосфи-
тов.
6. Тио (д и а л ки л проп иона т ы) характеризуются наличием
атома серы, связанного с группировкой —СН2—СН2—COOR.
7. Металлсодержащие соединения — различные соли
органических и неорганических кислот, оловоорганические соедине-
ния.
При классификации по областям применения (техническая
классификация) по характеру действия можно разделить стабили-
заторы на 6 групп. Деление это условно потому, что некоторые
соединения могут защищать полимер от нескольких видов старе-
ния.
1. Термостабилизаторы, или антиоксиданты, за-
щищают полимер от термической и термоокислительной деструк-
ции. Сюда относятся производные нафтиламина, дифениламина,
диарил- и алкиларил-п-фенилендиамина, дигидрохинолина; бенз-
имидазол, дифенилтиокарбамид, дитиокарбаматы, тио (диалкилпро-
пионаты), производные фенолов, фосфиты, оловоорганические со-
единения и некоторые другие.
2. Антиозонанты защищают резины от озонного и свето-
озонного старения. К этому типу стабилизаторов относятся диал-
кил- и алкиларил-п-фенилендиамины, 6-алкоксизамещенные дигид-
рохинолины, трибутилтиокарбамид, никелевые соли диалкилдитио-
карбаминовой кислоты и некоторые производные фенолов.
3. Светостабилизаторы защищают полимеры от дейст-
вия УФ-лучей и в целом от влияния естественных погодных усло-
вий. Сюда относятся производные гидроксибензофенона, гидрокси-
бензтриазола, серосодержащие оловоорганические соединения, со-
единения никеля, производные триацетонамина.
4. Противоутомители защищают полимерные материалы
(главным образом резины) от растрескивания при действии пере-
менных нагрузок. К таким стабилизаторам можно отнести произ-
водные дифениламина, дигидрохинолина и л-фенилендиамина.
5. Пассиваторы поливалентных металлов защищают поли-
меры от разрушающего действия металлических «ядов», главным
образом от примесей продуктов разложения катализаторов, ис-
пользуемых при полимеризации. Дезактивировать действие метал-
лов переменной валентности могут практически все производные
20
ароматических аминов; особенно эффективны в этом отношении
ароматические диамины с развитой системой конъюгированных
двойных связей, например дифенил- и динафтил-п-фенилендиамин.
6. Антирады защищают полимерные материалы от разру-
шающего действия у-излучения. Такими свойствами в большей сте-
пени обладают ароматические углеводороды с конденсированны-
ми ядрами, а в меньшей степени — фенолы и ароматические амины.
Наряду с химической и технической классификацией в прак-
тике применения стабилизаторов сложилось их деление на группы
по некоторым специфическим признакам.
1. Окрашивающие и неокрашивающие стабили-
заторы. При выборе стабилизатора для зашиты конкретного
полимера важно учитывать влияние добавки на цвет полимера в про- -
цессе изготовления и эксплуатации изделий. Стабилизаторы, окра-
шивающие полимерный материал, используют только для произ-
водства темных изделий и материалов, а неокрашивающие стаби-
лизаторы применяют для изготовления белых и светло-окрашенных
изделий.
Ароматические амины и их производные относятся к группе ок-
рашивающих стабилизаторов; производные гетероциклических ами-
нов в меньшей степени окрашивают полимер (дигидрохинолины),
а с некоторыми из них можно получить даже светлые изделия
(2-меркаптобензимидазол, 2-гидроксибензтриазолы). Производные
тиокарбамида, диалкилдитиокарбаматы, тио(диалкилпропионаты)
практически не окрашивают полимер (например, трибутилтиокарб-
амид является пока единственным неокрашивающим антиозонан-
том).
Фенольные стабилизаторы выгодно отличаются от аминных тем,
что большинство из них практически не окрашивает полимерные
материалы как при введении в них, так и после действия на них
УФ-излучения. Незначительное изменение окраски полимера, со-
держащего фенольные стабилизаторы, связано с тем, что под дей-
ствием высоких температур или УФ-лучей некоторые фенольные
стабилизаторы могут превращаться в окрашенные соединения. Од-
нако причины неодинакового влияния близких по структуре фено-
лов на окрашивание полимеров до сих пор не выяснены. В полиме-
рах, загрязненных следами металлов переменной валентности, в
присутствии производных фенолов может возникнуть желтая, серая
или розоватая окраска. То же справедливо и для гидроксиаромати-
ческих кетонов, которые можно рассматривать как производные
фенолов.
В результате анализа многочисленных данных можно сделать
отдельные практические выводы. Одноядерные алкилфенолы (2,6-
ди-трет-бутил-4-метилфенол) обычно не окрашивают полимер, пе-
реход от 2,2'- к 4,4'-бисфенолам при одних и тех же заместителях
в орто-положении к гидроксилу и одинаковой структуре мостика
приводит к резкому повышению влияния стабилизатора на окраску
резин. Способность 4,4'-бисфенолов к окрашиванию усиливается
21
при наличии заместителей типа трет-бутила в орто-положении к
гидроксильной группе. Известны промышленные фенольные ста-
билизаторы, например 4,4'-метиленбис(2,6-ди-трет-бутилфенол),
которые нельзя использовать для стабилизации светлых каучуков
и защиты белых и цветных резин от старения в связи с высокой
окрашивающей способностью. Отсутствие теоретических представ-
лений о механизме превращения стабилизаторов в окрашенные со-
единения не позволяет с достаточной достоверностью прогнозиро-
вать окрашивающие свойства и эффективность новых соединений.
Фосфорсодержащие стабилизаторы типа фосфитов сами по се-
бе не окрашивают полимер, но продукты их гидролиза — замещен-
ные фенолы — могут в определенных условиях, как сказано выше,
изменять окраску полимера. Поэтому фосфиты нужно применять
только в условиях, исключающих их гидролиз.
Среди оловоорганических соединений только серосодержащие
могут вызывать легкое пожелтение поливинилхлоридных компози-
ций. Соли карбоновых и неорганических кислот (бариевые, кадми-
евые, цинковые) обычно позволяют получать белые и светло-окра-
шенные изделия из поливинилхлорида.
В некоторых случаях возникновение окраски может быть вызва-
но взаимодействием стабилизатора с другими компонентами поли-
мерной композиции.
2. Токсичные и нетоксичные стабилизаторы. Рас-
ширение областей применения полимеров, использование полимер-
ных изделий в медицинской и пищевой промышленности, для из-
готовления игрушек и т. п. требует применения нетоксичных стаби-
лизаторов. Токсичные стабилизаторы можно использовать только
для полимеров технического назначения. Нетоксичные стабилиза-
торы применяют в композициях, непосредственно соприкасающих-
ся с пищевыми продуктами или с организмом человека.
Ароматические амины относятся к токсичным соединениям, ге-
тероциклические соединения аминного типа обычно мало токсич-
ны, а некоторые производные 2-гидроксибензтриазола разрешены
для полимеров, контактирующих с пищевыми продуктами. Произ-
водные тиокарбамида и дитиокарбаматы характеризуются средней
токсичностью, а производные тиодипропионатов являются практи-
чески нетоксичными стабилизаторами.
Производные фенола характеризуются низкой токсичностью,
поэтому многие из них в СССР и за рубежом разрешены органи-
зациями здравоохранения для контакта с пищевыми продуктами, а
2,4,6-три (4-гидрокси-3,5-ди-трет-бутилбензил) мезитилен разрешен
для защиты детских сосок. Фосфиты относятся к малотоксичным
веществам. Оловоорганические стабилизаторы в ряде капитали-
стических стран разрешены для упаковочных пленок. Оловоорга-
нические соединения, содержащие серу, характеризуются более
высокой токсичностью. Токсичность солей органических и неорга-
нических кислот зависит главным образом от природы катиона.
Все свинцовые соли характеризуются выраженной токсичностью;
22
стеараты кальция и цинка разрешены для контакта с пищевыми
продуктами.
В целом санитарно-гигиенические требования значительно ог-
раничивают выбор стабилизатора, пригодного для контакта с орга-
низмом человека.
3. Летучие и нелетучие стабилизаторы. Интенсифи-
кация режимов переработки полимеров, ужесточение условий экс-
плуатации и повышение требований к стабильности свойств изде-
лий из полимерных материалов вызвали необходимость при выборе
стабилизатора учитывать его летучесть.
Взаимосвязь летучести и структуры стабилизаторов рассмотре-
на в работе [24]. Отмечено, в частности, что способность к обра-
зованию водородных связей, обеспечивающая межмолекулярное
взаимодействие, в известной степени определяет летучесть стаби-
лизаторов. Имеющиеся в молекуле фенола или амина заместители,
экранирующие НО- или HN-группу, создают препятствия к обра-
зованию водородных связей. В качестве примера приводятся два
изомера и данные о скорости их миграции (скорость улетучивания)
из полимера:
Высокая летучесть 2,6-ди-т’рет-бутил-4-метилфенола — одного из
известных и эффективных стабилизаторов — ограничивает сферу
его применения, особенно тогда, когда возможно действие высоких
температур (при переработке полимера) или действие умеренных
температур, но в течение длительного времени (сушка полимера).
Для процессов высокотемпературной переработки полимеров ис-
пользуют стабилизаторы группы трис- или тетрафенолов, обладаю-
щие низкой летучестью.
Номенклатура стабилизаторов. Химические названия стабили-
заторов в большинстве случаев сложны, громоздки и неудобны для
практического использования. Кроме того, одно и то же соединение
часто имеет несколько названий, что серьезно затрудняет инфор-
мационный поиск и выбор стабилизатора при разработке новых
полимерных материалов.
Это положение усугубляется конкуренцией между фирмами,
производящими стабилизаторы, когда для сохранения в тайне ис-
тинного состава и структуры стабилизатора одному и тому же со-
единению приписываются разные торговые наименования. Приме-
ром могут служить такие широко распространенные стабилизато-
ры, как Ы-фенил-2-нафтиламин и 2,6-ди-т,рет-бутил-4-метилфенол:
23
для каждого известно более 25 названий. Продолжающееся рас-
ширение ассортимента и стремление давать выпускаемым стаби-
лизаторам оригинальные наименования приводят к появлению все
новых и новых названий, перевод которых на русский язык ста-
новится весьма затруднительным. В названия включаются самые
разные сокращения — и указывающие фирму-изготовитель и да-
ющие представления о назначении стабилизатора. Пример — ста-
билизатор Santoflex AW (2,2,4-триметил-6-этокси-1,2-дигидрохино-
лин) фирмы Monsanto. В это название входит наименование фирмы
и указание на назначение стабилизатора — антифлексинг (т. е.
противоутомитель). Иностранные фирмы, поставляя продукты, со-
храняют на них маркировку со своими названиями или трансфор-
мируют их па язык страны-экспортера. На отечественных заводах-
потребителях эти обозначения входят в техническую документацию
большей частью без изменения, иногда своеобразно «русифициру-
ясь», а затем переходят на страницы специальных журналов, где
сегодня можно встретить такие названия, как Эджерайт уайт, Но-
нокс, Сантонокс. В литературе один и тот же стабилизатор встре-
чается под разными названиями, что затрудняет ориентировку.
Отечественные названия стабилизаторов пока тоже не имеют
единой системы. Наиболее распространенной в СССР является но-
менклатура, разработанная в НИИХИМПОЛИМЕР в 1966 г., ког-
да в справочнике «Вспомогательные вещества для полимерных ма-
териалов» [25] отечественные названия стабилизаторов впервые
были приведены в систему. В 1973 г. круг этих названий был зна-
чительно расширен [26].
В основу использованной в данной книге номенклатуры поло-
жено химическое название, общепринятое в органической химии.
Название стабилизатора складывается из сокращенных наимено-
ваний фрагмента, составляющего основу молекулы. Заместитель
обозначают начальной буквой с цифровым указанием его положе-
ния. Ниже приведены названия основных фрагментов, входящих
в состав широко применяемых в настоящее время стабилизаторов:
Основной фрагмент стабилизатора Название по пред- лагаемой номен- клатуре Основной фрагмент стабилизатора Название по пред- лагаемой номен- клатуре
Дифениламин Дифенам Бензофенон Бензон
Фенилендиамии Диафен Имидазол Идазол
Нафтиламин Нафтам Бензтриазол Беназол
Алкил фенол Алкофен Хинолин Хинол
Если в состав молекулы входят два или более радикала, то, как
принято в органической химии, перед названием добавляется соот-
ветствующая приставка, например бисалкофены, трисалкофены.
Названия основных классов стабилизаторов по предлагаемой но-
менклатуре будут формулироваться так.
Производные дифениламина — дифенамы: гидроксидифе-
ниламин— Дифенам О, диметоксидифениламин —Дифенам
МО, бис(1,1'-диметилбензил) дифениламин — Дифенам МБ.
24
Производные n-фенилендиамина — диафены: дифенил-н-
фенилендиамин— Диафен ФФ, динафтил-и-фениленди-
амин — Диафен НН, изопропилфенил-п-фенилендиамин—
Диафен ФП и т. д.
Производные нафтиламина — нафтамы: Ы-фенил-2-наф-
тиламин —Нафтам Ф2, Н-фенил-1-нафтиламин— Нафтам
Ф1, Ы-(4-гидроксифенил)-2-нафтиламин— Нафтам ОФ.
Одноядерные алкилированные фенолы — алкофены: 2,6-
ди-трет-бутил-4-метилфенол — Алкофен БП, 2,4,6-три-трет-
бутилфенол — Алкофен Б и т. д.
Алкиленбисфенолы — бисалкофены: 2,2'-метилен-бнс (6-
трет-бутил-4-метилфенол) — Бисалкофен БП, 4,4'-метилен-
бис(2,6-трет-бутилфенол) — Бисалкофен Б и т. д.
Трехъядерные фенолы — трисалкофены: 2,4,6-три (4'-гидр-
окси-З'.б'-ди-трег-бутилбензил) мезитилен—Трисалкофен БД,
1,1,3-(4'-гидрокси-5'-трет-бутил-2'-метилфенил) бутан — Трис-
алкофен БМБ и т. д.
Замещенные дигидроксидифенилсульфидов — тиоалкофе-
ны: 2,2/-тиобис(6-трет-бутил-4-метилфенол) —Тиоалкофен
БП, 4,4/-тиобис(6-грет-бутил-3-метилфенол) —Тиоалкофен
БМ и т. д.
Производные бензофенона — бензоны: • 2-гидрокси-4-мет-
оксибензофенон — Бензон ОМ, 2-гидрокси-4-октоксибензо-
фенон — Бензон 00 и т. д.
Производные хинолина — хинолы: 2,2,4-триметил-1,2-ди-
гидрохинолин — Хинол Д, 2,2,4-триметил-6-этокси-1,2-дигид-
рохинолин —Хинол ЭД.
Производные бензтриазола — беназолы: 2-(2'-гидрокси-
б'-метилфенил)бензтриазол — Беназол П, 5-хлор-2-(3/-трет-
бутил-2/-гидрокси-5/-метилфенил)бензтриазол — Беназол
ПБХ и т. д.
Производные бензимидазола — идазолы: 2-меркаптобенз-
имидазол — Идазол МБ, его цинковая соль — Идазол МБЦ.
Данная номенклатура, как и всякая другая, не лишена недо-
статков. Так, в отличие от отечественной номенклатуры красителей,
в предложенных терминах нет указания на область применения
стабилизатора, но, имея представление о принадлежности стаби-
лизатора к тому или иному химическому классу, можно определить
и его основные области применения. Названия не всех стабили-
заторов строго укладываются в предлагаемую схему. Од-
нако для большинства соединений эта номенклатура достаточно
четко отражает их химическое строение, названия кратки и удоб-
ны для повседневного применения и при маркировке готовой про-
дукции, однозначность позволяет легко находить их при информа-
ционном поиске. Предложенная номенклатура положена в основу
разрабатываемого общесоюзного стандарта названий добавок для
полимерных материалов.
25
1.3. ТЕНДЕНЦИИ РАЗВИТИЯ ПРОИЗВОДСТВА
СТАБИЛИЗАТОРОВ
Развитие производства стабилизаторов тесно связано с разви-
тием производства пластических масс, синтетических каучуков, ре-
зин и других полимерных материалов. В настоящее время сложил-
ся ассортимент стабилизаторов для защиты промышленных поли-
меров от основных видов старения, однако этот ассортимент не
полностью удовлетворяет возрастающим требованиям техники.
Необходимость создания новых стабилизаторов обусловлена
не только потребностями промышленности, связанными с интенси-
фикацией процессов переработки полимеров и повышением эксплу-
атационных требований! к материалам и изделиям из них, но и
ужесточением санитарно-гигиенических требований, изменением
сырьевой базы синтеза стабилизаторов или изменением рыночной
конъюнктуры.
Создание изопреновых каучуков, близких по свойствам к нату-
ральному, потребовало разработки эффективных неокрашивающих
стабилизаторов. Для полипропилена при многотоннажных поточ-
ных линиях его переработки в виде гранул (температура гранули-
рования 220—230 °C) понадобились эффективные при высоких
температурах стабилизаторы, не летучие и не влияющие на цвет
исходного полимера.
В связи с санитарно-гигиеническими требованиями идет посто-
янное обновление ассортимента аминных стабилизаторов. Еще 15—
20 лет назад N-фенил-М'-цнклогексил-п-фенилендиамин был заме-
нен на менее токсичный N-изопропил-М'-фенил-л-фенилендиамин, а
в настоящее время в ряде стран последний постепенно заменяется
на еще менее токсичные Ы-(1,3-диметилбутил)-Ы/-фенил-п-фенилен-
диамин и Н,Ы/-ди(1,3-диметилбутил)-н-фенилендиамин. Тенденция
создания и развития производства нетоксичных стабилизаторов со-
хранится и на будущее. Вместе с тем необходимо с осторожностью
относиться к появляющимся время от времени в литературе дан-
ным о вновь открытых токсических свойствах уже известных ста-
билизаторов. Такие сведения требуют тщательной проверки, по-
скольку они могут быть связаны с конъюнктурой рынка и конку-
ренцией разных фирм.
Основную часть себестоимости стабилизаторов составляет стои-
мость сырья, поэтому изыскание более дешевого сырья — важное
направление развития производства стабилизаторов. В производст-
ве фенольных стабилизаторов вместо дорогого и дефицитного 4-ме-
тилфенола все шире используется и будет использоваться фенол.
Разработаны способы получения на основе фенола тех стабилиза-
торов, которые раньше выпускали только на основе 4-метилфено-
ла,— 2,6-ди-трет-бутил-4-метилфенола, 2,2'-метиленбис (6-трет-бу-
тил-4-метилфенола). В производстве аминных стабилизаторов ани-
лин заменяет более дорогие ароматические аминосоединения. На-
пример, наблюдаются снижение производства 2,2,4-триметил-6-это-
26
кси-1 2-дигидрохинолина, получаемого из н-фенетидина, и расши-
рение производства несколько менее эффективного, но более деше-
вого 2,2,4-триметил-1,2-дигидрохинолина, получаемого из анилина.
Растет выпуск дешевых смесевых фенольных стабилизаторов —
продуктов алкилирования технических смесей фенола, крезолов и
ксиленолов смесями олефинов. Заметно возрастает производство
одного из самых дешевых фенольных стабилизаторов — смеси
а-метилбензилфенолов. Стабилизаторами этого типа частично за-
меняют более эффективные, но более дорогие метиленбисфенолы.
Например, для удешевления композиции вводят не 2, а 1,5 масс. ч.
дорогостоящего метиленбисфенола и добавляют 0,5 масс. ч. смеси
а-метилбензилфенолов. Таким же образом в полимерной компози-
ции часть 2-меркаптобензимидазола заменяют метилмеркаптобенз-
имидазолом, получаемым из более дешевого сырья — метил-о-фени-
лендиамина.
Рост потребности в стабилизаторах потребовал нового подхода
к организации их производства. Если раньше оно было связано
только с анилинокрасочной промышленностью, то новые производ-
ства развиваются и в нефтехимической промышленности (за ру-
бежом— фирмы Ethyl, Shell, Enny, UOP). Использование нефтехи-
мического сырья в синтезе стабилизаторов будет непрерывно воз-
растать. Наблюдается тенденция создания комплексов производств
широкого ассортимента стабилизаторов на базе единого сырья и
промежуточных продуктов. Важнейшими промежуточными продук-
тами в производстве стабилизаторов являются 2,6-ди-трет-бутилфе-
нол, 2-трет-бутил-4-метилфенол, N-нитрозодифениламин, 2,4-ди-
гидроксибензофенон. Комплексные производства большой мощно-
сти позволят существенно снизить себестоимость стабилизаторов.
В 1963—1976 годах ежегодный прирост объема производства
стабилизаторов в США составлял 8,1% при увеличении потребле-
ния пластических масс на 9,1%; на период до 1990 г. планируется
ежегодный прирост их производства на 9,1%. Высокими темпами
растет потребление стабилизаторов и других химикатов для каучу-
ков и резин. За 1970—1974 годы оно возросло в США на 39% (хо-
тя расход каучука увеличился только на 20%). По прогнозам спе-
циалистов, потребление антиоксидантов будет ежегодно увеличи-
ваться на 7—8%, а потребление антиозонантов на 4—5% [27]. Из
производных п-фенилендиамина наибольшее развитие получают
алкиларил-н-фенилендиамины с разветвленным алкилом, содержа-
щим 7 и более атомов углерода (Santoflex 13). Применение
2,2,4-триметил-6-этокси-1,2-дигидрохинолина уменьшается, зато его
более дешевый аналог — Ацетонанил — в последнее время получил
заметное развитие; наблюдается тенденция к снижению потреб-
ления продукта конденсации ацетона с дифениламином. Производ-
ные гидрохинона и тиобисфенолы несколько утратили свое значе-
ние для резиновой промышленности. 2-Меркаптобензимидазол и
его производные (синергисты с другими стабилизаторами) еще не
получили всеобщего признания, хотя эффективны и как пассива-
27
торы. Из неокрашивающих антиозонантов выпускают трибутилтио-
карбамид (фирма Monsanto) и ненасыщенные простые эфиры
(фирма Bayer), однако они малоэффективны, и ведутся активные
поиски неокрашивающего соединения, аналогичного по эффектив-
ности алкиларил-п-фенилендиаминам. В промышленности США в
значительных количествах применяют фенольные стабилизаторы
резин; возрастает потребление три (n-нонилфенил) фосфита [28].
Стабилизаторы полимерных материалов выпускают почти во
всех промышленно развитых странах. В 1977 г. в США их произ-
водили на 59 специализированных заводах, а еще на 40 установках
выпускали отдельные партии стабилизаторов. В 1976—1977 годах
были организованы новые производства (фирма Ciba — Geigy —
производные фенола; фирма Cyanamid — тиоэфиры); ожидается
расширение производства фенольных стабилизаторов фирмами Et-
hyl и Goodrich. Фирма Exxon разработала для полиолефинов и бу-
дет выпускать новые фенольные стабилизаторы — поликарбонаты с
низкой молекулярной массой на основе бисфенолов. Фирма Bell.
Labor, организует производство стабилизаторов для полиолефи-
нов— так называемых винилогов. Эти соединения содержат амин-
ные и фенольные группы, разделенные двойными связями, имеют
высокую температуру плавления и предназначены для кабельной
промышленности. Фирма Ethyl будет выпускать (по лицензии
японской фирмы Nippon Zeon) 2,6-дициклопентил-4-метилфенол —
стабилизатор для упаковочных пленок [29].
В СССР за последние 10 лет достигнуты значительные успехи
в увеличении объемов производства и расширении ассортимента
стабилизаторов. В настоящее время отечественная промышленность
располагает основным ассортиментом, необходимым для стабили-
зации промышленных полимеров. Широким фронтом ведутся ис-
следования и разработка технологии новых высокоэффективных
стабилизаторов, решаются вопросы создания многотоннажных
комплексных производств стабилизаторов на базе единого сырья
и промежуточных продуктов.
ЛИТЕРАТУРА
1. Основные направления экономического и социального развития СССР на
1981—1985 годы и на период до 1990 года. Проект ЦК КПСС к XXVI съезду
партии. М., Политиздат, 1980.
2. Эмануэль И. М., Денисов Е. Т. — Нефтехимия, 1976, т. 16, № 3, с. 366—
382.
3. Эмануэль Н. М. — Усп. хим., 1979, т. 48, вып. 12, с. 2113—2158.
4. Денисов Е. Т. — Усп. хим., 1978, т. 47, вып. 6, с. 1090—1118.
5. Эмануэль Н. М., Денисов Е. Т., Майзус 3. К-—Цепные реакции окисления
углеводородов в жидкой фазе. М„ Наука, 1965. 375 с.
6. Пивоваров А. П., Луковников А. Ф. — Химия высоких энергий, 1967, т. 2,
с. 220.
7. Гурвич Я. А., Зимин Ю. Б. — В кн.: Тезисы конференции по проблеме «Ста-
рение и стабилизация полимеров». М., Наука, 1970. с. 32.
8. Heller И. Blattmann Н. R. — Pure Appl. Chem., 1972, v. 30, № 1—2, p. 145—
163.
28
9 Шляпинтох В. Я. и др.— ДАН СССР, 1975, т. 225, № 5, с. 1132—1134.
Ю Розанцев Э. Г. Свободные иминоксильные радикалы. М., Химия. 1970.
216 с.
11 Шляпинтох В. Я. Фотохимические превращения и стабилизация полимеров.
М., Химия, 1979. 344 с.
12 Разумовский С. Д., Зайков Г. Е. Озои и его реакции с органическими соеди-
' иениями. М, Наука, 1974. 322 с.
13 Кузьминский А. С.. Кавун С. М., Кирпичев В. П. Физико-химические основы
получения, переработки и применения эластомеров. М., Химия, 1976. 238 с.
14 Гурвич Я. А., Маслова И. П. — В кн.: Синтез и исследование эффективности
химикатов для полимерных материалов. Тамбов, Тамбовская правда, 1969.
Вып. 2, с. 10—37.
15. Маслова И. П., Баранова А. С. Там же, с. 48—65.
16. Маслова И. П., Глазунова И. А., Пугачева Л. А. Там же, с. 82—107.
17. Маслова И. П. и др. Там же, с. 467—483.
18. Скрипко Л. А., Маслова И. П. — В кн.: Синтез и исследоваине эффективно-
сти химикатов для полимерных материалов. Тамбов, Тамбовская правда,
1969, вып. 3, с. 3—32.
19. Маслова И. П. и др. — В кн.: Синтез и исследование эффективности химика-
тов для полимерных материалов. Тамбов, Тамбовская правда, 1970, вып. 4,
с. 30—58.
20. Гурвич Я. А., Золотаревская Л. К., Ку мок С. Т. Фенольные стабилизаторы.
М., Изд. ЦНИИТЭнефтехим, 1978. 80 с.
21. Sagava S.—-Нихон тому кёкайси, 1969, V. 42, № 11, р. 1065—1084.
22. Эмануэль Н. М. и др. — Тестирование химических соединений как стабилиза-
торов полимерных материалов. М., Препринт. ИХФ АН СССР, 1972.
23. Гладышев Г. П., Цепалов В. П. — Усп. хим., 1975, т. 10, с. 1830—1850.
24. Темчин Ю. И. Канд. днсс. Воронеж, ВГУ, 1974.
25. Вспомогательные вещества для полимерных материалов. Справочник./Под
ред. К. Б. Пиотровского и К. Ю. Салнис. М., Химия, 1966. 176 с.
26. Химические добавки к полимерам. Справочник, М., Химия, 1973. 272 с.
27. Rubber World, 1976, v. 175, р. 45.
28. Kemper mann Т. — Kaut. u. Kunstst., 1978, В. 31 (4), S. 234—246; C.A., 1978,
v. 89, p. 60768.
29, Chem. Age., 1978, p. 18.
ГЛАВА 2
Производные аминов
Стабилизаторы, условно объединяемые в группу аминных, по
объему производства, ассортименту и областям применения зани-
мают ведущее место среди соединений, используемых в промыш-
ленности для повышения стабильности полимеров или изделий на
их основе к различным воздействиям окружающей среды. В 1978 г.
в мире выпускалось в промышленном масштабе около 100 азот-
содержащих стабилизаторов — примерно 600 торговых марок
(табл. 2).
Стабилизаторы аминного типа начали выпускать в промышлен-
ном масштабе для резин на основе натурального каучука, а потом
и для синтетических каучуков и пластических масс. До 1930-х го-
дов единственными представителями промышленных стабилиза-
торов были производные нафтиламина — альдоль-а-нафтиламин,
фенил-р-нафтиламин. В 1940-х годах на рынке появились произ-
водные н-фенилендиамина — диалкил- и алкиларил-м-фениленди-
амины для защиты резин (прежде всего, шинных) от озонной дест-
рукции. В 1950—1960-х годах получили признание алкилированный
дифениламин, продукт конденсации дифениламина с ацетоном,
производные дигидрохинолина, неокрашивающий антиозонант —
Таблица 2. Динамика потребления аминных стабилизаторов (в тоннах)
в капиталистических странах с 1970 по 1980 год
Стабилизаторы США Страны Западной Европы
1970 г. 1974 г. 1980 г. (прогноз) 1970 г. 1974 г. 1980 г. (прогноз)
Днфенил-л-феиилендн- амин 6 500 6750 7 000— 7 250 1750 2 100 2 930
Алкиларил-п-фенилен- диамины 17000 19 500 21 500— 22 500 13 500 17 С00 21 000— 22 000
Феиил-а- и фенил-₽-наф- тиламииы 2500 2200 — 10 000 5000
Днгидрохннолины 3000 4 000 6 500— 7 000 4 500 5500 6500— 7 000
Дифеииламины и др. 13 000 15 000 15 SOO- 16 000 6000 6700 9 600— 10 000
Итого 42.000 47450 50 500— 52 750 35 750 36 350 40 000-^- 42 000
30
трибутилтиокарбамид— и первый азотсодержащий светостабили-
3дТОр__2-(2/-гидрокси-5'-метилфенил)бензтриазол. В настоящее
время производные нафтиламина постепенно утрачивают свое зна-
чение; из производных п-фенилендиамина расширяется применение
менее'токсичных соединений с более высокой молекулярной массой
типа П-(1,3-Диметилбутил)-Ы/-фенил-п-фенилендиамина.
В основе промышленных способов получения аминных стаби-
лизаторов лежат реакции: араминирование, нитрозирование, ката-
литическое гидрирование, каталитическое восстановительное алки-
лирование, N-алкилнрование ароматических аминов, конденсация
аминов с карбонильными соединениями, тиоацилирование аминов.
Развитие производства стабилизаторов аминного типа зависит
прежде всего от цены и доступности исходного сырья. В связи с
этим наблюдается тенденция перехода от стабилизаторов на осно-
ве нафтил- и дифениламина к производным анилина — наиболее
дешевого из ароматических аминов. Вводятся новые комплексные
технологические линии производства стабилизаторов, начиная с по-
лучения анилина. По прогнозам специалистов, после 1980 г. про-
изводство и потребление антиоксидантов аминного типа будет еже-
годно увеличиваться на 7—8%; для антиозонантов эта цифра со-
ставит 4—5%.
2.1. СЫРЬЕ ДЛЯ ПОЛУЧЕНИЯ АМИННЫХ
СТАБИЛИЗАТОРОВ
Важнейшим видом сырья для производства стабилизаторов —
производных ароматических аминов — является анилин (аминобен-
зол). Сейчас 40% выпускаемого в мире анилина расходуется на
производство добавок к полимерным материалам, в частности фе-
нилнафтиламинов, дифениламина, 4-гидроксидифеннламнна,
2,2,4-триметил-1,2-дигидрохинолина, диаминодифенилметана,
М-изопропил-М'-фенил-н-фенилендиамина, М,М'-дифенил-/г-фе-
нилендиамина, дифенилтиокарбамида.
В настоящее время практически весь анилин получают газо-
фазным гидрированием нитробензола или газофазным аммоноли-
зом фенола. Экономические показатели первого процесса зависят
от цен на азотную кислоту и водород, для второго процесса оп-
ределяющими являются цены на фенол и побочный продукт—аце-
тон. Оба процесса базируются на бензоле:
31
Объем производства анилина в технически развитых странах
постоянно увеличивается. Так, в США с 1960 по 1974 г. он возрос
с 120 до 550 тыс. т. В дальнейшем темпы роста производства ани-
лина несколько уменьшились, но ожидается, что к 1990 г. объем
. его выработки в США составит 800 тыс. т. Расширение производ-
ства анилина обусловлено главным образом увеличением спроса
иа полиуретановые пенопласты.
п-Фенетидин (4-аминоэтоксибензол) — сырье для производства
2,2,4-триметил-6-этокси-1,2-дигидрохинолина — обычно получаю!
этоксилированием n-нитрохлорбензола с последующим восстанов-
лением. Катализаторами этоксилирования служат оксид меди иль
диоксид марганца. Более экономичный метод получения п-фенети-
дина — восстановление п-нитрофенетола водородом; в этом случае
можно снизить себестоимость сырья и в конечном итоге улучшить
технико-экономические показатели производства стабилизаторов.
п-Нитроанилин (1-амино-4-нитробензол)—важнейшее сырье
для выработки производных п-фенилендиамина. п-Нитроанилив
обычно получают аминированием п-нитрохлорбензола. Известен
также способ получения п-нитроанилина ацилированием анилина
с последующим нитрованием и омылением. Однако себестоимости
продукта, полученного по первому способу, примерно на 35% ни-
же, чем при втором. В настоящее время в большинстве стран п-нпт-
роанилин получают по непрерывной схеме аминированием п-нитро
хлорбензола в трубчатом реакторе.
Из о-нитроанилина (1-амино-2-нитробензол) получают такщ
стабилизаторы, как 2-меркаптобензимидазол и 2-(2'-гидрокси-5-
метилфенил)бензтриазол. Обычный метод получения о-нитроани-
лина — аминирование о-нитрохлорбензола, осуществляемое непре-
рывным способом.
п-Фенилендиамин (1,4-диаминобензол) —сырье для произведет
ва главным образом Й.М'-диалкил-п-фенилендиаминов. п-Фенилен
диамин обычно получают восстановлением и-нитроанилина перио-
дическим или непрерывным способом.
Вследствие дефицитности п-нитроанилина в последнее врем?
развиваются и другие методы получения n-фенилендиамина. Ис
способу фирмы AKZO (Нидерланды) п-фенилендиамин получаю!
перегруппировкой Гофмана из диамида терефталевой кислоты £
две стадии. Диамид хлорируют, а образовавшийся Ы,Ы'-дихлорди-
амид терефталевой кислоты обрабатывают щелочью. По способ)
фирмы Toye soda koge (Япония) м-фенилендиамин получают взаи
модействием дихлорбензола с аммиаком (аммонолиз) в присутст
вин соединений меди в качестве катализатора. В поисках боле<
дешевого и доступного сырья разрабатываются и другие методь
получения n-фенилендиамина, например аминирование п-аминофе
нола в присутствии кислотных катализаторов.
о-Фенилендиамин (1,2-диаминобензол)—сырье для произвол
ства 2-меркаптобензимидазола — обычно получают восстановле
нием о-нитроанилина.
32
«-Нитродифениламин широко применяется для выработки-
производных «-фенилендиамина (N-изопропил-М'-фенил-п-фенилен-
диамин, Ы-фенил-Н'-циклогексил-«-фенилендиамин и др.). В про-
мышленности его обычно получают аминированием «-нитрохлор-'
бензола анилином:
а-Нафтиламин(1 -аминонафталин)—сырье для получения та-
ких стабилизаторов, как N-фенил-а-нафтиламин, альдоль-а-нафтил-
амин. а-Нафтиламин обычно получают нитрованием нафталина с
последующим восстановлением нитронафталина. Метод получения
а-нафтиламина аминированием 1-хлорнафталина более прост и эко-
номичен, но этот метод можно применять только в том случае, ес-
ли в исходном 1-хлоранилине отсутствует 2-хлорнафталин, кото-
рый в тех же условиях превращается в канцерогенный р-нафтил-
амин. В настоящее время более прогрессивным считается способ'
получения а-нафтиламина каталитическим аминированием р-наф-
тола аммиаком в газовой фазе, что позволяет получать а-нафтил-
амин, практически не содержащий примеси канцерогенного р-изо-
мера. "
«-Аминофенол (4-аминогидрокСибензол)—сырье для производ-
ства Ы,Ы'-дифенил-«-фенилендиамина и «-гидроксифенил-р-нафтил-
амина — обычно получали омылением «-нитрохлорбензола с после-
дующим восстановлением. Более экономичен метод каталитического:
восстановления «-нитрофенола— улучшается качество «-амино-*
фенола, уменьшается количество отходов. «-Аминофенол 99,5%-ной
чистоты получают каталитическим гидрированием «-нитрозофенола
в органическом растворителе на катализаторах платино-палладие-
вой группы.
P-Нафтол (2-гидроксинафтиламин) —один из важнейших видов
сырья для производства таких стабилизаторов, как N/N-ди-р-наф-:
тил-«-фенилендиамин, N-фенил-р-нафтиламин, «-гидроксифенил-
Р-нафтиламин. р-Нафтол обычно получают сульфированием нафта-
лина с последующим щелочным плавлением нафталин-2-сульфокис-
лоты. Непрерывный метод получения р-нафтола щелочным плавле-"
нием натриевой соли нафталин-2-сульфокислоты в растворе щелочи
в проточном трубчатом реакторе под давлением позволяет полу-
чать р-нафтол с выходом 93—97%. Другой, наиболее распростра-
ненный метод получения р-нафтола — окисление 2-алкилнафтил-
аминов (метил- и изопропилпроизводных) до соответствующего
гидропероксида, который затем разлагают в присутствии кислоты
и получают р-нафтол с почти теоретическим выходом. Перспектив-
ным методом получения р-нафтола является также прямое окис-
ление нафталина.
Гидрохинон (1,4-дигидроксибензол) применяют для производ-
ства Й.Ы'-дифенил-п-фенилендиамина, «-гидроксидифениламина,
ди-трег-бутил- и ди-трег-амилгидрохинонов. Гидрохинон ' обычно
3—Ю57
33
получают окислением анилина до бензохинона с последующим вос-
становлением. Например, окисляют анилин активным оксидом мар-
ганца в присутствии сильной минеральной кислоты и восстанав-
ливают полученный бензохинон железным порошком или другими
восстановителями.
Более прогрессивный метод получения гидрохинона из п-изопро-
пилбензола отличается применением более дешевого сырья и мень-
шим количеством образующихся побочных продуктов. Технологи-
ческая схема включает стадии алкилирования бензола, окисления
образующегося изопропилбензола в дигидропероксид, расщепления
последнего на ацетон и гидрохинон с помощью серной кислоты,
очистки гидрохинона.
Фирмой Upjohn (США) разработан способ получения гидрохи-
нона из фенола и ацетона через дифенилолпропан, который на ка-
тализаторе разлагается на фенол и 4-изопропенилфенол. Фенол
возвращают в цикл, а 4-изопропенилфенол окисляют в гидрохинон .
и ацетон; ацетон тоже возвращают в цикл. По мнению специали-||
стов фирмы, этот способ должен быть экономичнее двух предыду-Я
щих. -1
Метод получения гидрохинона прямым окислением фенола пер Л
оксидом водорода освоен фирмами Rhone Poulenc (Франция) Л
Ube industries (Япония), а также некоторыми итальянскими фирЯ
мами в промышленном масштабе. Интересен также способ полуЛ
чения гидрохинона электрохимическим окислением бензола. ПерсЛ
пективным может оказаться получение гидрохинона из этилена Л
оксида углерода в присутствии водорода и катализатора. Этот спо-<
соб позволяет получать гидрохинон из доступного сырья с хоро-1
шим выходом, в одну стадию без образования побочных продуктов, I
в растворителе. Благодаря широкому применению гидрохинона во!
всем мире ведутся интенсивные поиски новых методов его полу- J-
чения, более экономичных и конкурентоспособных по сравнению с '
рассмотренными выше промышленными.
Изобутилметилкетон (4-метилпентанон-2) применяют для про-
изводства Ы-(1,3-диметилбутил)-Ы/-фенил-п-фенилендиамина и
М,М'-ди-(1,3-диметилбутил)-п-фенилендиамина. Изобутилметилке-
тон обычно получают из ацетона и водорода в жидкой фазе под
давлением в присутствии катализатора. Метод обеспечивает высо-
кне степень конверсии и селективность. Синтез осуществляют на
‘ бифункциональном катализаторе (например, палладий, осажден-
ный на фосфате циркония). При наличии доступного оксида мези-
тила изобутилметилкетон получают его гидрированием или элект-
рокаталитическим восстановлением. Изобутилметилкетон получа-
ют также из изопропанола контактированием в жидкой фазе на
катализаторе, содержащем ZnO, Сг20з и ZnCrO4, или на скелетных
Ni, Со и Си. Если в каталитической системе преобладает ZnO,
главным продуктом является изобутилметилкарбинол; в случае ис-
пользования ZnCrO4 и скелетного никелевого катализатора обра-
зуется изобутилметилкетон.
34
Циклогексанон — сырье для производства М-фенил-Ц'-цикло-
гексил-н-фенилендиамина — получают по методу Сабатье и Сан-
депана гидрированием фенола на никеле при 190—200 °C; при
этом образуется смесь циклогексанола и циклогексанона, содер-
жащая небольшие примеси фенола, который отделяют перегонкой.
Другой промышленный метод получения циклогексанона (так-
же в смеси с циклогексанолом) —окисление циклогексана в жид-
кой фазе в присутствии катализатора, например нафтената ко-
бальта.
2.2. РЕАКЦИИ АРАМИНИРОВАНИЯ-
АРОМАТИЧЕСКИЕ АМИНЫ
Реакция араминирования, приводящая к образованию вторич-
ных ароматических аминов общей формулы ArNHAr', лежит в ос-
нове синтеза большой группы стабилизаторов—-замещенных наф-
тиламина и дифениламина, некоторых производных п-фениленди-
амина. Араминирование проводят, действуя первичными аромати-
ческими аминами на ароматические соединения, содержащие реак-
ционноспособные амино- или гидроксигруппу.. В общем виде реак-
цию можно представить схемой
ArX + Ar'NH2 -> ArNHAr' + НХ
где X — ОН или NH2 (в редких случаях SO3H).
Впервые эту реакцию для получения вторичных ароматических
аминов применил Лэр, осуществивший в 1886 г. синтез дифенил-
амина по схеме:
2C,H5NH2 -> CeHBNH-CeH6 + NH3
В качестве катализатора он использовал хлрристый водород.
В присутствии значительных количеств хлористого водорода (его
можно вводить в форме солянокислого анилина) отщепляющийся
аммиак образует хлорид аммония:
ArNH2 + Ar'NHgCl -> ArNHAr' + NH4C1
Позднее было показано, что араминирование проходит в присутст-
вии каталитических количеств трихлорида фосфора, арилсульфо-
кислот [1—3], хлоридов железа и алюминия, иода и его соедине-
ний и некоторых других [4].
Применение катализаторов кислотного характера требует защи-
ты аппаратуры от коррозии. Поэтому большой интерес представ-
ляет катализ соединениями, не вызывающими коррозию [5]. При-
менение оксидов алюминия или титана, эфиров ортотитановой
кислоты позволило осуществить араминирование в газовой фазе
(о, 7]. В последнее десятилетие появились патенты, рекомендую-
щие активировать оксид алюминия различными кислотными аген-
тами. При этом заметно возрастает активность катализатора, а
срок его службы увеличивается до года.
Важное место в синтезе стабилизаторов занимает араминиро-
вание ароматических гидроксисоединений, позволяющее получать
труднодоступные ароматические амины, в частности производные
р-нафтиламина. Одним из первых синтезов, осуществленных этим
методом, был синтез N-фенил-р-нафтиламина, полученного Каро
из р-нафтола и анилина в 1879 г.:
Араминирование гидроксисоединений катализируется кислота-
ми— серной, соляной, фосфорной, борной, арилсульфокислотами.
Каталитическое действие оказывают и добавки агентов, способст-
вующих отнятию воды (например, хлоридов кальция или цинка
[8]). В ряде случаев араминирование удается осуществить в более
мягких условиях в присутствии солей сернистой кислоты. Эта ре-
акция, известная как реакция Бухерера, широко применяется в
синтезе промежуточных продуктов для красителей.
Замещение гидрокси- или аминогруппы на ариламиногруппу
представляет собой реакцию нуклеофильного замещения. Роль
кислотного катализатора в этой реакции до конца не выяснена.
Одна из гипотез предполагает, что в результате протонирования
ароматического кольца образуются аренониевые ионы, замещение
в которых протекает с большой легкостью [9]:
N-Фенил-р-нафтиламин впервые был получен Бухерером в 1875 г. араминн-
рованием 2-гидроксинафтойной кислоты анилином в присутствии бисульфита
натрия (в дальнейшем этот метод получил название бнсульфнтного). Практиче-
ского применения этот способ не нашел, несмотря на то что N-фенил-р-нафтнл-
амин образуется с выходом 60%.
В 1879 г. Каро разработал способ получения N-фенил-Р-нафтиламииа ара-
мннированием р-нафтола анилином нлн его гидрохлоридом под давлением при
200—210 °C без катализатора. Этот метод был положен в основу первой про-1
мышленной технологии получения N-феннл-Р-иафтиламнна, которая заключа-
лась в следующем.
Смесь р-нафтола и соли анилина нагревали до 230 °C и выдерживали при^
этой температуре в течение 4 ч, отгоняя воду, хлористый водород и Р-нафтол.
Затем в реакционную массу добавляли соляную кислоту, кипятили в течение
36
20 мнн и сливали на воду. Затвердевший продукт фильтровали и отмывали от
кислоты; осадок смешивали с раствором щелочи, кипятили 20 мин и сливали
на воду. Осадок фильтровали и отмывали от щелочи. Выход 88% от теоретиче-
ского (считая на гидрохлорид); т. пл. 103—104°C. После перегонки при 275—
385 °C и 133—266 Па получали N-фенил-р-нафтиламин с т. пл. 105—105,5 °C.
Затем в качестве катализаторов получения N-фенил-р-нафтиламина исполь-
зовали цинкат и хлорид алюминия, соляную кислоту, хлорид кальция. Позднее
было описано применение серной [10] и сульфаниловой кислот, толуолсульфокис-
лоты [2], бензолсульфокислоты [3], алюмосиликатов [4, 7], иода и его соединений
[10], эфиров титановой кислоты и оксидов титана [5], сульфатов. В последнее
время хорошие результаты получены в присутствии фосфитов [11], например
трнфенилфосфита. Выход N-фенил-Р-нафтиламина 96%; т. пл. 100—102°C.
В целом метод каталитического араминирования обеспечивает
получение N-фенил-р-нафтиламина с высоким выходом и хороше-
го качества. Так, в присутствии алюмосиликатов выход составляет
98,5%, в присутствии бензол сульфокислоты 99,47%, в случае и-то-
луолсульфокислоты 93%.
Разработан непрерывный процесс получения N-фенил-р-нафтил-
амина (Неозон Д) в газовой фазе при 320—375 °C на твердых
катализаторах [7, 12]. Катализаторы, активные в газовой фазе (ок-
сид алюминия, крупнопористый алюмосиликат, твердая фосфор-
ная кислота, а также кварц и силикагель, обработанные ортофос-
форной кислотой), активны и для жидкофазного процесса. Кон-
тактное араминирование р-нафтола осуществляют, пропуская пары
анилина и р-нафтола над катализатором. Продукты реакции, со-
держащие N-фенил-р-нафтиламин, анилин и р-нафтол, перегоняют
для выделения чистого неозона Д.
N-Фенил-р-нафтиламин с выходом «50% получают из натрие-
вой соли нафталин-2-сульфокислоты и анилида натрия [13]:
Этот метод позволяет получать N-фенил-р-нафтиламин, минуя ста-
дию производства р-нафтола, и представляется перспективным.
Другие методы [14, 15] имеют лишь препаративное значение.
Технологическая схема получения. В промышленности N-фенил-
Р-нафтиламин получают араминированием р-нафтола анилином в
присутствии солянокислого анилина или бензолсульфокислоты:
Технологический процесс получения Неозона Д состоит из сле-
дующих основных стадий: араминирование р-нафтола, нейтрализа-
ция катализатора, отгонка анилина, очистка Неозона Д, кристал-
37
Плав
Рнс. 2. Технологическая схема получения N-фенил-Р-иафтиламина:
J — аппарат для приготовления суспензии; 2, 12—конденсаторы; 3 —колонка; 4, 13 — сепа-
раторы; 5 — реактор; 6— сборник; 7, 9 — емкости; 8 — холодильник; 10— отстойник; 11 —
ректификационная колонна; 14 — хранилище; 15 — барабан; 16 — вибросито.
лизация [16, 17]. В суспензатор 1 (рис. 2) загружают анилин, на-
гревают его до 145—-150 °C и прибавляют расплавленный р-наф-
тол. Смесь перемешивают и передавливают сжатым воздухом в
реактор 5, куда предварительно дозируют необходимое количество
.бензолсульфокислоты (БСК) или солянокислого анилина. Реак-
ционную массу нагревают до 240—250 °C. При этой температуре
отгоняют реакционную воду вместе с анилином.
Смесь паров анилина и воды через колонну 3 поступает в кон-
денсатор 2. Конденсат принимают в сепаратор 4, где анилин отде-
ляется от воды. Анилин собирают в емкость 7, откуда его возвра-
щают на синтез, а воду (с растворенным в ней остаточным анили-
ном) через сборник 6 подают в колонну 11 на выделение анилина.
По окончании конденсации нейтрализуют катализатор, добавляя
щелочь в реактор, и отгоняют анилин через систему: холодильник
8 — емкость 9 — отстойник 10. Анилин из емкости 9 направляют в
емкость 7, а воду через отстойник 10 подают в колонну 11 на вы-
деление остаточного анилина. Смесь паров анилина и воды через
конденсатор 12 направляют в сепаратор 13, где анилин и воду раз-
деляют.
По окончании отгонки анилина передавливают плав Неозона Д
из реактора 5 в хранилище 14, где поддерживают температуру
130—150°С. Из хранилища Неозон Д подают на чешуирование на
барабан 15, охлаждаемый водой. Застывший плав срезают с ба-
рабана ножом, а чешуйки продукта после вибросита 16, где от-
деляются пыль и мелкие частицы, направляют на упаковку.
38
Обычно N-фенил-р-нафтиламин в виде примесей содержит ани-
лин и канцерогенный р-нафтиламин. Один из эффективных спосо-
бов очистки от примесей — обработка продукта органическими кис-
лотами или их производными [18]. Например, к N-фенил-р-нафтил-
амину, содержащему 0,002% Р-нафтиламина и 0,5% анилина, до-
бавляют 5% фталевого ангидрида и выдерживают в течение 50 мин
при 250°С. Остаточное содержание р-нафтиламина (14-2) 10-5 %, а
анилина (14-2) 10-3 % •
Недостатком товарного Неозона Д является его слеживаемость
при хранении (продукт способен при определенных условиях обра-
зовывать комки, монолит той или иной твердости [19]). Слежав-
шийся Неозон Д плохо распределяется в резиновых смесях и в
значительной степени теряет свою эффективность.
Показана [20] возможность получения Неозона Д, легко сма-
чиваемого водой, за счет введения в расплав поверхностно-актив-
ных веществ типа алкилсульфонатов, натриевых солей сульфиро-
ванных эфиров олеиновой или рицинолевой кислот или продук-
тов взаимодействия жирных спиртов с оксидом этилена. Легко
смачиваемый Неозон Д быстро образует суспензии, стабильные к
седиментации. Применение легкосмачиваемого Неозона Д при про-
изводстве изделий из латексов позволяет ускорить приготовление
водной суспензии, улучшить качество резин, продлить срок их экс-
плуатации.
Для улучшения транспортирования Неозона Д и непрерывного
его дозирования целесообразно применять гранулированный про-
дукт. Хороший способ гранулирования разработан в Японии (фир-
ма Yositomi) [21, 22]. Гранулированный Неозон Д получают, мед-
ленно добавляя его расплав к водному раствору гидрофильного
высокомолекулярного соединения или к смешанному растворителю
(водно-спиртовой раствор с добавками алифатического кетона и
диэтилового эфира). Смесь диспергируют и гранулируют до частиц
диаметром 0,1—5 мм. Технология получения гранулированного
Неозона Д разработана также в СССР.
Свойства и применение. N-Фенил-р-нафтиламин представляет
собой белый порошок; т. пл. 108 °C; т. кип. 395,5 °C. Растворяется
в ацетоне, бензоле, диэтиловом эфире, хлороформе, четыреххло-
ристом углероде, этилацетате, дихлорметане, горючем этаноле.
Горюч. Т. самовоспл. слоя пыли 605°C; для аэровзвеси 603 °C.
Нижний предел взрываемости аэровзвеси 26,4 г/м3; нижний предел
воспламеняемости 38 г/м3; минимальное взрывоопасное содержа-
ние кислорода 11 % (об.).
Мало летуч. Умеренно токсичен; ЛДбо= 15004-2000 мг/кг. По
Данным [23], в организме человека может превращаться в канце-
рогенный Р-нафтиламин.
N-Фенил-р-нафтиламин выпускают под многочисленными тор-
говыми наименованиями практически во всех развитых странах в
виде порошкообразных, гранулированных или таблетированных
продуктов, различающихся по температуре плавления:
39
Торговое наименование Страна, фирма Выпускная форма Т. пл., °C
Неозон Д СССР Порошок, че- шуйки 104,5—105
Accinox D Индия, Alkali Порошок 105
Age Rite Powder США, Cyanamid, Van- derbilt То же 106—107
Altofane MC Франция, Kuhlmann » 106—107
Antage B, D Япония, Kawaguchi » 106,103
Antigene D Япония, Sumitomo > >105
Antioxidant D Япония, Seiko > 105
Antioxidant PBN ЧССР » 105—106
Antioxidant 116 Англия, Anchor » 105
Antioxygene MC Франция, Mat. Col. » 107
ASM PBN ФРГ, Bayer » J05—106
Good Rite AO 3100 США, Goodrich » 106
Inibitore OD Италия, ACNA Monteca- tini » 107
Neozone D США, Du Pont > 106—107
Nocrac D Япония, Ouchi Shinko > 102
Nonflex D Япония, Seiko » 195—108
Nonox D Англия, 1CI, Monsanto Порошок, гра- нулы, таб- летки 107—108
PBN Англия, Monsanto Порошок 106
PBN wettable Англия, Monsanto Порошок, сма- чиваемый во- 107
PBN Франция, Soc. Prod. дой Порошок 106—107
PBN Нидерланды, Van Hasselt То же 104
PBNA США, Naugatuck — —
Rionox Франция, Man. Land. — 104
Spolen ЧССР Порошок 104—108
Stabilizator AR ПНР То же 105,5—106,5
STD США, Benzon > 107
Кроме того, известно до 30 торговых наименований смесевых ста-
билизаторов, составной частью которых является N-фенил-р-наф-
тиламин (Thermoflex A, Akroflex).
N-Фенил-р-нафтиламин является широко распространенным
стабилизатором синтетических каучуков — бутадиен-стирольных,
бутадиен-нитрильных, хлоропреновых, изопреновых, бутадиеновых,
бутилкаучука и др. Часто применяется в сочетании с другими ста-
билизаторами и термостабилизатором резин на основе НК и СК
общего назначения. Частично защищает резины от светоозонного
старения и разрушения при многократных деформациях.» Является
термостабилизатором полиэтилена, полиизобутилена и сополиме-
ров этилена с винилацетатом. Стабилизатор смазочных масел.
N-Фенил-а-нафтиламин
40
N-Фенил-а-нафтиламин впервые был получен араминированием а-нафгола
анилином в закрытом сосуде при 280—290 °C в присутствии хлорида кальция
в качестве катализатора, а также араминированием а-нафтиламина в запаянной
трубке при 240 °C в присутствии соляной кислоты. Последняя реакция легла
в основу промышленного способа получения. В дальнейшем в качестве катали-
заторов применяли металлический иод, иодистоводородную кислоту, подпетый
аммоний, сульфаниловую кислоту, алюмосиликаты [4]. Применение органических
кислот в качестве катализатора обеспечивало получение более чистого продукта,
так как побочный дифениламин либо вовсе не образовывался, либо получался
в незначительных количествах [1]. Все современные промышленные способы по-
лучения N-фенил-а-нафтиламина основаны на этом методе (метод Фирц-Да-
вида).
В последнее время хорошие результаты получены при использовании фосфи-
тов в качестве катализаторов [И]. Реакцию проводят в жидкой фазе при
200—230°C под давлением; выход N-фенил-а-нафтиламина 95%.
Известны способы получения N-фенил-а-нафтиламниа в присутствии твер-
дых катализаторов [24]. Один из них заключается в нагревании а-нафтиламина
с циклогексанолом и а-нитронафтиламином в присутствии платины на угле.
Процесс сопровождается дегидрогенизацией (ароматизацией) алициклического
углеводородного радикала, причем нитронафтила мин играет роль акцептора
водорода. По другому способу араминирование а-нафтиламина анилином на
твердых катализаторах проводят в газовой фазе над гелем алюминия при
300 °C [6]. Максимальный выход (84,3%) обеспечивается при соотношении ани-
лина к а-нафтнламину 10 : 14. Побочным является так называемый полимерный
продукт, который представляет собой, по-видимому, продукт поликонденсации
а-нафтиламина.
Технологическая схема производства. В промышленности N-фе-
нил-а-нафтиламин (Неозон А) получают араминированием а-наф-
тиламина анилином в присутствии сульфаниловой кислоты:
Технологический процесс получения состоит из следующих ос-
новных стадий (рис. 3): араминирование а-нафтиламина, фракци-
онная вакуум-разгонка, чешуирование. В реактор / через мерник
загружают анилин и расплавленный а-нафтиламин, а через люк—
сульфаниловую кислоту (катализатор). Реакционную массу на-
гревают до 180 °C. При этой температуре отгоняется вода, содер-
жащаяся в исходном сырье. Ее выводят вместе с парами анилина
через холодильники 2 и 3 и сепаратор 4. По окончании отгонки во-
ды поднимают в реакторе температуру до 240 °C и выдерживают
реакционную массу в течение 2 ч. Выделяющийся аммиак погло-
щают водой в скруббере 5. Водный раствор аммиака собирают в
сборник'7.
Плав Неозона А из реактора / направляют на вакуум-разгон-
ку в перегонный куб 8. Для лучшего разделения продуктов пере-
гонки на кубе смонтирована насадочная колонна. При 250°C и ос-
таточном давлении 96—98 кПа отбирают первую фракцию, содер-
жащую в основном а-нафтиламин, в сборник //. Затем с паром
‘отгоняют остатки анилина. Пары конденсируются в холодильнике
9. Конденсат в сепараторе 10 разделяют на воду и анилин; воду на-
41
правляют на биохимическую очистку, а анилин —в сборник И, от-
куда его вместе с а-нафтиламином возвращают в производство.
Неозон А собирают в сборник 12, откуда его подают на чешуи-
рование (аналогично Неозону Д). Отгоняют его при 310—350 °C.
Кубовый остаток, представляющий собой вязкую смолу, сли-
вают на воду при перемешивании в смеситель 13. Смола застывает
в виде каплевидных гранул. Их отделяют и направляют на сжига-
ние.
Свойства и применение. N-Фенил-а-нафтиламин представляет со-
бой кристаллический порошок желтого цвета; т. пл. 62 °C; т. кип.
226°C (при 2 кПа). Растворяется в ацетоне, бензоле, гептане, хло-
роформе, этаноле, этилацетате, уксусной кислоте, четыреххлори-
стом углероде.
Пыле-воздушные смеси взрывоопасны.
N-Фенил-я-нафтиламин выпускается во многих странах раз-
личного (по температуре плавления) качества, в основном в виде
порошка:
Торговое наименование Страни, фирма Выпускная форма Т. пл., «с С
Неозон А. Additin PAN СССР 'ФРГ, Bayer Кусочки, чешуйки 54,2
Altofane А Франция, Kuhlmann Порошок 55
Anti age PAN Нидерланды, Francon Donders То же —
Antigen A Япония, Dyestuffs » 50
Antigene PA Япония, Sumitomo » 50
Antioxidant PAN Англия, Monsanto » 50
Antioxygene A Франция, Mat. Col. » 57
ASM PAN ФРГ, Bayer » 50
Inhibitor OA Италия, ACNA Monte- catini » 56—57
Neozone A США, Du Pont > 50
Nocceler PA Япония, Ouchi Shinko > —
Nocrac PA Япония, Ouchi Shinko Порошок, кусочки 50
Nonox AN Англия, ICI Порошок 50
Nonoxol AN Англия, ICI Чешуйки 57—60
За рубежом выпускают также гранулированные смеси стаби-
лизаторов, составной частью которых является N-фенил-а-нафтил-
амин: чаще всего с И.М'-дифенил-п-фенилендиамином (АсгоПех,
Anox JO, Antigene FC, Nonflex PC) и с 2,4-диаминотолуолом (Anti-
gen С, Antioxygene CAS, Neozone С).
N-Фенил-а-нафтиламин является стабилизатором синтетических
каучуков — бутадиеновых, бутадиен-стирольных, изопреновых,
хлоропреновых. Для стабилизации резин чаще всего применяется
в смеси с ^№-дифенил-п-фенилендиамином и 2,4-диаминотолуолом.
Для резин на основе СКИ-3 более эффективен, чем N-фенил-р-наф-
42
Рис. 3. Технологическая схема получения N-фенил-а-нафтиламииа:
1 — реактор; 2, 3, 9 — холодильники; 4, 10 — сепараторы; 5 — скруббер; 6, 7, 11, 12 — сбор-
ники; 8 — перегонный куб; 13 — смеситель.
тиламин. В сочетании с М,М'-дифенил-п-фенилендиамином приме-
няется для стабилизации полиэтилена.
Кроме того, N-фенил-а-нафтиламин используют в качестве ста-
билизатора смазочных масел и противокоррозионных жидкостей
для двигателей внутреннего сгорания.
п-Гидроксифенил-^>-нафтиламин
n-Гидроксифенил-р-нафтиламин впервые был получен Бухерером в 1875 г.
араминированием p-гидроксинафтойной кислоты аминофенолом в присутствии
биосульфита натрия.
В промышленном масштабе п-гидроксифенил-р-нафтиламин
(Пара-оксинеозон) получают араминированием гидрохинона
Р-нафтиламином [8] в присутствии дегидратирующих катализа-
торов, обычно хлорида цинка:
43
Рис. 4. Технологическая схема получения п-гидроксифенил-Р-нафтиламина:
1 — реактор-кондеисатор; 2, 5 — приемники; 3 — промывной аппарат; 4 — перегонный куб;
6 — напорная емкость; 7 — барабан-кристаллизатор; 8, 10 — системы улавливания пыли; 9 —
мельница.
Выделяющуюся воду удаляют через колонку в виде азеотропной
смеси с высококипящим ароматическим или алифатическим угле-
водородом или исходным р-нафтиламином. В СССР его получают
араминированием р-нафтола л-аминофенолом в присутствии би-
сульфита или сульфита натрия:
Реакция проходит при сравнительно низкой температуре (135—
140 °C) и 0,3—0,4 МПа.
Технологическая схема получения. Пара-оксинеозон получают
конденсацией л-аминофенола с р-нафтолом в водном растворе сме-
си сульфита и бисульфита натрия. Реактор 1 (рис. 4) представля-
ет собой стальной эмалированный котел, снабженный приборами
для измерения температуры и давления, сигнализатором электро-
проводности, мешалкой, рубашкой для нагрева паром (6 МПа) и
охлаждения водой.
В реактор сначала загружают растворы сульфита и бисульфита
натрия, а затем через люк из бункеров — п-аминофенол и р-нафтол.
Герметизируют аппарат, подают в рубашку пар и при 135—140 °C
и 0,25—0,35 МПа проводят конденсацию в течение 12—14 ч. По
окончании реакции отстаивают реакционную массу и разделяют ее
на два слоя. Нижний слой — маточный раствор — принимают в
приемник 2. Верхний слой — плав Пара-оксинеозона — передают в
аппарат 3 на промывку водой.
44
Плав промывают водой в аппарате 3 вначале при 50—60 °C, а
затем при 135—140 °C и 0,25—0,35 МПа. Нижний слой — плав Па-
ра-оксинеозона— передают по обогреваемому трубопроводу на
обезвоживание, а промывные воды (верхний слой) объединяют с
маточным раствором в приемнике 2. Обезвоживание плава прово-
дят в стальном кубе 4 вначале при атмосферном давлении, посте-
пенно повышая температуру от 135 до 145 °C. Остатки воды уда-
ляют при 140—145°С насосом РМК-3 (на схеме не показан) при
остаточном давлении 40—80 кПа. Конденсат собирают в приемник
5. По окончании обезвоживания передают плав через напорную
емкость 6 на кристаллизацию.
Кристаллизацию проводят на вращающемся цилиндрическом
барабане 7 с поверхностью охлаждения 3,7 м2, делающем 3,5 обо-
ротов в минуту и имеющем ванну с обогревом. Плав подается тон-
кой струей в ванну; в процессе вращения барабана происходит
кристаллизация Пара-оксинеозона. Образующуюся тонкую пленку
срезают ножом в виде чешуек и направляют в заранее приготов-
ленную тару. Для улавливания пыли узел кристаллизации снаб-
жен системой 8, состоящей из циклона и вытяжного венти-
лятора.
Чешуированный Пара-оксинеозон размалывают на маятниковой
мельнице 9, улавливая пыль в системе 10, состоящей из рукавного
фильтра, вентилятора высокого давления и циклона. Размолотый
продукт упаковывают в бумажные мешки, вставленные в деревян-
ные барабаны.
Маточный раствор и промывные воды из приемника 2 фильтру-
ют от выпавшего продукта, который присоединяют к очередной
порции на стадии промывки. Фильтрат возвращают на конденса-
цию.
Свойства и применение. н-Гидроксифенил-р-нафтиламин пред-
ставляет собой мелкокристаллический порошок светло-серого цве-
та с фиолетовым оттенком; т. пл. 135 °C. Растворяется в ацетоне,
этиловом спирте, диэтиловом эфире, горячем растворе едкого нат-
ра; слабо растворяется в метаноле, хлорбензоле, бензоле, щело-
чах.
Горюч. Т. самовоспл. слоя пыли 605 °C; т. самовоспл. аэровзвеси
603 °C. Нижний предел взрываемости аэровзвеси 26,4 г/м3; нижний
предел воспламенения 38 г/м3; минимальное взрывоопасное содер-
жание кислорода 11% (об.).
Умеренно токсичен. ЛДбо = 2200-е-5200 мг/кг.
За рубежом л-гидроксифенил-р-нафтиламин в качестве стаби-
лизатора не применяют. В СССР он находит ограниченное приме-
нение как стабилизатор синтетических каучуков — бутадиеновых,
бутадиен-стирольных, бутадиен-нитрильных, хлоропреновых.
Может применяться в сочетании с другими стабилизаторами.
Защищает резины от термоокислительного и светоозонного старе-
ния. Пассивирует действие металлов переменной валентности. При-
меняется в сочетании с N-фенил-р-нафтиламином.,
45
Термостабилизатор полиформальдегида и полипропилена
(в смеси с 2-меркаптобензимидазолом), полипропиленового волок-
на. Стабилизатор минеральных масел.
Дифениламин
Дифениламин впервые был получен и идентифицирован в 1864 г. Хофманом
разложением красителя «анилиновый синий» при нагревании:
Первый метод синтеза дифениламина (1866 г.) заключался в автоарами-
нировании анилина при температуре кипения реакционной смеси в присутствии
соляной кислоты. Дифениламин получался в смеси с солянокислым дифенилами-
ном и анилином. Для его выделения обрабатывали смесь соляной кислотой и
разбавляли 20—30-кратным количеством теплой воды.
В Германии до 1940 г. дифениламин в промышленности получали продол-
жительным нагреванием анилина в присутствии катализатора (0,3% РС13)
в автоклавах при 300—325 °C и « 1 МПа. В дифениламин превращалось
45% анилина; выход составлял 92% на вступивший в реакцию анилин
{25]
При взаимодействии анилина с трихлоридом фосфора первоначально обра-
зуется фосфазосоединение
2/-VNHa + PCl3 —
\ / —«jrici
а затем гидрохлорид анилина [26]. Недостаток этого катализатора — образова-
ние самовозгорающегося фосфина РН3.
До 1950-х годов на заводах фирмы Du Pont (США) дифениламин полу-
чали нагреванием анилина в присутствии 2% воды и 0,5—3% хлорсодержащего
катализатора под давлением. При 275—325 °C выход дифениламина был почти
теоретический; очистку осуществляли фракционированием. Фирмой Du Pont
в качестве катализаторов были запатентованы хлориды металлов, а также ми-
неральные, карбоновые и сульфокислоты. На практике чаще всего применяли
NH4Cl+FeCl2. Позднее было найдено, что хлорное железо меньше корродирует
оборудование, а с начала 1950-х годов фирма Du Pont [25] и особенно фирма
Koppers в США внедряют в производство катализатор А1С13 [28—30]. Процесс
получения дифениламина оформлен непрерывно: температура 275—350 °C; ката-
лизатором служил комплекс А1С13 и гидрохлорида анилина, образующегося во
время реакции.
Хлориды металлов вызывают значительную коррозию аппаратуры и дают
большое количество сточных вод, осложняется разделение реакционной смеси,
а стадия араминирования в автоклаве трудно поддается переводу на непре-
рывный режим. Поэтому в настоящее время от применения хлоридов металлов
полностью отказались. Недостатками араминирования анилина в жидкой фазе
являются также осаждение катализатора па стенках реакторов и необходимость
его выделения из реакционной смеси.
Если в качестве сокатализатора к А1С13 или FeCl3 добавлять фосфорную
кислоту, снижается образование смолообразпых продуктов и уменьшается вре-
мя реакции [31].
46
В настоящее время разработан способ получения дифенилами-
на в газовой фазе при температуре выше 400 °C над катализато-
рами, содействующими отщеплению аммиака и содержащими в ка-
честве главной составной части А12О3 или (реже) TiO2 [32—34].
Для увеличения активности оксид алюминия обрабатывают рас-
твором соляной кислоты, сушат и прокаливают на воздухе при
500—550 °C [35, 36]. Отмечено, что число активных центров на
единицу поверхности А120з, обработанного соляной кислотой, при-
мерно на 103 больше, чем на необработанном у-А120з. На оксиде
алюминия, активированном борной кислотой [37—39] или трифто-
ридом бора [40], можно проводить процесс при более низких тем-
пературах (350—500°C), что повышает качество дифениламина и
увеличивает срок службы катализатора.
Реакция образования дифениламина в газовой фазе обратима;
оптимальная температура «460 °C. Выход дифениламина (на всту-
пивший в реакцию анилин) почти теоретический при степени пре-
вращения анилина 20—35% [41—43].
Преимущества газофазного способа — непрерывность процесса,
отсутствие коррозии и сточных вод, простота обслуживания уста-
новки и высокий уровень автоматизации процесса. Недостатки —
громоздкость схемы, частая регенерация катализатора и большой
расход электроэнергии.
Применение фторированных соединений бора в качестве ката-
лизаторов [44—46] повышает выход дифениламина и увеличивает
срок службы катализатора. Метод позволяет применять анилин,
содержащий воду; температура реакции 300—350 °C.
Разработаны методы жидкофазного получения дифениламина
из анилина. В качестве катализаторов рекомендованы гидросили-
каты, активированные кислотами.
Предложенный в 1973 г. в Японии [47] процесс получения ди-
фениламина на алюмосиликатных катализаторах запатентован в
США, ФРГ и Франции. Реакцию можно проводить периодически
или непрерывно в стационарном слое катализатора. Катализатор
получают, смешивая водный раствор Si-содержащего материала с
водным раствором Al-содержащего материала для образования
водного геля. Регенерируют катализатор, нагревая его при 450—
500 °C в токе воздуха. Анилин пропускают через слой катализато-
ра при времени контакта 0,5—3 ч и давлении 1,3—3,2 МПа. Ди-
фениламин выделяют перегонкой, анилин возвращают в процесс
без очистки.
Получение дифениламина араминированием фенола впервые
было осуществлено в 1880 г. (Мерц) — нагреванием комплекса
анилин+хлорид цинка с фенолом при 250—260 °C. Катализаторами
могут служить ароматические сульфокислоты [48], триарилфосфи-
ты [11], триарилфосфаты [49], соединения титана [5], активиро-
ванный оксид алюминия [50]. В присутствии сульфокислот, напри-
мер n-толуолсульфокислоты, температура араминирования состав-
ляет 290—310 °C. При использовании соединений, титана, например
47
Ti(OC3H7-zz3o)4, применяют такую температуру и давление, чтобы
реагенты находились в жидкой фазе. Мольное соотношение фенола
и анилина от 1 : 1 до 1 :2,5. В присутствии активированного оксида
.алюминия осуществляют процесс в газовой фазе при 380—480°C
и мольном соотношении фенола к анилину (0,5-ь-1) : 1. Для увели-
чения срока работы катализатора процесс ведут при степени кон-
версии фенола 30—40%• Непрерывный или периодический способ
получения дифениламина из фенола и анилина при 300—500 °C
под давлением осуществлен в присутствии Р2О5, фосфорных кис-
•лот, их кислых солей, триалкилфосфатов и триарилфосфатов при
азеотропной отгонке воды [51].
В мягких условиях, но с невысоким выходом дифениламин по-
лучается при араминировании бензола N-фенилгидроксиламином
[52]:
ЛЛ + HONH-fA -------->. /~A_nh_/~A
Дифениламин можно получать по Ульману конденсацией ацет-
анилида с галогенбензолом в присутствии карбоната калия и раз-
личных солей меди [53] (например, иодида меди) в среде нитро-
бензола с последующим омылением образовавшегося N-ацетилди-
фениламина раствором соляной кислоты:
Вг
Метод обеспечивает получение чистого дифениламина (т. пл. 54 °C;
из спирта) с выходом 60%. Фирмой Du Pont (США) предложено
осуществлять этот процесс без растворителя в одну стадию. Ва-
риантом синтеза дифениламина по Ульману является метод фир-
мы Dow (США)—взаимодействие хлорбензола и анилина в без-
водной среде в присутствии едкого натра, закиси меди и хлорида
калия при 235—240 °C.
В1961 г. фирмой Monsanto (США) был разработан [24] ори-
гинальный процесс получения дифениламина каталитическим вос-
становительным алкилированием нитробензола циклогексанолом;
катализатор — палладий на угле. Процесс сопровождается дегид-
рогенизацией (ароматизацией) алициклического углеводорода
(циклогексанол), а нитробензол играет роль акцептора водорода.
Реакцию контролируют по отгону воды. Дифениламин получают
с т. пл. 46—47 °C и выходом 97%.
Несколько меньше (84,5%) выход дифениламина по способу
фирмы Bayer (ФРГ) —дегидрированием N-циклогексилиденанили-
на в присутствии никель-хромового катализатора [54]:
48
Фирмой Snam Progetti (Италия) в 1975 г. был разработан и
запатентован во многих странах способ получения дифениламина
контактированием этого имина с дегидрирующим катализатором
(Ni, Pt, Pd, Cu+Cr) при 300—500 °C в газовой фазе.
Дифениламин можно получить декарбоксилированием дифенил-
амин-2-карбоновой кислоты при 180—210 °C в анилине [57], раз-
ложением фосфата анилина при 170—300 °C и разложением дифе-
нилбориодфторида в присутствии анилина [58], а также фотохи-
мическим синтезом при облучении N-алкилдифениламина УФ-све-
том в гексане [59].
Дифениламин с высоким выходом (93%) получен при обработ-
ке соли анилина солями ароматических сульфокислот [60]:
nh2
+NH2Na
- NH3
-f-CjjHgSOsNa ’
- NaaSO3
Несмотря на значительное число описанных методов, промыш-
ленным способом получения дифениламина во всех странах пока
остается араминирование анилина на разных катализаторах.
Технологическая схема получения [17]. Промышленный про-
цесс осуществляют (по непрерывной технологии) методом жидко-
фазной конденсации анилина при 300—340 °C и 1,5—2 МПа в при-
сутствии катализатора:
Выделяющийся аммиак отводят из системы. Наряду с целевой ре-
акцией протекает побочное образование смолистых веществ. Сте-
пень конверсии анилина 20—40%; селективность катализатора по
дифениламину 98%.
Технологический процесс (рис. 5) осуществляют в две стадии:
араминирование анилина в дифениламин под давлением и выде-
ление дифениламина из реакционной смеси.
Араминирование проводят по непрерывной схеме в автоклаве 7.
Свежий и возвратный анилин из емкости 3 через теплообменник 5
подают в автоклав. 50%-ный водный раствор катализатора (смесь
фторида и бифторида аммония с борной кислотой) из емкости 4
подают в низ автоклава по барботеру под слой реакционной смеси.
В автоклаве при 300—340 °C и давлении до 2 МПа происходит кон-
денсация анилина в дифениламин с выделением аммиака. Аммиак
вместе с парами воды и анилина отводят через дефлегматор 6,
емкость 8, холодильник 1 и приемник 2 и направляют на сжигание.
Реакционная смесь с 20—24% дифениламина из автоклава по
стояку поступает в емкость 8, где частично отделяют анилин дрос-
селированием. Из этой емкости дифениламин (»40%-ный) посту-
пает в ректификационную колонну 9, где при 102—120 °C в верхней
части, 215—240 °C в кубе и остаточном давлении 133—666 Па от-
гоняется анилин. Сырой дифениламин подают в ректификацион-
ную колонну 13 (температура в кубе 230—250 °C, в верху 200—
4—1057
49
Рис. 5. Технологическая схема получения дифениламина:
1, 10, 12— холодильники; 2, 11, 14 — приемники; 3, 4, 8 — емкости; 5 — теплообменник; 6 —
дефлегматор; 1 — автоклав; 9, 13 — ректификационные колонны.
205°C, остаточное давление 133—666 Па). Пары дифениламина
конденсируются в холодильнике /2; конденсат стекает в приемник
14 и подается на чешуирование (товарный продукт). Часть дифе-
ниламина подают на нитрозирование.
Следует иметь в виду, что в присутствии окислителей дифенил-
амин окисляется с образованием дифенилазотоксида
\ / —НО- \ / I
О-
который сам является эффективным стабилизатором.
Наряду с дифениламином образуются побочные продукты, име-
ющие более высокую температуру кипения, чем т. кип. дифенил-
амина, и концентрирующиеся в кубовом остатке. Среди них иден-
тифицирован карбазол. Общее количество смолообразных побоч-
ных продуктов доходит до 5% (считая на дифениламин).
Свойства и применение. Дифениламин представляет собой бе-
лый кристаллический продукт со слабым характерным запахом,
темнеющий на свету; плотность 1159 кг/м3 (при 20°C); т. пл.
54 °C; т. кип. 302 °C. Растворяется в ацетоне, бензоле, метаноле,
этаноле, сероуглероде, минеральных кислотах, диэтиловом эфире.
Горюч; нижний предел взрываемости паров в смеси с воздухом
0,7% (об.); т. самовоспл. 184°C, т. всп. 150°C.
Умеренно токсичен; ПДК 0,1 мг/м3.
Б0
Дифениламин более ста лет применяется для стабилизации по-
рохов и некоторых пищевых продуктов (рыбные консервы).
Эффективный стабилизатор многих полимерных материалов
(сополимеры формальдегида, полиолефины, эпоксидные полимеры,
поливинилхлорид), однако вследствие высокой летучести на прак-
тике применения не находит. Является эффективным ингибитором
фотодеструкции полихлоропреновой пленки и ингибитором поли-
меризации метилметакрилата.
Дифениламин является одним из основных видов сырья для
производства многих стабилизаторов полимерных материалов, кра-
сителей и пластических масс.
п-Гидроксидифенидамин
n-Гидроксидифениламин впервые был получен в 1883 г. Кальмом арамини-
рованием гидрохинона анилином:
NH.+HO—АД—ОН ——> NH—ОН
\ / \ / —нао \ / __/
Гидрохинон и анилин нагревали в течение 8—10 ч при 250^—260° С в присутствии
хлорида кальция и растворяли в соляной кислоте. Образовавшийся п-гидрокси-
дифениламин осаждали раствором ацетата натрия в виде желтоватых кристал-
лов, отфильтровывали, сушили и очищали перегонкой в токе водорода.
Большое влияние на этот процесс оказывают природа катализатора и тем-
пература. Уже Кальм отмечал, что с одним и тем же катализатором — хлори-
дом кальция — при 260°С преимущественно образуется и-гидроксидифенил-
амин, а при 290 °C — П.П'-дифенпл-п-фенплендиампн. Без катализатора эта
реакция протекает при более высокой температуре (300—320 °C) с преимущест-
венным образованием дифенил-п-фенилендиамина. Применение более кислотного
катализатора — хлорида цинка — приводит к снижению температуры до 200—
210 °C и также к образованию дифенил-п-фенилендиамина [8].
В патентах фирмы Du Pont (США) даны результаты изучения хлоридов
Zn, Fe, Al, Со, Ni, Cd, Mg, Hg, Ca, In и Ti в качестве катализаторов и отме-
чается, что лучшим является хлорид алюминия. Однако применение хлоридов
металлов, а также бисульфата калия, фосфорной кислоты, фосфатов металлов
[61], эфиров фосфорной кислоты [62, 63] и других кислотных катализаторов при-
водит к побочному образованию значительных количеств М,ЬГ-дифенпл-п-фени-
лендиамина и кислотных сточных вод и усложняет разделение реакционной сме-
си. Стадия, проводимая в автоклаве, не поддается переводу на непрерывный
режим.
В присутствии арплсульфокпслот (например, п-толуолсульфокислоты) п-гпдр-
оксидифениламин получают с выходом 98% и выше. В этом случае проводят
реакцию в присутствии органического растворителя (ксилол) и при темпера-
туре кипения реакционной массы [64].
В присутствии соединений титана и-гидроксидифениламин получают в жид-
кой фазе [5]. Смесь гидрохинона, анилина и сульфата титана, содержащего
50% Т1О2, 25% H2SO4 и 25% Н2О, нагревают в течение 4,5 ч при 185—250 °C.
Затем добавляют анилин и выдерживают 12 ч при 195—205 °C. Реакционную
смесь дважды промывают горячей водой и фильтруют. После перегонки получа-
ют п-гидроксидифениламин с выходом 36%. Наряду с этим образуются
М,М'-дифенпл-п-фенплендиампн (53,5%) и анилин (10%).
Получение п-гидроксидифениламина арамииированием. п-аминофенола
Изучено мало [65]. Хорошие выходы получены при использовании ароматических
сульфокислот, но сохраняются все недостатки, присущие кислотным катализа-
торам. Реакцию проводят в избытке анилина, побочным продуктом является
дифенил-п-фенилендиамин. Чтобы повысить выход n-гндроксидифениламина, пре-
рывают реакцию через 4—5 ч и подвергают смесь фракционной разгонке. Фрак-
цию, содержащую избыток анилина и непрореагировавший n-аминофенол, воз-
вращают в цикл, а n-гидроксидифениламин отделяют от менее летучего дифе-
нил-п-фенилендиамина.
n-Гидроксидифениламин может быть получен по реакции Ульмана нагрева-
нием п-аминофенола с бромбензолом в присутствии карбоната калия и иодида
меди [521:
Реакцию проводят в растворителе (амиловый или изоамиловый спирт, гли-
церин) и получают сравнительно чистый продукт.
Заслуживает внимания описанный в патенте фирмы Du Pont (США)
способ получения n-гидроксидифениламина каталитическим гидрированием в при-
сутствии фенола [661- В качестве катализаторов применяют мелко измельченный
палладий, платину или их оксиды в среде разбавленной серной кислоты в при-
сутствии четвертичных аммониевых соединений (хлорид или бромид тетраалкил-
аммония).
В промышленном масштабе л-гидроксидифениламин получают
по двум вариантам в зависимости от наличия сырья: 1) из анили-
на и п-аминофенола и 2) из гидрохинона и анилина.
Технологическая схема получения п-гидроксидифениламина из
анилина и д-аминофенола. Процесс протекает по схеме
NHj + HaN—<Л-ОН —Н4<>. NH—/~\-ОН
\=/ \=/ -nh3 W \-^/
Побочные реакции такие:
Технологический процесс состоит из следующих стадий: 1) при-
готовление суспензии л-аминофенола в анилине, 2) приготовление
водного раствора хлорида аммония, 3) араминирование и-аминофе-
нола анилином, 4) приготовление суспензии кальцинированной со-
ды в анилине, 5) дистилляция л-гидроксидифениламина.
Араминирование ведут в стальном эмалированном реакторе 4
(рис. 6), снабженном якорной мешалкой и рубашкой для нагрева-
ния жидким высокотемпературным теплоносителем (ВОТ). В ре-
актор при работающей мешалке принимают суспензию и-аминофе-
52
Рис. 6. Технологическая схема получения л-гидроксидифениламииа:
1 — аппарат для приготовления суспензии; 2 — дефлегматор; 3 — фазор азделитель; 4 — реак-
тор; 5, 1> 9 — холодильники; 6, 8, 10 — приемники; 11 — дистиллятор.
нола в анилине из аппарата 1, где предварительно от реакционной
смеси отгоняют воду. Массу в аппарате 4 нагревают до 185 °C и
выдерживают 14—20 ч, затем постепенно повышают температуру
до 209—212 °C и выдерживают еще 4 ч, после чего анализируют
реакционную массу. Готовую реакционную массу передают в ди-
стиллятор 11. Выход по стадии араминирования (реактор 4) со-
ставляет 70%
Выделяющиеся пары анилина, воды и аммиака проходят через
дистилляционную колонну (на схеме не изображена) и поступают
в дефлегматор 2. Там конденсируется анилин, который через фа-
зоразделитель 3 возвращается в реактор 4. Аммиак с примесью
анилина направляют в адсорбер, заполненный коксом.
Дистилляцию м-гидроксидифениламина проводят в дистилля-
торе И, куда при работающей мешалке принимают реакционную
массу из аппарата 4 и суспензию соды в анилине для нейтрализа-
ции хлорида аммония. Дистиллятор И герметизируют, создают
там разрежение и при работающей мешалке начинают фракцион-
ную разгонку. В процессе отгонки товарной фракции температура
застывания готового продукта начинает подниматься выше 62 °C и
затем постепенно понижается. При удовлетворительном анализе
n-гидроксидифениламин при ПО—120°С из приемника 10 направ-
ляют на розлив.
Технологическая схема получения д-гидроксидифениламина из
анилина и гидрохинона. Этот метод считают перспективным. В ка-
честве катализатора применяют сульфаниловую кислоту. Примене-
ние более или менее сильнокислотных катализаторов снижает вы-
ход n-гидроксидифениламина за счет увеличения выхода побочного
53
продукта — М,М'-дифенил-п-фенилендиамина. Схема аналогична
изображенной на рис. 6.
В реактор загружают анилин, гидрохинон и сульфаниловую
кислоту. Реакционную массу нагревают до 180—190 °C (до рас-
плавления), включают мешалку и продолжают нагревание. При
Г80—190 °C начинается реакция с отщеплением воды, что видно
по повышению температуры на выходе из дефлегматора. Воду (с
небольшим количеством анилина) отгоняют через колонну, деф-
легматор и холодильник в приемник. Температуру в реакторе в те-
чение 1,5 ч поднимают до 210 °C и еще за 13,5 ч до 220 °C. Затем
выдерживают 1 ч при 220—225 °C. Отогнанную воду и анилин раз-
деляют и направляют на рециркуляцию.
После охлаждения реакционной массы при перемешивании на-
чинают фракционную разгонку в вакууме (остаточное давление
2,7—3,3 кПа). Первую фракцию отгоняют при 205—240°C в кубе
и 100—207 °C в парах, вторую при 240—255 °C в кубе и при 207—
218 °C в парах, третью — товарную фракцию — при 255—270 и
218—225 °C соответственно. При отгонке третьей фракции темпе-
ратура затвердевания погона вначале растет и достигает 67—68 °C,
а затем начинает снижаться. При снижении температуры затвер-
девания до 60 °C переходят на прием четвертой фракции. Азотом
доводят давление до атмосферного и сливают готовый продукт в
барабаны. Четвертую фракцию отгоняют при 270—350 °C в кубе
и 225—270 °C в парах до прекращения отгонки. Выход л-гидрокси-
дифениламина составляет 70—75%
Свойства и применение. л-Гидроксидифениламин представляет
собой белый кристаллический порошок; т. пл. 74 °C; т. кип.
330 °C при атмосферном давлении и 216 °C при 1,6 кПа. Растворя-
ется в этиловом спирте, хлороформе, диэтиловом эфире, горячем
бензоле, разбавленных щелочах и кислотах; незначительно рас-
творяется в холодной воде и лигроине. На воздухе легко окисля-
ется.
Горюч; т. воспл. 217—219°C; т. всп. 192—194 °C; т. самовоспл.
580 °C. Взвешенная в воздухе пыль имеет нижний предел взрывае-
мости 6,25 мг/м3. Минимальное взрывоопасное содержание кисло-
рода 12% (об.).
Токсичен. Отравление возможно при вдыхании пыли и при по-
падании на кожу. ПДК в атмосфере рабочих помещений 0,5 мг/м3.
л-Гидроксидифениламин является стабилизатором синтетичес-
ких каучуков общего назначения. Он более эффективен, чем Нео-
зон Д. Обеспечивает термостабильность резин на основе натураль-
ного и синтетического каучуков; активность его повышается в
сочетании с феноло-формальдегидным полимером. Является инги-
битором полимеризации бутадиена, антиокислителем бензина, тур-
бинного и трансформаторного масел и топлив. л-Гидроксидифенил-
амин — один из важнейших видов сырья в производстве ряда ста-
билизаторов, органических красителей, фармацевтических препа-
ратов.
54
Nfl'-Дифенил-п-фенилендиамин
Впервые Кальм получил это соединение в 1883 г., нагревая в запаянной
трубке гидрохинон с анилином при 200—210 °C в присутствии хлорида цинка.
Наряду с дифенил-п-фенилендиамином образовывались значительные количества
л-гидроксидифениламина:
Продукт араминирования выделяли, обрабатывая реакционную массу разбав-
ленной соляной кислотой, а затем очищали кипячением с раствором едкого нат-
ра и дважды перекристаллизовывали из бензола. Температура плавления полу-
ченного таким образом М.М'-дифенил-гг-фенилендиамина составляла 152 °C.
Эффективными катализаторами кроме хлоридов кальция и цин-
ка [8, 67—70] являются хлориды алюминия [71] и железа [72—
75]. С хлоридом цинка выход N.N'-дифенил-н-фенилендиамина в
среднем составляет 84% (т. пл. 126—127°C), а в присутствии хло-
рида алюминия достигает 85% (т. пл. 133—134°C). На хлориде
железа М,М'-дифенил-и-фенилендиамин получен с выходом «38%
(т. пл. 134—140°C). Железо осаждают в виде нерастворимой соли
или в виде гидроксида.
Изучен [67—70] процесс получения М.М'-дифенил-л-фениленди-
амина взаимодействием анилина с гидрохиноном в присутствии
хлорида цинка при 185—250 °C. Реакцию проводили при мольном
соотношении гидрохинон : анилин : хлорид цинка, равном 1:2,13:
: 0,19, и 230 °C в среде технического дихлорбензола. Для сокраще-
ния времени реакции и подавления побочных процессов вводили
анилин в два приема. М,Ы'-Дифенил-/г-фенилендиамин образовы-
вался с выходом 92% (т. пл. 130°C). В тех же условиях, но с ис-
пользованием хлорида алюминия выход продукта составлял 69%
(т. пл. 138—140°C).
Таким образом, наиболее эффективными катализаторами из
хлоридов металлов являются хлориды цинка и алюминия, причем
‘[54] хлорид цинка более подходит для первой стадии
но-^Л-он+^^А но-АЛ—nh
а хлорид алюминия — для второй:
55
Хлориды металлов, как катализаторы араминирования, имеют
ряд недостатков: выше 200 °C в условиях реакции они разлагают-
ся с образованием хлорида аммония, который отгоняется из реак-
ционной массы вместе с анилином. Катализатор отделяется полно-
стью только после многократной отмывки водой, что приводит к
образованию большого количества сточных вод.
Применение соляной, серной, борной и фосфорной кислот,
фосфатов [61—63], сульфатов, оксалатов в качестве катализаторов
приводит к преимущественному образованию /г-гидроксидифенил-
амина. Триалкилфосфаты в меньшей степени способствуют образо-
ванию побочных смолистых веществ, чем другие перечисленные ка-
тализаторы [68], и выход М,М'-дифенил-п-фенилендиамина повы-
шается.
ЙТМ'-Дифенил-п-фенилендиамин (Диафен ФФ) получается из
гидрохинона и анилина в присутствии различных оксидов. Так, на
оксиде железа ведут процесс при 340 °C под давлением; выход про-
дукта 92% (т. пл. 132—138 °C) [72]. Есть также данные по при-
менению смеси оксидов молибдена и никеля [76]; носителем слу-
жит SiO2-bAl2O3, а процесс ведут при 300 °C в течение 4 ч под
давлением.
В последнее время появились сведения по получению N.N'-ди-
фенил-л-фенилендиамина в присутствии катализатора, который го-
товят, обрабатывая у-А12О3 10%-ным раствором борной [77] или
карбоновой кислоты [78].
Араминирование гидрохинона проводят еще и в присутствии
катализаторов, которые в качестве носителя содержат А12О3 пли
SiO2 [79]. Высокоактивной и высокоселективной каталитической
системой при араминировании, в том числе и в случае гидрохино-
на, является смесь оксидов титана и кремния [80]. Каталитическое
араминирование гидрохинона обычно осуществляют при 200—
300 °C и атмосферном или повышенном давлении. Реакцию удобно
проводить в присутствии растворителей, способных образовывать
азеотропные смеси с водой (толуол, бензол, ксилол, хлорбензол).
В 1940-х годах в Японии (фирма Seiko) М,М'-дифенил-/г-фени-
лендиамин получали полунепрерывным методом. Анилин и гидро-
хинон подвергали араминированию при повышенном давлении в
присутствии специально приготовленного катализатора (раствор
катализатора не стабилен при длительном хранении). Непрореаги-
ровавший анилин отгоняют, а Н.Н'-дифенил-н-фенилендиамин по-
сле очистки подвергают вакуумной перегонке и получают чистый
продукт.
С 1960-х годов на заводе фирмы lavata (Япония) получают
ЬкМ'-дифенил-и-фенилендиамин следующим образом. Гидрохинон,
анилин и катализатор смешивают в специальном аппарате. Раствор
подогревают до необходимой температуры и непрерывно подают
в реакторы. В каждом из трех реакторов поддерживают заданные
температуру и время контакта. Пары избыточного анилина и об-
разующейся воды удаляют через вентиль для регулирования
56
давления и конденсируют. Конденсат разделяют на две фазы и ани-
линовую фазу возвращают в цикл. В испарителе, понижая давле-
ние до атмосферного, из реакционной массы удаляют непрореаги-
ровавший анилин. Побочный продукт — дифениламин — конденси-
руют и собирают в емкости.
Оставшаяся реакционная масса содержит промежуточный
и-ги^роксидифениламин, Ы,К'-дифенил-п-фенилендиамин, катали-
затор и небольшое количество дифениламина. Эту смесь подверга-.
ют вакуум-разгонке. Первым отгоняется л-гидроксидифениламин,
который конденсируют. Затем отгоняется смесь л-гидроксидифе-
ниламина и Ы,М'-дифенил-л-фенилендиамина и, наконец, целевой
продукт — Ы.ЬК-дифенил-п-фенилендиамин.
Жидкий Н.Ы'-дифенил-п-фенилендиамин непрерывно подают в
аппараты для чешуировання, а л-гидроксидифениламин возвраща-
ют в реактор.
Другой способ получения Ь^Ы'-дифенил-и-фенилендиамина —
взаимодействие л-аминофенола и анилина — был описан в 1938 г.:
л-Аминофенол нагревают в автоклаве с хлоридом аммония в при-
сутствии избытка анилина. При 250—275 °C выделяется аммиак,
который выпускают через предохранительный клапан. Затем тем-
пературу поднимают до 300 °C и выдерживают до прекращения
выделения воды. Смесь охлаждают и обрабатывают щелочью. Хло-
рид аммония отфильтровывают, а фильтрат подвергают фракцион-
ной разгонке.
Нагреванием солянокислого л-аминофенола с избытком анили-
на при 210—220 °C и интенсивном перемешивании в запаянной
трубке получен Г^М'-дифенил-л-фенилендиамин с почти теоретиче-
ским выходом (т. пл. 141 °C). Процесс протекает практически без
побочных реакций [81]. В качестве катализаторов описано приме-
нение соляной кислоты, иода, железа и хлорного железа [82].
Реакция идет в избытке анилина через стадию образования и-гидр-
оксидифениламина:
+СбН5МН2
—н2о
+CaH5NH2
- nh3
В промышленном масштабе араминирование л-аминофенола
осуществляют следующим образом. В стальной автоклав с кера-
мическим вкладышем загружают л-аминофенол, анилин и хлорид
аммония. Реакционную массу при перемешивании нагревают до
230—240 °C. Образующиеся аммиак и воду, а также избыточный
57
анилин отгоняют из реактора через холодильник в соответствую-
щие приемники.
Как видно из приведенной схемы, араминирование л-аминофе-
нола идет в две стадии: вначале образуется л-гидроксидифенил-
амин, который, вступая в реакцию со второй молекулой анилина,
дает Ы,М'-дифенил-л-фенилендиамин. Обе эти реакции идут одна
за другой в автоклаве при 220—240 °C и 0,2—0,25 МПа с 10-часо-
вым выдерживанием.
В присутствии хлоридов протекает и побочная реакция:
NHa+ZnCl2 ---> NH2j-ZnCl2
При выделении и промывке М,М'-дифенил-л-фенилендиамина по-
бочный комплекс разлагается с регенерацией катализатора:
Z»CI, + 2HC1
2
-NHj-HCl
Поскольку араминирование гидрохинона и л-аминофенола про-
текает через стадию n-гидроксидифениламина, разработан про-
мышленный процесс получения Ы,Ы'-дифенил-л-фенилендиамина
непосредственно из л-гидроксидифениламина.
Еще один способ получения П,М'-дифенил-л-фенилендиамина—
восстановление л-аминоазобензола [63]. При этом образуется
смесь анилина и л-фенилендиамина, которая при многочасовом ки-
пячении с концентрированной соляной кислотой дает N.N'-дифенил-
л-фенилендиамин:
+НС1
—ын4а*
М,Ы'-Дпфенил-л-фенилендиамин получается также араминиро-
ванием 5-амино-2-гидроксибензойной кислоты анилином или соля-
нокислым анилином при 210 °C
—СОа; —НаО; —NH4C1
с образованием n-гидроксидифениламина в качестве побочного
продукта.
Интересен способ получения М,М'-дифенил-и-фенилендиамина
По Ульману из ацетанилида и л-дибромбензола с отгонкой реакци-
58
онной воды; Образующееся диацетильное производное подвергают
гидролизу:
2 NH-CO—СН3 + Вг—Вг ——
\ / 3 1 \ / —2НВГ
Ацетанилид, днбромбензол, безводный карбонат калия, медный
порошок и иодид калия нагревают в течение 15 ч в реакторе до
170 °C. При этой температуре через каждые 3 ч в реакционную
смесь добавляют толуол. Выдерживают несколько часов, затем
охлаждают реакционную массу до 60 °C, прибавляют воду и бен-
зол, отфильтровывают медный порошок. Из фильтрата с водяным
паром отгоняют бензол и избыток n-дибромбензола; остаток пере-
мешивают с этилацетатом и отфильтровывают диацетил-И.И'-днфе-
нил-л-фенилендиамин (т. пл. 152°C; из спирта).
Чистое диацетильное производное кипятят с концентрирован-
ной соляной кислотой и этиловым спиртом в течение 2—3 ч. Спирт
отгоняют, остаток сушат, полученный сухой продукт разбавляют
водой и кипятят в течение 1 ч. Образовавшийся М.М'-дифенил-п-
фенилендиамин кристаллизуют из спирта и получают продукт
(т. пл. 152°C) с выходом 60—65% [83].
По другому варианту М,М'-дифенил-л-фенилендиамин получают
кипячением (с обратным холодильником) при 140—150 °C антра-
ниловой кислоты с л-дибромбензолом в аллиловом спирте в при-
сутствии безводного карбоната калия, медного порошка и хлорида
меди:
При наличии соответствующих исходных соединений получение
М,1\Г-дифенил-л-фенилендиамина конденсацией по Ульману может
быть оформлено как промышленный процесс.
Есть и еще способы получения М.М'-дифенил-и-фениленди-
амина — смешение спиртового раствора л-нитрозодифениламина с
фенилгидразином, а также восстановление спирто-щелочного рас-
твора хинондианила (дифенил-л-азофенилен) цинковой пылью.
Ь^М'-дифенил-н-фенилендиамин получается наряду с азофенилом
при нагревании хинондианила с анилином.
С невысоким выходом («20%) N.N'-дифенил-л-фениленди-
амин получается из динатриевой соли бензолдисульфокислоты —
59
Вода.
Рис. 7. Технологическая схема получения Ф^М'-дифенил-п-фепилендиамина:
реактор-конденсатор; 2. 9 — холодильники; 3, 6, 10 — ирнемннкн; 4 — выделитель; 5 -
нутч-фильтр; 7 — емкость; 8 — перегонный куб.
нагреванием с анилидом натрия (по аналогии с методом щелоч
ного Плавления) [13]:
NaO3S-f3-SO8Na + 2 Q-NHNa
М.М'-Дифенил-п-фенилендиамин с т. пл. 100—102 °C получают
также каталитическим восстановительным алкилированием нитро-
бензола циклогексанолом в присутствии палладия на угле [24].
Технологическая схема получения (рис. 7). В реактор-конден-
сатор 1 (стальной котел с эмалированным вкладышем, снабжен-
ный якорной мешалкой, электрообогревом и змеевиком для обо-
грева или охлаждения паром или водой) загружают хлорид цинка
(в виде 49%-ного водного раствора) и полихлориды бензола. Кон-
денсатор герметизируют и отгоняют воду при остаточном давле-
нии не менее 53 кПа до ее содержания не более 0,3%. Пары воды
и полихлоридов бензола конденсируются в холодильнике 2 и сте-
кают в приемник 3. По окончании отгонки воды охлаждают массу
до 30—50 °C, останавливают мешалку и через мерник загружают
анилин, а через люк — н-гидроксидифеииламин. Конденсатор про-
дувают азотом, герметизируют, нагревают до 100—120 °C, вклю-
чают мешалку и при этих условиях выдерживают в течение 10—
12 ч. Затем смесь охлаждают до 90—100 °C, разбавляют водой и
передавливают в выделитель 4 на 7%-ный раствор соляной кисло-
ты, нагретый до 55—65 °C.
60
Суспензию в выделителе нагревают до 80±5°С и перемешива-
ют в течение 2 ч, после чего отстаивают. Верхний (водный) слой
по короткой трубе сжатым воздухом выдавливают в нутч-фильтр
5 для улавливания уходящего с раствором Диафена ФФ, который
затем возвращают в выделитель 4.
Оставшуюся в выделителе 4 суспензию Диафена ФФ в поли-
хлоридах бензола несколько раз промывают водой до нейтральной
среды. Последнюю промывную воду используют (после отделе-
ния суспензии Диафена ФФ) для приготовления солянокислого
раствора для следующей операции выделения. Промывные воды из
нутч-фильтра 5 отфильтровывают в приемник 6, откуда их по мере
надобности передают в емкость 7 для дополнительного отстаива-
ния от полихлоридов бензола. Отстоявшиеся полихлориды из ниж-
ней части емкости 7 сжатым воздухом передавливают в перегонный
куб 8.
Промытую в выделителе 4 суспензию Диафена ФФ сливают
на нутч-фильтр 5. Фильтрование ведут при остаточном давлении
53 кПа в приемник 6, откуда маточный раствор передавливают
сжатым воздухом в куб 8 для вакуум-перегонки. Пасту на нутч-
фильтре промывают полихлоридами бензола и отжимают до со-
держания летучих веществ не более 60%•
Выход готового продукта после сушки составляет 57,4% (от
теоретического по /г-гидроксидифениламину).
Свойства и применение. П.ЬУ-Дифенил-лг-фенилендиамин пред-
ставляет собой темно-серый порошок (т. пл. 152°C). Растворяется
в ацетоне, бензоле, хлороформе, этаноле, бутаноле, метаноле, про-
паноле, толуоле, ксилолах, анилине, горячем хлорбензоле, поли-
хлоридах бензола, этиленгликоле.
Горюч. Пыль пожароопасна. Аэровзвесь взрывоопасна; ниж-
ний предел ее взрываемости 15,6 г/м3 (по другим данным,
26,3 г/м3). Т. воспл. 870°C, т. самовоспл. пыли 600°C.
Умеренно токсичен, ЛДбо= 18500 мг/кг. Оказывает раздражаю-
щее действие на кожу, слизистые оболочки глаз и верхних дыха-
тельных путей.
М,П'-Дифенил-п-фенилендиамин выпускается во многих стра-
нах под разными названиями в виде темно-серого порошка или
гранул с т. пл. от 125 до 152 °C.
Этот стабилизатор плохо растворим в полимерах, поэтому це-
лесообразно применять его в сочетании с другими, лучше раство-
римыми стабилизаторами. За рубежом выпускается до 40 наиме-
нований готовых промышленных смесей П.П'-дифенил-п-фенилен-
диамина с фенил-р-нафтиламином. (Antioxidant 18, Nocrac HP),
с продуктом взаимодействия дифениламина с ацетоном (Flexami-
пе), с 2,2,4-триметил-1,2-дигидрохинолином (Perflectol), с фенил-
Р-нафтиламином и диметоксидифениламином (Thermoflex А).
М,М'-Дифенил-н-фенилендиамин — широко распространенный
стабилизатор синтетических каучуков общего назначения; защи-
щает резины на основе этих каучуков от термоокислительного ста-
61
I
Торговое наименование Страна, фирма T. пл., °C
Диафен ФФ СССР 133 (II сорт), 136 (I сорт)
Agerite DPPD США, Vanderbilt 145—152
Altofane DIP Франция, Kuhlmann 134
Antage DP Япония, Kawaguchi 144—145
Antigene P Япония, Sumitomo 140
Antioxygene DIP Франция, Mat. Col. 134
DPPD Англия, Monsanto 134
Good-Rite Caromax США, Goodrich 144
Hoboken США, Goodrich
Inhibitor OB Италия, ACNA Montecatini 125-130
JZF США, Naugatuck 138—152
Nocrac DP Япония, Ouchi Shinko 140
Nonflex H Япония, Seiko 145—150
Nonox DPPD Англия, ICI «130
Perflectol X США, Monsanto
Permanax 18 Франция, Rhone Poulenc 1 135
рения и разрушения при многократных дефдрмациях. Пассивирует
действие металлов переменной валентности. Термостабилизатор
полиэтилена и полиамидов. Ингибитор полимеризации эфиров не-
насыщенных карбоновых кислот. Стабилизатор ракетных топлив
и смазочных масел.
Д,Д'-Ди-р-нафтил-п-фенилендиамин
М.М'-Ди-р-иафтил-п-фенилеидиамин впервые был получен в 1889 г. Руфом
араминироваиием р-нафтола л-фенилендиамином. Реакцию проводили при мно-
гочасовом нагревании n-фенилендиамина с избытком р-иафтола и последующей
обработке расплава разбавленной щелочью, кислотой, этанолом и диэтиловым
эфиром. После двух-трехкратной перекристаллизации из горячего анилина был
получен продукт с т. пл. 235 °C; выход очищенного М,М'-ди-Р-пафтил-л-фепилеп-
диамипа 40%.
В промышленном масштабе N^N'-дн-Р-нафтил-п-феннлендиамни начали вы-
пускать с 1910 г. в Германии. Метод заключался в выдерживании п-фенилепди-
амина с р-нафтолом и 2—3 масс. ч. иода (катализатор) при 200 °C (до оконча-
нии выделения реакционной воды) и при 260 °C. Выход технического продукта
-был почти теоретический; выход очищенного продукта 88%.
М.П'-Ди-р-нафтил-п-фенилендиамин получали нагреванием р-нафтола
с и-фенилендиамином в присутствии металлического иода при 200 °C в тече-
ние 14 ч; плав измельчали в порошок. Выход составил 72%. Для очистки про-
дукта был применен метод Руфа однако обработка реакционной массы щелочью,
кислотой, этиловым спиртом и диэтиловым эфиром позволяла получать N.N'-ди-
Р-нафтил-п-фенилендиамин с т. пл. не выше 224 °C. В дальнейшем 1Ч,М'-ди-Р-
нафтил-п-фенилендиамин был получен в аналогичных условиях с катализатора-
ми и без них [70, 71, 84—87].
Получение Ы,Ы'-ди-р-нафтил-п-фенилендиамина сводится к ара*
минированию р-нафтола, взятого в избытке, л-фенилендиамином
при 200—300 °C. Присутствие катализаторов (НС1, H2SO4, Н3ВО3,
KHSO4, ZnCl2, AICI3, В2О3) позволяет вести процесс при темпера-
туре ниже 300 °C; в то же время повышение температуры до 300 °C
при расплавлении увеличивает выход продукта. Практическое осу-
ществление метода связано с трудностями при выгрузке и даль-
нейшей обработке расплава для выделения целевого продукта.
В зарубежной литературе выделение Ы,Ы'-ди-р-нафтил-л-фени-
лендиамина из расплава, застывающего в твердую массу при
200 °C, мало освещено. Есть указания на то, что в инертной атмо-
сфере (например, в атмосфере СО2) можно получить это соедине-
ние с т. пл. 234 °C, без специальной обработки [84].
По способу фирмы ICI (Англия) [88] N.N'-ди-р-нафтил-л-фе-
нилендиамин выделяют из расплава, добавляя его к подкисленной
воде, содержащей поверхностно-активное вещество, при 50—100 °C
и перемешивании. В качестве поверхностно-активного вещества ре-
комендуют применять натриевые соли сульфированных эфиров
олеиновой или рицинолевой кислот, продукты конденсации цетил-
стеарилового спирта с оксидом этилена. Получается легкосмачи-
ваемый порошок.
Фирмой Goodrich (США) выделение М,М'-ди-р-нафтил-л-фени-
лендиамина из расплава проводят так. Расплав выливают на ме-
таллическую поверхность для застывания, массу измельчают в по-
рошок, который затем смешивают"с растворителем (75% ксилола+
+25% метанола) и вновь нагревают; полученный расплав охлаж-
дают до 18—20 °C и фильтруют. Осадок промывают метанолом и
сушат при 60 °C и остаточном давлении 13,3 Па [87].
Предложено также выделять М,М'-ди-р-нафтил-л-фениленди-
амин, выливая расплав в этиленгликоль [89—91] или в полихло-
риды бензола [71]i.
Первый способ заключается в следующем. Полученный расплав
с температурой 300 °C при интенсивном перемешивании выливают
в этиленгликоль, предварительно нагретый до 150 °C. В результа-
те образуется суспензия М,М'-ди-р-нафтил-л-фенилендиамина в
этиленгликоле. Избыточный р-нафтол, непрореагировавший л-фе-
нилендиамин и образующаяся смола растворяются. Суспензию про-
дукта отфильтровывают, пасту промывают последовательно эти-
ленгликолем (150°С) и водой (95—100°С) и высушивают в ваку-
уме. Получают продукт с выходом 90—95% от теоретического
(в расчете на л-фенилендиамин); т. пл. не ниже 228 °C. Этиленгли-
коль регенерируют. Преимущества предложенного метода — воз-
можность получения достаточно чистого продукта без дополни-
тельной перекристаллизации из анилина или нитробензола, отсут-
ствие стадий дробления и размола, быстрота проведения и хорошая
воспроизводимость технологического процесса.
По второму способу выделение М,ЬГ-ди-р-нафтил-л-фениленди-
амина идет с применением растворителя и катализатора. В сталь-
63
ной сосуд загружают р-нафтол, /г-фенилендиамин, иод и техничес-
кий дихлорбензол. Реакционную массу в течение 6 ч нагревают
до 200—205 °C. При 160 °C начинают отгонять дихлорбензол и
воду. По достижении 200—205 °C ведут процесс при непрерывном
кипении еще 9 ч, после чего массу выливают в дихлорбензол, на-
гревают до 150 °C и при этой температуре размешивают 30 мин до
растворения продукта. Выкристаллизовавшийся М,М'-ди-р-нафтил-
л-фенилендиамин охлаждают и фильтруют. Осадок промывают
подогретым до 120 °C дихлорбензолом, из пасты отгоняют с паром
остатки растворителя, фильтруют и сушат при 100—120 °C. Выход
продукта в расчете на п-фенилендиамин достигает 89—90%; т. пл.
230 °C. Дихлорбензол подвергают разгонке в вакууме: сначала от-
гоняют дихлорбензол, а затем р-нафтол. Выход дихлорбензола
90% от загруженного, количество регенерированного р-нафтола
20%.
Описанную реакцию можно осуществлять и без катализатора.
При этом процесс протекает медленнее и выход снижается незна-
чительно, но несколько увеличивается смолообразование. Наи-
лучшие результаты достигнуты, когда в качестве катализатора
применяли л-толуолсульфокислоту или твердый хлорид цинка.
М,Ь1'-Ди-р-нафтил-л-фенилендиамин часто получается загряз-
ненным р-нафтолом и канцерогенным р-нафтиламином.
Один из способов очистки продукта от этих нежелательных примесей описан
в патенте [84]. 0-Нафтол, п-феиилеидиамии и В2О3 помещают в реакционный
сосуд и перемешивают смесь 4 ч при 232 °C. Расплав выливают в алюминиевый
чан, где ои охлаждается и затвердевает. Отвердевшую смесь измельчают, сме-
шивают порошок с ксилолом, охлаждают взвесь до 20 °C и фильтруют. Влажный ,
продукт промывают метаиолом, высушивают при 60 °C и 13,3 Па. Содержание
Р-иафтнламина 1,4-10~4%, содержание р-нафтола <0,05%; в продукте, выделен-
ном из ксилола, остается 1,7-10_‘% р-иафтиламина и <0,05% р-нафтола. .
Для облегчения транспортирования, непрерывного дозирования
М,№-ди->р-нафтил-л-фенилендиамина и улучшения условий работы i
с ним целесообразно применять гранулированный продукт. Хоро- !
ший эффект дает способ, по которому гранулирование осуществ- !
ляют, медленно добавляя расплав продукта к водному раствору
гидрофильного высокомолекулярного соединения или к смешанно-
му растворителю (этиловый спирт, алифатический кетон, диэтило-
вый эфир и вода). Смесь диспергируют и доводят размер частиц
до 0,1—5 мм.
Технологическая схема получения. В промышленности М,М'-ди-
р-нафтил-п-фенилендиамин (Диафен НН) получают араминиро-
ванием р-нафтола л-фенилендиамином:
64
Рис. 8. Технологическая схема получения М,К'-ди-Р-нафтил-п-феиилендиамииа:
1 — реактор; 2, 4 — холодильники; 3, 10, // — сборники; 5 — кристаллизатор; 5-друк-
фильтр; 7 —сушилка; 8 — мельница; 9 — ёмкость.
В стальной реактор 1 (рис. 8) загружают чешуврованный или
расплавленный р-нафтол и л-фенплендиамин и при перемешивании
нагревают реакционную массу до 220 °C. Выдерживают смесь в
течение нескольких часов, постепенно поднимая температуру до
270 °C. Затем массу нагревают до 300 °C и снова выдерживают.
Пары воды, выделяющиеся при реакции, конденсируются в холо-
дильнике 2; конденсат собирается в сборнике 3.
По окончании процесса полученный расплав Н,М'-ди-[3-нафтил-
л-фенилендиамина сливают в кристаллизатор 5 на нагретую до
120 °C смесь анилина с этиленгликолем. При перемешивании на-
гревают массу до 150рС и после 30 мин выдерживания охлаждают
до 30—40 °C. Суспензию готового продукта фильтруют на друк-
фильтре 6, снабженном поднимающейся гребковой мешалкой. Па-
сту промывают подогретым до 40 °C анилином и подогретым до
150 °C этиленгликолем, а затем горячей водой. Промывные воды,
анилин и этиленгликоль собирают в сборники 10 и 11 и направля-
ют на регенерацию. Пасту Диафена НН передают на вакуум-
гребковую сушилку 7 типа «Венулет» и после сушки и размола на-
правляют на упаковку.
Свойства и применение. Н,М'-Ди-|3-нафтил-л-фенилендиамин
представляет собой светло-серый порошок; плотность 1140—
1170 кг/м3; т. пл. 235 °C; т. кип. выше 400 °C (без разл). Раство-
5—1057 ' 65
ряется в кипящей уксусной кислоте, концентрированной серной кис-
лоте, анилине, ацетоне, бензоле, кумоле, нитробензоле, хлорофор-
ме, горячих полихлоридах бензола.
Горюч; т. воспл. 420 °C; т. самовоспл. 614 °C (гель) и 820 °C
(аэровзвесь); минимальное взрывоопасное содержание кислорода
при разбавлении пыле-воздушной смеси азотом 13,0% (об.). Ниж-
ний предел взрываемости 45 г/м3.
Токсичен; ЛДбо=2ООО мг/кг.
М.ЬГ-Ди-р-нафтил-п-фенилендиамин выпускается во многих
странах в виде светло-серого порошка под многими торговыми на-
именованиями, различающимися по температуре плавления:
Торговое наименование Страна, фирма T. пл„ °C
Днафен НН СССР 230
Agerite White Англия, Anchor 224—230
Antage F Япония, Kawaguchi 228—234
Antigene F Япония, Sumitomo >227
Antioxidant 123 Англия, Anchor 228—234
Antioxidant DNPD ASM DNP ГДР ФРГ, Bayer 220
DNPPD Франция, Soc. Prod. 220—237
Good-Rite AO 3120 США, Goodrich 225
Nocrac White Япония, Ouchi Shinko 225
Nonflex F, PF Япония, Seiko 225—230
Nonox CI Англия, ICI 228—235
Oxystop DNPPD, N Франция, SAFIC 230
Santowhite CI Англия, Monsanto >235
За рубежом выпускаются смеси антиоксидантов, составной ча-
стью которых является М,М'-ди-р-нафтил-л-фенилендиамин: с 2,5-
ди-трет-бутилгидрохиноном (Agerite White D), с фенил-р-нафтил-
амином (STD-Х), а также с фенил-р-нафтиламином и дифенил-л-
фенилендиамином (Nonflex К).
Н,№-Ди-р-нафтил-л-феннлендиамин — широко распространен-
ный стабилизатор синтетических каучуков общего назначения. Эф-
фективность повышается в сочетании со стабилизаторами феноль-
ного типа.
Термостабилизатор резин на основе натурального и синтетичес-
ких каучуков. Пассивирует действие металлов переменной валент-
ности.
Термо- и светостабилизатор полипропилена, пентапласта, поли-
оксипропилена, полиацетальдегида, высокомолекулярного полиок-
сиэтилена. Снижает скорость светодеструкции отвержденных эпок-
сидных покрытий. Термостабилизатор полипропиленового и поли-
амидных волокон, повышает усталостные свойства полиамидного
корда, но снижает адгезию корда к резине. Повышает стабиль-
ность химических волокон к атмосферным воздействиям. Ингиби-
тор полимеризации эфиров ненасыщенных карбоновых кислот.
Важнейший вид сырья при получении прочных красителей для
шерсти.
ЛИТЕРАТУРА
1. Фирц-Давид Г. Е., Бланжё Л. Основные процессы синтеза красителей. Пер.
с иемецкого/Под ред. С. В. Богданова и И. В. Фодимана. М., Издатинлит,
1957, с. 189—190.
2. BASF, пат. 848196, 1952 г. (ФРГ); С.А., 1953, V. 47, р. 5138.
3. А.с. 154862, 1962 г. (СССР).
4 Monsanto, пат. 3170956, 1963 г. (США); РЖХим, 1966, 24Н466.
5. Goodrich, пат. 2824137, 1955 г. (США); РЖХим, 1959, 79448.
6 Tsutsumi S., Yamamoto Н. — Technol. Repts, Osaka Univ., 1954, v. 4, p. 63—
66; C.A., 1956, v. 50, p. 271.
7. Иоффе И. И. и др. — Проблемы кинетики и катализа, 1960, № 10, с. 294—
297; РЖХим, 1960, 83986.
8. Sofragerm, пат. 1179775, 1959 г. (Франция); РЖХим, 1961, 19Л160.
9. Богданов С. В., Карандашева И. И. — ЖОХ, 1956, т. 26, с. 3365. *
10. Huaxue Shijie, 1959, v. 14, № 7, р. 343—344; РЖХим, 1960, 5512.
11. BASF, заявка 2544504, 1977 г. (ФРГ); РЖХим, 1978, 4Н172.
12. Добровольский С. В. и др. — В кн.: Сборник трудов НИОПиК. М., 1961.
Вып. 2, с. 148—150.
13. А.с. 498288, 1976 г. (СССР); РЖХим, 1977, ЗН215.
14. Ворожцов И. И. Основы синтеза промежуточных продуктов и красителей. М.,
Госхимиздат, 1955. 470 с.
15. Трунина Т. А., Маслова И. П., Скрипко Л. А,-—В кн.: Синтез и исследова-
ние эффективности химикатов для полимерных материалов. Тамбов, Тамбов-
ская правда, 1970. Вып. 4, с. 81—111.
16. Чекалин М. А., Пассет Б. В., Иоффе Б. А. Технология органических красите-
лей и промежуточных продуктов. Л., Химия, 1972. с. 214—215.
17. Гурвич Я. А., Ку мок С. Т. Химия и технология промежуточных продуктов,
органических красителей и химикатов для полимерных материалов. М., Выс-
шая школа, 1974, с. 156—157.
18. ICI, пат. 1385474, 1975 г. (Англия); РЖХим, 1976, 3H138.
19. Пестов Г. Н. Физико-химические свойства зернистых и порошкообразных хи-
мических продуктов. Л., Изд. АН СССР, 1947. 280 с.
20. Rubber, 1972, v. 154, № 12, р. 40а. Экспр.-инф. резино-техн, пром-ти, 1973,
№ 3, с. 31.
21. Yoshitomi, пат. 48-3210, 1973 г. (Япония); РЖХим, 1973, 22С525.
22. Yoshitomi, пат. 48-14650, 1973 г. (Япония); РЖХим, 1974, ЗС429.
23. Kummer R., Tordoir W. F. — Tijdschrift voor Sociale geneeskunde, 1975, B. 53,
№ 13, S. 415—419.
24. Monsanto, пат. 3219702, 1965 г. (США); РЖХим, 1967, 7Н145.
25. Ворожцов И. Н. — Хим. пром-сть, 1947, № 6, с. 20—25.
26. Grimmel Н. W., Guenther A., Morgan I. F. — J. Am. Chem. Soc., 1946, v. 68.
p. 539.
27. Du Pont, пат. 644938, 1950 г. (Англия); C.A., 1951, v. 45, p. 4742.
28. Koppers, пат. 2514430, 1950 г, (США); С. А., 1950, v. 44, р. 9478.
29. Koppers, пат. 2645662, 1953 г. (США); РЖХим, 1955, № 3, 4597.
30. Koppers, пат. 2656389, 1953 г. (США); РЖХим, 1954, № 22, 48974.
31. А.с. 16246 (НРБ); РЖХим, 1978, 22Н199.
32. Cyanamid, пат. 3079439, 1963 г. (США); РЖХим, 1965, 5Н161.
33. Cyanamid, пат. 3118944, 1964 г. (США); С.А., 1960, V. 60, р. 11939.
34. ICI, пат. 1439838, 1976 г. (Англия) .
35. Ogasavara S. е. а. — J. Chem. Soc. Japan, 1971, v. 74, № 3, p. 385—390-
РЖХим, 1971, 23Б1150.
36. Ogasavara S. e. a.— J. Catal., 1972, v. 25, № 1, p. 105—110; РЖХим. 1972.
20Б962.
37. Пат. 55583, 1973 г. (CPP); РЖХим, 1975, 17H209.
38. BASF, пат. 1187628, 1965 г. (ФРГ); РЖХим, 1967, 7Н147.
39. BASF, пат. 1219034, 1967 г. (ФРГ); РЖХим, 1968, 14Н199.
40. 1CI, пат. 1402707, 1972 г. (Англия); РЖХим, 1976, 8Н180.
41. Hoelscher Н. Е., Chamberlain D. F. — Ind. Eng. Chem., 1950, v. 42, p. 1558.
5* 67
42 Hoelscher H. E. — Ind. Eng. Chem., 1951, v. 43, p. 1828.
43. Vriens G. N„ Hill A. G. —Ind. Eng. Chem., 1952, v. 44, p. 2732.
44. Goodrich, пат. 3071619, 1963 г. (США); РЖХим, 1964, 17Н71.
45. А.с. 411743, 1978 г. (СССР); РЖХим, 1978, 22Н198; заявка 2503680, 1976 г.
(ФРГ); Изобр. за рубежом, 1976, вып. 24, № 16, с. 172.
46. Bayer, заявка 2521293, 1976 г. (ФРГ); С.А., 1977, v. 86, р. 55145.
47. Sinnippon rika, пат. 48-102340, 1977 г. (Япония); РЖХим, 1978, 5Н220.
48. Заявка 2042774, 1971 г. (ФРГ); приор. Швейцарии; С.А., 1971, v. 75, р. 5454.
49. BASF, заявка 2549305, 1977 г. (ФРГ); Изобр. за рубежом, 1977, вып. 24,
№ 9, с. 30.
50. Пат. 50-7061, 1975 г. (Япония); РЖХим, 1976, 10Н159.
51. Пат. 165293, 1975 г. (ЧССР); РЖХим, 1978, ЗН214.
52. Shionogi, заявка 101340, 1974 г. (Япония); С.А., 1975, V. 82, р. 97817.
53. Du Pont, пат. 2924620, 1960 г. (США); С.А., 1960, V. 54, р. 18434.
54. Bayer, заявка 2520893, 1976 г. (ФРГ); РЖХим, 1977, 17Н150.
55. Snam. Progetti, пат. 323716, 1971 г. (Австрия); приоритет Италии; С.А., 1971,
V. 81, р. 13245.
.56. Snam. Progetti, пат. 323717, 1971 г. (Австрия); приоритет Италии; С.А., 1971,
V. 80, р. 95463.
57. Bayer, пат. 1112990, 1962 г. (ФРГ); РЖХим, 1963, 8Н134.
58. Макарова Л. Г. — Уч. зап. МГУ, 1950, т. 1, вып. 132, с. 109.
59. Merck, пат. 3196097, 1965 г. (США); С.А., 1965, v. 63, р. 13317.
-60. Sugai Chem. Ind., заявка 29448, 1976 г. (Япония); С.А., 1976, v. 85, р. 108398.
61. Eastman, заявка 792837, 1950 г. (США); С.А., 1951, V. 45, р. 5184.
62. Eastman, пат. 2503712, 1950 г. (США); С.А., 1950, v. 44, р. 6434.
63. Eastman, пат. 2503778, 1950 г. (США); С.А., 1950, v. 44, р. 6435.
64. Universal, пат. 3450764, 1969 г. (США); РЖХим, 1970, 14Н211.
65. А.с. 249397, 1963 г. (СССР).
66. Du Pont, пат. 2780647, 1957 г. (США); РЖХим, 1959, 50646.
67. Киссин Б. И., Куракин Е. Н., Березин В. А. — В кн.: Синтез и исследование
эффективности химикатов-добавок. Тамбов, Тамбовская правда, 1969. Вып. 2,
с. 263—267.
68. Киссин Б. И., Сыпяк О. И. — Хим. пром-сть, 1961, № 4, с. 26.
69. А.с. 168710, 1967 г. (СССР).
70. Киссин Б. И., Куракин Е. Н. — Хим. пром-сть, 1965, Ks 2, с. 24—26.
71. Zdrojek Т., Ziolko М., Debowski А. — Pr. nauk. Inst. Technol. organicz. 1 twor-
zyw sztuczn. pwr., 1975, № 16, p. 343—349; РЖХим, 1975, 14H245.
72. Goodyear, пат. 3081348, 1963 г. (США); РЖХим, 1966, ЗН164.
73. Goodyear, пат. 3305584, 1967 г. (США); РЖХим, 1968, 10Н215.
74. Goodyear, пат. 3081349, 1963 г. (США); С.А., 1961, V. 55, р. 8351.
75. Goodyear, пат. 3927099, 1975 г. (США); С.А., 1976, v. 84, р. 73858.
76. Universal, пат. 3510518, 1970 г. (США); РЖХим, 1971, 5П295.
77. Mitsui, пат. 49-14738, 1974 г. (Япония); РЖХим, 1975, 1Н180.
78. Mitsui, пат. 49-14737, 1974 г. (Япония); РЖХим, 1975, 1Н179.
79. Mitsui, пат. 49-29176, 1974 г. (Япония); РЖХим, 1975, 5Н145.
' 80. Masayoshi Ytoh, Hideshi Hattori, Кого Tahabe.— J. Catal., 1974, v. 35, № 2,
p. 225—231; РЖХим, 1975, 11Б1443.
81. Пат. 883751, 1953 г. (ФРГ); С.А., 1958, v. 52, р. 11920.
82. Monsanto, пат. 3418373, 1968 г. (США); РЖХим, 1970, 9Н176.
83. Пат. 7467, 1960 г. (Япония); РЖХим, 1961, 24Л165.
84. Пат. 29983, 1964 г. (ГДР); РЖХим, 1965, 12Н147.
85. Пат. 56796, 1967 г. (ГДР); РЖХим, 1969, 12Н211.
86. Goodrich, пат. 3941843, 1976 г. (США); РЖХим, 1976, 23Н172.
87. Goodrich, пат. 4060551, 1977 г. (США); РЖХим, 1975, 14Н203.
88. ICI, пат. 1403231, 1972 г. (Англия); С.А., 1973, v. 79, р. 105001.
89. А.с. 140065, 1961 г. (СССР).
90. Тараненко А. С., Белоусова Г. А. — Хим. пром-сть, 1964, № 4, с. 32.
91. Белоусова Г. А., Тараненко А. С., Вайданич В. Г. — В кн.: Синтез и исследо-
вание эффективности стабилизаторов для полимерных материалов. Воронеж,
1964, с. 1976—1979.
68
2.3. РЕАКЦИИ НИТРОЗИРОВАНИЯ.
НИТРОЗОПРОИЗВОДНЫЕ ДИФЕНИЛАМИНА
Реакция нитрозирования лежит в основе получения целого ря-
да органических соединений, применяемых как добавки к полиме-
рам и как полупродукты для синтеза стабилизаторов. Нитрозо-
производные дифениламина могут быть получены нитрозировани-
ем дифениламина или фенола.
В производстве М-изопропил-М'-фенил-п-фенилендиамина (Диа-
фен ФП) промежуточными стадиями являются N-нитрозирование
дифениламина и его перегруппировка в /г-нитрозодифениламин.
n-Нитрозо дифенил амин обычно получают нитрозированием дифе-
ниламина.
Образование нитрозопроизводных дифениламина можно пред«
ставить схемой:
NO
Для нитрозирования применяют метанольные растворы нитрита
натрия и НС1. При этом нитрозируюгцим агентом считают нитро-
зилхлорид, концентрация которого определяется концентрацией
нитрита [1]. Агентом нитрозирования может быть нитрозацидий-
ион (H2NO2) или медленно образующийся из него нитрозоний-ион
[2]:
быстро -J-
H++HNO2 *==* h2no,
I медленно ।
H2NO2 \ *• NO + H2O
Авторы [5] рассматривают N-нитрозирование дифениламина
как кислотнокатализируемую реакцию, протекающую по механиз-
му нуклеофильного замещения Sn2 через стадию образования
нитрозациднй-иона, с которым взаимодействует затем дифенил-
амин:
NO
69
Предполагается, что наиболее медленной стадией, определяющей
скорость процесса, является образование промежуточного комп-
лекса А.
Кроме нитритов натрия ;[3—5] и калия для нитрозирования
дифениламина могут быть использованы алкилнитриты [4] и ок-
сиды азота, например NsOg [6] в растворе кислот, спиртов, бен-
зола.
Авторы [9] предложили следующую схему перегруппировки
N-нитрозодифениламина в п-нитрозодифениламин:
7Г-комплекс
Первая стадия — денитрозирование (реакция 2), вторая —электро-
фильное замещение в ароматическом ядре (реакции 3—5). Пред-
полагают '[9], что наиболее медленной стадией, определяющей об-
щую скорость процесса, является образование a-комплекса из
«-комплекса — реакция 4. Она имеет кислотнокаталитический ха-
рактер и протекает по ЗвЗ-механизму. Перегруппировку обычно
осуществляют в среде спирта (метанол, этанол), иногда в смеси
с хлорсодержащим растворителем и в присутствии НС1. Изучение
кинетики [10, 11]| показало, что реакция имеет первые порядки
по N-нитрозодифениламину и хлористому водороду.
Метод получения n-нитрозодифениламина из фенола путем его
нитрозирования, этерификации и араминирования анилином раз-
работан в 1960-х годах в исследовательском центре фирмы Hercu-
les (США) [12, 13]. Реакция представляется следующей:
70
+ROH
Реакция (I) нитрозирования фенола хорошо изучена, фенол лег-
ко и избирательно нитрозируется в лара-положение.
л-Нитрозофенол существует в двух таутомерных формах; в
форме оксима реагирует с иодистым метилом, щелочами, диазоме-
таном, гидроксиламином. В уксусной кислоте л-нитрозофенол пре-
вращается как типичное ароматическое нитрозосоединение, давая
л-гидроксиазобензол. л-Нитрозофенол проявляет свойства сильной
кислоты благодаря активированному фенольному гидроксилу.
Простые эфиры л-нитрозофенола легче араминируются анили-
ном, чем сам л-нитрозофенол [12, 13], и стадия этерификации
оказывается доминирующей при получении л-нитрозодифенилами-
на из л-нитрозофенола. л-Нитрозофенол реагирует (III) со спир-
тами в присутствии катализаторов кислотного характера, образуя
соответствующие л-нитрозофенилалкильные эфиры.
Выход л-нитрозодифениламина зависит от времени контакта
n-нитрозофенола с подкисленным раствором спирта. Более высо-
кий выход получается при выделении эфира из реакционной мас-
сы. Наибольшая степень превращения (96%) достигается в при-
сутствии этилового эфира л-нитрозофенола, соляной кислоты и эта-
нола в качестве растворителя. Спектроскопическими методами
был обнаружен л,л'-диалкоксиазоксибензол?
Этот метод технологически более прост и экономически целесооб-
разен для получения л-иитрозодифениламина, чем метод нитрозм-
Рования дефицитного и более дорогого дифениламина.
71
N -Нитрозодифениламин
NO
N-Нитрозодифениламин (дифенилнитрозамии) был получен Виттом в 1875 г.
при действии этилнитрита на раствор дифениламина в бензоле при 15—20 °C, а
также действием нитрита калия на раствор дифениламина в уксусной кислоте.
В дальнейшем ннтрозирование дифениламина под действием соляной кислоты
изучали многие исследователи [1, 5, 14—17]. Хорошие результаты дает иитрози-
роваиие в органических растворителях (спирты, бензол) и в присутствии серной
кислоты [18].
N-Нитрозодифениламин с теоретическим выходом образуется
при пропускании диоксида азота в раствор тетрафенилгидразина
в толуоле, нагретый до 90—95 °C:
При пропускании диоксида азота в раствор дифениламина в
безводном диэтиловом эфире образуется N-нитрозодифениламин,
а в растворе бензола реакция идет с преимущественным образова-
нием п-нитродифенилнитрозамина:
Отмечается [19]', что этим методом N-нитрозодифениламин полу-
чается без примесей соединений, содержащих серу или хлор, и мо-
жет быть использован для восстановительного алкилирования в
присутствии платинового катализатора.
Ннтрозирование дифениламина проводят в жидкой фазе при
пропускании жидкого амина и диоксида азота через колонну, за-
полненную кольцами Рашига [19]. N-Нитрозодифениламин можно
получить, прибавляя тетранитрометан в кипящий раствор N-метил-
дифениламина к смеси этилового спирта и пиридина:
ЛЛ_М_/~А +C(N.^ Z“Vn_ZA
\ж=/ I \=/ -CHsNOi | Х^/
CHS NO
72
рыделяющийся метилнитрит весьма нестабилен, он разлагается и
иитрозирует новые молекулы N-метилдифениламина.
Ннтрозирование можно проводить при помощи азотной кислоты
и ее эфиров, хлористого нитрозила, нитрозилсерной кислоты, окси-
дов азота и других соединений. Однако нитрит натрия и соляная
(реже — серная) кислота — самые простые и распространенные
нитрозирующие агенты.
Ннтрозирование дифениламина другими NO-содержащими со-
единениями изучено мало и не имеет практического значения. Об-
разование N-нитрозодифениламина наблюдали, в частности, при
действии нитрозогуанидина на дифениламин в водно-спиртовом
растворе соляной кислоты:
О—NH—-р HaN—С—NHNO > AA—N—+ С—NH.
Х==/ || \=/ | \=/ ||
NH NO NH
Описано получение N-нитрозодифениламина, когда дифенил-
амин обрабатывали л-толил-И-нитрозометилсульфамидом в тече-
ние 3 ч при 55—60 °C. Образовавшийся продукт извлекали диэти-
ловым эфиром и промывали водой:
—SOg-NH—СН3
В промышленности N-нитрозодифениламин обычно получают
нитрозированием дифениламина нитритом натрия в присутствии
соляной кислоты в органическом растворителе.
Технологическая схема получения. В стальной эмалированный
аппарат 1 (рис. 9) с якорной мешалкой и рубашкой для обогрева
и охлаждения загружают раствор дифениламина (ДФА) в 73—
76%-ном изопропиловом спирте и заливают 30%-ную соляную кис-
лоту. Массу охлаждают до 18°C и прибавляют 40%-ный раствор
нитрита натрия, поддерживая температуру 18—22 °C. Среда долж-
на быть кислой (содержать азотистую кислоту). После часового
выдерживания нейтрализуют смесь 20%-ным раствором NaOH,
разбавляют водой, охлаждают до 10—12 °C и фильтруют на нутч-
фильтре 2. Осадок промывают водой, разбавляют и расплавляют
в плавителе 3, где нагревают до 85—90 °C, и потом в аппарате 4
отстаивают расплав от воды.
Нижний слой — расплав N-нитрозодифениламина — в аппарате
обезвоживают при 90—95 °C и остаточном давлении 67—80 кПа.
73
Рис. 9. Технологическая схема получения N-нитрозодифениламина:
J — нитрозатор; 2 — нутч-фильтр; 3 — плавитель; 4 — сепаратор; 5 — холодильник; 6 — аппа-
рат для обезвоживания; 7 — приемник; 8 — барабанный кристаллизатор.
Конденсат из приемника 7 и верхний (водный) слой из отстойни-
ка 4 возвращают на нутч-фильтр 2.
Маточный раствор и промывные воды с нутч-фильтра направ-
ляют на регенерацию изопропилового спирта. Обезвоженный
N-нитрозодифениламин при 90—100 °C подают на чешуирование в
барабанный кристаллизатор 8. Выход готового продукта 96%.
Непрерывный процесс получения N-нитрозодифениламина дан поз-
же при описании схемы получения л-нитрозодифениламина
(стр. 79).
Свойства и применение. N-Нитрозодифениламин представляет
собой светло-желтый кристаллический порошок; т. пл. 67,6 °C;
т. затв. 65,8°C; плотность 1110 кг/м3 при 70°С. Растворяется в
бензоле, горячем этаноле, концентрированных щелочах и серной
кислоте.
Пожаро- и взрывоопасен; нижний предел взрываемости пыле-
воздушных смесей 5,2 г/м3; т. самовоспл. 970 °C.
Токсичен; ЛД5о=23О4 мг/кг; ПДК 3 мг/м3. Выпускается в ряде
стран под различными наименованиями:
74
— Торговое наименование Страна, фирма Выпускная форма T. пл., "С
ПиФенам Н СССР Чешуйки 64
Curetard А Англия, Monsanto Гранулы 65
piphene SD Франция, Kuhlmann Порошок 63—64
NDA Франция, Prochim .— —
Retarder AN Италия, ACNA Montecatini Порошок 63
Retarder J США, Naugatuck » 63—67
Sconce Япония, Ouchi Shinko » 62
Vulcatard A Англия, ICI Гранулы 65
Vulkalent ФРГ, Bayer Чешуйки 65—66
N-Нитрозодифениламин— широко распространенный замедли-
тель подвулканизации и структурирования резиновых смесей на
основе каучуков общего назначения. Эффективность повышается
в присутствии сульфенамидных ускорителей.
Ингибитор радикальной полимеризации латексов, стабилиза-
тор резиновых смесей. Недостатком является то, что он может
разлагаться с выделением оксидов азота (это приводит к получе-
нию пористых вулканизатов), вызывает окрашивание стабилизи-
рованных полимеров, выцветает на поверхности резин.
п-Нитрозодифениламин
NH—NO
Впервые я-нитрозодифениламин был получен в 1886 г. Фишером и Хеппом,
которые обнаружили, что при обычной температуре из раствора N-нитрозодифе-
ниламина в безводном этиловом спирте, насыщенном сухим хлористым водоро-
дом, образуется гидрохлорид л-нитрозодифениламина. При обработке гидрохло-
рида щелочными реагентами (Na2CO3, NH3) выделяется свободное основание.
На этой основе был разработан ряд методов получения я-нитрозодифениламина,
различающихся в основном растворителем, в котором проводится перегруппи-
ровка.
На заводе фирмы Hechst в Германии до второй мировой войны получали
я-нитрозодифениламин перегруппировкой N-нитрозодифениламина, растворенного
в четыреххлористом углероде, с помощью спиртового раствора соляной кислоты.
В США [18, 20] и ФРГ [21] я-нитрозодифениламин получают в среде неполярно-
го, не смешиваемого с водой растворителя (бензол, толуол или их смесь); выход
чистого продукта (т. пл. 145,1—146,1 °C) 96—98,5%.
Известно получение л-нитрозодифениламина в одну стадию,
без выделения N-нитрозопроизводного. Процесс осуществляют в
безводной среде, а в качестве растворителя применяют этиловый
спирт, к которому добавляют НС1 в виде раствора в безводном
Диэтиловом спирте. Нитрит натрия для перегруппировки добавля-
ют в порошкообразном виде. Предложено также [22] вести пере-
группировку в безводном бутаноле; выход продукта теоретический.
Перегруппировка происходит при наличии воды в реакционной
Массе и в присутствии галогенсодержащих соединений. Процесс
75
осуществляют, пропуская галогенсодержащее соединение, напри-
мер фосген, в суспензию дифениламина и нитрита натрия в спир-
те (метанол) [23]. Этот метод в 1950-х годах получил признание
и был запатентован во многих странах {24, 25]. В качестве гало-
генсодержащих соединений применяют ацетилхлорид, пропионил-
хлорид, бензоилхлорид, четыреххлористое олово, галогенпроизвод-
ные серы, бора, мышьяка, фосфора и кремния, оксигалогениды
фосфора, пентахлорид сурьмы, бензолсульфохлорид, тиэнилхло-
рид. В качестве растворителей можно применять низкомолекуляр-
ные алифатические спирты, содержащие 1—2% воды.
При взаимодействии хлорангидридов с водой, содержащейся
в спирте, выделяется галогеноводород, а растворитель обезвожи-
вается. Фактически реакция происходит в безводной среде под дей-
ствием хлористого водорода. Синтез осуществляют, пропуская га-
логенсодержащее соединение в суспензию дифениламина и нитри-
та щелочного металла в спирте.
Возможен и другой вариант. В спирт, насыщенный галогенсо-
держащим соединением, загружают дифениламин, а затем нитрит
щелочного металла. Расход хлорангидридов, например фосгена,
составляет 2—8 моль на 1 моль основания. При использовании
фосгена выход л-нитрозодифениламина составляет 90—95%, а вы-
ход хлористого тионила 85—90% от теоретического, Недостаток
способа — образование большого количества сточных вод, которые
необходимо очищать и обезвреживать.
По Фишеру и Хеппу, «перегруппировка» нитрозаминов заклю-
чается в том, что атом водорода бензольного ядра, находящийся
в пара-положении к нитрозаминогруппе, замещается на нитрозо-
группу в «пределах действия молекулярных сил связи»:
Хлористый водород присоединяется к I, образуя соответствующий
гидрохлорид, который под действием кислотных агентов превра-
щается в солянокислую соль л-нитрозометиланилина.
Мнение о том, что при перегруппировке нитрозаминов фактиче-
ски идет мономолекулярная реакция или внутримолекулярная пе-
регруппировка, долгое время оставалось общепринятым, хотя ряд
экспериментальных данных противоречил этому. В частности,
нельзя было объяснить заметного различия активности кислот: в
присутствии НС1 перегруппировка нитрозаминов идет довольно хо-
рошо, в присутствии H2SO4 — очень плохо, а с азотной кислотой
не идет совсем. Бромистоводородная кислота может отщеплять^
нитрозогруппу от молекулы нитрозамина с образованием гидро-’
бромида вторичных аминов. Все эти факты давали основание пред-’
полагать, что при превращении N-нитрозосоединения в л-нитро-’
зосоединение не может идти ни мономолекулярная реакция, нщ
76
внутримолекулярная перегруппировка. Позднее было показано, что
реакция протекает межмолекулярно. Под действием хлористого
водорода молекула N-нитрозодифениламина разлагается; при этом
в качестве промежуточных продуктов образуются дифениламин и
хлористый нитрозил. В результате их взаимодействия образуется
л-нитрозодифениламин [26]:
Предполагается [26], что превращение N-нитрозодифениламина в
дифениламин и NOC1 протекает медленно, а взаимодействие ди-
фениламина и NOC1 — быстро.
В ходе первой (медленной) реакции возможно образование
промежуточного соединения (типа «клетки») i[27]
(CeH^gN-NO
в результате разложения которого образуются дифениламин и
нитрозилхлорид.
Недостаточная стабильность N-нитрозодифениламина проявля-
ется не только в присутствии кислот (НС1, НВг). В нейтральной
среде (хлорбензол) при нагревании в отсутствие кислорода эта
молекула разлагается на радикалы [28]
NO
NO + N—
которые взаимодействуют
например:
между собой, образуя ряд продуктов,
—С вН6
^/^он—с,н5
(CeHB)1N-^2^-N-^2^-NH-CeH8
В присутствии кислорода (или воздуха) образуются еще нитро-
и динитродифениламин.
Для изучения механизма перегруппировки проводили реакцию
в присутствии HВ * * * * * * 15NO2 [29, 31]. Был сделан вывод, что перегруп-
пировка может быть межмолекулярной — следствием взаимодейст-
вия двух молекул нитрозамина между собой. Однако в других ра-
ботах [9, 32, 33] было показано, что перегруппировка Фишера'—
Хеппа носит внутримолекулярный характер и реакция имеет пер-
вый порядок по нитрозамину,
77
В работах [3, 10, 11, 34] изучена кинетика перегруппировки.
Оказалось, что реакция имеет первые порядки по N-нитрозодифе-
ниламину и хлористому водороду и общий второй порядок. Энер-
гия активации перегруппировки, определенная по уравнению Ар-
рениуса графическим способом, равна 72,1 кДж/моль [3]. Реакция
подчиняется соотношению Гаммета для кислотнокаталитических
реакций. Полученные данные подтверждают механизм электро-
фильного замещения.
В 1960-х годах предметом внимания химиков стал новый спо-
соб получения л-нитрозодифениламина— из л-нитрозофенола [35].
Способ заключается в нитрозировании фенола, этерификации
л-нитрозофенола и араминировании полученного соединения ани-
лином [7, 12, 13, 30]:
Первоначальные попытки провести замещение гидроксильной
группы фенола прямым араминированием л-нитрозофенола анили-
ном не увенчались успехом; исключение составили реакции в спир-
товых средах с кислотными катализаторами. Анилин взаимодейст-
вовал в этих условиях при комнатной температуре с л-нитрозофе-
нолом, образуя л-нитрозодифениламин, но выход его был низким
и нестабильным. Эфиры л-нитрозофенола вступают в эту реакцию
легче. Таким образом, получение эфиров стало важной стадией в
синтезе л-нитрозодифениламина. Удаление реакционной воды и
применение большого избытка спирта сдвигают равновесие в сто-
рону образования эфира. В качестве катализаторов этерификации
можно применять л-толуолсульфокислоту, серную кислоту, хлори-
стый водород и треххлористый бор; фосфорная и трихлоруксусная
кислоты менее активны.
Катализаторами араминирования могут служить соляная кис-
лота и моногидрат л-толуолсульфокис'лоты. Серная кислота дает
худшие результаты, потому что сульфат анилина практически не
растворяется в такой реакционной смеси. Уксусная кислота обес-
печивает достаточно высокую скорость реакции.
Способ получения л-нитрозодифениламина из л-нитрозофенола,
анилина и метанола без выделения л-нитрозоанизола дает выход
67% от теоретического (считая на л-нитрозофенол и 71,1% считая
на анилин). Катализатор — моногидрат л-толуолсульфокислоты
if 12, 13]. При ведении реакции в две стадии (с выделением л-нит-
розоанизола) выход л-нитрозодифениламина составляет 85,3—
93% в расчете на эфир; при одностадийном процессе выход равен
55% (в расчете на л-нитрозофенол) [35].
78
Рис. 10. Технологическая схема получения п-нитрозодифеииламина:
1 — нитрозатор; 2 — дозреватель; 3 — аппарат для насыщения; 4 — реакторы перегруппиров-
ки; 5, 6 — сепараторы; 7 — нейтрализатор; 8 — распылительная сушилка.
Таким образом, существуют два метода получения л-нитрозо-
дифениламина, каждый из которых имеет свои достоинства и не-
достатки [8]. Оба они обеспечены сырьем и по стоимости сырья
равноценны. Однако недостатками получения л-нитрозодифенил-
амина из фенола являются необходимость сушки взрывоопасного
л-нитрозофенола перед араминированием, неудовлетворительное
качество готового продукта (примесь л-аминофенола), сложность
регенерации непрореагировавшего нитрозофенола и большое ко-
личество кислотных сточных вод, образующихся на стадии синтеза
нитрозофенола.
Технологическая схема получения. Получение л-нитрозодифе-
ниламина осуществляют непрерывным способом, применяя на ста-
дии перегруппировки трихлорэтилен в качестве растворителя. Рас-
твор дифениламина (ДФА) в трихлорэтилене охлаждают в двух
параллельно работающих холодильниках (на рис. 10 не изобра-
жены) и подают в реактор 1 на ннтрозирование при интенсивном
перемешивании. Сюда же подают 19—21%-ную серную кислоту и
38—39%-ный раствор нитрита натрия. Ннтрозирование проводят
при 20—30°C. Из реактора 1 смесь перетекает в дозреватель 2.
При нормальном ведении процесса нитрозные газы не выделяют-
ся, так как при интенсивном перемешивании ннтрозирование про-
текает быстрее, чем разложение азотистой кислоты до оксидов
азота.
Реакционная смесь из дозревателя 2 перетекает в сепаратор 5.
Верхний, водно-солевой слой после нейтрализации раствором ед-
79
кого натра передают на распылительную сушилку 8. Нижний
слой — раствор N-нитрозодифениламина в трихлорэтилене — через
переливное устройство после дополнительной сепарации подают в
реакторы 4 на перегруппировку.
Это — эмалированные аппараты с мешалками и рубашками для
охлаждения. Перегруппировку проводят газообразным хлористым
водородом, растворенным в метаноле. Смесь последовательно пе-
реходит из одного реактора в другой, а затем поступает в нейтра-
лизатор 7—вертикальный стальной аппарат с мешалкой и рубаш-
кой для охлаждения. Температура в нейтрализаторе (204-30°C)
поддерживается автоматически. Там при взаимодействии л-нитро-
зодифениламина с 20%-ным раствором NaOH образуется натрие-
вая соль л-нитрозодифениламина.
Нейтрализованная масса из аппарата 7 переливается в отстой-
ник 6, где расслаивается. Нижний слой (трихлорэтилен) переда-
ют на установку экстрагирования этого растворителя, а верхний
слой — натриевая соль-—самотеком поступает в сборник и на вы-
деление товарного продукта. Часть раствора идет на получение
n-аминодифениламина или на получение производных л-фенилен-
диамина.
Свойства и применение. л-Нитрозодифениламин представляет
собой вещество зеленоватого цвета; т. пл. 143 °C. Растворяется в
горячем этаноле, диэтиловом эфире, хлороформе, растворах щело-
чей, горячем бензоле, ацетоне.
Горюч. Взвешенная в воздухе пыль взрывоопасна; т. воспл.
176°C; т. самовоспл. 416°С. Нижний предел взрываемости 57,5г/м3.
.Минимальное взрывоопасное содержание кислорода 11% (об.).
Умеренно токсичен; ЛД5о=1О75 мг/кг (для мышей).
л-Нитрозодифениламин является эффективным модификато-
ром и стабилизатором резиновых смесей, улучшающим некоторые
технологические и технические свойства резин. Модифицирующая
добавка в производстве каучука СКИ-3. Применение л-нитрозоди-
фениламина повышает стабильность этого каучука к термоокис-
лительной деструкции. л-Нитрозодифениламин является замедли-
телем вулканизации резиновых смесей и важнейшим сырьем при
получении стабилизаторов полимерных материалов и органических
красителей.
ЛИТЕРАТУРА
1. Schmidt Н. — Chemiker Ztg., 1962, В. 86, S. 809.
2. Hughes Е. D., Ingold С. К., Ridd J. H.—S. Chem. Soc., 1958, v. 77, p. 2324—
2326.
3. Кампель Л. П., Стрепихеев Ю. А., Наумова И. И. — В кн.: Основной органи-
ческий синтез и нефтехимия. Ярославль, 1976. Вып. 5, с. 81—86.
4. Большакова Л. Н. и др. — В кн.: Основной органический синтез и нефтехи-
мия. Ярославль. Вып. 1, 1974, с. 68—73. Вып. 6, 1976, с. 115—118.
Б. Большакова Л. Н. и др. — В кн.: Основной органический синтез и нефтехи-
мия. Ярославль, 1977. Вып.-7, с. 83—88
6. Hercules, пат. 3062887, 1962 г. (США); РЖХим, 1964, 17Н79.
80
.7. Hercules, пат. 3310583, 1967 г. (США); РЖХим, 1968, 18Н182.
Мартынов Н. В. и др. — Хим. пром-сть, 1972, № И, с. 24—27.
'д’ Беляев Е. Ю., Колянова 3. Ф., Логинова Г. И. — В кн.: Труды Сибирского
‘ технологии, ин-та, Красноярск. 1970, т. 43, с. 147.
Ц). Мартынов Н. В., Муратова Л. В., Пивоварова В. Ф. — Хим. пром-сть, 1976,
№ 9, с. 657.
11. Большакова Л. Н., Шеин В. Д., Уставщиков Б. Ф.— РЖХим, 1976, 19Ж172.
12 Hays J. Т., de Butts Е. И., Young Н. L.—J. Org. Chem., 1967, v. 32, № 1,
’ р. 153—158.
13 Hays J. T., Young H. L., Espy H. H.—J. Org. Chem., 1967, v. 32, № 1, p. 158—
162.
14. Wexman S.— Farmacia Chilena, 1946, v. 20, p. 299; C.A., 1947, v. 41, p. 405.
15. Nippon, пат. 3620, 1952 г. (Япония); C.A., 1954, v. 48, p. 4001.
16. Цымбал P. M., Бурмистров С. И., Лошкарев M. A. — В ки.: Труды Днепро-
петровского хим.-технологии, ин-та, 1956. Вып. 5, с. 171.
17. Лошкарев М. А., Бурмистров С. И., Цымбал Р. М. — Изв. вузов, Сер. хим.
технология, 1958, т. 6, с. 32.
18. Universal, пат. 3728392, 1973 г. (США); РЖХим, 1974, 4Н126.
19. Du Pont, пат. 772081, 1957 г. (Англия); С.А., 1957, v. 51, р. 14799. -
20. Cyanamid, пат. 3748302, 1973 г. (США); РЖХим, 1974, 13Н189.
21. Bayer, заявка 2211341, 1973 г. (ФРГ); РЖХим, 1974, 22Н189.
22. Gramor Chem., пат. 3429924, 1969 г. (США); С.А., 1969, V. 70, р. 87268.
23. Aniline, пат. 2495774, 1950 г. (США); С.А., 1952, v. 45, р. 650; пат. 2560892
и 2560893, 1951 г. (США); С.А., 1952, v. 45, р. 1588; пат. 2560894, 1951 г.
(США); С.А., 1952, v. 45, р. 650.
24. Aniline, пат. 820897, 1951 г. (ФРГ); Chem. Zbl., 1952, S. 5009.
25. Aniline, пат. 492270, 1953 г. (Канада); РЖХим, 1955, № 7, 12665.
26. Glazer I. е. а.—J. Chem. Soc„ 1950, р. 2657—2678; Chem. Zbl., 1953,
S 2897
27. Baliga В. T. — J. Org. Chem., 1970, v. 35, № 6, p 2031—2032.
28. Welzel P. — Bcr., 1971, B. 104, S. 808—822.
29. Steel G., Williams D. L. H.— Chem. Comm., 1969, p. 975—976.
30. Hercules, пат. 3340302, 1967 г. (США); РЖХим, 1969, 5Н136.
31. Williams D. L. H. — Tetrahedron, 1975, v. 31, № 10, p. 1343—1349; C.A., 1975,
v. 83, p. 8205.
32. Беляев E. Ю., Порай-Кошиц Б. A. — В кн.: Реакционная способность органи-
иеских соединений. Тарту, 1964. Т. 1, вып. 2, с. 204—213. '
33. Беляев Е. Ю., Никуличева Т. И. — В кн.: Реакционная способность органиие-
ских соединений. Тарту, 1970. Т. 7, вып. 2, с. 374.
34. Большакова Л. Н. и др. Основной органииеский синтез и нефтехимия, 1977,
. № 8, с. 83—87; РЖХим, 1978, 18Б1019; Шеин В. Д. и др. Там же, с. 87—94;
РЖХим, 1978, 17Н152.
35. Hercules, пат. 3107264, 1963 г. (США); РЖХим, 1965, 18Н89.
2.4. КАТАЛИТИЧЕСКОЕ ГИДРИРОВАНИЕ
АРОМАТИЧЕСКИХ НИТРОЗОСОЕДИНЕНИИ,
НИТРОСОЕДИНЕНИИ И АМИНОВ.
ДРУГИЕ СПОСОБЫ ВОССТАНОВЛЕНИЯ
Каталитическое восстановление водородом (гидрирование) аро-
матических нитросоединений впервые было проведено в 1871 г.
Процесс был осуществлен при пропускании паров нитробензола в
смеси с водородом через палладиевый катализатор. В 1897—
1914 годах были проведены систематические исследования катали-
тического восстановления нитросоединений и их производных на
никелевом катализаторе. Каталитическое гидрирование органиче-
6—1057 81
ских соединений в жидкой фазе при обычных температуре и дав-
лении проводили также в присутствии палладиевой черни и кол-
лоидной платины. В дальнейшем был расширен круг восстанав-
ливаемых соединений и предложен ряд новых веществ в качестве
катализаторов восстановления нитро- и нитрозосоединений. Было
установлено, что в газовой фазе (и в нейтральной или кислой
среде в жидкой фазе) нитросоединения, а также нитрозо-, азо- и
азогидроксисоединения превращаются в амины, обычно почти с
теоретическим выходом. В щелочной среде представляется возмож-
ным восстановить нитросоединения и до гидразосоединений [1].
При восстановлении нитро- и нитрозосоединений водородом в
присутствии катализатора реакции идут по схеме:
ArNO2
ArNO
+зн2
—2НаО
+2Н2
—э- ArNH,
—Н2О
Водород может содержать примеси азота (2—3%), оксида угле-
рода (0,1%) и следы диоксида углерода, но эти примеси практи-
чески не влияют на процесс. Поскольку восстановление водородом
широко применяют для производства ароматических аминов, ки-
нетика и механизм каталитического восстановления ароматиче-
ских нитро- и нитрозосоединений явились предметом многочислен-
ных исследований.
В промышленности применяют два способа каталитического
восстановления водородом: жидкофазный, когда восстанавлива-
емое соединение смешивают с твердым катализатором и затем под
давлением насыщают водородом, и газофазный, когда восстанав-
ливаемое соединение испаряют, пары смешивают с водородом и
смесь пропускают через слой катализатора.
Для реакций гидрирования применяют катализаторы трех ви-
дов: оксидные (медь-хромовые), металлы VII группы (например,
Со, Pd, Pt) и сульфидные. Для восстановления в псевдоожижен-
ном слое обычно используют катализаторы в виде порошков, для
процессов в твердом слое — зернистые.
Промышленные катализаторы жидкофазного гидрирования —
металлический никель или никель Ренея; в лабораторной практи-
ке, кроме того, применяют платину и палладий. Катализаторами
газофазного гидрирования являются активированные сплавы ни-
келя, алюминия, вольфрама, а также медь, нанесенная на оксид
кремния. Наиболее селективный катализатор восстановления аро-
матических нитросоединений в амины — медь: в ее присутствии
проходит восстановление только нитрогруппы, без гидрирования
ароматического ядра.
Роль катализаторов в процессах восстановления заключается
в активировании нитросоединения, нитрозосоединения и молеку-
лярного водорода. Последний, как известно, в отсутствие катали-
затора как восстановитель не действует.
82
Активность катализаторов увеличивают за счет добавок спе-
циальных промоторов. Большое значение имеет и степень измель-
чения катализатора; максимальное раздробление достигается при
осаждении каталитически активного вещества на носитель. Роль
промоторов и носителей полностью не выяснена. Известно, что те
и другие вызывают изменения структуры поверхности катализато-
ра, а носители, кроме того, значительно увеличивают его поверх-
ность. Предполагают, что с помощью промоторов происходит регу-
лирование электронных свойств катализатора и степени окисле-
ния активного компонента [2]. В качестве промоторов чаще всего
применяют оксиды и гидроксиды меди, марганца, бария и других
металлов, реже их соли. Для каждой пары катализатор — промо-
тор существует оптимальное соотношение, при котором катализа-
тор имеет максимальную активность. В качестве носителей приме-
няют различные пористые материалы—активированный уголь,
асбест, пемзу.
Металлические катализаторы, в том числе и смешанные, весь-
ма чувствительны к каталитическим ядам, которыми являются со-
единения, содержащие серу или галоген. Особенно легко отрав-
ляется медный катализатор, поэтому нитро- и нитрозосоединения
предварительно очищают от примесей. В ряде случаев применяют
комбинации катализаторов, которые обеспечивают разложение
сернистых соединений с образованием продуктов, не отравляющих
катализатор.
В процессе восстановления катализатор постепенно снижает
свою активность («утомляется»). Для восстановления первоначаль-
ной активности его подвергают регенерации.
Весьма перспективным представляется новый метод жидкофаз-
ного гидрирования [3], предусматривающий применение системы,
содержащей катализатор активирования молекулярного водорода
и катализатор переноса активированного водорода. В качестве пе-
реносчиков водорода рекомендованы различные хиноны и комп-
лексные соединения, способные к обратимому присоединению про-
тонов или электронов. Еще одним компонентом системы являются
обычные катализаторы гидрирования. Достигаемые при этом ско-
рости гидрирования значительно превышают скорость в отсутствие
катализатора переноса водорода, что делает возможной реализа-
цию процесса при обычных температуре и давлении [4].
Для гидрирования ароматических нитро- и нитрозосоединений
можно использовать разнообразные растворители: низкомолеку-
лярные алифатические спирты (метанол, этанол, изопропанол),
Циклические эфиры (диоксан, тетрагидрофуран), ароматические
углеводороды (бензол, толуол, ксилол). Особенно важен подбор
растворителей для периодических процессов гидрирования. Вос-
становление нитросоединений можно проводить и в тех органиче-
ских соединениях (морфолин, этилендиамин, пиперидин, диметил-
формамид), которые служат растворителями исходных нитросо-
единений [5].
6* 83
Механизм гидрирования в нейтральной и щелочной водно-спир-
товой среде исследован на примере нитробензола [6—8]; в каче-
стве катализатора применены никель Ренея или платина. Предло-
жена следующая схема превращения нитросоединения при гидри-
ровании:
Ч~Н2 4-^2 Ч"Н2
RNO. —> RNO --------► RNHOH г——> RNH,
—Н2О —НдО “
I—>rn=nr«— I
I
о
I
+н2 +н2
f RN=NR -----> RNH— NHR—
В условиях избытка водорода реакция идет через нитрозобен-
зол и фенилгидроксиламин до амина, поэтому важно проводить
гидрирование под давлением водорода (особенно при работе с ни-
келевым катализатором, с поверхности которого адсорбированный
водород легко вытесняется нитросоединением). Платиновый ката-
лизатор содержит прочно адсорбированный водород, не вытесня-
емый нитросоединением, поэтому скорость реакции зависит от ак-
тивности самого нитросоединения.
Добавка платины к никелевому катализатору сильно активи-
рует его, так как происходит сочетание свойств никеля (сорбирует
нитросоединения) и платины (сорбирует водород).
При гидрировании на никелевом катализаторе реакция идет
ступенчато. Это связано с уменьшением концентрации активного
водорода, вытесняемого с поверхности хорошо сорбируемым там
нитросоединением, а восстановление активного водорода на ката-
лизаторе идет медленно. Промежуточные нитробензол и фенил-
гидроксиламин, вытесняемые в раствор, реагируют друг с другом
с образованием азогидроксибензола, а он затем уже через азо- и
гидразобензол восстанавливается в анилин.
С развитием промышленных методов получения производных
оксифенилбензтриазола в 1960-х годах большое значение приобре-
ла реакция восстановительной циклизации о-нитроазосоединений
(восстановительное триазолирование о-нитроазокрасителей)
где R — остаток ароматического ядра, содержащего алкильные
или гидроксильные группы. В качестве восстановителей могут быть
использованы соединения серы: сульфид аммония или натрия,
гидросульфит натрия [11—14] в щелочной среде. С гидросульфи-
том выход достигает 82% считая на азокраситель :[12]. С невысо-
84
ким выходом («30%) бензтриазолы получаются при мягком вое»
становлении гидразингидратом [15—17]. Восстановление о-нитро-
азосоединений можно проводить и в присутствии триалкилфосфи-
тов (например, триэтилфосфита), получая конечный продукт с вы-
ходом до 85% f 18].
Более широко применяется восстановление о-нитроазокрасите-
лей цинковой пылью в солянокислой или щелочной среде [11, 19—
31]. Восстановление в солянокислой среде сопровождается частич-
ным восстановлением азокрасителя по азосвязи с расщеплением
до о-нитроанилина и соответствующего ариламина, что снижает
выход бензтриазолов. Поэтому наиболее приемлемым является
восстановление цинком в щелочной среде; выход достигает 75%
считая на азокраситель ,[28].
Известен также электролитический способ восстановления
о-нитро азосоединений [32], однако выход бензтриазолов в этом
случае не превышает 9—10%.
Мало изученным, но перспективным способом синтеза бензтри-
азолов, замещенных в положении 2, является жидкофазное катали-
тическое гидрирование о-нитроазосоединений. В качестве катали-
заторов применяют Pt, Ni, Со, Мо и их оксиды. Реакцию проводят
при 15—200 °C и 70 МПа. Среда — вода, полярные растворители
или их смеси с водой в присутствии щелочи [33—35].
Восстановление о-нитроазокрасителей протекает с непосредст-
венным образованием бензтриазолов
или через промежуточные бензтриазолоксиды, которые дальней-
шим восстановлением (хлоридом олова в соляной кислоте [36],
цинком в солянокислой или щелочной среде, гидразингидратом
в щелочной среде [37]) превращают в соответствующие производ-
ные бензтриазола:
О
В последние годы в химии и технологии стабилизаторов боль-
шое значение приобретает реакция каталитического гидрирования
ароматических аминов в ядро. Ароматические соединения обычно
85
гидрируются в более жестких условиях, чем олефины, альдегиды
или кетоны. Поэтому, например, не представляет трудности гид-
рирование двойной связи стирола C6Hs—СН=СН2 с сохранением
фенильной группы.
Такой активный катализатор гидрирования, как оксид рения,
в присутствии НС1 восстанавливает бензол лишь частично, а наф-
талин не восстанавливает совсем. Очень гладко присоединение во-
дорода к ароматическим соединениям идет в присутствии мелко-
раздробленного никеля. Еще легче гидрирование протекает в при-
сутствии мелкораздробленной платины при атмосферном давле-
нии или в присутствии скелетного никелевого катализатора под
давлением водорода.
Анилин гидрируется в циклогексиламин с трудом. Под давле-
нием водорода и в присутствии никеля при нагревании анилин
превращается в циклогексиламин, однако в результате замены
аминогруппы на водород одновременно может образоваться и цик-
логексан. При гидрировании анилина на оксиде кальция большую
роль играет наличие влаги в зоне реакции. Повысить выход ани-
лина и других циклоалифатических аминов можно за счет специ-
альной обработки катализатора [38]. Толуилендиамины гидриру-
ют до соответствующих гексагидротолуилендиаминов в присутст-
вии катализаторов — металлов VII группы на носителях (СоО на
СаО или на Na2CO3); одновременно с гидрированием происходит
дезаминирование толуилендиамина и образование изомеров метил-
циклогексиламина [39]. Реакции гидрирования ароматических
аминов протекают сложно и взаимосвязанно с реакциями изоме-
ризации, расщепления и (иногда) алкилирования в процессах га-
зофазного гидрирования. Селективность гидрирования повышается
при соответствующем подборе катализаторов.
Диаминодициклогексилметан — сравнительно новый стабили-
затор — получают гидрированием диаминодифенилметана. Процесс
гидрирования диаминов остается малоизученным. Такие простые
катализаторы, как никель Ренея, успешно применяемые для гид-
рирования одноядерных ароматических аминов, в случае диаминов
оказались неэффективными. Хорошие результаты при гидрирова-
нии диаминодифенилметана получены на кобальтовых и рутение-
вых катализаторах.
4-Аминбдифениламин
Впервые 4-аминодифениламин был получен в 1879 г. восстановлением
4-анилиназобензола цинковой пылью в среде уксусной кислоты и восстановле-
нием 4-анилиназобензол-4-сульфокислоты цинковой пылью в разбавленной уксус-
ной кислоте.
В настоящее время известно несколько методов получения
4-аминодифениламина.
86
1. Восстановление 4-аннлиназобензола или 4-анилиназобен-
зол-З'-сульфокислоты:
Удобнее использовать не 4-анилиназобензол, а его производные,
содержащие сульфогруппу. Действуя на 4-анилиназобензол-4'-суль-
фокислоту цинковой пылью в разбавленной уксусной кислоте, по-
лучают 4-аминодифениламин и сульфаниловую кислоту. 4-Амино-
дифениламин можно получать восстановлением 4-анилиназобен-
зол-З'-сульфокислоты гидросульфитом натрия в водной среде, а
также обработкой водно-спиртовым раствором йодистого водорода
[53]. Натриевую соль 4-анилиназобензол-З-сульфокислоты можно
восстановить до 4-аминодифениламина электрохимическим мето-
дом в содовом растворе при 95 °C на ртутном электроде. Способ 1
получения 4-аминодифениламина обычно не обеспечивает высоких
выходов и хорошего качества продукта: выход технического 4-ами-
нодифениламина не превышает 45—70%, содержание нераствори-
мого остатка доходит до 20%.
2. Гидролиз 4-нитродифениламин-2'-сульфокислоты с последу-
ющим восстановлением
или гидролиз 4-аминодифениламин-2'-сульфокислоты (нероловая
кислота):
Этот метод применяли для промышленного получения /г-аминоди-
фениламина в Германии на заводах фирмы I. G. Farbenindustrie
до второй мировой войны. Обычно процесс осуществляли гидроли-
87
зом нероловой кислоты 15%-ной серной кислотой при повышенном
давлении или 55%-ной серной кислотой при обычном давлении.
4-Аминодифениламин получался с довольно высоким выходом
(85—90%) и т. пл. 71—73 °C [40]. Недостаток метода — образо-
вание большого количества кислотных сточных вод и применение
дорогого дефицитного сырья (нероловая кислота). В настоящее
время метод не имеет промышленного значения.
3. Восстановление 4-нитродифениламина:
Восстановление 4-нитродифениламина по Ульману обычно прово-
дят в спиртовой среде, что осложняет технологический процесс.
В таких условиях 4-аминодифениламин получается с выходом 90%
в пересчете на 4-нитродифениламин. В патенте [41] описан ва-
риант этого метода, когда 4-аминодифениламин получают гидри-
рованием смеси 4-нитродифениламина с натриевым или калиевым
производным 4-аминодифениламина в растворе алифатического
спирта в присутствии 0,1—0,3% (масс.) катализатора (Ni, Со, Ru,
Pd, Pt) при 50—100°С и повышенном давлении.
Скорость восстановления нитросоединений в водной среде оп-
ределяется в основном степенью дисперсности образующейся ге-
терогенной системы, т. е. поверхностью фазового контакта. Даже
при очень интенсивном перемешивании скорость восстановления
в гетерогенной среде часто оказывается недостаточной: реакция
задерживается на промежуточных стадиях, создаются благоприят-
ные условия для образования стабильных азогидрокси- и азосо-
единений, разлагающихся с образованием смол. Восстановление
ароматических нитросоединений сульфидами в водной среде прохо-
дит более гладко в присутствии одно- или двухатомных фенолов,
арилкарбонатов или арилсульфонатов [42—44]. При этом повы-
шается выход амина и улучшается его качество. Механизм поло-
жительного влияния указанных добавок не выяснен и не пред-
ставляется единым для всех случаев.
Способ 3 получения 4-аминодифениламина до сих пор остается
малоизученным.
4. Взаимодействие форманилида с и-нитрохлорбензолом [45]:
88
Реакция требует присутствия меди и основания для нейтрализа-
ции выделяющегося хлористого водорода. Это —вариант класси-
ческого метода Гольдберга — нагревание анилина с п-нитрохлор-
бензолом в присутствии 10—20% (мольн.) иодида меди (или меди
и иодида калия) и небольшого избытка карбоната калия. Гольд-
берг показал, что с ацетанилидом реакция протекает быстрее, чем
с анилином. В течение ряда лет многие исследователи [46—56]
пытаются усовершенствовать метод Гольдберга и применить его
для промышленного получения аминов (использование формани-
лида, различных соединений меди и оснований, введение диметил-
формамида). Например, в случае форманилида образующийся
К-формил-4/'-нитродифениламин декарбонилируется во время реак-
ции, что исключает последующий гидролиз ацильной группы.
В настоящее время в ряде стран прорабатывается вопрос о целе-
сообразности промышленного внедрения этого метода.
5. Азосочетание 4-нитрофенилдиазопия с дифениламином с по-
следующим гидрированием образовавшегося азосоединения:
o8n——*
--> O2N—Z~A-N=N—
2 \ ' \ / \ / —H2O
--► H,N—NHs + H2N~^~y—
Азосочетание с активной солью диазония с последующим восста-
новительным расщеплением является стандартным методом для
введения аминогруппы в ароматическое ядро [57]. Этот метод ис-
следуют для промышленного внедрения параллельно с предыду-
щим [45]. В качестве побочного продукта образуется «-фенилен-
диамин.
Дифениламин не растворяется в воде, поэтому азосочетание
обычно проводят в органических растворителях [58]. Однако ис-
пользование таких растворителей для данного метода экономиче-
ски нецелесообразно. Обсуждается возможный вариант проведе-
ния азосочетания в нагретом растворе соляной или серной кислоты
(т. пл. дифениламина 53 °C) с эмульгирующим агентом. В раство-
ре серной кислоты азосочетание протекает намного быстрее.
Диазотируют «-нитроанилин в нитрозилсерной кислоте (или в
70—75%-ной серной кислоте) и при 25—30 °C вводят дифенил-
амин; кислотность поддерживают от 25 до 30%. Реакция протека-
ет очень быстро и смесь трудно поддается перемешиванию. Испы-
тан ряд поверхностно-активных веществ с целью улучшения пере-
мешивания; наиболее эффективным оказался н-октанол. Процесс
полностью заканчивается через 15—30 мин при обычной темпера-
туре [45]. Азосоединение промывают водой, высушивают, а затем
. кипятят с «-гептаном для удаления непрореагировавшего дифенил-
амина.
89
6. Гидрирование М-(4-иитрозофенил)-П-гидроксил амина [34,
35]: -
Метод разработан в 1970 г. в Англии (фирма ICI) в двух вариан-
тах.
По одному варианту получают 4-аминодифениламин гидриро-
ванием щелочной соли N-(4-нитрозофенил)-N-гидроксиламина, ко-
торая образуется при его смешении с КОН или NaOH. Реакцию
проводят в водной среде при 40—80 °C в присутствии катализато-
ра (Pd на угле) в количестве 0,1—2% (масс.) и растворителя
(этиловый спирт, толуол). N-(4-Нитрозофенил)-N-гидроксиламин
растворяют в 10%-ном растворе смеси щелочи и толуола и добав-
ляют катализатор. Раствор переносят в автоклав, в котором водо-
родом создают давление 3 МПа, нагревают до 60 °C и выдержива-
ют при этой температуре в течение 6 ч. Реакционную массу охлаж-
дают и фильтруют. Экстрагируют водный слой толуолом, удаляют
растворитель и получают кристаллический 4-аминодифениламин с
выходом 88% [59].
По другому варианту 4-аминодифениламин получают гидриро-
ванием N-(4-нитрозофенил)-N-гидроксиламин в присутствии ме-
таллов VIII группы (на носителях или без них) и с использовани-
ем спирта в качестве растворителя (пропанол, изопропанол, «-бу-
танол, изобутанол). Замена указанных растворителей другими
спиртами снижает выход 4-аминодифениламина. Реакцию можно
проводить в присутствии воды (пригодны, например, водные пас-
ты, содержащие исходный гидроксиламин). Смесь N-(4-нитрозофе-
нил)-N-гидроксиламина с катализатором (Pd на угле), суспенди-
рованную в изопропаполе, выдерживают в автоклаве 6 ч при
95 °C и начальном давлении водорода 10 МПа. Холодную реакци-
онную массу фильтруют и после отгонки растворителя получают
продукт, содержащий, по данным ПК- и тонкослойной хромато-
графии, 83% 4-аминодифениламина. Если вместо изопропанола
использовать м-изоамиловый спирт, выход 4-аминодифениламина
снижается соответственно до 32,5 и 45,8% [60].
Этот метод в промышленности пока не реализован, но при на-
личии сырья [N-(4-нитрозофенил)-N-гидроксиламин] его можно
считать перспективным благодаря простоте технологического
оформления, довольно высокому выходу продукта, отсутствию
сточных вод.
7. Араминирование фенола п-фенилендиамином в присутствий
катализатора [61]?
В 1930-х годах в качестве катализаторов были предложены сер-
ная кислота или катализаторы Фриделя—Крафтса. Синтез вели,
90
нагревая компоненты до 200—220 °C, выдерживая в течение 2 ч
при перемешивании и удаляя образующуюся воду. При этом по-
лучается большое количество кислотных отходов, а 4-аминодифе-
ниламин загрязнен побочными продуктами. В конце 1950-х годов
фирмой Goodrich (США) разработан более совершенный способ
|[61], по которому реакцию осуществляют в жидкой фазе в присут-
ствии эфиров титана в качестве катализаторов.
8. Окисление анилина и гидрирование продуктов окисления.
Это новый способ, предложенный фирмой ICI (Англия). Он за-
ключается в следующем [62]. Анилин подвергают окислению фер-
рицианидом щелочного металла в присутствии сильного основания
до образования продуктов, в молекулах которых содержится боль-
шое число бензольных ядер, связанных амино- или азогруппами.
Смесь обрабатывают восстановителем, способным расщеплять свя-
занную с ядром азогруппу, т. е. осуществляют гидрирование. В ка-
честве катализатора рекомендован палладий на угле. После гидри-
рования получают полиамины такой общей формулы:
Анилин, феррицианид калия и едкий натр растворяют в трихлор-
этилене, нагревают реакционную массу до 60 °C и выдерживают
несколько часов. При этом феррицианид восстанавливается до
ферроцианида. Полученную массу гидрируют в автоклаве (ката-
лизатор— Pd на угле) в течение 12 ч при 95 °C и 3 МПа. Как пре-
имущество метода, отмечаются его малостадийность и простота.
9. Восстановление 4-нитрозодифениламина:
+4Н
---->
—н2о
Процесс можно осуществить сульфидом натрия или аммония в
спиртовом растворе, нагреванием с алкоголятом калия в трубча-
том реакторе (при 120—125 °C) с цинковой пылью в ледяной ук-
сусной кислоте. Способ получения 4-аминодифениламина, предло-
женный фирмой Bayer (ФРГ), предусматривает проведение гидри-
рования в среде растворителей, не смешивающихся с водой (аро-
матические амины), в присутствии скелетного никеля [63]. Реак-
цию проводят при 90—110°С под давлением водорода 1—1,6МПа.
По способам американских фирм Rubber и Uniroyal [64, 65] для
гидрирования берут не гидрохлорид 4-нитрозодифениламина, а его
соль (обычно натриевую). В этом случае целесообразно применять
4-нитрозодифениламин в таутомерной форме оксима
/~V-N=
=NOH
91
так как оксим растворим в щелочи. В присутствии щелочи водород
гидроксильной группы замещается натрием:
Соль 4-нитрозодифениламина растворяют в воде в присутствии
органического растворителя, обычно изопропанола [66]. Реакцион-
ную смесь перемешивают при достаточно высокой температуре —
выше т. пл. 4-аминодифениламина (76°C). Отделяют органиче-
ский растворитель от воды и катализатора, подкисляют смесь со-
ляной кислотой и доводят раствор до щелочной реакции для выде-
ления 4-аминодифениламина.
В работе [67, 68] изучена сравнительная активность металли-
ческих катализаторов при гидрировании 4-нитрозодифениламина.
В кинетической области при обычных давлении и температуре ис-
следовано гидрирование в присутствии Ni, Rh, Pd, Ru и Ir. Найде-
но, что Ru и Ir не активны, a Ni, Rh и Pd по начальной удель-
ной активности и производительности располагаются в ряд
Ni<Rh<Pd, причем палладий активнее никеля в 140 раз. Опреде-
лены удельная активность и стабильность металлов платиновой
группы и никеля. Наиболее активным и стабильным к дезактиви-
рованию оказался палладий.
Сравнительно недавно опубликованы результаты электрохими-
ческого способа восстановления 4-нитродифениламина [69—71].
.Авторы полагают, что 4-нитрозодифениламин восстанавливается
аналогично 4-нитрозофенолу.
Из девяти описанных способов получения 4-аминодифенилами-
на технологически освоены два — гидролиз нероловой кислоты и
восстановление 4-нитрозодифениламина. Сравнительный анализ
других способов дан в работе [72]. Прогрессивным является вос-
становление Ы-(4-нитрозофенил)-К-гидроксиламина водородом на
катализаторах. Ниже рассмотрена технология получения 4-амино-
дифениламина восстановлением 4-нитрозодифениламина.
Технологическая схема получения. В реактор 1 (рис. 11) пода-
ют щелочной раствор 4-нитрозодифениламина и 24—26%-ный рас-
твор полисульфида натрия в трихлорэтилене (ТХЭ). Температура
восстановления 40—60 °C. Масса из реактора перетекает в дозре-
ватель 2, где поддерживают температуру 70—75 °C. Из дозревате-
ля через сепаратор 4 раствор 4-аминодифениламина поступает в
выпарной аппарат 5, где отгоняют метанол и отдувают трихлор-
этилен. Пары конденсируют и расслаивают в сепараторе 6. Воду
направляют на биохимическую очистку, а трихлорэтилен возвра-
щают в производство. Очищенный от трихлорэтилена 4-аминодифе-
ниламин промывают в аппарате 7 горячей водой (70—75 °C) и да-
лее через сепаратор 9 подают на дистилляцию. Сначала 4-амино-
дифениламин-сырец передают в испаритель 8, куда одновременно
подают 42%-ную щелочь (NaOH) для связывания сульфидов. В ис-
92
Рис. 11. Технологическая схема получения 4-амино дифенил амина:
/ — реактор; 2 — дозреватель; 3, 11 — холодильники; 4 — сепаратор; 5 — выпарной аппарат;
6, 9— сепараторы; 7 — промывной аппарат; 8 — испаритель; 10, 13— ректификационные ко-
лонны; 12, 14—приемники: 15 — кипятильники.
парителе 8 частично отгоняются вода и низкокипящие органиче-
ские примеси; их сжигают, а 4-аминодифениламин подают в ко-
лонну 10. Там окончательно отгоняют воду и низкокипящие при-
меси (их тоже сжигают). Очищенный 4-аминодифениламин в ко-
лонне 13 отгоняют от смолы, которую выводят из куба и
сжигают.
В техническом 4-аминодифениламине (АДФА), полученном вос-
становлением 4-нитрозодифениламина сульфидами, обычно при-
сутствуют нежелательные и трудноудаляемые соединения серы.
Это не позволяет применять такой 4-аминодифениламин во многих
промышленных процессах (прежде всего, в реакциях восстанови-
тельного алкилирования) из-за отравления гидрирующих катали-
заторов соединениями серы (известно, что катализаторы, содер-
жащие палладий и платину, отравляются даже следами сернистых
соединений). Примеси обычно представляют собой смесь неоргани-
ческих соединений серы (сульфиды) и свободной серы, хотя могут
присутствовать и некоторые органические вещества, например
арилмеркаптаны.
Полностью удалить эти примеси из 4-аминодифениламина обыч-
ными методами (перегонкой) чрезвычайно трудно, а иногда и
просто невозможно. При перегонке эти примеси удаляются лишь
частично: «35% загрязненного 4-аминодифениламина превраща-
ется в продукт, пригодный для алкилирования, но недостаточно
хороший и стабильный. Кроме того, при перегонке образуются
циклические сульфиды, по-видимому, за счет взаимодействия
4-аминодифениламина с серой. Следовательно, можно ожидать, что
Перегнанный 4-аминодифениламин (тем более технический) содер-
жит примеси, способные отравлять катализаторы, используемые
Для восстановительного алкилирования.
Существует несколько способов очистки 4-аминодифениламина
°т серы. В качестве примера можно привести способ фирмы Rub-
93
ber (США) [73]. Он заключается, в обработке технического 4-ами-
нодифениламина оксидом свинца (свинцовый глет) с последую-
щей перегонкой и выделением 4-аминодифениламина, не содержа-
щего серы. При нагревании с оксидом свинца неорганические
сернистые примеси превращаются в сульфид свинца, а арилмер-
каптаны— в меркаптиды свинца и арилдисульфиды.
Свойства и применение. 4-Аминодифениламин — кристалличе-
ский порошок белого, розоватого или светло-желтого цвета; плот-
ность 1125 кг/м3; т. пл, 76 °C; т. кип. 204—230 °C (при 133—
1580 Па). Растворяется в ацетоне, бензине, хлорбензоле, хлоро-
форме, этаноле, безводном диэтиловом эфире, разбавленной соля-
ной кислоте.
Горюч; т. воспл. 259°С; т. самовоспл. 601 °C. Взвешенная в воз-
духе пыль взрывоопасна. Нижний предел взрываемости 65,0 г/м3;
минимальное взрывоопасное содержание кислорода 12% (об.).
Умеренно токсичен; ЛД50=244 мг/кг для мышей и 819,6 мг/кг
для крыс.
4.-Аминодифениламин — важнейшее сырье для производства
многих стабилизаторов (производные м-фенилендиамина), краси-
телей и других органических веществ; обладает стабилизирующим
действием, но на практике не применяется вследствие высокой
летучести.
2- (2'-Гидрокси-5'-метилфенил) бензтриазол
Самые ранние сведения по синтезу этого вещества относятся к 1960 г. [74].
В основу процесса был положен метод окислительной циклизации о-аминоазо-
соединения сульфатом меди в аммиаке. 2-Гидрокси-5-метиланилин диазотируют
обычным путем, а полученную соль диазония сочетают при pH 4,04-4,2 с л<-фе-
нилендиамииом. Получают 2-гидрокси-2',4'-диамино-5-метилазобензол, который
подвергают окислительному триазолированию в присутствии сульфата меди
в аммиаке до образования 2- (2'-гидрокси-5'-метилфенил) аминобензтриазола
(Беназол П) с т. пл. 180—190 °C. Затем повторным диазотированием и после-
дующим кипячением с медным порошком в этаноле отщепляют аминогруппу и
получают целевой продукт с т. пл. 128—130 °C. Схему синтеза можно предста-
вить так: .
94
он
Аналогичный способ приведен в работе [75] с подробным описанием окисли-
тельной циклизации 2-гидрокси-2',4'-диамино-5-метилазобензола. Однако способ
окислительной циклизации не имеет промышленного значения из-за многостадий-
ности и нерентабельности.
В 1960 г. в Швейцарии был предложен способ получения замещенных в по-
ложении 2 бензтриазолов восстановительной циклизацией о-нитроазосоединений
[76, 77]. Это явилось основой промышленного способа получения 2-(2'-гидр-
окси-б'-метилфенил) бензтриазола (Tinuvin Р). В настоящее время известны два
варианта этого способа: восстановление цинком в щелочной среде (иногда в
присутствии не смешиваемого с водой растворителя) [11, 26, 78] и восстановление
гидросульфитом натрия в щелочной среде [11, 12, 26, 79]. Например, восстанов-
лением 2-гидрокси-5/-метил-2'-нитроазобензола цинковой пылью в водном эта-
ноле в присутствии аммиака получали целевой продукт с выходом 72% и
т. пл. 130—131 °C [26]. Восстановлением азосоединения гидросульфитом натрия
в щелочной среде получен продукт с выходом 82% и т. пл. 131 °C [26]. Восста-
новление цинком приводит к большому количеству отходов (цинковый шлам),
требующих утилизации, и выход продукта ниже, чем при восстановлении гидро-
сульфитом.
Перспективный способ восстановления азосоединения — каталитическое гид-
рирование, в последнее время все больше привлекающее внимание исследовате-
лей. Для получения 2-(2'-гидрокси-5'-метилфенил) бензтриазола в качестве ката-
лизаторов гидрирования изучены никель, медь, кобальт, платина, палладий, хром
н родий [33—35, 80]. В присутствии платины в спиртово-щелочной среде выход
Достигал 65—80%. В присутствии никеля, активироваииого молибденом в бен-
золо-щелочной среде (pH >10), выход продукта составлял 90%. Например, по
способу фирмы Uniroyal (США) 2-(2'-гидрокси-5'-метилфенил) бензтриазол по-
лучают жидкофазным каталитическим гидрированием азосоедииеиия при ком-
натной температуре и небольшом давлении водорода («0,1 МПа) в присутст-
вии щелочи. Средой служит вода, а катализатором — палладий на угле. Реакция
9Б
заканчивается за час, катализатор отделяют, фильтрат подкисляют и получают
продукт с выходом 92,5% и т. пл. 139—140 °C [34]. Из приведенного примера
видны преимущества каталитического гидрирования по сравнению с выше-
описанными методами.
Из других способов получения 2-(2'-гидрокси-5'-метилфенил)бензтриазола
следует отметить способ термического разложения 2-азидо-2'-гидрокси-5'-метш1-
азобензола [32]. Его получают сочетанием диазотированного о-азпдоанилина
с n-крезолом в щелочной среде с выходом 78% (т. пл. 73—76 °C; с разложе-
нием) и нагреванием в диоксане при 65 °C превращают в целевой продукт с вы-
ходом 99% и т. пл. 130—133 °C:
ОН
N3 ОН
N-N—
сн3
Этот способ практического значения не получил вследствие трудностей техииче]
ского оформления процесса.
В промышленности Беназол П получают способом, включаю-]
щим три стадии: диазотирование о-нитроанилина, сочетание полу-
ченного о-нитрофенилдиазонийхлорида с n-крезолом и восстанов-
ление в присутствии цинка гидроксиламина или гидросульфита.
В общем виде реакцию можно представить так:
ОН
СНз СН3
Первую стадию — диазотирование первичного амина — осуще-
ствляют приемами, сходными с N-нитрозированием вторичного
амина (стр. 72). Нитрозирующим агентом служит нитрозилхло-
рид NOC1 или динитротриоксид N2O3; последний образуется из азо-1
тистой кислоты при низкой кислотности среды:
2HNO, ONO— NO-f-H2O j
96
При повышенной кислотности (в среде концентрированной H2SO4)
такими агентами являются нитроцидий-ион, образующийся при
протонировании азотистой кислоты
+ +
hno2 + Н3О Н2о—NO + Н2О
или нитрозоний-ион NO.
Диазотирование можно представить схемой:
44- б— 4- 4*
RNH2 + N=-0---> RNH2—NO’ —rnh2—n=o —>
2 i 2 I -X 2 -h+
X X
+H*- +
---► RNH—N=O » RN=NOH ——*• RN=N
—H2O
X — Cl, ONO или
На практике реакцию осуществляют в присутствии нитрита нат-
рия и соляной кислоты при 2—3°С.
Азосочетание — реакция электрофильного замещения в арома-
тическом ряду; электрофильным агентом служит катион арилди-
азония ArNa:
Катионы диазония существуют только в кислой или слабощелоч-
ной среде, в связи с чем реакция азосочетания может идти лишь
в этих условиях. В случае фенолов (в данном случае /г-крезола)
лучше всего проводить реакцию в слабощелочной среде, посколь-
ку фенолят-ион намного легче атакуется электрофильным агентом,
чем сам фенол, из-за значительно более высокой электронной
плотности фенолят-иона:
7—1057
Восстановление о-нитроазокрасителя, образовавшегося в ре-
зультате азосочетания, протекает (в зависимости от условий) ли-
бо с непосредственным образованием бензтриазолов, либо через
промежуточное образование бензтриазолоксидов, которые восста-
навливаются в бензтриазолы. Механизм этих двух реакций изучен:
пока недостаточно.
9EF
О’Ншпрсашпин "
Рис. 12. Технологическая схема получения 2-(2'-гидроксифенил-5'-метилфенил)-
бензтриазола:
1 — аппарат для диспергирования; 2, 5, 8 — реакторы; 3, 6f 9, 11 — фильтры; 4 -> емкость;
7 — приемник; 10, J2 — смесители; 13 — перегонный куб; 14— холодильник; 15 — приемник.
Технологическая схема получения. В аппарат 1 (рис. 12) загру-
жают о-нитроанилин, воду и ОП-10. Массу перемешивают до по-
лучения тонкой суспензии и передают на диазотирование в реак-
тор 2, куда приливают соляную кислоту и при перемешивании
и охлаждении дозируют раствор нитрита натрия. Реакционную
массу после фильтрования на дисковом фильтре 3 принимают в
емкость 4, откуда направляют на азосочетание.
Азосочетание проводят в реакторе 5, куда загружают воду, рас-
твор соды, /г-крезол и к этой смеси дозируют раствор диазосоеди-
нения через мерник. Скорость дозирования определяется темпера-
турой, которая не должна превышать 5 °C. Полученную суспензию
азосоединения фильтруют на барабанном вакуум-фильтре 6. Пас-
ту принимают в приемник 7 и после перемешивания направляют
на восстановление в реактор 8.
Восстановление проводят в водной среде гидросульфита или
гидроксиламина. Раствор натриевой соли Беназола П фильтруют
на фильтре 9 и подают в аппарат 10 на выделение. Для выделе-
ния к раствору добавляют соляную кислоту до pH 10,54-11,9. Тех-
нический Беназол П отфильтровывают от маточного раствора на
барабанном вакуум-фильтре 11 и в аппарате 12 смешивают с рас-
творителем. Суспензию Беназола П сливают в перегонный куб 13,
где отгоняют воду. Азеотропная смесь паров растворителя и воды
конденсируется в холодильнике 14. Конденсат собирают в прием-
ник 15, где отделяют воду от растворителя. После отгонки раство-
рителя расплав Беназола П из куба 13 подают на вакуум-разгон-
ку и кристаллизацию. Готовый продукт сушат и расфасовывают.
98
Свойства и применение. 2-(2'-Гидрокси-5'-метилфенил)бензтри-
азол существует в двух кристаллических формах. Если его крис-
таллизовать из 95%-ного этанола при комнатной температуре, об-
разуются длинные иголки, плавящиеся при 100 °C (при быстром
нагревании). При кристаллизации из горячего 95%-кого этанола
получаются ромбические кристаллы с т. пл. 131,5—133 °C. Если
игольчатые кристаллы расплавить и охладить, то при повторном
нагревании они плавятся уже при 131,5—133 °C.
Беназол П — светло-желтый кристаллический порошок. Раство-
ряется в ацетоне, бензоле, толуоле, метилэтилкетоне, хлороформе,
ксилоле, стироле, этаноле, в водных щелочах.
Горюч; т. воспл. 220 °C; т. самовоспл. 925 °C для аэровзвеси,
555 °C для геля. Взвешенная в воздухе пыль взрывоопасна; ниж-
ний предел взрываемости аэровзвеси 20 г/м3; минимальное взры-
воопасное содержание кислорода 13% (об.).
Умеренно токсичен; ПДК 5мг/м3 (расчетная); ЛД5о=488Омг/кг
для мышей и 6500 мг/кг для крыс.
Беназол П является эффективным светостабилизатором поли-
стирола, поливинилхлорида, полипропилена, ацетата целлюлозы,
поливинилбутираля, полиамидов, лакокрасочных покрытий на ос-
нове перхлорвиниловых смол и ненасыщенных полиэфиров.
Выпускается в виде белого порошка в Швейцарии фирмой Ci-
ba — Geigy (т. пл. 131 °C) и в США фирмой Color (т. пл. 129—
132 °C).
4,4'-Диаминод1щиклогексилметан
Известны два метода получения 4,4'-диаминодициклогексилме-
тана: каталитическое аминирование 4,4'-дигидроксидициклогексил-
метана и каталитическое гидрирование 4,4'-диаминодифенилме-
тана.
1. Каталитическое аминирование 4,4'-дигидроксидициклогексил-
метана протекает по схеме [82—85]:
Реакцию обычно осуществляют в присутствии водорода в жидкой
фазе при повышенных температуре и давлении. В качестве ката-
лизаторов используют металлы VIII группы.
Впервые этим методом был получен диаминодициклогексилме-
тан в 1964 г. фирмой Du Pont (США) [82, 83]. Катализатором
служил рутений — свободный металл или в виде соединений, вос-
станавливающихся в процессе реакции. Наиболее активен рутений
на носителе (активированный уголь, SiO2, AI2O3, асбест, MgO, ки-
зельгур). Скелетные никель и кобальт менее активны. Реакцию»
7»
99
осуществляют при 275—325 °C под давлением в жидкой фазе. Ре- |
акцию можно вести и без растворителя, но более чистый продукт i
получается, если применять в качестве растворителя диоксан, тет-
рагидрофуран, диизопропиловый или дибутиловый эфир.
Если аминирование ведут в присутствии никелевых или кобаль-
товых катализаторов, необходимо во время реакции тщательно
обезвоживать систему. Для повышения выхода и степени конвер-
сии по способу фирмы Mitsui (Япония) смесь несколько раз воз-
вращают на реакцию с аммиаком [85]. Процесс оформляется тех-
нологически более сложно, но он оказывается более экономичным »
благодаря меньшей стоимости никеля по сравнению с рутением.
Как правило, 4,4/-диаминодициклогексилметан, получаемый
способом аминирования, содержит примеси аминоспиртов разного
строения. Количество нежелательных примесей можно уменьшить,
увеличивая продолжительность реакции.
Метод аминирования, предложенный фирмой BASF (ФРГ),
отличается от вышеописанного катализатором и условиями реак-
ции: оптимальная температура 170 °C [84]. В качестве катализато-
ра рекомендуется использовать смеси кобальта, марганца и фос-
форной кислоты в определенных соотношениях.
Реакцию осуществляют следующим образом. В автоклав загружают
4,4'-дигидроксид1щиклогексплметан и катализатор (91 % Со, 5,5% Мп и 3,5%
Н3РО4). Автоклав продувают водородом при «5 МПа и вводят туда аммиак.
Реакционную смесь выдерживают 10 ч при 170 °C, удаляя избыточный аммиак,
водород и воду. Для полного завершения реакции еще раз продувают авто-
клав водородом при том же давлении и снова вводят аммиак. Реакционную
массу нагревают до 170 °C и выдерживают в течение 10 ч. Затем охлаждают
смесь, снижают давление и растворяют реакционную массу в этаноле. Из полу- И
ченной суспензии отфильтровывают катализатор, а остаток перегоняют при Я
13,3 Па. Получают 4,4'-диаминодициклогексилметан с выходом 90% от теорети- Я
ческого. ,аН
4,4/-Дигидроксидициклогексилметан в промышленном масштабе Я
не вырабатывают, поэтому синтез 4,4'-диаминодициклогексилмета- Я
на целесообразно вести в одну стадию из 4,4'-дигидроксидифенил- Я
метана — доступного промышленного сырья. 4,4'-Дигидроксиди- Я
фенилметан гидрируют до 4,4'-дигидроксидициклогексилметана и Я
аминируют продукт гидрирования без выделения промежуточных Я
соединений. Такой одностадийный метод получения диаминодицик- Я
логексилметана может успешно конкурировать с промышленным Я
методом получения — гидрированием 4,4'-диаминодифенилметана. Я
2. Каталитическое гидрирование 4,4'-диаминодифенилметана .Я
осуществляют по схеме: - зЯ
HaN—^~~^-СН2—nhj H2N-/ \-СН2—/—NH2 Я
Это — промышленный способ, так как исходный 4,4'-диаминодифе- Я
нилметан в настоящее время выпускается во многих странах, а Я
его каталитическое гидрирование не представляет затруднений для Я
промышленной реализации. Я
дос Я
При детальном изучении реакции оказалось, что применение
никеля Ренея не дало ожидаемых результатов, поэтому основной
задачей оказался подбор оптимального катализатора для гидриро-
вания.
Впервые гидрирование 4,4'-диаминодифенилметана было осуще-
ствлено в начале 1940-х годов в Германии на оксиде кобальта,
промотированном оксидом калия и безводным карбонатом натрия.
В дальнейшем кобальтовым катализаторам было уделено самое
пристальное внимание [86—93]. На промотированном оксиде ко-
бальта ведут гидрирование при 210—230 °C и 15—30 МПа. В при-
сутствии металлического кобальта гидрирование протекает при бо-
лее высокой температуре (250°C), но при меньшем давлении (5—
10 МПа). При гидрировании 4,4'-диаминодифенилметана на ко-
бальт-кальциевых катализаторах при 270—290 °C и 20—30 МПа
полученный гидрированный диамин представлял собой смесь сте-
реоизомеров с т. пл. 39—43 °C и 85—95%-ным содержанием ами-
на (при расходе 5,5% масс, водорода) [86, 87]. Гидрирование
4,4'-диаминодифенилметана на оксиде кобальта, промотированном
щелочью, также дает смесь стереоизомерных 4,4'-диаминодицикло-
гексил метанов £89]:
цис, транс-
цис, цис-
Болыпое значение для проведения реакции на оксиде кобаль-
та, промотированном щелочью, имеет присутствие влаги в систе-
ме. На примере гидрирования анилина было показано, что повы-
сить выход циклоалифатических аминов можно за счет специаль-
ной обработки катализатора: перед гидрированием карбонат нат-
рия в смеси с другими компонентами катализатора тщательно вы-
сушивают при 100—130 °C.
В 1950-х годах было показано, что гидрирование 4,4'-диамино-
циклогексана можно успешно осуществить на катализаторах Ре-
нея—никеле или кобальте [92], а в 1967 г. в Японии был запатен-
тован способ получения 4,4'-диаминодициклогексилметана жидко-
фазным гидрированием 4,4/-диаминодифенилметана в присутствии
кобальта Ренея, содержащего карбонат натрия, и растворителя
[93]. Однако попытка прогидрировать 4,4/-диаминодифенилметан
при комнатной температуре в присутствии катализаторов (Pt, Pd
или Ni Ренея) в уксусной кислоте или этиловом спирте при повы-
шенном давлении оказалась малоуспешной [94]: в уксусной кис-
лоте при 50 °C выход 4,4'-диаминодициклогексилметана не превы-
шал 40%. Кобальтовые катализаторы дают возможность получать
101
диаминодициклогексилметан с более высоким выходом (50—60%),
чем на платине или палладии, но для промышленного процесса
этот выход тоже недостаточен.
Наибольший интерес представляет гидрирование 4,4,-диамино-
дифенилметана на рутениевых катализаторах, причем рутений
можно использовать в любой форме: в виде свободного металла,
оксидов, рутенатов Са, К, Na, Ba, Sr, Mg и Ag, перрутенатов калия
и натрия, хлоридов, сульфата, сульфида и других соединений [9,
10, 95—100]. Наилучшие результаты были получены на гранули-
рованном рутениевом катализаторе. Для гидрирования при низкой
температуре берут катализатор в количестве 1—5% от массы гид-
рируемого амина; при повышенных температурах количество ка-
тализатора понижается до 0,001%- При низких концентрациях
катализатора целесообразно применять рутений на носителях
(уголь, силикагель, цеолиты, оксид алюминия).
Интересно, что в зависимости от природы катализатора и тем-
пературы можно направить гидрирование в сторону образования
определенного изомера: в присутствии RujO путем низкотемпера-
турной гидрогенизации получают 4,4'-диаминодициклогексилметан
в основном в ^we-форме, а при 100—120 °C на том же катализа-
торе наблюдали преимущественное образование цис, цис- и цис,
транс-изомеров [95]. Повышение температуры и уменьшение коли-
чества катализатора от 2—5 до 0,1% сопровождается постепенным
увеличением содержания транс,транс-изомера, которое при 200 °C
становится максимальным [95]. Определить содержание каждой
модификации и разделить изомеры довольно трудно. Кроме того,
пока еще не выяснено, какой из изомеров диаминодициклогексил-
метана является наиболее активным стабилизатором. Поэтому в
большинстве случаев используют смесь стереоизомеров.
Следует отметить, что до 200 °C в присутствии рутения не на-
блюдается отщепления аммиака, следовательно, при гидрировании
на рутениевых катализаторах отпадает необходимость вводить ам-
миак, который обычно добавляют при использовании других ка-
тализаторов (добавление безводного аммиака, хотя и подавляет до
некоторой степени гидрирование, в то же время уменьшает дезами-
нирование и конденсацию, и в итоге выход 4,4'-диаминодицикло-
гексилметана повышается [95]).
Рутениевые катализаторы дорогие, но могут работать длитель-
ное время без заметного снижения активности, что особенно важ-
но при их промышленном использовании [95, 100].
В зависимости от соотношения изомеров 4,4/-диаминодицикло-
гексилметан имеет различные характеристики. При 93% транс,
транс-изомера т. пл. продукта равна 61 °C, а т. кип. 105°C (при
26,6 Па). При 80% транс, транс-изомера т. пл. продукта равна
57 °C. 4,4'-Диаминодициклогексилметан, получаемый двумя опи-
санными способами, имеет следующие характеристики: плотность
960 кг/м3; т- пл. 40—43 °C; т. кип. 120 °C при 10,7 Па и 128 °C при
16 Па; «0=1.5054. Растворим в этаноле (67,4% при 18°С).
102
4,4'-Диаминодициклогексилметан — эффективный стабилиза-
тор поливинилхлорида и пластичных смазок (эффективность зна-
чительно повышается в сочетании с тиодипропионатами); не изме-
няет цвет смазок.
4,4'-Диаминодициклогексилметан — перспективный отверди-
тель эпоксидных полимеров, позволяет заметно повысить эксплуа-
тационные свойства полимерных покрытий (особенно из электро-
изоляционных лаков и эмалей), является отвердителем полиурета-
нов и полиэфиров. Кроме того, 4,4'-диаминодифенилметан — эффек-
тивный ингибитор коррозии металлов, химический реактив и сырье
для производства полиизоцианатов.
ЛИТЕРАТУРА
1. Ворожцов И. И. Основы синтеза промежуточных продуктов и красителей.
М., Госхимиздат, 1955, с. 266—273.
2. Murakami Yojem — Chem. a. Chem. Ind., 1978, v. 31, № 4, p. 260—262;
РЖХим, 1978, 21Б1188.
3. Иванова P. Б., Хидекель M. Л.— ЖОХ, 1969, т. 39, № 11, с. 2393—2401.
4. Царева Р. С. и др. — Хим. пром-сть, 1973, № 10, с. 734—736.
5. Aniline, пат. 2894036, 1959 г. (США); РЖХим, 1961, 11Л165.
6. Сокольский Д. В., Шмонина В. П. — ДАН СССР, 1951, т. 78, с. 721.
7. Шмонина В. П. — Уч. зап. Казахского гос. ун-та, 1952, т. 14, вып. 2, с. 5.
8. Сокольский Д. В., Шмонина В. П. — В кн.: Сборник статей по общей хи-
мии. М, Изд. АН СССР, 1953. Т. 2, с. 1186—1194; РЖХим, 1955, № 13,
25823.
9. Behr L. С. е. а. —J. Am. Chem. Soc., 1946, v. 68, р. 1269.
10. Генкина Е. В. и др.-—Журн. ВХО им. Д. И. Менделеева, 1969, т. 14, № 4,
с. 475—476.
11. Пат. 119098, 1966 г. (ЧССР); РЖХим, 1968, 13Н16.
12. Пат. 119332 и 119333, 1966 г. (ЧССР); РЖХим, 1968, 9Н268. -
13. Пат. 128667, 1968 г. (ЧССР); С.А., 1969, v. 70, р. 115164.
14. Belusa J., Janousek Z., K.noflickova И.—Chem. Zvesti, 1974, v. 28, № 5,
p. 673—679; РЖХим, 1975, 13Ж311.
15. Bavin R. — Canad. J. Chem., 1958, v. 36, p. 238.
16. Balcon D., Furst A. — J. Am. Chem. Soc., 1953, v. 75, p. 4334.
17. Moore R„ Furst A. — J. Org. Chem., 1958, v. 23, p. 1504.
18. Cadojan J., Searle R. — Chem. Ind., 1963, v. 34, p. 1434.
19. Cyanamid, пат. 3159646, 1964 г. (США); C.A., 1965, v. 62, p. 7951.
20. Cyanamid, пат. 3230194, 1966 г. (США); РЖХим, 1967, 14С266.
21. J. R. Geigy, пат. 410968, 1966 г. (Швейцария); РЖХим, 1967, 23Н340.
22. J. R. Geigy, пат. 434280, 1967 г. (Швейцария); РЖХим, 1969, 6Н296.
23. J. R. Geigy, пат. 476745, 1969 г. (Швейцария); РЖХим, 1970, 9Н200.
24. Ciba — Geigy, пат. 529815, 1972 г. (Швейцария); РЖХим, 1973, 22Н246.
25. Kego, пат. 7853, 1963 г. (Япония); РЖХим, 1965, 9С295.
26. Пат. 119324, 1966 г. (ЧССР); С.А., 1967, v. 67, р. 64404.
27. Ghatge N. D., Vemekar S. Р. — Angew. makromol. Chem., 1971, В. 20, S. 165—
174.
28. Far v as M., Holcik J. — Chem. Prum., v. 17, № 10, p. 543—549.
29. BASF, заявка 2413005, 1975 г. (ФРГ); C.A., 1976, v. 84, p. 31086.
30. Eastman, пат. 4024153, 1977 г. (США); C.A., 1977, v. 87, p. 54108.
31. А.с. 157761, 1975 г. (ЧССР); РЖХим, 1977, 7Н178.
32. Hall J. H.—J. Org. Chem., 1968, v. 33, № 7, p. 2954—2956.
33. Пат. 26012, 1973 г. (Япония); РЖХим, 1974, 12Н196.
34. Пат. 3978074, 1976 г. (США); РЖХим, 1977, 11Н270.
103
35. Ciba — Geigy, заявки 2311010 и 2311011, 1977 г. (Франция); Изобр. за
рубежом, 1977, т. 24, № 2, с. 114.
36. Lauer W. М., Filbert IT. F., Ullyot G. E. — J. Am. Chem. Soc., 1947, v. 59,
p. 2584—2586.
37. Ciba — Geigy, заявка 2252341 (Франция); пат. 2454889 (ФРГ); пат. 75-88072
(Япония); пат. 822588 (Бельгия); С.А., 1975, v. 83, р. 131607.
38. ICI, пат. 969542, 1964 г. (Англия); РЖХим, 1966, 9Н118.
39. Mobay, пат. 3445516, 1969 г. (США); РЖХим, 1970, 15Н231. 1
40. Беркман Б. Е. Промышленный синтез ароматических нитросоединений и ами-
нов.М., Химия, 1964. с. 35.
41. IC1, пат. 947082, 1964 г. (Англия); РЖХим, 1965, 2Н100.
42. А. с. 181655, 1966 г. (СССР).
43. А. с. 319587, 1971 г. (СССР). .
44. Тимохин Г. А., Киссин Б. И.—Журн. ВХО им. Д. И. Менделеева, 1972, т. 17,
№ 4, с. 468—470.
45. Rondestvedt Ch. S. — J. Org. Chem., 1977, v. 42, № 10, p. 1786—1790; РЖХим, ,
1978, 5Ж140.
46. Bayer, пат. 1056619, 1961 г. (ФРГ); С. А., 1961, v. 55, р. 7354.
47. Nippon, пат. 9452, 1971 г. (Япония); РЖХим, 1971, 20Н221.
48. Ethyl, пат. 2742350, 1956 г. (США); РЖХим, 1958, № 9, 30272.
49. Rubber, пат. 2927943, 1960 (США); РЖХим, 1961, 14Л166.
50. Rubber, пат. 3055940, 1962 г. (США); РЖХим, 1964, 7Н99.
51. Universal, пат. 3121736, 1964 г. (США); РЖим, 1965, 21Н111.
52. Goodyear, пат. 3155727, 1964 г. (США); РЖХим, 1966, 8Н154.
53. Eastman, пат. 3277175, 1966 г. (США); РЖХим, 1968, 4Н226.
54. Universal, пат. 33113854, 1967 г. (США); РЖим, 1968, 14Н216.
55. Sumitomo, пат. 3435074, 1969 г. (США); РЖХим, 1970, 11Н266; пат. 6506527
(Нидерланды); пат. 1434005 (Франция); пат. 1103244 (Англия) приоритет
Японии, 1964 г.
56. 1С1, пат. 3065269, 1962 г. (США); приоритет Англии.
57. Schrdter R.— In: Houben — Weyl Methoden der Organischen'Chemie. 1957, i
В. II. Georg Thieme Verlag. ' ;
58. ICI, пат. 770299, 1957 г. (Англия); 1958, p. 7601.
59. ICI, пат. 1296211, 1972 г. (Англия); РЖХим, 1973, 14Н206.
60. ICI, пат. 1304525, 1973 г. (Англия); РЖим, 1973, 18Н148. |
61. Goodrich, пат. 2824137, 1958 г. (США); РЖХим, 1959, № 22, 79448. !
62. ICI. пат. 1440767, 1976 г. (Англия). I
63. Bayer, пат. 2355737, 1975 г. (ФРГ); РЖХим, 1976, 6Н167. I
64. Rubber, пат. 2974169, 1961 г. (США); РЖХим, 1962, 14Л173. |
65. Uniroyal, пат. 2974169, 1961 г. (США); пат. 835669, 1960 г. (Англия); С. А., (
1961, V. 55, р. 17578. (
*66. 1С1, пат. 935303, 1963 г. (Англия); РЖХим, 1964, 23Н185.
67. Кулишкин И. Т. и др. — В кн.: Сборник трудов Кузбасского политехи. ии-та,я
1976. Т. 81, с. 23—27; РЖХим, 1976, 18Б1218. Я
68. Кулишкин И. Т., Корнилова В. Н. — РЖХим, 1978, 17Б1418.
69. А. с. 517582, 1976 г. (СССР). Я
70. А. с. 582245, 1977 г. (СССР). Я
71. Сарсенбаева Г. М„ Авруцкая И. А., Фиошин М. Я- — Электрохимия, 1977.Я
т. 13, № 2, с. 290—293: РЖХим, 1977, 13Б1616. I
72. Мартынов Н. В. и др. — Хим. пром-сть, 1972, № 11, с. 24—27. я
73. Rubber, пат. 3248427, 1966 г. (США); РЖХим, 1967, 13Н216. 1
74. Nisso, пат. 19799, 1964 г. (Япония); С. А., 1965, V. 62, р. 14868. Я
75. Karvas М. е. а. — Chem. Priim., 1969, v. 19, № 3, р. 109—112. 1
76. Ciba—Geigy, пат. 410946, 1966 г. (Швейцария); РЖим, 1967, 24С553. 1
77. Ciba—Geigy, пат. 434279, 1967 г. (Швейцария); РЖХим, 1967, 2Н166. |
78. Редникова Т. А. и др.— В кн.: Тезисы докладов V Всесоюзной научно-тех-|
нической конференции по химическим добавкам для полимерных материалов.!
Тамбов, 1976, с. 23—24. я
79. Пат. 153696, 1974 г. (ЧССР); С. А., 1975, V. 82, р. 31329. Л
104 Я
80. Ciba—Geigy, заявки 2311012 и 2311013, 1977 г. (Франция); Изобр. за рубе-
жом, 1977, вып. 24, № 2, с. 115.
81. Сайкс П. Механизмы реакций в органической химии. Пер. с англ./Под ред.
Я- М. Варшавского. М., Химия, 1977. 284 с.
82. Du Pont, пат. 1019929, 1966 г. (Англия); РЖХим, 1967, 2Н151.
83. Du Pont, пат. 1420744, 1965 г. (Франция); РЖХим, 1967, 6Н86.
84. BASF, пат. 1272919, 1969 г. (ФРГ); пат. 3551485, 1970 г. (США); С. А., 1969,
V. 69, р. 51725.
85. Mitsui, пат. 49-39670, 1974 г. (Япония); РЖХим, 1975, 15Н137.
86. Кучинский В. Н., Г риз В. Е. — Нефтехимия, 1962, т. 2, Ns 4, с. 624—631.
87. Левин С. 3. и др.— В кн.: Сборник рацпредложений, внедренных в химиче-
скую промышленность. М., Изд. НИИТЭХИМ, 1963, вып. 9, с. 14—16.
88. Farbenindustrie, пат. 748457, 1952 г. (ФРГ); Chem. Zbl., 1953, S. 5578;
89. Пат. 718508, 1954 г. (Англия).
90. Пат. 113740, 1965 г. (ЧССР); РЖХим, 1967, 2Н184.
91. Mobay, пат. 3424792, 1969 г. (США); РЖХим, 1970, 9Н136.
92. BASF, пат. 954504, 1957 г. (ФРГ); РЖХим, 1958, 8916.
93. Mitsui Toachu, пат. 13095, 1970 г. (Япония); РЖХим, 1971, 10Н119.
94. Ciba Pharmacetical, пат. 2697728, 1954 г. (США).
95. Du Pont, пат. 2606924—2606928 (1952 г.); пат. 2494563, 1950 г. (США); С. А.,
1950, V. 44, р. 4498.
96. Upjohn, пат. 3591635, 1971 г. (США); РЖХим, 1972, 7Н133.
97. Upjohn, пат. 3856862, 1974 г. (США); РЖХим, 1975, 18П140.
98. ICI, пат. 1163895, 1969 г. (Англия); РЖХим, 1970, 12Н227.
99. Asahi, пат. 48-34735, 1973 г. (Япония); РЖХим, 1974, 15Н154.
100. Barkdoll А. Е. е. а. — J. Am. Chem. Soc., 1951, v. 73, р. 741; 1953, v. 75,
р. 1156—1159, 1238—1239.
2.5. КАТАЛИТИЧЕСКОЕ
ВОССТАНОВИТЕЛЬНОЕ АЛКИЛИРОВАНИЕ
АРОМАТИЧЕСКИХ НИТРОЗОСОЕДИНЕНИИ,
НИТРОСОЕДИНЕНИИ И АМИНОВ. N.N'-ДИАЛКИЛ-
И Н.И'-АЛКИЛАРИЛ-п-ФЕНИЛЕНДИАМИНЫ
Восстановительное алкилирование — процесс введения алкиль-
ной группы в органическую молекулу путем обработки амина, нит-
ро- или нитрозосоединения альдегидом, кетоном или спиртом в
присутствии восстановителя. Эта реакция, открытая в 1931 г.
Мейджером, в последние 20—30 лет получила широкое примене-
ние в синтезе и технологии производных /г-фенилендиамина. По
способу Мейджера смеси /г-нитроанилина с различными кетонами
жирного ряда подвергают восстановлению при обычной темпера-
туре в присутствии платины (катализатор); при этом образуются
вторичные 1Я,М'-диалкил-п-фенилендиамины: с ацетоном М,М'-ди-
изопропил-н-фенилендиамин, с метилэтилкетоном Ы,Ы'-ди-втор-бу-
тил-м-фенилендиамин.
В промышленности чаще всего реализуется восстановительное
алкилирование кетоном или альдегидом, так как их можно ис-
пользовать повторно, а спирт необходимо предварительно подвер-
гать дегидрогенизации до альдегида или кетона, что усложняет
процесс [1]. Реакция восстановительного алкилирования в присут-
ствии кетонов обычно приводит к вторичным аминам; даже при
105
избытке кетона значительные количества третичных аминов не
образуются [2]. Наоборот, альдегиды применяют главным обра-
зом для получения третичных аминов — NjN'-диалкил- или
Н.Ы.М'.М'-тетраалкил-п-фенилендиаминов и их смесей. Так, восста-
новительным алкилированием n-нитроанилина с альдегидом
(2-этилгексаналь) в среде метанола в присутствии никеля Ренея
и водорода при 1—1,5 МПа в течение 1 ч при повышенной темпе-
ратуре получен Ы,М'-ди(2-этилгексил)-п-фенилендиамин [3].
Восстановительное алкилирование спиртами изучено сравни-
тельно мало. В 1954 г. М.М'-диалкил-п-фенилендиамины были полу-
чены [4] восстановительным алкилированием n-нитроанилина пер- I
вичными или вторичными спиртами при 150—230 °C, давлении во- ;
дорода «10 МПа в присутствии катализаторов, содержащих ни-
кель или кобальт с добавками меди и марганца в качестве промо- ]
торов. Наиболее подробно эта реакция исследована на примере ]
восстановительного алкилирования /i-нитродифениламина н-окта- ’
нолом в присутствии никеля Ренея [5—7]!. Разработан способ по-
лучения М-октил-Ы'-фенил-п-фенилендиамина при 152 °C и атмо-
сферном давлении в жидкой фазе с отгонкой реакционной-воды и
изучена кинетика процесса.
Предполагают, что процесс состоит из четырех основных реак-
ций: 1) восстановление п-ннтродифениламина до п-аминодифенил-
амина, 2) дегидрирование спирта до альдегида, 3) алкилирование
n-аминодифениламина альдегидом с образованием Шиффова ос-
нования, 4) гидрирование Шиффова основания с образованием
М-октил-М'-фенил-п-фенилендиамина. Дегидрирование спирта мож-
но осуществить на никелевом катализаторе при «160°С [1]. Об-
разование Шиффова основания может протекать как в присутст-
вии алкилирующих средств (типа хлорида цинка или карбоновых
кислот), так и без них [8, 9]. Гидрирование Шиффова основания
на никелевом катализаторе протекает гладко при 160 °C с 86—
90%-ным выходом вторичного амина [4, 10].
При аналогичном исследовании восстановительного алкилиро-
вания 4-метил-4'-нитродифениламина алифатическими спиртами
было установлено [11], что скорость реакции зависит от приме-
няемого спирта: в случае первичного спирта процесс длится 2—
5 ч, а в присутствии вторичных спиртов до 10 ч.
Механизм восстановительного алкилирования ароматических
нитросоединений спиртами изучали [12] на примере восстанови- 1
тельного алкилирования м-нитродифениламина н-дециловым спир- |
том на никеле Ренея при 145 °C; на каждой стадии выделяли и I
идентифицировали промежуточные продукты. Изучена кинетика ’
реакции и сделан вывод, что оптимальная скорость гидрирования
n-нитродифениламина должна равняться скорости алкилирования .
амина спиртом, т. е. скорость подачи нитросоединения и спирта в
реакционную зону должна быть такой, чтобы скорость поглоще- '
ния водорода составляла 14,9 мл в минуту на 1 г катализатора. I
Максимально допустимая скорость при гидрировании п-нитроди-
106
фенил амина в жидкой фазе при 145 °C, когда обеспечивается хо-
рошая стабильность никеля Ренея, составляет 30 мл водорода в
минуту. На основании этих данных восстановительное алкилиро-
вание n-нитродифениламина н-дециловым спиртом на никеле Ре-
нея рекомендовано проводить в одну стадию и непрерывным спо-
собом.
Подробнее остановимся на восстановительном алкилировании
карбонильными соединениями, так как этот способ нашел про-
мышленное применение. Наиболее детально изучено получение
Н,М'-диалкил- и Ы.М'-алкиларилпроизводных п-фенилендиамина
алкилированием n-нитроанилина или п-фенилендиамина [2, 3, 13—
27], алкилированием w-иитрозо-, п-нитро- или п-аминодефинил-
амина кетонами [28—56] и алкилированием N-алкил-и-фениленди-
амина или N-алкил-и-нитроанилина (тоже кетонами) [57—63].
С алифатическими или циклоалифатическими альдегидами и
кетонами, содержащими от 2 до 30 атомов углерода в молекуле,
реакцию обычно проводят в жидкой фазе в атмосфере водорода
при 100—250 °C и 0,5—30 МПа в присутствии металлических
катализаторов. Процесс протекает в две стадии — сначала обра-
зуется азометиновое основание, которое затем восстанавливается
во вторичный амин:
RNOa-|-R'CHO jgo---
+hJ
* +Н2 i +Н2
RNO + R'CHO -->- RN=CHR' ->- RNH—CH2R
. —H2O
+H»|
RNHa-f-R'CHO —+“2
—H-2
Синтез азометинового основания проводят при 120—150 °C в при-
сутствии небольшого количества А1С13, ZnClj, CH3COONa, n-толу-
олсульфокислоты, других органических кислот (яблочная, винная)
или иода с непрерывной отгонкой выделяющейся воды. В качестве
растворителя применяют ксилол или толуол. Восстановление азо-
метинового основания идет в присутствии гидрирующих катализа-
торов (медь-хромовый, никель Ренея, Ni на Сг2О3 и др.) при 120—
140 °C и 8—12 МПа.
Недостаток метода — то, что реакция не останавливается на
стадии синтеза азометинового основания, а идет дальше с обра-
зованием продуктов более высокой молекулярной массы:
NH—N=C(CH3)R+CHS—COR >
---► ^—NH—N=C—CR=CR—СН8
СН3
В результате продукты имеют «размазанный» фракционный со?
став и гидрирование приводит не к индивидуальному вторичному
107
Рнс. 13. Кинетические кривые образова-
ния п-аминодифениламина (/) и п-изо-
пропиламинодифениламина (2).
амину, а к смеси аминов. Кроме
того, в продуктах присутствуют
непрореагировавший кетон и со-
ответствующий ему вторичный
спирт, которые также подверга-
ются гидрированию. Чтобы по-
давить нежелательные превра-
щения, рекомендуется вводить в
реакционную смесь небольшие
количества органических соеди-
нений серы — сульфатов, мер-
каптанов, сульфидов, тиокислот,
тиоэфиров [24, 42, 47, 48, 51, 53].
В качестве катализаторов восстановительного алкилирования
карбонильными соединениями изучены сотни различных веществ.
Оказалось [28, 47, 50, 64—66], что выход алкилированных арома-
тических аминов в присутствии сульфидов металлов выше, чем
на наиболее распространенных катализаторах — медь-хромовом,
никелевом, платине и палладии на носителях. Наибольший выход
продуктов алкилирования («99%) получен при использовании
сульфида платины на угле [28]. В присутствии сульфидов рения
и палладия в определенных условиях выход продуктов алкилиро-
вания составлял 96% [66]. Селениды и теллуриды металлов пла-
тиновой группы длительное время не теряют активности, не от-
равляются примесями сернистых соединений (не требуется очист-
ки водорода) и практически не вызывают побочных реакций [50].
В зависимости от технологического оформления процесса ка-
тализатор может находиться в виде неподвижного слоя (в трубча-
том реакторе), во взвешенном состоянии в реакционной зоне или
попадать туда как добавка к одному или нескольким реагентам.
Чаще всего восстановительное алкилирование осуществляют в
жидкой фазе.
При изучении кинетики восстановительного алкилирования
и-нитродифениламина ацетоном на гидрирующем катализаторе бы-
ло установлено [67], что процесс состоит из двух необратимых
последовательных реакций:
-н2о
Было сделано допущение, что первая стадия имеет первый поря-
док по n-нитродифениламину. График зависимости скорости обра-
+CH3COCHS;
+Н2 (^2)
108
зования /г-аминодифениламина и л-изопропиламинодифениламина
от времени [67] приведен на рис. 13. Полученные кинетические
данные были использованы для расчета технологической схемы
каскада реакторов и выбора режима процесса. Подтверждена пра-
вильность кинетического уравнения и установлена хорошая вос-
производимость процессов.
NJ^'-Ди-втор-бутил-п-фенилендисмин
СН3—CHj—CH—HN—NH—СН—СН2—СН3
СН3 СН8
NJV'-Ди-втор-бутил-и-фенилендиамин впервые был получен Мейджером
в 1931 г. восстановительным алкилированием п-нитроанилина или п-фениленди-
амина метилэтилкетоном в присутствии оксида платины при комнатной темпера-
туре. Описано также получение М,М'-ди-егор-бутил-п-фенилендиамина алкили-
рованием n-нитроанилина 1-метилпропаналем в среде метанола [3] и алкилиро-
ванием п-фенилендиамина втор-бутанолом в среде этанола [17].
Наибольшее число способов получения N/N-ди-втор-бутил-н-фе-
нилендиамина основано на восстановительном алкилировании
n-нитроанилина или л-фенилендиамина метилэтилкетоном [13—
16, 21, 24, 27, 48, 50, 54, 59, 66]:
h2n-(/ no2
+CHsCOC2H6; —ЗН2О
СН3—СН2—CH—HN-
I
сн3
сн3
H2N—NH>
+CHSCOC2H5; I -Н2О
Реакцию осуществляют при ПО—180°С, 0,7—20 МПа и мольном
отношении водорода к кетону от 20: 1 до 100: 1. Катализаторы —
смеси оксидов меди, хрома и бария [14, 54], меди и хрома [15,
54], серебра и палладия [27], никель на носителе и без него [69],
платина на оксиде алюминия [48, 68] или на других носителях
[13, 16, 24, 59], селениды и теллуриды металлов платиновой груп-
пы [50], сульфиды рения или платины [66], соли органических
кислот (преимущественно ацетаты) ,[21].
Г^М'-Ди-втор-бутил-л-фенилендиамин, наиболее известный за ру-
бежом под названием Topanol М, получают непрерывным восста-
новительным алкилированием л-нитроанилина метилэтилкетоном
при 160°C и 5 МПа в трубчатом реакторе [16]. Метилэтилкетон
и л-нитроанилин берут в соотношении 8: 1 при скорости подачи
водорода 0,5 ч-1 и его давлении 0,73 МПа. Органические реаген-
ты и водород вводят в верхнюю часть вертикального реактора и
пропускают через неподвижный слой катализатора — оксида алю-
миния, содержащего 0,3% платины и 0,3% фторорганического со-
109
единения (эту смесь предварительно обрабатывают водородом и
парами бензина при 465°C).
Поток полученных продуктов отбирают из нижней части реак-
тора (без понижения давления в системе) через холодильник и
отделяют охлажденные жидкие продукты. Водород возвращают в
цикл, а жидкие продукты пропускают в первую фракционную зо-
ну, где продукт алкилирования отделяют от более низкокипящей
фракции, содержащей непрореагировавший кетон и втор-бутанол.
Эту фракцию разделяют во второй фракционной зоне на кетон и
спирт; кетон возвращают в реактор. Фракцию, содержащую про-
дукт алкилирования, пропускают в третью фракционную зону, где
М.М'-ди-втор-бутил-я-фенилендиамин отделяется от п-фениленди-
амина и N-втор-бутил-п-фенилендиамина. Степень конверсии п-ни-
троанилина в Й,М'-ди-втор-бутил-п-фенилендиамин превышает
99%.
Свойства и применение. Ы,П'-Ди-втор-бутил-п-фенилендиамин
представляет собой прозрачную красную жидкость, темнеющую на
воздухе. Плотность 940—942 кг/м3; т. пл. 15 °C; т. кип. 295—
300 °C.
Токсичен; ЛД5о=250 мг/кг; раздражающе действует на кожу.
М,ЬГ-Ди-втор-бутил-п-фенилендиамин, (Диафен ББ) эффектив-
ный и широко применяемый ингибитор смолообразования автомо-
бильных бензинов, стабилизатор смазочных масел и топлив. Вы-
пускается в промышленном масштабе в США, Англии и ФРГ в
виде красновато-коричневой жидкости под разными торговыми
наименованиями:
Торговое наименование Страна, фирма | Торговое наименование Страна, фирма
Amoco 632 Antioxidant 22 Antioxidant М Gasoline АО-22 США, Amoco США, Du Pont Англия, ICI США, Du Pont Kerobit BPD Tenamene 2 TopanolM UOP 5 ФРГ, BASF , США, Eastman Англия, ICI США, UOP
Для повышения эффективности его' часто используют в сочета-
нии с другими стабилизаторами, а также выпускают промышлен-
ные марки смесевых стабилизаторов: с дисалицилиденметилдипро-
пилтриамином (Kerobit СОМ, ФРГ) —для повышения пассивиру-
ющего действия металлов переменной валентности; с 6-трет-бутил-
2,4-диметилфенолом (Topanol AN/М, Англия).
М.М'-Ди-втор-бутил-п-фенилендиамин защищает резины на ос-
нове натурального и синтетических каучуков от термоокислитель-
ного старения (на уровне Неозона Д) и от озонного старения (ус-
тупает Диафену ФП). Стабилизатор полипропилена и волокон на
его основе. На практике для стабилизации полимеров не исполь-
зуется из-за высокой токсичности.
М-Фенил-Ы'-циклогексил-п-фенилендиамин
Впервые №фенил-М'-циклогексил-п-фенилендиамин был получен и запатен-
тован в 1937 г. в США фирмой General Aniline Film, восстановительным алкили-
рованием n-аминодифениламина циклогексанолом или циклогексаноном в присут-
ствии никелевого катализатора и водорода. Полученный продукт плавился при
118—119 °C.
В 1939 г. аналогичный способ его получения был запатентован во Франции,
а в 1941 г. в Германии.
Известные способы получения N-фенил-М'-циклогексил-п-фени-
лендиамина можно разделить на 5 типов.
1. Гидрирование Ы,Ы'-дифенил-п-фенилендиамина при 5—
30 МПа на никелевом катализаторе:
Этот метод получения оказался для промышленности непригод-
ным, потому что не удается так подобрать условия реакции, что-
бы селективно прогидрировать одно ядро дифенил-п-фениленди-
амина; всегда получается смесь продуктов разной степени гидри-
рования. Кроме того, способ экономически нецелесообразен, так
как дифенил-п-фенилендиамин — дорогой реагент.
2. Восстановительное алкилирование смеси п-циклогексилнит-
робензола, N-циклогексил-п-фенилендиамина и циклогексанона:
Этот способ предложен фирмой Monsanto (США) в 1961 г. [57].
Циклогексанон нагревают с N-циклогексил-п-фенилендиамином и
/г-циклогексил-л-нитробензолом в присутствии палладия на угле
при 130—200 °C с отгонкой образующейся азеотропной смеси воды
с циклогексаноном. Реакционную смесь охлаждают и фильтруют.
Избыток циклогексанона отгоняют в вакууме и получают N-фе-
нил-ГК-циклогексил-л-фенилендиамин с выходом 82%- Т. пл. про-
дукта 117,6—118,6 °C (из н-гептана). По другому варианту этого
способа М-фенил-Ы'-циклогексил-п-фенилендиамин получают из
N-циклогексил-п-нитроанилина с выходом 36% [57].
Способ 2 промышленного значения не имеет, так как получае-
мый продукт загрязнен продуктами гидрирования.
3. Восстановительное алкилирование N-циклогексил-п-нитро-
анилина циклогексаноном [58]:
+нг
-няо
111
Реакцию проводят при 150—300 °C в присутствии оксидов, сульфи-
дов, хроматов, нитратов и манганатов металлов (Со, Сг, Си, Fe,
Mg, Ni, Os, Pt), иногда в смеси с соединениями других металлов:
Си с Ag, Сг или Zn, Ni с Al, Zn, Со или Си. Смесь N-циклогек-
сил-/г-нитроанилина, циклогексанона и катализатора (палладий
на угле) нагревают в атмосфере водорода и в течение 1 ч выдер-
живают при 161—174 °C, отгоняя образующуюся воду. Затем реак-
ционную массу охлаждают до 20°C и отфильтровывают. От фильт-
рата отгоняют легколетучие продукты, нагревая смесь до 200 °C в
кубе. Остаток перемешивают с петролейным эфиром; твердый про-
дукт отфильтровывают, промывают этим же эфиром и сушат на
воздухе. Получают Ы-фенил-П'циклогексил-п-фенилендиамин с вы-
ходом 36% и т. пл. ИЗ—118 °C.
Способ 3 промышленного значения тоже не имеет.
4. Восстановительное алкилирование N-нитрозо-п-нитродифе-
ниламина циклогексаноном '[41]:
Реакцию проводят в присутствии палладия на угле под давлени-
ем— вначале при комнатной температуре, а затем при 160 °C до
прекращения поглощения водорода. Выход Ы-фенил-Ы'-циклогек-
сил-п-фенилендиамина 78,9%; т. пл. 111 —114 °C (из петролейного
эфира).
5. Восстановительное алкилирование п-нитрозо-, n-нитро- или
n-аминодифениламина циклогексаноном (или циклогексанолом)
[30—32, 45, 50, 70, 71]:
На этом методе основано промышленное получение рассматри-
ваемого стабилизатора. Реакцию осуществляют в атмосфере во-
дорода при 80—250°С и 3—15 МПа в присутствии различных гид-
рирующих катализаторов — свободного никеля или его хромата.
смеси оксидов меди, хрома и никеля, а также платины, селенидов
и теллуридов кобальта и др.
Для промышленного получения И-фенил-Ы'-циклогексил-п-фе-
нилендиамина прорабатывались два варианта восстановительного
алкилирования — взаимодействие n-аминодифениламина с цикло-
гексанолом под давлением в присутствии гидрирующего катализа-
112
тора и водорода и взаимодействие n-аминодифениламина с цикло-
гексанолом при атмосферном давлении в присутствии никеля Ре-
нея с одновременной отгонкой образующейся воды.
Условия реакции, сырье и катализаторы в значительной степе-
ни определяют выход и чистоту получаемого стабилизатора. Один
из ранних технологических процессов получения N-циклогексил-
Ы'-фенил-л-фенилендиамина осуществляли следующим образом.
Циклогексанол загружали в реактор с мешалкой и змеевиком и
добавляли туда /г-аминодифениламин и никель Ренея. Реакцион-
ную массу нагревали до 180°C. При этой температуре отгоняли
образующуюся воду с небольшим содержанием циклогексанола,
а дистиллят передавали в сепаратор. Конец реакции определяли
по снижению давления в системе.
Реакционную массу охлаждали и фильтровали. Фильтрат сли-
вали на бензин и нагревали до 60 °C при включенной мешалке, за-
тем охлаждали до 20 °C и фильтровали вторично. Осадок на
фильтре трижды промывали бензином, смешивали с водой и спус-
кали в перегонный куб, где отгоняли бензин. Остаток охлаждали
до 20 °C, фильтровали на нутч-фильтре, промывали и сушили. Ото-
гнанный циклогексанол, содержащий примесь циклогексанона, на-
правляли на регенерацию. Отфильтрованный кристаллический
Ц-фенил-М'-циклогексил-л-фенилендиамин высушивали и упаковы-
вали. Выход 82% от теоретического.
В ГДР предложен несколько отличный от описанного метод
восстановительного алкилирования [72, 73], когда М-фенил-Ы'-цик-
логексил-/г-фенилендиамин получают алкилированием /г-аминоди-
фениламина циклогексанолом или циклогексаноном. Образующее-
ся на первой стадии основание Шиффа восстанавливают цинковой
пылью (в присутствии муравьиной кислоты) или железным порош-
ком при 80—90 °C (в присутствии этиленгликоля, глицерина или
гексантриола). Получают М-фенил-М'’-циклогексил-/г-фениленди-
амин с т. пл. 112—116 °C [73]. Химические превращения, приводя-
щие к образованию Ы-фенил-М'-циклогексил-/г-феиилендиамина,
представлены ниже:
Механизм реакции включает образование основания Шиффа или
гем-диаминосоединений с аминами и последующее восстановление
муравьиной кислотой:
\ I —н2о /
RNH2 + /С=О RNCOH R'N=C\
8—1057
113
На первую, гетеролитическую стадию оказывает влияние катали-
затор. На гомолитическое восстановление влияет температура.
Механизм восстановительного алкилирования /г-аминодифенил-
амина циклогексанолом можно объяснить следующим образом.
При нагревании с катализатором происходит дегидрирование цик-
логексанола в циклогексанон. Последний реагирует с /г-аминоди-
фениламином, образуя основание Шиффа или гидроксиамин, ко-
торые гидрируются в М-фенил-М'-циклогексил-п-фенилендиамин:
Из большого числа изученных катализаторов для этой реакции
лучшими оказались никель, осажденный на оксиде алюминия, и
скелетный никель. Менее эффективны никель, осажденный на као-
лине, и медь, осажденная на оксиде магния. В отношении расхода
более экономичен никель на А12О3, но он был чувствителен к за-
грязнениям и его приготовление связано с известными трудностя-
ми. Более выгодным оказался скелетный никель в дозировке 12—
13% (масс.) от количества n-аминодифениламина. Увеличение ко-
личества катализатора не повышает выход N-фенил-М'-циклогек-
сил-п-фенилендиамина и не улучшает его качество.
Описано алкилирование n-аминодифениламина циклогексано-
ном в органическом растворителе:
NH—nh2 + О=\~/
В качестве растворителей можно использовать алифатические
спирты С4—Cg, циклические спирты, например циклогексанол и
метилциклогексанол. На первой стадии в отсутствие воды обра-
зуется М-фенил-М'-циклогексилиден-п-фенилендиамин. Не выделяя
промежуточного продукта, проводят восстановление цинковой
пылью и 85%-ной муравьиной кислотой. Муравьиную кислоту до-
бавляют медленно после введения небольшого количества цинко-
114
вой пыли при 85—90 °C. При этом муравьиная кислота разлагает-
ся с образованием диоксида углерода, который интенсивно выде-
ляется и вместе с незначительным количеством растворителя уно-
сит всю реакционную воду. Реакция слабоэкзотермическая, закан-
чивается через 90 мин. Избыток муравьиной кислоты нейтрализу-
ют карбонатом натрия, а образующийся формиат цинка выделяют
в виде карбоната цинка. После этого вакуум-перегонкой отгоняют
растворитель. После фильтрования от цинкового шлама промыва-
ют реакционную смесь 50—70%-ным метанолом и отжимают;
М-фенил-ЬГ-циклогексил-п-фенилендиамин кристиллизуют. Выход
превышает 80%; т. пл. 112—-114 °C. Этот метод может быть оформ-
лен в непрерывном варианте по первой стадии и в полунепрерыв-
ном— по второй.
Периодический способ. В реактор с мешалкой загружают пе-
регнанный n-аминодифениламин и растворяют его при 90°C в ме-
тилциклогексаноле в течение 0,5 ч при перемешивании. Затем до-
бавляют циклогексанон и перемешивают смесь при 80—90 °C в
течение часа. После этого загружают цинковую пыль, а затем за
час добавляют необходимое количество муравьиной кислоты. Об-
разующийся диоксид углерода отсасывают вентилятором. Азео-
тропной отгонкой с циклогексаноном удаляют реакционную воду
с парами растворителя. Пары конденсируют в холодильнике, а
конденсат (смесь воды и метилциклогексанона) направляют в се-
паратор. После перегонки к реакционной смеси добавляют воду
(для растворения выделившейся натриевой соли), а реакционную
массу, нагретую до 100—110°С, отводят по обогреваемым трубо-
проводам через фильтр в кристаллизатор, где находится смесь ме-
танол-1-вода. Кристаллическую массу охлаждают до 20°C и через
2 ч отсасывают на нутч-фильтре. А1аточный раствор разбавляют
водой и выпускают в канализацию. Осадок на фильтре промывают
метанолом. Продукт сушат при 60—70°C в вакуумном шкафу и
размалывают на шаровой мельнице. Выход 78—83%.
Полунепрерывный способ. В реактор загружают п-аминодифе-
ниламин и добавляют метилциклогексанол. Реакционную массу
нагревают до 80 °C до полного растворения амина. Затем добав-
ляют циклогексанол и перемешивают в течение 1 ч при 90 °C. Пос-
ле этого в течение часа добавляют из мерника муравьиную кисло-
ту. Вентилятором отсасывают образующийся по реакции диоксид
углерода вместе с парами воды и растворителя. Воду и раствори-
тель через холодильник собирают в делительную воронку, где раз-
деляют на два слоя — водный (нижний) и органический (верхний).
После добавления всей муравьиной кислоты перемешивают ре-
акционную массу в течение часа при 90 °C, нейтрализуют карбона-
том натрия, промывают в течение 15 мин и отжимают на фильтр-
прессе (при подаче азота) в две промежуточные емкости. В вы-
парном аппарате при 90—110°С и «4 кПа отгоняют раствори-
тель. Реакционную смесь дозировочным насосом подают в тонко-
слойный выпарной аппарат. Образующийся в нижней части аппа-
8*
115
рата расплав нагнетают дозировочным насосом в кристаллизатор,]
где находится смесь метанол + вода. Пары растворителя конденсиЗ
руют в холодильнике, а дистиллят улавливают в мернике. Крис-1
таллическую массу охлаждают при перемешивании до 20 °C, neJ
рез 2 ч выгружают из центрифуги и промывают холодным метано-1
лом. Выход 80—86°/о- 1
Свойства и применение. М-Фенил-М'-циклогексил-л-фенилен-!
диамин (Диафен ФЦ) представляет собой светло-серый порошок,
темнеющий при хранении; т. пл. 118—119°С; т. кип. 232—235°C
(при 0,26 кПа). Растворяется в ацетоне, бензоле, метиленхлориде,
тетрахлорэтане, четыреххлористом углероде, хлороформе, этаноле,
этилацетате.
Умеренно токсичен; ЛД5о=39ОО мг/кг.
К-Фенил-М'-циклогексил-л-фенилендиамин является стабили-
затором синтетических каучуков общего назначения; пассивирует
действие металлов переменной валентности. Защищает резины на
основе натурального и синтетического каучуков от термоокисли-
тельного и светоозонного старения и разрушения при многократ-
ных деформациях. Является термостабилизатором полипропиленам
полиамидов, полиацетальдегида, полиформальдегида. Эффектив!
ный стабилизатор ракетных топлив. 1
М-Фенил-Ы'-циклогексил-и-фенилендиамин выпускают в США|
Японии и ФРГ:
Торговое наименование Страна, фирма Выпускная форма Т. пл., ®С .«
Anti gene GH Япония, Sumitomo Порошок 103—107 1
ASM 4010 ФРГ, Bayer То же 115
Antigene 6H Япония, Sumitomo 103—107
Ervo США, Vanderbilt » 103—110 1
Flexizone 6H США, Naugatuck — —
Ozozone 6H Япония, Seiko Порошок 1.05—110
Santoflex CP США, Monsanto То же >114
Super Santoflex США, Monsanto — —.
UOP 36 США, Universal Oil — - —
По эффективности этот стабилизатор уступает N-изопропил
N'-фенил-л-фенилендиамину и постепенно теряет свое значение.
Ы-Изопропил-1\,г'-фенил-п-фенилендиамин
V-NH——NH—СН(СН8)2
Первое упоминание о синтезе К-изопропил-И'-феиил-п-феиилендиамииа я
применении его в качестве стабилизатора относится к 1952 г., когда этот про
дукт был запатентован фирмой IC1 (Англия) как новое химическое соединений
[39]. Позднее его получали восстановительным алкилированием п-нитрозо-
п-иитро- или п-аминодифениламииа ацетоном в среде водорода на палладиевом
катализаторе, нанесенном на уголь, и взаимодействием фенилгидразииа с N-изо
пропил-п-нитрозоанилином по Фишеру и Вакеру. Методы синтеза и свойства
116
К-изопропил-Н'-фенил-п-фенилеидиамина были изучены в 1950—1960 годах,
когда многие зарубежные фирмы начали создавать его промышленное произ-
водство.
Известные способы получения М-изопропил-Ы'-фенил-п-фени-
лендиамина основаны на нескольких реакциях [74].
1. Араминирование N-изопропил-п-аминофенола анилином
[18]:
Но-С_/ nh-ch(chs)8 + £3-NH*
-NH—NH—СН(СН8)2
2. Араминирование /г-гидроксидифениламина изопропиламином
[18]:
NH-ОН + H2N—СН(СН8)2
---► ^-ЫН-/”Л-ЫН-СН(СН8)2
3. Алкилирование п-аминодифениламина изопропилбромидом
[75]:
Эту реакцию проводят при 55—120 °C в течение 2—3 ч в присут-
ствии бензилтриэтиламмония (катализатор). Полученный гидро-
бромид М-изопропил-М'-фенил-п-фенилендиамина в системе раство-
рителей вода+диэтиловый эфир нагревают и выдерживают в те-
чение 30 мин при 65—80 °C, затем охлаждают до 20 °C, обраба-
тывают ацетоном и нейтрализуют щелочью.
4. Восстановительное алкилирование М-(п-нитрозофенил)-М-фе-
нилгидроксиламина ацетоном [70, 76]:
М-(н-Нитрозофенил)-М-фенилгидроксиламин растворяют в аце-
тоне, смесь выдерживают в течение 1 ч при 60—150°С под давле-
нием водорода 5 МПа в присутствии палладия на угле, а затем
нагревают еще 6 ч при 95 °C. После удаления катализатора и от-
гонки растворителя получают М-изопропил-М'-фенил-п-фениленди-
амин с выходом 93%. Реакцию можно проводить в присутствии
117
воды, например в качестве исходного сырья можно брать водную]
пасту №(п-нитрозофенил)-№фенилгидроксиламина.
5. Восстановительное алкилирование нитро- и аминосоединений
в присутствии циклогексанона [57]:
H2N-(
+4Н2
—4HgO
NH—СН(СН3)2
Смесь N-изопропил-п-фенилендиамина, N-изопропил-п-нитроанили-
на и циклогексанона в присутствии палладия на угле нагревают
20 мин при температуре, повышающейся от 137 до 178 °C, отгоняя
выделяющуюся воду. Реакционную смесь охлаждают и фильтру-
ют; избыток циклогексанона отгоняют в вакууме (отгонку заканчи-4
вают при 175°С и 26,6 кПа) и получают 70%-ный N-изопропил-^
Ы'-фенил-м-фенилендиамин с примесью М-циклогексил-Ы'-фенил-, I
№циклогексил-№-изопропил- и М.Ы'-дифенил-п-фенилендиамина. I
6. Восстановительное алкилирование п-аминоазобензола ацето-j
ном [77]: ]
/) -J-3H2
;-N=N-/ \-NH2 -+- CHS—GO—СН3 - >
\ _ / —П2О, —NH3
•NH—e NH-CH(CH3)2
Реакцию осуществляют в жидкой фазе при 165°С под давлением Л
в присутствии медь-хромового катализатора. В качестве побочных]]
продуктов образуются N-изопропиланилин и N.N'-ди (изопропил)-]
n-фенилендиамин. Этот процесс менее экзотермичен, чем восстано- ,
вительное алкилирование п-нитрозо- или п-нитродифениламина. 1
7. Восстановительное алкилирование N-нитрозо-п-нитродифе-
ниламина ацетоном [41]:
—NO, + СН3—СО—СН3
NO
+2Н2
---->-
—HNO3
NH—СН(СНд)2
N-Нитрозо-п-нитродифениламин и ацетон подвергают восстанови-J
тельному алкилированию на палладии, нанесенном на уголь, под]
давлением водорода 8—10 МПа (3 ч при «20 °C и еще 6 ч при!
160°С — до прекращения поглощения водорода). К продуктам ре-Ц
акции добавляют ацетон, отфильтровывают смесь, упаривают
фильтрат и получают М-изопропил-Ы'-фенил-п-фенилендиамин с
выходом 65% и т. пл. 78—79 °C.
118
8. Восстановительное алкилирование п-нитро-, n-нитрозо- или
п-аминодифениламина ацетоном:
no2 —
NH—СН(СНа)2
Этот метод наиболее широко освещен в литературе [18, 30, 32, 33,
35, 36, 40—43, 46, 47, 49—51, 56, 64, 78] и является основой про-
мышленного получения описываемого стабилизатора. Реакцию
обычно осуществляют в жидкой фазе в атмосфере водорода при
100—250 °C и давлении от 0,5 до 30 МПа в присутствии различ-
ных гидрирующих катализаторов. Наибольший выход и чистота
конечного продукта достигаются при использовании никеля на ки-
зельгуре, модифицированного тиокарбамидом [51], селенидов ме-
таллов [50] и некоторых смесей оксидов металлов, причем соот-
ношение этих оксидов влияет на выход и качество продукта.
В целом метод получения Ы-изопропил-М'-фенил-д-феннленди-
амина восстановительным алкилированием н-нитро-, п-нитрозо-
или гг-аминодифениламина имеет существенные преимущества:
применяется доступное сырье; продукт получается с высоким вы-
ходом и хорошего качества. В то же время метод не лишен недо-
статков: процесс необходимо проводить под давлением; использу-
ется большой избыток алкилирующего агента (ацетон) по отно-
шению к амину, причем значительная часть ацетона гидрируется
в спирт и не может быть использована в последующих синтезах;
применяются дорогостоящие катализаторы; велика длительность
процесса (от 14 до 24 ч). Поэтому разработка методов синтеза
этого соединения по реакциям 3 и 4 [70, 76] (см. стр. 117) пред-
ставляет несомненный интерес.
В промышленности К-изопропил->Ы,-фенил-д-фенилендиамин
обычно получают непрерывным или полунепрерывным способом,
которые различаются применяемыми катализаторами и раствори-
телями. Процесс проводят на медном [40] и медь-хромовом [46]
катализаторах, полученных по патенту [44]. Известно использова-
ние и платиновых катализаторов [33, 41, 78]; для них требуется,
чтобы исходный n-нитродифениламин не содержал S и С1.
В качестве побочного продукта может образоваться И-(1,3-ди-
метилбутил)-И'-фенил-п-фенилендиамин
NH—СН—СНа—СН— СН8
~~ CHS СН3
119
который как стабилизатор более эффективен, чем N-изопропил-
N'-фенил-и-фенилендиамин. В некоторых процессах эту примесь
отделяют перегонкой, в других товарный стабилизатор выпускает-
ся с примесью этого побочного продукта.
Технологическая схема получения. N-Изопропил-М'-фенил-п-фе-
нилендиамин (Диафен ФП) получают восстановительным алкили-
рованием n-аминодифениламина ацетоном на медь-хром-железном
катализаторе:
0_nh-0-nh2 + chs^o-ch8
—NH—СН(СН8)2
Побочно идет гидрирование ацетона др изопропанола:
. +на
сн8—со-сн8 —►. сн3—сн—сн8
он
п-Аминодифениламин (АДФА) через подогреватель (на рис. 14
не изображен) и смесь ацетона с катализатором подают в авто-
клав 2; туда же поступает водород при 150 °C и »5,8 МПа. Из ав-
токлава 2, водород вместе с реакционной массой уходит в авто-
клав 4. Пары ацетона из автоклава 4 конденсируют и через ем-
кость 5 возвращают в производство, а водород с 20% азота сжи-
гают. В автоклавах 2 и 4 при 150 °C и 5 МПа протекает процесс
Рис. 14. Технологическая схема получения М-изопропил-М'-фенил-л-фениленди-
амина:
1 — смеситель для приготовления каталнзаторной суспензии; 2, 4 — автоклавы; 3, 10, 13 —
холодильники-конденсаторы; 5 — емкость; 6 — приемник; 7 — фильтр; 8 — испаритель; 9, 12 —
дистилляционные колонны; 11, 14 — приемники; 15 напорная емкость; 16, 17 — выносные
кипятильники.
120
восстановительного алкилирования с образованием л-изопропил-
аминодифениламина; при этом ацетон частично гидрируется до
изопропанола.
Реакционная масса после снижения давления поступает в при-
емник 6\ туда же подают на разбавление рециркулирующий аце-
тон. Разбавленную массу фильтруют на фильтре 7; катализатор
отводят в шламонакопитель, фильтрат подают в испаритель 8,
ацетон и изопропанол направляют на регенерацию, а полученный
Диафен ФП (сырец) —на дистилляцию.
В колонне 9 отгоняют легкую фракцию (н-аминодифениламин),
а кубовую жидкость подают в колонну 12. Там при 230 °C и оста-
точном давлении 2,66 кПа происходит дистилляция Диафена ФП.
Последний поступает в емкость 14 и далее в напорную емкость 15,
откуда подается на чешуирование и упаковку. Кубовый остаток
из колонны 12 сжигают.
Свойства и применение. М-Изопропил-М'-фенил-/г-фениленди-
амин представляет собой белый кристаллический порошок, розо-
веющий при хранении; плотность 1290 кг/м3; т. пл. 80,5 °C; т. кип.
366 °C. Растворяется в ацетоне, бензоле, диметилформамиде, диок-
сане, дихлорэтане, изопропаноле, метилэтнлкетоне, толуоле, пири-
дине, четыреххлористом углероде, хлорбензоле, этаноле, этилаце-
тате, диэтиловом эфире, в растворах кислот. Ограниченно раство-
ряется в циклогексане, ксилоле, этиленгликоле.
Горюч; т. самовоспл. 529 °C.
Умеренно токсичен. ПДК=2 мг/м3; ЛДбо=3030+280 мг/кг.
М-Изопропил-|М/-фенил-п-фенилендиамин выпускается в боль-
шинстве развитых стран под различными торговыми наименова-
ниями:
Торговое наименование Страна, фирма Выпускная форма Т. пл., °C
Диафен ФП СССР Чешуйки >73
Antigene ЗС Япония, Sumitomo Порошок >70
Antioxidant С ЧССР Чешуйки, >70
порошок
Antioxidant CD ГДР Чешуйки 75
ASM CD ФРГ, Bayer Порошок 75
ASM 4010 NA ФРГ, Bayer Чешуйки, 75
хлопья
Gyazone IP США, Cyanamid — —
Eastozone 34 США, Eastman Чешуйки —
Flexzone 3C США, Naugatuck Пластинки >70
Ipognox 44 ПНР — —
Monox ZA Англия, 1CI Кусочки —
Nocrac 810-NA Япония, Ouchi Shinko Порошок >70
Nonox ZA Англия, 1CI Кусочки >70
Ozozone 3C Япония, Seiko Хлопья >70
Permanax 115 Франция, Rhone Poulenc Порошок, 74
чешуйки (т. затв.)
Santoflex 36 Англия, Monsanto — —
Santoflex IP Англия, Monsanto Чешуйки >76
121
П-Изопропил-Ы'-фенил-п-фенилендиамин является эффектив-
ным стабилизатором синтетических каучуков общего назначения;®
пассивирует действие металлов переменной валентности при де-в
струкции каучуков. Важнейший стабилизатор резин (особенно®
шинных); защищает их от термоокислительного и светоозонного я
старения, от разрушения при многократных деформациях. Е
М-Изопропил-№-фенил-л-фенилендиамин — эффективный термо- Ж
стабилизатор полиэтилена, полистирола и полиамидов, но практи-а
чески для этих целей он не используется, так как окрашивает по- »
лимерные материалы. Ингибитор смолообразования в моторных и а
ракетных топливах.
М-(1,3-Диметилбутил)-№-фенил-п-фениленд11амин а
NH-CH—СНа—CH—СН3 <
СН3 СН3 i
К-(1,3-Димс1Илбутил)-Н'-фенил-п-фенилендиамин как стабилизатор впервые Я
был предложен в 1963 г. фирмой Monsanto (США) [79]. Позже на способ его -1
получения были выданы приоритетные патенты и другим фирмам [38, 80]. Это' 1
соединение оказалось невымываемым эффективным антиоксидантом, и антиозо-
нантом резин. 1
И-(1,3-Диметилбутил)-И'-фенил-д-фенилендиамин получают 1
восстановительным алкилированием /г-нитро-, n-нитрозо- или 4
n-аминодифениламина изобутилметилкетоном в присутствии гид- ;
рирующих катализаторов [28, 35, 38, 43, 50, 52, 53, 79, 80]:
Реакцию обычно осуществляют при 165—185 °C и 2,5—10 МПа.
В качестве катализаторов применяют: ацетаты металлов; никель,
модифицированный серой; смеси оксидов цинка, меди и хрома;
сульфиды платины; родий; селениды и теллуриды кобальта; слож-
ные многокомпонентные системы, содержащие никель, кислоту и
органические соединения серы.
Приведем несколько примеров таких синтезов.
В автоклав загружают n-аминодифениламин, изобутилметилкетон и палла-
диевый катализатор на угле. Автоклав продувают азотом и водородом, доводя
давление водорода до 3,5 МПа. Реакционную массу нагревают и выдерживают
при 180 °C и 2,8—4,2 МПа в течение 1,25 ч. Катализатор отфильтровывают, а
фильтрат концентрируют и перегоняют при 200 °C и остаточном давлении
«3,7 кПа. Выделенный МДКЗ-диметилбутилрЫ'-фенил-я-фенилендиамин иден-
тифицируют методом газовой хроматографии. Чистота его 99%; выход 98% от
теоретического [80J.
Фирма Monsanto (США) получает М-(1,3-диметилбутил)-1Ч'-фенил-я-фени-
лендиамин (Santoflex 13) восстановительным алкилированием я-аминодифенил-
амина изобутилметилкетоном. Выход выше 60%.
Существует так называемый одноступенчатый способ получения жидких
смесей М-алкил-№-фенил-п-фенилендиаминов фирмы Goodyear Tire (США) [52].
По этому способу в автоклав загружают «-аминодифениламин, изобутилметил-
кетон, изоамилметилкетон, никелевый катализатор и я-толуолсульфокислоту.
При «6,8 МПа пропускают водород; температура при этом повышается до
185 °C. При перемешивании смесь выдерживают 3 ч, охлаждают до 100 °C и
снижают давление до атмосферного. Катализатор отфильтровывают, а продукт
перегоняют при 190 °C и остаточном давлении 1,33 кПа и охлаждают. Получают
жидкий (при 22 °C) стабилизатор, содержащий 0,1% п-аминодифсниламина,
46,1% М-(1,3-диметилбутил)-М'-фенил-я-фенилендиамина и 48% М-(1,4-диметил-
амил)-М'-фенил-я-фенилен диамина.
Другой вариант получения «смесевого» стабилизатора запатентован фирмой
Kodak (Англия) [60]. Смесь анилина и циклогексиламина по каплям в течение
30 мин добавляют к смеси 1Ч-(1,3-димет11лбутил)-я-гпдроксианнлина, иода, же-
леза и хлорида аммония. Смесь перемешивают в токе азота при 215—220 °C
(для поддержания такой температуры добавляют ксилол), выдерживают 3 ч
при этой температуре, охлаждают, растворяют в тетрагидрофуране и фильтруют.
После упаривания получают смесь, содержащую 30% М-(1,3-диметилбу-
тил) -N'-фенил-я-фенилендиамина, 10% М,М'-ди(1,3-диметилбутил) -я-фениленди-
амина, 9% М,М'-дифенил-я-фенилендиамина, 8% М-фенил-М'-циклогексил-я-фени-
лендиамина и 5% М'-^.З-диметилбутил^М'-циклогексил-я-фенилендиамина.
Технологическая схема получения. Опытное производство
М-(1,3-диметилбутил)-N'-фенил-я-фенилендиамина (Диафен 13)
состоит из шести стадий: приготовление раствора я-аминодифенил-
амина в изобутилметилкетоне, восстановительное алкилирование,
разбавление катализата, очистное фильтрование, отгонка раство-
Рис. 15. Технологическая схема получения М-(1,3-диметилбутил)-М'-фенил-я-фе-
иилендиамина:
7 — аппарат для растворения; 2. 4, 8— холодильники; 3 — автоклав; 5 — аппарат для раа-
бавления; 6 — друк-фильтр; 7 — перегонный куб; S. 70. 17 — сборники.
123
рителя и вакуум-перегонка Диафена 13, ректификация смеси изо-
бутилметилкетона и изобутилметилкарбинола.
В стальной эмалированный аппарат 1 (рис. 15) с пропеллер-
ной мешалкой, рубашкой (для подогрева горячей водой) и обрат-
ным холодильником 2 загружают изобутилметилкетон и п-амино-
дифениламин. Смесь подогревают до 40—50 °C и выдерживают при
этой температуре до полного растворения п-аминодифениламина.
Полученный раствор передают на восстановительное алкилирова-
ние.
Процесс проводят в автоклаве 3 из нержавеющей стали с про-
пеллерной мешалкой и рубашкой для обогрева жидким ВОТ.
В автоклав принимают из аппарата 1 раствор п-аминодифенил-
амина и загружают оксидный железо-медь-хромовый катализатор.
Из автоклава вытесняют воздух азотом и затем водородом, вклю-
чают обогрев и начинают перемешивание. По достижении темпе-
ратуры 180 °C поднимают давление водорода до 5 МПа. При этой
температуре проводят восстановительное алкилирование, поддер-
живая давление в пределах 4—5 МПа. По окончании реакции вы-
ключают обогрев, снижают давление до 0,3—0,6 МПа и выдавли-
вают реакционную массу через холодильник 4 на разбавление.
В аппарат 5 из нержавеющей стали с мешалкой принимают
охлажденную массу из автоклава, а затем загружают рециркули-
рующую смесь изобутилметилкетона и изобутилметилкарбинола и
перемешивают. После перемешивания передают катализат на очи-
стное фильтрование, а в аппарат 5 загружают свежий изобутил-
метилкетон, которым промывают катализатор на друк-фильтре 6.
Фильтрат передают на вакуум-перегонку в куб 7, а катализатор
периодически направляют на сжигание.
В кубе 7 от раствора Диафена 13 отгоняют растворитель при
атмосферном давлении и ПО—170°C (в парах). Когда эта темпе-
ратура начнет снижаться, в системе создают разрежение (до оста-
точного давления 40—53 кПа), отгоняют остатки растворителя и
продукты с низкой температурой кипения. Затем в системе созда-
ют более глубокий вакуум (остаточное давление 0,4—0,6 кПа) и
перегоняют Диафен 13 при 210—230 °C в парах и при 230—250 °C
в аппарате. Пары Диафена 13 конденсируют в холодильнике 8.
Расплав продукта принимают в сборник 11, откуда сливают в хра-
нилище.
Свойства и применение. К-(1,3-Диметилбутил)-К/-фенил-п-фе-
нилендиамин представляет собой серый кристаллический порошок;
плотность 986—1100 кг/м3; т. пл. 50°С; т. кип. 212—224°C; т. затв.
40 °C. Растворяется в бензоле, ксилоле, метилизобутилкетоне, то-
луоле, этаноле.
Горюч; т. всп. 204,4 °C.
Мало токсичен; характеризуется наркотическим действием.
ПДК 2 мг/м3. ЛД50=3200+310 мг/кг.
Выпускается во многих странах под различными торговыми
наименованиями:
124
Торговое наименование Страна, фирма Выпускная форма Т. пл., °C
Antozite 67 США, Vanderbilt
ASM 4020 ФРГ, Bayer Порошок >45
Flexzone 7L Ipognox 45 США, Naugatuck ПНР Расплав 50
Nonox ZC Англия, ICI Расплав —
Permanax 120 Франция, Rhone Poulenc Гранулы, чешуйки 50
Santoflex 13 США, Monsanto Расплав, хлопья 44 (т. крист.)
UOP-588 США, Universal Oil Расплав 45—50
ЬР(1,3-Диметилбутил)-№-фенил-л-фенилендиамин — эффектив-
ный стабилизатор каучуков общего назначения; пассивирует дей-
ствие металлов переменной валентности; защищает резины на ос-
нове натурального и синтетических каучуков от термоокислитель-
ного и светоозонного старения и разрушения при многократных
деформациях. В значительно меньшей степени, чем N-изопропил-
N'-фенил-я-фенилендиамин, вымывается из резин водой: потери со-
ставляют 15—20% против 50% соответственно.
ЛИТЕРАТУРА
1. Universal, пат. 2969394, 1961 г. (США); РЖХим, 1962, 8М242.
2. Universal, пат. 797224, 1958 г. (Англия); приоритет США; С. А.; 1959, V. 53,
р. 2157.
3. Koei Chem., пат. 34323, 1972 г. (Япония); С. А., 1973, v. 78, р. 97300.
4. Пат. 87381, 1957 г. (ЧССР); РЖХим, 1960, № 20, 82023.
5. А. с. 168711, 1964 г. (СССР).
6. Мартынов Н. В., Белоногов К- И., Брюске Я. Э. — В кн.: Синтез и исследова-
ние эффективности химикатов для полимерных материалов. Тамбов, Там-
бовская правда, 1969. Вып 2, с. 247.
7. Скрипка Л. А. Там же, с. 270—278.
8. Rice R. G., Hehn О. У.— J. Am. Chem. Soc., 1955, v. 77, р. 4052.
9. Бебих Г. Ф., Гринберг А. £. — В кн.: Синтез и исследование эффективности
стабилизаторов для полимерных материалов. Воронеж, 1964, с. 184—190.
10. Roe A., Montgomery I. А.—J. Am. Chem. Soc., 1953, v. 74, № 4, р. 910—
912.
11. Дядченко А. И., Тараненко А. С. — В кн.: Синтез и исследование эффектив-
ности химикатов для полимерных материалов. Тамбов, Тамбовская правда,
1969. Вып. 2, с. 38—42.
12. Мартынов Н. В., Брюске Я- Э. Там же с. 251—259.
13. Universal, пат. 2779789, 1957 г. (США); Chem. ZbL, 1958. S. 8192.
14. Universal, пат. 2867604, 1959 г. (США); РЖХим, 1960, К» 9, 37186.
15. ICI, пат. 712100, 1954 г. (Англия); Ch. ZbL, 1955, S. 3968.
16. ICI, пат. 716239, 1954 г. (Англия); Ch. ZbL, 1955, S. 9908.
17. BASF, пат. 1155136, 1964 г. (ФРГ); РЖХим, 1965, 8Н146.
18. Du Pont, пат. 2734808, 1956 г. (США); РЖХим, 1958, 44649.
19. Eastman, пат. 2887369, 1959 г. (США); С. А., 1959, v. 53, р. 18467.
20. Goodrich, пат. 2975213, 1961 г. (США); РЖХим, 1962, 5Л138.
21. Goodrich, пат. 3135796, 1961 г. (США); РЖХим, 1966, ЗН161.
22. Monsanto, пат. 3481983, 1969 г. (США); РЖХим, 1970, 24Н169..
125
23. Pennwalt, пат. 2222353, 1974 г. (Франция); приоритет США; С. А., 1975, v. 82, 1
р. 16531.
24. Texaco, пат. 3384664, 1968 г. (США); РЖХим, 1969, 15П196.
25. А. С. 170998, 1966 г. (СССР).
26. Monsanto, пат. 2331594, 1977 г. (Франция); приоритет США.
27. BASF, пат. 1179947, 1965 г. (ФРГ); РЖХим, 1966, 11Н146.
28. Dovell F., Greenfield Н. — J. Am. Chem. Soc., 1965, v. 87, № 12, p. 2767—
2768; РЖХим, 1966, 7Б816.
29. Николаев Ю. T. и др. — Хим. пром-сть, 1967, т. 43, № 1, с. 20.
30. А. с. 230828, 1969 г. (СССР).
31. Пат. 113939, 1965 г. (ЧССР); РЖХим, 1967, 2Н183.
32. Пат. 119336, 1966 г. (ЧССР); РЖХим, 1968, 9Н213.
33. Пат. 151974, 1974 г. (ЧССР); РЖХим, 1975, 8Н228. |
34. Пат. 58572, 1969 г. (ПНР); РЖХим, 1970, 21Н114. 1
35. Пат. 63933, 1971 г. (ПНР); РЖХим, 1973, ЗН192. :
36. Никки, пат. 15601, 1968 г. (Япония); РЖХим, 1969, 22Н207. '
37. Seiko, пат. 49-17248, 1974 г. (Япония); РЖХим, 1974, 24Н193.
38. Engelhard, пат. 1066579, 1967 г. (Англия); приоритет США; С. A., J967. V. 67,1
р. 73323. 4
39. ICI, пат. 760315, 1956 г. (Англия); Chem. Zbl., 1960, S. 6325.
40. ICI, пат. 804113, 1958 г. (Англия); РЖХим, 1960, № 19, 78329. ]
41. ICI, пат. 817142, 1959 г. (Англия); РЖХим, 1961, 9Л166. |
42. ICI, пат. 1098236, 1968 г. (Англия); С. А., 1968, v. 69, 10188.
43. ICI, пат. 1241569, 1971 г. (Англия); РЖХим, 1972, 1Н119. I
44. Bayer, пат. 851053, 1952 г. (ФРГ). ч
45. Bayer, пат. 851344, 1952 г. (ФРГ); С. А., 1953, V. 47, 5437.
46. Bayer, пат. 1077667, 1960 г. (ФРГ); С. А., 1961, v. 55, 24678.
47. Rubber, пат. 1386654, 1964 г. (Франция); пат. 643911, 1965 г. (Бельгия); пат. 1
1064958, 1965 г. (Англия); пат. 6402424 (Нидерланды); приоритет США, 1963; 1
С. А., 1965, у. 62, р. 11733.
48. Universal, пат. 3209030, 1965 г. (США); РЖХим, 1967, 9Н172. J
49. Universal, пат. 3290376, 1966 г. (США); РЖХим, 1968, 11П203.
50. Universal, пат. 3538161, 1970 г. (США); РЖХим, 1971, 15Н214; пат. 3538162,1
1970 г. (США) ; РЖХим, 1971, 15Н215. 1
51. Goodyear, пат. 336684, 1968 г. (США); РЖХим, 1969, 14Н199.
52. Goodyear, пат. 3542691, 1970 г. (США); РЖХим, 1971, 14С475.
53. Goodyear, пат. 3739026, 1973 г. (США); РЖХим, 1974, 9Н168.
54. Eastman, пат. 2323948, 1944 г. (США); С. А., 1945, v. 38, р. 116.
55. Eastman, пат. 3163616, 1964 г. (США); С. А., 1965, V. 62, р. 6661.
56. Eastman, пат. 3216959, 1965 г. (США); РЖХим, 1967, 6С689.
57. Monsanto, пат. 3219702, 1965 г. (США); РЖХим, 1967, 7Н145; пат. 3219703. s
1965 г. (США); С. А., 1965, v. 62, р. 5227. _ ]
58. Monsanto, пат. 3219705, 1965 г. (США); РЖХим, 1967, 2Н185.
59. Monsanto, пат. 1475620, 1977 г. (Англия); РЖХим, 1978, 2Н158.
60. Kodak, пат. 1486641, 1977 г. (Англия); РЖХим, 1978, 7Н224.
61. Universal, пат. 753740, 1957 г. (Англия); С. А„ 1957, v. 51, 5827.
62. Universal, пат. 3504032, 1970 г. (США); РЖХим, 1971, 5Н179. . 3
63. Outi Sinio, пат. 38906, 1971 г. (Япония) ; РЖХим, 1972, 13С901. г
64. Dovell F. S., Greenfield И. — Ind. Eng. Chem, Prod. Res. Develop., 1962, v. 1, !
№ 3, p. 179—181; РЖХим, 1963, 10H114. I
65. Greenfield H., Dovel F. S. — J. Org. Chem., 1966, v. 31, № 9, p. 3053. 1
66. Ряшенцева M. А., Миначев X. Al., Чейдыш JI. C. — Изв. АН СССР, сер. хим.,
1968, № 7, c. 1601—1605. 1
67. Иоффе И. И. и др. — Кинетика и катализ, 1966, т. 7, № 5, с. 898—901.
68. Universal, пат. 3238177, 1967 г. (США); РЖХим, 1967, 15Н150.
69. Witco, пат. 3074908, 1963 г. (США); РЖХим, 1965, 22С273.
70. ICI, пат. 1295672, 1972 г. (Англия); РЖХим, 1973, 14Н207.
71. Universal, пат. 631334, 1963 г. (Бельгия); приоритет США; С. А., 1964, v. 60,
р. 14429.
72. Пат. 25793, 1963 г. (ГДР); РЖХим, 1964, 16Н77.
126
73. Пат. 29462, 1964 г. (ГДР); РЖХим, 1965, 15Н117.
74. Скрипко Л. А., Маслова И. П. — В кн.: Синтез и исследование эффективности
химикатов для полимерных материалов. Тамбов, Тамбовская правда, 1969.
Вып. 3, с. 3—32.
75. Пат. 62287, 1971 г. (ПНР); РЖХим, 1973, 4Н195.
76. ICI, пат. 1304525, 1973 г. (Англия); РЖХим, 1973, 18Н148.
77. ICI, пат. 770299, 1957 г. (Англия); РЖХим, 1960, № 3, 10317.
78. ICI, пат. 760315, 1956 г. (Англия); пат. 1055515, 1957 г. (ФРГ); С. А., 1060,
V. 54, р. 7650.
79. Monsanto, пат. 1390061, 1964 г. (Франция); приоритет США; С. А., 1965,
V. 62, р. 14914.
80. Uniroyal, пат. 3336386, 1963 г. (США); РЖХим, 1969, 3JI183.
2.6. АЛКИЛИРОВАНИЕ АРОМАТИЧЕСКИХ АМИНОВ
Алкилированием называется реакция замещения водорода ал-
кильной группой. В зависимости от того, какой водород (ядра или
аминогруппы) замещается, различают С-алкилирование и N-алки-
лирование.
Для синтеза стабилизаторов промышленное значение имеет ал-
килирование ароматических аминов в ядро (С-алкилирование).
Эту реакцию открыли Гофман и Марциус в 1871 г.; позднее было
установлено, что алкильные группы вступают преимущественно в
орто- и /zapa-положения к аминогруппе.
Предполагают [1], что образование С-алкилированных аминов
проходит через стадию N-алкилирования:
+н*
RNH2 + Aik ——> RNHAlk ---»- Aik—R'—NH2
—H*
Для выяснения механизма этой реакции Бекман и Карене под-
вергли солянокислый метиланилин нагреванию в присутствии хло-
ридов алюминия и цинка. Предполагалась возможность двух на-
правлений процесса:
При сплавлении солянокислого N-метиланилина с равным количе-
ством хлорида алюминия при 230—240 °C образуется коричневая
стекловидная масса, практически не содержащая аминов; сплав-
ление с хлоридом цинка в аналогичных условиях приводит к сме-
си равных количеств первичных и вторичных аминов.
При бимолекулярной реакции (второе направление) следова-
ло ожидать образования наряду с первичными также вторичных
и даже третичных аминов. При пропускании хлористого водорода
в раствор метиланилина в уксусной кислоте и уксусном ангидри-
де, трехчасовом нагревании, выдерживании при 150 °C и омылении
127
был получен только вторичный амин. На основании полученных
результате Бекман и Карене пришли к выводу, что рассматри-
ваемый процесс представляет собой бимолекулярную реакцию
(уравнение 2), т. е. сначала проходит разложение солянокислого
метиланилина на анилин и хлористый метил, а затем метильная
группа внедряется в ядро в орто- или пара-положение к амино-
группе согласно общим правилам замещения.
Исследования Бекмана и Каренса имели большое значение для
развития представлений о С-алкилировании ароматических ами-
нов— после этих работ начали вести алкилирование ароматиче-
ских аминов на катализаторах Фриделя — Крафтса, что позволи-
ло сократить время реакции и повысить выход алкиламинов.
Превращение свободных N-алкилированных аминов в С-произ-
водные легко осуществляется в присутствии галогенидов метал-
лов (Со, Zn, Cd, Мп) при температуре ниже 250°C. Его нельзя
объяснить наличием галогеноводорода, образовавшегося в резуль-
тате гидролиза или термического разложения соли, поскольку ре-
акция проходит в отсутствие воды и при сравнительно низких тем-
пературах.
По мнению Н. Н. Ворожцова [1], наиболее вероятна такая
схема:
C6H5NH2 + СаНбВг
|+Вг-
CeH8NH—СаН6 НВг —CelUNHj + QHb --------> CgHgNHg + СНа=СНа
NHg NH3
II I
Начальный акт — образование алифатического карбкатиона+С2Н5.
Внедрение этого катиона в ядро ароматического амина конкуриру-
ет с побочными реакциями — присоединением бром-аниона (с об-
разованием бромистого этила) и отрывом протона (с переходом
в олефин). Взаимодействием с олефином, а также изомеризацией
алкилкатиона при перегруппировке солей некоторых алкиланили-
нов можно объяснить образование гомологов анилина с иным
строением алкильного остатка.
В промышленности дифениламин алкилируют смесью олефи-
нов С?—Сэ, диизобутиленом, стиролом, а-метилстиролом и други-
ми олефинами в присутствии катализаторов Фриделя — Крафтса
(чаще всего А1С13). Продукты реакции часто содержат непрореаги-
128
ровавший дифениламин, особенно в случае применения малоактив-
ных олефинов. Применение алюмосиликатов [2, 3], природных
глин [4—6] и анилида галлия [7] в качестве катализаторов обес-
печивает большую селективность алкилирования дифениламина,
значительно упрощает схему переработки алкилата, позволяет из-
бежать образования токсичных сточных вод [2].
Кроме олефинов в качестве алкилирующих агентов использу-
ют спирты. Отличительная особенность алкилирования спиртами
состоит в том, что в условиях реакции не происходит перегруппи-
ровки углеродного скелета исходного спирта (как в случае оле-
финов). Это дает возможность ввести в молекулу дифениламина
алкильный остаток нормального строения. Для алкилирования ди-
фениламина могут быть применены и простые эфиры, например
диизопропиловый. Реакцию осуществляют при 265—300 °C в при-
сутствии ZnCl2.
Очистку технического алкилированного дифениламина обычно
осуществляют вакуумной разгонкой, трудоемкой и дорогостоящей
(в некоторых случаях на очистку алкилированного дифениламина
приходится до 30% общей стоимости стабилизатора).
В качестве стабилизаторов полимерных материалов и смазоч-
ных масел в последние годы успешно применяются смеси алкили-
рованных дифениламинов. Обычно применяют смеси орто- и пара-,
ди- и моноалкилированных дифениламинов вследствие трудности
их разделения на индивидуальные вещества.
4,4'-Диалкилдифениламин
Aik =0,4-0,
Впервые 4,4'-диалкилдифениламины были получены в 1887 г. Ллойдом взаи-
модействием n-алкилфенолов с бромидами цинка и аммония (или с хлоридами
цинка и аммония) при 320—350 °C за 40 ч.
Хорошие результаты при получении алкилированного дифениламина дают
взаимодействие соответствующего ацетанилида с бром- или иодалкилбензолом
в среде нитробензола в присутствии карбоната калия и катализатора (металли-
ческая медь с небольшим количеством иода) и последующее омыление образо-
вавшегося ацетильного соединения смесью этанола и концентрированной соля-
ной кислоты. Известен улучшенный вариант этого метода, когда алкилирован-
ный дифениламин получают взаимодействием алкилформанилидов с алкилзаме-
щенными бром- или иодбензолами в присутствии катализатора Ульмана и карбо-
ната калия при 170—240 °C с последующим выделением алкилированного дифе-
ниламина из реакционной массы [8, 9].
Алкилированный дифениламин может быть получен из п-алкилнитробензо-
лов и алкилбензола каталитическим восстановлением при повышенном давлении
в среде фтористоводородной кислоты [10] или каталитическим дегидрированием
соответствующих алкилированных дифениламинов (полностью или частично гид-
рированных) . Катализатор — никель-хромовый, содержащий сульфаты щелочных
или щелочноземельных металлов [11]. Этот способ позволяет получать алкили-
рованный дифениламин с повышенными степенью конверсии и селективности.
Все перечисленные способы получения алкилированного дифениламина не
нашли, однако, практического применения.
9—1057
129
Приемлемым для промышленности оказалось алкилирование
дифениламина в присутствии хлорида алюминия [12] и алюмо-
силикатных катализаторов [2, 13], причем в последнем случае зна-
чительно упрощается схема получения алкилированного дифенил-
амина и можно избежать образования токсичных сточных вод. Ал-
килат, получаемый при алкилировании дифениламина на алюмо-
силикатном катализаторе, не содержит смолистых примесей и не
требует поэтому специальной очистки (в отличие от алкилата, по-
лучаемого в присутствии хлорида алюминия). Оказалось, однако,
что кристаллические алюмосиликатные катализаторы (цеолиты)
несмотря на более развитую поверхность менее активны для ал-
килирования дифениламина, чем аморфные алюмосиликаты.
Алкилирование дифениламина смесью олефинов проходит по
схеме:
Реакцию осуществляют в присутствии хлорида алюминия при
140—160°С и под давлением; мольное соотношение олефин:дифе-
ниламин равно (2,25-4-3) : 1.
Дифениламин, хлорид алюминия и смесь олефинов нагревают
до 125—130 °C. При перемешивании выдерживают реакционную
массу до содержания в смеси «0,5% дифениламина, 8% моно- и
не менее 83% диалкилдифениламина. Реакционную смесь охлаж-
дают и при 85 °C выливают в раствор едкого натра. Выдержива-
ют «10 мин при 80—90 °C, а затем отделяют маслообразную
жидкость, промывают водой и перегоняют в вакууме. Полученный
диалкилдифенил амин кристаллизуют.
Стабилизатор можно выпускать и в виде маслообразной жид-
кости (для смазочных масел). В этом случае дифениламин, оле-
фины и хлорид алюминия кипятят при 130—150 °C в течение 5 ч.
Реакционную массу выливают в раствор едкого натра и промыва-
ют водой. Избыток олефинов удаляют вакуум-разгонкой при
130 °C.
Свойства и применение. 4,4'-Диалкилдифениламин — масля-
нистая красновато-коричневая жидкость; плотность 9669 кг/м3;
т. кип. 150—223°C (при 4 кПа); «р =1,5632. Растворяется в аце-
тоне, бензоле, бензине, сероуглероде, этаноле, этилацетате, эти-
ленхлориде.
Является стабилизатором различных латексов, синтетических
каучуков и резин на их основе, а также пассиватором металлов
переменной валентности. Нужно отметить, что применение его в
промышленности резиновых технических изделий понемногу сни-
жается вследствие неудобств при дозировании (жидкий продукт с
непостоянным составом). 4,4'-Диалкилдифениламин находит широ-
кое применение как антиокислительная присадка к смазочным
маслам и может служить термостабилизатором полиолефинов (по-
130
липропилен, полиэтилен, сополимеры этилена с пропиленом) и
ударопрочного полистирола, причем его эффективность значительно
повышается в сочетании с тио(дилаурилпропионатом).
4,4'-Ди(1,1,3,3-тетраметилбутил)дифенилам1ш
СН3 ч СН3
(СН3)3С—СН3—С-^^—Nil—С—CH2-G(CH3)3
сн3 сн3
4,4'-Ди(1,1,3,3-тетраметилбутил)дифениламин (4,4'-ди-грет - ок-
тилдифениламин) получают алкилированием дифениламина диизо-
бутиленом при высоких температурах и давлении. Катализатора-
ми служат хлорид алюминия [12, 14—21], природные алюмоси-
ликаты (кислотные глины) [6] и анилид галлия [7], в присутст-
вии которого алкилирование проходит наиболее селективно.
Реакционная смесь после алкилирования обычно содержит не-
которое количество непрореагировавших диизобутилена и дифенил-
амина, а также моноалкилированный дифениламин (дифениламин
и диизобутилен легко удалить перегонкой). Содержание указан-
ных компонентов можно изменять в зависимости от метода введе-
ния реагентов, их соотношения, условий реакции. Трудно отделяе-
мой примесью является монооктилдифениламин; он образуется в
значительных количествах и снижает антиокислительную актив-
ность стабилизатора. Поскольку выделение этого соединения —
процесс трудоемкий и дорогостоящий, в качестве стабилизатора
в большинстве случаев применяют смесь ди- и моноалкилдифе-
нил амина. Такая смесь представляет собой вязкую жидкость, не
кристаллизующуюся при хранении. Чистый 4,4'-ди-трет-октилди-
фениламин кристаллизуют из реакционной смеси метанолом или
диизобутиленом.
Исследовано [20] влияние некоторых параметров (температу-
ра, соотношение реагентов, длительность синтеза) на выход и со-
став алкилата, полученного при алкилировании дифениламина
диизобутиленом на хлориде алюминия. Установленные законо-
мерности графически представлены на рис. 16. Рассмотрение этих
графиков показывает, что скорость независимо от температуры
через 60 мин резко снижается, причем скорость образования моно-
алкилпроизводных практически равна нулю. С повышением темпе-
ратуры растет степень превращения дифениламина (при 120 °C она
составляет 90—95%), уменьшается количество полимерных про-
дуктов, а скорость образования моно- и диоктилдифениламина
возрастает. Выход монооктилдифениламина практически не зави-
сит от количества катализатора; наибольший выход соответствует
соотношению дифениламин : А1С13= 1 : 0,15. Максимальное пре-
вращение дифениламина достигается при мольном соотношении ди-
фениламин : диизобутилен = 1:3.
9* 131
Рис. 16. Зависимость состава алкилата от продолжительности алкилирования при
85—90 °C (а) и 120—125 °C (б) и мольном соотношении дифениламин : олефин ;
: А1С1з=1 : 3 : 0,15:
1 — дифениламин; 2 — монооктнл дифениламин; 3 — дноктилднфениламнн.
Оптимальными условиями, при которых степень превращения
дифениламина составляет 90—95%, являются НО—120°C, моль-
ное соотношение дифениламин : диизобутилен : А1С1з= 1 : 3 :0,5 и
продолжительность реакции 3 ч. В патентах [12, 14—19] условия
взаимодействия дифениламина с диизобутиленом в присутствии
А1С1з отличаются от оптимальных: в большинстве случаев реко-
мендуется проводить реакции при более высокой температуре
(130—155°С) в атмосфере азота под давлением 0,3—0,5 МПа.
Мольное соотношение дифениламин: диизобутилен обычно состав-
ляет 1 : (24-4). Получаемая смесь содержит не выше 80% 4,4'-ди-
грет-октилдифениламина.
При алкилировании дифениламина диизобутиленом в присутст-
вии хлорида алюминия алкилирование идет преимущественно в
пара-положение к аминогруппе. Свидетельством этому служат
изменения в спектре диизооктилдифениламина, где полоса погло-
щения при «825 см-1, характерная для пара-замещения, выраже-
на особенно ярко [21]. Полосы же в области 1500—1600 см'1, по-
добно спектру незамещенного дифениламина, очень интенсивны,
так как при пара-замещении сохраняется сопряжение неподелен-
ной электронной пары аминного атома N с бензольными кольцами.
При алкилировании дифениламина а-олефинами нормального
строения (н-октилен) алкилирование, по данным [21], идет пре-
имущественно в орто-положение. Например, интенсивность скелет-
ных колебаний связей С = С-ароматического кольца значительно
уменьшилась в спектре 4,4'-диоктилдифениламина, что свидетель-
ствует, по-видимому, о нарушении сопряжения бензольных колец
с неподеленной электронной парой азота в HN-группе, которая
блокирована орто-заместителем. Полоса 690 см-1 полностью отсут-
ствует, а единственная полоса поглощения при «745 см-1 указы-
вает на присутствие в бензольном кольце четырех атомов водоро-
да, что подтверждает наличие орто-заместителя.
132
Авторы патента [17] дают следующий анализ алкилата: 85%
орто- или .мега-изомера диоктилдифениламина, 9% орто- или ме-
та-монооктилдифениламина, 0,3% дифениламина.
4,4'-Ди-трет-октилдифениламин (Октамин) на практике полу-
чают алкилированием дифениламина диизобутилеиом в присут-
ствии алюмосиликатного катализатора:
Три-, тетра- и полизамещенные производные не образуются, а
замещение водорода алкильным радикалом происходит в основном
в лара-положение — содержание орто-изомера в алкилате всего
«3%- Общая степень превращения дифениламина на алюмосили-
катном катализаторе составляет 75—80%. Выход продукта 160—
170% на пропущенный дифениламин.
Алюмосиликатный катализатор без потери активности работа-
ет 20 суток. Потеря активности, по-видимому, связана с адсорб-
цией дифениламина и его алкилпроизводных на активных центрах
катализатора.
Технологическая схема получения. Процесс осуществляется не-
прерывным способом и состоит из 7 стадий: алкилирование дифе-
ниламина диизобутиленом, отгонка диизобутилена от алкилата-1,
отгонка дифениламина от алкилата-П, кристаллизация Октамина,
фильтрование суспензии, сушка пасты и регенерация изопропано-
ла (растворитель).
Алкилирование дифениламина проводят в двух алкилаторах,
работающих поочередно: в одном идет алкилирование, в другом
перегрузка катализатора. Сырье подают в алкилатор через смеси-
тель и теплообменник. Через каждые 12 ч реакционную массу ана-
лизируют: определяют показатель преломления и проводят раз-
гонку продукта. По остатку судят об активности катализатора.
Остаток анализируют методом тонкослойной хроматографии для
определения содержания непрореагировавшего дифениламина, ко-
торое не должно превышать 15—20%. При содержании остатка в
алкилате <35% и содержании дифениламина в нем >20% ката-
лизатор считают неактивным и выгружают из алкилатора.
От алкилата отгоняют диизобутилен в пленочном испарителе.
Диизобутилен через холодильник и приемник собирают в емкость,
а оттуда направляют на рециркуляцию. Алкилат из нижней части
Испарителя поступает в кристаллизатор, заполненный охлажден-
ным изопропанолом. Суспензию охлаждают в кристаллизаторе и
Выдерживают там в течение 3 ч, после чего азотом выдавливают
133
на фильтрование. После промывки изопропанолом отжимают пас-
ту и передают на сушку.
Свойства и применение. 4,4,-Ди-трег-октилдифениламин — бе-
лый кристаллический порошок; т. пл. 103 °C. Растворяется в бен-
золе, бензине, хлористом этилене, ацетоне.
Горюч; т. воспл. 265 °C; т. самовоспл. 498 °C; нижний предел
взрываемости 20 г/м3; минимальное взрывоопасное содержание
кислорода 10% (об.).
Умеренно токсичен.
За рубежом выпускается под различными торговыми назва-
ниями и в разных выпускных формах:
Торговое наименование- Страна, фирма Выпускная форма Т. пл., °C
Agerite Stalite S Irganox LO-1 Nonox OD Octamine США, Vanderbilt Швейцария, Ciba—Geigy Англия, ICI США, Naugatuck Порошок или гра- нулы Гранулы Воскообразное ве- щество 89—103 90 75—85
Октамин — стабилизатор бутадиен-стирольных, хлоропреновых
и бутадиен-нитрильных каучуков. Защищает резины от термоокис-
лительного старения и разрушения при многократных деформа-
циях; особенно эффективен для резин, на основе хлоропреновых
каучуков.
Антиоксидант полиолефинов и широко распространенный ста-
билизатор смазочных масел.
4,4'-Д и (а-метилбензил)дифениламин
4,4'-Ди (а-метилбензил)дифениламин впервые был получен в
1955 г. фирмой Bayer (ФРГ) .[22]. На заводе этой фирмы было
организовано и промышленное производство стабилизатора — ка-
талитическим алкилированием дифениламина стиролом. Алкили-
рование обычно осуществляют в безводной среде при 150—200 °C
в присутствии катализаторов Фриделя—Крафтса [14, 16]: чаще
всего с А1С1з, реже с ZnCl2. Давление — от атмосферного до
1,5 МПа; время реакции 0,5—10 ч. Известно также применение
алюмосиликатов, которые в силу своих слабых кислотных свойств
не вызывают коррозии аппаратуры [3].
134
4,4'-Ди (а-метилбензил) дифениламин (Антиоксидант ДДА) по-
лучают по схеме;
+ 2СНа=СН— \
В реактор загружают дифениламин и катализатор (гумбрин) и
нагревают смесь до 125 °C. В течение 3 ч под слой реакционной
массы при 125—130 °C подают стирол, затем выдерживают смесь
при этой температуре и интенсивном перемешивании. Реакционную
массу охлаждают до 105—ПО °C и отфильтровывают от катали-
затора. Катализатор промывают ацетоном, выгружают, сушат и
повторно используют. Промывные воды направляют на регенера-
цию катализатора.
Реакционную массу передают в другой реактор и нагревают
там до 90 °C. При остаточном давлении 2,66 кПа отгоняют фрак-
цию, выкипающую в пределах 60—80°C. Она представляет собой,
стирол, который без дополнительной очистки можно использовать
на последующих операциях. После отгонки стирола поднимают
температуру до 220 °C и при 0,266 кПа отгоняют вторую фракцию,
выкипающую в пределах 135—220 °C. Эту фракцию, содержащую
непрореагировавший дифениламин и моноалкилированный дифе-
ниламин, добавляют к основным реагентам в последующих опера-
циях. При дальнейшем повышении температуры при 0,266 кПа от-
гоняют фракцию, выкипающую при 220—320 °C; это — целевой
продукт. В нем содержится 60—70% 4,4/-ди(а-метплбензил)дифе-
ниламина, 10—12% 4-(а-метилбензил) дифениламина и 2—3% ди-
фениламина; остальное — продукты неустановленного строения.
Степень превращения дифениламина 80—90%; степень превра-
щения стирола 50—60%. Кубовый остаток представляет собой
смесь продуктов разной степени полимеризации.
Свойства и применение. 4,4'-Ди (а-метилбензил) дифениламин —
красновато-коричневая вязкая жидкость; плотность 1080—1090
кг/м3; т. кип. 190—320°С при «0,4 кПа; «р =1,6330—1,6400. Рас-
творяется в ацетоне, бензине, бензоле, четыреххлористом углероде,
этаноле, этил ацетате, метил енхлориде. Горюч.
Антиоксидант ДДА выпускается в ФРГ фирмой Bayer в виде
красновато-коричневой жидкости с т. кип. выше 300 °C или в виде
30%-ной водной эмульсии (Antioxidant DDA-EM).
4,4'-Ди (а-метилбензил)дифениламин — эффективный стабилиза-
тор различных синтетических каучуков; особенно пригоден для
бутадиен-стирольных и бутадиен-нитрильных каучуков. В виде
30%-ной водной эмульсии применяется для стабилизации латек-
сов. Защищает резины от термоокислительного и озонного старения
и разрушения при многократных деформациях; пассивирует дей-
135
ствие металлов переменной валентности; обеспечивает сополиме-
рам акрилонитрила хорошую стойкость к набуханию
озона (например, в сочетании с фентиазином).
и действию
4,4'-Бис (а,а-диметилбензил) дифениламин
4,4'-Бис (а,а-диметилбензил) дифениламин был получен алкилированием ди
фениламина а-метилстиролом в присутствии катализаторов — серной кислоты
[23] и алюмосиликатов [5].
Производство этого стабилизатора было организовано во Франции в конщ
1960-х годов на заводах фирмы Rhfine Poulenc [23]. Способ заключается
в следующем. Дифениламин алкилируют а-метилстиролом (соотношение 1 : 2) в
присутствии серной кислоты в инертном растворителе:
В реактор загружают дифениламин, растворенный в инертном растворителе, на
сревают до 50 °C и добавляют серную кислоту (катализатор) в течение 30 мин.
Реакционную смесь перемешивают 1 ч и добавляют а-метилстирол; смесь пере-
мешивают еше 2 ч и добавляют карбонат натрия в течение 50 мин при 75—80 °C
(для нейтрализации).
По окончании алкилирования и при pH 12 органический слой отделяют,
охлаждают до 15 °C и выделяют 4,4'-бис (а,а-диметилбензил) дифениламин с
т. пл. 97 °C. Такой способ имеет ряд существенных недостатков: большое коли-
чество кислотных сточных вод, необходимость нейтрализации реакционной смеси
Кроме того, получаемый продукт загрязнен примесями.
Лучших результатов удается достичь в присутствии природных алюмосили-
катов (глины) [4—6, 24] и хлорида алюминия [5, 6, 24].
По способу, разработанному фирмой Uniroyal (США), стаби-
лизатор получают следующим образом. В реактор загружают ди-
фениламин, катализатор (монтмориллонитовая глина) и бензол.
Реакционную смесь перемешивают при кипячении, отгоняя азео-
тропную смесь воды с бензолом при 130 °C. Затем медленно (за
20 мин) добавляют а-метилстирол. Смесь перемешивают в тече-
ние 4 ч при 130—135 °C и отфильтровывают от катализатора.
Фильтрат выливают в гексан, а выпавший осадок снова отфильт-
ровывают. Получают 4,4'-бис (а,а-диметилбензил) дифениламин с
выходом «75%. После двойной перекристаллизации из гексана по-
лучали продукт с т. пл. 101—102 °C.
Отечественными исследователями [4] при более высокой тем-
пературе алкилирования (130—200 °C) в присутствии зикеевской
опоки получен 4,4'-бис(а,а-диметилбензил) дифениламин с выходом.
136
84,2%. Катализатор применяют в количестве 5—10% от исходно-
го сырья. Преимущество алкилирования в присутствии зикеевской
опоки в том, что отпадает необходимость предварительной обра-
ботки (в присутствии монтмориллонита необходимо до реакции
удалить из катализатора излишнее количество воды путем азео-
тропной отгонки с растворителем). Применение глин вместо сер-
ной кислоты позволяет вести процесс практически без сточных
вод, отпадает стадия нейтрализации, стабилизатор получается
более чистый и с хорошим выходом.
Свойства и применение. 4,4'-Бис (ю,'а-диметилбензил) дифенил-
амин (Дифенам ДМБ) —белый порошок; т. пл. 102 °C (из гекса-
на). Растворяется в ацетоне, бензоле, трихлорэтилене, хлорофор-
ме, циклогексане, этаноле и воде (ограниченно).
Горюч. Токсичен.
Является широко распространенным стабилизатором различ-
ных синтетических каучуков; защищает резины на основе нату-
рального и синтетических каучуков от термоокислительного и све-
тоозонного старения. По действию близок к Неозону Д. Особенно
эффективен для хлоропреновых каучуков. Антиоксидант полиоле-
финов, нефтяных и синтетических смазочных масел. Отличается
стабильностью при 200—230°C и малой летучестью.
Выпускается во Франции фирмой Rhone Poulenc под названи-
ем Permanax 49 (светло-зеленый порошок; т. пл. >90 °C) и ис-
пользуется как стабилизатор различных синтетических каучуков.
ЛИТЕРАТУРА
1. Ворожцов Н. И. Основы синтеза промежуточных продуктов и красителей. М.,
Госхимиздат, 1955. 757 с.
2. Зарубина И. В. и др. — Химия и технология топлив и масел, 1975, № 8,
с. 4—5.
3. Goodyear, заявка 2621060, 1976 г. (ФРГ).
4, А. с. 443026, 1974 г. (СССР).
5. Uniroyal, пат. 3452056, 1969 г. (США); РЖХим, 1970, 15С704.
6. Uniroyal, пат. 3505225, 1970 г. (США); РЖХим, 1971, 4П302; пат. 3649690,
1972 г. (США); РЖХим, 1972, 24Н192; пат. 3666716, 1972 г. (США); РЖХим,
1973, 5Н200; пат. 3751472, 1973 г. (США); РЖХим, 1974, 11Н126.
7. Ethyl, пат. 3862233, 1975 г. (США); РЖХим, 1975, 20Н131.
8. Du Pont, пат. 2924620, 1960 г. (США); С. А., 1960, V. 54, р. 18434.
9. Scardiqlia F., Roberts J. — J. Org. Chem., 1958, v. 23, p. 629—631; Chem. Zbl.,
1960, S. 1456.
10. Weinmayr V. — J. Am. Chem. Soc., 1955, v. 77, p. 1762.
11. Bayer, заявка 2520893, 1976 г. (ФРГ); С. А., 1977, v. 86, р. 106158.
12. Cyanamid, пат. 3496230, 1970 г. (США); РЖХим, 1971, 6Н127.
13. А. с. 396359, 1971 г. (СССР).
14. Goodrich, пат. 2530769, 1950 г. (США); Chem. Zbl., 1951, S. 1663.
15. Goodrich, пат. 2776994, 1957 г. (США); Chem. Zbl., 1961, S. 10776.
16. Pennsalt, пат. 2943112, 1960 г. (США); РЖХим, 1962, 5П367.
17. Reichhold, пат. 3714257, 1973 г. (США); РЖХим, 1973, 24Н157.
18. Reichhold, пат. 3714258, 1973 г. (США); РЖХим, 1973, 22Н159.
19. Geigy, пат. 2017253, 1970 г. (ФРГ); приоритет Англии; С. А., 1971, V. 74,
р. 4472.
20. Зарубина И. В. — Химия и технология топлив и масел, 1973, № 5, с. 18—21.
137
21. Гаврилова Т. Е. и др. — Химия и технология топлив и масел, 1974, № 1, с. 50-ч
54.
22. Bayer—Mitt, Gummi Ind., 1955, № 22, р. 19—22; РЖХим, 1958, № 5, 16375.
23. Rhone Poulenc, пат. 1472132, 1967 г. (Франция); С. А., 1968, v. 68, р. 95483.
24. Uniroyal, пат. 3758519, 1973 г. (США); РЖХим, 1974, 18Н206.
2.7. КОНДЕНСАЦИЯ АРОМАТИЧЕСКИХ АМИНОВ
С КАРБОНИЛЬНЫМИ СОЕДИНЕНИЯМИ
Конденсация аминов с карбонильными соединениями представ-
ляет собой межмолекулярный процесс образования новой С-—С-
или С—N-связи, сопровождающийся элиминированием молекулы
НгО. Реакция обычно протекает в присутствии катализаторов»
которые не только ускоряют процесс, но образуют реакционноспо-
собные промежуточные продукты или связывают отщепляющуюся
молекулу Н2О, смещая равновесие в системе вправо. Действие ка-
тализаторов является комбинированным. Строгой специфичности
они не проявляют; условно их можно сгруппировать по типам
реакций — в соответствии с элиминируемой молекулой.
Реакции конденсации могут протекать без образования циклов
(нециклическая конденсация) и с образованием новых циклов
(циклическая конденсация).
Конденсация без образования циклов наблюдается при полу-
чении 4,^-диаминодифенилметана и альдоль-а-нафт,иламина; цик-
лизацией получают 2,2,4-триметил-1,2-дигидрох'инолин, 2,2,4-триме-
тил-6-этокси-1,2-дигидрохинолин, 5,5щиметилакридан.
Нециклическая конденсация ароматических аминов с аль-
дегидами подробно изучена на примере реакции анилина с форм-
альдегидом, приводящей к диаминодифенилметану.
Формальдегид используют в виде 40%-ного водного раствора
(формалин); катализатором обычно служит серная или соляная
кислота. При взаимодействии формальдегида с анилином в присут-
ствии соляной кислоты образуется 4,4,-диаминодифенилметан:
2/~A-NH2 + HCHO H2N—/"Д-СН.—
/ —Н2О \ / \ /
Кислота протонирует формальдегид; образовавшийся при этом
карбкатион атакует ароматическое ядро молекулы амина. В ре-
зультате получается гидроксибензиловый спирт, который в при-
сутствии кислоты легко взаимодействует со второй молекулой
амина.
Строение и состав продуктов конденсации арилмоноаминов
с формальдегидом зависят прежде всего от соотношения реаген-
тов и pH среды [1, 2].
При рН>7 реакция идет преимущественно в сторону образо-
вания соединений типа ArNH—СН2—NHR, которые не стабильны
в средах с рН<7. При pH 7 реакция направлена в сторону обра-
зования оснований Шиффа ArN = CH2, легко полимеризующихся
с образованием продуктов типа ангидроформальдегиданилина.
138
При проведении конденсации с эквимольными количествами ани-
лина и формальдегида в солянокислой среде (pH 4) основным
продуктом является полимерный ангидро-п-аминобензиловый
спирт. Он' при нагревании до 80 °C в избытке солянокислого ани-
лина дает 4,4/-диаминодифенилметан.
Механизм образования 4,4'-диаминодифенилметана окончатель-
но не установлен. Некоторые исследователи i[3] считают, что пер-
вичным продуктом конденсации является азометиновое соедине-
ние
NHa + HCHO N=CH2
\ / —Н2О \ /
и предлагается [3] следующая схема получения 4,4'-диамино-
3,3'-диметилдифени л метана:
н4
ArNHa + HCHO ~ArN=CH2
ArN=CH2 + ArNH3
-*• Ar—CH2NH—ArNHa------1
1 H2NAr—CH2—ArNHa
-* ArNH—CH2NHAr -------i
и
Ar — CeH4CH3
Основное направление образования диаминодифенилметана, по-
.мнению работы [3], — перегруппировка метиленди (фенилими-
на) (II). В работах же [4, 5] предложен механизм реакции, в осно-
ве которого лежит перегруппировка п-аминобензиланилина(1).
Взаимодействие ароматических аминов с высшими альдегида-
ми приводит к образованию «альдегидаминов» (основания Шиф-
фа). «Альдегидамины» были первыми стабилизаторами и ускори-
телями вулканизации резиновых смесей, но теперь утратили свое
значение. Обычно их называли по исходным веществам, вступаю-
щим в реакцию (бутиральдегиданилин, ацетальдегиданилин).
Химизм процесса на примере реакции анилина с масляным аль-
дегидом можно представить схемой:
С3Н7-СНО + CeH6NH2 —*• С3Н7—CH=N—СвН6
—rigU
Образовавшийся бутилиденанилин взаимодействует с одной «ли
с двумя молекулами альдегида:
4-С3Н,СНО +C3H7CHO
С3Н7— CH=N—С,Н6----—* С3Н7—СН=С—CH=N—СеН6 -------—►
—rigU | -flgO
сна—сн3
С3Н7—СН=С—СН==С—CH=N—свн3
СН3—СН2 СН2—сн3
13»
Масляный альдегид конденсируется в p-пропил^а-этилакролеин
2С,Н,—СНО —*• С3Н,—СН=С-СНО
—н2о ।
СН2—СН3
который может непосредственно взаимодействовать с анилином:
С3Н,—СН=С—СНО + C6H6NHS ——> С,Н,—СН=С—CH=N—CeHs
। —нао - ।
СН2—СН3 СН2—СН3
Альдегиданилины представляют собой смолообразные продук-
ты с «размазанным» фракционным составом, они неудобны для
применения. Единственным стабилизатором подобного типа, при-
емлемым (ограниченно) до настоящего времени, является альд-
оль-а-нафтиламин.
„Конденсация ароматических аминов-, и карбонильных соедине-
ний с образованием циклических продуктов наиболее подробно
изучена на примере так называемого хинальдинового синтеза, ко-
торый был открыт в 1881 г. Дебнером и Миллером:
НС1
+ 2RCH2- СНО
1
I
I
I
Образование хинальдина объяснялось следующими реакциями:
Более поздние исследования механизма циклизации диеновых ами-
нов показали, однако, что перегруппировка кротонилиденанилина
не основное направление хинальдинового синтеза.
Более вероятным считается механизм, включающий первона-
чальное образование кротонового альдегида из ацетальдегида и
последующее присоединение ароматического амина по двойной
углерод-углеродной связи с образованием р-ариламиномасляного
альдегида и его производных, циклизующихся в хинальдин:
140
Эта схема, однако, экспериментально не доказана: если а,р-непре-
дельные кетоны в отдельных случаях способны присоединять аро-
матические амины по двойным связям, то а,р-непредельные альде-
гиды типа кротонового в такие реакции не вступают.
Строение промежуточных продуктов изучали по реакции кон-
денсации ксилидинов с ацетальдегидом в кислой среде при комнат-
ной температуре. В этих условиях были выделены два изомерных
вещества, получивших название альдольных оснований:
(СН3)2С6Н3—N=CH—СН,—СН—СН3 (СН3)2С6Н3—NH—СН—СН,—СНО
ОН СН,
I п
Из этих двух возможных структур правильной была признана
структура II, а наличие двух изомеров объяснено стереоизомерией
трехвалентного азота. При дальнейшем исследовании альдольных
оснований была опровергнута структура II и показано, что альд-
ольные основания являются вторичными аминами, содержащими
гидроксильную группу.
Подробное изучение свойств альдольных оснований позволило
сделать вывод, что они представляют собой цис- и транс-изомеры
4-гидрокси-2-метил-1,2,3,4-тетрагидрохинолина
а реакция образования хинальдина была представлена схемой:
Одновременно с альдольными основаниями исследовали и дру-
гие промежуточные продукты реакции Дебнера — Миллера — ди-
меры этилиденариламинов. При взаимодействии эквимольных ко-
личеств анилина и ацетальдегида в кислой среде был получен
димер этилиденанилина с т. пл. 126 °C, которому была приписана
формула:
NH—СН—СН,—CH=N
сн3
v
Наряду с этим изомером диэтилиденанилина образуется и дру-
гой — с т. пл. 85—86 °C. Оба изомера (получившие название осно-
141
ваний Эйбнера — Экштейна) при нагревании с соляной кислотой
легко образуют хинальдины, а аналогичное основание из анилина
и пропионового альдегида в этих условиях дает З-метил-2-этилхи-
нолин. Полученные данные позволили сделать вывод, что образо-
вание димеров этилиденариламинов и их последующая циклизация
являются одним из направлений хинальдинового синтеза, которое,
однако, нельзя принимать за главное, так как при разложении
ди(этилиденанилина) получается лишь 22%-ный выход хиналь-
дина.
Легкость образования хинальдиновых производных из основа-
ний Эйбнера — Экштейна привела к выводу, что эти основания,
как и альдольные, имеют циклическую структуру и им- должны
соответствовать формулы [6, 7]:
По современным представлениям, альдольная конденсация в
кислой среде (на примере анилов ацетальдегида) проходит по
схеме:
+Н+ + +СН3—CH=N-CeHs
СН3—CH=N—С6Н5-----> СН3— CH—NH—С6Н5--------------
-н+
--►СНз—CH—СН2— CH=N—С6Н5 ч—*:
NH—С6Н5
СН3
Н NH— С6Н5
Широко распространенные стабилизаторы полимерных мате-
риалов— 2,2,4-триметил-1,2-дигидрохинолин и 2,2,4-триметил-
6-этокси-1,2-дигидрохинолин — получают конденсацией анилина и
n-фенетидина с ацетоном.
В изучении механизма взаимодействия первичных ароматиче-
ских аминов с ацетоном большая заслуга принадлежит Тунгу, ко-
торый экспериментально доказал строение продуктов этой реак-
ции в присутствии кислотных катализаторов [8]: получены и
идентифицированы 2,2,4-триметил-1,2-дигидрохинолин и мезитил-
оксиданил, из которых неизменно получались 2,2,4-триметил-6-
этокси-1,2-дигидрохинолин.
Таким образом, экспериментально доказано, что образование
2,2,4-триметил-6-этокси-1,2-дигидрохинолина проходит через обра-
зование оснований Эйбнера — Экштейна. Отмечается, что эти
основания находятся в состоянии равновесия, образуя исходный
n-фенетидин и 6-этоксимезитилоксиданил, который, гидролизуясь,
142
в свою очередь, может также давать исходный n-фенетидин и смо-
лообразные продукты.
Конденсация дифениламина с ацетоном впервые была изучена
Крейгом в конце 1930-х годов. По схеме Крейга, в солянокислой
среде реакция в зависимости от условий протекает с образованием
следующих продуктов:
+ —Н2О \_/ \___/ । \__/ \__/
CHS—со-сн3 сн3
III
Конденсация дифениламина с ацетоном во многом зависит от
условий, главным образом от температуры. При 120—150 °C реак-
ция идет с образованием 4,4/-дианилино-2,2'-дифенилпропана(1).
При 160—180 °C образуется главным образом п-изопропенилдифе-
ниламин(П). Выше 200°C происходит молекулярная перегруппи-
ровка промежуточного продукта II с образованием гетероцикличе-
ских соединений акриданового ряда. Практически уже при 260 °C
конденсация дифениламина с ацетоном завершается с образова-
нием 5,5-диметилакридана (III).
В патентных описаниях приводится состав веществ, полученных в резуль-
тате конденсации, однако экспериментальных доказательств предложенного
строения веществ обычно не бывает.
СН3
А
143
В результате нагревания при 150 °C избытка дифениламина с ацетоном
в присутствии кислотного катализатора получался 4,4/-днанилино-2,2'-дифенил-
пропан. Последний, выделенный из реакционной массы разгонкой, дает прн
дальнейшем нагревании с эквимольным количеством ацетона в нейтральной нлн
слабокислой среде при 140 °C вещество А (стр. 143) н высокомолекулярные со-
единения. В более кислой среде замещение идет больше в ядро, чем по амино-
группе. В присутствии 2 моль соляной кислоты получается вещество Б:
Б
Конденсацию дифениламина с ацетоном обычно проводят в
присутствии катализаторов (минеральные кислоты, галогены, га-
логениды тяжелых металлов) при температуре от 100 до 200 °C
под давлением в автоклаве или при атмосферном давлении, про-
пуская пары ацетона в расплав амина.
4,4'-Диаминодифенилметан
Впервые 4,4'-днаминодифенилметан был
4,4'-динитроднфенилметана хлоридом олова
дит с теоретическим выходом:
получен в 1872 г. восстановлением
в соляной кислоте. Реакция прохо-
4-8Н
- 4Н2О
В 1884 г. 4,4'-днамннодифенилметан был получен нагреванием смеси экви-
мольных количеств n-аминобензилового спирта и солянокислого анилина в вод-
ном растворе (реакция араминирования):
H2N-/~\-CH2OH+/^-NHg HCl ----•>
\ \ / —-гт2'~Л -HU
В то время способ практического значения не нашел. Однако позднее на
основе реакции араминирования был разработан оригинальный метод получения
диаминоднфеннлметанов, запатентованный во многих странах [42]. Процесс
осуществляли прн высокой температуре (300—400 °C) под давлением на оксиде
алюминия, содержащем небольшие количества (менее 1%) оксидов щелочных
металлов.
Известен ряд других реакций, приводящих к 4,4'-диаминодифеннлметану.
144
Нагревание 4,4'-диаминодифенилуксусной кислоты в присутствии SnCl2 и
соляной кислоты:
Гидролиз 4,4'-диаминодифенилметан-3,3'-дикарбоновой кислоты разбавленной
соляной кислотой при 200 °C:
HCI
- 2СО2
Восстановление щелочного раствора 4,4'-бис(4-аминобензил) азоксибензола
феррицианидом калия:
Взаимодействие солянокислого анилина с метилалем в водном растворе при
60—90 °C:
2 HC1 + (сн3О)2сна 0 _2H-g?
\ / —zcrigun, —,zrici
--> H3N—CHj—NH2
Взаимодействие анилина с метиленхлоридом в автоклаве в среде водного
аммиака при 120—250 °C в течение 0,5—3 ч, когда получается смесь первичных
полиаминов, содержащая до 40% диаминодифенилметана [13], и аминирование
дигидроксиднфеннлметана:
НО-/Л-СН2-/~Ч—ОН H2N-/~A_cH2Y^-NHa
\\ / —2HgO \ \ /
Из всех указанных методов практический интерес представляют восстанов-
ление диннтродифенилметана и аминирование дигндроксидифеннлметана. Они
позволяют получать 4,4'-диамнноднфенилметан без примесн изомеров.
Несомненный интерес представляет разработанный в последнее
время метод каталитического гидрирования 4,4'-динитродифенил-
метана на твердых катализаторах [9-П]. Восстановление дини-
тродифенилметана в диаминодифенилметан осуществляли электро-
литическим водородом в среде н-бутанола на скелетных Ni—Al-,
Со—А1-, Си—А1- и Fe—А1-катализаторах [9, 10]. Максимальный
выход диаминодифенилметана составлял соответственно 69,2, 60,5,
7,8 и 7,5%- Увеличение продолжительности процесса до 10 ч и бо-
лее позволило повысить выход амина. Например, на Ni—Al-ката-
лизаторе почти теоретический выход достигался при длительности
процесса 15 ч, а на Со—Al-катализаторе за 18 ч. При введении
До 5% хрома в состав скелетных Ni—Al- и Со—А1-катализаторов
Ю—Ю57
145
[10] выход диаминодифенилметана возрастает, а дальнейшее по-Ж
вышение количества хрома резко понижает выход.
Основательно изучено получение 4,4'-диаминодифенилметана
конденсацией анилина с формальдегидом, на основе которой раз-
работан промышленный способ синтеза этого продукта. Реакция
впервые была описана в 1884 г. Толленсом; он при действии ани-
лина на водно-метанольный раствор формальдегида получил кри-
сталлический продукт, названный им ангидроформальдегидани-jj
лином: «И
^A-N^ + HCHO —AA-N=CHa Я
\--/ ~Н2° \---/ W1
В 1889 г. фирмой Hochst (Германия) был заявлен первый патент
на получение диаминодифенилметана: ангидроформальдегидани-
лин, полученный по Толленсу, растворяли в избытке анилина
в присутствии солянокислого анилина.
Взаимодействие анилина с формальдегидом в кислой среде
[14-31] протекает с образованием первичных аминов разной сте-
пени конденсации. Из диаминов образуются 2,2'-, 2,4'- и 4,4'-ди-
аминодпфенил метаны; кроме того, получаются триамины, тетрами-
ны и другие продукты.
Характер и количество образующихся продуктов определяются
соотношением анилина к формальдегиду. При избытке анилина '
увеличивается содержание двухъядерных аминов с преобладанием
2,4'-изомера; при этом наблюдаются N-метилирование и смолооб- Г
разование. С уменьшением отношения анилин : формальдегид по- ;
нижается содержание диаминодифенилметана в получаемой смеси
аминов с одновременным образованием высших полиаминов [32].
Установлено, что последние могут превращаться в симметричные :
диаминодифенплалканы при нагревании в избытке солянокислого *
анилина [28]. ?
Количество соляной кислоты, взятой для конденсации анилина i
с формальдегидом, также существенно влияет на протекание про- |
цесса: с уменьшением соотношения анилин : кислота повышается «
содержание диаминодифенилметана с преобладанием 4,4'-изомера |
[32—34]. Фирма Tolochimie (Франция) [20] рекомендует прово- I
дить конденсацию анилина с формальдегидом при 90—100 °C
в присутствии малых количеств кислоты (0,15—1 моль на 1 моль '
формальдегида). Фирмой BASF (ФРГ) предложен способ получе- '
ния диаминодифенилметанов в присутствии лишь каталитических
количеств кислоты — от 0,001 до 0,46 г-экв на 1 моль формальде-
гида; оптимальной температурой считают 60—180 °C. После уда- ‘
ления избытка анилина получается чистый продукт с незначитель-
ной окраской и небольшим содержанием вторичных аминогрупп
[35, 36].
Характерная особенность смеси диаминодифенилметанов, полу-
ченных в присутствии малых количеств кислоты, — высокое содер-
жание 2,2'- и 2,4'-изомеров. В случае больших количеств кислоты
146
достигается высокий выход 4,4'-диаминодифенилметана без стадий
нейтрализации и осаждения соли [17, 25, 37].
Важным фактором, обусловливающим разнообразие продуктов
конденсации анилина с формальдегидом, является также многоста-
дийность этого процесса. Первая стадия протекает с образованием
так называемых предконденсатов (форконденсация); они стабиль-
ны только в нейтральных или щелочных средах и представляют
собой продукты, содержащие —СН2—iN-группы.
На последующих стадиях в кислой среде происходит перегруп-
пировка: вначале образуется продукт, содержащий группы
—СН2—СвН4— (первая стадия), а затем продукт с группами
—С6Н4—СН2—С6Н4— (вторая стадия). Схему получения 4,4'-ди-
аминодифенилметана упрощенно можно представить так:
—NHS + НСНО + H2N—\
форконденсация
-Н20
О Г/ \ Н+ (I стадия)
—NH-CH2—HN-" \ ----------------*•
N, N '-метилеидианилин
Н+ (11 стадия)
—NH-CH2—^-nh2 ------------------>
анилинобеизиламин
днаминодифенилметан
Форконденсацию и первую стадию перегруппировки обычно про-
водят в мягких условиях. Экспериментально доказано, что чем
выше температура на стадии форконденсации, тем больше получа-
ется диаминодифенилметана, обогащенного орто-изомером, а так-
же олигомерных аминов и N-метилированных продуктов [38].
Чистый 4,4/-диаминодифенилметан (96%-ный) получается при по-
ниженном давлении и температуре форконденсации не выше 40 °C
[39]. Вторую стадию перегруппировки проводят при температуре
до 100 °C [19, 32—34, 38, 40—43] с последующей нейтрализацией.
Процесс может быть оформлен периодически и непрерывно.
Соблюдать оптимальные соотношения реагентов для получения
максимального выхода 4,4/-диаминодифенилметана при периодиче-
ском оформлении процесса несложно, особенно для реакторов не-
большого объема. Стадию форконденсации проводят, медленно
добавляя формальдегид в хорошо перемешиваемую реакционную
массу.
При переходе к более экономичному, непрерывному способу
появляются трудности из-за забивания трубопроводов и теплооб-
менников коллоидными частицами, склонными к коагуляции. Эти
частицы отличны от продуктов обычного осмоления, возникающего
в начале реакции [14, 44—47]-. Чтобы исключить забивание аппа-
ратуры в условиях непрерывного процесса, фирма Upjohn (США)
10
147
предлагает применять раствор ангидроформальдегиданилина и
смешивать его с соляной кислотой в условиях турбулентного ре-
жима при температуре не выше 70 °C [14].
При конденсации анилина с формальдегидом в присутствии
кислоты получается смесь изомеров с большим или меньшим со-
держанием 2,4'- и 2,2'-диаминодифенилметана в зависимости от
условий реакции. Разделять изомеры с целью получения чистого
4,4'-диаминодифенилметана довольно сложно. Известно много
способов выделения целевого продукта:
1) нагревание смеси изомеров с непредельными соединениями
(например, с изопропенилбензолом, стиролом, аминостиролом) в
присутствии кислотного катализатора и инертного органического
растворителя [48];
2) нагревание смеси изомеров с гидроксилсодержащими соеди-
нениями (фенол, пирокатехин, резорцин) [49];
3) экстракция 4,4'-изомера, когда изомеры превращают в гид-
рохлориды [50—52]; их нагревают в течение нескольких часов
в хлорбензоле и после охлаждения выделяют твердый гидрохло-
рид, содержащий 99,3% 4,4/-диаминодифенилметана [50, 52];
4) экстракция 4,4'-изомера гидрофобным растворителем в при-
сутствии солей щелочных металлов, галогенидов, цианидов или
без них [26, 53—55];
5) выделение чистого 4,4'-изомера кристаллизацией из поли-
аминов [56]; метод заключается в медленном охлаждении рас-
плавленной смеси при перемешивании до температуры выше
т. затв., но ниже т. крист. 4,4'-диаминодифенилметана.
Наиболее удобна и экономически целесообразна, однако, очист-
ка вакуум-перегонкой. Иногда вакуум-перегонку проводят с добав-
кой ингибитора термического разложения (парафин, высококипя-
щее минеральное масло). Дистиллятом является 4,4'-диаминоди-
фенилметан (с остатками ингибитора); в кубе остаются полиамин
с ингибитором [57]. Чтобы снизить количество вторичных аминов
в полиамине, смесь перед нейтрализацией обрабатывают цинковой
пылью при 90—98 °C. Осадок отфильтровывают [58].
С 1960-х годов реакция конденсации анилина с формальдеги-
дом стала предметом еще более всесторонних исследований.
В связи с большими потребностями в 4,4'-диаминодифенилме-
тане особый интерес для промышленности представляют способы
его получения по непрерывной безотходной технологии. Такой про-
цесс разработан фирмой Upjohn (США) [29, 59], которая реко-
мендует проводить конденсацию в присутствии соляной кислоты
при 0—60 °C до исчезновения (по ЯМР) метилениминных групп
/N—СН2—N^. Для получения 4,4'-диаминодифенилметана высо-
кой чистоты решающую роль играет сочетание трех факторов: тем-
пературы смешения реагентов, количества воды и времени пере-
мешивания. При мольном отношении анилин : формальдегид : НС1 =
=2: 1 : 1,5 получается продукт, содержащий до 98% 4,4'-изомера,
148
Непрерывный процесс осуществлен на одном из заводов ГДР.
Все стадии проводят в проточных трубчатых реакторах, по темпе-
ратуре разделенных на четыре зоны. В первой зоне при 0—60 °C
смешивают водную солянокислую соль анилина с водным раство-
ром формальдегида; во второй зоне поддерживают температуру 20—
50 °C; в третьей зоне температура ниже 50 °C, но выше температу-
ры, при которой образуется твердая фаза; в четвертой зоне темпе-
ратура равна 80—100 °C, а время контакта составляет 100—
240 мин. Мольное соотношение анилин : формальдегид= (1,5—!
-г-4 : 1); на 1 моль анилина вводят 0,45—0,95 моль кислоты и
1,3—7,6 моль воды (не считая 2—15 моль воды, вносимой
с 1 моль НСНО). Общее количество воды составляет 4—7 моль
на 1 моль анилина.
Фирмой Du Pont (США) [18] разработан непрерывный способ
получения 4,4'-диаминодифенилметана с высоким выходом и хо-
рошего качества. При конденсации в реакционную зону до начала
стадии перегруппировки добавляют дополнительное количество
анилина. Реакцию осуществляют при соотношении солянокислого
анилина к формальдегиду от 2,8 : 1 до 4 : 1; температура в первой
зоне 38—52 °C, во второй 60—70 °C и в третьей 88—110 СС. Полу-
ченный продукт нейтрализуют щелочным агентом и очищают орга-
ническую фазу от нежелательных примесей. Получают диаминоди-
фенилметан с выходом 88,6% и ^96% 4,4'-изомера.
Окончательная особенность непрерывного метода, предложен-
ного фирмами Kaiser Aluminium (США) [45] и Nippon Soda (Япо-
ния) [60], — применение инертного растворителя (чаще всего
хлорбензола), что исключает накопление смолистых продуктов на
стенках реактора. Перемешивание исходных компонентов в турбу-
лентном режиме проводят при времени контакта от 15 с до 2 мин.
Во второй зоне при 80—150 °C реакционную массу выдерживают
в течение 0,5—2 ч. Обычно продукт реакции выделяют в виде
свободного основания, нейтрализуя смесь щелочным агентом. Отде-
лившийся органический слой отмывают водой от неорганических
примесей и отгоняют избыточный анилин.
Чтобы уменьшить коррозию аппаратуры и исключить стадию
нейтрализации, вместо соляной кислоты предложено применять
нейтральные соли органических и неорганических кислот в коли-
честве 0,2—10% [61—64]. Для ускорения стадии перегруппировки
добавляют так называемые промоторы, например NaCl, NaBr,
сильные сульфокислоты. Отмечается [63] необходимость соблю-
дать при этом рН<8. Известен и непрерывный процесс [65] в при-
сутствии нейтральных солей, запатентованный двумя фирмами —
Bayer и Mobay, однако этот способ позволяет получать диамино-
дифенилметан с высоким содержанием нежелательных 2,2'- и
2,4'-изомеров. Низкое содержание целевого 4,4/-изомера получает-
ся и в присутствии Н3ВО3, Н3РО4 и их солей [66, 67].
Кроме кислот в качестве катализаторов при конденсации ани-
лина с формальдегидом используют природные глины, синтетиче-
149
ские алюмосиликаты, сульфированные сополимеры дивинилбензо-
ла со стиролом. ‘
Использование этих твердых катализаторов имеет ряд преиму-
ществ по сравнению с применением минеральных кислот: отпадает
необходимость в коррозионностойкой аппаратуре, нет образования
большого количества сточных вод, исключается многостадийность,
сокращается процесс получения диаминодифенилметана. Правда,
диаминодифенилметаны, получаемые конденсацией анилина с форм-
альдегидом в присутствии глин, предварительно обработанных
кислотой, или синтетических алюмосиликатов, обычно получаются
с высоким содержанием 2,2'- и 2,4'-изомеров [68—70].
В последнее время удалось, однако, получить 4,4'-диаминоди-
фенилметан с выходом 64% на алюмосиликатном катализаторе
[71]. Для этого отделяют 2,2'- и 2,4'-изомеры и возвращают их на
конденсацию с формальдегидом. 4,4'-Диаминодифенилметан
с 80%-ным выходом получается на алюмосиликатном катализато-
ре в присутствии щавелевой кислоты [23] или В2О3 [30].
В присутствии сульфированных сополимеров дивинилбензола
со стиролом конденсацию анилина с формальдегидом проводят
при 70—150 °C в течение 20 ч; выход диаминодифенилметана со-
ставляет 80% [72]. Проведение реакции в отсутствие кислорода
(в токе азота, например) позволяет значительно повысить срок
службы катализатора [35, 72]. Анилин берут в избытке — до
10 моль на 1 моль формальдегида. Катализатор отфильтровыва-
ют, а фильтрат перегоняют. Катализатор можно использовать по-
вторно (до 600 ч), если хранить его в атмосфере азота.
В присутствии сульфированного полистирола проводят реакцию
при 190 °C и получают диаминодифенилметан с выходом 92,8%
и содержанием 2,4/-изомера до 33%; при повышении температуры
до 250 °C доля 2,4/-изомера увеличивается до 91% [45].
В последнее время диаминодифенилметан стали получать
в присутствии барийнитридных катализаторов [73] при температу-
ре конденсации 200 °C. В присутствии ВазМг—SisN-; получается
смесь 2,2'-, 2,4'- и 4,4'-диаминодифенилметана и высших гомоло-
гов; вторичных аминов образуется мало.
Как жидкие (кислотные), так и твердые катализаторы имеют
свои преимущества и недостатки, и в настоящее время интенсивно
развиваются оба направления процесса.
4,4'-Диаминодифенилметан, известный в СССР стабилизатор
под названием Диамет, получают конденсацией анилина с форм-
альдегидом в присутствии сильных минеральных кислот:
2HZN—+ НСНО НС 1 • H2N—C^-Z^-NHa • НС 1
—2НС1 ЫааСОз
150
В эмалированный реактор загружают 31%-ный раствор соляной
кислоты и воду. При перемешивании и охлаждении добавляют
анилин (по окончании загрузки анилина реакция среды должна
быть слабокислой). Затем реакционную массу охлаждают до 7—
10 °C и в течение часа из мерника добавляют формалин. К концу
загрузки формалина поднимают температуру реакционной массы
до 30 °C и при 30—35 °C выдерживают ее в течение 2 ч; затем на-
гревают смесь до 90 °C и выдерживают при этой температуре в те-
чение 10 ч. После этого из реакционной массы выделяют целевой
продукт (в виде основания) раствором кальцинированной соды.
Реакционную массу перемешивают в течение 2 ч при 90 °C, затем
охлаждают до 70 °C и передают в делительную воронку. Верхний
слой представляет собой темно-коричневое масло, застывающее
при охлаждении.
Диаминодифенилметан получают в смеси с анилином и продук-
тами разной степени конденсации. Для выделения 4,4'-изомера
сырой маслообразный продукт подвергают вакуум-перегонке. По-
лучают 4,4'-диаминодифенилметан с т. пл. 90—91 °C и содержа-
нием основного вещества более 99% (по диазотированию).
Свойства и применение. 4,4'-Диаминодифен.илметан — белое
кристаллическое вещество; т. пл. 89—92 °C; т. кип. 398 °C при
103 кПа (249—253°C при 2 кПа); т. затв. 91,1 °C. Растворяется
в ацетоне, этаноле, бензоле, диэтиловом эфире; ограниченно рас-
творяется в воде.
Токсичен; ЛД50=664ч-665 мг/кг; ПДК 1 мг/м3.
4,4'-Диаминодифенилметан выпускается во многих странах под
разными названиями:
Торговое наименование Страна, фирма Выпускная форма Т. пл., °C
DADM, DDM Epikure DDM NA-11 Resistox В Robac 4,4' Tonox Tonox D Франция, Man. Land Англия, Shell США, Du Pont США, Monsanto Англия, Robinson США, Naugatuck To же Хлопья Порошок Воскообразная масса Воскообразная ма.сса То же » >86 75—85 73—86 73 73 >86 (очи- щенная фор- ма)
4,4'-Диаминодифенилметан — эффективный стабилизатор ре-
зин на основе каучуков общего назначения. Защищает от термо-
окислительного и светоозонного старения, от разрушения при
многократных деформациях. Однако, как первичный амин, влияет
на вулканизацию — вызывает скорчинг, поэтому его значение по-
степенно падает.
Эффективный активатор вулканизации; его действие особенно
сильно проявляется в присутствии ускорителей класса тиазолов.
151
Ускоритель вулканизации хлоропрена, сшивающий агент для эпок-
сидных полимеров и полиуретанов.
4,4'-Диаминодифенилметан является мономером для синтеза
термостойких полимеров; это — полупродукт для органических
красителей, лаков и химических реактивов. Значение его все более
возрастает и в качестве сырья для получения полиизоцианатов.
Альдоль-а-нафтиламин
Промышленное производство альдоль-а-нафтиламина началось в Германии
с 1918 г. по патенту фирмы Farbenindustrie конденсацией а-нафтнламнна
с альдолем в мольном отношении 1 : (14-2) при температуре кипения реакцион-
ной смеси в присутствии соляной кислоты.
В дальнейшем способ был усовершенствован: альдоль-а-нафтиламин получа-
ли в присутствии органической кислоты (муравьиная, щавелевая) в среде рас-
творителя (этиловый спирт, бензол или его гомологи). Для повышения темпе-
ратуры плавления получаемого соединения в реакцию кроме альдоля вводили
и другие альдегиды, например ацетальдегид.
При конденсации ароматических аминов с альдегидами обра-
зуются соответствующие основания Шиффа (азометины); реакция
протекает гладко прн смешении веществ или их растворов. При
наличии функциональных групп в алифатической цепи альдегида
могут, в свою очередь, протекать побочные реакции. Например,
продукт конденсации анилина с альдолем алкилирует другую мо-
лекулу амина с образованием основания:
сн.,—сн—сн2—сн
I II
CeH5—NH N—С6НБ
При более длительном нагревании в присутствии соляной кислоты
возможна дегидратация и даже циклизация с получением хиналь-
диновых соединений (реакция Дебнера — Миллера).
Альдоль-а-нафтиламин, получаемый в промышленности (Аль-
нафт), представляет собой хрупкую смолу с т. пл. 60—70 °C. В ра-
боте [74] предложен способ получения альдоль-а-нафтиламина с
т. пл. не ниже 145 °C: конденсацию а-нафтиламина с ацетальдолем
проводят в присутствии соляной кислоты при 100—105 °C и моль-
ном соотношении 1 : 1 : 0,6 с последующей «разваркой» ллава в во-
де при 96—98 СС в течение 9 ч:
—NH2 + НОС—СН2—СН—СН3
ОН
----»-
—Н2О
152
Таким образом, в результате получается смесь продуктов разной
степени конденсации.
Ввиду сильной кислотности среды молекула амина превра-
щается в катион, что замедляет процесс присоединения. Реакция
проходит более полно [75], если а-нафтиламин взаимодействует
с ацетальдолем вначале в отсутствие катализатора, а затем
продукт присоединения дегидратируют, добавляя катализатор.
При этом получают продукт с т. пл. 145 °C и содержанием а-на-
фтиламина не более 1%. Расплав при кипячении с водой перехо-
дит в суспензию, которая хорошо фильтруется, давая порошкооб-
разный продукт.
Технологическая схема получения. Альдоль-а-нафтиламин
с т. пл. 60—80 °C получают конденсацией а-нафтиламина с
ацетальдолем в присутствии соляной кислоты:
EW + H__CH_CH3 N=CH—сн,—сн—сн3
/ I ~Н2° \=/ I
\ он ЛА он
Конденсацию проводят в реакторе-конденсаторе 5 (рис. 17). Туда
из мерника 2 загружают а-нафтиламин, включают холодильник 3
как обратный, нагревают смесь до 100—105 °C и подают через
мерник 1 смесь ацетальдоля с соляной кислотой в течение часа.
Затем холодильник 3 включают как прямой, нагревают реакцион-
ную массу до 130 °C и отгоняют воду, собирая конденсат в прием-
ник 4. По окончании отгонки воды сливают плав в приемник 7,
откуда расфасовывают продукт в тару.
Свойства и применение. Альдоль-а-нафтиламин является
смесью продуктов конденсации. По внешнему виду —это смола от
желтого до темно-коричневого цвета; плотность 1160—1170 кг/м3;
т. пл. от 60—70 до 80—100 °C. Растворяется в ацетоне, бензине,
хлороформе, формамиде, уксусной кислоте.
153
Альдоль-а-нафтиламин выпускается в разных формах в боль-
шинстве развитых стран:
Торговое наименование Страна, фирма Выпускная форма Т. пл., “С
Альнафт Agerite Resin AL Antage, A, C Antigen BC Antioxygcne AN, INC, ПД npc ASM АН и ASM AP Nocrac A, C Nonox S СССР США, Vanderbilt Италия, ACNA Mon- tecatini Япония, Kawaguchi Япония, Dyestuffs Франция, Mat. Col. ФРГ, Bayer Япония, Ouchi Shinko Англия, ICI Хрупкая смола Смола Порошок Смола, порошок Порошок Паста, порошок, смола Смола и порошок То же Порошок 60—70 80—100 . 145 >145 >145 60 и 145 >145 145
Горюч; не взрывоопасен. Т. воспл. 270 °C; т. самовоспл. 520 °C,
нижний предел взрываемости пыли, взвешенной в воздухе, 25 г/м3.
Ясно выраженным токсическим действием не обладает. Вслед-
ствие наличия в техническом продукте до 15,5% а-нафтиламина
проявляет канцерогенное действие.
Альдоль-а-нафтиламин — стабилизатор синтетических каучу-
ков и резин на их основе. Защищает резины от термоокислитель-
расфассбку
ного и светоозонного старения,
от действия многократных де-
формаций. Эффективный анти-
оксидант полиформальдегида и
полиэтилена; повышает термо-
стойкость клеев на основе на-
ирита; является активатором
при регенерации бутилкаучука,
вулканизованного смолами.
Несмотря на экономич-
ность продукта (простота его
получения) он постепенно те-
ряет свое значение из-за кан-
церогенности, окрашивания и
нестабильности состава.
Рис. 17. Технологическая схема полу-
чения альдоль-а-иафтиламина:-
1, 2 — мерники; 3 — холодильник; 4, 7 —
приемники; 5 — ре актор-конденсатор; 6 —
емкость.
154
Полимер 2,2,4-триметил-Г,2-дигидрохинолина
СН3
2,2,4-триметил-1,2-дигидрохинолин
(мономер)
2,2,4-Триметил-1,2-днгидрохинолин был получен в 1921 г. Кновенагелем кон-
денсацией анилина с ацетоном в присутствии иода.
Установление структуры 2,2,4-триметил-1,2-дигндрохинолина в течение дли-
тельного времени явилось предметом многочисленных исследований. Лишь
в 1950—1960-х годах с использованием современных методов была окончательно
доказана записанная выше структура [8, 76—80].
При конденсации анилина с ацетоном образуется смесь моно-
мера указанного строения и олигомерных продуктов разной степе-
ни поликонденсации. Оказалось, что олигомеры 2,2,4-триметил-1,2-
дигидрохинолина являются более эффективными стабилизаторами,
чем мономер [81]. Фирма Seiko kagaky (Япония) для увеличения
выхода олигомерного продукта разработала такой способ конден-
сации ацетона с анилином: в реакционную смесь вводят мономер
в присутствии соляной кислоты и ведут процесс в среде толуола
при 140 °C [82].
Обычно процесс получения полимера осуществляют в присут-
ствии иода в периодическом [83, 84] и непрерывном [85] вариан-
тах, причем отмечается [84], что в присутствии активаторов (ди-
хлорэтан, дихлорэтилен, трихлорэтилен) реакция проходит полнее.
В работе [85] обсуждается возможность проведения реакции тре-
мя способами в зависимости от агрегатного состояния реагентов.
Предпочтение отдают взаимодействию жидкого анилина с парами
ацетона. Оптимальными условиями считают температуру реакции
165—170 °C, температуру паров ацетона 56 °C, скорость подачи
паров 12,5 г/мпн на 1 кг анилина, расход катализатора (иод) 1,2—
3% от количества анилина [86, 87].
Хорошие результаты дает применение сульфаниловой кислоты
в качестве катализатора '[81, 84]; при этом получается преимуще-
ственно полимеризованный 2,2,4-триметил-1,2-дигидрохинолин.
В присутствии 0,01—0,02 моль сульфаниловой кислоты оптималь-
ная температура равна 170—175 °C. Наиболее эффективная ско-
рость подачи ацетона 0,45—0,6 моль на 1 моль анилина.
Описано получение 2,2,4-триметил-1,2-дигидрохинолина в при-
сутствии /г-толуолсульфокислоты [84, 87] и других ароматических
сульфокислот [83, 88], а также глины, активированной сульфокис-
лотами [83]. Для подавления полимеризации образующегося
2,2,4-триметил-1,2-дигидрохинолина рекомендуется добавлять
обычные ингибиторы, например изобутилпирокатехин [88]. В при-
сутствии сульфокислот выход 2,2,4-триметил-1,2-дигидрохинолина
колеблется от 30 до 88%; температура реакции 120—170 °C.
155
Советскими учеными i[89] разработан способ полимеризации
мономера для получения эффективного стабилизатора (Ацетон-
анил) — низкоплавкого (т. пл. 70—85 °C) однородного вещества.
Процесс осуществляют в две стадии. На первой проводят поли-
меризацию 2,2,4-триметил-1,2-дигидрохинолина в кислой среде до
получения форполимера (т. пл. 63—70 °C) и его нейтрализацию.
На второй стадии продолжают полимеризацию нейтрального фор-
полимера до получения полимера с т. пл. 70—85 °C. Гранулирован-
ный Ацетонанил получают, расплавляя продукт при 120—130 °C
и пропуская расплав через фильеры с последующим охлаждением
на транспортерной ленте.
Свойства и применение. 2,2,4-Триметил-1,2-дигидрохинолин—
-кристаллический порошок от желтого до коричневого цвета; пла-
вится не выше 120 °C; кипит, почти не разлагаясь, при 258—
259 °C. Растворяется в ацетоне, бензоле, хлороформе, этаноле, со-
ляной кислоте.
Полимеризованная форма 2,2,4-триметил-1,2-дигидрохинолина
в зависимости от метода получения и соотношения реагентов мо-
жет представлять собой вязкую жидкость, порошок или смолооб-
разное вещество с т. пл. от 60 до ПО °C.
Горюч; т. всп. 145 °C; т. воспл. 190 ₽С. Не взрывоопасен, горит
коптящим пламенем с большой скоростью. Пожароопасен в усло-
виях сушки. Пыль взрывоопасна (нижний предел взрываемости
15,6 г/м3).
Умеренно токсичен; ПДК в воздухе рабочей зоны 1 мг/м8.
Полимер 2,2,4-триметил-1,2-дигидрохинолпна
выпускается
под
разными наименованиями во многих странах:
Торговое наименование
Страна, фирма
Выпускная форма
Т. пл., «С
Ацетонанил
Agerite АК
Agerite Resin D
Antigene RD
Antioxidant 184
Antioxidant В
Antioxygene PS
Anox HB
Cyanox 12
Flectol А, В, H, Flakes
Good-Rite AO 3140
Nocrac 224
Nonflex RD
Nonox TQ
Permanax 45
Polnoks R
Santoflex R
СССР
Англия, Anchor
США, Vanderbilt
Япония, Sumitomo
Англия, Anchor
США, Goodyear Tire
Франция, Kuhlmann
Италия, Bozzetto
США, Cyanamid
Англия, Monsanto
США, Goodrich
Япония, Ouchi Shinko
Япония, Seiko
Англия, ICI
Франция, Rhone Pou-
lenc
ПНР
Англия, Monsanto
Гранулы
Порошок
Порошок, гранулы
Смола
Порошок
Вязкая жидкость
Чешуйкн
Хлопья, порошок
Порошок, вязкая жид-
кость, чешуйки
Гранулы
Порошок
Смола
Чешуйки
Порошок, чешуйки
Порошок
То же
70—85
114
100—120
70—110
>114
НО
114—120
75
70—100
83—86
<120
74
<114
156
Ацетонанил — широко распространенный стабилизатор различ-
ных синтетических каучуков — бутадиеновых, изопренового, бу-
тилкаучуков. Эффективность его повышается в сочетании с произ-
водными п-фенилендиамина. Эффективен для защиты резин от
термоокислительного и светоозонного старения; стабилизатор ра-
кетных топлив и смазочных масел; сырье для производства инсек-
тофунгицидов.
2,2,4-Триметил-6-этокси-1,2-дигид рохинолин
2,2,4-Триметил-6-этоксн-1,2-дигидрохинолнн был получен Киовенагелем
в 1921 г. длительным кипячением п-фенетидина с ацетоном в присутствии иода.
Выход (после перегонки) составил 29%. считая на вступивший в реакцию аце-
тон. Структуру выделенного соединения Кновенагель определил как «парафе-
нетидннацетонанил»:
С2НБО
N=C(CH3)2
Позднее соединениям, полученным конденсацией ароматических аминов
с ацетоном в присутствии кислотных катализаторов, была приписана 1,2-дигид-
рохинолиновая структура (см. предыдущий раздел), которая в последующих
работах с помощью физико-химических методов исследования была подтвержде-
на [76—78, 90].
Образование 2,2,4-триметил-6-этокси-1,2-дигидрохинолина про-
текает по схеме:
157
2,2,4-Триметил-6-этокси-1,2-дигидрохинолин получают конден-
сацией /г-фенетидина с ацетоном в мольном отношении 1 : (2-ь-З)
при 95—180 °C в присутствии катализаторов — иода [83, 84, 91—.
97], брома [91], НВг или HI [98, 99], ароматических сульфокис-
лот {100]. Условия реакции и природа катализатора заметно влия-
ют на выход и качество продукта. В присутствии иода реакция
обычно заканчивается через 3—4 ч при 140—160 °C. Процесс ведут
при атмосферном давлении, а иногда в автоклаве. Выход продукта
85—95%.
Разработан непрерывный процесс получения 2,2,4-триметил-6-
этокси-1,2-дигидрохинолина [84]. Катализатор берут в количестве
1—4% от массы /г-фенетидина и добавляют активатор—дихлор-
этан, дихлорэтилен, трихлорэтилен (13—18% от количества ката-
лизатора). При этом на 1 моль /г-фенетидина берут не менее
3,33 моль ацетона. Степень конверсии /г-фенетидина « 45%.
По способу японской фирмы lamamoto [95] 2,2,4-триметил-6-
этокси-1,2-дигидрохинолин высокой чистоты (98%-ный) получают,
нагревая /г-фенетидин, ацетон и иод в течение 8—10 ч при 140—
150 °C. Образующуюся воду и избыток ацетона отгоняют. Пары
ацетона обезвоживают в колонне на молекулярных ситах и воз-
вращают в реактор. К полученному 2,2,4-триметил-6-этокси-1,2-ди-
гидрохинолину добавляют равный объем воды и концентрирован-
ную соляную кислоту, перемешивают в течение 60 ч при 20 °C и
экстрагируют смесь бензолом. Выход 95%.
В присутствии HI или НВг конденсация /г-фенетидина с ацето-
ном протекает при более низкой температуре (100—ПО °C).
В смесь /г-фенетидина и 47%-ной НВг, нагретую до 105 °C, вводят
в течение 10 ч ацетон, отгоняя непрореагировавший ацетон и воду.
После охлаждения смеси до 80 °C добавляют толуол и воду и пе-
ремешивают 1 ч. Органический слой нейтрализуют 10%-ной ще-'
лочью, многократно промывают водой и перегоняют. Получают
фракцию с т. кип. 145 °C при 0,93 кПа и из нее 2,2,4-триметил-6-(
этокси-1,2-дигидрохинолин с т. кип. 145—165 °C. Из водного слоя j
выделяют бромистоводородную соль /г-фенетидина, которую вновь '
используют в реакции [98—99].
В присутствии ароматических сульфокислот (/г-толуолсульфо- :
кислота, бензолсульфокислота) для получения 2,2,4-триметил-6-1
этокси-1,2-дигидрохинолина берут значительный избыток ацетона,'
иногда до 15 моль на 1 моль /г-фенетидина [104]; выход продукта’
превышает 90% [101]. В присутствии ингибитора полимеризации
(например, изобутилпирокатехина) выход несколько повышается
(92%) [102].
С выходом более 95% 2,2,4-триметил-6-этокси-1,2-дигидрохино-
лин получается по способу японской фирмы Mitsubishi газофазным 1
способом [87]. /г-Фенетидин и /г-толуолсульфокислоту при пере-?
мешивании (500 об/мин) нагревают до 140 °C в атмосфере азота. :
В течение 3 ч потоком азота подают в смесь ацетон; пары ацето- :
на, содержащие воду, собирают в приемник. После прекращения
158
подачи ацетона добавляют толуол, нейтрализуют реакционную
смесь 10%-ным раствором NaHCO3 и промывают водой. Слой то-
луола отделяют; остаток перегоняют в вакууме. Выход 2,2,4-три-
*метил-6-этокси-1,2-дигидрохинолина 95,2%; т. кип. 135—140 °C
(при 79,8 Па).
Несколько отличается от описанного способ [105], когда вме-
сто ацетона проводят конденсацию н-фенетидина с изопропенил-
этиловым эфиром в присутствии эфирата фтористого бора (ката-
лизатор). В раствор n-фенетидина в бензоле вносят катализатор
и добавляют по каплям изопропенилэтиловый эфир. Смесь пере-
мешивают в течение 1 ч, промывают 10%-ной щелочью, сушат
сульфатом магния in выделяют 2,2,4-триметил-6-этокси-1,2-дигид-
рохинолин. Т. кип. 118—123°C (прн 133,3 Па); выход 64,5%.
В основу промышленных процессов получения 2,2,4-триметил-
б-этокси-1,2-дигидрохинолина (Хинол ЭД) положена улучшенная
реакция Кновенагеля [106].
Один из промышленных процессов осуществлен следующим об-
разом. В автоклав загружают n-фенетидин и при постоянном пере-
мешивании нагревают до 170—180 °C. Затем вносят 1/в часть рас-
твора иода в ацетоне и одновременно через ротаметр в нижнюю
часть автоклава со скоростью 70—80 кг/ч начинают подавать аце-
тон. Содержание воды в ацетоне определяют через каждый час.
При уменьшении содержания воды ниже 2% добавляют следую-
щую 7s часть раствора иода и так далее, пока не израсходуют весь
приготовленный раствор. После этого прекращают подачу ацето-
на и охлаждают реакционную смесь до 140—150 °C. Затем с целью
удаления легких фракций в течение 4—5 ч выдерживают смесь
в вакууме.
Очистку сырого продукта проводят в вакуум-дистилляционных
аппаратах непрерывного действия при 133—200 Па. Из резервуара
сырой продукт насосом подают через два подогревателя в пленоч-
ный дистиллятор. В первом поддерживают температуру 100—
120 °C, во втором 140 °C, в верхней зоне пленочного дистиллятора
160—180 °C, в нижней зоне 200—210 °C. Из пленочного дистилля-
тора смолообразный остаток стекает в приемник, а головной погон
отводят в колонну. Ректификацию проводят при флегмовом числе
(1 : 5)ч-(1 : 4), устанавливаемом по ротаметру. В донной части
колонны-ректификатора поддерживают температуру 160—180 °C,
а дистиллят отгоняют при 120—140 °C.
Конденсат, содержащий наряду с п-фенетидином 5—10% ко-
нечного продукта, собирают в приемнике, предназначенном для
«-фенетидина, и периодически подают в автоклав на конденсацию.
Готовый продукт через холодильник стекает в приемник.
Другой процесс получения 2,2,4-триметил-6-этокси-1,2-дигидро-
хинолина осуществляют по схеме, описанной ниже.
Обычно получаемый продукт наряду с 2,2,4-триметил-6-эток-
си-1,2-дигидрохинолином содержит п-фенетидин и различные ос-
нования Шиффа, образованные ацетоном или продуктами его
159
Рнс. 18. Технологическая схема получения 2,2,4-три метил-6-этокси-1,2-дигидро-
хинолина:
I — каплеотбойник; 2 — реактор-конденсатор; 3 — дефлегматор; 4 — насадочная колонна; 5
скруббер; 6 — испаритель; 7 — сборник; 8 — емкость.
уплотнения. Поэтому стараются заменять ацетон в этой реакции
на другие реагенты, менее склонные к полимеризации, — на изо-
пропенилэтиловый эфир [105], оксид мезитила или диацетоновый
спирт [87, 98, 99, 103]. Хорошие результаты получены с примене-
нием ингибиторов полимеризации ацетона [102].
Следует учитывать, что в перегнанном Хиноле ЭД может со-
держаться 2,4-диметил-6-этокси-1,2-дигидрохинолин [Ю7].
Технологическая схема получения. В реактор-конденса-
тор 2 (рис. 18) из мерника, установленного на весах, загружают
бензолсульфокислоту (БСК) и подают n-фенетидин. После загруз-
ки /г-фенетидина продувают реактор инертным газом, включают
мешалку и пускают в рубашку аппарата пары ВОТ, нагревая
реакционную массу до 165—170 °C. В это время готовят к работе
узел испарения ацетона. Испарение ведут в стальных кожухотруб-
ных теплообменниках 6, обогреваемых паром. Для улавливания
капель, уносимых с парами, после испарителя установлен кап-
леотбойник 1.
Ацетон подают на испарение дозировочным насосом из расхода
ной емкости. Перед началом операции емкость с остатками ацетон
на от предыдущей операции продувают инертным газом, принима-
ют регенерированный ацетон и из хранилища догружают необхо-
димое количество свежего ацетона. Включают дозировочный на-
сос и заполняют трубы испарителя ацетоном (уровень ацетона
160
поддерживают автоматическим регулятором). Подачу пара на
обогрев труб автоматически регулируют так, чтобы температура
паров ацетона в каплеотбойнике 1 была равна 68—80 °C. По до-
стижении избыточного давления паров ацетона 0,05—0,1 МПа и
при температуре в реакторе 2, равной 165—170 °C, начинают пода-
вать туда пары ацетона через ротаметр.
Смесь паров непрореагировавшего ацетона, воды и п-фенети-
дина из реактора поступает в дефлегматор 3. Там конденсируются
высококипящие вещества, которые через колонну 4 стекают обрат-
но в реактор. Температуру паров после дефлегматора поддержи-
вают автоматически в пределах 80—95 °C путем охлаждения
дефлегматора холодной водой. Из дефлегматора пары ацетона и
воды с небольшим содержанием п-фенетидина поступают в скруб-
бер 5 на улавливание ацетона. 3%-ный раствор ацетона собирает-
ся в емкости 8, откуда его подают на ректификацию.
При содержании /г-фенетидина в реакционной массе не более
2% подачу ацетона прекращают и в течение 10—15 мин через
реакционную массу пропускают сжатый инертный газ для отдув-
ки летучих веществ. После отдувки выдавливают массу из реак-
тора 2 инертным газом в сборник 7, где готовый продукт охлаж-
дают до 70—90 °C, и направляют на перегонку.
Свойства и применение. 2,2,4-Триметил-6-этокси-1,2-дигидрохи-
нолин представляет собой темную вязкую жидкость; т. кип. 169 °C
при «1,6 кПа.
Растворяется в бензоле, четыреххлористом углероде, этиловом
спирте, ацетоне, диэтиловом эфире, лигроине.
Горюч; т. всп. 93 °C; т. воспл. 167 °C; т. самовоспл. 387 °C.
Характеризуется умеренной токсичностью; ЛД5о~3000 мг/кг
(для мышей) и невысокой кумулятивной способностью. Слегка
раздражает кожные покровы и слизистые оболочки глаз; облада-
ет слабыми канцерогенными свойствами.
2,2,4-Триметил-6-этокси-1,2-дигидрохинолин выпускается во
многих странах в виде вязкой жидкости от желтого до красно-ко-
ричневого цвета:
Торговое наименование Страна, фирма Т. кип., °C
Хинол ЭД СССР 123—125 (при 0,26 кПа)
Anox W Италия, Bozzetto —
Antigene AW Япония, Sumitomo 60-65
ASM ЕС ФРГ, Bayer 125—135 (при«0,4 кПа) или 169 (при«1,6 кПа)
Ethoxyquin Испания —
Niflex D ВНР 133,5 (при 0,13 кПа)
Nocrac AW Япония, Ouchi Shinco —
Nonflex AW Япония, Seiko —
Permanax 103 Франция, Rhone Poulenc —
Pol у flex США, Naugatuck —
Н—1057
16Т
Под названием Antioxidant Premix 2,2,4-триметил-6-этокси-1,2-
дигидрохинолин выпускается с добавлением инертного наполните-
ля в виде порошка.
Хинол ЭД является эффективным стабилизатором различных
синтетических каучуков и широко применяется для защиты резин
на основе натурального и синтетических каучуков от светоозонного
старения и растрескивания при многократных деформациях. Эф-
фективность повышается в сочетании с N-изопропил-М'-фенил-
n-фенилендиамином. В последнее время наблюдается тенденция
заменять Хинол ЭД в этой комбинации на Ацетонанил, как более
дешевый и обеспеченный сырьем (n-фенетидин более дефицитен,
чем анилин).
Очищенный 2,2,4-триметил-1,2-дигидрохинолин является хоро-
шим антиоксидантом и применяется для консервирования кормов;
стабилизатор овощей, жиров, молока, рыбы и других пищевых
продуктов; способствует сохранению окраски красного перца. При
добавлении в корм защищает животных от недостатка витамина Е;
может быть использован в качестве гербицида, в частности для за-
щиты кожуры яблок.
Продукты конденсации дифениламина с ацетоном
В результате конденсации дифениламина с ацетоном образует-
ся смесь продуктов, применяемая в качестве стабилизатора поли-
мерных материалов.
Условия реакции (прежде всего температура) влияют на при-
роду и состав продуктов конденсации. Разделяют стабилизаторы,
полученные при низкой температуре (до 200 °C) типа Aminox
(продукт низкотемпературной конденсации) и полученный при вы-
сокой температуре (более 200°C) —типа BLE-25 (продукт высоко-
температурной конденсации дифениламина с ацетоном).
Низкотемпературная конденсация. Впервые конденсация дифе-
ниламина с ацетоном была осуществлена в 1930 г. фирмой Nauga-
tuck (США), запатентовавшей применение полученного продукта
в качестве антиоксиданта синтетических каучуков и резин на их
основе. В 1932 г. была проведена конденсация дифениламина с
ацетоном в автоклаве и при атмосферном давлении. В первом
случае катализатором служил Fel2-4H2O и продукт получался
в виде темной вязкой жидкости. Во втором случае осуществляли
многочасовое нагревание дифениламина с ацетоном в присутствии
HI и получали конечный продукт в виде твердой массы.
Наиболее подробно конденсация дифениламина с ацетоном
в различных условиях была изучена в конце 1930-х годов. Было
установлено, что при 120—150 °C реакция идет с образованием
4,4/-дианил ино-2,2-дифенилпропа на:
СН
162
При 160—180 °C получается главным образом 4-изопропенилдифе-
ниламин
который выше 200 °C превращается в 5,5-диметилакридан
Н3С СН3
Практически при 260 °C реакция завершается образованием 5,5-ди-
метилакридана. В качестве побочных продуктов образуются акри-
дан и его производные.
Процесс низкотемпературной конденсации проводят следующим
образом. Смесь дифениламина с ацетоном в присутствии катализа-
тора (иод, иодистоводородная кислота, н-толуолсульфокислота)
выдерживают в автоклаве при 150—154 °C в течение 10—15 ч; при
этом развивается давление свыше 1 МПа. Реакционную массу
после охлаждения нейтрализуют содой или щелочью и промывают
водой. Избыток непрореагировавших веществ удаляют вакуум-пе-
регонкой, а полученный в виде янтарной прозрачной смолы про-
дукт размалывают в порошок.
В качестве катализатора целесообразнее применять аромати-
ческие сульфокислоты, так как иод окисляет реакционную массу
(и конечный продукт получается темным), а серная кислота при-
водит к образованию сульфатов и других веществ, требующих
нейтрализации.
В конце 1940-х — начале 1950-х годов было установлено, что
продукт высокотемпературной конденсации — BLE-25, содержа-
щий в качестве основного компонента 5,5-диметилакридан, являет-
ся более эффективным антиоксидантом синтетических каучуков и
резин на их основе, чем Aminox. Поэтому производство стабилиза-
торов низкотемпературной конденсацией было прекращено и уси-
лия химиков были направлены на получение продуктов высокотем-
пературной конденсации (типа BLE-25 фирмы Naugatuck, США).
К началу 1960-х годов аналогичные стабилизаторы выпускались на
рынок уже в пяти странах под различными торговыми названиями.
Высокотемпературная конденсация. Конденсацию дифенилами-
на с ацетоном можно вести при 220—310 °C в избытке ацетона в
присутствии НС1, а также катализаторов, содержащих ионы гало-
генов (бром, иод, бромиды или иодиды тяжелых металлов — Sn,
Al, Си, Со, Bi, Sb, Fe, Mg, Zn, алкилгалогениды, галогенсодержа-
Щие органические кислоты, ацилгалогениды) [108—112]. Все эти
катализаторы позволяют получать готовый продукт с вязкостью
И* - 163
не выше 1 Па-с при 30°C. Вязкость стабилизатора имеет большое
значение в технологии его применения: подвижные продукты легко
подвергаются обработке при комнатной температуре и не требуют
предварительного подогрева.
В присутствии ароматических сульфокислот тоже можно полу-
чить продукт конденсации дифениламина с ацетоном, но он имеет
вязкость выше 1 Па-с при 30°C. Смесь дифениламина, ацетона и
бензолсульфодифенилимида выдерживают в автоклаве сначала 4 ч
при 250 °C, а затем 4 ч при 290 °C и давлении 6,5 МПа. Реакцион-
ную массу выливают в воду, отмывают от катализатора, упарива-
ют в вакууме при 70 °C и получают конечный продукт с вязкостью
1,12 Па-с [113].
Пиролизом продукта конденсации при 300—350 °C в присутст-
вии веществ, катализирущих саму реакцию [НО], можно увели-
чить вязкость продукта в 3,5 и более раз. Механизм пиролиза не
установлен, можно лишь предположить, что при этом происходит
молекулярная перегруппировка. Продукт конденсации дифенил?
амина с ацетоном, полученный в растворителе (чаще всего в изо-
пропаноле), тоже имеет повышенные вязкость и температуру плав-’
ления (178—191 °C) [111].
Интересен полунепрерывный способ конденсации, разработан-
ный фирмой Grant Chemical (США) и обеспечивающий получение
стабилизатора с вязкостью до 10 Па-с [112]. Конденсацию осу-
ществляют при 315—350 °C в присутствии иода или иодида желе-
за. Массовое соотношение ацетон : дифениламин берут в пределах
(0,44-0,7) : 1, а количество катализатора составляет 0,4—1,2% от
массы дифениламина. В автоклав загружают ацетон, дифениламин,
и иод, после чего герметизируют аппарат и помещают его в жид-
кий теплоноситель. Среда должна иметь достаточно большой объ-
ем, чтобы при погружении автоклава не было существенного пони-,
жения температуры. По изменению давления в автоклаве судят об
установлении равновесной температуры. Выдерживают смесь и
затем резко охлаждают реакционную массу, погружая автоклав
в теплоноситель, имеющий температуру 25 °C, на 5 мин. Из реак-
ционной массы отгоняют образовавшуюся воду и избыток ацетона ,
до достижения 170 °C. Затем снижают давление до 266,6 Па и про-
должают перегонку до 200 °C. К этому времени отгонка практиче-
ски прекращается, а температура в парах начинает расти до
100 °C. Ниже показано изменение вязкости получаемого продукта
в зависимости от условий реакции [112] (см. табл, на стр. 164).
Из представленных данных видно, что заданную вязкость стабили-
затора можно получить, варьируя условия реакции.
Следует отметить, что конденсация дифениламина с ацето-
ном — сложный процесс и до сих пор мало изученный. Условия
реакции, особенно температура, оказывают значительное влияние
на состав, свойства и эффективность получаемого стабилизатора.
Технологическая схема конденсации дифениламина с ацетоном.
В автоклав из нержавеющей стали через мерник загружают аце-
164
———" Массовое соотношение ацетои : дифенил- амин Количество катализатора, % (масс.) Время реакции, ч Температура. °C Вязкость продук- та при 30 °C, Па-с
0,40 0,4 6 315 0,5
0,52 0,4 6 320 2,0
0,55 0,4 6 315 2,3
0,52 0,1 '6 325 3,6
0,52 0,4 3 335 1,3
0,69 0,4 6 315 1,9
0,69 0,4 3 325 0,5
0,69 0,4 5 325 5,1
0,69 0,4 8 325 7,7
0.69 1,2 3 325 10,0
0,69 1,2 1 315 4,1
тон, а через люк — дифениламин и иод. Герметизируют аппарат,
включают мешалку и перемешивают реакционную массу в тече-
ние 20 мин до растворения дифениламина и иода в ацетоне. Массу
нагревают до 250 °C и выдерживают в течение 3 ч (в это время
давление в автоклаве за счет испарения реагирующих веществ
повышается до 3,5—4 МПа). После этого выключают обогрев и
охлаждают реакционную массу до 50 °C. Снижают давление; при
этом увлеченные пары ацетона конденсируются. Их охлаждают и
собирают в сборнике.
Через нижний спуск автоклава подают реакционную массу на
нейтрализацию в делительную воронку, снабженную мешалкой. Из
хранилища через мерник подают туда бензол (растворитель). За-
тем через мерник загружают 1%-ный раствор карбоната натрия,
перемешивают в течение 30 мин и дают отстояться. После отстаи-
вания верхний слой сливают в приемник. Первый отработанный
раствор карбоната натрия используют для следующей операции
нейтрализации, а после вторичного использования передают от-
работанный раствор на очистку [114]. Реакционную массу, остав-
шуюся после отделения раствора соды, дважды промывают водой
и передают в аппарат для отгонки ацетоно-бензольной смеси. По-
сле отгонки охлаждают реакционную массу до 50 °C и осущест-
вляют вакуум-сушку готового продукта.
Свойства и применение продуктов конденсации. Смесь, полу-
ченная при конденсации дифениламина с ацетоном, содержит 40—
45% 5,5-диметилакридана, 35—40% непрореагировавшего дифенил-
амина и «20% ароматических аминов неустановленного строе-
ния.
Она представляет собой темную вязкую жидкость; растворяется
в ацетоне, бензоле' и хлористом этилене; ограниченно растворяется
в бензине, этаноле и петролейном эфире.
Мало токсична; ЛДво = 23 000 мг/кг.
Горюча; при 280 °C горит желтовато-красным коптящим пламе-
нем; т. воспл. 181 °C (в открытом тигле); т. всп. 161 °C.
165
Смесь продуктов конденсации дифениламина с ацетоном выпус-
кается во многих странах в виде темно-коричневой вязкой жидко-
сти под разными торговыми наименованиями:
Торговое наименование Страна, фирма Плотность, КГ/мЗ
Accinox BL Индия, Alkali 1100
Agerite Superflex США, Vanderbilt 1100
Altofane PCL Франция, Mat. Col. 1100
Antigene BLE Япония, Sumitomo 1090
BLE-25 США, Naugatuck, Cyanamid 1094
KA 2002 ФРГ, Bayer -—
Naugatex 519 США, Naugatuck —
Nocrac В Япония, Ouchi Shinko —
Nonflex BA Япония, Seiko 1090—1100
Nonox BL Англия, ICI 1089—1100
Nonox BLN Индия, Alkali 1100
Permanax 47 Франция, Rhone Poulenc 1090
Santoflex DPA США, Monsanto ИЗО
Только один продукт — Altofane PCL-101 — выпускается фирмой
Mat. Col. (Франция) в виде порошка.
Смесь продуктов конденсации дифениламина с ацетоном еще
в 1960-х годах была одним из наиболее широко применяемых ста-
билизаторов различных синтетических каучуков и резин на их
основе (для резин на основе натурального каучука проявляет
меньшую эффективность). Она сообщает резинам стойкость к мно-
гократным деформациям, а резинам на основе хлоропрена — ста-
бильность к действию озона. В качестве стабилизатора СК и ре-
зин постепенно утрачивает свое значение, уступая более эффектив-
ным или менее окрашивающим соединениям.
Продукты высокотемпературной реакции дифениламина с аце-
тоном могут, однако, найти применение как антиозонанты хлоро-
преновых вулканизатов, как термостабилизаторы для темных ма-
рок полипропилена, поливинилхлорида и полиамидов (для послед-
них в сочетании с органическими солями меди).
ЛИТЕРАТУРА
1. Wagner Е. С. — J. Org. Chem., 1954, v. 19, № 12, р. 1862—1881.
2. Попов Л. К. и др. Методы получения диаминов — исходных веществ для
диизоцианатов. М., Изд. НИИТЭХИМ, 1972. 76 с.
3. Федорова О. Я., Аскаров М. А., Лосев И. П. —ЖОХ, 1957, т. 3, с. 775.
4. Ogata У., Kawasaki А. — Tetrahedron, 1964, v. 20, № 6, р. 1573—1578; РЖХим,
1966, 23Ж50.
5. Ogata У., Kawasaki A., Okumura N. — Kagaku Chemistry, 1966, v. 21, № 6,
p. 629—639; РЖХим, 1967, ЮЖ55.
6. Залукаев Л. П. — Изв. АН ЛатвССР, 1951, т. 1, с. 131.
7. Залукаев Л. П. — Изв. АН ЛатвССР, 1959, т. 3, с. 469.
8. Tung С. С. — Tetrahedron, 1963, v. 19, № 11, р. 1685—1689; РЖХим, 1965,
14Ж261.
9. Юкельсон И. И., Тищенко Т. Р., Легачева В. В. — Хим. пром-сть, 1971, № 2,
с. 11—12.
166
10. Тищенко Т. Р., Юкелъсон И. И. — В кн.: Химия и химическая технология по-
лимеров и органического синтеза. Воронеж, 1973, с. 8—11; РЖХим, 1974,
11Н168.
И Фрейдман Л. X., Руднева К. Г» Султанов А. С. — Изв. АН СССР, охн., 1954,
' с. 511-
12 Halson, пат. 3860650, 1969 г. (США).
13 А. с. 168015, 1963 г. (СССР).
14 Upjohn, пат. 3297759, 1957 г. (США); РЖХим, 1968, 15С229.
15. Upjohn, пат. 3676497, 1972 г. (США); РЖХим, 1973, 10Н143.
16 Upjohn, пат. 2049707 (ФРГ); пат. 1291908 (Англия); пат. 2065779 (Франция);
приоритет США; пат. 7000882 (ЮАР); С. А., 1971, v. 75, р. 5456.
17 ICI, пат. 1088205, 1967 г. (Англия).
18 Du Pont, пат. 3367969, 1968 г. (США); РЖХнм, 1969. 13Н235.
19. Du Pont, пат. 3476806, 1969 г. (США); РЖХим, 1971, 2Н160.
20. Tolochimie, пат. 1335124, 1963 г. (Франция); РЖХим, 1965, 1Н66.
21. Wingfoot, пат. 2683730, 1956 г. (США).
22. Allied, пат. 3163666, 1964 г. (США); РЖХим, 1966, 12Н159.
23. Jefferson, пат. 3971829, 1976 г. (США); РЖХим, 1977, 8Н148.
24. ICI, пат. 1167987, 1969 г. (Англия); РЖХнм, 1970, 13Н230.
25. Bayer, пат. 2227110, 1973 г. (ФРГ).
26. Bayer, заявка 2343658, 1975 г. (ФРГ); РЖХнм, 1975, 23Н197.
27. Monsanto, пат. 2818433, 1957 г. (США).
28. Monsanto, пат. 2974168, 1961 г. (США); РЖХим, 1962, 5Л137.
29. Upjohn, пат. 1181763, 1970 г. (Англия); приоритет США, 1967; С. А., 1970,
V. 72, р. 121159.
30. Пат. 57048, 1974 г. (ПНР) ; РЖХим, 1975, 17Н213.
31. Кучинский В. И., Г риз В. Е. — Нефтехимия. 1962, т. 2, № 4, с. 624—631.
32. А. с. 161346, 1975 г. (ЧССР).
33. Mobay, пат. 3260750, 1966 г. (США); РЖХим, 1968, 2Н215.
34. ICI, пат. 1228495, 1971 г. (Англия); РЖХим, 1971, 23Н167.
35. BASF, пат. 1179945, 1964 г. (ФРГ); С. А., 1964, v. 61, 16011.
36. BASF, пат. 1205975, 1965 г. (ФРГ); С. А., 1966, v. 64, 4991.
37. Bayer, пат. 1131877, 1962 г. (ФРГ); РЖХим, 1964, ЗС495. -
38. Kaiser Alum., заявка 2045420, 1971 г. (ФРГ); С. А., 1972, v. 76, 14078.
39. Bayer, пат. 2149998, 1973 г. (ФРГ).
. 40. Заявка 2426116, 1975 г. (ФРГ); приоритет Австрии, 1973; С. А., 1975, v. 82,
р. 125053.
41. Bayer, пат. 1643363, 1976 г. (ФРГ).
42. Bayer, заявка 2133870, 1973 г. (ФРГ); С. А., 1973, V. 78, 98282.
43. Sumitomo, заявка 50-16355, 1973 г. (Япония).
44. Mobay, пат. 3260751, 1966 г. (США); РЖХнм, 1968, 9Н104.
45. Kaiser Aluminium, пат. 3478099, 1969 г. (США); РЖХим, 1971, 1Н178.
46. Bayer, пат. 1267215, 1968 г. (ФРГ); С. А., 1968, v. 69, 18821.
47. Mobay, пат. 3277173, 1966 г. (США); РЖХим, 1968, 2Н221.
48. Upjihn, пат. 4008275, 1977 г. (США); РЖХим, 1977, 18Н137.
49. Upjohn, заявка 2719997, 1977 г. (ФРГ); приоритет США, 1976.
50. Upjohn, заявка 2719996, 1977 г. (ФРГ); приоритет США, 1976; С. А., 1978,
V. 88, р. 74185.
51. А. С. 386923, 1973 г. (СССР).
52. Upjohn, пат. 4029705, 1977 г. (США); РЖХим, 1978, 5Н221.
53. Bayer, пат. 1593865, 1975 г. (ФРГ); РЖХим, 1976, 8Н179.
54. Bayer, заявка 2238379, 1974 г. (ФРГ); РЖХим, 1975, НН213.
55. Du Pont, пат. 3899438, 1975 г. (США).
56. Минкин В. И. Канд, дне., Ростовский Гос. ун-т, 1959.
57. Пат. 66925, 1973 г. (ПНР); РЖХим, 1974, 8Н133.
58. А. С. 368281, 1973 г. (СССР).
59. Upjohn, пат. 3954867, 1971 г. (США); РЖХим, 1977, 1Н174.
60. Nippon, пат. 16691, 1970 г. (Япония); РЖХим, 1971, 12Н152.
61. ICI, пат. 1223039, 1971 г. (Англия).
167
62. ICI, пат. 1228562, 1971 г. (Англия); РЖХим, 1971.23Н168.
63. ICI, пат. 1229595, 1971 г. (Англия); РЖХим, 1971, 22Н191.
64. Mobay, пат. 3496229, 1970 г. (США); РЖХим, 1971, 6С393.
65. Bayer, Mobay, заявка 2549890, 1977 г. (ФРГ).
66. Asahi kasei, пат. 4625, 1965 г. (Япония) ; РЖХим, 1968, 14Н207.
67. А. с. 161352 и 161353, 1975 г. (ЧССР).
68. Jefferson, пат. 3362979, 1968 г. (США); С. А., 1968, v. 68, 59255.
69. Попов Л. К-, Ушакова М. Б., Толмачева Г. М.—ЖПХ, 1970, т. 43, № 12!
с. 2765—2767. '
70. Jefferson, пат. 2202500, 1972 г. (ФРГ); С. А., 1972, v. 77, р. Г26201.
71. Jefferson, пат. 3860637, 1975 г. (США); РЖХим, 1976, 2Н134.
72. BASF, пат. 1210872, 1973 г. (ФРГ); РЖХим, 1973, 18Н147.
73. Texaco, пат. 4053513, 1977 г. (США); С. А., 1978, V. 88, р. 6529.
74. А. с. 371206, 1973 г. (СССР). -
75. Егидис Ф. М., Рахманова А. С. — В кн.: Тезисы V Всесоюзной научно-техни-
ческой конференции по химикатам-добавкам для полимерных материалов
Тамбов, 1976, с. 13—14.
76. Johnson W. S., Buell В. G.—J. Am. Chem. Soc., 1952, v. 74, p. 4517—4520.
77. Craig D., Gregg C. — J. Am. Chem. Soc., 1953, v. 75, p. 2252.
78. Brown J. P. — Chem. and Ind., 1960, № 9, p 233.
79. Хидекель M. Л., Берлина В. Б., Разуваев Г. А. — В кн.: Труды совещания
по физическим методам исследования органических соединений и химических
процессов, Фрунзе, 1962, с. 170. , ,
80. Разуваев Г. А., Хидекель М. Л., Берлина В. Б. — ДАН СССР, 19Й, т. 145,
Ns 5, с. 1071—1074.
81. Залукаев Л. П. и др.— В кн.: Труды лаборатории химии высокомолекуляр-
ных соединений. Воронеж, Изд. В ГУ. 1962. Вып. 1, с. 80—89.
82. Seiko kagaku, пат. 49-34480," 1974 г. (Япония); РЖХим, 1975, 16С299.
83. Пат. 389908, 1973 г. (Испания); С. А., 1975, v. 82, р. 31270.
84. Пат. 3910918, 1975 г. (США); пат. 944135, 1974 г. (Канада); С. А., 1975, v. 82,
р. 43196; пат. 7102702 (ЮАР); С. А., 1972, v. 77, р. 88336.
85. Луговик Б. А., Юдин Л. Г.. Кост А. Н. — ЖПХ, 1975, т. 38, с. 216—220.
86. Brit. Chem. Eng, 1969, v. 14, № 4, p. 407.
87. Mitsubishi, заявка 52-116478, 1977 г. (Япония); РЖХнм, 1978, 15Н195.
88. Пат. 157370, 1970 г. (ВНР); РЖХим, 1972, 6Н196.
89. А.с. 398558, 1974 г. (СССР).
90. Elliott J., Jates P. — J. Org. Chem, 1961, v. 26, p. 1287—1289.
91. Monsanto, пат. 2748100, 1956 г. (США); РЖХим, 1958, 23429.
92. Пат. 149469, 1962 г. (ВНР); С. А, 1963, v. 58, р. 5646.
93. Пат. 55659, 1967 г. (ГДР); С. А, 1968, V. 68, р. 87203.
94. Пат. 51715, 1971 г. (РНР); РЖХим, 1972, 16Н259.
95. Yamamoto, пат. 50-29473, 1975 г. (Япония); РЖХим, 1976, 23Н188.
96. Roussel — UCLAF, заявка 2243679, 1975 г. (Франция); С. А, 1975, v. 8
р. 131485.
97. Заявка 2278338, 1976 г. (Франция); РЖХим, 1977, 70138.
98. Seiko, пат. 49-774, 1973 г. (Япония); С. А., 1973, v. 79, 105097.
99. Seiko, пат. 49-35628, 1974 г. (Япония); РЖХим, 1975, 15Н218.
100. Monsanto, пат. 1302647, 1971 г. (ФРГ); РЖХим, 1972, 11Н149.
101. Monter Sarabia, пат. 369261, 1971 г. (Испания); С. А., 1972, V. 76, р. 3714.
102. Пат. 161563, 1974 г. (ВНР); РЖХим, 1976, 20123; пат. 4296, 1972 г. (ВНР
С. А., 1972, v. 77, р. 75141.
103. Seiko, пат. 48-11103, 1973 г. (Япония); РЖХнм, 1974, 7Н165.
104. loshitomi, пат. 48-12744, 1973 г. (Япония); РЖХим, 1974, 9Н149.
105. А. с. 287943, 1971 г. (СССР).
106. Синтез органических препаратов. М., Издатинлит, 1953. Сб. 4, с. 194.
107. Mesraros е. а. — Magyar Kemai Folyoirat, 1968, v. 74, p. 482—484.
108. Naugatuck, пат. 2650252, 1953 г. (США); РЖХим, 1955, 10444; пат. 26572.
1953 г. (США); РЖХим, 1955, 24762; пат. 2663734, 1953 г. (США); п,
2666792, 1954 г. (США); РЖХим, 1955, 47221.
109. Rubber, Naugatuck, пат. 888615, 1953 г. (ФРГ); РЖХим, 1956, 20382.
168
HO Rubber, пат. 687233, 1953 г. (Англия); Chem. ZbL, 1953, S. 7421; пат. 698362,
' 1953 г. (Англия); РЖХим, 1956, № 2, 5190.
Ill Sumitomo, пат. 11363, 1964 г. (Япония); С. А., 1964, v. 61, р. 13237.
}12 Grant Chem., пат. 3417142, 1968 г. (США); РЖХим, 1970, 5Н236.
ИЗ Пат. 66624, 1973 г. (ПНР); РЖХим, 1974, 8Н153.
114 А. с. 175922, 1964 г. (СССР).
2.8. ТИОАЦИЛИРОВАНИЕ АМИНОВ.
ДИТИОКАРБАМАТЫ, ТИОКАРБАМИДЫ
и МЕРКАПТОБЕНЗИМИДАЗОЛЫ
При тиоацилировании аминов (взаимодействие с сероуглеро-
дом) в зависимости от условий реакции и природы амина получа-
ются различные соединения.
При взаимодействии сероуглерода с вторичными алифатически-
ми аминами (диметил-, диэтил-, дибутилам!ины) в водных раство-
рах гидроксида натрия образуются диалкилдитиокарбаматы нат-
рия
-f-NaOH
RsNH+CS2 > R,N—С—SNa
—Н2О ||
S
из которых реакцией обменного взаимодействия получают диал-
килдитиокарбаматы цинка, висмута и никеля — эффективные ста-
билизаторы резин. Опубликован ряд обзоров по методам синтеза
дитиокарбаматов [1—3].
Взаимодействие первичных аминов с сероуглеродом приводит
к замещенным тиокарбамидам:
2RNH, -I- CS2 ——* RNH—С—NHR
-h2s и
S
Этот метод может быть применен также для синтеза 1-монозаме-
щенных тиокарбамидов
+nh3
RNH2 + CS2 --> RNH-C-SH ——> TRNHCS]- [NHJ+ ----► RNH-C-NH,
II II II
s I s J s
несимметричных 1,3-дизамещенных тиокарбамидов
RNH—C— SH-RR'NH, ---> TRNHCS)- [NR'H3]+ ——> RNH—C—NHR'
II И 2 И
s L s J s
и 1,3,3-тризамещенных тиокарбамидов:
RNH—C—SH + R2NH * TRNHCSl- (NRiH,]+ ——* RNH—C—NRJ
|| li ~Hafe II
s L S s
Для синтеза стабилизаторов имеют значение взаимодействие
первичных ароматических аминов с сероуглеродом (синтез дифе-
169
нилтиокарбамида) и получение 1,3,3-тризамещенного тиокарбами-
да (1,3,3-три-н-бутилтиокарбамид). Взаимодействие аминов с се-
роуглеродом как частный случай получения производных тиокарб-^
амида подробно рассмотрено в обзорах [4, 5]. ’
Процесс получения тиокарбамидов обычно осуществляют в ор-'
ганических растворителях — этаноле, бензоле, диэтиловом эфире,
пиридине, четыреххлористом углероде. Синтез проводят в присут-
ствии катализаторов (сера, пероксид водорода, щелочь, пиридин,
пиридин с иодом, этилксантогенат калия). Сера — хороший ката-
лизатор, но возникают трудности при ее отделении от продуктов
реакции. Эффективным катализатором является также пероксид
водорода, однако и в этом случае выделяется свободная сера, ко-
торую трудно удалить.
Самые доступные катализаторы — едкое кали и едкий натр;
в их присутствии в качестве промежуточных продуктов образуются
тиу р а м дисул ьфиды
4-NaOH [О]
RNHa + CSa —-* RNH—С—SNa -->• RNH—С—S—S—С—NHR
а° Ц у ц
S . S S
и изотиоцианаты:
RNH—C-S—S-C—NHR > 2RNCS + HaS + S
II II
S S
Тиокарбамиды образуются на последующих стадиях:
RNCS+RNH2 —RNH—С—NHR
II
S
RNH-C—S—S-C-NHR —RNH—C-NHR
II II ~CS2 II
S S S
Если R — алкил, образование тиокарбамидов возможно только по
схеме I, так как алифатический изотиоцианат быстро перегруппи-
ровывается в соответствующий амин, не успевая вступить в реак-
цию с первичным амином.
Для ускорения реакции можно применять, как уже говорилось,
пиридин, который с сероводородом образует нестабильные продук-
ты. Лучших результатов удается достичь, если к раствору пири-
дина добавлять определенное количество иода, легко удаляемого
в виде пиридинийиодида:
2RNHa + CSa + Ig + 2CeI4N —-* RNH—C—NHR + 2CeHsN • HI
—b II
В ходе реакции раствор обесцвечивается, а пиридинийиодид осаж-
дается. Необходимо очень тщательно дозировать иод, так как его
170
избыток приводит к образованию соответствующего изотиоциа-
ната:
RNH, + CSa 4-12 + 2CeH6N ——> RNCS + 2CeH6N HI
—ь
При взаимодействии о-фенилендиамина с сероуглеродом в при-
сутствии щелочи образуется 2-меркаптобензимидазол [6, 7]:
NH2
+ CS2 + NaOH
NH2
;C-SH + Na2S2O3
NH
Имидазольное кольцо отличается высокой стабильностью к восста-
новлению: при каталитическом восстановлении различных бенз-
имидазолов гидрированию подвергается ароматическое, а не имид-
азольное кольцо.
В последнее время развивается метод получения 2-меркапто-
бензимидазола непосредственно из о-нитроанилина — взаимодей-
ствием с сероуглеродом в присутствии восстановителей.
Диалкилдитиокарбаматы металлов
" Alk^
/N—С—S—
A11Z ||
S
Me
п
Диалкилдитиокарбаматы металлов обычно получают взаимо-
действием диалкиламина с сероуглеродом в присутствии щелочи
с последующим обменом натрия па ион другого металла — обра-
боткой диалкилдитиокарбамата натрия соответствующими солями
соляной или серной кислоты (реже азотной). Процесс сопровож-
дается побочными реакциями с образованием окрашенных продук-
тов, а при получении и хранении диалкилдитиокарбаматов в при-
сутствии щелочи или аммиака иногда образуются сульфиды же-
леза в результате контакта реакционной массы с металлическими
поверхностями.
Для осветления таких растворов к ним добавляют небольшие
количества щавелевой или полифосфорной кислоты, поверхностно-
активные вещества [8, 9]. В присутствии поверхностно-активных
веществ можно получать диалкилдитиокарбаматы в виде эмуль-
сий, что особенно важно при их использовании в качестве стаби-
лизаторов. Пирролидон и его производные, а также тетрагидрофу-
ран являются хорошими стабилизаторами водных растворов диал-
килдитиокарбаматов аммония [10].
Например, диметилдитиокарбамат натрия получали с выходом
98% при медленном добавлении сероуглерода к водному раствору
едкого натра и диметиламина при 15°C [11]. При замене части
NaOH на Na2S удается исключить побочное образование изотио-
171
цианатов, тиокарбамидов и тиурамдисульфидов и получить более
чистые диалкилдитиокарбаматы. Замена натриевой щелочи на
карбонаты металлов позволяет сразу получить дитиокарбаматы.
Амины можно вводить в реакцию с сероуглеродом не только в
жидком, но и в газообразном состоянии совместно с газом-носите-
лем (азот, оксид углерода, воздух, водяной пар). При получении
диалкилдитиокарбаматов аммония газообразный аммиак барботи-
руют через смесь амина и сероуглерода [13] при комнатной тем-
пературе. Образующуюся соль непрерывно удаляют, экстрагируя
водой; это смещает равновесие вправо, сокращает процесс до 2—
5 ч и увеличивает выход аммониевой соли до 96%.
Интересен способ получения диалкилдитиокарбаматов щелоч-
ных металлов с помощью ультразвука, разработанный в Японии
[14]. Смесь сероуглерода, лаурилсульфата и воды облучают уль-
тразвуком с частотой 23 кГц и мощностью 150 Вт в течение 30 мин
для получения гомогенной эмульсии. Добавляют по каплям
40%-ный водный раствор диметиламина и 25%-ный раствор NaOH
в течение 20 мин. Получают бесцветный раствор диметилдитио-
карбамата натрия с выходом 97—98%.
Если однозамещенные производные алкилдитиокарбаминовой
кислоты легко выделить в виде бесцветных аморфных порошков,
то диалкилдитиокарбаматы на основе вторичных аминов часто
получаются в виде трудно кристаллизуемых масел. Их выделение
облегчается, если проводить реакцию в органическом растворителе
[12, 15, 16]: при получении аммониевых солей диалкилдитиокарб-
аминовой кислоты применяют вещества (ксилол, бензол, хлор-
бензол, толуол), растворяющие исходные компоненты и не раство-
ряющие образующуюся соль [13].
Цинковые и никелевые соли диалкилдитиокарбаминовой кисло-
ты получают взаимодействием натриевых или аммониевых солей
с хлоридом [11, 17, 18] или с сульфатом [13, 19, 20] соответствую-
щего металла. Диалкилдитиокарбаматы более тяжелых элементов
(висмут) обычно получают обменной реакцией диалкилдитиокарб-
амата натрия с нитратом висмута; дитиокарбаматы мышьяка по-
лучают взаимодействием диалкилдитиокарбамата аммония с рас-
твором АйгОз в концентрированной соляной кислоте [21]. Выход
диалкилдитиокарбаматов на стадии обменного взаимодействия
почти теоретический. Для улучшения фильтрования образующейся
соли в раствор добавляют поверхностно-активные вещества [17].
Диалкилдитиокарбаматы цинка, никеля и других металлов
можно получать в одну стадию в присутствии оксидов этих метал-
лов [15, 22], а диалкилдитиокарбаматы цинка — и в присутствии
Zn(OH)2 [23]. Едкий натр растворяют в воде, добавляют соляно-
кислый диметиламин (в виде водного раствора) и раствор
ZnSO4-7H2O, а затем медленно вводят сероуглерод. Реакционную
массу оставляют на несколько часов при 20 °C. Полученный
диметилдитиокарбамат цинка отфильтровывают, промывают водой
и сушат. Выход 94%; т. пл. 246 °C.
172
Антиокислительная активность диалкилдитиокарбаматов ме-
таллов зависит от их термической стабильности [18, 24]. Установ-
лено, что термическая стабильность понижается в ряду: Ni, Со, Zn,
Cd, Pb, Си, Sb, Bi, а длина алкильной цепи на термическую ста-
бильность влияет сравнительно мало. Ниже приведена темпера-
тура начала разложения (в °C) диалкилдитиокарбаматов некото-
рых металлов [24]:
Ди-н-амилдитиокарб- Диалкилдитиокарбама-
аматы ты цинка
никеля .... 255 диэтил- . . . 230
кобальта . . . 242 ди-я-бутил- . 234
цинка .... 233 ди-я-амил- . . 233
кадмия . . . 231 ди-я-октил- . . 242
свинца .... 229 диизобутил- . . 233
меди .... 192 диизоамил- . . 250
сурьмы . . . 179
висмута . . . 150
Разложение диалкилдитиокарбаматов металлов происходит
с выделением сероуглерода [25—27]; остаток представляет собой
сульфид металла [24]. При термическом разложении возможно
образование дитиокарбаматов, изотиоцианатов и олефинов [28,
29]. Исходя из состава образующихся продуктов предложена сле-
дующая схема термического разложения диалкилдитиокарбаматов
металлов [24]:
R2N—С—S—Me—S—с—NR, -----> CSa + R'CH=CH2 + R2NH + RNCS + MeS
II II
s s
R—алкил; R'-алкил, содержащий на два атома С меньше, чем у R
Энергия активации разложения диалкилдитиокарбаматов метал-
лов составляет 106—354 кДж/моль, а термическая стабильность
находится в прямой зависимости от энергии активации [24].
Технология получения солей дитиокарбаминовой кислоты не-
сложна.' В реактор загружают водный раствор диалкиламина,
раствор едкого натра и при 20—22 °C под слой жидкости подают
сероуглерод. Герметизируют аппарат и перемешивают реакцион-
ную массу в течение 20—30 мин. Давление в аппарате на короткое,
время может подняться до 0,01 МПа вследствие испарения серо-
углерода и диалкиламина. Реакционную массу, свободную от при-
меси сероуглерода, передают на очистное фильтрование, чтобы
удалить примеси нерастворимых солей железа.
Разработан технологический процесс получения диалкилдитио-
карбамата натрия по непрерывной схеме [30]. Дозировочными
насосами с постоянной скоростью в реактор вводят 50%-ный
NaOH, воду и раствор диалкиламина. Реагенты перемешивают
циркуляцией. Сероводород вводят в реактор противотоком, а об-
разующийся диалкилдитиокарбамат натрия выводят из верхней
части аппарата. Количество непрореагировавшего сероуглерода
доставляет 0,67 кг/ч; выделяющееся тепло снимают рециркуля-
< Цией сероуглерода и охлаждением.
173
Дибутилдитиокарбамат цинка
(C4I I9)2N—С—S—Zn—S—С—N(C4H8)2
II II
S S
представляет собой белый порошок с характерным запахом; т. пл.
112 °C. Растворяется в бензоле, четыреххлористом углероде, хлори-
стом метилене, сероуглероде. Ограниченно растворяется в этил-
ацетате и ацетоне; практически не растворяется в бензине, этаноле,
воде. Стабилен при хранении, мало токсичен.
Дибутилдитиокарбамат цинка выпускается во многих странах:
Торговое наименование Страна* фирма T. пл.. °C
Accelerator 77 США, Du Pont 95—108
Butasan Англия, Monsanto 95—108
Eutazate США, Naugatuck 95—100
Butyl Zimate США, Vanderbilt 104—108
Butyl Ziram Франция, Prochim 95—108
Nocceler BZ Япония, Ouchi Shinko 102
Robac ZBUD Англия, Robinson 106—110
Vulcafor ZNBZ Англия, ICI 95-108
Vulkacit LDB ФРГ, Bayer 105
ZDBC США, Du Pont 95—108
Дибутилдитиокарбамат цинка — эффективный стабилизатор
уретановых, хлоропреновых и других синтетических каучуков. Мо-
жет быть использован в рецептурах, применяемых для изготовле-
ния протекторов шин, резиновых технических изделий и изделий,
применимых в медицине и пищевой промышленности, для кабель-
ной изоляции, для самовулканизующихся клеев.
Обеспечивает высокотемпературную стабилизацию реактивных
топлив и смазочных материалов. Применяется как добавка, пони-
жающая износ металлических поверхностей при трении.
Дибутилдитиокарбамат цинка — широко известный ультраус-
коритель резиновых смесей на основе натурального и синтетиче-
ских каучуков и латексов. Не изменяет цвет изделий.
Дибутилдитиокарбамат никеля
(C4H9)2N—С—S—Ni—S—С—N(C4H8)2
II II
S S
представляет собой кристаллический порошок зеленого цвета; т. пл.
90 °C; т. всп. 65,5 °C. Растворяется в ацетоне и четыреххлористом
углероде, ограниченно растворяется в этаноле, не растворяется
в воде.
Относится к числу слабоокрашивающих стабилизаторов. Вы-
пускается (в основном в виде порошка) под различными назва-
ниями:
174
Торговое наименование Страна, фирма Выпускная форма Т. ПЛ., °C
Antage NBC Япония, Kawaguchi — 87—90
Antigene NBC Япония, Sumitomo Порошок 87—90
JO 4011, JO NBTC Италия, Bozzetto То же 87—90
NBC США, Du Pont » 88—90
Nilame Франция, Prochim Гранулы —
Robac Ni BuD Англия, Robinson Порошок 81—88
Van Hasselt BTN Нидерланды, CH То же 87—90
Дибутилдитиокарбамат никеля — эффективный стабилизатор
синтетических каучуков и резин на их основе. Повышает свето- и
озоностойкость; пассивирует действие металлов переменной ва-
лентности; увеличивает стабильность полиизоцианатов к действию
кислорода, света и тепла. Повышает теплостойкость сульфохлори-
рованного полиэтилена, фотохимическую стабильность полипропи-
лена, атмосфере- и термостойкость полиуретановых композиций
(используемых для производства пенопластов, клеев), а также
покрытий, эластичных нитей и других изделий. Обеспечивает вы-
сокотемпературную стабилизацию реактивных топлив; защищает
резины и пластические массы (полистирол) от действия у-излуче-
ния.
Диметилдитиокарбамат висмута
Г(СН3)2И—С—S—1 Bi
I S .3
представляет собой желтоватый порошок; т. разл. выше 230 °C.
Растворяется в бензоле, хлороформе и сероуглероде, не раство-
ряется в воде.
Относится к числу слабоокрашивающих стабилизаторов. Вы-
пускается в США фирмой Vanderbilt в виде порошка и гранул
(Rodform Bismate); т. пл. 230°C (с разложением).
Диметплдитиокарбамат висмута является стабилизатором га-
логенированных каучукоподобных сополимеров; защищает резины
на основе каучуков общего назначения от термоокислительной
деструкции; ускоряет вулканизацию резиновых смесей на основе
натурального и синтетических каучуков.
Свойства и эффективность диметилдитиокарбамата висмута
изучены пока недостаточно.
Замещенные тиокарбамиды
Из замещенных тиокарбамидов промышленное применение на-
ходят 1,3-дифенилтиокарбамид (стабилизатор поливинилхлорида)
и 1,1,3-три-н-бутилтиокарбамид (неокрашивающий антиозонант
резин).
175
1,3-Дифенилтиокарбамид
1,3- Дифенилтиокарбамид впервые был получен в 1848 г. Лаурентом взаимо-
действием анилина с сероуглеродом в спиртовой среде при нагревании, а
в 1873 г. было установлено каталитическое действие серы на эту реакцию.
В дальнейшем для ускорения процесса было предложено вводить в реакционную
массу в небольших количествах щелочь и пероксид водорода.
Реакцию анилина с сероуглеродом можно проводить в органических раство-
рителях (пиридин, нитробензол). Добавка пиридина и других органических осно-
ваний сокращает время реакции и повышает выход 1,3-дифенилтиокарбамида.
Известен способ получения 1,3-дифенилтиокарбамида из нитро- нли нитрозо-
бензола н сероуглерода в присутствии щелочи [31].
Есть и другие, не имеющие промышленного значения, методы получения
1,3-дифенилт1юкарбамида — перегруппировка тиоцианата дифениламина, взаимо-
действие анилина с тиофосгеном в среде пиридина или воды, гидролиз фенилизо-
тиоцианата [33], взаимодействие фенилизотиоцианата с ароматическими азомети-
нами [34].
Распространенным в промышленности способом получения ди-
фенилтиокарбамида является взаимодействие анилина с сероугле-
родом в присутствии щелочи:
Для более полного использования сероуглерода предложено вво-
дить в реакцию только 1 моль сероуглерода на 2 моль анилина, а
образующуюся соль тиоугольной кислоты разлагать бисульфитом
натрия:
4CeH5NH2 + 2CS2 + 2NaOH + 4NaHSO3 -> 2(CeHBNH)aCS + 3Na2S2O3 + 5H2O
। Выделяющаяся соль тиоугольной кислоты реагирует с анилином,
образуя дифенилтиокарбамид
2CeHBNH2 + Na2CS3 + 4NalISO3 -► (CeH6NH)2CS + 3Na2S2O3 + 3H2O
। Б этом случае сероуглерод используется полностью. Маточный
раствор в основном содержит тиосульфат натрия, который можно
। выделить (упариванием и кристаллизацией) и использовать как
товарный продукт в фотографии.
1 В качестве окислителя вместо бисульфита натрия можно ис-
пользовать нитрит натрия [32]:
! 2ArNH2 + 2CS2 + 2/3NaOH -f- 4/3NaNO3 ->
I 2(ArNH)2CS + Na2S2O3 + %NH3 + V3H2O
В этом случае расходуется в три раза меньше щелочи и окислите-
ля, чем при использовании бисульфита натрия.
176
Механизм реакции ароматических первичных аминов с сероуг-
леродом предложен Снедкером и Дроздовым; наиболее вероятной
признается следующая схема:
ArNH2 + CS2 + NaOH--ArNH—С—SNa + H2O
II
S
ArNH—C—SNa —
II
S
4-NaSH
ArN=C=S + NaSH
Na2CSs + ArNH2
При анализе массы, полученной при синтезе ди-о-толилтиокарб-
амида, были обнаружены о-толилизотиоцианат и .Na-соль тиоуголь-
ной кислоты [35]. Прибавляя (по каплям) бензольный раствор
о-толилизотиоцианата в кипящий бензольный раствор о-толуидина,
получили о-толилтиокарбамид, что подтверждает механизм, пред-
ложенный Дроздовым.
По мнению авторов работы [35], вторая стадия процесса мо-
жет протекать по схеме:
Na2CS3 Na2S + CS2
CS2 + 2ArNH2 ---> (ArNH)2CS + H2S
H2S + Na2S -->- 2NaSH
2NaSH + 4NaHSO3 ---► 3Na2S2O3 + 3H2O
Технологическая схема получения. В аппарат 1 (рис. 19), снаб-
женный рубашкой и мешалкой, загружают при перемешивании
Рис. 19. Технологическая схема получения дифенилтнокарбамида:
— реактор; 2 —сборник; 3 — холодильник; 4 — ловушка; 5 — барабанный вакуум-фильтр;
6 — транспортер; 7 — бункер; 8 — емкость.
12—1057
177
2%-ный раствор NaOH, воду и анилин. Останавливают мешалку |
и из мерника под слой жидкости загружают сероуглерод. Вклю- '
чают мешалку, а поскольку процесс конденсации экзотермичен,
температура реакционной массы в течение 10—15 мин после вклю-
чения мешалки поднимается до 45—55 °C. Через 30 мин в рубашку
подают пар и нагревают реакционную массу до 98 °C; давление
при этом поднимается до 0,1—0,2 МПа. Газы из реактора посте-
пенно сбрасывают в атмосферу через холодильник 3 и ловушку 4,
заполненную 20%-ным раствором NaOH. После снижения давле-
ния в реактор загружают при перемешивании 22%-ный раствор
бисульфита натрия; при этом поддерживают температуру 95—
98 °C. 1
Суспензию из реактора 1 с содержанием 180—230 г/л выдавли- 1
вают в сборник 2, а оттуда центробежным насосом подают на I
вакуум-фильтр 5. Во время фильтрования промывают пасту горя-
чей водой до содержания анилина 0,4% и до нейтральной реакции
промывных вод. Фильтрат и промывные воды собирают в ем-
кость 8, откуда фильтрат откачивают в цех получения тиосульфата
натрия. Вакуум в системе фильтрования создают вакуум-насосом
через барометрический конденсатор.
Паста, содержащая 23% влаги, срезается ножом и ленточным Л
транспортером 6 подается в бункер 7 с тарельчатым питателем. Ц
Свойства и применение. 1,3-Дифенилтиокарбамид представляет Я
собой белый порошок с сероватым оттенком; т. пл. 154 °C. Раство- Ц
ряется в ацетоне, бензоле, петролейном эфире, хлороформе, эта- Я
ноле, в разбавленных щелочах. Ограниченно растворяется в бен* Я
зине и воде.
1,3-Дифенилтиокарбамид—мало токсичное вещество; ЛД5о=|вИ
= 1500 мг/кг (для крыс).
Выпускается в большинстве развитых стран под разными топ^И
говыми названиями:
Тортовое наименование Страна, фирма T. пл., °C)
Тиокарбанилид СССР >148
Activit Бельгия, UNBE >148
Anchor acel Англия, Anchor >148
Ekagom LL Франция, Kuhlmann 148—150
Eveite TC Италия, ACNA Montecatini 149
Excellerex NCC США, Naugatuck «148
Interstab C 27 Нидерланды, Akzo >148
Soxinol A Япония, Sumitomo «148
Stabilisator C ФРГ, Bayer 150
Thip США, Monsanto 148 -
Vulcanol NCA США, Du Pont «148
1,3-Дифенилтиокарбамид — в прошлом широко распространен
ный стабилизатор поливинилхлорида и сополимеров винилхлорн
да, но в последнее время теряет свое значение и вытесняется 6i
178
лее эффективными стабилизаторами. Является эффективным
ускорителем вулканизации резиновых смесей на основе натураль-
ного и синтетического каучуков общего назначения.
1,1,3- Т ри-н-бутилтиокарбамид
(C4H9)2N—С—NH—С4П8
II
S
Впервые он был получен в 1904 г. взаимодействием бутилгорчич-
ного масла с дибутиламином, растворенным в холодном бутаноле:
с4н9он
C4H9NCS + NH(C4H,)2 --> С4Н,-NH-С-N(C4H9)2
II
S
Способы получения N-алкилированных тиокарбамидов можно
разделить на несколько основных групп '[5].
1. Взаимодействие органических изотиоцианатов с алифатиче-
скими аминами [36, 37]:
RNCS + R'R"NH--► RNH—С—NR'R"
II
S
Реакцию обычно проводят в мягких условиях с применением спир-
тов в качестве растворителей. В зависимости от природы амина
реакция может быть экзотермической (ди ал кил а мины, морфолин)
или эндотермической (дициклогексиламин, этиланилин) [34].
2. Взаимодействие щелочных тиоцианатов с солянокислыми
аминами. Образующиеся при этом моно- и дизамещенные аммо-
нийтиоцианаты при нагревании в течение нескольких часов пере-
группировываются в тиокарбамиды:
RNH, + НС1 + NH4SCN --> RNH—С—NHa + NH4C1
II
S
Реакцию, как правило, ведут в инертном органическом раствори-
теле (хлорбензол) или в водной среде. Выход N-замещенных тио-
карбамидов по этому способу не превышает 40% вследствие по-
бочного образования изотиоцианата:
RNH2 + 2НС1 + NH4SCN -► RNCS + 2NH4C1
Недавно разработан способ получения N-замещенных тиокарбами-
дов из тиоцианата на основе трифенилфосфина (C6Hs)3P(SCN)2
с выходом до 80% [38].
3. Из солей или эфиров алкилдитиокарбаминовой кислоты
[39]:
RNH—С—SNa 4- НС1 NHR 'R" ► RNH—C-SH • NHR'R" + NaCl
RNH—C-SH-NHR'R" —RHC—NR'R"
|| -H«S II
s s
12’
179
I
Процесс идет в две стадии: сначала образуются соли или эфиры
алкилдитиокарбаминовой кислоты, а их затем превращают одним
из указанных выше приемов в алкилдитиокарбамиды.
4. Взаимодействие сероводорода с алкилцианамидами и амина-
ми. Его обычно проводят, насыщая этанольный раствор соответст- |
вующим цианамидом, аммиаком и сероводородом [5] (аммиак иг- I
рает роль катализатора). Реакционную смесь кипятят или выдер- ;
живают при 100 °C:
RR'NCN + HgS -* RR'N—С—NH2 Jj
u -Я
Я
2RNH2 + HjS + СС14 -► RNH—С—NHR + 4НС1 Я
Этим способом можно получить моно- и 1,3-диалкйлтиокарбамид.
Взаимодействие сероводорода с первичными аминами в присут-
ствии четыреххлористого углерода происходит при небольшом дав-
лении [40].
5. Тиоацилирование алкиламинов сероуглеродом [42—49К
Этот метод является наиболее общим для синтеза Ы-алкилтиокарпИ
амидов, в том числе и для 1,3,3-тризамещенных: fl
CS2 + RNHa -> RNH-C—SH
I! J
S 1
RNH—C—SH + RaNH ► [RNH—C-SI" [RJNHa]+ ► RNH-C NRa + H2S
Реакцию проводят в водной или спиртовой среде (иногда с доба^И
лением щелочи) при 20—150 °C. Выход тиокарбамидов достигае^Н
95—98%. Ш
Из известных способов получения несимметричных алкилтио^Ч
карбамидов, в частности 1,3,3-три-н-бутилтиокарбамида, наиболь- >1
ший практический интерес представляют взаимодействие бутилизо- I
цианата с дибутиламином
C4H8NCS + NH(C4Hg)2 -> C4H9NH—С—N(C4Hb)2 Я|
ii
s Ш
взаимодействие натриевой соли бутилдитиокарбаминовой кислоты |
с гидрохлоридом дибутиламина 1
QHbNI I—С—SNa + НС1 • NHfC^Jj
S
---* C4H.,NH-C-SH-NH(C4H9)2 —-* C4H8NH-C-N(C4H8)2
II ~Has II
s S
180
и реакция бутил- и дибутиламина с сероуглеродом с последующим
дегидросульфидированием аммониевой соли дитиокарбаминовой
кислоты:
+cs2
C4HeNH2 + NH(C4H8)2 -> C4H8NH—С—-SHNH(C4H9)2 —*
|| —
s
---> C4H9NH—C-N(C4H9)2
II
s
В связи с дефицитностью сырья (бутилизоцианат) и несмотря
на простоту технологии при промышленном получении 1,1,3-три-
н-бутилтиокарбамида исходят из бутиламина, дибутиламина и се-
роуглерода.
Технологическая схема получения. В реактор загружают моно-
бутиламин, дибутиламин и воду. Смесь охлаждают до 10 °C, после
чего, не прекращая охлаждения и перемешивания, под слой реак-
ционной массы загружают сероуглерод (температура 10—15 °C).
Выдерживают смесь в течение 1 ч, под слой массы барботируют
азот, нагревают ее до 93—98 °C и выдерживают в течение 2 ч (вы-
деляющийся сероводород улавливают). После снижения темпера-
туры до 60 °C и прекращения перемешивания расслаивают реак-
ционную массу; нижний слой сливают в сборник. Полученный
три-н-бутилтиокарбамид представляет собой маслянистую жид-
кость желтого цвета, которую в реакторе промывают и сушат.
Свойства и применение. 1,3,3-Три-н-бутилтиокарбамид—мас-
лянистая жидкость желтого цвета со слабым характерным запа-
хом; «0 = 1,5159; d20 = 965 кг/м3. Растворяется в бензине, петролей-
ном эфире, сольвент-нафте, этаноле.
Мало токсичен; ЛД6о = 3000±280 кг/кг (для крыс); раздра-
жающим действием на кожу не обладает.
Выпускается в США фирмой Monsanto под названием Santo-
white TBTU.
1,3,3-Три-н-бутилтиокарбамид — неокрашивающий антиозонант
и стабилизатор (слабый) светлых и цветных резин на основе на-
турального, изопренового, хлоропренового и бутадиен-стирольных
каучуков. Вызывает подвулканизацию резиновых смесей. Пред-
ставляет интерес как ингибитор коррозии, как стабилизатор изо-
цианатов и полупродукт для их синтеза.
2-Меркаптобензимидазол
Он может существовать в двух таутомерных формах:
N NH
СХХ— схх
NH NH
181
Впервые был получен Лелманном в 1883 г. нагреванием рода-
нистоводородной соли (тиоцианат) о-фенилендиамина при 120—
130°C. Другой вариант этого метода—-нагревание о-фениленди-
амина с водным раствором тиоцианата калия. Метод получения
2-меркаптобензимидазола из тиоцианатов развития не получил:
сырье мало доступно, выход продукта невысок. Препаративное зна-
чение имеют способы получения 2-меркаптобензимидазола из гид-
рохлорида о-фенилендиамина и тиокарбамида (или дифенилтиф
карбамида) в пиридине, а также из о-фенилендиамина и тиофс
сгена в хлороформе или воде.
Практическое значение имеет способ тиоацилирования о-фени
лендиамина [50, 51] или о-нитроанилина сероуглеродом в присут
ствии восстановителя [41, 52]; на этой основе в Германии в
1930-х годах было организовано промышленное производство
2-меркаптобензимидазола.
В настоящее время промышленное значение имеют оба вариан
та — из о-фенилендиамина и из о-нитроанилина, в зависимости о]
ресурсов сырья.
Получение 2-меркаптобензимидазола из о-фенилендиамин:
проводят в среде изопропанола в присутствии гидроксида аммония
с почти теоретическим выходом [51] или в среде бензтиазола
в присутствии щелочи с выходом «98% [52]. Применение бензти-
азола в качестве растворителя позволяет проводить реакцию в го-
могенной фазе.
Синтез 2-меркаптобензимидазола проводят в водной среде
в присутствии H2S или NaSH [41]. Преимущество этого способа —
то, что 2-меркаптобензимидазол получают в свободном виде, а не
в виде щелочных солей, благодаря чему исключается одна техно-
логическая стадия — нейтрализация щелочных растворов, необхо-
димая в первом варианте.
Промышленное производство 2-меркаптобензимидазола состоит
из следующих стадий: восстановление о-нитроанилина сульфидом
натрия с получением о-фенилендиамина, взаимодействие о-фени-
лендиамина с сероуглеродом, выделение технического 2-меркапто-
бензимидазола бисульфитом натрия, очистка щелочных растворов
2-меркаптобензимидазола активированным углем и выделение очи-
щенного 2-меркаптобензимидазола серной кислотой:
Общую схему синтеза 2-меркаптобензимидазола можно предста-
вить так:
+ 2Na2S + 4CS2 + 6NaOH —>
182
N
SNa + 3NasS<jO3 + 5HgO
По окончании взаимодействия о-фенилендиамина с сероуглеродом
избыток последнего остается в реакционной массе связанным
(в виде тритиокарбоната натрия) и выделяется в свободном со-
стоянии при последующей обработке реакционной массы кислотой.
Для выделения целевого продукта обрабатывают реакционную
массу бисульфитом натрия при 97—98 °C в аппарате с открытым
выходом в атмосферу. При этом реакционная масса освобождает-
ся от избыточного сероуглерода. В осадке не содержится свобод-
ной серы, что дает возможность при дальнейшей очистке продукта
растворять его в щелочи, а не в растворе извести с последующим
осаждением 2-меркаптобензимидазола из раствора серной кисло-
той вместо соляной.
Свойства и применение. 2-Меркаптобензимидазол представляет
собой белый порошок с желтоватым оттенком; т. пл. 304 °C. Рас-
творяется в ацетоне, этаноле, этилацетате, петролейном эфире, ме-
тиленхлориде (ограниченно). Не растворяется в бензоле, бензине,
хлороформе, толуоле, четыреххлористом углероде, воде.
Умеренно токсичен; ЛД5О = 1250 мг/кг (для мышей).
2-Меркаптобензимидазол выпускается во многих странах под
разными названиями в виде порошка с желтоватым или кремо-
вым оттенком:
Торговое наименование Страна, фирма T. пл., °C
Age Resistor MB Италия, ACNA Montecatini 230
Altofane МТБ Франция, Pcchime 275
Antage MB Япония, Kawaguchi —
Antigene MB Япония, Sumitomo >285
Antioxidant MB ЧССР >270
Antioxygene MTB Франция, Mat. Col. 270—290
ASM MB, MBI ФРГ, Bayer 290
Nocrac MB Япония, Ouchi Shinko 280
Permanax 21 Франция, Rhone Poulenc 290—300
2-Меркаптобензимидазол находит широкое применение в каче-
стве стабилизатора различных синтетических каучуков — бута-
диен-стирольных, бутадиен-нитрильных, уретановых. Неокраши-
вающий антиоксидант резиновых смесей на основе натуральных и
синтетических каучуков; пассивирует действие металлов перемен-
ной валентности. Является эффективным термостабилизатором по-
лиэтилена, полипропилена, полиолефиновых волокон, акриловых
полимеров; предотвращает обесцвечивание эластичных пенополи-
уретанов при экзотермических реакциях, развивающихся в процес-
се формования; предотвращает полимеризацию бензальдегида.
183
Эффективность 2-меркаптобензимидазола значительно повы-
шается в сочетании с такими стабилизаторами, как пространствен-
но затрудненные фенолы, 4-гидроксифенил-2-нафтиламин и метил-
меркаптобензимидазол.
ЛИТЕРАТУРА
1. Thorn. G. D., Ludwig R. A. Dityocarbamates and Related Compounds. Amster-
dam — New York, 1962. 298 p.; РЖХим, 1963, 23Ж74.
2. Livio C. e. a. — Chim. Ind., 1966, v. 48, № 7, p. 689—695; C. A., 1966, v. 65,
p. 16886.
3. Walter W., Bode K. D. — Angew. Chem. Tnt. Edn., 1967, B. 6, № 4, S. 281—
293; C. A., 1967, v. 67, p. 634145.
4. Schroeder C.— Chem. Revs, 1955, v. 55, p. 181.
5. Мозолис В. В., Иокубайтите С. 77,— Успехи химии, 1973, т. 27, с. 1310—
1324.
6. Wright J. — Chem. Revs, 1951, v. 48, p. 397.
7. Пожарский А. Ф„ Тарнавский А. Д., Симонов A. M. — Успехи химии, 1966,
т. 35, с. 261—302.
8. Пат. 25628, 1974 г. (Япония); С. А., 1975, v. 82, р. 170127.
9. А. с. 183735, 1966 г. (СССР).
10. Тоуа Yuki, пат. 51-29126 и 51-29128,. 1976 г. (Япония); РЖХим, 1977, 140355,4
140356.
11. Issoire J., Musso L. — Mempotidres, 1960, v. 42, p. 427—443. C. A., 1961, v. 55,
p. 16419.
12. Рухадзе E. Г. и dp. — ЖОХ, 1969, № 11, c. 2545—2551.
13- Пат. 69651, 1973 г. (ПНР); РЖХим, 1975, 5H174.
14. Sumitomo, пат. 9009, 1965 г. (Япония); РЖХим, 1967, 23Н143.
15. Пат. 107070, 1963 г. (ЧССР); РЖХим, 1964, 18Н36. -
16. Пат. 135434, 1970 г. (ЧССР); РЖХим, 1971, 2011655.
17. А. С. 165705, 1964 г. (СССР).
18. Шхиянц И. В. и др. — Нефтехимия, 1969, т. 9, № 4, с. 616—619. —
19. Tripathy FL, Mahapalra G. N. — J. Inst. Chem., Calcutta, 1968, v. 41, p. 200—
203; C. A, 1970, v. 72, p. 43077.
20. Uniroyal, пат. 3674824, 1972 г. (США); РЖХим, 1973, 9С502.
21. А. с. 245075, 1969 г. (СССР).
22. А. с. 168684, 1952 г. (СССР).
23. Пат. 19114, 1963 г. (Япония) ; РЖХим, 1965, 20Н68.
24. Афаносова Г. П. и др. —Нефтехимия, 1971, т. И, № 6, с. 911—918.
25. Miller D. М., Latimer R. А. — Can. J. of Chem., 1962, v. 40, № 2, р. 246—255;
РЖХим, 1963, 6Ж40.
26. Bighi С., Saglietto б-— J. Chromatogr., 1965, v. 17, p. 13—22; РЖХим, 1966,
1Ж100.
27. Ляликов Ю. С., Китовская М. И.—Укр. хим. ж., 1969, т. 35, с. 719—725.
28. Takatni F. е. a.— Chem. Pharm. Bull., 1973. v. 21, p. 329—334, 594—599; 1974,
v. 22, p. 275—279; РЖХим, 1974, 18Б1004.
29. Madsen J. e. a. — J. Org. Chem., 1967, v. 32, p. 2054.
30. Пат. 113, 1975 г. (ГДР); РЖХим, 1976, 15Н121.
31. Olin, пат. 3636104, 1972 г. (США); РЖХим, 1972, 21Н123.
32. А. с. 185895, 1965 г. (СССР).
33. Martin D., Mucke W. — Monatsber. Dtsch. Akad. Wiss. Berlin, 1964, B. 6, № 5,
p. 362—363; РЖХим, 1965, 6Ж190.
34. Козлов H. С. и др. —Ж. орг. химии, 1969, № 12, с. 2195—2196.
35. Мартынов Н. В. и др. — В кн.: Синтез и исследование эффективности хими-
катов для полимерных материалов. Тамбов, Тамбовская правда, 1969. Вып. 2,
с. 326—331.
36. Kostir I. V., Loukota L., Vejdelek Z.-—Chem. Listy, 1946, v. 40, p. 281—282.
37. Синтезы органических препаратов. M., Издатинлит, 1949. Вып. 3, с. 318.
184
38 Tamura У. e. a. — Tetrahedron Letters, 1978, №20, p. 1753—1754; РЖХим,
' 1978, 23Ж126.
39 Hydrierwerke, пат. 1155432, 1964 г.; пат. 1183070, 1965 г. (ФРГ); РЖХим,
’ 1966, 11Н86, 24Н445.
40 Dunlop, пат. 1071874, 1963 г. (Англия); С. А., 1967, V. 66, р. 104724.
4L Bayer, пат. 1162377, 1971 г. (ФРГ); РЖХим, 1972, 7Н210.
42. Купченко В. И. и др.— В кн.: Синтез и исследование эффективвости химика-
тов для полимерных материалов. Тамбов. Тамбовская правда, 1969. Вып. 2,
с. 576—585.
43. А. с. 176289, 1966 г. (СССР).
44. Salzgitter, пат. 1173521, 1961 г. (Англия); приоритет ФРГ, 1966; С. А., 1970,
V. 72, р. 54821.
45. BASF, пат. 1910174, 1974 г. (ФРГ); РЖХим, 1975, 9Н144.
46. Pennsalt, пат. 3288684, 1966 г. (США); С. А., 1967, v. 66, р. 55069.
47. Firestone Tire, пат. 2849420, 1958 г. (США); РЖХим, 1960, 20334.
48. Пат. 13624, 1961 г. (Япония); С. А., 1962, v. 56, р. 12749.
49. Sanshin, пат. 50-25456, 1975 г. (Япония); РЖХим, 1976, 17Н123.
50. Синтезы органических препаратов. М., Издатинлит, 1953. Вып. 4, с. 295—
296.
51. Du Pont, пат. 3895025, 1975 г. (США); РЖХим, 1976, 9Н228.
52. Bayer, пат. 2202204, 1973 г. (ФРГ); РЖХим, 1974, 21Н234.
ГЛАВА 3
Производные фенола
Защитное действие соединений этого класса известно более ста
лет (первый патент был взят в 1870 г.), но широкое практическое
применение они нашли в последние 20—25 лет. Это связано с внед-
рением полимерных материалов во многие сферы деятельности че-
ловека — технику, здравоохранение, быт. Резко возросли требова-
ния к внешнему виду изделий из полимерных материалов и к их
санитарно-гигиеническим свойствам. Это вызвало необходимость
| производства полимеров, защищенных неокрашивающими и неток-
сичными стабилизаторами. Такими стабилизаторами и являются
производные фенола.
Фенольные стабилизаторы эффективно защищают от старения
полимеры разных классов, они мало влияют на цвет полимера и
позволяют получать белые или ярко окрашенные изделия. Многие
фенольные стабилизаторы нетоксичны и могут применяться в из-
делиях, контактирующих с продуктами питания, лекарственными
препаратами и другими биологическими средами. Этим и объяс-
няется широкое внедрение фенольных стабилизаторов в производ-
ство полимерных материалов во всех промышленно развитых стра-
нах [1, 2].
В настоящее время известно более 1000 производных фенола,
обладающих свойствами стабилизаторов полимерных материалов,
и число их постоянно возрастает. Примерно 80 соединений нашли
практическое применение и выпускаются в различном масштабе в
промышленности.
3.1. СЫРЬЕ ДЛЯ ПРОИЗВОДСТВА ФЕНОЛЬНЫХ
СТАБИЛИЗАТОРОВ
Основное сырье для производства фенольных стабилизаторов —
фенол и n-крезол (4-метилфенол).
В настоящее время в мире производится более 1 млн. т фенола
в год; 80% синтетического фенола вырабатывают из бензола через
гидропероксид изопропилбензола (кумольный метод). Этот метод
] был впервые разработан в СССР П. С. Сергеевым, Б. Д. Кружа-
ловым и Р. Ю Удрисом и нашел широкое применение во всем мире
благодаря высокой чистоте получаемого фенола и возможности
полной утилизации побочного продукта — ацетона. Гораздо' мень-
шее значение имеют другие способы получения фенола — щелоч-
ное плавление бензолсульфокислоты, газофазный гидролиз хлор-
186
бензола, окисление толуола, дегидрирование смеси циклогексано-
ла и циклогексанола.
На получение фенольных стабилизаторов расходуется 3—4%
всего выпускаемого фенола. Фенол, получаемый кумольным спо-
собом, полностью удовлетворяет требованиям к сырью для стаби-
лизаторов. Для их производства обычно используют свежепере-
гнанный фенол.
/г-Крезол получают в ограниченных количествах сульфирова-
нием толуола и щелочным плавлением п-толуолсульфокислоты.
Выделение /i-крезола из коксохимической дикрезольной фракции
не перспективно из-за ограниченности сырьевой базы.
Для производства индивидуальных стабилизаторов применяют
/г-крезол с содержанием целевого вещества не ниже 98%. н-Крезол
в течение многих лет служил основным сырьем для выработки
эффективных фенольных стабилизаторов; менее активные стаби-
лизаторы получали на основе фенола.
Развитие химии пространственно затрудненных фенолов и со-
здание промышленного процесса орто-алкилирования фенола изо-
бутиленом изменило структуру сырьевой базы фенольных стаби-
лизаторов. В результате исследований, выполненных в Научно-ис-
следовательском институте резиновых и латексных изделий,
Институте химической физики АН СССР, на Стерлитамакском
опытно-промышленном нефтехимическом заводе и в Научно-ис-
следовательском институте химикатов для полимерных материа-
лов, была показана возможность создания широкого ассортимента
эффективных фенольных стабилизаторов не из »-крезола, а из фе-
нола. Тем самым была открыта возможность производства
фенольных стабилизаторов на доступной сырьевой базе. Кроме фе-
нола и n-крезола в промышленном синтезе фенольных стабилиза-
торов применяют в сравнительно небольших количествах 3-метил-
фенол, резорцин, гидрохинон.
В качестве алкилирующих и конденсирующих агентов исполь-
зуют изобутилен, стирол, формальдегид, тиохлориды. Вспомога-
тельными веществами и растворителями служат хлоролефины,
спирты (метиловый, этиловый, изопропиловый), диметиланилин.
Все эти соединения производятся в промышленном масштабе в
больших количествах.
Изобутилен — мономер в производстве полиизобутилена и
сырье для получения изопрена — выделяют из бутан-бутиленовой
фракции газов пиролиза нефтепродуктов или получают дегидри-
рованием изобутана. Изобутилен для производства индивидуаль-
ных стабилизаторов содержит 98,5—99% основного вещества.
Стирол, широко используемый для производства полистирола
и каучуков, получают каталитическим дегидрированием этилбен-
зола. В 1970-х годах разработан процесс получения стирола окис-
лением этилбензола до метилфенилкарбинола и дегидратацией по-
следнего. Для производства стабилизаторов используют стирол,
предназначенный для производства полистирола.
187
Гидрохинон, который обычно вводят в стирол для ингибирова- У
ния его полимеризации при ректификации и хранении, практически 1'
не влияет на процесс алкилирования стиролом в производстве ста- •>
билизаторов.
Формальдегид, получаемый окислением метанола, используют |
в виде водного раствора — технического формалина. Примесь ме- «
танола в формалине обычно не влияет на синтез стабилизатора. ]
В некоторых случаях метанол предварительно отгоняют.
К фенольным стабилизаторам относятся соединения, защитное 3
действие которых связано с реакционной способностью фенольного а
гидроксила. В соответствии с современными представлениями д
о механизме действия стабилизаторов (см. гл. 1), молекула эффек- |
тивного фенольного антиоксиданта должна содержать фрагмент з
с пространственно\затрудненной гидроксильной группой [3]:
Молекула фенольного светостабилизатора содержит фрагмент’5)
о-гидроксифенилкетона:
По структуре фенольные стабилизаторы можно разделить н<
4 основные группы.
1. Одноядерные алкилфенолы — а) одноосновные
ОН
ОН
R, R', R"—алкнл, цнклоалкил. ара;
кил (одинаковые или разные); X-
фрагмент, содержащий группы OF
COOR, NHR, NR2f SR и др, (R = I
или н-алкил Су—Cig, а п = 0-г-З)
например:
ОН
СН8-(СН2)„-Х
он
С(СН3)8
(СН8)3С
СН3
2,6-ди-гиретп-бу ти л-4-метил-
фенол
CH^CHj-C-O-CieHg,
октадецил-3(3* .б'-ди-трет-бутил-
4-гидроксифенил) пропионат
О
188 -
R—алкил, циклоалкил, аралкил
например:
он
2»5-ди-треп-амилгидрохинон
2. Бисфенолы. В этих соединениях две молекулы алкилфенола
соединены мостиками различного строения (Y):
R и R'—алкил, циклоалкил, аралкил
(одинаковые или разные);
—S—, —CHaSH2—• и др.
(R—алкил Q—Cg)
В качестве примера приведем такие соединения:
2»2'-метиленбис(6-/пргт-бутил-4-метилфенол)
(СН8)3С
но—
(СН3)8С
С(СН3)3
•ОН
С(СН3)3
4,4'-метиленбис(3,5-ди-/прет-бутилфенол)
он
сн3—нс
I
н3с
он
сн—сн3
:н3
2,2'-тиобис(4-метил-6-<х-метил-
бензилфенол)
СН3
3. Трис- и полифенолы. В этих соединениях три или более
молекул алкилфенола соединены через метиленовый мостик или
через центральную группу атомов. Например:
189
он он он
(СНз)зС^к^С^^С^С/С<СНз)«
СН3 СН3 сн3
2,6-бис(2'-гвдрокси-3'-трет-бутилд
5'-метилбензил)-4-метилфенол ,
q »С(СН3)3
11
—СН2—O-C-CHj-CHj—" у-он
'Qch,,)^ 4
эфир тетра-3(3',б'-ди-трет-бутил- 4
гидроксифенил) пропионовой кислоты
и пентаэритрита
4. о-Гидроксифенилкетоны. В основе молекулы этих стабилиза-
торов лежит фрагмент с о-карбонильной группой:
2-гидрокси-4-метоксифенил-
гептадецилкетон
R—алкил' Ci—Cg или гидроксиалкил
Ci—Cg; R'—алкил С5—Ср?
Из большого числа химических реакций, используемых в синте-
зах фенольных стабилизаторов, важнейшими являются алкили-
рование фенолов, конденсация алкилфенолов с карбонильными
соединениями, конденсация алкилфенолов с тиохлоридами, ацили-
рование фенолов. Эти реакции — общие для синтеза большого
числа стабилизаторов. Реакции, имеющие частное значение, рас-
смотрены при описании синтеза отдельных соединений.
190
$ 2 АЛКИЛИРОВАНИЕ ФЕНОЛА
Эта реакция — одна из важнейших в синтезе фенольных стаби-
лизаторов. Путем алкилирования фенола и крезолов получают
простейшие стабилизаторы этого класса — одноядерные моноал-
килфенолы, а также диалкилфенолы, являющиеся промежуточны-
ми продуктами в синтезе фенольных стабилизаторов более слож-
ного строения.
Для получения стабилизатора в ароматическое кольцо вводят
алкильные группы, разветвленные на а-атоме углерода; наиболь-
шее практическое значение имеют реакции алкилирования фено-
лов олефинами и циклоолефинами в присутствии кислотных ка-
тализаторов.
Алкилирование фенолов изучено достаточно подробно [4—7].
В свете современных представлений о механизме электрофильного
замещения в ароматическом ядре, каталитическое алкилирование
фенола олефинами можно представить следующей схемой. Олефин
взаимодействует с катализатором НХ, образуя поляризованный
комплекс:
₽2С=СН2 + НХ ► R2C=CH2—НХ > R2C—СН3-Х
Крайним случаем поляризации комплекса «катализатор — олефин»
является образование карбкатиона:
RaC—СН3-Х ---> Ra(CH3)C + X-
Карбкатпон (свободный или в виде ионной пары) атакует моле-
кулу фенола, образуя с ней л-комплекс, в котором алкильная
группа может сравнительно легко перемещаться от одной части
ароматической молекулы к другой. При перегруппировке л-комп-
лекса в о-комплекс возникает ковалентная связь между вступаю-
щим алкильным заместителем и одним из атомов углерода арома-
тического кольца, причем электронное влияние гидроксильной
группы благоприятствует образованию о-связи с пара- или
орто-атомом углерода. о-Комплекс не стабилен; он легко перегруп-
пировывается в л-комплекс алкилароматического соединения, а
последний, отдавая протон, стабилизируется в алкилфенол:
c(ch3)r2
191
Несмотря на то что промежуточные соединения обладают низкой
стабильностью, существование большинства из них доказано. На-
пример, выделены и описаны трет-бутильный [8], а-метилбензиль-
ный [9] и другие карбкатионы [10, 11]. В некоторых случаях
удается выделить о-комплекс, образующийся при присоединении
карбкатиона к ароматическому кольцу [12, 13].
В качестве катализаторов при алкилировании фенола олефи-
нами применяют серную кислоту, арилсульфокислоты и катионо-
обменные смолы, реже — фтористый бор, алюмосиликаты, щавеле-
вую, фосфорную и полифосфорные кислоты.
При первых исследованиях алкилирования фенола олефинами
предполагали, что в этой реакции сначала образуется простой
эфир, который затем перегруппировывается в алкилфенол. Позд-
нее было показано, что алкильная группа может вступать непо-
средственно в ароматическое кольцо. В присутствии BF3 трет-бу-
тилфениловый эфир легко превращается в 4-трет-бутилфенол,
поэтому можно предположить [14], что эфир может образовывать-
ся как промежуточный продукт алкилирования [15], хотя алкил-
фениловые эфиры могут алкилироваться и непосредственно в ядро
[16]. Возможен [17] и механизм с прямой атакой кольца фенола
карбкатионом, а в работе [18] при рассмотрении механизма при-
соединения ароматических соединений к олефинам, указывается,
что «в случае фенолов это может происходить прямым или кос-
венным путем (через образование эфиров фенола)».
По-видимому, образование алкилфениловых эфиров и алкил-
фенолов протекает параллельно. В условиях кислотного катализа
эфиры легко превращаются в алкилфенолы:
Особенно легко эти превращения протекают у трет-алкиловых
эфиров фенола, например у трет-бутилового [14]. С повышением
температуры скорость превращения возрастает, поэтому в алкила-
те, полученном при низкой температуре, обнаруживается значи-
тельное количество эфиров. Алкилируя фенол и 2-трет-бутилфенол
изобутиленом при минус 10 — плюс 15 °C, можно получить
трет-бутилфениловые эфиры как основной продукт реакции [19].
В алкилатах, полученных при высокой температуре, эти эфиры
практически отсутствуют.
Для введения в молекулу фенола разветвленных алкильных
групп используют различные олефины и циклоолефины; для син-
теза стабилизаторов наибольшее значение имеют продукты алки-
лирования фенола и крезолов стиролом, изобутиленом и циклогек-
сеном.
Алкилирование фенола стиролом — одна из первых реакций
алкилирования фенола.. Катализатором служила серная кислота
192
Рис. 20. Кинетические кривые алкилиро- ПО
вания фенола стиролом в присутствии
разных кислот: «
! — трихлоруксусиая; 2 — щавелевая: 3 — ЛИ' § 80
хлоруксусиая; 4 — моиохлоруксусная; 5 —фу- 5
маровая; 6 — малеиновая; 7 — малоновая.
а 60
а
и
[17, 20—23]. Из алкилата, полу- § 40
чеиного при комнатной темпера- *
туре, за 2—3 суток были виде- §20
лены только монозамещенные. |
При увеличении количества сти- 0
рола наряду с моно- образуются
ди- и тризамещенные [20, 21].
Серная кислота активно катали-
зирует алкилирование, но вызывает и побочные реакции — поли-
меризацию стирола, изомеризацию и деалкилирование а-метил-
бензилфенолов [21, 24].
При алкилировании фенолов в присутствии фенолятов алюми- -
ния, цинка и некоторых других металлов образуются преимущест-
венно 2-а-метилбензилфенолы [25—28]. На катионообменной смо-
ле КУ-2 идет преимущественно пара-замещение [30—32]. В при-
сутствии каталитических количеств фосфорной кислоты алкилиро-
вание фенола и крезолов стиролом идет с высоким выходом [33],
причем образуются почти исключительно монозамещенные; увели-
чение соотношения фенол : стирол не повышает выход дизамещен-
ных. Описано также алкилирование фенола стиролом в присутст-
вии полифосфорной кислоты [34, 89], трехфтористого бора [35, 90]
и алюмосиликатного шарикового катализатора [36].
При исследовании алкилирования фенола стиролом в присут-
ствии карбоновых кислот была показана зависимость между ка-
талитической активностью исследованных кислот и их рКа [37].
На рис. 20 представлены кинетические кривые алкилирования фе-
нола стиролом в присутствии различных карбоновых кислот, а на
рис. 21 дана зависимость каталитической активности кислот, выра-
женной эффективной константой скорости превращения фенола
в а-метилбензилфенолы (/гЭф), от рК0. Наиболее активными ката-
лизаторами оказались трихлоруксусиая и щавелевая кислота
(константы скорости соответственно равны 10,4-103 и
7,4-10-3 л/моль). В присутствии щавелевой кислоты скорость ал-
килирования фенола стиролом настолько превышает скорость
полимеризации стирола, что полистирол в реакционной массе
практически отсутствует.
Ориентация алкильной группы при вступлении в ароматическое
ядро определяется электронодонорными свойствами гидроксиль-
ной группы: за счет сильного (+)мезомерного эффекта повышает-
ся электронная плотность в орто- и пара-положении к гидроксиль-
ной группе, и при перегруппировке л-коьщлекса в о-комплекс.
13—1057
1937
Рис. 21. Зависимость lg от
р/Са для разных кислот:.
1 — трихлоруксусная; 2 — щавеле-
вая; 3 — дихлоруксусная; 4 — ма-
леиновая; 5 — малоновая; 6 — моио-
хлоруксусиая; 7 — фумаровая.
алкильная группа фиксируется в орто- или пара-положении. При
алкилировании фенола разветвленными олефинами (особенно выс-
шими) вследствие стерических влияний легче идет вступление за-
местителя в пара-положение, чем в орто- [38].
Основным продуктом алкилирования фенола олефинами в при-
сутствии обычных катализаторов является 4-алкилфенол [39].
В табл. 3 приведен состав продуктов алкилирования фенола экви-
мольным количеством изобутилена [39]. Увеличение отношения
изобутилен : фенол приводит к образованию 2,4-ди- и 2,4,6-три-
трет-бутилфенола; при этом образуется лишь незначительное ко-
личество 2,6-ди-трет-бутилфенола.
Синтез 2,6-диалкилфенолов в течение длительного- времени был
сложной проблемой. Эти соединения образовывались в небольших
количествах как побочные продукты алкилирования фенола оле-
финами в присутствии кислотных катализаторов. В качестве пре-
паративного синтеза было предложено каталитическое дегалогени-
рование 4-галоген-2,6-диалкилфенолов [40, 41]:
ОН ОН
Взаимодействием фенола с олефинами при высокой температуре
без катализатора были получены в качестве основных продуктов.
Таблица 3. Состав продуктов алкилирования фенола изобутиленом
Катализатор Температура, °C Выход, %
общий 4-тре/п-бутил- фенола 2-mpem-6ymwi- и 2,4-дн-трет-» бутилфенолов
Комплекс H3PO4-BF3 100 96 81 15
Трехфтористый бор 100 96 83 13
Серная кислота 100 84 65 19
Фосфорная кислота 100 94 60 28
Алюмосиликат 130 93 75 20—25
Катионит КУ-2 юо 90 80 5—10
194
Таблица 4. Алкил- и диалкилфенолы, полученные реакцией
врто-алкилирования в присутствии 2-крезолята алюминия
Исходный олефив
Продукт реакции
Т. кип., °C Т. пл., °C
орто - Ал к ил иро в а иие фенола
Этилен 2-Этилфенол 119 (при 6,66 кПа) —
» 2,6-Диэтил фенол 135 38—39
Пропилен 2-Изопропилфенол 126 » 8
» 2,6-Диизопропилфенол 149 » 18
Бутилен 2-втор-Бутилфенол 135 12
» 2,6-Дп-втор-бутилфеиол 169 » —
Изобутилен 2-трет-Бутилфенол 136- -137 » —
» 2,6-Ди- трет- бути лфено л 161 » 39
2-Метилбутен-2 2-трет-Амилфенол 148 —
Диизобутилен 2(1,1,3,3-Тетраметилбутил) фенол 150 (при 2,67 кПа) —
Циклогексен 2-Циклогексилфенол 145 (при 1,33 кПа) 57
» 2,6-Дициклогексилфенол 205 » 76—77
о р то - А л к и л и р о в а н не 2-крезола
Этилен 2-Метил-6-этилфенол 124 (при 6,66 кПа) —
Бутилен 6-втор-Бутил-2-метилфенол 144 » —
Пропилен 6-Изопропил-2-метилфенол 135 » —
Изобутилен 6-трет-Бутил-2-метилфенол 141 » —
Стирол 2-Метил-6-а-метилбензилфенол 166 (при 1,33 кПа) —
а-Метилстирол 6-Кумил-2-метилфенол 180 » —
Циклогексен 2-Метил-6-циклогексилфенол 192 (при 6,66 кПа) 70,5
2-алкилфенолы; 2,6-дизамещенные образуются в небольших коли-
чествах [42].
В середине 50-х годов фирмами Bayer (ФРГ) [44] и Ethyl
(США) [26] был предложен способ алкилирования фенола олефи-
нами в присутствии фенолята алюминия. Основным продуктом
в этом случае является 2,6-диалкилфенол, образующийся с высо-
ким выходом. В табл. 4 приведены характеристики полученных
2-алкил- и.2,6-диалкилфенолов [43]. Таким путем можно получить
широкий ассортимент о-алкилфенолов, за что эту реакцию и на-
зывают орто-алкилированием.
Катализатор готовят, выдерживая металлический алюминий
с фенолом при 150 °C до прекращения выделения водорода; при
этом образуется фенолят алюминия: .
2А1 + 6СвН5ОН —> 2А1(ОС6Нб)3
Каталитическое действие фенолята алюминия связано с его спо-
собностью образовывать в среде фенола комплексную алюминий-
феноксикислоту:
А1(ОС6Н5)3 + С6Н5ОН --> [А1(ОСвНв)4]Н
При этом кислотность среды возрастает, о чем можно судить по
изменению pH от 4,4—4,6 для фенола до 1,0 для фенола, со дер-
13* 195
жащего 1% фенолята алюминия [43]. Алюминийфеноксикислота
взаимодействует с олефином, и вступление алкильной группы
в орто-положение к гидроксилу может происходить [43] либо не-
посредственно при перегруппировке этого комплекса, либо через
простой эфир. Однако при алкилировании фенолов разветвленны-
ми олефинами в присутствии фенолята алюминия (а также BF3)
трет-алкиловые эфиры в реакционной массе не обнаружены [45].
Авторы [46] полагают, что орто-алкилирование протекает через
образование простых эфиров и представляет собой ряд последова-
тельных стадий:
По их данным, эфир фенола стабилен в присутствии катализатора
орто-алкилирования до 100—ПО °C, и они рекомендуют вести про-
цесс при 140—150 °C. Реакция III идет через образование трет-
бутилового эфира 2-трет-бутилфенола, настолько нестабильного,
что его не удалось обнаружить. Позднее эти эфиры были выделе-
ны и исследованы [47]. Оказалось, что в присутствии фенолята
алюминия и фенола они легко разлагаются уже при 65—70 °C, и
их присутствие в реакционной массе практически не влияет на-
выход продуктов орто-алкилирования. Если орто-алкилирование'
вести при 100—ПО °C, 2-трет-бутилфенол и 2,6-ди-трет-бутилфенол
практически не изомеризуются в 4-изомеры. Не происходит и
превращения 2,6-дп-трет-бутилфенола в 2,4,6-три-трет-бутилфенол.
Общую схему орто-алкилирования можно представить совокуп-
ностью последовательных и параллельных реакций 1—6:
Наряду с основными продуктами орто-алкилирования образуются
и побочные — 4-трет-бутилфенол, 2,4-ди-трет-бутилфенол и
2,4,6-три-трет-бутилфенол, причем повышение температуры сверх
100 °C увеличивает выход побочных продуктов, например отноше-
196
Рис. 22. Температурная зависимость отношения констант скорости орто- (/ и 3)
и лара-замещения (2 и 4) при орто-алкилировании.
ние констант скорости реакций 1 и 2 при 90 °C равно 27,1, а при
130 °C всего 10,2.
На рис. 22 представлена температурная зависимость отношения
констант скорости реакций 1—4, параллельно протекающих при
орто-алкилировании. Из данных рис. 23 следует, что энергия
активации основных реакций орто-алкилирования (реакции 1 и 3)
значительно ниже, чем для побочных реакций 2, 4, 5 и 6, и увели-
чение образования побочных продуктов с повышением температу-
ры связано с различием энергий активации.
Те же закономерности наблюдаются и при алкилировании 2-ме-
тилфенола. Катализатор готовят, нагревая 2-метилфенол с алюми-
нием, и затем подают газообразный изобутилен в раствор 2-метил-
фенолята алюминия в 2-метилфеноле. Наряду с б-трет’-бутил-2-ме-
тилфенолом образуется 4-трет-бутил-2-метилфенол:
ОН
I
С(СН3)3
Линейный характер зависимости lg(£i//ea) от 1/7 (рис. 24) пока-
зывает, что различие в количестве образующихся изомеров, как
и в случае фенола, связано с неодинаковой энергией активации
реакций 1 и 2. При 160 °C и выше фенолят алюминия может вы-
ступать как катализатор деалкилирования [48], поэтому необхо-
197
Рис. 23. Зависимость 1g k от 1/7 для реакций 1—6, протекающих в процессе орто-
алкилирования фенола изобутиленом.
Энергия активации, вычисленная по углу наклона прямых /—6, равна: £i==I8 кДж/моль;
£2=49 кДж/моль; £3=13 кДж/моль; £4=48 кДж/моль; £б=40 кДж/моль; £6=б7 кДж/моль.
димо полностью удалять фенолят алюминия из алкилата перед
ректификацией смеси алкилфенолов.
Механизм реакций, протекающих при орто-алкилировании,
можно представить схемой:
1) образование феноксиалюминиевой кислоты:
Л1(ОСвНБ)8 + СвН6ОН
I
А1(ОС6Н6)3
2) образование комплекса «катализатор — олефин», в котором
объединены атакующая электрофильная частица и атакуемое аро-
матическое ядро:
+cr8=ch,
11(ОСвНь)3
3) образование л-комплекса и перегруппировка его в <т-комп-
лекс:
А1(ОС6Н5)3
I
А1(ОС6Н5)3
198
4) стабилизация <г-комплекса с образованием 2-трет-алкилфе-
нола
C(CH3)Ra
ОН
л выделением А1(ОС6Н5)з. При повышении температуры возраста-
ет вероятность разложения комплекса «катализатор — олефин»
с образованием карбоний-иона:
Последний атакует молекулу фенола по механизму обычного
алкилирования с преимущественным образованием пара-замещен-
ного. Этим и объясняются увеличение выхода пара-замещенных и
снижение выхода орто-замещенных при повышении температуры.
В первом патенте в качестве катализатора орто-алкилирования
предлагали металлы способные образовать с фенолом соли,—Na,
К, Li, Mg, Zn, Fe, Al, Си; позднее была обнаружена каталитиче-
ская активность фенолятов галлия [49, 50], титана [51, 52], цир-
кония, гафния, ниобия, тантала и ванадия [53], но практическое
значение нашел только фенолят алюминия.
За последние 20 лет способ орто-алкилирования был значи-
тельно усовершенствован. Предложены различные добавки к ка-
тализатору, например хлорид натрия [54], щелочные металлы.
Вместо металлического алюминия применяют его -различные со-
единения: оксиды [58, 59], галогениды [60—62], смешанные соли
[63—65], тиофеноляты [65] и некоторые другие [55, 56, 66, 67].
В качестве катализаторов орто-алкилирования описаны также
модифицированный алюмосиликат [68], диалкилсульфаты [69],
метансульфокислота [70], трифенилборат [71], полимерные алко-
голяты алюминия, продукты их взаимодействия с оксидом или
гидроксидом алюминия [72]. В работе
[73] показана возможность орто-алкили-
рования фенола стиролом и а-октиленом
в присутствии алюминиевой соли трет-
бутилфенолсульфокислоты и салицилата
алюминия.
Рис. 24. Температурная зависимость отношения
констант скорости орто- и пара-замещения при
орто-алкилировании 2-метилфенола изобутиленом
9т 1/7 (/) и ее полулогарифмическая анамор-
фоза (2).
199
Кроме фенолята алюминия практическое значение в синтезе
алкилфенольных стабилизаторов имеют и другие катализаторы___
серная кислота, п-толуолсульфокислота, катионообменные смолы
(например, КУ-2), щавелевая кислота.
Серная кислота — самый дешевый и очень активный катализа-
тор алкилирования. Несмотря на опасность коррозии аппаратуры,
побочные реакции и необходимость дополнительной обработки
алкилата, она все еще находит применение для периодических
процессов алкилирования в производстве малотоннажных стаби-
лизаторов. Необходимы дополнительные операции — нейтрализа-
ция реакционной массы щелочными агентами (сода или щелочь),
отделение образовавшейся соли и промывка алкилфенолов водой
перед ректификацией.
Если вязкость реакционной массы после алкилирования высо-
кая (как, например, при алкилировании стиролом), ее разбавляют
органическим растворителем (толуол). Это облегчает нейтрализа-
цию п отделение органических продуктов от водного раствора суль-
фата путем расслаивания.
n-Толуолсульфокислота может быть нейтрализована и отделена
либо теми же приемами, что и для серной кислоты, либо путем
перевода ее в аммониевую соль. Последняя не растворяется в
большинстве органических растворителей и может быть отделена
фильтрованием.
Катионообменные смолы используют в многотоннажных про-
цессах получения алкилфенолов. Этот катализатор позволяет ве-
сти алкилирование по непрерывной схеме и исключает нейтрали-
зацию реакционной массы. Вместе с тем необходима система реге-
нерации ионообменной смолы, а при этом образуются сточные
воды.
Щавелевая кислота практически не вызывает полимеризации
олефинов и других побочных реакций. Алкилаты, полученные с ис-
пользованием щавелевой кислоты, можно передавать на выделе-
ние алкилфенолов ректификацией без дополнительной обработки.
Особенно удобен этот катализатор при алкилировании фенолов
стиролом.
Реже применяют фосфорную и полифосфорную кислоты, фто-
ристый бор, активированные глины.
В настоящее время разработаны способы синтеза алкилфено-
лов с различным строением и расположением алкильных групп.
В производстве стабилизаторов практическое значение имеют
2,4-ди-, 2,5-ди-, 2,6-ди- и 2,4,6-триалкилфенолы. Диалкилфенолы
служат промежуточными продуктами в синтезе стабилизаторов
класса бис- и трисфенолов и одноядерных алкилфенолов более
сложной структуры. Триалкилфенолы используют непосредственно
как стабилизаторы. Иногда в качестве стабилизаторов применяют
смеси моно-, ди- и триалкилфенолов. Кроме того, алкилфенолы
служат исходными соединениями в синтезе фосфорсодержащих
стабилизаторов.
200
2 fj-Ди-трет-бутилфенол
(СН3)3С
ОН
3
2,6-Ди-трет-бутилфенол образуется в небольших количествах
как побочный продукт при алкилировании фенола изобутиленом
в присутствии обычных кислотных катализаторов, но его трудно
выделить из реакционной массы. Другой способ его получения —
алкилирование 4-галогенфенола изобутиленом и дегалогенирова-
ние полученного 4-галоген-2,6-ди-трет-бутилфенола [40]:
ОН
(CHJaC^^^CfCH,).
С выходом до 25% можно получить 2,6-ди-грет-бутилфенол алки-
лированием фенола изобутиленом без катализатора при 280—
350 °C и 15—30 МПа [42].
Основной промышленный способ получения 2,6-ди-трет-бутил-
фенола — алкилирование фенола изобутиленом в присутствии фе-
нолята алюминия.
Катализатор готовят, обрабатывая фенол металлическим алю-
минием, реже — алюминийорганическим соединением (например,
триалкилалюминием) [74]:
6СеН5ОН + 2Д1 —2А1(С6Н5О)3
—о о 2
ЗСвН5ОН + А1(С2Н5)з —> А1(С6Н5О)3
—оС2Нб
Алюминий реагирует с фенолом, растворяясь в нем, при 150—
160 °C, но, если в феноле есть небольшое количество фенолята
алюминия, растворение идет и при более низкой температуре.
Образование фенолята алюминия — экзотермическая реакция, и
в производственных условиях необходимо отводить выделяющееся
тепло. Фенол не должен содержать влаги, так как фенолят алю-
миния легко гидролизуется, теряя при этом каталитическую актив-
ность. Фенолят алюминия взаимодействует с избытком фенола,
образуя комплексную фенокспалюминиевую кислоту. Катализатор
вводят из расчета 5—10 г алюминия на 1 кг фенола; используют
также различные добавки — феноляты щелочных металлов [551,
хлорид натрия [54], пространственно затрудненные фенолы [75].
В качестве катализатора применяют комплексную феноксиалю-
201
миниевую кислоту, в которой часть феноксигрупп заменена на
2-трет-бутилфеноксигруппы [76].
Алкилирование ведут в автоклаве или колонном реакторе,
подавая изобутилен в расплавленный фенол, в котором растворен
катализатор. Для препаративного синтеза фенол с растворенным
в нем катализатором охлаждают в автоклаве до —10 °C, затем
подают жидкий изобутилен и нагревают смесь до заданной темпе-
ратуры [77]. Процесс орто-алкилирования существенно зависит
от температуры: выше 100—ПО °C возрастает скорость побочных
реакций и снижается выход 2,6-ди-т’рет-бутилфенола [26] (табл. 5).
Давление не оказывает существенного влияния на орто-алкилиро-
вание — процесс можно вести при атмосферном или при повы-
шенном давлении (в последнем случае возрастают растворимость
изобутилена в феноле и скорость алкилирования). Изобутилен
можно загружать сразу или постепенно пропускать через слой
фенола.
По окончании алкилирования катализатор дезактивируют, об-
рабатывая реакционную массу водой [78] или кислотой [79]. При
этом фенолят алюминия разрушается с образованием гидроксида
алюминия
2(СвН6О)3А1 + ЗН2О -> 2А1(ОН)3 + 6СвНБОН
который отделяют фильтрованием. Полнота разрушения катализа-
тора имеет большое значение для дальнейшей переработки алки-
лата — даже небольшая примесь фенолята алюминия может при-
вести к деалкилированию 2,6-ди-трет’-бутилфенола на стадии рек-
тификации. Гидроксид алюминия не обладает каталитическим
действием, и алкилат, в котором он присутствует, может быть
непосредственно передан на ректификацию [78].
В реакционной смеси находятся 2-трет-бутилфенол, 2,6- и
2,4-ди-трет-бутилфенол, 2,4,6-три-трет-бутилфенол и небольшое
количество фенола, не вступившего в реакцию. 2,6-Ди-трет-бутил-
фенол выделяют вакуум-ректификацией. Фенол и 2-трет-бутилфе-
нол можно возвратить на алкилирование, а остальные соединения
выделить, очистить и использовать для синтеза стабилизаторов.
Таблица 5. Влияние температуры на выход изомерных
трет-бутилфенолов при орто-алкилировании [26]
Температура, °C Степень конверсии фенола, % Выход /мрет-бутилфенолов, % от теоретического
2-изомер 4-изомер 2,6-изомер 2,4-изомер 2,4,6-изомер
90 . 98 11,2 78,6 1,0 9,2
110 98 12,2 — 75,6 1,0 П.2
ПО 98 19,0 — 59,2 6,3 15,3
160 98 27,2 —. 47,5 10,1 15,2
180 98 23,5 3,0 42,9 12,2 18,4
200 91 61,6 1,0 19,8 13,2 4,4
210 98 24,5 6,1 16,3 33,7 19,4
250 74 50,0 17,6 5,4 24,3 2,7
202
Рис. 25. Технологическая схема получения 2,6-ди-трет-бутилфенола:
/ — аппарат для приготовления катализатора; 2 — реактор алкилирования; 3 — аппарат для
разложения катализатора; 4 — фильтр; 5, 6, 7 — ректификационные колонны.
Смесь 2,4-ди-трет-бутилфенола и 2,4,6-три-трет-бутилфенола может
быть подвергнута каталитическому переалкилированию с фенолом;
полученный 2-трет-бутилфенол возвращают в процесс [80]. 2,6-Ди-
трет-бутилфенол дополнительно очищают водным раствором гид-
росульфита натрия [81] или гидроксида натрия с последующей
перегонкой [82].
Технологическая схема получения. Процесс получения 2,6-ди-
трет-бутилфенола может быть осуществлен по непрерывной или
периодической схеме. Основные стадии процесса — приготовление
катализатора, алкилирование фенола изобутиленом, разрушение
и, отделение катализатора, выделение 2,6-ди-трет-бутилфенола.
Технологическая схема получения 2,6-ди-трет,-бутилфенола
представлена на рис. 25. Фенол со склада поступает в аппарат 1,
куда загружают и металлический алюминий. Полученный там
раствор фенолята алюминия в феноле подают в реактор 2 на ал-
килирование изобутиленом. По окончании алкилирования переда-
ют реакционную массу (алкилат) в аппарат 3 для разложения
катализатора. Гидроксид алюминия, образовавшийся в результате
взаимодействия катализатора с водой, отфильтровывают, а алки-
лат подают на выделение 2,6-ди-трет-бутилфенола. В ректифика-
ционных колоннах 5, 6 и 7 отделяют сначала 2-трет-бутилфенол
и фенол, а затем 2,6-ди-трет-бутилфенол, 2,4-ди-трет-бутилфенол
и 2,4,6-три-трет-бутилфенол.
На стадии алкилирования выход 2,6-ди-трет-бутилфенола за
проход составляет 70—72% от теоретического при селективности
его образования из фенола «90% [83]. Побочные продукты алки-
лирования могут быть превращены в исходные вещества и возвра-
щены в процесс; в этом случае выход 2,6-ди-трет-бутилфенола по-
вышается до 95—97% [83].
203
Свойства и применение. 2,6-Ди-трет-бутилфенол — белое крис-
таллическое вещество; т. крист. 36,0—36,2 °C. Температура кипе-
ния заметно зависит от давления:
Т. кип., °C . . . 247 238 231 198 176 150
Давление, кПа . . 106,8 80 66,6 26,6 13,3 5,3
Плотность снижается от 918 кг/м3 при 50°C до 846 кг/м3 при 150°С.
Вязкость в интервале 50—150 °C уменьшается от 4,6 до 0,92 мм2/с.
Хорошо растворим в ацетоне, бензине, бензоле и диэтиловом эфи-
ре; в воде не растворяется.
2,6-Ди-грет-бутилфенол — важнейший промежуточный продукт
в синтезе стабилизаторов. Ниже приведены некоторые промыш-
ленные стабилизаторы, получаемые из 2,6-ди-грет-бутилфенола не-
посредственно или через промежуточные стадии:
2,6-ди-тр«т-бутил-4-метилфено л
(СН3)3С
С(СН3)3
но—
(СН3)3С С(СН3)3
4,4'-метнленбнс(2,&-ди-трет-бутилфенол)
ОН
(СН3)3С
сн2—осн3
2,6-ди-трет-бутил-4-метокси-
метилфенол
(СН3)3С
С(СН3)3
—он
С(СН3)3
4,4'-дигидрокси-3,3', 5,5'-тетра-
тргт-буталдифенил
О
II
—СНа—CHS—с—ОС18Н3,
2,4,6-три(4-гидроксибензил-3',5'-ди-трет-бутил)мезитилен
204
(CH3)3c
—CH2-S—HjC
-он
С(СН3)3
(СН8)3С С(СН3)3
ди(4-гидрокси-3,5-ди-/прет-бутил)бензилсульфид
2,6-Ди-трет-бутил-4-метилфенол
Это — один из первых фенольных стабилизаторов. Он был
предложен в конце 30-х годов сначала для защиты нефтепродук-
тов от окисления, а позднее для защиты полимеров.
Основной способ получения — каталитическое алкилирование
4-метилфенола изобутиленом:
Катализатором может служить концентрированная серная кисло-
та, олеум, серная кислота с добавкой глицерина, полифосфорная
кислота. Позднее были предложены катионообменные смолы [93],
алкилсульфокислоты [94], BF3 и его молекулярные соединения
[95], катионообменная смола, модифицированная BF3 [95], кре-
золят алюминия [96, 97].
Кроме изобутилена для алкилирования применяют смеси угле-
водородов, например бутан-бутиленовую фракцию [98, 99]. Так,
используя фракцию, содержащую 14—21% изобутилена, и ведя
процесс в присутствии серной кислоты, получают продукт, содер-
жащий 93—96% основного вещества [99]. Двухступенчатым алки-
лированием (в присутствии катионообменной смолы) смеси крезо-
лов пиролизными газами, содержащими 2,7—4,5% изобутилена,
получен 2,6-ди-77?е7’-бутил-4-метилфенол с выходом 50—60% счи-
тая на 4-метилфенол [100].
Вместо дефицитного 4-метилфенола используют крезольную
фракцию каменноугольной смолы, представляющую собой смесь
3- и 4-метилфенолов. Разделение их ректификацией затруднено
из-за близости температур кипения; один из способов разделе-
ния— перевод в трет-бутильные производные [101]. На стадии
205
моноалкилирования образуются соответственно 6-трет-бутил-4-ме-
тил- и 6-трет-бутил-З-метилфенол:
ОН ОН он он
З-Метилфенол алкилируется с большей скоростью [102]. На ста-
дии диалкилирования образуются соответственно 2,6-ди-трет-бу-
тил-4-метилфенол и 4,6-ди-трет-бутил-З-метилфенол, которые зя-
тем разделяют ректификацией (т. кип. при 2,66 кПа соответствен-
но 146,5 и 167 СС). По этой схеме получают чистый 3-метилфенол
каталитическим деалкилированием 4,6-ди-трет-бутил-З-метилфено-
ла [101]. В качестве катализатора алкилирования используют сер-
ный ангидрид [101], серную кислоту [102, 103], смеси ее с сульфа-
том железа [104] или с комплексами BF3 [105], катионообменную
смолу КУ-1 [106].
Получение 2,6-ди-трет-бутил-4-метилфенола объединяют с по-
лучением 6-трет-бутил-З-метилфенола. Для этого разделение про-
изводных 3- и 4-метилфенола ведут на стадии моноалкилирования
и затем дополнительно алкилируют изобутиленом 2-трет-бутил-4-
метнлфенол [107]. Принципиальные схемы переработки крезоль-
ной фракции представлены на рис. 26. Для повышения выхода
2-трет-бутил-4-метилфенола проводят переалкилирование смеси
4,6-ди-трет-бутил-З-метилфенола и монобутилкрезолов в присутст-
вии кислотного катализатора [108].
Технологическая схема получения. Промышленный синтез
2,6-ди-трет-бутил-4-метилфенола из 4-метилфенола ведут его алки-
Рис. 26. Принципиальные схемы переработки крезольной фракции:
с —с разделением продуктов моноалкилирования; б —с разделением продуктов диалкнли-
рования; 1 — реактор алкилирования крезольной фракции; 2 — ректификационная колонна;
4 — реактор алкилирования 2-трет-бутил-4-метилфенола; 4— реактор деалкилирования 4.6-ди-
трет-бутил-3-метилфенола.
206
Рис. 27. Технологическая схема получения 2,6-ди-трет-бутил-4-метилфенола из
4-метилфенола:
/ — реактор-алкилатор; 2 — промывной аппарат; 3 — нейтрализатор; 4 — сборник кислотных
промывных вод; 5 — сборник солевого раствора; 6, 7 — ректификационные колонны; 8—,кри-
сталлизатор; 9 — фильтр; 10 — сушилка.
лированием в присутствии /г-толуолсульфокислоты, катионообмен-
ных смол или серной кислоты (последний способ считают более
экономичным) [92, 109]. В качестве промежуточного продукта об-
разуется 2-т,рет-бутил-4-метил фенол.
Технологическая схема процесса в присутствии серной кисло-
ты представлена на рис. 27. В реактор 1 с мешалкой и барботе-
ром для подачи изобутилена загружают расплавленный 4-метил-
фенол, серную кислоту (катализатор; 2—5% масс.) и подают изо-
бутилен в мольном отношении к 4-метилфенолу (2,1-4-2,3) : 1. По
окончании алкилирования промывают реакционную массу водой и
нейтрализуют раствором соды; нейтрализованный алкилат пере-
дают на ректификацию. Основную фракцию, содержащую 92—96%
целевого продукта, перекристаллизовывают.
Алкилирование ведут по периодической или непрерывной [98,
110] схеме, при атмосферном или повышенном давлении. В ротор-
но-дисковом алкилаторе непрерывного действия целевой продукт
образуется с выходом 80% из бутилена и с выходом 67% из бу-
тан-бутиленовой фракции [98]. Его можно выделять из алкилата
без ректификации — обработкой водой и кристаллизацией из вод-
ного раствора спирта [92]: двухкратной кристаллизацией из вод-
ного метанола получают продукт 99,99 %-ной чистоты с т. пл.
69,3°C [Ш], а двухступенчатой кристаллизацией из бензина по-
лучают продукт с 98% основного вещества [112]. Другие раство-
рители, пригодные для кристаллизации, — гликоли, разбавленный
изопропиловый спирт, водно-ацетоновый раствор, гексан. Кристал-
лизацию ведут и непосредственно из алкилата (после нейтрализа-
ции), а маточный раствор, содержащий 30—38% 2-т’рет’-бутил-4-
метилфенола и 40—45% 2,6-ди-грет-бутил-4-метилфенола, направ-
207
ляют на повторное алкилирование, предварительно удалив приме-
си, мешающие кристаллизации [113].
Для получения продукта, не желтеющего при хранении, рас-
плавленный алкилат перед кристаллизацией очищают, нагревают
и пропускают через колонну с активированным углем, а затем
обрабатывают водяным паром [114]. Обработкой 1%-ным раство-
ром NaOH и двухкратной перекристаллизацией из этилового спир-
та в атмосфере азота получают бесцветный продукт, не имеющий
запаха; т. пл. 68,8—70,0 °C [115].
В связи с дефицитом 4-метилфенола в СССР разработан про-
цесс получения 2,6-ди-трет-бутил-4-метилфенола из фенола через
2,6-ди-трет-бутилфенол [83, 84]. Последний подвергают аминоал-
килированию и полученное основание Манниха восстанавливают:
ОН
СН2—N(CH3)S СН3
Аминоалкилирование ведут при 50—80 °C в среде метанола, ис-
пользуя смесь диметиламина и формальдегида либо продукт их
взаимодействия — тетраметилдиаминометаи
НСНО + 2NH(CH3)2
—н2О
(CH3)2N—СН2—N(CH3)2
+ (CHS)2N—СН2—N(CH3)2
(СН3)3С.
(С113;3^
—СН2—N(CH3)2 + NH(CH3)
К-диметил(4-гидрокси-3,5-ди-тр«т-бутилбензил)амин
Выход этого продукта ~ 97% от теоретического.
Ы-Диметил(4-гидроксн-3,5-ди-трет-бутилбензил)амин — белое кристаллическое
вещество; т. пл. 92 °C; плотность 878 кг/м3 при 100 °C; вязкость 0,24 мПа-с при
100 °C. Хорошо растворим в метаноле. Обладает высокой реакционной способ-
ностью [116].
Является антиоксидантом для белых и цветных резин. Под торговой маркой
•Ethyl Antioxidant 703 выпускается фирмой Ethyl (США).
При взаимодействии с водородом на сплавном катализаторе
(Ni-I-Al, Ni-j-Cr или Ni+Cu) при 140—160 °C происходит гидроге-
208
нолиз связи С—N с образованием целевого продукта:
(СН3)3С,
Н<
(СН3)3С
>-CH2N(CH3)s
(СН3)3С
+н2
—> но- -сн3 + NH(CHs)2
(СН3)3С
С целью уменьшения побочных реакций ведут гидрогенолиз в сре-
де органического растворителя [117] в проточном реакторе
с 4,5-кратным избытком водорода. Из исследованных катализато-
ров лучшие результаты по активности и селективности при дли-
тельной работе получены с Ni—Al—Ti-катализатором [117].
Технологическая схема получения 2,6-ди-трет-бутил-4-метилфе-
нола из 2,6-ди-трет-бутилфенола представлена на рис. 28. Она
состоит из следующих узлов: смешения диметиламина и форма-
лина; аминоалкилирования и выделения 2,6-ди-трет-бутил-4-диме-
тиламинометилфенола; гидрогенолиза; ректификации и перекри-
сталлизации готового продукта; регенерации растворителей.
Диметиламин и формалин предварительно смешивают; эту
смесь или продукт их взаимодействия подают в реактор 2 на ами-
ноалкилирование. Туда же поступают растворитель и расплавлен-
ный 2,6-ди-трет-бутилфенол. Аминоалкилирование протекает при
100 °C с высокой скоростью — за 30 мин достигается практически
полная конверсия 2,6-ди-трет-бутилфенола. Реакционная масса
поступает в колонну 3 для отделения растворителя, диметиламина
и низкокипяших побочных продуктов. 2,6-Ди-трет-бутил-4-диметил-
аминометилфенол (основание М.анниха) направляют в колонный
реактор 4 на гидрогенолиз.
Рис. 28. Технологическая схема получения 2,6-ди-трет-бутил-4-метилфенола из
?,6-ди-грет-бутилфеиола:
/ — смеситель: 2 — реактор амииоалкнлирования; 3— ректификационная колонна; 4 — ко-
лонна гидрогенолиза; 5 — емкость; 6 — ректификационная колонна; 7 — аппарат для рас-
творения; 8 — кристаллизатор; 9 — фильтр; 10 — сушилка.
14—1057
209
Гидрогенолиз ведут на твердом катализаторе при 140—160 °C
и избытке водорода. Образующийся диметиламин удаляется с из-
быточным водородом. Реакционную массу после гидрогенолиза на-
правляют на ректификацию в колонну 6. Сырой продукт отбирают
с верха колонны и подают на перекристаллизацию. Перекристал-
лизованный (из метанола или этанола) продукт поступает на
фильтрование, сушку и упаковку. Растворитель уходит на регене-
рацию. Кубовый остаток используют как антиоксидант нефтепро-
дуктов. Т. пл. целевого продукта 69,7—69,9 °C.
Этот метод производства 2,6-ди-тре7'-бутил-4-метилфенола ис-
пользует доступное и сравнительно недорогое сырье — фенол, фор-
малин, диметиламин, изобутилен. Промежуточные продукты могут
служить сырьем для широкого ассортимента стабилизаторов.
Сравнительные технико-экономические показатели получения
2,6-ди-трет-бутил-4-метилфенола из бензола через 2,6-ди-трет-бу-
тилфенол (способ 1) и из толуола через 4-метилфенол (способ 2)
приведены ниже (показатели рассчитаны на мощность производ-
ства 25 тыс. т в год):
Способ 1 Способ 2
Расход сырья на 1 т продукта, кг
толуол....................................’ — 1032
серная кислота............................. — 1260
едкий натр (50%-ный)....................... — 2431
изобутилен................................ 680 669
бензол.................................... 557 —
пропилен............................ 304 ' —
формальдегид . . •................... 396 —
Основные энергозатраты
пар, Гкал............................... 14,6 13,8
вода, м3.................................. >68 739
электроэнергия, кВт-ч............... 1513 830
Побочные продукты на 1 т целевого про-
дукта. кг
ацетон . . . ’................ 338 —
фенол...................................... — 57
2-метилфенол .............................. — 51
дикрезольная фракция....................... — 200
сульфат натрия............................. — 2С00
Удельные капиталовложения, руб. на 1 т
продукта................................. 445 1065
Себестоимость 1 т продукта, руб. . . 548 659
Приведенные данные показывают преимущество способа 1, уж<
осуществленного в СССР.
Описано также получение 2,6-ди-трет-бутил-4-метилфенола восстановление!
4-гидрокси-3,5-ди-грет-бутилбензилового спирта [118, 119]:
(СН3)8С< (CH3)SG
(СН3)3С/ (СН„)3С/
По способу [118] растворяют 4-гидрокси-3,5-ди-трет-бутилбензиловый спирт
в 90%-ной . уксусной кислоте, перемешивают, нагревают до 70—75 *С и постепен-
но добавляют гранулированный цинк. По окончании восстановления в вакууме
210
отгоняют уксусную кислоту; остаток обрабатывают водой и извлекают целевой
продукт толуолом. Выход «97% от теоретического. По патенту фирмы Shell
rj 19] восстановление ведут водородом на медь-хромовом катализаторе при по-
вышенном давлении и температуре до 200 °C. Эти способы не получили пока
практического применения, как н получение из 4,4'-метиленбис(3,5-ди-трет-бутил-
Аенола) — взаимодействием с метанолом в присутствии метилата натрия под
давлением 200 °C [120].
Свойства и применение. 2,6-Ди-трет-бутил-4-метилфенол вы-
пускают в виде кристаллов или чешуек. Сферические кристаллы,
обладающие высокой текучестью, получают, вводя расплавленный
2,6-ди-7’рет’-бутил-4-метилфенол в охлажденный до —5 °C 80%-ный
метанол [121]. Композицию с хорошей текучестью получают, ког-
да расплав продукта разбрызгивают в порошок силикагеля (раз-
мер частиц 3 мкм) [122].
2,6-Ди-трет-бутил-4-метилфенол — белое кристаллическое веще-
ство; т. пл. 70,0 СС; вязкость 3,22 мПа-с при 80 °C; плотность
1048 кг/м3; насыпная масса 654 кг/м3. Температура кипения замет-
но зависит от давления:
Т. кип., °C. ... 266 256 248 214 191 146,5
Давление, кПа . . 106,8 80 66,6 26,6 13,3 2,65
Растворим в ацетоне, этаноле, изопентане, бензоле, жирах,
сложных эфирах. В воде и 10%-ном едком натре не растворяется.
Под различными торговыми марками выпускается многими за-
рубежными фирмами:
Торговое наименование Страна, фирма T. пл., °C
Antigen BHT Япония, Sumitomo 69
Antioxidant 4 ЧССР 69
Antioxidant DBPC Франция, Organo Sintese —
Aiffioxidant KB США, Mobev Chemical >66
Antioxidant 29 США, Du Pont —
AO-11 США, Akron Chemical —
Annulex BHT Англия, Pearson —
BHT Англия, Bennett —
BHT-Raschig ФРГ, Raschig 69—70
BHT Япония, Seiko Chemical -—
BUT (пищевой) США, Koppers 69,4
Bisoxol 220 Франция, CdF 69,8—70
CAO-1 США, Aschland Chemical 68,8
CAO-3 США, Aschland Chemical 69,3
Ionol США, Shell 69,8
Naugard BHT США, Uniroyal Chemical 69,4 (т. затв.)
Nocrak Япония, Ouchi Shinko 69
Nonox TBC Англия, ICI 70
Embanox BHT (пищевой) Англия, May a. Baker 69,2
DBPC США, Koppers 68,7
Renadox DB ФРГ, Rein Chemie >66
Sustan BHT США, UDP Chemical 69—70
Tenox BHT США, Eastman Chemical Pro- 69—70
duction
Vulkanox KB ФРГ, Bayer >66
Permanax BHT Англия, Vulnax International 70
14* 211
Широко известен под названиями Ионол (торговая марка фир.
мы Shell) или сокращенным обозначением ВНТ (от англ, buthye-
hydroxytoluen). Крупнейшими изготовителями его за рубежом яв-
ляются [123] в США фирмы Aschland Chemical, Shell и Koppers, в
Японии Sumitomo, в Англии ICI.
2,6-Ди-77?ет-бутил-4-метилфенол применяют для защиты раз-
личных синтетических каучуков (бутадиен-нитрильные, бутадиено-
вые, изопреновые регулярного строения, уретановые) от старения.
Он защищает от термоокислительной деструкции полиолефины и
резины на основе натурального и синтетических каучуков. Широ-
ко используется в белых и цветных резиновых изделиях. Летучесть
2,6-ди-трет-бутил-4-метилфенола затрудняет его применение в по-
лимерах, перерабатываемых пли эксплуатируемых при высокой
температуре.
Эффективно защищает от окисления смазочные и трансформа-
торные масла, моторные топлива и другие нефтепродукты.
Мало токсичен; ПДК=50 мг/м3. Разрешен для применения в
полимерах, контактирующих с продуктами питания (в США разре-
шена дозировка до 5% от массы полимера).
Используется в качестве антиокислительной добавки к жирам
и маслам; разрешенные дозировки — до 0,02% (масс.).
Является радиопротектором.
2-трет- Б у тил-4-метилфено л
Это соединение образуется при каталитическом алкилирования
4-метилфенола эквимольным количеством изобутилена в присут-
ствии серной кислоты или катионитов:
В присутствии катионита получен продукт с выходом 61—65%
[124—127].
Детально исследовано [128] алкилирование 4-метилфенола изо-
бутиленом в присутствии H2SO4 и катионита КУ-2. При алкили-
ровании в присутствии 5% (масс.) H2SO4 при 75—85°C алкилат
содержал 75—76% 2-7/?ет-бутил-4-метилфенола, 22—23% 2,6-ди-
212
трет-бутил-4-метилфенола и 1,0—1,3% эфиров. Показана возмож-
ность получения 2-трет-бутил-4-метилфенола с высоким выходом
при алкилировании в присутствии КУ-2 на лабораторной установ-
ке непрерывного действия [129, 130]. При исследовании [131] 24
катионитов оказалось, что лучшие результаты дает Lewatit/SP-120:
выход 2-трет'-бутил-4-метилфенола составил 74,7%.
Температура существенно влияет на процесс алкилирования в
присутствии катионитов: при взаимодействии 4-метилфенола с эк-
вимольным количеством изобутилена в присутствии Aliasion CS
при 90 °C образуется 2-7'рет’-бутил-4-метилфенол; при 135—150 °C
идет замещение метильных групп и образуются 4-трет-бутилфенол
и 2,4-ди-т/?ет--бутилфенол [132].
Алкилированием в присутствии BF3-H3PO4 при 120°С получен
целевой продукт с выходом 40,9%; при этом образуется более 4%
трет-бутилового эфира 4-метилфенола [133]. В присутствии BF3
моноалкилирование с выходом 78—84% проходит при мольном
соотношении 4-метилфенол : изобутилен : BF3= 1 : (0,954-1,05) :
: (0,0014-0,006). Увеличение количества BF3 до 0,018 моль повы-
шает выход побочных продуктов [134]. Описано получение 2-трет-
бутил-4-метилфенола в присутствии тетрафосфорной кислоты в
реакторе колонного типа [135].
Получение 2-трет-бутил-4-метилфенола алкилированием кре-
зольной фракции рассмотрено в разделе «2,6-Ди-трет-бутил-4-мё-
тилфенол» (стр. 206). Для разделения 2-трет-бутил-4-метилфенола
и б-трет-бутил-З-метилфенола используют способность последнего
образовывать нерастворимый комплекс с диметилсульфоксидом
[136]. Смесь обрабатывают диметилсульфоксидом в среде инерт-
ного органического растворителя и отфильтровывают комплекс,
выпавший в осадок. В фильтрате остается 2-трет-бутил-4-метил-
<^енол.
Технологическая схема получения. Практическое значение име-
ет алкилирование в присутствии катионообменных смол или сер-
ной кислоты. Описана непрерывнодействующая пилотная установ-
ка для алкилирования под давлением [137]. Смесь жидкого изобу-
тилена и 4-метилфенола при 2—2,5 МПа подают в проточный ре-
актор с неподвижным слоем катионита; температура алкилирова-
ния 85—95 °C. Для повышения селективности и снижения деалки-
лирования берут некоторый избыток 4-метилфенола. Выбраны ус-
ловия, обеспечивающие получение целевого продукта с селектив-
ностью 85—92% по 4-метилфенолу.
Алкилирование в присутствии H2SO4 ведут следующим образом.
В реактор с мешалкой загружают расплавленный 4-метилфенол,
96—98%-ную кислоту (5% масс.) и при 80°C подают через барбо-
тер осушенный изобутилен в количестве 1,05 моль на 1 моль 4-ме-
тилфенола. По окончании алкилирования нейтрализуют реакцион-
ную массу содово-солевым раствором, отделяют органический слой
и перегоняют его в вакууме. Собирают целевую фракцию, кипящую
при 125—130°C (при 2,66 кПа). Для повышения выхода целевого
213
продукта побочные фракции, содержащие 4-метилфенол и 2,6-ди-
трет’-бутил-4-метилфенол, собирают и вводят в реактор перед по-
дачей изобутилена. Перемешивают в течение 1 ч при ПО °C, а за-
тем охлаждают до 80 °C и ведут алкилирование изобутиленом. Вы-
ход целевого продукта при этом существенно возрастает [138].
Развитие производства 2,6-ди-трет-бутил-4-метилфенола из фе-
нола открыло возможность получения 2-трет-бутил-4-метилфенола
реакцией каталитического деалкилирования:
ОН
+ СН2=СН—СН,
Свойства и применение. 2-трет-Бутил-1-метилфенол— белое
кристаллическое вещество; т. пл. 54—54,5 °C; плотность 941 кг/м^
при 55 °C. Температура кипения заметно зависит от давления: 1
Г. кип., °C. ... 233 224,6 218 1₽8 167 127,2 j
Давление, кПа . . 106,8 80 66,6 26,6 13,3 2,65
Растворяется в метаноле (486 г в 100 см3 при 20 °C) и бензине
(84 г в 100 см3 при 20°C).
Т. всп. 80°C; т. воспл. 109°C; т. самовоспл. 358°C.
Является промежуточным продуктом в производстве важней-
ших стабилизаторов группы бисфенолов. Ниже приведены некото-
рые промышленные стабилизаторы, получаемые на основе 2-трет-
4-метилфенола:
2, 2'~метиленбис(6-тр гт-бутил-4-
метилфенол)
2,2'-бутилиденбис(6-трет-бутил-4-
метилфенол)
2,2 *-тнобис(6-треп1-бутил-4-метилфено л)
214
6-трет-Бутил-З-метилфенол
ОН
Простейший способ его синтеза — каталитическое моноалкили-
рование 3-метилфенола изобутиленом:
ОН ОН
(CH^CsJ.
+ (сн3)2с=сн2—> Y £
Реакцию ведут в присутствии обычных кислотных катализаторов;
6-трет-бутил-З-метилфенол является основным продуктом моно-
алкилирования. При дальнейшем алкилировании образуется 4,6-ди-
трет-бутил-3-метилфенол.
Промышленное получение. В связи с дефицитом 3-метилфенола
промышленные способы получения 6-трет-бутил-З-метилфенола ос-
нованы на алкилировании изобутиленом крезольной фракции
(смесь 3- и 4-метилфенолов) с последующим разделением продук-
тов моно- или диалкилирования; получение его часто объединяют
с производством 2,6-ди-тре7’-бутил-4-метилфенола или 2-трет-бу-
тил-4-метилфенола (см. выше). При моноалкилировании крезоль-
ной фракции, содержащей 60% 3-метилфенола (80°C, катализа-'
тор — 3,5%-ная H2SO4), и последующем разделении получают це-
левой продукт с выходом «60% [139].
Для отделения целевого продукта можно использовать его спо-
собность к комплексообразованию, например с диметилсульфокси-
дом (комплекс состава 1:2). Этот комплекс выпадает в осадок
из раствора монобутилированной крезольной фракции в циклогек-
сане или октане; его отфильтровывают и разлагают, перегоняя с
водяным паром [140]. Смесь продуктов алкилирования крезольной
фракции изобутиленом можно обрабатывать 20%-ным раствором
NaOH с последующим подкислением или разбавлением водно-ще-
лочного слоя; при этом получена [141] смесь 6-трет-бутил-З-метил-
и 6-трет-бутил-4-метилфенола, содержащая 84% первого изомера.
Выше (стр. 205) было описано получение 2,6-ди-трет-бутил-4-
метилфенола из крезольной фракции. Образующийся при этом
4,6-ди-7’рет’-бутил-3-метилфенол каталитическим деалкилированием
превращают в 6-трет-бутил-З-метилфенол. Процесс ведут, пропус-
кая пары 4,6-ди-трет-бутил-З-метилфенола через слой алюмосили-
катного катализатора, нагретого до 260 °C [142]. Температура в
испарителе 190°C; давление в системе 2,66 Па. Пары, прошедшие
через слой катализатора, конденсируют; полученную смесь 6-трет-
бутил-3-метилфенола (64—65%) и 3-метилфенола (35—36%) раз-
деляют ректификацией.
215
Свойства и применение. 6-трет-Бутил-З-метилфенол— кристал-
лическое вещество; т. затв. 23 °C; т. кип. 121—122 °C (при
2,13 кПа); плотность 964 кг/м3 (при 25°C).
Служит промежуточным продуктом в синтезе стабилизаторов
группы бис- и трисфенолов.
а-Метилбензилфенолы
Этот стабилизатор — продукт каталитического алкилирований
фенола стиролом — представляет собой смесь 2-а-метилбензилфе^
пола (I), 4-а-метилбензилфенола (II), 2,6-ди(а-метилбензил)фено^
ла (III), 2,4-ди(а-метилбензил)фенола (IV) и 2,4,6-три(а-метил-
бензил) фенола (V):
Впервые а-метилбензилфенолы были получены в 1890 г.; в ка-
честве катализатора была использована серная кислота [143].
Описан также синтез а-метилбензилфенолов в присутствии галоге-
новодородов, фосфорной [144] и хлорной кислот [17], арилсульфо-
кислот [145], катионообменных смол [30], фтористого бора [35]
или его комплексов [146], щавелевой кислоты [147].
Синтез в присутствии серной кислоты или и-толуолсульфокисло-
ты ведут следующим образом. В реактор — эмалированный аппа-
рат с мешалкой и рубашкой — помещают толуол (растворитель),
фенол и катализатор. Смесь нагревают до 85—90°C и при сильном
перемешивании постепенно прибавляют стирол. По окончании ал-
килирования нейтрализуют алкилат раствором соды или щелочи
отмывают водой от минеральных солей, отгоняют растворитель,
отгоняют не вступившие в реакцию исходные вещества и при по-
ниженном давлении перегоняют целевой продукт. Последняя опе-
рация нужна, чтобы освободить продукт от полистирола и смоли-
стых примесей. В присутствии n-толуолсульфокислоты можно вести
алкилирование и без органического растворителя.
Из а-метилбензилфенолов различного строения, входящих в
состав технического продукта, наибольшей ингибирующей активно-
стью обладает 2,4-ди-(а-метилбензил) фенол; несколько уступают
216
ему 2,6-ди- и 2,4-6-три-(а-метилбензил) фенолы [85, 86]. С целью
повышения содержания в реакционной массе этих соединений изу-
чена кинетика алкилирования фенола стиролом в присутствии ща-
велевой кислоты, не вызывающей побочных реакций [87,88]. Про-
цесс состоит из ряда последовательных и параллельных реакций:
ОН
ОН
4-CeHs-€H=CH2
ОН
+ Свн5—СН=СН2
В табл. 6 представлены константы скорости этих реакций при
разной температуре и энергия их активации.
На основе этих исследований разработан способ получения тех-
нической смеси а-метилбензилфенолов [91]. Алкилирование ведут
в присутствии щавелевой кислоты в две стадии: сначала при 95 °C
и эквимольном соотношении фенол: стирол получают преимущест-
венно монозамещенные, а затем поднимают температуру до 135 °C
217
Таблица 6. Константы скорости и энергия активации реакций
при алкилировании фенола стиролом
Реакция k-104, лДмольмин) Е, кДж/моль
при 85 *С при 95 °C при 115 °C при 125 °C при 135 °C
1 0,26 3,6 5,5 6,1 6,5 18
2 0,32 4,7 1,5 8,0 8,4 22
3 0 0,98 1,7 2,5 4,5 48
4 0 0,83 1,5 3,9 6,9 64
5 0 1,8 15,4 18,5 22,6 90
6 0 0 0,6 1.9 3,25 • ПО
и вводят вторую порцию стирола. В результате получают смесь
а-метилбензилфенолов, содержащую более 60% дизамещенных.
Технологическая схема получения (рис. 29). В стальной эмали-
рованный реактор 4, снабженный рубашкой для нагрева и охлаж-
дения, мешалкой и ловушкой (для воды), связанной с обратным
холодильником, загружают щавелевую кислоту и расплавленный
фенол. Массу нагревают до 90—95 °C и при этой температуре по-
степенно загружают из мерника 1 первую порцию стирола. Пере-
мешивают 1 ч, нагревают до 135 °C и загружают вторую порцию
стирола. Реакционную массу перемешивают при этой температуре
и передают в колонну 5 для отгонки не вступивших в реакцию ис-
ходных веществ. Отгонку ведут при остаточном давлении 6,65—
13,33 кПа. Сначала отгоняют фенол (вместе со щавелевой кисло-
той), а затем стирол, которые возвращают в процесс. В кубе оста-
ется смесь а-метилбензилфенолов.
Свойства и применение. Смесь а-метилбензилфенолов выпуска-
ется в СССР под торговым названием Стабилизатор АО-20 (Аги-
Рис. 29. Технологическая схема получения смеси а-метилбензилфенолов:
1 — мерник; 2 — конденсатор; 3 — сепаратор; 4 — реактор; 5 — ректификационная колонна.
218
Таблица 7. Физико-химические свойства индивидуальных
а.метилбензилфенолов
— Соединение Т. кип., °C (прн 66,5 Па) Т. пл., °C Плотность, кг/м8 Относительная вязкость при 80 °C
2-а-Метилбензнлфенол 115—116 1084,5 12,3
4-а-Метилбензилфенол 128,0—128,5 57,7—58 — —
2,4-Ди (а-метилбензил) фе- 218,5—219,5 — 1090,3 по
НОЛ 2,6-Ди (а-метилбензил) фе- 178,5—179,5 — 1085,7 45,3
НОЛ 2,4,6-Т ри (а-метилбензил) фе- 238—239 — 1094,5 210
НОЛ
pQj\ 20) и представляет собой вязкую жидкость желтого или светло-
коричневого цвета; плотность 1080 кг/м3; пр =1,5500; вязкость 1—
6 Па-с при 25°C. Растворим в алифатических и ароматических уг-
леводородах, спиртах, ацетоне; в воде не растворяется.
Свойства индивидуальных а-метилбензилфенолов представлены
в табл. 7.
Смеси а-метилбензилфенолов под различными торговыми мар-
ками выпускаются многими зарубежными фирмами:
Торговое наименование
Страна, фирма
Acronox SP
Acrochem Antioxidant 16
Aceinox SP
Age Rite Spar
Anox 62
Antigen S
Antioxidant SP
Montacler
Naugard SP
Nocrac SP
Permanax SP
Stabilite SP
Wing-Stay S
США, Rubber Regenerating
США, Acron Chemical
Индия, Alkali
США, Vanderbilt
Италия, Bozzetto
Япония, Sumitomo
Англия, Anchor Chemical
США, Monsanto
США, Uniroyal Chemical
Япония, Ouchi Shinko
Англия, Vulnax International
США, Peichnold Chemical
США, Goodyer
Некоторые из этих фирм выпускают смесь а-метилбензилфенолов в
форме порошков (в смеси с инертными наполнителями) или в виде
легко эмульгируемых жидкостей.
Смеси а-метилбензилфенолов применяют для защиты полиоле-
финов, каучуков, латексов и резин на основе натурального и син-
тетических каучуков. Эти смеси не влияют на цвет полимера и
находят применение для защиты белых и цветных изделий. Осо-
бенно широко применяются для защиты латексных пенорезин.
Служат промежуточным продуктом в синтезе смеси фосфористо-
кислых эфиров а-метилбензилфенолов — стабилизатора Фосфит
П-24 [148].
219
2,6-Ди-трет-бутил-4-метоксиметилфенол
Этот стабилизатор получают взаимодействием 2,6-ди-трет-бу^
тилфенола с формальдегидом и метанолом в присутствии щелочи
{149]:
(СНз)3С
НО—
(СН3)3С
(СН3)3С,
4-НСНО + СН3ОН
-^сн2—осн.
Метанол является здесь и реагентом и реакционной средой. Целе^
вой продукт при охлаждении реакционной массы выпадает в оса-'
док, и его можно отделить фильтрованием.
Наряду с основной могут протекать и побочные реакции, при-
водящие к образованию других соединений, например 4-гидрокси-
3,5-ди-трет-бутилбензилового спирта или 4,4/-метиленбис(3,5-ди-
трет-бутилфенола) :
Последняя реакция особенно нежелательна, поскольку образую-
щийся метиленбисфенол (содержание которого в техническом про- 1
дукте достигает 15—18%) в полимерах дает желтую окраску и за-1
трудняет использование 2,6-ди-трет-бутил-4-метоксиметилфенола |
как неокрашивающего стабилизатора. Очистка от этой примеси
связана с потерями целевого продукта.
Технологическая схема получения. Разработан способ получе-
ния 2,6-ди-трет-бутил-4-метоксиметилфенола взаимодействием 2,6-
ди-трет-бутилфенола с формальдегидом в присутствии щелочного
буфера при pH 12,5-5-14,0 [150]. Таким буфером может быть смесь
220
>• Фильтрат''
на рецирну-
лящм
Рис. 30. Технологическая схема получения 2,6-ди-трет-бутил-4-метоксиметилфе
иола:
i — реактор; 2 — кристаллизатор; 3, 5 — фильтры; 4 — нейтрализатор.
гидроксида и ацетата натрия. При этом повышается селективность
реакции и уменьшается содержание побочных продуктов.
Схема получения 2,6-ди-трет-бутил-4-метоксиметилфенола пред-
ставлена на рис. 30. В стальной эмалированный реактор 1 с ме-
шалкой и рубашкой загружают метанол, формалин и катализа-
тор— метанольный раствор гидроксида и ацетата натрия. Переме-
шивают, нагревают до 60 °C и подают 2,6-ди-трет-бутилфенол.
Реакционную массу еще перемешивают при этой температуре и
передают на кристаллизацию. В аппарате 2 охлаждают массу до
0—^минус 5 °C и затем отфильтровывают образовавшуюся суспен-
зию. Осадок с фильтра 3 передают в аппарат 4 на нейтрализацию,
а фильтрат используют для рециркуляции или регенерации раство-
рителя. В аппарат 4 добавляют воду и кислоту до достижения ней-
тральной реакции, перемешивают и передают массу на фильтро-
вание.
Отфильтрованные кристаллы 2,6-ди-трет-бутил-4-метоксиметил-
фенола промывают и передают на сушку и измельчение. Если его
используют как промежуточный продукт в производстве других
стабилизаторов, заключительные операции можно не проводить.
Полученный продукт имеет т. пл. 96 °C и известен под торговой
маркой Агидол 42 (или стабилизатор АО-42).
Свойства и применение. 2,6-Ди-т’рет-бутил-4-метоксиметилфенол
белое кристаллическое вещество; т. пл. 101 °C. Технический про-
дукт иногда окрашен в слабо-желтый цвет. Хорошо растворяется
в бензоле, ацетоне, четыреххлористом углероде и этаноле, хуже—
в гексане и бензине; в воде не растворим.
Выпускается фирмой Ethyl (США) под торговым названием
Ethyl Antioxidant 762. Неокрашивающий стабилизатор изопрено-
221
вых, tfwc-бутадиеновых, бутадиен-стирольных и бутадиен-нитриль-
ных каучуков. Защищает от термоокислительной деструкции поли-
олефины и резины на основе натурального и синтетических каучу.
ков. Служит промежуточным соединением в синтезе высокоэффек-
тивных нелетучих стабилизаторов класса многоядерных алкилфе-
нолов.
4-Гидрокси-3,5-ди-трет-бутилбензиловый спирт
:н2он
Его получают взаимодействием 2,6-ди-трет-бутилфенола
формальдегидом в присутствии щелочи в среде вторичного или т]
тичного спирта:
Чтобы исключить побочную реакцию окисления, рекомендуете"
вести синтез в атмосфере азота [151]. Оказалось, что с повышени-
ем полярности растворителя растет скорость образования метилен-
бисфенолов и затрудняется выделение целевого 4-гидрокси-3,5-ди-
трет-бутилбензилового спирта [152]: с такими растворителями, как
метанол, этанол или изопропанол, последний взаимодействует с
образованием соответствующих эфиров. Лучшие результаты полу-
чены с применением безводного трет-бутанола. Выход целевого
продукта возрастает, если в качестве агента оксиметилирования
использовать параформ.
Предложен также способ получения 4-гидрокси-3,5-ди-трет-бу-
тилбензилового спирта каталитическим восстановлением соответст^
вующего альдегида: I
В качестве восстановителя применяют литийалюминийгидрид [151]
или водород в присутствии никеля Ренея [152]. Этот способ не
222
приобрел технического значения, как и синтез из 2,6-ди-трет-бутил-
4-метилфенола через хинонбромид [154]:
Технологическая схема получения. В реактор с мешалкой за-
гружают безводный трет-бутанол, параформ, 2,6-трет-бутилфенол
и гидроксид калия. Смесь перемешивают без нагревания -в тече-
ние 4—5 ч и передают на выделение целевого продукта. Выделе-
ние ведут, приливая реакционную массу при перемешивании к
большому объему воды. Выпавшие кристаллы отфильтровывают,,
промывают и высушивают.
Свойства и применение. 4-Гидрокси-3,5-ди-трет-бутилбензило-
вый спирт — белое кристаллическое вещество; т. пл. 140 °C. Рас-
творяется в бензоле, гексане, ацетоне, этаноле; в воде не растворя-
ется.
4-Г идрокси-3,5-ди-трет-бутилбензиловый спирт — стабилизатор
средней эффективности для каучуков, резин, полиолефинов. Не по-
лучил широкого распространения из-за высокой текучести. Явля-
ется-исходным соединением при получении стабилизаторов группы
трис- и тетрафенолов.
6-трет-Бутил-2-метилфенол
Его получают алкилированием 2-метилфенола изобутиленом в
присутствии. 2-метилфенолята алюминия при повышенном [43] или
атмосферном давлении [155]:
+ (СН3)2С=СНа ---*-
223
Катализатор готовят взаимодействием металлического алюми-
ния с 2-метилфенолом. Как и при реакции с фенолом, в избытке
2-метилфенола образуется комплексная 2-метилфеноксиалюминие.
вая кислота:
6СН3—С6Н4ОН + 2А1 --> 2(СН3—СеН4О)3А1-р ЗН2
(СН3—СвН4О)3А1 + СН3—С6Н4ОН --> Н[А1(ОС6Н4—СН3)4]
Растворение алюминия в 2-метилфеноле идет с меньшей скоростью
чем в феноле, и требует более высокой температуры (190 вместе
160°C для фенола). Алюминий берут в количестве 0,8—1,0%
(масс.) от 2-метилфенола.
Алкилирование ведут, постепенно пропуская газообразный изо-
бутилен через раствор 2-метилфенолята алюминия в 2-метилфено-
ле. Повышение температуры с 90 до 130 °C заметно ускоряет кон-
версию 2-метилфенола, но при этом возрастает скорость побочногс
образования 4-трет-бутил-2-метилфенола (см. рис. 24), поэтому
процесс целесообразно вести при 90—95 °C. По окончании алки-
лирования разрушают катализатор водой и в виде А1(ОН)3 отде-
ляют от алкилата. Целевой продукт выделяют ректификацией.
Технологическая схема получения. В аппарат для приготовле-
ния катализатора загружают 2-метилфенол и нагревают до 120—
130 °C, затем загружают металлический алюминий, нагревают дс
180—190 °C, выдерживают до прекращения выделения водородг
и передают на алкилирование. Алкилирование ведут в вертикаль-
ном цилиндрическом реакторе, снабженном приспособлениями для
равномерного распределения потока газа и змеевиками для нагре-
вания и охлаждения. 2-Метилфенол с катализатором охлаждают
до 90—95 °C и при этой температуре через испаритель подают га-
зообразный изобутилен. По окончании алкилирования к реакцион-
ной массе добавляют воду и нагревают до 100—105 °C для разло-
жения катализатора, отделяют образовавшийся гидроксид алю-
миния и передают алкилат на ректификадию. На ректификацион-
ной колонне сначала отбирают фракцию, представляющую собой
не вошедший в реакцию 2-метилфенол, а затем целевую фракцию
Свойства и применение. 6-трет-Бутил-2-метилфенол — бел»
кристаллическое вещество; т. крист. 29 °C; т. кип. 141—142 °C (npi
6,65 кПа). Растворяется в бензоле, гексане, бензине, петролейноь
эфире, ацетоне, этаноле, диэтиловом эфире; в воде не растворяется
Применяется как промежуточный продукт в синтезе стабили
заторов группы бис- и трисфенолов.
Эфиры $-(4-гидрокси-3,5-ди-трет-бутилфенил) пропионовой
кислоты
(СН3)3С
НО—/ р—СН2—СН2—COOR
(СН3)3с/
составляют важную группу стабилизаторов
224
;р-4(Гидрокси-3,5-ди-7’рет’-бутилфенил) пропионовая кислота мо-
jger быть получена из 2,6-ди-трет-бутилфенола— взаимодействием
с акрилонитрилом [156] и последующим гидролизом р-замещенно>
го пропионитрила:
(СН3)3С
(СН3)3С
(CH3)3G
+ ch2=chcn
+нао
НО— -СН2—ch2cn ---->
(СН3)3С
(СН3)3С
НО—. сн2—сн2—соон
н
(СН3)3С‘
Реакцию ведут в присутствии основного катализатора. К раствору
трет-бутилата калия в трет-бутаноле прибавляют 2,6-ди-трет-бу-
тилфенол и акрилонитрил, нагревают до кипения и выдерживают
в течение 8 ч. Отгоняют растворитель при пониженном давлении,
нейтрализуют остаток, диэтиловым эфиром извлекают (3-замещен-
ный пропионит^ил, отгоняют эфир и проводят гидролиз пропио-
нитрила.
Известен ряд других способов синтеза [157], причем для по-
лучения стабилизаторов наибольшее значение имеют способы, по-
зволяющие непосредственно в молекулу 2,6-ди-трет-бутилфенола
ввести остаток метакриловой кислоты:
(СН3)3С о (СН3)3С,
О
НО—\-сн2—сн2—с—осн,
О
+ сн2=сн—с—осн3
НО-
(СН3)3С (СН3)3(У ,
Этот синтез ведут в среде третичного спирта в присутствии основ-
ного катализатора [158]. В безводном трет-бутаноле растворяют
металлический калий, загружают 2,6-ди-трет-бутилфенол и метил-
метакрилат и перемешивают 18 ч при 50 °C. Затем отгоняют рас-
творитель (при пониженном давлении), нейтрализуют реакционную
массу и экстрагируют образующийся продукт диэтиловым эфиром.
После отгонки эфира перегоняют остаток при пониженном давле-
нии и собирают фракцию 125—130°C (при 13,3 Па), кристаллизу-
ющуюся при 63—64 °C. Перекристаллизацией из гексана или бен-
зина получают продукт с т. пл. 66,0—66,5°C.
Метиловый эфир р-(4-гидрокси-3,5-ди-трет’-бутилфенил) пропио-
новой кислоты (в литературе его иногда называют Метилокс)—-
белое кристаллическое вещество с т. пл. 67 °C. Он легко вступает
в реакцию переэтерификации и благодаря этому служит исходным
соединением в синтезе .ряда эффективных фенольных стабилизато-
15—1057
225
ров [3, с. 299; 158—163]. Переэтерификацию ведут, нагревая Ме-
тилокс со спиртом в присутствии основного катализатора (алкого-
лят натрия или калия, гидрид лития).
Взаимодействием Метилокса с октадециловым спиртом полу-
чают октадециловый эфир р-(4-гидрокси-3,5-ди-трет-бутил фе-
нил) пропионовой кислоты:
В реактор загружают октадециловый спирт и Метилокс и нагрева-
ют в атмосфере азота до 85—90 °C. Вносят катализатор и выдер-
живают при 85—90 °C и остаточном давлении 2,66 кПа до отгонки
95—98% метанола. Охлаждают, добавляют бензол и нейтрализу-
ют смесь слабой соляной кислотой. Отделяют органический слой,
промывают его водой, отгоняют растворитель и перекристаллизовы-
вают целевой продукт.
Октадециловый эфир р-(4-гидрокси-3,5-ди-трет-бутилфенил)-
пропионовой кислоты — белое кристаллическое вещество; т. пл.
55°C (технический продукт плавится в интервале 49—54°C). Рас-
творяется в гексане, бензоле, хлороформе, ацетоне, метаноле и
этилацетате; в воде не растворяется.
Мало токсичен; разрешен в ряде стран для защиты полимеров,
контактирующих с пищевыми продуктами.
Под торговой маркой Irganox 1076 выпускается фирмой Ciba —
Geigy (Швейцария).
Метилокс— неокрашивающий и нелетучий стабилизатор. При-
меняется для защиты от старения полипропилена, полиэтилена,
пленок и волокон из полиолефинов, сополимера этилена и пропи-
лена и тройного сополимера на их основе, поливинилхлорида, по-
лиэфиров, полистирола и различных сополимеров стирола.
Является исходным соединением в синтезе ряда эффективных
фенольных стабилизаторов. Структура некоторых из них приведе-
на ниже:
-(СН3)3С
_(CHs)sc
226
-(СН3)3С
но—
-(СН^С
-(СН3)3С
_(СН3)3С
о
II
—си2—сн2—с—о—сн2—сн2—
"(СН3)3С
но—
_(СН3)3С
о
II
—сн2-сн2—с—о-сн2-сн2—
N.
8
S
2
О /С(СН3)3
—СН2—О-С—СН2—Н2С—ОН
. С(СН3)3в 4
2,5-Ди-трет-бутилгидрохинон
ОН
_/L^/C(CH3)3
лУ
он
Его получают каталитическим алкилированием гидрохинона
изобутиленом, реже — трет-бутанолом [164].
Алкилирование гидрохинона описано еще в 1892 г., причем долгое время
обязательными условиями реакции считали отсутствие воды и большое количе-
ство катализатора — концентрированной H2SO4. Так, при алкилировании гидро-
хинона грет-бутанолом (мольное соотношение 2:1) в присутствии 70%-иой
H2SO4 был получен только 2-трет-бутилгидрохинон.
Позднее было показано, что 2,5-ди-трет-бутилгидрохинон может быть полу-
чен при алкилировании гидрохинона изобутиленом в среде 65—70%-ной H2SO4
при 25—45 °C [165] и при 100 °C [166]. Серная кислота служит и реакционной
средой и катализатором, причем при переходе от концентрированной кислоты
К разбавленной исключается образование окрашенных побочных продуктов.
2,5-Ди-трет-бутилгидрохинон был получен [167] в присутствии катионообмен-
Ной смолы КУ-2: гидрохинон растворяли в толуоле, вводили КУ-2 (в Н-форме)
И пропускали газообразный изобутилен при 80—90 °C.
Предложен способ [168] алкилирования гидрохинона в присутствии молиб-
дата никеля при 200 °C: изобутилен пропускают через расплавленный гидрохи-
нон, затем отфильтровывают катализат и отгоняют летучие продукты.
При исследовании алкилирования гидрохинона трет-бутанолом в присутствии
хлорида цинка оказалось, что наряду с С-алкилированием протекает и О-алкили-
рование — образуется 4-трет-бутоксифенол [169]. В качестве промежуточного
15* 227
продукта при алкилировании гидрохинона изобутиленом или трет-бутанолом
образуется 2-трет-бутилгидрохинон, превращающийся при дальнейшем алкили-
ровании в 2,5-ди-трет-бутилгидрохннон. При этом не наблюдается образования
2,6-ди-трет-бутилгидрохинона, т. е. орто-алкилирование здесь не идет.
2,6-Ди-трет-бутилгидрохинон получают также, исходя из 2,6-ди-трет-бутил-
фенола, который превращают в 2,6-ди-трет-бутил-4-бензохинон (например, через
нитропроизводное [170]), а затем восстанавливают [171].
При алкилировании 2-метилгидрохинона наряду с целевым 5-трет-бутил-2-
метилгидрохиноном в небольшом количестве образуется и 2,6-изомер. Ввести
трет-бутильную группу в 5-трет-бутил-2-метилгидрохинон и в 2,5-диметилгидро-
хинон не удается, что, по-видимому, связано с пространственными препятст-
виями.
Описано получение 2,5-ди-трет-бутилгидрохинона взаимодействием гидрохи-
нона с трет-бутилхлоридом [172]: последний медленно добавляют к раствору
гидрохинона и хлорида цинка в безводном диэтиловом спирте.
Практическое значение для получения 2,5-ди-трет-бутилгидро- ।
хинона имеет алкилирование гидрохинона изобутиленом в среде
разбавленной серной кислоты. В стальной эмалированный реактор
с мешалкой загружают 65%-ную серную кислоту, нагревают до
50 °C, добавляют гидрохинон, интенсивно перемешивают и затем
постепенно подают изобутилен (из расчета 2,6 моль на 1 моль
гидрохинона). Реакционную массу перемешивают еще 2 ч при |
50—60 °C, охлаждают до 20—25 °C и отфильтровывают продукт
реакции. Осадок промывают водой, 5%-ным раствором соды (для
удаления следов серной кислоты) и горячей водой (для удаления
остатков непрореагировавшего гидрохинона). Серную кислоту воз-
вращают в процесс. Готовый продукт высушивают в вакуум-сушил-
ке при 60—70 °C.
Свойства и применение. 2,5-Ди-трет-бутилгидрохинон— светло-
серое кристаллическое вещество; т. пл. 218 °C (т. пл. технического
продукта 212—213°C). Растворим в бензоле, хлороформе, ацето-
не, диэтиловом эфире, диоксане, этаноле (при нагревании); в воде
не растворяется.
Мало токсичен. Разрешен для стабилизации полимеров, кон-
тактирующих с пищевыми продуктами (кроме жиров и спиртсодер-
жащих); ПДК=3 мг/м3. JU
Под торговой маркой Nocrac NS-7 выпускается фирмой OuchM
Shinko (Япония). Я
Неокрашивающий стабилизатор синтетических каучуков и клеЯ
ев на их основе. Термо- и светостабилизатор полиолефинов и поли'Я
формальдегида. Термостабилизатор полиолефиновых волокон. Я
2,5-Ди-трет-амилгидрохинон мВ
ОН
228
Впервые ои был получен при взаимодействии гидрохинона с изоамиленами
в присутствии концентрированной серной кислоты [164[:
ОН ОН
-J- 2u30-CgHio
он он
Позднее этот синтез был осуществлен в присутствии разбавленной серной
кислоты; в качестве алкилирующего агента наряду с изоамиленами использова-
ли трет-амиловый спирт. В колбу с мешалкой и обратным холодильником поме-
щали 70%-ную H2SO4 (4 моль), растворяли в ней 0,5 моль гидрохинона, при
энергичном перемешивании добавляли (постепенно) 1,2 моль трет-амилового
спирта и медленно нагревали до 70 °C. После выдерживания отфильтровывали
осадок, нейтрализовали, промывали и высушивали.
В зависимости от условий алкилирования получали продукт с разной тем-
пературой плавления: при 70 °C в течение 30 мин получен продукт с т. пл.
165—70 °C, а при 40 °C (2 ч) —с т. пл. 178 °C.
2,5-Ди-трет-амилгидрохинон был получен [169] алкилированием гидрохино-
на 2-метилбутеном-2 в присутствии 85%-ной Н3РО4 и трет-амиловым спиртом
в среде 65 %-ной H2SO4 при 50 °C.
Описано получение 2,5-ди-трет-амилгидрохинона взаимодействием гидрохи-
нона с трет-амилхлоридом в присутствии хлорида цинка [157].
Практическое значение для получения 2,5-ди-трет-амилгидрохи-
нона имеет алкилирование гидрохинона изоамиленами в среде раз-
бавленной серной кислоты.
Технология получения. В эмалированный реактор с мешалкой,
рубашкой (для нагревания и охлаждения) и обратным холодиль-
ником загружают 70%-ную серную кислоту и нагревают до 45°C.
При этой температуре добавляют гидрохинон и перемешивают
30 мин. Затем в течение 20—30 мин при 45—47 °C подают свеже-
приготовленный раствор изоамилена в 70%-ной серной кислоте.
Мольное соотношение гидрохинон: изоамилен = 1 :2,4. Реакцион-
ную массу перемешивают 1 ч при 45—47 °C и затем 1 ч при 20—*
25 °C. Осадок отфильтровывают, промывают водой, раствором би-
карбоната натрия (для удаления остатков серной кислоты и гид-
рохинона), разбавленным раствором поверхностно-активного ве-
щества и высушивают.
Свойства и применение. 2,5-Ди-трет-амилгидрохинон — белое
или светло-кремовое кристаллическое вещество; т. пл. 179 °C; плот-
ность 1020—1080 кг/м3. Растворим в бензоле, хлороформе, ацето-
не; в воде не растворяется.
Мало токсичен. В США разрешен (в количестве до 5%) для
защиты полимеров, контактирующих с пищевыми продуктами.
В СССР разрешен для стабилизации полимеров, контактирующих
с пищевыми продуктами (кроме жиров и спиртсодержащих).
ПДК=1 мг/м3.
Под торговой маркой Santovar А выпускается фирмой Monsan-
to (Англия). В СССР известен под названием Дитаг.
Применяется для защиты синтетических каучуков, клеев на их
основе, полиолефинов и волокон на их основе.
229
ЛИТЕРАТУРА h
VI
1. - Фойгт И. Стабилизация синтетических полимеров против действия тепла н ||
света. Пер. с нем./Под ред. Б. М. Коварской. Л., Химия, 1972. 544 с. • ?
2. Гурвич Я. А., Золотаревская Л. К., Ку мок С. Т. Фенольные стабилизаторы. Ч
М., Изд. ЦНИИТЭнефтехим, 1978. 80 с. J|
3. Ершов В. В., Никифоров Г. А., Володькин А. А. Пространственно-затруднен- Я
иые фенолы. М., Химия, 1972. 352 с. Я
4. Friedel — Krafts and Related Reactions (ed. Olah G.). Intern. Publ., New York, .1
London, Sydney, 1963. V. 11, p. 75. Ж
5. Шуйкин H. И., Викторова E. A. —Успехи химии, 1960, т. 29, с. 1229.
6. Топчиев А. В., Завгородний Е. В., Паушкин Я- М. — Фтористый бор в орга-зИ
нической химии. М., Изд. АН СССР, 1956. Я|
7. Исагулянц В. И. — Хим. пром-сть, 1958, с. 93.
8. Olah G. е. а. — J. Am. Chem. Soc., 1963, v. 85, р. 1328. 1
9. Olah G., Porter R., Kelly D. — J. Am. Chem. Soc., 1971, v. 93, p. 464. 11
10. Olah G., Kuhn S. — J. Am. Chem. Soc., 1955, v. 80, p. 6535.
11. Olah G., Pittman C. — Adv. Phys. Org Chem., 1966, v. 4, p. 305. j
12. Lieser K., Pfluger C. — Ber., 1960, B. 93, S. 176. Jj
13. long J. — Rec. trav. chim., 1964, v. 83, p. 469. 4 j
14. Топчиев А. В. и dp. — Нефтяное хозяйство, 1954, т. 32, № 7, с. 65. ;
15. Топчиев А. В.— Избранные труды. Алкилирование. М., Изд. АН СССР, 1965, Ц
с. 141, 220. <]
16. Алисова Е. В., Завгородний С. В. — ЖОХ, 1964, т. 34, с. 3079. э-i
17. Foster G. — J. Chem. Soc., 1959, р. 2788. 'Я
18. Мюллер Е. Новые воззрения в органической химии. Пер. с нем./Под ред. Я]
А. Н. Несмеянова. М., Издатинлит, 1960, с. 239.
19. Гурвич Я- А., Старикова О. Ф., Стыскин Е. Л. — Хим. пром-сть, 1973, № 1,
с. 9.
20. General Electric, пат. 2394754, 1946 г. (США).
21. Bun-Hoi Ng., Bihan Н., Binan F. —J. Org. Chem., 1952, v. 17, p. 243.
22. Goodrich, пат. 2674340, 1954 г. (США); пат. 2714120, 1955 г. (США).
23. Goodyear, пат. 3035015, 1961 г.; пат. 2909504, 1959 г. (США).
24. Сергеев П. Г., Кружалов Б. Д. — Труды НИИСС, 1961 г. вып. 4, с. 89.
25. Bayer, пат. 944014, 1956 г. (ФРГ).
26. Ethyl, пат. 2831898, 1958 г. (США).
27. Stroh R., Seydel R., Hahn W. — Angew. Chem., 1957, B. 69, S. 699.
28. Ethyl, пат. 670530, 1966 г. (Бельгия).
29. Lukashov A. I. — Soviet Technology and Engineering Didest. Licences from
the USSR, vol. 1, № 1, 150 (1977).
30. Исагулянц В. И., Тишкова В. М. — В кн.: Труды Всесоюзного совещания по
комплексной химической переработке нефти и газа. М., Изд. АН СССР,
1956, с. 464.
31. Исагулянц В. И. — В кн.: Труды МИНХ и ГП им. Губкина, 1960, т. 28,
с. 68.
32. Boisselat L., Parc G. — Bull. Soc. chim. France, 1958, p. 856.
33. Гаджибалаев А. А., Горяев M. И. — Азерб. хим. ж., 1969, № 5, с. 86.
34. Паушкин Я. М. и др. — Нефтехимия, 1969, т. 8, с. 842.
35. Завгородний С. В., Зайцев Б. А., Ельчинов Д. П. — ЖОХ, 1960, т. 30, с. 2196.
36. Потоловский Л. А., Кукуй Н. М., Васильева В. Н. — В кн.: Труды ВНИИНП,
1970, вып. 3, с. 263—268.
37. Гурвич Я. А., Кумок С. Т., Стыскин Е. Л. — В кн.: Тезисы I Всесоюзного
симпозиума по органическому синтезу. М., Изд. АН СССР, 1974, с. 27.
38. Караваев Н. М. и др. — ДАН СССР, 1967, т. 173, № 4, с. 832.
39. Паушкин Я. М. Нефтехимический синтез в промышленности. М., Наука, 1966.
279 с.
40. Gulf Res. Dev., пат. 2459597, 1949 г. (США).
41. Hart Н„ Cassis F. — J. Am. Chem. Soc., 1951, v. 73, p. 3179.
42. Goldsmith E., Schlatter M., Toland W. — J. Org. Chem., 1958, v. 23, p. 1871.
43. Stroh R., Seydel R., Hahn W. — Angew.-Chem., 1957, B. 69, S. 699.
230
44. Bayer, пат. 944014, 1956 г. (ФРГ).
45. Подберезина А. С., Хейфец Л. А. —Ж. орг. хим., 1970, т. 6, с. 995.
46. Knapp G. G. е. а. — In: VII Congr. mund. petrol., Mexico, 1967, Preprint 39/1.
47. Гурвич Я. А. и др. — В кн.: Тезисы V Всесоюзной научно-технической конфе-
ренции по химикатам-добавкам для полимерных материалов, Тамбов, 1976,
с. 25.
48. Besan J. — Nehezvegyipori kut. inter, kozl., 1972, v. 4, p. 67.
49. Berny M, Perrin R. — C. R. Acad. Sc. Paris, 1967, v. 264, p. 1734.
50. Berny M. — Bull. Soc. chim. France, 1969, № 3, p. 973.
51 Koppers, пат. 3267155, 1966 г. (CHIA).
52. Koppers, пат. 3267152. 1966 г. (США).
53. Koppers, пат. 3331879, 1967 г. (США).
54. Bayer, пат. 1044825, 1961 г. (ФРГ).
55. S. A. Prod. Chimie, пат. 1331450, 1963 г. (Франция).
56. Stauffer Chem., пат. 3133947, 1964 г. (США).
57. Houben — Weyl. Methoden der Organishe Chemie, G. Thieme Verlag. Stutgart,
1955, B. 9, S. 211.
58. Bayer, пат. 1142873, 1963 г. (ФРГ).
59. Ethyl, пат. 3367981, 1963 г. (США).
60. Shell, пат. 2923745, 1960 г. (США).
61. Shell, пат. 895928, 1962 г. (Англия).
62. Ethyl, пат. 3185735, 1965 г. (США).
63. Koppers, пат. 3267154, 1966 г. (США).
64. Koppers, пат. 3267153, 1966 г. (США).
65. Shell, пат. 617913, 1964 г. (Бельгия).
66. Ethyl, пат. 655274, 1965 г. (Бельгия).
67. Compagnie Francaise de Produits chimiques shell, пат. 1398153, 1965 г. (Фран-
ция).
68. Shell, пат. 2874193, 1959 г. (США).
69. Shell, пат. 3116336, 1963 г. (США).
70. Shell, пат. 1008592, 1965 г. (Англия).
71. Monsanto, пат. 3177259, 1963 г. (США).
72. Jefferson Chem., пат. 3733365, 1973 г. (США).
73. Алисова Э. В., Пономарева Э. А. — В кн.: Вестник Киевского политехническо-
го ин-та, сер. хим. машиностр. и техиол., 1976, № 13, с. 32.
74. Ethyl, пат. 3555504, 1967 г. (США).
75. Пат. 1229480, 1971 г. (Англия).
76. Пат. 2345419, 1977 г. (Франция).
77. Володькин А. А., Ершов В. В. — Изв. АН СССР, ОХН, 1962, с. 342.
78. Bayer, пат. 1199277, 1966 г. (ФРГ).
79. Gupta S. D., Joshi S. С. — Chim. Age India, 1971, v. 22, № 8, p. 537.
80. Consolidation Coal, пат. 3461175, 1969 г. (США).
81. Shell, пат. 919428, 1963 г. (Англия).
82. Hochst, заявка 2039062, 1972 г. (ФРГ).
83. Гершанов Ф. Б. и др. — Промышленность СК, 1973, № 12, с. 6.
84. Пат. 3946086, 1976 г. (США).
85. Бройтман А. Я- и др. — Пласт, массы, 1963, № 4, с. 19.
86. Гурвич Я- А. и др. — В кн.: Тезисы докладов III Всесоюзной латексной кон-
ференции. М., 1968, с. 72.
87. Гурвич Я- А., Кумок С. Т., Стыскин Е. Л. — В кн.: Тезисы первого Всесоюз-
ного симпозиума по органическому синтезу. М., Изд. АН СССР, 1974, с. 27.
88. Кумок С. Т. и др. — Хим. пром-сть, 1971, с. 566.
89. Курашев М. В. и др. — Нефтехимия, 1970, т. 10, с. 840.
90. Курашев М. В. и др. — Нефтехимия, 1969, т. 9, с. 418.
91. А. с. 330151, 1972 г. (СССР).
92. Joklick О. — Prakt. Chem., 1959, № 12, S. 499.
93. Исагулянц В. И., Фаворская Н. А., Тишкова В. Н.—ЖПХ, 1961, т. 34,
с. 693.
94. Eastman, пат. 3082258, 1963 г. (США).
95. А. с. 347327, 1972 г. (СССР).
231
96. Bayer, пат. 944014, 1956 г. (ФРГ).
97. Bayer, пат. 1044825, 1959 г. (ФРГ).
98. Афанасьев И. Д. и др. — В кн.: Присадки к маслам. М., Химия, 1966. Т. 2,
с. 129.
99. Potlin Е., Vitca V. — Rev. chim., 1965, v. 16, p. 546.
100. Бодан А. И., Кахмар Б. В. — Нефт. и газ. пром-сть, 1963, № 3, с. 56.
101. Coalite-Chem. Prod., пат. 1370325, 1974 г. (Англия).
102. “ — ................................—
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
Trocsanyi Z.— Magy asvanyolai es foldgaskiserl inter, kozl., 1975, v. 16, p. 119
ICI, пат. 890959, 1953 г. (ФРГ).
Sumitomo, пат. 2974, 1969 г. (Япония).
Sumitomo, пат. 21414, 1968 г. (Япония).
Исагулянц В. И., Фаворская И. А. — В кн.: Труды МИНХ и ГП, 1960, т. 28,
с. 56.
Koppers, пат. 3519692, 1970 г. (США).
Koppers, пат. 3534111, 1970 г. (США).
Prinz И. — Chem. Techn., 1970, v. 22, р. 493.
Ceras у Disoluentes, пат. 393501, 1972 г. (Испания).
ICI, пат. 1323066, 1973 г. (Англия).
Сулимов А. Д., Кукушкин В. С. — Химия и технология топлив и масел, 1972
№ 2, с. 15.
Пат. 3940451, 1976 г. (США).
Пат. 2109347, 1972 г. (Франция).
Гудринисуне Э. Ю. и др. — В кн.: Химическая технология и химия. Рига
1973. Вып. 1, с. 72.
Воронцова В. И. и др. — В кн.: Тезисы докладов II Всесоюзной научно-тех
нической конференции по химикатам-добавкам для полимерных материалов
Тамбов, 1976, с. 36.
Захарова И. В. и др. — В кн.: Каталитические реакции в жидкой фазе. Ал
ма-Ата, 1974. Ч. IV, с. 774.
Soc. Anonim. des Prod. Chim., пат. 1344909, 1963 г. (Франция).
Shell, пат. 893896, 1962 г. (Англия).
Bataafse Petroleum, пат. 1119293, 1958 г. (ФРГ).
116.
117.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
118.
119.
120.
121. Yoshitomi, пат. 24055, 1974 г. (Япония).
Yoshitomi, пат. 16282, 1969 г. (Япония).
Modern Plastics, 1976, v. 6, № 9, р. 51.
ICI, пат. 731270, 1955 г. (Англия).
Boisselet A., Park G. — Bull. Soc. chim. France, 1958, № 6, p. 836.
Исагулянц В. И., Фаворская И. А. — В кн.: Труды МИНХ и ГП, 1960, вып. 28,
с. 56.
Белов И. С., Исагулянц В. И. —ЖПХ, 1964, т. 37, с. 1797.
Гринберг А. А., Гурвич Я. А., Рыбак А. И.—Хим. пром-сть, 1967, с. 202.
Гусев В. К. и др. — Нефтепереработка и нефтехимия, 1969, № 3, с. 15.
Гусев В. К. Канд. дисс. М., МИНХ и ГП, 1968.
Malinowski М. — Prz. Chem., 1975, v. 54, № 1, р. 38.
Kamis J. — Collection, 1964, v. 29, p. 923.
Карченко П. С., Завгородний С. B. — Укр. хим. ж., 1964, т. 30, № 3, с. 261
Вдовцева Е. А., Федорова Г. Ф. — В кн.: Синтез и исследование катализа
торов нефтехимии. Ташкент, Фан, 1973. Вып. 6, с. 215.
Пат. 117675, 1966 г. (ЧССР).
Koppers, пат. 3452104, 1969 г. (США).
Лебедев Е. В. и др. — Нефтепереработка и нефтехимия, 1971, № 5, с. 51.
135.
136.
137.
138. А. с. 245127, 1969 г. (СССР).
139. Weiberg М. — Rev. chim. (RSR), 1966, v. 17, № 11, p. 669.
140. Koppers, пат. 3452104, 1969 г. (США).
141. А. с. 343970, 1972 г. (СССР).
142. А. с. 194836, 1968 г. (СССР). 1
143. Пат. 49152, 1965 г. (ПНР).
144. Гаджибалаев А. А., Архипов М. И. — Изв. ВУЗов. Хим. и хим. технология
1965, т. 8, с. 469.
145. Cyanamid, пат. 3407147, 1968 г. (США).
232
146. Наметкин Н. С. и др. — Изь. АН СССР, сер. хим., 1975, т. 10, с. 2268.
147. А. с. 228694, 1968 г. (СССР).
148. Гурвич Я. А., Зимин Ю. Б. — Хим. пром-сть, 1969, № 3, с. 173.
149. Bayer, пат. 1071092, 1961 г. (ФРГ).
150. А. с. 395351, 1973 г. (СССР).
151. Coppinger G., Campbell Г.— J. Am. Soc., 1973, v. 75, p. 734.
152. Егидис Ф. M„ Кретинина E. С., Попов Л. K-— В кн.: Синтез и исследование
эффективности химикатов-добавок дли полимерных материалов. Тамбов,
1969. Вып. 2, с. 376.
153. А. с. 226603, 1968 г. (СССР).
154. Никифоров Г. А. и др. —Изв. АН СССР, 1962, с. 1836.
155. А. с. 216011, 1968 г. (СССР).
156. Geigy, пат. 3121732, 1964 г. (США).
157. Mathieson Alkali Works, пат. 596461, 1958 г. (Англия).
158. Geigy, пат. 3364250, 1968 г. (США).
159. Geigy, пат. 3330859, 1967 г. (США).
160. Geigy, пат. 3497549, 1970 г. (США).
161. А. с. 448197, 1974 г. (СССР).
162. Geigy, заявка 6610810, 1966 г. (Нидерланды).
163. Osaka Seiko Kokoge, пат. 48-26024, 1973 г. (Япония).
164. Eastman Kodak, пат. 2511193, 1950 г. (США).
165. Monsanto, пат. 2832808, 1958 г. (США).
166. Monsanto, пат. 1164423, 1964 г. (ФРГ).
167. Исагулянц В. И. и др.— В кн.: Труды МИНХ и ГП, 1964, т. 51, с. 122; ЖПХ,
1964, т. 37, с.2729.
168. Un. Shoe Machin., пат. 2572091, 1951 г. (США).
169. Po&pi&il Tainer L. — Collection, 1964, v. 29, p. 381.
170. Ethyl, пат. 3415850, 1968 г. (США).
171. Ethyl, пат. 3395160, 1968 г. (США).
3.3. КОНДЕНСАЦИЯ АЛКИЛФЕНОЛОВ
С КАРБОНИЛЬНЫМИ СОЕДИНЕНИЯМИ.
СИНТЕЗ МЕТИЛЕНБИСФЕНОЛОВ
Взаимодействие алкилфенолов с карбонильными соединениями,
в частности с альдегидами,— одна из важнейших реакций при син-
тезе стабилизаторов класса метиленбисфенолов.
Впервые реакцию фенола с альдегидом исследовал Байер еще в прошлом
веке; он показал, чт.о в присутствии кислотных катализаторов фенол взаимо-
действует с альдегидом с образованием высокомолекулярного, продукта поли-
конденсации.
Позднее было установлено, что фенол реагирует с альдегидами и в присут-
ствии оснований. Эти реакции нашли практическое применение в синтезе фено-
ло-альдегидных смол и на протяжении многих лет были объектом исследований,
обобщенных в монографиях [1—3].
По современным представлениям, взаимодействие фенола с
альдегидами представляет собой реакцию электрофильного заме-
щения, по-разному протекающую в условиях кислотного и основ-
ного катализа.
В присутствии кислотного катализатора альдегид протониру-
ется:
НСНО + Н+ CHgOH
233
Протонированный альдегид является электрофильным агентом,
способным взаимодействовать с реакционноспособными аромата-:
ческими соединениями. Реакция идет через стадии образования
л- и сг-комплексов, и в случае фенола ее можно представить схе-
мой:
Аналогично:
Реакция не останавливается на стадии образования гидроксибензи-
лового спирта: он реагирует с фенолом, причем эта реакция также
идет через образование карбкатиона, который атакует молекулу
фенола по схеме электрофильного замещения с образованием ме-
тиленбисфенола:
Образование гидроксибензилового спирта не является обязатель-
ной стадией в синтезе метиленбисфенола. В реакцию с фенолом
может вступить карбкатион, образовавшийся за счет отщепления
молекулы воды от о-комплекса [2, 4, 5]:
При конденсации фенола с кетоном, например с ацетоном, кислот-
ный катализатор протонирует кетон
СН3—СО—СН3 -f- Н+ ч=ь (СН3)2СОН
234
а образовавшийся карбкатион атакует молекулу фенола:
Полученный карбинол взаимодействует со второй молекулой фе-
нола, причем, как и в случае альдегида, реакция идет через соот-
ветствующий карбкатион:
Карбкатион может образоваться и непосредственно из сг-комплек-
са:
Таким образом, образование бисфенола из фенола и карбонильно-
го соединения в присутствии кислотного катализатора идет через
стадии протопировапия карбонильного соединения и электрофиль-
ной атаки молекулы фенола появившимся карбкатионом с образо-
ванием гидроксибензилового спирта (или атаки соответствующим
ему карбкатионом и взаимодействием последнего со второй моле-
кулой фенола).
По-иному протекает реакция в присутствии оснований. В этом
случае катализатор взаимодействует с фенолом, и возможность
взаимодействия альдегида и кетона с фенолом связана со значи-
тельным повышением нуклеофильности ароматического ядра при
235
переходе от фенола к фенолят-аниону, который легко образуется
в присутствии основания. Электрофильной активности молекулы
альдегида или кетона, несущей на карбонильном углероде частич-
ный положительный заряд, оказывается достаточно для взаимодей-
ствия с фенолят-анионом даже без добавления кислотного катали-
затора—’Происходит замещение оксиметильными или карбиноль-
ными группами:
В случае кетона процесс таков:
Гидроксибензиловый спирт или гидроксифенилдиметилкарбинол
вступает в реакцию со вторым фенолят-анионом, образуя соответ-
ствующий бисфенол.
В случае синтеза дифенилолпропана реакция, по-видимому,
идет через поляризованную хиноидную структуру [31] \
с-
Приведенные схемы не исчерпывают всего многообразия реак-
ций, протекающих при взаимодействии фенола с альдегидами. Так,
при взаимодействии двух молекул гидроксибензилового спирта мо-
жет происходить отщепление воды и образование дигидроксибензи-
ловых эфиров [4]:
ОН ОН
Эти эфиры при нагревании в присутствии кислоты или щелочи спо-
собны к отщеплению формальдегида и образованию дигидрокси-
236
дифенилметана [1, 2]:
+ нсно
Метиленовые мостики могут образоваться и в результате так на-
зываемой автоконденсации гидроксибензиловых спиртов [6—10]:
+ НСНО + н2о
В синтезе стабилизаторов альдегиды (реже кетоны) взаимодей-
ствуют с алкилфенолами, содержащими в орто-положении к гидр-
оксилу разветвленные алкильные группы; наибольшее значение
имеют реакции с 2,4- и 2,6-диалкилфенолами [11—13].
В течение длительного времени на эти реакции переносились
представления, сложившиеся при изучении реакций альдегидов и
кетонов с незамещенным фенолом. Между тем переход от фенола
к пространственно затрудненному диалкилфенолу связан не толь-
ко с уменьшением числа реакционных центров в ароматическом
ядре. В силу пространственных затруднений, испытываемых НО-
группой, такие фенолы могут проявлять реакционную способность,
отличающуюся от реакционной способности незамещенного фено-
ла [14].
При изучении зависимости между строением диалкилфенола и
его реакционной способностью в синтезе метиленбисфенолов ока-
залось [15], что 2,4- и 2,6-диалкилфенолы в реакции с формаль-
дегидом проявляют неодинаковую реакционную способность. На
Рис. 31. Кинетические кривые превращения изомерных ди-трет-бутилфенолов (а)
и трет-бутил-а-метилбензилфенолов (б) в бисфенолы при взаимодействии с форм-
альдегидом в условиях кислотного катализа (3%-ная H2SO4, 60 °C, растворитель—
бензол):
I — 2,4-ди-трет-бутилфенол; 2 — 2,6-ди-грет-бутилфенол; 3 — 4-грег-бутил-2-а-метилбеизилФе-
нол; 4 — 6-грег-бутил-2-а-метилбеизилфеиол.
237
Рис. 32. Кинетические кривые образования бисфенолов из 2,4-ди-трет-бутилфенола
и формальдегида (кривые 1—4) и из 2,6-ди-трет-бутилфенола (кривые 5—8) в
различных растворителях (3%-иая H2SO4, 60°C):
1, 5 — в бензоле; 2, 6 — в четыреххлористом углероде; 3, 7 — в н-октаие; 4, 8 — в беизиие.
Рис. 33. Кинетические кривые образования 2,2'-метиленбисфенолов из различных
2,4-диалкилфенолов и формальдегида (3%-ная H2SO4, 60 °C, растворитель — бен-
зол):
1 — из 2-трет-бутил-4-метилфенола; 2 — из 2,4-ДИ-тре7'-бутилфенол а; 3 — из 4-трет-бутил-2-
а-метилбензилфенола; 4 — нз 4-метил-2-а-метилбеизилфенола; 5 — из 2-<х,а-днметилбензил-
4-метилфенола.
рис. 31 представлены кинетические кривые реакции между форм-
альдегидом и изомерными ди-трет-бутилфенолами (и изомерными
трет-бутил-а-метилбензилфенолами) в присутствии серной кисло-
ты. На рис. 32 представлены кинетические кривые образования
бисфенолов при кислотнокаталитических реакциях формальдегида
с ди-трет-бутилфенолами в различных растворителях. Из приве-
денных данных следует, что в реакциях с формальдегидом в усло-
виях кислотного катализа 2,4-диалкилфенолы обладают значитель-
ной реакционной способностью и с высоким выходом образуют со-
ответствующие бисфенолы, а 2,6-диалкилфенолы в эту реакцию
практически не вступают.
Пониженная по сравнению с 2,4-изомерами реакционная спо-
собность 2,6-диалкилфенолов в некоторых реакциях электрофиль-
ного замещения отмечалась и ранее — при исследовании реакций
Конденсации с дихлоридом серы [12].
Исследование кислотнокатализируемых реакций между 2,6-ди-
.алкилфенолами и формальдегидом показало, что с увеличением
степени пространственного затруднения гидроксильной группы ско-
рость превращения диалкилфенола в бисфенол уменьшается: за
5 ч реакции выход бисфенолов составил для 2,6-диметилфенола
16% от теоретического, а для б-трет-бутил-2-метилфенола 11°/0;
2,6-ди-трет-бутилфенол практически не образует бисфенол.
На рис. 33 представлены кинетические кривые образования
2,2'-метиленбисфенолов взаимодействием формальдегида с раз-
238
личными 2,4-диалкилфенолами. Видно, что 2,4-диалкилфенолы об-
ладают высокой реакционной способностью в условиях кислотно-
го катализа: выход 2,2'-метиленбисфенолов через 4 ч реакции со-
ставляет 70—95% от теоретического.
Зависимость между строением диалкилфенола и его реакцион-
ной способностью, наблюдаемая в реакциях с формальдегидом,
присуща и другим альдегидам (рис. 34).
В реакционной массе, полученной при кислотнокатализируемом
взаимодействии 2-трет-бутил-4-метилфенола с формальдегидом,
обнаружен 5-трет-бутил-6-гидрокси-3-метилбензиловый спирт, ко-
торый расходуется на дальнейших стадиях. Этот спирт был полу-
чен направленным синтезом [16] и введен в реакцию с 2-трет-бу-
тил-4-метилфенолом; основным продуктом оказался 2,2'-метилен-
бис(6-трет-бутил-4-метилфенол). На основе этих данных и общих
представлений о протекании кислотнокатализируемых реакций
между фенолом и альдегидами [2, 3] реакцию между 2,4-диалкил-
фенолом и формальдегидом можно представить схемой:
НСНО + Н+ СН2ОН
+ СН2ОН-—>
При кислотнокатализируемой реакции формальдегида с 2,6-ди-
трет-бутилфенолом и 2-т’рет-бутил-б-а-метилбензилфенолом не уда-
лось обнаружить образования гидроксибензиловых спиртов.
Полученные результаты позволяют сделать вывод, что разли-
чие в реакционной способности 2,4- и 2,6-диалкилфенолов в кислот-
нокатализируемых реакциях с альдегидами существенно зависит
239
Рис. 34. Кинетические кривые превраще-
ния диалкилфенолов в бисфенолы в кис-
лотнокатализируемой реакции с альдеги-
дами (3%-ная H2SC>4, 60 °C, раствори-
тель — бензол):
1,2 — 2-7’рет'-бути л-4-метилфенол (/ — с масля-
ным альдегидом, 2—с валериановым альде-
гидом); 3, 4 — 2,6-ди-грет-бутилфеиол (3 —с
масляным альдегидом, 4-— с валериановым
альдегидом).
испытываемых гидроксильной труп-
от стернческих затруднений,
пой: повышение степени пространственного затруднения диалкил-
фенола снижает его реакционную способность. Это может быть
связано с понижением нуклеофильности диалкилфенола вследствие
некоторого уменьшения эффекта сопряжения НО-группы с арома-
тическим кольцом [14]. Следствием этого является низкая реак-
ционная способность пространственно затрудненных 2,6-диалкил-
фенолов в кислотнокатализируемых реакциях с альдегидами.
Превращение фенола в фенолят-анион сопровождается значи-
тельным повышением нуклеофильности, поэтому можно было пред-
положить, что в присутствии основных катализаторов реакционная
способность 2,6-диалкилфенола возрастает за счет повышения нук-
леофильности. На рис. 35 представлены кинетические кривые ре-
акции изомерных 2,4- и 2,6-диалкилфенолов с формальдегидом в
присутствии гидроксида натрия. Из приведенных данных следует,
что в реакции с формальдегидом в условиях основного катализа
2,6-диалкилфенолы проявляют высокую реакционную способность
и с большим выходом образуют 4,4'-метиленбисфенолы.
На основе общих представлений о взаимодействии фенолов и
формальдегида в щелочной среде [3] можно предположить, что
Рис. 35. Кинетические кривые превращения изомерных ди-трет-бутилфенолов (а)
и трет-бутил-а-метилбензилфенолов (б) в бисфенолы при взаимодействии с форм-
альдегидом в условиях катализа основанием (3,3%-ный NaOH, 60 °C, раствори-
тель — изопропиловый спирт):
1 — 2,6-ди-трет-бутилфенол; 2 — 2,4-ди-трет-бутилфенол; 3 — 6-грет-бутил-2-а-мётилбензилфе-
нол; 4 — 4-трет-бутил-2-а-метилбензилфенол.
реакции 2,6-диалкилфенолов с формальдегидом идут через обра-
зование соответствующего 4-гидроксибензилового спирта:
Анализ реакционной массы показал, что в ней действительно при-
сутствует 4-гидрокси-3,5-ди-т’рет'-бутилбензиловый спирт, который в
дальнейшем расходуется (рис. 36,а). Вторая стадия образования
бисфенола, по-видимому, протекает так:
Для проверки этой схемы синтезировали 4-гидрокси-3,5-ди-трет-
бутилбензиловый спирт (по специально разработанной методике
[17]) и исследовали его взаимодействие с 2,6-ди-трет-бутилфено-
лом в условиях катализа основаниями. Главным продуктом ока-
зался 4,4/-метиленбис(2,6-ди-трет-бутилфенол) (рис. 36,6), что
подтверждает справедливость приведенной схемы. Возможность
получения 4,4'-метиленбисфенола по этой схеме показана и в па-
тенте [18].
Рис. 36. Кинетические кривые превращений 2,6-ди-трет-бутилфенола при реакции
с формальдегидом (а) и 4-гидрокси-3,5-ди-трет-бутилбензиловым спиртом (б):
I — 2,6-ди-трет-бутилфенол; 2 — 4-гидрокси-3,5-ди-грет-бутилбензиловый спирт; 3 — 4.4'-мети-
леибис(2,6-дн-трет-бутил)фенол; 4 — ди(4-гидрокси-3,5-дн-грет-бутилбензпловый) эфир.
16—1057
241
2,4-Диалкилфенолы в этой реакции проявляют низкую реакци-
онную способность (см? рис. 35), что, по-видимому, можно объ-
яснить меньшей стабильностью о-хиноидного переходного состоя-
ния фенолят-аниона по сравнению с п-хиноидным. Большая реак-
ционная способность /шра-положения по сравнению с орто-положе-
нием незамещенного фенола в реакции с формальдегидом в
условиях основного катализа была уже отмечена А. Е. Порай-Ко-
шицем.
Таким образом, выбор условий синтеза стабилизаторов, отно-
сящихся к метиленбисфенолам (каталитическим взаимодействием
диалкилфенолов с альдегидами), в значительной степени опреде-
ляется строением и расположением алкильных групп в молекуле
диалкилфенола, причем синтез метиленбисфенолов из 2,4-диалкил-
фенолов целесообразно вести в присутствии кислотных катализа-
торов, а синтез из 2,6-диалкилфенолов — в присутствии оснований.
Этот принцип лежит в основе промышленных процессов синтеза
стабилизаторов класса метиленбисфенолов.
Первые стабилизаторы — продукты взаимодействия а-иафтола и альдеги-
дов— были предложены в 1922 г. [19]. Позднее были предложены в качестве
стабилизаторов метиленбис(Р-нафтол) и гидроксидифеиил, но эти соединения
были мало эффективными: в отличие от современных стабилизаторов этого типа,
гидроксильные группы в иих не были экранированы.
В начале 40-х годов фирмой ICI были синтезированы и предложены для
защиты резин гораздо более эффективные соединения — 2,2'-этилиденбис (4,6-ди-
метилфенол) и 2)2'-бутилиденбнс(4)6-диметнлфепол):
ОН ОН он он
[20] В начале 50-х годов появился 2,2'-метиленбис(6-трет-бутил-4-метилфенол]
ОН ОН
(СН3)3С
сн3 сн3
один из самых эффективных стабилизаторов этого класса.
В настоящее время в промышленном и опытно-промышленном
масштабе выпускается примерно 25 стабилизаторов класса мети-
лепбисфеполов. Основной способ их получения — взаимодействие
диалкилфенолов с альдегидами. В качестве кислотного катализа-
тора используют серную кислоту и /i-толуолсульфокислоту; описа-
но применение муравьиной кислоты, являющейся и реакционной
средой и катализатором [21], бутилфосфорпой кислоты [22], ок-
сида серы [23], хлористого водорода [24], катионообменпых смол
242
[25]. Основными катализаторами обычно служат гидроксиды нат-
рия или калия. Формальдегид вводят в- реакцию в виде водного
раствора (формалин), триоксиметилена или параформа, а другие
альдегиды и кетоны —в форме индивидуальных соединений.
Первые процессы синтеза метиленбисфенолов в условиях кис-
лотного катализа вели в отсутствие растворителя. Диалкилфенол
и катализатор (концентрированная соляная кислота) перемеши-
вали, нагревали и вводили альдегид. Кислоту вводили в количест-
ве 20—50% от массы диалкилфенола, а в отдельных случаях —
в эквимольном количестве. После добавления альдегида нагревали
реакционную массу до 80—90 °C и перемешивали в течение 1—2 ч.
Продукт реакции образовывал ком, который растворяли затем в
органическом растворителе, нейтрализовали, отделяли от раствора
минеральной соли, промывали и выделяли кристаллизацией.
Более технологично проведение синтеза в среде органического
растворителя, например гептана. Исходный диалкилфенол раство-
ряют в органическом растворителе и вводят кислотный катализа-
тор. Последний, как правило, не растворяется в системе углеводо-
род— диалкилфенол, поэтому необходимо непрерывно и интенсив-
но перемешивать реакционную массу для равномерного распреде-
ления катализатора в ней. К реакционной массе постепенно добав-
ляют альдегид и для завершения процесса перемешивают массу
в течение нескольких часов при повышенной температуре. Обра-
зующийся метиленбисфепол, как правило, меньше растворим в уг-
леводородах, чем исходный диалкилфенол, и выпадает в осадок.
Его отделяют, промывают, высушивают и измельчают. Для повы-
шения выхода целевого продукта проводят дополнительное выде-
ление метиленбисфенола из фильтрата. Для повторного использо-
вания фильтрат нейтрализуют и перегоняют.
В конце 1950-х годов был предложен новый технологический
способ получения 2,2'-метиленбисфенолов [26]. По этому способу
ведут реакцию между альдегидом и 2,4-диалкилфенолом в водной
среде в присутствии кислотного катализатора и поверхностно-ак-
тивного вещества.
Диалкилфенол диспергируют в воде в присутствии небольших
добавок поверхностно-активного вещества, стабильного в кислот-
ной среде при нагревании (например, алкилбепзолсульфоната или
динафтилметансульфоната натрия). К полученной дисперсии до-
бавляют катализатор (серная кислота) и при перемешивании по-
степенно вводят альдегид. Образующийся 2,2/-метиленбисфенол
выпадает в осадок в кристаллическом виде; после охлаждения ре-
акционной массы его отфильтровывают, промывают, сушат и раз-
малывают. Для получения целевого продукта в форме мелких кри-
сталлов добавляют в реакционную массу 1,5—2,5% органического
растворителя, не смешивающегося с водой (гептан, хлорбензол, ке-
росин). Добавка растворителя облегчает диспергирование диал-
килфенола и позволяет получить более чистый и светлый целевой
продукт. Воду для диспергирования берут в количестве 200—
16*
243
250 мл на 1 моль диалкилфенола. При проведении процесса по
этому способу можно смешать все реагенты, нагреть до 90 °C и
перемешивать в течение 1,5—2 ч при этой температуре.
Конденсация в водной среде в присутствии поверхностно-актив-
ного вещества и органического растворителя имеет ряд техноло-
гических преимуществ перед другими способами и в течение мно-
гих лет являлась основным промышленным способом получения
важнейших 2,2'-метиленбис(4,6-диалкилфенолов). Увеличение объ-
емов производства этих стабилизаторов и возросшие требования к
охране окружающей среды привели, однако, к переоценке этого
процесса: образовывалось значительное количество сточных вод,
загрязненных поверхностно-активными веществами, алкилфепола-
ми, минеральными солями и органическими растворителями.
С целью создания многотоннажного производства 2,2'-метилен-
бис(4,6'-диалкилфенолов) в СССР разработан способ их получе-
ния, основанный на использовании ацеталя (например, метилаля)
[27] в качестве конденсирующего агента:
ОН ОН ОН
R' I О' I СН2 | П*
^О+(СН.О>1СН.^КХУ'ХХ '
R R R
Ацеталь является одновременно и растворителем; при этом обра-
зование сточных вод исключается.
Развитие производства дифенилолпропана открыло возмож-
ность синтеза стабилизаторов класса метиленбисфенолов на его
основе. Их получают каталитическим алкилированием дифенилол-
пропана стиролом или изобутиленом в присутствии серной [28],
щавелевой [29] кислоты или фенолята алюминия [30]. Стабилиза-
тор представляет собой смесь дифенилолпропанов разной степени
алкилирования (преимущественно три- и тетразамещенных).
2,2'-Метиленбис(6‘трет-бутил-4-метилфенол)
ОН ОН
(Щ)3С
СН3 СН3
Это соединение — один из самых эффективных стабилизаторов,
нашедших широкое применение в технике. Один из первых спо-
собов его получения — конденсация формальдегида с 2-трет-бутил-
244
4 метилфенолом в присутствии концентрированной соляной кисло-
ты [20]:
ОН он он
(СН,)/\1 (СН3)8С.,1 C«»/IX.C(CHS)S
2 (Q+HCHO^ UlZ
iis ii, сн3
2-трет-Бутил-4-метилфенол и катализатор загружают в реактор,
перемешивают, нагревают до образования дисперсии и при интен-
сивном перемешивании добавляют 36%-ный раствор формальдеги-
да. Затем поднимают температуру до 80—90 °C и перемешивают в
течение 1—1,5 ч; при этом реакционная масса постепенно загусте-
вает. Добавляют гептан, растворяют реакционную массу и нейтра-
лизуют ее содой или щелочью. Органический слой отделяют от вод-
ного и охлаждают до 0—5 °C. Выпавший осадок отфильтровывают
и получают технический продукт с т. пл. 123—125 °C. По такой
схеме был получен 2,2'-метиленбис(6-траг-бутил-4-метилфенол) с
т. пл. 130—131 °C и выходом 77% от теоретического [34].
Синтез удобнее вести в среде органического растворителя, на-
пример алифатического углеводорода [32]. 2-трет-Бутил-4-метил-
фенол растворяют в гептане, вводят катализатор (соляная или
серная кислота) и небольшое количество поверхностно-активного
вещества (0,2% от массы растворителя). К раствору при переме-
шивании постепенно добавляют 36%-ный раствор формальдегида,
выдерживают 2—3 ч при 50—60 °C, охлаждают смесь и отфильтро-
вывают выпавший осадок. Затем переносят его в сосуд с мешал-
кой, куда загружают воду, гептан (3—4% от количества воды) и
поверхностно-активное вещество (0,05%), перемешивают в тече-
ние 1 ч и фильтруют. Получают продукт с т. пл. 125—128°С и вы-
ходом «87%.
Вместо водного раствора формальдегида используют параформ,
а воду, выделяющуюся в результате реакции, отгоняют [41]. Смесь
2-трет-бутил-4-метилфенола, бензола, параформа и концентриро-
ванной соляной кислоты нагревают при перемешивании до 77—
80°C, отгоняют воду и бензол, добавляют 60%-ный изопропанол и
нагревают до 80 °C. Выдерживают смесь несколько часов, а затем
охлаждают до 10 °C, отфильтровывают выпавший осадок и про-
мывают его водой. Получают целевой продукт с т. пл. 131,5—
133,5 °C и выходом 99% от теоретического.
Большой интерес представляет проведение этой реакции в вод-
ной среде [26, 33]. 2-трет-Бутил-4-метилфенол диспергируют в во-
де, содержащей серную кислоту и поверхностно-активное вещест-
во, и добавляют к дисперсии водный раствор формальдегида. По
Другому варианту 2-трет-бутил-4-метилфенол постепенно добавля-
ют к смеси остальных реагентов [35]. При этом продукт реакции
245
выделяется в кристаллическом состоянии. Его отфильтровывают
промывают и сушат.
Переход от органической среды к водной упрощает технологи-
ческое оформление процесса и исключает регенерацию раствори-
теля. Однако для получения кристаллического продукта требуется
очень строго соблюдать технологический режим (в частности, по-
степенно и равномерно прибавлять 2-7’рег-бутил-4-метилфенол к
смеси остальных реагентов). Отклонения приводят к комкованию
реакционной массы.
Этот недостаток устраняется, если в реакционную массу вво-
дить 1—3% органического растворителя — бензина или гептана.
Добавление уже 1 % гептана позволяет получить кристаллический
продукт просто путем смешения всех реагентов и постепенного на-
гревания смеси до нужной температуры. Увеличение количества
гептана до 2—3% позволяет получить целевой продукт в виде мел-
кого порошка, но дальнейшее повышение количества растворите-
ля снижает выход продукта. Синтез ведут при 70—80 °C. При этом
образуется водная эмульсия 2-трет-бутил-4-метилфенола и раство-
рителя, не смешивающегося с водой. Введение поверхностно-актив-
ного вещества повышает стабильность этой эмульсии.
Формальдегид и катализатор находятся в водном растворе, и
реакция, по-видимому, осуществляется на границе раздела фаз.
«Эмульсионная конденсация» (этот термин для таких реакций
предложен в работе [36]) лежит в основе большинства промыш-
ленных процессов производства 2,2/-метиленбис(6-грет’-бутил-4-ме-
тилфенола) [36—38].
Технологическая схема получения этого стабилизатора эмуль-
сионной конденсацией 2-т’/?ег-бутил-4-метилфенола и формальдеги-
да представлена на рис. 37. В реактор 2 (сталыюй эмалированный
Формалин
Очистку
Рис. 37. Технологическая схема получения 2,2'-метиленбис(6-трет-бутил-4-мет1
фенола) конденсацией 2-трет-бутил-4-метилфеиола с формальдегидом:
1 — мерник; 2 — реактор; 3 — нейтрализатор; 4 — фильтр; 5 — сушилка.
246
аппарат с мешалкой и рубашкой для нагрева и охлаждения) за-
гружают воду, бензин, ПАВ (сульфонол), серную кислоту и 2-трет-
бутил-4-метилфенол. Смесь нагревают при перемешивании до
gO°C. При этом образуется водная эмульсия 2-т'рат’-бутил-4-метил-
фенола и бензина. К ней постепенно добавляют формалин и пере-
вешивают еще 1—2 ч. Реакционную массу нейтрализуют раствором
едкого натра до pH 14-2 и отфильтровывают выпавший в осадок
мелкокристаллический продукт. Осадок промывают, высушивают
и измельчают.
Эмульсионная конденсация может быть осуществлена и по не-
прерывной схеме [39] в каскаде из нескольких вертикальных ци-
линдрических аппаратов с мешалками и рубашками для нагрева
и охлаждения. В первый реактор непрерывно дозируют воду, сер-
ную кислоту, поверхностно-активное вещество, водный раствор
формальдегида и расплавленный 2-трет’-бутил-4-метилфенол и под-
держивают там температуру 90 °C. Реакционную массу перекачи-
вают затем во второй реактор, причем суммарное время пребыва-
ния в обоих аппаратах составляет »1 ч. Затем реакционная мас-
са непрерывно поступает в аппараты для охлаждения; при 50 °C
ее перекачивают на центрифугу.
Пасту готового продукта промывают, отжимают и передают на
сушку и измельчение.
Чтобы повысить дисперсность готового продукта, в реакцион-
ную массу добавляют 0,5—5,0% (масс.) диметилолкарбамида, ко-
торый в условиях реакции образует продукт автоконденсации с
молекулярной массой 200—1000. Благодаря этому целевой продукт
в течение длительного времени сохраняется в высокодиспергиро-
ванном жидком состоянии [40]. Неагломерирующийся чистый про-
дукт с тонким и равномерным гранулометрическим составом по-
лучают, проводя конденсацию в присутствии диоксида серы вместо
серной кислоты. В качестве защитного коллоида используют поли-
виниловый спирт. Диоксид серы после реакции легко удалить на-
греванием [41, 42].
Способ эмульсионной конденсации позволяет получить 2,2'-ме-
тиленбис(6-трет-бутил-4-метилфенол) достаточно высокой чистоты;
дополнительная очистка может быть осуществлена перекристал-
лизацией из уксусной кислоты. Для получения стабилизатора в
форме сферических гранул при 140 °C вводят расплав в воду при
комнатной температуре [43].
Все способы получения 2,2'-метиленбис(6-трет-бутил-4-метил-
фенола) в водной среде приводят к образованию значительного ко-
личества сточных вод, загрязненных органическими растворителя-
ми, поверхностно-активными веществами, минеральными солями
и кислотами. В связи с возрастающими требованиями по охране
окружающей среды большой интерес представляет разработанный
в СССР способ получения 2,2/-метиленбис(6-т/2в7’-бутил-4-метилфе-
нола) с использованием метилаля в качестве конденсирующего
агента и реакционной среды [27]. Метилаль — продукт кислотно-
247
катализируемого взаимодействия формальдегида и метанола
НСНО + 2СН,ОН —* (СН3О)2СНа
—HgU
является широко распространенным растворителем. В присутствии
кислотных катализаторов он вступает в реакцию с 2-трет-бутил-4-
метилфенолом, образуя соответствующий метиленбисфенол. Реак-
ция идет через несколько промежуточных стадий; в упрощенном
виде ее можно представить схемой:
ОН ОН ОН
Процесс ведут в избытке метилаля. Образующийся метанол отго-
няют и направляют на синтез метилаля. Принципиальная схема
процесса представлена на рис. 38.
Дальнейшее развитие этого способа — объединение в одной
стадии селективного деалкилирования 2,6-ди-трет-бутил-4-метил-
фенола в 2-трет-бутил-4-метилфенол и конденсации последнего с
Выделяющийся изобутилен возвращают в процесс алкилировали
фенола, а метанол — на синтез метилаля.
Рис. 38. Технологическая схема^И
получения 2,2'-метиленбис(6- 3
т'рет-бутил-4-метилфенола) кон- |
денсацией 2-трет-бутил-4-метил- д
фенола с метилалем:
1 — смеситель; 2 — аппарат для я
конденсации; 3 — пленочная колон- j
на. -Я
248
Метилальный способ получения 2,2'-метиленбис(6-трет-бутил-
4-метилфенола) является перспективным при создании многотон-
нажного процесса производства того стабилизатора.
Свойства и применение. 2,2'-Метиленбис(6-т’рет’-бутил-4-метил-
фенол) —белое кристаллическое вещество; т. пл. 133 °C; плотность
1040 кг/м3. Растворяется в ацетоне, диоксане, этилацетате, хлоро-
форме, этаноле, бензоле, гексапе.
Мало токсичен и разрешен в СССР для применения в полиме-
рах, контактирующих с пищевыми продуктами, питьевой водой,
косметическими средствами.
В СССР выпускается под торговой маркой НГ-2246 (т. пл.
2^128°C); другие его названия, принятые в СССР,— Бисалкофен
БП, Агидол 2. За рубежом выпускается рядом фирм под разными
названиями:
Торговое наименование Страна, фирма T. пл., °C
Antigen MDP Япония, Sumitomo 120
Antioxidant 2246 Англия, Anchor Chemical —
АО-235 США, Acron Chemical 125—130
АО-21-Т США, Acron Chemical 127,5
Bisoxol О Франция, CdF 130
САО-5 США, Aschland Chemical —
ОАО-14 США, Aschland Chemical 129,5
МВР-5 Франция, Organo Syntese —
МВР-5Р США, Aceto Chemical —.
Nocrac NS-6 Япония, Ouchi Shinko — •
Plastonox 2246 США, Cyanamid 121—128
Synox 5LT США, Neville Synthese —
Synox 5 (чистый) США, Neville Synthese —
Vilkanox В KF ФРГ, Bayer >124
2,2'-Метиленбис(6-трет-бутил-4-метилфепол) эффективно защи-
щает от старения многие типы полимеров и практически не влияет
на их окраску. Нелетуч. Разрешен для применения в изделиях,
контактирующих с пищевыми продуктами и биологическими сре-
дами.
Применяется для защиты от старения бутадиеновых каучуков ,
регулярного строения, бутадиен-стирольных, бутадиен-нитрильных,
этилен-пропиленовых, хлоропреновых и хлорбутилкаучука, а так-
же для защиты резин на основе натурального и синтетических кау-
чуков. Дезактивирует действие меди и других металлов перемен-
ной валентности как катализаторов окисления каучука. Эффектив-
но защищает от старения полиэтилен, полипропилен и волокна на
его основе, поливинилхлорид, полиоксиметилен, полистирол. Ис-
пользуется для защиты от старения полиацеталей, полиамидов, по-
лиуретанов, полиэтилентерефталата, политетрагидрофурана, поли-
акрилонитрила,-*Эпоксидных полимеров.
249
Является эффективной антиокислителыюй присадкой к нефте-
продуктам, моторным топливам и смазкам, работающим в широком
интервале температур. Применяется для сохранения многих пище-
вых продуктов — жиров, фуражных кормов, витамина А.
2,2'-Метиленбис(6-трет-бутил-4-этилфенол)
ОН ОН
(СНз)3С^^/»^^>С(СН.),
СаН6 СаНа
Его получают конденсацией 6-трет-бутил-4-этилфенола с форм-
альдегидом в присутствии кислотного катализатора:
ОН ОН ОН
(СНз^С^.! +нсно
2 и и XX
। । ।
с2н6 сан4 С2Нв
Конденсацию можно вести без растворителя, в органическом рас-
творителе и в водной среде.
В органическом растворителе ведут реакцию следующим обра-
зом [32]. В колбу помещают 6-трет-бутил-4-этилфенол, концент-
рированную соляную кислоту, поверхностно-активное вещество и
растворитель — гептан. При перемешивании добавляют по каплям
36%-ный формальдегид. После 2 ч перемешивания выпавший оса-
док продукта отфильтровывают и промывают водой, к которой до-
бавлено 4% гептана и 0,25% поверхностно-активного вещества, пе-
ремешивают и отфильтровывают. Получают 2,2'-метиленбис(6-
трет-бутил-4-этилфенол) с т. пл. 123 °C и выходом 75—80% от тео-
ретического. Поскольку синтез ведут в органическом растворителе,
а промывку— в водной среде, требуется регенерировать раство-
ритель и очищать промывные воды.
Процесс упрощается, если синтез ведут в водной среде [26],
т. е. методом эмульсионной конденсации. В стальной эмалирован-
ный реактор с мешалкой и рубашкой загружают воду, гептан (5%
от количества воды), алкилбензол сульфонат натрия (1%),
97%-ную серную кислоту (2,5%), 6-трет-бутил-4-этилфенол и 37%-
ный формальдегид перемешивают и в течение 5 ч выдерживают
при 90 °C. Затем реакционную массу охлаждают до 20—25 °C и
нейтрализуют раствором едкого натра. Осадок стабилизатора от-
фильтровывают; кристаллы отмывают от сульфата натрия и вы-
сушивают. Получают продукт с т. пл. 118—122 °C и выходом, близ-
ким к теоретическому. Эффективность такого стабилизатора до-
250
статочно высока. Перекристаллизацией можно поднять температу-
ру его плавления до 126—127 °C.
Предложен способ получения 2,2'-метиленбис(6-трет-бутил-4-
уетилфенола) взаимодействием б-трет-бутил-4-этилфенола с ме-
тилалем в присутствии кислотного катализатора [27]. Этот способ
исключает образование сточных вод и упрощает процесс.
Свойства и применение. 2,2'-Метиленбис(6-трет-бутил-4-этилфе-
нол)—белое кристаллическое вещество; т. пл. 127°С; плотность
]100 кг/м3. Растворим в бензоле, бензине, хлороформе, этаноле,
ацетоне; в воде не растворяется.
Выпускается рядом зарубежных фирм под разными торговыми
марками:
Торговое наименование Страна, фирма T. пл.. °C
Antioxidant 425 Англия, Anchor Chemical 119—125
Antioxidant ТВЕ-9 Франция, Organo Synthese —
Endox 22 США, Akron -—•
Naruxol 25 США,* Naugatuck •—
Plastonox 425 США, Cyanamid 117—123
2,2/-Метиленбис(6-трет-бутил-4-этилфенол) —эффективный не-
окрашивающий стабилизатор синтетических каучуков — силоксано-
вых, уретановых, хлоропреновых, бутадиеновых и бутилкаучука.
Защищает резины на основе натурального и синтетических каучу-
ков и латексов от термоокислителыюй и частично от фотодеструк-
ции; особенно эффективен в тонких латексных изделиях (пленки,
нити). Защищает от старения полиэтилен.
2,2'-Метиленбис(4,6-диметилфенол)
ОН ОН
^снз
СН3 СН3
Этот стабилизатор получают конденсацией 2,4-диметилфенола
с формальдегидом в присутствии кислотного катализатора (в ор-
ганическом растворителе или в водной среде):
ОН ОН ОН
Н3С .1 Я5 ДЗН3
4-нсно-
—н»о
СН3 СН3
2.4-Диметилфенол, Соляную кислоту и водный формальдегид, взя-
тые в эквимольных количествах, перемешивают и выдерживают
251
при 90 °C в течение 2 ч. К образующейся густой массе добавляют
толуол. Раствор нейтрализуют, промывают до нейтральной реак-
ции и охлаждают до 3—5 °C. Выпавший осадок продукта отфиль-
тровывают [20].
Свойства и применение. 2,2'-Метиленбис(4,6-диметилфенол) —
белое кристаллическое вещество; т. пл. 127 °C; плотность 1100 кг/м3.
Хорошо растворим в метаноле, этаноле и хлороформе, меньше —
в ацетоне и трихлорэтилене; в воде не растворяется.
Под торговой маркой Permanax BMP выпускается фирмой
Vulnax (Англия). Технический продукт представляет собой белый
порошок, обработанный небольшим количеством масла, чтобы уст-
ранить пыление; т. пл. 123—126 °C.
2,2/-Метиленбис(4,6-диметилфенол) — неокрашивающий стаби-
лизатор резин на основе натурального, бутадиен-стирольного и дру-
гих каучуков общего назначения, вулканизуемых серой. j
2,2'-Изобутилиденбис (4,6-диметилфенол)
ОН он "
Это — один из первых стабилизаторов группы бисфенолов. Его
получают конденсацией 2,4-диметилфенола с изомасляным альде-
Если в качестве фенольной компоненты используют не индивиду-
альный 2,4-диметилфенол, а его смесь с 2,5-диметилфенолом, то по-
лучают смесь бисфенолов — 2,2'-изобутилиденбис(4,6-диметилфено-
ла) и 4,4'-изобутилиденбис(3,6-диметилфенола).
Конденсацию 2,4-диметилфенола с изомасляным альдегидом
ведут в среде органического растворителя в присутствии серной
или соляной кислоты. В стальной эмалированный реактор загру-
жают бензин, кислоту, 2,4-диметилфенол и перемешивают при 25—
30 °C. Затем (тоже при интенсивном перемешивании) за 1,5—2 ч
добавляют из мерника изомасляный альдегид. После подачи аль-
дегида поднимают температуру до 60—65 °C, перемешивают 3—4 ч
для завершения процесса и охлаждают реакционную массу. Вы-
павший осадок продукта отфильтровывают, промывают водой до
нейтральной реакции, высушивают и размалывают. Фильтрат, со-
252
держащий кислоту, бензин и не вступивший в реакцию диметилфе-
нол, передают па переработку: отделяют кислотный слой, а орга-
нический слой нейтрализуют содой и затем отгоняют бензин, ко-
торый возвращают в процесс.
Свойства и применение. 2,2'-Изобутилиденбис(4,6-диметилфе-
нол) — белое кристаллическое вещество; т. пл. 162 °C; плотность
1120 кг/м3. Растворим в четыреххлористом углероде, хлористом
метилене, ацетоне и этаноле; плохо растворим в алифатических
углеводородах; в воде не растворяется.
Известен под название Vulkanox NKF (торговая марка фирмы
Bayer, ФРГ). Эффективный неокрашивающий стабилизатор белых
и цветных резин на основе натуральных и синтетических каучуков.
Несколько уступает такому высокоэффективному стабилизатору,
как 2,2/-метиленбис(6-трет-бутил-4-метилфенол), но совершенно не
влияет на цвет полимера.
В аналогичных условиях получают 2,2/-н-бутилиденбис (4,6-ди-
метилфенол) — продукт конденсации 2,4-диметилфенола с «-мас-
ляным альдегидом:
Это — белое кристаллическое вещество; т. пл. 129 °C.
'2.,2'-Метиленбис(4-метил-6-а-метилциклогексилфенол')
Этот стабилизатор, появившийся в промышленности в конце
50-х годов, получают конденсацией 4-метил-2-а-метилциклогексил-
фенола с формальдегидом или параформом в присутствии соляной
кислоты [44]:
При конденсации в водной среде в присутствии поверхностно-ак-
тивного вещества выход метиленбисфенола не превышал 68% от
теоретического; т. пл. продукта 116 °C. При конденсации в среде
253
Рис. 39. Технологическая схема получения 2,2'-метилеибис(4-метил-6-а-метилцик;4
логексилфенола): 1
2 — мерники; 3 — конденсатор; 4 — реактор конденсации; 5, 7 — друк-фильтры; 6 — кри-|
сталлизатор; 8 — сушилка; 9 — емкость для фильтрата. J
бензина получен продукт с т. пл. 133 °C и выходом 45% от теоре- Я
тического. I
В СССР разработан [25] способ его получения, позволяющий Я
проводить на катионообменной смоле КУ-2 обе стадии синтеза: J
алкилирование 4-метилфенола а-метилциклогексаном и конденса- I
цию полученного диалкилфепола с параформом. Конденсацию ве- 1
дут без растворителя; его вводят лишь перед отделением катали- 1
затора от образующегося продукта. Максимальный выход целевого 'т
продукта («70%) получен в интервале 140—150°С; повышение
температуры приводит к заметной термической деструкции катио-
нита. Оптимальное количество катализатора—10% (увеличение
снижает выход и повышает образование смолистых побочных про-
дуктов). Получают стабилизатор с т. пл. 135—138 °C.
Технологическая схема получения 2,2'-метиленбис(4-метил-6-а-
метилциклогексилфенола) представлена на рис. 39. В реактор 4
(стальной аппарат с мешалкой, рубашкой для нагрева и охлаж-
деиия и обратным холодильником) загружают из мерника 4-метил- 1
2-а-метилциклогексилфенол, параформ (избыток 8—10% от теоре- 1
тического) и катионит КУ-2 в Н-форме. Смесь перемешивают, на- 3
гревают до 140—150 °C и выдерживают 1 ч. Затем снижают темпе- j
ратуру до 80—90 °C, вводят бензин, перемешивают, нагревают до 1
95—100 °C и фильтруют горячий раствор на друк-фильтре 5. Ка- |
тионит промывают на фильтре горячим бензином и возвращают в |
процесс. Фильтрат и промывные воды объединяют и передают на 1
кристаллизацию в аппарат 6. Кристаллизацию ведут, постепенно /
охлаждая раствор до 0°С. Кристаллический осадок отфильтро- 1
вывают, промывают холодным бензином, высушивают и измель- л
чают. j
Свойства и применение. 2,2'-Метиленбис(4-метил-6-а-метилцик- ]
логексилфенол)—белое кристаллическое вещество; т. пл. 140 °C; Ч
плотность 1170 кг/м3. Растворим в бензоле, толуоле, гептане, гек- )
254
сане, бензине, ацетоне, хлороформе, четыреххлористом углероде и
этаноле; в воде не растворяется.
Разрешен в ряде стран для применения в полимерах, контакти-
рующих с пищевыми продуктами.
Выпускается фирмой Vulnax Internation (Англия) под маркой
permanax WSP и фирмой ICI (Англия) под маркой Nonox WSP.
Неокрашивающий стабилизатор резиновых и латексных изде-
лий, полиолефинов, ударопрочного полистирола, полиэфиров.
4,4'-Метиленбис(2,6-ди-трет-бутилфенол)
Этот стабилизатор получают конденсацией 2,6-ди-трег-бутилфе-
нола с формальдегидом в присутствии основных катализаторов:
(CHS)SC
2 НО—
(СН,)3С
+НСНО
-Н20 '
2,6-Ди-т-рет-бутилфепол и формалин растворяют в безводном эта-
ноле, пропускают азот, нагревают до 50—55 °C и постепенно за-
гружают едкий натр. При охлаждении продукт выпадает в осадок
в виде блестящих бесцветных кристаллов с т. пл. 154 °C; выход
90% от теоретического [46].
Удобнее вести этот синтез, растворяя в этаноле 2,6-ди-трат-бу-
тилфенол и катализатор и постепенно вводя водный раствор форм-
альдегида [13, 47]. Едкое кали растворяют в изопропаноле и в
токе азота в раствор загружают 2,6-ди-трет-бутилфенол. Нагрева-
ют до 30°C, добавляют при перемешивании 37%-ный раствор
формальдегида, поднимают температуру до 60 °C, перемешивают
в течение несколько часов и охлаждают реакционную массу. От-
фильтровывают выпавший осадок, промывают его изопропанолом,
подкисленным НС1, охлаждают и высушивают. Получают продукт
с т. пл. 154—155 °C и выходом 87% от теоретического [47].
Этот стабилизатор образуется с высоким выходом при нагрева-
нии *2,б-ди-т’рет’-бутилфенола с параформом в концентрированной
муравьиной или уксусной кислоте [48]. Реакционную смесь нагре-
вают до кипения и кипятят с обратным холодильником 10 минут.
Выпавший осадок продукта отфильтровывают, а фильтрат упари-
вают при пониженном давлении и добавляют бензол. Выпавший
осадок еще раз отфильтровывают и вместе с первой порцией про-
дукта перекристаллизовывают из 95%-ного этанола. Получают
продукт с т. пл. 153,5—154,5 °C и выходом 96% от теоретического.
255
Описаны еще два способа получения стабилизатора. Осущест-
вляют взаимодействие 2,6-ди-трет-бутилфенола с 4-гидрокси-3,5-ди-
трет-бутилбензиловым спиртом. [49]:
Реакцию ведут в среде вторичного или третичного спирта в присут-
ствии щелочи. Едкое кали растворяют в трет-бутаноле, добавляют
2,6-ди-трет-бутилфепол и 4-гидрокси-3,5-ди-трет-бутилбензиловый
спирт, нагревают до 85 °C и выдерживают 1 ч. Охлаждают смесь до
20—25 °C, фильтруют выпавший осадок продукта, промывают его
и высушивают. Выход 98,5% от теоретического; т. пл. 151,5—
153,5 °C.
Стабилизатор образуется почти с теоретическим выходом и при
взаимодействии симметричного эфира 4-гидрокси-3,5-ди-трет-бу-
тилбензилового спирта с 2,6-ди-трет-бутилфенолом в присутствии
щелочи [50]:
(СН3)3С
НО—
(СН3)3С
С(СН3)3
-он
--->
—Н2О
С(СН3)3
(СН3)3С. С(СН3)3
---* НО-^у-СН2—он
(СН3)3с/ \С(СН3)3
Исходные соединения и каталитическое количество КОН растворя-
ют в безводном трет-бутаноле, нагревают и выдерживают 5 мин
при 80 °C. Затем охлаждают, добавляют воду и выпавший осадок
отфильтровывают.
Наибольший технический интерес представляет получение ста-
билизатора непосредственно из 2,6-ди-трет-бутилфенола в присут-
ствии основного катализатора. Эта реакция положена в основу про-
мышленного метода получения [45, 51, 52].
Технологическая схема получения 4,4'-метиленбис(2,6-ди-трет-
бутилфенола) представлена на рис. 40. В реактор 2 (вертикальный
256
Рис. 40. Технологическая схема получения 4,4'-метилеибис(2,6-ди-грег-бутилфе-
вола):
1 — мерннк; 2 — реактор-конденсатор; 3 — друк-фильтр; 4— емкость; 5 — кристаллизатор; б—*
фильтр.
цилиндрический аппарат с мешалкой, рубашкой, барботером для
подачи азота и обратным холодильником) загружают изопропанол,
каталитическое количество едкого натра и при пропускании азота
подают 2,6-ди-трет-бутилфенол. Содержимое реактора нагревают
до 30 °C и при этой температуре и непрерывном перемешивании и
пропускании азота прибавляют из мерника 1 формалин. Затем ре-
* акционную массу нагревают до 50—60 °C, выдерживают 1,5—2 ч,
охлаждают до 20—25 °C и передают на друк-фильтр 3. Кристалли-
ческий осадок продукта отфильтровывают, промывают изопропано-
лом и передают на перекристаллизацию. Фильтрат и промывные
воды направляют на регенерацию. Перекристаллизацию ведут в
аппарате 5 с мешалкой и рубашкой для охлаждения рассолом. Ту-
да загружают пасту технического продукта и бензин. Полученный
раствор постепенно охлаждают до минус 10 °C. Выпавший в оса-
док кристаллический продукт отфильтровывают, промывают ох-
лажденным бензином, отжимают и сушат. Бензиновый фильтрат и
промывные воды направляют па регенерацию. Выход перекристал-
лизованного продукта 70—75% от теоретического.
Свойства и применение. 4,4'-Метиленбис(2,6-ди-7-рет-бутилфе-
нскчг) — светло-желтое кристаллическое вещество; т. пл. 155—
155,5°C; т. кип. 289°C при 5,34 кПа; плотность 990 кг/м3. Раство-
рим в бензоле, толуоле, ацетоне, метилэтилкетоне; ограниченно
растворим в спирте; в воде не растворяется.
Мало токсичен. В СССР разрешен для применения в полиме-
рах, контактирующих с пищевыми продуктами. ПДК = 10 мг/м3.
В СССР выпускается под техническим названием ЛЗ-МБ-L
В США его вырабатывают под различными торговыми марками:.
17—1057
257
Торговое наименование Фирма T. пл., °C
Ethyl Antioxidant 702 Ethyl 154
Ethyl Antioxidant 728 Ethyl Жидкость
lonox 220 Shell 155
Стабилизатор синтетических каучуков, резин на основе нату-
рального и синтетического каучуков, полиолефинов. Окрашивает
резины в желтый цвет.
4,4'-Бутилиденбис(6-трет-бутил-3-метилфенол)
Этот стабилизатор получают конденсацией 6-трет-бутил-З-ме-
тилфенола с масляным альдегидом [66] в присутствии соляной
кислоты:
В колбу с мешалкой и обратным холодильником загружают диал-
килфенол, масляный альдегид и соляную кислоту, перемешивают и
нагревают в течение нескольких часов на кипящей водяной бане.
Реакционную массу охлаждают и экстрагируют продукт гептаном.
Органический слой промывают раствором соды и после охлажде-
ния фильтруют выпавшие кристаллы. После перекристаллизации
из смеси бензол + гептан получают продукт с т. пл. 207,5—208 °C
[67].
Реакцию проводят и в среде растворителя, например хлористо-
го метилена. В качестве катализатора используют серную кисло-
ту. Некоторый избыток альдегида повышает выход бисфенола, од-
нако избыток сверх 30% ведет к образованию побочных продуктов
неустановленного строения [68].
Если в качестве растворителя применять спирт, можно сокра-
тить продолжительность реакции. 6-трет-Бутил-З-метилфенол рас-
творяют в метаноле, вводят кислоту, нагревают и постепенно при-
бавляют масляный альдегид. По окончании реакции нейтрализу-
ют кислоту, экстрагируют продукт гептаном, кристаллизуют и
фильтруют. Получают белые кристаллы с т. пл. 212—213°С
[69]!.
258
Конденсация в спиртовом растворе положена в основу про-
мышленного получения стабилизатора. В реактор (стальной эма-
лированный аппарат с мешалкой, рубашкой для нагрева и охлаж-
дения и обратным холодильником) загружают б-траг-бутил-З-ме-
тилфенол, метанол и концентрированную соляную кислоту. Смесь
перемешивают и нагревают до 80—82 °C. При этой температуре
из мерника постепенно в течение I ч прибавляют масляный аль-
дегид и перемешивают еще 1 ч для завершения реакции. Затем
охлаждают массу до 30°C, обрабатывают ее последовательно во-
дой и раствором соды для удаления кислоты. Отделяют водный
слой, а к органическому добавляют бензин и охлаждают до минус
5 °C. Выпавший осадок кристаллического продукта отфильтровы-
вают, промывают бензином и передают на сушку и измельчение.
Свойства и применение. 4,4'-Бутилиденбис(6-трет-бутил-3-ме-
тилфенол) — белое кристаллическое вещество; т. пл. 2!3°С. Рас-
творим в метаноле, этаноле, ацетоне и диэтиловом эфире; в воде,
не растворяется.
Фирма Monsanto (США) выпускает его под торговой маркой
Santowhite Powder, фирма Yoshitomi (Япония)—под маркой Yos-
hinox ВВ.
Применяется для защиты от старения синтетических каучуков
и резин на основе натурального и синтетического каучуков. Защи-
щает полиэтилен, полипропилен, ударопрочный полистирол и по-
лиацетали от термо- и фотодеструкции. Стабилизатор полиамида и
полиамидных волокон.
ЛИТЕРАТУРА
Г. Кормак В. В. Методы высокомолекулярной органической химии. М., Изд.
АН СССР, 1953. 341 с.
2. Hultzsch К. Chemie der Phenolharze. Springer Verlag, Berlin, 1950.
3. Martin R. The Chemistry of Phenolic Resins. Chapman Holl, New York — Lon-
don, 1956.
4. Hultzsch K. — Angew. Chemie, 1948, В. A60, S. 179.
5. Leibnitz E., Naumann K. — Chem. Techn. (Berlin), 1951, B. 3, S. 5.
6. Reese I. — Kunststoffe, 1955, B. 45, S. 137.
7. Martin R. — J. Am. Chem. Soc., 1951, v. 73, p. 3952; пат. 2579329, 1951 г.
8. Ziegler E., Zigenner C. — Kunststoffe, 1949, B. 39, S. 191.
9. Bartel R., Plastica (Holland), 1949, B. 2, S. 247.
10. Freeman I., Lewis C. — J. Am. Chem. Soc., 1954, v. 76, p. 2080.
11. Гурвич Я. А., Маслова И. П. — В кн.: Синтез и исследование эффективности
химикатов для полимерных материалов. Тамбов, 1969. Вып. 2, с. 10.
12. Гринберг А. А., Гурвич Я. А., Фришман Т. А. — В ки.: Синтез и исследование
'эффективности химикатов для полимерных материалов. Тамбов, 1969, вып. 2,
с. 383.
13. Coffield Т. е. а. — J. Am. Chem. Soc., 1957, v. 79, р. 5019.
14. Никифоров Г. А., Ершов В. В. — Успехи химии, 1970, т. 39 с. 1369.
15. Гурвич Я- А., Старикова О. Ф., Стыскин Е. Л. — В кн.: Тезисы I Всесоюзного
симпозиума по органическому синтезу. М., Изд. АН СССР, 1974, с. 28.
16. А. с. 450791, 1974 г. (СССР).
17. А. с. 226603, 1968 г. (СССР).
18. Ethyl, пат. 2807653, 1957 г. (США).
19. Williams G. Е. — IRI Trans., 1956, v. 32, № 1, р. 43.
17'
259
20. Cyanamid, пат. 2538355, 1955 г. (США).
21. Пат. 2052116, 1971 г. (Франция).
22. Rubber, пат. 3318961; 3318962, 1967 г. (США).
23. Пат. 155266, 1969 г. (ВНР). <i
24. Садыхов Ш. Г. и др. — Азерб. хим. жури., 1972, № 4, с. Ill; Sumitomo, пат.,}
2350, 1967 г. (Япония). ;
25. Бугакова Л. П„ Медведев А. И., Михайлов В. В. — В ки.: Синтез и исследо--
вание эффективности химикатов-добавок для полимерных материалов. Там-.;
бов, 1969. Вып. 2, с. 393.
26. Cyanamid, пат. 2796445, 1957 г. (США).
27. Пат. 2335481, 1977 г. (Франция).
28. Monsanto, пат. 1184424, 1970 г. (Англия).
29. А. с. 184870, 1966 г. (СССР).
30. Пат. 153757, 1974 г. (ЧССР).
31. Верховская 3. И. Дифенилолпропан. М., Химия, 1971, 196 с.
32. Cyanamid, пат. 2834752, 1958 г. (США).
33. Cyanamid, пат. 2675366, 1954 г. (США).
34. Исагулянц В. И., Фаворская И. А.— В ки.: Труды МИНХ и ГП, 1960. Вып. 28,
с. 56.
35. Cyanamid, пат. 2796444, 1957 г. (США).
36. Гринберг А. А., Гурвич Я- А., Фришман Т. А.— Хим. пром-сть, 1967, № 5,
с. 19.
37. Белов И. С., Исагулянц В. И. — ЖПХ, 1964, т. 37, р. 1860.
38. Гусев В. К- и др. — Нефтепереработка и нефтехимия, 1969, № 3, с. 29.
39. Пат. 114369, 1965 г. (ЧССР).
40. Cyanamid, пат. 2968630, 1961 Г. (США).
41. Sumitomo, пат. 2350, 1967 г. (Япония).
42. Wein Г., Ocskay G. — Kem. Kozl., 1974, v. 42, р. 139.
43. loshitomi, пат. 14-109355, 1974 г. (Япония).
44. ICI, пат. 728290, 1952 г. (Англия).
45. Гушанская П. Г. и др. — В кн.: Синтез и исследование эффективности хими-
катов-добавок для полимерных материалов. Тамбов. 1969. Вып. 2, с. 351.
46. Kharasch М. S., /oshi В. S. —Л. Org. Chem., 1957, v. 92, р. 1435.
47. Ethyl, пат. 3175010, 1965 г. (США).
48. S. A. Prod. Chimiq., пат. 1331447, 1963 г. (Франция).
49. Shell, пат. 3091645, 1963 г. (США).
50. Shell, пат. 3053904, 1962 г. (США).
51. Афанасьев И. Д. и др. — В кн.: Присадки к маслам. М., Химия, 1966, с. 129.
52. Гушанская И. Г. и др. — В кн.: Совершенствование технологии производства ।
присадок. Киев, Наукова думка. 1976, с. 50.
53. Monsanto, пат. 2816945, 1957 г. (США).,
54. Firestone, пат. 2822404, 1958 г. (США).
55. Yoshitomi, пат. 40-16538, 1965 г. (Япония).-
56. Monsanto, пат. 2831897, 1958 г. (США).
3.4. КОНДЕНСАЦИЯ АЛКИЛФЕНОЛОВ
С ТИОХЛОРИДАМИ. СИНТЕЗ ТИОБИСФЕНОЛОВ
Продукт взаимодействия фенола и двухлористой серы — 4,4'-тиобисфенол —
был впервые получен в 1887 г. в среде органического растворителя:
2НО—Ч 4- SCla 2^ НО—S—/~\-ОН
Эта реакция 40 лет спустя была использована для синтеза сульфидов алкил-
фенолов, обладающих свойствами гербицидов и инсектицидов, а еще через не-
сколько лет — для синтеза первых стабилизаторов класса тиобисфеиолов. В ос-
нове этих способов лежит некаталитическое взаимодействие диалкилфеиола с дву-
хлористой серой в среде апротонного растворителя (галогенуглеводороды, али-
фатические и ароматические углеводороды, сероуглерод).
260
При взаимодействии с алкилфенолами атом серы вступает в
орто- или /гара-положение к гидроксилу, образуя соответственно
2,2'- или 4,4'-тиобисфенолы:
ОН он
Вступление атома S в места наибольшей электронной плотности
ароматического ядра и катионоидный характер серы в молекуле
двухлористой серы
(21-о,ов4 s+0»128 Cl-0.064
позволяют рассматривать взаимодействие фенолов и двухлористой
серы как реакцию электрофильного замещения [1]. Взаимодейст-
вие двухлористой серы и ароматического углеводорода протекает
только в присутствии катализатора, например хлористого алюми-
ния. Если в ароматическом кольце есть электронодонорные заме-
стители, реакция протекает в присутствии более мягких катализа-
торов (железо, хлориды железа) [1]. 2,4-Диал кил фенолы и 2-ал-
кил-4-хлорфенолы вступают в реакцию с двухлористой серой без
катализатора [2—4]. Обычные катализаторы Фриделя — Крафтса
промотируют хлорирование и в качестве катализатора этого про-
цесса рекомендован хлорид бора.
Большое практическое значение для защиты полимеров приоб-
рели стабилизаторы, получаемые взаимодействием 2,4- и 2,5-диал-
килфенолов [5—8], получаемых соответственно из 4- и 3-метилфе-
нола с двухлористой серой. Синтез тиобисфенолов из пространст-
венно затрудненных 2,6-диалкилфенолов идет с низким выходом.
Так, при взаимодействии 2,6-ди-т’рет-бутилфенола с двухлористой
серой образуется 4,4/-тиобис(2,6-ди-трв7’-бутилфенол) с выходом
3,5-5% [4, 9]:
(СН3)3С. (СН3)3С\ _______ ,С(СН3)3
* HO-QS
(СН3)3с/ (СН3)8С/ ^С(СН3)3
В де же время изомерный 2,4-ди-грет-бутилфенол легко образует
соответствующий тиобисфенол с выходом 80—85%: _
ОН ОН ОН
261
Проведенное одним из авторов этой книги исследование показа-
ло, что реакционная способность диалкилфенолов существенно за-
висит от строения и расположения алкильных групп. На рис. 41
представлены кинетические кривые образования тиобисфенолов из
различных диалкилфенолов. Видно, что пространственно затруд-
ненные 2,6-диалкилфенолы (кривые 6 и 7) практически не обра-
зуют тиобисфенолов в тех условиях, в которых 2,4-диалкилфенолы
образуют их с выходом 70—90%. Следовательно, диалкилфенолы,
проявившие низкую реакционную способность в кислотнокатали-
зируемых реакциях с альдегидами и при каталитическом алкили-
ровании олефинами [10], обладают низкой реакционной способ-
ностью и в реакциях с двухлористой серой.
Для пространственно затрудненных 2,6-диалкилфенолов, в част-
ности для 2,6-ди-трет-бутилфенола, описан ряд реакций электро-
фильного замещения: нитрование [11], галогенирование [12], азо-
сочетание [13]. В то же время известны примеры разрыва суль-
фидной связи в некоторых дигидроксидифенил- и дигидрокспди-
нафтилсульфидах указанными электрофилами с образованием со-
ответственно нитро-, галоген- и азофенолов и нитро-, галоген- и
азонафтолов [14], например:
Это показывает, что атакующая частица двухлористой серы яв-
ляется менее активным электрофилом, чем нитроний-, бромоний-
и диазоний-катион.
С другой стороны, ход реакции зависит от нуклеофильной ак-
тивности ароматического соединения (бензол в обычных условиях
не взаимодействует с двухлористой серой). Повышение степени
пространственного затруднения фенольного гидроксила снижает
реакционную способность 2,6-диалкилфенола при нитровании
[11], т. е. в реакции электрофильного замещения сильным элект-
рофилом. Можно предположить, что различие в реакционной спо-
собности 2,4- и 2,6-диалкилфенолов в реакциях с двухлористой се-
рой и низкая реакционная способность пространственно затруднен-
ных 2,6-диалкилфенолов связаны с неодинаковой степенью прост-
ранственного затруднения НО-групп: увеличение пространственно-
го затруднения снижает нуклеофильную активность ядра.
Это подтверждается резким возрастанием реакционной способ-
ности при переходе от 2,6-ди-трет-бутилфенола к 2,6-дп-трет-бутил*
феноляту натрия. Известно, что при переходе от фенола .к
феноляту заметно возрастает нуклеофильная активность аромати-
ческого ядра. 2,6-Ди-трет-бутилфенолят натрия, приготовленный
взаимодействием 2,6-ди-трет-бутилфенола с метилатом натрия, рас-
творяли в тетрагидрофуране и при 40 °C добавляли двухлори^тую
серу. На рис. 42 приведены кинетические кривые образования
26?
Рис. 42. Кинетика образования 4,4'-тиобис(6-трет-бутил-2-метилфенола) из 2,6-ди-
трет-бутилфенолята натрия (/) и из 2,6-ди-трет-бутилфенола (2).
Растворитель — тетрагидрофуран; температура 40 °C.
♦
Рис. 41. Кинетика образования тиобисфенолов из диалкилфенолов разного строе-
ния (40 °C, растворитель — ССЦ):
/ — из 2,4-диметилфенола; 2 — из 2-трет-бутил-4-метилфенола; 3—из 2,4-ди-трет-бутилфено-
ла; 4 — из 4-7рет-бутил-2-а-Метилбензилфенола; 5 — из 2,6-диметилфеиола; 6 — из 2-трет-бу-
тил-6-а-метилбензилфенола; 7 — из 2,6-ди-трет-бутилфеиола.
4,4'-тиобис (6-трет-бутил-2-метилфенола) -из 2,6-ди-трет-бутилфено-
лята натрия и из 2,6-ди-трет-бутилфенола. Видно, что 2,6-ди-трет-
бутилфенолят натрия образует тиобисфенол с выходом «50%),
причем реакция проходит в основном в первые 10—15 мин [15].
Добавляя каталитическое количество порошкообразного железа,
удается поднять выход целевого продукта до 60—65%.
При взаимодействии 2,6-ди-трет-бутилфенола с двухлористой
сёрой наряду с монотиопроизводным образуются ди- и политио-
производные. Например, при взаимодействии 0,1 моль 2,6-ди-трет-
бутилфенола с 0,066 моль двухлористой серы в присутствии ката-
литических количеств железа (растворитель — четыреххлористый
углерод, 20 °C, длительность 18 ч) после отгонки растворителя по-
лучают смесь 23% моно-, 11% ди-, 19% три- и 28% тетратиопроиз-
водных 2,6-ди-трет-бутилфенола. Длительным нагреванием со спир-
товой щелочью удается, перевести ди- и политиопроизводные в
4,4'-тиобис(2,6-ди-трет-бутилфенол) с выходом «80% [16].
Описано получение 4,4'-тиобис(2,6-ди-трет-бутилфенола) взаи-
модействием 2,6-ди-трет-бутилфенола со свободной серой в среде
органического растворителя в присутствии щелочи, взятой в экви-
мольном количестве к 2,6-ди-трет-бутилфенолу. Получают смесь
моно- и дитиопроизводных 2,6-ди-трет-бутилфенола. Целевой про-
дукт.можно выделить кристаллизацией с выходом «60%) [17].
Невысокая селективность образования 4,4'-тиобис (2,6-ди-трет-
бутилфенола) и сложность его выделения из реакционной массы
затрудняют разработку промышленного способа получения. Более
селективно протекает, по-видимому, реакция между 2,6-ди-трет-бу-
263
тилфенола S2CI2 и образуется дитиопроизводное:
(СН3)3С. (СН3)3С.__ С(СН3)3
/ 'К +S2CI2 /
2 НО—" \ НО—" S—S— —ОН
\ / —2НС1
(СН3)3С/ (СН3)3С С(СН3)3
При проведении этой реакции в присутствии каталитических коли
честв иода выход дитиопроизводного достигает 93% [18], а выхо
монотиопроизводного 4,8%.
4,4/-Дитиобис(2,6-ди-трет,-бутилфенол) окрашивает полимеры
желтый цвет и не нашел еще широкого применения как стабилиза
тор. Интересна возможность получения из него труднодоступны
меркаптофенолов с экранированной гидроксильной группой:
(СН3)3С С(СН3)3 (СН3)3С
Zn; 4-2Н
—ОН ---------> 2 НО-
но—
,-SH
(СН3)3С С(СН3)3 (СН3)3(У
При взаимодействии тйохлоридов с диалкилфенолами наряду с об
разеванием сульфидов протекает побочная реакция хлорирования
Так, при взаимодействии 2,6-ди-трет-бутилфенола (1,5 моль) с дву
хлористой серой (1 моль) при 40 °C в присутствии каталитическо
го количества иода после отгонки (при пониженном давлении) ле
тучих веществ получают продукт, содержащий 7,6% серы и 2,7’J
хлора и рекомендуемый как антиокислительная присадка к топли
вам и маслам [19].
При сплавлении алкилфенолов со свободной серой в присутст
вии небольших количеств едкого натра при 150—170 °C образуете:
смесь моно- и дитиобисфенолов, а также соединений, в молекул
которых сульфидным мостиком связаны три и более ароматически:
ядра [20]. Продукт реакции (хрупкая твердая смола; т. затв. 50
60 °C) обладает свойствами антиоксиданта. Использование сер:
вместо тиохлоридов в качестве сульфидирующего агента при син
тезе тиобисфенолов могло бы иметь большое техническое значе
ние: сера доступна, дешева, стабильна при хранении и безопасн
в обращении. Однако пока нет способов получать индивидуальны
тиобисфенолы сплавлением серы с алкилфенолами.
Другой путь синтеза тиобисфенолов без применения двухло
ристой серы — взаимодействие производных 4-хлор- или 4-бромфе
нола с сульфидом натрия [21]:
(СН3)3С
(СН3)3С
+Na2S
2 НО— — Вг ——>
—2NaBr
(СН3)3С
но-
С(СН3)3
он
(СН3)3С ХС(СН3)3
Реакцию ведут в водно-спиртовой среде при 75—200 °C и «2 МПа;
на 1 моль галогенфенола берут 0,5—0,8 моль сульфида натрия.
264
Этот способ мог иметь практическое значение до разработки про-
мышленного процесса орто-алкилирования, когда единственным пу-
тем получения 2,6-ди-трет-бутилфенола было алкилирование
4-хлор- или 4-бромфенола изобутиленом с последующим дегалоге-
нированием 4-галоген-2,6-ди-трет-бутилфенола.
В настоящее время основной промышленный способ получения
стабилизаторов группы тиобисфенолов — взаимодействие диалкил-
фенолов с двухлористой серой.
Важная стадия производства — получение двухлористой серы
при взаимодействии серы с хлором: S + С12—>5С1г. Газообразный
хлор пропускают через колонну с измельченной серой. Жидкая SC12
стекает вниз навстречу потоку хлора, что обеспечивает полноту хло-
рирования. Удобнее проводить эту реакцию в растворителе, напри-
мер в четыреххлористом углероде, а добавление иода ускоряет про-
цесс. В реактор загружают СС14, суспендируют в нем тонкоизмель-
ченную серу, добавляют иод и при 0±5°С и интенсивном переме-
шивании пропускают сухой хлор. Получают прозрачный раствор
SCI2 в СС14, который можно непосредственно использовать в синте-
зе стабилизаторов [22]. Другой способ получения SC12 — хлориро-
вание однохлористой серы.
S2C12 + С1а--> 2SC12
Эта реакция медленно протекает при комнатной температуре и за-
метно ускоряется при охлаждении до 0 — минус 5 °C. Хлорирова-
ние ведут в присутствии каталитических количеств иода, древесно-
го угля или пентахлорида сурьмы. S2CI2 является промышленным
продуктом — в отечественном производстве стабилизаторов принят
последний способ получения SCU.
Другой способ получения стабилизаторов группы тиобисфено-
лов — алкилирование 4,4'-тиобисфенола олефинами и арилолефина-
ми, в частности изобутиленом и стиролом [23—26]:
265
Реакцию ведут в среде толуола в присутствии обычных катали
заторов алкилирования — серной кислоты, арилсульфокислот, фе
нолята алюминия. 4,4'-Тиобисфенол суспендируют в толуоле, вво
дят катализатор и постепенно подают изобутилен. По мере алии
лирования тиобисфенол постепенно растворяется, а в конце прр
цесса суспензия полностью превращается в раствор. Удаляют ка-
тализатор, отгоняют растворитель и получают стабилизатор в виде
коричневой смолы (т. затв. 35—40°С). Смола представляет собой
смесь тиобисфенолов разной степени алкилирования; обычно в мо-
лекулу 4,4/-тиобисфенола вступают от 2 до 4 алкильных групп. .
Если в качестве катализатора использовать щавелевую кисло-
ту, синтез упрощается и применение растворителей исключаете»
[25]. В реактор загружают 4,4'-тиобисфенол и щавелевую кислоту
нагревают до 130 °C и при этой температуре и интенсивном пере-
мешивании постепенно вводят стирол. Реакционную массу переме-
шивают при 140—143 °C и затем отгоняют не вошедший в реакцию
стирол. В кубе остается светло-желтая прозрачная смола; это и есть
стабилизатор.
Существует ряд фенольных стабилизаторов, содержащих се-
ру,— в них атом серы связан с ароматическим ядром не непосред-
ственно, а через метиленовый мостик. При нагревании 2,6-ди-трет-
бутилфенола с формалином, диметиламином и сероуглеродом г
среде этанола в течение 3 ч образуется 4-гидрокси-3,5-ди-трет-бу-
тилбензил-М,М-диметилдитиокарбамат [27]:
(СН3)3С
HO— +HCHO-+NH(CH3)24-CS2
(СН3)3С
(СН3)3С
но-
СН2—S—С—N(CH3)2
II
s
(СН3)3О
Это соединение является довольно эффективным стабилизатором, а
при нагревании со щелочью и этантиолом-1,2 с высоким выходом
образует бисфенол с двумя атомами серы в мостике [27]:
(СН3)3С
2 НО— —CHj—S—С—N(CH3)2 + HS—С2Н4—SH + 2NaOH ——*
|| — 2HjO
s
(СН3)3С
(СН3)3С
С(СН3)3
НО— —CH2—S—CaH4—S—CH2—. ОН + 2(CH3)aNC—SNa
II
s
С(СН3)3
(СН3)3С
266
Аналогичным образом при взаимодействии этого карбамата с
сульфидом натрия в среде метанола получают с выходом, близким
к количественному, бис(4-гидрокси-3,5-ди-трет-бутилбензил) суль-
фид [28]:
(СН3)3С.
2 НО—/ СН2—S—CN(CH3)2 + Na2S ----►
S
(СН3)3С/ 5
+ 2(CH3)2NC-SNa
II
S
Большой интерес представляет получение стабилизатора такого
типа непосредственно из 2,6-ди-трет-бутилфенола — бис-4-гидрок-
си-3,5-ди-трет-бутилбензилсульфид может быть получен взаимо-
действием 2,6-ди-трет-бутилфенола с формальдегидом и сульфи-
дом натрия [29]:
Все эти реакции протекают в сравнительно мягких условиях с вы-
соким выходом целевых продуктов, однако широкого промышлен-
ного применения они до сих пор не нашли. Из различных способов
синтеза фенольных стабилизаторов, содержащих серу, наиболь-
шее промышленное значение имеет взаимодействие алкилфенолов
с тиохлоридами, в основном с двухлористой серой.
2,2'-Тиобис (6-трет-бутил-4-метилфенол)
ОН ОН
:н3 сн3
Этот стабилизатор — один из важнейших в группе тиобисфено-
лов. Его получают взаимодействием 2-трет-бутил-4-метилфенола с
двухлористой серой [30] в среде органического растворителя:
ОН ОН ОН
267
2-трет-Бутил-4-метилфенол растворяют в четыреххлористом угле-
роде и к раствору в течение 1 ч добавляют раствор двухлористой
серы в этом же растворителе; мольное соотношение диалкилфено-
ла и двухлористой серы равно 2: 1; температура реакции 15—20°C.
После прибавления двухлористой серы нагревают реакционную
смесь с обратным холодильником для удаления хлористого водо-
рода. Затем от реакционной массы отгоняют растворитель, а оста-
ток, представляющий собой вязкую смолообразную массу, раство-
ряют в петролейном эфире. После охлаждения раствора до минус
20 —минус 30°C отфильтровывают выпавшие кристаллы. Полу-
чают 2,2'-тиобис(6-трет-бутил-4-метилфенол) с выходом 47% от
теоретического и т. пл. 80—82 °C. После перекристаллизации из
петролейного эфира получают продукт с выходом 36% и т. пл.
83—84 °C.
Разработан [31, 32] способ получения 2,2'-тиобис (6-трет-бутил-
4-метилфенола) взаимодействием 2-трет-бутил-4-метилфенола с
двухлористой серой в среде бензина, насыщенного продуктом кон-
денсации. Это позволяет сразу получить кристаллический продукт
с т. пл. 80—82 °C.
Большую роль в процессе получения этого стабилизатора иг-
рает качество двухлористой серы. 2,2'-Тиобис(6-трет-бутил-4-ме-
тилфенол) — неокрашивающий стабилизатор и один из немногих
неокрашивающих антизонантов для резин. Присутствие в S2CI2
примесей других тиохлоридов приводит к образованию побочных
продуктов
п = (24-4)
окрашивающих полимер в желтый цвет и обладающих меньшей
эффективностью, чем тиобисфенол. Поэтому для получения стаби-
лизатора применяют свежеперегнанную двухлористую серу, при-
чем для повышения стабильности при перегонке добавляют в куб
трихлорид фосфора в количестве 1,0—1,5% от массы тиохлоридов.
Для конденсации с диалкилфенолом отбирают фракцию тиохло-
рида, выкипающую в интервале 59—61 °C.
Технологические схемы получения тиохлорида и 2,2'-тиобис(6-
трет-бутил-4-метилфенола) представлены на рис. 43 и 44.
Основные стадии технологического процесса таковы: хлориро-
вание однохлористой серы; перегонка двухлористой серы; конден-
сация 2-трет-бутил-4-метилфенола с тиохлоридом; фильтрование
технического продукта; перекристаллизация; фильтрование пере-
кристаллизованного продукта; сушка и измельчение; регенерация
растворителя.
268
Рис. 43. Технологическая схема полу-
чения двухлористой серы:
1,3 — мерники; 2 — реактор; 4 — ректифи-
кационная колонна; 5 — емкость.
Однохлористую серу из же-
лезнодорожной цистерны при-
нимают в емкость и насосом
через мерник 1 (рис. 43) пере-
качивают в реактор 2 для хло-
рирования, куда предвари-
тельно загружают кристалли-
ческий иод (1% масс, от коли-
чества тиохлорида). Хлор из баллона через ротаметр подают в
реактор 2, где с помощью охлаждающей рубашки поддерживают
температуру 0 — минус 5°C. По окончании хлорирования реакци-
онная масса поступает в куб ректификационной колонны 4, куда из
мерника 3 загружают трихлорид фосфора (1,3—1,5°/о от массы
хлоридов). Отгонку ведут при атмосферном давлении. Отогнанная
двухлористая сера (т. кип. 59—61 °C) собирается в приемнике, от-
куда ее перекачивают в емкость 5.
Конденсацию (рис. 44) ведут в стальном эмалированном реак-
торе 5 с мешалкой, рубашкой для нагрева и охлаждения и обрат-
ным холодильником, связанным с системой поглощения хлористо-
го водорода. В аппарат загружают бензин (или бензинсодержа-
щий фильтрат со стадии первого фильтрования) и расплавленный
2-трет-бутил-4-метил фенол, перемешивают и охлаждают до 10—
15°C. При этой температуре и интенсивном перемешивании посте-
пенно приливают из мерника 1 двухлористую серу (из расчета
1,05 моль на 2 моль диалкилфенола). Реакционную массу переме-
шивают 35—40 мин при 30 °C и затем 40—45 мин при 60 °C. Выде-
ляющийся хлористый водород через холодильник поступает в си-
стему поглощения.
Рис. 44. Технологическая схема получения 2,2'-тиобис(6-грег-бутил-4-метилфе-
нола):
1, 2, 3 — мериики; 4 — конденсатор; 5 — реактор; 6, 9 — кристаллизаторы; 7 — друк-фильтр;
<8 — емкость для фильтрата; 10 — центрифуга; 11 — сушилка; 12 — емкость.
269
Содержимое реактора охлаждают в кристаллизаторе 6 до ми-
нус 10 °C, перемешивают 1 ч и фильтруют на друк-фильтре 7.
Фильтрат направляют на рециркуляцию или регенерацию, а оса-
док передают в аппарат 9 для перекристаллизации, где его раство-
ряют в бензине и постепенно при перемешивании охлаждают до
минус 10 °C. Перекристаллизованный продукт фильтруют на цент-
рифуге 10, промывают охлажденным бензином, отжимают и пере-
дают на сушку и измельчение. Фильтрат собирают в емкость 8 и
возвращают в цикл.
Свойства и применение. 2,2'-Тиобис(6-трет-бутил-4-метилфе-
нол)—белое кристаллическое вещество; т. пл. 86 °C. Растворим
в бензоле, толуоле, гексане, бензине, хлорированных углеводоро-
дах, ацетоне, этаноле; в воде не растворяется.
Мало токсичен. Разрешен в СССР для использования в поли-
мерах, контактирующих с пищевыми продуктами, питьевой водой,
и косметическими препаратами. ПДК 3 мг/м3; ЛД50>10 г/кг.
В СССР известен под торговой маркой Тиоалкофен БП. Под:
маркой САО-6 выпускается фирмой Ashland (США).
2,2'-Тиобис (6-трет-бутил-4-метилфенол) — высокоэффективный
неокрашивающий антиоксидант белых и цветных резин на основе
натурального и синтетического каучуков; обладает свойствами
слабого антиозонанта и противоутомителя. Эффективно защищает
полиолефины от термоокислительной деструкции. В полиолефинах
проявляет свойства слабого светостабилизатора. Может применять-
ся в сочетании с техническим углеродом и фосфитами. Защищает
полиолефиновые волокна от старения.
2,2'-Тиобис(4-метил-6-а-метилбензилфенол)
Этот стабилизатор получают взаимодействием 4-метил-6-а-ме-
тилбензилфенола с двухлористой серой [51]:
Реакцию ведут без катализатора в среде органического раствори-
теля (бензин); массовое соотношение диалкилфенол : бензин= 1:1.
Изучение влияния количества двухлеристой серы и температуры
270
конденсации на выход и качество целевого продукта показало, что
конденсацию следует вести при температуре не выше 10 °C, а из-
быток двухлористой серы не должен превышать 5% от теоретичес-
кого [33, 34]. Продукт конденсации выпадает в осадок в виде кри-
сталлов с т. пл. 100 °C.
Технический продукт можно подвергнуть дополнительной очист-
ке [35]. Его растворяют в бензине, добавляют силикагель, кипятят
с обратным холодильником до полного растворения продукта и
фильтруют горячий раствор. После охлаждения фильтруют выпав-
ший кристаллический осадок и затем перекристаллизовывают его
из уксусной кислоты. Получают продукт, плавящийся при 110—.
112 °C.
Процесс получения 2,2'-тиобис(4-метил-6-а-метилбензилфенола)
ведут следующим образом. В реактор (стальной эмалированный
аппарат с мешалкой, рубашкой для охлаждения и обратным холо-
дильником, связанным с системой поглощения хлористого водоро-
да), загружают бензин и 4-метил-6-а-метилбензилфенол, перемеши-
вают и охлаждают до 0—10 °C. Свежеперегнанную двухлористую
серу прибавляют из мерника с такой скоростью, чтобы температу-
ра реакционной массы не превышала 10—12 °C. При этой темпера-
туре перемешивают реакционную массу еще 4 ч и оставляют на
10 ч для кристаллизации. Выпавший осадок отфильтровывают,
промывают бензином, отжимают и передают на сушку в вакуум-
сушилку. Сушку ведут при 60 °C. Фильтрат и промывные воды
собирают и передают на регенерацию.
Свойства и применение. 2,2'-Тиобис(4-метил-6-а-метилбензил-
фенол)—белое кристаллическое вещество; т. пл. 114 °C. Раство-
рим в бензоле, хлороформе, ацетоне, диэтиловом эфире; ограничен-
но растворяется в бензоле; в воде не растворяется.
Мало токсичен. Допущен к применению в пластических массах,
используемых в пищевой промышленности и водоснабжении.
Известен в СССР под названием Тиоалкофен МБП. Техничес-
кий продукт представляет собой белый кристаллический порошок
с т. пл. 99—103 °C.
Термо- и светостабилизатор поливинилхлорида. Термостабили-
затор полиолефинов и волокон на их основе. Эффективен в смеси с
фосфитами.
2,2'-Тиобис(4,6-ди-втор-амилфенол)
I I
С3Н7—СН СН—С3Н7
СН3 СН3
271
Этот стабилизатор получают взаимодействием 2,4-ди-втор-амил-
фенола с двухлористой серой [36]:
ОН
ОН ОН
втор-СъНц
2
+sci2
—2НС1
втор-СБН1:
етор-С6Нц
CjHu-emop
Реакцию ведут в среде органического растворителя или без него
в расплаве диалкилфенола. В последнем случае по окончании кон-
денсации освобождают реакционную массу от хлористого водоро-
да, нагревая при пониженном давлении или продувая инертным
газом. При синтезе этого стабилизатора не предъявляется высоких
требований к чистоте исходного 2,4-ди-втор-амилфенола — присут-
ствие в нем небольших количеств других втор-амилфенолов (на-
пример, 2,6-изомера) не сказывается на качестве технического про-
дукта. Последний выпускается в виде вязкой жидкости, которая
легко эмульгируется с казеинатом аммония и олеатом натрия и
вводится в латексные смеси.
Этот стабилизатор был разработан специально как неокраши-
вающий антиоксидант для латексных изделий, причем, по данным
[36], втор-амильные заместители обусловливают минимальное вли-
яние стабилизатора на стабильность латексных смесей при хране-
нии и обеспечивают получение неокрашенных (белых) резиновых
изделий.
Процесс получения стабилизатора ведут следующим образом.
В стальной эмалированный реактор с мешалкой, рубашкой и об-
ратным холодильником загруэкают расплавленный 2,4-ди-етор-
амилфенол и затем при интенсивном перемешивании постепенно
подают из мерника двухлористую серу (из расчета 0,57 моль на
1 моль 2,4-ди-втор-амилфенола). Температуру реакционной массы
поддерживают в пределах 25—30 °C. После прибавления двухло-
ристой серы перемешивают содержимое реактора еще 2 ч и для
отгонки летучих продуктов передают в аппарат, также снабжен-
ный мешалкой и рубашкой для нагрева и через поглотительную
•систему связанный с вакуум-насосом. Реакционную массу нагре-
вают до 100 °C и при остаточном давлении 10,6 кПа перемешивают
6—8 ч для удаления летучих продуктов. Для полного удаления
хлористого водорода продувают реакционную массу азотом. Про-
дукт реакции представляет собой вязкую жидкость коричневого
цвета.
Фирма Monsanto (США) выпускает жидкий продукт конден-
сации ди-втор-амилфенола с двухлористой серой под торговой мар-
кой Santowhite L. Он применяется для защиты белых и цветных
латексных изделий от старения.
272
Этот стабилизатор получают взаимодействием 6-трет-бутил-2-
метилфенола с двухлористой серой [4, 37]:
Реакцию ведут без катализатора в среде органического раствори-
теля [37]. 6-трет-Бутил-2-метилфенол растворяют в петролейном
эфире и при перемешивании добавляют в течение 5 ч раствор све-
жеперегнанной двухлористой серы в петролейном эфире. Раствор
прибавляют в два приема: первую половину подают при 15—20°C,
затем прекращают подачу, перемешивают реакционную массу при
20—28 °C в течение 30 мин и после этого при 24—28 °C добавляют
вторую половину раствора. Реакционную смесь перемешивают 1 ч
и отфильтровывают осадок кристаллического продукта. После об-
работки активированным углем и перекристаллизации (из цикло-
гексана) получают белые кристаллы 4,4'-тиобис(6-трет-бутил-2-ме-
тилфенола) с т. пл. 124—125 °C.
Эта реакция, в отличие от аналогичной реакции с 2,6-ди-трет-
бутилфенолом, протекает с высокой скоростью и хорошим выходом:
уменьшение степени пространственного затруднения НО-группы в
молекуле 2,6-диалкилфенола облегчает вступление этой молекулы
в реакцию электрофильного замещения с двухлористой серой.
Выход целевого продукта на стадии конденсации достигает 90—
95%. Мольное соотношение диалкилфенола и двухлористой серы
целесообразно брать 2:1,05. Для получения технического продук-
та в качестве растворителя используют бензин, причем двухлори-
стую серу вводят без растворителя [4].
Процесс ведут так. В стальной эмалированный реактор (с ме-
шалкой, рубашкой для нагрева и охлаждения и обратным холо-
дильником, связанным с системой поглощения хлористого водоро-
да) загружают 6-трет-бутил-2-метилфенол и бензин и перемешива-
ют в течение 30 мин. Затем из мерника в течение 2 ч прибавляют
свежеперегнанную двухлористую серу, поддерживая в реакторе
температуру 18—20 °C. Для завершения реакции перемешивают
смесь еще 2 ч при 20—25 °C и передают в кристаллизатор, тоже
снабженный мешалкой и рубашкой для охлаждения рассолом. Ре-
акционную массу в течение 3 ч при перемешивании охлаждают до
минус 5 — минус 10 °C и передают на фильтрование. Кристалличе-
ский осадок на фильтре промывают охлажденным бензином, отжи-
18—1057
273
1
мают и передают на перекристаллизацию из бензина. Перекристал-
лизованный продукт высушивают и измельчают.
Свойства и применение. 4,4'-Тиобис(6-трет-бутил-2-метилфе-
нол) — белое кристаллическое вещество; т. пл. 125 °C; плотность
1084 кг/м3. Хорошо растворяется в бензоле, толуоле, дихлорэтане,
этаноле, ацетоне; мало растворим в гексане и бензине; в воде не
растворяется.
Выпускается фирмой Ethyl (США) под торговой маркой Ethyl
Antioxidant 736. Неокрашивающий стабилизатор для белых и цвет-
ных резин на основе натурального и синтетических каучуков. В по- i
лимерных композициях, содержащих технический углерод, эффек-
™в— Й
4,4'-Т иобис(б-трет-бутил-З-метилфенол) |
С(СН3)3 С(СН3)3
Н3С СН3
Это один из первых стабилизаторов группы тио бисфенолов; его »
применение описано в начале 1940-х годов. Получают его взаимо- 5
действием 6-трет-бутил-З-метилфенола с SCh
без катализатора в среде органического растворителя. Продукт!
реакции выпадает в осадок; его отфильтровывают и высушивают!
В качестве исходного сырья для получения б-трет-бутил-З-метил-1
фенола обычно используют дикрезольную фракцию. Ее подвергают!
моноалкилированию и разделяют. I
Для синтеза можно использовать также неразделенную смесь]
трет-бутилкрезолов [44]. При конденсации с SC12 в петролейном!
эфире в присутствии каталитических количеств брома получают]
смесь тиобисфенолов, состоящую в основном из 2,2/-тиобис(6-трет-1
бутил-4-метилфенола) и 4,4'-тиобис (6-трег-бутил-З-метилфенола) J
Последний выделяют из этой смеси в чистом виде, а оставшуюся
смесь тиобисфенолов используют как смесевой стабилизатор.
Реакция 6-трет-бутил-З-метилфенола с SC12 протекает, по-види-
мому, менее селективно, чем реакция 2,4- или 2,6-диалкилфенола.
Как правило, в этих последних подвергается атаке электрофиль-
ным агентом одно свободное положение (соответственно 6 или 4);
в молекуле 6-трет-бутил-З-метилфенола атом серы может вступить
не только в положение 4, но и в положение 2. Известно, что 4,6-ди-
274
при действии SC12 легко образует тио-
он
С(СН3)3
грет-бутил-З-метилфенол
бисфенол [45]:
он
I SCI
-f-OUJg
----->
—2НС1
I Н8С СН3 |
(СН3)3С С(СН3)3
По-видимому, и при взаимодействии 6-трет-бутил-З-метилфенола с
SCI2 наряду с основной могут идти и побочные реакции образова-
ния 2,2'- и 2,4-тиобисфенолов:
ОН ОН ОН
Для очистки целевого продукта от этих примесей требуется допол-
нительная перекристаллизация.
Применение исходного диалкилфенола разной чистоты и воз-
можность протекания побочных реакций являются, по-видимому,
причиной появления различных марок этого стабилизатора. На-
пример, фирма Monsanto наряду с белым кристаллическим продук-
том Santonox R (т. пл. 161 °C) выпускает Santowhite Crystalls
(т. пл. 140°C) и Santowhite МК — жидкий продукт конденсации
6-трет-бутил-З-метилфенола с SC12-
Технологическая схема получения 4,4'-тиобис (6-трет-бутил-З-
метилфенола) представлена на рис. 45. В стальной эмалирован-
ный реактор 3, снабженный мешалкой, рубашкой для охлаждения
рассолом и нагрева паром и обратным холодильником, загружают
бензин и 6-трет-бутил-З-метилфенол. Смесь перемешивают и ох-
лаждают до минус 5 — минус 10°С. В мерник 1 заливают бензин,
засасывают в вакууме двухлористую серу, перемешивают азотом
и приготовленный раствор в течение 2 ч равномерно подают в ре-
актор. Массу перемешивают еще 4 ч при температуре не выше 0°С,
затем нагревают до 20 °C, выдерживают при этой температуре 6—
8 ч и передают на фильтрование. Выпавший осадок продукта от-
фильтровывают на центрифуге 4, промывают бензином, отжимают
и передают в барабанную вакуум-сушилку, а затем на измельче-
ние. Фильтрат и промывные воды направляют на регенерацию. По-
лученный таким образом продукт представляет собой светло-серый
кристаллический порошок с т. пл. 150—155 °C.
18’
275
Рис. 45. Технологическая схема получения 4,4'-тиобис(6-трет-бутил-3-метилфЗ^
нола):
1 — мерник; 2, 6 — Обратные холодильники; 3 — реактор; 4, 9 — центрифуги; 5 — аппарат для
растворения; 7 — друк-фильтр; 8 — кристаллизатор.
Для очистки подвергают технический продукт перекристаллиза-
ции в присутствии адсорбента [46]. В аппарат 5 с мешалкой, ру-
башкой для нагрева и обратным холодильником загружают бензин,
четыреххлористый углерод, силикагель и пасту технического про-
дукта из центрифуги 4. Смесь нагревают до кипения, перемеши-
вают 30 мин и, не охлаждая, передают на обогреваемый друк-
фильтр 7. Горячий раствор из приемника передают в кристаллиза-
тор 8. Раствор охлаждают до 15—20 °C и выпавший осадок кри-
сталлического продукта отфильтровывают на центрифуге 9, про л
мывают бензином, отжимают, высушивают и измельчают. Полуя
чают белый кристаллический порошок с т. пл. не ниже 161 °C. А]
Свойства и применение. 4,4/-Тиобис(6-трет-бутил-3-метилфеИ
нол) — белое кристаллическое вещество; т. пл. 162°C; плотностИ
1060—1120 кг/м3. Растворяется в этаноле, ацетоне, бензоле, хлороИ
форме, дихлорэтане, четыреххлористом углероде, бензине; в водД
не растворяется. В США разрешен для применения в полимерах»
контактирующих с пищевыми продуктами.
В СССР выпускается под названиями Тиоалкофен БМ (светло- ^
серый кристаллический порошок; т. пл. 156—158 °C) и Тиоалкофе«
БМЧ (т. пл. 161 °C). За рубежом производится рядом фирм: Д
Торговое наименование Страна, фирма T. пл., °c j
Antigene WX Япония, Sumitomo 152
Antigene WX-R Япония, Sumitomo 160
Antioxidant TBM-6 Франция, Organo Synthese —
Annullex PSA 10 Англия, Pearson —
Nocrac 300 Япония, Ouchi Sliinko 155
Santonox США, Monsanto 150
Santonox R США, Monsanto 161
Santowhite Crystalls США, Monsanto MO
TBM-6P США, Aceto Chemical —
276
4,4/-Тиобис(6-трет-бутил-3-метилфенол) —эффективный неокра-
щивающий стабилизатор резин на основе натурального и синтети-
ческих каучуков, особенно хлоропренового. Защищает полиолефи-
ны и волокна на их основе от теплового старения.
Бис(4-гидрокси-3,5-ди-трет-бутилбензил)сульфид
,С(СН3)
—СН2—S—Н2С-^ "—ОН
^С(СН3)
Известен ряд способов синтеза этого стабилизатора.
Например, его получают из 4-гидрокси-3,5-ди-трет-бутилбензил-
бромида взаимодействием с сульфидом калия [38, 39]:
(СН3)3«
С(СН3)3
(СН3)3С
+K2s
2 HO-Z/ xx-CH2Br НО— -CH2-S-H2C- —ОН
—2К,1эГ
(СН3)3С
(СН3)3С
С(СНз)8
Сульфид калия растворяют в безводном этиловом спирте и к рас-
твору при 0—5 °C и интенсивном перемешивании постепенно добав-
ляют 4-гидрокси-3,5-ди-трет-бутилбензилбромид. Реакционную
смесь перемешивают 1 ч, отфильтровывают осадок КВг и при по-
ниженном давлении осторожно отгоняют растворитель. Остаток
перекристаллизовывают (из этанола) и получают целевой продукт
с т. пл. 140—142°C и выходом 76% от теоретического.
Это соединение образуется с хорошим выходом при взаимодей-
ствии сульфида натрия с 2,6-ди-трет-бутил-4-метоксиметилфенолом
[40]:
(СНз)3С. (СНа)3С. ,С(СН3)3
2 и°-^^у-сн2-осн3 ho-^)-ch2-s-h.c-£^-oh
(CH3)3CZ (СН3)ЭС/ \С(СН3)3
Последний растворяют в метаноле и к полученному раствору до-
бавляют Na2S-9H2O. Смесь кипятят 30 мин, разбавляют метано-
лом и охлаждают до 0°С. Выпавший осадок продукта отфильтро-
вывают.
Еще один способ получения — хлорметилирование 2,6-ди-трет-
бутилфенола и взаимодействие полученного хлорметильного про-
изводного с Na2S в среде изопропанола [41]. Na2S вводят в виде
водного раствора, кипятят реакционную смесь в течение 2 ч и ос-
тавляют на двое суток. Получают целевой продукт с т. пл. 141—
143 °C.
277
Из 2,6-ди-трет-бутилфенола этот стабилизатор можно синтези-
ровать и через тиокарбаминовое производное, с высоким выходом
получаемое взаимодействием 2,6-ди-трет-бутилфенола с диметил-
дитиокарбаматом натрия и формальдегидом в кислой среде [28].
При нагревании тиокарбаминового производного с раствором Na2S
в среде метанола получают целевой продукт, который выпадает в
осадок. Выход 98% от теоретического; т. пл. 141—142°C.
Более простым путем описываемый стабилизатор можно полу-
чить взаимодействием 2,6-ди-трет-бутилфенола с формальдегидом ч
и сульфидом натрия в спиртовом растворе [29, 42]. По этому спо- Й
собу все реагенты растворяют в метаноле, через раствор пропуска- Ы
ют поток азота и кипятят реакционную смесь с обратным холо- й
дильником в течение 30 мин. После охлаждения кристаллический й
продукт выпадает в осадок; выход я» 90% от теоретического. И
Возможность осуществления синтеза непосредственно из 2,6-ди- |
трет-бутилфенола в одну стадию обусловливает практическую |
перспективность этого способа [43]: можно получить из соответст-
вующих 2,6-диалкилфенолов различные бис(4-гидрокси-3,5-диал-
килбензил) сульфиды:
(CHS)3C
\____ Ч-НСНО;
4-NaaS
—2NaOH
(СН3)3С
2 НО—
СН2—S—Н2С—,
С(СН3)3
ОН
СН3
сенБ—СН-СН3
2 НО—
сн3
свнБ—сн—сн3
СН.
4-HCHO;
+NaaS »
—2NaOH
НО-
СвНБ—СН—СН3
сн3
СН3
‘2
—ОН
сн3
бис (4-гидрокси-3,5-ди-трет-бутилбензил) -
। Процесс получения
. сульфида ведут так. В реактор (стальной аппарат с мешалкой, ру-
башкой и обратным холодильником) загружают метанол, форма- j
лин и сульфид натрия. Смесь перемешивают, подают 2,6-ди-трет- J
бутилфенол и перемешивают 30 мин при 20—25 °C. Затем подни- ;
мают температуру до 60—65 °C и перемешивают еще 30 мин. Реак- /:
ционную массу при перемешивании охлаждают до 15—20°C и пере-
дают на фильтрование. Выпавший осадок кристаллического про- 3
дукта отфильтровывают на друк-фильтре, промывают водой и пе-
редают в вакуум-сушилку (температура сушки 60—70°С).
Свойства и применение. Бис(4-гидрокси-3,5-ди-трет-бутилбен-
зил) сульфид —белое кристаллическое вещество; т. пл. 143 °C. Рас-
творим в большинстве органических растворителей; в воде не рас-
творяется. Технический продукт имеет т. пл. 139СС. В СССР из-
вестен под названием Стабилизатор ТБ-3.
278
НО—;
Применяется как стабилизатор синтетических каучуков (бута-
диен-стирольных, бутадиен-нитрильных), термоэластопластов, по-
лиолефинов.
ЛИТЕРАТУРА
1. Fugisawa Т. е. а. — Tetrahedron Letter, 1968, № 43, р. 4553.
2. Ethyl, пат. 3092585, 1963 г. (США).
3. Ethyl, пат. 3240705, 1966 г. (США).
4. Гринберг А. А., Гурвич Я. А., Фришман Т. А. — В ки.: Синтез и исследование
эффективности химикатов для полимерных материалов. Тамбов, 1969. Вып. 2,
с. 383.
5. Firestone, пат. 2581919, 1952 г. (США).
6. Редникова Т. А. и др. — Хим. пром-сть, 1967, с. 98.
7. Гринберг А. Е., Сорокин Э. С. — Хим. пром-сть, 1970, с. 499.
8. Гурвич Я- А. и др. — В кн.: Тезисы докладов на X Научной сессии по химии
сероорганических соединений. Уфа, Изд. БФ АН СССР, 1968, с. 10.
9. Ethyl, пат. 3069384, 1962 г. (США).
10. Гурвич Я- А., Кумок С. Т., Стыскин Е. Л. — В кн.: Тезисы I Всесоюзного сим-
позиума по органическому синтезу. М., Изд. АН СССР, 1974, с. 19.
11. Ершов В. В. — Докт. дисс. М., ИХФ АН СССР, 1965.
12. Ершов В. В., Володькин А. А. — Изв. АН СССР, ОХН, 1962, с. 730.
13. Никифоров Г. А., Гейдыш Л. С., Ершов В. В. — Изв. АН СССР, 1968, с. 1132.
14. Rege A., Airan J., Shah S. — J. Indian Chem. Soc., 1948, v. 25, p. 43.
15. Плетнева И. Д., Гурвич Я. А.. Гринберг А. Е. — В кн.: Тезисы XII Научной
сессии по химии и технологии сероорганических соединений, Рига, 1971, с. 136.
16. Sagani Chem., пат. 3678115, 1972 г. (США).
17. Sagani Chem., пат. 3835196, 1974 г. (США).
18. Consolidation Coal, пат. 3479407, 1969 г. (США).
19. Exxon Res. Eng., пат. 1443329, 1976 г. (Англия).
20. Юкельсон И. И., Федотова Л. В. — Хим. пром-сть, 1973, с. 416.
21. Ethyl, пат. 3449441, 1969 г. (США).
22. Пат. 10920,-1956 г. (Япония).
23. Ethyl, пат. 3060121, 1962 г. (США).
24. Гринберг А. А., Гурвич Я. А. и др. — В кн.: Синтез и исследование эффектив-
ности химикатов для полимерных материалов. Тамбов, 1969. Вып. 2, с. 407.
25. А. с. 193532, 1967 г. (СССР).
26. А. с. 189843, 1967 г. (СССР).
27. Rubber, пат. 3310587, 1967 г. (США).
28. Rubber, пат. 3260756, 1966 г. (США).
29. Rubber, пат. 3272869, 1966 г. (США).
30. Cyanamid, пат. 2841619, 1958 г. (США).
31. А. с. 188965, 1966 г. (СССР).
32. Гринберг А. Е., Сорокин Э. С. — Хим. пром-сть, 1970, с. 499.
33. А. с. 176292, 1965 г. (СССР).
34. Редникова Т. А. и др. — Хим. пром-сть, 1967, с. 98.
35. А. с. 273819, 1977 г. (СССР).
36. Monsanto, пат. 2776998, 1957 г. (США).
37. Monsanto, пат. 3107233, 1963 г. (США).
38. А. с. 197579, 1967 г. (СССР).
39. Брук Ю. А., Рачинский Ф. Ю. — ЖОХ, 1967, т. 3, с. 2174.
40. Ethyl, пат. 3652679, 1972 г. (США).
Ml. Ethvl, пат. 3065275, 1962 г. (США).
«42. U. S. Rubber, пат. 3322649, 1967 г. (США).
’43. Афанасьев И. Д. и др.— В кн.: Присадки к маслам. М., Химия, 1966, с. 129.
44. Пат. 1493941, 1967 г. (Франция).
45. Monsanto, пат. 2581919, 1952 г. (США).
46. Бурмистрова Р. С., Попова 3. Г. — В кн.: Синтез и исследование эффектив-
ности химикатов для полимерных материалов. Тамбов, 1969. Вып. 3, с. 70.
3.5. СИНТЕЗ СТАБИЛИЗАТОРОВ ГРУППЫ ТРИС-
И ТЕТРАФЕНОЛОВ
Стабилизаторы этой группы появились в конце 1950-х годов.
Создание их связано с развитием производства полиолефинов, в
первую очередь полипропилена и волокон на его основе, а также
других полимеров, перерабатываемых при высоких температурах.
Трис- и тетрафенолы имеют высокую молекулярную массу и соот-
ветственно пониженную по сравнению с одноядерными фенолами
и бисфенолами летучесть.
В молекулах стабилизаторов этой группы объединены несколь-
ко фрагментов, содержащих пространственно затрудненный фе-
нольный гидроксил. Исходными соединениями в синтезе являются
прежде всего 2,6-ди-трет-бутилфенол или его производные, имею-
щие реакционноспособную группу в лара-положении к гидроксилу:
<сн3)3с
но-
(СН3)3С
В отдельных случаях используют другие диалкилфенолы, напри-
мер 6-трет-бутил-З-метилфенол. Основные процессы в синтезе этих
стабилизаторов — рассмотренные выше алкилирование ароматиче-
ского ядра, конденсация алкилфенолов с альдегидами, переэтери-
фикация сложных эфиров.
Простейшие стабилизаторы группы трисфенолов содержат три
ядра, связанных метиленовыми мостиками:
а = сн2он. сн2осн3, СН2СООСН3,
СН2СН2СООСН3. CH2N(CH3)2,
СН2С1, ОН или SH
А
Трисфенолы такой структуры получают конденсацией диметил
ольного производного 4-алкилфенола с 2,4-диалкилфенолом [1];
» ОН ОН
280
2-трет-Бутил-4-метилфенол растворяют в гептане, добавляют со-
ляную кислоту, лаурилсульфат натрия и 2,6-дигидроксиметил-4-
метилфенол, перемешивают 15 ч при 25—30 °C и отфильтровывают
выпавший осадок. После кристаллизации из гептана получают (с
выходом 51 % от теоретического) белые кристаллы с т. пл. 154—
158 °C. Эту реакцию проводят и без растворителя, выдерживая ис-
ходные вещества с катализатором в течение 36 ч при 125°C [2].
После перекристаллизации получают (с выходом 21% от теорети-
ческого) белые кристаллы (т. пл. 174—175°C).
Другой путь синтеза таких трисфенолов — взаимодействие
4-алкилфенола с соответствующим гидроксибензиловым спиртом:
ОН ОН ОН ОН ОН
В этом синтезе исходят из 2,4-диалкилфенола, а гидроксибензило-
вый спирт не выделяют из реакционной массы. Например, 2,4-ди-
метилфенол, 36%-ный раствор формальдегида и катализатор на-
гревают в среде пропиленгликоля до кипения, кипятят с обратным
холодильником в течение 5 ч, загружают 4-метилфенол и кипятят
еще 3 ч. После выделения и перекристаллизации получают трисфе-
нол с т. пл. 184,5°C:
Взаимодействие 2,6-дигидроксиметил-4-метилфенола с 2,6-диал-
килфенолами получают трисфенолы иной структуры:
Синтез ведут, нагревая исходные соединения в присутствии кис-
лотного катализатора до 95—100 °C [4]. Трисфенол такой структу-
281
-w
ры получают также взаимодействием 4-метилфенола с 4-гидрокси-
2,6-ди-трет-бутилбензилхлоридом [5]:
Исходные вещества выдерживают при 95 °C в течение 24 ч, отго-
няя выделившийся хлористый водород. Затем добавляют бензол,
промывают водой и выделяют кристаллический продукт с т. пл.
130 °C. Этот же стабилизатор получают, используя в качестве ал-
килирующего агента 2,6-ди-трет-бутил-4-метоксиметилфенол [6].
Вторую группу трисфенольных стабилизаторов составляют со-
единения, в которых ароматические ядра присоединены к углево-
дородной цепочке RCH—СН2—СН—, где R = H, СН3 или С6Н5.
Такие стабилизаторы получают взаимодействием алкилфенолов с
ненасыщенными альдегидами, имеющими а,р-двойную связь [7],
а также с р-галогенальдегидами и р-гидроксиальдегидами [8]. Эти
альдегиды реагируют с алкилфенолами как бифункциональные со-
единения: присоединяют две молекулы алкилфенола по углероду
альдегидной группы и одну — по р-атому углерода:
Здесь одновременно протекают две реакции — конденсация альде-
гида с двумя молекулами диалкилфенола (с образованием мети-
ленбисфенола) и алкилирование третьей молекулы диалкилфенола
непредельным углеводородом (в случае p-гидроксиальдегида идет
282
алкилирование вторичным спиртом, в случае р-галогенальдегида—
алкилирование галогенуглеводородом). Обе реакции протекают в
условиях кислотного катализа, и на них распространяются рас-
смотренные выше закономерности замещения диалкилфенолов
слабыми электрофилами. 6-трет-Бутил-З-метилфенол легко вступа-
ет в реакцию с кротоновым альдегидом, образуя трисфенол, в то
время как попытки синтезировать в этих условиях трисфенол исхо-
дя из 2,6-ди-трет-бутилфенола успеха не имели. Это можно объяс-
нить на основе современных представлений о реакционной способ-
ности пространственно затрудненных фенолов в реакциях электро-
фильного замещения слабыми электрофилами [9, 10].
Взаимодействием 6-трет-бутил-З-метилфенола с акролеином по-
лучают 1,1,3-трис(5/-трет-бутил-4'-гидрокси-2,-метилфенил)пропан
ОН
а взаимодействием с коричным альдегидом—1,1,3-трис(5'-трет-бутил-
4'-гидрокси-2'-метилфенил)-3-фенилпропан:
Наибольшее практическое значение среди трисфенолов этой груп-
пы имеет 1,1,3-трис(5'-трет-бутил-4'-гидрокси-2'-метилфенил)бутан,
получаемый взаимодействием 6-трет-бутил-З-метилфенола с крото-
новым, p-хлормасляным или р-гидроксимасляным альдегидом.
283
Трисфенолы третьей группы имеют симметричную молекулу,
центральной группировкой которой чаще всего является аромати-
ческое ядро или шестичленный гетероцикл.
Симметричные трисфенолы с центральным бензольным ядром
получают циклической кротоновой конденсацией алкилзамещенных
4-гидроксибензофенона [11]:
' Большую группу трисфенолов составляют соединения, централь-
ной частью молекулы которых является мезитилен. Практический
интерес представляет алкилирование мезитилена реакционноспо-
собными 4-замещенными 2,6-ди-трет-бутилфенола. В ряду арома-
тических углеводородов мезитилен является одним из самых ак-
тивных нуклеофилов и легко вступает в реакции электрофильного
замещения. По мере вступления заместителя нуклеофильная актив-
ность замещенного мезитилена снижается, и для введения третьего
заместителя требуется применение избытка алкилирующего агента.
Первым алкилирующим агентом, использованным в синтезе трис-
фенолов этого типа, был 4-гидрокси-3,5-ди-трет-бутилбензиловый
спирт [12]. В условиях кислотного катализа он легко протониру-
ется. Образовавшийся карбкатион
(СН3)3С\ -
H0-Q4h2
(СН3)3с/
атакует молекулу мезитилена; происходит последовательное вступ-
ление заместителей в положения 2, 4 и 6 с образованием трехзаме-
щенного мёзитнлена:
284
С помощью 4-гидрокси-3,5-ди-трет-бутилбензилового спирта алки-
лируют не только мезитилен, но фенол и алкилфенолы, получая
при этом трис- и тетрафенолы различного строения [25], напри-
мер:
При исследовании алкилирования мезитилена 4-гидрокси-3,5-ди-
трет-бутилбензиловым спиртом было установлено, что в реакцион-
ной массе присутствует ди(4-гидрокси-3,5-ди-трет-бутилбензило-
вый) эфир
(СН3)3С /С(СН.,)8
но-ЛА-сна-о-нас-^~^-он
(СН3)3С С(СН3)3
который образуется в начальной стадии алкилирования, а потом
расходуется [13]. Этот эфир может быть непосредственно исполь-
зован как агент алкилирования мезитилена [14]. Таким образом,
285
алкилирование мезитилена 4-гидрокси-3,5-ди-трет-бутилбензило-
вым спиртом представляет собой ряд последовательных и парал-
лельных реакций, протекающих по приведенной ниже схеме:
4-ceH3(CHs)s (Н+)
Позднее было показано, что в качестве алкилирующего агента
можно применять другой эфир 4-гидрокси-3,5-ди-трет-бутилбензи-
лового спирта (метиловый эфир). Установлено, что 2,6-ди-трег-бу-
тил-4-метоксиметилфенол в кислотнокатализируемой реакции с
286
тезитиленом образует с высоким выходом 2,4,6-три(4-гидрокси-
3 й-ди-трет-бутилбензил) мезитилен [15] —один из важнейших ста-
билизаторов группы трисфенолов.
Взаимодействием 2,6-ди-грет-бутил-4-меркаптофенола и 2,4,6-
Три(хлорметил)метилмезитилена получают трисфенол, содержа-
щий в мостиковых группах атомы серы:
Другой содержащий серу трисфенол такого типа получают на основе
6-трет-бутил-2,4-диметилфенола [16]:
s
I
сна
За последнее десятилетие появились трисфенолы, в молекуле
которых центральной группировкой является шестичлениый гетеро-
цикл , с тремя атомами азота. Эти стабилизаторы получают глав-
ным образом на основе циануровой кислоты и ее производных.
Простейший из трисфенолов этой группы — три (4-гидрокси-3,5-дц-
трет-бутилбензил)изоцианурат [17]:
Более сложную структуру имеет трисфенол, получаемый на основе
гидроксиэтильного производного симм-триазинтриона и 4-гидрокси-3,5-
ди-трет-бутилпропионовой кислоты [18]:
А—о
(^На)2 (CHS)SC
/N—СО __/
ОС ^N—(СН2)2—-О—С—(СН2)2—/ V-OH
А
(СН2)2 (снз)зс
А—О
__/О(СН3)3
где А равно С—(СН2)2—/ >—ОН
II \=/
° 'С(СН3)3
Переэтерификацией метилового эфира 4-гидрокси-3,5-ди-трет-
бутилфенилпропионовой кислоты триэтаноламином получают трис--
фенол с центральным атомом азота [19]:
С(СН3)3
ЗНО-^Л-СН2-СН2-С-О-СНз + (HOCH2-CH2)3N ——
\ / || —ovnguri
/ о
С(СН8)з
288
С(СН3)3
но—сн2—сна—с—о—сн2-сн2—
о
N
С(СН3)3
3
Все эти трисфенолы — неокрашивающие стабилизаторы, предназ-
наченные для защиты полипропилена.
Тетрафенолы получают взаимодействием диалкилфенолов с ди-
альдегидами, например:
ОН
Тетрафенолы другой структуры получают, вводя три фрагмен-
та, содержащих пространственно затрудненный фенольный гидрок-
сил, в молекулу фенола или алкилфенола:
ОН ОН
ОН
ОН
19—1057
289
Тетрафенол, содержащий тиометиленовый мостик, получают
взаимодействием 4- (4'-гидрокси-3',5'-ди-трет-бутилбензилиден) -2,6-
ди-трет-бутилцикл’огексадиен-2,5-она-1 с H2S [20]:
О
ОН ОН
ОН он
Наибольшее практическое значение среди стабилизаторов группц
тетрафенолов имеет эфир 4-гидрокси-3,5-ди-трег-бутилфенилпро-ч
пионовой кислоты и пентаэритрита:
Этот стабилизатор получают переэтерификацией метилового эфи-
ра 4-гидроксифенил-3,5-ди-трет-бутилпропионовой кислоты пента-
эритритом в присутствии основных катализаторов [21].
На основе меркаптоацетата пентаэритрита и 4-гидрокси-3,5-ди-
трег-бутилбензилхлорида получают тетрафенол с атомами серы
в мостиковых связях:
С Г— CHj—о—С—CH2SNa] + 4С1Н.С—
II
О
14
С(СН3),
—4NaCl
C(CH3)S
290
,С(СН9)3
—сн2—о—с-сна—s—н
1,1,3-Трис(5'-трет-бутил-4'-гид рокси-2'-метилфенил) бутан
Этот стабилизатор впервые был синтезирован в 1959—1960 го-
дах взаимодействием б-трет-бутил-З-'метилфенола с кротоновым
альдегидом [7]:
ОН
ОН он
В качестве катализатора используют соляную или серную кислоту,
хлорид цинка; растворителем служит этиловый спирт или ледяная
уксусная кислота. 6-трет-Бутил-З-метилфенол растворяют в метано-
ле, добавляют соляную кислоту и затем при перемешивании при-
бавляют эквимольное количество кротонового альдегида. Реакцион-
ную массу перемешивают еще 15 мин и отфильтровывают выпав-
ший осадок, промывают его водой и перекристаллизовывают из то-
луола. Получают белые кристаллы с т. пл. 180—182 °C и выходом
82% от теоретического.
Вместо кротонового альдегида используют р-гидроксимасляный
альдегид [8]:
ОН
C(CHS)S
CHS—СН—СН2—СНО + 3
ОН HsC
-Н2О
19*
291
Метанол смешивают с концентрированной соляной кислотой, загру-
жают 6-трет-бутил-З-метилфенол, нагревают до кипения и в тече-
ние 1 ч добавляют p-гидроксимасляный альдегид. Затем отгоняют
метанол, воду и НС1, одновременно добавляя толуол, чтобы объем
реакционной смеси оставался постоянным. Раствор охлаждают до
25 °C, отфильтровывают кристаллический продукт, промывают его
толуолом и высушивают. Получают продукт с т. пл. 184—188 °C.
Технологическая схема получения. В основу промышленного
процесса получения стабилизатора положена конденсация 6-трет-
бутил-3-метилфенола с кротоновым альдегидом. Главные стадии —
приготовление раствора 6-трет-бутил-З-метилфенола в метаноле;
конденсация 6-трет-бутил-З-метилфенола с кротоновым альдегидом;
выделение сырого продукта и его очистка; регенерация раствори-
телей.
Процесс ведут так (рис. 46). В реактор 3 (стальной эмалиро-
ванный аппарат с мешалкой, рубашкой для нагрева и охлаждения
и обратным холодильником) загружают метанол, концентрирован-
ную соляную кислоту и 6-трет-бутнл-З-метилфенол. Смесь переме-
шивают и нагревают до 70 °C. К полученному раствору постепенно
добавляют из мерника 1 кротоновый альдегид, перемешивают в те-
чение 1 ч для завершения реакции, охлаждают до 20°C и переда-
ют на фильтрование. Пасту сырого продукта из друк-фильтра 4
Кротоновый
альдееид
Метанол
6-трет-Бутил-
3-метилфенол
Иоляная кислота
Рис. 46. Технологическая схема получения 1,1,3-трис(5,-трет-бутил-4'‘-гидрокси-2'-
метилфенил) бутана:
/, 6 — мерники; 2, 7 — конденсаторы; 3— реактор; 4, 9— друк-фильтры; 5, // — емкости;
8 — кристаллизатор; 10 — вакуум-сушилка.
292
передают на кристаллизацию в стальной эмалированный аппарат 8
с мешалкой, рубашкой для нагрева и охлаждения и обратным хо-
лодильником. Туда загружают пасту продукта и толуол, нагрева-
ют до кипения, а затем медленно охлаждают. Выпавший осадок
кристаллического продукта отфильтровывают на друк-фильтре 9,
промывают толуолом, охлаждают и передают на сушку. Фильтра-
ты, содержащие метанол и толуол, направляют на регенерацию.
Свойства и применение. 1,1,3-Трис(5'-трет-бутил-4'-гидрокси-2'-
метилфенил) бутан —белое кристаллическое вещество; т. пл. 188 °C.
растворяется в ацетоне, этаноле, диэтиловом эфире, этилацетате,
метаноле, бензоле и толуоле; в воде растворяется мало.
Мало токсичен; ЛД5о=16,1 мг/кг (для мышей).
Выпускается фирмой ICI под торговой маркой Topanol СА. Не-
окрашивающпй низколетучий стабилизатор, разработанный для за-
щиты полипропилена. Эффективность его повышается при сочета-
нии с тио(дилаурилпропионатом). Защищает полиэтилен, по-
ливинилхлорид, синтетические каучуки. Эффективно защищает по-
лимеры в присутствии меди и может быть применен в составах для
покрытия проводов и кабелей. В пластифицированном поливинил-
хлориде защищает не только полимер, но и пластификатор.
В США разрешен для применения в изделиях из ударопрочного
полистирола, контактирующих с пищевыми продуктами.
Этот стабилизатор впервые был получен алкилированием мези-
тилена 4-гидрокси-3,5-ди-трет-бутилбензиловым спиртом [12]
293
в присутствии кислотного катализатора (серная кислота). Мезити-
лен и 4-гидрокси-3,5-ди-трет-бутилбензиловый спирт растворяют в
хлористом метилене и к полученному раствору при охлаждении
постепенно добавляют серную кислоту. Реакционную массу пере-
мешивают в течение 3 ч и отделяют кислотный слой. Органический
слой промывают водой до нейтральной реакции, отгоняют хлори-
стый метилен и перекристаллизовывают остаток из изопентана. По-
лучают кристаллический продукт с т. пл. 200—207 °C.
Реакцию ведут и в присутствии других кислотных катализато-
ров— п-толуолсульфокислоты, трифторметансульфокислоты, бен-
золсульфокислоты [23]. Мезитилен, 4-гидрокси-3,5-ди-грет-бутил-
бензиловый спирт, катализатор и хлористый метилен загружают в
реактор, нагревают смесь и ведут процесс при т. кип. реакционной
массы, отгоняя образующуюся воду. Синтез ведут в течение 4—
4,5 ч, добавляя свежий растворитель. Затем реакционную массу
охлаждают, промывают раствором аммиака и водой, отгоняют рас-
творитель, а остаток дважды перекристаллизовывают из гексана.
Получают целевой продукт с т. пл. 239,5—240 °C.
Рис. 47. Технологическая схема получения 2,4,6-трис(4,-гидрокси-3,,5,-ди-трет-бу-
тилбензил) мезитилена:
/ — реактор; 2— сепаратор; 3— конденсатор; 4— аппарат для нейтрализации; Б — друк-
фильтр; 6 — ректификационная колонна; 7 — кристаллизатор; 8 — центрифуга.
294
По другому способу стабилизатор получают алкилированием
мезитилена 4-гидрокси-3,5-ди-трет-бутилбензилацетатом [24]:
ОН'
Исходные вещества растворяют в хлористом метилене, а затем при
охлаждении и перемешивании добавляют серную кислоту. По окон-
чании реакции отделяют кислоту, нейтрализуют и промывают ор-
ганический слой, отгоняют растворитель и выделяют целевой про-
дукт.
В СССР разработан способ получения этого стабилизатора ал-
килированием мезитилена 2,6-ди-трет-бутил-4-метоксиметилфено-
лом [15]. Последний растворяют в хлористом метилене, вводят ме-
зитилен, охлаждают до 0—минус 5 °C и при интенсивном переме-
шивании постепенно добавляют серную кислоту. По окончании
реакции отделяют кислоту, отгоняют растворитель и перекристал-
лизовывают продукт из метанола.
При алкилировании мезитилена 4-гидрокси-3,5-ди-трет-бутил-
бензиловым спиртом или его эфирами наряду с основной протека-
ет и побочная реакция автоконденсации с образованием 4,4'-мети-
ленбис (2,6-ди-трет-бутилфенола):
(СН3)3С
НО—,
—сн2—,
С(СН3)3
ОН
(СН3)3С
С(СН3)3
Повышение температуры ускоряет побочную реакцию, поэтому ал-
килирование ведут при 0+5 °C.
295
По мере вступления заместителей в- молекулу мезитилена его
нуклеофильная реакционная способность падает и скорость даль-
нейшего алкилирования снижается: даже при избытке алкилирую-
щего агента в конечном продукте присутствуют примеси ди- и сле-
ды монозамещенного мезитилена. Однако дизамещенный мезитилен
тоже является высокоэффективным неокрашивающим стабилиза-
тором, поэтому небольшая его примесь (2—3%) практически не
снижает эффективности трисфенола.
Реакция каталитического алкилирования мезитилена 2,6-ди-
грет-бутил-4-метоксиметилфенолом положена в основу промышлен-
ного получения 2,4,6-трис (4/-гидрокси-3',5'-ди-трег-бутил бензил) -
мезитилена [15,26].
Технологическая схема получения представлена на рис. 47.
В реактор 1 (стальной эмалированный аппарат с мешалкой, ру-
башкой для охлаждения рассолом и обратным холодильником) за-
гружают хлористый метилен, мезитилен, 2,6-ди-трет-бутил-4-меток-
симетилфенол и охлаждают до О °C. При этой температуре и ин-
тенсивном перемешивании постепенно добавляют 94%-ную серную
кислоту, перемешивают еще 30 мин и передают в сепаратор 2. По-
сле отстаивания отделяют сернокислотный слой, а органический
слой передают в аппарат 4 на нейтрализацию. Вводят нейтрализу-
ющий агент (аммиак), перемешивают еще 1 ч и фильтруют на
друк-фильтре 5. Фильтрат передают в колонну 6 для отгонки хло-
ристого метилена, который возвращают в цикл. Упаренный раствор
из куба колонны подают в аппарат 7 для кристаллизации, добав-
ляют метанол и отфильтровывают осадок. Пасту из центрифуги 8
передают на сушку и измельчение, а фильтрат направляют на ре-
генерацию метанола. Получают продукт с т. пл. 239—240 °C.
Стабилизатор известен в СССР под торговой маркой Агидол 40
или АО-40.
Свойства и применение. 2,4,6-Трис (4/-гадрокси-3/,5'-ди-трет-бу-
тилбензил) мезитилен — белое кристаллическое вещество; т. пл.
244 °C. Растворим в бензоле, хлоралифатических углеводородах;
плохо растворим в метаноле и этаноле; в воде не растворяется.
Обладает низкой летучестью. Стабилен при высоких температу-
рах.
Практически нетоксичен. Разрешен в СССР для применения в
полимерах, контактирующих с пищевыми продуктами.
Под торговой маркой lonox 330 выпускается фирмой
Shell (США), под маркой Ethyl Antioxidant 330 — фирмой Ethyl
(США).
Высокоэффективный неокрашивающий, нелетучий и нетоксич
ный стабилизатор полипропилена, полиэтилена, полиизобутилена;
полиформальдегида, полиуретанов, полиацеталей и поликарбона
тов. Защищает от термоокислительной деструкции полипропилене
вые, полиамидные и полиэфирные волокна, резины на основе нату
рального каучука и латекса. Предохраняет косметические и ле
карственные препараты от окисления.
296
Эфир 4-гидрокси-3,5-ди-трет-бутилпропионовой кислоты
и пентаэритрита
Этот стабилизатор получают переэтерификацией метилового
эфира 4-гидрокси-3,5-ди-т-рет-бутилпропионовой кислоты пентаэрит-
ритом [21]
в присутствии основного катализатора. В колбу, снабженную ме-
шалкой и соединенную с насадкой Дина — Старка, помещают пен-
таэритрит и гидрид лития (катализатор), нагревают до 220°С и
выдерживают 2—3 ч. Затем охлаждают до 50 °C, вносят метиловый
эфир 4-гидрокси-3,5-ди-грв7'-бутилпропионовой кислоты, нагрева-
ют до 185—190 °C и выдерживают еще 13 ч, удаляя выделяющий-
ся метанол из сферы реакции. Затем охлаждают реакционную мас-
су, добавляют бензол, нейтрализуют и промывают раствор. Отде-
ляют органический слой, отгоняют растворитель и перекристалли-
зовывают остаток из гексана. Получают кристаллический продукт
с т. пл. 119—122 °C.
Более технологично проведение этой реакции в среде органи-
ческого растворителя [21]. В реактор, снабженный обратным хо-
лодильником и мешалкой, помещают диметилсульфоксид, метило-
вый эфир 4-гидрокси-3,5-ди-трет-бутилпропионовой кислоты и пен-
таэритрит, перемешивают до образования гомогенной смеси и на-
гревают до 90 °C. При этой температуре перемешивают 1 ч, охлаж-
дают до 50 °C, вносят катализатор (гидрид лития) и перемешивают
до прекращения выделения водорода. Затем подключают реактор
к вакуум-системе и выдерживают при 85—90 °C и остаточном дав-
297
------------------------------------------------------------П
лении 2,66 кПа, пока не прекратится выделение метанола. Нейтра- 1
лизуют реакционную массу, выделяют продукт и перекристаллизо-
вывают его из гексана. Получают белые кристаллы с т. пл. 120—
123 °C.
Свойства и применение. Стабилизатор представляет собой бе-
лое кристаллическое вещество; т. пл. 124 °C. Растворим в бензоле,
ацетоне, хлороформе; ограниченно растворим в метаноле; в воде не I
растворяется. *
Мало токсичен. В ряде стран разрешен для использования в по-
лимерах, контактирующих с пищевыми продуктами.
Выпускается фирмой Ciba — Geigy под торговой маркой Irga-
пох 1010.
Эффективный неокрашивающий и нелетучий стабилизатор пш^|
липропилена, полиэтилена, волокна на основе полипропилена.
ЛИТЕРАТУРА М
1. Cyanamid, пат. 2819329, 1958 г. (США). 'II
2. Esso, пат. 3109829, 1963 г. (США). fl
3. Пат. 928169, 1963 г. (Англия). Я
4. Ethyl, пат. 3346648, 1966 г. (США). Л
5. Ethyl, пат. 3255255, 1965 г. (США). Я
6. Пат. 3845142, 1975 г. (США). 4
7. ICI, пат. 951935, 1964 г. (Англия). fl
8. ICI, пат. 1082204, 1967 г. (Англия).
9. Гурвич fl. А., Кумок С. Т., Стыскин Е. Л. — В кн.: Тезисы I Всесоюзного сим- Д
позиума по органическому синтезу. М., Изд. АН СССР, 1974, с. 19. 1
10. Гурвич fl. А., Старикова О. Ф„ Стыскин Е. Л. —Там же, с. 28. 1
11. Esso, пат. 3644538, 1972 г. (США). J
12. Shell, пат. 3026264, 1960 г. (США). >1
13. Гурвич Я. А. и др. — В кн.: Тезисы I Всесоюзного симпозиума по органиче-И
скому синтезу. М., Изд. АН СССР, 1974, с. 21. Я
14. А. с. 397503, 1973 г. (СССР). Я
15. Пат. 3845142, 1975 г. (США). И
16. Пат. 3704326, 1970 г. (США). Я
17. Antioxidant Good-rite 3114; Техническая информация фирмы Goodrich, Клив-Я
- ленд, 1977 г. Я
18. Antioxidant Good-rite 3125; Техническая информация фирмы Goodrich, Клив- к
ленд, 1977 г. -»
19. Geigy, пат. 3435065, 1971 г. (США). 1
20. Ethyl, пат. 3567682, 1971 г. (США). 1
21. Geigy, пат. 3364250, 1970 г. (США). . 1
22. Distillers Chem., пат. 3925488, 1976 г. (США). Л
23. Ethyl, пат. 3925488, 1976 г. (США).
24. Shell, пат. 6909355, 1969 г. (Нидерланды). fl
25. Shell, пат. 3053803, 1962 г. (США). 1
26. Пат. 859577, 1977 г. (Бельгия). . Л
5.6. АЦИЛИРОВАНИЕ ФЕНОЛОВ. I
СИНТЕЗ о-ГИДРОКСИФЕНИЛКЕТОНОВ Я
Особое место среди фенольных стабилизаторов занимают про- II
изводные фенола, содержащие в орто-положении к гидроксилу I
карбонильную группу. В отличие от алкилфенолов, эти соединения II
298 У
не являются антиоксидантами, но защищают полимеры от фото-
деструкции [1, 2].
Простейшими светостабилизаторами этого типа являются алк-
окси-2-гидроксибензофеноны и 4-алкоксифенил-2-гидроксиалкилке-
ТОНЬЦ
НО
R—н-алкил Ci—Св; R'—н-алкил С3—Ci7
Эти соединения могут быть синтезированы несколькими способа-
ми, но исходным сырьем во всех синтезах является резорцин.
По первому способу алкилируют резорцин галогеналкилами
или галогенсульфатами и получают диалкилрезорцин:
Для введения карбонильной группы ацилируют диалкилрезорцин
бензойной кислотой или ее хлорангидридом и получают 2,4-диалк-
оксибензофенон:
При нагревании последнего с А1С13 происходит селективное деал-
килирование о-алкоксигруппы с образованием 4-алкокси-2-гидрок-
сибензофенона:
RO НО
По той же схеме, используя вместо бензоилхлорида хлорангидри-
ды карбоновых кислот жирного ряда, получают 4-алкоксифенил-2-
гидроксиалкилкетоны, причем наибольший интерес представляют
производные карбоновых кислот с пятью и более углеродными ато-
мами в цепи [3].
Ацилирование и деалкилирование можно осуществить в одном
реакторе, не выделяя диалкоксибензофенона; при этом ацилиро-
вание ведут при 10—15 °C, а затем поднимают температуру до
80—90 °C. Селективность деалкилирования, установленная для
бензофенонов [4] и для жирно-ароматических кетонов [5], связа-
на со стерическими факторами, которые благоприятствуют взаимо-
действию А1С13, связанного в комплексе, с алкоксильной группой,
299
расположенной в орто-положении к карбонилу, что приводит к
деалкилированию:
Синтез стабилизаторов по этой схеме имеет практическое значе-И
ние в СССР и за рубежом. В качестве алкилирующего агента на>В
первой стадии используют диметилсульфат и алкилбромиды. Наи-’Я
большее значение имеют стабилизаторы, полученные на основе |
октилбромида. Чтобы удешевить стабилизаторы, вместо индиви- *
дуального С8Н17Вг используют технические смеси алкилбромидов, L
полученных из фракции синтетических спиртов С7—С9 нормально- Л
го строения. w
Получение стабилизаторов по схеме Ш
НО
имеет существенные недостатки. Алкилбромиды (дорогое и де-
фицитное сырье) используются здесь только на 50%, поскольку
при деалкилировании о-алкоксильной группы в присутствии AICI3
алкил отщепляется в виде RC1, являющегося, по существу, отхо-
дом производства. Для ацилирования и деалкилирования исполь-
зуется большое количество А1С13. При деалкилировании идут по-
бочные реакции, приводящие к загрязнению целевого продукта.
Второй путь синтеза стабилизаторов этой группы — ацилиро-
- вание резорцина с образованием 2,4-дигидроксибензофенона и се-
лективное алкилирование последнего в положение 4:
но но но
300
Первая стадия осуществляется взаимодействием резорцина с
бензойной кислотой или с ее хлорангидридом:
Эта реакция, открытая еще в прошлом веке, нашла практическое
применение лишь в последние 25—30 лет. Бензойную кислоту ред-
ко применяют для введения бензоильной группы, гораздо чаще ис-
пользуют ее хлорангидрид — хлористый бензоил. Синтез ведут в
среде хлорбензола или дихлорэтана; катализатором служит AICls-
Резорцин и катализатор вносят в хлорбензол, перемешивают, на-
гревают до 80—90°С и постепенно прибавляют хлористый бензоил.
Смесь выдерживают при этой температуре до завершения реакции,
а затем соляной кислотой разлагают катализатор, отделяют орга-
нический слой и выделяют целевой продукт. Важнейшее условие
синтеза — отсутствие влаги, поскольку А1С13 и хлористый бензоил
гидролитически нестабильны. Этим методом целевой продукт по-
лучают с выходом 94% от теоретического [40].
Вместо хлористого бензоила в качестве ацилирующего агента
используют бензотрихлорид:
НО НО
Эту реакцию можно рассматривать как алкилирование алкилгало-
генидом — подобно алкилированию хлористым бензилом
с последующим гидролизом продукта алкилирования. В качестве
реакционной среды используют водные растворы спиртов, уксус-
ной кислоты и других органических растворителей, смешивающих-
ся с водой. Резорцин растворяют в 35 %-ном метаноле и при 45 °C
добавляют к раствору бензотрихлорид; температура поднимается
до 50—55°C. Реакционную массу перемешивают еще 30 мин, ох-
лаждают и отфильтровывают выпавший осадок продукта; выход
301
97% от теоретического [38]. Добавки хлоридов Zn, Na и К ускоря-
ют процесс. Поскольку бензотрихлорид дешевле и доступнее хло-
ристого бензоила (сырьем для получения которого он является),
а технологическое оформление процесса с ним значительно про- I
ще (не нужны безводные растворители и . большое количество I
А1С13), этот способ получения 2,4-дигидроксибензофенона представ- I
ляется весьма перспективным. Л
Вторую стадию — алкилирование 2,4-дигидроксибензофенона — I
осуществляют взаимодействием с алкилгалогенидами или диметил- I
сульфатом. Гидроксильные группы в 2,4-дигидроксибензофеноне |
неравноцены. Находящаяся в орто-положении к карбонилу гидрок- I
сильная группа образует с ним водородную связь я
.ОН--О 3
НО—----------с—I
и вследствие этого менее реакционноспособна в реакции алкилиро- Е
вания. Из галогенидов наибольшее практическое значение для "
синтеза таких стабилизаторов имеют октилгалогениды. Синтез ве- ’
дут в растворителе в присутствии кислоты, связываемой акцепте- j
рами в форме хлорида или бромида:
НО но |
НО-^~^-С-^Л + С8Н17С1 -~ОНН > СвНиО-^Ч-С-^Л
\ / \ / —КС1, —rigO \ / \ / У
При нагревании 2,4-дигидроксибензофенона с CgHnCl в моноэти- :
ловом эфире этиленгликоля в присутствии Na2CO3 целевой про-
дукт образуется с выходом 83,4% [6]. В качестве растворителя
используют ацетон [7, 15] , циклогексанон [8], диметилформамид 1
или диметилсульфоксид [9], тетрагидрофуриловый спирт [10], али-
фатические спирты С4—С12 [И]. Синтез ведут при т. кип. реак-
ционной массы.
С высоким выходом получен целевой.продукт при алкилирова-
нии в водной среде с добавкой эмульгатора [12—14]. 2,4-Дигидр- d
оксибензофенон и октилбромид кипятят с водным раствором NaOH j
и добавкой додецилсульфоната натрия в течение 8 ч. Отгоняют с !
водой не вступивший в реакцию октилбромид и перекристаллизо- '
Бывают органический остаток; выход продукта 85% от теоретиче-
ского.
Интересен способ получения 2-гидрокси-4-октоксибензофенона j
в среде октилового спирта [11]. Спирт нагревают с К2СО3 и ок-
тилбромидом до 150 °C, за 4 ч добавляют раствор 2,4-дигидрокси-
бензофенона в октиловом спирте и перемешивают в течение 2 ч.
Охлаждают смесь, отфильтровывают осадок минеральных солей
и перегоняют продукт при пониженном давлении. Выход 98,3% от
теоретического. Добавка небольших количеств иодидов ускоряет
алкилирование [6, 7, 16].
302
Третий путь синтеза 2-гидрокси-4-алкоксибензофенонов — аци-
лирование моноалкиловых эфиров резорцина [17]. Их синтезиру-
ют взаимодействием монокалиевой соли резорцина с алкилброми-
дом [18]. Например, синтез монооктилового эфира ведут, посте-
пенно добавляя раствор КОН в этаноле к этанольному раствору
резорцина (мольное отношение КОН :резорцин=1: 1). К получен-
ному раствору монокалиевой соли резорцина добавляют такое же
количество резорцина (в виде этанольного раствора) и октилбро-
мид. Смесь нагревают до 72 °C и выдерживают 6—8 ч. Затем от-
фильтровывают выпавший осадок КВг, отгоняют спирт, а остаток
промывают водой и перегоняют при пониженном давлении. Выход
монооктилового эфира резорцина 70% от теоретического считая на
резорцин:
НО НО НО
A +KOH />—+с£Н1,Вг /Г~С
н°~О к°“О w-o
В присутствии А1С13 моноалкиловые эфиры ацилируются бен-
зоилхлоридом в орто-положение к свободной НО-группе, образуя
2-гидрокси-4-алкоксибензофеноны с выходом «80% от теоретиче-
ского [17]:
НО НО
С8н17о-^Х -pcic-^A
\\ / —riel \ / \ /
В реактор загружают дихлорэтан, монооктиловый эфир резорци-
на и бензоилхлорид, перемешивают и охлаждают до 0°С. При
перемешивании постепенно вносят А1С13 и перемешивают 1 ч при
10 °C, 6 ч при 20 °C и 2 ч при 30 °C. Затем постепенно приливают
реакционную массу к охлажденной смеси разбавленной соляной
кислоты и дихлорэтана. Органический слой отделяют, промывают
водой и перегоняют при пониженном давлении. Выход «80% от
теоретического. Вместо А1С13 в этом синтезе в качестве катализа-
тора используют порошок железа [19]. При 220 °C монометиловый
эфир резорцина за 3 ч образует 2-гидрокси-4-метоксибензофенон
с выходом 63%:
но но
Описано получение 2-гидрокси-4-алкоксибёнзофенона окислени-
ем производных дифенилметана [20]:
303
Для создания промышленного производства 4-алкокси-2-гидр.
оксибензофенонов наиболее целесообразен синтез через 2,4-дигидр-
оксибензофенон:
НО НО НО
В этом случае на основе одного промежуточного продукта, но
используя разные алкилирующие агенты, можно получить широ-
кий ассортимент фенольных светостабилизаторов.
Среди производных 2,4-дигидроксибензофенона имеется ряд
соединений, содержащих в молекуле два ароматических фрагмен-
та с о-гидроксильной группой и имеющих такую общую формулу:
Их получают взаимодействием 2,4-дигидроксибензофенона с раз-
личными бифункциональными соединениями," например с 1,3-ди-
хлоргидрином глицерина '[21].
Взаимодействием резорцина с салициловой кислотой получают
2,2',4-тригидроксибензофенон [22, 27], а его алкилированием —
2,2'-дигидрокси-4-алкоксибензофенон [26]:
НО он
Из соединений этого типа практическое значение имеют 2,2'-ди-
гидрокси-4-метоксибензофенон, выпускаемый фирмой Cyanamid
(США) под торговой маркой Cyasorb UV 24 [23], и 2,2'-дигидрок-
си-4-октоксибензофенон.
На основе салициловой кислоты получают ряд более простых
соединений, являющихся светостабилизаторами [24]. Например,
взаимодействием салициловой кислоты с фенолом в присутствии
РОС13 получают фениловый эфир салициловой кислоты (известный
под названием Салол), а взаимодействием с 4-т’рет-бутилфено-
лом — 4-7’рет-бутиловый эфир салициловой кислоты, выпускаемый
фирмой Dow (США) под торговой маркой Inhibitor TBS:
Салол Inhibitor TBS
Для промышленного производства 2-гидрокси-4-алкоксибензо-
фенона наибольшее значение, по-видимому, имеют способы их
304
синтеза через 2,4-дигидроксибензофенон. Вместе с тем продолжа-
ется совершенствование других способов синтеза. В частности,
фирма Bayer (ФРГ) разработала способ получения таких стаби-
лизаторов путем ацилирования диалкилового эфира резорцина
бензоилхлоридом в присутствии TiCU, причем селективное деалки-
лирование проводится без выделения промежуточного продук-
та [25].
2,4-Д игидроксибензофенон
Это соединение впервые было получено около 100 лет назад ацилированием
резорцина бензойной кислотой или хлористым бензоилом
Синтез вели в присутствии хлорида цинка, но выход целевого- продукта был
очень низким.
По другому варианту синтез вели в среде полифосфорной кислоты, исполь-
зуя в качестве катализатора безводный ZnClj и РС13 [29]. Здесь удалось суще-
ственно повысить выход целевого продукта, но для промышленного использова-
ния метод оказался неудобен из-за применения больших количеств катализатора
и растворителей; кроме того, здесь образуются в большом количестве сточные
воды, содержащие соединения фосфора и цинка. В качестве катализатора аци-
лирования резорцина бензойной кислотой предложен HF [31].
Близким к нему является способ получения 2,4-дигидроксибен-
зофенола перегруппировкой монобензоатрезорцина по Фрису [30] :
Эфир получают взаимодействием небольшого избытка резорцина
с хлористым бензоилом в среде инертного растворителя при 30—
35 °C; катализатор — ZnCl2. Растворяют эфир в хлорбензоле, до-
бавляют ZnCl2, нагревают до 120 °C и пропускают сухой НС1. Пос-
ле выделения получают продукт с т. пл. 142—4 °C.
С выходом 94% от теоретического 2,4-дигидроксибензофенон
Получают ацилированием резорцина бензоилхлоридом в присутст-
20-Ю57
305
вии А1С13 при 90 С в среде хлорбензола [39]. Этот способ может
представить практический интерес.
С высоким выходом 2,4-дигидроксибеизофенон получают по
реакции Геша взаимодействием резорцина и бензонитрила:
НО
Резорцин, бензонитрил и ZnCk (безводный) перемешивают в те-Я
чение 30 мин при 25—30 °C, затем пропускают НС1, перемешиваютЯ
8 ч при 50 °C и, добавляя воду, гидролизуют продукт реакции. Ле-Я
тучие вещества отгоняют с водяным паром, остаток охлаждают, 'Я
а образовавшийся осадок отфильтровывают. Выход 93% от тео- т
ретического считая на резорцин; т. пл. продукта 133—134 °C '[32]. \
Однако большая длительность синтеза и применение дорогого и {
дефицитного бензонитрила снижают промышленное значение ме-
тода.
Практический интерес представляет способ получения 2,4-ди-
гидроксибензофенона взаимодействием резорцина с бензотрихло-
ридом:
НО НО
При проведении реакции в спиртовой среде образуются побочные
продукты — эфиры бензойной кислоты, от которых целевой про-
дукт очистить трудно.
Было предложено
[53] вести
эту реакцию
в среде уксусной кислоты при 80 °C, а для уменьшения образова-и
ния побочных продуктов добавлять в реакционную массу ацетат1
натрия. Выход целевого продукта 88% от теоретического; т. пл.
141—141,5 °C. В качестве реакционной среды использовали раствор
уксусной кислоты [34], водный метанол [35, 38], воду с добав-
кой эмульгатора и водорастворимых хлоридов металлов [36], бен-
зол с добавкой метанола или этанола [37].
Сравнительно недавно предложен новый способ получения
2,4-дигидроксибензофенона из резорцина и фталевого ангидри-
да [40]:
НО
НО СООН
---->
—со2
306
но
фталевый ангидрид и резорцин выдерживают в течение 4 ч при
180°C, охлаждают до 120°C и нагревают затем 6 ч с 35%-ным
NaOH. После подкисления выпадает осадок 2-(2',4'-дигидрокси-
бензоил)бензойной кислоты. При нагревании последней в среде
хинолина с каталитическим количеством порошкообразной меди
получают 2,4-дигидроксибензофенон с выходом 40% от теорети-
ческого считая на резорцин.
Для очистки технический 2,4-дигидроксибензофенон, получен-
ный разными способами, растворяют в водном NaOH, при pH 7,5
обрабатывают 3—5% (от массы) гидросульфита натрия при
70—100 °C с последующим подкислением и фильтрованием очи-
щенного продукта. Получают светло-желтые кристаллы с т. пл.
144—145°C и выходом 93% [141].
В основу промышленного способа получения 2,4-дигидрокси-
бензофенона положено ацилирование резорцина бензоилхлоридом.
Технологическая схема получения 2,4-дигидроксибензофенона
представлена на рис. 48. В стальной эмалированный аппарат 2
с мешалкой, рубашкой и обратным холодильником загружают без-
водный хлорбензол, резорцин и хлористый алюминий. Смесь пере-
мешивают, нагревают до 75—80 °C и при этой температуре посте-
пенно прибавляют из мерника 1 бензоилхлорид. После подачи все-
го бензоилхлорида вновь перемешивают реакционную массу и пе-
Рис. 48. Технологическая схема получения 2,4-дигидроксибензофеиона:
/ — мерник; 2 — реактор ацилирования; 3 —ап-парат для выделения 2,4-дигидрокси дибензил-
фенола; 4 — друк-фильтр с поднимающейся мршалкой; 5 — емкость.
20* ЗЭ7
ред’ают на выделение 2,4-дигидроксибензофенона в аппарат Я
Выделение ведут, постепенно добавляя к реакционной массе
8%-ный раствор соляной кислоты. Образующуюся суспензию филь!
труют на друк-фильтре 4 с мешалкой, промывают, отжимают и
передают на синтез светостабилизаторов.
Свойства и применение. 2,4-Дигидроксибензофенон — белое
кристаллическое вещество; т. пл. 148°C. Растворяется в этаноле^
метаноле, диэтиловом эфире; ограниченно растворяется в воде. J
В СССР известен под названием, Бензон О. Под различными
торговыми марками производится рядом зарубежных фирм: <
Торговое наименование Страна, фирма T. пл , °C
Advastab 48 ФРГ, Advance 145
Inhibitor DHBP США, Eastman —
Usolvin PS США, Color 136—140
UV Absorber DHB ГДР, VEB Greiz-Dolau 144—147 4
Uvinul 400 США, Aniline 136—140
2,4-Дигидроксибензофенон является эффективным светостаби-
лизатором лакокрасочных покрытий и ацетатного волокна; менее
эффективен при стабилизации полиолефинов и ненасыщенных по-
лиэфиров.
Служит важнейшим промежуточным продуктом в синтезе ши-
рокого ассортимента фенольных светостабилизаторов. Ниже при
ведены формулы некоторых промышленных стабилизаторов, по-
лучаемых из 2,4-дигидроксибензофенона:
2-гидрокси-4-(2-этилгексокси)бензофенои
2-гидрокси-4-(3'-акрилоксипропокси-2-гидрокси)бензофенон
308
4-Алкокси-2-гидроксибензофенон
Этот светостабилизатор представляет собой смесь 2-гидрокси-
4-алкоксибензофенонов с алкильной составляющей Су—С9:
НО
Описываемый смесевой стабилизатор является, по существу,
аналогом высокоэффективного 2-гидрокси-4-октоксибензофенона,
но, в отличие от него, производится из сравнительно дешевой сме-
си алкилбромидов С7— С9 вместо дорогого и дефицитного октил-
бромида.
Стабилизатор получают двумя способами.
По первому способу алкилируют резорцин алкилбромидами,
перегоняют полученную смесь диалкилрезорцинов и передают на
ацилирование хлористым бензоилом в присутствии А1С1з. Эту реак-
цию ведут при 10—15 °C, а потом поднимают температуру до 80—
90 °C и проводят деалкилирование. Затем разлагают катализатор,
отделяют органический слой от водного и перегоняют целевой про-
дукт;
НО RO
4-NaOH ft V.
+ RBr —7-----RO—< - \ R = 0,-0,
\ / 1 — NaBf, —НаО \ /
НО
Ио
—С—/-Ч
Недостатки способа — высокий расход алкилбромидов и трудность
получения стандартного состава конечного продукта.
По второму способу исходным соединением служит 2,4-дигидр-
оксибензофенон, который алкилируют смесью алкилбромидов
С7Н15Вг—С9Н19Вг:
Алкилирование ведут избытком алкилбромидов в врдно-эмульсион-
ной среде в присутствии щелочи. После отделения водно-солевого
309
Рис. 49. Технологическая схема получения 4-алкокси-2-гидроксибензофенона:
1 — мерник; 2 — реактор алкилирования; 3 — сборник; 4 — аппарат для отгонки избытка ал-
килбромидов; 5, 7 — холодильники; 6 — промежуточная емкость; 8 — пленочный испаритель.
слоя и нейтрализации отгоняют избыток алкилбромидов и целе-
вой продукт.
Технологическая схема получения стабилизатора представлена
на рис. 49. В алкилатор 2 (стальной эмалированный аппарат
с мешалкой и рубашкой) загружают пасту 2,4-дигидроксибензофе-
нона, раствор едкого натра и эмульгатор (например, ОП-Ю).
Смесь перемешивают, нагревают и добавляют из мерника 1 ал-
килбромиды. Алкилирование ведут при 106—120°C. По окончании
алкилирования добавляют воду для растворения минеральных со-
лей, образующихся при алкилировании, перемешивают и после от-
стаивания отделяют водно-солевой слой.
Органический слой нейтрализуют небольшим количеством со-
ляной кислоты и передают в аппарат 4 для отгонки избытка ал-
килбромидов. Отгонку ведут, нагревая массу водяным паром че-
рез рубашку; отогнанные алкилбромиды направляют на рецирку-
ляцию, а целевой продукт охлаждают, передают в промежуточную
емкость 6, а оттуда дозируют в пленочный испаритель 8. Пары
целевого продукта из верхней части испарителя через холодиль-
ник поступают в сборник. Кубовый остаток, отобранный из ниж-
ней части испарителя, передают на сжигание. Выход целевого
продукта 80% от теоретического.
Свойства и применение. Продукт представляет собой вязкую
светло-оранжевую жидкость; т. пл. 140—260°C (при 533 Па); плот-
ность 1044—1069 кг/м3; Ид = 1,56004-1,5746. Растворим в метано-
310
ле, этаноле, дихлорэтане, диэтиловом эфире; в воде не растворя-
ется.
Светостабилизатор полиолефинов, полистирола, пентапласта,
поливинилхлорида и волокон на основе полиамидов и полиолефи-
нов.
2-Гидрокси-4-метоксибензофенон
НО
Этот светостабилизатор—один из простейших в группе 4-алк-
окси-2-гидроксибензофенонов — был впервые синтезирован путем
алкилирования 2,4-дигидроксибензофенона диметилсульфатом:
Здесь используется различие в реакционной способности 2- и
4-гидроксигрупп в молекуле 2,4-дигидроксибензофенона.
Синтез ведут следующим образом. К изопропанолу добавляют
взятые в эквимольном соотношении диметилсульфат и 2,4-дигидр-
оксибензофенон, перемешивают и нагревают до 35 °C. При этой
температуре в течение 3—4 ч прибавляют 30%-ный NaOH, под-
держивая pH реакционной массы в интервале 7,4-?-7,8. Затем вы-
держивают 1 ч при 35—40 °C, разбавляют водой, охлаждают до
15 °C и отфильтровывают выпавшие кристаллы 2-гидрокси-4-меток-
сибензофенона. После перекристаллизации (из изопропанола) по-
лучают продукт с т. пл. 64—5 °C [49]. В качестве алкилирующего
агента наряду с диметилсульфатом применяют метилбромид, ме-
тилхлорид и метиловые эфиры арилсульфокислот [43—47]. Реак-
цию ведут в присутствии гидроксидов или карбонатов палочных
металлов.
Другой способ получения 2-гидрокси-4-метоксибензофенона —
ацилирование диметилового эфира резорцина бензоилхлоридом с
последующим деметилированием [4, 48, 49]:
31 £
СН3О—
Ацилирование и деметилирование ведут в присутствии А1С13. Од-
нако в условиях деметилирования при 90—100 °C протекают и по-
бочные реакции, снижающие выход целевого продукта и затруд-
няющие его очистку. Для устранения побочных реакций добавля-
ют соляную кислоту [44], хлориды цинка, натрия, калия [28].
Вместо А1С13 в этом процессе используют и Т1С14 [25].
Бензоилхлорид и диметиловый эфир резорцина растворяют в
хлорбензоле, потом в течение 1 ч при 35 °C добавляют хлорид ти-
тана и нагревают до 120 °C. Выдерживают 1 ч, охлаждают реак-
ционную смесь до 70 °C и при этой температуре порциями добавля-
ют воду. Отделяют водный слой; от органического слоя отгоняют
часть растворителя, добавляют метанол и после охлаждения от-
фильтровывают выпавший осадок. Получают светло-желтые кри-
сталлы с т. пл. 69°C. Выход 78% от теоретического считая на ди-
метиловый эфир резорцина.
Для промышленного процесса выбран способ селективного ал-
килирования 2,4-дигидроксибензофенона диметилсульфатом.
Технологическая схема получения представлена на рис. 50.
В реактор 4 из нержавеющей стали с мешалкой, рубашкой и об-
ратным холодильником загружают изопропанол, 2,4-дигидрокси-
бензофенон (ДГБФ) и перемешивают до получения раствора. За-
тем загружают диметилсульфат, нагревают до 30 °C и при этой
температуре и интенсивном перемешивании постепенно приливают
раствор едкого натра. Для завершения реакции поднимают тем-
Рис. 50. Технологическая схема получения 2-гидрокси-4-метоксибензофенона:
Л 2t 3 — мерники; 4 — реактор алкилирования; 5 — аппарат для выделения; 6, 9 фильтры^
7 — аппарат для очистки; 8 — холодильник; 10 — емкость; 11 — аппарат для кристаллизации.
312
пературу до 50 °C и выдерживают 1 ч, а затем добавляют воду
и медленно охлаждают до 15 °C, Образовавшуюся суспензию от-
фильтровывают, осадок промывают изопропанолом и водой, отжи-
мают и передают на очистку. Растворение и очистное фильтрование
ведут, растворяя пасту технического продукта в водном изо-
пропаноле, нагревая раствор до кипения и фильтруя горячий рас-
твор через друк-фильтр. Фильтрат принимают в емкость с мешал-
кой и рубашкой и медленно охлаждают до 15—20 °C. Выпавший,
осадок кристаллического продукта отфильтровывают и передают
на сушку.
Свойства и применение. Это светло-желтое или белое кристал-
личество вещество; т. пл. 70 °C; плотность 1324 кг/м3; насыпная
масса 380 кг/м3. Растворимо в бензоле, стироле, четыреххлористом
углероде, диоктилфталате, этаноле, н-гексане; в воде практически,
не растворяется.
В СССР его производят под названием Бензон ОЛ4. Под раз-
личными торговыми марками выпускается рядом зарубежных
фирм:
Торговое наименование Страна, фирма Т. ПЛ., °C
Advastab 45 ФРГ, Advance 63
Cyasorb UV-9 США, Cyanamid 63—64,5
Ongrostab HMB ВНР 62—65
Uvinul М-40 США, Aniline 62
Uvistat 24 Англия, Ward 62,5—65
2-Гидрокси-4-метоксибензофенон является светостабилизато-
ром полистирола, пентапласта, поливинилхлорида, ацетобутирата
целлюлозы, а также волокон на основе полиамидов и полиолефи-
нов. Применяется как светостабилизатор лакокрасочных покры-
тий.
ЛИТЕРАТУРА
1. Луковников А Ф., Матвеева Е. Н. — В кн.: Старение и стабилизация полиме-
ров. М., Наука, 1964, с. 91—132. /
2. Гурвич Я- А., Золотаревская Л. К.', Кумок С. Т. — Фенольные стабилизаторы.
М., Изд. ЦНИИТЭнефтехим, 1978. 80 с.
3. А. с. 215930, 1968 г. (СССР).
4. А. с. 130507, 1960 г. (СССР).
5. Гурвич Я. А., Зимин Ю. Б. — Ж- орг. химии, 1969, т. 5, с. 1658.
6. Cyanamid, пат. 2142593, 1972 г. (ФРГ).
7. Corlische Chem. Works, пат. 1901482, 1970 г. (ФРГ).
8. Furukava Electr., пат. 1947920, 1975 г. (ФРГ),
9. Cyanamid, пат. 1194320, 1969 г. (Англия).
10. Witsubishi, заявка 117453, 1974 г. (Япония).
11. Пат. 121088, 1966 г. (ЧССР).
12. Pensylvania Ind. Chem., пат. 3697599, 1972 г. (США).
13. Furukava Electr., пат. 3639483, 1972 г. (США). \
14. Cyanamid, пат. 1145318, 1969 г. (Англия).
313
15. Eastman Kodak, пат. 2861053, 1958 г. (США).
16. Ferro, пат. 1167679, 1969 г. (Англия),
17. А. с. 218144, 1968 г. (СССР).
18. А. с. 172827, 1965 г. (СССР).
19. Земзина И. Н., Степаниченко И. А. — В кн.: Научные труды Ташкентского
ун-та, 1972, вып. 419, с. 180.
20. А. с. 370198, 1973 г. (СССР).
21. А. с. 277764, 1974 г. (СССР).
22. Cyanamid, пат. 3073866, 1963 г. (США).
23. «Plastics Additives», Cyanamid, USA, 1977.
24. Dow, пат. 3270046, 1966 г. (США).
25. Bayer, заявка 2209527, 1972 г. (ФРГ),
26. Cyanamid, пат. 2853521, 1960 г, (США),
27. Cyanamid, пат. 2854485, 1961 г. (США),
28. Aniline, пат. 2861104, 1958 г. (США).
29. Aniline, пат. 3073866, 1963 г. (США),
30. Пат. 3403183, 1968 г. (США).
31. Keppers, пат. 3403183, 1968 г. (США),
32. Du Pont, пат. 3371119, 1968 г. (США),
33. А. с. 248653, 1969 г. (СССР).
34. Пат. 335827, 1972 г. (СССР).
35. Furukava Electr., пат. 3639483, 1972 г. (США).
36. Furukava Denki, пат. 28431, 1973 г. (Япония).
37. Bayer, пат. 2208973, 1973 г. (ФРГ).
38. Пат. 93546, 1972 г. (ГДР).
39. Mutsubishi, пат. 88844, 1974 г. (Япония).
40. Nippon Steel, пат. 64251, 1975 г. (Японий).
41. Du Pont, пат. 3830845, 1974 г. (США).
42. Пат. 52944, 1967 г. (ПНР).
43. Ларин И. А., Матвеева Е. Н„ Кржуткина Л. В. —ЖОХ, 1962, с. 367,
44. Riedel Наёп, пат. 1139848, 1963 г. (ФРГ); РЖХим, 1964, 1ОН87,
45. Пат. 12852, 1966 г. (Япония).
46. Carl. Chem. Works, пат. 3632650, 1968 г. (США); С. А., 1970, v, 73, 16855.
47. Пат. 62052, 1966 г. (ПНР).
48. Cyanamid, пат. 2861053, 1958 г. (США).
49. Cyanamid, пат. 2892872, 1955 г. (США).
ГЛАВА 4
Эфиры фосфористой кислоты
Первые попытки получить полные алкиловые эфиры фосфори-
стой кислоты (триалкилфосфиты) типа (ИО)зР взаимодействием
треххлористого фосфора со спиртами окончились неудачей —
Раильтон (1854 г.) и Вихельхаус (1868 г.) приняли за триэтил-
фосфат полученный ими диэтилфосфит. Позднее А. Е. Арбузов
[1] предложил схему протекающих реакций и объяснил причину
образования диалкилфосфитов даже в присутствии избытка спир-
та. При добавлении спирта к треххлористому фосфору сначала
происходит ступенчатое замещение атомов хлора на алкоксиль-
ные радикалы:
4-ROH +ROH +ROH
РС1з R0PC'2 “wT (ВО)зрС1 —> (RO)3P
—riv.1 —rid — rid
Одновременно идет деалкилирование алкоксильных производных
хлористым водородом, причем деалкилируется почти исключи-
тельно триалкилфосфит:
+HC1
(ВО)3Р —Б7Г (рО)2РОН
Взаимодействие алкилдихлор- и диалкилхлорфосфитов, а так-
же диалкилфосфитов с хлористым водородом при комнатной и
более низкой температуре протекает значительно медленнее. Та-
ким образом, даже при соотношении ROH:PC13 = 3:1 основным
продуктом реакции является диалкилфосфит. Эти же закономер-^
ности были получены позднее при изучении реакции треххлористо-
го фосфора и бутилового спирта [2]. Получение и свойства низ-
ших фосфитов описаны в работах [1, 3, 4].
В качестве стабилизаторов полимерных материалов широко
применяются полные эфиры фосфористой кислоты; их синтез и
свойства наиболее подробно представлены в работах [5—10].
Полные эфиры фосфористой кислоты можно получать несколь-
кими методами.
1. Этерификация треххлористого фосфора соединениями, со-
держащими свободную гидроксильную группу [1—5, 19, 20, 35,
41,42]:
3ROH-|-PC13 —* (RO)3P
—arid
Это — реакция электрофильного замещения; ее скорость определя-
ется нуклеофильностью гидроксилсодержащего соединения. Реак-
ционная способность последних понижается в ряду: опирт>вода>
315
>фенол. Этерификация треххлористого фосфора алифатическими
спиртами осложняется взаимодействием выделяющегося хлористо-
го водорода с триалкилфосфитом, что приводит к получению диал-
килфосфитов (перегруппировка Арбузова). Образование диалкил-
фосфитов протекает через промежуточный комплекс, который ста-
билизируется, выделяя алкилхлорид:
4-НС1
(RO)3P ---> (RO)^ —* (RO)X
\ci “RCI X)
Поэтому получить триалкилфосфит по такой реакции затрудни-
тельно.
Если перегруппировка Арбузова проходит с меньшей ско-
ростью, чем этерификация, то диалкилфосфиты будут примесью к
триалкилфосфитам. Эти смеси трудно разделяются, так как по
физико-химическим свойствам диалкилфосфиты и соответствую-
щие им триалкилфосфиты нередко бывают близки. Если же пере-
группировка Арбузова проходит с большей скоростью, чем этери-
фикация, то для получения триалкилфосфитов с практически при-
емлемым выходом нужно добавлять к реакционной массе вещест-
ва, способные связывать галогеноводород.
В качестве акцепторов галогеноводорода применяют амины [7,
9, 15, 23, 25, 27, 29, 30, 40] (анилин, пиридин, триалкил амины, ди-
этиланилин), аммиак и аммиакаты водорастворимых солей щелоч-
ноземельных металлов [17], оксиды олефинов [19, 34], сополиме-
ры винилпиридинов с диенами [22]. Иногда галогеноводород уда-
ляют, продувая реакционную массу азотом [21, 31, 33]. На прак-
тике предпочтение отдают третичным аминам. Основное их пре-
имущество— хорошая растворимость в большинстве органических
растворителей, что позволяет связывать галогеноводород более
полно. Образующаяся галогеноводородная соль амина практически
не растворяется в органических растворителях, а это способствует
емещению равновесия в сторону образования целевого продукта.
Недостатками третичных аминов как акцепторов хлористого во-
дорода являются трудность их отделения от реакционной массы
(перед выделением целевого продукта) и необходимость регенера-
ции. Кроме того, некоторые из них окрашивают полимер, и поэто-
му фосфиты могут утратить свойства неокрашивающих стабили-
заторов.
Удобным и эффективным акцептором хлористого водорода яв-
ляется безводный аммиак, так как образующийся хлорид аммо-
ния можно легко отделить от реакционной массы. Однако аммиак
-взаимодействует с некоторыми триалкилфосфитами, осуществляя
их частичное или полное деалкилирование
+NHS
(RO)3P ---> ROH + (RO)2PNH2
+NHaCl
2(RO)SP------> (RO)2PNH2-J-(RO)2POH-|-ROH-|-RC1
.316
л поэтому имеет ограниченное применение. Чтобы исключить неже-
лательные реакции, предлагается вести этерификацию треххло-
ристого фосфора спиртами в присутствии аммиака, но не допус-
кать его избытка. Однако в промышленных условиях соблюдать
это требование практически невозможно.
Недавно предложены [22] гетерогенные акцепторы хлористого
водорода — сополимеры винилпиридина с диенами. Они дают об-
надеживающие результаты, так как легко удается отделить хло-
ристоводородную соль от реакционной массы и регенерировать
акцептор.
Синтез триалкилфосфитов взаимодействием алифатических
спиртов с треххлористым фосфором обычно ведут в среде ней-
тральных органических растворителей. Для этого чаще всего при-
меняют алифатические или ароматические углеводороды — гексан,
бензол [13, 14, 25], ксилол [16], фенилциклогексан [26]. Можно
использовать и хлорированные углеводороды [25, 35, 37].
2. Переэтерификация триарил (алкил) фосфитов алифатическими
спиртами [36, 37, 45—56]:
(ArO)3P-f-3ROH (RO)3P
Переэтерификацию чаще всего применяют для получения триал-
килфосфитов, но можно использовать ее и для получения высших
триалкилфосфитов из низших. Реакция, как правило, идет в при-
сутствии катализаторов — веществ основного характера (металли-
ческий натрий [46, 49, 50, 53] и калий [50], щелочноземельные
металлы [49], оксид кальция [46], оксид свинца [46], алкоголя-
ты и феноляты щелочных и щелочноземельных металлов [45, 46,
51], щелочи [56], гидриды и сульфиды щелочных металлов [50], t
цианиды [36], безводный карбонат кальция [46], органические
диэфиры фосфористой кислоты [47, 51]). Кроме того, в качестве
катализаторов можно применять амины с высокой температурой
кипения: C12H25NH2, C12H25NHCH3, С12Н25К(СНз)2,
С6Н5П (С2Н5)2, (СН2СН2ОН) 3N, хинолин [49].
Переэтерификация триалкилфосфитов (RO)3P алифатическими
спиртами R'OH (где Rz >R) протекает с одновременным образо-
ванием трех продуктов [45]:
—ROH
(RO)3P
+R'OH
—2ROH
—3ROH
(RO)2(R'O)P
(RO) (R'O)2P
(R'O)3P
Состав продуктов определяется избытком спирта.
3. Алкоголиз (фенолиз) три(М,Ы-диалкиламидов) фосфористой
кислоты спиртами [57—60]:
(R2N)3P + 3R'OH _3NHR-> (R'O)3P
317
Триалкилфосфиты при этом образуются почти с теорехическим
выходом. Реакцию обычно проводят при температуре выше т. кип.
выделяющегося вторичного амина. Из-за трудностей получения
три(Ы,М-диалкиламидов) этот способ не получил широкого приме-
нения.
4. Разложение комплексов триалкилфосфитов с галогенидом
меди [61]:
(RO)sP CuHal -. > (RO)SP
—v,oCli2iial2
Этот комплекс получают взаимодействием спирта, треххлористого
фосфора и галогенида одновалентной меди. Разложение комплекса
осуществляют термообработкой в вакууме или химическим пу-
тем (используя цинковую пыль, сульфид натрия, пиридин, циа-
ниды) .
5. Полные ароматические эфиры фосфористой кислоты могут
быть получены из хлорангидридов дифенил фосфористой или пи-
рокатехинфосфористой кислоты [Н]. В промышленном масштабе
эти эфиры почти исключительно получают взаимодействием спир-
тов или фенолов с треххлористым фосфором.
Триалкилфосфиты на полупроизводственных установках нача-
ли производить в США с 1953 г. [62]. Сырьем для их получения
служили спирты (этиловый, бутиловый, а главным образом окти-
ловый) и треххлористый фосфор. Вследствие коррозионного дей-
ствия РС13 и НС1 процесс вели в стеклянных аппаратах. Реакции
образования триалкилфосфитов сильно экзотермичны, поэтому про-
водятся при охлаждении.
Триалкилфосфиты — бесцветные жидкости с приятным запахом.
Термически нестабильны. На воздухе тоже разлагаются, а в при-
сутствии влаги гидролизуются. Скорость гидролиза фосфитов за-
висит от строения: с увеличением числа углеродных атомов в ал-
кильном радикала уменьшается способность к гидролизу триалкил-
и три (алкиларил) фосфитов. Полные ароматические эфиры фос-
фористой кислоты обычно более стабильны к гидролизу, чем сме-
шанные.
Три-н-октилфосфит
(С8Н17О)3Р
Три-м-октилфосфит [14—36, 38, 39, 49, 57, 61] и другие стаби-
лизаторы этого типа могут быть получены всеми реакциями, опи-
санными выше. Промышленные процессы основаны на этерифика-
ции треххлористого фосфора спиртом и различаются применяе-
мым органическим растворителем, акцептором хлористого водоро-
да и условиями реакции, обусловленными свойствами исходных
веществ и целевого продукта.
Технологическая схема получения. Три-н-октилфосфит получа-
ют этерификацией треххлористого фосфора к-октанолом в при-
318
сутствии триэтиламина:
РС13 + ЗС,Н17ОН + 3(C2Hb)3N -> Р(ОСвН17),4-3(СД)^.НС1
В реактор загружают н-октанол, триэтиламин и бензол. Смесь ох-
лаждают до минус 5 —минус 3°С и при перемешивании добав-
ляют раствор треххлористого фосфора в бензоле. Этерификацию
проводят при 0±2°С; время реакции «3 ч. Затем выдерживают
реакционную массу в течение 3 ч при 0±2°С, медленно нагревают
до комнатной температуры и выдерживают еще 2—3 ч. После за-
вершения реакции масса представляет собой суспензию три-н-ок-
тилфосфита, солянокислого триэтиламина и непрореагировавшего
октанола.
Фильтрованием отделяют осадок солянокислого триэтиламина
от бензольного раствора три-н-октилфосфита. Бензольный раствор
направляют на нейтрализацию и выделение три-н-октилфосфита,
а водный — на регенерацию триэтиламина.
Выход три-н-октилфосфита »85%. Потери продукта происхо-
дят в основном при промывке, так как нет четкого разделения
слоев вследствие эмульгирующего действия н-октанола, присутст-
вующего в реакционный массе.
Другой способ выделения три-н-октилфосфита — обработка ре-
акционной массы раствором едкого натра. После разделения и
промывки от органического слоя отгоняют сначала азеотропную
смесь бензола с водой, а затем смесь бензола, триэтиламина и
н-октанола. Три-н-октилфосфит содержит до 1,5% н-октанола ц
следы смолистых веществ, которые придают три-н-октилфосфиту
слабую окраску. Для получения бесцветного продукта его перего-
няют в вакууме.
Свойства и применение. Три-н-октилфосфит представляет собой
прозрачную жидкость; плотность 8937 кг/м3; ид = 1,4480. Смеши-
вается с органическими растворителями без ограничения; в воде не
растворяется. В водных эмульсиях (особенно в присутствии кис-
лот и щелочей) может частично гидролизоваться с образованием
фосфористой кислоты и октанола.
Три-н-октилфосфит применяется в основном как компонент сме-
севых стабилизаторов поливинилхлорида и сополимеров винилхло-
рида. Оказывает стабилизирующее действие на синтетические
каучуки и резины. Защищает полиуретаны от старения и действия
светопогоды, повышает теплостойкость полиэтилентерефталата и
акрплонитрил-бутадиен-стирольных сополимеров. Понижает окис-
ляемость каменноугольных масел. Область его применения огра-
ничена из-за склонности к гидролизу.
Три(2-этилгексил) фосфит
[С4НВ—СН—сн2—О—1 Р
СУТ; _1з
Три (2-этилгексил) фосфит получают методами, рассмотренными
выше ;[15, 17, 22, 23, 25—28, 30, 36, 49, 57, 61].
319
Технология получения. В стальной эмалированный аппарат за-
гружают 2-этилгексанол, триэтиламин и толуол. Реакционную мас-
су охлаждают до —5 °C и медленно добавляют раствор треххло-
ристого фосфора в толуоле при энергичном перемешивании; тем-
пература не должна быть выше О °C. Выдерживают в течение 2 ч
при 20 °C, охлаждают реакционную массу до 10 °C и добавляют
25%-ный раствор едкого натра. После разделения промывают ор-
ганический слой водой и перегоняют. Первая фракция с т. кип.
185—210°C (при 266 Па), представляющая собой смесь 2-этилгек-
санола с три (2-этилгексил) фосфитом, может быть использована
на повторных операциях.
Фосфитирование 2-этилгексанола и разложение солянокислого
триэтиламина описываются уравнениями:
4-РС1з
3C4HS—СН—СН.2ОН —ГС4Н8-СН-СН2-О-1 Р
| -—orlCl | I
6-гНв _ С2Н, J3
NaOH
HCl-HCsHsJsN —* (C2H6)3N-HC1 ---> (C2H5)3N + NaCl + Н2О
Свойства и применение. Три (2-этилгексил) фосфит представля-
ет собой бесцветную прозрачную маслообразную жидкость желто-
ватого цвета с неприятным запахом; плотность 900—905 кг/м3 при
20°С; л2Л = 1,4480; т. кип. 230°С при 1,33 кПа и 170—200°С при
133—266 Па. Содержит примеси моно- и ди (2-этилгексил) фосфи-
тов. Растворяется в большинстве органических растворителей,
гидролизуется водой, кислотами и щелочами.
Горюч; т. всп. 162 °C в открытом тигле и 129 °C в закрытом;
т. воспл. 229 °C; т. самовоспл. 247 °C; нижний предел воспламене-
ния 0,3% (об.).
Мало токсичен; при длительном воздействии обладает слабым
кумулятивным эффектом.
Три (2-этилгексил) фосфит чаще всего применяют как компо-
нент комплексных (смесевых) стабилизаторов поливинилхлорида
и сополимеров винилхлорида. Является стабилизатором синтети-
ческих каучуков, резин, полиэтилентерефталата и некоторых моно-
меров, в частности ароматических аминов. Область его примене-
ния ограничена из-за склонности к гидролизу.
Трифенилфосфит
Ю-Нг
Трифенилфосфит можно получить вышеописанными методами
[1, 16, 18, 31, 58, 59, 63—67]. Как правило, его получают этери-
фикацией треххлористого фосфора фенолом:
ЗС6Н6ОН 4- РС13 —-* (СЯН6О)3Р
—ОГ1С1
320
Технология получения. В стальной эмалированный реактор с
рубашкой, якорной мешалкой и ротаметром (для пропускания азо-
та) загружают фенол и нагревают до 40—50 °C без перемешива-
ния. После расплавления фенола включают мешалку и из мерника
подают треххлористый фосфор. Выделяющийся хлористый водо-
род поглощается в водных ловушках с образованием соляной
кислоты. По окончании подачи треххлористого фосфора выдер-
живают смесь при 40—50°C в течение 1 ч. Затем через нижний
спуск реактора подают осушенный азот; одновременно реакцион-
ную массу нагревают до 190—195 °C. Выдерживают при этой тем-
пературе в течение 2 ч; в это время из реакционной массы вместе
с хлористым водородом частично удаляется не вступивший в реак-
цию фенол, который собирают в приемник. Затем в реакторе со-
здают вакуум и при 160—165°C отгоняют оставшийся фенол, а ре-
акционную массу направляют на вакуум-разгонку. Товарный три-
фенилфосфит сливают в тару.
Свойства и применение. Трифенил фосфит представляет собой
маслообразную жидкость желтоватого цвета; плотность 1191 кг/м3
при 20 °C; «о =1,5905; т. пл. 25 °C; т. кип. 360 °C при атмосфер-
ном давлении, 200—201 °C при 0,8 кПа и 260°С при 1,33 кПа. Рас-
творяется в большинстве органических растворителей; не раство-
ряется в воде (гидролизуется).
Горюч; т. всп. 190 °C в открытом тигле и 92 °C в закрытом;
т. воспл. 329 °C; т. самовоспл. 487 °C.
Мало токсичен; обладает раздражающим действием и выра-
женными кумулятивными свойствами; ЛД50=1333 мг/кг (для мы-
шей); ПДК 1—2 мг/м3.
Трифенилфосфит выпускается под разными наименованиями;
выпускаемые продукты отличаются по плотности и температуре
плавления:
Торговое наименование Страна, фирма Плотность, КГ/мЗ т. пл., «С
Advamod ТРР ФРГ, Advance .
Ergochel Р-03 ПНР 1185
Irgastab ТРР ФРГ 1189 —
Mark 1440 Бельгия, Argus —. — । «
Mellite 310 Англия, Albright — —
Mod Epox США, Monsanto — •
Naftovin TP ФРГ, Kaustschuck 1170—1190 <
Ongrostab 37-P ВНР 1170—1190 23—24
Phosclere T 36 Англия, Pure Chem. 1184 21 (т. затв.)
Phosphan T Италия, Italcolloid 1180—1188 19—22
Phosphit TPP ГДР 1160—1180 23—24
Stabilizer ФРГ, Bayer —
Трифенилфосфит находит широкое применение для стабилиза-
ции синтетических каучуков; пассивирует действие металлов пере-
менной валентности. Вторичный термостабилизатор полиолефи-
21-1057 о2]
нов, полиорганосилоксанов, эпоксидных полимеров и полиэтилен-,
терефталата. Термо- и светостабилизатор эфиров целлюлозы.
Трифенилфосфит проявляет синергическое действие с феноля-j
тами, бензоатами и другими солями органических кислот. Го-1
товые к применению такие смесевые композиции выпускаются, на-'
пример, в Бельгии (фирма Argus) под названиями Mark XX,i
Lancro Q-189 и др. |
Три(п-нонилфенил) фосфит
Три (л-нонилфенил) фосфит может быть подучен одним из выше-
описанных способов [14, 24, 31, 32, 38].
Технологический процесс получения три (n-нонилфенил) фосфи-
та состоит из двух стадий — алкилирования фенола фракцией но-
ниленов (125—175 °C) и фосфитирования полученного л-нонилфе-
нола треххлористым фосфором:
А \ +СэН18 п л +РС13 г А \\ 1
он —> cshm-^J*-oh р
Алкилирование ведут в аппарате из нержавеющей стали с ме-
шалкой, холодильником и рубашкой (для обогрева) в присутствии
катализатора. В реактор загружают расплавленный фенол и фрак-
цию нониленов. Массу при перемешивании нагревают до 35—40 °C
и подают бензолсульфокислоту (катализатор). Через 0,5 ч после
загрузки катализатора нагревают реакционную массу сначала до
120—125 °C, а затем постепенно поднимают температуру до 175 °C.
По окончании алкилирования охлаждают реакционную массу, про-
мывают сырой л-нонилфенол горячей водой и передают на дистил-
ляцию.
Фосфитирование осуществляют в эмалированном аппарате с
мешалкой, обратным холодильником, барботером для подачи азо-
та и системой для поглощения выделяющегося хлористого водо-
рода. В высушенный реактор загружают л-нонилфенол и при 90 °C
начинают подавать треххлористый фосфор, который вводят в те-
чение 45—60 мин. Затем температуру постепенно доводят до
150 °C (за 1—1,5 ч) и выдерживают в течение 5—6 ч до прекра-
щения выделения хлористого водорода (его поглощают водой или
щелочью в течение всего процесса фосфитирования). После этого
охлаждают реакционную смесь до 80 °C и при этой температуре
через барботер подают осушенный азот для отдувки оставшегося
хлористого водорода. Непрореагировавший л-нонилфенол отгоня-
ют в вакууме (в специальном реакторе) при 240 °C. Оставшийся
в кубе три (л-нонилфенил)фосфит охлаждают до 100—110 °C и
сливают в тару.
Свойства и применение. Три (и-нонилфенил) фосфит представ-
ляет собой светло-желтую вязкую жидкость; плотность 970—
322
990 кг/м3. Растворяется в ацетоне, бензоле, лигроине, сольвент-
нафте, четыреххлористом углероде, этаноле. В воде и кислотных
средах гидролизуется.
Нетоксичен; ЛД50=23000 мг/кг. Разрешен в СССР для стаби-
лизации полимеров, контактирующих с пищевыми продуктами.
Три (п-нонилфенил) фосфит выпускается во многих странах под
различными названиями:
Торговое наименование Страна, фирма Плотность, КГ/м3 T. ПЛ., °C
Фосфит НФ СССР 980—990
Advamod TNPP ФРГ, Advance — —-
Advastab CH-55 ФРГ, Advance —
Altofane TNP Франция, Ugine Kuhlmann — —
Antigene TMP Япония, Sumitomo 970 105 (при 0,665 кПа)
Irgafos TNPP Швейцария, Ciba — Geigy 990 —
Irgastab CH-55 Швейцария, Ciba — Geigy Бельгия, Argus — —*•
Mark 329 960—980
Mellite 312 Англия, Albright 990 —-•
Phosclere P 194 Англия, Pure Chem. 990 —
Phosclere T 315 Англия, Pure Chem. 990 Меньше —5 (то затв.)
Phosphan E Италия, Italcolloid 1056—1062
Phosphit TNPP ГДР 985 ——
Polygard США, Naugatuck 990 —
Wytox 312 США, National Polychem. — —
Тринонилфенилфосфит находит широкое применение для стаби-
лизации синтетических каучуков. Предотвращает гелеобразование.
Повышает светостойкость полихлоропрена; предотвращает термо-
окислительную деструкцию поликарбоната. Является эффективным
термостабилизатором ударопрочного полистирола, полиуретанов,
поливинилхлорида, акрилонитрил-бутадиен-стирольных сополиме-
ров.
Три( а-метилбензил фен ил) фосфит
Три (га-метилбензилфенил) фосфит представляет собой смесь
полных эфиров фосфористой кислоты и продукта реакции фенола
со стиролом:
Известен единственный способ получения три («-метилбензилфе-
нил) фосфита — взаимодействие смеси а-метилбензилфенолов с
21
323
треххлористым фосфором [12, 21, 33, 34, 43]. Продукт фосфитиро-
вания имеет неоднородный состав, так как исходный алкил фенол
состоит из моно-, ди- и тризамещенных фенолов, получаемых при
алкилировании фенола стиролом. Кроме того, в готовом продукте
может присутствовать некоторое количество неэтерифицирован-
ных а-метилбензилфенолов.
По методу, принятому в ЧССР [34], смесь а-метилбензилфено-
лов нагревают при непрерывном перемешивании до 100 °C и мед-
ленно (в течение 1 ч) вводят треххлористый фосфор. В результате
реакции температура поднимается до 150°C и обильно выделяется
хлористый водород. После выдерживания смесь охлаждают до
100 °C и в течение 2 ч отдувают хлористый водород вначале диок-
сидом углерода, а затем оксидом этилена (для удаления следов
НС1). При других методах [12, 21, 33] для отдувки хлористого
водорода используют азот. Смесь а-метилбензилфенолов и трех-
хлористого фосфора перемешивают в течение 10 мин, нагревают
до 90 °C, выдерживают при этой температуре и нагревают реак-
ционную массу до 150°C, одновременно барботируя азот. Реак-
ционную массу перемешивают в течение 5 ч при 150 °C, нагрева-
ют до 180 СС и увеличивают скорость подачи азота для более пол-
ного удаления НС1. Оба способа обеспечивают получение три(а-ме-
тилбензилфенил)фосфита с 3—5% фосфора и 0,4—0,5% хлора.
Технологический процесс ведут следующим образом. В сталь-
ной эмалированный реактор с нижним спуском, рубашкой для обо-
грева, мешалкой и барботером для подачи азота загружают про-
дукт алкилирования фенола стиролом (смесь а-метилбензилфено-
лов). При перемешивании подают треххлористый фосфор. За 40—
60 мин нагревают реакционную массу до 90 °C и выдерживают
при этой температуре в течение 1 ч. Затем начинают пропускать
осушенный азот, одновременно нагревая массу до 150 °C, в тече-
ние 60—80 мин. При 150°C выдерживают в течение 5 ч, не прекра-
щая пропускать азот со скоростью 100—120 л/ч. Затем массу по-
догревают до 180 °C и выдерживают в течение 5 ч, увеличив ско-
рость подачи азота до 800—1000 л/ч. За эти 5 ч анализируют ре-
акционную массу на содержание хлора. Прн содержании хлора
не выше 0,4% массу, представляющую собой готовый продукт, ох-
лаждают и сливают в сборник.
При фосфитировании большую роль играют режим продувки
и скорость перемешивания. В среднем при загрузке треххлори-
стого фосфора в количестве, соответствующем теоретическому, в
продукт вводят 3,3—3,6% фосфора. При этом возможность образо-
вания продуктов неполного замещения треххлористого фосфора
практически сводится к минимуму, а содержание хлора опреде-
ляется количеством хлористого водорода, растворенного в про-
дукте.
Свойства и применение. Три (а-метилбензилфенил) фосфит пред-
ставляет собой вязкую светло-желтую жидкость; плотность
1050 кг/м3 при 20°С; =1,6130; т. затв. —5°С. Растворяется в
324
бензоле, толуоле, ацетоне, метаноле, этаноле, трихлорэтилене. В во-
де не растворяется. Гигроскопичен; под действием влаги разла-
гается.
Горюч; т. всп. 257°С (в открытом тигле); т. воспл. 321 °C; т. са-
мовоспл. 530 °C. Можно гасить распыленной водой со смачивате-
лями.
Относительно мало токсичен; ЛД50=5847—7340 мг/кг (для
крыс); 20000 мг/кг (для мышей).
В СССР известен под названием Фосфит П-24, в ЧССР под
названием Antioxidant 6.
Три (а-метилбензилфенил) фосфит применяется в качестве ста*
билизатора синтетических каучуков (СКС, СКН, СКЭП), которые
сохраняют начальные вязкость и растворимость в присутствии это-
го стабилизатора при многолетнем хранении. Улучшает светоста-
бильность полихлоропрена. Эффективный термостабилизатор по-
лиэтилена (высокой и низкой плотности), полипропилена и сопо-
лимеров этилена с пропиленом. Предотвращает термоокислитель-
ную деструкцию поликарбоната.
ЛИТЕРАТУРА
1. Арбузов А. Е. Избранные труды. М., Изд. АН СССР, 1952.
2. Gerrard W. е. а. — J. Chem. Soc., 1953, р. 1920.
3. Нифантьев Э. Е. Химия фторорганических соединений. М., Изд. МГУ, 1971.
4. Цветков Е. Н., Кабачник М. И. — В кн.: Реакции и методы исследования ор-
ганических соединений. М., Химия, 1964, т. 13, с. 269—419.
5. Кирпичников П. А., Мукменева Н. А. — В кн.: Старение и стабилизация по-
лимеров. М., Химия, 1966, с. 168—195.
6. Кирпичников П. А. и др. — В ки.: Труды КХТИ, 1965, вып. 34, с. 367—374.
7. Кирпичников П. А., Гурвич Я- А., Иванова М. В. — ЖОХ, 1964, № 3, с. 856—
857.
8. Кирпичников П. А., Мукменева И. А., Победимский Д. Г. — Там же, с. 280—
285; РЖХим, 1967, 19Ж370.
9. Кирпичников П. А., Попова Л. М., Ричмонд Г. Я. —ЖОХ, 1966, № 6, с. 1143—
1147.
10. Верижников Л. В., Кирпичников П. А., Ангерт Л. Г. — В кн.: Труды КХТИ,
1964, вып. 33, с. 287—292.
11. Гурвич Я- А. и др. — Хим. пром-сть, 1965, № 2, с. 20—21.
12. Гурвич Я. А., Зимин Ю. Б. — Хим. пром-сть, 1969, № 3, с. 13—20.
13. Гусев В. К. и др.—Химия и технология топлив и масел, 1968, № 11, с. 14—
17.
14. Мамедова П. С.—Азерб. нефт. хоз-во, 1963, № 2, с. 34—35; РЖХим, 1963,
16П243.
15. Shell, пат. 3255629, 1966 г. (США); РЖХим, 1967, 14Н128.
16. Пат. 31236, 1964 г. (ГДР); РЖХим, 1965, 11Н90.
17. Hochst, пат. 1185168, 1965 г. (ФРГ); С. А., 1965, V. 62, р. 10339.
18. Deutsche Advance, пат. 2007070, 1971 г. (ФРГ); С. А., 1972, V. 76, р. 3542.
19. А. с. 173234, 1963 г. (СССР).
20. А. с. 182151, 1966 г. (СССР).
21. А. с. 202131, 1967 г. (СССР).
22. А. с. 297641, 1971 г. (СССР).
23. Virginia — Carolina, пат. 2678940, 1954 г. (США); РЖХим, 1956, 44451.
24. Rubber, пат. 2733226, 1956 г. (США); РЖХим, 1957, 49427.
25. Monsanto, пат. 2859238, 1958 г. (США); РЖХим, 1960, 53762.
26. Monsanto, пат. 2865942, 1958 г. (США); РЖХим, 1960, 58117.
325
27. Stauffer, пат. 3068267, 1963 г. (США); РЖХим, 1964, 22Н63. • t
28. Olin Mathieson, пат. 3194827, 1965 г. (США); РЖХим, 1966, 18Н96.
29. Hooker, пат. 3271481, 1966; пат. 3281506, 1966 г. (США).
30. Monsanto, пат. 3309431, 1967 г. (США); РЖХим, 1968, 13Н144.
31. Monsanto, пат. 3514506, 1970 г. (США); РЖХим, 1971, 6С527.
32. Пат. 1271964, 1961 г. (Франция); РЖХим, 1962, 20Л52.
33. Goodyear, пат. 3244661, 1966 г. (США); РЖХим, 1968, 9С337.
34. Пат. 115828, 1965 г. (ЧССР); РЖХим, 1967, 19С241.
35. Sumitomo, пат. 8309, 1967 г. (Япония); С. А., 1967, v. 67, р. 733340.
36. Nixon, пат. 47-50091, 1972 г. (Япония); РЖХим, 1974, 2Н100.
37. А. с. 190893, 1965 г. (СССР).
Petrochemicals, пат. 803557, 1968 г. (Англия); РЖХим, 1961, 14П226.
39. Hooker, пат. 1075584, 1960 г. (ФРГ); С. А., 1961, v. 55, р. 15348.
40. Гильм Камай, Хасанов А. С.—Изв. вузов, химия и хим. технология, 1963,
№ 5, с. 799.
41. А. с. 488821, 1975 г. (СССР).
42. Пат. 76659, 1977 г. (СРР); РЖХим, 1978, 23Н150.
43. Hooker, пат. 3415907, 1968 г. (США); РЖХим, 1970, 12Н324.
44. Kuhlmann, пат. 2101275, 1972 г. (Франция); РЖХим, 1973, 10Н151.
45. Имаев М. Г, —ЖОХ, 1961, т. 31, № 6, е. 1770—1773; РЖХим, 1962, 10Ж340.
46. Montrose, пат. 2970166, 1961 г. (США); РЖХим, 1962, 1Л100.
47. Pure, пат. 1173763, 1969 г. (Англия); РЖХим, 1970, 17С466.
48. Weston, пат. 2961454, 1960 г. (США); С. А., 1961, V. 55, р. 25761.
49. Argus, пат. 3056824, 1962 г. (США); РЖХим, 1964, 17Н100.
50. Hooker, пат. 3101363, 1963 г. (США); РЖХим, 1965, ЗН65; пат. 3320337,
1967 г. (США); РЖХим, 1968, 18Н126.
51. Weston, пат. 3326939, 1967 г. (США); РЖХим, 1969, 15Н131.
52. Kuhlmann, пат. 1411831, 1965 г. (Франция); С. А., 1966, v. 64, р. 4942.
53. Yoshitomi, пат. 22092, 1968 г. (Япония); РЖХим, 1969, 19Н134.
54. А. с. 502006, 1976 г. (СССР).
55. Prod. Chimiques, пат. 1500821, 1967 г. (Франция); С. А., 1968, V. 69, р. 86604.
56. Sumitomo, пат. 95942, 1974 г. (Япония); С. А., 1975, V. 82, р. 111761; пат.
51-2455, 1976 г. (Япония); РЖХим, 1977, 3H130.
57. Bayer, пат. 1079022, 1960 г. (ФРГ); РЖХим, 1961, 18Л94.
58. Mitsunobu О. е. а. — Bull. Chem. Soc. Japan, 1966, v. 39, № 2, p. 214—219;
C. A., 1966, v. 64, p. 15780.
59. Burgada R. — Ann. Chemie, 1963. B. 8, № 5, S. 347—381; C. A., 1963, v. 59,
p. 15164.
60. A. c. 182157, 1960 г. (СССР).
61. Ministry of Supply, пат. 786770, 1958 г.; РЖХим, 1959, 46755; пат. 791590,
1958 г. (Англия); РЖХим, 1959, 46754.
62. Chem. Eng., 1953, v. 60, № 2, р. 126, 128—129; РЖХим, 1954, 35067.
63. Eng. Mater., 1964, v. 12, № 4, p. 73—78; РЖХим, 1964, 24C283.
64. Петров К. А. — ЖОХ, 1962, т. 32, № 3, с. 920—923.
65. Cherbuliez Е., Prince R., Rabinowitz J. — Helv. Chim. Acta, 1964, v. 47, № 6,
p. 1653—1659.
66. Hieber W., Duchatsch H. — Naturforsch., 1963, B. 186, № 12, S. 1132—1133.
67. Мукменева И. А., Кирпичников П. А., Пудовик А. И. — ЖОХ, 1963, т. 33,
№ 10, с. 3192—3196.
ГЛАВА 5
Эфиры тио(дипропионовой) кислоты
Эфиры тио (дипропионовой) кислоты и спиртов Сю—Сю—дидо-
дециловый [тио(дилаурилпропионат)], диоктадециловый [тио(ди-
стеарилпропионат)] и дитетрадециловый [тио (димиристилпропио-
нат)]—появились на мировом рынке в 1950-х годах и находят
широкое применение как синергисты фенольных стабилизаторов
для полиолефинов.
Известны три основных метода получения тио(диалкилпропио-
натов) в зависимости от исходного сырья — из пропилена, ацети-
лена и кетена. Эти методы можно представить
превращений [1]:
СН2=СН—СН3
схемой взаимных
сн=сн
сн2=с=о
ch2=chcn
СН2=СН—СОО—СН3
Лактон, тиолактои
S(CH2—CH2CN)2
cich2-ch2cn
CH2=CH-COOR ''
S /сн2—сн2—С—осн3
II
I \ о
I S(CH2—СН2—СС)ОН)2
С1СН2—сн2—соон '
//2
S(CH2—СН2—СООН)а
S(CH2—СНа—СООН)а
Этерификация или переэтерификация -*
ROOC—СН2—СН2—S—СН2—СН3—COOR
тио(диалкилпропионат)
Технико-экономическая оценка способов получения тио(диал-
килпропионатов) показала [2], что во всех случаях синтез осуще-
ствляют не менее чем в две стадии. На первой получают промежу-
точные соединения — тио (дипропионовую) кислоту, ее динитрил
327
'11
или низший диалкиловый эфир. На второй стадии получают ||
тио(диалкилпропионат); при этом превращение динитрила тио (ди- В*
пропионовой) кислоты и тио(диметилпропионата) в тио(диалкил-
пропионат) можно вести как с выделением, так и без выделения j|
свободной тио(диопропионовой) кислоты. - 3|
Синтез тио(дипропионовой) кислоты из лактонов, тиолактонов ®
и других производных пропионовой кислоты осуществляют путем Ж)
продолжительного нагревания их водных растворов в присутствии
едкого натра или сульфида натрия в зависимости от использу-
емого сырья [3—5, 6]. Этот метод применим также для получения
других тиокарбоновых кислот из соответствующих производных.
Получение тио(диалкилпропионатов) из метилакрилата может
быть осуществлено переэтерефикацией метилакрилата высшими
спиртами с последующим присоединением сероводорода к высше- Ям
му алкилакрилату Ж
+2ROH +HaS Ml
2СН2=СН—С—ОСН3 2СН2=СН—С—OR -----> ▼
II —2CH3UH II
о о
СН2—СН2—С—OR
II
или присоединением сероводорода к метилакрилату с последующей
переэтерификацией образовавшегося тио (диметилпропионата) выс-
шими спиртами [7—14]:
—СН2—СН2-С—OR
II
+H2S +2ROH
2СН3=СН—С—ОСН3------> SF—СН2—СН2—С—ОСН3'1 »ОТ
О О J2
---► S
Переэтерификацию метилакрилата высшими спиртами осуще-
ствляют при большом избытке метилакрилата, отгоняя азеотроп-
ную смесь метанола с метилакрилатом в течение 8—10 ч в при-
сутствии ингибиторов полимеризации. Присоединение сероводорода
к высшему алкилакрилату осуществляют, как правило, в среде
растворителя в течение 10—20 ч.
Присоединение сероводорода к метилакрилату происходит в
широком интервале температур, а при переэтерификации тио (ди-
метилпропионата) общая продолжительность процесса составляет
«4 ч.
В качестве агентов, содержащих серу, при получении тио (диме-
тилпропионата) кроме сероводорода могут быть использованы
сульфид натрия, гидросульфиды натрия или аммония, сероуглерод,
тиокислоты и другие соединения, содержащие ион S2-, однако
их применение затрудняется побочными реакциями (гидролиз
сложноэфирных групп в присутствии сульфида или гидросульфида
натрия) и необходимостью работы под давлением (при использо-
228
/
вании сероуглерода). В последнее время было показано, что хо- '
роших результатов можно достичь в присутствии свободной серы
[14] или при сочетании гидросульфида аммония с сероуглеро-
дом [10].
Присоединение сероводорода к метилакрилату обычно осуще-
ствляют в присутствии различных оснований или веществ щелоч-
ного характера — аммиака, триэтиламииа, метилата натрия, чет-
вертичных аммониевых оснований (трилон Б), цианида калия.
В присутствии гидросульфидов натрия или аммония и сероуглеро-
да реакция проходит без катализатора [10]. Главный недостаток
использования оснований —значительное количество сточных вод,
образующихся при промывке реакционной массы с целью удаления
катализатора. Присоединение сероводорода к метилакрилату пред-
ложено проводить в присутствии анионитов [11, 12]. Указанную
реакцию можно осуществить и с помощью веществ, генерирующих
радикалы (например, пероксидов), или под действием электромаг-
нитных излучений высокой частоты [15].
Таким образом, в зависимости от условий и применяемых ка-
тализаторов присоединение сероводорода к метилакрилату может
идти или по радикальному механизму или через промежуточное
активное соединение нерадикального типа. Присоединение серово-
дорода к метилметакрилату в присутствии оснований и анионитов
идет исключительно в p-положение. Скорость реакции возрастает
с увеличением рКа тиолов, рКв оснований, применяемых в качест-
ве катализаторов, и полярности среды [16]. Присоединение проис-
ходит последовательно; в качестве промежуточного соединения
образуется меркаптопроизводное метилакрилата (I) [17]:
H2S + CH2=CH—С—ОСН3 >
II
О
---► HSCH2—СН2-С-ОСН3 + СНа=СН—С—ОСН3 ----►
II ‘ II
О о
Получение высших тио(диалкилпропионатов) из тио(диметил-
пропионата) осуществляют переэтерификацией высшими спиртами
в присутствии катализаторов — кислот Бренстеда и Льюиса. Чаще
всего используют серную, фосфорную и ароматические сульфокис-
лоты, сильнокислотные катиониты, титансодержащие соединения,
изопропилат алюминия [4, 18]. Переэтерификация (как и этери-
фикация) обратима [19]:
S[—СН2—СН2— С—OCH3] + 2ROH ST—СН2—СН,—С— OR] +2СН3ОН
О
О _ 2 . 0-2
Ее равновесие можно сдвинуть вправо, удаляя метанол из зоны
реакции или вводя избыток этерифицирующего спирта.
329
Тио(диалкилпропионаты) могут быть получены из акрилонит-
рила:
1) взаимодействием последнего с безводным хлористым водо-
родом при охлаждении с последующим гидролизом (соляной кис-
лотой) образовавшегося (З-хлорпропионитрила и конденсацией .
p-хлорпропионовой кислоты с сульфидом натрия в щелочной среде а
и последующей этерификацией тио (дипропионовой) кислоты выс- •>
шим спиртом:
+2НС1 «Г4Н2О(НС1)
2CH2=CHCN ----> 2С1СН2—CHjCN —,)~
+Na2S +2R0H
---> 2С1СН2—СН2—СООН —> S(-CH2-C«2-COOH)2 —*•
---► S(—CH2—CH2—COOR)2
2) взаимодействием акрилонитрила с водным раствором суль-
фида натрия или с сероводородом с последующей экстракцией
образовавшегося тиодинитрила бензолом (или другим растворите-
лем) и омылением его концентрированной соляной кислотой до
тио (дипропионовой) кислоты и этерификацией последней [20—23]:
+Na2S
2CH2=CHCN -----*• S(—CH2-CH2CN)2
+4H2O(HC1)
- 2NH3 *
+2ROH Л
----> S(—CH2—CH2—COOH)2 —-> S(—CH2—CH2—COOR)2 H
— zX~12^ №]
По такому методу был получен первый стабилизатор этого клас- Я
са — тио(дилаурилпропионат) — в 1950 г. [22].
Более удобен способ получения тио(диалкилпропионатов) из 1
акрилонитрила, заключающийся в одновременном омылении и эте- 1
рификации тиодинитрила [16, 24, 25]. Этим способом в промыш- 1
ленном масштабе получают тио(дистеарилпропионат). Акрилонит-
рил смешивают с 17%-ным раствором сульфида натрия:
+н20 J
2CH2=CHCN + Na2S ——Sf-C^-CI^CN^
Реакционную массу охлаждают до 10—15 °C (тиодинитрил плавит-
ся при 24°C), после чего образовавшийся продукт выпадает в оса-
док. Его обрабатывают соляной кислотой и октадециловым спир-
том:
+н2о
S(—CH2—CH2CN)2 + НС] 4-ClgH37OH -> S Г-СН2-СН2—С-ОС17Н371
<• II
О J2
Органический слой, являющийся основным продуктом с примесью
спирта, отгоняют от водного и промывают водой. Готовый тио(ди-
стеарилпропионат) содержит до 10% спирта. Спирт не отделяют,
так как он не влияет на качество стабилизатора.
Вследствие высокой токсичности исходного акрилонитрила бо-
лее прогрессивным считается метод получения тио(диалкилпро-
пионатов) из метилакрилата.
330
Рис. 51. Технологическая схема получения тио(диметилпропионата):
1 — скруббер; 2, 9 — холодильники; 3 — реактор; 4 — фильтр; 5 — расходомер; 6, 10, 11 — при-
емники; 7 —перегонный куб; 8 — насадочная колонна; 12 — ловушка.
В промышленном масштабе во многих странах выпускаются
тио(дилаурилпропионат) и тио (дистеарилпропионат) и в меньших
объемах — тио(димиристилпропионат). Области их применения
одинаковы (стр. 332).
Технологическая схема получения тио(диалкилпропионата)
включает две стадии: получение тио(диметилпропионата) (рис. 51)
и получение тио(диалкилпропионата) (рис. 52).
В реактор 3 (рис. 51) загружают метилакрилат и катализатор
(анионит в НО-форме или триэтаноламин). Реакционную массу
нагревают до 60 °C и пропускают через нее сероводород (через
расходомер 5) до тех пор, пока она не будет иметь п о° = 1,47404-
4-1,4750. По достижении такого показателя преломления фильтру-
ют массу через фильтр 4 и собирают в приемник 6. Отходящие из
реактора 3 газы через холодильник 2 и водяной скруббер 1 сбра-
сывают в атмосфер^- Технический тио(диметилпропионат) из при-
емника 6 передают в перегонный куб 7, снабженный насадочной
колонной 8. Продукт собирают в приемнике 10, откуда подают на
переэтерификацию.
При получении тио(диалкилпропионата) в реактор 4 (рис. 52)
загружают, катализатор (TiCU), спирт и тио(диметилпропиоиат) —
ТДМП.’ Нагревают до 150—190 °C и через насадочную колонну /
и холодильник 2 отгоняют выделяющийся метанол, который после
ректификации в колонне 7 собирается в приемнике 9.
После отгонки метанола загружают в реактор влажный акти-
вированный уголь, перемешивают и передают реакционную массу
через фильтр 5 в приемник 6. Отсюда смесь поступает на перегон-
ку в куб 10, снабженный насадочной колонной //.'Здесь отгоняют
избыток спирта, который принимают в приемник 14 и снова ис-
331
12
2
Рис. 52. Технологическая схема получения тио(диалкилпропионата):
1. 11 — насадочные колонны; 2, 8, /2 — холодильники; 3, 6, 9, 13, /4 — приемники; 4 —реак-
тор; 5 — фильтр; 7 — ректификационная колонна; 10 — перегонный куб; /5 — ловушка.
пользуют в реакции. Готовый продукт из куба 10 передают на
расфасовку.
Тио(дилаурилпропионат)
S —СН2—СН2—С—ОС12Н2а
представляет собой белый кристаллический порошок с характер-
ным запахом; т. пл. 42 °C. Растворяется в ацетоне, бензине, бен-
золе, дихлорэтане, уксусной кислоте, этаноле, толуоле, эфире; не
растворяется в воде, глицерине, спиртах.
Практически нетоксичен; разрешен для контакта с пищевыми
продуктами.
Выпускается во многих странах в виде порошка (иногда чешуи-
рованным) с т. пл. 37—42 °C:
Торговое наименование Страна, фирма Т. пл., *С
Advastab PS-800 ФРГ, Advance 39—40
Anti gene TLP Япония, Sumitomo
Antioxidant LTDP США ФРГ, Англия 41,5
DLTP Япония, Yoshitomi 39,5—42
Irganox PS-800 Швейцария, Ciba — Geigy 39—40
Naugard DLTDP США, Uniroyal —
Negonox DLTP Англия, ICI 39,0
Nissan DLTP Япония, Japan Oil «—-
Plastonox LTDP США, Cyanamid 40,3
TPL США, Cyanamid —
332
Тио(дистеарилпропионат)
S Г—СНа—СНа—с—OC18HS7'
яредставляет собой белый кристаллический порошок с характер-
ным запахом; т. пл. 65 °C. Растворяется в ацетоне, бензоле, мета-
ноле, толуоле;
Практически нетоксичен. Разрешен для контакта с пищевыми
продуктами," однако поглощается организмом с большим трудом,
чем тио(дилаурилпропионат), и труднее выводится из организма.
Выпускается в меньших объемах, чем тио(днлаурилпропионат),
в основном в виде порошка:
Торговое наименование Страна, фирма T. пл., °C
Advastab PS-802 ФРГ, Advance 63—65
Antioxidant STOP США, Cyanamid 65
DSTP Yoshitomi Япония, Yoshitomi 63,5—68,5
Irganox PS-802 Швейцария, Ciba — Geigy 63—65
Nissan DSTP Япония, Japan Oil —-•
Plastonox STOP* США, Cyanamid 64,5
Rasmit SS Япония, Daiichi
• Чешуйки.
Тио(димиристилпропионат)
S —СН2-—СН2—С—ОСИН20
представляет собой бйТый кристаллический порошок; т. пл. 51 СС.
Растворяется в бензоле, хлороформе, дихлорэтане, толуоле; не
растворяется в воде, этиловом спирте, этилацетате.
Выпускается в ФРГ, Бельгии и Швейцарии:
Торговое наименование Страна, фирма Выпускная форма Т. пл., °C
Advastab PS-801 ФРГ, Deutsche Advance Порошок « 50
Carstab DMTDP Бельгия, Cinn. Milacron Чешуйки 51
Irganox PS-801 Швейцария, Ciba — Geigy Порошок «50
Применение тио(диалкилпропионатов). Все высшие тио(диал-
килпропионаты) являются антиоксидантами (разрушители гидро-
пероксидов) с пластифицирующими свойствами. Широко применя-
ются как синергисты к различным термо- и светостабилизаторам
(особенно к производным фенола) для полиолефиновых волокон.
333
Тио(диалкилпропионаты) повышают термостойкость лаков на ос-
нове инден-кумароновых смол и цветостойкость термореактивных
полимеров. Являются эффективными стабилизаторами синтетиче-
ских каучуков, жиров и масел. Тио(диалкилпропионаты) могут
найти применение как отвердители эпоксидных полимеров, а так-
же в качестве исходных веществ при синтезе полиэфиров.
ЛИТЕРАТУРА
1. Медведев А. И. Канд. дисс. М., ИХТИ, 1978.
2. Медведев А. И. — Хим. пром-сть, 1973, № 12, с. 896—898.
3. BASF, пат. 840996 и 840997, 1952 г. (ФРГ); Chem. ZbL, 1953, S. 287—288.
4. BASF, пат. 893501, 1953 г. (ФРГ); РЖХим, 1956, 13970.
5. BASF, пат. 917247, 1954 г. (ФРГ); РЖХим, 1956, 66177.
6. Nagakubo К- е. а. — J. Chem. Soc. Japan, Ind. Chem. Soc., 1959, v. 62, № 6,
p. 817—820; РЖХим, 1961, 2П116.
7. ICI, пат. 980219, 1965 г. (Англия); С. А., 1965, v. 62, р. 7645.
8. Matsuda Sumio, пат. 15170, 1965 г. (Япония); РЖХим, 1967, 20Н66.
9. Пат. 17368, 1965 г. (Япония); РЖХим, 1968, 5Н149.
10. Sankyo, пат. 1358019, 1974 г. (Англия); приоритет Японии; РЖХим, 1975,
9Н146.
11. Медведев А. И. и др. — В кн.: Синтез и исследование эффективности химика-
тов для полимерных материалов. Тамбов, Тамбовская правда, 1969, с. 91—93.
12. Медведев А. И. и др. — В кн.: Синтез и исследование эффективности химика-
тов для полимерных материалов. Черкассы, 1974, с. 87—93; РЖХим, 1974,
201181.
13. А. с. 449045, 1974 г. (СССР).
14. Witco, пат. 4052440, 1977 г. (США); С. А., 1977, v. 87, р. 200825.
15. Gerschbein L. L., Hurd С. D.—J. Am. Chem. Soc., 1947, v. 69, p. 241—242;
2328—2335; C. A., 1947, v. 41, p. 3431.
16. Медведев А. И., Игнатов В. А., Романченко T. С. — В кн.: Тезисы докладов
XIII научной сессии по химии и технологии органических соединений серы и
сернистых нефтей. Рига, Зинатне, 1974, с. 232.
17. Медведев А. И., Левин И. И., Романченко Т. С. — В кн.: Органические соеди-
нения серы. Часть I. Состав, промышленное получение, переработка, примене-
ние. Рига, Зинатне, 1976, с. 340.
18. Distillers, пат. 1352686, 1964 г. (Франция); приоритет Англии; С. А., 1964,
V. 61, р. 4224.
19. Каржев В. И. и др. — Хим. пром-сть, 1966, № 2, с. 103—105.
20. Paltin Е., Badiu I., Teodoru Е. — Rev. chim., 1966, v. 17, № 9, p. 627—531;
C. A., 1967, v. 66, p. 65036.
21. Kucsman A., Kremmer T. — Acta chim. Acad. Sci. Hung., 1962, v. 34, № 1,
p. 75—78; РЖХим, 1963, 13Ж124.
22. Gregor J. A., Pugh C. — J. Chem. Soc., 1950, p. 736—738; C. A., 1950, v. 44,
p. 6816.
23. Говорова В. M. — В кн.: Труды Кишиневского сельскохоз. ии-та, 1962, т. 26,
с. 175; РЖХим, 1963, 5Ж92.
24. Bombini — Parodi — Delfimo Soc., пат. 803226, 1968 г. (Италия); С. А., 1970,
v. 72, р. 42838.
25. Oesterreichische Sticks toff, пат. 292652, 1971 г. (Австрия); С. А., 1972, V. 77,
р. 6458.
ГЛАВА 6
Металлсодержащие стабилизаторы
Органические и неорганические соли кальция, бария, кадмия,
цинка, свинца, олова находят широкое применение для стабилиза-
ции поливинилхлорида. Мировая промышленность производит при-
мерно 150 наименований солей стеариновой, лауриновой, малеино-
вой, фталевой, салициловой, угольной, серной, фосфористой и крем-
невой кислот; насчитывается до 1500 торговых марок индивиду-
альных солей и более 100 марок смесевых композиций. Такой ши-
рокий ассортимент стабилизаторов этого вида объясняется не
столько созданием новых химических соединений или синергичес-
ких смесей, сколько растущей конкуренцией зарубежных фирм,
требующей непрерывного совершенствования стабилизаторов с
целью повышения их эффективности и конкурентоспособности
[1].
Ниже приведены данные за 1974—1978 годы по объему произ-
водства солей металлов, используемых для стабилизации поливи-
нилхлорида в США [2]:
Стабилизатор Объем производства, тыс. т
1974 г. Ь75 г. 1976 г. 1977 г. 1978 г.
РЬ-соедииения 10,4 6,4 8,2 8,8 9,1
Ва — Cd-соедииения 22,3 17,5 22,0 23,0 25,0
Са — Zn-соединения 1,4 1,0 1,3 1,4 1,47
Sn-органические соединения 7,5 5,8 7,0 8,0 9,0
Sb-соединеиия —• 0,23
Итого 41,6 30,7 38,5 41,2 44,8
Дальнейшее увеличение объема выпуска к 1990 г. обусловлено
возрастанием производства изделий из жесткого поливинилхлори-
да. При уменьшении доли РЬ-, Ва—Cd- и Ва—Cd—Zn-стабилиза-
торов ожидается рост применения оловоорганических и Са—Zn-со-
единений.
В ближайшем будущем темпы роста объемов использования
солей существенно увеличатся [3]: если ежегодный прирост их
производства в США в 1963—1976 годах составлял «8,1% при
335
общем увеличении потребления пластических масс на 9,1% в год,
то к 1990 г. относительный прирост объемов использования этих
стабилизаторов превысит прирост выпуска пластиков. Такое по-
ложение объясняется необходимостью снижения цен на пластиче-
ские массы за счет использования дешевых минеральных напол-
нителей.
В последнее время получили широкое распространение комп-
лексные соли металлов, которые можно подразделить на 1) про-
стые смеси солей, 2) соосажденные карбоксилаты металлов и
3) комплексные стабилизаторы.
Соосажденные карбоксилаты бария и кадмия совмещают хоро-
шую термостабильность, характерную для солей бария, с хоро-
шей светостойкостью, характерной для солей кадмия. В промыш-
ленном масштабе производятся соосажденные стеараты, лаураты
и рицинолеаты, часто с добавками антиоксидантов.
В настоящее время выпускается большая гамма комплексных
стабилизаторов, характеризуемых высокой эффективностью. Точ-
ный состав сложных комплексных стабилизаторов, вырабатыва-
емых за рубежом, обычно не раскрывается, хотя их с каждым го-
дом производится все больше и в разных выпускных формах [1].
6.1. СОЛИ СВИНЦА
Соли свинца являются в настоящее время наиболее распрост-
раненными стабилизаторами — акцепторами поливинилхлорида.
Преимущество свинцовых стабилизаторов состоит в их малой стои-
мости, однако их существенный недостаток — высокая токсичность,
поэтому имеется тенденция избегать применения порошкообраз-
ных форм. Это достигается гранулированием, обработкой поверх-
ности маслами, жирными кислотами или их солями, диспергиро-
ванием в пластификаторах, созданием жидких стабилизаторов.
Применяемый ассортимент стабилизаторов на основе свинца
можно разделить на три группы: неорганические основные соли с
высоким содержанием реакционноспособного РЬО; средние и ос-
новные свинцовые соли жирных кислот (стеараты, лауринаты);
смесевые стабилизаторы.
Строение большинства неорганических основных солей свинца
изучено недостаточно; часто в результате реакции образуются сме-
си солей с разной основностью. Основные соли свинца производят
в значительных масштабах, и наиболее целесообразен непрерыв-
ный способ получения. Однако разработка непрерывных процес-
сов получения неорганических солей свинца методом химического
осаждения требует решения ряда вопросов [4]; наиболее сущест-
венными являются сложность получения осадка с заранее задан-
ными свойствами и составом и автоматическое регулирование про-
цесса по pH среды.
Получение стабилизаторов-смазок — средних и основных свин-
цовых солей жирных кислот (стеариновая, лауриновая), часто на-
336
зываемых свинцовыми мылами, особых затруднений не вызывает.
Их получают обменным взаимодействием натриевых солей жир-
ных кислот с неорганическими солями свинца или взаимодействи-
ем жирной кислоты с оксидом свинца. В последнем случае при-
мольном соотношении кислота : РЬО = 2 :1 получают средние соли,
а при 2:3 получают основные соли свинца. Скорость реакции за-
висит от степени измельчения РЬО. Часто реакцию проводят в
присутствии катализаторов — растворимых в воде полярных кис»
лородсодержащих алифатических соединений (насыщенные спир-
ты или эфиры). Реакция протекает в сравнительно мягких усло-
виях с высоким выходом солей.
Получение соосажденных солей свинца изучено мало. Большое
распространение получили основные и средние соли, т. е. системы,
включающие все необходимые стабилизаторы и смазки. Примене-
ние сбосажденных солей позволяет упростить составление поли-
винилхлоридных композиций и заметно снизить общее количество
вводимых добавок, так как соосажденные компоненты часто про-
являют синергический эффект. По этой причине, а также вследст-
вие хорошей диспергируемое™ в полимере, они выгодны с эконо-
мической точки зрения. Общий состав смесевых свинцовых стаби-
лизаторов выражается формулой [1]:
лРЬО • PbAQ тРЬ(ЛС2)х • РЬ( АСЯ)Й - zH2O
где п, т и г—мольные доли компонентов; х и у — коэффициенты, зависящие
от основности соответствующего кислотного остатка; АС, — неорганический
двухосновный анион (сульфат, карбонат, фосфит), АСг — остаток алифатической
моно- или дикарбоновой кислоты (стеариновой, лауриновой) или ароматиче-
ской (фталевой, бензойной).
В промышленности выпускается широкий ассортимент соосажден-
ных солей свинпа. Наибольшую известность получили композиции
сульфата и стеарата свинца, фосфида и стеарата свинца, сульфата
и фосфита свинца.
В 1962 г. появилась новая группа свинцовых стабилизаторов —
жидкие; они легче диспергируются. Данные о составе жидких
стабилизаторов, к которым прежде всего относятся концентриро-
ванные растворы свинцовых солей в соответствующих растворите-
лях, обычно не публикуются.
По аппаратурному оформлению все процессы получения солей
просты и практически одинаковы. Поэтому в данной главе дается
принципиальная схема на примере получения силиката свинца;
для других солей описана лишь технология с указанием основных
параметров процесса.
Силикат свинца
PbO-SiOj
Силикат свинца получают обычно из нитрата или ацетата свин-
z ца осаждением силикатом натрия [5, 6]. Например, силикат свин-
22—1057
337
ца, по составу близкий формуле PbO-2SiO2, получают, смешивая
20%-ный раствор Pb(NO3)2 с раствором Na2SiO3, причем необхо-
дим избыток нитрата свинца сверх стехиометрического количе-
ства [5].
При взаимодействии ацетата свинца и силиката натрия в вод-
ной среде при pH 5,04-8,6 в конце реакции можно получить сили-
кат свинца с мольным отношением РЬО : SiO2= (0,5-4-1,5) : 1 [6].
Интересен способ получения микрокристаллического силиката
свинца взаимодействием свинцового глета с метакремиевой кисло-
той H2SiO3 или с ее солями в водном растворе (или в водной
эмульсии) алифатических жирных кислот или их солей в присут-
ствии катализаторов [7]. Полученный таким методом силикат
свинца характеризуется повышенной термостойкостью и может
применяться в меньших дозировках благодаря более высокой эф-
фективности.
Силикат свинца можно получить способом, в котором объеди-
нены стадии окисления свинца и получения силикатов [8]. Рас-
плавленный свинец равномерно (по каплям) поступает в нагретую
до 800—900 °C печь, заполненную шихтой, состоящей из рассчитан-
ных количеств SiO2, ZnO, буры и других веществ. Тепло, выделяю-
щееся при окислении свинца подаваемым в печь кислородом, спо-
собствует расплавлению компонентов шихты и облегчает их сме-
шение. При получении простых силикатов типа Pb-SiO2 предвари-
тельный разогрев печи не требуется. Для повышения температуры
в процессе изготовления сложных силикатов, обедненных свинцом,
рекомендуется часть SiO2 заменять Si. В нагретую печь с шихтой
можно подавать порошкообразный свинец, а в свободную печь —
смесь свинца с компонентами шихты. Однако в этом случае не
удается получить силикаты свинца однородного состава.
Технологическая схема получения. В промышленности силикат
свинца получают обменным разложением нитрата свинца и сили-
ката натрия (жидкое стекло):
Pb(NO3)2 + Na2SiO3 -► PbSiO3 + 2NaNO3
Пульпу силиката свинца приготавливают в реакторе путем взаи-
модействия непрерывно подаваемых (в определенном количестве)
растворов нитрата свинца и силиката натрия при перемешивании.
Получаемую суспензию силиката свинца непрерывно удаляют че-
рез переливную трубу реактора при перемешивании.
Установка (рис. 53) для непрерывного процесса, состоит из
следующих аппаратов: емкости 1 нитрата свинца, емкости 3 сили-
ката натрия и реактора 2 непрерывного действия. Реактор снаб-
жен пропеллерной мешалкой, паровым барботером и воронками
для приема жидкого стекла и нитрата свинца.
Отфильтрованные растворы нитрата свинца и силиката нат-
рия непрерывно подают центробежными насосами в соответствую-
щие напорные емкости, снабженные переливными линиями (для
338
Рис. 53. Технологическая схема получения силиката свинца:
1, 3 — расходные емкости; 2 — реактор; 4, 5 — барабанные вакуум-фильтры; 6 — емкость
промывных вод; 7 — сушилка; 8 — холодильник; 9 — приемник. *
создания постоянного напора перед диафрагмами) и вентилями,
регулирующими потоки. Общее количество нитрата свинца, по-
ступающего в реактор, должно быть таким, чтобы на выходе из
реактора раствор имел pH 6,7-?7,0, а избыток нитрата свинца в
пульпе не превышал 0,2 г/л. Температура в реакторе 2 поддержи-
вается автоматически в интервале 92—94 °C путем подачи острого
пара через барботер. Образующаяся суспензия силиката свинца
непрерывно сливается через карман в корыто вакуум-фильтра 4.
Промывку пасты силиката свинца осуществляют непрерывно,
методом репульпации с использованием пяти репульпаторов (на
схеме не изображены) и пяти вакуум-фильтров 5. Это дает воз-
можность удалить из пасты ионы ЫОз до содержания 0,01 % и ни-
же в пересчете на сухое вещество. Среду во всех репульпаторах
поддерживают нейтральной; в случае щелочной среды добавляют
необходимое количество серной кислоты. Пасту передают на суш-
ку в аппарат 7 и на упаковку готового продукта.
Свойства и применение. Силикат свинца представляет собой
бесцветный кристаллический порошок; плотность 6490 кг/м3;
Пд =1,9610; т. пл. 766°С. Реагирует с кислотами. В воде не рас-
творяется. Негорюч и невзрывоопасен.
Токсичен; ПДК 0,01 мг/м3. Выпускается в виде порошка:
Торговое наименование Страна, фирма Примечание
Силикат свинца Durostabe S 19 Lead Silikate BS 44514/54 Plumb Stavinor 110 Stavinor 120 СССР Англия, Durham Нидерланды, Chem. Fabrik США. Lead Франция, Rhone Poulenc Франция, Rhone Poulenc Содержит SiO2 J Возможно, ортосили- кат Двухосновный орто- силикат
22*
339
Силикат свинца обычно используют как наполнитель, улучша-
ющий термическую стабильность поливинилхлоридных компози-
ций и. их диэлектрические свойства. Позволяет получать полупроз-
рачные изделия, ярко окрашенные с помощью пигментов. Может
быть использован как термостабилизатор поликарбонатов и поли-
амидов. Производство его во многих странах хорошо налажено,
хотя он постепенно вытесняется основными свинцовыми солями,
смешанными или соосажденными Pb-стабилизаторами (например,
Tribase Е является сочетанием сульфата и силиката свинца).
Термостабилизирующие свойства силиката свинца могут быть
улучшены при использовании смесей ортосиликата свинца и сили-
кагеля. Добавки этих веществ облегчают диспергирование стаби-
лизатора и одновременно повышают светостойкость изделий.
Трехосновный сульфат свинца
3PbO-PbSO4H2O
Один из первых способов получения трехосновного сульфата
свинца — взаимодействие нитрата свинца с сульфатом натрия в
присутствии едкого натра [9].
Обычно трехосновный сульфат свинца получают взаимодействи-
ем оксида свинца и сульфата аммония в присутствии водных рас-
творов уксусной или азотной кислот [10]. Реакцию проводят при
одновременной сушке смеси. Оксид свинца и сульфат аммония
загружают в реактор, подогревают до 80 °C, при перемешивании
добавляют раствор уксусной кислоты и в течение 2 ч выдержива-
ют реакционную смесь при этой температуре. Если в качестве ка-
тализатора используют ацетат аммония, то выход трехосновного
сульфата свинца достигает 92—93% [11]. Процесс требует тща-
тельного контроля pH среды в пределах 9-4-10.
Технологически более удобным считается способ получения
трехосновного сульфата свинца взаимодействием оксида свинца
с серной кислотой [12—15]. Реакцию обычно проводят при pH
7,5-4-10 в присутствии уксусной кислоты и эмульгаторов (напри-'
мер, смесь стеариновой кислоты, этаноламина и аммиака). Приме-
нение эмульгаторов, однако, приводит к образованию сточных вод
и загрязнению продукта органическими примесями. Интересен спо-
соб получения трехосновного сульфата свинца в присутствии не-
большого количества сорбитового эфира жирной кислоты, прибав-
ляемого в конце реакции [14].
Большое влияние на выход и качество конечного продукта ока-
зывают дисперсность исходного оксида свинца и эффективное пе-
ремешивание реакционной массы. Например, трехосновный суль-
фат свинца очень хорошего качества получается из тонкого порош-
ка РЬО, который вводят в раствор серной кислоты, фильтруют об-
разовавшийся сульфат и сушат распылением [15].
Технологический процесс получения. В промышленности трех-
основный сульфат свинца получают взаимодействием оксида свин-
;340
ца с сульфатом аммония в присутствии ацетата аммония при
pH 94-10:
4РЬ0 + (NH4)2SO4--* 3PbO-PbSO4-H2O
Реакция протекает в гетерогенной среде, реагирующий оксид свин-
ца лишь частично переводится в раствор с помощью ацетат-ионов.
Образующийся трехосновный сульфат свинца тоже не растворим
в реакционной среде.
В реактор заливают воду, при энергичном перемешивании и
нагревании до 50—60 °C подают ацетат аммония, после чего не-
большими порциями начинают загрузку оксида свинца (pH не ни-
же 8). Затем очень медленно (в течение 2 ч) подают сульфат ам-
мония и водный раствор аммиака. Полученную густую белую мас-
су фильтруют, промывают водой до отсутствия сульфат-ионов и
сушат продукт при ПО °C.
Свойства и применение. Трехосновный сульфат свинца пред-
ставляет собой белый мелкодисперсный порошок с содержанием
свинца не менее 80%. Растворяется в щелочах, водных растворах
ацетата аммония.
Относится к числу наиболее эффективных свинцовых термоста-
билизаторов. Широко применяется при переработке поливинилхло-
рида в жесткие и пластифицированные изделия, особенно предна-
значенных для электроизоляции. Обеспечивает хорошую термо-,
свето- и водостойкость. Один из наиболее подходящих стабилиза-
торов для изделий, эксплуатируемых при низких температурах.
Трехосновный сульфат свинца не оказывает смазывающего дей-
ствия, поэтому его рекомендуется применять в сочетании со стеа-
ратами РЬ, Ва и Cd.
Является промышленным продуктом многих фирм. Выпускается
в виде тонкого порошка (иногда с модифицирующими добавками)
под разными названиями:
Торговое наименование Страна» фирма Содержа- ние Pb, % Примечание
Сульфат СО СССР so —,
Alaixol 22 Франция, Prod. Chim. — —»
Austrostab 133 Австрия, Bleibergen Bergwerks Union — Белый или светло- желтый порошок
Carbefix Р 10 Франция, Prod. Chim. —
Naftovin TV ФРГ, Kautschuk —- —>
Sicostab D-10; D-14, D-15 ФРГ, Siegle 87 Порошок
Stabilisator BS-53 ГДР — Модифицирован- ный
Stabilisator V-220, V-220 G США, Ferro —
Stabophor KSG ГДР Содержит стеарат свинца и диок- тилфталат
Stabophor 5011 NS ГДР Модифицирован- ный
341
Цвет изделий удается улучшить при сочетании трехосновного
сульфата свинца с оловоорганическими стабилизаторами. По
сравнению с другими свинцовыми стабилизаторами он плохо дис-
пергируется вследствие высокой плотности («7000 кг/м3), поэтому
его поверхность иногда модифицируют жирными кислотами, что
устраняет некоторые трудности стабилизированных композиций
при их переработке.
Основной карбонат свинца
2РЬСО3-РЬ(ОН)а
Это — стабилизатор, известный под названием белый свинец
из-за высокой пигментирующей способности.
Существуют две близких по составу и свойствам структуры ос-
новного карбоната свинца, образующихся при действии СО2 и
Н2О на свинец или оксид свинца,— гидроцеррусит 2РЬСО3-
•РЬ(ОН)2 и плюмбонакрит 6РЬСО3-РЬ(ОН)2-РЬО. Поэтому час-
то отмечаются расхождения в данных рентгеноструктурного ана-
лиза (интенсивность линий, параметры решетки) для основного
карбоната свинца (гидроцеррусит), применяемого в качестве ста-
билизатора.
Оба основных карбоната свинца получают, пропуская смесь
воздуха и диоксида углерода через суспензию РЬО в воде, к кото-
рой добавлено небольшое количество уксусной кислоты [16].
При взаимодействии раствора нитрата свинца (а также его
смеси с NH4C1) с карбонатом аммония при рН^7,1 из раствора
выделяется средний карбонат свинца РЬСО3. Взаимодействие тех
же растворов со смесью аммиака -и карбоната аммония приводит
к образованию вначале основных нитрата или хлорида свинца
(при тех же pH), которые в присутствии избытка осадителя раз-
рушаются и переходят в 2РЬСО3-РЬ(ОН)2— основной карбонат
свинца.
В работе [17] состав выделенных соединений подтвержден
данными рентгеноструктурного анализа, ИК-спектроскопии и тер-
мическими исследованиями и предложен такой механизм разло-
жения карбонатов свинца:
348—350 °C
2РЬСО3----------> РЬСО3 РЬО + СО,
2РЬСО3 РЬ(ОН)2 ------► 2РЬСО3 РЬО + Н,О
300—400 °C
3(РЬСО8 РЬО) --------► 2(РЬСО3-2РЬО) + СО,
450—470 °C
PbCO3‘2PbO ----------► ЗРЬО-f-CO,
(частично)
6PbO-f-O2 ---------► 2Pb3O4
ggO_59Q 0(2
РЬО(красн) --------► РЬО(желт)
880—885 °C
РЬО----------* Плавление
342
Выводы авторов [17] о характере разложения карбоната свин-
ца совпадают с -данными работы [18], полученными при терми-
ческом разложении природного гидроцеррусита в тех же усло-
виях.
При термическом разложении основного карбоната свинца
2РЬСО3-РЬ(ОН)2 происходит образование сначала 2РЬСОз*РЬО,
затем РЬСОз-РЬО, а в дальнейшем разложение протекает так же,
как и для карбоната свинца.
Один из наиболее ранних способов получения основного карбо-
ната свинца — взаимодействие PbSO4 с водными растворами солей
щелочных, щелочноземельных металлов или аммония (хлориды,
ацетаты, нитраты) и соответствующими количествами ВаСОз при
100—120 °C [19].
По способу фирмы Merk (ФРГ) [20] основной карбонат свин-
ца получают, смешивая водно-щелочной раствор ацетата свинца
с метанольным раствором карбоната аммония. Осаждение прово-
дят при pH 94-10,5 в присутствии избытка свинцовой соли. В реак-
тор загружают воду, ацетат свинца и немного 25%-ного раствора
NH4OH, добавлением которого поддерживают pH среды
«10.
При постоянном перемешивании подают в аппарат метанольный
раствор (NH4)2CO3 и водный раствор ацетата свинца в
соотношении 3:5 со скоростью 600 мл/ч. Через час образовавший-
ся осадок отфильтровывают и получают кристаллический карбо-
нат свинца состава РЬ(ОН)г- 1,67РЬСО3 с размером кристаллов
«10 мкм, хорошо распределяющийся в полимере.
Оригинальный способ получения основного карбоната свинца
предложен фирмой Monsanto (США) [21]. Водные растворы нит-
ратов свинца и магния со скоростью 1,21 кг/ч подают в реактор,
куда поступают одновременно природный газ и воздух (со ско-
ростью 3,12 и 79 кг/ч соответственно) для создания окислительно-
го пламени. Раствор впрыскивают в пламя; продукты горения ох-
лаждают, впрыскивая воду, частицы улавливают в скруббере, оро-
шаемом водой. Образовавшийся осадок фильтруют и сушат. Со-
держащий 2% MgO основной карбонат свинца восстанавливают
водородом при 400 °C. По этому способу получают трехосновный
сульфат свинца, идущий для изготовления изделий из металличе-
ского порошкообразного свинца. Однако этим способом без
введения нитрата магния можно получить высокодисперсный
основной карбонат свинца, пригодный и в качестве стабилиза-
тора.
В промышленном масштабе основной карбонат свинца полу-
чают из нитрата свинца.
Свойства и применение. Основной карбонат свинца представ-
ляет собой белый аморфный порошок; т. пл. (с разложением)
400 °C; плотность 6140 кг/м3. Растворяется в слабых растворах
угольной кислоты; не растворяется в воде (ни в горячей, ни в хо-
лодной) .
343
Токсичен, поэтому его рекомендуется применять в виде пасты.
Стабилен к действию света, но имеет существенный недостаток
при использовании в поливинилхлоридных композициях: в резуль-
тате реакции с НС1 выделяется СО2, вызывая порообразование
в материале.
Основной карбонат свинца выпускается в ряде стран под раз-
ными названиями:
Торговое наименование Страна, фирма Выпускная форма Содержа- ние РЬ, %
Карбонат О Austrostab 191 Basic Lead Carbonate Chromostab ВОК Euston White Lead Interstab L 3615 СССР Австрия, Bleibergen Нидерланды, Haagen СФРЮ США, Euston Нидерланды, Akzo Порошок То же » > Хлопья, по- рошок >80 84—86 54,7
Используют основной карбонат свинца для пластифицирован-
ных и (главным образом) для жестких.изделий из поливинилхло-
рида; применяется в производстве огнестойких композиций для
кабельной изоляции. Однако вследствие токсичности и указанных
недостатков производство и применение основного карбоната
свинца в последнее время сокращается.
Двухосновный фосфит свинца
2РЬОРЬНРО30,5Н2О
Главный компонент этого стабилизатора — фосфит свинцг
РЬНРО3 — получают при медленном добавлении оксида свинцг
к расплавленной фосфористой кислоте при 100 °C при действия
раствора фосфористой кислоты на водную суспензию РЬО прц
50 °C. При реакции НРО3 с РЬ(МОз)2 образуется, по-видимому
Pb(NO3)2-PbHPO3, который в воде разлагается на Pb(NO3)2 и не;
растворимый РЬНРО3. При добавлении Pb(NO3)2 к раствору
Н3РО3 выпадает в осадок кристаллический РЬНРО3; здесь, воз-
можно, образуется одноосновный фосфат свинца, однако в свобод-
ном виде он выделен не был. Процесс заканчивается образовани-
ем 2РЬО • РЬНРОз [22], но в целом образование двухосновного
фосфита свинца изучено мало.
В промышленном масштабе этот стабилизатор получают взаи-
модействием фосфористой кислоты с гидроксидом свинца и после-
дующим сплавлением с оксидом свинца или взаимодействием свин-
цового глета с фосфористой кислотой в присутствии уксусной кис-
лоты [23]. В реактор загружают дисперсию глета в воде, добав-
ляют 5 н. раствор уксусной кислоты и постепенно при перемеши-
344
вании загружают 15 н. раствор фосфористой кислоты. При этом
выпадает осадок двухосновного фосфита свинца. Его фильтруют,
промывают, сушат и растирают в порошок.
Свойства и применение. Двухосновный фосфит свинца пред-
ставляет собой белый кристаллический порошок; т. разл. >200°С.
Растворим в азотной кислоте; нерастворим в воде и органических
растворителях.
Выпускается во многих странах под разными наименованиями;
Торговое наименование. Страна, фирма Выпускная форма Содержа- ние РЬ, %
Austrostab 122 Австрия, Bleibergen —
Carbefix PR 12 Франция, Soc. Prod. Порошок —
Chromostab DOF СФРЮ То же —
Dutch Boy Dyphos США, Lead
Dyphos Англия, США, Япония ——
Haro Chem. PDF' Нидерланды, CFHA •—
Haro Mix PC-301, LK-103, LK-117 То же Модифицирован- ный ——
Interstab L 200 Нидерланды, Akzo То же 61
Interstab L 238, L 239 То же Порошок 82,5
Interstab L 3610 Порошок, хлопья 40,5
Naftovin D-61 ФРГ, Kautschuk Модифицирован- ный ——
Phosphit S2 СРР — ——
Stabilisator P-2NS ГДР Содержит связую- щую добавку
Stanclere DLP Англия, Pure Chem. — —
Vinstab Pb F4 Италия, Commer — —
Двухосновный фосфит свинца превосходит другие известные
свинцовые соли по светозащитному действию и обладает антиокис-
лительными свойствами. Улучшает внешний вид изделий из поли-
винилхлорида и придает им погодостойкость. Его используют в
небольших количествах и при сравнительно невысоких температу-
рах (<200°C). Часто применяют в смеси с трехосновным сульфа-
том свинца для улучшения окраски изделий и для стабилизации
цвета. Композиции с двухосновным фосфитом свинца по термо-
стойкости уступают композициям с трехосновным сульфатом свин-
ца вследствие меньшей реакционноспособности по отношению к
НО. Производство и потребление двухосновного фосфита свинца
в последние годы непрерывно увеличиваются.
Средний стеарат свинца
(СКНЗБ-СОО)2РЬ -
Средний (или нейтральный) стеарат свинца получают одним
из трех методов:
345
1) обменным взаимодействием стеарата натрия с нитратом
свинца:
2Cj,H36—COONa 4-Pb(NO3)a ~п> (С17Нз5-СОО)2РЬ
2) реакцией стеарата натрия с ацетатом свинца [24, 25]:
2С17Нз6—COONa + (СН3—СОО)2РЬ - > (С17НзБ-СОО)аРЬ
3) взаимодействием стеариновой кислоты с оксидом свинца в
присутствии катализатора [26] (насыщенные спирты или эфиры»7
например, монобутиловый эфир этиленгликоля):
2С17Нз6-СООН + РЬО —> (С17Нз5-СОО)аРЬ
"*It2v
Преимущество первых двух методов — то, что стеарат свинца,
получаемый осаждением из разбавленных растворов стеарата
натрия, более чистый, а недостаток — необходимость тщательной
отмывки стеарата свинца от нитрат- или ацетат-ионов, что приво-
дит к образованию большого количества сточных вод. При этих
методах нагревают раствор едкого натра в реакторе до 95—100 °C
и при постоянном перемешивании загружают туда стеариновую
кислоту. Реакционную массу охлаждают до 45—50°C и в течение
15 мин приливают 5%-ный раствор ацетата свинца. Полученный
стеарат свинца отфильтровывают и промывают водой до полного
удаления ацетат-ионов. Осадок высушивают при 80—90 °C и из-
мельчают [25].
Третий способ наиболее простой. Смесь стеариновой кислоты,
оксида свинца, воды и катализатора перемешивают на шаровой
мельнице в течение 16 ч при комнатной температуре. Осадок от-
фильтровывают и высушивают при 80°C. Получают стеарат свин-
ца с т. пл. 115,5 °C [26]. Недостаток метода — необходимость при-
менения катализаторов.
Одним из промышленных методов получения среднего стеара-
та свинца является первый способ — обменное взаимодействие
стеарата натрия с нитратом свинца.
Свойства и применение. Средний стеарат свинца представляет
собой белый порошок; т. пл. 115,7°C (с разложением); плотность
6940 кг/м3. Ограниченно растворяется в скипидаре, диоктилфта-
лате, дибутилсебацинате.
Токсичен.
Находит самое широкое применение в качестве стабилизатора
и смазки в сочетании с основными солями свинца. Выпускается
во многих странах в виде порошка под разными названиями.
Средний стеарат свинца плавится при температурах перера-
ботки поливинилхлорида и растворяется в нем. При комбиниро-
вании с кальциевыми, кадмиевыми и бариевыми солями жирных
кислот (особенно в присутствии эпоксидных полимеров) наблю-
дается высокий синергический эффект по термостабильности и цве-
346
Торговое наименование Страна, фирма Т. пл. (с раз- ложением), °C
Стеарат С СССР 200
41-ASL ФРГ, Barlocher —-
Austrostab 110 Е Австрия, Bleibergen —•
Bleistearat 5002 ГДР 95—120
Chromostab NOS* СФРЮ 300
Durostabe S 15 Англия, Durham —•
Dutch Boy DS-207 США, Lead —
Haro Chem P28G, P51, P51X Нидерланды, Haagen ——
Harshaw 102 США, Harshaw —
Hochst Pb S82S ФРГ, Hochst —-
Leadstar США, Lead —
Lead Stearate P Нидерланды, Haagen 96—100
Listab** 28. 41, 51 ФРГ, Kautschuk —
Pb-51 ASL, 41-52/ND ФРГ, Barlocher —
Stanclere NSL Англия, Pure Chem. —
Stavinor 10, 20 Франция, Rhone Poulenc —
Unistab 4810, 4815 Нидерланды, Uniliver . 107
* Содержит PbO.
** Высокое содержание Pb.
тостойкости материалов, улучшается прозрачность и текучесть
композиций.
Двухосновный стеарат свинца
2РЬО (С15Нз5—СОО)2РЬ
Двухосновный стеарат свинца получают теми же методами,
как и средний стеарат, при мольном соотношении стеариновой
кислоты к оксиду свинца 2:3. Проводят обменное взаимодействие
стеарата натрия и ацетата свинца [27, 28] или взаимодействие
оксида свинца со стеариновой кислотой в присутствии спиртов
[29] или других катализаторов [30]. В реактор загружают спирт,
стеарин (с кислотным числом «200 мг КОН/г) и перемешива-
ют до полного растворения стеарина. К этому раствору прибавля-
ют измельченный оксид свинца (в виде пасты в небольшом коли-
честве этилового спирта). При нагревании реакционную массу
выдерживают до получения требуемого кислотного числа.
Продукт отфильтровывают, промывают водой и сушат. Измель-
ченный и просеянный через сито с отверстиями 0,3 мм продукт
имеет белый цвет и кислотное число 0,9—4 мг КОН/г.
При разработке непрерывного процесса получения двухоснов-
ного стеарата свинца в СССР исходят из взаимодействия стеари-
новой кислоты с нитратом свинца [31]:
+6NaOH
2С17Нз5—СООН + 3Pb(NOs)2 —-р > 2РЬО (С1,Нз8-СОО)аРЬ
-4Н2О2’
Раствор стеарата натрия (3%-ный) обрабатывают 5%-ным рас-
твором РЬ(ЬЮз)2 при 75—80°C и полученную суспензию нагрева-
347
ют до 95—97 °C. Вместо стеарата натрия можно использовать рас-
творы солей синтетических жирных кислот Сп—С20, содержащих
стеарат натрия.
В двух раздельных аппаратах готовят раствор стеарата натрия (содержание
свободной щелочи 8,24 г/л, содержание стеарата натрия 30 г/л) и раствор нит-
рата свинца с концентрацией 50,8 г/л. Оба раствора вводят в реактор непрерыв-
ными потоками со скоростью 24 л в час на 1 л рабочего объема аппарата.
Реакционную массу энергично перемешивают при 75—80 °C. Образующуюся
суспензию непрерывно отводят сбоку, нагревают до 95—97 °C, выдерживают при
этой температуре 10 мин и охлаждают до 50—55 °C. Осадок отжимают на
вакуум-фильтре, промывают холодной дистиллированной водой и сушат в ап-
парате с псевдоожиженным слоем. Выход теоретический.
Указанный способ позволяет исключить стадии истирания и
просеивания продукта после сушки, так как двухосновный стеарат
сразу получается с хорошей дисперсностью.
Свойства и применение. Двухосновный стеарат свинца пред-
ставляет собой белый мелкодисперсный порошок; т. разл. >300°C.
Растворяется в обычных растворителях; не растворяется в воде.
Токсичен. Выпускается во многих странах в виде белого тонкого
порошка или гранул:
Торговое наименование Страна, фирма Выпускная форма Т. пл., ®С
Стеарат СО СССР Порошок (на стеа- риновой кисло- те), паста (на СЖК) >200
Alaixol 20 Франция, Comp. Rend — —.
Austrostab 112Е Австрия, Bleibergen — —
Bleistearat 5004 ГДР Порошок >180
Carbefix Pl 13 Франция, Prod. Chim. — —.
Chromostab DOS СФРЮ Порошок —
Dibasic Lead Stearate 641/52 DS 207 Нидерланды, Haagen — —.
США, Lead —
Durostabe S 15, S 16, S 61 Англия, Durham — —
Ergoterm DSO, SOP-13, SOP-14 ПНР Порошок или па- ста (с диоктил- фталатом) 180
Hochst PbS 80S ФРГ, Hochst «-Ч- —
Interstab L 3150, L 3151, L 3155, L 3156 Нидерланды, Akzo Порошок 107—300
Listab 50 ФРГ, Kautschuk — —
Stabinex C 18 — — —
Stab P 82 — •—
Stavinor 20 Франция, Rhone Poulenc —
Stearat S-2, S-5 СРР — —
Vinstab Pb 20 Италия, Commer — —
Широко распространенный стабилизатор композиций на осно-
’ ве поливинилхлорида и сополимеров винилхлорида, особенно для
кабельной изоляции.
Более эффективен, чем средний стеарат свинца, как термоста-
билизатор (особенно в присутствии фосфатных пластификаторов); -
иногда выпускается в виде пасты (например, с диоктилфосфатом).
Двухосновный стеарат свинца обладает смазывающим действием.
Для повышения термо- и светостабилизирующего действия при-
меняется в сочетании с одноосновным фосфитом свинца.
Двухосновный фталат свинца
2РЬОС6Н4(СОО)2РЬ
Впервые двухосновный фталат свинца был получен взаимо-
действием фталевой кислоты с ацетатом свинца при нагревании
в водной среде:
С6Н4(СООН)2 + (CHS—СОО)2РЬ ► С6Н4(СОО)2РЬ + 2СН3—СООН
По методу фирмы Lead (США) [32, 33] одноосновный, двухос-
новный и средний фталаты свинца или их смеси получают при
медленном взаимодействии водной суспензии оксида свинца с вод-
ным раствором фталевого ангидрида (или фталевой кислотой)
при перемешивании в интервале 75—95 °C и мольном соотношении
РЬО и фталевой кислоты 1: (1-у-З).
Отличительная особенность метода, предложенного англий-
ским отделением этой фирмы [34],— получение фталата свинца
смешением порошкообразных фталевого ангидрида и оксида свин-
ца в стехиометрических количествах. Смешение осуществляют с ми-
нимальным количеством воды, достаточным для достижения пасто-
образной консистенции. Время реакции 2—3 ч при 75—100 °C. Для
ускорения реакции рекомендуется добавлять кислотный катализа-
тор в воду перед введением ее в смеситель. При реализации этого
метода в промышленности сводится к минимуму количество обра-
зующихся сточных вод. В смеситель с зигзагообразной мешалкой
загружают сухой оксид свинца и мелкоизмельченный фталевый
ангидрид. Смесь тщательно перемешивают и добавляют воду, со-
держащую уксусную кислоту и смачиватель. Время реакции 2 ч
при 75—85 °C.
В СССР фталат свинца готовят взаимодействием фталевой
кислоты (которую получают растворением фталевого ангидрида
в воде) с оксидом свинца [35]. Фталевый ангидрид в течение Зч
при энергичном перемешивании добавляют к суспензии свинцового
глета в воде при 85—90 °C, регулируя скорость так, чтобы раствор
оставался нейтральным (после добавления всего ангидрида среда
должна быть слабокислой). Затем смесь перемешивают еще
30 мин, а образовавшийся осадок фталата свинца отфильтровы-
вают и промывают до нейтральной реакции и до отсутствия ионов
свинца в промывных водах. Выход продукта 97—98%.
Свойства и применение. Фталат свинца представляет собой бе-
лый тонкий порошок. Растворяется в азотной и уксусной кислоте;
в воде и органических растворителях не растворяется.
349
Производится в виде порошка (плотность «4600 кг/м3):
Торговое наименование / Страна, фирма
Фталат СО Alaixol 21 Carbefix Р-11 Dibasic Lead Phtalate 175/53 Dythal Ergoterm DFO Haro Chem. PDP, PDPS, PDPX, PDPXG Phtalat S2 Stabilisator BS-30 Vinstab Pb Fl СССР Франция, Prod. Chim. Франция, Prod. Chim. Нидерланды, Haagen США,. Lead; ФРГ, Metallgesellsch ПНР Нидерланды, Chem. Fabr. CPP ГДР Италия, Commer
Фталат свинца может быть использован для стабилизации по-
лиолефинов и поливинилацетата.
6.2. СОЛИ МЕТАЛЛОВ I—III ГРУПП
В настоящее время в промышленном масштабе выпускается
много солей металлов I—III групп (неорганических и особенно
органических). Основная функция этих солей — связывание НС1,
выделяющегося при переработке поливинилхлорида. От химиче-
ского строения стабилизатора (природа металла и кислотного ос-
татка) зависит характер разложения поливинилхлорида.
Как и соли свинца, их можно разделить на три группы:
1) фосфаты Na и Mg;
2) бораты Са, Cd, Ва и Mg;
3) карбоксилаты Са, Ba, Sr, Cd, Zn, Na, Li, Mg и Al (насыщен-
ных и ненасыщенных карбоновых и гидрокарбоновых кислот, преж-
де всего стеараты, лауринаты, салицилаты), а также смеси карб-
оксилатов металлов или комплексные (соосажденные) соли.
Фосфаты в настоящее время практически утратили свое значе-
ние как стабилизаторы поливинилхлорида. Признание и распро-
странение приобретают органические и комплексные соли, полу-
чение и свойства наиболее широко применяемых представителей
которых рассмотрены ниже.
Стеарат кальция
(С^Нзь-СООКСа
Стеарат кальция впер'вые был получен в 1923 г. взаимодейст-
вием эквимольных количеств хлорида кальция и стеарата магния
при нагревании в интервале 80—100 °C.
В промышленности стеарат кальция получают реакцией обмен-
ного разложения стеарата натрия с хлоридом кальция
2С1,Н35 COONa-f-CaCls _9Na^ (С17Н35—СОО)2Са
350
или взаимодействием гидроксида кальция с расплавом техниче-
ской стеариновой кислоты [36, 37]:
ЗСцНзв-СООН Ч-Са(ОН), —> (Cj7Hs6-COO)2Ca
—ngV
Предпочтительнее второй способ, так как при использовании хло-
рида кальция необходимо отмывать стеарат кальция от ионов
хлора, что приводит к образованию большого количества промыв-
ных вод (расходуется до 10 тыс. л воды на одну операцию филь-
трования) .
Технологическое оформление производства обычно не вызыва-
ет затруднений, однако специфика применения стеарата кальция
для стабилизации поливинилхлорида требует определенных вы-
пускных форм — стабильных эмульсий, растворов или высокодис-
персных порошков. Например, стабильные растворы стеарата
кальция, не выпадающего в осадок при добавлении в лаки на ос-
нове поливинилхлорида, получают, сплавляя стеарат с лаурилсар-
козином при 40—50 °C и добавляя к этому расплаву смесь раство-
рителей, содержащую толуол и тетрагидрофуран [38].
Интересен способ получения высокодисперсного стеарата каль-
ция на носителе, например на кизельгуре [39]. Суспензию кизель-
гура в растворе хлорида кальция смешивают с разбавленным рас-
твором стеарата натрия; на носителе осаждается стеарат кальция.
Жидкость отделяют, а продукт сушат и просеивают. Он представ-
ляет собой кизельгур, равномерно покрытый стеаратом кальция.
Технологическая схема получения (рис. 54). В реактор 2 в те-
чение 30 мин загружают стеарат натрия, нагретый до 92—95 °C,
и одновременно через раз-
брызгиватель подают 18 % -
ный раствор хлорида каль-
ция. Реакционную массу
перемешивают в течение
10 мин и отбирают пробу на
определение конца обменно-
го разложения. По оконча-
нии реакции подогревают
массу острым паром через
барботер до 95—98 °C и вы-
держивают в течение 1 ч.
Затем массу разбавляют во-
дой и размешивают в тече-
ние 20 мин, после чего при
75—80 °C передают на цент-
Рис. 54. Технологическая схема
получения стеарата кальция:
1 — мерник; 2 — реактор; 3 — центрифу-
га; 4 — аэрофонтанная сушилка.
351
рифугу 3. По окончании фильтрования отмывают осадок от ионов
хлора. Промытый стеарат кальция выгружают и сушат в аэрофон-
танной сушилке 4.
Свойства и применение. Стеарат кальция представляет собой
белый порошок; плотность 1035 кг/м3; т. пл. 175 °C. Растворяется
в бензоле, толуоле и других органических растворителях: в воде
не растворяется. Прн нагревании до 400 °C медленно разлагается.
Пожароопасен в обычных условиях; горит при 75°C. Пыль
взрывоопасна; нижний предел взрываемости 15,6—17,6 г/м3; т. са-
мовоспл. 825—830 °C. Тушить можно распыленной водой, в кото-
рую добавлен смачиватель.
Практически не токсичен. Разрешен для стабилизации поливи-
нилхлорида, идущего на изготовление тары для пищевых продук-
тов (не содержащих жиров), с ограничением дозировок.
Выпускается во многих странах в виде порошка:
Торговое наименование Страна, фирма Т. пл., °с
Стеарат К СССР >132
Calcium Stearate 26 956 Нидерланды, Haagen 145—150
Calcium Stearat 3001 ГДР 140—160
Durostabe S 14 Англия, Durham —
Elektrika A ФРГ, Barlocher
Harshaw 5-V-l США, Harshaw ——
Hochst Ca S 82s ФРГ, Hochst —
Nuostabe V 60 ФРГ, G. Siegle —
Stayrite 25 Нидерланды —
Stavinor 30 • Франция, Rhone Poulenc
Стеарат кальция является одним из основных стабилизаторов
для получения нетоксичных полимерных материалов, например ис-
пользуемых для упаковки пищевых продуктов; его также можно
использовать при переработке пластизолей поливинилхлорида.
Стеарат кальция как стабилизатор-акцептор НС1 обладает низ-
кой эффективностью (поливинилхлорид быстро изменяет цвет), по-
этому его чаще используют в синергических композициях с други-
ми стабилизаторами. Преимущества стеарата кальция — прекрас-
ные смазочные свойства, и он применяется в качестве смазки
для переработки многих пластических масс. Стеарат кальция
применяют как пластификатор при производстве карандашей, в
медицинской и парфюмерной промышленности, для изготовления
грампластинок. -
Стеарат цинка
(Cj,H35—COO)2Zn
Стеарат цинка был получен в 1934 г. при взаимодействии рас-
твора стеариновой кислоты с избытком оксида цинка. В том же
году стеарат цинка был получен в виде тонкоизмельченного по-
352
рошка тем же способом, но с применением растворителя (бутанол,
толуол, бутилацетат). Длительность реакции зависела от приро-
ды растворителя.
Для увеличения выхода продукта рекомендуется проводить ре-
акцию при повышенной температуре (120—150 °C) без раствори-
теля [40]. В расплав стеариновой кислоты при 85—100 °C и пере-
мешивании вводят стехиометрическое количество оксида цинка,
нагревают до 120—150 °C и выдерживают до прекращения выделе-
ния воды.
Чтобы получить высокодисперсный стеарат цинка, применяют,
оксид цинка, диспергированный в расплавленном стеарате цин-
ка [41]. Смесь оксида и стеарата перемешивают при 120°С до по-
лучения взвеси и постепенно добавляют расплавленный стеарин
при 125—130 °C. Продолжительность реакции 30—40 мин. Полу-
ченный прозрачный расплав распыляют форсункой при охлажде-
нии до получения тонкого порошка; часть образующегося стеара-
та цинка возвращают в цикл.
Другим промышленным способом является реакция обменного
разложения стеарата натрия с хлоридом цинка:
-Ь Zncij
2С13Н35-СОО№ —Г (C15H35-COO)2Zn
Чтобы получить водную дисперсию стеарата цинка, смешивают
раствор хлорида цинка с избытком стеарата натрия [42].
Вместо хлорида цинка можно применять и другие соли, на-
пример сульфат цинка [36, 43, 44]. К раствору ZnSO^-HgO в воде
добавляют при перемешивании NaOH и стеариновую кис-
лоту, расплавленную при 65—70 °C. При слабом перемешивании
подают еще немного воды (для исключения эмульгирования), до-
бавляют раствор Zn(OH)2 и отфильтровывают образовавшийся
стеарат цинка. Отмечается, что при добавлении небольших коли-
честв фталевой или малеиновой кислоты в реакционную массу
улучшается фильтруемость продукта.
Технологический процесс производства стеарата цинка подо-
бен процессу производства стеарата кальция.
Свойства и применение. Стеарат цинка — белый аморфный по-
рошок; т. пл. 140 °C. Растворяется в бензоле, метаноле, этаноле,
толуоле, эфирах, четыреххлористом углероде, кетонах; в воде не
растворяется.
Пожароопасен в условиях сушки. Пыле-воздушные смеси взры-
воопасны; нижний предел взрываемости 11,6—17,6 г/м3; т. само-
воспл. 900 СС.
Мало токсичен; специфичность действия в значительной степе-
ни определяется токсичностью цинка. Разрешен для стабилизации
изделий для поливинилхлорида, предназначенных для контакта ё
пищевыми продуктами (не содержащими жиров), и для поливи-
нилхлоридных труб в системах хозяйственно-питьевого водоснаб-
жения (с ограничением дозировок).
23—1057
353
В промышленности он выпускается в виде порошка:
Торговое наименование Страна, фирма Т. пл., °C
Стеарат Ц СССР >130
Chromostab ZnS СФРЮ 116—120
Haro Chem ZG, ZPR Нидерланды, Haagen* -—
Hochst Zn S 82s ФРГ, Hochst —
Interstab M 3160 Нидерланды, Akzo —•
Radia 1170 Бельгия, Oleofina 120
37-SLS ФРГ, Barlocher 117—121
Stearat Z СРР -—
Zinc Stearat 501, 501A ГДР 112—128
Zinc Stearate США 124—130
Стеарат цинка для стабилизации поливинилхлорида индиви-
дуально не применяется, поскольку имеет низкую эффективность,
но широкое применение находит в комбинировании с карбоксила-
тами кальция и (или) магния, с эпоксидированным соевым мас-
лом и др. Стеарат цинка используется преимущественно при по-
лучении нетоксичных композиций.
Может применяться в качестве смазки при переработке пласти-
ческих масс, как активатор вулканизации в резиновой промыш-
ленности, как антиагломератор в процессе водной дегазации при
производстве синтетических каучуков.
Стеарат кадмия
(C^-COO^Cd
В промышленности стеарат кадмия получают двумя метода-
ми— обменным взаимодействием нитрата или хлорида кадмия со
стеаратом натрия [45] или реакцией оксида кадмия со стеарино-
вой кислотой [46, 47]. Отличительная особенность получения стеа-
рата кадмия — необходимость катализаторов (например, диэта-
ноламин или алкилцеллозольв). Это дает возможность получать
бесцветный порошок стеарата кадмия. Температура реакции
60—90 °C.
Технологический процесс получения стеарата кадмия аналоги-
чен процессам получения стеаратов кальция и цинка.
Свойства и применение. Стеарат кадмия — белый порошок;
т. пл. 134 °C. Ограниченно растворяется в горячем бензоле и ски-
пидаре; в воде не растворяется. Стабилен на воздухе и трудно
разлагается при действии разбавленных кислот.
Токсичен.
Индивидуальный стеарат кадмия производится под разными
наименованиями во многих странах:
354
* Торговое наименование Страна, фирма Т. пл., *С
Стеарат КА СССР >85
Alaixol 11 Франция, Prod. Chim.
Austrostab 410 Австрия, Bergwerks —
Cadmium Stearate 4001 Нидерланды, Haagen 95—105
Chromostab CdS СФРЮ ——
Durostabe 13 Англия, Durham .—
Ergoterm SK ПНР 95—110
CD-16 SL ФРГ, Barlocher 104
Haro Chem. KPR Нидерланды, Chem. Fabrik —
Interstab M 3191 Нидерланды, Akzo —
Listab 16 ФРГ, Kautschuk —
Stabilisator SCD Франция, NYCO 102—105
Stayrite 20 Нидерланды —
Stavinor 50 Франция, Rhone Poulenc —
Stearat KD СРР —
Vinstab DOO 10 Италия, Commer —
Стеарат кадмия сообщает изделиям из поливинилхлорида
прозрачность и погодостойкость. Однако в качестве единственного
стабилизатора его применять не рекомендуется из-за резкого по-
чернения содержащего его полимера после кратковременной теп-
ловой обработки. Поэтому более широко распространены комп-
лексные кадмиевые стабилизаторы (твердые и жидкие), содержа-
щие добавки антиоксидантов, эпоксидных полимеров и пр.
6.3. ОЛОВООРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Оловоорганические соединения составляют важную группу ста-
билизаторов для поливинилхлорида, сополимеров винилхлорида и
некоторых других полимеров. Первый патент на их применение в
этом качестве был выдан в 1940 г. В последующие два-три года
были запатентованы и другие оловоорганические стабилизаторы,
часть которых сохранила свое значение до настоящего времени. .
Наибольшее практическое значение как стабилизаторы имеют
следующие соединения четырехвалентного олова:
R2Sn(OOCR')2 R2Sn(SR')2
диалкилдикарбоксилаты олова диалкилдиалкилмеркаптиды олова
(R—алкил С4—С8, (R—алкил С4—Се,
R'—алкил Ci—С17) R'—алкил С4—С12)
RaSn[—S—(CH2)n—COOR'h
диал кил димеркаптоалкил карбокси-
латы олова (R—-алкнл С4—Cg,
R'—алкнл Ci—Cg, «== 14-17)
Почти все промышленные оловоорганические стабилизаторы
являются производными ди-«-бутил- и ди-н-октилолова.
Промежуточными продуктами в их синтезе служат диалкил-
оловодихлориды R2SnC12 и оксиды диалкилолова RjSnO, синтези-
руемые несколькими способами [48—50].
23
355-
При взаимодействии алкилмагнийхлоридов с хлоридом олова
образуется тетраалкилолово. При нагревании последнего с экви-
мольным количеством хлорида олова происходит диспропорцио-
нирование с образованием диалкилоловодихлорида [51]. Послед-
ний при действии гидроксида натрия превращается в -оксид диал-
килолова:
Mg + RCl----► RMgCl
4RMgCl + SnCl4 --> R4Sn + 4MgCl2
R4Sn-f-SnCl4 ——► 2R2SnCl2
R2SnCl2 + 2NaOH -> R2SnO + 2NaCl + H2O
Чтобы упростить эту схему, предложено [52] вести синтез ди-н-
бутилоловодихлорида, минуя стадию диспропорционирования, —
взаимодействием рассчитанных количеств я-бутилмагнийхлорида
и хлорида олова;
2C4H8MgCi + S11CI4 ----► (C4Hs)2SnCl2 -J- 2MgCl2
При этом в качестве побочного продукта образуется некоторое
количество триалкилоловохлорида (C4Hg)3SnCl, но его не выде-
ляют, а взаимодействием с хлоридом олова переводят в дихлорид.
В реактор с мешалкой, обратным холодильником и рубашкой
для охлаждения и нагрева загружают ди-я-бутиловый эфир, ме-
таллический магний, часть я-бутилхлорида (15% от общего ко-
личества) и небольшое количество иода. Смесь нагревают до 65—
70 °C и после начала реакции постепенно прибавляют н-бутилхло-
рид, растворенный в ди-я-бутиловом эфире, перемешивают в те-
чение 2 ч при 75—80°C и затем охлаждают до 0°С. К охлажден-
ному раствору Гриньяровского реактива постепенно прибавляют
рассчитанное количество хлорида олова с такой скоростью, чтобы
температура не поднималась выше 15 °C. Перемешивают 1 ч при
15—20 °C и затем при охлаждении добавляют разбавленную соля-
ную кислоту. Органический слой отделяют, а водный экстрагиру-
ют ди-я-бутиловым эфиром. Экстракт присоединяют к органиче-
скому слою и отгоняют растворитель. Выход ди-я-бутилоловоди-
хлорида 79,5% от теоретического (считая на SnCl4). Если анализ
обнаруживает примесь монохлорида, добавляют расчетное количе-
ство SnCl4 и выдерживают 2 ч при 170—180 °C:
(C4H9)3SnCl -J- SnCl4 -*- 2(C4Hs)2SnCl2
Алкилоловогалогениды могут быть получены также взаимодей-
ствием хлорида олова с триалкилалюминием [53] с последующим
диспропорционированием по Кочешкову:
+SnCI4
SnCl4-f-R3Al 2_a1cj -----------* 2R2SnCl2
При непосредственном синтезе диалкилоловодихлорида
3SnCl4 + 2RsAl 3R2SnCl2
образуется смесь алкилоловохлоридов, из которой трудно выде-
лить целевой продукт. Высокая стоимость и пирофорность низших
гомологов ряда AIR3 затрудняют промышленное оформление этого
способа. Он может оказаться перспективным при использовании
высших, менее пирофорных алюминийалкилов, например триоктил-
алюминия.
Большой практический интерес представляет метод прямого
синтеза — взаимодействие алкилгалогенида с металлическим оло-
вом [48]:
2RHal -f- Sn-э- R2SnHal2
В первом описанном в литературе промышленном процессе
[54] проводили реакцию между н-бутилиодидом и порошкообраз-
ным оловом в присутствии 0,3% магния при 140—145°С:
2C4H9I + Sn —- (C4H9)2Snl2
Эту же реакцию ведут с другими алкилгалогенидами, например
с алкилбромидами [55]. В качестве катализаторов применяют вто-
ричные или третичные алифатические амины или первичный аро-
матический амин ,[56].
Предложена двойная каталитическая система, состоящая из
азот- или фосфорсодержащего органического соединения и иодсо-
держащего соединения [57]. Синтез ведут при 100—200 °C; дли-
тельность реакции 2—4 ч для алкилиодидов и 3—10 ч для алкил-
бромидов и алкилхлоридов. Выход смеси алкилгалогенидов олова
70—90% от теоретического.
Двойная каталитическая система предложена также для син-
теза октилоловохлоридов в среде спиртов [58]. В реактор поме-
щают оловянную фольгу, октанол, октилхлорид, катализатор (гек-
саметилфосфорамидхлорид) и иод. Смесь нагревают до 170—
185°C и выдерживают 4 ч. Затем отгоняют растворитель, катали-
затор и не вошедший в реакцию октилхлорид, добавляют соляную
кислоту, перемешивают при 100 °C и отделяют органический слой.
Выход октилоловохлоридов 94,8% от теоретического (в расчете на
Sn). Октилхлориды олова получают кипячением при 170°C смеси
гранулированного олова, октилхлорида и октилового спирта в при-
сутствии анилина или циклогексиламина [59].
Сравнительно недавно предложен способ прямого синтеза диал-
килдигалогенидов олова — взаимодействие олова с ненасыщенны-
ми соединениями и галогеноводородом [62]. Порошкообразное
олово смешивают с метилметакрилатом и диэтиловым эфиром,
насыщают безводным хлористым водородом и перемешивают 3 ч
при 20 °C. Получают с высоким выходом замещенный алкилолово-
хлорид:
СИ3ООС—СН=СН—СН., + Sn + НС1 ► [СН3ООС—СН2—СН(СНз)—]2SnCl2
Дибутил- и диоктилдибромиды олова получают радиационно-хи-
мическим взаимодействием жидких алкилбромидов с порошком
олова [60, 61].
357
Методы прямого синтеза ди алкилдигалогенидов олова являют-
ся весьма перспективными, работы в этом направлении ведутся
многими исследователями [63].
В литературе имеется мало сведений о процессах получения
алкилхлоридов олова, но, по-видимому, синтез через магнийорга-
нические соединения сохраняет пока практическое значение.
Гидролизом дибутил- и диоктилоловодихлорида получают с
высоким выходом соответствующие оксиды:
+кон
(C4H9)2SnCl2 —>- (C4Hg)2SnO
—I\C1
: +кон
(C8H14)2SnCl2 > (C6H15)2SnO
Гидролиз ведут действием гидроксида кадия или натрия в водной
или водно-спиртовой среде.
Диоктилоловодихлорид плохо смачивается водой, и его гидро-
лизуют в присутствии эмульгаторов. К водно-этанольному рас-
твору КОН добавляют ализариновое масло, нагревают до 80—
90°C и при сильном перемешивании добавляют расплавленный
диоктилоловодихлорид, перемешивают 30 мин, охлаждают и филь-
труют. Осадок промывают водой и спиртом и высушивают. По-
лучают оксид диоктилолова с выходом 96% от теоретического.
Предложено получать чистый оксид диоктилолова, ведя гидролиз
водной щелочью в присутствии жидкого углеводорода; кристалли-
ческий оксид выпадает в осадок из охлажденного углеводородного
слоя [64].
Дибутилоловодихлорид гидролизуют в спиртовом растворе при
50—60 °C. При добавлении раствора NaOH выпадает осадок окси-
да дибутилолова. Его фильтруют, промывают и передают на син-
тез стабилизаторов.
Д нал кил карбоксилаты олова
Для синтеза диалкилкарбоксилатов олова ведут взаимодействие
оксида диалкилолова с карбоновыми кислотами:
R2SnO + 2R'COOH ——* R2Sn(OOCR')2
—HgO
Для получения дибутилдилаурината олова в реактор загружа-
ют лауриновую кислоту, нагревают до 50—60 °C и при этой темпе-
ратуре и непрерывном перемешивании добавляют оксид дибутил*
олова. Мольное соотношение кислоты и оксида равно 2:1. Реак-
ционную массу перемешивают 2—3 ч при 60—70 °C и затем при
пониженном давлении отгоняют воду. В аппарате остается целе-
вой продукт; выход его 95—96% от теоретического. Процесс идет
так:
(C4H9)2SnO 2CUH23—СООН (C4H9)2Sn(OOC—С11Н23)2
358
Этот же стабилизатор получают взаимодействием дибутилоло-
водихлорида с лауринатом натрия
(C4Hg)2SnCla-f-COONa 2^ (C4H0)2Sn(OOC-C11Ha3)a
но второй способ, по-видимому, имеет меньшее значение.
Дибутилмалеинат олова получают взаимодействием оксида ди-
бутилолова с малеиновым ангидридом [48]:
СО
(C4Hs)2SnO + O
ОСО
/ \сн
(C4H9)aSr/ II
\ ^сн
ОСО
Малеиновый ангидрид растворяют в толуоле, при 70—80 °C
постепенно прибавляют эквимольное количество оксида дибутило-
лова и перемешивают до полного растворения оксида. Толуол от-
гоняют в вакууме, а в реакторе остается целевой продукт; выход
его 95—98% от теоретического. В качестве растворителя исполь-
зуют тетрагидрофуран и метилэтилкетон [65].
Этот же стабилизатор получают взаимодействием дибутилоло-
ва с малеиновой кислотой: малеиновый ангидрид растворяют в во-
де при 60 °C, добавляют оксид дибутилолова, перемешивают и
фильтруют выпавший осадок дибутилмалеината олова [66]. Этот
стабилизатор имеет преимущества перед полученным из малеино-
вого ангидрида.
Сравнительно недавно предложен способ получения «нестехио-
метрических» аддуктов оксида диалкилолова и карбоновых кис-
лот [67]. Тонкоизмельченные оксид дибутилолова (1,1—6 моль)
и малеиновый ангидрид (1 моль) перемешивают в течение 24—
48 ч. Полученный стабилизатор имеет более слабый запах, чем
синтезированный химическим путем дибутилмалеинат олова. Взаи-
модействием оксида дибутилолова с малеиновым ангидридом,
этерифицированным по одной карбоксильной группе гликолями,
получают дибутилмалеинаты, содержащие в молекуле несколько
атомов олова [68].
Нагреванием оксида дибутилолова с 3,3-бис (4-гидрокси-З',5'-ди-
трет-бутилфенил) масляной кислотой получают оловоорганический
стабилизатор с фрагментами пространственно затрудненного фе-
нола [69]:
359
Рис. 55. Технологическая схема получения диалкилкарбоксилатов олова:
J —реактор для получения тетрабутнлолова; 2 — аппарат для разложения; 3 — куб для от-
гонки тетрабутилолова; 4, 10 — аппараты для диспропорционирования; 5, 8 — реакторы для
получения дибутнлднлаурнната олова; 6 — куб для отгонки бензола; 7 — фильтр; 9 — реак-
тор получения оксида дибутнлолова.
Юн рекомендован для защиты полиолефинов.
В основе технологического процесса получения стабилизаторов
группы диалкилкарбоксилатов олова лежат следующие реакции:
RX----> RMgX ---► R2MgX2 ---* R2MgO --> R2Mg(OCOR')2
Технологическая схема получения дибутилдилаурината и дру-
гих диалкилкарбоксилатов олова представлена на рис. 55. Схема
предусматривает возможность получения целевых продуктов как
из оксидов диалкилолова, так и из диалкилдихлоридов олова.
В реактор 1, снабженный мешалкой, рубашкой и обратным хо-
лодильником, загружают магниевую стружку, бутилбромид, иод
(катализатор), бензол и диэтиловый эфир (10—-15% от количества
бензола) и постепенно нагревают до 60 °C. Через 20—30 мин на-
чинается бурная реакция, к концу которой магний растворяется.
Реакционную смесь охлаждают до 60—70 °C и прибавляют бен-
зольный раствор SnCl4. Перемешивают 3 ч при 70—80 °C и пере-
дают в аппарат 2 для разложения избытка магнийорганического
соединения.
Разложение ведут, добавляя к реакционной массе при переме-
шивании 10%-ный раствор НС1. Органический слой отделяют, от-
гоняют от него бензол и эфир, а тетрабутилолово в кубе 3 отгоня-
ют при пониженном давлении, отбирая- фракцию, кипящую при
165—170°C (при 0,266 кПа), и передают на получение дибутил-
оловодихлорида.
360
В реактор 10, снабженный мешалкой, рубашкой для нагрева и
охлаждения и обратным холодильником, загружают тетрабутил-
олово и при 180—190 °C и интенсивном перемешивании посте-
пенно добавляют хлорид олова. Полученный таким образом дибу-
тилоловодихлорид растворяют в этаноле или изопропаноле и пе-
редают на синтез оксида дибутилолова.
В аппарате 9 к спиртовому раствору дибутилоловодихлорида,
нагретому до 50—60 °C, при интенсивном перемешивании прибав-
ляют раствор едкого натра. Оксид дибутилолова выпадает в оса-
док; его промывают водой и в виде пасты передают на синтез
стабилизаторов (в реактор 8 загружают лауриновую кислоту и
при 50—60°C вводят оксид дибутилолова).
По другой технологической нитке (ап. 4, 5 и 6) ведут синтез
стабилизаторов исходя из дибутилоловодихлорида, полученного в
ап. 4. Его растворяют в бензоле и прибавляют раствор в ап. 5 к
бензольному раствору лаурината калия, полученному нагреванием
лауриновой кислоты с карбонатом калия. После отгонки бензола
Торговое наименование Страна, фирма
Advastab DBTL ФРГ, Advance
Austristab 20 S Австрия, Bergwerks
Carbcfix R-61, T-6 Франция, Prod. Chim.
Clarite N8-51 CILIA, Lead
E-101, E-112, E-114, E-115, E-117 Япония, Tokyo Fine Chem.
Embylizer-101, 107, 112, 114, 115, 117 To же
Embylizer L-101 »
Estabex L ФРГ, Oxydo
KS 1280 Япония
Mark SBTL Бельгия, Argus
Meister Z-l Швейцария, Meister
Meister Z-2 To же
Mellite 12 Англия, Albright
Mellite 133 To же
Naftovin SN/L ФРГ, Kautschuk
Niax D-22 США, Union Carbide
Ongrostab BL-T ВНР
Stabilisator DBL-19 ГДР
Stabilisator BTL ГДР
Stabilisator BTS ГДР
Stanclere DBTL Англия, Pure Chemical
Stanclere TL Нидерланды, Akzo
Stann 405A Япония, Sankyo
Stann 411 То же
Stann BI I. BL
Stann BU
Stann SB-58
Stann SN-240 A »
Stavinor 1200 SH Франция, Rhone Poulenc
Stavinor 1211 SN То же
Thermolite 12 США, Metal a. Thermit
TVS Tin-Lau Япония, Nitto
TVS TL-600, TL-700, TL-860 То же
361
(в кубе 6) и охлаждения фильтруют суспензию дибутилдилаури-
ната олова на фильтре 7.
Свойства и применение. Дибутилдилауринат олова — белое кри-
сталлическое вещество; т. пл. 24°C; т. кип. 215—220°С (при 0,4—
0,52 кПа). Растворим в бензоле, гексане, петролейном эфире, этил-
ацетате, хлороформе, четыреххлористом углероде. В воде не рас-
творяется, но постепенно гидролизуется.
Дибутилдилауринат олова под разными торговыми марками
выпускается многими зарубежными фирмами (табл, на стр. 361).
Дибутилдилауринат олова — термостабилизатор поливинилхло-
рида — эффективно защищает его при температуре переработки
до 165 °C. Придает композициям высокую прозрачность. Эффек-
тивность повышается при совместном применении с солями кад-
мия и бария.
Оловоорганические стабилизаторы,
содержащие серу
Оловоорганические стабилизаторы, содержащие серу, получают
взаимодействием оксидов диалкилолова с меркаптанами. Напри-
мер, при взаимодействии оксида дибутилолова с додецилмеркап-
таном получают дибутилдидоцецилмеркаптид олова [70]:
(C4H9)2SnO + 2C12H26SH (C4H9)2Sn(SC12H25)2
Синтез ведут, нагревая исходные соединения в среде толуола. Вы-
деляющуюся воду удаляют азеотропной отгонкой.
Другой способ получения таких стабилизаторов — взаимодей-
ствие алкилмеркаптана с диалкилдигалогенидами олова [71]:
(C4H9)2SnCl2 + 2C12H25SH —> (C4H9)2Sn(SC12H25)2
—rid
Эту реакцию ведут в присутствии алифатического амина или рас-
твора щелочи.
Наибольшее практическое значение как стабилизаторы имеют
производные тиогликолевых кислот. Их получают взаимодействи-
ем оксида диалкилолова с эфирами тиогликолевых кислот. Напри-
мер, нагревая оксид дибутилолова с изооктилмеркаптопропионатом
в среде толуола получают дибутилолово-S,S'-бис (изооктилмеркап-
топропионат) [72] з
(C4H9)2SnO-|-HSCH2-СН2-С-О-С8Н17 —>
|| —Н2О
О
SCH2—СН2—С—О—С8Н„1
II
О J,
Синтез ведут в среде толуола при 120 °C; выделяющуюся воду от-
гоняют. Взаимодействием оксида дибутилолова и изооктилмеркап-
тоацетата в бензольном растворе получают с высоким выходом
дибутилолово-S,S'-бис (изооктилмеркаптоацетат) [73].
' * (C4H9)2Sn
362
При реакции изооктилмеркаптоацетата с оксидом ди-н-октил-
олова получают ди-к-октилолово-S,S'-бис (изооктилмеркаптоаце-
тат) — жидкий нетоксичный стабилизатор поливинилхлорида [74].
Для подавления побочных реакций ведут синтез диалкилдимер-
каптоалкилдикарбоксилатов в присутствии агентов, связывающих
воду (сульфаты натрия, кальция, меди) [75].
Технология синтеза этих стабилизаторов близка к технологии
синтеза диалкилкарбоксилатов олова.
Среди новых стабилизаторов этой группы следует отметить
производные дитиогликолей или эфиров дитиогликолей и дикарбо-
новых кислот [76], например:
Г(С7Н]Б—О—С—СИ,—CH2S— )2Sn—S—сн2—СН2—О—С—Civ-
il I II
о сн3 о
Описаны и более высокомолекулярные оловоорганические ста-
билизаторы, содержащие серу и рекомендованные как малоток-
сичные стабилизаторы для упаковочных пленок из поливинилхло-
рида. Их получают взаимодействием оксида дибутилолова со
сложными эфирами, образованными меркаптокарбоновыми кисло-
тами и полиолами (полиолы могут быть частично этерифицирова-
ны карбоновыми кислотами С]—С]8) [77].
Для стабилизации упаковочных пленок предложен, например,
н-октилолово-5,5',8"-три (этилгексилмеркаптоацетат), получаемый
взаимодействием н-октилоловотрихлорида и этилгексилмеркапто-
ацетата [78]. В качестве «стабилизатора с незначительным запа-
хом» предложен продукт взаимодействия диметилметакрилатоло-
водихлорида и нонилмеркаптопропионата [79]:
ГСН3О—С—СН2—СН2—] Snr—S—СН2—СН2—С—OCsHj.l
II II
L о . з L о J2
В последние годы синтезирован ряд стабилизаторов, в молеку-
лу которых входят азотсодержащие гетероциклы [80—83], напри-
мер:
п = 54-293
Оловоорганические стабилизаторы широко используются для
стабилизации поливинилхлорида [1] и некоторых других полиме-
ров. За последние 10—15 лет выработка этих стабилизаторов рас-
тет высокими темпами, что связано в первую очередь с развитием
производства поливинилхлорида. Дальнейшее развитие выпуска и
применения этих стабилизаторов будет, вероятно, зависеть от ре-
шения двух проблема
363
1) разработка процесса получения дешевых промежуточных
продуктов (алкилхлориды олова и оксиды диалкилолова) или от-
крытие способов прямого синтеза стабилизаторов из олова или,
хлорида олова;
2) создание эффективных стабилизаторов, обладающих низкой
биологической активностью и безусловно пригодных для защиты
полимеров, контактирующих с продуктами питания и с организ-
мом человека.
ЛИТЕРАТУРА
1. Минскер К- С., Федосеева Г. Т. Деструкция и стабилизация поливинилхлорида.
М., Химия, 1972, с. 173—186.
2. Mod. Plast. Int., 1978, v. 8, № 9, p. 61.
3. Plast. World, 1978, v. 36, № 1, p. 15.
4. Вассерман И. M., Фомина Е. А. — ЖПХ, 1965,. т. 38, № 7, с. 1507—1513;
РЖХим, 1965, 24Л143.
5. Банных 3. С., Полякова Е. М. — В кн.: Труды УНИХИМ, 1954, № 1, с. 81—
84; РЖХим, 1956, № 2, 4347.
:6. Пат. 5668, 1953 г. (Япония); РЖХим, 1955, № 21, 20589.
.7. Midzusava, пат. 49-20474, 1974 г. (Япония); РЖХим, 1975, ЗТ66.
8. Goldschmidt, пат. 934885, 1955 г. (ФРГ); РЖХим, 1957, № 11; 38471.
9. Memoires Soc. Chem., 5 series, 1956.
10. Seikisui, пат. 6068, 1958 г. (Япония); РЖХим, 1965, 2Л95.
,11. А. с. 170929, 1965 г. (СССР).
12. Sakai, пат. 955, 1959 г. (Япония); РЖХим, 1960, № 5, 18499; № 20, 81619.’
13. Sugavara, пат. 9559, 1956 г. (Япония); РЖХим, 1960, № 2, 5658.
14. Пат. 2014, 1971 г. (Япония); РЖХим, 1971, 23Н78.
15. Nippon, пат. 38438, 1974 г. (Япония); С. А., 1975, v. 83, р. 11415.
16. Olby J. К. — J. Inorg. a. Nucl. Chem., 1966, v. 28, № 11, р. 2507—2512; РЖХим,
1967, 23Б355.
17. Андреева В. Н., Лимарев Т. Ф. — Ж- неорг. химии, 1970, т. 15, № 8, с. 2028—
2092; РЖХим, 1970, 23В35.
18. Warne S., Bayliss Р. — Am. Mineralog., 1962, v. 47, p. 1011—1023; C. A., 1963,
v. 58, p. 2275.
19. Goldschmidt, пат. 901414, 1954 г. (ФРГ); РЖХим, 1955, № 13, 26641.
20. Merck, пат. 1039046, 1959 г. (ФРГ); РЖХим, 1960, № 14, 57730.
21. Monsanto, пат. 3150927, 1964 г. (США); РЖХим, 1966, 5Л126.
22. Pulidori F., Gilli G., Traversa О. — Ann. chimica, 1966, v. 56, p. 1449—1462;
Chem. Zbl., 1967, № 48, S. 692.
23. Sugavara, пат. 3270, 1962 г. (Япония); РЖХим, 1964, 12С268.
24. Kikuti Si Kisei, пат. 29018, 1971 г. (Япония); РЖХим, 1972, 9С488.
25. Пат. 55965, 1973 г. (СРР); РЖХим, 1975, 8Н46.
26. Phillips Petroleum, пат. 3002935, 1961 г. (США); РЖХим, 1963, 6Н45.
27. Пат. 16973, 1962 г. (Япония); РЖХим, 1964, 17С259.
28. Пат. 3910, 1963 г. (Япония); РЖХим, 1964, 10С301.
29. Пат. 46413, 1962; РЖХим, 1964, 1Н18; пат. 46409, 1962; РЖХим, 1964, 1Н17;
пат. 57046, 1969 г. (ПНР) ; РЖХим, 1970, 17Н59.
30. Пат. 93880, 1977 г. (ПНР); РЖХим, 1979, 1Н66.
31. А. с. 215940, 1968 г. (СССР).
32. Lead, пат. 880137, 1953 г. (ФРГ); РЖХим, 1956, № 1, 2396.
33. Lead, пат. 1066898, 1967 г. (Англия); РЖХим, 1968, 2Н148.
34. Ass. Lead, пат. 981837, 1965 г. (Англия); РЖХим, 1966, 12Н171.
•35. Брудзь В. Г., Розина Д. Ш., Нестеренко Л. Т. — В кн.: Труды ИРЕА, 1958,
вып. 22, с. 139—141; РЖХим, 1960, № 2, 6003.'
36. Cyanamid, пат. 2945051, 1960 г. (США); РЖХим, 1961, 19Л72.
364
37. Sekisui, заявка 2331589, 1977 г. (Франция); С. А., 1978, v. 88, р. 51610
38. FMC, пат. 3607806, 1971 г. (США); РЖХим, 1972, 17С987.
39. Rene — Chimie Sante, пат. 1143139, 1957 г. (Франция); РЖХим, 1960, № 8
31483.
40. А. с. 174615, 1965 г. (СССР).
41. А. с. 169510, 1965 г. (СССР).
42. Phillips Petroleum, пат. 2849427, 1958 г. (США); РЖХим, 1960, № 6, 24760.
43. РЖХим, 1960, № 2, 7195.
44. Cyanamid, пат. 2809121, 1957 г. (США); РЖХим, 1959, № 21, 76888.
45. Prasad S., Ranganathachar Т., Krishnan V.— J. Indian Chem., Soc., 1958, v. 35,
№ 4, p. 267—268; РЖХим, 1959, № 4, 11286.
46. Sinagava, пат. 2860, 1962 г. (Япония); РЖХим, 1964, 13Н53.
47. Sankyo, пат. 36687, 1974 г. (Япония); С. А., 1975, V. 82, р. 125984.
48. Прохорова А. А., Ашбель Ф. Б., Паршина П. М. — Промышленные методы по-
лучения и области применения оловоорганических соединений. М., Изд.
НИИТЭХИМ, 1968. 82 с.
49. Кочешков К- А. и др. — В кн.: Методы элементоорганической химии. М., Изд.
АН СССР, 1968. 704 с.
50. Organometallic Compounds. New York, Springer Verlag, 1967. V. 2.
51. Кочешков К- A.-—В кн.: Синтетические методы в области металлоорганиче-
ских соединений. М., Изд. АН СССР. Сб. 5, 1947, с. 383.
52. Шевердина И. И. и др.-—Хим. пром-сть, 1962, с. 707.
53. Ethyl, пат. 3095433, 1963 г. (США).
54. Inabaz — Juki Gosei Kagaku, 1965, v. 23, p. 806.
55. Pure, пат. 3448130, 1970 г. (США).
56. Pure, пат. 1168456, 1969 г. (Англия).
57. Nitto, пат. 3745183, 1973 г. (США); РЖХим, 1974, 10Н98.
58. Sumitomo, пат. 52-5483, 1977 г. (Япония); РЖХим, 1978, 6Н139.
59. Пат. 148776, 1973 г. (ЧССР).
60. Верещинский И. В.—Журнал ВХО им. Менделеева, 1973, т. 18, с. 278.
61. Мерецкий В. Ю. и др. — Хим. пром-сть, 1968, с. 258.
62. Akzo, заявка 7503116, 1976 г. (Нидерланды); С. А., 1977, V. 86, р. 44403.
63. Элементоорганические соединения. Вып. 3. Прямой синтез органических соеди-
нений олова и германия. М., Изд. АН СССР, 1976. 127 с.
64. Nitto, пат. 48-7090, 1973 г. (Япония).
65. Tokyo Fine Chemicary, пат. 48-30622, 1973 г. (Япония).
66. Tokyo Fine Chemicary, пат. 29648, 1970 г. (Япония).
67. Metal a. Thermit, пат. 4032552, 1977 г. (США).
68. Ciba — Geigy, заявка 2549332, 1976 г. (ФРГ).
69. Yoshitomi, пат. 13302, 1978 г. (Япония).
70. Argus, пат. 1008908, 1958 г. (ФРГ).
71. Пат. 2726227, 1955 г. (США); С. А., 1956, V. 50, р. 6095.
72. Carl. Chem. Works, пат. 3478071, 1969 г. (США).
73. Теппесо, пат. 3562305, 1971 г. (США).
74. Metal a. Thermit, пат. 3657294, 1972 г. (США).
75. Nitto, пат. 3631082, 1971 г. (США).
76. Cincinnati, пат. 3979359, 1976 г. (США).
77. Kureha, пат. 1605463, 1976 г. (Франция).
78. Metal a. Thermit, пат. 1510973, 1978 г. (Англии).
79. Nitto, заявка 7835752, 1978 г. (Япония).
80. Ive, заявка 160277, 1975 г. (Япония),
81. Ive, заявка 157373, 1975 г. (Япония).
82. Заявка 25581, 1978 г. (Япония).
83. Заявка 25582, 1978 г. (Япония).
365
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Азотсодержащие стабилизаторы 19
Ы,Ы'-Алкиларил-п-фенилендиамины
105 сл.
Алкилфенолы 186 сл., 195
конденсация 233 сл.. 260 сл.
4-Алкокси-2-гидроксибензофенон
309 сл.
Альдоль-а-нафтиламин 152 сл.
Аминные стабилизаторы 21, 30 сл.
классификация 19 сл.
синтез 35 сл., 69 сл., 81 сл., 105 сл.,
127 сл., 138 сл., 169 сл.
сырье 31 j
4-Аминодифениламин 86 сл.
Амины ароматические 35 сл., 127 сл.,
138 сл.
Антиозонанты 14 сл., 20
Антиоксиданты (Термостабилизато-
ры) 12, 16 сл., 20, 188 сл.
Антирады 21
Беиазолы 25
Бензоны 25
Бисалкофены 25
Бис(4-гидрокси-3,5-ди-трет-бутил-
бензил) сульфид 277 сл.
4,4'-Бис (а,а-диметилбензил) дифе-
ниламин 136 сл.
Бисфенолы 189
2,2'-Бутилиденбис(6-трет-бутил-4-ме-
тилфенол) 214
4,4'-Бутилиденбис(6-трет-бутил-3-ме-
тилфенол) 258
2,2'-Бутилиденбис(4,6-диметилфе-
нол) 242
2-трет-Бутил-1-метилфенол 214
2-трет-Бутил-4-метилфенол 212 сл.
6-трет-Бутил-2-метилфенол 223 сл.
6-трет-Бутил-З-метилфенол 215
о-Гидроксибензофеноны 13
о-Гидроксибензтриазол 13
4-Гидрокси-3,5-ди-трет-бутилбензи-
ловый спирт 222 сл.
₽-(4-Гидрокси-3,5-ди-трет-бутилфе-
нил) пропионовая кислота, эфиры
224 сл.
Гидроксидифенил 242
п-Гидроксидифениламин 51 сл.
2* (2'-Гидрокси-5'-метилфенил) бенз-
триазол 94 сл.
4-Гидрокси-4-метокСибензофенон
311 сл.
п-Гидроксифенил-Р-нафтол 43 сл.
о-Гидроксифенилсульфоксиды 13
Диалкилдиалкилмеркаптиды олова
' 355
Диалкилдикарбоксилаты олова 355
Диалкилдимеркатоалкилкарбоксила-
ты олова 355
Диалкилдитиокарбаматы металлов
171 сл.
4,4'-Диалкилдифениламин 129 сл.
Диалкилкарбоксилаты олова 358 сл.
Ы.Н'-Диалкил-п-фенилеидиамины
105 сл.
Диалкилфенолы 195
2,5-Ди-трет-амилгидрохинон 228 сл.
4,4'-Диаминодифенилметан 144 сл.
4,4'-Диаминодициклогексилметан
99 сл.
Диафены 25
2,5-Ди-трет-бутилгидрохинон 227 сл.
Дибутилдилауринат олова 358 сл.
Дибутилдитиокарбамат цинка 174
Дибутилмалеинат олова 359 сл.
2,6-Ди-трет-бутил-4-метилфенол
205 сл.
2,6-Ди-трет-бутил-4-метоксиметил-
фенол 204, 220 сл.
Ы.ЬГ-Ди-втор-бутил-п-фениленди-
амин 109 сл.
2,6-Ди-трет-бутилфенол 201 сл.
Дибутилолово-S,S'-бис (изооктилмер-
каптоацетат) 362
Дибутилолово-S,S'-бис (изооктилмер-
каптопропионат) 362
2,4-Дигидроксибензофеион 305 сл.
Ди (4-гидрокси-3,5-ди-трет-бутил) -
бензилсульфид 205
4,4'-Дигидрокси-3,3',5,5'-тетра-трет-
бутилдифенил 204
4,4'-Ди (а-метилбензил) дифениламин
134 сл.
N- (1,3-Диметилбутил) -N'-феиил-п-
фенилендиамин 122 сл.
Ы-Диметил(4-гидрокси-3,5-ди-трет-
бутилбензил)амин 208
Диметилдитиокарбамат висмута 175
М.М'-Ди-р-пафтил-п-фепилендиамин
62 сл.
366
4,4'-Ди-7'р<?7'-октилдифе11иламин['4,4'-
Ди (1,1,3,3-тетраметилбутил) дифе-
ниламин] 131 сл.
Ди-н-октилолово-S,S'-бис (изооктил-
меркаптоацетат) 363
4,4'-Ди (1,1,3,3-тетраметилбутил) ди-
фениламин (4,4'-Ди-трет-октилди-
фениламин) 131 сл.
Дитиокарбаматы 169 сл.
Дифенамы 24
Дифениламин
нитрозопроизводные 69 сл.
продукты конденсации с ацетоном
162 сл.
1,3-Дифенилтиокарбамид 176 сл.
Д.Н'-Дифснил-п-фенилендиамин
55 сл.
Идазолы 25
2,2'-Изобутилиденбис (4,6-диметил-
фенол) 252 сл.
N-Изопропил-Ы'-фенил-п-фенилен-.
диамин 116 сл.
Карбонат свинца основной 342 сл.
2-Меркаптобензимидазол 181 сл.
Меркаптобензимидазолы 169 сл.
Металлсодержащие стабилизаторы 20,
335 сл.
а-Метилбензилфенолы, смесь 216 сл.
2,2'-Метиленбис(6-трет-бутил-4-ме-
тилфенол) 214, 242, 244 сл.
2,2'-Метиленбис(6-трет-бутил-4-этил-
фенол) 250 сл.
2,2'-Метиленбис(4,6-диметилфеиол)
251 с л.
4,4'-Метиленбис(2,6-ди-трет-бутил-
фенол) 204, 255 сл.
2,2'-Метиленбис(4-метил-6-а-метил-
циклогексилфенол) 253 сл.
Метиленбис(Р-нафтол) 242
Метиленбисфенолы 233 сл.
2,2'-Метилен бисфенол 243 сл.
4,4'-Метиленбисфенол 241
Метиловый эфир Р-(4-гидрокси-3,5-
ди-трет-бутилфенил)пропиоиовой
кислоты 225
Нафтамы 25
N-Нитрозодифениламин 72 сл.
п-Нитрозодифениламин 75 сл.
Озонное старение полимеров 14
Октадецил-3-(4'-гидрокси-3',5'-ди-
трет-бутилфенил) пропионат 204
Октадециловый эфир р- (4-гидрокси-
3,5-ди-трет-бутилфенил) пропионо-
вой кислоты 226
н-Октилолово-8,8',8"-три(этилгек-
силмеркаптоацетат) 363
Оловоорганические стабилизаторы 22,
355 сл., 362 сл.
Пассиваторы 20
Полифенолы 189
Продукты конденсации дифениламина
с ацетоном 162 сл.
Противоутомители 20
Светостабилизаторы 13 сл., 20, 188
Серосодержащие стабилизаторы 12,
19, 327 сл., 362 сл.
Силикат свинца 337 сл.
Стабилизаторы
азотсодержащие 19
аминные см. Аминные стабилиза-
торы
защита от фотодеструкции 13 сл.
классификация 19 сл.
летучие 23
металлсодержащие 20, 335 сл.
механизм действия 10 сл.
нелетучие 23
неокрашивающие 21
нетоксичные 22
номенклатура 23 сл.
окрашивающие 21
оловоорганические 22, 355 сл.,
362 сл.
развитие производства 26 сл.
серосодержащие 12, 19, 327 сл.,
362 сл.
свинецорганические 336 сл.
структура 15 сл.
токсичные 22
фенольные см. Фенольные стабили-
заторы
фосфорсодержащие 20, 22, 325 сл,
эффективность действия 15 сл.
Стеарат кадмия 354 сл.
Стеарат кальция 350 сл.
Стеарат свинца
двухосновный 347 сл.
средний 345 сл.
Стеарат цинка 352 сл.
Сульфат свинца трехосновный 340 сл.
Термостабилизаторы (Антиоксидан-
ты) 12, 16 сл., 20, 188 сл.
Тетрафенолы 280 сл., 289 сл.
Тиоалкофены 25
2,2'-Тиобис(6-трет-бутил-4-метилфе-
нол) 214, 267 сл.
4,4'-Тиобис(6-трет-бутил-2-метил-
фенол) 273
4,4'-Тиобис (6-трет- бутил-3-метилфе-
нол) 274 сл.
367
2,2'-Т иобис (4,6-ди-етор-ам илфено л)
271
2,2'-Тиобис (4-метил-а-метилбепзил-
фенол) 270
Тиобисфенолы 260 сл.
Тио(диалкилпропионаты) 20, 327 сл.
Тио(димиристилпропионат) 333
Тио (дипропионаты) [Эфиры тио(ди-
пропиоиовой) кислоты] 327 сл.
Тио(дистеарилпропионат) 333
Тиокарбамиды 169 сл., 175 сл.
Тиокарбанилид 178
Триацетонамин, производные 13, 14
1,1,3-Три-н-бутилтиокарбамид 179 сл.
2,4,6-Три(4-гидроксибензил-3',5'-ди-
трет-бутил) мезитилен 204
Три (а-метилбензилфенил) фосфит
323 сл.
2,2,4-Триметил-1,2-дигидрохинолии,
полимер 155 сл.
2,2,4-Триметил-6-этокси-1,2-дигидро-
хинолин 157 сл.
Три (n-нонилфенил) фосфит 322 сл.
Три-н-октилфосфит 318 сл.
Трифенилфосфит 320 сл.
Три-(2-этилгексил) фосфит 319 сл.
Трисалкофены 25
1,1,3 - Тр ис (б'-трет- бу тил- 4'- гидрокси-
2'-метилфенил) бутан 291 сл.
2,4,6-Трис (4'-гидрокси-3', 5'- ди-трет-
бутилбензил)мезитилен 293 сл.
Трисфенолы 189, 280 сл.
n-Феиилендиамины как антиозонанты
14
N-Фенил-а-иафтиламин 40 сл.
N-Фенил-Р-нафтиламин 36 сл.
Ы-Фенил-ЬТ'-циклогексил-п-фенилен-
диамин 111 сл.
Фенольные стабилизаторы 21, 23,
186 сл.
как антиоксиданты 12, 16 сл.
классификация 19
механизм действия 188
синтез 191 сл., 233 сл., 260 сл.,
280 сл., 298 сл.
структура 16 сл., 188 сл.
сырье 186 сл.
эффективность 16 сл.
Фосфит свинца двухосновный 344 сл.
Фосфиты (Эфиры фосфористой кис-
лоты) 315 сл.
Фосфорсодержащие стабилизаторы
20, 22, 325 сл.
Фотодеструкция полимеров 13
Фталат свинца двухосновный 349 сл.
Хинолы 25
2,2'-Этиленбис (4,6-диметилфенол) 242
Эфиры
тио(дипропионовой) кислоты
[Тио(дипропионаты)] 327 сл.
фосфористой кислоты (Фосфиты)
315 сл.
БОРИС НИКОЛАЕВИЧ ГОРБУНОВ
ЯКОВ АБРАМОВИЧ ГУРВИЧ
ИЗОЛЬДА ПЕТРОВНА МАСЛОВА
Химия
и технология
стабилизаторов
полимерных
материалов
ИБ № 869
Сдано в наб. 19.11.80. Подп. к печ. 29.01.81.
Т. 04827. Формат бумаги 60X90Vte. Бумага тип. № 2.
Гарн. литературная. Печать высокая.
Усл. печ. л. 23. Уч.-изд. л. 26,39. Тираж 2900 экз.
Заказ 1057. Цена -1 р. 60 к. Изд. № 1706.
Ордена «Знак Почета» издательство «Химия».
107076, Москва, Стромынка, 13.
Редактор Н. И. Урывалова
Художественный редактор
Н. М. Биксентеев
Художник А. К. Малкин
Техннческий редактор В. В. Лебедева
Корректор Г. М. Гольбиндер
Московская типография № 11 Союзполиграфпрома
при Государственном комитете СССР по делам
издательств, полиграфии и книжной торговли.
Москва, 113105, Нагатинская ул., д. 1.